/
























































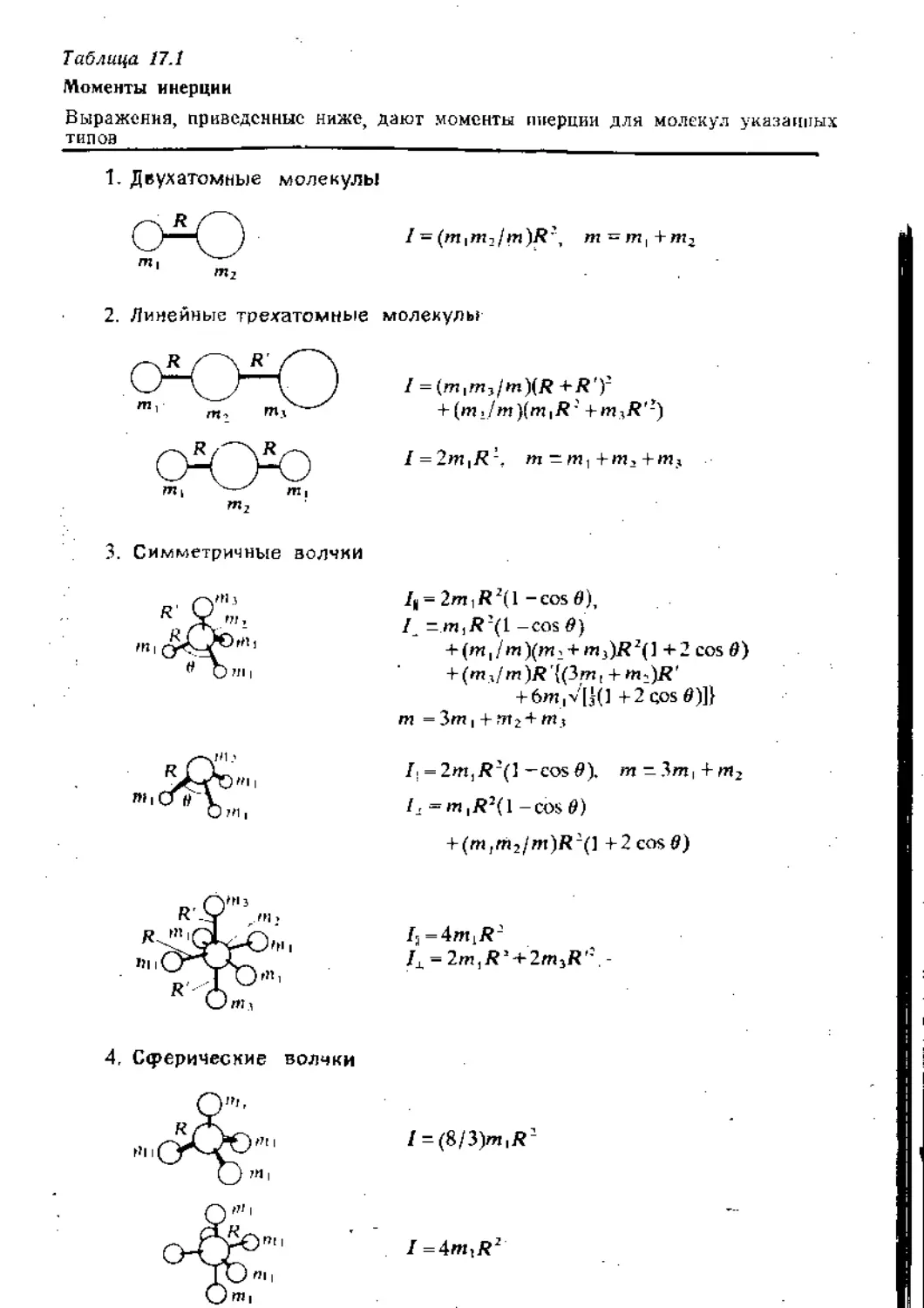













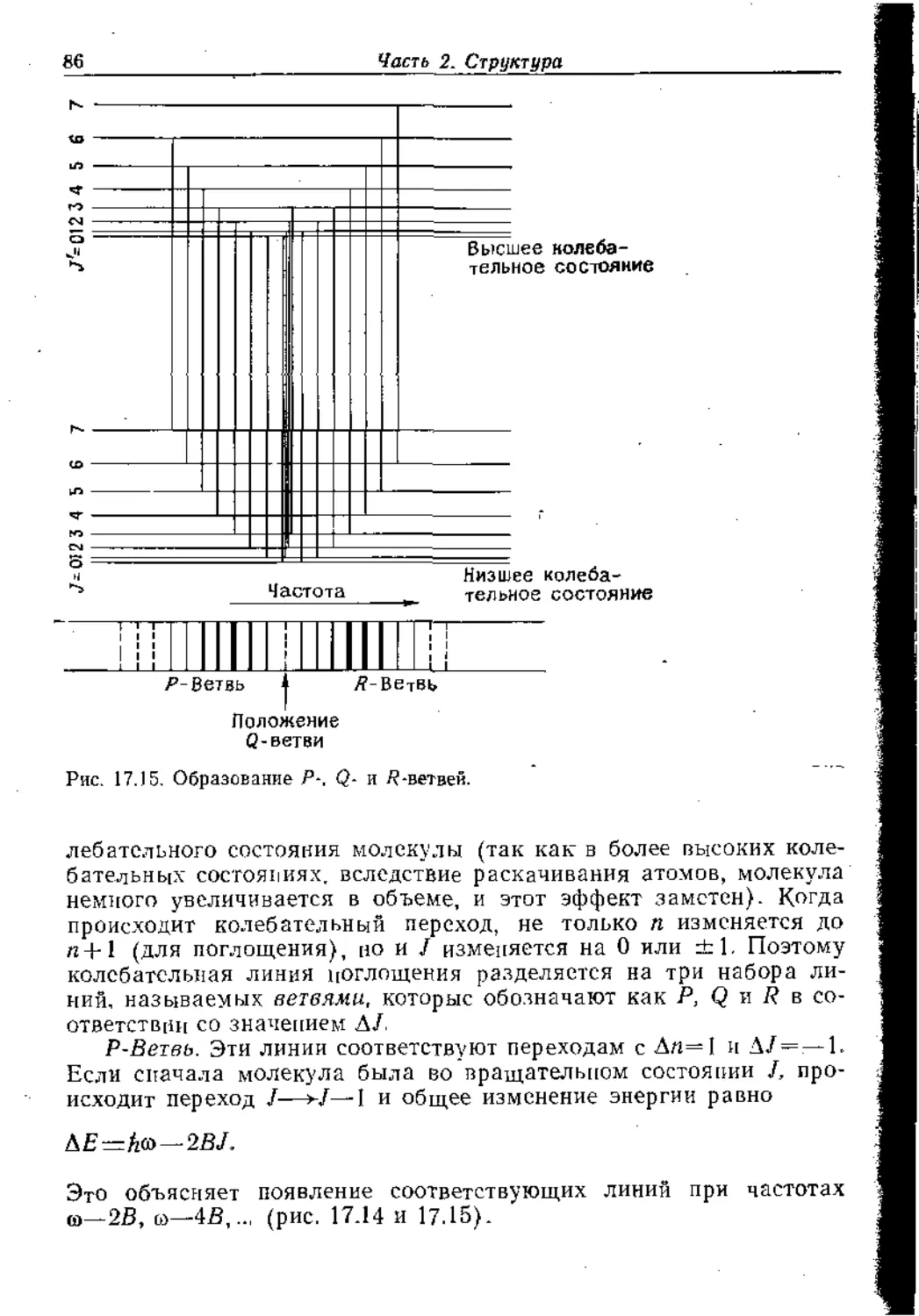

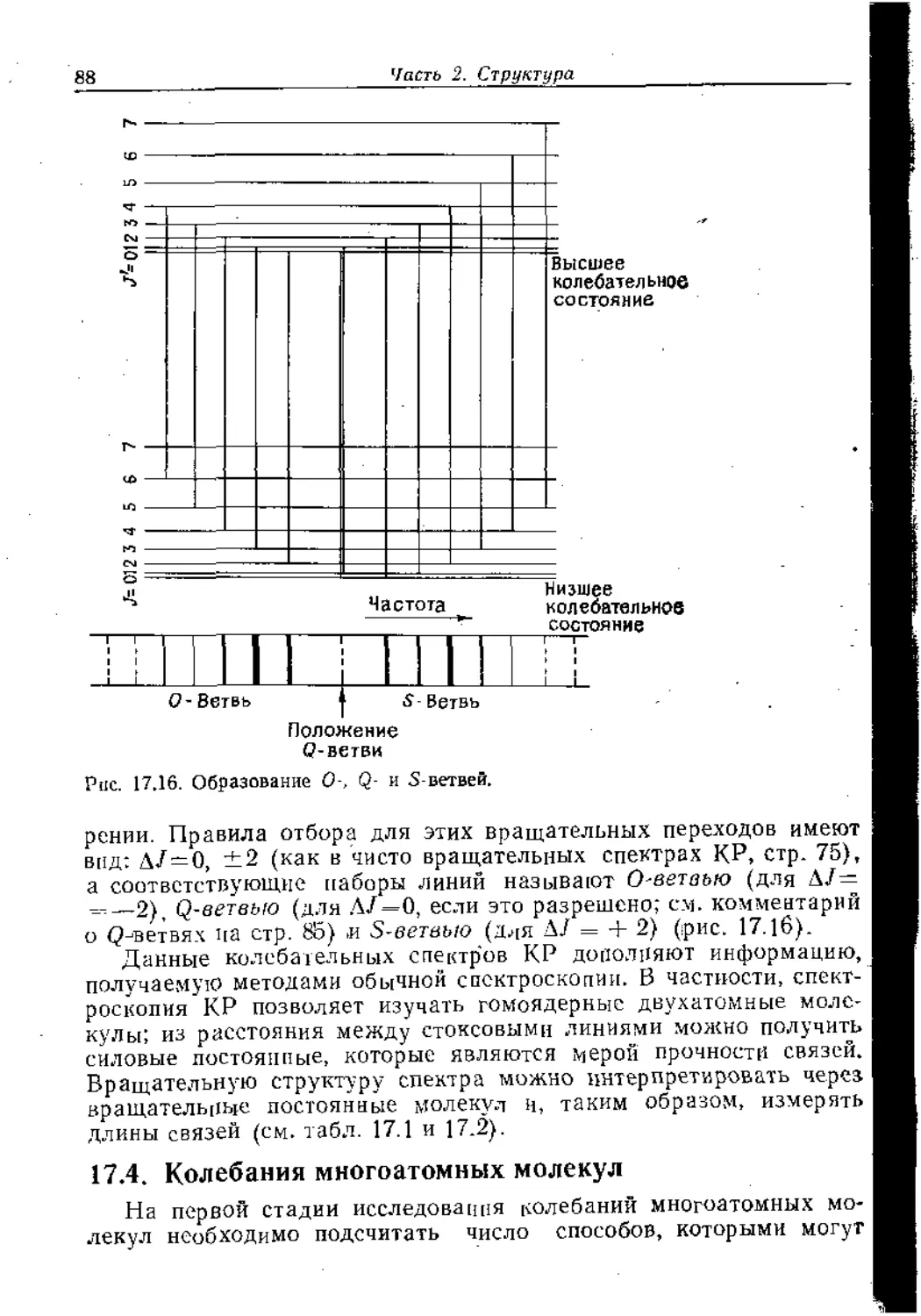




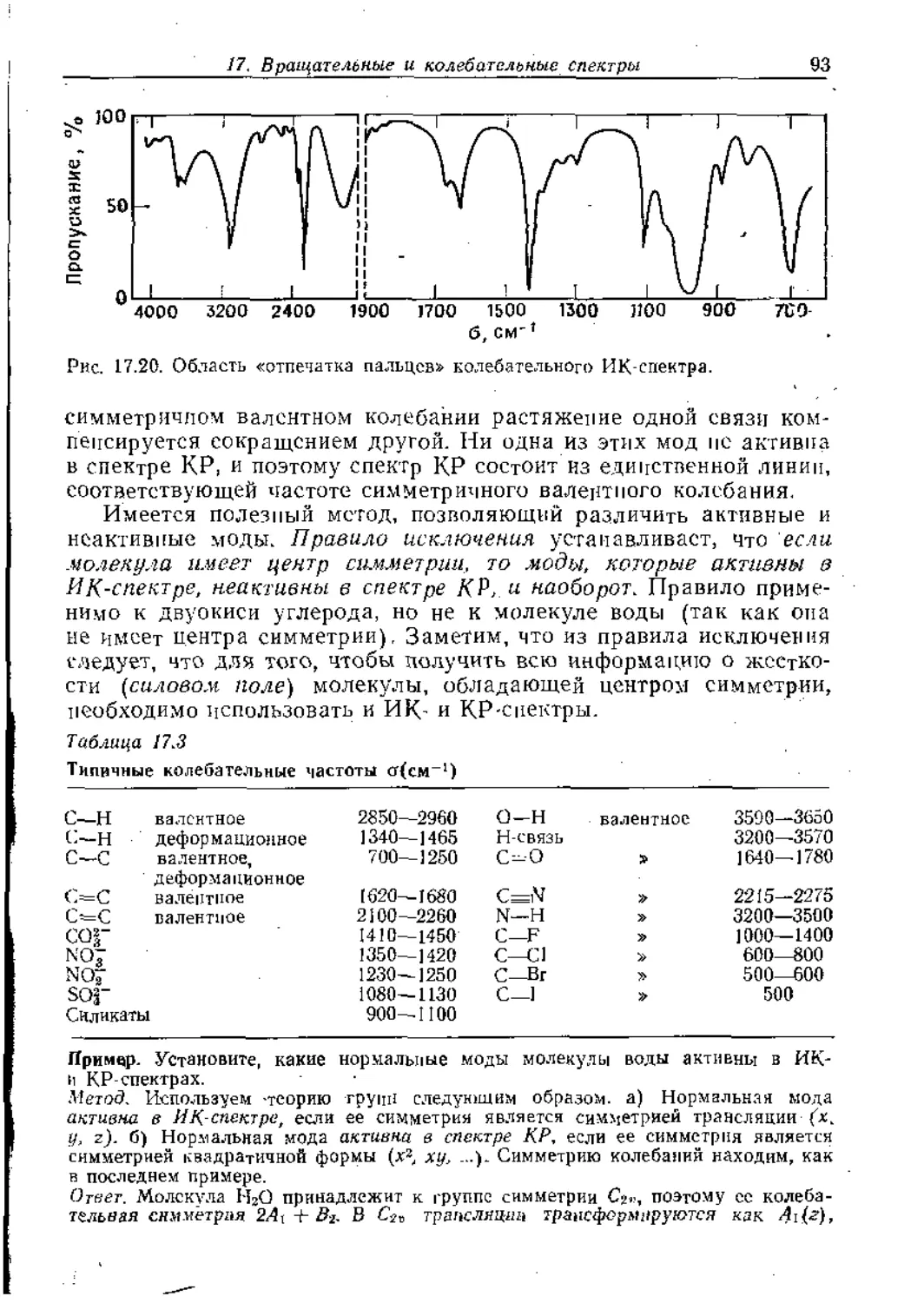





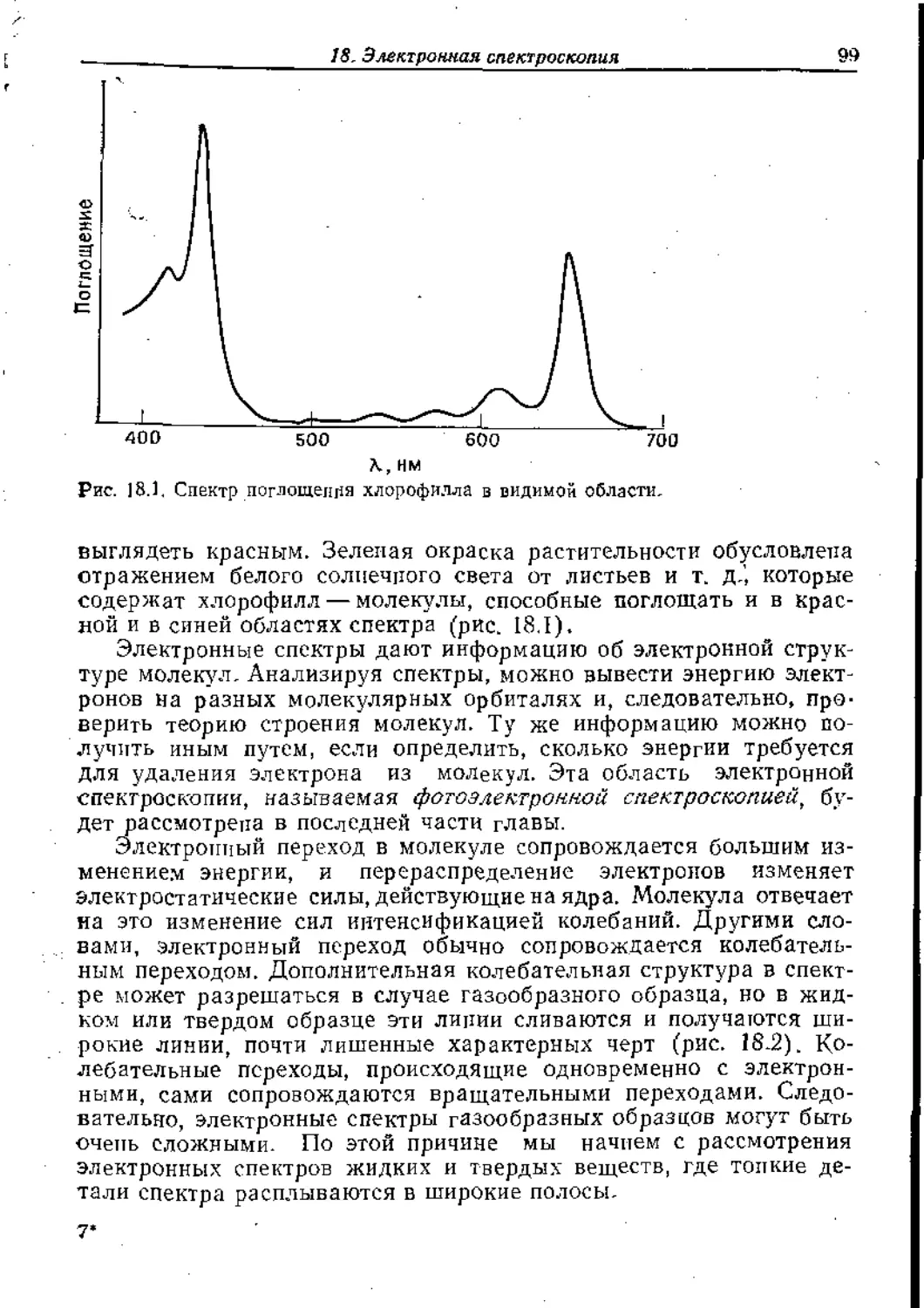
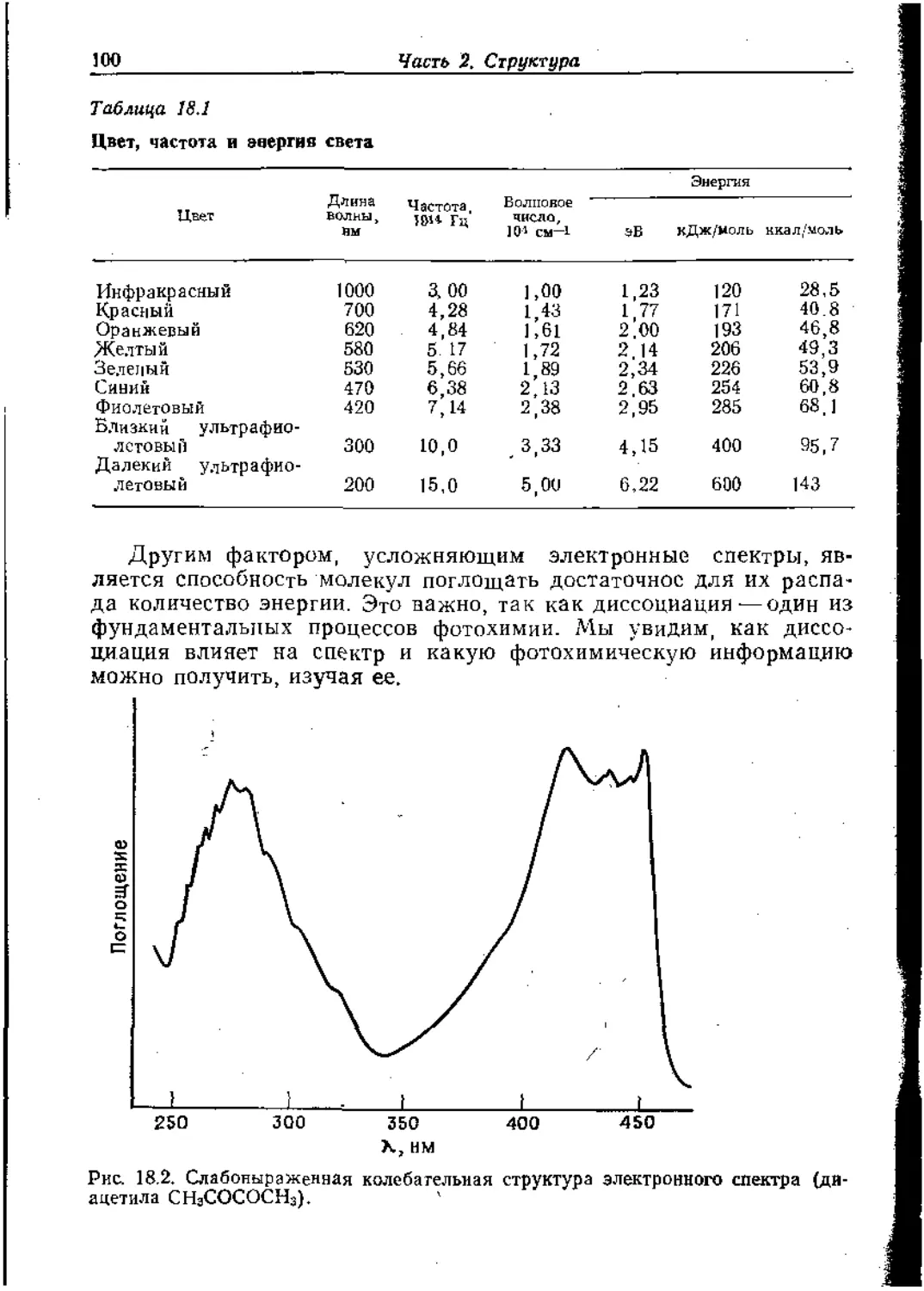




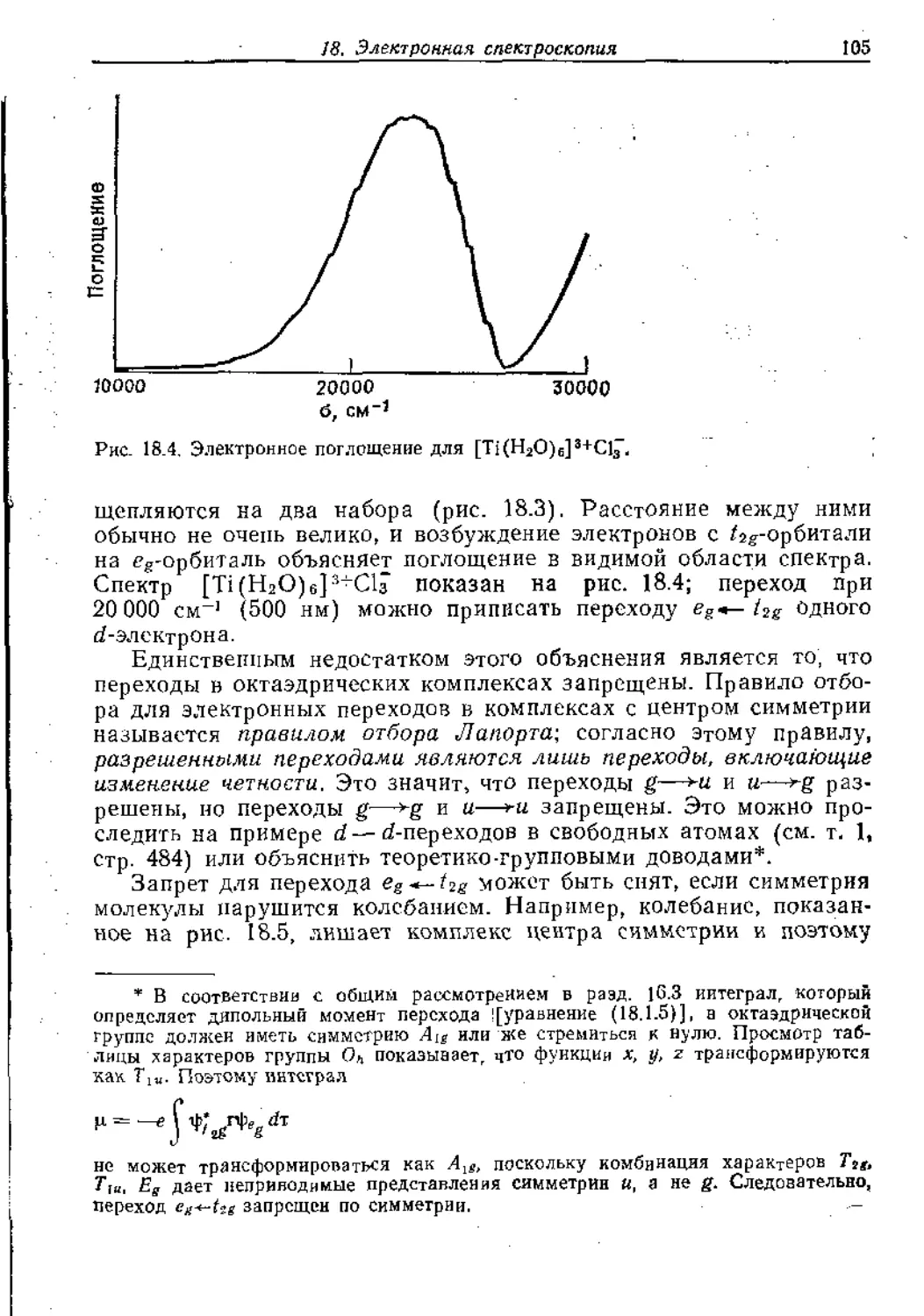

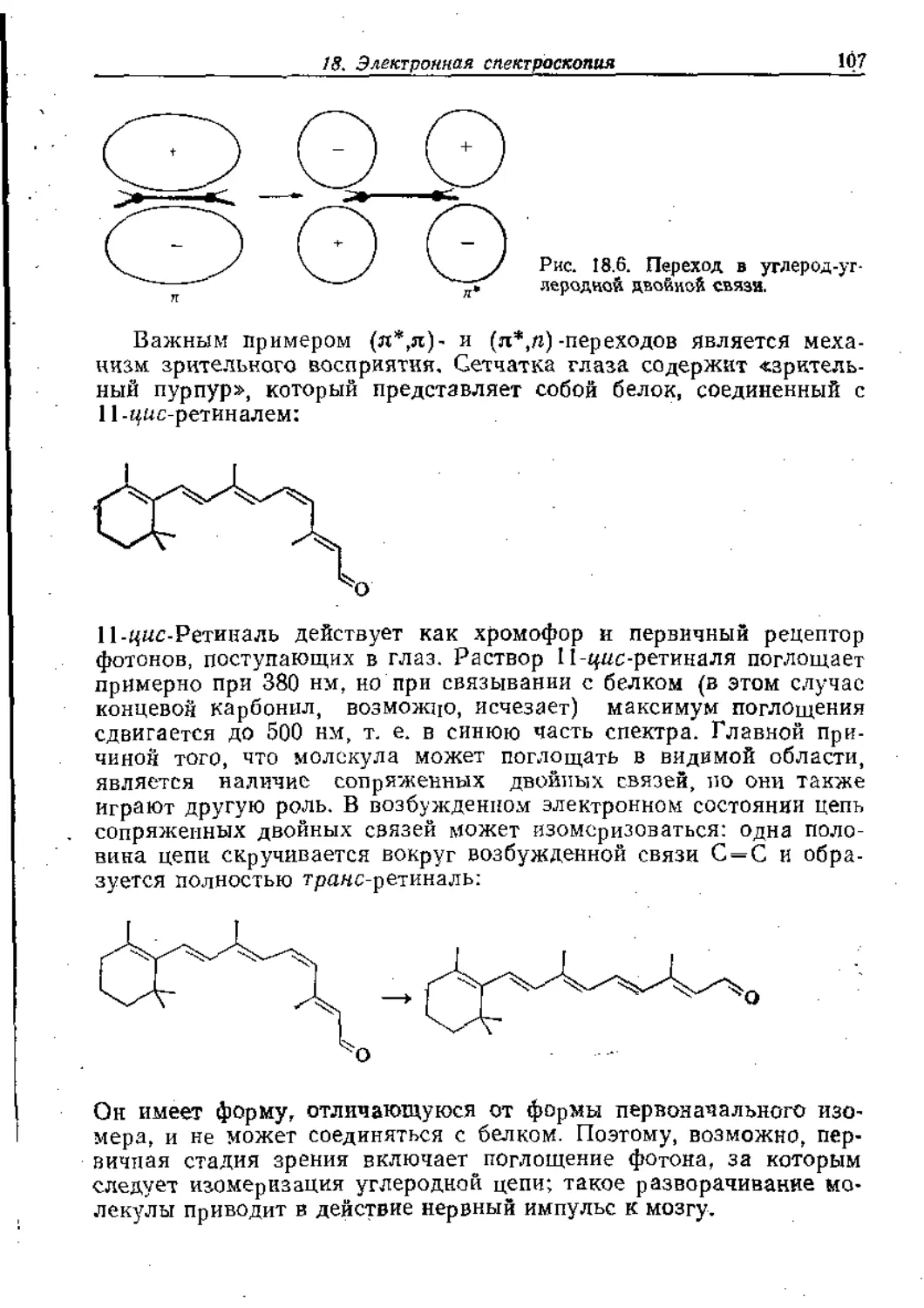

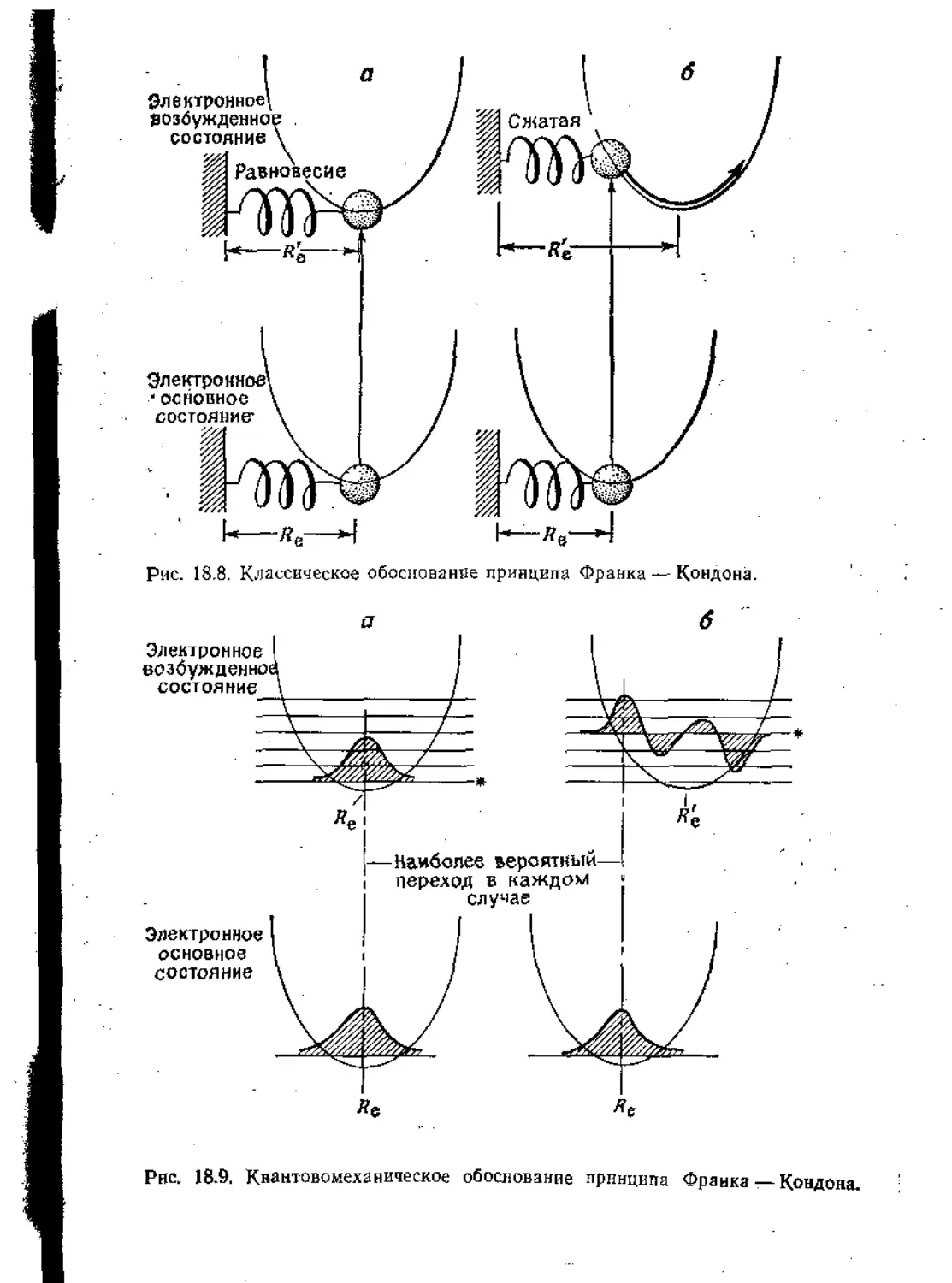



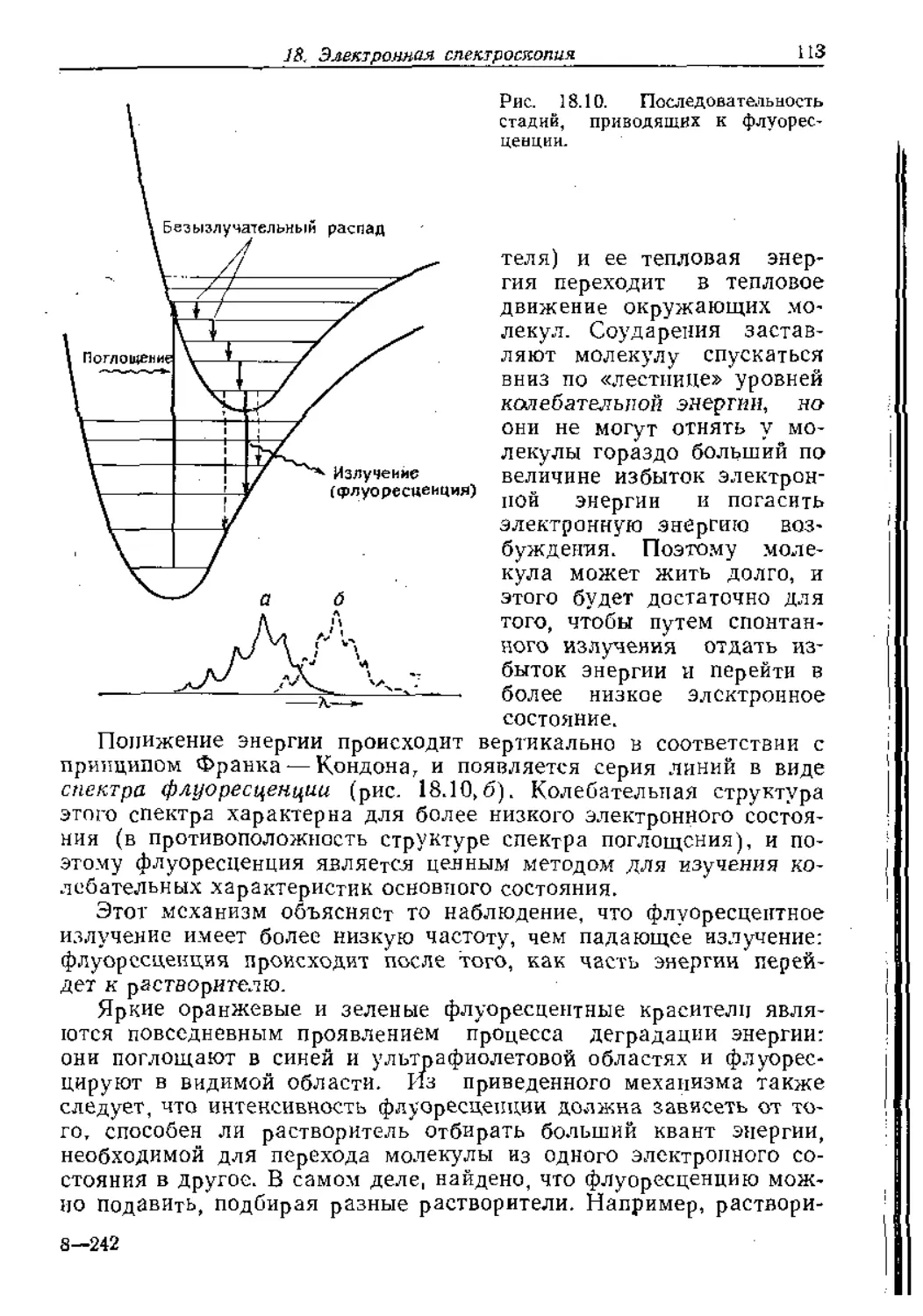
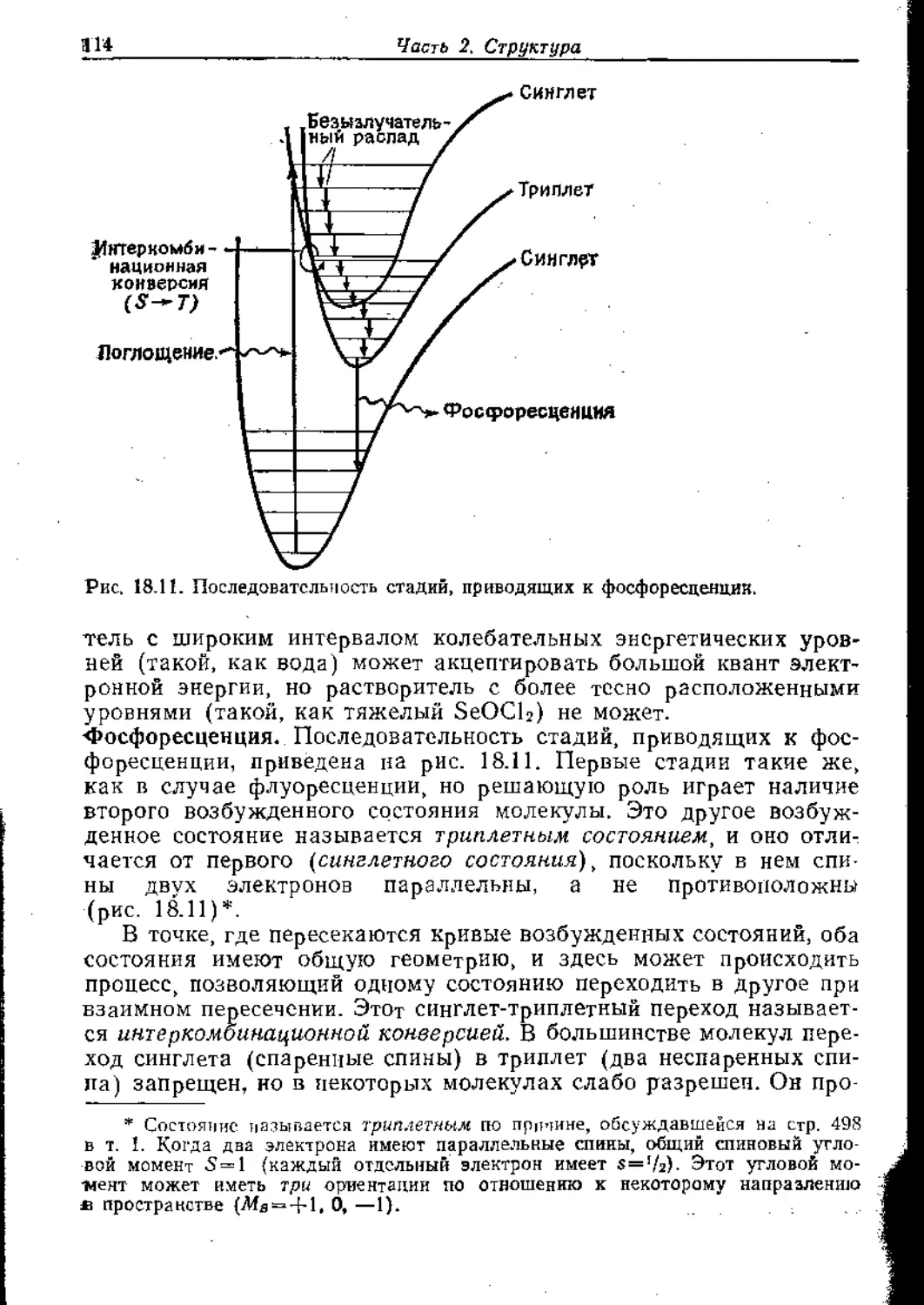

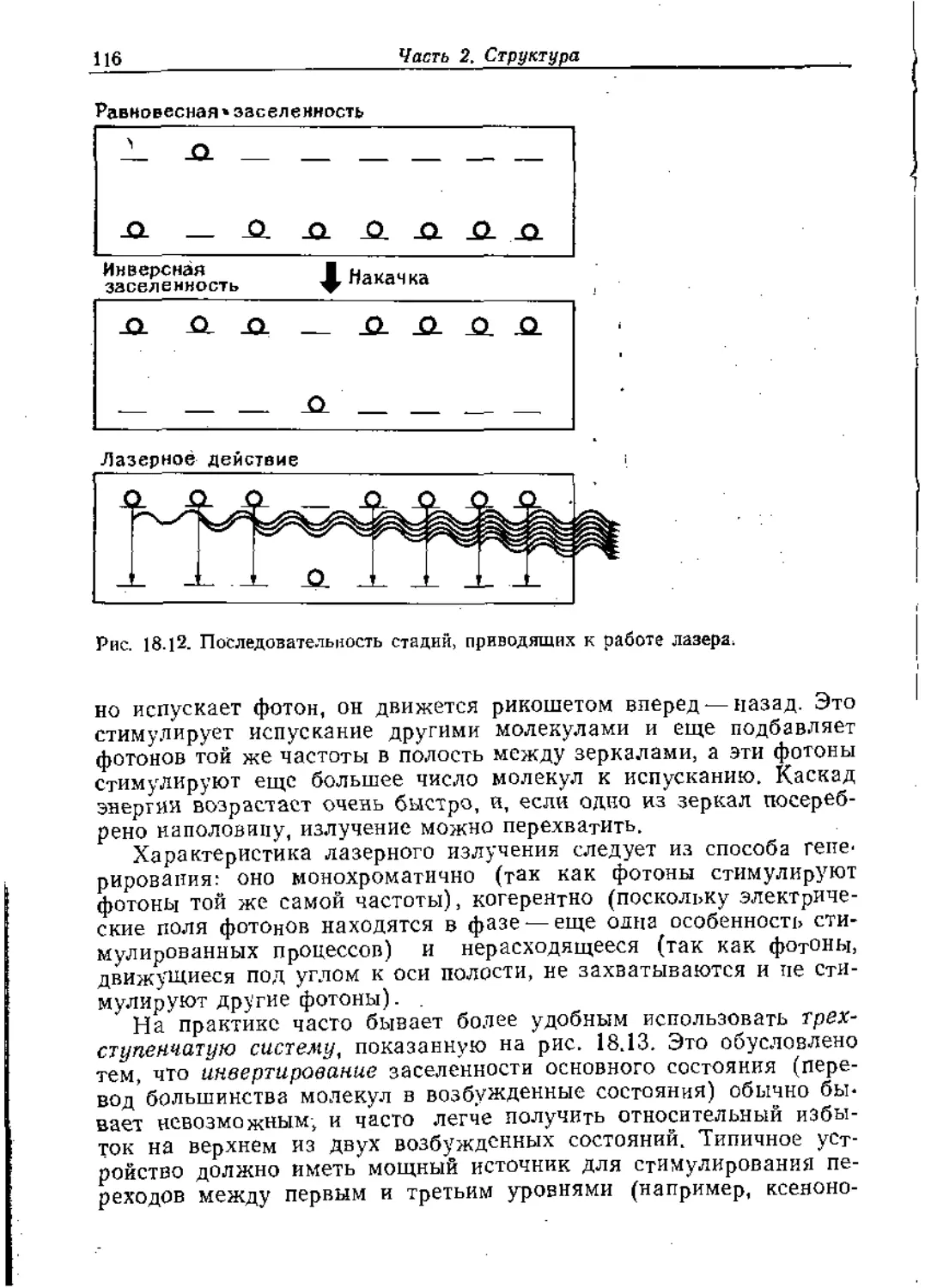

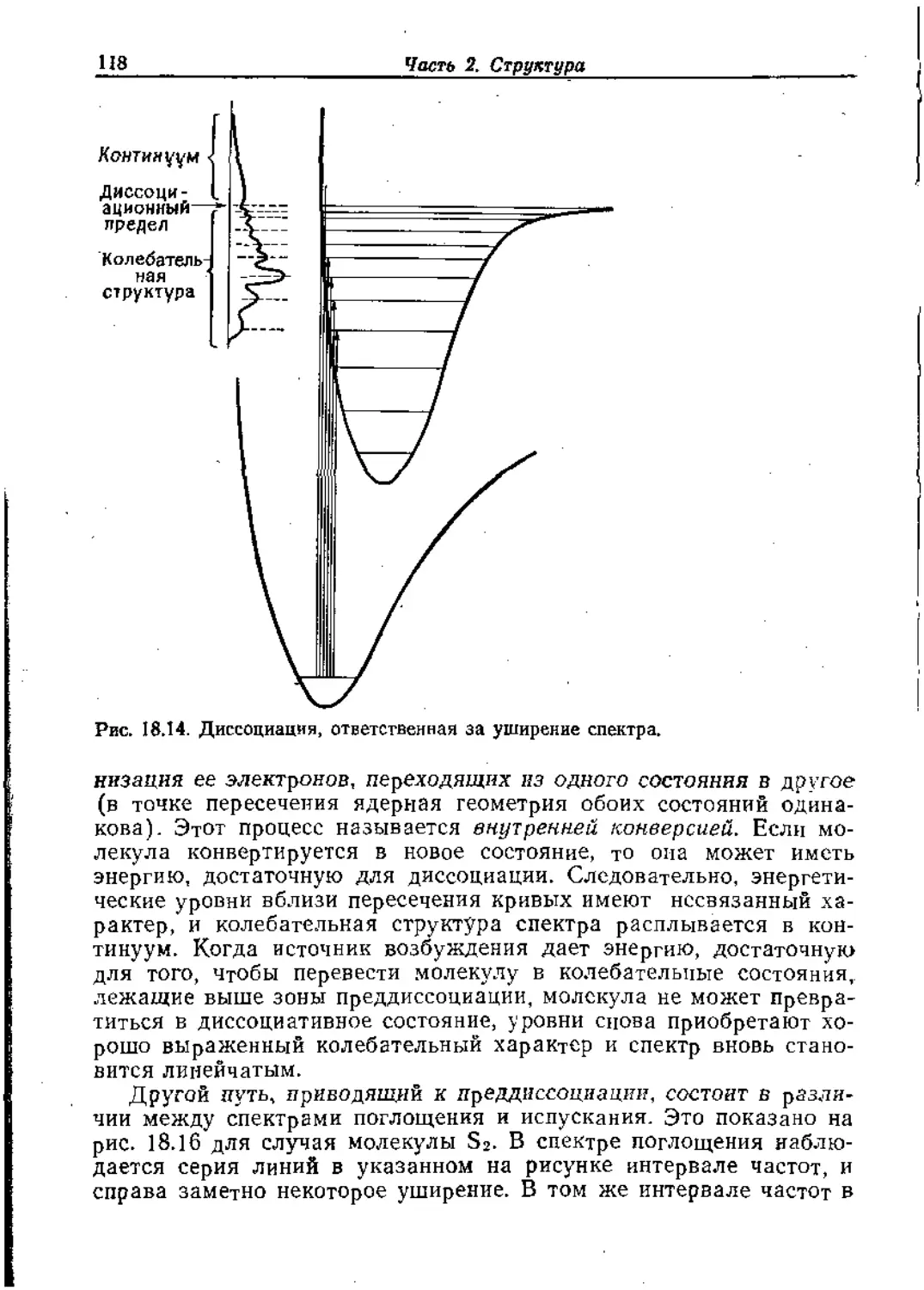




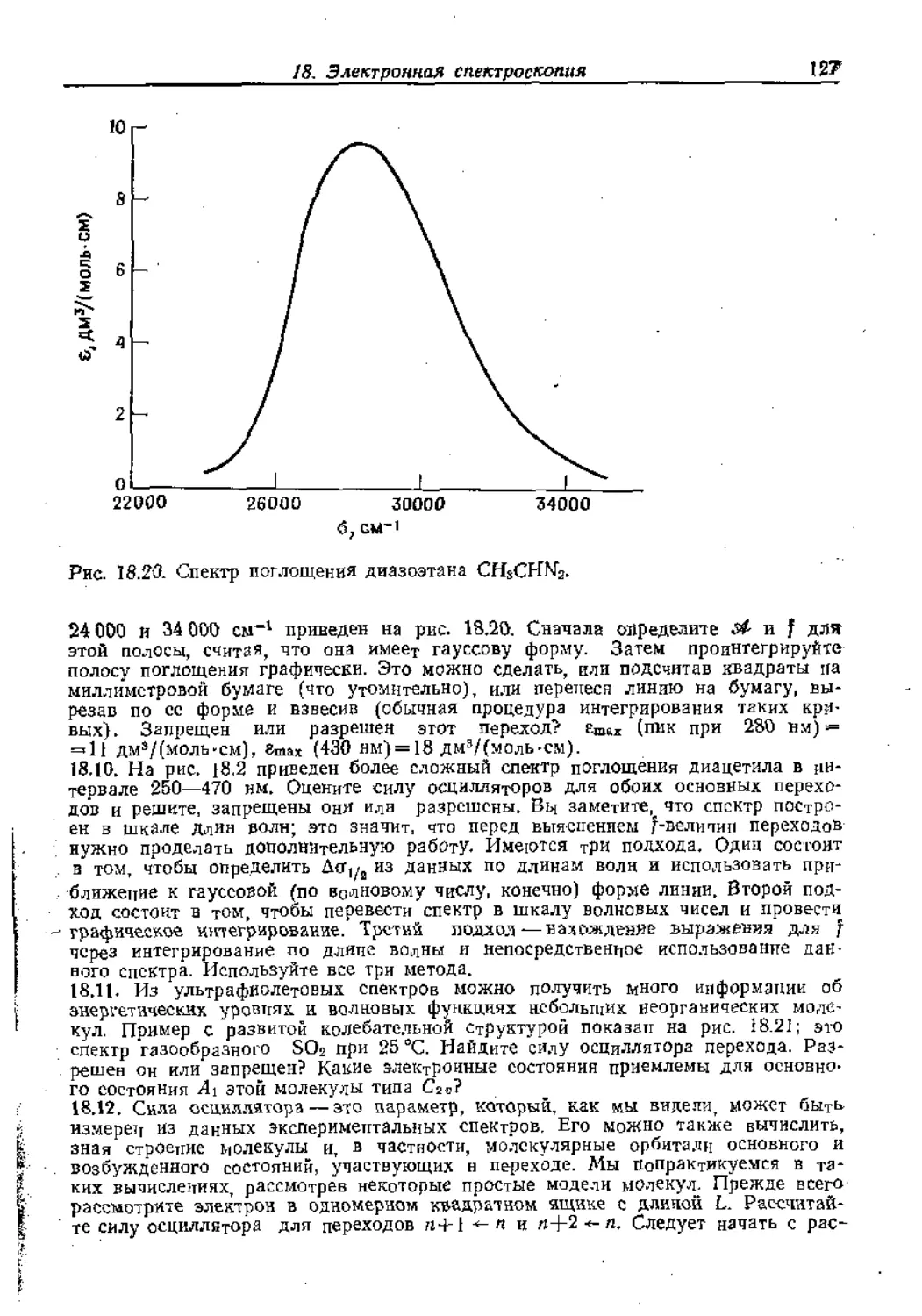
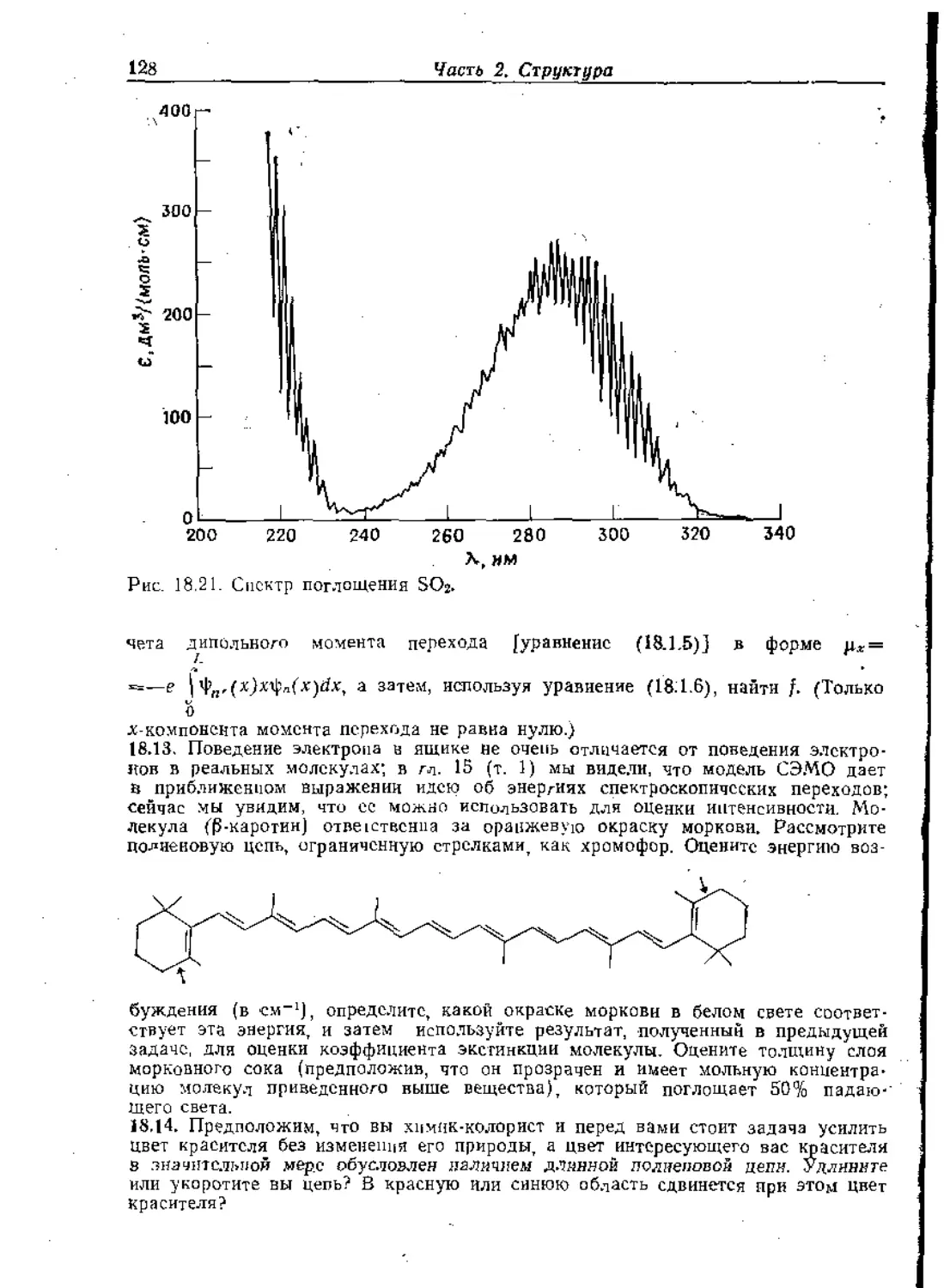
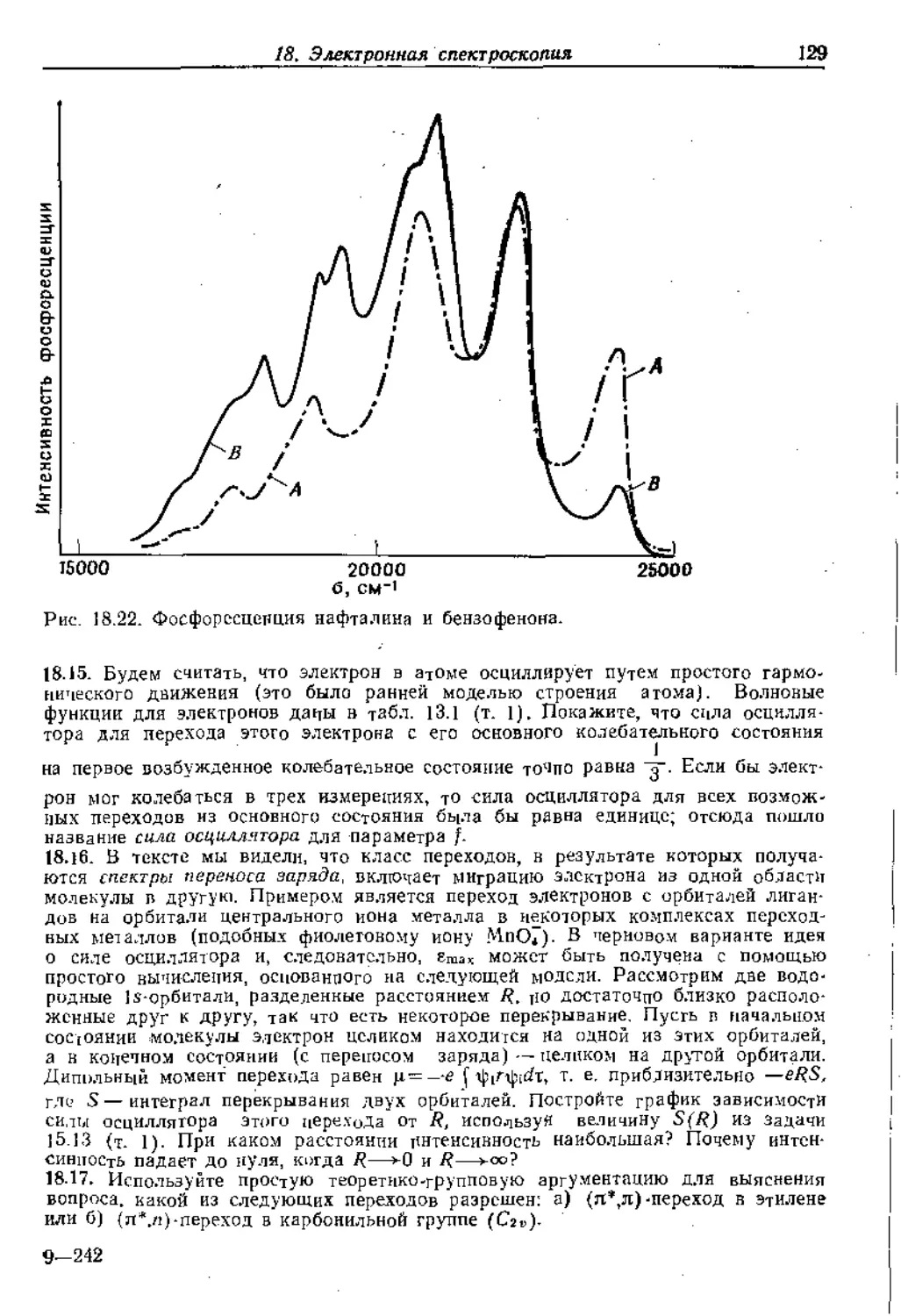




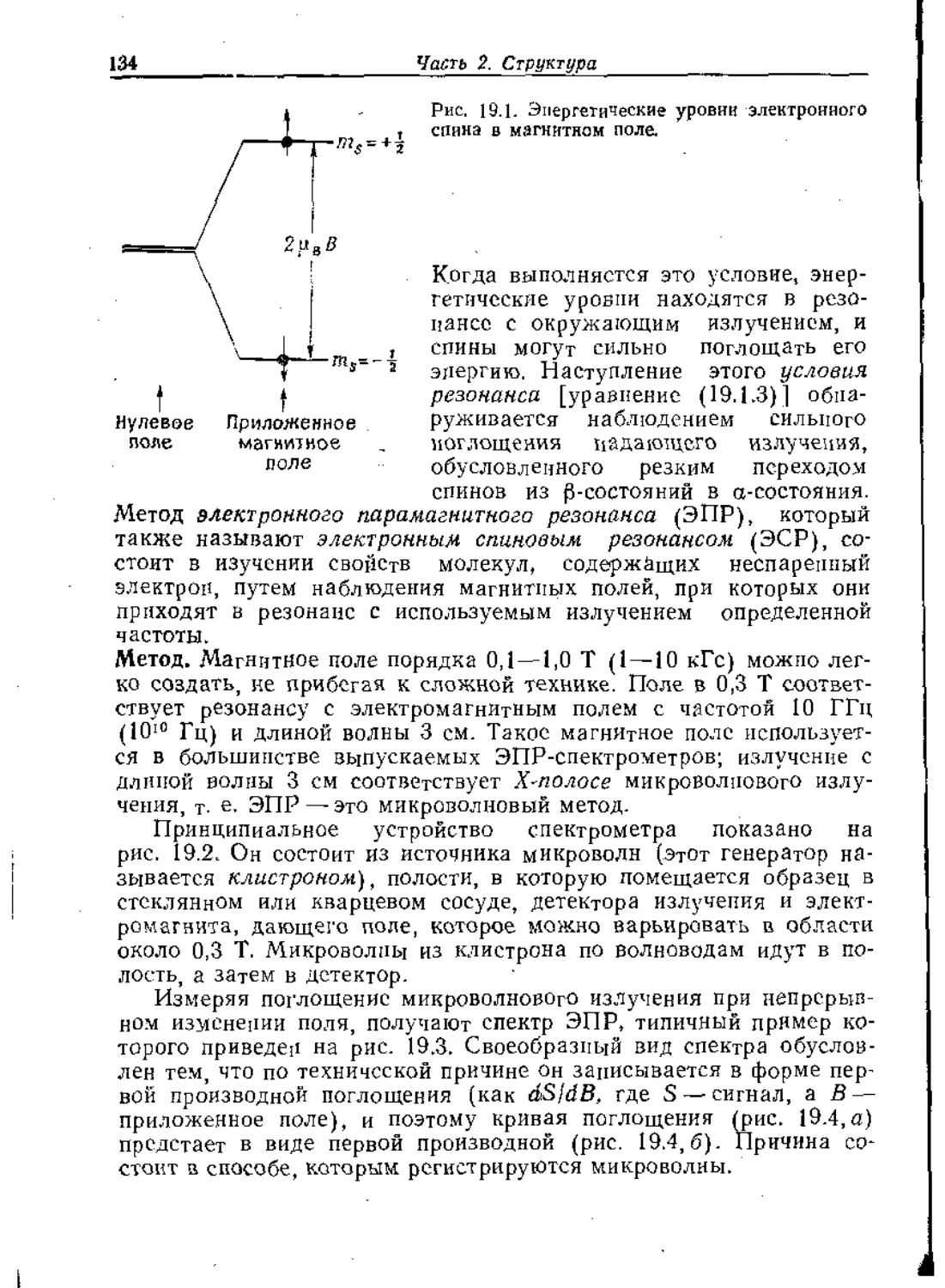

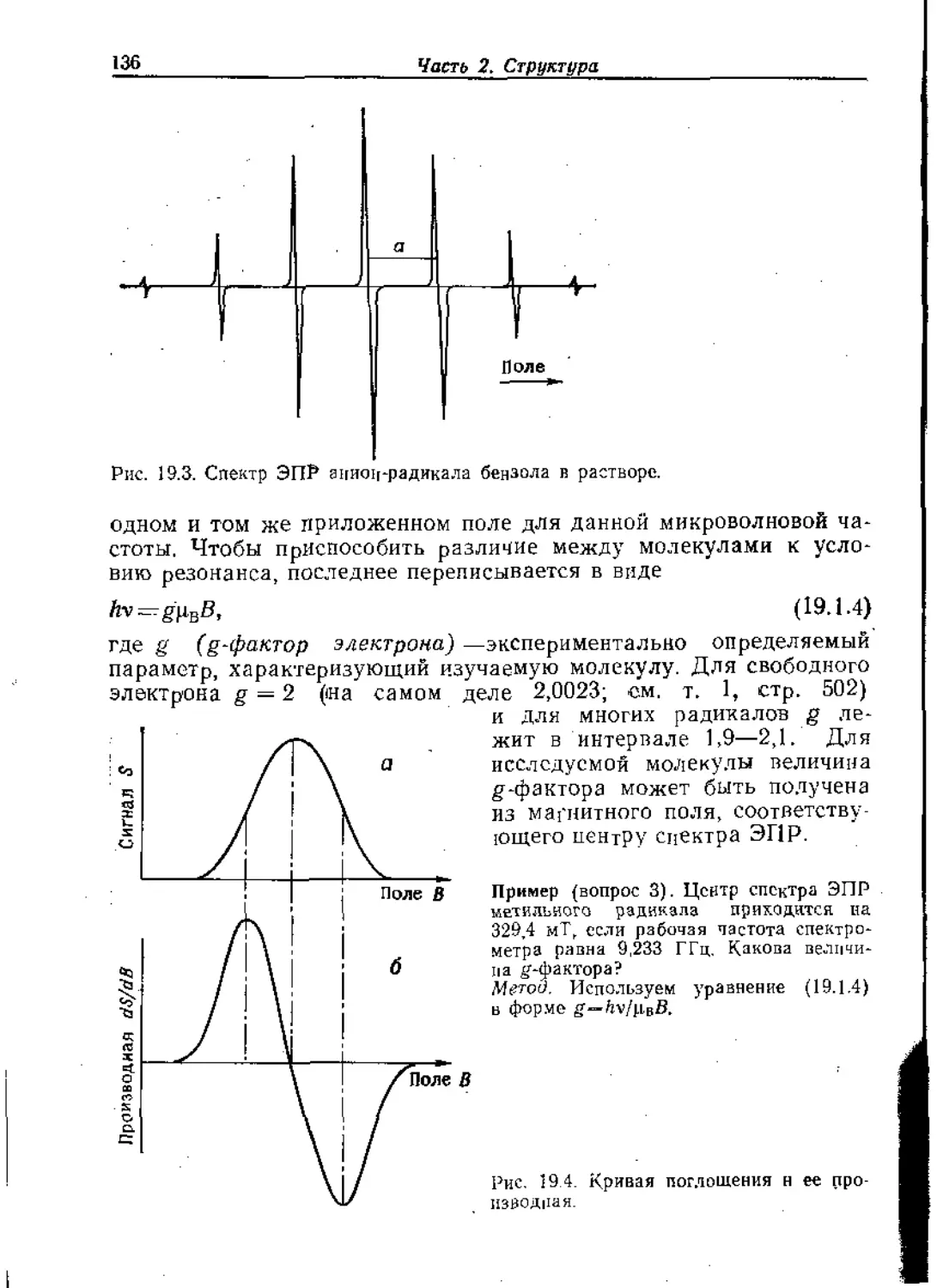









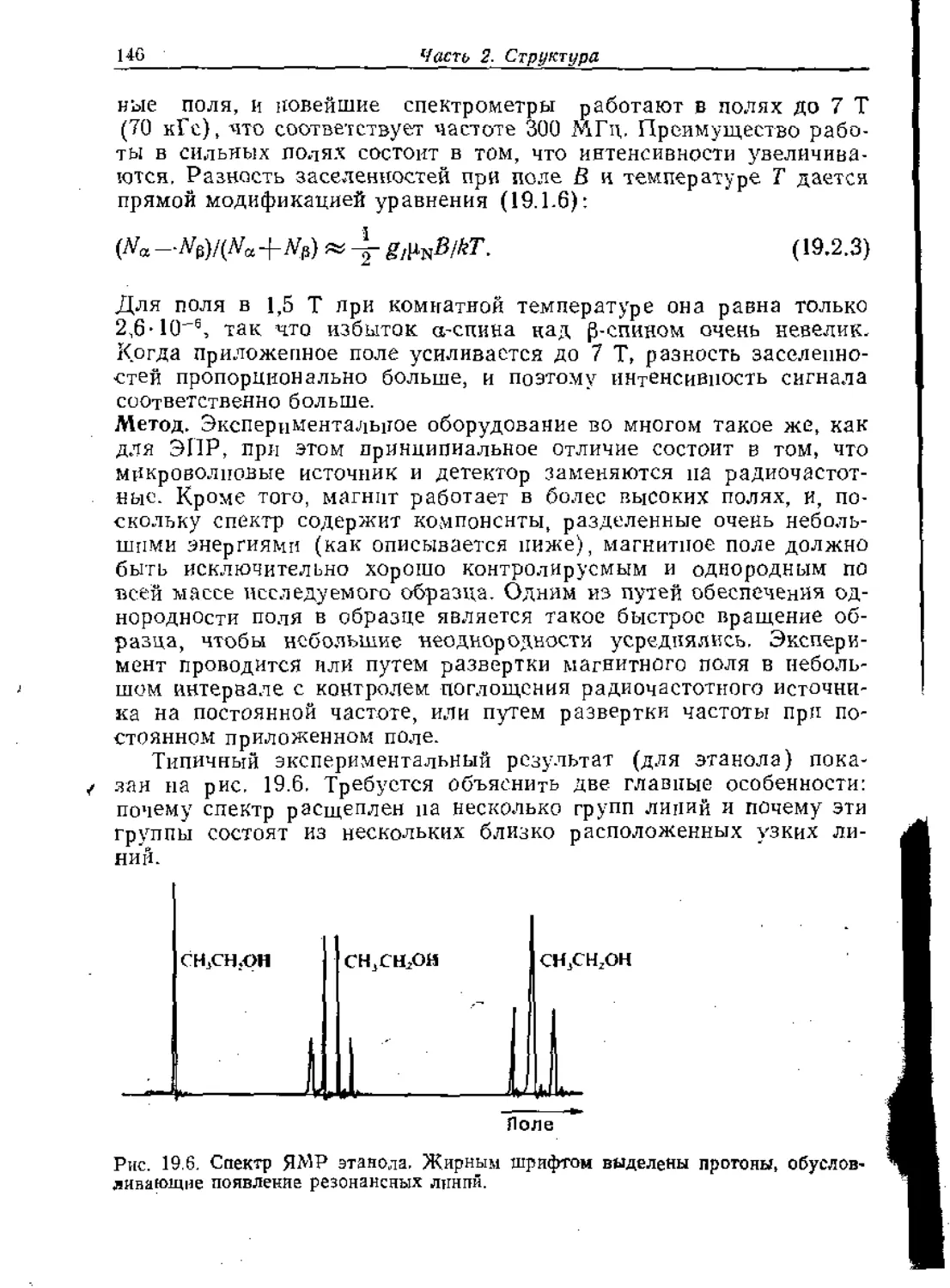

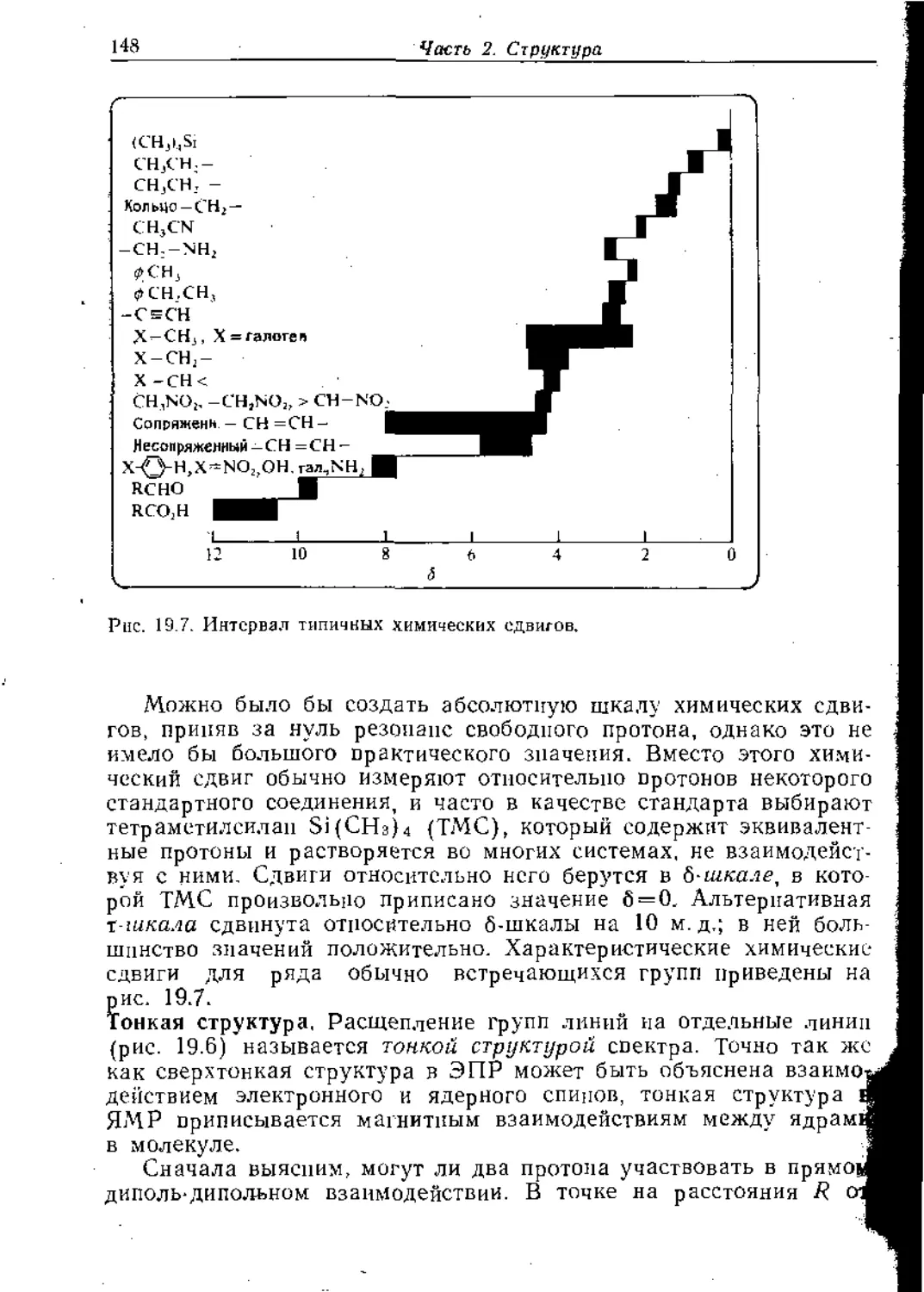
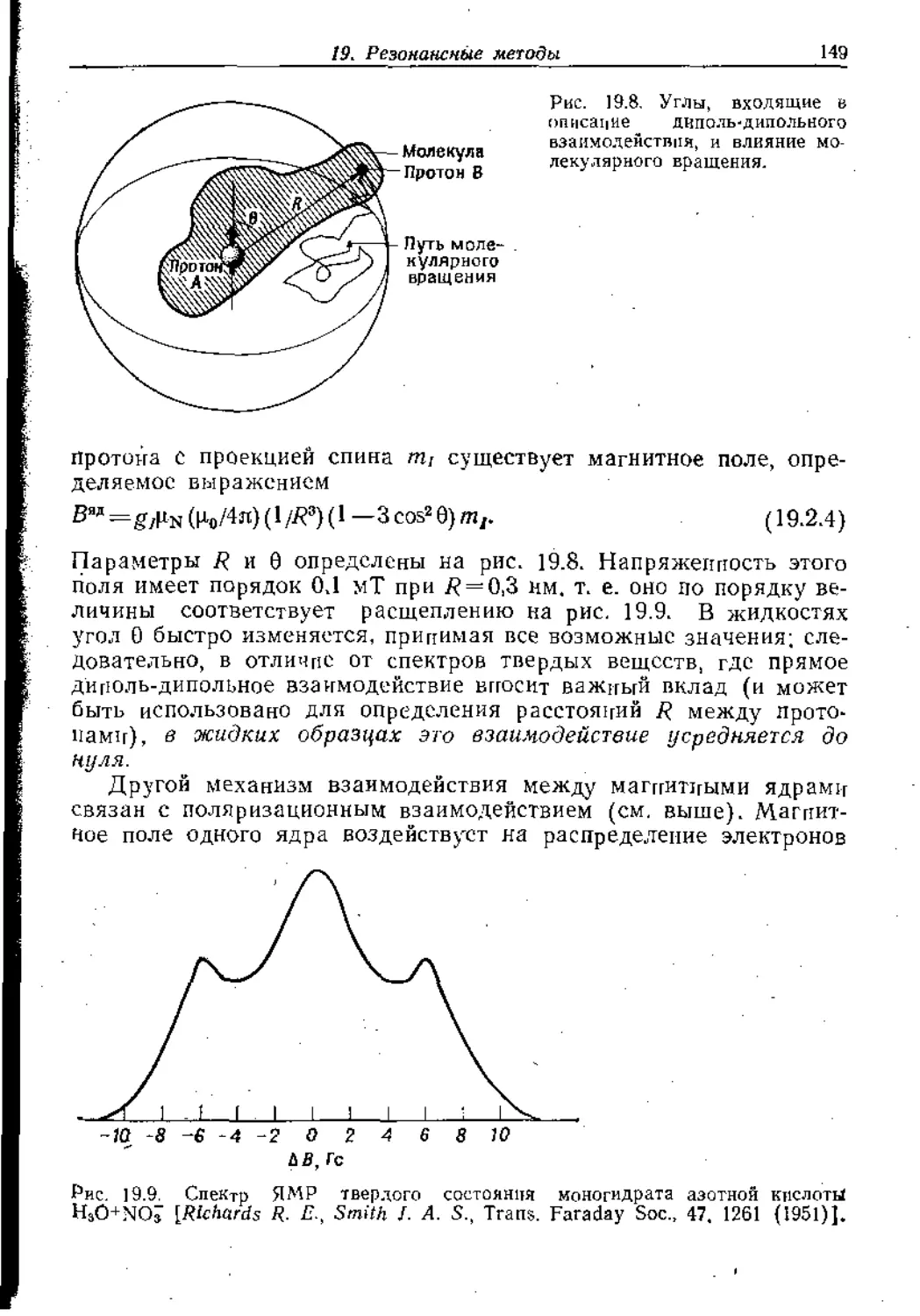




































































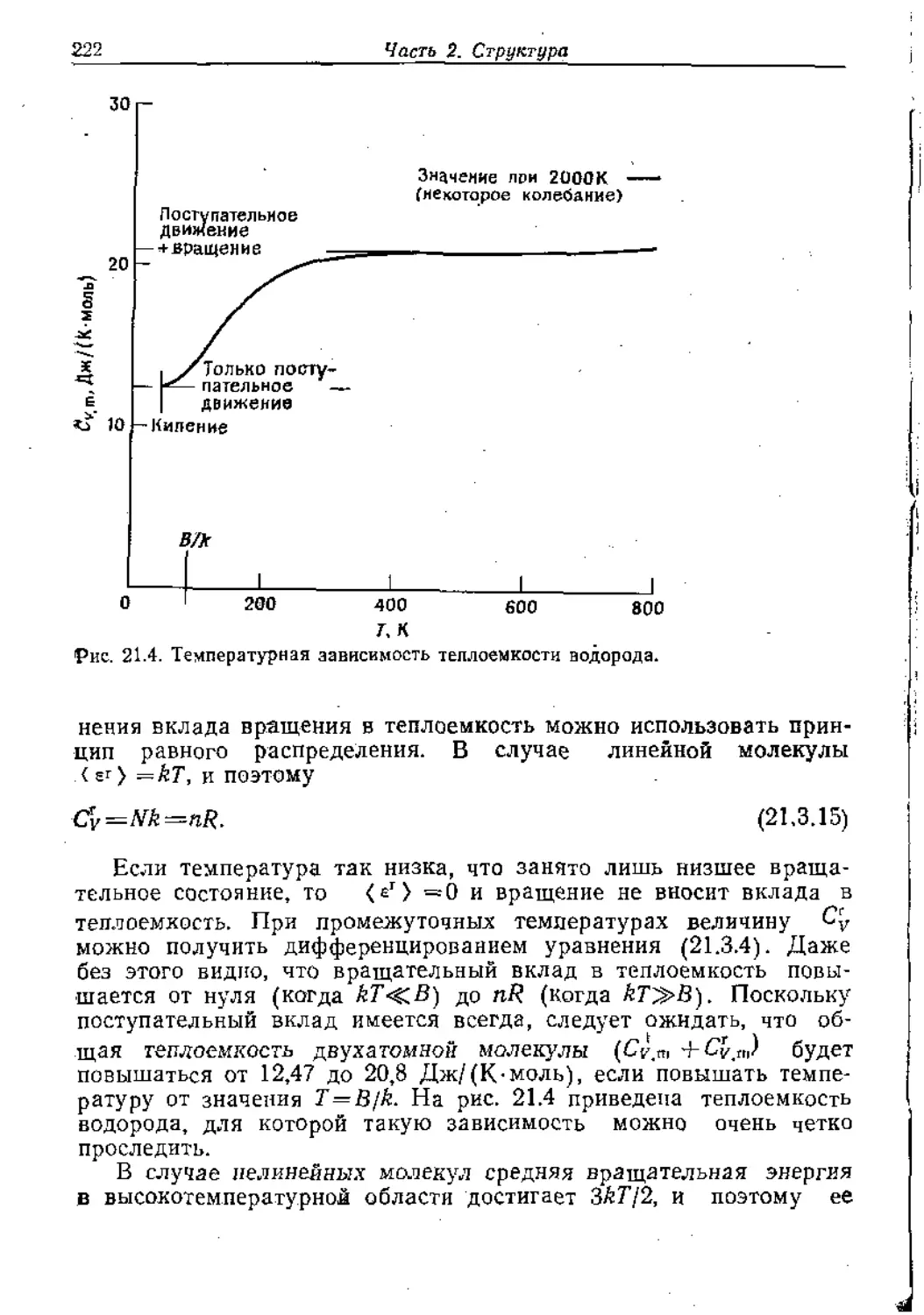

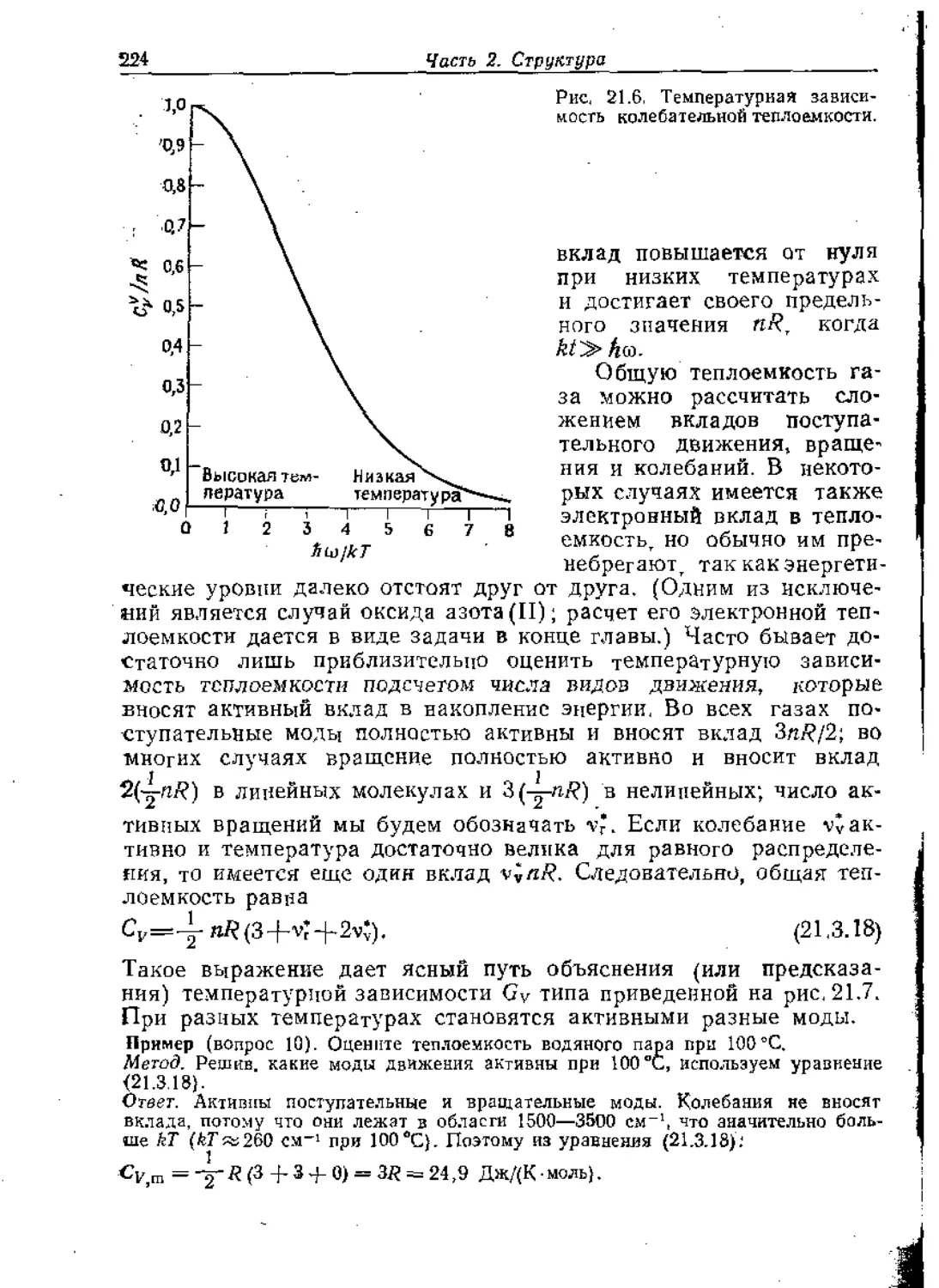



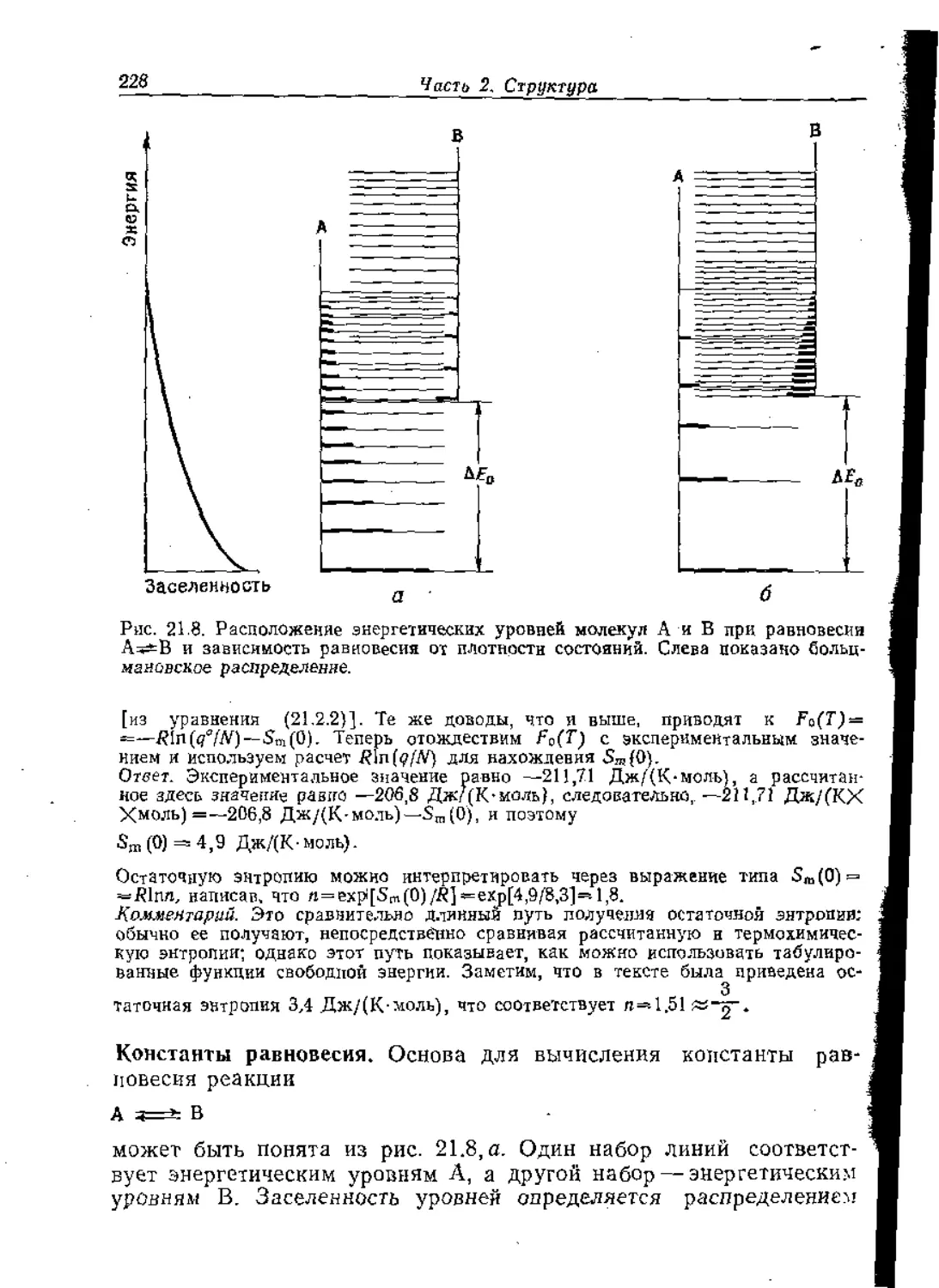


















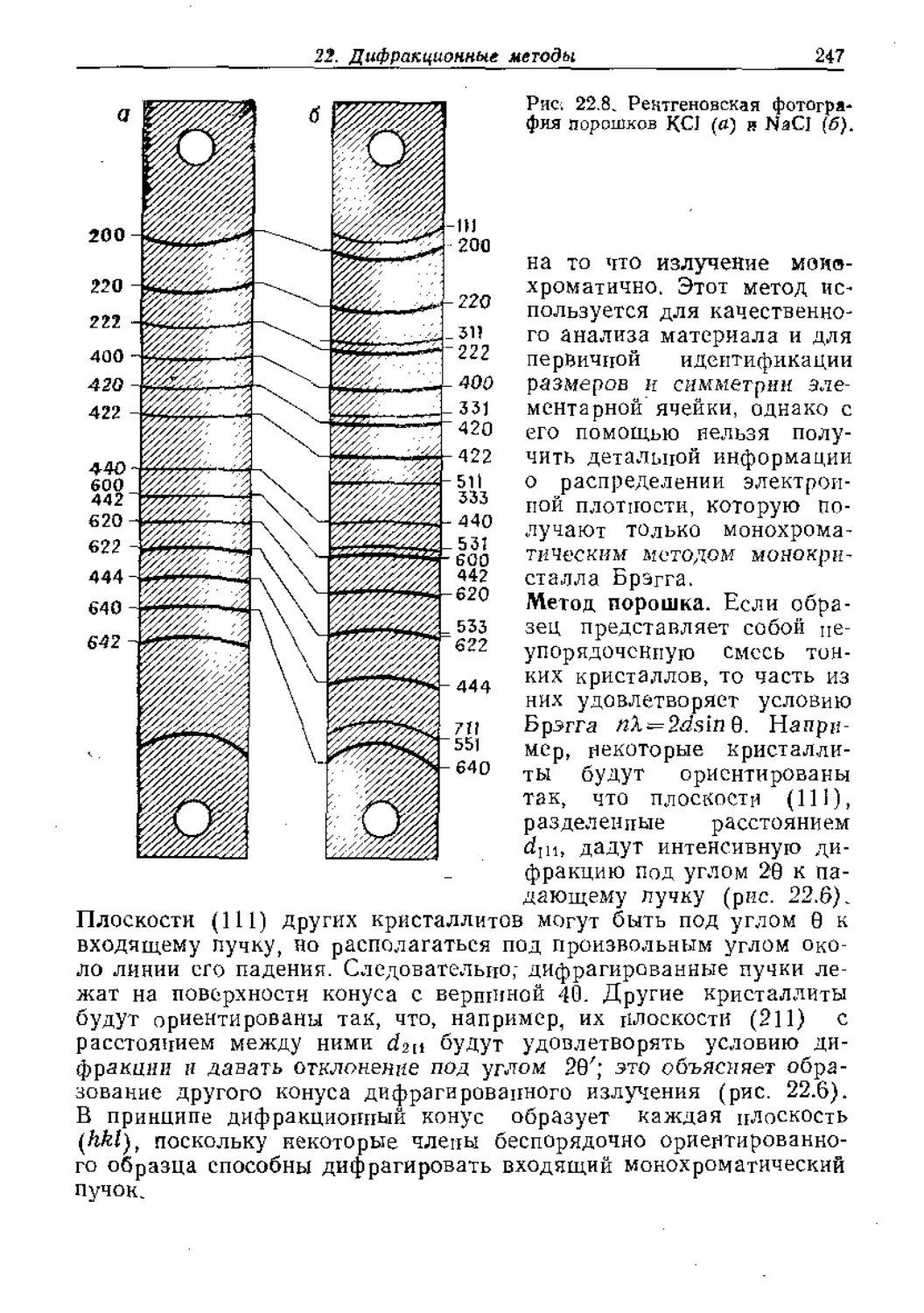







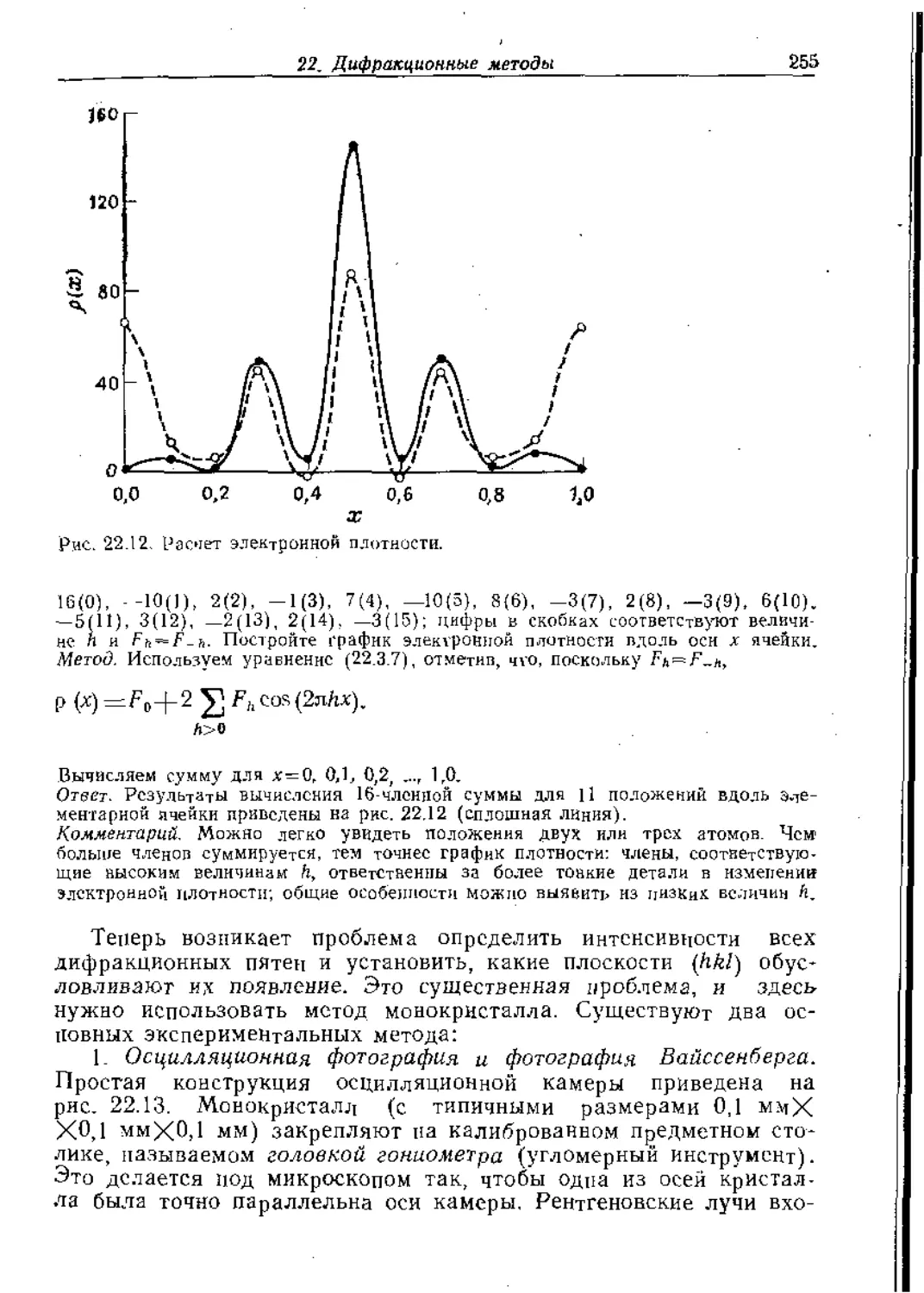





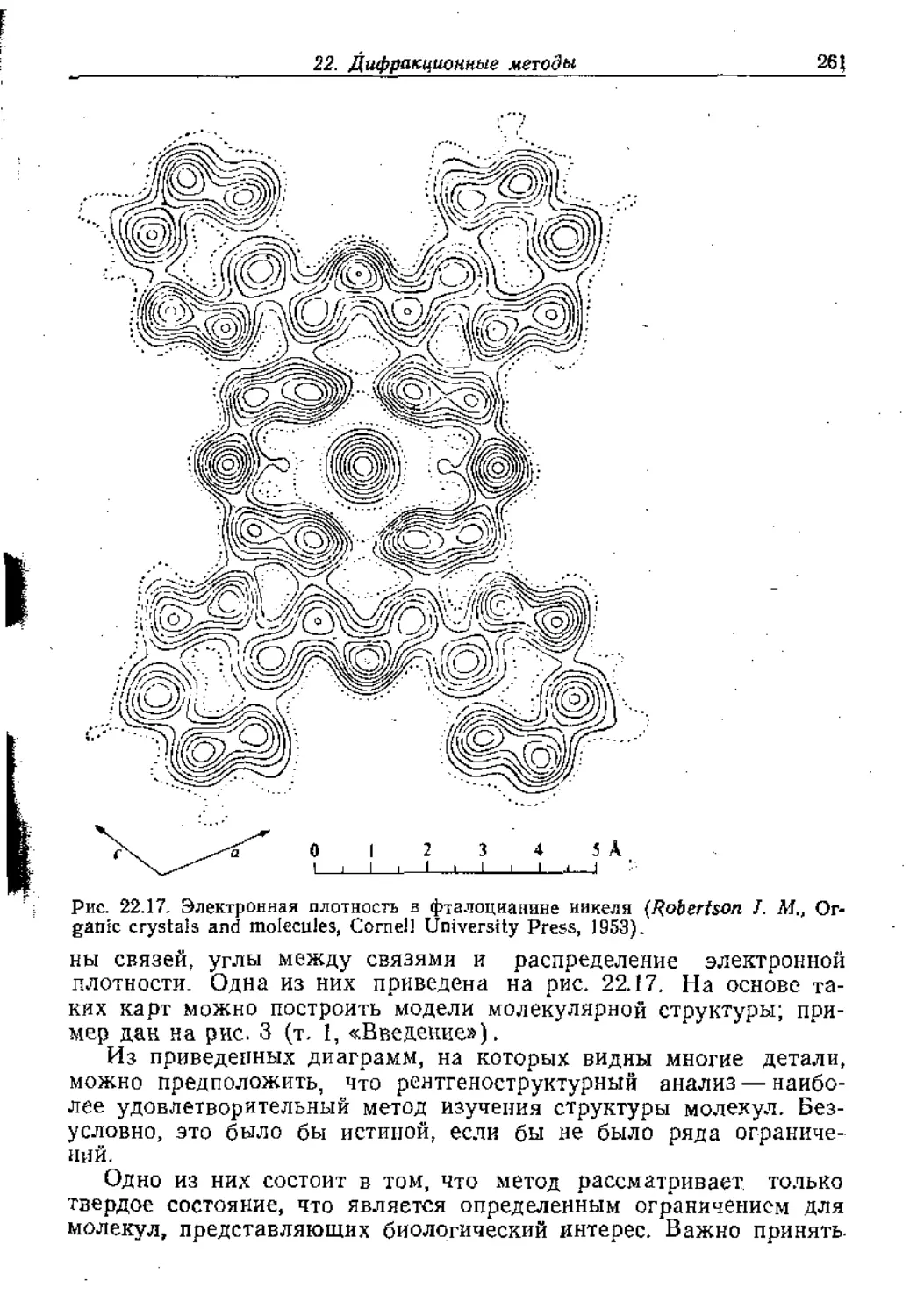


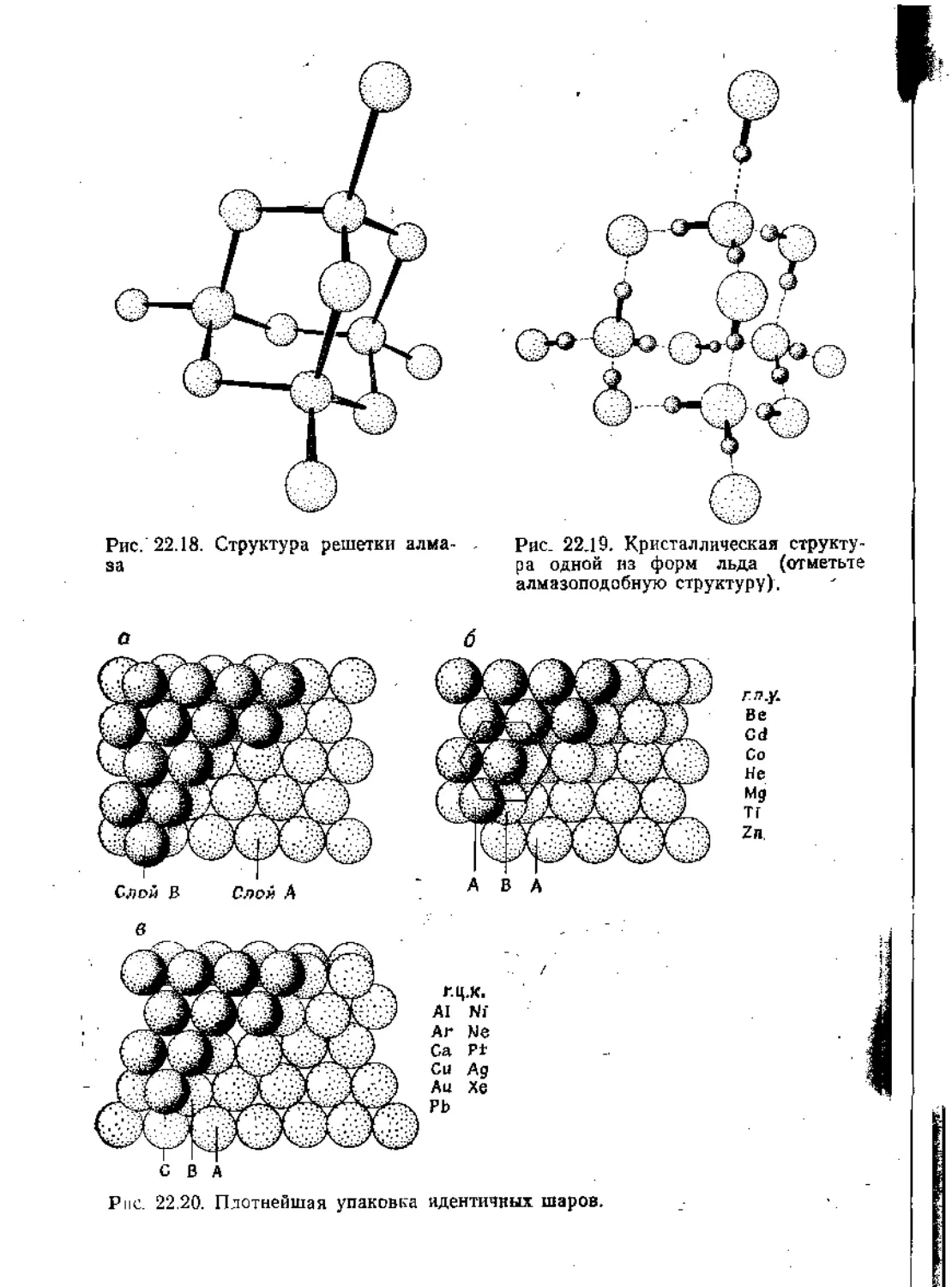

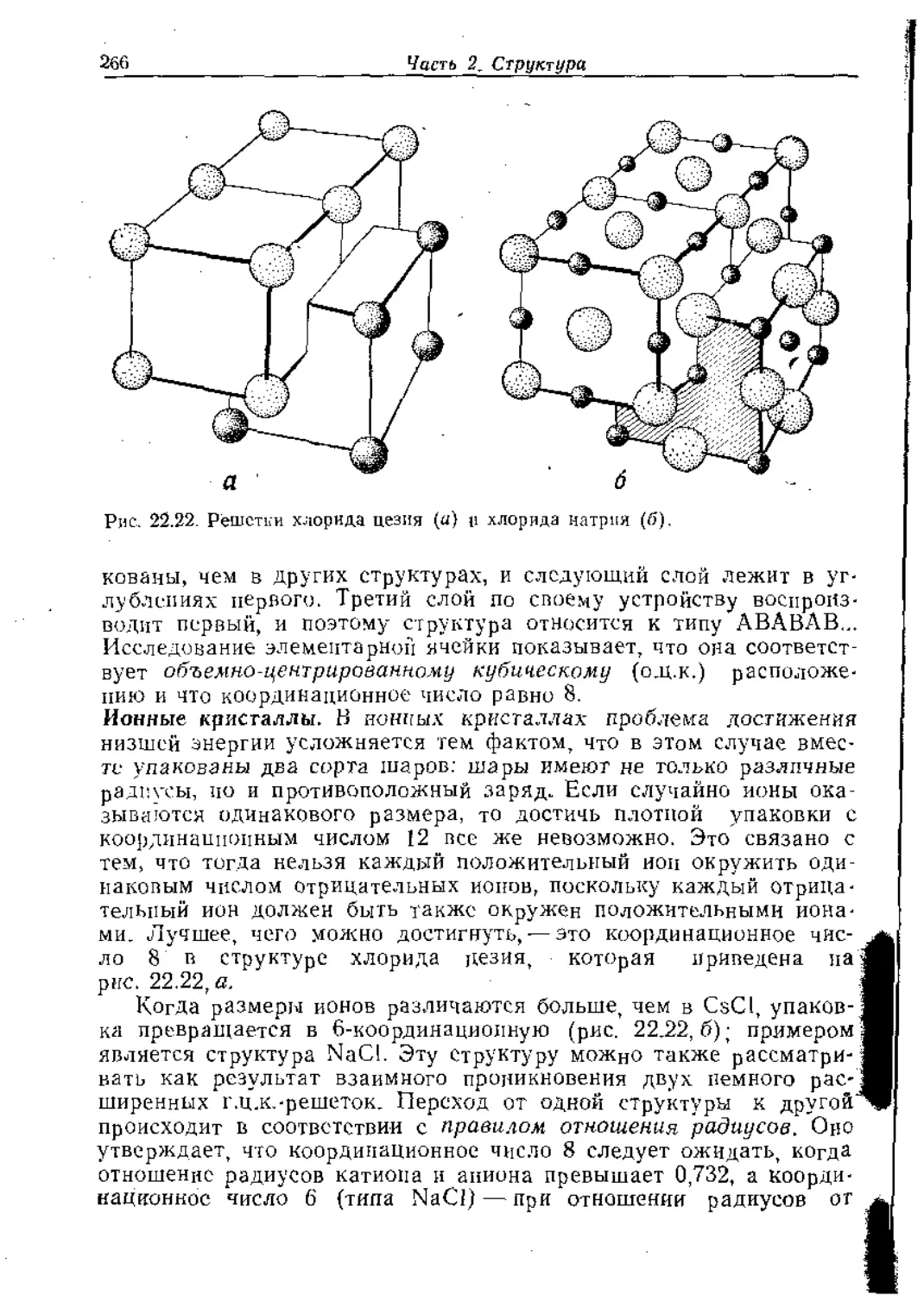
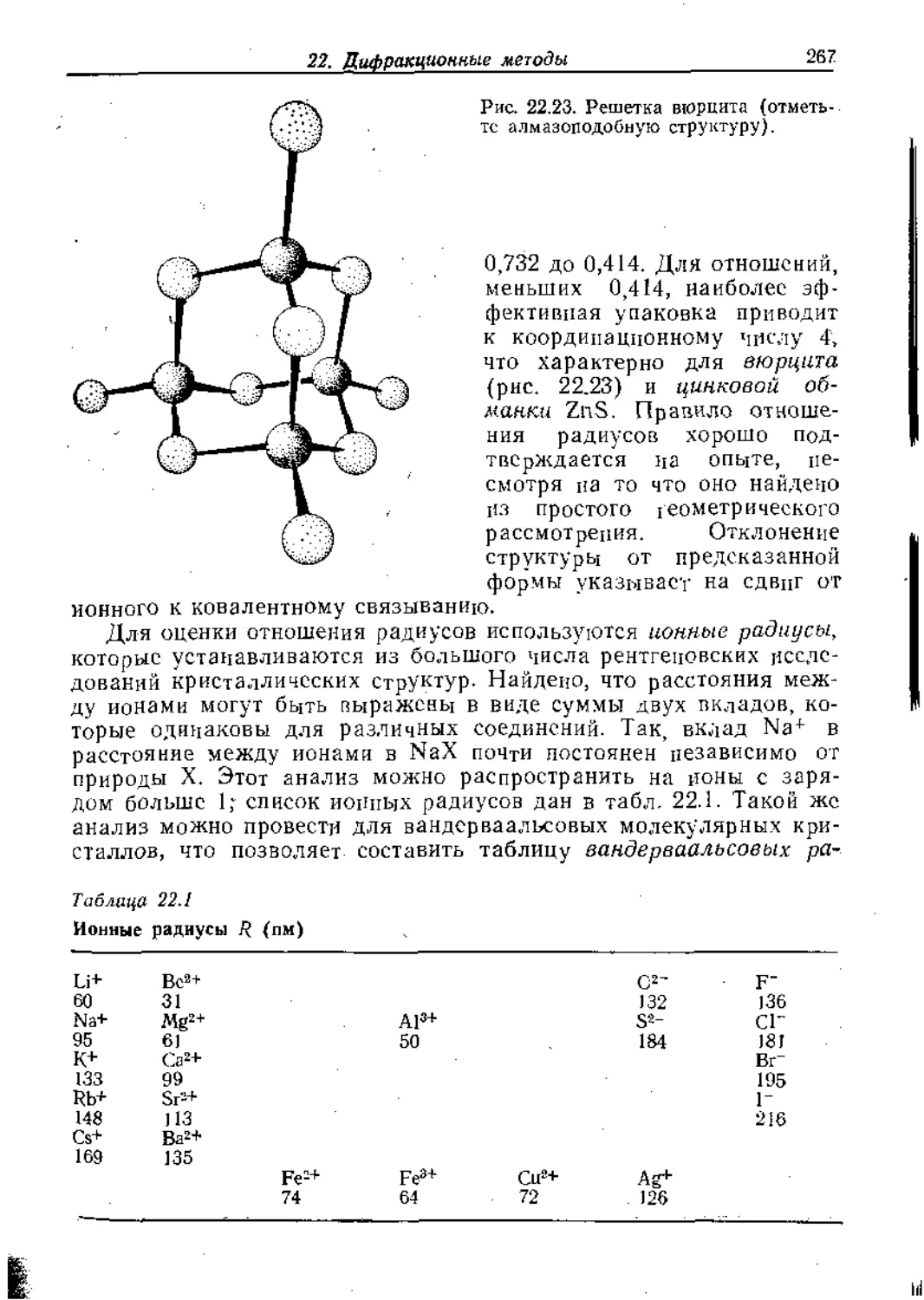

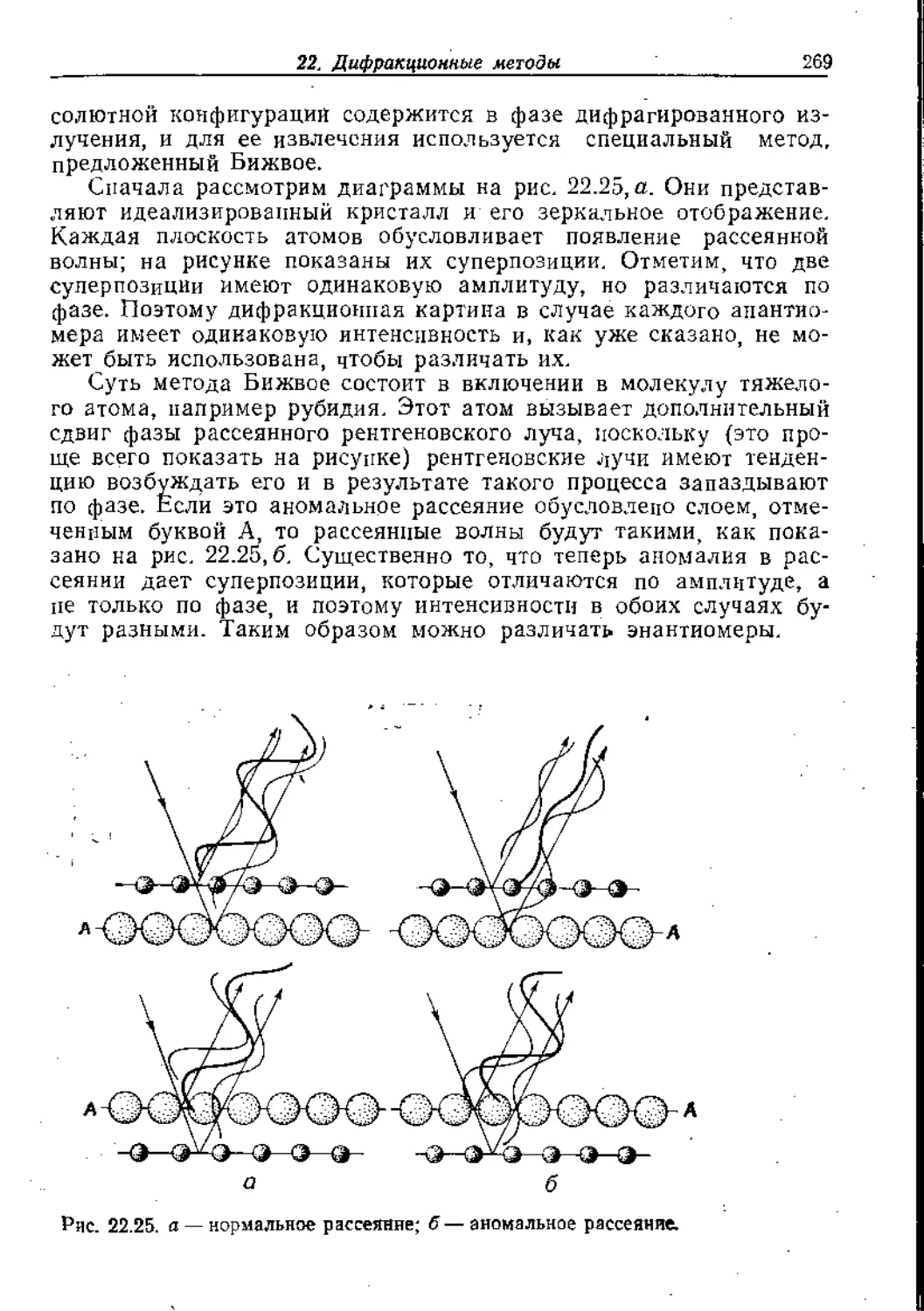


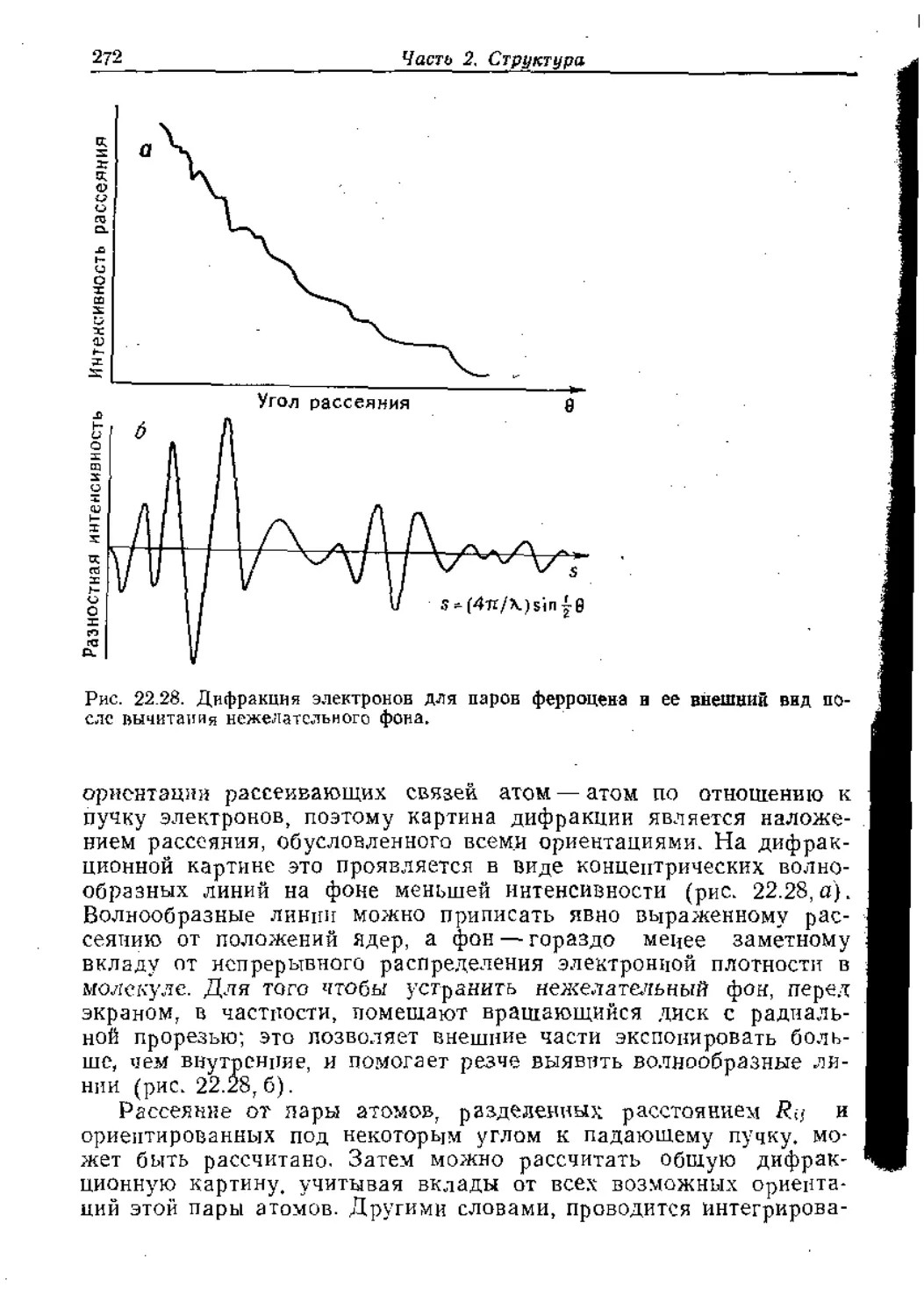


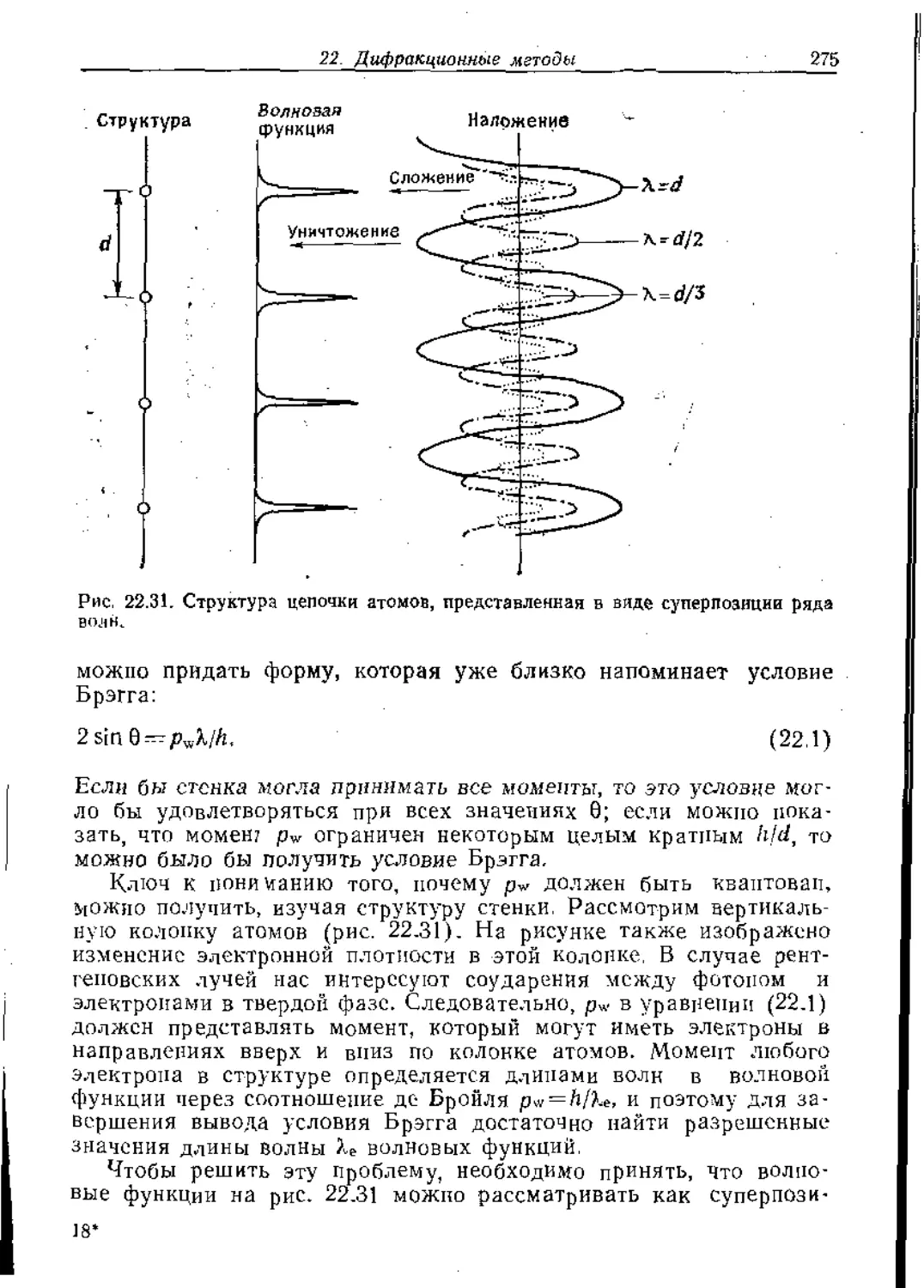
































































































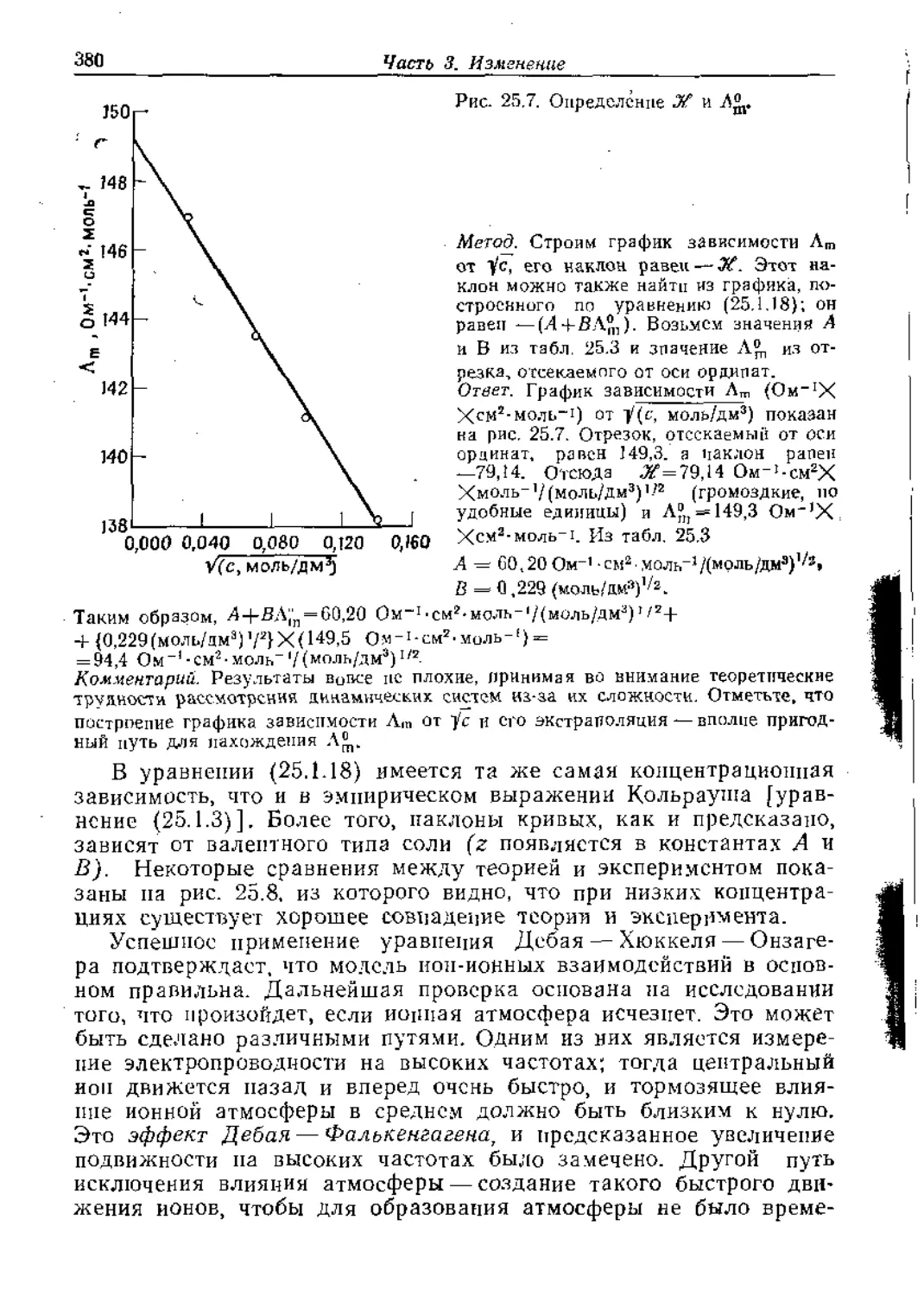
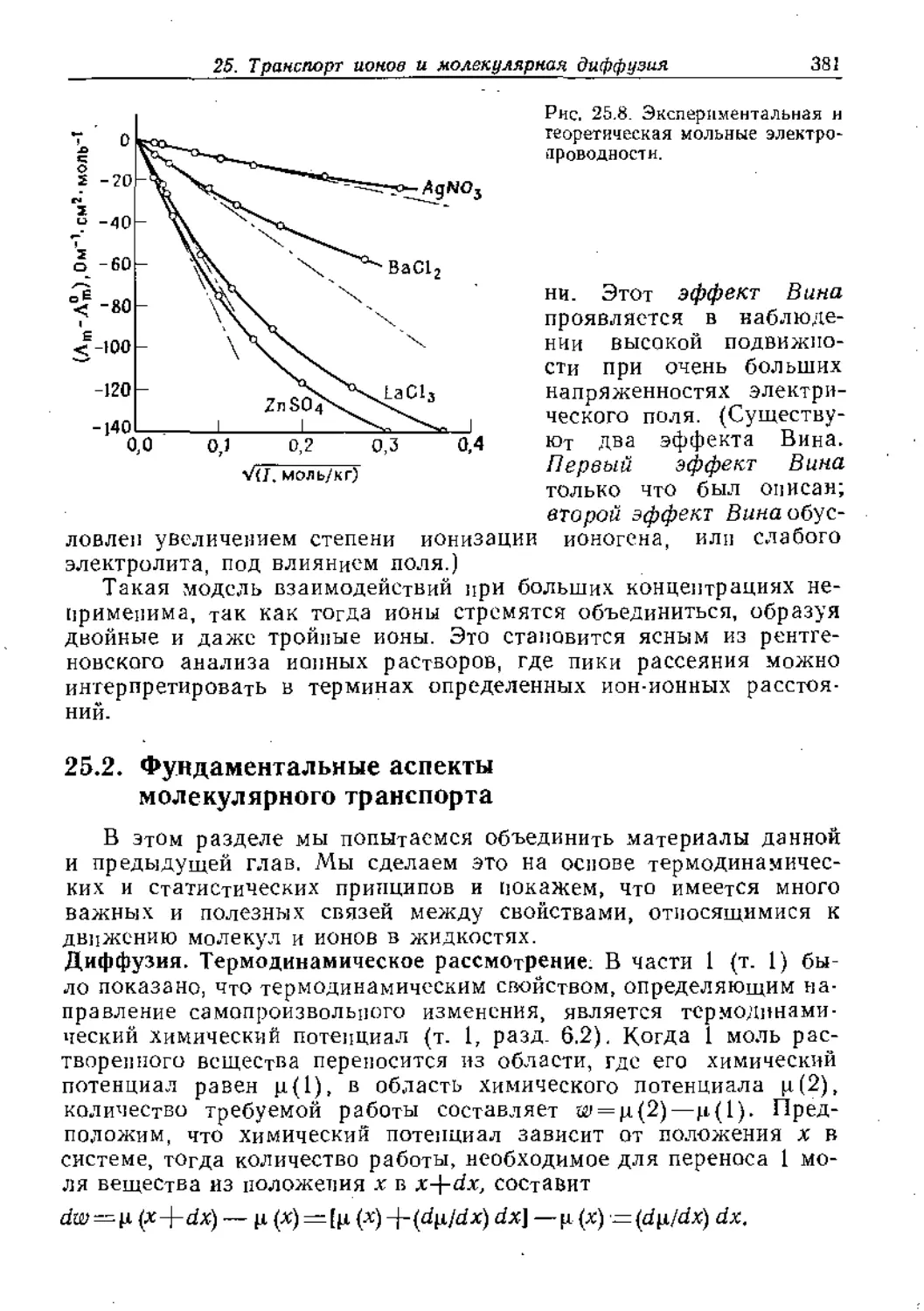






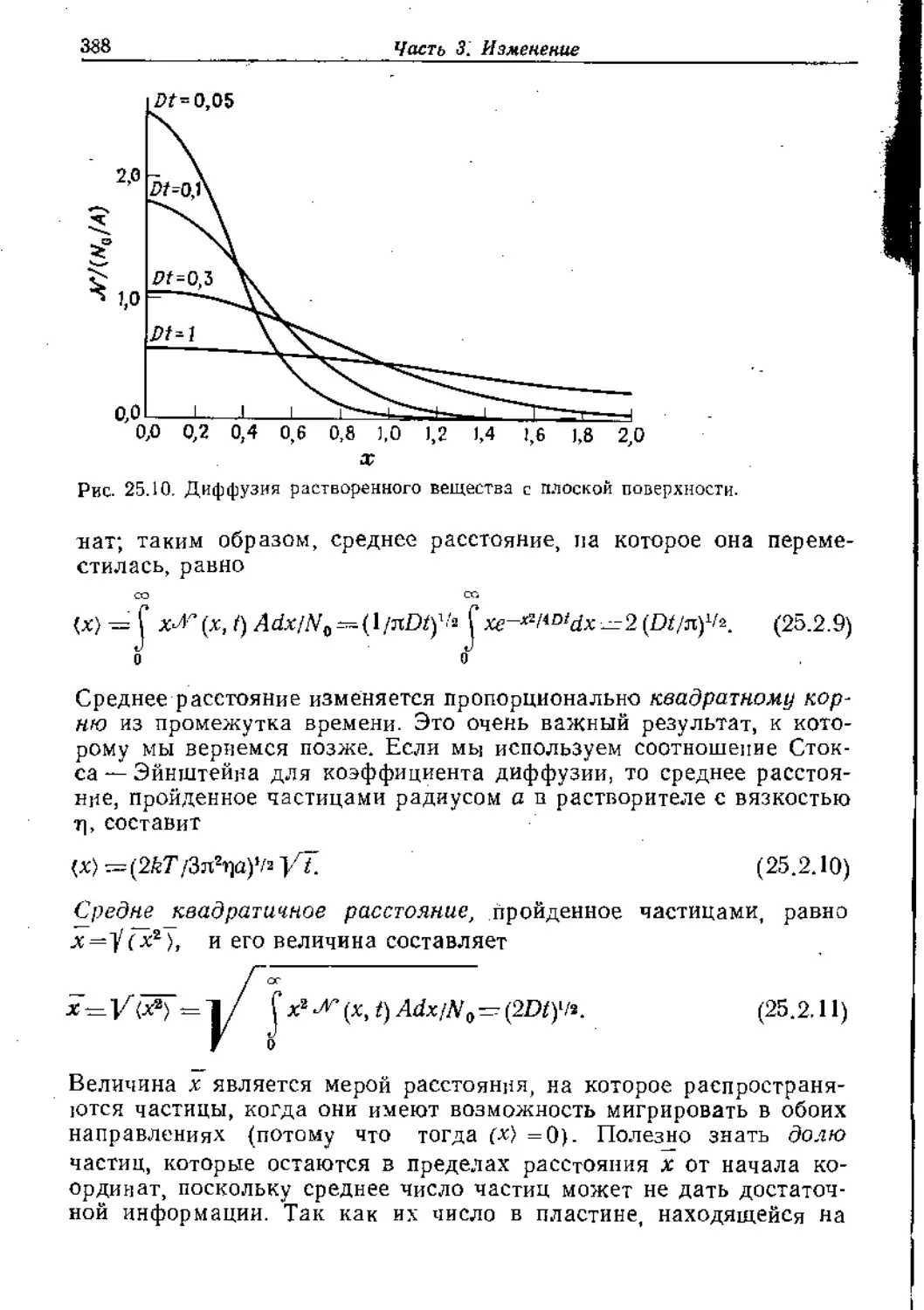

























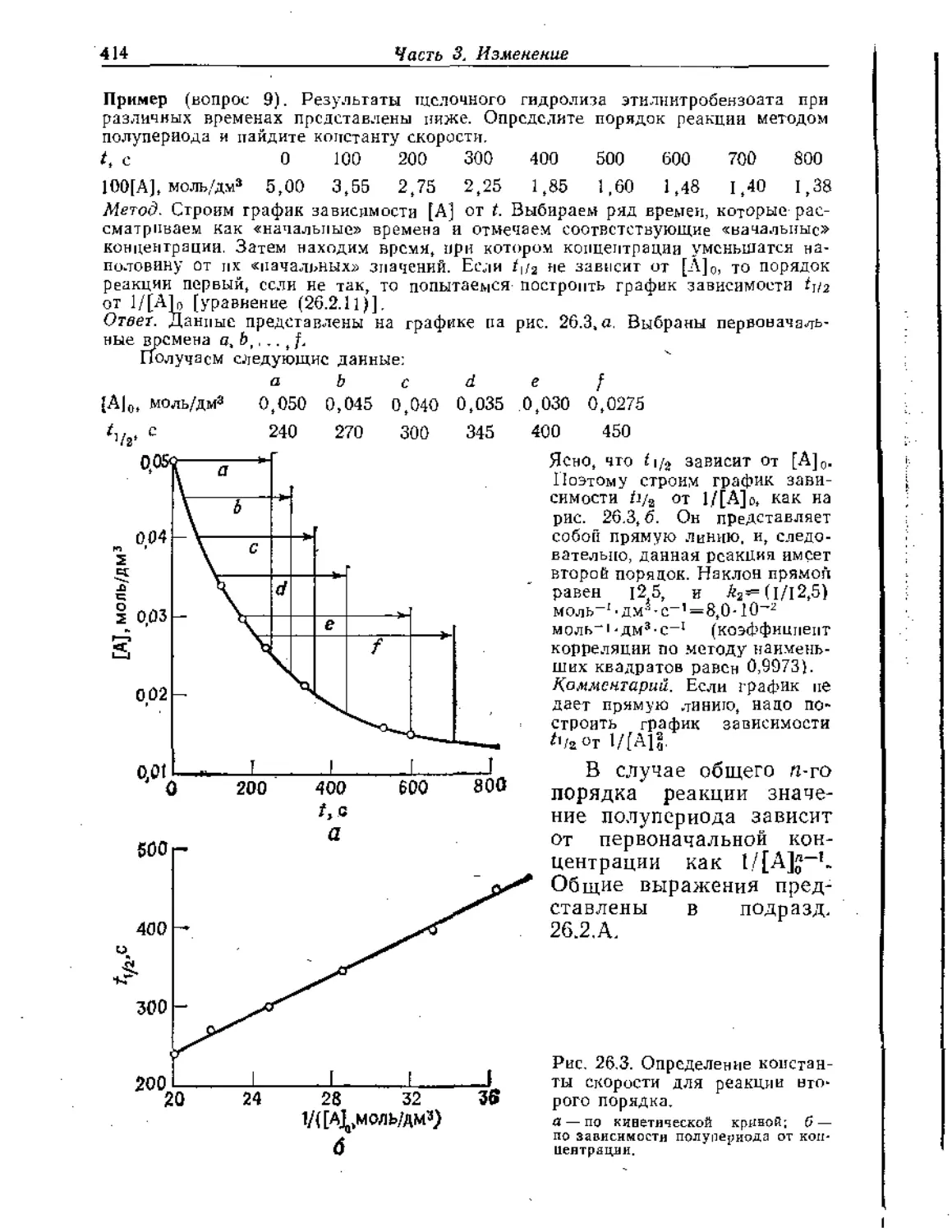

















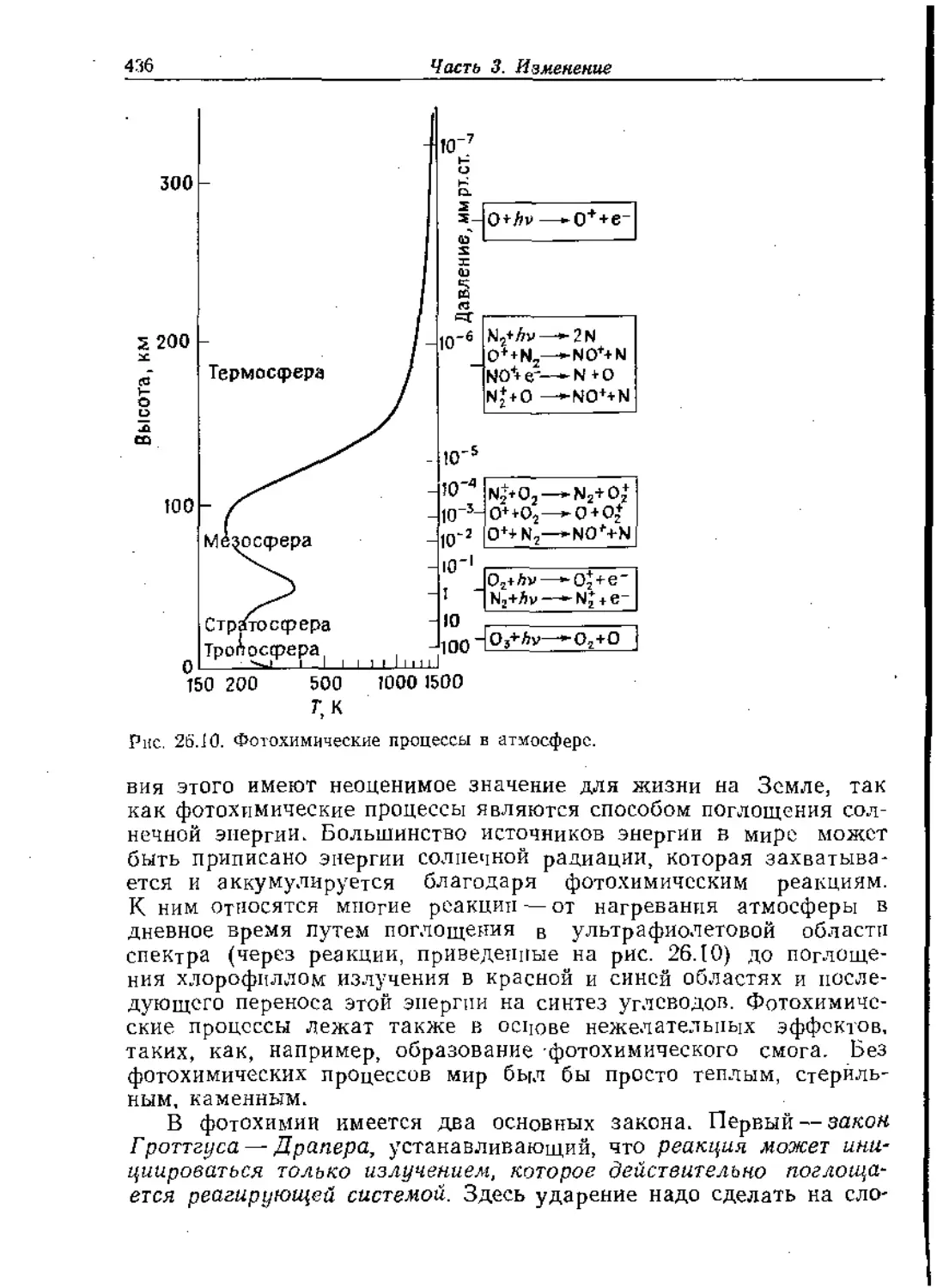





























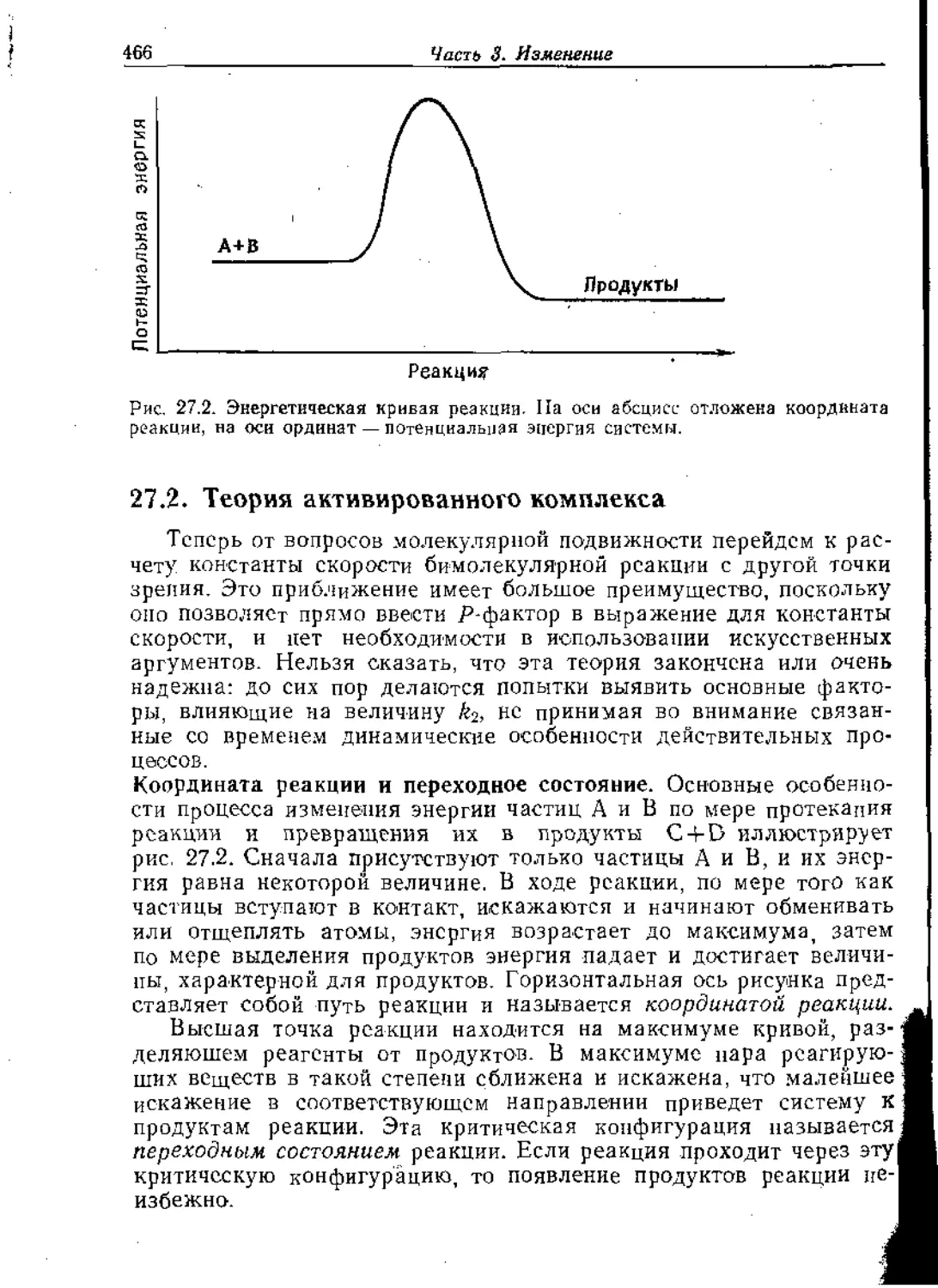






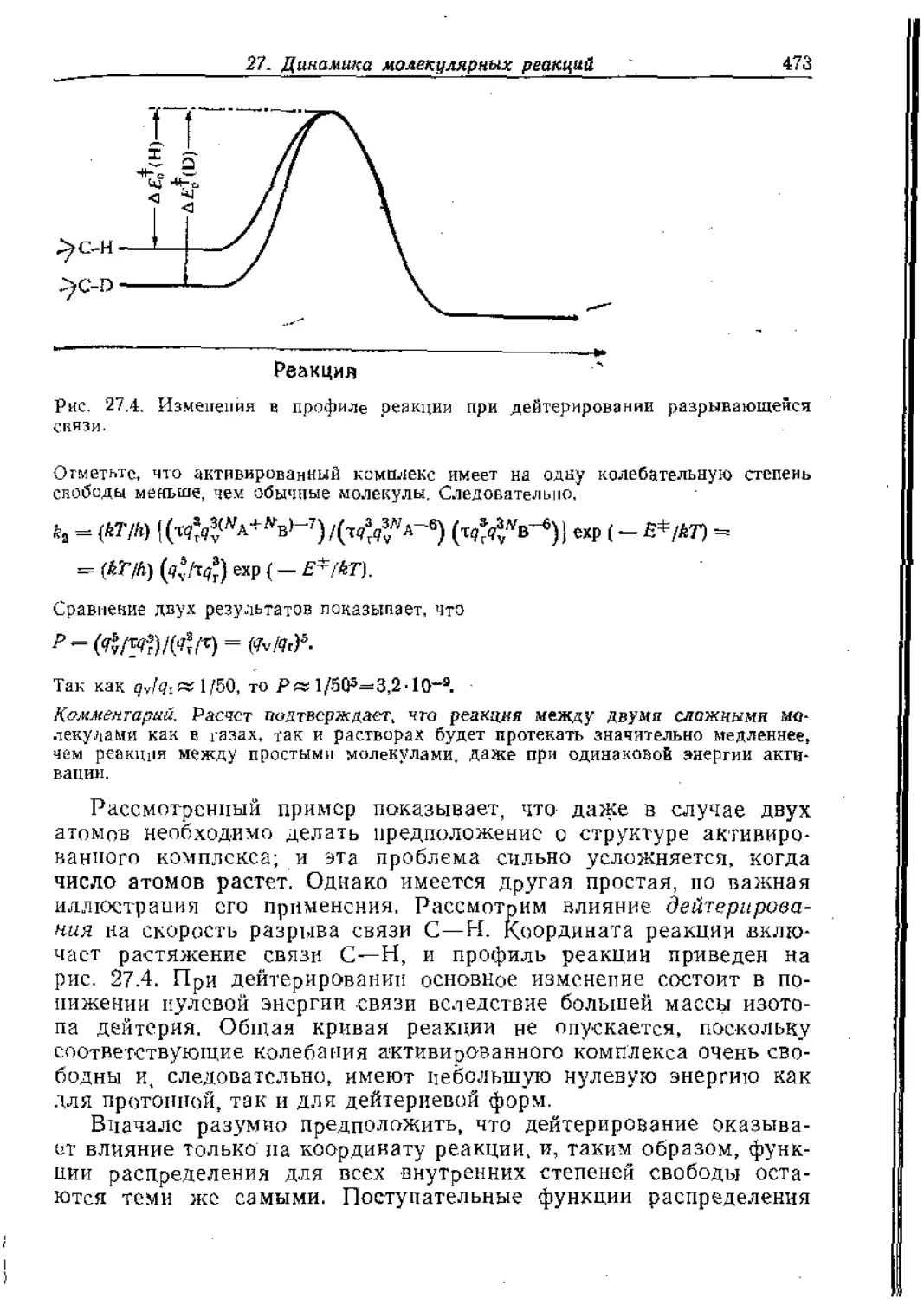




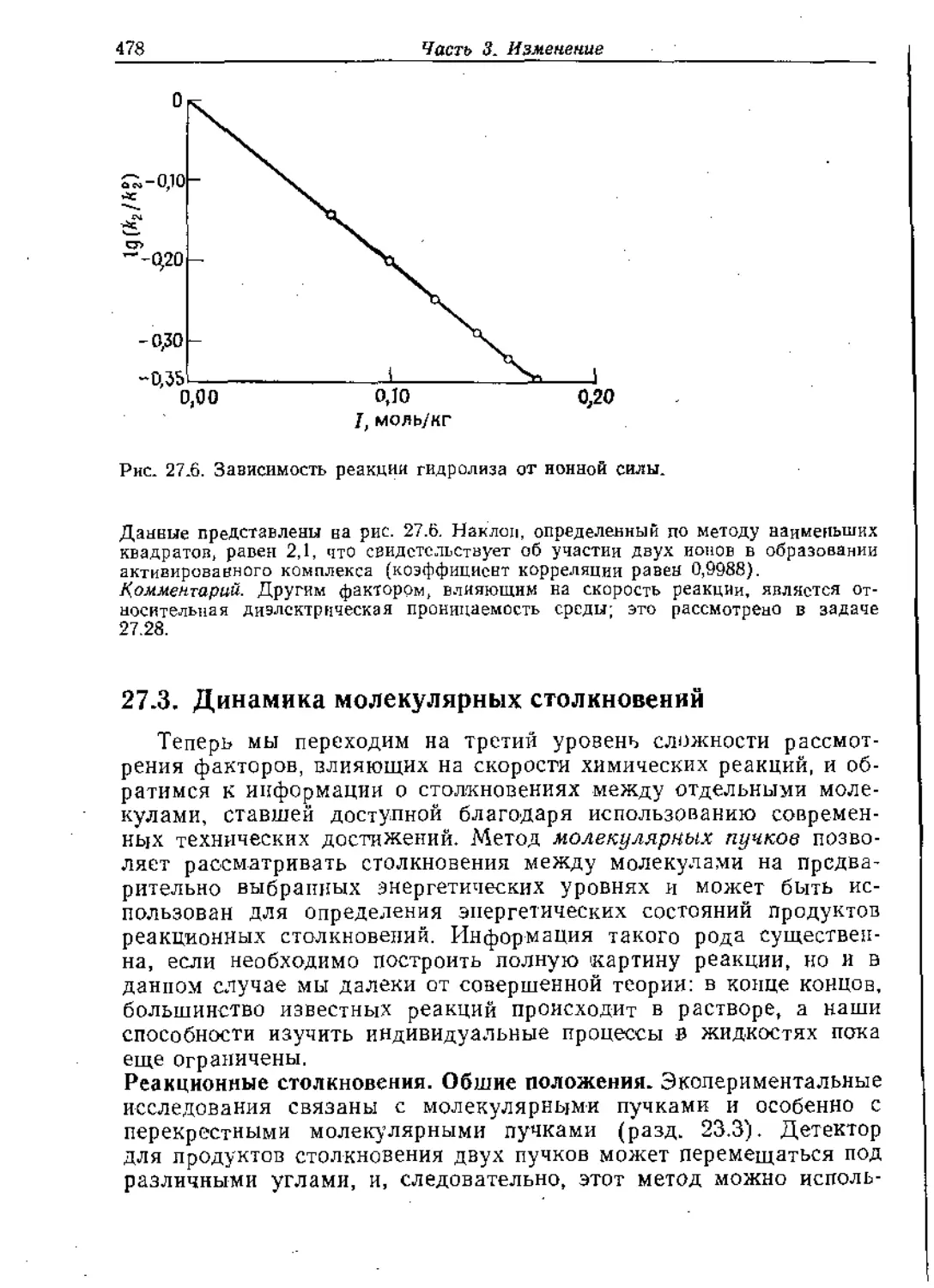


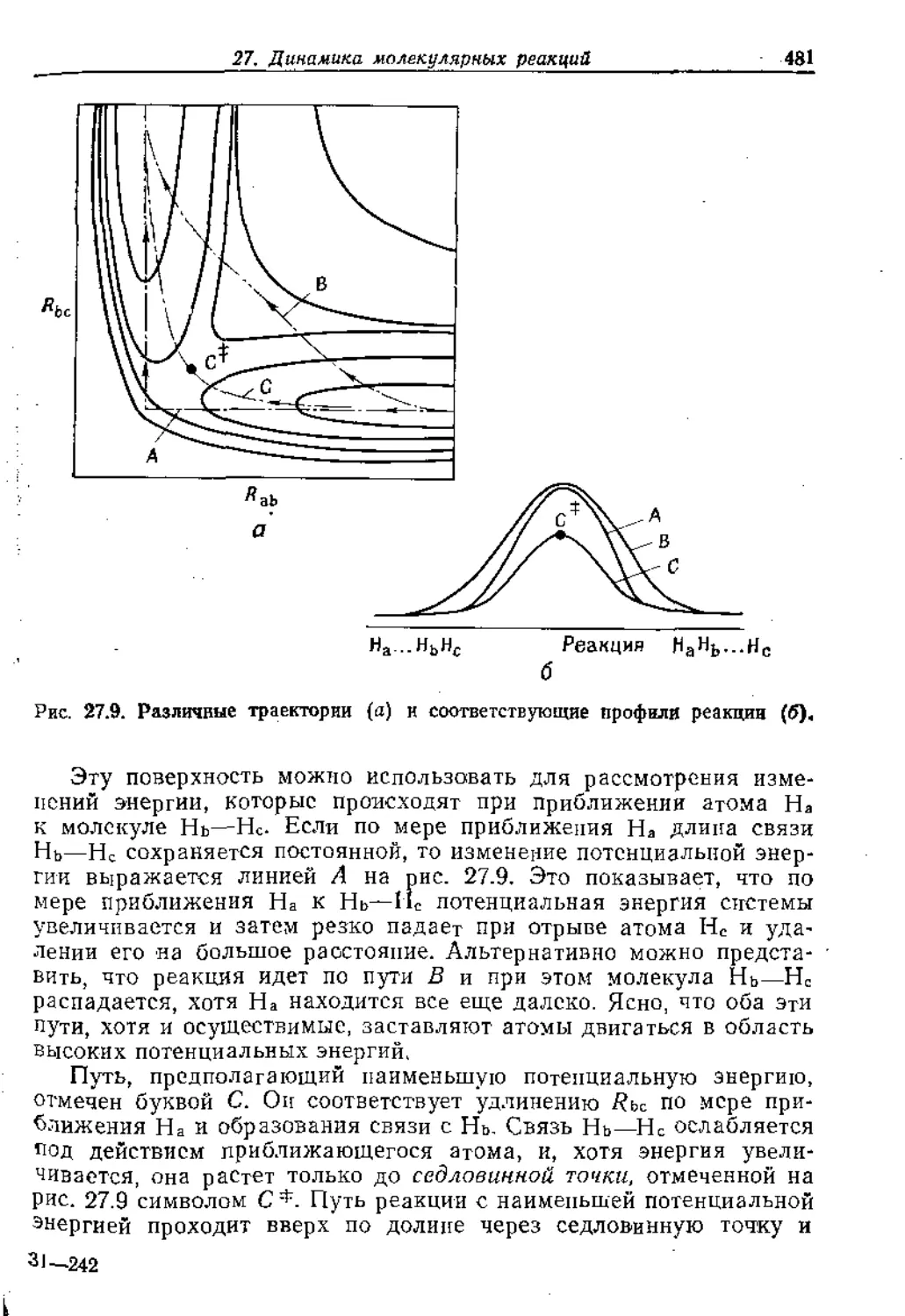














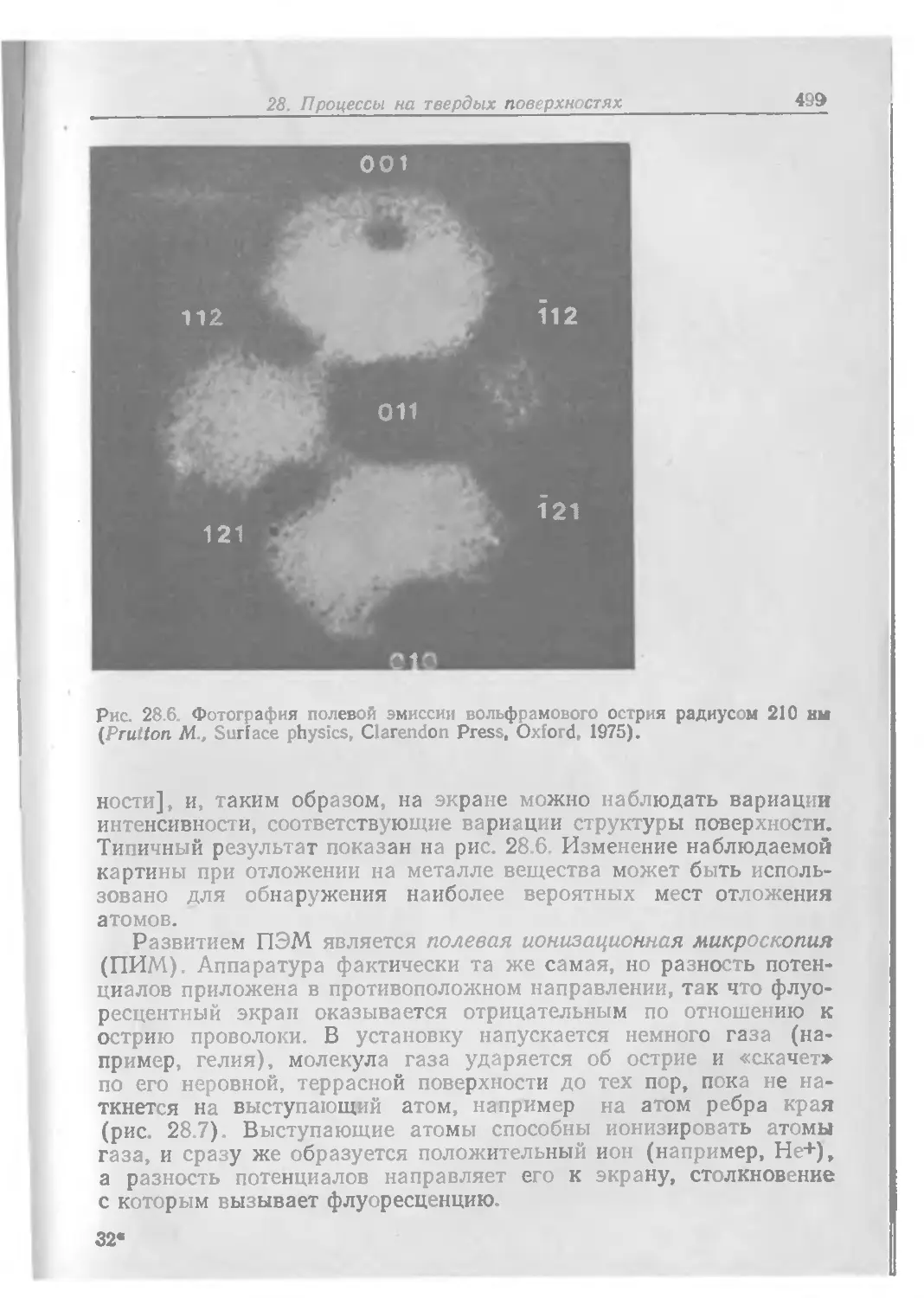

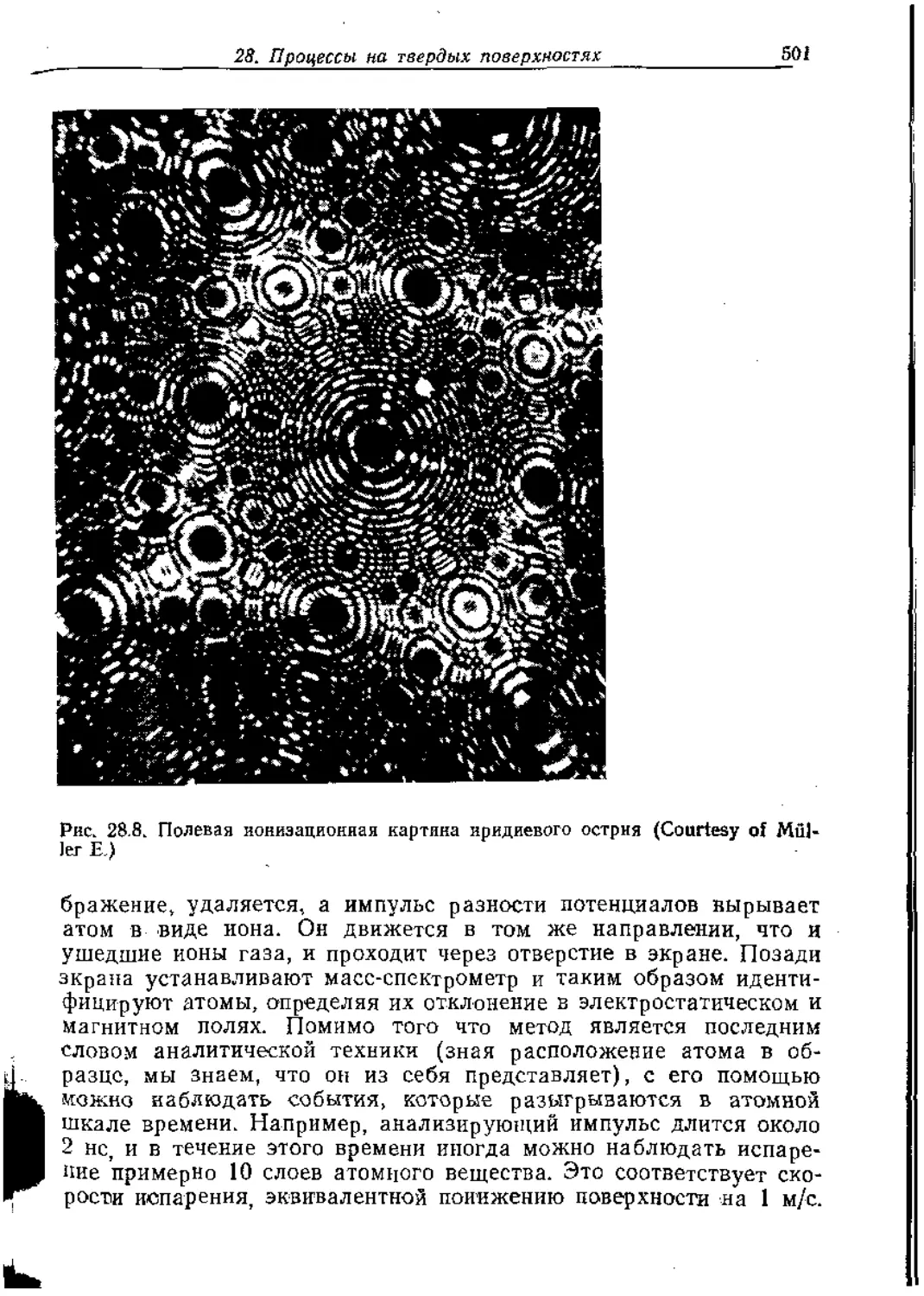



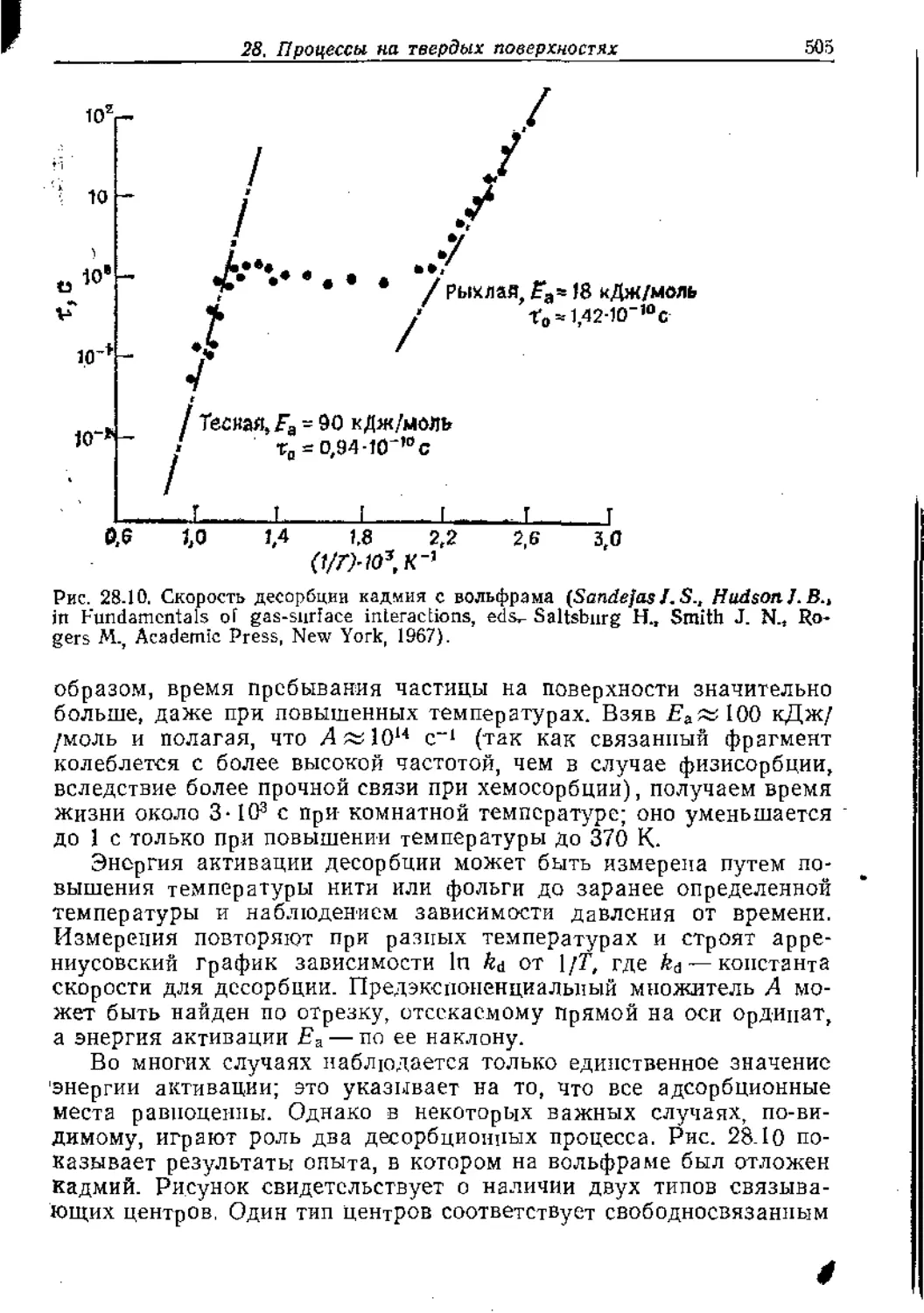



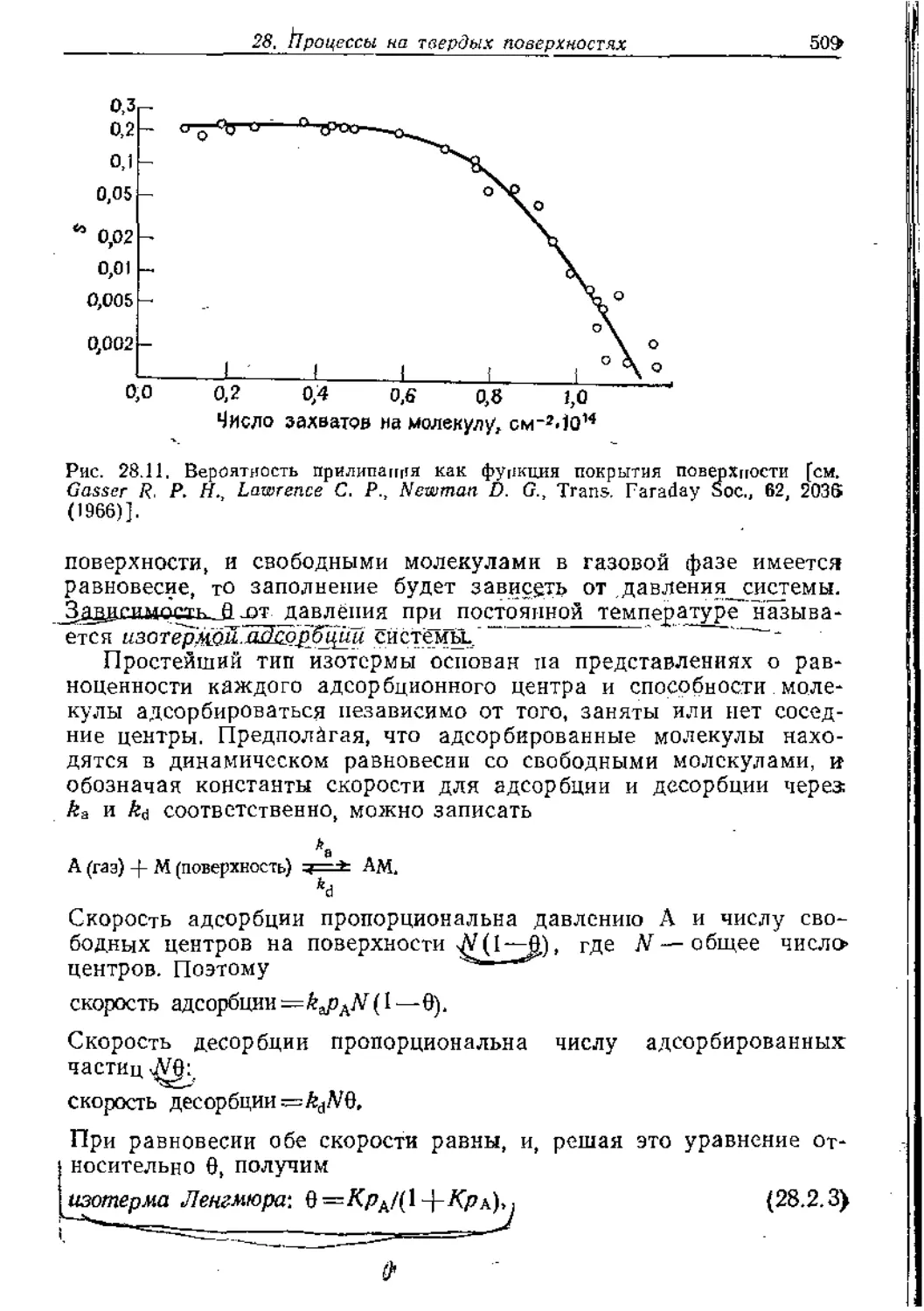
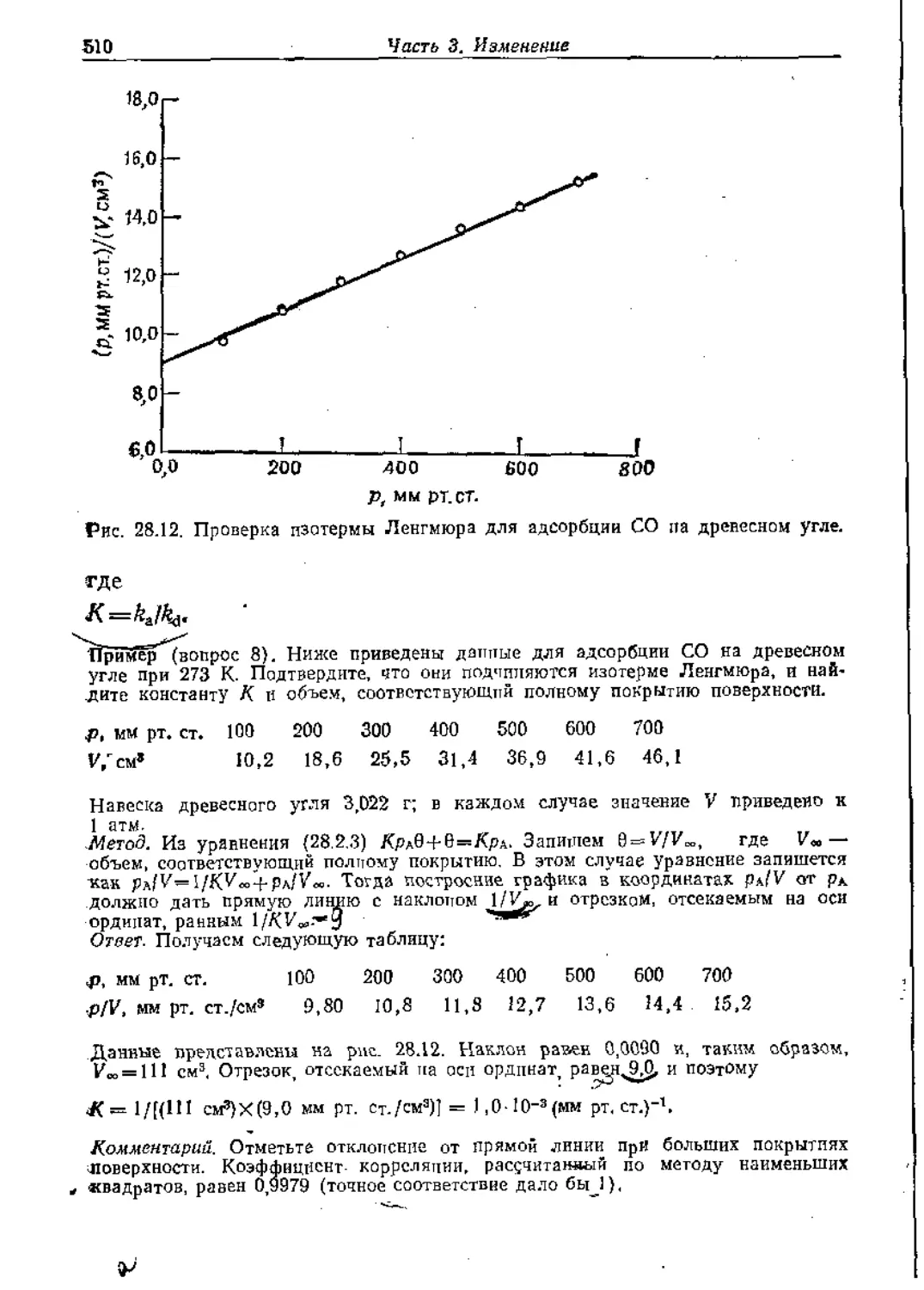
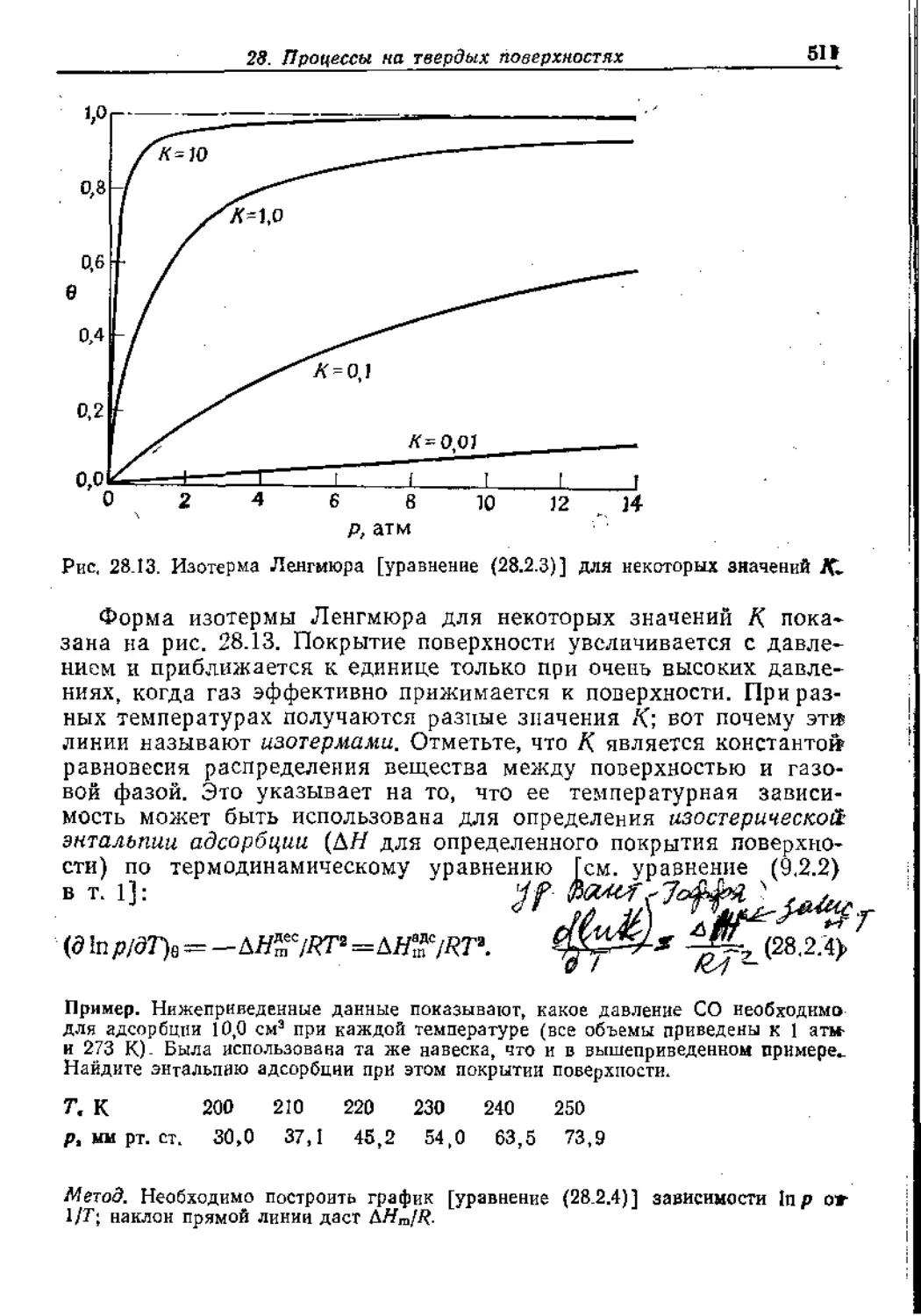
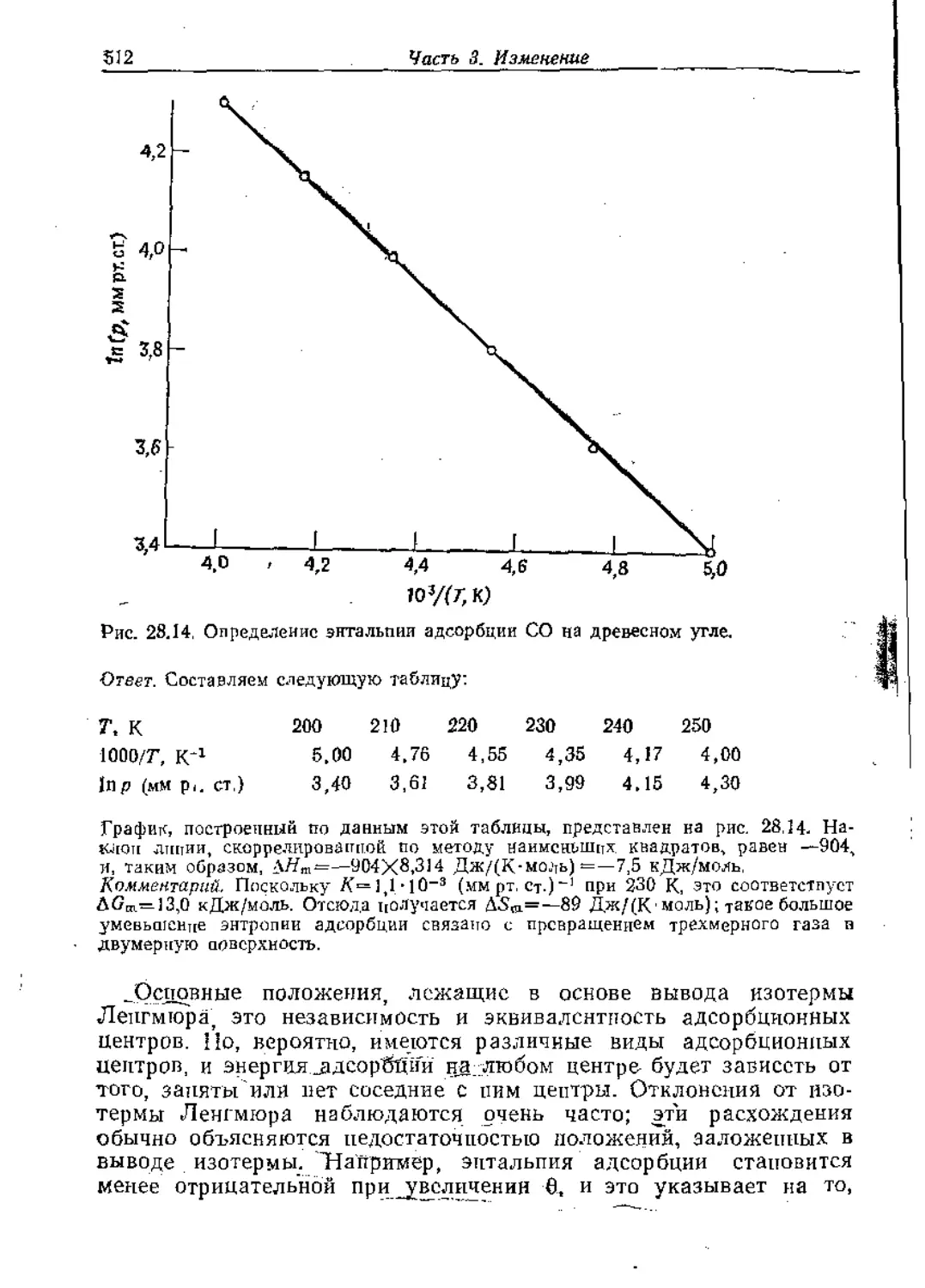
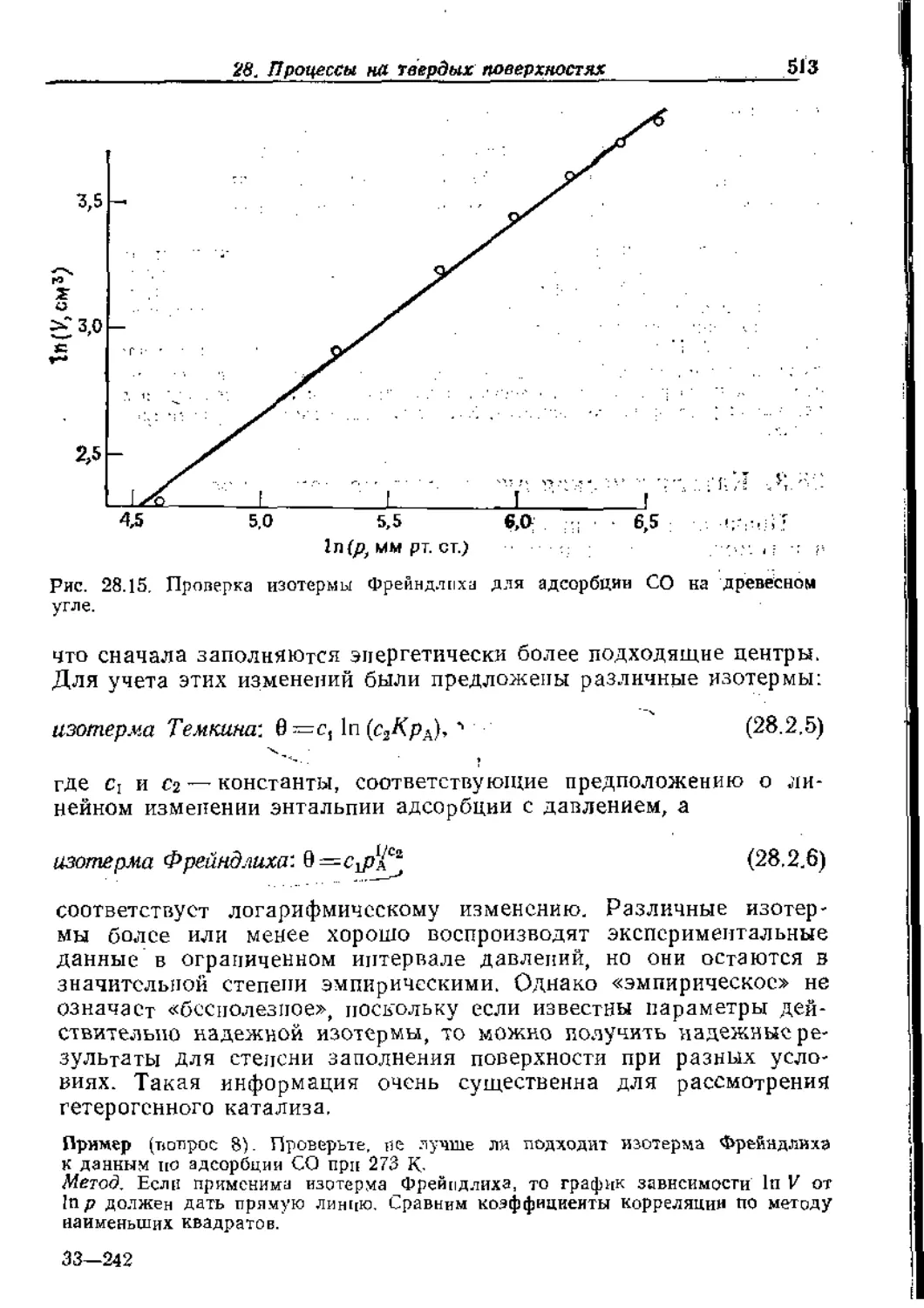




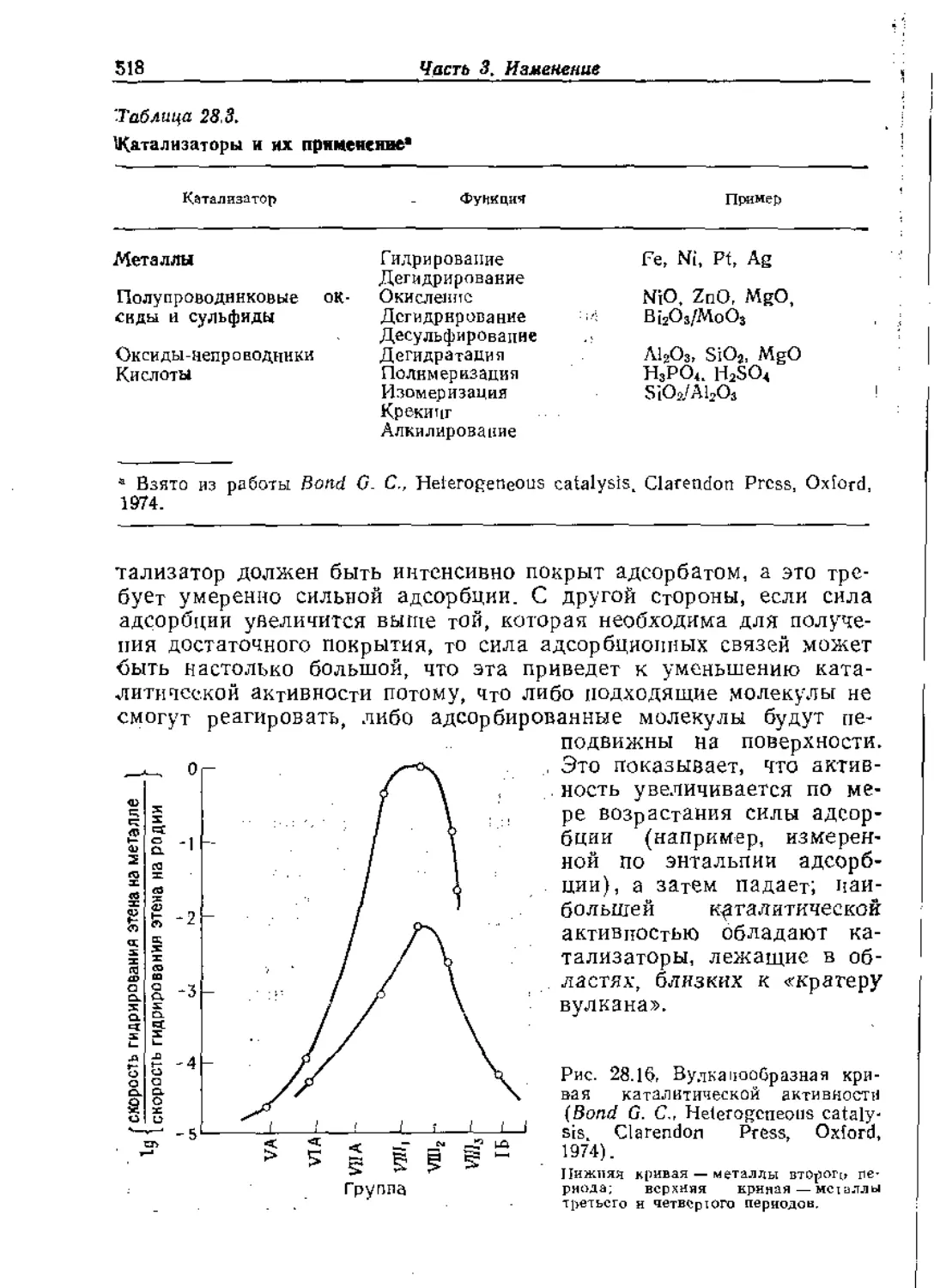











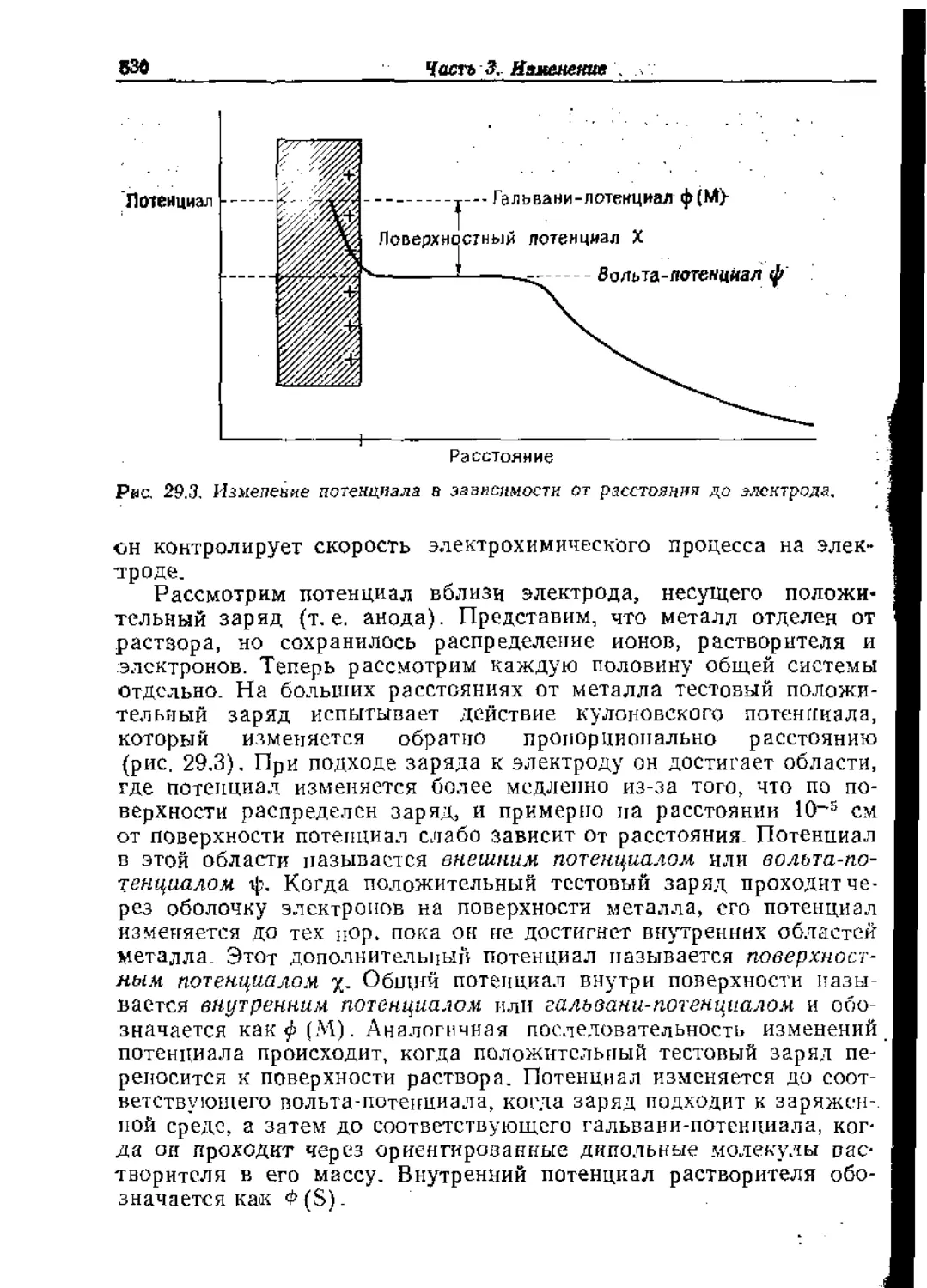







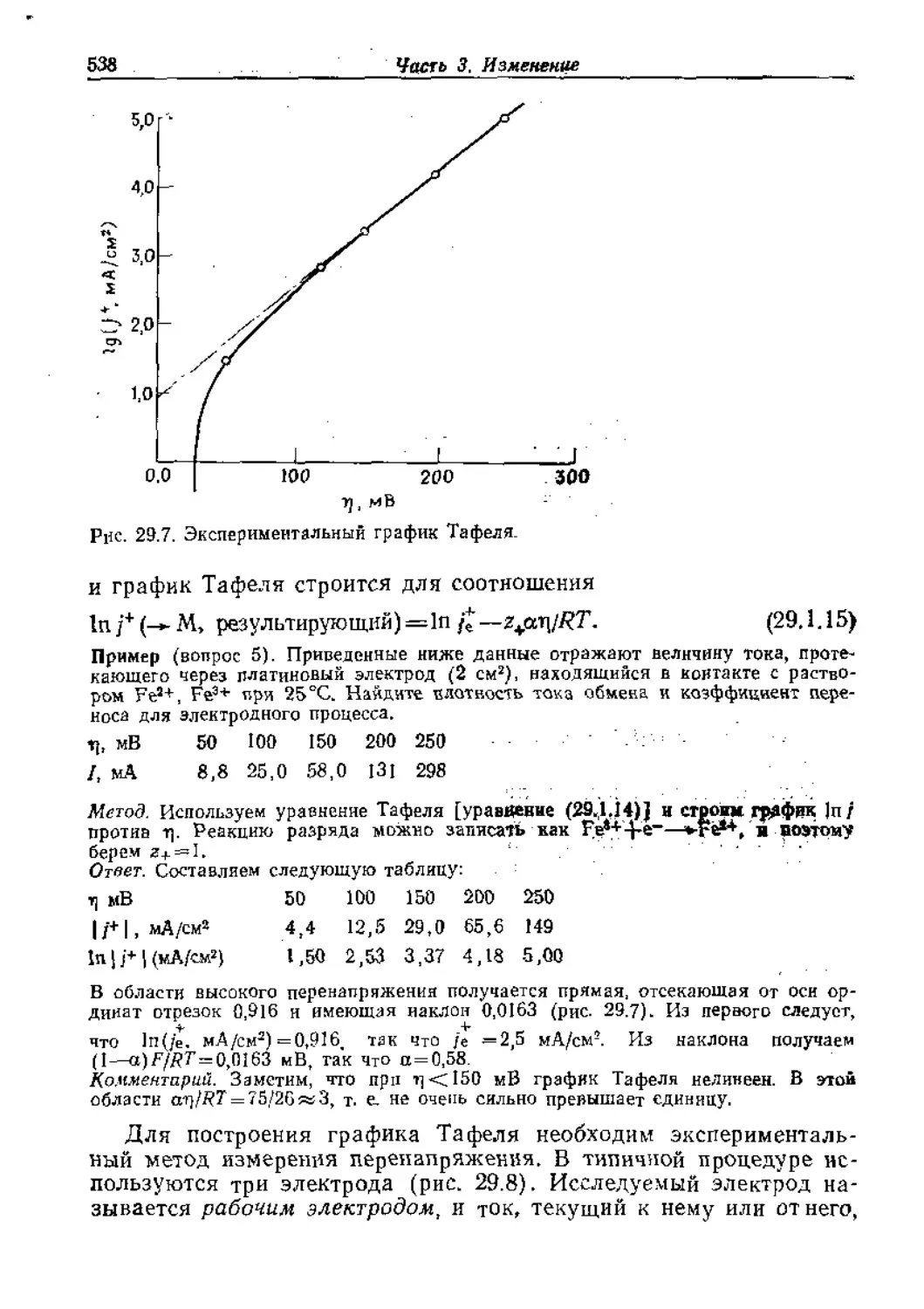




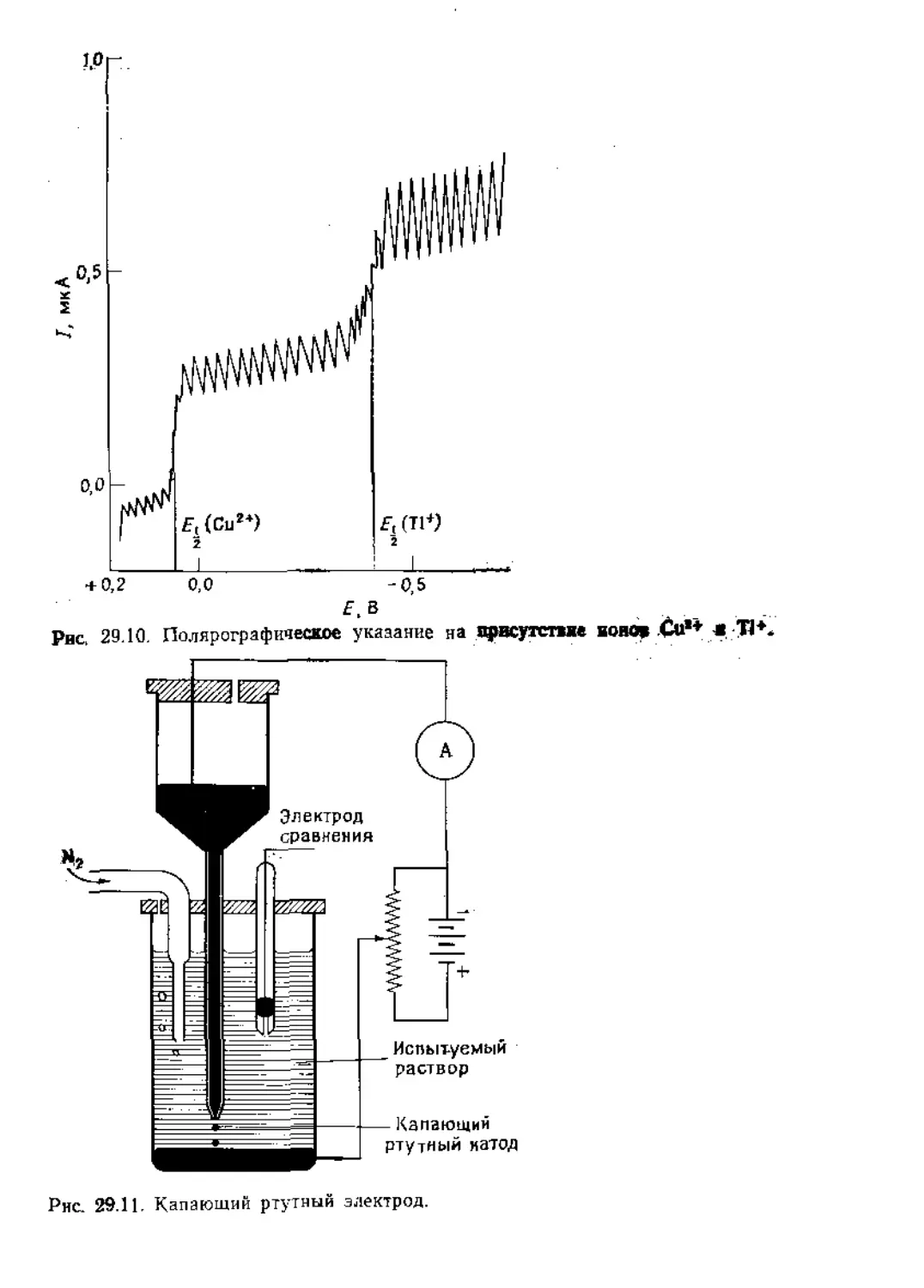





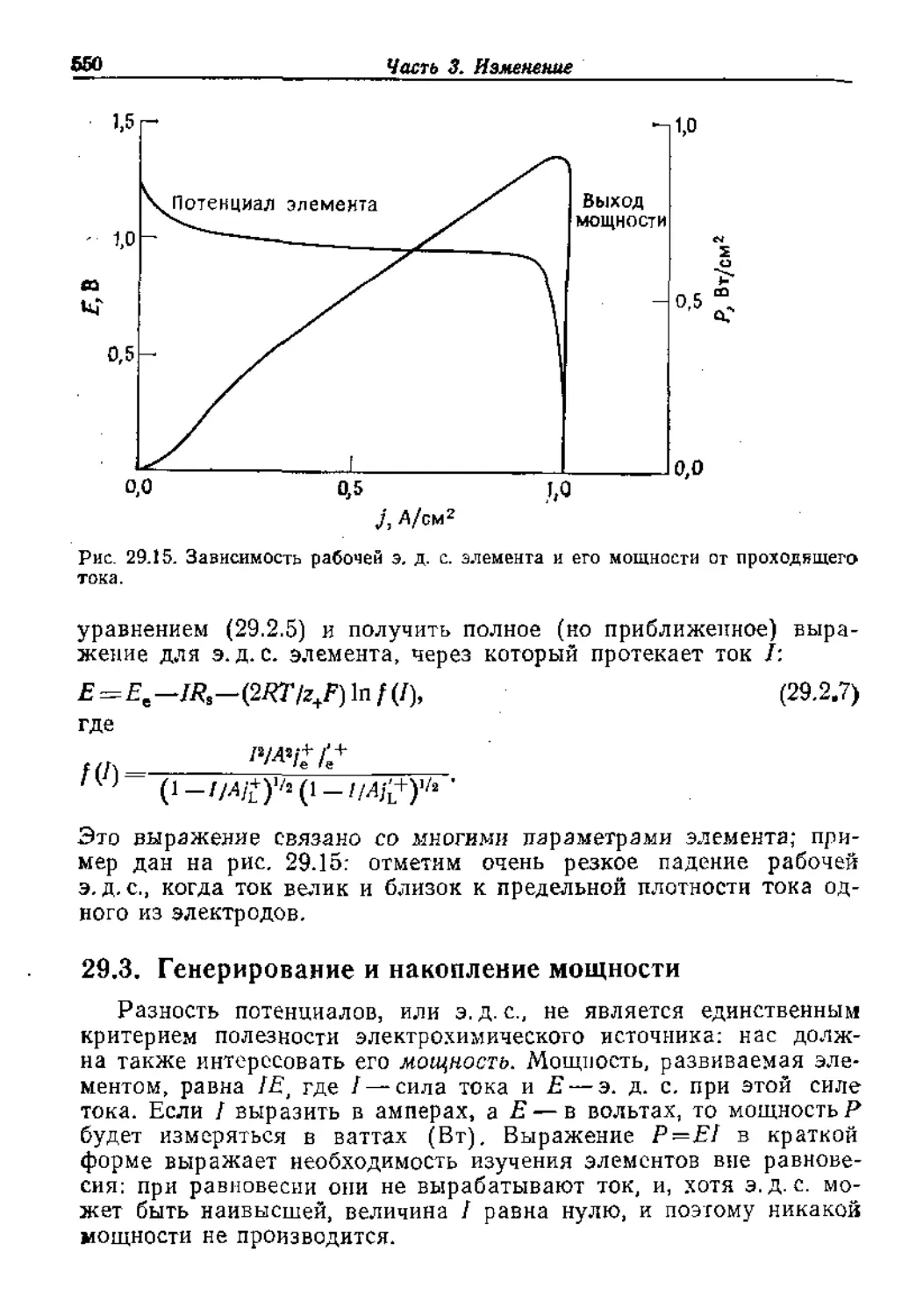





































Text
П. Эткинс___
ФИЗИЧЕСКАЯ
ХИМИЯ
P. W. ATKINS
Physical chemistry
Oxford University Press
П. Эткинс
ФИЗИЧЕСКАЯ
ХИМИЯ
2
Перевод с английского
доктора хим. наук К. И. Бутина
жму
ИЗДАТЕЛЬСТВО «МИР»
Москва 1980
УДК 541.1
Книга является монографией учебного характера
по физической химии, в которой излагаются практиче-
ски все важные вопросы этой области химии, начиная
с основ термодинамики и кончая квантовой теорией
строения молекул и физическими методами его иссле-
дования.
Имя автора хорошо знакомо советскому читателю
по другим его трудам (Спектры ЭПР и строение неор-
ганических радикалов.—М.; Мир, 1970; Кванты. Спра-
вочник концепций.— М.: Мир, 1977).
В переводе книга разделена на 2 тома. Во 2-м. то-
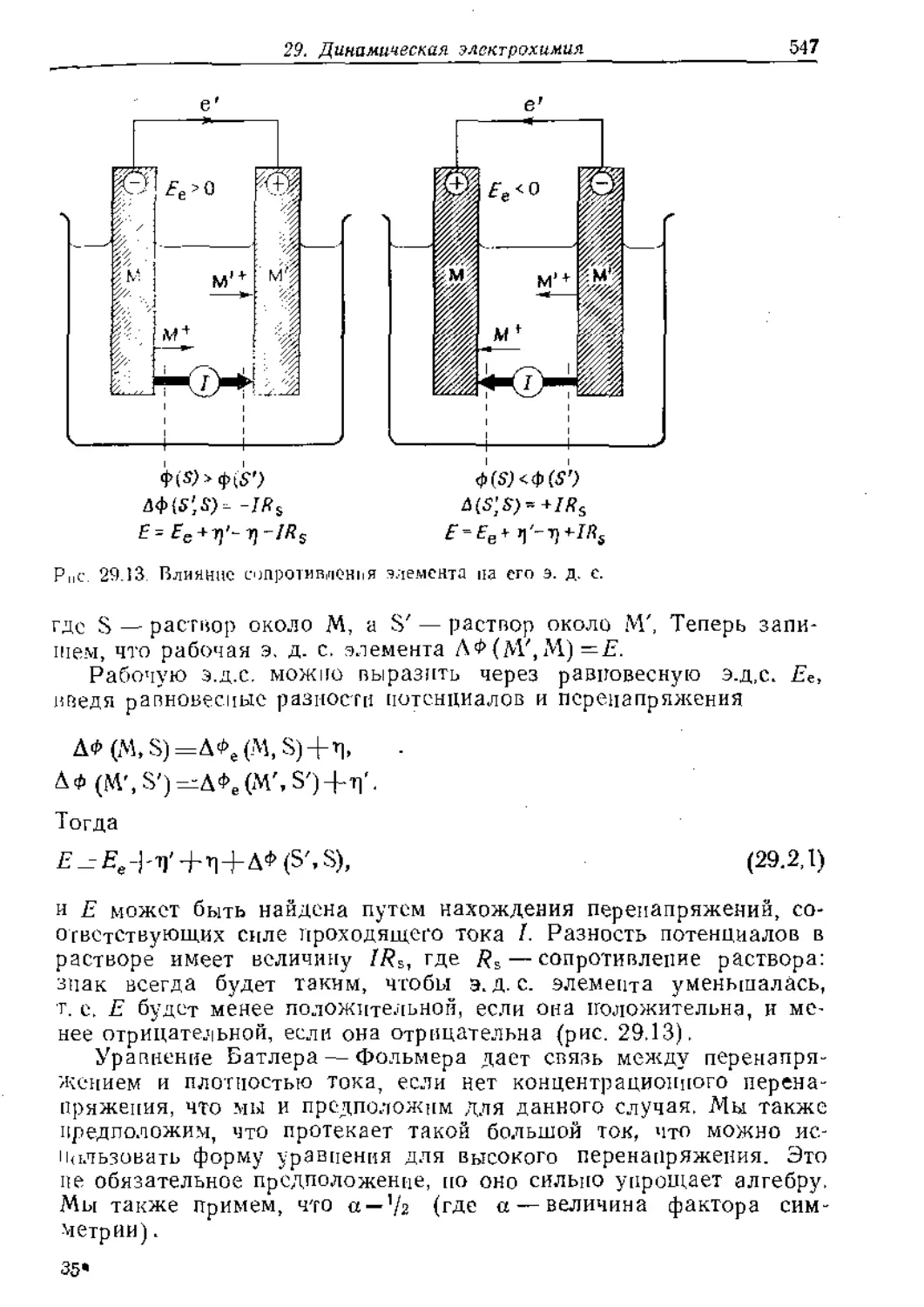
ме рассмотрены экспериментальные методы определе-
ния строения молекул (колебательная, вращательная,
электронная к резонансная спектроскопия, дифракцион-
ные методы), электрические и магнитные свойства мо-
лекул, симметрия молекул и кристаллов, статистичес-
кая термодинамика. В отдельной части даются сведе-
ния о кинетике процессов транспорта вещества, хими-
ческих л электрохимических реакций и о процессах на
твердых поверхностях.
Предназначена для студентов и аспирантов хими-
ческих и хнмико-технологических вузов, а также для
преподавателей физической химии в качестве пособия
при составлении планов лекций и семинаров.
Редакция литературы по химии
1805000000
„ 20503—484 ш
Э 041(01)—80 80
jtJb.: .С
© Р. W/ A’JHtW, $078
© Перевод на русский язык, «Мир», 1980
Часть 2. Структура
16 Симметрия. Описание и следствия
Изучаемые вопросы
После тщательного изучения этой главы вы сможете;
1, Дать определение операции симметрии и найти у данногс
тела ось симметрии, плоскость симметрии, центр симметрии илг
зеркально-поворотную ось (стр. 7—9).
2. Классифицировать молекулы по их точечной группе симмет-
рии (стр. 9).
3. Из известной группы симметрии молекулы вывести, может
ли она быть полярной или оптически активной (стр. 16).
4. Сформулировать групповое свойство (стр. 17) и матричное
представление операции симметрии (стр. 21).
5. Определить характер (стр. 22), неприводимое представле-
ние, прямую сумму (стр. 23, 24) и класс (стр. 22).
6. Вывести трансформационные свойства функций х, у, Z, х2
у2, ху, xz и т. д. (стр. 28).
7. Использовать таблицы характеров для решения вопроса:
когда интеграл должен стремиться к нулю? (стр. 31)-
8. Использовать таблицы характеров для отбора орбиталей
имеющих ненулевое перекрывание (стр. 34),
9. Использовать таблицы характеров для вывода правил от-
бора для спектральных переходов (стр. 36).
10. Использовать таблицы характеров для построения сим-
метризованных орбиталей (стр. 38).
11. Определить кристаллическую систему и класс кристалла
(стр. 40, 41).
12. Показать, что элементарная ячейка кристалла может иметь
вращательную симметрию только 1, 2, 3, 4, 6-го порядка (стр. 45).
13. Определить решетку Бравэ и различать примитивные, объ-
емно-центрированные и гранецентрированные элементарные ячей-
ки (стр. 45).
14. Определить пространственную группу, винтовую ось и
плоскость скольжения (стр. 46).
Введение
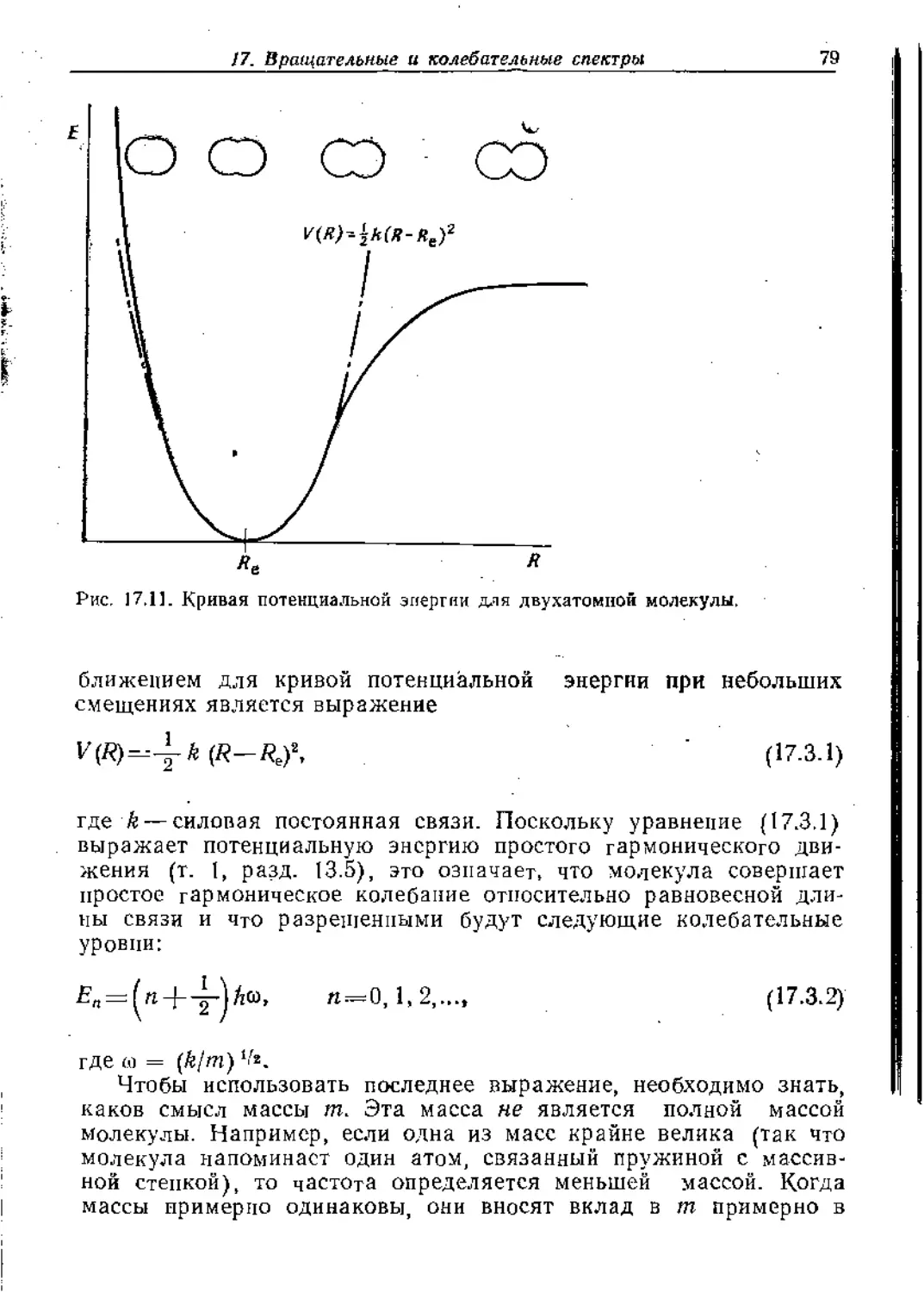
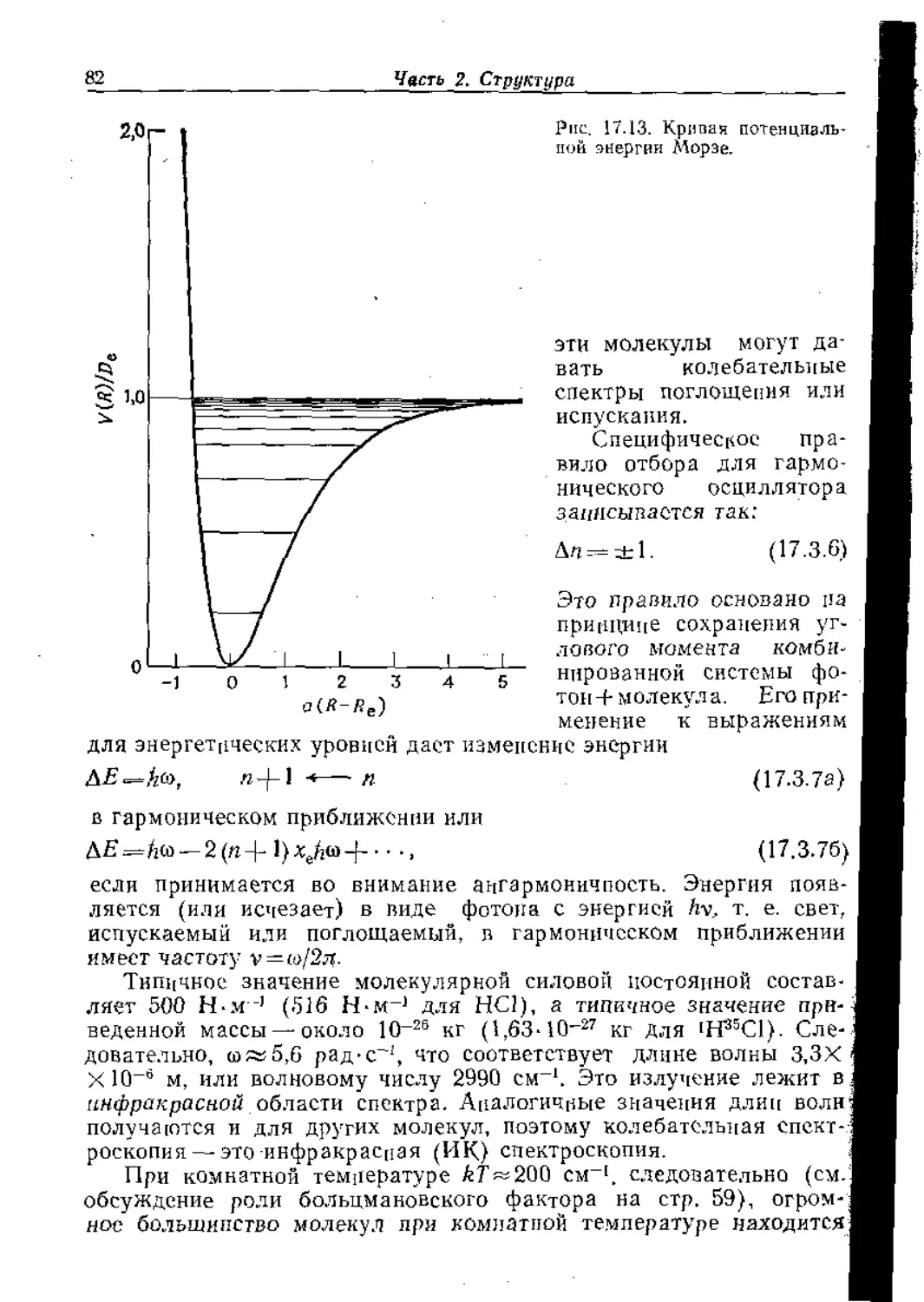
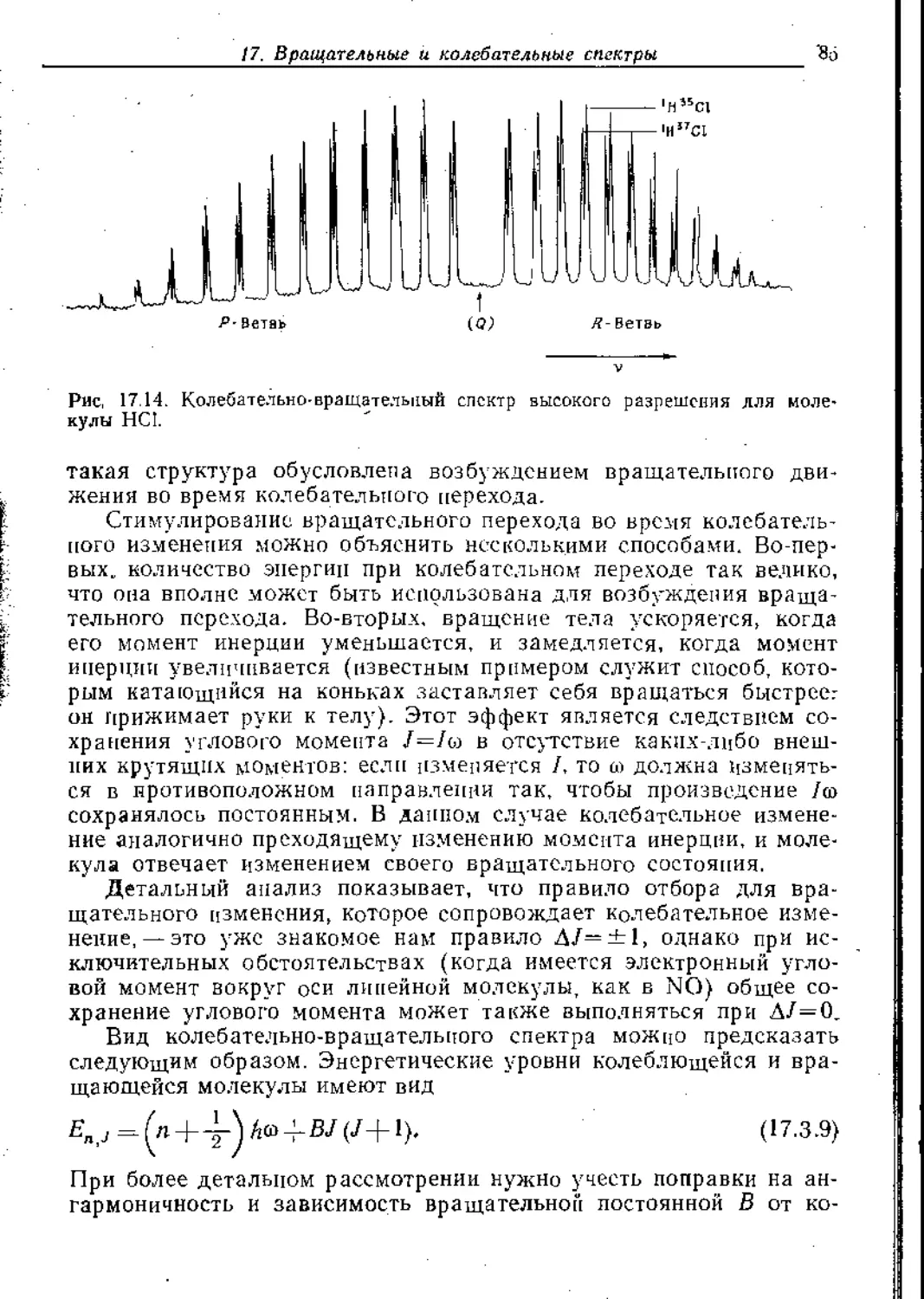
В предыдущей главе мы выяснили основополагающие кванто-
вомеханические причины, определяющие строение и форму моле-
кул. а в последующих главах мы увидим, как строение молекул
€
Часть 2. Структура
определяется экспериментально. Когда молекулы я ионы объеди-
няются в кристаллы, образуются пространные структуры с хо-
рошо выраженной симметрией. В данной главе мы сконцентри-
руем внимание на оценке формы и симметрии в общем смысле и
покажем, что симметрию молекул и кристаллов можно обсуждать
количественно. Один из результатов такого подхода — создание
системы классификации любой молекулы или кристалла в соот-
ветствии с их симметрией. Классификация, как бы полезна она
ни была, не является конечной самоцелью, н мы покажем, что та-
кой количественный подход позволяет объяснить ряд свойств мо-
лекул без детальных расчетов.
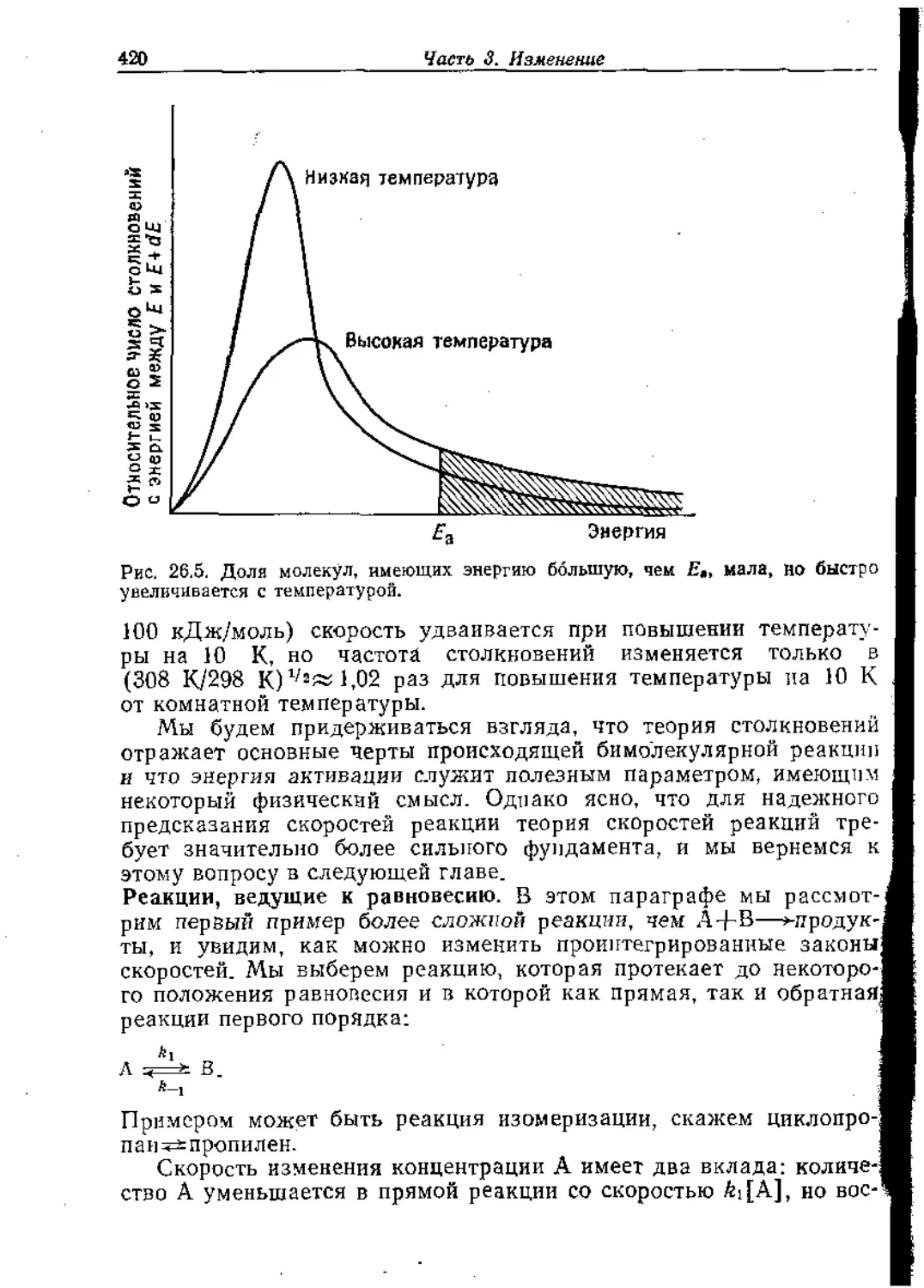
Количественное описание симметрии называется теорией
групп. В теории групп в основном рассматривается систематиче-
ское описание симметрии объекта в общем смысле, и о таком под-
ходе никогда не следует забывать. Тем не менее вследствие того,
что теория групп является систематической, ее правила можно
применять непосредственно, механически, и в некоторых случаях
получаются неожада/гяые результаты. В большинстве случаев эта
теория дает простой, прямой метод получения полезных заклю-
чений с минимальной затратой труда, и этот аспект здесь будет
особенно подчеркиваться.
I6.I. Элементы симметрии объекта
Некоторые объекты «более симметричны», чем другие. На-
пример, шар симметричнее куба, поскольку он выглядит одина-
ково при вращении на любой угол относительно диаметра, тогда
как куб выглядит одинаково, если только его повернуть на 90,
180 ли 270° относительно оси, проходящей через центры его гра-
ней (рис. 16.1), или на 120 и 240° относительно любой из четырех
осей, проходящих через противоположные углы. Аналогично мо-
лекула NH3 «симметричнее», чем Н2О,
поскольку ее вид не отличается от
первоначального после вращения ва
120 или 240° относительно оси, пока-
занной на рис. 16.2, тогда как вид
молекулы воды соответствует перво •
начальному только после вращения
на 180°. Объекты можно классифици-
ровать по их симметрии, найдя все
t Ось 2-го порядка
А Ось 3-го порядка
О Ось 4го порядка
Рис. 16.1. Некоторые элементы симметрии
куба.
16. Симметрия. Описание ц следствия
7
операции, которые приводят к одинаково выглядящему объекту;
они называются операциями симметрии или элементами симмет-
рии объекта. При таком подходе шар (сфера) следует отнести
к другой группе, чем та, в которую входит куб, а молекулу NH3 —
к другой группе, чем та, в которую входит молекула НаО.
Операции симметрии не ограничиваются вращением. Напри-
мер, шар имеет плоскость симметрии, которую можно представить
как зеркало, разрезающее шар на полушария. Отражение в этой
плоскости приводит к зеркальному отображению, не отличающе-
муся от оригинальной сферы, и это также является операцией
симметрии для шара (сферы). Эта зеркальная плоскость может
иметь любую ориентацию, проходящую через центр сферы. Одна-
ко в Н2О имеются только две зеркальные плоскости (рис. 16.3),
что вновь проясняет смысл выражения «высокая симметриям
сферы.
Для нашей цели, т. е. для обсуждения симметрии молекул,
достаточно рассмотреть пять категорий элементов:
1. Идентичность. Идентичность может показаться слишком
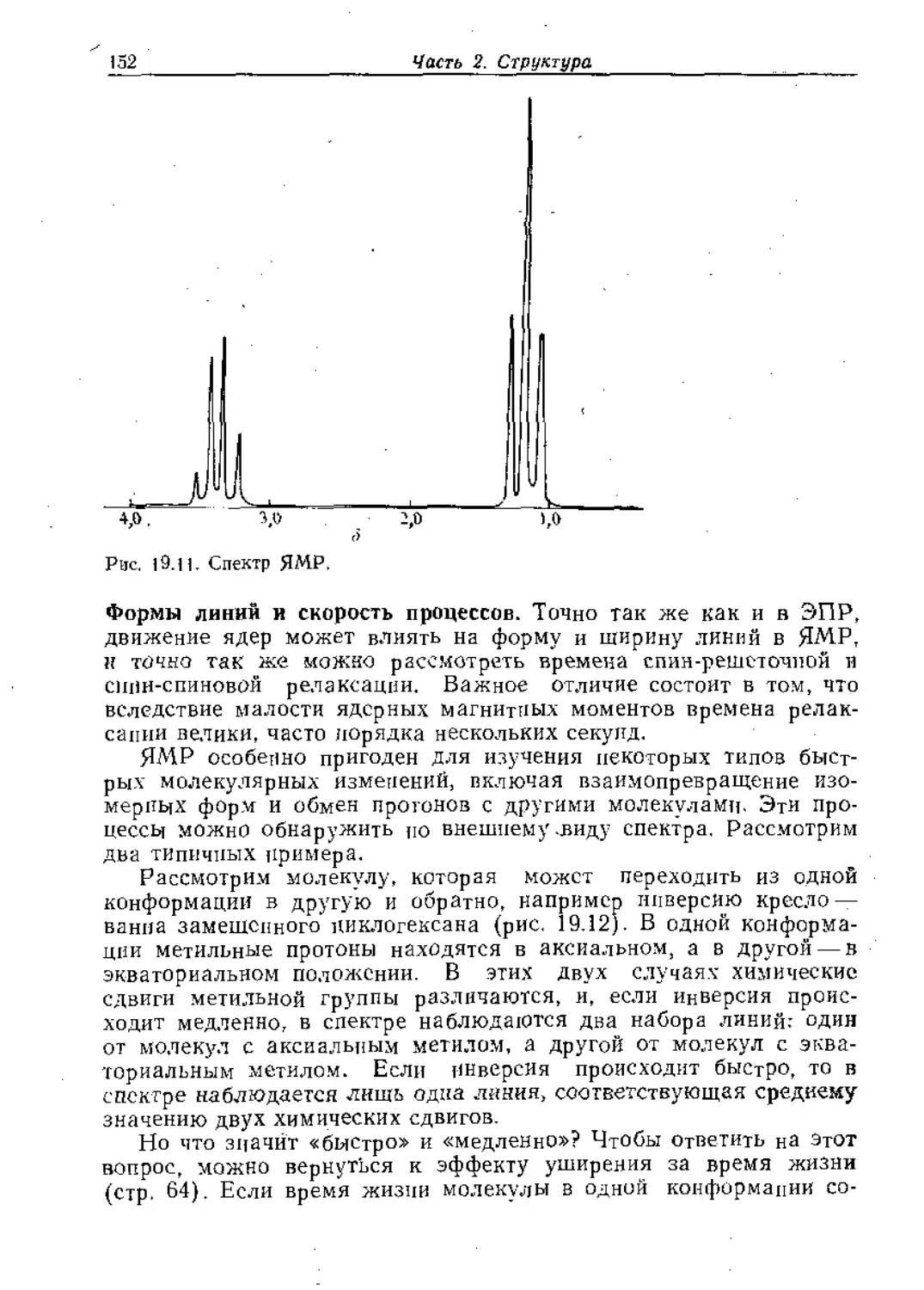
тривиальным элементом, так как при этой операции с объектом
ничего не делают. Поскольку каждый объект неотличим от са-
мого себя, если с ним ничего не делать, он обладает по крайней
мере этим элементом симметрии. Причина включения этого эле-
мента частично обусловлена желанием классифицировать каж-
дую молекулу по классу симметрии, поскольку некоторые моле-
кулы (например, CHClBrF) могут не иметь других элементов
симметрии. Другая причина техническая; она связана с форма-
лизмом теории групп. Элемент идентичности мы будем обозна-
чать через Ё.
2. Вращение вокруг оси симметрии. Если вращение на угол
36О°М приводит к неотличимой от первоначальной молекуле, то
говорят, что она имеет ось симметрии п-го порядка, а наличие
этого элемента обозначают через Сп. Так, молекула воды имеет
Рис. 16.2. Ось третьего порядка в ам-
(й) 15 второго порядка в во-
Рис. 16.3. Две зеркальные
плоскости воды.
8
Часть 2. Структура
Рис. 16.4. Диагональные зеркальные плоскости н их пример.
ось, симметрии 2-го порядка Сг, так как элементом симметрии яв-
ляется вращение на 180°=36072. Молекула аммиака имеет ось
3-го порядка Cs, так как вращение последовательно на 120° =
= 360°/3 пе изменяет ее вида. Куб имеет три оси Cit четыре оси
Сз и шесть осей С2, но даже эта высокая симметрия побивается
сферой, которая имеет любое число осей симметрии всех возмож-
ных порядков п— даже бесконечное число осей Сж, поскольку
вращение на бесконечно малый угол (ЗбО’М, п—*-оо) есть опера-
ция симметрии, и ось такого вращения может иметь любую ори-
ентацию.
3. Отражение в плоскости симметрии (зеркальной плоскости).
Если при отражении в зеркальной плоскости, проходящей через
молекулу, последняя остается неотличимой от начальной формы,
то говорят, что молекула обладает плоскостью симметрии, кото-
рая обозначается буквой сг. Зеркальные плоскости могут иметь
разную ориентацию по отношению к возможным осям симметрии
молекулы. Например, в Н2О имеются две зеркальные плоскости,
и каждая проходит через ось С2 (рис. 16.3). Когда зеркальная
плоскость проходит через главную ось симметрии (т. е. через ось,
которая имеет высшую величину п, если осей несколько), она
обозначается как сг„ (о означает «вертикальный»). В Н2О обе
плоскости являются ст?; в NH3 три плоскости оЕ.. Когда зеркаль-
ная плоскость перпендикулярна главной оси, так что о —горизон-
тальная плоскость, если Сп — кертнкзлънан ось, ее обозначают
как (Тд. Например, молекула бензола имеет ось и плоскость он
(а также некоторые другие элементы симметрии). Другая воз-
можность появляется, когда зеркальная плоскость вертикальна и
проходит через главную ось, но обладает еще одной особенностью:
делит пополам угол между двумя осями Сг, которые сами пер-
16, Симметрия. Описание и следствия
>9
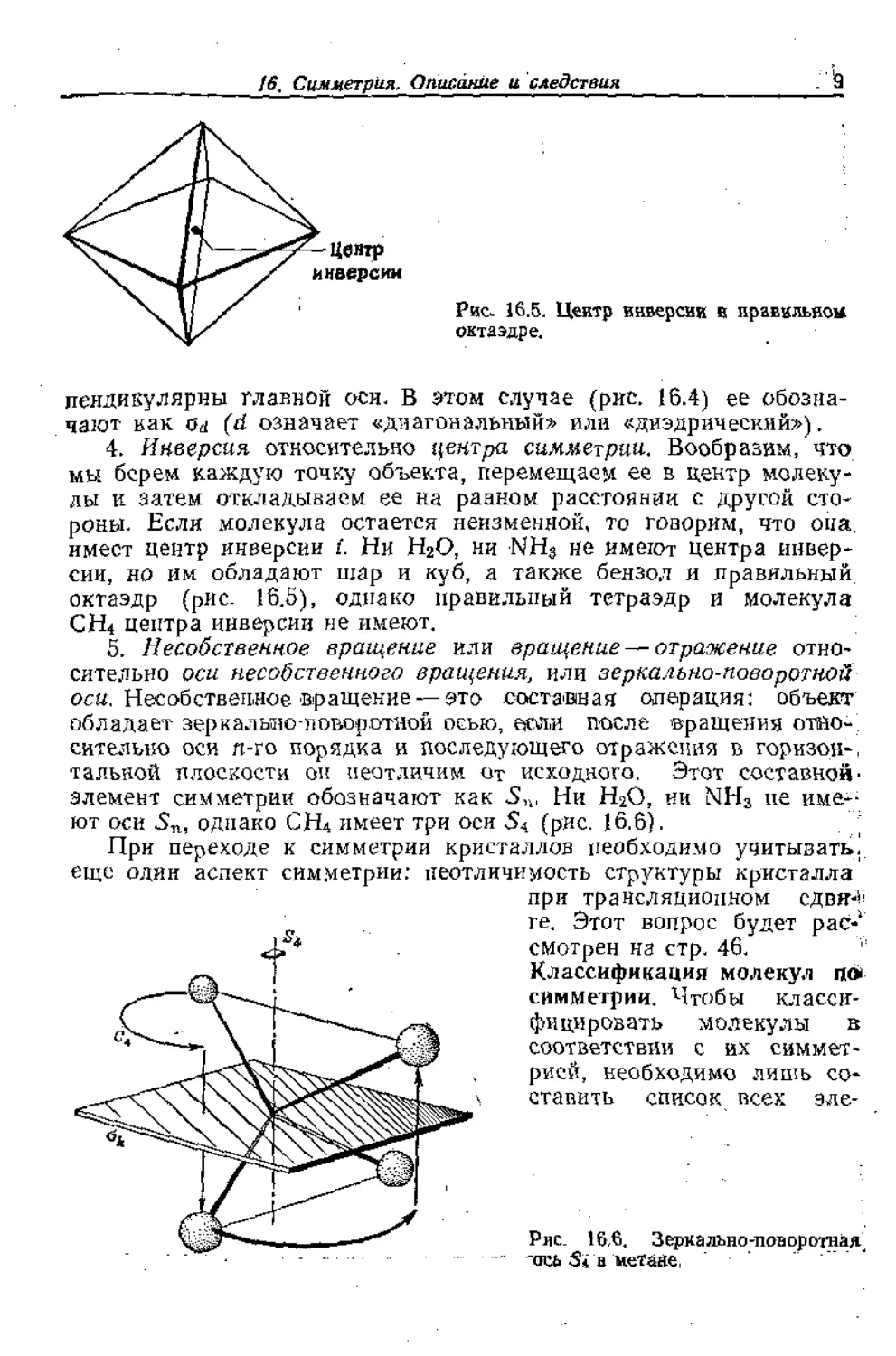
Рис. 16.5. Центр инверсии в правильной
октаэдре.
пендикулярны главной оси. В этом случае (рис. 16.4) ее обозна-
чают как Gd (d означает «диагональный» или «диэдрический»).
4. Инверсия относительно центра симметрии. Вообразим, что
мы берем каждую точку объекта, перемещаем ее в центр молеку-
лы к затем откладываем ее на ранном расстоянии с другой сто-
роны. Если молекула остается неизменной, то говорим, что она.
имеет центр инверсии i. Ни НаО, ни NH3 не имеют центра инвер-
сии, но им обладают шар и куб, а также бензол и правильный
октаэдр (рис. 16.5), однако правильный тетраэдр и молекула
СН4 центра инверсии не имеют.
5. Несобственное вращение или вращение — отражение отно-
сительно оси несобственного вращения, или зеркально-поворотной
оси. Несобственное вращение — это составная операция: объект
обладает зеркально-поворотной осью, если после вращения отйо-
сительно оси п-го порядка и последующего отражения в горизон-,
тальной плоскости он неотличим от исходного. Этот составной'
элемент симметрии обозначают как Ни Н^О, ни NH3 ие име-
ют оси Su, однако СНд имеет три оси 5< (рис. 16.6).
При переходе к симметрии кристаллов необходимо учитывать^
еще один аспект симметрии: неотличимость структуры кристалла
при трансляционном сдви^
те. Этот вопрос будет рас-1
смотрен на стр. 46.
Классификация молекул пей
симметрии. Чтобы класси-
фицировать молекулы в
соответствии с их симмет-
рией, необходимо лишь со-
ставить список всех эле-
Рлс. 16.6. Зеркально -поворотная,
'ось 3* в Метане, ’
10
Часть 2. Структура
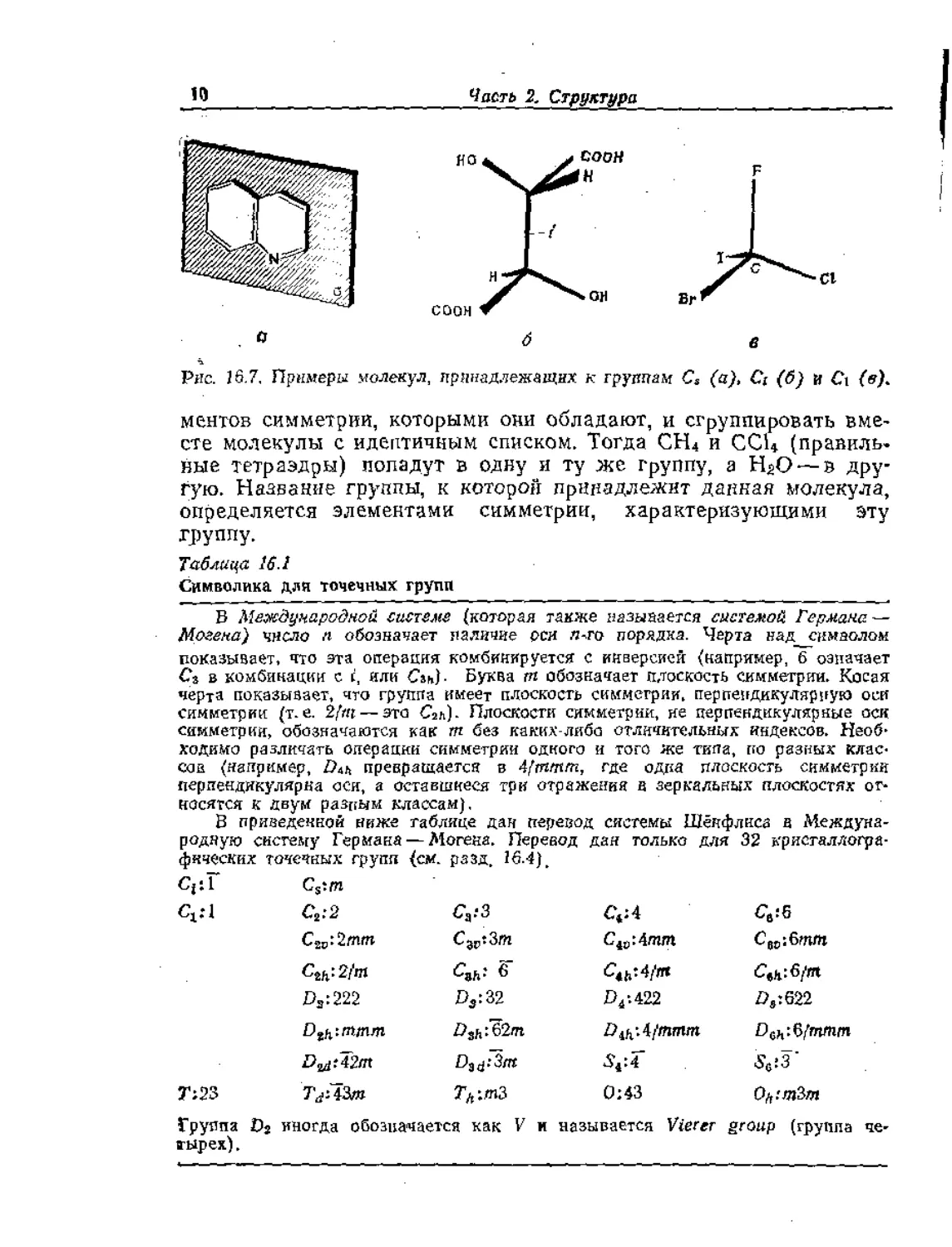
Рис. 16.7, Примеры молекул, принадлежащих к группам С3 (a), Ci (б) и Ct (в).
ментов симметрии, которыми они обладают, и сгруппировать вме-
сте молекулы с идентичным списком. Тогда СН4 и СС14 (правиль-
ные тетраэдры) попадут в одну и ту же группу, а Н^О — в дру-
гую. Название группы, к которой принадлежит данная молекула,
определяется элементами симметрии, характеризующими эту
группу.
Таблица 16,1
Символика для точечных групп
В Международной системе (которая также называется системой Германа —
Мргена) число п обозначает наличие оси п-.го порядка. Черта над симаолом
показывает, что эта операция комбинируется с инверсией (например, 6 означает
Сз в комбинации с i, нли Сж). Буква m обозначает плоскость симметрии. Косая
черта показывает, что группа имеет плоскость симметрии, перпендикулярную осп
симметрии (т.е. 2(щ — это Сал). Плоскости симметрии, не перпендикулярные оск
симметрии, обозначаются как m без каких-либо отличительных индексов. Необ-
ходимо различать операции симметрии одного и того же типа, по разных клас-
сов (например, превращается в Mmmm, где одна плоскость симметрии
перпендикулярна оси, а оставшиеся три отражения в зеркальных плоскостях от-
носятся к двум разным классам),
В приведенной ниже таблице дан перевод системы Шёнфлнса в Междуна-
родную систему Германа — Могена. Перевод дан только для 32 крисгаллогра-
фкческих точечных групп (см. разд. 16.4),
С,:1 Cs:m
Сх:1 С2.-2 Ca:3 C,:4 Ce:6
C2D:2mm Citi:4mm C№-.6mm
C2fl:2/m '--ah : 6 C<h:4/«
Пэ:222 Z?s:32 ZV-422 Z?s:622
Dgfr-.mtnm Z23h:62m
Оз^.-Зж Si-4 S6t3’
7423 7'^: 43m 0:43
Группа Dj иногда обозначается как V и называется Vierer group (группа
гырех).
16. Симметрия. Описание и следствия
11
Рис, 16,8. Пример молекулы, принадле-
жащей К группе Сг.
Используются две системы символов: система Шёнфлиса и
система Германа—Могена. Первая чаще употребляется при рас-
смотрении индивидуальных молекул, а последняя используется
почти исключительно при обсуждении симметрии кристаллов.
В следующих параграфах объясняется система Шёнфлиса; си-
стема Германа — Могена описывается в табл, 16,1,
1. Группы Cs, Съ Ci, Если молекула имеет плоскость симмет-
рии в качестве единственного элемента, кроме идентичности, то
она классифицируется как принадлежащая к группе С,. Приме-
ром является молекула хинолина (рис, 16.7,а). Если молекула
имеет в качестве единственных элементов идентичность и инвер-
сию, подобно мезовинной кислоте (рис. 16.7,6), то она принадле-
жит к группе С., Если молекула не имеет других элементов сим-
метрии, кроме идентичности (подобно CHFCIBr), то она принад-
лежит к группе Ct (рис, 16.7, в); такая символика объясняется в
следующем параграфе,
2, Группы Сп. Если молекула (или любой объект) имеет эле-
мент идентичности и ось симметрии п-га порядка, то она принад-
лежит к группе Сп (заметим, что Сп играет двоякую роль: это
обозначение одного из имеющихся элементов симметрии и обозна-
чение группы). Наименее симметричный объект обладает только
идентичностью Е в качестве единственного элемента симметрии.
Но такую молекулу можно рассматривать и как имеющую ось
Сь поскольку вращение ее на 360° также оставляет ее неизмен-
ной, Следовательно, такая молекула принадлежит к группе С),
Рис. 16.9. Примеры молекул, принадлежащих к группам Спь.
12
Часть 2. Структура
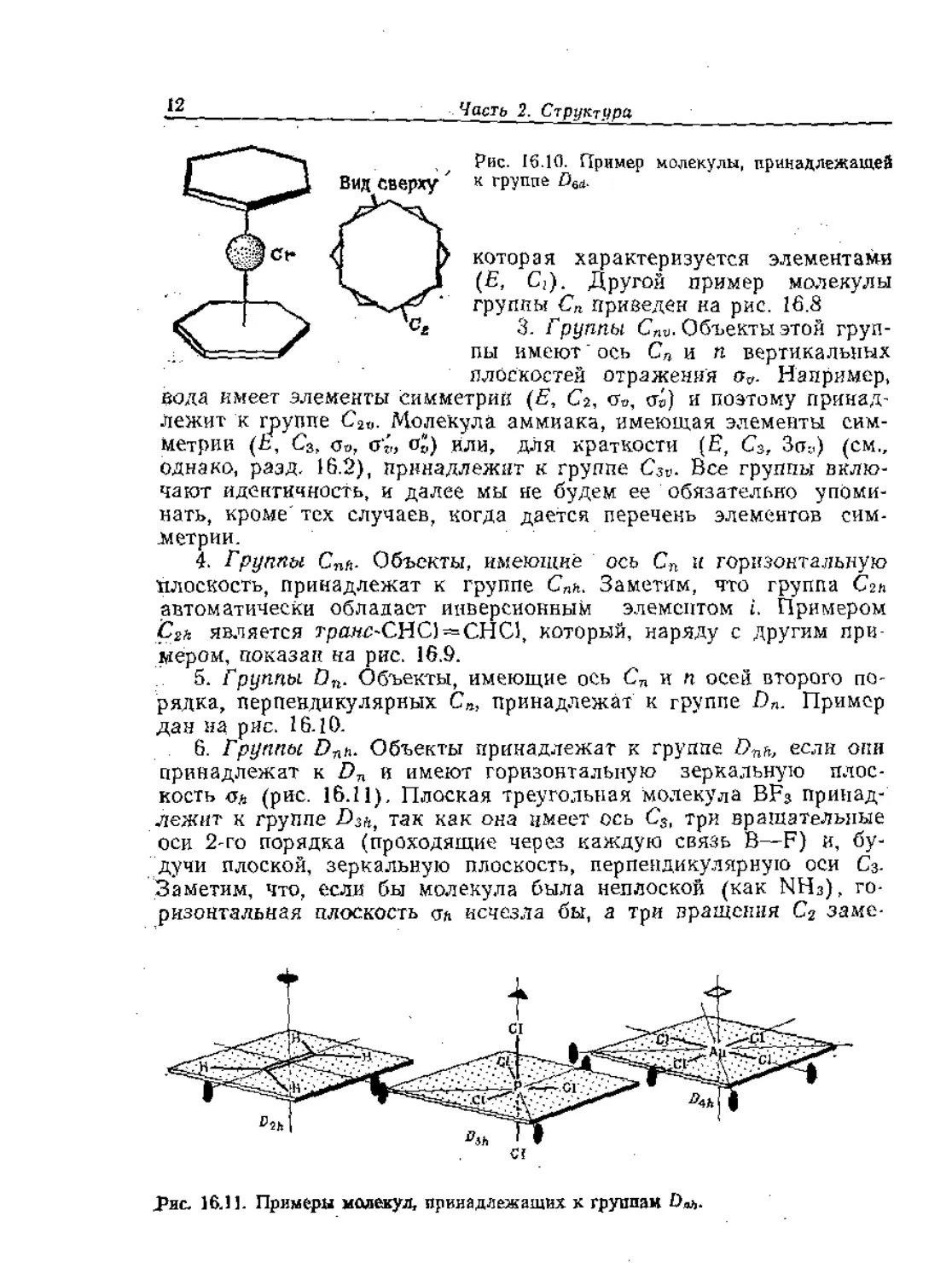
<Г" Рис. 16.10. Пример молекулы, принадлежащей
__1 Вид Сверху ' к группе Ом.
Шрс** м и которая характеризуется элементами
J I. _S (Е, С;). Другой пример молекулы
---—v ‘ группы Сп приведен на рис. 16.8
сх 3. Группы Спи. Объекты этой груп-
" - - г пы имеют ось Сп и п вертикальных
плоскостей отражения Например,
Пода имеет элементы симметрий (E, Ca, Go, ст») и поэтому принад-
лежит к группе С2И. Молекула аммиака, имеющая элементы сим-
метрии (6, С3, 0О, Gj-j °о) ИЛИ, ДЛЯ КратКОСТИ (E, С3, Зйу) (см.,
однако, разд. 16.2), принадлежит к группе C3v. Все группы вклю-
чают идентичность, и далее мы не будем ее обязательно упоми-
нать, кроме'тех случаев, когда дается перечень элементов сим-
метрии.
4. Группы Cnft- Объекты, имеющие ось Сп и горизонтальную
плоскость, принадлежат к группе Спи. Заметим, что группа Сгп
автоматически обладает инверсионным элементом 1. Примером
С2н является тронд-СНС) = СНС1, который, наряду с другим при-
мером, показан на рис. 16.9.
5. Группы Оп. Объекты, имеющие ось Сп и п осей второго по-
рядка, перпендикулярных Ся, принадлежат к группе Dn. Пример
дан на рис. 16.10.
6. Группы Z?nfl. Объекты принадлежат к группе Dnti, если они
принадлежат к Dn н имеют горизонтальную зеркальную плос-
кость о* (рис. 16.11), Плоская треугольная молекула BFa принад-
лежит к группе Е>зй, так как она имеет ось Cs, три вращательные
оси 2-го порядка (проходящие через каждую связь В—F) и, бу-
дучи плоской, зеркальную плоскость, перпендикулярную оси Сз-
Заметим, что, если бы молекула была неплоской (как NHa), го-
ризонтальная плоскость ой исчезла бы, а три вращения С2 заме-
.рис. 16.11. Примеры молекул, принадлежащих к группам Вал-
/6. Симметрия. Описание и следствия
13
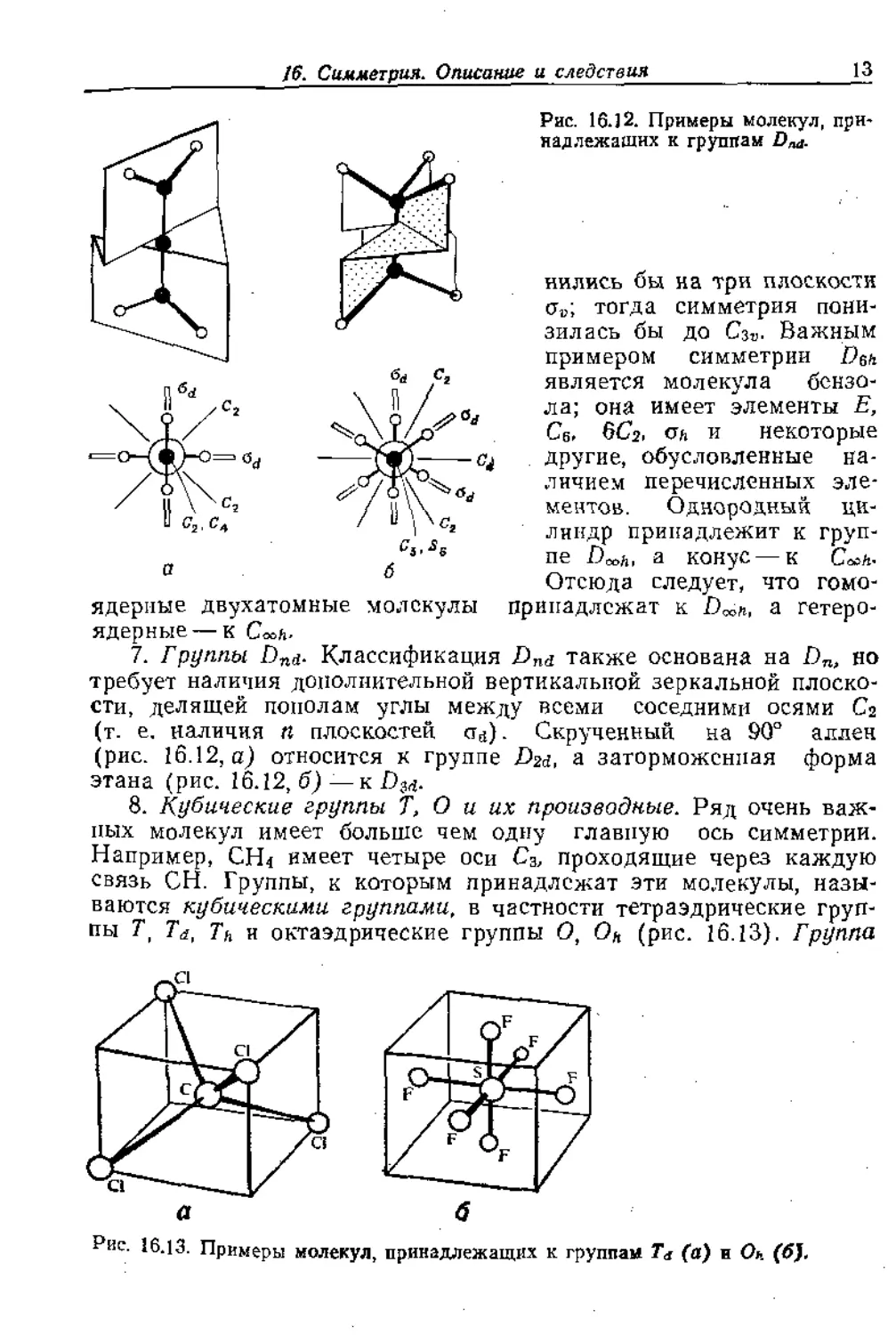
Рис. 16.12. Примеры молекул, при-
надлежащих к группам Dm-
нились бы на три плоскости
сгР; тогда симметрия пони-
зилась бы до Сзъ. Важным
примером симметрии Евк
является молекула бензо-
ла; она имеет элементы Е,
Св, 6Сг, ан и некоторые
другие, обусловленные на-
личием перечисленных эле-
ментов. Однородный ци-
линдр принадлежит к груп-
пе а конус — к С»*.
Отсюда следует, что гомо-
пгадлежат к а гетеро-
ядерные--- К Cosh-
7. Группы Dnd- Классификация Dnd также основана на Dn, но
требует наличия дополнительной вертикальной зеркальной плоско-
сти, делящей пополам углы между всеми соседними осями Са
(т. е. наличия п плоскостей ста). Скрученный на 90° аллен
(рис. 16.12, а) относится к группе Dzd, а заторможенная форма
этана (рис. 16.12, б) —к D-^.
8. Кубические группы Т, О и их производные. Ряд очень важ-
ных молекул имеет больше чем одну главную ось симметрии.
Например, СН4 имеет четыре оси С3, проходящие через каждую
связь СН. Группы, к которым принадлежат эти молекулы, назы-
ваются кубическими группами, в частности тетраэдрические груп-
пы 7, Та, Тн и октаэдрические группы О, Oh (рис. 16.13). Группа
Рис 16.13. Примеры молекул, принадлежащих к группам ТЛ (а) и Oh (6J,
14
Часть 2. Структура
GirfaHgaww)
(конус)
’ г=>ДпО0
Оий (ПЛОСКОСТЬ,-------
ИЛИ 6НП1Н________1
рзмида)
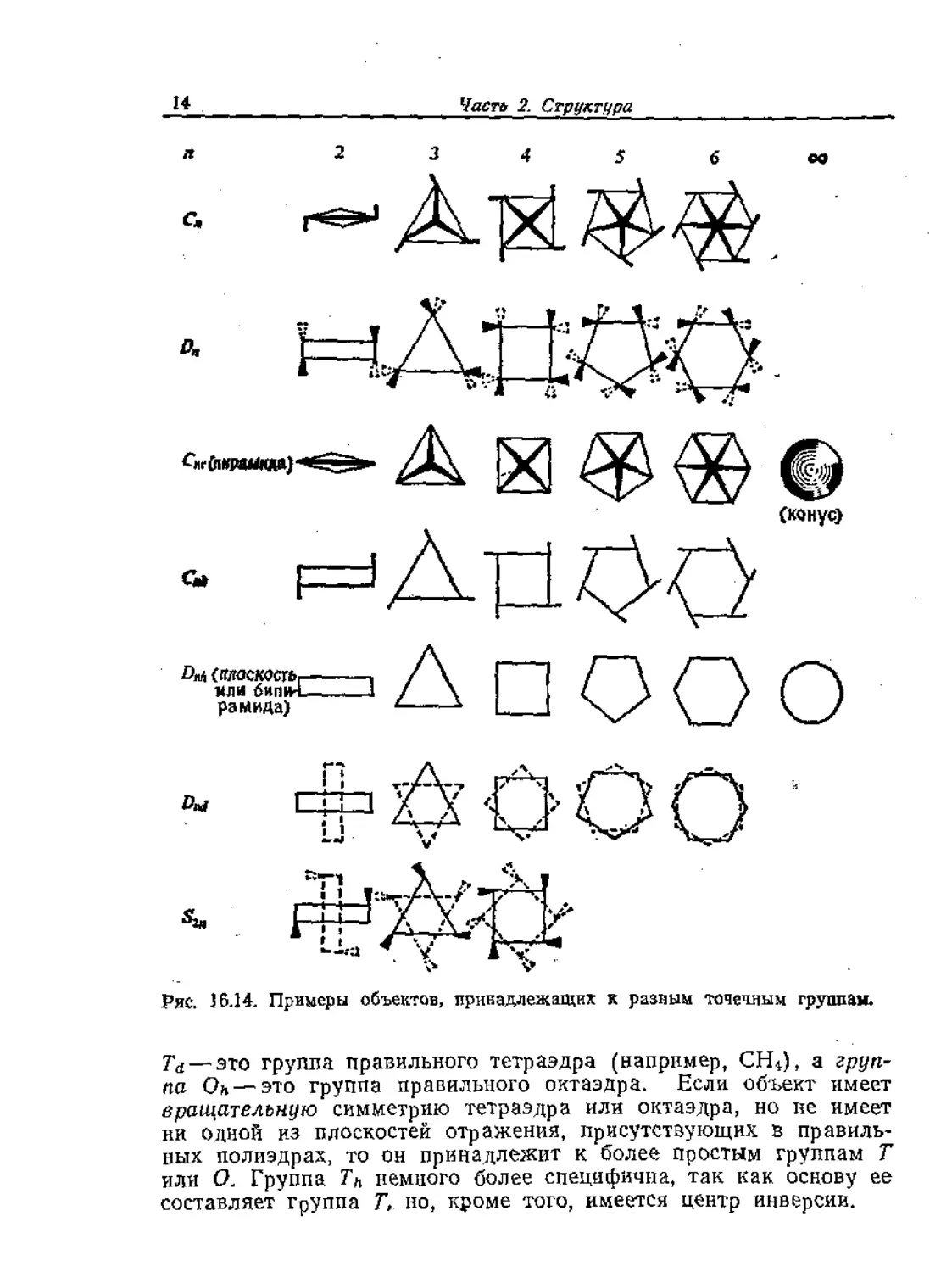
Ряс. 16.14. Примеры объектов, принадлежащих к разным точечным группам.
Td — это группа правильного тетраэдра (например, СН^), а груп-
па Он — это группа правильного октаэдра. Если объект имеет
вращательную симметрию тетраэдра или октаэдра, но не имеет
ни одной из плоскостей отражения, присутствующих в правиль-
ных полиэдрах, то он принадлежит к более простым группам Т
или О. Группа Th немного более специфична, так как основу ее
составляет группа Т, но, кроме того, имеется центр инверсии.
16. Симметрия. Описание и следствия
15
9. Группа полного вращения — это группа операций, свой-
ственных сферическому объекту. Атом принадлежит к 7?3, но ни
одна молекула не принадлежит к этой группе. Использование
следствий из 7?3-симметрии является очень важным путем при-
менения теоретике-групповых представлений к атомам.
Идентификация элементов симметрии, которыми обладает мо-
лекула, позволяет провести классификацию по группам. Эти
группы обычно называют точечными группами, чтобы отличить их
от пространственных групп, с которыми мы встретимся при рас-
смотрении трансляционной симметрии кристаллов. Во многих
случаях задача классификации облегчается сравнением структу-
ры молекулы с формами объектов, показанных на рис. 16.14.
Пример (вопрос 2). Определите точечную группу, к которой принадлежит сэнц-
вичевая молекула рутенацена (два заслоненных циклопентадненовых кольца).
Метод. Решите, не больше ли одной главной оси л-го порядка (при л 5=3) име-
ется в дайнам случае; если да, то переходите к кубическим группам. Если толь-
ко одна, ищите оси С2, перпендикулярные Сп. Если их нет, переходите к груп-
пам Си. Если их несколько, переходите к Затем найдите плоскости отра-
жения и центры инверсии и сделайте выбор между Опл, Dna, СПР1 S2n.
Ответ. Имеется ось Cs, но нет других осей с л^З. Имеется пять перпендику-
лярных осей Ct, каждая из которых переворачивает молекулу верхом вниз; сле-
довательно, выбираем Ds. Имеется горизонтальная плоскость, отражающая верх-
нее кольцо в нижнее; поэтому молекула относится к группе Dst..
Комментарий. Если бы молекула имела заторможенные кольца (как в ферро-
цене), то оси С2 еще присутствовали бы, но были бы смещены. Горизонтальной
плоскости не было бы, но имелись бы пять диэдрических плоскостей. Следова-
тельно, молекула принадлежала бы к группе Dsa.
Некоторые очевидные следствия симметрии. Как только становит-
ся известной точечная группа молекулы, можно сформулировать
некоторые следствия, касающиеся се свойств. Например, только
молекулы, принадлежащие к группам Сп, СПп, G и Cs, могут
иметь электрический дипольный момент, и в случае Сп и Сп» ди-
польный момент должен быть направлен вдоль оси вращения.
Это можно понять на основании следующего. Если молекула от-
носится к Сп, то она не может иметь распределение заряда, соот-
ветствующее дипольному моменту, перпендикулярному этой оси
(рис. 16.15, а); однако, поскольку группа ничего не говорит о
симметрии молекулы между «вершиной» и «дном» молекулы,
может существовать распределение заряда, дающее диполь вдоль
оси симметрии (рис. 16.15, б). Те же замечания относятся к груп-
пе Cnv, в случае которой отсутствует симметрия «верх—низ». Во
всех других группах, таких, как С3а, D и т. д., имеются операции
симметрии, соответствующие переворачиванию молекулы верхом
вниз. Поэтому, не имея дипольного момента, перпендикулярного
оси, такая молекула не обладает и дипольным моментом вдоль
оси; в противном случае переворачивание ее верхом вниз не было
бы операцией симметрии.
16
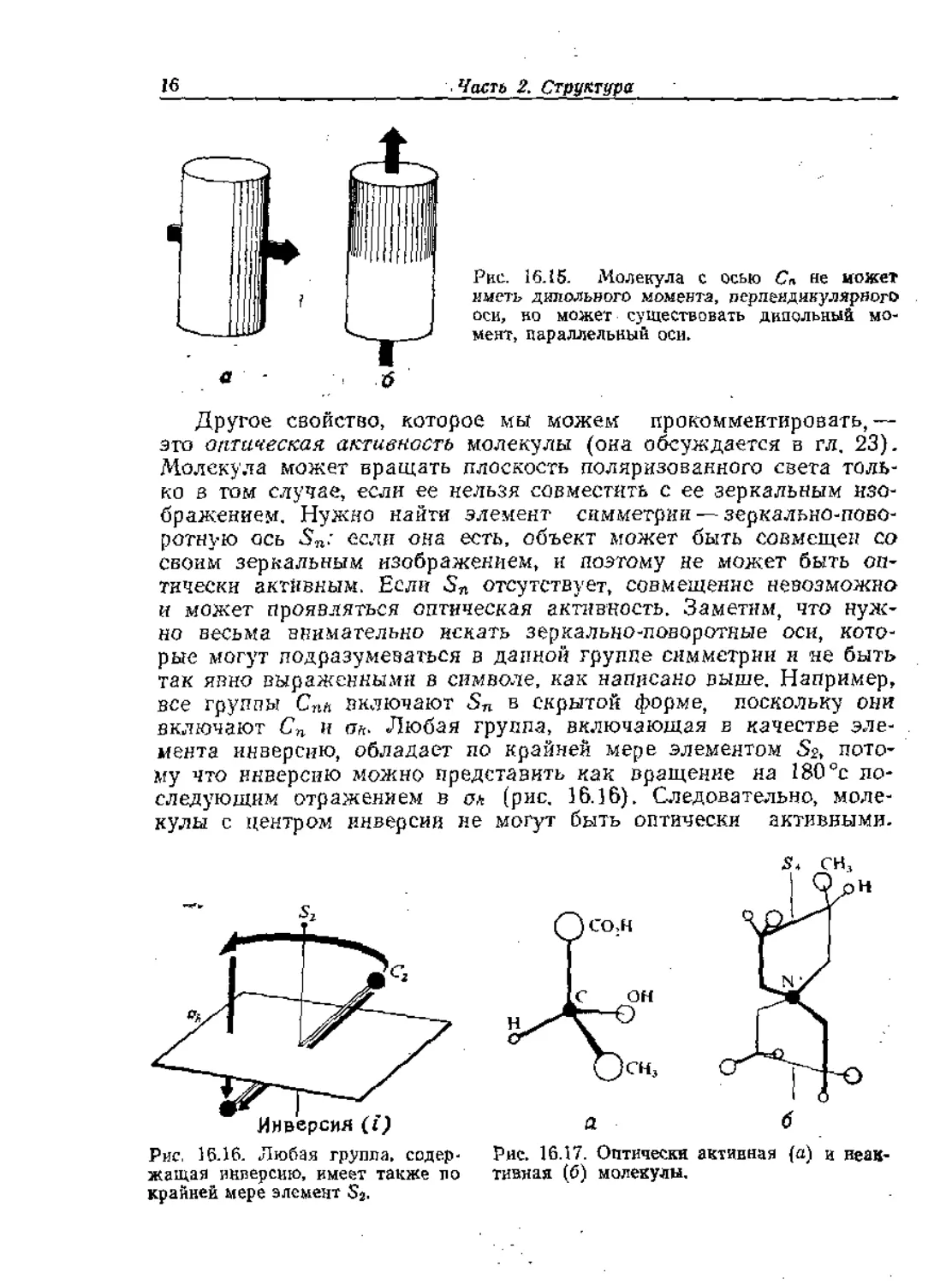
Часть 2. Структура
Рис. 16.15. Молекула с осью Сп не может
иметь дипольного момента, перпендикулярного
оси, но может существовать дипольный мо-
мент, параллельный оси.
Другое свойство, которое мы можем прокомментировать,—
это оптическая активность молекулы (она обсуждается в гл, 23).
Молекула может вращать плоскость поляризованного света толь-
ко в том случае, если ее нельзя совместить с ее зеркальным изо-
бражением, Нужно найти элемент симметрии — зеркально-пово-
ротную ось 5„.’ если она есть, объект может быть совмещен со
своим зеркальным изображением, и поэтому не может быть оп-
тически активным. Если отсутствует, совмещение невозможно
и может проявляться оптическая активность. Заметим, что нуж-
но весьма внимательно искать зеркально-поворотные оси, кото-
рые могут подразумеваться в данной группе симметрии и не быть
так явно выраженными в символе, как написано выше. Например,
все группы Спь включают Sn в скрытой форме, поскольку они
включают Сп и пл. Любая группа, включающая в качестве эле-
мента инверсию, обладает по крайней мере элементом Sa, пото-
му что инверсию можно представить как вращение на 180°с по-
следующим отражением в ол (рис, 16.16). Следовательно, моле-
кулы с центром инверсии не могут быть оптически активными.
Рис, 16.16. Любая группа, содер-
жащая инверсию, имеет также по
крайней мере элемент Sj.
Рис. 16.17. Оптически активная (а) и неак-
тивная (б) молекулы.
3 3 b C n
16. Симметрия. Описание и следствия
17
Оптически активная молекула приведена на рис. 16.17, а. Не
все молекулы без центра инверсии активны: например, если они
имеют симметрию S4j то элемент i отсутствует, во S4 означает от-
сутствие активности. Пример дан на рис. 16.17, б-
Если известна групповая структура, то можно проанализиро-
вать и другие свойства, но такая информация более глубоко
скрыта в типе группы. Чтобы ее получить, необходимо приступить
к более строгому анализу теории групп, но даже и в этом случае
мы лишь бегло коснемся поверхности этого очень тонкого и мощ-
ного метода. Теперь мы переходим от качественных к количест-
венным аспектам симметрии.
16.2. Группы, представления и характеры
Рассмотрим элементы симметрии молекулы Н2О (С2г,). Мож-
но мысленно произвести одну операцию симметрии (например,
вращение вокруг С2), а затем другую (например, Оц). Получен-
ная молекула будет идентична начальной, и поэтому последова-
тельность двух операций даст такой же эффект, как операция
идентичности Е. Символически Это можно записать в виде
Е—о;С2, (16.2.1)
что является стенограммой утверждения: «Операция С2 с после-
дующей операцией oi эквивалентна операции Е». В качестве
другого 11римера рассмотрим молекулу NH3. Хотя мы уже гово-
рили, что элементами симметрии являются Е, Сз, Зог,, это недо-
статочно точно. Вращение вокруг оси 3-го порядка по часовой
стрелке — операция симметрии, вращение против часовой стрел-
ки тоже операция симметрии. Фактически эти операции различ-
ны. Мы обозначим их как С3 (против часовой стрелки на 360°/3)
и С3 (по часовой стрелке на 360°/3). Таким образом, операция-
ми симметрии будут Е, Сз, Сз, ой, Оп или Е, 2С3, Зо^. Те-
перь очевидно, что С3 с последующей С3 есть идентичность:
Е = С;С3. (16.2.2)
Однако можно также отождествить С3 и последующую Сз (два
последовательных вращения на 120° против часовой стрелки,
т- с. „общее вращение на 240°) с Одним вращением на 120° по ча-
совой стрелке. Символически это записывается как
(16.2.3)
Далее предположим, что за С3+ следует Из рис. 16.18 видно,
ч/то эти операции можно заменить на единственную операцию
Символически
(16.2.4)
2~242 ZxnfocwuffK
18
Часть 2. Структура
б'
Рис. 16.18. Композиция вращения с последующим отражением.
Таблица всех этих комбинаций носит название таблицы группо-
вого умножения (для Сз-и см. табл. 16.2). Очень важной особен-
ностью всех этих результатов является то, что последовательные
серии операций симметрии можно всегда выразить как одну опе-
рацию симметрии данной группы. Это свойство называется груп-
повым свойством и является главным в теории групп. Набор опе-
раций образует группу, если они удовлетворяют групповому свой-
ству и некоторым другим условиям, перечисленным в под-
разд. 16.2.А. Все операции симметрии в случае молекул удовлет-
воряют этим условиям, и поэтому теория симметрии молекул на-
зывается теорией групп.
Таблица 16.2
Таблица группового умножения для Сзо
Первая —* Вторая 1 Б ^з" ^3 «в * %
Е Е Сз ав °; °;
ct с; Е °; Ср
ct Е £+ °;
°; Е £3
gd °C М Е С*
По са ct Е
16.2. А. Свойства групп. Группа — это набор объектов, или эле-
ментов (таких, как операции симметрии) G = {gi, £2,...,£дг}, под-
чиняющихся правилу комбинации, благодаря чему символ gig-
имеет строго определенный смысл (например, операция gi следу-
16. Симметрия. Описание и следствия
19
ет за операцией gj), который удовлетворяет следующим крите-
риям:
1. Набор включает элемент идентичности; этот элемент обыч-
но обозначают как Е, так что Egi = gtE—gt для всех элементов
набора.
2. Набор включает инверсию каждого его элемента; инверси-
ей gi (записывается g?1) является элемент, для которого
gig-1 =glgi = E.
3. Правило комбинации является ассоциативным, т. е. комби-
нация (gigj)gk — это то же самое, что gi(gjgk).
4. Комбинация любых двух элементов этого набора должна
сама быть членом набора, т. е. gigj=gk, где — член G. Это на-
зывается групповым свойством.
Заметим, что определение не требует, чтобы gigj = g;gi (за ис-
ключением специальных случаев определения Е и g7 J. Группы»
для которых gigj = gjgt, называются коммутативными или абе-
левыми группами.
Представление трансформаций. Выражения, подобные £' = СзСз»
выглядят как нормальное алгебраическое умножение, но на са-
мом деле они являются символическим способом записи того, что
происходит при проведении последовательных физических опера-
ций. Тем не менее им можно придать действительный алгебраиче-
ский смысл, и в этом состоит цель данного раздела. Если мы при-
дадим им алгебраический смысл, то это значит, что мы сможем
действовать с числами вместо абстрактных символов для опера-
ций, а действуя с числами, мы сможем получить количественные
выводы.
Рассмотрим молекулу С31), в которой s-орбитали связаны с
атомами так, как показано на рис. 16.19. Центральную s-орби-
таль обозначим как Sn, а другие как ад, sb и sc- Ясно, что при-
мер относится к молекуле NH3,
и об этом можно помнить; одна-
ко в действительности такое
расположение очень общее, так
как оно применимо к любому
предмету симметрии C3lJ. То, что
названо s-орбиталью, могло бы
быть любой другой функцией
Рис. 16.19. Орбитальный базис молеку-
лы С3р.
2*
20
Часть 2. Структура /
—------— —------------------г-------
объекта, например ^-орбитально (когда z параллельна оси сим-
метрии) или даже настоящими резиновыми мячиками.
Теперь рассмотрим, что произойдет с этими функциями, когда
к молекуле применяется операция симметрии. При операции
происходит следующее:
(SN, SA* «С, Sb) "* (SN> SA, SB,
Такую трансформацию можно выразить, используя матричное
умножение (обзор предмета дап в приложении к этой главе).
Можно найти некоторую матрицу, обозначаемую как D (Он), ко-
торая воспроизводит последнее соотношение:
/1 0 0 0)
(SN, SA,SC, Sa) — (sN, SA, Ss, Sc) Q Q Q |
'о 0 1 0/
(16.2.5)
Эта матрица D (щ>) называется представлением операции сь. Та-
кой же способ можно использовать, чтобы найти матрицы, кото-
рые воспроизводят другие операции симметрии. Например, эф-
фект операции Сз состоит в следующем:
(sN, sc, sa, sb) *- (sN, sA, SB, Sc),
и его можно представить матричным умножением
/1 0 0
(SN, sc, Sa, sb) — (SN) SA, SB. Q 0 0 1 0 J . (16.2.6)
1 0 0/
Эта матрица обозначается как D (Сз ). Операцию Сз, которая име-
ет эффект
(sn, sb, sc. sa) 4 (SN. sa> sb> Sc).
можно представить матричным умножением
11 0 0 01
C"n> ?b,Sc, Sa) ~ (Sn, Sa, Sb, ®c) 0 0 0 1 0 0 J. 06.15?
^0 0 1 0/
IS. Симметрия. Описание и следствия
э|
Эта матрица обозначается как D(Cs/ Матричное представление
идентичности Е оставляет функции (sNj S&, sB, sc) неизменными
и поэтому имеет вид
/1 0 0 0)
0 1 0 0
D(£) = 0 0 1 0
\о 0 0
(16.2.8)
что можно подтвердить наблюдением.
Теперь можно получить очень важное свойство этих матриц.
Использование правил матричного умножения для произведения
О (<т0) D(Ct) дает
Dfa)D(Cj)= 71 0 0 0\ /1 0 0 0\
/1 0 0 0\
= 1° 10 0 0 0 0 1 10 0
“10 0 0 1 0 10 0 “(оо 10
\о 0 1 о/ \о 0 1 0) \о 1 о 0/
Важность этого уравнения можно видеть при сравнении его
с уравнением (16.2.4); оно имеет точно такую же структуру:
<тс.Сд=сг(, сравнимо с D (oo) D (С£) — D (о„).
Какую бы группу элементов мы ни выбрали, матричные пред-
ставления перемножаются друг с другом аналогично. Следова-
тельно, полная таблица группового умножения воспроизводится
алгебраическим умножением матричных представлений. Набор
из шести матриц называется матричным представлением группы
Сз0, и это означает, что между символическими действиями дан-
ной группы и алгебраическими действиями с числами можно уста-
новить определенную связь.
Пример (вопрос 4). Рассмотрите четыре водородные ls-орбитали метана. Най-
дите матричные представления для операций С3 н 5< и подтвердите, что они
Удовлетворяют свойству группового умножения.
Метод Метан принадлежит к Гн. Операция С3 — это вращение вокруг связи
С—Н (например, С—Hi), и Поэтому при таком вращении три другие орбита-
ли превращаются одна в другую. При происходит вращение на 90° вокруг
биссектрисы угла СН2 (например, Н,СН2) и затем отражение в горизонтальной
Плоскости. Находим матрицы 4X4 которые воспроизводят эти изменения.
Ответ. При операции С3 (Hi, Н2, Н3, Н4)—'(Н,, Н4, Нг, Н3), а при S4 (Hlt Н2.
22
Часть 2. Структура
Н3, Н4)—>-(На, Н3, Н1, Нг). Эти изменения
матрицами:
выряжаются также следующими
/1 0 0 0 /о 0 1 0
Р 0 1 0 1 о 0 о 1
D(Cj)— I D(S<) = „
1 о 0 0 1 > Io 1 0 0
1° ] 0 о/ V 0 0 0
так как необходимо, чтобы (Hi, Н2, Нз, D (С3)= (Н>, Н*, Н2, Н») и .(Hi, (Ь,
Hs, H4)»(S4)=(H4, Нз, Нь Йз).
Чтобы проверить групповое свойство, записываем
1 о 0 0\ /0 0 1 °\ /° 0 1 °\
D(G)D(S<) = 0 0 1 0 ’о 0 ° 11 - 0 I 0 0
0 0 0 1 0 1 0 0 " 1 0 0 0
1° 1 0 0 0 о/ 0 0 >/
Новая матрица превращает первоначальный базис в (Н3, Н2, Hi, Н4), что соот-
ветствует действию ©а (плоскость проходит через Н2, Н4) на первоначальный
базис. Однако в группе C3S4=oa, т. е. групповое умножение воспроизводится.
Комментарий, Полученные матрицы зависят от выбранного базиса (в данном
случае — это четыре водородные ls-орбитали), Четырехмервый базис приводит
к четырехмер и ом у представлению.
Характер операций симметрии. Вообще говоря, вращения Сз и
Сз группы Сз имеют одинаковый характер: они различаются лишь
по направлению. Аналогично три отражения тоже имеют одинако-
вый характер, но отличающийся от вращения. Это замечание
можно выразить точным количественным соотношением.
Рассмотрение матричного представления Сз„ с использованием;
s-орбиталей в качестве базиса приводит к выявлению примеча-
тельного факта. Если сложить диагональные элементы каждого
матричного представления, то можно получить следующие числа:
D(E) D((£) D(C7) D(a0) D(op
x= 4 1 1 2 2 2
Выявляется, что матрицы, представляющие операции одного и
того же типа, имеют одинаковые диагональные суммы. Этот ре-
зультат имеет большие последствия. Можно назвать сумму диа-
гональных элементов матричного представления операции харак-
тером данной операции и обозначить ее через %. Говорят, что опе-
рации симметрии с одинаковым характером относятся к одному
классу.
Пример (вопрос 5). Каков характер операций Cit Sa и од в базисе, использо-
ванном для метана в последнем примере?
16. Симметрия. Описание и следствия 23
Метод. Обратимся к D-матрицам, приведенным в последнем примере, н вычис-
лим сумму диагональных элементов.
Ответ. Для Сэ:х (Са) = 1 + 0 + 0 + 0 = 1.
Для S4:x(S<) = 0 + 0q-0 + 0 = 0.
Для arf;X- х(C3S*J = 0+ I + 0 + t - 2.
Комментарий. Быстрое правило определения характера состоит в прибавлении
I всякий раз, когда атом остается неизменным прн операциях симметрии, так
как только эти атомы дают запись на диагонали [в некоторых базисах может
быть изменение знака: если (—{—)—’(---!•••), то на диагонали матрицы по-
является — 1, поэтому при расчете суммы берется —1]. Ясно, что для данного
базиса характер идентичности х(£)—4. Правило также дает %(С2)=0 для дан-
ного базиса.
Характер операции зависит от базиса, использованного для
составления матричного представления. Например, если вместо
рассмотрения набора из четырех s-орбиталей ограничиться лишь
sN, то никакие операции симметрии ее не изменят, и каждая опе-
рация симметрии приведет к тривиальной трансформации
sn*-sn- Поэтому каждую операцию можно представить одним и
тем же правилом: sn = sn1, где 1, конечно, можно рассматривать
как матрицу, но только в тривиальном смысле. Характер каждой
операции равен 1, так как каждое матричное представление есть
I, и поэтому таблица характеров будет иметь вид
D(£) О(Сф ЩС$ D(aa) D(o^ ОДО
Х= 1 1 1 11 1
Утверждение, что операции одного и того же класса имеют рав-
ные характеры, остается истинным, но этот пример служит для
того, чтобы подчеркнуть, что характеры разных классов могут
быть одинаковыми. Далее, очевидно, что, поскольку 1X1 = 1, мат-
рицы для этого базиса на самом деле воспроизводят всю табли-
цу группового умножения, но это воспроизведение тривиально и
практически неинформативно. По этой причине представление, в
котором каждый элемент выражен единицей, носит название не-
доверительного (unfaithful) представления группы.
Неприводимые представления. Хотя одна SN-орбиталь является
базисом для недоверительного представления группы, представ-
ление есть представление и не может быть отброшено как неин-
тересное. В следующих нескольких разделах будет показано, что
представление с 1 для каждого элемента является наиболее важ-
ным представлением для многих химических задач.
При базисе (sn, Яд, sb, scj матрицы относились к типу 4X4,
а представление было четырехмерным. Тем не менее рассмотре-
24
Часть 2. Структура
ние этого представления показывает, что каждая матрица имеет
вид
1
о
о
о
ООО
н операции симметрии никогда не перемешивают sn с другими
тремя функциями базиса. Отсюда вытекает, что базис можно
разбить на две части: одна из них содержит только Sn, а другая
(Sa, Sb, sc); $n, как мы видели, — это базис для Недоверительного
представления, а другие три орбитали — это базис для трехмер-
ного представления, состоящего из следующих матриц:
JXC5) Pfo) D(ai) »(^)
(1 о о\/о
О 1 0 1
О 0 1До
О 11/0
О 0 10
1 о|р
о
1 01/1 О 01/0 о 11/0 1 01
О 11 о 0101 01 о о|
о о]\о 1 оД1 о о]\о 0 1}
0 111
Отметим, что характеры и здесь еще удовлетворяют правилу
об операциях симметрии одного класса. Эти матрицы такие же,
как матрицы четырехмерного представления, за исключением то-
го, что отсутствуют первый ряд и первая колонка. Мы говорим,
что первоначальное четырехмерное представление приведено к
сумме (точнее, к прямой сумме) одномерного представления, свя-
занного с Sn, и трехмерного представления, связанного с (зд, sg,
sc). По общему смыслу это соответствует точке зрения, что цент-
ральная орбиталь Sjq играет иную роль, чем три другие.
Теперь эту ситуацию можно выразить в символах, записывая
одномерное представление как /ТЦ трехмерное — как н че-
тырехмерное — как Тогда приведение можно выразить фор-
мулой
Нельзя упускать из виду, что это лишь символическое обозна-
чение: оно не означает, что четырехмерные матрицы являются
суммой, в обычном смысле, одно- и трехмерных матриц. Однако
оно действительно означает, что четырехчленный базисный набор
может быть подразделен на два независимых базиса (один с од-
ним членом и другой с тремя) и что соответствующие представ-
ления являются одно- и трехмерными.
Представление т. е. набор матриц 1, 1, 1, 1, 1, 1, очевидно,
нельзя привести дальше, поскольку нет подходящей формы отбо-
16. Симметрия. Описание и следствия
25
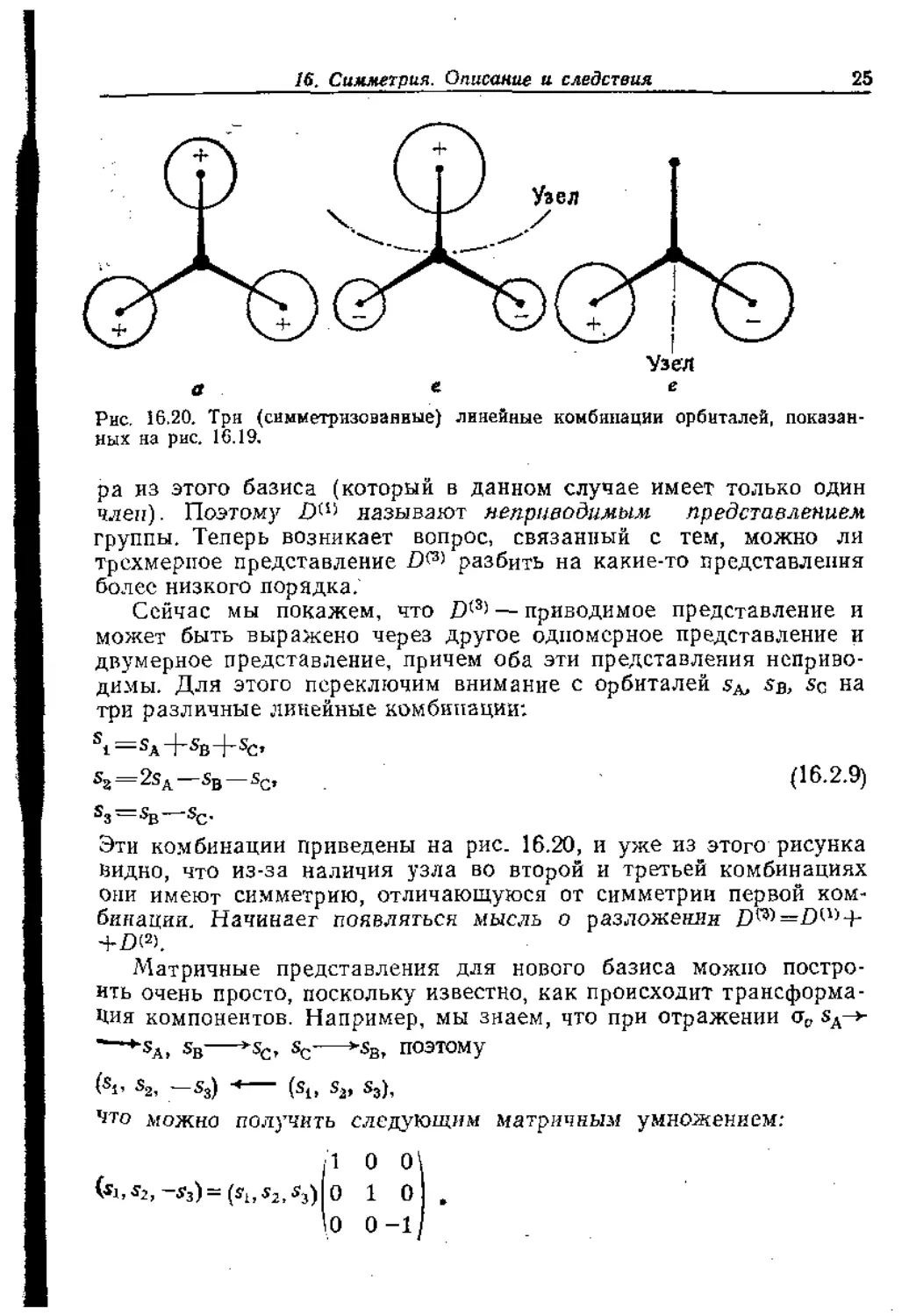
Рис. 16.20. Три (симметрнзованные) линейные комбинации орбиталей, показан-
ных на рис. 16.19.
ра из этого базиса (который в данном случае имеет только один
член). Поэтому называют неприводимым представлением
группы. Теперь возникает вопрос, связанный с тем, можно ли
трехмерное представление № разбить на какие-то представления
более низкого порядка.
Сейчас мы покажем, что Д(3> — приводимое представление и
может быть выражено через другое одномерное представление и
двумерное представление, причем оба эти представления неприво-
димы. Для этого переключим внимание с орбиталей Дв, Дс на
три различные линейные комбинации:
si=sA+sB+sc,
s3=2sA—sB—(16.2.9)
S3 — Sg—sc.
Эти комбинации приведены на рис. 16.20, и уже из этого рисунка
видно, что из-за наличия узла во второй и третьей комбинациях
Они имеют симметрию, отличающуюся от симметрии первой ком-
бинации. Начинает появляться мысль о разложении
+£)(2).
Матричные представления для нового базиса можно постро-
ить очень просто, поскольку известно, как происходит трансформа-
ция компонентов. Например, мы знаем, что при отражении % sA->-
*"sa> sB---->5Ь Sc”—^Sg, поэтому
(st, s2, — $3) -<— (S1, s2, s3),
Что можно получить следующим матричным умножением:
/1 0 0\
^2, — 5*3) = ($£, $2, Sj) I 0 1 0 ,
\0 0-1/
26
Часть 2. Структура
Таким образом, мы находим форму представления D (оо) в но-
вом базисе. Матричное представление операции Сз требует не-
много больше расчетов и связано с трансформациями
sb—sc—>sA. Подстановка выражений для sb s2, s3 дает
/ 1.3 I I \
g sa-r 2 S3’ 2 Я®] "* (si*s2>sa)>
и, следовательно, матричное представление можно получить из
/1 ° °\
(^1, — 5 J, + TS3, — 2S2 — 2^з) ~ (51, S2, ^з)! 0 ~| | .
хи 2 г <
t о 01 i
о i olio
о о i Io
3 •
Таким путем могут быть найдены полное представление и его ха-
рактеры:
, D(£) D(<?B D(CH O(<r„> D(4) D(<r3
0 o\/l 0 oWl 0 o\ll 0 o\ll 0 о I
4 4 P 4 2 b i o o 4 4 0 -i И
I Ч|\о -I -i)\o о -iHo -I q\o I if
о о 1 i t
Выявляется ряд важных особенностей. Во-первых, характеры
соответствуют принципу, касающемуся операций одного и того
же класса. Во-вторых, характеры совпадают с характерами для
первоначального трехмерного базиса. Это является иллюстраци-
ей положения, которое всегда верно: при линейных комбинациях
базисного набора характеры не изменяются. В-третьих, и это
наиболее важно, все представления в виде матриц 3X3 можно
записать в блок-диагон а л ьееой форме
/10 0
°
(о --
и комбинацию Si нельзя смешать с двумя другими никакой сим-
метрической трансформацией группы. Следовательно, можно сде-
лать следующее приведение:
= £><!> 4-
где Sj образует базис для того же представления 1, 1, 1, 1, 1, 1,
как и раньше, а — это двумерное представление на базисе
(ss, s3). Его матричным представлением является набор матриц
16. Симметрия. Описание и следствия ^7
2X2, образованных из представления 3X3, из которого изъяты
первый ряд я первая колонка:
D(E) D(CJ) DCCJ) О(<тс) D(ai')
y-J -1-10 00
Очень легко убедиться, что эти матрицы действительно образуют
представление группы, перемножая их попарно: они воспроизво-
дят оригинальную таблицу группового умножения.
Остается вопрос: можно ли найти какую-либо линейную ком-
бинацию s2j s3, которая приводит О(2) к двум одномерным пред-
ставлениям? Такой возможности нет: само № является непри-
водимым представлением группы С31>. Следовательно, можно сде-
лать вывод, что характеристики sn и Si по симметрии одинако-
вы: обе они являются базисом для одного и того же одномерно-
го неприводимого представления; пара же s3 имеет иную сим-
метрию, но s2 и s3 должны рассматриваться вместе именно как
пара. Эти особенности полностью согласуются с диаграммами
этих функций и их линейных комбинаций.
Списки характеров всех возможных неприводимых представ-
лений различных групп называются таблицами характеров. Для
группы Сзи таблица.характеров имеет следующий вид:
CSE | Е 2С3 Зоу,
Л 11 1
Л 1 1 • -1
£2—1 О
В заголовках колонок даны операции, которые характеризуют
группу. Указывать характер каждого элемента не обязательно,
поскольку характеры в каждом классе одни и те же, и поэтому
в колонках приведен класс операций; однако число 2 в 2С3 по-
казывает, что в этой группе можно осуществить два С3-враще-
ния. В левой колонке дано наименование неприводимого представ-
ления. Там, где мы использовали символ Д(1> для обозначения
одномерного представления, по соглашению применяется сим-
вол А. В этой группе возможны два одномерных представления, и
поэтому они обозначаются Аг и А2. Двумерное неприводимое
представление обозначается буквой Е; этот символ ие нужно пу-
тать с символом операции идентичности.
Пожалуй, наиболее удивительным в этой таблице является то,
что неприводимых представлений довольно мало, но Эти три пред-
28
Часть 2. Структура -
ставления исчерпывают все возможности. Из очень элегантной
теоремы теории групп следует, что
число неприводимых представлений равно числу классов.
В Сзи имеются три класса операций (три колонки в таблице ха-
рактеров), и поэтому указанные три неприводимые представле-
ния являются единственными неприводимыми представлениями
группы.
Все наши рассуждения основывались на группе Сз„, однако
сделанные замечания имеют совершенно общее значение, и для
любой группы можно составить список характеров возможных
неприводимых представлений. Эти таблицы характеров настоль-
ко полезны и важны, что некоторые из них даны в табл. 16.5 в
Конце главы. Мы должны рассмотреть еще один вопрос, а затем
покажем, как использовать таблицы характеров для решения
типичных проблем и почему эти таблицы настолько важны.
Трансформации других базисов. До сих пор внимание концентри-
ровалось на наборе орбиталей, и мы видели, что Sn и si ведут
себя иначе, чем s2 и % Теперь вместо этих объектов рассмотрим
координаты х, у, z с центром на центральном атоме. Пусть они
16. Симметрия. Описание и следствия
29
связаны с молекулой и преобразуются при трансформации моле-
кулы. При отражении ов получаем (рис. 16.21)
—х, у, z) <- (х, у, 2),
откуда можно записать
1-1 0 01
(-x,y,z) = (x,y>z) 0 1 0.
\ 0 0 1/
D(E) D(C3+)
1 о oi I Ч 4VT о|
О1о |1Ч Ч о
001/ 10 01/
* = 3 о
DCo-J Dfo-;)
Аналогично при вращении на 120° против часовой стрелки имеем
1 Ч -W3 of
(Чк+р/Зу, -^/3x-{yrz) = (x, y,z) 5^3 Ч О!.
1 0 0 1/
Полный набор представлений таков:
»(с3-)
~2 2>/3 0 \
Ь/з Ч о
О 0 1/
о
D(<r3
/-1 о oi 1 1 -1VT 01 / I о)
о 1 о - -1VT -? о НТз" Ч о
10 0 1/ \ о 01/ \ о 01/
" 1 1 1
Отсюда видно, что это трехмерное представление приводимо, по-
скольку все матрицы имеют блок-диагональную форму, из кото-
рой можно удалить части, содержащие z. Оставшееся двумерное
представление имеет следующие характеры:
2—1—1000
Из сравнения этих значений с таблицей характеров C3v ясно, что
(х, у) составляет базис для неприводимого представления Е.
Вывод из этой важной части анализа состоит в том, что z ведет
себя по-иному, чем образующие пару хну.
Пример (вопрос 6). Найдите, как трансформируются функции х, у, г в группе
30
Часть 2. Структура
Метод. Группа имеет элементы Е, Cs, cre, его. Найдем влияние каждого эле-
мента на указанные три функции и затем запишем матричное представление.
Определим блок-диагональную форму.
Ответ. При Е (х, у, г)—>(х; у, г); при Cj (х, у, г)—>(—х, —Д г); при <тв
(х, у. г)—>('—х, у, z)\ при По (х, у, z)—>fx, — у, z). Поэтому матричное пред-
ставление имеет нид
БАЗИС Е
<х, у, zj
' 1 О 01
0 10
0 0 1;
Это уже блок-диагональная
мерные п едет а в лени я:
Ог nv»
О О U.-1 О 0 \ I1 о
-1 О О 1 0 0 -1
О 1/1 О О 1 ) \ -о о
форма, и ее можно разбить на
0\
О
1/
следующие одио-
лг; 1 —1
1 -1
Z-. I 1
-1 1,
1 —1,
1 1.
Ларактерамн представлений являются сами числа (так как матрицы относятся
к тнпу 1 X 1)> и поэтому неприводимыми представлениями, связанными с х, у
и z, будут соответственно Bi, Bz и At.
Комментарий. Такую последовательность операций можно использовать всегда.
К счастью, для большинства групп представления, связанные с к, у и z, уже
табулированы, н через них всегда можно выразить трансформационные свой-
ства более сложных функций.
Неприводимые представления для х, у, z настолько важны,
что обычно они включаются в таблицы характеров. Тот же метод
можно также применить к другим простым функциям, и способ
трансформации квадратичных форм х1, ху, xz,...,z2, как правило,
также табулируется. Поэтому полная таблица характеров обычно
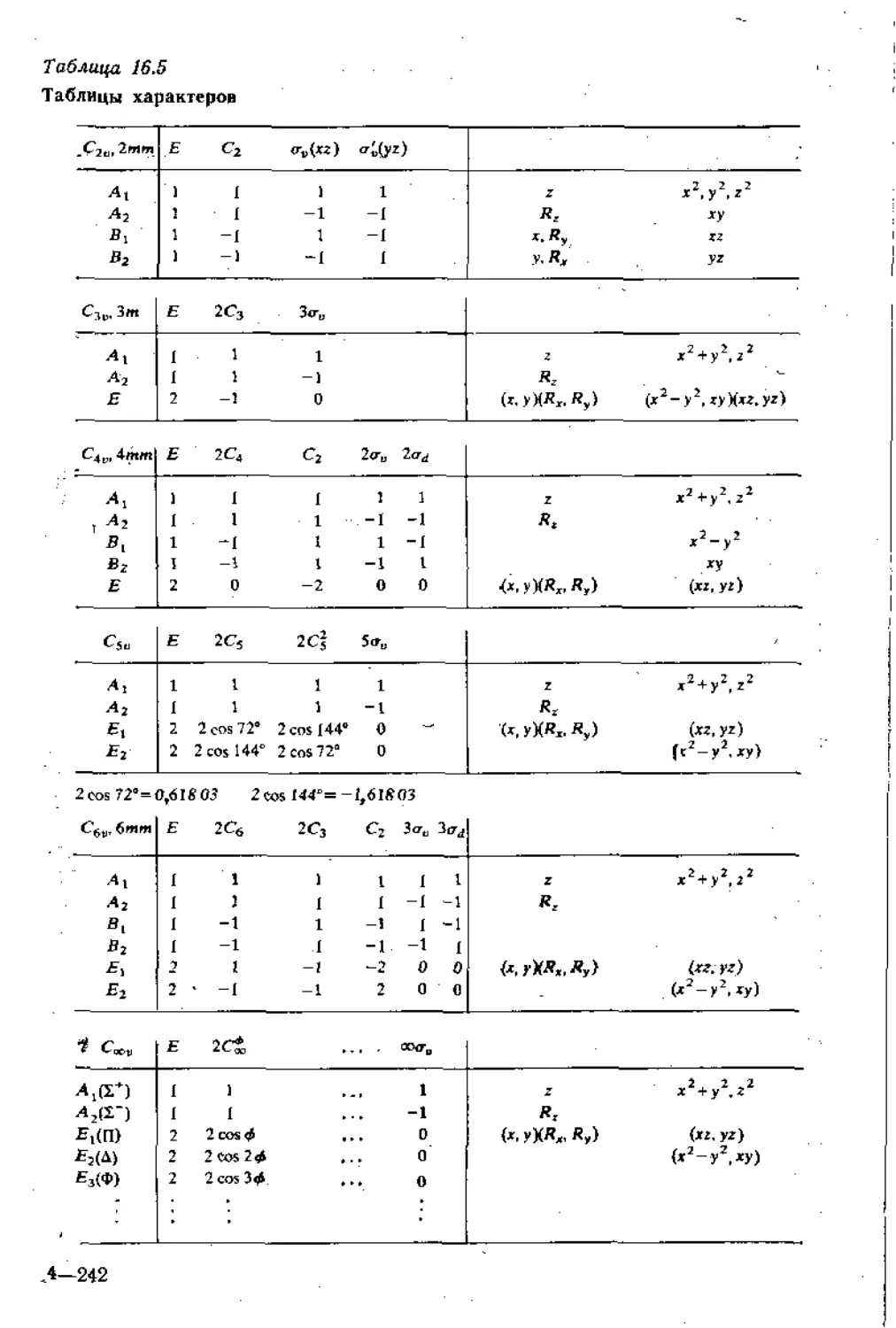
выглядит несколько похожей на следующую:
Сэо } Е 2С3 3aD
1 1
1 -1
А 1
Л, 1
z, х2 + у2 + z2, 2г2 — Xs — у*
Е 2
—1 0 (х, у) {xz, уг) (ху, х2 —у2)
Rz
(Rz, Ry)
Требуется объяснить лишь одну особенность этой таблицы:
символы Rx, Ry и Rz в последней колонке. Ими обозначается
вращение, и в таблице показано, как они трансформируются при
операциях данной группы. Легче всего их понять с помощью
картинок, как показано на рис. 16.22 для R& которое соответству-
ет вращательному движению относительно оси г. Ясно, что ни
-одна нз операций симметрии данной группы не трансформирует
вращательное движение относительно г во вращательное движе-
ние относительно х и у, и поэтому R? не смешивается с Rx и Rv,
16. Симметрия. Описание и следствия
31
Рис. 16.22. Трансформации вращений в молекуле Сзе.
т. е. связано с одномерным неприводимым представлением. Оба
вращения Сз превращают 7?г в само себя; таким образом, в каж-
дом случае трансформацию Rz^Rz можно записать как Т?2 = Т?г1,
и характером этого одномерного представления является %(С3) —
= 1. Однако отражение обращает направление вращения
(рис. 16.22), и поэтому при каждой из Ои-операций — Rz -*—Rz. Это
выражается как — RZ = R (—1), а значит, матричное представле-
ние будет—1, а его характер таким же. Следовательно, Rz связа-
но с базисом, имеющим характеры неприводимого представления
Л2. Вращения Rx и Ry визуально представить немного труднее, но-
иптуитивно очевидно, что они перемешиваются при симметриче-
ских трансформациях и поэтому оба связаны с Е.
Все таблицы характеров в конце главы даются в форме таб-
лицы для C3!Jj приведенной выше. Они показывают, как трансфор-
мируются различные функции и вращения. Почему они так по-
лезны, будет объяснено в следующем разделе.
16.3. Использование таблиц характеров
Хотя характеры — суммы диагональных элементов матриц;
представления — нс содержат всей информации, имеющейся в
матрицах, они все же содержат достаточно информации, чтобы
играть важную роль в химии. Одна из причин состоит в том, что
таблицы характеров позволяют с первого взгляда сказать, будет
ли интеграл равен нулю без детального его вычисления. Это эко-
номит много времени и позволяет быстро качественно оценить-
свойства молекул.
Интегралы, стремящиеся к нулю. Предположим, что нужно вы-
числить интеграл
/=J fi (г) f2 (г) dr,
гле fi и f2 — волновые функции, распространяющиеся на всю мо-
лекулу. Например, может быть одной атомной орбиталью, а
32
Часть 2, Структура
Рис. 16.23, Величина интеграла (т. е. площадь) яе зависит от системы коорди*
наг, выбранной для его вычисления.
/г — другой; тогда I будет интегралом перекрывания между ними
(см. т. 1., разд. 15.2), и, если бы мы знали, что он стремится к ну-
лю, мы могли бы сказать без колебаний, что ft и f2 не перекры-
ваются и не вносят вклада в связывание в молекуле. Ключевым
моментом является| то, что величина интеграла не зависит от
ориентации молекулы (рис. 16.23). В терминах теории групп мы
могли бы сказать, что I не изменяется пи при каких симметриче-
ских трансформациях молекулы. Следовательно, каждая транс-
формация по симметрии приводит к тривиальному изменению
/-«-/. Поэтому интеграл должен быть базисом для полностью сим-
метричного, одномерного неприводимого представления А; груп-
пы молекулярной симметрии.
Теперь предположим следующее: мы знаем, что каждая нз
функций fi и f2 является членом базиса для неприводимых пред-
ставлений. Пусть f; — член базиса для неприводимого представ-
ления D\, a fs — для другого неприводимого представления D2.
Как определить, каким образом трансформируется их произведе-
ние fif&? Это важно, поскольку, если Произведение f[f2 не транс-
формируется как Л|, интеграл должен исчезнуть, так как / есть
базис для Aj. Рассматривая таблицы характеров, можно сказать,
какие неприводимые представления охватываются fifa. Правило
состоит в следующем:
1. Выясняем характеры для неприводимых представлений,
охватываемых базисами, членами которых являются и f 2, и за-
писываем их в два ряда по порядку операций.
Например: Пусть ft будет s-орбиталью sn молекулы NH3, а
f2 — комбинацией s3 (рис. 16.20). В С3у первая отвечает характе-
ру А{, а последняя является членом базиса для Е. Следователь-;
но, используя таблицу характеров для С3и, записываем '
fi- 111. j
А: 2-10.
2. Перемножаем числа в каждой колонке и записываем их з]
том же порядке. (
IS. Симметрия. Описание и следствия
33
Например: Для приведенного случая получаем
flfz' 2 I 0.
3. Просматриваем получившийся таким образом ряд и видим,
можно ли его разложить на сумму характеров неприводимых пред-
ставлений. группы. Если эта сумма не включает Ah то интеграл
должен быть равен нулю.
Например: Числа, полученные для C3tl, можно всегда выра-
зить в виде Cix(Ai) -}-С2х(А2)-1-Сзх(Е), и интеграл должен исче-
зать при Cj=O. В данном примере характеры 2, —1, 0 являются
только характерами Е, и поэтому интеграл должен быть равен
нулю. Проверка формы этих функций показывает, почему это так:
ss имеет узел, проходящий через орбиталь (рис. 16.20).
Если бы мы взяли в качестве комбинацию $ь а в качестве
fj орбиталь sn (рис. 16.20), то, поскольку каждая орбиталь от-
носится к типу А], в обоих случаях их характеры 1,1,1 и произ-
ведение равно 1,1,1, т. е. само является Аь Следовательно, sN и
Si могут иметь ненулевое перекрывание*.
Тот же метод можно использовать, чтобы выяснить, обяза-
тельно ли исчезают интегралы вида
Z=pt (г) (г)/з (г) ^т-
Этот интеграл не должен изменяться пи при какой симметриче-
ской трансформации молекулы, и поэтому он должен быть пред-
ставлением А] подходящей группы. Таким образом, тройное про-
изведение /д/а/з должно содержать компонент с представлением
А[. Это можно определить, расширив уже описанную процедуру
перемножением трех наборов характеров друг с другом с после-
дующим рассмотрением, содержит ли произведение характеры для
А1. Пример этой процедуры дац ниже.
Пример (вопрос 7). Стремится ли к нулю интеграл fd^.xdy-dr в тетраэдричес-
кой молекуле?
Метод. Обратимся к таблице характеров Т±- Найдем характеры представлений,
которые охватываются d.:, х и dVi! затем образуем xdVz и d^xd^. Проверим,
включает ли последнее произведение представление Ai.
Ответ. Из таблицы следует, что х и являются членами базисов, которые
охватывают Т%, a d г=—член базиса, охватывающего Е (последнее следует из
того, что Зг2—г2=2гг—№—-у-). Составим следующую таблицу:
* Мы говорили, что интеграл должен, быть равен нулю или может быть
не равен нулю. Теорети ко-групповое рассмотрение позволяет сказать, когда ин-
теграл должен быть пулевым, но могут существовать другие причины, по ко-
торым иитетрал равен нулю, несмотря на то что симметрия позволяет ему быть
енулевым. В данном случае, например, орбиталь яц может быть настолько не-
ольшой. что она не будет распространяться на области, где Д не равна нулю
3—242
34
Часть 2. Структура
E 8C3 3C2 6S4 6dd
fs — dp? 3 0 —1 —1 1
fi = X 3 0 —I —1 1
fits 9 0 1 1 1
fl ~ 2 — 1 2 0 0
fifth 18 0 2 0 0
Эти характеры представляют собой сумму Л1+Л3-|-2£'+27'14-2Г2. Таким обра-
зом, интеграл не обязательно будет равен нулю.
Комментарий. Можно сделать три замечания; а) Этот интеграл встречается в
теории электронных спектров, и мы встретимся с ним вновь в гл. 18, б) Тот
же вывод можно было бы с де лить при рассмотрении dxv вместо dVI, так как dIV
также принадлежит к Г2. Более глубокое рассмотрение симметрии данной проб-
лемы показывает, что интеграл стремится к нулю, Будьте осторожны, посколь-
ку доводы, основанные на таблицах характеров, строго справедливы только
тогда, когда они показывают, что интеграл должен быть нулевым, в) Нахож-
дение суммы характеров, чтобы определить, включает ли она А,, может занять
много времени. Здесь есть простое правило; ултножьге характеры (18, О, ...) на
число элементов в заголовке каждой колонки (1, 8. и сложите полученные
числа (18+04-... =24}. Разделите на порядок группы (24). Это даст число (1),
показывающее, сколько раз At входит в приводимое представление.
Орбитали с ненулевым перекрыванием. Только что приведенные
правила позволяют сразу сказать, какие атомные орбитали мо-
гут иметь суммарное перекрывание в молекуле. Мы видели, что
интеграл перекрывания между Sn и линейной комбинацией s3 ра-
вен пулю в молекуле С3с, и поэтому центральная 2$-орбиталъ не
имеет суммарного перекрывания с этой комбинацией орбиталей.
Однако она может иметь ненулевое перекрывание с комбинацией
S), а значит, образование одной из молекулярных орбиталей NH3
можно рассматривать как результат перекрывания этих двух
орбиталей (рис. 16.24). Поскольку эта орбиталь (и соответству-
ющая разрыхляющая «аптисвязывающая») трансформируется
как Ai, она называется ai-орбиталью. Существует общее прави-
Рис, 16.24. Интегралы ненулевого перекрывания и приемлемые связывающие
орбитали в NHS.
IS. Симметрия. Описание а следствия
35
ло, согласно которому суммарное ненулевое перекрывание могут
иметь только орбитали одинакового типа симметрии, и поэтому
только эти орбитали могут комбинироваться связывающим и ан-
тисвязывающим способами.
Комбинации Sa и s3 имеют симметрию Е, Обладает ли атом
азота орбиталями, которые могут давать суммарное перекрывание
с ними? Интуиция подсказывает, что для этой цели должны под-
ходить рх- и ру-орбитали азота. Можно быстро проверить, так
ли это. У атома водорода 2рж-орбиталь имеет алгебраическую
форму
ф2рЖ=Лт*ехр(—г/2а0),
и хотя Орбитали атома азота не точно такие же, его 2рх-орбиталь
определенно имеет форму
где f(r) — функция расстояния по радиусу от ядра. Аналогично
для 2ру-0рбитали
Каким образом трансформируются эти орбитали в С3и? Посколь-
ку операции симметрии не влияют на радиус г, они не влияют
также на и трансформация управляется факторами х и у.
Но в C3tl функции х и у связаны с Е (см. табл. 16.5). Следова-
тельно, и ру-орбитали центрального атома азота имеют сим-
метрию Е и могут иметь ненулевое перекрывание с комбинациями
-s2- и Зз-орбиталей протонов. Образующиеся е-орбитали показаны
на рис. 16.24.
Сила метода может быть продемонстрирована вопросом; мо-
гут ли d-орбитали центрального атома принимать участие в свя-
зывании с атомами водорода? Достаточно знать, что независимо
от типа атома d-орбитали имеют следующую форму;
(Зг2—г2) / (f), dX2~y^(x* —у1) f (г),
^яегу/Д), d^yzfir), dX2^xzf(f).
Их симметрию можно взять из таблиц характеров, просто по-
смотрев, как трансформируются квадратичные формы ху, xz и
т- Д. Из таблицы для C3t, видно, что d7* имеет симметрию Аг, па-
ра dxy) —симметрию Е и пара (dxz, dyK) —тоже симмет-
рию Е. Отсюда сразу следует, что молекулярная орбиталь может
образоваться перекрыванием dz> -орбитали азота (или фосфора и
т- Д-) с комбинацией Si водородных орбиталей, а каждая из обе-
их Других пар d-орбнталей может перекрываться с двумя комби-
нациями s2 и $3.
3*
36' Часть 2. Структура
Хотя этот метод был проиллюстрировал для группы С3в, он
совершенно общий, и необходимость знать, как перекрываются
S-, р- и d-орбитали, является одной из Причин, почему функции х,
xz и т. д. включены в таблицы характеров.
Пример (вопрос 8). Четыре водородные Is-орбитали метана имеют симметрию
Ai + TV С какими орбиталями атома углерода они могут перекрываться? Что
было бы, если бы атом углерода имел доступные d-орбнтали?
Метод. Ищем симметрию Ai а Л у 5-, р- и d-орбита.чей.
Ответ. В комбинации A-L водородные орбитали могут перекрываться с s-орби-
талью углерода (симметрия А), а в комбинации Га —с тремя р-орбиталями
(которые охватывают симметрию Ts). Если доступны d-орбитали, то d4-, Аг-
и dji-орбитали имеют симметрию Тц, и поэтому они могут образовывать связи
и «антисвцзаз- с комбинацией Т2 воцородтгых орбиталей. Ни одна d-орбиталь не
трансформируется как Ль а следовательно, как dj2, так и dA.2_y2 являются не-
связывающими,
Комментарий. Молекулярные орбитали, образующиеся при перекрывании Ai—
—Л(, обозначаются символом а1р а орбитали, образующиеся при перекрыв а ни ни
Т2—Т?.— символом ti. Две несвязывающие d-орбитали (симметрия Е) можно
назвать е-орбиталями.
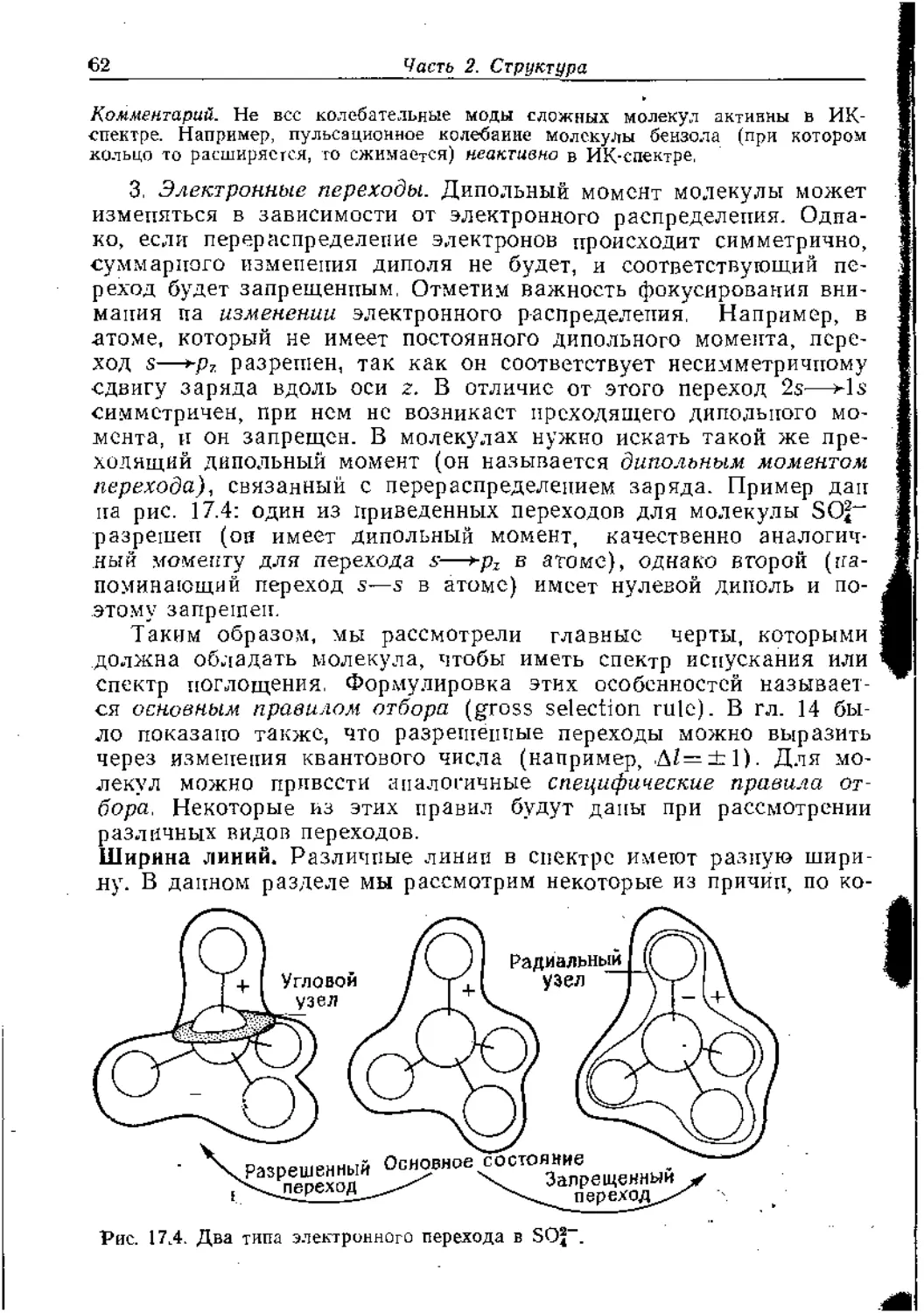
Правила отбора. В гл. 18 мы увидим, что интенсивность спект-
ральных линий, возникающих вследствие перехода молекулы
между некоторым начальным состоянием с волновой функцией
ф; и конечным состоянием с волновой функцией ф:, зависит от
величины, названной дипольным моментом перехода. г-Комно-
нента этой величины определяется соотношением
А ~ J ф^сфДт.
Чтобы установить, когда она равна нулю, формулируется прави-
ло отбора для данной молекулы. Дипольный момент перехода
имеет точно такую же форму, как интеграл, с которым мы уже
сталкивались: /— J fifzfzdr. Поэтому, если известна симметрия
состояний, между которыми происходит переход, можно сразу
сказать, будет ли разрешен данный переход. Дипольный момент
перехода имеет также х- и у-компопенты, определяемые анало-
гично; таким образом, чтобы быть уверенным, что определенный
перехюд не может произойти, мы должны исследовать, все ли три
компоненты ух, и рг равны нулю.
Метод можно проиллюстрировать, рассмотрев вопрос о том,
может ли молекула воды (Czv) дать линию в спектре, соответст-
вующую переходу электрона с агорбитали (рис. 16.25) на ^-ор-
биталь (также показана на рис. 16.25). Необходимо проверить
три возможности, по одной для каждой из компонент дипольного
момента перехода. Функция fa в J fifzfsdr-—Это х, у иди z, и из
таблицы следует, что они трансформируются соответственно как
16. Симметрия. Описание и следств ия
37
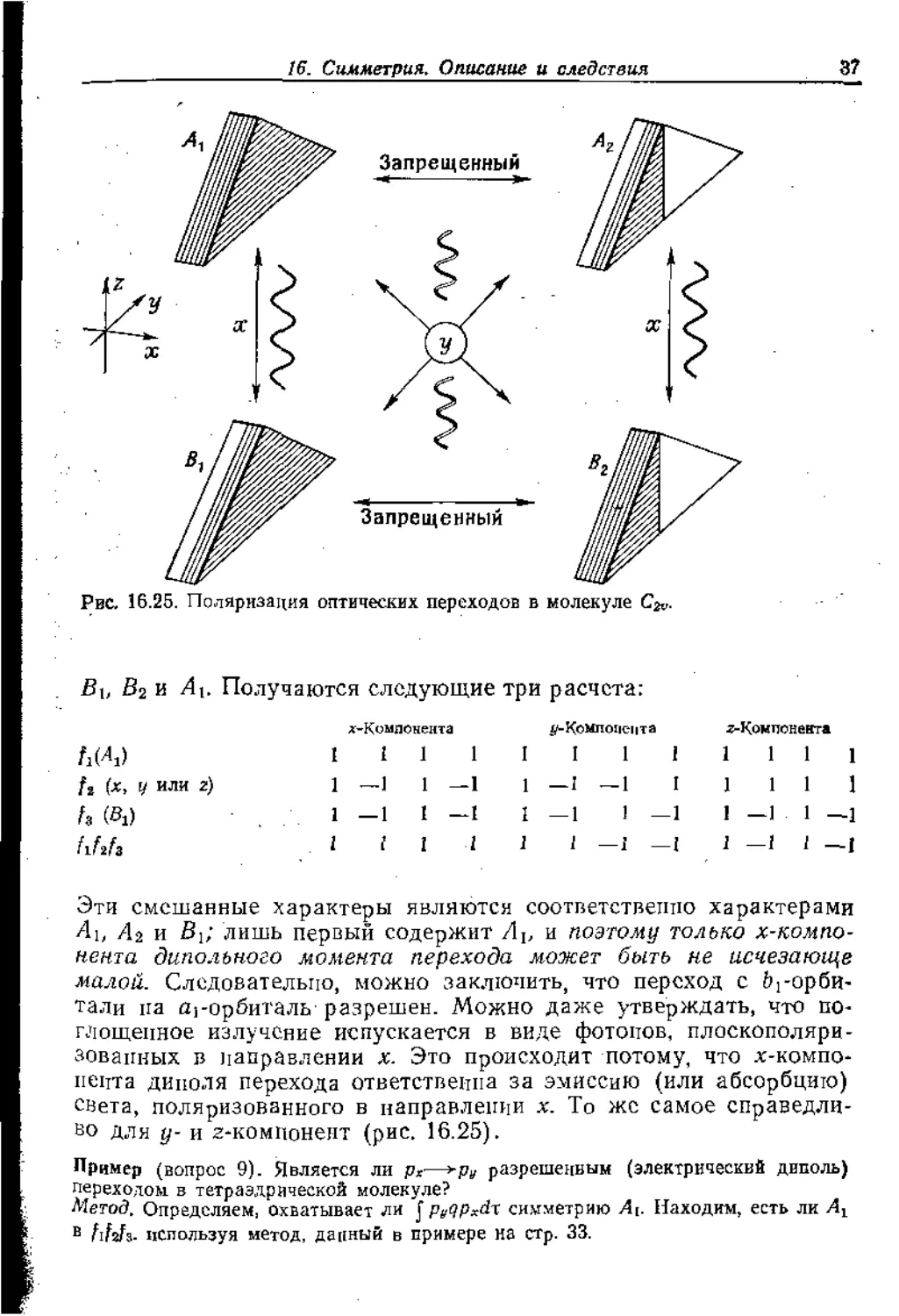
Въ в2 и Ль Получаются следующие три расчета:
^-Компонента ^-Компонента г-Конпонеита
А(Л) 1 1 1 1 I I 1 1 1 1 1 1
(х, у или г) 1 —1 1 -1 1 —I -1 I 1 1 1 1
(а №) 1 —1 1 —1 1—1 1 - -1 1 —1 1 —1
fififa 1 1 1 1 1 1 —1 - -I 1 —1 1 —1
Эти смешанные характеры ЯВЛЯЮТСЯ соответственно характерами
4ц Аз и В{; лишь первый содержит Лр и поэтому только х-компо-
нента дипольного момента перехода может быть не исчезающе
малой. Следовательно, можно заключить, что переход с ^-орби-
тали па aj-орбиталь разрешен. Можно даже утверждать, что по-
глощенное излучение испускается в виде фотонов, плоскополяри-
зовапных в направлении х. Это происходит потому, что х-компо-
пеита диполя перехода ответственна за эмиссию (или абсорбцию)
света, поляризованного в направлении х. То же самое справедли-
во для у- и z-компонент (рис. 16.25).
Пример (вопрос 9). Является ли р?—>р^ разрешенным (электрический диполь)
переходом в тетраэдрической молекуле?
Метод. Определяем, охватывает ли ( peqpxdx симметрию At. Находим, есть ли А1
в используя метод, данный в примере на стр. 33.
38
Часть 2. Структура
Ответ. Используя тот же метод, что и выше, записываем
3 0—1—1 1 fs=Py(T2l 3 0-1-1 1
П = г(Т2) 3 0-1-1 1 М2(9 27 0 -1 -Ч 1
Метод, описанный в комментарии к примеру на стр. 36, позволяет сказать, что
А[ входит в приводимое представление одни раз. Тот же вывод можно сделать
для q—x и следовательно, переход рь-—>-ру в тетраэдрической молекуле
разрешен по симметрии.
Комментарий. Более детальный анализ (рассмотрение самих представлений, а
не характеров, т. е. суммы диагональных элементов представлений) показыва-
ет, что эффективным является лишь z-переход. Таким образом, свет будет поля-
ризованным по оси г.
Этот тип анализа очень полезен в молекулярной спектроско-
пии, поскольку, если нам известно, что основное состояние
Cst,-молекулы имеет симметрию А, и При наблюдении ее спектра
обнаруживаем, что свет, испускаемый при возврате возбужден-
ного состояния в основное, поляризован п направлении х, мы.
знаем, что возбужденное состояние имеет симметрию В{. Наобо-
рот, если мы хотим возбудить молекулу в состояние Вь то мы
знаем, что необходимо применить x-полярцзованный свет. Эта
информация получается прямо из таблиц характеров, и требует-
ся лишь минимум вычислений.
Симметризованные орбитали. До сих пор мы просто говорили о
комбинациях атомных орбиталей, которые служили базисом для
различных неприводимых представлений. Например, мы говорили
о комбинациях st, s2, s3 в NH3 и об а{- и ^-орбиталях в 112О.
Теория групп является частью «машины», в которую вводятся
первоначальные орбитали (например, Sn, Sa, seg Sc), а па выходе
получаются орбитали разной симметрии. Такие орбитали называ-
ются симметризованными, поскольку они представляют собой
комбинации, «подогнанные» к симметрии молекулы.
Правила построения таких орбиталей состоят в следующем
(причины, по которым они «работают», можно выяснить из книг,
перечисленных в списке литературы). Составим таблицу, показы-
вающую, как каждая операция группы воздействует на каждую
орбиталь. Например, в NH3 исходные четыре орбитали ведут се-
бя следующим образом:
Первоначальный набор .?N SA Jg
16. Симметрия. Описание и следствия 39
Чтобы получить орбиталь некоторой симметрии, берем каждую
колонку в отдельности и производим следующее:
1. Умножаем каждый член колонки на характер соответству-
ющей операции.
2. Складываем все орбитали в каждой колонке с коэффици-
ентами, определенными согласно и. 1.
3. Делим эту сумму на порядок группы (число элементов).
В А; все характеры равны 1, и поэтому первая колонка дает
Sn+Sn+..- = 6s>;. Порядок группы равен 6, так что орбиталь сим-
метрии А[, которую можно построить из орбитали азота, есть
просто Sjr. Применяя тот же метод к колонке под заголовком 8д,
в качестве орбитали симметрии Аь которую можно построить из
трех водородных 1 s-орбиталей, получаем —|— (sa4-$b4-Sc4-Sa4-
Ч-Sc+sb) =-?г 6sa4-Sb-]-sc). Та же Aj-орбиталь получается при рас-
смотрении двух оставшихся колонок, и, следовательно, они не
дают больше информации. Тогда одна из молекулярных орбита-
лей общей симметрии А] (щ-орбиталь) точно является соответст-
вующей линейной комбинацией
+ СН (SA + SB + Sc) >
где коэффициенты Сц должны находиться решением соответ-
ствующего уравнения Шредингера и непосредственно не следуют
из симметрии задачи.
Предположим теперь, что мы делаем ошибку, пытаясь создать
молекулярную орбиталь симметрии А2. Предыдущий анализ по-
казывает, что данный базис дает только щ- и е-орбитали. Что мы
получим, применяя приведенные правила? Характеры для шести
операций равны соответственно 1, 1,1, —1, —1, — 1. Следователь-
но. сумма в первой колонке равна нулю, но опа также равна ну-
лю в остальных трех колонках. Таким образом, соответствующая
линейная комбинация симметрии Aj исчезает как для орбиталей
азота, так и для водородных орбиталей. Эти правила даже ис-
ключают возможность ошибки.
Когда мы пытаемся построить две е-орбитали, необходимо
проделать некоторые дополнительные расчеты, разрешенные пра-
вилами. Это всегда необходимо для представлений с размерно-
стью больше 1, поскольку правила основаны на свойствах харак-
теров. и, так как последние нс содержат всей информации самих
матриц, не всегда получаются однозначные результаты. В одно-
мерных представлениях характеры также являются представле-
ниями, и поэтому неоднозначность отсутствует. В самом деле, по-
скольку характеры являются суммами элементов, правила дают
сумму орбиталей, образующих базис для Е. Можно видеть, что
это происходит следующим образом. Характеры операций в пред-
ставлении £ равны соответственно 2, —1, —1, 0,0,0. Таким обра-
40
Часть 2. Структура
зом, колонка 5ыдает25м—Sn—Sn + 0 + 0 + 0—0. Другие колонки дают
-£-(2sa—«в—Sc), (2sb—sc—Sa) и (2sc—sa—sB)- Но для
двумерного представления три орбитали не могут быть базисом,
и на самом деле эти орбитали не являются линейно-независимы-
ми (любую из них можно выразить как сумму двух других). Ес-
ли взять разность двух последних, то мы получим -g- (зв—sc), и
поэтому в качестве пары е-орбиталей молекулы должны быть взя-
ты эта комбинация и первая из трех, написанных выше.
Метод, рассмотренный здесь для С3„, можно применить ко всем
молекулярным точечным группам, и, следовательно, данные пра-
вила позволяют построить любую требуемую симметризованпую
комбинацию.
16.4. Симметрия кристаллов
Уже давно предполагалось, что правильная внешняя форма
кристаллов означает регулярность внутреннего строения. Тот
факт, что кристаллы — это правильные симметричные тела, поз-
воляет предположить, что методы, описанные в данной главе,
должны быть основой как для их классификации, так и для бы-
строй оценки их физических свойств. Кроме того, поскольку кри-
сталлы построены из ионов, атомов или молекул, соединенных
друг с другом способом, обусловливающим внешний вид, или
морфологию (от греческого морфос — форма), кристалла, следу-
ет также ожидать, что теория групп дает путь обсуждения того,
как локальная симметрия молекул в кристалле может определять
его общую симметрию.
Кристаллы снаружи. Симметрия и классификация. Наблюдение
множества кристаллов приводит к заключению, что все они могут
быть отнесены к одной из семи правильных фигур. Эти основные
правильные фигуры называются семью кристаллическими систе-
Рис. 16.26. Кристаллы, принадлежащие к кубической системе (а) и моноклинной
системе (б).
16. Симметрия. Описание и следствия
41
Таблица 16.3
Семь кристаллических систем
(^нг.тема Симметрия
Кубическая 4 оси третьего порядка
Моноклинная 1 ось второго порядка
Ромбическая 3 оси второго порядка
Тетрагональная 1 ось четвертого порядка
Триклинная Нет осей симметрии
Гексагональная, включая ромбоэдра- 1 ось третьего порядка или 1 ось шесто-
чсскую (или тригональную) Jro порядка
мами. К какой системе принадлежит данный кристалл, опреде-
ляется измерением углов между его гранями и определением чис-
ла осей, необходимых для решения вопроса о принципиальных
особенностях его формы. Например, если необходимы три экви-
валентные и взаимно перпендикулярные оси, то кристалл при-
надлежит к кубической системе (рис,- 16.26. а). Если необходимы
две перпендикулярные, эквивалентные оси и третья неэквивалент-
ная ось, ле.перпендикулярная первым двум, то кристалл принад-
лежит к моноклинной системе (рис. 16.26, б). Системы и их ми-
нимальная симметрия даны в табл. 16.3.
Установление кристаллической системы точно не определяет
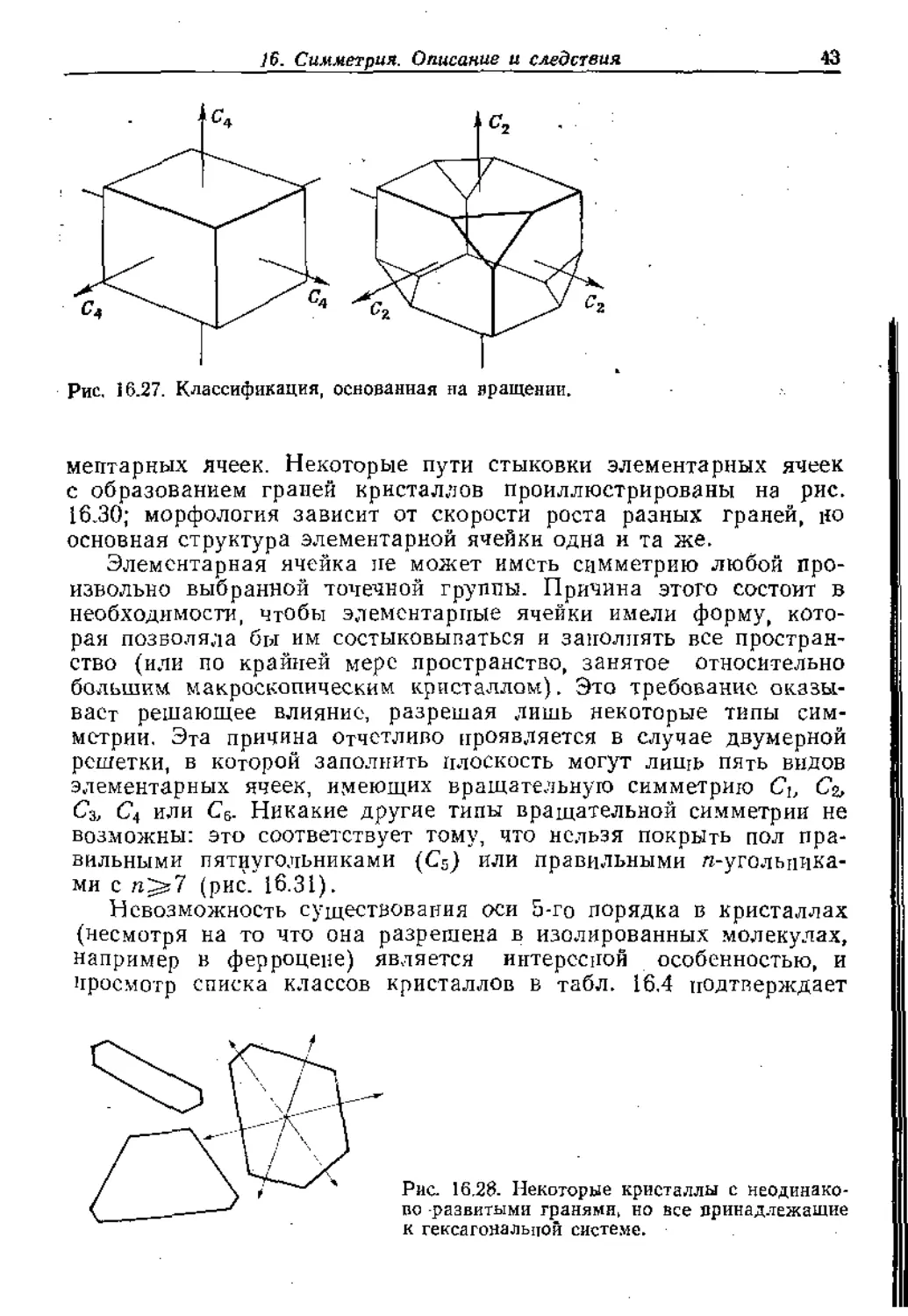
общую симметрию кристалла. Например, на рис. 16.27 показаны
два кристалла, принадлежащие к кубической системе, однако
общая симметрия второго ниже, чем первого. Это различие в сим-
метрии появляется, когда за основу принимаются операции вра-
щения: в то время как первый кристалл имеет ось 4-то порядка,
второй имеет лишь ось 2-го порядка. Первый кристалл имеет пол-
ную кубическую симметрию, а второй'—симметрию тетраэдра, и
поэтому они принадлежат к разным классам кристаллов. Все семь
кристаллических систем можно подразделить на ряд классов кри-
сталлов, если принять во внимание отражение, вращение и ин-
версию.
Точно так же как идентификация элементов симметрии моле-
кул позволяет классифицировать их по принадлежности к особым
точечным группам, элементы симметрии кристалла позволяют
классифицировать его в соответствии с точечной группой. В от-
личие от изолированных молекул, для которых возможно беско-
нечное число точечных групп, кристаллы могут принадлежать
только к 32 точечным группам. Эти 32 точечные группы являются
32 классами кристаллов. Каждая из семи кристаллических си-
стем может быть подразделена на несколько разных классов, а
32 класса могут быть распределены по семи системам, как пока-
зано в табл. 16.4. Кристаллографы номенклатуре Шёнфлиса
42
Часть 2. Структура
Таблица 16.4
7 кристаллических систем и 32 класса кристаллов
Систему Классы ,, . обозначение Обозначение Шенфлнса Гериана-Могена
1. Триклинная 2. Моноклинная 3. Ромбическая 4. Ромбоэдрическая 5. Тетрагональная Clr Ct 1, Г С\> Сз, Cafc m, 2, 2/m С№, D2, D2ft 2mm, 222, mmm C3, Cte, Ds, D3h, 5e 3, 3 m, 32, G2m, 3 C4, Civ, C4h, D3d, Dih> St 4, 4mm, 4/m, 422, 42 m. 4mmm, ~4
6. Гексагональная 6, 6|3z73, 622, бвж
7. Кубическая или правильная T, Td, Th, o, Oh 23, 43 m, m3, 43, m3m
(стр. 10) предпочитают систему Германа — Мотена, поэтому в
таблицу включен «словарь для перевода».
Важно подчеркнуть, что па данном этапе мы рассматриваем
эмпирические результаты морфологического анализа кристаллов.
Система и класс кристалла могут быть определены изучением его
внешнего вида и измерением углов между гранями. Единственная
трудность при этом состоит в том, что грани могут расти с раз-
ными скоростями, поэтому внешний вид кристалла может откло-
няться от идеальных форм (рис. 16.27; см. также рис. 16.28) и
отнесение к классу симметрии затрудняется наличием таких нару-
шений. Это, одиако, представляет собой техническую проблему,
на которой на данном этапе нет необходимости задерживаться.
Кристаллы изнутри. Решетки и элементарные ячейки. Кристалл
построен из множества ионов, атомов или молекул. Правильная
внешняя морфология означает, что кристалл образуется из не-
больших агрегатов, которые сами симметричны. Эти основные аг-
регаты (блоки), которые содержат несколько атомов или моле-
кул, называются элементарными ячейками решетки. Если пред-
ставить двумерный набор точек, аналогичный приведенному на
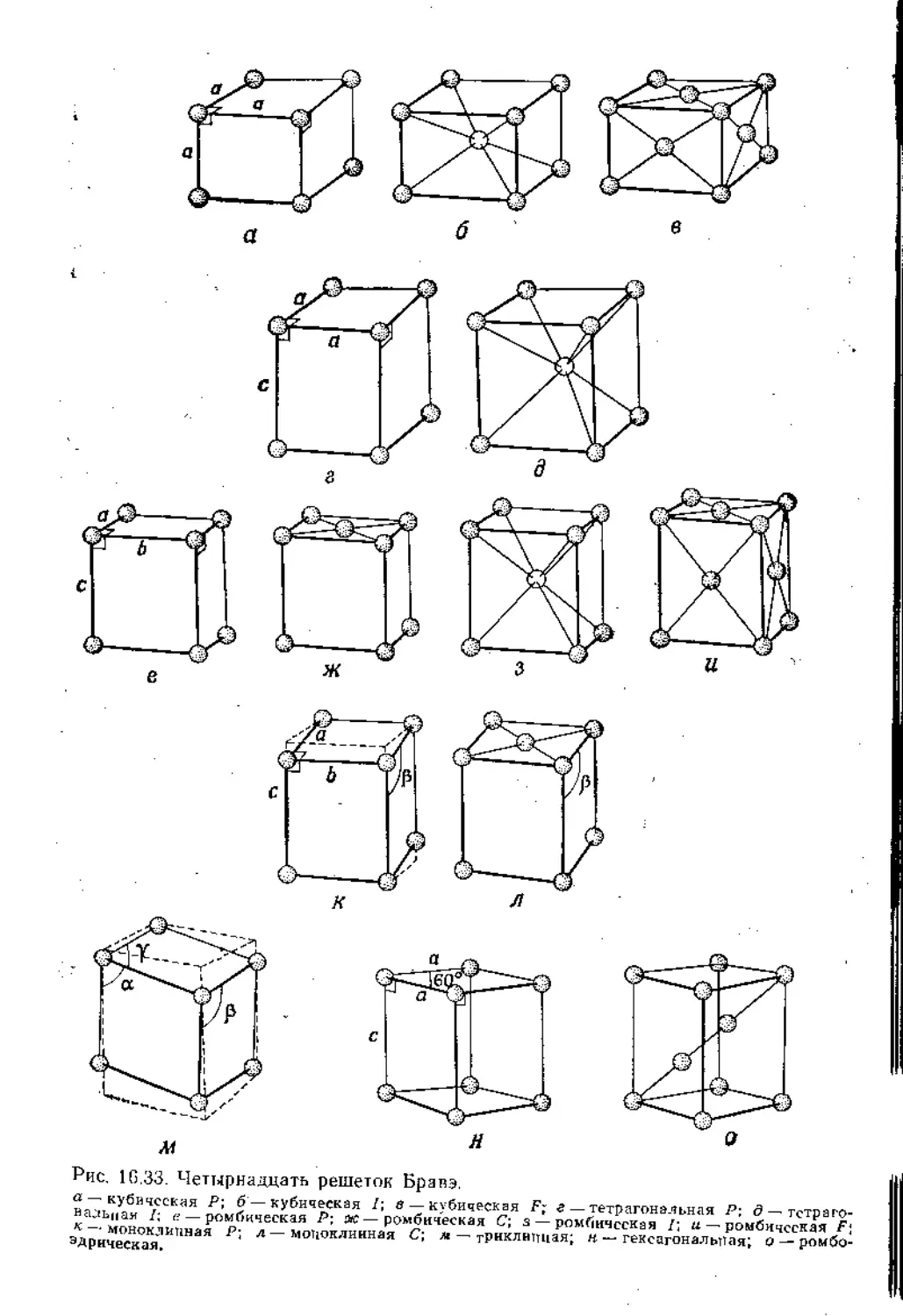
рис. 16.29, то легко видеть, что элементарные ячейки должны
иметь общую симметрию кристалла. Возьмем для примера куби-
ческую решетку (рис. 16.29, а). В целом кристалл имеет ось Сд,
перпендикулярную плоскости, и это означает, что элементарная
ячейка также должна иметь ось С4, поскольку в противном слу-
чае сам кристалл не содержал бы ее. Следовательно, нужно ожи-
дать, что морфология кристалла обусловлена симметрией эле-
16. Симметрия. Описание и следствия
43
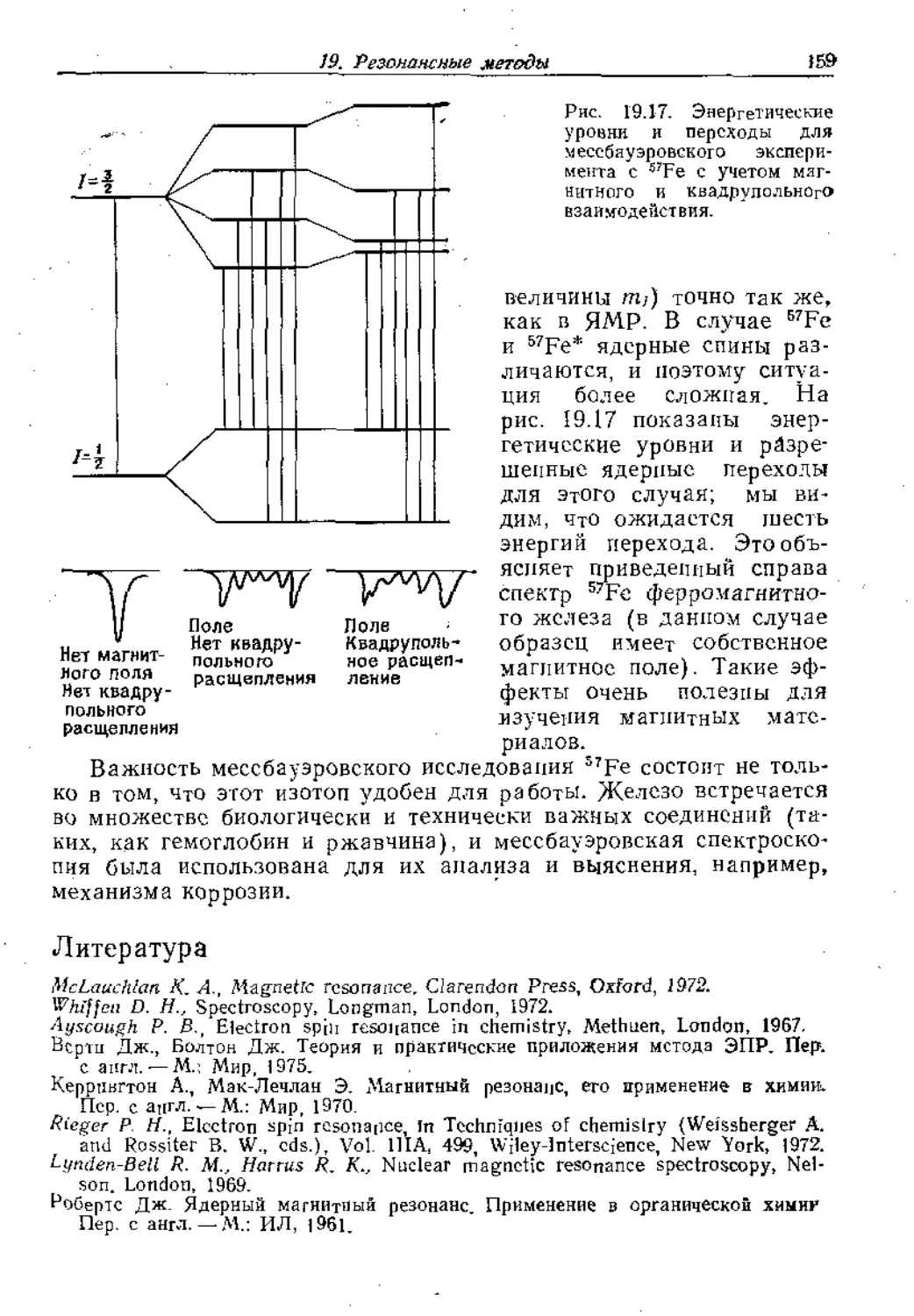
Рис, 16.27. Классификация, основанная на вращении.
мептарных ячеек. Некоторые пути стыковки элементарных ячеек
с образованием граней кристаллов проиллюстрированы на рис.
16.30; морфология зависит от скорости роста разных граней, цо
основная структура элементарной ячейки одна и та же.
Элементарная ячейка не может иметь симметрию любой про-
извольно выбранной точечной группы. Причина этого состоит в
необходимости, чтобы элементарные ячейки имели форму, кото-
рая позволяла бы им состыковываться и заполнять все простран-
ство (или по крайней мерс пространство, занятое относительно
большим макроскопическим кристаллом). Это требование оказы-
вает решающее влияние, разрешая лишь некоторые типы сим-
метрии, Эта причина отчетливо проявляется в случае двумерной
решетки, в которой заполнить плоскость могут лишь пять видов
элементарных ячеек, имеющих вращательную симметрию Cj, С&
С$, С4 или Св. Никакие другие типы вращательной симметрии не
возможны: это соответствует тому, что нельзя покрыть пол пра-
вильными пятиугольниками (С$) или правильными га-угольпика-
ми с п^7 (рис. 16.31).
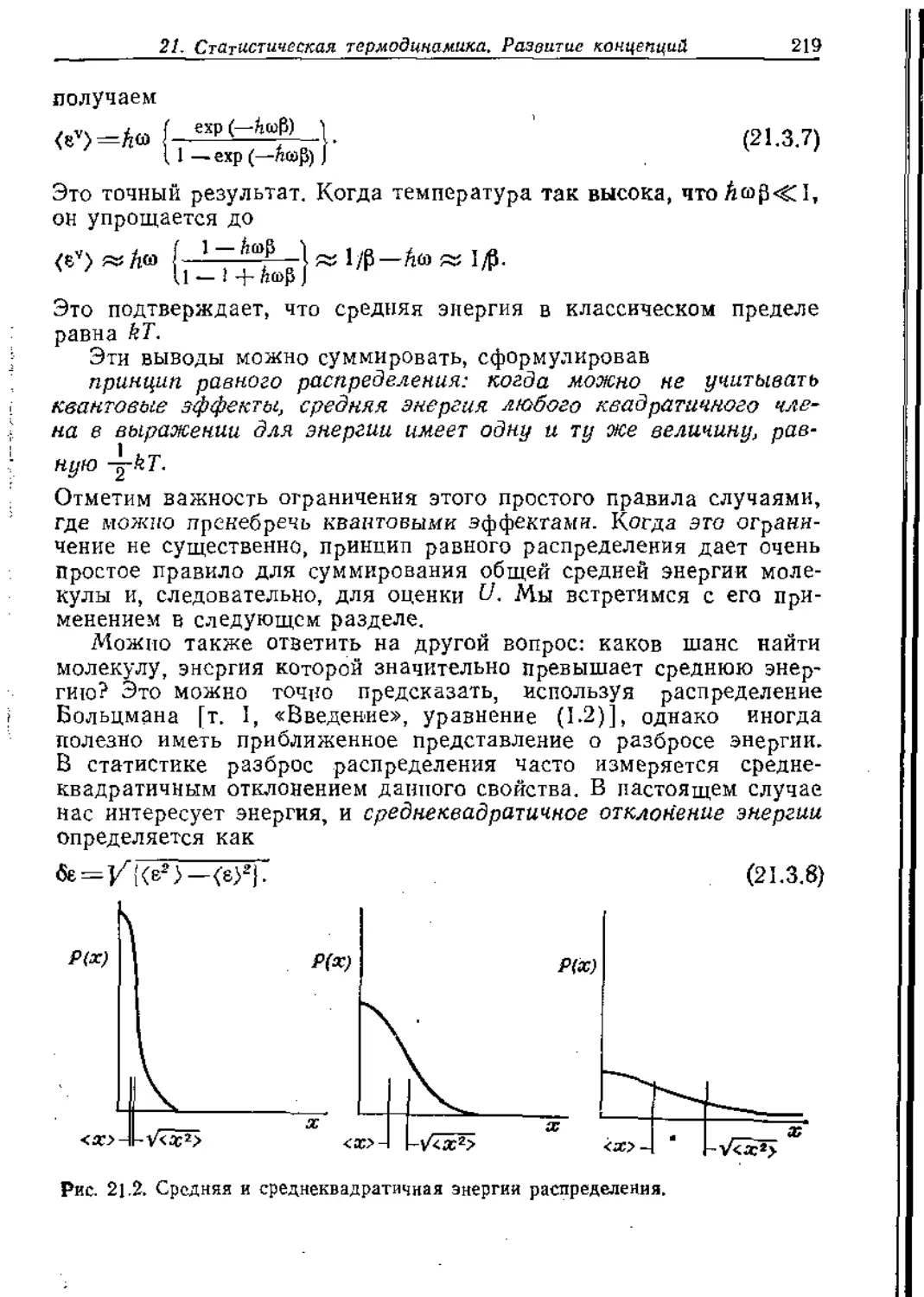
Невозможность существования оси 5-го порядка в кристаллах
(несмотря на то что она разрешена в изолированных молекулах,
например в ферроцене) является интересной особенностью, и
просмотр списка классов кристаллов в табл. 16,4 подтверждает
Рис. 16,28. Некоторые кристаллы с неодинако-
во -развитыми гранями, но все принадлежащие
к гексагональной системе.
44
7деть 2. Структура
Рис. 16.29. Две элементарные ячейки двумерной решетки.
отсутствие какой бы то ни было группы с осью СД Доказательст-
во ее отсутствия получается следующим образом. Рассмотрим
линейное расположение атомов, показанное на рис. 16.32. Можно
представить, что эта линия выделена из кристаллической решетки,
имеющей вращательную симметрию n-го порядка. Поскольку ре-
шетка имеет симметрию п-го порядка, кристалл можно вращать
вокруг атома Лз на уол 360°/п, и после такого вращения решетка
должна быть неотличимой от первоначального вида. Это означа-
ет, что в заполненной решетке должен быть атом в точке (узле)
и т. д. Такое же вращение можно осуществить вокруг любого
другого атома, и при этом будут воспроизводиться все узловые
точки кристалла (края кристалла мы игнорируем и рассматрива-
ем только бескопечно большую решетку). Вращение на —360°М
также воспроизводит узлы решетки точно таким же образом. На-
пример, вращение на —360°/п вокруг атома А2 выводит атом At
в узел решетки А -
Теперь мы подходим к основному аргументу. Рассмотрим рас-
стояние между узлами решетки на первоначальной линии; пусть
оно будет равно а, Узлы решетки А" и A't находятся на расстоя-
нии аД-2а cos (360°/п) Друг от друга и лежат параллельно перво-
начальной линии. Однако, если атомы имеют правильное, перио-
Рис, 16.30, Форма кристалла и стыковка элементарных ячеек.
16. Симметрия. Описание и следствия
45
Рис. 16.31. Пространство заполняется объектами Сл-снмметрии только при я=
= 1, 2, 3, 4, 6.
дическое расположение, расстояние между ними должно быть
целочисленно кратным а. Следовательно, величина cos(360o/'n),
которая не может превышать единицу, ограничена значениями
0(«=1, 2), -g- (п = 3, 6) и 1(л = 4). Это значит, что разрешены
только Сп с п=1, 2, 3, 4, 6.
Для трехмерного расположения должно быть найдено 14 ти-
пов элементарных ячеек, которые стыкуйтся друг с другом и за-
полняют пространство; они называются решетками Бравэ. Эти
решетки приведены на рис. 16.33. Решетки с узлами только в уг-
лах называются п римитивнымщ когда узел находится в центре,
решетки называются объемно-центрированными; когда решетки
содержат атом на гранях, они называются гранецентрированными.
Отметим, что 14 решеток Бравэ составляют семь групп (представ-
ленных каждой примитивной решеткой), и Эти правильные фигу-
ры точно такие же, какие соответствуют кристаллическим систе-
мам, Таким образом, мы выявили первую характерную особен-
ность структуры кристаллов: семь кристаллических систем отра-
жают существование семи правильных форм, которые могут быть
Плотно упакованы п заполняют пространство.
Это наблюдение можно распространить с кристаллических си-
стем на классы кристаллов. Наличие определенных классов кри-
сталлов указывает, что отдельные элементарные ячейка имеют
соответствующую симметрию. Таким образом, тетраэдрическая
морфология кристалла указывает па то, что элементарная ячей-
46
Часть 2. Структура
Рис 16,32. Причина, по которой разрешенное значение п равно только 3, 2, 3,
4, 6,
ка имеет тетраэдрическую симметрию. Теперь ясен смысл изуче-
ния морфологии кристалла: из визуальных наблюдений мы мо-
жем установить симметрию элементарной ячейки и, следователь-
но, способ, каким упакованы молекулы, образующие кристалл.
Однако действительное положение атомов еще нельзя определить;
устанавливается только симметрия их расположения, но даже это
является ценной информацией. Определение детальной Структуры
будет рассмотрено в гл. 22.
Пространственные группы. Расположение элементарных ячеек в
пространстве. Как стыкуются элементарные ячейки данной сим-
метрии? Например, кирпичную стену можно построить разными
способами, несмотря на то что элементарные ячейки (кирпичи)
одни и те же. Сложность состоит в том, что для кристалла проб-
лема трехмерная и элементарные ячейки не обязательно прямо-
угольные. Для решения этой проблемы кроме локальной сим-
метрии нужно учесть трансляционную симметрию. Она указыва-
ет на пространственную группу симметрии, относящуюся к бес-
конечному объекту, заполняющему пространство. Аналогично вра-
щению и т. д. точечных групп теперь необходимо рассмотреть три
другие операции симметрии, связанные с перемещением в прост-
ранстве. Первая из них — простая трансляция (рис. 16.34, а). Она
передвигает объект точно на некоторое расстояние по прямой ли-
нии. Вторая операция — винтовая ось, при которой происходит
скручивание на некоторый угол, составляющий часть от 360е,
вместе с трансляцией (рис. 16.34, б). Третья операция—плос-
кость скольжения — представляет собой трансляцию с последую-
щим отражением в плоскости, через которую проходит ось транс-
ляции (рис. 16.34, в). Эти элементы симметрии не могут быть про-
извольно скомбинированы с 32 классами точечных групп элемен-
тарных ячеек, и поэтому можно построить лишь ограниченное
Рис. lfi.33. Четырнадцать решеток Брапэ.
а — кубическая Р; б —кубическая I; S —кубическая F; г — тетрагональная Р; д — тстраго-
нальпая р г; — ромбическая Р; ж — ромбическая С; з — ромбическая р и — ромбическая Г;
* моноклинная Р; л — моноклинная С; я — трнклнттцая; н ~ гексагональная' о — ромбо-
эдрическая. '
48
Часть 2, Структура
Рис. 16.34. Трансляция, пинтовая ось н плоскость скольжения,
число пространственных групп. В действительности существует
только 230 пространственных групп, которые объясняют струк-
туру всех возможных кристаллов, Определение пространственной
группы-—сложная задача, связанная с детальным изучением внут-
реннего строения кристалла, но здесь пет необходимости рассмат-
ривать ее.
Свойства кристаллов. Значение группы симметрии молекул по-
зволяет сразу сделать вывод об их физических свойствах; то же
самое можно сказать о кристаллах. Например, хотя сами по себе
индивидуальные элементарные ячейки кристалла могут н не быть
оптически активными, поскольку отдельно взятые они совмести-
мы с их зеркальными отображениями (стр,' 16), кристалл, кото-
рый они составляют, может иметь оптическую активность. Следо-
вательно, чтобы определить, обладает кристалл оптической ак-
тивностью или нет, нужно проверить его симметрию и посмот-
реть, отсутствует ли зеркально-поворотная ось (стр. 16), Кварц,
например, имеет симметрию 32 (которая в системе Шёнфлиса
обозначается как Т) и является оптически активным.^ то же вре-
мя кальцит имеет центр симметрии (его симметрия 3m иди D3d),
и поэтому мы сразу заключаем, что он не будет оптически актив-
ным.
Приложение. Матрицы
Матрицы — это просто ряды и столбцы чисел со специальными
правилами комбинации их друг с другом. В общем, матрица мо-
жет иметь любую форму, но мы рассмотрим только квадратные
матрицы нХн.
Таблица 16.5
Таблицы характеров
_C2tl,2mm E C2 <rv(xi) o'Lfj’z)
At i I ] 1 x2,y2,z2
А? 1 I -1 -I jy
Bi 1 -I 1 -I X»IZ
В2 i -] -I I y- R* . yz
С3р, 3m E 2C3 3cr„
I 1 1 z x2 + y2,t2
а2 I 1 -] вг
Е 2 -1 0 (x2-y2,zy)(xz.yz)
С41„ 4mm E 2C4 Ct 2<r„ 2<t4
Л1 ] I I 1 1 z x^y2.z2
т I 1 1 -I -1
1 -I 1 1 -I X2-/
Вг I -1 1 -1 I xy
Е 2 0 -2 0 0 <x, у MR,. R,) (xi, yz)
Qu E 2Q 2 ci 5»„
At 1 1 1 1 z " x2 + y2,z2
At I 1 1 -1 Ry
£t 2 2 cos 72’ 2 cos 144“ 0 (x, yXR,. Ry) (xz, yz)
Et 2 2 cos 144" 2 cos 72° 0 h2-y2.xy}
2 cos ~2’=O,6IS0j 2 cos 144"=- 1,6 IS 03
C6tlT6mm E 2C6 2C3 C2 Зо-J, 31ZJ
I 1 ] 1 I 1 Z X2+y2,!2
At I J I I -I -1
By I -1 1 -I I -1
Bt I -1 I -1-1 I
2 1 -1 -2 0 0 (х,уХД„Лу} te.yz)
Et 2 • -I -1 2 0 0 (x2-y2,xy)
4 cxv E 2C* , . г 0OffB
A^) I ] 1 z X2+y2.Z2
a2O.~) I I -1 R;
^1(П) 2 2 cos 0 (x, уКЯ4, Ry) (XI, yz)
£;(A) 2 2 Cos 2^ 0 (x2-y2,xy)
Е3(Ф) 2 2 cos 3^ * 0
Л—242
50 Часть 2. Структура
Fd, 43m Е вс, зс2 {игл
Л] 1 1 1 1 1 х2 + у2 + Д
[ [ 1 -1 ’"1
Е 2 -1 2 0 0 (2zI-Jti-y2,xI->>2)
Г, 3 0 ” [ 1 -[ (R,, Ry, RJ
Ъ 3 0 -1 -1 [ (x.r.z) (ху.хг.и)
0.43 Е sc. зс2 6С4 6С2
I 1 1 [ 1 x2l-y2 + z2
1 [ i -[ -[
Е 2 -[ .2 0 (1
р 3 0 -[ 1 -I (X. Ry)
Т) 3 0 -1 -[ 1 (xy.xx,уг)
Квадратная п-мерная матрица состоит из л2 элементов.
^11 Ali3 ••• Min
.- Л13 ( Л!,, ... Л'1,
_Л^П£ Мп2 ... Л'1ЛП _
Элемент в ряду г и столбце с обозначается Мгс (г и с соответст-
вуют широте и долготе на географической карте). Две матрицы
равны, что записывается в виде M = lV, если только все соответст-
вующие элементы равны: Mn — Nrc для всех г и с.
Сложение двух матриц записывается как M + N = P и произ-
водится путем Р,с = ЛДе4-Агге (т. с. складываются соответствующие
элементы). Например, если
м /5 6\
и N =( ),
\7 8/
то
8\
12/
Умножение двух матриц записывается как MN = P и произво-
дится путем Prc=ZqMrqNqc. Это правило можно запомнить на ос-
нове следующей диаграммы;
Iff. Симметрия. Описание и следствия
51
Например, для приведенных выше матриц
.„к, (1 2\/5 6\ /1X 5—f-2 х 7 1х6 + 2х8\ /19 22\
\,3 4/\7 8/ \Зх5-|-4х7 3x644x8^ ^43 60 J
Отмстим, что в общем случае NM=^MN. В данном случае, напри-
/23 34\
мер, произведение NM— Xj 4gl. Это означает, что матричное
умножение некоммутативно.
Существует определенный тип матриц, которые имеют специ-
альные названия и свойства. Среди них отметим следующие;
Диагональная матрица — это матрица, в которой МГс = 0, кро-
ме М, для которых г —с. Например, М = g) — диагональная
/О 1\
матрица, а I 2 q I — нет.
Единичная матрица записывается как 1; это диагональная
матрица с ненулевыми элементами, равными 1. Следовательно,
, /1 0\
1=to 1/ — двумерная единичная матрица.
Транспонированная матрица М записывается как М; элемен-
ты этих матриц связаны следующим образом: Мге — М,:г. Так, ес-
„ /1 2\ ~ /1 3\ , .
Ли М=(з 41, то М=1 2 4J (меняются местами ряды и столб-
цы)-.
Матрица, обратная матрице М, записывается как М~!; это
матрица, которая удовлетворяет условию ММ"1 = М_1М = 1. Она
строится на основе следующего набора правил:
1. Находим детерминант (det М) матрицы М. Например, если
( 1 о\ 1121
М== \3 4/’ т0 det М|з 41 - 1X4—2ХЗ = —2. Если det М ра-
вен нулю, то матрица М является сингулярной и М"1 нс сущест-
вует (точно так же, как О-1 соответствует неопределенности в
52
Часть 2. Структура
обычной арифметике). Если det М^О, то обратная матрица су-
ществует, поэтому читайте дальше.
2. Записываем перестановку М матрицы М. Например, М =
3. Определяем JW, где Мг£~ минор (с соответствующим зна-
ком) матрицы Мге. Например, М'=1 И. (Вообще говоря, ми-
нор — это детерминант матрицы с вычеркнутыми рядом г и столб-
цом с.)
4. Тогда
инверсия матрицы Мопределяется как M_[ = M7deGM.
/ 4 _______________2\ /—
М-’ = (1/-2) [4 =
эта матрица удовлетворяет
IX
1 , Очень легко про-
~2 /
соотношению ММ [-
Например,
верить, что
-AVM-1.
Одно из важных применений матриц (кроме их роли, как пред-
ставлений операций симметрии, рассмотренной в этой главе) со-
стоит в решении системы уравнений. Предположим, что вы встре-
тились с системой из п линейных уравнений вида
^21^1 “Ь -f- М2пХ1г = С2
и хотите найти х. Выразим это уравнение через матрицу М и
матрицы 1Хп (столбцовые векторы)
в виде
Мх- с
это уравнение с помощью правила умножения мат-
(проверьте г_* ;; ;
риц). Поскольку М-!М=1, умножим обе части на М-1 и получим
Х-.МА'
(так как 1х —х). Это и есть решение системы уравнений; для
его получения необходимо лишь инвертировать матрицу, и дан-
ные выше правила говорят, как это нужно делать.
16. Симметрия. Описание и следствия
53
Литература
Weyl Н., Symmetry, Princeton University Press, Princeton, 1952.
Джаффе Г., Орчин М. Симметрия в химии. Пер. с англ. — М.: Мир, 1967.
Colton F. A., Chemical a plica lions с>5 group theory, Wiley, New York, 1971.
Hall L. H., Group theory and symmetry in chemistry, McGraw-Hill, New York, 1969.
Bishop D. M., Group theory and chemistry. Clarendon Press, Oxford, 1973.
Buerger M. Introduction to crystal geometry, McGraw-Hill, New York, 1971.
Atkins P. UK, Child At. S., Phillips C. S. C., Tables for group theory. Clarendon
Press, Oxford, 1970.
Задачи
16.1. Назовите точечные группы, к которым принадлежат следующие объекты:
тар, равнобедренный и равносторонний треугольники, незаточенный и заточен-
ный карандаши, трехлопастный пропеллер, роза, снежинка, стол, вы сами.
16.2. Запишите элементы симметрии следующих молекул и назовите точечные
группы, к которым они принадлежат: NO2 (изогнутая), CHaCl, CCljH, СН2=СН2,
ijuc-CHCl=СНС1, транс-СНС1 = СНС1, нафталин, антрацен, хлорбензол.
16.3. Сделайте то же самое для следующих молекул: CHsCHs (заторможенная),
циклогексан (кресло), ВгН5, СО2, Со(еп)^ («еп» означает этилендиамин; не об-
ращайте внимания на его детальную структуру), S8 (коропа),
16.4. Группа Cat содержит элементы Е. Сг, оь !. Составьте таблицу группового
умножения. Найдите пример молекулы, обладающей симметрией этой группы.
16.5. Группа D?k имеет ось С2, перпендикулярную главной оси 2-го порядка, а
также горизонтальную зеркальную плоскость. Покажите, что Эта группа должна
иметь также центр инверсии.
16.6. Какие из молекул в задачах 16.2 и 16.3 могут иметь постоянный электри-
ческий дипольный момент?
16.7. Какие из молекул в задачах 16.2 и 16.3 могут быть оптически активными?
16.8. Рассмотрим молекулу воды (которая имеет симметрию С211) и возьмем в
качестве базиса для построения молекулярных орбиталей дне водородные ls-op-
бнтали, а также 2s- и 2р-орбитали кислорода. Напишите матрицы 6X6, кото-
рые носпроизводят влияние операций симметрии группы на этот базис.
16.9. Используйте матрицы, выведенные в предыдущей задаче, чтобы подтвер-
дить, что они правильно представляют групповые умножения: a) Cta„ = vv' и
б) OvO'^Cs.
16.Ю. Матрицы (одномерные) D(C3) —I, D(C2) = 1 и D(CS) = 1, О(С2) = —1 яв-
ляются разными матричными представлениями для группы С61) в том смысле,
что они правильно воспроизводят умножение СзСг — Се при D(C6J, равном 1 в
первом случае и —I во втором. Подтвердите это, используя таблицу характе-
ров. Какими являются представления пв я Ол в каждом случае?
16.11. Одно из полезных качеств таблиц характеров состоит в том, что их мож-
но использовать для очень быстрого получения выводов с минимальной затра-
той работы. В качестве первого в серии упражнений по их использованию рас-
смотрите молекулу NO2 (С?„). Комбинация pxi—рХ2. где ри и —орбитали
Двух атомов кислорода (ось х перпендикулярна плоскости), относится к сим-
метрии Аз. Есть ли у центрального агома азота какие-нибудь орбатали, с ко-
торыми эта комбинация может дать суммарное перекрывание? Что будет н
ТОМ случае, если центральным атомом является сера?
16.12, Основное состояние NO2 имеет симметрию А,. В какие возбужденные со-
стояния можно возбудить эту молекулу, когда она поглощает свет (дипольным
переходом), и какую поляризацию света необходимо использовать?
16.13. Молекула С1О2, имеющая симметрию С2„, поймана в твердую ловушку.
Известно, что ее основное состояние имеет симметрию Bi (она содержит одни
электрон на рл-орбитали, выступающей из компактной основы молекулы, а р»
54
Часть 2. Структура
имеет симметрию Bi). Свет, поляризованный параллельно оси у. возбудил мо-
лекулу до более высокого электронного состояния. Какова симметрия этого со-
стояния?
16.14, Какие состояния а) бензола н б) нафталина могут быть достигнуты при
поглощении света их основными состояниями А>е и какова поляризация перехо-
дов?
16.15. Какие неприводимые представления группы Td охватывают водородные
ls-орбитади в метане? Имеет ли центральный атом s- и р-орбитали, которые
могут образовывать с ними связи? Могли ли d-орбитали углерода, если бы они
были доступны, играть роль в связывании (в метане?
16.16. Предположим, что метай искажен до: а) симметрии Сзо удлинением од-
ной связи и 6) симметрии С2„ развертыванием его по принципу действия нож-
ниц. Стали ли d-орбитали более доступны для связывания?
16.17. Найдите подходящие симметризоваипые комбинации базисов, использо-
ванных для Н2О в задаче '16.8, Рассмотрите кислород и водородную пару от-
дельно и установите, какая кислородная орбиталь и с какой комбинацией водо-
родных орбиталей может иметь суммарное перекрывание.
16.18. Вернитесь к задаче 16.8. и подтвердите, определив все матрицы, что: а) эле-
менты симметрии одного класса имеют одинаковый характер, б) представление
приводимое и в) базис охватывает + 2В3
16.19. /-Орбитали менее известны, чем а-, р~ и d-орбитали, одпако они играют
важную роль в химии лантаноидов. Несмотря на то что мы не знакомы с их
формой, мы все же можем сделать выводы об их свойствах с точки зрения
симметрии. Но какова их симметрия? Они имеют следующие алгебраические фор-
мы; z(5zs—Зг2)/(г), у(5уг— Зг2)/(г), Х(5х2—3ra)/(r), z(№—р2)/(г), у(х2—z?)f(r),
x(z2—уг')1(т) и xpz/(r). Какие неприводимые представления охватывают эти ор-
битали в молекулах симметрии: а) Сг„, б) Сз„. в) Td и г) О/,.
16.26. Рассмотрите лантаноидный пои в центре а) тетраэдрического и б) окта-
эдрического комплексов. На основе теории кристаллического поля определите,
какие наборы орбиталей будут взаимодействовать с семью /-орбиталями.
16,21. Какие из следующих переходов разрешены в а) тетраэдрическом и б) ок-
таэдрическом комплексах: 1) dxy ~<1гг, 2) dIF—’fxvT?
16.22. В тексте мы установили свойства симметрии вращения с помощью про-
стого картипочного аргумента. Теперь мы сделаем это несколько более фор-
мально. При таком подходе считается, что угловой момент по оси z определя-
ется как хрц—урх, где рх и Ру — компопедгьг линейного момента. Подтвердите,
что зга z-компонента углового момента является базисом для Ai и СД,.
16,23, На какие неприводимые представления преобразуются трансляции и вра-
щения в следующих группах: а) С21,, б) Сз» в) С9о. г) Он?
16.24. Реакция молекулы на электрическое поле управляется способностью пос-
леднего растягивать электроивую структуру (трансляционное искажение). В ре-
зультате такого растяжения появляется тенденция к подмешиванию возбуж-
денных состояний в основное состояние молекулы (так что р-орбиталн могут
подмешиваться в s-орбитали, и молекула «выпячивается» в ту сторону, где эти
две орбитали интерферируют конструктивно). Есть ли необходимость рассмат-
ривать смешивание р-, d- и /-орбиталей с s-орбиталью центрального атома
а) октаэдрических и б) тетраэдрических комплексов?
16.25. Магнитное поле имеет тенденцию искажать молекулу скручннарием ее
вокруг направления, по которому действует поле. Скручивающее искажение
имеет симметрию вращения. Какие состояния могут подмешиваться под влШг
пием магнитного поля к основному состоянию: a) NOs, б) октаэдрического
комплекса Ti3+ и в) бензола, когда поле перпендикулярно плоскости молекулы?
16.26. Теория групп дает быстрый путь определения, когда интеграл равен ну-
лю. Вот два примера. Интеграл sin 0 cos 0 в интервале, симметричном относитель-
но 0 = 0, равен нулю. Докажите это анализом симметрии, начав с /i = sin 0, /4^
= cos 0, показав, что /, и /2 охватывают разные неприводимые представления
группы Сь (или С„, или С;), и используя доводы, описанные в разд. 16.3. По-
16. Симметрия, Описание и следствия
55
чему этот интеграл может не быть равным нулю, когда интервал не симметричен
относительно начала координат?
16.27, Стремится ли к нулю произведение хуг, когда интегрирование проводвтся
по: а) кубу, б) тетраэдру и в) гексагональной призме (центр каждой фигуры
в начале координат)?
16.28, На диаграмме показаны три кристалла железного пирита (или «золота
глупцов») FeS;. Они очевь тщательно вычерчены, и, как принято, вид нх дает-
ся из пункта, который Лонсдейл (Lonsdale К., Crystals and X-rays, Bell, 1948)
описал «как если бы глаз смотрел свысока, находясь немного выше и слегка
справа, подобно благотворительному консервативному правительству». Убедитесь,
что все они имеют черты, которые делают их членами кубической системы. Но
к какому классу принадлежат эти кристаллы?
16.29, Ниже показан кристалл сфалерита (ZnS). К какой кристаллической сис-
теме и к какому классу он принадлежит?
16,30. Единственным элементом симметрии голубых кристаллов пентагидрата
сульфата меди является центр инверсии. К какой системе и к какому классу
они принадлежат?
16.31, Кристаллы сахарозы, как найдено, имеют ось 2-го порядка в качестве
единственного элемента симметрии. К какой системе они принадлежат?
16.32, При внешнем изучении обнаруживается, что кристаллы каменной соли
имеют три оси 4-го порядка, четыре оси 3-го порядка, шесть осей 2-го порядка,
три зеркальные плоскости и центр инверсии, К какой системе и к какому классу
принадлежат такие кристаллы?
16.33, Морфология кристаллов много говорит о внутренней симметрии, но даже
простые элементарные ячейки могут обусловливать появление большого числа
внешних форм (см. приведенную выше диаграмму для кристаллов пирита).
Чтобы провести эксперимент с разными формами, которые могут вырасти даже
из кубической элементарной ячейки, возьмите около 1 фунта кубических кусков
сахара и посмотрите, какие внешние формы можно сконструировать их Стыков-
16.34, Используя сахарные кубнкп, покажите, что в углу в других отношениях
совершенного куба можно сформировать треугольную грань. Рассчитайте угол
между нею н верхней плоской поверхностью.
17 Определение структуры молекул.
Вращательные и колебательные спектры
Изучаемые вопросы
После тщательного изучения этой главы вы сможете;
1. Перечислить основные особенности спектроскопии поглоще-
ния, испускания и комбинационного рассеяния (стр. 57).
2. Связать интенсивность спектральных линий с заселенностью
начального состояния [уравнение (17.1.2)].
3. Сформулировать основные правила отбора для вращатель-
ных, колебательных и электронных переходов (стр. 60—62).
4, Объяснить ширину спектральных линий через доплеровский
сдвиг и уширение за время жизни (стр. 62).
5. Вывести вращательные энергетические уровни молекул и
сфопмулировать смысл квантовых чисел / и К (стр. 67).
6. Применить вращательные правила отбора для объяснения
чисто вращательных спектров (стр. 72).
7. Сформулировать основные правила отбора для вращатель-
ных спектров комбинационного рассеяния (КР) и объяснить об-
разование стоксовых и антистоксовых линий (стр. 74).
8. Описать колебательные энергетические уровни двухатомной
молекулы с учетом и без учета эффектов ангармоничности [урав-
нения (17.3.2) и (17.3.5)]. '
9. Применить колебательные правила отбора для объяснения
колебательного спектра двухатомной молекулы (стр. 81).
10. Описать причину появления Р-, Q-, R-ветвей в колебатель-
но-вращательном спектре (стр. 84).
11. Перечислить правила отбора и объяснить колебательные
спектры К.Р двухатомных молекул (стр. 87).
12. Описать нормальные моды многоатомных молекул и под-,
считать их число (стр. 88). , J
13. Определить термины колебание, активное и неактивное в
ИК-спектре, и колебание, активное и неактивное в спектре KPi
и сформулировать правило исключения (стр. 92, 93).
Введение 1
Основная причина появления спектральных линий в случае
молекул та же, что и в атомной спектроскопии: когда молекул®
переходит с одного энергетического уровня на другой, избыто^
17. Вращательные и колебательные спектры 57
энергии испускается в виде фотона. Различие между молекуляр-
ной спектроскопией и атомной спектроскопией состоит в большем
разнообразии путей, по которым может изменяться энергия мо-
лекулы: энергия может меняться не только при электронных пе-
реходах, но и при изменении колебательных и вращательных со-
стояний молекулы. Изучение молекулярного спектра может дать
большую информацию о силе связей в молекуле, ее размерах и
форме.
Богатая информация, которую можно извлечь из данных мо-
лекулярной спектроскопии, по сравнению с атомными спектрами
получается за счет того, что молекулярные спектры имеют более
сложную структуру и их труднее интерпретировать. Мы увидим,
что, хотя можно получить чисто вращательный спектр, колеба-
тельный молекулярный спектр обычно состоит не только из линий,
обусловленных изменениями колебательных энергий, но и содер-
жит линии, соответствующие изменениям вращательных энергий,
а электронный молекулярный спектр (который рассматривается
в следующей главе) имеет структуру, обусловленную как колеба-
тельными, так и вращательными изменениями. Простейший путь,
позволяющий справиться с этой трудностью, состоит в том, чтобы
разобрать каждый тип энергетических изменений по очереди, а
затем выяснить, как одновременное возбуждение различных ти-
пов движения влияет на внешний вид спектров. Тем цс менее все
виды спектров связаны некоторыми общими особенностями, ко-
торые мы выясним прежде всего.
17.1 Общие особенности спектроскопии
Экспериментально спектры можно цолучить при помощи трех
основных методов: спектроскопии испускания (эмиссионной спект-
роскопии), спектроскопии поглощения (абсорбционной спектро-
скопии) и спектроскопии комбинационного рассеяния (раман-
спектроскопии).
В спектроскопии испускания молекула переходит из высокого
энергетического состояния в более низкое. Избыток энергии ис-
пускается в виде фотона, и экспериментатор наблюдает частоты,
соответствующие каждой энергии перехода. Если происходит пе-
реход из состояния с энергией Е" в состояние с энергией Е!, то в
спектре наблюдается линия при частоте v, которая определяется
соотношением
— (17.1.1)
Иногда это соотношение выражают через длину волны l = c/v
или через волновое число о = 1/Л; последнее обычно измеряется в
обратных сантиметрах (см-1). Переходы между этими величина-
58
Часть 2. Структура
1,43 1,61 1,72 1,89 2,13 2,38-Ю4см-'
Рис. 17.1. Электромагнитный спектр.
ми и их связь с различными областями электромагнитного спект-
ра представлены на рис. 17.1.
В спектроскопии поглощения экспериментатор наблюдает, ка-
кие частоты поглощаются при прохождении падающего излучения
через образец. Если поглощается свет с частотой т, то молекула
может быть возбуждена фотоном с энергией hv. Это возможно,
когда молекула из начального состояния с энергией Е' может
быть возбуждена в состояние с энергией Е", причем v, Е' и Е"
удовлетворяют соотношению (17.1.1). Например, если молекула
находится на низшем энергетическом уровне (в основном состоя-
нии) и поглощает голубой свет с длиной волны 470 нм (т. е. ка-
жется красной при наблюдении в белом свете), можно сделать
вывод, что возбужденное состояние, в которое она переходит, на-
ходится по энергии на 21 000 см-1 (2,6 эВ) выше основного со-
стояния. Спектроскопия поглощения и спектроскопия испускания
дают одинаковую информацию об энергетических уровнях моле-
кул, и в общем выбор между этими двумя методами определяет-
ся практическими соображениями.
Спектроскопия комбинационного рассеяния (спектроскопия
КР)—метод, в котором энергетические уровни молекул опреде-
ляются по рассеянию ими света. Когда падающий пучок опреде-
ленной частоты сталкивается с молекулами образца, в рассеянном
излучении появляются новые частоты, поскольку при столкнове-
17. Вращательные и колебательные спектры
59
нии фотоны могут приобретать или терять энергию. Если при
столкновении происходит возбуждение молекул светом, они отби-
рают часть энергии у фотонов, и поэтому рассеянное излучение
имеет более низкую частоту, чем частота падающего излучения.
Если молекулы уже возбуждены перед столкновением, они могут
отдавать энергию. В этом случае выходящие фотоны имеют повы-
шенную энергию, что проявляется в увеличении частоты рассеян-
ного излучения. Экспериментатор анализирует частоты рассеян-
ного излучения и затем интерпретирует их через молекулярные
энергетические уровни. Сдвиги частот могут быть очень неболь-
шими, и если падающий пучок не имеет строго определенной дли-
ны волны (т. е. не является монохроматическим), то их трудно
интерпретировать. В последние годы спектроскопия комбинаци-
онного рассеяния получила значительное развитие, так как став-
шие доступными лазеры позволили создать источники света, ко-
торые удовлетворяют этому требованию и являются настолько
интенсивными, что появилась возможность использовать меньшее
количество образца и более короткие экспозиции.
Интенсивность спектральных линий. Беглый взгляд на любой
спектр, помещенный в данной и двух последующих главах, пока-
зывает, что они содержат линии разной интенсивности: одни силь-
ные, другие слабые. Некоторые линии, которые можно было бы
ожидать, не проявляются вовсе: в таких случаях, несмотря на то,
что может быть известным, что молекула обладает энергетиче-
скими уровнями, находящимися на некотором расстоянии ЛЕ
друг от друга, поглощение или испускание при частоте \=ЛЕ/!1
не обнаруживается. Чтобы объяснить эти особенности, необходи-
мо рассмотреть, как интенсивность зависит от числа молекул, на-
ходящихся в разных состояниях, при получении спектра и на-
сколько сильно отдельные молекулы могут генерировать или по-
глощать фотоны.
Заселенность и интенсивность. Интенсивность линии в спектре,
обусловленной переходом молекулы из некоторого начального
состояния с энергией Е, в конечное состояние Ер зависит от числа
молекул, которые имеют данную начальную энергию. Это число
называется заселенностью состояния с энергией Ei. Когда обра-
зец из N молекул находится при температуре Т, число молекул с
энергией Ei определяется распределением Больцмана (т. 1, стр. 21):
N (Е;) ~ЛГ exp (—EJkT).
Другая линия в данном спектре может соответствовать переходу,
при котором молекула первоначально находилась в состоянии El.
Аналогично интенсивность этого перехода будет пропорциональ-
на ЛДЕ'(), т. е. заселенности состояния с энергией EJ. Следова-
тельно, интенсивности этих двух линий связаны соотношением
(El)=N (Ei)/N(El)=exp[-(Ei-El)/kT]. (17.1.2)
60
Часть 2. Структура
(В данном случае мы не учитываем того, что на интенсивность
могут влиять другие факторы, а не только заселенность началь-
ных уровней.)
Если Ei—E'i намного больше kT, то число молекул с энергией
Ei намного меньше числа молекул с энергией Е’:. В этом случае
интенсивность линии с начальным состоянием Ei намного мень-
ше интенсивности линии с начальным состоянием £j. Такую си-
туацию можно проиллюстрировать, рассмотрев вещество при ком-
натной температуре, когда kT~ 200 см'[, Первое возбужденное
состояние молекулы обычно лежит на несколько десятков тысяч
см-1 выше основного состояния, и поэтому в спектре поглощения
обычно наблюдаются линии молекул в начальном основном
электронном состоянии, так как другие состояния заселены незна-
чительно, Колебательные спектры вещества при комнатной тем-
пературе обычно тоже можно рассматривать как результат пе-
рехода с низшего колебательного состояния, поскольку первое
возбужденное колебательное состояние лежит на несколько сотен
см-1 выше основного состояния и лишь слабо заселено. В проти-
воположность этому вращательные молекулярные уровни обычно
отстоят друг от Друга только на доли см-1 и многие из них засе-
лены даже при комнатной температуре; поэтому во вращатель-
ных спектрах поглощения наблюдаются линии, обусловленные пе-
реходами молекул с целого ряда заселенных вращательных со-
стояний.
Можно сделать устройства, позволяющие перед регистрацией
спектра возбудить молекулы до искусственного, неравновесного
распределения заселенности. Например, при фотолизе, электри-
ческом разряде иди химической реакции могут получаться моле-
кулы в различных электронных и колебательных возбужденных
состояниях; тогда в спектрах наблюдаются линии, обусловленные
переходами, начинающимися с этих возбужденных состояний.
Правила отбора и интенсивность. С идеей правил отбора, кото-
рые определяют, будет лп атомный переход разрешенным или
запрещенным, мы встречались в гл. 14 (т. 1): разрешенные пе-
реходы проявляются в виде линий в спектре; запрещенные линии
отсутствуют, несмотря на то что имелись энергетические уровни
на подходящем расстоянии друг от друга. В данном разделе мы
разовьем эту идею в применении к различным типам переходов,
которые происходят в молекулах.
Основная идея состоит в том, что электромагнитная волна с
частотой v генерируется электрическим диполем, осциллирующим
с частотой V. Сейчас мы увидим, каким образом Эта картина при-
менима к различным типам эмиссионных или абсорбционных пе-
реходов (КР-цереходы будут рассмотрены позднее),
]. Вращательные переходы. Молекула, обладающая постоян-
ным дипольным моментом, при вращении напоминает наблюла-
17, Вращательные и колебательные спектры
61
Рис. 17.2. Вращающаяся полярная мо-
лекула выглядит как осциллирующий
диполь^
телю, находящемуся в плоскости вращения, флуктуирующий ди-
поль (рис. 17.2). Если молекула не имеет постоянного диполя,
наблюдатель флуктуирующего диполя не увидит. Следовательно,
испускать или поглощать излучение путем перехода между раз-
личными состояниями вращения могут только молекулы с посто-
янным электрическим дипольным моментом. Постоянный диполь-
ный момент можно рассматривать как «рукоятку», с помощью ко-
торой молекула может «разволновать» электромагнитное поле,
вызвав его осцилляцию (и наоборот для поглощения).
2. Колебательные переходы. Когда связи в колеблющейся мо-
лекуле растягиваются пли изгибаются, дипольный момент может
меняться. Этот осциллирующий диполь заставляет «дрожать»
магнитное поле, т. е. вызывает его осцилляцию, и в результате
может происходить испускание или поглощение излучения. Не-
которые колебания не изменяют молекулярный диполышй мо-
мент (например, растягивающее связь движение в гомоядерных
двухатомных молекулах; рис. 17.3) и поэтому не взаимодейству-
ют с излучением и сами нс излучают. Следовательно, возбуждать-
ся электромагнитным излучением или генерировать излучение
могут только те колебания, которые сопровождаются изменени-
ем дипольного момента.
Пример (вопрос 3). Объясните, какие из следующих молекул дают чисто вра-
щательный микроволновый спектр: COs, OCS, Na, С На=С На, бензол, вода. Ка-
кие на них могут дать колебательный спектр?
Метод. Выбираем молекулы с постоянным электрическим дипольным моментом.
Для решения второго вопроса подумаем, какие молекулы имеют колебательные
моды, которые изменяют дипольный момент (включая моды, изменяющие его
от первоначального нулевого значения).
Ответ. Постоянный дипольный момент имеют только OCS и НгО, поэтому толь-
ко эти молекулы могут дать чисто вращательный спектр. Все другие, за иск-
лючением N2, обладают по крайней мере одной колебательной модой, изменяю-
щей дипольный момент. Таким образом, все молекулы, за исключением Nj, мо-
гут дать колебательный спектр.
Рис. 17.3. Осциллирующая непо-
лярная молекула в отличие от по-
лярной не выглядит как осцил-
лирующий диполь.
62
Часть 2. Структура
Комментарий. Не вес колебательные моды сложных молекул активны в ИК-
спектре. Например, пульсационное колебание молекулы бензола (при котором
кольцо то расширяется, го сжимается) неактивно в ИК-спектре,
3, Электронные переходы. Дипольный момент молекулы может
изменяться в зависимости от электронного распределения. Одна-
ко, если перераспределение электронов происходит симметрично,
суммарного изменения диполя не будет, и соответствующий пе-
реход будет запрещенным, Отметим важность фокусирования вни-
мания па изменении электронного распределения, Например, в
атоме, который не имеет постоянного дипольного момента, пере-
ход s—*-р7. разрешен, так как он соответствует несимметричному
сдвигу заряда вдоль оси z. В отличие от этого переход 2s'—Из
симметричен, при нем нс возникает преходящего дипольного мо-
мента, и он запрещен. В молекулах нужно искать такой же пре-
ходящий дипольный момент (он называется дипольным моментом
перехода), связанный с перераспределением заряда. Пример дан
па рис. 17.4: один из приведенных переходов для молекулы SOj~
разрешен (он имеет дипольный момент, качественно аналогич-
ный моменту для перехода з—>-р. в атоме), однако второй (на-
поминающий переход s—з в атоме) имеет нулевой диполь и по-
этому запрещен.
Таким образом, мы рассмотрели главные черты, которыми
должна обладать молекула, чтобы иметь спектр испускания или
спектр поглощения, Формулировка этих особенностей называет-
ся основным правилом отбора (gross selection rule). В гл. 14 бы-
ло показано также, что разрешенные переходы можно выразить
через изменения квантового числа (например, ,Д/—±1). Для мо-
лекул можно привести аналогичные специфические правила от-
бора, Некоторые из этих правил будут даны при рассмотрении
различных видов переходов.
Ширина линий. Различные линии в спектре имеют разную шири-
ну. В данном разделе мы рассмотрим некоторые из причин, по ко-
рне. 17.4. Два типа электронного перехода в SO| .
17. Вращательные и колебательные спектры
63
торым линии поглощения и испускания уширяются так, что охва-
тывают интервал частот.
Одним из важных механизмов, приводящих к уширению спект-
ральной линии, является классическое смещение Доплера. Когда
объект, испускающий излучение с длиной волны к, удаляется от
наблюдателя со скоростью v, наблюдатель обнаруживает излу-
чение с длиной волны (1 + v/c) А, где с — скорость излучения
(с —скорость света, если излучение электромагнитное, но ско-
рость звука, если волновое движение звуковое). Объект, прибли-
жающийся к наблюдателю со скоростью v и испускающий излу-
чение с длиной волны А, неподвижному наблюдателю кажется
излучающим при длине волны (1—и/с)А. Скорости молекул мо-
гут достигать очень высоких значений (т. 1, стр. 23), и неподвиж-
ный наблюдатель фиксирует излучение от ансамбля быстро дви-
жущихся молекул в виде интервала длин волн. Некоторые моле-
кулы в образце движутся к наблюдателю, некоторые от него; не-
которые движутся быстро, некоторые медленно. Наблюдаемая
форма линий соответствует поглощению или испусканию, обус-
ловленному всеми сме-
щениями Доплера в дан-
ном образце.
Чтобы предсказать
форму спектральной ли-
нии, нужно знать отно-
сительное содержание
молекул, имеющих каж-
дую данную скорость.
Поскольку оно опреде-
ляется распределением
Максвелла (т. 1, стр. 23)„
форму линии можно рас-
считать для любой тем-
пературы. Распределение:
Максвелла представля-
ет собой колоколообраз-
пую гауссову кривую
(зависимость от е~х1), и
поэтому доплеровская
линия также имеет гаус-
сову форму (рис. 17.5).
Рис. 17,5. Доплеровское уши-
рение спектральных линий.
64
Часть 2. Структура
Ширина этой линии (ширина на полувысоте, см. рис. 17.5) дается
выражением
ДХ=2 (Х/с) (2АТ/т)*/»_ (17.1.3)
Она увеличивается с ростом температуры, так как при этом моле-
кулы имеют более широкий диапазон скоростей. Чтобы получить
максимально резкую линию (это одна из стадий получения спект-
ров, в которых линии имеют максимальное разрешение), нужно,
следовательно, пытаться работать с образцами при низких темпе-
ратурах. Другой практический аспект использования доплеров-
ского уширения состоит в его применении для определения тем-
пературы поверхности звезд путем измерения ширины их спект-
ральных линий.
Пример (вопрос 4). Солнце испускает Спектральную линию при 677,4 нм, кото-
рая была отождествлена с переходом в высокоионизированиом 57Fe; ее шири-
на 0,0053 нм. Какова температура на поверхности Солнца?
Метод. Делаем перестановку в уравнении (17.1.3). Используем значение
m(57Fc) =56,94X1.661 X Ю-г7 кг.
Ответ. Т = (.U/2A)2 (тс1/2 k) =
(0,0053 нм/2-677,4 нм)аХ(9,45-10-^ кг)х(2,998-10» м/с)9
= “ 2 (1,381 1Дж/К) In 2 ” .
= 6,8-Ю3К.
Комментарий. Заметим, что для данной температуры наибольшую ширину име-
ют линии с большой длиной волны.
Даже при низких температурах, при которых доплеровское
уширение пренебрежимо мало, спектральные линии не имеют ну-
левой ширины (т. с. бесконечной резкости), поскольку всегда
действует механизм, называемый уширением за время жизни.
Если решить уравнение Шредингера для системы, которая изме-
няется во времени, то окажется, что энергию невозможно точно
определить по квантовым уровням. Если система изменяет свое
состояние со скоростью 1/т (например, если ее время жизни рав-
но т), то энергетические уровни «размазываются» в некотором
интервале б£, где
6£^/г/т. (17.1.4)
Следовательно, если время жизни возбужденной молекулы при-
близительно равно т, то ее энергия, вместо того чтобы быть рав-
ной £, может принимать любое значение в интервале б£ вбли-
зи £. Энергию можно точно определить, только если состояние
имеет бесконечно большое время жизни. Это отношение между
уширением энергии и временем жизни напоминает соотношение
неопределенности Гейзенберга (т. 1, стр. 436) и является его
следствием.
17. Вращательные и колебательные спектры 65
Возбужденных состояний, имеющих бесконечное время жиз-
ни, не бывает, поэтому не бывает возбужденных состояний, име-
ющих точно определенную энергию. Следовательно, спектральные
линии всегда покрывают интервал частот. Короткоживущие воз-
бужденные состояния характеризуются широкими линиями; дол-
гоживущие возбужденные состояния характеризуются узкими ли-
ниями. Степень уширения можно оценить из уравнения (17.1.4),
Выразив 6Е в обратных сантиметрах н т в секундах, получим
Йо (см-1) ~ 5 10-11 (т, с)-1.
Следовательно, для времени жизни 10-10 с уширение будет состав-
лять — 0,5 см'Т; много это или мало — зависит от требуемого
разрешения. С примерами уширения за время жизни мы встре-
тимся в последующих разделах.
Время жизни возбужденных состояний определяется главным
образом двумя механизмами: стимулированным процессом испус-
кания и спонтанным процессом испускания.
Стимулированное испускание. Свет, омывающий исследуемый об-
разец, действует как стимулятор процесса эмиссии, точно так же
он может стимулировать абсорбцию (осциллирующее электриче-
ское поле переводит молекулу в более высокое или более низ-
кое состояние). Поэтому, если молекулы омываются светом под-
ходящей частоты, время жизни возбужденного состояния умень-
шается. Отсюда можно ожидать, что скорость дезактивации бу-
дет подчиняться уравнению
скорость стимулированного испускания — В (v),
где ’?Z(v) —плотность энергии падающего излучения на частоте
перехода. В известен под названием коэффициента Эйнштейна
для стимулированного испускания.
Спонтанное испускание. Название «спонтанное испускание» отра-
жает тенденцию всех возбужденных состояний сбрасывать энер-
гию и переходить на более низкий энергетический уровень даже в
отсутствие света. На самом деле этот процесс не «спонтанный» в
том смысле, что он не беспричинный, и следует рассмотреть его
механизм.
Объяснение процесса может быть найдено, если возвратиться
к рассмотрению излучения черного тела (т. 1, разд. 13.2). Там
было показано, что плодотворной моделью электромагнитного
поля является совокупность осцилляторов — по одному на каж-
дую возможную частоту света. В гл. 13 указывалось также, что
осциллятор не теряет всю свою энергию даже в том случае, ког-
да он переходит в самое низкое энергетическое состояние (т. 1,
разд. 13.5). Следовательно, даже в том случае, когда нет ника-
ких возбуждений (т. е. нет фотонов), электромагнитные осцилля-
торы не полностью «утихомирены». Эти случайные электрические
5—242
66
Часть 2. Структура
поля пулевого уровня могут вызывать колебание молекулы и сти-
мулировать ее к генерированию фотона. Поскольку первоначаль-
но в системе не было фотонов, мы рассматриваем процесс как
спонтанный.
Общая скорость дезактивации возбужденного состояния равна
скорость испускания — A 4-fi7Z(v),
где А — коэффициент Эйнштейна для спонтанного испускания.
Соотношение между спонтанным и стимулированным процесса-
ми зависит от их интенсивности и величины отношения AjB. Из
теоретического рассмотрения следует, что
А[В = 8лй (v/c)3, (17.1.5)
поэтому следует ожидать, что спонтанный процесс будет сущест-
вен при высоких частотах. Например, для того чтобы возбужден-
ный атом испустил фотон видимого света, требуется всего лишь
10-7 с, но для обращения магнитного момента ядра в магнитном
поле в 1 Т (тогда разность энергий будет только около 10-4см-1)
необходимо время ^10|Э с (это примерный возраст нашей Все-
ленной).
Пример. Свет ртутной лампы сконцентрирован в пучок с поперечным сечением
1 с.ч3 и пропущен через образец, содержащий возбужденные атомы ртути. Ка-
ковы о твоей тельные скорости спонтанной и стимулированной эмиссии при
254 нм, если лампа имеет мощность ИХ) Вт?
Метод. Рассчитаем отношение А/В. Тогда о тио си тельные скорости даются фор-
мулой AjBQi, где °U следует понимать как энергию в единице объема на еди-
ницу интервала частоты. Рассчитаем Ф/, рассматривая фотоны: лампа испускает
N фотонов в секунду на площади 1 смг. Каждый фотон движется со скоростью
с. Следовательно, число фотонов в единице объема будет равно К/(1 см2)-с.
Каждый фотон имеет энергию Av. поэтому плотность энергии составит
ATiv/c (1 см2). Если ширина спектральной лцийи dv, то энергия в единице о&ъ-
ема на единицу интервала частоты будет равна Nhvjc(l cm2)6v. Примем, что
fiv эквивалентна 1 см-1 (т. с. 6v~1010 Гн). Заметим, Что Nhv— энергия в се-
кунду-, или мощность лампы,
Nhv (100 Вт)
Ответ. 42- С{1 fiv = (2,998-10“ см/с)х(1 см2).х(!01с' Гц) в
= 3,34 10"1!1 Дж-см-3-с.
Поскольку
Л/В = 8пА (v/c)3 = 8nft (1 Д)э =
= 8лХ(6,626 10-34 Дж-с)Х(1/254-10’ см)3 =
== 1,02-Ю-18 Дж-см'3-с,
имеем
Л/Ва.' = (1,02-10-18 дж-см-3 с)/(3,34-Ю*1’ Дж-см-3-с) = 3,04.
Комментарий. Расчет показывает, что скорость спонтанной эмиссии в 3 раза
выше скорости стимулированной эмиссии. При другом подходе, применимом,
когда возбужденные атомы находятся в «горячем» окружении, 3!/ интерпрети-
руется как плотность энергии на единичный интервал частоты для излучателя—
черного тела — и используется уравнение Планка.
17. Вращательные и колебательные, спектры
67
Переход из возбужденного состояния в более низкое состоя-
ние может также стимулироваться и другими процессами. Одним
из наиболее важных является дезактивация при столкновении,
при которой энергия возбужденного состояния не излучается, а
переходит в движение молекулы, сталкивающейся с возбужден-
ной частицей. Этот путь дезактивации очень важен в жидкостях,
и позднее мы рассмотрим его более детально (разд. 18.4).
Все эти процессы приводят к одному и тому же эффекту. Не-
зависимо от метода дезактивации, если время жизни возбужден-
ного состояния уменьшается, линия в спектре имеет ширину, оп-
ределяемую уравнением (17.1.4). Если действуют несколько дез-
активационных процессов н каждый из них вызывает гибель воз-
бужденного состояния со скоростью соответственно l/iy, 1/т2 и
т. д., то общая ширина линии будет суммой индивидуальных уши-
рений:
б£^й(1/Т1+ 1/г2-|- • -.). (17.1.6)
17.2. Чисто вращательные спектры
Сначала мы рассмотрим вращательные энергетические уровни
молекул, а затем рассчитаем частоты разрешенных переходов,
применяя правила отбора. В финальной части мы предскажем
внешний вид спектра, приняв во внимание заселенность враща-
тельных уровней при некоторой постоянной температуре образ-
ца Т.
Вращательные энергетические уровни. Вращательные энергетиче-
ские уровни можно получить, решая уравнения Шредингера для
рассматриваемой молекулы. В этом случае имеется очень эффек-
тивный короткий путь. В его основе лежит тот факт, что, соглас-
но классическому выражению Для энергии тела, вращающегося
относительно некоторой оси х,
где — угловая скорость вокруг этой оси и — момент инер-
ции [два индекса у I появляются по технической причине: момент
инерции связывает реакцию (угловой момент) с ее движущим
стимулом (моментом вращения), и как реакция, так и ее сти-
мул имеют направление в пространстве; это технический пред-
мет, на котором здесь нам не Нужно останавливаться: рассмат-
ривайте просто этот индекс как чрезмерно раздутый символ];
Тело, свободно вращающееся относительно всех трех осей, имеет
энергию
& = ЛгаФЧ—2" —Г
5‘
68
Часть 2, Структура
Классический угловой момент тела с моментом инерции !хх и уг-
ловой скоростью {.оЛ- равен следовательно, выражение
для энергии через угловые моменты относительно каждой моле-
кулярной оси имеет вид
(17.2.1)
Это ключевое уравнение для остальной части данного раздела.
Квантовомеханические свойства углового момента были разобра-
ны в гл. 13; их можно подставить в это уравнение и получить
квантовомеханические свойства вращательной энергии.
Обсуждение удобно проводить в соответствии с типом рас-
сматриваемой молекулы. Мы рассмотрим сферические волчки, к
которым относятся молекулы со всеми тремя равными моментами
инерции (такие, как метан), симметричные волчки, в которых
два момента инерции одинаковы, но отличаются от третьего (та-
кие, как аммиак или хлористый метил), и линейные молекулы
(такие, как двуокись углерода и любая двухатомная молекула).
Энергетические уровни н вращательные спектры асимметричных
волчков, в которых все три момента инерции разные (такие, как
вода), очень сложны, и мы не будем их рассматривать. Все энер-
гии зависят от моментов инерции молекул; выражения для них
собраны в табл. 17.1.
Молекулы типа сферического волчка. Когда моменты инерции
lxx, 1уу и 7iZ одинаковы, выражение для энергии упрощается;
В=(1 /27) (Л+Л, 4- 7е)=Л/27.
Это классическое выражение; 7а представляет собой квадрат ве-
личины классического углового момента. Его можно превратить
в квантовое выражение, введя соотношение, устанавливающее,
что величина углового момента ограничивается значениями
[Ц1+ l)]VaA, где 7=0, 1, 2,... (т. 1, стр. 455). Следовательно,
энергия вращающейся молекулы типа сферического волчка огра-
ничена значениями
Ej=J(J-^l)W, 7=^0, 1,2............................. (17.2.2)
Множитель /г/2 7 обычно записывают как В и называют враща-
тельной постоянной молекулы. Тогда получается Простое выра-
жение
Ey = BJ(J+l), J=0, 1,2............................. (17.2.3)
Разность энергии между соседними вращательными уровнями
составляет
Ej~ (17.2.4)
Она уменьшается при возрастании момента инерции: большие мо-
лекулы имеют близко расположенные энергетические уровни.
Таблица 17.1
Моменты инерции
Выражения, приведенные ниже, дают моменты инерции для молекул указанных
типов
1, Двухатомные молекулы
1 = (пцгпт/rn)R',
2. Линейные трехатомные молекулы
3. Симметричные волчки
I = +/?')’
+ (т . + m ,/?'-’)
I = 2т,/?т - m, + m,+m5
= 2/тт 1Л J( 1 -cos 0),
2 - -cos 0)
+ (mj m)(m1 + m3)R2(] +2 cos 0)
+ (m4/m)/?'{(3m, +
+ 6ffl|<[}(l +2 №S 0)]}
m = 3m । + m2 + m}
Ii = 2т,Я~(1 —cos 0), m - 3m + m2
l,=m ,/?3( 1 -cos 0)
+ (m,m2/m)R’(] +2 cos 0)
fj=4m1/?2
= 2т,Я' + 2туК'\ - 4
4, Сферические волчки
I^(8/3)mtR-
I =4т,/г2
70
Часть 2. Структура
Численную величину расстояния между соседними уровнями мож-
но оценить, рассмотрев молекулу СС1$. Из длин связей и массы
атомов находим, что момент инерции составляет 4,85-Ю-45 кг-м5;
тогда В=1,15-10-24 Дж, или 0,058 см-1 (переводной коэффици-
ент был дан в г. 1, стр. 35). Поэтому расстояние между уровнями
составляет величину порядка 1,2 см-1, когда /^10, что соответст-
вует энергиям микроволновой области спектра. Подобные вели-
чины характерны и для других типов молекул, и по этой при-
чине чисто вращательные переходы изучаются в микроволновой
спектроскопии.
Молекулы типа симметричного волчка. В симметричных волчках
= примером является цилиндр. Момент инерции, па-
раллельный осп симметрии (/гг), мы будем обозначать /ц,- а м°'
мент инерции, перпендикулярный ей,-—/?_ ( = !хх~ /уу}. (На язы-
ке теории групп молекула типа симметричного волчка имеет по
крайней мере ось симметрии третьего порядка,)
Основное классическое выражение дает для энергии
Е = (1/2/ L) (^~-j-^|)_п(1/2/1|) А-
Ее можно выразить через величину углового момента J2 = /“+
прибавляя и вычитая /г/2/ |. •’
£=(1/2Л)(Л+Л+Л)-(1/2/1)Л+(1/2/11)^=
= (1/2Л)^+[(1/2/„)-(1/2/±))Л.
Чтобы получить квантовое выражение, квадрат классической ве-
личины /2 заменяют на квантовую величину Из кван-
товой теории также следует (т. I, стр. 456), что компонента угло-
вого момента по любой оси ограничена значениями KJi, где
Л = 0, ± 1,..., ±7. (В данном случае вместо М принято использовать
символ К.) Таким образом, компонента /г ограничена этими зна-
чениями. Следовательно, энергия вращающейся молекулы типа
симметричного волчка может принимать значения
£jk = BJ(J+1)+(4-B)№( (17.2.5)
I Л — U, ±1., itJ»
где В=й2/2/± и Л = й2/2/|].
Квантовое число К фигурирует в этом уравнении потому, что
энергия вращения зависит От того, как распределен общий угло-
вой момент. Если Л велико (близко к +7 или к —Z), то боль-
шая часть молекулярного вращения осуществляется вокруг оси
симметрии (рис. 17.6, а), по если оно равно нулю, все движение
представляет собой вращение «кувырком» (рис. 17.6,6). В пер-
вом случае энергия зависит главным образом от / 1Г, а во вто-
ром—только от lj_. Заметим также, что энергия зависит от №,
17. Вращательные и колебательные спектры
71
Рис. 17.0. Смысл квантового числа К.
так что состояния с одинаковыми значениями JK|, по противопо-
ложного знака имеют одинаковую энергию. Это отражает физи-
ческий вывод о том, что молекулы с противоположными знака-
ми К вращаются вокруг своих осей с одинаковой скоростью, но
в разных направлениях; однако способ вращения не может влиять
на энергию.
Пример (вопрос 5). Молекула аммиака — симметричный волчок (имеет ось С3)
с длиной связи 101^2 пм и углом между связями 107°. Найдите ее вращательные
энергетические уровни.
Метод. Рассчитываем вращательные постоянные Л к В. Моменты инерции мож-
но найти, используя соответствующие выражения из табл. 17.1.
Ответ. В обозначениях, принятых в табл. 17.1, имеем mi = 1.0078 X
X(1,6605-10“27 кг), т2= 14,0031Х(1,6605-10-27 кг), /?=101,2 пм, 0 = 107°. Следо-
вательно.
/ц = 2x0 ,6735-Ю-27 кг)х(Ю1,2-м)3х<1 — 107°) =
= 4,4300-10-^ кг-м3.
^2,2150-Ю-1" кг-м2 +
f(l,6735-Д0~8? кг)х(2,3252 Ю-аа кг)х(Ю1,2-Ю~и м)3Х(1 +2cos 107°)
+ | Зх(1,6735-10-3’ кг) + (2.3252 10-2а кг) ;
= 2,8003-10-*? кг м®.
Отсюда
Л = Й3/2/)( = (1,055 -Ю-34 Дж с)3/2 (4,4300 -10-4? кг м2) =
= 1,2562-Ю'23 Дж (эквивалентно 6,32 см-1).
72 Часть 2. Структура
= (1,055-Ю"31 Дж-с)й/2 (2,8003-1<Н- кг№) =
= J, 9873-1О'22 Дж (зк виза лен тао 10,00 см-1).
Таким образом,
EjKM (с.и -1) = 10,0W (/ + J) (6,32 ~ 10,00) № =
= 10,00J(J+ 1)-3,№,
где 3=0, 1, 2 .... и К=3, 3—1, ..., —3.
Комментарий. В случае /-Д энергии вращения молекулы вокруг се оси (Д’=3)
равна 16,32 см"!, но для вращения «кувырком» (Д=0) с тем же полным угло-
вым моментом (/= 1) требуется энергия, эквивалентная 20 см"1.
Линейные молекулы. В линейных молекулах (таких, как двух-
атомные молекулы, СО2 и С2Н2) суммарный угловой момент на-
правлен примерно по оси, перпендикулярной межатомной линии.
Отсюда следует, что можно использовать последнее уравнение
с A’sO. Поэтому энергетические уровни будут следующими:
£f^BJ(J-|-l), J —0,1,2,..., (17.2.6)
где И — п2/2/, а I — простой момент инерции (/±).
Причина, по которой угловой момент должен быть^направлен
примерно но оси, перпендикулярной межатомной липни, наибо-
лее ясно видна в случае двухатомных молекул. Чтобы точно ука-
зать положение обоих атомов, можно указать три координаты
каждого. Поскольку атомы могут изменять свое положение из-
менением любой одной из этих шести координат, говорят, что мо-
лекула имеет шесть степеней свободы. Имеется, однако, другой
путь для определения положения атомов. Можно установить по-
ложение центра масс молекулы (который имеет три координаты),
расстояние между атомами (одна координата) и ориентацию мо-
лекулы (еще две координаты). При этом получается тоже шесть
степеней снободы, но их физический смысл теперь более ясен. Три
степени свободы (движение центра масс) описывают поступа-
тельное движение молекулы как целого; еще одна (изменение
расстояния между атомами) соответствует колебанию. Остаются
только две степени свободы, и они соответствуют вращению мо-
лекулы, при котором происходит изменение ориентации ее оси.
Это движение перпендикулярно ее оси симметрии, и поэтому вра-
щательные степени свободы соответствуют движению относитель-
но осей, перпендикулярных оси молекулы.
Вращательные переходы. В предыдущих разделах мы установи-
ли вращательные энергетические уровни для некоторых важных
типов молекул. В данном разделе мы применим правила отбора и
определим, какие переходы могут проявляться во вращательном
спектре.
На стр. 62 было введено основное правило отбора: если мо-
лекула дает вращательный спектр испускания или поглощения,
17. Вращательные и колебательные спектры 73
опа должна иметь постоянный электрический дипольный момент.
Поэтому молекулы типа сферического волчка и гомоядерпые
двухатомные молекулы не могут дать чисто вращательный спектр.
Молекула типа симметричного волчка должна иметь дипольный
момент (если дипольный момент в принципе для нее возможен),
направленный вдоль ее оси симметрии (иначе она не имела бы
осевой симметрии). Следовательно, испускание или поглощение
света не может способствовать се переходу в различные состоя-
ния вращения относительно этой оси. Таким образом, можно ожи-
дать, что вращательные спектры будут давать только гетероядср-
ные двухатомные молекулы, другие полярные линейные молеку-
лы, а также полярные молекулы типа симметричного волчка, если
они вращаются «кувырком».
Специфическое правило отбора (которое формулирует, какие
изменения могут произойти в J и К) можно получить так же. как
для атомов (т. 1, стр 485). Поскольку фотон имеет спиновый
угловой момент, когда он выбрасывается из молекулы во время
вращательного перехода, угловой момент молекулы изменяется
компенсирующим образом. Фотон несст одну единицу углового
момента, и поэтому при переходе угловой момент молекулы дол-
жен измениться на одну единицу. Следовательно, получаем пра-
вило отбора
Д7-±1.
Мы уже видели, что свет
7 = 9 ----------------
8 -------г----------
7-------г----------
б------J------------
5 ----Н------------.
4 ---J_____________
3 —
не может ускорять вращение симмет-
ричного волчка вокруг оси симмет-
рии, и поэтому другое правило отбора
записывается как
ДК^О.
Применение этих правил к энергети-
ческим уравнениям приводит к выво-
ду, что для переходов J +1-^-7 разре-
шены следующие частоты поглоще-
ния:
Av=^2B(J-j-l), .7=0,1,2,.... (17.2.7)
Соответствующий спектр показан
на рис. 17.7. Наиболее существенной
особенностью является то, что оп со-
Рис. 17.7. Переходы, обусловливающие воз-
никновение чисто вращательного спектра.
74 Часть 2. Структура
стоит из серий линий с частотами 2В, 4В, 6В,... и расстоянием
между ними, равным 2В. При измерении расстояния между ли-
ниями получают В, и отсюда момент инерции, перпендикулярный
оси симметрии молекулы. Поскольку массы атомов в молекуле
известны, очень просто получить информацию о форме и разме-
рах молекулы. К сожалению, для молекулы типа А—В—С та-
кой анализ дает единственное значение />, хотя в ней имеются
две связи разной длины. Эту трудность можно преодолеть, ис-
пользуя изотопозамещепную молекулу, например А—В—С, и оп-
ределяя также ее момент инерции. Тогда, если считать, что
л('А—В)=К(А'—В), из этих двух измерений можно получить
как Л? (А—В), так и —С). Хорошим примером применения
этого метода является изучение молекулы OCS; ее расчету посвя-
щены задачи 17.23 и 17.24.
Пример (вопрос 6). Предскажите вид вращательного спектра молекулы аммиака.
Метод. Энергетические уровни были рассчитаны в предыдущем примере. Моле-
кула имеет постоянный электрический дипольный момент (вдоль осн Сз). Пра-
вила отбора следующие: Д/ = ±1,
Ответ. Энергия переходов /+1 К-*- К равна Л£ = 2В(7 + 1), 7 = 0, 1, 2, .. .
Следовательно, спектр состоит из серии линий при 20,00, 40,00, 00,00, 80,00 см-1
и т. д. Расстояние между линиями 23=20.00 см~!.
Комментарий. Нс все линии имеют одинаковую интенсивность. Одна из причин
этого состоит в том, что вращательные уровни заселены по-разному. Макси-
мальная интенсивность для образца при комнатной температуре наблюдается в
области 7 а-6, Другая причина состоит в том, что внешний вид спектра моди-
фицируется ориентацией спинов протонов, так как принцип Паули разрешает
лишь некоторые из вращательных состояний.
Вращательные спектры КР. При столкновении фотона с молеку-
лой он может забрать часть вращательной энергии молекулы и
выйти с более высокой частотой. Линии, которые образуются по
такому механизму, называют антистоксовыми линиями спектра
комбинационного рассеяния (спектра КР или раман-спсктра).
Фотон может также отдать часть собственной энергии молекуле,
стимулируя ускорение вращения, и выйти с более низкой часто-
той. Образующиеся по такому механизму линии называют сток-
совыми линиями. Пример наблюдавшегося в эксперименте спект-
ра дан на рис. 17.8.
Правила отбора для КР-процессов отличаются от нормальных
процессов поглощения или испускания. На первом месте стоит
основное правило отбора, согласно которому молекула должна
обладать анизотропной поляризуемостью. Смысл этого состоит
в следующем. Когда молекула помещена в электрическое поле,
она искажается под влиянием сил, которые действуют па электро-
ны и ядра. Степень искажения определяется поляризуемостью
молекулы: при высокой поляризуемости даже умеренное поде мо-
жет вызвать большое искажение. Например, атом ксенона иска-
жается намного легче, чем атом гелия, так как его внешние элект-
роны менее тесно связаны с центральным ядром. Мы говорим, что
17. Вращательные и колебательные спектры
75
10 СМ~’
Hg (253,6 нм)
Рис. 17.8. Чисто вращательный спектр КР для молекулы NSO.
атом ксенона более поляризуем, чем атом гелия. В случае атома
не имеет значения, в каком направлении действует поле, посколь-
ку искажение одинаково в любом направлении — поляризуе-
мость изотропна. То же справедливо для молекулы типа сфериче-
ского волчка. Однако в общем поляризуемость молекул зависит
от направления приложенного поля. Например, молекулу водо-
рода легче исказить, когда поле приложено вдоль направления
связи, чем когда оно приложено перпендикулярно связи. В этих
случаях мы говорим, что молекулярная поляризуемость анизо-
тропна.
Согласно основному правилу отбора, метан не может дать
вращательного спектра КР, тогда как молекула водорода и лю-
бая другая двухатомная молекула (и гомоядерная, и гетероядер-
пая) могут. В этом состоит главное значение вращательных спект-
ров КР: они позволяют изучать многие молекулы, не активные в
чисто вращательном микроволновом спектре.
Специфическое правило отбора для спектров КР имеет вид
комбинационное рассеяние: ±2.
Появление значения 2 можно объяснить несколькими способами.
Физическая причина состоит в том, что поляризация молекулы
возвращается к своей начальной величине дважды во время каж-
дого оборота. Поэтому кажется, что молекула вращается как бы
в два раза быстрее, чем при рассмотрении дипольного момента
(рис. 17.9). Это правило отбора не противоречит ранее приведен-
ным аргументам, основанным на спине фотона, равном единице,
и общем сохранении углового момента: в КРпроцессе участву-
ют два фотона, из которых один входит, а другой выходит.
Классическое объяснение необходимости анизотропной поля-
ризуемости и появления множителя 2 состоит в следующем. Если
молекула облучается светом с частотой «о, появляется индуци-
рованный дипольный момент, величина которого связана с ам-
плитудой электрического поля соотношением i
li(i)=<x,E(t)=aE0 cos aot.
76
Часть 2. Структура
Рис. 17.9. Поляризация молекулы возвращается в свое канальное состояние по-
сле вращения только на 180’.
Когда молекула вращается, се поляризуемость будет изменяться
(если опа анизотропна), и можно записать
а а0 | - Да cos 2coR?,
где «о — средняя поляризуемость и w — частота вращения. Под-
ставляя это выражение в предыдущее и применяя некоторые три-
гонометрические соотношения, получим
р (t) = ай£0 cos и0/ 4- Да£0 cos 2®R? cos =
= a0E0 cos 4“ 4“ A«£e [cos (to0 4- 2ша) 14-
4-cos(<o0—2<or) f].
Отсюда следует, что молекулярный диполь, индуцированный по-
лем, имеет компоненту,'осциллирующую при <оо, а также компо-
ненты, осциллирующие при wo + 2wr. Эти осциллирующие диполи
излучают,-и поэтому в рассеянном излучении появляются линии
с указанными частотами. Таким образом, КР-линии возникают
только при Да=4=0, следовательно, анизотропия необходима.
Когда молекула претерпевает переход с Д/=+2, рассеянное
излучение покидает молекулу, находящуюся -в более высоком
вращательном состоянии, и имеет более низкую частоту. Этот
переход обусловливает одну из стоксовых линий спектра. Зная
энергетические уровни молекулы, можно записать частоту таких
линий следующим образом:
стоксовы линии (Д2=^4-2): v=v0 — 2В (224-3) для J 4-2 <— J.
Эти линии появляются в низкочастотной области падающего све-
та с частотой Vo со смещениями 6В, 10В, 14В,... соответственно.
Интенсивность падает до нуля для линий, соответствующих высо-
ким значениям 2, поскольку эти состояния лишь слабо заселены
(рис. 17.10).
17. Вращательные и колебательные спектры
Рис. 17.10. Стоксовы и анти-
стоксовы линии во вращатель-
ном спектре КР.
Стоксовы линии Амт и с то к совы линии
Когда молекула стал-
кивается с фотоном па-
дающего света и претер-
певает переход с Д/=
= —2, фотон захватыва-
ет избыток энергии и вы-
ходит с более высокой
частотой. Эти переходы
объясняют аптистоксовы
линии в спектре:
аптистоксовы линии
(AJ — — 2): v =v0 -ф
-ф2В(2/—1)
для J —> J —2.
Эти линии сдвинуты на расстояния 6В, 10В, 14В,... соответственно
в высокочастотную область падающего света.
Пример (вопрос 7). Предскажите вид вращательного спектра КР газообразно-
го аммиака, когда он облучается монохроматическим излучением лазера с дли-
ной волны 336,732 им.
Метод. Молекула активна в КР, поскольку при вращении «кувырком» ее поля-
ризуемость, с точки зрения неподвижного наблюдателя, модулирует. Величина В
была рассчитана на стр. 72. Стоксовы и антистоксовы липни имеют частоты,
определяемые приведенными выше выражениями. Сделаем перевод в длины
волн.
Ответ. В = 10,00 см-1, ?.о = 336,732 нм, что эквивалентно 29697,2 см-1. Следо-
вательно, стоксовы липин будут проявляться при
.; ст (см -’) - 29697,2 — 20,00 (27 + 3) =
= 29637,2; 29597,2; 29557,2; 29517,2;...
или
X(нм) = 337,414; 337,870; 338,327; 338,786;...,
а аптистоксовы линии при
о (см-’) = 29697,2 + 20,00 (2/ — 1) =
= 29757,2; 29797,2;...
78 Часть 2. Структура
или
J. (нм) 336,053; 335,602;,..
Комментарий. Спектр будет иметь следующий вид: сильная центральная линия
при 336,732 нм, окруженная с обеих сторон линиями с возрастающей, а затем
убывающей интенсивностью (распределение интенсивности обусловлено эффек-
тами теплового заселения). Ширила полного спектра очень мала (около 500 см-'
при комнатной температуре), и поэтому длина волны падающего излучения
должна быть строго определенной.
Анализ структуры спектров КР состоит из измерения расстоя-
ний между соседними линиями в стоксовой и антистоксовой се-
риях и приравнивания их к 4В. Когда определена величина В,
можно найти момент инерции, перпендикулярный оси симметрии
молекулы, и, как и в случае микроволнового вращательного спект-
ра, момент инерции можно интерпретировать через длины связей
и углы. Как уже указывалось, метод КР может быть применен
как к полярным, так и к неполярным молекулам, если только они
имеют анизотропную поляризуемость. Сюда входят все молекулы
типа симметричного волчка и линейные молекулы; молекулы ти-
па сферического волчка исключаются.
17.3, Колебания двухатомных молекул
Этот раздел мы начнем с установления уровней колебатель-
ной энергии молекулы и затем применим соответствующие прави-
ла отбора, чтобы объяснить колебательный спектр. Вначале мы
рассмотрим колебания двухатомных молекул, в которых единст-
венным типом (модой) колебаний является растягивание н сжа-
тие связи, а затем перейдем к рассмотрению того, как влияет
на спектр одновременное возбуждение вращательных переходов.
В следующем разделе будет показано, что простое расширение
этого подхода позволяет рассмотреть колебания многоатомных
молекул.
Молекулярные колебания. Типичная кривая потенциальной энер-
гии для двухатомной молекулы приведена па рис. 17.11. Эта кри-
вая показывает, как изменяется потенциальная энергия молеку-
лы е изменением расстояния между ядрами, т. е. длины связи. На
кривой имеется минимум, соответствующий равновесной длине
связи в молекуле (,Ле). Слева от минимума энергия резко повы-
шается, т. е. два атома трудно «прижать» друг к другу; справа от
минимума кривая поднимается и затем переходит в горизонталь,
когда связь становится настолько длинной, что атомы разделя-
ются.
В области вблизи 7?е потенциальная энергия изменяется по
параболе, как показано на рис. 17.11. Поэтому приемлемым при-
j7. Вращательные и колебательные спектры
79
Рис. 17.1]. Кривая потенциальной энергии для двухатомной молекулы.
ближецием для кривой потенциальной энергии при небольших
смещениях является выражение
V(R)^±k(R-R^, ' (17-3-1)
где k — силовая постоянная связи. Поскольку уравнение (17.3.1)
выражает потенциальную энергию простого гармонического дви-
жения (т. 1, разд. 13.5), это означает, что молекула совершает
простое гармоническое колебание относительно равновесной дли-
ны связи и что разрешенными будут следующие колебательные
уровни:
= + п=0,1,2,..., (17.3.2)
где го = (А/m) V».
Чтобы использовать последнее выражение, необходимо знать,
каков смысл массы in. Эта масса не является полной массой
молекулы. Например, если одна из масс крайне велика (так что
молекула напоминает один атом, связанный пружиной с массив-
ной стенкой), то частота определяется меньшей массой. Когда
массы примерно одинаковы, они вносят вклад в т примерно в
80
Часть 2. Структура
Рис. 17.12. Отклонение истинных колеба-
тельных энергетических уровней от чисто
гармонической форм hl
равной пропорции. Этим свойствам удовлетворяет следующее вы-
ражение для т;
1/т = 1/^ + 1 /т^. (17.3.3)
Например, если объект 1 — кирпичная стена, то mi~ со и
l//n~l/ma; это значит, что т приблизительно равна щ2, т. е. мас-
се более легкого объекта. Масса т, определяемая с помощью
последнего уравнения, называется приведенной массой молекулы.
По физическому смыслу выражение для т совершенно оправ-
дано. Мы сейчас покажем, что это выражение можно строго вы-
вести. Рассмотрим две массы, тх и т2, колеблющиеся около об-
щего центра масс, который фиксирован. Центр масс определяется
соотношением mifa = m2R2, где fa и fa — расстояния точек 1 и 2
от центра масс в любой момент времени. Поскольку + =
где R— длина связи в данный момент времени, то
= (m2/(m1 + m-j) IR, Rs=[ml /(mt ф- ги%) I fa
На оба атома действует возвращающая сила, пропорциональная
расстоянию, на которое растянулась связь, R—Rc. Поэтому уско-
рения для каждого атома подчиняются соотношениям
(d^/di2) -^-k (R—RJ, m2 {d2Rjdt'i) =—k (R —Re).
Подставляя значение в первое соотношение или значение R?
во второе, получим уравнение
[ггьт^т^т^] (d2R/dt2) — —k (R — Rj,
которое представляет собой классическое уравнение движения
частицы с .массой
т =m1mi/(ral -ф т2),
совершающей простое гармоническое движение вдоль R с часто-
той и ₽ (А//пф/з:
6>R = ^(R-Pe).
17, Вращательные а колебательные спектры 81
Это выражение для m можно преобразовать в форму уравнения
(17,3,3), и, такие образом, введение приведенной массы будет
оправдано.
Энергетические уровни (17.3.2) дают лишь приближенную
оценку истинных колебательных энергий, так как они основаны
на параболическом приближении к истинной кривой потенциаль-
ной энергии. Когда колебания молекулы настолько интенсивны,
что при раскачивании атомы значительно смещаются от равно-
весной длины связи, потенциальная энергия отличается от пара-
болического приближения. Истинный потенциал менее тесно
ограничен расстоянием, чем парабола (см. рис, 17.Ц), и поэто-
му следует ожидать, что энергетические уровни при высоких
энергиях будут расположены ближе друг к другу, чем при низких
энергиях. Это иллюстрирует рис, 17.12.
Одни из подходов к вычислению энергетических уровней при
умеренных возбуждениях состоит в том, чтобы потенциальной
энергии придать математическую форму, которая была бы более
похожа на кривую на рис. 17.11, чем парабола. Особенно удачное
предположение было сделано Морзе, предложившим выражение
V (R) =De{l—-ехр [_а (Я-ОД }а, (17.3.4)
где Z?e — глубина минимума потенциальной энергии и а =
= -^-(i>(2zn/Z?eJФорма этой кривой приведена на рис. 17.13; не-
смотря на то что в этом случае имеются только два подгоняемых
параметра (k, которая входит в <о, и Ds)r ее форма является под-
ходящей, и можно достаточно хорошо описать ряд молекул. Че-
рез потенциальную энергию Морзе можно выразить уравнение
Шредингера и найти энергетические уровни. Их можно записать
как
(1 \ f 1 \2
п —(17.3.5)
где xe = ha2/fn(o — постоянная ангармоничности, Второй член вы-
читается из первого, что подтверждает конвергенцию (сходи-
мость) уровней при больших значениях квантовых чисел.
Колебательные спектры двухатомных молекул. Как ранее объяс-
нялось в этой главе, согласно основному правилу отбора, чтобы
колебание было активно в спектре, во время его осуществления
должен изменяться дипольный момент. В гомоядерпой двухатом-
ной молекуле растяжение связи между двумя атомами не изме-
няет дипольного момента от нулевого значения; следовательно,
гомоядерные двухатомные молекулы не будут давать колебатель-
ный спектр. В случае гетероядериой двухатомной молекулы при
изменении длины связи дипольный момент изменяется, и поэтому
6—242
82
Часть 2. Структура
«4-1 — п
Рис, 17.13. Кривая потенциаль-
ной энергии Морзе.
эти молекулы могут да-
вать колебательные
спектры поглощения или
испускания.
Специфическое пра-
вило отбора для гармо-
нического осциллятора
записывается так:
Др^±1. (17.3.6)
Это правило основано на
принципе сохранения уг-
лового момента комби-
нированной системы фо-
тон-f- молекула. Его при-
менение к выражениям
для энергетических уровней даст изменение энергии
(17.3.7а)
в гармоническом приближении или
Д£=йсо —2(п-|-1)хей(й-|—•, (17.3.76)
если принимается во внимание ангармоничность. Энергия появ-
ляется (или исчезает) в виде фотона с энергией hx\ т. е. свет,
испускаемый или поглощаемый, в гармоническом приближении
имеет частот)’ v —ы/2л.
Типичное значение молекулярной силовой постоянной состав-
ляет 500 Н-М ’1 (516 Н-м-1 для НС1), а типичное значение при-
веденной массы —около 10-26 кг (1,63-10~27 кг Для [Н35С1). Сле-
довательно, (о»5,6 рад-с~\ что соответствует длине волны 3,ЗХ
X 10-6 м, или волновому числу 2990 см-1. Это излучение лежит в
инфракрасной области спектра. Аналогичные значения длин волн
получаются и для других молекул, поэтому колебательная Спект-
роскопия — это инфракрасная (ИК) спектроскопия.
При комнатной температуре АТ — 200 см_[. следовательно (см.
обсуждение роли больцмановского фактора на стр. 59), огром-:
ное большинство молекул при комнатной температуре находится;
17. Вращательные и колебательные спектры
в основном колебательном состоянии. Таким образом, основным
переходом в ИК-спектроскопии поглощения является переход от
л = 0 к п = 1.
Если молекула образуется в некотором колебательно-возбуж-
денном состоянии, например при реакции
H, + F2----» 2HF*,
где HF* — «горячая», колебательно-возбужденная частица, то пе-
реход вниз (от п к л—1, от п—1 к н—2 и т. д.) можно наблю-
дать в ИК-спектре испускания. В гармоническом приближении
все эти переходы генерируют фотон одной и той же частоты, и
предсказанный спектр будет состоять из одной линии. Если же
учесть, что колебания могут быть слегка апгармоничпыми, будут
наблюдаться два изменения. Во-первых, энергии перехода будут
соответствовать уравнению (17.3.76) и уже слабо зависеть от п.
Следовательно, переходы с разных уровней будут генерировать
фотоны слегка отличающейся частоты, и в спектре появится не-
сколько линий.
Второе следствие ангармоничности проявится в том, что пра-
вило отбора (которое относится к гармоническому движению) бу-
дет немного нарушаться, ц наряду с переходами, для которых
Ап— ± 1 (первая гармоника, или фундаментальные переходы),
будут разрешены переходы с Дп=±2 (они называются вторыми
гармониками). На второй гармонике образуются фотоны с часто-
тами, определяемыми по формуле
hv=AE = 2kdi — 2(2/14-3)л'е/,®4- • • • (п+2 *—' п).
Появление линий, обусловленных переходами ца разные уровни,
позволяет определить хе и, следовательно, De.
Пример (вопрос 9). Инфракрасное испускание «горячей» молекулой HF прояв-
ляется в виде серии линий при 3958,38 см-’ (1—*-0), 3778,25 см”1 (2—*-1),
3598,10 см-1 (3—>-2) и слабой второй гармоники при 7736,63 см-1 (2—+).
Каковы силовая постоянная связи HF и энергия диссоциации молекулы-1
.Метод, к первым гармоникам (п+1—*-я) применяем уравнение (17.3.76). Вторая
гармоника нс дает дополнительной информации, но ее можно использовать для
контрольной проверки. Найдем со и Из кривой Морзе имеем сохе=(дг/20*.
Отсюда найдем De.
Ответ. Записываем: йса—2(л+1) йсохе. Далее, w=co0+«[-|-co2 и
<nxe= -j- («о—(Bi) =~2 (coi—ы2). Следовательно, <а= (3958,38 см-’ +3778,25 см-1-—
1
—3598,10 см-') = 4138,53 см”’ и toxe=-g-(3958,38 см-1—3778,25 см-1), или соле=
1
(3778,25 см-1-3598,10 см-1), что в среднем дает 90,07 см~1. Отсюда De =
=95 000 см-1, или 11,8 эВ.
Комментарий. Когда доступно большее число частот, лучше применять графиче-
ский метод (строится зависимость а>п от п), который дает результат, ближе со-
ответствующий истинной величине (^6,4 эВ). Связь между De и энергией дис-
социации молекулы D°(H_F) рассматривается ниже.
6’
84 Часть 2. Структура
Анализ колебательного спектра дает два типа данных: часто-
ту о и постоянную ангармоничности хе. Из частоты находят си-
ловую постоянную связи, которая является мерой се жесткости.
Из постоянной ангармоничности получают величину Ds для мо-
лекулы, т. е. энергию, необходимую для разрыва связи. Однако
к интерпретации смысла De нужно подходить с определенной осто-
рожностью. По определению, Z?e — глубина минимума потенци-
альной энергии молекулы, однако молекула должна обладать по
крайней мере энергией нулевого уровня fia> (см. рис. 17.13).
Следовательно, энергия разрыва связи, которая с термохимиче-
ской точки зрения рассматривалась в т. 1 (разд, 4.2), связана с
Do соотношением
D3{A-B)^De-|to. (17.3.8)
Это является ценным методом определения силы химических свя-
зей, в него введены улучшения, позволяющие преодолеть неточ-
ности, присущие анализу кривой Морзе. Многие данные по силе
связен в молекулах, приведенные в табл. 17.2, были получены
этим спектроскопическим методом.
Колебательно-вращательные спектры. Если проанализировать ко-;
лебательный спектр газообразной гстсроядерпой двухатомной мо-
лекулы при высоком разрешении, то оказывается, что каждая ли-
ния состоит из большого числа близко расположенных компонент
(рис, 17.14). По этой причине молекулярные спектры часто назы-
вают полосатыми спектрами в противоположность атомным спект-
рам, которые называют линейчатыми спектрами. Расстояние меж-
ду линиями в полосах часто имеет порядок 1 см-1; и, по-видимому,
Таблица 17.2
Свойства двухатомных молекул
Молекула Колебателен ая частота, см-1 Вращательная постоянная, см —1 Дляяа связи, ли Энергия диссоциациит эВ
*н£ 2321,8 29,8 106 2,651
1Н2 4395,2 60,809 74,16 4,476
“Н. 3118,4 30,429 74,16 4,553
W’F 4138,52 20.939 91,71 ^6,40
1Н35С1 2989,74 10,5909 127 460 4,430
*Н79Вг 2649,67 8,473 141,3 3,75
1H127J 2309,5 6,551 160,4 3,056
2359,61 2,010 109,4 9,756
1еО, 1580,361 1 44566 120,739 5,080
19Р2 829,1 [0 8828] 143,5 <2,75
^С1а 564,9 0,2438 198,8 2,475
17. Вращательные и колебательные спектры
Во
Рис, 17.14. Колебательно-вращательный спектр высокого разрешения для моле-
кулы HCI.
такая структура обусловлена возбуждением вращательного дви-
жения во время колебательного перехода.
Стимулирование вращательного перехода во время колебатель-
ного изменения можно объяснить несколькими способами. Во-пер-
вых.. количество энергии при колебательном переходе так велико,
что она вполне может быть использована для возбуждения враща-
тельного перехода. Во-вторых, вращение тела ускоряется, когда
его момент инерции уменьшается, и замедляется, когда момент
инерции увеличивается (известным примером служит способ, кото-
рым катающийся на коньках .заставляет себя вращаться быстрее^
он прижимает руки к телу). Этот эффект является следствием со-
хранения углового момента J=Ia в отсутствие каких-либо внеш-
них крутящих моментов: если изменяется /, то to должна изменять-
ся в противоположном направлении так, чтобы произведение /со
сохранялось постоянным. В данном случае колебательное измене-
ние аналогично преходящему изменению момента инерции, и моле-
кула отвечает изменением своего вращательного состояния.
Детальный анализ показывает, что правило отбора для вра-
щательного изменения, которое сопровождает колебательное изме-
нение,—это уже знакомое нам правило Л/= ± 1, однако при ис-
ключительных обстоятельствах (когда имеется электронный угло-
вой момент вокруг оси линейной молекулы, как в NO) общее со-
хранение углового момента может также выполняться при Л/=0.
Вид колебательно-вращательного спектра можно предсказать
следующим образом. Энергетические уровни колеблющейся и вра-
щающейся молекулы имеют вид
+ + (17.3.9)
При более детальном рассмотрении нужно учесть поправки на ан-
гармоничность и зависимость вращательной постоянной В от ко-
86
Часть 2. Структура
P-Ветвь | Я-Ветвь
Положение
Q-ветви
Рис. 17.15. Образование Р-. Q- и R-ветвей.
лебатсльного состояния молекулы (так как в более высоких коле-
бательных состояниях, вследствие раскачивания атомов, молекула
немного увеличивается в объеме, и этот эффект заметен). Когда
происходит колебательный переход, не только п изменяется до
«4-1 (для поглощения), но и J изменяется на 0 или ±1. Поэтому
колебательная линия поглощения разделяется на три набора ли-
ний, называемых ветвями, которые обозначают как Р, Q и R в со-
ответствии со значением АЛ
P-Ветвь. Эти линии соответствуют переходам с Дн=1 и AJ = —1.
Если сначала молекула была во вращательном состоянии J, про-
исходит переход J—1 и общее изменение энергии равно
Это объясняет появление соответствующих линий при частотах
го—2В, го—4В,.„ (рис. 17.14 и 17.15).
17. Вращательные, и колебательные спектры
87
Q-Ветвь. Это набор линий, обусловленный Дп=1 и Д/ = 0. Их
частоты даются формулой
hv=\E —fan
для всех значений J. Следовательно, Q-ветвь, когда она разреше-
на, представляет собой одну линию при частоте данного колеба-
тельного перехода. В реальных молекулах вращательные постоян-
ные колебательных состояний п и п+1 мало отличаются друг от
друга, и поэтому Q-ветвь проявляется в виде «грозди» очень тесно
расположенных линий. На рис. 17.14, там, где ожидается Q-ветвь,
виден пробел; это обусловлено тем, что она запрещена для моле-
кулы, с которой проводился данный эксперимент (НС1).
R-Ветвь. Переходы с Дп=1 и Л/ = +1 составляют /?-ветвь, и
их частоты даются формулой
hv = \E =Л(о-|-2В (/ + 1).
Эти линии смещены на 2В, 4В,... соответственно в сторону высо-
ких частот, как показано на рис. 17-15.
Расстояние между Р- и /?-ветвями колебательного перехода
дает значение вращательной постоянной, и поэтому из колеба-
тельного спектра можно получить длину связи в молекуле, и нет
необходимости снимать чисто вращательный микроволновый
спектр (хотя последний дает более точные результаты).
Колебательные спектры КР двухатомных молекул. Основное
правило отбора для проявления колебательного спектра КР связа-
но с поляризуемостью молекулы. В этом случае важный критерий
состоит в том, изменяется ли поляризуемость, когда молекула ко-
леблется. Если она изменяется, то колебание будет КР-активным.
Это значит, что молекула может обмениваться энергией с фотона-
ми облучения, которые затем рассеиваются с увеличенной часто-
той (если соударение стимулирует колебательное возбуждение).
Во время колебания как гомоядерныс, так и гетероядерные двух-
атомные молекулы расширяются и сокращаются и «контроль»
ядер над Электронами, а следовательно, и молекулярная поляри-
зуемость соответственно изменяются. Оба типа молекул могут
поэтому давать колебательные спектры КР.
Специфическое правило отбора состоит в том, что Дп=±1.
Линии в области высокой частоты падающего излучения являют-
ся антистоксовыми; им соответствуют Дп = — 1. Они обычно очень
слабы, поскольку в любом образце первоначально в возбужденном
колебательном состоянии находится Очень мало молекул, и поэто-
му только очень небольшое число соударений приводит к переносу
энергии от молекул к фотонам. Линии в области низкой частоты
являются стоксовыми; им соответствуют Дл = + 1. На эти линии
накладывается вторичная структура, которая возникает из-за од-
новременных вращательных переходов, возбуждаемых при соуда-
88
Часть 2. Структура
состояние
> I I —гд———।—1—
—i—i—I—----1------1--------------LJ_
О-Ветвь | Ветвь
Положение
Q-ветви
Рис. 17.16. Образование 0-, Q- и S-ветвей.
рении. Правила отбора для этих вращательных переходов имеют
вид: ДУ = О, ±2 (как в чисто вращательных спектрах КР, стр. 75),
а соответствующие наборы линий называют Оветаью (для ДУ —
= — 2), Q-ветвью (для ДУ=О, если это разрешено; см. комментарий
о Q-ветвях па стр. 85) и З-ветвыо (для ДУ = + 2) (рис. 17.16).
Данные колебательных спектров КР дополняют информацию,
получаемую методами обычной спектроскопии. В частности, спект-
роскопия КР позволяет изучать гомоядерные двухатомные моле-
кулы; из расстояния между стоксовыми линиями можно получить
силовые постоянные, которые являются мерой прочности связей.
Вращательную структуру спектра можно интерпретировать череа
вращательные постоянные молекул и, таким образом, измерять
длины связей (см. табл. 17.1 и 17.2).
17.4. Колебания многоатомных молекул
На первой стадии исследования колебаний многоатомных мо-
лекул необходимо подсчитать число способов, которыми могут
17. Вращательные и колебательные спектры
89
Рис. 17.17. Альтернативные
описания колебаний молеку-
лы двуокиси углерода.
ь; (1340 см ’ Ч
О0ОЧ34
(2340 см'1)
вверх вниз вверх
М66? см-1) ГЫ>7 см-'?
происходить их колебания. В двухатомниках имеется лишь один
способ колебания — растяжение связи, но в многоатомниках при
искажении молекулы могут растягиваться связи и изменяться уг-
лы. Линейная молекула (типа двуокиси углерода), построенная
из У атомов, может колебаться ЗУ—5 различными способами, а
нелинейная многоатомная молекула ЗУ—6 способами. 1Четод, по
котором у можно получить эти цифры, тот же, что и метод, при-
мененный на стр. 72 для расчета степеней свободы двухатомных
молекул, а аргументация состоит в следующем.
Рассмотрим нелинейную молекулу из .V атомов. Общее число
координат, необходимое для определения положения всех атомов,
равно ЗУ. Положение каждого атома выверяется изменением его
координат, н поэтому общее число степеней свободы равно ЗУ.
Как и в случае двухатомных молекул, эти степени свободно мож-
но выразить способом, имеющим больший физический смысл. На-
пример, три координаты необходимы, чтобы точно определить
центр масс молекулы, и поэтому эти три степени свободы соответ-
ствуют поступательному движению всей молекулы. Из оставшихся
ЗУ—3 степеней свободы три можно отождествить с вращательным
движением молекулы. Таким образом, ЗУ—6 степеней свободы от-
вечают движениям иным, чем поступательное и вращательное: они
отражают колебания молекулы. В случае линейной молекулы, ко-
торая может вращдться только вокруг двух осей, имеется три по-
ступательные и две вращательные степени свободы, и число коле-
бательных степеней свободы равно ЗУ—3—2 = ЗУ—5. Например,
вода — тред-атомная нелинейная молекула имеет три колебатель-
ные моды (и три вращательные); с другой стороны, СОг — линей-
ная молекула имеет 4 колебательные моды (и только две враща-
тельные) .
На следующей стадии нужно найти наиболее подходящее опи-
сание мод колебаний. Рассмотрим в качестве примера молекулу
СО2. Один из возможных выборов четырех мод колебаний СО2
показан на рис. 17.17 слева: мы выбрали валентное колебание од-
ной связи С—О, валентное колебание другой такой же связи и два
взаимно перпендикулярных деформационных колебания. Этот вы-
бор совершенно справедлив, однако он имеет серьезный недоста-
90
Часть 2. Структура
ток. Например, если возбуждено колебание одной связи С—О, то
движение атома углерода приводит в движение другую связь С—О,
и, следовательно, энергия колебания перетекает назад и вперед
между двумя связями.
Намного более простая ситуация получается, если принять, что
одна мода колебания представляет собой симметричное валентное
колебание обеих связей одновременно (v, на рнс. 17.17). При таком
способе атом углерода испытывает воздействие одновременно с
обеих сторон. Другая мода — ангиеа.имегричное валентное колеба-
ние -v.3, при котором одна связь растягивается, а другая сжимает-
ся. Эти моды движения стабильны в том смысле, что, если воз-
буждена одна, она не возбуждает другую. Они являются двумя
нормальными модами молекулы. Две другие нормальные моды —
два взаимно перпендикулярных деформационных колебания v2.
Все четыре взаимно независимы: любую моду можно возбудить, и
это не приведет к возбуждению других.
Пример (вопрос 12). В случае линейной симметричной трехатомной молекулы,
такой, как СО2, с атомными массами mit га2, mj частоты симметричного валент-
ного колебания (v,) и антисимметричного валентного колебания (v3) равны
(k!tni)1,2 и (А/m)1^2 соответственно, где 1/m—При колебании Vi
центральный атом неподвижен и частота определяется mt. При колебании Vj рас-
стояния, которые проходят внешние и центральный атомы, относятся как (l/rrt;) :
; (—: (1/wii). Если бы все три атома имели одинаковую массу, то цент-
ральный атом двигался бы в два раза быстрее, чем внешние (в v,). н это объ-
ясняет множитель 2 в выражении для т. В случае СО2 амплитуды движения на-
ходятся в отношении (1/16) : (—2/12) : (1/16), или 1 : (—2,67) : I, ц т=4,36.
Когда 12С замещен на 13С, частота V; остается неизменной, но v3 изменяется в
у'4,36/4,22= 1,02 раза, поскольку эффективная масса для моды у, меньше в
13СО2, нем в i2CO2, потому что центральный атом движется меньше, чем в 12СО2
[амплитуды находятся в соотношении (1/16);(—2/13):(1/16), гми 1:(—2,46): 11.
Четыре нормальные моды двуокиси углерода (и 3W—6 мод ли-
нейных многоатомных молекул) являются ключом к описанию ко-
лебаний молекул. Каждая независимая нормальная мода ведет
себя подобно независимому гармоническому осциллятору (если
пренебречь ангармоничностью), и поэтому каждая может быть
возбуждена до энергии (пр-р/зМ^сь где шр — частота нормальной
моды Q. Эта частота зависит как от силовой постоянной для
данной моды (например, связь часто больше сопротивляется рас-
тяжению, чем изгибу), так и от приведенной массы причем со-
отношение имеет вид usq-Приведенная масса для дан-
ной моды зависит от того, как много массы начинает «раскачи-
ваться» при колебании. Например, при симметричном валентном
колебании атом углерода в двуокиси углерода неподвижен, и при-
веденная масса для этой моды определяется массой двух атомов
кислорода; при антисимметричном валентном колебании движутся
все три атома, и поэтому все три атома вносят вклад в приведен-
ную массу для этой моды.
17. Вращательные и колебательные спектры
91
Рис. 17.18. Координаты атом-
ных смещений в
тана.
молекуле ме-
12). О пред е-
нормальных
Пример (вопрос
лите симметрию
колебаний молекулы метана.
Метод. Возможно 3-5=15 сме-
шений, из которых 15—6—9
относятся к колебаниям. Их
можно классифицировать по
теории групп. Простейшим пу-
тем является учет того факта,
что при операции симметрии а
диагональ матрицы преобразо-
вания вносят вклад только
координаты смещений, которые
остаются неизменными, и по-
этому их вклад в характер операции равен 1. Если координата и и аер тируется,
на диагнонали появляется —1. Сошлемся на рис. 17.18.
Ответ. Ддя операции симметрии Е никакие координаты смещений нс меняются,
н поэтому матрица преобразования — диагональ е 1 для каждого ненулевого
элемента; баанс 15-мерный (15 возможных смешений), следовательно, характер
Е равен %(Е) = 15. При операции С3 никакие смещения не остаются неизмен-
ными, и поэтому у(С3)=0. При операции Сг «-смещение центрального атома
остается неизменным, тогда как х- и р-с.мещения меняют знак, поэтому х(ба) =
=—1. При S4 «-смещение центрального атома обращается, и поэтому %(S)f =
=—1. При CFd «-смещения атомов С, Н3, Н4 остаются неизменными, поэтому
Х(<та)=3. Следовательно, характеры будут 15 0 —1 —1 3, что соответствует сим-
метрии колебаний А + Е + Л+ЗТУ. Три из этих смещений соответствуют общему
поступательному движению молекулы: они трансформируются как х, у, «; это
Та. Три смещения вращательные и трансформируются как 1\. Следовательно, ко-
лебания охватывают симметрию Д^Е+ЙТз.
Комментарий. Мы вскоре увидим, что такого рода анализ симметрии позволяет
легко установить, какие моды активны.
Колебательные спектры многоатомных молекул. Основное пра-
вило отбора состоит в том, что нормальная мода колебания долж-
на приводить к изменению дипольного момента, если опа спектро-
скопически активна. При симметричном валентном колебании дву-
окиси углерода дипольный момент остается неизменным (около
нуля), и поэтому эта мода неактивна в ИК-спектре. Антисиммет-
ричное валентное колебание и два деформационных колебания из-
меняют дипольный момент, и поэтому все три моды активны в
ИК-спектре. Активные моды подчиняются специфическому прави-
лу отбора: StiQ— ± 1, поэтому энергия фундаментального перехода
каждой активной моды равна Отсюда следует ожидать, что
в спектре проявится поглощение при частотах vq=ч)е/2д (или при
волновых числах vq/c). Из анализа спектра можно установить си-
ловую постоянную для всех активных нормальных мод и, следо-
92
Часть 2. Структура
(3652 ем ') (1595см-’> (3756 см'1)
Рис. 17.19. Колебательные моды молекулы воды.
вательно, построить картину эластичности разнил частей молеку-
лы. На рис. 17.19 приведены три колебательные моды молекулы
воды. Заметим, что преимущественно деформационное колебание
(уй) находится при значительно более низкой частоте, чем другие
моды; все они включают растяжение и сжатие связей. Связи часто
легче изогнуть, чем растянуть.
На эту простую схему накладываются усложнения, возникаю-
щие из-за ангармоничности и эффектов молекулярного вращения.
Очень часто образец является жидким или твердым, и молекула
имеет возможность вращаться только очень небольшое время до
столкновения с соседом, поэтому она изменяет свое вращательное
состояние очень часто. Поскольку в жидкости время жизни враща-
тельных состояний молекул очень небольшое, вращательные уров-
ни энергии плохо выражены (стр. 64). Соударения между моле-
кулами часто происходят со скоростью около 1013 с_|, и даже, ес-
ли предположить, что скорость перехода молекулы в другое вра-
щательное состояние составляет только 1 % этой величины, можно
легко определить уширение за время жизни (стр. 64), превышаю-
щее 1 см-1. Этот эффект затемняет вращательную структуру коле-
бательного перехода, и поэтому колебательные спектры молекул
в конденсированных фазах обычно состоят из широких линий без
вращательной структуры.
Одним из очень важных применений ИК-спектроскопин много-
атомных молекул в конденсированных фазах является ее исполь-
зование в химическом анализе. Колебательные спектры различных
групп в молекуле играют роль «отпечатка пальцев»; часто молеку-
лу можно идентифицировать путем изучения ИК-спектра и отне-
сения полос поглощения по таблице характеристичных колеба-
тельных частот (табл. 17.3). Это иллюстрирует рис. 17.20.
Колебательные спектры КР многоатомных молекул. Нормальные
моды колебания многоатомных молекул активны в спектре КР,
если они сопровождаются изменением поляризуемости. Например,
симметричное валентное колебание двуокиси углерода растяги-
вает и сжимает молекулу; при этом поляризуемость изменяется, и
эта мода активна в спектре КР. Другие моды колебаний двуокиси
углерода оставляют поляризуемость неизменной. Так, при анти-
17, Вращательные и колебательные спектры
93
100
Л
SO -
I:।И|1 * т I V ( .1
° 4000 3200 2400 1900 1700 1500 1300 1100 900 ТС О
б, СМ'*
Рис. 17.20. Область «отпечатка пальцев» колебательного ИЦ-спектра.
симметричном валентном колебании растяжение одной связи ком-
пенсируется сокращением другой. Ни одна из этих мод по активна
в спектре КР, и поэтому спектр КР состоит из единственной линии,
соответствующей частоте симметричного валентного колебания.
Имеется полезный метод, позволяющий различить активные и
неактивные моды. Правило исключения устанавливает, Что 'если
молекула имеет центр симметрии, то моды, которые активны в
ИК-спектре, неактивны в спектре КР, и наоборот. Правило приме-
нимо к двуокиси углерода, но не к молекуле воды (так как опа
не имеет центра симметрии). Заметим, что из правила исключения
следует, что для того, чтобы получить всю информацию о жестко-
сти (силовом поле) молекулы, обладающей центром симметрии,
необходимо использовать и ПК- и КР-спектры.
Таблица 17,3
Типичные колебательные частоты <т(см-1)
с—н валентное 2850—2960 О-Н валентное 3590—3650
с—н деформационное 1340—1465 Н-связь 3200—3570
с-с валентное, деформационное 700—1250 С-0 1640—1780
с=с валентное 1620—1680 с=,ч » 2215—2275
о=с валентное 2100—2260 N—Н » 3200—3500
сор 1410—1450 С—F 1000—1400
NO7 no? 1350—1420 С—С1 » 600—800
1230—1250 С—Вт » 500—600
sor Силикаты 1080—1130 900—1100 С—1 » 500
Пример. Установите, какие нормальные моды молекулы воды активны в ИК-
И КР-спектрах.
Метод. Используем -теорию групп следующим образом, а) Нормальная мода
активна в И К-спектре, если ее симметрия является симметрией трансляции (х.
у, г), б) Нормальная мода активна в спектре КР, если ее симметрия является
симметрией квадратичной формы (х2, ху, ...). Симметрию колебаний находим, как
в последнем примере.
Ответ. Молекула Н3О принадлежит к группе симметрии Сг„, поэтому ее колеба-
тельная сям-иётрля 2/41 + Bi. В С/л трансляции трансформируются как ,4 У л),
94
Часть 2. Структура
В2(у) и Вг(х); поэтому все моды ИК-активны. Квадратичные формы трансфер-
мируются как Ai, A?, Bir В2, и поэтому вес колебательные моды КР-актпвиы.
Комментарий. Колебания Л] поляризованы по осн г, а колебания В2— по оси у.
Нарисуйте формы ьолсбаний и подтвердите физическую справедливость сделан-
ного вывода.
Литература
Barrow G. М„ Introduction to molecular spectroscopy, McGraw-Hill, New York,
1962.
Banwell C. If., Fundamentals ol molecular spectroscopy McGraw-Hill, New York,
1972.
Уитли П. Определение молекулярной структуры. Пер. с англ. — М.; Мир, 19/0.
Brand J. С. £>., Speakman /. С., Molecular structure; the physical approach, Ar-
nold, London, 1960.
Whiffen D. IL, Spectroscopy, Longman, London, 1972.
Atkins P. W., Molecular quantum mechanics, Clarendon Press, Oxford, 1970.
King 0. W., Spectroscopy and molecular structure, Holt, Reinhart and Winston,
New York, 1964.
Biygare W. H., Microwave spectroscopy. In Techniques ol chemistry (Weissber-
gcr A. and Rossiter B, W., eds.) Vol, 111 A 439, Wiley-Interscience, New York,
1972.
Sugden T. M., Kenney C. If., Microwave spectroscopy of gases. Van Nostrand,
Loudon, 1965.
Townes C. K., Schawlow A. L., Microwave spectroscopy, McGraw-Hill, New York,
1955.
Gans P., Vibrating molecules. Chapman and Hall, London, 197].
Rao С. V. R-, Chemical applications of infrared spectroscopy, Academic Press,
New York, 1963.
Woodward L. A., Introduction lo the theory of molecular vibrations and vibratio-
nal spectroscopy, Clarendon Press, Oxford' 1972.
Anderson D. IL, Woodall If. B., Infrared spectroscopy, in Techniques of Chemistry
(Weissberger A, and Rossiter B. W., eds.). Vol. 11 IB, 1, Wiley-Interscience,
New York, 1972.
Вильсон E., Дешиус Дж., Кросс П. Теория колебательных спектров молекул.
Пер. с англ, —М.; ИЛ, I960.
Беллами Л. Инфракрасные спектры сложных молекул. Пер. с англ. — М.: ИЛ,
1963.
Герцберг Г. Колебательные и вращательные спектры многоатомных молекул.
Пер. с англ. — М.: ИЛ, 1949.
Szymanski Н. Raman spectroscopy. Plenum, New York, 1967.
Long D. A., Raman spectroscopy, McGraw-IIili, New York, 1977.
During J. K, Harris W. C., Raman spectroscopy, ]n Techniques of chemistry
(Weissberger A. and Rossiter B. W., eds.). Vol. ШВ, 85, Wiley-Interscience,
New York, 1970.
Гилсон T., Хендра П. Лазерная спектроскопия КР в химии, пер. с англ.—М.:
Мир, 1973.
Задачи
17.1. Какие из следующих молекул могут давать чисто вращательный спектр:
Н2, НО, СН4, СН3С1, CHjClj, Н2О, Н2О2, NH3, NH4Cl?
17.2. Какие из следующих молекул могут поглощать в инфракрасной области:
Hs, IIC1, COs, HjO, СНзСНз, СН4, CHsCl, n2, n3_?
17.3. Какие из следующих молекул могут давать чисто вращательный спектр
КР; На. НС1, СН4, СН3С1, СН3С12, СН3СН3, НаО, SF6?
17. Вращательные и колебательные спектры 95
17.4. Какие из следующих молекул могут давать колебательный спектр КР; Нг,
НС1, СН4, НгО, СО2, СН3СНз, SbF6?
17.5. Какова длина волны с учетом сдвига Доплера красного света (Л=660 нм)
уличного светофора, когда к нему приближаются со скоростью 50 миль/ч? При
какой скорости приближения свет светофора казался бы зеленым (Л = 520 нм)?
17.6. Было найдено, что линия 'lsTi84- в спектре удаленной звезды сдвинута с
654,2 до 706,5 нм, и уширена до 61,8 пи. Какова скорость удалении звезды и
температура се поверхности?
17.7. Микроволновая спектроскопия — очень точный метод, однако из-за ушире-
ния линий детали спектра могут не проявиться. Какова доплеровская ширина
(выраженная в долях длины волны перехода) для каждого вида перехода в
a) HCi, б) 1С1 при 298 К? Какова ширина вращательного (в МГц) и колеба-
тельного (в см-1} перехода в этих частицах? i[7J(1C1) =0,114 сМ-Ч-
17.8. Из соотношения определите время жизни состояния, которое
обусловливает появление линии с шириной а) 0,1 см-1, б) 1 см’1 и в) 100 МГц.
17.9. Молекула в жидкости претерпевает 1013 соударений каждую секунду.
Предположим, что; а) каждое соударение приводит к колебательной дезактива-
ции молекулы, б) одно из ста соударений приводит к этому. Какова ширина
(в см-1) колебательных переходов в этом возбужденном состоянии?
17.10. Число столкновений, которое претерпевает молекула в единицу времени
в газе с давлением р, равно }'2о(8/гГ/лт) ^LpjRT. Каково лимитированное соу-
дарениями время жизни возбужденного состояния молекулы, если считать, что
каждое соударение эффективно? Какова ширина вращательных переходов в мо-
лекуле НС1 (п=0,30 нм“) при давлении газа 1 атм? Каково должно быть дав-
ление, чтстбы уширение вследствие столкновении было менее существенно, чем
доплеровское уширение?
17.11. Каково относительное содержание молекул Хлора (о=564,9 см-1) в ос-
новном и первом возбужденном колебательных состояниях при а) 273 К,
б) 298 К и в) 500 К?
17.12. Какое значение имеет 1 в наиболее заселенном состоянии молекулы СН4
при комнатной температуре? Не забудьте, что д.тя каждого значения 7 имеется
27 +1 состояний с одинаковой энергией (В = 0,]14 см-1).
17.13. Каково значение 1 для наиболее заселенного состояния молекулы СН4
при комнатной температуре? Здесь есть маленькая хитрость: хотя энергетиче-
ские уровни даются таким же выражением, как для линейной молекулы, нужно
не забыть, что каждому значению 1 соответствуют (271)2 состояний. Почему?
Используйте значение В=5,24 см-*,
17.14. В этой главе несколько раз появлялась приведенная масса, п важно знать,
как ее рассчитывать. Рассчитайте (в кг) приведенные массы и моменты инер-
ции: a) IHS5Cl, б) 2Н35С1 и в) 'H^’CI. Используйте значение 7? = 127,460 пм.
17.15. Вращательная постоянная для молекулы NH3 равна 298 ГГц (гигагерц).
Рассчитайте расстояние между линиями в ГГц, cm_i и мм и покажите, что зна-
чение В согласуется с длиной связи N—11, равной 101,4 пм, и углом между свя-
зями Ю0°47'. (Информацию о моменте инерции возьмите из табл. 17.1,)
17.16. Был спроектирован космический прибор для поиска следов СО в атмо-
сфере Сатурна. Было решено применить микроволновую установку на вращаю-
щемся по орбите спутнике. Где будут лежать первые четыре перехода молекулы
12О60, если длина связи составляет 112,82 пм? Какое необходимо разрешение,
если желательно отличить спектр 12С'6О от 13С16О, чтобы выяснить относитель-
ное содержание этих двух изотопов углерода?
17.17. Линии вращательного поглощения для газообразного НС1 имеют следую-
щие положения (в CM-i) [Hausler R. L., Oetjen R. Л. J. chem. Phys., 21, 1340
(1953)]: 83,32; 104,13; 124,73; 145,37; 165,89; 186,23; 206,60; 226,86. Определите
момент инерции и длину связи в молекуле (для *Н35С1).-
17.18. Предскажите положение линий микроволнового поглощения для DC1
(,2Н35С1) на основе данных предыдущей задачи.
96
Часть 2, Структура
17.19. Одинаковы ли размеры молекул НС1 и DC1? Данные, полученные из вра-
щательной структуры ИК-спектров этих частиц, приведены ниже (в см-1)
[Mills I. М., Thompson Н. IP., Williams R. L., Proc. R. Soc., A 218, 29 (1953);
Pickworth !., Thompson H. IP., Proc, R. Soc.. A 218, 37 (1953)]:
J O i э a 4 5 6
ГД-ветвь 2906,25 2925,92 2944,99 2963,35 2981,05 2998,05 3014,50
l Р-ветвь — 2865,14 2843,63 2821,59 2799,00 2775,77 2752,01
aH3’C f R-нетвь 2101,60 2111,94 2122,05 2131,91 2141,53 2150,93 2160,06
I P-ветвь — 2080,26 2069,24 2058,02 2046,58 2034,95 2023,12
17.20. Покажите, что момент инерции двухатомной молекулы, состоящей из
двух атомов с массами тА и гон. при длине связи R дается выражением 1 =
=r[mAmB/(mx-|-mR)]R:!. Какой момент инерции имеют: а) ‘Нг б) |2712?
17.21. Чисто вращательный спектр HI состоит из серии линий, расположенных
ил расстоянии 13,10 см-1 друг от друга. Какова длина связи в молекуле?
17.22. С термодинамической точки зрения следует ожидать, что моногалогеииды
меди СиХ в газовой фазе существуют в основном в виде полимеров, и поэтому
возникают трудности при получении Спектров мономеров достаточной интенсив-
ности. Трудность преодолевается продуванием газообразного галогена над тон-
кой пластиной меди, нагретой до 1000—1100 К [Alanson Е. L., de Lucia F. C.t
Gordy IP., J. chem. Phys., 63, 2724 (1975)]. Для CuBr переходы c 7 = 13—14,
14 15, 15—16 происходят при 84421,34, 90449,25, 96476,72 МГц соответствен-
но. Найдите вращательную постоянную и длину связи для молекулы СиВг.
17.23. В микроволновом спектре ISO12C32S наблюдаются лицин поглощения при
24,32592; 36,48882; 48.65164; 60,81408 ГГц, Каков момент инерции молекулы?
Заметим, что длины индивидуальных связей из этих данных получить нельзя;
однако читайте дальше.
17.24. При изучении микроволновых спектров различных изотопозамещенных мо-
лекул OCS [Тошдеэ С. Н-, Holden A. bL, Merritt R. F., Pljys. Rev., 74, 1113
(1948)] наблюдались следующие переходы (в ГГц):
J—>J+1 1---------->2 2----->3 3------>4 4----->5
24,32592 36,48882 48,65164 60,81408
ЦО«С345 23,73233 47,46240
Используя выражение для момента инерции, приведенное в табл. 17.1, и пред-
полагая, что длины связей при изотопном замещении це изменяются, найдите
длины связей С—О и С—S в OCS.
17.25. Изменение вращательной постоянной (или момента инерции) связано с
положением, в котором произведено изотопное замещение, рядом уравнений,
известных как уравнения Крайтчмана (Kraitchman). Покажите, что, когда изо-
топное замещение произведено на расстоянии г по этой оси от первоначального
центра масс молекулы типа симметричного волчка, величины г и АВ (изменение
перпендикулярной вращательной постоянной) связаны соотношением (z, пм)2 =
= 5,05376-Ю1* (ЛВ, МГц)Д(В, МГц) (В', МГц) (ДАК)], где В и В' — вращатель-
ные постоянные до и после замещения соответственно и &M,=Mr(Mr'—Mr)!M't'
17.26. Микроволновые спектры различных изотопозамещенцых молекул ClTeF5
свидетельствуют о строгом соответствии этих молекул типу симметричного
волчка ILegon Л. С., J. Chem. Soc., Faraday II, 1973, 29). Четыре атома фтора
образуют квадрат, атом теллура находится точно над центром плоскости, кото-
рую они образуют, еще один атом фтора расположен ниже, а атом хлора выше
этой плоскости. Используйте выведенное в предыдущей задаче уравнение Крайт-
чмана лля определения длины связи Те—С1 и значения 0 (угла между горизон-
талью и экваториальными связями Те—F), предположив, что все четыре связи
Те—F имеют одинаковую длину. Используйте следующие данные; переход
17. Вращательные и колебательные спектры
97
11—10 в S5Cl126TeF5: 30711,18 МГц; в «Cl^TeF-j: 30713,24 МГц; в ’тСР“ТеР8:
; | 29990,54 МГц.
17.27. Моменты инерции линейных галогенидов ртути (П) очень велики, и, как
следствие, О- и S-ветви в колебательных спектрах КР мало структурированы,
i Тем не менее пики обеих по; вс можно различить, что было использовано для
» измерения врашатсльных постоянных (Clark R. 1. Н., Rippon D. М., J. Chem.
ж Soc., Faraday II, 1973, 1496). Зная значение У, соответствующее максимуму за-
Ж" селенности, покажите, что расстояние между пиками О- и S-ветвей дается соот-
Я ношением Плачека—Теллера 6v=(32 BkT/hc)^1. При указанных температурах
Ч? были получены следующие значения расстояний: HgC12 (282°С): 23,8 см-1;
HgBr2 (292°C): 15,2 см->; Hgl2 (292°C): 11,4 см-1. Рассчитайте вращательные
постоянные для этих молекул.
17.28. Галогениды водорода имеют следующие фундаментальные частоты коле-
баний (в см-'); HF (4141,3), Н35С1 (2988,9), Нв1Вг (2649,7), Н>271 (2309,5). Най-
дите силовые постоянные связей водород — галоген.
17.29. Из данных последней задачи предскажите фундаментальные частоты ко-
лебаний галогенидов дейтерия.
17.30. Кривая Морзе [уравнение (17.3.4)] очень полезна как простое представ-
ление кривой потенциальной энергии молекулы. При изучении RbH было най-
дено, что о=936,8 см”1 и хо=14,15 см-'. Постройте график кривой потенциаль-
ной энергии в интервале от 50 до 800 пм при 7?е = 236,7 пм.
17.31. При вра|1|ении молекулы связи в пей могут ослабляться. Это можно про-
иллюстрировать, построив график кривой потенциальной энергии с учетом кине-
тической энергии вращения молекулы. Таким образом, строится график зави-
симости V*(Я) = V(/?)-f-B(/?)/(/+!), где V —потенциал Морзе, B=h/2mR2 и
m — приведенная масса. Постройте эти кривые для молекулы на том же гра-
фике, который был построен в предыдущей задаче, взяв 7=40, 80 и 100. Отметь-
те, как вращение влияет на энергию диссоциации. '[Если использовать значение
В (/?.-) =3.020 см-1, расчет B(R) упрощается.]
17.32. Первые пять колебательных энергетических уровней для молекулы НС1,
как было найдено, находятся при 1331,84; 3917.44; 6398,94; 8776,34: 11049,6 см-1.
Какова энергия диссоциации этой молекулы?
17.33. Колебательным энергетическим уровням молекулы Nal соответствуют сле-
дующие волновые числа: 142,81; 427.31; 710.31* 991,81 см-', Покажите, что они
/ 1 \ / 1 у
удовлетворяют выражению [rt-|--g-icF—fI_i""2'l ха, н определите силовую посто-
янную, кулевую энергию и энергию разрыва связи.
17.34, Молекула НС1 имеет потенциальную энергию, которая очень хорошо опи-
сывается кривой Морзе с параметрами 7)е = 5,33 эВ, хег-=52,05 см-1 и п =
= 2689,7 см-1. Предположив, что при дейтерировании потенциал остается неиз-
менным, предскажите энергии диссоциации D° для: а) НС1, б) DC].
17.35. В выражениях д.тя Р-, Q- и R-ветвей (стр. 86) предполагается, что мо-
мент инерции молекулы в возбужденных состояниях такой же, как в основном
состоянии. Выведите выражения для положения линий, ре делая этого предпо-
ложения.
17.36. В Р-ветви спектра |Н35С] наблюдались линии при 2865,1; • 2843,6 и
2821,6 см-' для 7 = 1, 2 и 3, а в R-ветви — при 2906,2; 2925.9; 2945,0 и
2963,3 смдля 7=0, 1, 2, 3 [Mills 7. (И., Thompson Н. IP., Williams R. L.r Proc. R.
Soc., A218, 29 (1953)]. Найдите силовые постоянные и длины связи для верхне-
го и нижнего колебательных состояний.
17.37. В колебательном КР-спектре Э1С15 имеется серия стоксовых линий с рас-
стоянием между линиями 0,9752 см-1 и подобная серия антистоксовых .тиний.
. Какова длина связи в С12?
17.38, Какие из трех колебаний молекулы АВ2 активны в ИК- или КР-спектре,
если молекула; а) изогнута и б) линейна?
17.39. Рассмотрите колебательную моду, 'которая соответствует равномерному
расширению бензольного кольца. Будет ли она а) КР-активной, б) ИК-активной?
7—242
18 Определение структуры молекул.
Электронная спектроскопия
Изучаемые вопросы
После тщательного изучения этой главы вы сможете:
1. Определить коэффициент поглощения, коэффициент экстинк-
ции, оптическую плотность и силу осциллятора (стр. 101) и ис-
пользовать закон Бера — Ламберта для расчета интенсивности
света после прохождения через образец [уравнение (18.1.1)].
2. Объяснить окраску комплексов переходных металлов
(стр. 104).
3. Описать переходы, ответственные за поглощение карбониль-
ной группой и двойной связью С = С (стр. 106).
4. Сформулировать принцип Франка — Кондона и применить
его для объяснения колебательной структуры электронных пере-
ходов (стр. 108).
5. Описать механизм флуоресценции (стр. 112) и фосфоресцен-
ции (стр. 114).
6. Описать работу лазера (стр. 115).
7. Определить термины предел диссоциации, преддиссоциация и
внутренняя конверсия (стр. 117).
8. Описать основы фотоэлектронной спектроскопии (стр. 119) и
связать фотоэлектронный спектр с электронным строением моле-
кулы (стр. 122).
9. Объяснить, как можно использовать рентгеноэлектронную
спектроскопию в химическом диализе и для изучения внутренних
электронных оболочек молекулы (стр. 124).
Введение
Когда электроны совершают переходы между молекулярными
орбиталями, па это затрачивается энергия порядка нескольких
электрон-вольт (1 эВ х 8000 см"1), и поэтому испускаемые или по-
глощаемые фотоны принадлежат к видимой и ультрафиолетовой
областям спектра (который охватывает участок от 14 000 см-1 для
красного света до 21 000 см"! для синего и до 50000 см-1 для да-
лекого ультрафиолета; табл. 18.1). Изучение электронных спект-
ров поглощения и испускания молекул позволяет объяснить окрас-
ку веществ. Например, если некоторый объект содержит молекулы,
которые поглощают сипий свет, в отраженном белом свете он будет
18. Электронная спектроскопия
99
Рис. 18.1. Спектр поглощения хлорофилла в видимой области.
выглядеть красным. Зеленая окраска растительности обусловлена
отражением белого солнечного света от листьев и т. д.’, которые
содержат хлорофилл — молекулы, способные поглощать и в крас-
ной и в синей областях спектра (рис. 18.1).
Электронные спектры дают информацию об электронной струк-
туре молекул. Анализируя спектры, можно вывести энергию элект-
ронов на разных молекулярных орбиталях и, следовательно, про-
верить теорию строения молекул. Ту же информацию можно по-
лучить иным путем, если определить, сколько энергии требуется
для удаления электрона из молекул. Эта область электронной
спектроскопии, называемая фотоэлектронной спектроскопией, бу-
дет рассмотрена в последней части главы.
Электронный переход в молекуле сопровождается большим из-
менением энергии, и перераспределение электронов изменяет
электростатические силы, действующие на ядра. Молекула отвечает
на это изменение сил интенсификацией колебаний. Другими сло-
вами, электронный переход обычно сопровождается колебатель-
ным переходом. Дополнительная колебательная структура в спект-
ре может разрешаться в случае газообразного образца, но в жид-
ком или твердом образце эти лилии сливаются и получаются ши-
рокие линии, почти лишенные характерных черт (рис. 18.2). Ко-
лебательные переходы, происходящие одновременно с электрон-
ными, сами сопровождаются вращательными переходами. Следо-
вательно, Электронные спектры газообразных образцов могут быть
очеиь сложными. По этой причине мы начнем с рассмотрения
электронных спектров жидких и твердых веществ, где топкие де-
тали спектра расплываются в широкие полосы.
7*
100 Часть 2. Структура
Таблица 18.1
Цвет, частота и энергия света
Цвет Длина волны, НМ Частота, I&u Гц Волновое число, JQ4 см-1 Энергия
эВ кДж/моль ккал/моль
Инфракрасный 1000 3. 00 1,00 1,23 120 28,5
Красный 700 4,28 1,43 1 77 171 40 8
Оранжевый 620 4,84 1,61 2.00 193 46,8
Желтый 580 5. 17 1,72 2 14 206 49,3
Зеленый 530 5,66 1 89 2,34 226 53,9
Синий 470 6,38 2,13 2.63 254 60,8
Фиолетовый 420 7,14 2 38 2,95 285 68.1
Близкий ультрафио- летовый 300 10,0 3 33 4,15 400 95,7
Далекий ультрафио- летовый 200 15,0 5,00 6,22 600 143
Другим фактором, усложняющим электронные спектры, яв-
ляется способность молекул поглощать достаточное для их распа-
да количество энергии. Это важно, так как диссоциация-—один из
фундаментальных процессов фотохимии. Мы увидим, как диссо-
циация влияет на спектр и какую фотохимическую информацию
можно получить, изучая ее.
Рис. 18.2. Слабоныраженная колебательная структура электронного спектра (ди-
ацетила СН3СОСОСН3). '
18. Электронная спектроскопия
101
18.1. Мера интенсивности
В этом разделе мы установим экспериментальную меру интен-
сивности переходов. Рассмотрим уменьшение интенсивности, кото-
рое происходит, когда свет с интенсивностью / проходит через
пластинку образца толщиной dx, содержащую поглощающее ве-
щество в концентрации С. Уменьшение интенсивности пропорцио-
нально толщине, концентрации и интенсивности падающего света, и
поэтому можно написать
dl~—ojCIdx,
где коэффициент а называется коэффициентом поглощения и за-
висит как от изучаемой молекулы, так и от частоты света. Это
уравнение, которое можно переписать в форме
d In/ ——aCdx,
применимо к каждому слою, на который можно подразделить об-
разец. Чтобы получить интенсивность света /г, выходящего из об-
разца толщиной / при интенсивности падающего света /;, мы сум-
мируем все последовательные изменения:
Л i
j din/= — СojCdx.
Л о
Если концентрация по всей толщине образца одинакова, то С не
зависит от х, и это выражение интегрируется, давая
закон Бера—Ламберта: Ц—/[ — ехр(—аСГ). (18,1,1)
Видно, что интенсивность излучения падает экспоненциально с тол-
щиной образца и зависит как от концентрации поглощающих ча-
стиц, так и от их способности поглощать на данной частоте.
В большинстве работ в выражении закона Бера — Ламберта ис-
пользуются десятичные логарифмы, поэтому последнее уравнение
обычно записывается как
Л=Д 10-^ (18.1.2а)
или
Ig (Л/Л) = —еС/. (18.1.26)
В этих альтернативных формах е (которое равно а/2,303) назы-
вается коэффициентом экстинкции частиц, при данной интенсивно-
сти, а произведение еС1—оптической плотностью или поглощае-
мостью образца.
Промер (вопрос 1). Свет с длиной волны 256 нм проходит через кювету
толщиной 1 мм, содержащую 0,05 моль/дм3 раствора бензола. Интенсивность
света уменьшается на 16% ее начальной величины. Рассчитайте оптическую плот-
102
Часть 2. Структура
ность и коэффициент экстинкции образца. Каким будет пропускание через июне»
ту толщиной 2 мм? ,
Метод. Оптическая плотность и коэффициент экстинкции определяются по урав-
нению (18.1.26).
Ответ. 1g 0.16 = — 0,80.
8 = —(l/C01g(/f//i) =
= 0,80/(0,05 моль/дм3)Х(0,1 см) =
= 160 моль-1 • дм8 см-1.
Если толщина кюветы равна 2 мм, то
lg(4/6) = “(160 мольдм3-см-1)х(0,05 моль/д«-5)Х(0,2 см) = —1,60.
Следовательно, /1/6=0,03, т. е. выходящий свет теряет 3% своей начальной ин-
тенсивности.
Комментарий. Коэффициент экстинкции можно также выразить в моль-1-см5;
в данном примере это дает е=160 000 моль-'-см1.
Во многих случаях поглощение распространяется на интервал
частот, и поэтому указание коэффициента экстинкции для одной
частоты может не дать истинного значения интенсивности погло-
щения молекулярного перехода. Интегральный коэффициент по-
глощения—это сумма коэффициентов поглощения для всех час-
тот полосы:
,4---ja(v)dv. (18.1.3)
Он является истинной мерой общей интенсивности широкой спект-
ральной полосы. Л иногда выражают через безразмерную величи-
ну f — силу осциллятора перехода
f =(4m.eceafLer) Л, (18.1.4)
где £0 — фундаментальная постоянная (диэлектрическая проницае-
мость вакуума; см. т. 1, приложение к гл. 11). Получается, что
Таблица 18.2
Характеристики поглощения некоторых групп и молекул
Группе ’max- сы~1 \nax- НИ в. ДМЭ/{моль-см)
с=с 58 000 170 16 500
с=о 34 000 295 10
-NOi 36 000 278 10
48 000 210 10 000
_N=N— 28 800 347 15
Беизольное кольцо 39 000 255 200
50 000 200 6 300
55 500 180 100 000
Спг+(ач) 12 400 810 10
Си (NHS) 16 700 600 50
18. Электронная спектроскопия
103
для интенсивных переходов сила осциллятора около единицы.
Другим указанием на интенсивность перехода является макси-
мальная величина коэффициента экстинкции полосы. Ее записы-
вают как етах. Типичные значения для сильных переходов имеют
порядок 104—10s см-1-дм3-моль-1; это указывает на то, что в
I моль/дм3 растворе интенсивность света (подходящей частоты)
падает до 1/10 ее первоначальной величины при прохождении че-
рез раствор толщиной около 0,1 мм. Некоторые значения приве-
дены в табл. 18.2.
Пример. Переход в бензоле имеет коэффициент экстинкции 160 моль_,-дм3-см-1
(последний пример) и происходит в интервале около 4000 см-1. Оцените силу
осциллятора.
Метод. Используем уравнение (18.1.4), аппроксимируя площадь по кривой по-
глощения как высота Xширина.
Ответ. . а = 2,303в = 2,303х(160000 моль-1 см*) = 3,68-10® моль-1 см2.
°® = j"а (v^v = с J а ~ =
= (3,00'101|) см/с)X(3,68'105 моль-1-см2)X(4000 см"1) =
= 4,43-101в моль-1 • сма с-1.
Чтобы определить f, необходимо знать множитель
. 4Х(9,110-10-31 кг)Х(2,998-10® м/с)х [8,854-10-и Кл*/(м*-Н)]
4mt№e/U - (6,022-Ю2? моль'1)х(1,602-10-18 Кл)* “
( = 6,259-10-1В моль-с/м*.
Следовательно,
: /= (4,43 1015 моль-1-ма с-1)Х(6,3-10-« моль-с/м2) = 2,8-10-*.
Комментарий. Заметим, что f—безразмерная величина. В данном примере она
невелика; это указывает на то, что переход «запрещен».
Преимущество введения понятия «сила осциллятора» состоит в
том, что ее можно рассчитать теоретически. Мы уже видели, что
интенсивность любого спектроскопического перехода связана с су-
ществованием дипольного момента перехода (стр. 62). Этому па-
раметру можно дать точное определение через волновые функции
начальных и конечных состояний молекулы. Если эти волновые
функции обозначить соответственно ф; и фг, то дипольный момент
перехода представляет собой величину
р—-—е J ф^гф^т, (18.1.5)
и его значение можно рассчитать, если известны обе волновые
функции. Связь между моментом перехода и наблюдаемой интен-
сивностью выражается формулой
/=(8л’/3) (m^/йе*) | р. ,я. (18.1.6)
104
Часть 2. Структура
Следовательно, интенсивность линии можно оценить, если знать
волновые функции молекулы. Это важно как связь между теорией
и экспериментом.
Пример. Найдите величину диполя перехода в последнем примере.
Метод. Мы видели, что /=2,8-IO-3. Подставляем в уравнение (18.1.6) и нахо-
дим | |д.|. Переход осуществляется при 256 нм.
Ответ. Необходимо знать множитель
nL,,o . _____((6,626-IO-»* Дж-с)Х(1,602- 10-1В Кл)2]
3he /8л те — (З/вл2) | (9,110-10'*х кг) j —
=« 7,092-Ю-43 м* с-1-Кл3.
v-=cA = (3,0-J08 M/c)/(256 W-e м)= 1,2-JO16 Гц.
Тогда ;
| р [а == (7,09-10~4а м«-с-х Кл*)Х(2,8.10’®)/(1>2-101® Гц) = 1,7-IO-’0 Кл2 м3.
поэтому |р.] = 1,3-10-30 Кл-м.
Комментарий. Переведем дипольный момент перехода в дебан (D) по формуле
1D=3,3-10-30- Кл-м. Это дает р=0,40 D. Переход можно наглядно представить
как движение части электрона (около 40%) на расстояние, примерно равное
радиусу атома.
18.2. Хромофоры
В ряде молекул поглощение фотона можно связать с возбуж-
дением электронов небольшой группы атомов. Например, когда в
молекуле содержится карбонильная группа, обычно наблюдается
поглощение при 290 нм, хотя его точное положение зависит от при-
роды остальной части молекулы. Такие группы называются хро-
мофорами (от греческого «носители окраски»), и их присутствием
часто объясняется окраска веществ. Имеется несколько типов хро-
мофоров, но мы рассмотрим только три: ионы переходных метал-
лов, двойные связи (такие, как С = С) и неподеленные пары элект-
ронов.
Окраску комплексов переходных металлов обычно приписыва-
ют наличию низколежащих ^-орбиталей. В гл. 15 (т. 1) мы видели,
что при наличии лигандов d-Орбитали центрального атома рас-
Октаэдрический
комплекс (d')
s9-—
Тетраэдрический
комплекс (хР) .
~ I ‘г
18,3. Электронные переходы в комплек-
сах переходных металлов.
18. Электронная спектроскопия
105
б, СМ-1
Рис. 18.4. Электронное поглощение для [Ti(H2O)6]3+Cl3.
щепляются на два набора (рис. 18.3). Расстояние между ними
обычно не очень велико, и возбуждение электронов с /sg-орбитали
на е^-орбиталь объясняет поглощение в видимой области спектра.
Спектр [TifHaOjeF^Cla показан на рис. 18.4; переход При
20 000 см-1 (500 нм) можно приписать переходу eg-— tlg Одного
d-электрона.
Единственным недостатком этого объяснения является то, что
переходы в октаэдрических комплексах запрещены. Правило отбо-
ра для электронных переходов в комплексах с центром симметрии
называется правилом отбора Лапорта; согласно Этому правилу,
разрешенными переходами являются лишь переходы, включающие
изменение четности. Это значит, что переходы g—и u—~rg раз-
решены, но переходы g—+g и и—ш запрещены. Это можно про-
следить на примере d— d-переходов в свободных атомах (см. т. 1,
стр. 484) или объяснить теоретике-групповыми доводами*.
Запрет для перехода es^~t2g может быть снят, если симметрия
молекулы нарушится колебанием. Например, колебание, показан-
ное на рис. 18.5, лишает комплекс центра симметрии и поэтому
* В соответствии с общим рассмотрением в раэд. 16.3 интеграл, который
определяет дипольный момент перехода ([уравнение (18.1.5)], в октаэдрической
группе должен иметь симметрию А1е или же стремиться к нулю. Просмотр таб-
лицы характеров группы О* показывает, что функции х, у, z трансформируются
как Т1И. Поэтому интеграл
не может трансформироваться как A1St поскольку комбинация характеров Tig,
Ли. Eg дает неприводимые представления симметрии и, а не g. Следовательно,
переход eg+~t*g запрещен по симметрии. -
106
Часть 2. Структура
Рис. 18.5. Виброиный переход в октаэдрическом комплексе.
нарушает £,и-классификацию орбиталей. При отсутствии центра
симметрии d— d-переход разрешен, и поэтому комплекс способен
поглощать. Поскольку электронные d — d-переходы зависят от ко-
лебаний комплексов, они называются вибронными. На практике
переход по вибропному механизму разрешен лишь в малой сте-
пени, и поэтому d— d-переходы в октаэдрических комплексах
обычно слабые.
Другой путь, по которому может происходить поглощение све-
та, состоит в переходе электрона от лигандов к d-орбиталям цент-
рального иона или наоборот. В таком переходе с переносом заря-
да электрон движется на значительное расстояние; следователь-
но, момент перехода может быть большим и соответственно боль-
шой будет интенсивность поглощения. Эта мода_ хромофорной ак-
тивности характерна, например, для иона МпСЦ и объясняет его
интенсивную фиолетовую окраску (Х=420—700 нм).
В случае двойной связи С = С при поглощении л:-электрон под-
нимается на пустую разрыхляющую л*-орбиталь (рис. 18.6). По-
этому мы говорим о (л*,я)-переходе (что обычно читается как
«переход ци — пи со звездочкой»). Энергия перехода для несопря-
женной двойной связи составляет около 7 эВ, и поэтому она по-
глощает примерно при 180 нм, т. е. в ультрафиолетовой области.
Когда двойная связь является частью сопряженной цепи, энергии
молекулярных орбиталей лежат ближе друг к другу и (п*,л)-пе-
реходы сдвигаются в видимую область спектра. Очень скоро мы
приведем важный пример.
Переход, ответственный за поглощение карбонильных соедине-
ний, связан с неподеленными парами электронов, локализованных
па несвязывающих орбиталях кислорода. Один из этих электронов
может быть возбужден на пустую л*-орбиталь группы (рис. 18.7),
и тогда переход называется (я*,п)-переходом (читается «переход
эн — пи со звездочкой»). Типичные энергии поглощения лежат
около 4 эВ (290 нм), а поскольку (л*,п)-переходы часто запреще-
ны по симметрии, поглощение слабое.
18. Электронная спектроскопия
107
Рис. 18.6. Переход в углерод-уг-
лероднсЙ двойней связи.
Важным примером (л*,л)- и (л*,ц)-переходов является меха-
низм зрительного восприятия. Сетчатка глаза содержит «зритель-
ный пурпур», который представляет собой белок, соединенный с
11-грс-ретиналем:
фотонов, поступающих в глаз. Раствор I l-ifuc-ретиналя поглощает
примерно при 380 нм, но при связывании с белком (в этом случае
концевой карбонил, возможно, исчезает) максимум поглощения
сдвигается до 500 нм, т. е. в синюю часть спектра. Главной при-
чиной того, что молекула может поглощать в видимой области,
является наличие сопряженных двойных связей, по они также
играют другую роль. В возбужденном электронном состоянии цепь
сопряженных двойных связей может изомеризоваться: одна поло-
вина цепк скручивается вокруг возбужденной связи С = С и обра-
зуется полностью транс-ретиналь:
Он имеет форму, отличающуюся от формы первоначального изо-
мера, и не может соединяться с белком. Поэтому, возможно, пер-
вичная стадия зрения включает поглощение фотона, за которым
следует изомеризация углеродной цепи; такое разворачивание мо-
лекулы приводит в действие нервный импульс к мозгу.
108
Часть 2. Структура
Рис. 18.7. Переход в карбо-
нильной группе.
18.3. Колебательная структура
н принцип Франка—Кондона
Уширение электронных полос поглощения в жидких образцах
обусловлено неразрешенной колебательной структурой. Эта струк-
тура появляется вследствие того, что электронные переходы сопро-
вождаются изменениями колебательного состояния молекул; в не-
которых жидких образцах и в газах колебательная структура мо-
жет быть разрешена и использована для получения информации о
колебательных характеристиках молекул в возбужденном состоя-
нии. Проявление колебательной структуры в электронных спект-
рах можно объяснить на основе принципа Франка — Кондона.
Принцип Франка — Кондона основан на той точке зрения, что,
поскольку ядра намного более массивны, чем электроны, во время
перераспределения электронов ядра остаются в своих начальных
положениях. Электронный переход быстро создает плотность за-
ряда в новых местах, и эта плотность заряда оказывает воздейст-
вие на ядра. Такое воздействие заставляет их осциллировать, и
они раскачиваются вперед и назад по Отношению к первоначаль-
ным положениям. Поэтому равновесное положение ядер в началь-
ном состоянии молекулы становится точкой поворота направления
движения в возбужденном электронном состоянии. Это иллюстри-
руется на рис. 18.8.
В квантовомеханическом описании эта модель представлена в
несколько замаскированном виде и усовершенствована до такой
степени, что можно рассчитать интенсивность переходов на раз-
личные колебательные уровни электронно-возбужденной моле-
кулы.
Рассмотрим кривые потенциальной энергии молекулы
(рис. 18.9). Они относятся к различным электронным состояниям
одной и той же молекулы. Для каждого электронного состояния
Отмечены некоторые колебательные энергетические уровни и неко-
торые колебательные волновые функции; эти волновые функции
определяют, где могут быть найдены ядра. Перед тем как проис-
ходит поглощение, молекула почти наверняка находится в основ-
ном колебательном состоянии низшего электронного состояния, и
поэтому нужно рассматривать этот начальный уровень. Форма
Рис. 18.8. Классическое обоснование принципа Франка — Кондона.
Электронное
основное
состояние
а
Рис. 18.9. Кнантовомеханическое обоснование принципа Франка — Кондона.
110 Часть 2, Структура
низшей колебательной волновой функции показывает, что наибо-
лее вероятное расстояние между ядрами соответствует их равно-
весному расстоянию 7?е. На нем мы сконцентрируем свое внима-
ние, поскольку следует ожидать, что большинство переходов в
спектре будет происходить в тот момент, когда ядра находятся в-
данном положении.
Когда происходит электронный переход, молекула возбуждает-
ся в состояние, представленное верхней кривой. Согласно прин-
ципу Франка — Кондона, во время перехода ядернын остов остает-
ся неизменным, и поэтому можно представить, что энергия моле-
кулы возрастает по вертикальной линии, указанной на рис. 18.9..
Отсюда пошло выражение вертикальный переход, который исполь-
зуется для обозначения электронного перехода, происходящего без
изменения геометрии ядер.
При вертикальном переходе пересекаются некоторые колеба-
тельные уровни более высокого электронного состояния. Звездоч-
кой отмечен уровень, которому соответствует наиболее вероятное
расстояние между ядрами 7?е (здесь волновая функция имеет мак-
симальную амплитуду), а следовательно, это наиболее вероятный,
уровень, на который происходит возбуждение. Однако это не един-
ственный колебательный уровень, на котором может заканчивать-
ся возбуждение, поскольку соседние уровни также характеризуют-
ся высокой вероятностью нахождения ядер на расстоянии 7?в. Та-
ким образом, переход происходит на все колебательные уровни
этой области, но с наибольшей интенсивностью — на уровень с:
волновой функцией, наиболее благоприятно перекрывающейся с
волновой функцией начального колебательного состояния.
Из рис. 18.9 видно, что колебательная структура спектра зави-
сит от относительного смещения двух кривых потенциальной энер-
гии молекулы, и, если два состояния значительно смещены, это
способствует появлению длинной прогрессии колебаний (множест-
ва колебательных линий в спектре). Верхняя кривая обычно (но
не всегда) смещена в сторону удлинения связей, так как возбуж-
денные состояния, как правило, имеют больший разрыхляющий
(«антисвязывающий») характер, чем основные состояния.
Принципу Франка — Кондона можно придать количественную
форму, проанализировав дипольный момент перехода. Если на-
чальная волновая функция молекулы есть ipe.nfr, R), где в обозна-
чает электронное состояние, л—-колебательное состояние, г —
электронные координаты и R— ядерные координаты, и если конеч-
ная волновая функция есть (г, R), то дипольный момент пере*-
хода ъ'п' ел равен '
Р = J (г, R) (г, R) dx^dx^,
18. Электронная спектроскопия
111
где интегрирование проводится по всем электронным и ядерным
координатам. Комбинация электронной и ядерной (колебательной)
.волновых функций может быть выражена как произведение
-фв (г)ф„(Ю, где ф„ (R)—колебательная волновая функция (такое
разделение является приближением, основанным на приближении
Борна — Оппенгеймера; см. т. 1, разд. 15.1). Тогда последний ин-
теграл будет произведением двух интегралов:
I*=— е J фе< (г) гфе (г) с1гзл J ф„, (R) (R) (1тЯд.
Второй сомножитель отражает перекрывание между колебатель-
ными волновыми функциями конечного и начального состояний.
Поскольку интенсивность перехода связана с квадратом диполь-
ного момента перехода, следовательно, наибольшая интенсивность
•будет для перехода между колебательными состояниями, имеющи-
ми наибольшее перекрывание. Это является формальной основой
яредыдущего обсуждения. Можно оценить интегралы перекрыва-
ния, входящие в выражение для момента перехода, и таким путем
можно количественно оценить разделение интенсивностей.
Пример (вопрос 4). Рассмотрим переход из одного возбужденного электронного
состояния е длиной связи Re в другое состояние с длиной связи fit?. Интенсив-
ность определяется перекрыванием колебательных волновых функций для этих
двух состояний. Рассчитайте перекрывание между колебательными основными
состояниями каждого из двух электронных состояний и покажите, что интен-
сивность 0—0-перехода наибольшая, когда обе равновесные длины связей оди-
наковы.
Для простоты считайте, что силовые постоянные для двух электронных состоя-
ний одинаковы.
Метод. Интенсивность пропорциональна квадрату перекрывания между двумя
колебательными состояниями-, это множитель Франка — Кондона. Рассчитаем
J Фо(Л)фоО?)^Я., где фв— основное колебательное с ос то я пне более низкого
электронного состояния, а —основное колебательное состояние более высо-
кого электронного состояния. Волновые функции возьмем из табл. 13.1 (см. т. 1.)
Ответ, фо (R) =• (а/л1''2)1''2 ехр -g- a3 (R —Re)a
Ф5 (R) = (а/тЛ3)1'2 ехр [ — а2 (R - R')2 ,
где (Обе функции гауссовы; одна центрирована иа R,, другая —на
&е-) Перекрывание равно
т
S, = J’Ч’о (Я)’MR) = (aM1/2)J ехр -----£-a»(R — Re)3 —
(r-r;)4]</r.
Из этого нетрудно получить выражение, пропорциональное интегралу e~l2, ве-
личина которого равна л1^2. Поэтому интеграл перекрывания равен S=
112
Часть 2. Структура
=expl[—^-аг(/?е—а интенсивность пропорциональна S2=exp[——
—Она равна единице, когда Re=Kg, и уменьшается по мере того, как дли-
ны связей в этих двух состояниях начинают все больше отличаться одна ОТ дру-
гой.
Комментарий, В случае молекулярного брома /?в=228 пм, а для верхнего со-
стояния Ре = 266 нм. Приняв колебательную частоту равной 260 см-:, получим
S2=7-10_,°; следовательно, интенсивность 0—0-перехода составляет только 7Х
XJO-10 от величины, которая наблюдалась бы, если бы потенциальные кривые
располагались одна над другой.
Необходимо отметить, что расщепление между колебательны-
ми линиями в электронных спектрах поглощения зависит от коле-
бательных энергий возбужденного электронного состояния; отсюда
следует, что, кроме других упомянутых применений, электронные
спектры можно использовать для описания жесткости электронно-
возбужденных молекул.
18.4. Судьба электронно-возбужденных состояний
Теперь мы обратимся к процессам, которые Происходят после
поглощения света и образования электронио-возбужде.ч.чых состоя-
ний. Энергия возбуждения переходит в колебание, вращение и по-
ступательное движение соседних молекул. При такой термической
деградации энергия переходит в тепловую энергию окружающей
среды. Гораздо больший интерес Представляет, возможность уча-
стия возбужденных молекул в химических реакциях. Примером
является изомеризация хромофора в Зрительном пурпуре; общие
основы фотохимии будут рассмотрены в части 3. В этом разделе
мы коснемся главным образом излучательного распада,, который
состоит в способности молекул отдавать энергию возбуждения пу-
тем испускания фотона.
Различают два механизма излучательной дезактивации: флуо-
ресценцию и фосфоресценцию. При флуоресценции испускаемое
излучение сразу же прекращается, когда убирают возбуждающий
источник, но При фосфоресценции оно может Продолжаться долгое
время (даже часы, но, как Правило, секунды или доли секунды).
Такое различие означает, что флуоресценция — это мгновенное
превращение поглощенного света во вновь испускаемую энергию,
но при фосфоресценции энергия запасается в некотором «резер-
вуаре», откуда она медленно вытекает.
Флуоресценция. Последовательность стадий, из которых состоит
флуоресценция, приведена на рис. 18.10. Первоначальное погло-
щение света переводит молекулу па колебательные энергетические
уровни более высокого электронного состояния. Если записать
спектр поглощения, то он должен иметь вид, показанный на
рис. 18.10, а. В возбужденном состоянии молекула подвергается
соударвлчгям с ее окружением (например, с молекулами раствори-
JS. Электронная спектроскопия
ИЗ
Рис. 18.10. Последовательность
стадий, приводящих к флуорес-
ценции.
теля) и ее тепловая энер-
гия переходит в тепловое
движение окружающих мо-
лекул. Соударения застав-
ляют молекулу спускаться
вниз по «лестнице» уровней
колебательной энергии, но
они не могут отнять у мо-
лекулы гораздо больший по
величине избыток электрон-
ной энергии и погасить
электронную энергию воз-
буждения. Поэтому моле-
кула может жить долго, и
этого будет достаточно для
того, чтобы путем спонтан-
ного излучения отдать из-
быток энергии и перейти в
более низкое электронное
состояние.
Понижение энергии происходит вертикально в соответствии с
принципом Франка-—Кондона, и появляется серия линий в виде
спектра флуоресценции (рис. 18.10,6). Колебательная структура
этого спектра характерна для более низкого электронного состоя-
ния (в противоположность структуре спектра поглощения), и по-
этому флуоресценция является ценным методом для изучения ко-
лебательных характеристик основного состояния.
Этот механизм объясняет то наблюдение, что флуоресцентное
излучение имеет более низкую частоту, чем падающее излучение:
флуоресценция происходит после того, как часть энергии перей-
дет к растворителю.
Яркие оранжевые и зеленые флуоресцентные Красители явля-
ются повседневным проявлением процесса деградации энергии:
они поглощают в синей и ультрафиолетовой областях и флуорес-
цируют в видимой области. Из приведенного механизма также
следует, что интенсивность флуоресценции должна зависеть от то-
го, способен ли растворитель отбирать больший квант энергии,
необходимой для перехода молекулы из одного электронного со-
стояния в другое. В самом деле, найдено, что флуоресценцию мож-
но Подавить, подбирая разные растворители. Например, раствори-
8—242
314
Часть 2, Структура
Рис. 18.11. Последовательность стадий, приводящих к фосфоресценции.
тель с широким интервалом колебательных энергетических уров-
ней (такой, как вода) может акцептировать большой квант элект-
ронной энергии, но растворитель с более тесно расположенными
уровнями (такой, как тяжелый SeOCl2) не может.
-Фосфоресценция. Последовательность стадий, приводящих к фос-
форесценции, приведена на рис. 18.11. Первые стадии такие же,
как в случае флуоресценции, но решающую роль играет наличие
второго возбужденного состояния молекулы. Это другое возбуж-
денное состояние называется триплетным состоянием, и оно отли-
чается от первого (синглетного состояния), поскольку в нем спи-
ны двух электронов параллельны, а не противоположны
(рис. 18.11)*.
В точке, где пересекаются Кривые возбужденных состояний, оба
состояния имеют общую геометрию, и здесь может происходить
процесс, позволяющий одному состоянию переходить в другое при
взаимном пересечении. Этот синглет-триплетный переход называет-
ся интеркомбинационной конверсией. В большинстве молекул пере-
ход синглета (спаренные спины) в триплет (два неспаренных спи-
ла) запрещен, но в некоторых молекулах слабо разрешен. Он про-
* Состояние называется триплетным по причине, обсуждавшейся на стр. 498
в т. 1. Когда два электрона имеют параллелвные спины, общий спиновый угло-
вой момент 5=1 (каждый отдельный электрон имеет s='/2)- Этот угловой мо-
-мент может иметь три ориентации по отношению к некоторому направлению
е пространстве (Afs=+1, 0, —1).
18. Электронная спектроскопия
115-
исходит, когда молекула содержит тяжелый атом, поскольку его
сильное спнн-орбитальнос взаимодействие может обратить относи-
тельную ориентацию электронных пар.
Если молекула переходит в триплет, то она продолжает пере-
давать энергию в окружающую среду и при этом движется вниз
по триплетной лестнице колебательных энергий. Лестница кон-
чается па основном колебательном состоянии возбужденного три-
плета, и теперь энергия молекулы находится «в ловушке». Рас-
творитель не может изъять оставшийся большой квант энергии
электронного возбуждения, а молекула не может излучить эту
энергию, так как возврат в основное состояние включает запре-
щенный синглет-триплетный переход. Однако излучательный пере-
ход не совсем запрещен, поскольку1 проявляется спин-орбитальное
взаимодействие, нарушающее правило отбора. Поэтому молекула
может медленно излучать, и это излучение может продолжаться
долго после образования первоначального возбужденного состоя-
ния.
Приведенный механизм согласуется с тем экспериментальным'
наблюдением, что энергия кажется заключенной в некий резерву-
ар, из которого она медленно вытекает. Кроме того, из него сле-
дует (и это также подтверждается экспериментально), что фосфо-
ресценция должна быть наиболее интенсивной для твердых образ-
цов. Это обусловлено тем, что окружающие молекулы в твердом-
веществе менее эффективно сталкиваются с возбужденной молеку-
лой, и стадия интеркомбинационной конверсии имеет время, чтобы
осуществиться, так как синглетное возбужденное состояние теряет'
колебательную энергию и опускается ниже точки пересечения до-
статочно медленно. Из указанного механизма также следует, что-
фосфоресценция зависит от наличия тяжелого атома; это тоже
согласуется с экспериментом. Наконец, механизм предсказывает,
что из-за наличия неспаренных электронных спинов возбужденное
«резервуарное» состояние должно быть магнитным. Это было под-
тверждено наблюдением магнетизма фосфоресцентнцх возбужден-
ных молекул с применением чувствительных резонансных методов,,
описанных в следующей главе.
Работа лазера. Как флуоресценция, так и фосфоресценция явля-
ются способами возврата в основное состояние путем спонтанного1
испускания. Лазерное действие, как подразумевается в акрониме
лазер (Light Amplication by Stimulated Emission of Radiation, t. e.
усиление света стимулированным испусканием излучения), связа-
но со стимулированным процессом испускания.
Типичное устройство лазера показано на рис. 18.12. Большая
часть молекул образца некоторым способом возбуждается в более
высокое состояние. Например, для получения частиц в этом состоя-
нии можно использовать химическую реакцию. Образец помещает-
ся в полость между двумя зеркалами, и, когда молекула спонтан-
«*
116
Часть 2. Структура
Равновесная» заселенность
Рис. 18.12. Последовательность стадий, приводящих к работе лазера.
но испускает фотон, он движется рикошетом вперед-—назад. Это
стимулирует испускание другими молекулами и еще подбавляет
фотонов той же частоты в полость между зеркалами, а эти фотоны
стимулируют еще большее число молекул к испусканию. Каскад
энергии возрастает очень быстро, и, если одно из зеркал посереб-
рено наполовину, излучение можно перехватить.
Характеристика лазерного излучения следует из способа гене'
рирования: оно монохроматично (так как фотоны стимулируют
фотоны той же самой частоты), когерентно (поскольку электриче-
ские поля фотонов находятся в фазе —еще одна особенность сти-
мулированных процессов) и нерасходящееся (так как фотоны,
движущиеся под углом к оси полости, не захватываются и не сти-
мулируют другие фотоны). .
На практике часто бывает более удобным использовать трех-
ступенчатую систему, показанную на рис. 18.13. Это обусловлено
тем, что инвертирование заселенности основного состояния (пере-
вод большинства молекул в возбужденные состояния) обычно бы-
вает невозможным, и часто легче получить относительный избы-
ток на верхнем из двух возбужденных состояний. Типичное уст-
ройство должно иметь мощный источник для стимулирования пе-
реходов между первым и третьим уровнями (например, ксеноно-
18. Электронная спектроскопия
117
Лазерное действие
Когерентное
излучение
J Последующий
> термический
। распад
Накачка
IfSSSSSlSSSSSSlSSigliSSSit
Рис. 18.13. Трехступеичагая лазерная система.
вую лампу или другой лазер), который обеспечивает накачку, и
• тогда будет наблюдаться лазерное действие на средний, незасе-
• ленный уровень. Накачку можно производить или с помощью
мощной оптической вспышки, как в рубиновом лазере, или хими-
чески, как в химическом лазере
на + 5---► 2HF, 11F* ------> HF+фотон.
Другой метод связан с соударением лазерной молекулы с части-
цами, возбужденными радиочастотным разрядом. Он используется
в Не — Ne-лазере, где некоторые энергии возбуждения неона точ-
но соответствуют энергиям возбуждения атома. гелия. Поэтому
возбуждение неона и последующие соударения с атомами гелия
могут привести к возбужденным состояниям, если передастся
энергия.
Диссоциация и преддиссоциация. Другая судьба электронно-воз-
бужденных молекул состоит в фрагментации на два или большее
число компонентов. Диссоциация иллюстрируется на рис. 18.14.
Начало диссоциации можно обнаружить, наблюдая, где кончается
колебательная структура электронного спектра: это будет диссо-
циационный предел спектра. Определение диссоциационного пре-
дела является ценным методом определения энергии разрыва свя-
зей, Переходы к состояниям с еще большей энергией всегда при-
водят к распаду, и, поскольку частицы разделяются и не находятся
в связанной осцилляции, колебательная структура пропадает, и
линейчатая структура сменяется непрерывной полосой.
Однако в некоторых случаях колебательная структура исчеза-
ет, но затем восстанавливается при более высоких энергиях. Такое
поведение, которое можно назвать преддиссоциацией, можно ин-
терпретировать с помощью кривых молекулярной энергии, пока-
занных на рис. 18.15, Когда молекула совершает переход на коле-
бательные энергетические уровни в области, где одна кривая по-
тенциальной энергии пересекает другую, может произойти реорга-
118
Удсгб 2. Структура
Континуум <
Диссоци-
ационный—Л
предел
Колебатель-
ная
структура
Рис. 18.14. Диссоциация, ответственная за уширение спектра.
низания ее электронов, переходящих из одного состояния в другое
(в точке пересечения ядерная геометрия обоих состояний одина-
кова). Этот процесс называется внутренней конверсией. Если мо-
лекула конвертируется в новое состояние, то ома может иметь
энергию, достаточную для диссоциации. Следовательно, энергети-
ческие уровни вблизи пересечения кривых имеют несвязанный ха-
рактер, и колебательная структура спектра расплывается в кон-
тинуум. Когда источник возбуждения дает энергию, достаточную
для того, чтобы перевести молекулу в колебательные состояния,
лежащие выше зоны преддиссоциации, молекула не может превра-
титься в диссоциативное состояние, уровни снова приобретают хо-
рошо выраженный колебательный характер и спектр вновь стано-
вится линейчатым.
Другой путь, приводящий к преддиссоциации, состоит в разли-
чии между спектрами поглощения и испускания. Это показано на
рис. 18.16 для случая молекулы S2. В спектре поглощения наблю-
дается серия линий в указанном на рисунке интервале частот, и
справа заметно некоторое уширение. В том же интервале частот в
18. Электронная спектроскопия
119
Диссоциа-
ционный—*»
предел
Колебатель-Г
пая
структура I
Диффузная Г
область 1
Колебатель-
ная
структура
Рис. 18.15. Переходы, приводящие к преддиссоцнации.
спектре испускания наблюдается серия линий только при низких
частотах (слева), потому что молекулы в верхнем колебательном
состоянии, которое лежит вблизи пересечения типа, показанного
на рис. 18.15, могут осуществить переход в континуум и продис-
социировать прежде, чем произойдет испускание.
18.5. Фотоэлектронная спектроскопия
Анализ оптических спектров молекул включает определение
частоты поглощенного света .с последующей интерпретацией дан-
ных как переходов электронов между орбиталями. Это позволяет
выяснить картину относительного расположения орбиталей. Такая
же информация может быть получена совершенно иным путем: из-
мерением количества энергии, необходимого для удаления элект-
рона с некоторой орбитали. Эта энергия является потенциалом
120
Часть 2. Структура
Рис. 18.16. Спектр, указывающий на предднссоциацию
row R. Г., Canad. J. Phys., 47, 2423 (1969), with permission].
[Ricks J. M., Bar-
ионизации молекулы (т. 1, стр. 489). Сильносвязанные электроны
имеют высокие потенциалы ионизации, а слабосвязанные электро-
ны— низкие. Определив потенциалы ионизации электронов на раз-
личных орбиталях, можно получить картину орбитальных энер-
гий.
Метод измерения основан на выражении, с которым мы стал-
кивались в связи с фотоэлектрическим эффектом (т. 1, стр. 429).
Мы видели, что кинетическая энергия выбиваемых электронов
связана с частотой падающего излучения v уравнением сохране-
ния энергии
hv — Ф(М), (18.5.1)
где Ф (М) — работа выхода для данного металла. Если фотон с
частотой v соударяется с о'дной молекулой, а не с поверхностью
металла, то кинетическая энергия выбиваемого электрона опреде-
ляется выражением такого же типа:
ftv=-i-mev24-/{, (18.5.2)
в котором Ф (М) заменена на — потенциал ионизации электрона.
Индексом i обозначена орбиталь, занятая этим электроном в не-
ионизированной молекуле. Если падающий свет состоит из одной
18. Электронная спектроскопия 121
частоты, то выбиваемые электроны будут иметь раздую кинетиче-
скую энергию, которая зависит от занимаемой ими орбитали. Сле-
довательно, измеряя эту кинетическую энергию и зная v, можно
определить поте и пи алы ионизации. Выбиваемые электроны назы-
ваются фотоэлектронами, а определение спектра их энергий дает
начало названию Фотоэлектронная спектроскопия (ФЭС).
Экспериментально метод просъ. Преждё ~всего необходим ис-
тон ник излучения, который должен быть монохроматическим (что-
бы его частота была точно известна), мощным (чтобы каждый
фотон пес достаточную для ионизации энергию) и интенсивным
(чтобы получить хороший поток фотоэлектронов). Поскольку да-
же для внешних, валентных электронов потенциалы ионизации
составляют несколько электрон-вольт, необходимо работать в ульт-
рафиолетовой области спектра (при длинах волн меньше
''•'200 нм). Большинство работ выполнено с применением света,
генерируемого разрядом в гелии; это дает сильную линию при
58,4 нм, что соответствует энергии фотона 21,22 эВ. Если изучает-
ся энергия связывания электронов, находящихся гораздо глубже
в молекуле (т. е. сильносвязанные электроны остова), то, чтобы
высвободить их, пужпы фотоны с гораздо большей энергией.
В этом случае необходимо использовать рентгеновское излучение,
и метод называется рентгеноэлектронной спектроскопией (РЭС).
Типичными источниками рентгеновского излучения являются хром
(5400 эВ) и алюминий (1490 эВ). '
Пример (вопрос 8). При использовании света с длиной волны 58,43 нм, генери-
рованного переходом в молекуле гелия, стимулированным электрическим раз-
рядом. было найдено, что из молекулы N2 выбиваются электроны с кинетичес-
кой энергией 5,63 и 4,53 эВ. Каковы энергии связывания этих электронов в мо-
лекуле?
Метод. Используем уравнение (18.5.2); 1 эВ = 80&5 см-1.
Ответ. Свет с длиной волны 58,43 нм соответствует волновому числу (1/58,43Х
ХЮ-7 см) = 1,712-10s см-1 и, следовательно, энергии 21,22 эВ. Тогда из уравне-
ния (18.5.2)
а) 21,22 эВ = 5,63 эВ-J-у, или Ц =15,59 эВ,
б) 21,22 эВ = -у аЗ эВ -J- или 7; = 16,69 эВ.
Таким образом, энергии связывания равны —15,59 и —16,69 эВ,
комментарий. Более прочно связанные электроны выходят с более низкой кине-
тической энергией (и скоростью). Этот результат анализируется ниже.
Кроме ионизирующего источника необходимо иметь некоторый
метод определения кинетической энергии фотоэлектронов. Обыч-
но это делается пропусканием их между полюсами электромагни-
та или через электростатический анализатор, так как степень откло-
нения электронов зависит от их скорости. При некоторой силе поля
электроны, имеющие определенную скорость и, следовательно, оп-
ределенную кинетическую энергию, будут отклоняться по дуге и
попадать в детектор, установленный в соответствующем месте.
При изменении поля электроны разной энергии достигают детек-
122
Часть 2. Структура
Рис. 18.17. Фотоэлектрав-
ный спектрометр.
тора, и для каждой
силы поля можно из-
мерить поток электро-
нов. Теперь силу поля
можно связать с ки-
нетической энергией
электронов. Фотоэлек-
тронный спектр пред-
ставляет собой график
в координатах: электронный поток, падающий на детектор, — ки-
нетическая энергия или разность hv-----mv2 (которая прямо
дает I;). Установка схематически изображена на рис. 18.17.
Типичные ФЭ-спектры приведены на рис. 18.18. Верхний спектр
относится к газообразному азоту; потенциал ионизации возрастает
справа налево. Главные особенности спектра можно интерпрети-
ровать через молекулярно-орбитальную диаграмму, описанную в
гл. 15 (т. 1) и приведенную на рисунке. Если ре придавать значе-
ния детальной структуре спектра, то видно, что линии разделяют-
ся на три основные группы. Наименее прочно связаны электроны,
расположенные на высшей молекулярной орбитали 2pcrg; их потен-
циал ионизации 15,6 эВ (что соответствует кинетической энергии
выбиваемых электронов, равной,5,6 эВ, при использовании источ-
ника Не с энергией 21,22 эВ) является мерой глубины распрложе-
ния орбитали па диаграмме молекулярных орбиталей. Следующий
потенциал ионизации (следующая низкая серия линий) лежит при
16,7 эВ (кинетическая энергия 4,5 эВ) и соответствует удалению
2рЛк-электрона. Наиболее глубоко расположенные электроны, ко-
торые можно выбить При данном облучении, дают серию линий,
начинающуюся при 18,8 эВ (кинетическая энергия 2,4 эВ); их
можно рассматривать как занимающие 2йСи-орбиталь исходной
молекулы.
Тонкую структуру спектра можно интерпретировать через ко-
лебательное возбуждение иона. Если в процессе выброса электро-
на основное состояние молекулы переходит в колебательно-воз-
бужденное состояние иона, то часть энергии фотонов не переходит
в кинетическую энергию электрона и уравнение сохранения энер-
гии превращается в
(18.5.3>
18. Электронная спектроскопия
123
Импульсы 8 секунду Импульсы в секунду
Ряс, 18.18. Фотоэлектронные спектры N2 и СО.
где ДЕ^ь —количество энергии, затраченное на колебательное воз-
буждение иона.
Возвращаясь к спектру азота, мы видим, что выброс 2р<тя- и
2sou-электронов слабо стимулирует колебание, однако выброс
2рл„-электрона сопровождается значительным колебанием. Не-
большое колебательное возбуждение ожидается, если длины свя-
зей в молекуле и ионе подобны, по если они различаются, то сле-
дует ожидать значительных колебаний. Это обусловлено тем, что
при внезапном удалении электрона из молекулы может получить-
ся ион в сжатом или растянутом состоянии по отношению к его
новой равновесной геометрии. Таким образом, наблюдаемый
спектр N2 показывает, что ни 2роя-, ни 2хсгц-орбиталь не играет
роли в связывании в N2, но 2рли-орбиталь оказывает сильное влия-
ние на размер молекулы и поэтому играет важную роль в связи-
124
Часть 2. Структура
Рис. 18.19. Рентгеноэлектронный
спектр азид-иона.
вании. Обычное спектроско-
пическое исследование мо-
лекулы Na показывает, что
опа имеет колебательную
частоту 2345 см-1 (расстоя-
ние между двумя низшими
колебательными энергети-
ческими уровнями). При
удалении 2рсгй-электрона в
структуре ФЭ-спектра рас-
стояние между этими уров-
нями получается равным
2150 см это говорит о
том, что 2р<т?-электроны
имеют слабое влияние на
жесткость связи. В отличие от этого при удалении 2ряа-электро-
на колебательная частота понижается от 2345 до 1810 см-1, т. е.
данный электрон вносит значительный вклад в жесткость связи.
В РЭС энергия падающих фотонов настолько велика, что элект-
роны удаляются с тесно связанных уровней атомов остова моле-
кулы. В качестве первого приближения примем, что потенциалы
ионизации остова не чувствительны к связыванию между атомами:
электроны остова слишком прочно связаны, чтобы па них сильно
влияло изменение распределения валентных электродов, происхо-
дящее при образовании связи. Оказывается, что это в значитель-
ной степени верно, и, поскольку потенциалы ионизации внутренних
оболочек характеризуют индивидуальные атомы, а не всю молеку-
лу, определение РЭ-спектра дает линии, характерные для элемен-
тов, присутствующих в данном соединении или сплаве. В частно-
сти, потенциалы ионизации ls-орбитали элементов второго перио-
да имеют такие значения (в эВ): Li(50), Ве(ИО), В(190), С(280),
N(400), 0(530) и F(690). Обнаружение одного из этих потенциа-
лов ионизации в РЭ-спектре указывает на присутствие соответст-
вующего элемента. Такое применение в химическом анализе обус-
ловило для РЭС альтернативное название ЭСХА (электронная
спектроскопия для химического анализа).
Хотя в основном верно, что природа молекулы не влияет на
потенциалы ионизации внутренних оболочек, но это не совсем
так, и можно обнаружить небольшие, но заметные сдвиги, которые
18. Электронная спектроскопия
125
зависят от локального окружения атомов. Например, азид-ион Ns
дает РЭС, который расщеплен на две линии, отстоящие друг от
друга на 6 эВ (рис. 18.19), хотя область 400 эВ типична для
Is-электронов азота. Ион имеет строение N = N = N с распределе-
нием зарядов (—, +, —) вдоль линии атомов. Наличие отрица-
тельных зарядов на концевых атомах понижает потенциал иониза-
ции их внутренних оболочек, а положительный заряд на централь-
ном атоме повышает его: отсюда две линии в спектре. Такой ана-
лиз может дать ценную информацию о химически неэквивалент-
ных, но в других отношениях идентичных атомах, и вместе с мето-
дами резонансной спектроскопии, описанными в следующей главе,
может дать полезные данные об электронодонорной и электроно-
акцепторной силе заместителей.
Одна из наиболее важных особенностей ФЭС состоит в том,
что этот метод дает картину энергий молекулярных орбиталей в
молекулах, которая может подтвердить (или опровергнуть) их по-
рядок, рассчитанный по теории молекулярных орбиталей. Однако
при этом важно учесть два ограничения. Во-первых, отождествле-
ние потенциалов ионизации молекулы с энергией электрона на мо-
лекулярной орбитали является приближением (называемым тео-
ремой Купманса), и при детальном анализе необходимо очень
строго учитывать ряд осложняющих факторов. Во-вторых, хотя мы
рассматривали процесс ионизации для единичных электронов, в
действительности его следует рассматривать через общие состоя-
ния молекулы и иона. Однако это технические проблемы, и они
не могут подорвать мощь метода. Фотоэлектронный спектр дает
очень близкую к истинной картину энергетических уровней моле-
кулярных орбиталей и подтверждает основные закономерности
связывания в молекуле.
Литература
Эткинс П. Кванты. Справочник концепций. Пер. с англ. — И.: Мир, 1977.
Jaffee Н. Н., Orchin М., Theory and applications of ultraviolet spectroscopy, Wi-
ley, New York, 1962,
Gram F., Visible and ultraviolet spectroscopy, In Techniques of chemistry (Weiss-
berger Л. and Rossiter B. W., eds.), Vol. I1IB, 207, Wiley-Interscience, New
York, 1972.
Рао Ч. Электронные спектры в химии. Пер. с англ. — М.: Мир, 1964.
Whiffen О. Н., Spectroscopy, Longman, London, 1972.
Murrell J. N., The theory of the electronic spectra of organic molecules, Chapman
and Hall, London, 1971.
Герцберг Г. Спектры и строение двухатомных молекул. Пер. с англ. — М.: ИЛ,
1949.
Герцбсрг Г. Электронные спектры н строение многоатомных молекул. Пер. с
англ. — М.: Мир, 1969.
Atkins Р. W., Molecular quantum mechanics, Clarendon Press, Oxford, 1970.
Gaydon A. G., Dissociation energies, Chapman and Hall, London, 1952.
126 Часть 2. Структура.
King G. TP., Spectroscopy and molecular structure, Holt, Reinhart and Winston,
New York, 1964.
Lengyel B. A., Lasers, Wiley-1 nterscjence, New York, 1971.
Pressley R. J., Handbook ot lasers, Chemical Rubber Co., Cleveland, Ohio, 1971.
Haught Д, F., Lasers and their application to physical chemistry, Ann. Rev. Phys,
Chem.. 19, 343 (1968).
Wotherspoon h'., Osier Cr. K., Oster G,, The determination of fluorescence and
phosphorescence, in Techniques of chemistry (Weissberger A. and Rossi-
ter B. W., eds,). Vol. IIIB, 429, Wiley-Interscience, New York, 1972.
Калверт Дж., Питтс Дж. Фотохимия. Пер. с англ, — М.: Мир, 1968.
Birks Л В., Photophysics of aromatic molecules, Wiley, London, 1970.
Turner D, TP., Baker C., Baker A. D., Brundle C. R., Molecular photoelectron
spectroscopy, Wiley-Interscience, New York, 1970.
Бейкер Л., Беттеридж Д. Фотоэлектронная спектроскопия. Пер. с англ. — М.:
Мир, 1974.
Задачи
18.1. Для поглощения света бромом, растворенным я четыреххлористом углеро-
де, были получены следующие данные при использовании кюветы толщиной
2 мм. Каков коэффициент экстинкции брома при данной длине волны?
[Вт-.}, 0,001 0,005 0,010 0,050
Пропускание, % 81,4 35,6 12,7 3-10-3
18.2. В другом опыте та же кювета была заполнена 0,01 моль/дм3 раствором
бензола и длина волны спектрометра была изменена до 256 нм, где наблюдал-
ся максимум поглощения. При этой длине волны пропускание равнялось 48%
начальной интенсивности. Каков коэффициент экстинкции бензола при этой дли-
не волны?
18.3. Каковы оптические плотности двух образцов в двух предыдущих задачах?
18.4. Каким будет процент пропускания: а) 0,01 моль/дм3 раствора бензола и
б) 0,001 моль/дм3 раствора брома в четырех хлор истом углероде при толщине
кюветы: 1) 0,1 см, 2) 10 см и длине волны 256 нм?
18.5. Когда пловец пыряет на большие глубины, он входит в темный мир (в пря-
мом смысле). Приняв, что средний коэффициент экстинкции морской воды в
видимой области равен 6,2-10-5 дм3/(моль-см), рассчитайте глубину, на кото-
рой до ныряльщика будет доходить: а) половина интенсивности света на поверх-
ности и б) одна десятая этой интенсивности.
18.6. Полосы поглощения многих молекул в растворе имеют ширину па полу-
высоте, равную около 5000 см-1. Для таких случаев оцените (предполагая тре-
угольную форму пика) интегральный коэффициент поглощения для полосы, для
которой: а) Стах~1-104 дм3/(моль-см) и б) ета1»5-103 дм3/(моль-см). (Ши-
рина па полувысоте означает ширину полосы при e=Vsemax-)
18.7, Какова сила осцилляторов для переходов, описанных в предыдущей задаче?
18.8, При первом же взгляде на любой оптический спектр молекулы в растворе
видно, что форма линии лишь отдаленно напоминает треугольник. Во многих
случаях лучше предположить, что она является гауссовой функцией (пропорцио-
нальной е-*а), центрированной па волновом числе или частоте, соответствую-
щей Стах. Предположите такую форму линии и покажите, как выразить интег-
ральный коэффициент поглощения через величину Етм я Ао,^ (ШИРИНУ на по-
лувысоте). Оба параметра можно легко измерить, и поэтому мы получаем бы-
Стрыё метод определения как так и силы осциллятора полосы.
18,9, Быстрый метод —это хорошо, по насколько приемлемо предположение о
гауссовой форме линии? Спектр поглощения диазоэтана (CHsCHNa) между
18. Электронная спектроскопия
12?
Рис. 18.20. Спектр поглощения диазоэтана СН3СНМ3.
24 000 и 34 000 см”1 приведен на рис. 18.20. Сначала определите S4- и f для
этой полосы, считая, что она имеет гауссову форму. Затем проинтегрируйте
полосу поглощения графически. Это можно сделать, или подсчитав квадраты па
миллиметровой бумаге (что утомительно), или переггесн линию на бумагу, вы-
резав по сс форме и взвесив (обычная процедура интегрирования таких кри-
вых). Запрещен или разрешен этот переход? emai (пик при 280 нм) =
= 11 дм3/(моль-см), Втах (430 нм) = 18 дм3/(моль-см).
18.10. На рис. 18.2 приведен более сложный спектр поглощения диацетила в ин-
тервале 250—470 нм. Оцените силу осцилляторов для обоих основных перехо-
дов и решите, запрещены они или разрешены. Вц заметите, что спектр постро-
ен в шкале Длин волн; это значит, что перед выяснением f-вели чип переходов
; нужно проделать дополнительную работу. Имеются три подхода. Один состоит
в том, чтобы определить Д<г1(<2 из данных по длинам волн и использовать при-
ближение к гауссовой (по волновому числу, конечно) форме линии. Второй под-
ход состоит в том, чтобы перевести спектр в шкалу волновых чисел и провести
- графическое интегрирование. Третий подход—нахождение выражения для f
через интегрирование по длине волны и непосредственное использование дан-
ного спектра. Используйте все три метода.
18.11. Из ультрафиолетовых спектров можно получить много информации об
энергетических уровнях и волновых функциях небольших неорганических моле-
кул. Пример с развитой колебательной структурой показан на рис. 18.21; это
спектр газообразного SO2 при 25 °C. Найдите силу осциллятора перехода. Раз-
решен он или запрещен? Какие электронные состояния приемлемы для основно-
го состояния Л1 этой молекулы типа Civ?
18.12. Сила осциллятора—это параметр, который, как мы видели, может быть
измерен из данных экспериментальных спектров. Его можно также вычислить,
зная строение молекулы и, в частности, молекулярные орбитадн основного и
возбужденного состояний, участвующих н переходе. Мы попрактикуемся в та-
ких вычислениях, рассмотрев некоторые простые модели молекул. Прежде всего'
рассмотрите электрон в одномерном квадратном ящике с длиной L. Рассчитай-
те силу осциллятора для переходов п4-1 «-л в «+2 *- п. Следует начать с рас-
128
Часть 2. Структура
чета дипольного момента перехода [уравнение (18.1,5)] в форме р.г =
| фп,(х)хфл(х)йх, а затем, используя уравнение (18:1.6), найти f. (Только
6
х-компонента момента перехода не равна нулю.)
18.13. Поведение электрона в ящике не очень отличается от поведения электро-
нов в реальных молекулах; в гл. 15 (т. 1) мы видели, что модель СЭМО дает
в приближенном выражении идею об энергиях спектроскопических переходов;
сейчас мы увидим, что ее можно использовать для оценки интенсивности. Мо-
лекула (|5-каротин) ответственна за оранжевую окраску моркови. Рассмотрите
полиеновую цепь, ограниченную стрелками, как хромофор. Оцените энергию воз-
буждения (в см-1), определите, какой окраске моркови в белом свете соответ-
ствует эта энергия, и затем используйте результат, полученный в предыдущей
задаче, для оценки коэффициента экстинкции молекулы. Оцените толщину слоя
морковного сока (предположив, что он прозрачен и имеет мольную концентра-
цию молекул приведенного выше вещества), который поглощает 50% падаю-’
щего света.
18.14. Предположим, что вы хнмнк-колорист и перед вами стоит задача усилить
цвет красителя без изменения его природы, а цвет интересующего вас красителя
в значительной мере обусловлен наличием длинной полиеновой цепи. Удлините
или укоротите вы цепь? В красную или синюю область сдвинется при этом цвет
красителя?
18. Электронная спектроскопия
129
Интенсивность фосфоресценции
Рис. 18.22. Фосфоресценция нафталина и бензофенона.
18.15. Будем считать, что электрон в атоме осциллирует путем простого гармо-
нического движения (это было ранней моделью строения атома). Волновые
функции для электронов даны в табл. 13.1 (т. 1). Покажите, что сила осцилля-
тора для перехода этого электрона с его основного колебательного состояния
на первое возбужденное колебательное состояние точно равна -д-. Если бы элект-
рон мог колебаться в трех измерениях, то сила осциллятора для всех возмож-
ных переходов из основного состояния была бы равна единице; отсюда пошло
название сила осциллятора для параметра f.
18.16. В тексте мы видели, что класс переходов, в результате которых получа-
ются спектры переноса заряда, включает миграцию электрона из одной области
молекулы в другую. Примером является переход электронов с орбиталей лиган-
дов на орбитали центрального иона металла в некоторых комплексах переход-
ных металлов (подобных фиолетовому иону МпО4). В черновом варианте идея
о силе осциллятора и, следовательно, emai может быть получена с помощью
простого вычисления, основанного на следующей модели. Рассмотрим две водо-
родные ls-орбитали, разделенные расстоянием 7?. jio достаточно близко располо-
женные друг к другу, так что есть некоторое перекрывание. Пусть в начальном
состоянии молекулы электрон целиком находится на одной из этих орбиталей,
а в конечном состоянии (с переносом заряда)—целиком на другой орбитали.
Дипольный момент перехода равен р — —-е [ фр-ф^Фг, т. е. приблизительно —eRS,
где S—интеграл перекрывания двух орбиталей. Постройте график зависимости
силы осциллятора этого перехода от R, используя величину S(R) из задачи
15.13 (т. 1). При каком расстоянии интенсивность наибольшая? Почему интен-
сивность падает до нуля, когда R—>0 и R—><»?
18.17. Используйте простую теоретике-групповую аргументацию для выяснения
вопроса, какой из следующих переходов разрешен: а) (л*,л)-переход в этилене
или б) (л*.п)-переход в карбонильной группе (Сл>).
9—242
130 Часть 2. Структура
18.18. Спектр па рис. 18.22 имеет ряд особенностей, которые будут рассмотрены
в этой п следующей задачах. Остановимся на линии А. Это спектр фосфорес-
ценции бензофенона в твердом растворе в этаноле при низких температурах при
облучении светом с длиной волны 360 нм. Что можно сказать о ко.чеба юльных
энергетических уровнях карбонильного хромофора: а) в его основном электрон-
ном состоянии н б) в его возбужденном электронном состоянии?
18.19. Когда нафталин облучается светом с длиной волны 360 нм, он не поглота-
ет; однако линия 5 на диаграмме — это спектр фосфоресценции твердого рас-
твора смеси нафталина и бензофенона, и можно наблюдать компоненту, обус-
ловленную наличием нафталина. Объясните это наблюдение.
18.20. В спектре флуоресценции паров антрацена наблюдаются серия пиков уве-
личивающейся интенсивности с отдельными максимумами при 440, 410, 390 и
370 нм и далее резкий обрыв при более коротких длинах волн. В спектре погло-
щения наблюдается резкий подъем от нуля до максимума при 360 нм с «хво-
стом» пиков уменьшающейся интенсивности при 34а, 330 и 305 нм. Объясните
эти наблюдения.
18.21. Рассмотрим индикатор, такой, как бром феноловый синий, для которого
существует равновесие 1пНз=М1++1г1- (1пН гоже может иметь заряд), где 1пН
и 1п-—формы, поглощающие в разных частях спектра (так что при изменении
pH меняется окраска). Предположим, что О(1пН) и ) — оптические плот-
hocih образца индикатора, когда он находится в кислотной (1пЫ) и основной
(In-) формах. Пусть оптическая плотность па одной и той же длине волны для
смеси этих форм будет Л (mix). Покажите, что степень диссоциации а индикато-
ра дается соотношением [Л (mix)—D (InH)]/[O(In-)— О(1пН)], Далее покажите,
что pH раствора можно выразить через константу диссоциации Kin индикатора
и оптическую плотность £>(mix).
lS.22. При добавлении пиридина к раствору иода в четыреххлористом углероде
полоса поглощения при 520 нм смещается до 450 нм. Однако поглощение рас-
твора при 490 нм остается постоянным; это так называемая изобестическая точ-
ка. Используя метод предыдущей задачи и особенно соотношение между а и
Л (mix), покажите, что изобсстическая точка должна наблюдаться, когда погло-
щающие вещества находятся в равновесии.
18.23. Ниже приведены данные для коэффициента экстинкции (Г0—8 е,моль-|Х
ХДм3’СМ“!) раствора я-ни трофеи о ла в воде при разных pH [Biggs A. I., Trans.
Faraday Soc., 50, 800 (1954)]. Найдите значение рКа для ионизации нитрофе-
нола.
pH 4 5 6 7 8 9 10
317 им 9,72 9,72 9,03 5,55 1,81 1,39 1,39
407 нм — — 1,66 9,16 17,50 18,33 18,33
18-24. В молекуле кислорода особенно важный переход обусловливает возникно-
вение полосы Шумана — Рунге в ультрафиолетовой области спектра. Волновые
числа (см-1) переходов из основного состояния на колебательные уровни более
высокого электронного состояния (которое формально является 3SjT) имеют сле-
дующие значения: 50062,6; 50725,4; 513690' 51988,6; 52579.0; 531434- 53679,6;
54177,0; 54641,8; 55078,2; 55460,0; 55803,1; 56107,3; 56360,3; 56570,6. Какова
энергия диссоциации верхнего электронного состояния? Ответ на этот вопрос
следует получить экстраполяцией к точке схождения последовательности коле-
бательных уровней,
18.25. Известно, что возбужденное состояние молекулы О2, о котором шла речь
в предыдущей аадаче, диссоциирует на один атом кислорода в основном состоя-
нии и один возбужденный атом с энергией па 190 кДж/моль выше основного
состояния; этот возбужденный атом ответствен за многие фотохимические «про-
казы» атмосферы. Основное состояние кислорода диссоциирует на два атома
в основном состоянии. Объединив эту информацию С данными предыдущей за-
дачи, найдите энергию диссоциаций основного состояния кислорода.
18. Электронная спектроскопия
131
18.26. При распаде молекулы на атомы спиновый угловой момент сохраняется.
Какие атомные мульт и плети ости разрешены, когда на атомы распадается:
а) молекула кислорода и б) молекула азота?
18.27. На рис. 18.18 Приведены фотоэлектронные спектры N2 и СО. Припишите
линии определенным процессам ионизации и классифицируйте орбитали на свя-
зывающие (или разрыхляющие) и несвязывающие, учитывая степень развитости
колебательной структуры. Проанализируйте полосы в области 4 эВ с точки зре-
ния расстояния между колебательными уровнями ионов.
18.28. Фотоэлектронный спектр NO можно описать следующим образок
(Turner D. IP-, Physical Methods in Advanced Inorganic Chemistry (eds H.A.O.
Hill and P. Day), Wiley-lnterscrence, 1968). При использовании излучения He с
длиной волны 584 пм (21,21 эВ) наблюдается единственная сильная полоса
при 4,69 эВ и длинная серия из 24 линий, начинающаяся при 5,56 эВ и кончаю-
щаяся при 2,2 эВ. Более короткая серия из 6 линий начинается при 12,0 эВ и
кончается при 10,7 эВ. Объясните этот спектр.
18.29. Электроны с высшей энергией в фотоэлектронном спектре воды (при ис-
пользовании излучения Не с энергией 21,21 эВ) проявляются в области 9 эВ,
при этом наблюдается большое расстояние между колебательными уровнями,
равное 0,41 эВ. Полоса симметричного валентного колебания неионизированиой
воды находится при 3652 см~1. Какой вывод можно сделать о природе орбита-
ли, с которой выбивается электрон?
18.30. В том же спектре воды для полосы в области 7 эВ наблюдается Длинная
колебательная серия с интервалом между линиями 0,125 эВ. Полоса деформа-
ционного колебания воды лежит при 3652 см_|. Какой вывод можно сделать
о связывающих свойствах орбитали, занятой выбиваемыми электронами?
9
19 Определение структуры молекул.
Резонансные методы
Изучаемые вопросы
После тщательного изучения этой главы вы сможете:
1. Написать условие резонанса для электронного парамагнит?
кого резонанса {уравнение (19.1.4)],
2, Описать измерение ЭПР (стр. 134).
3. Объяснить смысл g-фактора и описать способ его измерения
(Стр. 136).
4. Описать причину сверхтонкого взаимодействия и сверхтон-
кую структуру спектра ЭПР (стр, 137).
5, Провести различие между диполь-дипольным (анизотроп-
ным) и контактным (изотропным) сверхтонкими взаимодействия-
ми (стр. 140).
6. Построить карту плотности неспаренного электрона в кольце
ароматических радикалов (стр. 142).
7. Рассчитать разности заселенностей спиновых состояний
электронов (стр- 142) и ядер (стр. 145).
8. Указать механизм процессов спин-решеточной и поперечной
релаксации (стр. 144).
9. Описать измерение ЯМР (стр. 146).
10. Объяснить смысл постоянной экранирования и химического
сдвига (стр. 147) и выразить последний в д- н т-шкалах.
11. Объяснить появление тонкой структуры в спектре ЯМР
(стр. 148).
12, Вывести скорости процессов из ширины спектральных ли-
ний и внешнего вида спектра (стр. 152).
13, Описать *эффект Мессбауэра и эксперимент по спектроско-
пии Мессбауэра (стр. 155).
14. Объяснить и интерпретировать изомерный сдвиг я электри-
ческое квадрупольное расщепление в мессбауэровском спектре
(стр. 157, 158).
Введение
С резонансом мы встречаемся, когда два маятника подвешены
на одной опоре, обладающей небольшой гибкостью. Если привести
в движение один из маятников, то другой будет приведен в колеба-
тельное движение благодаря движению общей оси, а энергия ко-
]9. Резонансные методы
133
лебания будет переходить взад-вперед между двумя маятниками.
Этот процесс наиболее эффективен в том случае, когда частоты
двух осцилляторов одинаковы. Условие сильного эффективного
парного взаимодействия вследствие одинаковой частоты называет-
ся резонансом’, говорят, что энергия возбуждения резонирует меж-
ду двумя спаренными осцилляторами.
Резонанс лежит в основе ряда повседневных явлений, например
настройки радио на слабые колебания электромагнитного поля,
вызванные удаленным передатчиком. В данной главе будут рас-
смотрены некоторые спектроскопические методы, использующие
резонанс. Их общая черта состоит в том, что система энергетиче-
ских уровней источника излучения точно определенной частоты
должна «подходить под пару» изучаемому осциллятору, и сильное
поглощение наблюдается тогда, когда выполняется условие резо-
нанса,
19.1. Электронный парамагнитный резонанс
Электрон обладает спиновым угловым моментом и вследствие
этого спиновым магнитным моментом (т. 1, разд. 14.3), Спин мо-
жет иметь две ориентации, обозначаемые как и и р, по отношению
к некоторому выбранному направлению, и Эти ориентации соответ-
ствуют проекциям углового момента msh, Это значит,
что спиновый магнитный момент может иметь две ориентации по
отношению к приложенному магнитному полю. При рассмотрении
эффекта Зеемана (т. 1, стр. 500—502) было показано, что энергия
электрона в магнитном поле ограничена двумя значениями в со-
ответствии с указанными двумя ориентациями. Эти значения та-
ковы:
Ems-=2p3tnsB, (19.1.1)
где В — приложенное магнитное поле, а цв — магнетон Бора
Отсюда следует, что при усилении магнитного поля энер-
гия электронов с а-спином (т, = -у) повышается, а энергия
электронов с ^-спином понижается (рис. 19.1). Разность энергий
этих двух спиновых состояний будет
Д£ = £1/2-£_1;2 = 2рвВ. (19.1.2)
Если образец облучается полем с частотой v, то иеспарснные
электронные спины имеют энергетические уровни, которые прихо-
дят в резонанс с излучением, если установить такое магнитное по-
ле, чтобы
(19.1.3)
134
Часть 2. Структура
Рис. 19.1. Энергетические уровни электронного
спина в магнитном поле.
Когда выполняется это условие, энер-
гетические уровни находятся в резо-
нансе с окружающим излучением, и
спины могут сильно поглощать его
энергию. Наступление этого условия
резонанса [уравнение (19.1.3) ] обна-
руживается наблюдением сильного
поглощения падающего излучения,
обусловленного резким переходом
спинов из (3-состояний в а-состояния.
Метод электронного парамагнитного резонанса (ЭПР), который
также называют электронным спиновым резонансом (ЭСР), со-
стоит в изучении свойств молекул, содержащих неспарепный
электрон, путем наблюдения магнитных полей, при которых они
приходят в резонанс с используемым излучением определенной
частоты.
Метод. Магнитное поле порядка 0,1 —1,0 Т (1 —10 кГс) можно лег-
ко создать, не прибегая к сложной технике. Поле в 0,3 Т соответ-
ствует резонансу с электромагнитным полем с частотой 10 ГГц
(Ю10 Гц) и длиной волны 3 см. Такое магнитное поле использует-
ся в большинстве выпускаемых ЭПР-спектрометров; излучение с
длиной волны 3 см соответствует Х-полосе микроволнового излу-
чения, т- е, ЭПР — это микроволновый метод.
Принципиальное устройство спектрометра показано на
рис. 19.2. Он состоит из источника микроволн (этот генератор на-
зывается клистроном), полости, в которую помещается образец в
стеклянном или кварцевом сосуде, детектора излучения и элект-
ромагнита, дающего поле, которое можно варьировать в области
около 0,3 Т. Микроволны из клистрона по волноводам идут в по-
лость, а затем в детектор.
Измеряя поглощение микроволнового излучения при непрерыв-
ном изменении поля, получают спектр ЭПР, типичный пример ко-
торого приведен на рис. 19.3. Своеобразный вид спектра обуслов-
лен тем, что по технической причине он записывается в форме пер-
вой производной поглощения (как dS]dB, где S — сигнал, а В —
приложенное поле), и поэтому кривая поглощения (рис. 19.4,а)
предстает в виде первой производной (рис. 19.4,6). Причина со-
стоит в способе, которым регистрируются микроволны.
19. Резонансные методы
135
Рис. 19.2. Спектрометр электронного парамагнитного резонанса (McLauch-
lan К. A., Magnetic Resonance, Clarendon Press, Oxford, 1972).
Образец может быть газообразным, жидким или твердым, од-
нако при работе с газами возникают сложности, которые здесь
не рассматриваются. Главное требование состоит в том, чтобы
молекулы образца обладали неспаренными спинами. Следователь-
но, ЭПР можно использовать для изучения свободных радикалов
(которые имеют один неспаренный электрон), образующихся в
ходе химических реакций или при фотолизе, комплексов многих
переходных металлов, а также молекул в триплетном состоянии
(два неспаренных электрона). Метод неприменим к обычным мо-
лекулам со спаренными спинами.
g-Фактор. Молекулы разных типов приходят в резонанс при раз-
личных значениях напряженности приложенного магнитного поля.
Это находится в кажущемся противоречии с уравнением (19.1.3),
из которого следует, «то все электронные спины резонируют при
136
Часть 2. Структура
Рис. 19.3. Спектр ЭПР анион-радикала бензола в растворе.
одном и том же приложенном поле для данной микроволновой ча-
стоты. Чтобы приспособить различие между молекулами к усло-
вию резонанса, последнее переписывается в виде
(19.1.4)
где g (g-фактор электрона) —экспериментально определяемый
параметр, характеризующий изучаемую молекулу. Для свободного
электрона g = 2 (на самом деле 2,0023; см. т. 1, стр. 502)
и для многих радикалов g ле-
жит в интервале 1,9—2,1. Для
исследуемой молекулы величина
g-фактора может быть получена
из магнитного поля, соответству-
ющего центру спектра ЭГ1Р.
Пример (вопрос 3). Центр спектра ЭПР
метильного радикала приходится на
329.4 мТ, если рабочая частота спектро-
метра равна 9,233 ГГц. Какова величи-
на ^-фактора?
Метод. Используем уравнение (19.1.4)
н форме g—hv/рвВ.
Рис. 19 4. Кривая поглощения н ее про-
изводная.
19. Резонансные методы
137
(6,620 10-34 Дж с)х(9,233’10s Гц)
Ответ. g = (9 274-10-34 Дж/Т)Х(0,3294 Т) “ 2-0026
Комментарий. Для многих органических свободных радикалов величина g близ-
ка к этой. Для неорганических радикалов она лежит В пределах 1,97—2,02. Для
ионов переходных металлов g-фактор часто лежит в более широком интервале
(например, 0—4).
Причина отклонения g от 2 состоит в том, что спиновой маг-
нитный момент электрона взаимодействует с локальным магнит-
ным полем, которое отличается от приложенного поля. Это обус-
ловлено тем, что приложенное поле может побуждать электроны
циркулировать по молекулярному каркасу, и такое орбитальное
движение создает небольшое дополнительное магнитное поле 6В.
Величина 6В пропорциональна самому В, и поэтому общее локаль-
ное поле B + SB можно записать как В—аВ, где о —некоторая
константа (отрицательный знак в В—а В следует из определения
о). Условие резонанса выполняется, когда
hv = 2рвВлок или hv — 2рв (В -]-6В) — 2 (1 — а) рвВ.
При сравнении с предыдущим уравнением получается, что g-фак-
тор равен 2—2а. Константа а может быть положительной или от-
рицательной в зависимости от тина молекулы, и поэтому g может
быть меньше или больше, чем 2. Типичными являются значения
2,00226 для атома водорода, 1,999 для NO2 и 2,01 для С1О2.
Величина g зависит от электронной структуры молекулы, так
как приложенное поле должно перемещать Электроны по молеку-
ле; следовательно, знание этой величины даст некоторую инфор-
мацию о Строении молекул. В химическом применении более важ-
ным является то, что g-фактор можно использовать для вспомога-
тельной идентификации частиц, которые участвуют в реакции,
идущей через свободные радикалы.
Сверхтонкая структура и сверхтонкие взаимодействия. Наиболее
важен тот факт, что спектр ЭПР имеет сверхтонкую структуру.
Сверхтонкая структура—это расщепление спектра на ряд линий
с центром в том месте, где наблюдается простой резонанс, который
мы обсудим в дальнейшем. Эта структура (типа, показанного на
рис. 19.3) является следствием наличия ядерных магнитных мо-
ментов.
Вначале рассмотрим роль одного протона в свободном радика-
ле. Известно, что протон обладает магнитным моментом. Этот не-
большой магнитный момент создаст вблизи протона магнитное
поле, и электронный спин испытывает действие комбинации при-
ложенного и ядерного полей. Поскольку спин протона равен -у
(как и электрона), его магнитный момент может принимать две
138
Часть 2. Структура
ориентации относительно приложенного поля. Одна ориентация
усиливает локальное поле электрона, другая уменьшает его. Если
обозначить г-компонеиту ядерного углового момента через mih,
= , то общее локальное поле выражается как
В^^В+ат,,
где а — некоторая константа, называемая константой сверхтонко-
го взаимодействия (мы не учитываем эффектов, приводящих к
сдвигу g-фактора). Если протон имеет одну ориентацию, то ло-
кальное поле изменяется до В + ~а, а если он имеет другую ори-
ентацию, то локальное поле изменяется до В—^-а.
Половина из общего числа молекул образца имеет протоны с
т.!— а другая половина — протоны с mi——Следовательно,
половина из общего числа молекул будет резонировать, когда при-
ложенное поле удовлетворяет условию
Av = 2р в (в 4-~- aj=2рвВ 4- рва
или ’
В — ~а,
а другая половина будет резонировать, когда приложенное поле
имеет величину, определяемую соотношением
ftv=2pB^B---aj
или
В —(ftv/2pB)-f-~-a.
Таким образом, вместо одной линии в спектре будут наблюдаться
две линии, находящиеся друг от друга по магнитному полю на
расстоянии а и равноудаленные от центра спектра; интенсивность
каждой из них равна половине общей интенсивности. Это разными
способами иллюстрируется на рис. 19.5.
Если неспаренный электрон подходит близко к ядру азота, то
спектр расщепляется на три линии одинаковой интенсивности.
Это связано с тем, что ядра 14N имеют спин, равный единице; по-
этому возможны три ориентации (гп/ — — 1, 0, 4-1), и каждую спи-
новую ориентацию имеет одна треть радикалов образца. В общем
для ядра со спицовым квантовым числом / спектр расщепляется
19. Резонансные методы
139
Резонанс
— Нет СТР
СТР на одном
протоне
Рис. 19.5. Сверхтонкое расщепление (СТР) па одном протоне.
на 2/4-1 линий одинаковой интенсивности, положение которых
дается формулой
/tv = m}=I, I— 1,..., —/. (19.1,5)
Если в одном радикале содержится несколько магнитных ядер,
то каждое вносит вклад в расщепление спектра. Например, при
наличии двух протонов спектр расщепляется на четыре линии; их
положение задается четырьмя значениями, при которых В удов-
летворяет условию резонанса
Л* = ^а2т/г; m/s^= ±Л-,
Константа сверхтонкого расщепления щ может Отличаться от as,
если атомы водорода в радикале не находятся в эквивалентных
положениях, поскольку несларенный Электрой может иметь боль-
шую плотность вблизи одного из ядер. Когда протоны эквивалент-
ны (так что а[ = йй, как в случае двух метиленовых протонов в
этильном радикале СН3СНг), линия с и т,г=—Ц- совпа-
1 1
дает с линиеи, для которой --------и т?г=2 , и поэтому спектр
является триплетом линий с соотношением интенсивностей 1:2:1.
Не трудно показать, что если радикал содержит N эквивалент-
ных протонов, то спектр расщепляется на jV-j-1 линий с бино&и-
140
Часть 2, Структура
альным распределением интенсивности, которое дается треуголь-
ником Паскаля:
Распределение интенсивности
N
0 1
111
2 12 1
3 13 3 1
4 1 4 6 4 1 и т. д.
На рис. 19.3 был приведен спектр отрицательного иона бензо-
ла; соотношение семи линий 1 : 6 ; 15 : 20 ; 15 ; 6 ; 1 подтверждает
наличие шести эквивалентных протонов в данном радикале.
Пример (вопрос 4). Радикал содержит одно ядро MN (7 = 1) со сверхтонкой кон-
стантой 1,03 мТ и два эквивалентных протона со сверхтонкой константой
0,35 мТ. Выведите форму спектра ЭПР.
Метод. Рисуем сверхтонкую структуру, обусловленную ядром HN, расщепляем ее
на одном нз протонов, а затем расщепляем полученный спектр ла другом про-
тоне.
Ответ. Ядро i4N дает три линии равной интенсивности на расстоянии 1,03 мТ
друг от друга. Каждая линия затем расщепляется на дублеты с расщеплением
0.35 мТ. Каждый из этих дублетов в дальнейшем расщепляется на дублеты,
отстоящие друг от друга на расстоянии 0,35 мТ: центральные линии каждой
пары дублетов совпадают и дают линию удвоенной интенсивности. Следователь-
но. в общем получаются три эквивалентных 1:2: 1-триплета с внутренним рас-
щеплением 0.35 мТ на расстоянии 1,03 мТ друг от друга. .
Комментарий. Этот метод всегда можно использовать для построения спектра.
Часто бывает быстрее нарисовать картины,' которые дают группы эквивалент-
ных прогонов (в данном случае два протона дают 1:2: 1-триплет), и пепо-
срсдстпепно^ наложить друг па друга эти картины.
Из анализа сверхтонкой структуры спектра можно получить
важную информацию о строении радикала (сверхтонкая структу-
ра— это своеобразный отпечаток пальцев). Более того, поскольку
величина расщепления зависит от распределения нссцарснного
электрона вблизи имеющихся магнитных ядер, измеренное расщеп-
ление можно также использовать для построения карты молеку-
лярных орбиталей, которые занимает песпаренный электрон. Но
прежде чем построить такую карту, мы должны знать, что опреде-
ляет величину константы сверхтонкого взаимодействия.
Сверхтонкое взаимодействие — это магнитное взаимодействие
между магнитным моментом песпаренного электрона и магнитным
ядром. Имеются два типа вкладов. Электрон на /7-орбитали не под-
ходит очень близко к ядру, и поэтому он испытывает действие маг- .
нитпого поля, которое можно рассматривать как возникающее из ;
точечного магнитного диполя. Это диполь-дипольное взаимодейст- 1
вие. В то же время s-электрон подходит к ядру так близко, что
19. Резонансные методы
141
приближенное представление последнего в виде точечного диполя
становится неверным, и начинает проявляться Другой тип взаимо-
действия — контактное взаимодействие Ферми. В обоих случаях
возникающие магнитные поля могут быть достаточно большими.
Например, электрон на 2р-орбитали атома азота испытывает дей-
ствие поля ядра 14N в 3,4 мТ (34 Гс). На ls-электрон атома водо-
рода действует среднее поле около 50 мТ (500 Гс), обусловленное
контактным взаимодействием, н-а 2х-электрон атома азота—поле
в 55,2 мТ, а на 2а-электрон атома фтора — поле в 1,7 Т (17 кГс).
В жидком образце диполь-дипольные взаимодействия усредняются
до пуля и остается лишь контактное взаимодействие. В твердом
образце сохраняются оба типа взаимодействия. Величину контакт-
ного взаимодействия можно интерпретировать через s-характер
неспаренного спина, а диполь-диполыгое взаимодействие — через
р-характер. Это дает ценную информацию о стру-ктуре молекуляр-
ной орбитали и гибридизации атомных орбиталей, занятых неспа-
ренным электроном (см. задачу 19.17). Величины констант сверх-
тонкого взаимодействия для атомов приведены в табл. 19.1.
К сожалению, внешний вид сверхтонкого спектра пока пол-
ностью не объяснен. 11а рис. 19.3 показан спектр отрицательного
иона бензола, в котором песпаренный электрон занимает л-Орби-
таль. Используется жидкий образец. Но каким образом может
осуществляться контактное взаимодействие электрона с протона-
ми, лежащими в узловой плоскости занимаемой Электроном орби-
тали? В ароматических ион-радикалах оно встречается часто и
является характерной особенностью спектров ЭПР органических
радикалов.
Такое взаимодействие объясняют поляризационным механиз-
мом сверхтонкого взаимодействия.' Согласно этому механизму, на
Электроны связи С—Н влияет магнитное поле, возникающее на
протоне, а Электроны связи С—Н могут в свою очередь влиять на
энергию соседнего неспаренного л-электрона. Это значит, что энер-
Габ.шца 19.1
Константы сверхтонкого взаимодействия для атомов
Изотоп Спин Изотропное взаимодействие, му Анизотропное взаимодействие, мТ
1н Чг 50,8 (1s) —
2Н 1 7,8 (Is) -—
13С 1/а 113,0 (2s) 6,6 (2p)
«N 1 55,2 (2s) 4,8 (2p)
19J? Чг 1720 (2s) 108,4 (2p)
Ч^ 364 (3s) 20,6 (3p)
а5С1 а/г 168 (3s) 10 (3p)
37С1 % 140 (3s) 8,4 (3p)
142
Часть 2. Структура
гия неспаренного электрона зависит от Ориентации спина протона,
и поэтому резонансная линия расщепляется на две. Расчет и экс-
перимент показывают, что возникающее таким путем дополнитель-
ное поле у электрона составляет около 2,8 мТ (28 Гс).
Если вернуться к спектру С6Н-“, то видно, что сверхтонкое рас-
щепление равно 0,47 мТ, т. е.-|- ‘2,8 мТ. Теперь эту величину
можно интерпретировать в свете поляризационного механизма.
Неспаренный электрон рассредоточен по кольцу и занимает л-ор-
биталь каждого атома углерода с равной вероятностью. Поэтому
следует ожидать, что поляризационное взаимодействие с каждым
соседним протоном будет составлять 1/6 от силы взаимодейстния
с целым электроном. Этот тип анализа дает мощный способ по-
строения карты распределения электронного спина вокруг колец
ароматических органических радикалов (и, следовательно, карты
молекулярной орбитали, которую он занимает). Его можно также
применять к гетероциклическим молекулам, чтобы исследовать
изменение электронного распределения под влиянием гетероатома.
Пример (вопрос 6). Спектр ЭПР иона (нафталин)* можно интерпретировать,
если учесть наличие двух групп из четырех эквивалентных протонов. Протоны в
а-положениях и.чеюг расщепление 0,490 лТ, а в jB-лоложениях — расщепление
0,183 мТ. Постройте карту плотности неспаренного спина вокруг кольца.
Метод. Рассмотренные в последнем параграфе результаты суммируются урав-
нением Мак-Коннелла a-e — Qp, где р — плотность неспаренного электрона на ато-
ме углерода, Q —константа и цц — константа сверхтонкого расщепления для
протона, связанного с углеродом. Для иона (бензол)’ 0,47 MT=Q-_g_j и поэтому
Q=2,8 мТ. Возьмем эту величину п для иона (нафталин) *
Ответ. Для «-положений р= (0,490 мТ/2,8 мТ) — 0,18. Для В-положений р=
= (0,183 мТ/2,8 мТ)=0,07.
Комментарий. Заметьте, что неспаренный электрон сосредоточивается в «-поло-
жениях. Для гетероциклов можно использовать приблизительно такое же зна-
чение Q.
Форма линий и релаксация. Осталось объяснить интенсивность и
форму резонансных линий в спектрах ЭПР. Наблюдаемая интен-
сивность пропорциональна разности заселенностей а- и fj-спиновых
уровней. Если бы заселенность была одинаковой, то приложенное
поле индуцировало бы переход такого же числа спинов из § в «
(поглощение энергии), как и из а в р (испускание энергии), асум-
марнос поглощение было бы равно нулю. Величину разности засе-
ленностей для образца при температуре Т можно рассчитать из
распределения Больцмана (т. 1, стр. 21). По энергии а-уровень
лежит на 2цвВ выше ^-уровня, и поэтому отношение заселенностей
равно
NoJN$ = ехр (—2у.вВ/кТ)я
19. Резонансные методы
143
Отсюда следует, что
•Лд-'Чх _ 1-exp(-2p.BW’) _
A₽ + Afa 1 +exp(-2[xBfWj
1 _(1 -2Ив6/ЛГ + .. .) ~
1 4" (1 "Ь + ...)
fwgEB/A7\ (19,1.6)
поскольку ‘Zy^BlkT мало. При комнатной температуре kT~
яй200 см-1, и в типичном случае (В—0,3 Т) 2цвВ эквивалентно
0,3 см'1. Следовательно, величина этого отношения равна только
7-Ю-4, т. е. заселенность не сбалансирована лишь на 0,07%. Мо-
жет показаться удивительным, что вообще наблюдается поглоще-
ние. Ведь при поглощении спины перекачиваются в более высокое
состояние, разность заселенностей уменьшается и, как можно ожи-
дать, вовсе исчезает. Таким образом, можно ожидать, что неболь-
шое первоначальное поглощение будет стремиться к нулю через
короткий промежуток времени после начала опыта.
Проблема слабого поглощения решается путем конструирова-
ния достаточно чувствительных спектрометров. С помощью со-
временных промышленных приборов можно определять малое ко-
личество спинов: до 1011. Это число выглядит большим, но оно так
не выглядит в мольном выражении: 10“ спинов соответствуют
10-12 молям. Эти цифры показывают, что ЭПР — очень чувстви-
тельный метод обнаружения свободных радикалов, значит, он
пригоден для обнаружения интермедиатов в химических реакци-
ях, а также слабого магнетизма фосфорссцеитпых триплетных со-
стояний (стр. 114).
Вторая проблема — исчезновение поглощения — Отпадает в
большинстве систем из-за свойств самих радикалов. Это обуслов-
лено тем, что существуют механизмы, возвращающие спины в низ-
шее энергетическое состояние. Эти механизмы называются процес-
сами релаксации. Если они достаточно быстрые, то первоначаль-
ное распределение заселенностей сохраняется, несмотря на то что
образец поглощает излучение, поскольку поглощенная энергия от-
дается спинами (которые таким образом переходят назад в £-со-
стоянце) и рассеивается в виде тепловой энергии. И только когда
мощность микроволнового излучения увеличивается (в этом слу-
чае скорость поглощения становится настолько большой, что про-
цессы релаксации не в состоянии перекрыть поток входящей энер-
гии), разность заселенностей ведет себя так, как описано первона-
чально; такое условие называется насыщением линии.
Наличие процессов релаксации влияет на ширину линий погло-
щения. Это связано с тем, что времена жизни спиновых уровней
при релаксации сокращаются, и поэтому их энергия не имеет точ-
ного значения; этот эффект называется уширением за время жиз-
144
Я
Часть 2. Структура
ни, он обсуждался на стр. 64. Теперь переходы входят в резо-
нанс в некотором интервале напряженности приложенного поля,
и линия уширяется. Такое уширение можно разложить на сумму
двух вкладов: один обусловлен спин-решеточной релаксацией,
которая называется также продольной релаксацией, а другой —
поперечной релаксацией. Скорость спин-пешсточной релаксации
выражается через время спин-решеточной релаксации Т\, а ско-
рость поперечной релаксации — через время поперечной релакса-
ции Т2. Вклад в уширение линии по каждому механизму состав-
ляет примерно й/Т. Для жидких образцов ТаЯ^Ю-8 и Tj/kIO’6 с,
так что первый механизм даст уширение приблизительно в 0,3 мТ
(3 Гс), а второй — около 0,003 мТ (30 мГс).
Продольный процесс Т\ является истинным процессом релак-
сации энергии. Он происходит вследствие того, что движение ра-
дикала вызывает флуктуации локальных магнитных полей, а эти
флуктуации могут стимулировать электроны к переходу в более
низкое энергетическое состояние с переносом избытка энергии к
решетке. Эффективность процесса определяется флуктуациями, и
поэтому изучение ширины линий является источником ценной ин-
формации о движении молекул в жидкостях. Изучение ширины
линий может показать, как быстро вращаются молекулы и проис-
ходит ли вращение преимущественно вокруг одной оси радикала.
Оно может также показать, что некоторые типы молекул враща-
ются путем последовательных небольших поворотов («5°), а дру-
гие при каждом повороте преодолевают почти 1 рад (57°).
Поперечный процесс Тг совсем иной. Его можно понять, при-
няв, что все радикалы в образце находятся в слегка различаю-
щихся магнитных условиях окружения. Это можно пояснить сле-
дующим образом. Хотя анизотропные взаимодействия (т. е. ди-
поль-дипольпые сверхтонкие взаимодействия) усредняются до
нуля, однако, если радикалы вращаются медленно, некоторые из
них остаются в одной ориентации значительное время и резони-
руют при подходящем значении напряженности приложенного
поля; другие радикалы остаются в других ориентациях и резо-1
нируют при иных полях- Отсюда следует, что радикалы образца!
резонируют в некотором интервале поля. Уширение резонансного!
поля проявляется как уширение линии, и это можно выразить че-|
рез эффективное время жизни Та. Изучение Т2 дает информацию!
о молекулярном движении, так как только быстрое вращение бо-1
лее эффективно усредняет анизотропное взаимодействие до пуля.1
19.2. Ядерный магнитный резонанс I
Основные принципы ядерпого магнитного резонанса (ЯМР):|
такие же, как для ЭПР, а главное отличие состоит лишь в томЛ
что в эксперименте контролируется обращение магнитных момен-
19. Резонансные методы
145
тов ядер. Каждое ядро со спином обладает магнитным моментом,
и по аналогии с Предыдущим разделом энергия ядра со спиновым
квантовым числом I (проекция пи') в магнитном поле В равна
(19.2.1)
где gf — ядерный £-фактор (см. табл. 19.2), a цм_ядерный маг-
нетон еп!2?Пр (перемена знака отражает различие в заряде элект-
ронов и ядер). Мы сконцентрируем внимание па ЯМР протонов,
которые имеют £/=2,79270. Отметим, что ядериый магнетон почти
в 2000 раз меньше, чем магнетон Бора, и поэтому ядерные момен-
ты почти в 2000 раз слабее, чем электронный момент. Это можно
понять, если учесть, что спиновые угловые моменты ядер почти
такие же, как для электрона (при I—моменты идентичны), но
массы ядер во много раз больше; таким образом, при классиче-
ском описании спина можно считать, что ядра вращаются во мно-
го раз медленнее, чем электроны. Это подразумевает более низкий
круговой ток и, следовательно, более низкий магнитный момент.
Условие резонанса для перехода протона из нижнего (а) в
верхнее (Р) спиновое состояние имеет вид
hv =-$- —------у £/g№ j =£/UNB, (19.2,2)
и подстановка соответствующих величин показывает, что при поле
в 1,5 Т (15 кГс) резонансная частота равна 60 МГц (60-10® Гц).
При таком поле ЯМР является радиочастотным методом. Сверх-
проводниковые магниты позволяют получить гораздо более силь-
Таблица 19.2
Свойства ядерных сливов
Изотоп (* —радиоактивный) Природное содержание, % Сини 1 Магнитный момент (ядерные магнетоны)3 Частота ЯМР при Г Т, МГц
—1,9130 29,165
Н 99,9844 72 2,79270 42,577
“Н 0,0156 1 0,85738 6,536
ан* 7г 2 9788 45,414
1 % 1,108 7г 0,70216 10,705
99,635 1 0 40357 3,076
’О 0,037 7г — 1 8930 5 772
100 7» 2,6273 40 055
3|р 100 7г 1,1305 17,235
3=8 0,74 0 64274 3 266
33С1 75,4 7г 0 82089 4,172
J7CI 24,6 72 0,68329 4,472
а Ядерцый магнетов имеет величину pN=£’A/2mjJ=5rO5r- Дж/Т;
10—242
14G
Часть 2. Структура
ные поля, и новейшие спектрометры работают в полях до 7 Т
(70 кГс), что соответствует частоте 300 МГц, Преимущество рабо-
ты в сильных полях состоит в том, что интенсивности увеличива-
ются, Рявность заселенностей при поле В и температуре Т дается
прямой модификацией уравнения (19.1.6):
(Мх — -^р)/(^а+^₽) ~2~ (19,2.3)
Для поля в 1,5 Т при комнатной температуре она равна только
2,6-10's, так что избыток а-спина над fj-спином очень невелик.
Когда Приложенное поле усиливается до 7 Т, разность заселенно-
стей пропорционально больше, и поэтому интенсивность сигнала
соответственно больше.
Метод. Экспериментальное оборудование во многом такое же, как
для ЭПР, при этом принципиальное отличие состоит в том, что
микроволновые источник и детектор заменяются па радиочастот-
ные. Кроме того, магнит работает в более высоких полях, и, по-
скольку спектр содержит компоненты, разделенные очень неболь-
шими энергиями (как описывается ниже), магнитное поле должно
быть исключительно хорошо контролируемым и однородным по
всей массе исследуемого образца. Одним из путей обеспечения од-
нородности поля в образце является такое быстрое вращение об-
разца, чтобы небольшие неоднородности усреднялись. Экспери-
мент проводится или путем развертки магнитного поля в неболь-
шом интервале с контролем поглощения радиочастотного источни-
ка на постоянной частоте, или путем развертки частоты при по-
стоянном приложенном поле.
Типичный экспериментальный результат (для этанола) пока-
/ заи на рис, 19.6, Требуется объяснить две главные особенности:
почему спектр расщеплен па несколько групп линий и почему эти
группы состоят из нескольких близко расположенных узких ли-
ний.
Рис. 19.6. Спектр ЯМР этанола. Жирным шрифтом выделены протоны, обуслов-
ливающие появление резонансных линий.
19. Резонансные методы
147
Химический сдвиг. Как и в случае ЭПР, резонирующий магнитный
момент взаимодействует с локальным полем, которое может отли-
чаться от приложенного поля, поскольку токи последнего могут
возбуждать электроны молекулы. В ЯМР локальное поле принято
записывать в форме
5ЛОк —(1—о)
где о называется постоянной экранирования. Эта форма связана
с предположением о том, что возбуждающие токи пропорциональ-
ны напряженности внешнего поля, так что общее поле, действую-
щее на протоны, имеет дополнительный вклад — аВ (такой же до-
вод приводился выше для g-фактОра). Дополнительное поле о В
называется химическим сдвигом группы протонов. Способность
приложенного поля индуцировать токи зависит От электронной
структуры молекулы вблизи интересующих лас протонов. Поэто-
му протоны химически отличающихся групп имеют различные по-
стоянные экранирования, и условие резонанса
Av=O*n(1— °) В
выполняется при разных значениях В для протонов в разном хи-
мическом окружении. Это позволяет понять общую структуру
спектра этанола, приведенного на рис. 19.6. Метильные протоны
образуют одну группу п входят в резонанс в соответствии с их
химическим сдвигом. Два метиленовых протона находятся в дру-
гой части молекулы; следовательно, они имеют Другой химиче-
ский сдвиг и входят в резонанс при ином значении напряженности
магнитного поля. Наконец, протон гидроксильной группы находит-
ся в другом магнитном окружении, характеризующемся еще од-
ним химическим сдвигом; он входит в резонанс при третьем зна-
чении напряженности приложенного магнитного поля. Отметим,
что различать, какая группа линий соответствует какой группе
Протонов, можно по их относительной интенсивности (площади под
кривыми поглощения). Групповые интенсивности находятся в От-
ношении 3:2: 1, так как имеются три метильных протона, два ме-
тиленовых протона и один гидроксильный протон.
Величина химического сдвига обычно выражается в миллион-
ных долях (м. д.), поскольку постоянная экранирования имеет
порядок величины 10-Е—10-s (т. е. внешнее поле в 1 Т возбуж-
дает дополнительное локальное поле примерно в 1 мкТ). В случае
ацетальдегида (Этаналя) метильный и альдегидный протоны на-
ходятся друг от Друга на расстоянии 6,9 м. д.; это указывает на
то, что в поле 1,5 Т на них действуют локальные поля, отличаю-
щиеся на 1,0-10-® Т (0,10 Гс). Другими словами, в опыте на ча-
стоте 60 МГц их резонансные частоты отличаются приблизительно
на 410 Гц.
10*
148
Часть 2. Структура
I
2 0
_____________________>
CHjCH.-
CHjC'H.’ -
Кол ьио —CH; —
CH3CN
-CH.-NH;
(>CHj
<? CH,CH,
-C^CH
X-CHj, Х = галогеп
X - CH; -
X -CH<
CH,NO.. -СН,КОг, > CH-NO,
Сопряжен» — CH =CH-
Несопряженный-СН=СН-
X^Q-H.X —NO;,OH. raa-,NH
RCHO
RCO.H
'I______________!______________1______________I______________1_
12 10 8 6 4
i
Рис. 19.7. Интервал типичных химических сдвигов.
Можно было бы создать абсолютную шкалу химических сдви-
гов, приняв за нуль резонанс свободного протона, однако это не
имело бы большого практического значения. Вместо этого хими-
ческий сдвиг обычно измеряют относительно Протонов некоторого
стандартного соединения, и часто в качестве стандарта выбирают
тетраметилсилап 31(СНз)4 (ТМС), который содержит эквивалент-
ные протоны и растворяется во многих системах, не взаимодейст-
вуя с ними. Сдвиги относительно него берутся в 6-шкалс, в кото-
рой ТМС произвольно приписано значение 6 = 0. Альтернативная
т-шкала сдвинута относительно 6-шкалы на 10 м. дт; в ней боль-
шинство значений положительно. Характеристические химические
сдвиги для ряда обычно встречающихся групп приведены на
рис. 19.7.
Тонкая структура, Расщепление групп линий на отдельные линии
(рис. 19.6) называется тонкой структурой спектра. Точно так же
как сверхтонкая структура в ЭПР может быть объяснена взаимо
действием электронного и ядерного спинов, тонкая структура :
ЯМР приписывается магнитным взаимодействиям междуг ядрам:
в молекуле.
Сначала выясним, могут ли два протона участвовать в прямо:
диполь-дипольном взаимодействии. В точке на расстояния 7? о
19. Резонансные методы
149
Молекула
Протон В
Путь моле- .
кулярного
вращения
Рис. 19.8. Углы, входящие в
описание диполь-дипольного
взаимодействия, и влияние мо-
лекулярного вращения.
Протона С проекцией спина mi существует магнитное поле, опре-
деляемое выражением
Вяд (Р-о/4л) (1 /Я3) (1 — 3 cos2 0) mt. (19.2.4)
Параметры Й и 0 определены на рис. 19.8. Напряженность этого
поля имеет порядок 0,1 мТ при /? = 0,3 нм. т. е. оно по порядку ве-
личины соответствует расщеплению на рис. 19.9. В жидкостях
угол 0 быстро изменяется, принимая все возможные значения; сле-
довательно, в отличие от спектров твердых веществ, где прямое
дицоль-дипольное взаимодействие вносит важный вклад (и может
быть использовано для определения расстояний между прото-
нами), в жидких образцах эго взаимодействие усредняется до
нуля.
Другой механизм взаимодействия между магнитными ядрами
связан с поляризационным взаимодействием (см. выше). Магнит-
ное поле одного ядра воздействует на распределение электронов
Рис. 19.9. Спектр ЯМР твердого состояния моногидрата азотной кислоту
H3O+NO; [Richards R. £, Smith Л A. S., Trans. Faraday Soc., 47. 1261 (1951)].
150
Часть 2. Структура
связи, и этот эффект может передаваться через соседние связи к
другим ядрам в молекуле. Подробный расчет подтверждает, что
этот механизм может объяснить расщепление, наблюдаемое для
жидких образцов.
Теперь мы можем несколько продвинуться в объяснении внеш-
него вида спектра. Основой для рассмотрения будет спектр эта-
нола.
Сначала рассмотрим метильные протоны. Очевидно, что для
эквивалентных протонов не будет наблюдаться никакой тонкой
структуры (они на самом деле взаимодействуют, но правила от-
бора для спектральных переходов запрещают любой переход, ко-
торый дал бы указание на энергию взаимодействия). Поэтому изо-
лированная метильная группа, например в СН3С1, даст единствен-
ную нерасщепленпую линию. Однако в этаноле рядом с метильной
группой находятся два метиленовых протона. Линия метильной
группы вначале расщепляется на две в соответствии с двумя ори-
ентациями одного из метиленовых протонов, а затем каждая ли-
ния снова расщепляется в результате взаимодействия со вторым
протоном. Таким образом, резонанс метильной группы расщеп-
ляется на три линии с соотношением интенсивностей 1:2:1
(рис. 19,10,«), Обычно расщепление обозначают буквой J.
Теперь рассмотрим резонансные линии метиленовых протонов.
Если бы они были изолированы, то они бы резонировали в одном
месте. Тонкая структура обусловлена их спин-спнновым взаимо-
6
рис. 19.10. Источник тонкой структуры резонанса С Из (а) и резонанса СН2 (б)
в этаноле. Темные н светлые кружки соответствуют протонам с противополож-
ным сил ном. Жирным шрифтом выделены резонирующие протоны.
IS. Резонансные методы
151
действием с тремя соседними метильными протонами. Из-за нали-
чия трех эквивалентных протонов получаются четыре линии с со-
отношением интенсивностей 1:3:3: 1 (рис. 19.10,6), и Это объяс-
няет наблюдаемую структуру. В общем N эквивалентных протонов
расщепляют соседнюю группу на Л7+1 линий с распределением
интенсивности в соответствии с треугольником Паскаля (см. вы-
ше).
Гидроксильный протон в этаноле расщепляет свои соседние
группы на дублеты, а сам расщепляется метиленовыми протонами
на 1 : 2: 1-триплет, и каждая линия очень слабо расщепляется на
1 : 3 : 3 : 1-квартет удаленными метильными протонами. Во многих
случаях расщепление, обусловленное гидроксильным протоном,
нельзя обнаружить; причину Этого мы обсудим в следующем раз-
деле.
Спектры ЯМР часто бывают гораздо более сложными, чем сле-
дует из приведенного простого анализа. Мы рассмотрели предель-
ный случай, когда химические сдвиги значительно больше спин-
спиповых взаимодействий; поэтому довольно просто сделать вы-
вод о том, какие протоны эквивалентны, а какие нет. Когда хими-
ческий сдвиг сравним с константами взаимодействия, возникает
намного более сложная ситуация, и для расчета всех энергетиче-
ских уровней и вероятностей переходов становится необходимым
применение ЭВМ. При этом очевидно еще одно преимущество ра-
боты в очень высоких полях. Химический сдвиг равен аВ и воз-
растает с увеличением напряженности поля; в то время спин-спи-
новое взаимодействие не зависит от приложенного поля. Таким
образом, когда поле увеличивается, расстояние между группами
сигналов с разными химическими сдвигами возрастает, а спин-спи-
новые взаимодействия остаются теми же самыми. Следовательно,
сложный спектр можно значительно упростить, работая с высоки-
ми полями; группы линий разрешаются, и путаница в энергетиче-
ских уровнях и вероятностях переходов уменьшается.
Пример (вопросы 10 и 11). На рис. 19.11 показан экспериментальный спектр
ЯМР. снятый на частоте 60 МГц.. Интерпретируйте его. Какие изменения про-
изойдут, если спектр будет снят на 30(1 МГц?
Метод. Ищем группы е характеристическими химическими сдвигами (рис. 19.7).
Объясняем топкую структуру с помощью тех же аргументов, как и для этано-
ла. Определяем значения йи/и идентифицируем соединения.
Ответ. Резонанс при д —3,4 соответствует протонам группы СН2 в простом
эфире; резонанс при 6=1,2 соответствует протонам группы СН3 в СН3СН2. Спин-
спиновое расщепление резонанса СН2 (квартет) характеризует соседство с груп-
пой: СН3, а спнн-спиновое расщепление резонанса СНз—наличие рядом распо-
ложенной группы СН2. Константа расщепления J=6,0 Гц (одинакова для каж-
дой группы). Соединение имеет формулу CHjCHsOCHsCIIs. На частоте 300 МГц
группы линий отстояли бы в пять раз дальше друг от друга, но расщепление
внутри их осталось бы таким же.
Комментарий. В бо.дсс низком поле химические сдвиги могут быть сравнимы с
расщеплением, дающим тонкую структуру. Тогда внешний вид спектра будет
более сложным, и для его анализа, как правило, будет необходима ЭВМ.
152
Часть 2. Структура
Формы линий и скорость процессов. Точно так же как и в ЭПР,
движение ядер может влиять на форму и ширину линий в ЯМР,
и точно так же можно рассмотреть времена спин-решеточпой и
сипн-спиновой релаксации. Важное отличие состоит в том, что
вследствие малости ядсрных магнитных моментов времена релак-
сации нелики, часто порядка нескольких секунд.
ЯМР особенно пригоден для изучения некоторых типов быст-
рых молекулярных изменений, включая взаимопревращение изо-
мерных форм и обмен прогонов с другими молекулами. Эти про-
цессы можно обнаружить по внешнему .виду спектра. Рассмотрим
два типичных примера.
Рассмотрим молекулу, которая может переходить из одной
конформации в другую и обратно, например инверсию кресло —
ванна замешенного циклогексана (рис, 19,12). В одной конформа-
ции метильные протоны находятся в аксиальном, а в другой — в
экваториальном положении. В этих Двух случаях химические
сдвиги метильной группы различаются, и, если инверсия проис-
ходит медленно, в спектре наблюдаются два набора линий: один
от молекул с аксиальным метилом, а другой от молекул с эква-
ториальным метилом. Если инверсия происходит быстро, то в
спектре наблюдается лишь одна линия, соответствующая среднему
значению двух химических сдвигов.
Но что значит «быстро» и «медленно»? Чтобы Ответить на Этот
вопрос, можно вернуться к эффекту уширения за время жизни
(стр. 64). Если время жизни молекулы в одной конформации со-
19. Резонансные методы
153
Рис. 19.12. Инверсия замещенного циклогексана.
ставляет п, то ее энергетические уровни можно определить толь-
ко с точностью до SEzzhlx;. Когда эта неопределенность энергии
сравнима с расстоянием между двумя линиями с разными хими-
ческими сдвигами, они сливаются в одну широкую линию. Следо-
вательно, если расстояние по частоте между двумя типами прото-
нов равно 6v, то спектр уширяется в одну линию при
оЕ ~^г> ftv, —ftva =/zfiv,
т. е. при hlxj^h&v, где Vj и v2— две резонансные частоты. По-
скольку /г=й/2л, этот критерий можно переписать в следующем
виде:
слияние структуры, если т,«^1/2л(К. (19,2.5)
Например, если химический сдвиг между двумя группами равен
100 Гц, то две линии сливаются в одну при времени взаимопре-
вращения меньшем, чем 2 мс.
Пример (вопрос 12). Группа NO в N,N-ди метили итрозамине (CHsRN—NO враща-
ется, в результате чего происходит чис-гранс-взаимопрсвращение метильных
групп. На частоте 60 МГц резонансы двух метильных групп находятся на рас-
стоянии 39 Гц. При какой скорости таутомеризации произойдет слияние резо-
нансов в одну линию при частоте спектрометра 60 МГц?
Метод. Используем уравнение (19.2.5).
Ответ. ту<:1/2л (39 Гц)—4,1 мс.
Комментарий. Если спектрометр работает на частоте 300 МГц, то слияние про-
изойдет при tj^0,82 мс. Такой более быстрый переход можно стимулировать
нагреванием образца; необходимое для этого изменение температуры можно
использовать для определения энергии активации таутомеризации (см. задачу
19.34).
Аналогично объясняется исчезновение структуры спектра в
растворителях, способных обменивать протоны с исследуемым об-
разцом. Например, протоны гидроксила спиртов могут обмени-
ваться с протонами воды. Когда происходит такой химический
обмен, молекула ROH с а-спиновым протоном (запишем се в виде
ROH(a)) быстро переходит в ROH(S), а затем в ROH(a), поскольку
протоны, отдаваемые молекулами растворителя во время обмена,
имеют неупорядоченную ориентацию епцнов. Следовательно, вме-
сто спектра, состоящего из вкладов молекул ROH(a> и ROH(^
(т. е. спектра, в котором линии расщеплены на дублеты в резуль-
тате спип-спинового взаимодействия с гидроксильным протоном),
154
Часть 2. Структура
будет наблюдаться одна нерасщепленная линия в среднем поло-
жении. Этот эффект наблюдается в том случае, когда скорость
химического обмена 1/тЪбм так велика, что неопределенность энер-
гии больше дублетного расщепления; поэтому слияние тонкой
структуры спектра будет ожидаться, когда
l/2nSv,
где 6v — расстояние по частоте в дублетах, которое изменяется
из-за обмена протонов. Поскольку такое расщепление часто очень
мало (около 1 Гц), для того чтобы в спектре исчезло расщепле-
ние, протон должен быть связан с одной и той же молекулой в те-
чение времени не более чем (1/2л Гц)«0,1 с. В воде ско-
рость химического обмена намного выше, и поэтому для спиртов
в водном растворе расщепление на гидроксильных протоках не
наблюдается. Когда применяют очень сухой спирт, скорость обме-
на гораздо ниже и расщепление можно обнаружить.
Важное значение эффектов химического обмена состоит в том,
что их можно использовать для изучения быстрых химических
процессов и выяснения зависимости их скоростей от условий ре-
акции (например, температуры и растворителя).
19.3. Мессбауэровская спектроскопия
Спектроскопия ядерного магнитного резонанса основана на
поглощении фотонов очень низкой энергии — их частоты имеют
порядок 60 МГц (а длина волны 5 м) и являются одними из са-
мых низких из используемых в спектроскопии. В противополож-
ность этому мессбауэровская спектроскопия представляет собой
другой предельный случай. В ней применяются фотоны y-лучей с
частотой порядка 1019 Гц и длиной волны около 100 пм (10-10 м).
Но несмотря на такое различие в энергиях фотонов в этих двух
видах спектроскопии, мессбауэровские линии так четко разреше-
ны, что для них наблюдается расщепление того же порядка вели-
чины, как и расщепление в ЯМР-
Испускание и поглощение у-лучей. Основой мессбауэровской
спектроскопии является резонансное поглощение фотонов у-лучей
ядрами. Обычно в качестве первичного источника изотопа железа
57Fe используется образец, содержащий 57Со. Изотоп 57Со распа-
дается медленно (период полураспада 270 сут) и образует S7Fe в
возбужденном ядерном состоянии; обозначим его S7Fe*. Этот изо-
топ железа распадается Очень быстро (период полураспада около
2Л0“7 с) и испускает у-лучи (рис. 19.13). Эти фотоны использу-
ются в эксперименте. Когда в другой части спектрометра фотон
сталкивается с ядром 57Fe в основном состоянии, он поглощается,
если его энергия соответствует разности энергий между 57Fe;s и
19. Резонансные методы
155
s7Co
\ 270 сут
\ 57 Fe*
Z-IO'7 с Резонансное
поглощение
у-лучи
, (v - 3,54018 Гц)
1
57Fe
Рис. 19.13. Переходы, приводящие к эффекту Мессбауэра на 57Fe.
57Fe (это стадия резонансного поглощения); однако он не будет
поглощаться, если указанные энергии не подходят друг к другу.
Причину, по которой этот резонанс очень четко выражен, мож-
но объяснить рядом факторов. Во-первых, S7Fe живет достаточно
долго, чтобы его энергия была хорошо определена. Время жизни
около 2-Ю-7 с соответствует неопределенности частоты примерно
2 МГц. Фотоны у-лучей, испускаемых 57Fe* (и поглощаемых E7Fe),
имеют частоту ~3,5-101й Гц (что соответствует энергии 14,4 кэВ);
следовательно, ширина линии составляет только ~ 10-12 от часто-
ты фотона. Однако дело этим не кончается, поскольку фотон с та-
кой высокой энергией обладает также значительным моментом
(определяемым как hv]c). Когда 57Fe* распадается и испускает
фотон, в результате отдачи получается девозбужденное ядро,
имеющее скорость около 102 м/с. Эта отдача оказывает вредное
влияние на чистоту частоты фотона, так как следует принять во
внимание эффект Доплера. Если источник фотонов движется со
скоростью v относительно наблюдателя, то его частота v сме-
щается на величину w/c (стр, 62). Даже при vtxl см/с фотоны
из 7'7Fe* будут испытывать сдвиг по частоте примерно на 100 МГц,
а этого достаточно, чтобы нарушить естественную ширину линии.
Указанный эффект отдачи преодолевается с помощью одной Су-
щественной особенности мессбауэровского опыта. Если ядра
57Fe* и 57Fe жестко связаны в кристаллической решетке, то кине-
тическая энергия поглощается всей массой образца. Следователь-
но, скорость отдачи будет чрезвычайно небольшой, доплеровский
сдвиг и уширение линии — незначительными. Эффект Мессбауэра
заключается в таком свободном от отдачи испускании у-лучей и их
резонансном поглощении.
156
Часть 2, Структура
Образец
движение
подставки
Рис. 19.14. Гамма-резонансный спектрометр.
Гамма-резонансный спектрометр. Практический аспект метода со-
стоит в следующем. Источником -у-лучей является тонкая фольга
из 57Со (рис. 19.14). Этот изотоп удобен тем, что он имеет под-
ходящее продолжительное время жизни и может быть постоянным
источником 57Fe* на несколько часов или суток, в течение которых
проводится эксперимент. Изучаемый образец помещается в подоб-
ную фольгу, которая размещается перед счетчиком. В приборах
можно использовать многие изотопы, однако чаще всего применя-
ются S7Fe и 1,9Sn. Остановимся на них, особенно на первом.
Резонансное поглощение происходит в том случае, когда уров-
ни ядерной энергии источника и поглотителя соответствуют друг
другу. Энергию испускаемых фотонов можно изменять, используя
эффект Доплера (теперь он играет полезную роль, а нс мешает).
Это достигается установкой источника на движущуюся подставку:
если она движется равномерно с некоторой контролируемой ско-
ростью, то доплеровский сдвиг фотонов можно контролировать с
высокой точностью. Дтя того чтобы подогнать частоту фотонов
к энергетическим уровням поглощающего вещества, нередко бы-
вает достаточно скорости в несколько мм/с. Можно использовать
винтовой двигатель, но очень часто фольгу двигают электромаг-
нитом, как диафрагму громкоговорителя.
Пример (вопрос 13). В мессбауэровском опыте с S7Fe* было найдено, что для
получения резонансного поглощения у-лучей нужно двигать источник по нап-
равлению к образцу со скоростью 2,2 мм/с, Каков сдвиг энергии (в МГц) меж-
ду образцом и источником?
Метод. Величина доплеровского сдвига равна vv/c. у-Лучи 57Fe* имеют энергию
14,4 кэВ; переведем ее в частоту (1 эВ = 8065 см-1).
Ответ. у — (14,4 кэВ)X(8065 см-'/эВ)Х(2,998-1010 см/с). Следовательно, Sv —
= (14,4 кэВ) Х(8065 см-1/эВ)Х(2,2 мм/с) — 25,5-106 с~‘, или 25,5 МГц.
Комментарий. Эта частота соответствует 8,52-10_< см-1. Примечательная осо-
бенность мессбауэровского эксперимента состоит в том, что подобные небольшие
сдвиги измеряются с использованием фотонов такой огромной энергии.
Поглощение происходит при движении источника с некоторой
скоростью; однако очень часто поглощение наблюдается при не-
скольких относительных скоростях. Первичным результатом мес-
сбауэровской спектроскопии является график зависимости числа
19. Резонансные методы
157
сосчитанных фотонов от скорости источника. Типичный спектр
приведен на рис. 19.15; положительные скорости соответствуют
движению источника по направлению к поглотителю, отрицатель-
ные скорости — от поглотителя. Положительные скорости указы-
вают, что для того, чтобы поглощающее вещество могло резониро-
вать, частоту фотона нужно увеличить; следовательно, расстояние
между уровнями 57Fe*—г-?ре больше в поглотителе (FeSO-t), чем
в источнике.
Информация, получаемая из спектров. Мы должны понять обе
особенности спектров. Во-первых, почему резонанс сдвинут отно-
сительно частоты источника? Во-вторых, почему спектр часто рас-
щепляется на несколько линий?
Изменение положения резонанса называется изомерным сдви-
гом. Радиусы ядер в возбужденном и основном состояниях немно-
го, но заметно различаются (при возбуждении в 57Fe* ядро 57Fe
сжимается примерно па 0,2%), и поэтому их электростатическое
взаимодействие с окружающими электронами при возбуждении
изменяется. Этот эффект значителен только для электронов s-ор-
биталей (которые могут близко приближаться к ядру); следова-
тельно, энергия ядер изменяется на некоторую величину, пропор-
циональную изменению радиуса ядра и s-электронной плотности
около ядра. Если первое записать как 6/?, а второе —как ф|(0),
то получим
6Е ~ (0) (У?.
Хотя 6/? для источника и поглотителя одинакова, s-электронная
плотность может быть разной (поскольку ядра 57Fe находятся в
158
Часть 2. Структура.
разном химическом окружении), и изомерный сдвиг определяется
формулой
~ {[Зрз (0)1поглотитель [Я-'s (0) Lie точи те!
Измерением изомерного сдвига можно оценить степень участия
s-орбиталей в связывании.
Например, в мессбауэровских спектрах соединений n9Sn на-
блюдается изомерный сдвиг примерно в 20 МГц относительно
119Sn в виде белого олова в случае ковалентных соединений Sn(11)
(таких, как SnCl2) и примерно в —50 МГц для ковалентных со-
единений Sn(lV) (таких, как SnCl4). Наличие в Sn(11) двух лиш-
них s-электронов приводит к более положительному сдвигу, чем в
случае Sn(lV). Очевидно, что этот метод дает прямой путь опре-
деления валентности элемента в соединении.
Структура мессбауэровского спектра возникает вследствие двух
основных взаимодействий. В ряде случаев (в частности для S7Fe)
ядерпые спины невозбужденного и возбужденного изотопов разли-
1 3
чаются. Например, /(E7Fe)=-g-, но /(S7Fe*) =Такое изменение
спина сопровождается изменением распределения заряда в ядре;
в то время как E7Fe имеет сферическое распределение заряда, за-
ряд в 57Fe* концентрируется на полюсах. При октаэдрическом
окружении, как в Fe(CN)^ , энергия ядра не зависит От его ори-
ентации (рис. 19.16,а), но если окружение имеет осевую симмет-
рию, как в Fe(CN)5NO2“, то энергия зависит от ориентации ядра
(рис, 19.16,6). Это объясняет электрическое квадрупольное рас-
щепление мессбауэровского спектра некоторых соединений. Его
анализ позволяет представить картину симметрии распределения
электронов вблизи ядер.
Ядра со спицом имеют магнитные моменты. Если они находят-
ся в магнитном поле, то их энергия зависит от их ориентации (от
Рис. 19.16. Квадрупольное ядро в регулярном окружении (а) и в искаженном
окружении (б).
]9. Резонансные методы
159
Ж
Нет магнит-
лого поля
Нет квадру-
польного
расщепления
Рис. 19.17, Энергетические
уровни и переходы для
мессбауэровского экспери-
мента с S7Fe с учетом маг-
нитного и квадрулольного
взаимодействия.
величины ш/) точно так же,
как в ЯМР. В случае B7Fe
и 57Fe* ядерные спины раз-
личаются, и поэтому ситуа-
ция более сложная. На
рис. 19.17 показаны энер-
гетические уровни и разре-
шенные ядерные переходы
для этого случая; мы ви-
дим, что ожидается шесть
Поле
Нет квадру-
польного
расщепления
Лоле -
Квадруполь-
нов расщеп-
ление
энергий перехода. Это объ-
ясняет приведенный справа
спектр 57Fc ферромагнитно-
го железа (в данном случае
образец имеет собственное
магнитное поле). Такие эф-
фекты очень полезны для
изучения магнитных мате-
риалов.
Важность мессбауэровского исследования S7Fe состоит не толь-
ко в том, что этот изотоп удобен для работы. Железо встречается
во множестве биологически и технически важных соединений (та-
ких, как гемоглобин и ржавчина), и мессбауэровская спектроско-
пия была использована для их анализа и выяснения, например,
механизма коррозии.
Литература
McLauchlan Л, A., Magnetic resonance. Ci аге л don Press, Oxford, 1972.
Whiffeu D. H., Spectroscopy, Longman, London, 1972.
Ayscough P. B., Electron spin resonance in chemistry, Methuen, London, 1967,
Верти Дж., Болтон Дж. Теория и практические приложения метода ЭПР, Пер.
с англ. — М.; Мир, 1975,
Керрпнгтон А., Мак-Лечлан Э, Магнитный резонанс, его применение в химии.
Пер. с ацгл. — М.: Мир, 1970.
Rieger Р. Н., Electron spin resonance In Techniques of chemistry (Weissberger A.
and Rossiter B. W., eds.). Vol. 11IA, 499, Wiley-Interscience, New York, 1972.
Lynden-Bell R. M., Harrus R. K., Nuclear magnetic resonance spectroscopy, Nel-
son. London, 1969,
Робертс Дж. Ядерный магнитный резонанс. Применение в органической химии
Пер. с англ. — М.: ИЛ, 1961,
160
Часть 2. Структура
Muller N.j Nuclear magnetic resonance, in Techniques of chemistry (Weissber-
ger A. and Rossiter B. W., eds.), Vol. IIIA, 599, Wiley-lnterscience, New York,
1972.
Попл Дж., Шнейдер В., Бернстсйн Г. Спектры ядерного магнитного резонанса
высокого разрешения. Пер. с англ. — М.: ИЛ, 1962.
Абрагам А. Ядерный магнетизм. Пер. с англ. — М.: ИЛ, 1963.
fferber # Н., Hazony К, Experimental aspects of Mossbauer spectroscopy, in
Techniques of chemistry (Weissberger A. and Rossiter B. W., eds.), Vol. HID,
215, Wiley-lniersciencc, New York, 1972.
Hill H. A. 0.. Day P. (eds.), Physical methods in advanced inorganic chemistry.
Interscience, London, 1968.
Задачи
19.1. В некоторых промышленных ЭПР-спектрометрах используется микроволно-
вое 8 мм-излучение. Какое магнитное поле необходимо в этом случае, чтобы вве-
сти электронный спин в резонанс?
19.2. Промышленный ЭПР-спектрометр может обнаружить резонанс образца, со-
держащего лишь 1010 спинов в 1 см3. Какая мольная концентрация соответствуй
ет этому пределу обнаружения? Предполагается известным, что образец содер-З
жит 2,5'1014 спинов. Сколько этих спинов при комнатной температуре находят<
ся в а-состоянии и сколько в p-состояниц? Л
19.3. Рассчитайте относительную разность заселенностей
Ч-А^) Для электронных спинов в магнитном иоле 0,3 Т (3 кГс) при а) 4 К
б) ЗОЮ К.
19.4. Центр спектра ЭПР атомарного водорода находится при 329,12 мТ, ссди
спектрометр имеет рабочую частоту 9,2231 ГГц (1 ГГц^1{Р Гц). Какова ве-
личина g-фактора для электрона в атоме водорода?
19.5. Для апион-радикала бензола g-фактор равен 2,0025. В каком поле вы ис-
кали бы резонанс на спектрометре, работающем при а) 9,302 ГГц и б) 33,67 ГГц?
19,6. Среднее значение g-фактора для стабильного радикала С1Ог равно 2,0102.
На какую величину дополнительного локального ноля указывает эго значение,
если приложено поле: а) 3400 Гс и б) 12300 Гс?
19.7. Треугольная молекула NO2 имеет один песпаренный электрон и может су-
ществовать в матрице или может быть синтезирована внутри кристалла при ра-
диационном распаде нитрит-ионов. В том случае, когда радикал жестко удер-
живается в определенной ориентации, магнитное иоле может быть приложено
в выбранных направлениях. Когда оно параллельно линии О—О, центр спектра
находится при 333,64 мТ, при рабочей частоте спектрометра 9,302 ГГц, но когда
поле направлено вдоль биссектрисы угла ONO, резонанс сдвигается до 331,94 мТ.
Какова величина g-факгора в обоих направлениях?
19.8. Спектр ЭПР атомарного водорода состоит из двух линий. В спектрометре,
работающем на 9.302 ГГц, одна линия появляется при 357,3 мТ, а другая — при
306,6 мТ. Какова константа сверхтонкого расщепления для этого атома?
19.9. В спектрах ЭПР радикала, содержащего два эквивалентных протона, на-
блюдагогся три линии с соотношением интенсивностей 1:2:1. Линии находятся
при 330,2, 332,5 и 334,8 мТ. Какова константа сверхтонкого расщепления для
каждого протона?
19.10. Для получения спектра, описанного в предыдущей задаче, был применен
спектрометр с клистроном, генерирующим микроволны с частотой 9,319 ГГц.
Каковы g-фактор радикала и константы расщепления в МГц? (Вы должны вы-
вести полезное соотношение: 0,1 мТ»2,8 МГц.)
19,11. Радикал, содержащий два неэквивалентных протона с константами рас-
щепления 2,0 и 2,6 мТ, дает спектр с центром при 332,5 мТ. В каком по.ле "ле-
жат линки сверхтонкой структуры и какова их относительная интенсивность?
19. Резонансные методы
161
19.12. Радикал, о котором Шла речь в предыдущей задаче, изомеризуется, и при
этом два протона обмениваются местами. При низких температурах спектр име-
ет вид, описанный выше. При комнатной температуре центральная пара линий
сливается в одну линию двойной интенсивности, и тогда получается спектр, опи-
санный в задаче 19.9. Какова скорость изомеризации, при которой спектр пере-
ходят из одного вида а другой?
19.13. Предскажите соотношение интенсивностей линий сверхтонкой структуры в
спектре радикалов ‘СИ3 и .CD3 (для 2Н 7=1).
19.14. Константа сверхтонкого расщепления для метильного радикала равна
0,23 мТ. Используя информацию тпбл. 19.2. предскажи]е расстояние между лц-
нин.ии сверхтонкой структуры в спектре радикалов ‘CDs. Какова общая ширина
спектра в каждом случае?
19.15. Предскажите внешний вид спектра ЭПР этильного радикала СН3СН2‘,
использовав о(СН2) =0,224 мТ и й(СНз) = 0,268 мТ для соответствующих про-
тонов. Какие измепения произойдут в спектре, сели метиленовые Протоны заме-
нить на дейтроны?
19.16. Анион-раднкал пора-динитробеизола можно получить восстановлением
динитробензола. Этот аннои-радикал имеет два эквивалентных ядра азота (/=
= 1) и четыре эквивалентных протона. Предскажите вид спектра ЭПР, исполь-
зовав a(N) =0,148 мТ и а(Н)=0,112 мТ.
19.17. Когда электрон занимает 2з-орбиталь азота, его сверхтонкое взаимодей-
ствие с ядром равно 55,2 мТ (это магнитное поле генерируется ядром и усред-
няе1ся по области s-орбнтали. где чувствуется его наличие). В то же время в
спектре NОд наблюдается изотропное контактное сверхтонкое взаимодействие,
равное 5,7 иТ. Какую часть времени неспаренный электрон КО2, Как можно
считать, занимает 2з-орбиталь азота?
19.18. Ядерное поле, воздействующее на электрон, занимающий 2р-ор5италь ато-
ма азота, имеет величину 3,4 мТ. В NO2 анизотропная часть сверхтонкого рас-
щепления имеет величину 1,3 мТ. Какую часть времени кеспаренныЙ электрон
проводит па 2р-орбитали азота в ЬЮ2?
19.19. Информаций двух предыдущих задач достаточно, чтобы построить карту
распределения неспаренного электрона по молекуле. Какова общая вероятность
того, что он будет найден в а каждом атоме кислорода? Какова гибридизация
центрального атома азота? Следует ли из этой гибридизации, что NO2 является
изогнутой молекулой?
19.20. ' При облучении неорганического твердого вещества у-лучами образуются
два разных радикала. Их ^-факторы равны 2,0102 и 2,0060. Каково расстояние
между их резонансными линиями (в мТ) При исследовании ЭПР-спектрометром,
работающим на частотах: а) 9,218 ГГц и б) 34,22 ГГц? Первый радикал содер-
жал три линии с расщеплением 0,60 мТ, а второй был 1 : 3; 3 : l-Квартетом с
расщеплением 0,50 мТ. Нарисуйте эскизы спектров при двух рабочих часютах.
19.21. Ниже приведены константь] сверхтонкого расщепления (в мТ), наблюдае-
мые для различных анион-радикалов. Используя значение для бензола и сим-
метрию, составьте карту вероятности нахождения несцаренного электрона на
2рл-орбитали Каждого атома углерода.
0,172
NO2
19.22. Радикал пиридина (I) имеет указанные константы сверхтонкого расщеп-
ления (в мТ) на протонах, для карбоксильного производного (II) эти констан-
11—242
162
Часть 2. Структура
ты также известны [Zemel Н., Fessenden R. W., J. Phys. Chem. 79, 1419 (1975)]:
Рассчитайте спиновую плотность пе спа ре иного электрона по кольцу.
19.23. При изучении движения ннтрокспдных радикалов в твердой клатратной
клетке при низких температурах было найдено, что сверхтонкое взаимодействие
электрона с ядром азота зависит от ориентации радикала в приложенном поле
(McConnell A. A., MacNicol D. D., Porte A. L., J. Chem. Soc.F 1971, А, 3516).
Когда поле было перпендикулярно связи, константа нзаимодействия изменялась
между 113,1 и 11,2 МГц, а когда оно было параллельно связи, константа взаимо-
действия была равна 14,1 МГц. При повышении температуры до 115 К на
внешний вид спектра начинало влиять движение вокруг параллельной оси. На-
сколько быстро вращается молекула в клатратной клетке при этой температуре?
(Эта проблема в дальнейшем рассматривается в задаче 27.24.)
19.24. Используя таблицу свойств ядер (табл. 19.2), предскажите магнитные
поля, при которых ядра 'Н, 2Н, '3С, |,1Nf ,!iF и 3|Р входят-в резонанс с радио-
частотным нолем частотой: а) 60 МГц и б) 300 Л4Гц.
19.25. Рассчитайте относительную разность заселенностей 6N/N для протона в
поле 0,3, 1,5 и 7,0 Т при а) 4 К и 6} 300 К_
19.26. Какое магнитное ноле необходимо для того, чтобы использовать ЭПР-
спсктромстр, работающий ва Х-полосе (9 ГГц), для наблюдения протонного
спектра ЯМР и ЯМР-спектрометр. работающий па 60 мГц, для наблюдения
спектра Э11Р?
19.27. Ученый исследует возможность нейтронного спинового резонанса. Он име-
ет в своем распоряжении промышленный ЯМР-спектрометр, работающий на
60 МГц. Используя данные табл. 19.2, рассчитайте поле, необходимое для ре-
зонанса. и оцените относительную разность заселенностей при комнатной темпе-
ратуре. Какое спиновое состояние расположено ниже?
19.28. Химический сдвиг метильных протонов в ацетальдегиде (этанале) В=>
=2,20 м. д., а для альдегидного протона 6—9,80 м. Д. Каково различие в ло-
кальных магнитных полях этих двух частей молекулы, если приложенное поле
сосгазляст: а) 1,5 Т и б) 7,0 Т?
19.29. Каково расстояние (в Гц) между сигналами метильного и альдегидного
протонов, если спектрометр работает па частоте: а) 60 МГц и б) 300 МГц?
19.30. Нарисуйте эскиз спектра ЯМР этаналя, используя приведенные выше
значения 6 и константу спин-спинового взаимодействия, равную 2,90 Гц. Ука-
жите относительную интенсивность линий. Как изменится внешний вид спектра,
если вместо 60 МГн его записать на 300 МГц?
19.31. В спектре гипса в твердом состоянии наблюдается прямое днполь-ди-
подьное магнитное взаимодействие, которое зависит от угла в соответствии с
выражением (19.2.4). Спектр можно интерпретировать, предположив, что на
один протон действует дополнительное магнитное иоле в 0,715 мТ, генерируе-
мое другим протоном- Каково расстояние между протонами в твераом веще-
стве?
19.32. В обычной жидкости молекулярное вращение усредняет прямое диполь-
дипольное взаимодействие до нудя. Молскуда, растворенная в жидкости опреде-
ленного типа, называемой жидким кристаллом (т. 1, «Введение», разд. 2). не во
псех направлениях может вращаться свободно, и поЭ10му диполь-дипольное вза-
имодействие может нс усредняться до пуля. Предположим, что молекула распо-
ложена так, что, хотя вектор, разделяющий два протона, может свободно вра-
щаться вокруг направления приложенного поля, его угловое положение изме-
няется только между 0 и Усредните выражение для диполь-днполыюгю
19. Резонансные методы
163
взаимодействия по этому ограниченному интервалу ориентаций и подтвердите,
что усреднение превращается в нуль, когда Отах может стать равным л (сво-
бодное вращение по всей сфере). Какова средняя напряженность локального
поля (диполя) для молекулы из предыдущей задачи, если она растворена в жид-
ком кристалле, который позволяет ей вращаться только до 0 шах = 30“?
19.33. Чтобы получить информацию о быстрых процессах, в ряде случаев ис-
пользуют ЯМР. Вот Пример типового применения. При изомеризации молекулы
две группы протонов делаются эквивалентными. При низких температурах, ког-
да взаимопревращение происходит медленно, одна группа резонирует при 6 =
-4,0 м. д., а другая— при 6=5,2 м. д. При какой скорости взаимопревращения
два сигнала сольются в одну линию, если спектрометр работает на 60, МГц?
19.34. То же соединение, что и в предыдущей задаче, было исследовано при
различных температурах с применением разных спектрометров. Было найдено,
что спектр на 60 МГц сливается в одну линию при 280 К, но на 300 МГц для
обеспечения такого эффекта температуру нужво поднять до 300 К. Оцените
энергию активации (т. 1, «Введение», разд. 3) для этого взаимопревращения.
19.35. Эффект Мессбауэра связан с испусканием у-лучей без эффекта отдачи.
Насколько важно внедрение ядер излучателя в массивную жесткую кристалли-
ческую решетку? Рассчитайте скорость отдачи: а) свободного атома 57Fe,
б) атома 57Fe, составляющего часть жесткого кристалла весом 100 мг. Для каж-
дого случая найдите доплеровский сдвиг для у-лучей с энергией 14,4 кэВ.
19.36. Мессбауэровский спектр Na4Fe(CN)6 состоит из одной линии, а спектр
NasFe(CN)5NO — из пары линий (каждая с половинной интенсивностью), сим-
метрично смещенных относительно собственного резонанса молекулы. Объясните
эти наблюдения.
19.37, Было найдено, что изомерные сдвиги в мессбауэровском спектре Aul,
АцВг и АиС1 соответственно равны 0,125, 0,143 и 0,161 см/с [Bhide V. G.,
Shencry G. К-, Multani М. S.s Solid State Common., 2, 221 (1964)]. Что можно
сказать rra основе этцх данных об ионной природе указанных молекул?.
11
20 Статистическая термодинамика.
Концепции
Изучаемые вопросы
После тщательного изучения этой главы вы сможете:
1. Определить понятие канонический ансамбль (стр. 166).
2. Объяснить термины термодинамический предел и эргодиче-
ская гипотеза (стр. 166, 167).
3. Определить понятие распределение и его вес (стр. 172, 173)
и рассчитать вес наиболее вероятного распределения [уравнение
(20.1.12)].
4. Определить каноническую функцию распределения [уравне-
ние (20.2.1)] и связать ее с внутренней энергией системы [уравне-
ние (20.2.4)].
5. Определить молекулярную функцию распределения [урав-
нение (20.2.5)] и связать ее с канонической функцией распределе-
ния для различимых [уравнение (20.2.6)] и неразличимых [урав-
нение (20.2.7)] молекул.
6. Рассчитать поступательную (трансляционную) функцию рас-
пределения свободной частицы [уравнение (20.2.9)].
7. Вывести величину параметра 0 = XjkT (стр. 183).
8. Объяснить смысл величины молекулярной функции распре-
деления (стр. 186).
9. Описать больцмановское распределение заселенностей [урав-
нение (20.2.15)].
10. Определить понятия теплота и работа с молекулярной точ-
ки зрения (стр. 189). х
11. Определить механико-статистическую энтропию [уравнение
(20.3.7)].
12. Сформулировать третий закон термодинамики (стр. 191).
13. Связать энтропию с канонической и молекулярной функ-
циями распределения [уравнения (20.3.10) и (20,3.11)] и рассчк
тать энтропию одноатомного газа [уравнение (20.3.14)].
Введение
Энергия атомов и молекул может принимать только дискрет-
ные значения.
В нескольких предыдущих главах было показано, как эту энер-
гию можно рассчитать, определить спектроскопически и связать с
20. Статистическая термодинамика. Концепции 165
формой и размером молекул. Следующий важный шаг состоит в
том, чтобы показать, каким образом, зная эти разрешенные энер-
гетические уровни, можно объяснить поведение вещества в массе.
В данной главе мы установим связь между квантовой теорией и
термодинамикой.
Ключевой стадией перехода от квантовой механики к термо-
динамике является точное выяснение того, что в то время, как
квантовая механика рассматривает детализированное строение и
движение молекул, термодинамика имеет дело с их усредненным
поведением. Например, давление, оказываемое газом, интерпрети-
руется как средняя сила, с которой молекулы действуют па еди-
ницу площади; чтобы определить давление, не обязательно знать,
какая молекула соударяется со стенкой в некоторый момент вре-
мени. Вовсе не обязательно следить за флуктуациями давления,
обусловленными тем, что в разные моменты времени со стенкой
соударяется разное число молекул, поскольку шанс заметить эти
флуктуации очень невелик — слишком мало вероятно, что вдруг
произойдет временное затишье в столкновениях или вдруг грянет
«буря столкновений». Статистическая термодинамика основана на
принципе, согласно которому термодинамически наблюдаемыми
являются средние молекулярные свойства, и с учетом этого прин-
ципа строится схема для их расчета.
В данной главе мы вначале рассмотрим, как подойти к коллек-
тивному поведению молекул, составляющих систему, и почему в
статистической термодинамике центральную роль играет концеп-
ция хранилища теплоты. Это приведет к отождествлению термо-
динамической внутренней энергии U с особым видом усреднения
по энергетическим состояниям системы, и, следовательно, можно
будет рассчитать первый закон термодинамики. Далее мы уви-
дим, что с помощью определенного подхода можно рассчитать эн-
тропию системы, т. е. будем иметь дело с основной концепцией
второго закона термодинамики. В сумме оба этих расчета озна-
чают, что всю термодинамику можно выразить через молекуляр-
ные свойства.
20.1. Системы, ансамбли и распределения
Мы рассмотрим некоторое количество обычного вещества (ко-
торое может быть газообразным, жидким или твердым) в трех
аспектах. Во-первых, в нем имеются молекулы (пусть в этот тер-
мин будут включены также атомы и ноны). Во-вторых, оно пред-
ставляет собой собственно систему. Такая же концепция исполь-
зовалась при термодинамическом рассмотрении (т. 1, гл. 2), но в
отличие от того рассмотрения теперь мы должны знать, что систе-
ма состоит из N молекул. Эта система имеет некоторый объем V,
166
Часть 2. С тру к тура
И путем контакта со значительно более обширным хранилищем
теплоты в ней поддерживается температура Т.
Третий аспект включает способ рассмотрения данного количе-
ства вещества, позволяющий проводить статистические расчеты.
Концепция, которую мы уже почти ввели, довольно искусственна
и хитроумна, но она составляет основу статистической механики.
Это идея об ансамбле. Подобно многим научным терминам, этот
термин имеет преимущественно свой обычный смысл, который чет-
ко определен и однозначен.
Чтобы построить ансамбль, мы сначала представляем себе си-
стему и гораздо более обширное хранилище теплоты, с которым
она находится в тепловом контакте, как изолированную единицу,
а затем формируем ансамбль, воспроизводя эту единицу много
раз. Каждое воспроизведение имеет точно такую же температуру,
а система внутри каждой единицы — точно такой же объем и* со-
став, Такое воображаемое собрание репликаций (копий) назы-
вается каноническим ансамблем. Ансамбль рассматривается как
набор воображаемых репликаций, и поэтому мы свободны в выбо-
ре числа членов Л"’, которое может быть так велико, как мы хотим,
пусть даже, когда это нужно, оно равно бесконечности*.
Отметим, что число Ж совершенно нс связано с (W — число
молекул в реальной системе; и!/'—число воображаемых повторе-
ний этой системы),
Термодинамический предел. Способ введения представления о ка-
ноническом ансамбле заключается в следующем. Мы очень мало
знаем о действительном состоянии системы в каждый момент вре-
мени, Если бы система была совершенно изолированной, то она
находилась бы в определенном квантовом состоянии и оставалась
бы в этом состоянии. Но система не изолирована; она находится в
контакте с хранилищем теплоты, и вследствие обмена с ним Энер-
гией получается система, распределенная по своим квантовым со-
стояниям. Мы не только не знаем состояния системы в момент ее
получения, но и нс можем рассчитать ее детальную эволюцию во
времени.
Ансамбль — это путь в обход «го^ы трудностей». Прежде все-
го определим среднее во времени свойство системы как измеряемое
термодинамическое объемное свойство. Так, средняя во времени
энергия системы будет отождествляться с ее термодинамической
* В статистической термодинамике рассматриваются два важных типа ан-
самблей. В микроканоническом ансамбле условие постоянной температуры заме-
нено из требование, чтобы все системы во всех членах ансамбля имели одина-
ковую энергию, т. е. каждая система сама по себе изолирована. В большем ка-
ноническом ансамбле объемы всех 'систем во всех единицах одинаковы, каж-
дая система находится в контакте с хранилищем теплоты, имеющим темпера-
туру Т (как в каноническом ансамбле), ио можно вообразить, что вещество спо-
собно переходить из каждой одной единицы в каждую другую, так что состав
каждой системы может флуктуировать.
20. Статистическая термодинамика. Концепции 167
внутренней энергией U. Затем предположим, что объемное свой-
ство будет иметь точно такую же величину, если вместо усредне-
ния во времени для одной системы мы усредним это свойство по
всем членам ансамбля в определенный момент времени. Введение
ансамбля равносильно переводу мудреной задачи зависимости от
времени в задачу, не связанную с такой зависимостью, а расчеты,
в которые пе входит зависимость От времени, гораздо легче осу-
ществить. Предположение о том, что Эти две процедуры усредне-
ния эквивалентны, называется эргодической гипотезой, а ее закон-
ность является предметом многочисленных дискуссий эрудитов.
Имеются два подхода к расчету свойств ансамбля, и, посколь-
ку в каждом из них подчеркивается своя точка зрения, мы рас-
смотрим оба подхода.
Первая точка зрения: роль хранилища. Для начала установим, как
найти вероятность того, что, если выбран данный член ансамбля,
для содержащейся в нем системы будет найдена энергия £,. Каж-
дая единица ансамбля изолирована, и поэтому, несмотря на то что
энергия может перетекать между системой и хранилищем, общая
энергия единицы ансамбля £Общ будет постоянной. Следователь-
но, если энергия системы равна £,, то энергия хранилища будет
£общ—£ь
Имеет смысл предположить, что вероятность Р(£,) того, что
система будет находиться в данном состоянии с энергией £,-, про-
порциональна числу способов, которыми хранилище может при-
способиться к размещению остатка энергии. Если в хранилище
энергия может разместиться №"(Еос>щ—Et) различными способа-
ми, т. е. если оно имеет №"(ЕОбщ—Е{) состояний энергии £общ—Eir
то
Р(£,-)=СГ'(£оещ-£г),
где С — некоторая константа. Только что сделанное утверждение
является одним из тех, что подчеркивают многие особенности ста-
тистической термодинамики: оно называется принципом равенст-
ва априорных вероятностей. Согласно этому принципу, пока нет
какой-либо иной информации, все возможности следует считать
равновероятными; в данном случае, если имеются указанные выше
состояния, все они могут быть заняты, и ни одно не имеет преиму-
щества перед другим (например, состояния, которые соответству-
ют интенсивному колебательному движению, или состояния с оди-
наковой энергией, которые соответствуют менее интенсивным ко-
лебаниям и более интенсивному поступательному движению, не
могут считаться предпочтительными).
Поскольку хранилище значительно больше, его энергия намно-
го превышает энергию £, системы. Поскольку £, много меньше, чем
Еобщ—£;, и поэтому много меньше, чем £ОбЩ, число состояний хра-
нилища с энергией £оащ—Е; можно связать с числом состояний с
168
Часть 2. Структура
энергией Ео6щ, используя разложение Тейлора. В самом деле, луч-
ше работать с логарифмами (которые изменяются менее резко,
чем сами величины), и тогда можно написать
\ /Lt>6,n
Другие члены ряда содержат Ei в более высоких степенях, и ими
можно пренебречь, так как Е, очень мала. Далее, дифференциаль-
ный коэффициент не зависит от состояния системы, а зависит
только от общей энергии системы и хранилища. Следовательно,
его можно записать как константу |к
. (20,1,1)
\ с /£общ
Это приводит к выражению
In IF (Еобщ Е;) = 1п W’ (Ео6щ) рЕ;
или
Г’(Еобщ-Е;)=Г'(Еобщ)^,
так что
Р(Ег)=СТГ'(Еобщ)е-ееС.
Число W'(EO(,m), свойство хранилища, не зависит от состояния си-
стемы, и поэтому его можно объединить с С и получить новую
константу С', Эта константа может быть определена из условия,
что общая вероятность нахождения системы в некотором состоя-
нии должна быть равна единице:
2E(E/)=C'2®’^-t
1 Z
откуда
с
е
Поэтому конечный результат для вероятности выбора из ансамбля
системы с энергией Ei имеет следующий вид:
Р(Ег)=е-₽сг (20.1-2)
L
Это чрезвычайно важное выражение называется каноническим
распределением.
Отмстим центральную роль, которую играет хранилище: пара-
метр р с размерностью 1/энергия [уравнение (20.1.1)] опреде-
ляется исключительно через хранилище теплоты, которое в свою
20. Статистическая термодинамика. Концепции 169
очередь характеризуется единственной величиной — его темпера-
турой Т. Позднее мы увидим, что р можно отождествить с \[kT.
Поскольку распределение определено через свойство хранилища,
можно ожидать, что оно будет иметь очень широкий смысл: в лю-
бой системе (независимо от ее строения, и как бы мала она ни
была), находящейся в контакте с хранилищем, имеющим темпера-
туру Т, распределение будет соответствовать уравнению (20.1,2).
Форма канонического распределения не совсем соответствует
той, которую можно предположить при беглом взгляде на урав-
нение (20.1.2). Нужно отметить, что это выражение применимо к
вероятности обнаружить только одно состояние с энергией Хо-
тя система мала в сравнении с хранилищем, она может быть до-
статочно большой, чтобы было много состояний с энергией, очень
близкой к Ei. Действительно, число состояний системы резко воз-
растает с энергией. Поэтому вероятность найти систему с данной
энергией Е, ио не с данным состоянием I равна Р(Е) W(Ey где
W — число состояний системы с энергией Е. W(E) резко возра-
стает; Р(Е) резко падает. Следовательно, их произведение являет-
ся функцией с резким максимумом в области некоторой средней
энергии.
Каноническое распределение можно рассмотреть глубже. На-
пример, средняя энергия системы представляет собой сумму
(Еу^Е^у^Е^ (20.1.3)
i i I
Поскольку мы согласились отождествлять среднюю энергию ан-
самбля с термодинамической внутренней энергией, это уравнение
позволяет вычислить U, если известны возможные энергии систе-
мы Ei. Это положение будет развито ниже, а в следующей главе
даны упрощающие правила и формулы.
Можно было бы подумать, что расчету поддаются только энер-
гии и аналогичные функции состояния (такие, как давление). Это
не так. Точную статистическую интерпретацию можно дать энтро-
пии системы, и уравнение (20.1.1)—лишь этап на пути к этому.
Из уравнения (6.1.3) (т. 1, гл. 6) следует, что
(dS/dU)v=\/T.
Уравнение (20.1.1) имеет точно такую форму, если р приравнять
к i/kT, a In W—к S/k. Эту связь мы установим в разд. 20.3 и по-
кажем, что энтропию можно вычислить из AlnlF. Тогда вся хими-
ческая термодинамика открывается для расчета на основе извест-
ных молекулярных энергетических уровней.
Вторая точка зрения: преобладающие распределения. Средняя
энергия системы определяется температурой Т хранилища. Иными
словами, между температурой и средней энергией имеется соот-
ветствие «один к одному». Это можно использовать, обратившись
170
Часть 2. Структура
rf*=20
I N.V.T 2N.V.T 3 X. г. т х.и.т N.V.T
N. V.T N.V.T+ t В-Л'.Н.Гч Геплота
*-
20
Рис. 20.1. Канонический ансамбль
(^=20).
к другой точке зрения на роль канонического ансамбля. Теперь
предположим, что мы игнорируем хранилища и рассматриваем
ансамбль как состоящий из повторений только одной системы,
и в то же время вообразим, что все члены ансамбля находятся в
тепловом контакте друг с другом (рис. 20.1) так, что другие чле-
ны по существу являются хранилищем теплоты для любого дан-
ного члена. Благодаря этому тепловому контакту энергия отдель-
ных членов может флуктуировать точно так же, как в ранее
описанном ансамбле, но, поскольку весь ансамбль изолирован, его
общая энергия постоянна и имеет величину ё. Общее число чле-
нов определено как Ж1, поэтому средняя энергия ё/.Л" также по-
стоянна п имеет величину { Е ). Следовательно, созданный та-
ким путем ансамбль полностью эквивалентен ранее описанному
ансамблю,, однако вместо того, чтобы точно определить его темпе-
ратуру и попытаться найти некоторую среднюю энергию, здесь мы
точно определяем среднюю энергию и обязательно приходим к по-
нятию, которое называется частной температурой.
Когда происходит флуктуация, энергия некоторых членов ан-
самбля может понизиться, но, чтобы компенсировать это, энергия
некоторых других членов должна возрасти. Невероятно, чтобы
энергия испускалась одновременно из большого числа членов и
накапливалась лишь в некоторых, поэтому большую часть време-
ни большая часть членов будет иметь энергию, очень близкую к
средней. Предположим, что мы находим наиболее вероятное рас-
пределение энергии и вычисляем все термодинамические свойства,
взяв среднее по всему ансамблю с таким распределением. Тогда,
поскольку наиболее вероятное, распределение намного более ве-
роятно, чем распределение, сильно отклоняющееся от него, можно
быть уверенным в получении приемлемого результата, Когда чис-
ло членов ансамбля становится очень большим, наиболее вероят-
ное распределение будет намного_более вероятно, чем отклоняю-
щиеся от него распределения, и в пределе, когда иГ бесконечно
20. Статистическая термодинамика. Концепции
171
велико, наиболее вероятное распределение подавляюще наиболее
вероятно. Этот предел, когда Ж -—>оо, называется термодинамиче-
ским пределом, и в этом пределе мы отождествляем усреднения
по всему ансамблю с термодинамическими свойствами реальной
системы.
Сказанное выше можно перевести в количественную форму
следующим образом. Средняя энергия Ж членов ансамбля с общей
энергией g равна
<£>=£Л,Г.
Предположим, что члены имеют некоторую энергию С7(0)4-£/, где
U(0) —некоторое начало, от которого измеряются все энергии, л
что эту энергию в некоторый момент времени могут иметь щ чле-
нов. Тогда общая энергия ансамбля будет
£ = Д/ (0)-j-£;}, . Г 2^11,,
i i
и поэтому
<£>=(1/ДД +
l
Термодинамическая внутренняя энергия является пределом этой
суммы при Ж—>оо, следовательно,
U—U (0) = (1/Д") ^П;£;, jT—► оо. (20.1.4)
t
Таким образом, расчет сводится к определению числа и,.
Наиболее вероятное распределение. До сих пор у лас не было не-
обходимости знать, какие значения может принимать щ; мы зна-
ем лишь, что средняя энергия должна иметь точно установленную
величину. Одним из путей для выяснения этого могло быть пред-
положение, что энергию cl''{U—U(0)} имеет лишь единственный
член ансамбля, а все остальные обладают меньшей энергией, ис-
ключая минимальную U(0). Такое распределение было бы чрез-
вычайно мало вероятным; действительно, имеются Ж‘ способов
его получения, так как любому из Ж членов можно отдать пред-
почтение. Другой способ распределения энергии состоит в том,
чтобы разрешить каждому члену иметь одну и ту же энергию
U~ V(0). Тогда среднее уже точно будет равно U—0(G), как тре-
буется, но будет существовать только один способ реализации
этого распределения, что гораздо менее вероятно, чем даже в пер-
вом случае. Необходимо найти распределение, которое может быть
осуществлено наибольшим возможным числом способов.
Мы подойдем к расчету, исключив столько сложных вопросов,
сколько возможно, и рассмотрим очень специфическую простую
172
Часть 2, Структура
систему. Предположим, что реальная система {и поэтому все ее
репликации) настолько проста, что она может быть найдена с
энергией (7(0), или U (0) + е, или (7(О)+2е и т. д. (рис. 20.2).
Напомним, что эти энергии являются общими энергиями состав-
ной системы. Более того, мы рассмотрим очень небольшом ан-
самбль, состоящий лишь из трех членов (Ж1 = 3), увеличим его до
пяти членов, затем до двадцати и, наконец, до бесконечности.
В каждом случае будем требовать, чтобы средняя энергия была
равна U (0) + е.
Ансамль из трех членов имеет общую энергию 3(7(0)+3е, если
средняя энергия его членов равна U (0) + е. Поскольку эта общая
энергия сохраняется, отдельные члены не обладают свободой вы-
бора своих состояний. Например, три члена не могут иметь рас-
пределение {(7(О)+Зе, (7(0) 4-е , (7(0) +2 }, так как общая энер-
гия будет превышать 3(7(0)+3е, В то же время возможны неко-
торые другие комбинации, и точно определенная общая энергия
может быть обеспечена такими распределениями, как {[7 (0) +€,
[7(О)+2е (7(0)}, пли {(7(0)+е, (7(0)+е, (7(0)т-е},или {(7(0),
(7(0), (7(0) +3 } и т. д.
Теперь возникает вопрос: следует или нет рассматривать все
разрешенные распределения как одинаково вероятные? Для отве-
та на него мы сформулируем принцип равенства априорных веро-
ятностей в форме: все распределения энергии, совместимые со
строго определенной общей энергией, имеют равную вероятность
20. Статистическая термодинамика. Концепции 173
осуществления. Это значит, что распределение {[7(0) +е, [/(О) +
+ 2е, [7(0)} не имеет преимущества перед распределением {£/(0),
[7(0), £7(0)-(-Зе} или {[7 (0) + е , [7(0)+е, [7(0) +е} или любой
другой комбинацией с той же самой общей энергией. Нет причин,
по которым одно ц.з этих распределений было бы предпочтитель-
ным, и тот факт, что статистическая термодинамика правильно
описывает явление, не дает оснований думать иначе.
Следующий этап состоит в подсчете числа распределений раз-
ных типов. Это легко сделать с помощью рис. 20.2, просто сосчи-
тав распределения. Поскольку нас не интересует, в каком члене
ансамбля реализуется конкретное состояние, приведенные на ри-
сунке результаты можно сформулировать, утверждая, что имеют-
ся шесть способов достижения распределения типа {[7(0), U (0) +
-1~е, £7(0) + 2е}, три способа достижения распределения типа
{[/(0), [7(0), [7(0)+3е} и только один способ достижения распре-
деления {[/(0) + ^, [7(0) -f-е, [7(0)+е}. Эти числа (6,3 и 1) на-
зываются весом распределений, и легко проверить, что они даются
формулой
^(п,^..^/!^^!... , (20.1.5)
где 11,, Иг,... — число членов с энергией [7(0), U(0) +е ,и т. д.
Например, в приведенном случае.#1 =3, мы можем сказать, сколь-
ко распределений имеют два члена на их низшем уровне (п,= 2),
ни одного члена на уровнях £7(0) +е или £7(0) -|-2е (112 — п3 —0)
и один член па уровне £7(0) +3 е (л4 = 1). Тогда, вспомнив, что
01=1, получим
(2, 0,0, 1,0.)--=3!/2!0!0! 110!... =
= 6/(2)(1)(1)(1)(1)...=3
в согласии с уже указанным числом. [Это выражение для веса
распределений получается из расчета числа способов, которыми Ж
различимых предметов могут быть размещены в ларях ( п,- пред-
метов в ларе i); предметы — это члены ансамбля, а разные лари
соответствуют разной энергии системы.]
Только что приведенный пример показывает, что один из ти-
пов распределения, в данном случае {£7(0), U(0) + е , [7(0) +2е},'
имеет наибольший вес, и поэтому вероятнее всего, что для ансамб-
ля будет найден этот тип распределения. Рассматривая усреднен-
ные свойства ансамбля, имеющего такое распределение, можно
ожидать, что получатся основные свойства реальной системы,
В случае Ж=3 наиболее вероятное распределение не особенно до-
минирует над другими, но его доминирование растет с ростом Ж.
Для ансамбля с пятью членами (Ж —5) и такой же средней
энергией (т. е, общая энергия равна 5[7 (0) + 5е ) веса распределе-
ний приведены на рис. 50.3. С помощью уравнения (20.1.5) легко
174
Часгб 2. Структура
%г(2,1,2)^зо Чеф,6,1,1)=2О
W(2,2,0,1)^30 W (3,1,0,0,1) *20
Рис. 20.3. Распределения для
5-членного ансамбля.
5г
4е
Щ
2-е
-е
!И. ОВД.
’ЙГ(4,0Д0,0Д)=5
%Г(1,3,1)=20
5г---—-
4е--------
Зг •------
2# _____
ЖЕЖ
4Ы(0,5>1
проверить, что ансамбль с
двумя членами на их низ-
ших уровнях, двумя члена-
ми на следующем уровне,
со следующим пустым уров-
нем и одним членом на
четвертом уровне можно
------------' получить 30 разными спо-
HBIEIWH, собами_ Следующие наибо-
—-----------— лее вероятные типы рас-
пределения могут быть об-
радованы только 20, 5 и 1 различными способами. На следующем
этапе (рис. 20.4) берем Л3 = 20. В этом случае наиболее вероятное
распределение можно достигнуть 9,8‘Ю9 способами, тогда как со-
седнее распределение (рис. 20.4)—только 3,7-JO3 способами. Те-
перь наиболее вероятное распределение сильно доминирует, и ес-
ли ограничиться ансамблем, имеющим такое распределение, то
ошибка в средних величинах будет небольшой.
Как найти наиболее вероятное распределение для данной
средней энергии (и, следовательно, температуры)? Число способов
достижения распределения данного типа, его вес, определяется
величиной W в уоавнении (20.1.5). Требуется найти набор чисел
Наиболее вероят-
-ное распре-----
деление
Я
В.
Л
Ж
Ж
J3_
Я
. ШШЛ Ш1Ш4
W 3,4,0,2,0,1,1)
= 1,71 JO®)
^(14,1,2,3,0,0,0,1)
= 4,SS-fO7
4317(10,5,3,2 I)
= 9,78-10 ’
%7(I2,3,4,0,0,2)
= 3,70-10®
Рис. 20.4. Некоторые из распределений для 20- ч ле иного ансамбля.
20. Статистическая термодинамика. Концепции 175
n*t nj,..., для которых W максимальна. Это эквивалентно опре-
делению условий (что, вероятно, сделать проще), при которых
/nW максимален; мы так и поступим. Поскольку выражение для
W — чисто статистическое выражение, не имеющее отношения
к расположению энергетических уровней реальной системы, мы
сейчас получим совершенно общие результаты, не ограниченные
простой системой, рассмотренной в предыдущих параграфах.
Поскольку W зависит от всех Ш, при изменении распределе-
ния таким образом, Что п, изменяется до ih + dtt?, In w изменяет-
ся согласно формуле
d (In (д 1п W/di^dn^ (20.1.6)
i
В максимуме это изменение стремится к нулю; однако имеется
одно осложняющее обстоятельство, которое препятствует решению,
получаемому простым приравниванием всех (d7Jf/dixк нулю.
Оно обусловлено следующим. Общее число членов ансамбля в
сумме не может измениться, и поэтому изменения drit должны
удовлетворять соотношению
<F=dnJ-7’dn3+-• =0 или^Ж1;=0. (20.1.7)
д
Это значит, что изменения dre в уравнении (20.1.6) не являются
независимыми, и поэтому это уравнение нельзя решить, приняв,
что отдельные члены стремятся к нулю. Кроме того, имеется еще
одно ограничение свободы изменения числа и,. Оно вызвано тем,
что любое изменение распределения (любое изменение И;) не
должно изменять общую Энергию ансамбля. Следовательно, если
какие-то th увеличиваются, то другие tt, должны уменьшаться,
чтобы общая энергия осталась постоянной. Это можно выразить
как
d<g 4- E2dnz-f- • - - =0 или ^Е-^п^О, (20.1.8)
t
что составляет второе ограничение свободы выбора п,.
Способ преодоления этих стесняющих обстоятельств был пред-
ложен Лагранжем и называется методом неопределенных множи-
телей,. Основа метода описана в приложении 20.А. Здесь же нам
необходимо лишь знать очень простое правило; величина, на кото-
рую наложено ограничение, должна быть умножена на некоторую
константу и затем прибавлена к главному вариационному урав-
нению [уравнение (20.1.6)]; тогда изменения апс рассматривают-
ся как независимые. Константы определяются в конце вычисления.
Применение метода очевидно из следующего. Два ограничения
176
Часть 2. Структура
умножаются на константы а и —р соответственно и затем при-
бавляются к уравнению (20,1.8)
d (In (din(iff/dni)d'tii-\~a.'£dni— “
£ . if
=2{(^W+«-^il dnt. (20.1.9)
i
Следуя правилу, все dn; можно считать независимыми. В этом
случае единственно удовлетворяющим условию d(}n Ж)=0 яв-
ляется требование, чтобы для всех i
(д in Чи'/дп^-^а—fiE^D при Hi—ttf. (20.1.10)
Чтобы решить последнее уравнение, используем приближение,
которое действительно очень хорошо выполняется для тех чисел,
с которыми мы должны иметь дело. Приближение Стирлинга за-
писывается в виде
для больших х: lnxiasxlnx—х. (20.1.И)
Подстановка этого приближения в выражение для 1п Ж дает
In — In (^Ч/Иу !и2! ...) =
= in — in (tty I n2*..,) =
— InJ^l — InnJ Sb
I
Sb {JC in JC — Л') — Jointly—n;) as’
as jc inj1'3—,ППЛ
/
так как сумма И/ равна . Поскольку JC—константа, дифферен-
цирование этого выражения по л, приводит к
(din Wnj « — !д(Пуinnj/dnj к,
j
~ — 2 ((dny/dn()iniiy+ti;(din Иу/бИ;)} ~
/
AS —{Inttj-h-lj,
потому что при П, не зависит от п;, т. е. (driy'dn;) = O; когда
j~i, этот дифференциальный коэффициент равен единице; мы так-
же использовали соотношение (din fty/dtti) — (1Mj) (dny/dnj.
В сравнении с 1п п; единицей можно пренебречь, и поэтому урав-
нение (20.1.10) превращается в
—1п Пе -ф-а—₽£\ = 0 j
20. Статистическая термодинамика. Концепции
177
или
ttt — exp (a—fifj). (20.1.12)
Конечный этап отыскания наиболее вероятного распределения
состоит в определении двух ковстапт аир. Заселенности должны
удовлетворять условию
2^ = .Г и {С7 —С7(0)}.
ii
Подставляя уравнение (20.1.12) в первое из этих уравнений,
получаем
2 exp (a —pEt) =е“ 2 exp (—₽£,) =Л*
i £
Отсюда
^=•^/{21^91—^1)},
i
и поэтому
ni/jT = ехр (— Ю/{2 ехР (20,1.13)
£
Подставляя последнее уравнение во второе условие, в принципе
можно найти величину константы р:
(1 /^о 2 n'—2 Ei ех₽ (—₽£<)/{ 2 ехР (—££;)}=и—v (°)- (20-1 14>
£ I I
Однако мы найдем иной путь для этого и тогда используем данное
уравнение для расчета средней энергии U. Но уже сейчас можно
предположить, что р — мера температуры ансамбля. Это следует
из того, что для всех щ существует только один параметр р
[уравнение (20.1.13)], а свойством, общим Для ансамбля в целом,
является температура Т. В дальнейшем мы увидим, что р — \Jk.T.
20.2. Функции распределения
В уравнении (20.1.13) мы достигли результата, являющегося
центральным в статистической термодинамике, так как он позво-
ляет установить наиболее вероятное распределение энергий для
ансамбля, а мы видели, что ансамбли с таким распределением
определяют термодинамические свойства системы.
' Каноническая функция распределения. Сумма ехр(— рЕ;) по энер-
• гиям членов ансамбля настолько важна, что она носит специаль-
i ное название канонической функции распределения Q:
каноническая функция распределения'. Q = 2 ехР(~р£;)- (20.2, Г)
| 12-242
178
Часть 2. Структура
Используя это обозначение, относительное содержание членов,
которые имеют энергию Et в ансамбле с наиболее вероятным рас-
пределением, можно выразить формулой
Pi=ttZ/^r=(l/Q)exp(—(20.2,2)
а среднюю энергию, т. с. термодинамическую внутреннюю энер-
гию, — формулой
U-U (0) = (1 /Q) 2 exp(-0Et). (20.2.3)
t
На основе последнего выражения можно предположить, что
для определения внутренней энергии кроме суммы Q необходимо
вычислить еще одну сумму. Это не так, потому что его можно пре-
вратить в форму, включающую только Q. Проведем следующие
преобразования. Сначала заметим, что
(d/d0)exp(—0£г) =—/Гг ехр(—
Тогда
U~V (0)=(i/Q> ^(—d/rfp)exp(—₽£,) =
i
= -(V/Q)(d/d₽)Sexp(-pZr()-
i
Здесь сумма — это функция распределения, так что
<7 — U (0) = — (1/Q) (dQ/dP) = —(d In Q/d₽).
Функция распределения зависит от объема системы (или от нали-
чия какого-либо приложенного поля), и поэтому производная по
Р на самом деле является частной производной при постоянном
объеме. Таким образом, конечное выражение, связывающее внут-
реннюю энергию с функцией распределения, имеет вид
U-U (dQ/dp)v = ^(d In Q/d₽)r. (20.2.4)
Последнее уравнение подтверждает, что для расчета внутрен-
ней энергии необходимо знать только функцию распределения.
В следующей главе мы расширим этот вывод и покажем, что все
термодинамические функции можно рассчитать, если известна Q.
В этом отношении она играет роль, очень похожую на роль волно-
вой функции в квантовой механике, которая содержит всю дина-
мическую информацию об отдельной системе. Аналогию можно
проследить еще ближе, если написать выражение для энергии в
форме
fU-U (O)]Q = -(dQ/dP)v
20, Статистическая термодинамика. Концепции:
1'79
и заметить, что оно напоминает связанное со временем уравнение
Шредингера (т. 1, подразд. 13.6.Б)
Hty=—(h/i} (dty/dt),
где гр — волновая функция, а И— оператор энергии для системы.
Молекулярная функция распределения. Хотя для иллюстрации мы
использовали простой тип ансамбля, до сих пор рассмотренные
системы были совершенно общими и не было необходимости знать,
вращаются ли молекулы, колеблются, движутся поступательно
или взаимодействуют друг с другом. Ограничившись рассмотрени-
ем лишь систем, состоящих из независимых молекул, можно до-
стичь сильного упрощения. Если мы хотим рассчитать термоди-
намические свойства реальных газов и жидкостей, то это будет
слишком большим ограничением; однако даже для систем с прс-
небрежимым межмолекулярным взаимодействием можно получить
много полезной информации, и мы ограничимся ими. Такое огра-
ничение будет смягчено в гл. 23, когда мы обратимся к реальным
газам.
В функции распределения Ei— общие энергии коллектива мо-
лекул, составляющих систему, В случае независимых молекул:
энергия системы равна сумме энергий всех отдельных молекул:
где — энергия молекулы 1 в системе с общей энергией Еь
Каноническая функция распределения имеет вид
Q = 2 ех? ’ +EJv)},
i
где суммирование производится по всем состояниям i системы. Все-
эти состояния можно охватить, если предположить, что все моле-
кулы входят в систему в разных собственных индивидуальных со-
стояниях (хотя ниже будет сделана важная оговорка). Следова-
тельно, вместо суммирования по коллективным состояниям i мы
суммируем по всем 'индивидуальным молекулярным энергетиче-
ским уровням et/- для молекулы 1, са/ для молекулы 2 и т. д. Это
приводит к выражению
(2^).
зI i
Если все молекулы одинаковы, то больше нет необходимости раз-
личать их энергетические уровни, и тогда последнее выражение:
сводится к виду
I i i з
12*
180 Часть 2. Структура
При введении молекулярной функции распределения
молекулярная функция распределения'. д — (20.2.5)
i
где сумма берется по состояниям отдельной молекулы, получаем
<2=^. (20.2.6)
Иногда некоторые молекулярные состояния обладают одинаковой
энергией; например, если и Е4=Е5=£в, то мы говорим, что
состояния с /=2, 3 образуют дважды вырожденную группу, а со-
стояния с /=4, 5, 6—трижды вырожденную группу. Члены вырож-
денных групп дают одинаковый вклад в сумму, и поэтому мы мог-
ли бы с полным основанием написать
<?=2^ехР<-₽6/)’
/
где gj — вырождение каждой энергии и / уже относится к разным
группам вырождения, а не к каждому индивидуальному и, воз-
можно, вырожденному состоянию. Мы не будем применять это
обозначение, хотя оно часто используется.
Пример. Напишите выражение для вращательной функции распределения линей-
ной гетеро яд ер пой молекулы.
Энергия вращающейся линейной молекулы равна =/?/('/+1J. 7=0, 1,
2, ... . Каждый / уровень имеет 2/-J-1 состояний, соответствующих различной
ориентации вращающейся молекулы; эти состояния отличаются квантовым чис-
лом Энергия не зависит от М?, а следовательно, каждый / уровень (2/-I-1)-
кратно вырожден. Поэтому суммируем или по У и М_г, или только по /. одна-
ко в последнем случае каждый член суммы умножаем на вырождение £Э=(2/+
+1).
Ответ. ? = 2 еХр т Ш =
S S ехр (J + Oi =
J—0 J
= 2 (2/ + l)exp [-pBJ(/+ 1)].
j=o
Последнюю строчку можно получить, если учесть, что g/=2/4-l.
Комментарий. Будьте внимательны: во вращающейся молекуле типа сферичес-
кого волчка вырождение уровня / равно (2J+1)2.
К сожалению, в одном очень важном случае вывод уравнения
(20.2.6) несправедлив. Рассмотрим этот случай. Если все моле-
кулы одинаковы и движутся свободно, то мы не можем отличить
одну от другой. Предположим, что молекула 1 находится в неко-
тором состоянии а, молекула 2 — в состоянии b и молекула 3 — в
состоянии с; тогда один член ансамбля имел бы энергию Еа + е& +
20. Статистическая термодинамика. Концепции
181
,+ ес. Этот член, однако, неотличим От члена, образованного пере-
водом молекулы I в состояние Ь, молекулы 2 в состояние с и мо-
лекулы 3 в состояние а, или от какой-либо иной перестановки. Это
значит, что в случае неразличимых молекул при переходе от сум-
мы по членам ансамбля к сумме по молекулярным состояниям
мы принимаем в счет слишком много состояний, и поэтому, запи-
сывая Q = qA, переоцениваем Q. Детальная аргументация очень
сложна (см. приложение 20.Б), но при температурах гораздо выше
абсолютного нуля поправочный множитель равен 1/Л7!, а следова-
тельно, для неразличимых молекул
Q^qN/NL- (20.2.7)
В каком же случае молекулы различимы и мы используем
уравнение (20.2.6), а в каком случае они неразличимы и мы при
меняем уравнение (20.2.7)? Прежде всего они должны быть оди-
наковыми частицами. Атом аргона [Никогда не будет [Неотличимым
от молекулы метана или атома неона. Однако идентичность—не
единственный критерий. Если идентичные атомы образуют кри-
сталлическую решетку, то каждый из них можно пометить систе-
мой координат, Идентичные молекулы, неподвижно удерживаемые
в решетке, различимы, и мы используем уравнение (20.2.6). В то
же время идентичные молекулы, свободно движущиеся в газе,
неразличимы, поскольку нет способов проследить индивидуаль-
ность отдельной молекулы. В этом случае мы применяем уравне-
ние (20.2.7). В дальнейшем мы рассмотрим следствие из этого
правила.
Поступательная функция распределения. В качестве Примера ис-
пользования формулы для q применим ее к идеальному одноатом-
ному газу в контейнере объемом V. Длина контейнера в х-направ-
лении равна X (и аналогично У, Z в двух других направлениях, так
что Xi'Z=Vi. Функция распределения для любой из молекул
имеет вид
9-=^ ехР(~₽еЛ
У
где в,- — поступательная энергия молекулы в контейнере, a j —
квантовое число, характеризующее состояние, Поскольку в/ — сум-
ма кинетических энергий в трех измерениях (Е;=е/(х)+е/(гу+бдг)),
функция 'распределения раскладывается па множители следующим
образом;
<7 =2 ехР 1 =
все /
= {2 ехР {2 ехр (-рЕди)] { ехр (~ред2>)} =
/(X) /(У) ,(Ю
182
Часть 2. Структура
Используя результаты рассмотрения энергетических уровней час-
тицы [т. 1, уравнение (13.4.4)], вводим выражение
^nx]=i2(h2/8mX2)t /=1,2,3,... ,
где m— масса молекулы. Тогда сумма, которую нужно определить,
равна
СО
Ях = 2 exp (—/Ча0/8т№).
/=i
В контейнере размером с типичный лабораторный сосуд поступа-
тельные энергетические уровни очень тесно расположены, и по-
этому сумма может быть заменена на интеграл
</x = Jexp (= j2h2fy8mX2)dj.
i
Расширение нижнего предела до /=0 вносит пренебрежимо малую
ошибку, но переводит интеграл в Стандартную форму. Это дости-
гается подстановкой х2=/2А2р/8тХ2, откуда dj= (8mX2/h-fiy/?dx:
qx =(8тХ2/йгР)1/2 J exp (—№) dx =
о
Такова молекулярная функция распределения для поступательно-
го движения по направлению х. Чтобы получить другие степени
свободы, нужно лишь заменить X на У или Z, и, следовательно,
функция распределения для движения в трех измерениях будет
иметь вид
q = (2nrnX2lh2^)1^(2nmY2/h2^(2nmZ2/h2^ — ^m/h^yc-V. (20.2.8)°
Пример (вопрос 6). Рассчитайте поступательную функцию распределения моле-
кулы водорода, заключенной в сосуд объемом 100 см3 при комнатной темпера-
туре.
Метод. Используем уравнение (20.2.8), предполагая, что р=1/АТ.
Ответ.
( 2лХ(3,348-10~а? кг)___________13/2 „
Я- |(6.626-10-34 Дж-с)2х[1/(298 К)Х(1,381 Ю-2» Дж/К)] J Х(
= {1,972-102° м-2}3/йХ(10-4 м3) -
= 2,769-102".
Комментарий. Эго показывает, что даже при комнатной температуре для такой
легкой молекулы термически доступно около 10гб квантовых уровней.
С некоторыми применениями этого простого выражения мы
встретимся в следующей главе. Пока же мы проиллюстрируем,
20. Статистическая термодинамика. Концепции
183
как рассчитать внутреннюю энергию газа, используя уравнение
(20.2.4) с Q = c/-v/iV‘. Сначала нужно найти (dQ/dfi)v-‘
(dQ/d^)v-^= (dqN(d^v/Nl =(Nq^-') (dq/df>)v/Nl,
Следовательно,
m- (V"1) WW)V!N\
<2 - (/>!) -
— —N(\(q)(dqld^)v. (20.2.9)
(Тот же результат получается, когда Q — qN.) На этой стадии вид-
но, что внутренняя энергия пропорциональна числу молекул, как
и ожидается для газа, состоящего из невзаимодействующих ча-
стиц. Кроме того, внутренняя энергия задается таким же выра-
жением, как в случае канонической функции распределения, но в
него входит множитель У — число молекул в системе.
Теперь выведем производную:
(Ж-4-{(1йТ И =(2™W/a V (-4
Следовательно,
Л'(2лт/Л2)3''2 V { - 4 р-Уа)
U—U(0) =-----------------=ЗУ/2В. (20.2.10)°
k 1 (2nm/V)3^ V (р/г) r v 1
Таким образом, если известна 0, сразу можно получить величину
внутренней энергии образца идеального одноатомного газа, со-
держащего У частиц.
Пример. Цакоаа теплоемкость одноатомного газа при постоянном объеме?
Метод. По определению Cr — (dUjdT)у. Предположим, что р=1/й7. Последнее
уравнение дает выражение для U.
Ответ. Cv = (ЗАГ/2) d (1/ty/dT « (ЗМ/2) d (kT)jdT =
= (3/2)Аг£=(3/2)л/?.
Поэтому мольная теплоемкость Cv,m = 3/?/2 = 12,47 Дж/(К-моль).
Комментарий. Это почти точно согласуется с экспериментальной величиной для
одноатомных газов. В более сложных молекулах вклад в U и, следовательно, в
Су вносят другие виды движения. Это рассматривается в следующей главе.
Параметр 0. Наиболее удовлетворительный метод отождествления
0 с 1/kT исходит из статистического определения энтропии (разд.
20.3). Однако уже сейчас можно сделать тот же самый вывод с
помощью расчета, который дает дополнительную практику в обра-
щении с функцией распределения и показывает, как эту функцию
можно использовать для расчета других величии, кроме внутрен-
ней энергии.
Мы рассчитаем давление некоторой общей системы и получен-
ное выражение применим к идеальному газу. Этот статистический
184
Часть 2, Структура
Рис. 20.5. Расчет средней силы, действующей в х-направлеини.
расчет дает р в виде функции параметра £, и затем, Чтобы свя-
зать р с температурой, можно использовать уравнение идеального
газа p=nRT[V.
Рассмотрим образец вещества с размерами X, У и Z и объемом
V. В свое время (т. 1, разд. 2.1 и 2.2) при обсуждении силы, рабо-
ты и энергии было выяснено, что работа по изменению размера X
на величину dX равна
dw = — FxdX,
где Fx — сила, противодействующая . Этому изменению. Поэтому
количество работы, проделанной по изменению размеров i'-го чле-
на ансамбля, составляет
dw^—F^dx или Fj.=—(dwJdX),
где F( — сила, противодействующая изменению i-ro члена. Проде-
ланная работа вызывает повышение энергии, следовательно,
dwL=dEi (рис. 20.5). Поэтому силу, противодействующую измене-
нию, можно записать так:
F^—tdEildX}.
Подобные выражения будут для у- и г-компонепт. Средняя сила,
с которой члены ансамбля действуют в х-направлении, равна
I
Давление, оказываемое образцом, — это сила на единицу площа-
ди, и, поскольку площадь грани образца, перпендикулярной х-на-
правлепию, равна }'Z= V/X, получается выражение для среднего
давления, оказываемого на эту грань системы:
/\=-
20. Статистическая термодинамика, Концепции 185
В термодинамическом пределе отношение характеризует
наиболее вероятное распределение, т. е. п’/Ж [уравнение (20.2.2)].
Следовательно, давление связано с функцией распределения фор-
мулой
рй =--(—X/1/Q) 2 ехр (—pFf) =
t
=(X/pVQ) 2 (д/дХ) ехр (-р£г) = (20.2.11)
: г
= (X/pVQ)(dQ/ax).
Это выражение применимо к любому веществу и дает давление,
оказываемое на плоскость, перпендикулярную оси х. В газе или
жидкости ориентация этой плоскости не имеет материальной осно-
вы и, как показано ниже, во всех направлениях давление одина-
ково.
На данном этапе применим это уравнение к идеальному газу
из Л' атомов. Каноническую функцию распределения можно заме-
нить па молекулярную функцию распределения с помощью соот-
ношения Q—q!4Nl', Это приводит к выражению
рх (Х/рИ9) (дд/дХ). (20.2.12)°
Подставляя упрощенную формулу поступательной функции рас-
пределения [уравнение (20.2.8)], получаем
=N V-} |<2“'л«>' =
=(У.¥/рК) (1/V) (дУ/дХ).
Поскольку V — XYZ, производная равна YZ, а это произведение
при умножении на X дает V. .Следовательно,
Такой же результат получается для ру и рг; это подтверждает, что
давление одинаково во всех направлениях, и поэтому опустим ин-
дексы х, у и z.
В случае идеального газа р = nRT/V. При сравнении этого вы-
ражения с последним сразу находим величину р:
/?-- нД>' пД>7'П, __ д/
откуда 4- < ’А
^ = L/RT—l/kT, (20.2.13)
Хотя это соотношение выведено для идеального газа, оно приме-
нимо к любому веществу (что будет подтверждено При рассмот-
рении энтропии); таким образом, с этого места мы можем писать,
что р — 1/kT, где бы параметр р нс появился.
186
Часть 2. Структура
Интерпретация функции распределения. Теперь можно получить
некоторое представление о смысле функции распределения. Возь-
мем молекулярную функцию распределения
9 = V ехр(—(20.2.14)
i
и начнем с рассмотрения интервала величин q.
Когда температура систему близка к абсолютному нулю, каж-
дый член суммы, за исключением одного, имеет форму е~х с
х—>оо, и поэтому все они стремятся к пулю. Исключение состав-
ляет член с е/ = 0, потому что тогда {eykT) =0 для всех значений
Т, включая 7’=0. Так как при Г = 0 уцелел только один член сум-
мы, равный 1, то, следовательно,
при^Т—0 9 = 1.
В качестве другого предельного случая рассмотрим такой, когда
Т настолько велика, что для каждого члена суммы ey/feT—>4). По-
скольку при х=0 е~х=1, каждый член суммы равен 1. Поэтому
сумма равна числу молекулярных состояний, которое в общем
бесконечно велико:
при Т--* оо q--► оо.
(В некоторых случаях система имеет только определенное число
энергетических уровней; тогда функция распределения характе-
ризуется верхним пределом, равным числу состояний.)
Как видно, функция распределения дает указание на число
состояний, доступных системе при данной температуре. При абсо-
лютном нуле доступно только основное состояние и 9=1; При са-
мых высоких температурах доступны фактически все состояния и
поэтому q стремится к бесконечности. Для промежуточных тем-
ператур необходимо учесть, что относительное число молекул в
состоянии е; равно (1/9)ехр(—Ej/kT). Это важное заключение
можно вывести несколькими способами. В простейшем из них от-
мечается, что средняя энергия может быть представлена в двух
формах. Если относительное число молекул в энергетическом со-
стоянии j равно Pj, а их действительное число равно Лу, то сред-
няя энергия е одной молекулы составляет
/ i
а внутренняя энергия всего образца точно в N раз больше:
20, Статистическая термодинамика. Концепции
187
Рис. 20.6. Расположение молекулярных энергетических
уровней, используемое для расчета молекулярной функции
распределения по уравнению (20.2.16).
Зс
2Е
О
Выражение для U дается также уравнением
-------7~ (20.2.10):
Е U— U(Q)^—N(l/q)(dq/dfyv=;
~ \ = (^/Я) 8/ exp (- pej.
Сравнение этих двух уравнений приводит к простому выражению
для относительного числа молекул в состоянии еу при температуре
Т. Оно называется распределением Больцмана:
расареде.гение Больцмана: Р}-=(1 /q) exp (— E^kT). (20.2.15)
Это выражение постоянно использовалось в предыдущих главах,
а здесь мы впервые дали его формальный вывод.
Способ, по которому функция распределения указывает на чис-
ло доступных состояний, можно проиллюстрировать рассмотрени-
ем простой молекулы, в которой расстояние с между энергетиче-
скими уровнями одинаково (рис. 20.6). Для такой молекулы вы-
ражение для зависимости функции распределения от температуры
выводится очень просто, поскольку
q — 1 -f-e-2₽е_|_ ... — 1 -(-(е-₽8) -}- (е~Ре)а-|_....
представляет собой геометрическую прогрессию, которую можно
точно просуммировать (нам знакомо разложение (1—х) ~J= (1+:
+ х + х2 + ...), и поэтому
е-Р8)-1. (20.2.16)
Следовательно, относительное число молекул в состоянии с энер-
гией в/ дается соотношением
p^Jl-e-Рг) е-рЕ/, (20.2.17)
и, подставив соответствующее значение температуры, можно по-
лучить численные значения как q, так и Р,. Это сделано на рис.
20.7, где показана заселенность состояний при разных температу-
рах и распределение молекулярных состояний отмечено численны-
ми значениями q, вычисленными по уравнению (20.2.16). При
очень низких температурах q близка к единице, как уже предпола-
галось, и единственной значимой заселенностью является низшее
состояние. При повышении температуры заселенность низшего
состояния понижается и постепенно заселяются более высокие
состояния. При этом значение функции распределения становится
188
Часть 2. Структура
3,0
9=1,05
ре=1,0
9 = 1,58
<7 = 1,99 д = 3,86
Рис, 20,7. Заселенность молекул
лирных состояний при разных
температурах.
больше единицы, и поэтому
оно дает общее представле-
ние о степени заселенности
уровней. При очень высоких
температурах заселенность
охватывает множество со-
стояний (на диаграмме некоторые из них значительно заселены)
и функция распределения имеет соответственно большее значение.
Пример (вопрос 9). Найдите относительное число молекул иода в основном, пер-;
вом возбужденном и втором возбужденном колебательных состояниях при ком-!
натиой температуре.
Метод. Колебательные энергетические уровни находятся на постоянном расстоя-
нии друг от друга, равном 214,6 см-1, и поэтому они соответствуют только что
описанному типу системы. Возьмем е = 214.6 см~' и отождествим } с колебатель-
ным квантовым числом п. Нулевое значение энергии можно определить как со-
стояние с к-0.
Ответ. Для будущих задач, так же как и для этой, будет удобным выразить
kT в см-'.
/6ТЛ, 1 - (! ,38] - ]023 Дж/К)Х(298 К) __
(й//Лс)гааК-(6 626 ]0_31 дж.с)Х(2г998 10'“ см/с) “
= 207,2 см-1.
В данном случае
= (214,6 см-’)/(207,2 см-’) = 1,036.
Тогда относительные заселенности даются выражением
Рп = (1 — гН3®) е-п₽е = 0,645ехр (—1,036п).
Отсюда /*0=0,645, /*,=0,229, 7*2—0,081 и т. д.
Комментарий. Связь в молекуле иода не сильная и атомы тяжелые, как ре-
зультат этого расстояние между колебательными энергиями мало, и даже при
комнатной температуре значительно заселено несколько колебательных уровне,'!.
Отметьте, как величина функции распределения (1,550) отражает это неболь-
шое, но значимое расширение заселенности.
20.3. Статистическая термодинамика и второй закон
До этого раздела мы рассматривали связь между функцией
распределения и концепцией внутренней энергии по первому за-
кону. Мы видели, что U можно рассчитать из функции распреде-
ления и ее температурной зависимости. Утверждение о том, что
30. Статистическая термодинамика, Концепции
18*
функция распределения содержит всю термодинамическую инфор-
мацию, можно принять только в том случае, если эту функцию*
можно использовать для рассмотрения энтропии — основной кон-
цепции второго закона.
Теплота, работа и статистическая энтропия. Когда в гл, 5 (т. 1)'
вводилась энтропия, она была представлена как мера распреде-
ления энергии. Поскольку функция распределения есть также ме-
ра распределения энергии, разумно предположить связь между
ними. Природу такой связи можно выявить, рассматривая связь
между внутренней энергией U, энергетическими уровнями и их.
заселенностью.
Для этого запишем общее выражение для изменения внутрен-
ней энергии системы. Так как
£7 — С/ (0) — (1ДГ) 2 Д" со,
i
го изменение dl' может возникнуть или вследствие модификации
энергетических уровней системы (так что £), энергия некоторого
члена ансамбля, изменяется на dEt), или в результате изменения
числа членов ансамбля, имеющих энергию Ei (так что И; изменя-
ется на dtti). Поэтому наиболее общим изменением будет
dV=. -(-(J/Д') (20.3.1).
£ Z
Из термодинамики известно, что для обратимого изменения
dC/=^o6p+rfwo6p; (20,3,2>
следовательно, должна быть связь между этими двумя величина-
ми. Такая связь устанавливается при условии, что при изменении
размера системы ее энергетические уровни изменяются; они не
изменяются, когда система нагревается (рис. 20,8), В то же вре-
мя при нагревании системы изменяется распределение энергии, но
каждый уровень сам по себе остается на том же месте. Отсюда
следуют равенства
^обР = (]/-Л (20.3,3>
Г
и
dt£,06p=(1M") (20.3,4>
i
Мы видим, что обратимый переход теплоты соответствует пере-
распределению заселенностей среди фиксированных энергетических
Уровней, а обратимая работа соответствует изменению самих уров-
нейг в то время как заселенности остаются теми же. Поскольку
(20,3.5>
190
Часть 2. Структура
Рис, 20.8. Различие между теплотой п работой.
приравниванием этих двух выражений для dq^p можно устано-
вить соотношение между изменением энтропии и изменением рас-
пределения в ансамбле:
TdS = (l/jT)^Eidnl. (20.3.6)
i
Теперь покажем, что такое же выражение для dS можно вы-
вести, если энтропию определить выражением
статистическая энтропия-. S—(k/j\cj hi W, (20.3.7)
взяв, как всегда, термодинамический предел :—н<х>. В этом вы-
ражении W— число распределений энергии в каноническом ан-
самбле, которое совместимо с данной общей энергией.
Подтверждение того, что это определение S приводит к урав-
нению (20.3.6) в термодинамическом пределе, начинается с уста-
новления факта, что УТ пренебрежимо мало отличается от числа
способов (веса) достижения наиболее вероятного распределения
(величина W'* была рассчитана в разд. 20.1). Изменение S
возникает вследствие изменения Ы'ЙТ'*, а изменение 1п^*—
вследствие изменения распределения энергии в ансамбле. Это зна-
чит, что можно написать
^5=(^/ж) d (In ®”*) = (£/ж) 2 (^ In ®“*/дП() dtij.
i
Поскольку число УТ* является весом наиболее вероятного распре-
деления, оно удовлетворяет уравнению (20.1.10):
(d 1л <Г*/дп£)4-а—=0,
20. Статистическая термодинамика. Концепции
19ft
Следовательно, 2t/i?i = 0, и последнее уравнение превращается в-.
as=(k/,r) 2 (-«+Р*ч)=(Фал 2 E A
I i
Это выражение точно такое же, как уравнение (20.3.6), если па-
раметр fj определить как 1/АГ.
Соотношение dS = dqO6p/T решает вопрос об идентичности тер-
модинамической и статистической энтропий, и термодинамический
критерий спонтанного изменения dS^>Q можно интерпретировать,
как сдвиг ансамбля к более вероятному распределению. Кроме
того, при понижении температуры системы величина и, сле-
довательно, S уменьшаются, так как с общей энергией совместимо
меньшее число распределений. При предельном значении Т, абсо-
лютном нуле, так что 1п^* = 0, поскольку с общей энер-
гией, равной нулю (относительно энергии нулевого уровня), сов-
местимо лишь одно распределение (каждый член ансамбля на
низшем уровне). Таким образом, когда Т достигает нуля, S стано-
вится равной нулю. Это согласуется с третьим законом термодина-
мики, по которому энтропия всех совершенных кристаллов при аб-
солютном нуле имеет тенденцию принимать одинаковое значение
(т. 1, стр. 163), однако мы его усовершенствуем для случая несо-
вершенных кристаллов (это будет сделано на стр. 225).
Выражение для энтропии можно перевести в очень простую
полезную форму, сделав несколько изменений и введя функцию
распределения. Поскольку нас интересует термодинамический пре-
дел, число членов ансамбля должно быть достаточно велико, что-
бы можно было применить приближение Стирлинга. Это приводит к
S = (W)
= (А/^Г)рп^1—
=(А/Л (ыГ In ыГ - — 2 лiln ni + 2 =
i i
— In .J’ — inilij.
i
Две части этого выражения можно- объединить, заменив на
произведение 1пЛ" и тогда
5 = k 2 In — (ъ/ЛГ) In ttj} =
r
= — A 2 (п.дЛ 1п(и;АЛ-
c
192
Часть 2. Структура
Отношение П;/Ж — относительное число членов ансамбля, имею-
щих энергию Е,-; обозначим его через pi. В этом случае энтропия
дается кратким выражением
(20.3.8)
i
Последнее выражение можно развить еще дальше и добиться
цели-—выразить энтропию через функцию распределения. В урав-
нении (20.3.7) W с пренебрежимо малой ошибкой можно заменить
на вес наиболее вероятного распределения, поэтому тц/Ж дается
выражением (20.1.13) для наиболее вероятного оаспределсния:
Pi==n;/^fi(l/Q)exp(—₽Ег), (20.3,9)
и так как
1пА = —₽Ег—InQ,
то
E = lnQ)--Ap^-t/(0)14-AlnQ,
С !
поскольку .сумма рг равна единице, а сумма ргЕ; — средняя энер-
гия, или, в термодинамическом пределе, внутренняя энергия U.
Уже было установлено, что p^l/AT, отсюда получается чрез-
вычайно важное выражение для механико-статистической энтро-
пии:
S-=[[/ —l/(0)|/T4-felnQ. (20.3.Ю)
В случае газа, состоящего из невзаимодействующих частиц, кано-
ническую функцию распределения можно заменить на ^А7№, в ре-
зультате чего получим
5 = [I/—V (0)]/.7'-| - kN In q — k in №,
и поскольку число молекул в образце N = nL почти всегда доста-
точно велико и приближение Стирлинга применимо, что выраже-
ние превращается в
5 = [17 — U (0)]/Т+«Е In q ~k (N in N—ЛГ)
или
5- [t/-U (0)]/T4-nE (In q — In N 4-1) (20l3.1 I)3
Мы уже видели, как рассчитывать внутреннюю энергию из функ-
ции распределениями поэтому последнее выражение представляет
собой точное уравнение для энтропии через молекулярные свойства.
Пример (вопрос 11), Рассчитайте колебательный вклад в энтропию иода при
комнатной температуре.
Метод. Колебательные уровни размещены равномерно на расстоянии 214,6 см-1
друг от друга. Поэтому используем у, определяемую уравнением (29.2.6) и рас-
20. Статистическая термодинамика. Концепции 193
считанную в предыдущем примере. Используем Sm=l[47m—Um(Q)]!T-{-Rlnq из
уравнения (20.3.10).
Ответ. Поскольку <?= (1—е-^Е)-1 и (7—У(0) =—N(llq) (dq/dtyv, имеем U—1/(0) =
=nLe[e"Pe/(l—е-^е}]. Иа предыдущего примера f}e= 1,036 и </=1,550. Следова-
тельно,
4/т — (7m (0) = (2,567 кДж/моль) х (0,355) X (1,550) — 1,413 кДж/мапь,
— Um ШТ = 4,741 ДжДК моль),
/? 1п</= [8,314 Дж/(К‘моль)] In 1,550 =
-=3,644 Дж/(К-М<эль).
Отсюда
5га = 4,741 ДжДК’Мйль) 3,644 Дж/(К-моль) = 8,385 Дж/(К’МОль).
Комментарий. Почему мы не использовали TV! в S? Потому что мы не имели
дела с поступательным движением; <V подсчитывается один раз, а не везде.
Этот вопрос рассмотрен в следующей главе.
Энтропия одноатомного газа. Очень простой пример только что
выведенного выражения можно получить, рассчитывая энтропию
газа, состоящего из невзаимодействующих атомов. Поскольку ато-
мы не взаимодействуют, для нахождения U и S можно использо-
вать функцию распределения <?, Если бы мы рассматривали взаи-
модействующие атомы, то мы должны бы были сохранить канони-
ческую функцию распределения. Это гораздо более сложная
проблема, и выводы будут дапы в гл. 23.
Для атомов единственной степенью свободы является поступа-
тельное движение, поэтому молекулярная функция распределения
имеет вид
q=^2mnf/i^Vt
а внутренняя энергия дается уравнением (20.2.9) как
U—= — (V/^(d?/dfl)v = 3Ar/2p^3ntf772. (20.3.12)°
Следовательно, энтропия определяется выражением
S = 3nRl2~\-nR {In (2nmkT /№fh- V — InnL-j-1) =
=n/?{lne3/2 + ln(2jimAT/A2)3^ V—InnL-J-lne} =
=,?№ {(2wnkT/h^ VinL), (20.3.13) °
которое представляет собой уравнение Сакку ра —Тетрода для
энтропии одноатомного идеального газа. Поскольку газ иде-
альный, V можно заменить на nLkTjp, что приводит к выражению
для энтропии как функции температуры и давления:
S=n£ln (kTlp)}. (20.3.14)°
Связь с материалом части 1 (т. 1) видна из того, что изменение
энтропии при изотермическом расширении идеального газа от Vi
13—242
194
Часть 2. Структура
до V; дается разностью двух выражений типа уравнения (20.3.13)
AS--S|—Si=n/?lnaVE—п/?1пйИ1 —nRln^/V;),
где аУ— величины под знаком логарифма. Это выражение точно
такое же, как выведенное на основании термодинамических аргу-
ментов, с помощью которых можно рассчитывать лишь изменение
термодинамических величин (т. 1, стр. 146).
Пример (вопрос 13). Какова величина поступательной энтропии газообразного
иода при комнатной температуре и давлении 1 атм?
Метод. Поступательная энтропия может быть получена, если рассматривать Г»
как одноатомный газ (прибавлением вращательного и колебательного вкладов
учитываются «внутренние» степени свободы).
Ответ, т(U = 2х 126,9х(1,6605-!0-2? кг) = 421,4-Ю"3’ кг,
так что
„ 2пх(4,2-14 10~» кг)Х(1,38Ы0-« Дж/К)х(298К)
2TtmkT/h^ =-----------(6,62бТо-31 Дж-Ер-------------=
= 2,482-КГ-3 м-2.
Из уравнения (20.3.14) следует, что
Зга = [8.314 Дж/(К-моль)] 1п[е',/зх(2,482.1033 м-^’^х
X (1,38М0-^ Дж/К)х(ЖК)((1,01325- 10s Н/мв] =
= [8,314 Дж/(К'М0ль)] In (1,935-10s) = 177,8 Дж/(К-моль).
Комментарий. Вращательную энтропию при комнатной температуре можно рас-
считать, как описано в следующей главе. Она равна 65,9 Дж/(К-моль). Общая
энтропия молекулы в газовой фазе при 1 атм и 298 К является суммой поступа-
тельного [177,8 ДжДК'Моль), колебательного [8,4 Дж/(К-моль) и вращатель-
ного [65,9 Дж/(К моль)] вкладов, т- е. равна 252,1 Дж/(К-моль).
Приложение 20.А, Неопределенные множители
Предположим, что мы желаем найти максимум (или минимум)
величины некоторой функции f, которая зависит от нескольких пе-
ременных Xi, Хг, ..., хп. Когда эти переменные претерпевают не-
большое изменение х,—>-Х; + Йх£, функция изменяется от f до
f+6f, где
б/=2 (df/dx^Sxi.
j=i
В точке стационарности (максимум или минимум) небольшое из-
менение Xi не приводит к изменению f, следовательно, 6f = 0;
д
е точке стационарности-. V (6Д'6хг)£х( = 0. (20.А.1)
20. Статистическая термодинамика. Концепции 155
Если все бх; независимы, то все произвольны, и поэтому по-
следнее условие удовлетворяется, когда все (dfldxi) —0. Это обыч-
ный путь определения максимума или минимума функции.
В некоторых представляющих интерес случаях изменения
бх; не являются независимыми. С такими примерами мы Сталки-
вались в данной главе: заселенности и(- менялись, но мы точно
знали, что их сумма г^Ч-паЧ-... оставалась равной фиксированно-
му числу Поскольку уже не являются независимыми, про-
стое решение (20.А.1) больше неприменимо. Мы получаем решение
следующим путем.
Пусть ограничение, связывающее переменные, будет записано
в виде
g(xltx2,...,x„)=0.
Например, в данной Главе мы имели Щ-р п2-|-... = Ж, что в нужной
нам форме записи превратитсяв£(1ц, 1Ь, ...) =—Ж-Иц-рп2 +... = 0.
Это ограничение верно всегда, и поэтому g остается неизменным
при изменении хи
п
^g=^^g/dXi)5Xi=0. (20.А.2)
(=[
Умножим это уравнение за некоторый параметр Л и прибавим к
уравнению (20.А.1):
[(df/tfxi)4-A.(dg/dxI)]&cI=0. (20.А.З)
i=l
Получается уравнение для одного бх, например для бхп, выражен-
ного через все остальные бх,-. Все эти 6x/(i= 1,2,..., п—1) незави-
симы и выбираются произвольно, так как на систему наложено
только одно ограничение. (Например, требование 6пп=—6Ttt —
—Sila—...—6ttn-j гарантировало бы, что ограничение на примере,
данном в этой главе, выполнялось бы несмотря на то, что 6ttj,
..., 6ttn_j выбирались бы произвольно.) Но здесь есть хитрость:
параметр Л, выбирается произвольно, причем так, чтобы коэффи-
циент в уравнении (20.А.З) обращался в нуль, г. е. А. выбира-
ется так, чтобы
(df/dxJ+4dg/dx„)=0. (20.А.4)
Тогда уравнение (20.А.З) превращается в
П—[
^=1
13*
196
Часть 2. Структура
Теперь п— 1 изменений fixj действительно независимы, и поэтому
решением является
(3/:/аг£)-|—% ^=0, г = 1,2,.,., я—1.
Но уравнение (20.А.4) имеет точно такую же форму, как это Урав-
нение; следовательно, стационарные величины находятся решени-
ем уравнения
(df/dx^-^/.fdg/dx^—O, все xt. (20.А.5)
В тексте был приведен пример с двумя ограничениями (и двумя
неопределенными множителями и или а и —£).
Множители никогда не могут остаться неопределенными. Один
подход состоит в решении уравнения (20.А.4) вместо введения его
в общую схему минимизации. В данной главе мы использовали
альтернативную процедуру, оставив Z. неопределенным до тех пор,
пока не было рассчитано молекулярное свойство, для которого
уже известен ответ. Так, коэффициент а был найден, когда мы
удостоверились, что rti+ »2+„.=Ж (а не какому-либо другому
числу), а коэффициент £ — на более поздней стадии при расчете
pV и приравнивании его к известной величине (nRT).
Приложение 20.Б. Квантовая статистика
Возможность приведения Q к q1'1 [уравнение (20.2.6)] зависит
от различимости частиц. Соответствующее уравнение можно полу-
чить, рассматривая молекулы как имеющие доступные энергети-
ческие уровни .Ej, вэ, ..., а обшее состояние системы как состоящее
из «1 частиц в состоянии ei, из Пй частиц в состоянии ег и т. д.
Тогда суммирование по состояниям системы будет эквивалентно
суммированию по числам занятости каждого молекулярного уров-
ня, допуская условие raI+«2’+.„=A. Общее число способов полу-
чения определенного распределения, в котором А частиц распре-
делены так, Что /11 из них занимают уровень Si, «й — уровень е2
и т. д., будет A/’l/zii 1/г21 (это как раз является выражением сорти-
ровки по ларям, использованным при обсуждении распределения
энергии среди различимых членов ансамбля, стр. 173), Поэтому ка-
ноническая функция распределения для системы из А различимых
молекул имеет вид
<2 = 2* (№/«1’«й!.-.)ехр{—-------------)}. (20.Б.1)
все п
где сумма берется по всем Пц п2,..., а «штрих» указывает, что
2л;- = А. Это выражение представляет собой точное разложение в
20. Статистическая термодинамика. Концепции 197
ряд (e'₽ei +е ₽®2 +..)*, что можно еще раз проверить, используя
разложение бинома
г '
и поэтому для различимых частиц
• ’)'V — 4N- (20.Б.2)
Однако идентичные частицы неразличимы, если они свободно
меняют свое местоположение, и тогда только что выведенное вы-
ражение непригодно. Когда частицы являются обычными молеку-
лами, они подчиняются определенному виду статистики, известной
как статистика Бозе—Эйнштейна, которую можно представить
следующим образом.
Когда ni молекул занимают энергетическое состояние ei, не
имеет значения, какие это молекулы из общего числа посколь-
ку молекулы неразличимы, существует только один индивидуаль-
ный способ получить П[ молекул в ei, «а — в р.а и т. д. Поэтому ка-
ноническая функция распределения имеет вид
<2вв (АГ) = S' ехР {-₽ («14 +«з4+ ••)]. (2О.Б.З)
все п
где BE означает Бозе — Эйнштейн, и сумма ограничена так, что
Sn/ = /V’. Это то же самое выражение, что и уравнение (20.Б.1), за
.исключением отсутствия весового множителя.
Величина Q очень сильно зависит от У, так как при увеличе-
нии /V на очень небольшую величину в сумму входит гораздо
больше слагаемых. Это дает ключ к искусному способу ее вычис-
ления. Если Q(;V) умножить на резко уменьшающуюся функцию
N, то произведение будет функцией с резким пиком. Поэтому за-
писываем произведение Q(N')e-a^', где e-“v'—резко уменьшаю-
щаяся функция. Если искусно выбрать а (функцию /V), то это
произведение даст резкий пик прй A|,z = /V’. Если произвести сумми-
рование по всем Л/7, то существенный вклад в сумму дадут только
слагаемые вблизи /V'=/V:
N’
dN представляет собой интервал величин АГ, которые вносят су-
. щественный вклад в сумму. Следовательно,
InZ — InQ(AT)—аУ-|-1пбЛГ, (20. Б. 4)
и в сравнении с другими, много большими членами последним
слагаемым можно пренебречь. Величина Z (главная функция рас-
198
Часть 2. Структура
) (2 П2) • • • =
пределения) выражается следующей формулой:
Z = S S' е^"ехр{—₽(«i8i+«2e2d---)) =
Л' все»
= S е~“"еХР { -₽ (”>е1 +«2е2Н-)] =
все г»
= S exp [—Ifa+ps^-Hoc + psajraa-l-]1. (20.Б.5)
все ft
Первая строчка получается из уравнения (2О.Б.З). Вторая строчка
означает, что, поскольку теперь берется сумма по всем величинам
N, ограничение Sn/ = N можно отбросить; суммирование по неогра-
ниченным величинам ni, Пг,... охватывает все значения, получен-
ные суммированием по ограниченным величинам с последующим
суммированием по ограничениям. Последняя строка — это просто
Z — g—(^+Pgl) П
»i «а
=( 1 ) (“j _a-<^+₽E21 ') ” ’ ’ (20.Б .6)
так что
In Z = — 2 In {1 ~e-(“+₽9). (20.Б.7)
l
Следовательно,
lnQBE=atf— 21n |J—(20.Б.8)
- /
Теперь мы должны связать а с каким-то свойством. Эта вели-
чина была выбрана так, чтобы Q(jV')e-«v' имела резкий пик при
JV'='V. Берем логарифмы и ищем максимум
(d/djV') {InQ (jV*) — aN'1^0 при JV’=jV.
Отсюда (заметим, что а есть функция :V)
{dlnQ(fl)/djV}—a(JV)=O.
Поскольку InQ (jV} = InZ(а)+аЛ\ получаем
dInQ/cW=a4-(da/дДГ) {dllnZ-j-aA/'J/da} — a-Ufda/cW) {(dinZ/daJ-j-Ar}.
Комбинация этого выражения с условием предыдущего уравнения
дает
(dinZ/da)-f-tf=O.
20, Статистическая термодинамика. Концепции
199
Но
lnZ=-21n[l-^a+^}.
I
поэтому после небольших преобразований получаем
*=£( ^ъ_т}:
/
(20.Б.9)
это условие можно использовать для определения а.
Хотя эти выражения можно преобразовывать и дальше, мы
имеем возможность проверить только их высокотемпературный,
классический предел. Когда доступны многие состояния, в уравне-
ние (20.Б.9) входит много слагаемых, поэтому каждое из них
должно быть мало. Это значит, что е“ + ₽в? ^>1. Когда это так,
^sh^rSe^s,,'=’"“
J У
Таким образом, в данном пределе
а = 1п(д/Л7). (20.Б.10)
Далее, в этом же пределе с применением приближения Стирлинга,
InQ да ctJV+ «
у
да ЛИп (д/Д^ф-ЛГ да
даЛИпд—ЛИпЛ/ф-Л/да
да fag"—In jV! (20.Б.11)
Именно таким путем мы получаем тождество Q^qN/№-, использо-
ванное в тексте.
Статистика Бозе—Эйнштейна — не единственный вид стати-
стики, применяемой в квантовой теории. Мы знаем, что такие ча-
стицы, как электроны, подчиняются принципу Паули (т. 1,
разд. 14.2), который запрещает в каждом состоянии находиться
более чем одному электрону. Ясно, что в этом случае статистика
Бозе — Эйнштейна не подходит, поскольку она позволяет любому
числу электронов занимать данное состояние. В статистике Фер-
ми-Дирака уровень может быть пустым или он может содер-
жать одну частицу (например, один электрон). Каноническая
функция распределения будет такой же, как в уравнении (20.Б.З),
поскольку электроны неразличимы, но здесь появляется еще одно
ограничение, а именно: подобно всем другим ru, щ = 0 или 1. Мож-
200
Удсгй 2. Структура
.2 — у gi—“+P£i) пЛ (у g—<<х+₽е2)
по использовать приведенный выше метод и показать, что уравне-
ние (20.Б.6) просто заменяется на
n2 j . . —
П1-=0,1 «2=0,1
= (1 -|_е-!<Х+₽Е1>) (1 -l-e-la + fel). . . ,
так что
1Л Qfd=aN+2 !п {1 +^(“+3г') 1 • (2OJ5.12)
Г
Можно легко проверить, что в классическом пределе Qfd = <ДО!,
т. е. для неразличимых частиц снова получается фактор У!.
Квантовая статистика существенна при обсуждении низкотем-
пературных явлений, таких, как сверхпроводимость и сверхтеку-
честь; интерпретация электрических и термических свойств элек-
тронов металлов основана на статистике Ферми — Дирака.
Литература
Casser R. Р, И., Richards IT. G., Entropy and energy levels, Clarendon Press,
Oxford, 1974.
Bent H. A., The second law, Oxford University Press, New York, 1965.
McClelland B. A, Statistical thermodynamics, Wiley, New York, 1973.
Nash L. K., Elementary statistical thermo dynamics, Addison-Wesley, Reading,
Mass., 1968.
Relf F., Fundamentals of statistical and thermal physics, McGraw-Hill, New York,
1965.
Hill T. L., An inlroduction to statistical mechanics, Add!son-Wesley, Reading,
Mass., 1960.
Wilks J., The third law of thermodynamics, Clarendon Press, Oxford, 1961.
Guggenheim E. A., Boltzmann’s distribution law. Interscience, New York, 1955.
Mayer 1, E., Mayer M. G., Statistical mechanics, Wiley, New York, 1940.
Tolman R, C., The principles of statistical mechanics, Clarendon Press, Oxford,
1938.
Miinster A., Statistical mechanics. Springer, Berlin, 1974.
Задачи
20,1, Ансамбль из 5 членов имеет среднюю энергию е04-е. Каждый член может
обладать энергией где j—целое число. Сколько имеется распределений,
соответствующих равномерному рассредоточению энергии по всем членам ан-
самбля?
20.2. В первой задаче мы рассчитали вес одного вида распределения, но имеет-
ся несколько других, более важных распределений. Составьте таблицу, озагла-
вив ее колонки энергией членов (от е0 до + 5е). и напишите под ними все рас-
пределения, совместимые со средней энергией eo-f-e. Начните, например, с. 4, 0,
0, 0, 0, 1 (только один член может иметь энергию ел-(-5е, а остальные четыре
должны тогда иметь энергию е0). Найдите вес каждого распределения [исполь-
зуя уравнение (20.1.5)]. Какое распределение наиболее вероятно?
20, Статистическая термодинамика. KoHijertijUu 201
20.3. В случае ансамбля с 9 членами мы достигаем области, когда средние чис-
ла как раз начинают иметь термодинамический смысл, но еще могут быть точно
вычислены. Составьте таблицу распределений для Л5=9 со средней энергией ец-1-
+е, как и выше.
20.4. Перед тем как определить веса распределений в ансамбле е Л°=9, взгляни-
те на составленную вами таблицу и догадайтесь (найдя «экспоненциальную»
форму), какое из распределений окажется наиболее вероятным (с наибольшим
весом). Теперь рассчитайте веса всех распределений (это не такая уж продол-
жительная задача, как могло бы показаться с первого взгляда) и найдите наи-
более вероятное распределение.
20.5. Наиболее вероятное распределение характеризуется температурой системы.
Но какова температура системы, которую мы сейчас рассматриваем? Она долж-
на быть такой, чтобы дать среднее значение энергии ео+е, причем системе раз-
решено иметь только энергию en-pje. Покажите, что температура системы мо-
жет быть получена иа графика зависимости In (ily/rts) от j, где И/— заселен-
ность наиболее вероятного распределения (в термодинамическом пределе). При-
мените этот метод к 9-членному ансамблю, чтобы увидеть, насколько близко
он соответствует термодинамическому пределу (считая график прямой лини-
ей). Примите, что расстояние между уровнями системы равно 50 см-1. Какова
температура?
20.6. «Температура» — это параметр, который имеет смысл только для наиболее
Вероятного распределения, и мы не должны ожидать, что для других распреде-
лений получится прямая линия. Выберите два других распределения для 9-член-
кого ансамбля, одно близкое к наиболее вероятному, а другое далекое от него,
я постройте график зависимости ln(tl//iu) от j.. Прямые лииии должны быть
очень плохими.
20.7. Рассчитайте каноническую функцию распределения для 9-членного ансамб-
ля при 160 (результат задачи 20.5), 155 и 165 К- Затем найдите градиент
dQjdfi при 160 К и таким образом подтвердите, что средняя энергия (которая
при этой температуре, как мы знаем, равна е0+е) составляет —(dlnQ/dJl). Все
несоответствия обусловлены вычислением температуры ансамбля.
20.8. Центральное место в статистической термодинамике занимает молекуляр-
ная функция распределения q '[уравнение (20.2.5)]; оставшиеся задачи посвя-
щены исключительно ей. В данной главе мы рассмотрели некоторые способы
расчета и преобразования молекулярной функции распределения, а в следующей
главе обсудим ее химические применения. Прежде всего сформулируйте, д.тя
какой системы при переходе от у к q существенно включение множителя ]/ЛЧ:
а) образца газообразного гелия, б) образца газообразной окиси углерода, в) то-
го же образна окиси углерода, цо замороженной до твердого состояния, г) па-
роп воды, д) льда, е) электронного газа, ж) электронного газа в металле.
20.9. Атом аргона заключен в кубический ящик объемом V. Какова его посту,
нательная функция распределения при а) 100 К, б) 298 К, в) 10 000 К и г) 0 К,
если сторона" ящика равна 1 см?
20.10. Обычно используемая форма поступательной функции распределения при-
менима только при огромном числе доступных энергетических уровней. Когда
обычное выражение становится неприменимым и мы должны обратиться к точ-
ному суммированию [уравнение (20.2.5)]? Какую температуру должен иметь
аргон в последнем примере, чтобы функция распределения уменьшилась до 10?
Какова точная величина функции распределения при этой температуре?
20.11. Несколько видов функции распределения можно рассчитать прямым сум-
мированием экспоненциальных слагаемых, используя спектроскопические данные
По молекулярным энергетическим уровням. Вначале рассмотрим атом теллура,
который имеет несколько низколежащих возбужденных состояний. Найдите
электронную функцию распределения для атомов при а) 298 К и б) 5000 К на
основе следующих данных из атомной спектроскопии; основное состояние (5-крат-
ио вырождено), 4751 см-1 (3-кратно вырождено), 4707 см-1 (однократно вы-
рождено), 10559 см-1 (5-кратно вырождено).
202
Часть 2, Структура
20.12. Каково относительное количество атомов теллура: я) в основном состоя-
нии и б) в состоянии при 4751 см-1 при двух температурах, указанных в пре-
дыдущей задаче?
20.13. Большинство молекул, с которыми мы встретимся, имеют электронные
состояния, расположенные по анергии настолько выше основного состояния, что
для описания их термодинамических свойств бывает необходимо рассматривать
только последнее. Однако имеется несколько исключений, одним из которых
является случай NO, для которого электронно-возбужденное состояние лежит
лишь на 121,1 см"1 выше основного состояния. Как возбужденное, так и ос-
новное состояния дважды вырождены. Рассчитайте и отложите на графике
электронную функцию распределения для NO от нуля до бесконечной темпера-
туры. Каково распределение заселенностей при комнатной температуре?
20.14. Какова средняя электронная внутренняя анергия молекулы NO при ком-
натной температуре? Ответьте на этот вопрос, вначале вывела выражение для
U при любой температуре (используйте пайдевную выше функцию распределе-
ния), а затем подставив величину 7=298 К.
20.15. Молекула Л имеет колебательные энергетические уровни при следующих
волновых числах относительно нулевого уровня: 213,30; 425,39; 636,27; 845,93;
1054,38 см*"1. Рассчитайте колебательную функцию распределения точным сум-
мированием выражения для q при а) 100 К и б) 298 К.
20.16, Канве относительные количества молекул иода находятся в основном и
двух первых возбужденных колебательных состояниях при двух температурах,
указанных в предыдущей задаче?
20.17. Какова средняя колебательная энергия молекулярного иода при этих двух
температурах?
20.18. В магнитном поле электронный спин может принимать две ориентации с
энергиями Найдите выражение для электр он-едино вой функции распре-
деления и средней энергии и графически выразите их в виде функции прило-
женного поля При 298 и 4 К- Какова относительная заселенность спиновых уров-
ней при этих двух температурах?
20.19. Система с тремя уровнями, такая, как ядро азота в магнитном поле, мо-
жет быть выражена в простой компактной форме. Выведите выражение для
функции распределения и средней энергии ядра азота (/=1) в магнитном поле
при 4 К. За нулевое значение энергии примите состояние Л1;—0.
20.20. Теперь можно проанализировать особенности системы с двумя уровнями.
Предположим, что некоторым искусственным образом мы сумели инвертировать
заселенности спиновых уровней (методы для этого действительно существуют,
и в них используются лазеры). Тогда а высшем энергетическом состоянии будет
больше электронов, чем в низшем. Это неравновесная ситуация, при которой
распределение заселенностей далеко от наиболее вероятного. Тем Це менее фор-
мально мы еще можем выражать отношение заселенностей через простой пара-
метр, который нам не запрещается назвать «температурой». Покажите, что эта
температура должна быть меньше абсолютного нуля. Какая «температура» соот-
ветствует; а) инверсии равновесной заселенности при 298 К. б) инверсии равно-
весной заселенности при 10 К и в) общей инверсии, когда все электроны будут
в высшем спиновом состоянии?
20.21, При каких обстоятельствах разрешается или имеет смысл говорить об от-
рицательных температурах для системы с тремя уровнями?
20.22. Теперь перейдем к соотношениям между энтропией и функцией распреде-
ления. Сначала кратко суммируем роль канонической функции распределения
Q. Какова энтропия канонической системы па примере 9-члепного каноническо-
го ансамбля из задачи 20.3?
20.23. Эта система рассматривалась также па примерах 3-, 5- и 20-членного ан-
самблей (стр, 172), Мы знаем также, как термодинамическая энтропия (в пре-
деле Л3—связана со структурой системы [уравнение (20,3.7)], Покажите,
что энтропии 3-, 5-, 7-, 9- и 20-членных ансамблей можно перевести в термоди-
намическую величину.
20. Статистическая термодинамика. Концепции
203
20.24. Рассчитайте электронный вклад в мольную энтропию атома теллура при
а) 298 К и б) 5000 К; используйте данные задачи 20.11.
20.25, Рассчитайте электронный вклад в мольную энтропию молекулы NO при
а} 298 К и б) 500 К; используйте данные задачи 20,13,
20.26. Рассчитайте колебательный вклад в мольную энтропию молекулы иода при
а) 100 К и б) 298 К; используйте данные задачи 20.15,
20.27. Выведите зависимость мольной энтропии коллектива независимых Элект-
ронных спинов от напряженности приложенного магнитного поля. Какая будет
предсказанная вами энтропия спинов при а) В=0, б) В=оо и какая будет рас-
считанная вами энтропия?
20.28. Предположим, что атомы аргона в количестве 1 моль вначале жестко за-
креплены в своих положениях, а затем им позволено двигаться свободно. Как
изменится энтропия?
20.29. Какова эвтропия разбавленного электронного газа при а) 298 К и
б) 5000 К? Возьмите Р=10 дм3.
20,30. Подтвердите, что статистико-термодинамическое выражение для энтропии
одноатомного идеального газа правильно объясняет зависимость энтропии от его
а) давления и б) температуры.
21 Статистическая термодинамика.
Развитие концепций
Изучаемые вопросы
После тщательного изучения этой главы вы сможете;
1. Рассчитать поступательный вклад в молекулярную функцию
распределения [уравнение (21.1.4)].
2. Определить число симметрии молекулы (стр. 207).
3. Рассчитать вращательный вклад в молекулярную функцию
распределения при низких температурах [уравнение (21.1.5)] и
высоких температурах [уравнения (21.1.7) и (21.1.8)].
4. Рассчитать колебательный вклад в функцию распределения
[уравнение (21.1.10)].
5. Рассчитать электронный вклад в функцию распределения
[уравнение (21.1.12)].
6. Связать с функцией распределения внутреннюю энергию
[уравнение (21.2.1)], энтропию [уравнение (21.2.2)], энтальпию
[уравнение (21,2.5)], функцию Гельмгольца [уравнение (21.2.3)]
и функцию Гиббса [уравнение (21.2.7)].
7. Связать давление с функцией распределения [уравнение
(21.2.4)].
8. Рассчитать среднюю энергию различных видов (мод) моле-
кулярного движения (стр. 216).
9. Сформулировать теорему равного распределения (стр. 219).
10. Использовать принцип равного распределения для расчета
теплоемкости (стр. 220).
11, Рассчитать теплоемкости при низких температурах [уравне-
ние (21.3.17)],
12. Определить остаточную энтропию и рассчитать ее для неу-
порядоченной системы (стр, 225).
13, Связать константу равновесия реакции с молекулярными
функциями распределения реагентов и продуктов [уравнение
(21.3.25)].
14. Использовать спектроскопические данные для расчета кон-
станты равновесия химической реакции (стр. 228),
Введение
В предыдущей главе были даны основы статистической термо-
динамики. Мы видели, Что центральной концепцией является
функция распределения Q и, когда она известна, можно рассчи-
21. Статистическая термодинамика. Развитие концепций 205
тать термодинамические функции U и 5. Функция распределения
осуществляет решающую связь между термодинамикой, спектро-
скопией и квантовой теорией, поскольку спектроскопические дан-
ные или молекулярные параметры, такие, как массы и силовые
постоянные, могут быть использованы для расчета ее величины
при некоторой температуре, а затем из Q могут быть вычислены
V и 5,
В данной главе методы статистической термодинамики будут
применены к рассмотрению проблем, значимых для химии. В ча-
стности, будут найдены способы расчета функции распределения
из набора спектроскопических данных или молекулярных констант.
Рассматриваемые здесь проблемы включают расчет теплоемко-
стей, энтропий и констант химических равновесий; в процессе об-
суждения будут открыты новые пути к пониманию смысла этих
параметров,
21.1. Как рассчитать функцию распределения?
В этом разделе мы ограничимся рассмотрением системы не-
взаимодействующих молекул, такой, как идеальный газ. Однако
мы возьмем молекулы, имеющие внутреннюю структуру, и поэто-
му энергию системы будем подразделять на поступательную, вра-
щательную, колебательную и электронную. Поскольку молекулы
не зависят друг от друга, <3 = ^ЛГ/Л^1; следовательно, можно сосре-
доточить свое внимание на расчете молекулярной функции распре-
деления q:
? = 2ехр(-рв;), $=l/kT. (21.1.1)
/
Прежде всего разделим энергию индивидуальной молекулы на
вклады различных мод движения. Если общую энергию молекулы
записать в виде суммы
В;=епост евращ Ц-ь™ 4-8элск,
то молекулярная функция распределения сведется к произведению
функций распределения для каждой моды:
<7- ехР<—₽е‘-₽£г — pev—0С) =
(пост,, вращ,,
кол,т элск.
состояния)
=2 ехр Sехр S ех₽ р®’) Sехр =
ПОСТ. вращ. кол. элск.
= (21.1.2)
Эта факторизация (разложение на сомножители) является лишь
приближением, поскольку моды движения не совсем независимы
206
Часть 2. Структура
Друг от Друга, однако многим целям она удовлетворяет. Ее боль-
шое преимущество состоит в том, что разные вклады в функцию
распределения можно исследовать независимо.
Поступательный вклад. Поступательная функция распределения
была выведена на стр. 181 [уравнение (20.2.8) ]
=(2jwi/AsP)3/s V= (2amkT/h*fh V. (21.1.3)°
Она применима к молекуле массой tn в сосуде объемом V. Внеш-
ний вид этого выражения, которое будет нам часто встречаться,
можно упростить, написав
Ч*=тУ, x=(2mnkT/h2f*. (21.1.4)’
Заметим, что величина q* повышается до бесконечности При уве-
личении температуры, так как становится доступным чрезвычайно
большое число квантовых состояний. Даже при комнатной темпе-
ратуре для молекулы кислорода в сосуде объемом 1DD см3
q^ 2- 103S.
Вращательный вклад. Один из методов расчета qT состоит в под-
становке экспериментальных значений вращательных энергетиче-
ских уровней в выражение для вращательной функции распреде-
ления и в последующем численном суммировании экспоненциаль-
ных членов. Одно лишь нужно помнить: некоторые вращательные
состояния могут соответствовать одинаковой энергии. Например,
в случае двухатомной молекулы состояния отличаются квантовы-
ми числами 7, Л1, однако энергия зависит только от J (стр. 72).
Поскольку для любого данного J существует 2/4-1 величии М,
для каждой величины 7 имеется 2/4-1 состояний с одинаковой
энергией. Тогда вращательная функция распределения запишется
как
9Г = 2 «хр (—£ (2J +!) ехр (—рЕ,). (21.1.5)
дм J
Пример (вопрос 3). Рассчитайте вращательную функцию распределения HCI при
комнаткой температуре.
Метод. Используем уравнение (21.1.5), определяя одно слагаемое за другим.
Вращательные энергетвческие уровни даются формулой £j=fiJ(/4-l), а В—
= 10,59 см-1.
Ответ. Нам потребуется знать, что B/kT=(U),5Q см-*)/(2О7,2 см'1) =0,051 И. Со-
ставим следующую таблицу: J 0 12 3 4 5 6 7 В 9 10
J (J-J-1) 0 2 6 12 20 30 42 56 72 90 ПО
е-Л/+1)В/4Г 1 0,903 0,736 0,520 0,360 0,216 0,117 0,057 0,025 0,010 0,004
274-1 13 5 7 9 И 13 15 17 19 21
Затем вычисляем сумму, как требуется по уравнению (21.1.5). Она равна 19,9,
следовательно, при данной температуре <7Г = 19,9.
21. Статистическая термодинамика. Развитие концепций
207
Комментарий, Заметим, что около десятка /-уровней заселено в значительной
степени. Позднее мы встретимся с приближением ч’ я* kT/B. В данном случае
это дает уг ~ 19,6, что очень хорошо согласуется с точной величиной н требует
затраты значительно меньшего труда.
Надо остерегаться включать в сумму слишком много состоя-
ний. Это тонкий нюанс; он связан с тем наблюдением, что, хотя гсте-
роядерные двухатомные молекулы (типа НС1) могут находиться
в состоянии с любым I, отдельная гомоядерная Двухатомная мо-
лекула может быть только с четными значениями J или только с
нечетными. К счастью, эту сложность легко учесть, если темпера-
тура так высока, что заселено много вращательных уровней, и
поэтому мы сосредоточим свое внимание на этом случае.
Вращательные энергетические уровни многих молекул лежат
достаточно близко друг к другу; следовательно, при комнатной и
более высокой температуре большое их число заселено. При ком-
натной температуре kT200 см-1, а вращательные постоянные
для молекул НС1, 12, СН4 и СО2 равны соответственно 10,6; 0,04;
5,2 и 0,39 см-1. Если мы не будем рассматривать более легкие мо-
лекулы, чем эти, и температуру ниже комнатной, то с хорошим
приближением будет заселено так много близко расположенных
вращательных состояний, что сумму в уравнении (21.1.5) можно
заменить интегралом.
На этом этапе мы должны принять во внимание тот факт, что
не все значения J могут реализоваться для каждой молекулы. Ес-
ли занято большое число квантовых уровней, то проблему можно
решить классически, и это приведет к четкому и простому реше-
нию. В случае гомоядерной двухатомной молекулы вращение на
180° меняет местами два эквивалентных ядра, и так как новая
Ориентация неотличима от первоначальной, то, чтобы избежать
подсчитывания неотличимых Ориентаций, мы должны разделить
интеграл на 2:
гомоядерные двухатомные молекулы: qr л; (2/4-1) exp {—fJEj] dJ.
о
В случае гетероядерной двухатомной молекулы вращение на 180°
приводит к отличимой ориентации (НС1—и поэтому мно-
житель не появляется. Эти выводы можно объединить, написав
со
9Г«(1/О) J (2J + l)exp(-p£j)dJ, (21.1.6)
о
где о — число симметрии, которое принимает значение 1 для гете-
роядерных двухатомных молекул и 2 — для гомоядерных двухатом-
ных молекул.
208
Часть 2. Структура
Такая же проблема возникает в случае мрЛекул других типов
симметрии, и для того чтобы исключить нейравильный подсчет
энергетических уровней, интеграл нужно откорректировать е уче-
том числа симметрии. В случае линейных молекул применимо по-
следнее выражение, потому что их вращательные свойства такие
же, как у двухатомных молекул: для СО2 о = 2, но для OCS о=1.
В более сложных молекулах неотличимыми могут быть более чем
две ориентации. Например, в то время как для Н2О ст=2, для
NH3 сг=3, а для СН2 = Сп2 о=4. Иногда число симметрии может
быть очень большим: например, в бензоле имеется 12 эквивалент-
ных ориентаций и о=12 (о способе определения сг см. раздел «За-
дачи») .
Можно взять интеграл в уравнении (21.1.6) и получить точное
выражение для вращательной функции распределения линейных
(включая двухатомные) молекул. Как найдено на стр. 72,
Е1=В1 (J +1), где В—вращательная постоянная молекулы, свя-
занная с ее моментом инерции соотношением B=h2!21. Следова-
тельно,
(f ъ (I/о) J (2J4-1) ехр {-0BJ (J+1)1 dJ.
о
Хотя этот интеграл и выглядит более сложным, его можно взять
без особых усилий, учитывая, что его можно также переписать как
qT ж (—1/(70В) [(d/dJ) ехр (— 0BJ (7ф-1)}] dj.
i
Интеграл производной функции есть сама функция, я поэтому
/ ж (—1/стрВ) ехр J— 1/стрВ.
о
Поэтому приближенная форма вращательной волновой функции
для линейной молекулы имеет вид
if ж kTlBo^IkTIffa, (21.1.7)
Приближенная вращательная функция для молекул других ти-
пов может быть найдена таким же путем, приводящим к
<7Г « (nVi/O) {(2/дАТ/Л1) (2/сАТ/Й1)]1'*, (21.1.8)
где 1а, 1в и 1с — их моменты инерции.
Пример (вопрос 3). Определите вращательную функцию распределения для Эте-
на при комнатной температуре.
Метод. Используем уравнение (21.1.8) с 0=4. Вращательные постоянные А =
=4,828 см-1, В==1,0012 см-1 и С—0,8282 см-1. Начинаем с выражения qT через
2/. Статистическая термодинамика. Развитие концепций
20»
А, В а С, где A^h^Hhc (Л выражено в волновых числах), а для В и С выра*
жения аналогичны.
Ответ. <?*' = (гЛ'а/о) [(kT/A/гс} (kT/Bhc)
При комнатной температуре АГ/йс=207,20 см-1, и поэтому gr=-j- (207,20 см'1)3''3 х
Х(л/4,828Х1,001X0,8282 см-а)1?2=660,8.
Комментарий. Этен достаточна большая молекула, его энергетические уровни
близки друг к другу; как следствие, даже? при комнатной температуре занято
много вращательных уровней.
Общий вывод из этого раздела состоит в том, что молекулы а
большими моментами инерции имеют большие вращательные
функции распределения. Это отражает близость друг к другу вра-
щательных уровней в больших, тяжелых молекулах и заселенно-
сти многих состояний при нормальных температурах.
Колебательные вклады. В случае двухатомных молекул колеба-
тельная функция распределения может быть определена подста-
новкой измеренных колебательных энергетических уровней в вы-
ражение
9"=2еХр(-М). (2!.1.9>
1
В многоатомных молекулах каждая нормальная мода колебания
(разд. 17.4) имеет свои собственные независимые энергетические
уровни,и поэтому
9V^V(I)^(2)...,
где ф(Л)— функция распределения Л’-й нормальной моды, Это
прямой метод для вычисления функция рас-дреде,тения из инфор-
мации, которую дает колебательная спектроскопия.
Если колебательное возбуждение не слишком велико, то при-
ближенно можно считать, что колебания являются простыми гар-
моническими, Тогда колебательные энергии даются выражением
Е1~ + / = 0,1,2,,., где к>= & — силовая постоянная
для колебания, а р— приведенная масса; это выражение обсуж-
далось в разд. 17-4. Если энергии измеряются от нулевого уровня,
то разрешенной величиной является Е] ^jhb>. Тогда использование
приближения гармонического осциллятора приводит к простому
выражению для колебательной функции распределения:
?У=2 ехР (~Мю₽) =2
I г
С этим рядом мы встречались на стр. 187 (что не случайно: ле-
стничное расположение уровней на рис. 20,6 точно такое же, как
14—242
.210
Часть 2. Структура
для простого гармонического осциллятора). Ряд можно суммиро-
вать таким же образом, что дает
^=4--------v-т-йГ* (21.1.10)
’ 1 — exp (—fitupj* 4 '
Когда молекула имеет несколько нормальных мод, общая колеба-
тельная функция распределения образуется умножением выраже-
ний, аналогичных этому, но с соответствующей величиной коле-
бательной частоты каждой моды.
Пример (вопрос 4). Волновые числа трех нормальных мод молекулы волы рав-
ны 3656,7. 1594,8 и 3755,8 см-1. Какова колебательная функция распределения
при а) 298 К. б) 1500 К?
Метод. Используем уравнение (21.1.10) для каждой моды. При 298 К kT/hc=
=207.2 см-1, а при 1500 К — 1043 см~1.
Ответ. Составляем следующую таблицу:
Мода: V1 *1
<Й, см-1 3656,7 1594,8 3755,8
17,65 7,70 18,13
(/i»/bT)isoo К 3,506 1,529 3,601
(298 К) 1.0000 1,0005 1,0000
<?; (1500 К) 1,031 1,277 1,028
Общая колебательная функция распределения' q=q‘(q'iq^. При 298 К ip'—1.0005.
при 1500 К ?v = 1.353.
Комментарий. Колебания молекулы воды имеют высокую частоту, и при ком-
натной температуре фактически все молекулы находятся в основном колебатель-
ном состоянии. Даже при 1500 К отклонение от этого невелико.
Во многих молекулах колебательные частоты так высоки, что
йкф;>1. Например, низкая колебательная частота для метапа
равна 1306 см-1, и поэтому при комнатной температуре йкф~6.
Обычно валентные колебания связей С—Н лежат в интервале
2050—2960 см-1, следовательно, при комнатной температуре
14. В этих случаях экспонентой ехр(—йсар) в знаменателе
выражения для Q4 можно пренебречь (например, е-®^ 0,002), и
колебательная функция распределения просто равна единице:
q4 ~ 1.
Электронный вклад. Расстояние между электронными энергетиче-
скими уровнями обычно велико, и поэтому все экспоненты
ехр(—ре®) очень малы, за исключением основного состояния, для
которого Еа=0. Таким образом, в большинстве случаев
4* = 1. (21.1.11)
Важным исключением из этого правила является случай, когда
атомы имеют вырожденное основное состояние (т. е. когда не-
сколько состояний атома имеет одну и ту же энергию). Если су-
21. Статистическая термодинамика. Развитие концепций
211
Рис. 25.1. Электронные состояния оксида азота(П).
щестнует g состояний, соответствующих одной энергии, так что
основное состояние g-кратно вырождено, то функция распределе-
ния имеет величину
<f=g. (21.1.12)
Например, щелочные металлы имеют дублетные основные состоя-
ния, и поэтому при отсутствии магнитного поля g~2 (что соот-
ветствует двум равным по энергии ориентациям неспаренного
электронного спина).
Некоторые молекулы тоже имеют электронно-вырожденные Ос-
новные состояния, а другие — возбужденные электронные состоя-
ния, очень близко расположенные к основному состоянию. Одним
из примеров является оксид азота (II) NO с электронной конфигу-
рацией ....л1 (см. диаграмму вт. I, стр. 521; расположите на ор-
биталях 15 электронов). л-ЭлектрОн может вращаться вокруг
межъядерпой оси двумя способами — по и против часовой стрелки,
и его спин может быть направлен так же, как его орбитальный
момент, или противоположно ему. Это дает четыре состояния, по-
казанные на рис. 21.1. Эти четыре состояния делятся на две груп-
пы; если орбитальный и спиновый моменты параллельны, то энер-
гия молекулы несколько больше, чем когда они противоположны
(из-за спин-орбитального расщепления; т. I, разд. 14.3). Первые
Два состояния составляют уровень, обозначенный йПз£, а два
Другие состояния —уровень 2П1/а. Эти обозначения не должны
нас беспокоить: важно то, что электронные энергетические уровни
NO такие, как показано на рис. 21.1; низшее состояние дважды
14*
212
Часть 2. Структура
вырождено, а возбужденный уровень лежит только на 121 см-1
выше него, и этот уровень также дважды вырожден.
Электронная функция распределения NO может быть получена
следующим образом. Если обозначить энергии двух уровней вщ
и ез/2 и записать ei/t=0 и ез/2 =6, то получим
< = 2 SJ ехР (—₽8/) =2 ехР (—₽8;/а)+2ехр (—₽е3/2) -=2(1 + е-₽6).
I (21.1.13)
При абсолютном нуле z/e = 2, поскольку на нижнем уровне находят-
ся только два состояния; при очень высоких температурах до-
стигает 4. При комнатной температуре 1/Р^200 см-1, и поэтому
<711?^2,8. Некоторые следствия такого поведения рассмотрены в за-
даче 21.31.
Общая функция распределения. Теперь мы имеем функции рас-
пределения для всех отдельных видов движения; для удобства они
собраны в пригодном для практического применения подразд.
21.1.А. Общая функция распределения есть их произведение.
21.1.А. Вклады в молекулярную функцию распределения
1. Поступательное движение
q' =tV, т = (2^mkT/hz)3 *^,
T(m-3) = I,8800.I026 (T,K)3''s Л#2,
ql°/N =т (kT/1 атм)=0,02562 (Т, Х?,ЛМ?*.
2. Вращение
а) Линейная молекула:
<f =(1/а) {2IkTjhz)=kTlBa =
=0,6952 (I/ст) (Т, К)/(В, см-1).
б) Нелинейная молекула:
<f =(л |/а/о) [(27Л£Г/Й2) (2/в£77Й2) (2/сйТ/й*)]1^ =
=(л*А/0) (kT}4L (I /АВС)1/® =
= 1,0270(1/ст)(Г,К)%/[(А, см-'ХЯ, см-’)(С, см"’)}1/®.
3. Колебание
^ = 1/П—ехр(—Йи/И1)].
4. Электронный вклад
где g — вырождение основного состояния (обычно термически до-
ступно лишь одно электронное состояние). При высоких темпера-
турах вычисляется точно.
2/. Статистическая термодинамика. Развитие концепций
213
Для удобства укажем некоторые значения kT, выраженные в
СМ“1:
При Т = 298 К kT/hc = 207,20
При Т = 500 К kT/hc = 347,65
При Т= 1000 к kT/hc — 695,30
При Т = 1500 К kT /he — 1042,95
Для двухатомной молекулы Общая функция распределения име-
ет вид
j—(21.1.14)
Соответствующее выражение для более сложных молекул можно
записать аналогично, перемножив частные функции, перечислен-
ные в подразд. 21.1. А. Общая функция распределения является
приближенной, так как предполагается, что врашательные уровни
близки друг к Другу, а колебания являются гармоническими. Эту
приближенность можно избежать, точно вычислив сумму на осно-
вании спектроскопических данных.
21.2. Как рассчитать термодинамические функции?
Б нашем распоряжении имеется метод расчета двух принци-
пиальных термодинамических функций — внутренней энергии U и
энтропии S;
l/-t7(O)=-(I/Q)(at?/3₽)v=-(<31nQ/S₽)v, (21.2.1)
S=[t7—L/(0)]/T4-AlnQ. (21.2.2)
Б случае независимых молекул эти уравнения упрощаются путем
подстановки Q=qN (различимые частицы) или Q = qNiNl (нераз-
личимые частицы). Все другие термодинамические функции мож-
но вывести из U и S; следовательно, мы имеем способ расчета
всех термодинамических функций. Действительные соотношения
можно установить на основании некоторых результатов, получен-
ных в части 1.
Прежде всего выразим через функцию распределения функцию
Гельмгольца А. Поскольку
А-U-TS
и при Т = 0 К А = £7(0), замена U и S сразу приводит к выражению
A-A(0)—ftTinQr (21.2.3)
Установив это соотношение, можно связать давление, оказываемое
системой, с ее канонической функцией распределения, используя
214 Часть 2. Структура
ту же аргументацию, что и в разд. 6.2 (т. 1):
р=—(дА/дУ)? =kT(dlnQ/dV)?. (21.2.4)
Этот результат совершенно общий и может быть использован для
реального газа, так же как для идеального газа (или жидкости).
Поскольку это уравнение связывает давление с объемом и темпе-
ратурой, оно дает очень важный способ определения уравнений
состояния реальных газов и соотнесения их с молекулярными взаи-
модействиями (которые должны быть введены в Q). То, что при
этом получается правильное уравнение идеального газа, можно
проверить подстановкой Q = qN/N\i
p^kT(}/Q) (dQ/dV)T—NkT (1/q) (dq/dV)T.
Так как от объема зависит только то это уравнение
преобразуется к виду
p=nLkT(I/9V7V) idV)T =
=ni?T(ilfqt)(dqt/dV)r-
=nRT
=nRT/Vt
что и требуется доказать. (Это можно рассматривать как еще
один способ вывода соотношения p = l/feZ.)
Поскольку как U, так и р могут быть связаны с Q, с ней мож-
но также связать энтальпию Н, а следовательно, найдеп способ
расчета этого важного термодинамического параметра.
Из определения H=U + pV, учитывая, что при 1 = 0 К 77= 17 (0),
получаем
77—77 (0) = — (д In (W)v + kTV (д In Q/dV)T. (21.2.5)
Набор термодинамических параметров можно завершить нахож-
дением способа расчета функции Гиббса G [уравнение (5.3,5),
т. 1]. Поскольку
G = 77—TS = U 4- р V—TS,
получаем
G —G щ Q + kTV (5 In Q/dV% (21.2.6)
В случае идеального газа (однако состоящего из молекул, име-
ющих структуру) это выражение значительно упрощается. Во-
первых, pv в определении G можно заменить на пЯТ‘, тогда вве-
дение выражений для U и S приводит к
G - G (0) kT In Q 4-л7?Т.
21. Статистическая термодинамика. Развитие концепций 215
Во-вторых, замена Q на qN/Nl дает
G —G (0) =—NkT In kT In ЛИA-nRT =
=— nRT In q+kT {N In N —N)+nRT,
так что
G—G(0)=— nRT\n{q/N), (21.27)°
где N — nL. Следовательно, молекулярную функцию распределения
можно непосредственно использовать для определения функция
Гиббса — основной функции химической термодинамики.
Приведенные важные соотношения собраны в подразд. 21.2.А.
21.2.А. Статистико-термодинамические соотношения. Через кано-
ническую функцию распределения Q:
Cr-[/(O)=-(5InQ/50)v,
3 =[£/—t7(0)J/T+ftlnQ,
p=kT {д In Q/dV)T,
И—Н (0) - — (д In Q/dp)v + kTV (д In Q/dV)T,
A-A(0)=~ kTinQ,
G—G (0)=— kT In Q + kTV (6 In Q/dV)T.
В случае Q^q:'/N\ (свободные, неразличимые частицы; внешние
моды движения, например поступательные)
U—и (0) - — N {din qffi)v,
S = [L/—t7{0)]/T+nR(In <7 — In V4-1).
G—G (0) =— nRT In {q/N).
В случае Q=qN (внутренние моды, например вращение)
I/1 _t/1 (0) = _Л’ (6 in q'/dfyv,
SI = [t7_L/(0)]/T + nRln</,
G1 — G'(0) = —
Вообще для неразличимых, невзаимодействующих частиц
Q = tfW/N I = [(<Г№1
Отсюда
So6m^s^5if t/общит д
Обозначения,- ex—внешний, i — внутренний.
216
Часть 2. Структура
Пример (вопрос 6). Рассчитайте величину функции свободной Энергии =
= [Gm(T)—Ят(0)]/Т для воды при 1500 К.
Метод. Gm(T} можно рассчитать по уравнению (21.2.7); тогда F0(T) =
=—R]n(q/N). Для стандартизации величины возьмем А = 7. и RT/[1 атм).
Колебательная функция распределения была рассчитана в примере на стр. 210.
Используем для вычисления других вкладов уравнения из подразд. 21.1.А.
Вращательные постоянные равны 27,8778; 14,5092 н 9,2869 см-1.
Ответ. Для поступательного вклада (подразд. 21.1.А)т= (0,02562) Х(18,015)37®Х
X(1500)67а, поэтому для поступательного вклада (<7%V)1 = 1,707 -10s. Для вра-
щательного вклада
qr = -TT X (1042,95 см~*)3/1 X (1/27,8778Х
X 14,5092-9,2869 см"3/72 = 274,8.
Мы уже видели, что для колебательного вклада
<?v= 1,353.
Поэтому общая функция распределения
9°/.V = (1,707 -10s) X (274,8) X (1,353) = 6,346 1010.
Следовательно,
Fo(1500) = —R In (tf/N) =
= —18,314 Дж/(К-моль)] In (б ,346 IO10) =-
= —206,8 Дж/(К'Моль).
Комментарий. Термохимическая величина составляет —211,71 Дж/(К-мольу.
Разница обусловлена предположением, что энтропия твердой воды при абсолют-
ном нуле равна нулю (см. ниже).
21.3. Применение статистической термодинамики
Теперь любое термодинамическое свойство можно вывести ис-
ходя из энергетических уровней молекул; термодинамика и спект-
роскопия объединены. В данном разделе мы укажем, как произ-
водить расчет для четырех важных проблем: принципа равного-
распределения, теплоемкости, остаточных энтропий и констант рав-
новесия. Этими вопросами применение статистической термодина-
мики никоим образом не ограничивается; другие проблемы будут
рассмотрены в части 3.
Средние энергии и принцип равного распределения. Очень часто
полезно знать среднюю энергию различных молекулярных мод,
когда молекула составляет часть системы при температуре Т. Ее
можно получить, используя выражения для термодинамической
внутренней энергии и факторизацию общей молекулярной функции
распределения:
.£/_[/(О)--^1/9)(ад5) =
=^(i/^V)(a7l9r9v/3p)=
21. Статистическая термодинамика. Развитие концепций_217
Поскольку [U—U(0y\{N — сумма средних энергий всех мод, т.е._
U-U (0) -N (<е‘) + (сг) + (ev) + (ве)}, (21.3.!)’
имеем
<Ет> = -(1/<Г)(^т/ф), (21.3.2)*
где m = t, г, v или е.
Средняя поступательная энергия молекулы может быть найде-
на из поступательной функции распределения. Чтобы показать
применяемый для этого метод, рассмотрим вначале одномерную
систему, а затем перейдем к трем измерениям. Поступательная
функция распределения для молекулы массой m выражается как
(2лт/рй2) */» А' (стр. 181), поэтому средняя поступательная
энергия равна
<е‘> =— (Pfta/2nm)^ (глт/й2)1/» ( —l- = 1/20 = ~~ АТ.
Тот же расчет для случая трех измерений приводит к
(et}=_|_Ar, (21.3.3)°
В обоих случаях Средняя энергия не зависит от массы молекулы
и размера сосуда. [Это согласуется с утверждением (стр. 174),
что внутренняя энергия идеального газа не зависит от занимаемо-
го им объема: (dU/дУ)т = 0-]
В классической механике кинетическая энергия Т частицы мас-
сой m связана с компонентами ее скорости соотношением
т=4'm^+4'm^+4'm^
Очевидно, что Среднюю энергию можно получить, если среднюю
величину каждого квадратичного члена принять равной -уАГ. Это
является обшим результатом, как мы увидим при рассмотрения
врашательиой и колебательной энергий молекулы.
Средняя вращательная энергия линейной молекулы равна
<ег>=—(1/^) (^г/5₽),
где
ef =2 <27+ П ехР (7+ ОЬ
т
Когда температура низкая, это выражение нужно вычислять по-
членно. В случае гетероядерной двухатомной молекулы все значе-
ния J вносят вклад в сумму; тогда
cf = 1 4- Зе-^в + 5е-б₽в -j-;
218
Часть 2, Структура
следовательно,
< (1 +3е-г^ + 5*-®^+ •••} ’ (21.3.4)
н средняя энергия падает до пуля, когда Т—>Z). Если температура
так высока, что занято много вращательных уровней, то мож-
но использовать приближенную форму функции распределения
(стр. 208):
<er) =-(cr/^/2Z) (27/оЙ3) (~1/ра)= 1/р=АТ. (21.3.5)
Классическое выражение для вращательной энергии линейной
молекулы имеет вид
(2-компоненты здесь нет, потому что она имеет нулевой момент
инерции относительно линии, соединяющей атомы), где I — мо-
мент инерции, а а>х и а у— компоненты угловой скорости. Видно,
что в классическом пределе средняя энергия kT может быть полу-
чена, если каждый из двух квадратичных членов равен Экс-
траполируя этот результат на нелинейные молекулы, для которых
разрешены три моды вращения, получаем, что средняя вращатель-
ная энергия должна быть равной Это можно подтвердить с
помощью уравнения (21.1.8).
Средняя колебательная энергия в классическом пределе (кото-
рый означает высокую температуру) может быть предсказана из
классического выражения для энергии
E=T+V=-i-m^+4^S’
в котором присутствуют два квадратичных члена. Отсюда можно
предположить, что средняя энергия равна 2(~kT). Но верно ли
мы приписываем равные количества энергии потенциальной и ки-
нетической энергиям? На этот вопрос можно ответить.точным рас-
четом. Точная функция распределения для гармонического осцил-
лятора имеет вид
(21.3.6)
и поэтому, так как
(д(Г/д&
21. Статистическая термодинамика. Развитие концепций
219
получаем
<ev>=Aco ( fecafi) 1 ’ (21.3.7)
(1 —ехр(—йш₽) J ' '
Это точный результат. Когда температура так высока, чтойш₽<С1,
он упрощается до
<sv> [ 1 ~ -] 1/Р-Йй) 1/0.
к 1 *— * ’t’ fttiJp J
Это подтверждает, что средняя энергия в классическом пределе
равна kT.
Эти выводы можно суммировать, сформулировав
принцип равного распределения: когда можно не учитывать
квантовые эффекты, средняя энергия любого квадратичного чле-
на в выражении для энергии имеет одну и ту же величину, рав-
ную -тт^Т-
Отметим важность ограничения этого простого правила случаями,
где можно пренебречь квантовыми эффектами. Когда эго ограни-
чение не существенно, принцип равного распределения дает очень
простое правило для суммирования общей средней энергии моле-
кулы и, следовательно, для оценки U. Мы встретимся с его при-
менением в следующем разделе.
Можно также ответить на другой вопрос: каков шанс найти
молекулу, энергия которой значительно превышает среднюю энер-
гию? Это можно точно предсказать, используя распределение
Больцмана [т. I, «Введение», уравнение (1.2)], однако иногда
полезно иметь приближенное представление о разбросе энергии.
В статистике разброс распределения часто измеряется средне-
квадратичным отклонением данного свойства. В настоящем случае
нас интересует энергия, и среднеквадратичное отклонение энергии
определяется как
220
Часть 2. Структура
Для краткости мы будем называть бе флуктуацией энергии мо-
лекул в образце. Если вдруг все молекулы находятся в одинако-
вом состоянии энергии е*, то средняя энергия была бы е*, а сред-
нее квадратов энергии е*2; в таком случае бе = О. Когда молекулы
распределены по многим состояниям с разной энергией, среднее
квадратов энергии не будет больше равно квадрату средней энер-
гии (рис. 21.2), и поэтому бе не будет равно нулю. Большая ве-
личина бе соответствует широкой заселенности уровней, а малая
величина — узкой заселенности.
Квадрат средней энергии равен (1/<?)2(dqjdfyy. Среднее этих
квадратов рассчитывается следующим образом:
<ег> =2 р^=Д1/<7) 2£?ехр(_ре;) =
= (V?) 2 (<W2) exp (—₽ej) =
i
= (!/?) (dWV
Следовательно, флуктуация энергии связана е функцией распре-
деления соотношением
бе> = (1/9) (<^/^-(1/^ (<W)v. (21.3.9)
Применение последнего выражения можно проиллюстрировать
на примере гармонического осциллятора. Поскольку точная функ-
ция распределения дается уравнением (21.3.6), прямое дифферен-
цирование приводит к
(21.3.10)
Зависимость этой функции от температуры показана на рис. 21.3;
отметим, что при абсолютном нуле бе = О, потому что тогда все
молекулы находятся в своих основных состояниях и лет разброса
энергии. При высоких температурах экспоненциальные члены мо-
гут быть разложены в ряд, и последнее выражение превраща-
ется в
бе яг 1/р=АТ.
Видно, что флуктуация энергии равна средней энергии, т. е. с уве-
личением средней энергии возбуждения флуктуация растет.
Теплоемкость. Теплоемкость при постоянном объеме Cv равна
Cv = (dU/dT)v. (21.3,11а)
Это выражение впервые встретилось в т. 1 [разд. 2.3, уравне-
ние (2.3.2)]. Иногда удобно записать Су в виде производной цо Р;
ехр — — Лир
1 — ехр (—АшВ)
бе
21. Статистическая термодинамика. Развитие концепций 221
Лир
Рис, 21.3. Средняя энергия и
флуктуация энергии набора
гармонических осцилляторов
при разных температурах.
так как р=1ДТ, то dp='
= —dTjkT21 и последнее
уравнение можно также за-
писать в виде
cv=—(i/feT®) (at//dp)v.
(21.3.116)
Внутренняя энергия для
поступательного, враща-
тельного, колебательного и
электронного вкладов равна
+ (ev)+<eU
и поэтому каждый вид движения вносит вклады в теплоемкость;
C^C^+Cv+Cv+C?. (21.3.12)
где
Су=N (д (е.ш>/дТ)у = - (21.3.1 За}
= —(У/йТг)(а<ет)/^)у, (21.3.136}
и т показывает вид движения (m = t, г, v или е). Теперь рассмот-
рим газ, состоящий из невзаимодействующих молекул, для кото-
рого нет вклада поступательной энергии, обусловленной межмоле-
кулярпым взаимодействием. Это значит, что мы рассматриваем-
идеальный газ, но позволяем молекулам иметь внутреннюю струк-
туру.
Температура всегда достаточно высока для того, чтобы сред-
няя поступательная энергия определялась принципом равного
распределения. Следовательно,
C\,=tf(d^-kT/dT^ (21,3.14)"
В случае одноатомных газов это будет единственным вкладом, и
поэтому должна наблюдаться теплоемкость, равная 3/?/2 =
— 12,47 Дж/(К-моль). Это прекрасно согласуется с эксперимен-
тальными значениями для редких газов (например, гелий имеет
теоретическую величину в интервале 2000 К).
Если температура достаточно высока, чтобы вращение моле-
кул можно было рассматривать как неквантованное, то для выяс-
222
Часть 2. Структура
Рис. 21.4. Температурная зависимость теплоемкости водорода.
нения вклада вращения в теплоемкость можно использовать прин-
цип равного распределения. В случае линейной молекулы
< er > =kT, и поэтому
С^=ЛГА=п/?. (21.3.15)
Если температура так низка, что занято лишь низшее враща-
тельное состояние, то <ег> =0 и вращение не вносит вклада в
теплоемкость. При промежуточных температурах величину Су
можно получить дифференцированием уравнения (21.3.4). Даже
без этого видно, что вращательный вклад в теплоемкость повы-
шается от нуля (когда АГ^В) до пН (когда АГ^>Й). Поскольку
поступательный вклад имеется всегда, следует ожидать, что об-
щая теплоемкость двухатомной молекулы (Сгл, 4-СуЛ1) будет
повышаться от 12,47 до 20,8 Дж/(К-моль), если повышать темпе-
ратуру от значения T=B[k. На рис. 21.4 приведена теплоемкость
водорода, для которой такую зависимость можно очень четко
проследить.
В случае нелинейных молекул средняя вращательная энергия
в высокотемпературной области достигает 3kTj2, и поэтому ее
21. Статистическая термодинамика. Развитие концепций
22»
Низкая температура
С
Высокая температура
Рис. 21.5. Теплоемкость затруднен-
кого ротора при высокой и низ-
кой температурах.
вклад в мольную теплоем-
кость имеет максимальную
величину 37?/2.
вклад в теплоемкость только
RT. ct = R
колебания вносят
достаточно высокой для их возбуждения. Если
Молекулярные
при температуре,
температура так высока или связи такие слабые, что для оценки
их средней энергии можно использовать принцип равного рас-
пределения, то каждая нормальная мода имеет <ev> сле-
довательно, каждая вносит в мольную теплоемкость вклад 7?,
В случае двухатомной молекулы существует лишь одна нормаль-
ная мода (растяжение и сокращение связи), и поэтому при вы-
соких температурах
(21.3.16>
Следует отметить, что вклад единственной колебательной моды
равен 7?, в то время как другие моды вносят вклад, равный толь-
ко -^-7?; это отражает два вклада — кинетический и потенциаль-
ный— в энергию гармонического осциллятора. С другим следст-
вием указанного большего вклада в теплоемкость мы сталкиваем-
ся в случае затрудненного вращения метильной группы, входящей
в состав- некоторой молекулы, например вследствие понижения’
температуры до такой величины, когда молекула находится в по-
тенциальной яме (рис. 21.5). Когда опа'достаточно «горячая», что-
бы вращаться, вклад в мольную теплоемкость молекулы состав-
ляет —7?. При более низких температурах вращение переходит в
колебание и та же самая мода вносит вклад 7?; значит, понижение
температуры приводит к увеличению вклада в теплоемкость выше
~g-7?, хотя полной величины 7? можно и не достигнуть.
Когда для оценки средней энергии возбуждения колебаний
Нельзя применить принцип равного распределения, следует ис-
пользовать точное выражение (21.3.7). Дифференцирование по Т
Приводит к
Cvv=nR
(Аи/йТ) exp I — ~2" Аа>Р
1 — ехр (—Ашр)
(21.3.17)
Cv.m — R-
Форма этой функции (рис. 21.6) показывает, что колебательный
224
Часть 2. Структура
Рис, 21.6, Температурная зависи-
мость колебательной теплоемкости.
вклад повышается от нуля
при низких температурах
и достигает своего предель-
ного значения nRT когда
А со.
Общую теплоемкость га-
за можно рассчитать сло-
жением вкладов поступа-
тельного движения, враще-
ния и колебаний. В некото-
рых случаях имеется также
электронный вклад в тепло-
емкость, но обычно им пре-
небрегают, так какэнергети-
от друга. (Одним из исключе-
ческие уровни далеко отстоят друг
ний является случай оксида азота (II); расчет его электронной теп-
лоемкости дается в виде задачи в конце главы.) Часто бывает до-
статочно лишь приблизительно оценить температурную зависи-
мость теплоемкости подсчетом числа видов движения, которые
вносят активный вклад в накопление энергии, Во всех газах по-
ступательные моды полностью активны и вносят вклад Зд/?/2; во
многих случаях вращение полностью активно н вносит вклад
2(-^-«г?) в линейных молекулах и 3(-^-пА*) в нелинейных; число ак-
тивных вращений мы будем обозначать т". Если колебание Vv ак-
тивно и температура достаточно велика для равного распределе-
ния, то имеется еще один вклад VyfiR, Следовательно, общая теп-
лоемкость равна
С __ 1
(21,3.18)
Такое выражение дает ясный путь объяснения (или предсказа-
ния) температурной зависимости Gv типа приведенной на рис, 21.7.
При разных температурах становятся активными разные моды.
Пример (вопрос 10). Оцените теплоемкость водяного пара прн 100°C,
Метод. Решив, какие моды движения активны при 100 °C, используем уравнение
(21.3.18).
Ответ. Активны поступательные и вращательные моды. Колебания не вносят
вклада, потому что они лежат в области 1500—3500 см-1, что значительно боль-
ше kT (kT«260 см-1 при 100°C). Поэтому из уравнения (21.3.18);
CVjtn =yR(3 4-34-0) = Зй = 24,9 Дж/(К-моль).
21. Статистическая термодинамика. Развитие концепций
225
7
2
Поступательное
— движение (t)
+ вращение (г)
+ колебание (v)
6
2
ес 5__________
— Поступатель-
ное движение
+ вращение
Поступательное
движение
3
2
2_х Атомы (t)
Диссоциация
Температура
Рис. 21.7. Общая зависимость теплоемкости молекул от температуры.
Комментарий. Экспериментальное значение равно 26,1 Дж/(К-моль). Расхож-
дение частично можно приписать приближенному характеру вращательной функ-
ции распределения.
Другой путь выяснения смысла величины теплоемкости начи-
нается с ее определения через молекулярную функцию распреде-
ления:
cv=—(1/АТ®)(W)v=(V/№) (4- v(-Ir)}v“
=(ЛГ/АТ®) {(!/<?) (d*q/d$2)y— (1/92) (Э<?/<Зр)г (d<7/dp)v]
Член в скобках — квадрат флуктуации энергии, поэтому
Cv=nR(te/KT)a. (21.3.19)
Это четкое выражение является точным, если предположить, что
молекулы независимы. При рассмотрении температурной зависи-
мости флуктуаций молекулярной энергии мы выяснили, что при
абсолютном нуле Se падает до нуля; отсюда можно предсказать,
что теплоемкость также будет равна пулю, что согласуется с экс-
периментом. При высоких температурах молекулярная энергия
флуктуирует в широком интервале, особенно если доступно много
близко расположенных энергетических уровней, и поэтому теп-
лоемкость растет. Этот анализ связывает теплоемкость с флуктуа-
циями в ансамбле молекул; величина флуктуации определяет го-
товность системы воспринимать термическую энергию.
15—242
226
Часть 2. Структура
Остаточная энтропия. Энтропию можно рассчитать из спектроско-
пических данных. Энтропию можно также измерить калориметри-
чески, как описано в гл. 5 (разд. 5.4, т. 1). Во многих случаях
получается хорошее согласие, но иногда экспериментально опре-
деленная энтропия ниже рассчитанной. Одно из объяснений ' со-
стоит в том, что экспериментатору не удалось обнаружить фазо-
вое изменение и поэтому он упустил из внимания член \Н[Т, где
&Н~ энтальпия перехода и 7’—его температура. Другое объясне-
ние в том, что в твердом веществе даже при абсолютном нуле
имеется некоторая разупорядоченность. Тогда энтропия твердого
вещества при абсолютном Нуле больше пуля и называется оста-
точной энтропией.
Источник и величину остаточной энтропии можно понять, рас-
сматривая кристалл, построенный из молекул АВ, где А и В
имеют сходные размеры. Различие в энергии между расположе-
ниями ...АВ АВ АВ АВ... и ...АВ АВ АВ ВЛ... может быть настоль-
ко небольшим, что в твердом веществе молекулы примут любую
ориентацию, т. е. будут неупорядоченны. Поскольку затвердевание
происходит не бесконечно медленно, неупорядоченное расположе-
ние может «заморозиться» и продолжать существовать при абсо-
лютном нуле.
Энтропию, обусловленную остаточной неупорядоченностью,
можно рассчитать на основе фундаментального выражения для
энтропии
в пределе Л":—н<». Предположим, что две молекулярные ориента-
ции равновероятны и что образец содержит N молекул. В одном
члене ансамбля одинаковая энергия может быть достигнута 2,v
различными способами (так как каждая молекула может нахо-
диться в двух ориентациях, а имеется N молекул). Ансамбль со-
стоит из ЛС членов, и каждый член может быть образован 2ЛГ
разными способами. Поэтому вес распределения — общее число
способов достижения одной и той же энергии — равен Сле-
довательно, энтропия равна
S = (&/.#) 1п (2")^ — k In = Afe In 2 In 2, (21.3.20)
где nL==N. Это выражение для молекул, которые при абсолютном
нуле могут принимать любую из двух ориентаций, предсказывает
значение остаточной мольной энтропии, равное Я1п2=5,7б Дж/(КХ
Хмоль). Если три ориентации энергетически эквивалентны (или
почти эквивалентны), то простая модификация вывода предсказы-
вает остаточную энтропию /?1нЗ и т. д. для других случаев.
Примером, где остаточная энтропия равна приблизительно
6 Дж/(К-моль), является твердый о^сид азота (II). Изучение ди-
21. Статистическая термодинамика. Развитие концепций 227
фракций рентгеновских лучей показывает, что молекулы NO сое-
динены друг с другом, образуя прямоугольный димер i . Этот
О—N
прямоугольник может принимать две ориентации в кристалле с
приблизительно равной энергией, и поэтому ожидается остаточная
энтропия около /?1п2. Молекула FCIO3 может принимать четыре
ориентации с приблизительно равной энергией, и поэтому можно
ожидать остаточную энтропию около /?1п4=11,5 Дж/(К*моль):
экспериментальное значение равно 10,1 Дж/(Д-моль). Поучитель-
ным примером, который раньше часто приводился для иллюстра-
ции полного согласия между теорией и экспериментом, является
окись углерода. Калориметрические измерения дали значение энт-
ропии 193 Дж/(К-моль), а спектроскопическая величина равна
197,9 Дж/(К-моль) при той же температуре. Разность в 5Дж/(КХ
Хмоль) не далека от /?1п2, и она была объяснена предположени-
ем о том, что молекулы СО могут принимать две ориентации в кри-
сталле, который мог иметь структуру типа... СО СО ОС СО СО
ОС... Однако недавно было показано, что в калориметрических
оцЫтйх не было замечено фазового изменения при низкой темпе-
ратуре, и поэтому на самом деле остаточной энтропии нет. Этот
пример подчеркивает, насколько нужно быть осторожным при по-
лучении и интерпретации экспериментальных данных. Возможно,
другие примеры остаточной энтропии со временем будут изъяты
подобным же образом.
В качестве последнего примера остаточной энтропии будет рас-
смотрен лед. Расхождение между калориметрическим и спектро-
скопическим определениями энтропии царов воды приводит к за-
ключению, что в структуре льда имеется неупорядоченность, со-
ответствующая значению остаточной энтропии 3,4 Дж/(Д'М0ль).
Это можно объяснить наличием водородных связей в кристалле
льда и тетраэдрическим расположением атомов водорода вокруг
каждого атома кислорода. Два нз атомов водорода соединены нор-
мальными, короткими сг-связями, а два другие — длинными водо-
родными связями. Разупорядоченность заключается в том, что две
из четырех связей короткие, и приближенный анализ этой пробле-
мы позволяет предсказать остаточную энтропию около J?ln-g-=
= 3,37 Дж/(Д-моль), что находится в хорошем согласии с экспе-
риментальным значением.
Пример (вопрос 12). На стр. 216 была рассчитана функция свободной энергии
Рц (1500) для воды, и там же бы.ад приведены экспериментальные значения
Функции. На основе расхождения между этими данными найдите остаточную
энтропию льда при абсолютном нуле,
Метод. Вместо предположения, что энтропия равна нулю, предположим, что она
имеет значение Sm (0). Тогда
Хц (П = $га (0) -у [l/„, - t/m (0)]/Т + k In Q
15’
228
Часть 2. Структура
Рис. 21.8. Расположение энергетических уровней молекул А и В при равновесии
А=^=В и зависимость равновесия от плотности состояний. Слева показано больц-
маневское распределение.
[из уравнения (21.2.2)]. Те же доводы, что и выше, приводят к Fa(T) =
=—7?!п—Sni(O). Теперь отождествим Ft>(T) с экспериментальным значе-
нием и используем расчет Pinдля нахождения SnJO).
Отеет. Экспериментальное значение равно —211,71 ДжДК-моль), а рассчитан-
ное здесь значение равно —208,8 Дж/(К-иоль), следовательно, —211,71 Дж/(КХ
Хмоль) =—206,8 ДжДК-моль)—5га(0), и поэтому
Sm (0) ~ 4,9 Дж/(К- моль).
Остаточную энтропию можно интерпретировать через выражение типа Sm(0) =
=Я1пп, написав, что п=ехр[5т(0)/Я] =ехр[4,9/'8,3]=-. 1,8.
Комментарий. Это сравнительно длинный путь получении остаточной энтропии:
обычно ее получают, непосредственно сравнивая рассчитанную н термохимичес-
кую энтропии; однако этот путь показывает, как можно использовать табулиро-
ванные функции свободной энергии. Заметим, что в тексте была приведена ос-
3
таточная энтропия 3,4 ДжДК-моль), что соответствует № 1.51
Константы равновесия. Основа для вычисления константы рав-
новесия реакции
А 4=*: в
может быть понята из рис. 21.8, а. Один набор линий соответст-
вует энергетическим уровням А, а другой набор — энергетическим
уровням В. Заселенность уровней определяется распределением
21. Статистическая термодинамика. Развитие концепций
229
Больцмана и не зависит от природы энергетических уровней. По-
этому можно предположить, что на оба набора энергетических
уровней распространяется одно и то же распределение Больцма-
на. Если расположение уровней А и В одинаково и А лежит ни-
же В, то диаграмма показывает, что в реакционной смеси при
равновесии будет преобладать А. Однако, если уровень В имеет
более высокую плотность состояний, как на рис. 21.8,6, несмот-
ря на то что этот уровень лежит выше по энергии, он может пре-
обладать в реакционной смеси, поскольку нас интересует общее
число молекул А и В в положении равновесия.
При некоторой температуре Т число молекул в некотором энер-
гетическом состоянии связанной системы равно
ЛГ(1/9)ехр (—₽Ег),
где А — общее число молекул. Общее число молекул типа А пред-
ставляет собой сумму ш этих молекул по энергетическим состоя-
ниям молекулы А;
Ад= Т. пг-=(^/?)2ехР(-₽еа)‘
iмолекул А а
Аналогично число молекул В в смеси является суммой по энерге-
тическим состояниям еь молекулы В:
Ав = пг=(ЛГ/9)2ехр(—
iмолекул В b
Сумма по состояниям А есть нс что иное, как молекулярная функ-
ция распределения для молекулы А(<?а):
^л—Л^д/?.
Сумма по состояниям В — это также функция распределения,
по не совсем такая, какие нам до сих пор встречались, потому что
энергии измеряются от основного состояния общей системы, ко-
торое в данном пршмере оказывается основным состоянием А.
К обычно принятому энергетическому минимуму легко перейти че-
рез соотношение = s& + A£o, где ЛАо— расстояние между низ-
шими уровнями А .и В (рис. 21.8). Тогда
Ав -=(N/q) exp (—рДА0) ехР (— ₽еб) =
ь
= (AtyB/9)exp(—рДА„).
Поэтому константа равновесия равна
А = Ав/Ад = (?в/9д) ехр (—рА Аи). (21.3.21а)
Это дает способ вычисления А просто из функции распределе-
ния, и следовательно, из спектроскопических данных или модеку-
230
7асгь 2, Структура
лярпых свойств, Форму выражения можно модифицировать так,
чтобы К была выражена через концентрации или давления. На-
пример, поскольку 9b=(t(?1)dV, где г?1 — функция распределения
внутренних мод, мы находим
Лс =(ЛГБ/У)/(ЛГД/У)={(^/(rrftj exp(-рДЕй), (21.3.216)
Тогда, поскольку ^л/Е-=пА£,/Е = рл/^Т,
КР = Рв!Рл ~1(^)в/Мл1ехр(— ₽Д£о)- (21.3,21b)
Содержание уравнения (21.3.21а) можно понять, рассмотрев
простой пример, в котором А имеет только один доступный при
данной температуре энергетический уровень (так что ^а = 1), а
В — большое число энергетических уровней на равном расстоя-
нии друг от друга (рис. 21,9). Функция распределения В будет
такой же, какая нам встречалась в гл. 20 [уравнение (20.2.16)],
и такой же, как для гармонического осциллятора, поэтому
рв=.(1—е-^)-*.
Если расстояние в мало по сравнению с kT, тоэто выражение упро-
щается до рц = й7'/е. Следовательно, если А и В разделены по энер-
гии на ДЕо, константа равновесия будет
К* (kT/t)exp(~\E0/kT), (21.3,22)
Этот результат можно использовать для изучения сдвига заселен-
ностей при уменьшении ДЕ0, но при постоянном е. Когда ДЕ0 вс-'
лика, доминирует экспоненциальный член и Л’~0, что означает
наличие в положении равновесия очень малого количества В.
Когда ДЕ0 мала, но еще положительна, значение Д’ может пре-
вышать единицу, что отражает предпочтительное образование В;
, , , ’в это обусловлено тем фактом, что В имеет
В большее число доступных состояний. Та-
кое же рассмотрение можно провести дли
случая, когда расстояния между уровням::
сохраняются одинаковыми, но меняется тем-
е псратура, При низких температурах экспо-
11 непцнальные члены почти равны нулю, по-
этому К~0, и система почти целиком со-
стоит из А.При высоких температурах экспо-:
&Ео )
А
Рис. 21.9. Модель для расчета равновесия.
21. Стат истин еская термодинамика. Развитие концепций
231
пенциальные члены приблизительно равны единице, а предэкспо-
непциальный множитель больше единицы, что указывает на пред-
почтительность В. Очевидно, повышение температуры благоприят-
ствует образованию В при равновесии просто потому, что стано-
вятся доступными очень многие состоянии В.
До сих пор нами не было установлено контакта с обычным,
химическим рассмотрением равновесия через функцию Гиббса и
химические потенциалы участников реакции, однако эти функции
были основой очень многих вопросов, обсуждавшихся в части 1
(т. 1). На базе выражения для функции Гиббса, приведенного в
предыдущем разделе, можно создать ключевую связь между спек-
троскопией и термодинамикой, и главная причина отступления от
темы, сделанного в этой части данного раздела, состоит в том, что-
бы дать беглый обзор основных соотношений.
В разд. 21.2 мы вывели выражение для функции Гиббса кол-
лектива независимых молекул
G~G(ty^—nRT]n(q/N).
Мы знаем связь между К? н стандартной функцией Гиббса для
реакции ДСт [т. 1, уравнение (9.1.15).]:
Ж = ~RT]nKp.
Необходимо лишь скомбинировать эти компоненты.
Во-первых, нам потребуется мольная функция Гиббса, поэто-
му возьмем N—L. Кроме того, нам желательно знать стандартное
значение, следовательно, молекулярная функция распределения
должна быть вычислена при давлении 1 атм; обозначим ее через
qa. Поскольку от давления зависит лишь поступательная функция
распределения (через входящий в нее объем К), для нахождения
Щ мы вычисляем (/ при V = RT/(l атм). Таким образом, для ком-
понента Л
Од,™—(Д,га(0)^-^Г1п(<Д /£). (21,3.23)
Тогда для реакции Лч^В констапта равновесия запишется как
’ 1п — Св,т — G\ „I — —RT |п (ув (0) — Ga,™ (0),
поэтому
*₽=№ /А )ехр{ -[G&>ra(0)-G\m(0)Wl, (21.3.24)
и, если Ов,1п (0)—0л,т (0) совпадает с t/в.т (0)—t/д.т (0), т. е.
Равно Д£о, получается приведенное выше выражение. Теперь
Уравнение для реакции в общем виде, например
АтВ c + D-1-F,
232
Часть 2. Структура
можно записать сразу через молекулярные функции распределе-
ния для всех пяти компонентов:
=(Чс ЧЬ <?f /Ча Чв L) exp (—AE0/kT), (21.3,25^
где Д£о — расстояние между энергиями нижних уровней А + В и
С + D + F.
Иллюстрацией применения этой формулы будет равновесие в
газовой фазе между двухатомной молекулой Хг и составляющи-
ми ее атомами:
Х2(газ) > 2Х(газ).
Константа равновесия дается выражением
Кр еХр(—Р°т /^?Г),
где Dm — мольная энергия диссоциации молекулы. Атомная функ-
ция распределения связана только с поступательным движением
и электронным вырождением g:
<fr =^xKtl, Vm=/?T/(1 атм).
Во-вторых, двухатомная молекула, кроме поступательного дви-
жения, имеет вращательные и колебательные степени свободы.
Поэтому
<?хг =^ха'/т<?х2<?х2.
Таким образом, константа равновесия равна
„ __ ёЧ^ехр(_П’5//?7’)
Ар
(21.3,26)
гМхг
В случае Naa^2Na известны значения всех параметров, и при
T^JOOO К мы находим, что Aps-2,4.
Пример (вопрос 14). Рассчитайте константу равновесия Кт. для равновесия
Na25±2\'a при 1000 К, используя следующие спектроскопические данные: £> =
=0,1547 ем-1, ы= 159,2 см-*, £>„=70,4 кДж/моль (0,73 эВ), Используйте для
натрия значение М,— 22,99, Атомы находятся в дублетном состоянии.
Метод. Используем выражения из подразд. 21.1.А, а затем подставляем в урав-
нение (21.3.26).
Ответ,
Поступательное движение Na2: т = (1,880- №6)Х(1000.45.98)'^ м'3 =
= 1,854-Ю33 м-3.
Вращение Naar q? = — (0,06952)X(1000/0.1547) 2247,
Колебание Na2: q- = 1/(1 — exp (— 159,2/695, 3)) = 4,89.
Электронная функция Ма2; 7е = g — 1.
Поступательное движение Na: т „ (1,880-Ю'-с)Х(1000-22,9)3’'2 м~3 =
= 6,553.№ м-А
Электронная функция Na: ф5 = g = 2.
21. Статистическая термодинамика. Развитие концепций 23?
Из уравнения (21.3.26):
Кр ~ екР =
атм)2 ехр (_D^/RY)
^т№2 (RE/1 атм) <?Na„ *?:ыав
(kTjl атм) Х4х (6,553 1032 м~3) ехр (—70,4/8,31)
= (1,854-iO33 м-3)Х(2247) Х(4,89) =
= (1,767-10« м-3)х(Л7/1 атм),
Поскольку
kT/'l атм = (1,381-I0-23 Дж/К)Х(1000К)/(1,01325-Ю6 И/ма) =
= 1,3629-Ю-25 м3,
находим, что
Кр = (1,767-Ю23 м-»)х(1,363-Ю-2-5 мЗ) = 2,408.
Комментарий. Заметим, что эта процедура приводит к К?. Если мы желаем вы-
разить константу равновесия через кои цент рацию, то необходимо изменить выра-
жение для К. (См. приведенное выше обсуждение.)
Литература
Nash L, К.. Elementary statistical thermodynamics, Addison-Wesley, Reading,
Mass., 1968.
McClelland B. Statistical thermodynamics, Wiley, New York, 1973,
Davidson №.. Statistical mechanics, McGraw-Hill, New York, 1962.
tint T. L., An introduction to statistical mechanics, Addison-Wesley, Reading,
Mass., 1960.
Rice О, K., Statistical mechanics, thermodynamics and kinetics, Freeman, San
Francisco, 1967.
Foailer J?. H.. Guggenheim E. A., Statistical thermodynamics, Cambridge Universi-
ty Press, 1965. (Имеется перевод 1-го издания: Фаулер Р., Гуггенгейм Э. Ста-
тистическая термодинамика. Пер, с англ.—М.: ИЛ, 1949.)
Munster A., Statistical mechanics, Springer, Berlin, 1974.
Задачи
21,1. Оцепите вращательную функцию распределения НС1 при а) 1 СЮ К,
б) 298 К и в) 500 К на основе высокотемпературного приближения.
2],2. Чисто вращательный микроволновый спектр НС1 имеет линии поглощения
При следующих частотах (в см-1): 21,19; 42,37; 63,56; 84,75; 105,93' 127,12;
148.31- 16949; 190,68- 2)1,87; 233,06; 254 24; 275,43; 296,62; 317,80' 33899;
360,18; 381’36; 402,55; 423,74; 444,92; 466,11; 487,30; 508,48. Рассчитайте враща-
тельную функцию распределения при а) 100 К и б) 298 К прямым суммирова-
нием.
21,3. Основываясь па высокотемпературном приближении, рассчитайте враига-
тединую функцию распределения молекулы воды при 298 К, используя следую-
щие вращательные постоянные: А ==27,878 см-‘, В=] 4,509 см-1, С=9,287 см-1.
При какой температуре справедливо высокотемпературное приближение?
21.4. Молекула метана представляет собой сферический волчок с длиной связи
109 пм. Рассчитайте вращательную функцию распределения при а) 298 К и
б) 500 X, используя в каждом случае высокотемпературное приближение (<т=-
234
Часть 2. Структура
21,5. Рассчитайте вращательную функцию распределения летала прямым сум-
мированием вращательных энергетических уровней.
21.6. Уравнение Саккура — Тетрода [уравнение (20.3.13)] дает теоретическую
энтропию одноатомного газа. Выведите соответствующее выражение для газа,
который может двигаться в двух измерениях. Отсюда найдите выражение для
мольной энтропии конденсации газа с образованием подвижной поверхностной
пленки. Как изменилась бы энтроиаяу если бы поверхностная пленка не была
подвижной?
21.7. Рассчитайте вращательную энтропию бензола, свободно вращающегося в
трех измерениях при 362 К. Его моменты янерцнн составляют 7л=2,93- 10-3® гХ
Хсм2, /в= fc= 1,46-10'3s гем2, а его число симметрии равно 12. Как изменилась
бы вращательная энтропии, если бы молекула была адсорбирована на поверх-
ности а могла бы аращаться только вокруг осн шестого порядка?
21.8. При экспериментальном изучении термодинамики адсорбции органических
молекул па графите при 362 К (Dollitnore D., Heal О. R.t Martin D. R.t J. Chem.
Soc., Faraday I, 1973, 1734) наблюдалось изменение энтропии в 111 Дж/(К-м<Мь),
когда поверхностное покрытие было небольшим, но это изменение падало де
52 Дж/(К*м«ль) в случае полного покрытия. Используя результаты двух преды-
дущих задач, предложите модель движения молекул бензола на поверхности,
21.9. Число симметрии. О можно рассчитать простым Подсчетом числа неразли-
чимых ориентаций молекулы, которые могут быть получены вращательными
операциями симметрии. Каково значение о в случае a) Nj, б) NO, в) бензола,
г) метана и д) хлороформа?
21.10. Рассчитайте при комнатной температуре энтропию С1О2 (изогнутая моле-
куда с неспареиныи электроном); угол ОС10: 118,5°; длина связи CIO: 149 им;
симметрия: С2„; электронное состояние: аВц
21.11. Рассчитайте и изобразите графически константу равновесия газофазной
реакции CDi-j-Hd^CHDa-j-DCi из следующих данных по колебательным энер-
гетическим уровням этих молекул: <г(СНЕ>3): 2993 131, 2142 (1), 1003 (3), 1291
(2), 1036 (2) см-i; a (CD4): 2109 (1), 1092 (2) 2259 (3) 996 (3) см-';
о (HClj: 2991 см-’, o(DCl): 2145 rad. Возьмите 300 К<Г=й1000 К. (Числа
в скобках указывают на вырождение.)
21.12. Обмен дейтерия между кислотой и водой является важным типом равно-
весия. Мы рассмотрим его на основании колебательных данных для Двух типов
молекул. Рассчитайте константу равновесия при а) 298 К и б) 800 К для га-
зофазной реакции обмена FUO+DCl^HDO+HCl на основе следующих колеба-
тельных и вращательных данных: о(П2О): 3656,7, 1,594,8, 3755.8 см~!; о (HDO):
2726,7, 1402,2, 3707,5 см-1; вращательные постоянные Н»О: 27,88; 14,51; 9,29 см-1;
HDO: 23,39; 9,102; 6,417 см-'; НС1: 10,59 см-'; DC1: 5,449 см"1.
21.13. Экспериментальная константа диссоциации для равновесия при
1000 К с экспериментальной точки зрения рассматривалась в задаче 9.22 (т. 1).
Спектроскопические данные для h таковы; 5=0,0373 см'1, 0=214,36 см-1,
Ое= 1,5422 эВ. Атом иода имеет основное состояние 2ES/ , что означает четырех-
кратное вырождение. Рассчитайте статистика -термодинамическую величину кон-
станты диссоциации при 1000 К.
21.14. Атомы иода имеют очень низко лежащие возбужденные состояния при
7603 rad (двукратное вырождение). При 2000 К опи значительно заселены. Как
их учет влияет на величину константы равновесия реакции, рассмотренной в
предыдущей задаче?
21.15. Теперь вернемся к более простой версии расчет;! (задача 21.13). Может ;
ли на константу равновесия атомизации заметно влиять магнитное поде? И селе- |
дуйте эту возможность, рассчитав влияние магнитного поля, на функцию pac-J
пределения атомов. Какое магнитное поле необходимо, чтобы сдвинуть положеЛ
ние равновесия на 1%?
21.16, Гармонический осциллятор в статистической термодинамике играет Осо б Л
важпукх роль (так же, как и в других областях), поскольку можно получите
краткие формы выражений для функций распределения и термодивамичесхихЧ
21. Статистическая термодинамика. Развитие концепций
235
свойств; кроме того, гармоническое движение обычно хорошо аппроксимирует
молекулярные колебания, и поэтому можно легко рассчитать колебательный
вклад н различные термодинамические свойства. Прежде всего выведите выра-
жение для внутренней энергии, энтальпии, энтропии, функции Гельмгольца и
функции Гиббса гармонического осциллятора и отложите их на графике в виде
функции
21,17, Функция свободной энергии Fo(T) равна [GmfT)—/7ш(0 К]/Г, и ее при-
менение было рассмотрено в гл, 9 (т. 1). Найдите выражение для Fo(T) гармо-
нического осциллятора.
21.18, Теперь используем расчет гармонического осциллятора. Рассчитайте коле-
бательный вклад при 1000 К в функцию свободной энергии: а) аммиака, который
имеет колебательные моды с частотами 3336.7 (1), 950,4 fl), 3443,8 (2). 1626,8
(2) см-1, и б) метана с колебательными модами 2916,7 (1), 1533,6 (2). 3018,9
(3). 1306.2 (3) см-1. Самый легкий метод состоит в использовании графов, по-
лученных в предыдущих задачах, и нахождении х для каждой моды, Числа в
скобках обозначают вырождение.
21-19. Функция свободной энергии очень удобна для расчетов равновесий при
условии, что имеются табулированные данные. Рассчитайте общую величину
Го (1000 К) для а) Н2, б) С12. н) NEL, г) N2 и д) NO, используя вращательные
и колебательные данные из табл. 17.2. Используйте B(NO) =1 7046 см-1 a(NO) =
= 1904 см-’.
21.20. Определите константу равновесия реакции N2+3H2₽r2NH3 прн 1000 К,
основываясь па данных предыдущей задачи.
21.2]. Хотя выражения, подобные dln$/rffi, и используются в статистической тер-
модинамике для формальных преобразований и получения лаконичных выраже-
ний для термодинамических параметров, они иногда доставляют больше беспо-
койства, чем пользы при их практическом применении. Если у вас есть таблица
энергетических уровней, т<Э часто удобнее непосредственно вычислять следую-
щие суммы: ^=Хехр(—ре,.). ^1'(в;//г7)схр(—fej, q‘ ‘ — S(в//йГ)2ехр(—fls,).
i i i
Чтобы увидеть, как используются эти суммы, прежде всего найдите выражения
U, S и Су через q, q~ ri q'".
21,22, Практическое применение можно еще больше упростить, если отделить
внутренние моды молекул от их поступательного движения. Покажите, что 17=
= (/й((С.,и-|-{Увнутр И S — ^цнешН-^вкутр, И выведите выражения ДЛЯ t/внутр, SBHy тр, Су гввутр
через q. q' и q "
21.23, Термодинамические свойства одноатомных газов можно рассчитать из из-
вестных электронных энергетических уровней, полученных из спектроскопии. Рас-
считайте электронные нклады в а) энтальпию НпГТ)—Нт((} К), б) функцию
свободной энергии Eof7) и в) электронный вклад в Су парой магния при 5000 К
из приведенных ниже данных. Используйте процедуру непосредственного сум-
мирования, примененную в двух последних задачах.
Состоя пне: 3Л s.S
Вырождение: 1 1 3 5 3 3
Энергия, см-1: 0 21850 21870 21911 35051 41197
21.24. Богатым источником данных по атомным энергетическим уровням явля-
ется сборник: Moore С. Е., Atomic Energy Levels. N. В. S. Crrc. No. 476 (1949).
Расчеты такого типа идеальны для программирования на ЭВМ или программи-
рованной счетной машине, особенно когда при рассматриваемой температуре до-
ступны многие электронные состояния. Если вы имеете доступ к такой счетной
Аащине, то рассчитайте значение Ей(Т) от комнатной температуры до 5000 К,
используя данные приведенной ниже таблицы, которая воспроизведена из сбор-
ника Мура. Если у вас нет такой счетной машины то вычислите F^fT) и Су при
3000 К-
236
Часть 2. Структура
Na]
Конфигурация Обозначение J Уровень Интервал
3s 3s S3 1 о 0,000
Зр 3psp° 1 9 16956,183
4s 4s SS 1 ’ T J_ 2 16973,379 25739,86 17,1963
3d 3d*D 1 2__ 29172,855
4р 4p2P’ 1 1 T 1 2 29172,904 30266,88 —0,0494
5 s 5s 2S 1 1 T ] V 30272,51 33200,696 5,63
43 4d2D 1 2 T 34548,754
5р 4fsf> 5p*F* 1 1 - 2 3T 1 2 34548,789 34588,6 35040,27 —0,0346
6s 6sES 1 1 ~T 1 35042,79 36372,647 2,52
21. Статистическая термодинамика, Развитие концепций___________237
Продолжение
Конфигурация Обозначение J Уровень Интервал
5J 5d2D 1 2 37036,781
5f2F° I —• | СМ -и | CM -и | —' Ol 0 37036,805 37057,6 —0,0230
5g 5g2G 4^ CO C, to | — to | — to 37060,2
6p"Pa 1 2 37296.51
7s 7s =4 1 1 — 2 1 2 37297,76 38012,074 1,25
&PD 38387,287
- 4 38387,300 —0,0124
21.25. Рассчитайте на ЭВМ электронный вклад в теплоемкость одноатомных па-
ров натрия при комнатной температуре и 5000 К.
21.26. Натрий кипит при 1163 К и пар содержит как мономеры, так и димеры.
Рассчитайте константу раньонесия димеризации при этой температуре и отно-
сительное содержание димеров в парах в точке кипения на основе функции энер-
гии Fa (1103 К) для атомов и молекул. (Молекула имеет синглетное электрон-
ное состояние, .8—0,1547 см-1 к <т=159 см-1.) Какой эксперимент нужно проне-
сти, Чтобы подтвердить предсказание этого вычисления?
21.27. Используя теорию равного распределения, предскажите вероятную тепло-
емкость следующих молекул при комнатной температуре: а) Ь, б) Нг, в) СН4,
г) бензола, д) водяного пара, е) двуокиси углерода.
21.28. Нарисуйте график Су.ш н виде функции x~ti<SjfkT для гармонического
осциллятора и предскажите теплоемкость аммиака и метана при а) 298 К и
б) 500 К (Колебательные данные см. в задаче 21,18.)
21.29. Фундаментальные частоты семи нормальных мод ацетилена (этина) имеют
238
Часть 2. Структура
значения; 612, 612, 729, 1974, 3287, 3374 см-1. Какова теплоемкость этого газа
при а) 298 К и б) 500 К?
21.30. Получите выражение для теплоемкости системы, в которой имеются толь-
ко два уровня, разделенные величиной Л. Нарисуйте температурную зависимость
в виде функции параметра Д/йГ.
21.31. Молекула NO имеет дважды вырожденное основное состояние и дважды
вырожденное электронно-возбужденное состояние, расположенное выше на
121,1 см-'1. Рассчитайте электронный вклад в теплоемкость этой молекулы при
а) 50 К, 6) 298 Кив) 500 К.
21.32, Может ли приложенное магнитное поле изменить теплоемкость молекулы?
Исследуйте попрею, выведя выражение для теплоемкости NO2 (молекула имеет
один несларепный электрон} в приложенном магнитном ноле. Какое изменение
теплоемкости вызовет поле в 5 Т при а) 50 К и б) 298 К?
21.33. Энергетические уровни метильпбй группы, связанной с большой молеку-
лой, даются выражением Для частицы на кольце до тех пор, пока она враща-
ется свободно. Каков высокотемпературный вклад такой свободно вращающейся
группы в теплоемкость и энтропию? (Момент инерции СНз равен 5.341Х
ХЮ-47 кг-m3).
21-34, Согласно принципу Паули, молекулярный водород существует в двух ви-
дах, отличающихся разной относительной ориентацией двух прогонных еннпов.
Если протонные спины противоположны друг другу, то этот вид называется
параводородом. Характеристикой1 такой молекулы является, то, что вращатель-
ное квантовое число может принимать только четные значения (7=0, 2, 4,
В образце чистого параводорода при низких температурах главный вклад в
теплоемкость вносит возбуждение от 7=0 до 7=2. Рассчитайте температурную
зависимость теплоемкости параводорода на основе того, что его вращательные
уровни (по эффекту) составляют систему с двумя уровнями (однако отметьте
вырождение состояния с 7 = 2), Исполвзуйте В = 60,864 см-1 и нарисуйте эскиз
кривой теплоемкости. Экспериментальная теплоемкость при низких температурах
в действительности проходит через максимум.
21.35, Скорость звука определяется теплоемкостью газа. Поскольку теплоемкость
можно рассчитать из молекулярных данных, следовательно, мы имеем способ
вычисления скорости звука. Нужно знать, что cs — где у = Ср/Ст и
m — мольная масса. Выведите выражения для Щ для а) идеального газа, состоя-
щего из двухатомных молекул, при высоких температурах (поступательное дви-
жение, вращение, но нс колебание) и б) для параводорода при низких темпера-
турах.
21.36. Используя результат предыдущей задачи, оцепите скорость звука в воз-
духе при комнатной температуре.
21.37. Какова остаточная энтропия кристалла, молекулы которого при абсолют-
пом нуле могут иметь а) 3, б) 5 и в) 6 ориентаций с одинаковой энергией?
21-38. Гексагональная молекула C6H«F(-n в принципе может иметь остаточиую
энтропию благодаря тому, что атомы водорода и фтора похожи. Какой могла
бы быть остаточная энтропия для каждого значения п и каждого изомера?
21.39. Термохимическая энтропия газообразного азота при 298 К обсуждалась
в примере в разд. 5.4 (т. 1), и было получено значение 192,06 Дж/(Кцмоль). На
основе вращательной постоянной В = 1,9987 см~' и колебательной частоты
2358 см-1 рассчитайте статистико-термодинамическую величину энтропии при
этой температуре. Что говорит эта величина о природе кристалла при абсолюг
ном пуле?
22 Определение структуры молекул.
Дифракционные методы
Изучаемые вопросы
После тщательного изучения этой главы вы сможете:
1. Вывести условие Брэгга для дифракции [уравнение (22.1.1)].
2. Определить плоскости кристаллической решетки с помощью
индексов Миллера (стр. 243).
3. Описать метод Лауэ, метод Брэгга и метод Дебая — Шерре-
ра реитгсноструктурного анализа кристаллов (Стр. 245).
4. Индексировать рефлексы (Отражения) и определить приро-
ду элементарной ячейки из систематических пропусков в дифрак-
ционной картине (стр. 248).
5. Связать структуру простых решеток с интенсивностью рент-
геновских рефлексов (стр. 250).
6. Определить структурный фактор [уравнение (22.3.4)] и свя-
зать его с интенсивностями и электронными плотностями [урав-
нение (22.3.7) ].
7. Объяснить смысл проблемы фазы (стр. 257).
8. Описать процедуру рентгеноструктурного анализа (стр. 260).
9. Описать гексагональную, кубическую и объемно-центриро-
ванную плотнейшие упаковки идентичных шаров (стр. 263).
10. Сформулировать правило отношения радиусов для ионных
кристаллов (стр. 266).
11. Нарисовать решетки каменной соли, вюрцита и хлорида це-
зия (стр. 267).
12. Объяснить, как можно определить абсолютные конфигура-
ции молекул (стр.268).
13. Указать, какую информацию можно получить из опыта по
дифракции нейтронов (стр. 270).
14. Описать опыт по дифракции электронов (стр. 271).
15. Использовать уравнение Вирля для вывода длин связей
и углов между связями (стр. 273).
Введение
Дифракция волн является основой нескольких мощных мето-
дов определения структуры молекул. Звуковые и световые волны
Дифрагируются объектами, имеющими размеры, сравнимые с
Длиной волны облучения, и, поскольку рентгеновские лучн имеют
240
Часть 2. Структура
длины волн, сравнимые с расстоянием между атомами в кристал-
лах (0,1 нм или 100 пм), при прохождении через кристаллические
решетки они претерпевают дифракцию. Из характера распределе-
ния интенсивности дифрагированных рентгеновских лучей можно
нарисовать детальную картину расположения атомов в молекулах,
даже таких сложных, как молекулы белков. Возможно даже оп-
ределить распределение электронной плотности в отдельных свя-
зях.
Электроны, движущиеся со скоростью 20 000 км/с (после уско-
рения разностью потенциалов 4 кВ), имеют длину волны 40 им, и
поэтому они также могут использоваться при изучении дифракции.
Нейтроны, генерированные в ядерном реакторе и затем замедлен-
ные до тепловых энергий, имеют примерно такую же длину вол-
ны и также интенсивно используются.
22.1. Общие особенности дифракции
Дифракционную картину можно рассматривать с точки зрения
или нолновой, или корпускулярной природы излучения. Оба ас-
пекта дают приемлемое объяснение природы дифракции и позво-
ляют интерпретировать дифракционную картину. Здесь приводит-
ся волновое объяснение, а корпускулярное описано в приложении
в конце главы. Волновая интерпретация дифракции основана на
конструктивной и деструктивной интерференции, которая происхо-
дит при наложении волн. Если в некоторой точке амплитуды на-
Деструктивная интерференция
, (темные места)
Рис. 22.1. Опыт Янга по щелевой дифракции.
22. Дифракционные методы.
241:
Рис. 22.2. Дифракция от набора!
плоскостей.
ходится в фазе, то они уси-
ливают друг друга, и в этом
месте интенсивность увели-
чивается; там, где амплиту-
ды находятся в противофа-
зе, они сокращаются, и ин-
тенсивность уменьшается.
Если волны исходят из общего источника, то их относительная
Йаза в некоторой точке зависит от длины пройденного ими пути,
ацример, дифракционная картина щелевого опыта Янга (рис.
22.1) может быть легко объяснена с помощью разности длин пу-
тей двух лучей, причем различные области на экране соответст-
вуют местам, где волны, идущие из двух щелей, находятся в фазе-
(светлые области) и в противофазе (темные области).
Теперь рассмотрим «стеллаж» из отражающих слоев, располо-
женных так, как показано на рис. 22.2. Разность длин путей двух
лучей, отраженных от двух соседних слоев, составляет
АВ + ВС = 2d sin 0,
где d—расстояние между слоями, 0 —угол, определенный на
рис. 22.2. Для многих выбранных значений 0 эта разность длин
путей не кратна целому числу длим волн, в этих случаях два луча
перекрываются вне фазы в интенсивность уменьшается. Другими
словами, зритель не мог бы наблюдать никакой интенсивности све-
та, если бы источник и детектор были размещены под углом 0.
Однако для некоторых углов разность длин путей АВ + ВС точно
кратна целому числу длин волн; амплитуды всех волн, отклоняе-
мых параллельными плоскостями, находятся в фазе, и интенсив-
ность значительна. Если длина волны равна Л, то такая конструк-
тивная интерференция происходит при АВ + ВС—п\, где п — це-
лое число. Таким образом, угол для конструктивной интерферен-
ции дастся выражением
условие Брэгга-. nX = 2dsin0. (22.1.1>
Пример (вопрос 1). Расстояние между слоями атомов в кристалле равно 404 пм.
При каком значении угла в дифрактометре, использующем рентгеновские луча
CuA'jx (длина волны 154 пм), будет наблюдаться рефлекс (отражение)?
Метод. Подставляем непосредственно в уравнение (22.1,1). Используем i^l
Для рефлекса первого порядка.
16—242
24!
Часть 2. Структура
Ответ. Из уравнения (22.1.1):
6 = sin-1 (b/2d) =
«sin-> (154/808) = sin-i (0,191) = lo°59'.
Комментарий. Рефлекс второго порядка произойдет при 22°27". При исиолыоаа-
нии рентгеновского излучения рефлекс первого порядка будет при 5°2';
заметим, что, чем короче длина волны, тем меньше дифракционный угол.
Условие Брэгга (названное так в честь пионеров рентгеногра-
фии; как отец, так и сын получили Нобелевскую премию за свои
исследования) является основным уравнением рентгеновской кри-
сталлографии. Его первое применение состоит в определении рас-
стояния между слоями в решетке Кристалла, так как, определяя
угол 0, при котором наблюдается максимальная интенсивность,
можно легко рассчитать d, если известна длина волны падающего
излучения.
22,2. Кристаллические решетки
Интерпретация дифракционной картины зависит от того, что
мы знаем о возможных расположениях атомов в пространствен-
ных решетках. В гл. 16 мы рассмотрели количественный аспект
этой проблемы, когда исследовали структуру кристаллов на осно-
ве симметрии их компонентов—элементарных ячеек, стыкованных
друг с другом. Теперь мы вернемся к проблемам измерения решет-
ки и локализации атомов е помощью дифракции рентгеновских
лучей.
Места расположения атомов или ионов обозначаются точками.
Если они объединены в кристалле, то их положение определяет
решетка. Элементарная ячейка — это выбранная небольшая пра-
вильная фигура, которая, постоянно повторяясь, образует полную
решетку. Ранее доказательство того, что кристаллы можно рас-,
сматривать как решетку атомных размеров, было получено из за-
кона рациональных индексов. Оп устанавливает, что отрезки, от-
секаемые плоскостями кристаллов на трех подходящим образом
выбранных осях, проходящих через кристалл, могут быть выраже-
ны как целые кратные трех основных измерений. Это очень ясно
указывает на конструкцию кристалла из однородных «строитель-
ных блоков», как показано на рис. 16.30 и как обсуждалось с точ-
ки зрения симметрии в гл. 16.
Обозначения плоскостей. Индексы Миллера. Плоскости, содержа-
щие узлы (точки) решетки, расположены так, что вызывают ди-
фракцию. Даже в прямоугольной решетке можно выбрать боль-
шое число различных плоскостей, отражающих расположение уз-
лов решетки (рис. 22.3), и поэтому важно иметь схему обозначе-
ний.
22. Дифракционные методы
243
-ч Вейсс-.(оо 1)
-J Миллер: (01)
Вейсс ; (Т1)
Миллер:(11)
Ь
Вейсс:(32)
Миллер:(23)
Вейсс; (И)
Миллер:(П)
Рис. 22.3. Индексы Миллера и Вейсса для некоторых
решетке.
плоскостей в двумерной
Скачала рассмотрим простую двумерную Прямоугольную ре-
шетку, образованную из элементарных ячеек со сторонами а и b
(рис. 22.3). На рисунке выбраны четыре набора плоскостей; ясно,
что их можно различить по расстояниям вдоль осей, па которых
эти плоскости нх пересекают. В одной из схем обозначений каж-
дый набор обозначается расстояниями по двум осям до точек пе-
ресечения. В этом случае четыре набора плоскостей могут быть
обозначены соответственно как (1а, 1Ь), (За, 2Ь), (—а, Ь) и (ооа,
16). Если мы согласимся всегда измерять расстояния вдоль осей
элементарной ячейки через длину ячейки в этом направлении, то
эти плоскости можно обозначить (1,1) (3,2), (—1,1) к (со, ^.Да-
лее, если нарисованная решетка является видом сверху на трех-
мерную прямоугольную структуру с длиной элементарной ячейки
в ^-направлении, равной с, то все три набора плоскостей пересе-
кают ось z при ею с, и поэтому полное обозначение плоскостей будет
{1. 1, сю), (3, 2, оо), (—1, 1, сю) ,и (оо, 1, сю). Этот тип обозначений
называется системой индексов Вейсса.
Обозначение «сю» неудобно, и, чтобы избежать его, можно
взять обратные величины индексов. Оказывается, что это имеет
другие преимущества, как мы сейчас увидим. Обратные величины
чисел Вейсса, не содержащие Дробей, называются индексами Мил-
лера. Например, плоскость (1 a, 1 Ь, сюс) выглядит как (1, 1, сю) в
системе Вейсса и как (НО) в системе Миллера. Следовательно,
плоскости (1 а, 1 Ь, сю с) на рис. 22.3 являются плоскостями (ИО)
кристалла. Аналогично плоскости (За, 26, сюс) на рис. 22.3 стано-
вятся плоскостями (3,2, сю) в системе Вейсса, плоскостями (-^. -у»
0), если брать обратные величины, плоскостями (2, 3, 0), если ис-
ключить дроби, и, наконец, плоскостями (230) в системе Мил-
лера. Индексы Миллера для плоскостей (сю, 1, сю) на рис. 22.3
имеют вид (0Ю). Отрицательные индексы [как в случае плоско-
стей (—1, 1, сю), показанных па рис. 22.3] записываются с чертой
над цифрой, например (110).
16*
244
7деть 2. Структура
Ряс. 22.4. Некоторые плоскости и их индексы.
Чтобы быстро попять, какие плоскости рассматриваются, нуж-
но помнить две особенности индексов Миллера. На рис. 22.4 при-
ведены некоторые важные плоскости. Во-первых, чем меньше чис-
ло h в индексе (&&/), тем плоскость более параллельна оси а. То
же применимо к А и оси Ь, а также к и оси с. Когда h равно /гу-
лю, отрезок, отсекаемый плоскостью от оси а, равен бесконечно-
сти, и поэтому плоскость параллельна оси й. Аналогично Т=0
указывает на плоскость, параллельную Ь, а ;=0 — на плоскость/
параллельную с. Во-вторых, система индексов Миллера не ограни-
чена кристаллами с ортогональными (перпендикулярными) осями
элементарных ячеек. На рис. 22.4 приведены также некоторые
индексы для Триклинной системы; они определяются тем же спо-
собом, что и выше, но оси уже не ортогональны (и по этой причи-
не они обычно называются а-, Ь- и с-осями кристалла, а не х, у
иг).
Одно из преимуществ индексов Миллера состоит в том, что в
кубическом кристалле расстояния между соседними (hkl) плоско-
стями выражаются просто через сами индексы h, kt I. С помощью
рис. 22.5 легко подтвердить, что в двух измерениях это расстоя-
ние равно
а в случае трех измерений расстояние между плоскостями (hkl}.
равно
<***,=(22.2.1)
22. Дифракционные методы 24J
Пример. Ромбическая элементарная ячейка имеет следующие параметры: а =
= 50пм (5А), Ь = 100пм (10 А), с= 150 пм (15 А). Каково расстояние между
плоскостями (123)?
Метод. Плоскости (hkl) разделены расстоянием dwt, которое определяется из
формулы l/d“w =(h/a)4-(k/b)*+(l/c)2.
Ответ. l/df23 - (1/50 пм)2-J- (2/100 пм)2 -J- (3/150 пм)2 =
= 3 (1/50 им)2.
Поэтому
Лт = (50 пм)/У"3 = 29 пм.
Комментарий. Использованное выражение является обобщением уравнения
(22.2.1) и применимо к любой ромбической (a—(J=y=90°) ячейке.
Это все, что нам необходимо знать об обозначении плоскостей
атомов. Теперь перейдем к применению дифракции рентгеновских
лучей для измерения расстояний day.
22.3. Рентгеновская кристаллография
Рентгеновские лучи образуются при бомбардировке металла
электронами с высокой энергией. Когда электроны проникают в
вещество, генерируются два вида рентгеновского излучения. Име-
ется непрерывный фон излучения, охватывающий интервал длин
волн. Этот континуум волн называется бремештралунг (по-немец-
ки Bremsse — чаща, кустарник, a Strahlung — луч). На этот конти-
нуум налагаются несколько резких пиков высокой интенсивности,
которые обусловлены взаимодействием электронов с электронами
внутренних оболочек атомов металла. В результате столкновения
выбивается электрон с внутренней электронной оболочки, и на это
свободное место падает электрон с более высокой по энергии обо-
лочки, испуская избыток энергии в виде фотона высокой энер-
гии.
На основании некоторых данных, подтверждающих, что недав-
но открытые рентгеновские лучи могли бы иметь длины волн, срав-
нимые с расстоянием между атомами в кристаллах, Лауэ пред-
положил (в 1912 г.), что они могли бы дифрагироваться при про-
хождении через кристалл. Это почти сразу же было подтверждено
Фридрихом и Кпиппингом; так был создан метод рентгеновской
кристаллографии.
' !
\ Ч •—Рис. 22.5. Расстояние между плоскостями решет-
' (М) ' ки. Иллюстрация для случая (21).
246
Часть 2. Структура
Рис. 22.6. Дифракция от двух
наборов плоскостей решетки монокристалла.
Метод Лауэ состоял в пропускании пучка рентгеновских лучей
с широкой полосой длин волн через монокристалл и в последую-
щей записи дифракционной картины на фотопластинке. В этом
методе не предусматривалась возможность ориентации монокри-
сталла таким образом, чтобы он действовал как дифракционная
решетка по отношению к единственной длине волны; однако, если
пучок содержит широкий интервал длин волн, условие дифракции
будет выполняться При любой ориентации, так как в пучке най-
дется излучение с длиной волны, удовлетворяющей соответствую-
щему условию Брэгга.
Модификация метода Лауэ Брэггом упростила дифракционную
картину использованием монохроматического пучка, Он образовы-
вался при пропускании пучка через металлическую фольгу, кото-
рая задерживала широкий интервал фоновых частот, но позволя-
ла проходить одному из высокоиптенсивиых монохроматических
пиков.
Ллвтернатива методов Лауэ и Брэгга (в которых используются
монокристаллы) была введена Дебаем и Шеррсром, а также Хил-
лом. Они применили монохроматическое излучение, но порошкооб-
разный образец. Поскольку порошок состоит из большого числа
мелких кристаллов, беспоря-
дочно ориентированных, часть
из них всегда удовлетворяет
условию дифракции, несмотря
—-Фотопленка
Конус джррагкрО'
ванных лучей
Образец
Падающий пучок
(направлен сверху
па страницу)
Рис. 22.7. Метод дифракции рентге-
новских лучей Дебая — Шеррера.
22. Дифракционные методы
247
Рис. 22.8. Рентгеновская фотогра-
фия порошков КС1 (й) и NaC] (б).
на то что излучение моио-
хроматично. Этот метод ис-
пользуется для качественно-
го анализа материала и для
первичной идентификации
размеров и симметрии эле-
ментарной ячейки, однако с
его помощью нельзя полу-
чить детальной информации
о распределении электрон-
ной плотности, которую по-
лучают только монохрома-
тическим методом монокри-
сталла Брэгга.
Метод порошка. Если обра-
зец представляет собой не-
упорядоченную смесь тон-
ких кристаллов, то часть из
них удовлетворяет условию
Брэгга лЛ = 2^з1л0. Напри-
мер, некоторые кристалли-
ты будут ориентированы
так, что плоскости (111),
разделенные расстоянием
б?ш, дадут интенсивную ди-
фракцию под углом 20 к па-
дающему лучку (рис. 22.6).
Плоскости (111) других кристаллитов могут быть под углом 0 к
входящему пучку, но располагаться под произвольным углом око-
ло линии его падения. Следовательно; дифрагированные пучки ле-
жат на поверхности конуса с вершиной 40. Другие кристаллиты
будут ориентированы так, что, например, их плоскости (211) с
расстоянием между ними d3[1 будут удовлетворять условию ди-
фракции и давать отклонение под углом 20'; это объясняет обра-
зование другого конуса дифрагированного излучения (рис. 22.6).
В принципе дифракционный конус образует каждая плоскость
(ЛА/), поскольку некоторые члены беспорядочно ориентированно-
го образца способны дифрагировать входящий монохроматический
пучок.
248
Часть 2. Структура
Метод Дебая — Шеррера проиллюстрирован на рис. 22.7. Пу-
чок монохроматических рентгеновских лучей входит в порошкооб-
разный образец, и относительно большая часть излучения прохо-
дит через порошок, не отклоняясь от первоначального направле-
ния, и выходит через отверстие в фотопластинке на другую сторону
прибора. Отклоненные пучки описывают конус и обнаруживают-
ся н виде дуг окружности на ленте фотопленки, намотанной
вокруг камеры. Типичная фотография порошка приведена на
рис. 22.8. Угол 0 для каждого конуса может отличаться от поло-
жения фотографического изображения. Если конус можно при-
писать какой-то конкретной плоскости (hkl)—это называется ин-
дексацией рефлексов, — то величина dhki может быть определена
из условия Брэгга.
Ключом анализа является индексация дифракционной картины.
К счастью, некоторые типы элементарных ячеек дают характер-
ную и легко распознаваемую картину линий, так что без труда
можно определить тин ячейки и ее размеры. В кубической решетке
элементарных ячеек размера а расстояние между плоскостями
(hkl) равно dJlk( = a/(/t2 + fe2-j-Z3)1-,,3; поэтому угол, При котором
дифрагируют плоскости (hkl) дается условием Брэгга как*
sin %kl -f-L k/a'j (22.3.1)
Чтобы предсказать дифракционную картину для кубической ре-
шетки, нужно лишь подставить в последнее выражение разрешен-
ные значения ht k и I. Это дает следующие значения sin2 0 (в крат-
ном отношении к-д-Х/п):
(hkl) (100) (НО) (111) (200) (210) (211) (220) (300) (221) (310)...
Л’ + йз + Р 1 2 3 4 5 6 8 9 9 10...
Отметим, что в этот ряд не входит число 7 (как и 15, и ряд дру-
гих чисел), потому что сумма трех квадратов целых чисел пе мо-
жет быть равной 7 (или 15 и т, д.). Следовательно, в картине
имеется серия систематических пропусков, которые характерны
для примитивной кубической решетки.
* Заметим, что использовано значение ге=1, и поэтому может показаться,
что мы обращаем внимание только на рефлексы первого порядка (ге=1) (наз-
вание «рефлекс» или «отражение» часто используется для обозначения дифрак-
ционного пятна). На самом деле это не так, поскольку рефлекс второго поряд-
ка (п=2), например от плоскости (ПО), происходит под тем же самым углом,
что и рефлекс от плоскости (220), удовлетворяющей условию duo По-
этому, принимая, что индексы (hkl) могут иметь все возможные значения, мы
автоматически учитываем рефлексы более высокого порядка.
22. Дифракционные методы 249
Пример (вопрос 4). Фотография дифракции порошка КС1 имеет линии при сле-
дующих расстояниях от центрального пятна (рентгеновские лучи Мо с длиной
волны 70,8 пм; камера радиусом 5,74 см): 13,2; 18,4; 22,8; 26,2; 29,4; 32,2; 37,2;
39,6; 41,8; 43,8; 46,0 (в мм). Индексируйте эти линии, определите тип элемен-
тарной ячейки и ее размеры.
.Мегод. Из уравнения (22.3.1) имеем sin20 = (л/2а)2(Л2Н-62+/2). Поэтому пере-
ведем расстояния,в 0, а затем в sin20, найдем общий множитель Л = (Х/2а)г, а
затем Л2+^24-12. ‘Выразим (ЛЫ). Для определения размеров ячейки из Л=
=(Х/2а)2 найдем а.
Ответ. Расстояние D от центрального пятна связано с дифракционным углом
(в радианах) соотношением Q=D/2R. Переведем 0 в градусы умножением на
360/2л. При /?=57,4 мм имеем 0 (градусы) = (0/57,4 мм) (360/2л)-2*й(мм),
и поэтому переход от D к 0 очень прост. Запишем следующую таблицу:
D, мм 13,2 18,4 22,8 26,2 29,4 32,2 37,2 39,6 41,8 43,8 46,0
6, град 6,6 9,2 11,4 13,1 14,7 16,1 18,6 19,8 20,9 21,9 23,0
100 sin30 1,32 2,56 3,91 5,14 6,44 7,69 10,2 11,5 12,7 13,9 15,3
Общий делитель равен 1 32/100; делим на него все данные, чтобы определить
Л24-А24-Р:
+ 1 2 3 4 5 6 8 9 Ю 11 12
и попытаемся выразить их в форме (hkl):
(hkl) (100) (ПО) (111) (200) (210) (211) (220) (300) (310) (311) (222)
Линин индексированы. Отметим пропуск между 6 и 8; это указывает на прими-
тивную кубическую ячейку, Иа (1/2о)2 =0,013 находим, что а=310 пм.
Комментарий. Ту же процедуру можно использовать для любого спектра по-
рошка. Позже мы увидим, что интенсивность линий дает дополнительную ин-
формацию. Отметим, что правильный выбор R экономит много работы.
Этот пример подтверждает только что приведенные особенно-
сти анализа и позволяет определить ячейки КС1. Если этот метод
применяется к очень похожим кристаллам NaCl, то получаемая
дифракционная картина сильно отличается от картины для КС1.
Причина состоит в том, что ионы К-1' и С1~, имеющие одинаковые
число и конфигурацию электронов, рассеивают рентгеновские лу-
чи почти одинаково, и поэтому, хотя решетка содержит два сорта
ионов, рентгеновские лучи рассеиваются так, как если бы эта ку-
бическая решетка была построена цз идентичных ионов. В слу-
чае NaCl рассеивающая способность ионов Na+ меньше, чем ионов
С1-, и приведенный способ анализа должен быть изменен, чтобы
учесть эффекты ионов двух типов в одном и том же кристалле.
Качественно эту Проблему можно рассмотреть, считая структуру
NaCl как бы состоящей из двух взаимно проникающих кубических
решеток; одна из ионов Na+, а другая из ионов СН Некоторые
рефлексы от решетки Na+ находятся в противофазе с рефлекса-
ми от решетки С1- и интерферируют деструктивно. Для других
ориентаций оба набора рефлексов находятся в фазе и появляются
’250
Часть 2. Структура
Рис. 22.9. Дифракция от кристалла, содержащего два сорта атомов.
интенсивные линии. Исчезновение рефлексов будет полным лишь
тогда, когда ионы имеют одинаковую рассеивающую способность,
но в случае NaCl исчезновение не полное и наблюдается чередова-
ние интенсивных и слабых линий. Это объясняет главные особен-
ности дифракционной картины NaCl, приведенной на рис. 22.8.
Природу дифракционной картины количественно можно рас-
смотреть па примере двумерной решетки, показанной на рис. 22.9.
Она состоит из двух взаимно проникающих решеток атомов тииаА
и В. Их рассеивающая способность измеряется величинами [ь и
/в; они называются длинами рассеяния атомов. Поскольку за про-
цессы рассеяния ответственны электроны, длина рассеяния гру-
бо пропорциональна атомному номеру.
Теперь рассмотрим единичную элементарную ячейку и ее об-
щий вклад в дифракционную картину. Возьмем ячейку, в которой
атом А находится в начале координат, а атом В — в положении
(xar yb). Когда падающий пучок имеет дифракционный угол 6 по
отношению к плоскости (hk) атомов А, он также имеет тот же
угол по отношению к плоскости (ЙА), содержащей атомы В
(рис, 22.9). Плоскости А и В отстоят друг от друга на некоторое
расстояние d'hk, и поэтому отражения от плоскости дают волны,
сдвинутые по фазе относительно волн, обусловленных плоскостью
А; рассмотрение рис. 22.9 показывает, что разность длин путей
между волнами, отклоненными плоскостями А и В, составляет
2dhk sin0. Так как 0 — дифракционный угол, то оп определяется
выражением 0=Л,/2йла; следовательно, разность длин путей для А
22. Дифракционные методы
251
и В равна d'hkK/dhk. С помощью простых тригонометрических до-
водов можно показать, что d'hk = (hx + ky)dhn, и поэтому разность
длин путей равна (hx + ky)h. Таким образом, А, В-фазовый сдвиг
равен [ (йхд-йу)%] (2л/Л), или 2n(hx + ky). В трех измерениях это
превращается в
А, В-фазовый сдвиг2л (hx-\-ky-(-lz), (22,3.2)
где атом В есть точка (ха, yb, zc) элементарной ячейки, а А на-
ходится в начале координат. (Последнее уравнение иллюстрирует
другой результат, который очень просто выражается при исполь-
зовании индексов Миллера.)
Когда фазы А и В отличаются на 180° (л радиан), амплитуды
двух волн сокращаются, и если атамы имеют равную рассеиваю-
щую способность, то интенсивность полностью исчезает. Напри-
мер, если элементарные ячейки являются объемно-центрирован-
ными, то каждая содержит атом в положении x = y = z=-^-, и поэ-
тому
А, В-фазовый сдвиг гь (Л —|—А—V-£).
Следовательно, все рефлексы, соответствующие нечетным значе-
ниям суммы /i + Аф!, пропадают, так как фазы смещены на 180°.
Это означает, что картина рассеяния от объемно-центрированной
кубической решетки может быть построена с учетом примитивной
решетки просто вычеркиванием всех рефлексов, для которых сумма
нечетна. Обнаружение этой картины в спектре порошка
(рис. 22.10) сразу указывает на объемно-центрированную решет-
ку. Подобный расчет можно сделать п для гранецептрированпой
решетки; эта картина линий также показана на рис. 22.10,
Если амплитуда волны, рассеянной от атомов А, равна (Л око-
ло детектора, то амплитуда волны, рассеянной от В, равна
fs ехр[2л/ (hx+kyd-lz) ] из-за дополнительного фазового сдвига.
Поэтому общая амплитуда около детектора равна сумме
F=fА+Fb е*р 12лг (hx -ф ky -ф /г)],
а интенсивность
4«~A*A=(Fa + Fb^P {2ziiT (hk!)} Р,
Поскольку интенсивность излучения пропорциональна квадрату мо-
дуля амплитуды волны. Для простоты мы использовали сокраще-
ние Т (hk!) ^=hx^~ky Ц-lz, Это выражение раскрывается в следую-
щую формулу:
Лы A+Fb-I-FaFb [ехр [2шТ (hkl)} +ехр [—2лГГ(hkl)}] ~
~ А А + 2fAfB cos {2лТ (hkl)}, (22.3.3)
252
Часть 2, Структура
Г.ц.к.(Л, fr.1 все четные или нечетные)
Рис. 22.10. Линии и пропуски в фотографиях- порошков,
что позволяет интерпретировать изменение интенсивности различ-
ных плоскостей в картине для порошка NaCl; длины рассеяния
атомов известны, и поэтому для определения Т (hkl) для каждого
рефлекса можно использовать структуру ячейки.
Определение кристаллической структуры. Если элементарная ячей-
ка содержит атомы с длинами рассеяния fi и имеет координаты
Xia, уф, Ztc, то общая амплитуда волны от плоскости (hk!) может
быть записана в виде простого разложения, которое уже приводи-
лось:
общая амплитуда = hexp (22,3.4)
(
(элемеитар. яч.)
Эта сумма называется структурным фактором и обозначается
Fhki, Поэтому интенсивность рефлекса от плоскости (hkl) кристал-
ла с такой составной элементарной ячейкой будет
^hki| Fhki Г- (22.3.5)
Теперь нужно выяснить общую идею изучения структурного
фактора Кристалла с помощью дифракции рентгеновских лучей.
В структурном факторе заложена структура элементарной ячейки,
так как он связан с природой присутствующих атомов (через fi)
и их положением (через hxi-^kyi-^lZi). Если интенсивности ди-
фракционных пятен можно интерпретировать через структурные
факторы, то получится правильная картина кристаллической
структуры,
22, Дифракционные методы
253:
Рис. 22.11. Расчет структурно-
го фактора.
Пример (вопрос 6). Рассчитай-
те структурные факторы для'
решетки каменной соли NaCl
[две взаимно проникающие
нраиеаен трир ом иные кубиче-
ские (г.н.к.) решетки].
Метод. Вначале рассмотрим ре-
шетку Na+. Элементарная ячей-
ка содержит атомы с координа-
тами (О, О, 0), (0, 1, 0), (0, 1,1)
и т. д. (рис. 22.11). Теперь рас-
смотрим решетку С1”; в ней ноны имеют координаты (0, Oh (^ Ь 0), (яр
ту) и т. д. Длину рассеяния для Na+ записываем как [на, а для С1~ — как:
fci- Отметим, что ионы на гранях подслепы между двумя элементарными ячей-
ками (используем ноны на ребрах — между четырьмя ячейками (использу-
1 1
ем ^-[), а ионы в углах—между восемью ячейками (используем -g- f). Применя-
ем уравнение (22.3.4), суммируя по всем 27 атомам, приведенным на рисунке..
Ответ. Для экономии места мы будем писать в каждой строке только несколь-
ко членов:
4-+4-еХР (2га'ИЧ-------ехР {2л[(Л-|-Й-Н)}-]------
—b4exp{2™(4 Zl+4 fe+ *)}] +
+/ci [ехР j2nl (4*h +"Г А + 4 *)} +
+ 4-еХр{2ж(4-4}Ч-------+4ехр |2ш (4
С учетом того, что ехр(2тЛ)=ехр(2лй) =ехр (2ni7) — 1, так как все h, k, 1—UR--
лые числа и ехр(2л()=1, это выражение значительно упрощается и дает
Fhki— fNaIl-hcos л Ц-cos(7г-]-0 л-|-cos (A-f-Z) л]-]-
+/ci [(—l)fi+^+t-]-eosfort-|-cos /jt-j-cos/гл].
Это выражение также можно упростить, поскольку совЛл = (—l)h, и поэтому:
Л.» =frUl+(- 1)й+*+(-l)ft+z+(- 1О +
+fci к- 1)й+А+1+(- V+(- !)* +
254
Часть 2. Структура
Такого упрощения достаточно для нашей цели. Отметим, что, если все hkl чет-
ные, fflJiJ = fP(a(l-bI-bl-bl)=fci(l-bl-bl + l) =4(f>-*+fci); если все hkl нечетные,
Fxx(=4(f№—fci); если один четный, другой нечетный или наоборот, Елм — О.
Комментарий, Заметим, что для всех четных hkl липин более интенсивные, чем
для всех нечетных hkl. Если f+=f- (как в КС1), то нее нечетные hkl имеют ну-
левую интенсивность, что соответствует систематическим пропускам для простой
кубической элементарной ячейки.
Этот анализ можно продолжить. Мы могли бы и не подразде-
лять элементарные ячейки па атомы, каждый из которых имеет
вклад, равный ft, в картину рассеяния (в ковалентных структурах
это даже невозможно сделать или возможно только как прибли-
жение). Вместо этого мы можем представить себе небольшой
объем dr, расположенный в точке г= (х«, yb, zc) в элементарной
ячейке, и в этом объеме — электронный заряд в количестве
р(г)йт. Рассеяние от этого объемного элемента пропорционально
числу электронов, которое он содержит, и поэтому вместо опре-
деления структурного фактора Fhkt суммированием по всем имею-
щимся атомам можно определить его интегрированием по всем
объемным элементам. Тогда уравнение (22.3.4) заменится па
следующее:
общая амплитуда ^Fhhl ~ J йто (г) ехр {2лi (hxky -|- lz)}. (22.3.6)
ЭЛЯ»г*11Тйр-
яч.
Это выражение сводится к уравнению (22.3.4), если ячейку можно
подразделить на идентифицируемые атомы, но является намного
более общим, так как оно позволяет исследовать электронную
плотность внутри молекул. Уравнение выглядит очень сложно, но
ему можно дать ясную физическую интерпретацию, если исполь-
зовать корпускулярную картину дифракции. Это объясняется в
приложении.
Мы измеряем интенсивности 1нм и из них получаем наблюдае-
мые структурные факторы Fhki- Реально мы хотим знать распре-
деление электронной плотности р (г). Последнее уравнение имеет
форму, которую можно инвертировать;
Р(г)=--2 FA«expi— 2ni(/ix-PAy+(z)]. (22.3.7)
hki
Это называется фурье-синтезом электронной плотности. Если для
всех рефлексов в кристалле известны структурные факторы, то
можно вычислить эту сумму и из нее найти электронную плот-
ность в точке г= (ха, yb, zc) и, следовательно, по всей элементар-
ной ячейке.
Пример (вопрос 5). Рассмотрим плоскость (ЛОО) кристалла, бесконечно длин-
ного по х-направлению. При проведении рентгеноструктурного анализа было
найдено, что структурные факторы для плоскостей имеют следующие значения:
22. Дифракционные методы
255
16(0), - -10(1), 2(2), -1(3), 7(4), —10(5), 8(6), -3(7), 2(8), —3(9), 6(10).
— 5(11), 3(12), —2(13), 2(14), —3(15); цифры в скобках соответствуют величи-
не А и Fh^F-ti. Постройте График электронной плотности вдоль оси х ячейки.
Метод. Используем уравнение (22.3.7), отметив, что, поскольку Fh — F^h,
Р (X) —Рь~Ь2 S Fh cos (2л7Я-
Л>0
Вычисляем сумму для х — 0, 0,1, 0,2, ..., 1,0.
Ответ. Результаты вычисления 16-членпой суммы для 11 положений вдоль эле-
ментарной ячейки приведены на рис. 22.12 (сплошная линия).
Комментарий. Можно легко увидеть положения двух или трех атомов. Чей
больше членов суммируется, тем точнее график плотности: члены, соответствую-
щие высоким величинам h, ответственны за более товкие детали в изменении
электронной плотности; общие особенности можно выявить из низких величин й.
Теперь возникает проблема определить интенсивности всех
дифракционных пятен и установить, какие плоскости (hkl) обус-
ловливают их появление. Это существенная проблема, и здесь
нужно использовать метод монокристалла. Существуют Два ос-
новных экспериментальных метода:
1. Осцилляционная фотография и фотография Вайссенберга.
Простая конструкция осцилляционной камеры приведена на
рис. 22.13. Монокристалл (с типичными размерами 0,1 ммХ
Х0,1 ммХО, 1 мм) закрепляют на калиброванном предметном сто-
лике, называемом головкой гониометра (угломерный инструмент).
Это делается под микроскопом так, чтобы одна из осей кристал-
ла была точно параллельна оси камеры. Рентгеновские лучи вхо-
•256
Часть 2. Структура
— Ловушка
— Кристалл
---Головка
гониометра
—- Пл емка.
Осциллирующий
механизм
Рис. 22.13. Осцилляционная рентгеновская камера.
.дят через щель в пленке, а дифракционные пятна регистри-
руются на пленке, намотанной вокруг цилиндра. Кристалл может
оказаться ориентированным в таком положении, что при первой
экспозиции ня для какой плоскости решетки не будет удовлетво-
ряться условие Брэгга; но, если его повернуть вокруг оси каме-
ры, ориентация плоскостей станет дифрагирующей. Например,
если кристалл вращать так, чтобы ось с была параллельна оси
камеры, то пятна появятся в том случае, когда условию Брэгга
будут удовлетворять плоскости (йАО), которые параллельны оси
с, но имеют различные расстояния dhka. Эти дифракционные пят-
лга лежат на окружности вокруг цилиндра в плоскости, проходя-
щей через падающий пучок: при этом получается одинарная слое-
вая линия. С каждой стороны этой экваториальной слоевой липин
находятся слоевые линии пятен, соответствующих дифракции от
плоскостей (hk 1) и (hk 1). Эти плоскости (hk 1) и (hk 1) образу-
ют угол с осью с и поэтому отклоняют пучок ввер_х или вниз. Слое-
вые линии за ними соответствуют (hk 2) и (hk 2) и г. д.
На практике кристалл вибрйрует в пределах примерно 10°, а
не всех 360°, и из расстояний между слоеввши линиями можно
очень просто определить расстояние между плоскостями с (или
расстояния по любой другой оси). Недостаток простой осцилля-
ционной фотографии состоит в том, что пятна (h, k) нельзя индек-
сировать (в случае Вращения вокруг оси с): можно идентифициро-
вать I из слоевой линии, но h и k идентифицировать нельзя. В ка-
мере Вайссенберга этот недостаток преодолен, потому что внима-
ние сконцентрировано на одинарной слоевой линии благодаря вве-
.дению в осцилляционную камеру экрана, как показано на
22. Дифракционные методы
257
Рентгенов--
сние лучи
(гониометр
установлен
внутри;
Осцилляция
кристалла
Пленка продоль-
ного движении
— Экран
(статический;
— Экран
(статический)
— Пленка (движется
вверх-вниз)
Д— -Этот слой
фотографируется
Рис, 22.14. Камера Вайссеиберга.
рис, 22.14. При вращении кристалла цилиндр из фотопленки дви-
жется вдоль его оси. Точки, которые проявились бы в виде оди-
нарной полосы, теперь распределены по всему цилиндру, и боко-
вое смещение пятна можно использовать для определения, от ка-
кой из плоскостей (Л, А) произошло отражение.
2. Четырехкружный дифрактометр. Наиболее тонким инстру-
ментом, использующимся в настоящее время, является четырех-
кружный дифрактометр (рис. 22.15). Кристалл, установленный на
головке гониометра (вероятно, после выверки в простой осцилля-
ционной камере), может вращаться вокруг трех осей. Детектор,
который обычно является электронным прибором, может также
вращаться вокруг одной из осей. Детектор работает или через
процесс ионизации (отклоненные рентгеновские лучи ионизируют
газ и позволяют проходить электрическому току), или как фото-
умножитель (электрон, выбитый фотоном из экрана, ускоряется
на другом экране, где он способствует появлению большого числа
электронов, и т. д.). Этот прибор можно непосредственно связать
с ЭВМ, с помощью которой можно контролировать углы, при ко-
торых выполняются измерения, и записывать интенсивность ди-
фракции в каждой точке наблюдения.
Проблема фазы. Из измерения мы получаем Fum, а затем со-
ставляем сумму в уравнении (22.3.7), чтобы найти р (г). К сожа-
лению, Ihki пропорциональна квадрату модуля | s, и поэтому
мы не можем сказать, должны ли мы использовать или
— в сумме, которая дает р(г). В действительности ситуация
Менее благоприятна, чем можно предположить, поскольку, если
17—242
258
Часть 2. Структура
мы записываем Ркы как комплексное число | Fhni 1 е’Ч’, где
|Fw| — действительная величина Рим, а ф — его фаза, из интен-
сивности мы можем Определить | Fhki |, но ничего ие можем ска-
зать о ф. Эта неопределенность полного вида Fdhi является непри-
ятной проблемой фазы рентгеноструктурпого метода. Необходимо
найти некоторые пути отнесения фаз к стр5гктурным факторам, в
противном случае сумму для определения р(г) нельзя вычислять,
и рентгеновский метод был бы бесполезным.
Пример (вопросы 6 н 7). Повторите решение предыдущего примера, но предпо-
ложите, что все структурные факторы после имеют одинаковую положи-
тельную фазу.
Метод. Повторим вычисление 16-члечной суммы, ио с положительными значени-
ями F;i после Л=6.
Ответ. Получающаяся электронная плотность графически представлена на рис.
22.12 пунктирной линией.
Комментарий. Этот пример подчеркивает важность проблемы фазы: разный вы-
бор фаз приводит к разной электронной плотности.
Проблема фазы до некоторой степени преодолевается различ-
ными методами, из которых основными являются следующие:
1. Метод проб и оишоок. Если элементарная ячейка проста, то
при установлении структуры можно руководствоваться догадка-
ми. Если предположить фазы Fhu и получить очевидно абсурдную
электронную плотность (например, перекрывающие атомы), то
нужно сделать другое положение, пока структура не станет прием-
лемой.
2. Изоморфное замещение. В сложные молекулы возможно
внедрять тяжелый атом без заметного нарушения формы кристал-
лической структуры; это называется изоморфным замещением
(«изоморфный» означает «той же формы»). Преимущество нали-
чия тяжелого атома состоит в том, что он имеет много электронов
и доминирует в рассеянии и основные особенности кристалдичс-
Рис. 22.15. Четырехкружный дифрактометр.
22. Дифракционные методы
259
Рис. 22.16. Элементарная ячей-
ка (а) и синтез Паттерсона
(б). Если центральный атом
заменен на более тяжелый эле-
мент, то для ячейки в синтез
Паттерсона показан па схеме г.
(5
ской структуры могут
быть выявлены при сов-
местном рассмотрении
двух структур. Затем,
изучая изменение интен-
сивности рефлексов тя-
желого атома под влия-
нием других атомов,
можно вскрыть более
тонкие детали структуры
и таким образом построить картину относительных фаз. Например,
если в присутствии другого атома в элементарной ячейке интен-
сивность пятна уменьшается, то фаза дополнительного вклада от-
рицательна относительно первого случая.
3. Синтез Паттерсона. Паттерсон исследовал возможность ис-
пользования интенсивностей непосредственно в фурье-синтезе
электронной плотности. Найдено, что, если Ны подставить вместо
Гьм в разложение в уравнении (22.3.7), полученная величина бу-
дет картой расстоянии между плотностями рассеяния (т. е. кар-
той расстояний между атомами в элементарной ячейке). Напри-
мер, если элементарная ячейка имеет структуру, приведенную на
рис. 22.16,«, то синтез Паттерсона дал бы карту, показанную на
рис. 22.16,6, где расстояние каждого пятна от начала координат
Дает ориентацию и расстояние между всеми парами атомов в ис-
ходной структуре. Недостаток этого метода состоит в том, что в
структуре из N атомов имеется —1) пар атомов, и поэтому
картина быстро усложняется. Тем не менее, если один из атомов
оказывается тяжелым, то он доминирует в карте Паттерсона, и
можно разгадать особенности, связанные с тяжелым атомом. На
рис. 22.16, в показано, что присутствие тяжелого атома влияет на
синтез Паттерсона. Этот метод можно использовать в сочетании
с методом изоморфного замещения.
4. Прямые методы. Для анализа распределения фаз в диф-
ракционном эксперименте существует статистический метод. В не-
17*
260
Часть 2. Структура
которых случаях можно показать, что некоторые фазы с большой
вероятностью сопутствуют друг другу, и поэтому фазы можно
обосновать статистически. Этот метод с некоторым успехом был
применен к центросимметричным кристаллам (где структурные
факторы будут реальными, следовательно, фазы имеют знак +
или —) и в настоящее время распространяется на более сложные
случаи. Проблема фазы могла бы исчезнуть, если бы фазы можно
было непосредственно измерить, а это может стать возможным,
когда будут созданы рентгеновские лазеры. Применение когерент-
ных рентгеновских лучей произвело бы революцию в рентгенов-
ской кристаллографии.
Методика структурного анализа. Метод связан с доступностью
монокристалла исследуемого вещества, и это привело к значи-
тельным усилиям при получении кристаллов больших биохимиче-
ски важных молекул, таких, как ферменты и белки. Для того
чтобы определить симметрию и оси кристалла, проводится ана-
лиз его внешнего вида. Затем можно провести индексирование
рефлексов, используя методы осцилляции или Вайссенберга, опп
санные выше (доступен также более точный прецессионный ме-
тод'). Кристалл, точно установленный на головке гониометра, по-
мещают в дифрактометр, например четырехкружный прибор уже
описанного типа. Затем определяют и записывают па печатаю-
щем устройстве ЭВМ интенсивность и положение всех дифрак-
ционных рефлексов.
На этой стадии имеются некоторые идеи о структуре распо-
ложения атомов, и поэтому в уравнение (22.3.4) можно подставить
известные факторы атомного рассеяния и оценить приблизитель-
ные значения структурных факторов. В свою очередь из этого
делается предположение о фазах для наблюдаемой Fhkt, следова-
тельно, наблюдаемые амплитуды можно скомбинировать с оценен-
ными фазами, что позволяет сделать первичный синтез электрон-
ной плотности. Рассчитанную таким образом электронную плот-
ность можно нанести на контурную диаграмму, и она покажет
положение атомов и, вероятно, некоторые атомы, вначале не вклю-
ченные в первоначальную оценку структуры. На основе этой струк-
туры можно рассчитать новый набор структурных факторов и
скомбинировать их фазы с наблюдаемыми амплитудами, что при-
ведет к лучшему синтезу электронной плотности. И такая проце-
дура продолжается до тех пор, пока при последующем цикле
вычислений структура не перестанет сильно изменяться.
Затем можно применить различные методы доводки результа-
тов. Например, атомы колеблются около их среднего положения
в структуре, и это расширяет их электронную плотность. Можно
оцепить величину этого эффекта и исключить его из карты элек-
тронной плотности. Таким образом строятся контурные карты элек-
тронной плотности, а из них можно определить равновесные дли-
22. Дифракционные методы
261
Рис. 22.17. Электронная плотность в фталоцианине никеля {Robertson J. М., Or-
ganic crystals and molecules, Cornell University Press, 1953).
ни связей, углы между связями и распределение электронной
плотности. Одна из них приведена на рис. 22.17. На основе та-
ких карт можно построить модели молекулярной структуры; при-
мер дан на рис. 3 (т. 1, «Введение»).
Из приведенных диаграмм, на которых видны многие детали,
можно предположить, что рентгеноструктурный анализ — наибо-
лее удовлетворительный метод изучения структуры молекул. Без-
условно, это было бы истиной, если бы не было ряда ограниче-
ний.
Одно из них состоит в том, что метод рассматривает только
твердое состояние, что является определенным ограничением для
молекул, представляющих биологический интерес. Важно принять.
262 Часть 2. Структура
во внимание ту возможность, что в природной среде биологически
важные молекулы могут быть в значительной степени развернуты-
ми и что в твердом состоянии на стереохимию молекул могут на-
кладываться ограничения. Нужно разработать методы, позволяю-
щие изучать молекулы in vivo с помощью рентгеновских лучей, од-
нако пока такие методы недоступны. В этом отношении часто бо-
лее удобен ЯМР, так как с его помощью можно изучать молекулы
в жидкой среде; использование специальных методик позволяет
изучать структуру ферментов в их естественном окружении. В ча-
стности, при изучении лизоцима методом ЯМР было показано, что
первоначально установленная с помощью рентгеновских лучей
структура содержит ряд ошибок, и это еще раз свидетельствует
о важности исследования сложных структур разными методами.
Другое ограничение (заставляющее дополнительно использо-
вать ЯМР) состоит в невозможности с помощью дифракции рент-
геновских лучей определить наличие атомов водорода в структу-
ре. Это связано с тем, что рассеяние рентгеновских лучей обус-
ловлено наличием электронов и рассеяние от атомов водорода
обычно маскируется другими атомами. При очень тщательной до-
водке Можно увидеть атомы водорода, но метод труден, и ЯМР
может оказать значительную помощь.
Наконец, стоимость полного рентгеновского анализа вещества
может быть значительной. В экономике рентгеновского анализа
следует учитывать исследование возможности получения подхо-
дящего кристаллического образца, а также большой объем счета
на машинах большой емкости.
22.4. Информация, получаемая
из рентгеноструктурного анализа
Связи между компонентами кристаллов могут быть разных ти-
пов. Простейшая из всех — ионная связь между заряженными ио-
нами: кристаллы удерживаются вместе кулоновским притяжением
между противоположными зарядами. Структура кристалла опре-
деляется геометрическими проблемами совместной упаковки попов
энергетически наиболее предпочтительным путем. Противополож-
ностью такого типа связывания является ковалентная связь, ког-
да атомы соединены Друг с другом химическими связями, имею-
щими определенную пространственную ориентацию. Стереохими-
ческие требования валентности более важны, чем простая геомет-
рическая проблема упаковки шаров; с помощью валентных связей
могут образовываться четкие обширные структуры. Превосход-
ным примером является алмаз, структура которого приведена на
рис. 22.18. Кристалл алмаза представляет собой одну огром-
ную молекулу, в которой каждый атом углерода соединен
зр3-гибридными связями с четырьмя соседними атомами в тетра-
22. Дифракционные методы
263
эдричсском расположении. Вещества, образующие такую решет-
ку, часто бывают жесткими и нереакциопноспособными.
Многие органические и некоторые неорганические молекулы
не являются ионными и не имеют свободных валентностей, чтобы
образовывать между собой ковалентные связи. При конденсации
в кристаллы эти вещества сохраняют свою индивидуальность и
притягиваются друг к Другу путем различных вандерваальсовых
взаимодействий того же типа, который обусловливает пеидеаль-
иость газов (т. 1, стр. 26). Эти взаимодействия могут быть ди-
поль-дипольным взаимодействием, если молекулы полярны, или
дисперсионным взаимодействием, возникающим из-за флуктуаций
во времени в их электронном распределении (разд. 23.2), если
молекулы неполярны. Тогда форма кристаллической структуры яв-
ляется отражением проблемы конденсации несимметричных объек-
тов в агрегат с минимальной энергией (в действительности с ми-
нимальной функцией Гиббса; т. 1, стр. 157), и предсказать пред-
почтительную форму очень трудно и почти невозможно. Пробле-
ма еще усложняется при наличии водородных связей, в которых
протон играет роль связи между двумя электроотрицательными
атомами. В некоторых случаях форма кристаллической структуры
определяется водородными связями, например для льда (рис.
22.19), однако в других случаях (например, для фенола) они ис-
кажают структуру, которая в основном определяется различными
вапдерваальсовыми взаимодействиями.
Наконец, существуют металлические связи, когда «море элек-
тронов» течет между катионами, имеющими определенное положе-,
ние, и связывает их вместе в жесткую структуру. В этом случае
кристаллическая форма в значительной мерс определяется спосо-
бом упаковки сферических катионов металла в упорядоченную-,
структуру.
Упаковка идентичных шаров. Кристаллы металлов. Простейшие
типы кристаллической формы ожидаются для металлов, посколь-
ку все «строительные блоки» являются идентичными и сфериче-
скими. Найдено, что большинство металлов кристаллизуется в ви-
де одной из трех простых форм, две из которых могут быть объяс-
нены размещением шаров в плотнейшую возможную упаковку
(рис. 22,20).
Плотноупакованный слой идентичных шаров может образо-
ваться, как показано на рис. 22.20, а, и в углублениях этой решет-
ки может уложиться аналогичный слой. Третий слой может раз-
меститься сверху этого слоя двумя различными способами. Если
он размещен так, Что воспроизводит первый слой (и поэтому при
продолжении этого процесса образуется слоистая структура, ко-
торую можно представить как АВАВАВ...), такая структура назы-
вается гексагональной плотнейшей упаковкой (г. п. у.) (рис.
22.20,6/ При альтернативном размещении третий слой распола-
Рис. 22.18. Структура решетки алма-
за
Рис_ 22.19. Кристаллическая структу-
ра одной из форм льда (отметьте
алмазоподобную структуру).
Рис. 22.20. Плотнейшая упаковка идентичных шаров.
22. Дифракционные методы
265
Рис. 22.21. Расчет эффективности упаковки.
гается так, что он не воспроизводит первый (тогда образуется
слоистая структура АВСАВС...); эта структура называется куби-
ческой плотнейшей упаковкой (к. п. у.) (рис. 22.20,6}. Рисунок
также показывает, что в случае к. п. у. возникают гранецентриро-
ванные элементарные ячейки, и поэтому к.п.у. можно также обо-
значить как г.ц.к.
Обе структуры (г.п.у. и к.п.у.) являются плотноупакованными.
Мерой этого является число атомов, окружающих один выбран-
ный атом: в каждом случае координационное число равно 12. Ме-
таллы, имеющие такие структуры, указаны на рис. 22.20.
Пример. Рассчитайте относительную часть гексагональной плотнейшей упаковки
элементарной ячейки, которую занимает пустое пространство.
Метод. Предполагаем, что ионы являются идеальными шарами с радиусом Я.
Обращаемся к рис. 22.2! и используем немного тригонометрии.
Ответ. Площадь основания элементарной ячейки (окрашенной в серый цвет)
равна 31?а^‘2/?= (2УЗ)7?г. Высоту получим так, как показано ниже. Следующий
слой атомов лежит выше точки В. Она находится на расстоянии P/cos ЗОэ=22?уЗ
ат центра соседнего нона. Центр ионз, лежащего сверху, находится на расстоя-
иии 2Д от только что указанного иона, т. е. он находится на высоте
К[(2/?2)—(2tf7'3j=]=2(=/3),/aJ?. Высота самой элементарной ячейки в два раза
больше, т. е. равна 4(!/$j1'fs^. Объем элементарной ячейки равен.
площадь)X (высота) = (2 /3) (2/3)1/е £ = (8 /2) R3.
На каждую элементарную ячейку приходится два полных иона, и поэтому на
долю каждого приходится объем, равный (4>'2)Я\ Объем сферического иона
равен (4/s)rt^3‘ Следовательно, объем заполненного пространства будет
^7з)л^-/(4у'2)^3=л/Зу'2=0,74; таким образом, пустое пространство занимает
Комментарий. Такие же доводы можно применять к другим ячейкам. В граяе-
центрированной кубической ячейке свободный объем составляет 26%, в объемно-
центрированной кубической — 32% и в простой кубической решетке — 48%. Эф*-
. Фиктивность упаковки равна соответственно 0,74, 0,68 н 0,52.
) В третьем виде расположения, характерного для обычных ме-
таллов (Ba, Cs, Fe, К, W), атомы первого слоя менее плотно упа-
266
Часть 2, Структура
Рис. 22.22. Решетки хлорида цезия (а) ц хлорида натрия (Я).
кованы, чем в других структурах, и следующий слой лежит в уг-
лублениях первого. Третий слой по своему устройству воспроиз-
водит первый, и поэтому структура относится к типу АВАВАВ...
Исследование элементарной ячейки показывает, что она соответст-
вует объемно-центрированному кубическому (о.ц.к.) расположе-
нию и что координационное число равно 8.
Ионные кристаллы. В ионных кристаллах проблема достижения
низшей энергии усложняется тем фактом, что в этом случае вмес-
те упакованы два сорта шаров: шары имеют не только различные
радпхеы, ио и противоположный заряд. Если случайно ионы ока-
зываются одинакового размера, то достичь плотной упаковки с
координационным числом 12 все же невозможно. Это связано с
тем, что тогда нельзя каждый положительный ион окружить оди-
наковым числом отрицательных ионов, поскольку каждый отрица-
тельный ион должен быть также окружен положительными иона-
ми. Лучшее, чего можно достигнуть, — это координационное чис-
ло 8 в структуре хлорида цезия, которая приведена на
рис. 22.22, а.
Когда размеры ионов различаются больше, чем в CsCl, упаков-
ка превращается в 6-координациоиную (рис. 22.22,6); примером
является структура NaCl. Эту Структуру можно также рассматри-
вать как результат взаимного проникновения двух немного рас-
ширенных г.ц.к.-решеток. Переход от одной структуры к другой
происходит в соответствии с правилом отношения радиусов. Оно
утверждает, что координационное число 8 следует ожидать, когда
отношение радиусов катиона и аниона превышает 0,732, а коорди-
национное число 6 (типа NaCl) — при отношении радиусов ог
22. Дифракционные методы
267
Рис. 22.23. Решетка вюрцита (отметь-
те алмазоподобную структуру).
0,732 до 0,414. Для отношений,
меньших 0,414, наиболее эф-
фективная упаковка приводит
к координационному числу 4,
что характерно для вюрцита
(рис. 22.23) и цинковой об-
манки ZnS. Правило отноше-
ния радиусов хорошо под-
тверждается на опыте, не-
смотря на то что оно найдено
цз простого геометрического
рассмотрения. Отклонение
структуры от предсказанной
формы указывает на сдвиг от
ионного к ковалентному связыванию.
Для оценки отношения радиусов используются ионные радиусы,
которые устанавливаются из большого числа рентгеновских иссле-
дований кристаллических структур. Найдено, что расстояния меж-
ду ионами могут быть выражены в виде суммы двух вкладов, ко-
торые одинаковы для различных соединений. Так, вклад Na+ в
расстояние между ионами в NaX почти постоянен независимо от
природы X. Этот анализ можно распространить на ионы с заря-
дом больше 1; список ионных радиусов дан в табл. 22.1. Такой же
анализ можно провести для вандерваальсовых молекулярных кри-
сталлов, что позволяет составить таблицу вандерваальсовых ра-
Iа б лица 22.1
Ионные радиусы R (пм)
Li+ Вс2+ С2~
60 31 132
Na+ Mg2+ Al34 S«-
95 61 50 184
K+ Ca2+
133 99
Rb+ Sr-+
148 J13
Cs+ Ba2+
169 135
Fe2+ Fe3+ Cu2+ Ag+
74 64 72 126
F"
136
СГ
181
Br"
195
1-
216
263
Часть 2. Структура
Рис. 22.24. Два оптически активных энантиомера виллой кислоты.
Ь-( + )-0инная кислота
диусов молекул и атомов. Эти данные представлены в табл. 22.2
и иллюстрированы графически во «Введении» (т. 1).
Абсолютная конфигурация молекул. Оптически активными моле-
кулами называются такие, которые не могут быть совмещены с их
зеркальным отображением (стр. 299). Хотя уже давно стало воз-
можным разделять оптические изомеры и измерять их одинаковое
и противоположное по знаку оптическое вращение, до появления
рентгеновской кристаллографии абсолютную стереохимическую
конфигурацию данного оптического изомера нельзя было устано-
вить. Теперь, например, можно сказать, что L-винная кислота
(рис. 22.24) является изомером, вращающим плоскость поляри-
зованного света по часовой стрелке (это обозначается знаком +),
а D-випцая кислота вращает против часовой стрелки (—). Рентге-
ноструктурный метод не тривиален, поскольку кристалл одного
изомера дает такую же картину интенсивности, как и кристалл его
зеркального отображения — его энантиомер. Информация об аб-
Таблица 22.2
Вандерваальсовы радиусы R (пм)
Н 120
N О F Ne
150 140 135 234®
320б
Р S Cl Аг
190 185 180 286®
383s
Se Вг
200 195
215
а Из вязкости газа.
й Из плотнейшей упаковки.
22. Дифракционные методы
269
солютной конфигураций содержится в фазе дифрагированного из-
лучения, и для ее извлечения используется специальный метод,
предложенный Бижвое.
Сначала рассмотрим диаграммы на рис, 22.25, а. Они представ-
ляют идеализированный кристалл и его зеркальное отображение.
Каждая плоскость атомов обусловливает появление рассеянной
волны; на рисунке показаны их суперпозиции. Отметим, что две
суперпозиции имеют одинаковую амплитуду, но различаются по
фазе. Поэтому дифракционная картина в случае каждого энантио-
мера имеет одинаковую интенсивность и, как уже сказано, не мо-
жет быть использована, чтобы различать их.
Суть метода Бижвое состоит в включении в молекулу тяжело-
го атома, например рубидия. Этот атом вызывает дополнительный
сдвиг фазы рассеянного рентгеновского луча, поскольку (это про-
ще всего показать на рисунке) рентгеновские лучи имеют тенден-
цию возбуждать его и в результате такого процесса запаздывают
по фазе. Если это аномальное рассеяние обусловлено слоем, отме-
ченным буквой А, то рассеянные волны будут такими, как пока-
зано на рис. 22.25,6. Существенно то, что теперь аномалия в рас-
сеянии дает суперпозиции, которые отличаются по амплитуде, а
не только по фазе, и поэтому интенсивности в обоих случаях бу-
дут разными. Таким образом можно различать энантиомеры.
Рис. 22.25. а — нормальное рассеяние; б— аномальное рассеяние.
270
Часть 2. Структура
22.5. Дифракция нейтронов
Нейтрон, генерированный в атомном реакторе и замедленный
до термических энергий путем ряда столкновений с некоторым
замедли гелем типа графита до тех пор, пока он не достигнет ско-
рости 3,9 км/с, имеет момент 5,2-10“24 кг-м/с и, следовательно,
длину волны 100 им. Это сравнимо с длиной волны рентгеновско-
го излучения, использующейся в дифракции рентгеновских лучей,
и поэтому можно ожидать аналогичные дифракционные явления.
Рассеяние рентгеновских лучей обусловлкно колебаниями элек-
тронов атома: когда падающее излучение заставляет их коле-
баться, они излучают. Рассеяние нейтронов — это ядерпое явле-
ние: нейтроны проходят через электронную структуру молекулы
и взаимодействуют с ядрами с помощью тех же сил, которые от-
ветственны за связывание в ядрах. Вследствие этого нейтроны
рассеиваются более равномерно всеми атомами (рентгеновские
лучи в большей степени рассеиваются тяжелыми атомами, бога-
тыми электронами). Поэтому дифракция нейтронов показывает
положения протонов в молекулярной структуре, тогда как рентге-
новские лучи выявляют их слабо.
Такое различие в чувствительности может вызвать заметный
эффект при измерении длин связей С—Н. Поскольку рентгенов-
ские лучи «чувствуют» накопление электронов,. слабые пики от
атомов водорода в опыте по дифракции рентгеновских лучей отра-
жают локализацию основной электронной плотности, а опа может
быть сдвинута по связи к атому углерода. Такие измерения дают
для длины связи С—Н значение около 80 пм. Однако рассеяние
нейтронов отражает лоложслие самих ядер, и лрл этом получается
длина связи С—Н, равная 308 пм.
Другим свойством нейтронов, которое отличает их от рентге-
новских фотонов, является присущий им спиновый магнитный мо-
мент. Этот магнитный момент может взаимодействовать с магнит-
ными полями ионов в кристаллической решетке (если они имеют
несцарепиые электроны) и изменять дифракционную картину.
В этом заключена другая причина, обусловливающая проведение
опытов по дифракции нейтронов, несмотря па то что источники
дороги, а пучок слаб (так как нужно отобрать монохроматический
луч с одной и той же скоростью нейтронов).
Простым примером такого магнитного рассеяния служит ди-
фракционная картина от металлического хрома. Решетка является
объемно-центрнрованпой кубической, и поэтому следует ожидать
серии систематических пропусков в дифракционной картине, ког-
да h-f-k-f-l— нечетное число (как описало для рентгеновских лу-
чей на стр. 250), На самом деле систематические пропуски в опы-
те по дифракции нейтронов не наблюдаются, поскольку структура
такова, что атомы в центрах кубов имеют магнитные моменты,
22. Дифракционные методы
271
Рве. 22.26. Две взаимопроникающие магнитные
решетки.
противоположные магнитным момен-
там атомов в вершинах, и структуру
лучше рассматривать как две взаимо-
проникающие решетки (рис. 22.26).
Так как чередующиеся слои по отно-
шению к нейтрону различны (по иден-
тичны для рентгеновских лучей), то
основа для систематических пропус-
ков теряется, и на дифракционной картине появляются пятна.
Дифракция нейтронов особенно важна для изучения таких маг-
нитно-упорядоченных решеток.
22.6. Дифракция электронов
С помощью разности потенциалов электроны можно ускорить
до точно известных энергий. Если ускоряющее напряжение равно
10 кВ, то они приобретают длину волны, равную 12 пм, и стано-
вятся пригодными для изучения дифракции молекул. При взаимо-
действии с зарядами электронов и ядер в молекуле электроны
сильно рассеиваются. Поэтому они не пригодны для изучения мас-
сивных образцов. Тем не менее они очень подходят для точного
измерения молекул в газовой
фазе и для изучения поверхностей и
тонких пленок.
Типичный прибор для дифрак-
ции электронов изображен па рис.
22.27. Электроны «выкипают» из
горячей нити слева и ускоряются,
проходя через разность потенциа-
лов. Затем они проходят через га-
зообразные пары образца к флуо-
ресцирующему экрану.
Газообразный образец пред-
ставляет собой все возможные
Рнс. 22.27. Прибор для дифракции электро-
нов.
272
Часть 2, Структура
Рис. 22.28. Дифракция электронов для паров ферроцена в ее внешний вид по-
сле вычитания нежелательного фона.
ориентации рассеивающих связей атом — атом по отношению к
пучку электронов, поэтому картина дифракции является наложе-
нием рассеяния, обусловленного всеми ориентациями. На дифрак-
ционной картине это проявляется в виде концентрических волно-
образных линий на фоне меньшей интенсивности (рис. 22.28, а).
Волнообразные линии можно приписать явно выраженному рас-
сеянию от положений ядер, а фон — гораздо мецее заметному
вкладу от непрерывного распределения электронной плотности в
молекуле. Для того чтобы устранить нежелательный фон, перед
экраном, в частности, помещают вращающийся диск с радиаль-
ной прорезью; это позволяет внешние части экспонировать боль-
ше, чем внутренние, и помогает резче выявить волнообразные ли-
нии (рис. 22.28, б).
Рассеяние от пары атомов, разделенных расстоянием Ra и
ориентированных под некоторым углом к падающему пучку, мо-
жет быть рассчитано. Затем можно рассчитать общую дифрак-
ционную картину, учитывая вклады от всех возможных ориента-
ций этой пары атомов. Другими словами, проводится интегрирова-
22, Дифракционные методы
273
ние по всем ориентациям, Это приводит к выражению
(0) (1-1-sin (5Ки)/5«пs=(4n/X)sin -J-0,
где X — длина волны электронного пучка и 0 — угол рассеяния.
Коэффициент электронного рассеяния [е — Это мера интенсивности
рассеивающей способности атомов. Если молекула состоит из ря-
да атомов с рассеивающей способностью Л, то общая интенсив-
ность связана с угловым изменением, которое определяется урав-
нением Вирля:
/(e)=S^^in(s^/s^r
(22.6.1)
Картина электронной дифракции дает расстояние не только
между связанными друг с другом парами атомов, но и между все-
ми возможными парами атомов. Поэтому анализ многоэлектрон-
ной молекулы может быть очень сложным. Если имеется лишь не-
сколько атомов, то пики можно проанализировать достаточно бы-
стро; анализ состоит в предсказании геометрии и расчете картины
интенсивности па основании уравнения Вирля, Лучший результат
считается истинной геометрией молекулы.
Мы опишем лишь одно из применений метода, которое пока-
зывает, как он дополняет рентгеновские измерения. При рентгено-
структурном анализе кристалла ферроцена выяснилось, что моле-
кулы имеют заторможенную конфигурацию (рис. 22.29, а). Однако
из газофазной дифракции электронов следует, что молекула име-
ет точно заслоненную конфигурацию и что атомы водорода откло-
нены по направлению к атому железа под углом примерно 5°. Это
показывает, как важно при теоретическом обсуждении молекуляр-
ной структуры отделять эффект кристаллической упаковки от соб-
ственно валентных сил-
Метод дифракции электронов позволяет получить точные дли-
ны связей и углы для разнообразного множества изолированных
молекул; некоторые данные приводятся в табл. 22,3, Применитель-
но к поверхностям этот метод часто называют методом дифракции
медленных электронов (ДМЭ), с его помощью изучают, например,
основной вопрос о конфигурации активных центров адсорбции. Из
18—242
Рис. 22,29, Структура ферроцена, оп-
ределенная в твердой фазе Дифрак-
цией рентгеновских лучей (а) и в га-
зовой фазе дифракцией электро-
нов (б),
274
Часть 2. Структура
Таблица 22.3
Типичные ковалентные радиусы 7? (пм)
н— 30 О— 66
с— 77 0= 57
с= 67
60 С1— 99
с= 69,5 (бензол) Вг— 114
N— 70 (амино) I— 133
N— 70 (нитрат) S— 104
Л = 65 (нитрат) s= 95 (сульфат)
изменения дифракционной картины можно получить картину цент-
ров адсорбции и связать ее со структурой чистой поверхности.
Приложение. Дифракция как свойство частиц
Основополагающим принципом рассмотрения дифракции как
свойства частиц является сохранение момента при соударении.
Мы начнем с вывода на этой основе условия Брэгга, а затем пе-
рейдем к обобщению, которое приведет к интерпретации уравне-
ния (22.3.7). В данном приложении используется ряд идей кван-
товой теории, рассмотренной в гл. 13 (т. 1). Цель состоит в том,
чтобы показать, пто фурье-синтез для дифракции рентгеновских
лучей можно четко интерпретировать физическим способом.
Рассмотрим упругое (сохраняющее поступательную энергию)
столкновение между частицей и неподвижной стенкой (рис. 22,30).
Сохранение линейного момента налагает два условия:
сохранение горизонтального момента: pcos0 — р' cos0',
сохранение вертикального момента: psin0 — pw—p'sinO',
где —конечный момент стенки, Поскольку соударение упру*
гое, величина момента частиц одинакова до ц после соударения,
и поэтому р' = р. Из первого условия получаем 0 = 0', а из второго
2psin0 = pw,
Согласно соотношению де Бройля
(13.2.6) (т, 1), момент частицы (в
случае фотона) можно выразить
как длину волны через p=-h!T, сле-
довательно, последнему уравнению
Рис. 22.30. Фотонная картина рефлекса.
22. Дифракционные методы
275
Рис, 22.31. Структура цепочки атомов, представленная в виде суперпозиции ряда
волн.
можно придать форму, которая уже близко напоминает условие
Брэгга:
2sin0 — py^/h, (22,1)
Если бы стенка могла принимать все моменты, то это условие мог-
ло бы удовлетворяться при всех значениях 0; если можно пока-
зать, что момен? pw ограничен некоторым целым кратным hid, то
можно было бы получить условие Брэгга.
Ключ к пониманию того, почему pv должен быть квантован,
можно получить, изучая структуру стенки, Рассмотрим вертикаль-
ную колонку атомов (рис. 22.31). На рисунке также изображено
изменение электронной плотности в этой колонке, В случае рент-
геновских лучей нас интересуют соударения между фотоном и
электронами в твердой фазе. Следовательно, р« в уравнении (22.1)
должен представлять момент, который могут иметь электроны в
направлениях вверх и вниз по колонке атомов. Момент любого
Электрона в структуре определяется длинами волн в волновой
функции через соотношение де Бройля p^ = h/Ke, и поэтому для за-
вершения вывода условия Брэгга достаточно найти разрешенные
значения длины волны волновых функций,
Чтобы решить эту проблему, необходимо принять, что волно-
вые функции на рис. 22.31 можно рассматривать как суперпози-
18‘
276 Часть 2. Структура
цию множества волновых функций с длинами волн d, d/2, d/3,....
Поэтому электроны в колонке атомов могут обладать моментами
h/d, 2h/d, 3h/dt следовательно, pw должен быть некоторым це-
лым числом, кратным h/d. Таким образом, при отражении фотона
от колонки атомов момент может измениться лишь па величину,
целочисленно кратную h/d, т. е. p^^nhjd.
Отсюда
2sin0 = [n(h/d)} (k/h)=nk/d,
что представляет собой условие Брэгга.
Волновая функция для электронов, приводящая к условию
Брэгга, является суперпозицией волн:
ф—cos (2лд/й) -Сcos (4ax/d)4- • • • - - 2 c°s {2/a?/d). (22.2)
!=!
Такая волновая функция — суперпозиция бесконечного числа гар-
монических волн — представляет собой серию резких выступов,
разделенных расстоянием d. В реальном кристалле электронная
плотность распределена в пространстве по-разному, и волновые
функции электронов имеют менее резко выраженные и менее ло-
кализованные пики. Нарушение гармоничности волн приводит к
уравнению, отличающемуся от приведенного выше простого урав-
нения, и поэтому разрешенные моменты также изменяются. В этом
состоит основной принцип изучения кристаллов с помощью ди-
фракции: дифракционная картина представляет собой моменты,
которые может иметь электронная структура, и разрешенные мо-
менты зависят от распределения электронной плотности.
Это положение можно развить следующим образом. Электроны
распределены с плотностью р(г), где г — некоторая точка в кри-
сталле. Эта плотность определяется электронной волновой функ-
цией, причем связь имеет вид р (г) =ф* (г)ф (г). Волновую функ-
цию ф можно разложить на суперпозицию гармонических волн, и,
если в разложение входит длина волны К, электрон имеет некото-
рую вероятность обладать моментом h/Х. Чтобы фотон отразился
от образца с изменением момента Ар (который в действительности
нужно рассматривать как вектор), электроны в кристалле долж-
ны быть способны претерпевать изменение момента Др в подходя-
щем направлении. Другими словами, если электрон первоначаль-
но описывается волновой функцией фр, то он должен быть спосо-
бен перейти в состояние фР+дР. Это разрешено, если общая вол-
новая функция ф содержит оба компонента.
Предположим, что общую волновую функцию можно записать
как
(г)=2м>,(г),
р
22. Дифракционные методы
277
где фр (г) — волновая функция, соответствующая определенному
моменту, а коэффициент сР определяет, в какой степени фР вносит
вклад в ф. Способность электронов образца принимать момент
Лр зависит от величины некоторого вклада фр в волновую функ-
цию п от величины вклада фр+др- Поэтому общая способность
принимать момент Лр, а следовательно, амплитуда рассеяния F на
некоторый угол, может быть выражена как сумма:
CpCp-j-Ap (22,3)
р
(мы допускаем возможность, что коэффициенты могут быть слож-
ными). Сумма берется потому, что нужно рассматривать переходы
р=>1Р+Лр от всех компонент фр.
Теперь необходимо найти величину коэффициентов ср. Волно-
вая функция для состояния с определенным моментом р в направ-
лении х имеет вид (т. 1, разд. 13.3)
фр(л)~ехр(!рл/Л).
Следовательно, уравнение, определяющее сР для этой одно-
мерной системы, записывается как
р
Если это умножить па ехр(—tp'x/fi^ и проинтегрировать по х от
одного края кристалла (где х=—ту- Д) до другого (где х = —Д)>
то получим
ь/а 1,-2
jtop (х) ср j dx exp [i (p—p') x/h]. (22.4)
-L/3 P —L!2
Обе части последнего уравнения можно значительно упростить.
При переходе от ячейки к ячейке Волновая функция ф(х) повто-
ряется, и поэтому вместо интегрирования по всему кристаллу до-
статочно проинтегрировать по элементарной ячейке и получить
общий интеграл умножением на число элементарных ячеек, Если
ячейка имеет размер а в х-направлении, то число ячеек будет
Д/а и левая часть последнего выражения превратится в
L/2 а
J top (х) ег-1Р'х/к =(L/a) Г top (х) r-to'x/k,
-д/а •
Интеграл в правой части уравнения (22,4) является суперпозици
ей большого числа волн, что дает нулевую амплитуду. Единствен'
278
Часть 2, Структура
ный случай, когда этого не происходит, соответствует р^р', так
как тогда интеграл равен
dx = E.
Следовательно, в сумму в правой части уравнения (22,4) из
всех р вносит вклад только р', и поэтому сумма упрощается до
LcP’, Теперь мы имеем простое выражение для коэффициента ср
(или, заменяя р' па р, для сР):
Jя
ср = (1 /а) J 7хф (х)
о
Интеграл этого вида называется фуръе-преобразованием периоди-
ческой функции ф(х).
Амплитуду рассеяния можно выразить через волновую функ-
цию ф(х). Подстановка последнего выражения в уравнение (22,3)
приводит к
а
F — (К/й2) j dxi|? (х) ехр {—ipxjh.) j dx\p* (хг) exp {£ (p-j-Ap) x’/h] =
p 0 0
a a
= (K/a2) 2 j <*x J (x) Ф* (x') exP {*P (x’ — x)/h} exp(iApx7/t),
p b о
где Д’—некоторый коэффициент пропорциональности. Теперь
можно получить сумму по р, так как она содержится только в
последнем экспоненциальном члене. Эта сумма /jexpftp (х'-~х)/А]
р
представляет собой суперпозицию волн с широким интервалом
длин волн; таким образом происходит аннигиляция, и сумма
стремится к нулю. Если, однако, х — х', то сумма нс стремится к
пулю, поскольку экспоненциальный член превращается в едини-
цу. Следовательно, единственным членом, который остается в при-
веденном выше выражении, является член с х=х', и поэтому
a
Г (Дщ) j" tfxvp (х) ф* (х) ехр ДДрХ/Й).
о
Видно, что ф (х)ф* (х) — электронная плотность в точке х, т. е.
р(х); а значит, мы получили важный результат, состоящий в том,
22, Дифракционные методы.
279
что рассеяние пропорционально фурье-преобразованию электрон-
ной плотности^
а
F ~ J dxp (х) ехр (гДрх/й).
О
Это уравнение уже близко напоминает уравнение (22.3,6).
Далее мы подставляем величину Др. Когда фотон с длиной
волны А соударяется с колонкой электронной плотности и откло-
няется на угол 26, изменение момента в х-паправлении равно
(2ft/Z)sin0= (4лй/Ъ)51п0. Если 0 расположен так, что наблюдается
(й00) -рефлекс, то, поскольку для данной решетки (1ыя=аЦ1,
sin6 —j./2dhoo = /r?./2n. Следовательно, величина Др для такого рас-
положения будет Др = 2лМ/а (й— индекс, п—постоянная Планка,
деленная на 2л), и амплитуда излучения от всей элементарной
ячейки, когда наблюдается (А00)-рефлекс, равна
л
Faoo Jdxp(х)ехр(|’2лйх/п),
b
а интенсивность этого излучения пропорциональна |Е|а.
Только что сделанные преобразования можно без труда рас-
пространить на трехмерную систему. Тогда электронная плотность
в элементарной ячейке с размерами а, й, с будет зависеть от х, у,
г, и амплитуда (W)-рефлекса будет равна
а Ь £
ООО
Это основное уравнение рентгеновской Кристаллографии, которое
идентично уравнению (22.3.6)
оно содержит х/а).
Таким образом, в данной
ческой структуры амплитуды
которые принимает решетка, а интегралы Фурье дают
электронной плотности, необходимый для того, чтобы найти при-
емлемые моменты.
(22.5)
(22.6)
ky
b
(за исключением того, что вместо х
интерпретации анализа кристалли-
рассеяния указывают на моменты,
анализ
Литература
Wells A. F., Tire third dimension in chemistry. Clarendon Press. Oxford, 1956
{reissued 1988).
Уитли П. Определение молекулярной структуры. Пер. с англ. — М.; Мир. 1970.
Гласкер Дж.. Трублад К. Анализ кристаллической структуры. Пер. с англ. — М.:
Мир. 1974.
Brand J. С. D., Speakman J. С., Molecular structure; the physical approach Ar-
nold, London, 1960.
280
Часть 2, Структура
Buerger N. J., Elementary crystallography, Wiley, New York, 1956.
Buerger Af. J., Introduction io crystal geometry, McGraw-Hill,. New York, 1971.
Lipscomb 117, V., Jacobson R, A., X-ray crystal structure analysis, in Techniques
of chemistry (Weissberger A. and Rossiter B. W., eds.), Vol. HID, 1, Wiley-
Interscience, New York, 1972.
Stoui G. H., Jensen L. H., X-ray structure determination, a praclical guide, Mac-
millan, New York, 1968.
Woolfson M. AL, An introduction io X-ray crystallography, Cambridge University
Press, 1970.
Bartell L. S., Electron diffraction hy gases, in Techniques of chemistry (Weissber-
ger A. and Rossiter B. W, cds.), Vol. HID 125 Wiley-Intersciciiccs, New York,
1972.
Rymer T. B., Electron diffraction, Chapman and Hall, London, 1970.
Sutton L. E. (ed.), Tables of interatomic distances and configurations of molecu-
les, Chem. Soc. Special Publication, No. 11, 1958 (Supplement, Special Publi-
cation, No. 18, 1965).
Wells A. E., Structural inorganic chemistry (4th ed.), Clarendon Press, Oxford,
1975.
Wycoff R. W. G.j Crystal structure (5 sections and supplements), Wiley-Inlerscien-
ce, New York, 1959.
Задачи
22.1. Первые несколько задач познакомят с расчетами, которые могут быть про-
изведены на простых решетках. Вначале нарисуйте расположение точек, соответ-
ствующее двумерной прямоугольной решетке, состоящей из элементарных ячеек
со сторонами а, Ь. Укажите плоскости со следующими индексами Миллера:
(10), (91), (UJ, (12), (23), (41).
22.2. Перестроите решетку так, чтобы ось b имела с осью а угол 60°. Укажите
те же плоскости, что в предыдущей задаче,
22.3. Рассчитайте расстояние между плоскостями (11) для решеток из преды-
дущих задач,
22.4. Плоскости кристалла проходят через оси кристалла в точках (2а, ЗЬ, с),
(а, Ь, с), (6а, 36. Зс), (2а, —-ЗЬ. —Зс). Каковы для них индексы Миллера?
22.5. Нарисуйте ромбическую элементарную ячейку и укажите плоскости (100),
(010), (001), (011), (101) и (Ц1).
22.6. Нарисуйте триклинную элементарную ячейку и укажите те же плоскости.
22,7. Каково расстояние между плоскостями с индексами (111), (211) и (100)
в кристалле, в котором кубическая элементарная ячейка имеет сторону 432 пм
(100 пм=1,00 А).
22.8. На заре рентгеновской Кристаллографии было крайне необходимо знать
длины волн рентгеновских лучей. Один метод состоял в измерении дифракцион-
ного угла для механически управляемой решетки, к которой рентгеновские лучи
подходили под углом скольжения. Другой метод состоял в оцевке расстояния
между плоскостями решетки из измерений плотности кристалла. Плотность
NaCl равна 2,17 г/см3, и При использовании -излучения палладия рефлекс
наблюдался при 6°0’. Какова длина волны рентгеновских лучей?
22.9. В своей книге «Рентгеновские лучи и кристаллические структуры» (которая
начиналась словами: «Прошло два года с тех пор, как-д-р Лауэ высказал
идею...») Брэгги привели ряд простых примеров рентгевоструктурвого анализа.
Например, они сообщили, что рефлекс первого порядка от плоскости (100) КС1
происходит при 5°23', но для NaCl — при 6°0’, причем применялись лучи с оди-
наковой-Длиной волны. Если сторона элементарной ячейки NaCl равна 564 пм,
то каков размер ячейки КО? Плотности КС1 и NaCl составляют соответственно
1,99 п 2,17 г/см3. Свидетельствуют ли эти величины о правильности проведен-
ного анализа?
22. Дифракционные методы
281
22.10. Монокристалл нитрата калия имеет ромбическую элементарную ячейку с
размерами а=542 пм, Ь=917 пм и с=645 пм. Рассчитайте дифракционный угол
для рефлексов первого порядка от плоскостей (100), (010) и (111) при исполь-
зовании Ка- излучен и я меди (154,1 пм).
22.11. Хлорид меди(1) образует кубические кристаллы с четырьмя молекулами
на элементарную ячейку. В фотографии порошка присутствуют только рефлексы
или со всеми четными индексами, или со всеми нечетными индексами. Какова
природа элементарной ячейки? Отметим, что на такой вопрос о форме элемен-
тарной ячейки можно Ответить, ничего не зная о длине волны излучения; она
определяется симметрией, а не размерами.
22.12. Фотография дифракции порошка вольфрама имеет линии с индексами
(ПО), (200), (211), (220), (310), (222), (321), (400),... . Каков тип данной эле-
ментарной ячейки?
22.13. Каменная соль (NaCl) образует кристаллы с простой кубической решет-
кой со стороной 564 пм. Каково расстояние между плоскостями (100), между
плоскостями (]11) и между плоскостями (012)?
22.14. Покажите, что расстояние между плоскостями (hkl) в кристалле с ромби-
ческой элементарной ячейкой со сторонами а. Ь и с дается формулой =
22.]5. Часто нам необходимо знать объемы элементарных ячеек. Покажите, что
объем триклинной элементарной ячейки со сторонами а, b и с и углами а, Р
и у равен
V=abc [ I —cos3 а — cos3 0—cos3 7 -|~2 cos a cos р cos т]хЧ
Отметим, что при соответствующем выборе углов и длин это выражение приме-
нимо к моноклинной и ромбической ячейкам.
22.16. Нафталин образует моноклинные кристаллы с двумя молекулами в каж-
дой элементарной ячейке. Стороны находятся в отношении 1,377 : ] : 1,436 и р =
= 122°49'. Удельный вес равен 1,152. Каковы размеры ячейки?
22.17. Этот тип расчета можно обратить и скомбинировать рентгеновский анализ
с измерениями плотности, чтобы найти число Лвогадро. Плотность LiF состав-
ляет 2,601 г/см3. (111 )-Рефлекс происходит под углом 8°44' при использовании
рентгеновских лучей от молибдена с длиной волн 70,8 пм. Найдите число Аво-
гадро.
22.]8. Сколько атомов находится в; а) примитивной кубической элементарной
ячейке, б) о. ц. к.-ячейкс, в) г. ц, к.-ячейке и г) элементарной ячейке решетки
алмаза?
22.19. Покажите, что максимальная часть доступного объема, которая может
быть заполнена твердыми шарами, равна; а) 0,52 для простой кубической решет-
ки, б) 0,68 для о. ы. к.-решетки ив) ft 74 для г, ц, к.-решетки. Эти числа представля-
ют собой эффективность упаковки решетки. Какой метод упаковки наиболее
экономичен для морской перевозки (твердых) апельсинов?
22.20. Какова эффективность упаковки для длинных, прямых цилиндрических мо-
лекул (или труб)?
22.21. Единственным известным примером металла, который кристаллизуется в
примитивную кубическую решетку, является полоний. Элементарная ячейка име-
ет размер 334,5 пм. Предскажите вид дифракционной картины при использова-
нии рентгеновских лучей с длиной волны 154 пи. Какую плотность полония вы
предскажите? Предположим, что полоний упакован в г- Ц- к.-решетку, но имеет
тот же атомный радиус. Какова была бы его плотность?
22.22. Расстояние между плоскостями (100) металлического лития равно 350 пм.
Его плотность составляет 0,53 г/см3. Какого типа его решетка; г. ц. к. или о. ц. к.?
22.23. Медь кристаллизуется в г. ц. к.-решетку со стороной 361 пм. Предскажи-
те вид дифракционной картины ее порошка при использовании рентгеновских
лучей с длиной волны 154 пм. Какова плотность меди?
282
Часть 2. Структура
a w
б Си -—4io°h—
Рис. 22.32. Дифракционная картина порошков вольфрама (а) и меди (б).
22.24. Коэффициент линейного термического расширения меди равен ] ,6] -10_; К-’
при комнатной температуре. Каково расстояние между углами (1 1 ])-рефлексов,
полученных при использовании рентгеновских лучей с длиной волны 70,9300 пм
при 100 ц 300 К? Какие другие изменения во внешнем виде дифракционной кар-
тины можно ожидать при нагревании образца?
22,25, Рассчитайте коэффициент термического расширения алмаза из тех дан-
ных, чгО при нагревании кристалла от 100 до 300 К (111 )-рсфлекс сдвигается
от 22°2'25"до 21°57'59", если используются рентгеновские лучи с длиной волны
154,0562 пм.
22.26. Хром кристаллизуется в о. ц. к.-решетку со стороной 288 пм. Предскажи-
те дифракционную картину его порошка при использовании рентгеновских лучей
с длиной волны 154 пм.
22.27, Оксид кальция имеет кубическую решетку, и элементарная ячейка со сто-
роной 480 пм содержит четыре молекулы СаО. Какова плотность кристаллов?
22.28. Дифракционная картина порошка КС! в камере радиусом 28,7 мм содер-
жит линии на следующих расстояниях от центрального пятна на пленке: 14,5:
20,6; 25,4; 29,6; 33.4; 37,]; 44,0; 47,5; 50,9; 54,4; 582; 62,1; 66,4; 78,1 мм.
Индексируйте эти линии. Использовалось излучение меди (]54 пм). Каков
размер элементарной ячейки?
22.29. В том же приборе при той же длине волны излучения LiF да,т дифрак-
ционные линии на следующих расстояниях от центрального пятна: J8,9; 22,1;
31,9; 38,3; 40,4; 48,9; 55,0; 58,0; 68,8; 83,3 мм. Сделайте вывод о природе и раз-
мерах элементарной ячейки.
22.30. Дифракционные картины порошков вольфрама (а) и медн (б) приведены
па рис. 2252. Обе были получены при использовании рентгеновских лучей с дли-
ной волны ]54 пм; шкала указана на рисунке. Определите тип кубической ячей-
ки в каждом случае и найдите расстояние в решетке. Каковы кристаллические
радиусы атомов вольфрама и меди?
22.31. Хлорид ртути(11) имеет ромбическую решетку, и при использовании
СнКа-иалучения (100)-, (010)- и (001)-рефлексы находятся соответственно при
7°25', 3Q28f п 1043'. Плотность кристалла равна 5,42 г/см3. Каковы размеры эле-
ментарной ячейки и сколько единиц HgCl2 она содержит?
22.32. Предыдущую задачу можно развить несколько дальше. Что говорят о сим-
метрии элементарной ячейки следующие четыре наблюдения: а) для (hkl)-реф-
лексов цс наблюдается систематических пропусков, б) (Л0/) пропускаются для
нечетных в) (hkO) пропускаются для нечетных k, г) для (Okl) не наблюдается
систематических пропусков.
22.33. Настоящий жемчуг состоит из концентрических слоев кристаллов кальци-
та, н которых тригональные осп ориентированы вдоль радиусов. Ядро искусст-
венного жемчута представляет собой кусочек перламутра, которому на токар-
ном станке придана форма шара, затем устрица откладывает концентрические
слои кальцита па этом центральном зерне. Предложите рентгеновский метод,
позволяющий отличать настоящий жемчуг От искусственного.
22. Дифракционные методы 283
22.34. Координаты (в единицах а) атомов в простой кубической решетке таковы:
(О, 0, 0). (0, 1, 0), (0, 0, 1), (0, 1, 1), (I, 0, 0), (I, 1, 0), (1, 0, I) и (J, 1, ]).
Рассчитайте структурные факторы Fkm на основе того, что все атомы одинаковы.
F определяется по уравнению (22.3.4).
22.35. Координаты атома А с длиной рассеяния /л в о. ц. к.-элементарной ячейке
такие же, как в предыдущей задаче, а атом В с длиной рассеяния /в лежит ирн
(др ~2~, ~2 ) Найдите коэффициент рассеяния Fhut и предскажите влд дифрак-
ционной картицы порошка в случаях, когда: a) fA = /, /в = 0, б) fa= ту/л и
в) А = Гв-Л
22.36. Обратимся к задаче 22.8. Почему (3J1 )-рефлекц намного менее интенсивен,
чем (222)-рефлекс? Вы должны обратить внимание на структурные факторы.
22.37. Синтез Паттерсона дает пятна с плотностью интенсивности, соответствую-
щей длине и направлению векторов, связывающих рассеивающие центры в эле-
ментарной ячейке. Нарисуйте эскиз картины Паттерсона, которая соответствует:
а) плоской треугольной молекуле ВЬ3 и 6) атому углерода в бензоле.
22.38. В таблице на стр. 284 приведены полученные на ЭВМ электронные плотно-
сти (в произвольных единицах) для одного сечения через элементарную ячейку
LiCN. С помощью визуального просмотра таблицы нарисуйте контуры, которые
указывают расположение групп Li и CN. Для контурных линий возьмите шаг
примерно в 20—30 единиц. (Данные заимствованы из кциги: Bijvoet J., K/olk-
meyer N. H., MacGiUary С. H., X-ray analysis of crystals, Butterworths, 1951.)
22.39. Канова длина волны электронов, ускоренных напряжением: а) 1 кВ,
б) 10 кВ и п) 40 кВ?
22.40. Какую скорость должны иметь нейтроны с длиной волны 50 пм? Какова
длина волны нейтронов, которые термализованы соударением с замедлителем
(тяжелой водой) при 300 К?
22.41. В качестве первой стадии для пэяакомленнл с настоящим элеятрояогрэфи-
ческим анализом возьмите уравнение Вирля [уравЕюнце (22.6.1)] и рассчитайте
ожидаемое распределение интенсивности дифракции электронов для молекулы
СС14 с неопределенной (до сих пор) длиной связи, но с известной тетраэдриче-
ской симметрией. Возьмите fc1 = 17f и fc = 6f и примите, что Я(С1,С1) =
= (8/3)l/J/?(C, С1). Постройте график зависимости Г/р от х= sR(C, Cl), н вы по-
лучите серию максимумов и минимумов, представляющую ожидаемую дифрак-
ционную картину.
22.42. В настоящем э-тектроиографическом исследовании СС14 были использованы
электроны, которые ускорялись напряжением 10 кВ. Положения максимумов и
минимумов были получены при следующих углах рассеяния. Максимумы: 3°]0'.
5’22', 7°54'; минимумы: 1°4б', 4°6\ 6°4(УГ 9°10'. Проверьте, подтверждает Ли эта
картина максимумов ц минимумов, что молекула является тетраэдрической (ср.
с задачей 22.41), а затем рассчитайте длину связи в СС14.
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 6 12 и 9 6 7 10
1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 7 20 39 42 32 16 10 12
5 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 6 23 60 99 107 86 40 16 17
d7 30 7 0 0 0 0 0 0 0 0 0 0 0 0 0 0 Ю 20 40 81 124 145 102 .32 42
jo 34 5 0 0 0 0 0 0 0 0 0 0 0 0 9 .;u 4t) 50 62 84 85 67 40 34 46
26 6 0 0 0 0 0 0 0 0 0 0 0 0 4 34 61 87 78 60 40 39 ?6 17 17 30
Q 0 1 О 0 0 0 0 0 « 0 c 5 3’3 79 104 90 57 14' S4 6 0 0 ' X
в 0 7 16 18 ]2 0 0 0 0 0 0 0 0 2 21 42 62 58 22 8 0 (1 0 0 0
0 2 21 42 62 58 22 8 0 0 (1 0 0 0 0 7 16 ]8 lj 0 0 0 (1 Cl 0 0
0 5 30 79 104 90 57 24 14 6 0 0 8 0 0 1 1 0 0 0 0 0 0 0 0 0
0 4 34 6l 87 78 60 40 39 26 >7 p 30 26 6 0 0 0 0 0 0 0 0 0 0 0
0 0 9 30 40 50 62 84 85 67 40 34 46 50 34 5 0 0 0 0 0 0 0 0 0 0
0 0 0 0 10 □0 40 81 124 145 102 32 42 47 30 7 0 0 0 0 .0 0 0 0 (1 0
0 0 0 0 6 23 60 99 107 86 40 l6 17 l6 5 0 0 0 0 0 0 0 0, 0 0 □
0 0 0 0 0 7 20 39 42 32 16 10 12 1 0 0 0 0 0 0 <1 0 0 0 0 0
0 0 0 0 0 0 6 12 II 9 6 7 I Cl 0 0 0 0 6 5 0 0 0 0 0 0 0
0 0 0 0 0 0 0 5 6 0 0 0 0 10 7 6 9 ll 12 6 0 0 0 0 0 0
C 0 0 0 0 0 0 0 0 0 0 0 i 12 Ю 16 32 42 39 20 7 0 0 0 0 □
0 0 0 0 0 0 0 0 0 0 0 5 16 17 16 40 86 107 99 60 23 6 0 0 0 0
0 0 0 0 0 0 0 0 0 0 7 30 47 42 32 102 145 124 81 40 20 10 0 0 0 0
0 0 0 0 0 0 0 0 0 0 5 34 50 46 34 40 67 85 84 62 51) 40 30 9 0 0
0 0 0 0 0 0 0 0 0 0 0 6 26 30 17 17 26 39 40 60 78 87 61 34 4 0
Q 0 0 0 0 0 0 0 0 1 1 0 0 8 0 0 6 14 24 57 90 ] 04 79 30 5 0
0 0 0 0 0 0 0 12 1ft 16 7 0 0 0 0 0 0 0 8 22 58 6? 42 21 2 0
0 0 0 0 0 8 22 58 62 42 21 2 0 0 0 0 0 0 0 0 12 18 16 7 0 0
8 0 0 6 14 j4 57 90 104 79 30' 5 0 0 Cl 0 0 0 0 0 0 0 1 1 0 0
30 17 17 26 39 40 60 78 87 61 34 4 0 0 0 0 0 0 0 0 0 0 0 0 6 26
46 34 40 67 85 84 62 50 40 30 9 0 0 0 0 - 0 0 0 0 0 0 0 0 5 34 5U
42 32 102 145 124 81 40 20 10 0 0 0 0 0 0 0 0 0 0 0 0 0 0 7 30 47
17 16 40 86 Ю7 99 60 23 6 0 0 0 0 0 0 0 0 0 0 tl 0 0 0 0 5 16
12 10 32 42 39 20 7 0 0 0 0 0 ^0 0 0 0 0 0 0 0 0 0 0 0 I
30 7 6 9 JJ 12 -6 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
23 Электрические и магнитные свойства
молекул
Изучаемые вопросы
После тщательного изучения этой главы вы сможете:
1. Определить дипольный момент молекулы (стр. 286) и поля-
ризацию диэлектрика (стр. 289).
2. Связать электрическую восприимчивость с относительной
диэлектрической проницаемостью образца [уравнение (23.1.5)].
3. Определить молекулярную поляризуемость [уравнение
(23.1.10)].
4. Связать постоянный дипольный момент с относительной про-
ницаемостью при помощи уравнения Дебая [уравнение (23.1.11)].
5. Связать показатель преломления образца с его поляризуе-
мостью и определить рефракцию [уравнение (23.1.16)].
6. Объяспитв основу оптической активности (стр. 299).
7. Определить причину и указатв величину диполь-дипольного
взаимодействия молекул [уравнение (23.2.1)] и взаимодействия
диполь — индуцированный диполь [уравнение (23.2.2.)].
8. Объяснить возникновение дисперсионных сил и использоватв
формулу Лондона для определения их интенсивности [уравнение
(23.2.3)].
9. Описать межмолекулярный потенциал Л еннарда-Джонса
[уравнение (23.2.5)].
10. Определить и рассчитать постоянную Маделунга [уравне-
ние (23.3.3)] и использовать ее для расчета внутренней энергии
ионных решеток.
И. Окисать образование молекулярного пучка (стр. 310).
12. Определить ударный параметр и дифференциальное сече-
ние рассеяния (стр. 311).
13. Объяснить термины ореольное и радужное рассеяние и опи-
сать, каким образом из опытов с молекулярными пучками получа-
ют информацию о межмолекулярных силах (стр. 313).
14. Описать связь между межмолекулярными силами и вира-
альными коэффициентами реальных газов (стр. 314).
15. Определить функции радиального распределения молекул
в жидкостях и описать их связь с термодинамическими свойства-
ми (стр. 317).
16. Объяснить температурную зависимость вязкости жидкости
[уравнение (23.3.10)].
286
Часть 2. Структура
Рис. 23.1. Условный выбор направления диполь-
ного момента.
17. Определить магнитную восприимчивость и намагничивае-
мость вещества [уравнения (234.1) и (234.4)]-
18. Указать причину парамагнетизма молекул с песпаренными
спинами (стр. 323).
Введение
В данной главе мы рассмотрим некоторые электрические и маг-
нитные свойства молекул и интерпретируем их через электронную
структуру молекул. Этими свойствами являются дипольный мо-
мент, поляризуемость и магнитная восприимчивость. Поляризуе-
мость связана с показателем преломления, оптической актив-
ностью и межмолекулярными силами; мы увидим, как Эти свойст-
ва могут быть связаны с молекулярной структурой.
23.1. Электрические свойства
Мы рассмотрим как постоянный электрический дипольный мо-
мент полярной молекулы, так и дипольный момент, инду-
цируемый в молекуле, когда опа находится в электрическом поле.
Приложенное поле искажает электронную структуру и обусловли-
вает возникновение дипольного момента, величина которого зави-
сит от напряженности поля и «отзывчивости» молекулы: эта «от-
зывчивость» представляет собой поляризуемость молекулы.
Постоянный дипольный момент. Если два заряда q и — q разделе-
ны расстоянием /?, то они составляют диполь с величиной qR. Кроме
величины диполь имеет направление, и принято считать, что он
направлен от отрицательно заряженного конца к положительному
(рис. 23.1). Дипольные моменты обычно выражаются в деба-
ях (D):
1D = 3,336. ПУ30 Кл-м
(эта единица названа в честь одного из пионеров изучения поляр-
ных молекул). Если заряд q имеет величину заряда электрона и
расстояние равно 100 пм (0,1 им, 1 А), то величина диполя будет
р = (1,6022 - НУ19 Кл) Х(1,0000-1010 м) = 1,6022- 1О ао Kt-m^4,8D.
Величины молекулярных диполвных моментов приблизителвно та-
кого порядка (но обычно несколько меньше).
23. Электрические и магнитные свойства молекул
287
Рис. 23.2. Сольватация иона полярным
растворителем.
Дипольные моменты использу-
ются для того, чтобы дать информа-
цию о структуре молекул, а также
чтобы проверить вычисленные вол-
новые функции. Однако на практи-
ке они важны для нахождения под-
ходящего растворителя для данного твердого вещества, так как
способность растворителя разрушать ионные кристаллы связала
с его способностью сольватировать ионы и уменьшать электроста-
тическое взаимодействие, удерживающее ионы в кристалле. В этом
процессе полярные молекулы Играют двойственную роль. Один
конец их диполя может электростатически притягиваться к иолу
противоположного знака, и это понижает потенциальную энергию
иона в растворе (рис. 23,2), Их роль состоит в трм, чтобы умень-
шить силу кулоновского взаимодействия между ионами в раство-
ре, Это получается следующим образом, Когда два иона находят-
ся на расстоянии г, потенциальная энергия их взаимодействия
пропорциональна 1/4пеоГ, если разделяющей средой является ва-
куум; однако, когда они помещаются в растворитель, энергия
уменьшается до 1/4лкоКгг, где Кт — относительная диэлектрическая
проницаемость (или диэлектрическая постоянная). Величина Л’г ча-
стично определяется полярной природой молекул растворителя и
может оказывать значительное влияние на силу кулоновского
взаимодействия. Например, вода имеет Кг = 78; это значит, что
кулоновский потенциал уменьшается почти на два порядка вели-
чины. Роль проницаемости растворителя в определении термодина-
мических свойств ионов в растворе рассматривалась в гл 11 (т. 1).
Определение дипольных моментов. Дипольные моменты можно из-
мерить несколькими способами. При исследовании вращательных
спектров молекулы в газовой фазе найдено, что, если па образец
действует сильное электрическое поле, линии в спектре сдвигают-
ся, Величина этого эффекта Штарка зависит от дипольного мо-
мента молекулы, и поэтому вращательные спектры могут быть ис-
пользованы для получения очень точных данных. Когда молекулу
нельзя исследовать вращательной спектроскопией (если опа очень
сложна, нелетуча или нестабильна в газовой фазе), измерение
обычно основано на определении относительной диэлектрической
проницаемости Л’г образца в массе. В следующих параграфах по-
казано, как можно измерить Кг и как ее связать, с дипольным мо-
ментом.
288
Часть 2. Структура
Нам необходимо дать определение емкости конденсатора. Рас-
смотрим вначале две плоскости, параллельные пластинам
(рис. 23.3). Заряды на пластинах равны q и —q, а их .площади
равны А, что можно выразить как ±аА, где о — поверхностная
плотность заряда (заряд на единицу площади). Если имеются за-
ряды, то между пластинами устанавливается разность потенциа-
лов ДФ. Емкость этого устройства определяется как
C = qih Ф.
Следовательно, в данном случае С = сгА/ДФ. Разность потенциалов
можно записать через плотность заряда ст и расстояние между пла-
стинами d. Прежде всего учтем стандартный результат элементар-
ной электростатики.- электрическое поле в области между пласти-
нами равно о/ео, если среда является вакуумом, но равно
o/еоКг, если она диэлектрик с относительной диэлектрической про-
ницаемостью Кг- Наличие поля с напряженностью Е указывает на
существование разности потенциалов* Ed между двумя пластина-
ми (например, при разности потенциалов 0,1 В между пластинами
на расстоянии 1 см устанавливается электрическое поле напряжен-
ностью 10 В/м), и поэтому в отсутствие диэлектрической среды
емкость равна
Со — a A/(ad/e0) — 80 A id,
но при наличии диэлектрика емкость равна
С = aAi(<rd/e0KT)=E0AKr/d.
Таким образом, измеряя относительную диэлектрическую прони-
цаемость, мы измеряем емкость в отсутствие и в присутствии об-
разца:
С/СО=КГ. (23.1.1)
Емкость легко измерить с помощью одного из стандартных экспе-
риментов (например, используя мост переменного тока); для это-
го имеются заводские приборы.
Следующий этап — установление связи между измеренной ве-
личиной Кт и молекулярным свойством. Это достигается ннтер-
q=<5A
q- в А
-q
Рис. 23.3. Поля (£) я разности
потенциалов в конденсаторе.
23. Электрические и. магнитные свойства молекул
289
I 1
РА - РА
! * Диполь РАЗ I
Рис. 23.4. Поляризация молекул, составляю-
щих среду, и соответствующий диполь.
претацией поляризации диэлектрика
через средний дипольный момент на
единицу объема. Смысл этой интер-
претации виден при рассмотрении рис.
23.4, который показывает, каким об-
разом отдельные диполи среды выст-
раиваются в приложенном поле и каким образом'на поверхности
диэлектрика возникает заряд: этот заряд — поляризация среды-—
противоположен заряду самих пластин и обусловливает понижение
электрического поля от ст/ео до о/во^С. Два заряда на противопо-
ложных гранях диэлектрика образуют диполь, и это является ос-
новой связи между поляризацией и средним дипольным моментом.
Следующая задача состоит в том, чтобы установить количествен-
ную связь между этими величинами.
Обозначим поляризацию, т- е. плотность заряда на поверхности
диэлектрика, через Р, так Что общий заряд на одной грани будет
РА, а на другой он равен —РА. Эти два заряда разделены рас-
стоянием d п поэтому составляют диполь величиной PAd. Объем
диэлектрика равен Adt и, следовательно, дипольный момент па
единицу объема диэлектрика будет PAd/Ad, или Р. Это подтверж-
дает, что поляризацию диэлектрика, т. е. индуцированный на еди-
нице площади заряд, можно рассматривать как средний диполь-
ный момент на единицу объема. Это указывает на два пути рас-
чета, который необходимо сделать: один состоит в нахождении
связи Р с измеряемой величиной Кг, а другой — в нахождении
связи Р с дипольным моментом отдельных молекул.
Чтобы связать Р с Кт, поступаем следующим образом. В от-
сутствие диэлектрика электрическое поле между пластинами
£o=io/eo. В Присутствии диэлектрика, но при таком же количестве
заряда на пластинах поле изменяется до Е. Имеются два способа
записи Е. Во-первых, Е можно выразить через относительную ди-
электрическую проницаемость:
Е =ст/е(/(г.
Во-вторых, можно предположить, что эффект среды состоит про
Сто в уменьшении о до ст—Р. Отсюда
£ = (СТ—
19—242
290
Уость 2. Crpyurtipa
Рис. 23.5. Локальное поле внутри ди-
электрика.
Исключая <т из этой пары уравнений, получим уравнение
Р---=ев(Кг-1)Е, (23.1.2)
которое связывает поляризацию и поле внутри диэлектрика.
Теперь сконцентрируем внимание на одиночной молекуле, на-
ходящейся где-то внутри диэлектрика. На эту молекулу действует
поле Е и поле, обусловленное наличием зарядов на поверхности
полости, которая ее окружает (рис. 23.5). Расчеты показывают,
что этот дополнительный вклад в общее локальное поле имеет ве-
личину если полость считать сферической, а среду непре-
рывной. Поэтому общее поле около молекулы будет
£* =Е = "Г Р (К+2)/«о (*r -1)- (23-1 -3)
Наконец, предположим, что поляризация диэлектрика пропор-
циональна полю, действующему на молекулу. Следовательно, если
коэффициент пропорциональности записать как safe, то лолучям
Р=ейуе£*. (23.1.4)
Величина "Хе называется электрической восприимчивостью, она
безразмерна. Если это выражение для Р подстелить в последнее
уравнение, то Е* сократится, и мы найдем соотношение, связываю-
щее восприимчивость и диэлектрическую проницаемость:
Хе=3(Кг-1да+2). (23.1.5)
Пример (вопрос 2). Емкость пустой ячейки 5,0 пФ. Когда она была заполнена
образцом камфоры, ее емкость стала 57,1 пФ. Какова относительная диэлектри-
ческая проницаемость н электрическая восприимчивость камфоры при комнатной
температуре?
Метод. Для нахождения К. используем уравнение (23.1.1), а для нахождения
— уравнение (23.1.5). Относительная диэлектрическая проницаемость воздуха
фактически равна единице.
Ответ. Из уравнения (23J.1) Лг=(57,1 пФ)/(5,0 пФ) = 11,4. Из уравнения (23.1.5)
1)/(11,4+2) = 2,33.
До умен тарий. Ниже мы продолжим этот пример. Отметим, что как Лг, так н
Хо безразмерны.
23. Электрические а магнитные свойства молекул
291
Восприимчивость зависит от природы молекул, так как она
определяет, каким будет приобретенный дипольный момент при
наличии некоторого электрического поля Е*. Даже если молекула
имеет постоянный дипольный момент р, в отсутствие приложенно-
го поля в жидком образце средний дипольный момент равен нулю,
поскольку их вращательное движение усредняет суммарный мо-
мент до нуля. При наличии поля некоторые ориентации энергети-
чески более предпочтительны, чем другие. В этом случае средний
момент образца отличается от нуля в степени, определяемой кон-
куренцией упорядочивающего влияния поля и разрушающего
структуру р азупор яд очивающего влияния теплового движения мо-
лекул в образце. Получающийся момент можно вычислить, ис-
пользуя распределение Больцмана [уравнение (20.2.15)] для об-
разца при температуре Т.
Энергия молекулы с дипольным моментом р, образующим угол
0 с электрическим полем £*, равна
_ р.Е* COS0.
Больцмана, то
(23,1,6)
Если эту энергию использовать в распределении
расчет среднего дипольного момента дает
Мсредн — С^)>
где х —iiE*/kT, Ж (х) — функция Ланжевена
Нас будет интересовать величина функции Ланжевена только при
небольших х, так как при приемлемых значениях температур и
дипольных моментов х много меньше единицы. Например, при
ц=-1 D и 7—300 К отношение pE*[kT превышает 0,01 только тог-
да, когда напряженность поля превышает 100 кВ/см; большинст-
во обычных измерений проводится при значительно меньших на-
пряженностях поля. Когда х<1, экспоненциальные члены в Ж (х)
можно разложить в ряд, и наибольшим по величине членом, сохра-
няющимся в разложении, является
Ж(х)=-1-х4----,
Поэтому средний молекулярный дипольный момент равен
Дсрсд, « №*/ЫТ. (23.1.7)
Последнее уравнение можно использовать при рассмотрении
данной проблемы. Если в единице объема содержится Л" моле-
кул, то общий дипольный момент на единицу объема в поле Е*
будет определяться соотношением
Е = .,Грсредн—/3kT. (23.1.8)
19*
292
Часть 2 Структура
Это соотношение имеет форму уравнения (23.1.4), и поэтому
можно отождествить с ц2/ЗеоАТ. Следовательно, относительная
диэлектрическая проницаемость раствора связана с постоянным
дипольным моментом молекул и температурой выражением
>р2/Зе0ЛГ=3 (/Q — 1 )/(/Сс 4- 2), (23.1.9)
Измерения Кг и плотности (для определения Ж) сразу дают ве-
личину дипольного момента. (Однако перед тем, как использовать
это выражение, прочитайте последующий текст.)
Поляризуемость. При помещении в электрическое поле неполяр-
ные молекулы могут приобретать дипольный момент. Это связано
с тем, что их электронное распределение становится искаженным,
а центры положительного и отрицательного зарядов, которые пер-
воначально совпадали, теперь разделяются. Величина этого инду-
цированного дипольного момента пропорциональна напряженности
поля (пока она не слишком велика), и можно написать
Р>1ндуц=;«ео£- (23.1.10)
Коэффициент пропорциональности а называется поляризуемостью
молекулы. Если прикладывается очень сильное поле, то индуциро-
ванный момент пропорционален Ег и коэффициент пропорциональ-
ности Р в ₽Е2 называется сверхполяризуемостью. Поскольку сверх-
поляризуемость становится важной лишь при очень высоких на-
пряженностях поля, таких, как в лазерных пучках, это усложне-
ние мы рассматривать не будем.
Если поляризуемость определяется уравнением (23.1.10), то
она имеет размерность объема. Это можно проверить простым
анализом размерности. Используя единицы СИ, находим
[а] —Ip]/[eol [Е] =(Кл-м)/(Ф/м) (В/м)=м’-Кл/(Кл/В) (В) = м3.
Очень часто ее выражают в см3 или А3 (см. табл. 23.1). Типичные
значения: 2,51 1СН4 см3 для Не, 1,32-10-28 для СС14. Объемы этих
двух частиц равны соответственно 40-10-24 и 230-10-24 см3; отме-
тим параллелизм между поляризуемостью и молекулярным объе-
мом.
Внутри образца на молекулу действует поле Е* и индуцирует-
ся диполь асоЕ*. Этот диполь вносит вклад в поляризацию среды,
равный Циидуц, и поэтому мы получаем другой вклад в форме
уравнения (23.1.4), но теперь Л==«<'"а. Таким образом, общая по-
ляризация среды равна
Р = е0..Г(аЦ-р2/ЗЕ0/?Г) Е*.
Если молекула не имеет постоянного момента, то остается только
Член с а, но, поскольку полярные молекулы также поляризуемы,
в уравнение для молекулы с постоянным моментом входят оба
23. Электрические и магнитные свойства молекул
293
Таблица 23.1
Дипольные моменты и поляризуемость
jjl, р a, 10 ai см»
н2 0 9,93
N, 0 22,1
соа 0 33,3
со 0,10 24.5
HF 1,91 16, 4J
НС1 1,08 33,1
НВг 0,80 45.4
HI 0,42 68.5
Н2О 1,85 18,6
NH3 1,47 27.8
СС14 127
СНС13 1,01 —
СН2С1а 1,57 81,4
СН9С1 1,87 103,4
сн4 —- 32,7
СН3ОН 1,71 40,6
СН3СН2ОН 1,68 —
С8Н6 —- 129,7
с8нЕсн3 0 36 —
о-СеН4(СН8)а 0,62 —
Не —- 2,5
Аг '— 20, 1
члена. Следовательно, в общем виде связь с суммарной относи-
тельной диэлектрической проницаемостью выражается соотноше-
нием
Д'(а+ц2аД) = 3(К-1)/|Д-гХ2), (23.1.11)
которое называется уравнением Дебая. Такое же выражение, но
без вклада постоянного дипольного момента, называется уравне-
нием Клаузиуса—Масотти.
Это уравнение можно выразить через плотность р и мольную
массу молекул М, записав Ж = р£/М. Тогда
а+^/Зе^Т^ЗСМ/^р) {(Кг - 1)/(Лг+2)}. (23.1.12)
Довольно часто это выражение записывают через молярную поля-
ризуемость Рщ неполярных молекул:
Рт = (М/р) [(Лг - 1)/(Кг+2)1. (23.1.13
Согласно уравнению (23.1.12), как поляризуемость, так и диполь-
ный момент молекул можно измерить, определяя температурную
зависимость относительной диэлектрической проницаемости и
плотность р. Если построить График зависимости правой части
Уравнения от 1/Т, то наклон линии дает ц2/ЗеоА, а отрезок, отсе-
294
Часть 2. Структура
каемый от оси ординат при 1/7 = 0,— поляризуемость о. Это обус-
ловлено тем, что при очень высоких температурах разупорядочи-
вающий эффект теплового движения заставляет постоянный ди-
поль вращаться настолько быстро, что его вклад в поляризацию
усредняется до нуля и остается один индуцированный диполь.
Этот индуцированный диполь располагается в направлении инду-
цирующего его поля, и поэтому он сохраняет это направление,
несмотря на то что сама молекула может вращаться; таким обра-
зом, он не усредняется до нуля тепловым движением и сохраняет
свой вклад в диэлектрическую проницаемость даже при самых
высоких температурах.
Пример (вопрос 4). Ниже приведены результаты серии измерений на образце
камфоры в той же ячейке, что и в предыдущем примере, при различных темпе-
ратурах. Используйте эти данные для определения дипольного момента и поля-
ризуемости молекулы.
t, °C 0 20 40 60 80 100 120 140 160 200
р, г/см3 0,99 0,99 0,99 0,99 0,99 0,99 0,97 0,96 0,95 0,91
С, пФ 62,6 57,1 54,1 50,1 47,6 44,6 40,6 38,1 35,6 31,1
Метод. Находим Кт при каждой температуре и (Кс—1 )/(Аг+2). Чтобы исполь-
зовать уравнение (23.1,12), умножаем па 3 MipL. Для камфоры 41= 152,23 г/моль.
Строим график зависимости правой части уравнения (23.1.12) от 1/Г: отрезок,
отсекаемый от оси ордипат, дает а и градиент р..
Ответ, При О °C, когда плотность равна 0,99 г/см3, имеем
3A?/pL — 3 (152,23 г/мольЖОгЗЮ3» моль-^Х^.ЭЭ г/см») - 7,659-10— см®.
При более высоких температурах эту величину можно получить, взяв необходи-
мые пропорции. Составляем следующую таблицу;
12,5 И,4 10,8 10,0 9,5 8,9 8,1 7,6 7,1 6,2
(Kt- 1)/(Аг+2) 0,793 0,776 0,766 0,750 0,739 0,725 0,703 0,688 0,670 0,634
(„) (3M/Lp).№ 6,07 5,94 5,87. 5,74 5,66 5,55 5,50 5,43 5,35 5,28
103/(7, К) 3,66 3,41 3,19 3,00 2,83 2,68 2,54 2.42 2,31 2,11
График приведен па рис. 23.6. Отметим, что отрезок, отсекаемый от ОСИ орди-
нат, равен 4,16-10_~ см3, и поэтому поляризуемость молекулы составляет 4,16Х
Х10~й см3. Градиент равец 5,26.10~20. Из уравнения (23.1.12) следует, что
(щ/3аой) -5.26-10-0 см3-К,
так что
р = /ЗХ(8,8510-12 Ф/м)х (1,38-10-21 Дж/К)Х(5,26-10~2» см®-К) =
= 4,39-10“®° Кл-м, или 1,32D.
Комментарий. Мы использовали Г=А®-с4/(кг-м2). Странным в этих результа-
тах является то, что камфора не плавится до 175 °C; следовательно, эти данные
показывают, что сферическая молекула вращается даже в твердом веществе.
Поляризуемость молекулы определяется силой, с которой ядер-
ные заряды контролируют электроны и препятствуют искажению
23. Электрические и магнитные свойства молекул
295
Рис. 23.6. График уравнения Де-
бая для камфоры.
их распределения приложен-
ным полем. Если молекула
содержит лишь несколько
электронов, то их распреде-
ление жестко контролирует-
ся зарядом ядра и поляри-
зуемость низкая. Если моле-
кула содержит большие
атомы с множеством элект-
ронов, то степень ядериого
контроля мала, распределе-
ние электронов более рых-
лое и поляризуемость боль-
ше.
Поляризуемость можно рассчитать из волновой функции, одна-
ко ее величину можно оценить, не прибегая к детальному расче-
ту. Рассмотрим атом с атомным номером Z. Он не имеет постоян-
ного дипольного момента, но диполь может быть индуцирован,
если приложить электрическое поле. Индуцирование дипольного
момента заключается в сдвиге электронного заряда внутри атома;
по иначе можно сказать, что атом в некоторой степени должен
возбуждаться. Можно предположить, что способность поля инду-
цировать дипольный момент будет пропорциональна отношению
энергии его взаимодействия с электронами к энергии, которую
необходимо затратить, чтобы возбудить их. Мгновенный диполь-
ный момент атома имеет порядок —ег, где г— его радиус, и по-
этому энергия взаимодействия с приложенным полем напряжен-
ностью Е порядка Если энергия возбуждения имеет порядок
половины величины потенциала ионизации /, то индуцированный
дипольный момент будет порядка —er(erEi-^I), потому что мгно-
венный дипольный момент — ег «заморожен» в атоме на уровне,
предопределенном соотношением двух энергий. Отсюда следует,
что поляризуемость будет порядка 2егг2Ды). Если имеется Z элек-
тронов, то можно считать, что
а «в 2Ze®rB/e0/.
Это выражение показывает, что поляризуемость на самом деле
увеличивается с возрастанием атомного номера, размеров атома
296
У деть 2. Структура
и легкости возбуждения атома, что подтверждается эксперимен-
тальными результатами.
Поляризуемость при высоких частотах. Показатель преломления.
До сих пор мы рассматривали молекулы в статическом поле. Ког-
да приложенное поле осциллирует, уравнение Дебая может уже
не соблюдаться, поскольку при очень высокой частоте постоянный
дипольный момент не в состоянии достаточно быстро переориен-
тироваться и выстраиваться в направлении постоянно меняющего-
ся направления Е. При таких высоких частотах постоянный ди-
польный момент не вносит вклада в поляризацию среды, и поэто-
му ок выпадает из уравнения Дебая. Так как для одного враще-
ния молекул в жидкости требуется 10-10 с, то дипольный вклад
стремится к нулю, когда относительная диэлектрическая прони-
цаемость измеряется при частоте выше 1012Гц (в микроволновой
области). Инвертируя этот аргумент, получаем возможность пред-
положить, что, наблюдая изменение поляризации и диэлектриче-
ской проницаемости при изменении частоты, можно измерить ско-
рость переориентации молекулы в жидкости. На этой идее основан
метод диэлектрической релаксации.
При микроволновых частотах вклад поляризуемости остается,
однако он модифицируется, когда частота достигает больших ве-
личин. Молекулярное искажение, вследствие которого возникает
индуцированный диполь, является геометрическим искажением по-
ложения ядер: в приложенном поле молекула изгибается и растя-
гивается. Когда частота облучения настолько высока, что она из-
меняется быстрее по сравнению со временем, необходимым для
изменения формы молекулы (это время приблизительно равно
времени, необходимому для одного колебания), вклад поляриза-
ции искажения исчезает (рис. 23.7). Следовательно, когда частота
облучения больше, чем частота колебания молекулы (в ИК-обла-
сти), поляризуемость падает до другого значения. При достаточно
высоких, оптических частотах только электроны довольно легко
реагируют на быстро меняющееся направление приложенного по-
ля, и в этой ситуации вклад вносит только электронная поляризуе-
мость.
При оптических частотах имеется очень простое соотношение
между относительной диэлектрической проницаемостью при час-
тоте v, A'r(v) к показателем преломления среды лг(м) на той же
частоте:
Kr(V)=n?(v). (23.1.14)
Поэтому, просто измеряя показатель преломления образца, мож-
но определить поляризуемость при оптических частотах
(табл. 23.2). Подставляя последний результат в уравнение Клау-
зиуса — Мосотти [уравнение (23.1.12)1, получим
a (v) - (ЗМ/£р) (л? -!)/(«“+2). (23.1.15)
23. Электрические и магнитные свойства молекул
297
1д(т,гц)
Рис. 23 7. Зависимость поляризации от частоты.
Этот результат часто выражают через молярную рефракцию /?га,
определяемую аналогично молярной поляризуемости:
/?гп = (М/р) [(п?-!)/(«!4-2)], (23.1.16)
так как тогда
a(v)^3/?,n(v)/L,
(23.1.17)
Эти выражения можно инвертировать для определения показате-
ля преломления среды из таблиц рефракций (таких, как
табл. 23.3) :
vm=MiP.
I V m — Ktr>. I
(23.1.18)
Пример (вопрос 5). Показатель преломления воды при 20°C равен 1,3330 для
света с длиной волны 589 нм. Какова поляризуемость молекулы на этой частоте?
Таблица 23.2
Показатели преломлении относительно воздуха при 20 °C
? -=434 нм 5S9 hv. 656 нм
Вода 1,3404 1,3330 1,3312
Вензол 1,5236 1,5012 1,4965
Четырех*пористый углерод 1,4729 1,4607 1.4579
Сероуглерод 1,6748 1,6276 1,6182
КС1 1,5050 1,4904 1.4873
KI 1,7035 1,6664 1,6581
298
Часть 2. Структура
Таблица 23 3
Молярные рефракции Рт (см’/моль) при 589 нм
II 1,100 Н-Н 2,08 С=О 3 42
С 2 418 С-Н 1,70 С—О—С 2,85
О (в ОН) I 525 С—С 1.21 С—О—II 3,23
О (в СО) 2,211 с=с 4,15
О (в—О—) 1,643 с=с 6,03
Не
0,50
L1+ Be35- оа- F- Ne
0,20 0,09 7 2,5 1,00
Mg2 + АР+ СГ Аг
0,50 0,29 0,1? 8,7 4,20
кл Са3+- Вг" Кг
2,2 1,35 12,2 6,37
Г Хе
18,5 10,42
Метод. Используем уравнение (23.1.15) с Л1= 18,015 г/моль и р=0,9982 г/см’.
Ответ. (nt - 1) /(п* + 2) = (1,3330а „ 1)/(1 ,ЗЗЗО3-р 2) = 0,206.
3MjpL =ЗХ(18,015 г/моль)/(6,022 102S моль-‘)Х(0,9982 г/см«) — 8,99-10~23 см®.
Отсюда
a = (8,99.J0-33 см3)Х(0,206) =Н,84- 10 аа смз
Комментарий. Повторение тех же расчетов для света с длиной волны 434 нм,
при которой показатель преломления равен 1,3404, дает а-=1,89-10-23 см3; моле-
кула более поляризуема ври более высокой частоте, так как входящий свет име-
ет большую энергию возбуждения.
Показатель преломления—-это отношение скорости спета в ва-
кууме к скорости света в данной среде, г. с, /гг = с/и. Причина, по
которой поляризуемость среды влияет на скорость, может быть
объяснена тем, что при распространении света в среде ее молеку-
лы искажаются, и поэтому в них индуцируется дипольный мо-
мент, который осциллирует с частотой падающего света. Этот ос-
циллирующий диполь генерирует излучение той же частоты, но
этот процесс отстает по фазе от волны распространяющегося све-
та. Это отставание по фазе соответствует снижению скорости све-
та в данной среде. Если молекулы неполяризуемы, свет не взаи-
модействует, и никакого отставания по фазе не происходит; если
молекулы сильно поляризуемы, взаимодействие сильное, и запаз-
дывание значительно. Свет высокой частоты несет достаточно
энергии для возбуждения молекул и более эффективен, чем свет
более низкой частоты. Поэтому при оптических частотах поляри-
зуемость возрастает с частотой падающего света. Это означает,
23. Электрические и магнитные свойства молекул
29»
Рнс. 23.8. Линейно-поляризованный свет рассматривается как суперпозиция двух
вращающиеся в противоположных направлениях компонент, которые в оптиче-
ски активной среде распространяются с разными скоростями.
что показатель преломления в оптической области увеличивается
с частотой. Это основа хорошо известного явления дисперсии све-
та призмой: голубой свет имеет больший показатель преломления,
чем красный, и его путь больше искривляется призмой. Из этого
явления пришло название дисперсия для обозначения изменения
показателя преломления или любого другого свойства с частотой.
Оптическая активность. Явление оптической активности, когда об-
разец вращает плоскость поляризации поляризованного светового
пучка, проходящего через него, можно связать с особым свойст-
вом показателя преломления. Плоскость поляризации будет вра-
щаться, если показатель преломления циркулярно (лево) поляри-
зованного света (щ) отличается от показателя преломления цир-
кулярно (право)поляризованного света («н). (Мы условимся, что
правополяризоваппый свет соответствует вращению электрическо-
го вектора по часовой стрелке с точки зрения наблюдателя, смот-
рящего навстречу пучку света; рис. 23.8.)
Тот факт, что оптическое вращение происходит при Пь#=/1к,
можно выявить из рис. 23.8. Перед входом в Среду пучок плос-
кополяризован под утлом 0 = 0° к некоторой оси. Плоскополяри-
зованный свет можно рассматривать как суперпозицию двух вра-
щающихся в противоположных направлениях циркулярнополяри-
зованных компонент (рис. 23.8). При вхождении в среду одна ком-
понента распространяется быстрее, чем другая, если показатели
преломления различаются, и при длине образца I разность во вре-
мени прохождения будет I/vr—1/Vl, где v — скорости компонент.
300
Часть 2, Структура
Рис, 23.9. Структура кварца.
Эго можно выразить через показатель преломления как
(nR—Пц)//с, поскольку n^cjv. Электрические векторы при выходе
из образца находятся в фазе с разными углами, и поэтому их на-
ложение ведет к плоскополяризованному пучку иод новым углом
60 к прежнему. Угол вращения равен
60= (>iR—nL) 2л/Д,
(23.1,19)
где —длина волны света.
Чтобы объяснить, почему показатели преломления для двух
поляризаций света отличаются между собой, мы должны объяс-
нить, почему поляризуемость молекулы связана с хиральностью
света. Полная теория оптического вращения довольно сложна,
однако одна из его допустимых интерпретаций состоит в том, что,
если молекула имеет спиральную структуру, ее поляризуемость
зависит от того, соответствует или нет электрическое поле зонди-
рующего излучения спирали молекулы. Если молекула имеет спи-
ральную структуру, то можно ожидать, что ее показатель прелом-
ления будет зависеть от хиральности света. Молекулы со спираль-
ной структурой не совмещаются со своим зеркальным отображе-
нием, и это является критерием для определения того, будет ли
молекула оптически активной (это обсуждалось при рассмотре-
нии симметрии в гл. 16).
Если оптически неактивные молекулы затвердевают в виде
спиральных образований, то можно также ожидать оптическую:
активность на основании того, что поляризуемость твердого веще-
ства зависит от хиральности света. Это подтверждается структу-
рой кварца, который состоит из длинных спиральных цепей SiOa
(рис. 23.9) и обладает сильной оптической активностью.
Изменение оптической активности с частотой света называется
дисперсией оптического вращения. Ее можно интерпретировать че-
рез изменения поляризуемости молекулы при разных частотах
света; она используется для исследования стереохимической кон-
фигурации больших молекул.
23. Электрические и магнитные свойства молекул
301
Рис. 23.10. Дипольные моменты молекул хлор замещенных бензолов.
Аддитивные свойства. Дипольный момент сложной молекулы до
некоторой степени можно описать в виде различных вкладов и по-
лучить общий дипольный момент их сложением (векторным сло-
жением). Это можно проиллюстрировать для случая молекул
хлор за мощенных бензолов (рис. 23.10). Монохлорбснзол имеет ди-
польный момент 1,57D, что обусловлено наличием атома хлора,
а п-дихлорбензол — нулевой момент, так как два равных, но про-
тивоположно направленных момента сокращаются. о-Дихлорбен-
зол имеет дипольный момент, направление и величина которого
являются результатом сложения двух дипольных моментов моно-
хлорбензола, расположенных под углом 60°. Момент .м-дихлорбен-
зола можно объяснить аналогично, но с углом 120° между вкла-
дами. Этот метод векторного сложения можно с большим успе-
хом применять для оценки дипольных моментов разнообразных
молекул.
Поляризуемость является приблизительно аддитивным свойст-
вом молекул, то же относится и к молярным рефракциям [урав-
нение (23.1.16) ]. Если известны величины молярных рефракций
ионов, то, складывая соответствующие рефракции, можно оце-
нить показатель преломления Кристалла. Такой же анализ можно
произвести и для групп в органических молекулах и таким путем
оценить показатель Преломления молекул в гомологическом ряду
-ft влияние введения различных групп. Рефракции ионов и различ-
ных групп приведены в табл. 23.3.
Пример (вопрос 5). Определите показатель преломления уксусной кислоты для
желтой ливни натрия.
Метод. Используем информацию табл. 23.3. Молекула CHjCOOH состоит из
302
Часть 2. Структура
2С+4Н+О (типа —О—)+О (типа 0= в С = О). Затем используем уравне-
ние (23.1.18).
Ответ. 2С + 4Н + (-О-) + (0=) = 2х(2,418 сма/моль) =
+ 4Х(1,100 см^/моль) + (1,525 сма/моль) +
+ (2,211 см’/моль) = 12,972 см3/моль.
Чтобы использовать уравнение (23.1.18), вычисляем
Vm = М/р = (60,05 г/моль)/(1,046 г/см3) ’ 57,41 см*/моль.
Поэтому из уравнения (23.1.18)
лт = [(57,41 + 25,94)7(57,41 — 12,97)]3/2= 137,
Комментарий. Экспериментальное значение также равно 1 т37, и поэтому метод
расчета пг очень хорош.
23.2. Межмолекулярные силы
В этом разделе мы рассмотрим силы притяжения между моле-
кулами, которые можно обнаружить при электростатических взаи-
модействиях, аналогичных рассмотренным в первой части этой
главы. Эти вандерваальсовы взаимодействия можно подразделить
на три типа: диполь-дипольное взаимодействие между полярны-
ми молекулами, взаимодействие диполь — индуцированный ди-
поль и, наконец, взаимодействие индуцированный диполь —инду-
цированный диполь, или дисперсионное взаимодействие. Ионные
твердые вещества связаны Простыми кулоновскими силами При-
тяжения взаимодействием, которое мы также обсудим.
Диполь-дипольное взаимодействие. Когда две полярные молекулы
находятся близко друг к другу, их диполи взаимодействуют, и по-
этому между ними действует сила. В жидкости или газе одна из
молекул вращается под всеми углами относительно Другой; та-
ким образом, можно было бы ожидать, что силы притяжения (ког-
да диполи расположены «голова к хвосту», рис. 23.11, а) будут
уравновешиваться силами отталкивания (когда диполи располо-
жены «голова к голове», рис. 23.11,6). Здесь нс учтено одно важ-
ное обстоятельство. Оно состоит в тенденции одного диполя вы-
страиваться в линию с другим диполем с образованием предпочти-
тельной конфигурации. В жидком растворе притяжательные ориен-
тации, будучи энергетически более выгодными, слегка преобладав
Ют над отталкивательными ориентациями диполей, и поэтому си^
лы притяжения слегка преобладают над силами отталкивания. |
Степень преобладания ориентирующего эффекта диполей паЛ
разупорядоченным тепловым движением можно рассчитать на ос|
нове распределения Больцмана. (Такой расчет напоминает рас®
чет, использованный для получения общей поляризации жидкого
образца, стр. 291.) Если провести расчет и принять приближение,
что энергия взаимодействия меньше kT (хорошее приближение
23. Электрические и миенитные свойства молекул
303
Рис. 23.11. Взаимодейстпие между дпумя полярными молекулами.
для большинства жидкостей), то можно найти среднюю энергию
взаимодействия двух молекул с постоянными дипольными момен-
тами pi и (1’:
¥(/?)== -Л (i^/(4TOoH ((!//?») (1/£Т)},
(23,2.1)
где R — расстояние между диполями. Важно отмстить, что эта
энергия обратно пропорциональна шестой степени R и обратно
пропорциональна температуре. Последнее отражает то положение,
что при высоких температурах тепловое движение будет нару-
шать взаимную ориентацию двух диполей.
Величину энергии взаимодействия можно оценить следующим
образом. Выражая дипольный момент в дсбаях, а расстояние в
нанометрах, получаем, что при комнатной температуре (Т—300К)
V (кДж/моль)
0Ч,Р)3 D)2
1025(7?, нм)в •
Поэтому энергия взаимодействия для коллектива молекул с
pstlD, находящихся на расстоянии 0,3 нм, равна примерно
— 1,4 кДж/моль.
Взаимодействие диполь — индуцированный диполь. Присутствие
полярной молекулы вблизи другой молекулы (которая сама может
быть полярной или неполярной) оказывает на вторую молекулу
поляризующее действие. Индуцированный диполь может затем
взаимодействовать с дипольным моментом первой молекулы, и обе
молекулы становятся связанными друг с другом. Величина эффек-
та зависит как от величины постоянного дипольного момента по-
лярной молекулы, так и от поляризуемости второй молекулы.
Поскольку индуцированный дипольный момент принимает то же па-
правление, что и индуцирующий диполь (рис. 23.12), эффект теп-
лового движения не нужно учитывать (потому Что он не может де-
зориентировать индуцированный момент от направления индуци-
рующего момента). Таким образом, эффект сохраняется даже в
том случае, когда полярная молекула свободно вращается вблизи
поляризуемой молекулы.
304
Часть 2. Структура
Рис, 23,12. Взаимодействие диполь — индуцированный диполь: полярная молекула
(темная стрелка) может индуцировать диполь (светлая стрелка).
Средняя энергия взаимодействия для двух молекул равна
V (Я)=-2 (^/16л%) (о,//?6), (23.2.2)
где Ц1 — постоянный дипольный момент молекулы 1, а а2 — поля-
ризуемость молекулы 2.
Величину энергии взаимодействия можно оценить из следующе-
го выражения:
Т7/ гт . i (jx,, D}2 (as. 10*21 см3)
V (кДж/моль) да j >е6 1()4 нм)е ,
Для молекулы с I D (например, НС1), соседствующей с моле-
кулой, поляризуемость которой ct ~ 120 -10г4 см3 (например, бен-
зола; табл. 23.1), энергия взаимодействия составляет около
0,8 кДж/моль при расстоянии 0,3 нм.
Взаимодействие индуцированный диполь — индуцированный ди-
поль. Рассмотрим две неполярные молекулы, находящиеся друг
от друга на расстоянии /?. Хотя они не обладают постоянными мо-
ментами, их электронные облака флуктуируют, и их можно рас-
сматривать как частицы, имеющие мгновенный дипольный момент,
который постоянно меняет свою величину и направление
(рис, 23.13). Предположим, что одна молекула быстро переходит
в электронную конфигурацию, которая дает мгновенный диполь
Pi. Этот диполь поляризует другую молекулу и индуцирует в ней
мгновенный диполь р3. Оба диполя связываются вместе, и поэто-
му молекулы притягиваются друг к другу. Хотя направление ди-
поля в первой молекуле будет продолжать изменяться, вторая
молекула будет реагировать на это изменение, и вследствие такой
корреляции эффект притяжения не усреднится до нуля.
Величина энергии зависит от поляризуемости цервой молеку-
лы, поскольку величина мгновенного диполя р, зависит от степени
контроля над внешними электронами со стороны ядерного потен-
циала. Опа также зависит от поляризуемости второй молекулы,
так как величина р3 связана со степенью наведения этого момен-1
та. Полный расчет сил притяжения между двумя индуцированны-1
ми диполями, т. е. дисперсионных сил, довольно сложен, однако!
23. Электрические и магнитные свойства молекул
305
Рис. 23.13. Взаимодействие
двух индуцированных диполей:
мгновенный диполь (заштрихо-
ванная стрелка) может инду-
цировать диполь (светлая
стрелка).
приемлемым приближением для энергии взаимодействия является
формула Лондона: V (R) ж — ) ("^ j' (23 2 3>
В этом выражении Л и /а — потенциалы ионизации двух молекул.
Из последнего уравнения можно оценить величину дисперсион-
ной энергии для двух молекул метана. Поскольку а~33-10-24 см3,
а 1^7 эВ (670 кДж/моль), получаем
V (кДж/моль) & -3,4- Ю-э/(/?, нм)6;
следовательно, V~—4,7 кДж/моль на расстоянии 0,3 нм (как в
жидкости). Грубая проверка этой величины по энергии испарения
жидкого метана дает значение 8,2 кДж/моль.
Формула Лондона применима также к взаимодействию между
поляризуемыми полярными молекулами, и поэтому общая энергия
взаимодействия между молекулами дается суммой уравнений
(23.2.1)—(23.2,3). Все три энергии Отрицательны, что означает
понижение энергии при сближении молекул, т. е. указывает на
силы притяжения между молекулами. Все три энергии изменяются
пропорционально К~й, и в этом состоит причина того, что энергии
межмолекулярного притяжения обычно записываются в форме
V (/?)=—Св//?в, (23,2.4>
где Cfi — некоторый коэффициент, зависящий от природы мо-
лекул.
Отталкивание и суммарное взаимодействие. Когда молекулы сбли-
жаются друг с другом, над силами притяжения начинает преоб-
ладать ядерпое и электронное отталкивание. Так, хотя два атома
гелия на больших расстояниях слабо притягиваются друг к Другу,
они не образуют стабильной молекулы Ней. Причины детально
объяснялись в гл. 15 (разд. 15.2, т. 1). Отталкивательные взаимо-
действия очень резко возрастают с уменьшением расстояния, и по-
этому следует ожидать, что межмолекулярная энергия будет опи-
сываться резко загибающейся вверх кривой типа приведенной на
20—242
306
Часть 2. Структура
Рис. 23.14. (12,6)-Потенциал Леннарда-Джонс*.
Поведение на коротких дистанциях очень сложно и
зависит
электронной структуры частиц. Тем не менее найдено,
хорошим приближением к суммарной кривой является
что очень
потенциал Леннарда-Джонса: У[R) — Cn/Rn—Ct/Re,
(23,2.5)
где н —большое целое число. Когда п= 12, мы говорим о (12,6)-по-
тенциале Леннарда-Джопса, и этот потенциал имеет совершенно
общее применение, так как его математическая форма очень удоб-
на. При малых Я величина /?12 намного меньше, чем J?fi, и поло-
жительный (отталкивательный) член CI2//?12 преобладает над от-
рицательным (притяжательным) членом (Д//?8. При больших рас-
стояниях справедливо обратное. Очень часто (12,6) -потенциал за-
писывают в более сжатой форме:
V (/?) .-^4е {(о//?)13 — (сг//?)в). (23.2.6)
В этой форме параметр е является глубиной минимума на кривой
(рис. 23.14), который соответствует /?е=2'/во. Типичные значения
приведены в табл. 23.4.
Коэффициенты Сц и С6 могут быть измерены эксперименталь-
но при помощи ряда способов, например при изучении несовершен-^
ства газов (отклонений от идеального поведения) и рассеяния мо-
лекул в молекулярном пучке; эти методы и их применение описы-
ваются ниже.
23. Электрические и магнитные свойства молекул
307
Таблица 23,4
Параметры (12,в)-потенцияла Деннарда-Джонса
{ъ/Ь}, к пм
Ne 34,9 278
Ar 119,8 340
Хе 221 410
н2 38 0 292
о2 118 346
N, 95,1 370
CJ3 257 440
Вг, СС)2 520 427
189 449
сн4 148 382
с,н4 205 423
23.3. Роль межмолекулярных сил
Силы, действующие между молекулами, удерживают их вме-
сте. Иногда их средние значения малы,, как, например, в газах, и
их наличие лишь слабо сказывается на подвижности молекул.
В кристаллах взаимодействие между атомами и ионами выражено
сильно, и это обусловливает жесткость структуры. Где-то между
газами и кристаллами лежат жидкости: кинетическая энергия мо-
лекул жидкости сравнима с потенциальной энергией их взаимо-
действия.
Ионные решетки. Зная расстояние между ионами в кристаллах,
можно оценить энергию связывания в ионных кристаллах. С тер-
мохимической точки зрения это было рассмотрено в гл. 4 (т. 1).
где было показано, что величину этой энергии можно определить^
построив цикл Борна—Хабера. Для этого пужцо измерить энер-
гию решетки (разность энергий между решеткой МА и газообраз-
ными ионами Мл и А~); в данном разделе мы посмотрим, как эту
величину можно оценить теоретически.
Ценность такого расчета зависит от того, насколько просты си-
лы, которые создают кристаллическую структуру ионной решетки.
Можно предположить, что энергия когезии почти целиком обуслов-
лена кулоновским притяжением между противоположно заряжен-
ными ионами и расстояние между ними в решетке определяется
конкуренцией между этой энергией (которая стремится сжать
весь кристалл в точку) и силами отталкивания, препятствующими
такому полному слиянию. Потенциальная энергия V для пары
попов с зарядами z(t> и z.e, разделенных расстоянием Яц, дается
соотношением
(23.3,1)
308
Часть 2, Структура
Потенциальная энергия отдельного катиона является результа-
том его взаимодействия со всеми другими анконами н катионами
решетки:
Vt+)^(zZ/4^o)22i//?+i,
t
где /?+?— расстояние выбранного катиона с зарядом z+e от иона
i с зарядом z,e. Если самое короткое расстояние между катионом
и анионом в кристалле раако к<г, то его можно взять за единицу
длины и записать £+< = p+j/?a; тогда
V(+) =(2+еа/4ле0/?0) (zs/p+i).
I
Аналогично можно выбрать отдельный анион и записать его по
тенциальную энергию как
V<-J=(z_ea/4rre0/?0) 2<zizP-i).
t
где p-i/?o — расстояние иона i от выбранного аниона. Поэтому
обшая энергия кристалла I моля вещества, обусловленная куло
новскими взаимодействиями между ионами, равна
V= L (И+»4- 0->) =(£г+г^/4лЕ^в) Л, (23.3.2
где
„ _ 1 у (&/*-) I
— 2 Zj p+i П" р_.
Множитель -g- в выражении для V возникает из-за того, что мь
не должны учитывать каждое ион-иопное взаимодействие дваждь
(что произошло бы, если бы мы сложили V4 и V~ и умножил]
сумму на число ионов L, присутствующих в кристалле). Величи
на Л играет особую роль в теории кристаллических решеток .
называется постоянной Маделунга. Ее важность обусловлена тем
что она зависит от тигга кристалла, а не ог размеров отдельно:
решетки. Опа вычислена для различных решеток, как следует и
данных табл. 23.5. Поэтому можно выбрать тип решетки и Прост*
через один параметр /?о, который определяет общие размеры ячей
ки, установить кулоновскую потенциальную энергию.
Расчет энергии данной кристаллической решетки теперь совер
шенно прост. Общая энергия равна сумме только что рассчитан
иой энергии притяжения и вклада сил отталкивания между иона
ми, которые начинают действовать, когда ноны прижимаются ДРУ
(23.3.3
53. Электрические и магнитные свойства молекул
309,
Таблица 23.5
Постоянные Маделунга
Решетка
Каменная соль 1,74756
CsCl 1,77667
Цинковая обманка 1,63805
В юр пит 1,64132
Флюорит 5,03878
Рутил 4,7701
Куприт 4,44248
Корунд 25,0312
к другу под влиянием кулоновского слияния. Этот отталкиватель-
ный член является сильным, но только на коротких расстояниях,
и его общую форму можно рассматривать как экспоненциальный
вклад интервала /?*. Тогда общая энергия решетки (для части-
цы М.А с 2+ = 1 и z_=—Д) будет
(^о)=—(Ее2/4л£0) +Д'ехр (—7?0//?*).
Минимальная величина этого выражения приходится на значение
Ro, для которого dU/dR0 = Qt и из этого условия можно опреде-
лить К:
Поэтому мольная внутренняя энергия равна
(i - R*/Ro). (23.3.4)
В этом выражении осталась только одна переменная R*—интер-
вал отталкивательного взаимодействия. Его можно определить из
сопротивления кристалла сжатию, т. е. из коэффициента сжатия
[т. 1, уравнение (3.2.12)], и поэтому энергию решетки можно вы-
числить, используя одно из доступных значений постоянной Маде-
лунга и измеренные значения Ro и R*. Экспериментальные значе-
ния приведены в табл. 23.6. Отметим, что уравнение (23.3.4) дает
внутреннюю энергию: энтальпия реакции
М+ (газ) -р А“ (газ)-► МА (тн.)
Таблица 23.6
Энергии решеток Л// (кДж/моль) при 25 °C
L1F — 1031
NaF -911,7 NaCl -772,8 NaBr —741,0
KF —810,0 КС! —702,5 КВг —678,2 KI —637,6
310
Часть 2, Структура
Селектор
скорости
Рис. 23.15. Прибор для получения молекулярного пучка.
очень легко может быть определена из pV — nRT для каждого га-
зообразного компонента и H=U-}-pV, —2RT.
Пример (вопрос 10). Рассчитайте внутреннюю энергию кристалла хлорида нат-
рия. Интервал отталкивательного взаимодействия R* составляет 32,1 пм, а рас-
стояние до ближайшего соседа — 282 пм.
Метод. Постоянная Маделунга для решетки каменной соли равна 1,747558; под-
ставляем се в уравнение (23.3.4).
Ответ.
( (6,022-10м моль-1)Х(1,602-10-1В Кл)^х(1,747558))
= [ 4лх(8,854-10-1й Дж-1-Кла-м-1)х(282-10-1г м)
/ 32,I \
X ( I — “рой- ) — —763 кДж/моль.
Комментарий. Способу определения 7?* посвящена задача 23.30. Эксперименталь-
ное значение —764 кДж/моль, т. е. совпадение превосходное.
Молекулярные взаимодействия в пучках. В последние годы в экс-
периментальном изучении межмолекулярных еил достигнут замет-
ный успех благодаря развитию метода молекулярных пучков. Мо-
лекулярный пучок—-это именно то, что означает данное название:
узкий поток молекул в пустом сосуде, в других отношениях. Пу-
чок молекул направлен к другим молекулам, и рассеяние, которое
происходит при соударении, связано с межмолекулярными силами.
Скорость молекул в пучке можно контролировать, и, таким обра-
зом, можно исследовать их поведение при столкновении е молеку-
лами-мишенями, имеющими разную кинетическую энергию.
Типичный прибор для исследования молекулярных пучков по-
казан па рис. 23.15. Устройство для выбора энергии часто пред-
ставляет собой набор колес с прорезями, которые вращаются на
пути луча и позволяют проходить через щели только молекулам,
имеющим подходящую скорость. Применяют также более сложные
устройства для генерирования молекул с желаемой скоростью; их
преимущество состоит в том, что возникает пучок с большей интен-
сивностью, чем та, которую можно получить, блокируя прохожде-
ние молекул, имеющих неподходящую скорость. Возможны также
23. Электрические и магнитные свойства молекул
311
Рнс. 23.16. Определение ударного парамет- Рнс. 23.17. Определение дифферен-
ра. _ цнального сечения рассеяния.
другие типы отбора: например, для отклонения полярных молекул
с различной ориентацией можно использовать электрические ноля
и таким образом получать пучок молекул, выстроенных в линию,
Моноэнсргетический пучок проходит через объем прибора.
В нем поддерживается высокий вакуум, так что молекулы пучка
не сталкиваются и, разупорядочивая свое движение, снова прихо-
дят в тепловое равновесие. Они проходят через газ-мишень, кото-
рый может находиться внутри камеры рассеяния при известном
давлении или сам может быть в форме молекулярного пучка, пере-
секающего путь первого пучка. Последний метод пересекаемого
пучка дает массу информации, поскольку в нем можно контроли-
ровать состояние как молекул-«мишеней», так и молекул-«снаря-
дов».
Столкновения рассеивают пучок молекул от их первоначалыю-
пого направления, и основная часть экспериментальной информа-
ции получается и,з угла рассеяния. Чтобы определить интенсив-
ность молекул, рассеянных под разными углами, применяют детек-
торы различных типов. Они могут состоять -из камеры, снабженной
чувствительным манометром, или представлять собой ионизацион-
ный детектор, в котором входящие молекулы сначала ионизируют-
ся, а затем обнаруживаются электрически. Можно определить со-
стояние возбуждения рассеянных молекул. Это может быть инте-
ресно, например, при определении вращательной или колебатель-
ной заселенности состояний частиц, после столкновения. В этом
случае для определения заселенности энергетических уровней мож-
но использовать колебательную или микроволновую спектроско-
пию.
При обсуждении результатов исследований с молекулярными
пучками основными являются два параметра: ударный параметр
b и дифференциальное сечение рассеяния do. Ударный параметр
представляет собой расстояние по вертикали между траекториями
соударяющихся частиц (рис. 23.16). Дифференциальное сечение
рассеяния — это мера рассеяния под разными углами. Рассмотрим
бесконечно малый телесный угол dQ при некотором угле Q
(рис. 23.17). Отношение числа молекул, рассеянных в этом телес-
312
Часть 2, Структура
ном угле, к их числу в падающем пучке является величиной da
при данном значении угла:
t _____ число молекул, рассеянных в dQ при Q
' ' число молекул в падающем лучке
Дифференциальное сечение рассеяния зависит от ударного па-
раметра и формы межмолекулярного потенциала. Это легче все-
го увидеть на примере столкновения двух жестких шаров
(рис. 23.18). Если ударный параметр равен нулю, то более легкий,
более подвижный шар имеет траекторию, которая приводит к ло-
бовому соударению, и поэтому вся интенсивность рассеяния будет
находиться в телесном угле d£i при 180°. Если ударный параметр
так велик, что шары не контактируют (6>/?л+/?в), то рассеяния
не произойдет и для всех углов сечение рассеяния будет равно
нулю. При Ь, большем нуля, но меньшем, чем /?а4-/?в, интенсив-
ность рассеяния распределяется в конусах вокруг прямого направ-
ления (рис. 23.18) и дифференциальное сечение максимально в
направлениях, лежащих на конусе.
Такое элементарное рассмотрение необходимо модифицировать,
чтобы оно включало соударение реальных молекул. Прежде всего
реальные молекулы не являются жесткими шарами, и картина
рассеяния зависит От радиальной составляющей их взаимодейст-
вия и От анизотропии (угловой зависимости), которая может на-
блюдаться, если молекула несферическая. Далее, рассеяние зави-
сит от относительной скорости подхода двух частиц друг к Другу,
__ поскольку очень быстрые
, ____________-___j = q молекулы могут проходить
через зону сильного взаимо-
действия без заметного от-
клонения, тогда как мед-
ленные молекулы того же
типа и с тем же ударным
параметром могут захваты-
ваться полем мсжмолску-
лярного потенциала, и от-
клонение может быть значи-
тельным (рис. 23.19). Это
означает, что зависимость
Рис. 23.18. Столкновение двух
жестких шаров с разными удар-
ными параметрами.
23. Электрические и магнитные свойства молекул
313
Рис. 23.19. Степень рассеяния
может зависеть от относитель-
ной скорости молекул, а так-
же от ударного параметра.
сечения рассеяния от относительной скорости и направления долж-
на давать информацию о силе и размерах межмолекулярпого по-
тенциала. Вот почему так важно уметь контролировать энергию
сталкивающихся пучков.
Вторая модификация принимает во внимание, что результат
столкновения контролируется не только классической, по и кван-
товой механикой. Главное отличие состоит в появлении волнопо-
добных явлений интерференции. Точно так же как в опыте Янга
по щелевой интерференции (разд. 22.1), изображая все классиче-
ские траектории, которые приводят молекулу От источника к де-
тектору, и затем позволяя траекториям интерферировать между
собой, можно получить некоторое представление о квантовой кар-
тине рассеяния.
Особенно важны два квантовомеханических эффекта. Молеку-
ла с определенным ударным параметром может приблизиться к
притяжательной части межмолекулярного потенциала таким об-
разом, что она отклонится по направлению к отталкивательной
«жиле» пучка (рис. 23.20) и затем выйдет из притяжательной об-
ласти и будет продолжать свое движение в прямом направлении
вместе с молекулами, которые имеют такие большие ударные па-
раметры, благодаря которым они не отклонились. Оба пути— для
неотклоненных частиц с большим b и для рассеянных в прямом
направлении с малым b — квантовомехапически интерферируют, и
интенсивность в прямом направлении изменяется. Это явление на-
зывается ореольным рассеянием, так как представляет собой то
же самое явление, которое объясняет оптический ореольный эф-
фект, при котором иногда можно наблюдать яркое гало, окружаю-
щее светящийся объект (примером ореола являются цветные
кольца вокруг тени самолета, отбрасываемой солнцем на облака;
они часто наблюдаются в полете). Другое явление состоит в на-
блюдении сильно увеличенного рассеяния в некоторых непрямых
Рис. 23.20. Интерференция путей,
приводящая к ореольному рассея-
НИЮ.
314
Vасть 2. Структура
Интерфери-
\рующие
пути
Рнс. Й3.21. Интерференция пу-
тей, приводящая к радужному
рассеянию.
люстрирован на рис. 23.21. Рассеяние
направлениях; оно назы-
вается радужным рассея-
нием, так как возникает
по той же причине, что и
оптическая радуга. Ос-
новной механизм проил-
под радужным углом йг
может возникнуть при взаимодействии трех траекторий, приведен-
ных на рисунке. Поскольку все три пути приводят частицу в одну
и ту же область пространства, между ними происходит интерфе-
ренция, и интенсивность рассеяния под углом йг увеличивается.
Подробное изучение всех данных по рассеянию не входит в
нашу задачу, но главное положение должно быть уяснено: распре-
деление интенсивности рассеянных молекул может быть связано
с межмолекулярным потенциалом,1 может быть построена деталь-
ная картина его радиальной и угловой зависимости. Одним из ус-
пехов такого подхода является определение параметра вандерва-
альсова взаимодействия Се [уравнение (23.2.4)] и проверка при-
менимости этого параметра в выражении Леннарда-Джонса [урав-
нение (23.2.5)]. Хотя обычно оказывается, что R~6-часть этого по-
тенциала является хорошим представлением притяжательного по-
тенциала, пропорциональность между отталкиванием и Д12 не
так строго выражена.
Несовершенство газов. Молекулярные пучки — не единственный
путь исследования межмолекулярных сил. Для определения меж-
молекулярпых потенциальных функций широко используется изу-
чение отклонений газов от идеального поведения, хотя по сравне-
нию с молекулярными пучками оно дает менее детальную инфор-
мацию. Теория и эксперимент обычно встречаются в вириальном
разложении, упоминавшемся в т. 1 (разд. 1.3). Оно имеет вид
pVjRT = 1 + В (Г)/У1п +С (Т)/П + • •, (23.3,5)
где В—второй вириалъный коэффициент, а С—Третий. Идеаль-
ный г?.з удовлетворяет уравнению pVm/RT = 1, и поэтому вирналь-
ные коэффициенты должны появляться из-за влияния межмоле-
кулярпых сил в реальном газе.
Связь между вириальиыми коэффициентами и межмолекуляр-
ным потенциалом в математической форме довольно сложна, но
принцип метода и выражение для В(Т) совершенно четкие.
23, Электрические и магнитные свойства молекул
315
В гл. 21, посвященной статистической термодинамике, мы видели
(разд. 21.2), что давление системы можно связать с функцией рас-
пределения при помощи уравнений
р = — (ЗА /dV)r, А—А (0) = —А7’ In Q. (23.3.6)
Первое из этой пары уравнений представляет собой термодинами-
ческое соотношение между функцией Гельмгольца А и давлением,
а второе выражает А через каноническую функцию распределе-
ния Q. При отсутствии межмолекулярных взаимодействий канони-
ческую функцию распределения можно выразить через число име-
ющихся молекул и функцию распределения отдельных молекул —
формулой Q = В разд. 21.2 мы видели, что это приводит к
p = nRT[V, При наличии межмолекулярных взаимодействий кано-
ническую функцию распределения нельзя так просто выразить,
поскольку молекулы не являются независимыми и их энергия за-
висит от расстояний между ними. Если межмолекулярная потен-
циальная энергия равна Г(^), то можно вывести уравнение для
канонической функции распределения, и, чтобы вывести выраже-
ние для В через Г(^), можно использовать уравнения (23.3,5)
и (23,3.6). Тогда получим
В(Т)^2лГр1— (23.3,7)
о
Видно, что, если V всегда равен нулю, В стремится к Нулю, как
это и происходит в случае идеального газа. В зависит [через
V(7?9] от расстояния между парой молекул, и, следовательно, вто-
рой вириальпый коэффициент можно интерпретировать на осно-
вании эффектов соударения пар молекул в реальном газе. Третий
вирпалыгый коэффициент зависит от относительного расположе-
ния трех молекул, и поэтому его роль состоит в том, чтобы учесть
эффекты кратковременного образования кластеров из трех моле-
кул.
Интеграл в последнем уравнении взять трудно, за исключением
некоторых случаев. Например, если истинный потенциал аппрок-
симирован потенциалом жестких шаров, в котором Г(7?) резко
возрастает от нуля до бесконечности, когда две молекулы контак-
тируют при R=dt то мы найдем
В (Г) =2nL J IFdR лЬсВ,
а
так как при R'>d подынтегральное выражение стремится к нулю,
а экспонента исчезает, когда V равен бесконечности (в интервале
0^7?^^). Еще задолго до этого (т. 1, разд. 1.4) мы видели, что
часть уравнения состояния Ван-дер-Ваальса, которая представля-
316
Часть 2. Структура
ет собой часть межмолекулярного взаимодействия, обусловленную
отталкиванием жестких шаров, соответствует второму вириально-
му коэффициенту точно в такой форме. Отсюда возникает вопрос:
можно ли найти потенциал, который, если рассчитаны вириальные
коэффициенты, даст полное уравнение состояния Ван-дер-Вааль-
са? Такой потенциал был найден; он содержит вклад жестких ша-
ров вместе с небольшим вкладом, обусловленным очень дально-
дсисгвующйм притяжением.
Важно то, что уравнение (23.3.7) дает связь между теорией и
экспериментом. Если можно определить второй вириальный коэф-
фициент экспериментально и воспроизвести его, выбрав подходя-
щее выражение для межмолекулярного потенциала и взяв инте-
грал, то можно найти форму межмолекулярного потенциала. Ча-
сто выбирают потенциал Леннарда-Джонса и варьируют парамет-
ры до достижения соответствия. Некоторые значения, полученные
таким путем, приведены в табл. 23.4.
Пример (вопрос 14). Возьмите потенциал Леннарда-Джонса для аргона в фор-
ме уравнения (23.2.6) с е/А= 120 К и а=340 пм н определите второй вириальный
коэффициент при 298 К.
Метод. Возьмем интеграл в уравнении (23.3.7). Сделаем это численно (техниче-
ский термин — численная квадратура), используя правило Симпсона. Правило
Симпсона состоит в том, чтобы взять подынтегральное выражение для равно-
мерного ряда точек (назовем эти величины ут) с интервалом h и затем образо-
вать сумму
(^/3) [Уо + 4 (уг Уз 4- + Ут-1) + 2 (Уг ф -ф ym_2) 4- ym].
Поскольку подынтегральная функпня возрастает от нуля до максимума, затем
пересекает нуль при 340 пм и имеет длинный чхвост» вплоть до нескольких
1000 пм, интегрирование целесообразно проводить в двух интервалах. Один ин-
тервал от Д=0 до 34о пм, а второй — от 340 до 5000 пм. При шаге 1 пм во
всем интервале нужно использовать программированную счетную машину, но для
второго интервала оправдано использование большего шага.
Отвег. Мы запишем два фрагмента подпой чисдопой таблицы величин подынтег-
рального выражения:
Я, пм 0 10 20 30... 340 440 540
Подынтегральное выраже-
ние, (пм)2 0 100 400 900... 0 —7,3-10 —2,88-10*
Тогда из правила Симпсона в первом интервале
1,1298010’ нм3,
а во втором интервале
j9/2raL = —0,51629-10’ пм»,
и поэтому полный интеграл имеет величину В/2л£—6,135-10е пм, так что В=
=2,321-1Ь“5 м3/моль, или 23,2 см3/моль.
Долей ей гарий. Расчеты, подобные этому, идеальны для ЭВМ или, как было ис-
пользовано в данном случае, для небольшого электронного счетно-решаюн1его
устройства. Два интервала отражают места, где преобладает отталкивание
>0), а затем притяжение (В<^0).
23. Электрические и магнитные свойства молекул
317
Несовершенство газов проявляется и другими путями; особен-
но полезным путем его изучения является измерение Транспорт-
ных свойств газов, в частности их вязкости. Подвижность моле-
кул в газе, которая определяет вязкость, зависит от пути их свя-
зывания друг с другом, и поэтому отклонение от идеальности дает
информацию о межмолекулярном потенциале. Результаты таких
исследований также включены в табл. 23.4.
Структура жидкостей. Сведения о ней. Отправной точкой при рас-
смотрении твердых веществ является строго упорядоченная форма
совершенных кристаллов, а при изучении газов — полностью хао-
тическое распределение частиц в идеальном газе. Отправной точ-
кой при исследовании жидкостей является то, что они занимают
промежуточное положение между этими двумя предельными слу-
чаями, т. е. в них в какой-то мере проявляется структура и в ка-
кой-то мере — хаос. Теоретическое описание и экспериментальное
изучение жидкостей находятся на ранней стадии развития, хотя
и был достигнут некоторый прогресс, и мы можем кое-что сказать
об их основных особенностях.
Молекулы жидкости удерживаются вместе межмолекулярным
притяжением, но их кинетические энергии сравнимы с глубиной
потенциальной ямы. В результате этого, несмотря па то что жид-
кости являются самостоятельной фазой (и отделены От твердых
веществ и газов фазовыми переходами), структура в целом оказы-
вается подвижной. В случае газов вириальное разложение дает
связь между теорией и экспериментом, и в случае жидкостей не-
обходима некоторая концепция, позволяющая сделать то же са-
мое. Концепция состоит в введении функций радиального распре-
деления. Наиболее важна функция парного распределения g(R),.
и она будет в центре нашего внимания.
Функция парного распределения дает вероятность того, что-
другая молекула будет найдена на некотором расстоянии R от не-
которой выбранной молекулы. Точнее, g(R)dR— вероятность то-
го, что молекула будет найдена в интервале dR на расстоянии R
от другой молекулы. В кристалле эта функция представляет со-
бой периодическое чередование резких пиков, соответствующих
точной определенности (при отсутствии дефектов и теплового-
движения), что атомы будут находиться в каждом узле решетки.
Эта регулярность продолжается неопределенно долго, и поэтому
говорят, Что кристалл имеет дальний порядок. При плавлении кри-
сталла дальний порядок исчезает, и уже для любой точки следует
говорить о некоторой вероятности нахождения рядом с ней второй
молекулы. Вблизи первой молекулы, однако, может сохраниться
остаточная упорядоченность, так как ближайшие соседи в перво-
начальном ее окружении могут еще в определенной степени со-
хранить свои исходные положения, и, если они вытесняются вновь
входящими молекулами, новые молекулы будут занимать их ос-
318
Часть 2. Структура
поводившиеся места. Еще можно обнаружить сферу ближайших
соседей на расстоянии Ri и, вероятно, за ней сферу следующих
ближайших соседей на расстоянии Rz от центральной молекулы.
Существование ближнего порядка означает, что функция парного
распределения вблизи центральной молекулы, по-видимому, имеет
колебательную форму с пиком при Ri, меньшим пиком при Rz и,
вероятно, еше несколькими пиками за ним.
Функция парного распределения (а при более точном описании
и другие функции распределения для триплетов, квадруполей и
т. д.) достаточно близка к истинному описанию структуры жидко-
сти. Тогда возникают вопросы: а) можно ли g(R) измерить экспе-
риментально? б) можно ли ее вычислить и, следовательно, исполь-
зовать для проверки теорий жидкой структуры? в) можно ли ис-
пользовать g(R) при рассмотрении свойств жидкостей?
функция парного . распределения может быть измерена экспе-
риментально с помощью дифракции рентгеновских Лучей. В гл- 22
мы виделд, что рентгеновские лучи дифрагируются на электрон-
ной плотности, имеющей определенное распределение, и что из
дифракционной картины можно определить структуру кристал-
лов. Жидкости тоже имеют структуру, хотя она и является лишь
локальной, и поэтому можно ожидать дифракционную картину в
виде пятен, которая должна иметь форму g(R). На самом деле
так и есть. Если бы распределение молекул было аморфным, бес-
форменным, то картина представляла бы собой одно большое пят-
но, но наличие определенных колец интерференции показывает,
что жидкость обладает некоторой структурой и что колебания
функции парного распределения распространяются на короткие
интервалы. Дифракционная картина может быть проанализирова-
на в основном тем же методом, что и для твердого вещества, а
для нахождения собственно функции парного распределения ис-
пользуется распределение интенсивности. Функция парного рас-
пределения для воды, полученная при разных температурах, при-
ведена па рис. 23.22; она безошибочно указывает па положение
оболочек с локальной структурой. Более подробный анализ пока-
зывает, что центральная молекула воды, по крайней мере в пер-
вой оболочке, окружена молекулами, расположенными в углах
тетраэдра. Это точно соответствует структуре льда (см. рис. 22.19),
и межмолекулярные силы, в данном случае длинные водородные
связи, достаточно велики, чтобы определить локальную структуру
вплоть до точки кипения-
Колебательный вид функции обусловлен двумя причинами. Ес-
ли бы даже молекулы были жесткими шарами без притяжатель-
ных взаимодействий, то была бы также получена колебательная
g(R) с сильным начальным пиком. Это показывает, Что одним из
факторов (иногда преобладающим), влияющих на структуру жид-
кости, является просто геометрическая проблема большого числа
23. Электрические и магнитные свойства молекул
319-
pW
2
Рис. 23.22. Функция радиального распределения жидкой воды при разных тем-
пературах [Norien Л. И., Danford М. L>., Levy Н. Л., Discuss. Faraday Soc., 43
97 (1967)].
шаров, собранных вместе. Потенциальная яма притяжения тоже-
играет роль, и ее эффект состоит в том, что она улавливает и со-
бирает молекулы в ее окрестностях. Одна из Причин трудности-
теоретического описания жидкостей состоит в том, что важны как
притяжательная, так и отталкивательная (связанная с жесткостью
скелета молекулы) части потенциала.
Если определена функция радиального распределения, то ее
можно связать с термодинамическими свойствами жидкости. На-
пример, внутренняя энергия, обусловленная межмолекулярным
потенциалом, дается интегралом
СО
М V М (23.3.8)
О
где Ж—число молекул в единице объема. Это выражение являет-
ся усреднением межмолекулярного потенциала V(R) взвешенного
ио вероятности g(R)dR того, что пара молекул будет найдена на
расстоянии R друг от друга. Аналогично давление дается интегра-
лом
р =jr-kT — (л-г /6) j1 g (R) [R (dV/dR) ]dR, (23.3.9)
b
где величина в квадратных скобках R(dVldR) называется вири-
алом. Поскольку первый член справа — это давление, оказывае-
мое в тех же условиях идеальным газом, и поскольку р для жид-
320
Часть 2. Структура
кости той же плотности значительно меньше, чем для газа вблизи
точки кипения, интеграл справа должен почти сократиться с пер-
вым членом. Такой очень тонкий баланс между двумя членами
является еще одной причиной, почему расчеты для жидкостей так
трудны.
Другая, более существенная трудность состоит в предположе-
нии, что взаимодействие между тремя молекулами можно выра-
зить в виде суммы взаимодействий между тремя парами. На са-
мом деле найдено, что межмолекулярные энергии так просто не
суммируются, потому что значительный вклад в энергию вносят
тройные взаимодействия, представляющие собой разность между
истинной межмолекулярной энергией трех молекул и попарной
их суммой.
Еще одна трудность заключается в том, что молекулы могут
иметь сильно анизотропное взаимодействие. Предельный случай
представляют длинные, тонкие молекулы типа м-азоксиапизола
О
При плавлении этого твердого вещества выше точки плавления
сохраняется несколько проявлений эффектов дальнего порядка,
благодаря чему, будучи жидким (т. е. имея неполный дальний по-
рядок), вещество в некоторых отношениях является в то же вре-
мя и кристаллом (т. е. имеет некоторые
элементы дальнего порядка) и поэтому
называется жидким кристаллом. Один
тип сохранившегося дальнего порядка
дает смектическую фазу (от греческого
слова «мыльный»), В этой фазе молеку-
лы располагаются параллельно в виде
слоев (рис. 23.23,а). Другие вещества и
некоторые смектические жидкие кристал-
лы при повышенной температуре не об-
ладают слоистой структурой, но сохраня-
ют параллельную ориентацию; это нема-
тическая фаза (от греческого слова «иг-
лы» или «игольчатые кристаллы»)
Рис. 23.23. Расположение молекул в смектической
(о) и нематической (б) фазах жидкого кристалла.
23. Электрические и магнитные свойства молекул
321
Таблица 23.7
Вязкость жидкостей т| [кг/(м с) 10-31 при 25 °C
Вода’ 0,8904 Бензол 0,601
и-Гептан 0,224 Ртуть 1,53
Циклогексан 0,895 Чстыреххлор истый угле- 0,909
____________ род
а Вязкость воды два всего интервала температур, в котором ова жидкая, с ошибкой ме-
нее 1% определяется выражением
, , . ч 1,37023 [</, °C)—20J -I- 8,36*10-4 [(/, °C)—20р
k(H20/^) =--------------- ,09 + <л .с>---------
Псрснод из кг/(м’с) в сантипуазы (сП) осуществляется умножением на 10* (так что для
воды I сП).
(рис. 23,23,6), Жидкие кристаллы имеют сильно анизотропные
оптические, электрические и магнитные свойства; они используют-
ся, например, для воспроизведения чисел в счетно-решающих уст-
ройствах и часах.
При рассмотрении одной лишь g(R) не принимается во внима-
ние динамическая структура фазы, однако подвижность жидкостей
является одной нз главных их характеристик, Для определения
зависящих от времени свойств локальной структуры Жидкостей
существуют различные экспериментальные методы: один из наи-
более эффективных — метод неупругого рассеяния нейтронов. Это
один из видов дифракции нейтронов, но в опыте главная задача
состоит в том, чтобы определить, сколько энергии отдает или при-
нимает нейтрон при прохождении через жидкий образец. Процесс
изменения энергии можно интерпретировать через детали движе-
ния молекул в жидкости.
Значительно более «земным» является измерение вязкости
(табл, 23.7), Поскольку для продвижения из одной точки в дру-
гую молекула должна терять своих ближайших соседей, она долж-
на приобрести минимальную энергию, чтобы быть способной
двигаться. Вероятность того, что она сможет приобрести эту энер-
гию, дается распределением Больцмана и пропорциональна
ехр(—Ea/RT). Поэтому следует ожидать, что температурная за-
висимость подвижности жидкости будет выражаться этим экспо-
ненциалом, т. е. фактором Больцмана. Так как вязкость »] обрат-
но пропорциональна подвижности, то
ex'p(EjRT), (23.3.Ю)
следовательно, ожидается уменьшение вязкости с увеличением
температуры; рис. 23.24 показывает, что это справедливо. Здесь
вновь величина £а зависит от межмолекулярных сил, однако проб-
лема расчета очень сложна, так как, чтобы одна молекула двига-
лась, несколько других необходимо закрепить в их положениях.
21—242
322
Часть 2. Структура
Рис, 23.24, Температурная зависимость вяз-
кости воды.
Итак, мы подошли к явлениям,
зависящим от времени; однако из-
менение— это предмет рассмотре-
ния в части 3, и вследствие необык-
новенно трудных, пока еше не раз-
решенных проблем мы отложим об-
суждение структуры и свойств жид-
костей и в следующих главах опять
перейдем к более простым вопро-
сам.
23.4. Магнитные свойства
Магнитные свойства молекул
аналогичны их электрическим свой-
ствам в двух смыслах. Во-первых,
некоторые молекулы обладают постоянным магнитным дипольным
моментом. Во-вторых, магнитное поле индуцирует дальнейший
вклад в общий магнитный момент. Аналогом поляризации явля-
ется намагничивание М, а степень намагничивания, обусловленная
магнитным полем напряженностью Н, называется магнитной вос-
приимчивостью
М =-ин.
(23.4,1)
Плотность магнитного потока обозначается как В; она связана с
напряженностью приложенного поля и намагничиванием формулой
в=и0 (И 4-Я)=(1 _|_Zm) н,
(23.4,2)
где р.о — фундаментальная постоянная, называемая проницаемо-
стью вакуума; она имеет величину 4л-10~7 EI/А2, или 4л-10“7 ДжХ
ХКл_2-с2 -м’1.
Если в систему, где плотность потока равна В, поместить маг-
нитный диполышй момент Цт, то он будет иметь энергию
— pmBcos0, (23.4.3)
где б—угол между ним и направлением магнитного потока. Ве-
личину В можно считать мерой числа магнитных силовых линий,
проникающих через среду, и энергия диполя определяется этой
плотностью, Плотность силовых линий увеличивается, если М при-
бавляется к Н (тогда Хт положительна), но уменьшается, если М
23. Электрические и магнитные свойства молекул
323
Весы
— образец
Рис. 23.25. Устройство весов Гун для опре-
деления магнитной восприимчивости.
противоположно Н (тогда х™ отри-
цательна). Вещества, для которых
%т положительна, называются па-
рамагнитными, а вещества, для ко-
торых Хт отрицательна, называют-
ся диамагнитными.
Поляризация — это электриче-
ский дипольный момент на еди-
ницу объема. По аналогии, М — магнитный дипольный момент на
единицу объема. Молекулы могут обладать постоянным моментом
Цт. В этом случае магнетизм имеет вклад, пропорциональный
Цш/ЗАГ, как и в случае электрического поля [уравнение (23.1.8)].
Приложенное поле может индуцировать магнитный момент, вели-
чина которого зависит от молекулярной намагничиваемости £. По-
этому можно написать уравнение, аналогичное уравнению
Zm(ё + l^/3kT), (23.4.4)
и, следовательно, магнитную восприимчивость можно связать с
намагничиваемостыо, постоянным моментом и температурой.
Часто восприимчивость измеряют на весах Гу и. Этот прибор
состоит из чувствительных весов, с которых свисает образец в фор-
ме узкого цилиндра (рис. 23.25). Образец висит между полюсами
магнита. Если образец парамагнитен, то его энергия внутри маг-
нитного поля понижается, и поэтому появляется сила, смещающая
образец в поле. Если образец диамагнитен, то его энергия меньше
снаружи поля, и поэтому некоторая сила выталкивает его из поля.
Эта сила пропорциональна восприимчивости; следовательно,
определяя положение равновесия весов, можно найти Хт. Обычно
прибор калибруют по образцам с известной восприимчивостью.
Типичная парамагнитная восприимчивость имеет порядок 10-3
(отмстим, что эта величина безразмерная в используемой нами
системе единиц), а типичная диамагнитная восприимчивость со-
ставляет около (—) 10-5 (табл. 23.8).
Постоянный магнитный момент. Постоянный магнитный момент
может быть определен из измерений восприимчивости построением
графика зависимости хт от l/Т. Часто выполняется закон Кюри
/.„—А-ГС/Т, (23,4.5)
сравнение которого с уравнением (23.4.4) позволяет установить,
что наклон С равен Жр,0р,^/ЗА.
21
324
Часть 2. Структура.
Таблица 23.8
Магнитные восприимчивости Хт'Ю®
Вода —12,97 CiiSO4.5H2O 4-1460
Бензол —54,8 СоС12-6Н2О -1-9710
Циклогексан —68,1 FeFs-3HaO 4-7870
Четыреххлорвстый углерод -66,1 FeFj 4-13760
Причиной постоянного магнитного момента может быть неспа-
ремный спин электронов в молекулах. Мы уже видели (т, J,
Стр. 502), Что электронный спин взаимодействует с приложенным
полем, как если бы он имел магнитный момент, пропорциональный
его угловому моменту;
ИШ-2?Л (23.4.6)
где уе — гиромагнитное отношение — е/2те. Следовательно, вели-
чина этого .момента равна
= > 2^8 (S+1)A2,
так как квантовомеханическая величина углового момента s рав-
на [s(s+1) Если ввести магнетон Бора (т. 1, стр. 500)
рв=е«/2те (Ив=9,274 10-мДж/Т),
то получится
Ип=2ив[8(8 + 1)^ (23.4.7)
Если в молекуле имеется несколько электронных спинов, то они
могут комбинироваться, образуя общий электронный спиновый
угловой момент с величиной, определяемой квантовым числом S.
В этом случае постоянный момент имеет величину
l*m=2|*B[S(S4-l)]^ (23.4.8)
Следовательно, вклад в магнитную восприимчивость составляет
Хт=4^ИоНав5(5-Ь1)/ЗАТ. (23.4.9)
Эта величина положительна, и поэтому постоянный спиновый мо-
мент вносит вклад в парамагнитную восприимчивость системы.
Отметим, Что вклад в восприимчивость при высоких температурах
стремится к нулю, так как тепловое движение разупорядочиваеп
ориентацию индивидуальных спиновых магнитных моментов и па
магпичивапие по этому пути исключается.
Пример (вопрос 18). Рассчитайте парамагнитную восприимчивость комплекс:
металла с тремя неспаренными электронами.
Метод. Используем уравнение (23.4.9); три неспаренных электрона дают S =
=%. Плотность образца равна 3,24 г/см3 в 44=200 г/моль (44 — молекулярная
масса).
23. Электрические и магнитные свойства молекул
325
<Х1Х1)ФФ®
Рис. 23.26. Расположение спинов в ферро-
магнитном (а) и антиферромагнитном (6)
веществах.
Ответ. Сначала вычисляем
LpjM — (6,022 1025 моль-i) х (3,24 г/с№)/(200 г/моль) =
= 9,76-10!\см-3, или 9,76-10®? м~3.
Тогда
4х(4Л'10-7Н/А2)х(9,274-10-»*Дж/Т)3Х(15/4)Х{9,76 1О2’м-«) по , .
Ьч— Зх(1,381 10-23 Дж/К)Х(298R) ~ 1,28 10’ '
Комментарий. Отметим, что магнитная восприимчивость безразмерна, положи-
тельна (парамагнитна) и имеет порядок величины 10-э. Диамагнитная восприим-
чивость безразмерна, отрицательна и имеет порядок величины 10-6.
При низких температурах в парамагнитном твердом веществе
иногда может произойти фазовый переход в состояние, в котором
на больших участках спины ориентируются параллельно. Такое
кооперативное расположение обусловливает очень сильное намаг-
ничивание образца; это явление называется ферромагнетизмом
(рис. 23.26, я). В других случаях при кооперативном эффекте бо-
лее предпочтительной может быть ориентация с чередованием
спинов (рис. 23.26,6), Это называется антиферромагнетизмом, о
котором уже упоминалось в связи с рассеянием нейтронов
(разд. 22.5). Переходная температура для ферромагнитного пере-
хода называется температурой Кюри, а для антиферромагнитного
перехода — температурой Нееля,.
Индуцированные магнитные моменты. Наложение магнитного по-
ля искажает распределение электронов в молекуле, однако в то
время как электрическое поле растягивает молекулу и отодвигает
друг от друга положительный и отрицательный центры, магнит-
ное поле скручивает молекулу. Искажение молекулы скручивани-
ем эквивалентно стимулированию кругового движения тока, а
этот ток обусловливает возникновение магнитного момента, кото-
рый обычно противоположен направлению приложенного поля: в
этих случаях восприимчивость диамагнитна. В ряде случаев ин-
дуцированный момент имеет то же направление, что и приложен-
ное поле, и тогда восприимчивость парамагнитна.
Большинство молекул обладает диамагнитной восприимчиво-
стью. Это вызвано тем, что протекание парамагнитного тока за-
висит от доступности низколежащих возбужденных электронных
326
Часть 2. Структура
состояний молекулы, а они не всегда имеются. Протекание диамаг-
нитного тока происходит в основном состоянии молекулы, и, сле-
довательно, не требуется наличия возбужденных состояний; поэто-
му такой процесс является гораздо более общим. Когда доступны
возбужденные состояния, индуцированный парамагнетизм можно
отличить от спинового парамагнетизма, так как ему соответст-
вует член g в уравнении (23,4,4) и поэтому он не зависит от тем-
пературы. Не зависящий от температуры парамагнетизм (НТП)
является следствием стимулирования орбитального движения в мо-
лекуле, и его нельзя путать с постоянным моментом, который обус-
ловлен спинами.
Резюмируя Эту ситуацию, можно сказать, Что все молекулы
имеют диамагнитную компоненту их обшей восприимчивости, но
ее превосходит спиновый парамагнетизм, если молекула содержит
неспаренные электроны. В некоторых случаях НТП достаточно ве-
лик и молекулы становятся парамагнитными, несмотря на то что
их электроны спарены.
Литература
Уитли П. Определение молекулярной структуры. Пер, е англ. — М.: Мир, 1970,
Hill Н. A. Q.j Day Р. (eds.), Physical methods in advanced inorganic chemistry,
Interscience, London, 1968.
Smith J. W., Electric dipole moments, Butterworths, London, 1955.
Smyth C. P.. Dielectric behaviour and structure, McGraw-Hill, New York, 1955.
Smyth C. P„ Determination of dipole moments, in Techniques of chemistry (Weiss-
berger A, and Rossiter B. W., eds.), Vol. IV, 397, Wiley-Interscience, New York,
1972.
Vaughan IP, E., Smyth С, P., Powles /. G., Determination ot dielectric constant
and loss, in Techniques of chemistry (Weis sb er ger A, and Rossiter B. W., eds.),
vol. IV, 351, Wiley-Interscience, New York, 1972,
McClellan A. L., Tables ol experimental dipole moments, Freeman, San Francisco,
1963.
Chu. В., Molecular forces, Wlley-Tnferscrencc, New York, 1967.
Селвуд П. Магнетохимия. Пер. с а^гл, — М.: ИЛ, 1958.
Schieber AL М., Experimental magneto ch emi str у, Wiley, New York, 1967.
Malay L. N., Instrumentation and techniques for Measuring magnetic susceptibility,
in Techniques of Chemistry (Weissberger A. and Rossiter B, W,, eds.). Vol. IV,
431, Wiley-Interscience, New York, 1972.
Earnshaw A., Introduction to magnet о chem is try, Academic Press, New York, 1968.
Goodenough J. B., Magnelism and the chemical bond, J n terscience, New York,
1963.
Fluently M. A. D., Lawley К. P., Molecular beams in chemistry, Chapman and
Hall, London, 1974.
Ross J. (ed.), Molecular beams; Adv. chem. Phys., 10 (1966).
Pryde /. A., The liquid state, Hutchinson, London, 1966.
Rowlinson J. S., The structure of liquids, in Essays in chemistry (Bradley J, N.,
Gillard R. D. and Hudson R. F., eds.), 1, 1; Academic Press, London, 1970.
Гиршфельдер Дж., Кертис Ч. и Др. Молекулярная теория газов и жидкостей.
Пер. с англ. — М.: ИЛ, 1961.
Dymond J. И., Smith Е. B.t The virial coefficients of gases; a critical compilation,
Clarendon Press, Oxford, 1969.
23. Электрические и магнитные свойства молекул
327
Heller W., Curme Н. G., Optica! rotation; experimenlal techniques and physical
optics, in Techniques of chemistry (Weis sb er ger A, and Rossiter B. W., eds.),
Vol. 111C, Wiley-In terscience, New York, 1972.
Задачи
23.1. Дипольный момент толуола равен 0,4 D; оцените дипольные моменты трех
ксилолов. Какой ответ можно считать безошибочным?
232. Дипольный момевт воды равен 1,84 D. Рассмотрите его как результат сло-
жения двух связевых диполей под углом 104,5° и предскажите дипольный мо-
мент перекиси водорода, как функцию азимутального угла между группами ОН
(считайте, что угол ООН равен 90°). Экспериментальный дипольный момент ра-
вен 2,13 D. Какому углу он соответствует?
23.3. Каковы напряженности полей, действию которых подвергаются атомы и
молекулы? Пусть молекула воды (p=l,84D) подходит к иону. Какова предпоч-
тительная ориентация молекулы, когда этот ион является анионом? Рассчитайте
напряженность электрического поля, действующего на ион, когда центр диполя
воды находится на расстоянии: а) 1,0 пм, б) 0,3 нм и в) 30 нм от его центра.
Выразите ответ в В/м.
23.4. Молекула воды ориентирована электрическим полем напряженностью
1,0 кВ/м и с одной стороны к ней медленно подходит атом аргона (а = 2,0Х
X1(HJ см3). На каком расстоянии молекула воды остановит приближающийся
атом аргона и ориентируется в направлении его приближения?
23.5. Относительная диэлектрическая проницаемость газообразных галогенидов
водорода дается выражением Кг = 14-Д7 в интервале 0<3, С<^300, а величины
Д7 приведены ниже (V — относительный удельный объем, равный единице при
273,15 К и 1 атм, т. е. 7=7/273,15 К). Каковы их дипольные моменты и статиче-
ские поляризуемости?
t, °C 0 100 200 300
10»Д(НС1) 4,3 3,5 3,0 2,6
105Д(НВг) 3,1 2,6 2,3 2,]
]03Д(Н1) 2,3 2,2 2,1 2,1
23.6. Поляризуемость молекулы воды на оптических частотах равна 1,59Х
ХЮ.-2’ см3, Определите показатель преломления воды. Экспериментальное значе-
ние равно 1,33. Как можно объяснить расхождение?
23.7. Относительная диэлектрическая проницаемость водяного пара при давле-
нии 1 атм дается выражением Кг=1,00785—1,6-10-4 (t °C —140) в интервале
140—150 °C. Каков дипольный момент и поляризуемость молекулы? На основе
этих данных определите величину Кг для жидкой воды. Экспериментальное зна-
чение при 20 °C равно 80. Почему имеется расхождение?
23.8. Относительная диэлектрическая проницаемость хлороформа была измерена
в интервале температур и были получены следующие данные:
I, °C —80 —70 —60 —40 —20 0 20
Кг 3,1 3,1 7,0 6,5 6,0 5,5 5,0
р, г/см3 1,65 1,64 1,64 1,6] 1,57 1,53 1,50
Точка плавления; —64 °C. Объясните эти результаты и найдите поляризуемость
и дипольный момент молекулы.
328
Часть 2. Структура
23,9, Относительная диэлектрическая проницаемость метанола (т. пл, —95 °C)
приведена ниже. Какую информацию о молекуле можно получить из этих дан-
ных?
t, °C —185 —170 —150 —140 —ПО —80 —50 —20 0 20
Кг 3,2 3,6 4,0 5,1 67 57 49 42 38 34
23.10, Покажите, что для газа показатель преломления (Он близок к еди-
нице) сняаан с давлением соотношением 1+ (а/йГ)р, Решая обратную задачу,
покажите, как получить поляризуемость из измерения показателя преломления
газа.
23.11, Показатель преломления бензола постоянен (1,51) от 0,4 до примерно
0,55 ГГц (заметьте, что это микроволновые частоты), но затем наблюдается се-
рия его колебаний между значениями 1,47 и 1,54, В указанном интервале частот
показатель преломления толуола больше (около 1,55), но в том же месте, как
и в случае бензола, также наблюдаются колебания и дополнительные колебания
между значениями 1,52 п 1,56 вблизи 0,4 ГГц. Объясните эти наблюдения.
23,12, Дипольный момент хлорбензола равен 1,17 D, а его поляризуемость со-
ставляет 6,1-10“23 см3. Определите его относительную диэлектрическую прони-
цаемость при комнатной температуре, приняв плотность ранной 1,1732 г/см3.
23.13. Определите поляризуемость редких газов на основе потенциалов иониза-
ции и радиусов (табл. 4,8 в т. 1 и табл, 22,2), Экспериментальные значения при-
ведены в габл. 23.1.
23.14. Используя уравнение (23,1.15), вычислите и изобразите на графике по-
казатель преломления ноды между 0 н 100°С с учетом следующих данных по
плотностям;
t, °C 0 - 20 40 60 80 100
р, г/см3 0,99987 0,99823 0,99224 0,98324 0,97183 0,95841
Примите а=1,88-10-23 см3.
23.15. Каков показатель преломления водяного пара при 100 °C и давлении
1 атм? Оцените показатель преломления кристаллов СаС12, NaCl и Аг из данных
табл. 23.3. Плотности равны соответственно 2,15, 2,163 и 1,42 г/см3.
23.16. Иногда вместо самой поляризуемости приводится молярная поляризуе-
мость Pm- Она определяется как Pm = La/3. Как°ва ее ра3мерноств? Как она
должна зависеть от плотности? Покажите, что она связана с Л’г и плотностью
образца о уравнением Масотти Рт= (Л4/р) (Кг— 1)/(Дг-1-2), Поэтому как
(К.-—1 )/(Лг+2) зависит от плотности?
23,17. В сноей классической книге «Полярные молекулы» Дебай опубликовал не-
которые результаты ранних измерений молярной поляризуемости аммиака. Из
приведенных ниже данных найдите дипольный момент и поляризуемость моле-
кулы.
Г, К 292,2 309.0 333,0 387,0 413.0 446,0
Рга, смз/моль 57,57 55,0! 51,22 44,99 42,5] 39,59
23.18. Показатель преломления аммиака при давлении 1 атм и 273 К равен
1.000379 (для желтой линии натрия). Какова молярная поляризуемость газа при
а) 273 К и б) 292.2 К? Объедините эти данные со статической молярной поля-
ризуемостью при температуре 273 К и из этих двух измерений получите вели-
чину дипольного момента молекулы,
23.19. Показатель преломления аргона при I атм и 298 К равен 1,0028]. Опреде-
лите напряженность электрического поля (в В/м), действующего на другой атом
при их соударении. jj
23. Электрические ц магнитные свойства молекул
329
23.20. Покажите, что средняя энергия взаимодействия 1 моля атомов диаметром
d, взаимодействующих с потенциальной энергией вида —дается выраже-
нием U=—' гдс V — объем, в котором они находятся, и все кластер-
ные эффекты игнорируются. Отсюда найдите связь между вандерваальсовым
параметром а (т. К разд. J.4) и Cs. Исследуйте логичность этого заключения
в случае аргона.
23.21. . Уравнение (23,3.7) дает выражение для второго вириального коэффпииен-
та В через меж молекулярный потенциал V'(R_). Предположите, что атомы рас-
положены па расстоянии наибольшего приближения d, а снаружи этого ннтер-
вала ови притягиваются друг к другу потенциалом вида —C-JR6. Предположите
далее, что, когда атомы не находятся в контакте, К/йГ настолько мала, что экс-
поненциал е~с можно аппроксимировать с помощью 1 —х. Найдите выражение
для В через Се и d.
23.22. Соотнесите В с поляризуемостью атомов газа, связав Се с а. Затем опре-
делите величину В при 298 К для аргона, взяв а—2,0-] О-33 см3, 7—15,8 эВ и
— d=286 пм.
23.23. Везде в данной главе мы полностью игнорировали влияние гравитацион-
ных сил между молекулами. Оправдано ли это? Оцените их относительную важ-
ность для двух атомов аргона, находящихся почти г. контакте. Возьмите грани-
тациониую постоянную G=6,67-10“11 Н-м!/кг!.
2324. Плотность энергии когезии определяется как — UjV, где (7—средняя пе-
тенцнальная энергия притяжения в образце и V — его объем. Покажите, что эту
величину можно записать как —-g-TT2 J V(R)dx, где Л'"— число молекул в едн-
вице объема и К(7?)— потенциальная энергия их притяжения, причем интеграл
охватывает интервал от d до бесконечности. Далее покажите, что плотность энер-
гии когезии при однородном распределении молекул, удерживаемых вместе вандер-
ваальсовым притяжением вида —Са/7?6, дается выражением (2л/3)
где р—плотность твердого вещества, a Мг— относительная масса молекул.
23.25. Плотность энергии когезии приблизительно равна энтальпии испарения па
единицу объема. Определите мольную энтальпию испарения четырех хлор и стог о
углерода на основе того, что ряа1,594 г/см3 и молекулярная поляризуемость
составляет 1,27-]0-33 см3.
2326. Еще в гл. 6 (т. 1) мы видели, что для вандерваальсовых газов (dUjdV)T=
^rfia’V2. Если отождествить U со средней энергией когезии, то можно испо-ц>зо-
вать это выражение для нахождения соотношения между а и С6. Найдите это
соотношение, а затем выразите критические постоянные через d и Cs,
2327. Оцените критические постоянные Для СС1( на основе его поляризуемости.
2328. Воспользовавшись результатами двух предыдущих задач, покажите, что
плотность энтропии испарения газа в критической точке равна 97?р/]6Л1г.
23.29. В уравнении (23.3.3) определена постоянная Маделунга. Одним из про-
стых случаев, когда сс- можно рассчитать без большого труда, является случай
чередующихся положительных и отрицательных ионов, разделенных расстоянием
R.. Каково значение постоянной для такого расположения?
23.30. Для определения внутренней энергии ионного кристалла по уравнению
(23.3.4) требуется знать параметр К*. Ето можно получить, зная изотермический
коэффициент сжатия кристалла; в дацной задаче используется это соотношение.
Прежде всего с помощью термодинамического уравнения состояния (dU/3V)r —
— TfapidTy.—p, решенного для абсолютного нуля, покажите, что 1/к пропорцио-
нальна dHJIdV2, а затем выразите /?* через V, х и 7?0. (На определенной стадии
вы, возможно, сочтете полезным написать V' = c7?^, где с — константа, которую
в дальнейшем можно исключить.)
23.31. Постройте цикл Борна—Хабера для определения энергии когезии КС!
И вычислите ее из данных гл. 4 (г. 1). Примите, что ДЯсу6л(К) =82,6 кДж/моль.
23.32. Рассчитайте энергию когезии кС1 из уравнения (23.3,4), используя я=
= 1.1-!Q-S атм-' для изотермического коэффициента сжатия и р=1,984 г/сд’.
330
Часть 2. Структура
23.33. Ионные радиусы Na+ и F“ равны соответственно 95 и 136 пм. Какова
энергия когезии решетки NaCl? Используйте Д*л29 пм.
23.34. Ионные радиусы Cs+ и С1“ равны соответственно 167 и 181 пм. Какова
энергия когезии кристаллической решетки? Что было бы, если бы эгн ионы на-
ходились: а) в решетке NaCl и б) в структуре вюрпита? Используйте /?* =
=40 пм.
23.35. Важным методом получения информации о межмолекулярных силах яв-
ляются молекулярные пучки. Эта и следующая задачи представляют собой очень
простое введение в сложную задачу разбора богатой информации, которую они
несут, и выделения из нее требуемых данных. Рассмотрите столкновение между
атомом типа жесткого шара радиусом и массой mi и бесконечно тяжелым
сферическим атомом радиусом Д2, который также является непроницаемым ша-
ром. (Представьте, например, что это Ни 1). Постройте график зависимости
угла рассеяния 0 ог ударного параметра Ь. Расчет производите на основе про-
стых геометрических соображений.
23.36. Характеристики рассеяния атомов связаны с энергией соударения. Ситуа-
цию можно промоделировать следующим образом. Предположим, что оба атома
всегда ведут себя как непроницаемые жесткие шары, по эффективный радиус
тяжелого атома зависит от относительной скорости его приближения к легкому
атому. Предположим также, что эффективный радиус тяжелого атома зависит
от скорости о как Т?2ехр{—о/о*), где в* — некоторая константа. Б таком случае
при низких скоростях подхода к легкому атому эффективный радиус тяжелого
атома будет и будет меньше, когда скорость приближения больше. Возьмите
ударный параметр fr=~g-A?2 я постройте график зависимости угла рассеяния от
а) скорости и б) кинетической энергии приближения.
23.37. Молярная диамагнитная восприимчивость молекулы может быть очень
просто рассчитана из волновой функции вещества. Например, для атома Она
равна —Л"(е2р0/6те) (и8). Какова диамагнитная восприимчивость атома водо-
рода? Какова его парамагнитная восприимчивость при комнатной температуре?
Какова общая восприимчивость?
23.38. Какова магнитная восприимчивость при комнатной температуре молекулы:
а) с Двумя неспарснными электронами и б) с пятью неспаренными электронами?
Какова восприимчивость в обоих случаях при 4 К?
23.39. Напряженность поля лигандов влияет на число неспареивых d-электронов
в комплексах переходных металлов. На основе предположения, что вклад в вос-
приимчивость вносят только спиновые магнитные моменты (приближение <гтоль-
ко спияз, spin-only approximation), составьте таблицу ожидаемой парамагнитной
восприимчивости октаэдрических (^-комплексов (п=1—>-]0) для слабых и силь-
ных полей лигандов (см. задачу 15.29 в т. 1).
23.4Й. Как мы видели в гл. 20 и 21, молекула NO специфична в том смысле, что
она имеет термически доступные электронно-возбужденные состояния. Она содер-
жит также иеспаренный электрон и поэтому, как ожидается, будет парамагнит-
ной. На самом деле ее основное состояние непарамагнитно. Эго связано с тем,
что магнитный момент, обусловленный орбитальным движением электрона вокруг
молекулярной оси, компенсируется спиновым магнитным моментом. Первое воз-
бужденное электронное состояние (при 12,1 см-1) парамагнитно, поскольку ор-
битальные моменты не компенсируются, а складываются со спиновым моментом,
и магнитный момент равен двум магнетонам Бора. Поскольку высшее состояние
термически доступно, парамагнитная восприимчивость молекулы имеет особую
температурную зависимость даже при комнатной температуре. Рассчитайте па-
рамагнитную восприимчивость NO и постройте график ее зависимости от темпе-
ратуры. Какова ее величина при 298 К?
Часть 3. Изменение
24 Молекулы в движении.
Кинетическая теория газов
Изучаемые вопросы
После тщательного изучения этой главы вы сможете;
1. Строго определить кинетическую модель идеального газа
(стр. 332).
2. Использовать кинетическую теорию для расчета давления,
оказываемого идеальным газом [уравнение (24.1.2)].
3. Определить среднюю величину дискретного [уравнение
(24.1.5)] и непрерывного [уравнение (24.1.7)] распределений.
4. Использовать вероятностные факторы для вывода распреде-
ления молекулярных скоростей по Максвеллу—Больцману [урав-
нение (24,1,11)] и по Максвеллу [уравнение (24.1.13)].
5. Рассчитать среднюю скорость [уравнение (24.1.15)], сред-
неквадратичную скорость [уравнение (24.1.14)] и наиболее веро-
ятную скорость [уравнение (24,1.16)] молекул газа.
6. Определить сечение столкновения (стр. 345) и рассчитать
частоту столкновений [уравнение (24.2.5)] и среднюю длину сво-
бодного пробега [уравнение (24.2.7)] в газе.
7. Рассчитать частоту столкновений молекул газа с поверхно-
стью [уравнение (24.2.9)].
8. Объяснить термин транспортное свойство (стр. 348) и опре-
делить поток (стр. 349).
9. Сформулировать и использовать первый закон диффузии
Фика [уравнение (24.3,1)].
10. Рассчитать скорость истечения газа через отверстие, сфор-
мулировать закон Грэма и использовать метод Кнудсена для оп-
ределения давления пара (стр. 352).
11. Вывести закон Фика и рассчитать коэффициент диффузии
для идеального газа на основе кинетической теории [уравнение
(24.3.8)].
12. Рассчитать коэффициент теплопроводности [уравнение
(24,3,10)] и вязкость [уравнение (24.3.12)] из кинетической тео-
рии,
13, Описать, как измеряется вязкость газа (стр. 359).
331
332
Часть 3. Изменение
Введение
В этой главе рассматривается поступательное движение сово-'
купности атомов и молекул в газообразном состоянии и выясняет-
ся, как можно объяснить свойства газов на основе постоянного
поступательного движения частиц, составляющих эту совокупность/
Сконцентрируем внимание на свободном поступательном движении,
молекул и не будем учитывать взаимодействия между ними. Это'
означает, что мы будем изучать кинетическую теорию газов. Пол-;'
ная проблема, учитывающая движение молекул под влиянием их
взаимодействия Друг с другом, рассматривается динамической
теорией; некоторые ее аспекты (простые столкновения атомов)
были затронуты в гл. 23, другие встретятся в гл. 27.
Элементы кинетической теории газов в общих чертах были
намечены в главе «Введение» в т, 1. Там мы видели, что пред*
ставлепне об идеальном газе, как об объекте, состоящем из сово-
купности частиц, позволяет рассчитать некоторые его свойства. Это
будет отправной точкой для более подробного рассмотрения в на-
стоящей главе, и, Прежде чем следовать дальше, мы снова обсудим
это положение. В данной главе мы заострим внимание на отдель*
ных доводах, использованных во введении, укажем на недостатки
расчетов и, Продолжая изложение,, выведем простые выражения
для некоторых особо интересных свойств идеальных газов. В ча-
стности, мы сконцентрируем внимание на транспортных свойствах
газов: сюда относятся такие свойства, как теплопроводность, вяз-
кость и диффузия, которая проявляется в переходе некоторого
свойства из одной области системы к другой.
Кинетическая теория базируется на трех предположениях.
1. Газ состоит из множества частиц с массой m в непрерыв-
ном беспорядочном движении.
2. Частицы имеют пренебрежимо малые размеры в том смыс-
ле, что их диаметры значительно меньше среднего расстояния
между ними.
3. Частицы не взаимодействуют друг с другом, пока их столк-
новения имеют характер упругих столкновений. Упругое столкно-
вение означает, что общая поступательная кинетическая энергия
сталкивающейся пары одинакова до и после столкновения: энер-
гия ни одной из Сталкивающихся частиц не переходит во враща-
тельную, колебательную или другие виды энергии.
Столкновения приводят к тому, что частицы постоянно меняют
свою скорость и направление. Число столкновений одной частицы
в единицу времени, частота столкновений Z, играет важную роль
в обсуждении транспортных свойств и химических реакций в га-
зовой фазе. Мы рассчитаем ее значение на основе кинетической
теории. Среднее расстояние, которое проходит каждая частица
между столкновениями, называется средней длиной свободного
24. Кинетическая теория газов
333
пробега она играет существенную роль при рассмотрении транс-
портных явлений потому, что указывает, как долго молекула со-
храняет некоторое свойство перед столкновением- Средний сво-
бодный пробег является также мерой среднего расстояния между
частицами, и его можно использовать в качестве критерия приме-
нимости кинетической теории к реальным системам* Например,
если мы знаем, что молекула реального газа имеет диаметр d, то
кинетическая теория даст хорошее описание его свойств, если
d/k мало (d/A<Cl), ио она не будет применима, если это не так*
Совокупность частиц постоянно также сталкивается со стенками
содержащего их сосуда, и столкновения происходят так часто, что
стенки практически подвергаются действию постоянной силы. Си-
ла па единицу площади — это давление, и, таким образом, с по-
мощью кинетической теории можно качественно объяснить давле-
ние газов. Теперь покажем, что с помощью кинетической теории
все эти свойства можно также рассчитать количественно.
24.1. Основные расчеты
Перевод кинетической теории газов на количественную основу
включает два основных типа расчетов* Первый приводит к выра-
жению для давления, а второй состоит в выводе детальной кар-
тины распределения скоростей молекул.
Давление, оказываемое газом. Рассмотрим систему, представлен-
ную на рис, 24*1* Когда молекула с массой пг сталкивается со
стенкой, перпендикулярной оси х, составляющая ее количества
движения вдоль оси х изменяется от mvx до —fnvx, другие состав-
ляющие остаются неизменными. Общее изменение количества
движения при каждом столкновении составляет величину [2тох|*
Число столкновений за промежуток времени Д/ равно числу мо-
Результирующая
сила
лекул, способных достичь
стенки за это время. Рас-
стояние, которое может
пройти молекула, двигаю-
щаяся со скоростью us,
за время Д/, равно
| Ух 1Д/, и, таким, об-
разом, все молеку-
лы, находящиеся в
Рис. 24.1. К расчету давле-
иияг оказываемого газом.
334
Часть 3. Изменение
пределах расстояния|| Д£ от стенки, натолкнутся на стенку, ес-
ли они будут двигаться по направлению и ней. Если поперечное
сечение сосуда имеет площадь А, все молекулы, находящиеся в
объеме А |еж| Д/, достигнут стенки (если они перемещаются по на-
правлению к ней). Если Ж — число молекул в единице объема, то
число молекул в интересующем пас объеме равно ЖЛ | ид| Д/. Око-
ло половины из их числа движется вправо, другая половина —
влево, так что среднее число столкновений в интервале времени
Д/ равно -уЛМ | | Д(. Общее изменение количества движения за
этот промежуток времени равно
изменение количества движения ~
(число столкновений) X (изменение количества движения
при столкновении) - А ) | Д<j 2m ( vx | j —
=m^ Avx&f,
и скорость изменения количества движения равна т-Л”Аих. Соглас-
но закону Ньютона, скорость изменения количества движения есть
сила, и, таким образом, сила, действующая на стенку со стороны
газа, равна тЖ/к4. Давление — это сила, действующая на еди-
ницу площади, поэтому давление равно m./Ti’’. (поскольку пло-
щадь стенки равна А).
Не все молекулы движутся с одинаковой скоростью, н поэто-
му определяемое давление является средним значением только
что рассчитанной величины. Если среднее значение vx записать
как ( , то придем к выражению
р=^т(о®).
Поскольку движение молекул беспорядочно, средняя величина
будет такой же, как в направлениях у и z.
<ц£)=(ф = (гф.
Скорость одной молекулы определяется следующим выражени-
ем: о2 = ож-|-^4'и > поэтому среднюю скорость Vs можно записать
как
(о2) -- (^) -h (ф + (ф =3 (»?).
Мы будем записывать среднее значение и2 как с2:
с2 = (^2) (24.1.1)
и называть с2 средним квадратом скорости молекул. Квадратный
корень из этой величины называется среднеквадратичной скоро-
24. Кинетическая теория газов
335
стью, с = У<иа) . Учитывая высказанные замечания, получим ко-
нечное выражение для давления
p~-^mc\ (24,1.2)°
Это одно из ключевых уравнений кинетической теории. (Кружок
над номером уравнения, как и в части 1, означает, что примене-
ние уравнения ограничивается идеальным газом, причем в этой
главе уравнения для идеального газа получаются из кинетичес-
кой теории.)
Плотность Ж (число молекул в единице объема) можно за-
менить на У/V, где У — истинное число имеющихся молекул и
V — общий объем системы. Выражая У через число Авогадро L
(N=nL), получим
рУ=-^-пСтс\ ’ (24.1.3)°
В главе «Введение» (т. 1) было показано, что это выражение пе-
реходит в уравнение идеального газа, если принять, что с2 про-
порциональна абсолютной температуре Т. Теперь мы встанем на
альтернативную точку Зрения и выведем это уравнение, рассмат-
ривая распределение скоростей согласно кинетической теории. Сна-
чала мы рассмотрим средние величины свойств, а затем специаль-
но обратимся к случаю средних значений компонентов скоростей.
В таком случае для описания средних значений молекулярных ско-
ростей (и любых других свойств) необходимо знать только один
неизвестный параметр р, и можно вывести, что рК~1/р. Посколь-
ку из эксперимента известно, что pV^nRT, мы можем получить
значение этого единственного неизвестного параметра р. Опреде-
лив его, мы сможем использовать этот метод для получения сред-
них значений целого ряда свойств и развить кинетическую теорию
с учетом столкновений и транспортных процессов.
Средние значения и распределения. Предположим, что мы хотим
найти Среднее значение свойства X, которое может принимать лю-
бые значения Хь Х%, ..., Xz (их мы назовем возможными исхода-
ми наблюдений), и вот в серии из У измерений мы находим, что
X, встречается У\ раз, встречается У2 раз и т. д. Среднее зна-
чение ( X ) свойства X является взвешенной суммой всех резуль-
татов, деленной на общее число наблюдений:
, уч _ УЛ + + • • • + МгХг _ + Л'2Ха + ... + NZXZ _
JV1 + JVa+ ... +Уг N
z
(24.1.4)
<=1
336
Часть 3. Изменение
Формула для средней величины может быть выражена другим пу-
тем, если ввести вероятность получения результата Xi в экспери-
менте, В данном примере вероятность того, что исходом наблю-
дения является Xi, равна Afj/X, и то же можно сказать для всех
других исходов. Обозначим эти вероятности Pi (так что Pj — ^’j/N
и т, д., и в общем виде Pi~Ni/N). Тогда средняя величина может
быть записана как
<Х)=2РЛ. (24.1.5)
i
где суммируются все возможные исходы. Заметим, что из опреде-
ления Pi
(24.1.6)
i t
Теперь допустим, что исход эксперимента может принять одно
из значений непрерывного ряда величин. Например, это справед-
ливо в случае, когда нас интересует Средний рост индивидов на-
селения или средняя скорость молекул газа. Тогда определение
среднего значения должно быть видоизменено.
Разделим на части непрерывный ряд возможных исходов (рис.
24.2). Будем считать за 1 каждый исход, который попадает в пре-
делы данного отрезка. Например, рассмотрим отрезок длиной ДХ,
начинающийся от X. Если в серии из 300 экспериментов какой-то
исход, лежащий в этом отрезке, получается в 6 экспериментах, мы
напишем Х(Х)=6, Если общее число экспериментов равно X, ве-
роятность того, что исход, полученный в любом одном эксперимен-
те, лежит в интервале от X до Х-|-ДХ, определяется как Х(Х) =
— N(X)/N, что в данном случае составляет 1/50. Значение Х(Х),
и поэтому значение Р(Х), пропорционально длине отрезка от X ;
до Х+ДХ (если ДХ мало). Мы определим это, написав Р(Х) = ,
=/(Х)ДХ.
Итак, мы перевели непрерывную задачу в дискретную с по-
мощью приближения, которое группирует вместе различные исхо-
ды экспериментов, как описано выше. Это приближение в самом
24, Кинетическая теория газов 337
деле справедливо, и сейчас мы будем продолжать изложение так
же, как и выше,
Так же как при рассмотрении настоящего дискретного случая,
мы можем рассчитать приближенное среднее значение X, взяв его
значение для каждого отрезка, умножив его на вероятность Р(Х)
того, что наблюдения лежат в этом отрезке, и затем суммируя по
всем отрезкам;
<Х)« 2 хр(х) = 2 Х/(Х)ДХ.
отрезки отрезки
На данном этапе это является лишь приближением, поскольку X
может значительно изменяться по ширине отрезка. Это соотноше-
ние может быть сделано точным, если каждый отрезок будет бес-
конечно малым, так что X можно будет считать неизменным в
пределах этого отрезка. Поэтому заменим конечный отрезок ДХ
бесконечно малым отрезком dX. Это даст возможность выразить
сумму через интеграл
(Х)=^ Xf(X)dX. (24 Л 7)
Это очень важное выражение: оно лежит в основе изложения ма-
териала этой главы.
Интервал интегрирования в последнем уравнении охватывает
все возможные значения наблюдаемого свойства X, Например, ес-
ли необходимо определить средний рост h индивидов, популяции,
его пределы будут От 0 до оо:
{h}^hf(h)dh, (24.1.8а)
с
потому что рост может быть только положительным. Если бы мы
рассматривали среднюю скорость частицы, соответствующий ин-
теграл выразился бы следующим образом:
<*Д> = J vj(y^dvx, (24.1.86)
поскольку х-составляющая скорости может иметь любой знак не-
зависимо От направления частицы,
Функцию f(X) называют распределением свойства X, Из его
определения P(X)—f(X)XX мы видим, что оно дает вероятность
того, что исход лежит в интервале от X до Х-фДХ. В Границах бес-
конечно малого отрезка dX функция f(X) дает вероятность того,
что исход лежит в бесконечно малом интервале от X до X-j-dX.
Например, функция f(h) характеризует вероятность того, что из-
меренный рост лежит в интервале от h до n±dh, а распределение
22—242
338
Часть 3. Изменение
/(^.t) — вероятность того, Что х-составляющая скорости находится
в интервале От vx до Так, если /(180 см) =0,12 см"1, то
это значит, что вероятность того, что индивиды имеют рост в ин-
тервале 180—181 см, будет приблизительно 0,12, а для интервала
180—182 см вероятность будет приблизительно 0,24 (отметим, что
этот интервал в действительности не бесконечно мал, но прибли-
зительно это так). Наоборот, если /(200 см) =0,001 см-1, это оз-
начает, что вероятность роста в интервале 200—201 см будет толь-
ко около 0,00 L
Другое свойство вероятностных распределений, которое нас ин-
тересует, связано с вопросом одновременного рассмотрения не-
скольких типов свойств. Например, может возникнуть необходи-
мость определения вероятности того, что система имеет как зна-
чение Х[ некоторого дискретного свойства X, так и значение У,
некоторого другого дискретного свойства У. Если эти свойства не-
зависимы друг от друга, то вероятность того, что система имеет
как значение Хг свойства X, так и значение У, свойства У, будет
Р^У^Р^Р^),
где Р(Хг) и P(Yi)—отдельные вероятности. Например, если ве-
роятность для человека родиться мужчиной составляет 0,495, а
вероятность человека (мужчины или женщины) быть левшой рав-
няется 0,110, то вероятность того, что при случайном выборе из
группы людей попадается мужчина левша, рав^а 0,110'0,495 =
=0,054, или 1 из 18,5. Однако этот расчет оказался бы неправиль-
ным, если бы левшой мог быть только мужчина.
Те же приемы можно применить к непрерывным свойствам. Ес-
ли вероятность того, что X лежит в интервале dX от X, есть
f(X)dX, и вероятность того, что независимое свойство находится в
интервале от У до Уф-^У, равняется f(Y)dY, тогда общая вероят-
ность того, что X лежит в интервале от X до dX и У попадает в ин-
тервал от У до У-j-d}', будет произведением вероятностей:
f(X)f(Y)dXdY. Например, вероятность того, что молекула имеет
компоненту скорости в интервале от до vx±dux и компоненту
скорости vy в интервале от vy до vy-\-dvv, будет равна
f(vx)f(Uy)dv^dvv, потому что эти скорости независимы друг от дру-
га (за исключением некоторых сложных случаев).
Распределение молекулярных скоростей. Теперь мы в состоянии
перейти к рассмотрению функции распределения для компонент
скорости частиц в идеальном газе с точки зрения кинетической
теории. Три компоненты скорости не зависят одна от другой, п
поэтому вероятность F(yx, vy, vx)dvxdvydvz того, что молекула будет
иметь скорость с компонентами соответственно в интервалах от t-’x
до Охфл/гло от vy до Wi/T-duj, и от vz до vz-[-dvz, является произве-
дением отдельных вероятностей для каждой компоненты:
F (рх, vz) dv^Vy dv2 =f (vx) f (vy) f (уг) dv^dv^.
24. Кинетическая теория газов
339
Теперь допустим, что вероятность того, что компоненты скорости
молекулы будут расположены в определенном интервале, не зави-
сит от ориентации направления ее полета, но зависит от скоро-
сти. Так, например, вероятность того, что компоненты скорости
имеют значения км/с, v# = 2 км/с, t>. = 3 км/с (и поэтому ско-
рость, равную 714 км/с), будет такой же, как вероятность того, что
эти компоненты скорости имеют значение 2, 3 и 1 км/с соответст-
венно (скорость та же) или имеют любые другие направления дви-
жения, при которых скорость равна "/14 км/с. Это означает, что
функция распределения F(vx, vy, и?) может зависеть только от
скорости Vj где о2 — о* 4-^+^, но не от индивидуальных компо-
нент. Поэтому F можно записать как функцию F(v* _|_ц| -фиД), и
тогда последнее уравнение будет выглядеть следующим образом:
Р (^-W+ - f (уя) f (уи) f (vz).
Этому уравнению удовлетворяет только экспоненциальная функ-
ция (так как еа-’-ь —е“е6), и поэтому запишем
f(ox)=-K ехр(± &’),
где Л и — константы. Это единственная функция, удовлетворя-
ющая уравнению, и поэтому можно записать
f (vx) f (vu) f (^) = №exp (± 5^4 ^4 ^J) - F (^4-t£+
что и требовалось доказать. Двойственность знака + в показа-
теле экспоненты можно ликвидировать, руководствуясь физичес-
ким смыслом. Вероятность очень больших скоростей должна быть
крайне мала, поэтому необходимо брать отрицательный знак.
Значение констант К и £ можно установить на основании сле-
дующих двух доводов. Во-первых, общая вероятность того, что
компонента скорости лежит в интервале —оо оо, должна
равняться 1 (должна же быть некоторая скорость в этом интер-
вале), Поэтому
+со
J f (их] dvx = 1.
Подстановка экспоненциальной формы f(x) приводит к
+» н-м
J f (ух) dvx=к Jexp (— М) dvx (л/£)’Д
Поэтому К— (£/л)1/2.
Значение £ может быть получено расчетом среднеквадратичной
скорости молекул с последующим использованием уравнения
(24.1,3), чтобы приравнять ее к величине pV, которую можно из-
мерить. Мы сконцентрируем внимание на расчете ( V* ) , зная, что
22*
340
Часть 3. Изменение
другие компоненты имеют то же среднеквадратичное значение.
Из общего выражения для средней величины (при Х = У»)
можно написать
-|-оо -f-co
<i£) = [u2J(vx)dux^/n)4> ft£exp(— &fydvx.
Интеграл с правой стороны является стандартным (табл. 24.1; он
может быть очень просто получен из интеграла, входящего в вы-
шеприведенный расчет Л) и имеет значение (п/£3) v’.
Следовательно,
<$=т(тя/?)’/!=4- ,
*
Поэтому среднеквадратичная скорость с2 = ( и2 ) — (+ v* + >1
равна
(24.1.9)
Объединяя этот важный результат с уравнением (24.1.3), полу-
чим
nLmiZ.
о *
Таблица 24.1
Интегралы хлехр(—ах2)
00 а
1„ — J х" ехр( — ax2)dx j
° 1
n= 0 1 2 3 4 J
1 1 3 ч
Л> = —хМа vW)*'1 I
Z 4 о .;'3
. z ’
Интеграл (2/Ул) f ехр(—xs)dx называется функцией ошибок и обозначается
о
eri z. Эта функция имеется в многочисленных таблицах; ниже даются некоторые
ее значения:
z] =0 0,2 0,4 0,6 0,8 0,9
erf z =0 0,2227 0,4284 0,6039 0,7421 0,7969
z =1,0 1,2 1,4 1,6 1,8 1,9
erf z = 0,8427 0,9103 0,9523 0,9763 0,9891 0,9928
24. Кипеть чес кая теория газов 341
Но известно, что идеальный газ подчиняется уравнению
pV=nRT=nLkT.
Приравнивая эти два уравнения, можно найти £, связанную с тем-
пературой системы;
±nLtnfc=nLkT, где (24.1.10)°
Поэтому полная форма распределения скоростей выразится сле-
дующим образом:
f (vx) —(m/2nkTy>!> ехр ( —i- mfykTj. (24.1.11)°
Как видно из последнего уравнения, его правая часть имеет фор-
му распределения Больцмана [см. уравнение (1.2) в т. 1] для ча-
стиц с кинетической энергией пгУх, двигающихся вдоль оси х, и,
таким образом, это уравнение можно вывести также через выра-
жение Больцмана. По этой причине распределение f(vx) называ-
ют распределением молекулярных скоростей Максвелла—Больц-
мана, отмечая как вклад Максвелла (он первым получил его), так
и Больцмана (он строго его доказал).
Пример (вопрос 5). Навеска цезия нагрета в печи до 500 °C. В одной из стенок
имеется небольшое отверстие, и атомы выходят из нее н виде атомного пучка.
Какова их средняя скорость?
Метод. Это одномерная задача. Поскольку только атомы, двигающиеся вправо
(х>0), будут попадать в пучок, можно написать Средняя скорость
равна ( vx ), и поэтому можно использовать уравнение (24.1.86), но интегри-
ровать только от 0^=0 до Используем уравнение (24.1.11) для f(vx),
СО
видоизменив его так, чтобы стало 1; для этого просто умножим
о
правую часть уравнения (24.1.11) на 2.
Ответ. Из уравнений (24.1.86) и (24.1.11)
ею
(v,)= Jv^(»x)dtU =
о
со
о
= 2 (т/2л^Л1/!! (kT/m) = V pkT/wri).
Для цезия при 500 °C
( (2/л) X (1,381 • 10-«> Дж/К) X (773 К) у/,
™ “ [ (132,9)х(1,6605-I0-s?кг) ) ~ 1'=>>Ьм/с-
342
Часть 3. Изменение
ем Трехмерной задачи. Вероятность
Рис. 24.3. Распределение молеку-
лярных скоростей.
Комментарий. Это средняя ско-
рость выходящих атомов: диапа-
зон скоростей может быть очень
большим, и в атомполучевой аппа-
ратуре обычно используют селек-
тор скорости (см. рнс. 24.5).
Перед обсуждением сущ-
ности распределения Макс-
велла-Больцмана мы за-
кончим вывод рассмотрени-
ем Трехмерной задачи. Вероятность того, что скорость имеет ком-
поненты, лежащие в элементе объема dvxdv,.dv, в точке (vx, иу,.
Vz), равняется j
dF fe- vd G'J f (^) f (Уг) duxdvydv, =
~(ml2nkT)3^ exp (— tnv1 j2kT} dvxdvydvs.
(24.1.12)°
Предположим, что нас в большей степени интересуют скорости
молекулы и не интересует направление их движения. Вероятность
того, что молекула имеет скорость V, независимую от направле-
ния, является суммой всех вероятностей, выраженных последним
уравнением по всем ориентациям скорости. Сумма всех объемных
элементов dvxdvydvz, получающаяся при движении точки (уч, vv,
и2) по всей поверхности сферы постоянного радиуса v, представ-
ляет собой объем сферической оболочки радиусом v и толщиной -
dv (рис. 24.3). Этот объем составляет 4nti2do. Поэтому вероятность-;
того, что скорость лежит в интервале от v до v~\-dv (независимая
от направления движения), равняется
dF (у) — 4л (m/2nkT)s't v2 ехр(— ти*/21гТ) dv. (24.1.13)°.-
Это важное выражение представляет собой распределение скоро--
стей Максвелла.
Пример (вопрос 5). Какова средняя скорость атомов цезия внутри печи в по-
следнем примере?
СО
Метод, Средняя скорость получается из выражения с= J uF(o)dv, где F(v)
о
правая часть последнего выражения.
24. Кинетическая теория газов
343
Рис. 24.4. Распределение скоростей по Максвеллу и его зависимость от темпе-
ратуры.
Ответ. Иа последнего выражения
са со
с - j* vF (о) dv = 4 л (т/2лАТ)3'2 j rfl ехр (—тсД/2А7) do =
о о
= 4л (m/2nAT)3^ (ikT/m)2 = (SkT/ят)1^.
Для цезия при 500 °C
- f 8Х(1,381 10-23 Дж/К)Х(773К)у/г . ’
с ~ [ лХ(132,9)Х 1,6605-10'27 кг) J — э ,и М
Комментарий, Заметьте, что эта средняя величина значительно выше скорости
одномерного атомного пучка в последнем примере.
Некоторые свойства распределения Максвелла были рассмот-
рены в главе «Введение» (т. 1). Основные особенности показаны
на рис. 24.4; они состоят в том, что при более высоких температу-
рах интервал скоростей увеличивается и самая вероятная ско-
рость, отмеченная на рисунке как с*, по мере повышения темпе-
f а туры сдвигается в сторону более высоких значений. (См. рис.
1 на стр. 24 в т. 1, из которого также очевидно следует, что при
одной и той же температуре скорость более легких молекул выше,
чем у более тяжелых.)
344
Часть 3. Изменение
Рис. 24.5, Определение молеку-
лярных скоростей.
Необходимо очень точно различать несколько путей нахожде-
ния средней скорости молекул. Среднеквадратичная скорость с
представляет собой квадратный корень из средней величины и2;
c=(3feT//n)'/s. (24.1.14)
Средняя скорость с — это среднее из скоростей, рассчитанных с
использованием распределения Максвелла по уравнению (24.1.13):
со
с = (у) ~ J* vF (у) de — (8kT/jim.yn
о
(24,1.15)
Наиболее вероятная скорость с* представляет собой скорость, при
которой кривая распределения Максвелла Проходит через макси-
мум :
(24.1.16)
Они различаются небольшими числовыми множителями (сяй
« 1,225 с*, 1,128 с*).
Распределение Максвелла наблюдалось в многочисленных пря-
мых и косвенных экспериментах, так что можно считать, что урав-
нение (24,1.13) действительно справедливо, Скорости молекул не-
посредственно могут быть измерены с помощью селектора скоро-
стей типа показанного на рис, 24.5, Вращающиеся диски имеют
щели, которые позволяют проходить только молекулам, движу-
щимся через пих с определенной скоростью, и число молекул м<
жет быть определено подсчетом их в детекторе. В косвенном мен
де используется эффект Доплера па длины волн света, испуска'
мото движущимся объектом; этот эффект рассматривался в раз,
17.1. Типичные средние скорости для некоторых молекул прив!
дены в табл. 24.2,
Таблиц а 24.2
Средние молекулярные скорости с (м/с) при 25 °C
Не 1254 н2 1766 Н„О 591 с.н. 284
Аг 396 n2 474 NH3 608 Hg 170
ог 443 - СО2 378 Воздух 466
С1. 286
24, Кинетическая теория газов
345
24.2. Столкновения
. Теперь мы рассчитаем частоту межмолекулярных столкнове-
ний. Основные особенности расчета были намечены в общих чер-
тах во введении. Здесь мы резюмируем этот расчет и затем улуч-
шим его.
Межмолекулярные столкновения. Мы считаем, что произошел
«удар» всякий раз, когда центры двух молекул находятся в преде-
лах некоторого расстояния d друг от друга, где d можно рассмат-
, ривать как их диаметры (рис. 24.6). Проще всего считать, что все
атомы, кроме того, который нас интересует, неподвижны, а затем
• исследовать, что происходит, когда подвижный атом перемеща-
ется в газе со средней скоростью с в течение времени Д/. Во время
этого перемещения подвижный атом движется в «трубке столкно-
вения» с сечением с = ш/2, длиной сД/ и, следовательно, объемом
осДЕ Величина сг называется сечением столкновения. Число моле-
кул, центры которых расположены внутри этого объема, равно
осД/Ж, где.#*— число молекул в единице объема, и, таким обра-
- зом, число регистрируемых ударов составляет стсДМ". Следователь-
но, число ударов р единицу времени (частота столкновений) равно
ос#*.
Неточность такого расчета возникает -из-за сделанного пред-
положения,_что все молекулы, кроме одной, неподвижны. В этом
уравнении с на самом деле должна быть средней относительной
скоростью сталкивающихся молекул. Если принять это в расчет,
то с необходимо заме-
нить на ]!3с, и тогда
частота столкновений
выразится следующим
образом:
, Г t/z/ //'
2^--|/ 2осЛГ/К (24.2.1)°
Это число столкнове-
ний, которое претерпе-
вает одна молекула.
Рис. 24.6. Сече пне столкнове-
ния и трубка столкновения.
346
Часть 3. Изменение
Для определения общего числа столкновений молекул необходимо z
умножить на (множитель — получается вследствие того, что
столкновения А...А' и А'...А надо рассматривать только как одно
столкновение). Поэтому число столкновений в единице объема в
единицу времени равно
ZA1^^zX/V=ac(N/V)2/]/2: (24.2.2)
Значение с, которое нужно здесь использовать, уже было рассчи-
тано по уравнению (24.1.15)» и поэтому можно написать, что
zaa (4kT/nm)Vi(NJV)^ (24.2.3)°
Пример (вопрос 6). Мы продолжаем исследовать свойства атомнолучевой аппа-
ратуры, рассмотренной в двух последних примерах. Сколько столкновений в се-
кунду совершает один атом Cs внутри лечи? Сколько столкновений в секунду
совершают все атомы внутри печи объемом 50 см3?
Метод. Используем уравнение (24.2.1) для столкновений одного атома и урав-
нение (24.2.2) для общего числа столкновений. Соотнесем NjV с давлением сле-
дующим образом: Nj\'—nLiV—p!kT. Давление паров Cs при 500 °C равно
80 мм рт. ст. Для поперечного сечения используем d=54() пм (5,40 А); с возь-
мем да последнего примера.
Ответ. Из уравнения (24,2.1)
z- 2l^ac(p/kT),
а = лй2 = лХ(540-10-12 м)2 = 9,2Д0-1Вм2,
р = (80/760)х(1,0133-105Н/ма) = 1,07 Ю‘Н/мг.
Поэтому
2^2Х(9,2- 10-1В ма)Х(351 м/с)Х(1,07-10< Н/м2) „ 4 6.
(1,381 Ю-23 Дж/К)Х(773 К)
Из уравнения (24.2.2), написанного для Zc3cE = -jj-zfp/ftT), имеем
I
-щ- X (4,6 1 Ове-1) х (1,07 х 104Н/м2)
J______________________—--------= 2,31-10«2 с-4 м-’.
(1,381 ДО-23 Дж/К)Х<773К)
Таким образом, в печи объемом 50 см3 число столкновений в секунду равняется
l,16-102S.
Комментарий. Столкновения будут продолжаться в атомном пучке, и это при-
водит к его расширению; за селектором скорости столкновения приводят к тому,
что распределение скорости будет соответствовать распределению Больцмана,
если только пучок не слишком слаб.
Если нас интересует частота столкновений молекул разных ве-
ществ, анализ следует несколько видоизменить. В первую очередь
относительная скорость равняется (8 ЛТ/лц)1'2, где ц— приведен-
24. Кинетическая теория газов
347
нал масса сталкивающихся частиц с массами шА и шв соответст-
венно:
1/р.= 1/тА+ 1//пв или p==rnAmB/(tnA^-mBp (24.2.4)
Если имеется УА и jVb молекул каждого вида, общая скорость
столкновений в единице объема равняется
(24.2.5)°
Численные величины частот столкновений могут быть очень боль-
шими. Например, при комнатной температуре для азота, исполь-
зуя значение d ж- 280 пм, получим Z ~ 5-Ю34 с-’-м-3, или 5Х
Х1028 с-1-см-3. Некоторые типичные сечения столкновений Приве-
дены в табл. 24.3.
; Таблица 24.3
‘ Сечения столкновений а(им2)
Не 0,13 Н3 0,15 СОа 0,66
Ne 0,17 Н3 0,31 НаО 0,23
Аг 0 26 о, 0 27 Воздух 0 30
Кг 0,32 CJ2 0 43 Hg 0,41
Расчет частоты столкновений позволяет определить выражение
для средней длины свободного пробега К. Если молекула двигает-
ся с относительной скоростью с и ее частота столкновений равна
z, то время свободного пробега между столкновениями равняется
1/г и между столкновениями молекула проходит расстояние с/г.
Следовательно, Средняя длина свободного пробега выражается
просто:
?.=с/г= l/K2otW- (24.2.6)°
Ее можно выразить через давление газа. N=nLt где п — число мо-
Xлей газа; используя уравнение идеального газа njV^pIRT, полу-
5 чаем
A=(l/><2&> (/?T/Zp)=(^25) (kT/p). (24.2.7)°
Видно, что Средняя длина свободного Пробега обратно пропорцио-
нальна давлению.
Пример (вопрос 6). Рассчитайте среднюю длину свободного пробега атомов Сз
в печи при 500 °C из последнего примера.
Метод. Используем уравнение (24.2.7) с /? = 80 мм рт. сг., <т = 9,2-10-!9 М“.
Ответ. Из уравнения (24.2.7)
А —
(1,38-10-®» Дж/К)Х(773К)
(9,2- 10-1Вмг)х (1,07- 104 5 * * *Н/мв)
= 766 10’9м, или 766 нм.
348
Часть 3. Изменение
Комментарий. Средняя длина свободного пробега равняется приблизительно
1400 атомным диаметрам, и поэтому мы находимся в тех пределах, где кинети-
ческая теория, вероятно, дает надежные результаты.
Столкновения со стенками и поверхностями. Подсчет числа столк-
новений с плоской поверхностью, имеющей определенную площадь,
довольно прост и лежит в основе многих расчетов, проводимых при
изучении транспортных свойств. В простейшем варианте аргумен-
тация выглядит следующим образом.
Рассмотрим стенку площадью Л, перпендикулярную осих. В со-
суде приходится Ж молекул па единицу объема (Ж =ЛГ/У). Пусть
молекула имеет скорость vx, лежащую между 0 и Ц-оо; за период
Ы произойдет ее столкновение со стенкой, если она находится в
пределах расстояния vxAt от стенки (когда молекула имеет ско-
рость vX/ лежащую между 0 и —со, она движется в противополож-
ном направлении). Поэтому все молекулы в объеме AvxAt) движу-
щиеся в прямом направлении, столкнутся со стенкой за промежу-
ток времени At. Следовательно, общее число Столкновений (в сред-
нем) за это время составит:
со
число столкновений j vj(vx)dvx.
о
Этот интеграл легко может быть рассчитан при использовании точ-
ной формы распределения скоростей:
J v*f(vx) dvx=(m/2nkTyn j* vx ехр ( — mziftkT) dvx=. (kTflwriyh.
о 6
Таким образом, число столкновений в единицу времени па еди-
ницу площади составит;
Z^^{kTj2nm}'^ N/V = 4- ^NlV- (24.2.8)°
Плотность числа частиц N[V можно выразить через давление, на-
писав NlV=nLjV—plkT, и поэтому
Zw = -^-pefkT ~ p/(2mnkT)'^. (24.2.9)°
При атмосферном давлении (1,01 -105 Н/м2) и температуре 300 К
стенки сосуда испытывают приблизительно 3-1023 столкновений
в 1 с на 1 см2.
24.3. Транспортные свойства
Переход вещества из одной области в другую является приме-
ром транспортного свойства. Например, если газ заключен в со-
суд, который сообщается с областью низкого давления через ма-
&
24. Кинетическая теория газов 349
ленькое отверстие, то газ будет вытекать через это отверстие до
тех пор, пока давление не сравняется с обеих сторон сосуда (этот
процесс называется истечением). Если газ имеет постоянную плот-
ность, цо не постоянный состав, молекулы диффундируют по си-
стеме до тех пор, пока состав не Станет везде одинаковым. Напри-
мер, диффузия радиоактивного изотопа в нерадиоактивцый гав
представляет собой диффузионный процесс, поскольку один тип
молекул перемещается в среде из других молекул.
Транспортные процессы происходят в газах, жидкостях и твер-
дых телах. Соль растворяется в растворителе, и вся система Ста-
новится однородной в результате процесса диффузии. Два твер-
дых вещества, находящиеся в контакте, диффундируют друг в
друга с очень малой скоростью, если только температура не ста-
новится достаточно высокой.
Транспортные процессы не ограничиваются только переходом
масс. При нагреве одной части системы термическая энергия пе-
реходит через систему до тех пор, пока температура не выравни-
вается. Термическая проводимость представляет собой переход
термической энергии. Электрическая проводимость представляет
собой переход заряда или посредством перемещения электродов,
как в металлах, или посредством перемещения ионов, как в ион-
ных растворах или ионизированных газах. Вязкость — это транс-
портное свойство более тонкого качества, и, как мы увидим да-
лее, ома связана с транспортом количества движения.
В этом разделе мы рассмотрим некоторые общие вопросы
Транспорта л затем рассчитаем скорости некоторых Процессов на
основе кинетической теории газов. Эта тема развивается также в
следующей главе, где будут рассмотрены транспортные процессы
в жидкостях и особенно движение иопов.
Поток. Если вещество, энергия, заряд или количество движения
переносятся из одной области системы в другую, бывает необходи-
мо измерить скорость этого переноса. Она дается величиной, на-
зываемой потоком, который представляет собой количество свой-
ства, проходящего через единицу площади в единицу времени. Ес-
ли происходит перенос массы, поток выражается в кг/(м2-с); если
переносится энергия, поток должен иметь размерность Дж/(м2-с)
и т. Д. Обозначим поток символом J.
Экспериментальные наблюдения по скоростям различных транс-
портных процессов показывают, что поток — это свойство, обычно
пропорциональное градиенту какого-либо свойства системы. На-
пример, скорость диффузии (т. е. переноса массы вещества) про-
порциональна градиенту концентрации:
j (масса) ~ doT/dz.
350
Часть 3. Изменение
(масса) >0
Рис. 24.7. Поток частиц вниз по
градиенту концентрации.
а скорость тепловой диффу-
зии (перенос энергии) про-
порциональна градиенту
температуры:
(тепловая энергия) dT/dz.
Индекс у потока указывает, что поток свойства вдоль оси z про-
порционален градиенту вдоль этой оси.
Мы будем рассматривать I как вектор, положительный знак
указывает на то, что поток направлен в сторону увеличения z
(вправо на рис. 24.7), а отрицательный знак — на то, что поток
направлен в сторону уменьшения z (влево на рис. 24.7). Поток
массы направлен вдоль градиента концентраций (вниз на рис.
24.7), и, таким образом, если djV’idz отрицательно (концентрация
уменьшается вдоль оси г; рис 24.7), Л будет положительным (по-
ток вправо на рис. 24.7). Поэтому константа пропорциональности
в выражении потока массы должна быть отрицательной. Если ее
обозначить через —D, где D — коэффициент диффузии, то получим
первый закон диффузии Фика: Л (масса) = — D{d^/dz). (24.3.1)
Передача тепловой энергии происходит по температурному гради-
енту, и те же рассуждения приводят к выражению
/г (тепловая энергия) = —H(dT/dz), ' (24.3.2)
где и — коэффициент теплопроводности.
Связь между потоком количества движения и вязкостью мо-
жет сначала показаться странной, по она может быть объяснена
следующим образом. Рассмотрим жидкость между двумя парал-
лельными пластинами (рис. 24.8). Для того чтобы передвинуть
Рис. 24.8. Транспорт количест-
на движения через градиент
скорости.
24, Кинетическая теория газов
351
Рис. 24.9. Транспорт, рассмат-
риваемый при расчете вязкости
газа.
верхнюю пластину относительно нижней, необходимо приложить
силу в направлении оси х, и для того, чтобы движение поддержи-
валось, эта сила должна сохраняться. Скорость жидкости изменя-
ется от 0 (около нижней неподвижной пластины) до z>x (около
верхней пластины), и в области ньютоновского потока скорость из-
меняется па одинаковую величину при переходе от одной плоско-
сти к другой. Наблюдения показывают, что сила, требуемая для
поддержания постоянной скорости верхней пластины, пропорцио-
нальна площади пластины (А) и градиенту скорости dvjazf —
= ox/d):
необходимая поддерживающая сила ~ A idv jdz), (24.3,3)
или
F£lA ==t) (dVjJdz), (24.3,4)
где т] — коэффициент вязкости (или просто вязкость).
Теперь сконцентрируем внимание на одном из слоев, движущем-
ся с постоянной скоростью; па такие слои мы можем мысленно
разделить жидкость (рис, 24.9). В этом сдое молекулы движутся
с некоторой скоростью v'x, и, таким образом, каждая из них имеет
количество движения, равное /ни), Между слоями происходит по-
стоянный обмен .молекулами. Молекулы из выше расположенных
слоев обладают большим количеством движения (они находятся
в более быстро движущихся слоях), а молекулы в сдоях, располо-
женных ниже, имеют меньшее количество движения. Результиру-
ющий поток количества движения па единицу площади интересу-
ющего пас тонкого слоя равен I (количество движения вдолв оси
х), и для того, чтобы поддерживать это изменение количества дви-
жения, требуется какая-то сила в направлении оси х; отсюда воз-
никает связь между вязкостью и потоком количества движения.
Это можно проиллюстрировать следующим образом. Если один
поезд обгоняет другой и пассажиры бросают апельсины из своих
купе в купе другого поезда, апельсины из быстро идущего поезда
сообщат свое первоначальное количество движения поезду, иду-
щему по параллельному пути железной дороги, и будут способст-
352
Часть 3. Изменение
вовать ускорению медленно идущего поезда путем столкновений с
поперечными стенками купе (или с пассажирами). В противопо-
ложность этому апельсины из медленно идущего поезда стремятся
замедлить быстрее идущий поезд. Если быстро идущий поезд не
разовьет слишком большую скорость, то переброс достаточно
большого числа апельсинов приведет к тому, что оба поезда ста-
нут двигаться с одинаковой скоростью.
Все приведенные до сих пор рассуждения являются совершен-
но общими и могут применяться к транспорту в жидкостях и твер-
дых веществах так же хорошо, как в газах. Теперь мы рассчита-
ем коэффициенты х и т] для конкретных примеров кинетичес-
кой модели газов.
Скорости истечения. Когда газ при давлении р и температуре Т
отделен от вакуума небольшим отверстием, скорость истечения
молекул равна скорости, с которой молекулы сталкиваются с пло-
щадью, занимаемой отверстием. Эта скорость выражается урав-
нением (24.2.8). Если обозначить площадь отверстия через Ло, то
число молекул, которые выходят в единицу времени, составит
Z^A^pA^mkT)1^. (24.3.5)°
Скорость истечения обратно пропорциональна корню квадратно-
му из относительных молекулярных масс (это закон истечения
Грэма). Это выражение лежит в основе метода Кнудсена опреде-
ления относительных молекулярных масс или, если они известны,
давления паров твердых веществ. Таким образом, если твердое ве-
щество имеет давление паров р и помещено в сосуд, где име-
ется небольшое отверстие, скорость потерн массы из сосуда про-
порциональна р, и, таким образом, р может быть определено из
уравнения (24.3.5), если контролировать потерю массы.
Пример (вопрос 10). Прибор для получения атомного пучка, использованный
в предшествующем примере, был применен для измерения давления паров Cs
при 500 °C. Диаметр отверстия в стенке равнялся 0,5 мм, и в течение 100 с по-
теря массы составила 385 мг. Каково давление паров цезия при этой темпера-
туре?
Метод. Используем уравнение (24.3.5) в виде бт = ZwABttn, так что р =
= (2^kT/m)'P(6m/Aax).
Ответ. Из только что приведенного выражения
_ ( 2лХ (1,38-10-гз-773 Дж))’/3 ( (0,385-10^ кг) ) . 2
р^\ 132,9х (1,6605 10-27 кг) ] ( л (0,25 10-3 м)2х(100с) J “ «.ивши-п/м .
Комментарий. Выразим давление в мм рт. ст., разделив на 1,0135-10s (Н/м2)/атм
и умножив на 760 мм рт. ст./атм, это дает 81,2 мм рт, ст.
При расчете скорости истечения предполагается, что длина от-
верстия значительно короче, чем средний свободный пробег моле-
кул газа. Если оно настолько длинное, что молекулы претерпева-
ют множество столкновений внутри этой трубки, то транспорт бо-
лее похож па вязкий поток и скорость истечения определяется вяз-
костью. Эта сторона вопроса будет рассмотрена на стр, 359.
Скорость диффузии. Теперь необходимо определить величину ко-
эффициента диффузии для газа и дать теоретическое обоснование
закона Фика [уравнение (24.3.1)].
Мы должны показать, что поток частиц пропорционален гра-
диенту их концентрации. Чтобы сделать это, рассмотрим образец
газа, показанный па рис. 24.10, в котором концентрация изменяет-
ся в направлении z. Поток рассчитывается определением общего
числа молекул, проходящего в единицу времени через воображае-
мое окно площадью А, расположенное перпендикулярно оси z.
Ключом к этому расчету является средняя длина свободного про-
бега молекул. Можно представить, что молекула появляется из
некоторого пункта, где плотность частиц .V'(z); это справедливо
только в том случае, если пункт Z расположен на расстоянии не
большем, чем некоторая средняя длина свободного пробега. Если
это расстояние больше, молекула, вероятно, столкнется с другой
молекулой.
Молекулы, проходящие через воображаемое окно, перемеща-
ются приблизительно па одну среднюю длину свободного пробега,
и, таким образом, средняя концентрация в той области, из которой
они приходят, составляет приблизительно (—?.)« М (0)—
—Z (йЖ/Уз) о, где индекс 0 указывает, что градиент определен у от-
верстия (где 2 = 0). Так как число попаданий в окно с левой сто-
23— 242
354
Часть 3. Изменение
роны за Промежуток Д/ в среднем равно —ЛЧ—Ъ)сАЫ, поток сле-
ва направо, соответствующий определенной концентрации с левой
стороны, составит
J (слева —* направо) а? (—X) с.
Но молекулы проходят также и справа налево, В среднем молеку-
лы, которые проникают через окно, исходят из пунктов, находя-
щихся справа на расстоянии X, и, следовательно, средняя концент-
рация молекул, проходящих через окно справа, составит Ж(Х)
Л’(О) -\-'K{dAC!dz)e- Поэтому поток справа налево равен
J (справа —* налево) (X) с.
Поток из более концентрированной области слева преобладает
над обратным потоком, и, таким образом, общий поток положи-
телен (слева направо) и имеет величину
Л = -Г 4 И (°) 1 (d^/d2)a] - (0) + л (d^/dz)0]} с-
= •—j-Zc((M7dz)0.
Потоке пропорционален градиенту концентраций в соответствии с
первым законом Фика;
Jz=-D(d^/dZ)0.
Этот расчет дает также выражение для коэффициента диффузии,
поскольку £) может быть определен как -g-Ac. Однако, необходимо
Короткий
полет(дез
столкновения)
Длинный
полет (столк-
новение в
полете)
Некоторое расстояние
от плоскости
помнить, что этот расчет очень груб
и в действительности представляет
собой лишь пример того, как пост-
роить приближенную модель рас-
сматриваемого процесса и грубо
оцепить порядок величины коэффи-
циента диффузии. Один из аспектов,
который должным образом не учи-
тывается в этой модели, иллюстри-
рует рис. 24.11. Он показывает, что,
Рис. 24.11. Некоторые молекулы проходят
более длинный путь, прежде чем пересекут
плоскость.
24. Кинетическая теория газов
355
хотя молекула может начинать свое движение очень близко к ок-
ну, она может совершить длинный путь, прежде чем пройдет че-
рез окно. Поскольку молекула совершает длинный путь, вероят-
ность столкновения до того, как она пройдет окно, высока, и, та-
ким образом, эту молекулу следует причислить к множеству
молекул, которые претерпевают столкновение. Можно предста-
вить, какие значительные затраты труда при расчете вносит учет
этого эффекта, тем не менее конечный результат будет таким же,
как уже выведенный нами, за исключением дополнительного мно-
жителя 2/з- Этот множитель отражает уменьшение потока вслед-
ствие столкновений, которые испытывают частицы, проходящие
более длинный путь.
В результате такой модификации должна получиться:
4=------(24,3,6)°
D=-^ic. (24.3.7)°
Поскольку средняя длина свободного Пробега уменьшается с уве-
личением давления и средняя скорость увеличивается с температу-
рой, коэффициент диффузии также зависит от этих факторов. Из
уравнений (24.1.15) и (24.2.7) ясно, что
D=~ (1/21-'2о)^71/р)(8^Г/лт)1/2=-±-(1/ар)(А:3Гз/тлу/31 (24.3,8)°
где <т —поперечное сечение столкновений ad2.
Теплопроводность, В случае теплопроводности переносится поток
тепловой энергии, и целью расчета является, с одной стороны, объ-
яснение эмпирических наблюдений, свидетельствующих о том, что
тепловой поток пропорционален Градиенту температуры, и, с дру-
гой стороны, вывод выражения для коэффициента теплопроводно-
сти [уравнение (24.3.2)]. Некоторые экспериментальные значения
к Представлены в табл. 24.4.
Таблица 24.4
Транспортные свойства газов
103 ДжЛсм-сК) 273 К 105 ц, КГ/(Ы-с)
273 К 293 К
Не 1,44 1,87 1,96
Ne 4,60-10-г 2,99 з, 14
Аг 1,79Ю-а 2,08 2,23
н2 1,75 0,85 0,89
О2 2,43-10-1 1,91 2,03
N3 2,39. К)-1 1,67 1,76
СО2 1,42-Ю-1 1,38 1,47
Воздух 2,41 10-1 1,71 1,82
23
356
Часть 3. Изменение
Предположим, что каждая молекула песет количество энергии
е. Когда она проходит через воображаемое окно, она транспорти-
рует это количество энергии. Скорость же переноса молекул пред-
ставляет собой поток, определяемый уравнением (24.3.6). Поэтому
поток тепловой энергии равен:
Л (теплота) =еЛ (вещество) —- —Хее (d-^/dz)0=
= —-g- Xr(d(£/dz)0,
где g = в Л"—плотность энергии (количество энергии на единицу
объема). Это соотношение показывает, что поток энергии опреде-
ляется градиентом энергии. Градиент энергии можно связать с
температурным градиентом через теплоемкость образца. Если обо-
значить мольную теплоемкость при постоянном объеме через Су,т,
то градиент плотности энергии выразится как
(dg/dz)(l = (n/V) Crm (dT/dz)0.
Тогда поток определяется выражением
Л (теплота) — -—Хс (п/V) CVm (dT/dz)0,
и, таким образом, коэффициент теплопроводности будет равняться
x^-L-XZCr.n/n/V). (24.3.9)°
Важной особенностью этого уравнения является то, что и не за-
висит от плотности, поскольку средняя длина свободного пробега
обратно пропорциональна n!V. Ясно, что
x=^ZCv(24.3.10)°
Из этого выражения видно, что х не зависит от давления. Физи-
ческой причиной этой независимости от давления является следу-
ющее. Теплопроводность велика в том случае, когда транспорт мо-
жет осуществляться большим числом молекул {отсюда множитель
п в уравнении (24.3.9)], однако при большом числе молекул их
средние свободные пробеги уменьшаются, и, следовательно, моле-
кулы не могут переносить энергию на большие расстояния: эти
два эффекта уравновешиваются. Экспериментально установлено,
что теплопроводность не зависит от давления, за исключением тех
случаев, когда давление очень мало; тогда наблюдается линейная
зависимость. Причина этого состоит в том, что при очень низких
давлениях средний свободный пробег больше, чем размеры прибо-
24, Кинетическая теория газов
,357
ра, и, следовательно, длина пробега, т. е. расстояние, па которое
транспортируется энергия, определяется размером сосуда, а не
наличием других молекул. Транспортируемая энергия пропорцио-
нальна числу переносчиков, но длина пути не зависит от их числа,
следовательно, при этих условиях n/V~р.
Пример (вопрос 12), Рассчитайте теплопроводность воздуха при комнатной
температуре.
Метод. Используем уравнение (24.3.10). Мольная теплоемкость может быть при-
5
пята равной (см. обсуждение рис. 21.7). Примем, что средняя относительная
молекулярная масса воздуха что достаточно точно; примем также, что
о ж 0,28 пм2. Рассчитаем ь' из уравнения (24.1.15).
Ответ. Из уравнения (24.1.15)
( (8/п)Х(1,38Ю--3 Дж/К)-(298К) )1 -'г__
с"| 30х(1,66 10-2' кг) / -4Ь9 м/с'
Затем из уравнения (24.3.10)
1 5
-д-Х(459 м/с);-—х [8,31 Дж/(К-моль)|
К ~ У 2^(0,2SJ О-^м2) х (6,022 ДО23 моль-i) = 13,3'!0 ЛМК-мс).
Комментарий. Если бы температурный градиент был порядка ДО К/см, тогда
тепловой поток равнялся бы [13,3-10—2 Дж/(К-м-с)] X (10 К/см), или 13,3 Дж/
/(м2Хс), или 1,33-Ю-3 Дж/(ем2-с).
Вязкость газов. Мы уже видели, что коэффициент вязкости опре-
деляется скоростью транспорта количества движения. Молекулы;
движущиеся вниз из быстрого слоя в медленный, передают своему
новому слою с z — 0 количества движения hwx(A), а молекулы;
движущиеся вверх, передают своему новому слою количество дви-
жения mvx(—А). Если плотность молекул везде равна ,4", число
ударов о воображаемое окно (рис. 24.12) составляет <Я’с на еди-
ницу площади в единицу времени. Для верхних молекул средняя
.r-компонента количества движения равна mvx(K)-mvx(0j -}-
-ym\(dvx/dz)<i, так как в среднем они появляются из плоскости)
находящейся на расстоянии А от окна. Нижние молекулы имеют
количество движения mvx(—А) — mvx(Q)— m^dv^dz)^ Поэтому
общий поток количества движения вдоль оси х равен :
Z, (количество движения вдоль л)-Джс|[tnvx (0) —mA (d^/dz)u] -
— (0) -(-mA (dvd2)ol}
=-----m-^Kc (dVjJdz)v
358
Часть 3, Изменение
Сравнение этого выражения с уравнением (22.3.4), определяющим
коэффициент вязкости, и замена множителя Ц- на приводит к
т) —-^-irikcL (njV).
(24.3.11)°
Так же как л теплопроводность, вязкость не зависит от давления.
Этот вывод можно пояснить введением выражения для среднего
свободного пробега:
т) = (n/V)/2^aL (n/V) = ^)1/2 тс/а. (24.3.12)°
Физическая причина в этом случае та же, что и в случае тепло-
проводности; чем больше молекул осуществляет транспорт количе-
ства движения, тем меньше их достигает цели вследствие укоро-
чения среднего свободного пробега, Следует также отметить, что,
согласно последнему уравнению, вязкость увеличивается с темпе-
ратурой, так как с ~ Т1;'2. Этот необычный результат получается по-
тому, что при высоких температурах количество движения транс-
портируется через данную площадь быстрее, и поэтому для под-
держания движения слоев газа надо увеличить необходимую для
этого силу [для примера из подразд. «Поток» (см. выше) это от-
вечает ситуации, когда при более высоких температурах происхо-
дит о^мсн большим количеством апельсинов между поездами].
Такое свойство вязкости газов сильно Отличается от свойства, ха-;
рактерпого для жидкостей, которые по мере повышения темпера-
туры текут более легко; различие происходит из-за того, что на
24. Кинетическая теория газов
359
вязкость жидкости преобладающее влияние оказывают внутримо-
лекулярные силы: для того чтобы жидкость могла течь, молекулам
необходима энергия для отрыва от своих соседей, и это легче осу-
ществляется при высоких температурах.
Эксперименты подтверждают, что вязкость не зависит от дав-
ления в очень широких пределах. Согласно данным для аргона
От 10 3 до 102 атм, представленным на рис. 24.13, а, вязкость по-
стоянна между 0,01 атм и приблизительно 50 атм. Уравнение
(24.3.12) предсказывает, что вязкость зависит от Т'1г. Это прибли-
зительно подтверждается данными для аргона, приведенными на
рис. 24.13,6. Прерывистая линия рассчитана для о=22-10-16 см2,
что соответствует атомному диаметру около 260 пм (ср. с вандер-
ваальсовым диаметром, найденным из упаковки атомов в твердом
аргоне и равным 335 пм), Согласие вполне приемлемое и дает
правильный порядок величины. Точного совпадения нс может быть
из-за приближенности расчета и неучена того факта, что молеку-
лярные столкновения являются динамическими процессами и в
действительности связаны с природой межмолскудярных взаимо-
действий.
Для определения вязкости используются два основных метода.
Один из них основан на наблюдении скорости торможения крутиль-
ных осцилляций (колебаний) набора дисков, помещенного в газ
(рис. 24.14). Анализ движения этой системы показывает, что амп-
литуда колебаний уменьшается до 1/е от первоначального значе-
ния за время 2/а/илсг4г], где I — момент инерции набора дисков,
s — расстояние между дисками, а — их радиус и п — их число.
Другой метод основан на формуле Луазейля для потока жид-
кости через трубку радиусом R:
360
Часть 3. Изменение
Ряс, 24.14. Метод колеблющихся дисков для опреде-
ления вязкости газа
где V протекший объем, р\ и р^ — давле-
ния у каждого конца трубки длиной I и
Ро — давление, при котором измеряют объ-
ем. Для определения вязкости измеряютг
скорость потока при известной разности
давлений и затем используют это уравне-
ние.
Пример (вопрос 13). Опыт с потоком Пуазейля по-
ставлен для измерения вязкости воздуха прп 298 К;
проба протекала через трубку длиной 1 и с диа-
метрлм отверстия 1 мм. Копен трубки е более вы-
соким давлением находился при 765 мм рт. ст., а конец с более низким давлени-
ем— при 760 мм рт. ст. Объем был измерен при последнем давлении. Через
трубку за 100 с прошла проба объемом 90,2 см3. Какова вязкость воздуха при
этой температуре?
Метод. Используем последнее уравнение. Выразим давление в Н/мг.
Ответ.
—рз = (7С5а_760а)х(10135.10Б Н/м2)2/(760)2 = 1.356-10е Н-./мЦ
п.Х(0.5-10-э м?
- J6x(1 M)X(J О135.]О5 H/Mij ~ Н 1 -и5.
dV/dt = (90.2 см3)/(100 с) = 9,02- 10~г м®/с.
24.ЗА. Транспортные свойства идеальных газов
Свойство Тра । icnopTH ру емая Прос тая к и нс ти ч еск а и материя теория Уточненная теория Единицы
Диффузия
Вещество
Проводимость
Термическая
энергия
Вязкость Количество
движения
1 - 3 _
D = —' Ас = Ас м2/с
3 16
= кТс/ЗроУ2~
х = тЛ’= (25я/64)Х Дж/(К
3 * ХМ’С)
-ъ /У хАсСг>гл^
tj = G1 ’ кг/(н<
= тс/ЗаУ 2
Л”— плотность числа частиц: /rV—nLfV=p!kT.
К — средаяя длина свободного пробег*; A=J/<?
24. Кинетическая теория газов
361
Поэтому
(1,211-10-1» H^ m5)x(1,356 10s H3/W)
t] =---------------------------------------------
,82-10-5 кг/(м.с).
(9,02-10-’ ма/с)
Комментарий, Теоретическая величина, полученная из кинетической теории по
уравнению (24.3.12), составляет 1,9-10-5 хг/(м-с), т. е. согласие очень хорошее.
Вязкость часто выражается в сантипуазах или (для газов) в ми крону азах.
J с!1 = 10—3 кг/(М'С); таким образом, вязкость воздуха составляет 182 мкП.
Некоторые результаты этих измерений представлены в табл.
24.4, а теоретические формулы (с точными выражениями, полу-
ченными из более точных расчетов) собраны в подразд. 24.3.А.
Литература
Каигтстп В7., Kinetic theory of gases, Benjamin, New York, J 966.
Hildebrand J. 14., An introduction to kinetic theory, Reinhold, New York, 1963.
Loeb L. B., Kinetic theory of gases, Dover, New York, 1961.
Kennard E. H., Kinetic theory of gases, McGraw-Hill, New York, 1938.
Present R. D., Kinetic theory oi gases, McGraw-Hill, New York, 1958.
Jeans J, H., 1 n trod uc I io,ti to the kinetic theory of gases, Cambridge University
Press, 1940.
Гиршфельдер Дж., Кертис Ч. и др. Молекулярная теория газов и жидкостей.
Пер. с англ, — М/. ИЛ, 1961.
Bird R. В.. Stewart W7. К, Lightfoot Е. N., Transport phenomena, Wiley, New
York. 1960.
Задачи
24.t . Необходимо иметь представление о численных значениях физических пара-
метров, рассматриваемых при обсуждении газов, и первые несколько задач дают
некоторую практику. Первым шагом является знакомство с численными значе-
ниями расстояний. Какова средняя длина свободного пробега в воздухе (о яг
гг-0,26 им2) цри 25 "С и а) 10 атм, б) 1 атм, в) 10-® атм?
24.2. При каком давлении средняя длина свободного пробега аргона становится
сравнимой с размером содержащего его пузырька объемом 1 см’? Возьмите
о.я-0,26 нм2.
24.3. При каком давлении средняя длина свободного пробега аргона становится
сравнимой с размером самих атомов?
24.4. На высоте 20 км температура 217 К и давление 0,05 атм. Какова средняя
длина свободного пробега молекул азота (of гд 0,31 им2)?
24.5. Теперь обратимся к временной шкале событий в газе. Сколько столкновений
совершает один атом аргона в 1 с при температуре 298 К и давлениях: а) 10 атм,
б) 1 атм, в) 10’ ° атм?
24,6. Каково обшее число молекулярных столкновений в 1 дма аргона при тех
же условиях, что и в задаче 24.5?
24.7. Частота молекулярных столкновений является важной величиной для хр-
мии атмосферы. Сколько столкновений в секунду сделает возбужденная молеку-
ла азога ил высоте 20 км? (См. задачу 24.4.)
24.8. Рассчитайте числа столкновений в 1 смэ воздуха при 25 °C и 1 атм: а) меж-
ду молекулами кислорода, б) между молекулами кислорода и азота. Примите
А’(Оз) »148 пи я 4?(Na) 158 пм.
24,9. Сколько столкновений в 1 см3 в рубашке сосуда Дьюара, где оставшийся
воздух имеет давление 1,2 мм рт. ст.?
.362
Часть 3. Изменение
24.10. Какова средняя скорость: а) атомов гелия, б) молекул метана при 1) 77 К,
2) 298 К и 3) 1000 К?
24.11. Какова средняя кинетическая энергия поступательного движения
(в кДж/моль) а) молекул азота и б) молекул иода в газе при 300 К и 1 атм?
24.]2. В эксперименте по измерению скоростей молекул с помощью вращающих-
ся дисков, имеющих щели, аппарат состоял из 5 коаксиальных дисков диамет-
ром 5 см на расстоянии 1 см друг от Друга; прорези в кольцах были смещены
на 2° между соседними дисками. Относительная интенсивность I пучка атомов
криптона для двух различных температур и для ряда скоростей вращения бы-
ла следующей:
V, Гц 20 40 80 100 120
I (40 К) 0,846 0,513 0,069 0,015 0,002
/ (J 00 К) 0,592 0,485 0,2]7 0,119 0,057
Найдите распределение молекулярных скоростей при этих температурах и про-
верьте, соответствует ли оно теоретическому предсказанию.
24.13. Распределение скоростей Максвелла — Больцмана [уравнение (24.1.И)]
было выведено из вероятностных доводов. Но, как было сказало, его можно вы-
вести и из распределения Больцмана как такового. Сделайте это.
24.14. Специально сконструированный селектор скорости принимает пучок моле-
кул из печи при температуре Т, но блокирует проход молекул, имеющих скорость
выше, чем средняя. Какова средняя скорость выходящего пучка? (Отметьте, что
это в основном одномерная задача.)
24.15. Мы столкнулись с двумя путями рассмотрения средних величин: один—
из дискретного распределения и второй — для непрерывного распределения. Вот
как можно проверить их на практике. С помощью радиолокаторов измеряется
время прохождения автомобилей под мостом в обоих направлениях. Их скорости
(миль/ч; в скобках дано число автомобилей в группах) при движении на восток
(В) и на запад (3) имели следующие значения: 50 В (40), 55 В (62), 60 В
(53), 65 В (12), 70 В (2), 50 3 (38), 55 3 (59), 60 3 (50), 65 3 (10),
70 3 (2). Каковы: а) средняя скорость автомобилей в каждом направлении,
б) общая средняя скорость и в) среднеквадратичная скорость?
24.16. Население состоит из людей следующего роста (в футах и дюймах,
в скобках число индивидов в группах): 5’5" (J), 5'6" (2), 5?7" (4), 5'8'' (7),
5’9" (]0), 5’10" (15), 5’11" (9), 6'0" (4), 6'1" (0), 6'2" (1). Каковы: а) сред-
ний рост и б) среднеквадратичный рост?
24.]7. Каково соотношение молекул, имеющих: а) бблыпую и б) меньшую по
сравнению со средней скорость при температуре 7?
24.18. Каково отношение содержания в газе молекул, имеющих скорость в п
раз больше наиболее вероятной, к содержанию молекул, имеющих наиболее ве-
роятную скорость? Этот расчет можно использовать для определения относи-
тельного содержания наиболее реакционноспособных молекул (что важно для
рассмотрения реакций). Рассчитайте это отношение для п=3 и п—4.
24.19. Какова скорость утечки, газа с поверхности планеты, имеющей радиус R?
Каково ее значение: а) для Земли (К=6.37-10s и, £=9,80 м/сг) и 2) для Марса
(A'=3,38-10s м. Шмарс/тзсмля =0,108)? При каких температурах водород, гелий
и кислород имеют средние скорости, равные скоростям их утечки? Какова доля
этих молекул, имеющих скорость, достаточную для у’течКи при температурах:
I) 240 К, 2) 1500 К? Расчеты такого типа очень важны при рассмотрении со-
става атмосфер планет.
24.20. Манометр был связан с сосудам, содержащим двуокись углерода под не-
большим давлением. Газ имел возможность вытекать через небольшое отверстие,
и через 52 с манометр показал, что давление упало е 75 до 50 см. Когда экспе-
римент был повторен с азотом (Мг=28), аналогичное уменьшение давления про-
изошло за 42 с. Какова относительная молекулярная масса двуокиси углерода?
24. Кинетическая теория газов
363
24,21. Специально изготовленный баллон электрической лампы содержит аргон
под давлением 50 мм рт. ст. и имеет вольфрамовую нить радиусом 0,1 мм и дли-
ной 5 см. В рабочем положении газ вблизи нити имеет температуру около 1000 °C.
Сколько столкновений происходит с нитью каждую секунду?
24.22, Космический корабль, имеющий внутренний объем 3 м3, столкнулся с ме-
теоритом, в результате чего образовалось Отверстие радиусом 0,1 мм. Если дав-
ление кислорода внутри корабля было первоначально 0,8 атм и температура
298 К, то сколько времени потребуется, чтобы давление упало до 0,7 атм?
24.23. Ячейка Кнудсена даст возможность измерять давление паров мало.четучих
твердых веществ, используя выражение для частоты столкновений с площадкой.
Взвешенное количество вещества нагревается внутри сосуда, в стейке которого
имеется небольшое отверстие. Уменьшение массы за некоторый промежуток вре-
мени можно связать с давлением паров при температуре эксперимента, Если
Ат — потеря массы за время 1 через круглое отверстие радиусом R, найдите вы-
ражение, связывающее давление паров р с Ат и t.
2424, Ячейка Кнудсена была использована для определения давления паров
германия при 1000 °C. За 2 ч потеря массы через отверстие радиусом 0,50 мм
составила 4,3-10-2 мг. Чему равно давление паров германия при J000°C?
24.25. При изучении каталитических свойств поверхности титана очень важно
защищать поверхность от загрязнений. Рассчитайте число столкновений молекул
кислорода с 1 см2 поверхности при давлении: а) J атм, б) 10-6 атм и
в) JO-10 атм при температуре 300 К, Оцените число столкновений, происходящих
каждую секунду с одним атомом поверхности. Этот расчет подчеркивает, что очень
низкие давления — непременное условие для изучения свойств чистых поверхно-
стей. Примите, что наименьшее расстояние между соседними атомами равно
29] „и.
24.26. Ядро as?Bk (берклий) распадается, образуя а-частнпы, которые присоеди-
няют электроны и образуют атомы гелия. Его период полураспада равен 4,4 ч.
Навеска в 1 мг помещается в сосуд объемом 1 см3, который непроницаем для
а-лучей, по в стенке которого тем не менее имеется отверстие радиусом 0,002 мкт
(это не совсем парадокс). Какое давление гелия будет внутри сосуда черед
а) 1 ч и б) 10 ч?
24.27. Со сферическим Стеклянным сосудом Диаметром 10 см связана трубка ра-
диусом 3 мм, В то время, когда сосуд находится при 300 К, трубка имеет тем-
пературу 77 К (оца помещается в жидкий азот). Сначала газ в сосуде был влаж-
ным; парциальное давление паров воды составляло J мм рт. ст. Рассчитайте вре-
мя, необходимое для конденсации воды в трубке и уменьшения парциального
давления до ]0-5 мм рт. ст.
24.28. Атомный пучок в некотором приборе может получаться при использова-
нии: а) кадмия и б) ртути. Источником служит печь, нагретая до 380 К, в ко-
торой имеется небольшая щель с размерами 1 см'КИ}-’ см. Давление паров кад-
мия составляет 1.0-10-3 мм рт. ст.. э давление паров ртути при этой темпера-
туре J138 мм рт. ст. Чему’ равен атомный поток в пучках?
24.29. В реакции Н2+Ъ—>2111 происходит столкновение различных частиц, имею-
щихся в реакционной смеси (это рассмотрено более детально в гл. 26). Рассчи-
тайте частоты столкновений для а) Н2-|-Нъ б) Ir-Нз И в) Н24-1з в газе с пар-
циальным давлением обоих компонентов 0,5 атм при температуре 400 К.
24.30. Какова вязкость воздуха при а) 0 К, б) 298 К и в) 1000 К? Примите что
яявО.28 им2
24.31. Рассчитайте теплопроводность: а) аргона и б) гелия при 300 К и
10~3 атм. Газы находятся в кубическом сосуде со стороной 10 см, одна стенка
имеет температуру 310 К, температура противоположной стенки 295 К. Какова
скорость теплопередачи от одной стенки к другой?
24.32. Вязкость двуокиси углерода была измерена сравнением скорости ее про-
текания через узкую длинную трубку (формула Пуазсйля) с соответствующей
скоростью для аргона. Для одного и того же перепада давлений одинаковые
объемы двуокиси углерода и аргона проходят трубку соответственно за 55 и 80 с..
364
Часть 3. Изменение-
Вязкость аргона при 25°C равна 208 мкП [2,08-10-5 кг/(мс)]; какова вязкость
двуокиси углерода? Оцените диаметр молекулы двуокиси углерода.
24,33. Рассчитайте теплопроводность аргона [Cv,m — 12,5 Дж/(К-моль), ояа
яь0,26 нм2] и воздуха [CV1Itl = 21,0 Дж/(К-моль), <тss 0,28 нм2] при комнатной
температуре.
24.34, Каково соотношение теплопроводностей газообразного водорода при 300
и 10 К? Будьте внимательны!
24,35, В двойных оконных рамах расстояние между стеклами 5 см. Какова ско-
рость теплопередачи из теплой комнаты (25 °C) к внешнему пространству
(—10 °C) через окно площадью 1 и2? Какова должна быть мощность нагревателя,
чтобы компенсировать потерю тепла?
24,36. Рассчитайте коэффициент диффузии D для аргона при 298 К и а)' 10-6атм,
б) I атм, в) 100 атм. Каков поток газа, обусловленный диффузией, если в труб-
ке установлен градиент давления, равный 0,1 атм/см?
24.37. Скорость роста капель свицпа из пара была изучена в лабораториях одной
нефтяной компании {Homer /. В., Pro there Л., J. Chem. Soc., Faraday Trans., 1,
1973, 673). Фактически весь свинец конденсируется за 0,5 мс от начала опыта,
И концентрация в газовой фазе была не больше чем около 3 10|S атом/см3. Най-
дите выражение для скорости роста радиуса сферических частиц, Возьмите 7 =
= 935 К и сделайте предположение, что каждый атом сталкивается с растущей
поверхностью.
24.38. В результате космологической катастрофы произошло разрушение огром-
ного количества материи где-то во вселенной, и поэтому инерционная масса всех
оставшихся атомов уменьшилась на 25%. Можно ли обнаружить это событие,
исследуя свойства идеального газа? Останется ли неизменным уравнение pV=
25 Молекулы в движении.
Транспорт ионов и молекулярная диффузия
Изучаемые вопросы
После тщательного изучения этой главы вы сможете;
1. Определить электропроводность [уравнение (25.1.1)] и моль-
ную электропроводность [уравнение (25,1.2)] растворов н объяс-
нить, как они измеряются.
2. Описать зависимость мольной электропроводности сильных
электролитов от концентрации (стр. 369).
3. Вывести и применить закон независимой миграции ионов
Кольрауша [уравнение (25.1.4)].
4. Сформулировать и применить закон разведения Оствальда
[уравнение (25.1-5) ] для мольной электропроводности слабых
электролитов.
5. Определить скорость дрейфа [уравнение (25.1.9)] и под-
вижность [уравнение (25.1,10)] ионов.
6. Связать мольную электропроводность с подвижностью ионов
[уравнение (25.1.14)].
7. Определить число переноса иона и описать его измерение
(стр. 377).
8. Указать факторы, влияющие на подвижность ионов, н обос-
новать уравнение Дебая — Хюккеля— Онзагера (стр. 379).
9. Описать эффект Вина и эффект Фалькенгагена (стр. 380).
10. Определить термодинамическую силу [уравнение (25.2.1)].
11. Вывести закон диффузии Фика, используя химический по-
тенциал (стр. 382).
12. Вывести и использовать соотношение Эйнштейна, связыва-
ющее подвижность с коэффициентом диффузии [уравнение
(25.2.4)].
13. Вывести и исполызовать соотношение Нернста — Эйнштей-
на, связывающее мольную электропроводность с коэффициентами
диффузии ноновфуравпение (25-2.5)].
14. Вывести и использовать соотношение Стокса — Эйнштейна,
связывающее вязкость с коэффициентом диффузии [уравнение
(25.2.6)].
15. Вывести уравнение диффузии [уравнение (25-2.7)] и исполь-
зовать его для описания диффузии растворяемого вещества в рас-
творитель.
305
366
Часть 3. Изменение
16. Выразить диффузионное движение частицы как одномер-
ное беспорядочное движение (стр. 390).
17. Вывести и использовать соотношение Эйнштейна — Смолу-
ховского, связывающее длину и время скачка с коэффициентом
диффузии [уравнение (25.2.15) ].
Введение
В гл. 24 мы исследовали простой тип молекулярного движе-
ния; молекулы двигались свободно, как газ. Эту главу мы начнем
е рассмотрения того, что может с первого взгляда показаться сов-
сем другой проблемой, а именно движения заряженных ионов в
растворе под влиянием электрического поля, создаваемого парой
электродов. Когда среднее расстояние между ионами велико, как
в очень разбавленных растворах, их движение определяется (не
учитывая соударений с молекулами растворителя) приложенным
электрическим полем и их суммарное движение, представляет со-
бой равномерный дрейф по направлению к одному или другому
электроду. До тех пор. пока мы не вникаем в детали движения,
общий дрейф можно рассматривать очень просто. Мы также дол-
жны учесть роль растворителя и других ионов, по и это можно
сделать довольно просто, если не слишком углубляться в анализ
движения.
Кроме того, имеется очень глубокая связь между движением
ионов и движением незаряженных молекул в жидкостях и газах.
'Эта связь может быть выражена термодинамически, она обсуж-
дается во второй половине главы. Представленное здесь термоди-
намическое рассмотрение основано па материале части 1 книги,
однако первые разделы этой главы не связаны с нею,
25.1. Транспорт ионов
Прямым доказательством существования ионов в растворе яв-
ляется то, что раствор может проводить электрический ток. Это
обусловлено движением положительно заряженных ионов (катио-
нов) и отрицательным электродам и отрицательно заряженных
ионов (анионов) к положительным Электродам.
Миграция ионов представляет собой такой же транспортный
процесс, какие рассматривались в предыдущей главе. Основные
два различия транспорта ионов и транспорта в газах состоят в
том. что движение ионов происходит под влиянием внешнего элек-
трического поля и поддерживается растворителем. Наличие поля
облегчает описание усредненного движения ионов, В присутствии
растворителя и при взаимодействии ионов между собой детальное
описание становится очеять трудным. Оба этих фактора будут рас-
25. Транспорт ионов и молекулярная диффузия
367
Рис. 25.1. Ячейка для измерения электропро-
водности, образующая одно плечо моста пере-
менного тока.
смотрены, но мы не будем останавли-
ваться на детальных расчетах, лежа-
щих в основе теоретических моделей.
Движение ионов. Экспериментальные
данные. Простейший путь изучения
движения ионов в растворе — изуче-
ние их электропроводности. т. е. спо-
{/ собности проводить электричество.
Определение электропроводности
включает измерение электрического
сопротивления, и стандартный метод
состоит в подключении ячейки (тина
показанной па рис, 25.1) в одно плечо
и исследовании точки равновесия, как
это объясняется в обычных руководствах по электричеству. Глав-
ная трудность метода сопряжена с необходимостью использовать
переменный ток. Постоянный ток вызвал бы реакции электролиза
на электродах, и поэтому природа раствора вблизи электродов из-
менялась бы в ходе измерений; это называется поляризацией элек-
тродов. Применение переменного тока исключает электродную
реакцию, поскольку та поляризация, которая происходит, когда
ток течет в одном направлении в первом полуцикле, уничтожает-
ся, когда ток течет в противоположном направлении в следующем
полуцикле. Обычно используется частота около 1 кГц.
Сопротивленце веществ увеличивается с их длиной I и умень-
шается с сечением A: R~l/A. Коэффициент пропорциональности
называется удельным сопротивлением и записывается как р. Тогда
Удельная электропроводность (проводимость) у.— это величина,
обратная удельному сопротивлению, поэтому
R =-(1 /и) IIА или х — 1./RA.
(25.1,1)
Поскольку сопротивление измеряется в омах (Ом), единицей % яв-
ляется Ом~1-м—1 иди Ом_,-см—*,
Если измерено сопротивленце образца, то удельную электро-
проводность можно рассчитать, зная размеры ячейки.. Тем не ме-
нее это ненадежная операция из-за многочисленных осложнений,
368
Часть 3. Изменение
включающих присутствие паразитных токов в ячейке. Ячейку
обычно калибруют по образцу с известной электропроводностью.
Если сопротивление стандартного раствора (часто водного рас-
твора КС1) равно /?* в определенной ячейке, а сопротивление ис-
следуемого образца равно R. в той же самой ячейке, то, обозна-
чив известную стандартную удельную электропроводность через
v.*, можно определить электропроводность образца как к —
= (/?*//?)х*.
Удельная электропроводность зависит от количества присутст-
вующих заряженных носителей (ионов), и поэтому ее обычно
представляют как мольную величину. Если концентрация равна
с (в молях на единицу объема), то мольная электропроводность
выразится следующим образом*:
Лга=х/с. (25.1.2)
Иногда используется эквивалентная электропроводность. Тогда
электропроводность выражают числом отдельных переносимых за-
рядов, Например, в растворе NaCl каждый поп переносит единич-
ный заряд в каждом направлении, и, таким образом, эквивалент-
ная электропроводность будет такой же, как и мольная электро-
проводность. В противоположность этому в CuSO4 каждый ион пе-
реносит заряд, равный двум, и в этом случае эквивалентная элект-
ропроводность равна половине мольной электропроводности. Мы
всегда будем использовать мольную электропроводность.
Пример {вопрос 1). Ячейка содержит 0,10 моль/дм3 водного раствора КС1,
который при этой концентрации имеет мольную электропроводность J29 Ом-'Х
Xсм2 мольИзмеренное сопротивление составляет 28 44 Ом. Когда ту же ячей-
ку заполнили 0,05 моль/дм3 водным раствором NaOH, се сопротивленце стало
31,6 Ом. Найдите мольную электропроводность водного раствора NaOH при
этой концентрации.
Метод. Калибруем ячейку, используя v. = K!R, где R — измеряемое сопротивление,
К _ постоянная ячейки и я--электропроводность. Определим К из данных для
КС1, затем найдем и для NaOIl. Наконец, перейдем к мольной электропровод-
ности.
Ответ. Электропроводность раствора КС1 равна
х = (129 Ом-1 см3.mo4bj)X(0,10 моль/дм3) = 12,9 Ом-1 (см2/дм3) =
= 1,29-10-- Ом-1-см-'.
K = (l,29 J0-2 Ом-1см-|)Х(28,44 Ом) —0,367 см-1.
Следовательно, для раствора NaOH
K,!R — (0,367 см-^дЗКб Ом)^0,01]6 Олг'-см-1.
* Чаще всего для Лщ используются следующие единицы: Ом-Нем2-моль-1,
но в некоторых случаях необходимо затем выразить Лт в концентрационных
единицах. Если х выразить в Ом-1 - см-1, с — в моль/дм3 и отношение
v./c умножить па J000, то получим Лш в Ом-'-см2•моль-1. Действительно,
Лт (Ом-^см^моль-Ч
JOOO(x, Om-J-cm-i)
(с, моль/дм3)
25. Транспорт ионов и молекулярная диффузия
369
Рис. 25.2. Зависимость мольцой
электропроводности от концен-
трации.
Поэтому
Лщ = (0,0116 Ом-] х
Хсм-ОДО.Об моль/дм3) =
•>=232 Ом-1см2ирль“1.
Комментарий. Использование
ячеек, калиброванных по стан-
дартным растворам, имеющим
близкую электропроводность,
является хорошей идеей.
Как показывают измерения мольной электропроводности, рас-
творы электролитов по своему поведению делятся на два класса.
К первому классу относятся вещества, электропроводность кото-
рых слабо зависит От концентрации растворенного вещества. Как
видно из рис. 25.2, для приведенных растворов мольная электро-
проводность незначительно увеличивается при уменьшении кон-
центрации. Такие растворенные вещества называют сильными
электролитами (или, точнее, ионофорами). С уменьшением кон-
центрации растворенного вещества мольная электропроводность
растет до определенного предела, который называют мольной элек-
тропроводностью при бесконечном разбавлении Д’..
Кольрауш, выполнив целый ряд измерений на сильных электро-
литах, показал, что мольные электропроводности разбавленных
растворов с концентрацией с подчиняются уравнению
( 5.1.3)
где X — константа, которая зависит в большей степени от типа
соли (например, МА, М2Л и т. д.), чем от специфических особен-
ностей составляющих ее ионов.
Кольрауш установил также, что для любой соли величина А?п
может быть представлена в виде суммы вкладов отдельных ионов.
Если мольные электропроводности катиона и аниона при бесконеч-
ном разбавлении равны соответственно и ?Л, то закон независи-
мой миграции ионов выразится следующим образом:
Л®, =v^l+v_^, (25.1.4)
где v+ и v_ — числа молей катионов и анионов, необходимых для
образования 1 моля соли (например, v+ — v-= 1 Для NaCl н CuSO4,
по хм-=1, v._=2 в случае MgClg), Этот простой вывод, который
24—242
370
Часть 3. Изменение
можно понять, если представить, что в бесконечно разбавленном
растворе ионы ведут себя независимо друг от друга, позволяет
найти электропроводность любого раствора ионофора, используя
данные табл. 25.1.
Таблица 25.1
Ионная электропроводность при бесконечном разбавлении Л^(Ом _| см2 .моль-1)
при 25 °C
Катионы Анионы
н+ 349,8 OH- 197,6
Li+ 38,69 CI- 76,34
Na+ 50,11 Br-- 78,4
К+ 73,50 I- 76,8
Ag+ 61,92 NO3 71,4
Mg2/ 106,12 ch3co; 40,9
nh: Ca2+ Cu2+ Zna+ Bs2+ ' 73,4 119,00 108 106 127,28 SOjf- 159.6
Пример (вопрос 3). Найдите мольные электропроводности водных растворов
LiBr и BaCla при бесконечном разбавлении при 25 °C.
Метод. Воспользуемся законом независимой миграции ионов [уравнение (25.1.4)]'
и данными табл. 25.1.
Ответ. Для LiBr
JA ~ 38,69 Ом-1-см2-моль-1,
-г
= 78,4 Ом-1 сн2-моль-1,
лга= *’--г^-= 117.* Ом-1-см2.моль-1.
Для ВаС12
I* = 127,28 Ом-*1-см2-мол»*1,
д т ’
Х°_= 76,34 Ом-1-см2 моль-1,
А®, =Л,“.-[-2?.^_ = 279,96 Ом-i-см3-моль-1.
Комментарий. Если бы задача состояла в определении эквивалентной электро-
проводности, то ее значение для LiBr было бы 117,1 0м-1-см2.экв-!, а для
ВаС!3 — 139,98 Ом-1-см3.зкв-1, так как в последнем случае на каждый моль
движущегося вещества переносится двойное количество зарядов.
В случае второго класса электролитов— слабых электролитов
(или ионогенов) — между мольной электропроводностью и концен-
трацией наблюдается заметная зависимость. Величина Ат остается
небольшой вплоть до очень низких концентраций, по достижении
25. Транспорт ионов и молекулярная диффузия
371
которых она резко поднимается до значений, сравнимых с моль-
ной электропроводностью сильных электролитов. Это также иллю-
стрируется данными, представленными на рис. 25,2, Такое поведе-
ние может быть обусловлено существованием равновесия между
ионизированной и неионизированной формами ионогена
МА М+ + А“>
которое сдвигается вправо по мере увеличения разбавления. Элек-
тропроводность связана с числом ионов в растворе и зависит от
константы равновесия диссоциации, Если степень ионизации обоз-
начить через а, то константа равновесия равна
Л -(см+, моль/дм3) (сА_, моль/дм3)/(смд, моль/дм3) =
—(ас, моль/дм3) (ас, моль/дм3)/[(1—а) с, моль/дм3) —
= {а3/(1—а)) (с, моль/дм3),
где с—концентрация добавленного ионогена, а Смд — концентра-
ция неионизированной формы, Электропроводность зависит от
степени ионизации, так как концентрация ионов пропорциональна
а. Если измеренная мольная электропроводность равна Лю, то ее
отношение к мольной электропроводности полностью ионизирован-
ного раствора Лт записывается как
Лт ^=аЛт,
таким образом, мы приходим к закону разведения Оствальда
1 моль/дм3), (25,1.5)
связывающему концентрацию с электропроводностью. Это выра-
жение может быть использовано для определения константы рав-
новесия диссоциации, если измерить Л,п и рассчитать Лт из урав-
нения (25,1,4), Его можно также представить в виде
Лт- 4 (^Лт/С) ( (1 +4<№ - 11. (25,1,6)
что дает точную зависимость мольной электропроводности от кон-
центрации (концентрация с выражается в моль/дм3).
Пример (вопрос 4). Сопротивление 0,01 моль/дм3 водного раствора уксусной
кислоты было измерено в той же ячейке, что и в примере на стр, 368, и оказа-
лось равным 2220 Ом. Найдите значение константы диссоциации Л\, значение
рЛ'а н степень диссоциации кислоты при этой концентрации.
Метод, Найдем из величины постоянной р.ченкя, определенной в указанном
прнмере (К=-0,367 см-1)- Найдем Лга из данных табл. 25.1, Для нахождения Д'а
используем уравнение (25.1.5), затем определим рКа = — 1g Ка. Наконец, степень
диссоциации получим нз выражения Л(п=аЛт.
Ответ. Из табл, 25.1 имеем A’j (СН3СООН) — A“(H+)+V(CH3COO~) = (349,8+
4-40,9) Ом-1-см2-моль-1 —390,7 Ом-1'см2-моль-1. Наблюдаемая электропровод-
24*
372
Часть 3. Изменение
пость и=Л'//?== (0,367 см-1)/(2220 Ом) = 1,65-10-4 Ом_1-см-1. Поэтому наблюдае-
мая мольная электропроводность равна
Лт = (1,65-Ю-1 Ом-1 см-1)/(0,01 "моль/дм3) = 16,5 Om-i.см2-моль-1.
Степень диссоциации а=Л(п /Лш« Л ;п/Л®*= 0,0422. Затем из уравнения
(25.1.5)
Л'а = {(0,0422)V(l —0,0422)] х0,01 = 1,86 -10-&.
Поэтому
рЛ'а — lg (1,86-10-*) = 4.73.
Так как. « — 0,0422, то 4.22% кислоты диссоциировано.
Комментарий. Необходимо внести .некоторые поправки (например, па то, что
Лга была отождествлена с А“п). Термодинамическая величина рД'а может быть
получена из повторных измерений при различных концентрациях и экстраполя-
ции к нулевой концентрации.
Существование ионон в растворе подтверждается также изме-
рениями величин, характеризующих коллигативцыс свойства раз-
бавленных растворов (т. 1, разд. 8.3), в частности осмотического
давления. Это доказательство было рассмотрено в гл. 8 (т. 1).
Приведенное выше экспериментальное доказательство может
быть использовано для объяснения структуры ионных растворов и
пути движения в них ионов. Наша задача состоит в том, чтобы
выяснить, почему ионы различаются по подвижности, почему ионы
имеют различные значения л* и почему -электропроводность рас-
творов сильных Электролитов зависит от концентрации ио закону
Кольрауша [уравнение (25.1.3)].
Подвижность ионов. В растворе на ион действует множество сил.
Одной из них является беспорядочная бомбардировка ионов рас-
творителем. Поскольку и направление, и сила этих столкновений
иеупорядоченны, они це вызывают общего движения ионов в од-
нородном растворе. Если появятся градиенты концентраций, то
ионы будут перемещаться из областей высоких концентраций в
области низких концентраций. Сейчас мы не будем учитывать это
обстоятельство, так как мы имеем дело с однородными раствора-
ми, но вернемся к нему в разд. 25.2, Ионы подвергаются также
действию электрических сил. Когда два электрода на расстоянии
I друг от друга находятся при разности потенциалов Ай, между
электродами имеется градиент потенциала с величиной А^Д. По-
ложительные ионы двигаются вниз по градиенту, отрицательные
ионы —вверх по градиенту (рис. 25.3). Это движение может быть
интерпретировано как результат электрических сил, действующих
на ионы, причем величина силы пропорциональна градиенту по-
тенциала.
Количественно эти идеи могут быть выражены следующим об-
разом. Отметим, что, если заряд иона равен ze (где z положите-
25. Транспорт ионов и молекулярная диффузия
373
Рис. 25.3. Катион в градиенте потен-
циала.
лен для катионов и отрицате-
лен для анионов), в электри-
ческом поле Е на ион действу-
ет сила
F—-zeE. (25.1.7)
Электрическое поле является отрицательным градиентом потенци-
ала:
Е — —dp!dx.
(25.1.8)
Это Соответствует предшествующему обсуждению, потому что,
если разность потенциалов в ячейке составляет градиент
и поле —ЬфП, то сила равна
F =~—zeb$H.
Если = отрицателен (^r и — потенциалы правого
и левого электродов), то сила F положительна для положительных
ионов (2 положителен) и, таким образом, заставляет их передви-
гаться в направлении более отрицательного электрода (рис. 25-3);
для отрицательных ионов можно сделать обратное заключение
(z отрицателен).
Сила, действующая на ион, ускоряет его, но, так как он прохо-
дит через растворитель, его тормозит сила трения. Поэтому он ус-
коряется только до некоторой определенной скорости, которая за-
висит от напряженности приложенного поля и вязкости раствори-
теля. Эта предельная скорость называется скоростью дрейфа иона
в растворе и обозначается символом v. Если предположить, что
формула Стокса для замедляющей силы вязкости, действующей на
сферический предмет радиусом а в растворителе с вязкостью т],
применима даже в микроскопическом масштабе (независимые до-
казательства подтверждают, что это часто дает правильный поря-
док величины), то сила трения будет зависеть от скорости как
блацу. Постоянная скорость дрейфа устанавливается в том случае,
когда ускоряющая сила zeE уравновешивается замедляющей си-
лой; эта значит, что и поэтому
и геЕ/6лат\. (25.1.9)
Поскольку скорость прохождения тока определяется скоростью
дрейфа, можно ожидать, что электропроводность будет уменьшать-
ся с увеличением вязкости раствора и размеров иона.
374
Часть 3. Изменение
Рис. 25.4. Механизм электропроводности в воде.
Экспериментальные результаты подтверждают первое из этих
предположений, но не второе. Например, мольные электропровод-
ности ионов щелочных металлов увеличиваются в ряду от Li+ к
Cs+ (их значения приведены в табл. 25.1), хотя хорошо известно,
что в этом ряду размеры ионов заметно возрастают (ионные ра-
диусы, полученные из данных ио дифракции рентгеновских лучей,
приведены в табл. 22.1). Это противоречие можно объяснить, при-
няв в расчет явление сольватации ионов в растворе, Молекулы
растворителя группируются вокруг иона и увеличивают его эф-
фективный радиус (который называют гидродинамическим радиу-
сом), Небольшие ионы являются источником более сильных элект-
рических полей, чем большие ионы (это выводится из электроста-
тической теории, согласно которой электрическое поле на поверх-
ности сферы радиусом 7? пропорционально ze/R2), и, таким обра-
зом, сольватация небольших ионов происходит в большей степени.
Небольшие ионы имеют больший гидродинамический радиус, мень-
шие скорости дрейфа и, следовательно, низкую электропровод-
ность.
Однако протон, хотя и очень мал, имеет очень большую моль-
ную электропроводность, равную 350 Ом'4-см2 • моль^1. Можно
предположить, что протон должен быть сильно сольватирован, тем
не менее он способен быстро передвигаться в растворе. В этом
случае нельзя применить гидродинамический довод, приводящий
к идее о вязком торможении, поскольку действует другой меха-
низм. Этот механизм учитывает как малую массу протона, так
и то, что структура воды содержит развитую систему водородных
связей. Считается, что вместо одного протона, двигающегося в
растворе, существует эффективное движение протона, включаю-
щее образование и разрыв связей вдоль длинной цепочки молекул
воды (рис. 25.4). Электропроводность определяется скоростью, с
которой протон может «прыгать» между соседними молекулами
воды, а это в основном лимитируется скоростью, с которой моле-
кулы воды могут переориентироваться, чтобы принять или отдать
протоны соседним молекулам.
25. Транспорт ионов и молекулярная диффузия
375
Скорость дрейфа иона имеет определенное направление и ве-
личину. Мы обозначим ее величину как $+, так что s±=|u±|. Ско-
рость дрейфа пропорциональна напряженности приложенного по-
ля; константу пропорциональности называют подвижностью иона
и±:
s , =и±Е. (25,1.10)
Из этого определения следует также, что подвижность соответ-
ствует скорости иона в электрическом поле с единичной напря-
женностью (например, 1 В/м). Позже мы увидим, что типичные
подвижности находятся в пределах (4—8) -10“3 мг/(с-В). Это
указывает, что падение потенциала на 10 В, происходящее в рас-
творе на участке 1 см (так что £=103 В/м), приведет к скорости
дрейфа в пределах (4—8) • 10~5 м/с, или (4—8) -10—2 мм/с. Эта
скорость может показаться небольшой, но, если ее выразить в мо-
лекулярной шкале, она соответствует столкновению иона в каж-
дую секунду приблизительно с 100000 молекул растворителя. По-
движности ионов представлены в табл. 25.2, а их определение опи-
сано на стр. 394,
Использование значений подвижности ионов состоит в том, что
они связывают рассчитываемые величины с измеряемыми. По-
движность может быть рассмотрена как с точки зрения динамики
ионов, так и в связи с электропроводностью раствора. Перейдем
теперь к выявлению этих связей.
Таблица 25.2
Подвижность ионов в водных растворах и [10_4 смг/(с-В)] при 25 °C
Катионы Лниоцы
н+ 36,25 он- 20,48
L1+ 4,01 F- 5,70
Na+ 5,19 С1- 7,91
К+ 7,62 Вг- 8,13
W 7,92 I- 7,96
Ag+ 6,42 no; 7,40
NH^ 7,67 со?- 7,46
Cas+ 6,17 so3- 8,27
Сп3+ 5,60 сн3со; 4,24
La3+ 7 21
Вначале найдем связь между подвижностью и электропровод-
ностью. Возьмем раствор соли Mv+2lv_ концентрации с (в молях
на единицу объема); такой раствор содержит ч±сЕ катионов и
v—cL анионов на единицу объема. Катионы имеют заряд а ани-
376
Часть 3, Изменение
А
Рис. 25.5. К расчету потока за-
ряда.
омы — заряд 2-е. Преж-
де всего рассмотрим ка-
тионы и их движение че-
рез воображаемое окно
площадью А, располо-
женное в растворе меж-
ду электродами (рис.
25.5). За промежуток
времени At все катионы,
которые имеют скорость дрейфа s+, в пределах расстояния s+AZ
и, следовательно, в пределах объема 5+Л1Л пройдут через это ок-
но, и их число составит (s+AtA)v+cL. (Такие же расчеты проводи-
лись для газов в предыдущей главе, например в разд. 24.1.) Каж-
дый ион несет заряд z+e, и поэтому поток положительного заряда,
т. е. заряд на единицу площади в единицу времени, равен
J (заряд) = (s^ ДМ) v+cLz+e/AAt — v+s+zi,ceL.
Ток переносится также анионами, они перемещаются в противопо-
ложном направлении и несут противоположный заряд. Их пере-
мещение должно увеличивать ток. Поэтому общий поток равен
J -^v+s_Lz+ceL-\-v_s_ | z_ | ceL = -*
• -•(«+v+zJ.-|-u_v_|z_|)cF£', (25.1,11)
где скорость дрейфа, согласно уравнению (25.1.10), заменена под-
вижностью, a eL — числом Фарадея F (1F — 9,65• 10Л Кл/моль).
Величина J представляет собой поток заряда: ток через пло-
щадь А равен I — JA. Если напряженность электрического поля Е
создается разностью потенциалов А ф, действующей через раствор
длиной I, то поток равен | Аф | Д, откуда
/ =(vrutz_¥ -\-v.u_ | 2_ |) ср А ] Аф \/1.
Однако ток и разность потенциалов связаны с электропроводно-
стью законом Ома:
/—| A’k\lR-— и I Аф IАЦ.
Сравнение этих двух выражений позволяет выразить электропро-
водность в виде
x = (v u | z_ |) cF.
(25.1,12)
25. Транспорт ионов и молекулярная диффузия
377
Отсюда следует, что мольная электропроводность равна
Am=(v+u+z+-|-v_u_ | z_ [)F, (25.1.13)
и для 1 : 1-соли с z+ = |z_| =z получим
(25.1.14)
Теперь видно, что электропроводность отдельных ионов может
быть связана с их подвижностью: .
Л± = и± | z± | F. (25.1,15)
Это и есть связь между подвижностью и электропроводностью
ионов, которую мы искали. Измеряя электропроводность и исполь-
зуя два последних уравнения, можно найти подвижность. На стр.
384 мы увидим, как подвижность связана с размером ионов и вяз-
костью раствора.
Пример (вопрос 6). Каковы подвижности иопов Н+ и SO4- в водном растворе
при бесконечном разбавлении?
Метод. Используем уравнение (25.1.15). Мольные электропроводности при 25°C
приведены в табл. 25.1.
Ответ. !/+(Н+) — (349,82 Ом-1 см2-моль-1)/(9,649'10* Кл/моль) =3,625- 10-Э cms/(c X
ХВ); и. . (SO**) = (159,6 Ом-1-см2-моль-|)/2Х(9,649'1(НКл/моль) =8,270-10-4 см2/
/(с-В).
Комментарий. Заметьте, что подвижность протона приблизительно в 5 раз больше
подвижности иона SO2- (и в 10 раз больше подвижности иона Li+). Это верно,
так как поп SO2- очень подвижен из-за того, что он лишь слабо сольватирован.
Доля общего тока, переносимая каждым типом ионов, прямо
связана с их подвижностью, и, если ионы подвижны, они перено-
сят большую часть общего тока. Относительная доля переноси-
мого тока называется числом переноса, которое (для 1 : 1-соли)
определяется как
Очевидно, что /+-Н- = 1. Теперь можно вернуться назад и выразить
t+ через мольные электропроводности ионов; используя K± — zu±F
(для 1 : 1-соли), получим
Х+ + Л_
или
X±-1-Amf±, (25,1,17)
Следовательно, если имеется независимый путь измерения чисел
переноса, то можно найти электропроводность отдельных ионов и
затем их подвижность. Некоторые применяемые методы кратко
рассмотрены в приложении.
378
Часть 3. Изменение
Рис. 25.6. я —ионная атмосфе-
ра, когда ионы ие имеют сум-
марной подвижности; б—ис-
кажение ионной атмосферы,
возникающее при движении
нона.
Электропроводность и взаимодействие ионов. Остается объяснить
зависимость мольной электропроводности в разбавленном раство-
ре от концентрации в степени [уравнение (25.1.3)]. В гл. 11
(т. 1), посвященной структуре ионных растворов, мы встречались
с зависимостью термодинамических свойств растворов от с'бц Мы
цидели, что ионы делятся на группы по своим зарядам, а не по
специфическим особенностям, и эти свойства могут быть количе-
ственно использованы с точки зрения воздействия ионной атмосфе-
ры (т. 1, разд. 11.2), окружающей данный ион.
Не вдаваясь в подробности той главы, можно сформулировать
основные качественные особенности поведения ионов и развить их
для настоящей динамической задачи.
Каждый ион стремится окружить себя ионами противополож-
ного заряда. Образуется небольшой кластер, но это имеет важное
значение вследствие дальнодействующих кулоновских сил. Обла-
ко заряда называют ионной атмосферой, и в среднем оно сфериче-
ски-симмстрично. Присутствие облака влияет на электростатичес-
кую энергию иона и благодаря этому на его термодинамические
свойства.
Если внешнее электрическое поле приложено к раствору ионов,
каждый из которых окружен небольшой атмосферой противопо-
ложного заряда, то возникает динамическая ситуация, и мы долж-
ны рассматривать облако как структуру в движении. Первое свой-
ство, на котором мы остановимся, связано со временем, необходи-
мым для создания и разрушения атмосферы. Ионы группируются
не бесконечно быстро, и, следовательно, если ион движется в рас-
творе, атмосфера не успевает полностью образоваться по фронту
его движения и не успевает полностью разрушиться в его «кильва-
тере» (рис. 25.6). Общий эффект состоит в смещении центра заря-
да атмосферы на небольшое расстояние позади движущегося иона.
Так как заряды иона и атмосферы противоположны, то движение
иона замедляется. Это называется релаксационным эффектом, по-
скольку образование и разрушение атмосферы является одним из
типов релаксации, приводящим к равновесному распределению
ионов.
25. Транспорт ионов и молекулярная диффузия
379
Другой эффект, который необходимо учитывать,— электрофо-
ретический эффект. По мере движения в растворе центральный ион
испытывает вязкое торможение, и это определяет скорость его
дрейфа и, следовательно, его электропроводность. Когда ионы не
бесконечно удалены друг от друга, они сталкиваются друг с дру-
гом, и эффект усиливается ионами противоположного заряда, каж-
дый из которых имеет свою группу молекул растворителя. Это
увеличивает вязкое торможение, уменьшает скорость дрейфа и,
следовательно, электропроводность.
Количественная формулировка этих эффектов далеко не проста,
но принято приближение, подобное софистике теории статических
растворов Дебая —Хюкксля. Его результатом явилось уравнение
Дебая — Хюккеля — Онзагера для мольной электропроводности.
Оно имеет форму
Лт^ЛаП1 -(А+ВЛ'ОсЧ (25.1.18)
где Л и В—константы, зависящие от электрических свойств и
температуры раствора. Для симметричного электролита
j _ zeF / 2z-e-L „ е-гф / 2z~e~L х* 1^
Я ~ Зли \ ’ ° ~~ 24га>0КгА:Г ' пе0МТ ) ’
где Кг — относительная диэлектрическая проницаемость среды
(т. 1, табл. 11.1) и <7^0,59 для 1 : 1-электролита. Их численные
значения для некоторых растворителей (только применение их к
воде не связано ни с какими ограничениями в расчете) приведены
в табл. 25.3.
Таблица 25.3
Константы Дебая — Хюккеля — Онзагера при 25 °C
Растворитель А В
Вода 60,20 0,229
Метанол 151,1 0,923
Этанол 89,7 1,83
Ацетонитрил 22 9 0,716
А выражается в Ом-| смг-моль“|/(моль/дм,1 V®, а В —в (моль/дм’] 1/з.
Пример (вопрос 8). Мольная электропроводность водных растворов КС1 изме-
няется с концентрацией так, как показано ниже. Найдите константу Кольрауша
для этого 1 : 1-электролита и подтвердите, что ее значение определяется из
уравнения Дебая — Хюккеля — Онзагера.
с, моль/дм® 0,001 0,005 0,010 0,020
Am, Ом-1-см2-моль-’ 146,9 143,5 141,1 138,2
1
380
Часть 3. Изменение
Рис. 25.7. Определение Ж и Л®
Метод. Строим график зависимости Лт
от Ус, его наклон равен — X. Этот на-
клон можно также найти из графика, по-
строенного по уравнению (25.1.18); он
равен — (А Ч-ВЛ®),). Возьмем значения А
и В из табл. 25.3 и значение Л®, из от-
резка, отсекаемого от оси ординат.
Ответ. График зависимости Лт (Ом-1Х
Хсм!-моль~1) от У (с, моль/дм3) показан
на рис. 25.7. Отрезок, отсекаемый от оси
ординат, равен 149,3. а наклон рапен
—79,14. Отсюда 3^ = 79,14 Ом-1-см2Х
Хмоль-,/(моль/дм3),?г (громоздкие, по
удобные единицы) и Л’, = 149,3 Ом-’Х,
Хсм2-моль_1. Из табл. 25.3
А — СО, 20 Ом-1 см2, моль-1/(моль/дмэ)'^>
Й = 9,229 (моль/дм3)’/2.
Таким образом, Д+ВА['П = 60,20 Ом 1'смг-моль '/(моль/дм3)1/2+
-1- (0,229 (моль/дма) ’/2}Х( 149,5 0м-1-см2' моль-') =
= 94,4 Ом“!-см3-моль-7(моль/дм3)1/2-
Комментарий. Результаты вовсе не плохие, принимая во внимание теоретические
трудности рассмотрений динамических систем из-за их сложности. Отметьте, что
построение графика зависимости Am от Ус п сто экстраполяция — вполне пригод-
ный путь для нахождения Л^,
В уравнении (25.1.18) имеется та же самая концентрационная
зависимость, что и в эмпирическом выражении Кольрауша [урав-
нение (25.1.3)]. Более того, наклоны кривых, как и предсказано,
зависят от валентного типа соли (z появляется в константах А и
В). Некоторые сравнения между теорией и экспериментом пока-
заны иа рис. 25.8, из которого видно, что при низких концентра-
циях существует хорошее совпадение теории и эксперимента.
Успешное применение уравнения Дебая — Хюккеля— Онзаге-
ра подтверждает, что модель иоп-ионных взаимодействий в основ-
ном правильна. Дальнейшая проверка основана па исследовании
того, что произойдет, если ионная атмосфера исчезнет. Это может
быть сделано различными путями. Одним из них является измере-
ние электропроводности на высоких частотах; тогда центральный
ион движется назад и вперед очень быстро, и тормозящее влия-
ние ионной атмосферы в среднем должно быть близким к нулю.
Это эффект Дебая — Фалькенгагена, и предсказанное увеличение
подвижности иа высоких частотах было замечено. Другой путь
исключения влияния атмосферы — создание такого быстрого дви-
жения ионов, чтобы для образования атмосферы не было време-
25. Транспорт ионов и молекулярная диффузия
381
Рис. 25.8. Экспериментальная и
теоретическая мольные электро-
проводности.
ни. Этот эффект Вина
проявляется в наблюде-
нии высокой подвижно-
сти при очень больших
напряженностях электри-
ческого поля. (Существу-
ют два эффекта Вина.
Первый эффект Вина
только что был описан;
второй эффект Вцнаобус-
ионогена, или слабого
ловлей увеличением степени ионизации
электролита, под влиянием поля.)
Такая модель взаимодействий при больших концентрациях не-
применима, так как тогда ионы стремятся объединиться, образуя
двойные и даже тройные ионы. Это становится ясным из рентге-
новского анализа ионных растворов, где пики рассеяния можно
интерпретировать в терминах определенных ион-ионных расстоя-
ний.
25.2. Фундаментальные аспекты
молекулярного транспорта
В этом разделе мы попытаемся объединить материалы данной
и предыдущей глав. Мы сделаем это на основе термодинамичес-
ких и статистических принципов и покажем, что имеется много
важных и полезных связей между свойствами, относящимися к
движению молекул и ионов в жидкостях.
Диффузия. Термодинамическое рассмотрение. В части 1 (т. 1) бы-
ло показано, что термодинамическим свойством, определяющим на-
правление самопроизвольного изменения, является термодинами-
ческий химический потенциал (т. 1, разд. 6.2). Когда 1 моль рас-
творенного вещества переносится из области, где его химический
потенциал равен ц(1), в область химического потенциала ц(2),
количество требуемой работы составляет w = ц(2) —р(1). Пред-
положим, что химический потенциал зависит от положения х в
системе, тогда количество работы, необходимое для переноса 1 мо-
ля вещества из положения х в x-\-dx, составит
dw — ц (x+dx) — ц (х) = [ц (х) ^(dpjdx) dx] — р (х) = (dp/dx) dx.
332
Часть 3. Изменение
(Строго говоря, производные должны быть частными производны-
ми и перенос должен осуществляться при постоянных давлении и
температуре; подробнее об этом см. в т. 1, разд. 6.3.) В классиче-
ской механике количество работы, требуемое для перемещения
предмета на расстояние dx против силы F, равно
da)—. —fdx.
Сравнивая последние два уравнения, мы видим, Что градиент хи-
мического потенциала ведет себя как сила. Поэтому запишем
&——(dp/dx). (25.2.1)
В действительности нет никакой силы, заставляющей молекулы
двигаться в сторону уменьшения химического потенциала; это осу-
ществляется благодаря их естественному движению, определяемо-
му как следствие второго закона термодинамики, и стремлению
к максимальной энтропии; тем не менее, как мы увидим далее, та-
кие иллюзорные представления об эффективных термодинамичес-
ких силах могут быть очень полезны.
В растворе с концентрацией с химический потенциал идеально-
го растворенного вещества составляет (т. 1, разд. 8.5)
р=р°Ц-7?Г1п (с, моль/дм3) (25.2.2)°
Если концентрация зависит от положения, то Действующая термо-
динамическая сила равна
& — —(d-dXj [р° -4-7?Г 1п (с, моль/дм3)] = — (7?Г/с) (dc/dx), (25.2.3)°
так как не зависит от положения, a dinf/dx= (\/f)dj/dx.
Из последнего уравнения можно вывести закон диффузии Фи-
ка [с термодинамической точки зрения поток пропорционален гра-
диенту концентрации; уравнение (24.3.1)]. Предположим, что по-
ток является реакцией молекул на действие некоторой силы. Если
сила на 1 моль равна F, то сила на единицу объема присутству-
ющих молекул составит су. Если предположить, что поток веще-
ства /(вещество) пропорционален приложенной силе, то J(ве-
щество) Но эффективная сила выражается уравнением
(25.2.3), и поэтому
J (вещество) —RT (dc/dx) —kT (d-^/dx),
и в соответствии с законом Фика мы получаем, что поток пропор-
ционален градиенту концентрации (Л'’ —платность числа молекул,
Л'' =cL).
Более удобно рассмотреть другие аргументы и интерпретиро-
вать поток частиц как произведение v^ — vcL, где о— средняя
25, Транспорт ионов и молекулярная диффузия 383
скорость частиц и с — их мольная концентрация. Тогда закон
Фика
=J = — D (d^/dx) = —DL (dc/dx)
можно записать как
cv = —D (dc/dxj,
или, используя уравнение (25.2.3),
у —(D/с) (dc!dx)^(D/kT)j.
Следовательно, в ответ на единичную силу молекулы диффундиру-
ют со скоростью дрейфа, равной величине DlkT. Однако известно,
что подвижность иона связана с электрической силой, действую-
щей на него. Так как подвижность определяется через s = tiE и
так как электрическая сила равна ezE, то из этого следует, что
$ ~иЕ—(и/ze) (ezE) —.(u/ze)
следовательно, скорость дрейфа под действием единичной силы
равна (ujze). Природа силы значения не имеет, поэтому две ско-
рости дрефа (D[kT)& и (и[ге)$ можно отождествить друг с дру-
гом, тогда
uiez=DikTt
и мы приходим к очень важному результату, известному как
соотношение Эйнштейна: D—ukT/ez, (25.2.4)°
которое связывает коэффициент диффузии и подвижность.
Последнее уравнение можно преобразовать дальше в двух на-
правлениях. Прежде всего оно может быть использовано для уста-
новления связи между мольной электропроводностью и коэффици-
ентами диффузии ионов D+ и D~. Записав (для 1: 1-солей), что
Am ~zF (ut-\-u.)^(z2eF/kT) (D+^-D_),
мы, таким образом, приходим к соотношению Нернста— Эйнштей-
на
ЛП1--=(^Рда(Д++П_). (25.2.5)°
Одним из применений этих выражений является Определение
коэффициентов диффузии ионов из измерений электропроводно-
сти, Другим — расчет электропроводности на основе моделей ион-
ной диффузии (см. ниже).
384
Часть 3. Изменение
Соотношение Эйнштейна, кроме того, может быть использова-
но для установления связи между подвижностью и вязкостью.
Комбинируя выражения
s=ezE/6лт\а и s~uE
(первое из них относится к скорости дрейфа, определяемой урав-
нением (25.1.9), можно написать
и ~ е?1&т\а.
Поскольку соотношение Эйнштейна имеет вид u — ezDjkT, прирав-
нивая эти два выражения и комбинируя их, получим соотношение
Стокса — Эйнштейна
D = kT/6w\a, - (25.2.6)°
которое связывает коэффициент диффузии и вязкость жидкостей.
Важной особенностью этого вывода является то, что коэффици-
ент диффузии не зависит от заряда Диффундирующей частицы, и
поэтому последнее соотношение также может применяться в пре-
деле к бесконечно малому заряду или к нейтральным молекулам.
Следовательно, соотношение Стокса — Эйнштейна можно исполь-
зовать для расчета коэффициентов диффузии из измерений вязко-
сти. Однако не нужно забывать, что Это приближение основано
па предположении, что формула Стокса применима для вязкого
торможения, Некоторые коэффициенты диффузии приведены в
табл. 25.4.
Таблица 25.4
Коэффициенты диффузии D(10-sl м2/с) при 2а °C
12 в гексане 4,05 N2 в CCI4 3,42
12 в ССЦ 3,42 Os н ССЦ 3,82
12 в бензоле 2,13
CCU в н-геетане 3 17 Аг в СС14 3,63
Глицин и воде 1,05 Н2О в воде 2,26
Глюкоза в воде Ионы в воде: 0,67
Н+ 9,31 С1- 2,03
L1+ 1,03 Вг- 2,08
Na+ 1,96 I- 2,05
Пример (вопросы 12, 13 и 14), Найдите коэффициент диффузии, мольную элек-
тропроводность и эффективный гидродинамический радиус иона SOj~ в воде
при 25 °C.
Метой. В примере на стр. 377 было найдено, что подвижностьиопа равна 8 27ОХ
Х10-4 см2/(с-В), Свяжем подвижность с D уравнением (25.2.4). Затем использу-
ем уравнение (25.2.5) для связи D (теперь напишем О_) с (вклад аниона
в Лт) Рассчитываем эффективный радиус а_ из уравнения (25.2.6). Вязкость
25, Транспорт ионов и молекулярная диффузия 385
воды при 25°C равна 1,00 сП [],00-10-3 кг/(м-с)]. Напомним, что Дж = Кл-В
и В/А=Ом.
Ответ. Из уравнения (25.2.4)
[8,270-10-4 сма/(с-В)]Х(1,381 I0-23 Дж/К)х(298К)
О_ —п_й7’/ег= 2х(1,602-10-‘9 Кл)
1,062-10*® см2/с.
Из уравнения (25.2.5)
, 22х(9,649-104 Кл/молъ)2Х(1,062-10-®см2/с)
[8,314 Дж/К моль)]X(298 К) = 159,60м-‘см -моль- .
Из уравнения (25.2.6)
________________________(1,381 10~33 Дж/К) X (298 К)_____
й_-Я//Олт]Р-- 6лХ [1 00 ]0-э кг/(м-с)[х(1,062- 10-6см2/с) —
— 2,056-10-19 м, или 206 пм, или 2,06 А.
Комментарий. Длина связи в SO4' равна 144 пм, И, таким образом, рассчитан-
ный здесь радиус (радиус сферы, представляющей молекулу) оказывается впол-
не вероятным и совместимым с низкой степенью сольватации.
Некоторые экспериментальные подтверждения этих идей следу-
ют из измерений электропроводности, так как по эмпирическому
правилу Вальдена величина произведения ЛтД должна быть при-
близительно постоянной для одинаковых ионов в различных рас-
творителях. Поскольку A,n~w и 1/г], теоретическая основа это-
го правила ясна. Применимость правила Вальдена к различным
растворителям не ясна, так как они сольватируют ионы в разной
степени, и поэтому эффективный гидродинамический радиус ионов
зависит От природы растворителй: как а, так и т] изменяются с
изменением растворителя.
Диффузия как зависящий от времени процесс. Закон Фика и его-
аналоги для транспорта других физических свойств (разд. 24.3)
относятся к потоку при постоянном градиенте. Поэтому они опи-
сывают процессы, не зависящие От времени. Например, они свя-
зывают диффузионный поток тепла с постоянным градиентом тем-
пературы или поток частиц с постоянным градиентом концентра-
ции, который поддерживается вводом частиц (газа или раство-
ренного вещества) в одну область и выводом их из другой. Мы об-
ратимся теперь к обсуждению зависящих от времени диффузион-
ных процессов, в которых к некоторому моменту времени устанав-
ливается некоторое распределение концентраций или температу-
ры и т. Д-, а затем происходит рассредоточение без пополнения
градиента (например, когда слиток металла быстро нагревается
с одного конца и тепловая энергия распространяется по слитку или.
когда слой растворяемого вещества распространяется по поверх-
ности растворителя и распределение концентраций в растворе из-
меняется по мере того, как вещество растворяется).
25—242
386
Часть 3. Изменение
Рис. 25.9. Диффузия вещества в
некоторую область.
Для того чтобы тракто-
вать диффузию как процесс
во времени, рассмотрим диф-
фузию вещества, но все до-
воды легко можно приме-
нить к другим свойствам.
Сконцентрируем наше вни-
мание на небольшой пласти-
не системы, занимающей
пространство от х до х-1-Дх, с поперечным сечением площадью А
(рис. 25.9). Пусть концентрация при х и времени t будет Л’(х, t);
тогда благодаря потоку с левой стороны увеличение концентра-
ции внутри пластины (объемом ЛДх) составит
д-Л” (х, t)/dt~J{x, t)A/AAx =J(x, t) &х,
так как JA — число частиц, которые входят в окно площадью А в
каждый единичный интервал времени. С правой стороны окна так-
же имеется поток; если этот поток равен /(х-фДх), то благодаря
ему концентрация внутри пластины изменяется со скоростью
дЛ*(х, f)/dt = — J (х-ф Дх, /) Л/ДДх= — 7(х-ф Дх, /)/Дх,
Отрицательный знак появляется потому, что, когда поток идет
вправо, концентрация в пластине уменьшается (Z положителен).
Поэтому общая скорость изменения концентрации составит
(х, tydt — J (х, /)/Дх—/(х-ЬДх,/)/Дх.
Теперь потоки можно связать с градиентами концентраций в
каждом окне. Используя закон Фика, можно написать
J ~-(х, t)—J(x-)-Дх, f) = { —D [&^(x, t)/dx]\ —
-{-р^(х+дх,о/^п=-п
-ф( ) Дх 1 = DfcP- J" (x, f)/<9xa] Дх,
25. Транспорт ионов и молекулярная диффузия 387
Подставляя последнее выражение в уравнение для скорости из-
менения концентрации в пластине, получим
уравнение диффузии-. (х, i)ldt\ — D (х, /)/дхг[. (25.2.7)
Иногда его называют вторым законом Фика.
Несколько слов об общей форме этого уравнения. Мы видим,
что скорость изменения концентрации пропорциональна кривизне
(второй производной) зависимости концентрации от расстояния.
Если концентрация изменяется быстро от точки к точке, то ско-
рость, с которой происходит изменение концентрации во времени,
соответственно большая. Если производная равна пулю, то кон-
центрация во времени не изменяется. Например, если концентра-
ция с расстоянием падает линейно, то концентрация в любой точ-
ке остается постоянной, поскольку приток концентрации уравнове-
шивается оттоком. Уравнение диффузии можно рассматривать как
математическую формулировку интуитивного представления о
том, что природа имеет естественную тенденцию устранять «мор-
щийы» в распределении.
Уравнение диффузии — это дифференциальное уравнение, и
поэтому, чтобы получить его решение, необходимо точно опреде-
лить граничные условия. Это можно проиллюстрировать на специ-
фическом примере растворителя, в котором растворенное вещест-
во находится в одной плоскости. В кулевой момент времени гра-
ничные условия таковы, что все молекулы (Уо) растворенного ве-
щества сконцентрированы на плоскости yz при х = 0. Решением
уравнения диффузии при таком начальном условии будет
(х, t) = {У0/Л (nZ)/)l/2| ехр(— x2/4Dt), (25.2.8)
в чем можно убедиться непосредственной подстановкой. Резуль-
тат, полученный в разные моменты времени, показан на рис. 25.10;
ясно, что концентрация молекул распределяется по веществу во
времени. Значение уравнения диффузии состоит в том, что с его
помощью можно предсказать концентрацию в любой точке систе-
мы в любое время.
На основе уравнения диффузии и, в частности, с помощью
только что описанного простого решения можно объяснить ряд
важных особенностей процесса диффузии. Например, может воз-
никнуть вопрос, каково среднее расстояние, на которое распро-
страняется растворенное вещество к моменту времени t. Число мо-
лекул в пластине толщиной dx в плоскости х составляет
(x.t )Adx, и поэтому вероятность того, что любая из Лго молекул:
находится в пластине, равна ^(х, i)Adx/Nf). Если молекула нахо-
дится в пластине, то она прошла расстояние х от начала коордит
25*
388
Часть 3. Изменение
Рис. 25.10. Диффузия растворенного вещества с плоской поверхности.
пат; таким образом, среднее расстояние, иа которое она переме-
стилась, равно
СО
(х) — J х^ (х, /) Adx/N0 = (1
о в
xe_Jc2^D/dx — 2 (ЕМ/л)1^. (25.2.9)
Среднее расстояние изменяется пропорционально квадратному кор-
ню из промежутка времени. Это очень важный результат, к кото-
рому мы вернемся позже. Если мы используем соотношение Сток-
са — Эйнштейна для коэффициента диффузии, то среднее расстоя-
ние, пройденное частицами радиусом а в растворителе с вязкостью
т], составит
(х) = (2feT /Злг,па)’/2 ]//. (25.2.10)
Средне квадратичное расстояние, пройденное частицами, равно
х=У(х2), и его величина составляет
Л = /(Х®) =
х2 Л'’ (х, t) Adx/N0—(2Dt)4i.
(25.2.11)
Величина х является мерой расстояния, на которое распространя-
ются частицы, когда они имеют возможность мигрировать в обоих
направлениях (потому что тогда (х) =0). Полезно знать долю
частиц, которые остаются в пределах расстояния х от начала ко-
ординат, поскольку среднее число частиц может не дать достаточ-
ной информации. Так как их число в пластине, находящейся на
25. Транспорт ионов и молекулярная диффузия
389
расстоянии х, равно (xt t)Adx, то число частиц во всех пласти-
нах перед первой пластиной, находящейся на расстоянии х, явля-
ется суммой (интегралом)
ЛЦх < М) = j dx (х, f) Adx _= (0,68.. .) (tf0/4),
о
где х заменяется на (2Z>0’/2 и интеграл определяется численно*.
Из него следует, что доля молекул, находящихся в области
^х, равна 0,68. Следовательно, больше 2 * */з молекул концентриру-
ется около первоначального положения и только 32% переходит
за х (не забывайте, что значение х растет со временем).
Уравнение диффузии может быть решено для более сложных
случаев, например когда ионы постоянно генерируются на плос-
кости электрода, погруженного в раствор, или когда ионы осаж-
даются на электроде и уходят из раствора. Расчеты, подобные
этим, играют важную роль при обсуждении скоростей реакций на
электродах (гл- 29).
Диффузия. Статистическое рассмотрение. Интуитивная картина
механизма диффузии состоит в том, что частицы движутся неболь-
шими скачками и постепенно мигрируют из их первоначального по-
ложения. Мы построим модель диффузии па основе того, что ча-
стицы могут перескакивать на расстояние d за время т. Это озна-
чает, что расстояние, проходимое молекулой за время t, составит
(t^x)d. Это не означает, что частицы будут находиться на таком
расстоянии от первоначального. Направление скачков различно в
каждом случае, и во внимание необходимо принимать общее рас-
стояние диффузии. Мы упростим обсуждение, предположив, что
частицы движутся по прямой линии, а именно по координате х,
но мы не должны забывать, что в реальной системе частица мо-
жет двигаться в трех измерениях. Кроме того, ограничимся рас-
* Интеграл может быть упрощен подстановкой y=xj2(Dt)'^, тогда он пре-
вратится в
Af(x<V) = (MMfnD/)1/2} j* dxe-xl‘wt =
О D
z
Оказывается, что интеграл ^2/jt7'2) e~s 6' dy является стандартной математичес-
6
кой формой, известкой как функция ошибок и записываемой erf z. Стандартные
таблицы этой функции вполне доступны (табл. 24.1), и нужное нам значение
erlp/a)1^ равно приблизительно 0,68.
390
Часть 3. Изменение
смотрением модели, в которой частицы с равной вероятностью мо-
гут двигаться на расстояние d вправо или влево. Это называется
одномерным беспорядочным движением.
Наша задача — установить вероятность нахождения молекулы
па расстоянии х от начала координат за время I. В течение этого
промежутка времени будет сделано i/x скачков: запишем n = t!x.
Многие из этих скачков были сделаны вправо, многие — влево.
Если гщ— число скачков вправо и щ. —число скачков влево, то
можно написать не только общее число скачков п=п^-{-Пъ, но и
общее пройденное расстояние x = nRd—nLd.
Вероятность нахождения на расстоянии х после п скачков дли-
ной d представляет собой вероятность того, что из п скачков nR
произойдет вправо, nL — влево и —n-L = xld.
Каково общее число возможностей для левых или правых скач-
ков? Поскольку каждый скачок может происходить в любом из
двух направлений (влево или вправо), это число равно 2”.
Сколько имеется путей для осуществления из п скачков Hr
скачков вправо? Их такое же число, как и число способов выбора
nR предметов из общего их числа п независимо от последователь-
ности: h!/hr! (н—Лц)! Это можно проверить в случае 4 скачков и
определить число вероятностей осуществления 2 скачков вправо.
В этом случае имеется 24 возможных последовательностей скачков:
LLLL LLLR LLRR LRRR RRRR
LLRL LRLR RLRR
LRLL LRRL RRLR
RLLL RLLR RRRL
RLRL
RRLL
Ясно, что существует 6 путей для осуществления 2 скачков впра-
во и 2 влево, которые рассчитываются из выражения 4!/2!2!=6.
Следовательно, вероятность того, что частица будет вначале ко-
ординат после 4 скачков, равна 6/16. Вероятность того, что части-
ца будет находиться при x = 4d, составляет 1/16, так как, для того
чтобы быть там, все 4 скачка должны быть направлены вправо, и
существует только один путь для осуществления этого.
Возвращаясь теперь к общему случаю, мы видим, что вероят-
ность нахождения частицы на расстоянии х после п скачков, каж-
дый из которых имеет длину dJ равна
р (х) ~n!/nR! (п— nR)I 2"
25. Транспорт ионов и молекулярная диффузия
391
при h = Hr+hl и x/d. — ti^—nL. Это означает, что
пя = ~(n-[-x/d), n—nR=--~(n—x/d).
Следовательно,
P(x)=n! /{[4-(ra+s)]l[4-(tt— s)l!2Л
(25.2.12)
где s—x/d.
Это выражение, по-видимому, нс похоже на вероятностное рас-
пределение Гаусса в отличие от уравнения (25.2.8), и поэтому оно
выглядит так, как будто модель беспорядочного движения, лежа-
щая в основе диффузионного процесса, совсем неправильна. Это,
однако, не так: последнее уравнение идентично распределению Га-
усса, если мы исследуем предел, при котором число скачков ста-
новится очень большим.
Алгебраические преобразования этого уравнения основаны на
приближенной формуле для факториалов больших чисел, впервые
использованной в гл. 20 (разд. 20.1). Когда N — большое число,
возможно использовать приближение Стирлинга
In Nl « (N + 4-1 In N —Л' + 1п (2л)’/г. (25.2.13)
Даже когда А довольно мало, это выражение достаточно точно.
Например, вместо 10! —3,629-10® оно дает 101якЗ,60-Ю6; чем
больше чисел включается в это выражение, тем больше мы мо-
жем быть уверены в правильности получаемого результата. Взяв
логарифмы в уравнении (25.2.12) и затем учитывая, что п велико,
приходим (после многочисленных алгебраических преобразований)
сначала к
In Р In n! —In
гг In 2
и затем к
In Р ял In (2/лп)1-'» —(n-f-s-H I) In (I+s/n) —
— 4-(n —s-l-I) In (I—s/n).
Если допустить, что s/n мало (так что х не должно быть большим
расстоянием от начала координат), то можно использовать при-
ближение In (I+y) и получить
In Г In (2/лп)'/а —8®/2п
392
Часть 3. Изменение
ИЛИ
Р » (2/лп)'/2 ехр (•— s2/2«),
которое уже имеет гауссову форму.
Теперь, заменив s на x/d и п на t/x, получим
Р (х, t) — (2т/лП1;2 ехр (— х2т/2/йа). (25.2.14)
Это выражение точно имеет форму i)/jV0, задаваемую урав- ,
нением (25.2.8) в виде решения уравнения диффузии. (Некоторые
различия обусловлены предположением, что частицы мигрируют ’
в двух направлениях от х = 0 и могут находиться лишь в дискрет-
ных точках, разделенных расстоянием d, вместо предположения о
нахождении частицы в любом месте непрерывной линии.) Поэто-
му можно быть уверенным, что диффузию можно интерпретиро- 3
вать как результат очень большого числа маленьких скачков в |
случайных направлениях. Это также указывает на область, в ко- ’
торой уравнение диффузии не выполняется: мы не должны его
применять к очень короткому времени, за которое частицы успеют
сделать только несколько скачков.
В конце концов можно использовать идентичную форму двух
распределений, чтобы получить еще одно выражение для D. Срав-
нение двух экспонент позволяет написать
%Р/т^4О,
и поэтому мы приходим к соотношению Эйнштейна — Смолухов-
с кого
D— ~ d?/x. (25.2.15)
Пример (вопрос 57). Предположим, что при движении в водном растворе ион
SOI- в каждый момент времени перескакивает на расстояние, примерно равное
его собственному диаметру. Как часто ион меняет положение?
Метод. В последнем примере были найдены коэффициент диффузии, равный
1.06-10-5 см2/с, и эффективный радиус, равный 206 пм. Находим т из уравне-
ния (25.2.15).
Ответ. Из уравнения (25.2.15) имеем
т= d"i'2D = (2-206 Ю-12 м)г/2Х(1,06-10-’ м2/с) = 8,01 • 10-че.
Комментарий. Большой, тяжелый нон SO’- проходит расстояние, равное его
диаметру, за 8-10—11 с- Если представить, что за один скачок ион проходит рас-
стояние, равное диаметру молекулы воды (150 пм), то на это потребуется
4,2.10-и с. ;
Соотношение Эйнштейна — Смолуховского является централь- j
пым связующим звеном между микроскопическими величинами I
размера (d) и скорости (1/т) молекулярного скачка и макроскопи- I
ческими величинами коэффициента диффузии и вязкости [через I
соотношение Стокса — Эйнштейна (25.2.6) ]. Таким образом, об- I
суждение, пройдя полный цикл, возвращается к свойствам газов. I
25. Транспорт ионов и молекулярная диффузия 393
Если d/т интерпретировать как среднюю скорость диффундирую-
щих молекул, а длину скачка d назвать средней длиной свободно-
го пробега и обозначить через X, то уравнение Эйнштейна—Смолу-
ховского выразится как £)=-|*Хс, т. е. будет таким же, как урав-
нение для коэффициента диффузии из кинетической теории газов.
Это показывает, что диффузию идеального газа можно также ин-
терпретировать как беспорядочное движение со Средней длиной
свободного пробега X.
Краткое описание общей ситуации. Эту главу мы начали с рас-
смотрения различных аспектов движения ионов в растворе. Мы
видели, что электропроводность может быть выражена через под-
вижность ионов, а также что любую частицу можно рассматри-
вать как движущуюся под влиянием эффективной силы $, если
химический потенциал частицы изменяется в зависимости от ее
положения; эта сила была выражена как —dp/dx [уравнение
(25.2.1)].
Выражение термодинамической силы приводит к первому зако-
ну диффузии Фика. Мы видели в общих чертах, что, если части-
ца подвергается действию единичной силы, она приобретает ско-
рость дрейфа, равную D/kT. Это выражение приводит к соотноше-
нию Эйнштейна [уравнение (25.2.4) ], связывающему D и подвиж-
ность, и к соотношению Нернста— Эйнштейна [уравнение
(25.2.5), связывающему электропроводность и D (см. ниже под-
разд. 25.2.Л). Введение силы трения Стокса приводит к соотноше-
нию Стокса — Эйнштейна [уравнение (25.2.6) ], связывающему D
и вязкость и применяемому для молекул любого заряда (включая
нулевой). Затем были выведены уравнения для зависящих от вре-
мени диффузионных процессов и найдено основное уравнение диф-
фузии [уравнение (25.2.7)]. Решения этого уравнения можно полу-
чить, если представить диффузионный процесс состоящим из ряда
небольших скачков с длиной d, происходящих с частотой,I/т; ре-
шения уравнения будут одинаковыми, если представить как
коэффициент диффузии D: в этом случае мы получаем соотноше-
ние Эйнштейна—Смолуховского [уравнение (25.2.15)]. На осно-
вании этого соотношения можно интерпретировать вязкость, ион-
ную подвижность, электропроводность н вообше диффузионные
процессы с точки зрения микроскопических, динамических пара-
метров d и т.
25.2 А, Транспортные свойства в растворах
Соотношение Эйнштейна — Смолуховского между скачком на рас-
стояние d и временем скачка т:
D=d!/2T.
394
Часть 3. Изменение
Соотношение Стокса — Эйнштейна между коэффициентом диффу-
зии D и вязкостью раствора -q:
D =kT/6лт]а.
Соотношение Эйнштейна между коэффициентом диффузии и под-
вижностью ионов tu;
D± ~u±kT/ег —ti+RT/zF.
Соотношение Нернста — Эйнштейна между коэффициентом диффу-
зии и электропроводностью ионов К±:
D±.
Приложение. Измерение чисел переноса
Ниже кратко излагаются три метода, используемые для изме-
рения чисел переноса ионов. С помощью этих чисел определяются
электропроводности отдельных ионов и их подвижности.
1. Метод движущейся границы. Пусть MX — интересующая пас
соль. Нальем раствор MX в нижнюю половину узкой вертикальной
трубки. Выберем соль NX, в которой N менее подвижен, чем М;
приготовим раствор NX и нальем его в ту же самую узкую трубку
так, чтобы отчетливо была видна граница раздела с раствором со-
ли MX. Пропустим ток I в течение времени t. Ионы X- <JOyT дви-
гаться по направлению к аноду (вниз), а ионы М+ и N+y— к като-
ду (вверх). Количество катионов, переносящих это количество
(JtlF) электричества, равно t+(ltlz+F), если заряд Zt.. Если их
концентрация составляет с, то изменение объема будет t+(It]z+cF).
Но если поперечное сечение трубки равно 5 и расстояние, на кото-
рое смешается граница раздела, составляет х, то этот объем так-
же равен х5. Поэтому t+(Itiz+cF) — xS, и, исследуя смещение гра-
ницы для некоторого интервала времени, найдем t±.
Пример (вопрос 7). Числа переноса Н+ и SO3- были намерены в эксперименте
с движущейся границей. Прибор состоял из трубки с отверстием 6,40 мм, со-
держащей водный раствор серной кислоты с концентрацией 0,015 моль/дм3. Был
пропущен постоянный ток силой 1,23 мА, и граница сместилась следующим об-
разом:
f, с 40 80 120 160 200
х, мм 0,860 1,722 2,586 3,450 4,309
Найдите н i .
Метод. Используем уравнение, выведенное выше.
Ответ. — (ScF/l) (x/i) —
= tix(3,20- 10-3 м)2х(0,015 моль/дм3)х
X (9,65 10* 1 К.т/мо ль) X (1 /1,23 10-э А) х
X (х/0 = 3,786 104 м-1 с (x/i) =
= 37,86](х, мм)/(/т с)].
25, Транспорт ионов и молекулярная диффузия 395
Составим следующую таблицу:
t, с 40 80 120 160 200
104(л, мм)/(/, с) 215 215,3 215,5 215,6 215,5
Средняя величина: 0,02154 мм/с. Поэтому 1+(Н+) =0,815 и f-(SO|“) = 1—0,815 —
= 0,185.
Комментарий. Эти результаты могут быть 1|С|Ю.чьзованы для определения по-
движности и электропроводности отдельных ионов.
2. Метод Гитторфа. Ячейка делится на три отделения, и через
нее проходит электричество в количестве И. Количество It/z^F
катионов разряжается на катоде, но в катодной области движутся
катионы в количестве t^Hlz^F). Общее изменение количества ка-
тионов вблизи катода составляет —(It[Z+F)-\-t+(ltlz+F) =—(1—
-—tj\ltiZi.F) =—t~(ltjz+F). Поэтому, измеряя изменение состава в
катодном отделении, получим I- — число переноса анионов. Анало-
гично изменение состава анионов у анода равно —t+(it/\z~\F).
3. Измерение э.д.с. Э. д. с. элемента с переносом, имеющего
электрод, обратимый по отношению к анионам, связана с э.д.с.
элемента, в котором протекает та же суммарная реакция, но без
переноса, следующим образом: Et = 2t+E (аргументы те же, что и
использованные в методе Гитторфа). Поэтому соберем два эле-
мента и сравним их э. д. с.
Литература
Shedlovsky Т., Shedlot'sky L., Condiictimetry, in Techniques of chemistry (Weiss-
berger A. and Rossiter B. W., eds.) Vol, IIA, 163, Wiley-Interscience, New
York. 1971.
Spiro M., Determination of transference numbers, in Techniques of chemistry
(Weissbergcr A. and Rossiter B. W., eds.), Vol. IIA, 205, Wiley-Interscience,
New York, 1971,
Madnnes D. A., The principles of electrochemistry, Dover, New York, 1961.
Харнед Г., Оуэн Б. Физическая химия растворов электролитов. Пер. с англ. —
М.: ИЛ, 1952.
Friedman Н. L., Ionic solulion theory, Wiley-Interscience, New York, 1962,
Робинсон P., Стокс P. Растворы электролитов. Пер. с англ. — М.: ИЛ, 1963.
Fuoss К- М., Aecascina F. Electrolytic conduclance, Wiley-Interscience, New
York, 1959.
Fortum. G., Treatise on electrochemistry, Elsevier, Amsterdam, 1952.
Dunlop P. J., Stell B. J., Lane J. E., Experimental methods for studying diffusion
in liquids, gases and solids, in Techniques of chemistry (Wcissbcrger A. and
Rossiter B. W., eds.). Vol. 1V, 205. Wiley-Interscjeiice, New York, 1972.
Jost U7., Diffusion in solids, liquids and gases, Academic Press, New York, 1960.
Задачи
25.1. Электропроводность часто измеряют, сравнивая сопротивления ячеек, запол-
ненных исследуемым раствором и некоторым стандартным раствором, таким,
как водный раствор хлористого ка.чня. Электропроводность воды равна 7,6Х
XI0-* Ом^-см^1 при 25 °C, а электропроводность водного раствора КС1
396
Часть 3. Изменение
(0,1 моль/дм3) равна 1,1639-Ю-2 Ом'-ем'1. Ячейка имеет ...сойротивление
33,21 Ом, когда она заполнена 0,1 моль/дм3 раствором КС1, к 300 Ом, когда за-
полнена 0,1 моль/дм3 раствором уксусной кислоты. Какова мольная электропро-
водность уксусной кислоты при этих концентрации и температуре?
25.2. Обычно при измерениях электропроводности записывают n = ClR, где /?—
измеренное сопротивление образна в ячейке и С — постоянная ячейки, являющая-
ся характеристикой данной ячейки. Поскольку z = ///?.4. С = 1/Д. Как /, так п А
непосредственно определить трудно, и поэтому обычно ячейку калибруют по рас-
творам с известной электропроводностью. В некоторых следующих задачах мы
будем ссылаться на «ячейку», при этом мы всегда будем иметь в виду «ячейку»
из данной задачи, и по атому сейчас найдем ее постоянную. Водный раствор
КС! СИ?20 моль/дм3) имеет мольную электропроводность 138,3 Ом-1-см2-моль-1.
ЛС£щрэтивление ячейки равно 74,58 Ом. Найдите постоянную ячейки С.
у25^ Сопротивления ряда водных растворов NaCl, образованных последователь-
ным разбавлением образца, были измерены в ячейке. Были иайлены следующие
сопротивления-
c(NaCl), моль/дм3
/?, Ом
0,0005 0,001 0,005 0,010 0,020 0,050-
3314 1668 342,1 174,1 89,08 37,14
Проверьте, подчиняется ли мольная электропроводность закону Кольрауша
[уравнение (25.1.3)], и найдите значения мольной электропроводности при еско-
вечном разбавлении. Определите константу Ж. (Теоретическое значение перед-
ней будет рассчитано в задаче 25.23.)
25.4* Используйте значение X (которое должно зависеть только от типа, но не
от конкретного ионного состава соли) из предыдущей задачи и информацию
о том, чю X3 (Na+) =50,1 Ом'1-см2-моль'1 п ?.°(1_)=76,8 Ом-1-см2-моль-1, для
нахождения: а) мольной электропроводности, б) Электропровода оста -и В; сопро-
тивлении ячейки, содержащей 0,0] моль/дм3 раствора Nal.
25,5. Мольные электропроводности бесконечно разбавленных растворов КО,
КМО3 и AgNO3 равны соответственно 149,9, 145,0 п 133,4 Ом-1 -см2-моль-' (при
25°С). Какова мольная электропроводность бесконечно разбавленного раствора
AgC! при этой температуре?
25,6. Было найдено, что электропроводность насыщенного раствора AgCl в воде
при 25°C равна 1,887-10-s-Ом-1 -см-1. Используя выводы предыдущей задачи,
найдите растворимость и произведение растворимости при Этой температуре.
25.7. Мольные электропроводности бесконечно разбавленных водных растворов
ацетата натрия, соляной кислоты и хлорида натрия равны соответственно 91,0,
425.0 и 128,1 Ом_'1-см2-моль'1. Какова мольная электропроводность бесконечно
разбавленного раствора уксусной кислоты?
25.8, Было найдено, что сопротивление 0,020 мощь/дм3 раствора уксусной кисло-
ты в ячейке равно 888 Ом. Какова степень диссоциации кислоты при этой кон-
центрации?
25,9. Чему равен pH кислоты в предыдущей задаче? Рассчитайте его двумя спо-
собами. Сначала пренебрегите- коэффициентами активности. Затем рассчитайте
гие-цз предельного закона Дебая — Хкжкеля [уравнение (J 1.2.11) в т. 1].
ШЦОЛЗакон разведения Оствальда выражается уравнением (25.1.5). Для того
чтобы проверить его применимость и возможности его использования, были из-
мерены сопротивления водных растворов уксусной кислоты при 25 °C в ячейке
н получены следующие результаты:
с, моль/дм3 0,00049 0,00099 0,00)98 0.01581 0,06323 0,2529
И, Ом 6146 4210 2927 1004 497 253
Мольная электропроводность при бесконечном разбавлении была найдена в за-
даче 25.7. Постройте соответствующий график и покажите, что мольная элек-
25. Транспорт ионов и молекулярная диффузия
397
тропроводпость, во величине меньшая, чем примерно 0,01, подчиняется уравне-
нию (25.1.5}. Найдите значения константы диссоциация кислоты Л. и рЛ..
*-25.11. Какова мольная электропроводность, электропроводность и сопротивление
(в ячейке) 0,040 модь/дм3 раствора уксусной кислоты?
25.12. Электропроводность ионов Li+, Na+ н К+ равна соответственно 38,7, 50,1
и 73,5 Ом-1-см2-моль-1. Какова подвижность этих ионов?
с 25.13." Каковы скорости дрейфа ионов в предыдущей задаче, если разность по-
тенциалов 10 В приложена к ячейке диаметром 1 см? Как долго ион будет пе-
редвигаться от одного электрода к другому? В измерениях электропроводности
используют переменный ток. Каково смещение ионов: а) в см и б) в диаметрах
растворителя (примерно 300 пм) при наложении потенциала 1кГц?
25.14. Число переноса иона в смеси определяется как доля общего тока, перено-
симая ионом. Покажите, что это определение приводит к уравнению (25.1.16)
н случае раствора одной 1 : 1-соли. Затем покажите, как отношение двух чисел
переноса trT I" для двух катионов в смеси зависит от их концентраций </, с"
и подвижностей и', и".
Подвижности ионов Н+ и С1" при 25 °C в воде равны соответственно 3,63\
XI0~3 и .7,9] •10-'’ см2/(с-В). Какую долю общего тока переносят протоны в
10-3 моль/дмэ растворе соляной кислоты? Какую долю этого тока будут они
переносить, если к кислоте добавить NaCl так, чтобы его концентрация была
1,0 моль/дм3? Определите, как концентрация и подвижность влияют на транс-
порт тока.
25.ПИ В методе Гитторфа д.тя определения чисел переноса (см. приложение) че-
рез раствор проходит изнестное количество электричества и контролируется из-
менение состава в области, близкой к электроду. На каждый фарадей электри-
честна, проходящего через раствор КС1, число молей КС], переходящих из анод-
ного отделения в катодное, равно Т+. В опыте, проведенном в серебряном куло-
нометре с набором ячеек, содержащих водный 0,04 моль/кг раствор КС], выде-
лилось ]87,2 мг серебра. После прохождения этого количества электричества бы-
ло найдено, что ] 18,42 г раствора из 250 см3 катодного отделения содержало
165,3 мг КС1, в то время как 114,11 г раствора из 250 см3 анодного .отделения
содержало 159,8 мг КС], Проба образца весом 112,62 г из центрального отде-'
ления перед опытом содержала 185 мг К^1, а после опыта аналогичная проба
весом 115,66 г содержала ]91 мг КО. Каковы числа переноса ионов К+ и СХ?
25.12/ Числа переноса могут быть также измерены методом движущейся грани-
1\ьг (см. приложение). В частном эксперименте с KCI прибор состоял из трубки
с отнерстием 4,146 мм и содержал водный раствор КС] с концентрацией
0,02] моль/дм3. Был пропущен постоянный ток силой 1,82 мА, и граница пере-
мещалась следующим образом;
t, с 20 40 60 80 100
х, мм 0,64 1,28 1,92 2,54 3,18
Найдите числа переноса ионов К+ и С1“,
25.18. Из данных предыдущей задачи найдите подвижность ионов К+ ,и С1_
. в водных растворах, а затем их электропроводность.
25.19. Третьим методом измерения чисел переноса и, следовательно, подвижности
иопон янляется измерение э. д, с. Покажите, что э. д. с. .элемента Ag, AgC] |
| НС] (с,) | HCl(cs) | AgCl, Ag с переносом (Ei) связана с э. д. с. элемента без
переноса (Е) следующим образом: E\ = 2t±E. Пользуясь теми же доводами, по-
кажите, что для электродов, обратимых по отношению к катионам, э. д, с. опре-
деляется формулой Ег=2СД
25.20. Для элемента, описанного в предыдущей задаче, при с1 = 0,01 моль/дмэ
И сг = 0,02 моль/дм3 измеренная э. д. с. была равна —29.2 мВ. Каковы числа пе-
реноса п подвижности ионов Hh и С]-?
,25.21? Протон в воде имеет аномальную подвижность, но в жидком аммиаке ве-
дет себя нормально. Для того чтобы исследовать этот вопрос, был использован
398
Часть 3. Изменение
метод движущейся границы Для определения числа переноса иона ЬчН4 в жидком
аммиаке (аналогично нона НзО+ в жидкой воде) при —40 °C {Baldwin. J
Ewans J., Gill J. В.. J. Chem. Soc. (A), 1971. 3389). Постоянный ток силон 5 мА
пропускали 2500 с, и в течение этого времени граница между но Дидом ртутн(П)
и иодидом аммония в аммиачных растворах переместилась на 286.9 мм
в 0,01365 модь/кг растворе и на 92,03 мм в 0,0425а моль/ь'г растворе. Рассчитай-
те число переноса NH4 при этих концентрациях н объясните подвижность про-
тона в жидком аммиаке.
25.22. Электропроводность очень чистой воды равна 5,5-10-$ Ои-’-см< Чему
было бы равно сопротивление образца этой воды в ячейке из задачи 25.2? Ка-
кова константа диссоциации воды? Чему равны значения pKw и pH для чистой
поды? (Подвижности Н+ и ОН- возьмите иэ табл. 25.2)
В задаче 25.3 было найдено экспериментальное значение .Ж" в выражении
Кольрауша для электропроводности NaCl. Рассчитайте значения А и В в урав-
нении Дебая — Хюккеля — Онзагера (25.1.18) и найдите теоретическое значе-
ние Ж
25.24. Был приготовлен разбавленный раствор перманганата калия в воде при
298 К,- Раствор залили в горизонтальную трубку длиной 10 см и сначала наблю-
дали линейный градиент интенсивности от фиолетового раствора слева (где кон-
центрация была 0.10 моль/дм3) до красного справа (где концентрация была
0,05 моль/дм3). Каконы величина ц знак термодинамической силы, действующей
на растворенное нещество: а) вблизи левой плоскости сосуда, б) в середине и
в) вблизи правой плоскости? Определите силу на моль и силу на молекулу в
каждом случае.
25.25. Коэффициент диффузии для сахарозы в воде при 25 °C составляет 5.2Х
х 10-s см!/с. Предположим, что перманганат в предыдущей задаче был заменен
сахарозой, и у нас есть способ (например, какой?) контроля его концентрации.
Чему равна скорость дрейфа сахарозы в трех точках (слева, в середине, сира*
ва)? Как влияет различие в скоростях дрейфа на распределение концентраций?
Нарисуйте эскиз распределения концентраций при различных временах. Чему
равна скорость дрейфа спустя бесконечно большое время?
25.26. Рассчитайте эффективный радиус молекул сахара (сахарозы) в воде при
25 °C на основе его коэффициента диффузии, равного 5,2-10~6 см2/с. и вязкости
Вольц равной 1,0 сП [ 10^ кг/(м-с)].
25.27. Коэффициент диффузии молекулы иода в бензоле равен 2,13-10-5 см?/с.
Сколько времени потребуется молекуле, чтобы перескочить на расстояние одного
молекулярного диаметра (это приблизительно яп.тяется фундаментальной дли-
ной скачка для модели прямолинейного движения).
25.28. Чему равно среднеквадратичное расстояние, преодолеваемое; а) молеку-
лой иода в бензоле и б) молекулой сахарозы в воде при 25 °C за время 1 с?
25.29. Сколько'времени в среднем потребуется молекулам цз предыдущей задачи,
чтобы мигрировать п точку, лежащую на расстоянии; а) 1 мм н б) 1 см от их
начального положения?
25.30, Рассчитайте коэффициенты диффузии и эффективные гидродинамические
радиусы катионов щелочных металлов в воде из их подвижностей при 25 °C
/табл. 25.2).
25.31. Рассчитайте приблизительное число молекул воды, которые увлекаются ка-
тиолами и предыдущей задаче. Ионные радиусы даны в табл. 22.]. z
25,32. Ядерпый магнитный резонанс может быть использован для определения
подппжносги молекул в жидкостях. Серия измерений коэффициентов диффузии
метана в четыреххлористом углероде приводит к следующим значениям: 2.05Х
ХЮ-5 см2/с при 0°С и 2,89-10-5 смг/с при 25°C. Какую информацию вы можете
получить о подвижности метана в четырех хлор истом углероде?
25,33. Подтвердите, что уравнение (25.2.8) является решением уравнения диффу-
зии с точными начальными значениями.
25.34. Концентрированный раствор сахарозы был вылит в цилиндр диаметром
5 см. Возмите его концентрацию, исходя из того, что 10 г сахара растворено в
25. Транспорт ионоя и молекулярная диффузия 399
5 см3 воды. Затем поверх этого слоя раствора осторожно, без встряхивания был
налит 1 дм3 воды. Игнорируйте гравитационные эффекты и обратите внима-
ние только на диффузионные процессы. Найдите концентрацию на расстоянии
5 см от нижнего слоя через а) 10 с и б) 1 год,
25.35. Уравнение диффузии справедливо, когда в интересующий нас промежуток
времени происходит много элементарных стадий; однако расчет беспорядочного
движения позволяет рассматривать распределение как для коротких, так и для
продолжительных времен. Используя уравнение (25.2.) 2), рассчитайте вероят-
ность нахождения в 6 скачках от начала координат (г. е. при x=6d) после
а) 4, б) 6 и в) 12 скачков.
25.Зв. Напишите программу для расчета Р(х) для одномерного беспорядочного
движения и рассчитайте вероятность нахождения при x=fid для n=6,10,14,.... 60.
Сравните численные значения с аналитическими значениями в пределе боль-
шого числа скачков. При каком значении п расхождение не более 0,] %.
25.37. В ccpnii экспериментов по перемещению шариков каучукового латекса ра-
диусом 2,12-10"5 см средние квадраты перемещений после выбранных проме-
жутков времени имели в среднем следующие значения:
t, с 30 60 90 120
Ю»(х2>,см3 88,2 113,5 128 144
Эти результаты впервые были использованы для нахождения значения числа
ЛвогаДро (очень утомительная для глаз процедура, так как нужно определить
такое большое число!). Однако В настоящее время имеются гораздо лучшие пу-
ти определения числа Авогадро, а слелоъзлелъпо, эти данные могут быть ис-
пользованы для нахождения других величин. Найдите эффективную вязкость
воды при температуре этого эксперимента (25°C).
26 Скорости химических реакций
Изучаемые вопросы
После тщательного изучения этой главы вы сможете:
1. Описать, как определяются концентрации реагирующих ве-
ществ (стр. 402).
2, Выразить скорость реакции через концентрации (стр. 404)
и истинную скорость через степень превращения [уравнение
(26.2,1)].
3. Определить константу скорости реакции (стр, 406).
4. Определить порядок реакции (стр. 406).
5. Использовать метод начальных наклонов для определения
порядка реакции (стр. 407).
6. Проинтегрировать зависимость концентрации от времени
для реакций первого [уравнение (26,2.6)] и второго порядка
[уравнение 26.2,9)].
7, Определить порядок из зависимости концентрации от вре-
мени (стр. 409).
8. Описать метод выделения Оствальда и определить реакцию
псевдопервого порядка (стр. 412).
9. Определить полупериод реакции и связать его с порядком
реакции (стр. 413).
10. Различать порядок и молекулярность реакции (стр. 416).
И. Описать зависимость Аррениуса для простых реакций и оп-
ределить их энергию активации и предэкспоненциальный множи-
тель (стр, 417).
12, Объяснить зависимость Аррениуса для случая бимолеку-
лярных газофазных реакций (стр, 418).
13. Решить уравнения для скорости равновесных реакций (стр.
420) и связать константу равновесия с константами скорости про-
стых реакций [уравнение (26.3.4)],
14, Решить уравнения для скорости последовательных реакций
(стр. 422),
15. Описать и подтвердить приближение стационарности и оп-
ределить лимитирующую стадию реакции (стр, 424).
16. Вывести уравнения Для скоростей реакций, включающих
стадию предравновесия (стр. 425).
17, Описать теорию Линдемана для реакций первого порядка
(стр. 428).
26. Скорости химических реакций 401
18. Написать цепную реакцию и объяснить термины: иницииро-
вание, развитие цепи, ингибирование, обрыв цепи и перехватчик
радикалов (стр. 432).
19, Решить уравнение для скорости цепной реакции (стр. 432).
20. Объяснить механизм взрывов (стр. 434) и существование
первого и второго взрывных пределов (стр. 435).
21. Сформулировать законы Гроттгуса— Драпера и Эйнштей-
на— Штарка (стр. 436).
22. Определить и измерить квантовый выход фотохимической
реакции [уравнение (26.4.3)].
23. Вывести и решить уравнения для скорости фотохимической
реакции (стр. 438).
24. Определить термины: катализ, кислотный катализ и основ-
ный катализ — и объяснить способ их действия (стр. 439).
25. Описать флеш-фотолиз и струевые методы для быстрых ре-
акций (стр. 442).
26. Указать основы релаксационных методов, методов темпе-
ратурного скачка и скачка давления для изучения быстрых реак-
ций и связать время релаксации с константами скорости (стр.445).
Введение
В данной главе мы рассмотрим, как происходят химические
реакции с точки зрения изменения их скорости и как скорость
реакции зависит от температуры и концентрации присутствующих
веществ.
Имеются две причины для изучения скоростей реакций. Во-
первых, практически очень важно знать, как быстро реакционная
смесь достигнет равновесного состояния: скорость может зависеть
от ряда контролируемых факторов, таких, как температура, дав-
ление, присутствие катализатора, и в зависимости от наших целей
мы можем проводить реакцию с оптимальной скоростью. Напри-
мер. в промышленном процессе экономичным является проведение
реакций с большими скоростями, но не настолько большими, что-
бы произошел взрыв. Наоборот, в биологических процессах более
подходящим может быть медленное протекание реакции с убыст-
рением или замедлением, когда требуется, чтобы проявился ка-
кой-либо вид активности.
Во-вторых (и это тесно связано с первой причиной), изучение
скоростей реакции помогает исследованию механизмов реакций.
Термин «механизм» в контексте имеет два значения. Одно из них—
разложение химической реакции на ряд элементарных стадий.
Например, мы можем представить, что реакция взаимодействия
водорода и брома Протекает через ряд Стадий, Предполагающих
разрыв Вгг на атомы брома, взаимодействие одного из этих ато-
мов с Н2 и т. д. Установление -всех элементарных стадий приводит
26—242
402
Часть 3. Изменение
к установлению механизма реакции. Другое значение термина ме-
ханизм связано с самими элементарными стадиями и касается их
детальной природы. В этом смысле «механизм» объясняет, что
происходит, когда атом брома Приближается и атакует вращаю-
щуюся, колеблющуюся молекулу водорода.
Первый тип анализа механизма является центральным вопро-
сом классической химической кинетики, и мы остановимся на нем
в этой главе. Второй тип анализа, называемый химической дина-
микой, не был развит до тех пор, пока в результате технического
прогресса для изучения индивидуальных молекулярных столкнове-
ний не был привлечен ранее недоступный метод молекулярных
пучков (см. следующую главу). Линия раздела между химичес-
кой кинетикой и химической динамикой резко не выражена: на
основе кинетического анализа строятся грубые модели индивиду-
альных стадий реакций, и мы увидим это в данной главе.
26.1. Эмпирическая химическая кинетика
Основными данными химической кинетики являются зависи-
мости концентраций реагирующих веществ и продуктов реакции
от времени. Метод, выбираемый для контроля концентраций, за-
висит от природы участвующих в реакции веществ и от скорости
реакции.
1Многие реакции заканчиваются (например, достигают термо-
динамического равновесия) через несколько минут или часов и
могут контролироваться классическими методами. Чаще всего вы-
бирают один из следующих методов:
1. Изменение давления. Реакция в газовой фазе может идти с
изменением давления, и тогда ее протекание можно контролиро-
вать, регистрируя изменение давления во времени. Примером яв-
ляется разложение оксида азота (V) NaOg:
2N2OS (газ) ——* 4ЬЮ2 (газ) -[- О2 (газ).
На каждый разложенный моль NaOs образуется 5/2 молей газооб-
разных продуктов, и, таким образом, в ходе реакции общее дав-
ление в системе увеличивается. Этот метод неприменим для реак-
ций, которые протекают без изменения давления, и для реакций
в растворе.
2. Спектроскопия. Спектроскопический анализ смеси может
применяться в том случае, когда не происходит изменения давле-
ния. Например, реакция
Н2 (газ) Вга (газ) -*- 2НВг (газ)
может изучаться по контролю интенсивности поглощения видимо-
го света бромом.
26. Скорости химических реакций 403
3. Поляриметрия. Когда оптическая активность смеси изменя-
ется в ходе реакции, она может контролироваться измерением уг-
ла оптического вращения. Этот метод является исторически важ-
ным, поскольку его применение к гидролизу сахарозы было пер-
вым значительным исследованием скорости реакций (Вильгельми,
1850).
4. Электрохимические методы. Когда во время протекания ре-
акции изменяется число или природа ионов, присутствующих в рас-
творе, ход реакции может контролироваться измерением электро-
проводности раствора. Очень важный класс реакций представля-
ют реакции па электродах, и мы рассмотрим их в гл. 29.
5. Другие методы. Другие методы определения состава вклю-
чают масс-спектрометрию и хроматографию. Для применения
этих методов небольшое количество реакционной смеси извлека-
ется из реакционной системы в определенные промежутки време-
ни от начала реакции и затем анализируется.
Имеются три способа Применения этих аналитических методов;
1. Настоящий анализ во времени. В этом методе состав систе-
мы анализируется в ходе реакции.
2. Гашение. В этом методе реакцию прекращают спустя неко-
торое время после ее начала, и затем состав анализируют Любым
подходящим методом. Гашение обычно достигается внезапным сни-
жением температуры, но метод подходит только для медленных
реакций, поскольку в этом случае за время охлаждения смеси
реакция пройдет в очень малой степени.
3. Струевой метод. В этом методе растворы реагирующих ве-
ществ смешиваются при поступлении их в камеру (рис. 26.1). Ре-
акция происходит в то время, когда полностью смешанные рас-
творы проходят через выводную трубку, и наблюдение состава в
различных местах трубки (например, спектроскопически) эквива-
лентно наблюдению реакционной смеси в различное время после
смешивания. Этим методом могут изучаться реакции, которые за-
канчиваются за несколько миллисекунд, но основным недостатком
этого метода является то, что необходимы очень большие объемы
растворов. Метод был улучшен, и его модификация — метод оста-
новленной струи (разд. 26.5)—нашла широкое применение.
Было найдено, что скорости химических реакций очень силь-
но зависят от температуры ц реакции подчиняются закону скоро-
стей Аррениуса, согласно которому скорость реакции пропорцио-
нальна ехр(—EJRT), где £а—энергия активации реакции- Вы-
вод из этого наблюдения для экспериментальных исследований за-
ключается в том, что температура реакционной смеси должна под-
держиваться постоянной по возможности в продолжение всей
реакции, в противном случае значение наблюдаемой скорости ре-
акции будет не имеющим никакого смысла средним значением ско-
ростей при различных температурах. Это необходимое условие
26
404
Часть 3. Изменение
Рис. 26.1. Аппарат, используемый в струевом методе.
накладывает серьезные требования на экспериментальную уста-
новку. Например, газофазные реакции часто осуществляют в со-
суде, находящемся в контакте с массивным металлическим бло-
ком, а жидкофазные реакции, включая струевые реакции, должны
осуществляться в эффективном термостате.
Основной вывод из этих опытов заключается в том, что скоро-
сти химических реакций зависят от состава реакционной смеси и
в большинстве случаев экспоненциально зависят от температуры.
В последующих разделах мы остановимся на этом более подробно.
26.2. Скорости реакций
Предположим, что интересующая нас реакция имеет вид
А -у в —Р (продукты)
и концентрации веществ А, В и Р равны [А], [В] и [Р] соответ-
ственно. Скорость реакции может быть выражена как скорость
изменения концентрации любого из этих веществ. Таким образом,
скорость образования продукта Р равна d[P]/d/, а скорость исчез-
новения А /[А]/Л. В данном случае
d jPj/df — dIAj/Л = — d(Bl/Л.
так как на каждую исчезнувшую молекулу А должна исчезнуть
и одна молекула В и при этом образуется одна молекула Р. Лю-
26. Скорости химических реакций 405
бая из этих производных может служить для определения скоро-
сти реакции; необходимо только следить за правильностью знаков.
Для реакции,в которой
А4-2В ---> 3C + D,
положение менее ясное. В этом случае концентрация В изменяет-
ся в два раза быстрее, чем концентрация А, а концентрация С
увеличивается в три раза быстрее, чем уменьшается концентра-
ция А. Для скорости реакции можно выбрать любую производ-
ную по времени, и они будут связаны между собой следующим об-
разом:
— d [А]/Л =----d [BJ/rft = 4- d [C]/d/ ^d [D]/d/.
Трудность заключается в том, что мы не можем просто говорить о
«скорости» реакции, не указывая точно, что под этим подразуме-
ваем: необходимо сообщить, чтб именно мы выбрали для выра-
жения скорости:
скорость =d [\]/dt или скорость=d [С]idt,
и обязательно написать химическое уравнение реакции. В этом
случае не будет никакой неопределенности.
Полное и краткое определение скорости конкретной реакции
может быть дано через степень превращения £ реакции. Эта вели-
чина была введена при термодинамическом обсуждении равнове-
сия (см. т. 1, разд. 9.1), где мы видели, что для реакции АЧ-2В:—>-
—>3C + D —d£ представляет количество прореагировавшего ве-
щества А, количество прореагировавшего В равно —2d£ и т. д.
Истинная скорость определяется как
истинная скорость =di/dt, (26.2.1)
т. е. как скорость изменения степени превращения. Это определе-
ние упрощается до элементарного выражения для скорости ре-
акции при введении соответствующих значений стехиометрических
коэффициентов*.
* Для точной интерпретации истинной скорости требуется, чтобы реакция
A 2В ----► ЗС 4- D
была записана в форме
0 = —Л —2B4-3C4-D
пли в общем виде
0=vaA-J-vbB--[-vcC4-vdD-|- . , . ,
где коэффициенты vq будут положительными для продуктов и отрицательными
для реагирующих веществ (например, va=—1. vb=—2, vc=3, vD— 1). Тогда
изменение количества А равняется VacZL и скорость его изменения будет
\'лд^д[ или (скорость изменения [А])—у.ч (истинная скорость).
406
Часть 3. Изменение
Законы скоростей и константы скорости. Измеряемая скорость ре-
акции часто бывает пропорциональной концентрации реагирую-
щих веществ в некоторой степени. Например, может быть найде-
но, что скорость реакции А+2В—*-3C-)-D зависит от концентра-
ции реагирующих веществ следующим образом:
скорость = — d [А]/Л = й[А] {В], (26.2.2)
где А —некоторая константа, нс зависящая от концентрации, но
обычно зависящая от температуры; ее называют коэффициентом
скорости или константой скорости. Скорость другой реакции с той
же стехиометрией (т. е. также выражаемой уравнением А+2В—>.
—*-3C-)-D) может подчиняться следующим уравнениям:
скорость = — d[A]/d/ — k‘ [А] (В[2 (26.2.3)
или
скорость = — d [A]/ttt=k" [А][В]. (26.2.4)
Установление закона скорости необходимо по трем причинам.
Первой причиной является возможность предсказания скорости
реакции при данном составе смеси и экспериментальном значении
константы скорости. Вторая причина—это то, что рассмотрение
закона скорости предполагает установление механизма реакции и
Приемлемый механизм должен соответствовать наблюдаемому за-
кону скорости. Третья причина связана с классификацией реакций
по различным «порядкам». Порядок реакции — это степень, в ко-
торой концентрация компонента входит в уравнение скорости, а
общий порядок — это сумма показателей степеней всех концент-
раций. Это означает, что реакция, подчиняющаяся закону скоро-
сти, выражаемому уравнением (26.2.2), является реакцией перво-
го порядка по веществу А, первого порядка по В, с общим вторым
порядком. Реакция с законом скорости, выраженным уравнением
(26.2.3), также является реакцией первого порядка во А, но вто-
рого порядка по В и общего третьего порядка. Реакция не обяза-
тельно имеет целые порядки; закон скорости, представленный
уравнением (26.2.4), соответствует половинному порядку реакции
по А и общему порядку, равному трем с половиной.
В большинстве случаев законы скорости отражают стехиомет-
рию реакции, но важно отметить, что это не всегда так: иногда
вакон скорости, полученный экспериментально, не может быть вы-
веден просто из уравнения реакции. Например, изомеризация цик-
лопропана в пропилен
является реакцией первого порядка с законом скорости
скорость[пропилен1/Л=^1 {циклопропан],
26. Скорости химических реакций 407
но разложение оксида азога(У), соогветствующее уравнению
2N2Os (газ) ->- 4NO2 (газ) 4- О2 (газ).
следует закону скорости
скорость — — d [N2O6]/d/ =kL [N2O6]
и поэтому также является реакцией первого порядка. Окисление
оксида азота(П) следует уравнению
2NO (газ) + О2 (газ) ---* 2NO2 (газ),
и было найдено, что по закону скорости
скорость —d [NO2J/d/=fe} [NO]2 [О2]
является реакцией третьего порядка. В отличие от этого катали-
тическое окисление SO2 в избытке кислорода имеет ту же самую
стехиометрию
pi
25Ог (газ) 4- О3 (газ) -ь 2SOs (газ),
по закон скорости совсем Другой:
d [SO3]/rf/=k [SO2] [SO,,]-1/».
Эти замечания указывают на три проблемы. Мы должны знать,
как получается закон скорости и значения констант скоростей из
кинетических данных. Затем мы должны попытаться найти меха-
низм, соответствующий данному закону скорости. И наконец, мы
должны попытаться рассчитать значения констант скоростей и
объяснить их температурную зависимость.
Определение закона скорости: интегральные уравнения. Имеется
несколько методов для определения закона скорости из кинетиче-
ских данных, представляющих зависимости концентрации от вре-
мени. Прямым методом является измерение начальных наклонов.
Например, предположим, что в реакции между А и В закон ско-
рости имеет вид
— d [Aj/d/ =А> [ А]« [В]&,
где а и Ъ — порядки реакции, которые нужно измерить. В начале
реакции, когда концентрации А и В равны [А]о и [В]о, скорость
-(d[A]/d()Ha4=A[A]S[B[*,
поэтому
lg { -(d [A[/d/)„a4j =lgA-!-alg [AIo + big [B[o,
и из наклонов графиков зависимости начальных скоростей от раз-
личных начальных концентраций А и В определяются порядки
а и b, a IgA представляет собой отрезок, отсекаемый на оси орди-
нат. Это иллюстрируется следующим примером.
408
Часть 3. Изменение
Пример (вопрос 5). Исследована реакция рекомбинации атомов иода в газовой
фазе в присутствии аргона, и найдены порядки реакции методом наклонов. При
различных концентрациях атомов I и Аг были найдены следующие скорости об-
разования молекуляря&го иода по реакции I+1+Аг—-^12+Аг:
[1]0, моль/дм1 1,0-10-’ 2,0-10'3 4,010-’ 5,0-10-3
(JlD/diJo, моль/дм1-с) з] 8,7-10-4 3,48 АО-1 1,39-10-“ 3,13-10-*
б) 4,35-10-3 1,74- IO-* 6,96-10-2 1,57-10-1
в) 8,69-10-3 3,47-10-2 I,38-10“' 3,13-ю-1
При згом концентрации аргона равны: а) 1,0-10“3 моль/дм’, б) 5,0-10“3 моль/дм3
и в) 10,0.10-3 моль/дм3 соответственно. Найдите порядки реакции по концентра-
циям иода и атомов аргона и константу скорости.
Метод. Постройте графики зависимости 1 gid[I2]/dt)□ от lg[I]o для данной кон-
центрации [Аг] и от [Аг]о для данной концентрации [1]о. Отрезки, отсекаемые
от оси ординат, дают 1g А, наклоны определяют порядки.
Ответ. Наклоны равны 2 (для графика зависимости от lg[I]o) и 1 (для графика
зависимости от [Аг]о). Поэтому реакция будет иметь второй порядок по I, пер-
вый по Аг и общий третий порядок. Отрезок, отсекаемый на оси ординат, равен
9,94, поэтому й3=8,7-109 дм6/(моль2-с).
Комментарий. Реакция третьего порядка соответствует столкновению трех частиц.
Третья частица поглощает некоторое количество энергии сталкивающихся ато-
мов I и, таким образом, предотвращает диссоциацию только что образовавшихся
молекул пода.
К сожалению, начальный наклон не дает возможности опреде-
лить полный закон скорости, поскольку в сложной реакции про-
дукты реакции сами по себе могут быть Промежуточными веще-
ствами в других стадиях. Например, в случае синтеза НВг в ис-
тинный закон скорости входит концентрация НВг, но сначала НВг
отсутствует, и изучение первоначальной скорости в зависимости
от количества водорода и брома дало бы неполную информацию
и ввело бы в заблуждение относительно полного механизма реак-
ции. Поэтому закон скорости должен соответствовать данным, по-
лученным в ходе всего проведения реакции. Поскольку законы
скорости являются дифференциальными уравнениями, выражаю-
щими скорость через изменения концентраций на любой стадии
реакции, интегрирование дает выражение для текущих концентра-
ций в любое данное время. Разные законы скорости дают разные
зависимости концентраций от времени, и поэтому истинный закон
скорости может быть найден путем подгонки различных расчетных
концентраций наблюдаемым.
Простейший закон скорости получается для реакции первого
порядка:
—d[k\idt=kt [Al
(26.2.5)
Его можно преобразовать в выражение
— (l/[Al)dA = fe1d/I
26. Скорости химических реакций 409
которое может быть непосредственно проинтегрировано. Если при
/ = 0 концентрация А равна [А]о, а при каком-то более позднем
времени t она равна [A] t, то получим
[А]( . t
j (~l/[A])d[A]^jM
R]o О
ИЛИ
-(ln[Ah-ln[A]0)=^,
Это выражение может быть преобразовано в два полезных урав-
нения:
1п([А](/[А]0) = -/М (26.2.6а)
и
[А]( = [А]оехр( —М- (26.2.66)
Второе из этих двух уравнений показывает, что концентрация А
падает со временем экспоненциально со скоростью, определяемой
Й1. Первое из этих двух уравнений показывает, что именно надо
отложить на графике для подтверждения справедливости уравне-
ния первого порядка и как определить значение ki.‘ если отложить
на осях In [А];/[А] 0) и время, то наклон прямой линии даст—kj.
Это иллюстрируется приведенным ниже примером, а некоторые
экспериментальные значения kj даны в табл. 2б. 1.
Таблица 26.1 Кинетические данные для некоторых реакций первого порядка
Реакция Фаза i, ’С £[. с—1
N2Os > NO3 -г NOg Газ3 25 3,14-10—ь 6,! ч 55 1,42-10-3 8,2 мин
С„Н6 > 2СН3 Циклопропан г- пропен N2OS —> NOs-(-NOs NsOa > NO2+ NO, Газ3 700 5,46-0}-* 21,2 мни Газ3 500 6,71-10-* 17,2 мин Раствор в HNOj 55 9,27-10-5 125 мин Раствор в Вг, 55 2,08-10-3 333 с
3При высоком давлении.
Пример (вопрос 7). Парциальное давление ааометана CHsNsCHj при 600 К за-
висит от времени; результаты даны ниже. Подтвердите, что разложение
410
Часть 3. Изменение
CH3N2Clh—*-CH3CH3+Ns является реакцией первого порядка по азометану и
найдите значение констант скоростей при данной температуре.
t, с 0 1000 2000 3000 4000
p(CH3N2CH3), мм рт. ст. 8,2’10-» 5,72-Ю 2 3,99-10-» 2,78’10-» 1,94-10-»
Метод. Постройте график аавнсимости ln(p/pfl) от t. Для реакции первого по-
рядка получите прямую линию, ее наклон равен —kj.
Ответ. Данные представлены на рис. 26.2. График дает прямую линию с накло-
ном —3.6-10-1, и, таким образом, fe[=3,6-10-4 с. Коэффициент корреляции гра-
фика, рассчитанный по методу наименьших квадратов, равен 1,000.
Комментарий. Газофазные реакции первого порядка имеют некоторые специфи-
ческие черты, рассмотренные ниже (см. подразд. «Реакции первого порядка»
и разд. 26.3).
В случае реакций второго порядка необходимо различать зако-
лы скорости вида
— d[A]/d/=A2[A]a
и
-d[A]/<tt=MA][B],
В первом случае интегрирование сразу дает
lA], t
J (- 1/{А]й) (Aj==
IAIo О
Следовательно,
O/IAJ,) — (I/[A]0)=feaZ,
(26.2.7а)
(26.2,76)
(26.2.8а)
что можно преобразовать в
(26.2.86)
Первое из этих уравнений пока-
зывает, что именно необходимо
отложить на графике для провер-
ки выполнения уравнения второ-
го порядка: должна получиться
прямая линия, если отложить
l/[A]f против t, а ее наклон даст
константу скорости k2. Второе из
этих уравнений позволяет пред-
сказать концентрацию А в лю-
Рис. 26,2. Определение константы скоро-
сти для реакции первого порядка.
26. Скорости химических реакций
411
бое данное время от начала реакции, если известна константа
скорости и первоначальная концентрация А.
Второй тип реакции второго порядка описывается уравнением
(26.2.76), которое можно проинтегрировать в конкретном случае,
если между концентрациями А и В имеется определенное соотно-
шение. Это зависит от стехиометрии реакции; рассмотрим для про-
стоты случай А + В—(-продукты. Если начальные концентрации А
и В равны [А]о и [В]о, то в какой-то момент времени, когда кон-
центрация А уменьшится до [А] 0 — х, концентрация В будет
[В] —х, так как исчезновение каждой молекулы А влечет за собой
исчезновение одной молекулы В. Это означает, что
— [A\/dt — [А] [В] (|А]0 —х) (!В]0 —х).
Но поскольку
d [A]/dt=d ([A]0—x)/d/-^~ dx/dt
(потому что [А]о постоянна), уравнение скорости может быть за-
писано следующим образом:
dx!dt=([А]о — х) ([В(о — х)
и решено для х как функции t.
Интегрирование последнего уравнения может быть проведено
таким образом: приняв, что при ^ = 0 х = 0, получим
г dx
J ([Ab - х) ([В|„ - х)
о
г/ и 1 1
J ( [А]о - [BJ0 J [(IА]о - х) ([В]о - х)
о
[А]о
[A lo xi
[вь
[В1о - xt
Это выражение может быть упрощено, если объединить два ло-
гарифма и учесть, что [А];=[А]о—Xt и [В](= [В]о—х<. Тогда
<26-2-9>
Для подтверждения выполнения уравнения второго порядка в хо-
де протекания реакции график зависимости правой части этого
выражения от времени должен представлять прямую линию.
В этом случае константа скорости может быть определена из на-
клона этой прямой. Некоторые экспериментальные значения kz
представлены в табл. 26.2.
412
Часть 3. Изменение
Таблица 26.2 Кинетические данные для некоторых реакций второго порядка
Рея кция Фаза t, *с *2. ДмЗДнпль-с)
2NOBr -—> 2NO-U Вг2 Газ 10 0,80
2NO2 > 2NO -РО2 300 0,54
Н*+1в г 2HI 400 2,42-10~2
Da + HCl >- DH+DCI 600 1,4110-1
i +I - ► i2 23 7.10s
i+r > i2 Гексан 50 18I№
CH3C1 +CH3O' Метанол 20 2,29’IO-6
CH3Br СНдО^ - у 20 9,23-10-5
H++ OH- * H2O Вода 25 1,5.100
Только что описанные технические приемы могут быть распрост-
ранены на другие законы скорости, и некоторые результаты пред-
ставлены в подразд. 26.2.А. Проинтегрированные выражения ско-
ростей быстро усложняются, но часто их можно упростить, исполь-
зуя метод выделения. Оствальда. Он основан на приближении о
небольшом изменении концентрации реагирующего вещества в
ходе реакции, если оно присутствует в значительном избытке.
Если скорость выражается законом
-d [А]/^=^[А] [В]
и В присутствует в значительном избытке, то концентрация [В]
будет практически постоянной, т. е. [В] «|[В]0, и может быть
включена в константу скорости, давая новую константу k[ =
~/?2[В]0. Тогда закон скорости упрощается до
--d [A]/dt ^k[[A].
Это закон скорости псевдопервого порядка. Его интегрированная
форма уже была выведена, и константа скорости псевдопервого
порядка может быть найдена из приведенного ранее уравнения
(26.2.6). Таким же образом закон скорости третьего порядка в
виде *
d [Р]/^=^[А]3 [BJ
может быть превращен в закон псевдопервого порядка, если взять
компонент А в значительном и фактически постоянном избытке,
так чтобы скорость подчинялась закону
diPV^=fe;iBi,
25. Скорости химических реакций 413
или в реакцию псевдовторого порядка, если будет в избытке ком-
понент В, Этот технический прием выделения по очереди вклада
различных компонентов может оказать большую помощь в уста-
новлении механизма сложных реакций.
Полупериод. Простым индикатором скорости химической реакции
является время, за которое концентрация реагирующего вещества
уменьшается наполовину первоначального значения; это так на-
зываемый полупериод реакции, который обозначается Полу-
период зависит от начальной концентрации характерным образом
для реакций различных порядков, и поэтому его измерение дает
указание на порядок.
В случае реакции первого порядка время, за которое концент-
рация А падает от [А]о до-^-[А]о, может быть рассчитано из
уравнения (26,2.6а)
—Vi/a=ln (4- [А ]0/[А]0) =1п-|“ — — 1п 2
или
(1/fei) In 2, (26.2.10)
Это уравнение дает два преимущества. Во-первых, ti/2 легко
может быть определен из графика зависимости [A]t от времени,
и, таким образом, можно быстро измерить константу скорости ре-
акции первого порядка. Во-вторых, Л/2 для реакции первого по-
рядка нс зависит от концентрации. Это означает, что. если кон-
центрация на некоторой произвольной стадии реакции равна [А]',
то за время (1/fei) In 2 концентрация уменьшается до4[А]'. Неко-
торые значения полупериодов представлены в табл. 26,1.
Для реакций более высоких порядков независимость полупе-
риода от концентрации отсутствует. Например, уравнение (26,2.8а)
может быть использовано для предсказания полупернода реакции
второго порядка. Заменяя /=6/а и [А]( = -у [А]о, получим
— 1 /(&2 [А]о). (26.2.11)
Ясно, что значение h/2 зависит от начальной концентрации: чем
выше начальная концентрация, тем меньше надо времени, чтобы
она уменьшилась наполовину. Это заключение указывает на дру-
гой путь проверки выполнения уравнения второго порядка: если
определить hr2 для серии различных первоначальных концентра-
ций, то график зависимости A/s от 1/[А]о должен дать прямую ли-
нию. Если получится прямая, то константа скорости реакции вто-
рого порядка может быть определена из наклона прямой.
414
Часть 3. Изменение
Пример (вопрос 9). Результаты щелочного гидролиза этнлнитробензоата при
различных временах представлены ниже. Определите порядок реакции методом
полупериода и найдите константу скорости.
/, с 0 100 200 300 400 500 600 700 800
100[А], моль/дм® 5,00 3,55 2,75 2,25 1,85 1,60 1,48 1,40 1,38
Метод. Строим график зависимости [А] от t. Выбираем ряд времен, которые рас-
сматриваем как «начальные» времена и отмечаем соответствующие «начальные»
концентрации. Затем находим время, при котором концентрации уменьшатся на-
половину от их «начальных» значений. Если 6/2 не зависит от [Л]о, то порядок
реакции первый, если не так, то попытаемся построить график зависимости 6/2
от 1/[А]о [уравнение (26.2.11)].
Ответ. Данные представлены на графике на рис. 26.3, а. Выбраны первоначаль-
ные времена а, 6,, ,. , f.
Получаем следующие данные:
е /
0,030 0,0275
400 450
Ясно, что П/а зависит от [А]о.
Поэтому строим график зави-
симости Ц/а от 1/[А]о, как на
рис. 26.3, б. Он представляет
собой прямую линию, и, следо-
вательно, данная реакция имеет
второй порядок. Наклон прямой
равен [2 5, и Л2= (1/12,5)
моль-' дм?-с_,=8,0-10-2
моль-1 -дм3 с-1 (коэффициент
корреляции по методу наимень-
ших квадратов равен 0,9973).
Комментарий. Если график не
дает прямую линию, надо по-
строить график зависимости
Л/г°т VfAl®.
В случае общего п-го
порядка реакции значе-
ние полупериода зависит
от первоначальной кон-
центрации как 1/[A]J-’.
Общие выражения пред-
ставлены в подразд.
26.2.А.
Рис. 26.3. Определение констан-
ты скорости для реакции вто-
рого порядка,
а—по кинетической кривой; б—
по зависимости полупериода от кон*
центряции.
I
26.2.А. Проинтегрированные законы скорости
Порядок Реакция Закон скорости
0 А > Р dx/dt = kfj
1 А >- Р dx/dt — fej [А]
2 А Р dx/dt = AJAp
А + В >- Р dx/dt = £S[A] [В]
А + 2В ► Р dx/dt = ks[ A] [B]
А Р с аутокатализом dx/dt = 6И[А[ [P]
3 А + 2В > Р dx/dt -^[A] [Bp
п>2 А — ► Р dx/dt = А’П[А]Л
Проинтегрированная форма
'1/2
V — х для х [А]о
А/ = In
’ [А]о |
. [Д1 о — x]J
[А]о/2^о
(1п 2)/^!
kJ= —------—------
[А]а([А]0 — х)
t = Г 1 1 1п HAMtBlo-x) 1
1 [В]о— [А]о J 1 ([А]о —X) [В]о /
, - I 1 I In / »41°([ В]0-2х) 1
t-UBel-2fAloJlnt ([А[„ —х)[В]0 J
w = [_____!___1 1П _tA[o(tp]0 + x> )
I [АЬ- х ) ([А]о —х) [Р]о J
[А[д — [В]о | I
[А[01В]о (2[А]0 + [В]„) + 12[А]0 - [В]о J '
х ( [А1„([В]о-2х)|
I ([А]0-х) [B]J
_ 1 If 1 \п~~’ ( Д \ fl~1
«— 1 1\ [АЬ — х / V [А]» /
2"-i — 1
(п — 1) ЫА^-1
416
Часть 3. Изменение
Краткое содержание общего подхода. Кинетическое исследование
реакции позволяет установить закон скорости и в соответствии с
ним измерить константу скорости, часто при нескольких темпера-
турах. Первой стадией является идентификация всех продуктов
реакции и исследование образования в ходе реакции промежуточ-
ных веществ. Затем для выяснения роли каждого компонента (по
очереди) и определения порядка реакции по каждому из них мож-
но использовать метод выделения. Методы начальных наклонов и
выделения дают указания на порядок, и первой проверкой явля-
ется установление соответствующей концентрационной зависимо-
сти полупериода. Если эти два измерения согласуются друг с дру-
гом, то для проверки найденного порядка реакции и подтвержде-
ния его сохранения в ходе реакции и нахождения значения кон-
станты скорости может быть использован график зависимости
концентрации от времени, соответствующий порядку реакции [т. е.
графики, выражаемые уравнениями (26.2.6а) и (26.2.8а)].
26.3. Объяснение законов скоростей
Теперь мы перейдем ко второй части анализа кинетических
данных и попытаемся объяснить наблюдаемые законы скоростей
специфическими механизмами реакций. Большинство реакций мо-
жет быть разбито на ряд стадий, которые являются или мономо-
пекулярными реакциями, в которых отдельная молекула распада-
“ ется на части или переходит в новую конфигурацию, или бимоле-
кулярными реакциями, в которых пара молекул сталкивается и
обменивается энергией, атомами пли группами атомов. Мы уви-
дим, что реакцию некоторого установленного порядка обычно мож-
но представить несколькими мономолекулярными или бимолеку-
лярными стадиями, и, таким образом, в общем случае порядок
реакции будет отличаться от молекулярности отдельных стадий.
Порядок является эмпирической величиной и получается из зако-
на скорости; молекулярность характеризует предложенный меха-
низм.
Простые реакции. Хотя порядок реакции не обязательно совпада-
ет с ее молекулярностью, большой круг реакций второго порядка
может быть объяснен на основе бимолекулярных столкновений.
Мы назовем эти реакции простыми. По таким одностадийным ме-
ханизмам бимолекулярных столкновений идет большинство про-
стых гомогенных реакций, димеризация алкенов и диенов и такие
реакции, как
СГ131 + CHjCHjONa -> СИ3ССН„СН3 + N*al
в щелочном растворе. Эти реакции записываются в виде
А + В--->- продукты,
26. Скорости химических реакций. 417
и их скорости зависят от того, насколько часто частицы А и В
сталкиваются друг с Другом. Эта частота столкновений пропор-
циональна концентрации как А, так и В, и, таким образом, ско-
рость реакции пропорциональна [А][В]. Это значит, что закон
скорости имеет вид
— d[A\/dt = k2 [Aj [В],
ц поэтому кинетика соответствует второму порядку.
Интерпретация закона скорости очень сложна, частично из-за
того, что выражение скорости реакции второго порядка может
также получаться из более сложной реакционной схемы, чем про-
стые бимолекулярные столкновения. В дальнейшем мы встретим-
ся с такими примерами. Сейчас мы подчеркнем, что если реак-
ция является простым процессом бимолекулярных столкновений, то
ее кинетика должна быть второго порядка, но если кинетика бу-
дет второго порядка, то реакция может быть и более сложной.
Истинный механизм может быть определен только с помощью при-
менения к системе детальных чувствительных методов и исследо-
ванием как побочных, так и промежуточных продуктов, появляю-
щихся в ходе реакции. Например, таким путем было показано, что
реакция
Н2+ Г2^ 2HI
протекает по сложному механизму, хотя в течение многих лет, не
имея достаточно точных доказательств, эту реакцию приводили в
качестве прекрасного примера простой бимолекулярной реакции,
в которой атомы обмениваются друг с другом при столкновении.
Доказательства механизма в химической кинетике меньше всего
напоминают математические доказательства и больше похожи на
доказательства в кодексе законов.
Температурная зависимость скоростей простых реакций. Скорос-
ти большинства реакций с повышением температуры увеличивают-
ся, Очень удобно запомнить следующее правило: скорость прибли-
зительно удваивается на каждые 10 К увеличения температуры.
Имеются исключения, но большинство простых реакций подчиня-
ется этому правилу. Например, для гидролиза метилацетата кон-
станта скорости при 35°C в 1,82 раза больше константы скорости
при 25 °C, а для гидролиза сахарозы При том же увеличении тем-
пературы скорость изменяется в 4,13 раза. Температурная зависи-
мость константы скорости выражается уравнением, предложенным
Аррениусом:
k2~A ехр(— EJRT). (26.3,1)
Два параметра этого выражения — предэкспоненциальный мно-
житель А (который нс зависит или почти не зависит от темпера-
27—242
418 ____________________Часть 3. Изменение________________________
туры) и энергия активации Еа — могут быть определены из графи-
ка зависимости In k2 от 1/Г:
Infe2—-1пЛ — EjRT, (26,3,2)
где Отрезок, отсекаемый па оси ординат, равен А, а наклон —EJR.
Это так называемый аррениусовский график, и считается, что ре-
акции, дающие в этих координатах прямую линию, проявляют
аррениусовское поведение. Найденная энергия активации может
быть использована для предсказания того, как изменяется скорость
реакции с температурой.
Пример (вопрос II). Скорость реакции разложения ацетальдегида (этаналя),
имеющего второй кинетический порядок, была измерена в интервале температур
700—850 К,; константы скорости приведены ниже. Найдите энергию активации
и предэкспонецциальный фактор.
Т, К 700 730 760 790 810 840 910 1000
моль-1.дм3-С-1 0,011 0,035 0,Ю5 0,343 0,789 2,17 20,0 145
Метод. Строим график зависимости in (k в единицах мо-чь-1 дм3-с^1) от 1/Г
(Т в единицах К) [уравнение (26.3.2)] и анализируем его, как описано выц!е.
Ответ. (См. рис. 26.4.) Отрезок, отсекаемый на оси ординат, равен 26,95 и по-
этому А =5,06-10'1. Наклон составляет _2.207-I04, и, таким образом,£а= (2,207Х
ХЮ4 К)Х[8,314 Дж/(К-моль)] =184 кДж/моль. (Коэффициент корреляции по
методу наименьших квадратов равен 0,9986.)
Комментарий. Это веевма типичная реакция, для которой повышение температу-
ры на 10°С приводит к удвоению скорости (фактически, при изменении темпера-
туры от 700 до 710 К скорость увеличивается в 1,6 раза). Может быть, более
удобно при построении графика использовать lg k2 вместо In k2. Тогда наклон бу-
дет равен Ея/2,303/?. Заметьте, что при использовании в эксперименте узкого
температурного интервала на аррениусовеком графике точки I/Г расположены
на небольшом участке и для нахождения In А требуется далекая и поэтому не-
надежная экстраполяция.
Форма аррсниусовского выражения может быть получена из
простого рассуждения. Если предположить, что константа скоро-
сти реакции второго порядка является результатом бимолекуляр-
ной реакции, то необходимы два условия протекания реакции.
Прежде всего молекулы должны встретиться друг с другом, т. е.
должно произойти их соударение или столкновение*. Обозначим
через Z скорость, с которой происходят эти соударения (или столк-
новения) в единице объема. Сначала рассмотрим реакцию в га-
зовой фазе; в этом случае Z можно рассматривать как частоту
столкновений (соударений; см. разд. 24.2). В газе при давлении
1 ат.м частота столкновений равна приблизительно 1028 с_(-см-3
даже при комнатной температуре.^ Если бы наличие столкновений
было единственным фактором, необходимым для протекания ре-
акции, то все газовые реакции заканчивались бы за 10-9 с, но это
* Автор называет по-разному столкновения молекул в газе (collision) и в
жидкости (encounter).— Прим, перев.
26. Скорости химических реакций
419
Рис. 26.4. Арренпусоаский гра-
фик. для разложения СН3СНО,
не так. Более того, часто-
та столкновений зависит
от квадратного корня из
температуры [уравнение
(24,2.5) ], и поэтому, пред-
сказывая абсурдно невер-
ную скорость, мы также
предсказываем абсурдно
неверную температурную
зависимость.
Другим необходимым условием для осуществления реакции яв-
ляется то, что молекулы при столкновении должны обладать до-
статочной энергией. Слабое столкновение не ведет к реакции,
столкновение должно быть сильным. Если предположить, что для
осуществления реакции столкнувшиеся молекулы должны иметь
по крайней мере энергию Еа, то частота столкновений должна
быть умножена па долю молекул, сталкивающихся по крайней
мере с энергией ЕЙ. Эта доля определяется распределением Больц-
мана :[см. уравнение (1,2) в гл. «Введение», т. 1] и равна
ехр(—EJRT) для данной системы при температуре Т. Из этого
следует, что предсказанная температурная зависимость выража-
ется следующим образом:
скорость =Z expi —EjRT'i.
При обычных температурах доля энергетически достаточных
столкновений очень мала (рис. 26.5), и, таким образом, данная
модель предсказывает скорость, которая значительно меньше, чем
величина Z. Болес того, модель предсказывает экспоненциальную
температурную зависимость, поскольку доля энергетически доста-
точных столкновений экспоненциально увеличивается с темпера-
турой. Единственным затруднением на этой стадии обсуждения
является то, что частота столкновений сама по себе зависит от
температуры, в то время как в соответствии с экспериментальными
результатами предэкспонеццнальный множитель Л, вероятно, не
должен зависеть от температуры. Фактически экспоненциальная
температурная зависимость значительно, сильнее, чем зависимость
Z от квадратного корня из температуры, и экспериментально об-
наружить отклонения от экспоненциальной формы очень сложно.
Например, для типичных энергий активации (около 50-—
27*
420
Часть 3. Изменение
Рис. 26.5. Доля молекул, имеющих энергию большую, чем Е,, мала, но быстро
увеличивается с температурой.
100 кДж/моль) скорость удваивается при повышении температу-
ры на 10 К, но частота столкновений изменяется только в
(308 К/298 К) 1,02 раз для повышения температуры на 10 К
от комнатной температуры.
Мы будем придерживаться взгляда, что теория столкновений
отражает основные черты происходящей бимолекулярной реакции
и что энергия активации служит полезным параметром, имеющим
некоторый физический смысл. Однако ясно, что для надежного
предсказания скоростей реакции теория скоростей реакций тре-
бует значительно более сильного фундамента, н мы вернемся к
этому вопросу в следующей главе.
Реакции, ведущие к равновесию. В этом параграфе мы рассмот-
рим первый пример более сложной реакции, чем АДВ—^продух-:
ты, и увидим, как можно изменить проинтегрированные законы!
скоростей. Мы выберем реакцию, которая протекает до некоторо-1
го положения равновесия и в которой как прямая, так и обратная]
реакции первого порядка: 1
А ч—»- В. I
й-i I
Примером может быть реакция изомеризации, скажем циклопро-1
пан пропилен. I
Скорость изменения концентрации А имеет два вклада: количе-1
ство А уменьшается в прямой реакции со скоростью АДА], но вое-’
26. Скорости химических реакций
421
полняется в обратной реакции со скоростью A-t[B], Поэтому об-
щая скорость изменения [А]
d = — k, [AJ+^JB],
Если первоначальная концентрация А равна [А]о и в начальный
момент В отсутствует, тогда в любой момент Времени [A]-j-[B] =
= [A] q. Поэтому
d [Al/di = -kL [A]-HA_t ([A]„-[A])= —(6, + M [A] + ^ [A]c.
Решение этого дифференциального уравнения первого порядка
приводит к
[A],=IА]о + (26.3.3)
[ i "-1/ J
Свойства этого решения соответствуют тому, что и следовало ожи-
дать. Например, если A_i = 0 (пет обратной реакции), последнее
уравнение превращается в уже найденное уравнение первого по-
рядка [уравнение (26.2.6)].
Каково конечное состояние системы?
Пусть t будет бесконечно большим. Тогда
[AU^fAy^ + fe.),
[Bjco^tAIo—[ А]00=А-1 [Ay^+Ai).
По истечении этого очень долгого времени система придет в рав-
новесие, и соотношение концентраций будет определяться констан-
той равновесия К. Из этого следует, что
Х^([В]/[А])е=[В100/1А[оо=А1/А_1. (26.3.4)
Таким образом, мы приходим к очень важному соотношению меж-
ду константой равновесия и константами скорости простой реак-
ции. Практическая важность этого вывода заключается в том, что,
измерив одну из констант скоростей и константу равновесия, Дру--
гую константу скорости легко вычислить.
Такие же расчеты можно сделать для другого типа элементар-
ного равновесия. Например, в случае простой бимолекулярной
реакции второго порядка
, ^2
^ + В ч—* C4-D
^“2
/Г__( [С] [DJ ] _ [Р]со_______
I W [BJ ]е- [А]ж fe-s
(26.3.5)
Заметьте, что мы делаем упор на простой реакции: вывод о том.
Что соотношение К = АП/А_П применимо для простой одностадийной
Реакции, вовсе не означает, что оно обязательно применимо для
422
Часть 3. Изменение
любой реакции второго порядка, которая может представлять со-
бой последовательность нескольких стадий.
Последовательные реакции и стационарное состояние. Некоторые
реакции протекают через образование интермедиата, например
две последовательные реакции первого порядка
7г 1
А----► В ----> С.
Такой реакцией является распад радиоактивного семейства (пе-
риоды полураспада указаны для каждой стадии):
и- р-
> «Жр ——> 3g?Pu.
вл 23,5 мин 5,2 Н 2.35 сут 9
Характеристика этого типа реакций может быть получена состав-
лением уравнений скорости образования и разложения всех трех
продуктов. Разложение А протекает со скоростью &i[A]
d [A]/d/= — [Л]. (26,3,6a)
Промежуточное соединение В образуется в результате разложения
А, но расходуется в реакции образования С:
d lB]/di=ki [AJ — k{ [B[. (26.3.66)
Наконец, С образуется из В по реакции первого порядка:
d[С] jdt = [В [. (26.3.6в)
Предположим, что первоначально присутствует только А, и его
концентрация равна [А]о-
Первое из этих трех уравнений для скорости соответствует экс-
поненциальному распаду, и поэтому концентрация А изменяется
следующим образом:
|А], = [А]оехр(-М. (26.3.7а)
Если это выражение подставить в уравнение для В при условии,
что [В]о —0, то получим
|В].^[А]„{ (26.3.76)
График этой функции представлен на рис. 26.6. Он показывает,
что концентрация промежуточного продукта В увеличивается от
нуля до максимума и затем падает до пуля, в то время, как кон-
центрация А падает, а концентрация С в смеси возрастает. Кон-
центрация С может быть получена подстановкой решения для В
и последующим интегрированием. Это довольно просто, поскольку
в любой момент времени концентрации всех веществ связаны
между собой:
[Н-г1В]ЖСЬ=[А]0.
26. Скорости'химических реакций
423
рис. 26 6. Концентрации реагентов, интермедиатов и продуктов для двух после-
довательных реакции первого порядка.
и) ft,=10 ft{; б) k-, =o,i fei.
Это означает, что [С]; может быть прямо получена из решений
уравнений для [А] и '[В]:
[С]<=[А]0{1 + ( yk[e-k^ (26.3.7в)
Графически это выражение также приведено па рис. 26.6, и мы
видим, что концентрация продукта растет до тех пор, пока опа не
достигнет конечного значения [А]о, когда все вещество А превра-
щается в С.
В этом расчете необходимо обратить внимание на несколько
обстоятельств. Во-первых, три уравнения (26.3.7) указывают, как
анализировать реакционную схему, состоящую из двух последова-
тельных реакций первого порядка: найдены методы для измере-
ния концентраций промежуточного вещества и продукта и предло-
женный механизм подтвержден проверкой справедливости этих
Уравнений, Если они выполняются, то можно найти значения
констант скоростей.
Во-вторых, этот расчет поясняет также смысл понятия «лими-
тирующей стадии». Предположим, что константа скорости k\ на-
много больше тогда каждая образующаяся молекула В быстро
превращается в С. В таком случае скорость образования С почти
Целиком определяется скоростью образования промежуточного ве-
щества В. Это подтверждается анализом уравнения (26.3.7в) для
случая k’ когда .значение ехр(—k’ti\ много меньше ехр(—k^t)
и поэтому им можно пренебречь. Тогда
fCh [А )0 (1 +( k^k[ )ГА]0 (1 -е-^) (26.3.8)
424
Часть 3. Изменение
при условии, что в знаменателе мы пренебрегаем константой ki,
поскольку опа мала по сравнению с Л'. Как и следовало ожидать,
скорость образования С зависит только от меньшей из констант
скоростей. По этой причине стадия с наименьшей константой ско-
рости называется лимитирующей стадией реакции.
Если скорость образования В много больше, чем скорость его
Превращения в С, то лимитирующей стадией будет реакция прев-
ращения В в продукты. Только что описанные рассуждения в слу-
чае, когда A'Cfei, Приводят к
[С], « [Aj0(l-е~Ы‘), (26.3.9)
и, как и следует ожидать, скорость реакции будет определяться
константой наиболее медленной лимитирующей стадии общей ре-
акции. Эту последовательность, когда за быстрой стадией идет
медленная, можно сравнить с тем, когда широкая дорога, состоя-
щая из 6 полос, переходит в однополосную дорогу на мосту: в
этом случае скорость движения будет определяться скоростью про-
хождения моста.
В-третьих, из расчета следует, что его математическая слож-
ность быстро нарастает с усложнением реакционной системы. Уже
сейчас можно предположить, что реакция, состоящая из многих
стадий, приведет к очень трудным уравнениям. Можно ли найти
приближение, основанное на настоящих точных решениях, которое
приведет к значительно более простым уравнениям?
Предположим, что k{ значительно больше k}. Это будет так, ес-
ли промежуточное соединение очень реакционноспособно. В таком
случае уравнение для [В] t [уравнение (26.3.76)] показывает, что
[В]# меньше концентрации А примерно в ki/k[ раз. Если А реаги-
рует очень медленно, концентрация В остается в течение долгого
времени одинаковой и небольшой. Поэтому для разумно большого
промежутка времени в ходе реакции приблизительно соблюдается
соотношение
d[B]/A^ 0.
Исключение составляют самое начало реакции (когда концентра-
ция В увеличивается от 0) и ее конец (когда концентрация В
должна уменьшиться до 0). Предположение о том, что основная
часть реакции с реакционноспособным промежуточным продуктом
происходит при его постоянной концентрации, называется приб-,
лижением стационарности. Приближение стационарности сильно
упрощает обсуждение кинетических схем. Поскольку промежуточ-
ное соединение находится в стационарном состоянии, уравнение
скорости для В [уравнение (26.3.66)] можно упростить до
fejAl— feJIBl^O,
26. Скорости химических реакций
425
поэтому
[В] « (/г,//г') [А].
Подставляя это выражение в уравнение скорости образования
продуктов {уравнение (26.3.6в)], получаем уравнение
4 [C]/d/=*;[BI « k. [А],
которое ясно показывает, что С образуется из А по реакции пер-
вого порядка, и решение этого уравнения может быть записано в
обычной экспоненциальной форме.
Такой же метод может быть использован для упрощения дру-
гого типа последовательных реакций, когда интермедиат находит-
ся в равновесии с реагирующими веществами, т. е. в так называ-
емом предравновесии. Рассмотрим реакционную схему в общей
форме
A -j- В ч—* (АВ) ->- С,
*-1
где (АВ) — некоторое промежуточное соединение. Предположим,
что (АВ) превращается в С очень медленно по Сравнению со ско-
ростями его образования и обратного разложения на А и В. Тог-
да в первом Приближении в уравнении скорости для концентра-
ции (АВ) можно пренебречь константой и, таким образом,
d Ъ [А] [В] - А.! [(АВ)].
Если промежуточное соединение находится в стационарном состоя-
нии, дифференциальное уравнение превращается в алгебраическое:
[A] [B]~-A.i[(AB)l=0
или
[(AB)]=(M-i) IA] [В].
Как мы уже видели, отношение прямой и обратной констант ско-
ростей реакций для простой реакции равно константе равновесия
К. Поэтому
[(АВ)[=К [А[ [В].
Скорость образования продукта С тогда выражается следующим
образом:
d IQ/di^kiK [А] [В]. ,(26.3.10)
Следовательно, реакция имеет общий второй порядок.
На двух примерах покажем, каким образом можно объяснить
механизм реакции, включающей последовательные стадии с пред-
426
Часть 3. Изменение
равновесием. Первый пример — окисление оксида азота(П), явля-
ющееся реакцией третьего порядка:
*3
2NO + Оз ----> 2NOa
Один из способов объяснения кинетики третьего порядка включа-
ет предположение о том, что реакция тримолекулярна. Это, одна-
ко, потребовало бы одновременного столкновения трех молекул,
а такие явления очень редки. Более того, скорость реакции умень-
шается с увеличением температуры. Это противоречит нормально-
му поведению реакций и указывает па то, что схема реакции со-
стоит из нескольких стадий,
Для того чтобы увязать эти замечания, исследуем механизм,
состоящий из предравновесия
NO + NO < N2O2 (константа равновесия К)
и бимолекулярной реакции
NaO2+O2 ---> 2NO£.
Применяя приближение стационарности (которое требует, чтобы
скорость второй реакции была значительно меньше, чем скорости
реакций, ответственных за предравновесие), получим
[NsOJ=K[NO]a.
Бимолекулярная реакция — реакция второго порядка, поэтому ско-
рость образования NO2 выразится следующим образом: -
d [NOj/dt =k2 [N2O21 [O3] fNO]2 [OJ.
Это закон скорости третьего порядка, что и получалось в опыте,
и константа скорости может быть записана как —Причи-
на аномальной температурной зависимости также может быть
проанализирована, Вероятно, k2 ведет себя нормально и увеличи-
вается с температурой, однако при повышении температуры рав-
новесие между NO и N2O2 сдвигается влево (Л уменьшается), и
это изменение достаточно сильно, чтобы значение уменьша-
что /С уменьшается с температурой, поскольку
лось. (Мы знаем,
реакции димеризации являются экзотермическими; см. термодина-
мическое обсуждение в т. 1, разд, 8.1.)
Другой пример предравновссной реакции — механизм действия
ферментов, предложенный Михаэлисом и Ментен в 1913 г. Обозна-
чим фермент буквой Е, а вещество, на которое он действует, суб-
страт, буквой S. Тогда общая реакция выразится как
E-f-S -----P-j-E,
и в процессе превращения S в продукт Р сам
неизменным. Опыты показывают, что скорость
фермент остается
образования
про-
26. Скорости химических реакций
427
дукта зависит от концентрации фермента, и, хотя суммарная ре-
акция выглядит просто, S—►‘Р, механизм се должен включать
стадии с участием фермента. Наиболее простой механизм следую-
щий:
^2
Е S (ES) —> Р 4- Е.
1
В этой схеме (ES)—некоторое связанное активное соединение
фермента и субстрата, которое может превращаться в продукты
по реакции первого порядка с константой скорости k\, или разла-
гаться, давая исходные вещества, по реакции первого порядка с
константой скорости k-i. Тогда скорость образования продукта вы-
разится следующим образом:
d [P[/dt=k [(ES)].
Для того чтобы решить это уравнение, необходимо знать концент-
рацию связанного субстрата. Поэтому записываем следующее
Уравнение скорости:
d [(ES)]/d/ =k. [Е] [S] - k_, [(ES)] -[(ES)].
образование
Из E, S
разложение разложение
До E, -S до продукта
реакции Р
Тогда приближение стационарности дает
^2 [Е] [S] [(ES)J k± [(ES)] ~ 0,
Откуда
[(ES)] «ME] [Sj/^+^O.
Величины [E] и [S] представляют собой соответственно концент-
рации свободного фермента и свободного субстрата- Если [E]q—
общая концентрация фермента, то.мы можем написать, что [Е] +
+{(ES)] — ТЕ]0, и эта величина постоянна в ходе реакции. По-
скольку добавляется только небольшое количество фермента, об-
щая концентрация субстрата приблизительно равна концентрации
Несвязанного субстрата, [S] + [ (ES)] ~ [S]. Поэтому
[(ES)] ([Е]о-](ES)]) [Sj/^+fe-O
Или
fea[E]0 [S]
ki + k-i 4- kz [S]
Отсюда следует, что скорость образования продукта
d[P\idf^
Wz [E[q [S[ . [E[o [S[
4* 4~ [S] [S] ’
(26.3.11)
428
Часть 3. Изменение
где /См — константа Михаэлиса:
/См =(&( -\-k_-i)/k2.
Согласно уравнению (26.3.11), скорость ферментативной реакции
линейно зависит от количества добавленного фермента, а также
от количества присутствующего субстрата.
Реакции первого порядка. В данном разделе, суммирующем эмпи-
рические наблюдения химической кинетики, показано, что ряд
газовых реакций следует закону скорости первого порядка. Закоп
скорости второго порядка часто может быть сведен к процессу би-
молекулярных столкновений, но как объяснить первый порядок
реакции в газовой фазе? Трудность состоит в том, что нужно объ-
яснить кинетику первого порядка, несмотря на то что молекула,
вероятно, приобретает энергию, необходимую для изменения ее
формы, при столкновении с другими молекулами. Нужно найти
схему реакции, которая приводит к форме fei[A], ио которая при
этом включает бимолекулярные процессы, необходимые для на-
копления в молекуле достаточной энергии.
Первое успешное объяснение реакций первого порядка было
сделано Линдеманом в 1922 г. Он предположил, что молекула А
сталкивается с другой молекулой А и одна из Них становится
возбужденной .за счет другой. Тогда первоначальный бимолекуляр-
ный процесс в схеме запишется как
А А -----> А* + А.
Возбужденная молекула А* не сразу теряет свою энергию, но мо-
жет потерять ее, если столкнется с другой молекулой А.
Таким образом, происходит также реакция
А* 4- А -А 4- А.
Имеется также возможность того, что возбужденная молекула
сможет сама по себе распасться или перейти в новую конфигура-
цию и при этом высвободить свою энергию, давая продукты реак-
ции мономолекулярцого разложения:
А* ---> Р.
(Предполагается, что активация А-]-Р и дезактивация A*-f-P не
играют роли, хотя в действительности они могут вносить вклад.)
Из предыдущего обсуждения можно ожидать что если образова-
ние А* является быстрым, то лимитирующей стадией в общей схе-
ме должно быть мопомолекулярное разложение и кинетический
первый порядок может появляться из этой суммарной схемы.
Скорость образования продукта выразится как
d [Р]/Л [А*].
26. Скорости химических реакций
429
Это уравнение показывает, что в выражение для скорости должна
входить концентрация активных молекул. Эта концентрация вы-
ражается с помощью уравнения скорости образования активных
молекул, поэтому, применив приближение стационарности, полу-
чим
d [А*Ж=А, (А]2- А.2[А*] [А] - kt [A*J=O,
Это даст
гд*1 ____
(26.3.12)
Только что полученное выражение, по-видимому, не приведет
ц кинетическому уравнению первого порядка. Однако мы должны
помнить, что еще ничего не было сказано об относительных скоро-
стях отдельных стадий реакции. Если скорость дезактивации столк-
новений А*+А значительно больше скорости мономолекулярного
разложения, то получим
k.2 [A*] J А] » ki [А*] или й.а [А] » kt.
В этом случае закон скорости
d \P]/di ж [А]2/А_а [А] (^2/й_а) [Д] (26.3,13)
соответствует кинетике первого порядка. Этот вывод согласуется
с предполагаемым механизмом: если k\ мала и лимитирующей
стадией является мономолскулярное разложение, то можно ожи-
дать кинетику первого порядка,
Достоинство механизма Линдемана заключается в том, что он
может быть проверен, Если сильно уменьшить давление А, усло-
вие А_2[А] выполняться не будет, а вместо него будет
&_a[A]<C&i, При таких низких давлениях закон скорости превра-
тится в
d[P]/d/~fea]Ap,
(26.3,14)
и вместо кинетики первого порядка будет кинетика второго по-
рядка, Физической причиной этого изменения является то, что при
низких давлениях лимитирующей стадией становится бимолеку-
лярное образование возбужденных молекул, Это изменение поряд-
ка реакции было подтверждено экспериментально, что показано
па рис. 26.7 для скорости изомеризации тродс-СаН2Е>2.
Более тщательной проверкой модели Линдемана должно быть
исследование количественных характеристик при изменении пер-
вого порядка на второй: действительно ли порядок реакции изме-
няется с давлением так, как это предсказывает модель? На это
430
Часть 3: Изменение
0,0 0,5 1,0 1,5 2,0
моль/дм3)
Рис, 26.7. Зависимость скорости реакции
первого порядка — изомеризации транс-
CsH2D^ от давления (Pilling Л1. F„ Reac-
tion kinetics, Clarendon Press, Oxford,
1975).
можно ответить следующим образом. Во-первых, закон скорости
[уравнение (26.3.13)] можно записать так:
d [P]/dt =ЛГ! [А], (26.3.15)
где йеп—эффективная константа скорости, которая выражается
следующим образом:
Р6-316)
Это выражение можно преобразовать в
(26.3.17)
и, таким образом, трафик наблюдаемой константы скорости
от 1/[А] должен дать прямую линию. Типичный результат пока-
зан на рис. 26.8. Он показывает выраженную кривизну, соответст-
вующую большим значениям йен при высоких давлениях (низкие
значения 1/[А]), чем можно ожидать при экстраполяции резуль-
татов, полученных при низких давлениях. Это значит, что мономо-
лекулярная стадия не входит в реальный механизм.
Одна из причин расхождения состоит в том, что модель не
учитывает того факта, что перед началом реакции, возможно, не-
обходимо особое возбуждение молекул, а бимолекулярное столк-
новение дает лишь общее, неспецифическое возбуждение. Напри-
мер, в реакции первого порядка изомеризации циклобутена
ключевая стадия включает растяжение одной из связей и проис-
ходит тогда, когда эта связь сильно возбуждена колебательно. Од-
нако при столкновении, ведущем к возбуждению, энергия возбуж-
дения разделяется поровну по четырем связям, и, таким образом,
реакция изомеризации происходит только в том случае, если име-
ется достаточно времени, чтобы энергия возбуждения накопилась в
ключевой связи. Отсюда следует, что нужно провести различие
между возбужденной молекулой А*, у которой имеется энергия,
26. Скорости химических реакций
431
достаточная для реакции, по эта энергия распределена по несколь-
ким связям, и активированным состоянием молекулы А*, у кото-
рой возбуждение особое, и молекула подготовлена для реакции.
Таким образом, мономолекуляркая стадия механизма будет иметь
вид
fel Jei
А* ----► Р
вместо одной стадии с константой скорости k}. Если учесть это в
схеме механизма, то несоответствие между предсказанной и экспе-
риментальной зависимостью кец от давления в значительной сте-
пени уменьшается.
26.4. Сложные реакции
Большая часть реакций, имеющих технологическое и биологичес-
кое значение, состоит из сложной последовательности стадий или
протекает с достаточной скоростью только в присутствии катали-
затора. В этом разделе мы покажем, как идеи, развитые в пре-
дыдущих разделах, могут быть применены к рассмотрению таких
процессов.
Цепные реакции. Многие реакции в газовой фазе протекают через
ряд стадий, включающих свободные радикалы. Эти молекулярные
фрагменты могут атаковать молекулы и давать новые радикалы,
например, отщепляя атом водорода в реакции
•сн3 + СН3СН3-------------------------> сн4 + -ch2ciis
f/(p, мм рт. ст.)
(свободный радикал
обычно записывается с
точкой, обозначающей его
неспаренный электрон).
Полученные таким обра-
зом радикалы, в данном
случае этильный радикал
• СНзСН3, могут реагиро-
вать дальше, давая дру-
гой радикал при атаке
молекулы или встречаясь
и соединяясь с другим
свободным радикалом.
Эта последовательность
Рис, 26.8. Проверка механизма
Линдемана (разложение NsOj),
432
Часть 3. Изменение
реакций (последовательность радикалов, дающих радикалы) но-
сит название «цепной реакции».
В общем случае цепная реакция может быть разложена на че-
тыре стадии. На стадии инициирования из обычных молекул обра-
зуются радикалы. Например, атомы хлора могут образовываться
в реакции С12—>2С1. Механизм таких стадий может включать
простое столкновение или, в случае более сложных молекул, про-
цесс типа процесса Линдемана, в котором молекула приобретает
достаточную энергию при столкновении с другой молекулой и за-
тем теряет ее в последующей мономолекулярной стадии. Альтер-
нативно. радикал может получаться при поглощении фотона, на-
пример, под действием солнечного света: C12-|-ftv=2Cl. Атаку сво-
бодных радикалов на другие молекулы, в результате чего получа-
ются новые радикалы, называют стадиями развития цепи. Ранее
приведенный пример, когда метильный радикал атакует этан и ге-
нерирует этильные радикалы, относится к стадиям развития цепи.
Другие примеры мы рассмотрим далее. Если радикал атакует мо-
лекулу продукта, эта стадия называется ингибированием.
Цепь радикальных реакций заканчивается стадией обрыва це-
пи. Цепи могут обрываться различными путями. Например, может
произойти димеризация
CHsCHf + *CH2CHS --> СН9СН3С11гСН3
или реакция со стенками реакционного сосуда. Некоторые моле-
кулы реагируют очень быстро со свободными радикалами, и, если
их ввести в систему, они способны остановить реакцию. Молекула
оксида азота(Н) NO является одним из очень эффективных пере-
хватчиков радикалов и «тушит» реакцию, если ее ввести в систе-
му, что является основанием предполагать цепной механизм.
Цепная реакция часто, но не всегда ведет к сложным законам
скорости. В качестве первого примера рассмотрим реакцию водо-
рода с бромом.
Общая реакция имеет вид
Н2 4- Вг„ -► 2НВг,
и эмпирический закон скорости записывается следующим образом;
•инад/л—(26.4.1)
Это выражение сложное, и оно, несомненно, предполагает слож-
ный цепной механизм. Был предложен механизм, включающий сле-
дующие стадии:
инициирование: Вг2 ->- 2Вг'.
Ч
развитие цепи: Вг‘-]-Н2 ->• HBr-f-H*,
26. Скорости химических реакций
433
Н* 4- Вг3 —НВг + Вг\
4
ингибирование: Н’+НВг -->- H2-f-Br’,
*е
обрыв цепи; Вг" -f-Вг" ->- Вг,-
Для того чтобы па основании этого механизма получить закон ско-
рости, мы должны найти скорость образования НВг. Поскольку
НВг образуется в реакциях «Ь» и «с», но удаляется в реакции
«б», общая скорость образования равна
d [НВг\ldt = [Вг] [Н214-fec [Hl [BrJ [H[ [HBrj.
Для того чтобы раскрыть это выражение, мы должны найти кон-
центрацию атомов брома и водорода. Самым простым было бы
найти два уравнения скорости для концентраций [Н] и [Вг] и уп-
ростить их, используя приближение стационарности:
d [H[/d/=kb [BrJ [HJ — kc [HJ [Br2]—kd [HJ [HBrJ ж 0,
d [Br]/d/ = 2fea [Brg] —[Br[ [Hj-l-feJH] [Br2J +
+^[Н1[НВг]-2йе[Вг}г«0,
Эти два уравнения могут быть решены для [Н] и [Вг] и резуль-
тат можно подставить в уравнение для d[HBr]/d/. В этом случае
получим
d [HRri/d/_(^а/^е) [Н2] [Вг2] да 4 2)
d [ НВг]/€Й - i {£d [НВг]/4 [Вга]) ' 1 ‘ '
Это уравнение имеет тот же вид, что и эмпирический закон скоро-
сти, и Две наблюдаемые константы скорости могут быть идентифи-
цированы как
=2kb{kjk^, k"=k,jkc.
Некоторые цепные реакции имеют простую кинетику, из чего
еще раз следует, что к данным химической кинетики необходимо
подходить осторожно. Например, дегидрирование этана до этиле-
на в соответствии с общей реакцией
СН,СНз ---> СН8 = СНа + н2
подчиняется кинетическим закономерностям первого порядка:
d [СН8 = СНз]/^ KWdW7’ [СН3СН3],
и очевидной интерпретацией является осуществление реакции по
типу процесса Линдемана. Однако дальнейшие эксперименты по-
казали, что во время реакции образуются свободные радикалы, и,
таким образом, необходимо найти более сложный цепной меха-
28-242
434
Часть 3. Изменение
низм. Последовательность реакций, известная как механизм Рай-
са — Герцфельда:
ка.
инициирование: Сн3СН3 -->- 2СН3* ,
Ч
развитие цепи: СН3 + СН3СН3 --> СН4 -J- СН3СН2 ,
ch3chs —> сна = сн2+н-,
н* дспэсн3 —на + сн3сн^,
ке
обрыв цепи: Н* + CHaCHi ----->- СН3СН3,
может быть проанализирована уже описанным способом с уста-
новлением соответствующих уравнений скоростей и применением
приближения стационарности для всех радикальных частиц. В ре-
зультате получим уравнение
d [СНа=СН21/^ = (МЛз/2М1/2 [СН3СН31,
которое соответствует наблюдаемой кинетике первого порядка.
Взрывы. Некоторые реакции протекают со взрывами. Имеются
две главные причины, ведущие к взрыву, которые зависят как от
условий, так и от типа реакции.
Основной причиной для термического взрыва является экспо-
ненциальная зависимость скорости реакции от температуры. Если
энергия экзотермической реакции не может быть отведена, то тем-
пература реакционной системы увеличивается и реакция ускоря-
ется. Это приводит к выделению тепла с громадной скоростью,,
оно уже цс может быть отведено, и, таким образом, реакция про-
должается еще быстрее.,., катастрофически быстро.
Другой основной тип взрыва связан с цепной реакцией. Если
имеются стадии, которые увеличивают число радикалов в системе,
скорость реакции может каскадно приводить к взрыву. Примером
разветвленной цепной реакции является реакция взаимодействия
водорода с кислородом. Хотя общая реакция очень проста:
2На+О2------- 2нгО,
ее механизм очень сложен и полностью еще не объяснен. Однако
известно, что она протекает по свободнорадикальному цепному
механизму и переносчиками цепи (радикалами, образующимися на
Стадиях развития цепи) являются Н*, О*. НО* и НО2. Некоторые
из стадий приведены ниже:
инициирование: Н.-уО, --»- НСу-ДЙН*.
развитие цепи: Н2 НО2 -->- нО* д. Н2О,
Н2 + НО* --»- Н* + Н2О,
Н- + О2 ---► НО* д- О* (разветвление),
О*4-Н2 ----►Но*+Н* (разветвление).
26. Скорости химических реакций
435
Две последние реакции являются реакциями разветвления, по-
скольку в них происходит увеличение числа переносчиков, и, сле-
довательно, скорость реакции может очень быстро увеличиваться:
это приводит к взрыву (сопровождающемуся характерным зву-
ком) при поджигании смеси водорода и кислорода.
Возможность взрыва зависит от температуры и давления реа-
гирующей системы; области взрыва для водорода и кислорода ука-
заны на рис. 26.9. При низких давлениях система находится вне
взрывных пределов, и даже при поджигании взрыва не происхо-
дит. При этих низких давлениях переносчики цепи, образующиеся
в реакциях разветвления, могут достигать стенок сосуда и там со-
единяться друг с другом и отдавать избыток энергии (хотя эффек-
тивность этого процесса зависит от химического состава стенок).
При увеличении давления система проходит через первый (ниж-
ний) взрывной предел (если температура превышает 400°C). Те-
перь, когда смесь поджигают, происходит взрыв, так как перенос-
чики цепи, прежде чем они достигнут стенки, успевают прореаги-
ровать и реакции разветвления цепи могут приводить к взрыву.
При дальнейшем увеличении давления система проходит через
второй (верхний) взрывной предел в область, где реакция проте-
кает без взрыва. Теперь давление настолько высокое, что радика-
лы, образующиеся в реакции разветвления, соединяются в газе, а
молекулы реакционной смеси способны поглощать избыток энер-
гии. Если давление повышается еще дальше, система проходит че-
рез третий взрывной предел, и реакция вновь становится взрыв-
ной. По-видимому, эта об-
ласть соответствует терми-
ческому взрыву.
Фотохимические реакции.
Одним из способов иниции-
рования цепной реакции мо-
жет быть поглощение света:
этот способ был упомянут в
начале разд. 26.4 в связи с
реакцией С12—>2С 1. Боль-
шинство реакций, как цеп-
ных, так и непеппых, может
быть инициировано погло-
щением фотона, н последст-
Рис. 26.9. Области взрыва для ре-
акции H2-f-O2.
28*
436
Часть 3. Изменение
Рис. 26.10. Фотохимические процессы в атмосфере.
вия этого имеют неоценимое значение для жизни на Земле, так
как фотохимические процессы являются способом поглощения сол-
нечной энергии. Большинство источников энергии в мире может
быть приписано энергии солнечной радиации, которая захватыва-
ется и аккумулируется благодаря фотохимическим реакциям.
К ним относятся многие реакции — от нагревания атмосферы в
дневное время путем поглощения в ультрафиолетовой области
спектра (через реакции, приведенные на рис. 26.10) до поглоще-
ния хлорофиллом излучения в красной и синей областях и после-
дующего переноса этой энергии на синтез углеводов. Фотохимиче-
ские процессы лежат также в основе нежелательных эффектов,
таких, как, например, образование фотохимического смога. Без
фотохимических процессов мир был бы просто теплым, стериль-
ным, каменным.
В фотохимии имеется два основных закона. Первый — закон
Гроттгуса— Драпера, устанавливающий, что реакция может ини-
циироваться только излучением, которое действительно поглоща-
ется реагирующей системой. Здесь ударение надо сделать на ело-
26. Скорости химических реакций
437
вс поглощается: если свет просто проходит через систему, он не
инициирует реакцию. Этот закон легко объяснить с современной
точки зрения, поскольку молекула приобретает энергию для реак-
ции поглощением фотонов с энергией fiv (см. т. 1, разд. 13.2). Да-
лее, поскольку процесс поглощения соответствует захвату одного
фотона рецептивной молекулой, получается обоснование для зако-
на Эйнштейна — Штарка. Согласно этому закону, молекула, ответ-
ственная за первичный фотохимический процесс, поглощает один
квант.
Из закона Эйнштейна — Штарка не обязательно следует, что
при поглощении одного фотона молекула даст только один про-
дукт: если далее следует цепная реакция, то поглощение одного
фотона может привести к образованию в качестве продукта не-
скольких молекул. Отношение числа реагирующих молекул к чис-
лу поглощенных фотонов называется квантовым выходом реакции
ф число реагирующих молекул 4
число поглощенных фотонов ’ 1 /
Например, при фотолизе HI светом с длиной волны 254 нм про-
исходит процесс
НГ-f-ftv -> Н* Г,
H’ + hi —* н2+г,
!•+!• ----> 12.
и, так как поглощение одного фотона приводит к распаду двух
молекул HI, квантовый выход равен двум. В цепной реакции
квантовый выход может быть очень большим, обычно порядка
Ю1. В этих случаях цепная реакция действует как химический
«усилитель» начальной Стадии поглощения.
Пример (вопрос 22). При образовании этилена из ди-н-проиилкстона (гсптан-4-
она) квантовый выход при облучении светом с длиной волны 313 пм равен 0,21.
Сколько молекул этилена в секунду и молей в секунду образуется, если образец
облучается 50 Вт-источником света С длиной волны 313 нм в таких условиях,
что весь свет поглощается образцом?
Метод. Рассчитываем число фотонов, испускаемых лампой в секунду; все они
поглощаются, и поэтому умножением на квантовый выход получаем число рас-
латающихся молекул.
Ответ. Энергия 313 цм-фотонов равна
ЛсД =(6,626- 10‘м Дж-с) >Д2,998-1ОВ м/с)/(313-10“9м)=:
=6,35- Ю'^Дж.
Так как 1 Вт=1 Дж/с, то лампа в 50 Вт испускает (1 с)Х(50 Вт)/(б,35Х
Х10-[° Дж) =7,88 10ls фотонов в 1 с. Число молекул этилена, образующихся за
это время, равно (7,88-10i9)XO,21 -1,65-КГ9, или 2,75-10-5 моля.
Комментарий. Заметим, что квантовый выход зависит от Длины волны света.
438
Часть 3. Изменение
В качестве примера кинетической схемы, основанной на фото-
химической активации, рассмотрим синтез НВг. Вместо Вг2—>2Вг,
т. е. стадии инициирования термической реакции, фотохимическая
реакция описывается схемой
Bra + ?iv - -> 2Вг,
И скорость процесса пропорциональна числу поглощенных фото-
нов (это является следствием объединенных законов Гроттгуса —
Драпера и Эйнштейна — Штарка). Если интенсивность поглоще-
ния равна /яьс. то скорость стадии инициирования, определяется
выражением
(d [Вг]/^)иниц = 2^/аЬ5, (26,4.4)
и поэтому 2fea [ Вг2] в схеме термической реакции (стр, 432) нужно
заменить’ на 2&аЛь5, (Заметим, что скорость инициирования про-
порциональна /abs, а не /abs[Br2]: количество поглощенного света
Дьс уже учитывает концентрацию брома (Вг2), а ее нс нужно учи-
тывать дважды,) Следовательно, скорость образования НВг в фо-
тохимической реакции можно найти путем такой замены в уравне-
нии (26.4.2):
а гнтъ-1 26 5
d [HBrJ/dZ — i + >
и поэтому можно предсказать, что скорость будет увеличиваться
пропорционально корню квадратному из количества поглощенного
света. Это подтверждается экспериментально.
Пример (вопрос 22). Число поглощенных фотонов можно определить с помощью
химической актинометрии. Тщательно выполненньге эксперименты показали, что
при 300 нм квантовый выход для разложения оксалата уранила равен 0,570.
В специальном опыте предварительное пропускание света через пустую кювету
привело к разложению 6,201-10"3 моля оксалата за 1 ч. Когда кювету заполни-
ли ацетоном и время облучения увеличили до 10 ч, было найдено, что разложи,
лось 1,40-10-3 моля ацетона, и свет, который прошел через кювету и не погло-
тился, разложил 8,631-Ю-2 моля оксалата. Каков квантовый выход для разло-
жения ацетона?
Метод. Найдем число фотонов, необходимое для разложения двукратного коли-
чества оксалата: первый опыт дает собственную скорость, и поэтому поглощен-
ное количество может быть получено из второго опыта.
Ответ. Число фотонов, необходимое для разложения 6 201-10*’ моля оксалата,
равно (6,201 10-3 моля)Х (6,022-1023 моль-1)/(0,570) =6,551 -102[. Отсюда поток
фотонов равен (6,551 - 102[)/(2Х60Х60 с)Х9ф099-10 с_[. Поэтому число падаю-
щих фотонов за 10 ч составляет 3,276-ГО22. Число непоглощенных фотонов во
время 10-часового облучения равно (2,631- 10 s моль) X (6,022 1023)/(0,570) =2,78Х
Х1022, Таким образом, число поглощен пых фотонов составляет 3.276- 10=2— 2,78Х
ХЮ22 = 4,96-1021. За тот же интервал времени разлагается (1140 -10-3 моль) X
X (6.022-1023 моль-1) =8 43-IO2’ молекул ацетона. Поэтому квантовый выход рх
век Ф= (8,43-1020)/(4,96-1О2') -0,17.
Комментарий. I моль фотонов называют эйнштейн. Когда квантовый выход ра-
вен 0,17, 1 эйнштейн 300 нм-фотонов приведет к разложению 0,17 моля ацетона.
26. Скорости химических реакций
439
В ряде реакций .молекула реагента не поглощает, поскольку ее
электронный спектр поглощения находится за пределами спект-
рального интервала падающего света. Это может быть в том слу-
чае, когда падающий свет охватывает очень узкий интервал, на-
пример когда он генерируется в лазере или в ртутной либо гелие-
вой разрядной лампе. Тем не менее в таких случаях фотохимиче-
ская реакция может происходить, если в смеси присутствует части-
ца, которая может как поглощать свет, так и передавать свою
энергию потенциально реакционноспособной молекуле. Этот про-
цесс называется фотосенсибилизацией.
Родь атомов ртути в синтезе формальдегида из окиси углеро-
да и водорода является примером фотосенсибилизации. Когда
смесь окиси углерода и водорода, содержащая следы паров ртути,,
облучается светом ртутной разрядной лампы, атомы ртути воз-
буждаются поглощением света 254 нм. Возбужденные атомы стал-
киваются с другими молекулами, и столкновение между атомом
ртути и молекулой водорода достаточно эффективно для того,
чтобы последняя получила энергию, достаточную для ее диссо-
циации. Это инициирует радикальную реакцию. Обшая схема при-
ведена ниже.
первичное поглощение: Hg-[-Av -> 11g’,
перенос энергии: Hg* + Н2 ----> 2Н" -J- Hg.
реакция: Н* 4- СО------> НСО",
НСО*+Н3 ----«-НСНО4-Н-,
2НСО* --->- HCHO-f-CO.
При димеризации формильных радикалов образуются также неко-
торые количества глиоксаля ОПС—С.НО.
Фотосепсибилизация играет также важную роль в кинетике
реакций в растворах, и в качестве ловушек падающего света и
переносчиков его к некоторым потенциально реакционноспособным
частицам часто используются молекулы, содержащие карбониль-
ные группы, такие, как бензальдегид (С6НвСНО) и бензофенон
(дифенилметанон, С6Н5СОС6Н5)-
Катализ. Если энергия активации высока, то "лишь небольшая до-
ля сталкивающихся молекул имеет энергию, достаточную для того,
Чтобы произошла реакция, а если она низка, то реагирует боль-
шая часть молекул, и поэтому константа скорости будет большой.
Из этого следует, что если каким-либо образом можно снизить
энергию активации, то реакция должна протекать быстрее. А’а-
тализатор — это вещество, которое заставляет реакцию протекать
быстрее, снижая энергию активации лимитирующей стадии (ката-
лизатор не может изменить положение равновесия, см. т- I,
разд. 9.2; он только увеличивает скорость достижения равновесия).
Некоторая мера эффективности катализаторов может быть по-
440
Часть 3. Изменение
лучела при рассмотрении изменений энергии активации различных
реакций, которые обусловлены наличием катализатора. При разло-
жении перекиси водорода в отсутствие катализатора энергия акти-
вации равна 76 кДж/моль, и при комнатной температуре разложе-
ние протекает очень медленно. Если добавить небольшое количе-
ство иодида, происходит та же самая реакция, но с энергией акти-
вации, равной лишь 57 кДж/моль, и, таким образом, при комнат-
ной температуре (когда RT ^2,5 кДж/моль) константа скорости
лимитирующей стадии увеличивается в
k (катализатор) ехр [—57 (кДж/моль)/2,5 (кДж/моль)]
k (без катализатора) ехр [—76 (кДж/моль)/2,5 (кДж/моль)]
« е7’6 я* 2000 раз.
Этим объясняется выделение пузырьков кислорода при комнатной
температуре при добавлении иодида к перекиси водорода. Более
существенное изменение энергии активации происходит при добав-
лении ферментов к биохимическим системам. Фермент является
биологической молекулой, которая обладает большой эффектив-
ностью. Это иллюстрируется изменением энергии активации в реак-
ции гидролиза сахарозы от 107 кДж/моль в присутствии ЕР до
36 кДж/моль при добавлении небольшого количества фермента
сахаразы. Такое изменение энергии активации соответствует изме-
нению значения скорости на 22 порядка.
Можно выделить два класса катализаторов. Первый — гомо-
генные катализаторы, которые имеют ту же фазу, что и реакцион-
ная смесь. Чаще всего реакционная смесь является жидкостью, и
в ней растворяют катализатор. Ко второму классу относятся гете-
рогенные катализаторы, находящиеся в другой фазе по сравнению
с реакционной системой: обычно реакционная смесь является жид-
костью или газом, а катализатор — твердым веществом. В этой
главе мы ограничимся рассмотрением гомогенных катализаторов,
а обсуждение гетерогенных катализаторов продолжим после рас-
смотрения структуры и свойств поверхностей (гл. 28). Принцип нх
действия, заключающийся в понижении энергии активации, в каж-
дом случае одинаков, но осуществляется различными способами.
Некоторые представления о способе действия гомогенных ката-
лизаторов могут быть получены при рассмотрении кинетики реак-
ции разложения перекиси водорода, катализируемой бромом.
Общая реакция выглядит следующим образом:
2Н2О3 >- 2Н2О + О2.
Обычно сама по себе общая реакция пе дает никаких представле-
ний о действительном механизме, и в данном случае, как и во всех
других каталитических реакциях, катализатор не входит в урав-
нение общей реакции, однако он должен быть учтен при рассмот-
26, Скорости химических реакций
441
релин механизма. Предполагается, что реакция проходит через
следующие стадии:
Вл + 2Н2О —НОВг + 11:,О+ + Вг-,
н3о+ + нсон----> H0OHJ + H2O,
НООН^+Вг- ------ НОВгЧ-HjO.
Суммарный результат двух последних реакций можно выразить
уравнением
kg
НООН -I- НдО+ + Вг- —-> НОВг + 2НаО,
и именно эта стадия является лимитирующей скорость реакции.
Если НОВг уже образовалась, она может атаковать оставшуюся
перекись по реакции
НООН + НОВг --> НаО+ 4- 0,4- Вг-.
Эта стадия быстрая, поэтому скорость реакции зависит от ka, и
закон скорости записывается следующим образом:
-d[HaO2I/<fc=6a [НаОа1 [Н3О+] [Вт-)
в соответствии с наблюдаемой зависимостью скорости от концент-
рации бромид-ионов и ионов водорода (pH).
Важным примером гомогенного катализа является кислотный
и основный катализ. Ряд органических реакций протекает через
один из этих процессов, а иногда и через оба. При кислотном ка-.
тализе происходит переход протона к субстрату
ВЦ + к----- в- 4- НХ+,
и затем реагирует НХ+. Это является первичным процессом при
сольволизе сложных эфиров, кето-енольной таутомерии и инвер-
сии сахарозы. При основном катализе происходит переход прото-
на от субстрата к катализатору:
ХН + в —+ X- + ВН+,
и затем реагирует X-. Эта реакция является основой изомеризации
и галогенирования органических соединений, а также реакции
Клайзеиа и альдольной конденсации. Эффективность катализато-
ров зависит от их силы как кислот и оснований (в общем виде Это
обсуждалось в разд. 12.4 т, 1). Важным положением изучения кис-
лотного и основного катализа является соотношение между актив-
ностью и силой кислот (или оснований), характеризуемой значе-
ниями рКя (или рА'ь; см. т. 1, разд. 12.4). Это основной мост меж-
ду физической и органической химией и одна из основных обла=
стен исследования физической органической химии,
442
Часть 3. Изменение
26.5. Быстрые реакции
Все реакции являются быстрыми. По крайней мере, индивиду-
альные стадии реакций, в которых молекулы перегруппировывают-
ся по мономолекулярному механизму, или переход атомов при би-
молекулярных столкновениях происходят в атомной шкале време-
ни и завершаются менее чем за 10-9 с. Низкая скорость обшей
реакции обусловлена тем, что молекулы активируются или встре-
чаются друг с другом с низкой скоростью, однако и общая ско-
рость может стать очень большой, если система быстро получает
энергию активации. Это происходит, например, в случае взрыва.
Что наглядно показывает, насколько быстрыми могут быть молеку-
лярные процессы.
В последние годы были достигнуты большие успехи в разработ-
ке методов изучения быстрых реакций, которые заканчиваются ме-
нее чем за 1 с (и часто даже меньше), и в современной химиче-
ской кинетике упор делается на изучение процессов, происходящих
за очень короткое время. С помощью специальной лазерной тех-
ники теперь имеется возможность наблюдать процессы, происходя-
щие за несколько пикосекунд (1 пс = 10-12 с). При таких коротких
временах .химия процессов полностью превращается в физику про-
цессов.
В настоящее время вполне доступными стали методы контроля
и измерения констант скоростей быстрых реакций. К ним относят-
ся флеш-фотолиз, струевые и релаксационные методы. Другие ме-
тоды используются для исследования зависимости скорости реак-
ции от состояния колебательного возбуждения молекул Или от
энергии столкновения молекул и от скорости, с которой энергия
переходит из одной формы (например, колебательной) в другую
(например, вращательную). Для этой цели применяются также
4>леш-фо то л из, ударные грубы и молекулярные пучки. В этом раз-
деле мы ограничимся рассмотрением первой группы методов, что-
бы выяснить, как они могут быть использованы для получения ки-
нетических данных, другой класс методов рассмотрен в следующей
главе.
Флеш-фотолиз. Применение метода флеш-фотолиза связано с при-
менением вспышки света, действующей в течение короткого вре-
мени на начальной стадии фотолиза. Состав реакционной смеси
затем контролируют спектроскопически (например, по поглоще-
нию в ультрафиолетовой или видимой областях спектра или по
магнитному резонансу), при этом наблюдают переход возбужден-
ных реагентов в стабильные продукты. В ранее применявшихся
методах фотолитические вспышки получали при разряде батарей
конденсаторов через газ.
Эти методы применяют и в настоящее время, однако, поскольку
вспышка длится около 10-4 с, нет возможности наблюдать за про-
26. Скорости химических реакций 443
цессами, происходящими в очень короткое время. Основой новых
методов послужила лазерная техника, позволяющая работать в
наносекундной (10-9 с) шкале времени. Импульс лазера длится
1 нс, и, таким образом, первичная стадия поглощения завершает-
ся за этот интервал времени.
Следующей задачей является нахождение метода определения
концентраций и состава непосредственно после вспышки. Если
используется спектроскопия, то проблема сводится к тому, чтобы
обеспечить вспышку белого света продолжительностью порядка
наносекунд в данное точно контролируемое время после фотоли-
тической вспышки. Один из методов состоит в отражении фото-
литической вспышки от зеркала, помещенного па небольшом рас-
стоянии от образца. За 1 нс свет проходит около 30 см, и поэтому,
располагая зеркало на некотором подходящем расстоянии от об-
разца, с помощью отраженной вспышки-можно зарегистрировать
спектр в некоторое выбранное время после фотолитической вспыш-
ки. Однако отраженная вспышка является монохроматической, и
поэтому нужно найти какие-то способы расширения спектральной
области. Это можно сделать введением в раствор флуоресцентного
соединения. Импульс возбуждает соединение, и оно затем испуска-
ет флуоресцентное излучение в некотором интервале частот.
Наносекундный флеш-фотолиз так же, как его более медлен-
ный предшественник, был применен ко многим задачам. Как при-
мер Этого метода, рассмотрим его применение к фотолизу молекул
галогена и их последующей рекомбинации. Первоначальная вспыш-
ка генерирует из молекул X? атомы 2Х'. Единственной причиной
гибели атомов является их рекомбинация, но для образования Ха
из атомов необходимо отводить избыток энергии (энергию диссо-
циации), иначе вновь образованные молекулы будут сразу же рас-
падаться на атомы. Одним из путей отвода избытка энергии яв-
ляется участие в столкновении третьей частицы:
X- -у х- -f-М -»- Ха4- М*.
Третьей частицей М может быть стенка сосуда или какое-нибудь
добавленное вещество, возможно инертный газ, Столкновение трех
частиц в газе—явление довольно редкое, однако есть газ, кото-
рый очень эффективен в данном случае: это оксид азота (II) NO.
Мы уже рассматривали реакцию третьего порядка, в предравнове-
сни которой принимает участие NO (стр. 426). Такой же процесс
Происходит и здесь, поэтому можно предположить, что NO соеди-
няется с атомом галогена (вспомпнте также, что NO является хо-
рошим тушителем свободных радикалов), давая NOX. Затем
с NOX сталкивается второй атом галогена и происходит замеще-
ние NO с образованием Ха. NO действует как «прилипающая»
третья частица. Если наблюдать спектр фотолизированного галоге-
444
Часть 3. Изменение
Рис. 26.11. Установка для метода остановленной струи.
на немедленно после вспышки тСприсутствии NO, то NOX можно
обнаружить спектрально.
Струевые методы. Основная идея струевого метода описана
в разд. 26.1. Как было указано, недостаток этого метода заклю-
чается н необходимости использования больших объемов раство-
ров, содержащих реагирующую систему. Это особенно необходимо
для быстрых реакций, так как для того, чтобы картина реакции
распределялась по длине трубки, поток должен быть быстрым. Со-
временная модификация этого метода—метод остановленной
струи — нс требует большого количества материала благодаря
тщательной отработке конструкции смесительной камеры. Уста-
новка приведена на рис. 26.11. Два раствора очень быстро сме-
шиваются впрыскиванием материала в некоторую полость. Полость
имеет плунжер, который движется назад, когда втекает жидкость.
Поток прекращается, когда плунжер останавливается, и реакция
продолжается в тщательно перемешанных растворах. Состав смеси
анализируется но времени, обычно спектроскопически. Основной
особенностью установки является то, что в ней происходит тща-
тельное смешивание небольших количеств реагентов за очень ко-
роткое время, а затем с помощью электронных приборов регистри-
руется протекание реакции во времени.
Возможность контроля за поведением небольших образцов по-
зволяет использовать этот метод для изучения биохимических про-
цессов, и была проделана большая работа по изучению кинетики
действия ферментов. Мы видели [см, уравнение (26.3.11)], что
модель действия фермента Михаэлиса — Ментен приводит к зако-
ну скорости в форме
d [P]/dt-keJi
где
26. Скорости химических реакций- 445
Выражение для эффективной константы скорости может быть пре-
образовано в
Wf = V^ + (KM№i)/lS]. (26.5.1)
Из этого следует, что график зависимости 1/йегг от 1/[S] должен
дать константу скорости ki и константу Михаэлиса—Ментен Кгл,
однако он не может дать значения индивидуальных констант A-i
и kt, которые входят в Дм- Задача может быть решена методом
остановленной струи, при использовании которого скорость обра-
зования комплекса фермент — субстрат может быть найдена из-
мерением его концентрации после смешивания фермента и суб-
страта. Зная и k\ и комбинируя их с известным значением Дм,
находим
k-y —&!
Релаксационные методы. Термин «релаксация» указывает на воз-
врат системы к равновесию. В применении к химической кинетике
этот термин означает, что некоторое внешнее воздействие сдвига-
ет положение равновесия реакции (обычно очень быстро) и реак-
ция релаксирует в новое положение равновесия.
В качестве примера рассмотрим простое равновесие, включаю-
щее реакции первого порядка в обоих направлениях:
k’i
А - - > В.
Верхние штриховые индексы при константах скоростей «привязы-
вают» их к данным условиям, например к некоторой температуре.
Скорость изменения концентрации А:
d [A]Jdt= — [AI+feLj [В]. (26.5.2)
Когда система находится в равновесии, d[A]/d£ равно 0. Если
концентрации А и В при равновесии обозначить как [А]е' и [В]ё,
то получим
k{ [AK=feLi [В]е.
Теперь предположим, что состояние системы внезапно изменилось
(например, температура поднялась), так что константы скорости
превратились в и Концентрации А и В вес еще на мгнове-
ние имеют старые равновесные значения, но система больше не
Находится в равновесии. Затем концентрации изменяются до зна-
чений, соответствующих новым равновесным условиям, которые
теперь определяются следующим образом:
Ai [А]е=й_1[В]е,
(26.5.3)
446
Часть 3. Изменение
и скорость, с которой достигается новое равновесие, зависит от
значений новых констант скоростей. Обозначим отклонение [А]
от нового положения- равновесия через х, тогда [А]/=Л'/+[Л]е.
При / — 0 (сразу же после скачка изменения условии) [А]о=[А]ё;
следовательно, концентрация А изменяется следующим образом;
d [A]/df — —kt (х(-|-(A]e)-1-Al, (— [B]e)— (^ -H-i)xt
[вследствие сокращения двух членов, включающих равновесные
концентрации; уравнение (26.5.3)]. Поскольку производная по вре-
мени в левой части равна dxjdt* это простое дифференциальное
уравнение первого порядка имеет решение
xt —х0 ехр(— f/т), (26.5,4) .
где 1/т — ki + k-j. Из него' следует, что концентрация А (и В) ре-
лаксирует в новое равновесие со скоростью, определяемой суммой
двух новых констант скоростей. Константа равновесия при новых
условиях равна К — Aj/A_I( поэтому, измерив ее значение и время
релаксации, можно найти Aj и k^.
Одним из наиболее важных релаксационных методов является
метод температурного скачка. Равновесие нарушают внезапным
изменением температуры и контролируют изменение концентра-
ции во времени. Одним из способов такого повышения темпера-
туры является разряд электрического тока через образец, которо-
му придана электропроводность добавлением ионов, Подобрав
подходящие конденсаторы, можно осуществить температурный
скачок в 5—10 К примерно за Ю-7 с.
Пример. Батарея конденсаторов разряжает 50 кВ через 10 см3 водного раствора
за 20 мкс. Рассчитайте повышение температуры, если сопротивление раствора
40 Ом.
1 1
Метод. Мощность, рассеянная в виде тепла, равна За время т
энергия, рассеянная в виде тепла, составит-у- V^r/R. Если С — теплоемкость об-
разна, то повышение температуры равно -^V^t/CR. Примем, что СХ—78 Дж/(КХ
Хмоль).
Ответ. Из только что полученного выражения следует:
(50-103В)2Х (2 JO-Sc)
ДГ— 2х(40 0м)Х(Ю/18)Х(78 Дж/К) =14К
(количество воды в образце равно приблизительно 10/18 моля).
Комментарий. Чтобы получить желаемый скачок температуры, можно изменить
размеры н сопротивление образца. •<
Важное применение метод температурного скачка нашел в опре-
делении скорости реакции
Н3О+ + он- ----► 2Н2О.
26. Спорости химических реакций
447
Температурный скачок изменяет число ионов, находящихся в рав-
новесии, и поэтому электропроводность раствора. Было найдено,
что при комнатной температуре время релаксации равно т=40 мкс,
что соответствует А2~1,4-10п дм3/(моль-с), и, следовательно, эта
реакция является самой быстрой из известных.
Пример (вопрос 26). Равновесие реакции релаксирует за
37 мкс при 25 °C. Найдите константы скорости прямой и обратной реакций.
Метод. Находим выражения для т через fei (прямая реакция первого порядка)
и k2 (обратная реакция второго порядка). Делаем это так, как было указано
выше, но приближенно считая, что отклонение от равновесия так мало, что х*
можно пренебречь. Связываем k2 с ki через константу диссоциации А4 = 1,0Х
ХЮ-14. Находим k2. а затем Аг,
Ответ. Скорость прямой реакции равна fei[H2O], а обратной ЫН+][ОН_]. Сле-
дуя только что приведенному рассуждеигпо, получим
1/т = + й2([Н+]е 4- [ОН-]С).
Константа равновесия реакции равна
к _ [Н+[ (моль/дм3) X FQH-} (моль/дм3) Kw________
~ [Н3ОJ (моль/дм3) ~ [Н2О] (моль/дм3)
= 1,0-W’14/55,5 — 1,8- 10-1а.
При равновесии й1[Н2О]е=А2[Н+]о[ОН-]е, и поэтому
ki = k2K (моль/дм3).
Отсюда
1/т = Й2 {к (моль/дм3) + [Н+]е + (он- |е} =
— (Я4- v'^Kn } (моль/дм3) = 2-10-’&2(моль/дм3).
Поэтому
А2 = 1/(37 Ю-Эс)X (2-10’’ моль/дм3) « 1,4 -1011 моль-1- дм3-с-1.
Из этого следует, что
= (1,4 1011 моль-1 дм3'С-1)Х (1,8-10-16) моль/дм3 = 2,4- 10’5c_1.
Комментарий. Заметим, что необходимо следить за размерностью: К и К»- без-
размерные, k2 выражается в моль—дм3-с-1 и —в с_[.
Положение равновесия реакции зависит от температуры, если
энтальпия реакции отлична от пуля (см. т. 1, разд. 9.2), поэтому
метод скачка температуры в принципе подходит ко всем таким
Случаям. Равновесные концентрации также зависят и от давления,
если объем системы во время реакции изменяется. Из этого сле-
дует, что метод скачка давления также является возможным. Это
так, однако равновесие менее чувствительно к изменению давле-
ния, чем к изменению температуры, и поэтому этот метод исполь-
зуется менее широко.
Релаксационные методы развивались различными путями, и
один из методов связан с наблюдением поглощения системой уль-
тразвукового излучения. Другие релаксационные методы основаны
448
Часть 3. Изменение
на магнитном резонансе, они были описаны в гл. 19. Диэлектри-
ческая релаксация были описана в гл. 23.
Литература
Laidler R. I., Chemical kinetics, McGraw-Hill, New York, 1965.
Gardiner It7. C., Bates and mechanisms of chemical reactions, Benjamin, New
York, 1969.
frost A. A., Pearson R. G., Kinetics and mechanism, Wi]ey, New York, 1961.
Maccoll A., Homogeneous gas phase reactions, in Techniques of chemistry (Le-
wis E. S., ed.}, Vol. VIA, 47, Wiley-Intcrscience, New York, 1974.
Bunnett J. F., Kinetics in solution, in Techniques of chemistry (Lewis E. S., ed.).
Vol. VIA, 129, Wiley-Interscience, New York, 1974.
Bamford С. H., Tipper C. F, (eds,), Comprehensive chemical kinetics (Vols. 1—
16), Elsevier, Amsterdam, 1969—1976.
Wayne R. P., Photochemistry, Butterworths, London, 1970.
Wells С. H. J., Introduction to molecular photochemistry, Chapman and Ha]],
London, 1972.
Калверт Дж., Питтс Дж. Фотохимия. Пер. с англ. — М.: Мир, 1968,
Porter G., West М. A., Flash photolysis, in Techniques of chemistry (Ham-
mes G. G., ed.). Vol. VIB, 367, Wiley-Interscience, New York, 1974.
Bradley J. A',. Fast reactions, Clarendon Press, Oxford, 1974.
Chance В. B., Rapid flow methods, in Techniques of chemistry (Hammes G. C.,
ed.), Vol. VIB, 5, Wiley-]nterScience, New York, 1974.
Hammes G. G., Tempera lure-jump methods, in Techniques of chemistry (Ham-
mes G. G., ed.). Vol. VIB, 147, Wiley-Interscience, Ncwr York, 1974.
Knoche W., Pressure-jump methods, in Techniques of chemistry (Hammes G. G.,
ed.). Vol. VIB, 187, Wiley-Interscience. New York, 1974.
Noyes R. Af., Photostationary methods, in Techniques of chemistry (Hammes G. G.,
ed.), Vo]. VIB. 343. Wiley-lnlerscience. New York, 1974.
Benson S. W., O’Neal И. E., Kinetic data on gas phase unimolecular reactions,
NSRDS-NBS-21, US Department of Commerce, Washington D. C., 1970.
Trofman-Dickenson A. F., Milne G. S., Tables of bimolecular gas phase reactions,
NSRDS-NBS-9, US Deparlrnent of Commerce. Washington D. C., 1967.
Задачи
26.1. Константа скорости для реакции первого порядка разложения N^Os имеет
значение 4,8-10“'’ с~г. Чему равен полупериод реакции? Начальное давление
500 мм рт. ст. Какое будет давление через а) 10 с и б) 10 мин после начала
реакции?
26.2. Если концентрации выражены: а) в моль/дм3 и б) через давление в атм,
то каковы будут единицы для констант скорости Аз и А3 реакций второго в
третьего порядков?
26.3. Полуцвриод радиоактивного распада '4С равен 5730 голам (он испускает
В-.лУти с энергией 0,16 МэВ), Археологический образец содержал древесину, ко-
торая имела только 72% НС по отношению к живым деревьям. Каков его воз-
раст?
26.4. Одной из опасных последствий ядерных взрывов является образование
S0Sr п его внедрение в кости вместо кальция. Этот изотоп испускает р-лучи с
энергией 0,55 МэВ и его полупериод распада равен 28,1 лет. Предположим, что
1 мкр ’“Sr был поглощен новорожденным. Сколько останется его к а) 18 годам
и б) 70 годам?
26.5. Константа скорости реакции второго порядка AcOEt-|-NaOH(aq)—*-
—*AcONa+EtOH (где АсО — ацетатная группа) равна 0,11 моль-'.дм3-с-'. Ка-
26. Скорости химических реакций 449
кона будет концентрация через а) 10 с и б) 10 мигг после добавления этилаце-
тата к едкому натру, если начальные концентрации [NaOH] =0,05 моль/дма и
[АсОЕЦ — 0,10 моль/дм3?
26.6. В разд. 26.2 выведено интегральное выражение закона скорости второ-
го порядка [уравнение (26.2.76)] для реакции A-J-B—>Р. Найдите соответст-
вующую интегральную форму для случая, когда стехиометрия следующая: 2А-|-
+ЗВ-^Р.
26.7. Найдите интегральную форму закона скорости третьего порядка —d[A]/dl—
=—Jfe3[A]2[B] для реакций со стехиометрией 2A-J-B—^-продукты, если реагирую-
щие вещества в начальный момент находятся в стехиометрических соотношениях.
26.8. Повторите последнюю аадачу при условии, что количество В в 2 раза боль-
ше А.
26.9. Найдите выражение для полупериода реакции, приведенной в задаче 26.7-,
для временно а) когда концентрация А уменьшится наполовину от первоначаль-
ной, б) когда концентрация В уменьшится наполовину от первоначальной,
в) когда степень превращения реакции достигнет величины g—-g-.
26.10. ' В аутокаталитической реакции скорость прямой реакции увеличивается, и
иногда очень сильно, по мере образования продуктов, а затем, когда реагенты
прореагируют, реакция внезапно прекращается. Это может быть продемонстри-
ровано рассмотрением простой реакции А—>-В, имеющей следующий закон ско-
рости:—d[A]/dr=fej[A][B]. Решите это уравнение относительно В, взяв началь-
ные концентрации Ли В равными [А]о и [В]о соответственно.
26.11. Покажите, что Для реакции n-го порядка по A t,/j"o 1/[А]о-г.
26.12. Покажите, что соотношение ii/j/tm, где (Ца — полупернод и 6/»— четверть
периода (время, за которое концентрация А уменьшится до 3/4 ее первоначаль-
ной величины, принимая, что может быть записано как функция толь-
ко п и поэтому может использоваться для быстрой оценки порядка реакции.
26.13. Состав жидкой фазы реакции 2А—*-В исследовался в зависимости от вре-
мени спектроскопическим методом, и были получены следующие результаты:
мин 0 10 20 30 40
[В], моль/дм3 0 0,089 0,153 0,200 0,230
Киков порядок реакции? Чему равна величина константы скорости?
26.14. В опыте по исследованию стабильности замещенных аллильных ра-
дикалов (Trenwtth А. В.. J. Chem. Soc., Faraday I, 1973, 1737) в реакции
СНзСН(ОН)СН = СН2—*-Н2О+СНа = СНСН — СН2 контролировали скорость об-
разования воды. При 810 К были получены следующие результаты:
t, МИН 0,5 1,0 1,5 2,0" 2,5
Е, сма 1,0 1,4 1,6 1,7 1,8
—объем полученной воды. Найдите порядок и константу скорости. Молекулы
С4П6 образуются не так быстро, как вода, объясните почему.
26.15. Опыт, описанный в последней задаче, был повторен при нескольких тем-
пературах, и были получены следующие значения коцстянт скоростей:
71. К 773,5 786 797,5 810 810 824 834
* 1,63 2,95 4,19 8,13 8,19 14,9 22,2
’Зс’му равны энергия активации и предэкспоненциальный множитель реакции?
(Найдите единицы размерности и величину k, используя предыдущую задачу.)
26.16. При нагревании в газовой фазе при 500“С циклопропан изомеризуется в
пропилен. Реакцию исследовали газохроматографическн при различных началь-
29-242
450
Часть 3. Изменение
ных давлениях ра. Через определенное время t конечное давление циклопропана
равнялось р.
р0, мм рт. ст. 200 200 400 400 600 600
t, с 100 200 100 200 100 200
р, мм рт. ст„ 186 173 373 347 559 520
Чему равны порядок и константа скорости реакции при этих условиях?
26.17. Реакция второго порядка в газовой фазе записывается как 2А—*В, при-
чем и А, и В являются газами. Найдите выражения для зависимости общего дав-
ления от времени в реакционной системе. Пусть начальное давление, когда В
отсутствует, равно р0; постройте график зависимости р/рв от x = pakit. Какое
время необходимо для того, чтобы давление изменилось наполовину по отно-
шению к его конечному значению? Какова степень протекания реакции к этому
времени?
26.18, Газофазную реакцию 2А—»-В контролировали измерением общего давле-
ния в зависимости от времени. Были получены следующие результаты:
I, с 0 100 200 300 400
р, мм рт. ст. 400 322 288 268 256
Каков порядок реакции и значение константы скорости? За какое время реакция
пройдет па 99,99%?
26.19. Радиоактивный распад ядерного семейства был рассмотрен на стр. 422.
Рассчитайте относительное содержание нуклидов в зависимости от времени, ре-
зультаты представьте графически.
26.20. В изолированном государстве Ма.ТЬтузии были приняты строгие законы
как в отношении женитьбы, так и в отношении рождения. Штрафы были так ве-
лики, что ни один муж не изменял своей жене, и в каждой семье рождался один
сын и одна дочь. У их детей, живущих по тем же законам, рождались дети с
такой же средней скоростью. Найдите и решите законы скорости для этого об-
щества.
26.21. Присоединение галогеноводородов к олефинам сыграло основную роль в
исследовании механизмов органических реакций. В одном из современных иссле-
дований [Haugh М. J., Dalton D. R., J. Amer. Chem. Soc., 97, 5674 (1975)] ис-
следовали реакцию при высоких давлениях хлористого водорода (до 25 атм) и
пропилена (до 5 атм) при различных температурах; количество образующегося
2-хлорпропана определяли методом ЯМР. Покажите, что если реакпия А-|-В—*Р
протекает за короткое время Af, то концентрация продукта подчиняется закону
]Р]/[А] =^т+п]А]г"-1 [В]'ЧД(, если реакция имеет порядок m по А. и п по В. В се-
рии опытов отношение [циклопропан]/[пропилен] не зависело от концентрации
пропилена, но отношение [циклопропан]/{НС1] при постоянном количестве про-
пилена зависело от [НС1] и для значения 100 ч (которое является неболь-
шим во временнбй шкале реакции) это отношение увеличивается от 0 до 0,05,
0,03, 0,0] для р(НС1) = 10, 7,5, 5,0 атм соответственно. Каковы порядки реакции
по отношению к каждому реагенту?
26.22. Покажите, что закону скорости реакции последней задачи соответствует
следующий механизм:
2НС1 =р=* (НС1)г Къ I
НС1 + СН3СН - - СН2 7—* комплекс 1
(НС1)2 + комплекс ---» СН3СНС1СН3 + 2НС1 k2, медленно, 1
Какие дополнительные опыты можно провести для проверки этого механизма?
26.23. В опытах, описанных в последних двух задачах, изучали зависимость ско?
26. Скорости химических реакций
451
рости реакции от обратной температуры, и общая скорость реакции при 70°С
составляла приблизительно одну треть скорости при 19 °C. Рассчитайте кажу-
щуюся энергию активации и энергию активации лимитирующей стадии, если эн-
радити двух равновесных стадий были порядка —10,5 кДж/моль.
^6^24ДКонстанты скорости для реакции второго порядка атомов кислорода с
о№тическими углеводородами были измерены в работе: Atkinson R., Pitts J.N.,
J. Phys, Chern., 79, 295 (1975), В реакции с бензолом константы скорости
(в моль-1-дм3-с-1) равны 1,44-107 при 300,3 К, 3,03-107 при 341,2 К и 6,9 10т
при 392,2 К- Найдите предэкспоненциальнын множитель и энергию активации
реакции,
26.25, При изучении аутоокислення гидроксиламина (Hughes М. N., Nicklin Н. G.,
Shrimanker К., J. Chem. Soc., A, JL971., 3485) найденная константа скорости
Анабл, входящая в закон скорости —сДМН2ОН]/^(=Анавл[НН2ОН][О2], имела сле-
дующую температурную зависимость:
t, "С 0 10 15 25 34,5
Ю4 Анабл, с-1 0,237 0,680 1,02 2,64 5,90
Какова энергия активации реакции? Такой же анализ проведите н задаче 26,28.
26.26. Равновесная реакция А^±В имеет первые порядки в обоих направлениях.
Найдите выражение для концентрации А в зависимости от времени, если пер-
воначальные количества А и В равны [А]о и [В]о соответственно. Каков конеч-
ный состав системы?
£26,27—43 задаче 26.16 изомеризация циклопропана рассмотрена в ограниченном
интервале давлений. Для проверки механизма Лщщемана, предполагающего
реакции первого порядка, необходимо получить данные при низких давлениях.
Они были получены в работе: Pritchard Н. О., Sowden R. G., Trotman-Dicken-
son A. F,, Proc, Roy. Soc., A, 217, 563 (1953).
p, мм рт. ст. 84,1 11,0 2,89 0,569 0,120 0,067
104*ieff. c-1 2,98 2,23 1,54 0,857 0,392 0,303
Проверьте применимость теории Линдемана к этим данным [см. уравнение
26.28. Кинетика аутоокисления гидроксиламина в присутствии ЭДТА (этилен-
диаминтетрауксуспой'кислоты! была изучена в интервале концентраций гндрок-
сид-ионов от 0,5 др 3,2 моль/дм3 (ссылка дана в задаче 26.25). Реакция проте-
кает по механизму
быстро
NH2OH + OH--------->- NH2O”-|-HaO,
Медленно
NHaO~ О2 ----------** продукты
с законами скорости
~d [(NHaOH)]/d( =Ана6л [(NHaOH)J [OJ,
- d [(NHaOH)i/d( =kt [NHaO-J [OaJ,
где [(NH2OH)1 — общая концентрация гидроксил амина, т. е. [NHSOH] +[NHaO-l
11 ^=Анаб.,'// (/ — доля гидр оксид амина, присутствующего в виде NH2O~). Пока-
жите, что график зависимости 1/^Навл от [Н+] должен представлять прямую ли-
нию и что из его наклона можно определить константу кислотной диссоциации
NrljOH.
29*
452
Часть 3. Изменение
26.29. Данные, представленные ниже, относятся к реакционной системе, описан-
ной в последней задаче. Найдите рА'а для гидроксиламина и количество присут-
ствующего NHjO” при каждой концентрации ОН--
[ОН~], моль/дм® 0,50 1,00 1,6 2,4
10* йна6л, с-1 2,15 2,83 3,32 3,54
26.30. Механизм Райса — Герцфельда для дегидрирования этана был изложен на
стр. 434, и было отмечено, что он ведет к кинетике первого порядка. Подтвер-
дите это и найдите приближения, которые ведут к закону скорости, приведен-
ному на стр. 433, Как можно изменить условия, чтобы реакция имела другие
порядки?
26.31. Для термического разложения ацетальдегида (этаналя) был предложен
следующий механизм;
CHjCHO -----►*СНэ+*СНО k„,
•СН3 + СН3СНО ----СН4+‘СН2СНО
*СН2СНО----->СО+‘С113 kc,
•СН3+ -СИ,-----»• СН5СН, йщ
Найдите выражение для скорости образования метана и скорости расходования
ацетальдегида.
26.32. Число фотонов, падающих на образец, может быть о;1рсделено различными
методами, одним из классических методов является химическая актинометрия.
Разложение щавелевой кислоты (СООН)2 в присутствии сульфата уранила
(UO2)SO4 протекает через следующие стадии;
uqT-Mv —(Vol*)*,
(Uoij+y + (соон)2 —> ucl* + н,о + со2 + со
с квантовым выходом 0,53 при данной длине волны. Количество щавелевой кис-
лоты после облучения может быть определена титрованием (КМпО4) и получен-
ная степень превращения может быть использована для нахождения числа па-
дающих фотонов. В специальном опыте актинометрический раствор состоял из
5,232 г щавелевой кислоты в 25 см3 воды (вместе с солью уранила). После 5-мн-
нутного облучения полученный раствор протитровали раствором КМпО4 в кон-
центрации 0’212 моль/дм3, и для полного окисления остающейся щавелевой кис-
лоты потребовалось 17,0 см3 KMnQ4. Чему равна скорость падения фотонов при
длине волны опыта? Выразите ответ в единицах фотон/с и эйцштейн/с, где эйн-
штейн—это 1 моль фотонов,
26.33. Интенсивность флуоресценции или фосфоресценпии возбужденной молеку-
ла М зависит от эффективности некоторых конкурирующих химических процес-
сов тушения. Рассмотрим газофазную флуоресценцию возбужденной молекулы М,
генерируемой в реакции M+frvj—при интенсивности поглощенного света /а.
За поглощением следует тушение M*+Q—это реакция второго порядка
с константой скорости конкурирующая с флуоресцентным процессом М*—»•
—имеющим константу скорости и интенсивность флуоресценции
Покажите, что эта схема приводит к соотношению Штерна — Фольмера
l/Zf^tl/yfl + fMrW-
связывающему интенсивность с концентрацией тушителя. Таким образом, соот-
ношение k4/ki может быть определено нз графика зависимости от концентра-
ции тушителя. Кроме того, покажите, что, если измерять скорость затухания
флуоресценции, то можно найти непосредственно
26. Скорости химических реакций
453
26.34. При облучении бензофенона ультрафиолетовым спетом он возбуждается
в синглетное состояние. Этот синглет быстро превращается в триплет, который
фосфоресцирует- Триэти.тамин NEta является для триплета тушителем. В опыте
с использованием метанола в качестве растворителя интенсивность фосфоресцен-
ции изменялась с концентрацией амина так, как это показано ниже. Эксперимент
по флеш-фотолизу также показал, что в отсутствие тушителя полупернод флуо-
ресценции равен 2,9-10-7 с. Чему равно значение k4?
[Q], моль/дм3 0,001 0,005 0,010
/ft произв. ед, 0,41 0,25 0,16
26.35, Найдите выражение для скорости исчезновения частицы Л в фотохимиче-
ской реакции со следуюпщм механизмом: а) инициирование светом с интенсив-
ностью I, А—»-2R‘, б) развитие цепи, A-|-R‘—»-R‘ + B, константа скорости &Р;
в) обрыв цепи, R’-f-R’—*-R2, константа скорости kt. Отсюда покажите, что изме-
рения скорости дадут то.тько комбинацию Ар и kt, если достигается стационар-
ное состояние, но они обе могут быть получены, если стационарное состояние
не достигается. Последнее лежит в основе метода вращающегося сектора (см. ли-
тературу).
26,36, Фотохимическое хлорирование хлороформа в газовой фазе следует закону
скорости d[CC14]/d/=fcI(2[Cl2] ‘Л/’1’2. Придумайте механизм, который приведет
к этому закону скорости при очень высоких давлениях хлора.
26.37, Обычное рассмотрение равновесий неприменимо, если реакция происходит
при поглощении света. В этом случае стационарные концентрации продуктов и
реагирующих веществ могут сильно Отличаться От равновесных величин: это
называется фотостационарным состоянием. Например, предположим, что в реак-
ции А—*-В происходит поглощение света, и скорость поглощения равна k,: [А]/а,
а обратная реакция В—*-Л является бимолекулярной реакцией второго порядка,
происходящей со скоростью fe[B]2 Какова фотостаццонарная концентрация В?
Почему фотостационарное состояние отличается от равновесного состояния?
26.38. Когда антрацен растворяют в бензоле и облучают ультрафиолетовым све-
том, он димеризуется. В концентрированных растворах димеризация происходит
с большим квантовым выходом (и небольшой флуоресценцией), но в разбавлен-
ных растворах флуоресценция дезактивирует возбуждение молекул и процесс
димеризации протекает с низким квантовым выходом. Рассчитайте зависимость
квантового выхода от концентрации, основываясь на такой последовательности:
A-l-Av—»-А*, А*-(-А—»-А2, Л*—t-A-J-Ziv,. Объясните наблюдения.
26,39. Изучение реакций сгорания связано со знанием концентраций атомов во-
дорода и гидроксильных радикалов. Измерения в струевой системе с использова-
нием ЭПР для обнаружения радикалов дали следующую информацию о реак-
циях:
Н4-ЫОа ------>- ОН + NO, #2 = 2,9-1010дм3/(моль'С),
ОН + ОН -----> Н2О + О, = 1,55-10* дм3/(моль-с),
О + ОН ------» Оа + Н, k£ — 1,1 • 10to дмЗДмолЬ'С).
(Bradley J. N., Hack IP., Hoyermann Wagner H. G., J. Chem. Soc. Faraday I,
1973, 1889). Ha основе начальных концентраций атомов водорода и NO2, равных
4.5-10-и 5,6-10“10 моль/см3 соответственно, рассчитайте и постройте кривые
зависимости концентраций О, О2 и ОН от времени в интервале 0—10 нс.
26.40, При изучении реакции между атомами кислорода и хлора струевым ме-
тодом при высоких давлениях хлора (Bradley I. N., Whytock D. A., Zaleski Т.А.,
Chem. Soc., Faraday I, 1973, 1251) получен прямолинейный график зависимости
11 [О]о/[0] от расстояний вдоль струевой трубки, где [О]о — концентрация кис-
лорода при нулевом давлении хлора. Используя скорость струи, равную 6,66 м/с,
454
Часть 3 Изменение
и данные, приведенные ниже, найдите константу скорости реакции О+С12—>
—*-С1О4-С1.
Расстояние, см 0 2 4 6 8 10 12 14 16 18
1п [О]0/[О] 0,27 0,31 0,34 0,38 0,45 0,46 0,50 0,55 0,56 0,60
[О]0 — 3,3-10-’ моль/дм3, [С12] = 2,54- 10"г моль/дм3,
р = 1,70 мм рт.ст.
26.41. В подраэд. «Релаксационные методы» разд. 26.5 мы рассмотрели релакса-
цию равновесной реакции первого порядка. Покажите, что реакция Аз^В^-С
первого порядка в прямом направлении и второго в обратном также релаксирует
экспоненциально при небольших отклонениях От равновесия. Найдите выражение
для времени релаксации через k, и k2.
26.42. Начальная скорость выделения кислорода при действии фермента на суб-
страт была измерена для ряда концентраций субстрата; данные приведены ниже.
Каково значение констант Михаэлиса для реакции?
[S]. моль/дм3 0,050 0,017 0,010 0,005 0,002
Скорость, мм3/мин 16,6 12,4 10,1 6,6 3,3
27 Динамика молекулярных реакций
Изучаемые вопросы
После тщательного изучения этой главы вы сможете:
1. Описать теорию столкновений бимолекулярных газовых ре-
акций (стр. 457).
2. Рассчитать константу скорости реакции второго порядка из
теории столкновений [уравнение (27.1.2)],
3, Определить и измерить P-фактор и сечение реакции
(стр, 459),
4. Различать реакции в растворах, контролируемые диффузией
и контролируемые активацией (стр. 461).
5. Связать константу скорости реакции второго порядка с ко-
эффициентом диффузии [уравнение (27.1.5)] и вязкостью [урав-
нение (27.1.6)].
6. Объяснить термины активированный комплекс, координата
реакции и переходное состояние (стр. 466).
7. Описать теорию активированного комплекса (стр. 466).
8. Вывести уравнение Эйринга для скоростей реакций [урав-
нение (27.2.7)].
9. Использовать уравнение Эйринга для расчета константы
скорости [уравнение (27.2,8)] и величины кинетического изотопно-
го эффекта [ уравнение (27.2.9)].
10, Определить функцию активации Гиббса, энтропию актива-
ции и энтальпию активации (стр. 474).
11, Объяснить сущность и определить величину кинетического
солевого эффекта [уравнение (27.2.16)].
12. Указать, как молекулярные пучки используются для изу-
чения реакционных столкновений (стр, 478),
13. Изобразить поверхность потенциальной энергии простой ре-
акции (стр. 479).
14, Описать информацию, получаемую из измерений углового
распределения продуктов (стр. 484).
15. Различать поверхности притяжения и отталкивания и объ-
яснить, как они контролируют энергию, необходимую для реакции
(стр. 485).
456 Часть 3. Изменение
Введение
Теперь мы находимся у самого сердца химии. Здесь мы рас-
смотрим детальное поведение молекул в критические моменты ре-
акции. Происходят изменения макроформы, энергии, по величине
близкие к энергиям диссоциации, перераспределяются ио связям,
старые связи разрываются н образуются новые. Скорость, с кото-
рой молекулы обмениваются атомами или группами атомов, или
скорость перехода простой молекулы в другую изомерную форму,
зависит от сил, действующих в высшей точке реакции, а они в
свою очередь зависят от детального расположения всех заряжен-
ных частиц, входящих в состав молекул, участвующих в данной
стадии реакции.
Можно представить, что количественное предсказание скорос-
тей реакций и расчет константы скорости k-; бимолекулярной реак-
ции второго порядка являются чрезвычайно сложной задачей. Тем
не менее, как в большинстве сложных вопросов, общие черты
могут бытв установлены довольно просто, и лишь тогда, когда
требуются более глубокие знания о деталях, возникают трудности.
В этой главе мы рассмотрим три приближения, используемые при
интерпретации и расчете fe2. Простейшее приближение делается в
теории столкновений, в которой внимание ограничивается главным
образом определением скорости соударений, энергии которых до-
статочно велики. Mbi рассмотрим некоторые недостатки теории
столкновений и увидим, что она может бвтть улучшена более по-
дробным рассмотрением природы образующихся при столкновении
частип; это теория активированного комплекса. Несмотря на то
что эта теория имеет серьезные недостатки, одним из путей, улуч-
шающих описание, является экспериментальное рассмотрение от-
дельных .молекулярных столкновений, происходящих в действи-
тельных реакциях. Современный технический прогресс делает это
возможным; использование молекулярных пучков позволяет де-
тально исследовать путь реакционных столкновений и создать мо-
дели динамики молекулярных реакции.
Понимание факторов, влияющих на значение fe2, полезно как
для технологии, так и для теории. Одним из важных технологи-
ческих применений является разработка химических лазеров;
в разд. 18.4 мы видели, что если молекулы привести в возбужден-
ное состояние, а затем заставить их когерентно испускать энер-
гию, то можно получить лазерный эффект. Исследования, выпол-
ненные с помощью молекулярных пучков, показали, что продукты
некоторых реакций находятся в определенных возбужденных со-
стояниях. Это значит, что энергия таких термических реакций мо-
жет быть прямо превращена в когерентное электромагнитное из-
лучение. Создание подходящих условий для действия химического
лазера зависит от дсталвного знания всех скоростей предполагав-
27, Динамика молекулярных реакций 457
мых процессов, а эта сведения могут быть получены из наблю-
дений и теорий, описанных ниже. Другое применение связано с
возможностью увеличения скоростей химических реакций за счет
перевода реагирующих веществ в особые энергетические состоя-
ния. Например, некоторые реакции идут быстрее, если энергия их
молекул сконцентрирована 1га колебательных модах, а тге па по-
ступательном движении. Если поступательная кинетическая энер-
гия может переходить в колебательную, то эти реакции могут
осуществляться быстрее. Предсказание таких реакций связано с
пониманием того, как в реакции выделяется энергия и каким пу-
тем происходит перераспределение связей в сталкивающихся мо-
л екулах.
27.1. Молекулярные столкновения
Химическая реакция зависит от столкновения двух частиц. Мож-
но представить, что в газовой фазе А ударяется в В со скоростью,
равной частоте столкновений, а прореагируют они или нет, зависит,
кроме других факторов, от того, сколько энергии выделяется при
столкновении. Мы не можем так легко говорить о «соударении»
в растворе, поскольку движение частиц относительно друг Друга
является диффузионным и растворитель препятствует ггх свобод-
ному перемещению. Тем не менее скорость, с которой потенциаль-
но реакционноспособные части вы сталкиваются друг с другом,
может быть рассчитана и связана с коэффициентами диффузии
растворенных частиц. В дачном разделе мы сконцентрируем свое
внимание тга скорости этих двух типов столкновений и посмотрим,
как они влияют на скорость реакции.
Теория столкновений. Идеи, лежащие в основе теории столкнове-
ний бимолекулярных газовых реакций, были изложены в преды-
дущей главе (разд. 26-3). Мы видели, что, если средняя частота
столкновений равна Zab, скорость реакции зависит от се значения,
умноженного на долго столкновений; последние происходят с энер-
гией, большей некоторого порогового значения Еа, называемого
энергией активации реакции:
скорость-----d (AA/V)/d/=ZABexp(— EjRT).
Часюта столкновений может быть рассчитана на основе кинети-
ческой теории газов (гл. 24)
= ц (8ЬТ/щ^ (Na/V) (Ab/V),
где о—сечение столкновений, которое для жестких сферических
частиц с радиусами RA и RB равно л(7?,$ 4-/?в)2 (разд. 24.2), ц —
приведенная масса сталкивающихся частит а Ад и Ав — число
каждой части и ы в объеме. Поскольку концентрация обычно выра-
жается в лголях, запишем NA/V~L [А] и точно такое же соотно-
458
Часть 3. Изменение
шение для В, где L—число Авогадро и [А]—число молей Л в
единице объема; тогда приходим к выражению
— d [А1/Л = а(8АТ/лц)‘/2£ [A] [BI ехр (-Еа//?Г).
Поскольку константа скорости второго порядка определяется из
уравнения скорости
А) — d [A]/d^Aa [A] ]Bj,
из теории столкновений получаем следующее выражение для ks:
=о£ (вАТ/лц)1/* ехр (—£а//?Г), (27.1.1)
которое качественно'Аюгласуется с наблюдаемой температурной
зависимостью аррениусовского типа:
А2-АехР(— EJRT). (27.1.2) |
Это несомненно подтверждает, что теория столкновений отражает
основные особенности механизма реакции, а именно то, что мо-
лекулы должны столкнуться (предэкспоненциальпый множитель)
и что прореагируют только те молекулы, энергия столкновения
которых достаточно большая (экспоненциальный множитель).
Однако одного качественного совпадения недостаточно, и нам
нужно выяснить, корректна ли теория количественно. Энергия ак-
тивации является сложным свойством молекул, и поэтому трудно
ожидать хорошего совпадения экспериментальных и расчетных
величин. В то же Время предэкопонепциальный множитель хорошо
определен и, если не обращать внимания на некоторые сомнения
относительно того, что подразумевается под сечением столкнове-
ния, зависит только от массы сталкивающихся частиц и поэтому
легко может быть рассчитан.
Простейшая операция состоит в том, чтобы отождествить это
ссчспие с сечением простых нереакциопных столкновений. Оно
может быть рассчитано из таблиц молекулярных радиусов или
определено экспериментально из измерений свойств реальных га-
зов (гл. 23). В табл. 27.1 сравниваются некоторые расчетные зна-
чения А с экспериментальными, полученными из аррениусовского
графика завнеимости Info от 1/Т. Хорошее согласно между теорией
и экспериментом наблюдается лишь в одном случае, в большинст-
ве же случаев теория и эксперимент не совпадают. Некоторые
Пред экспоненциальные множители по порядку 'величины меньше
рассчитанных: это свидетельствует о том, что столкновения с до-
статочной энергией не являются едипственнЫ'М критерием осуще-
ствления реакции и необходимо принимать во внимание другие
факторы, такие, как взаимная ориентация сталкивающихся частиц.
Второй недостаток заключается в том, что одна из реакций в таб-
лице имеет больший предэкспоненциальный множитель, чем пред-
сказывает теория столкновений. По-видимому, это указывает на
27. Динамика молекулярных реакций
459
Таблица 27,1
Энергии активации и преддяспоиенциальные
множители для газофазных реакций
Реакция А, дмЗ '(моль-с) Еа, кДж/моль Р—о*/а
эксп.
2NOC1 >- 2N0-J- С12 1,0- low 6,3101в 103,0 0,16
2NO, ——>- 2NO-}- О2 2,0-10й 4,0-10“ 111,0 5-Ю-2
2СЮ С12 4- Оа 6,3-107 2,5-10м 0,0 2,5-10-3
К+Вг2 ч- КВт 4-Вг 1,0-1012 2,1-104 0,0 4,8
Н2 + С2Н4 ► С2Нв 1,24-10’ 7,3-1011 180 1,7-10-’
то, что реакция происходит быстрее, чем сталкиваются молекулы!
Для того чтобы разрешить этот явный парадокс, можно напомниты
что рассчитанное значение А зависит от выбранного значения се-
чения, а приведенные в таблице значения основаны на использо-
вании сечений нереакциониых столкновений. Может случиться,
что расстояние, на которое молекулы сближаются для реакцион-
i ного столкновения, будет отличаться от расстояния, которое ведет
просто к отклонению от направления движения.
Оба эффекта — необходимая ориентация и требуемое расстоя-
; ние — могут быть учтены путем замены сечения столкновений о
в уравнении (27.1.1). на сечение реакции о*. Более принято выра-
жать сечение реакции ч^рез^Рчрактор, Tax' что ц*=.Ро; тогда кон-
станту скорости можно записат^глЦд'ующим образом;
= Р [сгА(8&77л|л)1/з) |ехр(— E.JRT)]. (27.1.3)
Из этого выражения видно, что на Ай влияют три фактора. Второй
фактор — транспортное свойство, показывающее, насколько быстро
движутся частицы относительно^ друг друга. Третьим экспоненци-
альным фактором является энергетический критерий, регулирую-
щий долю столкновений, которые имеют энергию, достаточную
для протекания реакция. Первый фактор Р включает локальные
свойства реакции; необходимые ориентации частиц и детали того,
насколько они должны сблизиться, чтобы произошла реакция.
Пример (вопрос 3). Рассчитайте P-фактор для разложения NO2 по реакции
2NQ2—>2NO+O;. если известно, что предэкспоненциальный множитель равен
2.0-109 моль-* 1-дм3-с-1.
Метод Используем уравнение (27,1.3) с Мг=46 и а=0,10 нм2. Возьмем темпе-
ратуру 500 К. Находим Р из Р=о*/о, где а* — значение, при котором по тео-
рии столкновений получается на и лучшее совпадение экспериментальных значе-
ний А с теоретическими.
460
Часть 3. Изменение
Ответ. Теоретический предэкспоненциальный множитель равен
А = a*L (8й7/лр.)1/а «
= ч*Х(6,022-1023 моль-^х
8х(1,381 • Ю-83 Дж/К)х(500К) | V»
nxf~-46^Х(1,66 10~27 кг) |
= (4,09- 10se моль-1 м с-1) а*.
Экспериментальное значение А равно 2,0 -103 моль-1 -дм3-с- *, или 2,0-10® моль 1X
Хм’-с”1; следовательно,
с* = (2,0-10® моль-1-м3 c-i})(4,09-102® моль-1-м-е-1) =
= 4,9 Ю-21м3, или 6,910-3 нм2.
Так как с=0,10 нм3, то Р=0,05.
Комментарий. Грубо говоря, чем сложнее сталкивающиеся молекулы, тем меньше
значение Р.
Теория была бы совсем хорошей, если бы мы смогли рассчи-
тать стерический фактор Р. В некоторых случаях это можно сде-
лать. Возьмем в качестве примера реакцию КЧ-Вг2, для которой
Р=4,8, что указывает на «аномально» большое сечение реакции:
о*^4,8о. Было высказано предположение, что реакция протекает
по гарпунному механизму. Это блестящее название обосновано
следующим механизмом реакции: атом К приближается к моле-
куле Вг2, и, когда они сближаются достаточно близко, электрон
(гарпун) переходит к Вг2. Вместо двух нейтральных частиц теперь
образуются два иона, между которыми имеется кулоновское при-
тяжение: это веревка гарпуна. Под ее влиянием ионы начинают
двигаться вместе (между ними находится веревка), начинается
реакция, появляются КВг+Вг. Гарпун расширяет сечение реак-
ционного столкновения, и, взяв с* как среднее из значений для
простых столкновений К + К и Вг3Ч-Вг2г мы занижаем скорость
реакции.
Этот качественный аргумент объясняет значение Р>1, однако
мы можем продолжить оценку его истинной величины. Расчет свя-
зан с оценкой расстояния между атомом К и молекулой Вг2, энер-
гетически наиболее благоприятного для перехода электрона от
одной частицы к другой. Необходимая энергия состоит из трех
вкладов. Первый вклад вносит потенциал ионизации, требуемый
для удаления электрона из атома К; он равен ЦК)- Когда элек-
трон переходит к Вг2, образуя Вг^-, энергия выделяется, и степень
ее понижения соответствует сродству электрона к молекуле
£а(Вг2). Третий вклад обусловлен кулоновскими силами притя-
жения между двумя ионами. Если расстояние между ионами рав-
но R, то электростатическая энергия составит— е2/4лгор. Если
электрон движется от одной нейтральной частицы к другой, нахо-
27, Динамика молекулярных реакций 461
дящейся на расстоянии R, то общее изменение энергии приблизи-
тельно равно
Д£ = I (К) — £а (Brs) -е2/4лео£;
/(К) больше, чем £д(Вг2), и Л£ становится отрицательной только
тогда, когда R уменьшается до величины меньше некоторого кри-
тического значения £*, определяемого равенством
ег/4ле0£* / (К) — £Л (Вга).
Когда частицы находятся на этом расстоянии, гарпун выстрели-
вает, и поэтому сечение реакции может быть выражено как
a^—nR*2. Это в свою очередь означает, Что P-фактор равен
Р —o*/o=--R*3/d2 « [/ (К) — £а (Вга)]}2,
где d = R (К) +£ (Вг2). Так как /(К)=4,3 эВ, £Л(Вг2) =3 эВ и
dft;310 пм, то получаем Р~12, что умеренно хорошо согласуется
с экспериментальным значением 4,8.
Этот пример указывает па два обстоятельства. Прежде всего
Р-фактОр нельзя считать совсем бесполезной величиной для вве-
дения в уравнение, поскольку в некоторых случаях он может быть
рассчитан. Другим обстоятельством является обратное утвержде-
ние: многие реакции значительно более сложные, чем реакция
К + Вгг, и мы не можем надеяться на такой простой путь расчета
Р. Желательно иметь более точную теорию для р, в которой не
нужно было бы отгадывать его значение.
Реакции в растворе. Диффузионный контроль. Расчет скоростей
реакций в растворе также может быть сведен к расчету скоростей
столкновений и необходимой энергии. Теперь молекулярное дви-
жение является диффузионным вместо беспорядочного, но понятия
энергии активации и стерических требований остаются. Некоторые
измеренные энергии активации приведены в табл. 27.2. В данном
разделе мы продолжаем рассмотрение частоты столкновений.
Таблица 27.2
Энергии активации и предэкслоненпиальные множители для реакций к растворе
Реакция Растиорите-ть Л, ДМЗ/(М0ЛЬ-с) Ед, КДж/МОЛЪ
СН3С1 +CHSO- Метанол 1 . 76-1012 100,4
СН3Вг + сн3о- 1,74-Ю1» 91,3
снз1 + сн3о- » 2,01-10J3 91,8
CgHjBr4* (С2Нб)й5 Бензиловый спирт 1,40-iO'i 106,6
CH3I+CeHsN(CH3}2 7,08-1О6 60,3
462
Часть 3. Изменение
Столкновения в растворе имеют совсем другую природу, чем
в газах. Молекулы должны прокладывать себе путь через раство-
ритель, и поэтому частота столкновений значительно меньше, чем
в газе. Тем не менее появляется другой важный фактор. Посколь-
ку молекула очень медленно мигрирует в область возможного ре-
акционного столкновения с другой частицей, она также очень мед-
ленно мигрирует от нее. Иными словами, участники пары столк-
новения задерживаются в непосредственной близости друг от
друга гораздо дольше, чем в газе, и поэтому вероятность проте-
кания реакции намного увеличивается. Кроме того, энергия акти-
вации реакции в растворе значительно более сложная величина,
чем в газе, так как каждая сталкивающаяся пара окружена рас-
творителем и энергия определяется всеми взаимодействиями.
Эту сложную ситуацию можно разделить на более простые
части. Прежде всего составим кинетическую схему общего про-
цесса, принимая во внимание наряду с реакцией две стадии миг-
рации. Предположим, что скорость образования пары столкнове-
ния пропорциональна концентрациям компонентов А и В:
*d
А 4-В ---> (АВ), d[(Ab}\ldt = [BJ.
Как мы увидим, kd определяется коэффициентами диффузии час-
тиц. Сталкивающаяся пара может распадаться без реакции или
реагировать, давая продукты- В обоих процессах предполагается
участие растворителя, но, поскольку он присутствует в большом
и постоянном избытке, нет необходимости точно учитывать его
концентрацию (как говорилось в предыдущей главе, и распад,
и реакция пары столкновения являются стадиями псевдопервого
порядка). Поэтому общую схему можно записать в виде
А -у В > (АВ) ----> продукты Р.
<<1
Стационарная концентрация (АВ) может быть найдена из урав-
нения скорости
dJ(AB)j/d/ = fcd [А] [В]-[(АВ)]-k, )(AB)f —О,
которое дает
[(АВ)] = (М^-а+М [А] [В],
и поэтому общая скорость выразится следующим образом:
d|P]/di=*i[(AB)l = (^:^r)[Aj[B].
Общий закон скорости имеет второй порядок, и константа скоро-
сти второго порядка равна
A2=^IAd/(A_d-|-A1). (27.1.4)
27, Динамика молекулярных реакций
463
Рис. 27.1. Поток через сферическую поверхность и результирующая концентра-
ционная кривая. Реакция происходит в радиусе R*.
Теперь можно отметить два предельных случая. Если скорость
распада пары столкновения значительно меньше скорости, при ко-
торой из нее образуются продукты, т. е. ^-а<£^ь то выражение
для £2 выглядит так:
k2 As k^/ki —kA.
В этом пределе скорость реакции определяется скоростью диффу-
зии частиц в среде; это так называемый предел, контролируемый
диффузией, когда реакция контролируется диффузией. В дальней-
шем мы увидим, что характерным признаком такой реакции яв-
ляется величина константы скорости порядка 109 дм3/(моль-с) или
больше. Поскольку соединение частиц с нссп арен ними спинами
происходит с очень небольшой энергией активация, реакции ради-
кальной и атомной рекомбинации часто контролируются диффу-
зией.
Другой предел связан с высокой энергией активации стадии
реакции, и тогда В этом пределе, контролируемом акти-
вацией, общая константа скорости выразится следующим образом:
где К — константа равновесия для частиц А, В и (АВ). В этом
случае скорость зависит от накопления энергии в паре столкнове-
ния в результате ее взаимодействия с молекулами растворителя.
Мы можем найти скорости реакций, контролируемых диффу-
зией, расчетом скорости, с которой молекулы диффундируют вмес-
те. Простейшим приближением является рассмотрение неподвиж-
ной молекулы А, помещенной в растворитель, содержащий также
молекулы вещества В. Рассмотрим сферу радиусом г, окружаю-
щую неподвижную молекулу. Чему равен поток молекул вещест-
ва В через поверхность сферы (рис. 27.1)? Так как поток J веще-
ства равен количеству вещества, проходящего через единицу пло-
щади в единицу времени (разд. 24.3), то общий поток У через сфе-
464
Часть 3. Изменение
рическую поверхность площадью 4лг2 составит У = 4лг2/. Согласно
первому закону диффузии Фика [уравнение (24.3.1)], поток про-
порционален градиенту концентраций Arf[B][dr, и поэтому
/ = 4№£ (DBd [B]/rfr),
где £>в — коэффициент диффузии для молекул вещества В.
Концентрация вещества В на любом расстоянии от А может
быть найдена интегрированием последнего уравнения. Нам необ-
ходимы следующие сведения: 1) когда гйэоо, концентрация В бу-
дет той же самой, что в объеме раствора, т. е. [В]; 2) общий по-
ток через оболо-чку будет одинаковым независимо от ее расстоя-
ния от Л, поскольку никакие молекулы не разрушатся до тех пор,
пока молекулы А и В не соприкоснутся, поэтому У будет постоян-
ным, не зависящим от г. Следовательно,
[В] оо га
j d [В] =J( f/inr2DBL)dr=(f/4aDBL) pl/ra)dr.
[B]r r 'r
Tаким образом,
[В]— [В],=//4лг£>в^
"и концентрация изменяется обратно пропорционально расстоянию.
Теперь предположим, что на некотором критическом расстоя-
нии R* молекулы А и В «соприкоснутся», начнется реакция и В
будет исчезать, т, е. предположим, что при r = R* [В]н =0. При
подстановке в последнее уравнение получим выражение для об-
щей скорости потока по направлению к А:
/ = 4.ч^*ПвТ[В].
Скорость реакции, контролируемой диффузией, определяется ско-
ростью потока частиц В к молекуле вещества А; если концентра-
ция вещества А равна L[A], то общая скорость реакции в образце
выразится следующим образом: 4nR*£)BL2[A] [В]. Предположение
о том, что молекулы А неподвижны, а движутся только молеку-
лы В, конечно, нереально, но его легко исправить, если коэффи-
циент диффузии Ов заменить на О = £)_«.+Оц. Тогда константа ско-
рости реакции, контролируемой диффузией, затгишется
^=Ad = 4^R*DL. (27.1.5)
Последнее выражение можно преобразовать дальше, используя
соотношение Стокса — Эйнштейна, связывающее коэффициент
диффузии с вязкостью среды (стр. 394) (подразд. 25.2.А):
DA=kT/6rn]RA, Л в == nr]RB.
где и — эффективные гидродинамические радиусы А и В.
'Так как Это соотношение не очень точное, то, ограничив рассмот-
27. Динамика молекулярных реакций
465
рение случаем, когда R&~Rs^~-R*t мы внесем лишь небольшую
дополнительную ошибку; в этом случае
k2^8LkT/3r\=^8RT/3x\. (27,1.6)
Отметим, что радиусы сокращаются, и, таким образом, в этом
приближении k2 не зависит от частиц, участвующих в реакции.
Пример (вопрос 5). Рассчитайте константу скорости реакции второго порядка
для рекомбинации атомов иода в гексане при комнатной температуре.
Метод. Используем уравнение (27.1.6). Вязкость растворителя равна 0 326 сП
[3,26.10-* кг/(м-с)] при 298 К.
Ответ. Из уравнения (27.1.6)
= (8/3)х [8,314 Дж/(К-моль)]х(298 К)/[3,26-10"* кг/(м-с)[ =
= 2,0-10’ моль-1-м».с-1, или 2,0101° моль-1 дм®>с-1.
Комментарий. Экспериментальное значение равно 1,3-1010 моль-1-дм®-с-1; с уче-
том данного приближения это очень хорошее совпадение.
Относительно реакций, контролируемых диффузией, .можно сде-
лать еще два замечания. Первое касается температурной зависи-
мости таких реакций. Поскольку вязкость растворителя подчиня-
ется уравнению р ^Aexp'Ea/RT) [т. I, гл. «Введение», уравнение
(2-1)], k2 имеет температурную зависимость аррениусовского типа:
Е, аэ (S^T/ЗЛ) ехр (— EjRT).
Второе замечание состоит в том, что многие реакции, контролируе-
мые диффузией, являются реакциями между ионами. Если ионы
имеют одинаковый заряд, то можно предположить, что скорость
столкновений будет меньше, чем рассчитанная в данном разделе,
но больше, если ионы имеют противоположные заряды. Влияние
заряда можно учесть, если уравнение (27,1,5) заменить на урав-
нение
^inPR^DL, (27,1.7)
где фактор Р равен
р _ 7 \ 7__________I___________\
4ле(|Л'г7?’й7 J — 1 )’
В этом выражении Л'г — относительная диэлектрическая проницае-
мость растворителя, a гА и —заряды частиц. Как и было уста-
новлено экспериментально, это выражение предсказывает, что ско-
рость должна зависеть от относительной диэлектрической прони-
цаемости среды. Заметим, что последнее уравнение является еще
одним примером расчета Р-фактора.
30—242
466
Часть 3. Изменение
Рис. 27.2. Энергетическая кривая реакции. На оси абсцисс отложена координата
реакции, на оси ординат — потенциальная энергия системы.
27.2. Теория активированного комплекса
Теперь от вопросов молекулярной подвижности перейдем к рас-
чету константы скорости бимолекулярной реакции с другой точки
зрения. Это приближение имеет большое преимущество, поскольку
оно позволяет прямо ввести P-фактор в выражение для константы
скорости, и пет необходимости в использовании искусственных
аргументов. Нельзя сказать, что эта теория закончена или очень
надежна: до сих пор делаются попытки выявить основные факто-
ры, влияющие на величину k?, нс принимая во внимание связан-
ные со временем динамические особенности действительных про-
цессов.
Координата реакции и переходное состояние. Основные особенно-
сти процесса изменения энергии частиц А и В по мере протекания
реакции и превращения их в продукты C + D иллюстрирует
рис, 27.2. Сначала присутствуют только частицы А и В, и их энер-
гия равна некоторой величине. В ходе реакции, по мере того как
частицы вступают в контакт, искажаются и начинают обменивать
или отщеплять атомы, энергия возрастает до максимума, затем
по мере выделения продуктов энергия падает и достигает величи-
ны, характерной для продуктов. Горизонтальная ось рисунка пред-
ставляет собой путь реакции и называется координатой реакции.
Высшая точка реакции находится на максимуме кривой, раз-
деляющем реагенты от продуктов. В максимуме пара реагирую-
щих веществ в такой степени сближена и искажена, что малейшее
искажение в соответствующем направлении приведет систему к
продуктам реакции. Эта критическая конфигурация называется
переходным состоянием реакции. Если реакция проходит через эту
критическую конфигурацию, то появление продуктов реакции не-
избежно.
27. Динамика молекулярных реакций
467
Чтобы яснее представить идею о координате реакции и пере-
ходном состоянии, необходимо привести какой-нибудь пример,
рассмотрим подход атома водорода к молекуле фтора и для про-
стоты возьмем случай, когда атом водорода подходит в направ-
лении связи F—F. На большом расстоянии энергия системы равна
энергии изолированных атомов и молекул. Когда атом подходит
к месту, где его орбитали начинают перекрываться с орбиталями
фтора, связь F—F начинает растягиваться, энергия увеличивает-
ся и образуется новая связь между атомом водорода и одним ато-
мом фтора. Атом водорода подходит ближе (если он имеет доста-
точную энергию), при этом связь F—F растягивается, а связь F—Н
становится сильнее. В некоторой точке энергия такой составной
молекулы, активированного комплекса, имеет максимальное зна-
чение, и молекула подготовлена для движения через переходное
состояние. Бесконечно малое сжатие связи F—Н выводит ее из
переходного состояния: другая связь F—F затем растягивается
дальше и разрывается. Координата реакции представляет собой
развитие такой последовательности событий.
В действительности атомы водорода приближаются к связи
F—F под любым углом, и точно установить координату реакции
довольно трудно. Поэтому мы рассматриваем координату реакции
просто как указание на то, что при образовании активированно-
го комплекса участвующие молекулы искажаются, т. е. на те из-
менения, которые происходят с реагентами при переходе в акти-
вированный комплекс и через него в продукты реакции. Однако
мы можем быть уверенными, что в переходном состоянии коорди-
ната реакции соответствует некоторому растяжению связей, и ос-
новной чертой теории активированного комплекса является утвер-
ждение, что имеется простое, специфическое колебание, которое
переводит активированный комплекс в продукты реакции.
Образование и распад активированного комплекса. Теория активи-
рованного комплекса представляет реакцию между двумя части-
цами А к В как протекающую через образование активированного
комплекса (АВ)*:, который превращается в продукты с некоторой
скоростью Скорость расхождения А, т. е. скорость реакции,
равна скорости превращения активированного комплекса в продук-
ты реакции:
rfIPj/dZ—fe* [(АВ)*Ь (27.2.1)
Возникают два вопроса: как найти и как найти концентрацию
активированного комплекса? Последняя, по-видимому, пропорцио-
нальна концентрации реагирующих веществ; позднее будет точно
показано, что
[(АВ)*]-^Д-Ф [A] [В], (27.2.2)
30*
468
Часть 3. Изменение
где К*— некоторый коэффициент пропорциональности*. Из этога
следует, что скорость реакции равна /гфА+[А] [В], и, таким об!
разом, константа скорости простой бимолекулярной реакции равн®
Aj=AW=. (27.2.3)
Коэффициент £4= можно связать со свойством активированного
комплекса следующим путем. Критерием перехода комплекса в
продукты реакции является прохождение его через критическую
конфигурацию — переходное состояние. Если он переходят через
него, образуются продукты. Движение однажды образованного
активированного комплекса вдоль координаты реакции характе-
ризуется растяжением некоторых соответствующих связей: поэто-
му прохождение активированного комплекса через переходное со-
стояние может быть идентифицировано с особой колебательно:!
модой.
Если частота критического колебания комплекса равна v, то
частота перехода через переходное состояние также равна т. На
самом деле возможно, что не каждое колебание приведет к пере-
ходу молекулы через переходное состояние; это связано с тем,
что критическая конформация молекулы имеет сложную зависи-
мость от расположения всех атомов в комплексе, и иногда общая
конформация может не быть подходящей для образования про-
дуктов. Возможно, что в разрушении активированного комплекса
играет роль также вращение, и поэтому переходное состояние
может также зависеть от вращательного состояния комплекса. Для
учета этих факторов предположим, что скорость перехода через
истинное переходное состояние пропорциональна колебательной
частоте у вдоль координаты реакции, и запишем
(27.2.4)
где х — так называемый трансмиссионный коэффициент, во мно-
гих случаях он приблизительно равен единице.
Следующий этап — расчет концентрации активированного ком-
плекса. Принимается такое приближение: о природе комплекса
ничего не известно и, в частности, ничего не известно о заселен-
ности энергетических уровней. Не считая это большим недостат-
ком, можно его использовать как конструктивное правило. Так
* Я* считается безразмерной, несмотря на то что это уравнение следовало
бы записать в виде
[(АЕ)т| (Моль/дм®) = Д'* ([AJ, моль/дм®) ([ВJ, моль/дм®),
ИЛ 4
1(АВ)Ф] = Д'* [А] [В]' (моль/дм3).
Такая запись очень громоздка, н поэтому зтн единицы просто будем держать
в памяти. Еще раз о них будет упомянуто в примечании к уравнению (27.2,7)-
27. Динамика, молекулярных реакций
469
Рис. 27.3. Энергетические уроини реагентов н актиниро-
± ванного комплекса,
(АВ)Т
--- кек ничего не известно о распределении энер-
7-=^ =— гин между различными формами активиро-
---=— ванного комплекса, то предположим, что все
_ I распределения энергий, согласующиеся с дан-
1 ной общей энергией, равновероятны. В этом
. . . . можно распознать гипотезу о равенстве апри-
———~орных вероятностей, используемую при пост-
~----------° роении статистической термодинамики
. __ (разд, 20.1).
, Это правило иллюстрируется на рис. 27.3.
L Нанесем на схему уровни энергии реагентов
Л + в--------------и активированного комплекса. Мы хотим
знать число молекул на уровнях, соответст-
вующих (АВ)=*=, по отношению к числу моле-
кул на уровнях, соответствующих А и В, при этом никаких указа-
ний о предпочтительности отдельных уровней или идентичности
энергетических уровней не имеется. Это в точности соответствует
вопросу, который мы рассматривали для статистико-термодинами-
ческого приближения при расчете констант равновесия, и рис. 27.3
по существу является тем же самым, что и рис. 21.8. Поэтому от-
вет на вопрос может быть взят из текста к рис, 21.8 и константы
равновесия связанной с функциями распределения трех пред-
полагаемых частиц. Мы имеем
£р =(рф, атм)/(рА, атм)(рв, атм) =
--=\Lq%eq?} ехр(—NE^fkT). (27.2.5)
Это уравнение по существу совпадает с уравнением (21.3.25);
здесь q*— молекулярная функция распределения для активиро-
ванного комплекса, qA и — молекулярные функции распреде-
ления для реагентов и Д£— разность энергий между комплексом
11 реагентами (рис. 27.3). Необходимо выразить константу равно-
весия через концентрации, а нс через Др. Эти два параметра лег-
ко связать, так как [X] — n^/V и tix — pV/RT. Поэтому
Кр —[(n^RT/V), a.4M]/[(nART/V), атм] [(пв/?Т/Й), атм] =
--{1(АВ)*]//[А]]В]}(атм//?7)-
£К ’ ?О(АВ)ФЬ моль/лмэ)/(IА], моль/дм3) ([В], моль/дм3)} х (моль/дм3) х
«В' X(атм/ЯГ) —R^/(RT, дм3-атм/моль).
470
Часть 3. Изменение
Отсюда следует, что
K*=(RT, дмэ-атм/моль) {Lq*e/qlqv} ехр (—Д£*/АГ).
Поскольку молекулярная функция распределения может быть рая-
бита на часть, связанную с поступательным движением q\ и часть,
связанную со всеми внутренними движениями молекулы q\ и по-
скольку первая часть всегда выражается в форме
?‘е = (2ятА71/Ла)8^ = tVS,,
мы также имеем 9c = ^lrVni. Более того, VS, =7?Т/(1 атм), поэтому
K*=(LRT, дмэ-атм/моль) {(т</)*/(т<Д)(т^в)} X
х(1 атм//?Т)ехр( —А£*Ж)=^
= (£, моль/дм3) {(т^)*/(т^д)(т?в)} ехр(— AEf/RT).
На этом месте хорошо бы остановиться и определить, что уже
сделано. Для того чтобы найти скорость бимолекулярного процес-
са, необходимо знать две вещи; концентрацию активированного
комплекса и скорость его прохождения через переходное состояние.
Последняя связана с колебательной частотой вдоль соответствую-
щей и критической координаты. Первую можно найти на основе
следующего простого предположения; нет причин при рассмотре-
нии распределения энергии отдавать предпочтение одному типу
частиц или одному типу энергетических уровней. То же самое вы-
ражение для константы скорости может быть получено в предпо-
ложении о том, что в ходе реакции активированный комплекс на-
ходится в равновесии с реагентами. Исходя из этого, мы сразу
получим уравнение (27.2.2), считая, что X*— константа равнове-
сия, и используя обычное статистико-термодинамическое выраже-
ние, и, таким образом, мы придем к уравнению (27.2.5). Хотя этот
путь очень короткий и прямой, основное предположение о том.
что скорость процесса включает равновесие, часто является кам-
нем преткновения для его принятия. Применение подхода, осно-
ванного па наименьшем предубеждении, хотя это в конечном счете
и эквивалентно приближению о существовании равновесия, ука-
зывает более ясно на природу сделанного приближения и откры-
вает пути улучшения модели. Например, данная модель может
быть улучшена, если считать заселенность уровней активирован?
ного комплекса таковой, что многие колебания сохраняют распре-
деление заселенности, которое они имели в реагентах.
Для того чтобы закончить обсуждение, обратим внимание на
внутренние функции распределения активированного комплексам
q1*. Уже было установлено, что колебание выводит активирована
ный комплекс из переходного состояния и превращает его в npoJ
27. Динамика молекулярных реакций 471
дукты. Функция распределения для этих особых колебаний имеет
вид {1—ехр (—hv/k.T}}~], где v — частота колебания (та же самая
частота, которая определяет fe*). Эта частота значительно меньше,
чем нормальная молекулярная колебательная частота, так как дви-
жение связано с распадом активированного комплекса. В данном
случае экспоненциал в функции распределения можно разло-
жить в ряд и оставить лишь главный член ряда; при этом он сво-
дится к kTfhv. Поэтому можно записать, что
— (kT
где — функция распределения для всех остальных внутренних
энергий активированного комплекса.
Объединяя все части расчета, получим
k2 Д 4- = zv
где
К-4- —(А, моль/дм3) |(т^)*/(т^)А (т^)в) ехр (—А£+//гГ). (27.2.6)
Или, если исключить неизвестную частоту v, придем к уравнению
Эйринга*
ki = v.(kTlh)K*. (27.2.7)
Два последних уравнения являются точными выражениями для
расчета скорости простой бимолекулярной реакции по первичным
параметрам.
Как использовать уравнение Эйринга. Возможность применения
уравнения Эйринга для расчета скорости реакции зависит от воз-
можности расчета функций распределения участвующих в реакции
частиц. Обычно функции распределения могут быть рассчитаны
иди на основании спектроскопических данных об энергетических
Уровнях, или путем использования различных приближенных вы-
ражений (подразд. 21.1. А). Действительная трудность состоит в
определении функций распределения активированного комплекса.
Спектроскопически обычно найти их нельзя, а расчет функций
распределения зависит от предположений, сделанных о структуре,
размерах и форме активированного комплекса.
В качестве первого примера расчета констант скорости возь-
мем простой случай столкновения двух неструктурированных час-
тиц А и В, даюших активированный комплекс, который может
* Как следует из примечания на стр. 468, это уравнение имеет вид
(дм3/моль) = х
472
Часть 3. Изменение
рассматриваться как двухатомная молекула. Затем эта двухатом
ная молекула распадается до каких-то продуктов реакции-
Поскольку реагенты являются неструктурированными «атома
ми», единственный вклад в их функции распределения дает посту
нательное движение:
Яа— ?а =(2лтАй77Л3)а/а V, так что
тА =(2лтААГ/Л2)’'2.
То же самое выражение можно написать для по с
Ша- Активированный комплекс является двухатомной
ГЦ в
вместо
с массой тлв = тд + тв и моментом инерции /Ав-
Здесь
молекулой
имеется
только одна колебательная мода, но она соответствует координа
те реакции и поэтому не входит в ^!4=. Отсюда следует, что функ
ция распределения активированного комплекса имеет вид
(т^)Ф^т* (2/авАЭД, т* = (2лтАВВД)3/й-
Поскольку момент инерции /дв связан с приведенной массой цАВ
и длиной -связи двухатомной молекулы соотношением 7ав = ЦлвА':
выражение для константы скорости можно записать как
(kTjh) {(т</)=*7гатв} ехр {—bEflkT} =
--=(8kTехр {—ДЕ'Ф/АТ},
и, заменив ил:/?2 па о* (сечение реакции),
мы
к
приходим точно
такому же выражению, какое получается из простой теории столк
новеннй [уравнение (27.1.1)].
Пример (вопрос 9).
Найдите
порядок
величины
Р-фактора
для реакции между
двумя нелинейными молекулами.
Метод. Используем уравнение Эйринга дважды; сначала для неструктурирован
ных молекул, а затем для двух молекул с внутренней структурой (так что ош
могут и вращаться, и колебаться). Отношение двух результатов рассматривается
как Р. Для опенки порядка величины предполагаем, что поступательные функ
дни распределения такие же, как вращательные и колебательные. Молекула из А
атомов имеет 3 поступательные. 3 вращательные в (ЭЛ!—6/ колебательных сте
пеней свободы.
Ответ. В отсутствие внутренних степеней свободы сталкивающихся молекул
W)a = т, W)b =т, —i9r.
поэтому
k2 - (kT[h) (т?г/п) ехр (— E^/kT) —
~ 77/0(9?/т) ехр (— E^ikT}.
При наличии внутренних степеней свободы
(Т90а = (т^) =^*9^8^,
(T9i)* .
21. Динамика молекулярных реакций
473
Реакция
Рис. 27.4. Изменения в профиле реакции при дейтерировании разрывающейся
связи.
Отметьте, что активированный комплекс имеет на одну колебательную степень
свободы меньше, чем обычные молекулы. Следовательно,
= (И’/Л) {(т^^а+^вЬ7)/^3^-6) ехр (-£*/АП =
= (йГ/й) ('ZvA'Zr) ехР (—
Сравнение двух результатов показывает, что
Р = = С^/?гЛ
Так как gv/<h»l/50, то 1/50®=3,2.10~s.
Комментарий. Расчет подтверждает, что реакция между двумя сложными мо-
лекулами как в газах, так и растворах будет протекать значительно медленнее,
чем реакция между простыми молекулами, даже при одинаковой энергии акти-
вации.
Рассмотренный пример показывает, что даже в случае двух
атомов необходимо делать предположение о структуре активиро-
ванного комплекса; и эта проблема сильно усложняется, когда
число атомов растет. Однако имеется другая простая, по важная
иллюстрация его применения. Рассмотрим влияние дейтерирова-
ния на скорость разрыва связи С—И. Координата реакции вклю-
чает растяжение связи С—Н, и профиль реакции приведен на
рис. 27.4. При дейтерировании основное изменение состоит в по-
нижении нулевой энергии связи вследствие большей массы изото-
па дейтерия. Общая кривая реакции не опускается, поскольку
соответствующие колебания активированного комплекса очень сво-
бодны и, следовательно, имеют небольшую нулевую энергию как
Для протонной, так и для дейтериевой форм.
Вначале разумно предположить, что дейтерирование оказыва-
ет влияние только па координату реакции, и, таким образом, функ-
ции распределения для всех внутренних степеней свободы оста-
ются теми же самыми. Поступательные функции распределения
474
Часть 3. Изменение
изменяются при дейтерировании, но масса остальной части моле-^
кулы обычно намного больше, что делает это изменение незначи-
тельным. Величина ДЕ* изменяется вследствие того, что низшее
состояние реагентов ниже для дейтерированного соединения, а низ-
шее состояние активированного комплекса остается одинаковым:
изменение ДЕ^ равно изменению пулевой энергии связи С—Н:
ДЕ* (D)—ДЕ* (Н) « 4 h {а (С—Н)—и (С-D)} «
где for — силовая постоянная, ар — приведенные массы.
Мы можем сделать вывод, что константы скорости для неден-
тсрированных и дейтерированных молекул должны находиться в
соотношении
(D)/^(H) ^exp J-g- (hkfykT} [(l/pCD)^ —(1/рсн)17а1}. (27.2.9)
Это свидетельствует о том, что разрыв связи С—Н при прочих
равных условиях должен происходить в 7 раз быстрее, чем разрыв
связи С—D. Как мы видели, причиной этого является большая
нулевая энергия связи С—Н и, следовательно, меньшая энергия
активации.
Термодинамические аспекты. При выводе выражения для fe2 было
указано, что тот же самый результат может быть получен, если
сделать предположение о том, что активированный комплекс на-
ходится в термодинамическом равновесии с реагирующими веще-
ствами. Эта аналогия может быть продолжена на языке термоди-
намического равновесия.
Прежде всего функция активации Гиббса может быть выра-
жена следующим образом:
— 7?Т1пЛФ=ДС% (27-2.10)
Тогда для константы скорости получим
(kT/h) ехр (—bG*/RT). (27.2.11)
Поскольку функция Гиббса связана с энтропией и энтальпией со-
отношением G = H~TS, мы приходим к энтропии активации AS-'
и энтальпии активации ДЯ* через
AG* - ДЯ* - ТДЗ*. ' (27.2.12)
Подстановка этого выражения в k2 приводит к
k2 ==< (kTjh) ехр (Д5™/Е) ехр ( —
(27,2.13)
27. Динамика молекулярных реакций
475
Только что выведенное уравнение весьма напоминает закон ско-
рости Аррениуса, и очень заманчиво отождествить предэкспонен-
циальный множитель А с z(fe?7/i)exp (AS*//?). Однако при этом
мы пренебрегаем тем фактом, что Д/f* не обязательно равна Еа.
Например, в бимолекулярной реакции для лимитирующей скорость
стадии £а = А//* + 2/?7'. Отсюда следует, что
А ж /-(feT/A)exp(AS^//?). (27.2.14)
Это выражение связано с приближением теории столкновений,
в которой А определяется числом столкновений в газе, а столкно-
вения приводят к уменьшению энтропии (поскольку при столкно-
вении происходит объединение двух частиц и поэтому уменьшение
неупорядоченности системы). Более того, столкновения с четко
определенной относительной ориентацией соответствуют даже
большему уменьшению энтропии, чем простые столкновения, и,
таким образом, энтропия активации должна быть более отрица-
тельной и соответствовать значению Р меньше единицы. Поэтому
энтропия активации соответствует как частоте столкновений Z,
так и стерическому фактору Р в простой теории столкновений.
Пример (вопрос 10). Рассчитайте энтальпию, энтропию и функцию активации
Гиббса для реакции второго порядка разложения двуокиси азота 2NO2—>-2N0-j-
+ О2 при 500 К.
Метод. Энергия активации раина 111 кДж/моль, а Л = 2,0-1019 моль_|-дм3-с~!.
В бимолекулярной реакции для лимитирующей скорость стадии энергия актива-
ции £а связана с энтальпией активации следующим образом: Ea — &fI^ + 2RT
(это установлено в задаче 27.26). Энтропия получается из уравнения (27.2.13),
а функция активации Гиббса—из уравнения (27.2.12). Вспомним (см. примеча-
ние па стр. 471), что k3 в уравнении (27.2.13) должна рассматриваться как
(дм3/моль) и точно так же А н уравнении (27.2.14) — как А (дм3/моль).
Ответ.
А Я* = £а — 2RT = 111 кДж/моль —2Х [8,314 Дж/(К-моль)[Х
Х(500 К) = ЮЗ кДж/моль.
Из уравнения (27.2.14)
«*=/?{-2 1п(ЛД/ЙГ)} =
„ „ ,( Г(6,626-Ю-34 Дж-с)х(2,010йс-1)-|)
- [8,314 Дж/(К-моль)] ^—2 In (Цзврю-иДж/Юх^ООК) ]| “
—[8,314 Дж/(К-моль)] { — 2 In (1,92-10-4)} = —88 Дж/(К -моль).
Тогда из уравнения (27.2.12)
AG* = (ЮЗ кДж/моль) — (500 К)Х [—88 Дж/(К• моль)] = 119 кДж/моль.
Комментарий. Отметьте большую отрицательную энтропию активации. Значение
•4, полученное из простой теории столкновений (пример с Р=1 на стр. 459), со-
ответствует значительно меньшей энтропии активации [—63 Дж/(1\-моль)].
476
Часть 3. Изменение
Реакции в растворе. Применение теории активированного комплек-
са к реакциям в растворе очень затруднено вследствие участия
растворителя в активированном комплексе. Трудно надеяться на
то, что мы сможем рассчитать функции распределения активиро-
ванного комплекса, окруженного молекулами раствори теля. Мы
примем эту трудность, но выйдем из безнадежного положения
путем использования термодинамического подхода к /ь.
Закон скорости связан с концентрациями реагирующих веществ
А и В, а константа равновесия К — с активностями молекул. По-
этому
Ав[А][В1=йФ[(АВ)+]
нужно объединить с
КФ = а (АВ)+/а (А) а (В) =?(АВ)Ф [(AB)*J/?a [А] ?в [В],
где а — активности и у — коэффициенты активности. Это дает
йг=йФК* {?л?в/?{ав)Ф] [?л?в/Т£Ав)ф}. (27.2.15)
где /ф — константа скорости, когда все коэффициенты активности
равны единице.
Следующим этапом может быть установление соотношения
между коэффициентами активности и ионной силой раствора. Для
этого используем предельный закон Дебая — Хкжксля (11.2.11).
выведенный в гл. 11 (т. 1):
lg?i =— Аг?(Л моль/кг)1^,
и тогда
lgA2 = lgfe° — A {za+zes —(zA+zB?} (Z, моль/Кг)У* =
—1g 2А2а?в (Л моль/кг)1^. (27.2.16)
Используется тот факт, что если заряды реагентов равны и zr„
то заряд активированного комплекса равен za + Zb-
Последнее уравнение показывает, Что константа скорости ион-
ной реакции должна зависеть От ионной силы: в этом состоит ки-
нетический солевой эффект. Если ионы имеют одинаковый знак,
то увеличение ионной силы (например, добавлением инертных
ионов) приводит к увеличению константы скорости. Это можно
понять, если представить образование одного высокозаряженного
активированного комплекса из двух ионов меньшего заряда: про-
цессу благоприятствует большая ионная сила, так как она способ-
ствует взаимодействию нового иона с его более плотвой ионной
атмосферой. Наоборот, ионы противоположного заряда реагируют
медленнее в растворах с большой ионной силой. Это связано с
тем, что при образовании активированного комплекса заряды реа-
27. Динамика молекулярных реакций
477
Рис. 27.5. Экспериментальная провер-
ка кинетического солевого эффекта.
гирующих веществ компенси-
руются, а это понижает взаи-
модействие с ионной атмосфе-
рой.
Кинетический солевой эф-
фект может быть проверен экс-
периментально, если взять ио-
ны различных зарядов и пост-
роить графики зависимости
lg от квадратного корня из
ионной силы. Предэкспоненци-
альный множитель А известен
(для воды при комнатной тем-
пературе он равен 0,509), и
поэтому наклоны графиков
можно сравнить с теоретиче-
ским значением, равным
1,019вдгв; рис. 27.5 показыва-
ет хорошее совпадение теоретических и экспериментальных дан-
ных. Кроме того, эта теория может быть использована для сужде-
ния о природе активированного комплекса в реакции: найдя за-
висимость от ионной силы, можно определить заряды участ-
вующих частиц. Следующий пример иллюстрирует это.
Пример (вопрос 11). Константа скорости щелочного гидролиза комплекса
[Co(NHs)sBr]2+ зависят от ионной силы раствора так, как это показано в таб-
лице. Что можно сказать о природе активированного комплекса в определяющей
скорость стадии?
I 0,005 0,010 0,015 0,020 0,025 0,030
ВД 0,718 0,631 0,562 0,515 0,475 0,447
Л1ето<5. Строим график зависимости 1g от 1'А Наклон прямой будет равен
1.02?Ацв. Иов ОН- имеет za = —1, комплексный ион имеет zB=-j-2, и поэтому,
если получится наклон —2,04, можно заключить, что определяющая скорость
стадия включает образование комплекса из двух ионов.
Ответ. Составляем следующую таблицу:
1 0,005 0,010
УТ 0,071 0,100
—0,014 -0,20
0,015 0,020 0,025 0,030
0,122 0,141 0,158 0,173
—0,25 —0,29 —0,32 —0,35
478
Часть 3. Изменение
Данные представлены на рис. 27.6. Наклон, определенный по методу наименьших
квадратов, равен 2,1, что свидетельствует об участии двух ионов в образовании
активированного комплекса (коэффициент корреляции равен 0,9988).
комментарий. Другим фактором, влияющим на скорость реакции, является от-
носительная диэлектрическая проницаемость среды; это рассмотрено в задаче
27.28.
27.3. Динамика молекулярных столкновений
Теперь мы переходим на третий уровень сложности рассмот-
рения факторов, влияющих на скорости химических реакций, и об-
ратимся к информации о столкновениях между отдельными моле-
кулами, ставшей доступной благодаря использованию современ-
ных технических достижений. Метод молекулярных пучков позво-
ляет рассматривать столкновения между молекулами на предва-
рительно выбранных энергетических уровнях и может быть ис-
пользован для определения энергетических состояний продуктов
реакционных столкновений. Информация такого рода существен-
на, если необходимо построить полную картину реакции, но и в
данном случае мы далеки от совершенной теории: в конце концов,
большинство известных реакций происходит в растворе, а наши
способности изучить индивидуальные процессы в жидкостях пока
еще ограничены.
Реакционные столкновения. Обшие положения. Экспериментальные
исследования связаны с молекулярными пучками и особенно с
перекрестными молекулярными пучками (разд. 23.3). Детектор
для продуктов столкновения двух пучков может перемещаться под
различными углами, и, следовательно, этот метод можно исполь-
27, Динамика молекулярных реакций
479
зовать для определения углового распределения продуктов. Более
того, если детектор в состоянии различать энергетические состоя-
ния продуктов, то можно определять эти состояния сразу после
реакции. Так как молекулы во входящих пучках могут находиться
в разных энергетических состояниях с различными ориентациями
(разд, 23.3), то можно изучать зависимость конечного энергети-
ческого состояния, углового распределения и вероятности реакции
для нескольких первоначальных состояний сталкивающихся мо-
лекул.
Мощным методом, позволяющим рассмотреть конечное энерге-
тическое распределение в молекулах, является инфракрасная хеми-
люминесценция. Если продукт образуется с неравновесным рас-
пределением колебательных состояний (например, в первом воз-
бужденном колебательном состоянии), то он переходит в основное
состояние, испуская инфракрасный свет. Изучая интенсивность ли-
ний в ИК-спектре испускания, можно
15 ----------------- найти все колебательные состояния
14
и
12
и
ю
9
8
(рис. 27.7).
Одна из наиболее важных концеп-
ций, развиваемых при обсуждении
пучков, —поверхность потенциальной
энергии реакции. Способ применения
этой концепции можно проиллюстри-
ровать на примере столкновения меж-
ду атомом и молекулой водорода. По-
верхность потенциальной энергии
представляет собой график изменения
энергии, происходящего в реакции
Н+На—^Н2 + Н.
С самого начала мы сталкиваемся
4 _-------------—
з —-------------------
2 --------------------
I •-------------------
0 --------------------
с главной трудностью; необходимо
точно определить 6 координат поло-
жений молекулы Н2 по отношению к
атому водорода; следовательно, диаг-
рамма изменений энергии должна
иметь 7 измерений. Это очень сложно,
и, таким образом, главная трудность
выявляется сразу. Более подробный
анализ показывает, что приближение
атома вдоль оси Н—Н требует для
Рис. 27.7. Инфракрасная хемилюминесценция
продукта (СО) реакции O+CS—>-CO-pS..
480
Часть 3, Изменение
Рис. 27.8. Поверхность потенциальной энергии (а) и соответствующая контурная
диаграмма (б) линейной реакции НЦ-Нз.
реакции меньше энергии, чем любой другой подход, и поэтому
сейчас мы обратим внимание на линейные столкновения. Теперь
для определения изменений расстояний между атомами во время
реакции потребуется только два параметра: один из них /?аь —
расстояние На—Нь, а другой /?ьс — расстояние Нь—Нс.
В начале столкновения /?аь бесконечно велико и /?Ьс равно рав-
новесной длине связи в Н2; к концу столкновения (если реакция
происходит) /?ьс бесконечно велико и /?аь имеет равновесное зна-
чение для молекулы Н2. Энергия трех атомов зависит от их отно-
сительного положения и может быть найдена обычными молеку-
лярно-структурными расчетами. График зависимости энергии от
/?аь и /?ьс представляет собой поверхность потенциальной энергии
этой реакции (рис. 27.8,а). Поскольку построение трехмерной диа-
граммы— довольно трудная задача, обычно строится контурная
диаграмма (рис. 27.8,6).
Если /?аь — постоянная и большая величина, то изменения энер-
гии, которые происходят с изменением соответствуют энергии
изолированной молекулы водорода, когда ее длина связи изменя-
ется. Сечение через поверхность при будет по типу таким
же, как кривая потенциальной энергии молекулы, с которой мы
встречались в гл. 15. (т. 1). С краю диаграммы, когда близко
к бесконечности (это соответствует случаю, когда молекула На
образована из атомов НаНь, а атом Нс находится далеко), про-
филь поверхности имеет форму кривой потенциальной энергии
изолированной молекулы На—Нь.
27, Динамика молекулярных реакций
481
Рис. 27.9. Различные траектории (а) и соответствующие профили реакции (б).
Эту поверхность можно использовать для рассмотрения изме-
нений энергии, которые происходят при приближении атома На
к молекуле Нь—Нс. Если по мере приближения На длина связи
Нь—Нс сохраняется постоянной, то изменение потенциальной энер-
гии выражается линией А на рис. 27.9. Это показывает, что по
мере приближения На к Нь—lie потенциальная энергия системы
увеличивается и затем резко падает при отрыве атома Не и уда-
лении его на большое расстояние. Альтернативно можно предста-
вить, что реакция идет по пути В и при этом молекула Нь—Нс
распадается, хотя На находится все еще далеко. Ясно, что оба Эти
пути, хотя и осуществимые, заставляют атомы двигаться в область
высоких потенциальных энергий,
Путь, предполагающий наименьшую потенциальную энергию,
отмечен буквой С. Он соответствует удлинению /?ьс по мере при-
ближения На и образования связи с Нь. Связь Нь—Нс ослабляется
ПОД действием приближающегося атома, и, хотя энергия увели-
чивается, она растет только до седловинной точки, отмеченной на
рис. 27.9 символом Сф. Путь реакции с наименьшей потенциальной
энергией проходит вверх по долине через седловинную точку и
31—242
482
Часть 3, Изменение
далее вниз ко дну другой долины, при этом Нс удаляется н новая
связь На—Нь сокращается до ее равновесного значения.
Круг вопросов, исследуемых с помощью молекулярных пучков,
может быть расширен за счет рассмотрения поверхностей потен-
циальных энергий реакций. Во-первых, для того чтобы путь от
реагентов до продуктов реакции осуществился по линии С, при-
ближающийся атом должен подойти с кинетической энергией, до-
статочной для того, чтобы подняться на поверхность потенциаль-
ной энергии до седловинной точки. Изменяя скорость приближаю-
щегося атома и определяя энергию, при которой происходит реак-
ция, можно проверить точность расчета или оценки поверхности
потенциальной энергии. Во-вторых, если приближающийся атом
имеет избыток кинетической энергии, то система может переско-
чить седловинную точку и пойти путем, который приведет к обра-
зованию сильно колеблющихся молекул продукта. Исследуя сте-
пень колебательного возбуждения продуктов, можно построить
поверхность потенциальной энергии в непосредственной близости
от седловинной точки и, возможно, во всем пространстве.
Другой вопрос, на который можно ответить, состоит в следую-
щем. Что лучше для осуществления реакции: когда частицы имеют
большую поступательную кинетическую энергию или когда энер-
гия вводится в комплекс в форме колебательной энергии? Напри-
мер, какая траектория на рис. 27.10 более эффективна для реак-
ции; траектория Cj, в которой молекула Нь—Нс находится перво-
начально в колебательно-возбужденном состоянии, или траекто-
рия, которая начинается на дне долины и соответствует постепен-
ному растяжению связи?
Каким образом данные, получаемые при исследовании молеку-
лярных пучков, связаны со значением константы скорости А2?
Связь основана на пони-
мании того, ЧТО k-i ЯВдЯ
ется статистической вели-
чиной. В обычных газо-
фазных реакциях проис-
ходят столкновения ча«
стиц с различными энер!
гиями и в различных ко!
лебательных и враща^
тельных состояниях. КаЖ-
Рис. 27.10. Удачные и неудач-
ные столкновения.
27. Динамика молекулярных реакций
483
Рис. 27.11. Изменения энергии для различных углов атаки: H+Hj (п),С1+Н1 (б).
дое отдельное столкновение можно представить в виде траектории
на поверхности потенциальной энергии реакции. Некоторые из
этих траекторий будут удачными (CJ и Ci на рис. 27.10), а неко-
торые неудачными (С3 и С4) или потому, что столкновения не
имеют достаточной энергии, или потому, что энергия не так рас-
пределена. Реакция в среднем проходит через все эти возможные
траектории, и, таким образом, для вычисления мы должны рас-
считать большое число траекторий и затем взять среднее значе-
ние. Основным методом, позволяющим это сделать, является ме-
тод Монте-Карло: он состоит в том, что выбирают произвольные
начальные условия для исследуемого образца, но так, чтобы обра-
зец находился при термическом равновесии.
Некоторые результаты исследований с помощью молекулярных
пучков. Здесь мы ответим на вопросы, поставленные в предыду-
щем разделе, и покажем, как изучение столкновений и расчет по-
верхностей потенциальных энергий могут пролить свет на ход ре-
акций.
1. Можно ла рассматривать линейное приближение как путь
с наименьшей энергией"? На рис. 27.11,а показаны результаты
расчета изменений энергии при атаке атомом водорода молекулы
водорода под различными углами; в каждом случае связи релак-
сируют до оптимальной длины, Как предполагалось при построе-
нии поверхности потенциальной энергии на рис. 27.8, при линей-
ной атаке энергия активации будет наименьшей. Тем не менее мы
31«
484
Часть 3. Изменение
должны осознать, что другие линии атаки также возможны и ели
вносят вклад в общую скорость.
В противоположность этому рис. 27.11,6 показывает измене-
ния потенциальной энергии, которые происходят при атаке атомом
хлора молекулы 111. К реакции ведут только те подходы, которые
происходят в пределах конуса с полууглом 30°, окружающего ат >м
водорода. Следует отметить связь этого расчета с P-фактором в
простой теории столкновений: не каждое столкновение приводи; к
реакции, потому что не каждое столкновение происходит под углом
атаки, лежащим в реакционном конусе.
2. Что определяет угловое распределение продуктов'? Если
столкновение «прилипчивое», т. е. такое, когда при столкновении
частицы вращаются вокруг Друг друга, продукты будут появляться
во всех направлениях, потому что молекулы «нс запоминают», в
каких направлениях они двигались до столкновения. Вращение
происходит за время 10-12 с, и если Столкновение происходит мень-
ше чем за 10 12 с, то комплекс не имеет времени для вращения
и продукты разлетаются в определенных направлениях. Например,
при столкновении К с 12 большинство продуктов разлетается в
прямом направлении*. Это согласуется с гарпунным механизмом
(разд. 27.1), где столкновения происходят на длинном расстоянии.
Наоборот, столкновение К с метилиодидом CHgl приводит к ре-
акции только в том случае, если частицы приближаются очень
близко друг к другу. Это напоминает столкновение К с кирпичной
стеной, так как образующийся К1 отлетает рикошетом в обратном
направлении. Наблюдение анизотропии в угловом распределении
продуктов дает указание на расстояние и ориентацию приближа-
ющихся частиц, необходимые для реакции, и свидетельствует о
том, что реакция должна завершаться менее чем за 10-12 с.
3. Какую энергию лучше иметь при столкновении: поступатель-
ную или колебательную? Некоторые реакции очень чувствительны
к тому, в каком виде у сталкивающихся частиц запасена энергия:
в колебательном или поступательном. Например, если две молеку-
лы Н1 сталкиваются с энергией, в два раза превышающей энер-
гию активации реакции, то никакой реакции не происходит, пото-
му что вся имеющаяся энергия является поступательной и не пе-
реходит в колебательную энергию. Реакция F + НС1 “Cl + HF
* Это, конечно, хитрый прием. В работах по молекулярным пучкам обычно
ссылаются на направление в системе координат центра масс. Началом коорди-
нат является центр масс сталкивающейся пары молекул, и столкновения про-
исходят в том случае, когда молекулы находятся в начале координат. Путь по-
строения координат центра масс и перевод карты событий в них в лаборатор-
ную систему координат включают слишком много деталей, чтобы их здесь рас-
сматривать. Необходимо лишь запомнить, что термины «прямой» и «обратный»
должны интерпретироваться нетрадиционным путем.
27. Динамика молекулярных реакций
485
происходит в 5 раз быстрее, если HCI находится не в основном
колебательном состоянии, а в возбужденном состоянии.
Причина этого может быть найдена при рассмотрении поверх-
ностей потенциальной энергии для других реакций, кроме Н + И2,
Два особо важных случая представлены па рис, 27.12. Для по-
верхности, показанной на рис, 27.12, а, высшая точка траектории
реакции наблюдается рано; эта поверхность называется поверх-
ностью притяжения. В противоположность этому на рис. 27.12,5
представлена поверхность, где седловинная точка появляется поз-
же на пути реакции; это поверхность отталкивания.
Рассмотрим сначала поверхность притяжения. Если исходная
молекула колебательно-возбуждена, то столкновение с другой час-
тицей можно изобразить тра-
екторией С: она препятствует
проникновению в область ре-
акции и мешает движению к
седловиниой точке. Однако ес-
ли та же самая энергия нахо-
дится в виде простой поступа-
тельной энергии, то система,
когда происходит столкнове-
ние, будет стремиться двигать-
ся по траектории, близкой к
С*. Ясно, что в этом случае
траектория реакции направля-
ется через седловинпую точку
и далее в область, соответст-
вующую продуктам. Можно
заключить, что реакции, соот-
ветствующие потенциальной
поверхности притяжения, про-
текают более эффективно, ес-
ли энергия системы находится
в виде поступательного движе-
ния. Более того, потенциаль-
ная поверхность показывает,
что после прохождения через
седловинную точку траектория
круто спускается в долину
продуктов и затем перекаты-
. п Рис. 27.12. Поверхности притяжения
о Поверхность отталкивания и отталкивания.
48С>
Часть 3. Изменение
вается от одной ее-стороны к другой, пока не опустится на дно до-
лины, где частицы разделяются. Другими словами, когда реакция
имеет поверхность притяжения, продукты остаются в колебатель-
но-возбужденном состоянии. Причина такого поведения заключа-
ется в том, что продукты образуются в начале реакционного пу-
ти, причем образуются с длинной связью. Быстрое отделение ухо-
дящей частицы оставляет молекулу продукта с растянутой связью,
и поэтому он «разражается» колебаниями.
Теперь обратимся к поверхностям отталкивания (рис. 27.12,6).
На траектории С энергия столкновения находится в основном в
поступательном движении. По мере приближения частиц друг к
Другу их потенциальная энергия растет. Траектория поднимается
к противоположному краю долины, а затем идет назад по направ-
лению к области реагентов. Это соответствует неудачному столк-
новению даже в том случае, когда общая энергия достаточна для
протекания реакции. По другой траектории, т. е. по С*, часть об-
щей энергии связана с колебанием молекулы. Колебание вызывает
изменение траектории от одной стороны долины к другой по мере
того, как система подходит к седловинной точке с приближением
другого реагирующего вещества. Этого движения достаточно для
того, чтобы система обошла угол и поднялась к конечному склону
и седловинной точке. Отсюда система попадает в долину продук-
тов. Можно ожидать, что в этом случае молекула продукта нахо-
дится в невозбужденном колебательном состоянии. Другим Путем
рассмотрения процесса является такой, при котором продукты не
разделяются до тех пор, пока не образуются достаточно прочные
новые связи,— фактически до конечного равновесия разделения.
Уходящие частицы получаются на последней стадии и уносят из-
быток энергии. Примером реакции с потенциальной поверхностью
отталкивания служит реакция Н + С12—s-HCl-i-Cl, и критерием ее
осуществления является необходимость колебательной энергии.
Информация этого подраздела может быть суммирована сле-
дующим образом. Поверхность притяжения соответствует реакции,
которая дает продукты в колебательно-возбужденном состоянии
и Протекает наиболее эффективно, если энергия столкновения на-
ходится в виде относительного поступательного движения реаген-
тов. Поверхность отталкивания указывает на то, что реакция про-
текает наиболее эффективно, если энергия возбуждения привно-
сится в столкновение в виде колебательного движения, и затем
продукты получаются в основном колебательном состоянии. Ре-
акция с поверхностью притяжения, идущая в одном направлении,
будет реакцией с поверхностью отталкивания в обратном направ-
лении.
4. Можно ли рассчитать траектории, показывающие реакцию
в развитии? Четкую картину реакции можно получить, используя
классические методы расчета траекторий атомов, участвующих в
27. Динамика молекулярных реакций 487
Рис. 27.13. Рассчитанные траектории для реакционных столкновений между Л
и ВС.
химической реакции. На рис. 27.13 представлены результаты клас-
сического расчета положений трех атомов в реакции АЦ-ВС-—>•
—>-АБ + С, На горизонтальной координате отложено время, а на
вертикальной — межъядерное расстояние. Этот рисунок ясно пока-
зывает колебание исходных частиц и приближение атакующего
атома. Сама по себе реакция — перегруппировка атомов—проис-
ходит очень быстро. Вновь образованная молекула находится в ус-
тойчивом гармоническом колебании, в то время как отщепившийся
атом удаляется.
Хотя этот тип расчета позволяет судить о том. что происходит
во время реакции, недостатки его налицо. Во-первых, реальнг ?
газофазные реакции происходят в широком интервале скоростей
и при различных углах атаки. Во-вторых, движение атомов, элек-
тронов и ядер подчиняется квантовой механике. Представление
о траектории используется все меньше и меньше и заменяется
применением волновой функции сначала для реагирующих ве-
ществ, а затем для продуктов реакции. Тем не менее знание этих
недостатков не затмевает того факта, что современные достиже-
ния в молекулярной динамике позволяют проникнуть внутрь ре-
акции.
Литература
Pilling М. J. Reaction kinetics, Clarendon Press, Oxford, 1974,
Johnstone H. S„ Gas phase reaction rate theory, Ronald. New York, 1966.
Marcus P- A., Activated complex theory; current status, extensions and applica-
tions, in Techniques of chemistry (Lewis E. S., ed.), Vol, VIA, 13, W!ley-fn-
terscience, New York, 1974.
Бенсон С. Основы химической кинетики. Пер. с англ. — М..; Мир, 1964.
488
Часть 3. Изменение
Bamford С. Н., Tipper С. F, (eds.). Comprehensive chemical kinetics (Vois. I—
16), Elsevier, Amsterdam, 1969—1976.
Fluendy M. A. D., Lawiey K. P.r Molecular beams in chemistry, Chapman and
Hall, London, 1974.
Ross /. (ed.), Molecular beams; Adv. chem. Phys., 10 (1966),
Jordan J. Б., Mason E. A.r Amdur I., Molecular beams in chemistry in Techniques
of chemistry. Vol. HID, p. 365, Wiley-Interscience, New York, 1972.
Levine R. D., Bernstein R. S., Molecular reaction dynamics. Clarendon Press, Ox-
ford, 1974.
Глесстон С., Лейдлер K-, Эйринг Г. Теория абсолютных скоростей реакций. Пер,
с англ, — М.: ИЛ, 1948.
Меландср Л. Изотопные эффекты в скоростях реакций. Пер. с англ..—М.: Мир,
1964.
Задачи
27.1. Рассчитайте частоты столкновений z и Z в газообразном а) аммиаке (Ак
яй150 пм) и б) окиси углерода (R~ 120 пм) при комнатной температуре и дав-
лении 1 атм. Насколько увеличатся (в %) частоты столкновений, если темпера-
туру поднять на 10 К при постоянном объеме?
27.2. В интервале dE при значении энергии Е распределение Больпмана даег
значение числа молекул, пропорциональное ехр(—Е/kT)dE, Каков коэффициент
пропорциональности? Чему равна доля молекул, имеющих энергию по крайней
мере £а?
27.3. Теория столкновений связана со знанием доли молекулярных столкновений,
происходящих по крайней мере с пороговой энергией Еа. Чему равна эта доля,
когда: а) £я = 10 кДж/моль и б) £а = 100 кДж/моль при 200, 300, 500, 1000 К?
27.4. Рассчитайте увеличение (в %) доли молекулярных столкновений в предыду-
щей задаче прн повышении температуры на 10 К?
27.5. В реакции димеризации метильных радикалов экспериментальный предэкс-
тюненциальный множитель равен 2,4-10'° дмя/(моль-с). Чему равны: а) сечение
реакции и б) Р-фактор для реакции,
27.6. В задаче 26.24 был определен предэкспоненциальный множитель. Рассчитай-
те P-фактор и сечение о* для реакции атомов кислорода с бензолом.
27.7. Двуокись азота образуется в бимолекулярной газофазной реакции 2NO+
Ч-О2. Температурная зависимость константы скорости второго порядка дана ни-
же. Чему равны Р-факюр и сечение реакции?
7, К 600 700 800 1000
см»/(моль-с) 4,6-10г 9,7-103 1,3-Ю5 3,1-10®
27.8. Диаметр метильного радикала приблизительно равен 380 пм. Какова мак-
симальная константа скорости реакции второго порядка для рекомбинации ра-
дикалов при комнатной температуре?
27.9. 10% образца этана объемом 1 дм3 при комнатной температуре и давлении
I атм диссоциирует на метильные радикалы. Чему равно минимальное время
для рекомбинации на 90%.
27.10. Рассчитайте значения контролируемых диффузий констант скоростей для
молекул в а) воде, б) н-пеитане и в) н-децилбензоле. Вязкости равны соответ-
ственно 1,00-10-’, 0,22-Ю-3, 3,36-10—3 кг/(м-с) (1,00 сП, 0,22 сП, 3,36 сП).
27.11, Чему равно значение P-фактора в уравнении (27.1.7) для ионов в воде
при 25 °C? Найдите выражение для 1 : Пионов: а) одинакового заряда н б) про-
тивоположного заряда —и примите, что 7?^ 300 пм.
27.12. Одним из наиболее существенных результатов, полученвых в этой главе,
является уравнение Эйринеа [уравнение (27.2.7)]; важно познакомиться с его
применением. Сначала рассчитайте фактор kTIh при а) 0'С, б) 25 °C и в) 1000’С.
27. Динамика молекулярных реакций 489
27.13. Затем рассчитайте порядки величин функций распределения, входящих з
выражение для скорости. Установите порядок значений т, уг, 7® Для типич-
ных молекул. Проверьте, что при столкновении двух неструктурированных мо-
лекул порядок величины предэкспоненциального множителя будет таким, как
предсказывает теория столкновений.
27.14. Используя порядки величины функций распределения, полученных в пре-
дыдущей задаче, рассчитайте Г-фактор для реакции А+В—>-Р, где А и В —
нелинейные трехатомные молекулы.
27.15. Катализируемое основаниями бромирование ннтрометана-<33 в воде при
комнатной температуре протекает в 4,3 раза медленнее, чем бромирование недей-
терированного вещества. Объясните это различие. Возьмите Ат (С—Н) ~450 Н/м.
27.16. Предскажите порядок величины изотопного эффекта на относительные ско-
рости замещения: а) Ш и SH (трития), б) 16О и ,аО. Увеличивается ли зго раз-
личие при повышении температуры? Возьмите Ai(CH)«550 Н/м и A((CO)rs
т 1750 Н/м.
27.17. Главная трудность при применении теории активированного комплекса (и,
нужно согласиться, в его изображении) заключается в необходимости выбора
структуры активировав и от о комплекса и в выборе для соответствующих связей
в нем определенных значений прочности и длины. Следующий пример дает неко-
торое представление об этих трудностях, но позволяет получить числовые резуль-
таты для реакции, представляющей определенный интерес. Рассмотрим атаку Н
на Dj, которая является одной из стадий реакция H2+Da. Предположим, что Н
приближается к ГИг сбоку и образует комплекс в виде равнобедренного треуголь-
ника. Возьмите расстояние Н—D на 30% больше, чем в Нг (74 пм), и расстоя-
ние D—D на 20% больше, чем в Н2. Пусть критической координатой будет асим-
метричное валентное колебание, при котором одна связь Н—D растягивается,
а другая укорачивается. Пусть все колебания будут находиться приблизительно!
при 1000 см’1. Рассчитайте А2 для этой реакции при 400 К, используя экспери-
ментальное значение энергии активации, равное ~35 кДж/моль,
27.18. Теперь измените модель активированного комплекса в предыдущей задаче,
представив, что он линеен. Используя те же рассчитанные длины связей в мо-
лекулах и частоты колебаний, найдите для выбранной модели.
27.19. Ясно, чго в Двух последних задачах существует большая свобода в вы-
боре параметров моделей активированного комплекса. Гели у вас имеется до-
ступ к вычислительной машине, то составьте и просчитайте программу, которая
позволяет подходящим образом изменять структуру комплекса и параметры, и
обратите внимание на модель (или больше чем ца одну модель), которая дает
значение близкое к экспериментальному значению, равному 4-10s дм9/(моль-с).
27,20. Какова энтропия активации для столкновения между двумя практически
Неструктурированными частицами при 25°C?
27.21. Рассчитайте энтропии активации для реакции H2-FD2, используя модели,
описанные в задачах 27,17 и 27.18.
27.22. Уравнение Эйринга можно также применять к физическим процессавг-
В качестве примера рассмотрим скорость диффузии атома, «прикрепленного»
к поверхности твердого тела. Предположим, чго для передвижения с одного ме-
ста на другое необходимо преодолеть вершину барьера, где атом сможет клас-
сически колебаться в вертикальном и в одном горизонтальном направлениях, но
колебание вдоль другого горизонтального направления приведет его в соседнее
Место. Найдите выражение для скорости диффузия и рассчитайте его для диф-
фузии атомов вольфрама на поверхности вольфрама (£а=60 кДж/моль). Пред-
положим, что частоты колебаний в переходном состоянии а) те же самые а
б) составляют половину от величины для адсорбированного агома. Чему равно>
значение коэффициента диффузии D при 590 К? (Возьмите расстояние между
центрами равным 316 пм и vss 10п Гц.)
27,23. Предположим теперь, что адсорбированная мигрирующая частица является
сферической молекулой и может свободно как вращаться, так и колебаться на
|Верщине барьера, но при адсорбции на некотором центре частица может только
490
Часть 3. Изменение
колебаться. Как это повлияет на коэффициент диффузии? Рассмотрите молекулу
метана. (Обратите внимание на один из наиболее важных аспектов теории ак-
тивированного комплекса: если мы принимаем беспристрастное (любое) прибли-
жение частиц друг к Другу, то мы предсказываем одно значение скорости, но
если мы хотим построить модель с некоторым определенным приближением их
друг к другу, то нельзя допускать, что молекула вращается даже на вершине
барьера.)
27.24. Исследование с помощью электронного парамагнитного резонанса показы-
вает, что нитроксидный радикал в твердой матрице вращается с частотой, при-
близительно равной 1.0-108 Гц при 115 К (см. задачу 19.23). Используйте урав-
нение Эйринга для расчета функции активации Гиббса для вращения.
27.25. Предэкслонеяциальный множитель для газофазного разложения озона при
низких давлениях равен 4,6-Ю12 дмэ/(моль-с), а энергия активации равна
10,0 кДж/моль. Каковы; а) энтропия активации, б) константа скорости при
“25 °C и в) функция активации Гиббса?
27.26. Какова связь между и £а? Покажите, что для бимолекулярной газо-
вой реакции Е, -iH*+2RT.
27.27. Скорости термолиза различных цис- и траяс-азоалкаков были измерены
в интервале температур для выяснения механизма реакции. В этаноле неустой-
чивый цис-азоалкан разлагался со скоростью, определяемой по выделению азота,
и на основании этого были найдены константы скорости, приведенные ниже
[Engel Р. S.. Bishop D. J. Amer- chem. Soc., 97, 6754 (1975)]. Найдите эн-
тальпию, энтропию, энергию и функцию активации Гиббса.
t, °C —24,82 —20,73 —17,02 —13,00 —8,95
lO’-fet, с-1 1,22 2,31 4,39 8,50 14,3
27.28. Если активированный комплекс образуется из заряженных ионов z'e и
г"е, которые в комплексе находятся на некотором характеристическом расстоя-
нни’Т?*, то функция активации Гиббса будет содержать член, пропорциональный
z'z'7/?*Kt, где Кг —относительная диэлектрическая проницаемость растворителя.
Найдите выражение для зависимости константы скорости от К.,. (Отметьте, что
эта задача не затрагивает вопросов, касающихся ионной силы.)
27.29. Только что приведенная модель может быть проверена на основе следую-
щих данных. Бромфеиоловый синий обесцвечивается при добавлении ОН”; реак-
ция контролируется стадией, которую можно записать как В!-+ОН"—*ВОН3-.
Реакция между азодикарбонатным ионом (А2") н Н+ имеет следующую опре-
деляющую скорость стадию; А2-+Н*—*-АН~. Обе реакции осуществляются в
растворителях с различной относительной диэлектрической проницаемостью; ре-
зультаты приведены ниже. Подтверждают ли эти данные модель и расчет, при-
веденные в предыдущей задаче?
Реакция бромфенолового синего:
Кт Я 60 65 70 75 79
lgfes —0,987 0,201 0,751 1,172 1,401
Реакция азодикарбонатного иона:
К, 27 35 45 55 65 79
lgkt 12,95 12,22 11,58 11,14 10,73 10,34
27.30. При экспериментальном исследовании бимолекулярной реакции в водном
растворе была измерена константа скорости второго порядка при различной ион-
ной силе; результаты приведены нцже. Известно, что однозарядный ион участву-
27. Пинсмика молекулярных реакций
491
ет в лимитирующей скорость стадии. Чему равен заряд другого участвующего
иона?
/, моль/кг 0,0025 0,0037 0,0045 0,0065 0,0085
ka, дм’/(моль-с) 1,05 1,12 1,16 1,18 1,26
27.31. Покажите, что интенсивности молекулярных пучков до и после прохожде-
ния камеры длиной I, содержащей инертные рассеянные атомы, связаны следую-
щим образом: 7(7) = /(0)ехр(—Л’.а!), где а —сечение столкновений и Л3,—
плотность рассеянных атомов.
27.32. В эксперименте с молекулярным пучком для измерения сечения столкнове-
ний было найдено, что интенсивность пучка CsCl уменьшается на 60% при про-
хождении через C1-[2F2 при 10~s мм рт. ст.; когда мишенью был аргон при том
же самом давлении, интенсивность уменьшилась только на 10%. Каковы отно-
сительные сечения этих двух типов столкновений? Почему одно сечение иного
больше другого?
27.33. Общие сечения для реакций между атомами щелочных металлов и моле-
кулами галогенов приведены ниже {Levine R. D., Bernstein R. В., Molecular reac-
tion dynamics, Clarendon Press, Oxford, 1974, p. 72). Объясните данные на ос-
нове гарпунного механизма (разд. 27.1).
а*, (нм)’ Cl, Bra I,
№ 1,24 1,16 0,97
К 1,54 1.51 1,27
Rb 1,90 1,97 1,67
Сз 1,96 2,04 1,95
28 Процессы на твердых поверхностях
Изучаемые вопросы
После тщательного изучения этой главы вы сможете:
I. Описать рост кристаллических поверхностей (стр. 493).
!' 2. Объяснить важность винтовой дислокации (стр. 494).
3. Указать методы изучения чистых поверхпюстей (стр. 497).
4. Описать полевую эмиссионную микроскопию и полевую
ионизационную микроскопию (стр. 498).
5. Различать физическую адсорбцию и хемосорбцию (стр. 502).
6. Определить и измерить энергию активации десорбции
(стр. 505).
7. Определить вероятность прилипания и степень заполнения
(стр. 507).
8. Вывести и использовать изотерму адсорбции Ленгмюра
{уравнение (28.2.3)], изотерму Темкина [уравнение (28.2.5)] и
изотерму Фрейндлиха [уравнение (28-2.6)].
9. Выразить скорость каталитической реакции, используя изо-
терму адсорбции (стр. 516).
16. Описать каталитические процессы: гидрирование (стр. 519),
окисление (стр. 520), крекинг (стр. 520) и риформинг (стр. 520).
Введение
Процессы на поверхностях определяют большую часть сторон
повседневной жизни, включая саму жизнь. Даже если мы ограни-
чим наше внимание процессами только на твердых поверхностях,
важность и повсеместность этих процессов вряд ли может быть
преувеличена. Процессы на твердых поверхностях влияют на раз-
витие промышленности либо конструктивно, как в катализе хими-
ческих процессов, либо деструктивно, как в коррозии. Мы уже упо-
минали о пути распространения нервных импульсов (см. т. 1,
разд. 11-4); эта активность, так же как действие других органов
и клеток тела, зависит от свойств биологических мембран. Биоло-
гические мембраны — органические поверхности, которые регули-
руют активность тела, и чем больше они изучены, тем ближе мы
подходим к пониманию физической сущности жизни.
Фундаментальным динамическим аспектом поверхности явля-
ется ее роль как области, где твердое тело формируется, испаряет-
28. Процессы на твердых поверхностях
Рис. 28.1. Дефекты поверхности.
ся, растворяется. Поэтому мы начнем краткое рассмотрение с ро-
ста кристаллов и структуры простых поверхностей. Затем рассмот-
рим пути загрязнения поверхностей при отложении на них
посторонних веществ. Адсорбция — первая стадия гетерогенного
катализа, и мы обсудим эту промышленно важную тему. Среди
систем, связанных с процессами на поверхностях, особо важное
значение имеют электродные: они рассмотрены в следующей главе.
28.1. Рост и структура поверхностей
В этом разделе мы увидим, как расширяются поверхности и
растут кристаллы. Это даст нам картину структур, ответственных
за катализ. Мы увидим также, как можно контролировать поверх-
ностные свойства.
Как растут кристаллы. Наиболее легко форму кристаллической
поверхности можно представить, сравнив ес с булыжной уличной
мостовой. Атом газа, сталкивающийся с поверхностью, можно изо-
бразить в -виде мяча, подпрыгивающего на булыжнике. При под-
прыгивании атом теряет энергию, но потенциальные ямы поверх-
ности — углубления между булыжниками — очень неглубокие, и это
Позволяет атому отскакивать от поверхности прежде, чем он поте-
ряет достаточное количество кинетической энергии и осядет в од-
ной из них. Такая же ситуация наблюдается для ионного кристал-
ла, находящегося в растворе: энергетически маловероятно, чтобы
ион в растворе мог оторваться от сольватирующих молекул и
«воткнуться» в определенное место на поверхности.
Картина изменяется, когда поверхность имеет дефекты,
т. е. когда имеются уступы из-за наличия неполных слоев атомов
и.'ш ионов. Типичным дефектом поверхности является край между
двумя в других отношениях плоскими террасами (рис. 28.1). Этот
краевой дефект может сам по себе быть дефектным: у него могут
быть изгибы.
Прежде чем атом осядет па террасе, он несколько раз отска-
кивает, пока отскоки не приведут его к краю или углу, образован*
494
Часть 3. Изменение
ному изгибом. Вместо взаимодействия с одиночным атомом на
террасе он может теперь взаимодействовать с несколькими атома-
ми, и это взаимодействие может быть достаточно сильным, чтобы
остановить и задержать атом. Подобно этому, в случае выделения
ионов из раствора потеря энергии сольватации компенсируется
значительно большим кулоновским взаимодействием между при-
ближающимся ионом и несколькими ионами в непосредственной
близости от поверхностного дефекта.
Это обсуждение показывает, что для выделения и дальнейшего
роста Кристаллов необходимы поверхностные дефекты. Но одного
этого недостаточно. По мере протекания процесса оседавия по кра-
ям к изгибам непрерывной поверхности наступает время, когда
более низкая терраса заполняется полностью. На этой стадии по-
верхностных дефектов нет, и, таким образом, рост приостанавли-
вается. Для продолжения роста необходимы такие дефекты по-
верхности, которые сами возникают в процессе роста кристаллов
и не исчезают в процессе роста. Такие дефекты можно обнару-
жить, рассматривая виды дислокаций, которые существуют в объе-
ме кристаллов.
Дислокации кристаллических решеток, которые могут иметь
различные формы, ответственны за механические свойства твердых
тел; ковкость и хрупкость. Такие дислокации появляются при нару-
шении правильности упаковки решетки атомов или ионов. Это
может произойти по разным причинам. Одной из возможных при-
чин является то, что рост кристаллов происходит так быстро, что
атомы не имеют достаточно времени, чтобы занять положения с
низшей потенциальной энергией и удержаться в этом положении
до отложения следующего слоя атомов. Другой причиной может
быть присутствие примесного атома, который в непосредственной
близости от себя искажает решетку.
Искажение решетки, называемое винтовой дислокацией, пред-
ставлено на рис. 28.2, изображающем срез в кристалле, когда ато-
мы слева от среза высту-
пают на расстояние эле-
ментарной ячейки. Теперь
элементарные ячейки
кристалла образуют не-
прерывную спираль вок-
руг конца среза — винто-
Рис. 28.2. Вивтовая дислока-
ция.
28. Процессы на твердых поверхностях
495
Рис. 28.3. Спиральный рост.
а — иодид кадмия (Rosenberg Н. М., The Solid State. 1975); б — я-парафии (Jennings В R
Morns V. J., Atoms in contact. Clarendon Press, Oxford, 1974) (Courtesy of H. F. Kay)’
вой оси. Круговое движение вокруг внутреннего края среза идет
по спирали к вершине кристалла. Там дислокация выходит к по-
верхности, где она имеет форму спиральной лестницы от дна края
до его вершины (рис. 28.2).
Поверхностный дефект, образованный винтовой дислокацией,
является краем, возможно, с несколькими изгибами, на котором
может происходить рост. Приближающийся атом ложится в из-
гибы лестницы, и вновь образующиеся ряды снова образуют край-
496
Часть 3. Изменение
Рис. 28.4. Противовращение соседних винтовых дислокаций приводит к террас*
ным плоскостям.
под углом к первоначальному положению края. По мере роста
кристаллов край вращается вокруг винтовой оси и не препятству-
ет росту. Поэтому рост может продолжаться неограниченно.
По всей длине края рост может происходить с неодинаковой
скоростью, и вместо полного покрытия поверхности новыми атома-
ми последовательное вращение приводит к постепенному образо-
ванию кристаллов в виде спирали. Может происходить образова-
ние нескольких слоев, и, таким образом. Края спирали могут круто
обрываться на высоте многих атомов. Этот тип роста грани кри-
сталла с образованием четко различимой спиральной поверхности
наблюдается часто (рис. 28.3,6).
Развитие спиральных выступов может также привести к обра-
зованию ряда плоских поверхностей, что делает грань кристалла
похожей на ступенчатую пирамиду (рис. 28.3, а). Это свидетель-
ствует о том, что рост происходит по соседним винтовым дислока-
циям слева направо; на рис. 28.4 показана последовательность
построения. Осаждающиеся слои на краях соседних дефектов
сталкиваются, и рост продолжается внутри V-образной впадины.
Здесь рост обусловливает появление двух краев, и оба они делают
еще одно полное вращение, а затем снова сталкиваются. Этот про-
цесс не обязательно происходит одинаково по всей длине грани,
и, таким образом, плитки могут последовательно уменьшаться в
размерах.
Террасы, образованные такими способами, могут заполняться
при дальнейшем отложении по их краям, и в результате могут
получаться более или менее плоские кристаллические грани. Ско-
рость роста часто различна для различных кристаллических гра-
28. Процессы на твердых поверхностях 497
ней, и внешний вид кристалла определяется наиболее медленно
растущими гранями. Это заключение можно легко понять из диа-
граммы на рис. 28-5. Мы видим, что хотя горизонтальная грань
вырастает наиболее быстро, Опа «перерастает свое существова-
ние», а медленно растущая грань продолжает свое существование.
Экспериментальные методы изучения поверхностей. Мы ограничим-
ся изучением металлических поверхностей, откачанных в хорошем
вакууме: в следующей главе мы увидим, какие методы использу-
ются для изучения поверхности раздела фаз металл — раствор.
Изучение чистых поверхностей обязательно требует чистых ус-
ловий. Как показывают простые вычисления, это означает гораздо
больше, чем очистку образца и осторожное с ним обращение.
В гл. 24 кинетическая теория газов была использована для вывода
выражения [уравнение (24.2.9)] для числа столкновений в еди-
ницу времени на единицу площади вещества, когда давление газа
равно р;
Zw «: pl{2nmkT)^. (28.1.1)
Это уравнение полезно выразить в форме
Zw (см-8, с’1) «3,51 • 10ир (мм рт. ст.)/[Т (К) Мг]1/« «
® 2,03-10а1р(мм рт. ст.)//Мг при 298К, (28.1,2)
где Мг — относительная молекулярная масса газа. Для воздуха
(Мг «29) при атмосферном давлении (р«760 мм рт. ст.) частота
столкновений довольно высока и равна 3-1025 см“2*с-1, а посколь-
ку 1 см2 металлической поверхности содержит около 1015 атомов,
то на каждый атом в каждую секунду будет приходиться около
1010 ударов- Даже если только некоторые из этих столкновений
приведут к тому, что молекулы газа останутся на поверхности,
действительное время, в течение которого поверхность остается
чистой, будет очень небольшим.
Уменьшение давления газа уменьшает частоту столкновений,
и если это уменьшение достаточно велико, то поверхность в тече-
ние долгого времени будет оставаться незагрязненной. Обычная
высоковакуумная система работает При давлении около 10_fi мм
рт. ст., и частота столкновений в этом случае составляет только
Рис. 28.5. Более медленно растущие грани определяют внешний вид кристалла.
32—242
498
Часть 3. Изменение
~ 1016 см-2-с-1, что соответствует одному удару на поверхностный
•атом каждую 0,1 с, но даже это слишком много для большинства
экспериментов. Техника сверхвысокого вакуума предназначена для
достижения обычным путем давлений до 10'9 мм рт. ст., а при
использовании специальной техники — ниже 10~10 мм рт. ст. Та-
кие давления уменьшают частоту столкновений до 10*1 см-2-с-1
или до 10’° см-2-с-1 соответственно, что отвечает одному удару
на каждый поверхностный атом в течение 103—105 с, т. е. в тече-
ние нескольких часов.
Типичная аппаратура УВВ предполагает прогрев всей откачи-
ваемой части при 200—300 °C в течение нескольких часов для обез-
гаживания Стенок установки. Для того чтобы избежать влияния
смазки, все краны и затворы делаются металлическими. Катали-
затор обычно используется в виде тонкой фольги, нити (волоска),
тонкой проволоки. Для очистки металлической поверхности ката-
лизатор нагревают пропусканием через него тока или бомбарди-
ровкой ускоренными газообразными ионами. Это необходимо де-
лать очень осторожно, так как ионная бомбардировка может при-
вести к разрушению поверхностной структуры и превращению ее
в аморфное скопление атомов.
Структура поверхности может быть изучена различными мето-
дами. Расположение поверхностных атомов может быть определе-
но дифракцией, если используемое излучение имеет низкую про-
никающую способность. Как указано в разд. 22.6, электроны с
низкой энергией имеют слабую проникающую способность и пре-
терпевают сильную дифракцию на поверхностных слоях атомов:
этот метод известен как дифракция медленных электронов. Он
особенно часто используется для сравнения поверхностей до и
после адсорбции газов. Отражение видимого света также может
быть использовано для получения некоторой информации о по-
верхности, даже если его длины волн значительно больше, чем
используемые для дифранции. Метод эллипсометрии основан на
исследовании изменений поляризации света, отраженного от по-
верхности, и используется для определения степени покрытия по-
верхностей примесями и толщины адсорбированных слоев.
Но наиболее эффективные результаты получаются с помощью
двух методов, близко связанных Друг с другом. В полевой эмис-
сионной микроскопии (ПЭМ) используется образец в виде нити с
вытравленным заостренным концом. Нить помещается в откачан-
ный сосуд с флуоресцентным экраном. При наложении большой
разности потенциалов электроны вылетают из образца по направ-
лению к экрану. Электрон, выходящий из образца, на пути от кон-
ца нити к экрану ускоряется и дает вспышку света в том месте, где
он ударяется об экран. Легкость выхода электронов из металла
зависит от его поверхностной структуры [и, в частности, от работы
выхода (см. т. 1, разд. 13.2), которая зависит от природы повевх-
28. Процессы на твердых поверхностях
49»
Рис. 28 6. Фотография полевой эмиссии вольфрамового острия радиусом 210 нм
(Prutton М., Surface physics, Clarendon Press, Oxford, 1975).
ности], и, таким образом, на экране можно наблюдать вариации
интенсивности, соответствующие вариации структуры поверхности.
Типичный результат показан на рис. 28.6. Изменение наблюдаемой
картины при отложении на металле вещества может быть исполь-
зовано для обнаружения наиболее вероятных мест отложения
атомов.
Развитием ПЭМ является полевая ионизационная микроскопия
(ПИМ). Аппаратура фактически та же самая, но разность потен-
циалов приложена в противоположном направлении, так что флуо-
ресцентный экран оказывается отрицательным по отношению к
острию проволоки. В установку напускается немного газа (на-
пример, гелия), молекула газа ударяется об острие и «скачет»
по его неровной, террасной поверхности до тех пор, пока не на-
ткнется на выступающий атом, например на атом ребра края
(рис. 28.7). Выступающие атомы способны ионизировать атомы
газа, и сразу же образуется положительный ион (например, Не+),
а разность потенциалов направляет его к экрану, столкновение
с которым вызывает флуоресценцию.
32«
-500
Часть 3. Изменение
Р.че 23.7. Явление, на котором осЕсовайо представление поверхности с помощью
ПИМ.
Разрешающая способность метода ПИ'М зависит от попереч-
ного движения ионов газа, которое можно уменьшить охлаждени-
ем острия примерно до 20 К, если требуется разрешение порядка
атомных размеров. Это значит, что можно разрешить положения
индивидуальных атомов, и примером служит рис, 28.8: неболь-
шие светлые пятна соответствуют отдельным атомам на террасах
острия. Эта удивительная картина показывает мощь метода ПИМ,
однако не следует забывать об его ограничениях. Прежде всего,
па разных атомах ионизация происходит в неравной степени, и
многие атомы на поверхности, даже некоторые атомы на ребрах,
выступают недостаточно для того, чтобы вызвать ионизацию, и по-
этому не приводят к пятнам на флуоресцентном экране. Кроме
того, образец должен иметь форму острия и быть изготовленным
из материала, достаточно прочного, чтобы выдерживать очень вы-
сокие электрические поля, которые имеют тенденцию разрушать
его структуру. Несмотря на эти ограничения, метод примечателен
своей способностью давать картину положения отдельных атомов.
Метод ПИМ настолько тонок, что позволяет идентифицировать
Индивидуальные атомы, вкрапленные в поверхность образца, чи-
стого в других отношениях. Полевая ионизационная микроскопия
атомного зонда является последним словом хирургии. ПИМ-изо-
бражение адсорбированного атома совмещают с отверстием на
флуоресцентном айране. Газ, с помощью которого получают изо-
28. Процессы на твердых поверхностях
501
Рис, 28.8. Полевая ионизационная картина иридиевого острия (Courtesy of Mul-
ler Е.)
бражение, удаляется, а импульс разности потенциалов вырывает
атом в виде иона. Он движется в том же направлении, что и
ушедшие ионы газа, и проходит через отверстие в экране. Позади
экрана устанавливают масс-спектрометр и таким образом иденти-
фицируют атомы, определяя их отклонение в электростатическом и
магнитном нолях. Помимо того что метод является последним
словом аналитической техники (зная расположение атома в об-
разце, мы знаем, что он из себя представляет), с его помощью
можно наблюдать события, которые разыгрываются в атомной
шкале времени. Например, анализирующий импульс длится около
2 нс, и в течение этого времени иногда можно наблюдать испаре-
ние примерно 10 слоев атомного вещества. Это соответствует ско-
рости испарения, эквивалентной понижению поверхности на 1 м/с.
502
Часть 3, Изменение
28.2. Адсорбция на поверхностях
Теперь мы обратимся к исследованию того, как посторонние!
вещества «прилипают» к поверхности {адсорбируются) и затем!
принимают участие в реакциях.
Физисорбция и хемосорбция. Молекулы могут адсорбироваться на!
поверхности двумя способами. В дшзисорбииц. (сокращение физи-
ческой адсорбции) взаимодействие между поверхностью и адсор-
бированной молекулой (стр. 302") является вандерваальсовым. Это
ляльно действующее но гпябое т^чнмппрйгтпиа, н количество энер-
гии, выделяющееся при фи.чигпрбпии, по величине такого же по-
рядка, как энтальпия конденсации. Эта энергия может поглощать-
ся в виде колебаний решетки и рассеиваться в виде тепла, и моле-
кула, подпрыгивающая по неровной поверхнасти, теряет свою,
кинетическую энергию и прилипает к поверхности. Теплота, выде!
ляющаяся при физисорбции, может быть определена путем изме!
рения повышения температуры проволоки или фольги; наиболеЯ
часто наблюдаются изменения энтальпии около 20 кДж/моли
{табл. 28.1). Этих энергий недостаточно для разрыва связи, »
Таблица 28И
Энтальпия адсорбции —ДЯ, кДж/моль
HS С,н* НИЭ NS Ol 1
Физисорбция Хемосорбция 8,4 33 38 21 21
на угле 209 71 — 406
на Fe 134 285 188 167 314
таким образом, в физисорбции молекула сохраняет свою индиви-
дуальность, хотя она может быть растянута и изогнута из-за бли-
зости поверхности.
Физисорбированная молекула колеблется в неглубокой потен-
циальной яме, к так как связывающая энергия мала, то молекула
может сама себя «стряхнуть» с поверхности. Следователвно, можно
считать, что молекула остается на поверхности только очень ко-
роткое время, прежде чем возвратиться в газ. Можно предпола-
гать, что скорость отрыва следует закону типа Аррениуса с кон-
стантой скорости, выражаемой уравнением
^десор*чня ~ А ехр (
28. Процессы на твердых поверхностях
503
.« величина ДУ&де^ддбция может Лыть отождествлена со средним вре-
менем ^кизди т молекульща поверхности. Тогда
гай тоехр(Еада,
(28.2.1)
где то=1/Д. Подставляя Еа«25 кДж/моль (энергию, которая долж-
на быть отобрана у решетки для осуществления десорбции) и по-
лагая, что предэкспоненциальпый множитель А имеет тот же по-
рядок, что и частота колебаний слабой связи молекулы с поверх-
ностью {порядка 1012 с'1), найдем время жизни, равное прибли-
зительно 10~в с при комнатной температуре. Времена жизни по-
рядка 1 с получаются только тогда, когда температура понижа-
ется приблизительно до 100 К.
В хемосорбции (сокращение химической адсорбции) молекулы
удерживаются на поверхности в результате образования химиче-
ской, и обычно ковалентной, связи. Энергия связи в данном случае Г
значительно больше, чем в физисорбции, обычные значения нахо- /
дятся в области 200 кДж/моль (измерены, как и раньше, контро-/
лем повышения температуры образца известной теплоемкости)/
Хемосорбированная молекула может разорваться за счет ненасы-
щенных валентностей поверхностных атомов и таким образом по-
терять свою индивидуальность. Существование молекулярных ос-
колков на поверхности, образующихся при хемосорбции целых
молекул, является одной из причин проявления поверхностью ка-
талитической активности и причиной, объясняющей, почему остав-
шаяся часть этого раздела посвяшена хемосорбции.
Хемосорбированные частицы. Если хемосорбция протекает само-
произвольно, то опа должна 'быть экзотермической. Эте—мджно ~
объяснить следующим образом. Когда частица адсор бирустсяца"^
поверхности, ее поступательная свобода уменьшается, и .поэтому
процесс сопровождается уменьшением энтропии. Так какДС —
— Д//~7~ТД'‘э''.ч^_-ч|;;:Ч1!т, что АН должна быть отрицательной, чтобы
—бы'йа^отрицатёльной. а^1ТгтщцателБН0^П31иененйе Лнталътщ»--
со о fSETCfB у ет экзотермическому проц&ёсу^ (см. т. 1. разд. 4.1). Ос- >
певным критерием для разл^чдямеждухемосорбцией^дФизисорб - я
1Ц{еЙ- служит величина
ции редко является более отрицательной, чем приблизительно/!
—25 кДж/моль, в то время как АЯт для хемосорбции обычно зна-1/
чительно более отрицательна и иногда более отрицательна, чем|
—400 кДж/моль (табл. 28.1). /
Знание изменения энергии, происходящего при подходе частиц,
позволяет сделать выводы об их скоростях. По мере приближения
молекулы к поверхности ее потенциальная энергия уменьшается.—.
поскольку она~"фй5НЧескГ~0сорбируется- Диссоциация на фраг-
мейтя-в случг!ё Рбд5рода~имкот;ДДмолекул углеводородов проис-
ходит по мере того, как молекула переходит в хемосорбированное
состояние. Пока ее связи растягиваются, энергия системы растет,
504
Часть 3. Изменение
Рис. 28.9. Профиль потенциале.- 4
пой энергии хемосорбции. ’
ДН(Р) — энтальпия физисорбция,
ДЯ(С) — энтальпия хемосорбции»
энергия активации хемосорб-
ции.
затем она резко падает, поскольку молекула приходит в хемосор-
биррняннпр спстояние, и СВЯЗИ ЧАСТИН с__по-всрхностыо достигают ~
^полной силы. Даже если молекула нс диссоциирует при хемисорб-”
ции, все же вероятно, что ^энергия увеличивается, поскольку на
поверхности происходит -перегрупдйровка ерпзйй^ёжду~ато^гшт^--
необходимая, для осуще£твления_^кемосдрбции приблйжаКппейся
молекулы^ Поэтому в обоих случаях можно Предположить суще*
ствование"'энергетического барьера, разделяющего физисорбиро-
ванное и хемосорбированное состояний (рис. 28.9). Этот барьер,
однако, не очень велик, он н^ может быть выше энергии частицы,
взятой отдельно. В этом случае-хемосорбция из газа не является
активационным процессом и, вероятно, протекает очень„быстро:
адсорбций многих., газов на очень чистых металлических поверх-
ностях безактивационнаТТ?“некоторых случаях барьер между фи-
зисорбнрованным и хемосорбированным состояниями может-быть
больше энергии свободной молекулы, тогда для протекания хе-
мосорбции требуется по крайней мере-минимальная энергия: в этом
случае процесс является активационным и протекает более мед-
ленно, чем безактивационная хемосорбция.
Такое рассмотрение свидетельствует о том, что скорости не
являются хорошим критерием для различия физисорбции и хемо-
сорбции. Хемосорбция может протекать быстро, если энергия ак-
тивации мала или равна нулю, но может быть медленной, если
энергия активации больше нуля. Физисорбция обычно протекает
быстро, но, по-видимому, может быть и медленной, если адсорбция
протекает на пористой поверхности.
_ Песпрgхемосорбированных частиц всегда является актива- .
цибн ным процессомГ так Т?а^1аст^11ам2^о5ходдмд^3еднятьёя~соД^
пмя___я*ццДмлспрбттыи .9тя активации значительно боль-
<ие7чем для десорбции из физисорбированного состояния, и, таким ;
*
28. Процессы на твердых поверхностях
505
Рис. 28.10. Скорость десорбции кадмия с вольфрама (Sandejas J. S., Hudson}. В.,
in Fundamentals ol gas-surface interactions, eds^ Saltsburg H., Smith J. N., Ro-
gers M., Academic Press, New York, 1967).
образом, время пребывания частицы на поверхности значительно
больше, даже при повышенных температурах. Взяв fa^lOO кДж/
/моль и полагая, что Д~10и с“* (так как связанный фрагмент
колеблется с более высокой частотой, чем в случае физисорбции,
вследствие более прочной связи при хемосорбции), получаем время
жизни около 3-1№ с при комнатной температуре; оно уменьшается
до 1 с только при повышении температуры до 370 К.
Энергия активации десорбции может быть измерена путем по-
вышения температуры нити или фольги до заранее определенной
температуры и наблюдением зависимости давления от времени.
Измерения повторяют при разных температурах и строят арре-
ниусовский График зависимости In ke от 1/Г, где fed — константа
скорости для десорбции. Предэкспоненциальвый множитель А мо-
жет быть найден по отрезку, отсекаемому Прямой на оси Ординат,
а энергия активации Е?.— по ее наклону.
Во многих случаях наблюдается только единственное значение
'энергии активации; это указывает на то, что все адсорбционные
места равноценны. Однако в некоторых важных случаях, по-ви-
димому, играют роль два десорбционных процесса. Рис. 28.10 по-
казывает результаты опыта, в котором на вольфраме был отложен
кадмий. Рисунок свидетельствует о наличии двух типов связыва-
ющих центров, Один тип центров соответствует свободносвязанным
506
Часть 3, Изменение
атомам Cd, они десорбируются при температурах порядка 450 К
с энергией активации 18 кДж/моль. Второй тип атомов удержива-
ется на поверхности до тех пор, пока температура не поднимется
до 700 К, и требует энергию активации 90 кДж/моль. Объяснение-
такого поведения заключается в том, что более прочно связанные-
атомы Cd прикрепляются прямо к поверхности вольфрама, а ме-
нее сильно связанные атомы являются слоем или слоями, лежащи-
ми выше этого основного монослоя. Обычно хемосорбция не может
превышать моиослойного покрытия, потому что углеводород, на-
пример, не может хемосорбироваться на поверхности, уже покры-
той углеводородом или его фрагментами. Многослойная хемосорб-
ция может происходить при отложении одного металла на другом^
Другим примером системы, имеющей две энергии активации,
является адсорбция нормального газа. Окись углерода десорбиру-
ется с вольфрама в двух температурных интервалах. Первый ин-
тервал соответствует температурам 400—600 К и энергии актива-
ции 120 кДж/моль, а второй-температуре 800—1400 К и энергии
активации 300 кДж/моль. Для объяснения в этом случае предпо-
лагают существование двух типов центров на поверхности металла,
связывающих СО. На одном из этих центров молекулы газа свя-
зываются простой линейной связью М—СО, а на другом, сильнее
связывающем, образуется мостиковая связь СО с двумя поверх-
№
ностными атомами в виде рС = О.
м/
Когда мы перейдем к рассмотрению каталитической активности,
увидим, что некоторые реакции происходят путем миграции двух
адсорбированных частиц по поверхности до тех пор, пока они не
встретятся. Катализатор не смог бы выполнять свою функцию,
если бы связывал частицы в виде диссоциированных фрагментов
и эти фрагменты связывались настолько сильно, что они были бы
неподвижны. Подвижность частиц на поверхности зависит от глу-
бины ямы хемосорбции, и этот процесс является активационным.
Энергия активации диффузии по поверхности не обязательно та
же самая, что и энергия активации десорбции, так как адсорби-
рованные частицы могут проходить через «долины» между потен-
циальными пиками. Более того, преобладающую роль могут Играть
дефекты структуры, потому что может быть так, что движущимся
частицам легче перескакивать по террасе, чем катиться по дну
края, и они могут задержаться в вакансиях на разных плоских
террасах. Диффузия может протекать на одной грани кристалла
легче, чем на другой, и, таким образом, подвижность молекул на
поверхности может зависеть от того, на каких плоскостях решетки
происходит диффузия.
Метод ПИМ используется в двух очень изящных методах опре-
деления диффузионных характеристик частиц на поверхностях.
28. Процессы на твердых поверхностях 507
Один метод включает отложение адсорбата на группе поверхност-
ных атомов при низких температурах и получение картины ПИМ
этого начального состояния. Затем температуру поднимают и на-
блюдают, как атомы выходят с различных поверхностей с различ-
ными скоростями. В модификации этого метода получают изобра-
жение отдельного атома; температуру повышают, а затем через
определенный интервал снижают. Получают новое изображение н
определяют новое положение атома. Последовательность картин
позволяет судить о перемещении атома по поверхности. Поскольку
среднее перемещение за время t равно где D— коэффи-
циент диффузии (см. разд. 25.2), величина D и его температурная
зависимость могут быть найдены для различных типов адсорбата
на различных участках поверхности. Диффузия является актива-
ционной и следует выражению Аррениуса
D =D0 ехр (— EJRT), (28.2.2)
причем предэкепоненциальный множитель Do и энергия активации
изменяются от одной грани кристалла к другой. Типичные значе-
ния энергий активации для атомов вольфрама находятся в интер-
вале 57—87 кДж/моль, а предэкспоненциальные множители име-
ют величину, примерно равную 3,8- 1О~Г см2-с_[.
Степень адсорбции; изотермы адсорбции. Скорость покрытия по-
верхности адсорбатом зависит от ряда факторов. Одним из них,
как мы видели, является энергия активации, ио, как правило, она
или мала, или равна нулю и редко играет преобладающую роль.
Другим фактором является частота столкновений, но при нормаль-
ных условиях она очень велика и становится медленной во вре-
менной шкале нормальных опытов только в условиях УВВ. Доми-
нирующий фактор может быть найден с помощью следующих до-
водов.^Когда молекула сталкивается с поверхностью, она может
удержаться на поверхности только в том случае, если ее энергия
рассеется в термические колебания лежащей под ней решетки и
это произойдет достаточно быстро. Иначе молекула просто отско-
чит от поверхности или будет перекатываться по поверхности до
тех пор, пока не дойдет до ребра, откуда молекула может вернуть-
ся в газовую фазу. Доля столкновений с поверхностью, приводя-
щая к адсорбции, называется вероятностью прилипания s:
скорость адсорбции молекул поверхностью
скорость столкновения молекул с поверхностью '
Поскольку знаменатель этого выражения может быть рассчитан
из кинетической теории, если известно давление, а числитель мо-
жет быть определен при измерении скорости изменения давления,
вероятность прилипания легко определить. Значения л изменяют-
ся в широких пределах. Например, при комнатной температуре
окись углерода на поверхностях некоторых переходных металлов
508
Часть 3. Изменение
имеет S в пределах 0,1-—1Д а азот, адсорбируясь на рении, дает
s< I0-2, что свидетельствует о том, что для адсорбции одной моле-
кулы должно произойти более ста столкновений.
Пример. Расгччпайте время покрытия 10% центров поверхности вольфрама (100)
азотом при 298 К, когда давление равно 2,0-10*э мм рт. ст., а вероятность при-
липания составляет 0,55.
Метод. Начнем с расчета числа атомов на единицу поверхности (100); постоян-
ная элементарной ячейки о. ц. к,-решетки равна 316 нм. Затем найдем Zw из
уравнения (28.1.1).
Ответ. Расстояние между центрами атомов на грани (100) равно 316 пм. Поэто-
му каждый атом занимает (316)s ни2 поверхности. Из этого следует, что число
центров на 1 м5 поверхности равно I м’/(316-10-В * * * 12 м)2=1,00-10г®. Частота
столкновений выражается уравнением (28.1.1); сначала найдем р:
р == (2,0- 10а мм рт. ст.)х(1,0133-№ Н-2)/(7б0 мм рт. ст.) = 2,7-10-? Н/м2.
Тогда
2 _________2,7-1(Т'Н/м2__________________
{2лХ28Х(1,66-10-2?кг)X(1,381 10^ Дж/К)Х(298 K))r/a “ j
= 7,810“м-2-с-1. 1
Поэтому время покрытия 10% от 1,00-1013 * центров равно |
i = (1,00-КЯц-2)/(7,8-1015м-2е-1-)Х(0,55) = 233с.
Комментарий. Покрытие полной поверхности будет длиться более чем 233 с, так
как вероятность прилипания падает с увеличением покрытия.
Вероятность прилипания зависит от доли свободной поверх-
ности и по мере заполнения поверхности падает до небольших
величин. Степень покрытия поверхности играет важную роль в
адсорбционных исследованиях и обычна выражается через степень
заполнения 9, выражаемую следующим образом:
q _ число заполненных адсорбционных мест
“..^число доступных адсорбционных мест ’
В простой модели поверхностных эффектов предполагается^ что
вероятность прилипания пропорциональна (i—9)—доли свобод-
ной поверхности, что обычно запит-ынаетс.я кяк._^яд%£1—ft),
где Да — .вероятность прилр1пан.ияи1а_£оведщен11(з .ццедой поверхно-
Результаты, приведенные па рис, 28.11, не соответствуют это-
му простому выражению, так как сначала, пока величина покры-
тия поверхности не достигнет 6-1013 молекула/см2, а остается
близким к s0, а затем резко падает до небольших значений, Веро-
ятно, объяснение такого поведения заключается в том, что снача-
ла сталкивающаяся молекула не хемосорбируется, а перекатыва-
ется по поверхности до тех пор, пока не подойдет непосредственно
к пустому центру.
Возникает вопрос, как степень заполнения зависит от давления
/газа над поверхностью. Если между молекулами, находящимися на
28, Процессы на твердых поверх костях
50»
Рис. 28.11. Вероятность прилипания как функция покрытия поверхности [см.
Gasser R, Р. Н. Lawrence С. Р., Newman D. G., Trans. Faraday Soc., 62, 2035
(1966)].
поверхности, и свободными молекулами в газовой фазе имеется
равновесие, то заполнение будет зависать от ,давления__системы.
Завдсимость^Д лзт давления при постоянной температуре называ-
ется системы.. - —*
Простейший тип изотермы основан па представлениях о рав-
ноценности каждого адсорбционного центра и способности моле-
кулы адсорбироваться независимо от того, заняты или нет сосед-
ние центры. Предполагая, что адсорбированные молекулы нахо-
дятся в динамическом равновесии со свободными молекулами, и
обозначая константы скорости для адсорбции и десорбции через:
йа и соответственно, можно записать
А (газ) М (поверхность) т—AM.
Скорость адсорбции пропорциональна давлению А и числу сво-
бодных центров на поверхности -jV(1 --Д), где N—общее число
центров. Поэтому
скорость адсорбции=йарАЛг(1—9).
Скорость десорбции пропорциональна числу адсорбированных
частиц jVg:.
скорость десорбции =AdA9,
При равновесии обе скорости равны, и, решая это уравнение от-
носительно 9, получим
изотерма Ленгмюра-. 9=Л^а/(1+^а)>. (28.2.3)
510
Часть 3. Изменение
Рис. 28.12. Проверка изотермы Ленгмюра для адсорбции СО па древесном угле.
ГДе
K=kJ^.
Пример (вопрос 8). Ниже приведены данные для адсорбции СО на древесном
угле при 273 К. Подтвердите, что они подчиняются изотерме Ленгмюра, и най-
" I объем, соответствующий полному покрытию поверхности.
дите константу К и
мм рт. ст. 100 200 300 400 500 600 700
IV см* 10,2 18,6 25,5 31,4 36,9 41,6 46,1
Навеска древесного угля 3,022 г; в каждом случае значение V приведено к
1 атм.
JWeroS. Из уравнения (28.2.3) Крл0+6=КрА. Запишем 0 = V/V„, где V« —
объем, соответствующий полному покрытию. В этом случае уравнение запишется
как рл(К=1/КК„4-рЛ/УТогда построение графика в координатах Pa(V от р*.
должно дать прямую линию с наклоном _1/Ку> и отрезком, отсекаемым на оси
ординат, ранным
Ответ. Получаем следующую таблицу:
,р, мм рт. ст. 100 200 300 400 500 600 700
p/V, мм рт. ст./см® 9,80 10,8 11,8 12,7 13,6 14,4 15,2
Данные представлены на рис. 28.12. Наклон равен 0,0090 и, таким образом,
К„ = 111 см3, Отрезок, отсекаемый па осп ординат, равен ^9,0, и поэтому
,К=1/[(111 см®)Х(9,0 мм рт. ст./см3)] = 1,0-10-3(мм рт, ст.)-1.
Комментарий. Отметьте отклонение от прямой линии при больших покрытиях
поверхности. Коэффициент- корреляции, рассчитанный по методу наименьших
жвадратов, равен 0,9979 (точное соответствие дало бы 1),
-28. Процессы на твердых поверхностях
51»
Рис, 28.13. Изотерма Ленгмюра [уравнение (28.2.3)] для некоторых значений JC
Форма изотермы Ленгмюра для некоторых значений /( пока*
зана на рис. 28.13. Покрытие поверхности увеличивается с давле-
нием и приближается к единице только при очень высоких давле-
ниях, когда газ эффективно прижимается к поверхности. При раз-
ных температурах получаются разные значения К', вот почему этц
линии называют изотермами. Отметьте, что /( является константой
равновесия распределения вещества между поверхностью и газо-
вой фазой. Это указывает на то, что ее температурная зависи-
мость может быть использована для определения изостерической
энтальпии адсорбции (ДН для определенного покрытия поверхно-
сти) по термодинамическому уравнению [см. уравнение (9,2.2)
(д In р/дТ)в = -ДЯ£7№ = ДЯ?Жа. (28.2.'4>
т
Пример. Нижеприведенные данные показывают, какое давление СО необходимо
для адсорбции 10,0 смэ при каждой температуре (все объемы приведены к 1 атм-
и 273 К)- Была использована та же навеска, что и в вышеприведенном примере..
Найдите энтальпию адсорбции при этом покрытии поверхности.
Т. К 200 210 220 230 240 250
р, мм рт. ст. 30,0 37,1 45,2 54,0 63,5 73,9
Метод. Необходимо построить график [уравнение (28.2.4)] зависимости [пр or
1/7; наклон прямой линии даст
512
Часть 3. Изменение
Рис. 28.14, Определение энтальпии адсорбции СО на древесном угле.
Ответ. Составляем следующую таблицу:
Т, К 200 210 220 230 240 250
1000/Т, К*1 5.00 4.76 4,55 4,35 4,17 4,00
Inр (мм р,. ст,) 3,40 3,61 3,81 3,99 4.15 4,30
График, построенный по данным этой таблицы, представлен на рис. 28,14. На-
клон лштии, скоррелированной по методу наименьших квадратов, равен —904,
И, таким образом, ДЯщ=—904X8,314 Дж/(К-мо.’ть) = —7,5 кДж/моль,
Комментарий. Поскольку К=1,1 -10“3 (мм рт, ст.)~’ при 230 К, это соответствует
AGm=13,0 кДж/моль. Отсюда получается ASra=—89 Дж/(К' моль); такое большое
уменьшение энтропии адсорбции связано с превращением трехмерного газа п
двумерную поверхность.
ДОсповные положения, лежащие в основе вывода изотермы
Ленгмюра, это независимость и эквивалентность адсорбционных
Центров. Цо, вероятно, имеются различные виды адсорбционных
центров, и энергия „адсорОДйи на любом центре будет зависеть от
того, заняты или пет соседние с ним центры. Отклонения от изо-
термы Ленгмюра наблюдаются очень часто; эти расхождения
обычно объясняются недостаточностью положений, заложенных в
выводе изотермы. "Например, энтальпия адсорбции становится
менее отрицательной при увеличении 0, и это указывает на то,
28. Процессы на твердых поверхностях
1п(У,см3)
Рис. 28.15. Проверка изотермы Фрейндлиха для адсорбции СО на древесном
угле.
изотерма Фрейндлиха: Q=c1PaC1‘
что сначала заполняются энергетически более подходящие центры.
Для учета этих изменений были предложены различные изотермы:
изотерма Темкина: 0 ~ci In (с2КрА), ' (28.2,5)
. . ♦
где С[ и Сг — константы, соответствующие предположению о ли-
нейном изменении энтальпии адсорбции с давлением, а
(28.2.6)
соответствует логарифмическому изменению. Различные изотер-
мы более или менее хорошо воспроизводят экспериментальные
данные в ограниченном интервале давлений, но они остаются в
значительной степени эмпирическими. Однако «эмпирическое» не
означает «бесполезное», поскольку если известны параметры дей-
ствительно надежной изотермы, то можно получить надежные ре-
зультаты Для степени заполнения поверхности при разных усло-
виях. Такая информация очень существенна для рассмотрения
гетерогенного катализа.
Пример (вопрос 8). Проверьте, вс лучше ли подходит изотерма Фрейддлиха
К данным но адсорбции СО при 273 К.
Метод. Если применима изотерма Фрейндлиха, то график зависимости In V от
1пр должен дать прямую линию. Сравним коэффициенты корреляции ПО методу
наименьших квадратов.
33—242
514
Часть 3. Изменение
Ответ. Составляем следующую таблицу:
р, мм рт. ст. 100 200 300 400 500 600 700
Inр (мм рт. ст.) 4,61 5,30 5,70 5,99 6,21 6,40 6,55
1пУ (см3) 2,32 2,92 3,24 3,45 3,61 3,73 3,83
Данные представлены на рис. 28.15. Коэффициент корреляции по методу наи-
меньших квадратов для прямой линии равен 0,9968, т. с. дальше от единицы,
чем соответствующий коэффициент для изотермы Ленгмюра (0,9979), и, таким
образом, в этом случае последняя изотерма лучше описывает опытные данные
в рассмотренном ряду давлений.
Комментарий. Изотерма Фрейндлиха часто используется при изучении адсорбции,
на жидких растворов. Тогда она записывается как w — ctc^ ^2, где w — масса ад-
сорбированного растворенного вещества на единицу массы адсорбата и с —кон-
центрация растворенного вещества. Это применение изотермы рассмотрено в за-
дачах.
28,3. Каталитическая активность на поверхностях
Причина гетерогенной каталитической активности та же, что
и в гомогенном катализе: катализатор ускоряет реакцию, снижая
энергию активации лимитирующей сталии. Поэтому, хотя он и не
смещает определяемый термодинамическим равновесием состав
реакционной смеси, он увеличивает скорость, с которой такой со-
став достигается (табл. 28.2).
Таблица 28.2
Энергии активации для каталитических реакций
Реакция Катализатор £а- кДж/моль
2Н1 > Н2-Ма Нет 184 Ац 105 Pt 59
2NH3 > N2+3H2 Нет 350 ! W ' 162 ।
Катализ, В большинстве случаев основой каталитической актив-
ности служит хемосорбция (иногда физисорбция), приводящая к
тому, что одна из реагирующих молекул находится в форме, в
которой опа может очень легко вступать в реакцию. Часто это
происходит потому, что хемосорбция сопровождается Диссоциа-
цией. В таком случае молекулярные фрагменты могут удаляться
с поверхности Другой приближающейся молекулой или переме-
щаться по поверхности до тех пор, пока не произойдет столкно-
вение с каким-нибудь другим адсорбированным молекулярным
фрагментом.
28, Процессы на твердых, поверхностях 515
Примером каталитического действия является гидрирование
олефинов. В этом случае олефин адсорбируется с образованием
двух связей у соседних атомов углерода:
На той же поверхности адсорбируются атомы водорода. В ходе
реакции происходит столкновение, и одна из поверхностных свя-
зей разрывается.
СН3СН2 СНЛСН2СН2 СН3СН2 СН3
> СН-СН2 + Н ЧСН2 или СН
гПттптТп гТп гггптгт Ьтт? гггггг^тттттгп
Затем другой атом водорода приближается к соседнему центру,
образует связь с адсорбированной молекулой, и, таким образом,
происходит термодинамически благоприятное образование пол-
ностью гидрированного углеводорода.
Доказательством двухстадийной схемы является существова-
ние в реакционной смеси различных изомерных олефинов. Это
связано с тем, что, перекатываясь по поверхности, углеводородная
цепочка может снова хемосорбироваться:
СН3СН2 СН,СН2 уСНз Н СН^ zCHs
\н-СН2+Н СН + CH—CH
/V'TTTTTT'VvT “V" “* П! i !T! I /1! 1:1i I >7/i ! i I II ill 1П :
Таким образом, при десорбции образуется СН3СН = СНСН3.
Если бы оба атома водорода реагировали одновременно, новый
олефин не смог бы образоваться.
Некоторые реакции протекают вследствие столкновений моле-
кул из газовой фазы с фрагментом молекулы ца поверхности.
В этом случае скорость образования продукта пропорциональна как
давлению неадсорбцрованного газа рЕ, так и степени покрытия по-
верхности адсорбированным газом А (0а). Поэтому скорость вы-
разится следующим образом:
А 4- В --* продукты, — dpfjat = ksp^fi А,
где k5 —• константа скорости для поверхностной реакции. Эта конс-
танта может быть значительно большей, чем для некаталитиче'
33’
5i6
Часть 3. Изменение
ской реакции, потому что она относится к реакции молекулы В с
молекулой А, которая уже оторвана вследствие хемосорбции.
Последнее уравнение может быть преобразовано далее, когда
известна изотерма адсорбции А. Если в интересующей нас области
давлений вещество А подчиняется изотерме Ленгмюра, то выра-
жение для скорости можно записать как
—dp^dt^ k&p&Kpk!(\-J-АрД
При больших давлениях А, когда наблюдается почти полное за-
полнение поверхности, скорость приблизительно равна так
как лимитирующей стадией является реакционное столкновение
молекул В с плотно покрытой поверхностью. При уменьшении
давления А, допустим, вследствие реакции значение Ард невели-
ко, скорость равна /гкАрАрв, а степень заполнения становится кри-
тической.
Пример (вопрос 9). Разложение фосфина РП3 на вольфраме является реакцией
первого порядка при низких давлениях и нулевого порядка при высоких давле-
ниях. Объясните эти наблюдения.
.Метой. Реакция записывается следующим образом: Р113—^-продукты. Ее ско-
рость пропорциональна 0 — степени заполнения поверхности фосфином. Выра-
зим 6 из изотермы Ленгмюра и возьмем нижние и верхние пределы давлений.
Ответ. Уравнение скорости записывается так;
— dpldt = k^^ kcKp/(l + Kp), ' ' ‘ :
где р—давление фосфина.
Когда 1 (при низких давлениях), — dpjdi^ kcKp. Это закон скорости
первого порядка, что и требовалось доказать. Зависимость давления фосфина от
времени выражается формулой р(0)ехр(— АСА7).
Когда др»1 (при высоких даиленвях), —dp/d/явfecKp/Kp = fc. Тогда ско-
рость реакции не зависит от давления фосфина, и это соответствует реакции
нулевого порядка.
Комментарий. Очень немногие гетерогенные реакции имеют первый порядок.
В задачах мы увидцм, как рассматривают скорости реакций, в которых продук-
ты адсорбируются ц тшгябируют действие катализатора.
Многие каталитические реакции, н том числе гидрирование оле-
финов, включают столкновения между фрагментами, уже адсор-
бированными па поверхности, и являются реакциями второго по-
рядка по степени заполнения поверхности. Их скорости можно
представить следующим выражением:
А 4-В —.*- продукты, —= 650A0Q.
Степени заполнения Од и 0В можно выразить через изотермы со-
ответствующих молекул, и если мы упростим задачу, предположив,
что как А, так и В подчиняются изотерме Ленгмюра, то получим
®а — АаРа/О+^лРа + АвРД . .
в = ^в/,в/(^ + АаРа-гАв/^б) .
28, Процессы на твердых поверхностях________
с Лл^^а/jfed ДЛЯ частиц А и точно так же для частиц В. Это зна-
чит, что зависимость скорости от давления выразится формулой
^Ра.!^=вЛ’аРвА Д- ^лРх ХвРв)!«
Если адсорбция частиц подчиняется другим изотермам, то необхо-
димо включить их.
Эти выражения скоростей являются приближенными, но опи
показывают, что зависимость от давления для каталитических ре-
акций может быть очень сложной даже для простых реакций.
Кроме того, константы, входящие в изотермы, зависят от темпера-
туры, и, таким образом, температурная зависимость скорости мо-
жет быть настолько сложной, что опа не подчиняется аррепиусов-
ской зависимости.
До сих пор при обсуждении скорости предполагалось, что ns-
крытие поверхности находится в равновесии с несвязанным веще-
ством. В этом случае адсорбция происходит настолько быстро,
что она не является определяющей скорость стадией. В таких слу-
чаях общая температурная зависимость определяется температур-
ной зависимостью константы &s и адсорбционных коэффициентов
Ад. Поскольку /Д является константой равновесия, то в случае-
экзотермической реакции он уменьшается с увеличением темпера-
туры, и степень заполнения поверхности также уменьшается.
Независимо от того, какой из этих эффектов преобладает, увели-
чение константы kg (которая может представлять подвижность
частиц на поверхности) зависит от природы как поверхности, так
и адсорбированных частиц.
В некоторых случаях покрытие поверхности не находится в
равновесии с объемной фазой, и тогда скорость реакции может
определяться скоростью поступления частиц к поверхности. Осо-
бенно часто это встречается в таких каталитических реакциях, где
объемной фазой является жидкий раствор, поскольку для диффу-
зии реагентов к поверхности металла необходимо время,
Примеры каталитических процессов. Хотя вся современная хими-
ческая промышленность зависит от методов получения, выбора и
применения катализаторов, все же в этом разделе мы попытаемся
указать скорее широкое применение катализаторов, чем детали их
технологического применения (см, табл. 28.3).
Выбор катализатора зависит от того, что необходимо сделать,
не будет ли он Отравляться побочными продуктами или примеся-
ми в реакционной смеси, и во многом определяется экономически-
ми расистами, связанными с его стоимостью и временем исполь-
зования.
Активность катализатора зависит от силы хемосорбции таким
образом, как это представлено на «вулкакообразной» кривой
(рис. 28.16). Для того чтобы иметь достаточную активность, ка-
518
Часть 3. Изменение
'.Таблица 28.3.
катализаторы и их применение*
Катализатор
Функция
Пример
Металлы
Полупроводниковые ок-
сиды н сульфиды
Ок си ды - неп р о во дни ки
Кислоты
Гидрирование
Дегидрирование
Окисление
Дегидрирование
Десульфирование
Дегидратация
Полимеризация
Изомеризация
Крекинг
Алкилирование
Fe, Nt, Pt, Ag
NiO, ZnO, MgO,
Bi2O3/MoO3
A13O3, SiOs, MgO
H3PO4. H2SO4
SiO2/Al2Oj
a Взято из работы Hoad G. C., Heterogeneous catalysis. Clarendon Press, Oxford,
1974.
тализатор должен быть интенсивно покрыт адсорбатом, а это тре-
бует умеренно сильной адсорбции. С другой стороны, если сила
адсорбции увеличится выше той, которая необходима для получе-
ния достаточного покрытия, то сила адсорбционных связей может
быть настолько большой, что эта приведет к уменьшению ката-
литической активности потому, Что либо подходящие молекулы не
смогут реагировать, либо адсорбированные молекулы будут не-
подвижны на поверхности.
Это показывает, что актив-
ность увеличивается по ме-
ре возрастания силы адсор-
бции (например, измерен-
ной по энтальпии адсорб-
ции), а затем падает; наи-
большей каталитической
активностью обладают ка-
тализаторы, лежащие в об-
ластях, близких к «кратеру
вулкана».
Рис. 28.16, ВудкаиооОразная кри-
вая каталитической активности
(Bond G. С., Heterogeneous cataly-
sis. Clarendon Press, Oxford,
1974).
Нижняя кривая — металлы второго ив’
риода; верхняя кривая—металлы
третьего и четвертого периодов.
28. Процессы на твердых поверхностях
519
Для адсорбции газов подходят многие металлы, и общий ряд
адсорбционной силы следующий: О2, СгНз, СаЕЦ, СО, Н2, СО2, N2.
Причем обычно кислород является наиболее сильно адсорбирую-
щейся молекулой, а азот — наименее. Некоторые из этих молекул
адсорбируются диссоциативно (например , водород). Переходные
металлы, такие, как железо, ванадий я хром, показывают силь-
ную активность в отношении всех этих газов, но металлы, подоб-
ные марганцу и меди, не способны адсорбировать азот и двуокись
углерода и только слабо адсорбируют водород. Металлы, находя-
щиеся слева в периодической системе (такие, как магний и ли-
тий), способны адсорбировать только наиболее активный газ —
кислород. Эти закономерности суммированы в табл. 28-4.
Таблица 28.4
Хемосорбционная способности
O2 с2н2 C2H1 co H2 co2 Na
Ti, Сг, Mo Fe + 4- 4- Ч- 4- 4- 4-
Ni, Со + ' 4- + 4- 4- + ——
Pd, Pt 4- 4- _L 4- -—-
Мп, Си 4- + + 4- i ‘— —-
Al, Au 4- _—, _ —-
Li, Na, К 4- 4- — — —- —- —-
Mg, Ag, Zn, Pb + — — — — —’
*+ сильная хемосорбция. ± слабая хемосорбция, — отсутствие хемосорбця и.
Гидрирование. Главным промышленным применением реакций
гидрирования является образование пищевых жиров из расти-
тельных и животных масел. Сырые масла, полученные из такого
сырья, как соевые бобы, имеют структуру СН3(ОгС1?'') —
—CH(O3CR'/)-—СЩОгСК"'), где R', R" и R^1" — длинные углево-
дородные цепочки с несколькими двойными связями. Присутствие
большого количества двойных связей вредно, так как делает мас-
ла чувствительными к атмосферному окислению, придает маслу
прогорклый вкус. Геометрическая конфигурация цепей, содержа-
щих жесткие двойные связи, обусловливает Тот факт, что масла:
являются жидкостями, а во многих случаях (например, для при-
готовления бутербродов) твердый жир не только значительно
лучше, но и часто просто необходим. Контролируемое частичное
гидрирование масла на катализаторе, подобранном таким обра-
зом, чтобы гидрирование было неполным и чтобы не происходила
изомеризация цепи, широко используется для получения пищевых
жиров. Сезонное изменение числа Двойных связей в маслах не
520
Часть 3. Изменение
оказывает влияния на промышленный процесс, так как реакция
'гидрирования при этом не облегчается.
'Окисление. Каталитическое окисление также широко иснользу-
“Стся в промышленности и во все возрастающей степени для конт-
роля загрязняющих стоков. Хотя в некоторых случаях желатель-
но добиться полного окисления, например для удаления окислов
азота из выхлопных газов двигателей, иногда необходимо непол-
ное окисление. Например, полное окисление пропилена в двуокись
углерода и воду убыточно, но мягкое окисление в акролеин
(СНЙ=СНСНО) является начальной стадией важных промыш-
ленных процессов. Точно так же процессы контролируемого окис-
ления этилена в этанол, этаналь (ацетальдегид) и, в присутствии
уксусной кислоты или хлора, в винилацетат (СН2=СНСООСНз)
или винилхлорид (СН2 = СНС1) являются первоначальными ста-
диями действительно важных химических производств.
Не все реакции, упомянутые в последнем параграфе, протека-
ют под влиянием простых металлических катализаторов, хотя
платина в контроле за выхлопными газами и палладий в образо-
вании винилацетата являются двумя исключениями. Некоторые
процессы протекают на различного вида твердых оксидах. Физи-
ческая химия этих сложных поверхностей очень часто неизвестна.
Некоторые вопросы могут быть рассмотрены на примере окисле-
ния пропилена в акролеин. Оно осуществляется на поверхности
молибдата висмута, которая адсорбирует пропилен в форме ал-
лильного радикала с отрывом атома водорода. Кислород поверх-
ности, взаимодействуя с аллилом, дает акролеин, который затем-
десорбируется. Атом водорода с поверхностным кислородом обра-
зует воду. На поверхности остаются заряженные оксидные ионы,-
которые присоединяют молекулы кислорода из атмосферы. Эти
молекулы кислорода хемосорбируются па поверхности в виде ок-
сидных ионов, и, таким образом, катализатор восстанавливается.
В реакциях такого типа происходят по-настоящему драматиче-
ские изменения поверхности, и под влиянием таких стрессовых
условий некоторые материалы могут разрушаться.
Крекинг и риформинг. Многие небольшие органические молекулы,
используемые при получении различных химических продуктов, от
фармацевтических до взрывчатых веществ п от душистых вешеств
До полимеров, получаются из нефти. Химические реактивы полу-
чают не только при каталитическом окислении, гидрировании, хло-
рировании и полимеризации; небольшие углеводородные фрагмен-
ты могут сами отрываться от длинной цепочки углеводородов, сжа-
тых в земной коре. Каталитическая деструкция углеводородных
цепей до меньших, более летучих фрагментов называется крекин-
гом-, крекинг часто осуществляется па алюмосиликатных катализа-
торах. Катализаторы способствуют образованию карбониевых ио-
нов, которые являются неустойчивыми, распадаются и изомеризу-
28. Процессы на твердых поверхностях
521
ются в более высоко разветвленные цепи. Эти разветвленные моле-
кулы сгорают в двигателях внутреннего сгорания мягче и эффек-
тивнее, чем их лилейные изомеры, и являются основой высокоокта-
новых топлив.
Каталитический крекинг в значительной степени вытесняется
каталитическим риформингом, в котором используется двухфунк-
ционалъный катализатор, такой, как смесь платины и обработан-
ного оксида алюминия. Платина обладает металлической функ-
цией и осуществляет дегидрирование и гидрирование. Оксид алю-
миния обладает кислотной функцией и способствует образованию
карбоииевого иона. Протекающие на этом катализаторе процессы
ясно показывают те сложности, которые необходимо объяснить
для полного понимания хода промышленной реакции и подбора
лучшего катализатора.
Первой стадией является хемосорбция на платине длинной
цепочки углеводорода: в этом процессе отрывается сначала один,
затем второй атом водорода ц образуется олефин. Затем олефин
мигрирует к кислотному центру, принимает протон й прикрепля-
ется к поверхности в виде карбоииевого иона. Этот карбонпсвый
иоц может подвергаться различным реакциям. Он может разры-
ваться на два карбонисвых иона, изомеризоваться в более высоко
разветвленные структуры (разветвленные карбопиевыс иоцЫ более
стабильны, чем прямые цепные иоиы) или подвергаться различ-
ным реакциям замыкания кольца. Затем он теряет протон, выры-
вается с поверхности и мигрирует как олефин (возможно, через
газ) к металлической части катализатора, где гидрируется. В кон-
це концов получается богатый выбор более мелких молекул, ко-
торые можно вывести из реакции, разделить, например, ректифи-
кацией и затем использовать как сырье в других процессах.
Литература
Bond G. С,, Heterogenous catalysis: principles and applications, Clarendon
Press, Oxford, 1974.
Bond G. C., Catalysis by metals, Academic Press, New York, 1962.
Hayward D. O... Trupnell M. B. W., Chemisorption, Butterworths, London, 1964'.
Adamson A. IF., Physical chemistry of surfaces, Wiley-Interscience, New York-
1967.
Somoriai G.r Principles of surface chemistry, Prentice-Hall. Englewood Cliffs
N.J., 1972.
Ross S.r Oliver J. P., On physical adsorption, Interscience, New York, 1964.
BoudaH M., Burwell R. L., Mechanism of geterogeneous catalysis, in Techniques
of chemistry (Lewis E. S., ed.). Vol. VI(A), 963 Wiley-Interscience New York.
1974.
Задачи
28.1. Рассчитайте частоту молекулярных столкновений на 1 см* поверхности в
сосуде, содержащем: а) водород, б) пропан при 25 °C. когда давление равно;
I) 1 мм рт ст., 2) 10 мм рт. ст.. -
522
Часть 3. Изменение
28.2. Известно, что частота столкновений на единицу площади не особенно ин-
формативная величина, если нельзя пересчитать число столкновений на один по-
верхностный атом. Это может быть сделано при использовании некоторых ма-
териалов из гл. 22. Никель имеет гранецентрированную кубическую решетку с
элементарной ячейкой, сторона которой равна 352 пм. Чему равно число атомов
На I см: поверхности, образованной плоскостями; а) (100), б) (НО), в) (111)?
28,3. Вольфрам кристаллизуется в объемно-центрированной кубической форме с
элементарной ячейкой со стороной 316 пм. Рассчитайте число атомов на 1 см®
на тех же трех поверхностях, как и в последней задаче. В вольфрамовых нитях
Преобладают плоскости (ПО) и (100). Рассчитайте среднее число атомов на еди-
ницу площади.
28.4. Переведите частоты в задаче 28.1 а частоты столкновений на один атом
Поверхности, как в двух последних задачах.
28.5. Как долго атом водорода останется на поверхности при 298 К, если энер-
гия активации его десорбции равна: а) 15 кДж/моль, б) ]50 кДж/моль? Возьми-
те Тэ= 10_|3 с.
28.6. Как долго те же атомы будут оставаться на поверхности при 1000 К?
,28.7. Чему будут равны соответствующие времена в двух последних задачах,
если водород заменить на дейтерий?
28.8. Время, в течение которого атом кислорода остается адсорбированным на
поверхности вольфрама, равно 0,36 с при 2548 К и 3,49 с при 2362 К- Найдите
энергию активации десорбции. Чему равен предзкспоненциальный фактор для
этих плотно хемосорбированных атомов?
.28.9. Хемосорбция водорода на марганце является активационным процессом, но
только в очень слабой степени. Тщательные измерения показали, что она проте-
кает на 35% быстрее при 1000 К, чем при 600 К- Какова энергия активации
;хемосорбции?
'.28.10, Отложение атомов и ионов на поверхности зависит от их способности
удерживаться и поэтому от изменений энергии, которые при этом происходят.
В качестве иллюстрации рассмотрим двумерную квадратную решетку (предста-
вив ее себе поставленной на страницу,—Иерее.) с одновалентными положитель-
ными и отрицательными нонами, находящимися на расстоянии 200 пм друг от
друга, и рассмотрим катион, который подходит к верхней террасе такого рас-
положения атомов сверху страницы. Рассчитайте Прямым суммированием куло-
новское взаимодействие этого иона, когда он находятся в пустом узле решетки,
непосредственно над анионом. Теперь рассмотрите ступеньку подъема в той же
решетке, н пусть приближающийся ион войдет в угол, образованный этой сту-
пенью я террасой. Рассчитайте кулоновскую энергию для этого положен л я и оп-
ределите вероятную точку посадки отлагающегося катиона.
28.11. Изотерма Ленгмюра довольно хорошо описывает адсорбционное поведение
газов на различных поверхностях, и достоинство изотермы состоит в ее значи-
тельной простоте. Всего несколько задач в этой главе будет посвящено этой изо-
терме, и они дадут представление о тех расчетах, которые могут быть сделаны.
Сначала покажите, что если двухатомный газ адсорбируется в виде атомов, то
изотерма Ленгмюра будет иметь вид 0=УКр/(1+УЛ».
"28.12. Как проверить изотерму Ленгмюра? Один путь — это построить график
.зависимости р/0 от р, ко 0 ке всегда можег быть известна. Покажите, что
график зависимости р/Уа от р. где V, —объем адсорбированного газа (приве-
денный, так же как и ниже, к стандартным температуре н давлению), должен
дать прямую линию, из которой могут быть определены как Va — объем, соот-
ветствующий полному покрыгию, так в константа К. Покажите также, что лря
небольших покрытиях поверхности для проверки изотермы Ленгмюра необходи-
мо построить график 1п(0/р) от 0. Наклон прямой линии должен равняться
— 1. Каков должен быть наклон прямой на графике зависимости In 1'а/р от
Уа при малых покры-гнвх?
28.13. Ниже представлены данные по хемосорбции водорода на порошке меди
при 25 °C. Подтвердите, что они подчиняются изотерме Ленгмюра. Затем найди-
28. Процессы на твердых поверхностях
523
те значение К. для адсорбционного равновесия и адсорбционный объем, соответ-
ствующий подлому покрытию.
р, мм рт. ст. 0,19 0,97 1,90 4,05 7,50 11,95
Ка, см3 0042 0,163 0,221 0,321 0,411 0,471
28.14. Предположим, что озон адсорбируется на определенной поверхности и со-
ответствии с изотермой Ленгмюра. Как можно воспользоваться зависимостью
давления от степени заполнения для того, чтобы различить адсорбцию: а) без
диссоциации, б) с диссоциацией ла O-j-Oj, в) с диссоциацией на O-f-O-j-O?
28.15. Изотерму Фрейндлиха можно записать как Vs=cIp1^2, где С[ и с% — кон-
станты. Для адсорбции метана на 10 г сажи при 0°С были получены приведен-
ные ниже данные. Какой изотерме, Ленгмюра или Фрейндлиха, лучше соответст-
вуют эти данные?
р, мм рт. ст. 100 200 300 400
Va, см3 97,5 144 182 214
28.16. Окись углерода адсорбируется на слюде; данные при 90 К представляй.'
ниже. Определите, какая изотерма, Ленгмюра или Фрейндлиха, лучше описывает
эти данные. Каково значение К? Взяв^общую поверхность равной 6,2-1(0
рассчитайте площадь, занимаемую каждой адсорбированной молекулой.
р, мм рт. ст. 100 200 300 400 500 600
Уа, см3 0,130 0,150 0,162 0,166 0,175 0,180
28.17. Какой объем окиси углерода адсорбировался бы на слюде при 90 К и дав-
лении 1 атм?
28.18. Проектировщики нового промышленного завода для стадии фторирования
бутадиена предполагали использовать катализатор с кодовым названием CR-1.
При исследовании они сначала определили форму адсорбционной изотермы.
Объем адсорбированного бутадиена на 1 г CR-1 при 15°С зависел от давления
так, как это представлено ниже. Подходит ли изотерма Ленгмюра к этому интер-
валу давлений?
р, мм рт. ст. 100 200 300 400 500 600
Уа, см3 17,9 33,0 47,0 60,8 75,3 91,3
28.19. Подтвердите уравнения на стр. 516 для степени покрытия поверхности,
в присутствии двух адсорбированных газов.
28.20. Фтор адсорбируется на CR-2 в соответствии с изотермой Ленгмюра,, а бу-
тадиен адсорбируется в соответствии с изотермой Фрейндлиха с Cs=2. Выведите;
выражение для скорости реакции, предполагая, что фторирование зависит отч
столкновений атомов F с бутадиеном на поверхности катализатора. Пр ед по л о-’
жите, что адсорбпия двух частиц происходит на центрах различного тип№
28.21. Изотерма Б рунауэра — Эммета — Теллера (БЭТ) может быть записана (ес-
ли пренебречь небольшой поправкой) как
(р/К)р°/(р°—p)-i/v^4-/j/va,
где ра — давление паров жидкого адсорбата. Покажите, что эта изотерма nBpfc-
ходит в изотерму Ленгмюра, если р° значительно больше р. Проверьте примени-.
мость этой изотермы к данным задачи 28.18 (р°=1490 мм рт. ст. при l-5-°G).:
28.22. Газообразный азот адсорбируется на древесном угле в количестве
0,921 см$/г при давлении 4,8 атм и температуре 190 К, но при 25(1 К та же ве~
524-
г/асть 3. Изменение
личина адсорбции достигается только тогда, когда давление увеличивается до
32 at-м. Чему равна мольная энтальпия адсорбции азота на древесном угле?
28.23. В опыте по адсорбции кислорода на вольфраме было найдено, что один
и тот же объем кислорода десорбируется через 27 мин при 1856 К, 2 мин при
1978 К и 0,3 мин при 2070 К. Какова энергия активации десорбции? Сколько
прс.меии потребуется для десорбции того же количества при а} 298 К и
б) .3000 К?
28.24. Данные для десорбции кадмия с вольфрама Показаны на рис. 28.10. Най-
дите .•.качения энергии активации и пред экспоненциального множителя для де-
сорбции. Связан ли больший предэкслоненциальный множитель с более сильно
адсорбирующим центром или нет?
28.25. _ Аммиак ввели в сосуд под давлением 200 мм рт. ст. Было найдено, что
при 856 °C на вольфрамовом катализаторе изменение давления, равное 59 мм рт. ст.,
происходит за 500 с, а 112 мм рт. ст, — за 1000 с. Какой порядок имеет ката-
литическое разложение? Объясните результаты.
28.26. В некоторых каталитических реакциях образующиеся частицы могут адсор-
бироваться более сильно, чем реагирующий газ. Например, Это происходят в
случае каталитического разложения аммиака па платине при 1000 °C. При нс-
£ледоиаиян кинетики процесса такого типа в первую очередь докажите, что
скорость разложения аммиака должна следовать уравнению — rfp(NH3)/d?=
= fecP(NH3)/rfp(H2) в пределе очень сильной адсорбции водорода. Начинайте с то-
го, что покажите следующее: если газ J адсорбируется очень сильно и его дав-
ление равно p(J), то доля свободных центров приблизительно равна l/Xp(J).
^8.27, Решите уравнение скорости для каталитического разложения аммиака на
платине и покажите, что график зависимости F(t)= от G(t) =
= (.P^Pa)/t, где р —давление аммиака, должен датЬ прямую линию, откуда мож-
но определить /гс. Проверьте закон скорости на основе данных, приведенных ни-
же, и найдите Ас для реакпии.
2, С 0 30 60 100 160 200 250
р, мм рт. ст. 100 88 84 80 77 74 72
28.28. Теперь предположим, что замедляющий газ адсорбируется слабее н что
необходимо использовать полную форму изотермы Ленгмюра. Предположим, что
реагирующий газ не адсорбируется. Выведите закон скорости — dpldt— fccp/(l +
+-Кр’), где р—давление реагирующего газа и р' — давление продукта. Проин-
тегрируйте закон скорости для простого разложения А—*-В+С, где В адсорби-
руется" и ингибирует процесс, а С не адсорбируется.
28.29. Примером такого типа реакции служит разложение N2O на платине при
750 °C. Образующийся в ходе реакции кислород в этом случае сильно адсорби-
руется и ингибирует катализатор. Подтвердите, что данные, приведенные ниже,
соответствуют проинтегрированному закону скорости, и определите К. н Для
реакции.
/, с 0 315 750 1400 2250 3450 5150
р, мм рт. ст. 95 85 75 65 55 45 35
28.39. Хотя вандерваальсово притяжение между индивидуальными молекулами
изменяется пропорционально R~e, взаимодействие молекулы с соседним твердым
телом (скопление молекул) изменяется пропорционально R~3, где /? —вертикаль-
ное расстояние над поверхностью. Докажите справедливость этого утверждения.
28.31. Рассчитайте энергию притяжения между атомом аргона и поверхностью
твердого аргона и постройте график зависимости энергии от расстояния. Оттал-
кивающее взаимодействие — это парное взаимодействие, и поэтому используйте
значение потенциала Леннарда-Джонса, приведенное в табл, 23,4. Рассчитайте
равновесное расстояние атома аргона над поверхностью, (В более точных расче-
28. Процессы на твердых поверхностях 525
тах необходимо принять во внимание силы между тремя телами; здесь нх не
принимайте но внимание.)
28.32. Адсорбция растворенного вещества па твердом теле нз жидкостей часто
подчиняется изотерме Фрейндлиха. Проверьте применимость этой изотермы к
следующим данным для адсорбции уксусной кислоты на древесном угле при
25 °C и найдите значения параметров гу и с2.
[кислота], моль/дм3 0,05 0,10 0,50 1,0 1,5
wa, г 0,04 0,06 0,12 0,16 0,19
(и>а — масса адсорбированного вещества на единицу массы древесного угля).
Проверьте, не лучше ли адсорбция уксусной кислоты на древесном угле подчи-
няется изотерме Ленгмюра.
28.33. Теперь рассмотрим применение изотермы Гиббса [уравнение (8.6.5)] к ад-
сорбции газов. Покажите прямыми аргументы и, аналогичными использованным
в гл. 8 т. 1. Что объем адсорбированного газа на единицу площади твердого ве-
щества, Га/а, связан с давлением газа следующим образом: lza =— (ojRT) (dpfd In р),
где р. - химический потенциал адсорбированного газа.
28.34. Если известна зависимость химического потенциала гааа от степени запол-
нения поверхности, то изотерма Гиббса может бы,ь проинтегрирована, и полу-
чится такое же соотношение между Va в р, как в обычной изотерме адсорбции.
Например, предположим, что изменение химического потенциала газа при его
адсорбции выражается, как dp =—ez(RTj<3}dVa, где с3 — константа пропорцио-
нальности; покажите, что в этом случае изотерма Гиббса приводит к изотерме
Фрейндлиха.
28.35. В конечном счете, сделав полный круг, возвращаемся к изотерме Ленгмю-
ра. Найдите выражение для dp, которое при подстановке в изотерму Гиббса даст
изотерму Ленгмюра.
29 Динамическая электрохимия
Изучаемые вопросы
После тщательного изучения этой главы вы сможете:
1. Описать структуру электрического двойного слоя (стр. 528)
и определить внешний потенциал и внутренний потенциал
(стр. 530).
2. Связать скорость электродного процесса с разностью потен-
циалов на поверхности раздела и фактором симметрии [уравне-
ние (29.1.3)].
3. Определить перенапряжение электрода [уравнение (29.1.11)]
и плотность тока обмена (стр. 536).
4. Вывести уравнение Батлера — Фольмера для плотности тока
на электроде [уравнение (29.1.12)].
5. Вывести формы уравнения Батлера — Фольмера для низко-
го перенапряжения и высокого перенапряжения (стр. 537) и ис-
пользовать график Тафеля для получения плотности тока обмена
и параметра симметрии (стр. 537).
6. Описать экспериментальное измерение перенапряжения
(стр. 538).
7. Провести различие между поляризуемым и неполяризуемым
электродами (стр. 539).
8. Описать концентрационную поляризацию и диффузионный
слой Нернста (стр. 540) и вывести выражение для плотности пре-
дельного тока [уравнение (29.1.21)].
9. Объяснить основы полярографии и использование вращаю-
щегося дискового электрода (стр. 542).
10. Рассмотреть отложение металлов и выделение газов при
электролизе (стр. 545).
11. Объяснить, как ток влияет на потенциал элемента
(стр. 546), и связать получаемую мощность с протекающим током
[уравнение (29.2.7)].
12. Описать действие топливных элементов (стр. 551).
13. Дать перечень факторов, входящих в понятие накопления
мощности (стр. 553).
14. Определить коррозионную разность потенциалов и корро-
зионный ток (стр. 557).
15. Описать процессы, протекающие при коррозии (стр. 556),
п имеющиеся методы пассивации и защиты (стр. 559).
29. Динамическая электрохимия ЬДТ
Введение
В этой главе мы от свойств поверхности раздела газ — твердое
вещество перейдем к процессам, которые происходят на поверх-
ности металла, находящейся в контакте с раствором ионов. Одним
из наиболее важных примеров процессов такого типа служит пе-
репое заряда между двумя материалами; основная часть электро-
химии связана именно с такими процессами, а исследование эф-
фективных форм генерирования и накопления мощности основано
на знании электродных реакций и свойств поверхности раздела
металл — раствор.
Экономическая эффективность электрохимических реакций на
поверхностях почти не поддается исчислению. Большинство сов-
ременных методов генерирования электричества экономически
невыгодно, и развитие топливных элементов до промышленно
удобных форм могло бы революционизировать производство и по-
требление энергии. В настоящее время мы производим энергию
неэффективно и с ее помощью производим товары, которые раз-
рушаются коррозией. Каждая стадия этой расточительной после-
довательности могла бы быть улучшена, если бы мы больше зна-
ли о процессах, которые протекают на электродах.
Краеугольным камнем этой главы является рассмотрение про-
блемы, насколько быстро могут разряжаться ионы па электродах.
Мерой этой скорости служит плотность тока (сила тока на едини-
цу площади) па электроде, и главная цель нашего рассмотрения
будет состоять в раскрытии факторов, которые влияют ла нес.
При равновесии поток заряда от электрода равен потоку заряда
к электроду, и разность потенциалов на межфазной границе элек-
трод— раствор имеет равновесное значение. Когда ячейка рабо-
тает, имеется результирующий ток, и разность потенциалов отлича-
ется от равновесной на величину, называемую перенапряжением.
Мы выведем уравнение, связывающее силу тока с перенапряже-
нием, л одним из практических выходов будет знание э.д. с. эле-
мента, когда он действительно производит ток. Создание опреде-
ленного перенапряжения (соединение электродов с внешним ис-
точником э.д. с.) приводит к появлению плотности тока; в связи с
Этим мы рассмотрим электроосаждение металлов.
29.1. Процессы на электродах
В гл. И т. I объяснялось, как возникает разность потенциалов
на поверхности раздела, и было показано, как эту разность мож-
но рассмотреть в терминах термодинамики. В данном разделе мы
вернемся к обсуждению этой разности потенциалов, но рассмот-
рим системы, которые не находятся в равновесии, а производят
528
Часть 3, Изменение
Внешнял плоскость
Гельмгольца.
Рис. 29.1. Внешняя плоскость Гельмгольца для
положительно заряженного электрода.
ток, и системы, в которых электрохими-
ческие процессы, такие, как электро-
осаждение и разложение, находятся в
действии.
Двойной слой на поверхности раздела.
Разность электрических потенциалов воз-
пикает иа поверхности раздела между
электродом и окружающим раствором
(см. т. 1, разд, 11.3). Если нас интересу-
ют свойства равновесной системы, то пет
необходимости выяснять структуру разделения зарядов. Однако
сейчас нас интересует прохождение тока между электродом и рас-
твором, а описание этой динамической ситуации зависит от выбо-
ра модели микроскопической структуры поверхности раздела,
Разность потенциалов па поверхности раздела возникает из-за
разделения зарядов. Если электроны уходят из электрода и вос-
станавливают катион в растворе, то электрод приобретает поло-
жительный заряд, а раствор утрачивает свою электронейтраль-
ность и становится локально слегка отрицательно заряженным.
Распределение избыточного отрицательного заряда раствора огра-
ничивается областью около электрода, где имеется аттрактивное
взаимодействие с положительным зарядом последнего. Наиболее
простой моделью ближайшего к электроду пространства является
покрытие положительно заряженной поверхности электрода по-
крывалом Отрицательных зарядов соседней поверхности раствора.
Такая пара заряженных «покрывал» называется электрическим
двойным слоем. (Если электрод имеет отрицательный заряд, то
полярность двойною слоя обращается.)
Более детальную модель двойного слоя можно построить, ос-
новываясь на относительном расположении ионов в растворе.
Проще всего предположить, что сольватированные ионы распреде-
ляются по поверхности электрода и удерживаются па некотором
удалении от нее только благодаря наличию сфер гидратации
(рис. 29.1). Положение «покрывала» из ионного заряда можно
определить как плоскость, проходящую через сольватированные
ионы; опа называется внешней плоскостью Гельмгольца. В этой
модели Гельмгольца не учитывается разрушающее двойной слой
тепловое движение, которое ломает и диспергирует жесткую
29. Динамическая электрохимия
529
«стену» заряда. Подобный эффект рассматривается в модели
Дебая — Хюккеля для ионной атмосферы противоположного за-
ряда, окружающей ион (см. т. 1, разд. 11.2); разница состоит в
том, что центральный ион заменен па плоский заряженный элек-
трод. Распределение заряда можно найти так же, как для модели
Дебая — Хюккеля, только гораздо легче, так как поверхность
плоская. На самом деле подобная модель Гуи— Чепмена для
диффузного двойного слоя опередила модель Дебая — Хюккеля
на тринадцать лет и, вполне возможно, стимулировала ее возник-
новение.
Здесь мы не будем останавливаться па выводе Гуи —Чепмена*
но проиллюстрируем их результаты. На рис. 29.2 показано, как
локальные концентрации у электрода катионов и анионов различ-
ных зарядов отличаются от концентрации в массе раствора: вбли-
зи электрода группируются ионы противоположного заряда, и
легко также видеть, что ионы одинакового заряда отталкиваются.
Это, в частности, показывает, как ошибочно использовать коэф-
фициенты активности, которые служат макроскопическими харак-
теристиками раствора, при обсуждении термодинамических
свойств ионов, находящихся очень близко к электроду.
Ни модель Гельмгольца, пи модель Гуи — Чепмена не дают
точного представления о структуре двойного слоя. В первой
переоценивается жесткость ионного окружения, а в последней пе-
реоценивается подвижность ионов.
Модель Штерна объединяет
ионы, ближайшие к электроду,
кость Гельмгольца, но снаружи
Избыточный отрицательный заряд
| Избыточный положительный заряд
обе эти модели. В этой модели
выстраиваются в жесткую плос-
этой
плоскости ионы распределе-
ны, как в модели Гун —
Чепмена.
Наиболее важным
свойством двойного слоя
является изменение по*
тенциала вблизи элект-
рода. От этого потенциа-
ла зависит скорость, с
которой заряды могут пе-
редвигаться с одной сто-
роны поверхности разде-
ла на другую, и поэтому
34—242
Рис. 29.2. Модель двойного
слоя Гун — Чепмена.
530
Часть 3. Изменение \
Рве. 29.3. Изменение потенциала в эааислмостя от расстояния до электроде.
он контролирует скорость электрохимического процесса на элек-
троде.
Рассмотрим потенциал вблизи электрода, несущего положи-
тельный заряд (т, е. анода). Представим, что металл отделен от
раствора, но сохранилось распределение ионов, растворителя и
электронов. Теперь рассмотрим каждую половину общей системы
отдельно. На больших расстояниях от металла тестовый положи-
тельный заряд испытывает действие кулоновского потенциала,
который изменяется обратно пропорционально расстоянию
(рис. 29.3), При подходе заряда к электроду он достигает области,
где потенциал изменяется более медленно из-за того, что по по-
верхности распределен заряд, и примерно на расстоянии 10~5 см
от поверхности потенциал слабо зависит от расстояния. Потенциал
в этой области называется внешним потенциалом или вольта-по-
тенциалом ф- Когда положительный тестовый заряд проходит че-
рез оболочку электронов на поверхности металла, его потенциал
изменяется до тех пор. пока он не достигнет внутренних областей
металла. Этот дополнительный потенциал называется поверхност-
ным потенциалом х. Общий потенциал внутри поверхности назы-
вается внутренним потенциалом или гальвани-потенцналом и обо-
значается как Ф (М). Аналогичная последовательность изменений
потенциала происходит, когда положительный тестовый заряд пе-
реносится к поверхности раствора. Потенциал изменяется до соот-
ветствующего вольта-потенциала, когда заряд подходит к заряжен-
ной среде, а затем до соответствующего гальвани-потенциала, ког-
да он проходит через ориентированные дипольные молекулы рас-
творителя в его массу. Внутренний потенциал растворителя обо-
значается как <f>(S).
29. Динамическая электрохимия
531
Рис. 29.4. Профиль функции Гиббса для разряда нона.
Если вновь соединить раствор и электрод (но без изменения
структуры распределения заряда), то разность потенциалов меж-
ду точками в массе металла будет равна Д^-(М, S) = ^-(М.) —
— Ф (S); это разность гальвани-потенциалов. Эта разность потен-
циалов являлась основой при обсуждении электродных потенциа-
лов в гл. 11 и 12.
Скорость переноса заряда. Чтобы катион разрядился (восстано-
вился) ка электроде, он должен сбросить сольватирующие моле-
кулы и промигрировать через двойной слой в катод. Если не было
разности потенциалов на границах слоя, то изменение функции
Гиббса для такой последовательности событий напоминает изобра-
женное на рис. 29.4; скорость определяется функцией активации
Гиббса ДО* и выражается уравнением Эйринга [см. уравне-
ние (27.2.7)]:
М)—(й7/й)ехр !—Дб^(~* М) ..I (29.1.1)
Знак «+» у константы скорости указывает, что рассматривается
катион, а стрелка обозначает движение по направлению к элек-
троду М, как указано на рис. 29.4.
Когда разность потенциалов на границах двойного слоя изме-
няется от нуля до Д Ф (М, S), константа скорости изменяется. Если
катион с зарядом z+e движется из массы раствора, где потенциал
равен ^(S), к металлу, где потенциал равен ^(М), то общее из-
менение функции Гиббса равно г+еДФ(М, S). Поскольку для про-
цесса движения 1 моля катионов в заменяется на фарадей, то
Г = еЛ. Если ^(М) <Ф (S), этот вклад в Дб отрицателен: катионы
имеют тенденцию двигаться в область более отрицательного по-
тенциала. Однако проблема состоит не в общем изменении функ-
ции Гиббса от одной стороны фазы к другой, так как мы больше
не рассматриваем равновесные свойства. Мы хотим знать измене-
34*
532
Часть 3. Изменение
Потен циад--——
Внешняя
плоскость
Гельмгольца
Рис. 29.5. Электрический потенциал
вблизи двух заряженных плоских по-
верхностей-.
иие функции Гиббса в пере*
ходном состоянии, влияние ко-
торого на AG* изменяет ско-
рость.
В связи с этим необходима
модель двойного слоя, так как,
хотя относится к общей разности, нам нужно найти выраже-
ние для потенциала в некоторой промежуточной точке. Мы возь-
мем простую жесткую модель Гельмгольца и будем рассматривать
двойной слой как две жесткие четко выраженные заряженные пла-
стины. Нахождение потенциала в области, ограниченной двумя
заряженными пластинами, является стандартной задачей электро-
статики; известно, что потенциал от одной границы к другой па-
дает линейно (рис. 29.5). Поскольку разность потенциалов во всем
двойном слое является гальвани-потецциалом Д^ (М, S), остает- :
ся лишь проблема локализации положения переходного состояния.
Если G достигает максимальной величины па половине пути ;
между двумя пластинами, то разность потенциалов между поло- '
[ j
жением переходного состояния и раствором равна -^-Д^ (М, S)
(рис. 29.6,а). Поскольку переходное состояние может находиться
не точно на половине пути между двумя плоскостями, модель
приспосабливают к этому введением фактора симметрии, или ко-
эффициента переноса, а, обозначающего долю расстояния между
двумя плоскостями, где локализовано переходное состояние
(рис. 29.6,6) (эксперименты показывают, что часто а примерно
равен 0,5). Потенциал в области переходного состояния равен
поэтому разность потенциалов относительно раствора состав-
ляет ф*—^(S) =аД^(М, S), и, следовательно, функция Гиббса
для активации в случае движения к электроду определяется как
Дб*(—М)^Дб*(-* M)+z+FaAfi(M,S) (29.1.2)
(черта добавлена к символу, чтобы обозначить наличие некото-
рого электрического потенциала; вспомним ц и ц). Поэтому при
наличии разности потенциалов константа скорости будет равна
F (— М) = (kT/h) ехр {- ДЙ " (— М)//?Г} =
—F (-» М) ехр д/хД|£ (М, S) F/RT\.
(29.1.3)
29. Динамическая, электрохимия
533
а
ПЛОСКОСТЬ
Гельмгольца
плоскость
Гельмгольца
плоскость
Гельмгольца
плоскость
Гельмгольца
6
Рис. 29.6. Модификация профиля реакции, обусловленная изменением разности
потенциалов.
Если значение (М, S) отрицательно (как в данном случае), то
функция активации Гиббса уменьшается и катион быстрее подхо-
дит к электроду (&+>&+).
До сих пор мы не учитывали ту возможность, что катионы мо-
гут уходить с электрода в массу раствора. При отсутствии какой-
либо разности потенциалов константа скорости для этого процес-
са может быть выражена через функцию активации Гиббса для
движения от электродной поверхности:
А+(— М) = (АТ/й)ехр{ — AG^(*~ M)/J?T). (29.1.4)
Если имеется разность потенциалов, то уход катионов ингибиру-
ется (когда потенциал способствует отложению), и новая функ-
ция активации Гиббса отличается от старой на величину, опреде-
ляемую разностью потенциалов между переходным состоянием и
электродом ^(М). Используя ту же модель, что и выше, раз-
ность потенциалов можно выразить как —(1—а)Д^(М, S),
(рис. 29.6, б), и поэтому новая функция AG* равна
АЙ(*- M)=AG*(*~ М)—z+F(l—a)A^(M,S), (29.1.5)
а новая константа скорости
(— M) = (AT/ft)exp{-AG™(*- М)Ж} =
= &+(-*— М) ехр {а+ (1 —а) ДФ(М, 8)Г//?Т}- (29.1.6)
Если Д$ отрицательна, как в данном случае, то новая константа
скорости для ухода катиона будет меньше старой.
534
Часть 3. Изменение
Некоторое представление о величинах изменения констант
скоростей под влиянием изменений разности потенциалов ца элек-
тродах можно получить, сравнивая £+(—*-М) и £+(—*-М) для раз-
ности потенциалов —1 В. Из уравнения (29.1.3) находим (пр^
300 К, приняв а = -^-и 2,+=1)
k+lk¥ = ехр
— 1 В) X (9, Б49 104 Кл/моль)
[8,314 Дж/(К моль)] X(300 К)
-ехр(+19) « 3-10®.
Такое огромное изменение константы скорости для очень неболь-
шого (и легко достижимого) изменения условий показывает, на-
сколько важны электрохимические методы контроля за скоростя-
ми реакций. Частичное объяснение причины такого большого из-
менения можно получить, выразив разность потенциалов через
электрическое поле, действующее на ион в двойном слое. Раз-
ность потенциалов 1 В в двойном слое толщиной 10-9 м
(несколько молекулярных диаметров) соответствует электриче-
скому полю 109 В/м, или 10 МВ/см. Едва ли вызывает удивление,
что при таком изменении электрического поля происходят огром-
ные изменения констант скоростей. Электрохимия с внешней сто-
роны кажется «слабосильной», но более глубокое исследование
процессов, происходящих на молекулярном уровне, показывает,
насколько велик электрический стресс при переходеэлектронов от
ионов к электроду или при отрыве ионов от электрода.
С помощью константы скорости k+ можно найти ток на элек-
троде. Рассмотрим область снаружи около двойного слоя, где
среднее число катионов на единицу равно «‘(снаружи). Число
катионов, проходящих через единицу площади двойного слоя по
направлению к электроду в единицу времени, равно о4 (снаружи) X
Х&+(—>М). Поскольку каждый катион несет заряд сила
тока на единицу площади в единицу времени, т. е. плотность тока,
по направлению к электроду определяется как
/+ (-> M)=z+eo+ (снаружи) k+ (~* М).
(29.1.7)
(Размерность /— заряд на единицу площади в единицу времени;
поэтому, выражая заряд в кулонах, длину в метрах и время В-
секундах, получим плотность тока в Кл/(м2*с), или А/м2, по-
скольку 1А—1 Кд/с.) Если число катионов на единицу площади
с внутренней стороны двойного слоя равно о’1- (внутри), то име-!
ется ток в направлении от электрода, и его плотность равна
/+(-*- М)=^еА+(*~ М) о+(внутри)..
29. Динамическая электрохимия
535
Результирующий ток по направлению к электроду равен разности
этих двух плотностей тока:
/+ (-* М, результирующий) = z^e J о+ (снаружи) (-► М) —
—й+ (*- М) о+ (внутри)}. (29.1.8)
Позднее мы используем соотношение /+(—*-М, результирую-
щий) =—)+(-«-М., результирующий), где последний член является
результирующим потоком от электрода.
Зависимость результирующей плотности тока от разности по-
тенциалов на поверхности раздела получается подстановкой в это
уравнение уравнений (29.1.3) и (29.1.6):
У+(-*~М, результирующий) =
=г+е (снаружи) й+ (-*- М) ехр [ — г^аД^ (М, S)5//?T] —
— А+ (*-М) сг+ (внутри) ехр [г+ (1 —а) Д^ (М, S) F/RT]}. (29.1.9)
Внешний вид этого уравнения можно упростить в том случае, ког-
да электрод находится в равновесии с раствором. Разность потен-
циалов для поверхности раздела будет тогда равной ДФе(М, S),
т. е. величине, которая появлялась при термодинамическом рас-
смотрении электродных потенциалов (часть 1). Однако при равно-
весии результирующий ток равен нулю, так что плотность тока,
направленного от электрода, равна плотности тока, направленно-
го к нему:
j t (-► М, рез ультирующий) =/£(-»- М) —/£ (М) = 0, . . (29.1.10)
где
j е (— М)=z+eA+ (— М) о* (снаружи) ехр [ — z+czA^e (М, S) F]RT\,
(*- М)=z+efe+ (-•- М) о+ (внутри) ехр [z+ (1 —ос) Д^е (М, S) F/RT\.
Если символом т| обозначить отклонение действительной разности
потенциалов от равновесного значения:
т]=Д^(М,5)—Д^(М, S), (29.1.11)
то результирующий ток в неравновесных условиях будет связан с
плотностями тока при равновесии соотношением
/+ (-► М, результирующий) —£ (-»- М) ехр [ — zjx.t\F/RT] —
— it (*- М) ехр [z+ (1 — а) .
536
Часть 3. Изменение
Но при равновесии обе плотности тока равны [уравнение
(29.1.10)], и поэтому стрелки, обозначающие направление, можно
опустить. При этом мы получим
уравнение Батлера — Фольмера-
;+(-»-М, результирующий) =
—-ft {ехр [— z+aT]F/^T] — ехр [z+ (1 — a) /RT]}. (29.1.12>
Это важное уравнение будет центральным в последующем изло-
жении.
Перенапряжение. Величина плотности результирующего тока свя-
зана с двумя факторами. Во-первых, она связана с равновесной
плотностью тока je для транспорта через двойной слой в каждом
направлении (когда поверхность раздела электрода имеет рав-
новесную разность потенциалов). Эта величина называется плот-
ностью тока обмена и зависит от деталей строения двойного слоя,,
например от концентрации и подвижности ионов с каждой сторо-
ны: чем более подвижны ионы на поверхности раздела, тем боль-
ше плотность тока обмена. Некоторые экспериментальные значе-
ния приведены в табл. 29.1. Другим фактором является т] — от-
клонение разности потенциалов от равновесного значения. Это
настолько важная величина, что ей дано специальное наименова-
ние: она называется перенапряжением.
Таблица 29.1
Плотности тока обмена и факторы симметрии при 25 °C
Реакция Электрод i* . A/CU2 a
Hl i е- На Pt 790 10-' —
. Ni 6,3-10-e 0,58
Pb 5,01‘10-12 —
Hg 0,79-10'13 0,50
резгц_е- Fes+ Pt 2,5-10-3 0,58
Се4+-|- е- <. * Се34- Pt 39,8-10-® 0,75
Содержание уравнения Батлера — Фольмера можно выяснить,
исследуя две его предельные формы. Когда перенапряжение очень
мало (меньше 0,01 В), экспоненциал можно разложить в ряд,ис-
29. Динамическая электрохимия.
537
пользуя выражение х и пренебрегая высшими членами.
Тогда
у+ (ч- М, результирующий) яз /1 ([ l—(z+aT]F/7?T)[ —
-1 l+z+ (1 - a) t]F/RT)} ^~ilzj]F/RT. (29.1.13)
В этом случае плотность тока пропорциональна перенапряжению,
и поэтому при низких потенциалах поверхность раздела ведет
себя как омический проводник (сила тока пропорциональна раз-
ности потенциалов). Отметим, что если перенапряжение отрица-
тельно (так что разность потенциалов более отрицательна, чем
при равновесии), то поток иатиолов направлен к электроду, но,
если перенапряжение положительно, величина /+(—>-М, результи-
рующий) отрицательна, и поэтому величина /+('*~М, результи-
рующий) положительна, т. е. поток направлен от электрода.
Пример (вопрос 2). Плотность тока обмена для электрода Pl|Нг, H+(aq) равна
Д,79 мА,'см2. Какой ток течет через стандартный электрод общей площадью
5 см2, когда разность потенциалов на поверхности раздела 5 мВ, температура
25°C н активность протона равна единице?
Детой. Равновесный потенциал равен нулю, и поэтому перенапряжение равно
5 мВ. Подставляем его в уравнение (29.1.13), а затем переводим плотность тока
в силу тока. Используем /?T/.F=25,68 мВ.
Ответ. Из уравнения (29.1.13)
j* (—* М, результирующий) = —(0,79 иА/см®)Х(5мВ/25 68 мВ) = —0,154 мА/см2.
Для площади 5 си2 общий ток равен —(5 см2)Х(0,154 мА/см2), т. е. —0,77 мА.
Комментарий. Отрицательный знак показывает, что поток направлен от электро-
да. Отметим, что 2+arjf/R7'=0,097<gi, и поэтому применимо линейное прибли-
жение.
Когда перенапряжение велико (больше 0,1 В), уравнение Бат-
лера— Фольмера может выражаться разными предельными фор-
мами. Если большое перенапряжение положительно, первый экспо-
ненциал в уравнении (29.1.12) много меньше второго и им можно
пренебречь. Тогда
С (-* М, результирующий) яр — Д ехр [z+ (1 — ос)
так что
ln]j+(-*M, результирующий)) д-1пjt+z+(1 ~а) (29.1.14)
Из графика зависимости логарифма плотности тока от перенапря-
жения — графика Тафеля. — можно определить плотность тока
обмена по отрезку, отсекаемому от оси ординат, и параметр сим-
метрии а по наклону прямой. Когда перенапряжение велико, но
отрицательно, первый экспоненциал доминирует над вторым:
/+ (-* М, результирующий) « ехр (— z+ai]E/7?7’)i
538
Часть 3, Изменение
Рцс. 29.7. Экспериментальный график Тафеля.
и график Тафеля строится для соотношения
1п/+(-^М, результирующий) =1п/£—z^ar\/RT. (29.1.15)
Пример (вопрос 5). Приведенные ниже данные отражают величину тока, проте-
кающего через платиновый электрод (2 см2), находящийся в контакте с раство-
ром Fe2*, Fe3+ при 25°С. Найдите плотность тока обмена и коэффициент пере-
носа для электродного процесса.
tj, мВ 50 100 150 200 250
/, мА 8,8 25,0 58,0 131 298
Метод. Используем уравнение Тафеля [уравнение (29,1.14)] и строим график |п/
против т]. Реакцию разряда можно записать как Fe*+-)-e_—*-Fe*+, и поэтому
берем z+ = I. ... .
Ответ. Составляем следующую таблицу:
Т] мВ 50 100 150 200 250
|/+ |, мА/см2 4,4 12,5 29,0 65,6 149
In 1 j+ I (мА/см2) 1,50 2,53 3,37 4,18 5,00
В области высокого перенапряжения получается прямая, отсекающая от осн ор-
динат отрезок 0,916 н имеющая наклон 0,0163 (рис. 29.7). Из первого следует,
+• "Ь
что 1п(/е, мА/см2) =0,916 так что /е =2,5 мА/см’ Из наклона получаем
(l--a)f/7?7’=0,0163 мВ, гак что а=0,58.
Комментарий. Заметим, что при т]<;150 мВ график Тафеля нелинеен. В этой
области ат)//?7 = 75/26я;3, т. е, не очень сильно превышает единицу.
Для построения графика Тафеля необходим эксперименталь-
ный метод измерения перенапряжения. В типичной процедуре ис-
пользуются три электрода (рис. 29.8). Исследуемый электрод на-
зывается рабочим электродом, и ток, текущий к нему или от него,
29. Динамическая электрохимия
539
Рис. 29.8. Устройство для измерения перенапряжения.
берется от внешнего источника. Если его площадь А, а сила
тока 1, то плотность тока на поверхности раздела равна 1/А. Раз-
ность потенциалов на границе раздела непосредственно измерить
нельзя, однако можно измерить потенциал относительно электро-
да сравнения. Рабочий электрод и электрод сравнения с помощью
потенциометра уравновешиваются так, что через эту половину
цепи не течет никакой результирующий ток. Изменение количе-
ства тока, протекающего через рабочий электрод, вызывает изме-
нение потенциала рабочего электрода. Обычно новый потенциал
относительно электрода сравнения находят с помощью потенцио-
метра со шкалой.
График Тафеля приведен на рис. 29.7: в области высоких пере-
напряжений наблюдается очень хорошая линейность, и из накло-
на и отрезка, отсекаемого от оси ординат при экстраполяции,
можно найти фактор симметрии и ток обмена электрода; некото-
рые значения приведены в табл. 29.1. Оба параметра дают пред-
ставление о строении двойного слоя (например, из фактора сим-
метрии можно найти положение максимума функции Гиббса) и о
подвижности ионов на поверхности раздела. Найдено, что плотно-
сти тока обмена изменяются в широком интервале, от
1,3 • 10-ls А/ома для электрода Н2(газ) |Hg до 1,6 - 10—3 А/см2
дли электрода Fe2+, Fe3+|Pd.
Электроды, которые имеют высокие плотности тока обмена,
называются неполяризуемыми; электроды, имеющие низкие плот-
540
'Часть 3. Изменение
ности, называются поляризуемыми. У первых двойной слон на-
столько подвижен, что ионы могут быстро приспособиться к изме-
нению тока, так что устанавливается почти постоянная межфаз-
ная разность потенциалов, В этом состоит одна из причин, почему
каломельный и илатиповодородный электроды так широко ис-
пользуются в равновесной электрохимии.
Другие аспекты отклонений от равновесия. Одним из предположен
ний при выводе уравнения Батлера — Фольмсра является постои
янство концентрации ионов о'1'(внутри) и <т+(снаружи), когдИ
электрод не находится в равновесии. При высоких плотностях токЯ
это предположение несправедливо, поскольку диффузия ионов
электроду из массы раствора не может достаточно быстро попол!
нять ионы на внешней плоскости Гельмгольца, и, следовательно^
число ионов, пересекающих двойной слой, не так быстро увели-
чивается. Это явление называется концентрационной поляриза^
цией, а разность потенциалов, обусловленная градиентом кон-
центрации, носит название концентрационного перенапряжения.
Концентрационное перенапряжение можно оценить следующим
образом. При равновесии, когда нет результирующей плотности
тока, разность потенциалов двойного слоя связана с активностью
ионов в растворе уравнением Нернста [см.т. I, уравнение (12.1.2)]:
Д^е (М, S) =. (М, S) -г (RT/z^F) In амт.
Если система пе находится в равновесии, концентрация с наруж-
ной стороны двойного слоя изменяется; здесь активность изменя-
ется до пм+> и поэтому потенциал изменяется до
Дф (М, S) — ДФ° (М, S) + {RTiz+F} In йм+.
Концентрационное перенапряжение цс, возникающее при таком
изменении активности,
if = ДФ (М, S) — ДФе (М, S) = {RT/Z+F) In (ам7«м0•
Мы уже видели, что при использовании активностей возникают не-
которые трудности (в данном случае имеются градиенты концент-
рации и растворы часто являются концентрированными), и поэто-
му мы просто проигнорируем коэффициенты активности и будем
работать с концентрациями См+. Тогда концентрационное перена-
пряжение будет равно
4c = (RT/z.F] In (с^г/с^). (29.1.16)
Следующая задача состоит в Оценке концентрации точно с на-
ружной стороны двойного слоя. Отклонение сг от концентрации
в массе зависит от величины ионного коэффициента диффузии;
если ионы подвижны и D велик, то локальная концентрация может
быть почти такой же, как в массе, несмотря на то что проходит
большой ток. . . . . , д
29. Динамическая электрохимия
54»
Простейший подход к расчету с' основан на диффузионном за-
коне Фика (стр. 387). Он связывает поток частиц с градиентом
концентрации:
J(— М) •= — D (дДДдх). (29.1.17)
Если с выражается в молях на единицу объема, тощ'’— концентра-
ция в числе частиц на единицу объема, лл=сЛ. Для простоты пред-
положим, что концентрация катионов имеет такую же величину,,
как в массе раствора, вплоть до расстояния от внешней плоско-
сти Гельмгольца, а затем линейно уменьшается до с' в самой
плоскости. Такой диффузионный слой Нернста показан на
рис. 29.9. Следовательно, градиент концентрации равен
(дЛПдх) “(др1'—.дД/Х = (см+—см+) (£/Х),
и поэтому поток, пересекающий единицу площади в единицу вре-
мени, определяется как
J(—► М) ~ — (7лО/Х) (с,м+ —См+)-
Этот поток катионов соответствует плотности тока
Г (_>M)=2+eJ (-^М),
(29.1-18)
и поэтому объединение последних двух уравнений приведет к вы-
ражению с' через плотность тока у двойного слоя:
см+=См* — (X/2+FD) f (^ М).
(29.1.19>
Подстановка в уравнение Нернста для разности потенциалов дает
выражение для концентрационного перенапряжения при наличии
плотности тока /+(—*~М):
vf={RT/F) In {I “(;+X/z+cM+F7)jj.
раствора онныи
слой Нернста
Внешняя
плоскость
Гельмгольца
(29.1.20)
Максимальная скорость,
диффузии через слой
Нернста получается в том
случае, когда градиент
наиболее крут, т. е. ког-
да с'— 0. Такое значение
концентрации будет &
том случае, когда каж-
дый ион, который диф-
фундирует через слой,
Рис. 29.9. Диффузионный слой.
Нернста.
542
Часть 3. Изменение
проходит через активационный барьер (через двойной слой) и
подходит к электроду. Более быстрого потока, чем в этом случае,
быть не может, и поэтому предельная плотность тока дастся урав-
нением (29.1.18) при с'=0.
щ (^ М) = (z+FD/X)Cm+- (29.1.21)
Пример (вопрос 8). Оцените величину плотности предельного тока на электроде,
в котором ионы меди имеют концентрацию 0,01 моль/дм3.
.Метод. Используем уравнение (29,1.21). Возьмем к=0,5 мм (типичная величина).
Вычислим D из ионной электропроводности Х+ = 108 Ом“'-смг-моль-1 н уравне-
ния (25.2.5).
Ответ. Из уравнения (25.2.5) Х+= (г!/72/!??") £)+, так что подстановка этого вы-
ражения и уравнение (29.1.21) приводит к
it (_>. М) = (z+FD+A)cM+ = (RT^/ZjFX) см(-.
Следовательно,
Jl (-► М) = (ЮЗ- Ю~4 Ом-1-м=.моль-1)Х(25,7 мВ)Х
Х(0,01 моль/дм®)/(2-0,5 мм) =
= 2,78 А/ма, или 0,278 мА/см4.
Комментарий. Это означает, что в данном растворе ток к электроду в I смг пе
может превысить 0,28 мА. Если раствор размешивается или если поверхность
электрода движется (как в полярографии), то расчет предельного тока сильно
усложняется.
Величина плотности предельного тока зависит от заряда и
коэффициента диффузии ионов, и определение плотности предель-
ного тока служит основой аналитического метода полярографии,
применяемого в случаях, когда нужно установить состав раство-
ра. Метод заключается в повышении потенциала между двумя
электродами и определении тока. При увеличении разности потен-
циалов сила тока возрастает до тех пор, пока не достигнет вели-
чины предельного тока, характеризующего ион в растворе. Если
присутствует несколько ионов, то сила тока может возрастать,
образуя последовательность предельных значений, и ионы иден-
тифицируются по их потенциалам полуволны (рис. 29.10 и
табл. 29.2). Величину предельного тока можно использовать для
определения концентрации ионов. Одна из проблем этого метода-
загрязнение поверхности электрода. Его можно избежать, ис-
пользуя капающий ртутный электрод (рис. 29.11), где электрод
постоянно регенерируется в виде капель, образующихся па конце
капилляра, которые затем падают. Осцилляции на полярографи-
ческой кривой объясняются ростом капли, т. е. ростом поверхно-
сти электрода.
Теоретический анализ капающего ртутного электрода далеко
не прост, поскольку он меняет размеры и форму. Тем нс менее
можно сделать некоторые достаточно приемлемые приближения и
1.0
+ 0,2 0,0 -0,5
Е. В
Рве, 29.10. Полярографическое указание на Присутспие ново* Си1* д TI*.
Рнс. 29.11. Капающий ртутный электрод.
544
Часть 3. Изменение
Таблица 29.2
Потенциалы полуволны для ионов в растворе 0,1 моль/кг КС1 при 25 °C
+1/(, В (относительно каломельного электрода)
Cd-+ —0,599 Fe3+ 0
Co'-ii- -1,4 Fe'-+ —1,3
С++ -0,91 ТГ —0,46
Cu2+- +0,04 Zn-+ —0,995
получить уравнение Ильковича для /ь. Из него следует, что
Ji, со/^з; такая своеобразная зависимость от времени отражает
конкуренцию между ростом капли и предельным током. Пробле-
ма сложной гидродинамики потока к растущему электроду нс воз-
никает и случае вращающегося дискового электрода (рис. 29.12).
Электрод представляет собой небольшой плоский диск, установ-
ленный на вращающейся оси. Вращение его поверхности создает
устойчивый гидродинамический поток, который заставляет раствор
циркулировать по поверхности. Картину потока довольно легко
вычислить, и предельный ток связать с угловой скоростью диска.
Для изучения кинетики электродных процессов можно ис-
пользовать различные
электрода. Одна из них
электрод
модификации вращающегося дискового
состоит в наложении импульса потенциа-
ла и последующем наблюдении роста си-
лы тока до предельного значения. Дру-
гой модификацией является дисковый
электрод с кольцом, где центральный
вращающийся диск окружен электродом
в форме узкого кольца. При вращении
диска раствор по линиям' потока перено-
сится к диску, а затем отбрасывается
к кольцу. Анализ потенциалов двух
электродов и знание времени, необходи-
мого для того, чтобы продукты, образо-
вавшиеся на диске, перешли к кольцу и
там были зафиксированы, дают очень
подробную информацию о скоростях пе-
реноса электрона и других процессах в
растворе.
Рис. 29 12. Вращающийся дисковый электрод.
29. Динамическая электрохимия
Мб
29.2. Электрохимические процессы
Данный выше материал можно резюмировать следующим об-
разом. Центральной концепцией является перенапряжение — от-
клонение разности потенциалов на электроде от равновесного зна-
чения. Перенапряжение связано с током, текущим через электрод,
и зависит от кинетики электродного процесса.
Для того чтобы вызвать протекание тока через ячейку, на-
пример, когда нужно осадить одни металл на другом, необходимо
создать разность потенциалов, превышающую равновесную вели-
чину (чтобы получить результирующий ток), а количество проте-
кающего тока и, следовательно, количество осадка или выделения
зависит от перенапряжения. Количественно зависимость тока от
перенапряжения дастся уравнением Батлера — Фольмера [см.
уравпепие (29.1.12)]. Если ток обмена /е мал, как для выде-
ления водорода па ртутном электроде, то, чтобы получить значи-
тельное выделение, необходимо большое перенапряжение; если jt
велик нз-.за того, что барьер прохождения через двойной слой
мал, то заметное выделение или отложение может происходить
даже при небольших перенапряжениях. Это происходит в случае
водорода па платине.
Ток протекает также в том случае, когда элемент использу-
ется для генерирования энергия. Тогда разность потенциалов эле-
мента отличается от величины, которая наблюдается, когда эле-
мент работает обратимо, будучи сбалансированным против неко-
торого внешнего потенциала. Различие между разностью потен-
циалов, наблюдаемой, когда протекает определенный ток, и равно-
весной разностью потенциалов, — когда ток не протекает (рав-
новесная э.д, с_), является также перенапряжением элемента. Ойо
возникает при смещении ионов близко к обоим электродам эле-
мента, исчезновении локальной концентрации ионов в соседстве с
двойными слоями и омическом падении потенциала от одного
электрода к другому в результате тока, проходящего через ячейку.
Как рассматривать выделение и отложение на электродах. Сжо-
рость выделения газа или отложения металла можно опенпть из
уравнения Батлера — Фольмера и таблиц токов обмена. Ток обме-
на сильно зависит от природы электрода, и, поскольку оно меня-
ется во время отложения одного металла на другом, мы рассмот-
рим лишь простейшие случаи отложения металла на том же са-
мом металле и выделения газа.
Беглый взгляд на табл, 29.1 показывает, каков интервал токов
обмена для электрода металл | водород. Наиболее слабые токи об-
мена наблюдаются для свинка и ртути, и равновесный ток в
10-2 А/смй соответствует обмену монослоя атомов (около
1016 см-2) за 100 —1000 с. В противоположность этому ток обме-
35—242
54§
Часть 3. Изменение
на на платине составляет около 10-3 А/см2, и поэтому монослой
образуется и разрушается за 10-11 с. Для данного перенапряже-
ния результирующие токи протонов по направлению к этим элек-
тродам отличаются в 109 раз.
Пример (вопрос 10). Стандартный электродный потенциал для электрода СсР+,
Cd равен —0,403 В. Плотность тока обмена электрода Н+, IlafPt равна
0,79 мА/см2. Можно ли отложить кадмий на платине из растворов, содержащих
единичные активности Cd2+ и Н+?
Метод. Металл будет отлагаться, если потенциал более отрицателен, чем —0,403 В;
водород выделяется, когда разность потенциалов раппа 0,0 В. Разность потен-
циалов упадет до —0,403 В, только когда плотность тока превысит juexp(zax\FIRT)
др и ц = —0,403 В. Возьмем а=1/2, г+(Н')“1. Находим п.тогйосгь тока, Если
для возникновения потенциала —0,403 В необходима лишь небольшая плотность
тока, кадмий будет отлагаться.
Ответ. /+=(0,79 мА/ем2)ехр(0,403 В/2Х0,257 В) =2,01 А/ем2.
Разность потенциалов падает до —0,403 В, только когда плотность тока дости-
гает 2 А/ем2; при меньших плотностях тока преобладает выделение водорода.
Комментарий. При отложении па никеле плотность тока обмена равна 3,2 А/ем2,
и поэтому для падения потенциала до значения, при котором кадмий будет от-
лагаться на никеле, достаточно плотности тока всего 8,1 мА/ем2.
Ток обмена зависит также от того, какая грань кристалла слу-
жит электродом. Для отложения меди на медном электродеграпь
(100) имеет ток обмена 1-Ю-3 А/см2, и поэтому при некотором
перенапряжении она растет в 2,5 раза быстрее грани (111),для
которой плотность тока обмена 4 -10 ’4 А/см2. Иначе результирую-
щей плотности тока в 10 мА/см2 для грани (100) можно достиг-
нуть при перенапряжении —125 мВ, но для грани (111) требу-
ется перенапряжение —185 мВ; для создания той же результи-
рующей плотности тока на грани (110) необходимо только
—85 мВ.
Как сила тока влияет на потенциал элемента. Можно ожидать,
что э. д. с. элемента уменьшится при пропускании тока, потому
что в Этом случае элемент не будет действовать обратимо и смо-
жет производить работу меньше максимальной. Это и было обна-
ружено, и связь между перенапряжением и током позволяет оце-
нить этот эффект количественно.
Рассмотрим элемент М|М'- (aq) ]] M/4-(aq) | М'. Не будем учи-
тывать все усложнения, возникающие из-за наличия жидкостных
соединений. Если ток не идет, э, д, с. такого элемента определяется
разностью электродных потенциалов £(М+|М) и £(№'+ |М'). Если
ток течет, разность потенциалов между электродами можно запи-
сать через разности потенциалов на поверхностях раздела п раз-
ность потенциалов в самом растворе:
ДФ (М', М) г= Ф(М')— Ф (М) —
= Ф (М') —Ф (S') -|-Ф (S') —Ф (S) -|-Ф (S) — Ф (М) =
=ДФ (М', S')••]-ДФ (S', S) — ДФ (М, S),
29. Динамическая электрохимия
547
-Ms
d(S» = +//fs
Е= Ee+1\'~1\~IR$
A = fe +
Рнс 29.13 Влияние сjnpOTMBiriCHiiя
элемента па его э. д. е.
где S— раствор около М, a S'— раствор около М', Теперь запи-
шем, что рабочая э. д. с. элемента АФ (М', М) — Е.
Рабочую э.д.с. можно выразить через равновесную э.д.с. Ее,
введя равновесные разности потенциалов и перенапряжения
ДФ(М,Й)=ДФе (M,S) + n>
А Ф (М', S') —ДФе (М', S') 4-т]'.
Тогда
Е Ее-]-т]' + т]+ДФ (S', S),
(29.2,1)
и Е может быть найдена путем нахождения перенапряжений, со-
ответствующих силе проходящего тока Z. Разность потенциалов в
растворе имеет величину Z/?s, где /?s — сопротивление раствора:
знак всегда будет таким, чтобы э.д.с. элемента уменьшалась,
т. е. Е будет менее положительной, если она положительна, и ме-
нее отрицательной, если она отрицательна (рис. 29.13).
Уравнение Батлера — Фольмера дает связь между перенапря-
жением и плотностью тока, если нет концентрационного перена-
пряжения, что мы и предположим для данного случая. Мы также
предположим, что протекает такой большой ток, что можно ис-
пользовать форму уравнения для высокого перенапряжения. Это
не обязательное предположение, по оно сильно упрощает алгебру.
Мы также примем, что а —'/г (где а —величина фактора сим-
метрии) .
35»
548
Часть 3. Изменение
Рис. 29.14. Ток, индуцируемый элементом.
В элементе, показанном на рис.
29.14, имеется спонтанный ионный ток
по направлению к правому электроду
М', поскольку £е>0 и М'4 восстанав-
ливается на М. Поэтому перенапряже-
ние т/ отрицательно. При высоком от-
рицательном перенапряжении резуль-
тирующая плотность тока по направ-
лению к электроду М' дается урав-
нением (29.1.12) как
/,+ (->М', результирующий) /^ехр( — г'^]' F/2RT),
и поэтому для данной плотности тока перенапряжение равно
^^-(2RTlz'F)in(j'+jj's+). (29.2.2)
Поток катионов всегда направлен от другого Электрода М,и
поэтому его потенциал должен быть более положительным, чем
при равновесии, что соответствует положительному перенапряже-
нию, Из уравнения (29,1.12).
/+ (*—М, результирующий)—/+(->-М, результирующий) яв
Sfjtfixp(zj\F/2RT),
так что
il^(2W+F)In(f//t). (29.2.3)
Если площади электродов равны А и А' и сила общего тока
равна I (одинакова для обоих электродов), то можно записать:
/'+(—>-М') — IjA', j* ( -t-М) =1/А. Следовательно, полное выраже-
ние для рабочей э. д. с. будет иметь вид
Е^Ее — (2RT/z'rF) In (J/Л'/е4) - (2RT/z+F) In {f/Ajt) -fRs. (29.2.4)
Этому выражению можно придать более компактную форму,
сделав некоторые допущения. Предположим, что ионы М-4 н М/4
имеют одинаковый заряд (z+ = z'l_) н что площади двух электро-
дов одинаковы. Тогда
E^-Ee-(47?T/z+O14 КМ VW51 (29.2,5)
Пример (вопрос 11). Рассчитайте э. д. с. работающего элемента Zn|ZnI+ (aq)ll
||Cu2+(aq) |Сп. когда через электрод, каждый площадью 2 см2, протекает ток
39. Динамическая электрохимия
549
силой 0,5 А. Активности ионов a(Zn2+) =0,2, a(Cu2+) = 0,l и внутреннее сопро-
тивление ячейки 0,05 Ом.
Метод. Используем уравнение (29.2.5). Возьмем jt(Zn3"'") as jefCti2+) = l мА/см?.
Найдем Ее из уравнения (11.3.6).
Ответ. Из уравнения (11.3.6)
Et (Си2+ Си) = (Cu2+, Си) + (RT/2F) In а (Си3 +) =
1
<= 0,337 В (0,0257 В) In 0,1 = 0.307В.
Ee(Zn2+. Zn)= —0,763 В-у- (0,0257 В) In 0,2 = —0,784 В.
Поэтому
Ее = Ее (Си2+, Си) — Ей (ZrrC Zn) = 1,091 В.
Тогда из уравнения (29.2.5) при (RT/F) =0,0257 В
Е= 1,091 В —(0,0257 В) Щ ((0,5 А/2 см2)/[(1 мА/ем2) (I мА/ем2)]1/2} —
— (0,05 0м)Х(0,5А)= 1,091 В — 0,142 В — 0,025В-= 0,924 В.
Комментарий. Скоро мы увидим, как рассчитывать мощность на выходе элемен-
та. Это главным образом IE, и поэтому в данном случае она равна 462 мВт.
Последнее выражение показывает, что, когда протекает ток,
положительная э. д. с. элемента уменьшается. Это можно попять,
если учесть роль тока положительных попов в транспорте элек-
тричества через элемент. Когда ток таков, что ноцы движутся
вправо, электроны во внешней цепи движутся вправо (рис.29.14).
Такое течение тока нельзя скомпенсировать полностью потоком
катионов через двойной слой, и поэтом}’ первоначально положи-
тельный потенциал становится менее положительным. Аналогич-
но для другого электрода отбор электронов вынуждает катионы
уходить через двойной слой в раствор: это ингибируется барье-
ром, и поэтому потенциал становится менее отрицательным. Сле-
довательно, когда проходит ток, общая разность потенциалов эле-
мента уменьшается.
До сих пор мы пренебрегали концентрационным перенапряже-
нием, однако оно также уменьшает э. д. с. элемента. Для рассмот-
рения этого эффекта мы используем простую модель диффузион-
ного слоя Нернста, а для расчета цс на каждом электроде можно
использовать уравнение (29.1.20), Сразу же можно записать из-
менение э.д. с., возникающее из-за этого эффекта:
Е =Ee-v(RT/z+F) ТП ((1 -I/Ajfi (1 -I/Ajt)}, (29.2.6)
где jl и jC — предельные плотности тока па обоих электродах
[уравнение (29.1.21)]. Этот результат можно комбинировать с
36—242
S50
Часть 3. Изменение
Рис. 29.15. Зависимость рабочей э. д. с. элемента и его мощности от проходящего
тока.
уравнением (29.2.5) и получить полное (но приближенное) выра-
жение для э.д.с. элемента, через который протекает ток Г.
E = E'~I£ls-(2XT/z+F) In /(/), (29.2.7)
где
/а/д«/+/' +
f (h =--------— le le____.___
Это выражение связано co многими параметрами элемента; при-
мер дан на рис. 29.15: отметим очень резкое падение рабочей
э.д.с., когда ток велик и близок к предельной плотности тока од-
ного из электродов.
29.3. Генерирование и накопление мощности
Разность потенциалов, или э.д.с., не является единственным
критерием полезности электрохимического источника: нас долж-
на также интересовать его мощность. Мощность, развиваемая эле-
ментом, равна /£', где / — сила тока и Е — э. д. с. при этой силе
тока. Если / выразить в амперах, а Е — в вольтах, то мощность Р
будет измеряться в ваттах (Вт). Выражение Р=Е1 в краткой
форме выражает необходимость изучения элементов вне равнове-
сия: при равновесии они не вырабатывают ток, и, хотя э.д.с. мо-
жет быть наивысшей, величина / равна нулю, и поэтому никакой
мощности не производится.
29. Динамическая электрохимия
551
Выражение для мощности, вырабатываемой элементом, очень
легко получить, умножая уравнение (29.2.7) на /:
Р=- (2RTI/z+F) In/ (/), (29,3.1)
Первый член этого уравнения представляет собой мощность, ко-
торая была бы получена, если бы элемент вырабатывал свою рав-
новесную э.д. с., даже если он вырабатывает ток; второй член—
это мощность, бесполезно рассеиваемая в виде тепла в электролите
в результате его сопротивления; а третий член — это поправка,
учитывающая понижение разности электродных потенциалов в
результате протекания тока. Форма зависимости мощности от
силы тока иллюстрируется на рис. 29.15, где приведены кривые
э.д.с. — ток. Отметим, что, по крайней мере в этом случае,мак-
симальная мощность получается как раз перед тем, как концент-
рационная поляризация быстро подавляет э.д.с. Информация та-
кого рода существенна, если необходимо найти оптимальные усло-
вия эксплуатации электрохимического устройства и улучшить его
характеристики.
Генерирование мощности в топливных элементах. Развитие совре-
менной электрохимии и техники привело к созданию компактного
источника мощности, в котором для получения электричества ис-
пользуется топливо без применения тепловых установок, таких,
как котлы, турбины и генераторы. Топливные элементы могут
быть гораздо более эффективными, чем обычные тепловые источ-
ники; они уже применялись в космических аппаратах и могут стать
основой безвыхлопных двигателей для транспорта.
Топливный элемент действует точно так же, как обычный
электрохимический элемент, за исключением того, что материалы,
которые нужно окислить л восстановить на электродах, хранятся
снаружи элемента и не являются конструктивной его частью.
Основным и важным примером топливного элемента является во-
дородно-кислородный элемент, приведенный на рис. 29.16. Если
электролитом служит кислота, то на катоде (приемнике электро-
нов из внешней цепи) идет реакция
Ог + 411+ + 4е- -> 2НаО,
а на аноде (источнике электронов для внешней цепи)—реакция
2Не---> 4Н+4-4е-.
Равновесная э.д.с. элемента равна (1,229 В)—(О В) = 1,229 В,
но, поскольку нас интересует мощность, мы должны исследовать
характеристики элемента ток — э.д.с.
Пример (вопрос 12), Рассчитайте э. д, с, пропаново-кислородного топливного
элемента, работающего обратимо.
Метод. Происходит реакция С3Н8+5О2—>3СОН-4 Н2О, и стандартная функция
Гиббса для реакции равна —2108 кДж/моль. Как написано, реакция включает
36’
Ъ52
Часть 3. Изменение
перенос 20 электронов. Это число можно получить, если реакцию выразить как
комбинацию 5 О2+20 Н+4-20 е_—*-ЮН2Ои С3Н9+6 Н2О—*-20 е~+3 СО2+20 Н+.
Выражаем величину AGm так, чтобы опа соответствовала переносу 1 моля элек-
тронов, а затем используем уравнение (12.3.1).
Ответ. Для переноса 1 моля электронов AG°m = (—2108 кДж/моль)/20=
= —105,1 кДж/моль. Тогда из уравнения (12.3.1):
£° = — (-1,054-105 Дж/моль)/(9,649-104Кл/моль) = 1,092 В.
Комментарий. Энгальпия сгорания при 25°С равна 2220 кДж/моль, и поэтому
в элементе, работающем с идеальной эффективностью, в реакции выделяется не-
которое количество теплоты (это количество составляет 2220 кДж/моль—
—2108 кДж/моль = 112 кДж/моль).
Одним лз преимуществ системы водород —- кислород является
большой ток обмена водородной реакции. К сожалению, для кис-
лородной реакции ток обмена равен всего лишь 10-10 А/см2, и
это предел тока, который можно получить от элемента. Один из
путей обхода этой трудности состоит в использовании каталити-
ческой поверхности (чтобы увеличить плотность тока обмена) и
очень большой площади поверхности электрода, чтобы получить
большой ток даже при небольшой плотности тока. В элементе,
изображенном на рис. 29.16, электроды изготовлены из титана,
покрытого платиной, а элек-
тролитом является катионо-
обменная смола.
Были разработаны дру-
гие типы топливных элемен-
тов, в которых используют-
ся различное топливо и тех-
нические приспособления
для увеличения плотности
тока обмена (табл. 29.3).
Последнего можно достнг-
нуть, выбирая, например,
ловерхность электрода с по-
лезными каталитическими
свойствами или нагревая
весь элемент для улучше-
ния диффузионных характе-
рце. 29.16. Водородпо-кис.тород-
ный топливный элемент. .
29. Динамическая электрохимия
553
Таблица 29.3
Термодинамические характеристики
некоторых возможных топливных элементов
Реакция АС^. кДж/моль АНт. кДж/м иль л, в ЕЭ
на + -у О2 > Н2О —237,2 —285,9 1,229 0.83
СН4-)-2О2 СО,- 2НаО —818,0 —890,4 1,060 0,92
3 СН3ОН ог * СО2 4-2НаО —706,9 —764,0 1,222 0,93
С+О2 ► соа —137,3 —ио.о 0.712 1,24
а ° ° гл— мера относительной эффективности электрических и ' тнческах ресурсов. гермтесцих энерге-
ристик и скорости пересечения двойных слоев. Хотя обычно окис-
ляющим топливом является кислород (потому что ojr легкодосту-
пен), топливо для анодной реакции можно выбрать из широкого
разнообразия материалов. Особенно важной группой анодных топ-
лив являются углеводороды. В топливных элементах углеводо-
род—-воздух обычно используют платиновые электроды, а в каче-
стве электролита — фосфорную кислоту; элементы работают при
температурах около 100 СС. В настоящее время доступны модели,
которые могут производить около 0,1 Вт на 1 см2 поверхности
электрода.
Накопление мощности. Электричество, генерируемое в одной
установке, можно накапливать в другой и затем передавать к ме-
сту использования. Один из примеров—аккумуляторная батарея
в автомобиле (хотя в нем имеется также генератор), другими
примерами являются перезаряжаемые батареи в магнитофонах,
электробритвах и т. Д. Эти примеры указывают па требуемыеха-
рактеристики: масса автомобильного аккумулятора относительно
не столь велика, но он должен давать сильный ток в течение ко-
роткого периода; в противоположность этому масса батарей в
магнитофонах, электробритвах и электрических вентиляторах со-
ставляет значительную часть общей массы, по они требуют уме-
ренных или небольших постоянных токов на долгий период.
Элементы, накапливающие электричество, работают как обыч-
ные элементы, когда они вырабатывают электричество, ио их топ-
ливо образуется внутри их при пропускании тока от внешнего
источника, В этом отношении они противоположны обычным эле-
ментам, топливо в которые добавляется во время их производства,
554
Часть 3. Изменение
и топливным элементам, которые снабжаются топливом извне во
время работы элемента. Мы рассмотрим лишь два из множества
доступных приборов и сконцентрируем внимание на свинцовом ак-
кумуляторе и никель-кадмиевом элементе.
Свинцовый аккумулятор — старый прибор, но один из наибо-
лее подходящих для стартера автомобиля. Во время зарядки про-
исходит катодная реакция восстановления Pb2_f и его отложение
в виде свинца на свинцовом электроде:
РЬг+ 4- 2е- -► РЬ.
Эта реакция происходит вместо восстановления кислотного рас-
твора до газообразного водорода, поскольку последний имеет низ-
кий ток обмена на свинце.
Анодной реакцией, которая происходит во время зарядки,Слу-
жит окисление РЬЙ+ до РЬ4+; последний гидролизуется до РЬОз
и отлагается на электроде в виде оксида;
РЬ2++2НЙО ----> PbOs 4- 4Н+ 4- 2е-.
При разрядке элемента эти две реакции идут в обратных направ-
лениях. Токи обмена для реакций велики, и поэтому разрядэле-
мента может происходить быстро; вот почему свинцовый аккуму-
лятор может при необходимости давать токи большой силы.
Во многих изделиях, когда требуется небольшой, устойчивый
ток, применяется никель-кадмиевый элемент. Один электрод по-
крыт Cd(OH)2, другой — Ni(OH)2, а электролитом является КОН.
При перекачке электронов в кадмиевый электрод происходит вос-
становление:
Cd(OH)a4-2e- -► Cd 4- 2ОН-.
В то же время электроны уходят из никелевого электрода, что
индуцирует реакцию
2Ni(OH)2 + 2ОН- -> 2N"iOOH 4~ 2Н2О 4-2е~.
Зарядка заканчивается, когда произойдет превращение всего до-
ступного количества Cd(OH)a и NifOHh- При разрядке элемента
реакции идут в обр-атпом направлении; электроны уходят с кад-
миевого электрода и принимаются никелем.
29.4. Коррозия
Может показаться чрезмерно пессимистическим заканчивать
книгу разделом о разрушении материалов коррозией, являющейся
полным триумфом второго закона термодинамики. Тем не менее
все знают о важности борьбы с коррозией, и понимание ее основ-
ных принципов по крайней мере дает некоторые указания на то,
как ее можно проконтролировать, хотя мы не можем надеяться на
окончательную победу. Поскольку годовые убытки от коррозии
составляют около 1% национального продукта, организация борь-
29. Динамическая электрохимия
555
бы с коррозией может иметь значительный экономический эффект.
Термодинамика коррозии. Термодинамический признак вероятно-
сти коррозии очень просто сформулировать па основе равновесной
электрохимии (гл. 12). Реакция М+М'+—*-М_4-М' имеет тенден-
цию к самопроизвольному протеканию вправо, если
£s (М’*|М')>£е(М+]М),
где Е—-(равновесные) электродные потенциалы двух полуреак-
ций М+4-е~*^М и Одной из наиболее важных полу-
реакций является
— Fe^+4-e' ч—> 4"Fe- ^(Fe2+lFe) = -Q-^02B>
поскольку если эта полуреакция идет в паре с другой полуреак-
цией, то она ответственна за разрушение стальных изделий. В по-
следующем изложении мы часто будем отождествлять М с Fe.
Часто окисление М—4W обусловлено восстановлением прото-
нов или кислорода:
е кислом растворе
(а) Н+ + е- 4” Н»‘ £е <Н+ । Нг) = 0;
(б) Н+ + О, + е- ^H,O, E’(H+.OS>= 1,23 В;
в щелочном растворе
1 1
(в) -у Н2О+ -5- О, + е- ч=»: CH- ££(O21OH-J = 0,401 В.
Поскольку все три электродных потенциала больше E°(Fez+|Fe),
все они могут обусловливать коррозию железа. В противополож-
ность этому электродные потенциалы так называемых бла-
городных металлов, например золота, очень положительны
(E°(Au3+jAu) =+1,50 В], и поэтому такие металлы не корроди-
руют во влажном воздухе.
Отметим, что мы привели стандартные электродные потенциа-
лы; когда концентрации ионов отклоняются от единичной актив-
ности, электродные потенциалы могут быть совершенно другими
и поэтому коррозия может стать термодинамически возможной.
В частности, электродные потенциалы двух кислородных реакций
(а) и (б) зависят от pH следующим образом:
Ее (а) -£°е' (а) — (RT/F) In сн+ = (—0,059 В) pH,
Ее (б)=Е1 (б)—(RT/F) 1п ан+=( 1,23 В) — (0,059 В) pH,
и, следовательно, возможность протекания ряда реакций окисле-
ния зависит от pH среды.
556
Часть 3. Изменение
Рис. 29.17. Модель коррозионной системы.
Пример (вопрос 14). Будет лк корродировать сосуд из малоуглеродистой стали
при контакте с раствором, имеющим pH 3,0?
Метод. Электродный потенциал для кислотной коррозии —(0,059 В) -pH. Ис-
пользуем тот критерий, что сосуд корродирует, если концентрация Fe3+ превыша-
ет Ю-6 моль/дм3. Найдем Е(Fe24- [ Бе) для этой концентрации и решим, будет ли
Е(Н+| HJ7>£(Fe2+ |Fc) при установленном значении pH,
Ответ. £(Н+ (На) = (-0,059 В)ХЗ,0=— 0,177 В.
i(Fe2+[Fc) = (—0,440 В) (0,059 В) 1g Ю”" = —0,6! 7 Вл —0,6В,
Поскольку водородный потенциал превышает потенциал железа, сосуд будет
корродировать при концентрации выше 10~6 моль/дм3.
Комментарий. Коррозия по этой реакции перестанет быть термодинамически вы-
годной, только когда pH Станет выше 10,1,
Кинетика коррозии. Термодинамическое рассмотрение коррозии
интересно лишь в том смысле, что оно показывает, будет ли дан-
ное сочетание металлов (пли металл в данном окружении) иметь
тенденцию к коррозии. Однако больший интерес вызывает кинети-
на коррозии. Придя к выводу, что система нестабильна, мы долж-
ны .знать, насколько быстро она будет корродировать, а зная это,
мы должны посмотреть, можно ли уменьшить скорость коррозии.
В оставшейся части Этого раздела мы рассмотрим некоторые фак-
торы, которые влияют па скорость реакции коррозии, имеющей
термодинамическую тенденцию к протеканию.
Модель коррозионной системы приведена па рис. 29.17; в ка-
чество ее можно взять каплю слегка подкислен ной или подщело-
ченной воды, содержащую некоторое количество растворенного
кислорода и находящуюся в контакте с металлом (М) с включе-
нием примеси (М7). Эта модель показывает, что коррозионная си-^
стема по существу представляет собой электрохимический эле-
мент с короткой цепью, при этом электродами являются М и М',
электролитом служит капелька воды и цепь замыкается кон-
тактом М и М'.
Разность потенциалов между М и раствором равна ДФ(М, S),
а разность потенциалов между М' и раствором равна Д0(М', S).
Поскольку элемент имеет короткую цепь и, следовательно, омиче-
29. Динамическая электрохимия 557“
ским сопротивлением можно пренебречь, между М и М' нет раз-
ности потенциалов, т. е.
0=ДФ (М,1Ч')=-Ф (М)— Ф (М') =
= Ф (М)— Ф (S)+* (S)— Ф(5')+Ф (S')—Ф (М’) =
= ДФ (М,5)+ДФ (S, S') — ДФ (M'.S'J,
где S — раствор вблизи М, a S' — раствор вблизи М'. Предполо-
жим, что разность потенциалов между одной частью раствора и
другой пренебрежимо мала (например, капелька может быть очень
небольшой), т. с. ДФ(5, S')=0. Следовательно, при протекании
коррозии разность потенциалов иа межфазной границе металл —
электролит будет такой же, как на поверхности примесь — элект-
ролит. Эта общая разность потенциалов называется коррозионной
разностью потенциалов и записывается как ДФС,
Коррозионную разность потенциалов можно выразить через пе-
ренапряжение на каждой поверхности раздела, Записав перена-
пряжение на М через ц, а на М' через if, мы получим
ДФ (М,5) = 114-ДФе(М,5),
ДФ (АГ, S) —ц'-j-ДФе (AV,S)
и
ДФ (М,5)=ДФ (M',S) —ДФС.
Позднее мы встретимся с разностьюДФе(М, S)—ДФе(М', S): это
нс что иное, как разность равновесных электродных потенциалов
£е(М+|М)~£е(М'+|М').
Скорость коррозии можно измерить по общему току ухода
ионов М+ с поверхности металла — току коррозии Iе. Поскольку в
любом Случае ток ухода М должен быть направлен к М', ток кор-
розии равен также общему току ионов М4" по направлению к при-
месному включению. Следовательно, можно написать
/С=/(^М)-/(*М').
Приняв, что площади металла и примеси, покрытые капелькой
воды, равны А и А', ток коррозии можно записать также через,
плотность тока:
1С = 1 (-«- М) —/+ (-*-М, результирующий) А —
— ! (—>!') (->№', результирующий). (29.4.1>
Отсюда
/с — |'{AA'j+ -М', результирующий)/+(*-М, результирующий).
(29.4.2>
558
Часть 3, Изменение
Плотность тока при соответствующем перенапряжении можно вы-
разить через уравнение Батлера — Фольмера. Для простоты пред-
положим, что перенапряжения достаточно велики, и можно исполь-
зовать экспоненциальную форму уравнения (29.1.12). Вид урав-
нения упрощается, если взять и = -у- и предположить, что заряды
на ионах М+ и М/+ оба равны единице, т. е. z+ = z^=I. Все эти
упрощения можно не делать, но это приведет к более сложным
выражениям, не дающим ничего нового.
В результате упрощений получим, что
j+ (-«- М, результирующий) = /1 ехр (t]F/2/?7') =
— /е ехр (АфС/2/гТ) ехр[ —А Фе (М, S)/2/fT],
j+ (-* М', результирующий) = /Г ехр (— i}'F/2RT) —
=» /е+ ехр (— АФC'2RT) ехр [А Фе (М', S)/2/?7']
Мы близки к концу вывода, так как эти два уравнения можно
объединить, используя уравнение (29.4.1) для нахождения потен-
циала коррозии;
ехр (А Ф C//?T) ехр {(F/2/?T) [А Ф е (М', S)+А Ф е (М, S) 1}.
Это выражение можно подставить в уравнение (29.4.2) и полу-
чить конечное выражение для тока коррозии;
Д=(ЛЛ')1^ (Й'+)1/з ехр {(F/4RT) )£е (М'+ | М') -Ее (М+ | М)]}. (29.4.3)
Из этого приближенного выражения можно вывести некоторые
заключения. Во-первых, скорость коррозии зависит от площадей
поверхности металла и примеси: если примеси нет, то Л'=0 и мы
должны ожидать, что коррозии не будет. Отсюда следует триви-
альный, однако часто эффективный, метод замедления скорости
коррозии: нужно покрыть поверхность пленкой, например крас-
кой, так, чтобы не было площадей примеси, открытых для влаж-
ной окружающей среды. При прочих равных условиях (т. е. для
реакций коррозии с подобными токами обмена) скорость больше,
когда Ее(М'+|М') значительно больше Ее(М+|М); таким образом
быстрая коррозия может происходить, если металл и примесь
имеют силищ отличающиеся электродные потенциалы.
Роль токов обмена в определении скорости коррозии можно по-
нять при рассмотрении случая железа в контакте с кислым рас-
твором. Термодинамически железо может быть Окислено реакци-
ями (а) и (б) (стр. 555), причем последняя реакция термодина-
мически предпочтительнее в том смысле, что она в паре с реак-
цией железа дает большую разность электродных потенциалов.
29. Динамическая электрохимия 559
Тем не менее для нее ток обмена на поверхности железа равен
лишь 10-14 А/см2, тогда как ток обмена для выделения водорода
на железе равен 10~6 А/см2. Поэтому последняя реакция происхо-
дит быстрее, и железо корродирует благодаря реакций выделения
водорода в кислом растворе. Кроме того, ток обмена зависит от
pH раствора (как и электродные потенциалы), и действительно
очень часто кислотная коррозия зависит от pH.
Ингибирование коррозии. Для ингибирования или предотвращения
коррозии применяют ряд методов. Покрытие поверхности неко-
торым непроницаемым слоем, например краской, может предот-
вратить доступ влажного воздуха. К сожалению, такая защита
оказывается гибельной для поверхности металла, если из-за де-
фектов поверхности краски открывается доступ влажного воздуха.
Кислород получает доступ к открытой поверхности металла и мо-
жет в этих местах оставлять свои электроны, которые расходу-
ются но краям крошечных отверстий в краске, куда доступ кис-
лорода более затруднен. Теперь коррозия происходит под краской,
и степень опасности может быть значительно выше, чем можно
предположить при нечастом осмотре пятен или царапин на крас-
ке. Почти каждый владелец автомобиля страдает от этого аспек-
та электрохимии; аналогично коррозия наносит большой ущерб в
случае погруженных в воду железных труб или свай.
Другая форма поверхностного покрытия получается гальвани-
зацией: так, железные объекты покрывают цинком. Поскольку
электродный потенциал реакции -4- Zn2+ + e_^~ Zn равен
—0,76 В, т. е. более отрицателен, чем потенциал восстановления
железа, коррозия цинка термодинамически более предпочтитель-
на и железо сохраняется. В противоположность этому лужение
железа оловом приводит к очень быстрой коррозии железа, если
поцарапать слой олова; потенциал восстановления олова равен
“0,14 В, и это вызывает окисление железа, Некоторые оксиды
стабильны кинетически в том смысле, что они адгезируются к ме-
таллической поверхности и образуют непроницаемый слой: по этой
причине алюминий стабилен на воздухе, несмотря на то что по-
тенциал его восстановления сильно отрицателен (—1,662 В). Такой
тип пассивации можно рассматривать как резкое уменьшение тока
обмена из-за герметической и ишяпьц поверхности.
Еще одним методом защиты является изменение потенциала
материала путем нагнетания электронов. Тогда можно расходовать
эти избыточные электроны на реакцию восстановления кислорода,
и нет необходимости в переходе М в М+. Одним из путей обеспе-
чения избыточного запаса электронов служит катодная защита.
Она достигается соединением материала с имеющим более отри-
цательный электродный потенциал металлом, таким, как магний
(“2,363 В). Магний действует как жертвенный анод, снабжая
560
Часть 3. Изменение
Рис. 29.18. Катодная защита от коррозии,
а— с жертвенным аладоя; б — с приложенным током.
своими собственными электронами железный материал и разру-
шаясь в этом процессе (рис. 29.18, а). Заменяемый время от вре-
мени блок магния обычно стоит гораздо дешевле, чем корабль,
здание или трубопровод, ради которых им и жертвуют. Другим
методом снабжения электронами является катодная защита с ис-
пользованием тока (рис. 29.18,6). В этом случае электроны бе-
рутся от внешнего источника, и это исключает необходимость для
железа отдавать собственные электроны. Однако в обоих случаях
второй закон термодинамики неумолимо приводит к конечному
разрушению. Все, что мы можем сделать, —это замедлить егодей-
ствие, умерить его влияние и не тратить зря время па борьбу с
его могуществом.
Литература
Hills G. J., Electrode processes, in Essays in chemistry (Bradley J. N., Gil-
lard R. D., Hudson R. F„ eds), 2, 19, Academic Press, London, 1971.
Bockris J. O.'M., Reddy А. К, AL, Alodern electrochemistry, Plenum, New York,
1970.
Albert) IP. J., Electrode Kinetics, Clarendon Press, Oxford, 1974.
Korium G., Treatise on electrochemistry, Elsevier, Amsterdam, 1965.
Hush N. S., Reactions of molecules at electrodes, Wiley-Interscience New York,
1971.
Wawzonek S., Potentiometry; Oxidation-rednciion potentials, in Techniques of
chemistry (Weissberger A. and Rossiter B. N,, eds.), Vol. IfA, 1, Wiley-Inter-
sciencc, New York, 1971.
Bockris 1. O.'AL, Srinivasan S. IV., Fuel cells; lltetr electrochemistry, McGraw-
Hill, New York, 1969.
Задачи
29.1. В данной главе ключевым является уравнение Батлера — Фолъмера, и по-
этому в нескольких первых задачах мы рассмотрим некоторые способы, с по-
мощью которых его можно использовать и обращаться с ним. Вначале примите,
1 ь
что и постройте график плотности тока /+/;е как функции перенапряже-
ния т) |1рц комнатной температуре для а) однозарядного катиона, б) двухзаряд-
но, о ка гиона.
_______29. Динамическая электрохимия______________________561
29.2. Основная трудность электрохимии состоит в правильном выдерживании
знаков. Попрактикуйтесь в этом, выведя уравнение Батлера — Фольмера для
тока анионов по направлению к электроду. Другими словами, найдите выраже-
ние для р(—>-М, результирующий).
29.3. Повторите задачу 29.1, но для анионов вместо катионов.
29.4, Типичная плотность тока обмена для разряда Н+ на платине равна
0,79 мА/см2 при 25 ’С. Какова плотность тока на электроде, когда его перена-
пряжение? а) 10 мВ, б) 100 мВ, в) —5,0 В?
29.5. Плотность тока обмена для электрода Pt|Fe5+, Fe2+ равна 2,5 мА/см2.
Стандартный электродный потенциал 4-0,771 В. Вычислите силу тока, текущего
через электрод площадью 1 см2, в виде зависимости от потенциала электрода.
Для обоих ионов возьмите активность, равную единице.
29.6. Предположим, что установлен потенциал электрода 1 В. Рассчитайте ток,
который течет через указанный выше электрод при отношении концентрации
[Fe2-]/[Fe33] в интервале от 0,1 до 10,0.
29.7, Какое перенапряжение необходимо для поддержания тока 20 мА на элек-
троде PtlFe3-1-, Fe2^, когда концентрации обоих ионов равны 0,1 моль/дм3?
29.8, Сколько электронов или протонов транспортируется через двойной слой
в каждую секунду, когда электроды Pt|H2, П+, PtjFeH, Fe?+, Pb|H2, Н+ нахо-
дятся в равновесии? (Плотность тока обмена в последнем случае 5,0-10~>2 А/см2;
в каждом случае возьмите площадь 1 см2.)
29.9, Чтобы получить величину этих параметров в атомной шкале, оцените, сколь-
ко раз в секунду одиночный атом на поверхности принимает участие в событии
переноса электрона.
29,10, Каково эффективное сопротивление поверхности раздела электрода, когда
Перенапряжение мало? Выясните его Для следующих электродов площадью
I см2: a) Pt, Н2|Ц!, б) Hg, Н2[Н+. (Данные в табл. 29.1).
29.11, Если a=i/s. поверхность раздела электрода не может выпрямить перемен-
ный ток, так как кривая плотности тока симметрична относительно а=0 (зада-
ча 29.1). Когда и^1/», величина плотности тока зависит от знака перенапряже-
ния, и поэтому можно получить некоторую степень выпрямления: это называется
фарадеевским выпрямлением. Предположим, что перенапряжение изменяется как
Л (t) =i]o cos ыЕ Найдите выражение для средней силы токд (усредненной по цик-
лу) для любых а и подтвердите, что средняя сила тока равна нулю при и=</2.
В каждом случае работайте в пределе небольшого q о, однако вы должны беде те
работать со вторым порядком по T\aF/RT.
29.12. Рассчитайте среднюю плотность постоянного тоКа для водородно-плати но-
вого электрода с а = 0,38, когда перенапряжение изменяется между ±10 мВ
при 50 Гн.
29.13, Теперь предположим, что перенапряжение находится в области высоких
перенапряжений все время, несмотря на то что оно осциллирует. Какой волно-
вой формы будет ток через поверхность раздела, если т| изменяется линейно и
периодически (как зубья пилы) между т|_ и Т|+? Примите, что а=‘/г-
29.14. Тафелевская зависимость (стр. 537) дает среднее значение цлопЩстц тока
обмена и коэффициента переноса а для электродов, и поэтому из нее получают
информацию, необходимую для обсуждения электродных процессов. В опытах
с электродом Pt|H2, П+ в разбавленной Н2ЗСЦ наблюдались приведенные ниже
значения плотности тока. Найдите а и для электрода.
1ф мВ 50 L0O 150 200 250
/+, цА/см® 2.66 8,91 29,9 100 335
29.15. Как бы зависела плотность тока на этом электроде от перенапряжения
при том же наборе величин, но противоположного знака?
29.16. По той же причине, что и в задаче 29 2, выведите подходящую форму та.
фелевской зависимости для днижения анионов по направлению к электроду для
обоих знаков перенапряжения.
562
Часть 3. Изменение
29-17- Установите, что происходит, когда платиновый электрод в водном растворе,
содержащем как ионы меди, так и ионы цинка в молярных концентрациях, ис-
пользуется в качестве катода в ячейке для электролиза.
29.18. Каковы минимальные (определяемые термодинамикой) потенциалы, при
которых может отлагаться из водных молярных растворов: а) цинк, б) медь?
Каковы соответствующие потенциалы, когда концентрации равны
0,01 моль/дмэ?
29.19. Каковы условия, позволяющие металлу осаждаться из водных кислых рас-
творов до того, как будет происходить интенсивное выделение водорода? Почему
из водного раствора нитрата серебра может осаждаться серебро? Почему кадмий
может осаждаться из водного раствора сульфата кадмия? (Перенапряжение для
выделения водорода на кадмии составляет около 1 В при плотности тока
1 мА/см1.)
29.20. Плотность тока обмена для разряда Н + па цинке составляет около 5Х
XI0”11 А/см2. Может лц цинк осаждаться из водного раствори Соли пинка еди-
ничной активности?
29.21. Стандартный потенциал электрода Zn1+JZn равен —0.763 В. Плотность
тока обмена для разряда Н+ на платине 0,79 мА/см2. Может ли цинк отлагаться
на платине? (Возьмите единичные активности.)
29.22. Может ли магний осаждаться на цинковом электроде из кислого раствора
единичной активности?
29.23. Найдите выражение для плотности тока на электроде, где скорость про-
цесса контролируется диффузией и известно т]с. Нарисуйте эскиз /+//t как функ-
ции т|с. Какие изменения произойдут, если рассматривать токи анионов?
29.24. Предельная плотность тока для реакции 13-р2е~—ьЗ!“ на платиновом
электроде составляет 28,9 мкА/см2, если концентрация КЬ равна 6,6 X
Х10-4 моль/дм3. Коэффициент диффузии 13 равен 1,14-10-’ см2/с. Какова тол-
щина диффузионного слоя?
29.25. Предельная плотность тока в уравнении (29.1.21) определяется через ион-
ный коэффициент диффузии но D+ может быть связан в с ионной электро-
проводностью 7^ (Стр- 383). Выведите выражение для предельного гена через
к+т концентрацию с+ и толщину диффузионного слоя 1.
29.26. Ионная электропроводность Fe2^ равна 40 Ом‘'1-см1-моль~[. Предельный
ток ла платиновом электроде площадью 40 см1, погруженном в раствор хлорида
железа(П) при 25°C, был измерен при разных концентрациях; результаты при-
ведены ниже. Какова толщина диффузионного слоя при каждой концентрации?
(Должна получиться совершенно типичная толщина.) Можно лв придумать не-
зависимый неэлектрохимический путь измерения 1?
[FeCIa], моль/дм3 0,250 0,125 0,063 0,031
/, мА 215 107 49 23
29.27. Стандартные электродные потенциалы евннца и олова составляют —126 и
—136 мВ соответственно, и для их осаждения требуется небольшое перенапряже-
ние. Какими должны быть их относительные концентрации, чтобы быть уверен-
ным в одновременном нх осаждении из смеси?
29.28. Стандартные электродные потенциалы серебра, меди и цинка составляют
799, 337 и —763 мВ соответственно. Когда они находятся в смеси с цианид-
ионами, нх можно осадить одновременно. Объясните Это наблюдение.
2959. Теперь перейдем к вопросу о генерировании мощности. Прежде всего най-
дите максимальную (обратимую) разность потенциалов никель-кадмиевого эле-
мента и максимально возможный выход мощности при прохождении тока силой
100 мА.
29. Динамическая электрохимия 563
29.30. Рассчитайте термодинамический предел для э.д.с. топливных элементов,
работающих на а) водороде п кислороде, б) метане и воздухе. Используйте ин-
формацию по функции Гиббса цз табл. 9.1.
29,31. Оценка выхода мощности и э. д. с. элемента в условиях его работы очень
трудна, однако некоторые необходимые параметры собраны в приближенном
уравнении (29.2.7). В качестве первого этапа обращения с этим уравнением опре-
делите все величины, которые зависят от концентрации ионов. Выразите Е через
концентрацию и электропроводность ионов, находящихся а элементе.
29,32, Оцените параметры из последней задачи для Zn [ZnSOi(aq) ||CuSO<(aq) |Cu.
Примите, что электроды площадью 5 см2 разделены расстоянием 5 см< Не прини-
майте во внимание ни разность потенциалов, ни сопротивление жидкостного со-
единения. Возьмите концентрацию 1 моль/дм3 и пренебрегите коэффициентами
активности. Отложите на графике Е как функцию протекающего тока.
29.33. На том же графике отложите выход мощности элемента. Какой ток соот-
ветствует максимальной мощности?
29.34, Рассмотрите элемент, в котором ток имеет активационный контроль. По-
кажите, что для максимальной мощности ток можно оценить, откладывая
’й(^Де) в Щ—с21 против / (здесь , а ст и с2— константы), и найдите
точку пересечения кривых. Проведите этот анализ для элемента из задачи
29.32, пренебрегая всеми концентрационными перенапряжениями.
29.35, Если медь и малоуглеродистая сталь находятся в контакте (как в плохо
спроектированной системе водоснабжения), то какой металл будет иметь тенден-
цию к коррозии?
29.36, Найдите зависимость от pH для электродного потенциала щелочной реак-
ции коррозии [реакция (в) па стр. 555). Реакция Н2 имеет более
отрицательный потенциал при любом pH. Почему же тогда ова играет роль в
коррозии?
29.37. Какие из следующих металлов имеют термодинамическую тенденцию кор-
родировать при pH 7; Fe, Си, Pb, Al, Ag, Сг, Со? В качестве критерия коррозии
примите концентрацию иона металла по крайней мере 10“в моль/дмэ.
29.38. Найдите pH, при котором каждый из металлов из предыдущей задачи бу-
дет корродировать.
29.39. Оцените величину тока коррозия д.тя участка цапка площадью 0,25 см®
в контакте с аналогичной площадью железа в водной среде. Плотности' токов
обмена равны 10“е А/см2.
Ответы к задачам
Глава 16
16.1. йз; СХ; Dai', D.xh; С.^; Da,; С5; Вел; C4tJ; Cs. 16.2. С?„; Сз1;; Сзг;
Dih', Cih', Czh’, Dih\ Dih', Civ- 16.3. Daa', Dad', Dzh', D^h', Da", D^. 16.4.
тря»с-С11С1=СПС1. 16.5. С2оЛ = i. 16.6. \O2, CH3CI. CCI3H, rfuc-CIICl =
= CIIC1. CellsCl. 16.7. Со(еп)Г. 16.12. x^Bf, y^B2; z^Ax. 16.13.
Л2. 16.14. а) Й1 (x, y}-, Ai (2), 6) Bi (x); B2 (у); Л1 (2). 16.15. Л(+
+T2; да; tZxy, d,-, dvl трансформируются как T%. 16.19. а) 2Л;, 2Bi, 2B2
б) Л-. ЗЕ. в) Z2. Tt, Ait г) Т1Ы, Т2и, Аги. 16.20. а) Тг, Tt, Ai, б) 7Х,
Т2и, А2и. 16.21. а) 1, 2, б) 2. 16.23. а) ЦЛ^В^Вг); г(Л2+В1+В2),
б) £(Л1+£); г(Л2+В), в) ЦЛт+й^; г(Л24-ЕГ1)) г) 1(Л«); r(Tie)-
16.24. а) Только р и /, б) да. 16.25. а) Л1—>-Л2, В(, В2, б) Е ГТ2,
в) AiS. 16.27. а) Да, б) нет, в) да. 16.28. E,i, ЗС2, 4С3, Зо; Oh (лгеЗт).
16.29. Е, ЗС2, 4С3. 6н; кубическая, Td (43 m). 16.30. Триклинная;
Ci (1). 16.31. Моноклинная. 16.32. Кубическая; ОьУпЗт). 16.34. Ф =
= 5444'.
Глава 17
17.1. НС1, СНзС1, СН2С12, Н20, Н2О2, NH3, NH4C1 (газ). 17.2. НС1. СО2,
Н2О. СНз—СНз, СН4, GH3CI, N7. 17.3. Н2, НС1, СН3С1. С1ЬС12
СИзСНз, Н2О. 17.4. Все. 17.5. 0.999999925-660 нм; ^6.36 1СГ м/с
(иди —7,02-Ю7 м/с при относительном рассмотрении). 17.6. 2,397Х
X1U4 км/с; 8,351-Ю3 К. 17.7. a) 2,l-10~6; 0,67 МГц; 0,006 см”1,
б) 9.7-10-7; 3.3-10-3 МГц; 3,7 10~4 см-'. 17.8. а) бД-Ю"1' с.
б) 5.3-10-12 с, в) 1,6 10-9 с. 17.9. а) 53 ем-1, б) 0,53 см”1. 17.10.
t = 1/Z^2.29-10-!0 с; 696 МГн; 0,73 мм рт. ст. 17.11. а) 0.051,
6) 0.065, в) 0,197, 17.12.30. 17.13.6. 17.14. а) 1,6266 10-27 кг;
^б^б-Ю-^кг-м2, б) 3,1623-Ю-27 кг; 5,1375-Ю"47 кг-м2, в) 1.6291Х
ХЮ-27 кг; 2,6466 IO-47 кг-м2. 17.15. 596 ГГп; 19,9 см-!; 0,2,>2 мм,
0,083 мм, ... (уменьшение разделения на Х-шкале). 17.16. В (12С16О)/
/Лг=1.9318см-1; В(13С16О)jhc=1.8466см-1. 17.17. 2,728-Ю"47 кг-м2;
129.5 пм. 17.18. 42.86. 53,56, 64,16. 74,77, ... см"1. 17.20. a) 4.60J6X
ХЮ-” кг-м=. б) 7,4943-Ю-43 кг-м2. 17.21. 160 5 им. 17.22.
Ответы к задачам
565
0,10057 пм; 218 им. 17.23. 1,37998- 1СН5 кт-м2. 17.24. К(СО) =
= 116.28 им; ff(CS) = 155,97 пм. 17.25. См. Kraitchman J., Amer. J.
Phys.,' 21, 17 (1953). 17.26. 215 пм; Г45'. 17.27. 0,0459 см-';
0,0184 см-'; 0,0103 см-'. 17.28. 967,1 Н/м; 515,6 Н/м; 411,8 Н/м;
314,2 Н/м. 17.29. 3002,3 см”1; 2143,7 смН; 1885,9 см"1; 1640.1 см-'.
17.32. 4,13 эВ. 17.33. 93 80 Н/м; 3,4 эВ: 3,38 эВ. 17.34. а) 5,16 эВ;
б) 5,21 эВ. 17.36. 480,7 Н/м; 128 км; 130 пм. 17.37. 198,9 им. 17.39.
а) Да, б) нет.
Глава 18
18.1. 449 см'1-мош,-1-дм3. 18.2. 159 см-1 -моль-'-дм3. 18.3. 0,090,
0/149, 0.896. 4,52; 0,32. 18.4. а) 69%; 1,3-10-’4%, б) 90,2%;
0,0032%. 18.5, а) 87.6 см, б) 291см. 18.6.а) 3,5-10'8дм3(моль-см-с),
б) 1,7-10'7 дм3/(моль - см с). 18.7. а) 0,219, б) 0,011.18.8. Л— 2,4515 сХ
ХетлхАп,/з. 18.9. ^=3,32-10's дм3/(моль-см-с); /—2,08- Ю-4; запре-
тен. 18.11. 4,74- 1СН; запрещен; А г, В\, В2. 18.12. \ (64/3+) X
Х[«3(«-Н)2/(2^4-1)3]; /„^,(+2 = 0. 18.13. 7350 см '; инфракрасное
ноглощсш/е; 4,3-106 моль-'-дмй-см-;, 7-10 7 см. 18.14. Удлинили; го-
лубой. 18.16. Я/«о = 2,10380. 18.17. а) Разрешен, б) запретен. 18.21.
Используйте Kin= [11+] 4 «/(1—а) 1 . 18.23. 7,1 (7.9 при pH 9). 1824.
0.818эВ. 18.25. 5,09 эВ. 18.26. а) 3+1. 5+3, б) 4+4, 2+2. 18.28. 16,52
эВ-монпзацня 2рсг-эле ^тронов; 15,65 эВ-иоыизация 2рл-электро-
нов; 9.21 эВ-иопизацпя 2/зл*-электронов.
Глава 19
19.1.1,34 Т (13,4 кГс). 19.2. 1,66-10"" моль/дм3; А% = 1,2492- 10м;
(Vu-1,2509- 1014. 19.3. а) 0,0503, б) 0,0007. 19.4. 2,00224. 19.5.
а) 331,9 мТ, б) 1,201 Т, 19.6. а) 13,4 Гс б) 48,5 Гс. 19.7. 1,9920;
2,0022. 19.8. 50,7 мТ. 19.9. 2.3 мТ. 19.10.’ 2.0025; 64.5 МГц. 19.11.
330,2 мТ, 332,2 мТ, 332,3 мТ,334,8 мТ. 19.12. т^9,4-10~9 с. 19.14.
0,69 мТ; 0/г2 мТ. 19.15. Квартет (1:3:3:1) триплетов (1:2:1); трип-
леты, замещенные (1:2:3:2:1) -квинтетами. 19.17. 10%. 19.18. 38%.
19.19. 048: 0,52: 3,8; да. 19.20. а) -6,697-Ю“4 Т, б) —2/86-КГ3 Т.
19.23. 1,6-10 9 с. 19.24. а) 1 409Т; 9,(81 Т- 5,605 Т; 19,504 Т- 1,498Т;
3.481 Т. б) 7,046 Т- 45.904 Т; 28.026 Т: 97.522 Т; 7,490 Т; ’17.407 Т.
19.25. а) 7,662-1(Г5 Н; ЗЛИ-НГ4 Н; 1,788-10 3 II. б) 1,022-10-° Н;
5,108-10"6 II; 2,384-i0-s II. 19.26. 211 Т; 2,14 мТ. 19.27. 2,06 Т.
—4 84-10-®; Р(и= — '/2) нижнее, 19.28. а) —11,4мцТ, б) — 53,2мкТ.
19.29. а) 456 Гц, б) 2,28 кГц. 19.31. 158 пм. 19.32. 77,6 мкТ. 19.33.
Когда время жпэни каждого изомера ^72 мс. 19.34. 56 2 кДж/моль.
19.35. а) 81,3 м/с: б,5б-101П Гц, б) 7,69-10-2ft м/с; бД- Ю’11 Гц. 19.37.
Ионностр Ди1<АпВг< AuCl.
566 Ответы к за дачам
Глава 20
20.1. Одно. 20.2. (4,0,0,0,0,1), 5; (3,1,0,0,10). 20- (3,0 1 100) 20-
(2,2,0,1,0,0), 30; (2,1,2,0,0,0), 30; (1,3,1,0,0,0), 20; (0,5,0,0,0,0), 1;
наиболее вероятны (2,2,0,10,0) и (2 12,0,0,0) совместно. 20.4.
(4,2,2,1,0,0,0,0,0,0) с весом=3780. 20.5. -100 К. 20.8. а), б), г), е);
для ж) нущно зпать принцип Паули. 20.9. а) 4,76 102S б) 2 45 1026
в) 4,76-Ю’8, г) 1- 20.10. Пока Ь2/тХ2кГ^}; 3,5-10 15 К- 7 53.
20.11. а) 5,00, б) 0,25. 20.12. а) 1,00; 0,80, б) 6,5-10-”; 0,12’. 20.13.
64% витией, 36% верхней. 20.14. 0 518 кДж/моль. 20.15. а) 1,0488,
б) 1,5553. 20.16. а) 0.9535; 0,0450; 0,0021, б) 0,6430- 0,2298; 0 0826.
20.17. а) 126,3 Дж/моль. б) 1,395 кДж/моль. 20.18. 0,904; 0.999.
20.19. 9=1+е~х+е-2х: (е)— - £пц|ХВ[ (1 .+е-2х)/(1Хе-х+е-а-х)'[,
где x=gTjllvBp. 20.20. а) —298 К, б) —10 К. в) -О К. 20.22.
S/k—0,92. 20.24. а) 13,38 Дж/(К-моль), б) 18,07 Дж/(К-моль). 20.25.
а) 11,2 Дж/(К моль), б) 11,4 Дж/'(К моль). 20.26. а) 1,661 Дж/(КХ
Хм°лв), б) 8,352 Дж/(К-моль). 20.28. 155 Дж/(К-моль).
Глава 21
21.1. а) 6,56. б) 19,0, в) 32,8. 21.2. а) 6,9051, б) 19.8893. 21.3. 43,10;
7’>40К. 21.4. а) 36,3. б) 78,9. 21.5. 36,6055 (при 298 К); 79.1716
(при 500 К). 21.6. Д5т = тп[(п/Г)(А2р/2лте)Ч- 21.7. 89 Дж/(КХ
Хмоль) • —62 Дж/(К-моль). 21.9, а) 2, б) 1, в) 12, г) 12, д) 3.
21.10. 256 Дж/(К-моль). 21,12. а) .3,886, б) 2,4(4. 21,13. 3,15-Ю-3-
21.14. Умножение на 1,004. 21.15,7,45 Т. 21.16.17—(7(0)-Я—Я(0) =
= Л'Аще'х/(1—е-*): S — Л7, [же =7(1 — е-1) — In (1 — е-х) ]; .4—.4(0) —
= G-6(0)»Ж1й(1-е%. 21.17, Я1п(1—е-*). 21.18. а) -4,31 Дж/
'/(К-моль), б) —6,52 Дж/(К-моль). 21.19. а) —136 Дж/(К-моль),
б) —232 Дж/(К-моль), в) —203 Дж/(К-моль), г) —198 Дж/
/(К-моль), д) -217Дж/(К-моль). 21.20. 4,16-10-7. 21.21. U = nRTX
X((Tfq); ^ = ^я[(гл/)+1п(е<7/Дг)]; Сг = кЯ[(гЛ1)-(<77?)2]- 21-22-
/7внутр = URT внутр /^виутр) ' ^внугр- ^Г(?7?) онутр -|-1 вдВЯу rp] i
Су,внутр = nR[q"/q} внутр— (я'/ч) в“У1Р ] .
21.23, а) 4 322 кДж/моль б) —0.138 Дж/(К-моль), в) 5.405 Дж/
/(К-моль).’ 21.24. -5 771 Дж/(К-моль); 0,060Я = 0,499 Дж/
/(К-моль). 21.26. 8,674; z(Na2) =0,095; ж(Ка)=0,905. 21.27
а) 21 Дак/(К-моль), б) 21 Дж/(К-моль), в) 25 Дж/(К-моль),
г) 25 Дж/(К-моль), д) 2.5 Дж/(К-моль), е) 21 Дж/(К-моль). 21.29.
а) 35,72 Дж/(К'Моль), б) 46,44 Дж/(К-моль). 21.30. Cv,m/R =
=x2e~xl(l-j-e-1)2 где гс=А//с7'. 21.31. а) 2,94 Дж/(К-моль), б)
0,654 Дж/(К-моль), в) 0,244 Дж/(К-моль). 21.32. а) 3,72-10 2 Дж/
/(К-моль) б) 1 06 10—3 Дж/(К-моль). 21.33. 4.16 Дж/(К-миль):
15.1. Дж/(К-моль). 21.35. a) cs = (1.10 RT/m) Чб) es=[5(l+a)X
ХЯ7,/(3+2а)ж]7.21.37. а) 9,13 Дж/(К-моль), б) 13,4 Дж/(К-моль),
Отпеты к задачам
567
в) 14,9 Дж/(К-моль). 21.39, 191,4 Дж/(К-моль); нулевая остаточ-
ная энтропия.
Глава 22
22.3. l/d2=l/a2 + l/fc2; sin2 60/й2= 1/й2+1/а2-1/аЬ, 22,4. (329),
(til). (122) (3*“), 22.7. 249 пм; 176 им; 432 пм. 22.8. 58,9 пм.
22.9. 628 им; да. 22,10. 8“Ю'; 4°49'; Ц°46'. 22.11. Граггепентрпро-
ваннан кубическая. 22.12. Объемно-центрировавиая кубическая.
22,13. 564 пм; 326 пм; 252 пм. 22.16. 834 пм ; 606 пм; 870 пм. 22,18.
а) 1 б) 2, в) 4, г) 8. 22,19, г.ц.к, 22.20. 0,9069. 22,21, 0=1347'.
18°59', 23’28' 9,32 г/см3; 13,3 г/см3, 22,22, о.ц.к. 22,23, 0 = 21°41',
2545', 37’06!, ...; 8,97 г/см3. 22.24. —3'45",- расплывчатые линии.
22,25. р[16 = 4,8-10-5 КД рЛ11Н= 16-10-' К-1, 22,26, 6 = 22’00',
32"00', 40’00', ... 22,27, 3,37 г/см3, 22,28, 312 пм. 22,29, г.ц.к.; а =
= 412 пм. 22.30. а) о.ц.к.; а —316 пм, б) г.цж.; а = 361 пм. 22.31.
597 пм; 1270 пм; 434 пм; 4. 22.32. а) Примитивная, б) (010) — пло-
скость скольжения с трансляцией, равной '‘/г длины диагонали, в)
(001) — плоскость сь'ольжопп» с трансляцией, равной ’А аксиальной
длины вдоль Ь, г) (100) — обычная плоскость симметрии. 22.23. См.
Bijvoet J. М., X-ray analysis of crystals, Butterworths, London, 1951,
p. 56, 22.39. a) 38,8 пм, 6) 12,26 пм, в) 6,132 пм. 22.40. 7912 м/с;
145 пм, 22.42. 177 пм.
Глава 23
23.1. 0 (п-кснлол); 0,7 D (о-ксилол); 0,4 D (.«-ксилол); величина
для п-ксилола из симметрии. 23.2. p=(3,00 D) cosVa 0 — 90°. 23.3.
а) 1,11- 10s В/м, б) 4,09-103 В/м, в) 4,09-103 В/м. 23,4. 2,26пм. 23.5,
НО : 1,03 D, 4,4-10“23см3; HBr:0,80 D, 4,6-10“23 см3; Н1 : 0,36 D
7,0-10-23 см3. 23.6. 1,34, 23,8. 1,49-10-22 см3; о,9 D. 23.9, а=
= 1,74-Ю-22 см3; ц = 0,32 D, 23,12. 21, 23,15. 1,00018; 1,65; 1,60; 1,24.
23.16. мольД-см3; не зависит от плотпости; пропорциональна плот-
ности. 23.17. 1,59 D; 2,49-10'23 см3. 23.18. а) 5,661 см3/моль. б) та-
кая же; 1,578 D, 23.19, Г=-(3,75-Ю9 Н)/(Я, пм)7, 23.20. а=
- (2л/3) L2 (Сс/<73); а(Аг)=0 019 Дж-м3/моль3. 23.21. В(Т)яз
« (2/3)nL&[t-(C6/kTd*]. 23,22. С6л;31а2/64л2; 228 см3/моль.
23.25. 1,6 кДж/моль, 23.26. Гт,к = (л/2)£о!3; рк— (8/9л) (Сс/с?9);
7^= (32/27)С6/Ы6. 23.27, ГГП|К~ 285 см3/моль; рк = 6,29 агм; Т,—
= 58 К. 23,29. 1,38629, ... 23,30. Я* = (2/5)К0+ (18гг/5)е01?g Гт/
/х£е2 JK’ 23.31. —709 кДж/моль. 23.33. —919 к-Дж/моль. 23.34.
—628 кДж/моль. 23.37. -1,2204-Ю-9; 2 1542-Ю^7; 2,1420-10 7,
23,38. а) 6,96-Ю4; 5,19-Ю-2, б) 3,04-Ю3; 0,225,
Глава 24
24.1. з) 11,0 им, б) 110 пм, в) 110 мм. 24.2. 1,10-Ю'6 атм. 24.3.
568
Ответы к задачам
217 атм. 24.4. l,35-10-G м. 24.5. а) 3,60-10’° с"1, б) 3,60-109 с"[,
в) 3,60-Ю3 с-!. 24.6. а) 4.43-Ю33 с-’-м-3, б) 4,43-Ю3’ с-’-м”3,
в) 4,43-Ю’9 с-'-м"3. 24.7. 3,00-Ю8 ел'. 24.8. а) 229-Ю27 с ',
б) 1,90-102? с-1. 24.9. 9,9-Ю22 с-1. 24.10. а) 638 м/с; 1256 м/с;
2301 м/с, 6) 319 м/с; 628 м/с: 1150 м/с. 24.11. а) 3,74 кДж/моль,
б) такая же. 24.14. (рг> = 0,47 {2кТ/лт) ’< 24.15. а) 1,75 миль
в вас, б) 56,2 миль в час, в) 56,4 миль в час. 24.16. б'О’А/', б) 5'9[/г".
24.17. а) 0,39, б) 0,61. 24.18. п2ехр(1-и2); 3,02-Ю^; 4,9-10-6. 24.19.
V(2iz?J; Ц,2 км/с; 5,0 км/с; 11900 К, 23600 К, 188900 К на Земле;
243() К, 4810 К, 38500 К иа Марсе; 2,3-10-27, '--О, '"Ю на Земле при
240 К; 1,3-10“°, 7,4-Ю’", —0 на Марсе при 240 К, 2,6-10“4,
9,1-10-’ ~0 па Земле при 1500 К; 0,26; 0.046; 7,9-10”4 на Марсе
при 1500 К. 24.20. 43. 24.21. 2,45 10” с'1. 24.22. 1 09-Ю5 с (30 ч).
24.23. р= (Am/St) (2nkT/m) 24.24. 7,3-Ю“3 Н/м2 = 7,2-Ю“® атм=
= 5,5 10“5 мм рт. ст. 24.25. а) 2,72-1023 с_|-см-2- 23'10® с-1 б)
2,72-Ю17 с-’-см-2; 230 с-/ в) 2,72-Ю’3 с ’-см-; 0.02 с"1. 24.26.
а) 0,61 мм рт. ст., б) 0,18 мм рт. ст. 24.27. 1,44 с. 24.28. а) 1,7-10’4
с б) 1,4-Ю20 с О 24.29. а) 1,83-Ю34 м^-с1, б) 6,5.3- Ю3^ M“3-c“',
в) 6,1-Ю34 м-3'С 24.30. а) Газа пет, б) 1,89-10“5 кг-м/с, в) 3,46Х
ХЮ-5 кг-м/с. 24.31. а) 7,49-10”3 Дж/(К-м-с); 1,1-10“2 Дя«/с, б)
473-10-2 Дж/(К-м-с); 7,1 -Ю“2 Дж/с. 24.32, 1,38-Ю-5 кг/(м-с);
190 пм. 24.33. 2,24-Ю’2 Дж/(м-с-К); 4,11-Ю-2 Дж/(м-с-К). 24.34.
9,1. 24.35. 29 Дж/с; 29 Вт. 24.36. а) 14,6 м2/с- 5,95-10-” моль/(см2Х
Хс) б) 146-1O-5 м2/с; 595-Ю''7 миль/(см2-с). в) 1,46-Ю-2 м2/с;
5,95-Ю 19 моль/(см2-с). 24.37. dr/dt= (S^f/Lp) (Mmff772n.) (J,9X
ХЮ-4 см/с, или <74 им за 0,5 мс.
Глава 25
25.1. 5,3 Ом"’-см2-моль'Г 25.2. 0,2063 см”. 25.3. 76,5 (Ом'”-см2Х
Хмоль’-1)/(моль/дм3). 25.4. а) 119,2 OirJ-си2-моль-’, б) 1Д92Х
ХЮ-3 Ом”-см в) 173,1 Ом. 25.5. 138,3 Ом-1-см2-моль-'. 25.6.
1,36-10-5 моль/дм3; 1,86-10“’°. 25.7. 387,9 Ом“г-см2-моль-”. 25.8.
0,03. 25.9. 3 22; 3,2.3. 25.10. 1,86-Ю-®; 4,73 . 25.11. 8,28 Ом-’-см2Х
Хмоль-1; 3,31-10 4 Ом”’-см-1; 623 Ом. 25.12. 4.01-Ю'4 см2/(с-В);
5,19-Ю“4 см2/(с-В); 7,62-Ю“4 см2/(с-В). 25.13. Lj+;4,01-ДН см/с,
~4 мип, 1,0‘Ю-6 см. 34 диаметра; Nar:5,19-10-3 см/с, -'-З мин,
1,3-10-® см 43 диаметра; К|:7,62-Ю“3 см/с, ~2 мин, 1,9-Ю-6 см,
63 диаметра. 25.14. t'jC=c+ «+,< и+. 25.15. 0,82; 0,003. 25.16. 0,48;
050. 25.17. 0 48, 0 52. 25.18. 7,46-Ю”4 см2/(с-В); 8,08-Ю-4 см7(с-В);
72,0 Om“j-см2-моль“); 78,0 Om“j-см2-моль-'. 25.20. 0,82; 0,18; 3,61Х
ХЮ'3 см2/(с-В); 7,92-10'4 см2/(с-В). 25.21. 0,407. 25.22. 3,75 МОм;
1,01-10”4; 14,0; 7,0. 25.23. 60,4 Ом"1 -см2-моль"1/(моль/дм3) 7»; 0,13/
/(моль/дм3)’.2; 76,8 Ом-1 - см2- моль-1/(моль-дм3) . 25.24. а) 1,24 -Ю4
Н/моль; 2,06-10"20 Н/молекула, б) 1,65-104 Н/моль; 2,75-10“2 ° Н/мо-
Ответы к задачам
569
легсула, в) 2,48*104 П/модь; 4,12*Ю-20 П/молекула. 25,25, а) 2,60 нм/
/с, б) 3.47 нм/с. в) 5,19 нм/с. 25,26, 420 пм. 25,27. 2.1 *10“” с, 25,28. а)
6,5‘Ю-5 м, б) 3,2*10-= м. 25,29, а) 235 с 962 с, б) 2.35* 104 с; 9,62Х
ХЮ4 с. 25,30 Ь%1,03*10%м%. 212 пм;’1\’а+:1,33* 10~5 см%, 161 tlM;
К+:1.96*10-5 см2/с, Ц2 пм; Rb+: 2,04* 10-5 см2/с, 107 пм. 25,35. а)
0, б) 0,016, в) 0,054. 25.37. 1,2*Ю“3 г;г/(м*с).
Глава 26
26.1. 1,4* 103 с; а) 504ммрт.ст., б) 688ммрт.ст. 26.2. а) дм3/(мольХ
Хе); дм6/(моль2*с), б) атм-!*с-’; атм-2*с,-[. 26.3. 2716 лет. 26.4. а)
0.64 мкг, б) 0,18мкг. 26.5. а) 0,09 моль/дм3, б) 0,05моль/дм3 (AcOEt).
26,6. 26.7. 26,8. 26.10. См. подразд. 26.2.А. 26.12. — (2"-'-1)/
/[(4/з)п-1— 1]. 26.13. Первый; 5,57 *10-4 с-‘. 26.14. Первый; 1,4*10 *
с~!; вероятно реагирует, 26,15, 241 кДж/моль; 1,1*1013с_г. 26,16, Пер-
вый; 7,2* 10%-’. 26,17, р= t= l/Po^'al
5— '/г. 26,18. Второй; 1.61 *10“3 (мм рт, ст.)-’*с_[; 3.1*106 с. 26,21,
Третий rro HCL; первый по пропану, 26.22, [НС1]3[СН3СН = СН2].
26,23, —18 кДж/мо.иъ,* 3 кДж/моль. 26.24. 1,14* Ю‘° дм3/(моль* с);
16,7 кДж/моль, 26,25, 66 кД?к/моль. 26,28, 1/Апабл= 1/Аг+ [H+j/fejAj.
26,29. рКа—13,7. 26.31. <7[СНД/<7/ —[СН3СНО] ;’а
d[CHsCHO]/d<=— Аа [СНзСНО] -Щк-JlkA ч* [СН3СНО] Ч 26.32,
1.9*1020 фотон/с; 3.1*10-4 ейпштсйп/с. 26.34. о.1*10® моль/(дм3*с).
26.36. Cl%Av—>2С1; С1+СПС13—^ССЕД-ПС!* СС13+С12^СС14-|-С1;
2ССЕ+СЕ—->2СС14. 26.37. (kjk?)^ [А]Х 26.38. Ф=2[А]/
/{(Аг/Аг) + [А]}. 26.40. 4,96*10= дм3/(моль* с). 26.42. 0,01 моль/дм3.
Глава 27
* 27.1. а) 6,0*Ю9 с-’; 7,4*Ю28 с-’*см-3; 1.8%, б) 3,0*109 сг[; 3,7*1028
с !*см-3: 1,8%. 27.2. ЛГР; ехр(-р%). 27.3. а) 2.44*10-3; 1.81*10%
9,02*10% 3,00*10% б) 7,62*10^ = ; 3.87*10“’% 3.57*10“l!; 5,98*10%
27.4. а) 30,1%: 13,4%; 4,8%; 1,2%; б) 301%; 134%; 48%; 12%.
27.5. а) 4,3*10-20 м2,' б) 0,14. 27.6. 0,20; 0,026 нм2. 27.7, 0,04;
4,0* 10-2’ м2. 27.8, 2,5*1011 дм3/(моль* с). 27,9. 4,4 нс. 27.10. а)
6.61*109 дм3/(моль*с), б) 3,00* 10’° дм3/(модь*с), в)1,97*109 дм3/
/(моль*с). 27.11. а) 0,24,6) 2,62. 27.12. а) 5,6915*Ю’3 с% б)
6,2124* 1012 с% в) 2.6528* 10гз с~‘. 27.13. т£ Ю29 дм% ?вр~ 102—10%
<?кол«1; 7эл«1. 27.16. а) к2 {Т)/кг (Н) = 1/20, б) *2[СО(1)]/
/к2 [СО(16) ] = 1,2; нет. 27.20. —47 Дя;/(К*моль). 27,21.-59 Дж/(КХ
Хмоль); —60 Дж/(К*моль). 27.22. Ar ~ (v3/v#,2)exp(—рД£); а) 2,7Х
Х10-” см2/с, б) 1,1*10~г9 см2/с. 27.24. 9,6 кДж/моль. 27.25. а)—19
Дж/(К*моль), б) 5,8 кДж/моль. в) 8,2*16’° дм3/(моль*с). 27.27. 88,2
кДж/моль; 17,8 Дж/(К*моль); 90,7 иДж/моль; 82.9 кДж/моль. 27.30.
(того же знака, как первый ион). 27.32. сЦСЙгЕ-ф'сДАг) =4,9.
37—242
570
Ответы к задачам
Глава 28
28.1. а) 1.44* J021 см^-с"1; 1,44-10*» см^-с.-1, б) 3.06-Ю20 см-2-с'1;
3,06-101а см-г-с-< 28.2. а) 1,61 • 1015 см"2, б) 1,14- Ю!3 с*г2; в) 1,86Х
ХЮ15 см-2. 28.3. 1,00-1015 см-2; 1,41-Ю15 см“г; 1.16-1015 см”2;
1,20-1015 «г3. 28.4 •'-Ир атом-1-с"1 для На прп 1 ми рт. ст.;
~105 атом-1-с-1 для пропана прп 1 ммрт.ст.: ~10-1 атом-1 -с-1 для Иг
при 10-7мм рт. ст,; ~10-г атом-1 -с 1 для пропана при 10-7 мм рт. ст.
28.3. а) 4,2-Ю-11 с, б) 1.9-101® с (600000 лет). 28.6. а) 6,1-10-’® с,
б) 7,3-10_6 с. 28.7. При 298 К. td=1,80 тц; при 1000 К, Тн=1.52 тц-
28.8. 611 кДж/моль; 1 07-Ю-’3 с. 28.9. 3,7 кДж/моль. 28.12. p/Vn =
=p/H+l/KVS; In (0/р) »]пК-0, если 0^1;-1/Г®. 28.13.0,52
(мм рт. ст.)~1, 0,48 см3. 28.16. Ленгмюр; 0,017 (мм рт. ст.)-’; 0,13 нм2.
28.17. 0,18 см®. 28.18. Нет. 28.20. к2р’^1 [Ц- (Крр)А{. 28.22.
—12,7 кДж/моль. 28.23.700 кДж/моль; а) 2,1-10104 мин, б) 50 мкс-
28.24. Тесная: 180 кДж/моль, 3-10-11 с; рыхлая: 64 кДж/моль, 1,1 X
Х10-й с. 28.25. Нулевой. 28.27. 17(f) =fcc-|-F(f): 0,02 мм рт. ет./с.
28.28. G(t) = (fcc/K) + {(l+X'pn)/A‘po}F(t). 28.29. 0,033 (ммрт.ст.)-1,
4Д.-10-4 с '.28.31. 382 пм. 28.32. 0,16 ; 2,4. 28.35. <7р'= (ЯГП/а) X
Хй[]п(1-0)]=-{(ЛГ/(т)/(1-6)}с1Иа.
Глава 29
29.2. /“(.V, результирующий) = 17 {ехр (| z_|paF/ff7) — ехр(— | z-|r|X'
Х(1-«)Г/НТ)}. 29.4. а) -0.31 мА/см\ б) -5,41 мА/см2. в)
1.39-1042 мА/см2. 29.7. -107 мВ. 29.8. 4,93 ДО15 см"2-с-’; 1.56-Ю1&
см'2-с-’; 3.12-Ю7 см~2-с-!. 29.9. 3,8 с"1; 12 с.-’; 2,4-10~® ст1. 29.10.
UT/Sjt. z+F; а) 32.5 Ом. б) 3,3-1610 Ом. 29.11. <(1/4) (2а—
-1) • (г+Л/ЙТ)2/:^. 29.12. -7,2мкА. 29.14. 0,38; 0.78мА/см2. 29.17.
Когда Е падает ниже 0,34 В, Си осаждается до тех пор, пока сохра-
няется предельная плотпостг, тока; затем разность потенциалов пада-
ет, пока не начпет осаждаться ццпк при Е ниже —0,76 В. 29.18. а)
—0,76 В; —0.82 В, б) 0,34 В; 0,28 В. 29.20. Протекает только 0,14 мА/
/см2, что позволяет пренебречь выделением водорода, пока потенциал
ас упадет до —0,76 В, поэтому iuihi; может осаждаться. 29.21. Будет
протекать 2,2-10-’ .А/см2. понтону происходи г питеяситгое выделе-
ние водорода, прежде чем сможет отложиться цинк. 29.22. Должен
быть тон 5,3-10® А/см2, который вызовет плтепснвлое выделение во-
дорода: поэтому магний не будет отлагаться. 29.23. j~~=jt (1 —
— expfFpc/ffT’)}. 29.24. 0.25 мм. 29.25. /2 = (ЯГ/z+F) (Х+/Х)с,г .29.26.
0,23 мм при всех концентрациях. 29.27. a(Sn^/^Pb2-) ^2.2. 29.30*
а) 1,23 В, б) 1,060 В. 29.35. Ее. 29.39. 5,6 мкА.
Приложение
Полезные соотношения
RT (при 298,15 К) =2,4789 кДж/моль
RT/F (при 298,15 К)-0,025693 В
Т, К; 100 298 500 1000
Дем-1: 69,1 206 346 691
hdk= 1,43878-10-2 м-К
1 атм = 1,01325-105 Н/мг
1 мм рт, ст. = 133,322 Н/м2
1 эВ= 1,602189-НТ19 Д?к= 1000 см'1 = 1,986-10-20 Дж =
= 96,486 кДж/моль = =11,96 кДж/моль = 0,1240 эВ
= 8065,5 см'(
1 кал (термохимическая) =4,184 Дж
1 дсбай —3„33564 • 10-30 Кл-м
1 Н= 1 Дж'м = 105 дин
1 Дж= 107 эрг
1 Вт=1 Дж/с
Префиксы
П у [I мк м с пико нано микро милли сапти 10-12 10-9 Ю-З JQ-2 Фундаментальные постоянные д деди 1О”1 к М Г кило мега гига 10s 10s 109
Скорость света с Заряд протона а е Число Фарадея F — Le Постоянная Больц- мана k Газовая постоянная R-Lk Постоянная Планка h h~h/2n Число Авогадро 1. 2,997925-10« м/с 1,60219-10~19 Кл 9,64846-ю4 Кл/моль 1,38066-10-23 Дж/К 8,31441 Дж/(К-моль) 1,98717 кал/(К-моль) 8.20575 -10-2 дм6-атм/(Кх ХМОЛЬ) 6.62618-10-34 Дж-с 1,05459-10-34 Дж-с 6,02205-1023 моль-1
а Заряд электрона равен —t).
37:
572
Приложение
Единица атомной
массы
Масса электрона
Масса протона
Масса нейтрона
Проницаемость ва-
куума
Восприимчивость ва-
куума
Магнетон Бора
Ядерный магнетон
Гравитационная по-
стоянная
Радиус Бора
ц = 10“3 кг/
/(L-моль)
те
т?
тп
ео = Ц^С2
Ио
Нв = е^/2/пв
PN = e^/2/np
G
«о
1,66057-Ю”27 кг
9,10953 -10-31 кг
1,67265-10“2Гкг
1,67495-ю-5’ кг
8,854188-Ю”12 Кл2/(Дж-м)
4я-10-7 Дж-с7(Кл2*м)
9,27408-10-54 А-м5
5,05082-10“27 А-м2
6,6720-10'11 Н-м2/кгг
5,29177-10"11 м
Массы (/м, а. е. м.)а и природное содержание (в % )6 некоторых изотопов
И
'Н(99,985)1,0078
Н(0,015)2,0141
Г Li В 1 c
sLi(7,42)6.O151 '’BOS,78)10,0129 I2C(98,89) 12.0000
Lj(92,58)7,0160 nB(80, 22)11,0931 I3C(1,11)13,0034
ь. - ... > I __ J
Na 15Na(100)22,9898
N 1 I4N(99,63)14,0031 I4N (0,37) 15,0001 k > 0 ,6O(99,76)15,9949 ”0(0,037)16.9991 . ISO(0,204)l 7,9992,
P !IP(100)30,97 38 s > S 3:S(95,0)31,9721 '3S(0,76)32,9715 ,’S(4,22)33,9679 36S(0,014)35,9671 j
He 3He(0,00013)3,0160 “НеО 00)4,0026
F 1 ieF(l 00)18,9984 Ne loNe(90,92)19,9924 :INc(O,25 7)20,9938 55Nc(8,82)21.9914
Cl 45C1 (7 5,53)34,9688 37CI (24,4)36,9651 * > Ar 16Ar(0,337)35.9675 зяАг(0,063)37,9627 inAr(99,60)39,9623
Br 79Br(5o,54)78,9183 SIBr(49,46)80.9163 ь - - . Kr 7sKr(0,35)77,9204 soKr(2,27)79,9164 eIKr(l 1,56)81,9135 ®3Kr(ll, 55)82,9141 SJKr(56,90)83,9115 Rt,Kr( 17,37)85,9106^
I
J27I(100)126,9045 '
‘ 1 а. е. м. = 1,6605-10~27 кг.
6 Числа в скобках.
Периодическая таблица
IA JIA IIIA IVA VA VIA VIIA VIII IE HE HIE IVE VE VIE VUE 0
iH 1.00S 2He 4,003
sLi 4 Be 9,012 В 10,81 X 12,01 7N 14,01 80 16,00 19,00 n)Ne 20,18
uNa 22,90 liMg 24,31 (зА1 26,98 ijSi 28,09 isP 3(1,97 ihS 32,06 l7CI 35.45 isA 19,95
ioK 39, 111 2oCa 40,08 21SC 44p96 2zTi 47,90 23V 5ii,94 24СГ 52,00 isMn 54,94 »Fe 55,85 2?Co 58.93 2«Ni 58,71 2vCu 63,55 juZn 65,17 3iGa 69,7^ 32 Ge 72,50 33 As 74,92 .uSe 78,96 «Вг 79,90 ЗбКг 83,80
37Kb R\4? 3sSr 87,62 ,19V 8H,9l 4(]Zr 91,22 4lNb 92,91 j;MO 95,94 4ДС 98,91 44Ru 101,1 45Rb 102,9 4,Pd 106,4 4?Ag 107,9 48Cd 112,4 49ft! lU,S srjSn 118,7 slSb l2l,8 «Те 127,6 126,9 54Xe 131,1
ssGs 132,9 5hB« 137,3 5?Ьй 138,9 ?2Hf 178,5 7 Да l8t>,S 74W 183,9 7sRe 186,2 76Os 190,2 7>1Г 192,2 7sPt 19S; 1 79 AU 197,0 S!>Hg 2 Oil,6 H.Tl 204,4 азРЬ 207,2 83®’ 209,0 84PO (210) 85ai (210) fi6Rn (222)
a?Fr (22.1) 68 Ra 226,0 69 Ac (=27}
Лаятаноиды 5?Lfl 138 9 .s»Ce 1411,1 5!lPr 140,9 6WNd 144>3 fiiPm (147) 15(1,4 6.3Е11 1-52,0 6jGd 157,3 65Tb 158,9 6* Dy 167,5 Л7Н0 164,9 6ft Er 167,3 6pTw> l6B,9 7^Vb 173,0 ZjLU 175,0
Актиноиды 89 At (2271 wTh 212,11 4iPa 231,0 42U 238.0 ^3Np 237 0 94₽U (5421 Am (24.31 9fiCm (24Я) 9?Bk {2471 98d (251) yyEs (254 l inciFni (253} iuiMd (256) 102N0 (254) io?Lw (2571
Предметный указатель
Абсолютная конфигурация молекул 268
Адсорбция на поверхностях 502
Акролеин 52Ъ
Активированный комплекс 467
Активность 476
каf^литическая 514
оптическая 299
Альдольная конденсация 441
А^г^р^очгечносгь
Анноны 366
Ансамбль 165
бскпьшой канонический 166
канонический 166, 170
мцкроканоимческий 166
Антиферромагнетизм 325
Аррс^;«усощ.'хий гр^фях 4JS
Атомный зонд 500
Аутокатализ 415
БазИс 22
Батарея 553
Биномиальное распределение иитепсивнп-
стн |39
Больцманевскос распределение заселенно-
стей JB7
Вероятность прилипания 507
Вес распределения [73
Весы -П/а 323
Встиь
Q 86-86
О 88
7’ 86
Я 86
S 88
Вещества
диамагнитные 323
парамагнитные 323
Вз а им оде н ст ни «
диполь-дипольное 14Я 302
диполь — н>1Дупнрован||Ь|й ди31О4-гь 303
кулоновское 287
между двумя индуцированными диноли-
мн 304
спин-спи но вое 150
Взаимодействия
|1<ждсрваальсовы 302
тройные 320
Взрывной предел'
второй 435
первый 435
третий 435
Взрывы 434
Винная кислота 268
Винтовая дислокация 494
Влриальяые коэффициенты 314
Вклад
пре sta тельный
колебательный 209
поступательный 206
эЛ &кт ро и 11 ы i) ? 10
Возбужденная молекула 430
Восприимчивость
диамагнитная 330
мпгидтняя 322, 324
ппрнмагнитная 524
электрическая 290
Время
поперечной релаксации 144
^и«-решст<1Чцой релаксации L44
Выделение газов 545
Вырождение 180
Вязкость 349, 351, 360, 464
газов 357
жидкостей 321
Гальванизация 559
Г армоцнки
вторые 83
первые 83
Гашение 403
ГпдриронаНие 515, Ь19
Гидродинамический радиус 374
Г Г гн ном етр 255
График Тафедя 637
Групповое свойство 13, 19
AaH.ietiHe 213, 333
жидкости 319
системы 183
Дебай 286
Дегидрирование этана 433
Дезактивация при столкновении 67
Дейтерирование 473
Десорбция 501
Детерминант 5i
Дефекты поверхности 493
Диамагнетизм 323
Динамика молекулярных реакций 456
Динамическая электрохимия 526
Диполь 286
Дниольпын мом?нт 285, 287, 293
магнитный 323
перехода 37, 62
trocTOMfiiiHfi 61, 286
средниЛ 289
электрический 15
Дисперсия 299
оптического вращении 300
Диссоциация ц7
Дифракции 240
нейтронов 270
рентгеновских лучей 245
электронон 271
медлевиых 273. 498
Диффузионный слой Нернста 541
Диффузия 381, 385, 389
Дли но
полны света 300
рассеяния атомов 250
связи 34
Доплеровское уширение спектральных ли-
нии 63
Емкость 288
Жертвенный аноа. 559
Жидкий кристалл 320
нематический 320
смектический 320
576
Предметный указатель
Закон
£>ера — Ламберта 101
Греттгуса— Драперр 436
истечения Грзла 352
Кюри 323
независимой миграции ионов Кольраби
369
разведения Оствальда 371
рациональных ннДексоа 242
скоростей .Лрренцуся 403
Эйнштейна — Штарка 437
Закины
диффузии Фй/сд 382
второй 387
первый 350
скоростей 4О6, 408, 4 [5
термодинамики
второй 188
третий 191
Заселенность 59
инверсная 116
молекулярных сосгояней 183
равновесная [16
Зрительное восприятие 107
Идентичность 7
Измерение а. д. с. 395
Изомерный сдвиг 157
Изоморфное замещение 258
Изотерма 511
адсорбции 509
— Элл era — Теллера 523
Ленгмюра 509
Тздкича 513
Фрейндлиха 513, 523
Инверсия 9
циклогексена [52
Ингнбироваттие 432
коррозии 659
Индексация рефлексов 248
Индексы
Вейсса 243
Л4цллс.*р« 242
Инициирование 432
Интегралы 340
стремящиеся к нулю 31
Интенсивность
рентгеновских рефлексов 249
спектральных линий 59
Инфракрасная хемилюминесценция 479
Ионная атмосфера 378
Испускание
у-лучей [S4
спонтанное 65
стимулированное 65
Истечение 349
Кальцит 48
Камера
Вайссенберга 257
осцилляцнонпчя рентгеновская 266
Карбонильная группа [06—108
Карбонильное соединения 106
Катализ 439, 5!4
кнслотный 441
основный 441
Катализатор 439. 518
гетеро! ецрый 440
гомогенный 440
двухфуккциональцыА 521
Катионы 366
Катодная защита 559, 569
Квантовая статистика 196
Квантовые числа 70
Квантовый выход 437
К*арц 48, 300
Кинетика коррозии 556
Кинетическая модель идеального гааа 332
Кинетическая теория газов 331
Класс кристалла 22, 41, 42
Клистрон 134
Колебание
активное в ИК-спектре 91
в спектре КР 92
валентное*
антисимметричное 90
симметричное 90
неактивное в ИК-спектре 91
в спектре КР 93
Колебания
ди уха томных молекул 78
метана 91
многоатомных молекул 88
Колебательные частоты 84, 93
Количество движения 333, 357
Коммутативные {абелевы) группы 19
Комплексы переходных металлов 104
окраска 104
Конверсия
внутренняя 118
кнтеркомбкнациоц^ая 114
Константа
Михаил ад 428
равновесия 228, 421, 476
сверхтонкого взаимодействия 138, 14)
скорости 406. 458
реакции 406
Координата реакции 466
Координаты 28
центр3 масс 484
Коррозионная ряэцость потенциалов 557
Коррозионный ток 557
Коррозия 554
Коэффициент
активности 476
вязкости 35с
диффузии 850, 354. 384, 464
ПОГЛОЩЕНИЯ 101
интегральный 102
теплопроводности 350, 356
трансмиссионный 468
65, 6$
экстинкции 101
Крекинг 520
Кристаллическая решетка 242
вюрцита 267
хлорнДа натрия 247, 266
Неяия 266
Кристаллическая система 40, 4J
кубическая 41
моноклинная 41
Кристаллическая структур,! 252
льдр 264
Кристаллы
ионные 266
металлов 263
Лимптирующая стадия реакции 424
Линии
аитнетоксовы 74, 77
стоксовы 74, 76
Лгжение 55?
Mei гнетши
5орн 324
ядертшй J 45
Предметный указатель
577
Магнитные свойства молекул 322
Магнитный момент
и н ду цнро^л и н h| 11 325
1|остояннм11 323
спиновый J33, 324
ядерный 137
Матрица 48
блок-днагонадьная 26, 29
диагональная 51
единитпяя jj
кнадрзтная 50
сингулярная 51
транспонированная 51
Мера интенсивности переходов ЮГ
Мессбауэровский спектр FeSO* [57
Метод
Гщмсное 259
Брэгги 246
вращающегося сектора 453
выделения Остялльда 412
Гитторфп 395. 397
движущееся границы 394, 397
Дебня — Шеррери 248
Кмг/JceHu 352
колеблющихся дисков 360
Лог/э 246
молекулярных пучков 47S
Morfrc-JG?p.rjQ 483
неопределенных множителей [75, 194
остапозленной струи 444
порошка 24?
прецессионный 260
проб ц ошибок 258
скачка Давлении 447
т«мпераТурного скачка 446
Методы
Дифракционное 239
доводки 260
прямые 259
редок JjjCtfwe 232
релаксационные 445
Струевые 403, 444
Методы контроля реакций
изменение Давления 402
масс-сиектромстрНя 403
поляриметрий 403
спектроскопия 402
хроматография 403
электрохимические 4ОЗ
гарпунный 460
Поляризационный (Ц
Райса — Гер^фелыЗа 434, 452
реакции 401
Микроскопия
Полевая ионизационная 499
эмиссионная 498
Множитель
Прсдэкснонеициа.тнный 417, 459, 4111
— fC&ud&aa Hl
Модель
Гельмгольца 528
Гун Йеллена: 529
Штесна 529
Молскч’лы
линейные 68. 7?
неразличимые [8[
различимые 18[
тина симметричного волчка 70
сферического нолике 63
Молекчлярносуь реакции 416
Молекулярный пучок 310, 478
Молекулярный транспорт 38[
Молярная рефракция 297
Моменты инерции 69
Морфология 40
Накачка 117
Намагяичнвасмость 323
Намагничивание 322
Начальные наклоны 407
Ненулевое церекрывапне 34
Hevnpyroe рассеяние нейтронов 321
(Гормальпые моды 90
Обозначения плоскостей 242
Обрыв Цепи 4З2
Одномерное беспорядочное движение 390
Окисление 520
Операции симметрии 7
Опыт Янга по щелевой дифракции 240. 241
Орбитами 34, 35
Снмметризованные 38
с ненулевым перекрыванием 34
Осаждение металлов 545
Ось
винтовая 46
зеркальяо-поворотцвя 9
симметрии 7
Относительная Диэлектрическая проанцвС’
мость 287. 289
Огтя0аняс 395
Пэря^Дгнстиам 323. 326
Параметр Р 183
Пассивация 659
Перенапряжение 527, 536
концентрационное 549
экспериментальное измерение 538
элемента 545
Переносчики цепи 434
ПерахВатчНК радикалов 432
Переход
вертикальный j J0
внброннЕл'1 ] 05
запрещенный 69
разрешенный 60
с переносим ээряда Ю&
(Л*, Л1 ]66
п) 106
Переходы
вращательные 60, 72
колебательные 6S
электронное 62, 98
Плоскость
Г ельмгвльца
внешняя 528
внутренняя 533
зеркальца^ 8
симметрии 8
скольжения 46
Плотнейшая упаковка 263
Iекснтональная 263
кубическая 255
объемно-центрированная 266
Плотность
млгцитцого потока 322
неспаренного электрона 142
оптическая 101
TOK.T 527. 534
обмена 536
предельного 5-12
электронная 254, 276
энергии когеанн 329
Поверхности
кристаллические 493
методы изучения 497
Првтяження 485
отталкивания 485
чистые 497
578
Предметный указатель
Поверхностная плотность заряда 288
Поверхность потенциальной энергии 479
Поглощение у-л у чей [54
.Подвижность
жидкостей 32 [
ионов 372, 375
молекул на поверхности 508
Показатель преломления 296
Поле
.тональное
м а га [itiidc [37, [47
электрическое 290
силовое 93
Полоса Шумана—Рунге [30
Лолулериод реакции 413
Поляризация
диэлектрика 289
искаженна 29$
концентрационная 540
э-тектродон 367
Поляризуемость 286, 292t 293
анизотропная 74
изотропная 75
мо.тярваи 293, 338
при высоких частотах 2^6
электронная 296
Поляриметрии 403
Т[олярогрифия 542
Порядок
ближний 318
дальний 317
Порядок реакции 406, 418
второй 41)6
общ [Hi 466
первый 406
половинной 406
третий 406
Постоянная
ангармоничности 8[
враи|ательнея 68, 85
Маделунга 308, 309
экранировали* 147
ячейки 368 , 396
Потенпип-т
ппешннй (Вольта) S3I)
внутренний (Галкали) БЗО
нонизаалн 1 [9
</7<ш нарда-Джонса 306
поверхностный 530
полуволны 542, 544
химический 38[
элемента а46
Поток 349, 463
тнь^да 376
11ЫГ)ТОНОИСКИЙ 351
Провидя отбора 36„ 6р, 74, 87
вращательные 72
колебательное 8р 9[
специфические 62, 73, 75
Правило
Вальдема 3&5
исключения 93
отбора
Лапорта 105
основное 62, 72
отношет|ня радиусов 266
<'«лтсоН17 316
Преаднссоциация 1 [7
Предея
диссоциационный 117
классический 218
термодинамический 168, 171
Предравновесие 425
Представление 20
мхи личное 21
неприводимое 23
приводимое 25
Приближение
линейное 483
стацноварвости 424
Стирлинга 175. 391
Приведенная масса 80, 90, 346
Принцип
Пццли у4
рлнеистна априорных вероятностей 167
равного распределения 216, 2[9
Франка — Кондона 1U8
Проблема фазы 257
Проводимость
термическая 349
электрическая 349
Проницаемость вакуума 322
Пространственная группа 15, 46
Профиль потенциальной зкергки кемозарО'
ции 504
Процессы
каталитические 517
электродные 527
.электрохимические 545
Прямая сумма 24
Работа [39
лазера 115
Радиусы
рэндерваальсовы 268
ионные 267
ковалентные 274
Развитие цени 432
Разложение
азометана 409
ацетальдегида 418
фосфина 5i6
Развость
заселенностей ИЗ
потенциалов Гальвана 531
Распад
безызлучательный J13
излучательный Г[2
радиоактивный 422
Распределение [66
каноническое ]68
наиболее вероятное [70, 171
Распределение скоростей 338, 342
Максвелла 342
Максвелла—^ольцлана 341
Рассеяние
аномальное 269
магнитное 270
нормальное 269
Ореоль]|Ое 3[3
радужное 314
Расстояние
среднее 387
среднеквадратичное 388
Реакции
бимолекулярные 416
быстрые 4^2
п растворе 476
второго порядка 410, 4’2
ктоиомолекулярные 416
нулевого порядка 5 [6
первого порядка 408, 409, 428
гсосдсд<№;гг(*дъюле 422
простые 116
Нсевдопервого порядка 412
третьего порядка 408, 426
ферментативные 426
фотохимические 435
цепные [3 [
Реакция
аэоднкарбонатного понн 499
Предметный указатель
579
аутокаталитическая 4-Ю
бромфсцо^|омого синего 490
водорода с бромом 432, 438
с кислородом 434
К/шнзе/ш 441
контролируемая активацией 463
диффузией 463
поверхностная 515
Резонанс 133
нейтронный Спиновый [62
электронный парамагнитный 133
ядерный магнитный 144
Релаксация [43
диэлектрическая 296
нс>переЧйая I44
енцп-ренгеточкая (продольная) ] Н
Р снтг с нов с ка я кри с т fi л ло гр аф и я г? 1л
PeHTyeRbCTfryxirypHhiii а на л на 266
Ретиналь
полностью транс 107
11-/4^ М7
Рефлексы
второго порядка 248
первого порядка 248
PcijsefitH 42
£>/?ыв.9 45, 4?
граЕ|Сце;|грнровац|||4с 45
ионные 266, 367
магнитно-уноридоченные 271
оНт.еМ||о-ЦС||грировлн1|ыс 45
примитивные 45
Риформинг 520
Рост кристаллов 493
Рутеноцен 15
Сверхвысокий вакуум 498
Сверхполяризуемость 292
Сверхтонкое взаимодействие [37
ДИ[К>Л Ь-АНПО ЛЬ||ОС 140
контактное Ферми i ll
С ве г
линейио-поляризоваппый 299
циркулярам лево)поляризованный 299
циркулярное прево) поляризованный 9’J9
Свинцовый аккумулятор 554
Свободные радикалы 431
Свойства
аддитивные 301
двухатомных молекул 81
Коллигативные 372
кристаллов 48
транспортные *132, 348, 3,56, 360, 393
пДерных Синцов 145
Связь
водоргцДная 203
двонв^н [06
ионная 262
ковалентная 262
мсгадлнчссквп 263
мостиковая 506
Ссчспне
рассеяния, дифференциальное Зп
релкцни <59
столкновения 345. 347
Сила
осциллятора 162, 129
термодинамическая 382
Силы
Дисперсионные 304
межмодек^лярные 302. 3(4
Симметрпя 5
вращательная i4
кристаллов 40
орбиталей 36
трансляционная 46
Синтез
/7дтг4?рс/.)ч.а: 259
формальдегида 439
Система |65
Германа — Модема 10, 11
координат центра масс 484
Шёнфлиса lO, Н
Систематические пропуски 248
Скорость
десорбции 505
диффузии 353
Дрейфе 373
звука 238
изменения количества движения .444
истечения 352
пеги иная 4115
каталитической реакции Ы6
коррозия 557
молекул гзза 334, 344
наиболее вероятная 314
переноса яарида 531
рсакпии 464
среднеквадратичная 334, 344
средння 344
утечки 362
электродных процессов 530
Слияние Структуры [53
Слоевая линия 2&6
Сложение матриц 50
Смещение Доллерд 63, 155
Сольватация 374
Соотношение
Иернста — ЭДмшгейнд 383, 394
/7л«чека — Теллера 97
Сгояса - 3£гншгщ}«д 384. 394
А/тедоа — Фольмера 452
Эйнштейна 383, 394
сМишгсинд — 302, З'Л
Состояние
активированное 431
возбужденное [12
осионНос 58
переходное 466
синглетное IН
стационарное 422
сринлет)|ос ] [4
фотостационарное 463
Спектр
бензофенона 129
диапстила LX)
испускания 57
нафталина 129
поглощения л8
хлорофилла 99
флуоресценции 1]^
электромагнитный 58
ЭПР ;щкон-ра,|ик ал а бензола [36, 142
ЯГЧР этанола 146
Сп&ктримргр
гаммч-резои«Н1С'1Ь|й ]56
Фотоэлектронный 122
ЭПР 135 1
Спектроскопия 57
испускании 57
ком()ияационнсп'о рассеянии 58
мессбауэровская Ю4
микроволновая 76
58
реггтгепо.члектронкам ]2[
фотоэлектронная 99, Ц9
электронная 99
Спектры
вращательН]1Ге 287
И К 82, 93
колебательно-пр лШ-ртелЫШе 84, ЯР?
колебательные
580
Предметный указатель
двухатомных молекул 81
лгиогоа томных молен ул 91
линейчатые 84
переноса заряда 129
полосатые 84
Спектры КР 68
вращательные 74
колебательные
двухатомных молекул 87
многоатомных молекул 92
Спиральный рост 495
Способность
рассеивающая 251
хсмосорбцяоннак 5Т9
Среднеквадратичное отклонение 219
Средняя длина свободного пробега 332,
347
Средняя энергия 216
вращательная 2] 7
колебательная 218
поступательная 217
системы 169
Статистика
£оае ~ Зйнштейня 197
Ферми — Дирака т99
Сглтистико-термодннамичсскпс соотноше-
ния 215
Статистическая термодинамика 164, 204
Степень
адсорбции 507
заполнения 508
ыонизацпн 371
превращения 105
свободы 72т 89
Столбцовые векторы 52
Столкновения 345т 418
меж молекулярные 345т 457
со стенками в поверхностями 348
Структура
групповая ]7
жидкостей 317
кварца 300
колебательная т08
решетки алмаза 264
сверл'гошсая 137
спиральная 300
тонкая 148
фталопизцииа никеля 26т
хлорида калия 219
Субстрат 426
Таблица группового умножения 18
Таблицы характеров 27, 31
Температура
Кюри 325
Пееля 325
системы 20]
Теорема /О/ллдиса 125
Теория
активированного комплекса 466
групп 6, 18
Линдсло^а 428
столкновений 457
Теплоемкость 220, 224
затрудненного ротора 223
при ниэкЕтх тем перату пах 223
Теплопроводность 350, 355
Теплота т89
Термическая деградация 112
Термический взрыв 434
Термодинамика коррозии 555
Термодинамические свойства системы у 71
Топленные элементы 551, 553
Точечная группа симметрии 10-45
Точка
иэобестическая 130
поворота J03
седловикная 48]
Транспорт ионов 366
Трансформационные свойства функция 28
Треугольник Паскаля т40
ТрехСЕупенчатая лазерная система т16
Угловое распределение продуктов 479, 484
Ударный параметр 311
Удельное сопротивление 367
Узел
радиальный 62
угловой 62
Умножение матриц 20, 50
Уравнение
Батлера — Фольмера 536
Бирля 273
Дебад 293
Дебая — Хюккеля — Онзагера 379
диффузии 387
Лдмсоеича 544
Кладощ/са — Лосоттн 293
.-Иан "Коннелл а 142
И ОСО ГТ к 328
Саякира — Тетрода т93
Шредингера 179
Зйранга 471
Уравнения Крайтчлана 96
Условие
Брэгга 241. 276
резонанса 133, 136
Уширение за время жизни 64Т Т43
Фактор
S 136
У 459. 465, 472т 475
симметрии 532, 536
структурный 252
Фарадеевское выпрамление 561
Фермент 425
Ферромагнетизм 325
Ферроцен 15, 272, 273
Физисорбция 502
Флеш-фотолиз 442
Флуктуации т?0, 220, 225
Флуоресценция 112
Форма линий 142, 152
Формула
Лондона 305
Пуазейля 359
Стокса 373
Фосфоресценция 1]4
Фотография
Байссенберг.а 255
осцилляциейпая 255
Фогонна1Е картина рефлекса 274
Фотосенсибилнзацня 439
Фотохимические процессы в атмосфере 435
Фотоэлектроны 12]
Функами радиального распределения 317
Функция
акгинацин Гиббса 474, 532
Гельмгольца 213
Гиббса 2Т4
.Танждеека 291
ошЕ]бок ЗЮ, 389
первого распределения 317
распределения г 77. »86, 205
вращательная 206
каноническая 177
колебательная 209
молекулярная 180
общая 212
Предметный указатель
581
пестунательная 1st, 206
электронная 210
Фурье-преобразование 278
Фурье-си hi ез 264
Характер 22
операций симметрии 22
Характеристики поглощения 102
Хемосорбированные частицы 503
ХемосорбЦитЕ 602, 503
Химическая актинометрия 438, 462
Химическая динамика 402
Химический обмен 163
Химический сдвиг 147
Хлорбензол 301
Хлорофилл 09
Хромофоры 104
Центр
инверсии 9
симметрии 9
Цинкован обманка 267
Частота столкновений 332, 345, 418, 457
Четность 105
Четырехкружный дифрактометр 257
Число
волиопое 57
координационное 265
переноса 377, 394, 397
симметрии 207,234
Шприца спектральных линий G2
Эйнштейн 438
Экниналентяые Протоны т39т i50
Эксперимент по спектроскопии Мессбауэра
155
Электрические свойства молекул 286
Электрический двойной слой 528
Электрическое квадруполь ное расщепление
158
Электрод
вращающийся дисковый 541
дкекольуй с кольцом 644
капающий ртутный 542
ненвляризуемый 539
Поляризуемый 540
рабочий 638
сравнения 539
Электролиты
сильные {ионофоры) 369
слабые (ионогены) 370
Электропроводность 367, 378
воды 374
ноянан 370
мольная 368, 377
удельная 367
эквивалентная 368, 370
Элементарная ячейка 42ь 242
грансцентрированная 45
объсмно-це|1тРнРОва иная 45
примитивная 45
Элемент
никель-кадмиевый 564
с переносом 395
Элементы симметрии 6
Эллигеометрия 498
Энантиомер 268
Энергетические уровни
вращательные 67
колебательные 80
Энергия
эктинацни J53. 403, 418, 457, 459, 461
Десорбции 504
катализа 514
внутренняя 178, 2j3. 399
диссоциации 84
потенциальная 79
Морзя 81
разрыва связи 84
решетки 309
среднеквадратичная 219
Эдтальгтоя 224
адсорбции 502
изостерическая 511
активации 474
Энтропия 213
активации 474
одноатомного газа 193
остаточная 225
системы 169
статнстнческап 189
Эргодическая гипотеза j67
Эффект
Вина 381
второй 381
первый 38т
Дебел — Фалысенгаге^а 380
кннетнчсскнй изотопный 474
солевой 476
Мессбауэра j55
релаксационный 378
Штерна 237
электрофоретический 379
Эффективность упаковки 265, 281
Ячейка
для измерения электро г, роводиостн 367
363
Содержание
ЧАСТЬ 2. СТРУКТУРА.................................................. 5
16. Симметрия. Описание и следствия.................................. 5
Изучаемые вопросы..............................................
Введение ......................................................... "
16.1. Элементы симметрии объекта.................................. «
16.2. Группы, представления и характеры......................... 17
16.2. Использование таблиц характеров.............................^1
16.4. Симметрия кристаллов.....................................
Приложение. Матрицы............................................
Литература................................................
Задачи ........................................................... м
17. Определение структуры молекул. Вращательные и колебательные
спектры.............................................................. 56
Изучаемые вопросы.................................................56
Введение..........................................................56
17.1. Общие особенности спектроскопии.............................57
17.2. Чисто вращательные спектры..................................67
17.3. Колебания двухатомных молекул...............................76
17.4. Колебания многоатомных молекул..............................88
Литература.....................................................
Задачи.........................................................
18. Определение структуры молекул. Электрпниая спектроскопия 98
Изучаемые вопросу.................................................98
Введение..........................................................98
18.1. Мера интенсивности.........................................'01
18.2. Хромофоры..................................................104
18.3. Колебательная структура и принцип Франка—Кондона . . 108
18.4. Судьба электронно-возбужденных состояний..................112,
18.5. Фотоэлектронная спектроскопия..............................119
Литература...................................................... 125
Задачи...........................................................126
19. Определение структуры молекул. Резонансные методы 132
Изучаемые вопросы................................................132
Введение.........................................................132
19.1. Электронный парамагнитный резонанс.........................133
19.2. Ядсрный магнитный резонанс.................................144
19.3. Мессбауэровская спектроскопия..............................154
Литература..................................................... 159
Задачи...........................................................160
20. Статистическая термодинамика. Концепции..........................164
Изучаемые вопросы................................................164
Введение........................................................ 164
20.1. Системы, ансамбли и распределения..........................165
20.2. Функции распределения......................................177
20.3. Статистическая термодинамика и второй закон . . . . 188
Приложение 20.Л. Неопределенные множители........................194
Приложение 20 Б. Квантовая статистика............................196
Литература.......................................................200
‘ Задачи.............................................................200
Содержание
583
21, Статистическая термодинамика. Развитие концепций
Изучаемые вопросы............................................
Введение ................................................
21.1. Как рассчитать функции распределения? ....
21.2. Как рассчитать термодинамические функции?
21.3. Применение статистической термодинамики ....
Литература . ............................................
Задачи ..................................................
22. Определение структуры мплекул. Дифракционные методы
Изучаемые вопросы........................................
Введение.................................................
22.1. Общие особенности дифракции........................
22.2. Кристаллические решстки...........................
22.3. Рентгеновская кристаллография.....................
22.4. Информация, получаемая и.з рентгеноструктурного анализа
22.5. Дифракция нейтронов...............................
22.6. Дпфракпия электронов . ..........................
Приложение. Дифракция как свойство частиц................
.Литература..............................................
Задачи ..................................................
23, Электрические и магнитные свойства молекул...............
Изучаемые вопросы........................................
Введение.................................................
23.1. Электрические свойства............................
23.2. Межмолекулярпые силы..............................
23.3. Роль межмолекулярнык сил..........................
2.3.4. Магнитные свойства................................
Литература..........................................
Задачи . ...........................................
ЧАСТЬ 3. ИЗМЕНЕНИЕ..........................................
24. Молекулы в движении. Кинетическая теория газов
Изучаемые вопросы.......................................
Введение................................................
24.1. Основные расчеты..................................
24.2. Столкновения......................................
24.3. Транспортные свойства.............................
Литература..............................................
. Задачи ......................... .........................
201
204
204
20.5
213
216
233
233
239
239
239
240
242
245
262
270
271
274
279
280
285
285
286
286
ЭД2
307
322
326
327
331
33i
331
332
333
345
348
361
361
25. Молекулы в движении. Транспорт ионов и молекулярная диффузия 365
Изучаемые вопросы...............................................365
Введение........................................................366
25.1.. Транспорт ионов..........................................366
25.2. Фундаментальные аспекты молекулярного транспорта . . . 381
Приложение. Измерение чисел переноса............................394
Литература......................................................395
Задачи......................................................... 395
26. Скорости химических реакций....................................400
Изучаемые вопросы . 400
Введение..................................................... 40l
26.1. Эмпирическая химическая кинетика 402
26.2. Скорости рсакинй . . 404
584
Содержание
26.3. Объяснение законов скоростей ............................! 416
26.4. Сложные реакции..........................................j ,431
26.5. Быстрые реакции..........................................
Литература......................................................4ч8
Задачи......................................................... 448
27. Динамика молекулярных реакций....................................455
Изучаемые вопросы...............................................455
Введение........................................................456
27.1. Молекулярные столкновения.................................457
27.2. Теория активированного комплекса..........................466
27.3. Динамика молекулярных столкновений...................... 478
Литература.................................................... 487
Задачи..........................................................488
28. Процессы па твердых поверхностях................................492
Изучаемые вопросы............................................. 492
Введение........................................................492
28.1. Рост и структура поверхностей ............................493
28.2. Адсорбция на поверхностях.................................502
28.3. Каталитическая активность ца поверхностях.................514
Литература......................................................521
Задачи........................................................ 521.
29. Динамическая электрохимия.......................................526
Изучаемые вопросы...............................................526
Введение........................................................527
29.1. Процессы на электродах ...................................527
29.2. Электрохимические процессы................................545
29.3. Генерирование и накопление мощности.......................550
29.4. Коррозия ...........................,.....................554
Литература......................................................560
Задачи .........................................................560
Ответы к задачам................................................564
Приложение . ...............................................; 571
Предметный указатель.............................................575
ЯБ Xs 1983
А. Эткинс
ФИЗИЧЕСКАЯ ХИМИЯ
ТОМ 2
Ст. научи, редактор С. Оганесян. Научн. редактор И, Беленькая. Мл. научи, редактор
Н. Котовщнкова. Художник Е. Урусов. Художественный редактор М. Кузьмина. Технический
редактор И. Крепдеяева. Корректор К. Еодаництгая
Сдало в набор 29.03.80. Подпясяяо х печати 1.09.80. Формат ООРсЙО'/тв. Бумага типографская
№ 2. Гарнитура латинская. Печать высокая. Объем бум. л. 18.25. Усл. печ. л. 36 50. Уч-над.
л. 38.38. Изд. № 3/0468. Тираж [2000 зкз. Зак. 242. Цена 2 р. 90 к.
ИЗДАТЕЛЬСТВО <МИР*. Москва, 1-й Рижский пер.. 2.
Моспоасдая типография Mr 11 Союаполптрафпрома гтрп Государственном комитете СССР по
дедам издательств, полиграфии в книжной торговли. Москва. II3jO5. Нагатинская ул., д. 1.
Отсканировал Семенюченко Владимир
chem_vova@mail.univ.kiev.ua
vova2002@mail.ru