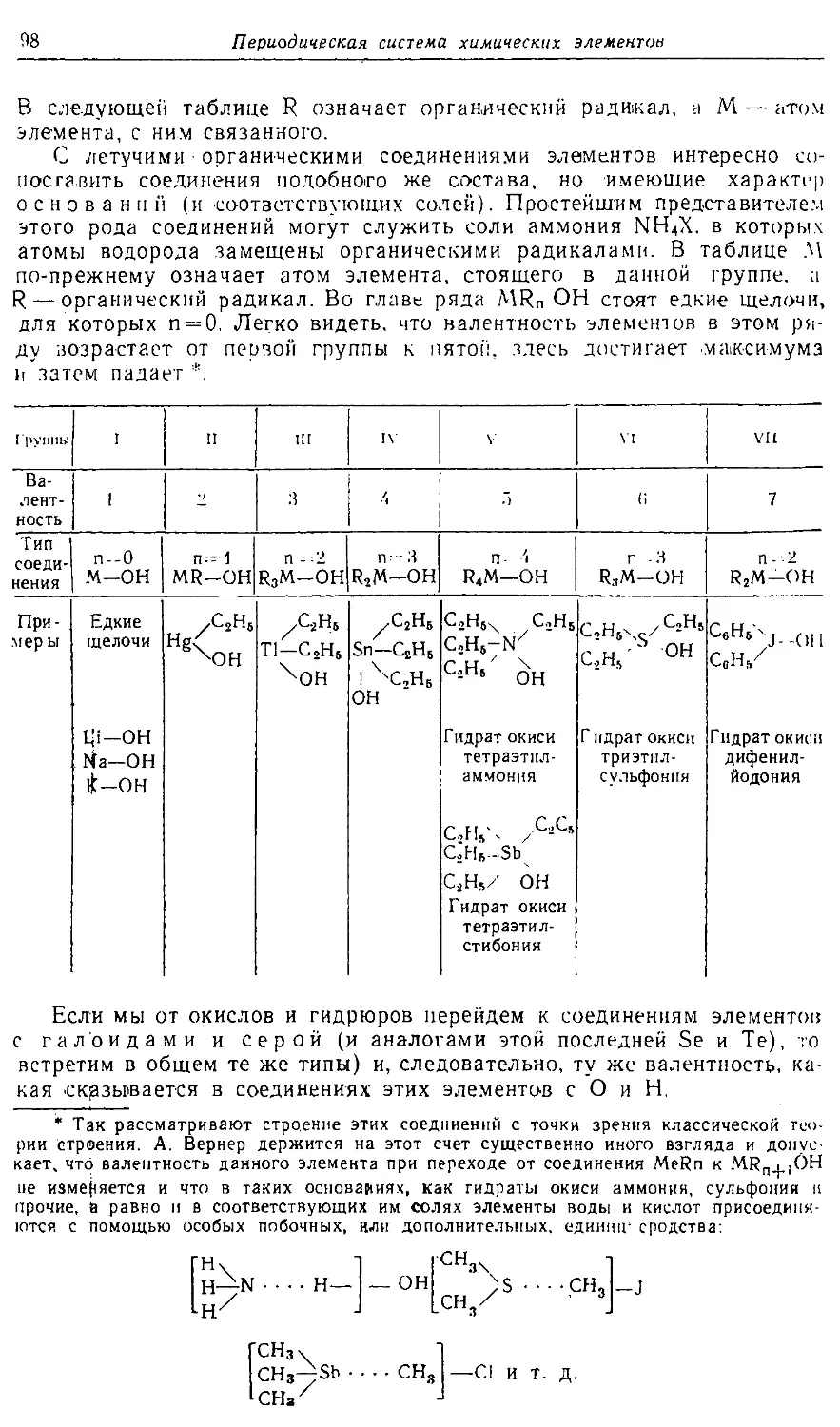
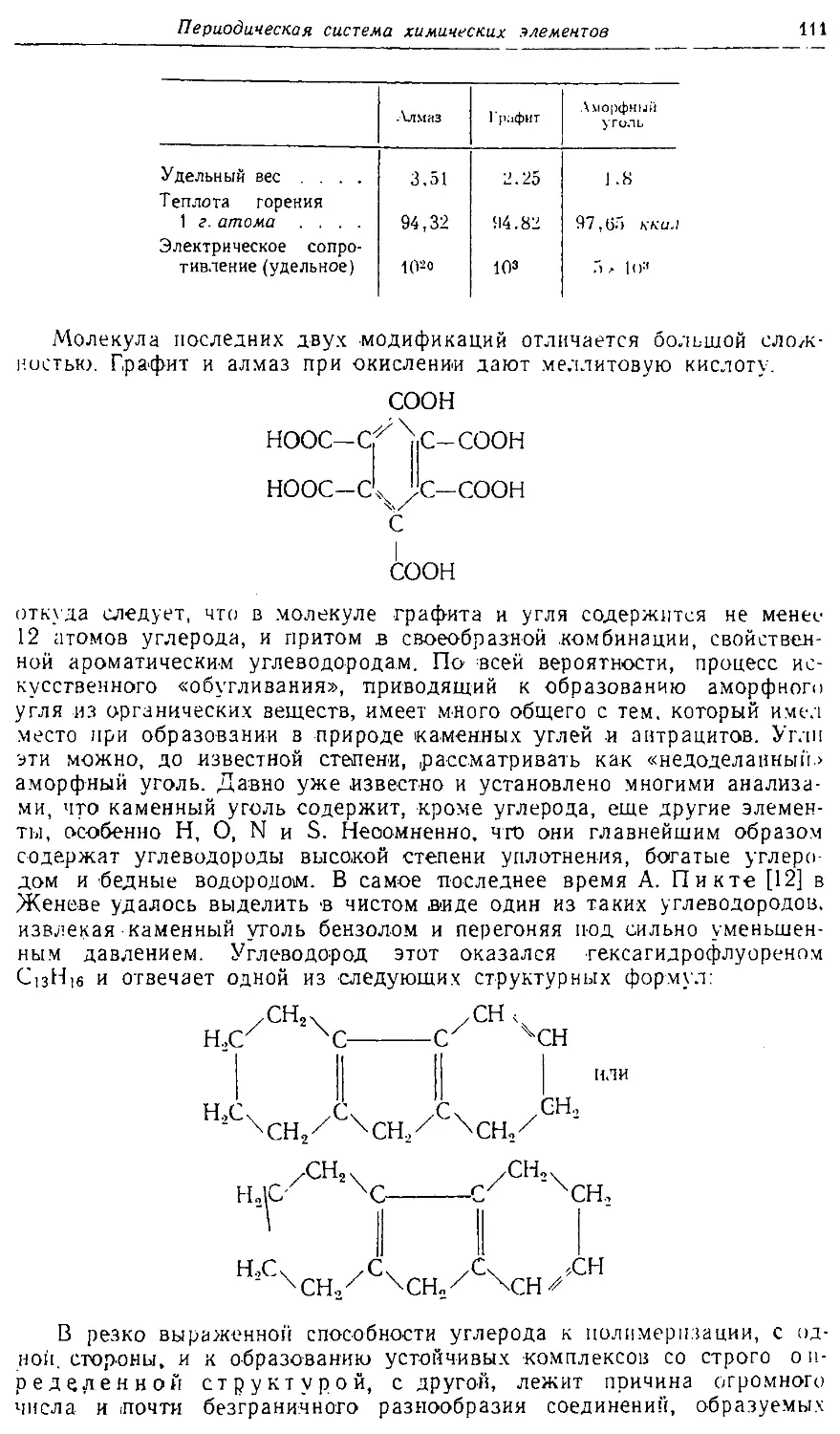
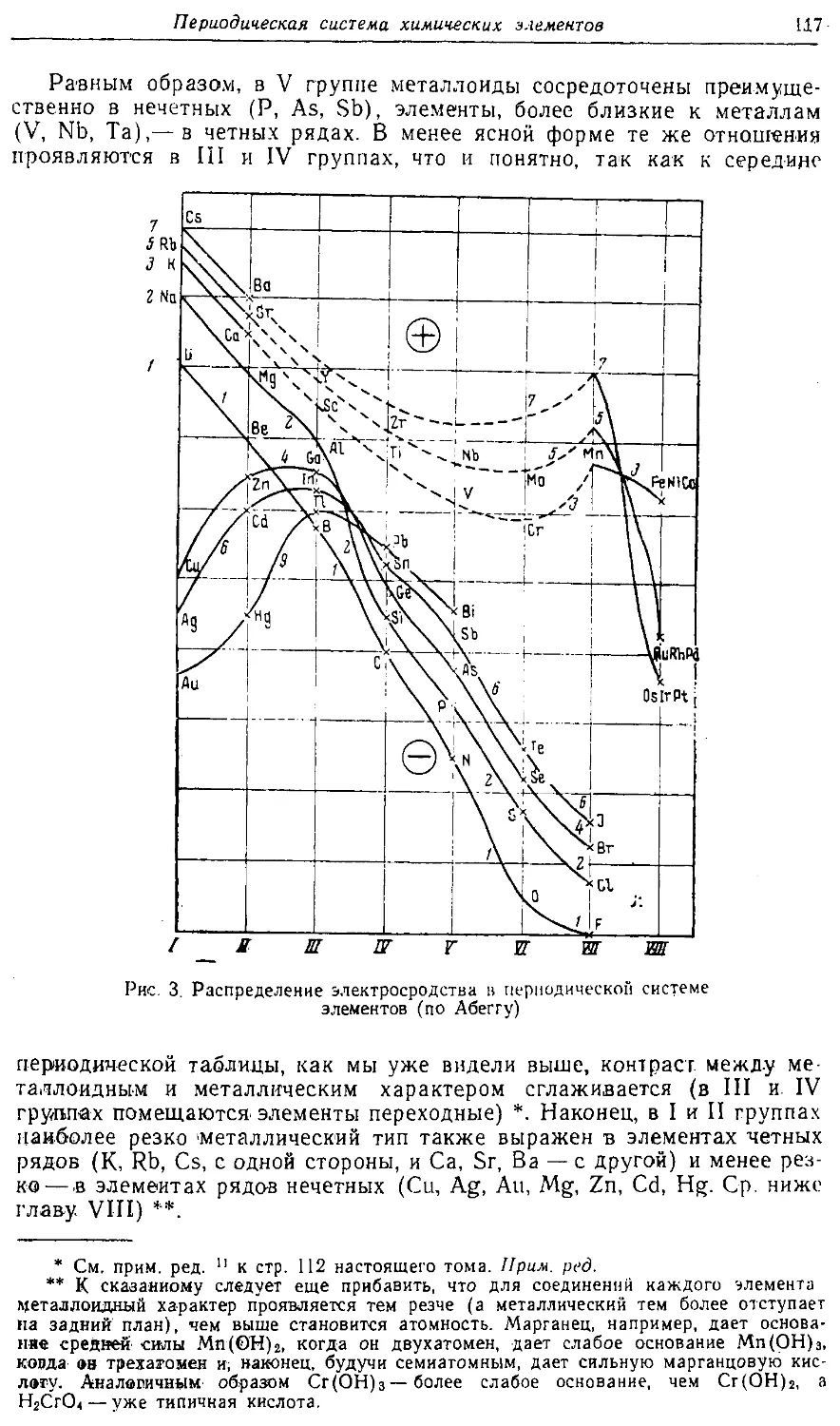
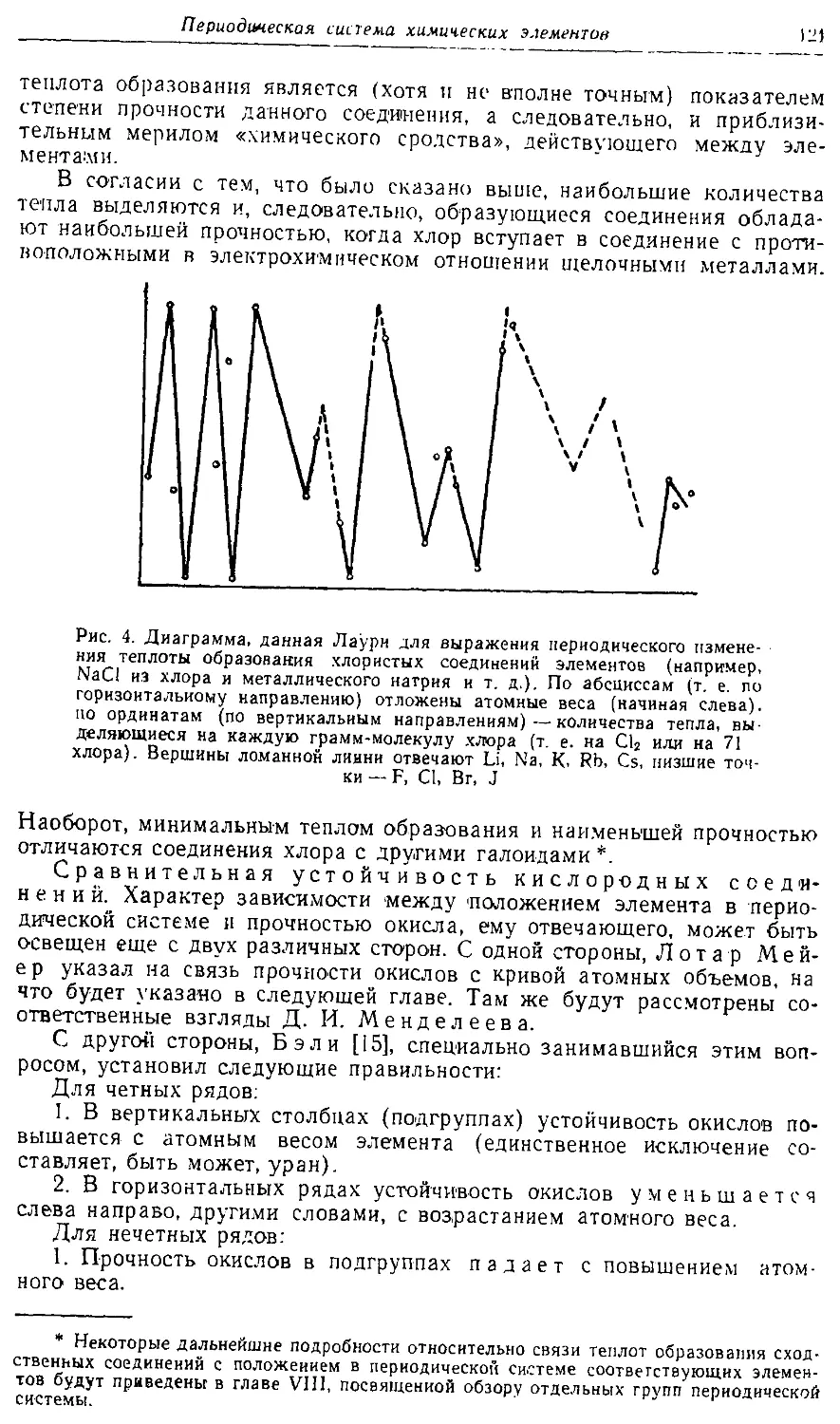
/






































































































































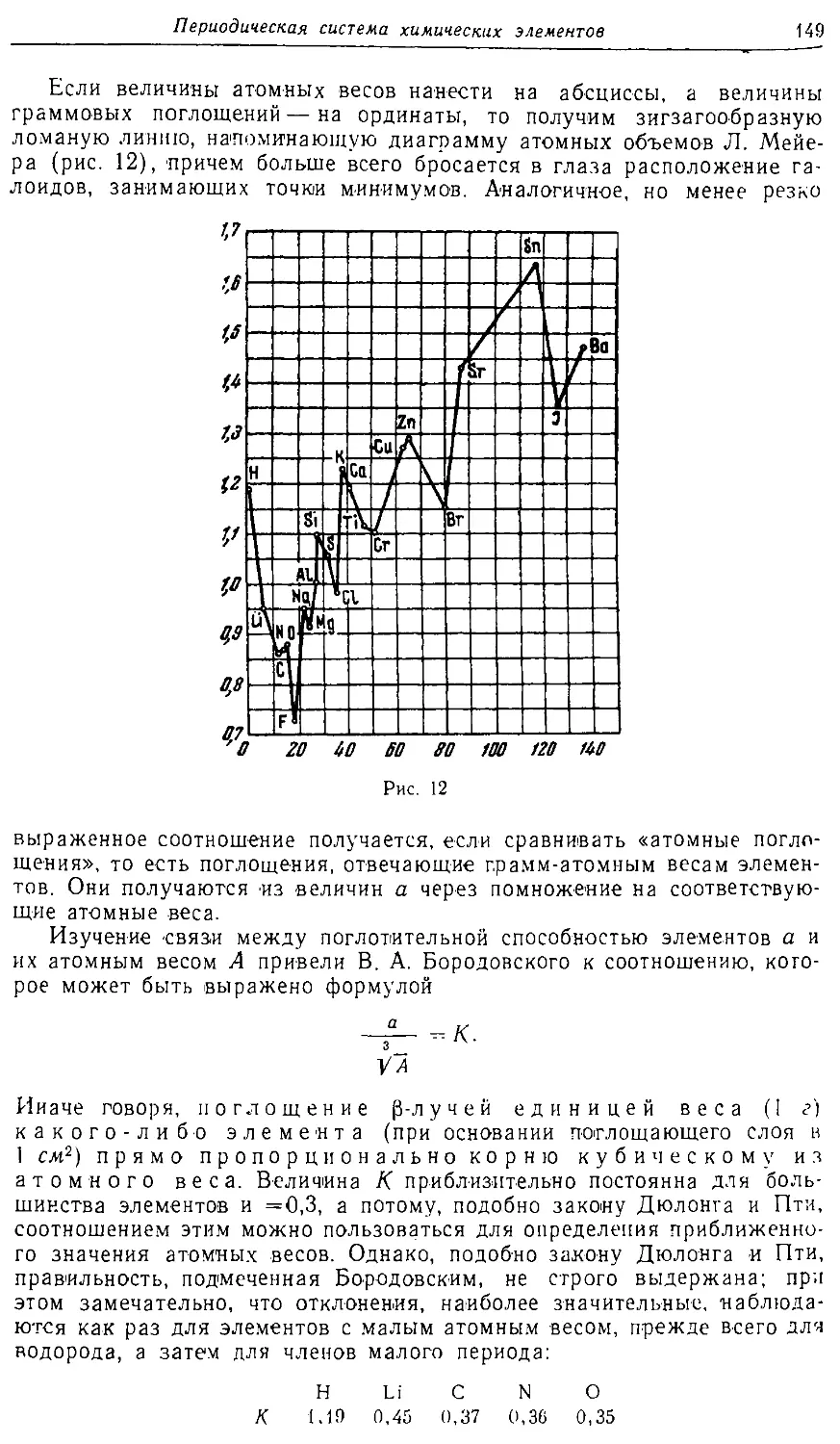
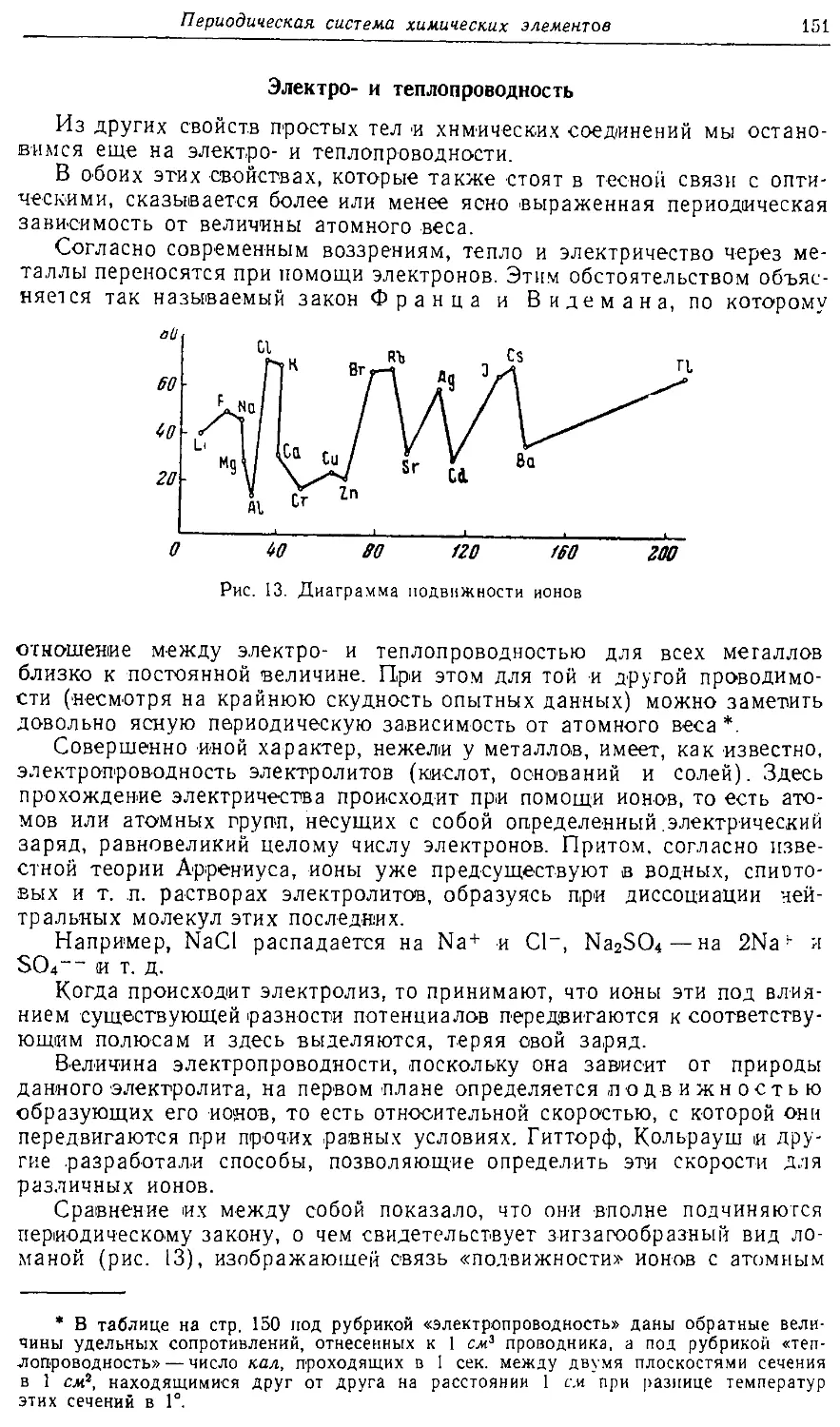















































































































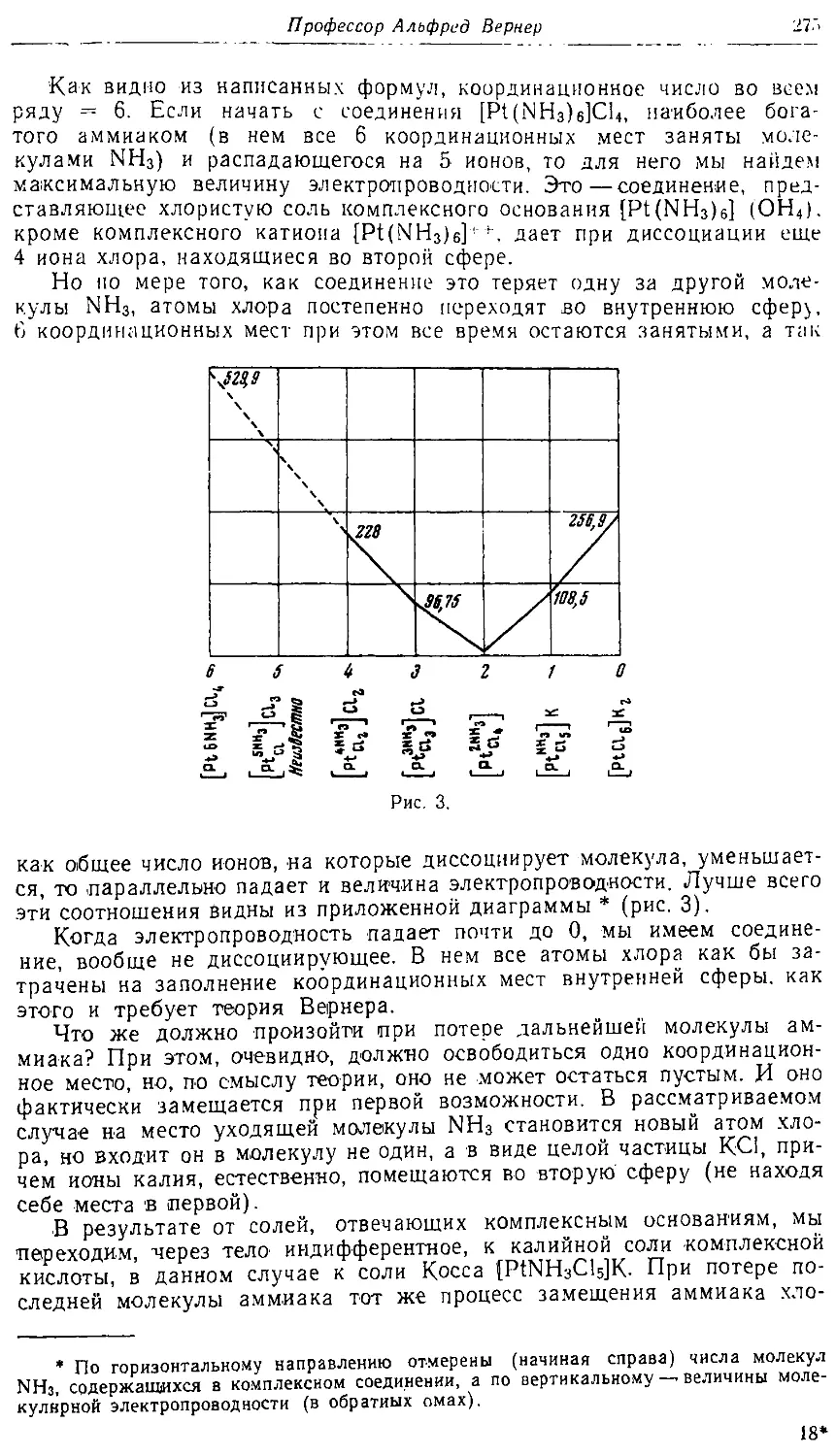
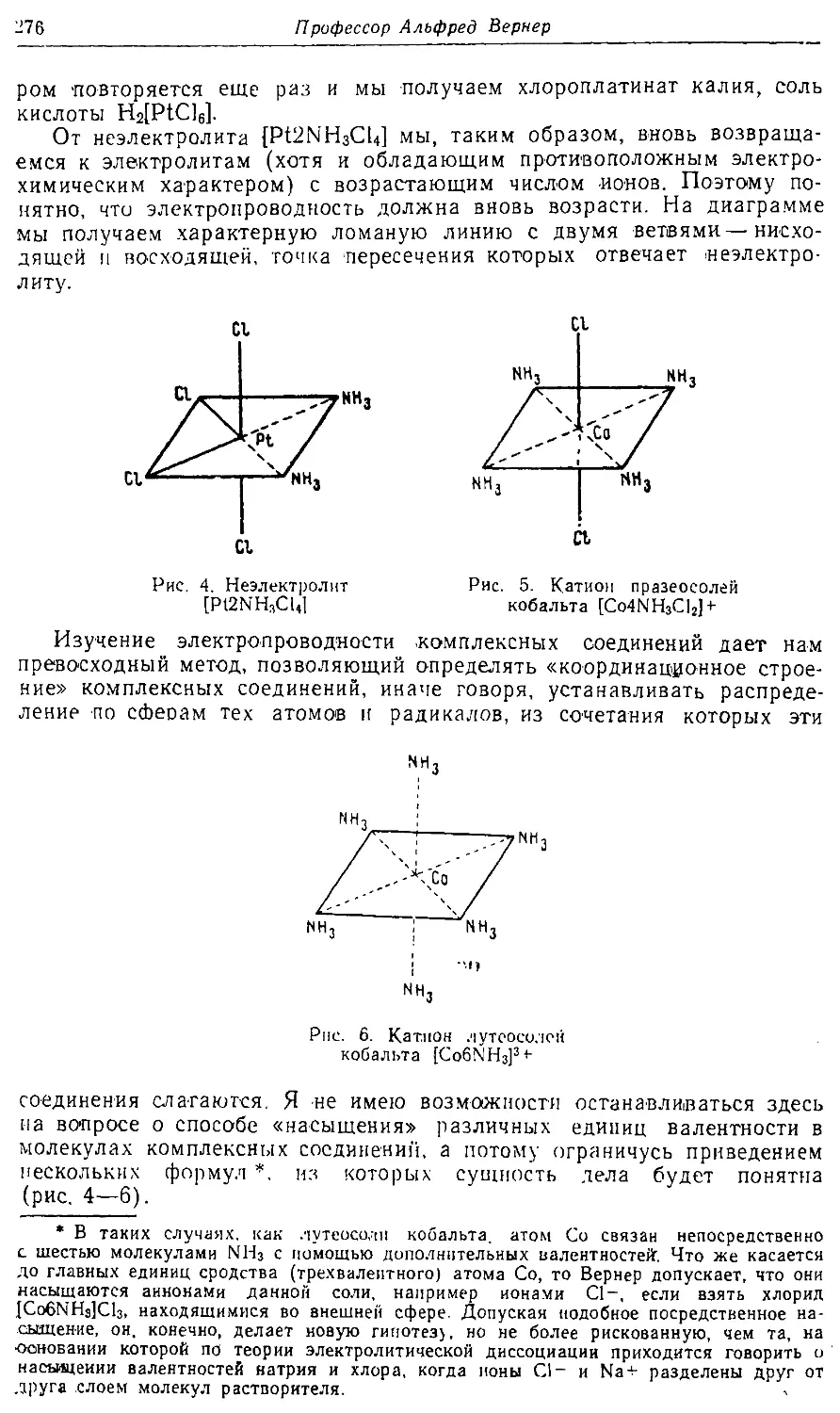




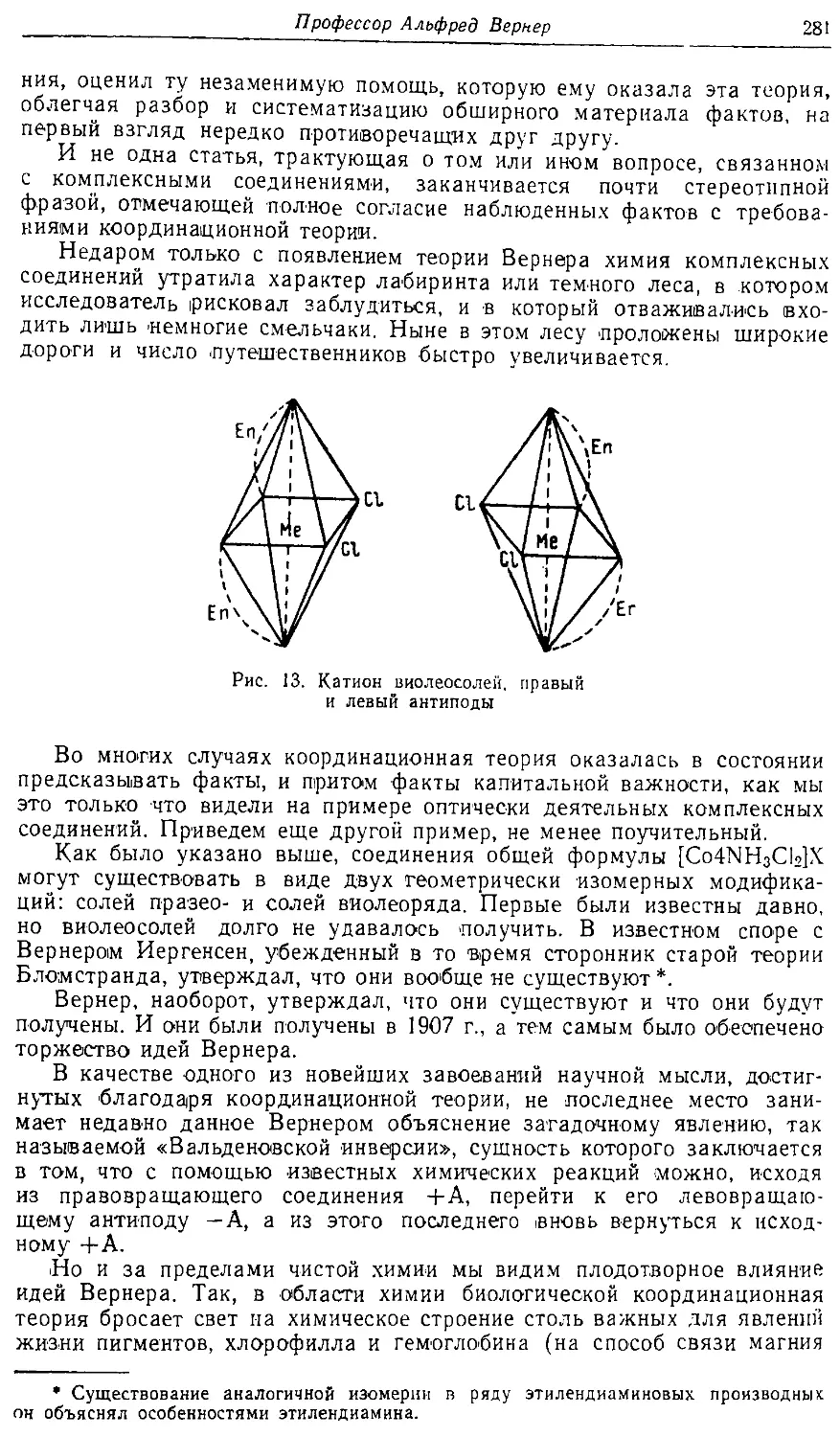




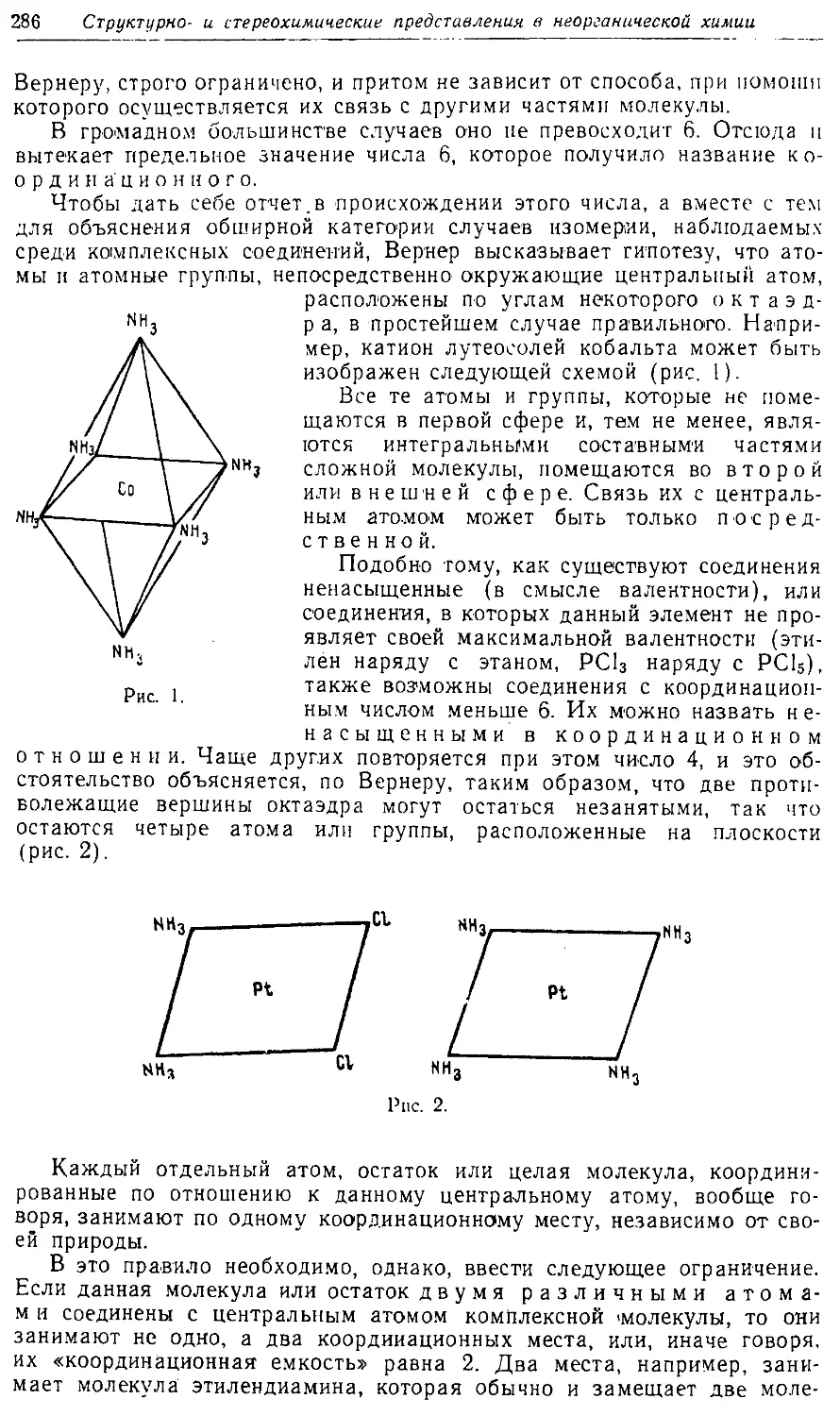

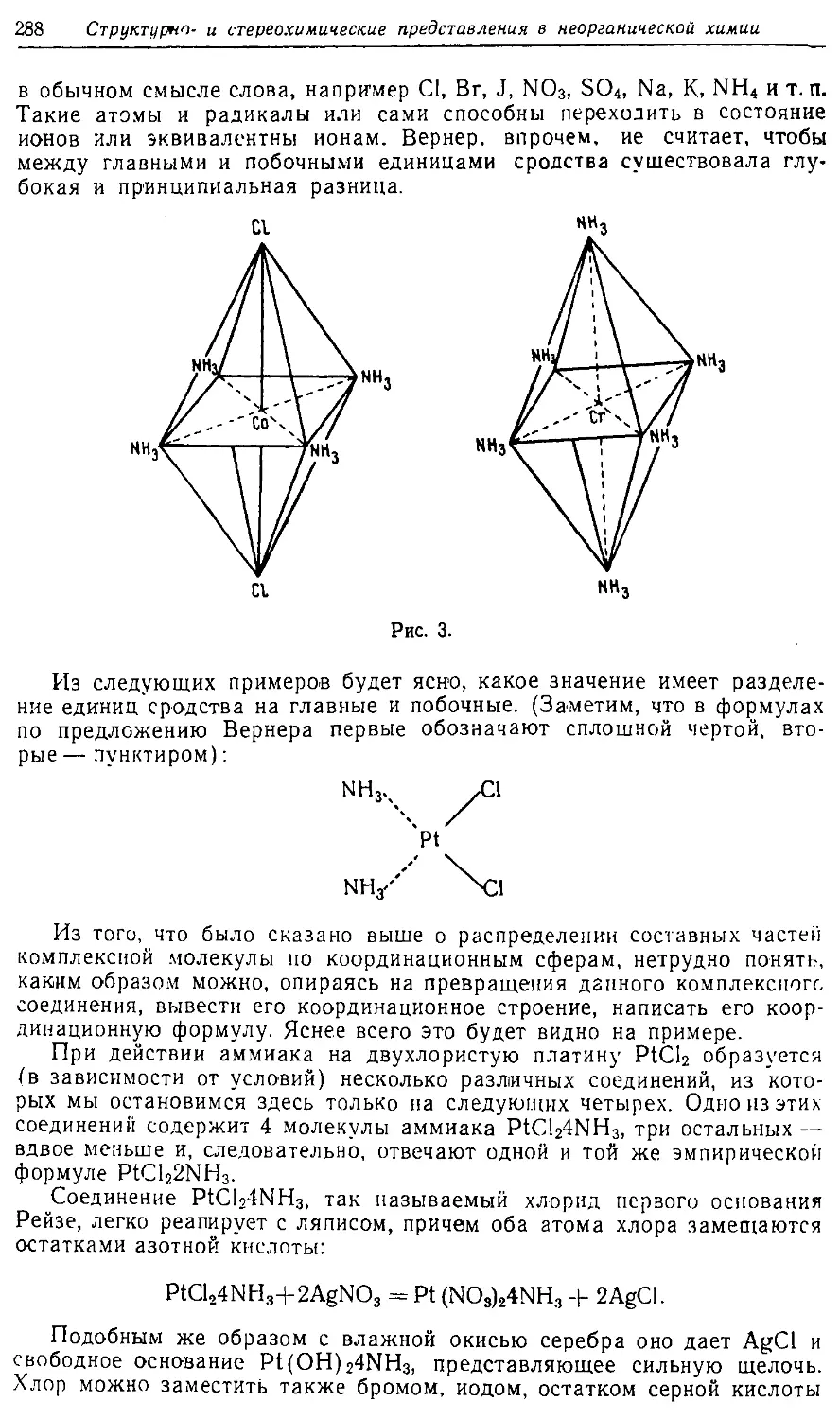


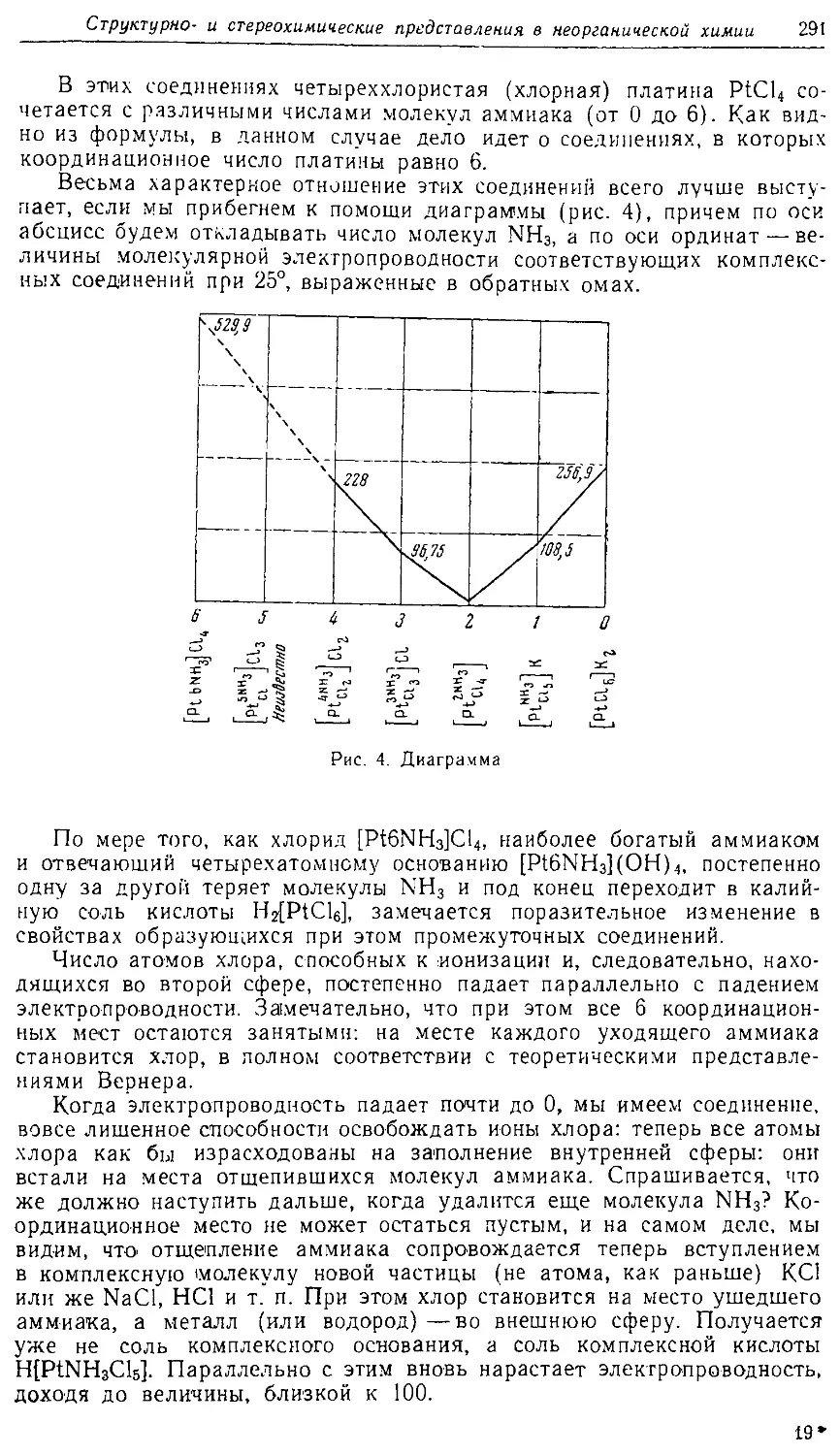





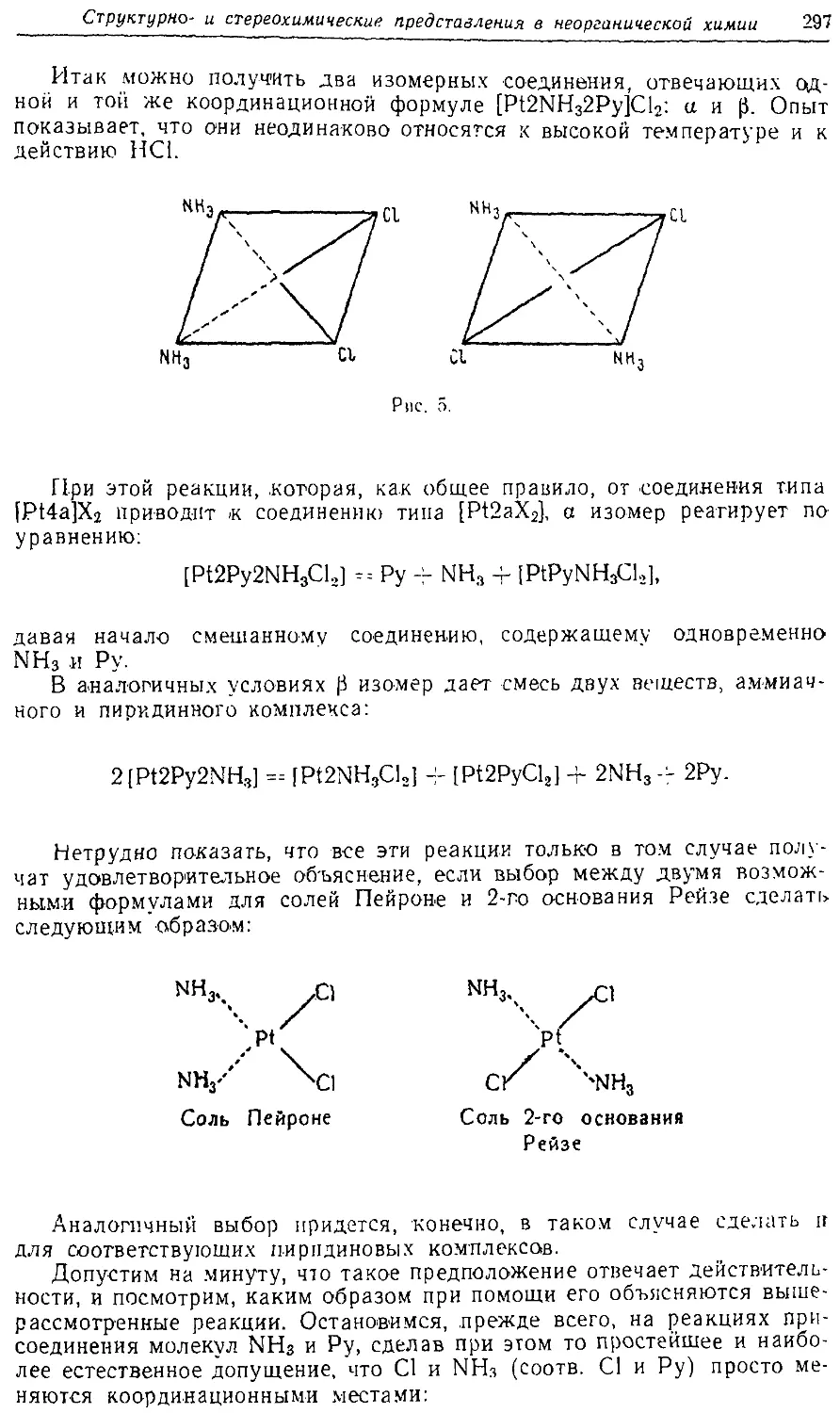
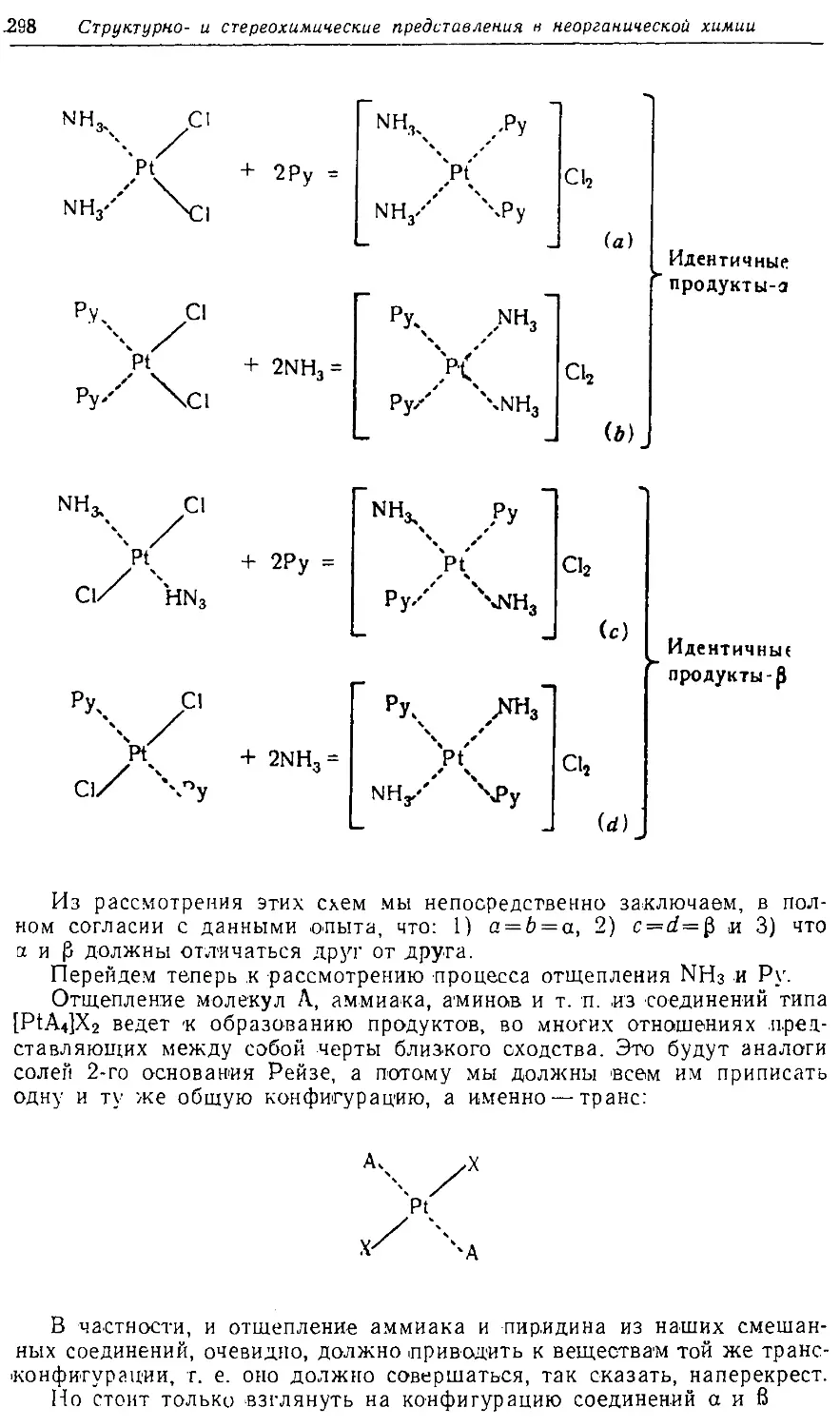

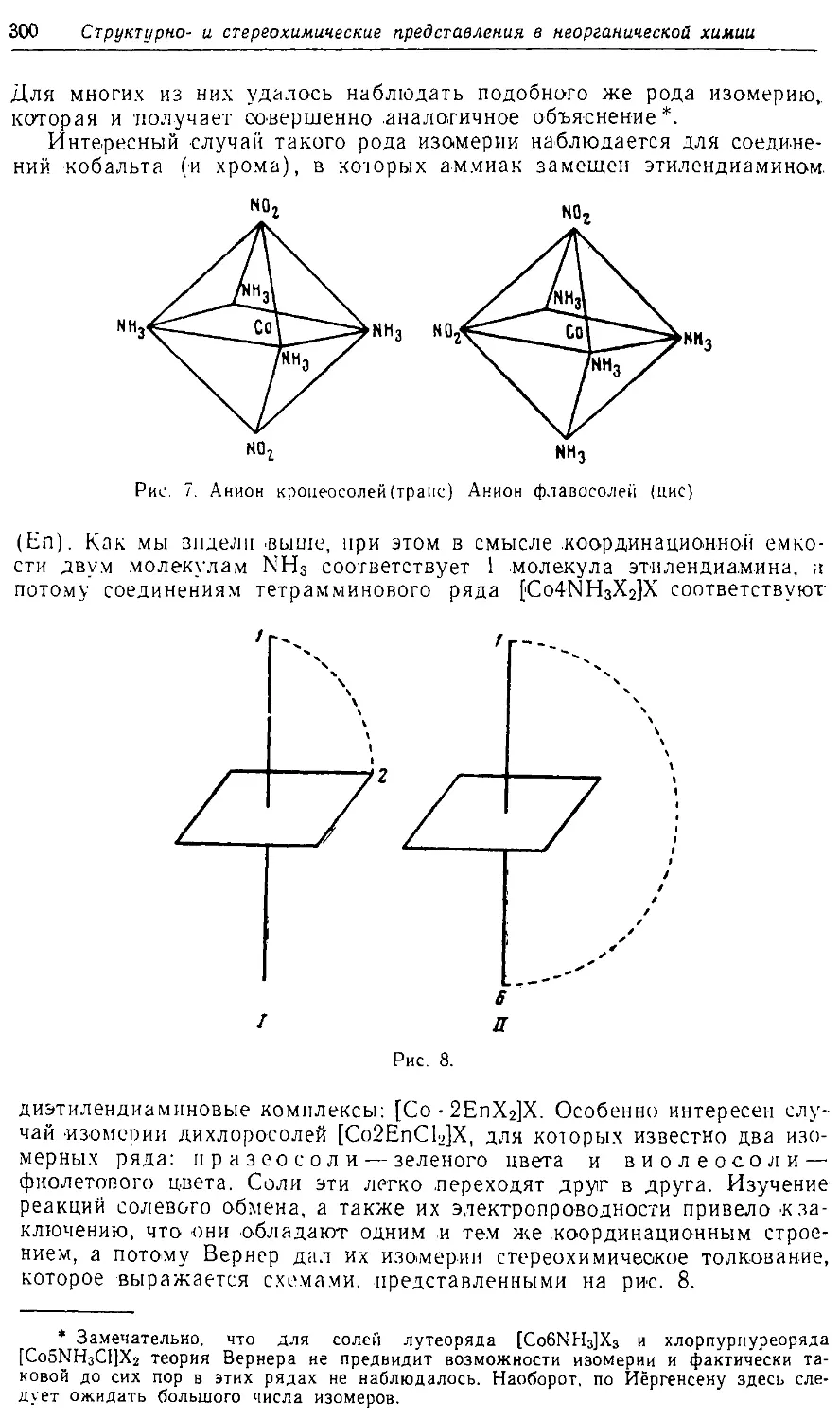

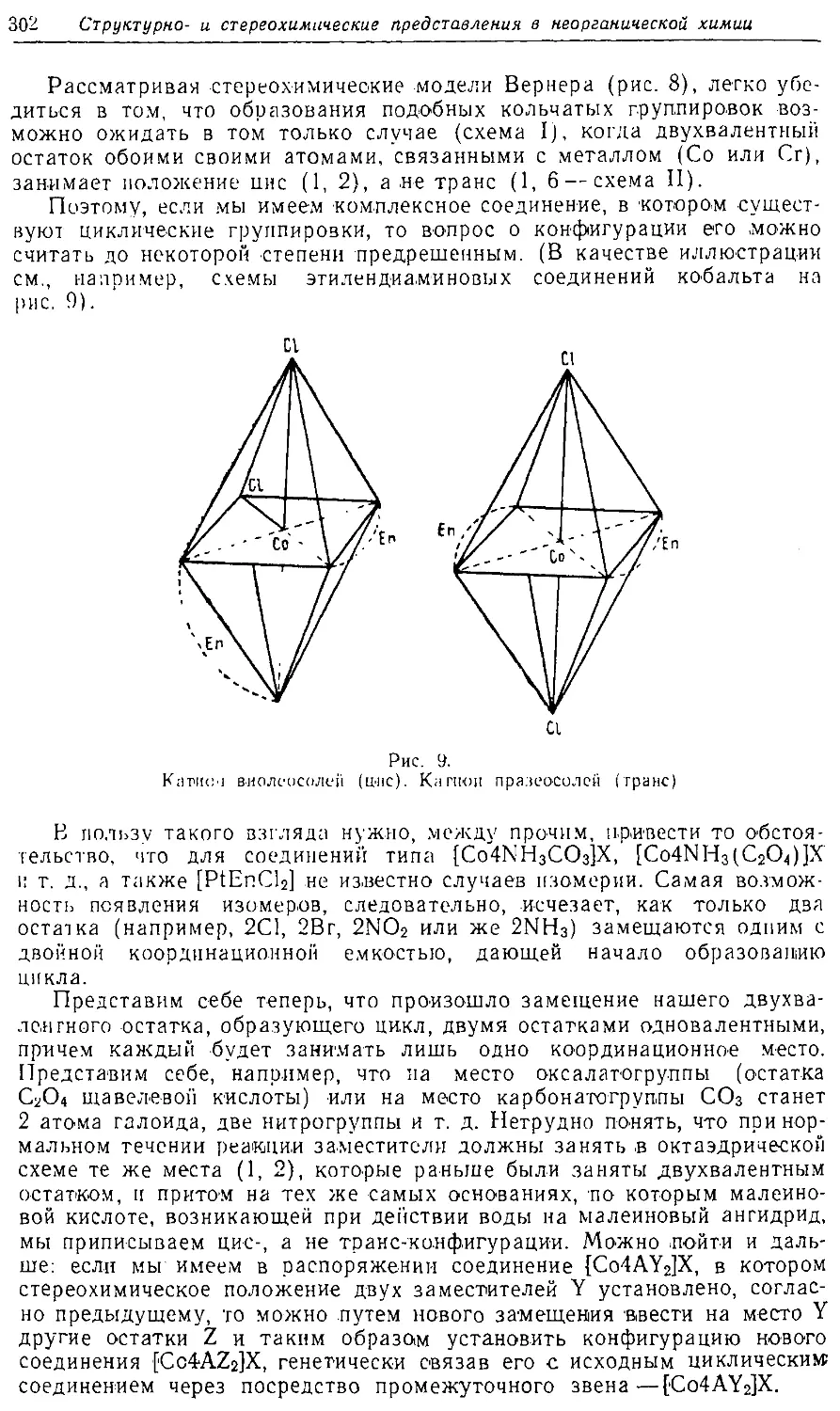


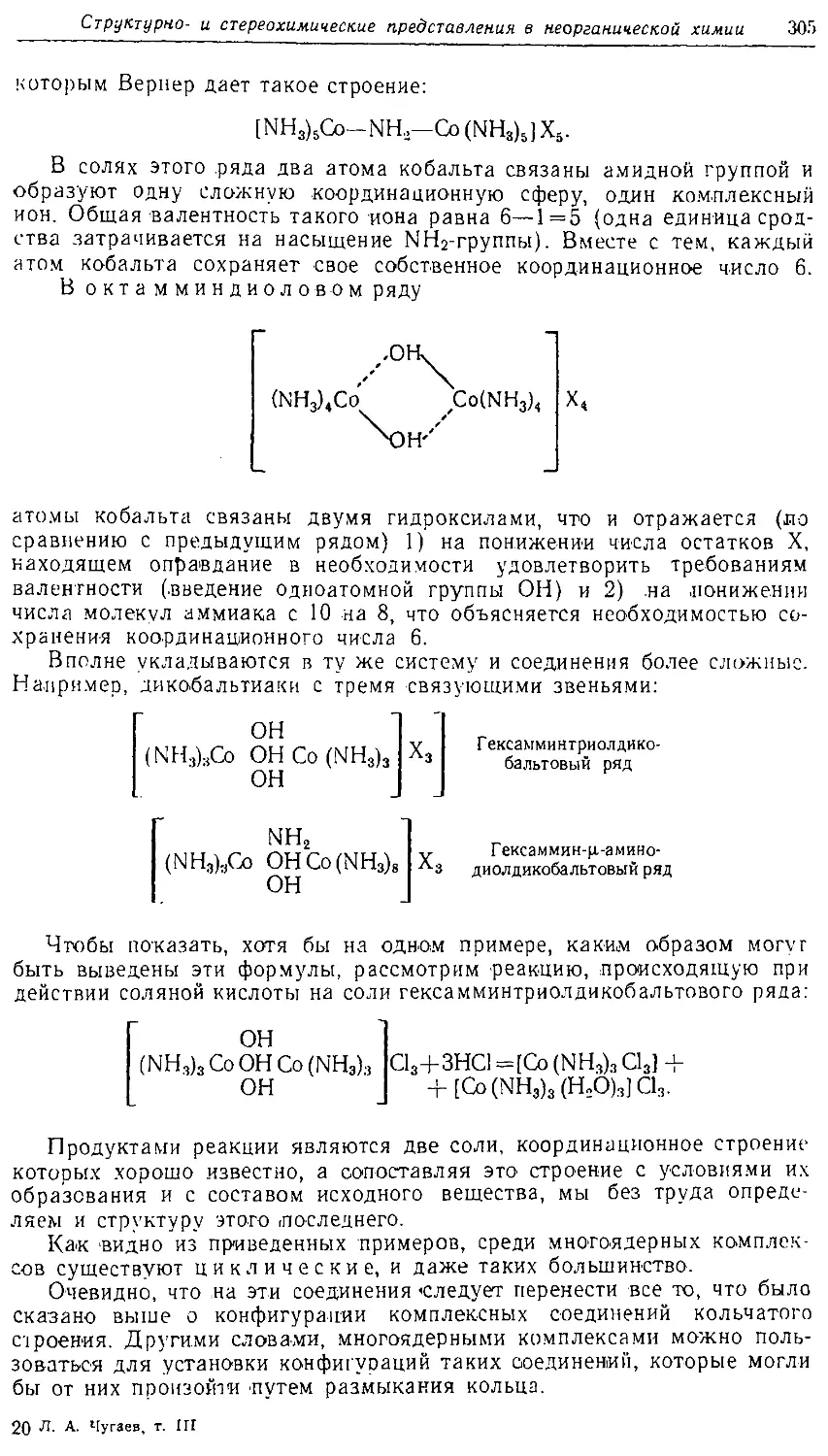
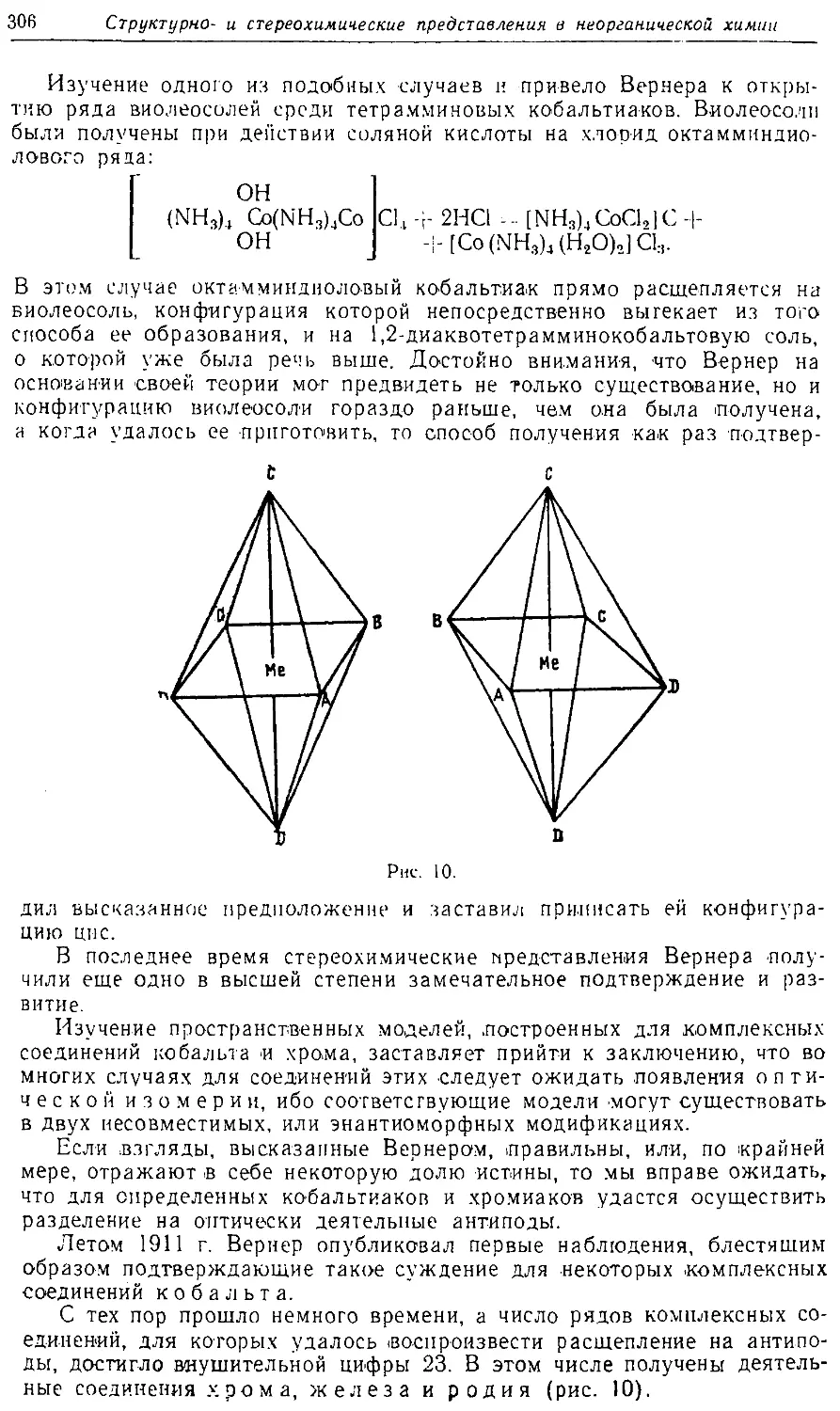
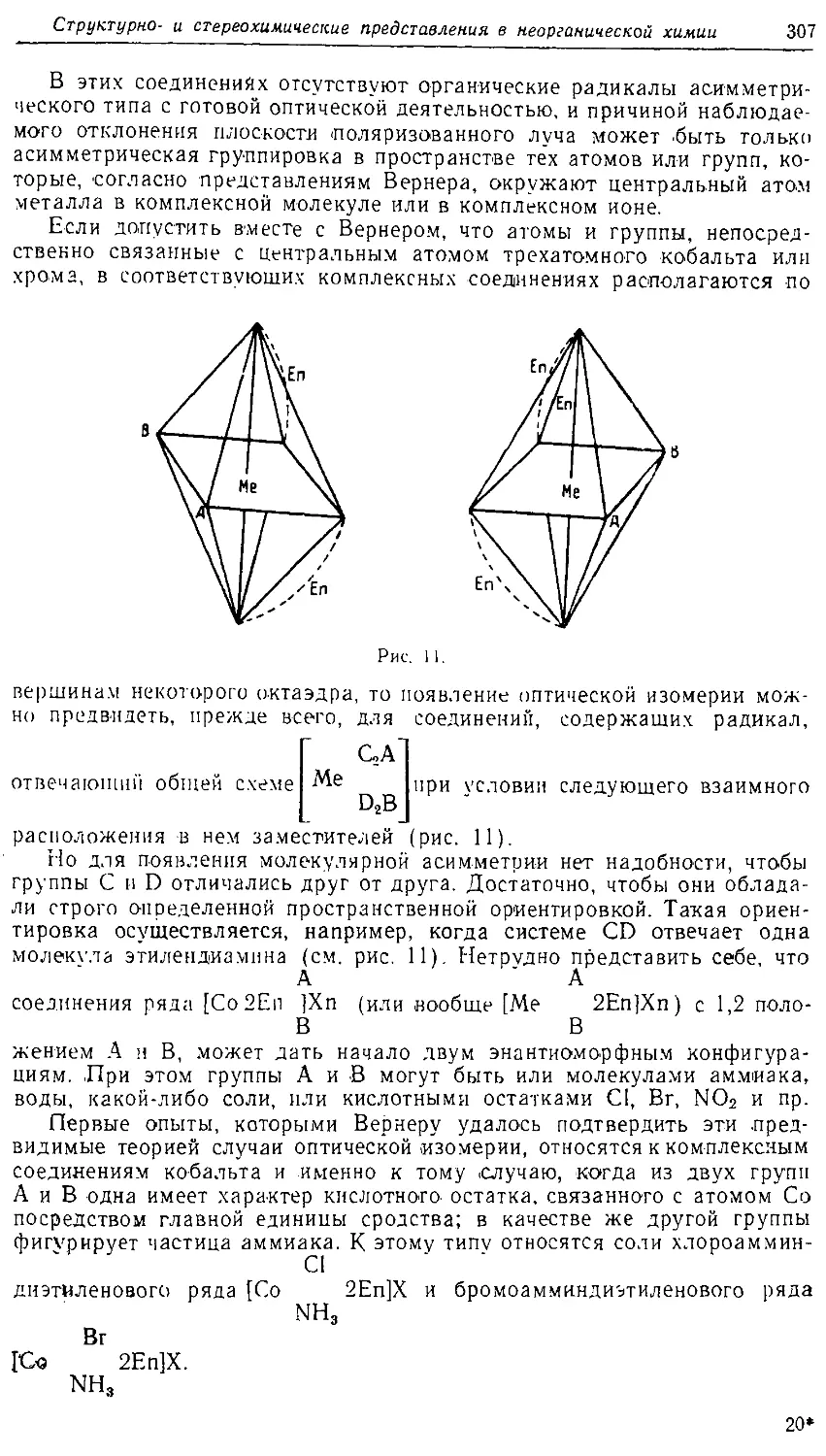



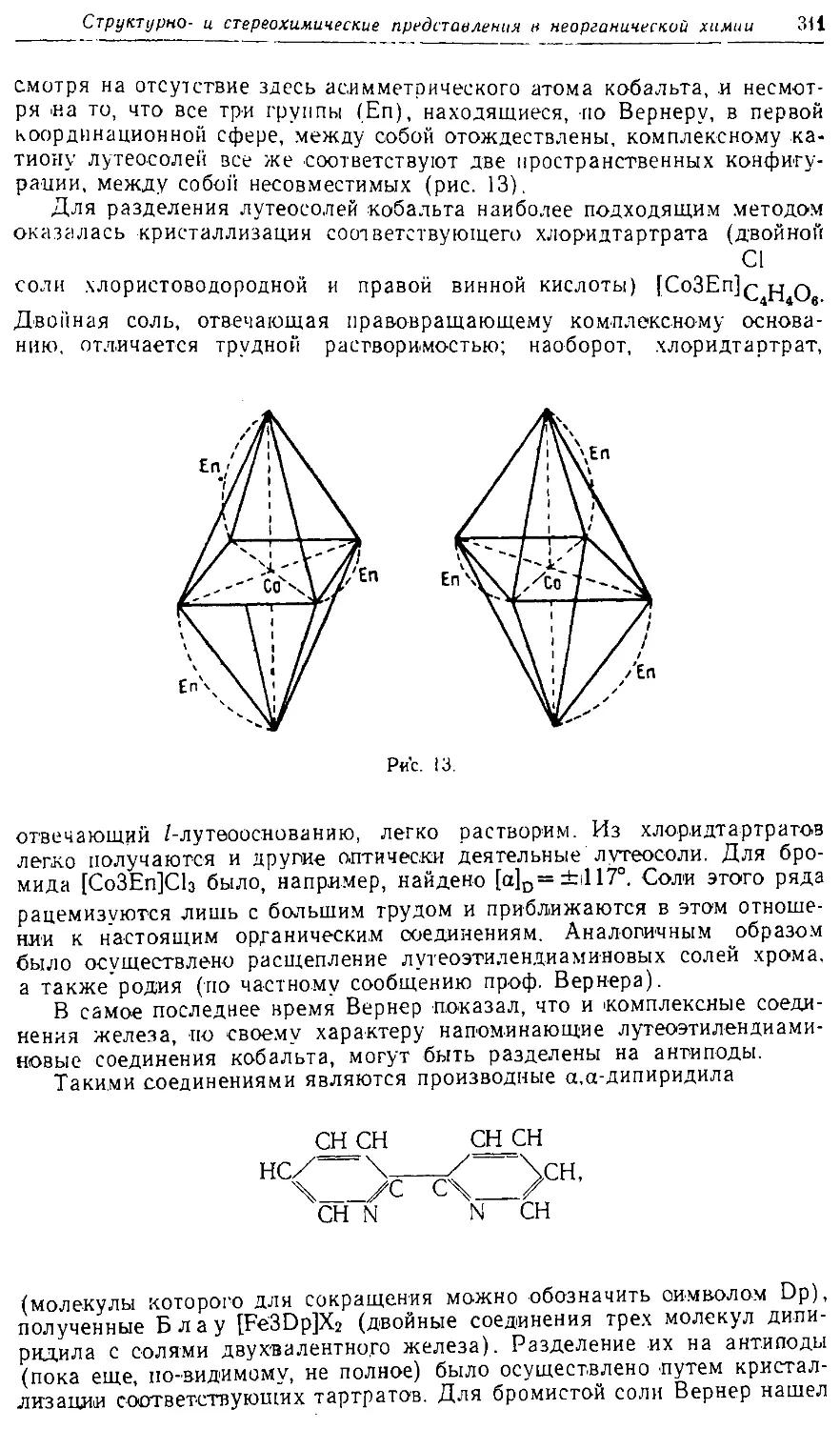



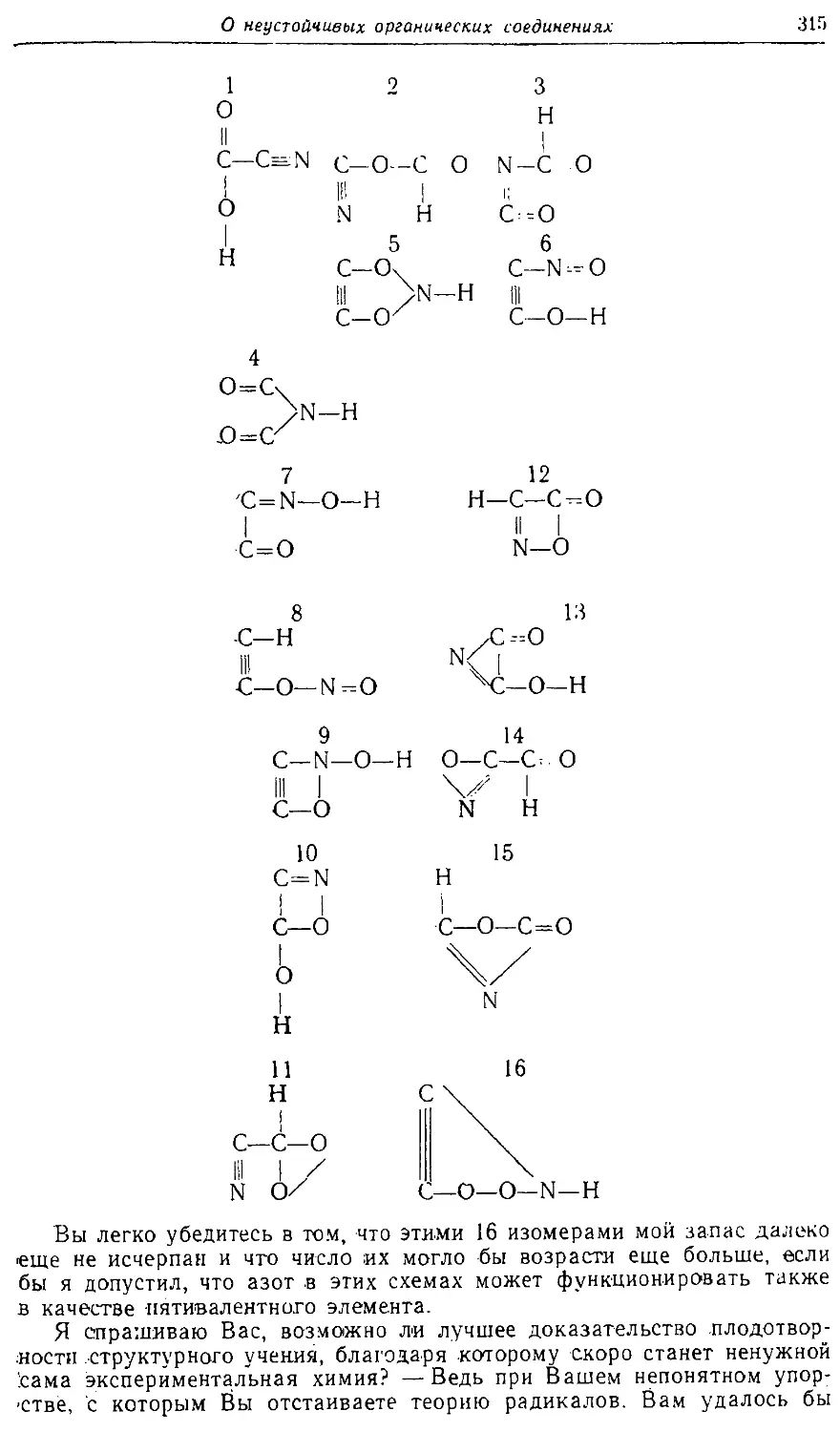















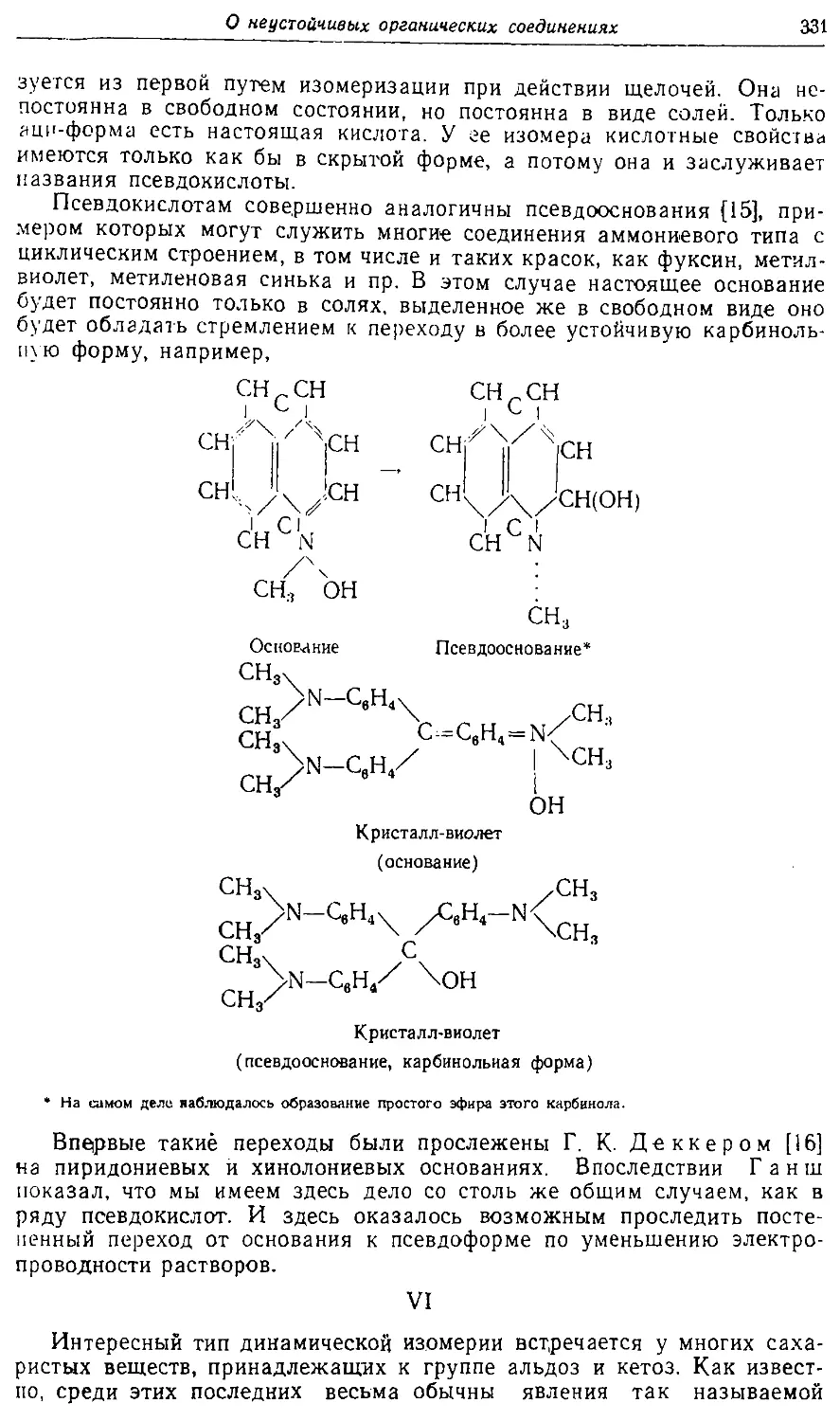


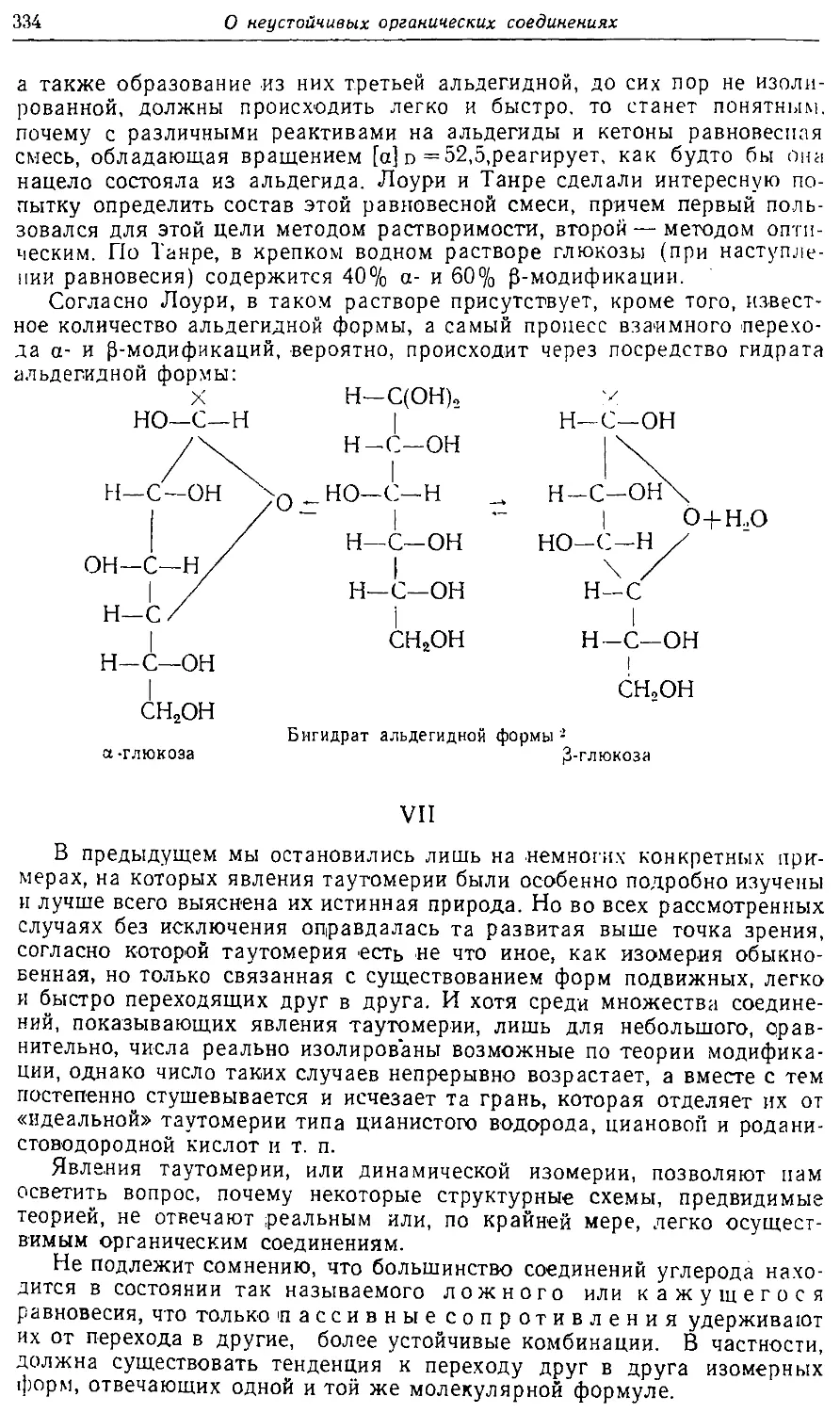

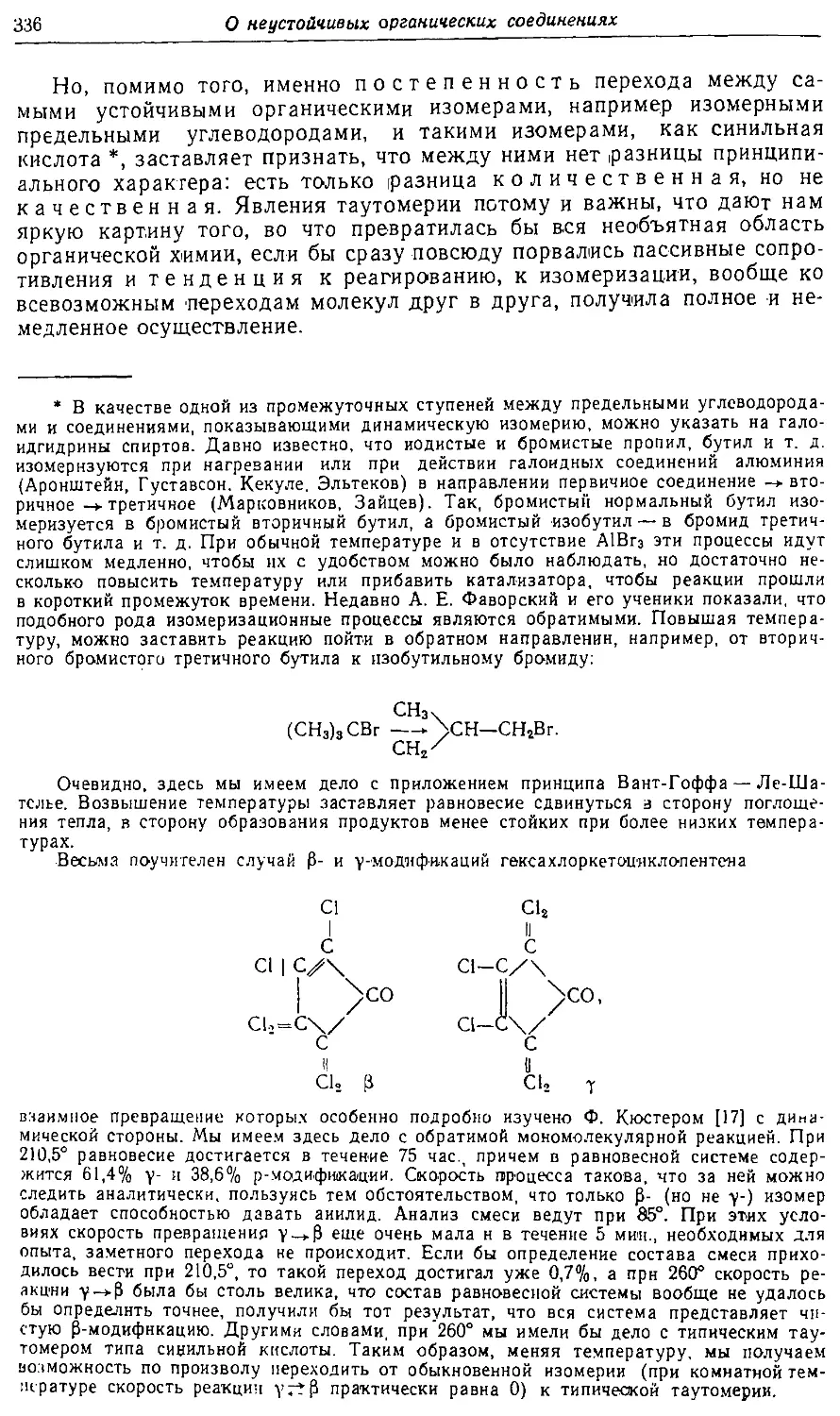



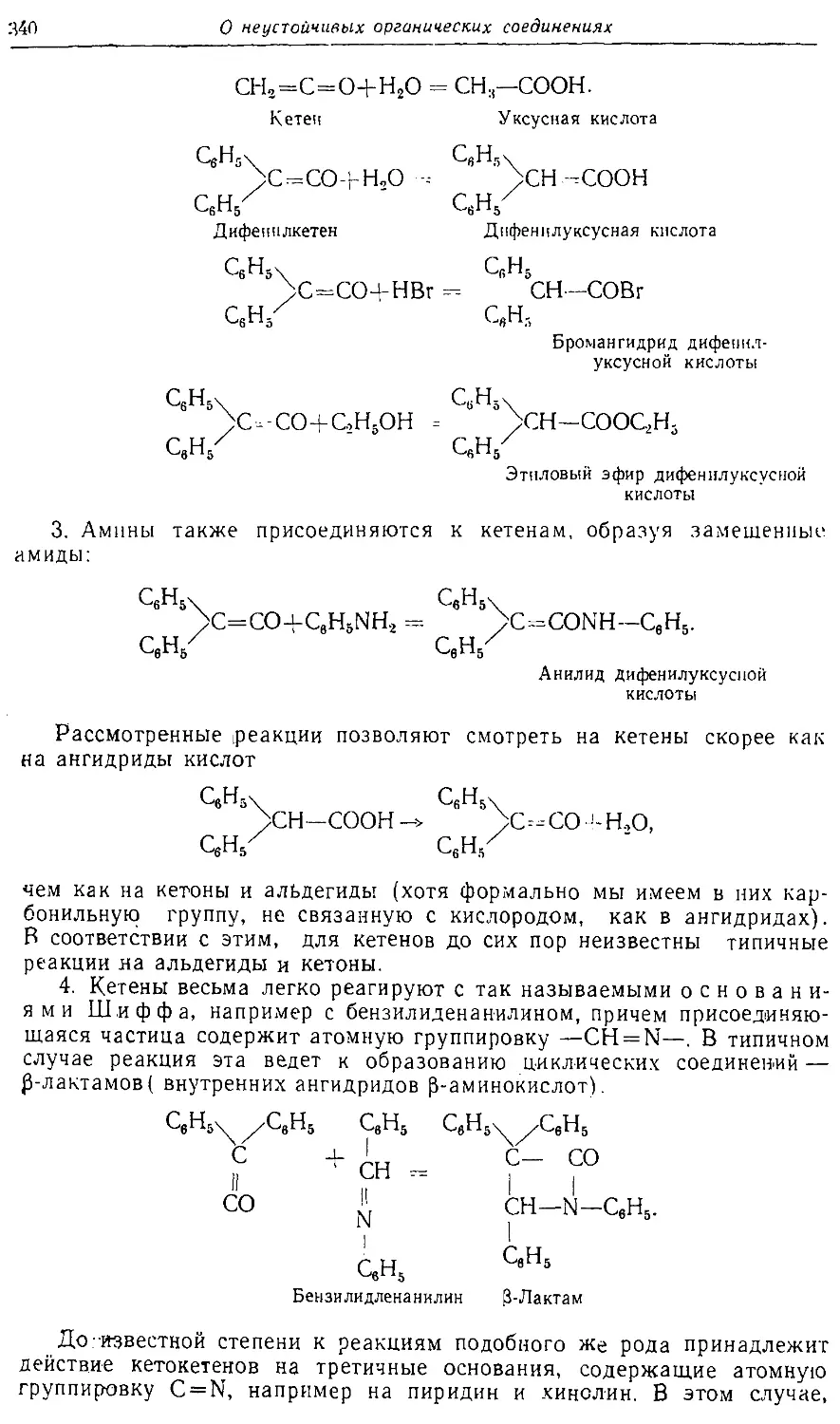





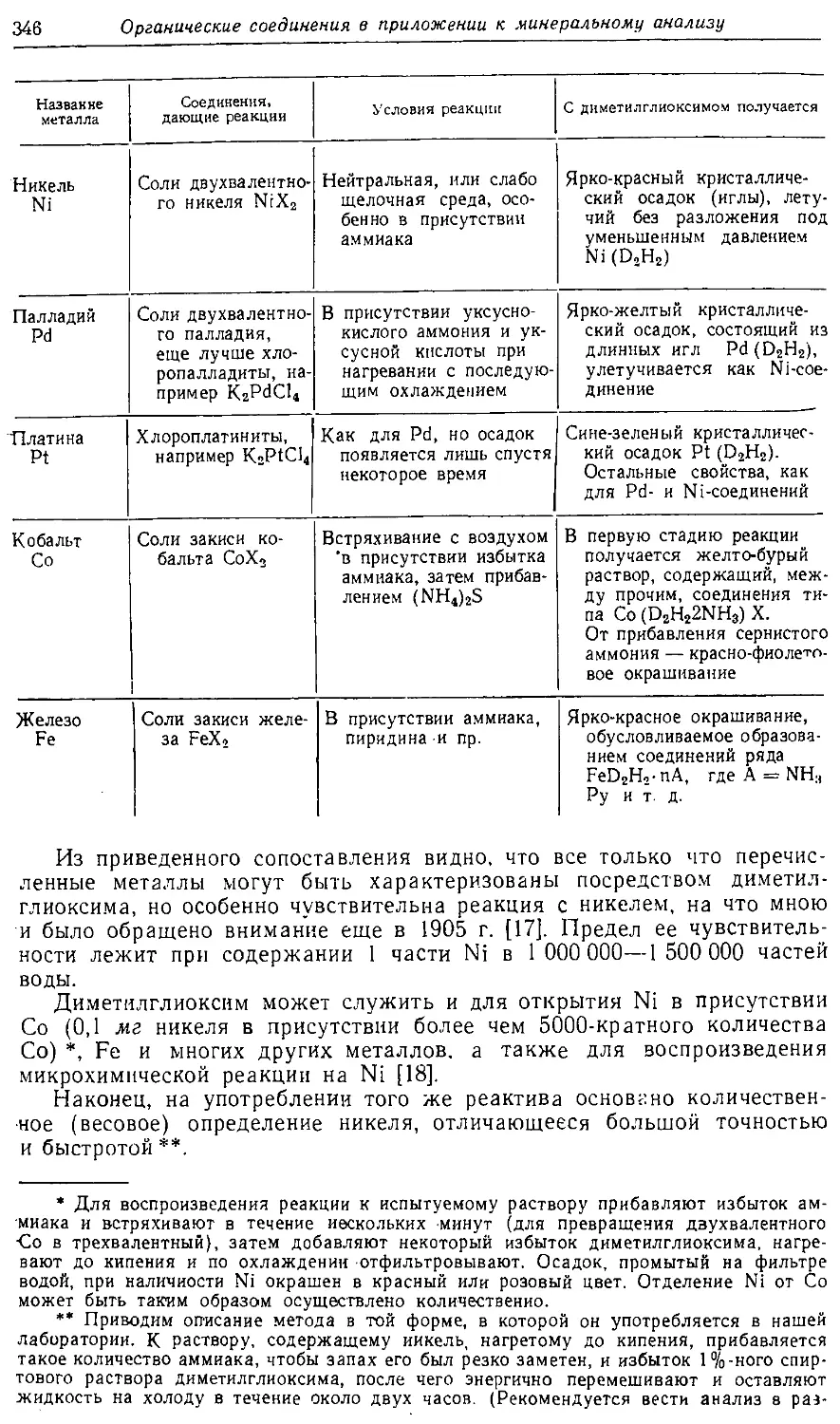












































































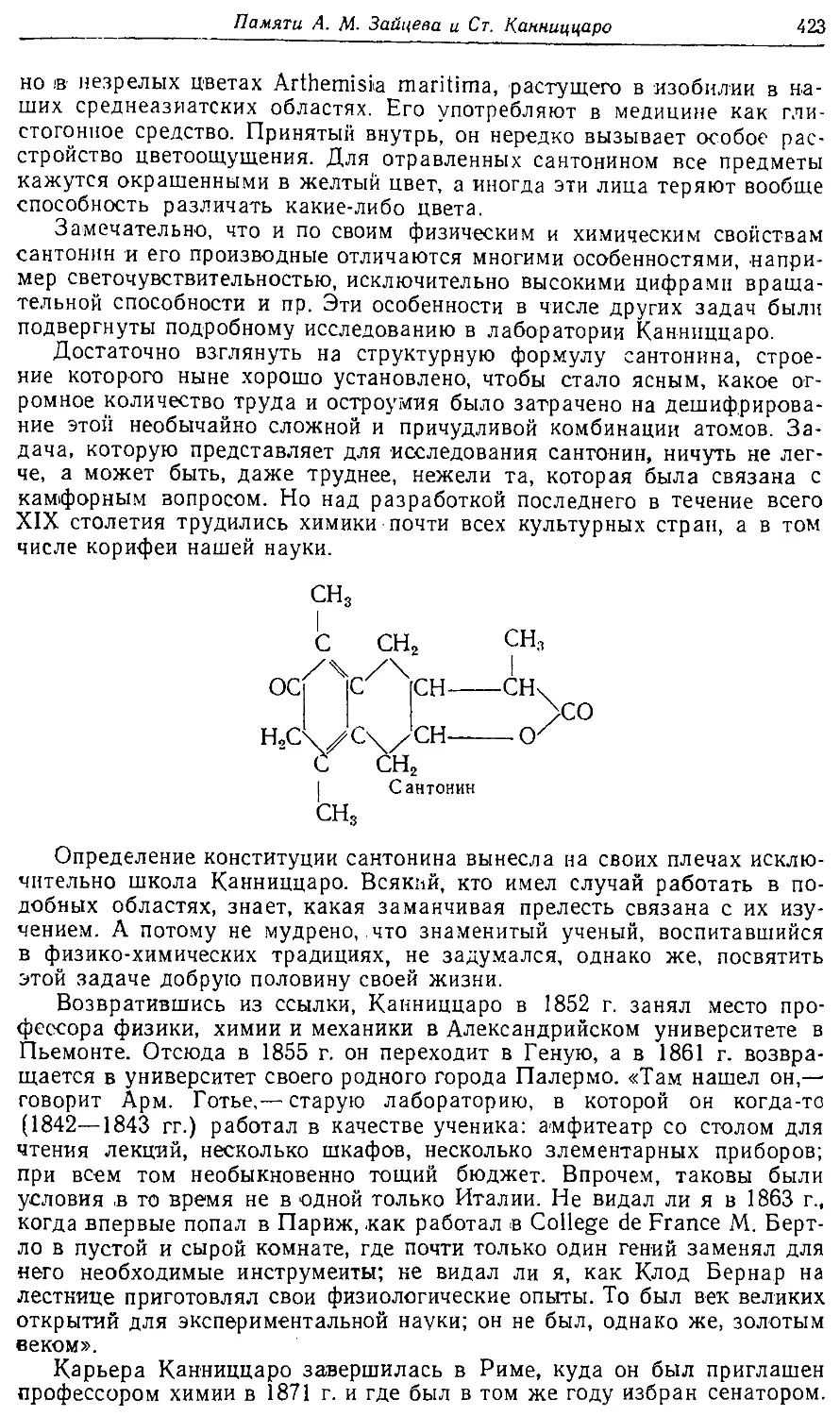



























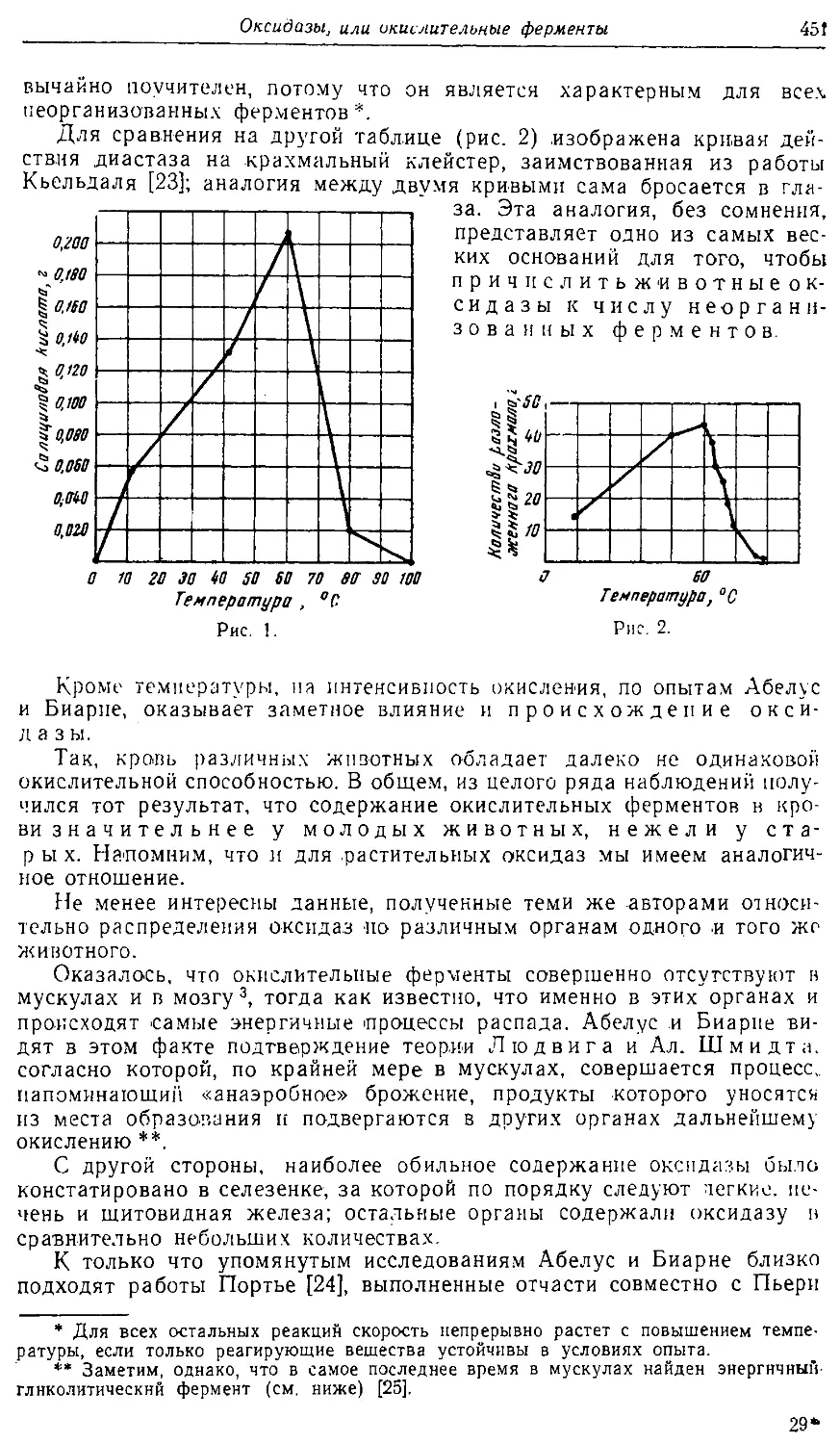













































Text
ААЧУГАЕВ
ИЗБРАННЫЕ
ТРУДЫ
комиссия по изданию трудов
Л. А. ЧУГАЕВА
Академик И. И. Черняев (председатель)
Член-корр. Академии наук СССР В. В. Лебединский
Член-корр. Академии наук СССР \И. К. II шеницын
Доктор хим. наук О, Е. Звягинцев
Доктор хим. наук Г. В. П игу ле в с кий
Канд. хим. наук ?. В. Шендерецкая
Канд. хим. наук И. А. Федоров (секретарь)!
Ответственный редактор III тома
Доктор хим. наук О. Е. Звягинцев
ОТ КОМИССИИ ПО ИЗДАНИЮ ТРУДОВ
Л. А. ЧУГАЕВА
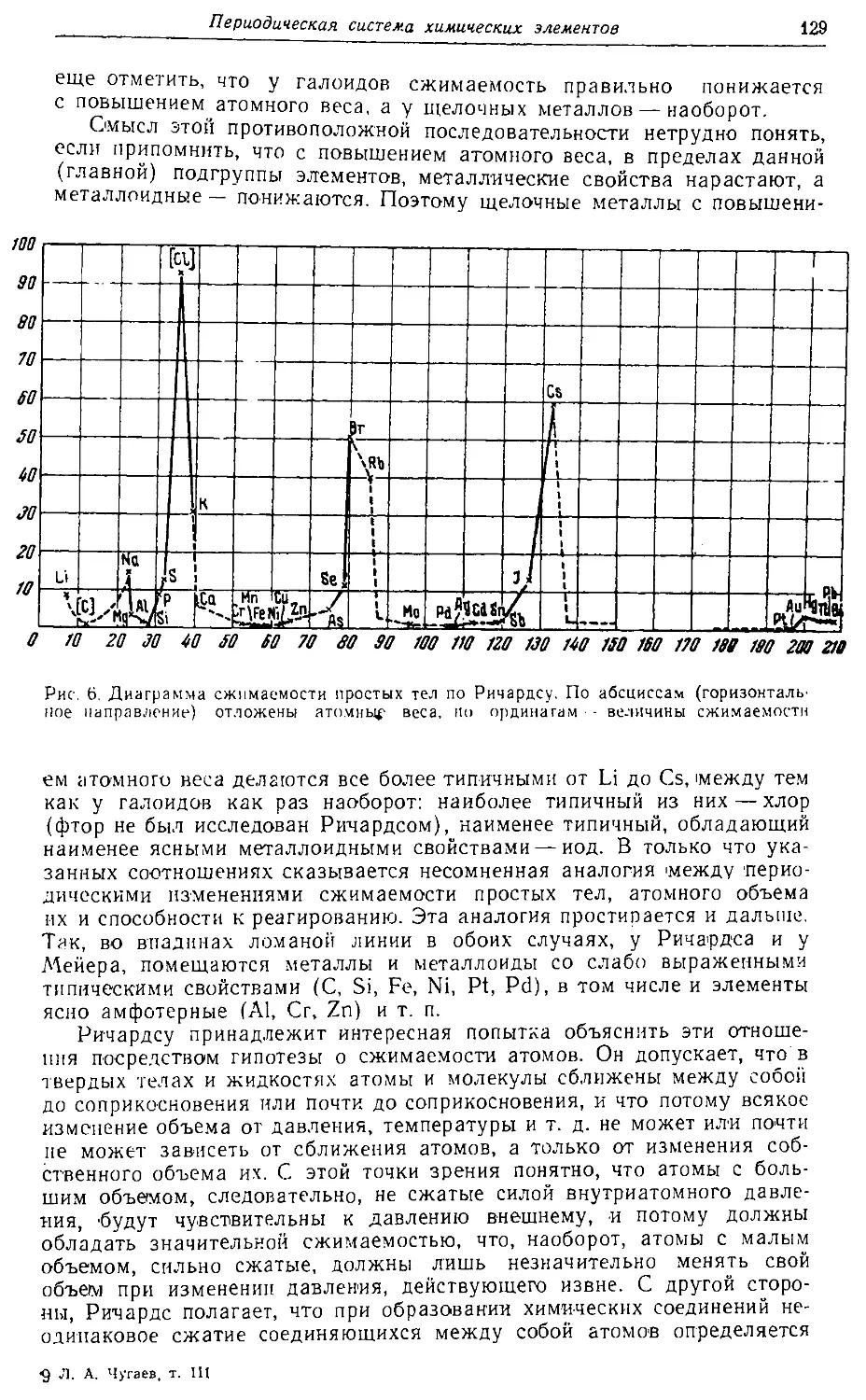
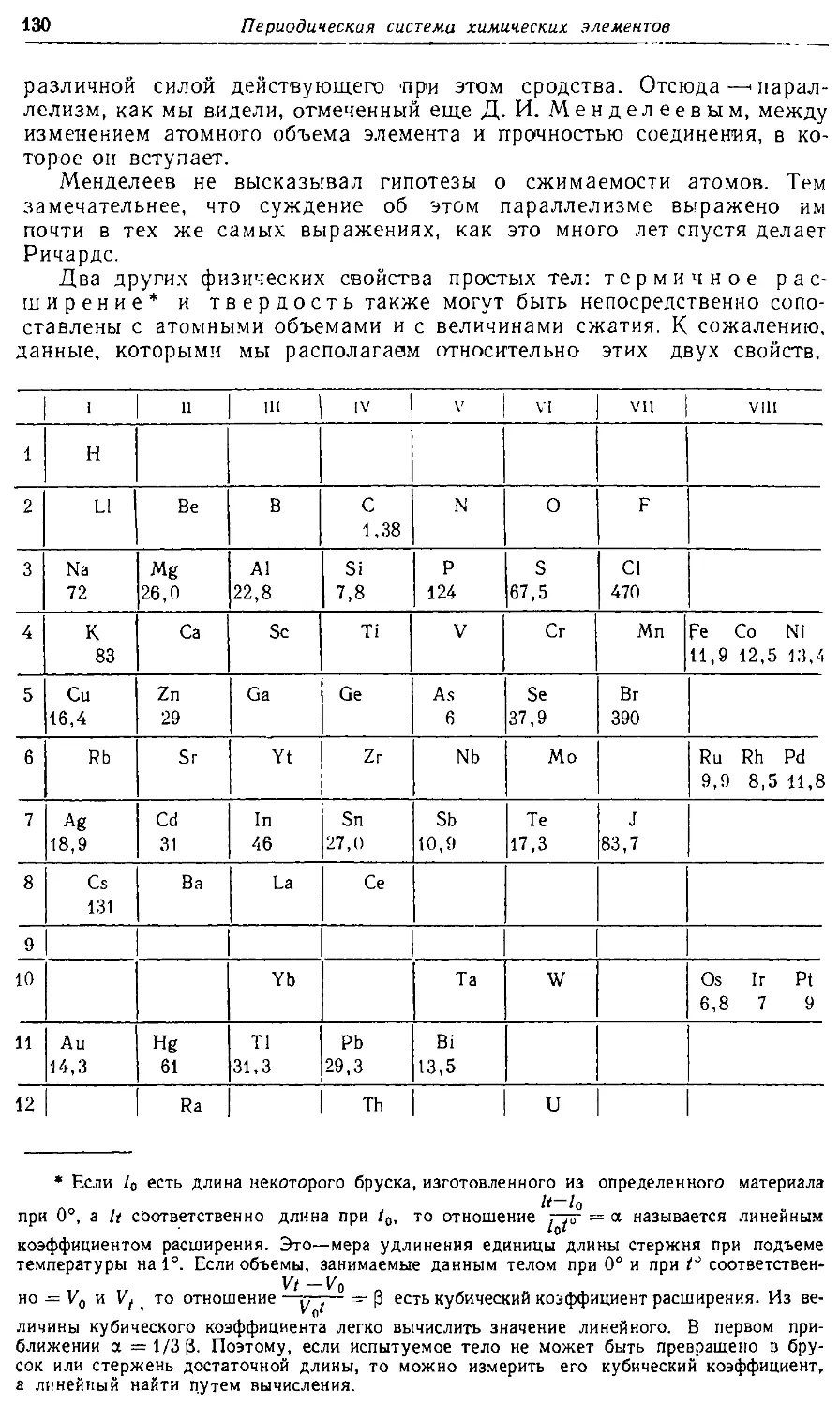
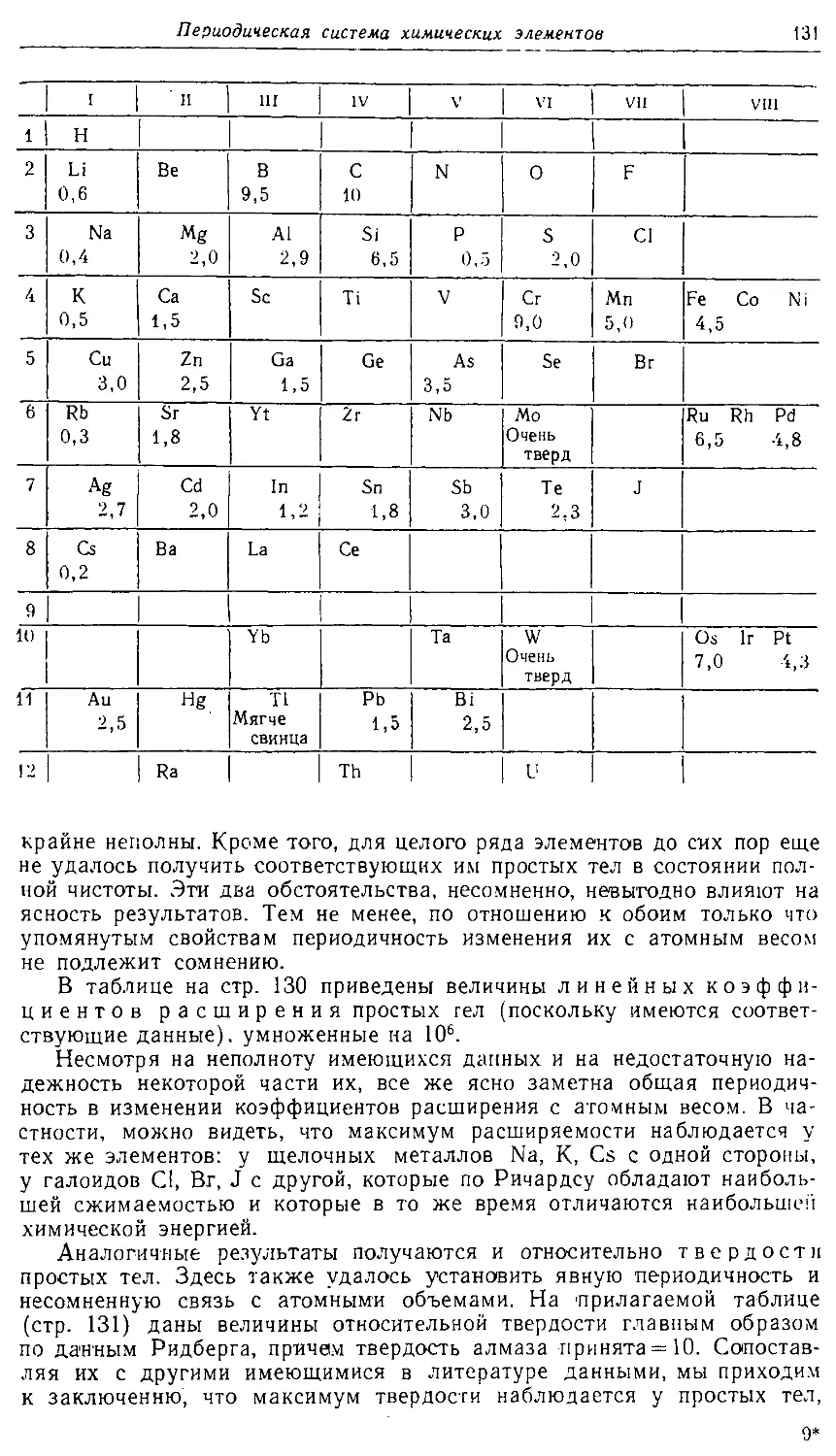
Третий, заключительный, том избранных трудов выдающегося
советского ученого профессора Льва Александровича Чугаева
содержит работы общего характера, относящиеся к тем вопросам
химии, которые интересуют химиков всех направлений:
периодический закон, стереохимия, валентность, а также
научно-популярные статьи, не утратившие интереса в настоящее время.
Кроме того, в третий том включены вводные лекции, читанные
Л. А. Чугаевым в высших учебных заведениях при начале
изложения курсов неорганической и органической химии; эти лекции
могут слукить образцом педагогического такта и глубины научной
мысли.
Перу Л. А. Чугаева принадлежат некрологи некоторых ученых,
характеризующие их выпукло, кратко и в то же время тепло
и-сердечно. Эти некрологи как известных ученых (Ст. Канницаро.
А. М. Зайцев), так и менее известных, но сделавших заметный
вклад в науку, комиссия также сочла нужным включить в издание.
Комиссия поместила в третьем томе работы Л. А. Чугаева по
биохимии, не вошедшие в первые два тома, составленные по
тематическим признакам.
Тексту «Трудов» комиссия предпослала краткую биографию
Л. А. Чугаева, написанную его учеником профессором Г. В. Пи-
гулевским, и статью о нем, составленную также учеником и
сотрудником Л. А. Чугаева — членом-корреспондентом Академии
наук СССР В, В. Лебединским.
Текст Л. А. Чугаева сохранен полностью; исключение
составляют опечатки и явные ошибки, которые исправлены.
В конце тома "помещены примечания комиссии, разъясняющие
некоторые места текста, ссылки на них в тексте помечены
цифрами над строкой.
К тому приложен список трудов Л. А. Чугаева, составленный
О. Е. Звягинцевым, которым были использованы списки, ранее
составленные Э. X. Фрицманом и М. В. Лазаревой.
4
От комиссии
Труды Л. А. Чугаева в третьем томе расположены в
хронологическом порядке. Только труды по биохимии и некрологи вынесены
в конец тома.
Комиссия выражает благодарность члену-корр. АН СССР
Б. М. Кедрову, профессорам Г. Л. Селиберу, Н. А. Фигуровскому и
доценту Н. С. Никитиной, а также сотрудникам Института общей
и неорганической химии АН СССР за ценные советы, оказавшие
помощь при отборе и редактировании статей третьего тома.
В подборе материалов к третьему тому избранных трудов
Л. А. Чугаева и в редактировании некоторой части текста
принимали участие ныне покойные члены-корр. Академии наук СССР
В. В. Лебединский и Н. К. Пшениіцын. Однако им не суждено было
довести это дело до конца.
Г. В. ПИГУЛЕВСКИЙ
ЖИЗНЬ И ДЕЯТЕЛЬНОСТЬ Л. А. ЧУГАЕВА *
Лев Александрович Чугаев родился 5 октября 1873 г. Отец его
Александр Фомич Чугаев был довольно видным московским педагогом.
Александр Фомич получил высшее образование на
физико-математическом факультете Московского университета. Свою педагогическую
деятельность он начал в Рязани, а затем преподавал в гимназии во
Владимире естественную историю. Вскоре А. Ф. Чугаев переехал в Москву,
где продолжалась его долгая педагогическая деятельность. Кроме
естественной истории, он преподавал в московских гимназиях физику и
математику. В последние годы жизни Александр Фомич вел занятия .в
учительской семинарии военного ведомства. Мать Л. А. Чугаева — Анна
Дмитриевна, урожденная Глики, по первому мужу Ипатьева, была
образованной женщиной. Мать Льва Александровича рано умерла от
туберкулеза. Близкое участие в нем приняла подруга матери Софья
Ивановна Мейцингер, полюбившая его, как сына, и заменившая ему
мать. О ней Лев Александрович сохранил самые теплые воспоминания.
Среднее образование Лев Александрович получил в нервом
кадетском корпусе, куда поступил в 1885 г. Он учился прекрасно, однако
физические упражнения ему были тягостны. У юноши было стремление
продолжать образование в университете. С этой целью, после окончания
кадетского корпуса (1889), он сдал экзамен по древним языкам и
поступил в 8-й класс 4-й гимназии, которую отлично окончил.
Восемнадцати лет Л. А. Чугаев был принят на естественное
отделение физико-математического факультета Московского университета.
Отец его умер, и всю заботу о нем приняла на себя Софья Ивановна.
В ту эпоху в университете не было специального химического
факультета, и студент, поступивший на естественное отделение, слушал
общие биологические дисциплины. Пытливого и любознательного юношу
больше привлекали такие науки, как химия и физика. Профессорами в
Московском университете в то время были такие крупные ученые, как
Иван Александрович Каблуков, Владимир Васильевич Марковников, и
начинал свою деятельность Николай Дмитриевич Зелинский.
Свою первую научную работу «О триметоксиглютаровой кислоте»
Лев Александрович выполнил под руководством Н. Д. Зелинского.
В 1895 г. после четырехлетнего пребывания в стенах университета
Л. А. Чугаев блестяще его кончил.
Окончив университет, Л. А. Чугаев поступил в Бактериологический
институт (при Московском университете) на должность помощника
прозектора.
* При составлении биографического очерка были использованы, кроме имеющихся
а литературе сведений (Сб. речей и докладов, посвященных памяти Л. А. Чугаева.
Л., НХТИ, 1924; И. И. Черняев. Лев Александрович Чугаев (по личным
воспоминаниям). Усп. химии, 1945, № 14, 330; Э. X. Ф р-и ц м а н, Светлой памяти Льва
Александровича Чугаеза. Изв. по изуч. платины, 1929, вып. 7, 179), рукописные материалы
М. С. Григорьевой-Сканави, В. В. Лебединского, Л. X. Баталина и краткая
автобиография Л. А. Чугаева.
t)
Г. В. Пигулевский
Директором института был Георгий Норбертович Габричевский,
человек широких взглядов, предоставивший начинающему ученому
благоприятные условия для работы.
В институте Л. А. Чугаевым были выполнены первые
самостоятельные работы по изучению действия ядов на бактерии, по физиологии
фосфоресцирующих бактерий, л о синтезу соединений, близких к гемо-
пирролу. Из этой же лаборатории вышли -и первые его работы по химии
терпенов и оптической деятельности органических соединений, создавших
ему славу отличного химика-органика.
Что побудило Льва Александровича войти в эту область химии
природных соединений, столь отличную от тематики его учителя?
Мы попытаемся ответить на этот вопрос.
Лев Александрович через всю свою жизнь пронес увлечение
проблемами, связанными с жизненными процессами. Об этом свидетельствуют
его первые работы биохимического характера, а впоследствии и статьи,
посвященные химии брожения, эволюции органических соединений в
живой природе,-образованию нефти и т. д. Большая тема оптической
деятельности органических соединений, которой он занимался всю жизнь,
в какой-то степени также связана с биохимическими проблемами. Не
случайно поэтому, выбрав специальность химика-органика, он избрал
область органической химии, имевшую близкое отношение к природным
соединениям, вырабатываемым живыми организмами,— химию терпенов.
Химия терпенов давно служила предметом, интересующим многих
исследователей. С этой трудной областью связаны имена таких ученых, как
Био, Дюма, Кагур, Бертло, Гладстон, Тильден и др. Но расцвет химии
терпенов начался тогда, когда на арену научной деятельности выступили
Вагнер, Флавицкий, Ваалах, Аскан, Бредт, Чугаев, Компа, Наметкин,
Земмлер и др. Особенно много развитие химии терпенов обязано
представителю старшего поколения русских химиков Егору Егоровичу
Вагнеру, давшему знаменитый метод изучения структуры органических
соединений окислением марганцевокислым калием, основанный на
преобразовании сложной молекулы в простую путем постепенных переходов, не
сопровождающихся изомеризацией. Вагнеру мы обязаны выяснением
строения пинена, камфена, лимонена, терпинеола и терпина. Одним из
животрепещущих вопросов в химии терпенов было установление
генетической связи между терпенами и спиртами. Задача заключалась в том,
чтобы найти гладкий переход от спирта к углеводороду без
изомеризации.
Лев Александрович, ознакомившись с литературой ло химии
терпенов, решил заняться этим вопросом. Уже в 1899 г. он сделал сообщение
в Отделении химии Русского физико-химического общества: «Новый
способ получения непредельных углеводородов». В сообщении излагаются
основы ксантогенового метода. Был найден метод, который дал
возможность получать углеводород реакцией, не сопровождающейся
изомеризацией или рацемизацией. Дальнейшее применение метода в полной мере
подтвердило первоначальные предположения и привело к открытию
новых углеводородов: а- и р-туйена, борнилена, изолимонена.
Значение этого открытия не ограничивается лишь фактом
получения новых терпенов.
Проблема взаимоотношения борнеола и камфена, разрешенная
Е.Е.Вагнером, не получила, однако, всеобщего признания. Недоставало
одного звена в этой цепи: получения из борнеола углеводорода,
структурно с ним связанного. Открытие Чугаевым борнилена, сделанное почти
одновременно с Вагнером, было решающим, так как дало недостающее
звено.
Жизнь и деятельность Л. Л. Чугаена
t
В 1903 г. Л. А. Чугаев блестяще защитил диссертацию и получил
степень магистра химии за «Исследования в области терпенов и
камфоры». Это исследование сразу принесло автору известность. После
защиты диссертации Лев Александрович был приглашен в качестве
профессора в Московское техническое училище, где он работал до 1908 г.
В Техническом училище Льву Александровичу приходилось
выполнять педагогические обязанности по двум кафедрам: органической и
неорганической химии.
^Годы пребывания в Техническом училище были заполнены
неутомимой творческой деятельностью, но Лев Александрович круто изменил
характер своей научной работы. Он оставил, в основном, область
терпенов, где так удачно разрешил ряд крупных задач, и перешел к другой
тематике, относящейся к комплексным соединениям. Этот переход не
был случайным.
Льву Александровичу приходилось, как мы уже упоминали, нести
педагогические обязанности и по кафедре неорганической химии. К
своим поручениям он отнесся со всей присущей ему добросовестностью и
ответственностью. Он глубоко вошел в эту область знания. Как
известно, в те годы, примерно в 1893 г., появилась выдвинутая Вернером новая
координационная теория комплексных соединений. Л. А. Чугаев быстро
оценил достоинство новой теории, открывающей возможность
приложения идей стереохимии к неорганическим соединениям. Каждый
металл — комплексообразователь, по словам Чугаева, служит как бы
своей собственной миниатюрной органической химией, в которой как в
зеркале отражаются черты его химической индивидуальности.
По мнению Чугаева, комплексные соединения по своим свойствам
и превращениям обнаруживают удивительную аналогию с соединениями
углерода, рассматриваемыми в органической химии. «Мы имеем здесь,—
говорит Чугаев,— то же самое разнообразие типов, те же градации
в степени подвижности атомов, например, галоидных, в молекулах, те
же, наконец, явления изомерии, как известно, вообще крайне редкие
среди соединений неорганической химии. С этой точки зрения
представляется крайне интересным и важным приступить к систематическому
изучению этих соединений экспериментальными и теоретическими
приемами, которые оказали уже нам огромные услуги в области
органической химии».
Этими словами Лев Александрович объясняет тот логический путь,
который привел его от органической химии к исследованиям в области
химии комплексных соединений.
Переходя к изучению химии комплексных соединений, Чугаев ставил
своей задачей проследить не только самый процесс образования, но и
выявить те причины, которые обусловливают устойчивость этого класса
соединений, их реакционную способность.
В связи с этим Лев Александрович предпринял широкие
исследования комплексных соединений с различными металлами в качестве
центрального атома и различными внутрисферньШи заместителями, как
органическими, так и неорганическими.
Первоначально Чугаев обратил внимание на комплексные
соединения различных металлов (меди, никеля и серебра) с такими
органическими молекулами, как органические имиды и комплексные соединения
никеля, палладия, меди и железа с диоксимами. В результате он
пришел к выводу, что наибольшей способностью к образованию
комплексных соединений обладают аммиак и первичные амины, наименьшей
же— третичные амины. Вторичные амины в этом отношении занимают
промежуточное положение. Гетероциклические амины с пиридиновым
8
Г. В. Пигулевский
кольцом по способности образовывать комплексные соединения
примыкают к первичным аминам.
Эти же работы Чугаева привели его к открытию «правила циклов»,
сводящегося к тому, что циклообразование значительно повышает
устойчивость комплексного соединения, в особенности в том случае, когда эти
соединения содержат во внутренней сфере пятичленный цикл. Лев
Александрович подметил, что этот цикл всегда замыкается только в цис-по-
ложении.
Работы Чугаева по изучению комплексных соединений с диоксимами
привели к открытию в 1905 г. в высшей степени чувствительной реакции
на никель и па палладий с диметилглиоксимом. Эти исследования
легли в основу работ целого ряда иностранных и отечественных ученых
и создали новую эпоху в аналитической химии. Мы знаем, что это
направление в аналитической химии в наши дни стало весьма
плодотворным.
В связи с этим Чугаев должен быть назван пионером в области,
создавшей действительно новую эпоху в аналитической химии,— области
применения органических реактивов к методам химического анализа,
так как только после его работ по комплексным соединениям с а-диок-
симами это направление аналитической химии получило должное
развитие в мировой науке.
К периоду пребывания в Техническом училище относится и
опубликование исследования, посвященного качественному и количественному
определениям гидроксильных групп. Это метод, известный как метод
Чугаева — Церевитинова, вошел в практику лабораторий органического
анализа.
В 1907 г. Петербургский университет предложил Чугаеву занять
кафедру неорганической химии. Лев Александрович долго колебался. Ему
не хотелось порывать связь с местом, где удалось так много сделать. Но
Петербургский университет открывал перед ним широкие перспективы
научной деятельности. Лев Александрович дал согласие, и 24 марта
1907 г. был издан приказ о зачислении 34-летнего ученого профессором
Петербургского университета. 11 сентября состоялась его вступительная
лекция «Эволюция вещества в мертвой и живой природе». В лекции
были затронуты основные вопросы неорганическое химии и химии
природных соединений. Лектор показал, с одной стороны, картину эволюции
знаний о химических элементах и развития учения о их взаимном
родстве, а с другой — эволюцию химических соединений на земле, в
особенности биохимическую эволюцию, идущую параллельно с
морфологической.
Лев Александрович был прекрасным лектором. Его живые, полные
глубокого содержания лекции привлекали молодежь. Количество
желавших работать под его руководством беспрерывно возрастало,
достигнув в 1913 г. невиданной цифры — 25 человек.
В 1909 г. Лев Александрович был приглашен в Петербургский
технологический институт. Здесь Чугаев с присущей ему энергией, кроме
чтения курса по органической химии, поставил научно-исследовательскую
работу.
В Петербургском университете Лев Александрович развивает работы
в области химии комплексных соединений, главным образом
платиновых металлов.
Причиною выбора Чугаевым для научных исследований именно
области комплексных соединений платиновых металлов послужило то
обстоятельство, что, как известно, эти металлы дают многочисленные
весьма устойчивые комплексные соединения, при исследовании' которых
Жизнь и деятельность Л. А. Чугае?а 9
можно не остерегаться тех самопроизвольно протекающих процессов
изомеризации, которые так характерны для комплексных соединений
других металлов, например кобальта и хрома. Можно было
рассчитывать на примерах комплексных соединений платиновых металлов
лучше изучить динамику внутренней сферы и легче вскрыть те
закономерности, которые лежат в ее основе.
Исследования комплексных соединений именно платиновых
металлов представляли интерес еще и потому, что они могли наметить
новые пути открытия и выделения этих металлов в чистом состоянии из:
природного сырья.
Изучая комплексные соединения платиновых металлов, Чугаев
получает соединения платины с органическими сульфидами, тиоэфирами,
нитрилами, изонитрилами, гидразином, гидроксиламином и т. д. Он
установил строение изомерных соединений двухвалентной платины с
органическими сульфидами, исследовал изомерию комплексных соединений
с нитрилами, гидразином, гидроксиламином и т. д.
В петербургский период жизни Л. А. Чугаев продолжал, хотя и в
меньшей степени, работы в области органической химии. Эти
исследования отчасти явились продолжением его трудов по терпенам, отчасти
затрагивали другие природные соединения (холестерин, смоляные
кислоты и т. д.).
Выше мы упоминали, что Лев Александрович в молодые годы
опубликовал работы по оптической деятельности органических соединений.
Интерес к указанной проблеме его никогда не покидал. Л. А. Чугаев
установил связь между оптическою деятельностью соединений и их
химическим строением («правило удаления», принцип постоянства
молекулярного вращения). Его выдающиеся исследования в области
вращательной дисперсии дали исключительно важные результаты. Льву
Александровичу удалось наблюдать эффект Коттона на индивидуальных
соединениях, им синтезированных. Чугаев открыл новый тип аномальной
вращательной дисперсии, обусловленной совместным присутствием в
молекуле двух асимметрических центров, вызывающих парциальное
вращение противоположных знаков с различными коэффициентами
дисперсии. Этот новый тип аномальной дисперсии Лев Александрович
назвал внутримолекулярной дисперсионной аномалией. В своих последних
работах он предсказал и доказал экспериментально, что и в
соединениях, содержащих один асимметрический центр, можно наблюдать
явления аномальной дисперсии.
Во всех этих исследованиях Лев Александрович исходил из
существования глубокой связи между электронным спектром поглощения и
оптической деятельностью.
Нельзя не отметить той большой популярности, которой пользовался
Лев Александрович за границей. Его приглашают принять участие в
международных конгрессах, где он работает в ряде комиссий, по
приглашению Британской ассоциации и Фарадеевского общества читает
доклады и обзоры. Он переписывается с крупнейшими европейскими
химиками Рамзаем, Вернером, Оствальдом, Галлером и другими.
В 1913 г. к Л. А. Чугаеву обращаются как «к одному из отличнейших
химиков Европы» за отзывом о работах Паттерсон и Макензи по
оптической деятельности и стереохимии. В 1915 г. к нему поступает запрос
о кандидатурах на Нобелевскую премию по химии. Круг деятельности
Л.А.Чугаева не замыкался научно-исследовательской и педагогической
работой. Его влекло к широкой популяризации дорогих ему идей. Лев
Александрович написал монографии по различным вопросам химии:
<0 химическом строении комплексных соединений», «Периодическая
10
Л В. Пигулевский
система элементов», «Открытие кислорода и теория горения» и т. д.
Кроме того, им напечатан ряд популярных статей по химии,
бактериологии и т. д. в ряде журналов: «Научное обозрение», «Природа» и др.
Лев Александрович, глубоко переживавший судьбы родины, отдавал
ей все силы. Во время первой мировой войны он участвовал в съезде
по борьбе с лекарственным голодом и по мобилизации отечественной
химии. Он был одним из основателей и активным руководителем
Института прикладной химии и опытного завода при этом учреждении; с
самого начала войны работал в Комиссии при Академии наук по изучению
естественных производительных сил России.
После Великой Октябрьской социалистической революции Л. А. Чу-
гаев энергично берется за организацию Института по изучению платины
и других благородных металлов при Академии наук. Директором этого
института он остается до конца жизни. Кроме того, Лев Александрович
был членом президиума и ученым секретарем Института прикладной
химии. В 1917—1921 гг. он принимает деятельное участие в организации
завода химически чистых реактивов и препаратов, участвует в
совещании по реформе университетов и т. д.
Кипучая деятельность Л. А. Чугаева оборвалась внезапно.
Отправившись в Вологду, чтобы перевезти свою семью в Петроград, он заболел
брюшным тифом. Истощенный организм не смог преодолеть болезни.
23 сентября 1922 г. Льва Александровича не стало.
Л. А. Чугаев умер 49 лет от роду, в полном расцвете таланта. Его
смерть была тяжелой потерей не только для нашей страны.
Международная семья химиков, выдающимся членом которой он был, понесла
тяжелую утрату.
Сейчас, когда со смерти Чугаева прошло почти 40 лет, можно
объективно оценить вклад, внесенный Львом Александровичем в химическую
науку. Его идеи, которые он не успел претворить в жизнь, с успехом
осуществляются его учениками. Реакции, им открытые, служат в
настоящее время предметом глубокого изучения. Нам, ученикам, дорог
светлый образ учителя, который был столь беззаветно и горячо предан науке.
*«8а*
В. В. ЛЕБЕДИНСКИЙ]
ЗНАЧЕНИЕ РАБОТ Л. А. ЧУГАЕВА В РАЗВИТИИ
ХИМИИ*
Русская химическая наука горда такими именами, как М. В.
Ломоносов, Д. И. Менделеев, А. М. Бутлеров, К. К. Клаус, Н. С. Курнакоов,
С. В. Лебедев, Л. А. Чугаев. Имена этих ученых, связанные с
величайшими открытиями и достижениями в области химии, известны всему
миру. На значении в развитии химии работ одного из славной плеяды
русских химиков — Льва Александровича Чугаева — я хочу
остановиться в настоящем докладе.
Л. А. Чугаев своими работами в области неорганической и
органической химии стяжал себе мировую славу. Как бывший профессор
Ленинградского государственного университета он явился одним из
ближайших преемников Д. И. Менделеева, кафедру которого занимал в
течение 14 лет (с 1908 по 1922 г.). В продолжение всей своей научной
деятельности Лев Александрович свято хранил традиции своего великого
предшественника и высоко держал знамя русской науки.
Исключительная эрудиция и многогранность научной деятельности Л. А. Чугаева
особенно близко напоминают облик и деятельность великого творца
периодического закона Д. И, Менделеева.
Лев Александрович Чугаев принадлежал к числу тех ученых,
которые целиком отдавали себя служению науке. Личная жизнь для него,
казалось, не существовала: он редко развлекался, редко и мало
отдыхал. В дни, свободные от лекций, он с утра и до позднего вечера
занимался экспериментальной работой.¦ Несколько раз в день Лев
Александрович обходил своих учеников и сотрудников в лаборатории,
интересовался ходом их экспериментальных работ и вел беседы на научные
темы. Наскоро позавтракав или пообедав, он снова спускался в
лабораторию к своему любимому делу и здесь, у лабораторного стола,
оставался до позднего вечера.
Своей любовью к науке и научным энтузиазмом Л. А. Чугаев
заражал и нас, своих учеников и сотрудников. Мы часто оставались в
лаборатории до поздней ночи, а иногда даже и до утра. Часто, бывало, среди
ночи Лев Александрович в ночном халате приходил к нам и
расспрашивал о ходе работы; он проявлял нетерпение, ему хотелось скорее и
скорее идти вперед по намеченному пути.
Лев Александрович показывал пример исключительной
работоспособности и много работал не только в лаборатории, экспериментально,
но и дома, ночью, за письменным столом, среди книг и журналов. Огонь
в его кабинете горел неизменно до 4—5-часов утра. Можно сказать, что
Лев Александрович сам горел на работе. Беззаветное служение науке
и горячо любимой родине, связанное с почти полным отказом от личной
жизни, было характерной особенностью его научной деятельности.
* Доложено 111 Совещанию по химии комплексных соединений 13 ноября 1944 г.
в Москве.
12
В. В. Лебединский
Первоначальные работы Л. А. Чугаева были посвящены вопросам
биологической химии и бактериологии. Им были произведены
исследования по действию ядов на бактерии, по физиологии фосфоресцирующих
бактерий, по синтезу соединений, близких к гемипирролу и др.
Исследуя действие ядов на микроорганизмы, Л. А. Чугаев ставил себе целью
выявить зависимость между химическим строением вещества и его
ядовитыми свойствами. Эти работы показали, что присутствие альдегидной
группы в органическом соединении обусловливает его ядовитость,
причем эта ядовитость понижается с увеличением молекулярного веса
альдегида и еще в гораздо большей степени — при переходе от альдегида
к соответствующему кетону или ацеталю, а также при введении в
частицу альдегида гидроксильиых групп. Введение же фенильной группы
или галогена в алифатическую часть молекулы альдегида или кетона
значительно повышает их ядовитые свойства.
Интересны работы Л. А. Чугаева по изучению жизнедеятельности
фосфоресцирующих бактерий. Он ставил себе задачей выяснить вопрос
о том, существует ли какая-нибудь зависимость между концентрацией
сахара в питательной среде и интенсивностью свечения
фосфоресцирующих бактерий. Эти работы показали, что лишь очень малые
концентрации (около 0,1%) углеводов оказывают благоприятное влияние на рост
и свечение бактерий. Продолжая дальше эти работы, Лев
Александрович в 1898 г. дал новую теорию дезинфицирующего действия оксидазы.
Почти одновременно появились работы Л. А. Чугаева и в области
чисто органической химии. Он изучал терпеновые углеводороды и
открыл новый, ксантогеновый метод получения терпеновых углеводородов.
Этот метод ценен тем, что он дает возможность осуществлять переход от
спиртов к соответствующим непредельным углеводородам и таким
образом получать терпеновые угловодороды в чистом виде, без примеси
изомерных слединений. Сущность этого метода заключается в том, что
спирт, подлежащий превращению в непредельный углеводород, после
переведения его в алкоголят подвергается действию сероуглерода. При
этом образуется соответствующий ксантогенат, например:
C10N19ONa+CS2 ->С10Н19—О—C=S.
\
S—Na
Полученный ксантогенат при взаимодействии с йодистым метилом
или с диметилсульфатом превращается в соответствующий
ксантогеновый эфир:
Сі0Н19—О—C=S+CH3J -* С1оН19—О—C=S+NaJ.
\ \
S—Na S-CH3
Последний при осторожном нагревании разлагается с образованием
соответствующего углеводорода:
С10Н19—О—C-S — C10H18+COS+CH3SH.
\
S—СН3
Ксантогеновый метод Л. А. Чугаева дал ему возможность не только
получить впервые оптически и структурно однородные изомерные мен-
тены, недеятельный пипен и другие углеводороды, но и осуществить
разделение изомерных алкоголей, получающихся совместно при синтезе и
встречающихся в природных продуктах, и, наконец, обогатить науку
открытием ряда новых углеводородов, как, например, а- и р-туйены,
туйан, борнилен, изолимонен и др. Получением борнилена из борнеола
Значение работ Л. А. Чугаева в развитии химии
13
Л. А. Чугаев показал, что взгляды прежних исследователей на процесс
дегидратации борнеола неправильны. Ксантогеновый метод, наконец,
дал возможность Льву Александровичу ближе подойти к вопросам,
связанным со строением холестерина, и получить новые углеводороды—<х-
и {3-холестерилены, дающие при гидрировании холестан, и, наконец,
показать, что одна из смоляных кислот — пимаровая С20Н30О2— содержит
только одну двойную связь.
Я не имеют возможности здесь останавливаться на других работах
Л. А. Чугаева в области органической химии, например, посвященных
вопросу о составе некоторых природных эфирных масел и выделению из
них индивидуальных химических соединений и т. п. Я хочу лишь
отметить, что уже из этого краткого обзора мы видим, что работы Л. А.
Чугаева в области органической химии являются крупнейшим вкладом
русской науки в сокровищницу мировой химической науки, они не
только дали в руки исследователей упомянутый метод получения терпеновых
углеводородов, но и обогатили науку открытием ряда новых соединений
и внесли ясность в крайне сложную область органической химии —
химию терпенов. Эти работы, наконец, дали толчок к развитию новых
исследований в области физико-химических (оптических) свойств
некоторых классов органических соединений.
Еще в 1898—1902 гг. Л. А. Чугаев выполнил исследования по
изучению зависимости величины угла вращения плоскости поляризации
света от гомологии и указал при этом на появление максимума в
молекулярном вращении («правило Чугаева»). Эти работы Л. А. Чугаева
по изучению оптических свойств органических соединений получили
новое и более широкое развитие после того, как он открыл свой
«ксантогеновый» метод, давший ему возможность располагать исключительно
чистыми, оптически деятельными соединениями.
Как известно, еще в 1838 г. Био открыл явление так называемой
аномальной вращательной дисперсии, сводящееся в основном к тому, что
для бесцветных, оптически деятельных веществ величина угла вращения
плоскости поляризации света не возрастает, а наоборот, падает в связи
с изменением длины волны проходящего света; часто для такого рода
соединений дисперсионная кривая имеет некоторый максимум. Био
объяснял аномальную вращательную дисперсию явлением оптической
суперпозиции, обусловленной в свою очередь неоднородностью
исследуемого вещества.
В 1916 г. Л. А. Чугаев открыл новый вид аномальной вращательной
дисперсии, близкой к аномалии типа Био, но обусловленной не
неоднородностью исследуемой смеси, а наличием в молекуле однородного
вещества двух противоположно вращающих атомов углерода.
Продолжая исследования окрашенных соединений и изучая
параллельно абсорбционные спектры и вращательную дисперсию на
примере некоторых сложных эфиров, Л. А. Чугаев со своими
учениками А. Г. Огородниковым, Г. В. Пигулевским, В. М. Пастаноговым,
А. А. Глебко, В. В. Лебединским и др. показал, что явление Коттоиа,
заключающееся в том, что максимум дисперсионной кривой лежит в
непосредственной близости к полосе поглощения в видимой части спектра,
должно быть распространено также и на вещества, у которых
абсорбционные полосы лежат в ультрафиолетовой части спектра.
Одним из практических результатов этого рода работ Л. А. Чугаева
явился предложенный им метод анализа жидких смесей с оптически
деятельными компонентами, основанный на измерении вращательной
дисперсии. Эти работы послужили толчком к развитию цикла работ
М. А. Ракоузина по исследованию нефти оптическими методами.
14
В, В. Лебединский
Работы Л. А. Чугаева в области исследования оптических свойств
органических соединений получили широкое признание не только у нас.
но и далеко за пределами нашей родины. Для иллюстрации можно
отметить, что в 1914 г. Л. А. Чугаев был приглашен президиумом Фарадеев-
ского общества в Лондон для прочтения доклада «Об аномальной
вращательной деятельности».
Л. А. Чугаев создал вокруг себя крупную школу химиков-органиков,
среди которых следует указать такие имена, как Ф. В. Церевитинов.
Г. В. Пигулевский, В. А. Фомин, Н. А. Шлезингер, А. А. Глебко и др.
Многие из этих ученых впоследствии создали свои школы, ценными
работами завоевали широкую известность и тем упрочили славу своего
учителя.
Я позволю себе перейти к работам Л. А. Чугаева в области
комплексных соединений и хотя бы вкратце остановиться на рассмотрении
достигнутых им в этой области результатов, с тем чтобы показать, какое
значение имели эти работы в развитии нашей науки.
Близкая аналогия между органическими углеродсодержащими
соединениями и комплексными соединениями с металлическим
центральным атомсмг как в смысле наличия у них пассивных сопротивлений и
прочности связи внутри молекул, так и проявления ими изомерии,
естественно, не могла не привлечь внимания Льва Александровича, и,
действительно, мы видим, что Л. А. Чугаев скоро переходит к исследованию
комплексных соединений и до конца своих дней продолжает работать
главным образом в этой области. Недаром Лев Александрович избрал
эпиграфом к своей книге «О химическом строении комплексных
соединений» следующие слова Д. И. Менделеева: «Можно думать, что
некоторые особенности реагирования органических соединений (например,
трудное, не солеобразное, реагирование продуктов мегалепсии, малая
скорость многих реакций и т. п.) зависят от тех же причин, какие ведут
к образованию «комплексных» соединений. Для предстоящих периодов
химии здесь, по моему мнению, содержится не одна из важнейших и
интереснейших задач, могущих осветить и смутно понимаемую область
растворов, и природу сил, управляющих образованием соединений».
По мнению Л. А. Чугаева, «комплексные соединения по своим
свойствам и превращениям обнаруживают удивительную аналогию с
соединениями углерода, рассматриваемыми в органической химии».
Первоначальными объектами исследований Л. А. Чугаева в области
комплексных соединений были соединения меди, никеля и серебра с
органическими имидами, а также соединения никеля, меди, железа,
кобальта, платины и палладия с а-диоксимами. Эти работы Л. А. Чугаева
имеют громадное значение в развитии учения о свойствах и строении
комплексных соединений. Лев Александрович показал, что имиды дикар-
боновых кислот, обменивая один атом водорода на металл, дают
комплексы, в свою очередь способные вступать в соединения с молекулами
аммиака и аминами жирного ряда, образуя при этом характерные и
достаточно прочные комплексные соединения. Исследование диоксиминов
показало, что из различных диоксимов только а-диоксимы образуют
действительно стойкие внутренние комплексные соединения благодаря
замыканию цикла. Это привело Л. А. Чугаева к выводу, что главным
образом пятичленный цикл является наиболее стойким («правило
циклов» Л. А. Чугаева). Указанная закономерность послужила отправным
пунктом для дальнейшего развития работ в области стереохимии
комплексных соединений и дала возможность одному из его учеников
(А. А. Гринбергу) впоследствии предложить метод определения
конфигурации комплексных соединений платины, основанный на способности
Значение работ Л. Л. Чугаева в развитии химии
15
оксалатного остатка замыкать пятичленный цикл в случае цис-положе-
ния замещаемых аддендов и давать биоксалаты в случае их
транс-конфигурации.
Исследование диоксиминов привело Л. А. Чугаева к открытию
исключительно чувствительной реакции на никель, дало возможность
предложить метод количественного определения железа и легло в основу
работ иностранных химиков Брунка, Атака, Вундера и Тюрингера по
методике аналитического определения никеля и палладия.
Не ограничиваясь исследованиями комплексных соединений меди,
никеля, кобальта и других неблагородных металлов, Л. А. Чугаев очень
скоро расширил круг своих исследований и перешел к широкому и
планомерному изучению комплексных соединений платиновых металлов.
Интерес Льва Александровича к исследованию комплексных
соединений платиновых металлов объясняется тем обстоятельством, что, по
его словам, «...каждый элемент платиновой группы является как бы
своей собственной, миниатюрной органической химией».
Мы видим, что Л. А. Чугаев был приверженцем и апологетом
координационной теории Вериера. • Своими работами он исключительно
много способствовал развитию и укреплению этой теории.
Исследование комплексных соединений платиновых металлов
особенно привлекало Льва Александровича еще и потому, что платиновые
металлы как типичные комплексообразователи дают наиболее
характерные и прочные комплексы. Поэтому можно было рассчитывать на
примерах этих соединений лучше изучить динамику внутренней сферы и легче
вскрыть закономерности, которые лежат в ее основе. Изучение
комплексных соединений платиновых металлов должно было дать в руки
исследователя новый ключ к более полному познанию химической
индивидуальности этой крайне ценной группы металлов и одновременно с этим
наметить новые пути к методике их разделения и получению их в чистом
виде.
Развивая свои исследования в области комплексных соединений
платиновых металлов, Лев Александрович изучает соединения платины
с органическими сульфидами, тиоэфирами, изонитрилами, нитрилами,
гидразином, гидроксиламином и т. д. Эти работы Л. А. Чугаева имеют
фундаментальное значение в развитии учения о строении комплексных
соединений вообще и комплексных соединений платиновых металлов в
частности. Лев Александрович установил строение изомерных
соединений двухвалентной платины с органическими сульфидами и
изонитрилами, исследовал изомерию комплексных соединений платины с
нитрилами, гидразином, гидроксиламином и т. д. Ему удалось наблюдать
чрезвычайно интересное явление молекулярных перегруппировок, а
также явления деполимеризации в ряду комплексных соединений
двухвалентной платины; он изучал явления окисления комплексных соединении
платины различными окислителями, синтезировал соединения
«трехвалентной» платины; синтезировал предсказанный теорией Вернера,
по отсутствовавший до тех пор член ряда аммиачных соединений
четырехвалентной платины —пентамминхлорид (Pt(NH3)5Cl]Cl3 и т. д. По
постановлению IV Менделеевского съезда в 1925 г., уже после смерти
Л. А. Чугаева, эта соль, ввиду ее важного значения в развитии учения
о строении комплексных соединений, была названа «солью Чугаева».
Л. А'. Чугаев впервые получил так называемые «сверхкомплексные»
соединения, в которых молекулы готовых координационно-насыщенных
комплексных соединений взаимно сочетаются друг с другом с
образованием нового класса сложных соединений, природа которых в настоящее
время так настойчиво изучается нашими учеными.
16
В. 5. Лебединский
Не только платина, но и ее спутники — палладий, иридий, родий и
осмий — также привлекли к себе внимание Л. А. Чугаева. Он изучил
диоксимины палладия и родия, гидразиновые соединения иридия,
производные осмиамовой кислоты, комплексные соединения осмия с
уротропином и тиомочевиною и т. п. Им с очевидностью установлена кислотная
природа четырехокиси осмия, открыта чувствительная реакция на осмий
с тиомочевиной, исключительно чувствительная реакция на платину с
к&рбиламином на иридий с малахитовой зеленью .и т. д.
Даже беглый просмотр и почти простое перечисление работ и
достижений Л. А. Чугаева в области неорганической химии заняли достаточно
много места. Необходимо добавить, что Л. А. Чугаев создал вокруг
себя крупнейшую школу химиков-неоргаников, которые собственными
исследованиями и исследованиями своих учеников способствовали
дальнейшему развитию неорганической химии вообще и химии комплексных
соединений в частности. Эта школа Л. А. Чугаева, школа русских
химиков-неоргаников, среди которых необходимо упомянуть академиков
И. И. Черняева, В. Г. Хлопина, А. А. Гринберга,
членов-корреспондентов В. В. Лебединского, Н. К- Пшеницына и других, завоевала
себе широкое признание не только в нашей стране, но и далеко за ее
пределами. Русская, «чугаевская» школа химиков-неоргаников является
в настоящее время одною из крупнейших в мире ячеек, ведущих
систематические исследования в области химии комплексных соединений.
Я должен еще остановиться на его работах, имеющих прикладной
характер, и на его деятельности как основателя и директора
Института по изучению платины и других благородных металлов.
Мировая война 1914—1918 гг. не могла, конечно, не коснуться
Льва Александровича, не могла не втянуть его в свою орбиту. В его
лаборатории велись работы но изготовлению ряда дефицитных
реактивов и лекарственных веществ.
Лев Александрович принимал горячее участие также в организации
и работах Института прикладной химии и опытного завода. На
опытном заводе по его инициативе и под его руководством были
организованы и велись работы по изготовлению (впервые в нашей стране)
красного фосфора, фосфорного ангидрида, хлористого хромила,
металлического мышьяка и т. п. Наконец, Л. А. Чугаев деятельно участвовал и в
работах Пищевого института.
В связи с возросшей нуждою отечественных химических заводов в
больших количествах контактной массы для производства серной
кислоты, по предложению Химического комитета при Главном
артиллерийском управлении, Л. А. Чугаев и его ближайшие ученики В. Г. Хлопин
и В. В. Лебединский разрабатывали методы аффинажа платины и
некоторых ее спутников.
Когда в 1916 г. при Академии наук по инициативе академика
В. И. Вернадского возникла Комиссия по изучению естественных
производительных сил России (КЕПС), Л. А. Чугаев оказался снова в первых
рядах русских ученых, откликнувшихся на призыв Комиссии. На ее
совещаниях Л. А. Чугаев горячо выступал по вопросу об отечественной
платине и ратовал за то, чтобы сырая платина и ее остатки не
вывозились за границу в непереработанном виде, выступал с предложением
об установлении государственной монополии на добычу и переработку
платины, и, наконец, предложил создать в России Государственный
институт по изучению платины и других благородных металлов.
Основными задачами этого Института, по его мнению, должны были быть
следующие:
1. Всесторонее научное-исследование металлов платиновой группы;
Значение работ Л. А. Чугаева в развитии химии М
изыскание новых полезных сплавов, образуемых металлами платиновой
группы.
2. Систематическое изучение комплексных соединений с
приложением к этой области различных физических и физико-химических
методов исследования, отыскание новых химических функций и новых
классов комплексных соединений, новых общих реакций образования и
превращения этих соединений, общих правильностсй и надежных методов
для определения их строения и конфигурации.
3. Разработка и усовершенствование методов аффинажа и анализа
платиновых металлов.
4. Изыскание методов использования относительно бедных платиной
отвалов и пород.
Только после Великой Октябрьской социалистической революции, в
1918 г. при Академии наук был создан Институт по изучению платины
и других благородных металлов. Л. А. Чугаев стал во главе этого
института. Он был первым его директором. В состав ученого совета
института в то время входили: Н. С. Курнаков, В. И. Вернадский и Ф. Ю. Ле-
вннсон-Лессинг. Членами института были: И. И. Черняев, В. Г. Хлопин,
Э. X. Фрицман, В. В. Лебединский, Н. И. Подкопаев и Н. И. Степанов.
Делопроизводителем Института был Н. К. Пшеницын.
Институт по изучению платины был любимым детищем Л. А.
Чугаева. Он жил с ним одной жизнью, радовался его успехам и
огорчался его невзгодам.
По инициативе Л. А. Чугаева с 1920 г. институт стал издавать свой
специальный журнал «Известия Института по изучению платины и
других благородных металлов». Впоследствии этот журнал продолжал
выходить под наименованием «Известия сектора платины и других
благородных металлов».
Совершенно неожиданно для всех, в самом разгаре своей кипучей
деятельности, еще в сравнительно молодом возрасте, 49 лет, 23 сентября
і922 г. Лев Александрович Чугаев скончался. Институт по изучению
платины осиротел. Осиротела дружная семья молодых исследователей,
стремившихся в-перед по научному 'пути, указанному любимым учителем.
Но жизнь требовала продолжения начатой работы. Перед
Институтом стоял еще ряд неразрешенных научных задач и вопросов. Институт
возглавил новый директор — академик Н. С. Курнаков.
Льву Александровичу не суждено было дожить до момента тесного
сближения Института платины с отечественной промышленностью. Уже
после смерти Л. А. Чугаева, в 1922 г. промышленные организации
обратились к институту со своими практическими нуждами и запросами,
связанными с необходимостью срочной разработки методики аффинажа
и анализа платиновых металлов. В связи с этим в институте были
созданы специальные аффинажная и аналитическая комиссии. Работа
Института платины вошла в новое русло, мысль Л. А. Чугаева о тесном
сочетании теории с практикой начала осуществляться. Работники
института, вооруженные теорией и широко знакомые с химией платиновых
металлов, смогли использовать свой научный опыт и в сравнительно
короткий срок оказать существенную помощь развитию отечественное
платиновой промышленности. Те, кто знает достижения института в этой
области, видят, насколько был прав Л. А. Чугаев, когда говорил, что
«...каждый научно-обоснованный вывод или сближение, каждая
закономерность, каждый точно установленный факт, касающийся хи-м-ии
-платиновых металлов, рано или поздно будет иметь свой практический
эквивалент». И действительно, благодаря работам Института по изучению
платины наша молодая платиновая промышленность очень скоро прочно
2 Л. А. Пугаем*. і. Ill
18
В. В. Лебединский
встала на ноги и смогла с успехом конкурировать с иностранными
промышленными организациями.
Подвожу итоги тому, что оказано о значении работы Чугаева в
развитии химии.
Имя Л. А. Чугаева широко известно не только в нашей стране, но
и далеко за ее пределами. Еще в 1913 г. -к Л. А. Чугаезу, как кі одному
из «отличнейших химиков Европы», обратились английские ученые с
просьбой дать отзыв о работах Паттерсона и Макензи по оптической
деятельности и стереохимии в связи с выдвижением их в качестве
кандидатов на кафедры органической и неорганической химии в г. Дэнди.
Его мнение запрашивалось также при выборе кандидатур на
Нобелевскую премию по химии. В нашей стране значение работ Л. А. Чугаева
было отмечено присуждением ему премии имени В. И. Ленина.
Л. А. Чугаев как выдающийся лектор всегда привлекал на свои
лекции громадное число слушателей и сумел в сердцах многих из них
зажечь огонь любви к науке и к своей родине. Л. А. Чугаев был
прекрасным популяризатором науки: не только лекции для широких кругов
слушателей, но и его научно-популярные книги и статьи в значительной
мере способствовали распространению и развитию химических знаний.
Смерть Л. А. Чугаева как тяжелая утрата была отмечена не только
в нашей стране, но и за ее пределами.
В американском химическом журнале «Chemical Reviews» за 1943 г.
был рассмотрен вопрос изучения платиновых металлов. Автор обширной
статьи Р. Джилкрист относит к числу ведущих исследователей
в этой области прежде всего русских ученых и в первую очередь
Л. А. Чугаева и его учеников, отмечая, что максимальное '«исло
научных статей (28%) по химии платиновых металлов за последние
25 лет является вкладом русских ученых. Мы знаем, чго ближайшие
ученики и сотрудники Л. А. Чугаева продолжали дело своего учителя.
Сперва их работа велась в Институте платины, а затем после слияния
его в 1934 г. с Химической лабораторией Академии наук и с Институтом
физико-химического анализа м переезда в Москву — в стенах Института
обшей и неорганической химии Академии наук СССР (ИОНХ). Ученики
Л. А. Чугаева в свою очередь создали свои школы и обогатили
химическую науку новыми открытиями. Они синтезировали громадное
количество новых классов и типов комплексных соединений, подвергли их
систематическому изучению, открыли ряд новых фактов и
закономерностей. На основе этих теоретических изысканий стало возможным
разрешить ряд практических вопросов аффинажа и анализа платиновых
металлов и тем самым выполнить заветы учителя — Л. А. Чугаева
и способствовать развитию отечественной платиновой
промышленности.
Дело, созданное Л. А. Чугаевым, не погибло,— оно растет и
развивается: его мысли и мечты претворяются в жизнь.
А. И. Лцкаиіук, В. В. Лебединский, Г. В. Пигулевский, П. Я. Теару,
И. И. Черняев, И. И. Жуков, Н. А. Данилов.
): A.M. Шпанион, К. Я. Л уте, Э. X. Фрицман, М. С. Сканави-Г ригорьева,
Л. А. Чу гаев, Н. К. Пшеницын, Я. Г. Кох
(фото 1920/21 г.)
ПРЕДМЕТ И ЗАДАЧИ СОВРЕМЕННОЙ ХИМИИ
Вступительная лекция, читанная в Московском техническом цчилище
8 октября 1904 г.
Сборник речей и докладов, посвященных памяти Л. А. Чцгаеви
Л., НХТИ, 1924, стр. 125
Savoir c'est prevoir. priivuir cVst pouvnir*
Вступая p. первый раз на эту кафедру, я естественным образом
испытываю потребность развить зам мою точку зрения на те основные
задачи и вопросы, вокруг которых сосредоточен в настоящее время
центральный интерес нашей химической науки. Эта точка зрения,
очевидно, не может не отразиться на том направлении, которого я буду
придерживаться в предстоящей для меня педагогической деятельности,
а потому понятно, что среди лиц, собравшихся в эту аудиторию, может
быть не мало таких, которые желали бы услышать сегодня нечто в роде
моего научного profession de foi **. И я постараюсь ознакомить с ним
моих слушателей, насколько позволит короткое время, находящееся в
моем распоряжении.
Господа, химия, как и всякая другая научная дисциплина на пути
своего развития проходила через ряд последовательных фаз. Каждая
из этих фаз отличается от предыдущей и последующей как по
объему фактических знаний и ценности теоретических обобщений, ее
характеризующих, так и по сущности тех вопросов, тех сторон изучаемого
предмета, на которые в известный момент направляется всеобщее
внимание.
То, что занимало по преимуществу одно поколение химиков,
представляло для них злобу дня, предмет усиленных работ, ожесточенных
споров—через несколько десятилетий отходило на задний план, теряя
свой жгучий интерес и уступая место новым вопросам и задачам,
которые становились на место старых в силу естественного хода эволюции.
Так последовательно сменялись интересы алхимии и иатрохимии.
теории флогистиков и последователей Лавуазье, так всеобщий интерес к
изучению химического действия гальванического тока и господство
теории Берцелиуса должны были уступить место возрастающему
значению процессов металепсии и вытекающей из них теории типов и т. д..
и т. д. Словом, каждая эпоха имела свой особый лозунг, который может
считаться ее характеристикой.
Какие же интересы, какие руководящие идеи могут считаться
лозунгом современного нам состояния химии?
Чтобы дать посильный ответ на этот вопрос, нам придется с некото^-
рой общей точки зрения бросить беглый взгляд на судьбы химической
науки.
* Знать это значит предвидеть, предвидеть это значит мочь (франц.). Прим. ред,
** Изложение убеждений (франц.). Прим. ред.
•>*
'Si
Предмет и задачи современной химии
Всякого рода явления, происходящие в природе, в том числе и
химические, могут быть рассматриваемы с двух различных сторон: с
материальной и энергетической.
Когда происходит любой банальный химический процесс, например,
когда горят дрова в печи, или когда палочка цинка растворяется в
кислоте гальванического элемента, то, что нас прежде всего поражает,
это глубокое изменение в материале. Вещество дерева превращается
в газообразные продукты, оставляя незначительное количество золы;
цинк и кислота также перестают существовать, как таковые: образуется
газообразный горючий водород и бесцветная легко растворимая соль.
Такова материальная, или, если хотите, морфологическая сторона
химических явлений.
Но одновременно с этими материальными изменениями, мы знаем,
происходит и нечто другое: горящие дрова выделяют тепло и свет;
палочка цинка, растворяясь в кислоте, дает начало электрическому току.
Мы, стало быть, имеем дело с другою энергетической стороной того же
самого химического процесса.
Исторически материальная сторона химических явлений первая
обратила на себя внимание исследователей. Ее почти исключительно
имели в виду алхимики и иатрохимики. Под ее влиянием Бойль, пер-
ный взглянувший на химию глазами философа и ученого,
определил нашу науку, как отрасль знания, имеющую своей задачей изучать
состав тел.
Такое почти исключительное увлечение материальным, или, как я
позволю себе назвать его, морфологическим направлением
химии, продолжалось в течение очень долгого времени, можно сказать
вплоть до наших дней.
И хотя первый проблеск представлений о химической энергии виден
еще в теории флогистона, а первая замечательная попытка учесть
количество тепла, выделяющегося при химических процессах, была
предпринята в том же XVIII столетии Лавуазье и Лапласом, однако прошло
более 50 лет, пока был открыт и применен к хим-и'и закон сохранения
энергии, и прошло без малого целое столетие, пока были прочно
установлены основные законы химической энергетики.
В то же самое нремя материальная сторона химических
явлений, на первых порах легче дававшаяся в руки исследователей,
успешно изучалась главным образом благодаря двум могущественным
рычагам: химическому анализу, на помощь которому позднее явился
синтез, и атомистической теории с ее широким и
многосторонним развитием.
Началось с того, что, вооружившись весами, Лавуазье, по
справедливому выражению Либиха, положил конец царству Аристотеля.
Принципы вечности вещества и неизменяемости
элементов, вместе с основными стехиометрическими законами,
заложили прочный фундамент для повой, как тогда говорили, антифлоги-
¦стической химии и ознаменовали собой период количественных
.изыскан и й.
Имена Дальтона, Пру, Дэви, Гей-Люссака и Берцелиуса навсегда
останутся связанными с этим знаменательным периодом.
В то же время атомистическая теория, еще так недавно
праздновавшая свой славный вековой юбилей, сделала возможным связать
основные законы стехиометрии в стройную систему, разработка и
совершенствование которой продолжались в течение целого столетия.
Разрешение одной из основных задач атомистики: определение сос-
'п ава химической' частицы завершилось в 50-х и 60-х годах, после
Предмет и задачи современной химии
•±\
всеобщего признания принципа Авогадро, обоснованного замечатеіь-
ными работами Лорана и Жерара и подкрепленного выводами
кинетической теории газов.
Непосредственным следствием отсюда была прочная установка
атомных весов, которая в свою очередь тесно связана с историей столь
важного по своим последствиям периодического закона Менделеев:!.
И другая не менее существенная и глубокая задача атомистики не
замедлила выступить на сцену: явилась потребность проникнуть
умственным взором в самые недра химической молекулы, уяснить себе ее
внутреннее строение.
После того, как самая возможность такой постановки вопроса,
поддерживаемая Берцелиусом и Либихом и опровергаемая Жераром, была
с особенной настойчивостью выдвинута, благодаря вновь открытому
явлению изомерии, заманчивая идея получила блестящее
осуществление в структурной теории, созданной нашим выдающимся
соотечественником А. М. Бутлеровым.
Примененная главным образом к углеродистым соединениям
структурная теория создала для них необычайно стройную и
законченную систему.
Допуская неодинаковое распределение атомов внутри химической
частицы, она позволила дать рациональное объяснение множеству
явлений изомерии и сделать большое число блестящим образом
оправдавшихся предсказаний. Знаменитая Кекулевская теория бензола является
наиболее ярким примером достигнутых в этой области результатов.
Наконец, и беспримерные успехи синтеза, особенно ознаменовавшие
собою последние годы развития органической химии, в значительной
степени также обязаны теории строения.
Но едва ли не самым поразительным результатом, к которому
привело дальнейшее развитие этой теории, явилось учение о
геометрическом расположении атомов внутри молекулы, созданное в 70-х годах
минувшего века В. Гоффом и Лебелем.
Это учение, получившее от В. Мейера название стереохимии,
не только дало возможность объяснить и предсказать целый ряд
случаев особого весьма тонкого вида изомерии, не только сделалось
незаменимой рабочей гипотезой, особенно при исследовании оптически
деятельных соединений (например, сахаристых веществ в руках Ф.
Фишера), но что особенно замечательно,— впервые далс возможность
связать оптическую деятельность жидкостей с их молекулярным строением.
Поистине достоин удивления тот логический ход мысли, который,
исходя из совершенно различных премиссов*: из структурно-химических
представлений, с одной стороны, и физической теории света — с другой,
пришел в принципе к одному и тому же выводу относительно характера
и свойств той среды, которая вызывает явления круговой поляризации.
В этих удивительных теориях.мы еще недавно видели высшее
развитие того направления химии, которое мы называли морфологическим.
Но события самых последних лет как будто развертывают перед
нами еще более широкие -перспективы, обещают еще дальше раскрыть
перед нами -интимное строение вещества.
Открытие радиоактивных веществ с их поразительной
способностью испускать из себя различного рода «лучи» и потоки
распыленной материи и еще более поразительной способностью давать
начало эманации, особому газу, самопроизвольно, на наших глазах
переходящему в другие элементы, между прочим в гелий, все это
* От французского premisse — логическая посылка. Прим. ред.
*>•>
Предмет и задачи современной химии
колеблет казалось бы достаточно прочно установившееся со времен
Лавуазье и Дальтона представление о постоянстве химических
элементов и о неделимости атомов.
Новые факты вызывают к жизни новые теории, допускающие, что
самые атомы имеют известную структуру, подлежащую изучению.
Невольно приходит в голову аналогия с теми атомными комплексами или
радикалами, которые, подобно давно известному аммонию или
сравнительно недавно открытому иодонию, обладают резко выраженной
индивидуальностью, определенными электрохимическими
свойствами и во многих отношениях напоминают настоящие
элементарные атомы. Не без основания они и были названы синтетическими
элементами. Кто знает, не найдем ли мы со временем ключ к
разъяснению этих аналогий и не окажется ли план постройки этих заведомо
искусственных атомов аналогичным тому плану, по которому построены
из более простого материала атомы «настоящих» элементов.
И вот,г как будто обещая нам помочь в разглядывании этих почти
неизмеримо малых объектов, пока доступных только умственному взору,
является открытие Зидентопфа и Зигмонди, уже давшее возможность
различать соответственно вооруженным глазом ультра
микроскопические тельца, по своим размерам приближающиеся к частицам коллоидов.
Как далеко поведет нас за собой опыт в этих новых областях, едва
открытых для науки, мы, конечно, не знаем, но во всяком случае 'перед
нами развертывается широкое поле для морфологических изысканий.
Только что намеченные общие выводы и теоретические
представления, возросшие на почве изучения материальной стороны химических
явлений и тесно связанные с атомистической гипотезой, в течение
большей части истекшего XIX века оставались почти единственными
обобщениями, на которые опиралось изложение обоих отделов, обычно
принимаемых в нашей науке: химии минеральной иорганической.
Однако за последние годы положение дела резко изменилось.
В короткое время, главным образом в течение последних 25—30 лет,
возникла, можно сказать, из самого ничтожного зачатка новая отрасль
знания — физическая химия.
Развиваясь, что называется не по дням, а по часам, она в самое
короткое время достигла необычайных размеров и превратилась в
стройную самостоятельную дисциплину. Центральное место в этой
дисциплине занимает именно та другая сторона химии, о которой я
упоминал в начале лекции — учение о химической энергии или
химическая энергетика.
Два могущественных двигателя: закон сохранения энергии
и принцип К а р н о - К л а у з и у с а, или так называемый первый
и второй принципы термодинамики, оказали особенное содействие вновь
народившемуся учению.
Прежде всего, как следствие первого принципа термодинамики,
возникло понятие об особой химической энергии, энергии потен-
циональной, и в этом отношении подобной всемирному тяготению.
В то же время, отчасти на почве, подготовленной законом сохранения
энергии, возникло учение о диссоциации и о химическом
равновесии, которым мы обязаны Девиллю и Бертло, а затем и
трории действующих масс Гульдберга и Вааге.
Далее определились законы, связывающие запас и состояние энергии
в системе тел с теми процессами, которые могут возникнуть в этой
системе.
Сначала явилось несовершенное по форме правило
наибольшей работы Бертло, потом дополненный и обобщенный Вант-
Предмет и задача современной химии
23
Гоффом и Ле-Шателье закон, лежащий в основании всей химическом
статики. Независимо от этого В. Гиббс выступил со своим правилом
фаз, давшим столь плодотворные результаты в руках Розебома и Вант-
Гоффа.
Почти одновременно с этим были выяснены Гельмгольцем и Нерн-
стом условия, при которых совершаются реакции в гальванических
элементах и переход химической энергии в электрическую.
На той же почве — приложения к химии принципов термодинамики—
возникла и грандиозная по замыслу, замечательная по смелому
полету мысли теория растворов Вант-Гоффа. завершенная гипотезой
электролитической диссоциации Арреннуса.
Наконец, в короткое время разрослась и область химической
динамики. Возникло представление о скорости реакций. Константы
реакций были связаны с константами химических равновесий. Вант-
Гофф показал возможность, наблюдая количественно течение реакции
во времени, делать заключение о ее порядке, определять числовые
коэффициенты химических уравнений.
Была Тс<кже изучена зависимость течения химической реакции от
температуры, от среды, в которой она протекает, и многое
другое...
Уже из этого беглого и, конечно, далеко не полного перечня можно
видеть, какое большое число вопросов затронуто и частью разрешено в
положительном смысле современной физической химией. Эта последняя
не только является поэтому равноправной отраслью химии, наряду с
химией минеральной и органической, но ее выводы и законы по
необходимости должны повсюду находить себе место прн обзоре различных
отделов химической систематики.
С этим требованием непременно должно ныне считаться
преподавание общей химии, в противном случае оно роковыім образом будет
отсталым.
Я спешу, однако, сделать одну существенную оговорку, ибо иначе
я рискую остаться непонятым.-
Ни ів 'каком случае не следует думать,-что принципы химической
энергетики призваны заменить то, что дала нам раньше другая,
морфологическая ветвь теоретической химии.
Громадное большинство выводов, сделанных в этой последней
области, остается в полной силе и поныне. В частности и атомистическая
теория, вопреки мнению, высказываемому в последнее время некоторыми
слишком тенденциозными авторами, продолжает оставаться
незаменимым орудием исследования, «неистощимым рогом изобилия», как весьма
метко определил ее значение известный физико-химик профессор Нернст.
Мало того, в химии существуют целые области, в которых химическая
энергетика с ее современным термодинамическим основанием не нашла
себе и, как кажется, по существу дела не может найти применения. Для
этих областей метод морфологический является и теперь еще почти
единственно возможным.
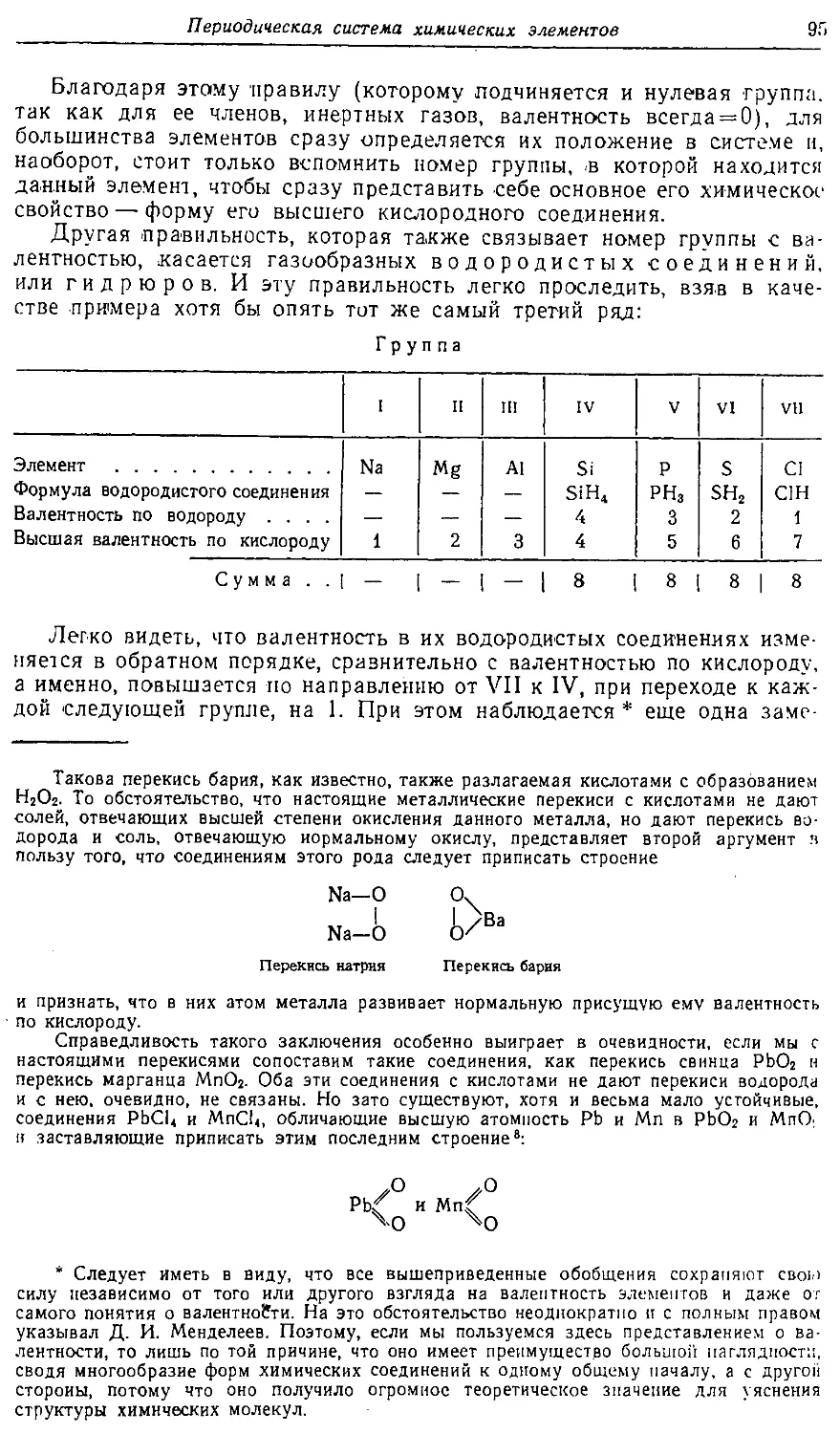
На первом месте следует здесь поставить явления изомерии, столь
широко распространенные среди органических соединений и, конечно,
играющие немаловажную роль в жизни организованного мира.
Эти явления, очевидно, не могут быть объяснены с точки зрения
термодинамики, ибо эта последняя учитывает только общий запас
энергии* данного вещества (или системы тел), но не распределение ее
* Точнее говоря, изменение этого запаса, происходящее при химических
превращениях.
24
Предмет и задачи современной химии
внутри химической частицы. Между тем, весьма вероятно, что в
большинстве случаев именно знание такого распределения могло бы
способствовать выяснению вопроса о природе и законах изомерии.
К той же области по -существу относится и обширная область
явлений катализ а.
Ввиду огромной важности этих явлений, мы должны войти здесь в
некоторые подробности.
Один и тот же химический процесс может совершаться с весьма
различной скоростью. Возьмем для примера гремучий газ: смесь двух
объемов водорода и одного объема кислорода. Если предоставить эту
смесь самой себе при обыкновенной температуре, ее объем не будет
заметно меняться в течение даже очень долгого промежутка времени.
Образование воды как будто не происходит. Тем не менее, исходя из
теоретических соображений, 'предполагают, что на самом деле хими-
4eqKnft процесс все же имеет место, но только протекает с ничтожно
малой скоростью. Нужны были бы десятки, может быть сотни лет,
чтобы можно было его заметить с 'помощью доступных нам средств.
Но возьмем тот же гремучий газ и оставим его в соприкосновении с
коллоидальным раствором платины (с так называемой жидкостью Бре-
дига). Теперь соединение наших газов уже несомненно совершается,
хотя івсе еще весьма медленно. В течение не слишком короткого
времени можно заметить сокращение объема газовой смеси, образование
малолетучей при низкой температуре воды.
Наконец, возьмем еще раз тот же гремучий газ -и приведем одну
ничтожнейшую часть его в соприкосновение с раскаленным телом, с
кусочком губчатой платины. Тогда соединение кислорода с водородом
произойдет с огромной скоростью, оно покажется нам почти
моментальным, мы будем иметь взрыв с его разрушительными последствиями и
выделением большого количества тепла.
Итак, одна и та же простая реакция (соединение кислорода с
водородом), которую химия изображает одним уравнением:
2Н2 j-02-2HoO,
на наших глазах совершается с тремя скоростями, резко
различающимися между собой:''одна из этих скоростей неизмеримо мала,
средняя по величине лежит в пределах наших измерительных средств,
третья, наконец, уже чересчур велика, чтобы ее можно было определить
обычным путем.
Между тем, одно и то же тело — вода — является единственным
продуктом, образующимся во всех трех процессах: одно и то же
количество энергии выделяется нашей системой, по которому бы из трех
возможных путей она ни двигалась к состоянию своего равновесия:
2НгО. И что особенно важно—термодинамика в согласии с опытом
требует, чтобы реакция прошла до конца независимо от
господствующих условий.
При всем том оказывается, что скорость процесса всецело
зависит от катализаторов — губчатой платины или жидкости Бреди га.
Подобного рода явления совершенно не предусматриваются
термодинамикой, лежат, так сказать, вне ее контроля.
На этом основании их в последнее время стали сравнивать с
процессами трения.
Следует, однако, иметь в виду чисто формальный характер этой
аналогии, в особенности же громадную разницу в значении этих двух
существенно разнородных явлений.
Предмет и задачи современной химии
2Гу
Если трение плохо приготовленной или заржазленной машины
составляет докучливое и по большей части неприятное обстоятельство,
вызывающее ненужную трату полезной энергии, то замедление и
ускорение химичеікой реакции при помощи катализаторов, помимо
глубокого чисто химического интереса, играет еще огромную роль н
экономике живой природы.
Вспомним, что огромные запасы органической материи,
накопленной в разной форме'на поверхности земли: живые и мертвые растения
и животные, все деревянные постройки, запасы угля и нефти и пр. и
пр., вспомним, что все это составляет сплошную массу горючего
материала, законами термодинамики рано или поздно обреченного на
соединение с кислородом воздуха, с которым этот материал постоянно
соприкасается.
И если наша земля не сделалась еще ареной грандиозного пожара,
перед которым бледнели бы все ужасы когда-либо пережитых
человечеством катастроф, то это только потому, что какие-то неведомые для
нас молекулярные механизмы подобно шлюзам и плотинам сдерживают
игру химического напряжения.
А между тем это напряжение готово перейти в действие от
повышения температуры в самом ограниченном районе, от малейшей искры,
а еще легче и притом даже в присутствии большого
количества воды, при участии подходящих катализаторов.
И это последнее обстоятельство имеет огромное значение,— на сей
раз для развития и поддержания жизни на нашей планете.
Известно, что всякий организм непрерывно расходует запасы
химической энергии, постоянно пополняемые им извне.
Органические вещества, носители этой энергии, или окисляются
при действии кислорода воздуха, например у высших растений и
животных, или распадаются интрамолекулярно —- у многих
микроорганизмов и при этих процессах скрытая энергия постоянно освобождается.
Но все такие процессы становятся возможными только в присутствии
особых катализаторов — химических ферментов. Если бы не было этих
ферментов, органическая жизнь или бы вовсе заглохла, или бы
совершалась с такой медленностью, что по сравнению с возрастом любого
организма Мафусаилов век показался бы одним мгновением.
Есть что-то величественное в этом двойном механизме, с помощью
которого природа оберегает мир живых веществ от одинаково
гибельного посягательства огня и времени.
И, конечно, изучение этого механизма, его устройства и действия
составляют одну из величайших проблем науки.
Мы видели, что те два направления, по которым до сих пор
развивалась химия, направление материальное и энергетическое, 'принесли
обильные плоды, каждое в соответствующей области химических
явлений. Оки имеют поэтому одинаковое право на самостоятельное
существование.
Но очевидно, что между этими двумя направлениями, касающимися
двух сторон одной и той же реальности, должна существовать глубокая
и тесная связь.
Мне кажется, что каталитические процессы, явно как и явления
изомерии, принадлежат именно к той области нашей науки, где обоим
направлениям суждено рано или поздно встретиться, соединить свои
усилия.
Химические молекулы представляют собой механизм, в который
вложен известный запас энергии. Чтобы постигнуть действие этих ме-
26
Предмет и задачи современной химии
ханизмов, в одинаковой степени необходимо знать их внутреннее
устройство и распределение в них энергии.
Быть может, методы, пригодные для решения этой заманчивой
проблемы, найдутся, когда мы сумеем отыскать и придать более тачную
математическую форму той связи, которая-существует между
различными «морфологическими» константами вещества (как, например,
атомный вес, удельный объем и пр. и пр.) и воспользуемся ею для
дальнейшего развития химической энергетики.
* * *
Наша беседа приближается к концу. Мне остается остановиться в
немногих словах еще только на одной стороне современной химии,
которая невольно напрашивается на обсуждение в настоящую минуту,
когда я вижу среди моих слушателей столько представителей
разнообразных отраслей практической деятельности. И, конечно, я не могу
окончить этой лекции, не затронув, хотя в общих чертах, вопроса о
взаимных отношениях между наукой и техникой.
Я должен сознаться, что никогда не стоял близко лицом к лицу с
техникой, но зато судьба заставила меня провести всю мою
предыдущую научную деятельность в учреждении, задачи и интересы которого
близко соприкасаются с областью медицины.
Вы, может быть, поймете теперь, почему вопрос о связи между
наукой и ее практическими приложениями никогда не представлялся для
меня праздным, что, наоборот, он возникал у меня неоднократно и
настойчиво.
Ведь врачи и техники, несмотря на все различие их специальностей,
в сущности люди, близкие по характеру своей деятельности .
Задача техники — изыскивать и применять на деле всевозможные
культурные средства для удовлетворения потребности человека и
преимущественно его физической натуры. А задача врача — изыскивать и
применять средства для лечения и предупреждения болезней.
И тот и другой — и врач и техник — когда-то в стародавние временя
шли ощупью, эмпирически — и не случайное, конечно, обстоятельство,
что успехи того и другого были тогда в одинаково жалком состоянии.
Но вот пришла наука, положительная наука XIX века, и картина
¦сразу изменилась.
Медицина, благодаря гению Пастера, получила в свои руки
власть над жизнью и смертью множества людей: достаточно вспомнить
о блестящих успехах хирургии и сывороточного лечения заразных
болезней.
Не менее блестящие успехи техники, завещанные нам веком пара
и электричества, вам, конечно, известны еще лучше чем мне, и о них
не приходится здесь напоминать.
Поистине, говоря словами одного из наших выдающихся
физиологов: «сорок лет теории дали человечеству то, чего не могли дать
сорок веков практики».
Двойной практический урок, вынесенный человечеством из этой
долговременной истории медицины и технологии, не пропал даром. Во всех
культурных странах престиж науки поднялся до небывалой высоты.
Практические деятели перестали встречать насмешками
представителей чистого знания, как это было в старое время.
Наука естественным образом сделалась неотъемлемым и притом
основным элементом технического (и медицинского) образования.
Убеждение в этом, особенно глубоко привившееся в Германии,
проникло и к нам.
Предмет и задачи современной химии
27
Доказательством уіого обстоятельства является существование
академического учреждения, в стенах которого мы находимся в
настоящую минуту. А красноречивым свидетельством тех забот, которые
прилагаются у нас к возможно рациональной постановке научного
преподавания, служат обширные и хорошо обставленные лаборатории,
имеющиеся в нашем распоряжении.
Итак, я заключаю, что в наше время существование техники, помимо
науки, есть вещь невозможная. И если мы желаем иметь образованных
и дельных техников, то. необходимо дать им, наряду со специальными
практическими сведениями, солидную научную подготовку.
Но этого мало. Если мы хотим уберечь нашу технику от застоя,
обеспечить ей известный самостоятельный прогресс, то мы должны
всеми силами заботиться о том, чтобы создать людей, способных научно
работать и двигать вперед чистое знание.
Этими требованиями, по моему мнению, и определяются те рамки, в
которых должна укладываться деятельность преподавателя химии в
учреждении, которое подобно нашему, имеет своей задачей служить
рассадником техники в самом широком смысле этого слова.
А залог успеха в этом отношении, как показал пример Западной
Европы, лежит прежде всего в добром согласии и единении между
деятелями чистой науки и представителями различных технических
специальностей. Недаром же гласит классическое изречение:
Concordia res parvae crescunt
discordia ma.ximae diiabuntur.*
* В согласии и малые дела растут, в несогласии и великие дела разрушаются
*лаі.}. Прим. ред.
ЭВОЛЮЦИЯ ВЕЩЕСТВА В МЕРТВОЙ И ЖИВОЙ ПРИРОДЕ
Вступительная лркціля, читанная в С.-Петербургском университете
11 сентября 1908 г.
Сборник речей и докладов, посвященных памяти Л. .4. Чугасни.
Л., НХТИ. 1924, стр. 1S7
Мысль о том, что все существующее, весь видимый мир возник путем
постепенного развития, путем эволюции из одного общего, сравнительно
простого начала, принадлежит к числу величайших обобщений, к
которым пришел человеческий гений.
В неясной форме эта мысль возникала еще в умах философов
древнего мира. В известной поэме Лукреция Кара «De rerum natura» *
содержится уже изумительное по форме пророчество ее будущего
блестящего развития.
Но только в течение минувшего XIX столетня вслед за появлением
Канто-Лапласовской гипотезы о происхождении мира, в особенности
же после опубликования знаменитой теории Дарвина, эволюционный
принцип делается достоянием положительной науки, получает широкое
распространение и, можно сказать, становится краеугольным камнем
всего нашего научного миросозерцания.
За последние годы с особенной настойчивостью выдвигается вопрос
об эволюции вещества и тех его качественных разновидностей,
изучение которых составляет задачу химии.
Этому вопросу я и имею в виду посвятить настоящий очерк.
I
Химик постоянно и б окружающей природе, и в лаборатории
наблюдает превращения вещества из одной формы в другую. Блестящие
металлы на влажном воздухе окисляются: железо ржавеет, медь
покрывается зеленым налетом. С другой стороны, оба металла могут быть
вновь получены из окислов и других соединений, в которых они
содержатся.
В нашем собственном организме постоянно происходят
разнообразные химические метаморфозы. Вещества пищи перевариваются,
превращаясь в растворимое состояние, затем после всасывания они
попадают в ткани организма и здесь окисляются, сгорают, превращаясь в
более простые продукты: воду, угольную кислоту и пр. Из этих
продуктов растительный организм вновь воссоздает те самые сложные
химические соединения, из которых состоит наша пища.
Естественно возникает вопрос: что же эта изменчивость материи —
имеет ли она где-нибудь самой природой вещей поставленные границы
или наборот — она по существу беспредельна? Ответ на этот вопрос
был дан в конце XVIII столетия французским ученым Лавуазье.
* О природе вещей. Прим. ред.
Эволюция вещества в мертвой и живой природе
29
Лавуазье точными опытами установил, что изменяемость вещества
ограничивается существованием химических элементов. Элементы
не изменяются, по крайней мере в условиях наших опытов. Количество
и качество их остается без перемены при всех химических
превращениях, а эти превращения сводятся к разнообразным перемещениям и
сочетаниям элементов.
Казалось бы, таким образом вопрос должен был быть решен
бесповоротно. Но когда стали ближе знакомиться со свойствами элементоз,
особенно изучать эти свойства с количественной стороны, то
мало-помалу явились серьезные основания думать, что и самые элементы
изменяемы, быть может даже способны превращаться друг в друга или
возникать из каких-либо иных неведомых субстанций, что мы только не
знаем и не умеем осуществить условий, необходимых для подобного
рода превращений. Явилось представление об общности
происхождения элементов- -и ?десь впервые в применении к химии было
произнесено слово эволюция.
О кровном, так сказать, взаимном родстве элементов догадывались
еще древние. Мы находим зачатки этого учения у атомистов Л ей кип-
па и Демокрита, а затем у Аристотеля. На той же мысли в
средние века алхимики строили свои несбыточные планы получения
золота из неблагородных металлов.
В течение минувшего XIX века мысль о единстве материи вновь
возродилась и получила более прочное обоснование. Каждый элемент
обладает некоторой весьма характерной и важной для него величиной —
атомным весом. В отношении этих именно весов или кратных им
элементы вступают в химические соединения, образуя сложные тела.
Сравнивая соотношения между атомными весами и другими
свойствами элементов, нашли замечательные закономерности, изучение
которых завершилось открытием периодического закона.
Д. И. Менделеев расположил все элементы в порядке
возрастающих атомных весов и показал, что при этом элементы обнаруживают
лишь периодическое чередование в своих свойствах и сами собой раз-
мещаклтя в стройную систему по естественным группам, между
членами которых замечается ряд закономерных отношений и аналогий.
Каждый элемент получает свое определенное месю; большая часть его
физических и химических свойств определяется его положением в системе
с логической необходимостью. Па этом основании Д. И. Менделеевым
могли быть предсказаны три новых, дотоле неизвестных элемента,
галлий, скандий и германий, которые и были вслед открыты в
различных редких минералах.
Периодический закон, связывающий все известные нам элементы
а одно органическое целое, невольно наводит на мысль об общности их
происхождения *.
Периодическая система представляется нам как бы грубыми
набросками того общего плана, по которому могли образоваться элементы из
того или иного единого начала.
Мысли, подобные только что изложенным, впервые с полной
ясностью были высказаны в 1887 г. У. Круксом в его остроумной речи:
«О генезисе химических элементов». Им же впервые была проведена
отдаленная, конечно, аналогия между предполагаемым генезисом и
происхождением организмов по Дарвину и Уоллесу.
* Еще в начале XIX столетия гипотеза об общем происхождении ^лементои была
высказана Проутом на основании совершенно иных соображений.
.40
Эволюция вещества в мертвой и живой природе
Капитальный факт, имеющий отношение к разбираемому нами
вопросу о 'происхождении элементов, был открыт при изучении так
называемых катодных лучей, испускаемых отрицательным полюсом
(катодом) при прохождении электрического разряда через Круксову трубку.
Лучи эти, как давно уже предполагал открывший их Крукс, оказались
потоками чрезвычайно быстро несущихся и необычайно мелких
материальных частиц, заряженных отрицательным электричеством.
В последнее время удалось определить массу этих частиц * как
электронов, которая оказалась О'Коло 1/2000 массы водородного атома,
удалось установить величину их заряда.
При этом получился тот замечательный результат, что величины эти,
а также все прочие доступные изучению свойства электретов,
совершенно не зависят от материала, из которого сделан катод. Между тем едва
ли уже можно сомневаться в том, что электроны представляют из себя
не что иное, как вещество катода в состоянии тончайшего распыления1.
Далее, те же электроны могут образоваться из различных металлов
и при совершенно иных условиях, например под влиянием
ультрафиолетовых лучей, очень высокой температуры — и во всех случаях свойства
их оказываются одни и те же.
Многие радиоактивные вещества испускают так называемые -fl-лучіи
которые стоят весьма близко к катодным и, подобно этим последним,
являются потоками необычайно быстро движущихся электронов.
Притом, несмотря на существенно различное происхождение, между
электронами Р-лучей и электронами, испускаемыми катодом лучей Круксо-
вой трубки, не удалось подметить существенного различия, если не
считать различия в скорости передвижения.
Если, таким образом, на электроны, при современном состоянии
наших знаний, следует смотреть как на мельчайшие частицы вещества,
доступные изучению, и потому происходящие через распыление самых
разнообразных элементарных тел, то естественно поставить себе вопрос, не
эти ли самые электроны являются тем общим материалом, теми
первичными кирпичами, из которых построены атомы наших элементов.
До некоторой степени ответ на этот вопрос подготовляется теми
опытами, с помощью которых удалось в последнее время фактически
доказать, или, по крайней мере, сделать весьма вероятной превращаемость
некоторых элементов друг в друга.
Эти поразительные факты тесным образом связаны с изучением
радиоактивных тел, открытых Беккерелем и супругами Кюри.
Исследования, особенно Резерфорда, показали, что существует целый
ряд радиоактивных элементов, имеющих преходящее существование, так
называемых метаболонов.
Элементы эти способны последовательно превращаться друг в друга
с весьма различной скоростью и притом с выделением огромного
количества энергии **.
Каждому такому элементу свойственна определенная долговечность,
колеблющаяся от немногих минут *** до миллионов лет. Она может
быть с достаточной точностью выведена из наблюдений над
постепенным спадением радиоактивности данного элемента.
* На. основании последних исследований можно предполагать, что самая масса
электронов имеет электромагнитный характер.
** По вычислениям Резерфорда, 1 <" радия на пути своего превращения в свиней
(см. таблицу) выделяет 389 000 ккал, т. е. количество тепла, равное тому, которое
развивается при сгорании 500 кг ('/г т) каменного угля. И это еще небольшая часть
того чапаса энергии, который заключается в радии!
*** В настоящее время известны изотопы, живущие доли секунды. Прим.. ред.
Эволюция вещества в мертвой и живой природе
Нлэпанкг мет.іболон.і
I іолонииная прол"іжли-.іь-
WKTU ЖИ.ШИ (ВГСМИ. В 104(41111
которого разрушается ГЮ1^
данного мстаболоніп
Уран . .
Уран X .
Ионии
Радий
і
Эмо нация
і
Радий А
і
Радий В
і
Радий С
і
Радий D
і
Радий Et
і
Радий Е2
і
Полоний
I
Свинец (?)
(5 800 поо 0(H) лет
2_ дня
Чтобы дать хотя некоторое представление о результатах этих
исследований, мы приведем таблицу, заимствованную из работ Р е.ч ер-
фор да (см. таблицу на стр. 31).
В этой таблице стрелки показывают последовательность, в которой
совершаются превращения метаболонов. Как видно, половинная про
должительность жизни этих «удивительных» элементов колеблется
между громадной величиной— 6 800 000 000 лет и совершенно ничтожной —
3 мин.
Самое превращение от
урана до (гипотетического)
свинца совершается с
переходом не менее как через
один промежуточный
продукт.
С другой стороны, Рам-
зай и Соддн нашли, что при
хранении э м а н а ц и и
газообразного метаболона,
испускаемого радмем (см.
таблицу), образуется гелий,
один из благородных газов
так называемой нулевой
группы периодической
системы с атомным весом = 4.
По мере того, ,как
эманация теряет свои
радиоактивные свойства, часть ее
превращается в гелий.
Последние опыты Р а м-
зая и Камерона
показали, что в присутствии
воды эманация радия вместо
гелия дает неон, другой член
той же нулевой группы с
более высоким атомным ве-
сом = 20. Наконец, в
присутствии солей меди
получается третий член нулевой
группы — аргон, с
атомным весом = 40.
Рамзай наблюдал также, что в присутствии солей меди эманация
дает еще литий, но этот результат в последнее время оспаривается
г-жой Кюри, на основании предпринятых ею специальных проверочных
опытов *.
Благодаря этим открытиям, мысль о превращаемости элементов друг
в друга впервые приобретает под собой экспериментальную почву и
выходит из стадии догадок.
Существование радиоактивных веществ, в ничтожных количествах
рассеянных на поверхности и в недрах земли, ставит перед нами вопрос,
не прошли ли и другие известные нам элементы, так сказать, через
радиоактивную стадию развития; быть может, только эта стадия давно
уже отошла для них в область истории. Невольно представляется, что
2Ш\ лет
3,8 дней
.'1 мин.
_(> мин.
1Я мин.
¦'и> лет (?)
О дней
і.8 дней
1VO дней
* Наиболее прочно установленным в настоящее время можно считать факт
превращаемости эманации в гелнй 2.
№
Эволюция вещества в мертвой и живой природе
перед нами приподнялся только маленький уголок завесы, скрывающей
.от нас тайны того грандиозного процесса, с помощью которого
образовались элементы.
Не следует ли смотреть на радиоактивные элементы, как на
остатки не совсем еще закончившегося эволюционного процесса, на
последние, готовые потухнуть головни гигантского костра, которые к счастью
для науки удается еще наблюдать в настоящее время?
Я сравнивал бы радиоактивные элементы с дошедшими до нас
ископаемыми, или пожалуй, еще лучше — с теми вымирающими видами,
последние экземпляры которых так ценятся зоологами, как живые
свидетели когда-то имевших место процессов развития среди органических
форм.
Нельзя не заметить здесь того замечательного обстоятельства, что
но многих горных породах найдены в настоящее время следы
радиоактивных веществ. Притом во многих случаях, особенно в минералах,
содержащих уран, удалось установить, что различные радиоактивные
части их находятся между собой в постоянном количественном
отношении. Особенное значение имеет здесь отношение между содержанием
урана, радия и свинца. Допуская, что между этими составными
частями достигнуто радиоактивное равновесие, и зная содержание в данном
минерале различных радиоактивных элементов, а также среднюю
продолжительность жизни этих последних, можно составить себе
приблизительное представление о возрасте данного минерала.
Так, по опытам и вычислениям Болтвуда, древность ряда
исследованных им первозданных горных пород колеблется -между 400 и 2000
миллионов лет. Для минерала аутенита, наоборот, является вероятным
более позднее происхождение и т. д.
Легко понять, какое значение этот только что нарождающийся метод
может получить в будущем для минералогии и геологии.
Подводя итоги только что сказанному, мы видим, что в настоящее
время предположение о происхождении элементов от одного общего
начала путем процесса эволюции может защищаться как научно
обоснованная гипотеза.
Английский физик Дж. Дж. Т о м с о н сделал даже попытку
составить представление о предположительной структуре атомов
различных элементов.
При этом он опирается на гипотезу, согласно которой атомы всех
элементов состоят из быстро движущихся электронов, и приходит к
возможности построить структурные схемы элементов, отвечающие
требованиям периодического закона.
Наш соотечественник Н. А. Морозов делает еще один шаг дальше.
Исходя из подмеченной им аналогии между химическими элементами,
-с одной стороны, и некоторыми углеродистыми соединениями
(углеводородами) и радикалами — с другой, он не только развивает теорию,
позволяющую ему, подобно Томсону, строить структурные схемы
элементов, но и высказывает предположение относительно последовательности
тех этапов, через которые должны были пройти элементы на пути
своего развития.
Но здесь мы подходим к самым крайним граням положительного
знаніия и вступаем в область смелых, хотя и остроумных догадок,
проверка и обоснование которых принадлежат будущему.
Эволюция вещества в мертвой и живой природе 33
II
До сих пор у нас шла речь исключительно об эволюции химических
элементов. Спрашивается теперь, нельзя ли также говорить об
эволюции химических соединений? На первый взгляд между этими
двумя типами эволюции— существенная принципиальная разница.
В громадном большинстве случаев мы не имеем возможности
вызвать или наблюдать образование новых элементов, подобно, тому, как
в природе за кррогкий период человеческой жизни не происходит
образования новых видов растений или животных.
Иначе обстоит дело с химическими соединениями. И в окружающей
природе, и в живых организмах, и в лаборатории химика постоянно
происходят и производятся синтезы из элементов и разнообразнейшие
превращения химических соединений, начиная от простейших и кончая
наиболее сложными *.. Эти превращения совершаются в сравнительно
короткие промежутки времени и по своей малой продолжительности
доступны человеку.
Тем не менее и здесь можно говорить об эволюции вещества, хотя
и в несколько ином смысле, нежели тот, в котором мы говорили выше,
об эволюции элементов. Всякому химику хорошо известно, с какими
затруднениями сопряжено искусственное получение, синтез сложных
органических соединений. Такой синтез при воспроизведении его
лабораторным путем совершается постепенно, через целый ряд
последовательных стадий, начиная от элементов и простейших соединений и переходя
к соединениям все более и более сложным.
В организме ныне живущих растений и животных, очевидно,
существуют главные условия для таких синтезов и иных превращений
сложнейших органических соединений.
Эти условия, по всей вероятности, весьма разнообразны: частью они
физического характера, в таком случае связаны с морфологическим
строением тфото'плазмы клеток и тканей; частью и, вероятно, главным
образом, они сводятся к факторам химического характера, к присутствию
в организме ряда готовых реагентов, необходимых для осуществления
тех или иных превращений.
Особенно важную роль, по-видимому, играют в этом отношении
весьма распространенные в живых организмах неорганизованные
ферменты или энзимы, служащие специфическими катализаторами, т. е.
возбудителями для множества химических реакций.
Но каковы бы ни были все эти условия, они, очевидно, могли
сложиться только постепенно, а история их развития, по всей вероятности,
была самым тесным образом связана и переплетена с
морфологической стороной развития органического мира.
Можно думать, что без этой истории, без длинного ряда
антецедентов ** едва ли бы одно из тех многочисленных белковых соединений,
углеводов, жиров, алкалоидов и множество других веществ, из которых
построены организмы животных и растений, могло получиться на земле
в сколько-нибудь значительном количестве, если, конечно, исключить
при этом вмешательство сознательной деятельности человека.
Таким образом, мы вправе сказать, что, вероятно, эволюция
организмов, в широком смысле этого слова, была в то же время эволюцией
* Самая трудная область органического синтеза — область белковых веществ
успешно разрабатывается в настоящее время в лаборатории Берлинского университета
проф. 3. Фишером и его учениками.
** Предшественников. Прим. ред.
3 Л. А. Чугаев, т. III
34
Эволюция вещества в мертвой и живой природе
и тех форм веществ, тех соединений, которые входят в состав живой
протоплазмы и которые в ней претерпезают всевозможные химические
превращения. И если в настоящее время в любом организме в течение
короткого времени на наших глазах разыгрывается ряд сложнейших
синтезов, то это происходит только благодаря заранее подготовленной
псчве, подобно тому, как вообще процесс онтогенетического развития
всякого организма (от зародыша до взрослого состояния) делается
возможным только благодаря предшествовавшему филогенетическому
развитию целого вида.
К сожалению, сравнительное изучение организмов, стоящих на
различных ступенях развития в биохимическом отношении, до сих пор еще
не было произведено сколько-нибудь систематическим образом.
Однако и среди того крайне неполного и отрывочного фактического
материала, который находится в нашем распоряжении, можно найти
немало указаний, свидетельствующих об известном параллелизме хода
морфологической эволюции организмов.
Прежде всего нетрудно найти родственные черты в химическом
составе самых разнообразных организмов — черты, сближающие все
живые вещесггва в одно неразрывное целое.
В этом отношении на первом плане заслуживает упоминания тот
общеизвестный факт, что все организмы, начиная от бактерии или
амебы и кончая человеком, с одной стороны, высшими растениями — с
другой, содержат в своем составе три типичных класса органических
соединений: белковые вещества, углеводы и жиры.
Этому обстоятельству обыкновенно не придают того значения,
которое оно по справедливости заслуживает, указывая на общие условия
происхождения живой протоплазмы в самых разнообразных живых
организмах.
Мы сейчас увидим, что этот вывод находит себе полное
подтверждение и с другой стороны.
Одним из характернейших признаков типичных растений является
способность ассимилировать углерод из воздуха при помощи хлоро-
ф ильного зерна.
Все типичные зеленые растения содержат хлорофилл, хотя
некоторые группы организмов, несомненно принадлежащие к растительному
царству, и лишены этой особенности, например грибы.
С другой стороны, высоко развитые представители животного
царства (позвоночные животные) характеризуются присутствием в
форменных элементах их крови (красных кровяных тельцах) особого
пигмента — гемоглобина. Гемоглобин, подобно хлорофиллу растений,
играет у этих животных весьма важную роль в процессе обмена
веществ, хотя и совершенно иную, нежели хлорофилл *.
Интересные исследования Ненцкого и Мархлевского
несколько лет тому назал установили тот в высшей степени важный
факт, что хлорофилл и гемоглобин суть вещества родственного харак-
* При участии хлорофилла поглощаемая растением из воздуха углекислота С02
распадается на кислород, уходящий в атмосферу, и на углерод, усвояемый растением
и идущий для целей синтеза. Процесс этот сопровождается накоплением в растении
солнечной энергии. Роль гемоглобина сводится к тому, что это красящее вещество
поглощает кислород в то время, когда кровь проходит по капиллярам, окружающим
легочные пузырьки (альвеолы). При этом темная венозная кровь переходит в алую —
артериальную. Последняя разносится по всему телу и отдает поглощенный кислород
тканям, в которых за счет его происходят процессы окисления сложных органических
соединений.
Таким образом, гемоглобин крови является как бы запасным резервуаром длі>
кислорода, потребляемого организмом.
Эволюция вещества в мертвой и живой природе 35
тера. Один из продуктов распада молекулы хлорофилла — ф и л л о
цианин и один из продуктов распада молекулы гемоглобина — г е м а-
тин при глубоком восстановлении дают одно и то же соединение —
летучее тело— гемопиррол. Из гемопиррола, полученного путем
восстановления филлоцианина, удалось получить у р а б и л и н, типичный
пигмент мочи человека, появляющийся в ней при кровоизлияниях и
некоторых других болезненных процессах и образующийся из
гемоглобина.
Из своих исследований Ненцкий и Мархлевский заключают, что в
молекуле хлорофилла и гемоглобина должно содержаться одно общее
ядро*, по своей химической природе сближающее оба пигмента с
красками индиговой группы.
Существование генетического родства между хлорофиллом и
гемоглобином отражается и на общем химическом характере обоих
пигментов. Еще недавно оно получило блестящее подтверждение в
исследованиях немецкого химика Вильштеттера, который показал, что
чистый хлорофилл содержит значительные количества (1%) магния
в крепко связанном состоянии и, по-видимому, в состоянии, весьма
близком к тому, в котором гемоглобин крови содержит железо.
Совокупность всех этих данных дает нам полное основание
предполагать, что хлорофилл и гемоглобин произошли в процессе
филогенетического развития организмов из одного общего зачатка. Можно
даже догадываться о природе этого зачатка. В молекуле белковых
веществ современных нам животных и растений недавно открыто,
благодаря работам Гопкинса и Коля, а также Эллингера
присутствие той самой индольной группировки атомов, которая, как мы
видим, лежит, по всей вероятности, в основе молекулы хлорофилла и
гемоглобина.
При некоторых болезненных состояниях организма вещества,
содержащие эту группировку, отщепляются от молекулы белка и попадают
затем в мочу. Из такой мочи легко можно (например, при действии
хлорного железа) получить индиго.
Вот в этой-то индольной группировке, которая, по всем вероятиям,
входила б состав белковых веществ у тех первобытных организмов, из
которых путем эволюционного процесса создались с течением
времени растения и животные, в этой группировке, согласно Иенцкому, и
следует искать общего химического родоначальника хлорофилла и ге-
моглобина.
Если родственный химический характер хлорофилла и гемоглобина
генетически сближает их между собой и тем самым подтверждает
кровное родство между растительным и животным царством, то, с другой,
стороны, можно привести ряд фактов, доказывающих наличность
особенной химической близости между представителями более тесных
систематических групп растений и животных
В качестве примера укажу на условия распределения в
растительном царстве таких соединений, как алкалоиды, глюкозиды, терпены.
У близких в систематическом отношении видов обыкновенно
встречаются и родственные в химическом отношении представители только что
указанных классов соединений.
К сожалению, число фактов этого рода, которыми мы располагаем,
сравнительно не велико, что объясняется и новостью дела, и почти
полным отсутствием систематических исследований в этом направлении.
К тому же эти исследования ввиду сложности тех соединений, кото-
* Т. е. группировка атомов.
3*'
В6
Эволюция вещества в мертвой и живой природе
рых они должны касаться, представляют большие экспериментальные
трудности.
Весьма убедительные доказательства в пользу существования
тесной связи между морфологическими признаками живых организмов
и химической природой их компонентов были найдены за последние
годы Мей ер сом, Уленгутом и др. при помощи особой открытой
ими биохимической реакции.
Бактериологами давно установлена способность животного
организма специфически реагировать, так сказать, откликаться на повторное
введение под кожу или в кровь ядовитых продуктов, вырабатываемых
бактериями. Так, при иммунизации лошади возрастающими дозами
дифтерийного яда, так называемого токсина, организм ее постепенно
делается невосприимчивым к большим количествам этого яда, и в то
же время в крови ее (в тканях) появляется и начинает циркулировать
особого рода противоядие — антитоксин. Этим противоядием, как
известно, пользуются .с успехом для лечения дифтерии (сывороточное
лечение или серотерапия).
По отношению к каждому микробному яду получается
соответствующее противоядие, годное только для нейтрализации данного токсина
и не действующее на другие аналогичные ядовитые продукты.
Эта способность заставлять организм вырабатывать противоядия
присуща не одним только ядовитым продуктам жизнедеятельности
микробов, но отчасти также и другим ядам, например ядам змей,
некоторым продуктам растительного происхождения: рицину,
входящему в состав семян клещевины, абрину, ядовитому началу красивых
семян растения abrus praecatorius и многих других.
Организм откликается также на введение в него некоторых
ферментов, например сычужного фермента, свертывающего молоко,
вырабатывая вещества, препятствующие действию этого фермента
Несколько лет тому назад Мейерс показал, что введение в
организм кролика различных белковых веществ обусловливает
образование в крови его особых антител, продуктов, вызывающих
образование осадка в растворе соответствующего белка. Как и в других
подобных случаях, кровь подготовленного таким образом животного
освобождают от кровяных телец и фибрина и полученную кровяную
сыворотку употребляют для опытов.
Эта реакция оказалась так же специфичной, как и все предыдущие.
Сыворотка данного животного вызывает осадок в- растворе только
того белкового вещества, которое вспрыскивали этому животному.
Вслед затем немецкий ученый Уленгут показал, что совершенно
аналогичные специфические осадки получаются, если для
вспрыскивания употреблять не изолированные в возможно чистом состоянии
белковые вещества, а просто кровь или кровяную сыворотку тех или иных
животных.
Так, если одному кролику повторно вспрыскивать, например (под
кожу и т. п.) кровь морской свинки, другому — кровь человека,
третьему— кровь голубя, то сыворотка каждого такого кролика будет
давать осадки (даже при разведении большими количествами воды) с
кровью того и только того животного, от которого бралась кровь для
иммунизации.
К сожалению, до сих пор почти ничего неизвестно относительно
природы тех веществ, которые вырабатываются в организме в ответ
на введение белков, инородной крови и других продуктов
органического происхождения.
Можно только догадываться, что вещества эти близко стоят к бел-
Эволюция вещества в мертвой и живой природе
37
ковым, а самое выпадение осадка, вероятно, относится к той группе
процессов свертывания, которые столь характерны для коллоидов.
Но как бы то ни было, едва ли можно сомневаться, что только что
упомянутые явления самым тесным образом связаны с
химическими процессами, которые совершаются в организме в ответ
на'введение инородных веществ, главным образом белкового характера.
Исследования Уленгута представляют большой интерес уже потому, что
дают нам в руки новый замечательный метод для распознавания
отдельных видов животных, даже сравнительно близко стоящих друг к
другу.
Этим обстоятельством уже воспользовалась судебная
медицина, и во многих государствах Запада способ Уленгута официально
признан ценным вспомогательным средством при судебно-медицинской
экспертизе.
Дело в том, что вещества, находящиеся в крови различных
животных и обусловливающие появление осадков со специфическими
сыворотками, являются довольно устойчивыми и долго сохраняются,
например, в засушенных кровяных пятнах и проч. без существенного
изменения.
Если такое кровяное пятно извлечь водой или лучше слабым
раствором поваренной соли и затем полученную жидкость испытать в
отдельных пробах путем прибавления к ней сыворотки от ряда кроликов,
иммунизированных кровью человека и различных животных, то
появление осадка прямо укажет, кому принадлежит подозрительная кровь.
Целый ряд преступлений был открыт 'при содействии этого метода.
В одном случае убийства, которому предшествовала кража овец,
Уленгут с помощью своего метода безошибочно установил в
кровяных пятнах присутствие человеческой и овечьей крови. Тот же
самый метод позволяет, например, распознавать присутствие
лошадиного мяса в колбасе.
До такой степени тонкости и чувствительности может доходить эта
биохимическая проба!
Но для занимающего нас вопроса еще больший интерес
представляет то обстоятельство, что при помощи метода Уленгута может быть
не только констатировано различие, поставлен диагноз между
отдельными видами животных, но и установлено кровное родство
между организмами, близко стоящими друг к другу.
Дело в том, что при взаимодействии специфической сыворотки с
кровью того вида, от которого была взята кровь для иммунизации,
быстро получается обильный осадок, если же для реакции взята кровь
животного другого, но близкого вида, то осадок получается
медленнее и притом менее обильный. Можно даже без труда заметить,
что интенсивность реакции убывает по мере того, как мы берем для ее
воспроизведения кровь животного все больше и больше удаляющегося
от того вида, которому соответствует специфическая сыворотка.
Указанным путем удалось, например, подтвердить родство курицы
с голубем, дикой свиньи с домашней, собаки с лисицей, удалось
усыновить постепенную градацию от овцы к козе и быку.
Но самые замечательные результаты были получены в этом
направлении английским ученым Нутталем при изучении специфической
реакции v человека и обезьян
Сыворотка полученная от кролика, которому раньше
впрыскивалась кровь человека, показывала специфическую реакцию и притом
почти одинаковой силы с человеческой кровью и с кровью трех
человекообразных обезьян: орангутанга, гориллы и шимпанзе.
38
Эволюция вещества в мертвой и живой природе
Слабее получилась реакция с кровью других обезьян Старого
Света и еще слабее с кровью обезьян Нового Света; у лемуров ее вообще
не удалось констатировать.
Мы приходим, таким образом, при помощи биохимической реакции
Уленгута к той же самой родословной обезьяны, которая была
установлена зоологами на основании признаков чисто морфологического
характера.
Вместе с тем оправдывается известный вывод, к которому в свое
время пришел Гексли, относительно места, занимаемого человеком
в животном царстве.
«Критическое сравнение организмов и их видоизменений в
пределах группы обезьян,— говорит знаменитый английский зоолог,—
приводит к одному и тому же результату: анатомические различия,
отделяющие человека от гориллы и шимпанзе, менее значительны, нежели
различия, существующие между человекообразными и высшими
обезьянами».
На основании результатов, полученных Нутталем, то же самое
можно сказать и о различиях биохимического характера.
Но исследования Уленгута и Нутталя представляют интерес и еще
с одной стороны.
Биохимическая реакция Уленгута позволяет отличать в организме
животных, во-первых, белковые вещества, общие весьма обширным
группам, и, во-вторых, вещества, показывающие самую крайнюю
степень индЛидуализации.
Между белковыми веществами, входящими в состав хрусталика
глаза, Уленгуту не удалось констатировать заметного различия для
всех отделов позвоночных, кроме рыб. Только белки хрусталика рыб
обнаружили резкое отклонение.
Кровь всех млекопитающих показывает, правда, необычайно
слабую, но все же заметную реакцию с сывороткой кролика,
иммунизированного кровью одного какого-нибудь позвоночного животного
(mammalia reaction).
С другой стороны, в крови даже различных индивидуумов
одного и того же вида находятся белки, которые при
благоприятных условиях (с помощью очень сильных специфических
сывороток) иногда удается отличить друг от друга по различной
интенсивности биохимической реакции
Таким образом, в живом организме существуют белковые вещества,
относительно которых можно думать, что их происхождение должно
быть отнесено к весьма различным эпохам: происхождение одних
теряется в глубокой древности, другие, наоборот, как бы еще не
сложились окончательно и видоизменяются на наших глазах.
Быть может, от этих поразительных фактов, если только они
подтвердятся дальнейшими исследованиями, можно ждать освещения
одного из самых темных и вместе с тем самых глубоких вопросов
биологии— вопроса о наследственности.
Если в нашей крови циркулируют вещества, до известной степени
представляющие химическую базу индивидуальных различий между
отдельными особями человеческого рода, и если эти же или
родственные им вещества констатированы и в других частях нашего организма,
то невольно является вопрос, не стоят ли эти вещества в ближайшем
отношении к тем, с помощью которых передаются по наследству
характерные особенности данного индивидуума. Быть может, этими
веществами являются ферменты, столь распространенные в самых
разнообразных организмах и, очевидно, игравшие первостепенную роль зо
многих явлениях жизни.
Эволюция вещества в мертвой и живой природе
39
Но здесь мы опять подходим к крайним пределам положительного
знания, за которыми начинается безграничная область фантазии — и
нам пора остановиться.
Фантазия, несомненно, является одним из могущественнейших
рычагов науки.
С гениальным полетом фантазии связано много величайших
событий, и не без основания на этом настаивал в одной из своих блестящих
речей знаменитый английский физик Дж. Тиндаль.
Но для фантазии естествоиспытателя существуют самой природой
поставленные границы.
Как сказочный Антей терял свою силу всякий раз, как утрачивал
связь с землей, так рискует потерять путеводную нить и заблудиться
в дремучем лесу естествоиспытатель, если даст слишком далеко увлечь
себя полету фантазии и покинет твердую почву фактов.
Позвольте поставить на этом месте точку и в заключение этой бе-
.седы высказать надежду, что мне удалось хотя сколько-нибудь
осветить перед вами обширную и в значительной степени еще непонятную
область эволюции вещества.
В нашем обзоре мы коснулись только двух основных случаев этой
химической эволюции: эволюции элементов в «мертвой» природе и
эволюции химических соединений в живых организмах.
Я полагаю, что если к этим двум основным группам явлений
присоединить еще процессы химической эволюции, совершающиеся
на поверхности и в недрах нашей планеты, химические процессы,
благодаря которым образовались и ныне выветриваются первозданные
горные породы, благодаря которым произошли каменный уголь, нефть
и множество других ископаемых; если мы прибавим сюда те еще
несравненно более многообразные и сложные химические превращения,
которые, протекая на небесных светилах, продолжают в течение
неисчислимых периодов времени медленно, но неуклонно изменять их
химический состав, то мы не можем не прийти к заключению, что
принцип эволюции является всеобъемлющим не только в приложении к
наукам биологическим, но и по отношению к химии.
Х8$8»«-!
СОВРЕМЕННЫЕ ЗАДАЧИ ОРГАНИЧЕСКОЙ ХИМИИ
Вступительная лекция, читанная в Технологическом институте
18 ноября 1909 г.
Сборник речей и докладов, посвященных памяти Л. А Чугаева.
Л.. И XT И, 1924, стр. 153
Когда говорят о происхождении органической химии, как отдельной
отрасли знания, о причинах, по которым соединения углерода
трактуются, как обособленный самостоятельный отдел химической науки,
обыкновенно прибегают к помощи истории и вызывают призрак
жизненной силы, который заставлял химиков старого времени считать
созидание сложных органических соединений из элементов за
исключительную привилегию живых организмов.
Теперь мы все хорошо знаем, что жизненная сила давно сделалась
мифом, по крайней мере в глазах химиков, что привилегии на
искусственное получение множества органических препаратов давно уже
перешли в руки предприимчивых немецких фабрикантов.
Историческая причина, в силу которой химия соединений углерода
в свое время обособилась от остальной химической науки, таким
образом, перестала существовать, а между тем и до сих пор
органическая химия продолжает оставаться самостоятельной научной
дисциплиной.
Спрашивается, имеем ли мы здесь дело с простым пережитком
прошлого, с своего рода традицией, или на это есть особые достаточно
веские основания?
Обыкновенно принято отмечать несколько таких оснований и между
ними на первом плане указывать на огромное число и
разнообразие органических соединений.
Число теоретически возможных соединений углерода, можно
сказать, безгранично. Число известных и описанных превосходит общее
число соединений всех остальных элементов, кроме углерода. Это
число давно уже считается сотнями- тысяч. Разнообразие органических
соединений почти беспредельно.
Это подавляющее число, это огромное разнообразие стоит прежде
всего в очевидной связи с двумя особенностями, характеризующими
углерод как химический элемент: 1) с его резко выраженной
способностью к полимеризации, к образованию сложных
комплексов, содержащих по нескольку атомов углерода в молекуле, и 2) с
его способностью давать начало многочисленным изомерам.
Под изомерами мы разумеем вещества, обладающие совершенно
одинаковой молекулой, в смысле качества и числа входящих в нее
атомов, и тем не менее глубоко различающиеся друг от друга по
свойствам. Таковы соли гремучей и циановой кислоты — исторически
первые примеры, на которых установлено самое понятие об изомерии. Оба
ряда солей отвечают одной и той же формуле, но между тем как соли
гремучей кислоты сильно взрывают и обладают страшной разруши-
Современные задачи органической химии
41
тельной силой, соли циановой кислоты вовсе лишены этой
особенности.
Обе эти отличительные черты органических соединений нашли себе
наглядное выражение в теории химического строения,
получившей з области органической химии и свое начало, и наиболее
последовательное и плодотворное применение.
Эти особенности органических соединений вызвали к жизни ряд
своеобразных вопросов и задач, ряд методов исследования и
теоретических представлений, только отчасти приложимых к изучению
соединений, образуемых другими элементами. В результате получилось, чго
органическая химия сохранила и по сие время то обособленное
положение, которое ей как бы пророчески было указано историей.
В тесной связи с только что отмеченными особенностями углерода,
отражающимися на его соединениях, находится еще одна
отличительная черта этого элемента, не всегда в достаточной степени оттеняемая,
а между тем едва ли не важнейшая из всех.
На этой именно черте я хотел бы сегодня несколько ближе
остановить ваше внимание, сделав ее исходным пунктом для того, чтобы
формулировать те главные вопросы, вокруг которых сосредоточены в
настоящее время интересы органической химии или же, как мне
кажется, будут сосредоточены в ближайшем будущем.
Но прежде всего позвольте мне сделать несколько предварительных
замечаний о химическом равновесии.
Мы говорим, что система тел находится в химическом равновесии,
когда с течением времени ничего в составе этой системы не меняется.
В состоянии химического равновесия находится, например, чистая
вода, сохраняемая при комнатной температуре; водяной пар при 2500°
и- нормальном давлении, если около 4% его находится в состоянии
диссоциации на кислород и водород.
Приложение принципов термодинамики к химии дает нам общее
начало, указывающее направление, в котором происходят химические
процессы. Это будет направление, отвечающее наибольшей возможной
потере свободной энергии. Когда эта потеря произойдет, наша
система придет в состояние равновесия.
Рассмотрим теперь сосуд, наполненный гремучим газом при
обыкновенной температуре. Всем известно, что могут пройти целые годы и
мы не будем в состоянии заметить какого-либо сокращения газового
объема; самые чувствительные методы исследования не позволят нам
уловить хотя бы следы образовавшейся воды.
Состояние системы с течением времени не меняется; следовательно,
мы имеем дело с состоянием равновесия — таково, казалось бы,
должно быть наше логическое заключение.
Но мы хорошо знаем, что стоит только проскочить маленькой
искорке, и тотчас же с огромной скоростью совершается реакция,
сопровождаемая взрывом, а если вместо искры взягь коллоидальный
раствор платины (так называемую жидкость Бредига), то та же реакция --
превращение гремучего газа з воду — произойдет с измеримой
скоростью. В обоих случаях совершится процесс, необходимость которого
предуказуется законами термодинамики, процесс, приводящий к
состоянию равновесия.
Что же представляет из себя состояние гремучего газа при низкой
температуре? Это не будет состояние химического равновесия в
настоящем смысле слова, а между тем наша система, очевидно, обладает
одним существенным признаком такового: состояние ее видимым
42
Современные задачи органической химии
образом не изменяется во времени. Такого рода состояние системы
можно, пользуясь термином Дюгема, назвать состоянием ложного
равновесия*.
Происходит ли в данном случае полная остановка химического
процесса, как это принимает Дюгем, иными словами, имеет ли скорость
реакции значение нуль или, наоборот, значение это не равно, а только
весьма близко к нулю, реакция не останавливается, а лишь идет с
ничтожно малой скоростью?
Не так легко решить, которое из этих предположений отвечает
действительности.
На основании некоторых соображений можно, однако, думать, что
более правдоподобным является второе предположение, допускаемое
Оствальдом и Нернстом.
В таком случае мы не замечаем течения реакции между
составными началами гремучего газа (позвольте употребить это грубое
сравнение) по причине, аналогичной той, по которой мы не заметим (в
короткое время) падения тяжелого тела в какой-либо очень густой
жидкости, например в асфальте, другими словами, в жидкости с очень
большим внутренним трением.
Нагревая асфальт, уменьшая его внутреннее трение, мы не
создаем нового процесса, а только увеличиваем скорость
происходившего ранее. Аналогичным образом возрастает скорость
реагирования в гремучем газе от возвышения температуры или от
каталитического действия платины (в других случаях, например, в случае
смеси хлора с водородом, также от действия света и т. п.)
На скорость химического процесса можно смотреть прежде всего как
на результат взаимодействия двух противоположных факторов:
движущей силы химической реакции и другого фактора, который
можно назвать химическим или пассивным
сопротивлением (Г и б б с).
Скорость реакции прямо пропорциональна движущей силе и
обратно пропорциональна сопротивлению. Здесь невольно приходит в
¦голову аналогия с известным законом Ома, согласно которому сила
тока прямо пропорциональна электродвижущей силе (разности
потенциалов на полюсах) и обратно пропорциональна сопротивлению
проводника.
Движущая сила реакции определяется потерей свободной энергии,
которую испытывает данная система, а величина этой потери в
некоторых случаях может быть выведена из данных опыта, например из
определения электродвижущей силы обратимых гальванических
элементов, из термических данных, по методу, недавно указанному
Нернстом и пр.
Таким образом, часто можно бывает теоретически предсказать
направление, в котором должна пойти реакция в той или иной системе
тел **, а в иных случаях, при наличности всех необходимых данных,
даже точно определить ее количественные результаты.
Но при всем том мы совершенно бессильны на основании*
принципов термодинамики сделать какое-либо заключение о скорости, с
которой будет протекать наша реакция, и это именно потому, что
вовсе не осведомлены о природе химического сопротивления.
* В состоянии ложного равновесия могут находиться как более или менее сложные
системы тел, так и химические индивидуумы, например гремучий газ или черный порох
с одной стороны, хлористый азот, сероуглерод, нитроглицерин — с другой.
** Этого результата очень часто можно достигнуть гораздо проще на основании
приблизительно верного (главным образом для низких температур) правила
наибольшей работы Бертло.
Современные задачи органической химии
43
Мы^ догадываемся, что оно очень мало, когда процессы идут с
огромной скоростью, практически моментально (взрыв гремучего газа,
реакции между ионами в солевых растворах); что оно очень велико,
когда наступает явление ложного равновесия, о котором мы только
-что говорили.
Само собой разумеется, что наступлению ложного равновесия
должна благоприятствовать и незначительная двигательная сила реакции.
Но уже на примере гремучего газа мы нидим, что весьма значительная
двигательная сила не мешает наступлению ложного равновесия. С
другой стороны, можно указать и такие случаи, когда при незначительной
двигательной силе реакция совершается почти моментально.
Мне кажется, что уже из этих немногих намеков, которыми по
необходимости приходится здесь ограничиться, для всех станет ясно,
какое огромное поле для будущих исследователей представляет эта
область химии, включающая учение о скорости химических процессов и
о химическом сопротивлении.
И вот для изучения и разработки эгой области, по-видимому, как
раз химия углеродистых соединений представляет наиболее
благодарную почву, наиболее богатый фактический материал.
Влияние химического сопротивления встречается, правда, и среди
соединений всех остальных элементов, и гремучий газ является в этом
отношении одним из весьма ярких примеров; однако мы не найдем ни
одного другого элемента, который бы мог конкурировать в этом
отношении с углеродом.
В области органической химии мы встречаемся с самыми
.разнообразными типами ложных равновесий буквально на каждом шагу, и я
постараюсь показать, что в этом обстоятельстве следует видеть ту
доминирующую черту, которая налагает печать на всю химию
углеродистых соединений.
Выше мы отметили две особенности углерода, проявляющиеся в
природе его соединений: во-первых, способность к полимеризации,
влекущая за собой существование гомологических рядов, в
которых мы принимаем цепеобразное соединение между атомами
углерода, и, во-вторых, способность давать начало изомерам, соединениям.
в которых предполагается неодинаковое распределение (тех же самых)
атомов внутри химической частицы.
Спрашивается, составляет ли самая способность атомов данного
элемента к взаимному сочетанию особенность, присущую одному
только углероду; является ли такою же специфическою особенностью
этого элемента способность давать начало изомерам? — Отнюдь нет.
Факты говорят совершенно обратное.
Мы знаем существование полимерных молекул кислорода 02 и
озона Оз, серы Se--S2, фосфора Р4 и т. д.; мы знаем существование таких
соединений, как перекись водорода Н202, азотистоводородная кислота
N3H, гидразин N2H4> жидкий и твердый фосфористый водород,
полисернистые соединения водорода, политионовые кислоты и многие
другие соединения, в молекуле которых не только содержится п-о
нескольку атомов кислорода, азота и фосфора, но и приходится принять
существование такого же взаимного (цепеобразного) сочетания этих
элементов, которое мы допускаем для соединении углерода.
Для соединений некоторых элементов, особенно платины, кобальта
и хрома, известны также случаи изомерии.
Таким образом, в обоих отношениях принципиальной разницы
между углеродом и остальным]! элементами, по-видимому, не существует.
44
Современные задачи органической химии
Нет разницы качественной, но зато есть огромная разница
количественная.
Явления полимерии и изомерии среди соединений других элементов,
составляющие сравнительно редкие исключения, для соединений
углерода совершенно обычны. И причину этой разницы следует прежде
всего искать в огромном распространении среди углеродистых
соединений явлений ложного равновесия
Легко понять, что если бы химия углеродистых соединений не
изобиловала случаями ложного равновесия, то мы не имели бы никакой
возможности изолировать и изучить в отдельности гомологи и
изомеры., в огромном количестве представленные во всех классах
органических соединений. Не подлежит сомнению, что гомологи или
изомеры не могут обладать одинаковой степенью устойчивости. Вообще
говоря, каждый член гомологического ряда стремится частью
распасться на более простые молекулы, частью полимеризоваться в более
сложные. Точно так же каждый изомер, отвечающий определенной
молекулярной формуле, будет иметь тенденцию, по крайней мере отчасти,
перейти в другие изомерные формы. Такие процессы фактически и
происходят, когда, благодаря подъему теміпературы или присутствию
катализаторов, устраняются химические сопротивления и процессы
могут идти сравнительно быстро.
В эту категорию относится прежде всего большая часть так
называемых пирогенетических реакций, изучению которых положено
начало классическими работами Берт л о.
Если бы такого рода процессы протекали и при низких
температурах, то, очевидно, ни один гомолог, ни один изомер не мог бы быть
получен в чистом состоянии, ибо он всякий раз на глазах
экспериментатора и вопреки его желанию переходил бы в равновесную систему
или же, в более редких случаях, в одну наиболее устойчивую
модификацию.
Конечно, дело не ограничивается рядами гомологов и изомеров.
Только что сказанное относится к многочисленным изологическим
рядам, и вообще ко всему разнообразию возможных типов комбинаций
между углеродом и теми немногими элементами, которые обычно
входят в большинство органических соединений (Н, О, N, отчасти S, Р и
др.).
Таким образом, мы с полным правом можем сказать, что явлениям
ложного равновесия химик обязан прежде всего возможностью
индивидуализировать, очищать органические
соединения самых разнообразных типов, а в частности, наблюдать
многочисленные случаи гомологии, изомерии, изологии и т. д.
Понятно теперь, что благодаря ложным равновесиям сделалось
возможным развитие и самого учения о химической структуре.
Прежде всего, потому, что толчком к развитию теории химического
строения послужило именно констатирование случаев изомерии и
гомологии. Во-вторых, потому, что структурная теория не ограничилась
выводами числа изомерных форм для каждой молекулярной
формулы, но дала еще руководящую нить для экспериментального решения
вопроса: какому реальному телу соответствует
каждая данная структурная формула.
Опытная же установка структуры в отдельных случаях стала
возможной только благодаря умелому пользованию явлением ложного
равновесия.
Существует целый ряд реакций, в которых молекула органического
соединения претерпевает только частичное вполне определенное изме-
Современные задачи органической химии
-45
нение, например замена водорода или гидроксила хлором, остатком
органической кислоты и т. п. При этом вся остальная часть молекулы,
составляющая некоторый сложный радикал, остается в
первоначальном состоянии.
Пропильная группа, например, не изомеризуется в изопропильную,
ацетиленовый остаток не уплотняется в бензольный и т. д., хотя бы
такое превращение и сопровождалось потерей свободной энергии и
переходом системы в более устойчивое состояние. Очевидно, и здесь
можно говорить о специальном случае равновесия, касающегося
некоторой части органической молекулы.
Правда, далеко не все реакции, в которых участвуют органические
соединения, удовлетворяют указанному условию. Опыт показал, что
процессы, протекающие при высокой температуре в присутствии
реагентов сильно кислого характера (например, сильных кислот,
галоидных соединений фосфора, вообще веществ, легко дающих сильные
кислоты во время реакции), а также и сильных оснований, сравнительно
часто сопровождаются нежелательными побочными реакциями,
например изомеризацией, т. е. перегруппировкой атомов, молекулу
образующих, полимеризацией и т. п.
Отсюда явилось, особенно за последние годы, стремление искать
реакций, свободных от этих недостатков, протекающих, в
нейтральной среде и при возможно низкой температуре. Особенно
следует отметить выдающиеся заслуги в этом отношении покойного
нашего соотечественника проф. Е. Е. Вагнера, автора известного
метода окисления непредельных соединений с помощью перманганата.
Потребность в подобного рода реакциях особенно остро ощущается
при исследовании таких групп органических соединений
(непредельных, особенно терпенов и их производных и др.), представители котот
рых отличаются значительной изменчивостью, склонностью к
процессам изомеризации, уплотнения и т. п.
Мы увидим, что реакции, протекающие в возможно умеренных
условиях, и в другом отношении заслуживают серьезного внимания и
тщательной разработки.
Вообще же можно сказать, что только широкое и умелое
использование случаев ложного равновесия в применении к изучению
органических соединений позволило во множестве случаев осуществить
основную, ставшую классической, задачу органической химии: сначала
шаг за шагом, вплоть до последних кирпичей, разобрать здание
химической молекулы, потом из этих кирпичей, опять в надлежащей
последовательности, воссоздать, синтезировать ту же молекулу из
продуктов ее разрушения и на этом двойном пути; а и ал и з а и синтеза
составить себе представление о ее внутренней архитектуре.
Развиваясь в этом именно направлении, органическая химия стала
par excellence* той областью знания, о которой Бертло имел
возможность сказать, что «la chimie сгёе son objet», что она сама создает
себе объект для исследования. Блестящие успехи органического
синтеза, о которых я упоминал в самом начале этой лекции, в значительном
степени обусловлены возможностью каждый химический процесс
останавливать, так сказать, на полдороге для того, чтобы получить те
или другие (хотя бы и малоустойчивые) продукты, имеющие
теоретическое или практическое значение: уясняющие или проверяющие наши
структурно-химические представления; ьещества, возникающие внутри
живых организмов или имеющие то или иное техническое применение.
* По преимуществу (франц.). Прим. ред.
46
Современные задачи органической химии
Так было выяснено химическое строение многих красящих веществ,
сахаристых веществ, алкалоидов, терпенов, углеводородов нефти...
Многие из них были получены синтетически, в том числе и такие,
которых в природе никогда не было и, вероятно, никогда не будет найдено.
С полным правом можно сказать, что в этой области человек не
только научился подражать живой природе, но во многих случаях
успел далеко оставить ее за собой.
Эта, так сказать, созидательная работа органической химии'
далеко еще не закончена и продолжается в настоящее время. Ряд
интереснейших, частью весьма трудных задач стоит на очереди, как-то:
синтез многих природных красящих веществ, в том числе на первом
плане хлорофилла и гемоглобина, терпенов, алкалоидов,
полисахаридов и пр.
Недавно один из самых выдающихся представителей этого
направления Э. Фишер приступил к монументальным исследованиям,
касающимся наиболее сложной и трудной задачи синтетической химии,—
выяснению конституции и синтеза белковых веществ, как
известно, составляющих основной материальный субстрат жизненных явлений.
В осуществлении этих задач огромную поддержку оказало и
продолжает оказывать то расширение начал структурной теории, в виде
представления о пространственном расположении атомов в молекуле,
которым мы обязаны Вант-Гоффу и Ле-Белю и которое известно под
именем стереохимического учения. Подобно тому как
структурная теория все огромное множество органических соединений
классифицирует в стройную систему, указывая для каждого свое
определенное место, так и стереохимическое учение довершает в указанном
отношении построение системы, обоснованной на теории строения, и
тем самым дает драгоценную руководящую нить для исследования
в тех областях, где возможны случаи пространственной изомерии.
Особенно выдающийся пример плодотворного приложения
стереохимии мы имеем в классических исследованиях Э. Фишера над
сахаристыми и белковыми веществами.
На последних ступенях органического синтеза наша наука
соприкасается еще с одним важных вопросом — с вопросом о пределах
возможного усложнения вещества.
Со времен алхимии и до наших дней не перестают интересоваться
вопросом о существовании нижнего предела превращений
вещества, о пределе его упрощения. Еще очень недавно мы были если
и не вполне убеждены в существовании такого предела, то все же
считали весьма вероятным, что мы имеем его в элементарных атомах.
Ныне этот взгляд сильно поколеблен удивительными фактами,
добытыми при изучении радиоактивных веществ и их превращений.
Границы делимости материи расширились на наших глазах.
Электрон, атом электричества, заступает место атомов химических
элементов. От науки будущего можно ожидать еще много интереснейших
открытий в этой все еще слишком мало изведанной области.
Но если мы еще мало знаем о пределе упрощения вещества, то
еще менее знаем о пределах его усложнения.
До сих пор химики не встречали препятствий в своих попытках
усложнять молекулы органических соединений, но, кто знает, быть
может мы уже не так далеки от этого предела. Быть может, поблизости
от этого предела понятие о молекуле сольется с представлением о тех
сравнительно крупных частичках вещества, которые, будучи
распределены в той или другой жидкости, образуют так называемые
коллоидальные растворы, а понятие о химическом сродстве как о силе, обуслов-
Современные задачи органической химии
47
ливающей существование наших химических индивидуумов, сольется с
понятием о тех силах, под влиянием которых образуются
адсорбционные и другие так называемые неопределенные соединения.
Выяснение всех этих вопросов принадлежит будущему, но едва ли
можно сомневаться, что органической химии будет принадлежать
существенная роль при их постановке и разрешении.
Говорят, что исключениями часто подтверждается правило. И в
данном случае, основная отличительная черта, характеризующая
соединения углерода и определившая направление, в котором развилась и
сформировалась органическая химия, особенно оттеняется по сравнению
со случаями, в которых органические соединения частью теряют свою
инертность и обнаруживают отсутствие или слабость пассивных
сопротивлений.
Сюда относятся так называемые таутомерные соединения,
показывающие как бы двойственную натуру, например одновременно
функцию кетонов и алкоголей, нитрозосоединений и оксимов и т. д.
Случаи этого рода, изучение которых составляет одну из
интереснейших задач современной органической химии, объясняются
существованием в каждом данном веществе смеси двух (иногда и большего числа)
изомерных форм, легко переходящих друг от друга.
В некоторых случаях эти формы могли быть изолированы, часто,
однако, равновесие между ними устанавливается настолько быстро,
что мы получаем смеси таутомеров, которые невозможно разделить.
Таковы, например, ацетоуксусный эфир и его аналоги, (3-дикетоны,
многие хиноны и их производные и, т. п. В этих явлениях тесная связь
между возможностью наблюдать явления изомерии н наличностью случаев-
ложного равновесия, существованием пассивных сопротивлений до
очевидности ясна.
Ту же самую мысль можно иллюстрировать примерами до
известной степени противоположного характера. Существуют некоторые-типы
соединений «минеральной» химии, для которых хорошо известны случаи
изомерии (и даже, по всей вероятности, стереоизомерии), близко
напоминающие изомерию органических соединений. Сюда в особенности
относятся азотистые (частью также сернистые) соединения некоторых
тяжелых металлов, особенно платины, кобальта и хрома.
Наибольший интерес представляют соединения, в которых соли
упомянутых металлов находятся в сочетании с аммиаком; это так
называемые комплексные металло-аммиачные соединения, или
аммиакаты.
Весьма замечательно, что наличность изомерии среди этих
соединений совпадает с появлением у них других характерных черт органических
соединений и прежде всего той пассивности и инертности, которую эти
последние так' склонны проявлять в химических превращениях. Ярким
примером этой особенности упомянутых комплексных соединений
является не только большая прочность связей, при помощи которых
удерживаются ими молекулы аммиака, но и тот особый характер, которым
нередко обладают в этих соединениях атомы галоида и вообще
кислотные остатки. Они не только не ионизированы, но и с крайней
медленностью переходят в состояние ионов, уподобляясь в этом отношении
галоиду органических соединений, особенно связанному с неокисленным
углеродом (например, в С2Н5С1, С2Н5ВГ, CH3J и пр.).
Весьма замечательным представляется также огромное число и
разнообразие типов химических соединений, которое обнаруживается
среди аммиакатов и близких к ним производных только что упомянутых,
металлов. Для примера укажу, что по данным А. Вернера, относя-
48
Современные задачи органической химии
щигмся. к 1906 г. число таких аммиакатов общей формулы MeXn(NH3)m
(где Ме = металлу) выражается приблизительной цифрой 1700.
Таким образом, там, где соединения, обычно слывущие под именем
«неорганических», по тем или иным причинам, обнаруживают особую
склонность к состояниям ложного равновесия, они роковым образом
приобретают ряд других основных черт органических соединений;
увеличивается и степень сложности соединений, и их число, и
разнообразие возможных комбинаций, наблюдаются явления изомерии...
Область комплексных соединений кобальта, хрома и платины уже
теперь представляет из себя как бы органическую химию в миниатюре.
с ее (соответственным образом, правда, видоизмененными) задачами,
методами исследования и теоретическими представлениями.
Эта область, еще только недавно сделавшаяся предметом более
широкого и систематического исследования, главным образом
благодаря работам цюрихского профессора А. Вернера, полна глубокого
интереса и обещает принести обильные плоды в будущем.
Мы только что говорили о тех специальных случаях, где столь
распространенные среди органических соединений пассивные
сопротивления ослабевают, давая начало явлениям таутомерии.
Не менее важное значение имеет изучение тех общих факторов,
под влиянием которых эти сопротивления, эти состояния ложного
равновесия могут быть нарушены для любого органического
соединения, так что временно задержанные химические процессы
становятся возможными.
Мы видели, что существует два могущественных фактора этого
рода: .возвышение температуры, к которому примыкает
действие света, и участие в реакции катализаторов.
Ближайшее изучение этих факторов составляет одну из насущных
задач органической химии.
Мы уже видели, что пользование высокой температурой для
ускорения химических реакций должно применяться с большой
осмотрительностью, особенно в тех случаях, где мы желаем использовать наши
результаты для того, чтобы изучить строение или осуществить
планомерный синтез того или другого органического соединения.
И если тем не менее возвышение температуры является одним из
самых обычных средств, которыми мы пользуемся в наших
лабораториях для того, чтобы увеличить скорость того или другого химического
превращения, то следует всегда иметь в виду, что при этом, особенно
если дело идет об очень высоких температурах (300—500° и выше),
легко может наступить слишком грубое нарушение пассивных
сопротивлений (нередко связанное и с нарушением истинного равновесия, в
смысле закона Вант-Гоффа). В результате получается глубоко идущее
разрушение первоначальных химических молекул, взамен которых
нарождаются новые, соответствующие более устойчивому состоянию
системы. Таковы, например, многочисленные пирогенетические реакции.
Изучение таких далеко идущих и, по большей части, весьма сложных
превращений, едва ли может способствовать выяснению постепенного
хода отдельных превращений под влиянием повышения температуры.
В некоторых случаях, однако, удается наблюдать переходы' более
простого характера. Сюда относятся, например, некоторые процессы
изомеризации: превращение пинена в дипентен (Бертло и др.), ТРИ_
метилена в пропилен (Танатар) и особенно весьма интересные
случаи изомеризации галоидопроизводных, исследованные А. Е.
Фаворским. Подобно возвышению температуры, во многих, хотя, конечно,
далеко не во всех, случаях действует освещение лучами той или иной
Современные задачи органической химии
49
длины волны. И в этом случае действие двоякое: с одной стороны,
только ускоряется реакция, устраняются пассивные сопротивления,
с другой — происходит перемещение самого равновесия. По-видимому,
примером первого рода влияния будет действие света на смесь хлора
с некоторыми углеводородами; примером второго рода —
фотохимическое разложение углекислоты в хлорофильном зерне, а в области
минеральной химии недавно наблюденное Кеном перемещение
равновесия в системе 2S02-h02^2S03 под влиянием лучей ртутной
кварцевой лампы с очень короткой длиной волны. Среди органических
соединений, вне всякого сомнения, число возможных фотохимических
процессов того и другого рода весьма велико. Но заманчивая область эта,
достойная внимательного и всестороннего изучения, пока еще весьма
мало разработана. О вероятном значении ее для органической химии
будущего можно отчасти уже судить по результатам недавних
интересных исследований Чамичиана и Зильбера, затем Штоббе,
Д. Бертлои др.
Как и следовало ожидать, огромное значение получило приложение
к органической химии процессов каталитических. Здесь на одном
из первых мест следует поставить русскую школу А. М. Бутлерова.
Начиная с замечательного синтеза метиленитана путем
конденсации формальдегида при действии извести, начиная с этой исторически
первой попытки синтезировать сахаристое вещество и кончая
известными исследованиями А. Е. Фаворского над изомеризацией
ацетиленовых углеводородов под влиянием алкогольной щелочи, в
лаборатории Бутлерова им самим и его учениками осуществлен целый ряд
превращений органических веществ, протекающих под влиянием
катализаторов. Но самые выдающиеся исследования, касающиеся
разработки каталитических процессов в органической химии, связаны с име
нем покойного Г. Г. Густавсона, впервые обратившего внимание
химиков на удивительные «каталитические» свойства галоидных солей
алюминия. Известный метод синтеза ароматических соединений Фри-
деля и Крафтса, недавно получивший новое приложение в поли-
метиленовом ряду благодаря интересным исследованиям Н. Д.
Зелинского, ведет свое начало из этого именно источника; в качестве
дальнейших наиболее ярких примеров каталитических реакций укажем на
синтез Клаіізена и К и е в е н а г е л я, на метод гидрирования
С а б а т ь е и С а н д е р е н а, на многочисленные и разнообразные
каталитические реакции, открытые и изученные В. Н. Ипатьевым, на
окисление перекисью водорода по Феитону и Руффу и пр.
Вопрос о катализе приводит к постановке дальнейшей проблемы
органической химии.
Давно уже один известный физиолог высказал мысль, что рано или
поздно физиологическая химия должна сделаться частью
каталитической. Не подлежит сомнению, что растения и животные, в недрах
которых происходят и сложнейшие синтезы и глубочайшие процессы
распада, осуществляют эти процессы не теми путями, какими это делаем
мы в наших лабораториях. Прижизненные процессы совершаются при
низкой температуре и отсутствии энергичных реагентов, ибо высокая
температура и энергичные реагенты являются ядами для живой
протоплазмы.
Со времен Доберейнера, Берцелиуса и Шенбенна
аналогия биохимических реакций с каталитическими не перестает
останавливать на себе внимание химиков и биологов.
Ныне, особенно после работ Э. Фишера, Э д. Бух не р а,
К р. Г и л л я, Г. Т а м м а н а, В. И. П а л л а д и н а. Дюкло, Берт-
4 Л. А. Чугаев, т. III
50
Современные задачи органической химии
рана, Баха, Эммерлинга и др., мы должны приписать роль утих
прижизненных катализаторов ферментам или энзимам. Под
влиянием ферментов происходят и процессы прижизненного окисления
(дыхания), и процессы анаэробного распада органических соединений,
и процессы синтетические. После только что упомянутых исследований,
особенно же после того, как Бредит и своих коллоидальных
растворах платины и других металлов дал нам прототип
неорганических ферментов, с особой настойчивостью в органической химии
возникла новая задача: искать катализаторов, работающих, подобие»
настоящим ферментам, при низкой температуре, в «умеренных»
условиях, и таким образом стремиться к осуществлению
биохимических процессов. К каталитическим реакциям подобного рода
относятся, например, процессы гидрирования органических соединений
в присутствии мелко раздробленных или коллоидальных металлов
платины и палладия (Фокин, Вильштоттер, П а а л ь), окисление
перекисью водорода в присутствии солей железа, отчасти реакции
К невена геля и пр.
Следуя этому пути, немецкий ученый Шаде недавно до известной
степени подошел к искусственному осуществлению процесса спиртового^
брожения, как бы подражая деятельности Бухнеровской зимазы.
При каталитическом разложении сахара, с одной стороны (при
действии щелочи), можно получить муравьиную кислоту, с другой (с
переходом через молочную кислоту)—одновременно муравьиную кислоту
к уксусный альдегид. Шаде показал, что муравьиная кислота
обладает способностью в присутствии мелко раздробленного
металлического родия восстановлять уксусный альдегид в этиловый спирт, окисляясь,
при этом в углекислый газ:
СНз-СНО+Н—СООН=СНя—СН2ОН-|-СО«.
Таким образом, в результате ряда каталитических реакций сахар
превращается в те самые продукты, как и под влиянием живых
дрожжей в процессе алкогольного брожения.
К той же самой области относятся исследования Эйлера и
особенно Б р е д * г а над асимметрическим катализом.
Названным авторам недавно удалось при помощи оптически-
деятельных катализаторов осуществить искусственное получение активных
соединений из недеятельных, подобно тому как это было известно и
раньше, но только для реакций, протекающих под влиянием
ферментов *.
Есть еще одна сторона, одно направление, в котором развивается и,
конечно, будет продолжать развиваться органическая химия. И это
направление ее, хотя отчасти, может быть также поставлено в связь
с той основной чертой органических соединений, которую мы здесь все
время подчеркивали.
Сущность этого направления заключается в стремлении
воспользоваться для разъяснения вопросов и задач органической химии
успехами сравнительно молодой еще отрасли пашей науки — химии
физической.
На самом деле мы все чаще и чаще пользуемся услугами как
физико-химических методов исследования, так и тем теоретическим освеще-
* Особенно характерным случаем последнего рода процессов является синтез
деятельного нитрила миндальной кислоты, недавно осуществленный Розе и талером
ил синильной кислоты и бензойного альдегида в присутствии эмульсина.
Современные задачи органической химии
:л
наем химических процессов, которым мы обязаны физихо-химии. В этом
отношении химия углеродистых соединении, благодаря неисчерпаемому
разнообразию как форм вещества, так и типов его превращений,
представляет богатейший материал для исследования.
Недаром органические соединения были призваны сыграть
выдающуюся роль в истории открытия многих основных физико-химических
законов и теоретически важных обобщении. Назовем только закон
действующих масс и все вообще учение о химическом равновесии,
законы химической кинетики, теорию растворов вообще и в частности
законы, касающиеся упругости пара бинарных смесей, и многие другие..
Достаточно будет упомянуть здесь о превосходных исследованиях
М. Б е р т л о, В и л ьгел ь м и, Д. П. Коновалова, С. А р р е н и у с а.
В. Hep н с т а, В. Оствальда, П. И. В а л ь д е н а и др.
И физико-химия не осталась в долгу перед химией органической:
не говоря уже о таких способах исследования, как криоскопия и
эбулиоскопия, которые вошли в обиход всякой химической
лаборатории и оказали органической химии неоценимые услуги, измерения
электропроводности, рефракции, термических
констант, вращательной способности, спектров
поглощения, магнитных свойств год от году все больше и больше
завоевывают себе право гражданства в органических лабораториях.
Исследования этого рода начались уже давно (назовем хотя бы
имена Ко п п а, Ш и ф ф а, Л о ее е и а, Г о р ст м а н а. В. М е и е р а.
К о н о и и и к о r а. Б р юля, Перки н а, Н. А. М е и ш у т к и и а
и многие другие). Но особенное оживление наблюдается в этом
отношении за последнее время, к которому относятся обширные
исследования Ганша и его учеников над диазосоединениями, псевдокислотами
и псевдосснованиями, кинетические исследования Гольдшмидт а,
работы Гартлея, Б эли и других английских химиков над спектрами
поглощения органических соединений. Интересные работы Ф. Г ю и,
П. И. Вальдена, Д. А. Хардина над оптической деятельностью.
Паскаля над магнитными свойствами и мн. др.
Кинетические исследования, по моему мнению, в особенности
имеют право рассчитывать на большую будущность. Мы видели, что
значительная часть современных задач и интересов органической химии
прямо ил'Н косвенно связана с господствующими среди соединений
углерода ложными равновесиями. Но весьма вероятно, с другой стороны,
что самая теория состояний ложного равновесия, до сих пор еще
загадочных, теория катализа, наконец вообще выяснение тех факторов,
которые, помимо химической силы, определяют скорость химического
процесса, должно последовать именно при изучении органических
соединений н их превращений.
Энергетика позволяет нам, подводя общий итог энергии,
выделяющейся при химических превращениях, учитывать потерю свободной
энергии телами, участвующими в реакции. Но ведь этот процесс
превращения энергии химической в другие виды ее совершается при
содействии определенных механизмов, каковыми являются химические
молекулы. О конструкции этих механизмов и о распределении в них
энергии энергетика ничего не может сказать. Первыми данными о
природе этих механизмов, хотя пока еще в форме догадок и
проблематических построений, мы обязаны органической химии. Быть может, ей
же суждено будет сделать и дальнейший шаг, осветить вопрос, как эти
механизмы действуют, привести в связь их конструкцию со скоростью
химических превращений. Не указывают ли нам на эту возможность
те случаи стереохимииеского препятствия (Sterische Hinde-
4*
52
Современные задачи органической химии
rung"), которые в большом числе известны для органических
соединений. Быть может, ценные услуги окажет здесь изучение физических
свойств органических соединений.
В предыдущем я сделал попытку осветить перед вами некоторые,
как мне кажется, особенно важные, из числа текущих проблем
органической химии, исходя из одной коренной особенности органических
соединений, по-видимому, тесно связанной с самой природой
углеродного атома.
Подведем итоги. Наряду с тем оснозным руслом, по которому
направлялось главное течение органической химии в эпоху ее наиболее
блестящего расцвета (во второй половине прошлого века), наряду с
направлением, задачи которого определяются проведением в жизнь
начал структурной теории и стереохимического учения, причем
наиболее могущественными методами исследования являются химический
анализ и синтез, наряду с этим направлением за последние годы в
нашей науке нарождаются новые течения, возникают новые методы,
намечаются новые задачи.
С одной стороны, тщательно отыскиваются и изучаются случаи,
в которых искусственно нарушаются состояния ложного равновесия
органических соединений: явления таутомерии, изомеризации, катализа.
С другой, задачи и методы исследования органической химии успешно
распространяются на химию «минеральную», на так называемые
комплексные соединения, изучение которых разрастается в новую
обширную область.
Ищутся условия для осуществления реакций при низкой
температуре и вообще в умеренных условиях, в подражание процессам,
протекающим з живом веществе. За синтезом органических соединений
естественным образом следует осуществление биохимических процессов.
Наконец, все более и более является потребность приложить к
области органических соединений обобщения и методы работы
современной физической химии.
На этом последнем пути, быть может, последует разгадка самых
темных вопросов химической кинетики: вопросов о природе катализа,
о пассивных сопротивлениях, о ложном равновесии.
Прежде чем заключить настоящую беседу, я хотел бы коснуться
еще одного вопроса, сказать несколько слов об отношении нашей науки
к химической технологии, и это, конечно, прежде всего потому,
что химия вообще и органическая в частности является теоретическим
базисом всего вашего технического образования.
Мы не можем здесь говорить о какой-либо еще одной новой
задаче органической химии, ибо задача пауки заключается только, в
познании истины, в познании природы в широком смысле этого слова.
Зато давно уже'культурное человечество пришло к убеждению, что
без прогресса в науке не может быть и прогресса в
области техники.
Я могу только засвидетельствовать перед вами, что именно успехи
органической химии создали з течение второй половины минувшего
века обширнейшую индустрию красящих веществ, фабрикацию
искусственных медикаментов, ароматических продуктов, взрывчатых
веществ, словом, целый ряд производств, которыми на первом плане
вправе гордиться химическая технология.
В наше время, когда каждый месяц приносит десятки, если не сотни
новых патентов, промышленные деятели Запада чутко прислушиваются
к каждому новому шагу, сделанному в области чистой науки. Ни одно
Современные задача органической химии
53
сколько-нибудь заметное химическое исследование не проходит оез
того, чтобы оказать более или менее значительного влияния па успехи
техники.
Мало того, крупные химические фабрикаты, особенно в Германии,
содержат на свои средства целые обширные, прекрасно обставленные
лаборатории с большим (доходящим до сотни и более) кадром научно-
подготовленных химиков. Эти химики занимаются чисто научными
исследованиями и обыкновенно вовсе не соприкасаются с производством
Там, на Западе, долгим опытом пришли к заключению, что
знание природы практически необходимо, а истина,
от которой как из рога изобилия, исходят практические применения,
часто приходит оттуда, откуда ее всего менее ожидали.
Наше отечество в этом, как и во многих других отношениях, как
известно, является отсталым. Мы не имеем здесь возможности входить
в обсуждение сложных причин этого прискорбного явления. Мы можем
только констатировать один из его результатов.
Несмотря на огромные природные богатства *, па изобилие
каменного угля, нефти, железных руд и других полезных ископаемых, наша
химическая промышленность едва существует.
Конечно, мы имеем здесь сложное явление, зависящее не от одной
только причины.
Но я твердо верю, что одним из залогов будущего успеха нашей
химической индустрии должно быть вселение в наше общество вообще,
особенно же в среду промышленников достодолжного уважения
к науке.
В частности от вас, будущих инженеров, химиков-практиков, будет
в значительной степени зависеть укрепление, и притом не на словах
только, а на деле, этой уверенности в реальной пользе, в настоятельной
необходимости научной химии для техники, укрепление ее в широких
промышленных кругах.
Но для этого вы сами должны проникнуться любовью и интересом
к основной для вас отрасли знания — химии. Вы не должны бояться
потратить время на серьезную научную подготовку: это время
окупится для вас сторицей. Вы должны теперь, пока не поздно, возможно
глубже окунуться в эту науку, возможно глубже заразиться ее
интересами, пежить ее жизнью.
В этом ваша будущая сила, залог вашего успеха в предстоящей для
вас практической деятельности.
Я же счастлив, что найду в вашей среде подготовленную почву в
смысле любви, интереса и уважения к науке вообще и в частности к
органической химии. Я счастлив, что мне придется здесь продолжать
дело, вслед за устройством новой лаборатории, так прекрасно начатое
трудами моего предшественника А. Е. Ф а в о р ско г о и его учеников.
* Из этих богатств только одна кавказская нефть сделалась предметом более
обстоятельных исследований со стороны русских ученых благодаря фундаментальным
работам Д. И. Менделеева, Бенльштейпа и Курбатова, особенно же
В. В. Марковннкова и его учеников, М. И. Коновалова, Н. М. К и ж н е р и
а в последнее время благодаря систематическим и многосторонним пл\-іедо,нашим
Н. Д. Зелинского, которому (а -равно и его ученикам) мы обязаны, между прочим.
синтезом многих нафтенов. Не в русских химиках, конечно, лежит причина такого
недостаточно внимательного отношения к научной обработке естественных богатств
родной страны, она лежит в том общем равнодушии, с которым обычно встречаются
результаты подобных исследований, в отсутствии поддержки моральной и
материальной со стороны как правительства, так и общества (за весьма немногими
исключениями).
ПЕРИОДИЧЕСКАЯ СИСТЕМА ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
ПОСОБИЕ ПРИ ИЗУЧЕНИИ ХИМИИ
Л. Л. Чугаев
Издательство «Образование», СПб , 1913 г.
ПРЕДИСЛОВИЕ
Около двух лет тому назад книгоиздательством «Образование» была
выпущена периодическая система химических элементов в виде
стенной таблицы, с приложением составленной мною краткой брошюры о
периодическом законе. Настоящая книга является как бы вторым
переработанным и значительно (более чем вдвое) расширенным изданием
той брошюры. В ней сделана попытка резюмировать в существенных
чертах современное состояние вопроса о периодическом законе. От
других сочинений, преследующих сходные задачи (например, УепаЬГя,
Rudorf'a, Garrett'a), составленная мною книга отличается прежде всего
двумя особенностями: приведением сравнительно подробного
систематического обзора отдельных групп периодической системы и
рассмотрением основных методов определения атомного веса. Полагаю, что
соответствующие главы (I, VIII и IX) окажутся не бесполезными, особенно
для приступающих к изучению химии.
С другой стороны, необходимость удержать размеры книги в
известных пределах заставила (сократив первоначально намеченный план)
вовсе отказаться от рассмотрения вопроса о генезисе химических
элементов, от воспроизведения и критической оценки бесчисленных вариантов
периодической системы, а также разбора многочисленных попыток найти
закон, выражающий связь между атомными весами элементов. Я
решился на эти сокращения, помимо других соображений, между прочим,
потому, что вопросы эти рассмотрены в недавно появившейся на русском
языке книжке сэра А. Тильдена «Химические элементы», на которую и
сделаны ссылки в соответствующих главах. В то же время нельзя
не признать, что попытки «усовершенствовать» периодическую систему
Д. И. Менделеева, а также найти точные числовые соотношения между
атомными весами элементов до сего Бремени не привели еще к сколько-
нибудь определенным результатам, а потому казалось возможным
оставить эту сторону дела без рассмотрения, не нарушая цельности
изложения.
Особое внимание было обращено на тщательную проверку и подбор
приводимого цифрового материала.
В заключение считаю приятным долгом выразить свою
благодарность Б. С. Швецову за существенную помощь, оказанную мне
составлением части VII главы, посвященной вопросу о соотношениях между
строением линейчатых спектров химических элементов и
периодическим законом.
J1. Чугаев
Периодическая система химических элементов
55
ИСТОРИЧЕСКОЕ ВВЕДЕНИЕ
Периодический .чакон был открыт Д. И. Менделеевым в 1869 г.
Поводом к этому открытию, одному из самых блестящих в области
теоретической химии за весь истекший XIX век, послужило, как известно,
составление классических «Основ химии», предпринятое в промежуток
времени между 1868 и 1870 гг. Для составления руководства нужно
было избрать определенную систему или классификацию элементов.
Существовавшие в то время системы не могли удовлетворить
Менделеева, и потому он стал искать новую, более совершенную. В этих поисках
им руководила предвзятая идея, плодотворность которой не замедлила
обнаружиться. Заключалась она в том, что атомный вес есть
коренное и наиболее глубокое свойстве к а.ж д о г о
элемента.
«Понятие о химических элементах,— говорит Менделеев в своих
«Основах»,— и о том, что мы считаем атомным их весом, принадлежит
к числу таких же основных во всем естествознании, как и понятие о
массе или количестве вещества». «По смыслу всех точных сведений о
явлениях природы,— пишет он в другом месте,— масса вещества есть
именно такое свойство его, от которого должны находиться в
зависимости все остальные свойства, потому что все они определяются
подобными же условиями или таковыми же силами, какие действуют,
определяя вес тела, он же прямо пропорционален массе вещества. Поэтому
ближе или естественнее всего искать зависимости между свойствами и
сходствами элементов, с одной стороны, и атомными весами, с другой»*.
Руководствуясь этой идеей, Менделеев и расположил элементы в
порядке возрастания их атомных весов, причем периодическая
повторяемость свойств выступила со всей очевидностью.
В 1869 г. Менделеев выпустил в свет первый набросок своего закона,
в котором, однако, заключались уже все его основные черты.
Через два года (1871 г.) периодическая система созрела
окончательно и вылилась в ту классическую форму, которая почти без перемен
сохранилась и до настоящего времени.
Великие научные открытия так же, как и крупные исторические
события, никогда не появляются внезапно, неожиданно, но
подготовляются упорным трудом предшествующих поколений, долго вынашиваются
выдающимися умами, прежде чем вылиться в окончательную форму
Поэтому не удивительно, что в известный момент, когда та или иная
идея, так сказать, носится в воздухе, хотя бы в неясной и
неопределенной форме, почти одновременно появляются два, иногда и большее число
ученых, которые чутьем гения схватывают эту идею, извлекают ее из
лабиринта противоречивых фактов, ходячих теорий, предвзятых мнений
и придают ей необходимую ясность и законченность выражения.
Так было с открытием дифференциального исчисления, подавшим
повод к знаменитой полемике между Ньютоном и Лейбницем из-за
приоритета. Так было с теорией происхождения видов путем естественного
подбора, теорией, связанной с именем Дарвина и Уоллеса. Так было и
с историей периодического закона, ибо Дмитрий Иванович Менделеев
имел также своих Ламарков и Уоллесов в лице де-Шанкуртуа, Нью-
лэндса и Л. Мейера.
В сущности, зарождение периодического закона началось вслед за
тем, как, с одной стороны, Лавуазье, следуя пути, указанному Р. Бой-
лем, установил понятие о химических элементах, а с другой —
* См. «Основы химии», 2-е изд., 1932, т. 2, стр. 59. Прим. Ред.
5G
Периобическая система химических элементов
Дж. Дальтон ввел в химию понятие об атоме и атомном весе1. Тогда
стали сличать между собой химические элементы, искать между ними
сходства и различий, и представляется вполне естественным, что в
качестве одного из сравниваемых свойств был избран атомный вес.
Несомненно, что одной из важнейших причин, побуждавших искать
правильности и соотношений между атомными весами элементов, было
появление знаменитой гипотезы Проута, согласно которой вес элементы
произошли путем прогрессивного сгущения одного простейшего,
именно,—водорода.
Определенные числовые соотношения были прежде всего найдены
для групп сходственных элементов. Так, Деберейнер (начиная с
1817 г.— окончательно в 1829 г.) установил так называемый «закон
триад», по которому аналогичные элементы могут быть сгруппированы
по три в так называемые триады, причем атомный вес среднего члена
такой группы близок к полусумме атомных весов крайних элементов.
Так,
Cl+J 35,5-j-127 D /0ПЧ Са+Ва 40+137
_ ._- _ ^Вг (8U), ^ = —2
= Sr(87) и т. д.
Позднее Петтенкофер, Дюма и другие указали на то
обстоятельство, что между аналогичными элементами часто наблюдаются
постоянные разницы, напоминающие по своему характеру постоянное
нарастание молекулярного веса в гомологических рядах органических
соединений, соответствующее осложнению молекулы на группу СНг-
Таковы, например, серии элементов:
Li Na К Rb Cs
7 23 39 85* 133*
Разница 16 16 46 48
(3x15,3) (3X16)
Mg Ca Sr Ba
Разница 16 47 50
(3X16) (3X17)
* Рубидий ц цезий были открыты позднее.
Значительно ближе к периодическому закону подошел Бегюйер
де-Шанкуртуа в 1862 г. Не ограничиваясь сопоставлением
небольших групп близких элементов, он сделал попытку построить систему,
объединяющую все элементы и устанавливающую закономерную связь
между их атомными весами. Шанкуртуа располагает все элементы по
винтовой линии (vis tellurique), навернутой на поверхность прямого
цилиндра под углом 45° к его оси. Направляющая окружность цилиндра
разделена на 16 частей (16 = атомному весу кислорода), атомные же
веса элементов откладываются по его образующей и пропорциональны
длине спирали между началом кривой и данной точкой. При этом за 1
принимается Vie доля окружности (Vie атомного веса О или атомный
вес Н).
При таком расположении сходные элементы часто, но не всегда
попадают на одну и ту же образующую цилиндра (из числа 16
«главных» образующих, проведенных через деления окружности), а атомные
веса их А могут быть приблизительно выражены общей формулой:
А = /і+16т, где т — целое число. Так, например, 0 = 16; S = 16+16X
XI =32; Se-16+16X4 = 80; Те= 16+16x7= 128 и т. д.
Периодическая система химических элементов
о t
іаким образом ясно выступает периодическое чередование сжміств.
Точки кривой, отвечающие элементам, де-Шанкуртуа зовет .\ а р а к-
терными и считает, что «зависимость между свойствами различных
тел проявляется в простых геометрических отношениях между их
характерными точками» *.
Ясно, что в этой системе заключается уже зародыш периодического
закона. Но система де-Шанкуртуа дает обширный простор произволу.
С одной стороны, в среду элементов-аналогов попадаются нередко
элементы совершенно посторонние. Так, за кислородом и серой между S и
Те попадает титан; Мп попадает в число аналогов Li, Na и К; железа
помещается на одну образующую с Са и т. д.
С другой стороны, та же система дает два места для углерода:
одно — для С с атомным весом 12, другое — отвечающее атомному
зесу 44.
Еще ближе, чем де-Шанкуртуа, подошел к периодическому закону
Ньюлэндс в своем законе октав. Первые попытки Ньюлэндса
найти соотношения между атомными весами элементов и их свойствами
относятся к 1863 году, но только в 1865 г. он сформулировал
окончательно свою классификацию элементов в следующей таблице.
н
Li
G1
Во
С
N
О
IF 8 С1 15 С
2 Na 9 К 16
3 MglO Са 17
4 А1 11 Сг 18
5 Si 12 Ті 19
6 Р 13 Мп 20
7 S 14 Fe 21
о и Ni
Си
Zn
V
In
As
Se
'''1
23
24
25
26
27
28
Br
Rb
Sr
Се и La
Zr
Di и Mo
Ro и Ru
2\) Pd
30 Ag
31 Cd
32 V
33 Sn
34 Sb
35 Те
36
37
38
39
40
41
43
Ba
1
Cs
и V
Та
W
Nb
Au
42
44
45
'.6
47
48
49
Pt
и Ir
Tl
Pb
Th
Hg
Bi
Os
54
53
54
56
52
55
51
Цифры, стоящие против знаков соответствующих элементов,
представляют номера, определяющие порядок следования друг за другом их
эквивалентов. В большинстве случаев элементы расположены в
системе в порядке этих именно чисел и только в последнем столбце
замечаются значительные отступления. Несмотря на то, что довольно
большому числу элементов отведено неподобающее место, основные
черты закона октав в таблице Ньюлэндса ясно выступают: элементы
аналогичные, относящиеся к одной и той же естественной группе,
располагаются в горизонтальных строках, причем между
последовательными номерами таких элементов, например, между Li, Na, К (2, 9,
16) постоянная разница = 7. Таким образом, соотношения между
членами такой естественной группы элементов 'напоминают те, которые
существуют в музыке между соответствующими нотами последовательных
октав.
Несомненно, что Ныолэидсу удалось бы избежать очень многих
противоречий и что система была бы много ближе к современной
периодической таблице, если бы он, подобно Менделееву и Мейеру, вместо
эквивалентов, воспользовался атомными весами, величины которых
были только что установлены Жераром к Канниццаро. Но и в
вышеприведенной форме таблица Ньюлэндса, бесспорно, является одним из
любопытнейших документов из истории развития периодического закона.
* Развернув поверхность цилиндра, мы будем иметь на оси абсцисс окружность
круга, на оси ординат —образующую; спираль же разделится на ряд прямолинейных
параллельных друг другу отрезков. Если через две характерные точки, расположенные
на двух соседних отрезках, провести прямую, продолжив ее до пересечения с
остальными отрезками, то получится часть некоторой новой «вторичной» спирали, по которой
расположатся элементы, разницы между атомными весами которых являются
кратными уже не 16, а какой-либо другой величины, и т. д.
'58 Периодическая система химических элементов
Странная судьба постигла автора закона октав. Когда в 1866 г. он
решился выступить со своей системой элементов перед Лондонским
Химическим Обществом, он встретил скептическое, даже насмешливое
отношение. Один из присутствующих (профессор Фостер) спросил Нью-
лэндса, не пробовал ли он расположить элементы в алфавитном порядке
и не нашел ли при этом каких-либо правильностей. Так далеки были
современники открытия периодического закона от восприятия идей, под
влиянием которых гри года спустя (1869 г.) в химической систематике
произошел крупнейший переворот, который только знает история нашей
науки.
Из только что сказанного видно, что хотя Шанкуртуа и Ньюлэндса
следует считать предшественниками Д. И. Менделеева в деле построения
периодической системы, однако, ни тот, ни другой не придали своей
классификации элементов тон стройности и законченности, а главное,
не вывели из нее тех необычайно важных последствий, которые дали
право периодическому закону запять место среди основных и
важнейших законов природы.
В заключение этой небольшой исторической справки нам необходимо
коснуться той доли участия в созидании периодического закона, которая
принадлежит немецкому ученому Лотару Мейеру, и сопоставить ее
с тем, что было сделано Д. И. Менделеевым.
Если внешним поводом для появления периодического закона для
Д. И. было составление «Основ химии», то такой повод был и у Лот.
Мейера, который за 6 лет до Менделеева, в своих «Moderne Theorien
d'er Chemie», книге в своем роде весьма замечательной, дал очерк
руководящих взглядов химии того времени. В этом первом издании
«Теоретической химии» Мейера некоторые желают видеть определенный уже
зародыш периодического закона.
Чтобы быть возможно объективным, я -приведу здесь (в переводе) те
строки, на которые опираются притязания немецкого химика на
приоритет *.
Своеобразные закономерные соотношения, которые давно уже
найдены М'ежду атомными весами различных элементов, неоднократно,
особенно за последние годы, давали повод для обсуждения вопроса, не
являются ли наши атомы сами сочетаниями атомов высшего порядка,
другими словами, группами атомов, или молекулами (следует ссылка
на руководство Гмелина, на работы Петтенкофера и Дюма). На самом
деле такой взгляд сам по себе представляется весьма правдоподобным,
гак как соотношения между атомными весами родственных элементов
вполне аналогичны, например, тем, которые 'существуют между
молекулярными весами в некоторых рядах органических соединений,
обладающих сходственной конституцией **.
Li =7,03 Метиловый спирт СН40 Метил CH:t 15
Разница 16,02 СЫ, СН.> 14
Na — 23,05 Этиловый спирт С2Н80 Этил С2Н5 29
Разница 16,08 CR, СН2 14
К ¦= 39,13 Пропиловый спирт С3Н80 Пропил С3Н7 43
* Стр. 135—139 оригинала. Перепечатано и «Ostwalds Klassiker der exakien Wissen
schaften» № 68. В этом издании (под редакцией К. Зейберта), кроме выдержки из
напечатанного Л. Мейером (а частью из неизданного) материала о периодическом законе,
приведены важнейшие статьи Д. И. Менделеева (на немецком языке), из которых
особенно ценной является знаменитая статья, напечатанная в 1871 г. в Либиховских
Анналах.
** В этих сопоставлениях нет ничего оригинального, так как они встречаются и у
более іранних исследователей, особенно у Дюма.
Периодических система химических элементов
50
Естественно допустить, что разница межд\' атомными весами **тих
металлов, как и в случае только что приведенных, а также сходных с
ними органических соединений или радикалов, определяется разницей
в составе их так называемых атомов. Последние не были бы в таком
случае величинами неделимыми, а представляли бы, в свою очередь,
соединения атомов высшего порядка, следовательно, сложные
радикалы. Аналогия их с теми радикалами, которые уже ныне считаются
сложными, получила бы с этой точки зрения вполне естественное
объяснение.
Численные соотношения, подобные только что приведенным,
встречаются неоднократно. Различные авторы, занимавшиеся этим
предметом, представили эти соображения весьма разнообразной формы.
В нижеследующей таблице такие соотношения даны для шести
хорошо охарактеризованных групп элементов.
Разница
Разница
Разница
Рн.шпца
Разница
Четырсхатомиыс
—
0-12,0
16,5
Si -118,Г»
80,1
—
89,1 ,, г-
2
Sn—117,6
89,4=2-44,7
РЪ-207,0
TpL'xaTOMiiuc
—
N И,о'.
10, Ш)
р ;н.о
41,0
As 75,0
45,6
Sb = 120,6
87,4-2-43,7
Ві-208,0
Двухатомные
—
0 -16,00
16,07
S -32,07
46,7
Se=78,8
49,5
Те- 128,3
—
Одно-
атомные
. _
F ¦ 10,0
1(5.46
С1 -35,46
44.51
Вг-.79Д)7
4(5,8
J-126,8
—
Одіюатомнііс
Li- 7,03
16,02
Na-23,05
16,08
К-39,13
7і6,3
Rb-85,4
47,6
Cs=--133,0
(71-2.35,5)
(Tl-204?)
Двухатомные
(Be--9,3?)
(14,7)
Mg-24,0
16,0
Ca-40,0
47,6
Sr--87,(5
49,5
Ba^l37,l
—
«Мы видим, что первая (соответственно первая и вторая) разница
в каждом вертикальном столбце повсюду близка к 16. Исключение
представляет разница между атомными весами магния и бериллия, причем
последний неизвестен с достоверностью. Обе следующие разницы
колеблются около 46; последняя приблизительно вдвое больше, именно =
= 87—90, если только мы и здесь отвлечемся от атомного веса таллия,
для которого (равно как и атомного веса цезия, предварительно
определенного с веществом, едва лишь открытым), быть может, принимается
величина, несколько ниже истинной». Далее Менер указывает, что
между атомными весами- Ті, Zr, Та и Мо, V, W (причем для Та принимается
137, 6 вместо 182, а для V— 137 вместо 51) разница близка к 46 и что
подобная же разница встречается в рядах следующих сходственных
элементов: Fe, Ru, Pt; Ni, Rh, Ir; Co, Pd, Os, Zn, Cd, Hg, Cu, Ag, Au
(между первыми двумя членами каждого ряда, например, Cd и Zn
разница близка к 46, а между последними —вдвое больше: Hg—Cd =
= 2X44,2).
Этими сопоставлениями исчерпывается все то, что было опубликовано
Лот. Мейером до появления (в 1869 г. в I томе журнала Русского физи-
60
Периодическая система химических элсментоь
ко-химического общества) первой публикации Д. И. Менделеева о
периодическом законе*.
При всем желании видеть здесь существенно больше, чем было
выражено Петтенкофером и Дюма, с одной стороны,
провозвестниками учения о валентности Франкландом, Кекуле и их
последователями, с другой, едва ли приходится. Шаикуртуа и особенно Ныолэндс
несмотря на значительно большее число возражений, которые могут
быть сделаны по поводу отдельных пунктов их классификации, во
всяком случае, ближе, нежели Мейер в 1864 г., подошли к самой сущности
периодического закона, ибо и тот и другой располагали элементы в
порядке возрастающих атомных весов (или эквивалентов), и оба
констатировали при этом периодичность в изменении их свойств.
В исторической последовательности за только что упомянутыми
теоретическими соображениями Л. Мейера следует первая статья
Д. И. Менделеева, которой предшествовало появление в печати
первого наброска периодической системы на отдельном листе, разосланном
Д. И. многим химикам в начале 1869 г.
I III III IV V VI
Ті=50 Zr=90 ? = 180
V=51 Nb=94 Та=182
Сг-52 Мо=96 W=186
Мп=55 Rh=104,4 Pt=197/j
Fe=56 Ru=104,4 Ir=198
Ni=Co=59 Pd=106,6 Os=l99
H=l Cu=-63,4 Ag=-108 Hg=200
Be=9,4 Mg=24 Zn=65,2 Cd=ll2
B=ll Al-27,4 ?=68 Ur=116 Au=197
C=12 Si = 28 ?=70 Sn=118
N=14 P=31 As=75 Sb=l22 Bi-210
0=16 S=32 Se=79,4 Te=128 ?
F=19 Cl=35,5 Br=80 J= 127
Li=^7 Na=23 K=39 Rb=85,4 Cs=133 Tl = 204
Ca=40 Sr=87,6 Ba=137 Pb=207
?=45 Ce=92
?Er=56 L<> = 94
?Yt=60 Di=95
?In=75,6 Th=118 (?)
Этот набросок вошел и в упомянутую выше статью. Размеры
последней не позволяют нам воспроизвести ее целиком, а потому мы
ограничимся приведением одного только ее резюме:
«1. Элементы, расположенные по величине их атомного веса,
представляют явственную периодичность свойств.
2. Сходственные по химическим отправлениям элементы
представляют или близкие атомные веса (подобно PtT Ir и Os) или последова-
* В цитированном уже издании «Ostwalds Klassiker» № 68 приводится еще
набросок системы элементов, сохранившийся после смерти Лот. Мейера и относящийся к
1868 г. Однако и в этом наброске заключается лишь немногим больше того, что
опубликовано им же в 1864 г. Мы вовсе не встречаем здесь выражения основней идеи
периодического закона, а лишь указания на правильности в нарастании атомных
весов сходственных элементов, которые расположены в 15 (или 16) группах, причем
странным образом А1 находится одновременно в одной группе с Fe, Rh, Ir и с Со, Pd.
Os; таллий со щелочными металлами со знаком (?), бор вовсе не включен в систему
и г. д. Сам Лотар Мейер не ссылается на этот набросок и не опубликовал его.
ПериоОинеския система химических элементов
61
тельни и однообразно увеличивающиеся (подобно К, Rb, Cs).
Однообразие такого увеличения в разных группах укрывалось от
предшествовавших наблюдателей, потому что они при своих сличениях не
воспользовались выводами Жерара, Реньо, Канниццаро и других, установивших
истинную величину атомного веса элементов.
3. Сопоставление элементов или их групп по величине атомного веса
соответствует так называемой атомности их и, до некоторой степени,
различию химического характера, что видно ясно в ряде: Li, Be, В, С,
N, О, F и повторяется в других рядах.
4. Распространеннейшие в природе простые тела имеют малый
атомный вес, а все элементы с малым атомным весом характеризуются
редкостью свойств. Они поэтому суть типические элементы. Водород, как
легчайший элемент, по справедливости, избирается как самый топический.
5. Величина атомного веса определяет характер элемента, как вели-
-чина частицы определяет свойства сложного тела, а потому при
изучении соединений должно обращать внимание не только на свойство и
количество элементов, не только на их взаимодействие, но и на вес их
атома. Оттого, на-пример, соединения S и Те, С1 и J и т. п. при сходстве
представляют и различия весьма ясные.
6. Должно ожидать открытия еще многих неизвестных простых тел,
например, сходных с А1 и Si, элементов с паями 65—75.
7. Величина атомного веса элемента иногда может быть исправлена,
зная его аналогии. Так, пай Те может быть не 128, а 123—126.
8. Некоторые аналогии элементов открываются по величине веса их
атома. Так, уран оказывается аналогом бора и алюминия, что и
оправдывается сличением их соединений. Д. И. добавляет, что цель его статьи
в том, чтобы «обратить внимание на отношение в величине атомного
веса несходных элементов».
Нельзя не признать, что вся сущность периодического закона и все
главнейшие следствия, из него вытекающие, уже заключаются в этих
немногих строках, да в таблице, их сопровождающей. Из всех выводов
Д. И. не подтвердилась только указанная в п. 8 аналогия U с В и Аі, да
остается под сомнением, должен ли атомный вес Те быть > или <J.
Следующий 1870 год принес статью Лот. Мейера [1], которую
особенно поучительно сопоставить с предыдущей статьей Менделеева.
Л. Мейер в начале своей статьи цитирует главный вывод Д. И.
в следующих выражениях: «Благодаря более правильному определению
атомных весов различных элементов сделалось возможным все
достаточно известные элементы расположить в одну систему. Недавно
Менделеев показал, что подобное расположение можно получить, просто
расположив все атомные веса в ряд без какого-либо произвольного
выбора, в порядке их возрастания, затем разбив этот ряд на отрезки и
присоединив последние друг к другу без изменения порядка.
Следующая таблица в существенных чертах тождественна с той, которую дает
Менделеев» *.
Из этой выдержки ясно видно, что до появления статьи Менделеева
самая сущность периодического закона— периодическое чередование
свойств элементов при расположении их в порядке возрастания атомных
весов — оставалась совершенно чуждой Л. Мейеру, что эти выводы были
впервые почерпнуты им из статьи Дмитрия Ивановича.
Существенно новой в статье Мейера является его кривая
объемов, представляющая, действительно, весьма важное и интересное
* Полагаем возможным воздержаться от приведения этой таблицы ввиду того,
что о ней говорит сам автор.
62
Периодическая система химических элементов
развитие периодического закона. В остальном Лот. Мейер не идет дальше
Менделеева, если не считать некоторых высказываемых им
теоретических соображений относительно возможного генезиса элементов.
Справедливость требует уже указания на то обстоятельство, ч:о
Мейер еще в 1864 г. принимал (правда со знаком ?) для Be атомный вес
9,3, а в таблице 1870 г. для индия — атомный вес 113,7, по-видимому,
руководясь при этом требованиями правильности, касающейся
закономерного нарастания атомного веса у элементов-гомологов. В статье
1870 года он указывает (вслед за Д. И.), что целый ряд элементов по
своим свойствам не укладывается в систему (особенно не подчиняется
закону объемов), если им приписать общепринятые в то время
атомные веса. Но, указывая на это обстоятельство, Мейер тем не менее
заключает свою статью следующими словами, которые в высшей степени
характерны для его отношения к периодическому закону: «Было бы
поспешно изменять доныне принятые атомные веса на основании столь
непрочного исходного пункта. Вообще в настоящее время на
подобного рода аргументы нельзя ни слишком сильно полагаться, ни
ожидать от них столь же определенного решения вопроса, как от
определения теплоемкости или плотности пара».
В противоположность этой скептической точке зрения, Д. И.
Менделеев видел в периодическом законе настоящий закон природы,
«новую точку опоры, которую следовало утвердить или отвергнуть». А
потому, вслед за открытием периодического закона, он решился
предпринять (1869—1871 гг.) исправления старых атомных весов,
неверно определенных, прочное установление
атомных весов элементов мало изученных и, наконец,
предсказания элементов неведомых, со всеми
свойствами их разнообразных соединений.
Список документов, относящихся к* истории открытия
периодического закона, завершается монументальной статьей Дмитрия Ивановича,
напечатанной в Либиховских Анналах за 1871 г. В статье этой
заключается: 1) текст периодической таблицы в ее окончательной и поныне
общепринятой форме, 2) закопченная формулировка и очень подробное
обоснование периодического закона, 3) все существеннейшие указания
относительно исправления атомных весов, неверно определенных, и
установление атомных весов элементов, мало изученных, и, наконец,
4) предсказат-ше трех неизвестных элементов экабо-ра, экалюминия
и экакремния со всеми их свойствами.
Едва ли после всего сказанного нужно еще что-либо добавлять для
того, чтобы бесповоротно и окончательно ответить на вопрос: кого
следует считать истинным творцом периодического закона.
Глава I
АТОМНЫЙ ВЕС И МЕТОДЫ ЕГО ОПРЕДЕЛЕНИЯ
В основе периодического закона лежит понятие об атоме и об
атомном весе. С определения этих понятий мы и начнем наше изложение.
Понятие об атоме было внесено в кимию около 1803 г. Джоном
Дальтоном1, учителем в Манчестере, а ближайшим к тому поводом
послужила возникшая потребность дать наглядное объяснение
поразительным по своей простоте законам, которым подчиняется состав
(весовой) химически однородных, или индивидуальных, веществ.
Законы эти носят название стехиометрических.
Периодическая система химических элементов {}?>
1. Закон постоянства состава, или закон Пру:
Всякое химически однородное тело заключает
в своем составе одни и те же элементы в одних и тех
же весовых отношениях.
Так, вода — на одну весовую часть водорода содержит 8 весовых
частей кислорода (точнее на 1,008 весовую часть Н 8 весовых частей О);
углекислый газ — на 3 весовые части углерода 8 весовых частей
кислорода и т. д.
¦2. Закон эквивалентов (Рихтер) и кратных
отношений (Дальтон).
Если сопоставить между собой весовые отношения, в которых
элементы соединяются между собою, и отнести их к одной определенной
единице, например к весовой единице водорода, то мы получим для
каждого элемента некоторое число, носящее название его
эквивалента. В отношении этих ч и с о л, или их крат н ы х, э л е-
менты сочетаются между собой при образовании
химических соединений.
Для примера приводим эквиваленты нескольких элементов (в
круглых цифрах), отнесенные к водороду как к единице:
Н О N Na К Са С1 А1
1 8 4,8 23 39 20 35,5 9
Для объяснения этих фактов, в особенности же закона простых и
кратных отношений, Дальтон и предложил свою атомистическую (или
атомно-молекулярную) гипотезу, основные положения которой
общеизвестны.
Гипотеза эта принимает, что все химические элементы состоят из
атомов, не изменяемых в условиях наших обычных химических
превращений. Атомы одного и того же элемента совершенно тождественны
между собой во всех отношениях. Наоборот, атомы различных
элементов отличаются друг от друга 'прежде всего по присущей им массе или
по весу. Из атомов различных элементов через сближение образуются
системы более сложные, атомы высшего порядка, как их называл
Дальтон, молекулы, или частицы, как называют их в настоящее
время.
В каждой такой молекуле атомы, ее образующие, удерживаются
друг около друга некоторыми силами притяжения. С другой стороны,
как атомы, так и молекулы находятся в состоянии постоянного
движения (смотря по обстоятельствам, поступательного, колебательного или
вращательного).
Все молекулы химически однородного тела совершенно
тождественны между собой, то есть каждая из них состоит из одних и тех же
элементарных атомов в постоянном и неизменном числе.
Строго говоря, это последнее положение молекулярно-атомистичо
ской теории не может быть признано абсолютно верным после того, как
стали известны такие ассоциированные жидкости, как вода (также
спирты, некоторые кислоты и пр.), в которых приходится допустить
одновременно существование, наряду с молекулами Н2О, более
сложных (НгОЬ, которые, однако, диссоциируют при переходе в пар. Для
уксусной кислоты СНзСООН удвоенные молекулы существуют и в
парообразном состоянии и переходят в простые только при достаточно
высокой температуре (250°). В таких случаях между простыми и
ассоциированными молекулами при данных физических условиях
(особой температуре), и притом весьма быстро, устанавливается строго оп-
-64
Периодическая система химических элементов
ределенное состояние равновесия. Мы имеем, таким образом, жидкость
или газ, во всех отношениях обладающие характером химического
индивидуума. Можно, однако, представить себе такие условия, в
которых скорость перехода двойных молекул в простые и обратно сделается
крайне малой. В таком случае было бы возможным получить в
отдельности два тела, два разных соединения с молекулярным весом М и 2М.
Для ассоциированных жидкостей этого пока еще не удалось достигнуть.
Но есть другого рода случаи, в которых химически индивидуальное
тело также представляет собой равновесную систему двоякого рода
молекул. Таковы таутомерные соединения, например ацетоуксусный
эфир в жидком состоянии, представляющий смесь двух модификаций
СН3—СО—СН2—СОО—С2Н5 и СН3—С=СН—СОО—С2Н5
!
Кетонная форма ОН Энольная форма
легко и быстро переходящих друг в друга.
Отделение этих модификаций друг от друга недавно удалось
Л. К'нор ру 12], который, работая при очень 'Низких температурах,
тем в достаточной мере уменьшил скорость взаимного превращения
обоих таутомеров.
Согласно только что сказанному, 'понятие атом мы определим как
наименьшее количество элемента, могущее
входить з состав химической молекулы. Под молекулой
же мы будем подразумевать наименьшее количество
простого или сложного тела, способное к
реагированию и вообще к самостоятельному существованию.
Если мы представим себе, что капля воды подвергается
механическому дроблению на части все меньшей и меньшей величины, то в
пределе такого мысленного процесса мы получим молекулу воды. Несмотря
на все наши старания, эту молекулу не представилось бы возможным
разделить на частицы воды, еще более мелкие. Но если бы нашли агент,
способный разделить молекулу воды, то имели бы дело с
химическим процессом, в результате которого получилась бы уже не вода, а
продукт ее разложения: кислород и водород. Допускают, что при этом
молекула дробится на атомы элементов, причем эти атомы могут
соединяться друг с другом, образуя молекулы простых тел, состоящие из
двух или же из большого числа однородных атомов.
Мы представляем себе, например, молекулу воды Н20 *, состоящую
из двух атомов водорода и одного атома кислорода. На эти атомы
частица воды и может быть разложена (напри-мер, действием
электрического тока). Мы увидим ниже, что существуют факты, заставляющие
нас признать, что раз выделившись, атомы Ни О сейчас же
соединяются попарно, так, что процесс разложения воды в окончательном виде
можно представить следующим уравнением:
2Н20=2Н2+02.
С точки зрения атомистической теории стехиометрические законы
получают весьма простое объяснение. Если каждому атому присущ
* При достаточно высокой температуре жидкая вода состоит из простых молекул
Н20. Таков же состач и водяного пара (см. ниже).
Периодическая система химических элементов
65
строго определенный вес и если, с другой стороны, атомы различных
элементов могут сочетаться только в определенном относительном
числе (например, на 1 атом одного элемента А —два атома другого В),
то ясно, что должен прежде всего иметь место закон постоянства;
что, с другой стороны, элементы могут входить в соединение друг с
другом или в отношении их атомных весов (если атомы соединяются
один на один), или в отношении чисел их кратных (если, например, на
п атомов одного элемента вступает в соединение m атомов другого),
а отсюда само собой вытекают законы эквивалентов и кратных
отношений.
Дж. Дальтон которому, как уже было упомянуто, принадлежит
заслуга ввести в область химии атомно-молекулярную гипотезу, сделал
первую попытку определить и атомные веса элементов. Невозможность
определить абсолютные веса атомов заставила его остановиться на
сравнении веса атома данного элемента с весом атома другого, конечно,
каждый раз одного и того же элемента, причем этот последний вес
условно принимается за единицу. За такую единицу, вслед за
Дальтоном, еще недавно принимали вес атома водорода. Ныне, по некоторым
соображениям, большинство химиков предпочитает пользоваться
другой величиной, весьма близкой к предыдущей, а именно Vie атомного
веса кислорода. Если мы, например, говорим, что атомный вес серы
=^32, а атомный вес натрия =23, то это значит, что атом серы весит в
32, а атом натрия в 23 раза более, нежели атом водорода (или чем
:/іб часть веса атома кислорода) 2.
Дальтон выводил атомные веса непосредственно из тех весовых
отношений, в которых соответствующие элементы образуют химические
соединения. Атомный вес кислорода для него был, например, близок к
8, так как в воде на одну весовую часть водорода содержится 8
весовых частей кислорода. При этом он допускал, что молекула воды
имеет простейший возможный состав: 1 атом Н и 1 атом О. Иначе
говоря, понятие «эквивалент» он отождествлял с понятием «атомный
нес». Но ясно, что в таком отождествлении заключается элемент
произвола.
Предположим, что какой-нибудь элемент А образует соединение с
водородом, в котором на п атомов Н приходится m атомов А. Тогда
химическая формула этого соединения будет Нп А т. Назовем атомный
вес элемента А через а (при Н = і), а его эквивалент, то есть его
весовое количество, соединяющееся с весовой единицей водорода, через Е.
Тогда ясно, что — ™= Е, и потому а=Е — • Другими словами,
эквивалент равен атомному весу только в том случае, когда в молекуле
данного соединения на п атомов водорода приходится столько же
атомов другого элемента, то есть когда дробь ~=1- Вообще же для того,
чтобы от эквивалента, который может быть найден непосредственно из
(чіыта, перейти к атомному весу, необходимо з=нать величину
множителя—-, а для этого, в свою очередь, надо знать состав молекулы
того соединения, из количественного анализа (или синтеза) которого
найден эквивалент (иными словами, необходимо знать число атомов,
данную молекулу составляющих).
Определение состава молекулы химических соединений сделалось
возможным после того, как Гей-Люссак в 1808 году открыл закон,
которому подчиняются объемы реагирующих веществ в газообразном
состоянии
вг) Л. А. Чугляв, т. III
66
Периодическая система химических элементов
3. Законы объемов Гей-Люссака:
Вещества, вступающие в соединение, занимают
в газообразном состоянии при одинаковых
условиях температуры и давления равные или
соизмеримые между собою объемы. Объем
образующегося при этом соединения в газообразном состоянии
и при тех же условиях температуры и давления
находится в простом отношении к объемам
составляющих.
Итак, реагирующие между собой вещества и продукты реакции
занимают в газообразном состоянии соизмеримые объемы. Но ведь
в реакцию вступают молекулы, и молекулы же возникают при
реакции. Поэтому весовые количества участвующих в реакции веществ
должны относиться, как веса соответствующих молекул или как
кратные их, а отсюда, в свою очередь, следует, что и объемы,
занимаемые молекулами, должны находиться в
простых кратных отношениях между собой. Каковы эти
отношения?
Ответ на этот вопрос был дан итальянским ученым Амедео Аво-
гадро в 1811 г. Авогадро сделал простейшее из возможных
предположений, допустив, что в равных объемах газов и паров при
одинаковой температуре и давлении заключается
одинаковое число молекул, независимо от природы данного
вещества. Или иначе: в газообразном состоянии при одинаковой
температуре и давлении молекула любого вещества занимает один и тот же
строго определенный объем. Это положение, получившее название
гипотезы Авогадро, первоначально не обратило на себя внимания
ученых и только начиная с сороковых годов, после знаменитых в истории
химии работ Жерара иКанниццаро получило огромное влияние
на дальнейшее развитие химических теорий.
В настоящее время гипотеза Авогадро настолько прочно обоснована
(между прочим, при помощи кинетической теории газов), что ее часто
называю? законом.
Посмотрим теперь, каким образом гипотеза Авогадро позволяет
определить число атомов, составляющих ту или иную молекулу и дает
необходимый материал для вычисления того фактора, на который надо
помножить эквивалент данного элемента, чтобы от него перейти к
соответствующему атомному весу. Для этого целесообразно остановиться
на каком-нибудь конкретном примере. В качестве такого примера мы
возьмем воду.
Нам известно: 1) что вода содержит (в круглых цифрах): на одну
весовую часть водорода 8 весовых частей кислорода; 2) чго два объема
водорода, соединившись с одним объемом кислорода, дают два объема
водяного пара.
Из первого следует, что 8 есть эквивалент кислорода. Из второго
заключаем, согласно закону Авогадро, что:
2 молекулы водорода + 1 молекула кислорода = 2 молекулы водяного пара.
Сколько атомов кислорода и водорода содержится в одной молекуле
этих газов, мы не знаем, но ясно, что ни в одной молекуле меньше, чем
по одному атому содержаться не может (так как атомы неделимы)
Если остановиться на том простейшем допущении, что молекулы Н и О
содержат по одному только атому, то ясно, что формула воды была
Периодическая система химических элементов
67
бы Н20, а уравнение, выражающее образование воды из Н и О, имело
бы такой вид:
2Н + О = НоО.
Переводя это уравнение на объемные отношения, и, опять-таки
вспоминая закон Авогадро, мы, однако, получим:
два объема Н и один объем О дают один объем Н20.
тогда как фактически
два объема Н и один объем О дают два объема водяного пара.
Возникающее противоречие легко можно устранить, если, вслед за
Авогадро и Жераром, мы предположим, что молекулы кислорода и
водорода содержат не по одному атому, а по два, соответственно
формулам 02 и Н2. Тогда формула воды останется прежняя: Н20, а
образование ее из кислорода и водорода выразится уравнением:
2Н2 +03 =2Н20,
2 объема 1 объем 2 объема
находящимся в согласии с фактически наблюдаемыми объемными отно-
шениями.
Предположение о том, что молекулы водорода и кислорода
содержат по два атома: Н2 и 02, вовсе не является произвольным, и прежде
чем идти дальше, мы остановимся на его логическом обосновании.
Мы видели, что, согласно закону Авогадро,
2 молекулы водорода + 1 молекула кислорода дают 2 молекулы воды.
В каждой молекуле воды, очевидно, содержится не менее одного
атома О. Но две молекулы воды образовались из одной молекулы
кислорода. Следовательно, в этой последней должно заключаться не менее
двух атомов О. Простейшая формула «Свободного кислорода» должна
быть Ог.
Легко также показать, что число атомов в молекуле кислорода
должно быть кратным двум соответственно формуле 02. В самом деле,
если допустить, что в молекуле кислорода содержится m атомов,
причем т — число нечетное, то мы получим такое соотношение:
1 молекула От + 2 молекулы водорода дают 2 молекулы воды.
Следовательно, в одной молекуле воды содержится V20m, иными
словами, дробное число атомов кислорода (например, 27г атома О при
т = 5), что, очевидно, невозможно.
То же самое можно вывести для водорода, если рассмотреть процесс
образования хлористого водорода. Опыт показывает, что:
один объем хлора и один объем водорода образуют два объема
хлористого водорода.
А потому:
одна молекула хлора + одна молекула водорода дают две молекулы
хлористого водорода.
Повторяя прежде рассуждение, мы заключим, что так как каждая
молекула водорода (и каждая молекула хлора) дает начало двум
молекулам хлористого водорода, то в молекуле водорода (а равным
68
Периодическая система химических элементов
образом и в молекуле хлора) не может содержаться менее двух
атомов, что соответствует формуле Нг (и СЬ) *.
Нетрудно также убедиться в том, что число атомов в молекуле ъидо-
рода (и хлора) должно быть четным, так как в противном случае
пришлось бы допустить, что в одной молекуле содержится дробное
число атомов водорода (и хлора). Поэтому молекула водорода может
только отвечать формуле H2n, где п есть какое-либо целое число (а
молекула хлора—формуле СЬп).
Итак, молекулы водорода и кислорода содержат четное число
атомов этих элементов. Спрашивается, возможно ли допустить, что они
состоят не из большего числа атомов (Н4, 06, Ов и т. д.).
Ответ на этот вопрос вытекает из следующих соображений.
Сопоставим объемный состав для целого ряда водородистых и
кислородных соединений, взятых в газообразном состоянии, и притом
таким образом, чтобы ясно было видно, из скольких объемов Н и О
образуется один объем соответствующего соединения:
Нашу таблицу можно было бы увеличить еще огромным числом
новых примеров, но при этом мы не нашли бы ни одного случая, в котором
* Что молекулы таких газов, как кислород, водород, азот, хлор и т. п. состоят не
менее чем из двух атомов, может быть доказано еще иным путем. Из кинетической
теории следует, что для всех газов, молекулы которых состоят из одного атома,
отношение ~^-~ между теплоемкостью при постоянном давлении и теплоемкостью при
постоянном объеме должно быть близко к 1,666. Такой вывод блестящим образом
оправдался на парах металлов, например ртути, для которых из чисто химических
соображений необходимо было прийти к заключению, что молекула их состоит из
одного только атома.
Кундти Варбург, а за ними и другие исследователи подтвердили, что в этом
ср
случае отношение -тг~ действительно = 1,666. Между тем, для частиц, заведомо
состоящих из нескольких атомов, отношение это меньше (как того требует и
кинетическая теория). В частности, это имеет место и для таких газов, как Н2, 02 и т. д.
Газ Кислород Водород Азот
g- МО Ml 1,41
'V
Замечательно, что для химических соединений, молекулы которых на основании
ср
химических соображении считаются состоящими из двух атомов, отношение уг-
было найдено как раз близким к величине 1,4 (например, для окиси углерода СО
{? = 1,40).
Су
Наоборот, для газов более сложного состава оно значительно ниже:
Формула С02 С2Н4 С2Нв СНС13
СР
-гг- 1,30 1,25 1,18 1,15
Ср
Величина отношения -q-, найденная для водорода и кислорода, таким образом
указывает на то, что молекула этих газов состоит именно из двух атомов.
Периодическая система химических элементов
69
Соединение
Формула
Число объемов водорода,
заключающихся в одном
объеме данного соединения
Хлористый водород ....
Бромистый водород ....
Йодистый водород .....
Синильная кислота ....
Азотистоводородная кислота
Вода
Аммиак
Метан
Этан
хіч
Vi
1
IV*
2
з
Соединение
Формула
Число объемов кислорода,
заключающихся в одном
объеме данного соединения
Вода
Этиловый спирт ..
Этиловый эфир .
Уксусная кислота
Двуокись азота .
Закись азота . .
Серный ангидрид
Н20
С2НвО
(С2Н5)20
С2Н4О2
N02
N20
Vi
Vi
1
1
Vs
один объем химического соединения образовался бы меньше, нежели
7г объема газообразного водорода или кислорода.
Другими словами, необходимо признать, что неизвестно ни
одного соединения, молекула которого содержала
бы в себе меньше, нежели У2 молекулы О или Н.
А отсюда, в свою очередь, следует, что не существует ни
одного факта, который заставлял бы нас думать, что
в молекулах.кислорода и водорода содержится более,
чем по два атома этих элементов.
Итак, в молекулах кислорода и водорода содержится не менее двух
и не более двух атомов, то есть именно по два атома, и потому
молекулы эти должны выразить формулами 02 и Н2 в согласии со
сделанным допущением.
После всего сказанного мы можем уже считать формулу воды Н20
логически обоснованною и, опираясь на нее, продолжать цепь наших
рассуждений.
Непосредственным следствием установления молекулярной формулы
воды является возможность определить атомный вес кислорода при
Н = 1. Мы видели, что для этого необходимо эквивалент данного
элемента, для кислорода — 8, помножить на некоторый фактор, который в
данном случае равен 2 (см. стр. 67). Атомный вес кислорода поэтому ='16.
Несравненно важнее, однако, что опираясь на состав молекулы воды
и на закон Авогадро, можно установить общий метод, пригодный
для определения атомных весов самых разнообразных элементов.
Согласно закону Авогадро, в равных объемах различных газов
заключается одинаковое число частиц. Предположим, что мы имеем ряд
сосудов равного объема, наполненных различными газами (или
парами) при одинаковой температуре и давлении. Тогда в каждом из них
70
Периодическая система химических элементов
будет заключаться одно и то же число, скажем, молекул. Пусть вес
газа в первом сосуде будет = Рь во втором — Р2, в третьем — Я3 и т. д.т
вес же молекулы первого газа Ми второго — М2 и т. д.
Очевидно, что для каждого газа, разделяя общий вес его на вес
одной молекулы, получим число молекул его в данном объеме, число,
которое должно быть одинаковым для всех газов n~Ny а следовательно,
Мі Мг М3 ' ' ' ' • \1>
Ясно, что объем сосудов может быть каким угодно. Сущность
написанного соотношения от этого не изменится. Изберем же этот объем
таким образом, чтобы в нем вмещалась при данной температуре и
давлении (например, при 0° и 760 мм рт. ст.) ровно одна весовая единица
водорода. Тогда в каждом сосуде будет заключаться столько весовых
единиц данного газа, сколько единиц в числе, выражающем плотность
этого газа, отнесенную к водороду (то есть отношение между весом
газа к весу водорода в том же объеме). Обозначая плотность по
водороду нашего первого газа через Du второго через D2 и т. д., мы можем
переписать уравнение (1) следующим образом:
Ліі іМ2"М3",,,л'" ^'
где К — некоторая 'постоянная величина.
Следовательно, можно сказать, что отношение плотности
газа или пара по водороду к его молекулярному весу
есть величина постоянная для всех химически
однородных веществ и не зависит от природы этих
последних.
Каково же значение этой величины? Очевидно, вполне .достаточна
определить это значение для одного и притом для любого вещества, и
этим самым оно будет установлено раз навсегда, разумеется, если
только верен закон Авогадро.
Для определения величины N надо знать две величины:
молекулярный вес данного вещества и плотность его пара, отнесенную к водороду.
Первая величина только что была определена нами для воды,,
молекулярный вес которой, очевидно, —2X1 + 16=18. Плотность
водяного пара по водороду легко определяется из опыта. Для воды она ока-
залась = 9.
Из этих данных непосредственно следует, что для воды, а стало
быть, и для любого другого химически однородного тела, способного
существовать в газообразном состоянии,
К = М = ~2Ит
М = 2D .. . (3)
Другими словами, молекулярный вес равен удвоенной
плотности пара по водороду*.
D 1
Нетрудно понять, что постоянная величина (V2) в выражении л? ~ Т"
только в том случае сохраняет значение (соотв. уравн. 2) числа молекул в известном
объеме газа, если единицей для выражения D и М служит вес атома водорода.
Плотность газа есть тогда вес того объема данного газа, который занимает 1 атом Н„
а М есть молекулярный вес того же газа, выраженный равным образом в водородных
единицах. Смысл уравнения (3) тогда можно выразить так: если мы мысленно выде-
*
Периодическая система химических элементов 71
С помощью этого соотношения, введенного в науку Ш. Жераром,
с большой легкостью может быть установлен молекулярный вес любого
летучего химического соединения.
Из опыта, например, мы находим, что некоторый объем
газообразного аммиака весит в 8,5 раз больше равного ему объема водорода.
Отсюда по формуле (3) находим для молекулярного веса амми-ака
2X8,5=17.
Другими словами, мы нашли, что молекула аммиака в 17 раз
тяжелее атома водорода.
Предположим теперь, что мы задались целью определить атомный
вес азота — элемента, который в аммиаке NH3 связан с водородом.
Опыт показывает, что в NH3 находится 82,4% азота. Остальное (до 100)
приходится на водород. Но молекула аммиака в 17 раз тяжелее атома
водорода. Спросим себя, сколько весовых частей азота содержится в
17 весовых частях, то есть в молекуле аммиака. По простой пропорции
100 _ 17
^Г~4 ^ V" получаем: 14. На долю водорода остается 17—14 = 3, что
отвечает трем атомам Н. Если в молекуле аммиака содержится только
один атом азота и формула его будет NH3, то, очевидно, 14 и
будет = атомному весу азота.
Но ясно, что эти 14 единиц могут отвечать 2, 3 и т. д. атомам, тогда
аммиак выражался бы формулами N2H2, N3H3 и т. д., а атомный вес
14 14
азОта выражался бы числами -9-1-3" и т- Д-
Чтобы избегнуть возникающей таким образом неопределенности, что
мы сейчас проделали с аммиаком, повторим для целого ряда других
•соединений, содержащих азот. Получим, например, такую таблицу:
Соединение
Содержание азота,
Молекулярный вес
Вес азота
в молекуле
Аммиак
Окись азота
Закись азота
Синильная кислота ....
Азотистеводородная кислота
82,4
46,8
63,7
51,9
97,7
17
30
44
27
43
14
14
28
14
42
Мы видим, что существуют соединения, в молекуле которых
содержится больше 14 весовых частей азота, но нет ни одного такого, в
котором заключалось бы меньше; 14, значит,.есть наименьшее весовое
количество азота, содержащееся в молекуле различных соединений этого
элемента. Другими словами, 14 и есть атомный вес азота.
лим' объем, который приходится на долю 1 атома (газообразного) водорода, то в этом
объеме поместится 7г молекулы любого газообразного соединения (или простого тела).
Понятно также, что если вместо атома водорода мы возьмем за единицу 1 г водород*.
то уравнение (3) будет означать, что объем, в котором содержится 1 грамм (1 г-атом)
газообразного водорода (точнее 1,008 о) вмещает 7г г-моль любого газообразного
вещества. По новейшим данным, первая единица (вес атома Н) в 7Х1023 меньше второй
(1 г). Иными словами, в каждой грамм-молекуле содержится 7Х1023 молекул 3.
В настоящее время в согласии с ныне общепринятой единицей атомных весов
(Vie О) за единицу плотности газов принимают не плотность водорода, а '/іе
плотности кислорода, иначе говоря, условно считают, что плотность кислорода= 1б2.
Практически разница между новой и старой единицей, конечно, не велика, и принципиально
она Rpna нисколько не меняет.
72
Периодическая система химических элементов
Отсюда вытекает следующее общее правило для определения
атомных весов. Берут ряд летучих соединений данного элемента А и для
каждого из них определяют:
1) плотность пара по водороду, а из него молекулярный вес М и
2) процентное содержание Р элемента А.
Затем по пропор-ции-р- = ^—рассчитывают, сколько весовых единиц
X элемента А содержится в молекулярном весе данного соединения.
Наконец, из ряда значений X, отвечающих взятым для исследования
соединениям, выбирают наименьшее, которое и считают
атомным весом элемента А.
Закон Авогадро не вполне точен, особенно для газов и паров,
далеких от идеального состояния. Поэтому и значения молекулярных весов,
полученные при помощи этого закона из плотностей пара летучих
соединений, будут лишь приблизительно верны. А отсюда понятно, в свою
очередь, что лишь приблизительно верны будут и атомные веса элементов,
если мы будем их вычислять, точно следуя только что указанному
приему. Каким образом могут быть получены значения атомных весов,
возможно близкие к истине?
Химический анализ позволяет во многих случаях весьма точно
определять процентный состав химических соединений, из которого
непосредственно выводятся эквиваленты элементов, образующих эти
соединения, а из этих последних, как мы видели выше, путем помноже-
ния на некоторый фактор может быть определено и точное значение
атомного веса. Притом, согласно закону простых и кратных отношений и
по смыслу атомистической теории, этот фактор может быть только
небольшим целым числом.
Предположим, например, что эквивалент азота (по водороду) и^
точных анализов оказался равным 4,67. Атомный вес азота поэтому мог
бы быть = 4,67, или 4,67X2 = 9,34, или 4,67X3=14,01, или 4,67x4=18,68
и т. д. Необходимо сделать выбор между этими величинами, или, что
одно и тоже, между факторами 1, 2, 3, 4 и т. д.
Вот здесь-то решающую роль имеет та приблизительная величина
атомного веса, которая может быть найдена, опираясь на закон
Авогадро. Так как для азота она всегда окажется близкой к 14, то точное
значение атомного веса должно быть 14,01.
Если для целого ряда элементов, для определения фактора, на
который нужно помножить эквивалент, чтобы от него перейти к атомному
весу, возможно пользоваться определением плотности газов летучих
соединений, то для других элементов этот путь оказывается
неприменимым, потому л,и, что соединения их трудно летучи; потому ли, что они
неспособны переходить в парообразное состояние без разложения. В
таких случаях пользуются другими вспомогательными приемами, на
которые мы укажем здесь в самых общих чертах.
1. Вместо того, чтобы определить молекулярный вес тел в
парообразном состоянии, можно воспользоваться для той же цели их
разбавленными растворами. Вант-Гофф показал, что на
растворенное состояние вещества распространяются простые законы газового
состояния: законы Бойля, Гей-Люссака и Авогадро. Для разведенных
растворов, как для газов, близких к идеальному состоянию,
справедлива формула pv — r\RT, в которой п есть число грамм-молекул
растворенного тела, распределенное вол, р — осмотическое давление,
аналогичное газовому, Т — абсолютная температура и R — постоянная
величина, численно равная газовой константе. Опираясь на эги законы
и измерив осмотическое давление (или же, что удобнее, величины,
Периодическая система химических элементов 73
связанные с ним простыми соотношениями — понижение
температуры плавления и повышение температуры
кипения*) раствора определенной концентрации вещества А в
растворителе В, легко можно вычислить молекулярный вес А. Для этого
необходимо только, чтобы вещество А растворялось в В, не подвергаясь при
этом химическим изменениям.
Покажем, каким образом применяется для подобных определении
так называемый криоскопический способ, основанный на
понижении температуры замерзания растворов.
Если чистый растворитель (например вода) замерзает при
температуре Го, а раствор, содержащий р г вещества в 100 г растворителя,— при
t, то согласно закону, установленному Раулем и Вант-Гоффом,
понижение t0—t будет пропорционально числу
грамм-молекул N растворенного тела, приходящихся на 100 г (вообще
на какое-либо определенное число граммов) растворителя. Это можно
выразить следующим уравнением:
tQ—t = KN . ... (I)
в котором К есть некоторое постоянное число (фактор
пропорциональности). Если неизвестный молекулярный вес растворенного тела назовем
Р
через М, то очевидно, что /V = -^, а потому наше уравнение можно
написать:
t0-t = K-^... (Н)
Р можно назвать весовой концентрацией, N — молекулярной
концентрацией нашего раствора. Полагая Р = М, получим ^о—t = K, а потому ясно,
что величина К", в сущности, выражает не что иное, как понижение
температуры замерзания, или, как .иначе говорят, депрессию такого
раствора, который в 100 г растворителя содержит ровно 1 г*моль
растворенного тела. Величину эту называютмолекулярным
понижением или молекулярной депрессией.
Она имеет строго определенную величину для каждого данного
растворителя (например для воды —18,7, для бензола —50, для уксусном
кислоты — 39) и не зависит от природы растворенного тела. Нетрудно
видеть, что по уравнению (II) легко определить молекулярный вес
неизвестного вещества, если только известна величина К для данного
растворителя. В самом деле, величина Р нам будет известна, если только
мы приготовим раствор определенной концентрации, а депрессию to — /
узнаем из опыта.
Молекулярное понижение К может быть определено двумя
различными путями: или 1) теоретически по формуле, выведенной Вант-Гоффом.
„ 0.02Г2
где А,— скрытая теплота плавления данного растворителя, а Г —его
абсолютная температура плавления, или же 2) опытным путем, к чему
прибегают, когда неизвестно значение X. Для этой цели берут вещество.
молекулярный вес которого заранее известен, приготовляют раствор
его в данном растворителе и определяют температуру замерзания, как
* Раствора по сравнению с чистым растворителем.
л
Периодическая система химических элементов
полученного раствора, так и чистого растворителя. Тогда в уравнении
(II) нам будут известны величины /о, ty Р и М> а потому легко вычислить
значение К Из ряда подобных опытов берут среднее, которое и
считают за молекулярное понижение данного растворителя.
Подробности, относящиеся до подобного рода определений, равно
как и соответствующие теоретические разъяснения, читатель найдет в
руководствах по физической химии.
2. Дюлонг и Пти в 1819 г. открыли закон, согласно kotopomv
атомная теплоемкость, то есть произведение из теплоемкости простого
тела в твердом состоянии на соответствующий атомный вес есть
величина постоянная, близкая к 6,4. Если для какого-нибудь элемента,
эквивалент которого = ?, возникает сомнение, верно ли выбран (между
возможными величинами: Е, 2Е} ЗЕ и т. д.) его атомный вес, то стоит только
определить теплоемкость С данного элемента, выделенного в свободном
состоянии (точнее, простого тела), и разделить на нее величину 6,4,
чтобы получить — —приблизительную величину искомого атомного
веса.
Закон Дюлонга и Пти не строго точный. Для многих элементов
отклонения от него составляют 10%, иногда значительно больше.
Особенно сильны уклонения у некоторых элементов с малым атомным весом.
Be В С Si
Атомная теплоемкость . . .3,7 2,8 1,9 4,6
С повышением температуры теплоемкость, вообще говоря, растет и
для упомянутых четырех элементов постепенно приближается к
нормальной величине. В -самое последнее время Hep нет [3] показал, что по
мере приближения к абсолютному нулю теплоемкость весьма быстро
надает и доходит до ничтожно малой величины, как это особенно резко
видно на примере алмаза:
Абсолютная температура ... 40° 86,5° 92° 205° 220°
Атомная теплоемкость .... 0,00 0,03 0,03 0,62 0,72
Заметим еще, что В. Я. Курбатов предложил относить атомные
теплоемкости к температурам, представляющим одну и ту же
(например 7з) долю абсолютной температуры плавления соответствующих
простых тел при атмосферном давлении. При этом получается несколько
лучшее согласие между величинами атомных теплоемкостей отдельных
элементов.
Нейман, Реньо и Копп нашли, что закон постоянства атомной
теплоемкости, вообще говоря, сохраняет свою силу и для химических
соединений в твердом состоянии. Разделяя молекулярную теплоемкость
{произведение теплоемкости данного соединения на его молекулярный
вес) на число атомов в молекуле, получим среднюю теплоемкость
атома, которая во многих случаях близка к 6,4; например, для хлористых
соединений МС1 она в среднем=6,4, для МС12 — 6,2, для йодистых
соединений MJ — 6,7 и т. д. Это положение известно под именем закона
Неймана. Для некоторых элементов наблюдаются, однако, более или менее
резкие уклонения. Например, атомная теплоемкость кислорода в
окислах, хотя и остается довольно постоянной, но близка не к 6,4, а к 4.
Из только что сказанного понятно, что в некоторых случаях законом
Неймана, как и законом Дюлонга и Пти, можно пользоваться для
проверки величины атомных весов. В хлористом кальции, например, 20 г Са
находится в соединении с 35,5 г хлора. Если формула этого соединения
СаСі, то молекулярный вес его равен 40+35,5=75,5. Для формулы же
Периодическая система химических элементов
75
СаС12 — молекулярный вес — 40 + 35,5 X 2 =111. Пусть Q есть
определенная из опыта теплоемкость хлористого кальция. Тогда для
молекулярной теплоемкости получим, если верно первое предположение — 75,5xQ
и если верно второе—111 X Q. Соответственно величины для средней
атомной теплоемкости будут:
75,5 х Q lit >: Q
2 и—"
Теоретически атомная теплоемкость должна быть близка к 6,4.
Фактически это оправдывается только для второго предположения, но не
для первого, а потому атомный вес Са близок к 40'(а не 20),
соответственно формуле хлористого кальция СаС12.
3. Изоморфизм. В 1820 г. М и ч е р л и х установил замечательное
соотношение между составом молекулы вещества и его
кристаллографическим характером. Соотношение это получило название закона
изоморфизма, а сходные в кристаллографическом отношении вещества
название изоморфных.
Критериями изоморфизма обыкновенно считают: 1) сходство
кристаллической формы, иногда практически доходящее до полного
тождества, 2) способность изоморфных веществ выделяться из раствора в
смешанных кристаллах, содержащих компоненты в различных
пропорциях, и 3) способность одного изоморфа служить «затравкой» для
другого. Последнее означает, что если кристаллик вещества А, изоморфного
с В, внесен в пересыщенный раствор этого последнего, то вместо того,
чтобы раствориться, он делается центром, около которого нарастает
кристалл вещества В.
Из этих трех критериев наименьшее значение имеет первый. Очень
часто вещества, кристаллизующиеся совместно и играющие друг для
друга роль затравок, а потому изоморфные, кристаллизуются каждое
в отдельности в различных формах и даже в различных
кристаллографических системах. Такие вещества носят название изодиморфных.
Закон, установленный Мичерлихом, может быть формулирован
таким образом, что вещества изоморфные обыкновенно (но не
всегда) обладают сходственным составом молекул. При
этом часто (но, опять-таки, не всегда) в молекулах изоморфных веществ
AnBmCq..., AnBmDq... содержатся одинаковые числа атомов одних и тех
же элементов (например, по п атомов элемента А и по m атомов
элемента В) и одинаковые же числа атомов, принадлежащих элементам,
между собой аналогичным (С и D).
Хорошим примером изоморфных веществ могут служить квасцы,
представляющие «двойные соли» сульфатов трехвалентного металла
(А1, Fe, Сг и другие) и одновалентного (К, Rb, С г, также NH4)
в комбинации с 24 молекулами кристаллизационной воды:
A12(S04)3K2S04-24H20, Cr2(S04h(NH4hS04-24H20, Fe2(S04)3K2S04 ¦
- 24Н20, Fe2 (S04)3 (NH4)2S04 - 24H20.
Здесь мы имеем дело с веществами, молекулы которых отвечают
совершенно аналогичному составу. В двух примерах калийных квасцов все
различие сводится к замещению двух атомов А1 двумя же атомами во
многих отношениях сходного с А1 железа, а в аммонийных квасцах мы
имеем в одном случае Сг2, а в другом — два атома, опять-таки,
аналогичного с Сг железа. Остальные части молекул тождественны.
Подобное же отношение представляют соли КН2Р04 и KH2As04,
изучение которых в свое время привело Мичерлиха к открытию закона
изоморфизма.
76
Периодическая система химических элементов
Каким образом можно воспользоваться законом Мичерлиха для
определения атомных весов?
Предположим, что требуется определить атомный вес железа, для
которого мы знаем (из анализа ряда его соединений) только эквива-
лент='28. Атомный вес Fe может поэтому быть= или 28, или 28X2 = 56,.
или 28X3 = 84 и т. д. Рассмотрим две соли, относящиеся к ряду купо-
росов: FeS04-7H20 и ZnS04-7H20. Опыт показывает, что они стоят
друг к другу в отношении изоморфизма. Согласно закону Мичерлиха,.
мы вправе поэтому ожидать, что они будут обладать сходственным
составом молекулы. Допустим, что нам известен состав молекулы серно-
цинковой соли ZnS04. Если обе соли изоморфны, то это позволяет
думать, что и серножелезистая соль будет обладать аналогично
составленной молекулой: FeSC>4. Анализ этой соли дает такие результаты
(в процентах):
S О Fe
21,05 42,11 36,84
Если в молекуле ее содержится один атом S и четыре атома О и
если нам известны атомные веса S=32 и 0=16, то из приведенных
цифр анализа легко вычислить, какое весовое количество железа
содержится в молекуле FeSO.*, другими словами, -приходится на 32 весовые
части (один атом) серы и на 16x4 = 64 весовые части (четыре атома)
кислорода. Мы получим 56 весовых частей Fe.
S О Fe
32 64 56
Но это будет именно то весовое количество, которое должно
отвечать одному атому Fe, раз формула серножелезистой соли = Ре504.
Таким образом, вопрос о выборе между рядом возможных значений для
атомного веса железа решен в пользу величины 56.
4. Наконец, весьма важным вспомогательным средством для
определения атомных весов является периодический закон, о чем
будет идти речь ниже.
С тех пор, как Дальтон сделал первую попытку определить атомные
веса химических элементов, разработка этого вопроса не переставала,
занимать наиболее выдающихся химиков, начиная от Берцелиуса„
Дюма, Стае а, Жерара, Канниццаро, Мариньяка,
Д. И. Менделеева и кончая Морлеем, Ричардсом, Г ю и и
другими, работы которых относятся к новейшему времени.
Сознание огромной важности дела возможно точного определения
атомных весов, этих основных постоянных химии, заставило с 1900 г.
образовать при Немецком химическом обществе международную
комиссию, которая систематически просматривает и подвергает критическому
анализу все вновь появляющиеся исследования по определению
атомных весов и ежегодно публикует сводную таблицу атомных весов,
которые следует считать в данную минуту наиболее достоверными. Мы
приводим такую таблицу, опубликованную на 1912 г. (стр. 77).
Ныне одну сторону задачи — установку приблизительного значения
атомных весов, или, иначе говоря, выбор ф а кто р а, на который следует
помножить эквивалент, чтобы получить атомный вес, можно считать
окончательно решенной для подавляющего большинства известных
элементов. Зато и до настоящего времени деятельно продолжается
разработка другой стороны задачи, касающейся определения точных
значений атомных весов (или эквивалентов), путем планомер-
Периодическая система химических элементов
77
ТАБЛИЦА АТОМНЫХ ВЕСОВ
1912 г.
Ag
А1
Аг
As
Au
В
Ва
Be
Ві
Вг
С
Са
Cd
Се
С1
Со
Сг
Cs
Си
Dy
Ег
Ей
F
Fe
Ga
Р
Pb
Pd
Рг
Pt
Ra
Rb
Rh
Ru
S
Sb
Sc
Se
Si
Sa
Sn
серебро
алюминий
аргон
мышьяк
золото
бор
барий
бериллий
висмут
бром
углерод
кальций
кадмий
церий
хлор
кобальт
хром
цезий
медь
дисорозий
зрбий
европий
фтор
железо
галлий
фосфор
свинец
палладий
празеодим
платина
радий
рубидий
родий
рутений
сера
сурьма
скандий
селен
кремний
самарий
олово
107,88
27,1
39,88
74,96
197,2
11,0
137,37
9,1
208,0
79,92
12,00
40,07
112,40
140,25
35,46
58,97
52,0
132,81
63,57
162,5
167,7
152,0
19,0
55,84
69,9
31,04
207,10
106,7
140,6
195,2
226,4
85,45
102,9
101,7
32,7
120,2
44,1
79,2
28,3
150,4
119,0
Gd
Ge
Н
Не
Hg
In
Ir
J
К
Кг
La
Li
Lu
Mg
Mn
Mo
N
Na
Nb
Nd
Ne
Ni
Nt
0
Os
Sr
Та
Tb
Те
Th
Ti
Tl
Tu
U
V
w
Xe
Yt
Yb
Zn
Zr
гадолиний
германий
водород
гелий
ртуть
индий
иридий
йод
калий
криптон
лантан
литий
лютеций
магний
марганец
молибден
азот
натрий
ниобий
неодим
неон
никель
нитон
кислород
осмий
стронций
тантал
тербий
теллур
торий
титан
таллий
тулий
уран
ванадий
вольфрам
ксенон
иттрий
иттербий
цинк
цирконий
157,3
72,5
1,00,
3,99
200,6
114,8
193,1
126,92
39,10
82,92
139,0
6,94
174,0
24,32
54,93
96,0
14,01
23,00
93,5
144,3
20,2
58,68
222,4
16,000
190,9
87,63
181,5
159,2
127,5
232,4
48,1
204,0
168,5
238,5
51,0
184,0
130,2
89,0
172,0
65,37
90,6
но задуманных исследований, опирающихся на ряд количественных
анализов и синтезов.
С течением времени в прежних определениях (например, Стаса),
еше недавно считавшихся образцом точности, открывают ранее
оставшиеся незамеченными погрешности и научаются их избегать.
Совершенствуются и орудия эксперимента (измерительные приборы, особенно
весы) и, что особенно важно, самые методы работы. Подвергаются
критике и совершенствуются старые методы и предлагаются новые,
свободные от прежних недостатков. Особенно замечательны систематиче-
78
Периодическая система химичес * их элементов
ские исследования этого рода, которые ведутся в настоящее время
Ричардсом в Гарвардском университете в Америке .
Некоторое представление об общем плане подобного рода
исследований могут дать нижеследующие указания.
Как уже было упомянуто, в качестве единицы для атомных весов в
настоящее время принята 1/\б * атомного веса кислорода. Поэтому
естественно, что определение состава кислородсодержащих соединений
должно давать основной материал для определения атомных весов. По
некоторым соображениям чисто практического свойства широкое
пользование кислородными соединениями для только что указанной цели
началось гораздо раньше, чем вошла в силу новая международная
единица (Vie О). При этом, однако, оказалось мало удобным определять
атомный вес того или другого элемента непосредственно на основании
состава его окисла RnOmj так как в этом случае по большей части не
представляется возможным для одного и того же элемента осуществить
несколько независимых друг от друга контролирующих определений.
Поэтому Мариньяк и Стае, а еще раньше Берцелиус и
другие избрали несколько элементов как бы в качестве посредника между
кислородом и всеми остальными. Атомные веса этих элементов; серебра,
натрия, калия, хлора, брома и иода были определены с наивозможной
точностью по отношению к кислороду, а затем уже представлялось
возможным для любого элемента определить его атомный вес по
отношению к одному из шести «образцовых» **.
Для разрешения первой части задачи в качестве кислородных
соединений элементов Ag, Na, К, С1, Вг и J избирались хлорноватые, бром-
иоватые и йодноватые соли, состав которых отвечает общей формуле
МеХОз (где Me — металл, а X — галоид). При нагревании соли эти по
большей части гладко разлагаются по уравнению:
2МеХ03 = 2МеХ + 302,
а потому, если воспроизвести это разложение строго количественно
(определив сколько МеХ получается из определенной навески МеХ03),
то представляется возможность установить весовое отношение между
МеХ и 30, а следовательно, и молекулярные веса галоидных солбй МеХ
по отношению к кислородной единице. Таким образом был установлен
с большой точностью молекулярный вес следующих соединений:
AgCl KC1 NaCl
AgBr Kar
AgJ
Дальнейшим шагом по пути к определению атомных весов
упомянутых шести элементов является установление отношения между
серебром и галоидными солями КС1, КВг, NaCl. Это может быть достигнуто
посредством титрования определенного количества каждой из этих
солей раствором ляписа AgN03, причем этот последний получается
растворением навески чистейшего серебра в тщательно очищенной азотной
кислоте и содержит строго определенное весовое количество Ag в
единице объема.
Из полученных таким образом данных уже можно вывести атомный
вес серебра относительно кислорода. В самом деле, если а г КС103 дают
* См. прим. ред.2 к стр. 65.
** Во -всех этих случаях дело идет об определении точных значений атомных весов.
Приблизительные же их значения, а также молекулярные формулы тех соединений,
состав которых служит для вычисления атомных весов, предполагаются уже
известными.
Периодическая система химических элементов
79
Ь г КС1 и, следовательно, а—Ь г кислорода, то очевидно, что молеку-
лярный вес КС1 или М = а_ь -. Если затем при титровании оказалось,
что на с г КС1 идет объем раствора AgN03, содержащий d г Ag, то на
М г хлористого калия пойдет X = -Мх d или 3><16х6х^ г серебра
с (а — Ь) с г г
Ясно, что X и будет=атомному весу Ag (ибо AgN03+KCl==-KN03+AgCl)
Дальнейшая задача заключается в определении атомных весов
галоидов: хлора, брома и иода. Для этой цели можно воспользоваться
различными приемами. Например, можно взять навеску чистейшего
серебра и, действуя на него хлором, нацело перевести ее в AgCl.
Увеличение в весе при этой реакции будет отзечать присоединившемуся
количеству хлора. Можно также, отвесив серебро, растворить его в азотной
кислоте, осадить разведенной соляной кислотой и взвесить тщательно
промытый и высушенный осадок AgCl. Существует еще ряд других
модификаций этого основного приема, сущность которого сводится к тому,
что мы узнаем, какое весовое количество хлористого серебра отвечает
данному весовому количеству чистого металлического серебра. Если
атомный вес серебра X известен, то легко вычислить, сколько грамм
хлора У требуется для того, чтобы соединиться с X г Ag в AgCl.
Очевидно, У и будет весом хлора.
Понятно, что различные приемы, которые могут быть пущены в ход
для определения У, позволяют подвергать взаимному контролю
получаемые результаты.
Очевидно также, что если мы знаем атомный вес хлора, то стоит
только вычесть его из молекулярного веса КО, чтобы получить атомный
вес калия, и что соответствующим образом может быть также получен
атомный вес натрия. Понятно далее, что прием, послуживший для
определения атомного веса хлора, пригоден для остальных двух галоидов
брома и иода, и что, наконец, зная атомный вес Вг, а с другой
стороны, молекулярный вес КВг, можно другим путем получить атомный
вес калия и, следовательно, проверить результат, полученный, исходя
из КС1 и С1.
Если, таким образом, представляется возможным определить
атомные веса Ag, К, Na, С1, Вг и J относительно кислорода, то, опираясь на
эти цифры, всегда можно найти подходящий прием для решения
соответствующего вопроса для всякого другого элемента. Если,
например, требуется определить атомный вес азота, то можно исходить из
некоторой навески чистого серебра, количественно перевести ее в азот-
носеребряную соль, формула которой AgN03 предполагается, конечно,
известной. Вычитая из веса полученного ляписа вес заключающегося и
нем кислорода и серебра (атомные веса этих элементов уже известны),
получим вес азота, содержащегося в данной навеске, а пересчитывая
этот вес на грамм-молекулу AgN03 (или на г-атом Ag), найдем и
атомный вес N.
Приведем некоторые подробности такого количественного синтеза
AgN03 по опытам Ричардса и Форбса. (В сущности, опыты их
имели задачей определение атомного веса серебра, но если последний
предполагать известным, то можно, рассуждая в обратном порядке, из
экспериментальных данных этих авторов вывести атомный вес азота.)
В своих опытах Р. и Ф. пользовались двумя реагентами — азотной
кислотой и серебром, кроме того, еще водой и воздухом (для удаления
влаги продуванием из аппаратов). На очистку этих веществ было
обращено особое внимание. Вся работа была проведена в кварцевых
so
Периодическая система химических элементов
и платиновых сосудах, до которых прикасались только платиновыми
петлями и крючками. Таким образом, было гарантировано отсутствие
примесей, происходящих как от разрушения сосудов (например, стеклянных),
так и от соприкосновения с внешними объектами. Азотная кислота
тщательно очищалась повторной перегонкой, и для опыта бралась
только средняя порция дистиллята. Серебро, которого было взято
несколько образчиков различного происхождения, очищалось особенно
тщательно.
Один из употребленных для этой цели приемов заключался в
следующем. Серебро растворялось в чистой азотной кислоте, осаждалось в
виде хлористого серебра, а это последнее редуцировалось до металла
при помощи щелочного раствора чистейшего виноградного сахара.
Затем следовало новое растворение полученного серебра в азотной
кислоте и повторная (до шести раз) кристаллизация AgN03 в платиновых
сосудах из азотной кислоты, перегнанной через платиновый
холодильник. Продукты последней кристаллизации вновь подвергались редукции
до металлического серебра, на этот раз с помощью муравьинокислого
аммония, и серебро сплавлялось в сосуде из чистой окиси кальция.
Затем освобожденное от извести серебро опять растворялось в HNO3,
осаждалось электролитически, вновь сплавлялось в атмосфере
электролитического водорода и сушилось в разреженном пространстве при 150°.
Вода подвергалась повторной перегонке сначала с КМп04, потом
с небольшим количеством серной кислоты. Очистка воздуха достигалась
пропусканием его через колонку с бусами, смоченными
концентрированной серной кислотой с небольшой примесью К2СГ2О7, затем через
крепкий раствор едкого кали, через слой твердого КОН, через
накаленный платинированный азбест и еще раз над кусочками едкого кали.
Операция растворения серебра в азотной кислоте производилась в
кварцевой колбочке с длинным горлом. Предварительно колбочка
сушилась при 250р в токе сухого чистого воздуха и взвешивалась после
остывания в эксикаторе, разумеется, с поправкой на пустоту. После
стояния на воздухе вес ее немного увеличивался, но всегда на одну и
ту же величину, так что повторные взвешивания в течение нескольких
дней давали разницу всего около 0,00003 г.
В кварцевой колбе взвешивалось серебро, а затем приливалось
потребное количество чистейшей HN03, свежеперегнзнной в платиновом
аппарате. Растворение серебра производилось при 40—50° в замкнутом
пространстве — в большом эксикаторе, стенки которого, по окончании
опыта, испытывались на отсутствие Ag (которое могло бы попасть на
них вместе с мелкими брызгами), раствор ляписа выпаривался досуха,
остаток сплавлялся, в горячем еще состоянии помещался в эксикаторе,
защищенный от действия света черным сукном, и тщательно
взвешивался.
Полученное таким образом азотнокислое серебро было
действительно чисто. Сумма возможных в нем примесей не превышала
.0,0015%.
Определение атомных весов
непосредственно из плотностей пара
летучих соединений
За последние годы, благодаря усилиям лорда Рэлея, Д. Бертло,
.Л е д ю к а и особенно женевского профессора Г ю и, все больше и
больше входит в употребление метод определения атомных весов, до
.известной степени противоположный тому, о котором шла речь на
Периодическая система химических элементов
81
предыдущих страницах. Вместо того, чтобы сначала путем весового
анализа точно определить эквивалент данного элемента и затем
(приблизительную) величину плотности пара, сторонники нового
направления поступают как раз наоборот. С наивозможно большей точностью
они определяют именно плотность пара летучего соединения данного
элемента или отвечающего ему простого тела, состав молекулы
которых предполагается известным на основании прежних (рсотя бы и
менее точных) определений.
На частном примере лучше всего будет объяснить сущность этого
метода. Предположим, что требуется определить точное значение
атомного веса азота. Для этой цели можно взять тщательно очищенный
газообразный азот и с возможной точностью определить его плотность по
водороду. Помножив найденную величину на 2, мы получим вес
молекулы азота, а так как последняя состоит из двух атомов, то атомный
вес прямо будет = найденной плотности газообразного азота,
отнесенной к водороду. Вместо свободного азота можно взять одно из его
газообразных соединений, например окись азота N0. Если плотность ее
из опыта =16,005 (при плотности 0=-16) и, следовательно,
молекулярный вес =30,01, то очевидно (при 0=16), что атомный вес азота =
= 30,01 —16=14,01. Все такие выводы, однако, основаны на законе Аво-
гадро. Между тем, закон этот для реальных (не идеальных) газов
лишь приблизительно верен. Поэтому, если мы желаем определить
точный атомный вес азота, в экспериментально найденное значение
плотности надо внести соответствующие поправки.
Если через М обозначить молекулярный вес данного тела, через L —
вес литра его в газообразном состоянии при 0° и 760 мм рт. ст., то мы
будем иметь такое соотношение:
22,41 XL
1 + Я '
Величина 22,41 есть объем в л, который занимает при 0° и
760 мм рт. ст. 1 г-моль идеального газа, а X есть дробь (обычно
значительно меньше единицы), обозначающая ту долю, на которую
содержание данного (не идеального) газа в 22,41 л выше или ниже
теоретического (то есть выше или ниже одной грамм-молекулы). Очевидно.
величина X и составляет ту поправку, которую необходимо внести, так
как ясно, что числитель дроби 22,41 XL, то есть произведение веса литра
данного газа на объем, занимаемый грамм-молекулой идеального газа,
должен быть равен весу грамм-молекулы данного газа в
предположении, что закон Авогадро применим к данному слу-
ч а ю< Величина 22,41 XL, очевидно, будет меньше истинного
молекулярного? веса, когда X отрицательна (этот случай имеет место только для
водорода и гелия), и будет больше, когда X положительна. Для
определения значения X было "предложено несколько различных методов. Один и:<
них основывается на применении уравнения Ван-дер-Ваальса,
(p + -Jj(*-b)~*7\
константы которого а и- b могут быть легко определены путем опыта.
С величиной X константы эти связаны с помощью уравнения;
1-1-Х -(1-!-а)(1-Ь).
6 Л. А. Чугаев, т. III
82
Периодическая система химических элементов
Чтобы дать понять о важности результатов, которые 'могут быть
получены при помощи этого метода, остановимся на определении
атомного веса азота.
В своих классических исследованиях над атомными весами Стае
(в 1865 г.) пришел к заключению, что атомный вес азота= 14,045. Эта
величина была найдена как среднее из ряда превосходно сходящихся
результатов (14,043; 14,048; 14,046; 14,044), полученных посредством
независимых друг от друга методов. Стае считает, что уже величина
N=14,03 является весьма мало вероятной, а величину 14,02 он
называет «materiellement impossible» *. До последнего времени цифра Стаса и
не возбуждала ни в ком ни малейших сомнений.
Тем большее значение имело в данном случае применение нового
метода, основанного на определении плотности азота и его
газообразных соединений.
Приведем здесь результаты, полученные за последние годы в
женевской лаборатории профессора Гюи.
Газ, плотность
котогого была
измерена
N2
N,
N20
N20
NO
NHS
Найденный
атомный вес
14,009
14,006
14,006
14,007
14,008
14,012
Среднее . .14,008
Таким образом, в среднем получается величина, близкая к 14,01,
которая по Ста су, является абсолютно неприемлемой. Эта цифра
была подтверждена затем и путем химического анализа, так что в
настоящее время она является общепринятой. Очевидно, что в
прежних определениях Стаса заключалась довольно значительная
погрешность.
Метод Дж. Дж. Томсона
В самое последнее время Дж. Дж. Томсон [4] опубликовал в
высшей степени замечательный метод, пригодный для определения
атомных весов сильно разреженных газов, находящихся в
ионизированном состоянии.
Как известно, в круксовой трубке при прохождении разряда
одновременно возникают двоякого рода лучи: отрицательные, или
катодные, и положительные, или каналовые. Первые представляют потоки
весьма быстро несущихся корпускул (отрицательных электронов),
вторые— потоки более тяжелых частиц, заряженных +электричеством и
распространяющихся с меньшей скоростью. Исследование этих частиц
показало, что они представляют из себя положительные ионы, свойства
которых, а прежде всего отношение между массой и зарядам,
определяются природой газа, через который проходит разряд.
Томсон пропускает пучок положительных лучей через узкое
отверстие в катоде и подвергает его совместному действию электрического и
* Физически невозможной (франц.)- Прим. ред.
Периодическая система химических элементов
83
магнитного поля, которые расположены по отношению к лучу таким
образом, что первое отклоняет его в горизонтальном, а второе
(магнитное) — в вертикальном направлении. Пучок лучей направляется на
фотографическую пластинку. До тех пор, пока оба поля бездействуют, он
оставляет на пластинке след в виде точки или пятна, скажем в О
(рис. 1). При одновременном действии обоих полей это пятно переме-
местится в Р. Очевидно, мерой отклонения под влиянием
электрического поля будет линия ON, а мерой отклонения магнитного — линия
PN(PN±ON).
Известно, что влияние обоих полей может быть выражено
следующим образом [5]:
Р
е Лж, ,. е
РМ -.-. A—, ON---- в
tnv mv-J
где А и В — некоторые постоянные величины,
определяемые силой электрического и магнитного полей,
размерами трубки и пр., е —заряд, m — масса положительной
частицы и у — ее скорость. Соединяя оба предыдущие
выражения, імы имеем:
m
Л* ON Л N
е В PNn~'
Рис.
Поэтому, зная величину отклонения, а
следовательно, и величину ON и PN, мы будем знать также отношение mje.
Постоянные Лив могут быть определены посредством специальных приемов,
указанных Томсоном. Впрочем, нет надобности и знать эти величины,
если задача исследования заключается только в сравнении величин
mje для различных сортов движущихся частиц. Если наш пучок лучей
будет состоять из нескольких разно-родных частиц с различными
отношениями mje, то мы получим соответствующее число точек Р. Но на
самом деле получается (на фотографической пластинке после
проявления) не ряд точек, а ряд кривых. Для каждого сорта движущихся частиц
отношение mje остается постоянным, а потому в условиях опыта посто-
янно и отношение дту~, а это значит, что геометрическое место точек Р,
отвечающих данному сорту положительных частиц, будет представлять
собой некоторую параболу. Число таких парабол равно числу
типов частиц. Причина же, по которой частицы каждого данного типа
дают не отдельную точку, а целую кривую, заключается в том, что не
все они распространяются с одинаковой скоростью, а потому и мера
отклонения их различна (более быстро двигающиеся отклоняются
слабее).
Изучая проявленную пластинку и сосчитывая число парабол, можно
прямо определить число различных типов, а измерение парабол дает
материал для вычисления соответствующих величин.
Если признать, как доказанное, то положение, что каждая частица
может нести с собой один или несколько (во всяком случае только
целое число) зарядов, равновеликих — электрону (но противоположных
по знаку), то мы, конечно, легко можем, зная из опыта отношения
между величинами mje для различных положительных частиц, найти и
отношения между их массами. Опыт показывает, что этим
путем прямо получаются эквиваленты (или величины, им кратные) тех
элементов, которые содержатся во взятом для исследования газе,
притом с точностью (до 1%), прямо поразительной для подобного рода
определений. Так, взяв азот, приготовленный из воздуха, Томсон мог
84
Периодическая система химических элементов
констатировать следующие положительные частицы (наиболее резкие
из соответствующих парабол видны на рис. 2).
Рис. 2
Н+ Н2+ Nf<" C+ Nf N2* Ar+ Hg++ Hg4-
1 1,99 6,80 11,40 13,95 28,1 39 100 198
Числа второй строки выражают весовые количества ионов,
отвечающие единице заряда (одному электрону), причем за единицу веса взят
вес атома водорода. В верхней строке число знаков + указывает, со
сколькими зарядами связан данный атом или молекула*. Интересно
сопоставить с этими данными результаты, полученные при анализе
азота, приготовленного из химического соединения (NH4NO2). Здесь
нельзя было констатировать аргона, но были найдены по-прежнему ионы:
С" N^ N*" N2'" Hgf+
6,1 7,02 14,01 27,9 100 " Т' А*
В высокой степени интересны результаты, полученные Томсоном при
анализе кислорода: 0+, 0"*"+, О24", Оз+, Об+. Кроме атомов и молекул
кислорода и озона, снабженных положительным зарядом, здесь
получилось указание на присутствие молекул «надозона» 064, аналогичных
S6, принадлежащих новой аллотропической модификации, кислорода,
до сих пор не полученной.
Не меньший интерес представляют результаты, полученные
Томсоном лри исследовании метана СН4. В этом случае было, между прочим,
констатировано присутствие положительно заряженных молекул СНь
СН2 и СНз, представляющих -из себя «свободные радикалы»,
изолирование которых, составлявшее мечту химиков старого времени, еще
недавно считалось неосуществимым. Такие радикалы обладают
«свободными», или «ненасыщенными» единицами сродства.
Н Н
I I I
-С—Н -С—Н -С—Н
н
Этим обстоятельством обыкновенно объясняют неспособность их к
существованию. Мимолетное образование таких радикалов, однако,
нередко и ныне допускается при химических превращениях органических
соединении **.
* Следы паров ртути, кероятно, попали и прибор из ртутного насоса.
** В последнее время Гимберг. Шлимк и другие сделали весьма вероятным, что в
некоторых особых случаях свободные радикалы в такой же мере реальны, как,
например, кетонная и эиольная модификации таутомерных соединений и пр. Классическим
примером может служить углеводород, ірнфенилмстил, в растворенном, состоянии даю-
лііііі начало равновесной системе *г':
(СвНв>:,<: • С(СзНл)^2(с$нв)яс-
Периодическая система химических элементов
8Г>
Из приведенных немногих примеров видно, что положительными
понятии могут быть как заряженные атомы, так и целые молекулы.
Томсон заметил, что наряду с положительными ионами одновременно
появляются л отрицательные, хотя и в меньшем количестве. В самом
деле, против серии парабол, отвечающих частицам положительным,
появляются слабые контуры других парабол. Они ориентированы в
обратном порядке, по сравнению с положительными, и в отличие от этих
последних, всегда отвечают отдельным атомам, но не молекулам.
Анализ этих парабол также привел к возможности вычислить
приблизительное значение соответствующих атомных весов. Так, при
исследовании окиси углерода СО, наряду с положительными ионами С+4\ 0+ +
С4", N+, 0+, СО+, СОг+ и прочими были найдены отрицательные ионы
С]2~ И Оіб".
В азоте из воздуха было найдено;
и - с- о-
1 11,2 15,2
Не подлежит сомнению, что этот новый и оригинальный метод,
созданный Томсоном, представляет огромный таг вперед в деле
химического анализа по точности, полноте и степени чувствительности (здесь
¦достаточно 7юо миллиграмма вещества), далеко оставляющий за
собой даже спектральный анализ. Этот метод позволяет проникать, так
сказать, в самые недра химической системы, улавливать состояние
участвующих в ней индивидуумов в каждый данный момент времена.
Для нас, конечно, он особенно интересен, потому что непосредственно
дает числа, являющиеся материалом для определения атомных весов.
Как справедливо замечает Дж. Дж. Томсон, он, вероятно, окажет в
будущем важные услуги при изучении таких мало доступных и быстро
изменяющихся газов, как эманации, и других, легко летучих
радиоактивных продуктов. Здесь, конечно, особенно ценною является
возможность делать ряд определений последовательно одно за другим и,
таким образам, шаг за шагом следить за историей изменяющегося
вещества.
Глава II
ОБЩЕЕ ПОНЯТИЕ О ПЕРИОДИЧЕСКОМ ЗАКОНЕ*
Возьмем ряд элементов, расположенных в порядке возрастания их
атомных весов, начиная с лития (водород мы пока оставим в стороне)
и кончая фтором. Мы увидим, что свойства этих элементов,
отвечающих им простых тел и их сходственных соединений сначала будут
изменяться с полной постепенностью от лития до фтора:
Li Be В С N О F
Na Mg Al Si PS CI
К Са Sc Ti V Cr Mn,
но затем сразу замечается скачок, и от типичного галоида — фтора мы
переходим к столь же типичному щелочному металлу натрию. Натрий
во многих, отношениях сходен с литием, которым начинается
выписанный нами ряд, а потому мы и подписываем Na под Li. Подвигаясь
далее, мы встретим последовательно элементы: магний — аналог
бериллия, алюминий —аналог бора, кремний-—аналог углерода, фосфор —
аналог азота, серу—аналог кислорода и хлор—аналог фтора.
Каждый из элементов этой второй строки естественным образом располо-
8G
Периодическая система химических элементов
жится над соответствующим членом первой. Притом, от натрия к
хлору мы, как и в первой строке, будем иметь постепенное изменение
свойств. Если мы пойдем еще дальше, то опять наткнемся на резкий
скачок: от галоида хлора к щелочному металлу калию. Калий, как
аналог натрия и лития, расположится непосредственно под ними, а затем
пойдет новый ряд постепенно изменяющихся в своих свойствах
элементов от К до Мп, из которых каждый найдет своего аналога в
соответствующем вертикальном столбце.
Из этого беглого обзора элементов с атомными весами, лежащими
в пределах от 7 до 55 (из них мы пока умышленно пропустим два: неон
Ne = 20,2 и аргон Аг = 39,88), уже ясно видна основная идея
периодического закона, сущность которого может быть выражена следующим
образом:
Свойства элементов изменяются периодически в
зависимости от величины их атомного веса, или
иначе, являются периодической функцией атомного
веса*.
Чрезвычайно важно отметить резкое отличие зависимости,
существующей между атомными весами элементов и их свойствами, от
таких периодических функций, как г/ = sin х> #=tg х и т. п. Эти последние
предполагают непрерывное изменение одной переменной величины
при постепенном изменении другой (независимой). Между тем,
центральный пункт периодического закона как раз заключается в наличности
резких скачков между отдельными элементами и особенно резких
между отдельными периодами (см. ниже). Мы имеем б этом случае
функцию не сплошную, но разрывную.
В целях ближайшего ознакомления с сущностью периодического
закона и выяснения того, каким образом и на каких свойствах
элементов сказывается существование периодичности, нам необходимо теперь
обратиться к рассмотрению самого текста периодической системы. Эта
система обычно резюмируется в виде таблицы, форма которой стала
классической (другая форма той же таблицы дана на стр. 87) **.
В этой таблице прежде всего следует различать:
1) горизонтальные ряды. Это те самые ряды: Li—F, Na—С1 и т. д.,
о которых мы говорили в самом начале этого очерка. В пределах
каждого ряда свойства элементов (и соответственно соединений)
постепенно изменяются с нарастанием атомного веса. Ряды принято
обозначать арабскими цифрами.
2. Из рядов составляются периоды. Периодом называется серия
элементов, начинающихся с элемента А, принадлежащего к I группе
и оканчивающаяся непосредственно перед элементом Аі — ближайшим
аналогом А. Для первых двух рядов обозначения: «период» и «ряд»
фактически совпадают. Соответствующие периоды носят название м а-
л ы х.
Но далее в периодической системе мы встречаем периоды иного
рода: так называемые большие. Эти последние составляются из двух
* Периодической функцией называется такая зависимость (математическая) одной
переменной величины от другой переменной же величины х, при которой величина у
периодически получает одно и то же значение всякий раз, когда величина х
увеличивается или уменьшается на одно и то же постоянное число а. Если вообще у~{(х},
го для того, чтобы функцию эту можно было назвать периодической, должно
существовать соотношение: f{x+a)=f{x). Число а называется периодом функции. Например,
-чп х есть периодическая функция угла ху так как возвращается к прежнему значению
всякий раз при нарастании х на 2 я.
** Приведенная Л. А. Чугаевым таблица периодической системы относится к 1906 г.
Прим, ред.
Периодическая система химических элементов
87
м
о
ее
А
О
V
S
f-
X
я
«
Я)
с
с
;>»
о.
, 2
к « Ь
§"8 8
8Й
3
Я
X J3
н н
я
ag
*= 5
со еп
а*
U
в)
3
с
с
а.
и
О
:м
см
NT
СМ
со
см
f-
ю
со
го
см
^vl
•*
О
С J
чН
00
СМ
со
,
1
г^
го
,*
г—
со
см
t—
•ч-ч
1!
•О
>
О
«,
ел
со
ю
см
**
о
NP
іО о
00 eg
03 ^
ел
см -¦» ~I- 2
і.О
^ ^ -гч 77 |, СМ СМ СМ
HiUli.il
о^а<Хна,еэ
^ <fl га со о
XUCQ J U
ю со
ел vr со
г>- о
00 0
.стг^00чг00огді0^
о <о іл о - -.Г -р^ -*•-¦.,... ^
- - ~ _ _ 1 - ^^CD^CMvrooi>CO
СМЮГ^о^г-1 rt CD О) О С'ОО'-с-іч-'ч-иСМСМСМ
о
СО
* сх
со о
00
¦^й^ ^ °
N -Z. %
л-
X
"> О ?Г _ СО Np Vf СО N N CD eg
^ ^ О ^ -и 0 о 05 ^j m ОЮСОСГ.іЛОі^^Оі
сГ оГ ^ S3 со ^ см" v**" ю со" со f2 *ъ Як ?! ** °^ ел* л1
ш
о
с*
о.
X
3
з:
CD
X
3
X
4>
S
а;
СП
см
со
*-. ^ °3 ,« ;~ *-* ь- с о о •- з с " ^ wjcu^t-
О —. ~ ~
^* 1—(
г-1. 3 г^ !> г> <-« '-,
"> ^н ~
^ > >
05 Л О-
о-о
NOc°.^.oo^co
s я а а я я й й s
іі I! II II II II II II
СП Vf<
СЛ СЭ -«н
со со оэ
¦*"• о л
О О О О О <N
„" <N ^ СО ОЗ О
— *н «Н *Ч чч СМ
ej^uzo^i
U
со
о
о
-ч —' Е > ^ ^
CD
ел
со
н—< н-4
С ^ > > о
Высшие
¦олеобраз-
іые окислы
Газообраз
ные
водородные
соединения
«о «о ** « «
« «
с*г сг: ^ с* о
88
Периодическая система химических элементов
рядов каждый (причем в середине больших периодов находятся
элементы VIII группы — см. ниже). Таких периодов три: первый (по
общему счету третий) состоит из 4-го и 5-го рядов, второй (по общему
счету четвертый)—из 10-го и 11-го. Таким образом, всего имеется
5 периодов: 2 малых и 3 больших (из последних один неполный). На
остальные периоды, буде вообще можно говорить об их существовании
(8-й -и 12-й ряды), имеются только намеки.
3. Групп ы. Вертикальные столбцы, в которые, как мы видели,
сами собой располагаются друг под другом элементы-аналоги,
образуют группы. Таких групп в настоящее время мы знаем 9. В то время, к
которому относится открытие периодического закона, их было известно
всего 8. Из этого числа в первых семи (1—VII) элементы
располагаются по той общей и простой схеме, которая нами уже была указана в
основных чертах. При этом из каждого ряда в данной группе
находится только по одному представителю. VIII группа показывает
отклонение от этой схемы: в ней каждый ряд представлен тремя элементами.
Мы увидим, что она и во многих других отношениях является, до
известной степени, как бы аномальной. Нулевая группа составляет
сравнительное новшество в периодической системе. Появление ее было
следствием открытия так называемых «благородных» газов (гелия,
неона, аргона, криптона и ксенона).
4. Рассматривая распределение элементов по группам и рядам
периодической системы, легко отметить в этой последней еще одну
важную особенность: существование в ней характерных четных и
нечетных рядов. В пределах каждой группы периодической системы
нечетные ряды 3-й, 5-й, 7-й и т. д. образуют как бы одну подгруппу,
характеризующуюся чертами более тесной и близкой аналогии своих
членов. Подобную же вторую более тесную подгруппу образуют в
пределах каждой группы элементы рядов четных (4-й, 6-й, 8-й и т. д.).
Мало того, во всей периодической системе элементы четных рядов
характеризуются некоторыми общими свойствами, отличающими их от
элементов, относящихся к рядам нечетным: то же самое можно сказать
¦и об этих последних. Так, только элементы нечетных рядов обладают
способностью образовать газообразные водородистые соединения
(например: SiH4, РН3. H2S, ИС1, НВг и т п.) и (по большей части летучие)
металлорганические, то есть соединения, в которых атом
металла связан с органическими радикалами: СИ3, С2Н5 и прочие.
Таковы Zn(CH:ih, Zn(C2Hs)?> HgfCaHsb. Sn(C2Hs)4 и прочие. Элементы
четных рядов (кроме некоторых типических — см. ниже) подобных
соединений не образуют. Далее можно указать на то, что элементы четных
рядов в большинстве случаев -магнитны, тогда как элементы рядов
нечетных— диамагнитны (о других отличиях, характеризующих
элементы четных рядов по сравнению с нечетными, "см. далее). В результате
получается такое впечатление, как будто вся совокупность химических
элементов образует (по крайней мере) две серии, хотя и связанные
между собой, но в то же время сохраняющие известную степень
самостоятельности. Периодическая система является как бы результатом
совмещения или вложения двух отдельных систем одна в другую.
Такой взгляд был, действительно, высказан некоторыми авторами.
Если, с одной стороны, четные ряды противополагаются рядам
нечетным, то, с другой стороны, при внимательном изучении
периодической системы бросается в глаза еще и другое подразделение. Можно
заметить, что в каждой группе обозначаются элементы, для этой
группы особенно характерные. К таким элементам
относятся, во-первых, хотя и не всегда вполне отчетливо, элементы второго и
Периодическая система химических элементов №
третьего рядов, а затем в группах I—IV элементы четных, а в
остальных трех—IV—VII— элементы нечетных рядов.
Главные подгруппы
2.
3.
4.
5.
6.
7.
8.
11.
12.
I
Li
Na
К
Rb
Cs
II
Be
Mg
Ca
Sr
Ba
Ra
III
В
A\
Sc
Yt
La
IV
С
Si
Ti
It
Ce
Th
V
N
P
As
Sb
Bi
VI
О
S
Se
Те
VII
F
CI
Br
J
Сюда же можно отнести и всю нулевую группу. Таким образом,
получается целая серия, заключающая элементы, наиболее
характерные, и, так сказать, нормальные для данной группы периодической си-
стемы. Подгруппы, образующие эту серию, иногда называют
главными подгруппами; подгруппы же, включающие все остальные элементы,
противополагают им и называют побочными.
Элементы этих побочных групп образуют другую самостоятельную
серию:
Побочные подгруппы
I II III IV V VI VII VIII
4.
5.
6.
7.
10.
И.
12.
Си
Ag
Аи
Zn
Cd
Hg
Ga
In
Tl
Ge
Sn
Pb
V
Nb
Та
Cr
Mo
W
и
Mn
Fe
Ru
Os
Co
Rh
Ir
Ni
Pd
Pt
Из дальнейшего мы увидим, что в группах I—VI элементы побочных
подгрупп отклоняются от основного характера каждой группы тем
резче, чем ближе эта группа к краям периодической таблицы. По
некоторым признакам к побочным подгруппам примыкает вся восьмая
группа.
5. Уже в своих первых статьях о периодическом законе Менделеев
указал, что элементы ряда
Li, Be, В, С, N, О, F,
обладающие наименьшими атомными весами, «отличаются резкостью
свойств». Элементы эти стоят как бы особняком, причем каждый из них
сильнее других элементов той же группы отклоняется от свойств
для этой группы характерных*. Менделеев назвал эти элементы
* Так, фтор вовсе не соединяется с кислородом7 и не дает соединений, в которых
его можно было бы считать семиатомным, водородистое соединение его представляет
сравнительно слабую кислоту (другие галоидоводороды — кислоты весьма сильные)
и притом показывает ассоциацию, то есть образует молекулы H2F2, чего также нет для
других галоидоводородов. Подобным же образом, кислород не образует таких соеди-
нейий, в которых бы проявлялась высшая атомность -«-6, свойственная VI группе-.
Для азота «аномальными» свойствами можно считать неустойчивость и эндотермичноеть
его кислородных соединений, а равно и соединений с галоидами (NC13, NJ?) и т. ,т.
90
Периодическая система химических элементов
типическими. К ним отчасти примыкают элементы третьего ряда
и между ними в особенности Na, Mg, А1 и Si, также
показывающие уклонения от некоторых общих правильностей. Совершенно
особняком стоит водород, занимающий место 1—I *: Менделеев говорит о нем,
что «как легчайший элемент, он, по справедливости, избирается, как
самый типический».
К числу замечательных особенностей типических элементов
принадлежит также большое распространение их в природе, что было
отмечено Менделеевым еще в 1869 г. В противоположность этому, элементы
с высокими атомными весами обыкновенно принадлежат к числу
редких (золото, платиновые металлы, радий и пр.)
По вычислению Кларка, около 50% по весу нашей планеты (той
части ее, которая доступна для исследования: воздушной оболочки,
воды морской, океанов и пр. и земной коры) приходится на долю
кислорода и около 49% —на долю элементов: Si, A1, Fe, Ca, С, Mg, Na, К
и Н. Более полные (грубо приблизительные, конечно) данные
содержатся в следующей таблице:
Элементы
В земной
коре,
%
В воде
океанов и пр..
%
В атмосфере.
В совокупности
доступных исследованию
частей земною шара
содержится,
Кислород
Кремний
Алюминий
Железо .
Кальций
Магний .
Натрий .
Калий
Водород
Титан
Углерод
Хлор
Бром
•Фосфор
Марганец
Сера . .
Бар ий
Азот . .
Хром
47,29
27,21
7,81
5,46
3,77
2,68
2,36
2,40
0,20
0,33
0,22
0,01
0,10
0,08
0,03
0,03
0,01
0,01
85,79
0,05
0,14
1,14
0,04
10,67
0,002
2,07
0,08
0,09
23,00
77,00
49,98
25,30
7,26
5,08
3,51
2,50
2,28
2,23
0,94
0,30
0,21
0,15
0,09
0,07
0,04
0,03
0,02
0,01
100,0
100,0
100,0
100,0
Для многих типических элементов, принадлежащих к числу металлоидов, характерна.
далее, устойчивость соединений их с галоидами по отношению к воде. Это имеет место
для ССЦ, СВг4, CJ4, NCb, NJ3, SF6, SOF2, SO2F2 и пр. (галоидные соединения других
металлоидов легко и нацело разлагаются водой). Наконец, типические элементы
представляют многочисленные случаи отступления от правила четных и нечетных рядов
(см. ниже).
* Положение водорода в I группе связано с тем обстоятельством, что атомность
его принимается за 1 и, кроме того, с его резко выраженным электроположительным
характером (см. главу IV). Некоторые химики (О. Массой и другие) считают, однако,
более правильным помещение его в VII группу над галоидами. В пользу такого мнения,
в числе других соображений, приводится, что молекула Н2 состоит из двух атомов,
как С12 и Вг2, в отличие от одноатомных молекул Na, К и пр. Точно также весьма
низкая температура кипения водорода — 253° больше приближает его к галоидам, нежели
к щелочным металлам.
Периодическая система химических элементов
91
Из всех многочисленных свойств, присущих элементам,
периодический закон особенно ярко и характерно отражается на следующих
двух: 1) на формах соединений, образуемых элементами, а
следовательно, и на валентности этих последних и 2) на
электрохимической природе, или, иначе говоря, на «металлическом и
металлоидном характере элементов. Ввиду этого, мы и начнем
более детальное изучение периодического закона с этих именно двух
пунктов.
Глава III
ПЕРИОДИЧЕСКИЙ ЗАКОН И ВАЛЕНТНОСТЬ
Как известно, различные формы химических соединений проще
всего выражаются валентностью элементов, их образующих.
Под валентностью (или атомностью) принято
подразумевать число, показывающее со сколькими
атомами водорода (или иного элемента с валентностью^ 1) соеди-
няется или сколько таких атомов способен
замещать атом данного элемента. Каждую единицу
валентности или сродства принято символически обозначать черточкой
<или точкой), присоединяемой к знаку того или другого элемента.
Число таких черточек и указывает нам наглядно на его валентность:
Кислород
двухатомный
Азот
трехатомный
С
/\
Углерод
четырехатомный
-S-
Сера
шестиатомная
Если мы пожелаем с помощью этих «единиц сродства» дать
наглядную (ко символическую, конечно) картину взаимного их «насыщения»
в молекулах химических соединений, то получим так называемые
структурные формулы, которые выражают «способ химической
связи элементарных атомов в частице» (А. М. Бутлеров). Таковы,
например, следующие формулы:
Н—С1
Хлористый
водород
н
\
о
н-
Вода
/н
Н
Аммиак
¦\
н
н
Сернистый
водород
О
°v°-H
с/ с/°хо—н
Серный ангидрид Серная кислота
0 = N
0=N
\
/
\
О
О
о
Азотный ангидрид
о
0=С1-0—Н
о
Хлорная кислота
о
\
N—О—Н
о/1
Азотная кислота
/О-Н
А1^0-Н
О—Н
Гидрат окиси алюминия
02
Периодическая система химических элементов
С точки зрения современной электронной теории, валентность
данного атома (или радикала) измеряется числом электронов, которое
он может потерять или, наоборот, воспринять извне. Яснее всего эти
отношения обнаруживаются при рассмотрении электролитов. Когда мы
имеем раствор хлористого натрия в воде, то, согласно теории Аррениу-
са, часть нейтральных молекул NaCl находится в состоянии
диссоциации на ионы. При этом предполагают, что нейтральный атом натрия
теряет один электрон, что соответствует приобретению им единицы
(96 500 кулонов на грамм-атом, или 1,5хЮ~19 кулонов на 1 атом)
положительного заряда. Электрон этот переходит на нейтральный атом
хлора, делает этот последний ионом и сообщает ему равный, но проти-
поположный по знаку, то есть отрицательный заряд=1. В. Р а м з а и
предлагает называть электрон символом Е. Тогда электролитическая
диссоциация хлорнстхуго натрия выраз-ится таким уравнением:
NanE — ClmE = Na (n — 1) E + CI (m + 1) E.
При этом n означает общее число электронов, содержащееся в
нейтральном атоме Na, a m такое же число для нейтрального атома С1.
Если вместо хлористого натрия взять хлористую медь, в которой
металл двухвалентен, то при ее электролитической диссоциации
(ионизации) происходит распад
СиС12 = Си + 2С1
на три иона, причем ион меди получает заряд, равный двум единицам
положительного электричества. На языке теории электронов это значит,
что атом меди теряет два электрона, а каждый из двух атомов хлора
приобретает по одному. При ионизации хлорного железа FeCl3 атом
железа теряет три электрона, из которых каждый присоединяется к
атому хлора, и т. д.
С этой точки зрения силы, действующие между атомами и
удерживающие их друг около друга в данной молекуле, имеют электрический
характер. Они действуют между электроном, присоединивпимся к
атому, скажем, хлора, и положительно заряженными атомами Na, Cu, FV
И Т. Д.
При этом каждому электрону и каждой единице положительного
заряда соответствует Фарадеевская трубка, символам которой и
является, таким образом, черточка, соединяющая в наших формулах
единицы сродства атомов, образующих молекулу.
Фарадеевская трубка всегда имеет определенное направление,
которое считается от положительно заряженного тела к
отрицательному. Поэтому, в сущности, и для единиц сродства в структурных
формулах может быть указано направление, как это видно из следующих
примеров:
Н+—С1 Н+ \- Н+ \-
нсі н+/— н+л-и
H2S Н20
Заметим еще, что все только что приведенные соображения на
основании некоторых аналогий могут быть распространены и на
молекулы неэлектролитов, хотя эти последние не подвергаются электролизу,
а потому для них и нет возможности прямым путем определить знак
и число единиц заряда, связанного с каждым данным атомом.
Периодическая система химических элементов
93
Так, мы имеем основание считать фосфор электроположительным в
РСЬ и в РСЦ, а хлор в этих соединениях электроотрицательным; серу
электроположительной в S03, а кислород — электроотрицательным.
Поэтому формулы эгих вещестз с «направленными» единицами сродства,
по всей вероятности, должны иметь такой вид:
pci» a-- +pcu
0-.-+S+
+ ^-0
SO
3
Из приведенных формул (стр. 91) непосредственно видно, что
валентность данного элемента легко может быть выведена не только из
состава молекулы соединения его с водородом (или иным одновалент
ным элементом), но и с элементом многоатомным. Так, например,
атомность серы = 6 легко вывести из формулы серного ангидрида.
Из тех же структурных формул можно вывести и еще одно
заключение, для нас в данную минуту особенно важное. Валентность есть
величина переменная. Для серы, например, она может быть и 2
(в SH2) и 6 (в S03 и H2S04), для хлора и 1 (в НС1) и 7 (в НС104),
для азота 3 (в NH3) и 5 (в \:205 и HN03) и т. я.
В настоящее время во-прсс о том, -может ли валентность данного
элемента изменяться (хотя и в узких пределах, и притом, конечно,
всегда оставаясь небольшим целым числом), решен окончательно в
положительном смысле*. Но в 60-х, 70-х и даже 80-х годах минувшего века
было много приверженцев учения о постоянной валентности и на этой
почве велись оживленные споры. Решающую роль в этом отношении
как раз сыграл периодический закон.
Возьмем третий ряд элементов, начинающийся натрием и
заключающийся хлором, и напишем для каждого из этих элементов
главнейшие кислородные соединения.
Достаточно одного взгляда на эту табличку, чтобы сразу бросилась
в глаза поразительная правильность. Для первых четырех групп
(постоянная) валентность данного элемента по кислороду прямо равна
номеру группы, в которой он находится; для остальных трех трупп, где
валентность меняется, номеру группы равно ее высшее значение.
Эта правильность, ясно выраженная для 3-го ряда, оказывается
верной и для всей периодической системы, ибо, возвращаясь от VII
группы к I, от хлора к калию, мы, после резкого скачка, «встретим
щелочной металл .калий, дающий окись Кі>0 и потому одноатемный; за ним
следуют двухатомный кальций Са (окись СаО), трехатомный скандий
Sc(Sc2Oa), четырехатомный титан Ті(ТіОг), ванадий V — пятиатомный
* С точки зрения современных взглядов на валентность и на конструкцию
химических атомов возможность переменной валентности является вполне понятой, ибо и:.
числа тех относительно свободных электронов, которые могут переходить с одного
іітеша на друігой и, таким обра лам, давать начало элементарным силовым трубкам,
при известных условиях не все могут проявлять свою активность. Вполне понятно
также, что число электронов, участвующих в соединении атомов между собою, может
зависеть от индивидуальной природы этих атомов, их структуры и пр.
94
Периодическая система химических элементов
Номер
группы
Элемент
Структурная
формула
Валентность
I
Na
Na/U
1
п
Mg
Mg=0
2
Ш
AI
A<°
A1>°
A4o
IV
Si
4
V
p
>°
0
p =o
>
P =0
li
0
5 (3)
VI
s
0=S/"°
6 (4)
VII
CI
CL
/°
CK
0
ll/°
iN
о
7(1)
h высшем солеобразном окисле V^Os, шестиатомный хром, дающий
СгОз, семиатомный марганец, которому отвечает марганцевая кислота:
О
НМп04 или 0=\Мп—ОН и т. д.
О
Таким образом, -мы .получаем общее правило, согласно которому
номер группы (периодической системы), в которой находится
данный элемент, численно равен высшей .валентности
этого элемента но кислороду, поскольку она вытекает из
состава оолеобразных или солеобразующих * окислов.
* Оговорка, заключающаяся в слове «солеобразный», имеет в виду исключить из
рассмотрения вещества перекисного характера, представляющие аномальное
отношение.
По составу этих соединений непосредственно нельзя делать заключение о
валентности образующих их элементов. Причина этого лежит в том обстоятельстве, что в
перекисях приходится принять присутствие атомов кислорода, непосредственно
связанных между собой. Одним из оснований для такого предположения является то
обстоятельство, что перекиси часто получаются при самоокислении (аутоксидации)
различных (простых и сложных) веществ при действии свободного кислорода. Так.
по Таубе, перекись водорода получается, когда водород in statu nascenrii действует
на молекулу кислорода согласно следующему уравнению:
н о о-н
+ II - і
н о о—н
Таким образом, в перекись водорода молекула кислорода перешла как бы
целиком. Поэтому перекисям, генетически связанным с перекисью водорода, Таубе дает
название голоксидов (от греческого слова, означающего «целый»). Такова, например,,
перекись натрия, получающаяся при пропускании сухого воздуха над нагретым
металлическим натрием
2Na + 02=Na202
м с разнсденными кислотами дающая перекись водорода:
Na202+2HCU--H2OH-2NaCl
Периодическая система химических элементов
95
Благодаря этому правилу (которому подчиняется и нулевая группа,
так как для ее членов, инертных газов, валентность всегда = 0), для
большинства элементов сразу определяется их положение в системе и,
наоборот, стоит только вспомнить номер группы, .в которой находится
данный элемент, чтобы сразу представить -себе основное его химическое
свойство — форму его высшего кислородного соединения.
Другая -правильность, которая также связывает номер группы с
валентностью, касается газообразных водородистых соединений,
или гидрюров. И эту правильность легко проследить, взяв в
качестве примера хотя бы опять тот же самый третий ряд:
Группа
Элемент
Формула водородистого соединения
Валентность по водороду ....
Высшая валентность по кислороду
I
Na
1
П
Mg
2
Ш
со 1 1 >
IV
Si
SiH4
4
4
V
P
PH3
3
5
VI
S
SH2
2
6
VII
CI
CIH
1
7
Сумма .. | — | — | — | 8 | 8 | 8 | 8
Легко видеть, что валентность в их водородистых соединениях
изменяется в обратном порядке, сравнительно с валентностью по кислороду,
а именно, повышается по направлению от VII к IV, при переходе к
каждой 'Следующей группе, на 1. При этом наблюдается* еще одна заме-
Такова перекись бария, как известно, также разлагаемая кислотами с образованием
Н2Ог. То обстоятельство, что настоящие металлические перекиси с кислотами не дают
солей, отвечающих высшей степени окисления данного металла, но дают перекись
водорода и соль, отвечающую нормальному окислу, представляет второй аргумент в
пользу того, что соединениям этого рода следует приписать строение
Na-О Оч
I I >Ва
Na-0 0/
Перекись натрия Перекись бария
и признать, что в них атом металла развивает нормальную присущую ему валентность
¦ по кислороду.
Справедливость такого заключения особенно выиграет в очевидности, если мы с
настоящими перекисями сопоставим такие соединения, как перекись свинца РЬ02 и
перекись марганца Мп02. Оба эти соединения с кислотами не дают перекиси водорода
и с нею, очевидно, не связаны. Но зато существуют, хотя и весьма мало устойчивые,
соединения РЬСЦ и МпС14, обличающие высшую атомность РЬ и Мп в РЬ02 и МпО:
и заставляющие приписать этим последним строение8:
Pbf и Mnf
^о ^о
* Следует иметь в виду, что все вышеприведенные обобщения сохраняют свою
силу независимо от того или другого взгляда на валентность элементов и даже от
самого понятия о валентности. На это обстоятельство неоднократно и с полным правом
указывал Д. И. Менделеев. Поэтому, если мы пользуемся здесь представлением о
валентности, то лишь по той причине, что оно имеет преимущество большой наглядности,
сводя многообразие форм химических соединений к одному общему началу, а с другой
стороны, потому что оно получило огромное теоретическое значение для уяснения
структуры химических молекул.
•96
Периодическая система химических элементов
чательная правильность: ;в четырех последних группах (IV—VII)
сумма валентностей по водороду и (высшей) по кислороду есть величина
постоянная и — 8. Значение этого эмпирического правила, а в
частности, фигурирующего в кем числа 8, до сих пор не вполне еще ясно, но
нужно думать, что здесь лежит ключ к уразумению многих
соотношений (особенно электрохимических) между отдельными элементами.
По теории, высказанной Абеггом, каждому элементу свойственна
двоякая валентность, иначе творя, двоякие единицы сродства:
единицы нормальной валентности и контравалентности,
причем общее число их для каждого атома = 8, так что:
нормальная валентность всегда = 8 — конгравалентность. Единицы
валентности обладают обратным знаком (в электрохимическом
отношении) по сравнению с единицами контравалентности и притом
значительно сильнее этих последних, а потому природа данного элемента
определяется главным образом его нормальной валентностью. Далее, Абегг
допускает, что численное значение и знак тех и других единиц сродства
в пределах периодической системы правильно изменяются согласно
следующей схеме:
Группы периодической системы
Контравалентность
I
+1
—7
II
+2
-6
ш
+3
—5
IV
±4
V
-3
+5
VI
—2
VII
—1
+7
¦С точки зрения этой теории в таких соединениях, как NaCl, KBr,
MgSO<, ZnCh, АіСіз, ТіСЬ, Zn(C2H5)2, Hg(C2H5)2> А1(С2Н5)3 и т. д.,
атомы металлов раззивают свою положительную нормальную
валентность; в НС1, HBr, HJ, HoS, H2Se, Н3Р, H3As и т. д. атомы металлоидов
проявляют также нормальную, но уже отрицательную валентность.
Положительная конгравалентность 'проявляется в высших кислородных
соединениях галоидов, элементов группы серы и т. д., например, в С1207,
НС104, НСЮз, НЛОз, HJ04 и пр.; наоборот, соединений, в которых
проявлялась бы отрицательная контравалентность, например для
щелочных металлов, с достоверностью не известно, так что в отношении
«•силы» контравалентности в периодической системе резко сказывается
отсутствие симметрии. Быть может это обстоятельство объясняется той
известной особенностью положительного электричества, в силу которой
связь его с весомой материей гораздо теснее; чем соответствующая
связь («электросродство», см. гла.ву IV) отрицательного электричества,
или электрона.
Заметим еще, что в новейшее время Дж. Дж. Том сон сделал
интересную попытку привести в езязь постоянство суммы обоих родов
валентности у различных элементов с предложенной им теорией
конструкции элементарных атомов.
Гораздо меньше, чем об упомянутых выше гидрюрах элементов IV-
VII групп .периодической системы, знаем мы о водородистых
соединениях первых трех групп. Полученные соединения здесь по большей
части нелетучи и нерастворимы без разложения (кроме очень мало
изученного водородистого бора), а потому и молекулярный вес их с
достоверностью неизвестен. Для некоторых из них возбуждает сомнение даже
самый факт их самостоятельного существования. Тем не менее,
по-видимому, и в данном случае для каждой группы периодической системы
Периодическая система химических элементов 97
существует определенный тип гидрюра, а следовательно, определенная
валентность по водороду, как это видно из следующих примеров:
I группа I] группа III группа (IV группа)
LiH СаН2 LaH2 (ThH4)
NaH SrH2
KH BaH2
RbH
CsH
Очевидно, что водородистые соединения, сюда относящиеся,
принадлежат к существенно иному типу, нежели газообразные гидрюры
элементов, принадлежащих к IV—VII группам. Во-первых, они
образованы металлическими элементами -четных рядов (кроме Na), во-вторых,
они представляют твердые нелетучие или трудно летучие тела;
в-третьих, валентность, .проявляемая в них -соответствующими
металлами, нарастает от I группы к III <и равна валентности тех же
металлов по кислороду, а -потому, .в-четвертых, в этом случае сумма
валентностей данного элемента по О и -по Н в сумме .не может быть равна 8.
К гидрюрам* этого второго типа принадлежит и недавно полученный
водородистый торий.
Ввиду того, что валентность по водороду является постоянной дли
В'Сех элементов данной группы, образующих водородистые соединения,
для этих последних так же, как и для высших окислов, можно дать
общие схемы, выражающие состав их молекул и отвечающие каждая
определенной группе периодической системы. Тогда получим:
Л"» группы
Форма высшего солеобразного
Форма высшего водородистого
соединения
I
RH
п
RO
RH2
ш
R303
RH3
IV
ROa
RH4
V
RH3
VI
ROa
RH2
VII
Ra07
RH
Группы
Валентность
Тип
соединения
Примеры
I
1
RM
NaC2H5
11
2
П---.2
ям
Be(C2H6)2
Zn(C.2H5),
Cd(QH6y
Hg(C2H5)2
111
3
n-3
R3M
B(C2Hs)<j
А1(С2Н5)з
IV
4
R4M
C(C2H6)4
Si(CH5)4
Sn(C4He)4
Pb(CH5)4
V
5
n-3
R:iM
N(C2H5h
P(C>H6);J
As(C3H6)3
Sb(C4H5)3
Ві(С4НьЬ
VI
6
n=2
R,M
0(QH5)2
S(C2H&)2
¦Se(C>H5)2
Te(C,H5)2
VII
7
.
RM
FC,H6
CIG.H5
BrC*H5
JC.Hft
Тип водородистых соединений в точности воспроизводится в
соответствующих летучих металлорганических и других аналогичных
-соединениях элементов с органическими радикалами (СН3, С2Нг„ С6Н& и т. п.).
* В настоящее время соединения элементов с водородом называют гидридами.
Прим. ред.
7 Л. А. Чугасв, т. Ш
08
Периодическая система химических элементов
В следующей таблице R означает органический радиікал, а М — атом
элемента, с ним связанного.
С летучими ¦ органическими соединениями элементов интересно
сопоставить соединения подобного же состава, но имеющие характер
основании (и соответствующих солей). Простейшим представителем
этого рода соединений могут служить соли аммония NH4X. в которых
атомы водорода замещены органическими радикалами. В таблице М
по-прежнему означает атом элемента, стоящего в данной группе, а
R — органический радикал. Во главе ряда MRn ОН стоят едкие щелочи,
для которых п = 0. Легко видеть, что валентность элементов в этом
ряду возрастает от первой группы к пятой, здесь достигает максимума
и затем падает *.
Группы
лентность
Тип
соединения
I
1
п--0
М-ОН
и
•>
MR-OH
ш
3
п-2
R3M-OH
IV
/*
n-3
R2M-OH
V
Г)
п- \
R4M—ОН
VI
(і
п М
RaM—ОН
VII
7
п---2
RSM—ОН
Примеры
Едкие
щелочи
Ці-ОН
Ма-ОН
t-OH
Hg
/
\
С2Н6
ОН
Т1-С2Нб
^ОН
/С2Н6
Sn—С2Н5
I хс2н6
он
С2Н5ч
C2H5~N
с,н
;П5
/
\
с,н
>чь
он
Гидрат окиси
тетраэтил-
аммония
4-2115 ч /
сх5
C,Hft-Sb
СоН5/ ОН
Гидрат окиси
тетраэтил-
стибония
С2Н6*
С2Н5
\S
/QH5
он
Гидрат окиси
триэтмл-
сульфония
СбНб\і--ОИ
с0н/
Гидрат окиси
дифенил-
йодония
Если мы от окислов и гидрюров перейдем к соединениям элементен
с галоидами и серой (и аналогами этой последней Se и Те), то
встретим в общем те же типы) и, следовательно, ту же валентность,
какая сказывается в соединениях этих элементов с О и Н.
* Так рассматривают строение этих соединений с точки зрения классической
теории строения. А. Вернер держится на этот счет существенно иного взгляда и
допускает, что валентность данного элемента при переходе от соединения MeRn к MRn . jOH
не изменяется и что в таких основаниях, как гидраты окиси аммония, сульфония и
прочие, & равно и в соответствующих им солях элементы воды и кислот
присоединяются с помощью особых побочных, клн дополнительных, единиц1 сродства:
Н\
h-)n • -.. н
LH
он
сн
3\
сн.
S СН.
—J
СН:
\
CHs-^Sb
1СН:
/
СН,
¦С1 и т. д.
Периодическая система химических элементов
Ш
Для примера лри-ведем следующий ряд:
Na
NaCl
NaF
Na2S
Mg
MgCla
MgF,
MgS
Al
A1C1,
A1F3
A12S,
Si
SiCl4
SiF4
SiS2
P
PC15
PCI3
PF6
PA
P,S3
S
SC14
SClo
SFe
SO,
so.
Отметим еще, что степень постоянства .валентности элементов, а
также сравнительная степень устойчивости соединений, отвечающих
различной валентности, довольно правильно изменяются в пределах
периодической системы.
Наиболее постоянной валентностью отличаются элементы, стоящие
в левой (электроположительной, см. следующую главу) части
периодической таблицы, именно принадлежащие к I—III группам, а среди них
особенно элементы четных рядов. В нижеследующей табличке собраны
все элементы упомянутых трех групп, для -которых до сих лор с
достоверностью известны * только соединения, отвечающие нормальной для
этой группы валентности (причем элементы нечетных рядов заключены
в скобки):
Li
(Na)
К
Rb
Cs
Be
(Mg)
Са
(Zn)
Sr
Ba
В
(Al)
Sc
Yt
La
Ra
Элементы нечетных рядов тех же (первых) трех групп
обнаруживают большее разнообразие валентности, причем это разнообразие в
общем возрастает с повышением атомного веса. Особенно это
бросается в глаза во II группе, где единственным представителем элементов с
валентностью, .по-видимому, изменчивой (см. главу VIII) является
ртуть, образующая соля закиси и окиси HgX и Н^Хг, а затем в III, где
галлию соответствует ряд непостоянных, легко окисляющихся солей
* Существование изученных Гюицем, Л. Веле.ром и другими нестойких
(разлагаемых водой и пр.) субгалоидных соединений состава CaCl, CaJ, CaF • BaCl и пр.
вовсе не доказывает, что в них Са и Ва одноатомны. При нелетучести этих соединений
и невозможности определить вес их молекулы вполне допустимо, что формулы их
сложнее, чем вышенаписанные, а тогда вполне возможно, что в них не металлы, а
атомы галоида проявляют высшую валентность (как, например, иод в JC13 и т. п.), что
соединения эти (подобно перекисям, комплексным соединениям, например периодидам
и т. п.) нельзя отнести к нормальному типу и что по их составу нельзя без оговорок
делать заключение о валентности входящих в них элементов .^считая Са и Ва одно-
атомными); на это указывает и то обстоятельство, что к числу субгалоидных
соединений относятся и производные одноатомных металлов, например, серебра: Ag2J и пр.
Здесь уже весьма вероятно, что высшую валентность.проявляет не металл, а галоид.
Так называемые недокиси, то есть низшие кислородные соединения металлов,
например, Na40, К46, Cs^O, а также Cs70, Cs702 и другие [6J, водой -разлагаемые с
выделением водорода и гидратов окисей NaOH, КОН, CsOH, еще менее изучены. В иных
случаях сомнительно даже самое их существование, а потому делать какие-либо
определенные выводы о проявлении в них металлическими элементами низшей
валентности по меньшей мере-преждевременно.
7*
ТОО
Периодическая система химических элементов
ОаХг и другой ряд постоянных солей типа GaX3 с нормальной для
III группы валентностью. В этой же -груше мы встречаем для металла
индия три типа солей InX, іпХг, іпХз, соответственно одно-, двух- и
трехатомному индию, причем, однако, опять-таки, соединения первых
двух ти'пов непостоянны и с водой легко переходят в 1пХ3 (например,
ЗіпС1 = іпС13 + 2іп). Наконец, для таллия известны типы Т1Х и Т1Х3,
но здесь уже первый тип отличается значительно большим
постоянством, чем второй, отвечающий трехатомному таллию.
В IV группе непостоянством валентности отличаются как элементы
четных, гак и нечетных рядов. В V—VII группах разнообразие типов
соединений и, следовательно, валентности достигает максимума.
Ванадий, молибден и вольфрам, быть может, -представляются наиболее ло-
учительными в этом отношении:
VO
v,o3
v2o.t
v2o5
VC12
VC13
VC14
VOC13
VF5
MoO
Mo203
Mo02
Mo205
M0O3
MoCl2
M0CI3
MoCl4
MoCl5
MoOCl4
MoF6
При этом интересно отметить, что повсюду в четных рядах наиболее
устойчивыми являются соединения высшего типа, следовательно,
такие, в которых данный элемент проявляет максимальную валентность
по отношению к элементам электроотрицательным, особенно к
галоидам и кислороду. Соединения, в которых элемент обладает низшей
валентностью, обыкновенно легко переходит в соединения высшего
предельного типа. Исключения представляют типические элементы N, О
и F, как известно, вообще .показывающие много уклонений от общих
правильностен, а отчасти также хром и марганец, для которого
наиболее устойчивы соединения типа МпХг *.
С другой стороны, в нечетных рядах периодической системы, с
повышением атомного веса элемента (в пределах данной группы),
устойчивость соединений с высшей валентностью падает, появляются и
делаются все более и более устойчивыми соединения низших типов. Мы
уже видели, что это имеет место во II и III группах. Так, для ртути
'появляется тип HgX; от галлия к талл'ию возрастает устойчивость типа
МеХ. То же самое замечается и в нечетных рядах IV группы; для Si
известен один только тип RX4. Оло-во образует два типа БпХг и SnX*,
довольно близких по степени устойчивости. Для свинца уже тип РЬХ4 **
отличается заметно меньшей устойчивостью, нежели тип РЬХ2. Особенно
это относится к соединениям с галоидами, ибо здесь соединения типа
РЪХі едва известны, а двухгалоидные соединения РЬС12, РЬВг2, РКЬ
отличаются большой устойчивостью.
Упомянутая -правильность еще сохраняет свою -силу в пределах
V группы, 'как видно, например, из следующих сопоставлений:
* Впрочем, и для этих двух элементов переход к высшим типам СгХз, СгОз, МпХз
и Мп207 происходит сравнительно легко, М.п(ОН)2, например, окисляется уже в
присутствии кислорода, воздуха.
** Мы имеем здесь в виду главным образом соединения с кислородом и
галоидами. 'Для -металлорганичвских соединений свинца дело обстоит как раз наоборот.
.'Устойчивые соединения здесь принадлежат как раз к типу RX*. например, РЬ(С2Н5)4.
Периодическая система химических элементов
101
Р203 малоустойчивое соединение Р205 весьма устойчивое соединение
As2Os устойчивое соединение, довольно As205 мало устойчивое соединение
легко окисляющееся
Sb203] устойчивые соединения, лишь с Sb205 мало устойчивое соединение
> трудом окисляющиеся
°4®з) Ві205 очень мало устойчивее соединение
РС13 окисляется на воздухе, легко при- РС15 довольно устойчивое соединение
соединяет хлор
AsCl3 \ труднее присоединяют хлор AsCl5 очень мало устойчивое соединение
> (особенно AsCl3)
3 * SbCl5 мало устойчивое соединение
ВіСіз устойчивое соединение. Не окисля- ВіС16 не существует
ется на воздухе и не присоединяет хлора
При .переходе к VI группе повышение атомного веса уже далеко
навсегда благоприятствует проявлению низшей валентности. Так,
например, в ряду галоидных соединений при переходе от серы .к селену и
теллуру прочность представителей высшего типа RX4: SCI4, SeCl4, TeCLj
заметно возрастает.
Наконец, в VII группе для галоидов наблюдается отношение,
совершенно обратное тіо сравнению с элементами предшествующих групп.
Здесь, вообще говоря, устойчивость соединений повышается при
переходе от низшего типа к высшему, а в пределах
данной группы такое относительное повышение устойчивости
увеличивается по мере возрастания атомного веса.
Так, среди кислородных соединений низшего типа наиболее
устойчива НСЮ и наименее HJO, СігО — взрывчатое тело. Вг^О и ЛгО вообще
неизвестны 9. С другой стороны, из высших кислородных соединений
наиболее устойчивы йодистые. Это особенно следует из сравнения
кислот НСЮз, HRr03 и HJ03, а также НС104 и Ш04.
Таким образом, в нечетных рядах периодической системы, по
большей части с повышением атомного веса, замечается преобладание нич
шей валентности над высшей и только VII и отчасти VI группа пред
ставляют обратное отношение.
В четных -рядах такой правильности не наблюдается, но зато здесь
почти всегда высший и, следовательно, нормальный для
данной группы тип отличается наибольшим
постоянством.
Необходимо иметь -в -виду, что все вышеприведенные соотношения
справедливы только для температур, не слишком далеких от
обыкновенной. При очень высоких температурах они часто подвергаются
весьма существенным изменениям. Бильц [7] считает -вообще валентность
функцией температуры и давления. Из своих опытов, сделанных с
окислами м-еталлоз, он приходит к заключению, что с повышением
температуры валентность данного металла по отношению к кислороду падает
(иными словами, уменьшается устойчивость высшего окисла,
увеличивается отвечающая ему дисеоциатхионная упругость кислорода). Так.
например, выше 1010° при давлении кислорода = 7ь атмосферы медь
уже неспособна функционировать в качестве двухвалентного
элемента, сохраняя, однако, одновалентность, а >выше 1670° становится как бы
аналогом элементов нулевой группы. Весьма наглядно эти отношения
выступают, если представить их графически, вычертив «изобары»
валентности.
В заключение этой главы, трактующей о связи между типами
соединений, образуемых элементами, и периодическим законом, уместно
102
Периодическая система химических элементов
коснуться еще вопроса об изоморфизме. Как уже было указано
в I главе, соединения, построенные по одному и тому же типу и
заключающие сходственные элементы, особенно наклонны к изоморфизму,
а потому мы вправе и по отношению к этому последнему ожидать
известной закономерной связи с периодическим законом. Такая связь на
самом деле существует. Лучше всего ее можно передать, воспроизведя
здесь таблицу, данную Ретгерсом[8] в 1895 г. В вертикальных
столбцах сопоставлены элементы, 'которые способны (с известными
ограничениями) замещать друг друга, атом за атом, в сходственных
соединениях, причем образуются вещества между собой изоморфные.
Достаточно бросить беглый взгляд на эту таблицу, чтобы бросилась в
глаза очень большая близость ее к периодической таблице элементов *.
Валентность
1
н
F
€1
Вг
J
і
Li
Na
К
Си
Rb
Ag
Cs
Au Hg
TI
0
Be
Mg
Ca
Zn
Sr
Cd
Ba
Pb
3
В
Al
Sc
Ga
Yt
In
La Ce,
Di Sa
Er Yb
4
С
Si
Ti
Ge
Zr
Sn-
3
N
P
V
As
Nb
Sb
Та
Bi
о
0
S
Cr Mo
Fe Co Ni
Se
Mo Ru Rh
Pd
Те
W Os Ir Pt
Th U
Глава IV
ПЕРИОДИЧЕСКИЙ ЗАКОН И ЭЛЕКТРОХИМИЧЕСКИЙ ХАРАКТЕР
ЭЛЕМЕНТОВ
Давно уже установилось подразделение всех известных элементов
на два основных разряда, или, вернее, ти>па: на тип металлов и на
тип металлоидов. И в свободном виде, как простые тела, и в виде
соединений металлы и металлоиды характеризуются совокупностью
строго определенных признаков.
Металлы хорошо проводят тепло и электричество, притом, как
'правило, тем лучше, чем ниже температура **. В соответствии с этим стоит
* В верхней строке указана валентность элементов в тех соединениях, для
которых наблюдается изоморфизм. Знак Di, очевидно, стоит для сокращения вместо
Рг и Nd.
" ** В самое последнее время Кеммерлинг-Оннес в Лейдене проследил
отношение некоторых металлов к электрическому току вплоть до самых низких
температур, которые могут быть ныне осуществлены при помощи жидкого гелия, кипящего
и пустоте. Электрическое сопротивление золота при температуре кипящего гелия
практически обращается в нуль. Для ртути были получены следующие результаты.
Некоторая нить жидкой ртути показывала при 273° абс. (0°С) сопротивление =172,7 ом.
Путем экстраполяции можно было вычислить, что сопротивление ее в твердом
состоянии при той же температуре (реализация соответствующего опыта, очевидно,
невозможна) было бы = 39,7 ом. Между тем, при 4,3° абс. сопротивление той же нити оказа-
Периодическая система химических элементов
10.4
их непрозрачность для света, и вообще для электромагнитных
колебаний, а вероятно, также и то обстоятельство, что молекула металлов,
в парообразном и растворенном состоянии, состоит из одного только
атома (отсутствие наклонности к полимеризации).
В отличие от металлов, металлоиды худо проводят тепло и
электричество (многие из них — типичные диэлектрики) іи прозрачны для света
Молекула их всегда состоит из нескольких атомов (N2, O2, 03, S8, P4,
As4 и .пр.). Они в большей степени, нежели металлы, наклонны к
образованию аллотропических модификаций * (углерод, бор, фосфор, сера,
кислород и пр.) и никогда не обладают, подобно металлам, в заметной
-степени такими свойствами, как тягучесть, ковкость и вязкость.
Различию в физических свойствах простых тел отвечает и различный
характер соединений, образуемых металлическими и металлоадными
элементами. Весьма .резко это сказывается прежде всего на
кислородных соединениях.
Окислы металлов имеют основной характер; с водой дают гидраты
окисей, представляющие основания.
Наоборот, окислы металлоидов являются кислотными
ангидридами и с водой дают кислоты. Чем типичнее металлоид, тем,
вообще говоря, более сильной кислоте он даег начало, тем в большей
степени эта последняя электролитически диссоциирована. Наоборот,
чем типичнее металл, тем сильнее соответствующее ему основание.
Галоидные соединения металлов имеют характер солей и
водой не разлагаются (NaCl, KCI). Галоидные соединения металлоидон
(РСЬ, РСіз) суть галоидангидрнды. Вода разлагает их нацело на НХ
(НС1) и на кислоту, отвечающую данному металлоиду.
лось равным 0,086 ом, т. е. 0,0022 величины, вычисленной для 273° абс. и для твердого
состояния. При дальнейшем понижении температуры на 1,3°, то есть при 3° абс, со-
1
противление падает до ЗХЮ-6 ом и, таким образом, составляет уже только ^аааапла
¦часть своей первоначальной величины. Это ничтожно малое сопротивление затем не
претерпевает существенного изменения при дальнейшем падении температуры до
1,5° абс. По-видимому, на кривой, выражающей зависимость сопротивления Hg от
температуры, имеется перелом между температурой плавления твердого водорода- и точ
кой кипения гелия.
* В типичных случаях аллотропии свойства отдельных модификаций, физические
к хиіміючеокие, резко отличаются между собой. Переходы этих модификаций друг в
друга сопровождаются резким тепловым эффектом. Ярким примером этого может
служить случай кислорода О2 и озона 03. В газообразном состоянии озон в Vfa раза
плотнее «молекулярного» кислорода. В жидком состоянии озон темно-синего (почти
черного), кислород—бледно-голубого цвета. Спектры поглощения резко различны. Озон
выделяет иод из йодистого калия (2Ю-Ь0з+Н2О= т24-2КОН+02), превращает PbS
в PbSO*, индиго в изатин и вызывает множество других процессов окисления, которые
или вовсе не совершаются или совершаются крайне медленно при действии
обыкновенного кислорода. При образовании 1 г-моль (48 г) озона из кислорода поглощается
34,5 ккал [7].
Металлы почти никогда не показывают столь резко выраженной аллотропии.
(Олово составляет одно из редких исключений.) Известные для них модификации
встречаются почти всегда у металлов, мало типичных, и обычно лишь в незначительной
степени отличаются друг от друга, особенно по химическим свойствам. Теплотный эффект
перехода одной модификации в другую вообще невелик, иногда ничтожен. Типичным
примером могут служить две модификации железа, известные под именем а и р и
отличающиеся друг от друга почти исключительно по своим магнитным свойствам. Feu,
постоянное ниже 770°, сильно магнитно, железо Р, устойчивое между 770—900°, лишено
магнитных свойств. Несколько больше отличается от предыдущих модификаций третья
постоянная выше 900°, так называемое у-железо. Оно лишено магнитных свойств,
подобно р-модификации, но образование его сопровождается заметным сжатием. Кроме
того, при образовании его из р электрическое сопротивление перестает возрастать с*
температурой.
104 Периодическая система химических элементов
Металлы менее типичные и в зтам отношении занимают середину
между типичнейшими металлами и металлоидами. Таковы, например,
Си, Zn, Fe и прочие. Соли их подвергаются частичному гидролизу -и
обнаруживают наклонность к образованию -основных солей, а также
комплексных соединений и гидратов. Не типичным 'металлам отвечают и
здесь не типичные металлоиды, например В, С, Si. Они также дают
начало солям, наклонным к частичному гидролизу, легко переходящим в
кислые и .комплексные -соли.
В соответствии с только что сказанным стоит и не одинаковое
электрохимическое отношение 'металлов и металлоидов, в свое
время положенное Берцелиусом в основу его знаменитой
электрохимической теории.
Металлы имеют характер по преимуществу электроположительный,
металлоиды — электроотрицательный. Между тем как атомы металлов
образуют катионы (Na4, К.ч% Са++, Zn+", Fe+ + + и т. д.) атомы
металлоидов, или сами но -себе или в комбинации -с другими элементами
(особенно с кислородом), лают начало анионам (С1~, Вг~, С104", S04~~,
Р04 , ASO4 и пр.). Поэто-му электролиз солей .всегда происходит
так, что на катоде (первично) выделяется металлический, а на аноде —
металлоидный ион *.
С другой стороны, чем типичнее -металл, тем, вообще говоря, он
менее «благороден», то есть тем легче он окисляется на воздухе,
разлагает воду, вообще подвергается действию металлоидов и их соединений.
Еще Вольта нашел, что если из па,ры металлов составить
гальванический элемент, то более благородный из них всегда становится анодом,
менее -благородный — катодом. Так возник электрохимический ряд
напряжения:
К Mg Zn Cd Fe Sn Pb Cu Hg Pt Au
В этом ряду каждый последующий член становится анодом по
отношению к одному из предыдущих и, кроме того, способен вытесняться
этим последним из его солей**. Так, например, медь вытесняется
цинком, а сама, в свою очередь, вытесняет серебро.
CuS04 + Zn = ZnS04 + Cu.
2AgN03-rCu(N03)2-|-2Ag.
Эти отношения яснее всего выражаются при помощи представления
об электросродстве, примененного к химической систематике Абеггом
и Бодлендером. Электросродство — это сродство атома к
электрическому заряду — положительному, если образует катион;
отрицательному, если дело идет об образовании аниона. При этом ион
рассматривается как нечто вроде соединения атома с электричеством.
С точки зрения электронной теории (Томсо-н и другие) отрицательные
ионы (анионы) образуются из атамо.в (или атомных групп) •через
присоединение одного или нескольких электронов. Сила, удерживающая в
этом случае электрон, и будет мерой электросродства. Положительные
ионы, согласно той же теории, образуются через потерю данным
атомом электронов. Поэтому атомы, их образующие, обладают
(минимальным электросродством и притом тем меньшим, чем легче они переходят
* Исключение составляют такие сложные ионы, как аммоний NH4+, по характеру
своему играющие роль металла.
** Между положением металлов в ряду напряжения и характером гидратов их
окисей нет, однако, полного параллелизма.
Периодическая система химических элементов 105*
в состояние ионов. Поэтому, например, у атома натрия электросрадство
очень мало, то-гда как, например, оно весьма значительно у хлора.
Если признавать существование положительных электронов, то
пришлось бы сказать, что у -натрля существует весьма .большое сродство
к положительному электрону, подобно тому как у хлора—к
отрицательному. В этом смысле можно говорить о положительном и
отрицательном электросродстве 10.
Мерой электросродства может служить величина потенциала
разложения или тесно связанная с ней величина электролитической
упругости растворения, отвечающая данному иону. Можно представить себе
вместе с Нернстом, что каждое вещество, растворяющееся в
жидкости, обладает определенным давлением, или упругостью
растворения (аналогичным упругости насыщенного пара жидкостей).
Давление ---это есть та сила, под влиянием которой частацы
растворимого тела досылаются в пространство, подобно тому, как всякая
летучая жидкость насыщает пространство, развивая определенную
максимальную упругость пара. Ясно, что всякое вещество будет растворяться
в данной жидкости лишь до тех пор, пока -парциальное осмотическое
давление его не достигнет величины упругости растворения. С этой
точки зрения, можно говорить и об электрической упругости
растворения как о силе, с которой атомы данного металла
изгоняются из металлического электрода в раствор, переходя одновременно
с этим в состояние ионов. Наперекор этой силе, назовем ее через Рг
очевидно, должно действовать осмотическое давление р данного рода
ионов, которые уже содержатся в растворе.
Здесь, очевидно, могут быть три случая: или 1) Р—ру или 2) Р>рг
или 3) Р<р.
В первом случае не будет тенденции ни к поступлению ионов из
металла в раствор, ни к осаждению металла из раствора. Электрод
останется электрически нейтральным. Что должно произойти во втором и з
третьем случае, легче всего будет понять, если стать на точку зрения
электронной теории.
Если Р>р> то, очевидно, будет существовать сила (не
уравновешенная осмотическим давлением), заставляющая нейтральные атомы
металла переходить в раствор в виде ионов (катионов). При этом
каждый атом металла теряет один или несколько электронов, а эти
последние, оставаясь на электроде, сообщают ему отрицательный заряд.
Между металлическим катодом и раствором возникает разница
потенциала соответствующего знака *.
Если, наоборот, Р<р> то будет иметь место тенденция к переходу
катионов в нейтральное состояние, к превращению их в атомы металла,
который и будет осаждаться на катоде.
При этом, очевидно, каждый .катион (например Na+, Zn++) будет
соединяться с электронами, заимствуя их из 'металлического электрода и
потому этот 'последний будет получать положительный заряд.
Между электродам и раствором и здесь также должна возникнуть
некоторая разница потенциалов, но обратного знака по сравнению с
предыдущим случаем.
Разность потенциалов между металлическими электродами и
раствором электролита пропорциональна работе, .которую можно .получить
в одном случае, которую надо затратить в другом, когда происходит
перенос электрического заряда, связанного с одним эквивалентом,—
* И вместе с тем так называемый «двойной электрической слой», приостанавливаю*
щнй, выделение металла в самом начале. См. выноску на стр. 106.
106
Периодическая система химических элементов
с электрода на .раствор электролита, другими словами, когда возникает
или порывается связь между атомом и электроном. Неряст показал, что
эта величина ?, .которая может быть определена из опыта
(концентрационные элементы), находится в следующем простом отношении с
величинами Р и р *:
Е = -у- lg ¦— .
In ^ р
В этом уравнении, которое выводится с помощью термодинамики,
R — газовая константа, Т — абсолютная температура, /=96540 кул и
п—валентность данного иона.
Если электродные потенциалы условиться относить к растворам
определенной, например нормальной, как это обыкновенно делают,
концентрации соответствующих ионов, то можно (с некоторыми, впрочем,
оговорками) прямо считать их мерой электросродства.
То, что было сказано о металлических ионах, относится mutatis
mutandis ** и к ионам металлоидным. И в этом случае, если Р>р, то
нейтральные атомы имеют тенденцию к переходу в состояние ионов.
Только ионы эти образуются из атомов через присоединение электронов, а
потому соответствующий электрод лишается части своих электронов и
заряжается 'положительно. Между электродом и раствором,
содержащим ионы, -возникает разница потенциалов, обратная по знаку
сравнительно с той, которая возникает в соответствующих случаях (Р>р)
между металлом и раствором его соли.
Понятно также, что обратные отношения должны иметь место,
когда Р>р.
В нижеследующей таблице приведены электродные потенциалы для
некоторых катионов и анионов, взятых в нормальном растворе.
Величины их выражены в вольтах, причем в скобки заключены те из них
(менее достоверные), которые не могли быть определены -прямым -путем,
а найдены посредством вычисления.
К
Na
Ва
Sr
Са
Mg
А1
Мп
Zn
Cd
(+2,9)
(+2,54)
(+2,54)
(+2,49)
(+2,28)
(+2,26)
+1,00
+0,80
+0,493
+0,143
Tl
Со
Ni
Sn (двухатомное)
Pb
н
Си (двухатомная)
As
Ві
Sb
+0,045
-0,045
—0,049
—0,08?
—0,129
—0,277
—0,61
—0,62?
—0,67?
—0,74?
Ag
Pd
Pt
Au
J
Br
О
CI
S04
F
-1,048
-1,07?
-1,14?
—1,35
-0,797
—1,270
—1,396
-1,694
-1,9
—2,24
Fe (двухатомное) +0,06 Hg (двухатомная) —1,03
Знак + указывает на то, что при соприкосновении раствора с
электродом первый электризуется положительно, а второй— отрицательно.
Знак — указывает на противоположные отношения.
Электродные потенциалы часто называются 'потенциалами
.разложения, так как, вычитая из потенциала катиона А потенциал аниона В, мы
как раз получим .величину электродвижущей силы, необходимой для
разложения электролита АВ, другими словами, для того, чтобы
каждый из ионов А и В перевести в нейтральное состояние. Так, например,
* Все эти рассуждения справедливы только для обратимых электродов (см.,
например, [10]).
**
Внося соответствующие изменения (лат.). Прим. ред.
Периодическая система химических элементов
107
для электролиза хлористого цинка необходима электродвижущая
сила = 0,493—(—1,694) =2,20 в\ для электролита СиС12—0,61 —
— (—1,694) = 1,09 в и т. д.
Понятие об электросродстве явилось чрезвычайно важным опорным
пунктом для химической систематики, главным образом потому, что с
ним мог быть связан ряд других свойств электролитов и прежде всего
растворимость, степень электролитической
диссоциации и спосо бность -к о 6d а зов а н и ю комплексных
соединений.
Связь между электросродством и растворимостью станет ясной из
следующих соображений, которые мы заимствуем из статьи Абегга и
Бодлендера: «Образование ионов есть следствие большего стремления
атомов и атомных групп соединяться с электрическими зарядами, чем
между собою. Количество'находящихся в растворе ионов растет
непрерывно с увеличением .концентрации; граница увеличения этого
количества .находится 'при концентрации, при которой раствор насыщен. Мы
можем предполагать, что граница эта лежит тем выше, чем больше
стремление частей электролита превращаться в ионы. Если одна
из составных частей имеет очень большое стремление
образовать и о н ы , то она принудит к этому и другую часть, хотя
бы сама по себе эта последняя обладала незначительным
электросродством. В ряду солей с общим компонентом растворимость будет
увеличиваться с возрастанием стремления ,к ионизации переменного
компонента. На самом деле 'мы находим, что эти предположения
подтверждаются во многих случаях. Так, Бодлендер показал, что в ряду
йодистых, бромистых, хлористых, сернистых металлов и их водных окисей
растворимость увеличивается вместе с ^лектро-сродством, измеренным
через потенциал разложения».
Еще легче понять, что должна существовать зависимость между
электросродством и степенью диссоциации электролитов. Впрочем,
необходимо заметить, что зависимость эта не гак «проста, как может
казаться с первого взгляда, ибо способность электролитов переходить в
состояние ионов, с одной стороны, определяется «сродством» этих
последних к электрическому заряду, к электрону, но, с другой, зависит
также от степени прочности связи между компонентами молекулы,
способными к ионизации, друга ми словами, от свободной энергии
образования недиссоциированных молекул. Этим объясняется, например, то
обстоятельство, что несмотря на явное падение (отрицательного)
электросродства галоидов от хлора к иоду, способность соответствующих
водородистых соединений (НС1, ИВг, HJ) к электролитической
диссоциации приблизительно одинакова. Можно думать, что ослабление
электросродства здесь компенсируется увеличивающейся в том же
порядке (от С1Н к JH) легкостью распада частиц НХ (ом. ниже) на овои
составляющие.
Наконец, те же теоретические представления позволяют предвидеть
уже известный из предыдущего факт, что незначительное
электросродство хотя бы одного из ионов, входящих в состав данной молекулы,
благоприятствует образованию из .нее комплексных соединений.
Отсылая за подробностями к оригинальным статьям Абегга и Бодлендера
[11], ограничимся здесь указаниями на одно замечательное сближение.
Если электросродство, представляющее primum movens * к переходу
частей молекулы в состояние ионов, можно рассматривать как сродство
атома (или группы атомов) к электрону, то величина его ceteris рагі-
* Первое движущее начало (лат.). Прим. ред.
108
Периодическая система химических элементов
bus* должна увеличиваться с объемом атома (шги группы).
Действительно, оказывается, что как раз элементы с большим объемом атома
(см. главу VI) (щелочные металлы с одной стороны, галоиды —с
другой) являются энергичными иоиообразователями и обладают
значительным электросродством. Наоборот, элементы с малым атомным объемом
(например металлы VIII гругшы) лишь в ничтожной степени способны
сами 'по себе образовать ионы, электросродство -их весьма
незначительно. Но как раз такие элементы обладают повышенной способностью к
образованию комплексных соединений. При этом они весьма чаете
переходят в сильно изо-низирован-ные молекулы, привлекая к себе новые
молекулы, насыщенные и способные к самостоятельному
существованию, В результате из атома с .малым объемом образуются атомные
группы, обладающие, вообще говоря, большим объемом -и в этом
обстоятельстве .можно видеть одну из основных причин увеличения
электросродства, а следовательно, и повышения способностей всей молекулы
к .ионизации. Так, например, неспособная к ион-изаци-и молекула PtCb
привлекает к себе 4ЫНз или 2КС1 и дает соединения і[Р14ЫНз]С12
и [PtCl4]K2, молекулы которых сильно диссоциированы с образованием
комплексных ионов: Pt4NH3++ и (2С1~), PtCl4 и (2К+). Здесь дело
происходит так, как будто бы тенденция атома к переходу в иониздрс •
ванное состояние представляла побудительную причину происходяіш
го усложнения простого элементарного атома до иона комплексного.
Кроме '.приведенных выше отличий -металлов от металлоидов, ести
еще одно, весьма характерное. Оно заключается в неодинаковом
отношении тех и других к водороду. Металлоиды образуют вполне
определенные водородистые соединения, газообразные при обыкновенной
температуре или (НгО, HF) летучие жидкости. Наоборот, гидрюры
.металлов там, где они существуют, сравнительно мало характерны и по
большей части нелетучи без разложения (см. выше главу III).
В периодической системе -можно заметить три направления, по
которым совершается постепенное и правильное изменение
металлического характера элементов в металлоидный, электроположительного в
электроотрицательный.
1. Если в каждом ряду периодической системы мы будем
двигаться слева направо, то есть от I группы к VII**, то в начале строки -мы
встретимся с типичным металлом, в конце ее — со столь же типичным
металлоидам, а между ними, вообще говоря, найдем элементы с
промежуточными свойствами. Для иллюстрации прежде всего опять
воспользуемся третьим рядом.
Na Mg Al Si Р S CI
NaOH Mg(OH)2 Al(OH)3 Si (OH)4 H3P04 H2S04 HCIO,
Как простые тела, первые три члена этого ряда Na, Mg и Лі
представляют из себя металлы, 'последние четыре (Si, Р, S, СЛ) нельзя
не отнести к металлоидам. Весьма резко .постепенность в изменении
свойств сказывается на характере ооединений этого ряда элементов.
Окислы и их гидраты. Гидрат окиси натрия NaOH есть
типичное и весьма сильное основание, легко растворимое в воде и в растворе
почти сполна диссоциированное на ионы: Na^ и ОН". Гидрат окиси
магния Mg(OH)2 — основание более слабое,— в воде мало раствори-
* Пр,и прочих разных условиях (лат.). Прим. ред.
** Восьмую и нулевую прушту мы здесь не рассматриваем. Этим двум грузинам
ниже посвящена отдельная глава (IX).
Периодическая система химических элементов
№
мое. Такой раствор, однако, еще окрашивает лакмус в син-ий цвет. При
диссоциации Mg(OH)2 дает ионы Mg+f и 20Н~. Еще -более слабое
основание представляет гидрат окиси алюминия А1(ОН)3, -практически
нерастворимый в воде и не изменяющий окраски лакмуса. Ионы А1+++
и ОН~ могут существовать одновременно лишь в лсчезающе малой
концентрации.
Если гидрат окиси алюминия представляет из себя весьма слабое
основание, то ой же обладает и свойствами очень слабой ік и с
л о ты. Между тем как гидраты окисей натрия и магния образуют соли
только с кислотами (например, NaCl, NaN03, Na2S04, MgCl2, MgS04
¦и пр.), Аі(ОН)з, с одной стороны, образует соли с кислотами
(например, А1С13, Ala(S04)3, проявляя при этом стойства основания, а с другой
стороны, функционирует, как алюминиевая кислота, или
/° /0Н
А1С ^А1^-ОН-Н20,
ЮН \0Н
ибо растворяется в щелочах и даег с ними соли, алюминаты (КАЮ2,
NaA102). Таким образом, гидрат окиси алюминия может образовать
двоякого рода ионы: катионы А1+ + + и анионы А102~, то есть является
представителем класса так называемых амфотерных электро-
л ит о в.
Это обстоятельство характеризует алюминий, как элемент
переходный от металлов к металлоидам.
Если мы пойдем дальше, то начиная с гидрата кремнезема (SiOj)
и кончая гидратом высшего окисла хлора (С1207), будем уже
встречать вещества с исключительным характером кислот, причем сила этих
кислот будет постепенно возрастать. Кремневая кислота Si(OH)4,
H2Si03 принадлежит к числу весьма слабых, ортофосфорная — к числу
средних по силе, хлорная — к числу наиболее сильных кислот, какие
только известны.
Таким образом, от гидрата окиси натрия к хлорной кислоте мы
имеем прогрессивное ослабление основных и затем нарастание кислотных
свойств.
Галоидные соединения. Те же самые отношения резко
проявляются на природе галоидных соединений рассматриваемого ряда
элементов. Сильному основанию — гидрату окиси натрия
соответствует хлористый натрий — NaCl, соль, растворимая в воде и при этом
не изменяющаяся (не подвергающаяся гидролизу), а потому и
показывающая нейтральную реакцию (например на лакмус). Хлористый
магний MgCl2 сохраняет еще характерные черты соли, но водный раствор
его уже частью подвергнут гидролизу (с образованием основных солей),
иапри-мер,
/ОН
MgCl, f HoO-Mg +HC1 или Mgh4-2Cr i-H^OH"-
Чі
/ОН
= Mg f-H^-СГ
Чі
Известно, что водный раствор MgCU не может быть вышрен без раз
ло<же>*ия, ибо лри этом выделяется часть НО, а остаток всегда
содержит примесь основной соли.
110
Периодическая система химических элементов
Хлористый алюминий представляет уже соль, весьма -сильно гидро-
лизованиую. Водный раствор его окрашивает синий лакмус в ясно
красный цвет. Наконец, галоидные соединения последних членов нашего
ряда представляют типичные галоидангидриды кислот, легко и нацело
разлагаемые водой (например, РСі5 + 4Н20 = НзР04 + 5НС1).
Общий вывод из рассмотрения всего ряда можно формулировать
еще и таким образо-м, что положительное электросродство, способность
удерживать + заряд постепенно падает слева направо от Na к А1;
затем появляется постепенно возрастающее стремление удерживать
отрицательный заряд.
Следующие (приблизительные) значения потенциалов разложения
вполне подтверждают только что сказанное:
Na Mg Лі Сі
г2,54 , 2,26 - 1,00 -1,694
С точки зрения теории А бег га и Бодлендера, весьма
поучительно также сравнение водородистых соединений рассматриваемого
пам.н ряда
SiH4 РН3 SH2 C1H.
В этом ряду тенденция элементарного атома переходить в состояние
аниона, другими словами, стремление атома к .соединению с
электроном, явно возрастает от водородистого кремния к хлористому
водороду. На одном конце ряда мы имеем нейтральное тело SirHU с едва
заметными зачатками кислотных свойств, которые проявляются в
существовании силицидов*, (Mg2Si). Затем идет РН3, кислотные свойства
которого зее еще едва заметны. Но зато следующий член ряда
—сероводород— ясная, хотя и очень слабая кислота. Наконец, хлористый
водород принадлежит к числу типичнейших и сильнейших кислот:
H2S НС1
Константа электролитической диссоциации при 25° . . . Г>.7> 10~8 4,77
То, что сейчас было выяснено для третьего ряда, в общем
подтверждается и для других рядов, ка-к -в этом легко убедиться при
внимательном просмотре соответствующих мест таблицы.
Так, во 2-м (типичном) ряду
Li, Be, В, С, N, О, F
литий — элемент с ясно выраженными металлическими свойствами.
аналог натрия. Гидрат окиси его LiOH — типичная щелочь. Соли с
сильными кислотами (LiCl, LiN03) в водном растворе почти -не гидр о-
лизованы и т. д. У бериллия металлические свойства сильно понижены.
Ве(ОИЬ — слабое основание. Соли бериллия сильно гидролизованы.
В боре уже преобладают металлоидные свойства (две
модификации— кристаллический и «аморфный»- бор); гидрат окиси В(ОН)з—
слабая кислота, но еще заметны следы металлических. У следущего
члена — углерода ясно заіметны только черты металлоида. Всем
известны три аллотропических модификации этого элемента: алмаз, графит
и аморфный уголь, резко .различающиеся друг от друга по физическим
свойствам.
* Силициды (и фосфиды) типичных металлов разлагаются водой и кислотами.
Разложение магниевого соединения применяется для получения SiH«.
Периодическая система химических элементов
111
Удельный вес . . . .
Теплота горения
1 г. атома . . . .
Электрическое
сопротивление (удельное)
3,51
94,32
1(120
2.25
94.82
103
Аморфным
уголь
1.8
97,15") кки.і
Г), 1 < і»
Молекула последних двух модификаций отличается большой
сложностью. Графит и аЛіМаз при окислении дают меллитовую кислоту,
соон
НООС-С^ХС~СООН
НООС—Сч /С—СООН
с
і
соон
откуда следует, что в молекуле графита и угля содержится не менее
12 атомов углерода, и притом в своеобразной .комбинации,
свойственной ароматическим углеводородам. По :всей вероятности, процесс
искусственного «обугливания», приводящий к образованию аморфного
угля из органических веществ, имеет много общего с тем, который имел
место при образовании в природе каменных углей -и антрацитов. Угли
'^ти можно, до известной степени, рассматривать как «недоделанный^
аморфный уголь. Давно уже известно и установлено многими
анализами, что каменный уголь содержит, кроме углерода, еще другие
элементы, особенно Н, О, N и S. Несомненно, что они главнейшим образом
содержат углеводороды высокой степени уплотнения, богатые
углеродом и 'бедные водородом. В самое последнее время А. Пикте [12] в
Женеве удалось выделить ів чистом виде один из таких углеводородов,
извлекая-каменный уголь бензолом и перегоняя под сильно
уменьшенным давлением. Углеводород этот оказался гексагидрофлуореном
СізНіб и отвечает одной из следующих структурных формул:
Н.>С
/СН2^
су Чн
нх
сн2/ чсн2/ \сн2/
хн
или
сн.
/СН2ч
нх- чс
г
сн.
Н,СЧ /Сч /Сч ХН
¦ чсн2/ \сн2/ \сн^
В резко выраженной способности углерода к полимеризации, с
одной, стороны, и к образованию устойчивых комплексов со строго
определенной структурой, с другой, лежит причина огромного
числа и лочти безграничного разнообразия соединений, образуемых
М2
Периодическая система химических элементов
этим элементом (органических). Эга особенность углерода находится
в теснейшей связи с его положением в .периодической системе. Он
принадлежит кэлементам переходным11, то есть к таким, в которых
не развиты с резкостью ни металлические (в данном случае
отсутствующие), ни металлоидные свойства. Поэтому углерод, вступая в
соединение с другими элементами, весьма мало склонен к образованию
ионО'В, а потому реакция между органическими 'Соединениями вообще
протекает крайне медленно, а последним обстоятельством, в свою
очередь, объясняется возможность существования большого числа ноли-
меризованных и изомерных форм, подолгу сохраняющих свою
индивидуальность и не лереходящих друг в друга.
Металлоидный характер углерода особенно резко сказывается на
электрохимической природе его соединений. С водородом он дает тела
или с индсферентными (СН*, СНз — СНз) или со слабо кислыми
свойствами (ацетилен СН=СН и его гомологи). Высшее кислородное
соединение С02 является ангидридом угольной кислоты Н2СОз (весьма
слабой и дающей щелочные соли, сильно гидролизованные, например
Na2C03). Ряд соединений со свойствами кислот получается далее при
/°
наличности в частице группы —С\ (карбоксила), иначе говоря,
хОН
когда водный остаток связан с окисленным углеродным атомом.
Таковы кислоты уксусная СН^СООН, щавелевая СООН—СООН,
янтарная СООСН2—СН2—СООН « т. д.
Последние три члена ряда N, О и F являются типичнейшими
металлоидами. Соответствующие простые тела газообразны при
обыкновенной температуре. Для кислорода известны две (а по последним данным,
даже три) модификации: «молекулярный» кислород 02 и озон Оз.
Усиление металлоидных свойств и «сродства» <к отрицательному заряду от
N к F, между прочим вытекает из сравнения водородистых соединений:
NH3 OH2 HF.
В аммиаке и гидраз-ине кислотные свойства* сказываются только в
виде намеков. Водород аммиака может быть замещен некоторыми
металлами, но получающиеся при этом соединения, нитриды (амид
натрия, нитрид кальция, Ca3N2, магния Mg3N и 'прочие) разлагаются уже
водой. В воде, представляющей -из себя типичный амфотерный
электролит, кислотные свойства хотя также очень сла<бо выражены, но все же
резче выступают, нежели в аммиаке (сказываясь в существовании
таких соединений, как КзО, Na20, NaOH, КОН и прочих). Наконец,
кислотные свойства фтористого водорода выражены уже вполне
определенно.
Отношения, в общем подобные только что рассмотренным, встретим
мы и в 4-м ряду, который начинается калием и кончается марганцем:
К Са Sc Ті V Сг Мп.
Как принадлежащие к четному ряду (см. ниже), элементы эти все,
в большей или меньшей степени, отличаются металлическим
характером. Но это общее свойство их наиболее резко .выражено у крайнего
* Об основных свойствах аммиака мы здесь не говорим, так как они, собственно,
принадлежат не NH3, как таковому, а продукту его гидратации NH<OH и соответствую-
'цнм солям, в которых содержится комплексный ион NH4 + .
Периодическая система химических элементов
113
члена слева— калия, который не только в свободном состояния
представляет типичный металл, но и дает сильную щелочь КОН, соли с
сильными кислотами (КО, KN03), не разлагаемые водой и т. д. Эти
металлические свойства постепенно слабеют при переходе к Са и Sc.
Наоборот, металлоидный характер, с ясностью появляющийся у титана,
постепенно усиливается к марганцу. Это обстоятельство ясно видно лри
сличении гидратов соответствующих окислов. От титана до марганца
они представляют свойства кислот:
Ті (ОН), Н2СЮ4 Н2Мп04
HV03 Н2Сг207 НМп04
Но тогда как титановая кислота принадлежит -к слабейшим,
марганцовая НМп04 примыкает к наиболее сильным (к соляной, азотной
и т. п.), а ванадиевая и хромовая по своей силе занимают середину
(причем хромовая явно сильнее ванадиевой).
Аналогичные переходы наблюдаются в 5-м ряду (от Си к Se и Вг),
в 6-м (от Rb к Мо), в 7-м (от Ag к J) и т. д. В последнем—12-м
ряду с открытием радия ясно -видна та же законность, ибо радий —
типичный щелочноземельный металл, примыкающий к барию, а уран
дает, с одной стороны, соли, отвечающие слабым основаниям и(ОН)4
и U02(OH)o, а с другой,— соли полиурановых кислот, например
н2и*о7.
Заметим, однако, что если з качестве критерия металлического и
металлоидного характера элементов остановиться на электросродстве,
то от вышеприведенного 'простого правила сказываются некоторые
уклонения, характер которых ясен из приводимой ниже диаграммы Абег-
га (см. стр. 117).
2. Обратимся теперь ко второму направлению, в котором
также совершается закономерное изменение электрохимического
характера элементов в системе. Это— вертикальное направление
в пределах каждой подгруппы, тф-ичем элементы главных .и
побочных подгрупп надлежит сравнивать между собой отдельно.
По мере того, как в пределах каждой из главных
подгрупп возрастают атомные веса (отдельно в четных и
нечетных рядах), понижается металлоидный и повышается
металлический характер соответствующих элементов.
Чтобы иллюстрировать эту правильность, мы опять остановимся на
одном конкретном примере — на элементах V группы, принадлежащих
к нечетным рядам: Р, As, Sb и Ві. В самом деле, уже при сравнении
свойств простых тел, образуемых этими элементами, мы не можем не
заметить, что висмут по внешнему виду, по хорошей электро- и
теплопроводности обнаруживает характерные признаки металлического
состояния, чего нельзя сказать о фосфоре, особенно его желтой
модификации. Мышьяк и сурьма занимают промежуточное положение, с яв-
ньш, однако, возрастанием металличности по направлению от As к Ві.
Как известно, для мышьяка хотя и существует металлоидная желтая
модификация, аналогичная желтому фосфору (и, -подобно этому
последнему, не проводящая тока и растворимая в CS2), но она весьма
мало прочна, сохраняется без изменения только лри низкой
температуре в отсутствии света и легко переходит в другие модификации, из
которых .наиболее устойчива металлическая. Еще менее устойчива
металлоидная модификация (также желтая) для сурьмы. Для висмута она
вообще неизвестна.
Молекула фосфора в парообразном состоянии состоит из четырех
атомов <?4) и лишь с трудом подвергается упрощению (диссоциации)
3 Л. А. Чугаев, т. Ш
114
Периодическая система химических элементов
при высокой температуре; точно так же и молекула мышьяка (As4); но
п-ри 'переходе к сурьме и к висмуту устойчивость сложной .молекулы
падает.
В кислородных соединениях фосфора сказываются ясно
кислые свойства. Эти свойства слабеют при переходе к сурьме; у
висмута они почти исчезают. Мышьяковая кислота H3As04 слабее фосфорной;
мышьяковистая НзАэОз слабее фосфористой Н3РО3. Кислородные
кислоты сурьмы H3vSb04, НБЬОз, H4Sb207 принадлежат к числу слабейших.
Щелочные соли их гидролизованы в очень сильной степени.
Галоидные соединения фосфора суть хлорангидр-иды кислот,
нацело разлагаемые водой. Таковы РС13, РВгз и прочие. Галоидные
соединения висмута суть соли слабого основания Ві(ОН)3. Мышьяк и
сурьма и в данном случае занимают среднее положение, причем у
галоидных соединений сурьмы, например SbCl3, солевой характер
сказывается заметно резче, нежели у соответствующих соединений мышьяка
(AsCl3) •
Водородистые соединения этой группы элементов
(газообразные) также обличают постепенное нарастание металлического
характера от фосфора к висмуту, ибо устойчивость их пад-ает по мере
возрастания атомного веса. Водородистое соединение висмута 12 вообще
неизвестно. Весьма ясно это отражается на термических данных —
теплотах образования водородистых соединений, которые *мы приводим
в ккал.
NH3 РН3 AsH3 SbH3
+ 12,2 +4,3 —36,7—81,8
Аммиак и фосфористый водород — соединения экзотермичные,
хотя и образуются с небольшим выделением тепла.
AsH3 и особенно SbH3 — соединения с резко выраженным
эндотермическим характером.
В качестве другого примера остановимся на VII группе периодичен
ской системы. Легко видеть, что хотя металлоидный характер и
выражен в достаточно резкой степени у всех галоидов
F CI Br J
однако у иода, как простого тела, уже появляются в зародыше
металлические свойства, проявляющиеся и в металлическом блеске и в
заметной электропроводности. ,
Постепенное падение металлоидных свойств от F к J сказывается
и на сравнительной прочности молекул F2, С12, Вг2 и J2.
Наименее устойчивой к жару оказывается молекула иода. При
температуре около 1500° молекул J2 практически уже не существует: они
распадаются на атомы J (В. Мейер). Между тем из молекул хлора при
той же температуре, по Пиру, диссоциировано менее 1%, и даже при
2337°^-только 50%. Бром, по опытам В. Мейера, занимает среднее
положение между иодом и хлором, а диссоциация фтора пока вообще не
наблюдалась. Таким образом, от фтора к иоду прочность молекулы Х2
падает: молекула X, состоящая из одного атома, что как раз типично
для металлов, становится все более и более устойчивой.
В том же самом направлении постепенно изменяется и степень
прочности водородистых соединений.
Фтор с водородом соединяется нацело, со гвзрывом даже при низкой
температуре и в темноте; хлор только на свету или же при нагревании;
Периодическая система химических элементов 19 Г»
бром и иод — только при нагревании или при действии катализаторов,
медленно, без взрыва, и притом не нацело, ибо здесь реакция явно
ограничена пределом.
В соответствии с этим, реакция образования HF сильно экзотермич-
на, а затем тепло образования НХ постепенно падает к HJ, для
которого оно близко к нулю.
Приводим теплоты образования галоидоводородо-в в ккал:
HF HC1 HBr HJ
+38,9 -\-22 -1-12 из газообразного брома -J-4 из газообразного иода
— — +8,4 из жидкого брома —4 из твердого иода
Таким образом, и в этом отношении типичнейшим металлоидом
является фтор, наименее типичным — иод.
Далее в ряду элементов: фтор, хлор, бром, иод каждый
последующий вытесняет предыдущий из соответствующего водородистого
соединения НХ 'или из соответствующей соли, например:
2KJ + CU = 2KCI + Ja
В тесной связи с этим стоит и правильное изменение
электросродства
F CI Br J
Потенциалы разложения . . . —2,24 — 1,694 —1,27 —0,797,
направление которого указывает на то, что от фтора к иоду падает
сродство атома галоида к электрону.
Совершенно аналогичное постепенное изменение свойств оды
наблюдаем в VI группе для серии
О S Se Те
Особенно ясно выступает здесь падение степени устойчивости
водородистых соединений, как это, между прочим, вытекает из следующих
термических данных:
Тепло образования водородистого Н20 H2S H2Se H2Te
соединения, ккал +68,4* +4,7 —4,8 —35
Затем сила кислот типа H2RO4, образуемых тремя последними
элементами данной группы, весьма заметно падает от серной кислоты к
теллуровой.
H2S04 принадлежит к числу сильных кислот. Соли ее с металлами
щелочей и щелочных земель (K2SO4, Na2S04, BaS04) не показывают
гидролиза. Наоборот, теллуровая кислота Н2Те04 относится к числу
слабых и соответственные соли ее подвергнуты сильному гидролизу.
В первых группах периодической системы для типичных металлов
(особенно в четных рядах) мы замечаем совершенно аналогичные
соотношения. Укажем на ясное возрастание металлических свойств с
повышением атомного веса в сериях: К, Rb, Cs и Са, Sr, Ba.
В серии Са, Sr, Ba повышение основных свойств резко сказывается,
между прочим, в степени устойчивости гидратов окисей. Как общее
правило, чем сильнее основание, тем крепче удерживается в нем вода.
Приводим, по данным Джонстона, величины упругости диссоциации р
(водяного пара) гидратов окисей Са, Sr и Ва для различных
температур:
* Теплота образования приведена для жидкой воды.
8*
116
Периодическим .систем,', химических элементов
р, мм рт. ст. Са (ОН:,) Sr (ОН), Ва (ОН)..
31, П 408° 524и 710°
92 448° 597° 787°
526 527° 742° 951°
Из этих данных видно, что определенная упругость диссоциации
достигается при самой низкой температуре для извести и при самой
высокой— для барита.
Можно также отметить, что в солях Са наблюдается слабо
выраженный гидролиз, способность к образованию основных солей. При
переходе к стронцию и барию эта способность видимым образом
понижается.
Цифры потенциалов разложения і'к сожалению, вполне точные
данные не известны) указывают также на повышение положительного
электросродства в направлении от Са к Ва.
Вышеприведенное правило, касающееся повышения металличностп
элемента с нарастанием атомного веса, не распространяется в общем
виде на побочные подгруппы. В следующих подгруппах:
Си Ag Au
Zn Cd Hg
Al Ga In Tl
мы наблюдаем понижение положительного
электросродства с повышением атомного веса. Наоборот, побочные подгруппы
IV, V и VI групп показывают «нормальное» отношение, подобное тому,
которое характерно1 для главных подгрупп (см. также главу VIII).
3. Третье направление, по которому происходит изменение
металлоидной и металлической натуры элементов, связано с
существованием четных и нечетных рядов..
. , В пределах каждой группы, при прочих равных условиях,
принадлежность данного элемента к четному ряду благоприятствует
.проявлению в нем металлического типа. Принадлежность к
нечетному ряду оказывает обратное влияние
В VII группе единственный (не считая фтора) элемент четного ряда,
марганец, как раз проявляет, в противоположность галоидам
(которые помещаются в нечетных рядах), ясно выраженный характер
/металла (в свободном состоянии образует металл, подобный железу); гидрат
закиси его Мп(ОН)2 представляет основание, дающее соли с
кислотами: MnCl2, MnS04 и прочие. В VI группе в нечетных рядах
помещаются типичные металлоиды: сера, селен и теллур; в четных — элементы,
в которых, наряду с металлоидным характером, сказывается и
металлический: Сг, Mo, W и U.
Хром, например, с одной стороны, образует хромовый ангидрид Сг03
и ряд солей, отвечающих хромовой кислоте Н2Сг04 и надхромовой
Н2Сг207; все эти соединения обличают металлоидный характер хрома
и имеют своих аналогов среди соответствующих соединений серы:
Сг03 КХгО; К2Сг.07
S03 K.SO, K2S,67
Но, с другой стороны, в свободном виде хром имеет все свойства
металла (блеск, электропроводность и пр.). Далее, он образует два
основания: гидрат закиси и гидрат окиси Сг(ОН)2 и Сг(ОН)3, которым, в
своюочередь, отвечают два ряда солей: СгС12, <і одной стороны, СгС!3,
Cr(S04)3, и т. д.—с другой. В большей или меньшей степени
аналогичными свойствами обладают Mo, W и U.
Периодическая система химических элементов
117-
Равным образом, в V группе металлоиды сосредоточены
преимущественно в нечетных (Р, As, Sb), элементы, более близкие к металлам
(V, Nb, Та),—в четных рядах. В менее ясной форме те же отношения
проявляются в III и IV группах, что и понятно, так как к середине
Ж
Рис. 3. Распределение электросродства в периодической системе
элементов (по Абеггу)
периодической таблицы, как мы уже видели выше, контраст, между
металлоидным и металлическим характером сглаживается (в III и IV
грудинах помещаются элементы переходные) *. Наконец, в I и II группах
наиболее резко металлический тип также выражен в элементах четных
рядов (К, Rb, Cs, с одной стороны, и Са, Sr, Ва - с другой) и менее
резко—в элементах рядов нечетных (Си, Ag, Аи, Mg, Zn, Cd, Hg. Ср. ниже
главу VIII) **.
* См прим ред. и к стр. 112 настоящего тома. Прим. ред.
** К сказанному следует еще прибавить, что для соединении каждого элемента
металлоидный характер проявляется тем резче (а металлический тем более отступает
на задний план), чем выше становится атомность. Марганец, например, дает
основание средне* силы Мп(?Н)2, когда он двухатомен, дает слабое основание Mn(UM)3.
хонда он трехазгомен и, наконец, будучи семиатомным, дает сильную марганцовую
кислоту. Аналогичным образом Сг(ОН)3 —более слабое основание, чем Lr(OH)2, a
Н2Сг04 — уже типичная кислота.
118
Периодическая система химических элементов
Подводя итог всему, сказанному выше о соотношении между
положением элементов в периодической системе и их электрохимической
природе, мы должны прийти к заключению, что наиболее
электроотрицательный элемент, типичнейший металлоид, должен находиться в
правом верхнем углу, а наиболее электроположительный элемент,
типичнейший металл, должен занять место в противоположном, то есть в
левом, нижнем углу периодической таблицы. На самом деле, первое
место занято фтором, второе — цезием 13, двумя элементами, которые по
праву должны считаться антиподами в электрохимическом отношении.
¦В заключение этой главы пр-иводим, по данным Абегга, диаграмму
(рис. 3), на которой наглядно (в условном масштабе) представлена
общая картина распределения электросродства (положительного 'и
отрицательного) среди элементов -по отдельным группам периодической
системы. Из рассмотрения этой таблицы можно видеть, імежду прочим,
и размер уклонений от правильностей, касающихся постепенных
изменений в металлическом -и металлоидном характере элементов, в
зависимости от их атомного веса [13].
Глава V
СПОСОБНОСТЬ ЭЛЕМЕНТОВ
К ОБРАЗОВАНИЮ ОПРЕДЕЛЕННЫХ ХИМИЧЕСКИХ СОЕДИНЕНИЙ.
СПЛАВЫ
Термохимические соотношения. Если формы соединений
элементов и их электрохимические свойства правильно изменяются в
пределах периодической системы по определенным направлениям, то
аналогичные правильности можно отметить и по отношению к
способности элементов образовать между собой определенные соединения.
Как известно, далеко не каждая пара элементарных (и вообще
химически однородных) тел способна к химическому взаимодействию.
Одни тела реагируют энергично и образуют прочные соединения;
другие обладают лишь незначительной тенденцией к взаимному сочетанию;
у третьих эта способность совершенно отсутствует. Подобные факты
еще очень давно заставили химиков говорить о существовании
«избирательного» сродства.
Еще во времена Дэви и Берцелиуса (в течение первых десятилетий
минувшего столетия), а частью даже и раньше, было замечено, что
характерные и прочные химические соединения образуются элементами,
резко расходящимися по своим свойствам, .и что при этом
первенствующую роль играет их электрохимическая противоположность.
Наипрочнейшие бинарные (двойные, из двух элементов состоящие) соединения
образуются типичными металлами и водородом, с одной стороны,
типичными металлоидами, с другой. Таковы HF, НС1, NaF, NaCI, Н20 и
т. п. Таким образом, первое правило, которое можно здесь
формулировать (правда, лишь в самых общих чертах), заключается в том, что
элементы, способные к образованию 'прочных (экзотермических)
соединений, следует прежде всего искать в возможно удаленных друг от друга
группах периодической таблицы (исключая, конечно, VIII -и нулевую
группы).
За последнее десятилетие, особенно благодаря систематическим
исследованиям двух школ---профессора Г. А. Таммана в Геттингене
и профессора Н. С. Курнакова в Петербурге, было исследовано, по
отношению к способности образовать соединения (хотя бы и нестойкие)
большое число бинарных систем (систем из двух химических эл?мен-
Периодическая система химических элементов
119
тов). При этом было обращено особое -внимание на сплавы, то есть
на системы, образуемые металлическими элементами, вообще близко
стоящими друг к другу, а потому и несклонными к образованию
прочных и характерных соединений.
По большей части, даже в тех случаях, когда компоненты сплава
заведомо вступают в химическое соединение друг с другом, свойства
их претерпевают дашь незначительное -изменение, и прежде всего
сохраняются характернейшие свойства металлов: «металлический» блеск,
непрозрачность для света и вообще для электромагнитных волн,
хорошая тепло- 'И электропроводность. В сплавах натрия со ртутью,
например, сохраняются и только что упомянутые физические свойства обоих
металлов, и химическая природа каждого из них, например 'не
утрачивается способность натрия реагировать с водой и т. п. Между тем
известно, например, что натрий образует со ртутью следующие
соединения: Na3Hg, Na5Hg2> Na3Hg2, NaHg, Na12Hg13, NaHg2, NaHg4-
Своеобразный характер подобных соединений, которые могут быть
названы интер металлически ми, особенно резко выступит, если
сопоставить их с соединениями тех же металлов натрия и ртути с
неметаллическими элементами, например с галоидами.
Если прибавить, что нередко соединения, образуемые металлами,
отличаются малой прочностью, то станет понятным, что обнаружить
существование таких соединений (прежде они часто и оставались
незамеченными) должно было представлять особые трудности. И
действительно, для систематического их отыскания пришлось
воспользоваться новейшими физико-химическими методами исследования,
особенно методом термическим, разработкой которого мы обязаны Л е-
Ш ателье, Курнакову, Тамману и другим.
На основании 'материала, добытого такого рода исследованиями,
Г. Та мм а и [14] устанавливает следующие правильности касательно
способности элементов попарно вступать в соединения.
1. Элементы одной и той же 'подгруппы
периодической системы не способны к химическому
соединению друг с другом. Если отвлечься от двух первых (малых)
периодов (типических элементов), то правило это является весьма общим.
Наиболее обстоятельно справедливость его доказана для следующих
естественных групп*:
Си Zn Ge As
Ag Cd Sn Sb
Au Hg Pb Bi
2. Если какой-либо элемент способен вступать в
соединение с одним из элементов данной
подгруппы, то он соединяется и со всеми остальными; и
наоборот, если он не способен соединяться с одним
из элементов подгруппы, то эта способность
отсутствует у него и по отношению к остальным еечленам.
Чтобы* иллюстрировать степень'приложимости этого второго
правила, укажем, что для бинарных комбинаций из 17 различных элементов
с металлами подгруппы
Си Ag А и
известны только два исключения: РЬ образует два соединения с Au, но
не образует таковых ни с Ag, ни с Си.
* Исключение составляет нестойкое соединение BrJ.
120
Периодическая система химических элементов
Точно так же из группы Zn Cd Hg из комбинаций с 15
металлическими элементами исключение наблюдается только в двух случаях:
Т1 не образует соединений с Cd и Zn, но соединяется с Hg. С другой
стороны, Sn соединяется с Hgf и с Cd, но не с Zn.
Таким образом, между членами вышеупомянутых подгрупп по
отношению к способности образовать определенные соединения существует,
несомненно, родство «... как между членами гомологического ряда
углеродистых соединений, которые подобным же образом или все реагируют
с каким-либо посторонним телом или же, наоборот, все л-ишены
способности вступать в реакцию». Весьма интересно, что из второго правила
Та'ммана значительно больше исключений наблюдается для
элементов со свойствами переходными — частью металлическими, частью
металлоидными, например, для элементов
As Sb Bi
или для
Ge Sn Pb
Элементы, образующие эти группы, таким образом, уже не
напоминают собой настоящие гомологические ряды.
Тамман сближает это обстоятельство со спектральными свойствами
элементов, образующих различные подгруппы периодической системы.
По Кайзеру и Рунге (см. главу VII), как раз спектры элементов
Zn Cd Hg
представляют между собой полнейшую аналогию, и то же самое
относится к подгруппе меди
Си Ag Au
Элементы эти дают спектры, разлагаемые на серии линий.
Наоборот, спектры Sb и Bi, Sn и РЬ столь глубокого сходства не
показывают; линии, их образующие, не могли быть разложены на серии.
Теплота образования. Только что приведенные правильности
частью выступают в более .рельефной 'форме, еслУі сравнивать между
собой те изменения, например тепловые, объемные и т. п., которые
происходят, когда один какой-нибудь элемент последовательно вступает в
сочетание с целым рядом других. До сих пор подобное сравнение
удалось провести лишь для немногих рядов соединений, главным образом,
для галоидных (рис. 4) и кислородных. Но зато полученные
результаты могут быть выражены в количественной форме и потому самая связь
данного свойства с атомным весом, его периодические изменения,
сказываются с полной отчетливостью.
Таким образом, например, Карнелл'И, Лаури и другие
показали, что теплоты образования галоидных соединений элементов и их
окислов изменяются периодически с атомным весом.
Это ясно видно из следующего сопоставления, в котором даны
количества тепла (*в ккал), выделяемые или поглощаемые, когда 1 г-моль
хлора С12 (71 г) соединяется с соответственным весовым количеством
простого тела, отвечающего данному элементу (см. таблицу на стр. 122).
Периодическое нарастание теплоты образования всего нагляднее
выразится, если его представить на диаграмме, которая для галоидных
соединений воспроизведена на рис. 4.
Сравнение количеств тепла, выделяемых (или поглощаемых) при
образовании сходственных соединений, особенно интересно потому, что
Периодическая система химических элементов
121
теплота образования является (хотя и не вполне точным) показателем
степени прочности данного соединения, а следовательно, и
приблизительным мерилом «химического сродства», действующего между
элементами.
В согласии с тем, что было сказано выше, наибольшие количества
тепла выделяются и, следовательно, образующиеся соединения
обладают наибольшей прочностью, когда хлор вступает в соединение с
противоположными в электрохимическом отношении щелочными металлами.
Рис. 4. Диаграмма, данная Лаури для выражения периодического
изменения теплоты образования хлористых соединений элементов (например,
NaC! из хлора и металлического натрия и т. д.). По абсциссам (т. е. по
горизонтальному направлению) отложены атомные веса (начиная слева).
но ординатам (по вертикальным направлениям) — количества тепла,
выделяющиеся на каждую грамм-молекулу хлора (т. е. на С12 или на 71
хлора). Вершины ломанной линии отвечают Li, Na, К. Rb, Cs, низшие
точки — F, CI, Br, J
Наоборот, минимальным теплом образования и наименьшей прочностью
отличаются соединения хлора с другими галоидами*.
Сравнительная устойчивость кислородных
соединений. Характер зависимости между 'положением элемента в
периодической системе и прочностью окисла, ему отвечающего, может быть
освещен еще с двух различных сторон. С одной стороны, Л отар Мей-
ер указал на связь прочности окислов с кривой атомных объемов, на
что будет указано в следующей главе. Там же будут рассмотрены
соответственные взгляды Д. И. Менделеева.
С другой стороны, Б эли [15], специально занимавшийся этим
вопросом, установил следующие правильности:
Для четных рядов:
1. В вертикальных столбцах (подгруппах) устойчивость окислов
повышается с атомным весом элемента (единственное исключение
составляет, быть может, уран).
2. В горизонтальных рядах устойчивость окислов уменьшается
слева направо, другими словами, с возрастанием атомного веса.
Для нечетных рядов:
1. Прочность окислов в подгруппах падает с повышением
атомного веса.
* Некоторые дальнейшие подробности относительно связи теилот образования
сходственных соединений с положением в периодической системе соответствующих
элементов будут приведены в главе VIII, посвященной обзору отдельных групп периодической
системы.
122
Периодическая система химических элементов
Элемент
н
Li
В
С
N
0
Na
Mg
Al
Si
P
S
CI
к
Ca
Mn
Fe
Co
Ni
¦Cu
Zn
Его
окисел
H20
Li20
в203
co2
N20
—
NasO
MgO
A1203
SiOo
PA
so2
CI20
K20
CaO
MnO
FeO
CoO
NiO
CuO
ZnO
Теплообразование
+68,4
+143
+90,9
+48,5
—
—17,7
+100,7
+143,3
+130
+92,3
+73,9
+38,6
—17,9
+86,8
+152
+90,8
+64,6
+63,8
+59,7
+40,8
+85,4
Хлористое
соединение
HC1
LiCI
всія
CC14
—
C120
NaCl
MgCl2
A1C13
SiCI4
PCI5
OoV^Jq
—
KC1
CaCI2
MnCl2
FeCl2
CoCl2
NiCl2
CuCI2
ZnCU
Теплообразование
+44
-4-194,1
+62,2
+ 14,1
—
—17,9
+ 19,7
+ 151,0
+107,5
+6'.
+42
+ 14,3
—
+208,1
+ 190,4
+112,0
+82,1
+76,5
+74,5
+65,8
+97,2
Элемент
As
Se
Br
Rb
Sr
Pd
Ag
Cd
Sn
Sb
Те
J
Cs
Ba
W
Pt
Au
Hg
Tl
Pb
Bi
Его
окисел
As206
Se02
—
Rb20
SrO
—
Ag20
SnO
—
—
J2O5
Cs20
BaO
W03
—
—
Hg20
TI20
PbO
Bi203
Теплообразование
+43,9
+28,5
+83,5
+ 139,6
—
+5,9
65,7
+73,8
—
—
+9
+82,7
+ 125,9
+65,5
—
—
+22,2
+42,2
+50,3
+6,6
Хлористое
соединение
AsCl2
SeCl2
BrCl
—
SrCl2
Pdcu
AgCl
CdCIs
SnCl4
SbC]a
TeCl4
JC1
—
BaCl2
—
PtCl4
AuCl
HgCl
T1C1
PbCl2
BiCl3
Теплообразование
+47,5
+22,2
+1,4
+ 195,7
+40,5
+58,8
+93,2
+63,6
+60,9
+38,7
+ 11,6
—
+197,1
—
+29,7
+11,6
+62,6
+97,2
+82,8
+60,4
2. В горизонтальных рядах прочность окислов сначала повышается
от I группы к IV, а затем снова падает к VII.
Таким образом, в этом, как и во многих других отношениях, между
свойствами элементов четных и нечетных рядов сказывается известная
противоположность. К этим правильностям примыкают результаты,
полученные профессором П. Г. Мели ков ым и его учениками, и из
них особенно профессором Л. В. Писа ржевским, относительно
условий образования и степени прочности перекисей и надкислот*.
Приводим здесь важнейшие из выводов, к которым пришли названные
авторы.
1. Прочность перекисей и надкислот в пределах каждой подгруппы
периодической системы, вообще говоря, повышается с возрастанием
атомного веса.
Иллюстрацией этого вывода могут служить, например, следующие
данные Д е- Фор кр а на и Л. В. Писаржевского:
По Де-Форкрану
Теплота образования из'окиси МеО и О, ккал .
Температура, при которой упругость диссоциации---760, °С
СаОз Sr02
+4,11
+1
+ 13,07
+598
ВаС2
+18,36
+951
По Л. В. Писаржевскому
Zr(OH)4+0-Zr(02H) (ОН), -21,786 ккал
Се(ОН)4+0=Се(02Н) (ОН), -20,392 ккал
Th(OH)4+0=Th(02H) (ОНЬ -14,29 ккал
Соответствующая литература весьма полно собрана и критически освещена
Лисяржевским в сочинении «Перекиси и надкислоты*. Одесса, JO02
Периодическая система химических элементов
123
Можно также указать, что по исследованиям профессора Меликова
надтанталовая кислота значительно превосходит по степени прочности
кислоту надванадиевую.
2. Наиболее прочные перекиси и надкислоты образуются
элементами четных рядов. Примерами особенно прочных перекисей могут
служить отвечающие щелочным и щелочноземельным металлам. Среди
более устойчивых надкислот укажем отвечающие таким элементам, как
Mo, W, U.
Сравнительная устойчивость гидратов окисей.
Чтобы привести еще один пример зависимости, существующей между
положением элемента в периодической системе и степенью прочности
образуемых им соединений остановимся на температурных условиях,
при которых различные гидраты окислов теряют воду. Вопрос этот был
предметом подробного исследования Карнелли и Уокера [16], которые
пришли к следующим заключениям:
1. В четных рядах элементам, принадлежащим к одной и той
же группе периодической системы, соответствуют гидраты окислов тем
более стойкие (то есть теряющие воду при тем более высокой
температуре), чем выше атомный вес данного элемента.
Для иллюстрации этого правила можно привести, по позднейшим
данным Джонс тона [17], упругости диссоциации (в мм рт. ст.)
гидратов окисей щелочноземельных металлов для различных температур
(в °С):
Упругость
диссоциации, мм Са (ОН)2 Sr (ОН)» Ва (ОН)->
31,5 408 524 710
92 448 597 789
526 527 742 951
К аналогичному результату приводит сличение теплот образования
гидратов окисей Ме(ОН)2 из окиси МеО и воды (в ккал):
Са(ОН)2 Sr(OH)2 Ba(OH)2
15,54 17,7 23,3
2. В нечетных рядах, наоборот, в пределах каждой группы
периодической системы стойкость гидратов (поскольку показателем ее
является температура полной дегидратации) падает с повышением
атомного веса.
Для примера приведем температуры полной дегидратации
оснований, отвечающих элементам нечетных рядов II группы:
Mg (ОН).. Zn (OH)2 Cd (OH)2 Hg (ОН)2
630° 585° 385° Неустойчива при
обыкновенной температуре *.
Для гидратов окисей элементов, принадлежащих к одному и тому
же периоду, температура дегидратации падает от начала периода
к середине, а затем снова повышается.
Сопоставление всех этих правильности позволяет заключить, что
и стойкость гидратов окисей элементов является периодической
функцией атомного песа.
* Карнелли и Уокер дают, однако, для температуры полной дегидратации
Hg(OH)2 175°.
124 Периодическая система химических элементов
Глава VI
ФИЗИЧЕСКИЕ СВОЙСТВА ПРОСТЫХ ТЕЛ И СОЕДИНЕНИИ
Объемные отношения
В предыдущих главах мы рассмотрели важнейшие химические
свойства элементов, поскольку ход изменения их с атомным весом
подчиняется закону периодичности. Весьма характерные соотношения
обнаруживаются также при изучении физических свойств простых
тел и сходственных соединений элементов.
Лотар Мей ер на первых порах обратил внимание на
периодичность изменения свойств простых тел. Особенно важные результаты
были им получены при изучении объемных отношений.
Есл'И удельный вес простого тела, отвечающего данному элементу в
твердом или жидком состоянии, будет d, то взяв і/rf, получим объем*,
в котором заключается 1 г данного простого тела, а помножив эту
величину на атомный вес А данного элемента, получим атомный
об ъ е м.
Очевидно, эта величина будет выражать в куоических сантиметрах
объем, занимаемый 1 грамм-атомом данного элемента, взятого в виде
простого тела.
Атомные объемы и были избраны Л. Мейером, как величины,
наиболее наглядно выражающие степень заполнения пространства
различными элементами.
Понятно, что, сравнивая между собой атомные объемы простых тел,
мы встречаемся с некоторым принципиальным затруднением.
Затруднение это, с одной стороны, связано с фактом существования для
многих элементов по нескольку аллотропических модификаций,
обладающих различными свойствами. В частности, и удельный вес этих
последних нередко бывает различен (например, удельный вес желтого
фосфора 1,83, а красного — 2,15), а потому различен будет и атомный
объем данного элемента, в зависимости от того, с какой из его
аллотропических модификаций мы имееім дело. Кроме того, неизвестно, при
каких температурах эти объемы являются сравнимыми между собой.
Отсюда возникает некоторая неопределенность, но, к счастью, влияние
побочных обстоятельств, по-видимому, не особенно велико и не
затемняет общей картины.
Соотношения между атомными объемами и атомными весами
элементов нагляднее всего выступают, если прибегнуть к графическому
методу изображения. На приложенной диаграмме (рис. 5) по абсциссам
отложены атомные веса, по ординатам — атомные объемы. Каждый
элемент будет тогда отвечать определенной точке. Соединив между собой
эти точки, мы получим историческую «кривую», или, точнее, ломаную
Лот. Мейера, зигзагообразный вид которой прямо говорит нам о
наличности периодического изменения атомных объемов. Всматриваясь в
построение этой кривой, мы видим, что она как бы слагается из пяти
отдельных взмахов, постепенно и довольно правильно повышающихся
с нарастанием атомного веса. Соответственно этому, мы имеем пять
восходящих и пять нисходящих ветвей, разделенных точками
максимумов и менее резко выраженными областями минимумов.
При этом сходственные элементы, находящиеся в одной и той же
группе (соответственно подгруппе) периодической системы, и на кри-
* Если удельный вес. относить к воде при 4°, то, очевидно, он будет равен числу,
показывающему, сколько грамм данного тела содержится в объеме, равном 1 см3.
ііериодическая система химических элементов
125
вой расположены аналогичным образом. Так, пяти наивысшим точкам
кривой отвечают щелочные металлы: Li, Na, К, Rb, Cs, обладающие,
следовательно, максимальным объемом атома, тогда как во впадинах
помещаются металлы более благородные, отвечающие слабым
основаниям и главным образам принадлежащие к VIII группе. Они
обладают наименьшим объемом атома.
Рис. 5. Кривая атомных объемов Лотара Мейера
По теории Абегга и Бодлендера, атомный объем элемента стоит в
прямо-м отношения к его электросродству. С другой стороны, согласно
той же теории, элементы с малым электросродством должны обладать
склонностью к образованию комплексных соединений, между тем как у
элементов, электросродство которых значительно, должно наблюдаться
обратное. Опыт дает результаты, вполне согласные с этим заключением,
так как ацелочные металлы, которым соответствует максимальный
атомный объем, почти вовсе лишены способности к образованию
комплексов, между тем 'Как эта способность особенно сильно развита у
металлов VHI ігрутіпы.
На восходящих ветвях кривой Л. Мейера помещаются типичные
металлоиды (например F, С], Вг и J, S, Se и Fe), на нисходящих —
электроположительные элементы, металлы II группы (Са, Sr, Ва и т. д.).
По отношению к ряду других свойств на-блюдаются аналогичные
правильности. Так, мы находим все элементы (простые тела) летучие (или
газообразные) и легкоплавкие на восходящих ветвях; трудноплавкие —
частью на нисходящих ветвях, частью в областях минимумов и т. д.
Позднее Бэл и и другие обратили внимание на то, что атомные
объемы сходственных элементов в кривой Л. Мейера находятся на одной
прямой.
Особенного внимания заслуживают соотношения, подмеченные
Л. Мейером между степенью прочности окислов и их положением на
126
Периодическая система химических элементов
кривой объемов. «Отношение элементов к кислороду,— пишет он в
своем известном сочинении «Moderne Theorien der Chemie» [18],—
прочность их окислов получает наглядное выражение в связи с кривой
атомных объемов. Все элементы, расположенные на отрезке кривой между
каждым максимумом и следующим минимумом (немного дальше, чем
этот последний), весьма прочно удерживают кислород, и потому лишь
с трудом восстановляются из окислов. Элементы, лежащие дальше, до
следующего максимума, наоборот, восстановляются легко. Это видно-
из следующего сопоставления:
Трудно восстанавливаются
I
Li
Na
К
Rb
CS
II
Be
Mg
Ca
Sr
Ba
III
В
Al
Sc
Yt
La
Yb
IV
С
Si
Ti
Zr
Ce
Th
V
p
V
Nb
Di
Та
VI
Сг
Mo
W
LI
Восстанавливаются
со средней легкостью
VII
VIII
Mn Fe Co Ni
H
Ru
VIII
Легко восстанавливаются
II
III
IV
V
VI
Rh
Ir
Pd
Pt
Cu
H
Au
Zn
Cd
Hg
Ga
In
Ti
Sn
Pb
N
As
Sb
Bi
О
s
Se
Те
vir
F
CI
Br
J
Граница не всегда падает на одну и ту же группу. Существуют
также переходные элементы, особенно принадлежащие к группе железа,
и водород, который, вероятно, также занимает положение, близкое
к минимуму (кривой объемов)».
Обсуждая те же объемные отношения, Д. И. Менделеев становится
на более широкую точку зрения. Он не ограничивается рассмотрением
одних только простых тел и останавливается на вопросе о том, как
изменяется * объем атома в зависимости от того, с какими другими
атомами он находится в соединении.
Идея, руководящая Д. И., ясно выражается в том факте, что у тех
сравнительно легких простых тел, которые легко и часто реагируют,
объем атома наибольший: «у Na 24, у К 45, у Rb 57, у Cs 56 (по
новейшим данным 71), у галоидов около 27, у тех же простых'тел, которые
трудно вступают в реакции, средний объем атома мал: для С в виде
алмаза менее 4, в виде угля — около 6, у Ni и Со менее 7, у Ir, Pt —
около 9...»
«Для того, чтобы выяснить отношения между объемами простых
тел и их соединений,— говорит далее Д. И.,— приводим засим
плотности (столбец S) и объемы (столбец M/S) некоторых из известных
высших солеобразных окислов, располагая их в том же порядке (по
величине атомного веса элементов), как простые тела. Для удобства
* Если мы возьмем молекулярный вес соединения М и разделим его на плотность
dt то получим объем молекулы, из которого, в свою очередь, можно, вывести объемы
атомов, молекулу образующих.
Периодическая система химических элементов
127
сравнения объемы окислов даны, считая во всех их по два атома
элемента, соединенного с кислородом. Например, плотность А12О3 = 4,0; вес
А12Оз=102; объем А1203 = 25,5.
Зная, что объем А1=11, видно, что при образовании окиси алюминия
22 его объема дают 25,5 объема окиси, а потому для 03 остается 3,5
объема, или на 1 атом кислорода приходится только 1,2 объема. По
отношению к удельным весам и объемам высших солеобразных окислов
можно заметить некоторую периодичность, но особенно важно обратить
внимание на то, что объем щелочных окислов меньше объема металла,
в них содержащегося, что и выражено в последнем столбце, дающем
эту разность на один атом кислорода. Так, 2 атома Na или 46 его
объемов дают 24 объема ЫагО и около 37 объемов 2NaOH, то есть
кислород и водород, распределившись в среде натрия, не только не увеличили
расстояние его атомов, но сблизили их, стянули силою своего большого
сродства, и можно думать, что это произошло в силу сравнительно
малого взаимного притяжения атомов натрия. Такие металлы, как А1 и
Zr, соединяясь с кислородом и образуя окислы со слабою солеобразо-
вательною способностью, почти не изменяют своего объема, т. е.,
вычитая из объема окисла объем металла, получаем малый остаток, но
обычные металлы и металлоиды, а особенно при образовании
кислотных окислов, всегда дают при окислении приращение объема, то есть
атомы расступаются, чтобы вместить кислород, ъ них не сжатый, как
в щелочах, а потому он легко сравнительно выделяется и его энергия
выступает».
Итак, атомные объемы простых тел, а равно и изменение объемов
при образовании химических соединений, находятся в
непосредственной связи с химической активностью элементов и с прочностью
образуемых при этом соединений.
н2о
Li20
Ве202
В203
С204
N208
NasO
Mg202
А1203
Si204
и *ш
р2об
SoOe
і v О
С1207
к2о
Са202
Sr203
Ті204
V206
Cr2Oe
Си20
Zn202
Ga^Og
5
1,0
2 0
3,06
1,8
1,6
1,64
2,6
3,5
4,0
2,65
2,39
1,96
?1,92
2,7
3,25
3,86
4,2
3,49
2,74
5,9
5,7
?5,1
M/S
18
15
16
39
55
66
24
23
26
45
59
82
95
35
34
35
38
52
73
24
23
36
Объем
кислорода
?—22
—9
+2,6
+ 10,0
+10,6
?+4
-22
-4,5
+1,2
+5,2
+6,2
+8,7
1 *
+6
-35
-8
? 0
+3
+6,7
+9,5
+9,6
+4,8
+4
GejA
As206
Sr202
Yt203
Zr204
Nbo05
Mo2Oe
Ag20
CdaOa
In203
Sn204
Sb205
Te2Oe
Ba202
La203
Ce204
Ta205
W2Oe
Hg202
Pb204
Th204
5
4 7
4,1
4,7
5,0
5,5
4,7
4,4
7,5
8,0
7,18
7,0
6,5
5,1
5,7
6,5
6,74
7,5
6,8
11,1
8:9
9,86
M/S
44
56
44
45
44
57
65
31
32
38
43
49
68
52
50
50
59
68
39
53
54
Объем
кислорода
+4,5
+6,0
—13
?—2
0
+6
+6,8
+11
+3
+2,7
+2,7
+2,6
+4,7
—10
+1
+2
+4,6
+8,2
+4,5
+4,2
+2
28
Периодическая система химических элементов
Можно указать на целый ряд других физических свойств, частью —
простых тел, частью-—их сходственных соединений, которые
находятся в теснейшей связи с атомными объемами и потому имеют
аналогичное с этими последними значение. На этих свойствах мы теперь и
остановимся.
Наиболее полные и отчетливые результаты были получены
относительно сжатия простых тел под влиянием сильного давления,
благодаря весьма обстоятельным исследованиям американского ученого
Ричардса [19]. Мы приводим здесь окончательную сводку, данную
этим авторам, причем с цифрами сжимаемости, соответствующими
повышению давления на 1 мегабару *, сопоставлены атомные веса
соответствующих элементов (в круглых цифрах) <и атомные объемы.
Элемент
Li
с
Na
Mg
Al
Si
P
S
CI
К
Ca
Cr
Mn
Fe
Ni
Cu
Литий
Углерод
Алмаз
Графит
Натрий
Магний
Алюминий
Кремний
Фосфор
Фосфор красный
Фосфор желтый
Сера
Хлор
Калий
Кальций
Хром
Марганец
Железо
Никель
Медь
Средняя сжимаемость
(среднее изменение
единицы объема,
приходящееся на 1 мегабару),
умноженная на 106
8,8
—
0,5
3,0
15,4
2,7
1.3
0,16
—
9,0
20,8
12,5
95
31,5
5,5
0,7
0,67
0,40
0,27
0,54
! Атомный вес (в
округленных цифрах)
7 "
12
—
—
23
24,4
27,1
28,4
31
—
—
32,1
35,5
39,1
40,1
52,1
55,0
55,9
58,7
63,6
Атомный объем (
13,1
—
3,4
5,4
23,7
13,3
10,4
11,4
.—
14,4
16,6
15,5
25
45,5
25,3
7,4
7,7
7,1
6,7
7,1
Элемент
Zn
As
Se
Br
Rb
Mo
Pd
Ag
Cd
Sn
Sb
J
Cs
Pt
Au
Hg
TI
Pb
Bi
Цинк
Мышьяк'
Селен
Бром
Рубидий
Молибден
Палладий
Серебро
Кадмий
Олово
Сурьма
Иод
Цезий
Платина
Золото
Ртуть
Таллий
Свинец
Висмут
Средняя сжимаемость(сре-
днее изменение единицы
объема, приходящееся на
1 мегабару).
умноженная на 106
1,5
4,3
11,8
51,8
40
0,26
0,38
0,84
1,9
1,7
2,2
13
61
0,21
0,47
3,71
2,6
2,2
2,8
і
Атомный вес (в I
округленных
цифрах) |
65,4
75
79
79,9
85,5
96
107
107,9
112,5
119
120
127
132,9
195
197
200
204
207
208
1
Атомный объем f
9,5
13,3
18,5
25,1
56
11,1
9,3
10,3
13,0
16,2
17,9
25,7
71
9Д
10,2
14,8
17,2
18,2
21,2
При просмотре этих данных Ричардса, особенно же
соответствующей диаграммы (рис. 6), всего более бросается в глаза то
обстоятельство, что наивысшие (выдающиеся) точки зигзагообразной ломаной
линии занимают и, следовательно, обладают максимальной
сжимаемостью, элементы, наиболее склонные к реагированию и по химической
природе своей наиболее противоположные друг другу, а именно,
щелочные металлы, с одной стороны, и галоиды, с другой. Можн^
* Мегабара=0,987 атм и соответствует «давлению одной мега-дины на 1 смЧ. Лри-
зеденные цифры выведены из опытов, при которых давление изменялось в предала*
100—500 мегабар.
Периодическая система химических элементов
129
еще отметить, что у галоидов сжимаемость правильно понижается
с повышением атомного веса, а у щелочных металлов — наоборот.
Смысл этой противоположной последовательности нетрудно понять,
если припомнить, что с повышением атомного веса, в пределах данной
(главной) подгруппы элементов, металлические свойства нарастают, а
металлоидные — понижаются. Поэтому щелочные металлы с
повышениям
90
80
70
60
30
40
J0
20
16
Li
- -• —
Вл
[с
NQ
\ г
ы
Я]
1
L
і
¦
г
і
ЬСа
К
Ип
Си,
Se
^
іг
f
t
1
Но
Pd^Cdj;
Cs
/I
і і
у
А
\
\
и,—
1
щ
8 10 20 J0 40 60 SO 78 80 90 WO 116 126 /JO M НО 160 170 189 188 280 218
Рис 6. Диаграмма сжимаемости простых тел по Ричардсу. По абсциссам
(горизонтальное направление) отложены атомные веса, по ординатам- величины сжимаемости
ем атомного неса делаются все более типичными от Li до Cs, 'между тем
как у галоидов как раз наоборот: наиболее типичный из них —хлор
(фтор не был исследован Ричардсом), наименее типичный, обладающий
наименее ясными металлоидными свойствами — иод. В только что
указанных соотношениях сказывается несомненная аналогия 'между
'периодическими изменениями сжимаемости простых тел, атомного объема
их и способности к реагированию. Эта аналогия простипается и дальше.
Так во впадинах ломаной линии в обоих случаях, у Ричардса и у
Мейера помещаются металлы и металлоиды со слабо выраженными
типическими свойствами (С, Si, Fe, Ni, Pt, Pel), в том числе и элементы
ясно амфотерные (А1, Cr, Zn) и т. п.
Ричардсу принадлежит интересная попытка объяснить эти
отношения посредством гипотезы о сжимаемости атомов. Он допускает, что в
твердых телах и жидкостях атомы и молекулы сближены между сооои
до соприкосновения или почти до соприкосновения, и что потому всякое
изменение объема от давления, температуры и т. д. не может ил'И почти
не может зависеть от сближения атомов, а только от изменения соО-
ственного объема их. С этой точки зрения понятно, что атомы с
большим объемом, следовательно, не сжатые силой внутриатомного
давления -будут чувствительны к давлению внешнему, и потому должны
обладать значительной сжимаемостью, что, наоборот, атомы с малым
объемом, сильно сжатые, должны лишь незначительно менять свои
объем при изменении давления, действующего извне. С другой
стороны Ричарде полагает, что при образовании химических соединении
неодинаковое сжатие соединяющихся между собой атомов определяется
«9 Л. А. Чугаев, т. III
130
Периодическая система химических элементов
различной силой действующего -при этом сродства. Отсюда—»
параллелизм, как мы видели, отмеченный еще Д. И. Менделеевым, между
изменением атомного объема элемента и прочностью соединения, в
которое он вступает.
Менделеев не высказывал гипотезы о сжимаемости атомов. Тем
замечательнее, что суждение об этом параллелизме выражено им
почти в тех же самых выражениях, как это много лет спустя делает
Ричарде.
Два других физических свойства простых тел: термичное
расширение* и твердость также могут быть непосредственно
сопоставлены с атомными объемами и с величинами сжатия. К сожалению,
данные, которыми мы располагаем относительно этих двух свойств,
1 1
1
2
3
4
5
6
7
8
9
10
И
12
Н
L1
Na
72
К
83
Си
16,4
Rb
Ag
18,9
Cs
131
Аи
14,3
и
Be
Mg
26,0
Ca
Zn
29
Sr
Cd
31
Ba
Hg
61
Ra
III IV
В
Al
22,8
Sc
Ga
Yt
In
46
La
Yb
Tl
31,3
с
1,38
Si
7,8
Ti
Ge
Zr
Sn
27,0
Ce
Pb
29,3
Th
V
N
P
124
V
As
6
Nb
Sb
10,9
Та
Bi
13,5
VI
О
S
67,5
Cr
Se
37,9
Mo
Те
17,3
W
U
VII
F
CI
470
Mn
Br
390
J
83,7
Vlll
Fe Co Ni
11,9 12,5 13,4
Ru Rh Pd
9,9 8,5 11,8
Os Iг Pt
6,8 7 9
* Если /0 есть длина некоторого бруска, изготовленного из определенного материала
при 0°, a It соответственно длина при /0, то отношение j-пг = а называется линейным
iQl
коэффициентом расширения. Это—мера удлинения единицы длины стержня при подъеме
температуры на 1°. Если объемы, занимаемые данным телом при 0° и при t°
соответственно .= Vq и Vt ^ то отношение—y-~t— — (3 есть кубический коэффициент расширения. Из
величины кубического коэффициента легко вычислить значение линейного. В первом
приближении се =1/3 р. Поэтому, если испытуемое тело не может быть превращено в
брусок или стержень достаточной длины, то можно измерить его кубический коэффициент,
а линейный найти п.утем вычисления.
Периодическая система химических элементов
131
1
2
3
4
5
6
7
8
9
It)
11
12
I
Н
Li
0,6
Na
0,4
К
0,5
Си
3,0
Rb
0,3
Ag
2,7
Cs
0,2
Аи
2,5
|
' Н
Be
2,0
Ca
1,5
Zn
2,5
St
1,8
Cd
2,0
Ba
Hg
| Ra
ш
В
9,5
Al
2,9
Sc
Ga
1,5
Yt
In
1,2
La
Yb
11
Мягче
свинца
IV
С
10
Si
6,5
Ti
Ge
2г
Sn
1,8
Се
Pb
1,5
Th
V
N
P
0,5
V
As
3,5
Nb
Sb
3,0
Та
Bi
2,5
|
VI
VII
0
s
2,0
Cr
0,0
Se
Mo
Очень
тверд
Те
2,3
W
Очень
тверд
V
F
CI
Mn
5,0
Br
J
VIII
Fe Co Ni
4,5
Ru Rh Pd
6,5 4,8
Os Ir Pt
7,0 4,3
крайне неполны. Кроме того, для целого ряда элементов до сих пор еще
не удалось получить соответствующих им простых тел в состоянии
полной чистоты. Эти два обстоятельства, несомненно, невыгодно влияют на
ясность результатов. Тем не менее, по отношению к обоим только что
упомянутым свойствам периодичность изменения их с атомным весом
не подлежит сомнению.
В таблице на стр. 130 приведены величины линейных
коэффициентов расширения простых гел (поскольку имеются
соответствующие данные), умноженные на 106.
Несмотря на неполноту имеющихся данных и на недостаточную
надежность некоторой части их, все же ясно заметна общая
периодичность в изменении коэффициентов расширения с атомным весом. В
частности, можно видеть, что максимум расширяемости наблюдается у
тех же элементов: у щелочных металлов Na, К, Cs с одной стороны,
у галоидов CI, Br, J с другой, которые по Ричардсу обладают
наибольшей сжимаемостью и которые в то же время отличаются наибольшей
химической энергией.
Аналогичные результаты получаются и относительно твердости
простых тел. Здесь также удалось установить явную периодичность и
несомненную связь с атомными объемами. На 'прилагаемой таблице
(стр. 131) даны величины относительной твердости главным образом
по данным Ридберга, причем твердость алмаза принята=10.
Сопоставляя их с другими имеющимися в литературе данными, мы приходим
к заключению, что максимум твердости наблюдается у простых тел,
0*
ш
Периодическая система химических элементов
обладающих минимальным атомным объемом, то есть, у таких, для
которых атомы (и молекулы) особенно сильно сжаты, сближены. Наоборот,
минимум твердости встречается у щелочных металлов, для которых
атомный объем наибольший, для которых, следовательно, заполнение
пространства атомами элементов является, так сказать, наиболее
рыхлым. Если кривую твердости нанести на диаграмму, то можно заметить
еще существование вторичных максимумов (As, Sb, Bi — все элементы
V группы) и минимумов (Ga, In, Tl — элементы III группы) *.
Температуры плавления и кипения. Мы заключим эту
глазу рассмотрением температур плавления и кипения, так как в этих
свойствах сказывается не только явственная периодичность, но и
подобно свойствам, раньше рассмотренным,— более или менее резко
заметная связь с кривой объемов Мейера. Систематическими
исследованиями, сюда относящимися, мы обязаны особенно Т. Карнелли.
90 /00 fW JZO /30 /40 /50 760 /70 /80 /90 200 210
Рис. 7. Кривая плавкости простых тел. По абсциссам отложены (начиная слева)
атомные неса элементов от 0 до 210, по ординатам (кверху)—температура плавления
соответствующих простых тел
На рис. 7 изображена диаграмма плавкости простых тел. Подобно
кривой Мейера, она имеет зигзагообразный характер. Во 2-м и 3-м
рядах с повышением атомного веса сначала наблюдается резкий
подъем (от Li к С, от Na к Si), а затем резкое падение плавкости. В
больших периодах наблюдается сначала довольно сильный подъем, потом
медленное понижение в пределах соответствующего четного ряда; в
следующем нечетном ряду того же периода кривая плавкости сначала
падает (от Си к Ga, от Ag к In), проходит через минимум, потом резко
повышается и, наконец, опять падает.
Численные значения температур плавления простых тел приведены
ъ таблице, которая дана в VIII и IX главах.
В следующей таблице, стр. 133, приведены (главным образом, по
данным Карнелли) точки плавления галоидных соединений элементов,
выраженные в градусах абсолютной шкалы. Верхняя цифра всюду
соответствует хлористому, средняя — бромистому и нижняя — йодистому
соединению. Для сравнения выбраны следующие типы галоидных
соединений CR —элемент данной группы, X — галоид).
Группа I II III IV V VI VII
Тип RX RX2 RX3 RX4 RX3 RX2 RX
Несмотря на неполноту данных, которыми мы располагаем,
периодичность чередования температуры плавления проявляется достаточно
ясно, если сравнивать отдельно хлориды, бромиды и иодиды элементов.
Весьма интересно также отметить, что переходя в каждой группе
* Можно заметить, еще что согласно Теплеру (Wied, Ann., 53; 343), изменение
объема при плавлении простых тел также является периодической функцией атомного
.веса.
Периодическая система химических элементов
133
Температуры плавления галоидных соединений элементов
Li
870
820
719
Na
1088
1031
934
К
1073
995
958
Си
683
777
901
Rb
983
956
915
Ag
733
701,6
829
Cs
904
—
894
Аи
RX
Be
870
—.
—
Mg
981
971
—
Ca
1077
948
904
Zn
535
667
719
Sr
1116
903
750
Cd
814
844
677
Ba
1195
1085
973
_~
Hg
561
517?
526
Ra
RX2
В
—
—
316
Al
—
363
398
Sc
Ga
348
—
—
Yt
In
La
Yb
Tl
RX,
Li
186
К
62,5
Rb
38,5
Cs
26,5
С
250
364
373
Si
184
260
393
Ti
250
306
420
Ge
—
—
417
Zr
Sn
240
303
419
Ce
Pb
Th
1093
RX4
Na
95,6
Си
1080
Ag
960
Au
1062
N
P
161
232
334
V
As
257
205
419
Nb
Sb
345
365
439
—
Та
Bi
503
480
712
RX3
Be
1430°
Ca
780
Sr
800
Ba
850
Mg
635
Zn
419
Cd
320
Hg
-39
О
s
Cr
Se
Mo
Те
448
553
433
—
W
и
RX,
F
CI
198
280
298
Mn
Br
298
266
309
—
J
—
—
386
_
RX
134
Периодическая система химических элементов
от элемента четного ряда к следующему по порядку нечетному, мы и
для простых тел и, поскольку позволяют о том заключить имеющиеся
данные, также для сходственных соединений, получим
попеременное повышение и понижение 'плавкости. Для примера
сопоставим точки плавления простых тел (в °С — I и II групп
периодической системы).
Такого рода правильность, при графическом изображении дающая
зигзагообразную кривую, представляет особый интерес, потому что
совершенно аналогичное соотношение мы находим в гомологических рядах
многих органических соединений. В ряду, например, одноосновных кар-
бо'новых кислот СпН2п02, двухосновных ми-слот, гомологов щавелевой
и пр. температуры плавления попеременно повышаются и понижаются
при переходе от членов с четным числом атомов углерода к следующим
за ними членам с нечетным числом атомов. Мы имеем здесь новый и
притом весьма любопытный аргумент в пользу существования глубокой
аналогии между гомологическими рядами органических соединений
и элементами одной и той же группы периодической системы.
Температуры кипения галоидных соединений элементов *
Li
Na
К
Си
1173
—
—
Rb
Ag
Cs
Аи
—
RCI
Be
Mg
Ca
Zn
1003
923
—
Sr
Cd
1173
1083
985
Ba
—
Hg
580
598
622
Ra
RC12
В
290
363
583
Al
453
543
623
Sc
Ga
490
—
—
Yt
In
La
Yb
Tl
—
RC13 J
С
350
462
—
Si
332
426
563
Ti
409
503
633
Ge
359
—
628
Zr
Sn
387
474
568
Ce
Pb
Th
RC14
N
334
—
—
P
349
448
—
V
As
405
493
477
Nb
Sb
496
548
674
—
Та
Bi
720
726
—
rci3 !
0
278
—
—
S
337
—
—
Cr
Se
Mo
Те
597
612
—
—
W
и
RClj
F
CI
239
286
373
Mn
Br
286
332
390
—
J
373
390
460
—
| -
RCI
* Первая цифра, стоящая под знаком каждого элемента, относится к хлористому, вторая — к
бромистому, а третья —к йодистому его соединению.
Периодическая система химических элементов
135
О температурах кипения простых тел имеющиеся сведения
еще более скудны, нежели о тачках плавления (ср. главу VIII, а
также приложение). Однако и здесь сразу бросается в глаза
периодичность и в то же время параллелизм с кривой Мейера. Аналогичные
заключения можно вывести из таблицы, помещенной на стр. 134, в
которой собраны температуры кипения хлоридов, бромидов я иодидов,
выраженные в градусах абсолютной шкалы и расположенные в том
же порядке, как и цифры предыдущей таблицы.
Глава VII
ОПТИЧЕСКИЕ, ЭЛЕКТРИЧЕСКИЕ И МАГНИТНЫЕ СВОЙСТВА
ПРОСТЫХ ТЕЛ И ХИМИЧЕСКИХ СОЕДИНЕНИИ
Из всех оптических 'Свойств, присущих химическим элементам и
стоящих в связи с .периодическим законом, простейшим и вместе с тем
важнейшим по своему теоретическому значению является характер спектров
испускания газов и паров в раскаленном состоянии.
Спектры элементов и периодический закон.
Отличительной особенностью спектров газообразных веществ, по сравнению с
твердыми и жидкими, является их «линейчатое» строение. Каждая
отдельная линия такого газового спектра соответствует существованию
в молекуле определенного колебания, а обилие этих линий (например,
по Кайзеру и Ру-нге, число спектральных линий железа
простирается до 5000) указывает на .необычайную сложность колеблющейся
системы.
Если бы строение этой системы (атома или молекулы), а также
величины и направления сил, действующих между ее отдельными
колеблющимися частями (электронами), были известны, то теоретически
явилось бы мыслимым составление уравнения, связывающего между собой
все возникающие в системе колебания; однако на -практике, ввиду
сложности задачи, это встретило бы непреодолимые математические
затруднения. Поэтому искомое уравнение до сих пор было выведено только
для простейших случаев, каковыми являются колебания однородного
стержня, пластинки, кольца «и т. -п., .причем оказалось, что в
колеблющейся системе, наряду с основным, может возникать целый ряд
добавочных колебаний, которые, по аналогии с известным'И акустическими
явлениями, можно назвать о бе рт.о н а ми. Тем не менее нельзя
сомневаться в том, что подобное уравнение должно существовать для каждой
колеблющейся системы и, если мы не можем вывести его теоретически,
то у нас остается опытный путь изучения отдельных колебаний и
установления между ними эмпирической зависимости. Нахождение
подобной зависимости может, наоборот, дать некоторые косвенные указания
относительно строения колеблющейся системы.
Прилагая сказанное к спектрам, мы должны допустить в них
существование целого ряда, так называемой «серии» — линий, длины волн
которых или соответствующие им числа колебаний будут связаны
определенным соотношением.
Прежде всего по аналогии со звуковыми колебаниями пытались
обнаружить в спектрах существование гармонических обертонов, то
есть найти серии таких линий, длины волн которых относились бы, как
ряд простых целых чисел. Однако Шустер на основании теории
вероятностей установил полную безнадежность подобных ¦попыток. Таким
образом, пришлось обратиться к отысканию зависимостей,
выражающихся более сложными формулами.
136
Периодическая система химических элементов
Первая из формул подобного рода была предложена Бальмером
для спектра водорода и имеют следующий вид:
ГС2
к = к
jt2 —4
где X—длина волны водородной линии, выраженная в единицах
Ангстрема, то есть в десятимиллионных долях миллиметра, а
К—постоянная величина, равная 3647,20. Подставляя .вместо я в формулу Бальме-
ра последовательные целые числа, -начиная с 3 и кончая 15, мы будем
получать соответствующие значения для длин волн водородной серии.
Насколько хорошо совпадение между вычисленными таким образом и
наблюденными значениями, показывает следующая таблица, в которой
первый столбец содержит последовательные значения я, второй и
третий столбцы дают величины Я, определенные Эмсом и вычисленные
по формуле, а последний столбец показывает разницу между
результатами вычисления и непосредственного
измерения.
Как видно из таблицы, отклонения
вычисленных от наблюденных величин
не превосходят ошибок измерения.
Впоследствии, благодаря работам
Геля, Деландра, Пике ринга, и
особенно Эвершеда, была
тщательно изучена ультрафиолетовая часть
водородного спектра, к которой формула
Бальмера оказалась также вполне
применимой. Так, например, для л = 31
вычисленная длина волны получилась
равной 3661,35, в то время как
наблюденная равна 3661,31, то есть разница
между ними достигает только 0,04
единицы Ангстрема.
В приложении к другим элементам
формула Бальмера потребовала
некоторых изменений. Еще ранее Гарт ли показал, что многие
закономерности в строении спектров обнаруживаются проще, если сравнивать
между собой не длины волн X, а обратные им величины А,-1,
пропорциональные числам колебаний. Так, в спектрах многих элементов
встречаются серии группировок из двух или трех линий, так называемых
«дуплетов» и «триплет о в». По Г а ртл и, между числами коле-
баний соответствующих линий дуплетов и триплетов существует
постоянная разница, как это видно из прилагаемого примера дуплетов
серебра:
я
3
4
5
б
7
8
9
10
11
12
13
14
15
X
наблюденная
6564,97
4862,93
4342,00
4103,11
3971,40
3890,3
3836,8
3799,2
3771,9
3751,3
3735,3
3722,8
3712,9
1
вычисленная
6564,96
4862,93
4341,90
4103,10
3971,4
3890,3
3836,7
3799,2
3771,9
3751,4
3735,6
3723,2
3713,2
Разница
+0,01
0,00
+0,10
+0,01
0,0
0,0
+0,1
0,0
0,0
-0,1
—0,3
-0,4
—0,3
I дуплет . .
разница. . .
II дуплет. .
разница. . .
III дуплет. .
1 -я линия
18031,5
2424,8
20456,3
9103,3
29559,6
Разница
920,8
—
920,2
—
920,8
2-я линия
18952,3
2424,2
21376,5
9103,9
30480,4
Чтобы формула Бальмера прямо давала вел'ичииы X \ ее нужно
написать в перевернутом виде:
1 ___ J 4_
Т ~ К Кп'
= А + Вп~2
Периодическая система химических элементов
137
Прибавляя ^юда еще член, содержащий л~4, мы получили формулу
Кайзера и Рунге:
Г1 = А + Вк-2 + Ся'4,
в которой Л, В и С будут постоянные величины, определяемые для
каждой серии линий кз опыта.
Рид бе р г пользуется для вычисления серий несколько иным
выражением:
X-l = A + B(m + v.)-*,
где Л, В, \і — эмпирические константы, a m принимает значения ряда
последовательных целых чисел.
И формула Кайзера и Рунге, и формула Ридберга
оказались весьма пригодным-и для вычисления серий, причем в одних случаях
лучшие результаты дает первая, а в других — вторая*.
Насколько хорошо совпадение между вычисленными и
наблюденными -величинами, показывает следующий пример одной из серий лития:
я
4
5
6
7
8
9
10
И
\ наблюденная
3232,77
2741,39
2562,60
2475,13
2425,55
2394,54
2373,9
2359,4
X, вычисленная по
формуле Кайзера
и Рунге
3232,77
2741,39
2562,60
2475,33
2425,56
2394,25
2373,2
2358,2
Разница
0,00
0,00
0,00
-0,20
-0,01
+0,29
+0,7
+1,2
Спектры большинства элементов содержат по нескольку серий, к
каждой из которых приложимы вышеприведенные формулы.
Так, спектры щелочных металлов состоят из шести отдельных сер.ий,
которые .попарно группируются в три серии дуплетов. Одна из этих
серий была названа «главной», а двум другим Ридберг дал
названия «диффузной» и «резкой» серии; Кайзер и Рунге те же
самые серии назвали именами «первой побочной» и «второй
побочной».
В спектрах Mg, Са (рис. 8), Sr, Zn, Cd и Hg также содержится по
шести серий, но, в отличие от щелочных металлов, они группируются в
две серии триплетов.
Между отдельными сериями одного и того же -спектра наблюдается
определенная закономерная зависимость.
Так, дуплеты главной серии в спектрах щелочных металлов по мере
увеличения я все теснее и теснее сближаются друг с другом, и, начиная
с некоторого определенного я (для Na я = 5, а для К я = 8), уже не
могут быть наблюдаемы раздельно. Для я=°о обе слагающие серии сли-
* Формула Ридберга может быть приведена к следующему виду:
то есть отличается от формулы Кайзера и Рунге только степенью и в третьем члене
разложения.
138
Периодическая система химических элементов
ваются в одну, вследствие чего константа А в формулах для их
вычисления имеет одну и ту же величину.
Далее, дуплеты каждой из побочных серий обладают постоянной
разностью в числах колебаний v, одинаковой для обеих серий, с
соответствующей разностью у первого дуплета главной серии. Поэтому
константы В и С в формулах для вычисления обеих слагающих каждой из
1
1
1 1 1
[
_1,
1 1 1!
ill
||
1
1
1 1 ,
1
1
1 і
1 li III, II11
II .
1і h 1і і,
III II
1 ... .
1
1
1
і , і
1
,1 ¦
III
II Din
11 i
1 1 К
1 1 1 1 1 II
1
Рис. 8. Сравнительная таблица спектров кальция и магния с содержащимися
в них сериями и с остаточными линиями
побочных серий имеют одинаковые значения, а константы А в тех же
формулах отличаются на постоянную величину v.
Наконец, для обеих побочных серий константы А имеют одинаковые
значения.
Серии триплетов в спектрах щелочноземельных металлов имеют
характер «побочных серий. Поэтому константы Б и С сохраняют
неизменные значения в формулах для вычисления всех трех слагающих каждой
такой сложной серии; константы же А отличаются друг от друга на
величины vi и V2, где vi означает разницу в числе .колебаний первой и
второй, a V2 — второй и третьей линий каждого триплета. Наконец,
константы А соответственно равны у обеих серий триплетов.
Ридберг указал на существование следующей зависимости
между отдельными сериями одного и того же спектра. Если написать его
формулу для первой линии главной серии и для второй линии второй
побочной серии:
1~г^А — В(гтг + \х)~2
и
XV1 -=Аг- В(пг + а)"»,
го оказывается, что
А = 5(1 + 0)-*
и
Периодическая система химических элементов
139
Последняя закономерность, независимо от Ридберга была
найдена Шустером, который формулировал ее следующим образом: число
колебаний первой линии главной серии равно разнице между числами
колебаний последних линий, главной и побочной серий.
Серии линий до сих пор были обнаружены в спектрах следующих
элементов: Li, Na, К, Rb, Cs, Си. Ag, Mg, Ca, Sr, Zn, Cd, Hg, Al, In,
Tl, Mn, He, Ne, O, S, Se.
Однако следует отметить, что далеко не .все лиши спектра удается
разложить >в серии, как это видно на рис. 8 и -из "примера следующей
таблицы:
Кроме серий, в
строении спектров была
обнаружена и другая
закономерность: определенная
группировка линий по
нескольку раз повторяется
в спектре, нередко с
почти постоянной разностью
в числах колебаний.
Подобные группировки были
найдены в спектрах Sn,
Pb, As, Sb, Bi, Cu, Ag, Pd,
Pt, Ru, Rh и других
элементов. Рис. 9 изображает такую повторяющуюся группу линии в
спектре сурьмы.
Несмотря на чисто эмпирический характер найденных до сих пор
иравильностей, вряд ли можно сомневаться в том, что строение всех
спектров подчинено определенному закону, который остается пока
неизвестным.
элемента
Калий . .
Литий . .
Натрий
Рубидий .
Цезий . .
Цинк . .
Мйгний
Кадмий
Линии,
разложенные
в серии, %
100
100
100
100
100
80
64
50
Название
элемента
Кальций .
Ртуть . .
Серебро .
Стронций
Медь . .
Золото . .
Барий . .
Линии,
разложенные
в серии, %
34
27
26
20
6
4
0
I я I ге I гв | за \ зг I м I m \п\и\и\н \іи \й
I
Б
?
Рис. 9. Повторяющиеся группы линий в спектре сурьмы
Вместе с тем, можно уже a priori утверждать, что строение
молекулы должно известным образом отразиться на характере -испускаемого
ею спектра, и что между спектром элемента -и его атомным весом
должна существовать определенная зависимость.
Первая попытка в этом направлении принадлежит ЛекокдеЬуа-
б одр а ну, который на примере щелочных-и щелочноземельных
металлов показал, что спектры химически сходных элементов обладают и
сходным строением, причем соответственные линии таких спектров по
мере увеличения атомного веса, перемещаются к красному концу.
Последнюю зависимость он пытался даже выразить формулой, однако
неудачно.
140 Периодическая система химических элементов
Далее, заслуживает внимания попытка Л е к о к а определить из
спектроскопических наблюдений атомный вес элемента. Сущность его
приема легче всего выяснить на числовом примере:
Соответственные линии
А1
Ga
In
3952
4101
4306
1-я
разность
140
205
2-я
разность
56
Вариация
56
14і)
0,37584
Атомные веса
1-я
разность
27,5
69,0
113,5
42,4
43,6
2-я
разность
1 ''
Вариацни
1
7Т7-0,0283
42,4
(\ QTf Q/
Частное из обоих вариации '",, 0.,- = 13,28 сохраняет, .по Л е коку,
для всех групп периодической системы постоянное значение, которым
можно воспользоваться для определения атомного веса. Применяя этот
прием к германию, Л еко к составил следующую таблицу, в которой он
сопоставляет измеренные им линии этого элемента с.соответственными
линиями кремния и олова:
Соответственные линии
Si
Ge
Sn
4010
4453
5077
1-я
разность
443
624
2-я
разность
181
Вариация
181
443 ~
0,4051
Атомные веса
28
X
118
1-я
разность
jr—28
118—д-
2-я
разность
146—2л*
Вариация
146—2л-
х—28 Л
Так как
1,4051
К
= 13,82, то для атомного веса германия Лек о к
получает величину * = 72,32, то есть тот же самый результат, который был
впоследствии получен Винклером из непосредственных измерений.
Однако это блестящее совпадение следует рассматривать, как
исключительно счастливую случайность. Эмс проделал подобное
вычисление с целым рядом других элементов, причем брал для сравнения
заведомо соответственные линии, чего нельзя сказать относительно
линий Лекока, и пришел к отрицательным результатам.
Первая вполне научная попытка установить зависимость между
строением спектра -и атомным весом элемента принадлежит Га р тли,
который на целом ряде примеров показал, что спектры сходных
элементов гомологичны друг другу, и что в пределах каждой группы
увеличению атомного веса соответствуют определенные закономерные
изменения в характере спектра. Хотя большинство замеченных Га р тли пра-
вильностей носит качественный характер, однако ему удалось устано-
Периодическая система химических элементов
141
вить и некоторые количественные соотношения. Так, например, он
показал, что триплеты в спектрах Mg, Zn и Cd обладают одинаковыми
разностями в числах колебаний.
Заключения Гартли в большинстве случаев были подтверждены
Эмсом, который, кроме того, обнаружил существование одинаковых
разностей между числами колебаний первых линий в триплетах цинка
и кадмия:
Zn 581,0 263,1 141,4 84,5
Cd 586,7 263,7 140,8 84,3
Очень важная законность была открыта Ридбергом, который
нашел, что для каждой группы сходных элементов разности между
числами колебаний дуплетов (v) или триплетов (vi и V2) почти
пропорциональны квадратам атомных весов (а), как это видно из
нижеприведенной таблицы:
Элементы
Na
К
Rb
Cs
V
17
57
234
545
V
325
381
322
309
Элементы
Си
Ag
Аи
V
249
921
3817?
V
а'-
622
794
981
Элеыент)»і
Mg
Са
Sr
Vi
41
106
394
Vl
а-
713
638
517
20
52
187
V;
и2
337
325
244
Элементы
Zn
Cd
Hg
Vl
389
1171
4<Ш
a-
918
929
1161
v=
190
542
1768
v-
u2
444
432
441
Элементы
Al
In
Tl
V
112
2213
7795
V
a-
1534
1721
1879
Элементы
О
s
Se
Vi
3,7
18,2
103,7
Vi
a1
144
177
166
v2
2,08
11,1
44,1
v.
a8
81
107
70
Далее оказалось, что если по оси абсцисс откладывать атомные ве-
са, по оси ординат выражения у. то полученная кривая будет иметь
периодический характер, отчасти напоминающий кривую атомных
объемов Л. Мейер а.
Несколько иную форму зависимости открыл Ра мед ж, который
тщательно сравнивал между собой соответственные линии в спектрах
сходных элементов. Если числа колебаний этих линий принять за
ординаты, а квадраты атомных весов —за абсциссы, то полученные точки
расположатся на одной прямой. Только в некоторых местах, а именно
между Na и К, а также между Mg и Zn полученные прямые будут иметь
переломы.
Наконец, Рунге и Прехт принимают, что атомные веса
химически сходных элементов -пропорциональны некоторой степени разности в
142
Периодическая система химических элементов
числах колебаний обоих линий соответственных дуплетов. Показателем
степени при этом всегда является правильная дробь, в случае щелочных
металлов равная xh-
Таким образом, если логарифмы атомных весов и логарифмы
разностей в числах колебаний дуплетов отложить по двум координатным
осям, то для каждой группы химически сходных элементов полученные
точки расположатся на одной и той же прямой, как это показывает
рис. 10.
W
/5 20 25 JO 35
У и с/т о колебаний
Рис. 10. Логарифмы разностей в числах
колебаний
Рунге и Прехт применили найденное ими правило к
определению атомного веса радия, однако получили для него чересчур большую
величину—258 вместо 225, найденной г-ой Кюри из непосредственных
наблюдений.
Таковы :в общих чертах результаты -всех более или менее серьезных
попыток найти зависимость между спектрами элементов и их атомными
весами. Если закон этой зависимости окончательно еще не найден, то
все же сделанное до сих пор с несомненностью указывает на
существование такого закона, а также на то, что, подобно остальным физическим
к химическим свойствам, спектры элементов являются
периодической функцией их атомного веса.
Цвета аналогичных соединений. Из других о-итических
свойств довольно правильные изменения в периодической системе
показывает цвет сходственных соединений, как на это было обращено
внимание Карнеллю, Шютце и другими. Есл-и какой-нибудь элемент
А последовательно вступает >в соединение с элементами В, С и т. д.
одной и той же-подгруппы 'Периодической системы, притом построенные
по одному общему типу, то, по мере возрастания атомного веса
элементов В, С, ..., цвет соответствующих соединений изменяется в
определенном порядке: от белого (бесцветного) постепенно к желто-зеленому,
желтому, оранжевому, красному и черному.
ZnS белого цвета
CdS желтого цвета
HgS красного или черного цвета
MgO белого цвета
ZnO желтоватого цвета
CdO коричневого цвета
HgO красного цвета
AgCl
AgBr
AgJ
P.2S3
As2S3
Sb2S3
NaJ
CuJ
AgJ
AuJ
белого цвета
очень бледно-желтого цвета
бледно-желтого цвета
бесцветный
желтого цвета
оранжевого цвета
белого цвета
почти бесцветный
бледно-желтого цвета
золотисто-желтого цвета
Периодическая система химических элементов
143
Эта прав-ильность для твердых тел * (конечно, только в самых
общих чертах), быть может, объясняется тем обстоятельством, что для
ряда сходственным образом построенных молекул, по мере увеличения
их массы, удлиняется собственный период их колебания, от которого,
в свою очередь, зависит положение области абсорбции (например,
полосы), а следовательно, и цвет данного тела. Вещества с наиболее
коротким собственным периодом будут соответствовать абсорбции в
ультрафиолетовой части спектра, и потому они должны быть бесцветны.
По мере удлинения периода, абсорбция будет переходить в область
видимого спектра и постепенно подвигаться к красному концу.
Соединение должно быть окрашено в цвет, дополнительный к поглощенному.
Поэтому появление абсорбции в крайней фиолетовой части спектра
вызовет окрашивание вещества в зеленовато-желтый цвет
(дополнительный к фиолетовому), а затем, по мере ее передвижения в область
наименее преломляющую, цвет будет изменяться в желтый, зеленый,
оранжевый и красный. Когда все лучи спектра будут сильно абсорбированы,
вещество будет казаться черным.
Более общее отношение между цветом элементов в соединениях и их
атомным весом было указано американским ученым Кери Ли [21]
(Carey Lea). Если принять вместе с Аррениусом, Оствальдом
и другими, что цвет раствора электролита слагается аддитивно из
цвета его ионов, то можно, сравнивая между собой цвет (и спектры
поглощения) различных электролитов, делать заключение о цвете
соответствующих ионов и о влиянии на окраску элементов, эти ионы
образующих. Следуя этому пути, Кери Ли мог разбивать все элементы на три
категории или типа: 1) образующие только бесцветные ионы, 2)
образующие только окрашенные ионы и 3) элементы переходного характера,
то есть образующие то бесцветные, то окрашенные ионы, в зависимости
от проявляемой ими валентности (степени окисления). При этом
оказалось, что упомянутые типы сменяют друг друга периодически, по мере
возрастания атомного веса элементов, что можно представить
следующей схемой (рис. 11).
Интересно также отметить, что окрашенные 'ионы образуются почти
исключительно парамагнитными элементами [22].
Эквиваленты рефракции (лучепреломления). Световые
волны с неодинаковой скоростью распространяются через различные
(прозрачные для них) тела. Если скорость света в пустоте или в свободном
эфире ( = 300000 км/сек) .принять за единицу, то распространение света
в данной среде можно характеризовать относительной скоростью vt
которая всегда менее единицы (кроме некоторых тел с аномальной
дисперсией), или же, как это является общепринятым, обратной величиной
— ==л, носящей -название показателя преломления. Последний всегда
V
больше единицы <и показывает, во сколько раз скорость света в пустоте
больше, нежели в данном теле.
Как известно, мерой показателя преломления служит отношение
между синусами угла падения (из пустоты на данное тело) и угла
преломления (в данном теле) sin '¦ ¦¦ = л,что и дает возможность определить
sin V
его величину из опыта.
Показатель преломления, вообще говоря, изменяется в зависимости
от условий, в которых .находится данное тело, а прежде всего от темпе-
* Если допустить, что в твердых телах каждая молекула колеблется, как целое,
около некоторого положения равновесия.
144
Периодическая система химических элементов
ратуры, давления и агрегатного состояния. Поэтому для сравнения луче-
преломляющей способности различных тел между собой необходимо
выбрать такую функцию показателя преломления, которая от
вышеуказанных факторов не зависит (другими словами, для которых =0 и
dp
' ф^ = 0). Наиболее известны две таких функции. Одна из них, просгей-
п—і
шая, предложена Гладстоном в 1858 г. и имеет вид —j- (я —
показатель преломления и d — плотность при одной и той же
температуре t). Другая, предложенная в начале 80-х годов, независимо друг от
й
CD
•?.
о
¦х.
а. 5
m о
32-38
GrMnFeCoN.
/ ^ Ohpatu
^ I ^ UOtibl
аі
и to e
65-30 103-fOb №-133
Ru Rh Pd
аз W
. OH paw.
5r /.. uonbt
/'к
r-J со
Бес?етніне
JJOtitR
Веса
М-168 132-136
PrNdSmEr QslrPtAu
*1
бесцбетнЬіе
ио/tbt
Рис. 11
друга Г. Лорентцем л Л. Лоренцом, отличается большей
сложностью
п2
1
- X—. Зато она имеет интересное физическое значение, имен-
л2+ 2 d
но, выражает («в -первом приближении) ту долю общего объема,
занятого веществом (независимо от его агрегатного состояния), которая
приходится на д о л ю самих молекул, как таковых (следовательно,
за вычетом, межмолекулярных пространств). В этом обстоятельстве
заключается одна из причин, по которой в настоящее время почти
исключительно употребляется формула Лорентца—Лоренца, или, как ее
часто называют, п2-формула. Впрочем, опыт показал, что обе формулы, и
старая и новая, лишь приблизительно удовлетворяют основному
условию— независимости от температуры, давления и агрегатного
состояния.
Так как іи -показатель преломления, и обе только что упомянутые
функции его изменяются от длины волны света, то для достижения
сравнимых результатов величину п измеряют для какой-нибудь
определенной спектральной линии, например, для желтой D натрия (или же для
ряда таких линий).
Оба выражения: Гладстона и Лорентц—Лоренца дают так
называемое удельное преломление. Помножение соответствующих
величин на молекулярный вес вещества М дает величину
молекулярного преломления:
d n-+2 d
Ближайшее исследование молекулярного преломления для ряда
различных веществ показало, что свойство это является приблизительно
аддитивным. Другими словами, для каждого химического элемента
можно'Вывести некоторую постоянную (-или колеблющуюся в узких
пределах) величину, так называемую атомную рефракцию; зная же
•атомные рефракции элементов, образующих данное соединение, и моле-
Периодическая система химических элементов
145
кулярную формулу этого последнего, легко можно теоретически
вычислить его молекулярную рефракцию, если атомную рефракцию каждого
элемента помножить на число атомов его в данной молекуле и сложить
полученные таким образом величины. Если, следовательно, мы имеем
соединение AnMmCpDq ..., образованное элементами А, В, С, D...,
атомные рефракции которых суть гА. /"в, rc, rD..., то молекулярная
рефракция такого соединения MR выразится следующим образом:
МЯ - пгА + пігв + prc -i- qrD ....
Для выяснения связи между лучепреломляющей способностью
элементов и их положением в системе можно сравнивать или непосредственно
молекулярные-рефракции'их сходственных соединений или вычисленные
значения атомных рефракций.
Первый прием .пригоден только в очень узких границах, например,
для элементов, находящихся в одной и той же группе периодической
системы и образующих соединение одного и того же типа. Таковы,
например, для галоидных солей калия:
"*-1 х J^_ KC1 КВг KJ
п*+Ч d 11,25 14,70 21,25
из сравнения которых видно, что молекулярная рефракция возрастает
от хлора к иоду.
Гораздо общее второй прием, но зато он страдает тем существенным
недостатком, что атомная рефракция многих элементов изменяется
иногда в довольно широких пределах, в зависимости от природы того
соединения, из которого она вычислена, и особенно от валентности, которую
проявляет при этом данный элемент. Отсюда возникает некоторая
неуверенность при выборе величин атомной рефракции для сравнения.
Тем не менее, сравнение все-таки возможно. Расположив элементы з
¦периодическую систему и подписав под каждым из них
соответствующую атомную рефракцию, мы заметим ясную периодичность, особенно
резко сказывающуюся в верхней (более полной) части таблицы. К
сожалению, данные применительно к п2 очень неполны, а потому мы
ограничиваемся приведением (стр. 146) таблицы атомных рефракций,
вычисленных по формуле Гладстона и относящихся к линии а-водорода.
Для хлора, брома и йода низшая цифра относятся к органическим соединениям
типа RX, высшая — неорганическим солям. Для марганца и железа вчяга цифра,
вычисленная из рефракции закисных солей Fe". Для соединения Fe"'r = 20,1, для Мп в пер-
манганатах г = 2б,2. Для хрома взятая цифра относится к трехагомиому хрому (СгХ3).
В солях хромовой кислоты атомная рефракция хпома = 23.
Из таблицы видно, что в пределах каждой группы рефракция
довольно -правильно возрастает с атомным весом. В горизонтальных рядах
она, начиная от нулевой группы, возрастает, около середины проходит
через максимум и затем снова падает к VII группе.
В связи с этими правильностями не лишенным интереса представляется следующее
эмпирическое соотношение, установленное Гладстоном между атомной рефракцией г
п — \
данного элемента (вычисленного по формуле —-— х М) и его эквивалентом Е:
гхУЕ =/С, где Л* есть постоянная, значение которой для одноатомных элементом =
= 1,01, а для 2-, 3-, 4- и 5-атомных = 1,30.
В самое последнее время .весьма интересные соотношения между
атомными рефракциями элементов были открыты Кётберсоном и
Меткольфом [23] *. Соответствующие измерения были сделаны для
* Дальнейшие исследования [24, 25] и др. вполне подтвердили вышеуказанные
правильности, особенно для нулевой группы.
10 Л. А. Чугасв, т. III
146
Периодическая система химических элементов
линии D нагр-ия над парами простых тел и расчислены по формуле
Гладстона.
В этих опытах определенное количество газа (соответственно твердого и жидкого
тела, переходящего в пар при высокой температуре) помещалось в кварцевую трубку,
замкнутую плоскопараллельными кварцевыми же пластинками, а самые измерения
производились с помощью интерференционного рефрактометра Жамена. Объем трубки,
а следовательно, и плотность газа d в условиях опыта были известны. Определив по-
т—1
казатель преломления /л, можно было вычислить удельную рефракцию —-—. Най-
а
денные таким образом величины перечислялись на нормальную плотность (Standard
density do), выбранную таким образом, чтобы при этом в единице объема данного газа
заключалось столько же атомов, сколько атомов водорода содержится в том же объеме
при 0° и 760 мм рт. ст. Очевидно, что мы должны иметь соотношение:
m — 1 и —1 m —1 d
== Л или — f
d d0 (i—l d0
0 I II
He
0,47
Ne
0,89
Ar
3,68
—.
Кг
5,51
—
Xe
9,0
—
—
Li
3,6
Na
4,65
К
8,0
Cu
11,7
Rb
11,4
Ag
13,1
Cs
15,6
—
Au
25,1
—
Be
6,6
Mg
6,9
Ca
10,1
Zn
9,9
Sr
13,3
Cd
13,9
Ba
16,1
—
Hg
21,5 и
19,8
__
in
В
4,8иЗ,5
Al
9,5
Sc
Ga
14,75
Yt
17,t>
In
17,4
La
19,8
—
11
21,6
. .,
IV V VI VII
С
4,6
Si
7,1 и
5,8
Ti
25,0
Ge
Zr
21,9
Sn
27,6
Ce
20,0
—
Pb
26,7 и
24,5
Th
28,7
N
4,8
P
18,4
V
24,7 (?)
As
15,0
Nb
Sb
24,5
—
Та
Bi
32
О
3,25 и
2,5
S
13,5 и
16,0
Сг
15,4
Se
26,8
Mo
Те
—
W
—
___,_
F
0,6—0,9
CI
10,0 и
10,7
Mn
11,5
Br
15,2 и
17,0
—
J
24,4 и
27,2
—
—
-
VIII
—
—
Fe
11,7 и 19,9
Co Ni
10,9 11,0
—
Ru Rh Pd
— 23,9 22,7
—
—
Os Ir Pt
- 31,9 33,5
—
__
Периодическая система химических элементов
147
где ц — коэффициент преломления газа при-нормальной плотности. Отношение межд^-
обеими плотностями определится по формуле:
cIq 0,00009хатомный все элемента объем трубки, см*
— /^
d атомный вес водорода вес газа в трубке, г
ц—1
Понятно, что если мы вместо удельных рефракций будем сравнивать
величине
ны ц—1, то как раз будем иметь величины, пропорциональные атомным рефракциям.
соответствующих элементов. Понятно, что величины эти непосредственно несравнимы
с атомными рефракциями элементов, вычисленными из опытов с жидкостями, в которых,
плотность относится не к водороду, а к воде при 4°.
В следующей таблице даны относительные величины атомной
рефракции, умноженные на 106.
Не
144x1/2
N
297
Р
299x4
As
258x6
Sb
О |
270
S
275x4
Se
261X6
Те
249хЮ
F
192
CI
192x4
Br
187x6
J
192x10
Ne
137
Ar
142x4
Kr
142x6
X
138x10
Легко видеть, что в нулевой группе, а также в трех подгруппах
периодической системы величины рефракций «гомологических» элементов
(приблизительно) относятся между собой, как одни и те же лростые
целые числа 1 :4:6: 10 (более значительные отклонения имеются для
As и Те). В других подгруппах периодической системы величины
рефракции элементов, однако, по-видимому, не подчиняются столь
простому закону, например, для паров цинка, кадмия и ртути были получены
следующие результаты:
Zn Cd Hg
2060 2675 1866
Вышеприведенные простые отношения, по всей вероятности,
находятся в связи со структурой атомов, которые для членов каждой груп-
лы должны представлять известные общие черты. С этой точки зрения
большой интерес представляют результаты, полученные при изучении
дисперсии элементарных газов и паров. Оказалось, что для этих
газов связь между коэффициентом преломления \х и числом колебаний
(в единицу времени) л0, отвечающим собственному (-свободному)
периоду данного газа, и числом колебаний! п = — 1. употребляемого для
измерения света, хорошо выражается формулой Зеллмейера \х—1 =
N
—, ^ где дг есть .неКоторая постоянная величина, характерная для
каждого газа. Согласно теории, развитой Друде, величина N
пропорциональна числу электронов в единице объема вещества и,
следовательно, должна находиться в тесной связи с валентностью.
Интересно, что для водорода, кислорода и азота величины N
выражаются числами 1,692, 3,397 и 5,0345x1027, которые относятся как
1:2:3, то есть как характерные валентности этих элементов.
10*
148
Периодическая система химических элементов
Вопрос этот, представляющий очень большую теоретическую
важность, настоятельно требует дальнейших исследований.
К только что рассмотренным оптическим свойствам элементов и ах
соединен-ий примыкают чрезвычайно интересные результаты,
полученные Краузером и особенно В. А. Бородовским [26],
исследовавшими поглощение (3-лучей радия весомой материей.
Бородовский предлагает сравнивать различные вещества по
способности поглощать |і-лучи с металлического алюминия *, определяя
толщину слоя данного вещества, вызывающего то же самое ослабление
пучка р-лучей, как и алюминиевая пластинка, толщина которой = 1.
Таки'М образом, поглощение может быть выражено «в терминах
алюминия». Чтобы исключить при этом влияние плотности испытуемого
вещества, поглощение относят к тому случаю, когда 1 г этого вещества
распределен равномерным слоем по 'поверхности в 1 см2.
Способность материи поглощать (3-лучи оказалась свойством
аддитивным, независимым от того состояния, в котором находятся
поглощающие элементы. Так, например, безразлично, находятся они ъ виде
простого тела или входят в состав какого-нибудь однородного химического
соединения, раствора, смеси и т. л. Поэтому поглощающая способность
отдельных элементов может быть вычислена из соответственных
величин, найденных для их соединений, по правилу смешения.
Сравнение найденных таким образом величин 'поглощения а
показывает, что они периодически изменяются с нарастанием
атомного веса, как это ясно видно из следующего сопоставления.
^ЛА-
мент
н
Li
С
N
О
F
Na
Mg
Al
Si
S
CI
К
Граммовое
поглощение a,
в терминах
алюминия
1,10
0,95
0,86
0,87
0,88
0,72
0,95
0,91
1,00
1,09
1,05
0,98
1,23
Атомное
поглощение
a ¦ Л
1,19
6,70
10,27
12,19
14,05
13,68
21,84
22,13
27,1
30,73
33,67
34,7
48,09
f—*
VT
1,19
0,45
0,37
0,36
0,35
0,27
0,33
0,32
0,33
0,36
0,33
0,30
0,36
Элемент
Са
Ті
Сг
Си
Zn
Вг
Sr
Sn
J
Ва
Pt
Pb
Граммовое
поглощение a,
в терминах
алюминия
1,21
1,11
1,10
1,27
1,29
1,15
1,42
,63
1,3 5
1 ,47
>1,60
2 ,06
Атомное
поглощение
ах.4
48,51
53,60
57,2
80,5
84,3
91,9
124,0
194,0
171,3
201,5
>312,0
426,7
a К
з *
VT
0,35
0,31
0,29
0,32
0,32
0,27
0,33
0,33
0,27
0,29
0,28
0,35
* Опыт показал, что если при прочих равных условиях изменять хотя
6ы^ и в широких пределах, толщину слоя t испытуемого вещества, то отношение между
ней и толщиной а пластинки алюминия, дающего тот же эффект, остается практически
постоянным и независимо от скорости р-лучей. Например, в одном опыте для воды
было найдено:
/ 0,04 0,10 0,14 0,18 0,21
a 0,0117 0,0354 0,0494 0,0631 0,0742
a 0,293 0,354 0,353 0,351 0,353
t
Периодическая система химических элементов 149
Если величины атомных весов нанести на абсциссы, а величины
граммовых поглощений —на ординаты, то получим зигзагообразную
ломаную линию, напоминающую диаграмму атомных объемов Л. Мейе-
ра (рис. 12), причем больше всего бросается в глаза расположение
галоидов, занимающих точкіи минимумов. Аналогичное, но менее резко
а
v
із
і*
V
to
?
ff?
'О 20 60 60 80 tOO 120 ?О
Рис. 12
выраженное соотношение получается, если сравнивать «атомные
поглощения», то есть поглощения, отвечающие грамм-атомным весам
элементов. Они получаются -из величин а через помножение на
соответствующие атомные веса.
Изучение связи между поглотительной способностью элементов а и
их атомным весом А привели В. А. Бородовского к соотношению,
которое может быть выражено формулой
VA
Иначе говоря, поглощение |5-л учей единицей веса (1 г)
какого-либо элемента (при основании поглощающего слоя в
1 см2) прямо пропорционально корню кубическому из
атомного веса. Величина К приблизительно постоянна для
большинства элементов и =0,3, а потому, подобно закону Дюлонга и Пти,
соотношением этим можно пользоваться для определения
приближенного значения атомных весов. Однако, подобно закону Дюлонга и Пти,
правильность, подмеченная Бородовским, не строго выдержана; при
этом замечательно, что отклонения, наиболее значительные,
наблюдаются как раз для элементов с малым атомным весом, прежде всего для
водорода, а затем для членов малого периода:
Н Li С N О
К 1.10 0,45 0,37 0,36 0,35
150
Периодическая система химических элементов
Интересно еще отметить, что галоиды отличаются наименьшей,
щелочные и щелочноземельные металлы — наибольшей величиной кон
ста-нты:
К
? С1 Вг
0,27 0,30 0,27
Na
0,33
К
0,36
Mg
0,32
J
0,27
Са
0,35
Sr
0,33
Крайние группы периодической системы обнаруживают таким
образом противоположные свойства. Поглощение р-частиц радия больше для
электроположительных, нежели для электроотрицательных элементов,
что, по всей вероятности, объясняется тем обстоятельством, что Р-часги-
цы сами являются носителями отрицательного электрического заряда.
Элемент
¦Литий
Углерод
Натрий
Магний
Алюминий
Кремний .
Калий . .
Кальций .
Железо
Кобальт .
Никель
Медь . .
Цинк . .
Мышьяк
Стронций
Палладий
Серебро .
Кадмий .
Индий . .
Олово . .
Сурьма
Теллур
Тантал
Осмий . .
Платина . .
Золото
Ртуть . .
Таллий
Свинец . .
Висмут
•
Температура,
0
0
0
0
0
—
0
16,8
0
20
0
0
0
0
20
0
0
0
0
0
0
19,6
—
0
0
0
0
0
0
0
Электропроводность
рх 10*
11,9
0,8
21,1
24,47
22,46
0,88
15,05
13,3
10,37
10,3
14,42
59
18,0
2,86
4,03
9,79
65
14,41
11,95
9,346
2,71
4,66
6,06
10,53
9,4
47,4
1,06
5,68
5,04
0,924
Температура,
°С
і.
—
—
0—100
0
—
.—
—
0
—
¦is
18
18
—
—
18
18
0
—
0
0
—
—
—
18
18
0
0
—
18
Теплопроводность
і
—
—
0,3760
0,3435
—
—
—
0,1665
—
0,1420
0,8915
0,2653
—
—
0,1683
1,006
0,2200
—
0,1528
0,0442
¦—
—
—
0,1664
0,7003
0,0148
—
0,0836
0,0177
Отношение
теплопроводность х 10~2
электропроводность
,.
—
—
1,53
1,53
—
—
—
і,б
—
1,46
1,51
1,43
—
—
1,72
1,55
1,53
—
1,63
1,63
—
—
—
1,85
1,49
1,43
—
1,66
1,9
Положение
элемента
в системе
1/2
IV/2
і/з
п/з
ш/з
IV/3
1/4
И/4
VIII/4
VIII/4
VIII/4
1/5
И/5
V/5
И/6
VIII/6
1/7
И/7
III/7
IV/7
V/7
VI/7
V/10
VIII/10
VIII/10
1/11
И/11
Ш/11
IV/11
V/11
Периодическая система химических элементов
151
Электро- и теплопроводность
Из других свойств простых тел и химических соединений мы
остановимся еще на электро- и теплопроводности.
В обоих этих свойствах, которые также стоят в тесной связи с
оптическими, сказывается более или менее ясно -выраженная периодическая
зависимость от величины атомного веса.
Согласно современным воззрениям, тепло и электричество через
металлы переносятся при помощи электронов. Этим обстоятельством
объясняется так называемый закон Франца и Видемана, по которому
он
60
$0
20
1 —'
>т
¦ L'M5V
к
Xtt
Br,
Си /
Zn
¦
Л л* А
Sr с* BQ
i .
гі
»¦
О
40
80
/20
too
200
Рис. 13. Диаграмма подвижности ионов
отношение между электро- и теплопроводностью для всех металлов
близко к постоянной величине. При этом для той и другой
проводимости (несмотря на крайнюю скудность опытных данных) можно заметить
довольно ясную периодическую зависимость от атомного веса*.
Совершенно иной характер, нежели у металлов, имеет, как известно,
электропроводность электролитов (кислот, оснований и солей). Здесь
прохождение электричества происходит при помощи ионов, то есть
атомов или атомных групті, несущих с собой определенный.электрический
заряд, равновеликий целому числу электронов. Притом, согласно
известной теории Аррениуса, ионы уже предсуществуют в водных, спиото-
вых и т. л. растворах электролитов, образуясь при диссоциации
нейтральных молекул этих последних.
Например, NaCl распадается на Na+ и С1~, Na2S04 —на 2Nah и
S04— и т. д.
Когда происходит электролиз, то принимают, что ионы эти под
влиянием существующей разности потенциалов передвигаются к
соответствующим полюсам и здесь выделяются, теряя свой заряд.
Величина электропроводности, поскольку она зависит от природы
данного электролита, на первом-плане определяется л о дв и жн ость ю
образующих его ионов, то есть относительной скоростью, с которой они
передвигаются при прочих равных условиях. Гитторф, Кольрауш и
другие разработали способы, позволяющие определить эти скорости для
различных ионов.
Сравнение их между собой показало, что они вполне подчиняются
периодическому закону, о чем свидетельствует зигзагообразный вид
ломаной (рис. 13), изображающей связь «подвижности» ионов с атомным
* В таблице на стр. 150 иод рубрикой «электропроводность» даны обратные
величины удельных сопротивлений, отнесенных к 1 см3 проводника, а под рубрикой
«теплопроводность» — число кал, проходящих в I сек. между двумя плоскостями сечения
в 1 см*, находящимися друг от друга на расстоянии 1 см при разнице температур
этих сечений в 1°.
152
Периодическая система химических элементов
весом, а также следующее сопоставление (.подвижность ионов дана для
^=25):
F
CI
Вг
J
Ь'і/і
75,1
78,1
77,0
Li
Na
К
Rb
42,6
52,6
75,5
78,6
1/2
1/2
1/2
1/2
Mg
Са
Sr
Ва
62
66
67
68
Cs 78,8
Аналогичному закону подчиняются и температурные
коэффициенты подвижности ионов.
Магнитные свойства. В главе II уже была отмечена
замечательная противоположность между элементами четных и нечетных
рядов в отношении их матнитных свойств. Первые — магниты, вторые —
днамагниты.
Другая форма зависимости между магнитными свойствами простых
тел-и атомным весом была указана Бахметьевым и .выражена им
при помощи ломаной линии, несколько напоминающей линию объемов
Мейера. К сожалению, она касается далеко не всех элементов.
Было также обращено внимание на существование некоторой связи
между знаком магнетизма и атомным объемам. Парамагнитные
элементы по преимуществу обладают минимальным объемом атома.
Все эти правильности не могут почитаться строго выдержанными; ни
одна из них не свободна от исключений. Главную причину отсутствия
более определенных, особенно количественных, соотношений между
магнитными свойствами элементов и атомными весами следует искать
частью в отсутствии систематических исследований по этому предмету,
а главным образом, вероятно, в том, что препараты, 'бывшие в руках у
многих исследователей, далеко не представляют гарантий достаточной
чистоты. Между тем, в данном случае это особенно важно, так как для
слабо матнитных и диамагнитных тел даже малейшие примеси железа
и других сильно магнитных (так называемых ферромагнитных)
металлов достаточны, чтобы в корне подорвать надежность результата.
Поэтому и до сих пор для некоторых элементов даже их магнитный знак
остается сомнительным.
В последнее время'более тщательные исследования были
предприняты Паскалем, которому, между прочим, удалось установить, что, по
мере возрастания атомного веса в подгруппах периодической системы,
величины магнитной восприимчивости элементов правильно возрастают,
по крайней мере, в исследованных нечетных рядах.
Приводим следующие данные Паскаля [27] для атомной
восприимчивости, помноженной на ]07.
Фтор
-65,5
Лзот
—58
Кислород
-5,8
Хлор
—209,5
Фосфор
—274
Сера
—156
Бром
—319
Мышьяк
—457
Селен
—240
Иод
—465
Сурьма
—775
Теллур
—.489
Висмут
-4806
Знак — указывает на то, что все вышеприведенные элементы, как
относящиеся к нечетным рядам (и частью к типическому
ряду)—диамагниты.
Периодическая система химических элементов
153
Глава VIII
ОБЗОР ОТДЕЛЬНЫХ ГРУПП ПЕРИОДИЧЕСКОЙ СИСТЕМЫ
Как мы уже видели, в каждую группу периодической системы
попадают элементы, представляющие .между собой ясные черты аналолии,
причем аналогия эта проявляется с особенной резкостью, если отдельно
сравнивать элементы четных и нечетных рядов, или же отдельных
подгрупп периодической системы.
Мы видели также, что некоторые свойства элементов, особенно
электрохимические, с большой правильностью и 'постепенностью
изменяются по мере того, как в пределах каждой группы или подгруппы
периодической системы нарастает атомный вес. Наконец, 'несомненно, что
самое это нарастание атомных весов, в свою очередь, подчиняется
некоторому, хотя и не вполне точному закону, открытие которого
натолкнуло еще Петтенкофера и Дюма на мысль о сходстве между
естественными группами элементов и рядами гомологов органических
соединений. Сближение это вылилось, между прочим, в ряд попыток
представить себе строение и образование элементов из одного мл-и
нескольких 'родоначальников. Наиболее последовательно эта идея проведена в
остроумной книге Н. А. Морозова: «Периодические системы
строения вещества» (М., 1907). Н. А. Морозов представляет себе атомы всех
элементов образованными из трех основных прото-элементов: про
тоге ли я, протоводорода и ар х о ни я, наподобие того, .как
молекулы углеводородов, принадлежащих к различным по степени
удаления от предела рядам, образованы из атомов углерода и водорода. Не
входя здесь в изложение этой теории, не могу не отметить, что,
опираясь на нее, Н. А. Морозов мог еще около 1883 г. предугадать
существование нулевой группы периодической системы, о которой химики з
то время не имели ни малейшего представления*.
Резюмируя предыдущее, можно сказать, что 'каждый из числа
элементов «гомологов» представляет как бы новую вариацию на одну и
ту же основную тему.
Настоящая 'глава будет посвящена до некоторой степени развитию
этого общего положения — краткому обзору отдельных групп (и
подгрупп) 'периодической системы. При этом для каждой группы
выясняются как границы сходства образующих ее элементов, так и
правильность, с которой изменяются свойства соответствующих простых тел и
сходственных соединений.
Такой обзор позволит нам, между прочим, остановиться на
рассмотрении некоторых свойств элементов и их сходственных соединений,
которые не настолько полно изучены, чтобы можно было на основании
их вывести какую-либо правильность, охватывающую всю
периодическую систему, и которые поэтому не были нами затронуты в
предшествующих главах.
I группа. Щелочные металлы. Сюда относится, кроме
представителей четных рядов, еще натрий:
Li Na К Rb Cs
Элементы эти составляют главную подгруппу I группы и являются
носителями характернейших особенностей этой последней. Сходство
между ними является настолько глубоким и полным, взаимная связь
* Ныне взгляды Н. А. Морозова нашли себе последователей (например, в лице
Никольсона), и можно надеяться, что дальнейшее развитие их поведет к новым
важным результатам.
154
Периодическая система химических элементов
настолько тесной, что только группа галоидов может соперничать с ними
в этом отношении.
Сходство между отдельными щелочными металлами прежде всего
отражается на свойствах простых тел, обнаруживающих все черты
металлического характера. Щелочные металлы обладают характерным
блеском, хорошо проводят тепло и электричество и легко вытягиваются в
тонкую проволоку. Мы видели выше, что спектры испускания
раскаленных паров у представителей этой группы также показывают ,глубочай-
шее сходство. Не менее аналогичны и Химические свойства щелочных
металлов. Со ртутью они дают амальгамы; с другими металлами
образуют соответствующие сплавы, в которых могло быть констатировано
большое число типичных «интерметаллических» соединений. Среди этих
соединений наблюдается довольно постоянное повторение общих типов,
на что было особенно обращено внимание Н. С. Курнаковым и его
учениками. Для примера приведем следующие меркуриды: Li, Na, К,
Rb и Cs.
Li3Hg Na:tHg
LiHg NaHg KHg CsHg
LIHg, NaHg2 KHg2 RbHga CsHg2
LiHg3 — KHg3
NaHg4 - RbHg4 CsHg4
KHg6 RbHg, CsHge
KHg10 CsHg10
Все щелочные металлы чрезвычайно энергично разлагают воду уже
при обыкновенной температуре, выделяя водород и образуя едкую
щелочь:
2Na-f2H20-2NaOH + H2.
На воздухе они весьма легко окисляются и притом тем легче, чем
больше атомный вес. Металлический цезий от соприкосновения с
воздухом воспламеняется уже при обыкновенной температуре. Щелочные
металлы всегда одноатомны (ср. выноску на стр. 94—95).
Сходственные соединения их отвечают вполне аналогичным формулам:
LiCl
NaCl
КС1
RbCl
CsCl
Li2C03
Na2C03
КвСОя
RbXO.,
Cs2C03
LiOH
NaOH
кон
RbOH
GsOH
Li,0
NauO
K20
Rb,0
Cs20
Li2S
Na2S
K2S
Rb2S
^_*SoO,
часто показывают отношения изоморфизма и вообще обладают
аналогичными свойствами. Гидраты окисей представляют с и ль н ы е
основания (едкие щелочи); хлористые соединения (и вообще соли сильных
кислот) имеют нейтральную реакцию (отсутствие гидролиза); наоборот,
•-соли со слабыми кислотами, например угольной (карбонаты), борной
и т. д. показывают реакцию щелочную, что указывает на наличность
гидролиза.
Как уже было упомянуто, особенно близки между собой члены
«триады» К, Rb и Cs. Это резко сказывается, например, при сличении состава
перекисей:
Li202 Na202 К,04 Rb204 Cs204
Между тем, как перекиси Li и Na являются производными перекиси
водорода (которую и выделяют при действии воды или кислот: Na202 +
Периодическая система химических элементов
155
+ H2S04 = H202 + Na2S04), перекиси калия, рубидия и цезия имеют
другой, но также одинаковый состав, как бы отвечающий гипотетическому
высшему окислу водорода Н20414, на существование которого имеются
намеки (как и на существование одного или нескольких «надозонов»).
Физические свойства простых тел и сходственных соединений в
группе щелочных металлов правильно изменяются с нарастанием атомного
веса. Исключения чаще всего встречаются для лития, реже — для натрия.
Физические свойства простых тел сопоставлены в следующей
таблице.
Элемент
Литий Li
Натрий N
Калий К
Рубидий Rb
Удельный вес
0,534
0,972
0,862
1,53
1,87
Атомный объем
13
23,7
45,4
56
71
Температура
плавления, °С
186
95,6
62,5
38,5
26,5
Температура
кипения, °С
1400
878
757
697
670
Электрическое
сопротивление
при 0°, мком
8,6
4,3
6,2
11,6
18,1
Из этой таблицы видно, что по мере повышения атомного веса в ряду
щелочных металлов постепенно и правильно возрастают: удельный вес,
атомный объем и электрическое сопротивление. Наоборот, температуры
плавления и кипения столь же правильно понижаются.
Рис. 14.
В самое последнее время Л, Гакспилль [28] определил упругость
пара щелочных металлов и нашел, что она правильно повышается от
Na к Cs, как это 'видно из прилагаемой диаграммы, где температуры
нанесены по оси абсцисс, а величины соответствующих упругостей
пара — по оси ординат.
К этим данным можно прибавить еще термоэлектрические
свойства, недавно изученные Броневским и Гакспиллем [29]. На рис.
14 сопоставлены термоэлектрические коэффициенты ~^~
(^—электродвижущая сила при температуре t, соответствующая разнице темпе-
* В пределах температуры между —78 и.—183°.
156
Периодическая система химических элементов
ратуры между спаями) для пары из щелочных металлов+свинец,
в мкв:
— 4,16—0,0144 і
—11,33—0,0376 /
— 8,26—0,0302 t
+ 0,66—0,0010 t
К, Rb и Cs представляют и здесь правильный ряд, между тем как Na-
показывает уклонение.
О других физических свойствах щелочных металлов см. главы VI
и VII.
Переходим к постепенному изменению химических свойств
элементов щелочной подгруппы.
Хотя точное определение электролитических потенциалов в данном
случае невозможно, однако сравнение теплот образования солей и
других свойств сходственных соединений приводит нас к заключению, что
положительное электросродство правильно возрастает (сродство к
электрону падает) в ряду
Li Na К Rb Cs
от лития к цезию.
Крайнее положение лития по сравнению с остальными членами
группы особенно резко сказывается. Соли лития с сильными кислотами
очень легко растворимы в воде, часто расплываются на воздухе. При
выпаривании водных растворов хлористого лития LiCl ясно заметен
гидролиз: раствор приобретает слабо-щелочную реакцию*.
С другой стороны, соли лития со многими слабыми и средней силы
кислотами отличаются трудной растворимостью в воде, и в этом
отношении литий приближается к щелочноземельным металлам. Так, при
комнатной температуре в 1 л воды растворяется:
COOLi
I
COOLi 1,2 г-экв
Li2COa 0,36 г-экв
LiF 0,1 г-экв
Lit1P04 0,01 г-экв
Сравнительно малой растворимостью отличается и гидрат окиси
лития.
Как элемент с относительно слабыми
электроположительными свойствами, с несколько ослабленной тенденцией к переходу в
ионное состояние, литий один из всех щелочных металлов обнаруживает
ясно выраженную способность к образованию комплексов. Так, соли его
дают ряд соединений с аммиаком:
LiCl • NHS LiCl - 2NH9 LiCl. 3NH3 LiCl • 4NH3
и аминами **. Этим обстоятельством объясняется увеличение
растворимости солей лития в воде от прибавления NH3.
Соединения эти отличаются довольно значительной прочностью. Так,
аммиакат LiCI*2NH3 при 68,8° имеет диссояиационную упругость NH3
* Чтобы приготовить безводный LiCl, удаление воды производится в токе HCI.
** При этом весьма вероятно, что молекулы аммиака и аминов присоединяются
к і.і, образуя комплексный катион.
К
Rb »
Cs »
Периодическая система химических элементов
157
в 373 мм, а при 89,2° —980 мм, и только наиболее богатый аммиаком
LiCl • 4NH3 уже при 12,74° имеет упругость = 754 мм.
Равным образом у солей лития значительно сильнее, чем у солей
других щелочных металлов, выражена тенденция к образованию
гидратов.
В некоторых из числа только что указанных отношений с литием
могут быть сопоставлены другие щелочные металлы, и тогда
постепенное возрастание электроположительного характера от Li к Cs ясно
выступает.
Соли натрия еще обнаруживают, хотя и в весьма слабой степени,
способность присоединения аммиака. По Жоанни, существует,
например, непостоянный аммиакат NaCl • 5NH3, разлагающийся уже при
—24°, следовательно, несравненно менее устойчивый, нежели
аммиакаты LiCl. Для солей К, Rb и Cs аммиакаты вообще неизвестны.
Способность к образованию гидратов ясно уменьшается от Li к Cs.
Так, соли лития и натрия дают гидраты со следующими числами
молекул воды (на 1 молекулу соли):
LiC]
1 и 2
NaCl
2
Очень
непос-
тоянно
LiN03
1/2 и 3
NaN03
7 (?)
Очень
непостоянно
Li2S04
1
Na2S04
10,7 и 1
LiC104
1 и 3
NaC104
1 (?)
LiClO^
1/2
NaCI03
О
Для К, Rb и Cs все перечисленные соли кристаллизуются без воды.
Зато для солей калия, рубидия и цезия является весьма
характерной способность присоединять частицы кислот, солей слабых оснований
с сильными кислотами, вообще молекул электроотрицательного
характера, причем образуются соединения типа комплексных. Эта
способность резче всего выражена у рубидия и особенно у цезия.
Так, азотнокислые соли этих металлов присоединяют одну и две
молекулы HN03, образуя (аномальные) сравнительно прочные,
кристаллические при обыкновенной температуре, кислые соли, что в этом
случае особенно замечательно, так как HN03 — одноосновная кислота:
RbN03-HN03
CsNO,rHN03
RbN03.2HN03
CsN03.2HNO:t
Аналогичные соединения, но менее устойчивые, дает н KN03.
Подобным же образом хлористый рубидий и хлористый цезий
присоединяют молекулы Вг2, СШг, BrJ и т. д.
RbCl ¦ Вг.>
CsCl ¦ Вг,
RbCl-ClBr
CsCl-СЮг
RbCl-BrJ
CsCl-BrJ
При этом комплексообразование происходит за счет аниона соли, но
прочность комплекса, как целого, находится в зависимости от сильных
электроположительных свойств Rb и Cs. Калий имеет лишь малую
склонность к образованию подобных соединений. У натрия и лития она
не могла быть замечена.
К той же категории явлений относится образование двойных
солей, например квасцов, о которых еще Дюма говорил, что при
возникновении их сульфат щелочного металла нейтрализует сернокислый алю-
158
Периодическая система химических элементов
миний, хром или железо, как щелочь — кислоту*. Из щелочных
металлов квасцы образуют Na, К, Rb и Cs. Квасцы лития, крайнего члена
группы, до сих пор не получены и, может быть, неспособны к
существованию; для Na они мало устойчивы. Все квасцы кристаллизуются в
правильной системе и между собой изоморфны. Многие свойства их
правильно изменяются от Na к Cs, например, растворимость,
молекулярные объемы и т. д.
Me2S04Al2(S04)a-24HsO
Температура, °С . .
100 частей воды
растворяют, г
Na
13
46,7
К
20
15,1
Rb
17
2,27
с
17
0,619
Молекулярные объемы (в твердом состоянии)
MeaSO*Me---2<SO+)a -24 Н20
Аі
Fe
Cr
Na
549,9
К
546,9
557,1
550,8
Rb
562,3
573,3
561,7
Cs
579,7
579,3
581,8
С электрохимическими свойствами щелочных металлов поучительно
сопоставить образования их галоидных солей. Мы приводим их (в
ккал/г-экв) по новейшим данным Де-Форкрана**:
Li
Na
К
Rb
Cs
Cl
97,04
98,52
104,05
105,00
106,38
Br
87,06
90,45
97,48
99,21
101,38
J
71,25
76,50
85,21
87,45
90,40
F
119,98
111,40
108,80
107,95
106,58
1/2 O
71,40
50,35
43,40
41,75
41,35
Теплоты образования для хлоридов бромидов и иодидов
медленно, но правильно возрастают от лития к цезию и в то же время от иода
к хлору, так что максимальная величина соответствует OsGl, а
минимальная— LiJ.
Интересно, что для фтористых солей порядок изменения теплоты
образования обратный по сравнению с другими галоидами, и притом
тот же самый, что и для соответствующих окислов: максимальное
теплообразование соответствует литию, минимальное — цезию.
Аналогия между кислородом и фтором, несомненно существующая, несмотря
на кажущееся огромное различие между этими элементами, очень ясно
проявляется в этом отношении.
* Ныне обыкновенно на «двойные» соли, подобные квасцам, смотрят как на
весьма непрочные комплексные соединения и предполагают, что соединение молекул двух
солей, из которых каждая сама по себе насыщена, с точка зрения классической теории
валентности, происходит за -счет остаточных или побочных (А. Берн ер) единиц
сродства. В квасцах, вероятно, имеется непрочный комплексный анион AMSOih или
AI(S04)*
** Щелочные металлы, окислы и соли — в твердой состоянии, галоиды и
кислород — в газообразном [30].
Периодическая система химических элементов
159
Постепенность в изменении термических свойств соединений
щелочной группы можно видеть также, сличая количества тепла,
выделяющиеся или поглощаемые при растворении соответствующих галоидных
соединений и окислов в воде (1 г-экв в 2 л) *:
Li
Na
К
Rb
Cs
CI
+8,37
—1,30
-4,40
—4,50
-4,68
Br
+11,35
-0,30
—5,08
-5,96
-6,73
J
+14,76
+1,30
—5,11
—6,90
—8,25
F
-1,04
-0,60
+4,10
+5,80
+8,37
о
+15,38
+28,25
+37,50
+40,00
+41,60
Если позволительно считать тепло растворения (грубоприблизитель-
ной, конечно) мерой «сродства» данного вещества к воде, то мы должны
заключить, что это сродство падает в ряду хлористых, бромистых и
йодистых щелочных металлов от лития к цезию. В этом отношении мы
имеем полный параллелизм с теплотами образования соответствующих
галоидных солей. Отношение фтористых солей и окислов в этом случае
также является противоположным по сравнению с солями С1, Вг и J **.
Из других соединений щелочных металлов, среди которых может
быть обнаружено правильное изменение степени устойчивости,
заслуживают внимания углекислые соли. Мы сравним их на основании данных
Лебо для диссоциационных упругостей С02 (соответствующие урав-
нению МеоСОз^МегО + СОо) при различных температурах:
Na2C03
Rb2C03
Cs2C03
700°
1
740°
2
680°
4
820°
2,5
830°
6
805°
6
1010°
14
1020°
20
1000°
44
1100°
21
1080°
33 мм
1090°
90
1200°
41 м
1180°
157 м
Как видно из этих цифр, устойчивость карбонатов ясно понижается
от Na к Cs. Интересно отметить, что в группе щелочноземельных
металлов мы имеем как раз обратное отношение.
Вторую подгруппу I группы образуют тяжелые металлы
Си Ag Au
Как уже было упомянуто (глава И), как раз в крайних группах
периодической системы, следовательно, в I и VII, расхождение между
элементами четных и нечетных рядов, отклонение от признаков,
которые можно считать нормой для данной группы, достигает максимума.
Отсюда понятно, что медь, серебро и золото, образующие
нетипичную, или побочную подгруппу I группы периодической системы,
сохраняют лишь небольшое число признаков, для этой группы
характерных С другой стороны, металлы эти показывают много общего с
металлами VIII группы, что. и дало повод Д. И. Менделееву
отнести их в обе эти группы одновременно.
* De Forcrand Ioc. cit. й
** Сравнивая таплоты растворения хлористых, бромистых и йодистых солеи
одного и того же металла, можно видеть что в случае лития и натрия они возрастают
от хлора к иоду, для К, Rb и Cs —- наоборот.
160
Периодическая система химических элементов
Типичными для первой группы признаками этих элементов следует
считать прежде всего способность образовать ряд соединений, в
которых они являются одноатомными, как щелочные металлы:
Cu20 CuCl Ag,0 AgCl AgNO, Au.,0 AuCI
CuBr
CuJ
AgBr Ag2S04
AgJ
AuBr
AuJ
Притом между аналогичными соединениями серебра и щелочных
металлов наблюдаются отношения изоморфизма, например:
Ag,S04 Ag2Se04 Ag2S.,06 • 2Н20
Na2S04 Na2Se04 Na2SsOe - 2H«0
Li2S2Oe-2H20
Химическая природа серебра и в других отношениях сближает
элементы нечетных рядов I группы со щелочными металлами. Так, подобно
I подгруппа
Li Na К Rb Cs
Легкие металлы с максимальным объемом атома
II подгруппа
Cu Ag Au
Тяжелые металлы с минимальным
объемом атома
Удельный вес
Атомный объем
Li
0,534
13,1
Na
0,972
23,7
К
0,862
45,!Г)
Rb
1,53
56
Cs
1,87
71
Удельный вес
Атомный объем
Си
8,93
7,1
Ag
10,5
10,3
Ли
19,3
10,2
Простые тела легкоплавки
Валентность, как правило, — 1
Окислы стойки и трудно
восстанавливаются
Гидраты окислов — сильные
основания, с трудом теряют воду и легко в ней
растворяются
Соединения с галоидами, типа МеХ,
особенно с О, Вг и J легко растворимы в
воде
Сернистые соединения (Na2S, K2S и пр.)
легко растворимы в воде и подвержены
гидролизу
Весьма мало (только для Li) склонны к
комллексообразованию
Соли все бесцветны
Электросродство (положительное)
повышается с нарастанием атомного веса
Простые тела плавятся около 1000°
Валентность вообще переменная:
Си15 Ag Au
1 и 2 \ 1 и 3
Окислы нестойки и легко восстанавливаются
Гидраты окислов — сравнительно слабые
(кроме AgOH) основания, все трудно
растворимы в воде и очень легко теряют
воду
Галоидные соединения типы МеХ очень
трудно растворимы в воде (CuCl, AgCl,
AuCI)
Сернистые соединения Ag2S, Cu2S, CuS
и пр. очень мало растворимы в воде и
водой не разлагаются
Крайне склонны к образованию комплексов:
например, с галоидоводородами и
галоидными солями K2CuCl3, (NH4)2CuBr3,
HAuCljj, NaAuCl-j с аммиаком и аминами
[Cu6NH3] С13, [Cu4NH3] CI.,, Ag2NH3Cl2
С цианистыми солями:
K[Cu(CN)2], K2[Cu(CN)3],
K2[Cu(CN)4], K[Ag(CN)2], KAu(CN)2
и пр.
Соли Си и Аи окрашены; соли Ag
бесцветны
Электросродство (положительное)
уменьшается с нарастанием атомного веса
Периодическая система химических элементов 161
этим последним, серебро образует только один солеобразный окисел
Ag2016, подобно Na20. КзО и т. д. Гидрат окиси серебра, присутствие
которого принимают в растворах Ag20, представляет сильное (хотя и
уступающее едким щелочам) основание. В соответствии с этим, соли
серебра с сильными кислотами AgN03, Ag2S04 и пр. в водных растворах
не показывают гидролиза, и реакция их нейтральна. В некоторых
случаях наблюдается тенденция к образованию кислых солей, а сульфат
серебра Ag2S04 способен образовать квасцы, как сернокислые соли
типичных щелочных металлов.
Посмотрим теперь, какие характерные черты, сближающие между
собой элементы Си, Ag, Аи, в то же время отличают их от щелочных
металлов, составляющих первую подгруппу I группы (см. стр. 160).
II группа. Общей чертой элементов II группы следует считать
прежде всего преобладание в типичнейших образуемых ими
соединениях валентности = 2 и связанное с этим обстоятельством сходство в
составе аналогичных соединений, отвечающих типу МеХ2:
МеН2 MeO MeS MeCU MeS04 МеСОя
В электрохимическом отношении у элементов II группы ясно
выражен металлический, а у их окислов и у соответствующих
гидратов — основной характер.
Галоидные соединения, как и для элементов I группы, носят
характер солей; с металлами элементы II группы дают сплавы, причем
обнаружены характерные интерметаллические соединения.
Элементы II группы, подобно элементам I группы, довольно
отчетливо распадаются на две подгруппы, особенно начиная с 4-го ряда,
причем представители первых двух периодов ближе, хотя и не вполне,
примыкают к главной подгруппе:
Главная подгруппа Побочная подгруппа
(Be)
Легкие
металлы
(Mg)
Са Тяжелые J
Sr металлы [
Ва
Ra
Zn
Cd
В главной подгруппе особенно тесную естественную семью образуют
щелочноземельные элементы Са, Sr, Ва, к которым примыкает и вновь
открытый Ra (см. главу X). Бериллий и особенно магний по многим
своим свойствам близки также к подгруппе Zn, Cd, Hg.
Обособление подгрупп является в данном случае менее резким,
нежели в I группе, ибо между Са, Sr, Ва, с одной стороны, Zn, Cd и Hg, с
другой, не существует уже столь глубокого различия, как между
щелочными металлами и Си, Ag, Аи. Мы увидим, что это различие еще
более стушевывается при переходе к III группе.
Главная1 подгруппа (бериллий и щелочноземельные
металлы) характеризуется прежде всего постоянной (ср. выноску на стр. 95)
валентностью = 2.
BeCI2 MgCl, СаС12 SrCU BaCI2 RaCl,
Аналогия в типах соединений, образуемых элементами этой
подгруппы, распространяется далее и на состав соответствующих
комплексных соединений и гидратов, хотя, как будет указано ниже, далеко
не все типы, известные для одного из членов подгруппы, осуществимы
(веонее, доСтаточнр прочны! 'для других.
П Л. А. Чугаев, т. III
162
Периодическая система химических элементов
Сходственные соединения и здесь обладают аналогичными
свойствами (особенно для Са, Sr и Ва). Например, сульфаты CaS04, SrS04,
BaS04 (и RaS04) весьма трудно растворимы в воде, а галоидные
соединения типа RX2 (кроме фтористых), наоборот, легко.
Положительное сродство в главной подгруппе, особенно для Са, Sr,
Ва и Ra (четные ряды) значительно выше, чем для элементов побочной
подгруппы (нечетные ряды), как и следует ожидать по их положению
в периодической системе. Притом электросродство возрастает от Be к
Ва (между тем как в побочной от Ъа к Hg наблюдается обратное
отношение). В соответствии с этим, в главной подгруппе правильно
изменяется основной характер гидратов окисей: Ве(ОН)2 — очень слабое
основание, Mg(OH)2 — значительно более сильное, но еще весьма
склонное к образованию основных солей; средние соли, например MgCl2,
легко подвергаются гидролизу; Са(ОН)2, особенно же Sr(OH)2 и
Ва(ОН)2 представляют уже сильнейшие из числа известных
оснований; такие соли, как СаС12 и особенно SrCl2 и RaCl2, практически
не гидролизованы в водном растворе.
Проявление возрастающей силы оснований от Са(ОН)2 к Ва(ОН)2
можно усмотреть, между прочим, в постепенно повышающейся степени
прочности соответствующих карбонатов, диссоциация которых,
происходящая по схеме МеС03Х-МеО+С02, становится тем труднее,
чем выше атомный вес металла*.
Упругость С02 достигает 1 атм при следующих температурах:
Для СаС03 SrCO- BaC03
при 825° 1158° 1450°
MgC03, несомненно, разлагается еще легче, чем СаС03) а ВеС03
принадлежит к числу наименее прочных карбонатов, ибо разлагается при
температуре немного выше 100°.
Постепенное нарастание электросродства от Be к Ва сказывается
далее в том обстоятельстве, что бериллий весьма склонен к
образованию часто весьма прочных комплексных соединений,
например летучих, без разложения ацетилацетоната
(СН3 — С - СН — СОСН,)2
\
о—
и ацетата Ве40(СН3СОО)б [31]. Магний также образует большое
число комплексов **, но прочность их значительно меньше, чем у
бериллия. Большинство их легко разлагается водой.
Таковы соединения солей Mg с простыми и сложными эфирами,
спиртами, кислотами т. п. [32].
MgBr2 (C2H5OH)e MgJ2(CH3OH)6
MgBr2 [(QH2)20]2 MgJ2[(C5H5)20]2
MgBr2 (C6H5COOC2H5)o MgJ2(CH3COOC2H6)e
MgBr2 (CH3COOH)6 MgJ2(CH3COOH)
6
С величайшей легкостью образуются гидраты, а также двойные
соли. Последние непрочны и сполна разлагаются водой, Таковы зсем
известные соединения MgC^HzO, MgS047H20, MgCl2KC16H20
(карналлит), MgSO4K2SO46H20 (шёнит) и т. д.
* В ряду карбонатов щелочных металлов наблюдается, однако, обратное
отношение, если верны имеющиеся на этот счет литературные данные (см. стр. 159).
** Одним из таких комплексов магния по недавним замечательным исследованиям
Вильштеттера следует считать хлорофилл и некоторые его производные.
Периодическая система химических элементов
163
У солей кальция способность к образованию комплексов и двойных
солей заметно меньше, чем у магния, а затем к Sr и Ва она
претерпевает дальнейшее весьма резкое понижение и постепенно сходит на нет.
Способность образовывать гидраты, сильно выраженная у солей
кальция, также заметно понижается при переходе к Sr и Ва. Это
прежде всего сказывается в том обстоятельстве, что от кальция к стронцию
и барию уменьшается максимальное число молекул воды, способное
присоединиться к 1 молекуле соли с одноименным анионом. Следующие
примеры могут послужить в качестве иллюстрации.
MgCl212Н20
MgCl28H26
MgCl2 6H20
MgCi2 4Н20
MgCl2 2Н20
Mg (N03)2 9Н20
Mg (N03)2 7H20
Остальные гидраты
не могут быть
получены без примеси
основных солей
СаС12 6Н20
СаС12 4Н20
СаС12 2Н,0
SrCl, 6Н20
SrCl, 2Н20
ВаСі, 2Н,0
MgS0412Н,0
MgS04 7H26
MgS04 6Н20
MgS04 5Н20
MgS04 4Н20
MgS04 Н20
CaS04 2Н,0
2CaS04 Н20
SrS.04 безводный
BaSO, безводный
Са (N03), 4Н20
Са (N03), ЗН,0 (?)
Са (N03)2 2H20 (?)
Sr (N03)2 4Н20
и безводный
Sr (N03)2
Ва (N03)2 2HoO
(не устойчивый и безводный)
ВаС1.2 Н,0 Ва (N03),
Разным образом, сопоставляя между собой гидраты аналогичных
солей по степени их прочности, легко заметить, что последняя
правильно падает от Са к Ва, как это видно из постепенного повышения
величин упругости диссоциации, взятых для одной и той же температуры:
Упругость диссоциации в мм рт. ст. при 30°:
6,6
11,9
не известен (следовательно упругость диссоциации
значительно больше, нежели для гексагидратов
СаС12 и SrCl2).
Упругость диссоциации в мм. рт. ст. при 36,5°:
4,0
12,75
Рассмотрение элементов этой подгруппы мы закончим
сопоставлением некоторых, главным образом физических, свойств
соответствующих им простых тел и сходственных соединений.
Теплота образования галоидных соединений Са, Sr и Ва
(из твердых металлов и газообразных галоидов в ккал по Де-Форкрану)
СаС126Н20
.ЧС1,бН20
B-iGUWoO
СаС122Н20
ВаС122Н20
Са
г
Sr
2
Ва
2
F
119,59
118,51
114,29
С1
95,22
97,83
98,54
Вг
84,60
88,25
89,91
J
70,50
73,75
74,95
И*
164
Периодическая система химических элементов
оООб
Hdu вннэнні/ооо ojox
-DHdoirx ojOHHoiracim
ocd qxooHtfoaoduod
-ін^іге веиі-нэігЕаиаме
oOG8 и^и винэниНэоэ
ojoiondoirx олоннэіг
-ярішо оэя иічняігэіг ^
Do 'zI03W KHHOHHtf
-ЭО0 OJOXOHdOITX BHH
-эігяиіш edXiBdauwsx
Растворимость (в лг в 1 л водного раствора) при 18°
••
О
ел
ге
f-
то
•е-
<=;
?".
о
фтористого соединения
MeF,
vvyy g '0*H 3MJ.RQ
-ей a o3W gk иэизіо
BHHSdoeioed оішэі
BHHeaOSBdgo оігцэ?
Эо 'В1ГЭХ OJOJ-OOdU ВИН]
нуц)0 упнкоху
вігэі олохэ
-odu эоа^ипнч^И1^
Элемент
1
!
1
j? то
X э*
О «
CQ cj
J3 О)
н в:
я о
В* ffl _
*-< и <
га о
Си 03
Очень легко
растворяется
і
;
1430
4,9
1,85
Бериллий
Be =-9,1
1
1
36,5 г в 100 г воды
при 20°
87,4
1
143,3
635
14,0
1,74
см
со
CN
«'II
GO***
CD
1,97
774
2023
16,3
СО
152
780
25,4
1,58
о
§11
& то
58,2
2,645
873
114,3
117,3
29,8
139,6
800
со
2,5
СО
со
¦a t^"
SjCO
U\\
и
со
3,12
950
2,30
1605
35,6
125,8
850
со
со
3,8
Барий
Ва = 137,37
Периодическая система химических элементов 165
Растворимость некоторых солей Са, Sr и Ва при 18°, по Абеггу
(указано число грамм-молекул на 1 л воды);
N03 С103 ВЮ3 JO, CI Br J
Са 5,2 5,3 2,3 0,007 5,4 5,2 4,8
Sr 2,7 4,6 0,9 0,006 3,0 3,4 3,0
Ва 0,33 1,1. 0,02 0,001 1,7 2,9 3,8
S04 Сг04 С03 С204 F ОН
Са 1,5x10-2 ЗхЮ-2 1,3x10-4 0,4x10'* 0,0002 0,02
Sr 6x10-4 бхЮ-з 0,7x10-* 2,6x10'* 0,0009 0,06
Ва Ю-» 1,4x10-* 1,1x10-4 3,8x10-4 0,009 0,22
При этом замечаются обычные правильности: повышение плотности
и атомного объема металлов с нарастанием атомного веса,
повышением температуры плавления; теплота образования окислов RO
правильно понижается от СаО к ВаО, а теплота гидратации RO + H20
соответственно повышается.
Теплоты образования галоидных соединений изменяются в
зависимости от атомного веса металла совершенно аналогично тому, что
наблюдается в группе щелочных металлов. Для хлоридов, бромидов и
иодидов тепло образования возрастает от Са к Ва, между тем как в
ряду фтористых соединений изменение происходит в обратном
порядке, то есть в том же, что и для кислородных. Аналогичные
правильности наблюдаются и при сопоставлении свойств сходственных
соединений этих элементов: температур плавления, растворимости и т. п.
Побочная подгруппа II группы содержит три элемента: цинк,
кадмии и ртуть. По многим своим свойствам к ней примыкают и два
элемента главной подгруппы — Be и Mg. Все эти элементы двуатомны,,
кроме, быть может, ртути.
Ртуть — единственный элемент II группы, для которого известны
два ряда солеобразных соединений: МеХ2 и МеХ или Ме2Х2. Для ртути
это будут закисные и окисные соли. Отсюда, однако, вовсе еще не
вытекает с необходимостью заключение об одновалентности ртути в
HgCl, HgN03 и т. д. Наоборот, существует ряд данных, позволяющих
предполагать, что и в солях закиси ртуть является двухвалентной, что,
следовательно, формулы этих солей должны быть удвоены, например
С1 — Hg — Hg — С1 и прочие.
Продолжительный спор о валентности ртути долгое время был
связан с экспериментальным решением вопроса о составе молекулы
каломели. Плотность пара была найдена (выше 400°) отвечающей простои
формуле HgCl; но затем оказалось (В. Мей ер и другие), что в этих
условиях происходит диссоциация с образованием паров ртути и
сулемы *. Так как диссоциация эта могла произойти по одному из двух
уравнений:
2HgCl^HgCI2+Hg или
Hg2Cl2CHgCl2+Hg,
то вопрос оставался открытым. Существование двойных молекул в
парах каломели было доказано позднее опытами Бр. Бекера, который
* Одно из доказательств такой диссоциация заключается в том, что листовое
золото при внесении в пары каломели тотчас же образует амальгаму. Пары сулемы не
обладают этим свойством.
166
Периодическая система химических элементов
получил при полном отсутствии влаги плотность пара, отвечающую
формуле Hg2Cl2. Наконец, Огг на основании электрохимического
отношения солей закиси ртути приходит к заключению, что они образуют
ион Hg4+2 *•
Как уже было указано в главе IV, положительное электросродство
падает в серии Zn, Cd, Hg по мере повышения атомного веса. Кадмий
благороднее цинка, но менее благороден, чем ртуть. Поэтому из трех
названных металлов цинк наиболее способен к образованию
самостоятельных ионов. За ним следует по порядку кадмий и, наконец, ртуть.
Поэтому для сходственных солей этих металлов, например для
галоидных, степень диссоциации, а следовательно, и электропроводности
падает от Zn к Ист. В соответствии с этим от цинка к кадмию возрастает
склонность к образованию комплексных соединений.
Для цинка они хотя и существуют, но вообще мало прочны и по
большей части разлагаются водой. Более прочны комплексы,
образуемые кадмием, для которого особенно характерна доказанная еще Гит-
торфом способность к образованию аутокомплексных соединений.
Наконец, ртуть образует очень большое число (частью весьма
прочных) комплексных соединений с аммиаком, аминами, тиоэфирами и
т. п., а также комплексных солей, из которых особенно известны соли
кислот HHgJ3 и H2HgJ4 (двойные соединения йодистых металлов с
HgJ2).
В связи со способностью металлов II группы к образованию
комплексных соединений в широком смысле этого слова, нельзя не
обратить внимания на следующее различие, существующее в этом
отношении между обеими подгруппами.
С одной стороны, способность к комплексообразованию резче
выражена в побочной подгруппе, соответственно с более слабо
выраженным (положительным) электросродством элементов, ее образующих.
Особенно ясно это бросается в глаза при сопоставлении- элементов
четных и нечетных рядов:
Са, Sr, Ba, (Ra) и Zn, Cd, Hg.
С другой стороны, в самом характере комплексов, происходящих от
элементов обеих подгрупп, нельзя не заметить существенной разницы.
Быть может, резче всего эта разница проявляется в способности солей
металлов побочной подгруппы сочетаться с органическими сульфидами
R2S, R—S—(СН2)П—S—R (где R — органический радикал) и
некоторыми другими сернистыми соединениями. Элементы главной подгруппы
лишены такой способности, но зато они образуют характерные
соединения с водой (гидраты) и органическими кислородсодержащими
соединениями (спиртами, кислотами, эфирами и т. п.).
Это различие приобретает интересное освещение, если его
сопоставить с неодинаковым характером простейших сернистых и кислород-
пых соединений (производных сероводорода), образуемых элементами
обеих подгрупп. Между тем, как элементы побочной подгруппы дают
характерные и прочные сульфиды: ZnS, CdS, HgS, характеризующиеся
малым произведением растворимости (см. ниже в таблице), сернистые
соединения элементов главной подгруппы BeS, MgS, CaS, SrS, BaS
непрочны, мало характерны, обладают высокими величинами
произведения растворимости и легко разлагаются кислотами. Вода также их
легко гидролизирует.
* Заметим, что Д. И. Менделеев до самого последнего времени считал ртуть з
солях закиси одноатомной и придавал этим последним простые формы: HgG, HgNOs
и т. п.
Периодическая система химических элементов
167
Совершенно обратное отношение представляют окислы
соответствующих металлов. Здесь наибольшей прочностью отличаются
соединения элементов главной, и наименьшей — соединения элементов
побочной подгруппы; термические данные дают этому отношению хорошую
иллюстрацию (см. стр. 164 и 169).
Таким образом, те именно элементы, которые способны образовать
прочные и характерные сульфиды (бинарные соединения с серой),
дают и характерные комплексы с тиоэфирами и другими сернистыми
органическими соединениями; наоборот, металлы, окислы которых
прочны и характерны, легко сочетаются с кислородсодержащими
органическими соединениями.
Ныне для объяснения химического строения молекул комплексных
соединений многие (особенно А. Берн ер) прибегают к гипотезе по*
бочных единиц сродства, допуская, что этими последними
удерживаются в молекуле целые частицы, сами по себе способные к
самостоятельному существованию, например HsO, NH3, C2H5NH2, (C2H5)2S, KG,
HNO2 и т. п. Только что приведенное сближение заставляет считать,
что между побочными и главными единицами сродства одного и того
же элемента (а также элементов, представляющих близкую аналогию)
должна существовать тесная связь,— вывод, яснее всего понимаемый с
точки зрения электронной теории валентности.
Характерной чертой побочной подгруппы II группы следует еще
считать способность к образованию летучих металлоорганических
соединений, например, Zn(CH3h, Cd(CH3b, Hg(CH3h*.
Целый ряд физических и химических свойств элементов
рассматриваемой подгруппы и их аналогичных соединений правильно и
постепенно изменяется с возрастанием атомного веса. Так, повышаются
удельные веса и атомные объемы, понижаются температуры плавления и
кипения простых тел; понижается теплота образования, а также
растворимость окислов и галоидных соединений и произведение
растворимости сульфидов (см. таблицу на стр. 169) и проч.
III группа. Положение III группы в периодической системе
определяет максимальную и притом диминирующую валентность ее
представителей — 3.
* Бериллий по способности образовывать летучие металлоорганические соединения
(например, Ве(С2Н5)2) примыкает к Zn, Cd и Hg. Наоборот, Mg летучих соединений
не дает, но образует смешанные соеди-нетшя
СН3 , ХВг
л т. и., легко вступающие в комплексное соединение с простыми эфирамн^ (Гриньяр).
По Бекману, аналогичным образом ведет себя и кальций, образующий, например,
нелетучее тело
XL2H5
Са^
М
¦но не дающий летучих соединений типа CaR2. Мы видим здесь еще одну черту,
сближающую Mg с Са, между тем как бериллий в данном отношении примыкает к под-
rpvnne Zn, Cd, Hg.
Заметим, однако, что другие черты сближают -с цинком также и магнии. Сюда
относится особенно изоморфизм некоторых солей. например. MgS047H20 и
2nS047H20.
168
ПериоОическая система химических элементов
В203 В(ОН)3 А120;* А1(0Н)3 Ga(OH)3 In(OH)3
ВСіз AICI3 GaCl3 InCl3
Sc203 Yt203 La203 Yb203
ScCl3 YtCl3 LaCl3 YbCI3
Число отступлений от «нормы» здесь, однако же, больше, нежели
в I и II группах.
Простые тела, кроме бора, сохраняют еще характер металлов.
Характер высшего солеобразования окисла R2O3 обычно слабоосновной
и в то же время слабокислотный, частью с преобладанием того или
другого. Так, для В2О3 преобладание получает характер кислотного
ангидрида, а для Ъг%Оъ резко преобладает характер основного окисла.
Главная и побочная подгруппы здесь еще менее резко
разграничены, чем во II группе.
Главная подгруппа III группы обнимает элементы
В АГ Sc Yt La (Yb)
(?)
•С достоверностью известны только такие соединения их, в которых
они являются трехатомными.
Об изменении электросродства в этом ряду мы можем судить
только по косвенным данным, так как электродные потенциалы здесь не
могут быть определены. Несомненно, однако, что с увеличением
атомного веса повышается основной характер окислов типа R2O3 и
гидратов И(ОНз), совершенно аналогично тому, что наблюдается в I и II
группах периодической системы.
Борному ангидриду В203 соответствует слабая борная кислота
В(ОН)з, у которой основые свойства имеются лишь в виде намеков.
Глинозем А120з —типичное соединение двойственного (амфогерного)
характера. Его гидрат А1(ОН)3 дает соли со щелочами так же, как с
кислотами. Гидрат окиси скандия Sc(OH)3—слабое основание. В
гидрате окиси иттрия Yt(OH)3 основные свойства уже значительно
повышены, а кислотные едва заметны, ибо она неспособна растворяться в
щелочах, а с другой стороны, вытесняет аммиак из солей аммония. Наконец,
гидрат окиси лантана La (ОН)3 принадлежит к числу сильных оснований:
он окрашивает лакмус в синий цвет и жадно поглощает СОг из воздуха.
Безводная окись лантана ЬагОз гасится подобно извести (Ьаг6з +
+ ЗН20 = 2Ьа(ОН)з), соединяясь с водой и выделяя значительное
количество тепла. Соли La с сильными кислотами почти не подвергаются
гидролизу в водном растворе [33]. Углекислая соль La2(C03)3
(образующая кристаллогидраты с 1, 3 и 8 молекулами Н20) характерна и
устойчива. Устойчивостью отличается также сернокислая соль
La2(S04)3, переносящая нагревание до :600° без заметного разложения.
Все это свойства, характеризующие гидрат окиси лантана как сильное
основание.
Можно еще указать на то обстоятельство, что из всех элементов
данной подгруппы способность образовать комплексные соединения
особенно резко проявляется у бора, для которого, кроме
общеизвестных борофторных кислот (HBF4 и других), полиборных (аутокомп-
лексных) кислот и т. п. существует еще большое ,число комплексных
соединений с органическими телами, содержащими гидроокислы,
особенно с многоатомными алкоголями и оксикислотами. У алюминия
способность к комплексообразованию значительно понижена. Между тем
* Положение алюминия является опорным (см. ниже).
Периодическая система химических элементов 169
Элемент
Цинк
Zn = 65,37
Кадмий
Cd = 112,4
Ртуть
Hg= 200,6
ее
Ч
О
О
о
сх
с
ffi
3
6,9
8,64
при 0° 13,5956
при 20° 13,546
ге
г;
Н
о
и
О
ь
U
о
а.
с
о
с:
3
г
о
<
9,5
13,0
14,8
я
я
ч°
со Ч
О. 0J
ё °
з- о
д> о.
Н с
419
320
-39
о
а
с
к
5
3
Я
«и
ге°
= Е
г о
г— (J
920
780
357
ГЗ
я.
о
Я
СО
8
а
0 .
85,0
65,7
20,7
, а
— '«г
о .
к у
2-g
§2
eg
97,2
93,2
53,3
Растворимость
(число граммов соли
на 100 і.
ч
о
С
о
51
368
при 20°
128,6
при 30°
7,4
при 20°
воды)
сч
Г"
О
о
о
3
ь
О
432
при 18°
85,3
при 10°
бхю-з
при 25°
Произведение
растворимости
сернистого
соединения MeS
От 5,0хЮ-2в
до 1.1Х10-2*
от 5,1хі0"29
до7,1хЮ-28
от l.OxlO-68
до7,7хЮ-*8
как комплексы бора по большей части довольно постоянны в присутствии
воды, соответствующие соединения А1 обычно образуются лишь в
отсутствие влаги, водой же легко разлагаются (например, многочисленные
двойные соединения с органическими веществами, полученные прежде
всего Г. Г. Густавсоном [34], а в последнее время Б. Н. Меншутки-
кым) *. Далее с повышением атомного веса от А1 к La и Yt способность
к комплексообразованию, несомненно, падает еще в более сильной
степени, как в том легко убедиться, просматривая известные соединения
этих элементов.
К побочной подгруппе III группы относят три элемента: Ga,
In и TI. Быть может, к ним же следует отнести и алюминий **. В числе
общих свойств, объединяющих между собой эти элементы, можно
указать на переменчивость валентности, о чем можно судить, например,
по составу известных хлористых соединений:
GaCU іпСіз ТіСіі
(Легко окисляется
и переходит в тип
GaX3
InCl.
С водой
разлагается на In и 1пС13
Соль слабого
основания
Т1(ОН)3
GaCl (?)
InCl
Tin J Соль сильного
ііЫ основания Т10Н
Далее между собой, а также с алюминием их сближает
изоморфизм сходственных соединений, особенно способность к образованию
квасцов, которая пока не доказана только для трехатомного таллия:
Al2 (S04)3 K2S04 24H20 In, (S04)3 K2S04 24H20
Ga2 (S04)3 K2S04 24H20
Положительное электросродство в .побочной группе падает от Ga
* Ряд статей в ЖРХО и «Известиях СПб. Политехнического института» за
последние годы.
** Д. И Менделеев считает алюминий ближайшим аналогом галлия и индия, а не
скандия, что соответствует положению в 3-м, т. е. нечетном, ряду.
170
Периодическая система химических элементов
к Т1, о чем можно судить по следующим приближенным значениям
потенциалов разложения;
Ga In Tl
+0,5 +0,17 +0,04
По-видимому, и здесь способность к образованию комплексов для
таллия является наибольшей, совершенно аналогично тому, что
наблюдается для ртути в периоде Zn, Cd, Hg,
В следующих таблицах сопоставлены главнейшие физические
свойства простых тел, образуемых элементами III группы.
Элементы главной подгруппы
Элемент
В
А1
Sc
Y.t
La
Yb
Атомный
вес
11,0
27,1
44,1
89,0
139,0
172,0
Температура
плавления
простого тела, °С
около 2300
655
1200?
1000?
810
Удельный вес
простого тела
2,5
2,6
2,5
3,8
6,15
Атомный
объем
4,4
10,4
17,G
23,4
22,6
Теплота
образования окиси, ккал
272,4
385,6
444,7
Элементы побочной подгруппы
Элемент
Ga
In
TI
Атомный
вес
69,9
114,8
204,0
Температура
плавления
простого тела,
°С
30
155
304
Плотность
простого тела
5,935
7,12
11,85
Атомный объем
простого тела
11,75
16,1
17,2
Из рассмотрения этих таблиц следует, что в обеих подгруппах с
повышением атомного веса элемента возрастают удельный вес и атомный
объем простого тела, а в подгруппе Ga, In, Tl правильно возрастает и
температура плавления. Зам-етим, что правильности эти нисколько не
изменятся, если во главе триады Ga, In, Tl поставить, следуя за
Менделеевым, А1.
IV группа. Элементы этой группы на первом плане
характеризуются высш-ей валентностью —4, которая яснее всего проявляется в
кислородных* и галоидных, а для С, Si и Ge также в водородистых
соединениях:
СОо Si02 ТЮо ZrO, Ce0217 Th02
ССЦ SiCl4 TiCI4 ZrCl4 СеОг Th04
СН4 SiH4 GeH4
Ge02 Sn02 РЬ02
GeCl4 SnCl4 PbCl4
* Такие соединения, как ТіОз и соответствующие гидраты, носят несомненный
характер перекисей. Титан, -по всей вероятности, сохраняет при этом валентность = 4 (см
стр. 94—95).
Периодическая система химических элементов 171
Кроме того, низшая валентность с достоверностью доказана у
следующих элементов: С (2 и 3) 18, Ті (2 и 3), Се (13), Sn (2) и РЬ (2).
Другая характерная черта элементов IV группы связана с их
электрохимической природой. Подобно элементам III группы, они являются
переходными, .или амфотерными, представляя одновременно черты
металлов и металлоидов. Можно, однако, отметить, что в этой группе
металлоидный характер несколько преобладает над металлическим,
особенно у элементов с малым атомным весом, тогда как в III группе
отношения скорее обратные.
В связи с амфотерным характером элементов IV группы стоит,
между прочим, то обстоятельство, что в ней находятся два элемента —
кремний и особенно углерод, из всех остальных выделяющихся по
своей .исключительной наклонности к полимеризации, к образованию
многочисленных и разнообразных соединений, содержащих по
нескольку атомов данного элемента в молекуле (см. выше стр. 111 — 112).
Сопоставляя между собой элементы IV группы,
Четные
ряды
Ті С j Типические
Zr Si I элементы ^е Г Нечетные
Се " \ ряды
( Th 1
мы заметим, что, как и в других группах периодической системы, они
распадаются на две подгруппы, причем, однако (;и это особенно
характерно для IV группы), расхождение между ними является слабо
выраженным. Поэтому сравнительно трудно ответить на вопрос, какую из
двух подгрупп следует признать главной и какую — побочной. Нелегко
также решить, к какой подгруппе следует отнести элементы первых двух
периодов: углерод и кремшш.
Между тем как по одним признакам они (особенно кремний)
ближе всего подходят к германию, олову, свинцу, образующим особенно
тесную подгруппу, по другим они приближаются к подгруппе Ті—Th.
На первой (по-видимому, более правильной) точке зрения [35],
стоял Д. И. Менделеев, а вторую поддерживал Лот. М е и е р.
Рассматривая физические свойства простых тел IV группы, мы
приходим к заключению, что углерод и кремний с равным основанием
могут быть поставлены во главе элементов, принадлежащих к четным
и нечетным рядам. К такому заключению приводит, например,
сравнение атомных объемов:
С
3,4-5,3
Si
12
Ті
13,6
10,7
Zr
21,8
Ge
13,2
Се
19,9
Sn
16,3
Th
20,9
Pb
18,2
и отчасти температур плавления (для кремния температура плавленая
с достоверностью неизвестна* и, вероятно, выше величины,
приведенной в таблице на стр. 173. В таком случае Si ближе к тугоплавким
Ті, Zr, нежели к легкоплавким Ge—Pb).
Некоторая неопределенность сказывается также и в свойствах
сходственных соединений элементов этой группы, физических и хими-
* По современным данным, температура .плавления кремния 1415°, температура
кипения 2400°. Прим. ред.
172
Периодическая система химических элементов
ческих, хотя здесь ясно перевешивают аргументы в пользу взгляда
Менделеева.
Так, хлористые соединения ССЦ и SiCl4 напоминают GeCU и SnCl4,
но отличаются от труднолетучих ZrCl4, CeCI4 (крайне неустойчиво) и
ТЬСЦ, Как известно, летучесть хлористого германия и
приблизительная точка кипения его, лежащая между точками кипения SiCl4 и SnCl4,
была угадана в свое время Д. И. Менделеевым. Между тем, по
температуре кипения ТіСЦ плохо укладывается в середину между SiCl4 и
ZrCI4 (|cp. табл.).
С другой стороны, углерод и кремний, подобно Ge, Sn и Pb,
образуют металлоорганические соединения, отвечающие одному и тому же
общему типу:
Si (СгЩі Ge(C2H5)4 Sn (С2Н5)4 РЬ(С2НБ)4
Температура кипения . . . .152° 160° 181° не перегоняется
без разложения
между тем как для титана, циркония, церия и тория такие соединения
неизвестны.
В частности, германий сближается с Si и С еще существованием
таких соединений, как гермзнийхлороформ, аналог кремнехлороформа
и хлороформа:
СНС13 • SiHCl3 GeHCl3,
а также (по Ганшу) вероятным существованием аналога муравьиной
кислоты РЮеОгН.
Наконец, можно еще отметить то обстоятельство, что в ТіСЦ (а
также ТіСЦ) гораздо яснее заметны признаки соли, а следовательно, в.
(ТіОН)4 и Ті(ОН)3 — признаки основания, нежели в соответствующих
соединениях германия (Ge02 растворяется в щелочах, давая соли
германиевой кислоты, но не растворяется в кислотах). Между тем
углерод и силиций являются наиболее электроотрицательными
элементами IV группы и потому, естественно, вслед за ними поставить именно
германий — обстоятельство, предсказанное в свое время Д. И.
Менделеевым.
От Ge к Sn и РЬ электроположительный характер правильно
возрастает. То же самое замечание касается и другой подгруппы, в которой
наиболее электроположительным элементом является торий.
Есть, однако, химические доводы, говорящие в пользу сближения
углерода и кремния с Ті, Zr и Th. Так, подобно этим последним, С и
Si отличаются малой склонностью давать соединения низших типоз,
и тем ясно отличаются от Ge, Sn и Pb, для которых соединения типа
RX2 легко образуются и сравнительно постоянны. К этому можно было
бы прибавить, что как раз для углерода (Гомберг, Шленк и другие)
и титана (а также для церия) существуют соединения, где элементы эти
являются трехвалентными. Для Ge, Sn и Pb такие соединения
неизвестны.
V группа. Элементы, образующие пятую группу периодической
системы, с достаточной отчетливостью разбиваются на две подгруппы,
причем к главной (металлоидной) примыкает, кроме элементов
нечетных рядов, один элемент 2-го типического ряда.
Максимальная валентность = 5 и вообще повторение некоторых
общих типов соединений, особенно RXs и RX3, составляет
характернейшую особенность, присущую всей группе.
Нередко представители обеих подгрупп сближаются и на почве
изоморфизма. Из множества примеров этого ряда приведем изомоп-
Периодическая система химических элементов
173
Элемент
Углерод
С =12,00
Кремний
Si = 28,3
Титан
Ті =48,1
Цирконий
Zr = 90,6
Церий
Се =140,25
Торий
Th = 232,4
Германий
Ge=72,5
Олово
Sn = 119,0
Свинец
РЬ = 207,1
Свойства простого тела
температура
плавления, °С
около 4000
(по Мак-Крэ)
1200-1600
1830
2350
623
1690
900
231
326,7
удельный вес
алмаз 3,51
графит 2,25
2,3-2,4
3,54
4,5
(по Гёнтеру)
4,15
7,04
11,1
5,47
7,29
11,37
атомный
объем
3,4
5,3
12
13,6
10,7
21,8
19,9
20,9
13,2
16,3
18,2
температура
кипения RC!4, °С
77
58
136
Сублимируется
около 440°
—-
—
86
114
—
физм трех соединений, в виде которых мышьяк и фосфор, с одной
стороны, и ванадий, с другой, встречаются в природе:
ЗРЬз (As04)2 PbClo 3Pb3 (P04)2PbGl2 ЗРЬ3 (V04)2 PbClo
Миметезит Пироморфит Ванадинит
Главную подгруппу образуют элементы:
N Р As Sb Bi
Из них высший тип с ясностью предстазлен в первых четырех,
особенно в кислородных и галоидных, соединениях (например
N205, HN03, PCI5, РВг5, As205, SbCl5) и только для висмута
представители этого типа не получены еще с достоверностью в чистом состоянии,
хотя, по-видимому, и существуют19.
Низший тип, отвечающий валентности 3, представлен для всех беч
исключения элементов подгруппы:
NH3 РН3 AsH
AsCl3
NCI
PCI
SbH3
SbCl3
N(CH3)3 P(CH3)3 As(CH3b Sb(CH3)3
N,0
2^3
PoO
2^Z
As„0
2^3
Sb>0
2^3
BiCl3
Bi (CH3)3
BU03
Заметим здесь, что азот из всех элементов данной подгруппы
отличается особенным богатством форм соединений и разнообразием
проявляемой валентности*. Это особенно резко сказывается в окислах
N20 NO N02 N203 N205
* В окиси азота валентность N, по-видимому.= двум N - О, в NO, - четырем
О ,N\ Nx
(\А/ Vе закиси, быть может,—трем/ Ц О, как в
\0 VN/
Thiele. Bed. Ber, 44, 2522.
\\ /NH). {-м-. впрочем,
N
174
Периодическая система химических элементов
Аналогия между элементами распространяется и на свойства
(физические и химические) простых тел и соединений сходного состава,
причем свойства эти в пределах данной подгруппы по большей части
подвергаются правильному и постепенному изменению без резких
скачков.
Как уже было указано в главе IV, основной чертой,
характеризующей порядок такого изменения, является повышение металлических и
понижение металлоидных свойств по мере повышения атомного веса.
В свойствах простых тел мы ясно видим такой постепенный переход
от типичнейших металлоидов азота и фосфора к металлам, каковы
сурьма и особенно висмут. То же самое можно сказать и о
сходственных соединениях. Водородистые соединения вое газообразны. Степень
устойчивости их падает с повышением атомного веса (см. стр. 115);
ВіНз уже не существует *. Водород в них с большей или меньшей
легкостью способен замещаться металлом. Продуктами такого
замещения являются, например, амиды натрия NaNH3, калия KNH2, азотю-
ры ** магния Mg-3N2, кальция Ca3N2 (образованием которых из N2 и
соответствующих металлов пользуются при добывании аргона из
воздуха), соответствующие соединения As и Sb (например, Mg3As2, Zn3As2„
Zn3Sb2) и т. д. В этих соединениях, которые водой и кислотами
разлагаются с образованием гидрюроз RH3 (Mg3N2 + 6H20 = 3Mg(OH)2 4-
-t-2NH3), очевидно, металл (Na, Mg...) играет роль
электроположительного элемента и таковую же роль, следовательно, играет водород
в соответствующих гидрюрах.
С другой стороны, присоединяя Н, гидрюры, RH3, способны
переходить в комплексный радикал RH4+, играющий роль катиона —
в солях аммония NH4X и фосфония РН4Х. Устойчивость таких
радикалов и соответствующих солей опять-таки быстро падает с повышением
атомного веса. Для As и Ві они уже неизвестны, но зато известны их
органические производные, замещенные арсонии и стибонии As(CH3)4X,
Sb(CH3)4X, которым замещение водорода органическими радикалами
сообщает повышенную устойчивость.
Кислородные соединения обоих основных типов R2O5 -и R2O3 имеют,
зообще говоря, характер кислотных ангидридов, а галоидные RXs и
RX3 — характер галоилангидридов, причем, как на это уже было в свое
время указано (стр. 114), с повышением атомного веса элемента R
постепенно в окислах начинает появляться основной характер
(особенно у Ві203), а в галоидных соединениях — характер солей (ВіС13). То
же самое относится к гидратам окислов. От типичных кислот HN02,
HN03, Н3Р03, Н3Р04 мы здесь постепенно переходим к
основному гидрату Ві(ОН)з, кислотные свойства которого едва заметны. В
соответствии с уменьшением силы кислот от азотной к сурьмяной
возрастает способность их к образованию сложных (комплексных) кислот.
Обзор элементов главной подгруппы мы закончим сопоставлением
некоторых физических свойств, отвечающих этим элементам простых
тел и аналогичных соединений. В общем и здесь наблюдаются
основные правильности, с особенной ясностью выступающие при сравнении
свойств водородистых и галоидных соединений.
Достойно внимания исключительное положение, занимаемое
азотом, элементом типического ряда. Особенно бросаются в глаза
отступления от общей правильности в изменении физических констант при
** В русской химической литерутаре тамие соединения называются
нитридами. Прим. ред.
* См. прим. ред.12 х стр. 114 настоящего тома. Прим. Ред.
Периодическая система химических элементов
175
сравнении водородистых соединений. Ясно видно, что аммиак обладает
аномально высокой температурой плавления, что совершенно
напоминает аналогичное поведение воды и фтористого водорода и указывает
на ассоциацию (образование сложных, например двойных молекул
(NH3)2 и т. д.) аммиака. С этим обстоятельством, без сомнения, стоят
в связи и сравнительно высокая диэлектрическая константа NH3
(22 при температуре кипения) и способность его, подобно воде,
служить для многих электролитов сильным ионизирующим растворителем.
Элемент
Азот
N =14,01
Фосфор
Р = 31,04
Мышьяк
As = 74,96
Сурьма
Sb = 120,2
Висмут
Ві =208,0
Свойства простого тела
Состав молекулы в
парообразном состоянии
N3
Р4 —^ Р-2
As4 —^ As.>
Sb выше 2070°
Ві выше 2070°
температура
плавления, °С
-211
+44,1
850 [3G]
+630
+269
температура
кипения, °С
-196
+287
Возгоняется
при +450°
при 1 атм
+1440
+ 1420
удельный вес
жидкий 0,80
при —196°
твердый 1,027
при -252,5°
красный (2,15)
2,34 при 15,5°
желтый 1,83
кристаллический
5,73
желтый, 3,88?
ромбоэдрический
6,70
9,78
атомный
объем
17,5
13,65
13,25
16,9
13,1
19,3
17,9
21,3
Водородистые соединения
Соединение
NH3
РНз
AsH3
SbH3
ВіНз
Температура кипения
при атмосферном
давлении
—33,5
—86,2
—55
—18
Температура
плавления, °С
—75
—133
—113,5
-88
Удельный вес
0,638 при 0°
0,744 при температуре
кипения
2,26 при —25°
Диэлектрическая
константа,
вычисленная для t -20.5°
15,6
2,9
1,8
Побочную подгруппу V группы составляют элементы:
V Nb Та
Сопоставление главнейших -форм образуемых ими соединений
обнаруживает наличность тех же главных типов, какие характеризуют эле-
176
Периодическая система химических элементов
менты главной подгруппы, и прежде всего
сти 6.
максимальной валентно-
v2o5
HV08
VF5
Nb205
NbCl5
NbF5
Ta206
TaCl5
TaF5
Соединения низших типов в большом числе и разнообразии
представлены для ванадия, и в этом отношении он напоминает азот (да
частью фосфор). При этом соединение четырех- и пятивалентного
ванадия почти одинаково прочны:
VO
vso4
VCl,
v2o3
V2 (S04)3
V2 (S04)3 K2S04 24H20
(ванадиевые квасцы)
vci3
VBr3
vo4
VO (S04)
VC14
VOCl2
Для Nb наиболее устойчивые соединения отвечают высшему типу
NbXs. Подчиненное значение имеют соединения трех- и, может быть,
двухатомного Nb. Что касается тантала, то для него с достоверностью
известен только один высший тип TaXs20.
Будучи элементами четных рядов, V, Nb и Та отличаются
преобладанием металлических свойств над металлоидными, хотя и эти
последние все же проявляются, особенно в соединениях высшего типа.
С 'повышением атомного 'веса кислотный характер окислов R2O5
ясно отступает на задний план и заменяется основным. Быть может,
особенно резко это сказывается на фтористых соединениях. Между тем
как VF5 имеет характер хлорангидрида .и при гидролизе дает фтор-
окись VOoF, для ниобия гидролиз уже не заходит столь далеко и
приводит к'фторокиси NbOF3, а для тантала соединение TaFs имеет
характер соли, лишь отчасти подвергнутой гидролизу.
Свойства простого тела
Элемент
Ванадий
V = 51,0
Ниобий
Nb = 93,5
Тантал
Та = 181,5
Температура
плавления, °С
1710
1950
2850
Удельный вес
5,7
7,2
10,4
Атомный
вес
9
13
17,4
VI группа. Б VI группе разделение на две подгруппы резко
выражено. К типичной подгруппе здесь прежде всего относятся элементы
нечетных рядов: сера, селен и теллур. Сюда же примыкает и кислород.
По своей химической природе — все это типичные металлоиды.
Зачатки металлических свойств, однако, заметны уже в селене и особенно
в теллуре.
Периодическая система химических элементов \11
Элементы четных рядов: хром, молибден, вольфрам и уран,
образующие вторую побочную подгруппу, представляют, наоборот, ясно
выраженные черты металлического характера, хотя соответственно
своему положению в правой части периодической системы, сохраняют
в соединениях высшего типа и характер металлоидный:
О S Se Те
Сг Mo W U21
Обе подгруппы объединяются общей максимальной
валентностью =6, которая не достигается только для одного кислорода
(подобно тому, как высшая валентность галоидов —7 не достигается для
фтора).
Главная подгруппа. Наиболее обычная (отрицательная)
валентность элементов, образующих эту группу = 2. Она проявляется
прежде'всего в следующих водородистых соединениях (гидрюрах):
Н20 H2S H2Se HoTe.
Затем в металлических окислах, сульфидах, селенидах и теллури-
дах, другими словами, в соединениях, которые можно рассматривать,
как продукты замещения водорода только что приведенных гидрюроз
металлами:
К20
СиО
А1203
KoS
CuS
А12Ьз
K2Se
CuSe
Al2Se3
КЛе
CuTe
А12Те3
Та же валентность проявляется в продуктах неполного замещения
водорода гидрюров НгХ металлами:
КОН KSH KSeH KTeH,
а равно и в аналогичным образом построенных органических
соединениях (спиртах, эфирах, меркаптанах, тиоэфирах и т. п.).
Другой довольно характерный тип соединений, образуемый
элементами этой подгруппы, отвечает валентности 4. Сюда относятся,
например, следующие кислородные и галоидные соединения:
S02 Se02 ТеО*
SC14 SeCl4 ТеСЦ,
а из органических соединений —- окиси тиоэфиров, например, (С2Н5)г>
>S = 0 и аналогичные соединения, отвечающие селену (C2H5h>Se=0
и теллуру (С2Н5)2>Те = 0, а также тиониевые, сульфониевые и теллу-
рониевые соли;
(C,H5)3S-J (C2H5)3Se-J (QH^Te-J.
К этим последним примыкают многочисленные так называемые
оксониевые соединения, изученные Фриде л ем, Колли и Т и к-
л ем, А. Байером, А. Е. Фаворским и другими. Простейшим
прлмеоом подобного рода соединений, в которых ныне многие
принимают четырехатомный кислород, -представляет соединение метилового
эфира с хлористым водородом, открытое Фриделем. Строение его
выражают такой формулой22:
сн,х лі
СН3Х ЧС1
12 Л. А. Чугаев, т. Ж
178
Периодическая система химических элементов
Высшая (положительная) валентность, свойственная VI группе,
достигается элементами S, Se и Те прежде всего в кислородных
соединениях
S03 Те03
H2S04 H2Se04 Hje04.
а из галоидных соединений—только во фтористых:
SFe SeFe TeFe.
Электрохимический характер элементов этой
подгруппы — резко выраженный металлоидный электроотрицательный. Сера,
селен и теллур, соединяясь с кислородом и водой, дают типичные
кислоты. Водородистые соединения их также обладают, хотя и слабыми,
но, несомненно, кислотными свойствами.
Кислород, во многих отношениях стоящий особняком, несомненно,
является типичнейшим электроотрицательным или
кислотообразующим элементом, что было отмечено еще Лавуазье и Берцелиу-
сом. Из многих фактов, подтверждающих это положение, укажем
здесь особенно на то обстоятельство, что энергия кислот обычно
повышается по мере накопления в них кислорода. Так, НС103 и НСЮ*
сильнее НСЮ, азотная кислота HNO3 сильнее азотистой HN02, серная
H2SO4 сильнее сернистой H2SO3 и т. д.
Из органической химии известно, что водород, связанный с
углеродным радикалом через посредство атома кислорода,
например, в СНз—О—Н, получает значительную подвижность и как бы
зачатки кислотных свойств, а если группа ОН связана с окислен-
ным углеродным атомом, например, в рН х ^„ то
кислотные свойства водорода становятся резкими.
По мере повышения атомного веса, от кислорода к теллуру
электроотрицательные свойства элементов понижаются, и постепенно
появляется характер металлический. Это заметно уже на свойствах простых
тел. Кислород — газ, при очень низкой температуре сжижающийся и
застывающий в прозрачный (голубой) лед, не обладающий и следами
металлических свойств. Сера во всех своих модификациях прозрачна
для света и является почти абсолютным изолятором. Среди
модификаций селена нет ни одной совершенно прозрачной, но зато есть
слабо проводящая электрический ток, причем проводимость эта
значительно возрастает от освещения. Прозрачность теллура еще
меньше, чем для селена, а электропроводность, наоборот, значительно
больше и приближается к металлической. Подобные же отношения
наблюдаются и в химических свойствах S, Se и Те. Так, сила кислот
ясно понижается от H2SO4 к H2Se04 и далее к Н2Те04, и в
соответствии с этим в том же порядке возрастает способность этих кислот к
комплексообразованіию.
С другой стороны, устойчивость летучих водородистых соединении
понижается, как это, между прочим, видно из следующего
сопоставления соответствующих т-егтлот образования*:
Н20 H2S H2Se НгТе
+58,1 +4,8 —24,6 —34,9
* Н20. H2S, H2Se и Н2Те — газообразный кислород и водород газообразный, S, Se
и Те — твердый. На стр. 115 теплота образования Н20 дана для жидкой воды, а для
hbSe ошибочно приведена величина — 4,8.
Периодическая система химических элементов
179
Приводим сопоставление физических свойств простых тел,
отвечающих элементам рассматриваемой подгруппы, и некоторых сходных их
соединений.
Свойства простого тела
Элемент
Кислород
0= 16,00
Сера
S = 32,07
Селен
Se = 7.9,2
Теллур
Те = 127,5
о
g
а
с
Кислород
Озон
моноклиническая
сера
ромбическая
Амфотер-
ный селен
Красный
кристаллический
Серый
кристаллический
—
U
№
іелькы
>>
1,17 при
-195,5°
1,96
2,07
4,3
4,5
4,8
6,25
2 е1
но
< О
13,7
16,4
15,5
18,4
17,6
16,5
20,4
и
Л О
мпсрат
явлени
??
-218
~
119
115
—
170—180
217
455
go
мпсрат
пения,
О) К
-182,9
-119
444,5
690
—1280
Цвет
Бледно-голубой в
жидком
состоянии
Темно-синий
в жидком
состоянии
Лимонно-
желтый
Красно-
коричневый
Красный
Серый
Серовато-белый с
таллическим
блеском
о О о
<35"gg
o2
Оз
bg — 02
Sea при
1440°
Молекулярный вес =
=120 при
2070°
Те*
Молекулярный вес =
166 при
2070°
Свойства водородистых соединений
Соединение
Н20
H2S
H2Se
Н2Те
Температура
кипения при
атмосферном
давлении. °С
+100
-60
-42
0
Температура
плавления, °С
0
.-83
-64
—48
Критическая
температура. ЭС
+365
+100
+137
Удельный вес
в жидком
состоянии при
температуре
кипения
0,958
0,87
2,12
2,57
Молекулярная
скрытая
теплота испарения,
кал
9650
4495
4670
Константа
Трутона Т
25,9
21,3
20,2
Просматривая эти таблицы, мы видим, что свойства простых тел и
их сходственных соединений в общем правильно изменяются от О к
Те. Однако во многих отношениях кислород занимает исключительное
положение (как элемент типического ряда), что оттеняет особенную
близость членов «триады» S, Se и Те*.
* Интересно отметить, что .по свойствам кислородных соединений селен не
занимает середины между S и Те (как бром не занимает среднего положения между
12*
т
Периодическая система химических элементов
Свойства фтористых соединений [37]
Температура плавления,
°С
Температура кипения . .
Критическая температура
Удельный вес
Молекулярный объем . .
SF,
—56
+62
+54
1 91
(при -50°)
76,5
SeFe
-34,5
-39
+72,35
2,51
(при —28°)
77,2
TcFfl
—36
—35,5
+83,25
3,025
(при —24°)
79,9
Особенность кислорода резче всего сказывается в аномальных
свойствах его окислов, особенно воды Н20. Вода не является кислотой,
подобно другим соединениям типа Н2Х, но представляет типичнейший
аморфный электролит, ибо при диссоциации дает как Н-, так и
ОН-ионы.
Вода является одним из энергичнейших ионизирующих
растворителей (растворенные в ней электролиты диссоциируют на ионы), что
стоит в связи с ее высокой диэлектрической константой (D=81), a
также, по -всей вероятности, с тем обстоятельством, что молекулы воды
ассоциируют, то есть уплотнены, образуя двойные (НгОЬ, а может
быть, и іболее сложные молекулы (Рамзай и Шильдс и др.)-
Ассоциацией воды объясняется ненормально высокая величина ее температуры
кипения, а также константы Трутона (частного от разделения
молекулярной скрытой теплоты испарения на температуру кипений по
абсолютной шкале).
Побочную подгруппу VI группы образуют металлы
Cr Mo W И
Характерной чертой, сближающей ее с главной, является прежде
всего высшая валентность=6, которая, например, проявляется в
следующих соединениях:
СгОз
Н2Сг04
СЮ2С12
МоОа
Н2Мо04
wo3
H2W04
W02Clo
WOCl4"
MoF6 WCle
Затем можно отметить, что высшие окислы типа R03 имеют здесь
характер ангидридов, а соответствующие гидраты — характер кислот.
Эти последние, а равно и соли их, не только построены по общему
ио8
Me2U207
U02 (N03),
UFft
CI и J). Для ¦иллюстрации приводим теплоты образования S02, SeOa и Те02 (в ккал,
по Микстеру).
SO,
70
Se02
63,5"
ТеО,
87,1
Отсутствие («ли малая устойчивость) Se03 (ТеОз известен) также указывает на
аониженное сродство Se к кислороду по сравнению с соседними (по лруппе)
элементами. Если бы, селеновый ангидрид существовал, то, ло данным Микстера, ему также
отвечал бы минимум теплоты образования среди окислов типа- R03, как видно из
следующих цифр:
S03
103
SeOs
48,8
Те03
83,6
Периодическая система химических элементов
181
типу, но и часто изоморфны со сходными соединениями главной
подгруппы (например, соли серной и хромовой кислот).
Отклонение от типичных свойств элементов главной подгруппы
выражается здесь прежде -всего .в появлении новых типов соединений,
отвечающих нечетной валентности 3 и 5. Таковы соединения ряда окиси
хрома Сг203, Сг(ОН)3, СгС13, Cr2(S04)3 и т. д. Таковы соединения
типа МоХ5 и МоХ3 (например, МоС13 и МоС15) и т. п.
В электрохимическом отношении, как уже было упомянуто выше,
следует отметить повышение металличности и электроположительного
характера по сравнению с членами главной подгруппы:
Элемент
Хром
Сг = 52,0
Молибден
Мо = 96,0
Вольфрам
W = 184,0
Уран
U = 238,5
Свойства простого тела
Температура
плавления, °С
1490
Выше белого
каления
2900
—
Температура
кипения, °С
2200
3200 (?)
около 3700
—
Удельный вес
7,0
8,56
19,1
18,7
Атомный
объем
7,4
11,1
9,6
12,8
VII группа. Все элементы этой группы, кроме марганца,
составляют ее главную подгруппу и еще в начале прошлого столетия были
выделены в естественную семью галоидов или солеродов. Из них три
(О, Вт и J23) принадлежат к нечетным рядам и один—фтор — к
четному, именно, ко 2-му (типическому) ряду.
Галоиды. Выделенные в свободном виде, как простые тела,
галоиды представляют типичные черты металлоидного характера:
прозрачность, отсутствие металлического блеска, плохую электро- и
теплопроводность. Один иод во всех этих отношениях немного
напоминает металлы.
Галоиды отличаются значительной химической активностью и
образуют соединения, частью весьма прочные, с большинством
металлоидов и особенно металлов. С этим в связи стоит то обстоятельство, что
они неизвестны в природе в свободном состоянии. Химическое
сродство С1, Вг и J подчеркивается тем обстоятельством, что они находятся
совместно главным образом в воде морей и океанов, причем
содержанке их резко падает от хлора к иоду.
Как уже было указано в главе IV, по мере повышения атомного
веса металлоидный характер галоидов несколько ослабевает, что
отражается (И на изменении свойств простых тел, и на природе
сходственных соединений.
Состав
молекул
F2
С12
Вга
h
Атомный
вес
19,0
35,46
, 79,92
( і26.,92
Температура
плавления. °С
—223
—102
¦ +114
Температура
кипения г °с
—187
—33,6
+63
+183
Критическая
температура, °С
+146
+302
+512
Удельный ьес
1,11 при-187°
1,47 при0°
3,19 при0°
4,93 при 10°
Цвет в жидком состоянии
Светло-желтый
Оранжево-желтый
Темно-красно-коричневый
Черно-сивий
182
Периодическая система химических элементов
Постепенное изменение физических свойств свободных галоидов
видно из прилагаемой таблицы (см. стр. 181).
По мере повышения атомного веса, правильно поднимается
температура плавления, температура кипения, критическая температура и
удельный вес. В то же самое время окраска становится все более и
более интенсивной.
Постепенно, хотя и весьма медленно, повышается и атомный
о бъе м:
F CI Br J
Атомный объем .... 17 25 25,1 25,7
В том направлении изменяются и магнитные свойствя, причем
от хлора к иоду увеличивается значение (отрицательной магнитной
восприимчивости) (см. стр. 152).
Степень прочности молекул F2, СЬ, Вг2, J2, как мы уже
видели, также правильно изменяется с повышением атомного веса
галоидов.
Способность галоидов подвергаться гидролизу в водном растворе,
по уравнению
х2+н2о-н+х+нхо,
изученная А. А. Яковкиным [38] и другими, сильно выраженная для
хлора, значительно слабее для брома и еще слабее для иода.
С той же постепенностью, как и свойства простых тел, изменяются
и свойства сходственных соединений, но здесь сходство более
бросается в глаза, чем различие, основная тема — более, нежели ее вариации.
Начнем с водородистых соединений
HF HC1 НВг HJ
Сходство между ними прямо поразительное (несколько более
отклоняется только фтористый водород, что и понятно, так как фтор —
элемент «типического» ряда). Состав их выражается совершенно
сходственными формулами. Все галоидоводороды — тела бесцветные,
газообразные при обыкновенной температуре (HF — весьма летучая
жидкость); чрезвычайно легко растворяются в воде, притом с
выделением большого количества тепла, что явно указывает на наличность
химического процесса. С водой они образуют постоянно кипящие
жидкости, которые хотя и обладают постоянным составом, однако
представляют из себя смеси, а не определенные соединения, как это было
показано Бино, Роско и другими. Вот некоторые свойства этих
постоянной кипящих смесей.
Температура кипения
при 760 мм, °С . .
Содержание галоидо-
водорода, вес. %
HF
120
35
ил
НО
20
ПС1
125
48
НВг
127
57
При перегоне под другим давлением также получаются (постоянно
кипящие смеси, но состав их будет уже иной. Так, тіо опытам Роско
Периодическая система химических элементов
183
и Дитмара, под давлением 7ю gtm гонится соляная кислота с
содержанием 23%, а под давлением 3 атм — с содержанием 18% НС1.
Эти данные и указывают на отсутствие здесь химически
индивидуальных -веществ.
В растворе галоидоводороды обнаруживают характер кислот: HF—
сравнительно слабой ки-слоты, остальные три (принадлежат к числу
сильнейших.
НС1 НВг HJ*
Константа электролитической диссоциации .... 4,77 6,27 5,82
Все соли этих кислот имеют вполне аналогичный состав (например
КС1, KJ, КВг, СаС12, CaBr2, CaJ2, А1С13, А1Вг3, AIJ3 и пр.) и часто
аналогичные свойства (например, AgCl, AgBr, AgJ) весьма трудно
растворимы в воде и отличаются светочувствительностью.
В связи с ослаблением металлоидного характера галоидов от фтора
к иоду, степень прочности как водородистых соединений (см. стр. 115),
так и сходственных солей, им отвечающих, правильно изменяется от
RF к RJ. При 600° абс. HJ диссоциирует в количестве 18,9%, между тем
как даже при 2000° абс. % диссоциации НВг составляет 6%, а для
НС1 — только 0,8% (Боденштейн).
Поучительны также следующие цифры теплот образования, в ккал
іпо Томсену и Де-Форкрану).
(К, X)
Vi (Са, X.)
(Ag, X)
У2 (Hg2i Х2)
V* №, Х2)
Vs (As, Хз)
F
108,80
119,59
—
—
—
—
С1
104,05
95,22
29,38
32,6
27,2
23,8
Вг
97,48
84,60
22,7
25,5
20,9
15,2
л
85,21
70,50
13,8
15,6
12,8
,4,5
Как и)для водородистых соединений (положительное), тепло
образования всюду падает от фтора к иоду.
Любопытно далее отметить, что степень прочности комплексных
аммиакатов,1 отвечающих металлам VIII группы, падает при переходе от
хлористого соединения к соответствующему йодистому, как это видно
из следующих примеров: по Изамберу [39] для PdJ24NH3
упругость диссоциации в 760 мм достигается при 110°, тогда как для
PdCl24NH3 для этого необходима температура 210°, то есть
значительно более высокая. По данным Жарри [40], упругость диссоциации в
миллиметрах ртути -при 0° для:
AgCl ЗШ3 AgBr 3NH3 AgJ 3NHS
262 605 Упругость очень велика,
соединение не могло быть
приготовлено
AgClCH3 NH2 AgBrCHaNH., AgJCH3NHa
9 11 35
Многие другие .свойства -сходственных соединений галоидов
изменяются в том же -порядке.
ч »¦ і ¦-.
* Вычислены *по формуле Рудольф и.
184
Периодическая система химических элементов
Температуры плавления и -кипения галоидо'водородов 'правильно
возрастают от хлора к иоду.
HF HC1 HBr HJ
Температура кипения при атмосферном
давлении, °С ." . . +19,5 —84,3 —69,9 —37,2
Температура плавления, °С —103 —111 —86 —51
Фтористый водород уклоняется от общей правильности, что и
объясняется его ассоциацией, на которую уже было указано выше.
Возрастают также скрытые теплоты испарения (.по данным Эст-
рейхера):
HCI HBr HJ
Молекулярные скрытые теплоты испарения 3600,3 3939,1 4331,8 ккал
Весьма характерно изменение растворимости в ряду галоидных
солей RX. Так, растворимость одних солей, например серебряных AgF,
AgCl, AgBr, AgJ и ртутных падает от фтора к иоду, между тем как
растворимость солей других металлов столь же правильно изменяется
в обратном порядке (соли фтористоводородной кислоты, впрочем,
иногда отклоняются от этого правила). В нижеследующей таблице
сопоставлены данные для растворимости некоторых солей галоидоводород-
ных -кислот. Приведенные числа указывают, сколько граммов данной
соли растворяется в 100 г воды (по большей части при 20°):
Вещество
AgF
AgCl
AgBr
AgJ
KF
KCI
KBr
KJ
NaF
NaCI
NaBr
NaJ
Растворимость
181,8
1,53x10-*
8,5xl0-e
3,53x10-'
92
34,2
65
144,2
4,4
35,8
90,3
179
Температура,
°C
15,5
20
20
20
18
20
20
20
18
20
20
20
Вещество
CaF2
CaCl2
CaBr2
CaJ2
BaF.>
BaCl.,
BaBr2
BaJo
HgF2
HgCU
HgBr2
HgJ2
Растворимость
1,6x10-3
74,5
143
204
0,163
35,7
104
198
—
7,4
0,61
6xl0-s
Температура,
°C
18
20
20
20
18
20
20
20
—
20
25
25
Наконец, правильно изменяются и свойства галоидозамещенных ор
панических соединений типа RX, как івидно из следующего примера:
Температура кипения при 760 мм,
°С
Удельный вес при 0°
Коэффициент вязкости т]х105 при
^}-0,0000323*
Молекулярное магнитное вращение
C,H,F
II it
С,Н7С1
+46,5
0,912
330
5,0?
С,Н7Вг
+71
1,3835
372
6,89
C,H7J
+102,2
1,783
404
11,08
' По Горпе и Роджеру [41] вязкость химических соединений рациональнее всего
сравнивать при температурах при, которых температурные коэффициенты их вакости одинаковы.
Периодическая система химических элементов 185
В составе и .свойствах кислородных соединений галоидов также
сказывается несомненная аналогия, но вместе с тем резче, нежели в
галоидоводородах, .появляется индивидуальность отдельных элементов.
Менее ясным делается и правильное изменение свойств.
Окисли
F* — — _
С1 С120 СЮ, -
Вг — —" —
J - jcv" J3o5
Кислоты
сі нею нею. нею, нсю4
Вг НВгО НВЮ2(?) НВгОз —
J HJO — HJO3 HJ04
Состав этих соединений только отчасти выражается сходственными
формулами. Кислородсодержащие соединения фтора до сих пор не
"получены ****. Равным образом для брома неизвестно ни одного окисла.
Больше сходства проявляется в составе кислородных кислот, среди
которых, однако, для брома отсутствует высшая НВг04. Общим свойством
¦кислородных соединений галоидов следует считать их малую
устойчивость и окислительные свойства. «Сродство» галоидов -к кислороду
видимым образом возрастает от фтора к йоду, то есть в обратном
отношении сравнительно со сродством кислорода *****, причем, однако, бром
не занимает промежуточного положения между хлором и иодом. На
последнее указывает не только отсутствие НВг04, но и, например,
следующие 'величины теплоты образования кислот НХОз в водном растворе
(Н, X, Оз, Ag)
HCIO3 НВгОз. HJ03 ккал
+ 23,94 +12,42 +55,80 (Томсон)
Марганец является единственным элементом24, принадлежащим
к четному (4-му) ряду в VII группе (если не считать «типического»
фтора) и, следовательно, к побочной подгруппе.
На ясно выраженные металлические свойства марганца уже было
обращено внимание выше. По своему положению между хромом и
железом марганец во многих отношениях 'Представляет сходство с этими
двумя элементами.
* В последнее время Г а л л о [42], действуя тихим электрическим разрядом на смесь
фтора с кислородом, наблюдал образование взрывчатого тела, по-видимому,
представляющего крайне нестойкий окисел фтора.
¦¦Бесцветная, относительно постоянная жидкость, кипящая при 82°.
¦¦¦Соединение J02 (или. J204), существование которого указано еще Миллоном
(в 1844 г.), недавно подробнее исследовано Паттисоном Мюром [43] (1909 г.);
представляет бледно-желтое кристаллическое тело удельного веса 4,2 при 10°, выше 130°
разлагается на иод и кислород; с едкими щелочами дает соли йодноватой кислоты,
6 КОН + 6Ю2 = 5KJ03 + KJ+3HaO. Кислота, отвечающая этому окислу, неизвестна.
¦¦¦* См. прим. ред. 7 к стр. 69 настоящего тома. Прим. ред.
***** Устойчивость кислот типа НОХ, наиболее бедных кислородом и, следователь
но, наиболее близких к НХ, понижается, как и для этих последних, с
возрастанием атомного веса, то есть от НОС1 к HOJ.
С120/*
186
Периодическая система химических элементов
С ними сближают его и металлические свойства «простого» тела, и
высокая точка плавления (1200°), и кипения (1900°), и .магнитность, и
электрохимический характер.
Способность давать, с одной .стороны, соли ряда МпХг и МпХз, а с
другой,— соли марганцовистой кислоты Ме2Мп04 также напоминает
соответствующие соединения хрома СгХг, СгХ3, МпСг04 л железа FeX2,
FeX2 и Me2Fe04 *.
С галоидами марганец всего больше сближает выошая форма
окисла Мп207 и соответствующая ему марганцовая кислота, хорошо
изученная в виде солей, например КМп04, Ba(Mn04b и пр. Соли эти
изоморфны с солями хлорной кислоты НС104. Далее, подобно
.кислородным соединениям хлора, соединение марганца МпОз и МП2О7 летучи,
обладают взрывчатыми свойствами и являются, как соответствующие
кислоты, сильными окислителями.
Все эти характерные свойства соединений марганца дают ему право
занять место в VII группе, несмотря на бросающуюся в глаза
парадоксальность такого -совмещения его с галоидами.
Глава IX
НУЛЕВАЯ И ВОСЬМАЯ ГРУППЫ ПЕРИОДИЧЕСКОЙ
СИСТЕМЫ
Две группы в периодической системе требуют особого рассмотрения:
восьмая и нулевая. Значение этих групп, несмотря на все глубокое
различие между ними, до известной степени аналогично.
Мы видели, что свойства элементов, а с особенной резкостью
валентность и электрохимический характер, постепенно изменяются в
•пределах каждого ряда от I груптіы к VII. В то время, когда возникал
¦периодический закон, между элементом VII группы, принадлежащим
к одному из нечетных рядов, и следующим по 'порядку элементом
I группы, принадлежащим к четному ряду, существовал резкий окачок,
ничем не .заполненный (например, от С1 к К, от Вг к Rb).
Понятно, что пробел между столь различными по свойствам
элементами, как галоид и щелочный металл, мог бы быть заполнен
постепенно, если бы между ними существовала целая -серия элементов с
•промежуточными свойствами, подобно тому, как это имеет место в
пределах каждого ряда (например, между Na и С1). Но такого перехода
здесь фактически не существует. Возможно, однако, представить себе
и другого рода переход. Межу двумя элементами —
электроположительным и электроотрицательные — можно представить себе нечто
среднее—элемент нейтральный. Чем должен быть этот элемент? Или
амфотерньш, подобно алюминию, то есть обладающим в одно и то же
время свойствами металла <и металлоида, но в значительно
ослабленном виде; или нейтральным в собственном смысле этого слова, точнее,
инертным, совершенно неспособным к реагированию.
Такими инертными элементами и оказались «благородные газы»:
гелий, неон, аргон, криптон <и 'ксенон25, открытые сэром
В. Р а м з а е м (первоначально совместно с лордом Рэлеем,
начиная с 1894 т.). В этих элементах, которые, несмотря на многочисленные
* Следует, впрочем, заметить, что и соединения типов RXs и RX<, а также
соответствующих гидратов до некоторой степени сближают соединения марганца с
галоидами. Например, МпОя по составу отвечает СЮз и JO2, М.П2О3 и соли МпХз —
нестойкому С120з, известному в виде гидрата НСЮ2, треххлористому иоду JCU и т. д.
Периодическая система химических элементов
187
попытки, не удалось ввести в химическое взаимодействие с другими
телами, противоположные электрохимические свойства ка.к бы нацело
погасили всякую химическую активность26. Благодаря именно этому
своему свойству инертные газы, вскоре же после того, :как были открыты,
нашли себе место в особой «нулевой» группе периодической системы,
выделенной по -предложению Эр р еры и Р а м з а я. Элементы этой
группы и некоторые свойства их сопоставлены в нижеследующей
таблице.
Элемент
Не Гелий
Ne Неон
Аг Аргон
Кг Криптон ....
Хе Ксенон
ЛтомныЙ
вес
3,99
20,2
39,88
82,92
130,2
Температура
плавления. °С
—250
—188
—169
-140
Температура
кипения, °С
—268,5
—243
—186,7
—151,7
—106,9 [44]
Удельный вес
в жидком
состоянии
0,15
1,24
1,213
2,155
Критическая
температура.
—268
—220
—117
— 63
+16,6
Легко видеть, что все физические свойства благородных газов
'Правильно изменяются с нарастанием атомного веса, то есть следуют
правилу, справедливому, в общих чертах, для любой естественной группы
элементов.
Замечательно, что -и относительные количества, в .которых эти
элементы находятся ъ природе (за исключением члена типического ряда
гелия), подчиняются тому же закону.
В 100 частях воздуха по объему содержится [45]:
аргона неона гелия криптона ксенона'27
0,93236 0,00181 0,00054 0,0000049 0,00000059 ч.
¦Отсутствие у этих элементов того, что мы называем химическим
сродством, делает понятным, что молекула их состоит из одного только
атома*, как это могло быть показано путем определения отношения
теплоемкости при постоянном давлении к теплоемкости при постоянном
объеме. Отношение это, как мы видели выше (см. стр. 68) для газов,
молекула которых состоит только из одного атома, как показывают,
теория и опыт (например, с парами ртути), = 1,666. Для благородных
газов (по методу Кундта, основанному на определении скорости
распространения звука в" газах), были получены величины, практически почти
совпадающие с вышеприведенной:
Не Ne Аг Кг Хе
1,652
1,67
1,642 1,659 1,684 1,666
Химическая инертность -благородных газов не препятствует им,
однако, растворяться в воде и других жидкостях. Растворимость
правильно возрастает (по опытам Эстрейхера и Антропова [46]) с повы-
* Сделать обратный вывод было бы неправильно, как это видно из того факта,
что молекула большинства металлов, в том числе наиболее активных в химическом
-отношении (Na, К, Rb, Cs), также состоит из одного атома.
i8S Периодическая система химических элементов
Не
Nc
Ar
Кг
Коэффициент растворимости
Температура минимальной
растворимости
О
50
0,0134
0,0226
около 10'
0,0114
0,1317
0°
0,0561
0,0343
около 40е
0,12
0,07
между
30-40°
0,2189
0,0878
40°
шением атомного веса. Исключение составляет, по-видимому, только
гелий. Весьма характерно, что с повышением температуры -кривая
растворимости сначала падает, а потом вновь .повышается.
Элементы восьмой группы периодической
системы расположены на перепутьи между к р а и н и.м (справа)
элементом четного ряда и следующим по .порядку элементом
нечетного ряда. Они, таким образом, заполняют середины
больших периодов.
Между тем .как при переходе от нечетных рядов к четным
скачок является весьма резким, в данном случае он значительно
смягчается. С одной стороны, в четных рядах, как было указано выше,
металлоидный характер вообще понижен, так что, например, крайний
справа член 4-го ряда, марганец, стоящий в VII группе, является еще
металлом, хотя и с не резко выраженными электроположительными
свойствами. С другой стороны, в нечетных рядах, по той же самой при-
ч-ине, ослаблены металлические свойства: от .марганца (VII группа) мы
¦переходим к меди, занимающей место © 5-м ряду и I группе. Между Мп
и Си вовсе нет той противоположности, как, например, между С1 и К,
а потому трех металлов VIII группы — железа, кобальта и никеля,
стоящих между ними, вполне достаточно для восполнения существующего
здесь пробела. Аналогичные отношения существуют и в других двух
рядах — б-м и 10-м, к которым примыкают элементы VIII группы.
О переходном характере VIII группы с особенной ясностью говорят три
элемента: медь, серебро и золото, которые одновременно могут
быть отнесены к I и VIII группам.
VIII группа заслуживает особого рассмотрения еще и по другим
причинам. Уже по самому расположению находящихся в ней
элементов, она отступает от других групп, ибо содержит по три (или -по 4)
элемента в каждом ряду:
Fe
Ru
Os
Со
Rh
Ir
Ni
Pd
Pt
[Cu]28
fAgl
[Au]
Затем высшая валентность по кислороду здесь лишь в виде
исключения— для двух элементов Ru и Os (в летучих соединениях Ru04 и
OsC^ достигает 8, как того требует периодическая система.
Если в R11O4 и OSO4 рутений и осмий действительно восьмиатомны,
то им следует приписать такие формулы строения:
О
II
0=Ru=0
О
II
0=:OS=--0
о
О
Периодическая система химических элементов
189
Собственно, окислы эти не -принадлежа! к числу солеобразных 29,
гак как им не отвечают ни кислоты, ни основания, ни соли -и в
растворе они обладают нейтральной реакцией и не проводят тока. Приводим
некоторые свойства этих во многих отношениях замечательных
соединений:
Ru04
Легко образуется при окислении рутения
Кристаллы желтого цвета с температурой
плавления 25,5°, легко сублимируют
при обыкновенной температуре. Пары
взрывают около 108°, При более
высокой температуре (выше 500°) пары
Ru04, наоборот, постоянны. Плотность
пара при 100° оказалась нормальной,
соотв. формуле Ru04
Легко разлагается, особенно на свету,
с образованием низших окислов
рутения. Щелочи дают соли рутеповой
кислоты H2Ru04
OsO*
Образуется при накаливании металла
осмия на воздухе или при действии
окислителей на другие соединения осмия
Бледно-желтая кристаллическая масса 80.
Плавится при 40°. Очень легко
сублимируется. При нагревании не
разлагается и обладает нормальной для
формулы Os04 плотностью пара.
Восстановители, различные органические
соединения, легко редуцируют уже при
обыкновенной температуре до низших
окислов Os или же до металла. При
действии щелочей в присутствии
спирта образуются соли осмиевой кислоты
H20s04
После недавних исследований А. Вернера [47] (1901 г.) можно
причислить к соединениям восьмивалентного осмия также так
называемую осмиа.мовую кислоту HOsC^N. Соли этой кислоты Жоли
получил при действии NH3 и щелочей на водный раствор OsO*. Вернер
показал, что при действии на калийную соль осмиамовой кислоты KOs03N
хлористого водорода получается соль нитрилопентахлоросмиевой
кислоты, откуда он выводит следующие формулы для осмиамовой кислоты
и для ее солей:
V°
q/ \NH
Осмиамовая
кислота
°\,'"
N
О
/°S\
ок
Калийная соль
(таутомерная форма)
Восымиатомность других элементов VII группы пока остается
весьма сомнительной и во всяком случае не может считаться
доказанной.
Ввиду своеобразного характера VIII группы полезно будет
привести главнейшие соображения, которые, наряду с вышеуказанными,
оправдывают ее существование, .и прежде всего ответить на два вопроса:
1) какими общими признаками характеризуются элементы VIII группы
и 2) какие правильности наблюдаются в пределах VIII группы .в связи
с распределением элементов, эту группу составляющих.
Ответ на первый вопрос можно резюмировать в следующих
пунктах.
1. Взятые в виде простых тел, все элементы VIII группы
представляют из себя металлы, отличающиеся серым цветом, очень высокой
температурой плавления, высоким удельным весом, незначительными и
очень близкими .между собой атомными объемами, «как видно из
следующего сопоставления:
190
Периодическая система химических элементов
Атомный объем
Температура плавления ....
Fe
7,1
1505
Со
6,9
1490
Ni
6,7
1450
[Си]
7,1
1080
Атомный объем
Температура плавления ....
Ru
8,3
1930
Rh
8,5
1940
Pd
9,3
1550
[AgJ
10,3
960
Атомный объем
Температура плавления ....
Os
8,5
2500
іг
8,6
2300
Pt
9,1
1750
[Au]
10,2
106231
2. Многие из металлов этой группы обладают способностью логло-
щать, а также, особенно при высокой температуре, пропускать через
себя водород. У палладия эта способность достигает наиболее
высокой степени (1 объем палладия поглощает до 900 объемов
водорода). Образование при этом определенных соединений (гидрюров) до
последнего времени не могло быть, однако, доказано с достоверностью,
что составляет противоположность с характерными гидрюрами
элементов I и II групп периодической системы.
С только что упомянутой особенностью металлов VIII группы, по-
видимому, находится в тесной связи способность ах в
мелкораздробленном состоянии действовать каталитически при различных поо-
цессах гидрогенизации и дегидрогенизации, следовательно, играть роль
переносителей водорода (Сабатье и Сендерен, Ипатьев,
Пааль, Фокин, Вильштеттер, Зелинский и другие).
3. Элементы VIII группы обладают весьма резко выраженной
способностью к образованию комплексных соединений, что, по
остроумной теории Аббегга и Бодлендера, стоит в связи с малым их
электросродством, или, иначе говоря, с незначительным
сродством атомов этих элементов к электронам. В свою очередь, малое
электросродство элементов VIII группы, согласно той же теории, стоит
в 'известном соотношении с малостью их атомного объема32.
Из числа комплексных соединений, образуемых элементами VIII
группы, особенно характерны и устойчивы металлоаммиачные,
например,
[Pt2NH3Cl2] [Ni (NH3)4]C12
[Pt4NH3]Cl2 [Pd4NH3]Cl2]
[Pt4NH3] (N03)2 [Co6NH3]Cl3
Затем двойные цианистые соли:
K2[Fe(CN)e] K2[Ni(CN)4)
K4 [Fe (CN),I Кз [Pd (CN)J
K,[Co(CN),]' K2[Pt(CN)4]
Периодическая система химических элементов
191
4. Многие .металлы VIII группы способны давать соединения с
окисью углерода, из которых некоторые обладают летучестью.
Наиболее известны летучие соединения никеля Ni(CO)4 и железа Fe(CO)5.
открытые М он дом, затем Fe(CO)4 и Fe3(CO)9, а также платины
(Шютценбергер) и палладия (Финк); в последних, кроме СО,
содержатся еще галоиды: PtCl2CO, PtCl22CO, 2PtCl23CO, PdCl2CO,
PdCl22CO, 2PdCl23CO. Недавно Мондом и его сотрудниками
приготовлены также соединения окиси углерода с кобальтом Со(СО)з "
[Со(С04)]2.
5. Все элементы VIII группы * дают о-крашенные соли, то есть
показывают избирательное поглощение в области видимого спектра.
Останавливаясь .на распределении элементов в пределах -самой VIII
группы, мы заметим, что и здесь сказываются обычные правильности:
элементы, расположенные друг над другом: Fe, Ru, Os; Co, Rh. Ir; Ni,
Pd, Pt в известных отношениях, а всего больше по типу образуемых
ими -соединений, представляют между собой особенно близкую
аналогию. С другой стороны, как -в только что упомянутых вертикальных
рядах, так и в рядах горизонтальных наблюдается правильное и
постоянное изменение свойств.
Для иллюстрации приведем несколько примеров. Расположенные в
одном -вертикальном столбце Ni, Pd и Pt дают двойные цианистые соли,
построенные іпо одному общему типу:
K2Ni (CN)4 K2Pd (CN)4 K2Pt (CN)4
To же самое можно оказать об образуемых ими аммиакатах
Pd2NH3Cl2 Pt2NH3Cl2
Ni (NH3)4CI2 Pd4NH3Cl2 Pt4NH3Cl2
а равно и о более сложных комплексах, например, о 'Соединениях Ni,
Pd и Pt с а-диоксимами — органическими соединениями общей
формулы **
R-C-C-Ri
II II
NOH NOH
Ni (D2H2) Pd (D2H2) Pt (D2H2)
Соединения эти, кроме одинакового состава (и строения), обладают
еще тем общим свойством, что улетучиваются без разложения.
Двойные цианистые соли и аммиакаты металлов, принадлежащих к
другому вертикальному ряду, Со, Rh, Ir, отвечают уже другому, но
также общему типу:
К3Со (CN)e K3Rh (CN)6 K3Ir (CN)e
[Co6NH3] Cl3 [Rh6NH3] Cls [Ir6NH3] Cl3
Соединения этих металлов с диоксимами также принадлежат к
иным типам, нежели для Ni, Pt и Pd, например для Со: [Co2NH3D2H2]Cl,
[C0NH3CID2H2], [CoD2H2(N02)2]K и т. д. Соединения эти нелетучие, без
разложения.
* Исключение представляет серебро, которое и по многим другим свойствам
ближе к I, чем к VIII груше.
** Для сокращения в следующих формулах молекула а-диоксима обозначена
символом DHa.
192
Периодическая система химических элементов
Можно также указать на то, что оба элемента, образующие окислы
типа Ru04,—рутений и осмий — находятся в одном и том же
вертикальном столбце; что также в общем столбце находятся металлы Си,
Ag, Аи, во многих отношениях особенно близкие между собой и
принадлежащие в одинаковой степени к I и VIII группам28, что члены первой
горизонтальной строки Fe, Со, Ni отличаются очень сильной парамаг-
нитіностью (ферромагнитны); что остальные члены VIII группы
(исключая Си, Ag и Аи), будучи связаны между собой как совместным
нахождением в природе, так и многими общими свойствами, образуют
естественную семью платиновых металлов (платина и ее спутники) и т. д.
С другой стороны, не менее бросается в глаза постепенность
в изменении свойств членов VIII группы. Так, температуры плавления
•правильно повышаются в вертикальных столбцах от Fe к Os, от Со к
іг, от Ni к Pd и столь же правильно понижаются в горизонтальных
рядах, от Fe к Ni, Ru к Pd и от Os к Pt.
Аналогичные градации наблюдаются и в химических свойствах.
Так, в вертикальных рядах способность металла восстановляться из
окислов, а также из большинства других соединений возрастает с
повышением атомного веса, металл делается все более и более
благородным, его электросродство понижается.
Равным образом правильно .изменяется степень устойчивости
соединений, отвечающих различной валентности. В горизонтальном ряду Fe,
Со, Ni заметно постепенное повышение степени устойчивости типа RX2
и понижения устойчивости типа RX?.
Соли закиси железа легко окисляются на воздухе и переходят в
более постоянные соли о.киси.
Соли закиси кобальта на воздухе окисляются только в присутствии
щелочей, особенно аммиака и некоторых аминов (этилендиамина).
В соответствии с этим, соли типа СоХ3 (например, СоС13, СоВгз) сами
по себе не существуют и постоянны только в виде комплексных
соединений лутеосолей, например [Со6ЫН3]Хз, розеосолей {Со5ЫН3Н20]Хз,
пурпуреосолей [Co5NH3Cl]Cl2, двойных цианистых солей [Co(CN)6]K3
и т. п. Наконец, для солей никеля известен почти только один тип №Х2.
Соли закиси Ni ни в кислом, ни в щелочном растворе не способны к
переходу .в соли окиси (NiX3).
Наличность подобных же постепенных изменений легко заметить и
в вертикальных рядах. Например, в ряду Ni, Pd, Pt возрастает
степень устойчивости высшего типа MX* (соответственно R2MX6), который
для Ni вообще неизвестен, для Pd мало постоянен (например, Кг[Р<іС16])
и весьма постоянен для Pt (хлороплатанаты, например КгРіСіб)-
Тип МХз, почти неизвестный для Ni и малопостоянный * для
палладия и платины (PtCl3, Me2PtCl5), для Ir отвечает наиболее -прочным
соединениям этого металла (соли комплексных кислот ЩігСіб],
Н3[1г(Ж)2)б] и т. п.).
К сказанному следует еще добавить, 'что электрохимический
характер металлов VIII группы довольно правильно изменяется в
зависимости от атомного веса.
В общем можно указать, что из всех этих металлов наиболее
электроотрицательным является осмий, который Сен-Клер Девилль
метко назвал «металлоидом платиновой труппы». К нему примыкает
* Лот. Вёлер [Щ приготовил (нагревая в струе хлора при 390—400° PtCU или
PtCU) треххлористую платину PtCU» образующую с хлористым цезием 'мало
постоянную соль CsaPtCIs, и, по-видимому, отвечающую комплексной кислоте HjPtCIi.
Подобные же, но еще менее -прочные соединения лолучены « для палладия.
Периодическая система химических элементов
193
рутений. Затем электроотрицательный характер понижается, а
электроположительный—возрастает: от Os через іг к Pt, от Ru через Rh к Pd.
от Os через Ru к Fe, от іг через Rh к Со, от Pt через Pd к Ni. Таки.м
образом, из шести платиновых металлов
Ru
Os
Rh
іг
Pd
Pt
осмий является наиболее электроотрицательным, а палладий —
наиболее электроположительным
Глава X
РАДИОАКТИВНЫЕ ЭЛЕМЕНТЫ
Когда (с 1896 г.) вслед за открытием Беккереля и супругов
Кюри, наука обогатилась целой новой областью радиоактивных
элементов и продуктов их распада (см. рис. 15), естественно возникает
ропрос, могут ли, и если могут, то каким образом, разместиться
в системе эти своеобразные тела, которые, будучи во многих
отношениях аналогичными нашим обыкновенным химическим
элементам, тем не менее резко отличаются от них способностью к
превращению в другие, нередко еще в большей степени изменчивые вещества.
Уран
\} = 238.5
Урон!
230,5
Средняя
продолжатель - |
л моего жизни \
( ) 8ж Шлёт \
Ост Г
Г 352 дня
Ионий
230,5
\
5*(Юй-103)
Д лет
Радий
228,U
Митом
Ht= 222.6
Оо
¦
(~)—О еС 2500лвт
t '
Q-*Otf 5,57 дня
*tf*0~Oa 4,3мим
38" О—р "-*»"•
і
Родий сЛ Г\+с>а 28JMUH.
2W.
Родий
218,Ь
Г. г t
25 дня
Радий F
'оо/іб
210,
Родий & s->^
соинеці ч—^
Z06.il
ч
ч*
\
N
Яктиний С )
V) у
РадиоактинииСу—О ос
Актиний X Г_)"*"0 *
6
6-
¦-О а
О а
Эманация
Актиний Д
Дктиний В С_)~*"* Р
Дктиний С, \/)*0 а
Актиний С2 С J-*0 #
(?) V
Актиний В О"*"*Р*?
Дктиний ?
(неиздестен)
6
гооии I- >*v
{полоний) ( )+0 ее 202 дня
210. U w
Средняя
продолжительность
жизни
?
28,1 дня
15 дней
5,6 сек.
fft8829cek.
52,1 мин.
3t1 aHjh
1
9,83 нин
*
Незотории I С*\
Средняя про-
доРжителоностб
жизни
Л 4*/8№лет
7,9 года
СУ»*Р+Г 8,
МезогорийЛ
Радиоторий С)~*0 ct
Торий X Q-KD ее
Змонация Г )-*0 ее
Горий A Q-~0 <х
\9 vac a
191гада
(?)
5,35 дня
76 сек.
8>203сек.
15, Jvaco
73 мин.
Торий Л (3~^*^*/ 4,5мин.
t
Торий Е Г~\
(неиздестенр-J
Рис. 15. Таблица радиоактивных рядов
Большой кружок является символом радиоактивного элемента, малый кружок - символом и-частицы
и. нпкенец, точка — символом электрона (корпускула). Материал для таблицы нгіят ім сводки Содди.
помещенное u Annual Reports on the progress of chemistry за 1911 r.
\\\ Л. А. Чугаев. т. Ill
104
Периодическая система химических элементов
Ответ на этот вопрос, по-видимому, приходится дать
утвердительный. Если для большинства радиоактивных элементов положение их
в системе не может еще считаться выясненным в сколько-нибудь
определенной форме, то тем яснее обстоит дело для следующих четырех,
которые все находят себе место в 12-м ряду.
Группа
12-й ряд
Атомный вес
и
Нитон *
Nt
2?Л
1
И
Радиіі
Ra
22ti,4
Ш
IV
Ториіі
Th
SXiA
\
YI
Уран
11
2:і8,5
VII
Из них два: уран и торий принадлежат к числу давно известных
элементов, нашедших себе место в системе вскоре же после
опубликования периодического закона. Новостью по отношению к ним является
только то, что они обладают радиоактивными свойствами и лишь
временным существованием *.
Радий, как общеизвестно, является аналогом бария, от которого
может быть отделен (дробной кристаллизацией солей) лишь с большим
трудом. Ввиду этого трудно было сомневаться в том, что радий
следует поместить во И группу периодической системы. Когда г-жа Кюри
определила атомный вес радия, то стало ясно іи 'положение его в 12-ом
ряду **. В числе многих других доказательств, свидетельствующих
об этом, приведем в следующей табличке соотношение между
атомными весами элементов, принадлежащих к четным рядам II группы.
В самое последнее время к числу
радиоактивных элементов,
относительно положения которых в
периодической системе можно высказаться с
некоторой уверенностью, присоединилась
еще газообразная эманация
радия ***, по поводу которой уже
несколько лет тому назад было сделано
предположение, что ее следует отнести
к нулевой группе, так как она
совершенно лишена способности вступать во
взаимодействие с другими телами33. Предположение это вполне
подтвердилось, когда с разных сторон были сделаны попытки определить
атомный вес эманации. Наиболее точные результаты получены В. Р а м-
заем и Греем [49] по весьма оригинальному методу. Ничтожное
количество эманации (меньше 0,1 мм3) заключалось в тончайшую
кварцевую трубочку (вес которой определялся в предварительном опыте).
Взвешивание производилось на особых весах, сделанных целиком из
сплавленного кварца. Чувствительность их достигала 1/г>ооооо мг- Весы
эти находились в замкнутом сосуде, из которого можно было
выкачивать воздух. Взвешиваемое тело уравновешивалось кварцевым
шариком, вес которого (кажущийся) менялся в зависимости от давления
Ряды
1-й
6-й
8-й
10-й
12-й
Лтомиыіі вес
Са
Sr
Ва
Ra
: 40,09
---87,63
= 137,37
—•
-226,4
Разница
47,.V*
49,74
89,03=2x44,52
* -По новейшиім исследованиям слабо радиоактивны также калий и рубидий.
** В настоящее время .радий .помещают в 10-й ряд .периодической системы.
*** Радон. Прим. ред.
Периодическая система химических элементов
195
Ряд
4-й
6-й
8-й
10-й
J 2-й
Л гимны» вес
Аг
Кг
X
Nt
-39,88
= 82,9
-: 130,2
—
-,223
Р.Ч ЗНИЦ.'і
02
43,02
47,3
8-2x16,4
воздуха (в силу закона Архимеда) и мог быть заранее вычислен для
каждого данного давления. Изменяя '^то последнее, можно было
привести весы в равновесие и по показанию манометра вычислить вес тела.
Опыты этого рода дали для плотности эманации по отношению к
водороду в среднем величину, бликую к 111,5, откуда молекулярный
вес=--223, а так как по данным прежних исследований представлялось
вероятным, что молекула эманации состоит только из одного атома, то
и атомный вес эманации должен быть близок к 223. Аналогичные
результаты были получены также Дебьерном іи другими путем
определения скорости истечения эманации через тонкие отверстия гго
способу Бунзена (по сравнению с кислородом и аргоном) [50].
По предложению В. Рамзая, эманация радия получила название
и и т о и a (Nt).
Если сопоставить атомные веса элементов четных рядов нулевой
группы, то получится следующая
табличка.
Мы видим, что в этом'случае
представитель семьи
радиоактивных элементов хорошо
укладывается в общую систему.
Заметим, что атомный вес Nt
очень хорошо согласуется с
теорией дезинтеграции Резер
форда и Содди, по которой атом
нитона должен происходить из
атома радия через потерю одного
атома гелия: Ra = Nt+He или 226,4 = 222,4 + 4. Совпадение между
теоретическим весом нитона (222,4) и опытным (223) нельзя не признать
превосходным.
Рамзай и Грей подвергли взгляд Резерфорда и Содди еще и
дальнейшей проверке. Взяв некоторое количество свежеприготовленного
нитона, они оставили его в течение месяца в запаянной кварцевой
трубке. Принимая во внимание краткий период жизни нитона (половинный
период=3,8 дня), можно было считать, что при этом практически
закончилось превращение его в гелий и нелетучий продукт, главным
образом радий D *. Трубка была тогда вскрыта и, после выкачивания из
нее газа, снова взвешена. Потеря веса дала весовое 'количество гелия,
образовавшегося из взятой для опыта навески эманации («при этом
приходилось извлекать часть гелия, окклюдированного в кварце). В одном
из таких опытов количество гелия оказалось = 35 миллионным долям
миллиграмма вместо 38, рассчитанных по теории распада.
Таким образом, радиоактивные элементы разнообразнейшей
степени прочности, начиная от урана, средняя продолжительность жизяи
(атома) которого выражается колоссальным числом 8000 000 000 лет,
и кончая нитоном, для которого эта величина составляет всего 5,6 дня,
находят себе место в периодической системе ** наряду с остальными
элементами, для которых неизменяемость в условиях земного
существования до недавнего времени считалась аксиомой. Понятно, какие
широкие горизонты открывает этот важный факт вместе со многими
другими, неожиданными наблюдениями, касающимися радиоактивных тел.
* іПо Резерфорду, превращение нитона и радий D должно сопровождаться
потерей трех атомов гелия (атомный вес RaD = 2lO,4).
** В какой мере удастся найти место в периодической системе также для
остальных элементов, <пока остается открытым вопросом [51].
13*
196
Периодическая система химических элементов
Возможность связать периодический закон с происхождением элементов
тем самым становится в ближайшую очередь (см. об этом В. А. Тиль-
лен. Химические элементы).
Глава XI
ПЕРИОДИЧЕСКАЯ СИСТЕМА КАК СРЕДСТВО
ДЛЯ НАУЧНОГО ИССЛЕДОВАНИЯ
«Система элементов,—пишет Д. И. Менделеев в одном из своих
мемуаров о периодическом законе,— имеет не одно только педагогическое
значение, является не одним только средством, облегчающим
запоминание разнообразных, систематически расположенных и связанных
между собой фактов, но имеет также и научноа значение, ибо открывает но
вые аналогии и тем самым намечает новые пути для познания
элементов». Менделеев сам не замедлил дать блестящее подтверждение
справедливости такого взгляда, установив для нелого ряда элементов
их положение в системе, а вместе с тем пролив новый свет на их
отношение к другим элементам. Но этого мало.
Как уже было указано выше (см. ^историческое введение), Дмитрий
Иванович, заметив, что целый ряд элементов не укладывается в
систему, ссліи им приписать атомные веса, общепринятые в то время, признал
необходимым их исправить, а с другой стороны, там, где был
известен эквивалент элемента, но атомный вес оставался сомнительным
(см. главу I), указал новый метод, для решения опорного вопроса,
опирающийся на периодический закон.
Так были исправлены или установлены атомные веса индия,
урана, платины, осмия, иридия, золота, титана, бериллия,
церия, иттрия.
Опыт блестящим образом оправдал все эти теоретические выводы
Менделеева.
Определение и исправление атомных весов «a основании
периодического закона теснейшим образом связано с точным установлением
места, которое должен данный элемент занимать в системе, результатом
всестороннего выяснения его прямых и отдаленных аналогий с
другими элементами.
На нескольких примерах легко будет уяснить сущность подобных
определений и исправлений. В то время, когда создавалась
периодическая система, оставалось спорным положение в системе элемента
бериллия, ибо он, с одной стороны, казался аналогом алюминия, с
другой, примыкал к магнию. Первая точка зрения насчитывала большое
число сторонников, хотя еще в 1819 г. русский химик Авдеев
убедительно доказал, что всего ближе Be стоит к Mg. Если бериллий — аналог
алюминия, то окись его должна отвечать формуле ВегОз как глинозем
{А1203), а так как в ней по аналшу на 16 г кислорода приходится
9,1 г Be, то атомный вес Be должен бы был быть=13,7 (ибо 3x16 г О
соответствует 9,1X3 = 27,3 г Be и это количество по формуле соответ-
27 3
ствует двум атомам; следовательно, вес 1 атома Be — -~ ==13,7).
Эта величліна почти совпадает с 14, то есть с атомным весом азота.
Соответствующее место в периодической системе занято, но если бы Be
и поместился на место азота в V или рядом с ким в VI группу, то по
аналогии следовало бы ожидать для него окислов Be2Os или Ве03<
которых на деле не существует. Если же мы предположим вместе с
Авдеевым, что бериллий, как аналог магния, дает окись ВеО, то атомный вес
іего будет = 9,!, if тогда для него как раз находится место во 2-ом ряду
Периодическая система химических элементов 197
и II группе периодической системы между Li и В. Многие свойства
соединений бериллия в соотношении со свойствами аналогичных
соединений его соседей по периодической системе вполне оправдывают такой
вывод.
Менделеев весьма наглядно иллюстрирует эти соотношения схемати- ¦
ческими пропорциями, .имеющими, разумеется, лишь качественное
значение.
Так, принимая вышеуказанное положение Be, мы можем написать:
1) Be: Li = B : Be. «На самом деле,—пишет Д. И.,— основные свойства
в ВеО гораздо слабее выражены, нежели в Li20, а в В203 еще слабее,
чем в ВеО. Хлористый бериллий летучее хлористого лития, а еще
больше того летучесть хлористого бора. 2) Be : Mg=Li : Na = B : AL Если
окись бериллия — менее энергичное основание, нежели MgO, то в
соответствии с этим основные свойства у Li20 слабее, чем v Na20, у ВоОз
слабее, чем у А1203. Поэтому гидрат окиси бериллия растворим в КОН.
По той же причине не имеет значения неполный изоморфизм между
солями ВеО и MgO, в кристаллической форме которых иногда
наблюдаются даже значительные отличия, ибо совершенно аналогичные
отношения существуют между солями Li и Na, между соединениями В
и А1. Так, например, фтористый бериллий растворим в воде, между тем
как фтористый магний — нерастворим, совершенно подобно тому, как
растворим в воде фтористый бор; а фтористый алюминий отличается
нерастворимостью. 3) Be : AI = Li : Mg=B : Si. Если, следовательно,
невзирая на различие в формулах окислов, окись бериллия во многих
отношениях напоминает глинозем, то сходство наблюдается также межлу
Li20 и MgO, между В20з и Si02. Если далее объем эквивалента ВеО =
— —-—=8,3 близок к объему эквивалента глинозема 1/зАі203==1/з—-1- —
3,05 4.0
= 8,5, то в этом случае подобное же совпадение наблюдается
между соответствующими соединениями Li и 1/гМё> между 7зВ203 и
72Si02». С тех пор, как были написаны эти строки, справедливость
мнения Менделеева относительно положения Be в системе и его
атомного веса получила новые подтверждения. Так, была
определена плотность пара хлористого бериллия, а также определен
молекулярный вес его ацетилацетоната. Полученные результаты вполне
установили двухатомность Be. Совсем недавно Галецкий (1909 г.)
показал, что тот же самый вывод вытекает из сравнительного определение
способности солей Be и других металлов осаждать из раствора
коллоидальный сернистый мышьяк. Опыт показал, что способность эта для
одно-, двух- и трехвалентных ионов выражается числами: 1 : 30 : 1650.
Для солей Be получились цифры, близкие к 30, подобно ионам MgM,
Са+ + и Ва + ^.
Индий представляет другой случай, на котором периодический
закон позволил установить атомный вес элемента, ранее неизвестный с
достоверностью. Индий соединяется с кислородом в -пропорции 37,7 : 8.
Отсюда эквивалент индия = 37,7. Каков его атомный вес? В то время,
когда создавалась периодическая система, для ответа на этот вопрос
не имелось еще достаточного материала, так как не были определены
ни теплоемкость индия, ни плотность пара летучих его соединений, ни
отношения его к другим элементам, связанные с изоморфизмом. Было,
однако, известно, что индий по некоторым свойствам походит на цинк.
Отсюда можно было предполагать, что окись индия построена
аналогично окиси цинка — из двух атомов: InO. Тогда очевидно (так как
198
Периодическая система химических элементов
состав ее 37,7X2 весовых частей In на 16 весовых частей О), что
атомный вес индия был бы 37,7X2 = 75,4. Такой атомный вес для индия
многие и принимали в то время.
Но в периодической системе место с атомным весом, близким к 75,
как раз занято мышьяком, ибо мы имеем в 5-м ряду элементы
Ge As Se
72,5 74,96 79,2
Поэтому In не может быть = 75,4. Остаются возможности: 1п = 37,7хЗ
или 37,7X4 и т. д.
Понятно, что если для атомного веса индия принять утроенный
эквивалент, то есть 37,7X3=113, то в периодической системе для него
как раз оказывается вакантное место в 7-м ряду и в III группе между
Cs—112 и Sn =119. Многие свойства индия и его соединений вполне
гармонируются с таким его положением в системе, что и было
высказано Д. И. Менделеевым еще в 1871 г.
Этот взгляд получил блестящее подтверждение в величине
теплоемкости индия, определенной Мен де л ее>в ы м (0,55) и Бунзеном
(0,57), а в особенности в открытой Реслером способности индия
давать квасцы, изоморфные с алюминиевыми, например 1пг(504)зХ
X (NH4hS04 • 24НгО. В этом 'последнем обстоятельстве резко
сказывается аналогия между In іи А1.
В только что упомянутых случаях установления истинных величин
атомного веса Д. И. Менделеев, на основании свойств элемента,
определил его положение в системе, которое, в свою очередь, давало*
указание относительно валентности этого элемента в характерных его
соединениях; зная же валентность, можно было определить и атомный вес,
раз было известно значение эквивалента (см. главу I). Случай
исправления атомного веса урана представляет дальнейший пример
аналогичного характера.
Иной характер носит исправление, предпринятое Д. И. Менделеевым
в VIII группе, где раньше для осім-ия, иридия, платины и золота
принимались такіие атомные веса:
Au Pt іг Os
196,2 196,7 196,7 196,8*
а по другим:
196,2 108 197 199—200.
Для Менделеева было ясно из рассмотрения свойств элементов VIII
группы в ее целом:
Fe Co Ni [Си]
Ru Rh Pd [Ag]
Os Ir Pt [Au],
что порядок нарастания атомных весов необходимо должен
совершаться в направлении от О? к Au. Иначе золото очутилось бы аналогом
рутения и железа, а осмий стоял бы в одном столбце с Ag и Си и, подобно
этим элементам, должен бы быть отнесенным в I группу. Все это,
очевидно, совершенно не вяжется с природой Os и Au. Менделеев поэтому
смело заяв-ил, что атомиые веса элементов As, Ir, Pt >и Au определены
* В.чято ил статьи Л отар а Мейера, относящейся к 1870 г.
Периодическая система химических элементов
19 9
неточно и нуждаются в пересмотре, что, во всяком случае, Os<Ir<Pt<
<Au. Все это вполне подтвердилось позднейшими определениями Зей-
бе р та и других.
Д. И. Менделеев не остановился на исправлении атомных весов
элементов известных. Ему принадлежит также,— и в этом, без сомнения,
самый блестящий результат, достигнутый благодаря периодическому
закону,— 'предсказание трех -н о в ы х, тогда еще неизвестных
элементов со всеми их физическими и химическими
свойствами. Элементы эти ныне носят название: галлия,
скандия и г е р м а н и я, по имени трех стран, в которых они были
впоследствии открыты. Менделеевым же они были предсказаны под именем
э к а а л ю м и н и я, экабора и экасилиция.
В 1875 г. Лекок д е-Б у а б о д р ан во Франции открыл галлий,
сернистое соединение которого содержится в виде ничтожной примеси
к цинковой обманке (ZnS), добываемой в Пиренеях. Свойства галлия
удивительно хорошо совпали с теми, которые были предсказаны
Менделеевым для экаалюминия.
Вслед за тем Нильсон в 1879 г., а за ним Клеве в Швеции
открыли другой редкий элемент—с кан д и и, который воспроизвел в
точности все предсказанное Менделеевым для экабора. Наконец, в 1886 г.
Кл. Винклер в Саксонии открыл экасилиций, получивший название
германия.
Какой путь привел Д. И. Менделеева к предсказанию новых
элементов и их свойств, лучше всего будет ясно из следующих слов, которые
мы выписываем из «Основ химии»: «Каждый элемент по периодической
системе имеет место, определяемое группою (означаем римской
цифрой) и рядом (цифра арабская), в которых он находится. Они
указывают величину атомного веса, аналогию, свойства и форму высшего
окисла, водородного и друлих соединений, словом, главные количественные
признаки элемента, хотя затем и остается еще целый ряд подробностей
или индивидуальностей, 'причину которых, быть может, должно искать
в небольших разностях величины атомного веса. Если в некоторой
группе находятся элементы Rb R2, Яз* а в том ряде, где содержится один из
этих элементов, например, R?, находится "перед ним элемент Q2, а после
него элемент Т2, то свойства R2 определятся как среднее — по свойствам
Ri, R2, Q2 и Т* Так, например, атомный вес R2:=1/4 (R1 + R3 + Q2 + T2).
Например, селен находится в группе с серой (S —32) и теллуром
(Те=127), а в 5 ряде перед ^ним стоит As = 75,0 и -после него Вг = 80.
Отсюда величина атомного веса селена = 74 (32-f 127 + 75 + 80) =78,5,
что близко к действительности, по которой Se = 79, без уверенности в
десятых долях, так что, быть может, 78,5 окажется еще ближе к
природе тела. Так можно определить и другие свойства селена, если бы они
не были известны. Например, As образует H3As, Br дает НВг;
очевидно, что селен, между ними находящийся, должен образовать H2Se, со
свойствами, средними между H3As и НВг. Самые физические свойства
селена и его соединений, не говоря об их составе, определенном
группою, могут быть с большой близостью к действительности определены
по свойствам S, Те, As и Вг. Таким образом, есть
возможность предугадать свойства неизвестных еще
элементов, особенно тогда, когда он окружен известными. Так,
например, на -месте IV—5, то есть в IV группе и 5 ряде, еще в 70-д годах
недоставало элемента. Такие неизвестные элементы можно назвать по
имени 'предшествующего известного элемента той же группы, прибавив
предварительно слог эк а, что значит по-санскритски один. Элемент
IV—5 следует за IV—'3, и это место занято кремнием или силицием, а
200
Периодическая система химических элементов
потому бывший неизвестным элемент назван мною э-какремнием или
зкасилицием и означен Es».
Как близко совпадение между свойствами элементов,
предсказанных Менделеевым и фактически открытых, лучше всего будет видно из
следующих сопоставлений.
Скандий
Предсказано Д, И. Менделеевым в 1871 г.
Экабор ЕЬ
Атомный вес 44
Окись экабора ЕЬ203
Удельный вес окиси 3,5
Сернокислая соль Eb2(S04h
Двойные сернокислые соли не будут
изоморфны с квасцами
В действительности оказалось (Клеве,
Нпльсон и другие)
Скандий Sc
Атомный вес 44,1
Окись скандия Sc203
Удельный вес окиси 3,864
Сернокислая соль Sc2(S04)3
Двойная соль 3K2SO4 • Sc2(S04)3
кристаллизуется в призмах
Галлий
Предсказано Д. И. Менделеевым в
1871 г.
Экаалюминий Еа
А томный вес должен быть близок к 68
Простое тело должно быть низкоплавко
Удельный вес его близок к 6,0
Удельный объем 11,5
Не должно окисляться на воздухе
Должно разлагать воду при краснока-
лильном жаре
Формулы соединений: ЕаСіз, Еа20з,
Ea2(S04)3
Должно образовать квасцы Ea2(S04h ¦
* Me2S04 • 24Н20, но труднее, чем А1
Окись Еа20з должна легко восстановлять-
ся и давать металл более летучий, чем
AI, а потому можно ожидать, что Еа
будет открыт путем спектрального
анализа
В действительности оказалось (Лекок-
де-Бауабодрон
Галлий Ga
Атомный вес 69,9
Тепло плавления свободного галлии 30J
Удельный вес = 5,96
Удельный объем 11,7
Слегка окисляется только при красном
калении
Разлагает воду при высокой температуре
Формулы соединений: GaCb, Ga20.i,
Ga2(S04)3
Образует квасцы Ga2(S04)3, (NFUhSO**
• 24Н20
Ga восстанавливается из окиси
прокаливанием в токе водорода; открыт при
помощи спектрального анализа
Германий
Предсказано Д. И. Менделеевым в 1871 г.
Экасилиций Es
Атомный вес 72
Удельный вес 5,5
Атомный объем 13
Формула окиси Es02
Удельный вес ее 4,7
Хлористое соединение EsCl4
Температура кипения его ниже \W
Удельный вес его 1,9
Металлоорганическое соединение
Es(C2H5)4
Точка кипения его 160^
Удельный вес 0,96
Найдено Кл. Винклером в 1886 г.
Германий Ge
Атомный вес 72,5
Удельный вес 5,409
Атомный объем 13,2
Формула окиси Ge02
Удельный вес ее 4.703
Хлористое соединение GeCl4
Кипит при 96°
Удельный вес его 1,887
Металлоорганическое соединение
Ge(C8H6)4
Точка кипения его 160°
Немного легче воды
Периодическая система химических элементов 201
Нельзя не признать, что совпадение прямо поразительное.
В гласе IX мы видели, что к числу элементов, предсказанных
(И. А. Морозовым) на основании 'периодического закона, следует
отнести и нулевую группу.
Соображения относительно необходимости существования инертных
элементов с нулевой валентностью были также развиты Ю. Томсеиом
вслед за открытием первых представителей нулевой группы. Томсен
также опирается при этом на периодический закон. Если свойства
элементов действительно являются периодической функцией атомного
веса, то необходимо иметь в виду, что в этом случае переход от
положительной величины к отрицательной (от I группы к VIII) и наоборот
может сопровождаться или постепенным прохождением функции через
нуль или резким скачком и переходом через оо. Третья возможность
исключена.
Первый случай соответствует постепенному изменению
электрического характера в пределах отдельных рядов периодической системы.
Чт о касается второго, то ему отвечал бы переход из одного ряда
в другой. Том'сен заключает, что при таком переходе можно
ожидать появления элемента, электрический характер которого
определяется величиной±оо, то есть :ілектрически индифферентный, и притом
с нулевой 'валентностью. На основании соображений, на которых мы не
будем здесь останавливаться, Томсен предполагает существование
элементов со следующими атомными весами:
Предсказано Томсеном
На самом деле
существуют
4
3,99
Гелий
20
20,2
Неон
36
39,88
Аргон
84
82,92
Криптон
132
130,2
Ксенон
212
223
Нитон
292
За исключением нитона, предсказанные атомные веса довольно близки
к истинным. В последнее время Д. И. Менделеев (1905) предложил
дополнить периодическую систему еще одним, нулевым, рядом. В этом
ряду 'И в нулевой группе Д. И, 'предвидит существование элемента х с
атомным весом ничтожно малым, между 0,00000096 и 0,000000000053,
и предполагает, что таким элементом является мировой эфир
физиков. Кроме того, Менделеев указывает еще на другое вакантное место
в 1-м ряду и 0 группе (над неоном) и предполагает, что оно должно
принадлежать элементу у с атомным весом, близким к 0,4, каковым
может быть короний, открытый в короне солнца.
Быть может, к числу оправдавшихся 'предсказаний гтридется
причислить случай эка м а р ганца, на возможное существование которого
указывал Д. И.. Менделеев. Недавно Френч (в Канаде) открыл новый
элемент в платиновой руде, по некоторым свойствам, по-видимому, по
хожий на марганец, отчасти напоминающий серебро. Возможно, что
этот элемент, если только факты, опубликованные Френчем (пока еще
в слишком общей и неопределенной форме), подтвердятся, займет место
в периодической системе между Мо и Ru (атомный вес около 99),
оставшееся до сего времени свободным *.
* Между W и Os Д. И. Менделеев считает возможным существование еще одного
элемента двимарганца с атомным весом, близким к 188. См. прим. ред. на
стр. 470 настоящего тома.
202 Периодическая система химических элементов
Глава XII
ОТСТУПЛЕНИЯ ОТ ПЕРИОДИЧЕСКОГО ЗАКОНА
Несмотря на огромное значение успехов, достигнутых в области
теоретической и систематической химии, благодаря периодическому
закону, несмотря на ширину перспектив, которые он открывает для науки
будущего, освещая самые глубокие философские проблемы, нельзя не
признать, что при ближайшем знакомстве с периодической системой
в ней открывается ряд отступлений, ряд случаев, указывающих 'как бы
на недостаток последовательности или выдержанности различных пра-
вильностей, периодическим законом определяемых. Мы остановимся на
некоторых из числа таких «отступлений».
В принципе место элемента в системе должно определяться его
атомным весом Между тем, есть ряд элементов, для которых атомный
вес указывает одно место в системе, а совокупность '.прочих свойств —
иное. Так, аргон должен бы стоять после калия (ибо его атомный
вес = 39,88, а для К = 39,10), но тогда бы он не подпал в нулевую
группу, куда, очевидно, относится. Так, кобальт (атомный вес 58,97) должен
бы стоять после никеля (атомный вес 58,7), а теллур (атомный вес
127,5)—после иода (атомный вес 126,92), тогда как по сумме свойств
очевидно обратное.
Эти несоответствия породили целую литературу. Вслед за
Д. И. Менделеевым многие искали ошибок в прежних определениях
атомных весов и делали новые, проверочные. Теллуру одно время
Б.. Б pay не р, Марквальди другие считали возможным приписать
атомный вес меньше иода, но позднейшие определения (особенно
Б р. Бэкера) этого -не подтвердили, и хотя в самое последнее время
Флинт в Америке вновь привел данные, указывающие на то, что
атомный вес чистого теллура должен быть меньше, нежели принимали
раньше, но результаты эти возбуждают большое сомнение и, во всяком
случае, вопрос далек еще от окончательного решения. Для пар Аг, К и Со,
Ni существующие данные, по-видимому, также не дают повода
сомневаться в наличности «отступления» *.
Далее, как уже было упомянуто, существование VIII группы в
периодической системе, несомненно, является некоторым осложнением в том
основном плане системы, согласно которому в каждой группе данный
ряд представлен только одним элементам.
Целая группа редкоземельных элементов, к числу которых
относятся: церий, празеодим, неодим, самарий, европий, гадолиний, тер-
* Исследования над абсорбцией вторичных лучей, испускаемых различными
металлами при шадении на них лучей Рентгена, .показало, что 'при нанесении на
диаграмму по оси абсцисс атомных весов элементов, испускающих лучи, а по оси ординат —
проницаемости какого-либо определенного тела для этих лучей, получается правильная
кривая. При этом, <по опытам ВагкГа и Sadler'a [52], никель только в том случае
попадает на кривую (с Си, Со и другими), если принять для него атомный вес 61,4, что
согласовалось бы с требованием периодического закона, по которому Ni>Co (ср.,
впрочем, [53]). Аналогичные результаты были получены при изучении поглощения
однородных рентгеновских лучей простыми телами. Все эти наблюдения, несмотря на их
несомненный интерес, только прибавляют новые факты к числу ранее известных,
заставляющих считать, что в естественной системе элементов Ni должен следовать за
Со, а не наоборот. Каких-либо прямых указаний на неоднородность ныне считаемых
чистыми никеля и кобальта в них не содержится.
Все попытки открыть в Со или в Ni какую-либо примесь до сих пор кончались
неудачей. Точно так же едва ли можно думать, что очистка аргона, а тем более калия,
была бы настолько недостаточна, чтобы извратить {порядок, в котором следуют друг
за другом их истинные атомные веса.
Периодическая система химических элементов
203
Пий, диспрозий, гольмий, эрбий, тулий, иттербий, лютеций, вовсе не
находят себе места в системе*. Атомные веса этих элементов лежат
между 140—178, а потому они должны бы разместиться между
лантаном и танталом. Но свойства их настолько близки между собой, что
распределение их по отдельным группам ¦не представляется возможным.
Чтобы выйти из затруднения, профессор Бог. Браун ер [54], один из
лучших знатоков этого вопроса, высказал предположение, что
«подобно тому, как в VIII группе по 4** элемента занимают -как бы одно
место в системе, так и приведенные элементы редких земель составляют в
системе узел или .пояс и стоят на месте IV—8, на котором до сих пор
стоял один церий». При этом предполагается в 8-м ряду переходить от
Се к Та, 8-й ряд тогда изобразится так ***:
11.1
1
(1
X
130
I
Cs
133
и
Ва
137
ш
La
139
IV
Се и т. д.
140-178
V
Та
181
VI
W
184
VII
?34
190
VIII
On іг, Pt
191, 193, 19Г.
При этом Се и т. д. будет означать (в соответствии с новейшими
успехами, достигнутыми при изучении редких земель):
Церий
Се
140,25
испрозий
Dy
Ш,Г>
Празеодим
Рг
140,6
Гольмий
Но
163,5
Неодим
Nd
144,3
Эрбий
Ег
167,7
Самарий
Sa
150,4
Тулий
Ти
168,5
Европий
Ей
152,0
Иттербий
Yb
172,0
Гадолиний
Gd
157,3
Лютеций
Lu
174,0
Для наглядности Браунер предлагает всю периодическую систему
представить в двух плоскостях, между которыми элементы редких
земель образуют как бы мостик, или пояс. «Подобно тому, как в
солнечной системе,— .пишет он по этому 'поводу,—целая группа астероидов
занимает одну зону ил-и одно место на эллиптическом пути, на котором,
судя по аналогии, должна бы была двигаться одна лишь планета,
также и в периодической системе целая группа редких земель может
занимать одно место, на котором должен бы был находиться один только
элемент. Если принять в соображение, что многие химики склоняются к
тому взгляду, что все наши элементы построены из одной и той же
первичной м'атер'ии, то можно представить себе, что при образовании
элементов редких земель сгущение первичной материи не достигло той
степени, как в случае других элементов, или вообще происходило здесь
несколько иным путем. Тогда столь близкие между собой элементы
* См. прим. ред. на стр. 471 настоящего тома. Прим. ред.
** Вернее, по три элемента. Прим. ред.
*** В типичнейших своих соединениях элементы редких земель трехитомиы --
таковы галоидные соединения RX3, окислы R203 и т. д. Но Браунер приводит и польз\
своей точки арения то обстоятельство, что некоторые из этих элементов образунп
окислы тала R02 (особенно Се, для которого известны также соединения CeF4, CeCI«
и т д.), не имеющие характера перекисей (к Рг и Nd степень устойчивости этих
окислов сильно -падает). Устойчивый высший окис-ел дает еще тербий. Состав его, шо-ви-
ди'мому, отвечает формуле ТЬ407. Интересно отметить, что свойства .редкоземельных
элементов 'изменяются -по большей части в порядке возрастания атомных весов,
причем, однако, для простых тел наблюдаются отступления: іплотпость металла Се не
укладывается в іряд, в температурах .плавления простых тел правильного
соотношения с атомным весом иообще незаметно.
Тербии
ТЬ
159,2
204
Периодическая система химических элементов
редких земель, обладающие в форме простых тел почти
одинаковыми атомными объемами, заняли бы одно место в периодической
системе...».
Несмотря на вес остроумие и заманчивость этих соображений, нельзя
не признать, что гипотеза Браунера * только подчеркивает факт нового
1
2
3
4
5
6
7
8
9
10
11
12
0
Не
4
Ne
2') 1
ЛГ
39, У
Кг
«2,0
X
130,2
1
н
1.00Н
Li
6, У
Na
К
Си
Rb
35. S
107,9
Cs
П2.8
Au
197,2
R20
It
Be
У,і
Mg
Ca
10,1
Zn
65,4
Sr
87,6
Cd
112,4
Ba
13 7,4
¦200
Ra
226,4
RO
III
В
11
A!
.'7,1
«4 r
4-1,1
Ga
69,9
b9
In
114.S
La
139
Yb
172
Tl
204
R-»o3
IV
с
12
Si
¦:.v>
Ti
4h,l
Ge
72.5
2r
Sn
119
140,3
Pb
207,1
Th
232,4
RO,
V
N
14
P
il
V
"1,1
As
75
Nb
9 1,5
Sb
120,2
Та
Bi
R,05
VI
О
Ih
s
32,1
Cr
V2
Se
:9,2
Mo
9b
Те
127,5
W
184
L
238,5
Ю,
VII
Г
19
CI
35,5
Mn
54,9
Br
79,9
J
126,9
R.O,
VIII
Fe Co \i
Ru Rh Pd
,101,7 102,'.' 106,7
Os lr Pi
19:),'' I9'i,l 19Ь.2
RO,
Рис. 16. Периодическая система Д. И. Мендлсева (старая таблица) зб
нарушения простоты и стройности системы по отношению к порядку
расположения в ней элементов.
Бели самый порядок расположения элементов не вполне выдержан
в духе простой и однородной схемы, то то же следует сказать о
чередовании свойств элементов, в зависимости от их положения в системе.
Так, далеко не строго выдержано постоянство «гомологической»
разницы атомных весов сходственных элементов. Во многих группах
Ретгерс 155], а также Бильту [56] предлагают поместить редкие земли
также в одной, но только не в IV, а в III группе. См. в выше цитируемой статье
Браунера критические замечания относительно этого предложения.
Периодическая система химических элементов
205
(особенно в VIII) ні> достигается высшее значение валентности в соле-
образующих окислах, которое должно бы отвечать данной группе.
Не строго, а лишь приблизительно подтверждаются правильности,
касающиеся соотношения между физическими и химическими
свойствами простых тел и соединений, образуемых элементами, и положение
этих последних в системе. Особенно свойства элементов «гомологов» не
всегда изменяются с той правильностью и постепенностью, которой
можно было бы ожидать. Так, бром далеко не во всех отношениях является
промежуточным звеном между хлором и иодом, селен — между серой и
теллуром, серебро — между медью и золотом, церий — между
цирконием и торием, мышьяк-- между фосфором и сурьмой и т. д. (см. VIII и
IX главы). Многие правильности, особенно связанные с существованием
четных и нечетных рядов, нарушаются существованием «типических»
элементов.
Весьма распространено мнение, что причина всех этих «недостатков»
периодического закона кроется в несовершенной форме, в которой он
выражен.
Отсюда длинный ряд попыток отыскать новые лучшие фор-
м ы, «усовершенствовать» периодическую систему. Нельзя, однако, не
признать, что ни одна из подобного рода попыток не
привела к цели и не дала чего-либо существенно нового,
по сравнению с тем, что было создано Менделеевым в 1869—1871 гг.*.
Ввиду этого невольно возникает некоторое сомнение в том, можег
ли вообще периодический закон, по самому существу дела, воспринять
ту простую и вполне точную форму, которую стремятся ему придать:
возникает вопрос, не заключается ли в нем лишь отражение того
плана, по которому совершалась эволюция существующих элементов,
отражение тех же сложных и многочисленных факторов, которые
оказывали влияние на ход этой эволюции.
Примечания и добавления**
К стр. 77. В только что вышедшей (сентябрь 1912 г.), международной
таблице атомных весов на 1913 г. ни одна цифра не подверглась изменению по -сравнению
с таблицей 1912 г., вновь включен только элемент гольмий с атомным весом 163,5***.
К стр. 82. Подробности относительно метода Дж. Томсона см. в Jahrbuch fur Ra
dioaclivitat und Elektronik, VIII, 198 (1911).
К стр. 106. Ввиду того, что относительные значения электродных
потенциалов легче определяются, нежели абсолютные, и обладают большей степенью
достоверности, что, кроме того, практически важны прежде всего относительные значения
этих потенциалов, часто, следуя за Нернстом, в электролитическом ряду напряжения
металлов потенциал водорода условно считается равным нулю, что нисколько не
отражается ни на порядке следования металлов, ни на разницах между потенциалами
любой пары. Величины электродных потенциалов в этом случае легко получить из
приведенных на стр. 106 чарез прибавление 4- 0,2770.
К стр. 132—135. Относительно связи между температурами кипения простых тел и
положением в периодической ситем-е можно указать на следующие правильности. У
металлических элементов в отдельных подгруппах первых четырех групп
температура кипения падаете повышением атомного веса, следовательно, летуче с ті.
возрастает. Такое отношение наблюдается в подгруппе щелочных металлов Li -- Cs
(см. стр. 134) и в подгруппе Zn, Cd, Hg (стр. 134). Серебро по Гринвуду кипит
(1956)° ниже меди (2310°), барий более летуч, нежели стронций и кальций (более
(очных данных, как и для многих других металлов, не имеется».
* Поэтому мы воздерживаемся здесь от -приведения этих попыток, игсылаи
интересующихся к цитированным ниже сочинениям В с и а б л я и Рудорфа.
** Сделанные Л. А. Чугаезым. Прим. ред.
*** Более поздние исследования дают величину атомного веса гольмия = 164,94.
Прим. ред.
206
Периодическая система химических элементов
Равным образом температура кипения А1 (1800°) выше, нежели у Т1 (1280°), а
в IV группе она падает от олова (2275е) к свинцу (1525').
Совершенно обратные отношения наблюдаются у типичных металлоидов,
образующих главные подгруппы V, VI и VII групп (см. стр. 134).
По-видимому, температура кипения повышается также и в подгруппе таких
«полуметаллов», как Сг (2200°), Мо (3200°?), W (3700°?) и U.
К стр. 135. Физические свойства простых тел и сходственных соединений
элементов, рассмотренные в VI и VII главах, вовсе не исчерпывают собою тех, на которых
в более или менее ясной степени -сказывается периодичность. Чтобы привести еще
несколько примеров, укажем, что подобное же отношение проявляется в диэлектрических
константах, в вязкости, в растворимости и пр. Некоторые из этих свойств
рассмотрены в VIII и IX главах. По опытам Барк л а и других, поглощательная способность
элементов по отношению к однородным первичным Рентгеновским лучам также
периодически изменяется с величиной атомного веса * (см. Jnhrbuch fur Radioakt.,
VII, I). To же самое относится и к химическим свойствам, каковы, например,
способность к образованию гидратов и подобных им соединений или способность простых
тел пли сходственных их соединений выступать в роли катализаторов при
определенных химических процессах. Такова, например, способность, свойственная некоторым
элементам (точнее, их галоидным соединениям), играть роль переносчиков (Ubertra-
цег) галоидов при реакциях замещения последними атомов водорода в органических
соединениях, например в бензоле и других углеводородах, особенно ароматического
ряда. Элементы, показывающие активность в этом отношении, занимают соседнее
положение в периодической системе (А1, Ga, In, Sb, Те, J). Наоборот, а I группе и
вообще в четных рядах среди легких элементов переносчиков не имеется. Насколько
далеко распространяется область влияния периодического закона, видно из того, что
периодическое чередование проявляется даже в свойствах физиологических (Полюта.
С. Боткин, Блек, Брентон и другие).
К стр. 155. Температуры кипения щелочных металлов даны по Руффу п Иогансе-
ну (1905—1906 гг.), удельные веса—по Ричардсу (см. стр. 128), по Гюнцу и Бронев-
скому (1909 г.) и Гакспиллю.
К стр. 157. LiCl дает, кроме кристаллогидратов с одной и двумя молекулами Н*0.
еще таковой лее с ЗНгО (А. Богородский).
К стр. 164. Температура плавления кальция дана по Руффу и Плато (1903 г.),
бария— по Гюнцу (1903 г.).
К стр. 176. Темлвратуіра плавления тантала дана по Пираші и Meiiepv (Chem.
Centr., 1911 г., II,' 1775). Больтон дает 2250° (Zeit. Elektr., U, 45).
К стр. 183. Прочность гидратов солей галоидоводородных кислот по большей
части (особенно для менее благородных металлов) повышается с повышением
атомного веса галоида. Приведем, например, температуры разложения трехиодных
гидратов (МС1 • ЗН20):
LiCl LiBr LiJ
—15" -і-4- -1-72°
К стр. 202. Бр. Бэкер нашел ошибку в опытах Флинта и, по-видимому,
убедительно показал неверность сто результатов.
БИБЛИОГРАФИЯ
В заключение настоящего очерка приведем список некоторых сочинений и статей,
посвященных периодическому закону.
Л.И.Менделеев. Основы химии (с 1869 г. вышло 8 изданий).
Его же. Два лондонских чтения. СПб., 1889.
Его же. Попытка химического понимания мирового эфира. СПб., 1905.
Loth. Meyer. Die modernen Theorien der Chemie, 5-е изд. в 1884 г. Ostwald's Kias-
siker der WLssensehaften, № 68 (1895). Статьи Л. Мейера и Д. И. Менделеева. Ср.
также L. Meyer. Berl. Ber., 26, 1230.
A. R. N е w I a n d s. The Discovery of the Periodic Low. London, 18н4.
F. P. V en able. The development of the periodic low.
H. А. Морозов. Периодические системы строения вещества. СПб.. 1907.
A.E.Garrett. The periodic low. London, 1909.
* Ср. с законом Мозли, связывающим частоту спектральных линий
характеристического рентгеновского излучения с порядковым номером элемента.
Периодическая система химических элементов 207
Л. Werner. Neuere Anschaunger auf dem Gebiete der organischen Chemie.
Braunschweig, 1909.
K.Wedekind und J. Lew is. Neue Atomgewichts-Kurven, 1910.
Отдельные статьи Д. И. Менделеева находятся в химическим отделе /Kvpua іа
Русского физико-химического общества: 1, 35, 60, 229: 2. 14; 3, 7. 25; 221, 284. 24К,
4, 7; 5, 119, 135; 7, 316; 11, 86; 13, 561; 18, 66, 434; 21, 233; 27, 69. 508. См. также
статью «Периодическая законность химических элементов» в Энциклопедическим
словаре Брокгауза и Эфрона. Критические замечания относительно
периодического закона см., например, у Berthelot. Origines de l'alchimie. 1885.
B.Brauner. Zeitschr. physik. Chemie, 70, 28.
Hinrichs. The elements of atom-mechanics. St. Louis. 1894.
ЛИТЕРАТУРА*
. 1. L. Meier. Ann. Chem.. 1870, Suppl, 7, 354.
2. LKnorr, O.Rothe, H.Averbeck. Ber., 1911. 44. 1138.
3. W.Nernst. Ann. Phys., 1911, 36, 395.
4. J.J.Thomson. Nature, 1 June 1911.
5. А А.Эйхенеальд. Электричество. М., 1908.
Г>. E.Ren gad е. С. R., 1909, 148, 1199.
7. W.Biltz. Z. phys. Chem.. 1909, 67, 561.
8. J.W. Retgers. Z. phys. Chem., 1895, 16, 577.
9. A. Kail an u. S. J a h n. Z. anorg. Chem., 1910. 68, 243.
10. П. T. M ю л л e p. Основные законы электрохимии. Перевод под ред. Н. А. Пуипнм.
СПб., 1911.
11. R. Abegg, G. Bodlander. Z. anorg. Chem.. 1899, 20, 453. (См. также А б е і /
и Бодл ендер. ЖРФХО. 1900, 32 (II), 119; R. A b e g g. Z. anorg. Chem.. 1904.
39, 330).
12. A. Pictet, LRamseyer. Ber., 1911, 44, 2486.
13. R.Abegg. Z. anorg. Chem., 1904, 39, 330.
14. >G. Та mm an. Z. fur Electrochem, 1908, 14, 798.
15. G.H.Bailey. J. Chem. Soc, 1894, 65, 315.
16. Т. С a r n e 11 e y, J.Walker. J. Chem. Soc, 1888, 53, 59.
17. J.Johnston. Z. phys. Chem., 1908, 62, 330.
18. L.Meier. Moderne Theorien der Chemie. 5 Aufh, Breslau, 1884, S. 607.
19. T.W.iRichards. Z. phys. Chem., 1908, 61, 100. 171, 183.
20. Tepler. Wied. Ann., 53, 343.
21. M.C.Lea. Z. anorg. Chem., 1895, 9, 312.
22. А. іИ. Горбов. Химические элементы. Ч. I, стр. 386.
23. C.Cuthberson, E.Pare Metcal fe. Phil. Trans, 204, 323; 207. 135.
24. Burton. Proc. Roy. Soc, 80, 390.
25. С and M. С u t h b e r s о n. Proc. Roy. Soc, A 81, 440; 83, 149; 84, 13.
¦26. В. А. Бородовский. Поглощение 8-лучей радия. Юрьев, 19.10,
27 M.P.Pascal. Ann. Chim. phys, {8], 19, I, (1910); 25, 289 (1912).
28. L.Hackspill. С R„ 1912, 154, 879.
29. L.Hackspil 1. C. R, 1911, 153, 814.
30. De Foreran d. Ann. Chim. Phys, 1911, 8, 24, 256.
31. A. Combes. C. R., 1894, 119, 1999; G. U г b a i n, H. L а с о m b е. С R„ 1901, 133.
874.
32. Б.Меншуткин. Об эфирах и других молекулярных соединениях бромистого м
йодистого магния. СПб, 1907.
33. В.Е.Павлов, Д.Герасимов. ЖРФХО, 1904, 36, 566.
34. Г..Г у ст а в сон. Органические соединения в их отношении к галоидным солям
алюминия. СПб., 188Э.
35. R.Abegg. Handbuch der anorgan. Chem, Ш, 2.
36. P.Jol ibois. С R, 3911, 152, 1767.
37. E.B.R. Prideoux. J. Chem. Soc. 1906, 89, 316.
38. А. А. Я ковки н. О гидролизе хлора. СПб., 1896.
39. 1 saber t. С. R, 1880, 91, 768.
40. R.Jarry. Ann. chim. phys, 1899. {7] 17, 327.
41. Т. E.Thorpe, J.W.Rodger. Phil. Trans, A, 1894. 185, 397.
42. G.Gallo. Chem. Zblt, 1910, 1, 1952; 1910, 11, 543.
43. M. M. P a 11 i s о n Muir. J. Chem. Soc, 1909, 95, 656.
44. H S. Patterson, R. S. Cripps, R. Why tl a w-Gr a y. Proc. Ro\. Soc. 1912,
8Q, A 579.
* Список литературы, вынесенной Я. А. Чугчевым в подстрочные примечания.
20S
Периодическая система химических элементов
45 Ch. Мои геи. Bull. Soc. chim., 1911, [4] 9, 1.
1fi. A.A nt го pof f. Proc. Roy. Soc, 19I0, 83, A 474.
47. A. Werner, L. Dink I age. Ber., 190I, 34. 2698.
48. LWohler. Ber., 42, 3958, 4100.
49. W.Ramsay. J. Chem. Soc, 1909, 95, 1073
50. A. Debierne. С R., 1910, 150, 1740.
51. A.Werner. Z. anorg. Chem., 1895, 8, 190; J. Thorn sen. Z. anorg. Chem., 1893.
9, 283.
52. Bark!, Sadler. Phil. Magaz., 1907.
53. К a ye. Phyl. Trans., 1908, A, 446.
54. Б. Браунер. ЖРФХО, 1902, 34, 143; В. В г а и п е г. Z. anorg. Chem.. 1902, 32, 1
55. Retgers. Z. fur physik. Chemie, 16, 650, 651.
56. В i 1 t z. Ber., 35, 562.
5X388»*
ВАНТ-ГОФФ И СУДЬБЫ СТЕРЕОХИМИИ*
«Новые идеи в химии», 1, СПб., 1912, стр. 9.
Имя великого ученого, памяти которого мы посвящаем сегодняшнее
заседание, неразрывно связано с одним из крупнейших поворотов мысли
в области теоретической химии и в особенности химии углеродистых
соединений. Вант-Гоффу мы обязаны введением в науку" учения о
пространственном расположении атомов, химическую молекулу
составляющих.. Это учение ныне разрослось в обширный и крайне* важный отдел
нашей науки, получивший (от Виктора Мейера) название стерео-
X 'И М И И.
Не могу скрыть некоторого смущения, которое я испытываю в
настоящую минуту, приступая к своей задаче —дать краткий отчет именно
об этой части многообразной деятельности Вант-Гоффа.
Между тем как, с одной стороны, основные положения стереохимии
всем известны и распространяться о них было бы, конечно, бесполезно
в этоім собрании, я, с другой стороны, за недостатком времени л-ишен
возможности входить в. какие-либо детали, ибо это потребовало бы
целого специального курса.
Я поэтому поневоле должен ограничиться немногим. Я начну.с
небольшой исторической справки, а затем попытаюсь наметить те
основные течения, те главные линии, по которым развивалась стереохимия
после того знаменательного момента, .когда вслед за первыми
гениальными работами Пастера ей был дан новый могущественный толчок.
Это было в 1874 г., когда теория химического строения не только
успела уже пустить крепкие корни и проникнуть в сознание большинства
химиков, .но успели также обнаружиться известные ее недочеты,
потребовались изменения и надстройки.
Одной из таких надстроек структурной теории, нисколько
не.уродующих здания, не нарушающих его стиля, а лишь дополняющих и
завершающих то, что было создано раньше, одной из таких надстроек и
является стереохимия.
Общеизвестно, что, кро-ме Вант-Гоф-фа, с историей стереохимическопо
учения . связано еще другое имя — французского химика Л е - Б е л я.
Независимо друг от друга высказали они оба одну и ту же по существу
основную идею, вывели из нее частью сходные результаты и
опубликовали на расстоянии какого-нибудь месяца: Ле-Бель — краткое
сообщение в журнале французского химического общества,.Вант-Гофф —
особую брошюру, появившуюся сначала на голландском (1874 г.), потом
на фрзнцузском языке (1875 г.) под названием «La chimie dans l'espa-
ee» ** и, наконец, в немецкой обработке Германна: «Lagerung der Atome
im Raume»*** (1877 г.).
*¦ Речь, прочитанная на торжественном заседании .Отделения химии-Русского
физико-химического общества 31 марта .1911 г., посвященном па-мяти.Вант-Гоффа. Прим. ред.
** Химия вліространстве (франц.). Прим., ред.
*** Расположение атомов в, пространстве (нем.). Прим. ред.
14 Л. А. Чугаев, т. III
210
Вант-Гофф и судьбы стереохимии
Когда какое-нибудь научное открытие обязано своим появлением на
свет двуіМ или нескольким ученым, работающим одновременно, перед
историком науки часто возникает трудная и но большей части
неблагодарная задача: выяснить долю участия и заслуги каждого из них в
данном открытии. Моя задача в этом случае значительно облегчается
тем обстоятельством, что между Вант-Гоффом и Ле-Белем никогда не
возникало недоразумений, полемики о приоритете, что они всегда
относились друг к другу с полным уважением и признанием обоюдных
заслуг. В качестве внешнего знака этого признания Вант-Гофф посвятил
Ле-Белю второе французское издание .своей брошюры, появившейся в
свет в 1887 г. под заглавием: «Dix annees dans 1'histoire d'une theorie» *.
До появления в печати своих первых сообщений Вант-Гофф и
Ле-Бель не обменялись по этому повод)1 ни единым словом, хотя оба
и работали в одной и той же лаборатории Ад. Вюрца в Париже.
Тем поразительнее кажется совпадение мысли. Но совпадения этого
рода в науке вовсе не составляют ^редкости. Наоборот, О'НИ появляются
почти всякий раз, когда известная идея, гак сказать, висит в воздухе,
как электричество перед грозой, висит, готовая принять определенные
формы, ожидая своего выразителя.
В таком положении находился закон сохранения энергии в 40-х годах
прошлого века, вопрос о происхождении видов в конце 50-х годов,
периодический закон — в конце 60-х.
Приблизительно в таком же положении находился вопрос о
пространственном расположении атомов в химической частице к моменту
зарождения стереохимического учения.
Достойно замечания, что исходная точка у обоих исследователей
была существенно различная. Для Ле-Беля это был вопрос о природе
молекулярной дисимметрии, о происхождении вращательной
способности орга-нических соединений. Для Вант-Гоффа это была прежде всего
недостаточность теории строения для удовлетворительного объяснения
некоторых случаев изомерии **. Для Вант-Гоффа вопрос вообще стоял
значительно шире. Задача Ле-Беля входила в него, как частный случаи.
Оба ученых почти одинаковым образом формулируют центральный
пункт стереохимического учения: род связи между молекулярной диемм-
метрией и оптической деятельностью.
Основные черты этой связи еще в I860 г. Л. Па-стер характеризовал
следующими словами: «расположены ли атомы (правой винной
кислоты) по спирали, закручивающейся вправо, или они расположены по вер-
шинам неправ-ильного тетраэдра? — мы этого не знаем. Но не подлежит
никакому сомнению, что мы имеем здесь дело с асимметрической
группировкой, не совместимой со своим зеркальным изображением».
Во времена Пастера *** учение о химической структуре, о структуре
* Десять лет исторического развития одной теории (франц.). Прим. ред.
** «У меня появилась эта мысль,— пишет Вант-Гофф,—за год перед тем (т. е.
» 1873 г.) в Утрехте после прочтения -работы Висли цен уса о молочной кислоте.
Следующая фраза этой статьи' врезалась у меня в памяти, и я воспользовался ею
влоследстви-и в качестве эпиграфа: «Факты заставляют нас объяснять различие
между изомерными молекулами, отвечающими одной и той же структурной формуле,
неодинаковым расположением их атомов в пространстве»».
*** Совершенно ошибочным является мнение, что Вант-Гофф и Ле-Бель только
дополнили и развили идеи Пастера, не внеся в них чего-либо существенно нового. Если
Пастер и указал в общих чертах на условия, которым должна удовлетворять
структура асимметрической молекулы, то он и в то время, когда специально занимался
вопросам о природе оптической деятельности, и много позднее, был очень дал-гк от
того, чтобы (придать своей идее конкретную форму. Отдельные указами я, которые мы
находим в его статьях, а также замечаниях то поводу работ других химиков,
например Юнгфлейша, свидетельствуют о том. что Пастер считал возможным разделение
Вант-Гофф и судьбы стереохимии
211
молекул сдза только зарождалось и потому пророческой мысли его н
течение целой четверти века суждено было остаться неиспользованной.
Но когда с возникновением структурной теории химия, в особенности
химия органическая, вступила в новый блестящий период своего
развития, вопрос о пространственном расположении атомов в молекуле сам
собою выступил на сцену и сделался очередным.
Бессмертная заслуга Вант-Гоффа и Ле-Беля заключается в том. что
они уловили ту особенность з строении молеклл, которая делает эти
последние асимметрическими.
Они подметили, или, вернее, угадали,— ибо фактов, на 'которые они
могли бы.опираться, в то время было слишком мало,— что в молекуле
каждого активного соединения существует атом углерода
асимметрический, т. е. связанный с четырьмя различными другими атомами
или радикалами.
Если эти последние распределены в пространстве, а не на плоскости,
то они должны лежать по углам некоторого неправильного тетраэдра.
В совокупности они образуют схему молекулы, которая лишена
плоскости симметрии и способна «существовать в двух несовместимых
между собой модификациях, каждая из которых тождественна с
зеркальным изображением другой (рис. 1).
Такая асимметрическая молекула и представляет элементарную
ячейку оптически деятельной среды, как раз удовлетворяющую условиям, о
которых говорил Пастер.
Но стоит только в этой тетраэдрической схеме два из четырех
радикалов 'сделать одинаковыми, и асимметрия исчезнет. По смыслу
гипотезы, стало быть, должна исчезнуть и оптическая деятельность
соответствующего соединения. Этот вывод вполне оправдался на опыте.
Мало того, по смыслу той же гипотезы, всякий раз, когда мы строим
асимметрическую молекулу (CR1R2R3R4) из симметрической (CR1R2R3R1)
через однократное замещение (радикала Ri радикалом R4), роковым
образом должна получиться смесь двух «антиподов», правого и левого,
в равных количествах. Причина этого понятна. Если в обеих
молекулах — «правой» и «левой» -- все атомы одни и те же, а разным образом
одинаковы расстояния и силы, действующие между ними, то молекулы
эти должны обладать одинаковой степенью устойчивости, а
следовательно, и тенденция к их образованию из одного общего материала должна
быть одинакова. Значит, одинаковы должны быть и шансы образования
молекул правых и левых, эти последние долж-ны образоваться в
равных количествах. Чтобы разделить их друг от друга, нужно 'Применить
специальные методы, которые и были открыты Пастером еще в 40-х и
50-х годах прошлого века.
Если в этих основных положениях теории Вант-Гофф и Ле-Беля.
можно сказать, шли pari passu*, и заслуги их должны быть
признаны одинаковыми, то вовсе нельзя сказать того же о- дальнейшем
развитии нового учения.
У Ле-Беля основная гипотеза 'высказана несколько расплывчато,
неопределенно. Вант-Гофф с проницательностью, присущей гению, сразу
почувствовал необходимость конкретизировать представление об асим-
иа антиподы таких органических соединений, как янтарная яислота, считал возможным
получение изомера недеятельного, благодаря внутренней компенсации (наподобие
мезовикной кислоты) у соединений с одним асимметрическим углеродом, например
у яблочной кислоты и т. п. Все это указывает на то, что теорая Вант-Гоффа и Ле-
Беля .представляет огромный шаг вперед в развитии стереохимического учения, по
своему значению едва ли много уступающий тому, который был сделан самим
Пастером.
* Ровным шагом, одинаково ' (лат.). Прим. ред.
14*
212
Вант-Гофф и судьбы стереохимии
метрическом углероде и о самой конструкции органических молекул.
Не опасаясь впасть в излишнюю схематичность, создать слишком
грубые модели, он строит свою тетраэдрическую схему и допускает, что
единицы сродства имеют определенную ориентировку в пространстве,
что они направлены к вершинам правильного тетраэдра, что существуют
как бы известные силы, заставляющие их расходиться на возможно
более далекие расстояния.
Пользуясь этой схемой и допуская свободное вращение около общей
оси двух тетраэдров, связанных между собой одиночной связью, Вант-
Гофф строит схемы для соединений с несколькими асимметрическими
углеродами, определяет число возможных изомеров, деятельных и
недеятельных. Эти схемы целиком легли впоследствии в основу
классических работ Эм. Фишера над сахаристыми веществами. У Ле-Беля на
псе это имеются только беглые намеки, общие указания, не принявшие
конкретной формы.
Еще резче и определеннее разница между взглядами обоих ученых
на природу изомерии соединений с этиленовой связью. Вант-Гофф из
своих, посылок выводит, что в такого рода соединениях центры тяжести
четырех радикалов, связанных с системой Л- \» должны находиться
в одной плоскости, что, следовательно, вся молекула симметрична и
совместима со своим зеркальным изображением, даже в том случае,
\с=с/
когда все четыре радикала I / \ ) различны между собой, что,
следовательно, оптическая деятельность в этом случае невозможна.
Ле-Бель становится на -более общую точку зрения. Для него
плоскостная конфигурация необязательна, оптическая деятельность возможна.
Он ищет ее и, по-видимому, находит, например, у цитраконовой кислоты:
СНз—С—СООН
н—с-соон
Но дальнейшие исследования не подтверждают этого результата.
Возможность оптической" деятельности у соединений с одной
этиленовой связью (при отсутствии; асимметрического атома углерода)
окончательно .опровергается. Таким образом, торжествует точка зрения
Вант-Гоффа..
Эта точка зрения и в ряде других случаев, например, для
соединений циклических, дала ряд важных результатов, прийти к которым было
бы невозможно, исходя из взглядов Ле-Беля.
'Первое время после ті>го; ка-к стереохимическая гипотеза ¦ увидела
свет, лишь очень немногие химики обратили на нее серьезное внимание.
Выдающийся немецкий ученый Герм. Кольбе, в то время
находившийся в периоде ожесточенной и известной в истории химии п->
своей резкости полемики, направленной портив теории строения, ее
провозвестников и защитников Кекуле, Байера, Фишера и др., Герм.
Кольбе, правда, отметил брошюру Вант-Гоффа, когда она (в 1877 г.)
появилась на немецком языке,-но отметил лишь для того, чтобы сделать ее
мишенью для своих обычных насмешек, в статье, поражающей своей
резкостью и-нетерпимостью. Чтение этой статьи невольно -переносит нас
к далекому прошлому/'к первой полонине истекшего века, когда Бер-
целиус -вел нещадную войну против сторонников теории-замещения и
унитарной системы, когда у одного из них — Лорана вырвалась по^п-
Вант-Гофф и судьбы стереохимии
2Y6
ная горечи фраза; «Je pardonne au dualisme ses injures, mais sa mauvai>e
foi, jamais» *.
Если Кольбе встретил теорию Вант-Гоффа -насмешками, то
большинство других химиков того времени на первых порах вовсе не обра-
гили на нее внимания. Характерно, что у нас в России А. М. Бутлеров,
всегда отзывчивый к каждому крупному успеху науки, который,
например, по вопросу о происхождении химических элементов далеко
опередил свое время, который один из первых у нас отметил и поднял
громадное значение для химии «лучистой матер-ии» Крукса,— не проявил
никакого интереса к зарождавшейся на его глазах стереохимии. В
последнем курсе лекций, читанных им в университете в 1884/85 учебном году,
мы находим только беглое упоминание имени Вант-Гоффа по поводу
вопроса об изомерии ванных кислот. На дальнейших страницах
несравненно больше места и внимания уделяется синтезу деятельных винных
кислот, осуществленному Юнгфлейшем. Нужно заметить, что синте.ч
этот представляет только некоторое дополнение к классическим опытам
Пастера и является не более как одним из сравнительно мелких
эпизодов в истории стереохимии К
В Германии прежде всего два химика — Вислиценус и Лан-
дольт поняли все огромное значение теории, развитой Вант-Гоффом
(и Ле-Белем) и отнеслись к ней с полным вниманием. Первого из
названных ученых к этому привели его собственные работы над молочными
кислотами. Ландольт в то время как раз занимался изучением
оптической деятельности органических соединений **.
Мало-помалу, однако, особенно со средины 80-х годов, отношение
химиков к новой теории изменяется. Одна за другой появляются
экспериментальные работы, ее подтверждающие. Заслуги Ле~Беля были уже
в 1881 г. официально признаны французской академией, присудившей
ему премию Джекера.
В 1886 г. умер Кольбе, и как бы по иронии судьбы кафедру его в
Лейпцигском университете заместил Вислиценус. Два других корифея
химической науки в Германии Ад. Байер и Эм. Фишер вскоре
тоже открыто перешли в лагеірь новаторов. Этим трем исследователям
стереохимия больше всего и обязана своими дальнейшими успехами и
проведением в жизнь ее основных принципов, притом в трех различных
направлениях.
Эм. Фишер воспользовался теорией асимметрического углерода для
формулировки изомерии сахаристых веществ. В своих классических
исследованиях он разработал не только приемы для определения стерео-
химической конфигурации отдельных индивидуумов, но также и
методы для планомерного синтеза оптически деятельных Сахаров и для
превращения одних изомеров в другие.
* Я прощаю дуализму его оскорбления, но его недобросовестность — никогда
(франц.). Прим. ред.
** Эм. Фишер в речи, посвященной (1911 г.) памяти Вант-Гоффа, вспоминает о том
впечатлении, которое брошюра «La chimie dans l'espace» произвела на него самого
и на его учителя Байера непосредственно после своего появления в свет (1875 г.). Он
вспоминает о том, как однажды Байер пришел в лабораторию с брошюрой
Вант-Гоффа в руках л со словами: «Вот опять появилась новая действительно хорошая мысль
в нашей науке, она, наверное, принесет богатые плоды». «Рассматривая -модели,--
продолжает Фишер,—и сравнивая их со структурными формулами, могли и мы,
молодые химики, сразу убедиться в преимуществах новой теории, которая с тех шор
часто бывала темой наших разговоров».
Из этих воспоминаний видно, что стереохимическая гипотеза приобрела себе
известное число сторонников непосредственно после того, как была опубликована, но
сторонники эти в течение ряда лет не высказывались открыто в печати. Их воззрения
и аргументы поэтому мало отражались на взглядах руководящих химических кругов
того времени.
214
Вант-Гофф и судьбы стереохимии
К этим исследованиям примыкают работы Викт. Мейера, а у пас в
России Н. Д. Зелинского и К. Бишофа.
Исследования Фишера над сахаристыми веществами (а в
последнее время над белками)— естественным образом заставили его
вернуться к заманчивой теме, которая в свое время увлекла и Пастера на
путь биологических изыскании. Давно было отмечено, что появление
оптически деятельных соединений в естественных условиях тесно
связано с процессами жизни. Пастер показал, что живые организмы,
особенно некоторые микроорганизмы, являются замечательно
чувствительными реагентами на деятельные вещества, так что с их помощью
удалось осуществить разделение недеятельных смесей и рацематов на
оптические антиподы. Фишер продолжил эти исследования,
распространив их на так называемые неорганизованные ферменты. Между
ферментом и стереохимической природой Сахаров и их производных
(например гликозидов), вообще веществ, на которые этот фермент действует,
была установлена теснейшая закономерная связь, которую Фишер
образно уподобил тому соответствию, которое существует между замком
и ключом, его отпирающим.
Избирательная в стереохимическом отношении жизнедеятельность
организмов получила, таким образом, естественное объяснение в стерео-
химической же специфичности тех -ферментов, которые в этих
организмах содержатся.
Вместе с тем Эмиль Фишер развил представление об
асимметрическом синтезе, т. е. о таком синтезе, который совершается при
помощи деятельных реагентов и приводит к образованию деятельных же
продуктов. Такой именно синтез должен происходить в растениях и
давать начало деятельному сахару и деятельному белку. Ныне
ученики и последователи Фишера (Мэк-Кензи, Бредиг и Фаянс, и др.)
осуществили примеры такого синтеза в лаборатории, а Розенталер
показал, что асимметрический синтез (например синтез нитрила,
деятельной миндальной кислоты) может быть осуществлен при содействии
естественных ферментов. Условия синтеза в живых организмах
выступают таким образом в новом свете.
Эти исследования, глубоко заманчивые по своим дальнейшим
перспективам, переносят нас в область биологии и красноречиво
свидетельствуют о том, что учение Вант-Гоффа и Ле-Беля распространяет
свой свет далеко за пределами чистой химии.
В другом направлении, не менее плодотворном, чем только что
упомянутые исследования Фишера, шли работы И. Вислиценуса, с одной
стороны, и Ад. -Байера — с другой.
Работы Вислиценуса были посвящены главным образом
изучению изомерии непредельных соединений. Вместе со своими
учениками он исследовал целый ряд сюда относящихся случаев: изомерию
кислот фумаровой и малеиновой, кротоновой и изокротоновой, ангели-
ковой и тиглиновой и пр., дал приемы для определения их конфигурации
и изучил условия образования этих соединений из соответствующих
предельных, равно как и превращений обратного характера.
Почти одновременно Ад. Байер, изучая циклические соединения
(полиметиленового ряда), установил для них важное понятие об
относительной асимметрии, влекущей за собой изомерию типа
цис — транс, и показал существование аналогии между этого рода
изомерией и изомерией непредельных соединений. С другой стороны,
опираясь на гипотезу Вант-Гоффа о «направленных единицах сродства,
Байер развил свою известную теорию натяжения, наглядно поясняющую,
почему наиболее устойчивы циклы пяти- и шестичленные. Всем хорошо
Вант-Гофф и судьбы стереохимии
215
известно, насколько плодотворной оказалась эта теория. Напомню среди
многих и важных ее приложений только одно: заключение, касающееся
числа возможных изомеров в бициклических соединениях, подобных
камфоре, в молекуле которой—-два асимметрических атома углерода,
а между тем фактически известно только два антипода: правая и
левая камфора. Другая пара антиподов, ожидаемая по теории, не
существует или отличается крайней неустойчивостью, и это обстоятельство
находит себе рациональное объяснение с точки зрения представлений,
развитых Байером.
Рис. 1
Здесь будет уместно коснуться одного в высшей степени
замечательного предсказания, которое было сделано Вант-Гоффом еще в 1877 г.
и которое блестящим образом оправдалось в самое последнее время.
Дело идет о соединениях, содержащих комбинацию двух двойных
связей «алленового» типа:
для которых Вант-Гофф без малого 40 лет тому назад предвидел
возможность получения их в оптически деятельном состоянии, несмотря
на отсутствие в их частице асимметрического атома углерода в
собственном смысле этого слова. Оставаясь на почве сво<их геометрических
представлений, Вант-Гофф указал на то обстоятельство, что каждому
из тел вышенатіисанной общей формулы соответствуют две
несовместимых конфигурации (рис. 1).
Продолжая быть последовательным, надо было заключить, что
подобные соединения должны обладать способностью к расщеплению на
оптически деятельные антиподы.
216
Вант-Гофф и судьбы стереохимии
В точности реализовать этот случай, к которому относятся
предсказания Вант-Гоффа, не удалось, правда, и до настоящего времени, но
зато оказалось возможным построить такую комбинацию атомов,
которая по смыслу стереохимического учения и в согласии с только что
сказанным об аналогии между этиленной и циклической связью,
совершенно аналогична алленовой группе С = С = С. Подобная комбинация
получится, если мы в алленовой группе одну этиленовую связь
заменим эквивалентной в стереохимическом отношении связью циклическом,
например кольцом гексаметилена.
К этому именно типу принадлежит метилоциклогексилиденуксусная
кислота
СН3\ /СН2—СН3ч /Н
y/ N^v-ch/" \хюн,
для которой совсем недавно Перкину, Попу и Валлаху удалось
пронести расщепление на оптические антиподы. Нетрудно видеть, что это
соединение в стереохимическом отношении совершенно подобно кис/юте
СН3\ /Н
W ХЮОН,
которая только по малой устойчивости своей, по-видимому, непригодна
для экспериментирования.
Таким образом, хотя и в несколько модифицированной форме,
предсказание Вант-Гоффа получило полное подтверждение. В этом
предсказании, сделанном еще в ту эпоху, когда, как мы только что видели,
основная идея стереохимии большинством химиков встречалась пли
прямо враждебно, или, в лучшем случае, безучастно, нам кажется
особенно достойной внимания поразительная смелость мысли, одна из
характерных признаков гения, та самая черта, которая немногими
годами раньше двинула Д. И. Менделеева на путь предсказания новых
неведомых химических элементов...
Наряду с многосторонним развитием стереохимии углерода
возникла, уже на наших глазах, отчасти также предвиденная Вант-Гоффом
стереохимия других элементов.
После того, как Ле-Бель дал первое экспериментальное
доказательство того, что асимметрический атом азота влечет за собой
появление оптической изомерии, подобная изомерия для несимметрично
замещенных аммониев была констатирована во многих случаях, главным
образом английскими химиками Попе и его учениками, Джонсом,
затем Ведекиндом и др.
За асимметрическим азотом последовали асимметрическая сер а,,
селен, олово, кремний, фосфор. Для каждого из этих
элементов получены асимметрические соединения, показывающие оптическую
деятельность. Приведем здесь лишь несколько таких соединений в
качестве примеров:
сн5
] G>H5\ /СНо—СО—СвН5
С,Н5-СН.—Ny-J " >S<
| ХСН, OV ХВг
СН2
I
СНо^СН
Вант-Гофф и судьбы стереохимии 217
СНЗЧ Х3Н7 СНЗЧ /СНо—СООН
/Sn\ /Se\
QH/ \j c6h/ хвг
Наиболее важными результатами мы и здесь обязаны английским
химикам, кроме упомянутых выше, особенно К и п п и н г у. затем
Смайльсу и др.
Из работ, относящихся к самому последнему времени, укажем еще
на открытые Мейзенгеймером легко получаемые и относительно весьма
устойчивые оптически деятельные модификации окисей несимметрично-
замещенных третичных аминов и фосфинов, например
СН3ч СН3\
С2Н5—N-0 С>Н5—Р=0
QH5 С6Н5
Наконец, совсем недавно А. Вернер получил оптически деятельные
комплексные соединения кобальта, хрома, железа и родия,
причем из строения этих соединений ясно, что единственным
источником их асимметрии является здесь пространственное расположение
атомов и атомных групп около атомов Со, Сг, Fe, Rh*.
Результаты всех только что упомянутых исследований дают нам дра-
во утверждать, что способность давать начало молекулярной
асимметрии и тесно связанное с нею появление оптической деятельности,
первоначально открытое для углерода, вовсе не составляет специфической
особенности этого элемента, что мы имеем здесь дело со свойством
более общим, быть может, даже универсальным для элементов с
валентностью, превышающей 3.
Распространение стереохимических представлений за пределы соб-
ственно-химии углерода совершалось еще и в другом направлении. Как
для углерода, так и для других элементов с течением времени были
открыты случаи изомерии, хотя и не сопровождаемые появлением
оптической деятельности, но тем не менее находящие себе единственное
возможное объяснение в неодинаковом геометрическом расположении
атомов, образующих соответствующие молекулы.
Такого рода случаи раньше всего были найдены для азота,
изомерия оксимов должна быть указана здесь на первом плане, затем
изомерия гидразонов, диазосоединений и некоторых других.
Исследования по стереоизомерии азота, исторически связанные с
именами В. Мейера, Гольдшмидта и Б е к м а н а, завершились
проведением в жизнь почти общепринятой в настоящее время теории
Ганша и Вернер а.
Напомним здесь, в качестве примера, хорошо известные схемы, об-*
ясняющие по этой теории изомерию оксимов.
С6Н5—С—Н СбН5—С—Н
II "
N—ОН НО—N
Бензальдоксим Бензальдоксим
син-форма анти-форма
В высшей степени замечательная попытка внести стереохимические
представления в области комплексных соединений, особенно
платины, кобальта и хрома, принадлежит А. Вернеру.
Попытка эта, как уже было упомянуто выше, недавно завершилась
открытием изомеров, оптически деятельных, и это обстоятельство еще более
* См. статью. Структурно-стереохимические представления в области
неорганической химии. Сб. Новые идеи в химии. 1, 1914, 2-е изд., стр. 103. Прим. ред.
218
Вант-Гофф и судьбы стереохимии
подкрепляет основательность гипотезы, высказанной Вернером около
20 лет тому назад, согласно которой целая группа явлений изомерии в
ряду комплексных соединений определяется пространственными
соотношениями.
Быть может, самой темной и трудной областью стереохимии
является вопрос о связи между величиной вращательной способности
вещества и составом его молекулы. После того, как заманчивая по своей
простоте попытка Ф. Гюи 2 подойти к его разрешению в общем виде
потерпела неудачу, мы, несмотря на множество исследований, в сущности
имеем лишь несколько эмпирических правил (например, связь величины
оптической деятельности с определенными атомными группами, с
двойной связью с ароматическими остатками, с положением их относительно
асимметрического углерода и пр.), но не имеем общей теории.
Интереснейший вопрос этот, по-видимому, еще слишком сложен для общего
разрешения. Несомненно, что последнее -в значительной степени
тормозится тем обстоятельством, что до весьма недавнего времени почти не
принимали во внимание ротационную дисперсию и сравнивали
вращения для одной лишь случайной точки на обширной
дисперсионной кривой (обыкновенно для желтого луча натрия). Влияние
температуры также далеко не всегда и не в достаточной степени принималось
во внимание.
Совершенно особняком стоит в стереохимии задача; выдвинутая так
называемой Вальденовской перегруппировкой,
названной так по имени академика П. И. Вальдена, ее открывшего, и
заслуживающей особого внимания потому, что в ней хотели одно время
видеть существенное возражение против самых основных устоев стерео-
химического учения. Перегруппировка эта позволяет нам
от одного из антиподов путем ряда реакций замещения перейти к
антиподу противоположному, а затем вновь вернуться к первоначально
взятому.
Так, при действии на обыкновенную левую яблочную кислоту пя-
тихлористого фосфора получается правая хлорянтарная кислота, а эта
последняя окисью серебра может быть переведена в правую яблочную;
правая яблочная кислота в свою очередь с РС15 дает левую хлорянтар^-
ную, от которой при помощи Ag20 можно вернуться к исходной левой
яблочной кислоте. Таким образом осуществляется своеобразный
«оптический круговой процесс».
сн2-соон сн2-соон сн2-соон
н—с—он —- сі—с—н —- (он)—с—н
I I I
соон соон соон
.Левая яблочная Правая хлор- Правая яблочная
кислота янтарная кислота кислота
сн2-соон
- Н—С—CI
I
соон
Левая хлор-
янтарная кислота
Вант-Гофф и судьбы стереохимии 219
Весьма вероятно, что разъяснение этого загадочного явления * будет
связано с крупным шагом вперед в области молекулярной механики.
В общих чертах основной принцип такого объяснения уже указан
независимо друг от друга А. Вернером и Э. Фишером, и хотя
исследования, сюда относящиеся, еще находятся в самом начале, но мы уже
теперь имеем право утверждать» что оптическая инверсия, открытая
Гі. И. В а льде ном, едва ли может считаться аргументом против сте-
реохимических воззрений.
Только что сделанных беглых указаний быть может достаточно для
того, чтобы оценить то многостороннее и в высокой степени
плодотворное влияние, которое оказала на химическую науку идея о
существовании связи между изомерией и расположением атома в пространстве,
чтобы представить себе, какому огромному числу исследований
первостепенной важности она дала начало.
Мы видели, что большинство этих исследований не было сделано
самим Вант-Гоффом. Но есть один отдел стереохимии, в разработке
которого он принимал непосредственное участие. Это вопрос о взаимном
отношении антиподов и рацематов, об условиях разделения этих
последних. Вместе с несколькими учениками Вант-Гофф дал
исчерпывающую теоретическую и опытную обработку этого важного вопроса,
выяснив при этом значение «точки перехода» (последняя впервые была
констатирована для перехода рацемата (QOebUNaNbUh • 2Н20 в смесь
соответствующих солей правой и левой винной кислоты 2C4H406NaNH4 *
• 4Н20 недавно скончавшимся выдающимся русским ученым Г. Н.
Вырубовым). Эти работы стоят в тесной связи с тем направлением, которое
мало-помалу все больше и больше захватывает деятельность Вант-Гоф-
фа, начиная с конца 70-х годов. Это — направление физико-химическое,
направление, в котором он проявил свою творческую фантазию не в
меньшей степени, чем при создании стереохимии, и поворот к которому
отмечается появлением в 1877—1881 гг. его обширного и
глубокомысленного сочинения: «Ansichten uber organische Chemie» **,
заключающего в себе и философию органической химии, и программу многих
дальнейших исследований в области химии физической. Но об этой второй
стороне деятельности Вант-Гоффа скажут другие референты на
сегодняшнем заседании.
Позвольте мне в заключение высказать несколько соображений
более общего характера, соображений, касающихся роли и значения
стереохимии в нашей химической науке.
Что стереохимические представления оживляли и одухотворяли в
течение нескольких десятилетий подряд научное исследование в
области не только органической, но и минеральной химии, это мы уже
видели. Но быть может сегодня, когда мы стоим перед скорбным
событием, перед концом земного существования автора одной из
остроумнейших химических теорий, уместно было бы задаться вопросом о ее
вероятном будущем. Говорят, что наши теории недолговечны, что они
редко живут больше четверти века. Для стереохимии роковой возраст
-прошел давно в 1899 г., и она еще существует. Но вправе ли мы ожидать
от нее дальнейших успехов, дальнейшего развития, движения
^вперед, или ей скоро суждено замереть вместе со структурной теорией, ее
взрастившей и с ней неразрывно связанной? Еще не так давно многие
* Решить с достоверностью, в какой стадии процесса совершается превращение
конфигурации, при действии РС15 ил.и AgjO, в настоящее время еще не представляется
огэможны-м.
** Взгляды на органическую химию. Прим. ред.
220
Вант-Гофф и судьбы стереохимии
химики готовы были обеим теориям читать отходную. Но, кажется,
именно в настоящее время мы переживаем момент, когда
неосновательность такого отрицательного отношения становится очевидной.
Было время, когда большинство химиков видело конечную задачу
своей науки в том, чтобы все химические превращения свести к
механике атомов.
В 90-х годах минувшего века, как известно, наступила резкая
реакция против такой точки зрения. Сама атомистическая теория прослыла
иллюзией, беспочвенной и наивной. Лозунгом нового направления в
теоретической химии была объявлена энергетика, а наиболее прочными
устоями его — первый и особенно второй принципы термодинамики.
Крайние сторонники такого взгляда * доходили до того, что считали
достоверными и вообще заслуживающими внимания только те выводы и
теоретические построения науки, которые могли быть выведены
термодинамическим путем.
Такая переоценка значения термодинамического метода находит
себе объяснение в тех действительно блестящих результатах, которые
были получены благодаря его приложению к теоретической химии.
Границы приложимости термодинамического метода стали особенно
ясны после неудавшейся попытки (1903 г.) приложить его к
истолкованию стехиометрических законов, которые ныне, как и во времена
Дальтона, единственное рациональное объяснение получают на
почве атомо-молекулярной теории.
Прошло еще несколько лет, и под давлением новых фактов наибо-ц^
убежденные противники атомистической теории, а также связанной с
ней кинетической теории вещества, открыто перешли в лагерь ее
сторонников.
Этими фактами были, с одной стороны, результаты, добытые
Перроном, Сведбергом и другими при изучении Броуновского молекулярного
движения, результаты, представляющие полное согласие с
требованиями кинетической теории, а с другой стороны, знаменитые опыты Ре-
зерфорда, Гейгера и др., позволившие, если не прямо видеть сс-частицы,
испускаемые радием, то во всяком случае иметь вполне осязательные
признаки существования этих частиц, убеждаясь или по отклонениям
электрометра или по вспыхиваниям искорки на цинковой обманке, что
перед глазами наблюдателя один за другим пролетают заряженные
атомы гелия.
В этих поразительных фактах нельзя не видеть явных признаков
возрождения атомистической теории.
Перед этой теорией — широкое будущее. Но будущее строится на
прошлом и настоящем. И мы не можем не связать грядущих успехов
атомизма с тем, что учение это дало для химии в прошлом, подобно
тому, как Ньютоново открытие закона тяготения не заслоняет заслуг
Коперника, Кеплера и Галилея, как теория Дарвина не заслоняет того,
что раньше сделали Ламарк и Жоффруа Сент-Илер.
В структурной теории и в ее логическом продолжении —
стереохимии мы имеем несомненный зачаток будущей механики атомов, о
которой некогда мечтали, в которой потом изверились и на которую вновь
постепенно устремляют взоры.
Современные рудиментарные теории не позволяют еще делать каких-
либо выводов количественного характера, и это, конечно, потому.
* Достойно внимания, что сам Вант-Гофф. которому область приложения
термодинамики к химию обязана более, чам кому-либо другому, никогда не /преувеличивал
ее значения и лишь пользовался услугами термодинамического метода там, гдг это
ему было необходимо.
Вант-Гофф и судьбы стереохимии
221
что нам неизвестны ни расстояния между атомами, ни величины и
характер сил, действующих между ними, ни характер их движений. Но
не подлежит сомнению, что известные черты конструкции молекул
подмечены верно и находят себе отражение в существующих теориях.
И когда атомистической теории суждено будет возродиться в
обловленной форме от соприкосновения с новыми областями фактов и
новыми обобщениями, то наряду со строителями атомистической теории,
наряду с Дальтоном и Авогадро, с Кекуле и Бутле ро-
в ы м,'придется поставить и имена творцов стереохимии, а между ними
на первом плане имя Вант-Гофф а; придется вспоминать об этой
поразительнейшей и плодотворнейшей попытке человеческого гения
проникнуть умственным взором далеко в область неизвестного за
пределы доступного нашим ограниченным чувствам.
Нам скажут, что стереохимия вся основана на песке, на
фантастической гипотезе — не больше. Со свойственным ему талантом Пуанкарэ
разовьет перед нами и докажет, что. для каждого фактически
наблюдаемого закона можно подобрать бесконечное число возможных схем
или объяснений. Пусть так! Но мы все же знаем и твердо верим, что
одно и только одно объяснение отвечает истине.
Задача гения — угадать эту истину возможно ближе и точнее.
История науки доказывает нам, что то, что еще вчера было гипотезой,
завтра может получить достоверность факта.
Если сегодня стало почти реальностью существование атомов, то
завтра .могут стать такой же реальностью удачно угаданные черты
молекулярной структуры.
Победоносное шествие науки никогда не выигрывало от излишнего
скептицизма, никогда не двигалось девизами, подобными пресловутому
«ioriorabimus» *. Позвольте же в день, посвященный памяти одного из
величайших представителей научной мысли на рубеже XVIII и XIX вв.,
выразить надежду, что не далеко то время, когда грядущий Вант-Гофф
установит прочные основания атомо-молекулярной механики.
* Не будем знать (лат.). Прим. ред.
ВАЛЕНТНОСТЬ
«Новые идеи в химии», сб. 3, СПб., 1913, стр. 1 Н7
Понятие о валентности, или атомности *, которому будет посвящено
настоящее собрание, принадлежит к числу понятий, которые, с одной
стороны, успели уже состариться, а с другой, не утратили еще своей'
юношеской свежести. О валентности заговорили впервые слишком
полвека тому назад, и в то же время связанные с нею теоретические
представления до сих пор еще находятся в периоде развития и как раз за-
последние годы существенно изменили тот облик, с которым успели
свыкнуться, по крайней мере, две генерации химиков.
О валентности ныне пишутся целые обширные трактаты, до такой
степени вопрос этот успел осложниться и расшириться. Им стали
интересоваться не только химики, но и физики, и это обстоятельство имело
своим последствием неожиданное освещение его с совершенно новой
стороны.
Я, конечно, не буду иметь возможность изложить современное
состояние учения о валентности во всей его полноте, ибо это потребовало
бы несравненно больше времени, чем сколько имеется в моем
распоряжении.
Мне придется поэтому ограничиться указанием на те главные
только пути и направления, по которым развивалось это учение.
Я напомню прежде всего основное понятие о валентности, затем
перейду к рассмотрению тех осложнений, которые с течением времени
были внесены в это понятие, и закончу обзором тех главнейших
попыток, которые были сделаны в разное время для того, чтобы ответить
на вопрос, в чем кроется самая сущность валентности, насколько
вообще подобные вопросы доступны нашему познанию.
С тех пор, как Джон Дальтон ввел в химию оказавшееся столь
плодотворным учением об атомах2, на химические соединения стали
смотреть, как на собрания множества химических частиц или молекул,
из которых каждая, в свою очередь, построена из атомов. Каждая такая
молекула может быть рассматриваема как механическая система,
отдельные части которой, т. е. атомы, удерживаются друг около друга
некоторыми силами притяжения, имеющими электрический характер,
как думал еще Берцелиус, скорее напоминающими силу всемирного
тяготения, как полагали другие.
Два свойства химических соединений, а следовательно, и молекул,
из которых они образованы, должна была объяснить или, если угодно,
описать атомо-молекулярная теория: состав и степень
устойчивости.
Степень устойчивости каждого рода молекул, о которой можно
судить по относительной легкости, с которой совершается превращение
их в другие молекулы, и по явлениям, сопровождающим такого рода
* Обозначения «валентность» и «атомность» (Valenz, Atomigkeit) суть синоші-мы.
В том же почти смысле употребляется немецкий термин «Wertigkeib, который
по-русски иногда передают словом «значность». В этам очерке я іпочти исключительно буду
пользоваться термином «валентность».
Валентность
223
лергходы, очевидно, должна находиться в зависимости от сил,
действующих между атомами (и от характера движения этих последних).
Этим силам давно уже дали несколько неопределенное, унаследованное1
еще со времен алхимии, обозначение «сродство», отмечая этим словом
и характерную их особенность, определяемую словом «избирательный».
Термин «химическое сродство» употребляется также в современной
физической химии, и притом в несколько ином смысле, допускающем
более точную формулировку и, что особенно важно, возможность
количественного измерения путем опыта. Мы признаем, что химическое
сродство является двигательной силой химических процессов и чго,
как таковое, оно измеряется величиной потери свободной энергии со
стороны системы, подвергающейся превращению.
Установлением законов, управляющих проявлениями химического
сродства, и выработкой методов для его количественного определения
с выдающимся успехом занимается отдел физической химии, известный
под именем химической механики.
Несомненно, что между двумя значениями, в которых мы только что
употребили слово «сродство», существует тесная связь, но сродство
современной химической механики и законы, ими управляющие,, до сих
пор еще не удалось перевести на язык «'механики атомов».
Больше того: можно сказать, что разработка химической механики
происходила при сравнительно малом и притом лишь косвенном участий
атомо-молекулярной теории *.
С законами химической механики можно было бы до известной
степени поставить в параллель так называемые стехио метрические
законы, управляющие составом химических соединений, ибо те и другие
суть законы «массовые» и, как таковые, должны в равной мере
отражать в себе то, что происходит в микрокосме атомов и молекул. Но
законность такого сопоставления существенно ограничивается тем
обстоятельством, что стехиометрические законы, в противоположность
законам химической механики, представляют ряд установленных
опытом положений, которые до сих пор удалось связать между собой
исключительно с помощью атомо-молекулярной теории. Только с
помощью этой теории и связанной с ней гипотезой Авогадро удалось,
исходя из этих законов, прийти к таким важным выводам, как к
установлению атомных и молекулярных весов и к определению числа
атомов, образующих каждую данную химическую молекулу.
Одним из важных теоретических последствий, вытекающих из
применения атомо-молекулярных представлений к истолкованию стехио-
метрических законов, и является учение о валентности, ближайшим
образом связанное с законом, носящим имя Дальтона**. Мы и
обратимся прежде всего к анализу этого закона.
* Рациональной разработке законов химической механики, несомненно, способстно-
вали установившиеся понятия об атомном и молекулярном весе, опирающиеся на
закон" Авогадро, но несравненно более значительную .роль при этом играла
термодинамика.
** Считаю не бесполезным привести здесь формулировку закона Дальтона,
данную самим автором: «Если имеются два тела А и В, склонные к соединению друг
с другом, то порядок, в котором могут образоваться соединения, следующий, начиная
с простейшего случая:
1 атом А + 1 атом В дают 1 атом С двойное (соединение)
1 атом А + 2 атома В дают 1 атом D тройное
2 атома А + 1 атом В дают 1 атом Е тройное
1 атом А + 3 атома В дают 1 атом F четверное
3 атома А + 1 атом В дают 1 атом G четверное „ и т. д.»
из New System of Chemical Philosophy. Manchester, 1808. Cp. Alembic Club Reprints^
№ 2, p. 30.
224
Валентность
Неделимость и вообще неизменяемость элементарных атомов в
условиях химических превращений является, очевидно, тем допущением,
которое прежде всего необходимо сделать, чтобы уяснить себе причину
возникновения целых чисел, выступающих в законе Дальтона. Но
столь же очевидно, что из такого допущения закон Дальтона вовсе еще
не вытекает с логической необходимостью. Это было ясно еще для
Берцелиуса [1], который писал в своем знаменитом «Учебнике химии»:
«Если бы даже было в достаточной степени доказано, что тела...
слагаются из неделимых атомов, то отсюда вовсе еще не следовало бы,
чтобы должны иметь место явления постоянных химических пропорций,
особенно те, которые наблюдаются в неорганической природе. Для этого
необходима еще наличность законов, регулирующих способы
сочетания атомов и определяющих границы таковых, ибо понятно, что если
бы неопределенное число атомов одного элемента могло соединяться с
неопределенным же числом атомов другого элемента, то существовало
бы бесконечное число соединений, образуемых этими элементами, и
разница в количественном составе этих соединений по своей
ничтожности не могла бы быть открыта даже с помощью точнейших опытов.
Очевидно, следовательно, что от этих именно законов зависят
химические пропорции».
В этих словах знаменитого шведского химика заключается целая
программа, осуществление которой продолжалось в течение большей
части истекшего столетия и до сего времени далеко еще не закончено.
На вопрос, заданный Берцелиусом, химия XIX века ответила учением
о валентности.
Как всякая крупная идея в науке, представление о валентности
имело своих предшественников. Таковым является прежде всего
теория типов, первоначально развитая знаменитым французским
химиком Дюма (1839 и 1840 гг.), после того, как им самим и Лораном
были установлены законы замещения.
По мысли Дюма, к общему химическому типу (или роду) относятся
соединения, обладающие сходственным составом молекулы и
аналогичными химическими свойствами. Это — соединения, которые могут быть
получены друг из друга путем замещения одних атомов другими.
Таковы, например, уксусная кислота С2Н4О2 и ее галоидозамещенные. Из
числа этих последних Дюма, как известно, получил в 1839 г. кислоту
трихлороуксусную С2НС1302, повторяющую до подробностей
химические свойства уксусной кислоты. На этом примере было
особенно ясно видно, что водород не только подвергся замещению хлором.
но что,- как того требовал закон замещения, вступившие в молекулу
атомы хлора заняли место и приняли на себя роль, принадлежавшую
прежде атюмам водорода. Позднейшие исследования показали, что
путем восстановления водородом in statu nascendi можно осуществить
обратный переход трихлороуксусной кислоты в уксусную, т. е.
воспроизвести замещение хлора водородом. Таковы были факты, заставившие
Дюма предположить, что соединения, относящиеся к одному
химическому типу, характеризуются не только внешним сходством формул, но и
аналогичным распределением атомов (или, как в то время говорили,
эквивалентов) в молекуле.
Параллельно с понятием о химическом типе Дюма, следуя за
Реньо, развил еще другое, более широкое представление о типе
механическом (или о химическом «семействе»). К такому типу
относятся вещества, хотя бы и несходные по своей химической природе
(например СН4і СНС13, CHBr3, CHJ8, НСООН, СИ3ОН), но которые
могут быть превращаемы друг в друга путем замещения одних атомов
Валентность
2'17і
другими или же группами таковых. Дюма сравнивал члены общего
типа с планетной системой, в которой можно без нарушения общего
плана ее производить замены одних планет другими.
Значительно позднее, чем Дюма (ч окончательной форме лишь в
1866 г.), подошел к теории тппоз другой знаменитый французский
ученый Жерар, притом руководствуясь существенно иными
соображениями. В противоположность Дюма, Жерар считал, что факты не
позволяют нам делать каких-либо заключений о структуре химической
молекулы. Все, что мы знаем о природе данного соединения, основано
на изучении его превращений, почерпается, так сказать, из его прошлого,
а не из настоящего. Поэтому для Жерара рациональная формула
химического соединения выражала не более, как сокращенную запись
возможно большего числа реакций, участником которых оно является.
В соответствии с этим общая формула данного типа имела для него
характер чисто эмпирической схемы.
«Мои типы,— пишет Жерар в своем классическом сочинении «Тгаі-
te de chimie organique» (Tome, IV, p. 586),—суть типы двойного
разложения (Жерар считал двойное разложение типичнейшей из химических
реакций). Вода в бесконечном числе двойных разложений может
обменивать свой кислород и водород на другие элементы (простые
радикалы) (или на группы элементов (сложные радикалы). Я отношу
поэтому к типу воды вещества, с которыми можно воспроизвести такого
рода замещение при условии, что образующиеся при этом продукты
находятся друг с другом в отношениях, сходных с теми, которые
существуют между веществами, получающимися через замещение других
радикалов одним из радикалов воды. Я произвожу, например, эфир от
типа воды, потому что можно путем двойного разложения заместить
в эфире кислород эквивалентным количеством хлора, брома, серы,
азота, причем образуются хлористый этил, бромистый этил, сернистый
этил, этиламин, и потому что продукты этих превращений находятся
друг к другу в таком же отношении, как хлористый водород к
бромистому водороду, к сернистому водороду, к аммиаку, т. е. к веществам,
возникающим при замещении кислорода воды хлором, бромом, серой,
азотом...
Производя данное тело от типа воды, я хочу выразить этим ту
мысль, что этому телу, на которое можно смотреть, как на окисел,
отвечает свой хлорюр, бромюр, сульфюр (сернистое производное), азо-
тюр и т. д., и что эти вещества способны к реакциям обменного
разложения, напоминающим обменные разложения, свойственные
хлористоводородной и бромистоводородной кислотам, сернистому водороду
и аммиаку. Тип есть потому единица для сравнения для всех
тел, которые, подобно данному (т. е. типическому), способны к
аналогичным реакциям двойного разложения или происходят из
таковых».
Несмотря на такую разницу во взглядах Дюма и Жерара,
развитые ими теории выражали, в сущности, одни и те же реальные
соотношения и только освещали их с разных сторон. Разногласия между
ними сохранились только на страницах истории.
В 50-х годах большинство химиков, державшихся «типических»
воззрений, вслед за Жераром признавало следующие три типа: тип
водорода Н2 или, что почти одно и то же, тип хлористого водорода
НС1, тип воды Н20 и тип аммиака NH3, к которому после открытий
Вюрца и Гофмана стали причислять весь обширный класс аминов.
В конце 50-х годов к этим типам присоединился еще четвертый тип
болотного газа СН4. Купер и Кекуле одновременно и независимо
If, -'1. Л. Чугагч», т. IIТ
226
Валентность
друг от друга показали, что к этому типу могут быть отнесены все
органические соединения.
Теория типов имела для своего времени большое значение в науке,
представляя, с одной стороны, превосходный принцип для
классификации химических соединений, а с другой — облегчая обзор взаимной
связи между отдельными соединениями, проявляющейся в их составе
и химических превращениях.
Но теория эта не давала еще ответа на упомянутый выше основной
вопрос, поставленный Берцелиусом. Чтобы резюмировать в виде
некоторой общей формулы способность атомов соединяться между собой в
определенных численных отношениях, надо было сделать еще один
шаг дальше. Этот шаг был сделан Эд. Франкландом в 1852 г.
Вот что пишет он в статье [2] «О новом ряде органических соединений,
содержащих металлы»:
-«При просмотре формул неорганических соединений даже
поверхностный наблюдатель будет поражен общей симметрией в их
конструкции. В особенности в соединениях азота, фосфора, сурьмы и
мышьяка сказывается тенденция этих элементов образовать соединения с
гремя или пятью эквивалентами других элементов, и именно при этих
пропорциях лучше всего удовлетворяются их сродства. Так, в группе
четверных соединений мы имеем *: іМОз, NH3, NJ3, NS3, Р03, РН3, РС13,
Sb03, $ЬИ3, SbCl3, As03, AsH3, AsCl3 и т. д., а в группе соединений,
составленных из пяти атомов,— NO5, NH40, NH4J, РО5, PH4J и т. д.
Не прибегая к какой-либо гилотезе для объяснения такой
симметрической группировки атомов, на основании только что приведенных
примеров нельзя не признать очевидным, что такого рода тенденция
или закон действительно наблюдается, и что вне зависимости от
характера присоединяющихся атомов соединительная способность
притягивающего элемента, если позволено будет употребить это выражение,
всегда удовлетворяется одним и тем же числом атомов».
Отдавая дань своим предшественникам, Франкланд замечает далее:
«По всей вероятности, проблеск этого закона у более сложных
органических групп привел Лорана и Дюма** к установлению теории
типов».
Несмотря на отчетливую ясность мысли, высказанной Франкландом,
взгляды его далеко не сразу привились в науке. Пятидесятые годы
были временем расцвета теории типов. Жераровские типы,
окончательно формулированные после появления статьи Франкланда, сначала
обратила на себя более внимания, чем соображения, высказанные
английским химиком. Поворотным пунктом в этом отношении следует
считать одновременное появление мемуаров Купера и Кекуле, в
которых впервые (1858 г.) была провозглашена четырехвалентность
углерода, а затем как из рога изобилия посыпались неисчислимые
последствия, вытекающие из этого положения для химии углеродистых
соединений. Теория валентности элементов делается общим достоянием
только в 60-х годах.
От этой исторической справки перейдем теперь к современному
определению понятия валентность. Я полагаю, что выражу взгляды
большинства химиков, если скажу, что под валентностью мы понимаем
относительное число атомов, которое способен непосредственно
* Формулы эпи отличаются от современных, потому что Франкланд пользовался
еще старыми атомными весами, бывшими в употреблении до Жерара.
** Франкланд не упоминает имени Жерара, типические воззрения которого в
окончательной форме тогда еще не сложились.
Валентность 227
удерживать один атом данного элемента, или которое он способен
замещать.
Само собою разумеется, что здесь необходимо должна быть принят::
какая-нибудь мерка для сравнения. Естественным образом за единицу
валентности избирают валентность атома водорода, а именно по той
простой причине, что в громадном большинстве химических соединении,
в состав их молекулы атомов водорода входит большее число
сравнительно с атомами других элементов.
Конечно, есть и исключения. Может быть, для всех напрашивается,
соединение N3H, где отношения как раз обратные. Можно и среди
углеродистых соединений найти подобные случаи. Но число их вообще
сравнительно незначительно. (Я имею в виду соединения простейшего,
бинарного типа, образованные только из двух элементов, ибо здесь
отношения наиболее простые.)
Итак, принимая за единицу валентности валентность водорода, мы-
должны будем определить валентность, как число, показывающее, со
сколькими атомами водорода способен соединяться непосредственно атом
данного элемента, или сколько атомов водорода он способен заместить.
Так, мы скажем, что водород одновалентен в НС1, кислород
двухвалентен в воде Н20, азот трехвалентен в NH3, а углерод четырехвалентен
в метане СН4.
Очевидно, что меркой валентности может служить не только
валентность водорода, но и валентность других одноатомных элементов,
например хлора, так же как и валентность любого двухатомного
элемента, например кислорода. Нужно тогда только произвести
соответствующий арифметический расчет.
Если из состава молекулы НС1 следует одновалентность хлора, то-
ясно, что, определив состав соединений ряда других элементов с хлором,
мы можем установить валентность последних. Так, состав молекулы
хлористого натрия NaCl приводит нас к заключению, что натрий
одноатомен: из формул СаС12> АіСіз, PCls мы заключаем о двухвалентности
кальция, о трехвалентности алюминия и пятивалентности фосфора.
К тому же самому заключению относительно валентности мы придем,
если в качестве «мерки» примем валентность кислорода = 2. Очевидно,
что в этом случае придется исходить из состава окислов, например
Na20, СаО, А1203і Р205 и т. д.
Наконец, к тем же числам валентности приводит нас и
рассмотрение процессов замещения. Так, валентности Na, Са, А1 могут быть
выведены из того факта, что атом первого из элементов замещает
(например, в кислотах) один, атом второго — два, атом третьего — три
атома Н.
Установив валентность для ряда химических элементов,
современная химия выражает особыми формулами строение или
структуру химических соединений и притом не только простейших,
бинарных, но и более сложных. Как известно, при этом число
валентностей, или, как иногда говорят, единиц сродства, у каждого атома
символически отмечается числом черточек, от него исходящих, а самые
формулы должны выражать собой «способ химической связи
элементарных атомов в частице», иными словами, должны не только указывать на
валентность отдельных атомов, но'и давать ответ на вопрос, с какими
другими атомами находится в непосредственной связи каждый из
атомов, данную молекулу образующих. Таковы, например, следующие
формулы:
15*-
228
Валентность
н\
Н—N
н'
Аммиак
С1
CI—Si-Cl
С1
Четыреххлористый
кремний
Ох
о--=сі-
о'
Хлорная і
--О
н\с/н
н н
Метан
°\я/°-Н
сг чо—н
Серная кислота
н н-
н
Н-С—О—Н
н
Метиловый спирт
^N—0—Н
Азотная кислота
II 0
1 /
-с—с—о-н
н
<ислота Уксусная кислота
Вскоре после того, как было внесено в науку понятие о валентности,
среди многих химиков упрочилось представление о ней, как о
величине постоянной для каждого данного элемента.
Такого взгляда придерживался в особенности немецкий ученый Август
Кекуле, которому мы так много обязаны широким применением
понятия о валентности в области органической химии.
«Атомность,—говорит в одном месте Кекуле,— есть одно из основных свойств атома, столь
же постоянное и неизменное, как самый атомный вес». Несомненно, что
представление о валентности, как о величине постоянной, является
простейшей из возможных в этом отношении гипотез. Если бы его можно
было поддерживать, го все взаимоотношения между числом различных
атомов, вступающих в химическое сочетание, оказались бы в
зависимости от одного ряда строго определенных чисел.
Однако с течением времени все больше и больше накоплялось
фактов, совокупность которых, в конце концов, заставила химиков оставить
учелие о постоянной валентности и признать, что валентность по
существу является величиной переменной.
Теперь мы знаем, что валентность не только может принимать
различные числовые значения для одного и того же элемента в различных
соединениях, но может варьировать и под влиянием внешних
физических условий. Теперь мы все принимаем, что хлор может быть и
одновалентным (в НС1), и семивалептным в CI2O7 и НС104, что сера,
двухвалентная в сероводороде H2S, четырехвалентная в сернистом газе S02 и
шестивалентиая в SF6, в S03 и в H2SO4. Однако учение о переменной
валентности далеко не сразу получило право гражданства в науке.
В числе химиков, которые с большей или меньшей настойчивостью
поддерживали эту новую точку зрения против Авг. Кекуле и его
сторонников, следует указать на Коль бе, Вюрца, А. М. Бутлерова,
особенно же на шведского ученого Хр. Бломстранда*. Весьма су-
* В сочинении Бломстранда «Die Chemic der Jetztzeit» (1869) мы находим почти
uce вариации валентности элементов, которые принимаются в настоящее Віремя (ом.
особенно сводку). Замечательно, что Бломстранд писал свою книгу до опубликования
периодического закона (предисловие помечено ноябрем 1868 г.) и потому не мог на-
Валентность
22U
щественным моментом в возгоревшемся на '^той почве споре было
открытие периодического закона Д. И. Менделеева (1869).
Желая .во что бы то пи стало удержать, согласно учению о
постоянной валентности, представление о хлоре, как об одновалентном
элементе, хлорной кислоте НС104. например, многие приписывали такого род;:
своеобразное строение:
Н—0—0-0--0--С1
Желание ограничить валентность азота и фосфора цифрой 3
аналогичным образом приводит к формулам Н--0--0—N = 0 для азотной и
Н—Оч
>р—о—он
Н—0х
для фосфорной кислоты и т. д.
Необходимость известных уступок, известного, так сказать,
смягчения догмы Кекуле была сознана многими химиками еще в конце 50-х
и особенно в 60-х годах и привела к появлению учения о постоянной
максимальной валентности. Согласно этому учению, которое было
особенно обстоятельно развито Эрленмейером, постоянной для
каждого элемента является его высшая валентность, проявляемая им
в так называемых насыщенных соединениях, в соединениях высших
типов. Существующие наряду с этими последними соединения низших
типов трактовались как ненасыщенные, причем допускали, что в них
часть единиц валентности остается ненасыщенной единицами
валентности других атомов. По этой причине такого рода соединения, например
ЫНз, РС13 и т. д., и способны к реакциям присоединения.
Эта точка зрения, в сущности, близка к нашим современным
представлениям, но сторонники ее, признавая ныне допускаемую высокую
валентность для одних элементов, отрицали ее возможность для ряда
других.
В 1869 г. Бломстранд, защищая семивалентность галоидов и
марганца в окислах типа R2O7 и в кислотах HR04, замечает, что
семиатомный хлор, вероятно, покажется большинству химиков абсурдом.
Для характеристики взглядов, господствовавших в то время, прибавим
еще, что Лот ар Мейер, один из передовых химиков той эпохи, в
1864 г. в 1-м издании своей книги «Die Modernen Theorien der Chemie»,
допуская валентность, равную 5, для некоторых элементов V группы
периодической системы, валентность 6 для Мо и W (а также для V!) и
т. д., считал возможным для галоидов допустить максимальную
валентность 3 «и, может быть, даже 4», и даже в 1872 г. во 2-м издании
той же книги сохраняет еще старые воззрения относительно
валентности серы, галоидов, марганца и пр. Для сернобариевой соли, например,
Л. Мейер считает возможным обсуждать одну из следующих
структурных формул, в которых сера является двухвалентной и в которой
допускается, как и у Кекуле, цепеобразиое сочетание атомов кислорода:
0_0ч X)—0-Ва--0—0\
S\ /Ва, или S\ /S
X)—0х Ч) - -О • -Ва -О—0/
ходнтьсяі под его влиянием. В соответствии со снішми взглядами на валентность, Блом
странд стал писать и структурные формулы таких соединений, как H2SO4, НСЮ* и
пр. так, как -их пишут ныне, отбросив прежние «цеиеобразные» формулы. Последние
были им, однако, сохранены для комплексных соединений, аммиакатов и гидратов,
что впоследствии и привело к -противоречиям.
230
Валентность
Против всех подобного рода формул много было бы можно
возразить, но нельзя привести окончательных и резких доказательств их
несостоятельности.
Гораздо, проще дело обстоит с такими соединениями, как пятихлори-
стый фосфор PCI5. Долгое время думали, что подобные соединения
являются «молекулярными». Действительно, легкость, с которой
происходит выделение хлора по реакции РСЦ = РСізЧ-СЬ, заставляет, казалось
•бы, думать, что здесь просто молекула хлора соединена с молекулой
треххлористого фосфора: РС13 * СЬ, а отсюда был уже только один шаг
до признания постоянной трехвалентности фосфора, не только там, где
это само собой очевидно, в РС13, РВг3, РН3, но и в РС15 (а также в РВг5),
¦где иначе пришлось бы признать валентность Р = 5.
Но когда оказалось, что разложение PCU не идет до конца, что мы
имеем здесь дело с обратимым процессом, что если мы в сферу
действия будем вводить избыток одного из продуктов
диссоциации, например РСіз, то реакция идет назад, и можно, следовательно,
снова заставить образовываться молекулы PCls из компонентов, притом
в газообразном состоянии, то стало очевидно, что первое предположение
должно было пасть; иначе пришлось бы допустить, что так называемые
«молекулярные» соединения существуют без разложения в
парообразном состоянии, а такого рода гипотеза представлялась весьма
маловероятной. Позднее был получен газообразный пятифтористый фосфор,
вполне постоянный и отвечающий формуле PFs.
Не менее поучителен случай хлористого аммония, NH4CI. Вещество
это, как известно, обладает плотностью пара, примерно вдвое меньшей
против величины, отвечающей формуле NH4C1 (13,4 .вместо 26,75).
В этом обстоятельстве думали сначала видеть исключение из закона
Авогадро. А когда оказалось, что в парообразном состоянии нашатырь
диссоциирует по уравнению NH4C1 = NH3 + HC1 (П еб а л ь, Та н), то
сторонники учения о постоянной валентности стали приводить этот
случай в качестве аргумента в пользу постоянства валентности азота,
равной 3, считая хлористый аммоний «молекулярным» соединением NH3
и НС1.
Б р. Бекеру удалось, однако, перевести в пар недиссоциирован-
иые молекулы NH4Ci. Необходимым условием для удачи такого опыта
является абсолютная сухость как самого испаряемого нашатыря, так и
всех сосудов, в которых ведется опыт. Переход в газообразную фазу
молекул NH4C1 соответствует случаю ложного (а не истинного)
равновесия, которое каталитически нарушается от присутствия даже следов
воды. Но для нас важно, что переход нашатыря в пар без разложения
все-таки возможен.
Под влиянием целого ряда подобных фактов представление о
постоянной валентности мало-помалу теряло под собой почву, по крайней
мере по отношению к большинству химических элементов. До
последнего времени, однако, оставалось не малое число элементов, для
которых представление это, казалось, сохраняло свою силу. Это в
особенности относится к углероду, для которого сохранение неизменной
валентности 4 особенно замечательно ввиду огромного числа
соединений, в которых она проявляется. Не случайно поэтому совпадение, что
.именно Август Кекуле, имя которого, как уже было упомянуто, так
тесно связано с проведением учения о валентности в органическую
химию, крепче других держался за признание постоянной валентности.
Он ясно понимал, что широкое допущение переменной валентности
сразу разрушило бы ту стройную систему, в которую ему удалось
привести десятки тысяч углеродистых соединений.
Валентность
231
Однако с течением времени оказалось, что и углерод далеко не во
всех случаях можно считать безусловно четырехвалентным.
Давно уже известна окись углерода СО, в которой волей-неволей
приходится признать углерод двухатомным.
Экспериментальными исследованиями американского ученого Нефа
установлен целый ряд фактов, говорящих в пользу того, что и в
некоторых других соединениях углерод должен быть признан
двухвалентным. Таковы особенно изонитрилы и соли гремучей кислоты.
Лет 10 тому назад другой американский ученый, Гомберг, выступил
сучением о трехвалентном углероде.
Он показал, что если взять соединение (СбН5)зС-С1,
представляющее метан, в котором три атома водорода замещены феиильными
группами, а четвертый — хлором, и если отнять хлор действием, например,
цинка, то получается чрезвычайно замечательное тело, названное трифе-
нилметилом. Оно обладает ясно выраженным непредельным характером,
способностью к реакциям присоединения. Так, уже под влиянием
кислорода воздуха трифенилметил окисляется и переходит в перекись, в
которой заведомо можно доказать, что между двумя атомами углерода
находится кислород: (С6Н5)зС~0—О—С(СбН5)з.
При действии галоидов он дает соединения типа (С6Н5)зС и пр.
Гомберг предполагал, что мы имеем здесь дело с свободным
радикалом, строение которого изображается формулой (СбН5)зС, причем
С является трехвалентным. Это открытие произвело в науке целую
сенсацию.
О тр.И'фенилметиле много спорили и в конце концов пришли, как
будто, к заключению, что в этом веществе все-таки углерод является
четырехатомиым, что этому соединению нужно придать удвоенную
формулу, и что его легкая способность к реакциям присоединения
объясняется только непрочностью связи между двумя его частями.
Первый раз на эту точку зрения стал іВ. В. Марковн и ков. Потом
она получила подкрепление со стороны А. Е. Чичибабина. Самые
последние исследования показали, что была права и та, и другая
стороны, что, в сущности говоря, большая часть молекул «трифенилмети-
ла» отвечает удвоенной формуле, но что в растворах наблюдается
частичная диссоциация, ведущая к образованию свободных радикалов
(СбНбЬС В последнее время для некоторых соединений, аналогичных
трифенилметилу, изученных немецким ученым Шленком и его
учениками, существование «свободных радикалов» с трехвалентным
углеродом удалось поставить вне всякого сомнения.
Если вместо фенила С6Н5 взять более сложный радикал—остаток
дифенила (С6Н5—С6Н4) і — и представить себе, что все фенильные
группы в «стрифенилметиле» замещены этими остатками, то удается полу-
R\
чить углеводород, который целиком отвечает строению типа •С—R,
и, следовательно, атом углерода должен быть тфизнан в этом
соединении" трехвалентным 3.
Заметим еще, что если в трифенилметиле заменить только две фе-
нильные группы этим радикалом, то получается соединение
промежуточного характера, которое хотя и в заметной степени, но не сполна
диссоциировано на простые радикалы.
Мы лмеем, следовательно, постепенный ряд переходов от такого
типа соединений, где С является типичным четырехвалентным элементом,
23 >
Валентность
к такому типу, в котором проявление четырехвалентное™ в
значительной степени парализовано, и выступают на сцену толькое три «единицы
сродства».
Опыты, опубликованные в самое последнее время, позволяют, по-
видимому, пойти еще дальше, раздвигая еще шире те рамки, ь пределах
которых может варьировать валентность углерода. Замечательно, что
опыты эти но самому своему характеру далеко выходят за пределы
органической химии.
Они 'были произведены знаменитым ф и з и к о м, сэром Д ж.
Дж. Томсоном при изучении тех явлении, которые имеют место при
прохождении электрического разряда через сильно разреженные газы
Том сон показал, что если применить особый метод исследования к
определению отношения между массой и зарядом летящих в Круксовой
трубке частиц,— он исследовал главным образом «каналовые» или
положительные частицы,— то является возможность пользоваться в
качестве индикатора фотографической пластинкой. Пучок
положительных частиц, подвергнутый одновременно действию электрического и
магнитного поля, попадая на пластинку, оставляет на ней ряд следов,
имеющих форму не точек или пятен, а целых кривых — парабол.
Промер этих парабол, из которых каждая соответствует определенному виду
летящих молекул, или «носителей» + заряда, дает возможность для
каждого из таких носителей определить отношение заряда к массе. Но
заряд всегда (по абсолютной величине) бывает или равным электрону,
или кратным ему. Понятно, что, зная для каждого вида летящих частиц
отношение—, мы, таким образом, будем знать и соответствующую ве-
личину m (или ей кратную).
Томсон пришел к этому замечательному результату, что если мы
наполним трубку весьма разреженными парами углеродистых соединений,
например метана СН4 и некоторых его производных, то получаются
такие отношения массы к заряду, что мы должны признать летящими не
целые молекулы, а свободные радикалы, или остатки.
Томсон приходит к заключению, что в таком газе существуют
свободные метильные группы, быть может, только на момент
рождающиеся в этом разреженном газе и так же скоро исчезающие из обращения.
Мимолетно появляются здесь и такие частицы, как СН2, СН и, наконец,
свободные атомы углерода *.
* Заметим, однако, что при всем огромном интересе, который связан с методом
Дж. Дж. Тамсона, в сущности, он не позволяет с уверенностью утверждать, что
«носитель», которому отвечает данная парабола, действительно имеет указанный вес.
тп
Дело в том, что метод дает только отношение ~, но не самую величину тп (атомный
или прупіповой вес). Вывод будет верен, если верно сделанное (предположение
относительно числа электронов, связанных с тп. Поясним это примером. Допуская, что и
роли носителей выступают радикалы СН3, СН2 и СН, Томсон принимает, что с
массами 15, 14 и 13 связано по 1 ( + ) электрону. Если, однако, допустить, что с ними
связано по 2 электрона, то іпридется удвоить значение тп (как пришлось удвоить массу
атома гелия, когда оказалось, что число зарядов на каждом атоме не I, а 2) и тогда
мы будет иметь вместо радикалов молекулы этана СНз—СН3, этилена СН2 —СН2 и
ацетилена СНЕгСН. И на самом деле Томсон неоднократно наблюдал (например, для
азота и ртути) случаи, когда с каждым носителем было связано, например, по 2 и
даже по 3 единицы заряда. Очевидно, здесь еще недостает дополнения к методу Том-
ссна, аналогичного тому, которое в свое время было сделано, благодаря закону Аво*
гадро, к первоначальному методу, которым пользовался Дальтон для определения
атомных весов химических элементов. Судя по данным Томсона, необходимые
контрольные данные отчасти могли бы быть получены на основании изучения снойства
парабол, отвечающих отрицательным носителям, ибо 'последние не склонны
принимать на себя более одного электрона.
Валентность
233
Таким образом, Томсону удалось, по-видимому, осуществить
понижение валентности углерода до 3, 2, 1 и даже до 0."
Замечательно, что Гомберг и Томсон пришли к возможности
понизить валентность углерода и получить «свободные радикалы»
существенно различным путем, первый — чисто химическим,
второй—физическим.
Получение радикалов жирного ряда и свободном состоянии было
старой мечтой химиков, для осуществления которой неоднократно
предпринимались попытки такими учеными, как Бунзеи, Франкланд,
Кольбе и др. Но все эти попытки оказались безуспешными *. Причина
неудачи, очевидно, лежала в той огромной тенденции, которая
существует к попарному сочетанию подобных радикалов. На самом деле при
всех попытках получить свободный метил образовался этан: 2СН3 =
=СНз—'СНз; опыты, которые должны были привести к метилену,
давали этилен: 2СНг< — СН2 : СН2 и т. д. Очевидно, необходим мощный
источник энергии, который мы имеем лр«и прохождении разряда через
трубку в опытах Дж. Томсона. чтобы обеспечить хотя бы
кратковременное существование таких нестойких частиц. Гомберг достиг того же
результата иным путем. Посредством соответственного подбора
радикалов, связанных с данным атомом углерода, он сделал невозможным
проявление его четырехвалентности, а вместе с тем создал условия,
обеспечивающие устойчивость и самостоятельное существование радикалов
R3C **.
Таким образом, есл-и результаты, полученные Гомбергом и его
последователями, представляют новое подтверждение того уже
высказанного выше положения, что валентность, проявляемая данным
элементом, 'является некоторой функцией от природы других элементов, с ним
связанных, то опыты Дж. Дж. Томсона подчеркивают другую сторону
дела — влияние физических факторов на валентность. На этом
последнем влиянии нам предстоит теперь остановиться несколько подробнее.
Из всех физических факторов, оказывающих влияние на течение и
на результат химических процессов, ближе всего изучено влияние
температуры. Факты, указывающие на то, что от температуры
определенным образом зависит и валентность, известны давно, но, сколько я
знаю, только Вант-Гофф в 1878 г. впервые обратил должное внимание
на это обстоятельство.
* Заметим здесь, что вовсе не исключена возможность того, что нам когда-нибудь
удастся искусственно понизить (например, прибегая к очень низкой температуре)
скорость полимеризации радикалов и получить новые индивиды органических соединений,
отвечающие формулам СНз, СН2) СН и т. д. Можно отметить как раз в качестве
одного из современных течений органической химии стремление использовать
«закаливание» нестойких химических соединений, возникающих, например, при высокой
температуре, путем быстрого их охлаждения. Доведенные до достаточно низкой темларатуры,
при которой скорость дальнейшего превращения этих легко изменяющихся соединений
ничтожна, они могут сохраняться в течение долгого времени.
Таким именно путе-м, т.- е. в сущности пользуясь методом холодной и горячей
трубки, который еще очень давно был введен в науку С. Клер Девиллем, Дильс
.=с=о
лолучил недокись углерода (X - а В и л ьс м о р —крайне нестойкое ве-
%=с=о
щестБО СН2=С = 0. Оба эти соединения, оказавшиеся простейшими представителями
класса кетенов, установлением которого мы обязаны замечательным исследованиям
Штаудянгера, необыкновенно легко уплотняются даже при сравнительно низких
температурах. Подобным же образом ныне Гарряес и др. получают из некоторых
терпенов (главным образом из лимонена) изощрен С5Н8 — углеводород, весьма
склонный ери полимеризации переходить в технически ценный каучук.
** О свободных радикалах см. прим. ред. 5 на стр. 459 настоящего тома.
234
Валентность
В последнее время вопросу этому было посвящено несколько работ
количественного характера. Мы остановимся ближе только на одной из
них — на работе Бил ьц а [3\.
Этот ученый изучал температурные условия диссоциации некоторых
химических соединений, между прочим — металлических окислов. Если,
например, взять в качестве объекта окись меди СиО, то оказывается,
что при постоянном давлении Р кислорода, ниже известной предельной
температуры tu разложения вообще не происходит. В этих условиях
•СиО постоянна, и медь сохраняет валентность, равную 2. Если при том
же давлении дойти до упомянутой предельной температуры, то
происходит диссоциация СиО на закись меди и кислород, причем CuO, C112O
и О2 будут находиться в равновесии (это будет как раз та температура,
при которой упругость диссоциации окиси меди сравняется с
господствующим парциальным давлением Р кислорода). Если перейти эту
температуру, то существование окиси М'еди сделается невозможным,
наступит область существования закиси меди, иначе говоря, меди
одновалентной. Возможность диссоциации Си20 на медь и кислород наступит, как
только упругость кислорода, отвечающего этому процессу, достигнет
господствующего давления Р. Температура t2, при которой это случится,
будет второй предельной температурой. Если мы поднимемся еще выше,
то, очевидно, и само существование СигО станет невозможным (ибо
теперь упругость диссоциации СигО будет превышать поддерживаемое
давление кислорода Р). Другими словами, наступит область
существования одной только металлической меди: валентность Си понизится до
О, и медь как бы уподобится элементам нулевой группы.
Так, например, при парциальном давлении Р, какое имеется в
воздухе (около 7б атм), первая предельная температура лежит при 1010°,
-а вторая (выше которой существует только металлическая медь)—при
1670°. Аналогичные соотношения были установлены и для окислов дру-
гих металлов. В следующей таблице приведены температуры перехода
•соединений высшего типа в низший для парциального давления
кислорода в 1 атм:
Pd02 PdO CuO CuaO PbQ2 Pb3Q4 PbO
PdO Pd Cu20 Cu Pb304 PbO Pb
t 180° 875° 1110° 1800° 390° 615° 2240°
Fe2Q3 Fea04 MnOa Mn2Q3
Fe304 Fe Mn203 Mn304
t 1470° 3025° 570° 1090°
Из опытов Бильца следует, что с повышением господствующего
парциального давления газообразного продукта диссоциации (кислорода)
предельные температуры повышаются. Из этого видно, что и
давление, подобно температуре, оказывает влияние на валентность. Влияние
это, однако, является лишь косвенным: оно определяется величиной
концентрации газообразного продукта диссоциации.
Условия изменения валентности от температуры и давления
наглядно изображены -на прилагаемой диаграмме, где тю оси ординат
отложены величины давления (в атм), а по оси абсцисс — соответствующие
предельные температуры. Мы видим (рис. 1), что вся площадь
диаграммы разбивается на три области, соответствующие условиям
существования СиО, Си20, Си*.
* Следует, однако, отметить, что резкие температурные границы, отмеченные Биль-
цем, характеризуют, в сущности, не валентность, как таковую, а условия химического
равновесия в унивариантных системах, к каковым относится случай диссоциации
твердого тела с одним летучим продуктом. Наоборот, когда дело идет о равновесных
Валентность
235
Итак, под влиянием различных факторов, как химических, так и
физических, валентность может принимать различные значения для одного
и того же элемента. Понятно, что, благодаря этому, существенно
осложняется как самое определение валентности, проявляемой тем или иным
элементом в каждом
отдельном случае, так и 2.0
приложение учения о
валентности к задачам
структурной химии.
Естественно
возникает вопрос, не теряет
ли вообще при таких
условиях понятие о
валентности своего
значения, своего raison
d'etre *.
История химии дала
вполне определенный
ответ на этот вопрос.
Представление о
валентности не исчезло
из науки. Можно
сказать наоборот, что оно
никогда не переставало
оказывать своего
благотворного влияния на прогресс химии, а за последнее время, несмотря
на возникшие новые осложнения, о чем речь впереди, интерес к
изучению вопроса о валентности особенно усилился.
700 800 900 WOO /Ш/Ш13001ШШ0 №0 ПВО 1800 №0 20001
Рис. 1.
системах, которые могут быть изучены в однородной, например газообразной, среде,
какова диссоциация пятихлористого фосфора (PC15j*PC1s+C12) или Фриделевского
соединения, образованного метиловым эфиром и НС1(СНз)гО • НС1^ (СНзЬО-ьНСі) и т. п.
когда, следовательно, вариантность систем > 1, то изменение валентности с
температурой (и давлением) происходит постепенно. Молекулы РС!5, например, при
повышении температуры мало-іпомалу уступают место молекулам РСЦ и CU. С другой
стороны, диссоциация карбонатов, например СаСОз, .-происходит совершенно по тому же
закону, как СиО, а между тем Са, С и О в .молекулах СаСОз, СаО/С02 проявляют
одну и ту же валентность. Таким образом, из опытов Бильца, так же как из более
ранних исследований, следует, что с повышением температуры понижается степень
устойчивости соединений, в которых данный элемент .проявляет свою высшую
валентность.
Заметим еще, что понижение валентности с температурой касается и -проявления
«дополнительных» и побочных единиц сродства, как это видно на диссоциации
многих пидратов и аммиакатов. Так как и здесь мы обычно имеем дело с системами
унивариантньгми, то -переход от одного типа к другому при определенном
парциальном давлении Н20 или NH3 совершается резко (скачками) при прохождении
температуры через определенную точку. Так, например, Бильц нашел следующие
предельные температуры лріи парциальной упругости NH3 в 1 атм для аммиакатов:
2AgC16NH3
2AgC13NH3
t 20°
CdCU6NH3
CdCl24NH3
/33°
2AgC13NH3
AgCi
68°
CdCla4NH3
CdCle2NH3
73°
CuCl36NH8
CuCl,4NH3
90°
CdClo2NH3
CdCl2NH3
355°
CuCl24NH3
CuCl22NH;1
140°
CdCl2NH3
CdCI2
338°
Для гидратов аналогичные отношения были установлены значительно раньше Ле
кером, Фровейном и многими другими.
* Разумное существование, смысл (франц.). Прим. рео.
236
Валентность
Если бы мы захотели выяснить причину этого на первый взгляд
странного обстоятельства, то нам пришлось бы прежде всего принять во
внимание, что при всех осложнениях, которым подвергалось учение о
валентности, изменению не подверглись столь характерные для нее
целые сравнительно небольшие числа, кратные чисел атомов,
непосредственно сочетающихся друг с другом. Потребность дать определенное
выражение этому чисто эмпирическому выводу из бесчисленного
множества фактов, очевидно, осталась. Притом, хотя определение
валентности для каждого данного случая и сделалось труднее, чем оно
представлялось для сторонников постоянной валентности, однако и в этом
случае выход из затруднения оказался возможным.
Это относится прежде всего к органическим соединениям, особенно
к соединениям предельного ряда, для которых огромное большинство
фактов удовлетворительно объясняются, если по старому признавать
углеводород четырехвалентным *.
Что касается вывода валентности отдельных элементов в
«минеральных» соединениях, то здесь проще всего дело решается в тех случаях,
когда данное (бинарное) соединение является электролитом и когд.ч,
следовательно, представляется возможным применять законы Фарадея.
При этом мерой валентности является число определенных единиц
электрического заряда, которые воспринимает данный атом, когда
переходит в состояние иона (см. ниже). Такой единицей является заряд
водородного иона. Таким путем, например, может быть установлена не
только одновалентность галоидов X в кислотах НХ (НС1, НВг и пр.)'
двухвалентность кислорода в воде Н20, серы в сероводороде H2S, но и
валентность многих металлов в их солях, например одновалентность
щелочных металлов, двухвалентность Mg, Са, Sr, Be, трехвалентпость
А1 и; Сг (в солях окиси) и т. д.
Валентность электрических ионов может быть определена еще и
другими методами. Один из таких методов основан на свойствах
коллоидов выделяться из раствора (свертываться) от прибавления
электролитов. В этих явлениях преимущественную роль играют катионы,
причем их свертывающая способность сильно возрастает с валентностью.
Так как для данного коллоидального раствора эквивалентная
концентрация ионов данной валентности, которая достаточна для свертывания,
колеблется в узких пределах, то можно прямо по величине этой
концентрации определить неизвестную валентность. Таким образом, например,
Галецкий определил валентность иона бериллия Be. По отношению
к некоторому раствору сернистого мышьяка AS2S3, свертывающая
способность которого для одно-, двух- и трехвалентных ионов выражалась
следующими числами: 1 :30 : 1650, опыты дали для солей бериллия
число, 'близкое к 30, и таким образом могла быть подтверждена
двухвалентность иона Ве++ і
Другой способ, основание которого было дано Нернстом, только
что получил развитие и весьма интересное применение со стороны Ге-
веши [4]. Этот метод основан па изучении процесса диффузии в
растворах электролитов. Нернст показал, что константа диффузии данного
электролита D связана с подвижностями ионов ни vc валентностью п
(например, для ВаС12 п = 2) следующим соотношением:
* Несколько сложнее обстоит дело с соединениями непредельными, где пине
некоторые химики (Гинрихсе-н, А. Е. Чичибабин и др.) принимают трех- .и
двухвалентный углерод вместо двойных и тройных связей между атомами углерода, допускаемых
классической тео-рией строения. Мы не будем в данную минуту касаться этого
интересного, но крайне сложного вопроса, тем более, что его предполагается затронуть
в одном из следующих выпусков «Новых идей в химии», который будет посвящен
главным образом теории химического строения и связанным с нею проблемам4.
Валентность
237
"п 'u4-v (для 18° постоянная /1=0.04185). Гевеши
воспользовался тем обстоятельством, что дело упрощается, если один из
конов находится в большом избытке, например, если мы имеем
хлористую соль какого-нибудь металла, скажем, ВаСіо в присутствии
большого количества НС1 (здесь в избытке находятся ионы С1). Для этого
¦случая (и для 18°) получается очень простая зависимость между
константой диффузии D, подвижностью катиона и и его валентностью
п • D - 0,02242 • - .
п
Пользяусь этим уравнением, Гевеши произвел ряд в высшей
степени замечательных определений для радиоэлементов. Измерение
концентрации солей этих последних при диффузии производилось с помощью
обычных методов радиологии. Благодаря крайней чувствительности
этих методов опыты могли быть сделаны с ничтожными количествами
вещества. Из полученных данных вычислялась величина D,
Подвижность катиона определялась при помощи метода переноса *.
Таким образом Гевеши и была определена валентность ионов,
образуемых 22 радиоэлементами. Мы еще будем иметь случай вернуться к
полученным при этом результатам.
В тех случаях, когда соединение данного элемента является
неэлектролитом, или когда дело идет о валентности, проявляемой элементом,
входящим в состав сложного иона (например, S в S0.1, С1 в С104 и пр.),
вывод осложняется гипотезой (впрочем, более чем вероятной), что
валентность данного элемента в ряду сходственных и генетически
связанных между собой его соединений остается постоянной. Так, допускают,
что валентность кислорода в его солеобразных окислах (например, в
СаО, А1203, S03, Р2О5) и их гидратах (в гидроксильной группе ОН)
остается равной 2 (т. е. той же, что в воде), что двухвалентность,
проявляемая серой в сероводороде, сохраняется в сульфидах (например, в
K2S, FeS, P2S5, As2S3 и пр.), что галоиды являются одновалентными не
только в галоидоводородных кислотах НХ, но и в хлорангидридах кислот
и вообще в галоидных соединениях, дающих при гидролизе кислоту НХ
(S02C12, S2CI0, PCI5, AsCl3, SiCl4), а также в продуктах, получаемых
путем металепсии из органических соединений (ССЦ, СНСЬ, СН3С1,
С2Н5Вг, С6Н5Вг и пр.) и т. д.
Было и ещ-е одно обстоятельство, способствовавшее укреплению
учения о валентности и самую изменчивость последней подчинившее
определенной закономерности. Таким обстоятельством было открытие
периодического закона.
Если Д.И.Менделеев был одним из главных виновников
ликвидации учения о постоянной валентности и признания высшей валентности
для целого ряда элементов (например, шестивалентности для S, Sc, Те.
Cr, W, U, семивалентности для галоидов и для марганца), то, с другой
стороны, созданная им периодическая система указала па огромное
обобщающее значение подобного более широкого представления.
Три правила регулируют соотношение между валентностью
элементов и положением их в периодической системе:
1. Номер группы, к которой относится данный элемент, равен
валентности его в высшем солеобразующем окисле.
2. Валентность элементов в их высших летучих водородистых
соединениях правильно понижается от IV группы к VII.
* Ср., например, Мюллер. Основные законы электрохимии. Перевод под ред.
проф. Н. А. Путина.
238
Валентность
3. При этом сумма высших валентностей по кислороду и по
водороду для каждого элемента равна постоянной величине 8.
К этим правильностям можно присоединить еще одну, но уже
далеко не столь строго выдержанную: в группах периодической системы,
отмеченных четными номерами, преимущественно повторяются четные
валентности, в остальных — нечетные*. Так, в I группе преобладает
валентность 1, во II—2, в III—3 и отчасти 1, в IV — 4 и 2, в V—5 и 3.
в VI—6, 4 и 2, в VII—7, 5, 3, 1.
Итак, если учение о валентности претерпело некоторое осложнение,
вследствие признания ее изменчивости, то, с другой стороны,
осложнение это, несомненно, искупается той закономерной связью, которая
при этом обнаружилась между валентностью элемента и его
положением в периодической системе.
Обратимся теперь к обзору эволюции представления о валентности-
за самое последнее время, за тот период развития хим-ии, который еще
не сделался достоянием истории. Нельзя не отметить, что именно за это
время и в самом объеме понятия о валентности, и во взглядах на ее
природу произошли особенно глубокие перемены. Произошло это под
влиянием огромного числа фактов, собранных за последние
десятилетия. Я, конечно, не буду здесь утруждать вашего внимания
сколько-нибудь подробным перечнем этих фактов и остановлюсь только на
немногих из них, которые кажутся мне наиболее характерными.
Давно известно, что соединения, образуемые парой элементов caete-
ris paribus **, тем прочнее и тем характернее, чем больше элементы
различаются друг от друга в химическом отношении, чем дальше они
отстоят друг от друга в периодической системе (по крайней мере, если
мы будем говорить о семи первых группах), чем в большей степени
отличаются элементы друг от друга в электрохимическом отношении.
Наоборот, элементы, близко стоящие друг к другу, в особенности
входящие в состав одной и той же группы периодической системы,— а тем
более одной подгруппы,— мало склонны, часто даже вовсе не склонны
к образованию химических соединений.
Параллельно с этим свойства тех соединений, которые образуются
из элементов мало сходных, противоположных друг другу, резко
отличаются от свойств компонентов. Что общего, например, между
поваренной срлью NaCl и ее компонентами: металлическим натрием и
газообразным хлором? Наоборот, чем ближе стоят друг к другу
соединяющиеся между собой элементы, тем менее отличаются свойства
соединений от свойств компонентов.
Замечательно, что в соответствии с этим стоит очень часто
нарушение основных признаков, или, по крайней мере, чисел валентности.
"Особенно резко это сказывается, когда мы исследуем взаимоотношение
металлов, когда мы изучаем сплавы.
* Однако, здесь нельзя не указать .и на большое число исключений. Так, в I
группе медь двухвалентна в солях окиси, в III — индий может быть двухвалентным в InCl?
и пр., в IV — известны соединения с трехвалентными С, Ті, Се, в V — четные
валентности возможны для N, Р, V, в VI — нечетные для Сг (особенно 3), Mo, W, в VII —
четные для хлора (СЮ2), йода (J02), марганца (МпО, Мп02, Мп03) и т. д. См. обзор
валентности, проявляемой различными элементами, в конце этой статьи. Заметим здесь.
что сам Д. И. Менделеев с большой осторожностью относился к учению о валентности
вообще и предпочитал говорить о тилах соединений, или сопоставлять числа
эквивалентов кислорода и пр., связанных с атомными весами элементов, подлежащих
сравнению. Сущность указанных ш іправсильностей от этого, однако, мало изменяется.
Несколько скептическое отношение Д. И. Менделеева к широкому пользованию
понятием валентность, быть может, объясняется именно тем обстоятельством, что
простейшая и, конечно, наиболее \гтоследовательная форма этого учения, данная Кекуле..
оказалась несостоятельной.
** При прочих равных условиях (лат.). Прим. ред.
Валентность
239
Здесь уже для простейших, бинарных соединений наблюдается
отклонение от тех цифр валентности, к которым мы привыкли, хотя
наряду с ними вовсе нередки и представители нормальных типов.
Существование образующихся в сплавах так называемых
интерметаллических соединений доказано с помощью физических
методов исследования. Между ними на первом плане стоит метод
термического анализа, разработкой которого и введением .в жизнь мы
обязаны главным образом усилиям трех выдающихся ученых: Ле-Ша-
т е л ь е в Париже, Н. С. Курнакова в Петербурге и Г. А. Т а м-
манна в Геттингене.
Для примера приведем перечень химических соединений,
образующихся при растворении щелочных металлов з ртути (амальгамы):
Na5Hg2
Li3Hg
LiHg
LiHg2
LiHg3
Na3Hg
NaHg
NaHg2
NaHg4
KHg
KHg2 RbHg2
KHg3
RbHg4
KHg6 RbHg6
CsHg
CsHg2
CsHg4
CsHg6
KHg10 CsHgl0
В других случаях наблюдаются аналогичные соотношения, как это
видно, например, из существования таких соединений, как Li2Sn5, KSn4,
NaCd5, NaZnu (?), CaZn10> CaZn4, Ca4Zn, Tl4Ca3, AuSb2, AuSn4 и мн. др.
Мы видим, что здесь индивидуальность соединяющихся элементов
сказывается в высокой степени, а вместо «нормальных», или, вернее,
общепринятых цифр валентности, выступают новые, частью совершенно
необычные *.
Правда, и здесь еще остаются целые числа и они все-таки не очень
велики.
Если бы мы захотели дать какое-нибудь объяснение всем этим
фактам, то нам пришлось бы допустить, что валентность элементов еще в
большей степени изменчива, чем мы принимали до сих пор, или же
существуют какие-то особые единицы сродства для тех случаев, когда
связываются элементы, особенно близкие друг к другу. То же самое
можно сказать о необъятной области так называемых комплексных
соединений, образчиком которых могут служить соединения,
образуемые двухлористой платиной PtCl2 с аммиаком. Известен целый ряд
таких соединений, в которых на одну молекулу PtCl2 приходится одна,
две, три и четыре молекулы NH3. И в этом случае, как и во множестве
других, не представляется возможным дать структурные формулы этих
соединений, если только не прибегать к помощи специальных гипотез.
На самом деле, всякий раз, когда делались попытки в этом
направлении, приходилось расширять представление о валентности или делать
к нему разные надстройки. Так, уже при первых попытках этого рода
(Бломстранд, Клеве, И ёргенсо н и др. {5]) было сделано
допущение, что во многих комплексных соединениях не только
проявляются высшие валентности (частью мало обычные, например, четырех-
атомность кислорода в гидратах) у таких элементов, как азот, кисло-
* Впрочем, делать какие-либо определенные выводы насчет валентности,
проявляемой «при этом Na, Hg, Cd, Zn и др., не оредставляется воз-можным, так как
молекулярный вес иитерметаллических соединений по большей части неизвестен.
240
Валентность
род, сера, галоиды, но и имеет место особое цепеобразыое сочетание
целых частиц, как то: NH3, Н20 и пр. наподобие того, как связываются
друг с другом атомы углерода в частицах органических соединений.
Строение соединения двухлористой платины с четырьмя молекулами
аммиака выражали, например, формулой I, а строение аналогичного по
химической природе хлористого лутеокобальтиака — формулой II.
I
/NHr-NHr С!
\NH3 \Н:| С1
II
/NH,—NH,--NH:r NHjr-CI
Со—NH3--C1
\\На—CI
Попытки эти оказались, однако, мало успешными и привели к ряду
противоречий.
Несравненно более глубокая, но вместе с тем и более удачная
перестройка в обычных представлениях о валентности была предпринята
в новейшее время А. Берне ром [6] в -его координационной теории и
покойным Р. Абеггом в его гипотезе о валентностях и
контрвалентностях. Взглядов этих ученых я коснусь здесь лиійь в самых
общих чертах, так как теория Вгрнера изложена мною в другом месте
[7], а гипотезе Абегга посвящены две отдельных статьи настоящего
сборника *.
В представлениях Абегга мы находим как бы отголосок старой
гипотезы Эрленмейера о постоянной для каждого элемента максимальной
валентности. По Абеггу, атому каждого элемента свойственно
определенное максимальное число единиц валентности и определенное число
единиц контрвалентности. Они противоположны по знаку. Притом
сумма тех или других для каждого элемента всегда равна 8.
Контрвалентностям (которые, вообще говоря, действуют слабее
валентностей) Абегг приписывает главную роль при образовании
комплексных соединений.
С другой стороны, и Верпе р (притом значительно раньше Абегга)
высказал предположение, что, помимо обыкновенно принимаемых
единиц валентности, которые он называет главными (Hauptvalenzen),
существуют еще другие, побочные (Nebenvalenzen). He определяя ближе
числа этих последних для каждого элемента, Вернер приписывает им
решающее значение при образовании комплексов, а именно, при
сочетании между собой готовых молекул, например, NH3, H20, HCL
(СгШЬО и пр. Наоборот, главными средствами, по Вериеру,
связываются .ионы (CI, Br, J, N03, Na, К, NH4) или такие атомы или остатки,
которые эквивалентны ионам (например, О, S, СН3, QH.-,, NH2 и пр.).
Таким образом, роль побочных валентностей Вернера близка к роли
контрвалентностей Абегга, с той только разницей, чго Вернер не приписывает
своим побочным валентностям электрического характера.
Абегг нигде не дает структурных форм соединений, опирающихся на
высказанные им представления **. Поэтому способы применения послед-
* Имеются в в-иду «Ноше идеи в химии», вып. 3. СПб., 1913. Прим. ред.
** Несомненной заслугой гипотезы Абегга является то обстоятельство, что о-на
представляет первую попытку выяснить значение того числа 8, которое «грает такую
Валентность
241
них на практике не всегда ясны. Наоборот, взгляды Вернера широко
использованы для формулировки строения и для систематики
комплексных соединений. Для иллюстрации приведем по Вернеру строение
упомянутых выше соединений PtCl24NH3 и CoCl36NH3 (побочные
валентности обозначаются пунктиром).
NH
NH.
Pt
NH,'
'NH-
CI,
NbL NH3 NH3
NH3---Co NH,
/f
NH.
CI.
Итак, факты заставляют значительно расширить значения
валентности и вывести их не только за те нормы, которые были установлены
школой Кекуле, но, по-видимому, и за те максимальные цифры,
которые вытекают из периодической системы элементов.
Чтобы покончить с обзором тех опытных данных, под влиянием
которых пришлось постепенно все более и более осложнять представление
о валентности, я хочу остановиться на одном ряде фактов, относящихся
к области органической химии.
Если мы к какому-либо непредельному соединению с двумя этилен-
ными связями, расположенными по соседству, например к бутадие-
н у. или д.и винилу
СН2=СН—СН=СН2
будем присоединять бром, то присоединение двух атомов последнего
произойдет, вопреки ожиданию, не к паре углеродных атомов,
связанных двойной связью, а по краям цепи и приведет в данном случае к
образованию дибромида
СНо—СН=СН—СН»
Вг
Вг
вместо ожидаемоего
СН2—СН—СН=СН2
I I
Вг Вг
Подобным же образом происходит присоединение водорода, напри
мер, к муконовой кислоте
СН=СН—СН=СН >СН2—СН=СН—СН2
СООН СООН СООН СООН
Мухоновая кислота Дигидромуконовая кислота
выдающуюся роль в периодической системе. Максимальная валентность
(соответствующая окислам типа R04) равна 8. Восьми же равна сумма валентностей элементов в их
кислородных и летучих водородистых соединениях.
16 Л. А. Чугаев, т. III
242
Валентность
Немецкий ученый Тиле (J. Thiele) -показал, что такой своеобразный
порядок присоединения к частицам органических соединений,
содержащим систему двух этиленовых связей >С = С—С=С< или так
называемую конъюгированную систему, представляет широко
распространенное, свойственное не только углероду, но и другим элементам,
явление.
Чтобы дать хотя некоторый отчет в причине такой аномалии, Тиле
высказал гипотезу, согласно которой всякий раз, когда два атома
углерода или иного элемента соединяются между собой при помощи двойной
связи, «затрачивается не вся сила сродства, отвечающая четырем
единицам валентности, а только часть ее». В результате при каждом атоме
углерода остается «ненасыщенная часть сродства», и таким образом
наряду с собственно валентностями такому атому присуща еще так
называемая парциальная валентность.
Последнюю в формулах изображают пунктиром:
С=С С=0 C=N N = N
Эти-то парциальные валентности, или, вернее, отвечающие им
остаточные силы сродства, составляют, по Тиле, primum movens * всяческих
реакций присоединения к непредельным соединениям.
Если теперь мы имеем конъюгированную систему, например в
бутадиене, то парциальные валентности должны существовать при каждом
из четырех атомов углерода.
12 3 4
СН2=СН—СН =СН2
Тиле, однако, принимает, что парциальные валентности остаются
свободными только при двух крайних атомах углерода 1 и 4, что, наоборот,
валентности соседних атомов 2 и 3 особенным образом насыщают друг
друга, образуя более прочную связь, которая в формулах изображается
скобкой:
12 3 4
СН2=СН—СН=СН2
Поэтому-то, полагает Тиле, присоединение брома, водорода и г. д.
происходит именно по краям цепи, где парциальные валентности еще
не насыщены.
Мы вовсе не намерены здесь входить в критический разбор гипотезы
Тиле, которая разделяется далеко не всеми химиками**, в которой, во
* Движущее начало (лат.). Прим. ред.
** Из русских химиков интересные попытки дать иное толкование механизма
реакций присоединения к -коиъюги-ро-ванной системе двойных связей были сделаны
И. И: Остромьгсленским и А. Е. Чіичибабиньгм. Мы надеемся вернуться к этой теме
ь одном из следующих сборников «Новых «ней в химии».
Валентность
243
всяком случае, можно видеть лишь очень грубое отражение, или символ.
действительности. Мы приводим здесь эту гипотезу вместе с фактами,
на которые она опирается, лишь в качестве примера дальнейшего
осложнения, которому подверглось представление о валентности.
В высшей степени поучительно, что три исследователя — Абегг, Бер-
нер и Тиле,—исходя из совершенно различных соображений, опираясь
на существенно различный фактический материал, приходят к
необходимости допустить наряду с основными или главными валентностями
еще другие, дополнительные, или побочные. Первоначальная простая
концепция повсюду оказывается недостаточной. За последнее время
аналогичные предположения в различных вариантах были высказаны
целым рядом других исследователей. Назовем только имена Кнёве-
нагеля, Шпигеля, Фринда, Байера, Кауфмана и др.
Мы не считаем возможным приводить здесь хотя бы вкратце всех
этих гипотез и построений и ограничиваемся указанием на них, как на
«знамение времени».
Перехожу теперь к третьей и последней части моего обзора, к
краткому рассмотрению тех -попыток, которые были сделаны в различное
время, для того, чтобы дать хоть какой-нибудь ответ на вопрос: что
собственно является причиной валентности?
При этом нам придется постараться дать отчет в источнике,
обусловливающем появление этих небольших целых чисел, которые для
валентности так характерны, и в то же время в изменчивости их, а с другой
стороны — в причине той расплывчатости понятия о валентности,
которая, с особенной настойчивостью выступает за последнее время.
Конечно, здесь мыслимы два течения, два основных направления
мысли. Можно себе представить, что силы, под влиянием которых атомы,
связанные в молекуле, удерживаются друг около друга, напоминают
силу тяготения (если только с ней не тождественны), что, подобно этой
последней, притягательная сила, обусловливаемая каждым атомом по
отношению к другим атомам, или сродство, действует совершенно
равномерно по всем направлениям. При таком предположении, разумеется,
особенно необходимо дать себе отчет и в причине тех разнообразных
фактов, которые обыкновенно объединяют словами «избирательное
химическое сродство», а с другой стороны, уяснить себе самое появление
целых, правильно повторяющихся чисел валентности.,'
Но возможна и совершенно иная точка зрения; МоЖно предположить,
что атомы по самой природе или конструкции своей обладают известной
полярностью, как это допускали многие еще задолго до появления
понятия о валентности, исходя из совершенно 'иных соображений. Тогда,
очевидно, число полюсов на каждом атоме и будет соответствовать
числу присущих ему единиц сродства, или валентности.
Мы рассмотрим прежде всего замечательную попытку обосновать
теорию валентности, сделанную с точки зрения первого взгляда на
природу атомного сродства.
Попытка эта принадлежит Вант-Гоффу и изложена в его
классическом сочинении «Ansichten fiber organische Chemie», 1878.
«Действие атомов,— пишет Вант-Гофф,—которое они оказывают друг
на друга, находясь на большом расстоянии, подобно тому, как в случае
притяжения небесных светил, может зависеть только от их массы и от
взаимного между ними расстояния. Форма их и движение значения не
имеют. Совсем иначе обстоит дело для тех малых расстояний, которые
имеются внутри .молекулы. Здесь влияние формы и движения (атома)
выступает на первый план, а простое проявление силы тяготения
оказывается затемненным * появляется сродство и валентность, появляется хи-
16*
244
Валентность
мическое действие. Хотя природа химических сил, по-видимому, и
противоречит коренным образом законам тяготения, однако (для ее
объяснения) нет надобности вводить какой-либо специальный принцип.
Наоборот, представляется возможным составить себе представление о
влиянии формы и движения на малых расстояниях, и оказывается, что
это влияние в общих чертах воспроизводит те изменения, которые
должно претерпеть тяготение, когда оно превращается в химическое
притяжение».
Останавливаясь прежде всего на влиянии формы атомов, Вант-Гофф
показывает, каким образом можно объяснить, почему некоторые
атомы, хотя и обладающие меньшей массой, «получают, так сказать,
предпочтение» сравнительно с другими, более тяжелыми, например, хлор
вытесняет бром и иод из их соединений и т. д. Причину этого
парадоксального факта он видит в том обстоятельстве, что в известных случаях,
когда возможно более тесное сближение между атомами
(следовательно, уменьшение расстояния между ними), атом с меньшей массой в силу
этого именно обстоятельства может обнаруживать несоответственно
большую притягательную силу по сравнению с атомом, масса которого
более значительна *.
Переходя собственно к понятию о валентности, Вант-Гофф выводит
его из следующих соображений. Нетрудно понять, что всякое
отступление атома от шарообразной формы должно иметь своим последствием,
что притяжение по известным направлениям, именно по тем, по которым
возможно более тесное сближение этого атома с другими, будет
сказываться сильнее, нежели по остальным направлениям. Мы получим, таким
образом, известное число направлений максимального действия, иначе
говоря, известное число единиц сродства или валентности. Вообразим
себе, например, атом, имеющий форму, представленную на рис. 3,
которую мы, простоты ради, будем рассматривать на плоскости. Ясно, что
* Представим себе вместе с Вант-Гоффом (рис. 2) случай взаимоотношения: 1)
между атомом А и атомом В и 2) между атомом А и атомом С, причем предполагается, что
плотность и масса всех этих атомов одинакова, и все различие сводится к их форме
(атомы А и В — шарообразны, атом С состоит из двух
соприкасающихся сфер). Обозначим массы этих атомов
через Мк> УИВ, Мс, радиусы соответствующих шаров —
через RAt RB и Rc и силы притяжения между А и
В — через /Сдв, а между А и С — через Kaq- Тогда
можно показать, что
Мк X Мв
Рис. 2.
причем С есть некоторый постоянный коэффициент. Предположим теперь, что
соотношение между размерами шаров таково, что, например, RA == 7RC. Тогда Клс > /Сдв
т. е. притяжение А к С сильнее, нежели А к В, хотя массы В и С одинаковы.
«Очевидно,— замечает Вант-Гофф, — что должны быть и такие случаи, когда меньшая масса
получает преобладание над большей».
Валентность
245
такой атом должен иметь три направления, отвечающие максимальному
притяжению ОА, ОВ и ОС, кроме того, три других аналогичных
направления Oa, Ob и Ос, имеющих второстепенное значение. Такой атом
должен, таким образом, функционировать то как шести-, то как
трехвалентный. Какую из этих валентностей (3 или 6) проявит в каждом отдельном
случае данный атом, будет в значительной степени
зависеть от природы его партнера.
Существенное влияние на проявление
валентности должно оказывать, по Вант-Гоффу, дви-жение
атома. В самом деле, достаточно предположить, что
атом равномерно передвигается по всем
направлениям (колеблется) около некоторого центра, чтобы
стало ясно, что такое движение будет иметь своим
последствием изменение внешней формы этого
атома, а отсюда проистечет соответствующее изменение
сродства и валентности. Останавливаясь на
простейшем, уже рассмотренном выше примере,
допустим, что атом ABC колеблется около некоторого центрального
положения. Если при этом атом удаляется от этого последнего на расстояние Ry
то часть пространства, занятая колеблющимся атомом, иначе говоря, его
внешняя форма будет отвечать фигуре AjCiBiaiCtbi (рис. 4). Понятно,
что одним из непосредственных следствий такого расширения контуров
атома будет увеличение расстояния
между ним и другими атомами, вследствие
ослабления действующих между ними
сил, проявляющихся в сродстве.
С другой стороны, направления
максимального притяжения, вследствие
движения атомов, не могут уже быть столь
резко выраженными. Увеличение
расстояния, на котором может происходить
взаимодействие между атомами, должно
иметь своим последствием прежде всего
прекращение функционирования тех
«второстепенных» валентностей, которые
отвечают направлениям Oa, Ob и Ос. По
мере того, как будет увеличиваться
интенсивность (амплитуда) колебания,
например, когда расстояние R превратится
в R2 и контуры атома в АзСгВгагСгЬг,
соответственно будут увеличиваться и вытекающие отсюда последствия.
«Атом будет мало-помалу утрачивать свой химический характер».
«Если принять во внимание,— говорит Вант-Гофф,— чго
интенсивность атомных колебаний обусловливается температурой, то
вышеизложенный взгляд приводит к выводу, оправдывающемуся на опыте, что с
повышением температуры должно сокращаться число валентностей и
ослабевать проявление сродства, иначе говоря, взаимодействие между
атомами должно постепенно приближаться к простому проявлению
тяготения. Известно, что существует некоторый верхний предел
температуры, за которым прекращается химическое влияние. Равным образом
известно, что в противоположных (температурных) условиях химические
явления до крайности осложняются. Без сомнения, это происходит по
той причине, что вступают в свои права валентности, до тех пор
остававшиеся незамеченными.
Рис. 4.
246
Валентность
Непосредственное следствие, вытекающее из только что
высказанных соображений, заключается в том, что и собрание атомов, т. е.
молекула, должно относиться к другим молекулам -подобно атому, только в
менее резкой форме. Молекуле также должны быть присущи и сродство,
и валентность, причем таковые хотя -и зависят от своеобразного
сочетания частей (т. е. атомов), но не принадлежат сами по себе этим
последним. Таким образом создается основание для суждения о молекулярных
соединениях».
Попытка Вант-Гоффа уяснить себе возможную причину полярного
характера единиц валентности, сколько мне известно, не получила
дальнейшего развития, если не считать гипотезы, высказанной В у н д е р-
лихом [8] относительно формы углеродного атома, из которой этот
автор старается вывести тетраэдрическую схему, положенную Вант-
Гоффом я основу стереохимии углерода.
Однако мысль о сродстве, как о «силе притяжения, действующей
равномерно из центра атома по направлению ко всем частям его
поверхности», не была оставлена и позднейшими авторами. Она легла, между
прочим, в основу гипотезы, впервые высказанной в 1891 г. [9] проф.
А. Вернером в Цюрихе.
Для Вернера валентность — не более, как эмпирически
находимое числовое отношение, в котором атомы соединяются
между собой. Это отношение зависит не только от природы данного
атома, но также и от природы тех атомов или групп, которые с ним
сочетаются. Нет .никаких предсуществующих отдельных единиц
валентности, способность же каждого атома удерживать около себя лишь
строго определенное число атомов других элементов находится в
зависимости от того обстоятельства, что, с одной стороны, силы притяжения
между атомами — иначе говоря, силы сродства — действуют заметно
лишь на малых расстояниях и что, с другой стороны, в непосредственной
близости от данного атома может поместиться только ограниченное
число других атомов или групп. Таким образом, валентность тесно
связана и с заполнением пространства, точнее,— с относительными
размерами сфер влияния атомов и атомных групп, сочетающихся между
собою.
Из этой гипотезы немного позднее (1893) выросла 'известная
координационная теория Вернера, о которой уже была речь выше [10].
В этой теории Вернер отделил понятие о сферах влияния атомов от
понятия валентности в собственном смысле слова, путем введения особого
представления о координационных сферах, а с другой
стороны—дополнил свое представление о валентности допущением
электровалентности в духе современной электронной теории.
Переходим теперь к электрохимическому взгляду на природу
валентности, получившему особенно важное значение за последние годы. Как
известно, возникновение этого взгляда относится еще к началу XIX в.
и связано с именами Дэви и Берцелиуса.
Когда Вольта расположил металлы в электрохимический ряд
напряжения, а с другой стороны, были открыты первые химические
действия тока, взаимная связь между химическими и электрическими
явлениями сделалась очевидной. Естественным отсюда следствием было
отождествление электрических сил с теми, которые обусловливают
химические процессы, которые удерживают атомы в молекулах.
На такую, именно точку зрения стал в начале XIX столетия Дэви.
Ее же полнее и определеннее развил Берцелиус и положил в основу
своей знаменитой электрохимической теории.
Для Берцелиуса атом каждого химического элемента был носителем
Валентность
247
электричества, и притом одновременно двух разноименных электричеств,
сосредоточенных на двух противоположных'полюсах. Смотря по тому,
какой полюс (по величине заряда) преобладает над другим,
соответствующий элемент получает более или менее резко выраженный
электрический характер, положительный или отрицательный. В зависимости от
этого элементы и располагаются по ряду напряжения, на одном конце
которого находятся щелочные металлы, на другом — галоиды. Теперь
мы знаем, что электрохимическая -природа элемента определяется не
величиной его заряда, а свойственным ему потенциалом (разложения),
но неправильная формулировка основной посылки не помешала Бер-
целиусу, опираясь на нее, развить теорию, охватившую большую часть
известного в то время фактического материала. Следуя Дэви, Берцелиус
принимал, что .при построении молекул из атомов между последними
действуют силы исключительно электрические. Поэтому прочнейшие
соединения суть те, которые образуются через сочетание элементов с
противоположными электрохимическими свойствами. Основная черта
системы Берцелиуса может быть охарактеризована словом «дуализм»,
ибо допускаемая им двойственная (биполярная) природа атомов
должна была отражаться и на природе соединений. Смотря по тому,
электрохимическая природа какого из сочетающихся атомов оказывалась
преобладающей, получаются соединения с электроположительными
(основными) или электроотрицательными (кислотными) свойствами; известь
СаО или серная кислота SO3 (серный ангидрид с современной точки
зрения); окись натрия Na20 или азотная кислота N^Os (ангидрид). При
действии тока основание направляется поэтому к — полюсу (катоду),
кислота — к + полюсу (аноду) и там выделяется.
Когда основания сочетаются с кислотами, противоположный
электрический характер тех и других выравнивается, а потому
образующаяся соль является соединением нейтральным. Она содержит в себе
кислоту и основание в одно и то же время. На эти два тела она и
разлагается при электролизе. Формула гипса будет поэтому (принимая
современные атомные веса) СаО • S03, чилийской селитры — КоО • N2Os и т. д.
В течение более двух десятилетий теория Берцелиуса оставалась
главной идейной руководительницей химической науки, объединяя около
себя большинство химиков того времени, <но уже к концу тридцатых
годов мы .видим ее переживающей кризис. Один за другим выступают
факты, ей противоречащие, и между ними на первом плане
открытые Дюма и Лораном законы замещения, или металепсии.
Мало-помалу на смену дуализму становится унитарное учение Же-
р а р а.
Центр тяжести интересов химической науки все более и более
перемещается в область органической химии, где электрохимические
отношения отступают на задний план. Взгляды Берцелиуса надолго
обрекаются забвению. Но в этих взглядах заключалось зерно истины, а
потому им суждено было возродиться. Случилось это, однако, только в
сравнительно недавнее время.
В 1881 г. Гельмгольц в своей знаменитой фарадеевской лекции
впервые обратил внимание на глубокую связь, существующую между
представлением о валентности я законами электролиза, открытыми Ф а-
радеем..
Фарадей показал, что пропускание (одного и того же)
электрического тока через ряд электролитов влечет за собой выделение продуктов
электролиза, или, как он их назвал, и о нов в (весовых) количествах,
л.ропорциональных их химическим эквивалентам и
протекшему времени.
248
Валентность
Эквивалент серебра равен его атомному весу, -или 108; эквивалент
железа в FeC^ (закисной соли) равен половине его атомного веси
— = 28, а в FeCI3 (окисной соли)—только третьей части его: -у = 18,7.
Поэтому, если при прохождении данного тока в течение определенного
времени из раствора AgN03 выделится 108 г Ag\ то при прохождении
гого же тока в течение того же промежутка времени из раствора FeCl2
выделится 28, а из раствора FeCU— 18,7 г железа. То же самое можно
выразить еще иначе. Чтобы посредством электролиза выделить 1 р.-экв
(например 1,008 г водорода) любого иона, необходимо через раствор
соответствующего электролита пропустить строго определенное
количество электричества. Последнее может быть определено путем опыта и
составляет 96 540 кул, >или 9654 электромагнитных единиц.
Как бы мы ни смотрели на механизм электролиза, невозможно
представить себе сущность этого явления иначе, как допустив, что
противоположные электрические заряды связаны с ионами, на которые
разлагается данный электролит. На этом предположении основаны все теории
электролиза от Гротгуса до Аррениуса. Совершенно ясно, что
закон Фарадея дает нам вполне определенный ответ на вопрос о
'величине заряда, связанного с каждым данным ионом. Этот заряд
всегда является кратным одной и той же строго определенной величины, и
он во столько раз больше нее, сколько раз эквивалент данного иона
содержится в соответствующем атомном (или, общее, пайном) весе,
иначе говоря,— сколько единиц в цифре валентности данного атома
или радикала.
Отсюда, естественно, мысль,— она впервые и была высказана Гельм-
гольцем,— что само электричество, подобно весомой материи, поделено
на определенные порции, на своего рода электрические атомы; что эти
атомы, подобно атомам элементов, неделимы в условиях наших опытов
и обладают известной индивидуальностью; что они могут быть связаны
с элементарными атомами или атомными группами (радикалами), но
при этом могут повторяться не иначе, как целое число раз, причем
число это есть не что иное, как валентность данного атома или
радикала. Такой электрический атом, по предположению английского физика
Стони, получил название электрона. Нетрудно вычислить и то
количество электричества, которое отвечает такому электрону.
Если мы возьмем грамм-ион -одновалентного металла или водорода
(1,008 г Н), то, по предыдущему, количество электричества, с ним
связанное, равняется 96 540 кул, или 9-654 электромагнитных единиц. Но в
грамм-атоме (или грамм-ионе) любого элемента, очевидно, содержится
всегда одно и то же число атомов (или ионов). Это число было
определено различными способами и оказалось равным 70X Ю22.
Если мы на это число, которое в то же время выражает и число
всех электронов, связанных с грамм-ионом нашего элемента, разделим
96 540 кул, то, очевидно, получим заряд, отвечающий одному электрону.
Это будет 1,4ХЮ~20 электромагнитных единиц.
Итак, валентность данного иона равна числу электронов, с ним
связанных. Естественно, возникает вопрос о причине этого удивительного
совпадения. Чтобы осветить этот вопрос, нам необходимо будет
заглянуть в самые недра элементарного атома и спросить себя, не можем
ли мы сказать что-либо о его структуре.
Современная наука располагает средствами, позволяющими отделить
электричество от весомой материи, в обычном смысле этого слова.
Необходимо оговориться, что это относится только к электричеству
отрицатель ном у. Связь положительного электричества с материей го-
Валентность
249
раздо прочнее и до сих пор ее не удалось преодолеть. Условия для
получения «свободного» электричества создаются, между прочим, в так
называемых круксовых трубках, наполненных разреженными газами,
при прохождении через них разряда. Исследования Крукса, Лена.р а
и особенно Дж. Дж. Томсона и его школы показали, что
возникающие 'В этих условиях так называемые катодные лучи суть потоки быстро
несущихся отрицательно заряженных частиц. Эти частицы, или
корпускулы, в условиях опыта отрывающиеся от катода круксовой
трубки, наделены некоторой массой, всегда одинаковой и
составляющей около 1/1800 массы водородного атома, а.также строго
определенным по величине зарядом. Ближайшее исследование их показало, что
по существу они ничем не отличаются от тех электронов, которые,
будучи связаны с весомой материей, образуют ионы электролитов, В
частности, величина заряда для тех и других совершенно совпадает.
Электроны, вполне тождественные с ними по своей природе,
получаются и при различных других условиях, например при накаливан-ии
различных тел до высокой температуры, при действии
ультрафиолетовых лучей на металлы, при многих химических процессах <и т. д. Так
называемые р-лучи, испускаемые некоторыми радиоактивными
веществами, также представляют из себя потоки электронов, несущихся с
огромной скоростью, которая может приближаться к скорости света.
Возникновение электронов с совершенно одинаковыми свойствами
при самых различных условиях и притом из самых разнообразных
химических элементов является одним -из поразительнейших фактов,
открытых современной физикой. Обладая ничтожной массой, электроны
могут возникать только в результате распада атомов химических
элементов и потому должны входить в состав этих последних. Химический атом
является, таким образом, сложной системой, образуемой через
сочетание электронов. Но одних ли только электронов? Ответ на этот вопрос
приходится дать отрицательный. Атомы химических элементов сами по
себе- нейтральны и обладают, как известно, значительной степенью
устойчивости. Между тем, все электроны являются носителями
отрицательного, следовательно, одноименного заряда. Через их сочетание
непосредственно нельзя себе представить образование системы устойчивой
.и притом электрически нейтральной. Кроме того, оказалось, что при
распаде нейтральных атомов в разреженных газах (ионизации)
одновременно с электронами возникают и положительно заряженные
носители электричества. Об этих носителях мы уже упоминали по поводу
новейших опытов Дж. Дж. Томсона. Они представляют из себя целые
атомы или атомные группы, несущие определенное число единиц+за-
ряда, из которых каждая по величине равна электрону. Дело
происходит так, как будто нейтральный атом распадался на электроны и на
положительно заряженную часть. Последняя, как показали опыты
Томсона, а также Герке и др., является главным носителем массы
первоначального атома или молекулы (уменьшение -массы, происшедшее
при потере электрона, ничтожно).
Опираясь на подобного рода факты, Дж. Дж. Томсон и построил
овою в высшей степени остроумную теорию конструкции атомов
химических элементов *.
Согласно этой теории, в каждом атоме имеется прежде всего
«сфера положительной электризации», а затем известное число электронов,
* Некоторые лодробностк об этой теории читатель найдет в I выпуске «Новых
идей в физике», где приведен отрывок из книги Томсона «Corpuscular theory of
matter».
:250
.Валентность
распределенных внутри нее или на ее поверхности, причем заряд
положительной сферы всегда равен сумме зарядов электронов, ею
удерживаемых. Томсон допускает далее, что электроны располагаются
симметрично концентрическими кольцами или слоями, образуя различные фигуры
равновесия, характер и степень сложности которых определяют природу
данного элемента. Условия равновесия в этих системах таковы, что одни
.ато'мы легко присоединяют один или несколько электронов извне и
таким образом приобретают соответствующее число единиц
отрицательного заряда, другие, наоборот, с большей или меньшей легкостью
теряют часть своих электронов. Понятно, что при этом каждый из
последних получает соответствующее число единиц 'положительного заряда.
Таким образом, не вводя новой гипотезы касательно структуры
положительного электричества, .мы получаем возможность уяснить себе, почему
и оно также распределяется (например, в а-частицах, в ионах
электролитов и пр.) порциями, наподобие электричества отрицательного, и
притом порциями, равновеликими электрону.
Вместе с тем получается возможность весьма наглядно представить
себе самое сочетание атомов, образующих молекулу. Вообразим себе,
например, атом натрия и атом хлора. Первый по своей структуре
отличается той особенностью, что легко теряет один из своих электронов,
атом же хлора, наоборот, способен присоединить к себе электрон.
Встреча между этими двумя атомами должна поэтому иметь своим
последствием 'переход одного электрона с Na на С1. Взаимодействие между
+ и — зарядами, следуя Фарадею, представляют себе в виде силовой
трубки, начало которой считают на положительном, конец — на
отрицательном 'полюсе. Электроны, находящиеся внутри атомов натрия и
хлора, очевидно, должны быть связаны с соответствующей
положительной сферой при помощи таких силовых трубок. Понятно, что такой же
силовой трубкой будет связан с + сферой атома хлора электрон,
перешедший на него с Na. Но этот электрон, в свою очередь, не потеряет
связи и с своим «материнским» атомом — натрия, с его положительной
сферой, а потому и здесь мы должны вообразить соответствующую
силовую трубку. В результате электрон окажется в роли связующего
звена, благодаря которому осуществится сочетание атомов Na и С1 и
образование молекулы поваренной соли NaCl. Сэр В. Рамзай* уподобляет
электрон как бы особому химическому элементу, обозначая его
символом Е, между тем как Абегги Бодлендер[11Д еще в 1899 г.
(выдвинули особое понятие об электросродстве, обозначая этим словом
сродство электрона к элементарному атому (или группе атомов).
Из предыдущего ясно, что число электронов, которое способен
потерять или присоединить данный ион, иначе, число + или — зарядов, с
ним связанных, должно быть равно его валентности. Для этого стоит
только допустить, что каждому электрону соответствует своя
«элементарная» силовая трубка, и что число таких трубок, исходящих от данного
атома, равно его валентности**. Так, одноатомный натрий с
одноатомным хлором связан только одной силовой трубкой, но в молекуле воды
от кислорода исходят две такие трубки, из которых каждая
протягивается к одноатомному водороду, и т. д.
Таким образом, валентность получает определенный знак, единица
* Взгляды В. Рамзая на этот предмет составляют предмет особой статьи,
которая будет помещена в следующем (4-м) сборнике «Новых идей в химии» (1914, стр. 148.
Прим. ред.).
** В такой приблизительно форме представление о валентности развито Дж. Дж.
Томсоком и В. Рамзаем. Более сложную теорию предложил Штарк. Подробности см.
в статье этого автора («Новые идеи в хим.ии», вып. 3. 1913, стр. 123. Прим. ред.}.
валентность
251
сродства—определенное направление. Другими словами, мы
возвращаемся к тому взгляду, который (с известными ограничениями,
конечно) был господствующим во времена Дэви и Берцелиуса и новейшим
выразителем которого явился покойный Абегг. Развиваемые здесь
представления особенно близко подходят к Абегговой гипотезе о
валентностях и контрвалентностях. Из теории Томсона вытекает, между
прочим, что один и тот же атом может обладать способностью то
присоединять электроны (проявлять — валентность), то их лишаться
{проявлять + валентность), в зависимости от условий, т. е. от природы
тех или других атомов, с которыми он встречается. Притом постепенное
и единообразное усложнение состава атома (увеличение числа
электронов в кольцах) влечет за собой периодическое изменение + и
— валентности, совершенно сходное с тем, которое наблюдается в
периодической системе (сумма + и — валентности равна постоянной
величине) элементов и которое воспроизводится в представлениях,
развитых Абеггом.
Само собой разумеется, что электронная теория валентности всего
проще прилагается к случаю электролитов, где и численное значение,
и знак валентности не могут возбуждать сомнения. Однако и в
большинстве других^случаев, когда дело идет о неэлектролитах, мы не
встречаем особых затруднений.
Так, еще во времена Берцелиуса было ясно, что металлоиды
электроположительны в своих кислородных соединениях, что галоиды
электроотрицательны в своих водородистых соединениях, а также в
соединениях с углеродом, азотом, фосфором, серой и т. п. *.
Обозначая стрелками направление единиц валентности, мы
получаем, например, такие структурные формулы:
-И
н—*-а
Хлористый водород
Nr* Н
Аммиак
Хлорная кислота
//
Серный
ангидрид
Н
н
Метан
Н
Н
Серная
кислота
Четыреххлористый
углерод
* Некоторые разъяснения по этому вопросу читатель найдет в статье Р. Абегга
.(«Новые идеи в химии», вып. 3, 1913, стр. 88. Прим. ред.).
252
Валентность
В числе других преимуществ электронной теории валентности нельзя
не указать на следующие два. С электронной точки зрения в
совершенно новом свете выступает старое химическое представление о двойной
и вообще кратной связи. Если мы не хотим отказаться от важных выгод,
связанных с признанием известной степени постоянства
валентности у некоторых элементов, то допущение такой связи является
необходимостью. Не говоря уже о непредельных органических соединениях,
весьма трудно представить себе структурные формулы таких
соединений, как С02, S03, CS2, Na2C03, Саб, CuS, H2S04, KC103, КС104 и пр.,
¦не прибегая к допущению, что атомы кислорода и серы в известных
случаях связаны двумя единицами валентности с атомами других
элементов (С, S, Са, Си, С1). Химики, в свое время как бы чутьем
угадавшие эту необходимость, создали символ кратной связи. Им недоставало
только реального представления, которое бы этому символу
соответствовало. Теперь этот пробел заполнен. Двойной связи отвечают две
отдельные Фарадеевские трубки, связывающие атомы А и В между
собой. Еще недавно скептически настроенные химики относились к
представлению о двойной связи с нескрываемой иронией. Ныне один из
величайших физиков нашего времени, сэр Дж. Дж. Томсон, приводя,
например, формулу этилена
Н /Н
н>С=С\н,
говорит о возможности перехода двух электронов с одного атома
углерода на другой.
Другое важное указание, идущее со стороны электронной теории,
освещает вопрос о молекулярных, или комплексных соединениях,
гидратах и т. п. Выше мы уже неоднократно упоминали о том, что в тех
случаях, когда, согласно классической теории валентности, мы имеем уже
дело с соединениями, вполне насыщенными, последние все-таки еще не
утрачивают способности присоединять к себе новые молекулы как
однородные с ними (ассоциация), так и неоднородные. Мы видели также,
что образование таких соединений высшего порядка породило
специальные гипотезы,—из числа которых мы разобрали лишь немногие,---
гипотезы об особых остаточных, или дополнительных единицах
сродства.
При этом особенно заслуживает быть отмеченным, что многие
факты заставляют считать подобные остаточные валентности, по крайней
мере, частью лишенными электрического или, вернее, полярного
характера, как это всего яснее высказано А. Вернером.
С этими гипотезами, выросшими из недр химии и продиктованными
назревшей потребностью, интересно сопоставить следующие
рассуждения Дж. Дж. Томсона, которые мы заимствуем из его известного
сочинения «Corpuscular theory of matter».
«Весьма важно и интересно,— говорит Томсон,— исследовать
природу сил, действующих между группами корпускул, и отношение их к
образованию химических соединений.
Рассмотрим сначала силы, действующие между двумя такими
группами, в простейшем случае, когда в центре сферы положительного
заряда находится один единственный корпускул. Вообразим себе две таких
системы, во всех отношениях тождественные между собой. Пока одна из
этих систем целиком расположена вне другой, между ними нет ни
притяжения, ни отталкивания.
Валентность
253
Наоборот, если сферы будут взаимно пересекаться (как на рис. 5),
го между системами будет существовать некоторое притяжение. Чтобы
и этом убедиться, рассмотрим действие системы А на систему В. Со
стороны Л не будет никакого действия на ту часть В, которая лежит
вне А, между тем как действие на ту часть положительного
электричества В, которая находится внутри А, выразится в притяжении к центру А,
так как внутри сферы сила, происходящая от корпускула, находящегося
в центре, больше той, которая обусловливается положительным
электричеством.
(3D
Рис. 5.
Корпускулы до тех пор остаются в центрах соответствующих сфер,
пока не приблизятся друг к другу на такое расстояние, что центр одной
сферы будет находиться внутри другой сферы. Когда наступит эта
стадия, произойдет вытеснение корпускул из их первоначальных
положений, и они уже теперь не будут находиться на прямой, соединяющей
оба центра.
В этом случае нет никакой разницы между зарядом обеих сфер. Мы
не можем сказать, что одна заряжена положительно, а другая —
отрицательно, и если бы эти сферы, первоначально соединенные, были бы
вновь разобщены, то обе оказались бы нейтральными, так как
положительное электричество в каждой из них уравновесило бы отрицательный
заряд корпускула. Итак, мы видим,— заключает Томсон,— что
возможны силы электрического происхождения, связывающие между собой две
системы, из которых ни одна не обладает электрическим зарядом».
Эти соображения ценны для нас по двум причинам. Они указывают
нам, во-первых, на возможность существования «остаточного сродства»,
обусловленного электронами, не подвергающимися переносу с одного
атома на другой, а во-вторых, на то, что сродство это может быть
лишено электрически полярного характера. В то же время понятно, что
мера его должна определяться внутренней природой взаимодействующих
атомов, другими словами, их специфической природой. Не в
подобных ли условиях взаимного влияния атомов следует видеть одну из
причин пресловутой «избирательности» химического сродства?
Возвращаясь к тем же «остаточным» силам, з другом месте своей
книги Томсон говорит, между прочим, следующее:
«Сила, действующая между двумя атомами, зависит также оі от
степени легкости, с которой корпускулы могут передвигаться внутри атома,
подобно тому, как сила притяжения между двумя телами, заряженными
положительно, более значительна в том случае, когда эти тела
являются проводниками, в которых электричество может двигаться, а
следовательно, возникает возможность электростатической индукции,—
нежели в том случае, когда эти тела — изоляторы, и электричество внутри
их неподвижно.
Если данный атом не насыщен, то это значит, что еще имеется
несколько корпускулов, которые обладают довольно значительной
степенью подвижности и потому могут при благоприятных условиях
254
Валентность
проникнуть в атом или покинуть его, и, таким образом, сообщить ему
высшую валентность. Можно, следовательно, ждать, что молекула,
содержащая ненасыщенные атомы, будет действовать со значительной
силой на другие молекулы, и что потому данный газ будет показывать
отступления от закона Бойля, будет легко переходить в жидкое состояние.
Но даже в том случае, когда все атомы в молекуле насыщены, и
корпускулы, обусловливающие валентность, подверглись переносу,
корпускулы все-таки могут еще сохранить некоторую подвижность, хотя бы
она и не была достаточна для выхода их из атома.
Нет надобности, чтобы эта подвижность была одинаковой для атомов,
принадлежащих различным элементам: она может даже иметь
неодинаковую величину для одного и того же атома, в зависимости от того,
проявляет ли он положительную или отрицательную валентность.
Другими словами: нет необходимости, чтобы притяжение, обусловливаемое
атомом, было исчерпано, когда удовлетворена его валентность; притом
«остаточное притяжение» может зависеть не только от природы атома,
но также іи от того, действует ли он своими положительными или
отрицательными единицами'сродства».
Итак, с точки зрения электронной теории существует широкая
возможность представить себе возникновение остаточных сил притяжения
¦между атомами, уже насыщенными в обычном смысле слова. Томсон
рассматривает эти силы, как 'источник притяжений между молекулами,
протяжений, от которых зависит так называемое молекулярное давление
жидкостей, отступление газов от идеальных законов, поскольку таковое
определяется константой а в уравнении Ван-дер-Ваальса, и пр.
Но едва ли что-либо можно возразить против допущения, что силами
того же порядка обусловливается во многих случаях образование
комплексных и других близких к ним соединений, что в них же следует
искать ключ к объяснению некоторых явлений, наблюдаемых в области
органической химии и не получающих объяснения с точки зрения
обычного представления о валентности. Весьма вероятно, что при этом
ценные услуги окажут соображения, касающиеся способности различных
атомов заполнять пространство и сближаться между собой на
расстояния, достаточные для проявления ощутимого взаимодействия. В этом
направлении уже теперь имеется ряд интересных попыток и сближений.
Назовем только работы Друде, Траубе, Ле-Ба, Барлова, Попа
и др.
Надо думать, что в связи с 'идеями А. В?рнера о координационном
числе работам этим суждено будет сыграть важную роль в дальнейшем
развитии наших представлений о валентности.
На этом я и позволю себе закончить настоящий очерк, по
необходимости очень неполный *. Боюсь, что попытка резюмировать итоги всего
того, что было сказано выше, в виде связной и последовательной
теории валентности, представит как бы синтез различных исторически
сменявших и дополнявших друг друга течений в этом сложном вопросе,
боюсь, что такая попытка был бы ныне почти безнадежной. Можно
сказать, что время для такого синтеза успело пройти и в то же время еще
не настало. В 60-х годах валентность представлялась очень простой кон-
* В настоящем очерке речь шла почти исключительно о валентности, как о
некотором числе «ли іряде чисел, характеризующих каждый данный элемент и
определяющих собой число атомов данного элемента, сочетающихся с другими элементаім-и
в молекулах химических соединений. Наоборот, мы почти совершенно не касались
вопроса о «сродстве» и о его отношении к валентности. Некоторые разъяснения по
этому вопросу, к которому редакция надеется вернуться в одном из следующих
сборников «Новых идей в химии», читатель найдет в статье Р. Абегга. (См. «Новые идеи в
химии», вып. 3, 1913, стр. 88. Прим. ред. -сборника).
Валентность
255
цепцией, и тогда, по крайней мере, для большинства химиков такой
синтез не представлял затруднений.
Мы видим, однако, что с тех пор много изменилось. Под влиянием
наплыва новых фактов -понятие о валентности утратило свои
первоначально резкие очертания.
Валентность, одно время считавшаяся постоянной для каждого
данного элемента, оказалась величиной переменной и притом способной
изменяться под влиянием ряда различных факторов. Затем оказалось
необходимым еще далее раздвинуть рамки теории, ввести представление
об остаточном сродстве, возник целый арсенал гипотез о скрытых,
побочных, дополнительных и иных единицах валентности.
С другой стороны, была обнаружена глубокая связь валентности.
с периодической системой и с новейшей электронной теорией.
В результате в-место цельной и законченной теории валентности мы
имеем скорее ряд отдельных набросков или схем, из которых каждая
удовлетворяет известному числу фактов, но оказывается непригодной
или недостаточно разработанной для объяснения остальных.
Приведение во взаимную связь всех этих схем, не
координированных между собой и часто друг другу противоречащих, или, что, быть
может, вернее, создание новой теории валентности на более широком
основании, теории, для которой прежние попытки должны послужить
в качестве сырого материала, есть дело, полагаю, не слишком
отдаленного будущего.
В заключение я считаю .полезным сделать беглый обзор тех
различных значений, которые принимает валентность известных нам
химических элементов в различных типах образуемых ими соединений. При
этом мы будем, .насколько это возможно, придерживаться периодической
системы Д. И. Менделеева. (См. таблицы. Цифры, стоящие под знаком
каждого элемента, указывают на валентность, ему свойственную в
различных случаях; 'под каждой такой цифрой приведены для примера
формулы соответственных соединений).
Элементы, -принадлежащие к нулевой группе -периодической системы
Не Ne Аг Кг Хе Nt5
0 0 0 0 0 0,
отличаются полным отсутствием какой-либо способности вступать в
соединение с другими элементами [11]*. Их валентность поэтому всегда
равна 0. Для первых пяти элементов отношение теплоемкостеи с
близко к величине 1,67,— обстоятельство, указывающее на то, что
молекула благородных газов состоит из одного только атома.
Из числа элементов, образующих I группу периодической системы,
щелочные металлы (главная подгруппа по Л. Мейеру) в соединениях
солеобразных — окисях, их гидратах и в солях, например в LbO, КОН,
КС1 и т. п.— всегда проявляют 'положительную валентность, равную 1.
Та же валентность проявляется и в водородистых соединениях (гид-
рюрах): LiH, NaH, КИ, RbH и CsH, существование которых в
настоящее время стоит вне сомнения, а также в соединениях металлоорганиче-
ских, например в ЫаСгНб-
Аномальную валентность щелочные металлы, по-видимому,
показывают лишь в некоторых сплавах, а цезий, может быть, также в некоторых
* См. прим. ред. к стр. 194 настоящего тома. Прим. ред.
256
Валентность
соединениях с кислородом, недавно изученных Рангадом *. Валентность
тяжелых металлов I группы (побочной подгруппы Л. Мейера) варьирует
в более широких пределах, продолжая оставаться положительной **.
Для меди, наряду с соединениями, в которых валентность
нормальна и равна 1 (ряд закиси), существуют соединения ряда окиси, где она
двухвалентна; золото проявляет в одних соединениях валентность 1,
а в других оно трехвалентно. Серебро с достоверностью известно только
в одновалентном состоянии6.
Во II группе периодической системы постоянную положительную (для
солеобразных соединений) валентность = 2 показывают Be, Mg, Ca, Sr,
Ва и Ra (элементы типичных и четных рядов, главная подгруппа Л.
Мейера); притом валентность и здесь, как правило, является положительной.
То же самое с некоторыми оговорками относится и к остальным
элементам II группы.
Для ртути, однако, в солях заки-си (HgCl или Hg2Cl2) некоторые
допускают одновалентность. Имеются также неясные намеки іна
^шествование одновалентного кадмия.
Для элементов III группы наиболее характерна положительная
валентность 3, но, как это видно из таблицы (стр. 259—262), она
является не единственной***.
В IV группе периодической системы валентность отличается еще
большим разнообразием, чем в III группе. У всех элементов IV группы,
кроме свинца и церия8, преобладает в смысле прочности
соответствующих соединений валентность 4. Для четырехвалентного свинца
постоянны главным образом органические соединения. Для церия в солях
* Таковы, например, недокиси Cs70, CS7O2» существование которых установлено
посредством кривых плавкости, но молекулярный вес неизвестен (Rengade, С. R.,
148, 1199).
** В «нтерметаллических со?динениях Си, kg и Аи могут проявлять
отрицательную валентность (контрвалентность, по Абеггу). Таковы куприды, аргентиды и ауриды
Н. С. Курнакова.
*** Для бора еще Франкланд допускал пятивалентность 7, например в соединении
Н СН3
I I
В (СН3)з NH3=H-N=B-CH?.
I I
Н СН3
Недавно Шток описал два летучих водородистых соединения бора ВчНю и В6Ні2, в
которых валентность В, во всяком случае, более трех. Если допустить, что в них В
пятивалентен, что согласовалось бы с правильностью, которой подчиняется состав
высших летучих гидрюров:
№ группы III IV V VI VII
[ВН6] СН4 NH3 ОН2 С1Н,
то строение тел B4Hi0 и ВбНі2 могло бы (предположительно, конечноі) выразиться
такими формулами:
Н Н Н Н Н Н
\/ \/ \/
Н—В В Н В
Н—В В Н Х,В ВГ
н/
н н н
\
н
н
н/ \/ чн
в
/\
ы н
Валентность
257
более постоянной является трехвалентная, в гидратах окислов —
четырехвалентная форма.
В V группе валентность еще более разнообразна, чем в
предыдущих. Наиболее характерной является валентность 5 и 3. Подчиненное
значение имеют цифры 2 и 4.
По мере повышения атомного веса элемента наблюдается
уменьшение степени разнообразия валентности, причем в четных рядах (Nb, Та)
получает пребладание высшая (5), в нечетных (особенно Ві)—
низшая (3) валентность. Наибольшее разнообразие типов встречается
у азота и ванадия.
Кроме гидрюров, их металлических субститутов * и органических
производных, в своих соединениях элементы этой группы проявляют
положительную валентность. Отрицательная валентность, равная 3,
проявляется в таких соединениях, как NH3, NH2CH3, N(CH3)3, РН3, PAg3 и пр.
В соединениях аммониевого типа, например NH4C1, PH4J и т. п.,
обычно считают N, Р и т. п. пятивалентными, принимая, что четыре
валентности данного элемента отрицательны, а пягая, удерживающая кис-
лотный остаток, положительна Н—N . Вернер полагает, однако,
н/ Чі
что атомы N, Р etc. при этом проявляют валентность=3, и что частицы
НХ и т. п. удерживаются дополнительными единицами сродства.
В VI группе характерная для нее высшая (положительная)
валентность 6 встречается у всех элементов, кроме кислорода, и
соответствующие соединения часто отличаются устойчивостью. Затем характерна
валентность 4, также, по большей части, положительная; наоборот,
валентность 2 в типичнейших случаях у гидрюров и их производных — окислов,
сульфидов, селенидов, теллуридов, также в соответствующих
органических соединениях— отрицательная. Наоборот, она электроположительна
в SC12, в гипотетической сульфоксиловой кислоте S(OH)29 и пр.
Нечетные положительные валентности 3 и 5 выступают у элементов четных
рядов Сг, Mo, W, U, с особенной резкостью у хрома в солях ряда окиси
СгХз. У S имеется намек на валентность 3 в нестойком соединении S2O3
(Вебер).
У кислорода нормальная отрицательная валентность всегда равна 2,
но в нестойких, так -называемых оксониевых соединениях многие (Фри-
дель, Колли и Тикль, Байер, А. Е. Фаворский и др.)
принимают, четырехвалентный кислород10, например, в соединениях простых
эфиров и спиртов с галоидоводородами.
Л\ СН3\ /Н
(СН3)2 0< )0(
ХС1, W ХС1 и пр.
В самых широких пределах варьирует валентность индия. Наиболее постоянны все-
таки соединения трехвалентного In(InCl и 1пС12 разлагаются уже при действии воды,
например ЗіпСі = 21п + 1пС1з). Трехвалентность In, первоначально отрицавшаяся,
доказывается, между прочим, изоморфизмом образуемых им квасцов, например,
In2(S04)3 (NH4hSCV24H20, с соответствующими квасцами А!, а также изоморфизмом
самих металлов In и А1; далее нормальным молекулярным весом ацетилоацетоната
1п(С5Н702)з (по кр.иоскопическому методу в бромистом этилене) и т. п*.
Во всей III группе для одного только таллия соединения, отвечающие низшей
валентности (I), отличаются большим постоянством, нежели высшие соединения типа Т1Х*
* Заместителей, Прим. ред.
17 Л. А. Чугаі.», т. Ill
258
Валентность
Соединения аналогичных типов» но отличающиеся большей
устойчивостью, образуют S, Se и Те, например (CH3bSJ, (CH3)3SeJ и
(СНз)зТеЛ. Во всех этих соединениях, по-видимому, три единицы
валентности следует считать отрицательными, а четвертую, удерживающую
галоид или вообще кислотный остаток,— положительной.
В VII группе периодической системы галоиды прежде всего
характеризуются отрицательной (нормальной, по Абеггу) валентностью, равной
1, проявляемой в водородистых соединениях, соответствующих солях,
а также в органических производных (галоидгидратах и пр.).
В таких "соединениях, как СЬО, НС10, НВгО, HJO, JC1 (для иода),
она становится положительной. Валентность фтора, по-видимому,
всегда равна 1 (если оставить в стороне дополнительные единицы сродства,
проявляющиеся в комплексных соединениях) и всегда отрицательна.
Высшие валентности, поскольку они свойственны галоидам CI, Br, J,
всегда положительны. Наибольшее значение имеет высшая валентность
7 (не проявляющаяся у Вг), затем 5. Подчиненное значение имеет
валентность 3 и 4. Впрочем, соединения трехвалетного иода весьма
характерны (JC13 и иодониевые основания).
Марганец, занимающий в VII группе особое положение (элемент
четного 4 ряда), сходен с галоидами по своей высшей валентности,
равной 7, которая проявляется в марганцевой кислоте НМп04, в ее
солях, которые изоморфны с солями хлорной кислоты, и в ангидриде
Mn2Q7> Кроме того, у марганца и у галоидов общими являются цифры
валентности. 3 и 4. Валентность марганца во всех случаях
положительна. ,
В VIII группе периодической системы высшая валентность, равная 8,
заведомо достигается для двух элементов Os и Ru в вы-сш-их окислах
OsO* и Ru04, в недавно открытом восьмифтористом осмии [12] OsF8 и,
по-видимому, также в осмиамовой кислоте
°\0s/°
c/^NH
Когда элементы этой группы проявляют свои «главные» единицы
сродства, валентность их положительна. Элементы эти отличаются высоко
развитой способностью к проявлению остаточной валентности.
У элементов редких земель, которые одни помещают в III, другие
в IV, а иные в VIII группу периодической системы:
Церий Празеодим Неодим Самарий
Се Pr Nd Sm
Европий Гадолиний Тербий Диспрозий Гольмий
Eu Gd Tb Dy Но
Эрбий Тулий Иттербий Лютеций
Ег Tu Yb Lu
преобладающая валентность равна 3. Наряду с ней существуют
соединения высших типов, особенно типа RX4, но нельзя считать
установленным (кроме церия), что в них данный элемент обладает валентностью 4
или вообще > 3.
В последнее время, главным образом благодаря работам Гевеши.
была определена валентность почти всех известных радиоэлементов по
Валентность
259
3
<
н
СО
—
и
3
<
«
и
3
<
2
а
3
<
-Р3
О
з
<
" UCJCJ
ев
сч
О да
та га
-хх
Г
X
J? U
хххх
ей
Р) с;
та ел
-5S
hf-
<
г
и
X
о
SX
о
U
си
3"
X
о
X
о.
СУ
ее
с
а.
- О °
_0Х
"*
3
о
>4
о о
=? 3
О CJ
-N
¦ ¦ *
и о
Л з
и и
2
ей
н
U
X
CJ
>х
о
U
О)
я*
X
еС
О
X
О.
4)
С
Я
с
с
а
5 с* у о о
*** тага ^х
да да ?
СЛ
¦о
г
X
U
— с
COO
с
_? X
й см а о о
^ "> 00
ииии
X
5 ~ о-той
С с с с;
NNNN
эХ
О
а
Г
X
е?
О
X
с
01
с
группа
Сеч та та
СО
та го
ОС
5 сч У % °
w та со то
и и о
се
г
- о я
та
2
та
се
W F.
^-.і Х^
ьо ьд од од ад
Л
L?
СС
^х
« см
ИЗ ~ и Ч.о
ей
СО
CN
О О
О)
да
О)
да
U9
X
о
^
да
CQ
с» еч,
тс
X ° е'
дадададасс
17*
260
Валентность
3
О)
н
и
X
и
о
и
4»
У
X
о
о.
О)
с
со
с
с
>>
о.
U
о
U
еч
СО
u о
х: х:
Н Е-
со X
U
1-1.
со
тг
о о
н
и о
н н
о
и
С)
«
^ ¦ ^
X
ею
<
О
2
II
и
*
<L»
и
а-
о
X)
а.
OJ
*г
"¦"
с»
и
XI
а
о
.с
а
X)
а
О
XI
CU
О
а
•О
d
и
X)
а
«5
0) Ui
и и
^Г с«
U О
t- U.
N Счі
і"М
*-і С4
С
ел
V
сч
и о
с с
ел ел
¦о
X
о о У
с: с с
ел ел ел
(М
GeCl
GeO
GeS
а
¦о
х
<и а> и ф
а а о а
W5
со со
04
3
о
о
X
о
»х
о
си
у
о
X
сх.
а>
с
СО
С
с:
>>
О.
XI
^
со
ю
и
XJ
2:
•а
О
О)
X)
2
->4
СО
1С
У)
X
у о х У
ёл ел ел ел
сч
со
CI
и
>
о
.-'
t
и
•*•"
п
ел
>
О
ем
>
и
О
>
и
о
о
>
о
>
о
>
10
о
о»
ю
со
X)
ел
і/Э
со
(Л
ю
О
и х Q. г
2 2 2 X
о
2
1С
о
5
« с»
о и
Ш CQ Ш
с
со
ю
X
U X ~ 4
х> х» хі хі
ел ел ел сл
CJ
U
б
см
U
X)
ел
X
Хі X) XI
ел ел ел
со со со w
< <: < <
* х
иг° у
ьо сл to
< < <
«О X? П « S
и Ч х У.
а а а а
и
а,
Q-
О
а
00
X
—э
X
а
¦«
иЭ
X
/"I
О О X
и о и
ю
о
О ^-
2 X
X 2
Валентность
261
* о
СО
О,
(Я
« О
Г) °
со
тг _?
00 W
о о
05
с» л
«о X
rt С ? Г?
U
и о
™» ч9»
О
С*
о
- -Ох
іО иэ из
о
X
сч
2
S
о
а:
о
V
т
X
<
о
S
о.
йі
с
ев
С
с
>»
о.
и
о
Ю
СО
см
CJ X
см
О
4)
« п
со
-7 «
и о
о о
п °
О О
Он
^ Q Q
о о о
? ? ?
U О
« о я.
и. и.
и a
и
о о
о о «
••,4 О
а: и to
о
X
6
с*
X
и
СО
CN
СО
сч
оо
со
ел
Ч1
С)
О)
Н
ц-
ш
н
О
0)
Н
«
С)
си
Ь-
ел
-7 «•
а о
си ч>
(У) 00
О
« О)
Ц, 00
00 X
2
2
«
о
X
«X
О
О
0)
У
X
г*
О
X
о.
1=
CQ
С
с
о.
и
— СО СО
СО і-
,j?co
? о
со
і^их
с*
О
со
<N 00 <Л»
ГС и
о о
оо оо
и. О
ОО 00
иэ
CQ
О
и
СО
О
с
СО
О О
с с
О
6 9
С S
? X
од
со
с»
и о
с с
О ,
с4 в»
С С
и
™
СО
^«
1Л
с-
о и % _?и
X ^ ОО X
О
о
X
С!
о
о
о
X
и. —
<j X
и.
х & о
262
Валентность
3
(Л
"ОС
<
3
<
т
~о^
сч U ел О
5 5 2
эт
о
со ._?*
і?:
ТЗ'
а.
<М
со
и'-?
-а О
а, -о
а
1 '
«
о
-о
Сі
^
J3
U
X
а
со U
О.
U
I—. «
а
CN
а ^УЕ
а а
и
»х
о
:?
о
*>
X
<=(
О
X
а.
4)
С
<0
с
с
>»
о.
о о о
о
со
5 z
О " О
сч
.с
ОС
ее
ио
.с д=
si
х: х:
ОТ 0J
СО
У ^ «г
и -
а»
qj а» а» а»
U. U. Ll U.
со
со
•Q
а
и
а-
«»
О
0)
а.
м
аі
?
Л
X
О О
с* —-
а>
Lb
ш
Ц_
3
СО
Г-
00
О
3
а?
ъб
^
О
KRu
О
3
от
г*-
04
со
•^
«
^—4
U
3
ОС
0Q
RuC
,
«
а
3
21
04
^
еч
ttf
іё>
RuCI
*—*
О
3
С*
іл
О
•v
со
00
*¦
а
to
о
о
(О
о
о*
^
о
(О
«о
Уо
&<3
00
Lb
to
оо
CM
со
о о
9.0 2.
tO СО Я!
о о ^
Валентность
263
Семейство урана и радия
а Р а а а+Р а а р р а Р Р
UrI—UX- UII- Io-*Ra >Nt- RaA- RaB- RaC,- RaO — RaD -* RaF -+ RaF
6 4 642 0? 1 2\a? 2 2 2
\
RaC2
Семейство тория
а ораа а а [і
Торий -*• мезо-1 -*¦ мезо- II -*¦ радио—^торийХ-* эманация-»• торий А-»- торий В -*¦ торий Сх -*
торий торий торий тория
4 2 3(?) 4 2 0 ? 1 2
Семейство актиния
и а а fS
Радиоактиний-> актиний X -*¦ эманация актиния-»- актиний А-*- актиний В-* актиний С3
4 2 0 ? 1 2
методу диффузии. Полученные результаты сопоставлены на стр. 263,
причем радиоэлементы расположены в порядке их превращения. Под знаком
элемента указана валентность, а над стрелкой, соединяющей два
последовательно идущих продукта распада,—характер выделяемой частицы
(а или Р).
Как это было впервые указано Содди, а затем подтверждено Ге
веши, сопоставляя изменение валентности, которое сопровождает
переход одного радиоэлемента в другой, с характером выделяющейся при
этом частицы, можно заметить следующий замечательный факт.
Выделение а-частицы из атома обыкновенно изменяет валентность на две
единицы, выделение (З-частицы — на одну единицу, и притом в
противоположную сторону. Этот результат находится в очевидной связи с тем
обстоятельством, что каждая а-частица несет два положительных
заряда, тогда как каждая р-частица соответствует одному отрицательному
(электрону).
Приведенная правильность, однако, не везде выдержана. Так, ей не
подчиняются радий В, торий В и актиний В, которые все одновалентны,
тогда как для них следовало бы ожидать четырехвалентности. Гевеши
полагает, что такое противоречие происходит оттого, что мы берем
всюду валентность, проявляемую данным электролитическим ионом,
тогда как следовало бы брать максимальную валентность, отвечающую
номеру группы периодической системы, к которой относится данный
элемент. Так, свинец образует двухвалентные ионы РЬ++, тогда как
максимальная его валентность равна 4.
Несомненно, что в правиле Содди и Гевеши мы имеем дело с
обобщением огромной важности, которое обещает в ближайшем будущем
пролить свет не только на механизм процессов радиоактивного распада
и на природу радиоэлементов, но и на самую сущность валентности.
264
Валентность
ЛИТЕРАТУРА
1. J.Berzelius. Lehrbuch der Chemie. 3 Aufl., B. V. Dresden — Leipzig, 1836, S. 31.
2. E. F г а п к I a n d. Phil. Trans., 1852, 417.
3. W. Biltz. Zeit. phys. chem., 1909, 67, 561.
4. G. v. Hevesy. Phys. Z., 1913, 14, 49.
5. C.W. Blomstrand. Chemie der Jetzzeit, 1869.
6. A. Werner. Neuere Anschauungen auf dem Gebiete der anorganischen Chemie.
2 Aufl. Braunschweig, 1909. (Русский перевод 1936 г. Прим. ред.).
7. Л. А. Чу гаев. Новые идеи в химии, 2-е изд. 1914, сб. 1, стр. 103.
8. Wunderlich. Configuration organischer Molekule. Wurzburg, 1886.
9. A Werner. Beitrage zur Theorie der Affinitat und Valenz. Zurich, 1891.
10. A.Werner. Neuere Anschauungen auf dem Gebiete der anorganischen Chemie.
2 Aufl., Braunschweig, 1909. (Русский перевод 1936 г. Прим. ред.). См. также
Новые идеи в химии, 2-е изд. 1914, сб. 1, стр. 106 и след.
11. R. A beg g, G. Bodlander. Z. anorg. Chem., 1899, 20, 496. Ср. также вторую
статью настоящего сборника (Новые идеи в химии, сб. III. 1913, стр.88. Прим. ред.).
12. О. Ruff, F. W.Tschirch. Вег., 1913, 46, 929.
ПРОФЕССОР АЛЬФРЕД ВЕРНЕР
«Природа», 1914, № 7—8, 805
Профессор цюрихского университета Альфред Вернер, труды
которого по химии увенчаны в текущем году премией Нобеля"! родился
12 декабря 1866 г. в Мюльгаузене в Эльзасе.
По окончании курса в цюрихском политехникуме в 1889 г. он
поступил ассистентом к проф. Лунге, известному технологу. Около того же
времени он работал в лаборатории хи-мии и в течение одного семестра
(1891 г.)— у Бертло в Париже по термохимии.
В 1890 г. Вернер защищает докторскую диссертацию, выполненную
под руководством Ганча. В 1893 г. он делается приват-доцентом,
в 1893 г. экстраординарным, а с 1895 г.—ординарным профессором
цюрихского университета.
В своих научных работах Вернер очень рано проявил
самостоятельность мысли, крупный и оригинальный талант.
Двадцатичетырехлетним юношей, едва покончив с докторской
работой, он уже выступает с оригинальной теорией стереоизом?рии
азотистых соединений, которая сделала бы честь и более опытному химику.
Достаточно сказать, что эта теория, экспериментально подтвержденная
Ганчем и его учениками, остается общепризнанной до настоящего
времени, что ей суждено было вытеснить теоретические воззрения, которых
держался такой знаменитый химик, как покойный Виктор Мейер.
Не прошло и года после опубликования этой теории, как Вернер
выступил с новой теоретической работой, в которой содержатся важные
соображения о природе валентности. Работа эта может считаться как
бы введением ко всей дальнейшей-научной деятельности Вернера, а
глубокие мысли, в ней заложенные, далеко опередила свое время. Только
за самые последние годы их начинают оценивать -по достоинству.
С 1892 г. начинается серия тех работ, беспрерывно продолжающихся
и до настоящего времени, которые составили Вернеру репутацию
первоклассного химика, теоретика и экспериментатора в одно и то же время.
Работы эти посвящены разъяснению химической структуры
соединений, относимых по старой привычке к области неорганической, или
минеральной, химии, а из этих-соединений прежде всего тех, которые
отличаются наибольшей степенью сложности, и ныне известны под именем
комплексных.
Судьбы этой огромной работы*, в которой принимало участие
большое число учеников и сотрудников Вернера, неразрывно связаны с
оригинальной теорией, основы которой были развиты им еще в 1892 и
1893 п\
* Некоторое представление о научной продуктивности проф. Вернера дает число
опубликованных им работ с 1890 по 1909 г. Оно достигает 103. За это время 109
молодых химиков приготовили под его руководством свои докторские диссертации. И вся
эта гигантская работа была выполнена в одной из самых жалких и старых
лабораторий, которую только можно себе представить!
Только в 1909 г. цюрихский кантональный университет, уступая настойчивым
представлениям Вернера, изыскал, наконец, средства на постройку новой и, нужно сказать,
прекрасной лаборатории.
266
Профессор Альфред Вернер
Эта теория, получившая название координационной, пролила луч
яркого света на одну из самых темных и трудных для исследования
областей химии. Она в течение слишком 20 лет не переставала служить -и
самому Вернеру, и многим другим ученым надежной путеводной нитью
в их исследованиях, а в самое последнее время увенчалась блестящим
открытием неорганических соединений, вращающих плоскость
поляризации света.
Поэтому естественным образом и мы должны будем уделить
координационной теории исключительное внимание, попытаться дать о ней
некоторое понятие, насколько это, конечно, представляется возможным,
при некоторой специальности темы, в тесных рамках настоящего очерка.
Но прежде всего необходимо сказать несколько слов о химических
теориях, появление которых на сцену хронологически предшествовало
развитию теории Вернера, и с которыми эта последняя находится в
неразрывной логической связи. Это — учение о валентности -и теория
химического строения, с одной стороны, и теория электролитической
диссоциации — с другой.
Теория химического строения допускает в каждой молекуле
наличность определенного плана, определенного порядка, в котором
отдельные атомы, данную молекулу образующие, распределены друг
относительно друга.
При этом число атомов тех или иных элементов, которые могут быть
непосредственно связаны с атомом данного элемента, ограничено
известным пределом. Мы говорим, что каждому элементу принадлежит
определенная (или варьирующая в узких границах) валентность.
Химическое строение каждого соединения наглядно изображается
при помощи так называемых структурных формул, в которых
непосредственная связь между атомами выражается черточками, соединяющими
между собой знаки (символы) этих элементов, а валентность каждого
.атома — числом черточек, к нему приписанных, например,
Вода Аммиак Метан (болотный газ)
H-°\sy° ? "
н-А н-с-с-о-н
н н
Серная кислота Винный (этиловый) спирт
В этих формулах валентность кислорода = 2, азота — 3, углерода -
4, серы — 6*. Валентность водорода условно принимается= 1. Мы
говорим, что каждая «единица валентности» ил-и «сродства» одного атома
«насыщает» такую же единицу валентности другого атома, с ним
непосредственно связанного.
* Максимальная валентность, свойственная данному элементу, не всегда
достигается, а потому, например, сера, шестивалентная в одних соединениях (SOs, H2S04),
обладает валентностью = 2 в сероводороде /S и в сернистом калии /S.
H\s к\.
/о и в сернистом калии
W К'
Профессор Альфред Вернер
267
Одним из главных импульсов для создания теории строения
послужило открытие явления изомерии.
Существуют соединения, при одинаковом химическом составе
различающиеся друг от друга по свойствам. Такие соединения называют
изомерными, или изомерами, а причиной их различия считают
неодинаковое распределение атомов в их молекулах, иначе говоря, их
неодинаковое химическое строение. Явления изомерии необычайно широко
распространены среди соединений углерода (органических). Наоборот, среди
так «называемых минеральных соединений они встречаются значительно
реже.
Теория химического строения была введена в науку и разработана
з конце 50-х и в начале 60-х годов Купером, Кекуле и А. М.
Бутлеровым *. Однако уже лет через 20 она оказалась недостаточной, и для того
чтобы объяснить ряд более тонких случаев изомерии, пришлось дальше
развить идею о химической структуре. Оказалось недостаточным
говорить только о порядке распределения атомов, пришлось перенести
формулы строений в пространство, создать геометрические схемы молекул.
Так возникла стереохимия, созданная в 1874 г. Ле-Белем и Вант-Гоф-
фом *.
Теория химического строения, дополненная и расширенная стереохи-
мичестсим учением, в течение нескольких десятилетий составляла и
продолжает составлять, так сказать, идейную основу современной
органической химии. Без помощи структурно-химических представлений
последняя обратилась бы в хаотическое собрание фактов, ничем не
связанных между собой и лишенных системы. Всякий дальнейший прогресс
в изучении углеродистых соединений был бы, несомненно, парализован.
Существенно иную картину представляет нам область «минеральной
химии». Здесь структурные идеи, по крайней мере в их классической
форме, далеко не о-казались в такой же степени плодотворными, как в
области химии углерода. Наоборот, здесь несравненно больше
значения получили представления электрохимические. Этим обстоятельством
объясняют исключительно крупные результаты, достигнутые в
минеральной химии под влиянием первой по времени электрохимической
теории Берцелиуса, а в наши дни — благодаря теории электролитической
диссоциации Сванте Аррениуса.
Характерная черта теории Вернера и до известной степени залог ее
успеха заключается в том, что в ней идея о химической структуре,
надлежащим образом обобщенная, удачно сочеталась с современными
электрохимическими представлениями.
Известно, что, -по учению Аррениуса, молекулы электролитов (т. е.
кислот, оснований и солей) в водном растворе подвергаются частичному
распаду, или электролитической диссоциации на ионы, т. е. на те
самые ** составные части, на которые всякий электролит разлагается под
влиянием тока. Притом ионы отличаются от обыкновенных атомов или
атомных групп тем, что всегда несут с собой электрический заряд,
определенный по знаку и по величине ***.
* Основы ее впрочем, были еще гораздо раньше заложены Пастером (см. ниже),
но он не мог придать своим идеям конкретную форму, так как в его время не
существовало еще современного представления о химическом строении.
** При электролизе (прохождении тока) ионы, по Аррениусу, передвигаются к
соответственным полюсам, выделяются на последних и здесь теряют свои заряд, а вместе
с тем перестают быть ионами.
*** Заряд иона всегда или равен определенной (неделимой) величине— единице
чаряда так называемому электрону, «ли же является кратным последнего. Пр.и этом
всегда'число электронов (на письме обозначаемое числом знаком + и —). связанных
с данным ионом, разно валентности последнего. Так, ионы натрия и хлора несут по
268 Профессор Альфред Вернер
Один из многочисленных аргументов, приведенных Аррениусом в
пользу своей теории, заключается в том, что разбавленные водные
растворы типичных электролитов ведут себя во всех отношениях именно
таким образом, как если бы в них содержались в свободном виде
электролитические ионы. Так, ряд физических свойств таких растворов
может быть представлен как простая алгебраическая сумма двух величин,
из которых одна характерна для катиона, другая — для аниона. Таковы
электропроводность, объем, цвет и т. д.
С другой стороны, и в химическом отношении ио>ны ведут себя
совершенно независимо друг от друга. Каждый ион, присутствие которого,
согласно теории Аррениу-са, следует допустить в данном растворе, дает
о себе знать целым рядом характерных химических реакций, которые
происходят так, как если бы других ионов не существовало.
Так, раствор хлорной меди СиС12 ведет себя, как содержащий, с
одной стороны, ионы Си++, с другой — анионы С1~ *.
Присутствие первых обнаруживает, например, появление черного
осадка сернистой меди CuS при пропускании сероводородного газа
(H2S)
Си++ + 2С1— + H2S = CuS + 2С1- + 2Н++
Присутствие вторых — появлением белого осадка хлористого
серебра (AgCl) от прибавления азотносеребряной соли, или ляписа (AgN03)
Си++ 4- 2С1— + 2Ag+ + 2N07 = Си++ + 2N07 + 2AgCI-
Притом всякая медная соль, например медный купорос (CuS04)r
азотномедная соль [Си(NO^b] и др., по теории при растворении в воде
распадающаяся с образованием медных ионов Си++, неизбежно
реагирует с сероводородом так же, как и СиСЬ, а с другой стороны, всякая
хлористая соль, дающая начало образованию анионов С1~ (например,
NaCl, КС1 и пр.), реагирует с ляписом, образуя осадок хлористого
серебра. В пользу самостоятельного существования ионов** в растворе
одной единице: Na+, С1~, ион меди —две Си+ + , ион алюминия и трехвалентного
железа по 3: А1 + + +, Fe+ + + и т. д.
В самом деле, по Фарадею, весовое количество каждого *иона, выделяющегося при
электролизе, пропорционально: 1) количеству прошедшего электричества и 2)
химическому эквиваленту данного иона. Но химический эквивалент содержится в атомном
(или групповом) весе иона столько раз, сколько единиц в валентности последнего, так
что каждой единице валентности (или «единице сродства») отвечает всегда одно и то
же количество электричества. Это количество, или заряд любого одновалентного иона,
например иона водорода, может быть вычислено из следующих данных.
Для выделения 1 грамм-иона (— 1 г-атому) водорода (1,008 г Н) требуется
96 540 кул. Но в грамм-ато:.:е любого элемента содержатся 6,2 X 102] атомов, а поэтому
заряд одного иена водорода, иначе говоря, одного электрона составит
- 9654° =1,5x10-"^.
6,2х 10*
Заметим, что современная физика приписывает самостоятельное существование только
отрицательным электронам2, допускает, что только отрицательное электричество
поделено на индивидуальные порции, подобные атомам весовой материи. Что касается
до положительного электричества, то его распределение по отдельным ионам,
подчиняющееся аналогичному закону, по-видимому, есть лишь косвенное следствие
атомарного строения электричества отрицательного.
* Иногда для обозначения + и- зарядов пользуются точками и черточками, но
я в интересах наглядности предпочитаю значки -f и — .
** Самостоятельное существование ионов в растворе, однако, органичено тем
обстоятельством, что раствор, как целое (или хотя бы очень малая порция его) всегда
электрически нейтрален. Поэтому всегда алгебраическая сумма -Ьи- зарядов,
связанных с отдельными ионами, в растворе находящимися, должна быть равна нулю.
Профессор Альфред Вернер 269
говорит и то обстоятельство, что все реакции, в которых участвуют
электролитические ионы, совершаются практически моментально. Наоборот,
многие реакции между органическими соединениями, водные растворы
которых не проводят тока и потому не содержат свободных ионов,
протекают сравнительно медленно. Такова, например, реакция между
ляписом и органическим соединением, хлористым этилом (С2Н5С1):
AgN03 + С2Н5С1 - QH5N03 + AgCI.
.В этом случае хлор, в отличие от хлора, содержащегося в NaCl или
-в СиС12—свободен, и -на вступление его в реакцию требуется
заметный промежуток времени.
В мою задачу, конечно, не может входить сколько-нибудь
обстоятельное рассмотрение современных структурных теорий, так же как
и теории Аррениуса. Недостаток места поневоле заставляет меня
ограничиться сделанными беглыми замечаниями.
Прежде чем перейти к теории Вернера, мне необходимо еще сказать
несколько слов о так называемых комплексных соединениях, ибо в
применении к ним, как уже было указано, теоретические воззрения
цюрихского ученого дали особенно важные результаты.
Под именем комплексных ныне известны сложные соединения,
образующиеся через сочетание между собой целых молекул, более
простых и в то же время «законченных», или насыщенных по своему
составу.
Таковы двойные и комплексные соли, которые можно
рассматривать, как результат сочетания двух отдельных солей. Таковы сложные
кислоты и основания, которые образуются через присоединение друг
к другу двух или же большего числа кислот и оснований. Таковы
гидраты, аммиакаты и т. п. соединения, образующиеся через сочетание
воды или аммиака с солями и т. д. Приводим несколько примеров.
PtCl42KCl PtCl24NH3
Хлороплатинат калия (комплексная соль, Хлорид первого основания Рейзе
образующаяся через сочетание хлорной (аммиакат двухлористой платины)
платины и хлористого калия)
CuS045H20 A1, (S04)3K2S04 24Н20
Медный купорос (гидрат серномедной Квасцы. Гидрат двойной соли
соли) (серноалюминиевая соль -f сернокалиевля соль)
'Степень прочности, с которой связаны между собой составляющие
комплексных молекул, может быть весьма различна*. В иных случаях
связь эта настолько слаба, что нарушается с величайшей легкостью.
Многие двойные соли, например квасцы, разлагаются на свои
компоненты уже при растворении в воде; раствор их реагирует как простая
смесь серноалюминиевой и сернокалиевой солей, которые могут быть
отделены друг от друга, например, посредством диффузии. В
соответствии с этим, свойства компонентов при образовании комплексного
соединения изменяются лишь в незначительной степени.
* Первоначально термин «комплексное соединение», по предложению В. Оствальда,
применяли только к прочнейшим соединениям, при образовании которых
индивидуальные свойства и реакции компонентов совершенно утрачиваются, но ізвиду от.>гстаии
резкой границы между такими соединениями и аналогично построенными, но кпайне
непрочными веществами, ныне принято понятие о комплексных соединениях употреблять
а более широком смысле.
270
Профессор Альфред Вернер
Это обстоятельство дало шов од некоторым химикам, например Ке-
куле, высказать -предположение, что мы имеем здесь дело не с
атомическими соединениями, где все атомы данной молекулы удерживаются
друг около друга с помощью определенных единиц сродства, или
валентности, а с так называемыми «молекулярными» соединениями.
Полагали, что в этих последних отдельные молекулы лишь слабо связаны
между собой остаточными силами сродства, что каждая из числа
сочетающихся между сабой молекул сохраняет при этом свой внутренний
распорядок, свою индивидуальность. Но такая точка зрения оказалась
несостоятельной.
Наряду с малоустойчивыми комплексными соединениями
существуют тела, аналогичные с ними во многих отношениях, но обладающие
выдающейся прочностью. При образовании таких комплексов свойства
их составляющих обычно утрачиваются и заменяются новыми, точь в
точь как это бывает при образовании любого «настоящего» типичного
химического соединения из простых тел.
Таковы, например, так называемые желтая и красная синильные
соли. Первую из них можно рассматривать как двойную соль
цианистого железа Fe(CN)2 ряда закиси (Fe двухвалентно) с цианистым
калием: Fe(CN)2 • 4KCN. Равным образом -на красную соль можно
смотреть, ;как на двойную соль цианистого железа ряда окиси Fe(GN)3
(Fe трехвалентно) с тем же цианистым калием: Fe(CN)3 ¦ 3KCN. Но и
в том, (И другом случае все физические и химические свойства,
характерные для компонентов, -совершенно отсутствуют. Соли закиси железа
(РеХг) характеризуются двухвалентными ионами Fe++, соли окиси
(FeX3) —трехвалентными ионами Fe+++. Цианистый калий
распадается на ионы К+ и CN~. Каждому из ионов FeH- + , Fe+ + + и CN-
свойствен ряд характерных .признаков. Так, в присутствии двухвалентных
ионов Fe++ едкое кали дает бесцветный осадок гидрата закиси
железа, чернеющий на воздухе; присутствие трехвалентных ионов Fe^4'4"
дает о себе знать по наступлению кровлно-красного окрашивания с
роданистым калием (KSCN). Наконец, в растворе, содержащем ионы
CN", раствор ляписа дает нерастворимый осадок цианистого серебра
(AgCN). Все эти реакции исчезают после образования комплексных
желтой и красной солей. Последние ведут себя как соли
четырехосновной ферроциановой кислоты H4Fe(CN)6, трехосновной феррициановой
H3Fe(CN)6, в которых содержатся весьма прочные (изомерные)
комплексные ионы Fe(CN)6"~ и Fe(CN)6 . О химической
самостоятельности этих ионов свидетельствуют собственные, им только свойственные
реакции. Например, желтая соль с солями окиси железа дает
нерастворимый осадок берлинской лазури Fe4[Fe(CN)6]3.
Можно привести очень большое число примеров (мы еще с ними
встретимся ниже) того, как образование комплексных соединений
сопровождается утратой или глубоким изменением многих свойств
составляющих. При этом исчезают, или, как говорят, маскируются, не
только реакции ионов, но и таких молекул, как вода, аммиак и т. л.
А так как между прочнейшими .комплексными соединениями и
упомянутыми выше «двойными» солями и т. п. телами, отличающимися
крайне малой устойчивостью, на деле существует ряд незаметных переходов,
то стало ясно, что строение тех и других должно подчиняться общим
законам. Представление о молекулярных соединениях, не приложимое
к одним, не может, следовательно, годиться и для других, а потому оно
и не удержалось в науке.
Мысль, что все комплексные соединения в широком смысле слова,
или, как называет Вернер, соединения высшего порядка, следует рас-
Профессор Альфред Вернер 271
сматривать под тем же самым углом зрения, что и соединения
«атомические», далеко не новая. Задолго до Вернера ее особенно подробно
развили скандинавские химики Бломстранд и Клеве, а за ними Иергеис-
сен. Это было в шестидесятых и семидесятых годах,—следовательно,
совпало с .периодом расцвета структурной теории, а потому вполне
естественно, что этой теорией и воспользовались для того, чтобы
формулировать строение комплексных соединений. При этом оказались,
однако, необходимыми вспомогательные гипотезы. Пришлось,
например, допустить, что атомы азота, кислорода, хлора не только обладают
более высокой валентностью, нежели в NH3, Н20, НС1, но и способны
к цепеобразному сочетанию друг с другом, подобно тому, каік это
-принимается для атомов углерода в органических соединениях. Приведем
несколько образчиков тех формул, которыми пытались выразить
строение комплексных соединений.
СК XI - С1—К
СК ХС1 = С1—К
Хлороплатинат калия
СК /NH3—NH3CI
Pt
о/ \шз—nh3ci
Соль Гро
/NH3—NH3— NH3—NH3CI
Со^-ГМНз—CI
[3-
rr— 1\Л3—v^l
\NH,—CI
Хлористый лутеокобальтиак
/NH3— NH3-NH3-NH3CI
Co^-NH3—CI
\N,0—CI
Хлористый розеокобальтиак
Помимо того, что предположения, на которые опираются подобные
формулы, являются произвольными и частью маловероятными,
последовательное проведение их привело к ряду противоречий и не дало
возможности объединить ком-плексные соединения в стройную систему.
Это обстоятельство и заставило Вернера стать на более широкую
точку зрения. Для него, так же как и для скандинавских химиков, не
могло быть речи о существовании особых молекулярных соединений.
Достаточно сказать, что эпиграфом для своего известного сочинения
«Neuere Anschauungen auf den Gebiete des anorganischen Chemie» *, он
избрал следующие слова Бломстранда, относящиеся к 1869 г.: «Одной
из главных задач новейшей химии следует считать атомистическое
истолкование таких соединений, которые раньше с большей или
меньшей определенностью считались молекулярными». Но при одинаковом
понимании цели Вернер избрал несколько иные пути для ее
осуществления. Для него структурная теория в том виде, в каком она нашла себе
столь удачное применение в органической химии, была лишь
специальной формой, частным случаем более общей и широкой теории. И надо
было формулировать эту последнюю для того, чтобы вместе с химией
* Новые воззрения в области -неорганической химии (нем.). Прим. ред.
¦212
Профессор Альфред Вернер
углерода охватить и химию всех остальных элементов. Так возникла
координационная теория.
В наиболее существенных чертах она опирается на две основных
посылки: на расширенное представление о валентности, на учение о
координационном числе и о координационных сферах.
Мы видели, что в структурной теории .каждому элементу
приписывается определенная валентность, определенное число единиц сродства,
способное варьировать лишь в узких границах. Когда элементарные
атомы образуют «насыщенную» молекулу, то все единицы сродства
«затрачены» или использованы, и способность молекулы к
дальнейшему присоединению исчезает. Большое число известных фактов,
противоречащих такому допущению, заставило Вернера высказать
предположение, что кроме обыкновенных, или главных, единиц сродства есть
еще другие, побочные или дополнительные. Последние играют
особенно важную роль при образовании соединений комплексных. При их
помощи соединяются между собой насыщенные молекулы, в которых все
главные единицы валентности уже насыщены *; например,
присоединяются молекулы воды и аммиака к молекулам солей, молекулы этих
последних сочетаются между собой и т. д. Наоборот, с помощью
главных единиц валентности (сохраняя прежний смысл этого слова)
соединяются друг с другом только атомы или радикалы, способные играть
роль ионов (CI, Br, J, Na, Н, ОН, Си) или равноценные ионам
(например С2Н5, СбНб). В формулах побочные единицы сродства Вернер
обозначает пунктиром.
Второе допущение, которое делает Вернер в своей теории,
опирается прежде всего на то им же отмеченное обстоятельство, что при
сравнении между собой комплексных соединений бросается в глаза
повторение немногих основных типов, из которых один, который можно
• обозначить схемой MeR6, имеет явно доминирующее значение. Именно
число молекул воды, аммиака и пр. в молекуле комплексного
соединения, число атомов галоида, вообще кислотных остатков, входящих в
состав комплексного иона, чаще всего равно шести, реже четырем,—
и тогда мы имеем дело с типом MeR4. При этом особенно
замечательно, что природа элементов, образующих данный комплекс, и их
валентность часто не имеет здесь существенного значения.
Для примера приведем комплексные ионы:
[Fe(CN)fl]—, [Fe(CN)e] , [PtClf]—,
[Со (NH8)e]+++, [Co (CN)e] типа
MeR6; [Pt (NH3)4r, [PtCI4]~
[Pt(CN)4]— типа MeRv
Эти любопытные соотношения заставили Вернера ввести
представление о координационном числе, оказавшееся в высшей степени
плодотворным В каждой комплексной молекуле один атом предполагается
занимающим центральное, если угодно, привилегированное положение.
Вокруг этого центрального атома и в непосредственной с ним связи
располагаются остальные компоненты данной молекулы (атомы,
радикалы и целые молекулы), окружая его как бы кольцом, или, точнее,
оболочкой и образуя так называемую внутреннюю' или первую коорди-
национн\7ю сферу, за пределами которой лежит вторая или внешняя
сфера. Последняя вмещает в себя все остальные части комплексной
молекулы. Число атомов или атомных групп, которые заключаются во
* Или, точнее, соединяются между собой атомы таких насыщенных молекул.
Профессор Альфред Вернер 273
внутренней сфере, ограничено (большее число не умещается) и в
огромном большинстве случаев не превосходит шести. Это число являет-
о* ™ Предельньш' И ВеРнеР назвал его координационным, так как
он считает, что атомы и радикалы первой сферы как бы
координированы около центрального атома молекулы (рис 1)
^tTvI'JZ*T Случаи' тогда Не все 6 «^РДИнационных мест»
заняты. Если свободны два, то получается молекула с
координационным числом 4 (упомянутый выше тип MeR4). "
Чтобы наглядно представить себе
происхождение этих ч<исел, а главное, уяснить себе
причину многочисленных случаев изомерии среди
комплексных соединений» о которых речь
впереди, Вернер развивает далее следующие
пространственные (стереохимические) представ- 2
ления. Он допускает, что центральный атом
Me молекулы находится в центре некоторого
(в простейшем случае правильного) октаэдра,
по вершинам которого располагаются
отдельные атомы и группы атомов R, находящиеся
в 1-й сфере (на рис. I обозначены цифрами). Рис. \.
Таким образом, получается самая общая
модель комплексной молекулы или иона типа
МеИб- Если представить себе, что из шести координационных мест два,
отвечающие противолежащим вершинам октаэдра, не заняты, то
получается плоскостная схема *, отвечающая молекуле типа MeR4 с
координационным числом 4. Какой реальный смысл имеет разграничение
первой и второй сферы? Ответ на этот вопрос дает другая особенность
комплексных соединений, обратившая на себя внимание Вернера.
Мы уже видели на примере желтой и красной соли, что ионы, а также
целые молекулы, могут терять свое индивидуальное существование, раз
они входят в состав комплекса. Подобно ионам циана, ионы галоидов,
например хлора С1"\ вообще кислотные
остатки (N03~\ S04~) могут переходить в прочно
связанное состояние (сходное с тем, в каком,
например, хлор находится в хлористом этиле . ^
С2И5С1). Те же кислотные остатки могут, одна- #
ко, сохранять и подвижный ионный характер.
Иногда в одном и том же соединении мы нахо- 2
дим часть галоида в крепко связанном и
часть— в подвижном состоянии. В соединена рис. 2.
ях Pt(NH3)4Cl2, Co(NH3)6Cl3 клор целиком
легко подвижен и осаждается ляписом. В пурпуреосоли кобальта
Co(NH3)5Cl2*Cl только два атома хлора имеют ионный характер и
осаждаются ляписом, а один прочно связан и входит в состав
комплексного иона{Со(ШзЬС1]-н+ и т. д.
Вернер показал, что все подобные факты (а их известно очень
много) могут быть приведены в стройную систему и поняты с одной общей
ючки зрения, если допустить, что поверхность раздела между
координационными офераш совпадает с той границей, по которой молекула
расщепляется на ионы при электролитической диссоциации.
Любопытно отметить, что такая гипотеза вполне гармонирует с
современными взглядами на сущность самого процесса диссоциации
электролитов на ионы.
* На ркс. 2 для наглядности сохранены те же цифры, что и на рис. 1. Места і
и б схемы остаются в схеме 2 вакантными.
18 Л. А.. Чугаев, т. Ш
274
Профессор Альфред Вернер
Когда Аррениус создавал теорию электролитической диссоциации,
он сравнительно мало занимался ролью растворителя, вопросом,
почему именно вода (потом прибавились и другие «ионизирующие»
растворители) способна вызывать диссоциацию электролитор. на ионы и
как она ее вызывает. Но с течением времени вопросы эти стали на
очередь и сделались предметом обсуждения. Ныне большинство химиков
допускает, что молекулы растворителя, внедряясь между составными
частями молекулы электролита, оттесняют друг от друга эти
последние, образуя с ними род соединения. В результате получаются ионы
«сольватированные», т. е. окруженные как бы роем молекул
растворителя, химически-с ними связанных (ионной атмосферой). С этой
точки зрения допускаемое Вернером внедрение, например, шести молекул
аммиака между тремя атомами хлора и атомом кобальта в
(нестойком) СоСіз можно рассматривать как процесс, предшествующий
электролитической диссоциации этой молекулы и как бы подготовляющий
диссоциацию.
Согласно теории Аррениуса, способность электролитов проводите
электрический ток обусловливается исключительно присутствием
свободных ионов. Поэтому мы вправе ожидать, что комплексные
соединения, неспособные к образованию ионов, а по Вернеру это будут
такие, у которых вся молекула сосредоточена в одной первой сфере, не
будут и проводить тока. С другой стороны, величина
электропроводности, очевидно, должна (при одинаковой степени диссоциации)
увеличиваться с увеличением оібщего числа ионов, на которые
распадается молекула данного комплексного соединения. Вернер нашел, что для
соединений, имеющих характер солей и распадающихся на один
комплексный ион и на п простых одновалентных ионов (всего,
следовательно, на п+1 ионов), величина молекулярной электропроводности
зависит 'почти только от числа п, если раствор взят достаточно
разбавленный.
Так, если 1 г-мол .вещества растворена в 1000 л воды, то при 25°
величина электропроводности колеблется в следующих сравнительно
узких грани-цах:
Общее число ионов, на Молекулярная электро-
которые распределяет- проводность, в обрат-
ся молекула ных омах
2 97—109
3 231—268
4 352—427
5 523
Этот важный результат позволяет дать весьма наглядную
иллюстрацию координационной теории. Хлорная платина PtCl4 образует
следующий ряд аммиакатов с постепенно убывающим содержанием NH3 от
шести молекул до 0.
[Pt (NH,)e]Cl4l [Pt^'j CI,. [Pt<N<?>*] CIt.
523 Неизвестно3 229
[Pt(Nc^]Cl, pt<Nc«;b],
96,8 Почти 0
[Pt^'] K, [PtCI,] K2
108,5 257
Под каждым членом этого ряда (фактически из них не хватает
только одного) подписана отвечающая ему величина электропроводности.
Профессор Альфред Вврнер 27."
Как видно из написанных формул, координационное число во всем
ряду = 6. Если начать с соединении [Pt(NH3)6]Cl4, наиболее
богатого аммиаком (в нем все 6 координационных мест заняты
молекулами NH3) и распадающегося на 5 ионов, то для него мы найдем
максимальную величину электропроводности. Это— соединение,
представляющее хлористую соль комплексного основания [Pt(NH3)6] (ОН4).
кроме комплексного катиона [Pt(NH3)6]'"h, дает при диссоциации еще
4 иона хлора, находящиеся во второй сфере.
Но по мере того, как соединение это теряет одну за другой
молекулы NH3, атомы хлора постепенно переходят во внутреннюю сфер>,
6 координационных мест при этом все время остаются занятыми, а так
\Щ9
как общее число ионов, на которые диссоциирует молекула,
уменьшается, то «параллельно падает и величина электропроводности. Лучше всего
эти соотношения видны из приложенной диаграммы * (рис. 3).
Когда электропроводность падает почти до 0, мы имеем
соединение, вообще не диссоциирующее. В нем все атомы хлора как бы
затрачены на заполнение координационных мест внутренней сферы, как
этого и требует теория Веірнера.
Что же должно -произойти при потере дальнейшей молекулы
аммиака? При этом, очевидно, должно освободиться одно
координационное место, но, по смыслу теории, оно не может остаться пустым. И оно
фактически замещается при первой возможности. В рассматриваемом
случае на место уходящей молекулы NH3 становится новый атом
хлора, но входит он в молекулу не один, а в виде целой частицы КО,
причем ионы калия, естественно, помещаются во вторую сферу (не находя
себе места в первой).
В результате от солей, отвечающих комплексным основаниям, мы
переходим, через тело индифферентное, к калийной соли комплексной
кислоты, в данном случае к соли Косса {PtNH3Cl5]K. При потере
последней молекулы аммиака тот же процесс замещения аммиака хло-
* По горизонтальному направлению от-мерены (начиная справа) числа молекул
NH3, содержащихся в комплексном соединении, а по вертикальному— величины
молекулярной электропроводности (в обратных омах).
18*
Ii76
Профессор Альфред Вернер
ром -повторяется еще раз и мы получаем хлороплатинат калия, соль
кислоты H2[PtCl6].
От неэлектролита {Pt2NH3Cl4] мы, таким образом, вновь
возвращаемся к электролитам (хотя и обладающим противоположным
электрохимическим характером) с возрастающим числом -ионов. Поэтому
понятно, что электропроводность должна вновь возрасти. На диаграмме
мы получаем характерную ломаную линию с двумя ветвями —
нисходящей и восходящей, точка пересечения которых отвечает
неэлектролиту.
Рис. 4. Неэлектролит
[Pl2NH*CU
CI
Рис. 5. Катион празеосолей
кобальта [Co4NH3Cl2]-*-
Изучение электропроводности .комплексных соединений дает нам
превосходный метод, позволяющий определять «координационное
строение» комплексных соединений, иначе говоря, устанавливать
распределение по ссЬерам тех атомов и радикалов, из сочетания которых эти
•\»j
nh.
Рис. 6. Катион лутоосо.юй
кобальта [Co6NH3]3+-
соединения слагаются. Я не имею возможности останавливаться здесь
на вопросе о способе «насыщения» различных единиц валентности в
молекулах комплексных соединений, а потому ограничусь приведением
нескольких формул *, из которых сущность дела будет понятна
(рис. 4—6).
* В таких случаях, как лутеосоли кобальта, атом Со связан непосредственно
с шестью молекулами NH3 с помощью дополнительных цалентностей*. Что же касается
до главных единиц сродства (трехвалентного) атома Со, то Вернер допускает, что они
насыщаются анионами данной соли, например ионами Q-, если взять хлорид
lCo6NHs]Cl3, находящимися во внешней сфере. Допуская подобное посредственное
насыщение, он. конечно, делает новую гипотез), но не более рискованную, чем та, на
•основании которой по теории электролитической диссоциации приходится говорить о
насыщении валентностей натрия и хлора, когда ионы CI- и Na+ разделены друг от
друга слоем молекул растворителя.
Профессор Альфред Вернер
277
Одна из главных заслуг теории Вернера заключается в том что
она дала в высшей степени наглядное объяснение изомерии
неорганических комплексных соединений, которая встречается особенно часто
у соединении платины, кобальта и хрома, и число известных случаев
которой быстро увеличивается на наших глазах.
В этом отношении координационная теория, бесспорно, далеко
оставляет за собой структурно-химические представления, которых лри-
держивалисьпредшественники Вернера.
Особенный интерес представляют те из случаев изомерии
комплексных соединений, которым Вернер дает стереохимическое толкование.
Рис. 7. Соль Нейроне
Рис. 8. Хлорид 2-го
основания Рейзе
объясняя 'причину различия между изомерами неодинаковым
расположением в пространстве атомов и остатков, образующих
соответствующие комплексные молекулы.
Несколько примеров дадут понятие о принципе, на который
опирается такое объяснение. Давно известны два изомерных платиновых
соединения, отвечающих формуле PtCl2 • 2NH3. Одно из них носит
название соли Пейроне (рис. 7), другое — хлорида 2-го основания Рен-
зе (рис. 8). Они различаются друг от друга по растворимости, по
оттенку (желтого цвета) и по другим свойствам, но имеют между собой
то общее, что почти не проводят тока и что атомы хлора, в них содеп*
жащиеся, лишь с трудом переходят в состояние ионов. По теории
Вернера, следовательно, о>ба атома хлора вместе с NH3 содержатся здесь
на 1-й сфере. «Координационное» строение обоих веществ одинаково:
[Pt(NHshCl2].
Но если мы припомним, что, по Вернеру, этим соединениям следует
приписать «плоскостную» схему, то легко понять, что изомерию их
можно объяснить неодинаковым расположением атомов хлора в
пространстве, как это непосредственно ясно из рассмотрения приводимых
схем.
Другой пример мы возьмем из ряда комплексных соединений
кобальта.
Существует два соединения, состав которых выражается общей
формулой CoCl34NH3*. Одно из них зеленого, другое — фиолетового
цвета. Различаются они и по многим другим физическим и
химическим свойствам, но координационное строение их и на этот раз оди-
* Вернер, вводя гипотезу б координационных сферах, предполагает, что простые
ионы С1-, Br-, N03~, К"*", Na+ и пр., связанные с тем или другим комплексным ионом
и находящиеся по отношению к нему во внешней сфере, как бы не занимают в
молекуле определенного положения, а потому и не могут быть причиной изомерии. Отсюда
понятно, что, например, лутеосоли кобальта [Co6NH3]X3, а также пурпуреосоли
[Co5NH3Cl]X2 не могут существовать в изомерных модификациях, а равно и на то, что
для солей [Co4NH3Cl2]X возможны только два ряда геометрических изомеров, как то
объяснено в тексте. Так на самом деле и есть. Между тем структурная теория Блом-
странда и Иергенсена во всех этих случаях предвидит большое число изомерных форм,
в действительности не существующих.
278
Профессор Альфред Вернер
наковое. И -в том, и в другом случае только один из трех атомов хлора
подвижен и способен 'переходить в ионное состояние; наоборот, два
других прочно связаны с кобальтом и образуют вместе с ним
комплексный нон, или, точнее, два изомерных комплексных иона [Со ¦ 4ЫН3е12]!.
От одного иена происходит целый ряд солей зеленого цвета, так
называемые лразеосоли, от другого — соли фиолетового цвета — виолео-
голи (рис. 35). Тем и другим отвечает общая координационная
формула [Сос'ЫНзЬСЩХ, в которой X обозначает какой-нибудь
(одновалентный) анион, например С1~, Вг~, ЫОз" и пр. Здесь причина изомерии
заключается, следовательно, в неодинаковом строении комплексных
ц \
Ы ««3
Рис. 9. Ли ион прамеосолой Р-ис. 10. Лн-шж виолеосолей
катионов. И на этот раз также оно 'получает наглядное стереохммиче-
ское объяснение.
Сущность последнего станет понятной, если 'Принять во внимание,
что для данного случая координационное число кобальта равно 6, а
потому следует исходить из «полной» октаэдрической схемы.
Очевидно, можно себе представить два случая: атомы хлора могут
располагаться или на противолежащих, или на смежных вершинах октаэдра
(рис. 9, 10).
Вернер показал, что первый случай отвечает тиразеосолям,
второй — виолеосолям.
Очень большое число случаев изомерии среди комплексных
соединений платины, кобальта и хрома получило рациональное объяснение
тіри свете только 'что упомянутых стереохіи.мических представлений.
Были также разработаны приемы, позволяющие для каждого данного
случая ответить на вопрос, какой именно схеме отвечает тот или
другой изомер.
Но этим еще далеко не исчерпываются открытия Вернера в области
стереохимии неорганических соединений.
До сих пор у нас шла речь об изомерии, которая хотя и
объясняется пространственными отношениями в химической молекуле, однако не
связана с появлением так называемой оптической деятельности, т. е.
способности изомеров вращать плоскость поляризации света *.
Между тем самой интересной группой изомеров, открытых и
изученных Вернером, является именно та, представители которой обладают
статической деятельностью. Долгое время этот вид изомерии был
известен только у органических соединений, для которых он был открыт
Пасгером в 40-х и 50-х годах прошлого века.
* В поляризованном луче световые колебания совершаются только в одной
определенной плоскости. Если поляризованный луч пройдет через слой оптически
деятельного, или вращающего, вещества, то после этого световые колебания будут проходить
уже в некоторой иной плоскости. Угол, на который плоскость колебаний отклонится от
і'иоей первоначальной ориентировки, называется углом вращения.
Профессор Альфред Вернвр 27у
Ныне известно большое число нар органических соединений,
тождественных по всем своим физическим и химическим свойствам, но
отличающихся по направлению, в котором они отклоняют плоскость
поляризации света (а также по форме кристаллов). Один из двух таких
«оптических» изомеров, или антиподов, как их называют, всегда
вращает на такой же угол вправо (по часовой стрелке), на сколько другой
влево (против часовой стрелки). Смешанные в равных количествах, они
дают недеятельную, невращающую смесь, так как парализуют один
действие другого*.
Гению Пастера обязаны мы и раскрытием той основной -причины,
которой объясняется появление вращательной способности у
определенных органических соединений и обусловленная этим их свойством
изомерия. Пастер первый высказал мысль, что молекулы антиподов
должны быть построены асимметрически, должны быть геометрически
несовместимы друг с другом, как правая рука или нога несовместима
с левой, или как винт с правой нарезкой несовместим с винтом,
имеющим левую нарезку.
Но правый антипод должен быть абсолютно идентичен с
зеркальным изображением левого. Поэтому оптическую изомерию часто
называют зеркальной.
Ле Бель и Вант-Гофф лет 30 спустя перевели общее положение
Пастера на язык структурной теории и -придали ему более реальную
форму, создав представление об асимметрическом атоме углерода **.
На долю Вернера выпала честь сделать аналогичный шаг по
отношению к комплексным соединениям минеральной химии.
В момент возникновения координационной теории деятельных
комплексных соединений известно не было, и Вернер, развивая свои
теоретические взгляды, всего менее мог считаться с возможностью их
существования. Тем поразительнее, что теория, имевшая в виду
объяснение совершенно иных фактов, оказалась в состоянии предсказать как
появление оптической изомерии в одних случаях, так и невозможность
ее в других.
В упомянутых выше празео- и виолеосолях и их аналогах,
отвечающих общей координационной формуле [Co^NHaRJX, где R (и также X)
обозначает какой-либо радикал, вместо каждой пары молекул аммиака
можно ввести гпо одной молекуле органического соединения этилен-
диамина NH2—СН2—СН2—NH2," которое мы для сокращения будем
изображать символом En. И в этом случае могут быть получены два
ряда солей для каждого данного R. Если R = C1, то это будут зеленые
празеосоли и фиолетовые виолеосоли, отвечающие формуле {Со2ЕпСЬ]Х
(рис. 11, 12). Изомерия их, как и для соответственных аммиачных
соединений, объясняется стереохимически.
* Деятельные вещества встречаются в природе в живых организмах (белковые
тела, крахмал, сахар, камфора), но их можно получить и искусственно, путем разделения
их недеятельных смесей. Методы, позволяющие это сделать, были открыты Пастером.
** Так называется атом углерода, связанный с четырьмя различными атомами или
радикалами.
Среди органических соединений основной особенностью строения молекулы, с
которой связано появление оптической деятельности, и является наличность одного или
нескольких асимметрических атомов углерода. Такова, например, молочная кислота
СНЗЧ .ОН
/ \
К ХООН
280
Профессор Альфред Вернер
Построив модели, отвечающие молекулам празео- и виолеосолей,
Вернер заметил, что для вторых можно представить себе два
пространственных расположения, которые будут между собой несовместимы и
будут относиться друг к другу как предмет к своему зеркальному
изображению.
Если іверен общий критерий молекулярной асимметрии, указанный
Пастером, то мы вирав-е предвидеть здесь существование двух
оптических изомеров, или антиподов.
На самом деле Вернеру удалось разделить на антиподы и .получить
в деятельном состоянии виолеосоли (Со2ЕпСЬ]Х (рис. 13) и ряд дру-
Рис. 11. Катион пра-
зеосолей
Рис. 12. Катион вио-
леосолсГі
гих солей общей формулы [Co2EnR2]X, в которых радикалы занимают,
как это ему удалось установить, положение на омежных вершинах
октаэдра.
У 'празеосолей и у их аналогов, у которых радикалы R
расположены на противолежащих вершинах, теория не позволяет предвидеть
оптической изомерии -и, в согласии с этим, ни для одно-го соединения
данной категории расщепление на антиподы не увенчалось успехом.
Вернер подверг расщеплению на антиподы большое число
комплексных соединений кобальта, хрома, железа, родия, и во всех
случаях получилось блестящее подтверждение тех выводов и
предсказаний, которые вытекают из координационной теории.
Не подлежит сомнению, что теория Вернера имеет свои уязвимые
пункты, что в отдельных частях своих она страдает неясностью и
недоговоренностью. Но подобными же недостатками отличается и
классическая теория строения, также как и вообще большинство физико-
химических теорий. Здесь, быть может, уместнее, чем где-либо,
сказать: La critique est aisee, Tart est difficile*. И не присутствием или
отсутствием тех или иных слабых сторон, неясностей и т. п.
определяется «а первом плане научная ценность той или другой теории, а
способностью этой последней предсказывать факты, направлять
исследование по пути к новым открытиям.
Удовлетворяет ли этим требованиям теория Вернера? Всякий, кому
приходилось за последние десятилетия принимать участие в
разработке глубоко заманчивой области комплексных соединений, без сомне-
* Критика —легка, искусство —трудно (франц.). Прим. ред.
Профессор Альфред Вернер
281
ния, оценил ту незаменимую помощь, которую ему оказала эта теория,
облегчая разбор и систематизацию обширного материала фактов, на
первый взгляд нередко противоречащих друг другу.
И не одна статья, трактующая о том или ином вопросе, связанном
с комплексными соединениями, заканчивается почти стереотипной
фразой, отмечающей полное согласие наблюденных фактов с
требованиями координационной теории.
Недаром только с появлением теории Вернера химия комплексных
соединений утратила характер лабиринта или темного леса, в котором
исследователь рисковал заблудиться, и в который отваживались
входить лишь 'немногие смельчаки. Ныне в этом лесу -проложены широкие
дороги и число .путешественников быстро увеличивается.
Рис. 13. Катион виолеосолей. правый
и левый антиподы
Во мнотих случаях координационная теория оказалась в состоянии
предсказывать факты, и притом факты капитальной важности, как мы
это только что видели на примере оптически деятельных комплексных
соединений. Приведем еще другой пример, не менее поучительный.
Как было указано выше, соединения общей формулы [Co4NH3Clo]X
могут существовать в виде двух геометрически изомерных
модификаций: солей празео- и солей виолеоряда. Первые были известны давно,
но виолеосолей долго не удавалось получить. В известном споре с
Вернером Иергенсен, убежденный в то время сторонник старой теории
Блоімстранда, утверждал, что они вообще не существуют*.
Вернер, наоборот, утверждал, что они существуют и что они будут
получены. И они были получены в 1907 г., а тем самым было обеспечено
торжество идей Вернера.
В качестве одного из новейших завоеваний научной мысли,
достигнутых благодаря координационной теории, не последнее место
занимает недавно данное Вернером объяснение загадочному явлению, так
называемой «Вальденовской инверсии», сущность которого заключается
в том, что с помощью известных химических реакций можно, исходя
из правовращающего соединения +А, перейти к его левовращаю-
щ?му антиподу —А, а из этого последнего вновь вернуться к
исходному +А.
Но и за пределами чистой химии мы видим плодотворное влияние
идей Вернера. Так, в области химии биологической координационная
теория бросает свет на химическое строение столь важных для явлений
жизни пигментов, хлорофилла и гемоглобина (на способ связи магния
* Существование аналогичной изомерии в ряду этилендиаминовых производных
он объяснял особенностями этилендиамнна.
282
Профессор Альфред Вернер
в первом и железа — во втором), а в совершенно другой области —
в области химической технологии та же теория дает интересное
освещение практически важному вопросу о протравном крашении.
Теория, позволяющая 'Предсказывать неизвестное, теория,
представляющая могущественный рычаг, двигающий научное исследование,
имеет право на всеобщее признание. Неудивительно поэтому, что число
противников координационной теории, -прежде многочисленных,
непрерывно уменьшалось, что наиболее убежденные из них постепенно
склонялись перед доводами Вернера, вернее, перед фактами.
Четыре года тому назад мне 'пришлось закончить монографию,
посвященную комплексным соединениям *, следующими словами: «То,
что уже добыто в науке при содействии и под влиянием теории Вернера,
по нашему мнению, обеспечивает за ней право считаться одним из
крупнейших завоеваний, сделанных за последнее время в области
минеральной химии».
Научные события последних лет, доставившие координационной
теории новые триумфы, вполне подтверждают справедливость этой оценки.
* «О химическом строении комплексных соединений», см. т. 1 настоящего издания,
¦стр. 317. Прим. ред.
X388S*
СТРУКТУРНО-И СТЕРЕОХИМИЧЕСКИЕ ПРЕДСТАВЛЕНИЯ
В ОБЛАСТИ НЕОРГАНИЧЕСКОЙ ХИМИИ
«Новые идеи в химии», J9J4, \, 103, изд. 2
Учение о пространственном расположении атомов внутри
химической молекулы, возникшее и развившееся на почве органической химии,
за последние годы нашло себе в высшей степени плодотворное
приложение в области так называемой минеральной или неорганической химии,
к которой, по старой традиции, относятся все простые и сложные тела,
не содержащие углерода.
Замечательная попытка в этом направлении была предпринята в
1893 г. известным цюрихским ученым проф. А. Вернером, а поводом
к ней послужила назревшая потребность дать наглядное объяснение
для многих случаев изомерии, наблюдаемых среди «неорганических»
соединений.
Явления изомерии, столь обычные среди соединений углерода,
неизвестны для большинства других элементов. По этой причине теория
строения, вызванная к жизни как раз открытием изомерии,
получила лишь ограниченное приложение в химии минеральной. Правда, мы
часто выражаем структурными формулами строение неорганических
соединений, например,
0 ОН Очх °Х
%/ оЪо^он >-он
</N)H 0/ °
Но такие формулы вовсе не имеют того глубокого научного
значения, как аналогичные формулы в органической химии, и это тем более,
что справедливость их не может быть выведена и доказана с той же
-степенью строгости, как для этих последних.
Существует, однако, обширная группа соединений минеральной
химии, для которых частью уже более полувека тому назад были
открыты случаи іизомерии. Это так называемые комплексные соединения, а
между ними па первом плане соединения трех элементов: платины,
кобальта и хрома.
Именем комплексных обычно обозначают такие, по большей части
сложные соединения, которые могут быть получены путем сочетания
целых молекул химических соединений между собой. Таковы гидраты,
аммиакаты, двойные соли и пр.
PtCl, 2NH3 СоС13 4NH3 CrCI3 6NH3
P1C124NH3 CoCl35NH3 CrCl35NH3
PtCl4 2NH3 CoCl3 6NH3 CrCl3 5NH3H,0
PtCl44NH3 CoCl35NH3H20 Cr(C204)3K3
PtCl4K2 CoCl3 4NH32H20
PtCl6 K2 Co (СМ)бКз
Pt (CN)4K2
284 Структурно- и стереохимические представления в неорганической химии
По мере того, как развивались наши сведения об этого рода
соединениях, постепенно возрастало и число известных среди них случаев
изомерии, а отсюда возникла естественная потребность приложить к их
формулировке основные принципы теории строения. Попытки в этом
направлении и были сделаны Б л о м ст р а н д о м. Клеве, Иерген-
сеном и др. До последнего времени, однако, они оставались мало
успешными.
Частью под влиянием возникших на этой почве затруднений, одно
время многие химики были склонны думать, что строение комплексных
соединений вообще не представляется возможным выразить при
помощи обыкновенных структурных формул, что в соединениях этих связь,
обычно непрочная, возникает между молекулами, их образующими, а
не между атомами. Их іи называли поэтому соединениями
молекулярными, в отличие от обыкновенных или «атомных».
В пользу такой точки зрения можно привести особенно то
обстоятельство, что гидраты, двойные соли, аммиакаты и т. п. соединения
очень часто весьма легко разлагаются на свои составляющие. Такие
двойные соли как квасцы разлагаются уже пр(и одном растворении в
воде. Многие кристаллогидраты солей, а также аммиакаты, теряют
Н20 и NH3 при незначительном повышении температуры и т. д.
Подразделение химических соединений на молекулярные и атомные
не могло, однако, удержаться в науке.
С одной стороны, не подлежит сомнению, что те и другие
подчиняются одним и тем же стехиометрическим законам: закону Пру, закону
эквивалентов и кратных отношений. С другой — между комплексными
соединениями очень много таких, которые характеризуются весьма
значительной степенью прочности. Таковы, например, аммиакаты
платины, кобальта, хрома, иридия, родия, многие двойные цианистые соли
и гтр. Соединения эти лишь с большим трудом разлагаются на свои
«молекулярные» компоненты (например на соди и аммиак и пр.) и,
кроме того, во многих отношениях напоминают соединения органические,
«атомный» характер которых никогда и никем не оспаривался.
Наконец, 'между прочнейшими комплексными соединениями и теми,
степень устойчивости которых весьма незначительна, существует ряд
незаметных переходов, что особенно ясно видно в ряду двойных солей,
где представителем одного крайнего типа могут служить квасцы,
разлагающиеся (например, на A12(S04)3 и на K2SO4) уже при одном
растворении в воде, а представителями другого — такие комплексные
цианистые соли K4Fe(CN)6, K3Fe(CN)6, K2Pt(CN)4 и пр., обладающие
выдающейся прочностью, не разлагаемые ни водой, ни действием
сравнительно высокой температуры.
Если поэтому первоначальные попытки внести в область
комплексных соединений структурно-химические представления не увенчались
успехом, то причина такой неудачи едва ли кроется в совершенно
обособленном характере этих соединений. Возникает вопрос, не следует ли
ее скорее искать в том обстоятельстве, что рамки классической теории
валентности и химического строения являются недостаточно широкими
для того, чтобы вместить новую область фактов.
На эту именно точку зрения стал А. Вернер, когда он в 1893 г.
подверг коренному пересмотру ходячие структурные представления и
формулировал свою известную координационную теорию,
позволившую ему объединить очень большое число минеральных
соединений в стройную и законченную систему. В этой системе весьма важное
место отведено отношениям стереохимическим, рассмотрение
которых и составит главное содержание настоящего очерка. Прежде чем.
Структурно- и стереохимические представления в неорганической химии 285
однако, остановиться на этой стороне воззрения Вернера, необходимо
вкратце ознакомиться с основными положениями его координационной
теории.
I
При разработке своей теории Вернер остановился, прежде всего,
на двух характерных особенностях комплексных соединений.
Одна из этих особенностей, касающаяся состава этих последних,
заключается в том, что сложная молекула их может быть построена 'по
одному из немногих основных типов. Число молекул воды, аммиака и
пр., присоединяющихся к молекуле металлической соли в гидратах
и аммиакатах, число отрицательных атомов или остатков, входящих в
состав комплексных солей, имеет строго определенное значение. Чаше
других повторяется 'число 6, затем 4.
Лишь в очень немногих случаях, составляющих исключение, число
таких молекул или остатков превышает 6. Число 6 имеет, таким
образом, как бы значение предельного.
Вторая особенность комплексных соединений, положенная в
основание 'Координационной теории, заключается в том, что кислотные
остатку, входящие в состав комплексной 'молекулы CI, J, N03, NO2, S04 .и
пр., могут обладать существенно различным характером. В одних
случаях остатки эти имеют характер типичных ионов (анионов),
отличаются легкой подвижностью, склонностью к реакциям солевого обмена
Таковы кислотные остатки в так называемых лутеосолях кобальта
и хрома:
СобЫНэРз Co6NH3(N03), • (Co6NH8)o(S04):1
Сгб NH3C13 Сг 6 NH3 (N03)s (Cr 6 NH3), (SO.,).,,
в тетрамминовых солях платины
Pt4NH3Cl2, Pt4NH3(N03),.
В других случаях, наоборот, кислотные остатки прочно
удерживаются комплексной ^молекулой, в обменное разложение вступают не
моментально, как анионы солей, но сравнительно медленно, вообще
скорее напоминают атомы и остатки, связанные с углеродом в
органических соединениях, например в C2H5CI, C2H5J, C2H5NO3 и т. п.
Существуют также и смешанные типы, где в одной и той же
комплексной молекуле, наряду с легко подвижными и, следовательно,
ионизированными кислотными остатками находятся* мало подвижные и
прочно связанные. Таковы пурпуреосоли кобальта, например Co5NH:iClCb,
Co5NH3Cl(N03)2 и пр., где один атом хлора мало подвижен, а два
других (в первой из написанных солей) иліи две группы N03 (во второй) -—
имеют характер ионов. В пурпуреохлориде 1 поэтому один атом хлора
на холоду не осаждается ляписом и вообще при реакциях обменного
разложения остается в связи с кобальтом, образуя сложный радикал
(катион) Co5NH3Cl.
Вернер предполагает, что в каждой комплексной молекуле один атом
¦(чаще всего металл, например Pt, Со, Сг, Си и т. п.) занимает
центральное положение, а около него уже группируются все остальные
атомы, атомные группы и целые молекулы. Это будет центральный
атом молекулы. Число атомов, групп и т. д., непосредственно
примыкающих к центральному атому и могущих поместиться в так называемой
Внутренней или первой координационной сфере, по
286 Структурно- и стереохимические представления в неорганической химии
Вернеру, строго ограничено, и притом не зависит от способа, при помощи
которого осуществляется их связь с другими частями молекулы.
В громадном большинстве случаев о-но не превосходит б. Отсюда и
вытекает предельное значение числа 6, которое получило название
координационного.
Чтобы дать себе отчет.в происхождении этого числа, а вместе с тем
для объяснения обширной категории случаев изомерии, наблюдаемых
среди комплексных соединений, Вернер высказывает гипотезу, что
атомы и атомные группы, непосредственно окружающие центральный атом,
расположены по углам некоторого
октаэдра^ простейшем случае правильного.
Например, катион лутеосолей кобальта может быть
изображен следующей схемой (рис. 1).
Все те атомы и группы, которые не
помещаются в первой сфере и, тем не менее,
являются интегральными составными частями
сложной молекулы, помещаются во второй
или внешней сфере. Связь их с
центральным атомом может быть только
посредственной.
Подобно тому, как существуют соединения
ненасыщенные (в смысле валентности), или
соединения, в которых данный элемент не
проявляет своей максимальной валентности
(этилен наряду с этаном, РС13 наряду с РСЬ),
также возможны соединения с
координационным числом меньше 6. Их можно назвать
ненасыщенными в координационном
отношении. Чаще других повторяется при этом число 4, и это
обстоятельство объясняется, по Вернеру, таким образом, что две
противолежащие вершины октаэдра могут остаться незанятыми, так что
остаются четыре атома или группы, расположенные на плоскости
(рис.2).
Pt
Каждый отдельный атом, остаток или целая молекула,
координированные по отношению к данному центральному атому, вообще
говоря, занимают по одному координационному месту, независимо от
своей природы.
В это правило необходимо, однако, ввести следующее ограничение.
Если данная молекула или остаток двумя различными атома-
м и соединены с центральным атомом комплексной молекулы, то они
занимают не одно, а два координационных места, или, иначе говоря,
их «координационная емкость» равна 2. Два места, например,
занимает молекула этилендиамина, которая обычно и замещает две моле-
Структурно- и стереохимические представления в неорганической химии 287
со-о—
кулы NH3. Равным образом, для остатка щавелевой кислоты I
СО—О—
координационная емкость равна 2, раз он обеими своими единицами
сродства связан с центральным атомом, как, например, в комплексной
соли [Со/СгС^ЫКз-
Распределение составных частей комплексной молекулы Вернер
связывает с той второй особенностью комплексных соединений, на
которую было обращено внимание выше. Компоненты первой, или
внутренней, сферы связаны непосредственно с центральным атомом, и
притом прочно, наподобие того, как хлор связан с углеродом в
органических соединениях.
Наоборот, компоненты внешней сферы, связанные с центральным
атомом через посредство других атомов или групп, легко подвижны,
склонны к ионизации.
Поэтому кислотный остаток, раз он не способен к переходу в
ионное состояние, должен находиться во внутренней сфере. Наоборот, все
кислотные остатки, а также атомы металлов, имеющие характер ионов,
должны быть ео ipso* отнесены во внешнюю сферу. Так, в
вышеупомянутых лутеосолях кобальта и хрома во внутренней сфере (около
центрального атома Со) должны находиться шесть молекул аммиака,
образующие (волесте с Со) комплексный катион Co6NH3. В хлорпурпурео-
солях [Co5NH3Cl]X2 ** во внутренней сфере помещаются 5 молекул
аммиака и атом хлора, а два остатка X — во внешней.
Существуют и такие случаи, в которых все составные части
молекулы находятся в первой сфере: ионизированные остатки отсутствуют.
Таковы, например, соединения [Pt2NH8ClJ, [Co(NH3)3(N02)3],fPtEnCU],
[CoD2H2NH3-Cl]*** и т. п.
Естественно спросить себя, каким образом представить себе по
теории Вернера природу тех связей или единиц сродства, которые
удерживают составные части комплексной молекулы. По весьма понятным
причинам, этот вопрос приобретает особенную остроту в приложении
к таким атомным группам, которые участвуют з образовании
комплекса в качестве составных частей и в то же время представляют из
себя .самостоятельно существующие молекулы. Таковы вода, аммиак,
амины, галоидные и цианистые соединения, соли и т. п.
Вернер предполагает, что присоединение таких частиц совершается
при помощи особых побочных или дополнительных
единиц сродства, существование которых он допускает наряду с
главными. Наоборот, при помощи этих последних соединяются
между собой атомы или радикалы, обладающие свободными «сродствами»
* Этим самым (лат). Прим. ред.
** В формулах Вернер предлагает отмечать границу двух сфер, заключая
внутреннюю в прямые скобки.
СНг—СНг
*** Символ En означает этилендиамлн I J , символ D2H2 введен для
coin Но In По
/СНз—С—С—СН:і
кращенного изображения двух молекул диоксима [ II II 1 МИНУС Два
\ _ NO NOH
атома Н.
288
Структурно- и стерео химические представления в неорганической химии
в обычном смысле слова, например CI, Br, J, N03, S04, Na, К, NH4 и т. п.
Такие атомы и радикалы или сами способны перехолить в состояние
ионов или эквивалентны ионам. Вернер. впрочем, не считает, чтобы
между главными и побочными единицами сродства сушествовала
глубокая и принципиальная разница.
Сі
nhJ
f X ч
f \,
/ Ч
f Ч
Г '
^ *• у
**¦ ** ш
r^7NH3
Со'
m
Рис. 3.
Из следующих примеров будет ясно, какое значение имеет
разделение единиц сродства на главные и побочные. (Заметим, что в формулах
по предложению Вернера первые обозначают сплошной чертой,
вторые— пунктиром):
NH34 XI
Pt
NH.
Из того, что было сказано выше о распределении составных частей
комплексной молекулы по координационным сферам, нетрудно понять,
каким образом можно, опираясь на превращения данного комплексного
соединения, вывести его координационное строение, написать его
координационную формулу. Яснее всего это будет видно на примере.
При действии аммиака на двухлористую платину PtCb образуется
(в зависимости от условий) несколько различных соединений, из
которых мы остановимся здесь только на следующих четырех. Одно из этих
соединений содержит 4 молекулы аммиака PtCl24NH3, три остальных —
вдвое меньше и, следовательно, отвечают одной и той же эмпирической
формуле PtCI22NH3.
^Соединение PtCl24NH3, так называемый хлорид первого основания
Рейзе, легко реагирует с ляписом, причем оба атома хлора замещаются
остатками азотной кислоты:
PtCl24NH3+2AgN03 = Pt (N03)24NH3 + 2AgCl.
Подобным же образом с влажной окисью серебра оно дает AgCl и
свободное основание Pt(OH)24NH3, представляющее сильную щелочь.
Хлор можно заместить также бромом, иодом, остатком серной кислоты
Структурно- и стереохимические представления ь неорганической химии 289
и т. п. Отсюда ясно, что оба атома хлора в соли Рейзе носят характер
ионов (анионов) и что группа Pt4NH3 представляет из себя
двухвалентный катион Pt4NH3, а потому координационная фор'мула хлорида
первого основания Рейзе будет {Pt4NH3]Cl2, формула "соответствующей
азотнокислой соли [Pt4NH3](N03)2, гидрата (Pt4NH3](OH)2 и т. д.
Из трех других упомянутых соединений два, довольно похожие друг
на друга (мало растворимые тела желтого цвета), трудно и медленно
реагируют с ляписом и вообще мало склонны к реакциям обменного
разложения. Очевидно, атомы хлора в них не ионизированы (или с
трудом переходят в состояние конов), и потому, по Вернеру, им следует
приписать координационное строение [Pt2NH3Cl2]; другими словами,
все составные части молекулы здесь помещаются во внутренней сфере,
в непосредственной связи с атомом платины. Вещества эти, однако,
различны во 'многих отношениях (по кристаллической форме,
растворимости, реакциям) ді получаются различным образом. Одно из них,
носящее название хлорида второго основания Рейзе, образуется при
отщеплении двух молекул NH3 от хлорида [Pt4NH3]Cl2 (при нагревании
последнего или при действии на него соляной кислоты). Другое,
носящее название соли Пейроне, получается при действии аммиака (в
сравнительно небольшом количестве) на раствор PtCl2 в соляной кислоте,
или же на двойную соль PtCl22KCl (хлороплатинит калия, см. ниже).
Оба эти соединения при действии избытка аммиака дают хлорид
первого основания Рейзе. Все говорит в пользу того, что они обладают
одинаковым 'молекулярным весом и, следовательно, изомерны. Мы увидим,
что их изомерия по Вернеру получает стереохимическое объяснение.
Наконец, последнее из интересующих нас платиново-а-ммиачных
соединений, отличающееся очень трудной растворимостью в воде и
зеленым цветом, носит название соли Магнуса. Судя по эмпирической
формуле, оно изомерно с двумя предыдущими, но легко показать, что
оно обладает вдвое более сложной молекулой.
Хлористый калий и другие хлористые металлы (NaCl, RbCl, MgCl2,
также NR;C1) легко соединяются с двухлористой платиной, давая
соединения типа PtCl2-2MeCl или PtCl2-MeCl2 и т. д.
Нетрудно показать, что при этом образуются соли некоторой
двухосновной комплексной кислоты, которой принадлежит координационная
формула H2[PtCl4]. В этой кислоте платина входит в состав аниона
PtCL, а водород играет роль катиона. Вышеприведенные «двойные»
соли двухлористой платины или хлороилатиниты должны быть
рассматриваемыми, как соли этой комплексной кислоты:
K2lPtCl4],Na,[PtCl4], MglPtCIJ.
Од,ин из весьма ярких фактов, говорящих в пользу такого взгляда,
заключается в том, что при действии ляписа на хлороплатиниты
вместо AgCl образуется серебряная соль той же комплексной кислоты
в виде нерастворимого осадка сёмож-но-красного цвета:
К2 [PtCl4] + 2AgN03 - Ag, |Pt Cl4] -г 2KNO;,.
Все хлороплатиниты дают эту характерную реакцию, и потому она
может служить для распознавания солей кислоты H2[PtCl.,"|. Мы
воспользуемся ею для уяснения структуры соли Магнуса.
Если эту последнюю подвергнуть действию ляписа, то моментально
зеленый цвет переходит в сёможно-красный, а исследование продуктов
превращения показывает, что оно выражается таким уравнением:
(PtCl22NH3)2 + 2AgN03 - Ag2[PtClt] -г [Pt 4NH:i] (NO:i),.
19 Л. А. Чугасв, т. III
290 Структурно- и стереохимические представления в неорганической химии
Ясно поэтому, что соль Магнуса есть продукт сочетания двух
комплексных ионов: РсС14 и Pt4NH3, что она есть хлороплатинит первого
основания Рейзе: [Pt4NH3]PtCl4. Верность такого заключения
подтверждается ее синтезом. Если слить бесцветный раствор соли первого основания
Рейзе с красным раствором какого-либо хлороплатинита, то немедленно
получается обильный осадок зеленой соли Магнуса:
[Pt4NH3] С12 + [PtCl4] К2 - [Pt4NH3] [PtCl4] -j- 2KC1.
Некоторые физ.ические свойства комплексных соединений
позволяют не менее наглядно характеризовать их координационное строение.
Как известно, по теории Аррениуса, электролитическая
проводимость солей (равно как оснований и кислот) обусловливается
ионами, на которые диссоциирует молекула электролита. Поэтому мы
вправе ожидать: 1) что комплексные соединения, составные части которых
целиком содержатся во внутренней сфере, не будут проводить
электрического тока и 2) что электропроводность комплексных соединений,
способных к ионизации, должна правильно изменяться вместе с числом
ионов, которые освобождаются пр;и диссоциации их молекул. На этом
принципе Вернер основал остроумный способ определения строения
комплексных соединений и имеете с тем наглядную иллюстрацию
координационной теории. Вместе со своими сотрудниками Миолати и Герти,
Вернер показал, что, если мы имеем какое-нибудь (достаточно
устойчивое) металлоаммиачное соединение или комплексную соль типа
+ - +
RX или RMe, где R — комплексный катион, R — комплексный анион,
X (не комплексный)— одновалентный кислотный остаток, a Me —
атом одноатомного металла или группа аммония и т. п., то можно
установить следующие средние цифры, около которых колеблется величина
соответствующей молекулярной электропроводности, причем каждая из
этих цифр соответствует числу ионов, на которые диссоциирует частица
данного комплекса (измерения относятся к температуре 25° и к
растворам, содержащим 1 г-моль вещества в 1000 л раствора).
Число ионов, на кото- Молекулярная злек-
рыс распадается моле- троироводкость, и об-
кула ратных омах
2 97—109
3 231—268
4 352—427
5 523
Очевидно, что по этим цифрам можно также хорошо определить-
координационное строение данной комплексной молекулы, как и с
помощью чисто химического метода, о котором шла речь выше.
Чтобы показать, насколько тесно связаны величины
электропроводности с химической природой комплексных соединений и числом ионов,
ими освобождаемых, мы остановимся на рассмотрении другого ряда,,
образуемого также соединениями платины, но только четырехатомной.
Pt (NH3)6
Cl4
(NH3)51
Pt
CI
CI я
(NH3)41
Pt
CI
Неизвестно2
(NH3)3
Pt
CI,
—
CI
(NH,)2~
Pt
ci4 J
Г (NH3)]
Pt
CI,
К
PtCl6
Структурно- и стереохимические представления в неорганической химии 291
В этих соединениях четыреххлористая (хлорная) платина PtCl4
сочетается с различными числами молекул аммиака (от 0 до 6). Как
видно из формулы, в данном случае дело идет о соединениях, в которых
координационное число платины равно 6.
Весьма характерное отношение этих соединений всего лучше
выступает, если мы прибегнем к помощи диаграммы (рис. 4), причем по оси
абсцисс будем откладывать число молекул NH3, а по оси ординат —
величины молекулярной электропроводности соответствующих
комплексных соединений при 25°, выраженные в обратных омах.
Рис. 4. Диаграмма
По мере того, как хлорид [Pt6NH3]Cl4, наиболее богатый аммиаком
и отвечающий четырехатомному основанию [Pt6NH3](OH)4, постепенно
одну за другой теряет молекулы NH3 и под конец переходит в
калийную соль кислоты HztPtCU], замечается поразительное изменение в
свойствах образующихся при этом промежуточных соединений.
Число атомов хлора, способных к ионизации и, следовательно,
находящихся во второй сфере, постепенно падает параллельно с падением
электропроводности. Замечательно, что при этом все 6
координационных мест остаются занятыми: на месте каждого уходящего аммиака
становится хлор, в полном соответствии с теоретическими
представлениями Вернера.
Когда электропроводность падает почти до 0, мы имеем соединение,
вовсе лишенное способности освобождать ионы хлора: теперь все атомы
хлора как бы израсходованы на заполнение внутренней сферы: они
встали на места отщепившихся молекул аммиака. Спрашивается, что
же должно наступить дальше, когда удалится еще молекула NH3?
Координационное место не может остаться пустым, и на самом деле, мы
видим, что- отщепление аммиака сопровождается теперь вступлением
в комплексную молекулу новой частицы (не атома, как раньше) КС1
или же NaCl, НС1 и т. п. При этом хлор становится на место ушедшего
аммиака, а металл (или водород) —во внешнюю сферу. Получается
уже не соль комплексного основания, а соль комплексной кислоты
HfPtNHaCls]. Параллельно с этим вновь нарастает электропроводность,
доходя до величины, близкой к 100.
19*
292 Структурно- и стере о химические представления в неорганической химии
Совершенно подобным же образом потеря последней молекулы а-м-
мнака приводит к образованию соли K^PtCU], т. е. хлороплатината
калия (вообще соли двухосновной хлороплатиновой кислоты H2[PtCl6]).
Как уже было упомянуто выше, еще до Вернера было сделано
несколько попыток дать рациональную формулировку комплексных
соединений с точки зрения теории строения. Наиболее замечательная
попытка этого рода принадлежит шведскому химику Бломстранду. Она
получила затем важное развитие со стороны Клеве и особенно Иёр-
генсена. Поучительно сопоставить взгляды этих авторов с теорией
Вернера. Пр(и этом преимущества последней выступят с достаточной
очевидностью.
Иёргенеен, частью следуя в этом отношении за своими
предшественниками, делает несколько вспомогательных гипотез, при помощи
которых получается возможность до некоторой степени примирить
особенности комплексных соединений с основными положениями
структурной химии. Из этих гипотез три имеют особенно важное значение.
1. Принимается, что некоторые элементы, участвующие в построении
молекул комплексных соединений, проявляют при этом высшую,
частью не совсем обычную для них, атомность. В частности,
допускается, что галоиды могут быть трех-, кислород — четырехатомным.
2. Принимается, что атомы хлора в двойных хлористых солях,
атомы азота —в аммиакатах, кислорода — в гидратах, обладают
способностью образовать цепеобразные сочетания наподобие атомов углерода
в органических соединениях, например
СК /С1 =С1—К /Н.О—С1
>Р<Г Со^-Шз—NH3—NHS— NHS—С1
CV XC1=CI—К \NH3—CI
Хлороплатинат калия Хлористый рспеокобальтиак
/NHs-Cl
Cr^-NH3—NH,—NH3— NH, -CI
\NH,-CI
Хлористый лутеохромиак
3. Принимается, что, когда атомы галоидов и вообще кислотные
остатки непосредственно связаны с металлом (Pt, Со, Сг и т. д.), то они
мало подвижны, не способны к ионизации; если же они связаны с
атомом металла через посредство молекул аммиака, аминов, воды,
сульфидов и т. п., то они находятся в подвижном состоянии л легко
подвергаются ионизации.
Не входя здесь в обсуждение того, насколько эти предположения
можно считать правдоподобными с общей точки зрения, посмотрим,
как они прилагаются к разрешению конкретных вопросов. Мы сделаем
это на примере ряда комплексных кобальтиаков, для которых
параллельно выпишем и координационные формулы.
В приведенном ряду мы замечаем соотношения, совершенно
аналогичные тем, которые мы констатировали выше для соединений
четырехатомной платины. По мере уменьшения числа молекул NH3,
приходящихся на 1 атом кобальта, постепенно <и здесь падает число
остатков X, способных переходить в состояние анионов. Когда число частиц
NH3 делается равным 3, мы наблюдаем как бы перелом в характере
вещества в кривой электропроводности. При б, 5 и 4 молекулах NH3
мы имеем дело с солью комплексного основания, при числе молекул
NH:j менее 3 — с солью комплексной кислоты. При трех молекулах,
Структурно- и стереохимические представления в неорганической химии 293
мы имеем тело индифферентное, не образующее ионов и почти не
проводящее тока.
Вес эти соотношения, непосредственно вытекающие из опыта,
находят себе наглядное выражение в координационных формулах Вернера.
Мы наблюдаем здесь, как и в выше разобранных комплексах платины.
как бы тенденцию к сохранению координационного числа 6. Этим
объясняется и постепенное ослабление ионизации вещества по мере
удаления NH3, и необходимость присоединения некоторой новой молекулы
(KN02), когда число это спускается ниже 3.
Переходя к формулам Иёргенсена, мы видим, что для трех первых
соединений, наиболее богатых аммиаком, они дают такое же
отчетливое объяснение фактов, как ,н формулы Вернера. Но уже в следующем
члене ряда формула Иёргенсена содержит один остаток N02, способный
к ионизации, что не согласуется с фактами. Точно так же совершенно
непонятно, почему соединение Co2NH3(N02h не только* не обладает
характером соли основания Со2МНз(Ж)2Ь(ОН), но и вообще-не способно
к самостоятельному существованию.
Только что отмеченные противоречия, равно как и большое число
друічх, аналогичных, не могли быть устранены Иёргеисеном, несмотря
на сделанные им в этом направлении попытки.
Входить в дальнейшие детали, сюда относящиеся,, едва ли было бы
уместно в этом кратком очерке. Впрочем, к критике воззрений
Иёргенсена * мы еще будем иметь случай вернуться при обсуждении стерео-
химических взглядов А. Вернера.
II
Среди довольно уже большого числа известных случаев изомерии
неорганических соединений Вернер различает несколько типов, из
которых для нас в настоящую минуту наибольший интерес представляет
тот, которому автор координационной теории дает стереохимические
объяснение. Но прежде чем перейти к этому типу изомерии, скажем
несколько слов о других.
Среди органических соединений мы различаем следующие виды
изомерии: 1) полимерию, 2) метамерию, 3) изомерию в тесном
смысле этого слова и 4) ** различные виды стереохимической
изомерии.
В сущности под те же самые категории імогут быть подведены и
известные случаи изомерии комплексных соединений минеральной химии.
Типичным примером полимерии может служить разобранный
нами выше случай соли Магнуса, с одной стороны, и соли Пейроне (или
2-го основания Рейзе), с другой:
[Pt4NH3]PtCl4 [Pt2NH.,Cl2|
Соль Магнуса обладает вдвое более сложной молекулой по
сравнению с оолью Пейроне, подобно тому как альдоль является лимером,
а паральдегид — тримером обыкновенного альдегида.
Случаям метамерии органических соединений, например
СН3СООС4Н9 и С2НбСООС3Н7, соответствуют среди комплексных со-
* Подробности, касающиеся критического обзора теории Вернера и Иёргенсена,
см. Л. А. Чу га ев. О химическом строении комплексных соединений. СПб.. 1910
(Л. А. Чу га ев. Избр. труды, т. 1, стр. 216. Прим. ред.)
** Таутомерия, в сущности, не представляет особого типа изомерии, так как
отличается от обыкновенной изомерии лишь по легкости взаимного превращения
изомерных форм.
294 Структурно- и стереохимические представления в неорганической химии
По ИСргенсеиу
1.
2
3.
¦і.
5.
NH3-X *
/
Со—NH.,—NH3—NH8—NH, —X
\
NH3—X
Лутеосоли
N02
/
Со—NH3-NH3—NH3—NH3—X
\
NH3—X
Ксантосоли
NO,
/
Со—NH3-NH3—NH3—NH3—X
\
N02
Кроцеосоли
N02
/
Co—NH3-NH3—NH3-N02
\
NO,
Триаминкобальтинитрит
NO 2
/
Co-NH3-NH3-N02-K
N02
Соль Эрдмана
1 Co—NH3—NH3—NO. ?
V N02 /
По Л. Борнсру
[Co6NH3] X:{
|Co5NH3(N02)]X2
|Co4NH3(N02)2]X
[Co3NH3(N02):J]
[Co2NH3(N02)4]K
Общее число
ионов, на которио
распадаются
молекулы
¦\
•}
0
• 1
* X —кислотный остаток, например CI, Br, J, N03, SCN и пр.
единений случаи, объединенные Вернером в категорию
координационной изо мер,и и. Сущность различия здесь сводится к
неодинаковому распределению компонентов комплексной молекулы по
координационным сферам.
Таковы, например, следующие пары солей:
С1
Со (NH3)4
N02
С1 и
Со
(NH3)4
CL>
N02,
где разница сводится к неодинаковому размещению атома хлора и
группы N02, или
[Cr(En]3] [Co(CN)6] и [Со(Еп)3] [Cr(CN)6],
в которых 3 молекулы зтилендиамина (для сокращения — Еп)
связаны в одном изомере с Сг, а в другом — с Со и т. д.
Структурно- и стереохимические представления в неорганической химии 295
Известная изомерия между фиолетовыми и зелеными солями
хрома относится в ту же категорию, как видно из следующих примеров:
[Сг(Н20)б]С13
Хлорный хром.
Фиолетовый
гидрат. Вводном
растворе все 3
атома хлора
ионизированы
Сг
№0),
сі
СіоНоО
(Н20)4
Сг
С1Н.0
Хлорный хром. Зеле- Хлорный хром.
ный гидрат Вернера Зеленый гидрат,
и Губсера. Только 2 Только 1 атом хлора
атома хлора легко легко ионизируется
переходят в состояние
ионов
Наконец, среди комплексных соединений наблюдаются и случаи
настоящей изомерии. Ниже мы остановимся на одном примере этого
рода, наиболее типическом и близко напоминающем по своеіму
характеру изомерию азотистых эфиров и нитросоединений. В соединениях,
в которых связь группы N02 осуществляется с Со через азот, она
отличается 'прочностью, как в нитросоединениях; наоборот, когда между
N и Со стоит кислород, связь легко рушится, и пріи действии кислот
выделяется HN02.
Обратимся теперь к тем случаям изомерии, которым Вернер, в
согласии с координационной теорией, дает стереохимическое
объяснение.
Простейший случай этого рода імы имеем в ряду соединений
двухатомной платины. Я говорю: простейший, потому что здесь число
остатков, помещающихся в первой координационной сфере, in maximo
равно 4 (соответственно координационному числу 4).
Давно уже известно, что соединения состава Pt2aX2, где под а
можно подразумевать молекулу аммиака, какого-либо амина, тиоэфира
R2S ц т. п., а под X—'кислотные остатки, существуют в двух
изомерных модификациях, которые различаются друг от друга по цвету,
растворимости и по химическим превращениям. Таковы, например,
упомянутые выше соли Пейроне и второго основания Рейзе, отвечающие
общей молекулярной формуле (Pt2NH3Cl2).
Сопоставим в качестве примера некоторые свойства этих двух
изомеров.
Соль Пейроне
Получается при действии NH3 на соли НгРКЗЦ:
(NH4)2 [PtCl4] + 2NH3 = [Pt2NH3Cl2] +- 2NH4Ci.
Кристаллический порошок оранжево-желтого цвета, 1 вес. часть
растворяется в 26 частях воды при 100° и в 387 частях при 0°.
Хлорид 2-го основания Рейзе
Получается при отщеплении NH3 из соли первого основания Рейзе:
[Pt4NH3]Cl, = [Pt2NH3Cl2] + 2NH3.
Микроскопические октаэдры 'бледно-желтого цвета. Одна вес. часть
растворяется в 160 частях воды при 100° и 4472 частях при 0°.
При действии пиридина оба вещества дают различные, но притом
изомерные между со-бой соединения, отвечающие общей
координационной формуле [Pt2NH32Py]Cl2. (см. ниже).
296 Структурно- и стерео химические представления в неорганической химии
Было сделано несколько попыток выразить структурными
формулами сущность этой изимерии. Согласно Бломстранду и Иёргенсену,
.строение солей Пейроне и Рейзе 2-го может быть -представлено
следующими схемами:
/NH3NH,C1 /NH3CI
Pt< Pt<
ХС1 XNH3C1
причем, однако, мнения ученых, придерживающихся такого взгляда на
строение изомерных ллатиниакатов, несогласны относительно того,
какая формула принадлежит соли Пейроне и какая соли Рейзе. Уже
этого разногласия, казалось бы, достаточно для того, чтобы усомниться
в обоснованности точки зрения Бломстранда и Иёргенсена. К этому
присоединяется еще следующее обстоятельство. Мы уже видели, что
общей обоим изомерам чертой является их ничтожная
электропроводность и малая ,подвижность в них галоида. Но вышенаписанные
формулы как раз не дают обтэяснений этой особенности, присущей солям
Пейроне 'и Рейзе. Наоборот, как уже неоднократно было указано, с точки
зрения координационной теория вполне понятно, почему атомы хлора
в этих соединениях не склонны к переходу в состояние ионов.
Самую изомерию солей Пейроне и Рейзе Вернер объясняет, исходя
из плоскостей схемы, которая, по его представлениям, должна отвечать
координационному числу -1. Легко видеть, что здесь по теории возможны
два и только два пространственных .изомера, как это видно из
следующих схем (рис. 5).
Чтобы сделать выбор между обеими формулами, Вернер отпирается
на следующие, факты, установленные Клеве иИёргенсеном.
Если на соль Пейроне подействовать пиридином * (в водном
растворе), то две молекулы его присоединяются іпо уравнению:
[Pt2NH3a>] + 2Ру - ^NH^PyjCU. (1)
То же самое соединение (обозначим его для краткости 'Через а)
получается при действии аммиака на пиридиновое соединение [Pt2PyCl2].
По своему происхождению и по свойствам оно втіолне напоминает соль
Пейроне:
[Pt2PyCl2] + 2NH3 = [Pt2Py2NH3] CI, (2)
Итак, (l) = (2)=a.
Если теперь подействовать -пиридином на (изомерную) соль 2-го
основания Рейзе, то .произойдет совершенно аналогичная реакция:
присоединятся 2 молекулы Ру, но образуется уже другое соединение,
изомерное соединению a: {Pt2Py2NH3]Cl2, мы обозначим его через |3.
То же самое соединение (3 получается при действии аммиака на
изомер соединения [Pt2PyCl2], отвечающий соли 2-го основания Рейзе и
образующийся через отщепление двух молекул пиридина из хлорида
[Pt4Py]Cl2.
* Пиридин
СИ
НС^^СН
мы для сокращения будем обозначать символом Ру.
НС
^
v/
сн
N
Структурно- и ствреохимические представления в неорганической химии 297
Итак можно получать два изомерных соединения, отвечающих
одной и тон же координационной формуле [Pt2NH32Py]Cl2: аир. Опыт
показывает, что они неодинаково относятся к высокой температуре и к
действию НС1.
Рис 5.
При этой реакции, .которая, как общее правило, от соединения типа
[Pt4a]X2 приводит -к соединению типа [Pt2aX2], а изомер реагирует по
уравнению:
[Pt2Py2NH3Cl2] - Ру ~ NH, -f iPtPyNH3Cl2],
давая начало смешанному соединению, содержащему одновременно
NH3 и Ру.
В аналогичных условиях |3 изомер дает смесь двух веществ,
аммиачного и пиркдинного комплекса:
2 [Pt2Py2NH,] - [Pt2NH3Cl2] + [Pt2PyCl2] + 2NH3 -r 2Py.
Нетрудно показать, что все эти реакции только в том случае
получат удовлетворительное объяснение, если выбор между двумя
возможными формулами для солей Пейрове и 2-го основания Рейзе сделать
следующим образом:
><
NH/ ХС1
Соль Пейроне
NH3,
Pt
/ N
СК 4NH3
Соль 2-го основания
Рейзе
Аналогичный выбор придется, конечно, в таком случае сделать и
для соответствующих пиридиновых комплексов.
Допустим на минуту, что такое предположение отвечает
действительности, и посмотрим, каким образом при помощи его объясняются выше-
рассмотренные реакции. Остановимся, -прежде всего, на реакциях
присоединения молекул NH3 н Ру, сделав при этом то простейшее и
наиболее естественное допущение, что О и NH3 (соотв. С1 и Ру) просто
меняются координационными местами:
.298 Структурно- и стереохимические представления н неорганической химии
NH
NH,''
'У.
Pt
С
С1
+ 2РУ -
NH1%
NH/
Pt
,Р* + 2NH,
Py''' NCI
.-Ру
\Py
С L
(a)
NH.
PyN ^.4
ч *
ч *
X
Py/ \NH,
a
(4)
Идентичные
продукты-а
NH
з. CI
/\ + 2Py
CI/ HN3
Py.
Pt
Cl
CI
/ \-
NH,
Py/
ч *
4 *
Pt
0 4
/ 4.
Py
4NH.
a
(O
+ 2NH,=
Py.
ч
^
NH/
W
0
0
\ 0
4 0
Pt
¦' \
\Py
Cl,
(d)
Идентичные
продукты-р
Из рассмотрения этих схем мы непосредственно заключаем, в
полном согласии с данными опыта, что: 1) а = Ь = а, 2) c = d=§ и 3) что
аир должны отличаться друг от друга.
Перейдем теперь .к рассмотрению процесса отщепления NH3 и Ру.
Отщепление молекул Л, аммиака, аминов и т. тт. из 'Соединений типа
[PtA4]X2 ведет к образованию продуктов, во многих отношениях
.представляющих между собой черты близкого сходства. Это будут аналоги
солей 2-го основания Рейзе, а потому мы должны всем им приписать
одну и ту же общую конфигурацию, а именно — транс:
А\ Л
\ /
Pt4
В частности, и отщепление аммиака и пиридина из наших
смешанных соединений, очевидно, должно приводить к веществам той же транс-
конфигурации, т. е. оно должно совершаться, так сказать, наперекрест.
Но стоит только взглянуть на конфигурацию соединений а и 8
Структурно- и стереохимические представления в неорганической химии ЯН)
.РУ
Pt
кнУ \ру
а
NH
Зч
»;
,Ру
Ру/ \NH3
Сі
чтобы стало ясно, что отщепление NH3 и Ру в только что указанном
порядке из соединения а как раз должно 'привести к образованию
смешанного соединения [PtNH3 • РуС13], между тем как соединение 3
должно быть при этом смесь двух тел: [РігРуСЬ] и [Р12ЫНзС12],
т. е. должно быть именно так, как наблюдается в действительности.
Теория Вернера приводит, таким образом, к
вполне определенному выбору между двумя
возможными конфигурациями для солей Пейроне
и Рейзе. Нетрудно убедиться в том, что
обратное предположение неизбежно привело бы к
противоречиям *.
Перейдем теперь к рассмотрению случаев
изомерии более сложных, в которых дело идет о
соединениях с координационным числом 6.
Примеров такого рода изомерии особенно много мы
встретим среди соединений кобальта и хрома.
Существует два ряда изомерных комплексных
кобальтиаков, отвечающих одному и тому же
координационному строению [Co4NH3(N02)2]X, но отличающихся друг от
друга по свойствам: цвету, растворимости, химическим превращениям.
Соли одного ряда носят название кроцеосолей, соли другого
называются ф л а в осо л я м и **.
В обоих рядах необходимо признать существование одинаковым
образом построенных, но притом изомерных катионов [Co4NH3(N02hL
составные части которых, согласно предыдущему (стр. 000) должны
разместиться во внутренней сфере по вершинам некоторого октаэдра,
которые мы отметим цифрами (рис. 6). Вернер объясняет эту изомерию,
допуская, что в кроцеооолях остатки NO2 располагаются на
противолежащих вершинах октаэдров -(1 и 6), тогда-как на флавосолях они
занимают смежное положение (1 и 2). Первое-положение обозначают
символом транс, второе — цис (рис. 7).
В противоположность Вернеру, Иёргенсен высказал предположение,
что изомерия кроцео- и флавосолей, а также некоторые другие
аналогичные случаи, обязаны своей ^причиной неодинаковому строению
группы N02. Но Вернер без труда мог доказать с помощью метода, о кото-
рам было упомянуто выше, неправильность такого взгляда. Строение
нитрогрушш в кроцео- и флавосолях оказалось одинаковым.
Известно большое число других комплексных соединений кобальта
и хрома, по составу молекулы напоминающих кроцео- и флавосоли.
* Более подробную критику взглядов Вернера и других авторов на сущность
изомерии платиновых соединений см. в цитированном уже сочинении Л. А. Чугаевэ
«О химическом строении комплексных соединений». СПб., 1910. Там же ом. и
литературу предмета. (См. Л. А. Чу га ев. Избр. труды, т. I, стр. 216. Прим. ред.)
** См. прим. ред. к стр. 285 настоящего тома.
300 Структурно- и стереохимические представления в неорганической химии
Для многих из них удалось наблюдать подобного же рода изомерию,,
которая и получает со-вершенно .аналогичное объяснение*.
Интересный случай такого рода изомерии наблюдается для
соединений кобальта (и хрома), в которых аммиак замещен этилендиамином.
¦г NH3
Рис. 7. Анион кроиеосолей(транс) Анион флавосолей (пне)
(Нп). Как мы видели .выше, при этом в смысле .координационной
емкости двум молекулам NH3 соответствует 1 молекула этилендиамина, а
потому соединениям тетрамминового ряда [Со4ЫНзХ2]Х соответствуют
/г^
6
Л
Рис. 8.
диэтилендиаминовые комплексы: [Со • 2ЕпХ2]Х. Особенно интересен
случай -изомерии дихлоросолей [Со2ЕпСЩХ, для которых известно два
изомерных ряда: п р а з ео с о л и — зеленого цвета и виолеосоли —
фиолетового цвета. Соли эти легко .переходят друг в друга. Изучение
реакций солевого обмена, а также их электропроводности привело
«заключению, что они обладают одним и тем же -координационным
строением, а потому Вернер дал их изомерии стереохимичеокое толкование,
которое выражается схемами, представленными на рис. 8.
* Замечательно, что для солей лутеоряда [Co6NH3]X3 и хлорпурпуреоряда
[Co5NH3Cl]X2 теория Вернера не предвидит возможности изомерии и фактически
таковой до сих пор в этих рядах не наблюдалось. Наоборот, по Иёргенсену здесь
следует ожидать большого числа изомеров.
Структурно- и стереохимические представления в неорганической химии 301
Иёргенсен в этом случае противопоставил схемам Вернера другое
объяснение, оказавшееся, однако, несостоятельным. Он допускал, что
изомерия празео- и виолеосолей зависит от присутствия в комплексной
молекуле этилендиамина, и думал, что замена 2Ёп четырьмя
молекулами NH3 устраняет возможность появления двух изомерных форм.
Долгое время, действительно, для дихлоротетрамминкобальтиаковых солей
-[Co4NH3Cl2]X был известен один только именно празеоряд.
Представители этого ряда обладают зеленым цветом, подобно празеосолям
этилендиамина, которые они и напоминают во многих отношениях,
отличаясь от них только неспособностью переходить в соответствующую вио-
леомодификацию. Однако четыре года тому назад Вернеру удалось
приготовить и соли виолеоряда [Co4NH3Cl2]X.
Это открытие явилось блестящим подтверждением целесообразности
стереохимических представлений, связанных с координационной
теорией. Воззрения Иёргенсена были тем самым окончательно
опровергнуты .
Последовательное проведение в жизнь стереохимических воззрений
Вернера привело его к необходимости найти приемы, которые бы
позволяли путем опыта определять конфигурации стереоизомерных
комплексных соединений.
Мы видели, каким образом вопрос этот решается для изомерных
соединений платины. Существенно -иной метод привел Вернера и его
учеников (из них особенно Пфейффера) к решению аналогичной
задачи в ряду изомерных комплексов кобальта и хрома. Они
воспользовались для этой цели приемом, совершенно аналогичным тому, с
помощью .которого в органической химии определяют конфигурацию
стереоизомерных соединений этиленового ряда.
Общеизвестно, что одно из главных оснований, заставляющих
приписать малеиновой кислоте конфигурацию цис, а фумаровой —
конфигурацию транс, заключается в том, что только первая из этих кислот
способна, и притом весьма легко, терять воду и переходить в
соответствующий ангидрид
Н—С—СООН Н—С—СО
| ^ZZ || V)+H20.
Н-С-СООН Н—С-СОУ
из которого она вновь легко получается путем гидратации.
Очевидно, что для определения конфигурации можно
воспользоваться как первой, так и второй реакцией, ибо, если процесс отщепления
воды должен легко совершаться только в том случае, когда Н и ОН
занимают цис-положеН'Ие по отношению друг к другу, то и обратно,
процесс присоединения Н и ОН к циклическому ангидриду должен
привести нормальным путем к кислоте, отвечающей конфигурации цис.
Понятно, что ;во .всех тех случаях, когда какой-либо радикал или
целая молекула занимает во внутренней сфере 2 -координационных
места, должна возникнуть циклическая группировка, как это видно,
например, из следующих схем, представляющих способ связи атомов Со
и Сг с молекулой этилендиамина и с остатками щавелевой (оксалато)
и угольной (карбонато) кислот:
CHo-NH* СО-Оч /Оч
> с°<0>-
СН2—NH/ СО-0
>Со
302 Структурно- и стереохимические представления в неорганической химии
Рассматривая стереохимические модели Вернера (рис. 8), легко
убедиться в том, что образования подобных кольчатых группировок
возможно ожидать в том только случае (схема I), когда двухвалентный
остаток обоими своими атомами, связанными с металлом (Со или Сг),
занимает положение икс (1, 2), а .не транс (1, 6 — схема II).
Поэтому, если мы имеем комплексное соединение, в -котором
существуют циклические группировки, то вопрос о конфигурации его -можно
считать до некоторой степени предрешенным. (В качестве иллюстрации
см., например, схемы этилен диа,миновых соединений кобальта на
рис. 9).
Сі
Рис. 9.
Катчкм виож'осолей (шіс). Юігиои празеосолсй {транс)
В пользу такого взгляда нужно, между прочим, привести то
обстоятельство, что для соединений типа [Со4МН3СОз]Х, [Co4NH3(C204)]X
и т. д., а также [PtEnCl2] не известно случаев изомерии. Самая
возможность появления изомеров, следовательно, исчезает, как только два
остатка (например, 2С1, 2Вг, 2N02 или же 2NH3) замещаются одним с
двойной координационной емкостью, дающей начало образованию
цикла.
Представим себе теперь, что произошло замещение нашего
двухвалентного остатка, образующего цикл, двумя остатками одновалентными,
причем каждый будет занимать лишь одно координационное место.
Представим себе, например, что на место оксалатогруппы (остатка
С2О4 щавелевой кислоты) или на место карбонатогруппы СОз станет
2 атома галоида, две нитрогруппы и т. д. Нетрудно понять, что при
нормальном течении реакции задіестители должны занять .в октаэдрической
схеме те же места (1, 2), которые раньше были заняты двухвалентным
остатком, и притом на тех же самых основаниях, по которым малеино-
вой кислоте, возникающей при действии воды на малеиновый ангидрид,
мы приписываем цис-, а не транс-конфигурации. Можно пойти и
дальше: если мы имеем в распоряжении соединение [Co4AY2]X, в котором
стереохимическое положение двух заместителей Y установлено,
согласно предыдущему, то можно путем нового замещения -ввести на место Y
другие остатки Z и таким образом установить конфигурацию нового
соединения [Cc4AZ2]X, генетически связав его с исходным циклическим;
соединением через посредство промежуточного звена—[Co4AY2]X.
Структурно- и стереохимические представления в неорганической химии 303
На самом деле, пользуясь подобного рода переходами, Вернеру и
Пфейфферу удалось установить конфигурацию большого числа
комплексных соединений кобальта и хрома. При этом карбонатосоли типа
[МеС034А]Х
оказались особенно пригодными в качестве опорных соединений*.
Приведем несколько примеров.
При действии на карбонатоэгилендиаминовые и аммиачные соли
ряда [Со4г\тНзСОз]Х разведенными кислотами легко заместить остаток
СОз двумя молекулами воды:
НЮ
[Co4NH3C03]X + 2НХ+Н20-
Co4NH:i
Хя+ СО,
НаО
Карбонатосоли Диаквосоль
При этом образуются соответствующие диа.квосоли, в которых, по
предыдущему, молекулы воды должны находиться в (1, 2) -положении
друг относительно друга. Соли того же ряда были ліолучены действием
разведенных кислот и на другие циклические соединения аналогичного
характера. Поэтому строение их может считаться твердо
установленным.
НХ>
Действуя на 1,2-диаквосоли ряда
Co4NH
той, легко перейти к динитритосолям
H2OJ
NO,
Co4NH.
NO,
X;, азотистой кисло-
X, неустойчивым и
легко разлагаемым кислотами. В этих солях поэтому N02-группа должна
иметь строение —О—N0 и должна быть связана с Со через кислород
Со—О—N0 (см. выше). По своему происхождению от 1,2-диаквосолей ди-
нитритосоли должны также отвечать конфигурации цис.
Этот вывод находит себе подтверждение в том обстоятельстве, что
при действии кислот последние вновь переходят в исходные
1,2-диаквосоли.
При хранении 1,2-динитритосоли претерпевают изомерное
превращение и обращаются в устойчивые по отношению к кислотам динитросо-
ли, которые оказались тождественными солями кроцеоряда
[Co2En(N02h]X, получившими свое название по аналогии с кроцеосо-
ля.ми, происходящими от аммиака (см. выше). Устойчивость -кроцеосо-
лей указывает на то обстоятельство, что в них группа нитро имеет стро-
• ^€ и связана с Со через азот, а их непосредственное об-
Х0
ение
разование из соответствующих 1,2-динитросолей заставляет и им
приписать конфигурацию транс. Все это можно представить
следующими схемами:
"(1) Н20 1 [(l)ONO Г(1)02Ы
Со(Еп)2 ХзТ! Со(Еп)2 Х- Со(Еп)2
(2) Н20 J [_(2)0N0 J (2)02N
1,2-диаквосоли 1,2-динитритосоли 1,2-динитросоли
X
* При этом, однако необходимо еще в большей степени, нежели в области'
органической химии, считаться с возможностью перегруппировок, с изомеризацией.
304 Структурно- и стереохимические представления в неорганической химии
С другой стороны, Вернеру удалось получить ряд диаквосолей,
изомерных с предыдущим/В этих новых солях положение молекул
воды должно, следовательно, отвечать тину транс (1, 6). Путем
превращений, совершенно аналогичных только что описанным, Вернеру удалось
перейти от 1,6-диа:К'восолей к соответствующим динитрито-, а от этих
последних—к динитросолям. Эти последние оказались идентичными с
кроцеосолями этилендиамина, которые были получены иным путем
много раньше. Таким образом могла быть установлена .конфигурация
другого стереоизомерного ряда, все члены которого ясно отличаются
по своим свойствам от ряда цис (1, 2):
(1) НоО
Со (Еп).,
(6) Н20
Л. о <;
(1) ONO
Со (Еп)о
(6) ONO
X
(1) O..N
Со (Еп).,
(6) O..N
X
1,6-диаквосоли 1,С-динитритосо;ш 1,6-динитросоли
Мы не будем здесь останавливаться на других многочисленных
случаях, для которых могла быть аналогичным путем установлена стерео-
химическая конфигурация отдельных комплексных соединений и
взаимная между ними связь. Но один случай заслуживает особого 'внимания,
и ¦потому мы коснемся его в немногих словах.
Выше было упомянуто, что Иёргенсен, Вер.нер и другие
исследователи комплексных соединений кобальта долго, но безуспешно пытались
¦получить для солей дихлоротетрамминового ряда {Co4NH3Cl2]X второй
ряд, изомерный давно известным кроцеосоля-м.
Путь, который привел Вернера к их открытию, в высшей степени
знаменателен как раз для оіценки того метода определения
конфигурации, который мы только что рассмотрели. Наряду с простейшими ко-
бальтиаками, в молекуле или, вернее, в отдельных ^ионах которых
содержится всегда один лишь атом кобальта., существуют и такие,
которым следует приписать и которым давно уже приписывали 'более
сложное строение. Химическая природа таких кобальтиаков была недавно
весьма обстоятельно выяснена Вернером, причем оказалось, что все они
построены по .некоторому общему плану, раскрытие которого и
позволило объединить их .в одну естественную группу.
Соединения эти характеризуются своеобразным сочетанием двух или
же большего числа атомов кобальта, связанных между собой, часто
весьма прочно особыми остатками или «мостками», как их зовет Вер-
нер, число которых может доходить до 6. В роли таких мостков могут
фигурировать атомные группы: ОН, NH, NH2, 02 и др.
Соединения подобного рода Вернер называет многоядерными.
Строение их * устанавливается путем изучения как условий их
образования, так и, в особенности, превращений, которым они подвергаются
при действии реагентов, например кислот.
Каждая формула многоядерного комплекса, построенная согласно
только что приведенному принципу, должна удовлетворять двум
требованиям: сохранению определенной валентности и неизменного
координационного числа (для кобальта — 6).
Одним из простейших примеров двуядерных кобальтиаков может
служить ряд ** декаммин-ц-амииодикобальтовых солей,
* A. Werner. Lieb. Ann., 1910, 375, 1.
** Группы, образующие связь между отдельными атомами Со {и других металлов,
образующих многоядёрные комплексы), Вернер обозначает буквой \х (meso); «оло-
йыми» он обозначает комплексы, в которых ро'ль мостка играет гидроксил.
Структурно- и стереохимические представления в неорганической химии
305
которым Вернер дает такое строение:
[NH3)5Co-NH,-Co (NH8)51 X5.
В солях этого .ряда два атома кобальта связаны амидной группой и
образуют одну сложную координационную сферу, один комплексный
ион. Общая'валентность такого иона равна 6—1=5 (одна единица
сродства затрачивается на насыщение NH2-rpynnbi). Вместе с тем, каждый
атом кобальта сохраняет свое собственное координационное число 6.
В октамминдиоловом ряду
,ОН>
(NH3)4Co Co(NH3)4
юн-
х4
атомы кобальта связаны двумя гидроксилами, что и отражается (до
сравнению с предыдущим рядом) 1) на понижении числа остатков X,
находящем оправдание в необходимости удовлетворить требованиям
валентности (.введение одноатомной группы ОН) и 2) .на понижении
числа молекул аммиака с 10 на 8, что объясняется необходимостью
сохранения координационного числа б.
Вполне укладываются в ту же систему и соединения более сложные.
Например, дикобальгиаки с тремя связующими звеньями:
ОН
(NH,),Co OHCo(NH3)3
ОН
NH2
(NH3)3Co OHCo(NH3)8
ОН
X;
X,
Гексамминтриолдйко-
бальтовый ряд
Гексаммин-р,-амино-
диолдикобальтовый ряд
Чтобы показать, хотя бы на одноім примере, каким образом могуг
быть выведены эти формулы, рассмотрим реакцию, происходящую при
действии соляной кислоты на соли гексамминтриолдикобальтового ряда:
ОН
(NH3)3CoOHCo(NH3),
ОН
Сіз+ЗНСЫОэ (Шз)з С13Н
+ [Со(Шз)з(НоО)з]С13.
Продуктами реакции являются две соли, координационное строение
которых хорошо известно, а сопоставляя это строение с условиями их
образования и с составом исходного вещества, мы без труда
определяем и структуру этого (Последнего.
Как 'видно из приведенных примеров, среди многоядерных
комплексов существуют циклические, и даже таких большинство.
Очевидно, что на эти соединения следует перенести все то, что было
сказано выше о конфигурации комплексных соединений кольчатого
строения. Другими словами, многоядерными комплексами можно
пользоваться для установки конфигураций таких соединений, которые могли
бы от них произойти 'Путем размыкания кольца.
20 Л. А. Чугаев, т. Ill
ЗОВ Структурно- и стереохимияеские представления в неорганической химии
Изучение одного из подобных случаев и привело Вернера к
открытию ряда виолеосолей среди тетрамминовых кобальтиаков. Виолеосолп
были получены при действии соляной кислоты на хлорид октамминдио-
ло'вого ряда:
ОН
(NH3)4 Co(NH,)4Co
ОН
С11Г2НС1 -[NH,)4CoCU|C
-j-[Co(NH3)4(HtO)2]Cl3.
В этом случае октаммиидиоловый кобальтиак прямо расщепляется на
виолеосоль, конфигурация которой непосредственно вытекает из того
способа ее образования, и на 1,2-диаквотетрамминокобальтовую соль,
о которой уже была речь выше. Достойно внимания, что Вернер на
основании своей теории мог предвидеть не только существование, но и
конфигурацию виолеосол-и гораздо раньше, чем она была лолучена,
а когда удалось ее -приготовить, то способ получения как раз подтвер-
Рис. 10.
дил высказанное предположение и заставил приписать ей
конфигурацию цн с.
В последнее время стереохимические представления Вернера
-получили еще одно в высшей степени замечательное подтверждение и
развитие.
Изучение пространственных моделей, .построенных для комплексных
соединений кобальта <и хрома, заставляет прийти к заключению, что во
многих случаях для соединений этих следует ожидать .появления
оптической изомерии, ибо соответствующие модели-могут существовать
в двух несовместимых, или энантиоморфных модификациях.
Если взгляды, высказанные Вернером, (правильны, или, по крайней
мере, отражают в себе некоторую долю истины, то мы вправе ожидать,
что для определенных кобальтиакоп и хромиаков удастся осуществить
разделение на оптически деятельные антиподы.
Летом 1911 г. Вернер опубликовал первые наблюдения, блестящим
образом подтверждающие такое суждение для некоторых комплексных
соединений кобальта.
С тех пор прошло немного времени, а число рядов комплексных
соединений, для которых удалось (воспроизвести расщепление на
антиподы, достигло внушительной цифры 23. В этом числе получены
деятельные соединения хрома, железа и родия (рис. 10).
Структурно- и стереохимические представления в неорганической химии 307
В этих соединениях отсутствуют органические радикалы
асимметрического типа с готовой оптической деятельностью, и причиной
наблюдаемого отклонения плоскости «поляризованного луча может 'быть только
асимметрическая группировка в пространстве тех атомов или групп,
которые, согласно представлениям Вернера, окружают центральный атом
металла в комплексной молекуле или в комплексном ионе.
Если допустить вместе с Вернером, что атомы и группы,
непосредственно связанные с центральным атомом трехатомного -кобальта или
хрома, в соответствующих комплексных соединениях располагаются -по
Рис. II.
вершинам некоторого октаэдра, то появление оптической изомерии
можно предвидеть, прежде всего, для соединений, содержащих радикал,
отвечающий общей схеме
Me
D,B
при условии следующего взаимного
расположения в нем заместителей (рис. 11).
Но для появления молекулярной асимметрии нет надобности, чтобы
группы С и D отличались друг от друга. Достаточно, чтобы они
обладали строго определенной пространственной op-иентировкой. Такая
ориентировка осуществляется, например, когда системе CD отвечает одна
молекула этилендиамина (см. рис. 11). Нетрудно представить себе, что
А " А
соединения ряда [Со 2Еп ]Хп (или вообще [Me 2Еп]Хп) с 1,2 поло-
В В
жением А и В, может дать начало двум энантиоморфным
конфигурациям. -При этом группы А и В могут быть или молекулами аммиака,
воды, какой-либо соли, или кислотными остатками CI, Br, N02 и пр.
Первые опыты, которыми Вернеру удалось подтвердить эти
предвидимые теорией случаи оптической изомерии, относятся к комплексным
соединениям кобальта и именно к тому .случаю, когда из двух групп
А и В одна имеет характер кислотного- остатка, связанного с атомом Со
посредством главной единицы сродства; в качестве же другой группы
фигурирует частица аммиака. К этому типу относятся соли хлороаммин-
С1
диэтиленового ряда [Со 2Еп]Х и бромоамминдиэтиленового ряда
NH3
ICo
Br
NH,
2En]X.
201
308 Структурно- и стереохимические представления в неорганической химии
Разделение их на антиподы было осуществлено п.ри 'Посредстве бром-
камферсульфонатов *, отличающихся неодинаковой растворимостью в
воде (d-бромкамферсульфонат d-комплексного основания в обоих
-случаях труднее растворим, нежели соответствующая соль 1-го основания).
Очищенные таким образом камферсульфонаты лепко могли быть затем
переведены посредством обменного разложения в оптически деятельные
бромиды, нитраты и другие соли.
В этих солях, очевидно, источник оптической деятельности может
заключаться только в асимметрическом расположении атомов и групп
около -центрального атома кобальта.
Приведем величины удельного вращения для красного света,
отвечающего фрауэнгО'феровой линии С.
Вг.>
С1
Со 2 En
NH3
[<*]с
Br.,
-+4:
Br
Со 2 En
NH:!
3° +46°
Соединения эти неожиданно оказались обладающими довольно
значительной степенью прочности. Между тем как оптически деятельные
органические основания, содержащие асимметрическую серу, селен,
олово, уже при хранении растворов весьма легко подвергаются
рацемизации с потерей вращательной -способности, у солей обоих только что
упомянутых оснований вращение в этих условиях сохраняется и не
исчезает даже при кратковременном кипячении растворов. В оптически
деятельной бром осоли ['СоВгЫНз * 2Еп]Хг можно даже заместить бром
молекулой воды и перейти к акзосоли с трехатомным катионом
[СоН20 • NH32En], и все же инактивиро-вания не происходит.
Совершенно анологичные случаи изомерии наблюдаются, когда обе
А
лтомных группы А и В в радикале [Ме2Еп ] имеют характер кислот-
В
ных остатков. Вернеру удалось осуществить разделение на антиподы
Со-2Еп
С1(1)
X. В этом
солей 1,2 хлорнитродиэтилендиаминового ряда
L NO.,(2)J
случае для оптических антиподов было найдено \a]D -j-200. Кроме того,
* При разделении на оптические антиподы органических оснований, особенно
содержащих асимметрические: азот, серу, селей и т. п., большую услугу оказали
следующие две сульфокислоты, происходящие от камфоры и шГедснные Ь употребление
английскими химиками, особенно Киппннгом и Попе:
СНо
I
S03H-CH-C СО
/
СНз
чсн3
сн2-с сн2
З-Камферсульфоновая кислота
СИ,
сн2- с—
-со
СН3-С-CH2-S03H
I
сн. сн
СНВг
ct-Бром-я-камферсульфоновая кислота
Структурно- и стереохимические представления в неорганической химии 301'
в деятельных 1,2-хлорнитросолях атом хлора может быть замещен родано-
NCS(1)-|
вой группой, причем получается ряд изороданатонитросолей Со2Еп X,
N0,(2) J
NCS
Со-2Еп
N02J
С1
в которых оптическая деятельность сохраняется. (Для
было найдено: [а]с = —50°; [a]D =—84° I.
Вернер показал далее, что для появления оптически деятельных изомеров
Г А^
нет необходимости в том, чтобы в радикале Ме2Еп
В
остатки А и В
были различны. Необходимо только, чтобы они занимали в схематическом
октаэдре положения 1,2 (цис). Поэтому, например, ряд флавосолей
производных этилендиамина [Co2En(N02)2]X легко поддается расщеплению.
Рис. 12.
Если в ранее разобранных случаях мы .имели дело с настоящим
асимметрическим атомом кобальта, так как стереохимичеокая схема
комплексного радикала [Со g 2Еп] представляет как бы сочетание
двух тетраэдров с четырьмя различными группами (Со, А, В и En), то
пріи условии тождества А и В дело существенно меняется, ибо здесь
нет более асимметрического кобальта в собственном смысле сло*ва.
Однако и в этом последнем случае молекула сохраняет
асимметрический характер. Схема, ее изображающая, 'может быть построена в
виде двух геометрически несовместимых -модификаций (рис. 12).
Вместе с тем не трудно убедиться в том, что когда остатки АА
занимают в октаэдрической схеме положение транс (1, 4), энантиоморф-
ных модификаций представить себе нельзя, а потому у таких
соединений нельзя ожидать и появления оптической деятельности.
Повторные по-пытки .получить <в оптически деятельном состоянии
N0,(1)
кроиеосоли [Со2Еп ]Х на самом деле приводили неизменно к отри-
N02(2)
цательным результатам. Наоборот, подвергая дро-бной кристаллизации
камферсульфоновую (или бромікамферсульфоновую) соль флаворяда
[Co2En(N02)2]X, Вернер без труда разделил ее на две части, различаю-
310 Структурно- и стереохимические представления в неорганической химии
щиеся друг от друга по растворимости. Одна из них дала с роданистым
натрием правый, другая — левый роданид, а из этих последних могли
быть приготовлены и другие соли обоих антиподов. Вращение для
хлористой -сол-и было найдено [u]D=±50°, для азотнокислой±41,5°, для
хлорнокислой ([Co2En(N02)2]C104) — ±39° и т. д.
Большой интерес представляет то обстоятельство, что при действии
нитритов (например, NaNCb) на соли вышеописанного активного 1,2-хлор
нитроряда [CoClN022En]X происходит гладкое замещение хлора ни-
трогруппой, причем получается деятельная флавосоль [Co2En(N02h]X,
отклоняющая плоскость .поляризации света на тот же угол, что и
флавосоль, -полученная непосредственным расщеплением соответствующего
рацемата (для хлорнокислой соли была получена величина [a]D=39°) *.
Таким образом можно считать доказанным, во-первых, что
разделение недеятельной флавосоли, а также хлорнитросоли на антиподы
было осуществлено нацело, и во-вторых, что при замещении хлора ни-
трогруппой не происходит рацемизации.
Подобно флавосолям этилендиамина, удалось разделить на 1,2-ди-
хлоро- или В'иолеосоли [Со2ЕпС12]Х. Полученное вращение оказалось
весьма значительным ([a]D около 200° для хлорида), но зато при
хранении раствора наблюдалась быстрая рацемизация. В одном опыте
вращение такого раствора через час после приготовления (при
комнатной температуре) уменьшилось более чем на половину, а через 24 часа
обратилось в 0. Такая быстрая утрата оптической деятельности виолео-
солями, без сомнения, объясняется сравнительно большой
подвижностью, которой отличаются «в них оба атома хлора, входящие в состав
комплексного катиона**.
Известно, что в водном растворе виолеосоли весьма подвергаются
гидролизу.
Совершенно аналогичные результаты были получены для виолео-
этилендиаминовых солей хрома [Сг2ЕпС12]Х, которые по многим своим
свойствам весьма близко напоминают виолеосоли кобальта. И «в этом
случае разделение удалось при помощи бромкамферсульфоновой
кислоты. Для хлорида [Сг2ЕпС12]С1 было найдено вращение [a]D=±183°,
для нитрата ±164°.
Рацемизация здесь .наступает так же легко и фыст-ро, как в случае
виолеосолей кобальта. Так, например, раствор хлорида потерял сполна
оптическую деятельность через 3 часа.
Таким образом, и для комплексных соединений хрома возможно
получение оптически деятельных изомероз, когда группы, связанные с
центральным атомом, дают начало двум энантиоморфным
конфигурациям.
Быть может, всего замечательнее третий тип комплексных
соединений, для которых Вернеру удалось доказать способность расщелляться
на опти-чеокие антиподы. Это тип лутеосолей кобальта и хрома, причем
все 6 координационных мест заняты тремя молекулами какого-либо
диамина, например этилендиамина (СоЗЕп]Х3.
Исследуя этот случай на моделях, легко убедиться в том, что, не-
* Хлорнокислая флавосоль, полученная из левого хлорнитрохлорида, вращала
плоскость поляризации влево же и, наоборот, флавосоль из правого хлорнитрохлорида
вращала вправо. Таким образом, чего-либо подобного Вальденовской инверсии здесь,
по-видимому, не происходит.
** Благодаря этой именно особенности виолео- (а, также празео-) соли при
растворении в воде не дают постоянной электропроводности, близкой к 100 единицам,
свойственной комплексным солям, распадающимся на 2 иона, но электропроводность
быстро возрастает с течением времени.
Структурно- и стереохимические представления в неорганической химии 311
смотря на отсутствие здесь асимметрического атома кобальта, .и
несмотря «а то, что все три группы (Еп), находящиеся, по Вернеру, в первой
координационной сфере, между собой отождествлены, комплексному
катиону лутеосолей все же соответствуют две пространственных
конфигурации, между собой несовместимых (рис. 13).
Для разделения лутеосолей кобальта наиболее подходящим методом
оказалась кристаллизация соответствующего хлоридтартрата (двойной
С1
соли хлористоводородной и правой винной кислоты) fCo3En]Cuo
Двойная соль, отвечающая правовращающему комплексному
основанию, отличается трудной растворимостью; наоборот, хлоридтартрат,
Рис. 13.
отвечающий /-лут?ооснованию, легко растворим. Из хлоридтартратоз
легко получаются и другие оптически деятельные лутеосоли. Для
бромида [СоЗЕп]СЬ было, например, найдено [a]D=±ill7°. Соли этого ряда
рацемизуются лишь с большим трудом и приближаются в этом
отношении к настоящим органическим соединениям. Аналогичным образом
было осуществлено расщепление лутеоэтилендиаминовых солей хрома,
а также родия (по частному сообщению проф. Вернера).
В самое последнее время Вернер показал, что и комплексные
соединения железа, по своему характеру напоминающие лутеоэтилендиами-
новые соединения кобальта, могут быть разделены на антиподы.
Таки.ми соединениями являются производные а,а-дипиридила
снсн сн^сн
НС/ \ -/ \СН,
\ /С с\ Л '
СН N N СН
(молекулы которого для сокращения мо-жно обозначить ои>мволо.м Dp),
полученные Блау [Fe'3Dp]X2 (двойные соединения трех молекул дили-
ридила с солями двухвалентного железа). Разделение их на антиподы
(пока еще, по-видимому, не полное) было осуществлено путем
кристаллизации соответствующих тартратов. Для бромистой соли Вернер нашел
312 Структурно- и стереохимические представления в неорганической химии
весьма значительное вращение [а] = —520° (для света Нернстовской
лампы). В водном растворе оптическая деятельность быстро падает. (Через
Уг часа — наполовину, через 36 часов исчезает совершенно) *.
Заканчивая этим блестящим подтверждением взглядов Вернера
настоящий очерк, .представляющий -попытку в возможно кратких чертах
изложить основные положения .координационной теории и особенно той
стороны ее, которая касается изомерии минеральных соединений, не
могу ,не отметить важного научного значения этой теории, в особенности
для систематики сложных неорганических соединений.
До сравнительно недавнего еще времени область комплексных
соединений, по своей запутанности, по противоречивости фактических
данных и отсутствию какой-либо общей руководящей нити казалась
совершенно безнадежной для систематической разработки. Благодаря
Вернеру и созданной им теории в огромной массе сырого материала
был впервые водворен порядок: множество отдельных соединений
получило место в стройной системе, в которой объединяющим началом
стали основные положения координационной теории.
Теория Вернера позволила «предсказать и новые факты, частью в
высшей степени поразительные, и продолжает служить наиболее
надежным руководителем при исследованиях над комплексными
соединениями **.
Но из ,всех сторон теории Вернера, бесспорно, самая замечательная
и плодотворная та, которая логически связана со стереохимическим
учением Вант-Гоффа и является непосредственным продолжением
дела, .начатого гениальным голландским химиком.
* Статьи Вернера об оптически деятельных кобальтиаках, хромиаках и т. п.
напечатаны в Berichte der Deutschen Chemischen Gesellschaft за 1911 и 1912 гг. См. также
классическое сочинение А. Вернера «Neuere Anschauimger auf dem Gebiete der anorga-
nischen Chemie», III Aufl., 1913. (Русский перевод 5-го издания «Ноаые воззрения
r области неорганической химии». Л., 1936. Прим. ред.).
** В 1913 г. работы А. Вернера увенчаны премией Нобеля.
О НЕУСТОЙЧИВЫХ ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
«Новые идеи в химии», 1914, вып. 6. стр. 105
I
В семидесятых и восьмидесятых годах прошлого XIX века среди
победного шествия теории химического строения А. М. Бутлерова,
прокладывавшей себе путь в области органической химии, полным диссонансом
с господствующим течением звучали голоса немногих староверов, упорно
отстаивавших традиции «реалистической» школы в химии и считавших
учение о взаимной связи между атомами в молекуле беспочвенной,
бесполезной и даже вредной фантазией. Всего меньше таких староверов
оставалось в Германии, где структурное учение быстро пустило глубокие
корни, но зато именно здесь жил и действовал самый ожесточенный
противник и самый беспощадный критик нового учения — Герман Кольбе,
профессор химии в Марбурге, а потом в Лейпциге.
Кольбе был выдающимся химиком своего 'времени. Наука обязана
ему многими важными открытиями, между которыми на первом плане
следует поставить синтез уксусной кислоты сначала из сероуглерода*,
а затем (совместно с Франкландом)—из йодистого метила,
установление ее строения, синтез салициловой ікислоты и ее аналогов,
электролиз кислот жирно-го ряда и многое другое. Но Кольбе не был только
экспериментатором. Он был также выдающимся теоретиком. В 1859 г.
он опубликовал замечательную теорию, в которой проводил ту мысль,.
что все органические соединения, содержащие элементы СН и О, могут
быть произведены из углекислоты через замещение кислорода
водородом и органическими радикалами Эта теория, -совмещавшая :в себе
черты теории радикалов Берцелиуса и теории типов, позволила Кольбе
совершить блестящее предсказание дотоле неизвестных вторичных и
третичных спиртов, а также кислот, и эти предсказания не замедлили
оправдаться (Фридель, А. М. Бутлеров).
Замечательно, что теоретические воззрения Кольбе, в сущности,
довольно близко .подходили к структурной теории. Тем удивительнее то-
упорство, с (которым он не хотел признать несомненных преимуществ
нового течения в органической химии. Шаг, на который никак не мог
решиться Кольбе, заключался .в том, чтобы от радикалов перейти
к атомам, расчленить молекулу на эти .последние и привести в связь
* При действии хлора на CS2 получается ССЦ, который при высокой температуре
восстанавливается водородом в тетрахлорэтилен СС12—СС12. При действии на
последний хлора и воды на свету, получается ССЬСООН. (Впоследствии Бессон [1] показал,
что превращение тетрахлорэтилена в хлорангидрид трихлоруксусной кислоты
происходит при действии кислорода на свету или озона через посредство промежуточного
ecu - ссі2
соединения \о^ * Наконец, трихлоруксусную кислоту легко восстановить-
водородом in statu nascendi в уксусную кислоту (Melsens).
:314
О неустойчивых органических соединениях
.возможный теоретически порядок взаимного распределения их с
наблюдаемыми фактами. Этот шаг он считал -ненаучным и опасным. По
его мнению, все, что можно было вывести из экспериментов, это
установить наличность е молекуле тех или иных радикалов и определить
.взаимное отношение между ними. Разумеется, еще более чуждым всему
мировоззрению Кольбе была всякая попытка выйти за пределы самой
•структурной химии и заняться построением молекулы в 'пространстве.
Известно, с какой злобной критикой обрушился Кольбе * на юного
основателя стереохимии Вант-Гоффа, когда знаменитая брошюра
«Lageriing der Atome im Raume» появилась на немецком языке.
Современники Кольбе, представители структурного учения, усвоили
себе привычку игнорировать его многочисленные полемические статьи,
переполненные резкостями, часто сплошным глумлением по адресу
противников (а .в числе этих противников были такие имена, как Кекуле,
Байер, Эм. Фишер, Эрленмейер, Вислиценус и др.), и, конечно, такое
отношение имело значительную долю основания. Критика Кольбе была
часто явно тенденциозной. Порой .казалось, что автор просто не хочет
дать себе труда понять те воззрения, которые он критикует. Однако
Кольбе недаром был выдающимся химиком-теоретиком. В своей
критике он нередко подчеркивал действительно слабые или недостаточно
•ясные стороны теории строения, на которые, быть может, слишком мало
обращали внимание, особенно в то время. На одной из таких сторон
структурной химии я и хочу остановиться >в настоящем очерке.
В 1877 г. в 'выходившем тогда под редакцией Кольбе «Journal fur
•praktische Chemie» напечатано «конфиденциальное письмо», якобы
исходящее от ученика Кекуле некоего доктора R..., в сущности
представляющее пародию, написанную самим Кольбе. В этом письме устами
наивного защитника структурной теории Кольбе говорит между прочим:
«Я не могу себе представить, чтобы учение Кекуле о взаимной связи
между атомами и о бензольном ядре скоро вышло из моды. Мне
кажется— как раз наоборот, так как (эти воззрения) оказались
неистощимым источником все новых и новых случаев изомерии. Я сам
занимался исследованиями относительно степени важности гипотезы Кекуле
и был поражен изобилием химических открытий, которые в ней
заключаются.— Вы качаете недоверчиво головой.— Посудите сами.
До сих лор довольствовались тем, что размещали в системе
химические соединения, открытые путем опыта, т. е. выясняли, следует ли их,
например,, отнести к орто-, мета- или пара-ряду. Кроме того, пытались
доказать, что для большинства производных бензола возможно только
ло три случая изомерии.— Я же как раз распространил последний во-
лрос — о числе возможных изомеров — на органические соединения
очень простого, состава, соединения, молекулы которых состоят из
немногих атомов, и был поражен большим числом и разнообразием
изомерных соединений, к открытию которых меня привел этот опыт,
соединений, которые едва ли даже нужно приготовлять, так -как их строение
и место в системе уже известны. Сейчас я приведу пример.
В поисках за соединениями, состоящими из четырех элементов
'разной валентности, притом таких, чтобы в их молекуле содержалось лишь
небольшое число атомов, я обратил внимание на цианоугольную
кислоту... Я спросил себя, сколько веществ, изомерных с этим соединением,
возможно представить себе согласно структурной химии. Вот
схематический ответ на этот вопрос:
* dp. 1-й выпуск «Новых идей в химии», 1, 1912, стр. 9 и сир. 212 этого тома.
Прим. ред.
О неустойчивых органических соединениях
31Г)
1
О
3
Н
С—C=N С-О—С О N-C О
О
н
N Н
5
С-Оч
111
С-0
4
;N—Н
7
'C=N—О—Н
I
с=о
>N—Н
0=0
б
C-N-0
III
С—О—Н
12
н—с—с-о
II I
N—О
8
-С—Н
111
С—О— N~0
13
N
-=0
—О—Н
9 14
С—N—О—Н О—С—О-О
С-0
10
C=N
С—О
I
О
I
н
11
н
N Н
15
Н
С—О—С=0
N
С—С—О
III I /
N О/
С—O-0-N—Н
Вы легко убедитесь в том, что этими 16 изомерами мой запас далеко
еще не исчерпан и что число их могло бы возрасти еще больше, если
бы я допустил, что азот в этих схемах может функционировать также
в качестве пятивалентного элемента.
Я спрашиваю Вас, возможно ли лучшее доказательство
.плодотворности .структурного учения, благодаря которому скоро станет ненужной
сама экспериментальная химия? —Ведь при Вашем непонятном
упорстве, с которым Вы отстаиваете теорию радикалов. Вам удалось бы
316
О неустойчивых органических соединениях
вывести всего четыре изомерных соединения того же состава, что циано-
угольная кислота:
Цианоугольная кислота yCN
чон
Циановокислый формил . CNOHCO
Карбонилформиламин . (СО)" ) м
НСО / iN
Оксимид (C2Oa)"HN».
II
Смысл того обвинения, которое в этих строках предъявляется
структурной теории, совершенно ясен. Рамки теории слишком широки. Число
возможностей, которые она предвидит, более числа изомерных форм,
фактически существующих.
Основательны ли эти нападки? Что могла на них ответить теория
строения и как. она отвечает на них ныне? Несомненно, что
принципиальный ответ был готов уже в то время, к которому относится
критика. Теория строения, определяя максимальное число изомеров,
возможных для каждой данной молекулярной формулы, ничего сама ло себе
не" может сказать об условиях образования и степени устойчивости
каждого из них. Поэтому легко может оказаться, что .многие из
структурных комбинаций, не реализованных в данный момент, окажется
возможным осуществить, когда будут открыты новые реакции, найдены
.подходящие условия для их получения. Возможно, что многие из таких
соединений окажутся даже весьма- устойчивыми. С другой стороны,
легко может случиться, что известная часть мыслимых по теории
структурных схем будет отвечать веществам, столь мало устойчивым, что
реальное существование их окажется невозможным, по крайней мере при
«обыкновенных» условиях наших опытов. Позволительно, однако,
надеяться, что, постепенно совершенствуясь, наши технические методы
исследования когда-либо позволят осуществить те условия, в которых
подобные соединения будут способны хотя бы к кратковременному
существованию. Тогда соответствующие им структурные схемы потеряют
свой гипотетический характер и вместо фантазии ,мы будем иметь дело1
с действительностью.
В семидесятых годах прошлого века соединений, оправдывавших
такую точку зрения, почти что не было известно, и потому Кольбе имел
некоторое основание не считаться с возражениями своих противников.
Но то, что не было возможно .во время Кольбе, было осуществлено
следующими генерациями химиков.
За три последних десятилетия органическая химия в интересующем
нас направлении сделала огромный шаг вперед. Был открыт длинный
ряд соединений с такими атомными 'комбинациями, с такими
химическими свойствами, которые можно было предвидеть или объяснить-
только с точки зрения структурной теории. Многие из числа этих
соединений, неожиданно даже для самих сторонников теории строенияг
оказались обладающими значительной, а иногда исключительно
высокой степенью устойчивости. Таковы, например, полиметиленовые
углеводороды и их многочисленные производные с 3, 4, 5, 6, 7 и 8 атомами
углерода в цикле, вслед за Фрейндом и Г. Г. Густ а в сон ом,
изученные особенно русскими химиками (Вреден, Марковнико в„
Коновалов, Демьянов, Зелинский, Кижнер и др.)- Как
известно, многие из этих углеводородов (особенно с 5- и 6-членнымк
О неустойчивых органических соединениях
317
циклами) оказались составными частями нефтей типа кавказской.
Другие, подобно циклооктотетраену.
сн/ \сн
СНч СН
сн-о-к
замечательному углеводороду, недавно полученному Вильштетте-
ром, стоят в родственных отношениях к естественным алкалоидам.
Благодаря .работам Е. Е. Вагнера, Бредта, Байера и др.
получила право гражданства идея о способности к существованию
углеводородов и других соединений с 2 -и 3 циклами в частице*, и такие
соединения были открыты в естественных продуктах, особенно в эфирных
маслах. Как -и следовало ожидать, новые типы соединений потребовали
и новых методоз для своего получения. Таковы, например, методы
Фрейнда, Густавсона, Сабатье, Ипатьева л др., позволяющие
осуществлять синтезы моноциклических полиметиленов .и их производных.
Методы получения аналогичных соединений с несколькими циклами
и до сих пор еще мало разработаны, хотя соединения эти и
синтезируются в растительном царстве и отчасти в животном (холестерин).
Исключение составляет только -процесс конденсации углеводородов
алленовой группы
СН2—С =СН., CH-t\
V-C-CH,,
сн/
Аллен Несимметричный диметилаллен
подробно изученный в превосходной работе С. В. Лебедева [2] и
приводящий к -ряду иолици-клических углеводородов частью весьма
своеобразного строения, например
СНо -С—СН.» СН2\
I I і >с-с—сн2
СНо—С-—СН2 СН2 | | /СН»
I " I СН,—С—С<
СНо—С—СНо СН*
¦2
12
СН.,
Шс с\а С С--СН.»
Тетрамер аллена Тример диметилаллена
Особенно велико число и разнообразие типов, осуществленных среди
соединений гетероциклически х. Хотя отдельные представители
последних были известны и во времена Кольбе, а для некоторых
(например, для индола, пиридина, хинолина) было тогда уже установлено
строение, однако лишь после открытия тиофена Виктором Me
пером (в 1883 г.) [3] началась широкая обработка этой области.
Перечислять здесь даже главнейшие типы гетероциклических соединений,
которые представлены и в продуктах жизнедеятельности растений и
животных (хлорофилл, гемоглобин, алкалоиды, пурины, отчасти белковые
вещества) и во многих технически важных препаратах (многие краски,
медицинские средства и пр.), нет ни надобности, ни возможности.
Достаточное число таких соединений приводится во всех, даже кратких
* В ароматическом ряду существование соединений с таким строением допускали
и раньше вслед за Эрленмейером, давшим известную формулу нафталина.
318
О неустойчивых органических соединениях
руководствах по органической химии (например, Н. А. Меншутки-
н а, Голлеманна, Шмидта и пр.).
Упоминать ли о том, что за тот же период времени были открыты
целые классы соединений, как оксимы (В. Мейер), гидразины и
гидразоны (Э. Фишер, Курциус), иодо-, иодозо- и иодоние-
вые соединения (Вильгеродт и Виктор Мейер) и многие
другие?
Все это опять соединения по большей части сравнительно
устойчивые. Но и при изучении малоустойчивых соединений были сделаны
крупные успехи. К началу 80-х годов такие случаи, как циановая кислота,
способная к существованию только при очень низких температурах,,
представляли редкое исключение. Между тем в наше время они стали
в своем роде типическими. В 1890 г. Кекуле получал мономерную
модификацию формальдегида СН20, быстро охладив переведенный в пар и
потому деполимеризованный параформальдегид в виде летучей
жидкости при комнатной температуре, подобно циановой кислоте, со
взрывом переходящей обратно в полимерную форму.
Недавно Гарриес показал, что подобным же образом могут быть
получены полимеры многих диальдегидов. Особенно поучителен случай
простейшего диальдегида глиоксаля СНО—СНО, в течение многих лет
(Де'бус, 1856 г.) известного только в виде бесцветного аморфного
полимера. Перегоняя последний в присутствии Р205 (для удержания вла
ги), Гарриес получил зеленого цвета пар, сгущающийся в желтую ле
тучую жидкость (температура кипения 50°), необычайно легко перехо»
дящую в бесцветный нерастворимый полимер параглиоксаль.
В этих приемах получения неустойчивых соединений, частью
представляющих модификации соединений давно известных, сказывается-
одна характерная черта современной экспериментальной химии вообще
и в частности органической — широкое пользование так называемым
принципом горячей и холодной трубки С. К. Девилля.
Если соединение А (или,.общее, некоторая система тел),
неустойчивое при данной температуре, имеет тенденцию к переходу в более
устойчивое состояние (систему) В с выделением тепла, то в согласии с
требованиями термодинамики (закон Вант-Гоффа — Ле-Ша-
телье) устойчивость его (А) должна повышаться с повышением
температуры. Поэтому, нагревая тело (или систему) В, мы при некоторой
достаточно высокой температуре достигнем практически полного
превращения его в А. При охлаждении, понятно, наступит обратная
реакция: А-*В. Возможно, однако, задержать этот обратный переход А в В,
применяя быстрое и сильное охлаждение. Скорость всякой реакции (а
значит, и реакции данного перехода) быстро падает с понижением
температуры, а потому при указанных условиях наше тело не успеет
подвергнуться превращению в В и сразу перейдет к температуре, при
которой скорость этого превращения близка к нулю. Мы будем иметь
наше тело в состоянии ложного или кажущегося равновесия.
Получение неустойчивых органических соединений, находящихся в
состоянии ложного равновесия, может быть осуществлено и чисто
химическим путем, подобно тому, как в минеральной химии получаются
такие тела, как окись и двуокись хлора, закись азота, хлористый азот и
пр. К подобным реакциям относится образование озонидов,
полученных Гарриесом при действии озона на непредельные соединения:
СНо СНо—0\
!! Ч03-.-= | " >0
СН2 СН2—0х
О неустойчивых органических соединениях 319
Озониды в большинстве случаев отличаются весьма малой
устойчивостью, а некоторые, например, озонид этилена, сильно взрывчаты.
В ту же категорию реакций относится получение соединения
полиацетиленовой группы (А. Байер, Ж. И. Иоцич), д и а з о с о-
единений, азидов, например взрывчатого цианазида CN—N3,
недавно описанное Дарзаном образование многих кет е н о в, реакции,
ведущие к выделению таутомерных модификаций, о чем еще будет
идти речь впереди * и многие другие.
Даже самый краткий обзор всех сюда относящихся фактов занял
бы гораздо более места, чем имеется в моем распоряжении. Поэтому
я предпочитаю остановиться с некоторой подробностью только на двух
объектах, которые, думается мне, более других пригодны для того,
чтобы осветить современное состояние вопроса о неустойчивых
органических соединениях и о той роли, которую они играют в структурной
химии. Я выбрал для этой цели обзор явлений таутомерии -и класса
кет о нов**. Обе эти темы и в других отношениях представляют
интерес современности, а потому сами'по себе заслуживают внимания.
Но даже того беглого перечня, который был дан на предыдущих
страницах, кажется достаточно для того, чтобы прийти к заключению,
что самые разнообразные, порой весьма причудливые комбинации
атомов в молекулах, совместимые с теорией строения, могут быть
реализованы. Имеем ли мы после этого основание сомневаться в
принципиальной возможности реализовать остальные?
III
Давно известно, что замещение водорода в синильной кислоте
углеводородными радикалами R(CH3, СгНэ, С&Н5 и пр.) дает два ряда
изомерных соединений: нитрилов и изонитрилов ***, что подобным
же образом единому роданистому водороду отвечают родановые
э ф и р ы и горчичные масла, а азотистой кислоте — нитросо-
единения и азотистые эфиры:
HCN( HSCN<
4R—N=C "I*—N=C:=:S
HNOo(' V0
*R—0—N=0
Для всех подобных случаев, впоследствии объединенных под общим
именем таутомерии (Лаар), или динамической изомерии
(Армстронг) **** характерно то, что появление изомерных форм связано
* Я не упоминаю здесь о той обширной уже группе химических соединений, к
которой принадлежит трифеиилметил Гомберга и его аналоги, ибо здесь мы имеем
область фактов, сказавшуюся до известной степени неожиданной в смысле самой
классической теории строения, поскольку она принимает валентность углерода, равную 4.
** Этот обзер по необходимости является далеко не полным. Я постарался кратко
охарактеризовать явления таутомерии на немногих примерах и установить і*е связь
с явлениями обыкновенной изомерии.
*** іВ изонитрилах ныне, вслед за Н э ф о м, многие химики считают углерод
двухвалентным.
**** Для обозначения различных разновидностей или тиіпов таутомерии
предложены и другие термины: десмотропия, мезотропия, аллелотропия и др.;
но, не представляя самостоятельного значения, они не вошли во всеобщее
употребление и едва ли не являются излишними.
320
О неустойчивых органических соединениях
здесь с замещением атома водорода в данном соединении на
органический радикал.
С точки зрения структурной теории этого обстоятельства, конечно,
объяснить нельзя, так как изомерия, возможная для дериватов, в той
же мере предвидится и для исходного вещества:
Н—C=N Н—S—C^N НО—N-0
Н—N=C Н—N-C-S H~W(
Очевидно, мы имеем здесь дело с одним из тех случаев
несоответствия между теорией и опытом, на которые указывал Кольбе.
Одна из первых попыток разобраться в этом противоречии
принадлежит А. М. Бутлерову, и замечательно, что мысли, развитые
знаменитым русским химиком, довольно близко подходят к современной
точке зрения на этот предмет.
Б 1877 г. в статье «Об изодибутилене» [4], обсуждая механизм
процессов отщепления воды от спиртов и уплотнения образующихся при
этом олефинов, Бутлеров говорит между прочим: «Следует думать, что
в смеси третичного спирта с разведенной серной кислотой уже и при
обыкновенной температуре находится некоторое количество
углеводорода, образовавшегося распадением.
Мы здесь имеем нечто подобное тому, что найдено Вислицениусом
для концентрированных растворов молочной кислоты, Вертело — для
растворов солей и т. п. При обыкновенной температуре химическому
равновесию в смеси соответствует малое количество углеводорода рядом
с большим количеством алкоголя (или отвечающей ему серновинноіі
кислоты); при повышенной температуре, наоборот, равновесие
устанавливается при большем количестве угловодорода и малом — алкоголя.
Если бы условия были таковы, что, во-первых, воссоединение продуктов
диссоциации вело к образованию не первоначального спирта, а его
изомера, и если бы, во-вторых, этот изомер, в свою очередь, при
существующих условиях, подвергался до известной степени диссоциации, то в
смеси находились бы, рядом с углеводородом, два изомерные спирта.
Мыслимо, что и без присутствия посредствующего реагента, каковым
является здесь серная кислота, частицы некоторых веществ, вследствие
постоянного распадения и воссоединения продуктов в новом порядке
постоянно изомеризуются, переходя от одного видоизменения в другое и
обратно. Для изомерных между собою частиц здесь происходило бы то
же самое, что происходит для частиц различного состава и что Пфаунд-
лер обозначил названием соперничества частиц (Concurrenz der
Molecule) (Poggend. Annal., 1874, Jubelband, стр. 189). В огромном
большинстве случаев мы имеем дело с веществами, в газообразной или жидкой
массе которых при обыкновенных условиях химическому равновесию
соответствует присутствие частиц одного известного определенного
химического строения в бесконечно большем числе, чем частиц других
строений, изомерных с первым, но невероятно, что в некоторых случаях
можно встретить и такие тела, масса которых постоянно заключает в
заметном количестве изомерные частицы различного химического
строения,—частицы, постоянно «соперничающие» между собою,
перегруппировывающиеся взаимно из одного строения в другое. В первом случае
можно смело говорить об определенном химическом строении тела, а во
втором —суждение о каком-либо одном определенном строении явится
бесполезным, потому что в массе вещества присутствуют частицы
различных, например двух, строений, и, при склонности частиц к перегруп-
О неустойчивых органических соединениях 321
гшровке, вся эта масса, понятно, будет подвергаться реакциям,
свойственным одному строению, или реакциям, свойственным другому
строению, смотря по натуре реагента, влиянию которого подвергается, смотря,
так сказать, по направлению действия этой реакции. Подобной
частицей двойственного строения будут, например, по-видимому, циановая
кислота, синильная кислота и т. п. С этой точки зрения представляются
бесполезными старания разрешить вопрос о том, гидратное или карба-
мидное строение свойственно циановой кислоте, нитрильное или карбил-
аминное строение имеет синильная кислота и т. п. Такой взгляд
исключает слишком абсолютные представления о химическом строении и
способен, мне кажется, уяснить некоторые явления, например появление
некоторых побочных продуктов в реакциях и т. п. Почти лишнее
прибавлять, что сказанное представляет приложение к представлению о
химическом строении тех динамических понятий, начало которым
положено Бертолле и которые все более и более захватывают и освещают
область химизма».
Итак, Бутлеров принимал, что в таутомерных соединениях
существует динамическое равновесие между двумя формами, между двумя
родами самостоятельно существующих молекул, причем относительная
пропорция тех и других зависит от природы соединения.
Эта точка зрения получает в дальнейшем прочное обоснование
благодаря работам В. Вислиценуса, Клайзена, К н о р р а, Брюля,
Димрота и др. в Германии и школы проф. Армстронга в
Англии. Основной характер этого рода изомерии хорошо выражается
термином динамическая изомерия, по почину Армстронга
общепринятым среди английских химиков *.
Параллельно с этим определенным и ясным воззрением в науку
проникло другое, более расплывчатое и менее доступное опытной проверке.
Было высказано предположение, что одна и та же молекула, благодаря
известному характеру движения атомов, ее образующих, может
проходить через различные состояния, непрерывно сменяющиеся во
времени, причем некоторые из этих состояний могут отвечать определенным
структурным формулам, которые мыслимы для данного соединения.
В первый раз подобного рода представления были высказаны
Авг. Кекуле и послужили ему для объяснения причины, по которой два
орто-положения в бензольном
* К ^той точке зрения, в сущности, примыкает, несмотря на кажущееся различие,
взгляд, высказанный Байером в 1883 г. по поводу изомерии, появляющейся в
производных изатина
СО /^\
QH/ ЧСО и СеН4 COR8.
но отсутствующей для самого изатина (REH);.
Байер предположил, что из двух возможных форм самого изатина одна,
псевдоформа, вследствие подвижности водородного атома является неустойчивой, а потому,
образуясь, немедленно изомеризуется в другую — устойчивую.
В сущности Байер принимает, что здесь равновесие сильно сдвинуто в одну
определенную сторону.
21 Л. А. Чугаев. т. Ш
322
О неустойчивых органических соединениях
ядре 1, 2 и 1, 6 фактически равноценны (1872 г.). При этом допускалась
непрерывная «осцилляция» двойных связей внутри кольца.
Позднее Лаар (1885 г.) * развил подробнее мысль Кекуле и
распространил ее на весь тогда уже довольно обширный круг явлений
таутомерии**.
Почти вся дальнейшая история вопроса о таутомерии представляет
развитие первого взгляда, согласно которому таутомерия есть
настоящая изомерия, но только изомерия подвижная, нередко мимолетная.
Однако между этим взглядом, как мы видели, ведущим свое начало
от Бутлерова, и между гипотезой Кекуле и Лаара часто не проводят
ясной границы***. Чтобы сделать разницу между ними более наглядной,
-полезно будет остановиться на каком-либо конкретном примере. Мы
воспользуемся для этой цели классическим случаем ацетоуксусного
эфира.
Соединение это, впервые полученное Гейтером в 1863 г., сразу
обратило на себя внимание способностью давать солеподобные
металлические производные. На этом основании Гейтер приписал ему формулу
оксикротонового эфира СН3 — С (ОН) = СН — СООС2Н5. Но когда
Франкланд и, особенно, Вислиценус со своими учениками
разработали общеизвестный метод «ацетоуксусных» синтезов и показали, что
углеводородный радикал R, входящий в молекулу при действии
галоидного производного RX на натряй-ацетоуксусный эфир, становится к
атому углерода (а не к кислороду), энольная формула Гейтера была
оставлена и заменена кетонной: СН3 — СО — СНг— СООС2Н5.
* Л а а р у принадлежит и наиболее полная классификация типов таутомерии в
структурно-химическом отношении {5]. Не имея в виду давать в этом очерке
систематического обзора таутомерных соединений, я остановлюсь на этой
классификации только в немногих словах. Соединения, у которых наблюдается таутомерия,
Лаар разделяет на два главных типа: диады и триады. В диадах два
многоатомных элемента X и Y соединены между собой кратной, например двойной связью*
а один из них, кроме того, связан и атомом Н, например X—Y—Н. Переход Н от X
к Y и обратно и вызывает таутомерию (пример Н—CErN и H = N = C). В триадах
имеется три многоатомных элемента X, Y и Z и, кроме того, атом водорода: X=Y—Z—Н
и Н—X—Y—Z. При этом валентность Y не может быть менее 3 (например N, С), а X1.
и Z -могут быть и двухвалентными (О, S и пр.). Триады, в свою очередь, Лаар делит
на б классов. В первых трех содержатся соответственно 1, 2 -и 3 атома углерода, в
четвертом—ни одного атома С; в пятом Y=K, а X или Z=C; в шестом Y=N, а X к
Z—С. Комбинация диады с триадой дает тип так называемых тетрад. Каждый класс
триад разделяется на ряд подгрупп. Уже из одного этого беглого перечня видно, как
велико разнообразие случаев таутомерии.
** іВ новейшее время английские химики, особенно Бэли и его ученики, сделали
попытку связать представление Кекуле и Лаара при помощи современной электронной
теории с явлениями избирательной абсорбции света в некоторых органических
соединениях. Такими соединениями были, с одной стороны, тела, показывающие кето-эноль-
ную таутомерию, а с другой — ароматические соединения и а-дикетоны. У последних
Бэли и Стюарт предположили особый вид таутомерии, которому дали название
изотропии. По мысли английских авторов, основной причиной появления полос поглощения
в спектрах всех этих соединений являются внутримолекулярные осцилляции атомов,
сопровождаемые непрерывным разрывом с возникновением связей. Не останавливаясь-
подробнее на этих представлениях, заметим, что они не оправдали первоначально
возлагавшихся на них надежд, по крайней мере, з той форме, в которой были высказаны.
*** Сам Лаар пишет по этому поводу: «По-видимому, из его (Бутлерова)
рассуждений вытекает, что он представляет себе процесс (перехода одного изомера в
другой) протекающим, по крайней мере в главных чертах, не внутри, а междумолеку-
лярно. Он ссылается при этом на теорию диссоциации Пфаундлера, тогда как я,
пожалуй, скорее мог бы сослаться на теории спектральных явлений Максвелла и
Видем-анна». В связи с представлениями' Лаара было предложено все случаи
таутомерии, для которых изолирование одной или обеих форм реализовано, не причислять
к собственно таутомерным соединениям, а обозначать новым термином, например
соединенмйми деамотро'шшм<и. Едва ли, однако, такая' классификация является
логичной.
О неустойчивых органических соединениях 323
Однако в 90-х годах ряд исследований, особенно Нэфа и Клай-
з е н а, показавших, что под влиянием других реагентов, например хлор-
угольных э фи ров СО. 9 могут быть получены дериваты, отвечаю-
4OR
щие формуле Гейтера, наряду с производными кетонного типа
ch^==ch-coor снз-со^сн~соок
р и со
СО R-o'
R-0
заставили химиков вновь изменить сложившийся взгляд и постепенно
прийти к заключению, что совокупность превращений ацетоуксусного
эфира может быть выражена лишь, принимая во внимание обе
формулы, старую и новую. Таким образом ацетоуксусный эфир оказался "в
числе соединений таутомерных.
С точки зрения гипотезы Лаара, все молекулы, образующие жидкий
эфир, должны быть признаны, по существу, одинаковыми. Но в каждой
молекуле совершается безостановочное колебание водорода, влекущее
за собой переход от кетонной формы в энольную и обратно,
//Н\\
СН3—СО—СН—СООС2Н5.
В каждый данный момент мы застанем известную часть молекул в
кетонной «фазе», другую — в энольной, третью (надо думать, далеко
преобладающую), в различных промежуточных фазах движения. При
таких условиях, очевидно, трудно себе представить возможность
изолирования какого-либо определенного рода молекул в отдельности. Чтобы
сделать картину еще более отчетливой, возьмем сравнение из области
стереохимии.
Для всякого вещества, отвечающего формуле CR1R2R3—CR4RsR6* как
известно, можно получить четыре оптически деятельные модификации.
Соответствующие этим последним схемы различаются по
пространственной ориентировке радикалов. Остановимся на одной из четырех
возможных здесь конфигураций:
Rj R-2 /Кз
с
с
I
Rd R(
R
Согласно теории, развитой Вант-Гоффом, такую молекулу можно
представить себе как систему двух схематических тетраэдров CRiR2R3 и
CR4R5R6, свободно вращающихся около оси СС (ргіпсіре de liaison
mobile) *.
* Принцип подвижной связи (франц.). Прим. ред.
21'
324
О неустойчивых органических соединениях
Если бы мы не допустили существования такого свободного
вращения, то пришли бы к необходимому заключению, что молекула, о
которой идет речь, а следовательно, и вещество, слагающееся из таких
молекул, способно образовать ряд новых геометрически изомерных
модификаций, соответственно различным относительным положениям обоих
тетраэдров. Только допустив свободное движение последних друг
относительно друга, мы устраним эту возможность, ибо тогда в каждый
данный момент времени в разных молекулах один тетраэдр будет занимать
относительно другого самые разнообразные положения, и положения
уги во времени будут непрерывно изменяться. До некоторой степени
аналогичную картину представляет, согласно гипотезе Лаара, ацето-
уксусный эфир и всякое другое вещество, показывающее явление
таутомерии *.
К существенно иным результатам приводит развитие взгляда,
намеченного Бутлеровым. Если таутомерное вещество представляет из
себя смесь, или, точнее, равновесную систему, образованную двоякого
рода молекулами, с большей или меньшей скоростью способными
превращаться друг в друга, по общим законам химических реакций, то
мыслимы условия, в которых эта скорость сделается достаточно малой
для того, чтобы разделение таутомерных модификаций и получение
каждой из них в отдельности стало осуществимым.
Действительность на многих примерах оправдала именно последнюю
точку зрения. Изолирование таутомерных модификаций удавалось
неоднократно, но самый замечательный (в то же время один из самых
последних в хронологическом порядке) случай этого рода как раз
составляет разделение ацетоуксусного эфира, недавно
воспроизведенное Л. Кнорром [6] и его учениками. Обыкновенный ацетоук-
сусный эфир представляет смесь энольной и кетонной модификаций со
значительным преобладанием последней. Если такую смесь растворить
в эфире или гексане и охладить до —78°, то выкристаллизовывается
чистая кетоформа. Энольная же модификация могла быть получена при
сн,—с=сн—сооад
разложении натриевого соединения LM
(взвешенного в петрилейном эфире) хлористым водородом при —78°.
Некоторые свойства этих соединений сопоставлены в следующей
таблице:
Кетонкая форма
Бесцветная жидкость, застывающая в
кристаллы при охлаждении
Температура плавления —39°
Удельный вес при 10°
Коэффициент преломления для D луча при
10° 1,4225
Температура кипения при давлении 2 мм
40—41°
Реагирует с бромом медленно, а с
хлорным железом при —40° не дает
окрашивания
Энольная форма
Бесцветная жидкость, застывающая в
жидком воздухе в аморфную
стекловидную массу
1,0119
1,4480
33°
Немедленно реагирует с бромом,
дает яркое окрашивание с хлорным
железом уже при —78°
* Разница сзоз,илась бы, конечно, к тому, что гипотеза подвижной связи касается
не структурной изомерии, а лишь стереохимической конфигурации.
О неустойчивых органических соединениях
325
Как видно, обе формы ясно различаются друг от друга. Заметим,
что энольная форма характеризуется способностью легче — иначе
сказать, непосредственно — давать окрашенные металлические
производные с солями железа, что вполне гармонирует с ее строением (с
присутствием ОН группы, соединенной с ненасыщенным
углеродом, что должно способствовать появлению кислотных свойств у
водорода).
При хранении, особенно в присутствии катализаторов, например
оснований, обе модификации претерпевают превращение и, в конце
концов, дают смесь, отвечающую состоянию равновесия. Превращение
кетоформы при отсутствии катализатора требует целых месяцев для
своего окончания, тогда как у энольной формы оно заканчивается в
10—14 дней. Различные методы исследования привели к согласному
результату, что в равновесной системе содержится 7—8% энольной
формы и 92—93% — кетонной.
IV
Ацетоуксусный эфир представляет частный случай обширной
группы таутомерных соединений, объединяемых одной общей характерной
чертой — присутствием атомных группировок:
ОН
-с=сн—со—
OHR
—С=С—СО-
Во многих случаях таутомерные модификации соединений,
принадлежащих к этой категории, были изолированы, а частью был
установлен и состав равновесной смеси, задолго до того, как в этом отношении
был исследован простейший случай, 'Представленный ацетоуксусным
эфиром. Здесь будет уместно остановиться вкратце на главнейших
методах исследования подобного рода таутомерных
соединений.
Всего чаще удавалось изолировать таутомерные модификации
посредством метода кристаллизации, но этим путем во многих случаях
можно бывает получить только одну модификацию. Если скорость
превращения одной таутомерной формы в другую значительна
(нарушенное равновесие быстро вновь устанавливается), то из жидкой смеси двух
таких форм при данной (достаточно низкой) температуре
выкристаллизовывается только одна — та, которая в этих условиях является менее
растворимой. При этом равновесную смесь можно рассматривать как
раствор одной из модификаций в другой (или же в смеси остальных).
Если система охладится до температуры, при которой такой раствор
станет пересыщенным по отношению к одному из таутомеров, то
последний и будет выделяться в кристаллическом состоянии. Как только
выпадет часть кристаллов, нарушится соотношение между
концентрациями обеих модификаций в жидкости, и немедленно недостающая часть
молекул выкристаллизовавшейся формы образуется за счет молекул
другой формы, и так продолжается до тех пор, пока все количество
вещества не выделится в виде кристаллов первой формы.
Н Н
\/
—СО—С—СО ±г^ —
R Н
\/
_СО—С—СО— —=±
326
О неустойчивых органических соединениях
В случае, если взаимный переход таутомерных модификаций
совершается с незначительной скоростью, очевидно, представляется
возможным, выделив из жидкой смеси часть одной из них путем
кристаллизации, тем самым обогатить остаток другой формой.
Совершенно аналогичным образом протекает кристаллизация, когда
обе таутомерные модификации выделяются из раствора исходной смеси
в каком-либо растворителе. Только условия равновесия при этом
соответственно сложнее.
Недостаточность чисто физического метода разделения пополняется
применением методов химических.
Когда из равновесной смеси выкристаллизовывается кетонная
форма, изоляция знольной формы удобнее всего осуществляется при
помощи метода, предложенного Л. Кнорром. Приготовляют натриевое
(или калийное) производное данного вещества, и это производное,
которое всегда происходит от энольной формы (содержит, например,
—С-СН—СО
группировку атомов | ), осторожно (при низкой темпе-
О—Na
ратуре) разлагают подходящей кислотой. Мы видели, что этот именно
метод позволял Кнорру получить энольную форму ацетоуксусного
эфира.
Комбинацией только что указанных методов были, например,
получены обе возможные модификации в следующих случаях кетоэнольной
таутомерии:
Формилфенилуксусный этил
сно
СН—СвН5
СООС2Н5
Кетонная форма жидкая,
окрашивается FeCl3
СН(ОН)
II
с—сйн
вА15
СООС.Н
211б
Энольная форма. Твердое тело,
температура плавления 70°;
не дает реакции с FeCls
Ацетилдибензоилметан
СН3
I
с=о
СН:
.СОС.Н
в115
хсос6н6
Кетонная форма. Температура
плавления 107—110°.
Нейтральное тело; не реагирует на
холоду с содой и не окрашивается
от FeCi3
СНз
I
С—ОН
!! /СОСвН5
¦\
сосн
6А15
Энольная форма. Температура
плавления 80—85°; при более
высокой температуре переходит в
кетонную формулу. Кислота,
выделяющая С02 из соды на
холоду, реагирует с FeCI3
О неустойчивых органических соединениях
327
СН*
Трибензоилметан [7J
СдН.
с=о
С—ОН
с
/
сосйн
6115
-сосн
<ЮС.Н
с
6х J 5
в115
Кетонная форма (а)
Рыхлая кристаллическая масса
(иголочки). Температура плавления
228—231°; 1 ч. растворяется более
чем в 300 см9 хлороформа при
комнатной температуре. Растворяется
постепенно в алкогольных щелочах,
но не в соде. С FeCl3 не дает
окрашивания
Весьма интересный случай предста
ного эфира, для которого, кроме трех
сос6н5
Энольная форма (?)
Плотная кристаллическая масса,
неясное плавление при 210—220°
с переходом в ос-форму; 1 ч.
растворяется в 6 смг хлороформа. Легко
растворяется в щелочах и 1%-ной
соде. С FeCI3 дает темно-красное
окрашивание
вляет таутомерия диацетилянтар
таутомерных модификаций
СН3—СО—СН—СООС2Н5
СНа—СО—СН-СООС2Н5
Кетоформа
СНа-С=СН-СООС2Н5
I 1
ОН |
СНз-СО—СН—СООС2Н5
Кетоэнольная форма
СН3—С=С—СООС2Н5
ОН |
СН3—С=С—СООС2Н5
I
он
Диэнольная форма,
возможны еще геометрические (и оптические) изомеры. Из семи воз
можных по теории недеятельных изомеров К. н о р р у [8] удалось полу
чить пять, со следующими свойствами:
Формы
Огереоизомерные \ $
кетоформы J т
Энол^ные формы? _
J a8
Температура
плавления, °С
90
68
Жидкость
21—22
31—32
Показатель
преломления прн 20°
—
1,46
1,4545
1,4392
Реакция с FeClj
Отсутствует
»
Бурое окрашивание
Фиолетово-красное
окрашивание
То же
Растворимость в
лигроине (точка кипения
50—60°) при 20°
1:122,5
1 -.15,.4
Смешиваются во
всех пропорциях
1:2,9
328 О неустойчивых органических соединениях
Не менее важную роль, чем методы выделения таутомерных
модификаций, сыграли при изучении динамической изомерии методы,
позволяющие определять количественный состав той
или другой смеси жидкой равновесной системы.
Наиболее общий из этих методов, лишь недацро разработанный
Куртом Мей ер ом [9], основан на титровании бромом в спиртовых
растворах. Бром присоединяется только к энольной форме, по месту
двойной связи, кетонная — остается нетронутой. Если реакцию вести при
сильном охлаждении и с соблюдением некоторых других
предосторожностей, то во время титрования положение равновесия не успевает
сместиться, и полученные результаты дадут нам правильную картину
состояния системы в момент равновесия.
Мы не останавливаемся на целом ряде других химических методов *
анализа таутомерных соединений в виду того, что они по большей части
недостаточно надежны. Главный недостаток их заключается в том, что
в условиях данной реакции равновесие, вообще говоря, смещается, и мы
получаем неправильное представление о первоначальном состоянии
системы.
От этого недостатка совершенно свободны ф -и з -и ч е с к ч е методы,
основанные на том, что при переходе одной таутомерной формы в
другую происходит изменение физических свойств. Чаще всего для этой
цели пользовались оптическими свойствами: рефракцией и
абсорбцией, естественной и магнитной вращательной
способностью.
Метод, основанный на определении рефракции, был применен раньше
других и до сих пор остается наиболее надежным. Известно, что
молекулярная рефракция MR, вычисленная по формуле Лоренца и Лорент-
ца М/?= — X "Г(где п — показатель преломления вещества, d — его
плотность при той же температуре и М — его молекулярный вес), не
зависит от температуры и притом является величиной, приблизительно
аддитивной для органических соединений. Иными словами, зная
атомные рефракции га, ^в, гс ... элементов А, В, С..., можно вычислить мо-
* Большая часть этих методов основана на применении веществ, реагирующих с
/N
гидроксильной группой, например, РС!5СбН5 — СОС1, диазометана CH2v ц ( пред-
ставляющего новейшее средство для метилирования (например, спиртов:
ROH+ |f \CH2=R~0—CH3+N3)
и пр. В тех случаях, когда энольная форма обладает свойствами кислоты.
было предложено судить о ее содержании в равновесной системе по
количеству аммиака, которое поглощает определенное количество испытуемого вещества.
Характерной качественной пробой на содержание энольной формы в равновесной смеси
является реакция с хлорным железом. При этом образуются комплексные соединения
(вероятно, относящиеся к категории так называемых внутренних комплексных солей),
окрашенные большей частью в различные оттенки фиолетового цвета.
Другой весьма интересной качественной реакцией на таутомерные модификации.
содержащие двойную связь, может служить желтое окрашивание с тетранитрометаном
C(N02)4 (И. И. Остромысленский). Эта реакция получается, однако, не только'со
всеми непредельными соединениями, но и с веществами, содержащими трех- и, по
видимому, также четырехчленные циклы в молекуле.
О неустойчивых органических соединениях
329
лекулярную рефракцию соединения ABC... по формуле М/? = ргА-|-
+ g/"B ~\-src ... Непредельные соединения, однако, не подчиняются этому
правилу. Так, двойной и тройной связи между углеродными атомами
отв-ечает определенный инкремент или избыток рефракции. Поэтому
при переходе, например, группировки —СО—CHL в —С=СН рефракция
ОН
должна измениться, и размер этого изменения можно заранее предвы-
числить. Таким образом Брюль [10]* пришел к заключению, что ацето-
уксусный эфир отвечает кетонной, а не энольной формуле. Для других
таутомерных соединений был получен противоположный результат.
Путем аналогичного, но более сложного расчета можно
воспользоваться для той же цели способностью веществ (оптически недеятельных
в естественных условиях) вращать плоскость поляризации света, когда
они находятся в магнитном поле. Применяя этот метод к случаю ацето-
уксусного эфира, Перкин пришел, в согласии с Брюлем, к тому
заключению, что вещество это обладает кетонным строением. Наоборот,
магнитное вращение ацетилацётона **
СН3—СО—СН2—СО—СН3 СН3—С=СН—СО—СН3
ОН
I II
отвечает энольной формуле (II). Наконец магнитное вращение,
найденное для ацетонощавелевого эфира СН3—СО—СНг—СО—СООС^Н5>
указывает на то, что -ему должно быть приписано строение
СН3—С=СН==С—СООСоН5.
I !
он он
Другими словами, в этом случае обе кетонные грушіы энолизиро-
ваны.
О применении к изучению таутомерии другого оптического метода,
основанного на исследовании спектров поглощения, см. в одном из
следующих сборников «Новых идей в химии»***.
Весьма любопытное качественное указание на присутствие в смеси
таутомеров энольной формы дает, согласно Друде [12], аномальная аб-
ссфбция электрических колебаний с короткой длиной волны. Такая
абсорбция свойственна вообще спиртовой функции (и отчасти кислотной).
Для примера укажем, что ацетонощавелевый эфир обладает сильным,
ацето-уксусный эфир, наоборот, едва заметным поглощением.
Чтобы дать некоторое представление о том, в какой зависимости
относительные пропорции таутомерных модификаций в равновесной
смеси находятся от строения вещества, приведем следующую табличку:
* Для желтой линии натрия, по Брюлю, молекулярная рефракция ацетоуксусиого
(этилового) эфира равна 31,92, между тем как кетонная форма требует величины 31,78,
а энольная СНз — С (ОН) = СН — СООС2Н5 — 32,72. Ныне расчеты Брюля признаны не
вполне точными. Ауверс, Эйзенлобр и др. внесли в них некоторые поправки, после чего
согласие выводов, полученных с помощью рефрактометрического и химического метода
(например метода Курта Мейера), оказалось вполне удовлетворительным.
** По новейшим исследованиям, содержание энольной фоимы в этом случае
близко к 80% [И].
***' Сборник не вышел. Лрим. ред.
;330 О неустойчивых органических соединениях
Содержание энольно іі
формы в жидкой смеси.
%
СН3-СО
Ацетилдибензоилметан /^~
(СвН6СО)3
Трибензоилметан (СвН6СО)3СН
Ацетоуксуснометиловый эфир СН3СО—СН2СООСН3 **,»
Ацетоуксусноэтиловый зфир СН3—СО—СН2
7,4
С2НвОСО
Бензоилуксуснометнловый эфир С6Н5—СО—СН2
СН3—ОСО
Бензоилуксусноэтиловый эфир С6Н6СО—СН2 90 ..
С2Н60-СО
Ацетилацетон СН3—СО—СН2
СН3—СО
Ацетондикарбоновоэтиловый эфир CHj—СО—СН2
СООСоН6 COOQHe
80,4
16,8
К кето-энольной таутомерии довольно близко примыкает обширный
класс явлений динамической изомерии, наблюдаемый среди
соединений, которые А. Ганш [13] назвал псевдокис л о тами и
псевдооснованиями.
Такие тела всегда появляются, в двух формах, из которых одна
является нейтральным телом, другая — или кислотой, или основанием.
Первичные и вторичные нитросоединения могут служить
превосходным примером псевдокислот. Сами по себе нитросоединения являются
нейтральными телами; растворы их не проводят тока и, следовательно,
не содержат ионов. Но известно, что при действии щелочей из нитро-
•соединений образуются соли. М. И. Коновалов, Ганш и Голлеманн [14]
одновременно показали, что при разложении этих солей кислотами
получаются изомерные тела, так называемые изонитросоедине-
н и я.
Систематическое исследование этих соединений, выполненное Ган-
шем и его учениками, показало, что они представляют собой кислоты,
действуют на индикаторы, дают соли со щелочами гораздо легче, чем
изомерные нитросоединения, из углекислых солей вытесняют СОз.
Растворы их проводят ток, но мало-помалу проводимость уменьшается и
¦параллельно происходит изомеіризация изонитросоединений,
превращение их в обыкновенные нитротела. С другой стороны, оказалось, что
изосоединения отличаются многими реакциями, свойственными эноль-
ным формам, например, дают характерные окрашивания с хлорным
железом, реагируют с фенилизоздианатом и пр.
Все это заставило Ганша прийти к заключению, что мы имеем здесь
дело с своеобразным случаем таутомерии. В одной из двух
модификаций (1) нитіротел
R_CH2—N02 R-CH-NOOH,
I II
которая является более устойчивой в свободном состоянии, кислотные
-свойства отсутствуют. Другая, так называемая аци-форма (II), обра-
О неустойчивых органических соединениях
331
зуется из первой путем изомеризации при действии щелочей. Она
непостоянна в свободном состоянии, но постоянна в виде солей. Только
аци-форма есть настоящая кислота. У ее изомера кислотные свойства
имеются только как бы в скрытой форме, а потому она и заслуживает
названия псевдокислоты.
Псевдокислотам совершенно аналогичны псевдооснования [15],
примером которых могут служить многие соединения аммониевого типа с
циклическим строением, в том числе и таких красок, как фуксин, метил-
виолет, метиленовая синька и пр. В этом случае настоящее основание
будет постоянно только в солях, выделенное же в свободном виде оно
будет обладать стремлением к переходу в более устойчивую карбиноль-
пую форму, например,
снгсн
I ^ I
снгсн
I с I
сн
//
\/\
сн
сн;
снх/ч/сн
c'hc,n
сн
СИ\/\/СН(ОН)
'\
СН, ОН
CHCN
СНз
Псевдооснование*
t^ClH4=N</
СН,
ОсноЕчДние
СН3\
ch,>n-c'h-'
СН3\ /~ """* У\ги
CH^N-C.H./
ОН
'б'Ч
Кристалл-виолет
(основание)
СН:
СН8
СНз
•СН.
сн,/
/>N-CeH44 /^-N\CH,
С
>N—СН*/ \0Н
Кристалл-виолет
(псевдооснование, карбинольная форма)
* На самом деле наблюдалось образование простого эфира этого карбинола.
Впервые такие переходы были прослежены Г. К. Деккером [16]
на пиридониевых и хинолониевых основаниях. Впоследствии Ганш
показал, что мы имеем здесь дело со столь же общим случаем, как в
ряду псевдокислот. И здесь оказалось возможным проследить
постепенный переход от основания к псевдоформе по уменьшению
электропроводности растворов.
VI
Интересный тип динамической изомерии встречается у многих
сахаристых веществ, принадлежащих к группе альдоз и кетоз. Как
известно, среди этих последних весьма обычны явления так называемой
332 О неустойчивых органических соединениях
мул ь тир от а ци и. Вращательная способность свежеприготовленных
растворов таких соединений в воде (и в некоторых других
растворителях) при хранении постепенно изменяется, пока не достигнет
предельной величины.
Так, для обыкновенной (правой) глюкозы начальное вращение в
водном растворе равно +113°, а конечное +52,5°. Для рамнозы
наблюдается даже перемена знака с переходом от —7° к +9,4°.
Достижение предельного вращения значительно ускоряется от
нагревания или от прибавления аммиака.
Очевидно, в растворе происходят какие-то химические процессы,
приводящие к изменению структуры, а быть может, и состава
растворенного вещества. Понятно, что уяснить себе сущность этих процессов
можно, только составив себе ясное представление о строении
сахаристых веществ. В качестве конкретного примера мы остановимся ближе
на глюкозе.
Общепринятая ныне формула глюкозы (и ее стереоизомеров: ман-
нозы, галактозы и пр.)
О
СН2(ОН)—СН(ОН)~СН(ОН)-СН(ОН)-СН(ОН)—С<^
н
была впервые предложена Р. Фиттигом в 1871 г. Но уже в 1883 г.
Тол лен с, а вслед за ним и некоторые другие химики, исходя из того
обстоятельства, что многие альдозы не дают таких типических реакций
на альдегиды, как окрашивание обесцвеченного сернистым газом
раствора фуксина и образование двойных соединений с бисульфитами,
предположили, что альдозам (а также кетозам) должно быть приписано
строение внутреннего ангидрида — окиси. Для глюкозы, например,
такое:
СН2(ОН)—СН(ОН)—СН—СН(ОН)—СН(ОН)—СН(ОН).
О
Предположение Толленса казалось, однако, противоречащим тому
обстоятельству, что глюкоза, равно как и другие альдозы, по отношению
к большинству характернейших реакций ведут себя, как типичные
альдегиды.
В особенности классические исследования Эм. Фишера заставили
большинство химиков склониться в пользу формулировки Фиттига.
По мнению Фишера, эта формула хорошо объясняет и явления мульти-
ротации. В частности, для глюкозы и для других Сахаров, содержащих
альдегидную группу, изменение вращения может имет> своей причиной
постепенно происходящую гидратацию альдозы с переходом от 'альде-
гидной группы — С<^ к ее бигидрату —НС(ОН)2. Однако
последующие работы Танре, а также английских химиков Армстронга и Лаури
показали, что явления мультиротации зависят от другой причины.
Вместе с тем оказалось, что обе формулы, предложенные для глюкозы (и
других сахаристых веществ), Фиттиговская « Толленсовская, имеют
право на существование, что мы имеем здесь своеобразный случай
динамической изомерии, причем дело еще осложняется стереохимическими
условиями. На почве этой динамической изомерии и получает вполне
О неустойчивых органических соединениях
333
удовлетворительное объяснение наблюдаемая изменчивость вращения
от времени.
Такая точка зрения была подготовлена работами самого Эм. Ф и ш е-
р а, которым была открыта способность многих альдоз (и кетоз) давать
со спиртами в присутствии хлористого водорода (как катализатора)
простейшие глюкозиды. Среди этих глюкозидов, которым Фишер
приписал циклическое строение, вполне отвечающее окисным формулам
Толленса, была обнаружена своеобразная изомерия. В частности, для
глюкозы были констатированы два ряда глюкозидов -а и Р, особенно
резко отличающихся по оптическим свойствам: а-глюкозиды,
отвечающие а-глюкозе, сильно вращают плоскость поляризации вправо, р-глю-
козиды—значительно слабее влево. Изомерии этой Фишер дает сте-
реохимическое объяснение, как видно из следующих формул,
отвечающих метилглюкозидам:
X X
сн3о—с—н
н-с -он
С-ОСН.
Н--С-ОН
Ни—С—Н
НО—С—Н
Н—С
СН,—ОН
Н—С—ОН
СН..ОН
[і-Глюкозид1
а-Глюкозид
іаіо + 157* [а]п-33(
Если принять для глюкозы альдегидную формулу, то при переходе
от нее к глюкозиду конфигурация около всех четырех асимметрических
атомов останется без изменения, но возникнет новый асимметрический
атом за счет альдегидной группы. Различие между а- и р-глюкозидом
и объясняется, по Фишеру, неодинаковым расположением в
пространстве атома Н и группы — ОСН3 вокруг этого нового асимметрического
углерода (в формулах он обозначен крестиками).
Очевидно, можно представить себе соответственно и две различные
конфигурации для исходного вещества глюкозы, если для последней
принять циклическую формулу Толленса.
Две изомерных модификации (безводной) глюкозы аа самом деле
приготовил Т а н р е, подвергая обыкновенную глюкозу кристаллизации
в различных условиях. Немедленно по приготовлении раствора обе
модификации вращают плоскость поляризации вправо, но одна (+113°)
значительно сильнее другой ( + 19°). С течением времени эта разница
выравнивается и обе модификации дают одну и ту же конечную
величину {чх]о =+52,5°, свойственную обычной глюкозе.
Армстронгу удалось показать, что при действии подходящих
энзимов на глюкозиды те из этих последних, которые принадлежат к
u-ряду (правовращающие), распадаясь, дают сильно вращающую
а-глюкозу. Наоборот, из левовращающих р-глюкозидов образуется сла-
бовращающая р-модификация глюкозы. Таким образом, связь между
изомерией глюкозидов и модификаций самой глюкозы была доказана.
Если принять во внимание, что взаимные переходы этих модификаций,
334
О неустойчивых органических соединениях
а также образование из них третьей альдегидной, до сих пор не
изолированной, должны происходить легко и быстро, то станет понятным,
почему с различными реактивами на альдегиды и кетоны равновесная
смесь, обладающая вращением [a] d =52,5,реагирует, как будто бы она
нацело состояла из альдегида. Лоури и Танре сделали интересную
попытку определить состав этой равновесной смеси, причем первый
пользовался для этой цели методом растворимости, второй — методом
оптическим. По Танре, в крепком водном растворе глюкозы (при
наступлении равновесия) содержится 40% а- и 60% (3-модификации.
Согласно Лоури, в таком растворе присутствует, кроме того,
известное количество альдегидной формы, а самый процесс взаимного
перехода а- и ^-модификаций, вероятно, происходит через посредство гидрата
альдегидной формы:
X Н-С(ОН)о У
но—с—н I н-с—он
н-с—он
\) _НО—С—Н
н—с—он
н-с-он
н—с—он
сн2он
Бигидрат альдегидной формы-
но-с-н
\ ,
Н-С
О+Н.,0
Н-С—ОН
I
СН.ОН
а -глюкоза
р-глюкоза
VII
В предыдущем мы остановились лишь на .немногих конкретных
примерах, на которых явления таутомерии были особенно подробно изучены
и лучше всего выяснена их истинная природа. Но во всех рассмотренных
случаях без исключения оправдалась та развитая выше точка зрения,
согласно которой таутомерия есть не что иное, как изомерия
обыкновенная, но только связанная с существованием форм подвижных, легко
и быстро переходящих друг в друга. И хотя среди множества
соединений, показывающих явления таутомерии, лишь для небольшого,
сравнительно, числа реально изолированы возможные по теории
модификации, однако число таких случаев непрерывно возрастает, а вместе с тем
постепенно стушевывается и исчезает та грань, которая отделяет их от
«идеальной» таутомерии типа цианистого водорода, циановой и родани-
стоводородной кислот и т. п.
Явления таутомерии, или динамической изомерии, позволяют нам
осветить вопрос, почему некоторые структурные схемы, предвидимые
теорией, не отвечают реальным или, по крайней мере, легко
осуществимым органическим соединениям.
Не подлежит сомнению, что большинство соединений углерода
находится в состоянии так называемого ложного или кажущегося
равновесия, что только пассивные сопротивления удерживают
их от перехода в другие, более устойчивые комбинации. В частности,
должна существовать тенденция к переходу друг в друга изомерных
форм, отвечающих одной и той же молекулярной формуле.
О неустойчивых органических соединениях
33S
С другой стороны, мы знаем, что по степени своей устойчивости
нзом-еры по большей части показывают лишь незначительную степень
различия между собой. На это, между прочим, указывает ничтожная
разница в соответствующих теплотах горения.
Можно думать поэтому, что и потеря свободной энергии при
переходе одного изомера в другой также незначительна. Дри таких условиях
становится весьма вероятным, что любое органическое соединение,
скажем, нормальный пентан, придя в состояние истинного равновесия,
должно превратиться в смесь изомеров (в данном случае трех
изомерных пептанов), не говоря уже о возможности образования высших и
низших гомологов (Вант-Гофф).
Если мы реально наблюдаем и можем изолировать большое число
изомеров, отвечающих общему составу, то лишь по той причине, что в
большинстве случаев имеем дело с ясно выраженными пассивными
сопротивлениями. В самом деле, когда условия реакции не
благоприятствуют проявлению пассивных сопротивлений, когда, например,
процесс совершается при очень высокой температуре или продолжается в
течение очень долгого (геологического) промежутка времени, то почти
всегда возникает пестрая смесь огромного числа изомеров и гомологов.
Такие условия имеются, например, при сухой перегонке дерева
или каменного угля или же при образовании нефти. В первых
двух случаях .первенствующую роль играет высокая температура, как
известно, ускоряющая все химические реакции, >в последнем, быть
может, на первый план выступает влияние времени.
Наоборот, когда пассивные сопротивления могут проявиться в
полной мере, многие реакции, в которых принимают участие органические
соединения, протекают гладко, нередко в одном лишь определенном
направлении, и продукты их легко изолируются в чистом виде.
Однако и в этом случае не все соединения ведут себя одинаково, и
в высшей степени замечательно, что разница между отдельными
случаями опять-таки определяется факторами, влияющими на степень
проявления пассивных сопротивлений. Состав соединений при этом играет
главную роль. Среди углеводородов, особенно предельных, если не все,
то большинство предсказываемых теорией формул отвечает реально
существующим индивидам. Но по мере того, как в молекуле
накопляются электроотрицательные элементы, например галоиды и
особенно кислород, возрастает число неосуществимых комбинаций,
г вместе с тем и число случаев динамической изомерии. Аналогичное
влияние оказывает и появление непредельных (кратных) связей. С этим
обстоятельством находится в очевидной связи то, что вступление в
молекулу таких элементов и атомных группировок (ненасыщенных)
нарушает электронейтральный характер, столь свойственный типическим
органическим соединениям, вносит в них электрическую полярность,
тем самым повышая их химическую активность, способность к быстрому
реагированию, нередко вызывая наклонность к электролитической
диссоциации.
Совершенно понятно, что среди подобных соединений наряду со
случаями таутомерии, допускающими хотя бы с трудом выделение и
чистом состоянии динамически изомерных модификаций, находится
немало случаев, в которых, невзирая на все попытки, вообще не удается
фактически констатировать, схватить эти модификации. Такова
динамическая изомерия синильной и роданистоводородной кислот и многие
другие. Это, однако, не значит, чтобы допущение изомеров здесь
являлось фантазией, чтобы попытки получить их должны были считаться
безнадёжными.
336 О неустойчивых органических соединениях
Но, помимо того, именно постепенность перехода между
самыми устойчивыми органическими изомерами, например изомерными
предельными углеводородами, и такими изомерами, как синильная
кислота *, заставляет признать, что между ними нет разницы
принципиального характера: есть только разница количественная, но не
качественная. Явления таутомерии потому и важны, что дают нам
яркую картину того, во что превратилась бы вся необъятная область
органической химии, если бы сразу повсюду порвались пассивные
сопротивления и тенденция к реагированию, к изомеризации, вообще ко
всевозможным 'переходам молекул друг в друга, получила полное и
немедленное осуществление.
* В качестве одной из промежуточных ступеней между предельными
углеводородами и соединениями, показывающими динамическую изомерию, можно указать на гало-
идгидрины спиртов. Давно известно, что йодистые и бромистые пропил, бутил и т. д.
изомеризуются при нагревании или при действии галоидных соединений алюминия
(Аронштейн, Густавсон, Кекуле. Эльтеков) в направлении первичное соединение -*.
вторичное -»- третичное (Марковников, Зайцев). Так, бромистый нормальный бутил изо-
меризуется в бромистый вторичный бутил, а бромистый изобутил — в бромид
третичного бутила и т. д. При обычной температуре и в отсутствие А1Вг3 эти процессы идут
слишком медленно, чтобы их с удобством можно было наблюдать, но достаточно
несколько повысить температуру или прибавить катализатора, чтобы реакции прошли
в короткий промежуток времени. Недавно А. Е. Фаворский и его ученики показали, что
подобного рода изомеризационные процессы являются обратимыми. Повышая
температуру, можно заставить реакцию пойти в обратном направлении, например, от
вторичного бромистого третичного бутила к изобутильному бромиду:
(СН3)зСВг *\СН—СН2Вг.
сн2/
Очевидно, здесь мы имеем дело с приложением принципа Вант-Гоффа — Ле-Ша-
телье. Возвышение температуры заставляет равновесие сдвинуться з сторону
поглощения тепла, в сторону образования продуктов менее стойких при более низких
температурах.
¦Весьма поучителен случай р- и умодиф-икаций гексахлоркетошіклопентена
С1 СЦ
с с
Сі | С/\ С1-С/\
Чсо
сі2=с\/ сі-с\/
^>со,
с с
II U
сі2 р сі2 г
взаимное превращение которых особенно подробно изучено Ф. Кюстером [17] с
динамической стороны. Мы имеем здесь дело с обратимой мономолекулярной реакцией. При
210,5° равновесие достигается в течение 75 час, причем в равновесной системе
содержится 61,4% у- и 38,6% р-модификации. Скорость процесса такова, что за ней можно
следить аналитически, пользуясь тем обстоятельством, что только fi- (но не у-) изомер
обладает способностью давать анилид. Анализ смеси ведут при 85°. При этих
условиях скорость превращения у-*Э еще очень мала и в течение 5 мин., необходимых для
опыта, заметного перехода не происходит. Если бы определение состава смеси
приходилось вести при 210,5°, то такой переход достигал уже 0,7%, а при 260° скорость
реакции y->P была бы столь велика, что состав равновесной системы вообще не удалось
бы определить точнее, получили бы тот результат, что вся система представляет
чистую ^-модификацию. Другими словами, при "260° мы имели бы дело с типическим тау-
томером типа синильной кислоты. Таким образом, меняя температуру, мы получаем
возможность по произволу переходить от обыкновенной изомерии (при комнатной
температуре скорость реакции уг?Р практически равна 0) к типической таутомерии.
О неустойчивых органических соединениях
337
VIII
Кетены {18]. Если при изучении тауюмерии мы столкнулись с
изомерами, очень близкими по степени устойчивости, притом легко
переходящими друг в друга и приводящими к равновесным системам, то в
кетенах, образующих новый, в высшей степени замечательный класс
органических соединений, недавно открытый (1905 г.) Штаудингером,
мы имеем пример малоустойчивых соединений иного рода. В этом
случае переход к устойчивой (обыкновенно полимерной) форме
сопровождается нередко значительным выделением тепла и притом является
необратимым (или, по крайней мере, возврат к неустойчивой форме
осуществляется лишь с трудом). Все указывает, с одной стороны, на
существенную разницу в степени устойчивости кетеиа и продукта, в
который он превращается, а с другой — на наличность, вообще говоря,
слабых, хотя в отдельных случаях весьма различных по силе пассивных
сопротивлений.
Кетены интересны не только по своей малой устойчивости и по
огромной способности к реагированию, но и потому, что в молекуле их
содержится совершенно своеобразная группировка атомов
которую, конечно, могла предусмотреть только теория строения. Однако
еще каких-нибудь лет 10 тому назад едва ли кто-нибудь им химиков
серьезно надеялся на возможность реализовать вещество, обладающее
строением кетена. Понятно после этого то общее внимание, которое
возбудило к себе открытие Штаудингера.
Основная реакция, с помощью которой Штаудингер впервые полу-
чил кетены, реакция, которая и в настоящее время является наиболее
общей и удобной для .получения этих соединений, первоначально была
им разработана для дифенилкетена (C6H5)2C = CO [19].
Бензиловая кислота (С6Н5)2С(ОН)СООН при обработке пятибро-
м истым фосфором дает бромангидрид дифенилуксусной кислоты
(СбН5ЬС • Вг—СО—Вг. Дифенилкетен был получен при действии на
это последнее соединение металлическим цинком в эфирном (или уксус-
но-этиловоэфирном) растворе
О
CtiIV
'эЧч'С— Вг--С-Вг -Zit =
С--С- О- -ZnBiv
Та же реакция позволяет получать и другие кетены. Так, из броман-
гидрида бром-изобут-ириновой кислоты получается диметилкетен.
СНЗЧ СНзЧ
;с-вг-совг -» )о=со.
сн:/ сн/
В некоторых случаях удавалось получить кетены из замещенных
малоновых кислот разложением (полимерных) ангидридов * этих
* Это соединение и до сих пор остается наиболее легко доступным и удобным для
экспериментирования кетеиом.
22 Л. А. Чугаеи. т. UI
338
О неустойчивых органических соединениях
последних. Так, получение диэтилкетена по этой реакции схематически
может быть выражено следующим образом:
СН\ /СООН С2Н5ч
/ \ /
с2н5 чсоон с2н/
Есть и другие способы получения кетенов, но они по большей части
не имеют общего характера, и мы не будем их здесь приводить.
Обзор главнейших до сих пор полученных кетенов и некоторых
физических свойств их содержится в прилагаемой табличке:
Кетен
Этилкетен
Фенилкетен
Диметилкетен
Диэтилкетен
Фенилметилкетен
Дифенилкетен
Дифеннленкетен
Метилвинилкетен
* Ни сама мал
/С0\
R2c< >о.
^сск
Температура
плавления, °С
Альдокетены
И2С=СО
ОгН5ч
сбн5ч
>с=о
—151
Кетокетены
СН3\
>С=СО
сн/
QH5V
)С-СО
СоНб^
СбН5ч
>с=со
сн3/
СвН6\
>с=со
| >С-СО
с6н4/
СН<>=^СНч
>с-со
си/
-98
—
—
0
90
оновая кислота, ни ее замещенные
Однако Эйгорну [20] ур
далось по;
Температура
кипения, °С
Цвет
—56
Бесцветный
+34
+91—92
+74 при
15 мм
+ 140 при
12 мм
—
Светло-желтый
Зеленовато-желтый
Светло-оранжевый
Оранжевый
Красный
Желтый
не дают нормальных ангидридов-
іучить поли
мерные ангидриды
/со\
(НгСч^ .,0)п. Для диэтилмалонового ангидрида в одном случае п было равно 4,.
а в другом— 12.
О неустойчивых органических соединениях
339
Кетены сами по себе распадаются на два подкласса —на альдо-
кетены RHC = CO, характеризующиеся присутствием незамещенного
водорода в группе
Н R
^С-СО, и на кетокетены, ^С-СО,
R R
где этот водород замещен радикалом R. Первые отличаются меньшей
устойчивостью и большей способностью к реагированию. С нашей точки
зрения, они поэтому представляют особый интерес. От полимеров,
образующихся из альдокетенов, нет возврата к исходным кетенам, между
тем как такой возврат для кетокетенов возможен. Кроме того,
кетокетены окрашены в различные оттенки желтого, оранжевого и красного
цвета, тогда как альдокетены в чистом состоянии бесцветны. Для кете-
нов прежде всего характерна способность к всевозможным реакциям
присоединения. Совокупность этих реакций не только позволяет
установить вне всякого сомнения присутствие в кетенах атомной
группировки С = СО, но и определить строение каждого кетона в
отдельности. У кетокетенов способность к присоединению выражена гораздо
резче, и самый процесс протекает, вообще говоря, более гладким
образом, нежели у альдокетенов, которые больше всего склонны к
полимеризации, отчасти приводящей к образованию весьма сложных
продуктов. Присоединение, как правило, совершается по месту двойной связи
между группой СО и неокислепным атомом углерода
4 С—СО.
Мы остановимся здесь только на нескольких важнейших случаях
реакций этого рода.
I. Кетокетены легко присоединяют свободный (молекулярный)
кислород и потому принадлежат к типичным самоокисляющимся
соединениям. Как всегда в таких случаях, по месту двойной связи
между двумя атомами углерода присоединяется целая молекула
кислорода 02. При этом образуются весьма нестойкие, нередко взрывчатые
перекиси, вероятно, по следующей общей схеме:
R О R4
:С:СО+
) С-СО
Rx I i
R О О-О.
При осторожном разложении этих перекисей получается кетон
R2CO и углекислый газ, например,
С2Н5ч С2Н5\
)С-СО = )СО+СН2.
С2Н/ | | С2Н5/
0-0
2. Ко всем кетенам без исключения присоединяется вода, галоид-
водороды и спирты, причем образуются в первом случае
свободные кислоты, во втором —их галоидангидриды и в третьем—сложные
эфиры. Например:
22*
340
О неустойчивых органических соединениях
сн,=с=о+н,о = сн:—соон.
Кетен Уксусная кислота
С6Н5\ 0,Н5ч
;>с=со-ьн,о - )сн-соон
с6н/ с6н/
Дифенилкетен Днфенилуксусная кислота
С6Н5\ CfiH5
>С-СО+НВг - СН—СОВг
сбн/ с,н5
Бромангидрид дифенил-
уксусной кислоты
СбН5\ QH54
\с-со+с2н5он = ^>ш-соос2н5
CeH5 QH5
Этиловый эфир дифенилуксусной
кислоты
3. Амины также присоединяются к кетенам, образуя замешенные
амиды:
QH5\ С6Н5\
pC=CO+CeH6NH2 - ^>C=GONH—CeH6.
С6Н5 С8Н5
Анилид дифенилуксусной
кислоты
Рассмотренные реакции позволяют смотреть на кетены скорее как
на ангидриды кислот
">сн-соон~> Ъе==са ±н*о,
сбн5 с6н5
чем как на кетоны и альдегиды (хотя формально мы имеем в них
карбонильную группу, не связанную с кислородом, как в ангидридах).
В соответствии с этим, для кетенов до сих пор неизвестны типичные
реакции яа альдегиды и кетоны.
4. Кетены весьма легко реагируют с так называемыми
основаниями Ш.иффа, например с бензилиденанилином, причем
присоединяющаяся частица содержит атомную группировку —CH = N—. В типичном
случае реакция эта ведет к образованию циклических соединений —
|}-лактамов( внутренних ангидридов р-аминокислот).
QH5\ /С6Н5 С8Н5 СбН5\ /С6Н5
с + ' с- со
11 сн ^ II
СО I" cH-N-QHb.
С6Н5 СбН*
Беизилидленанилин |3-Лактам
До-известной степени к реакциям подобного же рода принадлежит
действие кетокетенов на третичные основания, содержащие атомную
группировку С—N, например на пиридин и хинслин. В этом случае,
О неустойчивых органических соединениях
34t
однако, в реакции участвуют две молекулы кетена и образуется ше-
стичленное кольцо. Так, присоединение диметилкетена к
пиридину приводит к сложному основанию:
сн3 сн3
с со
сн / \С/Шз
СН\ >N~Ю ЧсН-
СН СН
С большею или меньшею легкостью к кетокетенам присоединяются
также соединения, содержащие атомные группировки C = S, N = 0 (нит-
розосоединения) и N = N (азосоединения), но мы не будем
останавливаться на этих реакциях.
К числу реакций присоединения кетенов относится и их
полимеризация, которая сводится к сочетанию между собою двух одинаковых
группировок >С = СО и протекает согласно схеме
R\ R\
>С=СО >С СО
R/j R/
.R
СО-С\ СО—С/
В результате образуются своеобразные дикетоны с тетраметилено-
вым циклом в частице. Тенденция к полимеризации у отдельных
кетенов весьма различна. Всего резче она выражена у альдокетенов, всего
слабее — у дифенилкетена.
Дикетены, получающиеся в результате полимеризации, в некоторых
случаях способны деполимеризоваться при нагревании с образованием
исходного кетена.
Эта реакция наблюдалась только у кегокетенов, и то не у всех. Легче
всего она протекает у продукта полимеризаций этилового эфира этил-
кетенкарбоновой кислоты именно при 200°. В других случаях она имеет
место только при о^ень высокой температуре (300—500°) или же и вовсі
не наблюдается *.
В заключение этого очерка остановлюсь в немногих словах на двух
представителях класса кетенов, у которых основная особенность этих
соединений — необычайно малая устойчивость и повышенная
способность к реагированию — выступает особенно резко, а именно на
простейшем из кетенов СИ2:СО и на своеобразной недокиси
углерода С302.
Кетен СН2: СО впервые получен в 1907 г. Вильсмором [21] при
пропускании паров уксусной кислоты или уксусного ангидрида над
накаленной платиновой спиралью.
СН8—СООН = =СН2 :СО+НА
Недавно Шмидлин и Бергман [22] нашли несравненно более
удобный способ получения этого любопытного вещества, состоящий в
пропускании паров ацетона через трубку, наполненную осколками
фарфора, при темно-красном калении.
СН3 -СО—СНз -СН2: CO-!-CHv
* В обоих случаях мы имеем дело с приложением принципа холодной и горячей
трубки Сен-Клэр Девилля.
М2
О неустойчивых органических соединениях
Присоединяя воду, кетен дает обратно уксусную кислоту, из которой,
как мы видели, может быть получен по Вильсмору, а с анилином прямо
образует ацетанилид CH3CONHC6H5 и, таким образом, является
удобным средством для введения в молекулу ацетильной группы (ацетили-
рования).
Кетен — бесцветный газ с неприятным едким запахом, крайне
непрочный, легко полимеризующийся и дающий при этом частью дикето-
со—сн2
циклобутан і | , частью смолообразные продукты. Особенно быстро
СН2 СО
полимеризация идет в жидком (сгущенном) кетене и в растворе. Она
начинается уже при —80° и очень быстро идет при 0°. Без изменения
кетен можно хранить только при очень низких температурах
(ниже —80°).
Замечательно, что появление едких паров кетена (а также
описываемой ниже недокиси углерода), несомненно, наблюдали раньше многие
химики, но, не применяя для их исследования низких температур, не
могли изолировать эти нестойкие соединения и доказать их
индивидуальность.
Недокись углерода. В 1906 г. Дильс и Вольф при действии
фосфорного ангидрида на малоновый эфир получили чрезвычайно
летучую и необыкновенно легко полимеризующуюся жидкость, состав
которой отвечает формуле Сз02. Авторы назвали ее поэтому недокисью
углерода (Kohlensuboxyd) и придали ей структурную формулу
о=с-с=с=о.
Позднее оказалось, что то же вещество удобнее может быть
получено из свободной малоновой кислоты при нагревании с Р2О5:
/СООН
/ ^с-о
СНо ,-, се; --2НХ)
\ хС-0
\соон
или же из хлорангидрида и диброммалоновой кислоты CBr2(COCIh
и цинка по способу Штаудингера.
Неустойчивость недокиси углерода такова, что для получения ее в
чистом состоянии приходится сразу сгущать выделяющийся газ [23] до
температуры жидкого воздуха (—180°). При более высокой температуре
С302, хотя и медленно *, полимеризуется, переходя в какой-то твердый
ближе неисследованный продукт, окрашенный в красный цвет. При
комнатной температуре полимеризация заканчивается в течение 2 суток.
Недокись углерода сгущается в жидкость при +7° и застывает 'При
— 107°. Она принадлежит к числу веществ, необыкновенно склонных к
химическим превращениям. С этим обстоятельством, вероятно, стоит в
связи и ее сильная ядовитость.
Открытие Дильса было встречено с понятным интересом. Недокись
углерода обратила на себя всеобщее внимание не только потому, что
она представляет из себя новый хорошо индивидуализированный окисел
углерода, что она отличается многими поразительными химическими
* Медленнее, нежели вышеописанный кеген СН2 = СО.
О неустойчивых органических соединениях
343
свойствами, но еще и потому, что открытие ее почти совпало по
времени с открытием кетенов, к которым она естественным образом
примыкает. Реакции недокиси углерода заставили признать в ней присутствие
двух кетенных группировок, а потому она и была признана за
простейший * дикетен.
Так она с двумя молекулами воды дает малоновую кислоту
ХО /СООН
С^ +2Н20=СН2/
с двумя молекулами анилина — малоанилид СИгСССЖНСбНбЬі
с двумя молекулами спирта — малоновый эфир СН2(СООС2Н5)2
и т. д.
Физические свойства недокиси углерода (величина ее молекулярной
рефракции) также гармонируют с принятым для нее строением (Дильс).
История кетенов позволяет надеяться, что, следуя путем,
аналогичным тому, который привел к открытию этого интересноги класса
органических соединений, будут получены еще многие другие категории
химически индивидуальных веществ, существование которых по теории
строения является возможным, но для которых еще не установлены ни
условия их образования, ни степень устойчивости.
ЛИТЕРАТУРА
1. A.Besson. С. R.. 1894, 118, 1347.
2. С. В. Л е б е д е в. Исследования в области полимеризации полиэтиленовых
углеводородов. ОПб, 1913.
3. V.Meyer. Die Thiophengruppe.
4. А.М.Бутлеров. ЖРФХО, ч. хим., 1877, 9, 68.
5. С. Laar. Ber., 1885, 18, 648; 1886, 19, 730; 1901, 34, 3516; Ann. Chem., 1877, 189, 77.
6. L. Knorr. Ber., 1911, 44, 1147.
7. L. С 1 a i s e п. Ann. Chem., 1896, 291, 25.
8. L. Knorr. Arm. Chem., 1897, 293, 70; 1898. 303, 133; 1899, 306, 332.
9. K.H.Meyer. Ann. Chem., 1911, 380, 212; Ber., 1911, 44, 2718; 1912, 45, 2843.
10. J.W.Bruhl. Ber., 1892, 25, 366.
11. W.H. Perk in. J. Chem. Sot, 1892, 61, 800.
12. Drude. Ber., 30, 940.
13. A. Hantsch. Ber., 1899, 32, 575.
14. A.Hantsch. Ber., 1896, 29, 699; 2251; M. Коновалов. Ber., 1896, 29, 2193;
A.F.HoIleman. Rec. Trav. Chim. Pays-Bas, 1895, 14, 129; 1896, 15, 366; 1897,
16, 162.
15. A.Hantsch, M.Kalb. Ber., 1899, 32, 3109.
16. H. Decker. J. prakt. Chem., 1892, 45, 161.
17. F.Kuster. Z. phys. Chem., 1895, 18, 161.
18. H.Staudi'nger. Der Ketene. Stuttgart, 1912.
19. H.Stau dinger. Ber., 1905, 38, 1735.
20. A. Einhorn. Ann. Chem., 1908, 359, 145.
21. N.T.Wilsmore. J. Chem. Soc, 1907, 91, 1938.
22. J. Schmidlin, M.Bergman. Ber., 1910, 43, 2821.
23. O. Die Is, B. W о 1 f. Ber., 1906, 39, 689.
He считая теоретически возможного 0=С = С = 0.
ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
В ПРИЛОЖЕНИИ К МИНЕРАЛЬНОМУ АНАЛИЗУ
Сборник, посвященный Клименту Аркадьевичу Тимирязеву
его учениками, М., 1916 г.
Отдельные органические соединения, как известно, давне уже стали
находить себе применение в минеральном анализе. Достаточно
напомнить употребление крахмала в качестве реактива на иод, таннина —
на окисное железо, винной кислоты на калий, бруцина и дифениламина
на азотную кислоту. Большую давность имеет также за собой
употребление некоторых органических красок в качестве И'нди'каторов при
объемном анализе, а равно и пользование такими органическими
растворителями, как спирт, хлороформ, сероуглерод и др.
Но до сравнительно недавнего времени число случаев такого
применения органических веществ для целей анализа все же оставалось
коайне ограниченным. Между тем, не подлежит сомнению, что именно
среди органических соединений, при их огромном числе и разнообразии
свойств, следует искать наиболее чувствительных реагентов, способных
обострить и довести до максимума даже самые тонкие и
незначительные различия между родственными элементами и их соединениями *.
Не подлежит сомнению, что до сих пор лишь малая часть
фактического материала, заключающегося в химии углеродистых соединений,
нашла себе применение к анализу, а потому можно только
приветствовать тенденцию к широкому и возможно всестороннему использованию
этого материала, которая все резче и резче обозначается за последние
годы. Об этом новом течении'в аналитической химии и о его
жизнеспособности до некоторой степени может, свидетельствовать длинный
список органических реагентов, нашедших себе применение к
минеральному анализу в течение последних десятилетий. Сюда относится,
например, нитрон (дифенилендианилодигидротрйазол), предложенный Бушем
[1] для открытия азотной кислоты и для количественного ее
определения; фуксиносернистая кислота, представляющая чувствительный
реактив на бром ;[2]; диоксивинная кислота, позволяющая открывать и
количественно определять натрий [3]; .бензидин и толидин [4], предложенные
для количественного определения и отделения кислот серной, вольфра-
* То же самое следует сказать о соединениях комплексных, которые по своему
характеру, а нередко (при наличности органического компонента) и по составу столь
близки к органическим. Применение отдельных комплексных соединений к анализу
так же старо, как применение соединений органических. Замечательно, что среди давно
вошедших во всеобщее употребление аналитических реакций большинство самых
чувствительных связано с применением комплексных соединений в качестве реагентов или
с образованием таковых при реакции. Сюда относятся, например, применение желтой и
красной соли для распознавания железа и меди, аммиака — в качестве реактива на
медь, родашетых солей — на окисное железо -и на кобальт (проба Фогеля), ніитропр'ус-
сидных солей —на серу (S~~) и пр. Многие характерные реакции, основанные на
употреблении органических соединений, в свою очередь связаны с образованием
комплексов (см. ниже).
Органические соединения в приложении к минеральному анализу 345
мовой, фосфорной и др.; нитрозонафтол [5], позволяющий количественно
осаждать кобальт в присутствии никеля; дициандиамидин — реактив на
никель [6]; 1,2-диамино-антрахинон-З-сульфоновая кислота —
чувствительный реактив на соли окиси меди [7]; ксантогеновокалиевая соль,
предложенная для определения меди [8] объемным путем; нитрозофе-
нилгидроксиламин (купферрон), позволяющий быстро и удобно
отделять железо от меди и никеля [9] и титан от алюминия {10];
салициловая кислота — реактив на окисное железо, пригодный для
колориметрического определения последнего; гематоксилин [11] и дифенилкарбазид
[12] —реактивы на медь; глицерин —на марганец [13]; диалкилтиокар-
баминовые соли — на железо и медь [14]; диоксималеиновая кислота —
на титан [15]; дитиощавелевая кислота — на никель и на палладий [ 16J
и многие другие. Кроме того, необходимо еще упомянуть о большом
числе различных вновь предложенных индикаторов (главным образом
для алкалиметрии и ацидиметрии).
В настоящей заметке я предполагаю ближе остановиться на
применении двух органических соединений, из которых одно, диметилглиок-
сим, было уже введено мною в аналитическую практику несколько лег
тому назад и теперь предлагается в качестве реагента на железо,
другое — третичный бутилкарбиламин вводится впервые, как реагент на
платину.
Диметилглиоксим
сн3—с—с—сн3.
II II
NOH NOH
Аналитическое применение диметилглиоксима основано на
способности его, присущей в равной мере и другим 1, 2 (или и)-диоксимам
R—С С—R
}\ II
NOH NOH
образовать с солями целого ряда тяжелых металлов VIII группы
периодической системы характерные, по большей части, весьма
устойчивые комплексные соединения, относящиеся к категории внутренних
комплексных солей и получившие название диоксиминов. Соединения
эти характеризуются присутствием своеобразной атомной группировки
і >Ме( і
R-C-NOI-Г NNOH=C—R,
в которой окснмидные группы частью обменивают свой водород на
металл и функционируют как кислотные остатки, частью играют роль,
аналогичную роли аммиака в металло-аммиачных соединениях.
В аналитическом отношении представляют интерес диоксимины
никеля, палладия, платины, кобальта и железа.
Диметилглиоксим, который из всех а-диоксимов наиболее пригоден
для аналитических целей, дает с солями этих металлов следующие
реакции *.
R С С R
* В дальнейшем молекулу диоксима / V 5! буду для сокращения
NOH NOH
изображать символом DHji причем особо отмечаются водороды оксимидных групп
(NOH) • Ру = пиридину.
346 Органические соединения в приложении к минеральному анализу
Название
металла
Никель
Ni
Палладий
Pd
"Платина
Pt
Кобальт
Со
Железо
Fe
Соединения,
дающие реакции
Соли
двухвалентного никеля NiX2
Соли
двухвалентного палладия,
еще лучше хло-
ропалладиты,
например K2PdCl4
Хлороплатиниты,
например КоРЮ4
Соли закиси
кобальта СоХ2
Соли закиси
железа FeXo
Условия реакции
Нейтральная, или слабо
щелочная среда,
особенно в присутствии
аммиака
В присутствии
уксуснокислого аммония и
уксусной кислоты при
нагревании с
последующим охлаждением
Как для Pd, но осадок
появляется лишь спустя
некоторое время
Встряхивание с воздухом
'в присутствии избытка
аммиака, затем
прибавлением (NH^S
В присутствии аммиака,
пиридина и пр.
С диметилглиоксимом получается
Ярко-красный
кристаллический осадок (иглы),
летучий без разложения под
уменьшенным давлением
Ni (D2H2)
Ярко-желтый
кристаллический осадок, состоящий из
длинных игл Pd(D2H2),
улетучивается как Ni-coe-
динение
Сине-зеленый
кристаллический осадок Pt (D2H2).
Остальные свойства, как
для Pd- и Ni-соединений
В первую стадию реакции
получается желто-бурый
раствор, содержащий,
между прочим, соединения
типа Co(D2H22NH3)X.
От прибавления сернистого
аммония —
красно-фиолетовое окрашивание
Ярко-красное окрашивание,
обусловливаемое
образованием соединений ряда
FeD2HrnA, r*eA = NH3
Ру и т. д.
Из приведенного сопоставления видно, что все только что
перечисленные металлы могут быть характеризованы посредством диметил-
глиоксима, но особенно чувствительна реакция с никелем, на что мною
и было обращено внимание еще в 1905 г. [17]. Предел ее
чувствительности лежит при содержании 1 части Ni в 1000 000—1500 000 частей
воды.
Диметилглиоксим может служить и для открытия Ni в присутствии
Со (0,1 мг никеля в присутствии более чем 5000-кратного количества
Со) *, Fe и многих других металлов, а также для воспроизведения
микрохимической реакции на Ni [18].
Наконец, на употреблении того же реактива основано
количественное (весовое) определение никеля, отличающееся большой точностью
и быстротой **.
* Для воспроизведения реакции к испытуемому раствору прибавляют избыток
аммиака и встряхивают в течение нескольких минут (для превращения дзухвалентного
¦Со в трехвалентный), затем добавляют некоторый избыток диметилглиоксима, нагре-
вают^ до кипения и по охлаждении отфильтровывают. Осадок, промытый на фильтре
водой, при наличности Ni окрашен в красный или розовый цвет. Отделение Ni от Со
может быть так-им образом осуществлено количественно.
** Приводим оггисание метода з той форме, в которой он употребляется в нашей
лаборатории. К раствору, содержащему никель, нагретому до кипения, прибавляется
такое количество аммиака, чтобы запах его был резко заметен, и избыток 1%-ного
спиртового раствора диметилглиоксима, после чего энергично перемешивают и оставляют
жидкость на холоду в течение около двух часов. (Рекомендуется вести анализ в раз-
Органические соединения в приложении к минеральному анализу 347
В последнее время был предложен совершенно аналогичный способ
для определения палладия и для отделения его от других металлов
платиновой группы (кроме платины).
Не останавливаясь подробнее на всех этих методах, так как они
уже описаны в литературе [19—25], перехожу теперь к применению ди-
метилглиоксима в качестве реагента на железо. Разработка сюда
относящейся методики произведена совместно с моим сотрудником,
оставленным при университете Б. П. Орелкиыым
Реакция с днметилглиоксимом и аммиаком на соли железа
получается в том лишь случае, если последние отвечают ряду закиси.
Наоборот, железо в трехвалентном состоянии, по-видимому, совершенно
лишено способности образовать типичные диоксимины. Поэтому, если
железо присутствует в виде соли окиси, то его следует предварительно
восстановить. Но присутствие восстановителя необходимо и в том
случае, когда железо находится в закисной форме, особенно если дело
идет о разбавленных растворах, так как в противном случае при
доступе воздуха легко могло бы произойти окисление и переход Fe++ в
Fe+++.
Подходящий восстановитель, нисколько не мешающий реакции,
удалось найти в гидразине N2H4. Для воспроизведения реакции к
раствору, в котором предполагается присутствие железа (в форме соли,
¦образующей Fe+H" или Fe+"h+ ионы), прибавляется небольшое
количество сернокислого гидразина, спиртовый раствор диметилглиоксима и
.аммиак в избытке. Раствор нагревают до кипения и затем охлаждают.
Слабое, но еще явственно заметное розовое окрашивание жидкости
наступает уже при содержании L части железа в 100 000 000 частей
воды. Таким образом, мы имеем здесь самую чувствительную из числа
цветных реакций на железо, в чем мы могли убедиться путем
непосредственных сравнительных опытов.
Присутствие щелочных металлов, а также магния (в 100-кратном
против железа количестве) не вредит реакции, но присутствие At и
Zn сильно понижает ее чувствительность.
Для количественного колориметрического определения железа опыт
ведут совершенно таким же образом, как только что описано, но с
соблюдением строго определенных условий относительно количества
реагентов, продолжительности нагревания и пр.
Удобно работать с объемом испытуемого раствора 100 смг. Гидра-
зинсульфат берется в количестве 1 г. По прилитии 5 см? насыщенного
при комнатной температуре диметилглиоксима, раствор нагревают до
кипения, прибавляют 10 cmz 25%-ного аммиака, продолжают кипятить
в течение 30 сек. и затем быстро охлаждают током воды из водопрово-
бавленном растворе и при избытке NH3, который не вредит. После прибавления диокси-
ма следует убедиться в том, жидкость сильно ли пахнет аммиаком, и в противном
случае добавить этого последнего. Недостаток NH? сильно замедляет последующее от-
фильтровьгвание осадка).
Образовавшийся ярко-красный осадок соединения
CHg-C = ONv /NO-C-CHg
>Ni(
сн2—c=noh/ Nsioh-c—сня
отфильтровывается на взвешенный фильтр (бумага Шлейхера и Шелля № Г>75),
вложенный в тигель Гуча или Нейбауера. До опыта с фильтром сушат до постоянного
веса при 120° и затем сохраняют в эксикаторе. Отфильтрованный (обычным путем с
помощью насоса) осадок промывается кипящей водой и сушится при 120° до
постоянного веса и по охлаждении в эксикаторе взвешивается. Сушение до постоянства веса
каждый раз требует от 1/2 до 3/4 часа времени.
348 Органические соединения, в приложении к минеральному анализу
да. Одновременно таким же образом при возможном соблюдении тех
же условий обрабатывается 100 см3 раствора, в котором содержание
железа заранее известно. Оба раствора, испытуемый и контрольный,
доводятся при комнатной температуре до определенного объема,
например до 125 of3, без промедления переносятся в колориметр, в
котором и производится сравнение интенсивности их окраски.
Описанный способ позволяет определять абсолютные количества
железа в несколько десятых долей миллиграмма (0,2—1 мг) с
точностью до 0,5%. В присутствии 100-кратного количества магния ошибка,
метода несколько больше и может доходить до 1%.
НОВАЯ ЧУВСТВИТЕЛЬНАЯ РЕАКЦИЯ НА ПЛАТИНУ
Карбиламины, или изонитрилы, образуют с комплексными солями
двухвалентной платины A42PtX4, например с хлороплатинитами K2PtCl4
три ряда соединений [26J, в которых карбиламин играет ту же роль,
что NH3 в платиново-аммиачных комплексах:
[Pt2RNCX2], [Pt4RNC] X2, [Pt4RNC] PtX4.
I II III
Соединения, принадлежащие к I типу, являются аналогами солек
Пейроне и 2-го основания Рейзе tPt2NH3X2], а соединения II типа
аналогичны солям 1-го основания Рейзе {Pt4NH3]X2, соединения III типа-
зеленой соли Магнуса [Pt4NH3][PtCl4].
При встряхивании растворимых хлороплатинитов, например КгРіСі*
или (NtVbPtCU с карбиламинами в присутствии воды образуются
соединения, отвечающие типу III:
2K2PtCl4+4RNC = [Pt4RNC]PtCl4+4KCl.
При действии избытка карбил амина соединения эти переходят в
раствор, образуя соединения типа II:
[Pt4RNC] PtCl4+4RNC-2 [Pt4RNC] Cl2.
Путем взаимодействия этих последних с растворимыми
комплексными солями типа [PtX4]Me2 можно вновь вернуться к двукомплексным
солям типа III, например:
[Pt4RNC] С12+К2 [Pt (CN)4] = [Pt4RNC] (Pt (CN)4)+2KC1.
На этих превращениях и основывается предлагаемый новый способ
открытия малых количеств платины. Для получения нужного реактива
сначала, исходя из третичнобутилового карбиламина (СНз)зСЫС,
приготовляется по предыдущему хлороплатинит [Pt4C4H9NC}PtCl4
(кристаллический порошок красного цвета, очень мало растворимый в воде), а из
него путем обработку некоторым избытком C4H9NC (для опыта
достаточно взять 0,005—0,012[Pt4C4H9NC]PtCl4) водны и р аствор хлор и да
[Pt4C4HsNC]Cl2. Последнюю реакцию необходимо вести при
охлаждении льдом и самый реактив (приготовленный непосредственно перед
употреблением) хранить в ледяной воде. В противном случае крайне
мало устойчивый комплексный хлорид [Pt4C4H9NC]Cl2 разлагается в
самый короткий промежуток времени*.
* Разложение это тесно связано с третичной природой радикала С4Н9,
обусловливается легкой омыляемостью, свойственной его производным. При хранении водного
раствора хлорида (Pt4CjH9NC]Cl2, еще легче при нагревании, происходит следующая
реакция:
[Pt4C4H0NC] Clo + 2Н20 - [Pt2C4H9NC] (CN) J + 2C<H9OH + 2HCI.
Органические соединения в приложении к минеральному анализу 349
Для того, чтобы с помощью этого реагента открыть платину, последняя
должна присутствовать в виде растворимой комплексной цианистой соли
M^[Pt(CN)4] (цианоплатинита). Прибавление раствора [Pt4C4H9NC]Cb
к раствору цианоплатинита вызывает образование осадка,
окрашенного в ярко-малиновый цвет и флуоресцирующего прекрасным зеленым
цветом. Реакция сводится к образованию двухкомплексного
цианоплатинита [Pt4C4H9NC]|Pt(CN)4] и выражается следующим равенством:
[Pt4C;H9NC]CU-h[Pt(CN4)]Ko-4Pt4C4H9NC][Pt(CN).I]+2KCl.
Если раствор цианоплатинита взять очень разбавленный, то осадок
остается взвешенным в жидкости. Чувствительность реакции такова, что
позволяет открыть платину в растворе, содержащем 1 вес. часть ее в
200 000 частей воды.
Если платина имеется в виде хлороплатината Me2PtCU, то
последний переводят в цианоплатинит посредством кратковременного
нагревания с цианистым калием. Избыток последнего необходимо разрушить,
так как он вредит реакции.
ЛИТЕРАТУРА
1. M.Busch. Berichte, !905, 38, 861; 1906, 39, 1401.
2. G.Denigc г. С. R., 1912, 155, 721.
3. Н. J.H.Fenton. J. Chem. Soc, 1898, 73, 178, Ср. id., 67, 48.
4. W.Muller. Berichte, 1902, 35, 1587; G.Knorre. Berichte, 1905, 38, 783.
5. M. I ii nski, G.Knorre. Berichte, 1885, 18, 699.
6. H. Grossman n. Berichte, 1906, 39, 3356.
7. R.Uhlenhuth. Chem. Ztg, 1910, 34, 887.
8. B. Oddo. Atti R. Acad. Lined., 1903, (5), 12, 435.
9. O. Baudisch. Chem. Ztg, 1909, 33, 1298.
10. I.Bel lucci, L. Grassi. Chem. Zbl, 1913, II, 716.
11. H.C.Bradley. Am. J. Soc, 1906, 22, 326.
12. P. С azeneuv е. С R., 1900, 130, 1561.
13. N.Tarugi, Gaz. Chim. Itaf, 1906, 36, I, 332.
14. M.Delepine. Bull. Soc. chim., 1908, 3, 652.
15. H.J.H. Fen ton. J. Chem. Soc, 1908, 93r 1064.
16. H.O.Jones, Robinson. J. Chem. Soc, 1912, 101, 932.
17. Л.А.Чугаев. ЖРФХО, 1905, 37, 243; Berichte, 1905, 38, 2520.
18. F.Emich. Lehrbuch der Microchimie. Wiesbaden, 1911, S. 97.
19. Л. А. Ч у г а е.в. Исследования в области комплексных соединений. М., 1906. (См.
Л.А.Чугаев. И:л>р. труды, т. I. М., Изд-во АН СССР, 1954, сгр. 10. Прим. ред.).
20. Л.А.Чугаев. Berichte, 1905, 38, 2520.
21. Л.А.Чугаев. С. R., 1907, 145, 679, 124G. (См. Л. А. Чу га с в. Избр. труды, т. I.
X, Изд-во АН СССР, 1954, стр. 156. Прим. ред.).
22. K.Kraut. Z. angew. Chem., 1906, 19, 1793.
23. O.Brunk. Z. angew. Chem., 1907, 20, 834.
24. H.W.Armit, A. H a r d e n. Proc. Roy. Soc, 1906, 77, В., 422.
25. M.W under, V. T h ii г i n g e r. Z. analyt. Chemie, 1912, 51, 740; 1913, 52, 101, 660,
661, 737, 740.
26. Л.А.Чугаев, П. Я-Те ар у. Berichte, 1914, 47, 568 и два сообщения в ЖРФХО
за текущий год (см. Л. А. Чу га ев. Избр. труды, т. I. М., Изд-во АН СССР, 1954,
стр. 432. Прим. ред.).
ОТКРЫТИЕ'КИСЛОРОДА И ТЕОРИЯ ГОРЕНИЯ
В СВЯЗИ С ФИЛОСОФСКИМИ УЧЕНИЯМИ ДРЕВНЕГО МИРА
Петроград, 1919 г.
ВВЕДЕНИЕ
Открытие кислорода принадлежит к числу величайших событиц в
истории развития человеческой мысли*. В частности, в истории
химической науки значение этого открытия усугубляется тем
обстоятельством, что по времени оно совпало с коренным переворотом, с
настоящей революцией (по выражению М. Бертло) в области химических
воззрений, с зарождением в конце XVIII в. новой, как тогда говорили,
антифлогистической, или французской, химии, причем оба эти события
находились между собой в состоянии глубокой и тесной связи, как
бы взаимно пополняя и освещая друг дрУга.
Если великий Лавуазье был творцом новых воззрений,
гениальным реформатором химической мысли, то англичанину Пристлею
обыкновенно приписывают честь открытия кислорода. К этому во всех
учебниках химии мимоходом добавляется, что Лавуазье имел
нескольких предшественников, которые, хотя и в менее ясной форме,
высказали подобные же мысли относительно сущности явлений горения и
состава воздуха, и что шведский химик Шееле (1742—1786 гг.) почти
одновременно с Пристлеем осуществил получение кислорода.
На самом деле история антифлогистической химии так же, как и
история открытия кислорода, гораздо длиннее и сложнее, чем это
обыкновенно принято думать, и богата многими интереснейшими
подробностями. И хотя отдельные эпизоды этой истории давно уже сталі>
известными химикам, более полная картина раскрылась сравнительно
недавно, после внимательного изучения ряда подлинных документов,
относящихся к различным эпохам далекого прошлого**.
В то время, к которому восходит начало этой истории, и даже
значительно позже не только развитие самой науки совершалось
несравненно более медленным темпом, чем ныне, но и сделанные открытия,
новые обобщения и теории за отсутствием удобных средств для
быстрых сношений, в частности, за недостатком специальных
периодических изданий лишь с трудом прокладывали себе дорогу и не всегда
становились известными более широким кругам современников; многие
из них просто затеривались и забывались, чтобы впоследствии
сделаться достоянием истории. Так было с вопросом, историческое
освещение которого составило предмет настоящего очерка. Над этим во-
* Либих сравнивает это открытие по его значению с открытием книгопечатания
Гуттекбергом и закона всемирного тяготения Ньютоном. (L i e b i g. Reden und Abhand-
lungen. Heidelberg, 1874, S. 14).
** В этих исторических исследованиях особенно видное участие приняли: М.
Бертло во Франции, С. Иёргенсен в Дании, Н о р денскь'ел ь д в Швеции,.
И. Кальбауми М. Шпетер в Германии» а у нас, в России, проф. Б. Н. М е н ш у т-
к и н. Трудами этих авторов я пользовался при составлении настоящей книги (см. ниже-
библиографию).
Открытие кислорода и теория горения
351
просом трудился целый ряд ученых и мыслителей, принадлежащих
почти всем культурным странам мира. Их деятельность обнимает
более чем тысячелетний период времени. Надо думать, что и сейчас,
несмотря на тщательные поиски первоисточников, имена многих деятелей
на этом поприще остаются еще неизвестными и ищут своего историка.
Чтобы уяснить себе причины необычайно медленного хода
развития наших знаний относительно состава воздуха и относительно его
участия в процессах горения, нужно обратить внимание на ту
обстановку, в которой приходилось работать действующим лицам этой
великой химической драмы. Можно указать на ряд факторов,
тормозивших решение вопросов, столь простых и легких, что в настоящее
время мы считаем их доступным-и пониманию детей. Но среди этих
факторов на первом месте и далеко впереди других нужно поставить-
своеобразные духовные тиски, в течение более тысячелетия сжимавшие
человеческую мысль, мешавшие ее свободному развитию и нередко
направлявшие ее наперекор простому здравому смыслу. Наука и
философия того времени (эти два слова, впрочем, означали почти одно и
то же) находились под неограниченным господством учений, в корне-
своем противоположных тем, которые лежат в основе современного
естествознания. Эти учения кажутся нам странными и искусственными,,
но во время своего возникновения они ознаменовали собой
несомненный прогресс в области философии, явившись противовесом наивному
миросозерцанию первобытного человека, безусловно, полагавшегося на
свидетельство своих органов чувств и не обладавшего искусством
подвергать эти свидетельства сопоставлению и критической проверке.
Основы философского учения, о котором я говорю, обычно
связываются с именем Аристотеля из Стагиры, но начало его на самом
деле относится к более ранней эпохе; его можно проследить еще в
философских системах древних индусов, египтян, вавилонян и других
пародов Востока, в учениях, сложившихся частью задолго до рождения
Стагирита. С достоверностью установлено, что сам Аристотель многое
заимствовал у своего учителя Платона и частью, подобно этому
последнему,— у Эмпедокла. На учениях этих философов нам
необходимо поэтому остановиться в дальнейшем.
I. ГРЕЧЕСКАЯ ФИЛОСОФИЯ. ПЛАТОН, ЭМПЕДОКЛ
В основе философии Платона (427—347 гг. до н. э.) лежит
представление о двух различных мирах, которые он противопоставляет один
другому: мир идей и мир чувственного опыта. Только мир идей
представляется ему истинно реальным, раскрывающим самую сущность
вещей. Наоборот, мир чувственного опыта является лишь собранием
теней или искаженных и изменчивых отражений действительности. Эту
мысль Платон в одном из своих диалогов поясняет следующим
красивым сравнением. Видимый кажущийся мир подобен пещере, в которую
заключены узники, крепко прикованные на цепях. Пещера ярко
освещена, но в нее ведет единственный вход, к которому пленники
обращены спиной. Все, что они могут видеть — это тени, отбрасываемые
предметами, проходящими мимо входа, и эти тени кажутся узникам
действительностью. В такие условия, говорит Платон, поставлены и
люди. Подземелье — это видимый мир; огонь, освещающий пещеру —
солнце; тени —наши ощущения, а знание теней —чувственное знание.
Только философия может освободить человека от сковывающих его
мысль цепей и дать ему истинное представление о действительности..
Такой действительностью является мир идей.
352
Открытие кислорода и теория го-рения
Что такое идея? Этим именем Платон обозначает все то, что есть
общего, неизменного, вечного в вещах, доступных чувственному опыту.
«Существованием идей обусловливается возможность родов, типов,
законов, которым подчиняется чувственный мир». Каждая идея есть
некоторый принцип, соединяющий в себе совокупность известных качеств,
сообщающий их тем или другим вещам и характеризующий эти
последние. Есть идея красоты, идея человека, животного, цветка...
Благодаря существованию этих идей возможно все доброе, все прекрасное;
возможно существование человека, животного, цветка... {1]. С течением
времени взгляды Платона на сущность идей подвергались изменениям.
В некоторых своих сочинениях он характеризует идеи как первообразы
реальных вещей, созерцая которые, творец создает эти последние, как
по определенному образцу. Согласно другим источникам, идеи скорее
являются «безусловными нормами, вечными пределами бытия».
Каким образом 'происходит явление идей в вещах видимого м.ира?
В чувственном мире, согласно Платону, іидеи никогда не находятся,
так сказать, в чистом виде. Они, правда, никогда не сочетаются между
собой по нескольку в одном и том же объекте, ибо это противно их
природе. Но в вещах чувственного мира есть нечто, сочетающееся с идеями,
изменяющее и, быть может, -искажающее их сущность, даже
извращающее их до противоположности. Поэтому Платон допускает
существование принципа, отчасти противоположного идеям. Представление об
этом щринци-пе и о связи его с идеями, как ,и представление о самих
идеях, довольно сильно -варьирует в различных сочинениях Платона,
в зависимости от той стадии эволюции мысли, которую в момент
создания данного диалога переживал философ. Одни комментаторы
думают, что этот принцип, который сам Платон обозначает, как
«несуществующее», принцип, составляющий, так сказать, внешнюю
оболочку идеи в каждой вещи, воспринимаемой чувствами, соответствует
современному понятию о веществе, и что именно ему Аристотель дал
впоследствии название vat] * материи. Другие вслед за Цел л ером хотят
видеть в этом принципе не что иное, как пространство. Как бы то ни
было, это нечто присутствует во всех вещах видимого мира. Оно по
природе своей «аморфно», т. е. лишено формы и воспринимает
последнюю только под влиянием идей. Оно подобно куску мрамора, из
которого только по мысли художника возникает статуя.
«Определенному, вмещающему в себя разновидности,— говорит
Платон в своем диалоге «Тимей»,— должна быть чужда какая-либо
ферма, подобно тому, как мазь, предназначенная для восприятия
благовонных эссенций, ранее очищается и по возможности лишается
всякого запаха, если кто хочет лепить фигуры из чего-нибудь мягкого, то
не оставит никаких форм и возможно лучше сгладит поверхность
материала. Таким же образом приличествует быть лишенным какой-либо
формы и Тому, которое предназначено для верного восприятия всем
существом своим — всего, в том числе и вечно Сущего».
Таким образом, ясно, что Платон признавал какой-то общий
субстрат, лежавший в основе всего материального мира, и если не может
считаться вполне установленным смысл, в котором он говорил об этом
субстрате, то можно утверждать, что большинство его современников
и особенно философов позднейшего времени были склонны понимать
этот субстрат как единое вещество, как первичную материю.
Наряду с этими представлениями, Платон включил в свою систему
и учение Эмпедокл а (490—430 гг. до н. э.) о четырех элементах,
* Сущности. Прим. реб.
Открытие кислорода и теория горения
353
хотя понимал он их не в том смысле, как знаменитый агригентский
философ *.
Эмпедокл видел в четырех элементах, за которые он считал огонь,
воздух, воду и землю, четыре «корня» всего существующего. Элементы
вечны; они качественно и количественно неизменяемы. Из них состоит
все: .и растения, и животные, и человек, и сами долговечные боги.
Элементы могут только слагаться друг с другом и вновь разъединяться.
Неодинаковым содержанием их обусловливается различие между
предметами. Так, кровь состоит .из четырех элементов в равных .пропорциях,
между тем, в костях содержится на половину огонь, на одну четверть
земля и на одну четверть вода. Филоссф сравнивает образование
различных вещей из элементов со смешением четырех основных красок,
которые на палитре художника дают бесчисленное множество тонов.
Элементы Эмпедокла чувствуют и мыслят, он наделяет их любовью
и ненавистью, чем определяется мера стремления к взаимному
сочетанию и разделению. Любовь и ненависть есть вообще два начала,
которые двигают миром, и самое возникновение вселенной делается
возможным благодаря их существованию.
Если ів элементах Эмпедокла нельзя не видеть смутного намека на
признание трех состояний материи: твердого, жидкого и газообразного,
то в его учении о любви и ненависти как об универсальных мировых
агентах заключается как бы предчувствие позднейшего представления
о силах притяжения и отталкивания. Самый выбор элементов и чіисло
их (4) у агригентского философа были, однако, по-видимому, до
известной степени случайными и, быть может, представляли лишь попытку
примирить между собой и воссоединить в одно целое учения различных
других философов, преимущественно ионийской школы.
Если Платон в своем учении о четырех элементах и следует за Эм-
педоклом, то содержание, которое он вкладывает в это понятие,
представляется существенно иным. Его элементы 'получают иное,
своеобразное логическое обоснование.
Один іиз путей, приводящих Платона к доказательству
существования элементов (телеологический), основан на уверенности в том, что
природа во всем стремится к прекрасному. «То, что возникло, должно
быть видимо и осязаемо. Но ничего не может быть видимо без огня,
не может быть осязаемо, не будучи твердым, и не может сделаться
твердым без (участия) земли». Так обосновывает философ
необходимость признания двух элементов — огня и земли. Дв? остальные —
воздух и воду, необходимо допустить как связующие звенья между землей
и огнем. Эту связь Платон считает «прекраснейшей».
Почему .именно вода и воздух должны быть посредниками и каким
образом они осуществляют связь между землей и огнем? Ответ на эти
вопросы мы дадим словами физико-химика и математика Дюгема [2],
недавно посвятившего обширное сочинение обзору космологии
философов древнего мира.
«Промежуточная величина между двумя другими и будет средняя
пропорциональная х, которая для современных нам алгебраистов
определяется равенством:
і _Л
д- "' г *
где заданы f и t, а х находится из равенства
* Эмпедокл происходил из сицилийского города Агригента.
23 Л. А. Чупев. і. 1И
354
Открытие кислорода и теория горения
Греки, чтобы представить себе это равенство, 'предавали ему
геометрическую форму, а именно, считали х стороной квадрата, равного по
площади с прямоугольником, стороны которого соответственно равны f и (.
Определение величины х .составляет задачу планиметрии, решение
которой, очевидно, было известно пифагорийцам. Квадрат х2,
равновеликий -по площади прямоугольнику, стороны которого равны f к t,
является средним между квадратами /2 и t2, стороны которых соответственно
равны / и t.
Если бы мир был плоской фигурой без толщины, то было бы
достаточно существование между землей и огнем одного только
посредника, который и играл бы роль средней пропорциональной между
двумя величинами. Но мир есть тело, распространенное в трех измерениях;
поэтому ©опросы, о которых идет речь, нельзя сравнивать с геометрией
в плоскости; аналогии следует искать среди задач о телах.
Итак, поставим вопрос из геометрии трех измерений,
представляющий распространение вопроса о средней пропорциональной. Мы
достигаем этого, подыскав к двум данным величинам / и t две средние а и е,
удовлетворяющие условию:
f _ а — е
а е t
Эти две величины а и е выясняются с помощью формулы:
з з
а = у72/ и е = Yft*.
Выраженные геометрическим языком по способу, обычному для
греков, эти формулы будут вполне соответствовать двум задачам
стереометрии: а—ребро куба, объем которого равен объему прямой
призмы, имеющей высоту, равную t, а площадь основания, равной квадрату
е — ребро куба, объем которого равен объему призмы, имеющей высоту
/ и площадь основания t2. Эти два куба а3 и ег будут сами средними,
между кубами fz и іг.
По аналогии с этой задачей Платон присоединяет к огню и земле
два промежуточных элемента: воздух и воду. Огонь, воздух, вода и
земля по отношению друг к другу будут находиться в отношениях,
подобных тем, в которых находятся величины /. а, еу t».
«По этой-то причине, говорит Платон, бог поместил между воздухом
и землей два промежуточных элемента; он установил между ними,
насколько это было возможно, одинаковое отношение так, чтобы воздух по
отношению к воде был тем же, что огонь для воздуха, и чтобы вода по
отношению к земле была тем же, что воздух для воды».
Можно признать изящество формы, в которую облечены эти
соображения, но чрезвычайно трудно понять, по крайней мере для
современного натуралиста, физика и химика, те соображения, которые
руководили Платоном при выборе именно этой странной аналогии. Какие
свойства воды и воздуха заставили его придать им именно такое, а не иное
соотношение между огнем и землей, почему эти соотношения ему
показались наиболее правдоподобными? На эти вопросы.мы напрасно стали
бы искать ответа в писаниях Платона. На них можно ответить разве
только с точки зрения психологической, можно понять или
почувствовать, почему в то время такой способ рассуждения о природе казался
возможным и даже необходимым.
Открытие кислорода и теория горения 355
Кроме вышеупомянутого, Платон дает еще другое (физическое)
обоснование своей гипотезы о четырех элементах. Во всяком случае, он
находится -под влиянием воззрений пифагорейцев, особенно Филолая.
Последний учил, что все тела образуются из окружающих их линий и
плоскостей, которые в свою очередь слагаются из чисел. В основе
четырех элементов лежат формы простейших многогранников: земле
соответствует куб, огню —тетраэдр, воздуху — октаэдр, воде — икосаэдр.
Кроме того, Филолай признавал еще пятый элемент — эфир,
которому приписывал форму додекаэдра. Эти представления станут отчасти
понятными, если припомнить, что по пифагорейскому учению «Числа
управляют М'иром»,— положение, в видоизмененной форме с тех пор
повторявшееся мыслителями разных времен, например Кантом, до
известной степени являющееся девизом и современного положительного
знания.
Платон принял эти представления 'пифагорейцев (кроме додекаэдра,
с которым он не связывает никакого определенного элемента), придав
им своеобразное толкование. Так, связь между землей и кубом, по его
мнению, определяется тем обстоятельством, что из всех геометрических
фигур именно куб всего больше отличается неудобоподвижностью—
свойство, как раз -присущее земле; между тем как огонь с его
заостренной формой (огненные «языки») уподобляется тетраэдру или
пирамиде. Подобные же рассуждения прилагаются и к остальным элементам.
«Огонь, земля, вода и .воздух суть тела — это понятно всякому. Но
тела имеют глубину, а глубина должна включать в себе сущность
'плоскости; плоская же боковая поверхность слагается из треугольников...».
Таким образом, Платон приходит к выводу, что элементы слагаются из
плоскостей, а эти последние в конечном счете — из треугольников. Из
всех возможных родов треугольников два являются наиболее
совершенными или «прекраснейшими». Один.—прямоугольный равнобедренный,
другой — также прямоугольный, но определяемый тем условием, что
один из его катетов равен половине гипотенузы. Из сложения этих
треугольников .получаются плоские фигуры, ограничивающие или
составляющие четыре элемента (равносторонний треугольник, квадрат и пр.).
Из этих отдельных треугольников слагаются, значит, и четыре
элемента, и -все тела мира. Эти представления Платона едва ли можно понять
иначе, как допустив, что постулируемые им элементы при
последовательном дроблении распадаются на правильные многогранники все
меньших и меньших размеров, но в каждом случае подобные друг другу
по форме, в роде того, как кристаллы, раскалываемые по плоскостям
спайности. Надо думать, что Платон представлял себе пространство,
ограниченное элементарными плоскостями, заполненным первичной,
неоформленной материей (Гомперц).
Принимая такую конструкцию элементов, Платон, очевидно, должен
был допустить способность их превращаться друг в друга-, ибо на самом
деле его треугольники, раз в них все слагается, могут ведь
группироваться различным образом. Какое-нибудь геометрическое тело,
например куб, может и образоваться из этих треугольников^ и вновь
распасться, а треугольники затем соединятся уже не в куб, а, скажем, в
октаэдр. Вот слова самого Платона, из которых видно, что он на самом
деле допускал превращаемость элементов друг в друга, видимо,
связывая ее с переходами между тремя состояниями вещества — твердым,
жидким и газообразным. «То, что в данный момент мы называем
водой, подвергается отвердению, мы это наблюдаем, и превращается в
землю, в камни; когда же/наоборот, вода делается более подвижной,
она превращается в пар или в воздух; горящий воздух .становится
23*
356
Открытие кислорода и теория горения
огнем; наоборот, сдавленный и угасший огонь принимает форму воздуха;
сжатый и сгущенный воздух образует облако и туман; однако и туман,
делаясь более компактным, делается текучей водой, а эта вода снова
становится землей и камнями».
Поэтому-то элементы Платона являются чем-то временным,
-преходящим. Он даже, называя их, избегает говорить «это» или «то», ибо
с этими местоимениями связывается представление о чем-то
пребывающем, неизменном. Он предпочитает говорить не «то», а «так»,
обозначая этим состояние, временно принимаемое бесформенным нечто.
II. АРИСТОТЕЛЬ, НЕОПЛАТОНИКИ
Из учения Платона, в котором, как можно заключить из
'Вышесказанного, много остается и недоговоренного, возникла система его
ученика Аристотеля. Но при наличности 'известного сходства между
учениями обоих философов наблюдается и весьма существенная разница.
Подобно Платону, Аристотель считает, что объектами истинного
познания может служить не сама вещь, а лишь общая сущность вещей,
не познаваемая чувствами, ибо наши чувственные представления
ошибочны и обманчивы, а доставляемое ими знание случайно и изменчиво.
Между тем истинное знание требует объекта неизмененного и
необходимого, как само знание. В отличие от Платона, он не признает,
однако, за «идеями» реального существования. Общее проявляется в
частном и с ним неразрывно связано, через него и познается. Единственна
реальными являются «частные» вещи. «Только единичным вещам можно
приписать полную метафизическую реальность. Родовые же понятия
вещей всегда суть качества вещей, общие многим вещам, в которых они
только и могут существовать» [3].
Лиэих в одной из своих речей [4] приводит іи подчеркивает
значение следующих слов, характеризующих взгляды Аристотеля: «Путь
философии тот же, что и путь всех других наук: прежде всего следует
собирать факты и знакомиться с вещами, с которыми факты (явления)
происходят; рассматривать следует не все факты сразу, но каждый из
них в отдельности и выводить отсюда заключения; если мы имеем
факты, то затем наша задача сводится к тому, чтобы связать их между
собой. Эти факты добываются путем чувственных восприятий; если эти
последние несовершенны, то таким же будет и познание, на них
основанное... Общие теоретические выводы мы можем получать только
посредством индуктивных заключений, которые (в свою очередь) могут
делаться на основании чувственных восприятий, эти же последние имеют
дело с частностями».
Из этих замечательных слов можно заключить, что Аристотель
гораздо ближе подходит к современной точ'ке зрения на* познание
видимого мира, нежели Платон с его отвлеченным миром идей. Однако на
самом деле, как в своей философской системе, так и при фактическом
изучении природы и при ее истолковании, «Стагирит» был далек от
сколько-нибудь последовательного проведения этих им же высказанных
взглядов. Слишком сильно было влияние на него философских
традиций того времени; недаром же он сделался пророком самого темного
периода в истории человеческих знаний — эпохи средневековой
схоластики.
Аристотель ;признает, что в основе всего сущего лежит вещество или
материя, которая, однако, становится реальностью лишь после того,
как воспринимает определенную форму. В этом восприятии веществом
некоторой фориы и заключается акт возникновения вещей.
Открытие кислорода и теория горения
357
Вещество Аристотеля существенно отлично от того представления,
которое мы связываем с этим словом .в настоящее время. Его материя —
только возможность воплощения известной формы (или идеи),
возможность появления «чувственной вещи». Так, мрамор делается статуей,
когда к -материи присоединяется форма ил*и идея статуи. Притом
понятие о материи относительно: самая статуя может трактоваться как
материя, как часть, служащая для украшения какого-либо здания, и
в данном случае форма или іидея относится уже к целому зданию.
Акт облекания материи .в известную форму, -иначе акт образования
вещей, доступных чувственному восприятию, Аристотель связывает с
движением, причем последнему слову придается расширительное
толкование. Всякое изменение, качественное или количественное, например
изменение геометрической формы, окраски, увеличение или уменьшение
размеров, -вплоть до возникновения и исчезания предмета, .и только,
как частный случай — перемещение в пространстве, подводится под
эту категорию. Мы теперь употребляли бы для обозначения этого
понятия слово — явление. Впрочем, как мы сейчас увидим, движению в
пространстве Аристотель приписывает здесь важное значение, хотя о«
и далек от механического истолкования природы в духе атомистов.
Всякое движение или явление ведет, по Аристотелю, к определенной
(разумной) цели. Начало целесообразности он считает мировым заю>
ном, объединяющим совокупность отдельных вещей и явлений, которые
и составляют природу*.
Подобно тому, как Платон считает, что его бесформенное начало
или несуществующее противодействует полноте воплощения идей в
вещах, так и Аристотель видит в материи принцип задержки, считает,
что она благодаря своей неопределенности препятствует полной
реализации формы и полному достижению цели.
У Платона (и Эмпедокла) Аристотель заимствует далее учение о
четырех элементах, или стихиях, но и этому учению он придает особую
окраску.
Все тела могут двигаться в пространстве. Эти движения могут
совершаться или по прямой, или по кругу, или слагаются из этих двух
родов движений. Но прямолинейные и круговые движения являются
самыми простыми и естественными, а потому они должны принадлежать
первоначальным телам, т. е. элементам. Прямолинейное движение
может происходить в двух противоположных направлениях
(подразумевается от центра и к центру, вверх и вниз), круговое— только в одном.
Поэтому круговое движение, которое сверх того совершается непрерывно
и безостановочно, является наиболее совершенным видом движения, а
тело, которому оно свойственно, должно быть само не имеющим
начала, вечно существующим. Таким телом Аристотель считает особый
-пятый элемент эфир, из которого построены небесные сферы и звездные
миры. Прямолинейное движение принадлежит, в противоположность
«божественному» эфиру, четырем земным элементам, причем
легчайший из них — огонь — движется снизу вверх, а тяжелейшая — земля — в
обратном направлении. Земля обладает только тяжестью, огонь —
только легкостью. Середину между ними занимают воздух и вода, которые
обладают тем и другим свойствами, но в разной степени: воздух
относительно легче и потому ближе стоит к огню, вода относительно
тяжелее и потому ближе к земле.
* По Аристотелю, существует только один «этот» мир, т. е. природа, причем
последняя не составляет особого существа или субстанции.
358
Открытие кислорода и теория горения
Пожалуй, несколько более понятным является другое обоснование,
которое Аристотель дает своим элементам. Из разнообразных свойств,
которыми обладают тела, он выбирает четыре: тепло и холод, сухость и
влажность. Эти свойства бросаются в глаза при первоначальном
знакомстве с внешним миром, при непосредственном прикосновении с ним,
при осязании. Они кажутся ему важнейшими, основными и наиболее
пригодными для характеристики тел. Каждое тело производит
ощущение теплоты или холода, сухости или влажности, с большим или
меньшим преобладанием того или другого из этих свойств. Из шести
возможных сочетаний этих свойств два противоположных, очевидно,
несовместимы, так как взаимно уничтожают друг друга: ни один предмет
не может быть теплым и холодным, сухим и влажным одновременно.
Остаются, следовательно, только четыре возможных сочетания, и их-то
Аристотель связывает с теми самыми четырьмя элементами, которые,
как мы видели, играли уже роль в предшествовавших -по времени
построениях Эмпедокла и Платона. Вот схема, дающая общую картину
этих сочетаний:
ОГОНЬ
сухость ( ^ тепло
земля J . воздух
холод \ ' влажность
вода
Элементы Аристотеля являются основными формами или
модификациями первичной материи, а потому способны превращаться друг в
друга. Они являются ближайшими составными частями различных тел,
в которых содержатся в весьма разнообразных пропорциях и которым
сообщают определенные свойства. Каждый элемент несет с собой,
кроме указанных выше свойств, еще ряд других, как бы вторичных свойств.
Так, с воздухом и водой связывают представление о прозрачности.
Прозрачность горного хрусталя, например, объяснялась присутствием в нем
воды. Наоборот, непрозрачность связывалась с землей. Когда
элементы входят в состав различных тел, то они исчезают, как
самостоятельные единицы, как бы сливаются в одно целое, приобретающее притом
определенную форму, но свойства их сохраняются в данном теле.
Элементы Аристотеля не тождественны с теми конкретными вещами
(огонь, воздух, вода и земля), имена которых им присвоены, но
находятся в некоторой внутренней связи с этими последними. Огонь
Аристотеля, например, есть то общее, содержащееся в горючих телах,
которое сообщает им способность гореть. При процессе горения элемент
огонь выделяется из данного тела, принимая конкретную форму
пламени, которая может быть неодинаковой в различных случаях. То же
самое можно сказать и о других элементах.
Нечего говорить, что «элементы» Аристотеля, а равно и его
предшественников, не имели ничего общего с тем значением, которое мы
придаем этому термину в настоящее время. Нечего говорить, что и самые
элементы, и 'их свойства, выразителями которых они должны были
явиться, были выбраны совершенно случайно, под влиянием
поверхностных, чисто субъективных впечатлений. Мы теперь хорошо знаем,
что ни огонь, ни воздух, ни вода, ни земля не являются простыми
телами. Земля есть понятие совершенно неопределенное, собирательное;
огонь даже нельзя назвать веществом в собственном смысле слова,
ибо этим именем мы обозначаем раскаленное состояние, в котором
могут находиться различные газы; воздух есть газовая смесь, и только
вода — сравнительно простое по составу вещество, химическое соедине-
Открытие кислорода и теория горения
359
кие кислорода и водорода... Все это окончательно выяснилось в течение
последней четверти XVIII в., эпохи, которая как раз завершает собой
историю вопросов, рассматриваемых в этом очерке. Для нас теперь
также совершенно понятно, что ощущение тепла и холода, сухости и
влажности, получаемое от соприкосновения с различными телами, всего
менее может служить для характеристики этих последних. Не говоря о
неопределенности этих свойств, они являются преходящими и как раз
не существенными для характеристики отдельных веществ.
Но не в одном только выборе веществ, которые должны почитаться
элементами, и основных свойств, эти вещества характеризующих,
заключается глубокая разница между точкой зрения Аристотеля и той,
на которой стоит современная наука.
На первый взгляд, разница в понимании того, что следует разуметь
под словом «элемент», не так велика. Подобно Аристотелю, мы видим
н элементах ближайшие составные части различных веществ. Если
Аристотель не считает элементы за конкретные тела, то ведь « наши
химические элементы — только «неизменяющиеся сущности
изменяющегося вещества» и становятся конкретными веществами лишь после
того, как выделены в свободном состоянии как простые тела. Если
Аристотель принимает, что тот или иной элемент может, освобождаясь
от сочетания с другими, принимать различные формы, то разве наши
элементы не показывают явления аллотропии, разве кислород (О) не
может существовать в виде «обыкновенного» кислорода (02) и озона
(Оз); углерод —в виде аморфного угля, графита и алмаза и т. д. Не
следует чересчур увлекаться этой аналогией, но в ней, без сомнения,
есть доля истины.
Таким образом, не в определении понятия «элемент» кроется
разногласие между Аристотелем и нашей точкой зрения. Это разногласие
лежит скорее в способе применения этого понятия к конкретным случаям.
Допуская присутствие того или иного элемента в данном теле,
Аристотель считал достаточным для мотивировки такого допущения
наличность в этом теле определенных свойств, соответствующему элементу
приписываемых. Наоборот, вовсе не считалось необходимым реально на
опыте доказать, что из данного тела на самом деле может быть
получена предполагаемая в нем составная часть—элемент, в нем якобы
содержащийся, и во всяком случае не считалось обязательным установить,
в каких именно формах этот элемент может появиться и почему эти
¦формы можно считать равноценными. Мы теперь утверждаем, что в
воде содержится водород и кислород, потому что обе эти разновидности
.вещества фактически могут быть из воды получены, хотя бы при
помощи электролиза, и при этом получаются со строго определенными
свойствами. Водород с достоверностью известен только в одной форме (Нг),
в .которой всегда и получается. Кислород известен, кроме обыкновенной
формы (Ог), еще в виде озона (Оз), и этот последний может (частью)
получаться при разложении воды. Но ведь точными опытами доказано,
что озон и кислород всецело, без потери, могут быть превращены друг в
друга (превращение 302—203 идет с трудом, но при соблюдении
известных условий все-таки возможно довести его до конца),
следовательно, мы вправе считать, что оба они, действительно, состоят из одного и
того же материала. Но о необходимости подобного рода строгих <и
точных доказательств даже не мечтали во времена Аристотеля.
Не что иное, как отсутствие требовательности к строгим
доказательствам каждого высказываемого положения было причиной того, что с
такой легкостью, руководясь только соображениями «разумности» или
-«красоты», допускали единство материи,, безграничную способность
360
Открытие кислорода и теория горения
вещества к изменению формы, держались за доктрину, сыгравшую такую
важную роль в судьбах химической науки.
Хуже всего было то, что экспериментальные методы исследования в
то время вообще не пользовались кредитом. Производить опыты
считалось делом, достойным раба, а не свободного гражданина. С другой
стороны, полагали, что природу можно гораздо лучше познать путем
умозрения и что результаты, полученные таким образом, несравненно
достовернее, ближе передают сущность идей, чем чувственный опыт. Мы
видели, что Платон в этом отношении шел гораздо дальше Аристотеля.
Но и Стагирит, когда от общих рассуждений переходил к частностям,
постоянно предпочитал вместо опыта апеллировать к принципу
целесообразности, в лучшем случае подкрепляя свои выводы случайными
наблюдениями. В противаположность современной науке, греческие
философы видели в природе разум, добро и зло, совершенство и
несовершенство. В их глазах вещи могли улучшаться и портиться.
Таким образом, возникал ряд критериев, чисто субъективных и
антропоморфных, дававших начало суждениям беспочвенным и
произвольным. К этому присоединялось еще отсутствие точности в
определении самых основных, с современной точки зрения, понятий и, как
следствие этого, неточность терминологии, переходившая в игру слов,
которая заменяла настоящее веское доказательство. Наконец, вся эта
'путаница слов и понятий усугублялась покровом мистицизма, которым
окутывали свое учение особенно позднейшие последователи Платона и
Аристотеля. Быть может, всего ярче эти черты выразились в
философских системах неоплатоников, развивших и дополнивших учение
Платона, а из них всего больше в системе Плотина, жившего между 204 и
236 гг. н. э.
Здесь будет достаточно указать, что, по учению этого философа, мир
представляет собой эманацию божества, единой первичной сущности,
вечной и непостижимой. Оно стоит выше мышления и выше бытия.
Порождая из себя все многообразие вещей, оно при этом не разделяется
и ничего не отдает от своего существа. Множественность вещей, их
качественное разнообразие философ объясняет своеобразными
уподоблениями. Эманация мира из божества подобна распространению света во
мраке. При этом от одного источника может получаться множество
различных отражений. С увеличением -расстояния свет ослабевает и,
наконец, совершенно затухает. Подобным же образом, рассуждает Плотин,
постепенно затухает эманация от первого начала. Высшее проявление
этой эманации есть дух, который порождает душу. С последней связана
и природа. Постепенно ослабевая, испускание божественного существа
переходит в материю, которая стоит на самой низшей степени и
обладает одними только недостатками. Она, учил еще Платон, дает
несуществующее; она мешает всему доброму, отличается полным
отсутствием добра, полнейшей скудностью и т. д. «Для Платона,—говорит Вин-
дельбанд [5],— из всего этого вытекает не только телеологическое, но
прямо-таки магическое воззрение на природу: всякое бытие есть
деятельность души; из чистой мировой души выделяются боги, духи
небесных светил, из природы — демоны; в таинственном взаимодействии
целого, все единичное настроение симпатически относительно этого целого
и может быть предугадываемо вещим чувством. Всякое 'Исследование
природы здесь устраняется, но зато широко раскрыта дверь всякой вере
и суеверию».
Такова была отрицательная сторона духовного наследия,
оставленного Платоном, Аристотелем и их 'последователями и в течение многих
веков оказывавшего сильнейшее влияние на историческое развитие че-
Открытие кислорода и теория горения
36і
іовеческой мысли. Если .в области естествознания это явление почти
ірекратилось к концу XVIII и началу XIX в., то в области отвлеченной
философии оно, можно сказать, почти докатилось до наших дней.
Говорил же еще Шопенгауэр о себе по сравнению с естествоиспытателем
как о «Монблане рядом с кротовой кучей». Читая эти слова, невольно
вспоминаешь горячую проповедь Д. И. Менделеева о «латинском
самообольщении» и о том огромном вреде, который оно несло человечеству.
Если бы я захотел подробнее документировать эту мысль и, в
частности, осветить, в какой мере господство учения греческих философов
и слепое поклонение их авторитету, соединенное с общим мистическим
настроением эпохи, путем систематической и непрерывной борьбы со
здравым смыслом мешало развитию правильных взглядов на сущность
химических процессов и особенно процессов горения, мне пришлось
бы написать целый том и изложить всю историю химических знаний в
течение периода времени, обнимающего без малого два тысячелетия. Но
это заставило бы меня далеко выйти из узких рамок настоящего
очерка, а потому мне остается только бегло указать на те формы, в которые
последовательно отливалось стремление внедрить «платоновские идеи и
аристотелевы элементы в живое тело экспериментальной химии. Нашу
науку спасло, вывело на правильный путь то, что является и в
настоящее время основным устоем положительной науки — эксперимент.
В свою очередь, делать эксперименты, изучать природу фактически, а
не умозрительно, заставлял могущественный рычаг, двигавший
алхимиками,— стремление к обогащению. Если вера в существование
философского камня, 'питаемая учением древних о единстве материи,
подкрепляла алхимиков в их -поисках за способами 'Искусственного получения
золота, то ясно, что эти поиски могли вылиться только в форму опытов
над различными веществами и их превращениями. А эти опыты
доставили тот фактический материал, который послужил для борьбы со
старыми предрассудками и в то же время для фундамента химической
науки.
Известный французский физико-химик Ле-Шателье неоднократно, ь
последний раз совсем недавно, развивал ту мысль, что крупные
открытия в науке являются чаще всего результатами стремления разрешить
те или другие вопросы, важные для техники, для промышленности. Эта
мысль в общей форме, без сомнения, преувеличенная и односторонняя,
особенно по отношению к современному фазису развития науки,
оказалась в значительной мере справедливой по отношению к алхимии. Здесь
инстинкт промышленника и купца медленно, но неуклонно побеждал
и мало-помалу вытеснил из его позиции античного философа и
средневекового схоластика.
III. АЛХИМИЯ И ИАТРОХИМИЯ
Алхимия, в собственном смысле слова, зародилась из столкновения
греческой культуры и ее философских систем с практическими знаниями
египтян, в течение долгого времени накоплявшихся в стране фараонов,
главным образом среди сословия жрецов. Это столкновение двух
культур произошло в эпоху процветания Александрии в течение первых
веков нашей эры. Из Александрии алхимические знания и навыки
проникли в Сирию, куда еще раньше греческая культура была занесена
благодаря походам Александра Македонского. От сирийцев, принявших
христианство, алхимия перешла к арабам, а эти последние занесли ее в
Европу главным образом через Испанию. В Европе она гвила себе
прочное гнездо начиная с XII—XIII столетия.
362
Открытие кислорода и теория горения
Каждый 'из этих четырех последовательных периодов развития
алхимии внес новые элементы в сумму фактического знания, открывая
новые вещества, ноівые приемы работы и т. д. Наиболее значительные
результаты, по-'видимому, дала греческая алхимия в течение
Александрийского периода. К этому времени относится деятельность Зосимы,
Синезия, Олимпиодора, Стефания и др.
Идейная сторона алхимии опиралась, как уже было сказано, на
философские системы Платона, Аристотеля и неоплатоников, к которым
присоединялись еще некоторые верования восточного происхождения.
Учение о четырех элементах стояло прочно, но, по-видимому, не имело
особенно большого 'практического значения. Зато весьма важную роль
сыграла уверенность в беспредельной способности вещества менять свое
состояние, принимать самые разнообразные формы. Что это именно
так, утверждали философы, и, казалось, подкрепляли уже известные в
то время факты химических (превращений.
Ближайшие задачи, которые поставила себе алхимия, заставили
обратиться к изучению свойств металлов и их превращаемости из одного
состояния в другое. Те фактические сведения, которые имелись в эпоху
Александрийскую, благоприятствовали применению основных
представлений греческих философов к решению алхимических проблем.
Большинство металлов не было известно в чистом состоянии,
главным образом потому, что, кроме золота, все они встречаются в природе
в виде руд, представляющих смеси (часто изоморфные), содержащие
сразу по нескольку металлов вместе. Разделять эти металлы друг от
друга тогда еще не умели, а 'потому при выплавке из руд получали почти
всегда сплавы, свойства которых, особенно цвет, плавкость, удельный
вес, твердость и т. п., менялись непрерывно и, казалось, безгранично,
от услоівий получения и от прибавления той или иной примеси в
различных пропорциях.
Смешение понятий еще усиливалось оттого, что каждый металл
рассматривали как понятие собирательное и различали несколько сортов
его. Так, считали, что существуют два сорта свинца — черный и белый,
причем под последним подразумевали не свинец, а олово, более или
менее загрязненное примесями. Было также известно несколько сортов
меди: 'красная, или кипрская, желтая (бронза, латунь), белая и т. д.
Эти названия до известной степени сохранились и до настоящего
времени. Под общим именем «серебра» также соединялись разнообразные
сплавы, часто вовсе не заключавшие настоящего серебра.
Еще у египтян сложилось представление, что в основе всех металлов
лежит некоторое первичное вещество, сообщающее им, как казалось,
наиболее характерное свойство — плавкость, затем ковкость и т. д.; и
за такое вещество считали легкоплавкий свинец, который обозначался
именем бога Озириса. В происхождении этого мифа не малую роль
играло общеизвестное содержание в свинцовых рудах подмеси серебра,
а иногда и золота. Естественно, что овинец мог прослыть как бы отцом
благородных металлов, которые могли быть получены из него путем
«улучшения». Впрочем, со свинцом смешивали в то время не только
олово, но и сурьму, а также некоторые сплавы.
Когда (около времени Пелопоннесских войн) стала известна ртуть
(можно думать, что она впервые была получена и привезена в Европу
карфагенянами), то свойство ее — жидкое состояние (так сказать,
абсолютная плавкость), соединенное с металлическим видом, произвело
настолько сильное впечатление, что в нее перенесли роль materia prima
или «души» металлов. Казалось, что стоит только фиксировать ртуть,
т. е. понизить ее летучесть и немного изменить цвет, чтобы получить
Открытие кислорода и теория горения
363
серебро. Она на самом деле была в глазах алхимиков жидким
серебром, и название argentum vivum * упрочилось за ней на долгое время.
Чтобы получить из нее золото, нужно было выкрасить ее в другой цвет.
Отсюда многочисленные поиски за подходящей краской, которую
алхимики наивно** представляли себе именно желтой. Поэтому пытались
кайти ее в таких веществах, как сера или сернистый мышьяк (ауритіиг-
мент As2S3, отличающийся, как известно, золотисто-желтым цветом).
Эта краска іи есть не что иное, как'пресловутый философский или
красный камень. Вот что, например, 'пишет Синезий [6], один из
выдающихся алхимиков IV в. н. э.: «Ртуть, значит, бывает разных сортов? — Да,
она бьивает разных сортов.— Но ведь она одна, как же она может быть
разных сортов? —Да, она бывает разных сортов, обладает большим
могуществом. Разве ты не слыхал, что было сказано Гермесу: «Медовая
струя (ртуть)—белая (серебро) и медовая струя желтая (золото)? —
Да, я слышал, как ему это было сказано. Но именно это я хочу понять,
Синезий, объясни мне.— Это действие, которое ты знаешь
(трансмутация).— Значит, ртуть различными путями принимает образ разных
тел? — Ты понял, Диоскорид. Так же, как воск принимает тот цвет, »в
который его окрашивают, так же точно и ртуть, о философ! Она
окрашивает в белый цвет все тела и притягивает их души, она
перерабатывает их кипением и завладевает іими. Имея к тому расположение и имея
в себе начало всего жидкого, она готова ко всякому изменению цвета,
когда подвергается трансформации. Она образует постоянную основу,
между тем как цвета не имеют собственного основания; или, лучше
сказать, ртуть, теряя свою собственную основу, делается изменчивой
через обработку металлических тел».
Чтобы понять смысл этих слов, необходимо иметь в виду, что
алхимики того времени признавали несколько разностей ртути, например
ртуть самородную и приготовленную из киновари, причем последняя
различалась по роду посуды (медная, свинцовая), в которой
'воспроизводились операции, служившие для ее приготовления. За особую
разновидность ртути считался также металлический мышьяк, из-за его
летучести и способности придавать белый цвет различным сплавам.
Когда говорили о ртути и о ее роли в трансмутациях, подразумевали,
следуя традиции греческих философов, некоторую гипотетическую
субстанцию, которую считали общим началом, или квинтэссенцией всех
разновидностей ртути, говорили, следовательно, о «едином во
множестве». Это общее начало, которое называли ртутью философов, старались
выделить или из предполагаемых разностей самой ртути, или из других
металлов путем отделения от примесей, наделенных противоположными
свойствами, а оіотому извращающих первичные свойства матери
металлов.
Перечень условий, которые считались необходимыми для того,
чтобы осуществить получение серебра и золота, нуждается еще в одном
дополнении. Характер эпохи требовал, чтобы в столь важных действиях,
как трансмутация, видная роль была отведена элементу таинственному,
мистическому. «Не подумай,— говорит Олимпиодор [7],— что достаточно
ручной работы; нужно еще вмешательство природы,— силы, стоящей
выше человека». На самом же деле для осуществления трансмутаций
* Живое серебро. Прим. ред.
** Впрочем, мысль о том, что свойства компонентов просто складываются, когда
происходит слияние последних (мы теперь сказали бы «аддитивны»), по отношению
к металлам могла иметь и то фактическое основание, что, как известно, металлы при
сплавлении по большей части лишь незначительно меняют свойства, во всяком случае
гораздо меньше, чем элементы, далеко стоящие друг от друга по своей природе.
364
Открытие кислорода и теория горения
считалось необходимым прибегать к молитвам и заклинаниям, к
особым магическим формулам. В тесной связи с этой таинственной
стороной алхимии стояла вера в существование какой-то неведомой, но
тесной связи между земной материей и небесными светилами. Это
верование, несомненно восточного происхождения, было не чуждо и
греческим философам.
Полагали, что каждому металлу (их считалось семь) отвечает одно
из семи светил, как бы управляющее его судьбами на земле. Именно,
золоту соответствоаало солнце, серебру — луна, меди — Венера,
железу—Марс, олову—Юпитер, свинцу — Сатурн и ртути — Меркурий.
Таким образом, сама по себе таинственная алхимия соединялась
неразрывными узами с не менее таинственной астрологией.
Понятно, что мистический и таинственный характер алхимии еще
более затуманивал и без того уже смутные представления того времени,
а желание сохранить в секрете якобы достигнутые успехи в области
трансмутации металлов заставляло авторов сочинений придавать
своему изложению преднамеренно темную форму каких-то таинственных
ребусов, анаграмм и т. п.
Наконец, изучение истории алхимии в сильной степени затрудняется
еще и тем обстоятельством, что многие авторы скрывали свое настоящее
имя, заменяя его псевдонимом, частью для того, чтобы прикрыть себя
авторитетом какой-нибудь общепризнанной знаменитости, частью, быть
может, для того, чтобы отвлечь от себя столь страшное в средние века
обвинение в колдовстве.
Так возникло сказочное имя Гебера. Настоящий Гебер (Джобер-
бен-Хайам эс-Суфи) жил, по-видимому, в VIII в. н. э., между тем как
ему приписывали ряд сочинений, безусловно относящихся к XIV—XV вв.
Та же самая судьба постигла полусказочного Гермеса Трисмеги-
ста (трижды величайшего), подлинные сочинения которого относятся,
вероятно, к III в. н. э., тогда как его именем пользовались алхимики
XIII и позднейших веков.
В XV в. появился ряд сочинений под именем Василия Валентина.
Это имя оказалось, однако, совершенно вымышленным, и открытия,
которые раньше ему приписывались, были сделаны другими и притом
различными лицами.
Все это было выяснено главным образом в 80-х годах прошлого
столетия знаменитым французским химиком М.Берт л о, в основу работ
которого легло тщательное сличение арабских и греческих текстов,
сохранившихся в виде неизданных рукописей, с латинскими сочинениями
позднейшего времени. Эти изыскания привели к заключению, что
исключительно выдающаяся роль в развитии алхимии, которую долгое время
приписывали арабам, сильно преувеличена. Арабы только продолжали
дело греков (и египтян) и их последователей сирийцев. Многие
открытия, которые приписывали арабам, сделаны были много позднее
алхимиками латинского Запада.
Во всяком случае, к тому времени, когда алхимия через посредство
арабов проникла и упрочилась в Европе (XIII в. н. э.), можно считать
установившимся представление о том, что в состав металлов входят в
различной пропорции два начала; кроме ртути, еще сера, причем эта
последняя, опять-таки, имеет значение до известной степени
символическое, как общий принцип, являющийся носителем определенных свойств,
присущих металлам, а прежде всего различным модификациям серы,
которые признавали в то время. Из этих свойств способность гореть и
летучесть стоят на первом плане. Содержание серы в различных
металлах ставили также в связи с тем фактом, что многие металлы способны
Открытие кислорода и теория горения 365
в известных условиях переходить в вещества красного цвета,
напоминающие киноварь. Сернистые соединения при этом соединялись в одну
группу с кислородными, как окись ртути, сурик, закись меди, окись
железа и пр.
Таких представлений, высказанных еще арабскими писателями,
придерживались знаменитые алхимики XIII в. Альберт Великий
(1193—1280), Роджер Бэкон (1214—1294), Арнольд Вилл анов-
ский (1235—1314), Раймунд Луллий (1236—1315) и ряд
писателей позднейшего времени, примерно до ct-редины XV в.
Только около конца XV (Василий Валентин) и начала XVI века
к двум компонентам металлов, а также ряд и других тел, к ргути и сере
присоединяется еще третий —соль, которая должна была олицетворять
собой твердость и нелетучесть. Так возникли знаменитые tria prima (три
сущности), учение о которых по времени почти созпадает с господством
гак называемого иатрохимического, т. е. врачебного, направления.
Родоначальником последнего является Теофраст Бомбаст Пара-
цельз (1493—1541), учивший, что задача химии заключается не в
искусстве делать золото, а в искусстве лечить болезни. «Каждое тело,—
пишет он,—образуется из трех субстанций, имена которых сера, мер-
курий и соль.— Если ты возьмешь в руки тело, то ты имеешь три
невидимых субстанции... Чтобы испытать это, возьми сначала дерево: уго
будет тело. Сожги его, тогда то, что будет гореть — это сера, то, что
будет дымить —меркурий, а то, что сделается золой — соль...».
IV. БОРЬБА СО СХОЛАСТИЧЕСКИМ НАПРАВЛЕНИЕМ.
РОДЖЕР БЭКОН. ВАН-ГЕЛЬМОНТ. РОБЕРТ БОЙЛЬ
Как мы неоднократно указывали, за все время своего
существования алхимия опиралась на философские учения Платона, Аристотеля и
неоплатоников. Эти учения, влияние которых, хотя и в более слабой
степени, продолжалось и в течение периода иатрохимии и даже периода
господства теории флогистона, как будто нарочно соединились с рядом
неблагоприятных обстоятельств для того, чтобы затруднить понимание
самых простых вещей -и отвлечь мысль от самого естественного их
объяснения. Нужно было окончательно, говоря словами Либиха, «порвать
с царством Аристотеля» для того, чтобы положить начало правильному
толкованию основных химических явлений. Это произошло только в
конце XVIII столетия.
Однако еще несравненно ра<нее стали появляться отдельные
выдающиеся мыслители, которые имели мужество, нередко навлекая на себя
обвинение в ереси и богоотступничестве (ибо Аристотелево учение были
одно время почти канонизировано), возвышать свой голос против
господствовавшего схоластического течения мысли и, в частности, против
основанных на этом течении объяснений химических процессов. Этим
новаторам никогда недоставало, однако, выдержки и
последовательности мысли для того, чтобы окончательно порвать с традициями
официальной школы, а главное для того, чтобы провести на практике
провозглашенные ими принципы. Для последнего им часто недоставало, и
фактических данных.
Первым из таких протестантов-новаторов был уже упомянутый выше
Роджер Бэтой, францисканский монах, прозванный «чудесным
доктором» (doctor mirabilis). Он громко возвысил свой голос против
мертвящего влияния авторитетов в защиту свободной науки, против
схоластического метода и в защиту метода экспериментального. «Но, кроме этих
наук, есть одна наука более совершенная, чем все другие, на которую
366
Открытие кислорода и теория горения
они опираются и которая чудесным образом всех их утверждает, она
называется наукой экспериментальной»,— пишет он.
Можно было бы подумать, что эти слова были написаны более чем
300 лет спустя однофамильцем монаха-алхимика знаменитым
Фрэнсисом Бэконом, лордом Веруламским, отцом опытной философии.
Отдавая должное значение опытному методу, Роджер Бэкон резко
осуждает и чистый эмпиризм; он видит з^лог правильного развития
химии в гармоническом сочетании двух направлений: спекулятивного и
практического. О спекулятивной алхимии он пишет, что это «наука
о происхождении вещей из элементов и о всех неодушевленных вещах:
об элементах и жидкостях, простых и сложных, о камнях обыкновенных
и драгоценных, о мраморах, о золоте и иных металлах; о
разновидностях серы, о солях и о чернилах; о киновари, сурике и других красках;
о маслах и горючих горных смолах и о бесконечном числе других вещей.
о которых ничего не сказано в книгах Аристотеля».
Но есть еще другая алхимия — опытная и практическая, которая
учит изготовлять благородные металлы и краски, и многое другое с
помощью искусства лучше и обильнее, чем с помощью природы. И потому
эта алхимия «своими делами (результатами) утверждает (снабжая
точными сведениями) спекулятивную алхимию, а также философию
природы и медицину».
В темный век царства темных людей такие светлые мысли могли быть
только мыслями гения. А между тем Роджер Бэкон, когда дело
доходило до конкретных воззрений алхимии, признавал четыре элемента
Аристотеля. «Элементов четыре: огонь, вода, воздух, земля и эти
элементы возникают из материи». Он признавал также ртуть и серу за
составные части всех металлов и верил в трансмутацию. Он сообщает
даже рецепт получения золота, причем в роли философского камня, по-
видимому, фигурирует киноварь. Его сочинения («Opus majus», «Opus
minus», «Opus tertius» и др.) уже по этому одному не были в состоянии
повлиять на господствующее тогда направление в алхимии; они
навлекли на него ряд гонений, продолжительное и неоднократное заключение
в тюрьмы.
Голос Бэкона прозвучал одиноко и в то время не нашел себе
подражателей. Его современники, как Альберт Великий, который не
только умел жить в мире с официальной церковью, который почитался
чуть ли не святым, и Арнольд Виллановский, подобно Бэкону
испытавший на себе тяжелую руку духовной власти, были очень далеки
от новаторских мыслей Роджера.
Только начиная с эпохи Возрождения вновь появляются люди,
решающиеся плыть против господствующего течения. До известной
степени к ним относится Парацельз *, о котором уже упоминалось выше*
как об основателе иатрохимии, и еще в большей степени живший без
малого сто лет спустя (1577—1614) Ван-Гельмонт.
Если уже Парацельз высказывал определенные сомнения в
истинности некоторых положений Аристотелевой философии и отрицал
возможность искусственного получения золота, если уже он, со
свойственной ему экспансивной энергией, провозглашал природу и опыт
единственными надежными руководителями и авторитетами, то
Ван-Гельмонт не только пошел в этом отношении значительно дальше, но, что
для нас особенно важно, своими экспериментальными работами в
сильнейшей степени способствовал открытию кислорода.
* О гениальном Леонардо да Винчи, который был почти современником Парацель-
за, будет упомянуто ниже, так как он принимал непосредственное участие в историй
открытия кислорода. -
Открытие кислорода и теория горения
367
Следующие слова могут служить для характеристики его отношения
к учениям Стагирита: «Различные школы так неизменно клялись в
верности Аристотелю, что его имя и сейчас еще считается нарицательным
для философа или для мудрого мужа. И тем не менее я пришел к
убеждению, что он был совершенно невежественным по отношению к природе,
и мне не трудно будет привести тому доказательства. Впрочем, я это
делаю ни для какой иной цели, -как для того, чтобы в будущем учителя
и слушатели по легковерию или по привычке не позволяли совращать
себя с прямого пути и чтобы, выбрав себе в путеводители слепца, они
вместе с ним не попадали бы в яму. В остальном я вовсе не хочу играть
никакой комедии, а против Аристотеля у меня горечи и досады не более,
чем против вещи, которой не существует» (очевидный намек на не
сущее схоластической философии).
Из четырех элементов Аристотеля Ван-Гельмонт сохраняет
только воду и воздух. Будучи мистиком от природы, он отказывается
признать огонь за элемент по той причине, что о его творении ничего
не говорится в библии. Но это, очевидно, не единственный мотив, как
видно уже из меткого определения, которое Ван-Гельмонт дает огню,
называя его «зажженным дымом» *. Что касается земли, то он отрицает
ее элементарную природу на том основании, что она «в конце концов
может быть превращена в воду и таким образом совершенно лишена
своего естества».
Ошибочные взгляды, мистические представления сплошь и рядом
уживаются уВан-Гельмонта с удивительно ясными, верными
мыслями и точно наблюдаемыми фактами. Он признает ртуть, серу и соль
основными принципами, считает, что они входят в состав воды, которая,
однако, сама является элементом. И наряду с этим мы находим у него
следующие слова: «Подобно тому, как вода остается водой по своему
существу и по веществу (по составу), превращаясь в снег, делаясь при
этом тверже и неподатливее, таким же образом она остается водой,
переходя в тончайший дым или газ...». В другом месте он говорит, что
ему удалось изготовить стекло, исходя из определенного количества
песка, затем «искусственно нарушить связь» и получить при этом то же
самое количество песка обратно. Он проводит также опыт с золотом,
которое после многократной, очень сложной (и не вполне ясной по
смыслу) обработки обращалось в красное летучее (?) масло, содержащее
ровно столько золота, сколько было взято первоначально для опыта.
«Мне помнится также,— говорит он далее,— что я облил серебряную
известь (т. е. окись серебра) кислым духом серы (серной кислотой),
который отдал серебру свою остроту и кислотность, превратил их (т. е.
остроту и кислотность) после отгонки в горечь желчи. При этом,
однако, серебро осталось без изменения по качеству и весу».
Таким образом, Ван-Гельмонт имел уже некоторое, хотя и смутное
представление о химическом индивидуальном теле, сохраняющем свое
«естество» при переменах состояния. С другой стороны, он как бы
предчувствовал закон сохранения элементов.
Особенно большое значение для развития химии имели работы
Ван-Гельмоита о газах. До него единственным известным газом был
воздух, и с ним смешивали все другие газы, образование которых
химикам постоянно приходилось наблюдать при различных химических
процессах.
* Впрочем, это определение принадлежит не ему. Его можно найти уже у Альберта
Великого, у которого оно мирилось с учением Аристотеля об огне, как об одном ич
четырех элементов.
368
Открытие кислорода и теория горения
Ван-Гельмонт впервые ввел в употребление самое слово «газ» для
¦обозначения родового понятия. Именно, говоря об испарении воды, он
указывает на необходимость различать образующийся при высокой
температуре (при кипении) водяной пар, легко сгущающийся в воду, от
«тончайшего дыма», образующегося на холоду, дыма, который он считал
постоянным и неспособным сгущаться в жидкость.
Чтобы обозначить особенности последнего, Ван-Гельмонт и вводит
новый термин газ, производя его из греческого хаоса. Образование
газа при испарении воды Ван-Гельмонт рисует себе в виде чрезвычайно
сложного процесса, в котором принимают участие все три компонента:
ртуть, сера и соль. Но в основе его представлений все же лежит
правильная идея о том, что сущность газообразования сводится к
тончайшему распылению материи. В одном месте он говорит даже о
«мельчайших неделимых», как бы намекал на современное представление о
молекулах. Ван-Гельмонт не ограничился этими теоретическими
соображениями. Ему принадлежит открытие и описание (насколько позволяли
тогдашние экспериментальные средства) двух новых газов, отличных
как от водяного пара, так и от атмосферного воздуха: углекислого газа
(СОг) под именем «лесного газа» и метана (СН4), под именем
«жирного» или «горючего газа». Ван-Гельмонту было известно, что его лесной
газ ядовит (животные в нем умирают), что при погружении в него
зажженная свеча ту>нет, что скопление его наблюдается в пещерах, что он
¦чходит в состав дыма, выделяющегося при горении угля, и образуется
такжо при брожении виноградного сока и пивного сусла.
Другой газ, открытый Ван-Гельмонтом, оказался горючим и был
обнаружен в толстых кишках, а также в продуктах разложения навоза.
Ни то-, ни другой газ не был, правда, получен в чистом состоянии. Для
этого Ван-Гельмонту недоставало подходящей техники, которая
впервые была разработана его современником Майовым (см. ниже) и затем
значительно усовершенствована уже в конце XVIII столетия. Те же
причины не позволили Ван-Гельмонту различить от углекислоты газы
(окислов азота), образующиеся при растворении серебра в азотной кислоте.
Но при всем своем несовершенстве, его работы имели большое, так
сказать, психологическое влияние на дальнейших исследователей. Им
впервые было высказа-но, что существует особая категория веществ, ряд
газов, качественно различавшихся между собой. Тем самым был дан
импульс к поискам за газами, еще неизвестными, был дан и повод
сомневаться в тождестве каждого наблюдаемого газа с обыкновенным
воздухом, в свою очередь, заставляло делать проверочные испытания,
позволяло избегать ошибок и приводило к новым открытиям.
Дела Ван-Гельмонта. конечно, гораздо больше принесли пользы для
опровержения схоластических предрассудков и для торжества нового
направления в науке, нежели его словесные выпады протиз учения
«язычника» Стагирита, которому, нужно сказать, противополагались взгляды,
пожалуй, не в меньшей степени полные мистического тумана.
Если мы перенесемся теперь мысленно к эпохе, еще более близкой
к нашему времени — ко второй половине XVII в., то встретимся с
деятельностью знаменитого Роберта Бойля (1627—1691), которому
наука обязана газовым законом, носящим его имя, и многими другими
замечательными открытиями. В области химии Бойлю принадлежит
великая заслуга указать для нее новые пути и новые методы. Он
настаивал на том, что химии гораздо больше приличествует опиоатьея на
физику, чем на медицину. В своем сочинении «The sceptical Chemist» * (по
* Скептический химик. Прим, ред.
Открытие кислорода и теория горения 369
латыни: Chymista scepticus) он обрушивается с ядовитой и остроумной
критикой на схоластическое направление и, в частности, на учение о tria
prima (ртуть, сера и соль). Для него задача химии заключается в том,
чтобы определять состав тел, иначе говоря, узнавать, какие вещества
на самом деле могут быть получены от каждого данного тела. В
соответствии с этим он изменил традиционное понятие об элементах, так много
препятствовавшее правильному развитию химии. «Я называю
элементами,—писал он (в «Sceptical Chemist»),--некоторые первоначальные
или простые тела, иначе говоря (ни с чем) не смешанные, которые, не
будучи в состоянии образоваться ни из других тел, ни друг из друга,
являются составными частями, из которых непосредственно слагаются или
на которые в конечном счете разлагаются совершенным образом
смешанные тела, т. е. химические соединения». Совершенно ясно также Бойль
различает между собой химические соединения от простых смесей,
разграничивая таким образом понятия, смешение которых не мало
способствовало спута-нности понятий химиков того времени.
Таким образом была подготовлена почва для переворота в
химическом мировоззрении, который наступил, однако, лишь спустя слишком
целое столетие после появления в свет (в 1661 г.) главного сочинения
Бойля.
Несмотря на все это, и Бойль, так же как и его предшественники,
не был в состоянии отрешиться от всех предрассудков своей эпохи. Так,
огонь он считал простым телом, притом субстанцией материальной,
весом.
Под влиянием такой предвзятой мысли он дает совершенно
неправильное объяснение замечательным опытам над изменением веса
металлов при их прокаливании в присутствии воздуха. Именно, он нагревал
различные металлы в запаянных сосудах, наполненных воздухом (что
позднее делали Ломоносов и Лавуазье). Он заметил при этом, что при
распаивании охлажденного сосуда, воздух врывался туда со свистом.
После того он взвешивал сосуд и наблюдал увеличение в весе. Отсюда
он заключает, что при прокаливании тонкая материя огня проникла
через стекло сосуда, присоединилась к металлу и утяжелила его.
Бойль пошел дальше своего предшественника Роджера Бэкона, но
ведь надо помнить, что их отделяют друг от друга четыре столетия.
И все-таки он не дошел до конца. Имея в руках ключ к окончательному
и бесповоротному решению задачи, он как будто нарочно уклоняется
в сторону от естественного и прямого пути. Здесь еще раз видна сила
господствующей системы, окаменелого в своей неподвижности
мировоззрения, характеризующего целую эпоху, отрешиться от которого
не во власти самых сильных и трезвых умов. Все наше предыдущее
изложение имело своей задачей осветить с этой точки зрения иего| лческин
ход открытия кислорода и теории горения, о котором будет идти речь
в последующих главах.
V. БЕХЕР И СТАЛЬ. ТЕОРИЯ ФЛОГИСТОНА
Во второй половине XVII в. в химии возникло новое научное течение.
хотя и далеко не свободное от предрассудков старого схоластического
направления, но во многих отношениях своеобразное и во всяком случае
весьма замечательное для того времени. Я имею в виду учение о
флогистоне, впервые выдвинутое Бехером (1635—1682), подробнее
обоснованное и развитое Ста л ем (1660—1734).
К тому времени, к которому относится деятельность этих химиков,
задачи нашей науки успели, как мы уже видели выше, значительно
24 Л. А. Чугаев, т. III
370
Открытие кислорода и теория горения
отойти от идеалов алхимиков. Бехер и Сталь были идейными потомками
иатрохимиков. Сталь был известным врачом и долгое время занимал
должность лейб-медика прусского короля. Нет ничего удивительного, что
и область химии, занимавшая этих ученых и их современников,
несколько сдвинулась в сторону от той, 'которая составляла предмет
исключительного интереса алхимиков, и притом расширилась. Центральное
место по возбуждаемому ими интересу заняли теперь всевозможные
реакции горения и обжига тел на воздухе и свойства продуктов, при этом
получающихся, т. е. кислот, оснований и солей, а с другой стороны —
реакции противоположного характера—'процессы восстановления.
Эти явления как бы заступили место процессов трансмутации, в
возможность которых благодаря длинному ряду неудач и разочарований
многие перестали верить. Вместо философского клмня мысль искала и
находила то общее, что придает телам горючесть. Это нечто в более или
менее смутной форме выдвигали и алхимики, и иатрохимики под именем
серы. Но роль последней в только что упомянутых процессах оставалась
неясной. Была брошена мысль, было произнесено слово, но не было
точно высказанной гипотезы, ни тем более последовательно проведенной
и продуманной теории.
Осуществить эту задачу и выпало на долю Бехер а и Сталя. Бехер
по своим общим воззрениям был одним из глашатаев нового
направления в науке, противником схоластики и сторонником
экспериментального метода. «Хороший перипатетик,— говорил он,—плохой химик»...
«Перипатетики держатся за слова, а мы — за факты».
В своих теоретических взглядах он, впрочем, еще довольно близко
примыкает к учению иатрохимиков. Разница, однако, заключается
в том, что для Бехера состав тел гораздо сложнее, чем думали раньше.
В своей «Подземной физике» он пишет: «Всякое подземное тело
является составным в большей или меньшей степени. Даже первоначальные
тела (элементы)—составные. Вообще во всем подлунном мире нет
ничего такого, что не было бы составным, только одно в большей, а другое
в меньшей степени. Тела составные в положительной степени
называются простыми; составные в сравнительной степени — сложными; в
превосходной степени — еще больше сложными».
Основными элементами минерального ил'И подземного царства Бехер
считает воду и землю. Но земля является как бы более отдаленным
компонентом различных тел, роль же ближайших составных частей
играют три различные земли: первая земля — плавкая или
стекловидная, вторая земля — жирная или горючая и третья земля — жидкая.
Эти земли напоминают, конечно, tria prima иатрохимиков и, подобно
этим последним, являются носителями определенных свойств. В
частности, жирная земля Бехера, являющаяся носителем горючести,
напоминает серу прежних химиков. Но, в отличие от своих предшественников
и в согласии со своими общими взглядами, Бехер считает серу телом
сложным. Ее горючесть обусловливается содержанием жирной земли, но,
'кроме того, в сере заключается еще другое кислотное начало, благодаря
чему она и может послужить источником получения серной кислоты, в
то время уже известной *. Для этого следует только освободить серу
от горючего начала.
В этих представлениях уже ясно видны основные черты будущей
теории, флогистона, которая была развита во всей своей полноте только
последователем Бехера —Сталем. В противоположность туманным и
противоречивым воззрениям алхимиков и некоторой недоговоренности,
* Открытие связано с эпохой Василия Валентина.
Открытие кислорода и теория горения
:ш
которую мы находим у Бехера, теория Сталя отличалась замечательной
для того времени ясностью и последовательностью. Terra pinguis *
Бехера превратилась у Сталя в ту огненную материю, или флогистон,
которая составляет центральную фигуру в нарисованной им картине
химических превращений. Развивая свои мысли, он начал с опытного
изучения состава серы в том смысле., как его понимал Бех?р. Сталь
-нашел, что серная кислота, потерявшая свою летучесть под влиянием
щелочи (от превращения в калийную соль K2SO4) и подвергнутая затем
действию такого горючего вещества, как уголь (при накаливании),
превратилась в серную печень (смесь многосернистых соединений калия
KsSn), из которой кислоты легко выделяли серу (например, K2S5 +
+ 2HC1 = 2KC1 + H2S + 4S). Он заключил отсюда, чго ему удалось
осуществить синтез серы из кислоты и угля и что, следовательно, мнение
Бехера о сложности серы, о том, что она составлена из двух различных
начал — кислотного и горючего, отвечает действительности. Тогда Сталь
пошел дальше-и смелым скачком мысли связал типичные процессы
горения с явлением обжига или кальцинации металлов. Последние при
этом процессе теряют не только свои типичные металлические свойства:
блеск, ковкость и пр., но и способность к дальнейшему изменению при
действии огня.
Сталь предположил, что общее горючее начало, находящееся в
различных телах, является причиной всех этих явлении. Когда тела горят,
оно выделяется, оно может дать начало огню. Когда лишенные горючего
начала вещества подвергаются действию тел, богатых этим началом,
происходит обратное явление. Так, металлические извести при
прокаливании с углем вновь приобретают все свойства, утраченные при
кальцинации.
Этому-то горючему началу Сталь и 'придал название флогистона.
Чтобы проникнуться представлением Сталя, надо заставить себя на
время совершенно забыть те понятия о процессах горения, ведущие свое
начало от Лавуазье, к которым мы привыкли со школьной скамьи,
которые теперь кажутся нам уже не истолкованием известных фактов,
а лишь -верной их передачей.
Мы теперь считаем, что когда горит сера, то она соединяется с
кислородом, образуя сернистый газ (SCb), который при известных
условиях может обогатиться кислородом воздуха и образовать серный
ангидрид (S03), который с водой дает серную кислоту или купоросное
масло (H2S04). Можно обратно отнять связанный с серой кислород, хотя
бы по тому способу, о котором упоминает Сталь. Тогда кислород от.серы
переходит к углю и вступает с ним в соединение (образуя углекислый
газ СОг), а сера освобождается. Равным образом, при обжиге металлов
последние соединяются с кислородом и образуют окислы. Медь,
например, превращается в черную окись меди (СиО), ртуть — в красную
окись ртути (HgO) и т. д.
Для нас сера по своему составу проще сернистого газа и серной
кислоты, металлы проще их кислородных соединений. Для пас образование
окислов серы и металлов обозначает усложнение вещества (синтез),
тогда как обратный процесс восстановления или разложения окислов
есть процесс упрощения (анализ).
С точки зрения теории Сталя дело должно обстоять как раз
наоборот.
Сера сложнее серной кислоты и металлы сложнее отвечающих им
«известок», ибо содержат, кроме некоторой земли, еще флогистон. Они
* Жирная земля. Прим. ред.
24*
372 Открытие кислорода и теория горения
геряют его при горении или обжиге и вновь приобретают при
прокаливании с углем. Горение есть процесс разложения, оно,'следовательно,
связано с упрощением вещества; наоборот, восстановление есть процесс
усложнения, процесс синтеза.
Все горючие вещества содержат флогистон, но особенно богаты и<м
те, которые сгорают почти без остатка. Таков уголь. Сталь даже как
будто еще дальше шел по пути сближения флогистона с углем, считая,
что все живые организмы, растения и животные содержат в своем
составе флогистон, что при горении этот последний переходит в воздух,
откуда вновь извлекается растениями.
Впоследствии, когда был открыт водород, многие химики были
склонны именно в нем видеть, так сказать, наиболее полное и
совершенное воплощение идеи флогистона.
Однако Сталь вовсе не отождествлял флогистон с каким-либо
реальным веществом. Флогистон представляется ему необыкновенно тонкой
и легкой землей, которая, однако, никогда не существует сама по себе,
а лишь в комбинации с другими субстанциями. Огонь и флогистон
также не одно и то же. Флогистон есть только начало огня, но не сам огонь.
В этих словах отчетливо сказываются пережитки схоластического
направления последователей Аристотеля.
Любопытны представления Сталя об огне и о самом процессе горе
ния. Огонь он не считает определенным веществом, а лишь собранием
телец, находящихся в сильнейшем вихревом движении. Но флогистон
способен приходить в состояние такого движения и принимать форму
огня лишь при наличности известных посторонних тел, лишь в
известной материальной среде, в которой он мог бы распространяться или
растворяться.
Роль такого растворителя и играет воздух, необходимость которого
¦для поддержания горения была хорошо известна Сталю, причем, как а
случае ограниченной растворимости солей в воде, некоторое количество
воздуха может воспринять или растворить лишь определенное
максимальное количество флогистона. Вот почему свеча, прогорев некоторое
время в закрытом пространстве, тухнет.
Нельзя не признать, что теория флогистона охватила обширный круг
важнейших химических процессов, дала им удовлетворительное по тому
времени объяснение, а главное, объединила разрозненные факты химии
в одно стройное целое. Можно сказать, что многие из числа известных
тогда химических явлений происходят действительно так, как если бы
флогистон на самом деле существовал и обладал свойствами, которые
ему приписывались. Нет поэтому ничего удивительного в том, что
учение Сталя быстро овладело умами и в течение целого столетия
оставалось господствующим, едва ли не общепризнанным, для многих превра-
гилось почти что в догму.
Блестящему успеху флогистона много содействовало и то
обстоятельство, что Сталь, до известной степени независимо от этой теории,
пришел к двум важным обобщениям и содержание их, так сказать,
органически влил в свое учение. Вот в каких выражениях говорит об
этих заслугах Сталя его научный противник Лавуазье: «Мы обязаны
Сталю двумя важными открытиями, которые не зависят ни от какой
системы, ни от какой-либо гипотезы, которые останутся вечными
истинами: во-первых, что металлы являются телами горючими, что кальцинация
есть настоящее горение... Второе открытие, которым мы также обтзаны
Сталю и которое является' еще более важным, заключается в том, что
способность гореть, быть воспламеняемым, может передаваться с одного
тела к другому. Если, например, смешать горючий уголь с пего; ючей
Открытие кислорода и теория горения
37;г
купоросной (серной) 'кислотой, то последняя превращается в серу, та
приобретает способность гореть, которую в то же время теряет уголь».
Весьма замечательно, что Сталю, несомненно, были известны многие
факты, которые, спустя почти целое столетие, привели к крушению его
теории. Как мы видели, ему была известна ограниченная способность
определенного объема воздуха поддерживать горение, он знавал также,
что при обжиге металлов происходит увеличение п весе. Но он
игнорировал эти факты или давал им превратное объяснение. Так, прибыль в
весе при кальцинации он, вслед за Кункелем, объяснял увеличением
удельного веса тела, происходящего при этом процессе вследств-ие
улетучивания легкого флогистона. Заметим, что еще Бойлю, на опыты
которого ссылается Сталь, было известно, что на самом деле переход
металлов в «известки» сопровождается не повышением, а понижением
-удельного веса.
По мере того как накоплялись новые факты, увеличивалось и число
возражений против теории Сталя. Его последователям приходилось
поэтому все более и более прибегать к произвольным, фантастическим
объяснениям, даже к софизмам, пополнять первоначальную теорию
рядом добавочных, часто противоречащих друг другу допущений. Теория
мало-помалу превращалась в запутанный клубок. В таком состоянии
застал ее Лавуазье.
VI. ПЕРВЫЕ НАМЕКИ НА СУЩЕСТВОВАНИЕ КИСЛОРОДА
Едва ли не первые намеки на существование кислорода и указания
на его роль в процессе горения мы находим в старинной китайской
литературе. В книге Маб-Khoa, относящейся к половине VIII в. и. э.,
развивается мысль, что все существующее слагается из двух начал. Одно
из.них — «янг» — является по своей природе совершенным, сильным и
преобладающим, другое —«ин» *— несовершенным, слабым и имеющим
подчиненное значение. Согласно этому принципу Mao-Khoa, составлен и
воздух. Он состоит из двух компонентов: совершенного воздуха,
который отвечает современному азоту, и несовершенного — одной из форм
«ина». Эта последняя отвечает нашему кислороду. Существует
несколько путей, позволяющих сделать воздух более совершенным, обогатить
его «янгом», т. е. азотом. Для этого можно подвергнуть его при
нагревании действию металлов, угля, серы и т. п. Все это формы, которые
может принимать совершенное начало —«янг». Накаленные па
воздухе, эти тела горят, причем они вступают в сочетание с «ином» воздуха,
а в этом последнем накопляется «янг» (азот).
Что касается до «ина» (кислорода), то, по мнению китайского
мудреца он никогда не получается в совершенно чистом состоянии. Тем не
менее образование его можно наблюдать при накаливании различных
каменистых пород. Названия этих пород приводятся в Mao-Khoa, но
точное значение их не могло быть установлено. Возможно, что одно из них,
Hh6-Siao означает селитру, и что таким образом китайским философам
VIII в было известно разложение селитры под влиянием жара,
сопровождаемое выделением кислорода. Возможно также, что вывод
относительно присутствия в селитре больших количеств кислорода, начала,
возбуждающего горения, был сделан китайцами, давно знакомыми с
порохом на том основании, что сера и уголь в соприкосновении с
селитрой горят интенсивнее, нежели на воздухе. Весьма любопытно, что
* По современным русско-китайским словарям кислород называется ян, водород
цин, азот — дань, селитра — сяо-ши. Прим. ред.1
374
Открытие кислорода и теория горения
автору Mao-Khoa было известно и присутствие кислорода в воде, притом
з весьма прочно связанном состоянии, что, как мы знаем, вполне
соответствует действительности.
Будущему историку химии выпадает на долю -найти связывающие
нити между эти'ми первыми указаниями на сложность воздуха, на
присутствие в нем кислорода, и между более близкими к нашему времени
данными, касающимися того же предмета. Пока ничем не заполненная
пропасть отделяет китайского философа VIII в. от эпохи Возрождения,
от Леонардо да Винчи (1452—1519), в котором гениальный
художник счастливо сочетается с великим ученым и выдающимся
инженером. Леонардо первый из европейцев, со свойственной ему
прозорливостью, не зная, конечно, о находках своих предшественников,
утверждал, что воздух, вопреки мнению древних, не является элементом,
что в воздухе заключаются два различных составных начала, так как
он при процессах горения, а также дыхания всегда расходуется, и
притом не всецело, а только отчасти.
«Огонь, — говорит он,— постоянно разрушает воздух, его питающий;
образовалась бы пустота, если бы не притекал новый воздух, чтобы ее
заполнить. Когда воздух не в состоянии, необходимом для того, чтобы
воспринять пламя, в нем не может существовать ни огонь, ни какое-
либо животное, земное или воздушное». Быть может, особенно
замечательно следующее место: «копоть в центре пламени свечи образуется
потому, что воздух, который входит в состав пламени, не может
проникнуть до середины. Он останавливается у поверхности пламени и здесь
подвергается превращению» *.
А. Гумбольдт в своем «Космосе» называет еще одно имя —
математика Кардана (Hieronimus Cardano, 1501 —1576), который
примерно спустя полстолетия после Леонардо (1553 г.) отметил (в
сочинении «De rerum subtilitate») увеличение в весе одного из металлов —
свинца при окислении. «Но,— замечает Гумбольдт,— приписал это
увеличение «удалению (из металла) небесной огненной материи, имеющей
способность облегчать тела»,— совсем в духе позднейшего мифа о
флогистоне».
Существенно новое и важное в вопросе о природе горения принес
только XVII век, вообще богатый научными открытиями. То, что дошло
до нас из сделанного в эту эпоху, связано с именами Гамера Поп-
пия, Лефевра, Жана Рея, Г у к а ив особенности Джона М а й-
о в а.
Л е ф е в р , один из наиболее авторитетных химиков-писателей той
эпохи (особенной известностью пользуется его сочинение «Traite de
chimie», появившееся в 1660 г.), цитируя опыты Гамера Поппия об
увеличении веса металлов при обжиге, останавливается подробнее на опыте
с сурьмой и констатирует, что если опыты вести при помощи
собирательной линзы, то получается явный привес, так что из 12 долей металла
получается пятнадцать долей белого порошка (т. е. окиси). Этот привес
Лефевр объясняет тем, что сурьма фиксирует рассеянное повсюду
начало, играющее, по его мнению, очень важную роль в экономии природы
*^Эта цитата яриведена из опубликованной Вентури (Ann. Chim. phys., 23, 150)
в 1797 г. части рукописей Леонардо. Для характеристики взглядов Леонардо на
методы естествознания, также опередивших свое время по крайней мере на три столетия,
приведем еще следующие слова: «Истолкователем тайн природы является опыт. Он
никогда не обманывает; обманывает иногда наше суждение, потому что опыт отвергает
ожидаемые результаты. Нужно обращаться к опыту, изменять его условия, пока мы не
сможем вывести из него общих законов, ибо это он приводит к верным законам. Вот
метод, который следует соблюдать при исследовании явлений природы>. Какая
противоположность с наставлением классической и псевдоклассической схоластики!
Открытие кислорода и теория горения
375
и обусловливающее переход металлов в нелетучее состояние. Дюма
не без основания видит в этом принципе смутное предчувствие
кислорода.
С экспериментальной стороны еще более замечательны опыты Жана
Рея (Jean Rey, род. в коаде XVI столетия в Bugnes, в департаменте
Dordogne, во Франции, умер в 1654 г.), который в 1630 г. тщательными
опытами установил прибавку в весе, испытываемую свинцом и оловом
при прокаливании, и пришел -к заключению, что привес происходит от
поглощения воздуха, который при этом подвергается сгущению.
^До Торричелли (1644) и до От то фон -Гер и ке (1602—1680)
Рей (правда, только теоретическим путем) пришел к выводу о весомости
воздуха и, вопреки господствовавшим тогда воззрениям, утверждал, что
при трансмутаци'И невозможно изменение веса тела без того, чтобы оно
поглотило или выделило из себя часть вещества, ибо «вес всех частей
каждого данного тела находится с ними уже в колыбели».
В двух отношениях Рей, однако, впал в заблуждение. Правильно
усматривая причину увеличения в весе металлов при прокаливании в
поглощении ими воздуха, он ошибочно предполагал, что акты
(химического) поглощения металлов в состав «извести» предшествуют
поглощению воздуха. Последнее явление он, по-видимому, рассматривал
скорее как физический процесс (уподобляя его удерживанию воды
песком), нечто в роде того, что в настоящее время называется
адсорбцией. Самому же процессу «кальцинации» он давал другое, довольно
туманное объяснение.
С другой стороны, опыты Рея были недостаточны для того, чтобы
сделать заключение о различии природы той части воздуха, которая
входит в состав извести, и той, которая остается в сосуде после
кальцинации. Итак, в этом отношении Рей по своим взглядам стоит ниже,
например, Леонардо да Винчи и других. Можно также указать, что Рею (как
и Лефевру) были известны опыты Гамера Поппия и некоторых других
лиц касательно увеличения в весе металлов при окислении. Во всяком
случае, сделанное ими оставило прочный след в науке и, между прочим,
оказало несомненное влияние на превосходные работы Майова, о
которых будет сказано ниже.
Нельзя не упомянуть здесь еще о взглядах, высказанных, правда,
бегло и как бы попутно знаменитым физиком Робертом Гуком
(1635—1703). В его сочинении «Micrographia» (1655 г.), главной темой
которого является изображение и описание разнообразных объектов,
видимых под микроскопом, между прочим говорится и о составе
воздуха. При этом автор высказывает мысль, что горение аналогично
процессу растворения.
На воздух ой смотрит как на универсальный растворитель
различных горючих субстанций. Это растворяющее действие он приписывает,
однако, не воздуху целиком, а лишь одной из двух его составных
частей- Последняя "по своей природе близка, а может быть, тождественна,
с газом, выделяющимся при накаливании селитры.
Селитру Гук считает телом более богатым этой растворяющей
составной частью, нежели воздух. Поэтому малого количества селитры
достаточно для сжигания сравнительно "больших количеств горючего
материала, между тем как воздух необходимо сравнительно часто
возобновлять для того, чтобы горящее тело не потухло. Этим же объясняется
интенсивный характер горения тел, соприкасающихся с расплавленной
селитрой.
376
Открытие кислорода и теория горения
VII. ДЖОН МАЙОВ
К сожалению, ни автор Ma 6-Khoa, ни Леонардо да Винчи, ни
Гук-не приводят описания тех наблюдений и опытов, на которые
опираются высказанные ими взгляды; опыты же Рея не давали разъяснения
по одному из главных и важнейших особенностей процессов горения.
Зато превосходное опытное подтверждение и широкое развитие
упомянутых взглядов мы находим в замечательных работах рано умершего
английского врача Джона Май о-в a (John Maiow), жившего (1645—
1679) в tv славную эпоху (вторая половина XVII в.), которая подарила
нам и Ньютона (1642—1726), и Бойля (1627—1691), и Оттона
фон Герике (1602—1686), и Гюйгенса (1629—1695), и
Паскаля (1623—1662), и Гука и много других великих деятелей в
области точного знания.
Сочинение Майова, -в котором он изложил результаты своих
исследований, «Tractatus quinque physico-medici», было опубликовано им в
Оксфорде на 29-м году жизни и представляет образец ясности мысли,
точности и убедительности эксперимента, особенно для того времени.
Вспомним, что Майов жил лишь незадолго до знаменитого В а н-
Гельмонта, воззрения которого, как мы только что видели, были
крайне запутаны и насквозь пропитаны мистицизмом.
Придя к заключению, что в воздухе находится составная часть,
поддерживающая горение, питающая огонь, Майов дал ей название «селит-
ряно-огненного воздуха» (spiritus nitro-aerius). В двойственности этого
названия уже заключается намек на то, что дело идет о начале, которое
содержится не только в воздухе, но и в селитре (nitrum), и в азотной
кислоте, происходящей из селитры. Spiritus nitro-aerius есть газ,
необходимый для горения, пишет Майов. Этот газ присутствует в воздухе,
ибо уже Бойль показал, что в безвоздушном пространстве горение
невозможно. Но поддерживающее горение начало не составляет воздуха
целиком, оно составляет только более активную часть его, которая
исчезает при горении, между тем как остающаяся часть воздуха но способна
к поддержанию огня. Поэтому, хотя воздух прежде и считался
элементом, но на самом деле на него следует смотреть как на сложное тело.
Spiritus же nitro-aerius скорее всего является элементом. Майов делает
даже попытку определить количественный состав воздуха и пользуется
для этой цели способностью окиси азота (он получал ее при действии
железных гвоздей на азотную кислоту) поглощать кислород из
воздуха и с водой давать азотную кислоту. Но опыт был слишком сложен,
чтобы результаты его могли быть в то время правильно
истолкованы.
Рядом опытов Майов доказывает присутствие кислорода в селитре
и в азотной кислоте. Вот некоторые из этих фактов, на которые он
опирается. Для горения смеси серы с селитрой, а также пороха не требуется
присутствия воздуха, так как горение происходит даже под водой.
Находящийся в селитре spiritus nitro-aerius содержится не в щелочи (кали),
а в летучей части, в кислоте, и притом при образовании селитры в
природе заимствуется из воздуха. На самом деле, если из селитряной
земли извлечь водой всю селитру, то через некоторое ьремя она снова
там образуется, если только в земле есть некоторое количество щелочи.
О присутствии кислорода в азотной кислоте Майов заключает на том
основании, что при действии ее на различные субстанции получаются те
же продукты, которые образуются и при горении этих субстанций на
воздухе. Так, из металлической сурьмы в обоих случаях получается
окись сурьмы. Очевидно, в обоих случаях сурьма соединяется со spi-
Открытие кислорода и теория горения 377
ritus nitro-aerius, чем и объясняется необходимое при этом увеличение
в весе. Важность последнего обстоятельства не ускользнула от
внимания Майова, и явления кальцинации были им совершенно правильно
истолкованы. В этом отношении Майов выгодно отличается от многих
химиков 'позднейшего времени, в том числе от Пр-истлея, а также от
Рея, исследования которого ему были известны. Подобно сурьме ведет
себя железо, которое, соединяясь с кислородом (и водой), образует
ржавчину, и сера, превращающаяся в серную кислоту. К подобного же
рода процессам Майов с замечательной проницательностью относит
окисление пива и вина, 'Ведущее к превращению их в уксус. Вообще,
заключает он, для того, чтобы имел место процесс горения, всегда
необходимо присутствие spiritus nitro-aerius, причем этот последний может
входить в состав горящей субстанции (смеси), примером чего может
служить порох; или он может заимствоваться из протекающего воздуха,
например в случае горения серы, или, наконец, может иметь место и то
и другое одновременно, как в случае растительных веществ.
Для Майова была также совершенно ясна глубокая аналогия между
процессами горения и дыхания. Путем прямого опыта он убедился в том,
что если зажечь свечу и заставить ее гореть под колпаком в
ограниченном объеме воздуха, отделенного водой от окружающей атмосферы
(прием н ныне постоянно употребляемый для этой цели), то через
некоторое время не только свеча тухнет, но и вообще оставшийся воздчх
коренным образом меняет свои свойства; горючие тела (например
камфора) перестают в нем гореть, животные не могут дышать и погибают.
С другой стороны, живая мышь, введенная под колокол с воздухом и
погибающая там через некоторое время, оставляет после себя воздух, в
равной мере непригодный для жиани и для горения, ибо животные в нем
погибают, а горящие тела—тухнут.
Наконец, Майов помещал под колоколом одновременно зажженное
тело и мышь. При этом горение и жизнь прекращались (при том же
объеме воздуха) примерно вдвое скорее, чем в том случае, когда каждое
явление происходило отдельно. Дело обстоит так, как если бы горящая
свеча и мышь конкурировали из-за ограниченного запаса огнетворного
начала. Иными словами, одна и та же материя поддерживает и
горение и жизнь.
В биологической части работы Майова заключается не мало и
других глубоких мыслей и ценных наблюдений. Алый цвет артериальной
крО'ВИ он приписывает содержанию в ней spiritus nitro-aerius. Ведь
черная венозная кровь делается алой прежде всего на поверхности
соприкосновения с воздухом, тогда как свежая, еще теплая артериальная
кровь чернеет под колоколом воздушного насоса, при этом пенится и
выделяется большое количество газа.
Образование теплоты в животном организме Майов также
приписывает spiritus nitro-aerius, точнее, акту соединения его с горючими
началами крови. Происходящий при этом процесс он сближает с выделением
тепла, которое наблюдается на свежей поверхности марказита
(разновидность двусернистого железа FeS2) и других -подобных минералов
через некоторое -время после соприкосновения их с воздухом. Вообще
он считает кислород источником всякого теплообразования и едва ли
не важнейшим из всех элементов, встречающихся в природе.
Опыты Майова и заключения, которые из них сделал автор,
отмеченные несомненной печатью гения, далеко опередили свой век. Для одних
они прошли почти незамеченными, другие, как Роберт Бойль, встретили
их с преувеличенной осторожностью, даже скептически, считая их
малодоказательными.
378
Открытие кислорода и теория горения
Одной из причин этого, на первый взгляд странного, обстоятельства,
по-видимому, следует отметить появление работы Майова в то самое
время, когда начинала развиваться теория флогистона, давшая
законченное объяснение целому циклу явлений и в то же время -не стоявшая
в таком резком противоречии, как взгляды Майо-ва, с живыми еще
традициями учения Аристотеля и алхимиков. С другой стороны,
теоретические представления Майова не охватывали достаточно широко
известных в то время химических процессов. Ему не хватало знания некоторых
существенных фактов. Особенно вредно сказывалось незнакомство с
условиями образования и со свойствами одного из самых обычных
продуктов горения — углекислого газа, хотя и открытого уже Ван-Гельмонтом,
но еще очень мало изученного.
Благодаря этому, например, оставалось непонятным, почему в одних
случаях горение сопровождалось уменьшением объема воздуха, в
других— нет. Равным образом отсутствовала ясность в понимании причин
изменения веса тел при горении и т. п.
Убедительная сила аргументации Майова, несомненно, много
выиграла бы, если бы он больше обращал внимания на весовые количества
реагирующих веществ и вообще производил бы количественный учет в
своих опытах, как это спустя сто лет делал Лавуазье. Не менее важным
является то обстоятельство, что, хотя Майов и провидел существование
кислорода, так сказать, чувствовал его присутствие в различных телах
и ясно понимал его роль во многих химических и биохимических
явлениях, однако, как это ни странно, он, по-видимому, даже не сделал
попытки изолировать свой spiritus nitro-aerius в чистом состоянии из
селитры, в которой, как он сам был уверен, должно было заключаться
особенно большое количество этого загадочного вещества.
VIII. МИХАЙЛО ВАСИЛЬЕВИЧ ЛОМОНОСОВ
Промежуток времени, равный почти целому столетию, отделяет
работы Майова от того славного в истории химии момента (середина 70-х
годов XVIII в.), когда благодаря Лавуазье с глаз химиков как бы спала
пелена и сразу сделалась очевидной естественная связь и соподчинение
длинного ряда фактов, частью давно известных, частью только что
открытых. На этот промежуток времени падает деятельность двух
выдающихся ученых, из которых работы одного английского химика — Блэ-
ка *—лишь косвенно касаются нашей темы. Мы поэтому можем
обойти их молчанием. Зато первого русского ученого — Михаила
Васильевича Ломоносова — мы вправе считать настоящим предшественником
Лавуазье, а потому на его научной деятельности необходимо
остановиться несколько подробнее.
До сравнительно очень недавнего времени Ломоносов был известен
главным образом как преобразователь русского языка и русской
грамматики, да как автор хвалебных од, по тому времени (середина XVIII в.)
составлявших явление выдающееся в нашей литературе. О трудах
Ломоносова поточным наукам,преимущественно по (общей и космической)'
физике и химии, знали очень мало даже специалисты и то большей
частью не придавали им особенно серьезного значения. Только
празднование 150-летнего юбилея со дня основания Ломоносовым первой pvc-
* Блэк (Joseph Black -род. в 1728 г., ум. 1799 г.) в своей знаменитой работе «On
magnesia alba» устанавливает химическую природу окиси магния (magnesia usta),
основной углекислой соли (magnesia alba) и сернокислой (горькой, Энсомской) соли
этого металла, выясняет взаимоотношение между этими телами и участие в составе
и образовании белой магнезии углекислого газа.
Открытие кислорода и теория горения
379
ской химической лаборатории при Академии наук оживило интерес к
этой стороне деятельности гениального русского ученого. Особенно много
мы обязаны в этом отношении профессору Б. Н. Меншуткину, который,
так сказать, реставрировал Ломоносова — физика-химика и" издал ряд
его сочинений, частью рукописных, нигде еще не напечатанных [8].
Важнейшие из этих последних были затем переведены на немецкий язык и
вошли в состав собрания «классиков» Оствальда (№ 178).
Ломоносов примерно за 25 лет до Лавуазье высказал правильный
взгляд -на сущность процессов горения и предвосхитил некоторые другие
важные истины, установленные затем знаменитым французским
химиком.
¦Уже -в 1748 г. Ломоносов пишет в письме к Эйлеру: «Нет никакого
сомнения, что частички воздуха, текущего постоянно над обжигаемым
телом, соединяются с ним и увеличивают его вес». В том же 1748 г. в
«Размышлениях о причине тепла и холода» («Meditationes de caloris et
frigoris causa») он обсуждает опыт Роберта Бойля с нагреванием
металлов в запаянных сосудах, о которых упоминалось выше, подвергает
критике точку зрения английского ученого и приходит к прямо
противоположному выводу относительно сущности данного явления: «Если не
ошибаюсь,— говорит он,— Р. Бойль первый опытами показал увеличение
веса при сжигании и объяснил, что части огня и пламени могут
делаться устойчивыми и весовыми. Но почти все опыты его доказывают лишь,
что увеличение в весе происходит от тяжелых частей пламени или
воздуха, текущего при обжигании вокруг накаливаемого тела. Так, если
пластинки металла сжигаются в пламени серы, то они увеличиваются
в объеме и весе, но -причина увеличения — кислота серы, которую по
освобождении от флогистона можно собрать под колоколом воздушного
насоса; она 'проникает в поры меди и серебра и, соединившись с ними,
увеличивает вес их... К накаленному телу или частичкам его, летающим
в воздухе (находящимся над телом), приходит во время обжигания
какая-то другая материя, увеличивающая вес окалины. Материя эта,
конечно, не та, которая считается свойственной огню, так как окалины,
удаленные от огня, даже на величайшем морозе не теряют в весе и не
показывают избытка теплоты. Я не вижу, почему в окалинах огненная
материя поступала бы несообразно со своей природой. Затем окалины
при восстановлении снова теряют излишек веса». Наконец, в 1756 г.
Ломоносов повторяет мысли Бойля и сообщает об этом следующее:
«Делал опыты в заплавленных накрепко стеклянных сосудах, дабы
исследовать, прибывает ли вес металлов от чистого жару. Оными опытами
нашлось, что славного Роберта Бойля мнение ложно, ибо без
пропущения внешнего воздуха вес сожженного металла остается в
одной мере».
К сожалению, точное описание этих опытов, в своей время
доложенных Академии, не дошло до нас, и даже краткая формулировка
выводов стала известна только в самое последнее время.
В своих сочинениях Ломоносов, правда, неоднократно упоминает о
флогистоне, но, критически сопоставляя различные места его статей и
рассуждений, можно прийти к выводу, что он в сущности относится к
огненной материи с явным несочувствием. Если он и пользовался при
случае теорией Сталя, то лишь потому, что она считалась в то время как
бы официальной (и что, отрицая ее, можно было повредить себе зо
мнении ученого мира). Последнее было особенно нежелательно для
Ломоносова в начале его академической карьеры*.
* Л. А. Чугаев здесь не совсем прав, так как М. В. Ломоносов еше в 40-х годах
неоднократно высказывался против принятой тогда теории флогистона. Прим. ред.
380 Открытие кислорода и теория горения
Если Ломоносов был, бесспорно, на правильном пути в понимании
явлений горения (о трудах своих предшественников, в частности Майо-
ва, он, очевидно, не знал), то этим далеко не ограничиваются его
заслуги перед химией и физикой.
За '11 год до Лавуазье он в совершенной форме высказал закон
сохранения материи: «Все перемены, в натуре случающиеся, такого
суть состояния, что сколько чего у одного тела отнимается, столько
присовокупится к другому. Так, ежели где убудет несколько материи,
то умножится в другом месте; сколько часов кто положит на бдение,
сколько же сну отнимает. Сей всеобщий естественный закон
простирается и в самые правила движения: ибо тело, движущее своей силой
другое, столько же оные у себя теряет, сколько сообщает другому,
которое от него движение получает».
Не укрылось от внимания Ломоносова и огромное значение для
развития химии количественных методов исследования или, говоря
общее, сближение химии с физикой и математикой.
В этом отношении Ломоносов оказался настоящим пророком, как
бы отцом современной физической химии. «Если кто хочет,—
пишет он в одном месте,— глубоко проникнуть в исследование
химических истин, то должен изучать механику. Так как знание механики
предполагает знание чистой математики, то стремящийся к
ближайшему изучению химии должен хорошо знать математику» («Элементы
математической химии», 1741 г. §§ 23 и 24). А вот что говорит Ломоносов
10 лет спустя (1751 г.) в своем «Слове о пользе химии» о времени, когда
химия будет действовать через геометрию, механику и оптику: «К сему
требуется весьма искусный химик и глубокий математик в одном
человеке. Химик требуется не такой, который только от одного чтения книг
понял сию науку, но который собственным искусством в ней прилежно
упражнялся; и не такой, напротив того, который, хотя великое
множество опытов делал, однако больше желанием великого и скоро
приобретаемого богатства поощряясь, спешил только к одному исполнению
своего желания и, ради того, последуя своим мечтаниям, презирал
случавшиеся в трудах своих явления и перемены, служащие к истолкованию
естественных тайн. Не такой требуется математик, который только в
трудных выкладках искусен, но который в изобретениях и в
доказательствах привыкнув к математической строгости, в натуре сокровенную
правду точным и непоползновенным порядком вывести умеет.
Бесполезны тому очи, кто желает видеть внутренность вещи, лишаясь рук к
отверстию оной. Бесполезны тому руки, кто к рассмотрению открытых вещей
очей не имеет. Химия руками математика, очами физическими по
справедливости называться может».
Эти слова, конечно, не могли быть оценены по достоинству
современниками, до такой степени далеки они от круга интересов и
понятий эпохи Ломоносова. Зато они сохраняют все свое значение в
настоящее время, как раз характеризующееся пышным расцветом
физической химии. Ломоносов из далекого прошлого каким-то изумительным
чутьем провидел не только возникновение этого важного отдела химии,
но даже те слабые или теневые стороны, которые могли обнаружиться
при неправильном и одностороннем развитии этой новой научной
дисциплины.
Если к сказанному прибавить, что Ломоносов был также
выдающимся физиком (особенно важно, что он был одним из первых, вполне
отчетливо высказавших основные принципы механической теории тепла
и кинетической теории вещества), то будет ясно, что в его лице Россия
дала миру истинного гения, и лишь случайное и несчастное для нас сте-
Открытие кислорода и теория горения
381
чение обстоятельств было причиной того, что великий «Архангельский
мужик» не довел и не мог довести до конца обширной программы работ,
многочисленных и важных опытов, столь блестяще задуманных, но
большей частью не получивших осуществления.
Главную причину этого нужно видеть в том обстоятельстве, что
современная Россия не оценила Ломоносова по достоинству. В нем
хотели видеть прежде всего стихотворца и ритора, тогда как он по
призванию принадлежал точным наукам — физике и химии. Между тем
к его физико-химическим работам относились со снисходительной
небрежностью, едва разрешая ему предаваться им в свободное от
официальных занятий, время, на что он горько жалуется в одном из своих
писем к Шувалову.
В результате о Ломоносове-химике основательно забыли, вернее,
никогда как следует и не знали его с этой стороны. Сделать переворот
в области химии выпало на долю Лавуазье.
IX. ШЕЕЛЕ, ПРИСТЛЕЙ, БАЙЕН
Если Лавуазье принадлежит великая заслуга установить
точнейшим образом истинную природу процессов горения, в основных
чертах столь ясно уже намеченную Майовым, тс Шееле и Пристлею
суждено было впервые увидеть воочию этот «spiritus nitro-aerius», этот
«жизненный воздух», в котором тела горят гораздо ярче, а животные
выживают больше, чем в том же объеме воздуха.
Внимательное сличение подлинных работ Шееле и Пристлея, а
также их переписки с Лавуазье и другими химиками, недавно сделанное
покойным Копенгагенским профессором Иёргенсоном, привело
последнего к заключению, что не Пристлею, а Шееле принадлежит приоритет
в деле открытия свободного кислорода.
Хотя знаменитое сочинение шведского химика «О воздухе и огне»
увидело свет только в 1777 г., однако теперь можно считать
установленным, что Шееле впервые получил кислород в промежуток времени
между 1771 и 1772 гг. и притом различными путями: во-первых,
нагреванием азотнокислой магнезии (2Mg[N03]2 = 2MgO-f2N204 + 02),
причем образующаяся одновременно с 02 двуокись азота поглощалась
известью; во-вторых, нагреванием селитры до высокой температуры
(2KNO3--=2KN02 + 02); в этом случае Шееле определенно говорит о
получении чистого «огненного», или «купоросного воздуха» без примеси
азотной кислоты и углекислого газа; в-третьих, Шееле наблюдал
образование кислорода при разложении углекислого серебра (2Ag2C03 =
= 4Ag + 2C02 + 02), причем для поглощения образующегося углекислого
газа, как и в предыдущем случае, служила известь. Описывая новый
газ, Шееле определенно отмечает, что он энергичнее поддерживает
явления горения, нежели обыкновенный воздух. В то время ему уже
было известно, что атмосферный воздух состоит из двух различных
газов, из которых только один, именно кислород, или купоросный
воздух, поглощается горящими телами (при горении фосфора, например,
в замкнутом пространстве, он наблюдал сокращение объема воздуха
wa 74—7б и образование безводной фосфорной кислоты, т. е.
фосфорного ангидрида P2Os); ему было известно, что тем же самым
свойством поглощать кислород из воздуха, притом без помощи нагревания,
обладает ряд других «флогистинизирующихся» тел, например
льняное масло, свежая кровь, аммиачный раствор закиси меди и др. Не
382
Открытие кислорода и теория горения
ускользнула от внимания Шееле и важная роль кислорода в акте
дыхания и много других замечательных особенностей этого газа.
Пристлей, который, как принято считать, впервые получил
кислород в 1774 г., а именно посредством нагревания окиси ртути, тогда
же заметил, что образующийся газ поддерживает горение лучинки
ярче обыкновенного воздуха и скорее всего напоминает закись азота.
Так как окись ртути, послужившая для приготовления кислорода,
была получена при помощи азотной кислоты (прокаливанием
азотнокислой ртути), то Пристлей сначала и предположил, что полученный
им «воздух» содержал какую-то примесь, происходящую из азотной
кислоты, а так как другой образчик кислорода, обладавший тем же
свойством, был получен им из сурика (РЬз04), приготовление которого
было также связано с употреблением азотной кислоты, между тем как
третий образчик был получен (раньше двух других) накаливанием
селитры *, то разнообразие источников, дающих новый газ, не было в
состоянии рассеять ошибки, в которую впал английский химик, тем
более, что он исходил из предвзятого мнения, будто «воздуха» чище
того, который имеется в атмосфере, вообще быть не может. В этом
убеждении он пребывал до марта 1775 г., когда случайно нашел, что газ,
оставшийся после действия (неполного) на кислород окиси азота, не
только не прекращает горения лучинки, но, наоборот, заставляет ее
еще ярче вспыхнуть. Далее, он нашел, что мышь живет в кислороде
дольше, чем в равном объеме воздуха. Если эти опыты не вполне еще
рассеяли его сомнения, то это, наконец, случилось, когда он убедился
в том, что для превращения определенного объема окиси азота
кислорода требуется в 4—5 раз меньше, нежели воздуха, что, следовательно,
кислород в 4—5 раз «чище» или «лучше» воздуха атмосферы. Только
после этого Пристлей придал своему газу особое название,
составленное в духе господствующих воззрений того времени. Он назвал
кислород «дефлогистинизированным воздухом».
Раз установив истину, Пристлей не замедлил понять важное
значение сделанного открытия и занялся тщательным изучением нового газа.
Он устанавливает, что кислород удельно тяжелее как воздуха, гак
и азота; что кислород дает более сильный взрыв, нежели с воздухом, и
что при этом наиболее благоприятное отношение объемов 2: 1. Он
выясняет роль кислорода при дыхании и предлагает пользоваться им для
очищения воздуха в жилых помещениях, а также применять его для
вдыхания при трудных болезнях. Он указывал на возможность
применять кислород в смеси с водородом для получения очень высоких
температур, например, для плавления платины и т. д.
Таким образом, Шееле и Пристлей шли частью разными
путями к одной и той же цели, один не знал о работах другого. Великая
заслуга их заключается в установлении ряда капитальнейших актов,
которые для науки того времени имели несравненно большее значение,
чем, например, в наши дни открытие радиоактивных веществ.
У обоих исследователей слабой стороной было теоретическое
освещение наблюденных ими явлений. Они были сторонниками теории
флогистона, которой оставались верными до конца дней своих. Они
являли собой блестящий пример того, как мысль, опирающаяся на
неправильные посылки, на предположения, заведомо расходящиеся с дей-
* Получение кислорода из селитры было осуществлено Пристлеем в 1771 г.,
но в то время он считал полученный газ за обыкновенный воздух, позднее (1774 г.)
видел в нем окись азота и лишь к 1775 г. признал в нем дефлогистинизированный
воздух, т. е. кислород.
Открытие кислорода и теория горения 383.
ствительностью, тем не менее может приводить к правильным выводам,
а главное — приводить к постановке новых важных опытов.
Нам необходимо назвать имя еще одного исследователя, который
принимал активное участие в открытии кислорода, хотя и не довел
своей работы до конца,—то был Пьер Б а йен (Pierre Bayen, 1725—
1798) родом^ из Шалона на Марне, по профессии военный аптекарь.
В начале 1//4 г., когда только еще начал;» складываться взгляды
Лавуазье на состав воздуха и на природу процесса горения (см. ниже),
Банен заметил, что окись ртути, приведенная в соприкосновение со
многими горючими телами, дает обильное образование газов.
Первоначально опыт был поставлен с серой и привел к сильному взрыву и к
образованию киновари. Так как для получения окиси ртути исходными
материалами служила азотнокислая сера, то Байен предположил, что
в окиси ртути оставалась часть азотной кислоты и что она-то и была
виновницей взрыва. Но окись ртути, приготовленная без помощи
азотной кислоты, дала тот же результат. С другой стороны, в смеси с
углем при прокаливании окись ртути привела к образованию газов и к
восстановлению до металлической ртути. Наконец, Байен наблюдал
образование газа (кислорода) и ртути при прокаливании окиси HgO
различного приготовления. Этот газ, не растворимый в воде, он считает
отличным от угольной кислоты.
Байен не подозревал, что в последнем случае он имел дело с
кислородом, хотя и показал, что новый газ при прокаливании в нем ртути
превращает ее в красную ртутную известь, или в Mercurius praecipitatum
per se * (HgO).
Всего интереснее то обстоятельство, что на основании своих опытов
Байен, в значительной степени независимо от Лавуазье, пришел к
выводу, что при кальцинации металлы вовсе не теряют своего
флогистона, а наоборот, поглощают некий газ, с которым приходят в
соприкосновение.
За счет этого поглощения и получается прибыль в весе. Таким
образом, Байона до известной степени можно считать причастным к
тому циклу открытий, которые связаны с именами Пр ист лея, Шееле и
Лавуазье. Но его работы не были закончены и сохранили значение
лишь в качестве любопытного документа из истории развития
человеческих знаний. При ярком свете гения Лавуазье труды Байена
казались мало заметной звездочкой, существование и значение которой
поэтому и не было в достаточной степени оценено, едва было замечено
современниками. Сам Байен очень скоро прекратил свои работы,
увидав, как сильно опередил его Лавуазье.
X. АНТУАН ЛОРАН ЛАВУАЗЬЕ
Общий характер и направление работ Лавуазье вполне
определились уже в одном из его первых научных исследований,
относящемуся к 1770 году.
Вопрос, который им тогда был поставлен на разрешение, был
старый вопрос о превращаемости воды в землю, связанный с учением о
четырех элементах и о единой первородной материи. Давно известный
факт, удостоверенный такими авторитетами, как Роберт Б о й л ь,
Маркграф, Жоффруа и др., заключался в том, что вода,
подвергаемая повторной перегонке, всегда оставляет некоторое количество
землистого остатка. Полагали, что эта «земля» образовалась за счет
воды. Ван-Гельмонт пришел к тому же заключению иным путем.
* Осажденная ртуть. Прим. ред.
.38-'і
Открытие кислорода и теория горения
Он выращивал ивовую ветку в большом глиняном сосуде с землей,
который до начала опыта был тщательно высушен и взвешен. Для
поливки служила только дождевая или перегнанная вода. Опыт
продолжался в течение 5 лет. За это время ивовая ветвь выросла в большое
дерево и увеличилась в весе с 5 до 169 фунтов, между тем вес земли в
банке (после высушивания) остался почти без изменения. Привес ивы
Ван-Гельмонт и относит на долю зелени, в которую перешла часть
воды, служившей для поливки.
Правда, уже Б у р г а в е, один из самых выдающихся химиков XVIII
столетия, высказал сомнение в правильности такого объяснения и
считал возможным предположить, что землистое вещество образуется не
из воды, а за счет воздуха, однако его мнение не разделялось другими
химиками.
Для объяснения наблюдавшихся фактов предлагали различные
объяснения. Иные полагали, что в образовании воды принимала участие
«материя огня», так как образование земли требовало нагревания;
другие (например, Ле-Руа) считали более вероятным, что земля уже
предсуществовала в воде в особой, чрезвычайно трудно отделимой
форме. Вопрос оставался неясным и неразрешенным.
Приступая к решению поставленной себе задачи, Лавуазье сразу
порывает со старыми предрассудками и без всякой предвзятой идеи
принимается за повторение опытов своих предшественников. «Я не
буду повторять,— пишет он в своем мемуаре,— то, что писали об
элементах философы первых столетий, и перехожу к тому, что более всего
интересует физиков,— я хочу говорить о фактах».
Он задается вопросом, откуда берется земля, количество которой
при повторных перегонках воды получается довольно значительное.
Чтобы решить вопрос, не предсуществует ли земля в воде, которая
подвергается перегонкам, он обращает особое внимание на чистоту
исходного материала и останавливается с этой целью на дождевой
воде, перегонка которой обеспечивается самой природой. А чтобы в
дождевую воду не попали посторонние примеси (например, пыль и т. п.
из воздуха или поверхности крыш), он собирает ее в большие
стеклянные, фарфоровые сосуды, начиная сбор после того, как дождь успеет
очистить атмосферу от пыли. Такая вода оказалась почти чистой
Выпаренная при низкой температуре, она оставила всего 0,3 доли
легкой, нерастворимой земли, да около 7і2 доли поваренной соли на
фунт. Удельный вес ее только на 0,00002425 превысил удельный вес
перегнанной воды.
Лавуазье констатирует, что под влиянием повторных перегонок,
несмотря на образование «земли», свойства воды, и прежде всего ее
плотность, не отличаются заметным образом.
«Я считал возможным,—говорит он,—заключить из этого опыта,
что одно из двух: или земля, выделяющаяся при перегонках, по своей
природе может находиться в растворе не увеличивая при этом
плотности воды, по крайней мере в такой степени, как это делают другие
вещества, или же, что эта земля не содержалась еще в воде, когда я
определил удельный вес, что она образовалась во время перегонки.
Чтобы с уверенностью решить вопрос, на каком из этих предположений
следует остановиться, ни одно средство не казалось мне более
достаточным, как повторение опыта в герметически запаянных сосудах,
принимая во внимание точный вес сосуда и воды, взятой для опыта. Если
при этом проникает через стекло и соединяется с водой материя огня,
то после многочисленных перегонок следует ожидать прибавления в
весе материалов, т. е. в общем весе воды, земли и сосуда. Физики ведь
Открытие кислорода и теория горения
385
знают, что материя огня вызывает увеличение веса тел, с которыми она
соединяется (очевидное указание на взгляд, развитый, как мы'видели,
Р. Б ой л ем).
Итак, вместо повторных перегонок воды, Лавуазье просто
нагревает определенное количество ее в замкнутом пространстве и
останавливается для этой цели на двугорлом «пеликане», старинном приборе
алхимиков, допускающем осуществлять нечто в роде нагревания с
обратно поставленным холодильником. Опыт длился в течение 101 дня.
Когда охлажденный пеликан был вновь взвешен, то вес его оказался
практически совпадающим с первоначальным (5 фунтов 9 унций 4
золотника и 41,75 доли вместо 5 фунтов 9 унций 4 золотников и 41,50
доли). Ничтожная разница (0,25 доли) всецело падала в пределы
возможных ошибок опыта.
Таким образом, участие «огненной материи» в образовании земли
оказалось исключенным.
Чтобы решить, действительно ли земля образуется из воды, или,
скорее, из стекла сосуда, подвергающегося растворению, Лавуазье
опоражнивает, тщательно высушивает и вывешивает свой пеликан. При
этом оказалась убыль в весе на 17,38 доли. Очевидно, это количество
стекла утрачено веществом сосуда. Лавуазье далее исследует воду,
извлеченную из пеликана, и обнаруживает, что она изменила свойства:
ее плотность возросла с і 000 000 до 1000 037. По испарении она дала
остаток земли, вес которого равнялся 203/5 долям, следовательно, лишь
немногим (на 3 доли) превышал потерю в весе пеликана. Было ясно,
что земля произошла из стекла, а не из воды. Небольшую разницу
между получаемым и действительным количеством земли Лавуазье
отнес на долю разъединяющего действия воды на стекло сосуда, в
котором она испарялась до получения сухого остатка. Таким образом,
вопрос о превращаемости воды в землю окончательно сошел со сцены.
Мы теперь знаем, что стекло, даже лучшие сорта его, при очень долгом
нагревании с водой понемногу разъедается, подвергается гидролизу.
В раствор сначала переходят преимущественно основные компоненты,
а потом начинают отстаиваться и частички кремнезема.
В этой работе Лавуазье обращает на себя внимание и
необыкновенная точная, количественная постановка опытов, в которой изящество
и простота сочетаются с множеством мелочных предосторожностей, с
полным учетом источников возможных погрешностей, ч замечательная
ясность мысли, логичность умозаключений, строгих в своей простоте
и не стесняемых никакими искусственными путами.
В историческом отношении интерес мемуара о природе воды
увеличивается еще тем обстоятельством, что здесь мы находим первое
указание на сложность состава воздуха. «Вероятно,—говорит
Лавуазье,— что воздух вовсе не является, простым телом, как это думали
первые физики» {9].
Вопрос о роли воздуха в явлениях горения и ставится Лавуазье в
ближайшую очередь. Первая серия опытов, предпринятых для его
разрешения, содержится в знаменитом сочинении «Opuscules physiques et
chimiques», опубликованном в начале 1774 г.
В одном из дошедших до нас лабораторном журнале (датированном
1772 г.) Лавуазье намечает программу своих «opuscules»* и развивает
соображения, вызвавшие постановку намечаемых им опытов. Для него
уже и в то время было ясно огромное значение того сложного и
запутанного цикла явлений, касавшихся поглощения воздуха и других га-
юв различными веществами, и процессов обратного порядка, сопро-
* Мелочи, мелкие произведения. Прим. ред.
25 Л. А. Чугаев, т. III
386 Открытие кислорода и теория горения
вождаемых освобождением газов. Некоторые в то время уже известные
факты, особенно знаменитые опыты Блэка над свойствами углекислого
газа, о которых Лавуазье говорит с большим уважением, позволяли
предвидеть качественное разнообразие газов, участвующих в подобных
процессах. Лавуазье ясно представляет себе громадность той работы,
которая потребуется для того, чтобы разобраться во всех этих
явлениях, изучение которых в то время едва только начиналось. «Но,—
пишет он,— важность предмета заставила меня приняться за всю
эту работу, которая, казалось мне, должна вызвать революцию в
физике и химии».
Начав с проверки и расширения опытов Блэка над углекислым
газом, над его отношением к извести и щелочам, Лавуазье
естественным путем переходит далее к явлениям обжига и восстановления
металлов. Гениальная интуиция заставляет его сблизить явления
последней категории с процессом поглощения углекислого газа (air fixe) *
веществами основного характера и с обратным процессом разложения
карбонатов при прокаливании.
«...Я начал подозревать, говорит он, что атмосферный воздух или
какая-нибудь упругая жидкость, содержащаяся в воздухе, способна во
многих случаях поглощаться металлами или соединяться с ними; что
от присоединения этого вещества зависят явления кальцинации,
увеличение в весе металлов при обращении их в известь и, может быть,
еще много других явлений, которым физики еще не дали никакого
удовлетворительного объяснения».
И вот, руководствуясь этой мыслью, Лавуазье подвергает
металлические окислы прокаливанию с углем; он наблюдает при этом
обильное выделение газа, в котором животные гибнут, а зажженные тела
потухают, который мутит известковую воду, словом, наблюдает
выделение углекислого газа, притом в количестве, раз в 1000 большем (по
объему), нежели, например, взятый для опыта сурик. А так как уголь
при прокаливании вообще не выделил, сурик же почти не выделил
газа, при одновременном же присутствии обоих тел наблюдалось
исчезновение угля и образование металлического свинца, то было ясно, что
выделяющийся газ должен был образоваться за счет того и другого
вещества: угля и сурика.
С другой стороны, Лавуазье изучает прокаливание (на солнечном
свету при помощи собирательной линзы (олова и свинца в замкнутом
пространстве) над водой' или над ртутью и констатирует при этом
уменьшение объема воздуха и прибавку в весе металла, причем вес
исчезнувшего воздуха близок к этой прибавке.
Эти опыты переносятся затем на металлоиды — на фосфор и серу,
и результаты получаются вполне аналогичные. Объем воздуха при
горении сокращается на Vs. Вес фосфора, обращающегося при горении
в белый порошок, увеличивается при этом в 2,5 раза. (Эти числа
довольно близки к истине, ибо 1 часть фосфора должна была дать 2,35
части фосфорного ангидрида Р2О5).
Лавуазье заключает из своих опытов, что каждый раз, когда
«металлическая известь переходит в состав металла, выделяется газ и,
наоборот, при кальцинации, когда металл переходит в известь,
соответствующее количество газа поглощается. В это время ни Лавуазье, ни
тем более Пристлей не представляли себе с достаточной ясностью
различие между углекислотой, азотом и кислородом. Это не помешало,
однако. Лавуазье в марте 1774 г. (в анонимной статье) указать
вполне определенно на противоречия, к которым приводит теория флоги-
* Сгущенный воздух. Прим. лед.
Открытие кислорода и теория горения 387
стона при попытках воспользоваться ею для объяснения наблюденных
им и другими химиками явлений. В том же 1774 г. он с большими
предосторожностями повторяет опыты кальцинации олова и свинца в
замкнутых сосудах и окончательно опровергает мнение Бойля, по
которому прибавка в весе при переходе металла в окись зависит от
фиксации огненной материи. Эти опыты с полной очевидностью показали, что
общий вес всего прибора в течение кальцинации не изменился.
Наконец, в том же 1774 г. в статье, напечатанной в Journal de
Physique, аббата Розье (извлечение из более подробного мемуара
Лавуазье) заключается уже определенно выраженный взгляд на состав
воздуха. «Воздух, освобожденный от части своей, способной
связываться, является как бы разложенным... Я вправе, как мне кажется,
утверждать, что воздух, насколько это возможно, очищенный и
освобожденный^ от всякого постороннего вещества, далеко не представляет
простого тела, как обыкновенно полагают, а, наоборот, должен быть
отнесен к ч=ислу смесей, а может быть, даже к числу соединении».
Окончательно состав воздуха был установлен Лавуазье в 1775 г.
не без влияния известия об открытии кислорода, только что
полученного от Пристлея. Но это открытие, тогда еще не ясно
формулированное, могло послужить для новой теории лишь в качестве сырого
материала. Подобно Майову, Лавуазье провидел существование кислорода
еще до открытия его, а потому, когда Пристлей во время пребывания
своего в Париже познакомил его с результатами своих опытов (полу-
-іение из окиси ртути при прокаливании такого газа, в котором
зажженные тела горят ярче, чем в воздухе), то семя упало на
подготовленную почву.
Несколько позднее (1777 г.) оно породило тот описанный во всех
учебниках классический опыт, который начинается с количественного
анализа воздуха с помощью металлической ртути и заканчивается
количественным" же анализом окиси ртути, регенерацией этой последней
и получением чистого кислорода.
Когда состав воздуха был установлен, перед Лавуазье открылась
дальнейшая задача, вполне соответствующая его широкому
обобщающему уму. Предстояло найти связующие нити между кислородом и
целым" рядом веществ, различных продуктов горения или окисления.
И Лавуазье не замедлил обнаружить в целом ряде таких веществ
содержание кислорода. Чтобы понять все огромное значение одной только
этой части работ Лавуазье, достаточно сопоставить результаты его
исследований относительно состава азотной и серной кислот с тем, что
думал об этом на основании своих опытов Пристлей. Проследив
качественно действие"некоторых металлов, и прежде всего ртути, на азотную
кислоту и затем разложение получающейся при этом соли (Hg(N03)2)
при нагревании вплоть до восстановления металлической ртути,
Лавуазье нарисовал ту картину этого ряда сменяющих друг друга
химических процессов, которая с несколькими поправками сохраняет свое
значение и до настоящего времени. Для него окись азота (N0),
образующаяся при действии ртути на азотную кислоту, есть не что иное
как азотная кислота, лишенная воды и части своего кислорода. Когда
ртуть растворяется в азотной кислоте, она именно и отнимает от
последней часть содержащегося в ней кислорода, лишая ее свойства
кислоты. Справедливость этого взгляда могла быть проверена синтезом
HN03 из окиси азота и кислорода в присутствии воды.
Пристлей держался совершенно обратного мнения. Он полагал, что
в кислороде заключается азотная кислота, и делал даже попытки
определить это содержание количественно, конечно, с отрицательными
25*
U88
Открытие кислорода и теория горения
результатами. Растворяя различные земли в азотной-кислоте-и.
прокаливая полученные нитраты, Пристлей получал газы,, которые были,
«лучше воздуха», и на этом основании он считал,, что. в них, содержится,
азотная кислота. Против этих явно ошибочных заключений энергично
протестует Лавуазье, отдавая, впрочем, должное опытной стороне
исследований Пристлея.
К аналогичным выводам пришел Лавуазье относительно состава;
серной, фосфорной и других кислот. Кислород стал для него «ргіпсіре
oxygene», «кислотвором», необходимой составной частью кислот
(водородных кислот НС1, НВг и HCN и т. п. тогда еще не знали). Таким
образом, кислород оказался общей составной частью целого ряда тел:
оснований, или известей, с одной стороны, кислот—-с другой.
Очевидно было, что он заключался в солях. Вещества., е которыми кислород
находился в сочетании, Лавуазье называл элементами или радикалами,
причем слову «элемент» он вслед за Бойлем приписывал условный,
чисто экспериментальный смысл «последнего предела, до которого
доходит анализ». «Все тела,— говорит он,— которые не могли быть еще
разложены, являются для нас элементами».
Видными моментами в развитии и укреплений .теоретических
взглядов Лавуазье, в корне противоречащих теории * флогистона, является1
установление им химического состава углекислого газа и воды.
Он констатирует образование углекислого газа при горячий угля
и алмаза и устанавливает, что в состав этого газа входят только доа
элемента, что углерод, содержится во многих природных телах
(органических) и что при сжигании последних он переходит в углекислый,
газ. При этом он выясняет глубокое различие между углекислотой и
азотом, между двумя газами, частичное сходство которых немало смущало*
прежних исследователей. Вместе с тем намечается и правильный взгляд
на сущность дыхательного процесса, близкая аналогия которого с
процессами горения и кальцинации в то время была уже отмечена
неоднократно.
Рядом блестящих опытов Лавуазье разъясняет, к чему сводится
химизм дыхания и чем отличается дыхание в воздухе от дыхания в
•чистом кислороде. Воробей, запертый в замкнутое пространство,
наполненное воздухом, погибает через определенный промежуток
времени. Остающийся газ занимает почти тот же объем*, что и
первоначально взятый воздух, но свойства его существенно иные. Этот газ не годен
для горения, так же как и для дыхания; окись азота не образует с ним
•бурых паров, поглощаемых водой. Все это свойства того газа — азота,
или «mofette» **, как его называл в то время Лавуазье, азота, который
остается после продолжительного нагревания или кальцинации ртути
и других металлов в замкнутом сосуде с воздухом. Но воздух,
оставшийся после опыта с воробьем, обладает еще одним свойством,
которого лишен азот: он дает осадок с известковой водой (образование уг-
леизвестковой соли по уравнению: Са(ОН)2 + С02 = СаСОз + Н20). Он,
однако, теряет это свойство после обработки избытком извести или ед->
кой щелочи. При этом часть газа — по Лавуазье около 7? — поглощает-
* Смысл этого обстоятельства выяснился в полной мере лишь значительно
позднее, именно после того как в основу химических воззрений (в 50 и 60-х годах) был
положен закон Авогадро. Тогда стало ясно, что при горении углеродсодержащих
веществ, на каждый объем затрачиваемого кислорода 02 должен выделяться равный
объем углекислого газа СОо по той причине, что из 1 молекулы 02 получается I
молекула С02, равные же числа молекул газов по закону Авогадро занимают одинаковые
объемы.
** Вредное испарение, ядовитый газ. Прим. ред.
Открытие кислорода и теория горения 38і?
ся, и новый остаток уже не дает мути с известковой водой, вообще этот
остаток ничем не отличается от азота. Смешанный с lU по объему
кислорода, он дает обыкновенный воздух. Вот тот ряд фактов, который
послужил Лавуазье для разъяснения роли углекислого rasa при
дыхании (аналогичные опыты были сделаны и с горением угля и
органических тел) и который лег в основу его замечательных исследований над
газообменом при дыхательном процессе, исследований, положивших
начало современной биологической химии.
Другое открытие, еще более важное для окончательной победы
учения Лавуазье над теорией Сталя,— это определение состава воды.
История этого открытия слишком сложна для того, чтобы она могла
быть предметом рассмотрения в этом очерке. Достаточно будет
сказать, что с фактической стороны главная заслуга здесь принадлежит
англичанину, лорду Кэвендишу, который получил в чистом
состоянии водород, изучил его свойства и произвел синтез воды. Последний
опыт, а также ряд других опытов, связанных с определением состава
воды, был произведен также во Франции Лавуазье, Менье и Мон-
жем, почти одновременно с работой Кэвендиша, и, по-видимому, не без
влияния этой последней. Но бесспорная великая заслуга Лавуазье
заключается в том, что он впервые правильно истолковал смысл этих
опытов *. Если для Кэвендиша водород или gaz inflammable ** был
почти чистым флогистоном, а вода и воздух — химическими
соединениями, содержащими флогистон, то Лавуазье сразу нашел для всех
этих тел место в своей системе. Для него кислород и водород были
элементами, вода — их соединением, а воздух — смесью кислорода с
третьим элементом — азотом.
Определение состава воды завершило собой цикл исследований,
которые были необходимы Лавуазье для обоснования своей теории,
кислородной теории горения, и для опровержения теории флогистона.
По своему обычаю, он не спешил делать заключения и в течение
долгого времени накоплял" факты и доказательства. Только в 1786 г. он
решил, что настало время подвести итоги и открыто выступить против
теории Сталя. И он сделал это-в своей знаменитой статье «Reflexions
sur la phlogistique» ***.
В этой статье Лавуазье начинает с указания на ту необыкновенную
простоту, с которой объясняется множество явлений, если допустить
участие в них кислорода, ргіпсіре oxygene. Он переходит затем к своей
главной задаче — критике теории флогистона и констатирует ее
беспомощность в ряде случаев, ее шаткость и неправильность. «Химики
сделали из флогистона,— говорит он,— начало туманное и неопределенное,
которое поэтому может быть приспособлено ко всем объяснениям, к
которым его желают применять: то он — начало весомое, то лишенное
веса; то это свободный огонь, то огонь, соединенный с землей; то ,он
проходит через поры сосудов, то последние становятся для него
непроницаемыми. Он объясняет одновременно едкость и ее отсутствие,
прозрачность и непрозрачность, окраску и отсутствие цвета. Это настоящий
Протей, ежеминутно меняющий свои фермы».
Приговор теории флогистона был подписан, а новое учение —«анти-
флогистическая» теория Лавуазье —с поразительной быстротой
завоевало себе всеобщее признание. К концу жизни Лавуазье ее авторитет
* Некоторая доля участия в разъяснении состава воды принадлежит такж^
Джемсу Уатту, изобретателю паровой машины.
** Воспламеняющийся газ. Прим. ред.
*** Размышления о флогистоне. Прим. ред.
390
Открытие кислорода и теория горения
стоял почти так же высоко, как лет за 15—20 перед тем авторитет
Сталя.
Историками химии не раз поднимался вопрос, в какой степени
открытия, приписываемые Лавуазье, принадлежат лично ему и в какой
мере —другим химикам, его предшественникам и современникам. Это,
в частности, касается открытия кислорода и теории горения.
Едва ли можно сомневаться в том, что Лавуазье по большей части
не были известны труды его предшественников. Это в достаточной
степени объясняется разбросанностью и малой доступностью научной
литературы того времени — обстоятельство, уже отмеченное нами в
самом начале этого очерка. Иногда Лавуазье узнавал о существовании
той или иной работы, в которой можно было видеть или смутное
предчувствие его обобщающих открытий, или фактический материал,
которым он мог бы воспользоваться слишком поздно, когда его работа
была уже закончена. Наконец, в тех случаях, когда он, хотя и знал о
существовании труда, близко подходившего к области его собственных
работ, но не останавливался на нем с достаточным вниманием, почти
всегда можно сказать, что этот труд мало повлиял на выводы, к
которым он пришел, а если повлиял, то лишь косвенным образом.
В таких случаях Лавуазье имел право сказать то, чго он говорил по
поводу использованных им опытов Пристлея, касавшихся состава
азотной кислоты. «Так как одни и те же факты привели нас к
диаметрально противоположным заключениям, то я надеюсь, что если уже меня
упрекают в позаимствовании доказательств у этого знаменитого
физика,, то у меня, по, крайней мере, не-будут оспаривать права на сделанные
мною выводы».
Иной раз то, что Лавуазье приходилось брать у других
исследователей, представляло до такой степени сырой и необработанный
материал, что почти нельзя было говорить о точно описанных фактах. Ведь
фактом, точно установленным фактом, в науке нельзя считать простую
передачу свидетельства наших органов чувств, да к тому же еще
изуродованную з основе неправильной интерпретацией. В таких случаях
легко понять, что Лавуазье, не только давший объяснение факту, но
впервые правильно его описавший, не мог не приписывать себе
существенной роли в самом его открытии.
Зато в тех случаях, когда Лавуазье говорил о своих действительно
идейных предшественниках, он неизменно отзывался о их работах с
полным уважением и признанием, не различая при этом своих
противников от союзников. Выше было уже указано, что Лавуазье отдавал
должное действительным открытиям Сталя. Также относился он к
работам Блэка и многих других химиков.
Некоторые ученые, занимавшиеся историей химии, особенно Фол fa-
rap д, Оствальд, а в последнее время Ш пет ер, напрягали много
усердия, чтобы обвинить Лавуазье в преднамеренном замалчивании
работ своих предшественников и конкурентов и тем набросить тень на
его нравственную личность и ума'лить его научные заслуги. В этом
неблагородном предприятии Фольгард зашел так далеко, что
признал Лавуазье не настоящим химиком, а лишь дилетантом. Оствальд
мимоходом заметил, что считает работы Майова в научном отношении
более ценными, нежели работы Лавуазье, а Шпетер, присоединяясь,
хотя и не в полной мере, к этим обвинениям, постарался с особенной
тщательностью собрать весь возможный обвинительный материал
против гениального французского химика.
К подобным обвинениям, в виду только что сказанного, едва ли
можно отнестить иначе, чем это сделал Гримо в своей биографии
Открытие кислорода и теория горения
391
Лавуазье, назвав их памфлетами, и следует даже прибавить,
памфлетами, не лишенными политического и национального оттенка.
Если нельзя отрицать —это достаточно ясно из предыдущего, что
в своей титанической работе по пересозданию химии, а в частности
по установлению новой теории горения, Лавуазье имел
предшественников, то это значит лишь, что сделанное им не избежало судьбы многих
других великих открытий. Но только Лавуазье оказался соединяющим
в одном лице то ясновидение, то терпение и настойчивость в
проведении своих идей до последних возможных пределов, которые отличают
истинного гения *.
Чтобы оценить относительное значение, так сказать удельный вес
его научного творчества, надо иметь в виду, что предшественники
Лапу азье сосредоточивали свое внимание на разъяснении различных
сторон процесса горения, на установлении отдельных фактов,
относящихся к этой области явлений. Для Лавуазье, наоборот, процессы
горения и роль кислорода были только частью, хотя и весьма
существенной частью, в начертанной им общей и цельной картине. Будучи
истинным философом, он охватывает сразу всю область химической науки;
он установил заново ее основные принципы и положил основание
целым новым направлениям или областям ее. Лавуазье положил начало
«эпохи количественных изысканий». Никто до него, кроме Ломоносова,
даже не предвидел огромного значения количественных методов для
изучения химических процессов, хотя употребляли их уже давно, и
только он фактически показал, что могут дать эти методы. Это
Лавуазье установил современное представление о химических элементах,
лишь в смутной форме предуказанное Бойлем; эго он экспериментально
подтвердил закон сохранения массы, который он, впрочем, подобно
некоторым из своих предшественников, считал самоочевидным. Это он
заложил основы учения о химической энергии (термохимия), а также
химии биологической.
К оценке творений Лавуазье как нельзя лучше подходят слова
Д. И. Менделеева: «Открытие закона природы принадлежит тому, кто
прежде других его ясно осознал, а не смутно только предчувствовал,
кто себя и других убедил в существовании этого закона рядом фактов
и умозаключений».
ЗАКЛЮЧЕНИЕ
Подобно тому, как люди, переживающие эпоху великих
политических потрясений, лишь с трудом и очень редко возвышаются до
совершенно беспристрастной оценки развертывающихся вокруг них событий,
* Многие считают, что гениальные люди превосходят остальных в способности
поддерживать внимание, направляя его в определенном направлении. По Бофону,
гениальность «c'est une longue patience» (это — долгое терпенье. Прим. ред.). С другой
стороны, вот взгляды Вольтера: «В сущности, не есть ли гении то же самое, что талант?
А что такое талант, как не предрасположение к успеху в том или другом деле?»
Но едва ли не наиболее метким является определение Джемса, который говорит, что
гения всего более отличает способность делать умозаключения необычайным путем,
достигающая громадного развития (яблоко Ньютона, висячая люстра Галилея и пр.).
Не без основания и терпении, в сосредоточенном внимании, отличающих гениев, он
видит лишь следствие первого основного признака. Гении потому сосредоточивают своп
мысли на долгое время, что способны соединять в одном мыслительном процессе и
связать в органическое целое большое число таких объектов мышления, которые для
обыкновенных смертных кажутся ничем не связанными между собой. Понятно, для
того, чтобы гениальная натура не оставалась бесплодной, чтобы она проявила себя
а действии, необходима наличность работоспособности, наклонности к напряженному
труду. Всеми этими качествами в высокой степени обладал Лавуазье.
392
Открытие кислорода и теория горения
их всецело увлекающих в своем бурном водовороте, так и деятели
науки, живущие в периоде оживленного спора из-за того или иного
воззрения, сами принимающие активное участие в борьбе мнений,
обыкновенно не могут отнестись вполне объективно к научному
значению той или иной теории, того или иного воззрения. В том и другом
случае справедливый приговор, основанный на вполне уравновешенных
суждениях, выносит только история. Так было и с теорией флогистона,
и с заступившим ее место учением Лавуазье, со старой и новой, или
антифлогистической, химией.
Если из наиболее убежденных сторонников теории флогистона и
неразрывно связанных с ним взглядов на процессы горения и на
химические процессы вообще, на состав тел, на химические элементы,
словом, на все крупные вопросы химии, одни долго, а иные до конца своих
дней не могли примириться с новым течением в науке, то сторонники
Лавуазье, пораженные величественной простотой внезапно
развернувшейся перед ними картины, без сомнения, впадали в другую крайность
и были склонны совершенно отрицать заслуги своих предшественников,
если не в смысле обогащения науки новыми фактическими
материалами, то в смысле истолкования и приведения в связь известных и вновь
открываемых фактов.
Мы отчасти уже видели, что такой взгляд является односторонним.
Теория Сталя дала все, что можно требовать от настоящей научной
теории. Она объединила в одно стройное целое ряд известных в то
время химических процессов и, соподчинив их друг другу, привела их
во взаимную логическую связь. Она дала возможность предвидеть
новые факты и вела выдающихся деятелей химической науки к новым
открытиям. Наконец, почти через столетие после ее падения
обнаружилось, что в самом существе этой теории заключается несомненное зерно
истины. Огненная материя, или флогистон, о которой, впрочем, смутно
говорили еще алхимики, получила как бы пророческое значение. Од-
лйнг (10], кажется, первый высказал мысль, что многие рассуждения
флогистоников можно всецело перевести на современный язык, если
заменить слово «флогистон» современным словом «энергия».
Во всех тех случаях, в которых Сталь и его сторонники
предполагали потерю флогистона горючим телом, мы знаем, что на самом деле
данная система теряет энергию; химическая потенциальная энергия
переходит в другие виды ее, например в энергию тепловую,
электрическую, световую. Таковы все явления горения и обжига металлов.
А когда происходит обратный процесс восстановления, когда по
учению Сталя должна происходить фиксация телом флогистона, мы
считаем теперь, что происходит поглощение энергии, переход ее в
скрытое, потенциальное состояние. Все экзотермические процессы были для
Стяля процессами разъединения, все эндотермические — процессами
соединения.
Гу же мысль мы находим у Гельмгольца, который в одной из
своих лекций [11] говорит: «Если флогистон Сталя перевести словом
скрытая теплота, то следует признать, что теоретические основы
его системы перешли в систему Лавуазье. Только Сталь еще не зняп
кислорода, чем были обусловлены некоторые ложные гипотезы,
например, относительно отрицательной тяжести флогистона».
Л и 0 и х идет еще дальше в сторону признания заслуг научных
предшественников Лавуазье из далекого прошлого, признавая долю
положительного значения за самой философией Аристотеля. До
известной степени нельзя не согласиться со справедливостью его доводов.
Правда, предыдущие страницы переполнены указаниями на вред, кото-
Открытие кислорода и теория горения
39*
рый Аристотелева схоластика принесла развитию науки. Но не надо
забывать, что виной тому был не столько сам Аристотель, сколько та
почва, на которую упало посеянное им зерно, сколько мрачная эпоха
застоя человеческой культуры, заживо хоронившая всякую живую
мысль, эпоха, из всего учения Стагиритг усвоившая только его
отрицательные стороны. Виноват был не столько сам философ, сколько err
комментаторы и последователи.
Отрицательное влияние Аристотеля настолько бросается в глаза
что за ним легко просмотреть оборотную сторону медали. Трудно
представить себе, что было бы со средневековой культурой, с прогрессом
науки в это время, если было бы устранено влияние греческого
философа, если бы погас этот единственный свет, который хорошо ли, худо ли
освещал в то время царившую непроглядную тьму. Мы видели, что
поиски алхимиков двигались побуждениями практического характера,
но одновременно с этим оказывала известное идейное влияние и
философия Стагирита, хотя бы его учение о едином веществе. Можно
сказать, что она далеко не была лучшим руководителем, какого можно бы
было себе представить. Но не лучше ли иметь хоть какое-нибудь
руководство, чем не иметь никакого, и невольно возникает мысль, не
остались ли бы средневековые монахи без помощи дошедших до них
обрывков из учений греческих философов в положении дикарей, для которых
время, потребное на ничтожное изобретение, измеряется многими
сотнями, если не тысячами, лет.
Можно сомневаться, могли ли быть при таких условиях открыты и
зафиксированы в известном, хотя и своеобразном порядке, те
многочисленные факты, которые оставила нам в наследие алхимия и иатро-
химия. Между тем ведь на этот материал опиралась вся позднейшая
эволюция нашей науки. А сколько нужно было таланта, подчас
граничащего с гениальностью, для того, чтобы при идейной и материальной
обстановке той эпохи точно установить какой-нибудь отдельный факт,
открыть новое тело или новое явление! И потому нельзя отрицать, что
по отношению к великим открытиям Лавуазье и его эпигонов в области
химии до известной степени справедливо то, что когда-то говорил о себе
великий Ньютон: «Если я видел дальше других, то потому, что стоял
на плечах гигантов».
БИБЛИОГРАФИЯ
Н.Кор p. Beitrage zu cler Chemie. Drittes Stuck. Braunschweig, 1875.
M. Berth e lot. Les origincs de l'alchimie. Paris, 1885.
M. Berthclot. Introduction a la chimie des anciens et du moyen ago. Paris, 1889.
M. Bertholot. La Revolution chimique Lavoisier. Paris, 1890.
Б.-Н. M ен ш у тки н. Ломоносов как физико-химик. ЖРФХО, 36, ч. И, стр. 77.
R. Ehrenfeld. Grundidee einer Entwickelungsgeschichte der chemischen Atomislik.
Heidelberg, 1906.
S. M. Die Entdeckung des Sauerstoffes. Stuttgart, 1909.
M. S peter. Lavoisier und seine Vorlaufer. Stuttgart, 1900.
Кроме того, следует указать на собрание сочинений Ваи-Гельмонта, Майова, Бон-
ля, Шееле, Лавуазье, Пристлея и других авторов, о которых идет речь в этом очерке.
Многие дошедшие до нас алхимические рукописи переведены и изданы М. Бертло а
Париже.
394
Открытие кислорода и теория горения
ЛИТЕРАТУРА*
1. А. Ф улье. История философии. СПб., 1901, стр. 68.
2. P.Duguem. Le Systeme du Monde. Paris, 1913, p. 29.
3. В.Виндельбанд. История древней философии. Изд. Ill, СПб, 1902.
4. J. Liebig. Reden und Abhandlungen. Leipzig u. Heidelberg, 1874, S. 314.
5. В.Виндельбанд. История древней философии. Изд. Ill, СПб, 1902, стр. 325.
6. M.Berthelot. Revue de Deux Mondes, 1893, 119, 315.
7. См. предыдущую ссылку.
8. Б.Н.Меншуткин. ЖРФХО, 1904, 36, II, 77, ИЗ, 159, 321.
9. A.L.Lavoisier. Oeuvres, 2. Paris, 1864, p. 6.
10. W.Odling. Chem. News, 1871, 23, 243, 256.
П. Н. L.HelmhoIz. Vortrage und Reden, 2. Berlin, 1884, S. 177.
* Приведены литературные ссылки, вынесенные Л. А. Чугаевьгм в подстрочные
примечания. Прим. ред.
Х38$8»
ЭВОЛЮЦИЯ УЧЕНИЯ О КАТАЛИЗЕ
«Новые идеи в химии», сб. 8. 1924, Петроград
В каждой области знания и в отдельных ее частях для правильной
опенки современного положения того или другого вопроса, той или
другой доктрины очень часто полезно бывает оглянуться назад на
пройденный путь, проследить ход эволюции идей от далекого 'прошлого и
до нашего времени. Любопытно и поучительно бывает узнать, как часто
мысль человека .повторяется с известными вариантами, уподобляясь
путнику, поднимающемуся на альпийскую вершину по спирально
извивающейся тропинке; от времени до времени перед ним открывается
один и тот же вид, но каждый раз в нем обнаруживаются новые черты,
новые подробности.
Так обстоит дело и с нашими теориями, этими незаменимыми
пособиями для .истолкования природы.
Возможность видеть перед собой каждый вопрос науки в
исторической 'перспективе представляет еще то преимущество, что благодаря
этому облегчается объективная оценка тех взглядов и теорий, которые
возникают на наших глазах и составляют последнее слово науки.
Наконец, внимательное изучение исторических документов, помимо
своего самостоятельного чисто исторического интереса, имеет и то
-важное значение, что среди этих документов можно найти немало вскользь
высказанных мыслей, неоцененных или непонятых и давно забытых,
равно как и отдельных наблюдений, которые могут иметь большое
значение в современном освещении.
Исходя из этой точки зрения-, я считал уместным >в настоящий
сборник <Новых идей в химии», посвященный одному из интереснейших
вопросов современной теоретической химии, вопросу о катализе, внести
очерк эволюции этого вопроса за период времени, простирающийся
примерно до 50-х годов минувшего века. Останавливаясь на отдельных
этапах этой эволюции, читатель найдет в зародыше большинство тех
течений мысли, тех теоретических взглядов на природу -катализа,
которые -составляют .предмет обсуждения и в настоящее время.
Если оставить в стороне некоторые бродильные процессы
(алкогольное, молочнокислое брожение), известные человечеству с незапамятных
времен, но выясненные по отношению к своей химической природе
только значительно позднее, то надо признать, что первые случаи заведомо
каталитических реакций, с ясностью установленные, были открыты в
конце XVIII века. Сюда прежде всего следует отнести забытое и вновь
сделанное в наши дни открытие разложения этилового спирта на воду
и эгилен 'в присутствии глинозема и глины. Это открытие, сделанное
в 1796 г. известными в истории науки четырьмя голландскими химиками:
Бондтом, Дейманом, Пэтсом Ва н-Т роствиком и Л а у-
веренбургом, было сообщено в заседании Института в Париже
28 фримера * 5 года республики гражданином Фуркруа. Приводим
* Фример (Frimaire) третий месяц республиканского года (с 21 ноября до 20
декабря). Прим. ред.
396 Эволюция учения о катализе
несколько выписок из этого сообщения, опубликованного в
следующем 1797 г. в Annates de Chimie [1].
«Маслородный газ можно получить также посредством пропускания
паров спирта или эфира через накаленную докрасна трубку,
наполненную кремнеземом или глиноземом, или просто через накаленную
глиняную трубку, ничем не наполненную. Он (маслородный газ) не образуется
при пропускании эфирных или алкогольных паров «и через накаленную
докрасна трубку без кремнезема или глиное?ма. ни через такую же
трубку, наполненную известью или магнезией. Горючий газ, который
получается в последнем случае, не может быть превращен в маслородный
путем пропускания через кремнезем или через глинозем».
«Амстердамские химики,— говорится далее в том же сообщении,—
помещали глиняную трубку в другую стеклянную и, накалив докрасна,
пропускали пары спирта или эфира; при этом происходит
образование маслородного газа так же, как и в там случае, когда в стеклянную
трубку введены куски глиняной трубки; они заключают отсюда, что
глина* способствует образованию этого газа». Еще далее категорически
подтверждается, что «...алкоголь и эфир должны непременно пройти
через накаленный докрасна слой глинозема или кремнезема в стеклянной
трубке, чтобы дать начало образованию маслородного газа. Газ,
образующийся из этих двух жидкостей при пропускании через стеклянную
трубку, не становится -маслородньш и в том случае, если его после
этого пропустить через глинозем или через кремнезем или через глиняную
трубку. Таким образом, это свойство, раз утраченное, не может быть
восстановлено...
Газ, обладающий способностью образовать (с хлором) масло,
теряет его при пропускании через накаленную докрасна трубку, при этом
выделяется уголь».
Эти опыты, поразительные по ясности постановки и по точности
выводов, особенно если принять во внимание, что в то время не был
разработан элементарный химический анализ и что почти единственным
методом, который служил авторам в качестве критерия о содержании
этилена в образующемся газе, была его «маслородность», т. е.
способность к более или менее полному превращению в маслянистую
жидкость— хлористый этилен — по реакции, которую мы ныне выражаем
хорошо известным уравнением
С1а+СНя=СНя=СН8С1—СНаС1.
Голландские химики ясно выразили мысль о специфическом
действии определенных веществ на изученную ими реакцию, и только вопрос
«отчего образование маслородного газа происходит при пропускании
эфира и алкоголя через глинозем, кремнезем или глину, а не через
стеклянную трубку», только этот вопрос, который, в сущности, и в на-
ше время остается еще нерешенным, они оставили без ответа.
Любопытно, что два химика той эпохи, Гехт и знаменитый В оке лен, касаясь
этого вопроса и пытаясь его осветить, повторили и несколько дополнили
опыты амстердамских ученых, но при истолковании своих результатов-
попали на ложный путь.
Они не допускали специфичности каталитического агента и хотели
видеть причину различия в неодинаково высокой температуре, которая
достигалась в опытах с глиноземом, кремнеземом и глиной, с одной
стороны, с известью и -магнезией или же с пустыми стеклянными
трубками— с другой. «Мы не думаем, чтобы можно было заклюгрть вместе
с голландскими химиками, что глинозем и кремнезем по своей природе
имели бы особую склонность к образованию маслородного газа и что-
Эволюция учения о катализе
397
бы это свойство отсутствовало у магнезии и у извести. Эти
неодинаковые результаты, без сомнения, обусловливаются тем, что сильнее
нагревали 'всякий раз, когда образовывался маслородный газ, нежели в
тех случаях, когда .получался простой углеводородный газ».
Теперь мы хорошо знаем, что для данной реакции специфичность,
и очень резкая, без сомнения, существует, как это особенно вытекает из
обширных исследований В. Н. Ипатьева.
Как справедливо замечает Фуркруа, «остроумные исследования
голландских химиков над маслородным газом .принадлежат к
небольшому числу таких, которые дают начало новым идеям. Эти исследования,
подобно тем, которыми мы обязаны им и которые касаются
разложения,' образования (recomposition) воды при помощи электричества, над
щелочными и металлическими сульфидами и т. д., -займут 'выдающееся
место з пневматической химии, с успехами которой они связали славу
своих работ и своих открытий».
Вторая в исторической последовательности и притом в высшей
степени замечательная работа о катализе, давшая первую рациональную
теорию .каталитических реакций, принадлежит французским ученым Де-
зорму и Клеману и относится к 1806 г. {2].
В противоположность забытой работе голландских химиков, она
часто цитируется, но лишь в общих чертах. Между тем ее значение для
развития учения о катализе так велико, а путь, по которому авторы
прошли к своей теории, настолько замечателен по своей ясности и
простоте, что я считаю полезным остановиться на этой работе несколько
подробнее и постараюсь изложить ее собственными словами авторов.
Дезорм и Клеман обратили свое внимание на способ, которым
в то время пользовались для фабрикации серной кислоты. Начиная,
примерно, с XV века, для этой цели было предложено пользоваться
сжиганием серы в присутствии некоторого количества (Vs—Vio по весу) се-
.литры, причем с 1846 г. стали входить в употребление свинцовые
камеры, куда пропускали продукты горения вместе с парами воды. В этом
виде способ был крайне мало рационален, ибо требовал большой
затраты на дорогую селитру при малой продуктивности. Тем не менее он
представлял собой значительное усовершенствование по сравнению с
ранее практиковавшимся способом получения серной кислоты,
унаследованным от алхимиков VIII века и заключавшемся в прокаливании
квасцов или железного купороса.
Клеман «и Дезорм начинают с разбора различных предположений,
которые высказывались относительно роли селитры в камерном
процессе. Из этих 'предположении одно заключалось в том, что -селитра
повышает температуру реакции; согласно другому, она доставляет
'нужный для сжигания серы запас кислорода. Первая гипотеза не
может быть поддерживаема, так как одновременно с прибавлением
селитры к сере примешивают глину, а также воду — то .и другое с целью
понизить температуру.
Обращаясь к второй гипотезе, авторы, как истинные последователи
•Лавуазье, производят количественный учет реакции и (зная содержание
кислорода в затрачиваемой селитре, в образующемся при горении серы
сернистом газе и, наконец, в окончательном продукте окисления
—серной кислоте) приходят к заключению, что количество кислорода,
нужное для окисления -сернистой кислоты в серную кислоту, может лишь
в Vs или даже только в 7ю своей части покрыться из запаса,
содержащегося в селитре *.
* Этот расчет был основан на неточных анализах того времени, и тем не менее,
несмотря на очень грубое приближение, полученных данных было достаточно для того.
398
Эволюция учения о катализе
«Итак,'— заключали авторы,— селитра служит для добывания
серной кислоты не так, как это предполагали. Если кислорода (селитры)
недостаточно для того, чтобы превратить сернистую кислоту в серную,
то тем более он не мог бы насытить кислородом серу бея
содействия атмосферного воздуха и замечательно, что в
кислоте, заключающейся в сернокислом калии, составляющем остаток
от сожжения, содержится кислорода больше, нежели сколько его могла
бы дать селитра *.
Если остаются еще какие-либо сомнения в правильности этих
-рассуждений, вследствие неуверенности в пропорции, в которой
употребляются действующие вещества, то они не замедлят рассеяться, когда
ясность правильной теории будет сопоставлена -с неопределенностью
первоначально высказанных мнений.
Если внимательно наблюдать за тем, как происходит сгорание обыч-
иой смеси серы, селитры и влажной глины, то можно заметить, что
азотная кислота не разлагается вполне и что большое количество
красно-бурого окисла азота проходит в свинцовую камеру вместе с
сернистой кислотой; цвет делает и-х ясно видными, и факт этот стоит вне
сомнения.
Вот наблюдение, которое дает ключ к верной теории и .приводящее
к выводам, с помощью которых можно найти ясное объяснение
процесса получения серной кислоты.
Для нас ясно, что при горении выделяется смесь окиси азота с
сернистой кислотой, парами воды и азотом, происходящим из
атмосферного воздуха; мы .можем та.кже предположить наличность некоторого
количества кислорода, ускользнувшего от действия серы. Это
'Предположение, в котором нет ничего невероятного, является единственным
пунктом, по отношению к которому может еще оставаться .некоторое
сом-нение. Но, согласно опыту, сделанному в этом направлении, оба
газа, сернистый и азотистый, не могут существовать б контакте, без
разложения второго и без превращения первого в серную кислоту. Это
и должно, очевидно, произойти после перехода газовой смеси в
свинцовую камеру.
Вдали от топки эта смесь оказывается при более низкой
температуре, которая обусловливает конденсацию части паров. Дождь, который
образуется при этом, увлекает с собой получившуюся серную кислоту...
После этого остаются азотистый газ, сернистая кислота .и атмосферный
воздух с меньшим содержанием кислорода. Азотистый газ неизбежно
превратится в кислоту, кото-рая сноъа подвергнется разложению за
счег второй порции сернистой .кислоты и т. д., до тех пор, пока эта
кислота или же атмосферный кислород или, наконец, то и другое
вещество не будут исчерпаны...
После 'превращения этой сернистой кислоты в серную в остатке
получается большое количество окиси азота іили азотистой кислоты, если
и начале было больше кислорода, чем сколько требуется для
(окисления) сернистой кислоты, а _может быть, еще и избыток кислорода,
оставшийся от насыщения обеих этих кислот.
Важно отметить, что основание азотной кислоты в количественном
отношении не должно изменяться, что после образования серной ки-
чтобы обосновать заключение авторов (Клеман и Дезорм принимали, что превращению
сернистой кислоты в серную, т. е. согласно тогдашним понятиям S02 в S03,
необходимо около у8 того количества О, которое заключается в S03. Точная величина^1^)-
* Это заключение неверно, так как основано на неточных анализах: 2 'молекулы
KN03, содержащие 6 атомов кислорода, образуют 1 молекулу K2SO4, содержащую
только 4 атома О.
Эволюция учения о катализе
39f>
слоты оно должно остаться тем же, в каком первоначально
образовалось из селитры.
На самом деле количество окиси азота или азотистой кислоты,
вероятно, немного меньше, нежели то,- что может образовать нитрат, так
как при горении температура может подняться слишком высоко, и
тогда может произойти полное разложение небольшой части азотной
кислоты. Мы говорим небольшой части -потому, что оіпыт обнаружил,
что полезно поддерживать весьма низкую температуру посредством
соответствующего увлажнения».
Авторы нигде не говорят о катализе, не обобщают своих заключений
на другие реакции аналогичного характера; вывод, который они делают
относительно роли окислов азота в камерном процессе, может служить
прототипом для объяснения всякой реакции переносного -катализа, как
часто говорят в наше время.
«Ит а к,— говорят Дезорм и Клеман,— азотная кіи с л от а
является не более как орудием полного окисления
серы; это ее основание, окись азота захватывает кислород юз воздуха,
чтобы -передать его сернистой кислоте в подходящем -состоянии».
«Ясно, что вода не является непосредственно необходимой для
получения серной кислоты, только смешение -воды с этой последней влечет
за собой выделение окиси азота; этот газ, 'будучи свободным, снова
захватывает кислород воздуха, находящегося в данном вместилище,
чтобы вно'вь ввести его в соединение с сернистой .кислотой. Водяной пар
имеет при этом двоякое значение: он обусловливает энергичное
движение в оставшихся газах и упомянутое уже выделение окиси азота.
Польза от него и была уже замечена, и потому вводят (в камеру) вместе с
газом из печи добавочное количество пара, не считая то, которое
происходит от увлажнения смеси.
Исходя из присутствия двуокиси азота совместно с сернистой
кислотой, мы проследили превращения, которым подвергаются эти два
вещества, опираясь на 'Совершенно достоверные факты: мы сделали только
одно предположение, а именно—допустили существование части
кислорода, еще свободного после прохождения воздуха над (горящей) серой.
Если это предположение может показаться сомнительным, то оно
перестанет быть таковым, если мы докажем путем опыта, что все
происходит так, как мы предвидели при условии, что наше допущение верно».
Далее следует описание опыта, который сделался классическим и
приводится во .всех, даже элементарных, учебниках.
«Смешивая -в .прозрачном сосуде вещества, которые мы считаем
необходимыми для данного .процесса, мы можем видеть, происходит ли
смена реакций таким образом, какіим мы ее себе представляли. Это
можно подтвердить, если в баллон .ввести сернистый газ, воздух и окись
азота в небольшом количестве, например 7і2о по весу от сернистого
газа. Можно видеть, как окись азота краснеет и распространяется в
пространстве; потом возникает белый дым и, подобно облакам,
клубится в баллоне и оседает на стенках з виде блестящих звездчатых
кристаллов *. После этих густых клубов серной кислоты (содержимое
* Впоследствии было выяснено, что кристаллы, образующиеся в этих условиях,
представляют так называемую нитрозилсерную кислоту HNS05, ангидрид серной и
азотистой кислоты:
f°
0-N-0—S=0
или так называемые камерные кристаллы. Водой она разлагается по уравнению.
2HNS06 + Н20 = N0 + N0-, + 2H2S04
400
Эволюция учения о катализе
баллона) делается прозрачным, и если в это время прибавить воды,
кристаллы кислоты плавятся с сильным выделением тепла, а окись азота,
освобождающаяся при этом, снова обращается в красно-бурую кислоту
(acide rutalant) и те же явления возобновляются сначала до тех пор,
пока не будет израсходован весь кислород, или пока не будет сожжена
вся сернистая кислота. Газы, остающиеся после реакции, .как раз те
самые, которые были нами указаны предположительно. В самом деле,
цвет азотистой кислоты появляется вновь почти что со своей
первоначальной силой; по окончании процесса нельзя заметить запаха
сернистой кислоты, но имеется много азота, и маслянистые капли серной
кислоты появляются на стенках баллона... Этот единственный опыт не
оставляет места никакому сомнению относительно теории фабрикации
серной .кислоты, которую мы дали и которая является не более как
простым следствием из фактов».
В следующих словах, которыми Дезорм и Клеман заканчивают свои
мемуар, они дают совершенно правильную оценку своих выводов,
главным образом в практическом отношении, в применении к проблемам
промышленности. «Эта теория,— говорят они,— подает нам надежду
найти тот же способ реагирования (mode (faction) и в других
химических операциях, по всей вероятности, мало выясненных; она позволяет
нам дополнить существующий способ разумными
усовершенствованиями. Форма свинцовых камер, конструкции дымоходов, по
необходимости, испытывают на себе влияние этой теории; но ее главным
благодетельным результатом надо считать почти полную экономию в
селитре».
На самом деле, -в области техники работа французских химиков
.получила огромное значение. Прямым следствием ее было введение двух
крупнейших усовершенствований в технику 'камерного процесса — башен
Гей-Л юссака (1830 г.) и Г л о в ер а (1860 г.)*, а еще раньше отказ от
крайне нерационального сжигания серы совместно с селитрой. Все эти
усовершенствования имеют одну глазную цель — уменьшить до
спайного предела потерю дорогой азотной кислоты при возможно
интенсивном введении реакции окисления SO3 в H2SO/A.
Странным'образом научное значение работы Дезорма и Клемана
долгое время оставалось непонятым, и в то время, когда обсуждались
всевозможные теории катализа, часто весьма фантастические, простая
и ясная химическая теория оставалась в тени.
Третья важная работа по катализу, на которую необходимо обратить
внимание, относится к 1811 г., она была сделана ъ С.-Петербурге
академиком Кирхгофом и касается гидролиза крахмала. Кирхгоф заме
тил, что при кипячении со слабой (1%-ной) серной кислотой крахмал
превращается вначале в особое камедеобразное вещество, которое мы
теперь называем декстрином, и затем в виноградный сахар.
При этом оказалось, что серная кислота не затрачивается при
реакции и не соединяется с сахаром, а потому может быть удалена из рас-
* Известно, что в башне Гловера горючие газы, получающиеся после горения серы
(или после обжига сорного колчедана FeS2, поднимаются снизу вверх, проходя через
слой подходящего пористого материала, постоянно смачиваемого дождем нитрозы
(раствор окислов азота в камерной серной кислоте). При этом достигается насыщение
газов окислами азота и одновременно некоторое сгущение еще разбавленной серной
кислоты. Из башни Гловера газы проходят в камеры, а оттуда — в башню Гей-Люссака,
где они также поднимаются вверх, встречая на своем пути поток камерной кислоты.
Отдавая последней окислы азота и превращая ее в нитрозу, газы становятся
отработанными и могут быть без вреда для производства выпущены в атмосферу.
Эволюция учения о катализе
401
твора по окончании осахаривания, например, с помощью извести (в
виде ^трудно растворимого гипса CaS04). Ряд дальнейших
исследователей (Де-Ла-Рив, Лемпадиус, Фогель, Браконно и др.) подтвердили и
расширили результаты, полученные Кирхгофом. Они показали, что не
только все сорта крахмала (опыты Кирхгофа были сделаны с
пшеничным) подвергаются осахариванию, но что и другие углеводы
подвергаются подобному же превращению. Особенно интересно наблюдение
Браконно (1819 г.), что даже такой необыкновенно прочный углевод,
как клетчатка, также подвергается гидролизу, а именно превращению
в виноградный сахар, хотя и с гораздо большим трудом, чем крахмал.
Было показано, что осахаривание .крахмала происходит без выделения
газов, что подобно серной кислоте, действуют и другие кислоты,
например соляная. Таким образом, была открыта обширная область
расщепления сложных углеводов, как было показано впоследствии,
протекающего при участии воды (гидролиз). Так, например, гидролиз
крахмала совершается согласно уравнению
(СвН10О6)п+пН20=пС6Н22Ос.
Гидролиз углеводов оказался частным случаем еще более общей
.категории реакций — гидролиза различных ангидросоединений, к числу
которых относятся, например, сложные эфиры, амиды, глюкозиды и пр.
Реакции эти оказались интересными в том отношении, что среди них мы
имеем большое число примеров катализа в гомогенной среде. С другой
стороны, многие из сюда относящихся явлений представляли
несомненную аналогию с рядом реакций, совершающихся в живых организмах.
Каталитический гидролиз крахмала и некоторых других углеводов
оказался чрезвычайно интересным также с технической точки зрения.
Расщепление крахмала с помощью серной кислоты легло в основу
важной отрасли химической промышленности—'производства патоки, а в
наши дни много работают над техническим использованием реакции
Браконно. Довольно давно уже применяют осахаренную целлюлозу (из
древесных опилок) в качестве материала для винокуренного
производства, а совсем недавно инженер А. А. Шмидт разработал остроумный
прием для 'Применения той же реакции к фабрикации чистого
виноградного сахара. На примере работ Дезорма, Клемана и Кирхгофа мы
видим, таким образом, какую огромную практическую пользу приносило
научное исследование каталитических процессов для разрешения
важных задач промышленности.
В 1818 г. известный французский химик Тенар открыл новое
соединение элементов О и Н — перекись водорода (Н2О2). С этим
замечательным соединением связан ряд наблюдений и систематических
исследований по катализу, и область эта, далеко не исчерпанная, продолжает
развиваться вплоть до нашего времени. Перекись водорода — вещество
крайне непостоянное; оно весьма легко разлагается с образованием
воды и с потерей половины своего кислорода.
2Н202=2Н20+02.
Эта реакция, сама по себе очень, медленно идущая при низкой
температуре, в сильнейшей степени ускоряется в присутствии
разнообразнейших посторонних веществ. Среди этих веществ особенно видное
место занимают благородные металлы: платина и ее спутники, серебро,
золото, затем ряд окислов, легко переходящих из одной стадии
окисления в другую, особенно окислы марганца и железа.
26 Л. А. Чугаев. т. III
402
Эволюция учения о катализе
Подавляющее большинство реакций этой категории должно быть
отнесено к области катализа гетерогенного.
Мы должны теперь обратиться к группе реакций, знакомство с
которыми оказало особенно сильное влияние на теоретические
представления относительно природы каталитических явлений в той форме, в
какой представления эти сложились во второй четверти прошлого
столетия. Реакции этой группы характеризуются тем, что в них роль
возбудителя или катализатора играют тяжелые металлы, преимущественно
благородные, а из них на первом плане платина и ее спутники, серебро,
в меньшей степени золото, а из неблагородных — никель, кобальт, меть
¦и некоторые другие, причем действие этих металлов заключается в
активировании обыкновенного газообразного (молекулярного Нг)
водорода и в сообщении ему свойств, близких к тем, которыми отличается
водород в так называемом состоянии выделения (in statu nascendi). Все
реакции, в которые входит молекулярный водород, в большей или
меньшей степени ускоряются вышеперечисленными агентами. При этом
особенно сильно действуют металлы в мелкораздробленном состоянии;
компактные металлы действуют подобным же образом, но только
значительно слабее.
Первое наблюдение, послужившее началом изучения этих реакций,
принадлежит знаменитому Гемфри Дэви. В 1817 г. в статье,
посвященной вопросу о природе платины, он описывал опыты,
показывающие, что нагретая платина, а также палладий обладают способностям!*
поддерживать медленное окисление горючих газообразных веществ.
Так, введенная в смесь кислорода и водорода или же в смесь паров
спирта, эфира, бензина с воздухом накаленная платиновая проволока
продолжает оставаться в состоянии красного каления, пока не будет
исчерпан, запас горючего материала. Дэви предложил пользоваться этим-
свойством платины для распознавания рудничного газа в
каменноугольных копях. Три года спустя (1820 г.) Эдмонд Дэви* обнаружил
подобное же каталитическое действие продукта, полученного
растворением сернистой платины в азотистой кислоте, выпариванием досуха
полученного раствора, кипячением остатка со спиртом и, наконец,
обработкой аммиаком. Продукт этот, бы-вший, по-видимому, смесью
платиновой черни с окислами платины, оказался обладающим способностью
в присутствии воздуха или кислорода окислять спирт в уксусную
кислоту. Вслед затем иенокий профессор Доберейнер [3] повторил и
расширил опыты Дэви. По его мнению, препарат Дэви, которому последний
дал название гремучей платины, представляет недокись платины.
Окисление спирта в уксусную кислоту оказалось идущим настолько
гладко, что могло быть прослежено количественно, и у Доберейнера
возникла даже мысль о техническом применении этой реакции. Смесь
паров спирта с кислородом под влиянием того же платинового препарата
сильно взрывает. Также оказалось, что. в иных уже условиях и с тем же
результатом (взрывом) происходит соединение составных частей
гремучего газа — кислорода и водорода — с образованием воды. Говоря о
реакции образования уксусной кислоты из спирта, Доберейнер замечает,
что «недокись платины при этом превращении алкоголя не
претерпевает никакого изменения и ею можно сейчас же вновь воспользоваться
для превращения нового, быть может, бесконечного количества
алкоголя». Особыми опытами было установлено, что окисление спирта в
уксусную кислоту в присутствии платинового препарата при
одинаковых прочих условиях не имеет места.
* Профессор химии в Корке, в Ирландии.
Эволюция учения о катализе
403
Продолжая свои опыты, Доберейнер заметил, что «гремучая
платина» способна поглощать большие количества водорода (100 г,
например, поглощают 15—20 кубических дюймов Н2). При этом препарат,
видимо, не изменяется, но при доступе воздуха или кислорода
происходит реакция, сопровождаемая раскаливанием, причем препарат теряет
не только водород, но и кислород и Превращается в металлическую
платину. По словам Доберейнера, при этом утрачивается способность
препарата возбуждать окисление спирта, но еще сохраняется
активность по отношению к смеси кислорода с водородом. Последнее
обстоятельство заставило Доберейнера испытать, не обладает ли подобной же
активностью мелкораздробленная платина вообще и, в частности, так
называемая губчатая платина, оставшаяся в виде серой массы
после прокаливания хлороплатината аммония (нашатырной платины).
«К моему большому удовольствию,—пишет он в письме к Гильберту,—
это предположение подтвердилось на опыте. Платиновая пыль из
нашатырной платины была завернута в белую фильтровальную бумагу и
приведена в соприкосновение с водородом; как и можно было ожидать,
не произошло никакого поглощения и вообще никакого видимого
взаимодействия. Тогда я подпустил атмосферного воздуха к водороду,
бывшему в соприкосновении с платиновой пылью, и тогда через несколько
мгновений произошла знакомая любопытная реакция. Произошло
изменение объема, и через 10 минут весь кислород введенного воздуха
оказался превращенным в воду. Затем водородный газ, находившийся в
соприкосновении с платиновой пылью, я смешивал с чистым
кислородом; сгущение обоих газов произошло настолько быстро и платиновая
пыль так сильно разогрелась, что бумага, в которую она была
завернута, сразу обуглилась. Эти опыты были повторены 30 раз в тот день
(27 июля 1823 г.), когда был открыт этот любопытный факт, и каждый
раз с теми же результатами».
Доберейнер считает, что наблюденное им явление «...должно быть
рассматриваемо как электрическое и что водород с платиной образует
электрическую цепь, в которой первый соответствует цинку». Выпуская
струю водорода на воздух и заставляя ее при этом приходить в
соприкосновение с губчатой платиной, Доберейнер наблюдал раскаливание
платины, а при сильной газовой струе — воспламенение водорода.
На этом явлении Доберейнер основал устройство своего
водородного огнива—остроумного приборчика, возбудившего, как и вообще
опыты Доберейнера, общее внимание химиков. Об этом свидетельствует,
например, заметка Дюлонга и Тенара, появившаяся в «Annales de
Chimie» в 1823 г. [4]. Узнав об открытии Доберейнера из текущей
прессы (Journal de Debats) и из личного сообщения Либиха, бывшего в то
время в Париже, они немедленно принялись за повторение опытов
немецкого химика «pour verifier un fait aussi surprenant» *.
Подтвердив результаты Доберейнера, они демонстрировали их в
заседании Академии и прибавили к ним ряд новых дополнительных
наблюдений. Так, они показали, что платина, превращенная в тончайшие
листки, действует подобно губчатой и вызывает взрыв гремучего газа.
С другой стороны, губчатая платина после сильного прокаливания
теряет способность к самораскаливанию в присутствии смеси кислорода
с водородом; соединение обоих газов в этом случае происходит
медленно при обыкновенной температуре. Другие благородные металлы,.
в том числе золото, серебро, палладий и родий, действуют подобно
платине и взятые даже в компактном виде заметно понижают температу-
* Для лроверки столь удивительного фа^та (франц.). Прим. ред.
26*
404
Эволюция учения о катализе
ру образования воды из компонентов. Дюлонг и Тенар нашли такжг,
что губчатая платина вызывает уже при обыкновенной температуре
соединение окиси углерода с кислородом*:
2СО + 02 = 2С02-
Наиболее интересными являются наблюдения Дюлонга и Тенарг,
указывающие на специфический характер наблюдаемых явлений. Так,
оказалось, что нагретая железная проволока разлагает аммиак на азот
и водород при температуре, при которой реакция не идет ни в
отсутствие катализатора, ни в присутствии платиновой проволоки. Наоборот,
железо совсем или почти совсем не возбуждает соединения водорода
с кислородом.
Во всех этих явлениях, по-видимому, особенно поразительным
казалось то обстоятельство, что даже столь индифферентные металлы, как
платина, способны вызывать также сильные эффекты, не претерпевая
притом заметного химического изменения.
Следующим важным этапом на пути развития наших знаний о
катализе надо считать появление мемуаров Митчерлиха и Берце-
л и у с а. До тех пор ряд частных случаев каталитических явлений
описывались и рассматривались отдельно друг от друга, без всякой
связи. Митчерлих первый, а вслед за ним Берцелиус объединили эти
факты, найдя в них общие черты, и подвели их под единую категорию.
Исходной точкой для соображений Митчерлиха послужило выполненное
им экспериментальное исследование над процессом образования
простого этилового эфира действием серной кислоты на винный спирт.
Реакция эта, эмпирически известная еще в XVI столетии, объяснялась
водоотнимающим действием купоросного масла. Но Митчерлих
показал, что это объяснение неправильно, так как вместе с эфиром
перегоняется вода (смесь из 65 ч. эфира, 18 ч. спирта и 17 ч. -воды). С другой
стороны, ряд энергично соединяющихся с водой веществ, например
едкий кали и едкий натр, не образуют эфира из алкоголя. «Из
произведенных фактов,— пишет Митчерлих,— вытекает, что спирт с серной
"кислотой при температуре около 140° распадается на эфир и воду.
Реакции разложения и соединения, которые происходят' таким образом,
встречаются очень часто, мы будем обозначать их, как реакцию
разложения, через контакт. Превосходным примером может служить
перекись водорода. Малейшие количества двуокиси марганца (Мп02),
мелко раздробленной двуокиси марганца, золота, серебра и других веществ
разлагают это соединение на воду и на кислородный газ, причем эти
вещества не подвергаются ни малейшему изменению; сюда же
относится распадение сахаристых веществ на алкоголь и углекислый газ,
окисление алкоголя в уксусную кислоту, распадение мочевины с водой на
угольную кислоту и аммиак. Вещества эти сами по себе не
претерпевают никакого изменения, но после прибавления крайне малых количеств
фермента, который при этом играет роль контактного вещества, при
известной температуре эта реакция происходит тотчас же. Переход
крахмала в крахмальный сахар при кипячении последнего с серной
кислотой совершенно аналогичен образованию эфира, но только при
образовании сахара разлагается вода, а составные части ее
сочетаются с составными частями крахмала, образуя новые соединения».
Из приведенной цитаты видно, что Митчерлих не только первый
объединял в одну группу столь разнородные по видимости явления, как
* Доберейнер -раньше наблюдал эти реакции для «гремучей платины» Дэви,
Эволюция учения о катализе
4U5
превращение спирта в эфир, разложение перекиси .водорода и осахари-
вание крахмала, подметив в них важные общие черты, но и указал на
существенную характерную особенность действующих -в «подобных
случаях возбудителей — неизменяемость их состава в течение процесса.
Мало того, он первый включил в круг этих явлений и процессы
ферментативные. Детальное развитие идей Митчерлиха относительно
«контактных» явлений мы находим в его статье, появившейся в «Annales de
Chimie et de Physique»* и ,в Анналах Поггендорфа за 1843 г.
На промежуток времени между появлением этих двух работ
Митчерлиха падают две другие статьи, чрезвычайно важные для истории
катализа. Одна из них принадлежит Б ер цел и усу, другая — Мих. Ф а-
радею. На этих статьях в интересах соблюдения не только
исторической, но и логической последовательности нам необходимо теперь
остановиться.
Статья Берцелиуса [5], получившая огромное значение для
дальнейшего развития вопроса о катализе, появилась в 1836 г. в известном
«Ежегоднике» под заглавием «Некоторые мысли об одной силе,
действующей при образовании органических соединений в живой природе,
но до сих пор незамеченной».
Значение этой статьи заключается не в приведении новых фактов,
ни даже в объяснении старых. В этом отношении Берцел.иус, пожалуй,
делает даже шаг назад по сравнению со своими предшественниками,
ибо совершенно не упоминает в своем обзоре о работе Дезорма и Кле-
мана и о том ясном и хорошо обоснованном объяснении, которое эти
химики дали для одной только, но зато .весьма тщательно изученной
имя реакции. Статья Берцелиуса важна потому, что в ней впервые
объединены в одно целое самые разнообразные явления, которые в
настоящее время мы называем каталитическими, указана тесная связь
между этими явлениями, а неизвестная причина, их вызывающая,
обозначена условным понятием каталитической силы. Таким
образом знаменитый шведский химик является автором -самого слова
«катализ». «Каталитическая сила,—говорит он,—по-видимому, заключается
г том, что то или другое вещество одним своим присутствием, а не
сродством* может разбудить дремлющие при данной температуре сродства,
ибо вследствие этого элементы какого-нибудь сложного тела
распределяются в новых соотношениях, благодаря чему достигается более
полная электрохимическая нейтрализация». Берцелиус делает при этом
оговорку, что, говоря о каталитической силе, он вовсе не думает давать
какое-либо объяснение ряду наблюденных явлений, он только хочет
подчеркнуть внутреннюю связь между этими явлениями; слово
«катализ» характеризует совокупность этих явлений наподобие того, как
слово «анализ» служит для обозначения совокупности реакций, служащих
для разделения составных частей тел с помощью обыкновенного
химического сродства.
Несомненно, что основное зерно этого объединения заключается уже
в вышеприведенной первой статье Митчерлиха, но только Берцелиус
поставил вопрос во всей его широте. Особенно замечательна та
проницательность, с которой он говорит об огромном значении
каталитических реакций для химии биологической. «Мы убеждаемся на опыте—
говорит он,—что, например, превращение сахара в углекислоту и спирт,
как оно имеет место при брожении под влиянием нерастворимого тела,
известного под названием фермента, который, правда, с более слабым
* Первоначально сообщение об этой работе Митчерлиха сделано з 1841 г. в Бер^
линсной Академии наук.
406
Эволюция ученая о катализе
эффектом может быть заменен фибрином, свернутым растительным
белком, творогом и другими подобными веществами, не может быть
объяснено химической реакцией между сахаром и ферментом,
подобным двойному разложению. Но при сравнении с отношениями,
известными в неорганической природе, оно ни с одним из них не представляет
такого близкого сходства, как с разложением перекиси водорода при
действии платины, серебра или фибрина. Таким образом, естественно
предположить аналогичное действие и для фермента». Далее по поводу
осахаривания крахмала под влиянием диастаза Берцелиус говорит:
«Отсюда вовсе не следует, чтобы этот каталитический процесс был
единственным в растительной жизни; наоборот, мы имеем полное
основание думать, что в живых растениях и животных среди тканей и
жидкостей происходят тысячи каталитических процессов, причем
появляется множество разнообразных химических соединений, для образования
которых из общего первоначального материала, из сока растений или
из крови, мы не могли найти понятной причины; эту причину мы в
будущем, быть может, найдем в каталитической силе организованной
ткани, из которой состоят органы животного тела».
Поставив на обширной и важной категории химических реакций
общую печать «катализа», Берцелиус обратил на них внимание
химиков, заставив рассматривать их под одним общим углом зрения.
Указав на важное значение катализа для явлений жизни, он сделался
одним из пионеров нового плодотворнейшего направления биологической
химии.
Фарадей в своей работе о катализе, в противоположность Берце-
лиусу, занимается не сопоставлением большого числа разнородных
явлений, а тщательным исследованием одного, именно каталитического
действия платины на реакцию образования воды из кислорода и
водорода. Многочисленными опытами он выяснил, что в этом случае
огромное значение имеет чистота поверхности платины.
Совершенно ничтожные и незаметные загрязнения, особенно налеты жира и т.п.,
возникающие от соприкосновения платиновой пластинки с самыми
разнообразными предметами, даже с воздухом, имеют своим последствием
частичное или даже полное инактивирование металла, утрату им
каталитической способности. Особенно вредно действует нецелесообразная
чистка, например, с помощью стеклянной бумаги, протирания тряпкой
и т. п. Все подобные приемы только способствуют покрытию пластинки
тончайшим слоем жира, предохраняющего металлическую поверхность
от непосредственного соприкосновения с газами. Еще резче сказывается
подавление каталитического действия, происходящее под влиянием
некоторых газов, например РН3, или летучих жидкостей. Подобного рода
влияние наблюдалось с водородом, полученным при действии кислот
на обыкновенное техническое железо. И в этих случаях также
образуется пленка, покрывающая поверхность платины. Дело идет о
явлении, подробно изученном много позднее (Бредиг) и известном под
названием «отравления» катализаторов. Исходя из этих опытов и из
общих соображений относительно явлений, происходящих на границе
между газом и поверхностью твердого тела, Фарадей приходит к
заключению, что при каталитическом действии платины играет роль
конденсация газов, происходящая на ее поверхности и сближающая между
собой молекулы этих последних.
Точка зрения, на которой стоит Фарадей, чисто физическая. В
рассмотренной 'Им группе явлений он не видит никаких проявлений
химизма. Возвращаемся теперь к работе Митнерлнха, опубликованной в
1843 году. В этой работе содержится обстоятельный, разбор большого
Эволюция учения о катализе
407
числа явлений, ныне относимых к области катализа, которых мы не
станем^здесь перечислять и, что особенно важно, попытка разъяснения
условий, при которых совершаются эти явления. «Прежде чем
исследовать,— говорит Митчерлих,— почему некоторые химические реакции
имеют место под влиянием веществ, которые учасгвуют в них только
своим присутствием, мы должны постараться отдать себе отчет в том,
каким образом ведут себя тела, которые не соединяются между собой
химически, когда те приведены друг с другом в соприкосновение».
Бесспорная заслуга Митчерлиха заключается в том, что он развил и
обобщил точку зрения Фарадея, указав на важное значение для
разнообразных контактных явлений влияния поверхности контактного
агента —обстоятельство, особенно выдвигаемое в ряде новейших
теорий гетерогенного катализа. При этом, однако, он до известной степени
избег односторонности и наметил более широкую точку зрения на
катализ.
В особенности он отмечает колоссальное развитие поверхности у
таких мелкораздробленных и мелкопористых тел, как губчатая платина,
как уголь и т. п. Он обращает внимание на то обстоятельство, что если,
например, объем всего только в 1 кубический дюйм наполнить сплошь
шариками молекулярных размеров диаметром в 1/10 000 000 дюйма,
расположивши их таким образом, чтобы прямые, соединяющие их
центры тяжести, были им перпендикулярны или параллельны друг
другу, то сумма поверхности таких шариков достигнет 218 166
квадратных футов. Подобным же образом для древесного угля, у которого
средний диаметр клеток в 1/2400 дюйма, вычисляется общая
поверхность приблизительно в 100 квадратных футов.
По отношению к углю Митчерлих приводит еще другой любопытный
расчет, опирающийся на следующий опыт. Взвешенный кусочек угля
кипятился некоторое время с водой и вновь взвешивался, после того
как вода заняла все поры. Величина привеса показала, что на долю пор
приходится около 5/8 всего объема угля. Отсюда и основываясь на
указаниях Аддами, что угольный ангидрид под давлением в
36,7 атм превращается в жидкость при 12°, и на опытах Соссюра,
согласно которым 1 объем угля поглощает 35 объемов СОг, Митчерлих
выводит, что в порах угля более 7з углекислого газа находится в
жидком состоянии на поверхности клеточных стенок. Он приводит целый
ряд других случаев конденсации на поверхности различных других
веществ из газов и из растворов, как-то: обесцвечивание окрашенных
жидкостей, углем, поглощение сивушного масла углем из неочищенного
спирта, увлечение растворенных веществ осадками, возникающими в
растворе, например NaN03, Ba(N03)2, BaS04, процесс кристаллизации,
наступающий преимущественно на поверхности готового уже кристалла.
С этими и рядом других подобных же явлений, ныне относимых к
категории абсорбции, Митчерлих сближает контактное действие
твердых порошков и пористых тел, например платины и угля, не считая,
однако, что причиной химического действия является одна только
конденсация.
«В некоторых случаях,—говорит он по этому поводу,— можно
легко объяснить химическое действие твердых тел на жидкости и на газы,
но в других случаях объяснение может оказаться затруднительным.
Возможно, что конденсация может быть целиком причиной явления для
веществ газообразных. Можно очень легко объяснить, таким образом,
взрыв, который Г. Тенар наблюдал при введении угля в смесь
сероводородной кислоты с кислородом; когда платиновая чернь вместе с
поглощенным ею кислородом вводится в соприкосновение с хлористово-
408
Эволюция учения о катализе
дородной кислотой, по Доберейнеру, образуется хлористая платина.
Легко понять, что в этом случае сжатый кислород легко может
соединиться с -водородом кислоты; однако сродство хлора к платине, по
необходимости, также приходит в эту реакцию.
Когда какой-либо газ находится в состоянии выделения, имеет место
явление, аналогичное тем, о которых мы только что говорили: газ часто
соединяется с веществом, с которым будучи при других условиях
приведен в соприкосновение, не подвергся бы никакому действию; так,
возможно, что в том случае, когда химическое сродство между двумя
газами слабо, конденсация заставляет их соединяться химически. Однако
сомнительно, чтобы следовало способность к соединению двух тел,
между которыми, как, например, между кислородом и водородом, действует
сильное химическое сродство, приписывать одной конденсации, хотя мы
и имеем основание допустить, что при различных физических
состояниях платина сгущает газы на поверхности. Известно, что платина в
листочках и в виде проволоки действует так же хорошо, как и в виде
губки или черни, но реакции происходят тем медленнее, чем меньше
применяемые поверхности».
Разбирая далее целый ряд каталитических процессов: окисление
спирта кислородом в уксусную кислоту в присутствии платины или ту
же реакцию в присутствии организованных ферментов, разложение
перекиси водорода щелочами и отсутствие такового при действии кислот,
.разложение бертолетовой соли при действии окислов меди и марганца,
осахаривание крахмала кислотами и ферментом, диастазом, инверсию
тростникового сахара, алкогольное брожение, Митчерлих всюду
отмечает, что в иных случаях, когда возбудителем является твердое тело,
эеакция, видимо, идет на его поверхности и влияние носит характер
контактного. С другой стороны, он в ряде случаев оттеняет специфичность
действия воз&удителя. Из следующих слов автора, которые находятся
в самом конце статьи, видно, что, признавая важное значение факта
сгущения газообразных и растворенных тел на поверхности контактного
агента, будь то мелко раздробленный металл или организованный
фермент, он придает важное значение и чисто химическому фактору.
«Химические отделения, происходящие под влиянием веществ, действующих
только через контакт, представляют большое сходство с теми, которые
возникают, когда, например, одно тело соединяется с другим и когда
соединение, образующееся при этом, обращает свое действие на третье
тело. Сернистая кислота имеет больше сродства к кислороду, чем
двуокись азота; однако сернистая кислота соединяется с кислородом только
в том случае, если оставить смесь этих двух газов в течение весьма
продолжительного времени, между тем как окись азота непосредственно
соединяется с газообразным кислородом с образованием азотистой
кислоты. Эта последняя уступает свой кислород сернистой кислоте и
вновь становится окисью азота. Кислород в азотистой кислоте
находится, таким образом, в таком состоянии, что может соединяться с
сернистой кислотой. Если смесь кислорода с сернистой кислотой привести в
соприкосновение с губчатой платиной; то оба газа вступят в соединение.
Действие платины и в этом случае значит то же, что и по отношению
к смеси кислорода с водородом. Все эти реакции, и в особенности
получение эфиров, и в частности (обыкновенного) эфира, приводят к
заключению, что химические разложения и соединения могут встречать
препятствия в относительном расположении атомов; но что сила
притяжения, которую- некоторые тела проявляют по отношению к атомам
веществ и которыми их приводят в соприкосновение, может изменить
их положение и, таким образом, вызвать химические реакции. Отноше-
Эволюция учения о катализе
Ш
ние газов к углю и в особенности к платиновой черни доказывает, что
это притяжение очень сильно лаже по отношению к телам различной
природы.
В 1839 г. появился мемуар Ю. Либиха —«Ober die ErscheinuTigen
der Gahrung, Faulniss und Verwesung und Ursachen» *, в котором
вопрос о каталитических реакциях вновь подвергается обсуждению.
Останавливаясь на превращениях органических соединений, Либих
рассматривает эти превращения как результат выравнивания сродства или
внутри молекул (органических атомов, как тогда говорили), или между
различными молекулами. В первом случае мы имеем дело с
реакциями,типом которых могут служить процессы брожения и гниения, во втором —
с реакциями одного порядка, с так называемым тлением. В обоих
случаях нарушается порядок, существовавший раньше в молекуле.
«Стремление элементов распределиться в порядке взаимного сродства с
образованием более простых и прочных сочетаний встречает препятствие со
стороны сродства радикала. Подавляющим сродством радикала
определяются особенности данного соединения, так как благодаря этому
сродству составные части удерживаются в определенном порядке.
Неодинаковое сродство элементов друг к другу стремится уничтожить эти
особенности. Органический атом, как это видно из предыдущего,
заключает в своей индивидуальной конституции причину, вызывающую его
собственное разрушение, как только стремление его элементов
распределиться в соответствии со специальными их сродствами, и дать начало
образованию простейших соединений и, следовательно, их химическое
различие получит преобладание над сродством радикала».
Лйбих указывает на различные случаи «...нарушения равновесия в
притяжении радикала к элементам или радикалам, с которыми он
связан, вызванного повышением химической разницы его элементов^.
В числе этих причин, наряду с влиянием состояния агрегаций,
связанного с действием теплоты, наряду с действием воды, по его мнению,
выступает и «...соприкосновение с третьим телом, которое при этом не
вступает ни в какое соединенно.
Интересно отметить, что, подходя к явлениям каталитическим, он
совершенно правильно оценивает основную причину явления: для него
ясно, что катализатор не создает, а только возбуждает реакцию, причем
влияние каталитических агентов он сравнивает с влиянием
температуры. «Повышенная температура,— говорит он по поводу ряда
гидролитических процессов,—во всех этих случаях может быть заменена
химическим сродством, все равно вступают или не вступают служащие для
того вещества в соединение с новым продуктом или с одним из его со-
ставных частей». Приводя в подтверждение своей мысли целый ряд
гидролитических реакций (как, например, гидролиз тростникового
сахара, крахмала, аллоксаны и др.), он замечает, что при осахаривании
крахмала, например, реакция в одинаковой мере может быть ускорена
повышением температуры и увеличением концентрации кислоты.
Действие возбудителя в этих случаях для Либиха — чисто химическое, хотя
и не вполне ясное по своему механизму, как это видно и в особенности
из следующих его слов: «При содействии предрасполагающего
сродства, при помощи кислот, щелочей и других тел во всяком случае
нарушается равновесие между элементами органического соединения, путем
обмена элементов образуются новые продукты, химические свойства
которых противоположны свойствам действующего вещества, и они
поэтому полагают передел его действию, вступая с ним в сочетание».
* С появлениях брожения, гниения и тления и их причины (нем.). Прим. ред.
410
Эвблюция учения о катализе
Переходя к явлениям гетерогенного катализа, Либих и здесь не
считает нужным говорить о специальной «каталитической» силе, но эту
группу явлений он выделяет в особую категорию, сближая их с
процессом брожения и гниения. Таким образом, он отказывается от того
широкого обобщения, которое заставило Берцелиуса и Митчерлиха
объединить все разнообразные случаи возбуждения реакции
материальными агентами, отметив в них при всем видимом различии черты
глубокого сходства, «Эти вещества,—-говорит Либих,— (перекись
водорода, многосернистый водород и др.) постепенно разлагаются сами по
.себе от малейшего -изменения температуры, и это постепенное
разложение делается моментальным от соприкосновения с мелкораздробленным
углем, платиной и бесчисленными другими твердыми телами, причем
эти последние не претерпевают изменения. Для объяснения этого
способа разложения искали особой причины, но без достаточного
основания. Если мы не желаем видеть в трении или в ударе каталитическую
.силу по отношению к гремучекислой окиси серебра и закиси ртути, по
отношению к гремучему серебру Бертолле,— или для окиси хлора
видеть эту силу в теплоте руки или в несколько более высокой
температуре, то эта сила не может действовать и при разложении многосерни-
-стого водорода и перекиси водорода. Разница заключается только в том,/
что разложение в одном случае происходит в твердом теле, в других
случаях —в газе, а в тех случаях, в которых усматривают действие
новой причины,—в жидкости».
В чем же заключается причина каталитических влияний этой
категории? Ответ на этот вопрос Либих дает в своей теории брожения и
гниения. По существу происходящих здесь реакций он справедливо
сравнивает их с.сухой перегонкой сложных органических соединений.
«Сухая перегонка,— говорит он,— есть горение, происходящее внутри
вещества, горение части углерода и водорода за счет всего
содержащегося в этом веществе кислорода или за счет части этого запаса, причем
в результате остальные элементы перегруппировываются в новые
соединения... Гниение (и брожение) можно было бы назвать сухой
перегонкой, которая происходит в воде, и притом при обыкновенной или же
немного ее превышающей температуре»."' Тление отличается от гниения
и брожения в том отношении, что это процесс окислительный, причем
нужный для 'него (свободный) кислород заимствуется из окружающей
среды. На все эти процессы мы теперь смотрим как на биохимические,
но Либих был другого мнения. Он считал, что возбудители этих
процессов, ферменты, не только не являются живыми существами, но что,
как раз наоборот, и это — сложные органические вещества,
находящиеся в состоянии непрерывного разложения, а следовательно, в состоянии
оживленного атомного движения. И вот этим разложением, этими
движениями фермент как бы заражает окружающую среду, распространяя
происходящие в нем самом химические процессы на вещества, с
которыми он приходит в соприкосновение.
Любопытно отметить, что в теории Либиха есть много общего с
первой теорией брожения, которую развили еще в XVII веке Виллис
(1659) и особенно Сталь, знаменитый'автор теории флогистона (в
своем сочинении «Zymotechnia fundamentalist). Согласно этой теории, при+
чиной брожения считалось оживленное внутреннее движение частиц
бродящего вещества, вызванное подобным же движением частиц фермента.
«Всякое • тело, приведенное в состояние' гниения,— говорит Сталь,- -
очень легко Передает это состояние другимтелам, еще не п<ідвер^е'ЕньБМ
порче.
Таким образом, тедо, в котором: уже возбуждено внутреннее движе-
Эволюция учения о катализе
411
ние, может с величайшей легкостью увлечь в подобное же внутреннее
движение другое тело, еще находящееся в покое, но по природе своей
способное к подобному движению... Роль фермента сводится лишь к
тому, чтобы сообщить свое движение аналогичным частям жидкости,
способной к брожению». Можно почти сказать, что теория Либиха
является переводом на язык начала девятнадцатого столетия мыслей Ста-
ля, высказанных в семнадцатом. Вот что говорит Либих в своем только
что цитированном мемуаре:
«Я хочу обратить внимание натуралистов на одну причину, которую
до сих пор не принимали в расчет и действием которой вызываются
метаморфозы и процессы разложения, которые обычно называют тлением,
гниением, брожением. Эта причина заключается в способности, которой
обладает тело, подверженное разложению или соединению, вообще
какому-либо химическому превращению,— вызывать то же самое явление
в другом теле, с ним соприкасающемся, или сообщать ему способность
подвергаться тому же изменению, которое оно испытывает само. Этот
способ действия лучше всего, можно уподобить горящему телу, при
помощи которого мы вызываем в других телах то же самое явление,
приближая к ним горящее тело. Способность тела, находящегося в
состоянии разложения или соединения, вызывать в других телах сродства,
которых раньше не существовало, или повышать сродства элементов,
составляющих это тело, до такой степени, что они (элементы) входят в
соединения, в которые они раньше не входили, есть своеобразное
свойство, особое проявление сродства, действующее наподобие
своеобразной силы. У горящего тела этим свойством является повышенная
температура». ЛиОих приводит ряд примеров, подходящих, по его мнению,
под эту категорию, как-то: способность серебра при сплавлении с
платиной сообщать этой последней свойство растворяться в азотной кислоте;
разложение Н2О2 окисью серебра, перекисями марганца и свинца,
которые при этом сами разлагаются или выделяют весь свой кислород
(Ag20) или же половину его (РЬОг, МпОг). Между прочим приводится
интересное наблюдение Т. Соссюра [6], что почва, а также
разлагающиеся органические продукты, как древесина, шелк, семена при
определенных условиях не только поглощают кислород и выделяют С02, но и
возбуждают своим присутствием образование воды из гремучего газа.
Нужно заметить, что приведенные Либихом примеры, по крайней
мере в части своей, с современной точки зрения, оказываются
неудачными: реакции между Н2О2 и такими окислами, как Ag20, вовсе не
относятся к каталитическим, именно потому, чт^ здесь оба участника
реакции необратимым путем изменяются, а наблюдения Соссюра, по
крайней мере в главной своей части, сведены на существование особых
бактерий, окисляющлх молекулярный водород. Конечно, мы и теперь
принимаем в них присутствие катализаторов.
Вообще нужно сказать, что взгляды Либиха, в целом не нашедшие
себе подтверждения, во многих случаях основаны на неточно
установленных и неправильно освещенных фактах. Этим объясняется,
например, его утверждение, относящееся к дрожжам, возоудителю
алкогольного брожения: «нерастворимое вещество, называемое ферментом,
не вызывает брожения. Если пивные или винные дрожжи тщательно
промыть прокипяченной и холодной дистиллированной водой так, чтобы
продукт все время оставался покрытым жидкостью, то остаток не
вызовет оольше орожения с сахарной водой. Эту способность
приобретают, однако, промывные воды; но они теряют ее через несколько часов
после соприкосновения с воздухом. Способность вызывать брожение,
которую проявляет растворимая в яоде часть дрожжей, не обусловли-
412
Эволюция учения о катализе
вается действием контакта... брожение вызывается растворенной частью
вследствие разложения, которому она подвергается». Если бы мы
не знали, что простым промыванием нельзя ни отнять у дрожжей
способности к брожению, ни извлечь из них фермент, обусловливающий
брожение, то можно было бы утверждать, что Либиху первому
принадлежит честь открыть зимазу — честь, всеми приписываемую Эд. Бухне-
ру. Впрочем, принимая во внимание возможность извлечь зимазу и.-і
особенным образом высушенных дрожжей, доказанную в последнее
время А. И. Л е б ед е в ы м, не исключается возможность, что Либих,
благодаря случайному стечению обстоятельств, имел дело с убитыми
дрожжами в подобном же состоянии, что в водный раствор у него
перешла действительно зимаза, которая, как это вполне естественно, в
условиях его опыта в сравнительно короткий промежуток времени утратила
свою активность. Отсюда, однако, еще далеко до вывода, что брожение
обусловливается саморазложением фермента.
Теория брожения Либиха довольно долго продержалась в науке и
пользовалась в свое время почти всеобщим признанием, с одной
стороны, благодаря его громадному авторитету, а с другой — благодаря
энергичной защите ее, с которой Либих выступал неоднократно и в
специальных статьях, и в популярной литературе со свойственным ему
несравненным остроумием и* полемическим талантом.
Защищая свои, взгляды против контактной теории катализа,
Либиху еще в большей степени приходилось отстаив-ать их против витальной
теории брожения, в свое время выдвинутой К а н ь я р-Л а т у р о м,
Тюрпэном,Шааном и др., а позднее, в особенности, Пасте-
р о м. По отношению к процессам брожения, гниения, тления, как мы
теперь хорошо знаем, вопрос существенно осложняется вмешательством
микроорганизмов. Странная судьба постигла изучение этой части
вопроса о катализе. Первоклассные исследователи этой области, казалось бы,
вопреки очевидности, были вовлечены в ряд роковых ошибок. Гей-Люс-
сак был первый, сделавший такую ошибку. В опыте, сделанном по всем
правилам химической, но не бактериологической
техники, он показал, что способная бродить, но еще не бродящая жидкость,
сохраняемая без доступа воздуха и стерильная, может быть приведена
в состояние брожения впуском небольшого количества воздуха или
кислорода.
Вывод из этого опыта казался ясным: наличность кислорода
является необходимым условием для -брожения. И этот вывод был сделан в то
время когда только что стал известен метод Апперта для
приготовления консервов путем их стерилизации и изолирования (запаиванием в
жестяных коробках, как бывает и ныне) от внешней среды. Гей-Люсса-
ку не только был известен этот способ, но самое исследование было
вызвано наблюдениями Апперта. На основании своих опытов он объясняет
тот факт, что консервы Апперта начинают разлагаться4вслед за
вскрытием коробки, когда кислород воздуха получает доступ к органической
материи.
С именем Либиха связан второй ряд недоразумений в этом вопросе.
Он категорически отрицал активную роль живых организмов в
процессах брожения, называя самую мысль об этом нелепою и преследуя ее
остротою своих сарказмов. Для характеристики его взглядов на этот
предмет дриведу небольшую выдержку из его знаменитых «Писем о
химии», в свое время пользовавшихся огромной и заслуженной
популярностью. «Что же касается мнения, что гниение животных веществ
происходит от микроскопических животных, то его можно сравнить с
мнением ребенка, который объясняет быстрое падение и течение Рейна
Эволюция учения о катализе 413
большим числом мельниц у Майнца, колеса которых с большой силой
передвигают воду к Бингену».
В1839 г., в самый год опубликования основного мемуара Либиха о
теории брожения в издаваемых им Анналах, появился отчет об
исследованиях Каньяр-Латура, доказывающих, что брожение вызывается
живыми организмами, и о проверочных опытах Тюрпена, по поручению
Парижской академии проверившего и подтвердившего эти исследования.
Этот отчет заканчивается заявлением Тюрпена, что брожение — это
«... чисто физиологическое действие, которое начинается и оканчивается
вместе с существованием микроскопических организмов, жизнь которых
прекращается вместе с полным истощением содержащего сахар
питательного материала, после чего этот материал собирается на дне сосуда
в виде слизи или в виде дрожжей».
Вслед за этой статьей в тех же Анналах Либиха напечатана
анонимная заметка, полная злого сарказма, как обнаружилось
'впоследствии, принадлежащая перу Вёлера и Либиха. Цель этой заметки, по
словам авторов, дать свою особую теорию брожения, могущую служить
новым доказательством того, как просты те средства, которыми
пользуется природа для того, чтобы вызвать чудеснейшие явления.
Следующая выдержка из этого своеобразного памфлета, характерная для
полемических приемов той эпохи, может дать понятие о его содержании [7].
«Пивные дрожжи, разболтанные с водой, разрешаются этим
инструментом особым микроскопом, изготовленным по указаниям самого
Эренберга, разрешаются в бесконечное число маленьких шариков,
поперечник которых едва достигает Veoo линии, и в тонкие нити,
которые, очевидно, являются разновидностью белка. Если эти шарики
перенести в сахарную воду, то можно видеть, что из яиц появляются живые
существа; яйца вздуваются, лопаются и из них образуются маленькие
животные, которые размножаются с поразительной скоростью и
совершенно непонятным образом. По форме своей эти животные
отличаются от всех до сих пор описанных 600 видов. Они имеют вид Рейндорфов-
ского перегонного куба (без холодильника).
Труба шлема является подобием сосущего хобота, который изнутри
покрыт тонкой, в V2000 линии, щетиной. Зубов и глаз незаметно.
Впрочем, можно іразличить желудок, кишечник, анус (в виде розовой точки)
и органы мочеотделения. С того момента, как эти животные
вылупились из яйца, они начинают поглощать сахарный раствор и явственно
видно, как он попадает к ним в желудок. Там он моментально
подвергается перевариванию, о чем онределеннейшим образом
свидетельствует выделение экскрементов. Одним словом, эти инфузории поедают
сахар, выделяют из кишечного канала спирт, а из мочеиспускательных
органов углекислый газ... Экскременты этих животных, выделяемые ими
за 18 часов, весят в 66 раз больше, чем сами животные».
Но ни блестящая логическая аргументация Либиха, ни его
саркастические выпады не были в состоянии противостоять неумолимой
логике фактов, выдвинутых гением Пастера, и прямой вывод из этих
фактов: «нет жизни — нет и брожения» не замедлил восторжествовать з
науке.
Сам Либих под давлением фактов, не отказываясь от своих
взглядов, до известной степени признал, однако, справедливость точки
зрения Пастера. В его последнем мемуаре «О брожении и об источнике
мускульной силы» мы находим такие слова: «Исходя из химической
точки зрения, от которой я не могу отказаться, жизненный акт есть
состояние движения и в этом смысле не находится в противоречии ни
со взглядами Пастера, ни с моим собственным».
414
Эволюция учения о катализе
Я не буду здесь входить в подробности продолжительного спора,
которые привели к этому результату, спора, в котором на одной
стороне вместе с Паютером стоял 'известный английский химик Тин да ль,
а на другой — наряду с Либихом — Пуше, Чарльтон, Бает пан
и др. Самый вопрос о природе брожения лишь косвенно касается темы
настоящего очерка, именно лишь постольку, поскольку игнорирование
роли микробов в процессах брожения, гниения и т. п. вносило путаницу
к неясность в самое представление о катализе. В основе мы не можем
не согласиться с Либихом в том отношении, что все подобные процессы
суть химические превращения. Мориц Траубе в 1858 г. первый дал
ясное выражение той мысли, что живые организмы являются только
носителями ферментов, вызывающих бродильные и им подобные процессы,
только посредниками, а не настоящими агентами в этих процессах. Эта
точка зрения в настоящее время является общепринятой, особенно
после открытия Эд. Бухнером (1896 г,) зимазы, фермента алкогольного
брожения. Но с чисто биологической точки зрения и точки зрения
приложения химии к биологии наличность внутри живых организмов
разнообразных катализаторов-ферментов, специфических по
вызываемому ими эффекту, представляет, конечно, огромное значение. И хотя
еще в 60-х годах минувшего века известный физиолог Людвиг
высказал мысль, что, вероятно, настанет время, когда физиологическая химия
сделается частью каталитической, однако только в наши дни это
предположение превратилось в уверенность и началось планомерное изучение
области знания, которое, быть может, более, чем какая-либо иная,
обещает пододвинуть нас к ответу на вопрос, что такое жизнь.
История развития вопроса о катализе в течение рассматриваемого
периода заканчивается двумя крупными исследованиями, из которых
одно принадлежит А. Вильям с он у, другое — Хр. Шенбейну.
Исследование Вильямсона касается вопроса о механизме
образования этилового эфира и простых эфиров вообще. В двух отношениях оно
получило чрезвычайно важное значение в науке. С одной стороны, мы
должны видеть в нем один из существеннейших подготовительных
шагов к созданию теории строения, ибо оно имело своим прямым
последствием выяснение молекулярного веса и взаимных отношений между
этиловым спиртом, этиловым эфиром, уксусной кислотой и рядом других
органических соединений. Эти вопросы как раз стояли на очереди и
возбуждали много споров между корифеями химической науки того
времени, особенно же между Берцелиусом, Дюма и Либихом. С другой
стороны, та же работа имела не менее важное значение для истолкования
типичной каталитической реакции, которая, как мы уже видели,
послужила Митчерлиху для защиты его контактной теории. Как уже было
указано, Митчерлих опирался при этом на тот факт, что при перегонке
серной кислоты со спиртом вместе с эфиром переходит также вода.Либих
держался другой, чисто химической точки зрения. Главным мотивом для
этого послужило его собственное наблюдение, что образующаяся, как
то было показано еще Магнусом в 1833 г. [8], при взаимодействии
серной кислоты со спиртом этилосерная кислота при нагревании до 127 —
140° разлагается на эфир и серную кислоту. Либих полагал, что
образование этилосерной кислоты происходит в тех частях жидкости, куда
попадают холодные капли постепенно приоавляемого спирта; когда
же образовавшееся соединение нагревается до более высокой
температуры, то происходит разложение, «эфир освобождается». В 1850 г. Грем
I9J доказал, что наолюдение Либиха было ошибочно и что чистая
этилосерная кислота не разлагается даже при 143". Зфир же получается
только при одновременном присутствии спирта.
Эволюция иченин о катализе
¦115
Это наблюдение и послужило отправной точкой для опытов Виль-
ямсона. Чтобы понимать руководившую им мысль, нужно помнить, что
в то время химикам не было еще ясно, что в молекуле эфира вдвое
больше атомов углерода, нежели в 'молекуле спирта. Так. Либих считал
эбир окисью радикала этила (С4Ню при тогдашних атомных весах)
и придавал ему формулу С4НюО, а спирт он считал гидратом эфира
С4Ню*Н20. Вильямсон получил симметричные и несимметричные
(смешанные) простые эфиры с одной стороны из алкоголятов и иодгидратов
C5H11ONa+CH8J=С6НцОСНа-Н Na J.
С другой стороны, через взаимодействие сериоэфирнон кислоты,
образующейся из соответствующего спирта, и H2S04 с новой молекулой
спирта, например
c5h11oh+h2so4-c6h11hso.1+h,o
C5H11HS04+C2H5OH-C5Hu-0-CHb+H2S04.
Очевидно, что образование смешанных эфиров было бы
невозможно, если бы приняли теорию Либиха. Вместе с тем опыты Вильямсона
выяснили и роль серной кислоты в этой группе реакций. Ясно, что,
вступая в реакцию со спиртом и вновь регенерируя, она играет ту же роль,
что окислы азота в фабрикации серной кислоты. Она является
типичным катализатором, действующим mutatis mutandis* по схеме Дезорма
и Клемана. Не дав общей теории катализа, Вильямсон своей работой
много способствовал укреплению чисто химической точки зрения на
природу этого рода явлений.
Обращаясь к работе Шенбейна, относящейся к 1857 году, мы
должны вспомнить, что ему принадлежит честь открытия озона, что он
является одним из пионеров учения об аллотропии. Это и отразилось
на взглядах, высказанных им на природу явлений катализа. Шенбейн
считал, что элементы могут являться в виде аллотропических разностей
не только когда они выделены в свободном состоянии, но и когда они
входят в состав химических соединений. Рассуждая последовательно,
мы должны признать, что это именно так. Ведь кислород \в воде и в
перекиси водорода, в окиси бария и s окиси серебра или в закиси азота —
все это как бы коренным образом различные вещества. Шенбейн,
однако, особенно в последующих своих работах, не шел так далеко, он
остановился в конце концов на том предположении, что кислород, в
частности, существует только в двух полярных формах О и О . В
процессах медленного окисления, например, фосфора, различных металлов
непредельных органических веществ кислорода и воздуха, как он
предполагал, наступает такая «химическая поляризация» нейтральных
атомов О с образованием поляризованных атомов О и О в равном числе.
При этом положительный кислород вступает в сочетание с водой,
образуя перекись водорода, а отрицательный идет на окисление фосфора,
металлов, скипидара и пр. Шенбейн отожествлял отрицательный
кислород с озоном, для положительного — он предложил новый термин а н-
тозон. Шенбейн считал, что эти две модификации кислорода
сохраняют свою химическую природу в соединениях, которые они образуют, и
эти соединения он соответственно называл о зон-идами и антозо*
н и д а м и.
* Внося соответствующие изменения (лат.). Прим. ред.
410
Эволюция мнения о катализе
Известно, что перекись водорода легко вступает в реакцию с озоном
.и со многими кислородными соединениями, разлагаясь и давая начало
образованию кислорода. Таковы, например, перекись марганца, соли
марганцевой кислоты и пр. При таких процессах, по Шенбейну,
происходит взаимная нейтрализация озона и антозона
Но0.6+0-20+Н20
Перекись «- Озон
водорода (антозонид)
Н20-0+МпО*0-МпО+20+Н20.
(озонид)
Здесь не место входить в критический разбор этих воззрений
Шенбейна, которые, хотя в значительной степени и не подтвердились
последующими исследованиями, однако, все же нашли отголосок в
теоретических взглядах новейшего времени (Ван т-Г о ф ф). Для нас эти
взгляды интересны потому, что делают понятным ход мысли, который
привел Шенбейна к своеобразному взгляду на природу катализа. По его
мнению, причиной каталитических воздействий является не
каталитическая сила и не саморазложение катализатора, а превращение под
влиянием этого последнего одних аллотропических модификаций химических
элементов в другие внутри подвергающихся разложению молекул. «Я
держусь потому того мнения, что наше проникновение в сущность
процессов брожения так же, как и во многие другие физиолого-химиче-
-ские процессы, только тогда выйдет из состояния неясности и получит
характер истинного знания, когда лучше и основательнее, чем ныне,
будет подвергаться исследованиям связь между аллотропизмом и
химизмом».
Несомненная и большая заслуга Шенбейна заключается в том, что
он первый обратил внимание на явление химич ее ко й индукции,
¦весьма близкое к катализу, впервые выделенное в особую категорию
Кесслерсм в 60-х годах прошлого столетия. Химическая индукция
заключается в наличности возбуждающего влияния одной реакции на
другую, так что вторая может идти vc заметной скоростью, только если
одновременно с ней имеет место первая. Таковы процессы самоокисления
и некоторые другие реакции, изученные Шенбейном, указавшим и путь
для сближения их с областью катализа.
В предыдущем была сделана попытка дать сжатую картину
истории развития учения о катализе в течение периода времени,
простирающегося примерно до середины прошлого века. Подводя итоги, можно
видеть, что в начале этого периода происходило -накопление отдельных
разрозненных фактов, нередко поражавших исследователей своим
неожиданным загадочным характером, но остававшихся без взаимной
связи и без рационального объяснения. В последнем отношении
единственным, правда, блестящим исключением является работа Дезорма
и Клемана, давшая по своему времени исчерпывающее объяснение
одной конкретной реакции. Затем наступил период обобщений и теорий,
обнимающий 30 и 40-е годы.
Благодаря Митчерлиху и Берцелиусу обширный, круг явлений был
подведен под одну общую категорию процессов каталитических или
контактных. Митчерлих, Фарадей, Либих сделали частичные попытки
выяснить причину, лежащую в основе катализа. За это время, в общем,
в этом вопросе наметились два основные течения мысли: физическое
и химическое. Первое течение подчеркивает доминирующее, если не
Эволюция учения о катализе
417
исключительное значение физической стороны дела и, в частности,
физической конституции катализатора. Второе течение выдвигает на первый
план химический элемент. Представители этого направления хотели
видеть в каждом катализаторе прежде всего химический агент,
наподобие того, как это имеет место по отношению к окислам азота в
камерном процессе.
Разрешения разногласий, существующих между сторонниками этих
двух взглядов, не дает и наука нашего времени, для которой многие
основные пункты в учении о катализе продолжают оставаться столь же
темными, как и во времена Берцелиуса и Митчерлиха, Фарадея и Либи-
ха. По-видимому, однако, уже намечается некоторый средний путь,
примиряющий крайние точки зрения. Причины, обусловливающие явления
катализа, по-видимому, многообразны; в них химическая и физическая
стороны могут играть роль одновременно, причем в разных случаях то
та, то другая выступает на первый план и играет доминирующую роль.
ЛИТЕРАТУРА
1. Lelievre et Pel 1 et i er. Ann. Chim.. 1797, 20, 48.
2. Desormer, Clement. Ann. Chim., 1806, 59, 329.
3. H. Dobereiner (L. W. Gilbert). Ann. Phys., 1822, 72, 193; 1823. 74, 269.
4. Dulong et Thenar d. Ann. Chim., 1823, 23, 440.
5. J. Berzelius. Jahresber. Forsch. Chem., 1836, 237.
6. N.T. S a u s s u r e. Bibliotheque de Gebeve Ferbiz. 1883, 360.
7. Das entrathsolte Deheimniss der geistiden Gahrung. Lieb. Ann., 1839, 29, 100.
8. G.Magnus- Lieb. Ann., 1883, 6, 152.
9. T.Graham. Chemical and physical researches Edinbure, 1876, p. 646.
27 Л. А. Чугаев, т. Ill
ПАМЯТИ А. М. ЗАЙЦЕВА и Ст. КАННИЦЦАРО
ЖРФХО, 1970, 42, вып. 7, 1318
Протокол заседания
Химического отделения Русского физико-химического общества
9 сентября 1910 г.
В течение последних месяцев всемирная наука понесла две тяжелые
утраты, в том числе одну для нас особенно близкую. Скончались:
председатель нашего отделения *, заслуженный профессор Казанского
университета Александр Михайлович Зайцев, один из старейших и в то же
время самых блестящих представителей русской химии, и знаменитый
итальянский ученый Ст. Канниццаро, профессор химии в Риме.
Ст. Канниццаро и А. М. Зайцев принадлежат к двум
последовательным генерациям химиков.
Канниццаро родился в 1826 г., всего на два года раньше А. М.
Бутлерова. Наш покойный председатель родился в 1841 г. и был одним из
первых представителей Бутлеровской школы.
Имя Александра Михайловича Зайцева неразрывно связано с тем.
славным периодом в истории русской химии, когда главным, а в одно-
время почти единственным, центром нашей науки была Казань. В
пятидесятых и шестидесятых годах минувшего столетия только начинал
создаваться другой такой центр в Петербурге, куда из Казани
переселился Н. Н. Зинин (для которого, однако, уже прошла самая блестящая
пора его научной деятельности), где только начинала восходить звезда
Д. И. Менделеева. Александр Михайлович был одним из последних
оставшихся в живых свидетелей той эпохи, традиции которой он с честью
хранил и поддерживал в течение всей своей жизни.
Подобно многим другим русским химикам, А. М. первоначально не
предполагал посвящать себя изучению химии, по-видимому,- не думал
даже сделаться естествоиспытателем. Подобно своему старшему
товарищу В. В. Марковникову, он поступил на камеральное ** отделение
юридического факультета, но, увлеченный обаятельной личностью
А. М. Бутлерова, решил избрать химию предметом своей специальности.
По окончании курса он отправляется за границу, работает у Кольбе
в Марбурге, у Вюрца в Париже и получает степень доктора философии
от Лейпцигского университета. Возвратившись на родину (1886 г.), он
занимает должность лаборанта при кафедре агрономической химии
Казанского университета. Затем в быстрой последовательности
появляются одна за другой его научные работы, доставившие ему степень
магистра (в 1868 г.) и доктора (в 1870 г.). В 1868 г. А. М. был назначен
доцентом, в 1870 г. экстраординарным, в 1871 г.—ординарным
профессором Казанского университета, в котором он заместил своего учителя
А. М. Бутлерова.
* Отделение химии Русского физико-химического общества. Прим. ред.
** Цикл наук, по преимуществу экономических, служащих для подготовки будущих
чиновников. Прим. ред.
Памяти Л. М. Зайцева и Ст. Канниццаро
419
Прекрасные работы Александра Михайловича Зайцева, конечно,
известны большинству здесь присутствующих. По традиции Бутлеровской
школы, они отличаются законченностью и изяществом
экспериментальной отделки. По той же традиции, почти все они касаются органической
химии и притом относятся к жирному ряду. Я ограничусь только
перечнем тех основных вопросов, которые затрагивались в этих исследованиях.
Первой крупной работой покойного было изучение реакции окисления
тиоэфиров, которое и привело его к открытию нового интересного
класса сернистых соединений — сульфоксидов, обладающих ясно
выраженным основным характером. Большое теоретическое значение этого
открытия станет ясным, если мы вспомним, что всего за два года
перед тем фон Эфеле открыл сульфониевые основания. Открытие
сульфоксидов явилось новым доказательством того, что атом серы,
становясь четырехвалентным, может приобретать основной характер.
Другая важная работа А. М., лежащая в основе его докторской
диссертации, дала новый усовершенствованный способ для превращения
кислот в алкоголи с тем же числом атомов углерода в молекуле. Путь,
избранный для этой цели, заключался в действии амальгамы натрия
на кислотные хлорангидриды в присутствии соответствующей
свободной кислоты. Первым плодом этой реакции было открытие нормального
бутилового алкоголя, в то время еще неизвестного.
При восстановлении хлорангидрида янтарной кислоты А. М.
сделано было важное фактическое открытие: был получен первый
представитель класса лактонов, вслед затем охарактеризованного немецким
ученым Фиттигом.
Со времени этой работы отыскание новых методов для получения
спиртов различной степени замещения не переставало занимать
внимание покойного. Нам всем хорошо знакомы эти методы, разработкой
которых в течение многих лет занимался A.M. Оригинальная и
плодотворная мысль, которой покойный воспользовался в широких размерах,
заключалась в применении для синтезов смешанных цинкорганических
соединений, которые, по большей части, возникают уже в самое время
реакции и, по мере образования, реагируют с тем или другим веществом
(кетоном, альдегидом, эфиром), содержащим карбонильную группу.
Вы знаете, что все эти «зайцевские» методы на наших глазах
подвергались весьма значительному усовершенствованию, благодаря
открытиям Барбье и Гриньяра. Теперь любой начинающий химик без
большого труда в течение нескольких часов путем «Гриньярджа» может
получить самые, разнообразные комбинации органических радикало-в. Но
ксхгда создавались цинікорганичеокие синтезы Зайцева, дело обстояло
вовсе не так благоприятно. Реакции не всегда шли гладко и не всегда
давали хорошие выходы. Для их воспроизведения нередко требовались
значительные промежутки времени, доходящие до недели и более.
И все-таки открытие этих реакций имело большое значение для
органической химии, ибо проложило путь для воспроизведения множества
синтезов, которые раньше были совершенно неосуществимы.
В середине 70-х грдов в промежутке между другими работами,
отчасти в связи с исследованием спиртов, А. М. впервые точно
формулировал правило, определяющее порядок отщепления галоидоводородных
кислот от галоидозамещенных углеводородов, и, таким образом,
дополнил то, что было сделано в этой области В. В. Марковниковым.
В 1885 г. появилась замечательная работа А. М. «О реакции
окисления олеиновой и элаидиновой кислоты марганцевокислым калием
в щелочном растворе». Этой работой началась серия исследований
из Казанской лаборатории, посвященных изучению высших жирных
27*
420
Памяти Л. М. Зайцева и Ст. Канниццаро
кислот, особенно непредельных. В изучение этой области А. М. с
течением времени все более и более погружался и посвятил ей, со своими
учениками, значительную часть второй половины своей научной
деятельности. И нельзя не признать, что почти всем наиболее крупным и
законченным, что сделано до сего времени для изучения этого отдела
органической химии, одинаково важного с научной и технической стороны, мы
обязаны школе покойного Александра Михайловича.
Я вовсе не имею даже отдаленной претензии излагать перед вами
биографию нашего покойного председателя; это, конечно, гораздо
лучше и полнее меня сделают его ученики, и потому я ограничусь здесь
приведенным беглым перечнем тех главных вопросов или областей химии,
с которыми были связаны [работы А. М., сделавшиеся классическими,
вопросы, для истории которых он оставил по себе крупный,
неизгладимый след.
Но и то немногое, чем я хотел бы помянуть перед вами славную
деятельность А. М., было бы слишком недостаточно и неполно, если бы я
не коснулся еще его* душевных качеств.
Я имел случай встретиться с А. М. всего два раза в жизни и то на
короткое время. Но достаточно было этого мимолетного знакомства,
кратковременной беседы с ним, чтобы простая, добрая душа его
оставила глубокое, неизгладимое впечатление. Все, что мне приходилось
слышать о покойном от его друзей, от его учеников, далеких и близких,
только подтверждает вынесенное мною личное впечатление. Все,
кому случалось так или иначе сталкиваться с А. М., единодушно
свидетельствуют, что он был на редкость добрый ;и отзывчивый человек. Он
был органически неспособен оставить без удовлетворения обращенной
к нему просьбы. К нему шли все за советом, за помощью, и он никому
не отказывал.
Эти редкие свойства души в связи с крупным научным именем и с
удивительными способностями ученого — педагога, руководителя
лабораторных исследований — сделали то, что около покойного всегда
группировалось много учеников. Между ними и профессором всегда были
самые теплые дружеские отношения; эти отношения сохранялись
надолго. Старые ученики после долгой разлуки приезжали к А. М., как к
родному. Так создалась Зайцевская школа, по численности своих
представителей и научному значению уступающая у нас, кажется, только школе
А. М. Бутлерова.
Многие из учеников А. М. Зайцева с честью занимают в настоящее
время кафедры химии в наших высших учебных заведениях. Но
особенно к двум из них, ныне уже покойным, невольно переносится
воспоминание в эту минуту. Всем вам известно имя И. И. Канонникова,
связанное с замечательными исследованиями по светопреломляющей
способности химических соединений, исследованиями, которые с полным
правом могут быть поставлены наряду с работами Гладстона, Ландоль-
та и Брюля, а может быть, даже отчасти и впереди их.
Есть другое имя, еще более громкое и дорогое для каждого из нас,
имя Е. Е. Вагнера, могучая личность которого и блестящие работы еще
так свежи у нас в памяти. Канонников и Вагнер, бесспорно,
составляют славу и гордость Зайцевской школы, в такой же мере как и всей
русской химии. И глубокая скорбь о кончине А. М. Зайцева усугубляется
для нас тем, что первые из учеников его покинули этот мир еще раньше
своего учителя.
Живой интерес к науке, к делам родного университета и особенно к
жизни своей лаборатории А. М. сохранил до самого конца своих дней.
Он умер в буквальном смысле слова на своем посту. До начала летних
Памяти Л. М. Зайцева и Ст. Канниццаро 421
каникул он работал в лаборатории, а в мае месяце состоял членом
физико-математической испытательной комиссии при Казанском
университете.
Тяжелая болезнь, которая свела А. М. в могилу, обнаружилась б
августа с. г. в день возвращения его из летней поездки на Рижское
взморье, йрирода этой болезни точно не могла быть установлена.
Известно только, что ближайшей причиной рокового исхода ее было
заражение крови. 19 августа в девятом часу вечера Александра
Михайловича не стало.
Другой знаменитый химик — Stanislao Cannizzaro, памяти которого я
считаю своим долгом посвятить несколько слов, сицилианец по
происхождению, родился в Палермо. Его жизнь, особенно в первой половине
своей, полная превратностей, представляет резкий контраст с жизнью
большинства ученых, которая, как известно, почти всегда протекает
столь тихо и спокойно, что только одни научные открытия представляют
в ней яркие события, нарушающие ее внешнее однообразие.
Подобно А. М. Зайцеву, Канниццаро не сразу избрал свою
специальность. Первоначально он думал посвятить себя медицине; его первые
юношеские работы относятся к области физиологии. Изучение этой
науки привело его к химии, подобно тому как оно почти в тот же самый
период времени сделало физиком знаменитого Гельмгольца. В 1845 г.
Канниццаро работал у известного физика Меллони и, благодаря этой
школе, у него навсегда запечатлелось убеждение в огромной важности
физических методов исследования для химии. Это убеждение
позволило ему несколько лет спустя сделать главное дело своей жизни.
Двадцатилетним юношей Канниццаро появляется в Пизе в
лаборатории Пириа, куда привлекал его пробудившийся в нем интерес к
химии. С 1846—1847 гг. он занимает у Пириа должность ассистента. Но
в этом последнем году вспыхнула революция, направленная против
неаполитанских Бурбонов. Пламенная любовь к своей родине и молодая
кипучая натура заставили Канниццаро на время покинуть лабораторию
и стать в ряды инсургентов в качестве артиллерийского офицера. Когда
революция была сломлена, Канниццаро должен был покинуть отечество.
Это обстоятельство, по-видимому, сыграло решающую роль в его
дальнейшем химическом развитии.
В 1849 г. мы видим его в Париже, работающим в лаборатории Шев-
реля в J-ardin des plarrtes *. Он находится в постоянном общении с
самыми выдающимися французскими химиками того времени: с Шеврелем,
Фреми, Клоэцом, особенно же со школой Дюма— с Кагуром, Вюрцем,
Пелиго, Малагути и др.
То было время, когда благодаря совокупным усилиям особенно
французских химиков только что рухнула электрохимическая теория
Берцелиуса, но на смену ей еще не успело создаться новой
общепринятой системы. Унитарная система Жерара привлекала к себе внимание
многих, но не была еще оценена по достоинству. Множество вопросов
еще ждало своего разрешения, и Канниццаро попал как раз в самый
разгар оживленных споров, связанных с построением нового здания
химической теории. Здесь, в Париже, очевидно, сложились основные
взгляды его на тот предмет, обработка которого, более чем что-либо
другое, прославило его имя в науке.
Как известно, Ж?рар в своей реформаторской деятельности до
известной степени остановился на полдороге. Проведя в жизнь закон
* Ботанический сад. Прим. ред.
4 22
Памяти А. М. Зайцева и Ст. Канниццаро
Авогадро, указав на его важность для определения молекулярных и
атомных весов, он для одних, сравнительно немногих элементов: С. О,
S, внес исправления в атомные веса — исправления, вытекающие, как
логическое следствие, из общего провозглашенного им принципа. Но для
других элементов, и притом для большинства металлов, отчасти по
недостатку данных, пришлось оставить прежние атомные веса, по
большей части совпадавшие с эквивалентами. Так продолжалось дело до
1860 г., когда химики всех стран съехались в Карлсруэ для того, чтобы
урегулировать путем 'международной конвенции выбор атомных весов.
Об этом конгрессе и о той выдающейся роли, которую играл на нем
Канниццаро, всегда вспоминал Д. И. Менделеев, между прочим и в
своих «Основах». О том же конгрессе вспоминает французский химик
Арм. Готье на нескольких страничках, недавно посвященных им
памяти Канниццаро. Он вспоминает, как после долгих бесплодных споров в
общем собрании конгресса, происходившем под председательством
Дюма, поднялся молодой итальянский химик и в горячей, блестящей
речи показал, каким путем можно было устранить все недоразумения
и разногласия. Канниццаро предложил тогда, в сущности, ту самую
систему атомных весов, которою мы пользуемся в настоящее время.
Дальнейшие изменения для некоторых элементов были внесены в нее
главным образом десять лет спустя Д. И. Менделеевым на основании
периодического закона.
Выбор фактора, на который следовало помножить эквивалент
многоатомного элемента (например, двухатомных Са, Sr, Ва), чтобы
получить его атомный вес, был сделан им частью на основании закона Дю-
¦лонга и Пти и точных цифр теплоемкости, незадолго до того
полученных Реньо, частью на основании плотности пара летучих металлоор-
ганических соединений или, наконец, на основании некоторых аналогий
между элементами.
Каждый из нас понимает, как громадна услуга, которую оказал
таким образом Канниццаро науке. Атомные веса, им предложенные,
скоро вошли во всеобщее употребление, а перед автором их с почетом
открылись двери почти всех европейских академий и химических
обществ.
Но имя Канниццаро связано и с другими крупными научными
работами. Еще во время своего пребывания в Париже (1851) он вместе с
Клоэцом открыл цианамид. Вслед за этим (1853) он открывает бензи*
ловый алкоголь, который получает с помощью общеизвестной реакции,
связанной с его именем,— при действии щелочи на бензальдегид. В этой
реакции одна частица альдегида окисляется в кислоту, другая
восстанавливается в алкоголь.
Чтобы оценить по достоинству это открытие, нужно вспомнить, что
бензиловый алкоголь был первым представителем ароматических
спиртов и что в то время, к которому относится первая публикация
Канниццаро об этом предмете, было вообще известно только четыре
алкоголя. Кроме винного спирта, еще метиловый алкоголь, изолированный
Дюма и Пелиго, эталь (Дюма), затем спирты, амиловый (Кагур), изо-
бутиловый (Вюрц). Открытие нового алкоголя, особеннО'принадлежаще-
го к неизвестному еще гомологическому ряду, представляло в то время
целое событие, нисколько не меньшее по своему значению, нежели
открытие нового элемента.
Вторую большую часть своей научной деятельности (с 1873 г.)
Канниццаро вместе со своими учениками посвятил разработке одного
частного вопроса или, точнее, изучению одной группы органических
соединений— производных сантонина. Сантонин встречается в природе, имен-
Памяти А. М. Зайцева и Ст. Канниццаро 423
но в незрелых цветах Arthemisita maritima, растущего в изобилии в
наших среднеазиатских областях. Его употребляют в медицине как
глистогонное средство. Принятый внутрь, он нередко вызывает особое
расстройство цветоощущения. Для отравленных сантонином все предметы
кажутся окрашенными в желтый цвет, а иногда эти лица теряют вообще
способность различать какие-либо цвета.
Замечательно, что и по своим физическим и химическим свойствам
сантонин и его производные отличаются многими особенностями,
.например светочувствительностью, исключительно высокими цифрами
вращательной способности и пр. Эти особенности в числе других задач были
подвергнуты подробному исследованию в лаборатории Канниццаро.
Достаточно взглянуть на структурную формулу сантонина,
строение которого ныне хорошо установлено, чтобы стало ясным, какое
огромное количество труда и остроумия было затрачено на
дешифрирование этой необычайно сложной и причудливой комбинации атомов.
Задача, которую представляет для исследования сантонин, ничуть не
легче, а может быть, даже труднее, нежели та, которая была связана с
камфорным вопросом. Но над разработкой последнего в течение всего
XIX столетия трудились химики почти всех культурных стран, а в том
числе корифеи нашей науки.
СН3
С СН2 СНз
ОС С СН СНч
со
НоС\ /С\/СН о/
с сн2
I Сантонин
СНз
Определение конституции сантонина вынесла на своих плечах
исключительно школа Канниццаро. Всякий, кто имел случай работать в
подобных областях, знает, какая заманчивая прелесть связана с их
изучением. А потому не мудрено, .что знаменитый ученый, воспитавшийся
в физико-химических традициях, не задумался, однако же, посвятить
этой задаче добрую половину своей жизни.
Возвратившись из ссылки, Канниццаро в 1852 г. занял место
профессора физики, химии и механики в Александрийском университете в
Пьемонте. Отсюда в 1855 г. он переходит в Геную, а в 1861 г.
возвращается в университет своего родного города Палермо. «Там нашел он,—
говорит Арм. Готье,— старую лабораторию, в которой он когда-то
(1842—1843 гг.) работал в качестве ученика: амфитеатр со столом для
чтения лекций, несколько шкафов, несколько элементарных приборов;
при всем том необыкновенно тощий бюджет. Впрочем, таковы были
условия .в то время не в одной только Италии. Не видал ли я в 1863 г.,
когда впервые попал в Париж, -как работал в College de France М. Берт-
ло в пустой и сырой комнате, где почти только один гений заменял для
него необходимые инструменты; не видал ли я, как Клод Бернар на
лестнице приготовлял свои физиологические опыты. То был век великих
открытий для экспериментальной науки; он не был, однако же, золотым
веком».
Карьера Канниццаро завершилась в Риме, куда он был приглашен
профессором химии в 1871 г. и где был в том же году избран сенатором.
424
Памяти А. М. Зайцева и Ст. Канниццаро
Здесь он создал прекрасную лабораторию и в течение 38 лет (он
умер 84 лет от роду) продолжал оставаться ее руководителем.
Увлекательная речь Канниццаро, энтузиазм к науке, не покидавший
его до глубокой старости, во все периоды его деятельности собирали
вокруг него большое число учеников и почитателей. Канниццаро создал
обширную школу. Можно сказать без преувеличения, что почти все по
большей части еще ныне действующие выдающиеся итальянские
химики являются его учениками, а самые молодые — учениками его
учеников. Мы назовем только имена Кернера, Патерно, Бертаньини, Чиамичк-
ана, Назини, Карнелутти, Пикчини, Амато, не считая многих других.
Имя Ад. Либена, впоследствии профессора в Вене, занимает в числе их
не последнее место.
Канниццаро умер 10 мая н. с. текущего* года, 84 лет от роду, вице-
президентом Итальянского сената, пробыв в течение 59 лет в
должности профессора, окруженный всеобщей любовью и уважением со
стороны многочисленных своих друзей и учеников.
* 1910 года. Прим. ред.
ПАМЯТИ В. А. БОРОДОВСКОГО
ЖРФХО, 1914, 46, 155
Протокол заседания
Химического отделения Русского Физико-химического общества
6 февраля 1914 г.
Еще одна тяжелая утрата постигла наше Общество, а вместе с тем и
всю русскую химию. 28 января с. г. скончался от рака желудка приват-
доцент СПб. Университета и научный сотрудник Главной Палаты Мер и
Весов магистр химии Василий Андреевич Бородовский. На нашем
горизонте он впервые появился в 1908 г. на первом Менделеевском съезде,
на котором им был прочитан доклад об энергии радия. Ближе
петербургские химики имели возможность познакомиться с В. А. в 1910 г.,
когда он в годичном собрании Физико-химического общества сообщает
о результатах своей превосходной работы о поглощении (3-лучей радия
различными телами. Вслед за тем Василий Андреевич сделался сначала
частым посетителем, а потом и -постоянным жителем Петербурга. Мы
слышали его высокоинтересные доклады по радиоактивности на втором
Менделеевском съезде и в нашем Отделении, а многие из здесь
присутствующих помнят также его блестящую вступительную лекцию, в которой
он ярко и выпукло обрисовал современное положение вопроса о
структуре атома в связи с учением о дезинтеграции. В. А. сразу завоевал
всеобщее уважение как талантливый ученый, глубокий знаток одного из
интереснейших вопросов современной науки и к тому же превосходный
лектор, а кто сталкивался с ним ближе, тот не мог не почувствовать
глубокой симпатии к этому человеку, открытому и честному, горячо
преданному интересам науки.
В. А. Бородовский родился 20 февраля 1878 г. в с. Бережнянах
Смоленской губернии в семье сельского священника. Отец его умер 48 лет
от роду, оставив огромное семейство (10 человек, из которых В. А. был
седьмым по счету) и только благодаря необыкновенной энергии матери
В. А. мог все-таки получить начальное, а потом и среднее образование.
9 лет от роду он поступает в Смоленское духовное училище, оттуда
переходит в семинарию, а по окончании последней в 1898 г. держит
дополнительный экзамен и вступает в число студентов
физико-математического факультета Юрьевского* университета.
По окончании университета в 1902 г. В. А. по представлению своего
учителя проф. Г. А. Таммана остается при университете, работает
сначала в Юрьеве, а потом в Геттингене, куда в это время проф. Тамман
был приглашен на кафедру общей химии. По возвращении в Юрьев он
получает место ассистента, сдает экзамен на магистра химии, а в
1908 г. получает заграничную командировку на 2 года. Он ставит себе
задачей изучение некоторых сторон явлений радиоактивности и
направляется с этой целью в Англию, где и работает сначала у сэра Джозефа
Томсона в К'эм'бридж'е, потом у проф. Э. Резерфорда в Манчестере.
* Ныне Тартуского (Эстонская ССР). Прим. ред.
426
Памяти В. А. Бородовского
Здесь им была 'подготовлена магистерская диссертация «О поглощении
0-лучей радия», которую он и защищал по возвращении на родину в
Москве (1910 г.). В последнее время В. А. получил место научного
сотрудника при Главной Палате Мер и Весов.
Такав сухой перечень этапов, по которым последовательно
проходил В. А. на своем коротком жизненном пути. В них, конечно, нет
ничего необычного, в этих этапах, через них проходило большинство
начинающих ученых. Только очень немногие, близко стоявшие к В. А., знали
или же только догадывались по отдельным отрывочным его рассказам,
какая поистине ужасная подкладка скрывается за этой ничего не
говорящей внешностью.
Существует глубоко поучительный документ, случайно попавший в
руки проф. Н. Г. Егорова и любезно представленный им в мое
распоряжение — автобиографический очерк, написанный самим покойным
В. А. для одного библиографического издания.
Это настоящий мартиролог и вместе с тем история той болезни,
которая свела В. А. в могилу. Едва ли часто даже у нас в России
доводится встречать ученых, которые в течение своей жизни так много и так
.систематически голодали, как приходилось голодать покойному Боро-
дрвекому. Голодать пришлось ему и в годы пребывания в духовном
училище, когда он по утрам «грыз казенный паек ноздреватой булки,
запивая холодной водой» и считал «особым счастьем», когда у
смотрителя училища, сыну которого он давал уроки, его иногда угощали
сладким чаем. «До этого я никогда не пил сладкого чая»,— замечает
он по этому поводу. Голодание продолжалось и по выходе из духовного
училища, но до особенно высокой степени оно дошло в течение первых
месяцев пребывания В. А. в университете. Он явился в Юрьев, имея
всего 75 рублей в кармане, из которых к тому же 50 руб. немедленно
пришлось внести в кассу университета. В это время В. А. вместо обеда
«выпивал два стакана молока с двумя баранками», после чего
чувствовал резкие боли в желудке.
Полуголодное существование не прекратилось даже после того, как
исполнилась заветная мечта В. А., после того как он получил
заграничную командировку.
Достаточно сказать, что ему выдавали всего 900 руб. в год. Всякий,
кто бывал за границей, а особенно в Англии, знает, что на эти деньги
нельзя прожить иначе, как впроголодь, особенно если принять во
внимание неизбежные расходы, связанные с проездом и с работами в
лаборатории. И вот В. А. снова не доедает и, вероятно, не досыпает, так
как для подкрепления своего тощего бюджета он вынужден был
одновременно с научной работой взяться за ремесло клерка и «за комнату»
(но без стола) надписывать для одного купца адреса на письмах,
предназначенных для русских клиентов.
За неплатеж денег В. А. однажды чуть не привлекли к судебной
ответственности (а неплатежи происходили от неаккуратной присылки
скудной казенной субсидии) и заставили его пережить тяжелые
душевные муки... Обратный путь в Россию В. А. совершает на грузовом
пароходе, опять за неимением средств.
Нужно ли еще подробностей? Такова была вся жизнь покойного,
полная терний, и эти тернии свели его в могилу. Впрочем, незадолго до
смерти судьба как будто ему улыбнулась. Управляющий Палатой Мер
и Весов проф. Н. Г. Егоров, познакомившись с В. А., проявил к нему
самое теплое участие, оценил его знания, его талант и любовь к науке и
пригласил его на только что учрежденную должность научного
сотрудника Палаты. Таким образом В. А. получал и материальное обеспече-
Памяти В. А. Вородовского
427
ние, достаточное для того, чтобы не думать о завтрашнем дне, и, что
для него было особенно важно, лабораторию и средства для работы над
радиоактивными веществами.
И В. А. со свойственной ему энергией и увлечением немедленно
принялся за работу. Но здоровье его уже было надломлено. Тяжелый
недуг с особенной силой стал развиваться с весны прошлого года.
Летний отдых мало принес пользы, а поездка за границу минувшей осенью
привела к окончательному заключению, что роковой конец неизбежен.
Красной нитью через всю жизнь В. А. Вородовского проходит
бескорыстная жажда знания, сначала безотчетная, потом отлившаяся в
определенные формы. В своей автобиографии В. А. такими словами xaj
рактеризует свое душевное настроение в тот юношеский период своей
жизни, когда он еще был учеником 5 класса семинарии: «Мне просто
хотелось много, много знать, чего я еще не знал, утолить свою жажду
истины». Около этого именно времени в его руки попадает
пожелтевший и прожженный том «Основ химии». Д. И. Менделеева, и чтением
этой книги определяется ею дальнейшая судьба. Он решил сделаться
химиком.
Провести это решение было, однако, не так легко. С одной стороны —
призрак материальной нужды и уговоры любимой матери и родственников
по окончании семинарии идти в священники, а с другой — соблазнитель;
ное предложение духовного начальства отправиться на казенный счет в
Академию.
В. А. пришлось выдержать тяжелую семейную сцену, но он остался
тверд.
Его решение не изменили ни соблазнительные предложения, ни
нужда, ни даже просящие глаза матери, перед которыми ему труднее всего
было устоять, которых он долго не мог забыть впоследствии.
Я не буду здесь говорить о научных работах покойного В. А. Их
коснется в своем слове М. М. Кучеров, я хотел бы только отметать, что
работы эти отличаются несомненной самостоятельностью и
оригинальностью. Новичок в вопросе о радиоактивных веществах, В. А. поехал в
Англию со своей собственной темой, и эта тема оказалась настолько
удачной и продуманной, что встретила сочувственное отношение со
стороны таких высокоавторитетных специалистов, как сэр Дж. Дж. Томсон
и Э Резерфорд. Мы знаем, какие интересные и важные результаты
дала ее разработка. За последние годы В. А. был занят изучением столь
важного для нашего отечества вопроса о радиоактивных веществах,
происходящих из русских месторождений. Об этой работе он уже сделал
интересное предварительное сообщение в нашем Обществе. Она должна
была лечь-в основу его докторской диссертации. Но судьба решила
иначе. Научная деятельность В. А. прервалась в самом ее разгаре.
ПАМЯТИ С. М. ТАНАТАРА
ЖРФХО, 1917, 49, вып. 7—8—9, 637
Протокол заседания
Химического отделения Русского Физико-химического общества
15 февраля 1918 г.
Севастьян Моисеевич Танатар родился в 1849 году. Среднее
образование он получил в Симферопольской гимназии, а высшее — в
Новороссийском * университете, который окончил по естественному отделении)
физико-математического факультета. После двухлетней заграничной
командировки покойный заинтересовался вопросом об изомерии фумаровой
и малеиновой кислот, сильно занимавшем видных химиков того времени.
Посвятив свои силы изучению этого трудного вопроса, он положил
добытые при этом экспериментальные данные в основу своей магистерской
диссертации «О строении фумаровой и малеиновой кислот». Одесса,
1880 г. Наиболее важным результатом этого исследования было
констатирование факта, что фумаровая и малеиновая кислоты при
окислении перманганатом дают две различные, притом изомерные диокси-
кислоты, которым автор придал название диоксифумаровой и диоксима-
леиновой. Эта работа покойного С. М. явилась, таким образом, предтечей
будущих классических работ А. М. Зайцева, с одной стороны, и
Е. Е. Вагнера — с другой. Вместе с тем она доставила науке факт
капитальнейшей важности, а именно имеющий существенное значение для
стереохимии. К сожалению, покойный не совсем правильно
интерпретировал полученные им опытные результаты. Он считал, что при
окислении обеих кислот водородные атомы, стоящие при двойной связи,
переходят в гидроксилы. Вскоре появившаяся работа Кекуле и Аншютца
дала неоспоримые доказательства неправильности этого мнения. Оксикис-
лоты на самом деле были богаче водородом, чем принимал С. М., и, как
мы теперь хорошо знаем, они оказались ранее известными стереоизмер-
ными модификациями винной кислоты, кислотами виноградной и мёзо-
винной. Работы над фумаровой и малеиновой кислотами продолжали
занимать С. М. в течение ряда последующих лет. Они доставили ему
материал для докторской диссертации «К вопросу о причинах изомерии
фумаровой и малеиновой кислот», Одесса, 1891.
Впоследствии С. М. перенес центр тяжести своих работ в другую
область— в область физической и неорганической химии. Однако он два
раза возвращался к органическим темам. В 1895 г. появилось его
известное исследование над превращением триметилена в пропилен под
влиянием высокой температуры. Результаты этой работы сначала
возбудили сомнение среди некоторых химиков, но потом точность опытов
С. М. была подтверждена М. Бертло и В. Н. Ипатьевым, который
нашел, что при содействии катализаторов можно значительно понизить
температуру изомеризационного процесса. Позднее, в 1908 г., С. М.
сообщил о ряде опытов, доказывающих, что превращение триметилена в
пропилен может также иметь место под влиянием химической индукции.
* Ныне. Одесский. Прим. ред.
Памяти С. М. Танатара
429
В области физической химии С. М. особенно охотно занимался
вопросами, связанными с теорией растворов, при изучении которых он
преимущественно пользовался термохимическими методами (процессы со-
леобразования в водных и неводных растворах и др.).
Его работы по минеральной химии весьма разнообразны.
Большинство их касается сравнительно малоустойчивых азотистых соединений:
азотноватистой и азотистоводородных кислот, гидроксиламина и
гидразина, реакций, ведущих к их образованию, и их характеристики. Ряд
работ С. М. был также посвящен изучению высших и низших степеней
окисления некоторых элементов; недокисей (Ві, Pb, Cd), с одной
стороны, и перекисей -и надкислот—с другой. В последнем отношении С. М.
как бы следовал за традициями Одесской лаборатории, где были
выполнены первоклассные исследования П. Г. Меликова и его учеников, а
из них особенно Л. В. Писаржевского.
Из других работ С. М. укажем еще на его исследование над ацетил-
ацетонами некоторых металлов и над солями элементов II группы
периодической системы, особенно бериллия.
В лице покойного С. М. Танатара сошел в могилу неутомимый
исследователь, всю жизнь продолжавший работу в лаборатории.
Последняя работа его была получена редакцией нашего журнала * всего
месяца за три до его кончины. Он умер скоропостижно 30 ноября 1917 года
заслуженным профессором Новороссийского университета, которому он
посвятил все сівои силы и где протекла воя его научная деятельность.
* Журнала Русского Физико-химического общества. Прим. ред.
ПАМЯТИ В. Е. ПАВЛОВА
(некролог)
«Природа», 1918, 1, 81
Владимир Евграфович Павлов был питомцем С.-Петербургского
университета. Ученик А. М. Бутлерова, в лаборатории которого он
сдавал свою первую научную работу, вслед затем ассистент и сотрудник
Д. И. Менделеева, он воспринял от своих знаменитых учителей горячую-
преданность науке, глубокую веру в ее научную творческую силу на всех
поприщах человеческой деятельности. Полный этой верой,
энергичный и сильный духом, В. Е. еще совсем молодым человеком вступил на
путь самостоятельной академической деятельности. Это было в 1885 г.,
когда он по рекомендации Д. И. Менделеева был избран в число
адъюнкт-профессоров Высшего московского технического училища по
кафедре аналитической химии. В этом учреждении, которому он отдал все
свои лучшие силы, протекла дальнейшая деятельность покойного до
самой его кончины.
Техническое училище, незадолго перед тем в корне преобразованное
и доведенное до уровня высшей школы, в то время далеко еще не успело»
освоиться с новыми для него академическими традициями. Даже во
внешнем облике его Осталось кое-что от бывшей ремесленной школы.
Так, студенты назывались еще воспитанниками, не утратила своей силы
ученическая система репетиций, которая, впрочем, удержалась в
обиходе училища еще в течение многих лет (до 1905 г.), и т. д.
Пережитки прошлого в большей или меньшей степени сказывались и
на постановке преподавания, особенно в химическом отделении училища.
Деятельность этого последнего, в противоположность механическому
отделению, уже тогда стоявшему на высоте и обладавшему крупными
научными силами (профессора А. В. Летников, Н. Е. Жуковский.
Е. П. Орлов и др.), можно сказать только еще начинала входить в
настоящую колею. В. Е. с самого начала пришлось потратить немало вре*
мени, труда и энергии на борьбу с устарелыми взглядами на постановку
учебного дела, в особенности на то, чтобы обеспечить надлежащую
высоту чисто научной химической подготовки будущих деятелей по
прикладной химии, которых должно было выпускать училище.
Наряду с серьезной постановкой практических занятий по химии,
занятий чисто учебного характера, покойный настаивал на важном
значении для студентов научных и научно-технических исследований тіод
руководством профессоров, занятий, которым он придавал не меньшее
значение, чем техническим проектам. Эта мысль, которую сначала многие
считали чуть не ересью, впоследствии получила всеобщее признание.
Привившись сначала в Политехнических институтах, она в начале 900-х.
годов проложила себе дорогу и в Техническое училище. Другая идея,
которую также горячо отстаивал покойный,— о необходимости создания
в Техническом училище особой специальной группы инженеров-химиков*,
с более обширным химическим и более сокращенным техническим обра-
Памяти В. Е. Павлова
431
зованием,— не получив осуществления при жизни В. Е., кажется, только
теперь начинает воплощаться в реальные формы.
Нельзя сказать, чтобы В. Е. в этой идейной борьбе остался
совершенно одиноким, но число лиц, ему сочувствующих и его поддерживавших,
особенно в первое время, было сравнительно невелико и среди них ему,
во всяком случае, должно быть отведено выдающееся место.
Текущая учебная и отчасти административная работа по
техническому училищу, в которую В. Е. окунулся со всем жаром молодости, не
мешала ему продолжать работу на научной ниве, успешное начало
которой было положено еще во время пребывания его в Петербурге. Его
первая научная работа относится к области органической химии и была
сделана в лаборатории А. М. Бутлерова. В. Е. открыл новый способ*
получения так называемых тетровых кислот (тетровой кислоты и
ее гомологов), только что перед тем открытых известным французским
химиком Демарсэ и возбудивших к себе общий интерес химиков своими
неожиданными свойствами. Продолжая далее свое исследование, В. Е*
исправил ошибку, вкравшуюся в работу Демарсэ, и впервые правильно
установил состав тетровых кислот.
Переселившись в Москву, он в течение нескольких лет продолжал
работать над этими интересными веществами, истинную природу которых
удалось раскрыть только в самое последнее время, но внимание его все
больше и больше привлекали вопросы аналитической химии,
преподавание которой сделалось его официальной обязанностью.
Глубокий и тонкий знаток химического анализа, он с особенной
любовью культивировал эту область химии, которой у нас в России
посвящено сравнительно мало внимания. Ему и его ученикам принадлежит
ряд ценных аналитических работ, посвященных новым методам
количественного анализа и усовершенствованию старых. К сожалению, большая
часть этих усовершенствований осталась неопубликованной, что
объясняется главным образом чрезмерной требовательностью автора к своим
результатам. Из работ, появившихся в печати, отметим ряд исследований
по оксидиметрии и, иодометрии, а также разработку нового
оригинального метода органического анализа в запаянной трубке мокрым путем
(с помощью хромовой и особенно йодноватой кислоты). Эти работы
были доложены на Московском съезде русских естествоиспытателей и
врачей в 1910 году. -
К педагогическому делу покойный относился необыкновенно
добросовестно и вдумчиво. Сознавая всю важность своего предмета для
будущих химиков-инженеров, он не допускал возможности, чтобы студент
покинул его лабораторию, не освоившись самым основательным образом
с важнейшими аналитическими методами. Особенно в течение первых
10—15 лет своей деятельности, когда число занимавшихся в лаборатории
студентов было невелико, В. Е. уделял очень много времени беседам с
практикантами, и если видно было, что студент плохо р-азбирается в том
или другом вопросе, то он неизменно получал приглашение к профессору
на дом, и здесь за чашкой чая велась оживленная и разнообразная
беседа, в которой помимо темы, послужившей ее ближайшим поводом,
затрагивалось также не мало других вопросов, как касающихся химии,
так и далеко выходящих за ее пределы. От многих учеников В. Е. я
слышал впоследствии, что они сохранили об этом времени, об этих беседах
самые теплые воспоминания.
В учебном комитете технического училища покойный всегда
примыкал к наиболее живому прогрессивному крылу, составляя оппозицию^
против всяческих темных сил, против веяний, исходившие из влиятельных
начальственных сфер. С чтими сферами он неизменно вел войну, оста-
432
Памяти В. Е. Павлова
ваясь на почве интересов науки и требований закона, отстаивая права и
достоинство коллегии, которыми он очень дорожил. И это в то время,
когда самое слово «автономия» находилось под запретом. От него не раз
приходилось слышать, что одна из главных причин нашего бесправия
кроется в пассивной покорности, с которой мы подчиняемся
начальственному произволу, не решаясь настойчиво противопоставить ему
требования закона.
На самом деле независимый и убедительный тон, в котором умел
говорить покойный, нередко импонировал начальству, и оно сдавалось
перед его настойчивыми аргументами. В 80-х годах, когда Техническое
училище еще находилось в ведомстве императрицы Марии, В. Е.,
бывший в то время секретарем Учебного комитета, пользовался в глазах
почетных опекунов большим авторитетом, чем назначенный помимо
желания коллегии директор.
За эти свои качества В. Е. пользовался всеобщим уважением своих
товарищей, и даже те из них, которые принадлежали к числу его
противников, в тяжелые минуты, переживаемые училищем, внимательно
прислушивались к его голосу. Не раз его выбирали в числе представителей
Учебного комитета в различные комиссии, междуведомственные
совещания и т. п., в которых разрешались важные вопросы общего характера,
затрагивавшие интересы Технического училища.
Как человек В. Е. отличался безупречной честностью и стойкостью
убеждений. Во всех своих поступках в личной и общественной жизни он
всегда оставался на строго принципиальной почве, не допуская никаких
компромиссов. Некоторые находили, что в этом отношении он даже
утрировал.
Убежденный демократ, он был им не на славах только, а на деле:
вел до крайности простой, почти спартанский образ жизни.
В течение многих и многих лет его можно было видеть в одном и том
же неизменном пальто-крылатке, которое выдержало и дожди, и
зимнюю стужу и которое он упорно отказывался заменить чем-нибудь
новым, более теплым и нарядным. У себя дома он часто и охотно принимал
простых людей; с каждым поддерживал знакомство, бывая у них и
оставаясь с ними на равной ноге, и все это просто, без всякой рисовки. Зато
он резко отрицательно относился к «демократам», которые истинный
демократизм подменивают показным и оперируют с помощью дешевых
демагогических приемов.
В своих отношениях к людям В. Е. был иногда резок и не стеснялся
говорить правду в глаза. Но это не мешало быть ему добрым и
отзывчивым.
Он всегда готов был помочь человеку, попавшему в беду, оказать ему
помощь словами и делом, не щадя времени и средств, будь это старый
товарищ или бывший ученик или студент, потерпевший аварию по
случаю беспорядков...
В 1910 г. В. Е. вышел из состава профессоров, но сохранил связь с
Техническим училищем, оставив за собой заведование лабораторией
количественного анализа. Но здоровье его к тому времени было уже
сильно подорвано, и болезнь, которая свела его в могилу, все более и
более подтачивала его силы. Тот, кто знал его только в течение этого
периода его жизни, с трудом мог составить себе представление о той
кипучей энергии, которую развивал В. Е. в былые годы.
28 сентября текущего года его не стало. Он умер от рака легких,
оставив по себе светлую память среди многочисленных своих друзей,
товарищей и учеников.
ПАМЯТИ С. С. КИЛЬТЫНОВИЧА
ЖРФХО, 1918, 50, 622.
Протокол заседания Химического отделения
Русского Физико-химического общества 24 ноября 1918 г.
7 октября 1918 г. в санатории «Рутка» близ Варшавы скончался от
туберкулеза легких оставленный при Петроградском университете по
кафедре химии Станислав Станиславович Кильтынович.
Покойный попал в наш * университет и в нашу лабораторию в 1916 г.,
в самый разгар войны, когда злая судьба, постигшая его родину,
заставила его покинуть Варшаву, в которой он оставил престарелых
родителей. К нам, на далекий негостеприимный север его влекло, кроме
нежелания сдаваться торжествовавшему свою победу врагу, еще в большей
степени жажда знания, острая потребность продолжить свое химическое
образование и работать научно в лаборатории.
Еще в Варшавском университете, в котором покойный провел свои
студенческие годы, им была выполнена первая экспериментальная
работа на тему проф. Н. А. Прилежаева **. Цель этой работы, которая
и была доведена до благополучного конца, заключалась в проверке
выводов, ранее полученных в Варшавской лаборатории при окислении
пинена по известному методу проф. Прилежаева — посредством над-
бензойной кислоты. Для большей убедительности на этот раз вместо
естественного пинена из скипидара был взят заведомо однородный пинен,
регенерированный из нитрозохлорида по способу Валлаха. Полученные
результаты вполне подтвердили прежние заключения Н. А. Прилежаева.
С переходом в нашу лабораторию С. С. сразу вошел в курс интересов,
занимавших меня и моих сотрудников, приняв деятельное участие в
работах по металлам платиновой группы. Несмотря на слабое здоровье,
он работал без устали, проводя в лаборатории все время с утра до
позднего вечера- Очень скоро он зарекомендовал себя как талантливый и
энергичный молодой химик, умелый экспериментатор, горячо преданный
делу науки.
Работа, выполнение которой было ему предложено, заключалась в
изучении комплексных соединений двухвалентной платины,
заключающих одновременно аммиак и группу нитро в своем составе***. Это
исследование было им закончено в сравнительно короткое время вполне
успешно и притом с необыкновенной тщательностью — чертой, которой
неизменно отличались все работы покойного.
Из полученных им при этом интересных результатов укажу здесь
только на следующие: была впервые точно установлена стереохимическая
конфигурация двух изомерных соединений, отвечающих общей формуле
* Ленинградский университет, лаборатория неорганической химии. Прим. ред.
** Н. Л. Прилежаев (1872—1944 гг.) профессор Варшавского, затем Минского
университета, действительный член Белорусской Академии наук, много лет 'работал
над реакцией окисления непредельных соединений. Прим. ред.
*** См. Л. А. Ч у г а е в. Избранные труды, т. I, стр. 531. Прим. ред.
28 Л. А. Чугаев, т. III
4М
Памяти С. С. Кильтьшовича
[Pt2NH3(NOi/)2]; был установлен факт легко протекающей изомеризации
одного из этих соединений (цис-модификации) в другой (в
транс-модификацию); было открыто и изучено новое комплексное основание,
аналогичное основанию Клеве, и получен ряд отвечающих ему солей общей
формулы [Pt3NHs(N02)]X, причем между прочим обнаружено, что нитрит
этого основания [Pt3NH3(N02)]N02 соединение весьма непрочное,
значительно легче разлагается при нагревании, нежели более богатый
аммиаком нитрит 1-го основания Рейзе [Pt4NH3](N02)2.
По окончании этой работы покойный продолжал изучение
комплексных платонитр'итов в различных направлениях. Между прочим, им было-
начато изучение интересных сложных кислот, соли которых были
получены еще Нильсоном, а затем Везэ при действии разбавленных кислот
на платонитрит калия. Вместе с тем покойный принял деятельное
участие в коллективной работе нашей лаборатории по изысканию новых,
методов для выделения металлов платиновой группы из шлиховой
платины и по приготовлению контактной массы.
Продолжению этой и ряда других работ, о которых мечтал покойный,
помешал, однако, подстерегавший его жестокий недуг. Открывшийся
туберкулез легких потребовал частых поездок в санатории сначала в
Финляндию, потом в Крым. Лабораторные занятия, естественно,
отодвигались- на второй план, а под конец стали совершенно невозможными.
С. С, естественно, отришлось довольствоваться кабинетной работой*
изучением литературы. За это время он очень много перечитал, особенна
по неорганической и физической химии. Результатом детального изучения
литературы до платине явилась составленная им обстоятельная
монография «О химических соединениях платины», которая ждет печати. Она1
должна войти, как часть, в коллективный труд, посвященный платине и
имеющий быть изданным Комиссией естественных производительных сил
России при Российской Академии наук. В самое последнее время С. С.
перевел с английского языка интересную книгу Сванте Аррениуса
«Quantitative Laws in biological Chemistry»*, затрагивающую ряд
животрепещущих вопросов современной биологической химии.
В высшей степени характерно для покойного, что он, едва
оправившись от очередного обострения болезни, уже начинал думать о работах
экспериментального характера. В одном из финляндских санаториев он
даже предпринял совместно с врачом, состоявшим во главе этого
учреждения, работу по обмену веществ при туберкулезе в связи с
возможностью лечить эту болезнь комплексными соединениями некоторых
тяжелых металлов.
Покойный С. С. был не только'талантливым и деятельным молодым
ученым, подававшим большие надежды, он был прекрасным человеком
с отзывчивой чуткой душой. Именно поэтому последние годы его жизни1
были для него настоящей пыткой.
К физическим страданиям его присоединялись тяжелые страдания-
нравственные. Он страдал за свое истерзанное отечество, которое горячо
любил, страдал из-за разлуки с родными, страдал от вынужденного
бездействия, будучи лишен возможности заниматься любимым делом^
работой в лаборатории. И эти нравственные страдания, без сомнения,
довершили ту разрушительную работу, которую сам по себе не успел
сделать физический недуг.
Не помогло продолжительное лечение на юге, не помогло и
возвращение в родную страну, где покойный провел последние дни своей жизни.
Надломленный организм не выдержал и С. С. не стало. Мир его праху..
* Количественные законы в биологической химии (англ.). Прим. ред.
ХЙ$8Х
ПАМЯТИ Ф. М. ФЛАВИЦКОГО
(некролог)
«Природа», 1У18, 2, 204
19 октября текущего года в Казани скончался заслуженный
профессор Флав'иан Михайлович Флавицкий, один из самых выдающихся
русских химиков за последние 40—50 лет. Покойный родился в 1848 г.; он
был питомцем Харьковского университета, в котором и окончил курс по
физико-химическому отделению физико-математического факультета,
после чего в течение 3 лет (1870—1873 гг.) работал в лаборатории
нашего знаменитого учителя А. М. Бутлерова в Петербургском университете.
За это время он успел выдержать магистерский экзамен и получить
звание приват-доцента (то и другое —в Харькове). В 1873 г. ой
поступил на службу в Казанский университет, где оставался до конца своих
дней, занимая сначала должность доцента, потом (с 1884 г.)
—<экстраординарного, 'затем (с 1888 г.) — ординарного и, наконец (с 1899 г.)—
заслуженного ординарного профессора. В 1875 г. он получил степень
магистра за исследование «Об изомерии амиленов из амилового
алкоголя брожения», в 1881 г.— степень доктора за диссертацию «О
некоторых свойствах терпенов и их взаимных отношениях».
Эти капитальные: труды оба отмечают собой первый период научной
деятельности профессора Флавицкого, посвященный работам в области
органической химии. Они сразу поставили их автора в ряду крупных
деятелей науки. В своей магистерской диссертации покойный впервые
точно выяснил химическую природу спирта, полученного еще Вюрцем
путем присоединения воды к амилену (CsHI0) из амилового спирта
брожения *. Ф. М. показал, что спирт Вюрца, получивший название ами-
ленгидрата, должен быть, вопреки мнению авторитетов того времени,
отнесен к числу третичных и что он обладает строением диметил-этил-
карбинола: (СНз)2С(С2Н5) -ОН. Вместе с тем Ф. М. было установлено,
что амиленгидрату отвечает непредельный углеводород — амилен с
температурой кипения 36° (триметил-этилен (СН3ЬС = СН • СН3), тогда как
амиловому спирту брожения отвечает изомерный амилен (изопропил-
этилен (CHahCH — СН = СН2) с температурой кмпения 25°. Переход
от амилового спирта брожения через амилен с температурой кипения
25° в амиленгидрат связан, следовательно, с изомеризацией, с
перегруппировкой атомов внутри химической молекулы**.
* Амиловый спирт брожения — главная составная часть так называемого сивуш-
ного масла.
** Если амиловый спирт брожения (СН3)2СН • СН2СН2ОН перевести в .иодор,
замещая группу ОН ибдом, а затем от последнего отнять йодистый1 водород при помощи
щелочи {(СНз)зСН- СН2*СН21 -> (CHS)2CH-CH = СН2)]. то получается амилен с
температурой кипения 25°, но если прямо отнимать воду из амилового спирта брожения
с помощью хлористого цинка при высокой температуре, то получается см.есь амиленов,
в которой есть оба углеводорода с температурой кипения 25° и с температурой
кипения 36°. Таким образом, при действии ZnCl2 на амиловый сігирт реакция течет
аномально, сопровождается изомеризацией. Этот важный факт также был. впервые
установлен проф. Флавицким
28*
436
Памяти Ф. М. Флавицкого
Докторская диссертация проф. Флавицкого по справедливости
считается классической. Она посвящена изучению химических реакций и
взаимоотношений в интереснейшем ряду органических соединений, так
называемых терпенов,— изомерных углеводородов, отвечающих одной
и той же эмпирической формуле СюНіб и входящих в состав скипидара
и многих других летучих (эфирных) масел растительного
происхождения.
Изучение этого класса веществ, которому посвятили свои силы
самые выдающиеся химики XIX и начала XX века (назовем только
имена Соссюра, Дюма, Девилля, Бертло, а в новейшее время Байера,
L. Е. Вагнера, Валлаха, Брехта, Земмлера и др.)» представляется
особенно интересным ввиду той легкости, с которой терпеновые
углеводороды и их производные переходят друг в друга (изомеризуются), и
благодаря тем в высшей степени своеобразным особенностям, которыми
отличаются такие переходы. К этому следует прибавить, что большая
часть терпенов (и их производных) обладает способностью вращать
плоскость поляризации света и что с этим обстоятельством связан
особый вид так называемой оптической изомерии, которая увеличивает
общее число возможных форм, отвечающих одной и той же
эмпирической формуле, и накладывает свою 'печать на самые процессы
изомеризации.
При таких условиях определение химического строения терпенов
(и их синтез) сделалось задачей необыкновенно трудной и, отчасти
именно благодаря этому, особенно привлекательной для исследователя.
В конце'70-х и в начале 80-х годов минувшего столетия химия
терпенов уподоблялась дремучему лесу, в который вело несколько
тропинок, по большей части, однако, терявшихся в чаще. Были известны
отдельные факты, были описаны отдельные химические превращения
терпенов, но между ними недоставало связи; одни данные
противоречили другим; нередко под различными именами описывались
соединения, между собой тождественные; классификация терпенов, в то
время уже намечавшаяся, главным образом, благодаря работам Бертло
и др., совершенно еще не была разработана. Чувствовалась
необходимость расчистить путь для дальнейших, более детальных и тонких
исследований. Разрешение этой задачи и выпало на долю покойного
Ф. М. Флавицкого и его современника английского химика проф.
Авг, Тильдена. Позднее теми же вопросами занимался О. Баллах в
Германии.
В это время химиками уже было ясно сознано, что терпены являются
непредельными углеводородами и что как таковые они способны
присоединять к себе молекулы свободных галоидов (С12, Вг2), галоидоводород-
ных кислот (НС1, HBr, HJ), -воды и т. п. При этом среди терпенов
намечались два типа. Представители одного оказались способными к
присоединению только одной молекулы галоидов, галоидоводородов, воды и т. д.,
представители другого присоединяют по две молекулы тех же соединений
(например, с НС1 одни дают тела состава СюНіеНСі, другие —
СюНіб2НС1, с бромом первые—СюН16Вг2— вторые СюНібВг4 и т. д.).
Со структурно-химической точки зрения, первые должны иметь в
'молекуле одну двойную (или этиленовую) связь, вторые — две таких связи.
В то время, о котором идет речь, эти терпены называли двухатомными
и соответственно четырехатомными. Различной «атомности» (т. е.
способности присоединять различное число атомов, например, хлора) отвечают
и различные физические свойства. Терпены двухатомные обладают
температурой кипения, близкой к 160°, и более высоким удельным весом,
тогда как терпены четырехатомные кипят около 180°, а удельный вес
ііх заметно ниже.
Памяти Ф. М. Фла?ицкого
437
К периой категории терпенов принадлежит, например, так
называемый пинен (две оптически деятельные разновидности которого,
правая и левая, входят в состав скипидара, русского и соответственно
французского); ко второй — лимонен, существенная составная часть
лимонного масла и многих других эфирных масел.
Ф. М. Флавицкому принадлежит 'прежде всего укрепление и
развитие этой классификации, устранение ряда нарушавших ее стройность
противоречий и затем установление весьма важных соотношений между
упомянуты-ми двумя категориями терпенов: «двухатомными» низкоки-
пящими, собственно «терпенами» и «четырехатомными» высококипящи-
ми,— «изотерпенами», как их называл покойный.
Особенно следует отметить, что ему первому удалось шаг за шагом
проследить последовательный 'переход от терпенов к изотерпенам,
притом в оптической деятельной их форме. Исходя из левого терпена
(/-пинена из французского скипидара) при действии на него серной
кислоты в водно-спиртовом растворе, он получил сначала третичный
спирт —левый гидрат терпена (продукт присоединения воды Сі0Ні6 +
-!-Н20 = СюНГ/ • ОН) или терпинеол, по современной номенклатуре, а
из него путем обратного отнятия воды — левый же изотерпен
(/-лимонен) . Происходящее здесь превращение аналогично рассмотренному
выше переходу от амилена, отвечающего амиловому спирту брожения
через амиленгидрат к изомерному амилену — триметилэтилену.
Вероятно, предвидение этой именно аналогии застазило автора приложить
реакцию, изученную им на простейшем примере, к более сложному случаю
терпенов.
В одной из дальнейших работ Ф. М. подобным же образом из
правого русского скипидара им был получен правый гидрат терпена и
правый изотерпен. Известный специалист по терпенам немецкий ученый
Земмлер, отмечая важность этих наблюдений, говорит, что названное
превращение «1st Flawitzki in genialer Weise gelungen...» * — похвала
редкая и весьма знаменательная в устах немецкого ученого.
Только что упомянутыми важными результатами далеко не
исчерпывается, однако, богатое содержание докторской диссертации Ф. М. Мы
находим в ней массу отдельных наблюдений, ценных мыслей,
сближений и выводов, которые и были использованы последующими
работниками в области терпенов.
Будучи осторожным и вдумчивым ученым, скорее классиком, чем
романтиком, по -известной классификации Оствальда, покойный не
спешил с далеко идущими более или менее рискованными выводами и:*
сделанных наблюдений и по большей части ограничивался схематиче
ской формулировкой заключений, непосредственно вытекавших из до
бытого экспериментального материала. Этим следует объяснить
причину, по которой в его работах по терпенам мы не находим ни одной
структурной формулы. И несмотря на это, Ф. М. и одновременно с ним
работавшему в Казани покойному профессору И. И. Канонникову
наука более «чем кому-либо иному, обязана подготовкой основного
положения терпеновой химии, гласящего, что терпены, подобные пинену и
камфену, являются бициклическими, т. е. содержат две кольцеобразных
системы атомов и одну двойную связь в молекуле, тогда как в
«четырехатомных» терпенах, например в лимонене, мы имели соединения
моноциклические, с одним кольцом и двумя связями» **.
* Удалось гениально Флавицкому. Прим. ред.
** Этот вывод находит себе выражение в структурных формулах трех важнейших
терпенов — п-ин?на и ка-мфена, с одной стороны, лн-моиена — с другой, честь
установления которых принадлежит нашему знаменитому химику покойному Егору Егоровичу
Вагнеру.
438
Памяти Ф. М. Флавицкого
К сожалению, Ф. М. вскоре после защиты докторской диссертации
покинул область терпенов, обработка которой была им начата с таким
выдающимся успехом. Его манили другие перспективы. Уже к концу
80-х годов центр тяжести его работ переносится из области
органической в область неорганической и физической химии, и этому
направлению он остается верен до самой смерти. Весьма возможно, что
внешним поводом для изменения характера работ покойного послужило
выпавшее на его долю (в 1884 г.) преподавание неорганической химии в
Казанском университете.
Работы Ф. М., относящиеся к этому второму периоду его
деятельности, касаются главным образам вопроса о природе растворов, а
отчасти также периодического закона. Будучи сторонником химической
точки зрения на растворы, Ф. М. стремился обосновать -и проследить эту
сторону дела на опыте. С этой целью, между прочим, он изучает, с
одной стороны, кристаллогидраты, соединения солей и др. веществ с
водой, выделяющиеся из растворов в твердом состоянии, с другой — так
называемые криогидраты, или эвтектические смеси.
Для систематики и рациональной формулировки кристаллогидратов
и других близких к ним «молекулярных» соединений (или соединений
«высшего порядка») он создает особую «теорию химических форм»,
опирающуюся, с одной стороны, на структурную теорию, с другой —
на периодический закон и на вытекающие из него значения
валентности элементов. При этом он широко 'пользуется высшими
валентностями таких элементов, как кислород, галоиды и т. п. в тех случаях, когда,
считая, -например, кислород двух-, а хлор одновалентным элементом,
невозможно построить структурную формулу того или иного гидрата,
двойной соли и т. п. Для проверки выводов этой теории покойным
вместе с рядом учеников и сотрудников (А. Я. Богородский, Д. С. Добро-
сердов, П. И. Кузнецов, Панфилов, А. М. Васильев и др.) были
предприняты систематические исследования над кристаллогидратами солей,
отвечающих сходственным элементам. Таним образом был собран
обширный и ценный материал и установлен ряд интересных тіравильно-
стей.
Ф. М. развил также своеобразные чисто химические 'представления
относительно природы криогидратов. На последние он смотрел как на
продукты распада определенных, но мало прочных гидратов, и видит
подтверждение этого взгляда в составе криогидратных смесей. Свои
общие идеи о природе растворов Ф. М. развил в ряде статей, из
которых последняя, наиболее законченная, появилась всего за 3 года до
кончины автора («Химическая теория растворов», 78 стр., Казань.
1914 г.). Взгляды, высказанные в этих статьях, частью вызвали
оживленную полемику; некоторые из них резко расходятся с
общепринятыми в настоящее время.
Но при оценке этой стороны работ покойного не следует терять из
вида, что вопрос о природе растворов принадлежит к старейшим и
труднейшим во всей теоретической химии и что здесь по сие время
много еще остается неразгаданного =и неясного. Еще не так давно так
называемая гидратная (химическая) теория растворов, допускающая
образование химических соединений между растворенным телам
(например, кислотой, солью) и растворителем (например, водой), теория,
защищавшаяся у нас Д. И. Менделеевым, а позднее Д. П.
Коноваловым, А. А. Яковкиным и др., вызвала к себе резко отрицательное
отношение со стороны руководящих химических кругов, особенно в
Германии. В настоящее время, однако, 'благодаря новейшим работам
П. И. Вальдена, Г. Джонса и др. огромное значение начал, вьгдвигае-
Памяти Ф. М. Флавицкого
4Ж>
мых этой теорией, является общепризнанным. А ведь горячим
сторонником ее, как мы видели, был как раз покойный проф. Флавицкий. Уже
из этого примера видно, как 'неосторожно и поспешно было бы делать
какие-либо окончательные выводы из разногласий, вызываемых
некоторыми взглядами Ф. М.
В связи с работами покойного о растворах стоит одно исследование
аналитического характера. Изучая реакцию между телами в твердом
состоянии и следуя в этом отношении по пути, указанному бельгийским
химиком Спринтом, Ф. М. разработал особый метод качественного
анализа, основанный на растирании испытуемого продукта с твердыми
реагентами — метод, годный для разведочных испытаний
предварительного характера.
К числу исследований Ф. М., связанных с периодическим законом,
как отчасти уже было указано, следует отнести систематические
исследования над 'кристаллогидратами сходственных солей.
В 1887 г. покойный сделал интересную попытку представить
количественные отношения, лежащие в основе периодического закона,
определенной математической функцией. При этом автор
останавливается на функции котангенса, исходя из того соображения, что
«...изменение характера элемента из металлического в неметаллический и
обратно происходит как при наименьших, так и при наибольших (например,
F—Na) значениях», а этим условиям удовлетворяет именно cotg.
Общая формула, выражающая зависимость свойств элементов от
атомного веса р, такова acotgzncp(p), где а некоторая постоянная величина.
Эта формула, дающая довольно наглядную картину (качественную)
реальных соотношений, правда, имеет в настоящее время только
исторический интерес, но достойно внимания, что подобная же формула была
дана через 8 лет (в 1895 г.) известным датским химиком Ю. Томсеном,
не знавшим о работе Ф. М.
Из отдельных работ Ф. М., носящих эпизодический характер,
укажем на открытый покойным интересный факт оптической деятельности
таннина (1890 г.), на ряд статей, посвященных вопросу о соотношении
между температурами кипения и некоторыми другими физическими
свойствами органических соединений, с одной стороны, и их
химическим строением — с другой и пр.
Перу покойного принадлежит оригинальный университетский курс
«Общая или неорганическая химия», выдержавший три издания (1-е
издание в 1894 г.).
Ф. М. принадлежит к тому ныне редкому тину ученых, для 'которых
все интересы жизни сосредоточены около науки, около лаборатории и
аудитории. За пределами этого тесного мирка для него в буквальном
смысле слова ничего не существовало. В течение многих лет, как
свидетельствует один из его учеников — А. М. Васильев,— он ни разу не
был в театре, ни в каких-либо собраниях.
Будучи человеком долга, он до щепетильности добросовестно
относился и ік своим педагогическим обязанностям, смысл которых он
понимал широко, как истинный университетский преподаватель. Покойный
оставрл после себя большое число учеников, из которых, сколько
известно пишущему эти строки, четверо занимают в настоящее время
кафедры химии: А. Я. Богородский—в Казанском, В. В. Курилов — в
Варшавском (ныне Ростовском) университетах, Д. С. Добросердов —
в Киевском и П. И. Кузнецов — в Донском политехническом инст/иту-
те. В течение всей своей долгой профессорской деятельности он почти
не пропускал лекций, я когда в 'прошлом году ему пришлось
пропустить две подряд, то у его товарищей и сотрудников по лаборатории
440
Памяти Ф. М. Флавицкого
сразу возникло тяжелое предчувствие. Необыкновенно добросовестно
относился он и к экзаменам и глубоко огорчался
неудовлетворительными ответами студентов.
В лице Флавиана Михайловича Флавицкого наука понесла тяжелую
утрату, потеряла крупную силу, исследователя с европейским именем,
неутомимо работавшего научно в течение всей своей жизни, несмотря
на скудность средств и на убогую обстановку лаборатории. В истории
русской науки его имя по праву займет место вслед за именами
знаменитых химиков Н. Н. Зинина, А. М. Бутлерова и А. М. Зайцева,
последовательно сменивших друг друга на кафедре химии славного своими
традициями Казанского университета.
ОКСИДАЗЫ, ИЛИ ОКИСЛИТЕЛЬНЫЕ ФЕРМЕНТЫ
Русский архив патологии, клинической медицины и бактериологии.
1899, 7, 322.
1
С тех пор как Лавуазье в конце прошлого столетия своими
бессмертными исследованиями положил начало изучению окислительных
процессов, непрерывно протекающих в живых организмах, был вызван к
жизни вопрос, который и до настоящего времени не может считаться
разрешенным.
Б своих знаменитых мемуарах [1], посвященных дыханию, Лавуазье
прямо приравнивает прижизненные окислительные процессы к горению.
Но в этой меткой характеристике скрывается и неизбежный парадокс,
перед которым наука нынешнего века стоит почти в таком же
недоумении, как и современники великого основателя химии. Дело в том, что
внутри живых организмов быстро и полно — до С02, Н20, NH3 и других
простейших соединений — превращаются такие вещества (жиры,
углеводы, белки), которые вне организма — в лабораторной практике и в
повседневной жизни — требуют для этой цели наличности высокой
температуры или сильно действующих окислителей (марганцевокислого кали,
хромовой кислоты и т. п.).
Что касается до свободного кислорода, то под его непосредственным
влиянием окисление наступает при обыкновенной температуре только
для весьма немногих соединений, например для белого индиго, для фос-
финов и некоторых металлоорганических соединений [2]. При этом если
температура не повышается от реакции и последняя не переходит в
настоящее горение, процесс никогда не доходит до возможного предела.
Так, белое индиго переходит в синее, а третичные фосфины в
соответствующие окиси, тогда как конечными (простейшими) продуктами
окисления были бы в этих случаях С02, НгО, Р2О5 и т. д.
На большинство же органических соединений, особенно
относящихся к предельному ряду, кислород воздуха вовсе не действует при
обыкновенной температуре, а когда эта последняя поднимается достаточно
высоко, наступает горение.
В живых организмах нет никакой возможности допустить
существование энергических окислителей или высокой температуры. То и другое
роковым образом разрушило бы неустойчивые соединения, образующие
основу живой протоплазмы, и быстро повлекло бы за собой смерть
организма. Если мы все же утверждаем, что в живых существах
происходит горение, то это верно лишь поскольку дело идет о конечных
продуктах совершающегося в них окислительного процесса. Наоборот,
механизм этого последнего нам неизвестен и, во всяком случае, имеет мало
общего с горением. Очевидно, организм обладает какими-то особыми
средствами, особыми условиями для того, чтобы при низкой
температуре и, по-видимому, с помощью веществ, обладающих сравнительно
незначительной способностью реагировать, достигать быстрого и полного
окисления самых стойких органических соединений.
442
Оксидазы, или окислительные ферменты
Каковы же эти средства, способны ли мы подражать этим условиям
и осуществить их вне организма — вот вопросы, которые в одинаковой
мере останавливают на себе внимание химика и физиолога, вопросы, с
разрешением которых связано понимание одного из главнейших
жизненных процессов.
Если оставить в стороне взгляды чисто виталистического характера,
которые не могут нас здесь серьезно занимать, то в истории
интересующего нас вопроса довольно ясно и отчетливо могут быть различены и
прослежены два направления, соответствующие двум взглядам на
процесс прижизненных окислений. В своем развитии эти взгляды меняются
в деталях, с разрастанием экспериментального материала меняется
их фактическая подкладка, но общий характер остается почти без
перемен.
Согласно одному из них, окисление происходит в
организме под влиянием кислорода воздуха, так или иначе
изменившего свои свойства, «активированного». Последнее название,
однако, далеко не все исследователи понимали одинаково. Так, Шен-
бейн, которого можно считать родоначальником разбираемого нами
взгляда, думал, что активированный кислород является в форме озона
[3], образование которого в организме он считал возможным.
Впоследствии, когда целым рядом исследований было выяснено, что
в организме нельзя констатировать и следов озона, что по своим
сильно окислительным и ядовитым свойствам он немедленно должен
разрушиться при соприкосновении с протоплазмой и в то же время
подействовать губительно на эту последнюю, Гоппе-Зейлер [4] выступил с
другой гипотезой. По его мнению, в организме под влиянием процессов
восстановления выделяется водород, который, действуя на частицу
кислорода Ог, разлагает и освобождает «активированный» атомистический
кислород, способный производить быстрое и сильное окисление. Однако
и эта гипотеза не выдержала критики и должна была пасть под напором
фактов. Против нее особенно говорит то обстоятельство, что многие
организмы, не только растительные, но и животные, могут в течение
значительного промежутка времени оставаться в живых в атмосфере,
лишенной кислорода, причем, однако, не наблюдается выделения даже
следов водорода. Между тем, образования этого газа следовало бы
ожидать, если бы теория Гоппе-Зейлера была верна. С другой стороны, Трау-
бе [5] показал, что водород в состоянии выделения (in statu nascendi) не
может активировать кислорода, а вызывает только образование
перекиси водорода, которое может быть представлено следующим образом:
Н О ОН
+ 1=1
н о он
Наконец, Рейнке [6] специально по отношению к растительному миру
допускает в организме существование перекиси водорода, которая
образуется под влиянием особых, легко окисляющихся веществ,
получивших название самоокисляемых. Перекись водорода в присутствии
особого возбудителя и вызывает окисление. Теория Рейнке также не
оказалась свободной от возражений. Не говоря уже о том, что относительно
самоокисляемых веществ, существование которых в организме, правда,
не подлежит сомнению, не было доказано, что они своим
присутствием способны вызвать окисление стойких органических веществ, по
опытам Пфеффера [7], а также Бокорни {8] перекись водорода не может
быть констатирована в живой клеточке.
Оксидазы, или окислительные ферменты 443
Итак, мы видим, что все попытки истолковать прижизненные
процессы окисления образованием в организме особой модификации
кислорода или его простейшего соединения, обладающего сильной
окислительной функцией, окончились неудачей.
Переходим теперь к другому взгляду, который ведет свое начало
от Морица Траубе [9] и который может быть назван теорией
ферме и то в. Учение Траубе, в первый раз изложенное в его классическом
сочинении «Theorie der Fermentwirkungen», изданном в 1858 г.,
замечательно как исторически первая плодотворная попытка объяснить
процессы, совершающиеся в организмах (особенно так называемые
бродильные), допуская существование одних чисто химических превращений.
По теории Траубе, окисление (или диализ, согласно его
терминологии) совершается в организме при помощи особых окислительных
ферментов (диалионов). Эти ферменты являются переносчиками кислорода
воздуха на вещества, подлежащие окислению (диалиты), причем
постоянно то разрушается, то вновь образуется нестойкое
соединение фермента с кислородом. Возможны и такие
случа-и, когда кислород берется не из воздуха, а отнимается ферментом
от какого-либо химического соединения.
При этом Траубе принимал, что фермент по степени своего
сродства к кислороду занимает среднее место между веществом, от которого
кислород заимствуется (молекулярный кислород или какое-либо
кислородсодержащее соединение) и тем веществом, на которое он
переносится.
Несмотря на сравнительно небольшое количество твердо
установленных фактов, которыми Траубе мог располагать для подтверждения
своих теоретических представлений, он с замечательной ясностью и
уверенностью предсказывал значение, которое может иметь в будущем его
учение об окислительных ферментах для понимания дыхательного
процесса. С особенной определенностью делает это он, когда ему
приходится в конце сочинения о ферментативном действии резюмировать
свои заключения. Вот его собственные слова: «Мы видим, что уже теперь
возможно дать для химизма дыхания чрезвычайно заманчивое
объяснение на основании наблюденных фактов. Мы доказали выше, что
красящее вещество крови представляет почти идеальный пример
переносчика кислорода. Возможно, что оно, будучи насыщено в легких
кислородом, прямо окисляет в капиллярах продукты, подлежащие дыханию.
Возможно также и то, что кислород раньше переходит к другому,
может быть, к целому ряду других ферментов и уже после этого
переносится на дыхательный материал. Если это так, то дыхание имеет много
наружного сходства с процессом брожения горячего индигового куба».
Дать более полное и точное определение вероятной роли
окислительных ферментов в дыхании, нежели то, которое содержится в этих
немногих строках, невозможно и теперь, несмотря на то, что прошло
сорок лет с тех пор, как они были написаны.
Я не могу покончить с воззрениями Траубе, не отметив его
отношения к виталистическим теориям.
Будучи учеником Либиха, он, однако, не разделял взглядов своего
гениального учителя на природу бродильных и жизненных явлений.
Известно, что знаменитый химик в области биологии был вовлечен в
роковую ошибку. Он признавал вмещательство жизненной силы там, где в
этом не представлялось никакой необходимости, и совершенно отрицал
участие организмов там, где они действительно входят промежуточным
звеном между чисто химическим процессом и его первоисточником —
организованной пылью, носящейся в воздухе.
444
Оксидазы, или окислительные ферменты
Траубе избегает обеих крайностей. Он не признает
«каталитических сил» Берцелиуса, как фразы, лишенной содержания, и в то
же время не отрицает возможного участия «грибов и инфузорий» в
процессах брожения. Но признание живого агента вовсе не приводит
его к необходимости допустить существование жизненной силы.
Принимая результаты опытов Шванна как факт и отвергая его
гипотезу, Траубе говорит между прочим: «Даже в том случае, если
бы все процессы гниения зависели от инфузорий и грибов,
здравомыслящий естествоиспытатель не преградил бы себе пути для дальнейшего
исследования подобного рода гипотезой; он просто вывел бы из фактов
то заключение, что в микроскопических организмах заключаются
химические вещества, которые и вызывают явления распада. Он сделал бы
попытку их изолировать, и если бы ему не удалось достигнуть этого без
изменения их свойств, он только бы пришел к заключению, что
употребляемые им средства изменяют химическую природу тех веществ, которые-
он стремился выделить».
В этих немногих словах с замечательной ясностью выражено
воззрение Траубе на химические процессы, протекающие в живых
организмах. Нельзя не заметить и той органической связи, которая
существует между его общими взглядами и приложением их к частному
случаю— к реакциям прижизненного окисления. Мы видим, что учение
о ферментах возникло в уме Траубе сразу во всей полноте и в этой
форме было высказано им уже в 1858 году. Оставалось только
надеяться, что данные опыта, недостаток которых давал себя чувствовать на
первых порах, постепенно заполняют фактические пробелы теории. Но
на деле вышло не так. Идеи Траубе далеко опередили свое время и
им суждено было надолго остаться забытыми. Если об его общих
биохимических воззрениях вспомнили почти только на днях по поводу
блестящего открытия Бюхнера, то немногим раньше получило
практическое осуществление и его учение об окислительных ферментах.
Последнее обратило на себя особенное внимание с тех пор, как по почину
Г. Бертрана [10] целый ряд исследователей принялся за изучение
окислительных ферментов растительного и животного происхождения.
Первый такой фермент был указан Шенбейном и затем изучен Траубе
в 1858 г. Он заключается в клубнях картофеля. Бертран в 1894 г. нашел.,
с своей стороны, в соке лакового дерева очень энергичный фермент,
который назвал лакказой.
Затем, отчасти совместно с Буркело, он подверг исследованию на-
присутствие окислительных ферментов или оксидаз целый ряд растений,
особенно высших грибов, и во всех обнаружил присутствие более или
менее энергичных ферментов.
Для животного -царства первое указание на присутствие оксидазы
принадлежит Шмидебергу и относится к 1881 г. [11]. Однако оно не было
истолковано в смысле Траубе. Позднее Жаке [12] подтвердил и
дополнил опыты Шмидеберга: в животном организме были констатированы
ферменты, частью только облегчающие окислительные процессы
(например, окисление бензилового алкоголя в бензойную кислоту), частью
даже дающие необходимое условие для их совершения (окисление
салицилового альдегида в салициловую кислоту). В последнее время
более подробные исследования, главным образом Абелус и Биарне [13],
выяснили многие частности этого рода процессов; в особенности вопрос
о локализации оксидаз подвергся детальной разработке. С другой
стороны, Портье показал [14], что оксидазы широко распространены в
животном царстве и встречаются в организмах, стоящих на самых
различных ступенях развития. В то же время он высказал весьма интересные
Оксидазы, или окислительные ферменты
445
соображения относительно физиологической роли окислительных
ферментов, как одного из средств борьбы организма с вредными внешними
влияниями. Наконец, опыты Лепина [15], подтвержденные целым рядом
авторов, особенно Краусом и Шпитцером, заставляют думать, что за
оксидазами, играющими лишь второстепенную роль в процессах
прижизненного окисления, скрываются еще другие окислительные ферменты,
способные стимулировать более глубокие процессы распада,
переносить кислород воздуха на более устойчивые соединения и
принимающие поэтому несравненно более видное участие в биохимических
явлениях, имеющих место в живом организме. Со всеми этими результатами,
достигнутыми при изучении окислительных ферментов, нам и
предстоит познакомиться в настоящее время.
II
Мы начнем наше изложение с общей теории переносчиков
кислорода2 и остановимся сначала на тех случаях, в которых
процесс переноса совершается веществами, вполне определенными и
известными со стороны их химической природы.
Представление о переносе одного химического элемента или
радикала на какую-либо другую частицу химического соединения ведет
начало от классических исследований Вильямсона [16], который показал,
что серная кислота в процессе фабрикации обыкновенного эфира играет
роль передатчика зтильной группы С2Н5 на остаток спирта — О—С2Н5.
В качестве примера передатчика кислорода мы рассмотрим прежде
всего закись марганца, недавно изученную в этом отношении Бертраном
[17]. Этот ученый показал, что в присутствии даже ничтожных количеств
солей закиси марганца кислород воздуха легко окисляет целый ряд
веществ (например, гидрохинон), которые сами по себе или вовсе не
окисляются, или окисляются весьма медленно. Механизм окисления
представляется при этом в следующем виде. Соли закиси марганца под
влиянием избытка воды разлагаются с выделением закиси марганца
МпО, которая легко переходит на воздухе в двуокись Мп02. Последняя
также легко уступает свой кислород веществам, подлежащим
окислению. При этом регенерируется закись марганца, которая опять дает на
воздухе перекись, и процесс протекает снова в том же порядке.
Способность закиси марганца служить переносчиком кислорода
¦объясняется, таким образом, попеременным присоединением и
отщеплением от нее атомов этого элемента, причем роль промежуточного звг-
на играет перекись марганца.
Другой пример, на который обратил внимание еще Т pay бе,
представляет окисление сахара под влиянием индиго в слабо щелочном
растворе. Щелочной раствор сахара (глюкозы) на воздухе окисляется и
буреет, но реакция совершается крайне медленно. В присутствии следов
индиго она, наоборот, протекает быстро, причем вся жидкость
(первоначально синяя) обесцвечивается и только на поверхности
соприкосновения ее с воздухом замечается синее кольцо. Процесс проходит
следующим образом: белое индиго отличается от синего присутствием двух
атомов водорода, которые обладают тем замечательным свойством, что
они легко фиксируются на синем индиго под влиянием восстановителей
и также легко опять отщепляются от белого при окислении. Последний
процесс, т. е. переход белого индиго в синее, происходит уже под
влиянием кислорода воздуха при обыкновенной температуре. Благодаря
этим-то двум «подвижным» атомам водорода индиго и может играть
446
0ксидазы1 или окислительные ферменты
роль переносчика кислорода. Синее индиго в присутствии сахара
разлагает частицу воды, причем водород Н2 соединяется с индиго, которое
обесцвечивается, а кислород идет на окисление сахара. Когда таким
образом все индиго перейдет в белое, на поверхности соприкосновения с
воздухом частицы индиго захватывают свободный кислород, причем и
образуется то синее кольцо, о котором мы уже упоминали. В свою
очередь, частицы синего индиго, встречаясь с частицами воды и сахара,,
снова восстанавливаются и таким образом процесс продолжается до-
окисления всего количества сахара, которое находится в растворе.
В предыдущем изложении мы познакомились с двумя случаями
переноса кислорода, из которых в первом деятельным агентом является
простое минеральное соединение, в другом — более сложное
органическое вещество. Мы могли бы присоединить к ним еще целый ряд
других, но я считаю возможным ограничиться приведенными примерами,.
на которых достаточно выясняется процесс переноса кислорода.
Мы видим, что этот процесс всегда обусловливается образованием
промежуточного соединения, в которое вступает кислород воздуха и
которое легко разрушается в присутствии вещества, сравнительно легко-
подвергающегося окислению.
Познакомившись с общей теорией переносчиков кислорода, мы
можем теперь перейти к настоящим окислительным ферментам,.
или оксид азам, для которых разобранные нами простейшие случаи
должны служить прототипами.
Мы начнем наше изложение с ферментов, которые были найдены в-
растениях. Познакомимся сначала с основными фактами.
Еще Шенбейн обратил внимание на то, что сок, вытекший из
пораненных частей некоторых растений (Senecio, Lactuca), обладает
способностью вызывать посинение гуаяковой тинктуры * и иодокрахмальной
бумажки. Так как известно, что та и другая реакция происходят под
влиянием многих окислителей и особенно характерны для озона, то
Шенбейн и допускал, что в организме образуется озон, как мы на это-
уже указывали в историческом обзоре. Однако дальнейшие исследования:
показали ошибочность мнения Шенбейн а.
Правильное толкование наблюденных им фактов было дано М. Т р а-
убе, который вместе с тем проверил и во многих отношениях дополнил
наблюдения своего предшественника. Опыты Траубе в простейшей
форме легко может повторить каждый, особенно с ферментом
картофеля. Если взять картофельный клубень, лучше всего в стадии
прорастания, сделать через него поперечный разрез и полить его
свежеприготовленной гуаяковой тинктурой, то очень быстро начинается посинение,
распространяющееся от периферии к центру. Замечательно, что она
впервые появляется и особенно энергично идет около ростков.
Внутренние части клубня, наоборот, обыкновенно вовсе не показывают реакции,
в чем легко можно убедиться, если осторожно вырезать часть
картофелины, так чтобы сок периферических частей не попал на поверхность
разреза.
Причину гуаяковой реакции, с точки зрения Траубе, следует
видеть в особом ферменте, который образуется в картофельном клубне,
особенно энергично в периоде прорастания и именно в тех частях его,
которые принимают ближайшее .участие в этом процессе.
* Раствор гуаяковой смолы в алкоголе. Действующее начало гуаяковой тинктуры,,
дающее реакцию посинения, составляет так называемая гуаяковая кислота, которая по
химическому характеру относится к многоатомным фонолам.
Оксидазы, или окислительные ферменты 447
Этот фермент может быть отделен от клеточек, в которых он
образуется.
Лучше всего удается получать его в растворе, если выжать сок из
растертых периферических частей картофельного клубня и
профильтровать его сначала через кисею, а потом через асбест при помощи
водяного насоса*. Из такого раствора фермент может быть осаждаем
алкоголем (разумеется, в смеси с целым рядом белковых, углеводистых и
многих других веществ) и высушен **. При этом он только отчасти
сохраняет способность растворяться в воде. Сухой препарат подолгу
сохраняется без изменения, и раствор, приготовленный из него, показывает
ясную гуаяковую реакцию. При нагревании до 73° картофельный
фермент разрушается (Тойнага) [18]. Точно так же действуют растворы
сулемы и формальдегида (5%-ного). По моим наблюдениям, фермент в
значительной степени задерживается порами Ш а м б ер л а н о вскога
фильтра — обстоятельство, указывающее на его высокий молекулярный
вес и на значительную сложность его частицы; кроме гуаяковой
тинктуры, оксидаза легко окисляет еще следующие вещества из класса
многоатомных фенолов: пирокатехин, гидрохинон и пирогаллол.
Продукты реакции окрашиваются при этом в бурый или черный цвет,
который и может служить указанием на присутствие фермента.
Замечательно, что в самом картофеле есть вещество, окисляющееся
при действии оксидазы. Присутствием его объясняется побурение,
наблюдаемое при соприкосновении с воздухом картофельного сока или
свежего разреза картофельного клубня.
Относительно условий среды на течение окислительных процессов,,
вызываемых картофельной оксидазой, необходимо заметить следующее.
При очень низкой температуре окисление крайне медленно и
значительно усиливается от умеренного нагревания. Точно так же
ускоряющим образом на окисление действует взбалтывание с воздухом,
обеспечивающее постоянный приток кислорода к реагирующей
жидкости.
Не менее заметное действие оказывает на течение реакции и
присутствие некоторых химических соединений. Именно, существуют такие
вещества, которым свойственно задерживать проявление
ферментативного действия оксидазы. Еще Шенбейн наблюдал, что гуаяковая
реакция не наступает, если к раствору фермента прибавить синильной:
кислоты или железного купороса (FeS04). Мне удалось также найти
несколько веществ, парализующих эффект оксидазы, к числу их относятся
фенилгидразин и гидроксиламин (NH2OH). Однако какого-либо
соотношения между задержкой окислительного процесса и химической
природой вещества, его вызывающего, до сих пор еще не найдено.
Еще обстоятельнее, нежели картофельный фермент, изучена
оксидаза лакового дерева — лакказа [19]. Сок этого растения
(Rhus vermcifera), а также других представителей семейства Anacard'ia-
се-ае представляет густую жидкость светло-серого цвета. На воздухе
он постепенно чернеет и делается нерастворимым в воде и в большей
части других употребительных растворителей. Такое свойство делает его*
драгоценным материалом для столярных работ и обеспечило ему
обширное применение под именем японского или китайского лака.
Исследования Гикорокуро Иошиды и в особенности Г. Бертрана [20] показали,,
что в свежем соке Rhus находятся два различных вещества. Одно из ник
* Этот способ давал мне особенно концентрированные растворы оксидазы.
** Лучше всего на тарелках слабообожженной глины..
44в
¦Оксидазы, или окислительные ферменты
растворимо в алкоголе и нерастворимо в воде; другое представляет как
раз обратные отношения. Первое, по-видимому, принадлежит к группе
многоатомных фенолов; оно получило название л а к к о л а. Второе
представляет окислительный фермент —л а к к аз у.
Лаккол сам не способен окисляться на воздухе. Но окисление
начинается немедленно, если к его эмульсии прибавить незначительные
количества лакказы. Кроме лаккола, фермент лакового дерева способен
вызывать окисление пирокатехина, гуаякола, гидрохинона, пирогаллола
и таннина, а также действующего начала гуаяковой смолы (дает
посинение с гуаяковой тинктурой).
Бертран констатировал, что при взбалтывании раствора одного из
только что перечисленных веществ с небольшим количеством лакказы
•в замкнутом пространстве происходит заметное поглощение кислорода
по мере того, как протекает реакция окисления.
Значительный интерес представляют исследования Бертрана над
соотношением между составом и структурой вещества,
подвергающегося окислению, и степенью легкости, с которой это окисление
совершается.
При сравнении окисляемости многоатомных фенолов оказалось, что
она возрастает с увеличением числа гидроксилов в их частице.
С другой стороны, из числа изомерных фенолов легче окисляются те,
в которых гидроксилы находятся в пара- или орто-положении друг к
другу. Наоборот, положение мета неблагоприятно для окисления.
Так, сравнение количества кислорода, которое в одно и то же время
и при прочих условиях поглощается в присутствии лакказы растворами
двуатомных фенолов, показало, что легче всего окисляется гидрохинон
СН _СН
ОН^ ^>ОН,
СН СН
несколько труднее пирокатехин
СН СН
ОН< >СН,
ОН СН
и особенно трудно резорцин
СН СН
он^ \:н.
СН ОН
Точно так же фенол С6Н5ОН (одноатомный) окисляется несравненно
труднее шестиатомного гексаоксибензола С6(ОН)6, а флорглюцин
(симметричный) труднее пирогаллола (рядового).
Чтобы покончить с лакказой, заметим еще, что она, подобно
картофельному ферменту, разрушается при нагревании до высокой
температуры (63°) и не диализирует через перепонку из растительного
пергамента (Портье).
Вообще лакказа и фермент картофеля во многих отношениях
сходны между собой, хотя и сомнительно, чтобы они были тождественны.
Существует еще третье местонахождение оксидаз, которое больше
других обратило на себя внимание исследователей. Некоторые высшие
грибы, особенно из родов Russula и Lactarius, обладают топ
замечательной особенностью, что они на изломе быстро чернеют от соприкос-
Оксидазы, или окислительные ферменты 449
новения с воздухом. В этих грибах Бертран и Буркело [21] нашли, что
так же, как в клубнях картофеля и в соке лакового дерева, здесь есть
еще два вещества, относящиеся друг к другу, как лакказа и лаккол.
Одно растворимо в воде, разрушается при нагревании, вызывает
посинение гуаяковой тинктуры и в то же время обладает способностью
окислять другое вещество, сообщая ему темную окраску. Последнее
растворимо в алкоголе и, вероятно, подобно лакколу, принадлежит к
классу многоатомных фенолов.
В предыдущем изложении мы для примера сравнительно подробно
описали несколько окислительных ферментов, встречающихся в
различных видах растений. Это именно те примеры, на которых развилось и
выросло самое учение об*оксидазах.
Во всех разобранных нами случаях наряду с ферментом мы
встречали всегда хромоген — вещество, способное окисляться при
совместном действии кислорода воздуха и оксидазы и принимающее при этом
резкую окраску, невольно обращающую на себя внимание
исследователя.
Но если бы окислительные ферменты органичивались в своем
распространении этими немногими случаями, мы, пожалуй, вправе были бы
смотреть на них, как на своеобразный курьез, быть может, лишенный
серьезного значения в экономии природы. Однако подробные
исследования, особенно последнего времени, показали, что оксидазы обладают
чрезвычайно широким распространением в природе и притом, как мы
увидим ниже, не только в растительном, но и в животном царстве.
Если испытать гуаяковой тинктурой целый ряд прорастающих
семян от самых разнообразных растений, то на всех разрезах мы
непременно получим резкое посинение. Точно так же, если ранней весной,
в период распускания листьев или немного раньше, мы выйдем в лес
или в поле, то вокруг нас буквально не найдется такого растения,
которое не показало бы на разрезе стебля более или менее ясную реакцию
на оксидазу. Этот факт был еще недавно отмечен Бертраном [20], и я
могу только подтвердить его на основании своих собственных наблюдений.
Повсеместное нахождение окислительных ферментов свидетельствует
о том, что они представляют отнюдь не случайное явление, что,
напротив того, присутствие их в организме связано с какой-либо существенной
физиологической функцией. При этом необходимо считаться с
возможностью следующих двух предположений. Или оксидазы сами по себе
играют определенную роль,— и тогда их присутствие не требует
дальнейших оправданий,— или, быть может, они являются только остатками,
так сказать намеками на существование других веществ, им
аналогичных, но с ними не тождественных и исполняющих в организме какую-
либо значительную функцию.
Кажется, факты говорят за то, что оба предположения верны и что
оксидазы, служа косвенным указанием на существование других, более
важных для организма ферментов, сами играют вполне определенную
роль в жизни организма.
Ниже мы постараемся дать доказательства в пользу такого мнения,
а пока обратимся к обзору тех успехов, которые достигнуты учением
об окислительных ферментах в области животного царства.
III
Хотя отрывочные наблюдения над окислительной способностью
некоторых жидкостей и органов животного организма известны уже
давно, систематические исследования в этом направлении начались только
29 Л. А. Чугасв, т. III
450
Оксидазы, или окислительные ферменты
с 80-х годов текущего столетия. Первые опыты принадлежат Шмидебер-
гу [11], который показал в 1881 г., что при пропускании через органы
(почки, легкие) крови, содержащей в растворе бензиловый алкоголь,
салициловый альдегид или бензиламин, вещества эти подвергались
постепенному окислению.
При этом бензиловый алкоголь С6Н5СН^ОН дает бензойную кисло-
,ОН
ту С6Н5СООН, салициловый альдегид СбН4Ч дает салициловую
хсон
/ОН
кислоту С6Н4Ч , а бензиламин С6Н5—СН2— NH2 сначала иод
хсоон
влиянием особого фермента переходит в бензиловый алкоголь (с
отделением NH3), а уже этот последний окисляется в бензойную кислоту.
Шмидеберг, кроме того, нашел, что кровь вне организма неспособна
окислять ни одного из только что перечисленных веществ, так что
источник окисления оставалось искать в том органе, через который кровь
пропускалась, а также сделал весьма вероятным предположение, что
органы способны проявлять свое действие и при нарушении их целости
(з измельченном состоянии).
Впоследствии Жаке [12] повторил и дополнил опыты Шмидеберга. Он
окончательно убедился в том, что окисление происходит при
пропускании крови через органы от особого фермента, заключающегося в этих
последних. Всевозможные воздействия, направленные к тому, чтобы
убить клеточки органа, как-то: замораживание, действие карболовой
кислоты, алкоголя, не говоря уже о механическом размельчении,— не
были ъ состоянии разрушить способности, присущей веществу органов
вызывать окисление бензилового алкоголя и салицилового альдегида.
Жаке удалось также приготовить раствор окислительного фермента в
воде, экстрагируя органы, -предварительно обработанные алкоголем, и
этот раствор, подобно самим органам, обладал способностью вызывать
окисление.
В качестве одного из аргументов в пользу ферментативной природы
окислительных процессов, возбуждаемых органами, может служить еще
и тот факт, также указанный Жа,ке, что органы теряют свою активность
после нагревания до 100°.
Еще раньше появления исследований Жаке Сальковский [22],
вопреки Шмидеберг у, нашел, что осадки могут быть консервативны
в крови, если только поставить опыт в особые условия, благоприятные
для реакции, а именно, если температуру поддерживать около 40° и
приводить все время кровь в мелкораздробленное состояние при
помощи пульверизатора.
Влияние температуры на интенсивность окисления, несомненно
вытекающее из вышеприведенных наблюдений, недавно было прослежено
количественно Абелус и Биарне [13]. Эти ученые исследовали быстроту
окисления салицилового альдегида в присутствии эмульсии из
селезенки при различных температурах. Оказалось, что интенсивность
окисления сначала повышается с температурой, достигает около 60° своего
максимума и затем быстро падает, обращаясь в нуль при температуре-
кипения воды.
Этот закон изменения скорости реакции, который может быть
иллюстрирован ходом кривой на приложенной диаграмме (рис. 1), чрез-
Оксидазы, или окислительные ферменты
45!
вычайно поучителен, потому что он является характерным для всех
неорганизованных ферментов *.
Для сравнения на другой таблице (рис. 2) .изображена кривая
действия диастаза на крахмальный клейстер, заимствованная из работы
Кьельдаля [23]; аналогия между двумя кривыми сама бросается в
глаза. Эта аналогия, без сомнения,
представляет одно из самых
веских оснований для того, чтобы
причислитьж'ивотныеок-
сидазы к числу
неорганизованных ферментов.
Is
"*.Я
**
? *
> *
§|
3*
56
kb
30
21)
W
О Ю 20 30 40 SO SO 70 8(Г 30 100 3 SO
Температура , °С Температура, °С
Рис. I. Рис. 2.
Кроме температуры, па интенсивность окисления, по опытам Абелус
и Биарие, оказывает заметное влияние и происхождение
оксида з ы.
Так, кроіпь различных животных обладает далеко не одинаковой
окислительной способностью. В общем, из целого ряда наблюдений
получился тот результат, что содержание окислительных ферментов в
крови значительнее у молодых животных, нежели у
старых. На-помним, что и для .растительных оксидаз мы имеем
аналогичное отношение.
Не менее интересны данные, полученные теми же авторами
относительно распределения оксидаз по различным органам одного .и того же
животного.
Оказалось, что окислительные ферменты совершенно отсутствуют н
мускулах и в мозгу3, тогда как известно, что именно в этих органах и
происходят самые энергичные процессы распада. Абелус и Биарие
видят в этом факте подтверждение теории Людвига и Ал. Шмидта,
согласно которой, по крайней мере в мускулах, совершается процесс,
напоминающий «анаэробное» брожение, продукты -которого уносятся
из места образования и подвергаются в других органах дальнейшему
окислению **.
С другой стороны, наиболее обильное содержание оксидазы было
констатировано в селезенке, за которой по порядку следуют легкие,
печень и щитовидная железа; остальные органы содержали оксидазу и
сравнительно небольших количествах.
К только что упомянутым исследованиям Абелус и Биарне близко
подходят работы Портье [24], выполненные отчасти совместно с Пьерн
* Для всех остальных реакций скорость непрерывно растет с повышением
температуры, если только реагирующие вещества устойчивы в условиях опыта.
** Заметим, однако, что в самое последнее время в мускулах найден энергичный-
гликолитический фермент (см. ниже) [25].
29*
452
Оксидазы, или окислительные ферменты
и касающиеся распределения іжсидаз з различных типах животного
царства.
Исследования Портье показали, что оксидазы широко
распространены среди животных, занимающих самые разнообразные ступени
иерархической лестницы от кишечнополостных до позвоночных. Этим
результатом подтверждается тот вывод, который мы сделали для растений,
что присутствие оксидаз в организме никоим образом не есть случайное
явление, наоборот, имеет глубокое основание в определенной, .присущей
им физиологической роли.
Из сопоставления своих результатов Портье делает еще одно
важное заключение, которое и приводит его к выяснению одного из
назначений оксидаз в организме. Он обратил (внимание на то обстоятельство,
что повсюду о с о б е н н о сильными окислительными
свойствами обладают лейкоциты. Так как почти все органы,
показывающие реакцию на оксидазы, содержат в большем или меньшем
количестве лейкоциты, то автор и полагает, что эти последние являются
единственными носителями окислительных ферментов и
что в каждом органе оксидазы содержатся постольку, поскольку в нем
находятся лейкоциты. С этой точки зрения понятно отмеченное нами
выше .значительное содержание оксидаз в селезенке и отсутствие их в
мозгу и в мускулах3,— в органах, в которых, как известно, лейкоциты
отсутствуют. Толкование, даваемое Портье такой локализации
окислительных ферментов, будет указано нами впоследствии.
Приведенными выше немногими примерами далеко не
исчерпывается все разнообразие тех окислительных процессов, которые происходят
под влиянием животных оксидаз.
К -процессу окисления бензилового алкоголя примыкает, по опытам
Поля, образование муравьиной кислоты из формальдегида в
присутствии вытяжки из печени.
Совершенно другого рода процессы, проходящие также под
влиянием окислительных ферментов, приводят к систематическому
образованию сложных красящих веществ путем окисления некоторых
ароматических аминов и фенолов.
Первый пример подобного рода реакций, указанный Вюрстером,
заключается в действии кислорода воздуха на ди- и тетраметилпарафени-
ленди амины.
Первоначально бесцветные растворы этих веществ в присутствии
вытяжек из органов, содержащих оксидазы (при нейтральной или
слабощелочной реакции), дают резкое синее окрашивание, которое и
является наглядным признаком происшедшего окисления.
Аналогичные реакции были позднее описаны Романом и Шпіитцером
[26]. К числу их относится образование и.н до фенолов, имеющее
место при взаимодействии эквимолекулярных количеств ароматического
диамина и фенола, например парафенилендиамина и а-нафтола.
Реакция, происходящая при этом, также сопровождается поглощением
кислорода, который может быть заимствован из какого-либо
соединения, легко его отдающего, и тогда реакция протекает быстро, иногда
почти мгновенно. Так, если к первоначально бесцветной смеси фенилен-
диамина и нафтола прибавить каплю раствора 'красной кровяной соли,
то жидкость немедленно .принимает темно-синюю окраску. Но окисление
может идти и на счет кислорода воздуха: тогда только оно протекает
несравненно медленнее, и требуются .целые дни для достижения тоге
самого эффекта, который под влиянием окислителей требует едва
заметного промежутка времени. П р и б авлени е к смеси раствора,
содержащего оксидазы или прямо эмульсии из органов, з н а-
ОксиОизы, или окислительные ферменты
453
чительно ускоряет процесс, и окрашивание наступает в этом
случае много раньше, нежели в пробе той же смеси, оставленной для
контроля.
В ту же категорию «синтезов при -помощи окисления» относится и
образование так называемых индаминов, которые происходят п,ра
действии друг на друга двух ароматических аминов. (Например, так
называемая зелень Биндшейдлера из диметиланилина и диметилпа-рафе-
нилендиамина.)
Относительно течения процесса и влияния на него окислительных
ферментов к индаминам относится все, что выше было сказано об
индофенолах.
Если до сих пор мы говорили почти только о таких реакциях,
возбуждаемых оксидазами, которые едва ли когда-нибудь имеют место в
живом организме, по крайней мере в нормальных условиях его
существования, то теперь нам 'предстоит коснуться процесса, несомненно,
играющего в организме чрезвычайно важную роль.
Лепин [15] первый доказал в 1890 г., что в крови находится особый
фермент, разрушающий глюкозу. Наблюдение это было затем
неоднократно подтверждено целым рядом исследователей (особенно Краусом
и Шпитцером), так что в достоверности его теперь едва ли возможно
сомневаться. Фермент Лепина, получивший название гликолитиче-
ского, представляет вещество чрезвычайно нестойкое, разрушающееся
уже при 55°. Наиболее энергичное действие он проявляет при 39°. Краус
показал, что при гликолизе поглощается кислород и освобождается С02.
а Шпитцер нашел, что реакция не происходит в отсутствие кислорода.
Таким образом, было доказано, что гликолитнческий фермент
представляет своеобразную О'ксмдазу. С другой стороны, целым рядом опытов
Лепина, Крауса [27], Шпитцер а [26] и др. было установлено, что
сыворотка крови не содержит фермента. Последний находится только в
форменных элементах, а именно в лейкоцитах. Гликолитнческий фермент
был найден также в тканях различных органов (Шпитцер) и может
быть извлечен оттуда, а равным образом, и из лейкоцитов, водным
раствором хлористого натрия. Этим обстоятельством, а также сохранением
гликолитических свойств крови, несмотря на прибавление ядовитых
веществ, убивающих форменные элементы, устраняется всякое участие
в гликолизе «чисто жизненных условий», и в то же время самый
процесс характеризуется как химическая реакция, вызываемая настоящим
неорганизованным ферментом.
Я позволю себе закончить обзор окислительных ферментов
животного происхождения одной крайне оригинальной разновидностью окси-
дазы, которая, по наблюдениям Р. Дюбуа [28], встречается и организме
некоторых животных (например, моллюска Pholas dactylus) и
обусловливает способность их светиться в темноте. Из светящихся органоз этих
животных автор выделил два вещества, из коих одно, растворимое
вводе, представляет фермент и обладает свойством разрушаться »іри
кипячении (люцифераза); другое (люциферин), растворимое в алкоголе,
способно окисляться под влиянием первого. В отдельности пи одно из этих
веществ не способно светиться, но стоит только слить вместе их
растворы, и тотчас появляется фосфорический свсг, ясно видимый в темноте.
Из целого ряда только что изложенных фактов нам кажется
возможным заключить, что животные оксидазы, подобно
растительным, обладают широким распространением в
АГЛ
Оксидазы, или окислительные ферменты
природе и способны возбуждать многочисленные и
разнообразные процессы окисления. При этом особенного
внимания заслуживает то обстоятельство, что среди этих процессов
находятся и такие, высокая важность физиологической р о-
л и которых является a priori весьма вероятной.
IV
В заключение нашего очерка нам остается еще остановиться ,на двух
вопросах 'первостепенной важности: на механизме окисления,
совершаемого оксидазами, и на их физиологической роли в организме.
Что касается до первого вопроса, то едва ли он может составить
затруднение ввиду ясной аналогии, существующей между оксидазами
и теми простейшими переносчиками кислорода, о которых мы уже
говорили.
По учению, ведущему начало от Траубе, оксидазы являются
соединениями, легко связывающими кислород и также легко его отдающими.
Именно это свойство и дает им возможность, подобно закиси марганца
или индиго, служить переносчиками кислорода. В пользу такой аналогии
особенно говорят новейшие наблюдения, которыми мы обязаны
Бертрану. Производя анализ золы, остающейся после сжигания лакказы,
этот исследователь'был поражен большим (до 2,5%) содержанием в ней
марганца, что и навело его на мысль, не играет ли марганец
существенной роли в химической функции оксидазы. Опыт показал, как мы
это уже видели выше, что соли закиси марганца4 и сами по себе могут
вызывать окисление гидрохинона и других многоатомных фенолов.
Из дальнейших исследований обнаружилось, что это окисление
еще усиливается при совместном действии
оксидазы и солей марганца. Есть -растения, например люцерна, окси-
даза которых очень бедна марганцем (менее 1/5000), и соответственно
этому окисляющая способность ее относительно слабая.
Последняя может быть, однако, значительно усилена прибавкой к
ферменту недостающего марганца. Вот цифры, взятые из работы
Бертрана, которые могут послужить прекрасной иллюстрацией «к сказанному:
Количество
поглощенного
кислорода, см9
Одна соль марганца . . 0,3
Одна оксидаза из люцерны 0,2
Оксидаза -|- соль марганца 6,3
Из целого ряда подобных опытов Бертран приходит к заключению,
что частица оксидазы слагается из двух элементов: из
минерального и органического. Первый обусловливает самую окислительную
функцию о.ксидазы; второй, по зсей вероятности, придает этой функции
определенное специфическое направление.
Теория Бертрана ,не только сближает простейшие неорганические
переносчики кислорода с оксидазами, но даже в известном смысле
прямо отождествляет их между собой, указывая на общие элементы
со-става тех и других, придающие их функции одинаковый характер.
Следует заметить, что мысль о том, что минеральные соединения
могут играть роль в процессах переноса кислорода, имеющих место в
организме, в принципе вовсе не новая.
Особенно в животном организме гемоглобин (содержащий железо)
уже давно невольно обращал на себя внимание своим своеобразным
Оксидазы, или окислительные ферменты
455
отношением к кислороду. Еще Траубе считал его идеальным
переносчиком .кислорода, хотя и не привел в пользу такого мнения прямых
доказательств. Позднее, однако, Гоппе-Зейлер [4] доказал, что кислород,
фиксированный гемоглобином, по своим окислительным свойствам
нисколько не .отличается от обыкновенного кислорода воздуха.
Под впечатлением такого отрицательного результата мысль о
возможном участии железа в прижизненном окислении надолго была
забыта. Позднее она опять вскользь была высказана Г. Бунге [29] в его
учебнике физиологической химии, и только недавно была развита более
подробно нашим соотечественником д-ром Сахаровым [30] по
поводу некоторых микроскопических реакций, которые ему приходилось
наблюдать в форменных элементах крови.
Наконец, в самое последнее время Шпитцеру [30] удалось
изолировать из органов ряд нуклеопротеидов *, содержащих железо (до 0,3%)
и обладающих способностью окислять салициловый альдегид и
вызывать синтезы индофенолов.
Если эти результаты подтвердятся, то, может быть, придется
признать за железом в животном организме роль, аналогичную той,
которую марганец играет в растительном.
Мы видим таким образом, что «минеральной» теории Бертрана,
вероятно, предстоит широкое применение в области учения об оксидазах.
Нам кажется, что в связи с некоторыми наблюдениями над другими
ферментами она обещает оказать .влияние и на еще -более обширный
класс явлений и послужить основанием для нового направления в эн-
зимологии, в которой на неорганические элементы до сих пор обращали
сравнительно мало внимания, считая их лишь бесполезными примесями
и не без некоторого пренебрежения соединяя их вместе под общим
названием «золы».
В применении собственно к окислительным ферментам теория
Бертрана имеет еще и то значение, что прямо указывает на источник, в
котором мы должны искать причину их ел е ц и ф и ч е с<ко г о х а р а к т е-
р а. Существование специфических отношений между ферментами м
теми веществами, на которые они способны действовать, как факт
известно уже давно, но особенно в последнее время оно было подчеркнуто
по поводу прекрасных исследований Э м. Фишера. По остроумному
замечанию этого ученого, между ферментом и веществом, на котором
проявляется его действие, существуют такие же отношения, как между
ключом и тем замком, для отпирания которого он предназначен.
Исследования последнего времени указывают на то, что оксидазы
различного происхождения не идентичны между собой и что,
по-видимому, между ними и веществами, на которые они способны переносить
кислород, имеет место такое же строго выдержанное соответствие,
какое существует и для всех остальных неорганизованных ферментов.
Так, Бертран нашел -в грибе Russula nigricans фермент, способный
вызвать окисление тирозина
/ОН NHo
С Н / ! "
хсн2-— с сорн
и названный лоэтому тирозиназой. Такой фермент был потом
констатирован еще только в крови ракообразных, а также в свекловице,
но его нет и следов в громадном большинстве других организмов. Зато
* Нуклеогьротеиды из различных органов показали далеко не "одинаковую степень
.активности.
456
Оксидазы, или окислительные ферменты
ферменты, заключающиеся в этих последних, вызывают другие
окислительные процессы, например, реагируют на гуаяковую тинктуру,
гидрохинон и другие многоатомные фенолы.
Подобным же образом, по опытам Поля [32], в животном организме
необходимо принять существование по крайней мере двух
окислительных ферментов, из которых один может вызывать только окисление
альдегидов в кислоты, а другой способствовать «синтетическому
окислению», которое приводит к образованию индофенолов, индаминон
и т. п. красящих веществ.
Не менее ясный пример специфичности оксидаз получается при
сопоставлении лакказы Бертрана с гликолитическим ферментом Лепина.
Из двух ферментов только последний обладает способностью окислять
глюкозу; лакказа, наоборот, лишена этой способности, как показали
специальные опыты, предпринятые в этом направлении Портье.
Итак, существование специфических отношений
между оксидазами и веществами, на которые он>и
действуют, можно считать доказанным, несмотря на
скудость относящегося сюда фактического материала, Но если причина
этого явления, действительно, лежит в природе органического радикала,
входящего в частицу фермента, то здесь открывается целое поле для
экспериментальных исследований.
Бертран показал, что можно «фабриковать» окислительные
ферменты, и теперь только дело времени-—обнаружить ту зависимость,
которая связывает состав и строение органических радикалов
«синтетических» ферментов с составом и строением веществ, могущих окисляться
под их влиянием *.
6 порядке нашего изложения нам остается еще выяснить, каково
физиологическое назначение тех о-ксидаз, которые нам
известны, и © каком отношении они стоят к іприжизненным процессам
окисления. При этом прежде всего необходимо иметь в виду тот факт,
что существуют весьма различные оксидазы. Мы займемся пока только
теми из них, которые наиболее изучены ,и типом которых может служить
лакказа Бертрана.
Целый ряд наблюдений, особенно из области растительной
физиологии, как будто говорит за то, что эти оксидазы не вызывают при жизни
организма именно тех процессов, для выполнения которых они,
«по-видимому, предназначены. Мы уже неоднократно указывали на то, что
обыкновенно в организме вместе с оксидазами встречаются и те
вещества, которые способны окисляться под их влиянием (многоатомные
фенолы, таннины и т. п.), но окислительный процесс, нередко
сопровождающийся изменениями цвета, наступает только на разрезе, на
пораненных местах того или другого органа и никогда не происходит при
полной неприкосновенности организма **.
С другой стороны, Пфеффер целым рядом опытов показал» что в
живой растительной клетке—ни в клеточном соке, ни в протоплазме —
нет даже таких сравнительно слабых окислителей, как перекись
водорода.
* Следует заметить, что трудность очистки оксидаз препятствовала до сих пор
определению их состава. Еще недавно Бертран считал, что они приближаются к
углеводам. В последнее время он считает их веществами альбуминозного характера.
** Следует, однако, заметить, что при жизни плазма никогда не смешивается с
клеточным соком и потому, если фермент содержится в первой, а хромоген в последнем,
то реакция между ними может и не обнаруживаться, особенно если продукты ее
быстро разрушаются по мере образования.
Оксидазы, или окислительные ферменты
457
Правда, отсюда еще нельзя заключить с достоверностью об
отсутствии в клетке окислительных ферментов, так как они вследствие
своего специфического характера могли и не проявить своего действия на те
реагенты (например, на цианин), которыми пользовался Пфеффер.
Однако, если и допустить, что оксидазы существуют в живом организме,
то вес же .роль их, как окислителей, может быть только крайне
ограниченной, и это именно потому, что большая часть оксидаз способна
окислять только .вещества, легко окислению поддающиеся. Поэтому в
лучшем случае оксидазы могут только дополнять действие других агентов,.
более энергичный эффект которых проявляется на окислении
соединений более стойкого характера.
С этой точки зрения широкое распространение оксидаз и особенно
то обстоятельство, что они обильнее всего встречаются в наиболее
энергично растущих органах, наталкіизает на мысль, не являются ли
оксидазы указанием на существование в организме особых, так сказать,
основных окислительных ферментов, более энергичных и более важных
в физиологическом отношении. А тот факт, что оксидазы становятся
особенно заметными при нарушении целости организма, заставляет
спросить себя дальше, не представляют ли они продуктов распада этих
основных ферментов, сохранивших окислительные свойства, но
утративших специфическую окраску.
Мне кажется, что в этом смысле можно провести некоторую
аналогию между отношением оксидаз обычного типа к нашим гипотетическим
ферментам и отношением обычных гидролитических ферментов
(диастаза, инвертина) к зим азе Бухнера.
Будущее покажет, не следует ли видеть в гликолитическом ферменте
Ленина первого -представителя этого нового типа ферментов. В пользу
возможности такого предположения говорит не только то
обстоятельство, что фермент Лепина окисляет сравнительно стойкое вещество, в то
же время столь свойственное организму,— глюкозу, но и меньшая
устойчивость его по отношению к высокой температуре, по сравнению с
обыкновенными оксидазадіи. И в этом отношении фермент Лепина
напоминает дрожжевую зимазу5.
Сам Лепин полагает, что .названный фермент и есть то самое
вещество, которое обусловливает окисление сахара в организме. Хотя это
мнение и оспаривается многими авторами, однако несомненно, что
существует какая-то внутренняя связь между интенсивностью распада
глюкозы в организме и содержанием в -крови гликолитического
фермента.
О существовании такой связи свидетельствует, между прочим, и тот
факг, приводимый Лепином, что глаколитическая способность крови
значительно «понижается при диабете, по сравнению с нормальным
состоянием организма, а также целым рядом других заболеваний
(pneumonia, uraemia, obesitas etc).
При значительном числе наблюдений едва ли можно здесь говорить
о простой случайности.
Главное возражение против взглядов Лепина, несомненно,
заключается в том, что процессы окисления в организме почти исключительна
совершаются в органах .и всего меньше -в крови. Таково, по крайней
мере, учение, общепризнанное в современной физиологии. Между тем,
по опытам Лепина и других, именно кровь обладает наиболее сильно
выраженной гликолигической способностью. Мне .кажется, однако, что
наблюдения над гликолизом вовсе не требуют, чтобы окисление
совершалось непременно в крови. Дело в том, что в органах при нарушении
их целости могут создаваться условия, влекущие за собой быстрое
458
Оксидазы, или окислительные ферменты
разрушение фермента, подобно тому, как это наблюдал Бухнер для своей
зимазы. С другой стороны, весьма возможно и то, что .в органах
фермент не накопляется без нужды в больших количествах, а постоянно
образуется по мере надобности, сообразно с количеством подлежащего
окислению сахара. И это .предположение имеет за собой также
многочисленные аналогии. По совершенно понятно, что при. таких условиях
количественное сравнение гликолитической способности органов и
крови не может дать ответа на вопрос о том, где 'Преимущественно
происходит окисление. Наконец, необходимо иметь в виду и то, что в
органах обеспечена несравненно большая поверхность соприкосновения
окисляемого вещества с кислородом воздуха, нежели в ,крови, и таким
образом интенсивность посмертного окисления, когда структура органа
нарушена, не может дать количественного понятия об .интенсивности
того же явления, «имеющего место при жизни организма. Ввиду
изложенных соображений мне кажется преждевременным категорически
высказываться против возможности участия гликолитического фермента
в прижизненном окислении глюкозы. По аналогии скорее можно было
бы ожидать, что фермент Лепина оправдает возлагаемые на него
надежды.
Итак, хотя наши современные сведения об оксидазах и дают почву
для многих заманчивых соображений, они все же не разрешают
основного вопроса о природе окислительных процессов в живом организме —
и здесь пока что мы не можем идти дальше одних предположений и
догадок.
Но теперь является другой вопрос. Если оксидазы (по крайней мерс
большинство :из них) играют подчиненную роль в явлениях горения (а
может быть, и вовсе в них не участвуют), то не предназначена ли им
какая-либо другая, если и более скромная, то, во всяком случае,
определенная роль в экономии природы.
Этот вопрос оставался открытым до самого последнего времени, и
всего два года тому .назад была сделана попытка его разрешить.
Изучая условия распространения окислительных ферментов, Портье
обратил внимание на следующие факты:
1) Оксидазы всегда находятся (в изобилии в молодых и нежных
органах, легко подверженных ранениям и другим вредным влияниям
извне.
2) Вместе с оксидазами обыкновенно встречаются вещества,
способные окисляться под их влиянием; окислительный процесс происходит
только при поранении организма и ведет (почти всегда к образованию
нерастворимого соединения, покрывающего поверхность раны как бы
коркой, предохраняющей ее от дальнейших повреждений.
Сопоставление этих фактов привело Портье к убеждению, что оксидазы
представляют своеобразное приспособление организма, предназначенное для
защиты -его от вредных внешних влияний.
С этим выводом согласуется указанное нами .выше наблюдение того
же автора, в силу которого оксидазы у животных сосредоточены
главным образом (а может быть, даже и исключительно) в лейкоцитах,
этих «существах», предназначенных для борьбы со всем тем, что
посягает на целость и безопасность организма.
В последнее время профессор Мечников [33] указывает на
возможную роль окислительных ферментов, заключающихся в лейкоцитах,
при разрушении токсинов бактериального происхождения, имеющем
место в организме во время борьбы его с заразным началом; ученик же
его Без редка [34] склонен приписывать такую же роль оксидазам
лейкоцитов при разрушении некоторых минеральных ядов.
Оксидазы, или окислительные ферменты
459
Если это предположение подтвердится, то мы будем иметь
замечательный случай деятельности лейкоцитов, в котором оксидазы игэают
роль пассивного, но могущественного орудия борьбы с заразой .и с
ядами на химической почве.
Мы в.идели, что учение об окислительных ферментах получило свое
начало благодаря стремлению понять процесс «внутреннего горения»
живых организмов, проникнуть в механизм этой грандиозной топки,
дающей организованной машине жизнь и движение.
Но, благодаря удивительному сплетению и многообразию тех путей,
которыми природа следует для выполнения своих целей, детище Трау-
бе совершенно неожиданно, далеко еще не исполнив своей задачи,
уклонилось в сторону, чтобы найти себе .применение в сфере борьбы за
существование. Дойдет ли оно .когда-нибудь до своей конечной цели?
Ответ на этот вопрос принадлежит будущему.
ЛИТЕРАТУРА*
1. A. L. L a v о і s і е г et A. S 6 g u i n. Sur la transpiration des animaux. Paris, 1790.
Cp. Oeuvres de Lavoisier. Paris, 1865.
2. E. F r a n k 1 a n d. Ann. Chem., 1849, 71, 213.
3. C.F. Sch onb ein. Z. Biologic, 1869, 1; J. pract. Chcmie, 1858, 75, 73.
4. F. Hoppe-Seyler. Med.-chem. Untersuchungen, I, S. 133; PHuger's Archiv 1878,
19; Z. physiolog. Chemie, 1878, 2, 149, 325, 427.
5. M.Traube. Ber. Chem., 1877, 10, 1984; 1882, 15, 222, 659, 2421, 2434, 2325, 2854;
1883, 16, 123, 463, 1201.
6. J. Reinke. Botanische Zeilung, 1883, 40, № 5—6. 30, 31, 57.
7. W. Pfeffer. Abh. mat. phys. klasse Kg. Sachs. Ges. Wiss., 1889, 15.
fi. Th.B okorny. Ber. Chem., 1888, 21, 1848; Pringsheim's Jahrbucher. Wiss. Bot.,
17, 347.
9. M.Traube. Theorie- der Fermentwirkungen. Berlin, 1858.
10. G.Bertrand. Bull. Soc. chim. de Paris, 1894 [3], 11, 674, 717.
11. Schmiedeberg. Archiv exp. Pathol., 1881, 14, 288, 379.
12. Jaguet. Archiv exp. Pethol., 29, 386; Compt. rend. Soc. Ыюі. 1892.
13. Abelous et Biarnes. Archives de physiolog., (5), 1894, 6, 591; 1895, 7, 195, 239;
1896, 8, 311; 1897, 9, 277; Compt. rend. Soc. biol., 1897.
14. P о г t i e r. Les oxydases dans la serie animale (these de doctorat). Paris, 1897.
15. R. Lepine et Barral. Compt. rend, de Г Acad. Sci., Paris, 1891, 112, 146, 411, 604;
189b ИЗ, 118, 729; R. Lepine et Metroz, Ibid., 1893, 177, 154; R. Lepine. Ibid.
1895, 120, 139.
16. A.Williamson. Ann. Chem., 1851, 77, 37; 1852, 81, 73.
17. G.Bertrand. Bull. Soc. chim., 1897, (3) 17, 753.
18. Цит. по Loew. The energy of the living protoplasm. London, 1896, p. 102.
19. Hikorckuro J о s h i d a. J. chem. Soc, 1883, 43, 472.
20. G.Bertrand. Arch, de physiol., 1896, (5), 8. Ann. chimie et phys., 1897, 12, 115.
21. С E. Bourquelot et С Bert rand. Compt. rend. Soc. biol., 1875; 1896; Сотр.
rend, de Г Acad., 1895, 121, 783; С. Е. Bourquelot. Compt. rend. Soc. biol., 1896.
22. E. Salkowsky. Z. physiol. Chem., 1882, 7, 115. Centr. f. medic. Wiss., 30, 849.
23. L.Kjeidahl. Meddeleser fra Carlsberg iaboratorie. 1879, № 2, 139.
24. Portier. loc. cit. P i e r i, P о г t i e r. Arch, de physiol. 1897, (5), 9, 60.
25. Lander, Brunton, Rhodes. Centr. f. Physiologie, 12, 353.
26. F. Rohmann, W. S p i t z e r. Ber., 1894, 27; W. S p i t z e r. Pflugers Archiv, 1895,
60, 303.
27. Kraus. Z. klin. Medicin., 21.
28. R.Dubois. Lecons de physiologie generale et comparee. Paris, 1898, p. 515.
29. G. В u n g e. Lehrbuch der physiolog. und pathol. Chemie. Leipzig, 1894, S. 257.
30. Г.П.Сахаров. Русский архив патологии проф. Подвысоцкого, 1897, 4, 38.
31. W.Spitzer. Berlin. KHn. Woch., 1898, № 37, 814.
32. A. Poehl. Archiv exp. Pathol., 38, 65.
33. іИ. Меч ников. Ann. Inst. Pasteur, 1898, 12, 263.
34. A.M. Безредка. Ann. Inst. Pasteur. 1898, 12, 305; 1899, 13, 49, 209, 465.
К ФИЗИОЛОГИИ ФОСФОРЕСЦИРУЮЩИХ БАКТЕРИЙ
(из химической лаборатории бактериологического института
Московского университета)
С таблицей микрофотографий
Русский архив патологии, клинической медицины
и бактериологии, 1900, 10, 352
1
Настоящая работа, имеющая целью пополнить некоторые пробелы
в наших знаниях относительно фосфоресцирующих микробов,
ближайшим образам касается следующих двух вопросов: отношения роста и
жизнедеятельности этих микробов к растворам солей и некоторых
явлений симбиоза фосфоресцирующих бактерий с другими микробами в
связи с условиями питания тех и других.
Фосфоресцирующие бактерии были открыты немецким физиологом
Пфлюгером в 1875 г. [1]. До того времени целый ряд явлений, так
называемого самосвечения, наблюдаемых в природе и нередко
представляющих необыкновенно красивое и эффектное зрелище, как-то:
свечение моря, фосфоресценция рыбы, мяса, иногда даже живых людей —
представляли совершенную загадку. На суеверную толпу явления эти
наводили ужас, зачастую порождая обвинения .в -колдовстве; ученых
исследователей они повергали в полное недоумение, заставляя делать
самые невероятные догадки относительно их происхождения.
Для того, чтобы дать хотя приблизительное понятие о тех
фантастических представлениях [2], которые возникали об этом предмете даже в
самых светлых умах прежнего времени, достаточно будет, например,
указать на то, что Аристотель приписывает свечение .моря содержа
нию соли в морской воде. Декарт видел причину этого явления в
особых вихрях, возникающих при волнении моря, Франклин объяснял
его электризацией .моря при трении о движущийся корабль или лодку,
а Роб. Бойль, этот трезвый мыслитель и замечательно точный
экспериментатор, после долгих бесплодных попыток раскрыть причину свечения
моря, хотел ее видеть в «каком-то неведомом космическом воздействии,
исходящем от земли или по крайней мере от нашей планетной системы»
После работ Пфлюгера свечение моря и аналогичные явления
фосфоресценции получили простое объяснение. Носителями зародышей
фосфоресцирующих бактерий являются морские рыбы. Пойманная и
сохраняемая на холоду такая рыба нередко через несколько дней
начинает светиться, и по всей поверхности ее тела, покрытой слизью,
разрастается богатая культура, фосфоресцирующих бактерий. Эту
светящуюся слизь можно смыть в морскую (или просто соленую) воду и
последняя получает тогда сама способность светиться. Очевидно, тот
же самый процесс происходит и в море, где всегда есть' много мертвой
рыбы и где, следовательно, постоянно существуют условия,
благоприятные для развития светящихся микробов.
К физиологии фосфоресцирующих бактерий
461
Мы имеем также основание предполагать, что и все остальные виды
свечения (например, свечение мяса и пр.), по всей вероятности,
объясняются инфекцией, ведущей начало от фосфоресцирующей рыбы*.
Первый фосфоресцирующий микроб, открытый Пфлюгером, не был
выделен :им в чистой культуре. Эта задача была выполнена несколько
позднее Людвигом, который и дал новому микробу название
Micrococcus Pfliigeri [3].
Впоследствии целым рядом других исследователей [4], между
которыми имена Фишера и Бейеринка [5] заслуживают особого
внимания, было выделено большое число фосфоресцирующих микробов,
главным образом из морской рыбы іи морской воды.
В настоящее время описано их более 20 видов морского
происхождения, хотя далеко не о всех можно сказать, что существование их
доказано с достаточной убедительностью.
Все фосфоресцирующие бактерии этой категории в морфологическом
отношении представляют значительное однообразие: по большей части
это короткие подвижные палочки, «из которых одни приближаются по
форме к коккам, у других, наоборот, длина заметно превосходит
ширину. Первые соответствуют типу В act. phosphorescens, вторые — типу
bacillus phosphorescens. Этог 'морфологический принцип является пока
единственным безупречным основанием классификации
фосфоресцирующих бактер.ий.
В биологическом отношении фосфоресцирующие бактерии морского
происхождения отличаются двумя характерными признаками:
во-первых, они являются -строгими аэроба.ми: их развитие и в особенности
свечение возможно только в 'Присутствии кислорода. По Бейеринку, они
являются даже одним из самых чувствительных реактивов на кислород.
Самый -акт свечения обусловливается, .по всей вероятности, особым
окислительным процессом, происходящим внутри бактериальной клетки.
Ближайшая природа этого процесса до сих пор остается неизвестной**.
Другая особенность этих микробов заключается в том, что они
требуют для своего развития сравнительно высокой концентрации соли в
питательной среде. Об этом предмете еще будет идти речь ниже.
Что ^касается до условий питания фосфоресцирующих бактерий, то
в этом отношении мы имеем замечательные исследования Бейеринка
[6], согласно которым среди светящихся бактерий наблюдаются два
различных типа литания.
Одни бактерии (Photobact. phosphorescens и Photobact. Pfliigeri)
требуют для своего развития и для успешного свечения двух органических
веществ: одного источника азота (пептон) и одного источника
углерода, в качестве которого лучше всего может служить глюкоза.
Другие бактерии (Photobact. balticum., Photobact. Fischeri, Photobact.
indicum и Photobact. luminosum) довольствуются одним органическим
соединением, а именно пептоном, который служит одновременно источ-
ником азота и углерода. В отличие от предыдущей, эта группа
фосфоресцирующих бактерий обладает способностью «пептонизировать белки
и, следовательно, разжижать желатину.
Наконец, следует сказать еще иеоколько слов об особой категории
фосфоресцирующих микробов, а именно о светящихся вибрионах,
выделенных из воды Эльбы во время Гамбургской холерной эпидемии
* Исключение представляет свечение гнилого дерева. По исследованиям Людвига
и др., причиной его являются особые грибки, один из которых получил недавно Кучер
в чистой культуре.
** Ср. мою статью «Оксидазы, или окислительные ферменты». Русск. архив
патологии за 1899 г. (Из-бр. труды, т. III, стр. 441 и далее. Прим. ред.).
462 К физиологии фосфоресцирующих бактерий
[7]. В морфологическом и биологическом отношении микробы эти
совершенно .напоминают холерный вибрион и даже являются патогенными
для морской свинки, как я мог в этом убедиться из личного опыта. И в
этом отношении фосфоресцирующие вибрионы стоят совершенно
особняком; все остальные светящиеся бактерии (кроме одного вида,
открытого Жкардом и патогенного для морских раков) совершенно лишены
болезнетворных свойств *.
После этих предварительных замечаний переходим к предмету
нашего исследования.
I
Как мы уже упоминали выше, фосфоресцирующие бактерии,
существование которых тесно связано с морем и с соленой морокой .водой,
требуют для своего развития и успешного проявления характерной для
них способности свечения присутствия -в питательной среде (все равно,
жидкой или твердой) значительных количеств (от 3 до 4%) ловареиноіЧ
или морской соли. На обыкновенных питательных средах, бедных
солями, эти м-икрабы показывают едва заметные следы .роста и совершенно
не дают свечения. Даже готовые, ярко светящиеся культуры быстро
теряют способность к свечению при разбавлении обыкновенной
стерилизованной водой.
гНе подлежит сомнению, что эта, отмеченная еще Пфлюгером,
характерная биологическая особенность светящихся бактерий морского
происхождения, или галофилия **, находится в тесной связи с
условиями их существования и -влияет одним из многочисленных случаев
.приспособления организма к окружающей среде (соленой воды морей).
В пользу такого заключения, между прочим, говорит и то
обстоятельство, что фосфоресцирующие микробы, выделенные из
обыкновенной речной воды, например описанные Дюнбаром и Кучером вибрионы»
не только не требуют для своего развития высокой концентрации соли
в питательных средах, но, наоборот, как показали мне специально
предпринятые в этом направлении опыты, -погибают на тех и.менно
субстратах, которые являются наиболее благоприятными для остальных гало-
фильных фосфоресцирующих бактерий. Таким образом, не подле-
жит сомнению, что самый акт фосфоресценции не стоит ни в какой
необходимой связи с галофилией.
В моих исследованиях я обратил внимание на вопрос о механизме
явлений галофилии и на изучение ближайших условий этого
своеобразного приспособления.
Естественно, в самом деле, спросить себя, в чем же заключается та
роль, которую играет в жизнедеятельности фосфоресцирующих бактерий
высокая концентрация соли в питательном субстрате. Очевидно, мы
можем себе представить две возможности: или хлористый натрий
действует на организм бактерий как таковой—и мы, следовательно, имеем
дело с явлением чисто химическим, или действие соли определяется
только осмотическими свойствами раствора и не зависит от ее состава;
оно будет тогда, по существу, физическим. Опыт должен был решить
в пользу того или другого предположения.
* Это обстоятельство может иметь значение на практике при санитарной оценке
светящейся ірыбы или мяса. Принимая в расчет,- что свечение всегда имеет место до
наступления процесса гнилостного разложения органического субстрата, тго-в-идимому,
следует признать фосфоресцирующие пищевые продукты вполне безвредными.
** Галофильными мы будем называть для сокращения таких микробов, которые
требуют высокой концентрации солей (а .может быть, и других соединений) в
питательной среде, подобно тому, как, например, микробы термофильные требуют для своего
развития высокой температуры.
Л' фіЩисуногиа фодфоресцирукщих бактерий 463
В качестве объекта я остановился на Photobact. phosphorescent
Beyerink^ бактерии,; свечение которой является, особенно ярким*.
Для тЬг.о, чтобы дать понять о световод-эффекте, которого можно
достигнуть с этим микробом,: укажу, что яри свете 1—2-суточных
желатиновых культур в темной 'Ком-нате^яегко можно читать крупный шрифт
и определять время По карманным часам. На .приложенной таблице
№ 1 изображается автофотогфафия нескольких ' культур, снятых в темной
комнате гіри помощ^их собственного света **.
Предварительные опыты показали, что эта бактерия прекрасно
растет и развивается на рыбьем бульоне с 3%-ного хлористого* натрия, а
также на среде нижеследующего состава:
Пептона . .
Аспарагина .
К2НР04 . .
NaCl ....
. 1000,0
. 10,0
5,0
. ¦ 1,0*
30,0
*- Среда должна иметь слабощелочную реакцию; при 22° свечении
наступает на другой или на третий день после посева.
В нижеприведенных опытах я и воспользовался этими двумя
средами, причем в каждой из них взамен хлористого натрия в отдельных
порциях прибавлялись различные другие соли с таким расчетом, чтобы
полученные растворы были изотоничны с 3%-ным раствором
поваренной соли. Привожу список испытанных мною соединений:
1. Морская соль;
2/Хлористый калин КС1;
3. Бромистый калий КВг;
4. Йодистый Калий KJ;
5. Азотнокислый калий KN03;
6. Азотнокислый натрий NaN03;
7. Уксуснокислый калий СНзСООК;
8. Хлористый рубидий RbCl;
9. Бромистый натрий NaBr;
10. Хлориоватокислый натрий №С10з;.
11. Хлориоватокислый калий КСЮз;.
12. Уксуснокислый натрий CH3COONa;
13. Азотнокислый аммоний NH4NO3.
* Микрофотография № 2 представляет эту бактерию (с желатинной" культуры),
снятую при помощи фотографического аппарата системы Лейтца.
** Позволяю себе сказать здесь несколько слов о многочисленных и очень
эффектных демонстрациях, которые могут быть воспроизведены в Photobact. phpsphorescens:
1) написав на желатине, разлитой и застывшей на ровной поверхности, буквы или
слова эмульсией из желатинных ' или агарных разводок Photobact. phosphorescens
и оставив при обыкновенной температуре на 1 или 2 суток, получаем надпись, ярко
светящуюся в темной комнате и видимую на далекое расстояние. 2) Бульонная
культура, налитая "в 'стаканчик или. лучше, в высокий цилиндр, через несколько минут
темнеет во всей своей массе вследствие поглощения кислорода бактериями, но стоит
только ее привести в движение (например, помешивая стеклянной палочкой), и струн
света постепенно распространяются вниз, пока вся жидкость не превратится в одну
светящуюся массу. 3) Быстрое потемнение жидкой культуры достигается еще
пропусканием тока H2S, встряхиванием с эфиром или хлороформом, прибавлением кислоты
или щелочи, формальдегида, гидроксиламина и других дезинфецирующих веществ.
Следует заметить, что все среды для фосфоресцирующих бактерий лучше всего (хотя
не обязательно, ом. в тексте) готовить из мяса рыбы (например, судака), причем рецепт
приготовления тот же, что и для мясного бульона (resp. агара и желатины), только
мясо заменяется ірыбой и .прибавляется 3% поваренной соли или морской соли.
Реакция должна быть слабощелочная. Небесполезно прибавление 7<— 7г7о аспарагина.
ч •, •
/ — автофогограмма культуры Photobacterium. phosphor esc ens, снятая в темной
комнате при экспозиции в 1 сутки; а — агарная культура; б, в и г — желатинные;
2 — микрофотография с Photobact. phosphorescens; 3—ауксанограмма Photobact.
phosphorescens, появившаяся под влиянием роста Sachar. membrane)'aciens;
снимок сделан при дневном свете; 4 — ауксанограмма Photobact. phosphorescens,
вызванная диффузией крепкого раствора глюкозы; снята при дневном свете;
5 — автофотограмма Photobact. phosphorescens, воз-никшая под влиянием роста
Sachar. ellipsoides. Снята в темной комнате при экспозиции в 1 сутки
К физиологии фосфоресцирующих бактерий
465
При^ выборе этих веществ я имел в виду, главным образом
определить, имеет ли какое-либо отношение к жизнедеятельности
фосфоресцирующих микробов индивидуальная природа ионов хлористого
натрия.
С этой целью я взял несколько солей, отличающихся от NaCl только
своим анионом. Сюда относятся: NaN03, NaBr, NaC103, CH3COONa;
затем две соли, отличающиеся от NaCl только катионом: КС1 и RbCl и,
наконец, ряд солей, не имеющих ни одного общего иона с хлористым
натрием: КВг, KJ, KN03, СН3СООК, КСЮ3, NH4N03.
Все питательные субстраты, приготовленные «согласно
вышесказанному, были распределены в Эрленмейерощские колбочки по 50 см5 в
каждую и после стерилизации заражались 'суточной культурой
Photobact. phosphorescens на рыбьем бульоне (по 3 капли на колбочку).
Для контроля было оставлено -в каждой серии опытов по две
колбочки, содержащие соответственные питательные среды с прибавкой
только 3% NaCl.
Все колбочки были поставлены в термостат при 22° и подвергались
исследованию сначала ежедневно, а потом через более
продолжительные промежутки времени в общем в течение трех месяцев. При этом
каждый раз наблюдался, во-первых, рост, насколько о нем можно было
судить по микроскопическому виду культуры, и, во-вторых,
производилось в абсолютно темной комнате оптическое исследование. При этом
необходимо непосредственно перед наблюдением хорошенько
встряхнуть испытуемые культурные жидкости, чтобы привести все части их
в соприкосновение с кислородом воздуха. Затем .колбочки помещаются
в ряд и определяется относительная интенсивность их свечения:
.распределив все серии на несколько групп, на основании такого
«предварительного наблюдения можно приступить к детальному сравнению колбочек,
составляющих каждую из таких групп. В конце концов определяется
степень свечения каждой колбочки по сравнению с контрольной и
отмечается порядок убывающей интенсивности света*.
Таким образом были произведены две серии опытов (с рыбьим
бульоном и іс искусственной средой вышеуказанного состава) по два раза
каждая.
Результат в обоих случаях получился один и тот же. Если
исключить из вышеприведенного списка веществ, употребленных взамен
хлористого натрия, две соли, а именно KJ и КО, обладающие, несомненно,
ядовитыми свойствами, как я имел в этом случай убедиться, то можно
сказать, что все остальные соли в большей или меньшей степени
способствуют росту Photobact. phosphorescens и поддерживают феномен
свечения. Сильнее всех и почти одинаково с хлористым натрием
действуют NaBr, KBr, NaN03f K.N03, Ш, КС103, NaC103. Слабее: NH4N03,
СН3СООК и СНзСООЫа **. На питательных средах без прибавки солей
микроб не развивается и свечение не имеет места, совершенно так же,
как и на растворах KJ и RbCl. Таким образом, факты не позволяют
сомневаться в том, что роль солей ,в явлениях фосфоресценции
обусловливается на первом плане осмотическими свойствами их -растворов, а
не их .химической природой, и поставленный нами вопрос разрешается
в определенном смысле.
' * Перед наблюдением необходимо пробыть несколько минут в темной комнате,
чтобы глаз успел отдохнуть и привыкнуть к распознаванию малых колебаний в
степени интенсивности света; лучше всего производить наблюдения вечером.
** Причина неодинаковой интенсивности действия этих двух групп солей, вероятно,
зависит от различной степени проницаемости протоплазмы бактерий для тех и других;
ближайшим изучением этого интересного вопроса я занят в настоящее время.
30 Л. А. Чугаев, т. Ш
466
Л' физиологии фосфоресцирующих бактерий
Что касается до самого -механизма действия солей на
жизнедеятельность фосфоресцирующих бактерий, то «ад-о думать, что мы имеем здесь
дело с теми условиями существования всякой живой 'Протоплазмы,
нарушение которых, как известно, ведет к явлениям плазмолизма *,
недавно изученным в .применении ,к бактериям Альфредом Фишером [8].
А если так, то характер действия раствора той или другой
(индифферентной) соли известной концентрации на фосфоресцирующую
бактерию будет определяться тем, изотоничен ли данный раствор с той
жидкостью, которая содержится в плазме бактерии. Если такой
изотонии не существует, то неизбежно наступает нарушение равновесия,
проявляющееся в изменении нормального объема содержимого клетки, а
также, по всей вероятности, в нарушении правильного течения
прижизненных химических процессов**.
II
Как мы уже указывали, Бейеринк в своих исследованиях по
биологии фосфоресцирующих бактерий отметил, между прочим, тот факт, что
Photobact. phosphorescens принадлежит к микробам, требующим для
успешного развития и полного проявления светящей способности, кроме
•пептона, -еще особого источника углерода, лучше всего в форме
углеводов.
Некоторые явления симбиоза фосфоресцирующих бактерий с
другими микробами, а также результаты вышеописанных опытов над
влиянием концентрации солей на явления фосфоресценции навели меня на
мысль о том, не существует ли аналогичной зависимости и между
концентрацией -сахара в питательной среде и интенсивностью
жизнедеятельности Photobact. phosphorescens. Опыты, предпринятые в этом
направлении, вполне подтвердили такое ожидание. Один из целого ряда
подобных опытов я позволю себе привести здесь в качестве иллюстрации
in extens.
Ряд колбочек, содержащих по 50 смъ раствора следующего состава ***:
Пептон .... 1,0%
К2НР04 ... 0,1%
Морская соль . 3,5%
NaOH до
слабощелочной реакции,
разделен на шесть серий по три в каждой. К колбочкам первой серии
прибавлено 0,1% глюкозы, ко второй — 0,2%, к третьей — 0,5%, к
четвертой— 1%, к пятой — 2% ,и к шестой — 0,25% аспараги-на. Три
колбочки оставлены были для контроля. 8.IV. 1899 г. был сделан засев по
3 капли бульонной культуры Photobact. phosph. в каждую колбочку.
Кроме контрольных колбочек, которые давали лишь едва заметный свет,
все остальные -в большей или меньшей степени засветились уже на
вторые сутки.
9. IV. 1899 г. Лучше всех светились колбочки I серии с 0,1%, II —
0,2% и III —0,5% глюкозы.
Слабее светились колбочки IV 'серии с 1% глюкозы и VI серии с
0,25% аспарагина; еще слабее — колбочки V серии с 2% глюкозы
* А в некоторых определенных условиях к не менее гибельному пла-смоптизу,
только что открытому тем же А. Фишером (Zeitsch. f. Hyg., 1900 г.).
** Существование таких нарушений не представится нам невозможным, если мы
вспомним, как относятся к растворам индифферентных солей многие белковые
вещества (особенно так называемые глобулины).
*** Для приготовления этого раствора была взята обыкновенная вода из
водопровода
К физиологии фосфоресцирующих бактерий
467
10. IV. 1899 г. Лучше всех светят уже колбочки VI серии с 0,25%
аспарагина, слабее—1 серии (0,1% глюкозы) и затем с убывающей
силой колбочки остальных серий до V — 2% глюкозы, свет от -которой
едва заметен.
Наконец, 13. IV. 1899 г. Лучше всех светят опять-таки колбочки VI
серии с аспарагином, значительно слабее — колбочки I серии с 0,1%
глюкозы.
Все же остальные колбочки о-к о нчательно потемнели.
Микроскопическое "исследование культур показало, что энергия роста
микробов, насколько можно было судить, идет .приблизительно параллельно
с силой свечения. Мы видим, таким образом, что жизнедеятельность
фосфоресцирующих микробов повышается в своей интенсивности лишь
при малом содержании .глюкозы в питательной среде. Наиболее
благоприятно действует, -по-видимому, 0,1%. При -концентрациях выше 1%
уже через сутки проявляется неблагоприятный эффект прибавки сахара.
С другой стороны, чем больше концентрация сахара, тем скорее слабеет
и совсем прекращается свечение. Легко показать, при помощи
лакмусовой бумажки, что это последнее явление стоит в связи с переменой
реакции субстрата из слабощелочной в кислую *. Очевидно, Photobact.
phosph., подобно многим другим микробам, образует кислоту из
глюкозы, и эта кислота играет роль ядовитого агента.
Те же явления, но только еще в более резкой форме, наблюдаются
и на твердых питательных субстратах. Здесь их легко
иллюстрировать весьма наглядно, если но обыкновенной питательной рыбьей
-желатине, зараженной зародышами Ph. phosph. и разлитой в чашку Петри,
провести черту крепким, например 20%-ным, раствором глюкозы. Тогда
через 2—3 суток мы увидим в темной комнате следующую картину**:
темный овал, соответствующий зоне с наибольшей концентрацией
глюкозы и ограничивающий его светлый ореол ярких .колоний,
выступающий на слабо освещенном фоне. Очевидно, этот ореол соответствует
образовавшейся вследствие диффузии зоне -с оптимальной для
жизнедеятельности бактерии концентрацией глюкозы. Светящемуся ореолу,
видимому в темноте, соответствует аналогичная кривая (ауксаног.рамма,
по Бейеринку) обильно разросшихся колоний, видимая на свету.
Картины, совершенно аналогичные только что описанным,
наблюдаются и -при симбиозе Bact. phosphorescens с другими .микробами и
именно с теми, которые способны разрушать глюкозу. В простейшем
случае, когда мы имеем дело с микробами, разрушающими сахар, но
не образующими из него сколько-нибудь значительных количеств
кислот,— к таким микробам принадлежат, например, дрожжи,—
получается наиболее жрная и эффектная картина. Рыбная желатина с 3,5%
морской соли и 1% глюкозы, засеянная большим количеством
зародышей Photobact. phosphorescens, разливается, как и в предыдущем опыте,
в чашку Петри и по застывшей поверхности проводится какой-нибудь
рисунок, например крест, разводкой Sachar. Mipsoides или Sachar. mem-
branefaciens (другие виды не были .мною испытаны). Через 2—3 суток
в темной комнате мы увидим яркий светящийся крест
(соответствующий месту разрастания дрожжей) на совершенно темном фоне***.
В другом случае, когда мы -имеем дело с микробом, разрушающим
глюкозу и образующим из нее кислоту, картина еще усложняется по-
* После подщелаадвания светящая .способность вновь восстанавливается, хотя
обыкновенно уже не с прежней интенсивностью.
** -На таблице фотография № 4 представляет снимок, сделанный ври дневном свете.
*** Фотография № 5 на таблице соответствует Sachar. ellips. и снята при
собственном свете ?той бактерии в темной комнате. Фотография № 3 представляет снимок
Sachar. membranefac, снятый при дневном свете (ауксанограмма).
30*
468
Л физиологии фосфоресцирующих бактерий
явлением черного креста посреди светящейся фигуры. По мере того; как
кислота, образующая бактерию, разрастается, черный крест постепенно
утолщается, а контуры светлой зоны отодвигаются все дальше и дальше
к периферии. Очевидно, что центральная часть креста является темной
вследствие накопления в ней кислоты, которая «постепенно
диффундирует в окружающую среду и таким образам расширяет область,
негодную для жизни Photobact. phosph.; что же касается до светлого ореола,
то он соответствует зоне, в которой содержание глюкозы понизилось под
влиянием диффузии в центральную часть и стало оптимальным, между
тем как кислота еще не успела туда проникнуть в достаточном
количестве. Из числа микробов, дающих только что описанную картину,
мною были исследованы Bact. Friedlaenderi и Bact. coli commune. С
последним опыт удается только при сравнительно небольшом (от 0,2 до
0.5%) содержании глюкозы в желатине. Так как тифозная палочка,
посеянная при тех же условиях на пластинчатую разводку Photobact.
phosph., не дает никакой светящейся фигуры *, то мы имеем здесь
эффективный и сравнительно простой в исполнении способ распознавания
Bact. typhi abd. от Bact. coli commune, микробов, которые, как
известно играют весьма важную роль в медицинской бактериологии.
ЛИТЕРАТУРА
l.E.Pfl tiger. Die Phosphoreseenz verwesender Organismen. Arch. ges. Physiologic
1875, 11, 222.
2. C.Bernoulli. Ueber das Leuchten des Meeres. Gottingen, 1803.
3. F.Ludwig. Micrococcus Pfltigeri. Hedwigia, 1884, № 3. Цит. по статье того же
автора в Ceritr. Bact, В. 2, № 13, 14, 1887, 2, 372.
4. B.Fischer. Ueber einen lichtentwickelnden im Meerwasser gefundenen Spaltrul;
Z. f. Hyg., 1887, 2, 54.
Ueber amen neuen lichtentwickelnden Bacillus. Centr. Bact., 1887, 3, 105, 137.
Bakterienwachstum bei 0°. Centr. Bact. 1888, 4, 89.
Die Bakterien des Meeres nach den Untersuchungen der Planktonexpedition. Kiel u.
Leipzig, 1894.
O. Katz. Zur Kenntniss der Leichtbakterien. Centr. Bact., 1891, 9, 157.
C.Eijkmann. Centr. Bact., 1892, 12, 656.
I.Foster. Centr. Bakt., 1886, 1892, 2, № 12; 12, 431.
G i а г d. Sur I'infection phosphorescence etc. Paris, 1889.
Также Soc. biol., 19 oct. 1889.
5. M. W. Beyerink. Les bacteries Iumineuses. Arch. Neerl., 1889, 23, 416; Le photobact.
luminosum. Ibid., 1889, 23, 401; Over lichtroedsel en plastisch voedsel van lich. bacte-
rien. Medd. konink. Akad. Wettens. 1890, (2), 7. Цитировано по реф. в Centr. Bakt.
6. M. W. В е у e r i n k, loc. cit.
7. Kutscher. Zur Phosphoreseenz der Eldvibrionen. Centr. Bakt., 1900, 18, 424.
Ein Beitrag zur Kenntniss der den Choleravibrionen ahnlichen Wasserbakterien.
Deutsche med. Woch, 1893, № 49.
8. A.Fischer. Die Plasmolyse der Bakterien. Ber. Sachs, kon. Ges. Wiss. Math.-Phys.
Kiasse, 1891, 43, 52; Vorlesungen uber Bakterien. Berlin, 1897; Untersuchungen iiber
Bakterien. Prinsheim's Jahrbucherwiss. Bota, 1895, 27, -1.
* Как известно, Bact. typhi abdom. гораздо менее энергично, нежели Bact. coli com'.,
перебраживает сахар с образованием кислоты.
ПРИМЕЧАНИЯ
Эволюция вещества в мертвой и живой природе
1 К стр. 30. Это не совсем точно: электроны нельзя считать веществом катода;
сии отщепляются от атомов веществе, из которых состоит катод.
2 К стр. 31. Исследования ряда ученых показали ошибочность опытов Рамзая
о возможности превращения эманации в неон, аргон и литий.
Периодическая система химических элементов
1 К стр. 56. В настоящее время доказано, что первоначально учение об атомах и
молекулах было создано М. В. Ломоносовым (1741—1760 гг.), но оно не было понято
и принято учеными того времени. В начале XIX века понятие об атоме было вновь
внесено в химию, конкретизировано Д. Дальтоном и получило общее признание.
2 К стр. 65. За единицу атомного веса до последнего времени принималась или
1/16 веса атома легкого изотопа кислорода-16 (физическая шкала) или 1/16 среднего
атомного зеса изотопов кислорода, равного по физической шкале 16,0044 (химическая
шкала). В 1961 г. Комиссия по атомным весам Международного союза чистой и
прикладкой химии приняла решение о переходе на новую шкалу атомных весов,
основанную на изотопе углерода-12. Основой относительных атомных масс (атомных весов)
служит Vi2 массы углерода-12. Необходимость в отдельных физической и
химической шкалах отпадает.
3 К стр. 71. Более точно: в одной грамм-молекуле содержится 6,023* 10й3
молекул (число Авогадро).
4 К стр. 84. Вопрос о степени сложности высших соединений кислорода в
настоящее время не вполне выяснен.
5 іК стр. 84. За последующие годы количество известных свободных радикалоз
значительно увеличилось. Так, в 1929 г. Ф. Пакетом был открыт свободный радакал
СН3. Далее был получен СН3—СН2, СНг. Свободные радикалы обладают большой
реакционней способностью, быстро соединяются друг с другом, так что существуют
в свободном состоянии только тысячные доли секунды. В настоящее время все яснее
становится значение радикалов при протекании химических реакций. Подробнее см.
А. Е. Чичи бабин. Основные начала органической химии, т. 1. М., 1954.
6 К стр. 85. Глаза II представляет в настоящее время в значительной степени
только исторический интерес.
7 К стр. 89. В настоящее время показано, что окись фтора F20 получается
медленным пропусканием фтора в 2%-ный раствор NaOH. Реакция идет, по-видимому,
по уравнению: 2F2+2NaOH=2NaF+H20+F20. Окись фтора сгущается при —145е
в интенсивно окрашенную желтую жидкость, затвердевает при —224°. При действии
тихого электрического разряда на смесь фтора с кислородом при температуре жидкого
воздуха получаются оранжевые кристаллы F2O2.
8 К стр. 95. В связи с изложенным различаем строения соединений типа Ва02 и
типа РЬ02 эти последние (PbQ*, Мр02) называются двуокисями, а не перекисями.
Перекиеным соединениям приписывается следующее строение
іЬ"[:6:6:]"Л
470 Примечания
9 .К стр. 101. Вернее, Вг20 крайне непрочное вещество, J20 — неизвестно.
10 'К стр. 105. В настоящее время доказано существование положительных
электронов (<поэитронав), однако положительно заряженные ионы нельзя рассматривать
как продукты присоединения позитрона к атому элемента.
11 К стр. 112. В настоящее время переходными элементами называются элементы,
атомы которых имеют незаконченные оболочки.
12 К стр. 114. Соединение висмута с водородом, открытое Пгнетом в 1918—4919 гг.,
представляет собой ядовитый газ, неустойчивый даже при комнатной температуре.
13 К стр. 118. В современной периодической системе в крайнем левом углу
помещается элемент франций Fr, атомный вес 223, порядковый номер 87 (открыт Перей,
1939 г.).
14 К стр. 155. В настоящее время, кроме указанных в тексте, известна еще черная
окись серебра AgO, которая получается путем анодного окисления Ag или действием
перманганата на Ag20 в щелочной среде.
15 К сгр. 160. Для меди известны еще соединения, отвечающие трехвалентной
меди. CU2O3 «красный порошок», при 400е' переходящий полностью в СиО. Со щелочами
образует нестойкие купраты Me[Cu(OH)J, где Me обозначает щелочной металл.
Известна также перекись меди Си02-
1в К стр. 161. Окиси AgO серебра отвечает двухфтористое серебро AgF2. Оно
представляет собой коричнево-черное вещество, плавящееся около 690е.
17 -К стр. 170. Л. А. Чугаев помещает церий и торий в IV группу, как это делал
Менделеев. В настоящее время церий относят к III группе к семейству лантанидов,
торий также выделяется в особое семейство актинидов. К четвертой группе
периодической системы относится также гафний Hf, открытый после смерти Л. А. Чугаева
(1923 г.).
18 К стр. 171. О трехвалентном углероде см. А. Е. Ч и ч и б а б и н. Основные
начала органической химии, т. 1. М., 1954, стр. 124.
19 К стр. 173. В настоящее время известны соединения пятивалентного висмута.
отвечающие солям метависмутовой кислоты NaBiOs и КВіОз- При обработке этих
солей HNO3 выделяется свободная метазисмутовая кислота НВіОз- Известен и ангидрид
этой кислоты ВігОб-
20 К стр. 176. Кроме ТаХб, известны низшие валентные формы этого элемента:
ТаСЦ, ТаСЬ, 2ТаСЦ • ТаОСі • ЗН20, при действии щелочей на ТаСі осаждается
зеленый Та(0Н)3. Термическим разложением ТаСЬ при 5О0—'600° может быть получен
ТаС12.
21 К стр. 177. Уран в современной периодической системе некоторыми авторами
включается в группу так называемых актинидов.
22 К стр. 177. В современных курсах по органической химии этому соединению
приписывается следующее строение:
>0-»Н
СН3Х
+
сі-
Оксояиевые соединения рассматриваются как продукты замещения атомов водорода
в ионе оксония НзО+.
23 К стр. 181. В 1940 г. Сегре и его сотрудниками был получен в результате
бомбардировки висмута ускоренными сс-частицами новый элемент VII группы астат
(№ 85), At. Вскоре изотопы астата были найдены и в лродуктах естественного распад г
радиоактивных элементов. Все изотопы астата весьма неустойчивы, период
полураспада самого долгоживущего =8,3 часа. По химическим свойствам астат сходен
с-галогенами, но обнаруживает и некоторые металлические свойства.
24 К стр. 185. В настоящее время, кроме марганца, в VII группе периодической
системы известны элемент технеций Тс (43) и рений Re (75). Технеций был впервые
Примечания
471
(1937 г.) получен Сегре и Перье путем бомбардировки ядер молибдена дейтронами.
По химическим свойствам технеций является аналогом марганца и рения.
25 К стр. 186. К группе благородных газов относится также радон Rn № 86
(прежнее название: нитон, эманация радия).
26 К стр. 187. В природе благородные газы, действительно, не образуют
химических соединений, но при высоком давлении (до 150 атм) они соединяются с водой,
давая гидраты (Аг6Н20). Гіри низких температурах благородные газы образуют
твердые растворы с гидридами.: НгБ, НС1, а также с S02l СО2 н т. д..
27 К стр. 187. По современным данным, содержание криптона = 0,0001, ксенона —
0,000006, радона —б-Ю-18.
28 К стр. 188. В современной периодической системе Си, Л# п Аи помещаются
только в 1-й группе.
29 К стр.. 189. О кислотных свойствах Os04 смотри Л. А. Ч у г а е в. Избранные
труды, т. I, стр. 592.
30 К стр. 189. В настоящее время установлены две модификации для четырехоки-
си осмия — а и р.
31 К. стр. 190. В современных справочниках приводятся следующие температуры
плавления: для Ru 2450°, Rh 1966°, Pd 1555", Os 2500°, Ir 2454°, Pt 1773°.
32 К стр. 190. Теория Абегта и Бодлендера в настоящее время устарела.
33 'К стр. 194. Для радона в 1936 г. (Б. А. Никитиным) был:і открыты соединения
молекулярного состава Rn6H20; Rn2C6H5OH.
34 К стр. 203. В современной периодической системе на этом месте находится
элемент рений (Re).
2Г К стр. 204. Все известные радиоактивные элементы в настоящее время хорошо
укладываются в периодическую систему элементов. В отличие от формы
периодической системы, которую дает Л. А. Чугаев, все редкоземельные элементы принято
помещать в III группе в 6-м периоде в одной клетке, а элементы, начиная от актиния,
в одной клетке 7-го периода той же III группы. Плеяда редкоземельных элементов
.названа лантанидами, плеяда тяжелых элементов — актинидами.
Вант-Гофф и судьбы стереохимии
1 К стр. 213. Было бы правильнее сказать, что А. М. Бутлеров не занимался
специально вопросом стереохимии. Однако он определенно протестовал против попыток
считать пространственное расположение атомов принципиально непознаваемым.
2 К стр. 218. О работах Г юн см. т. II настоящего'издания, стр. 355.
Валентность
1 К стр. 222. Статья «Валентность» представляет собой изложение основ учения
о валентности. По своему содержанию статья преназначена для преподавателей
средней школы, студентов первых курсов, интересующихся историей химии. Для лиц,
желающих познакомиться с современным состоянием учения о валентности, можно
рекомендовать университетские курсы общей и физической химии и соответствующие статьи
в БСЭ.
2 К стр. 222. В настоящее время считается, что учение об атомах и молекулах
было впервые введено в химию М. В. Ломоносовым, но его идеи не получили широкой
известности в науке.
3 JK стр. 231. По существующим взглядам, в трифенилметиле углерод надо признать
трехвалентным. См. например, А. Е. Чичибабин. «Основные начала органической
химии». М., 1954, т. 1. стр., 12!4.
4 'К стр. 236. О взглядах А. Е. Чичибабина по этому вопросу см. А. Е. Ч и ч<иб а-
б и н. Исследования по вопросу о трехвалентном углероде. М., 1912.
5 К стр. 255. Нитон (Nt) в настоящее время называется радоном (Rn).
6 К стр. 256. В некоторых соединениях серебро проявляет высшую валентность
472
Примечания
(двух- и трехвалентно), но соединения эти неустойчивы и научены мало. См. прим.
12 к стр. 259 этой статьи.
7 К стр. 256. В настоящее время считается, что бор в таких соединениях
трехвалентен. Для бороводородов принимается еще наличие водородной связи, получаются,
таким образом, цепи:
Н
/ \
\ -'' V
в в
8 К стр. 256. В современной периодической системе церий относят к III группе.
Этот элемент начинает семействе лантанидоз.
9 К стр. 257. Сульфоксиловой кислоте чаще -приписывают удвоенную формулу:
H4S2O4. Известна окись серы S202, отвечающая следующей структурной формуле:
0=S
o=s
10 .К стр. 257. Оксониевые соединения в настоящее время рассматриваются как
продукты замещения атомов водорода в ионе оксония НзО+:
СНз\ "¦ +
>о-*н
си/
сі-
11 К стр. 259. В современной периодической системе последним элементом I группы
является франций (Fr). Свойства его покз слабо -изучены. Бго природный изотоп
Fr223 является продуктом а-распада актиния-227.
12 іК стр. 259. Для меди .известно еще соединение Си20з (кристаллическое вещество
красного цвета). Получается окислением при действии перекиси водорода на
охлажденный раствор Си (ОН) 2 в едком натре. Для серебра в настоящее время, кроме Ag20,
получена окись AgO; кроме того, некоторыми исследователями предполагается
существование Ag40, Agi03 и Ag203.
13 К стр. 260. Церий в современной периодической системе помещается в III группе
в семействе лантанидов. Торий выделяется в семейство актинидов. Для кремния в
настоящее время известно более бедное кислородом соединение SiO, получаемое
восстановлением кремнезема Si02 при высоких температурах (1500°).
14 К стр. 260. После олова в IV группе современной периодической оистемы
помещается гафний. Он не упоминается у Л. А. Чугаева, так как был открыт в 1923 г.
О свойствах этого элемента см. учебники химии.
15 К стр. 260. Висмут образует с водородом мало устойчивое соединение ВіНз.
16 К стр. 261. Уран в современной периодической системе некоторыми авторами
выделяется в особое семейство актинидов.
17 К-стр. 261. Кроме перечисленных Л. А. Чугаявым элементов, к седьмой группе
периодической системы принадлежат открытые в последнее время элементы: технеции
(Тс) (43), рений (Re) (75), астат (At) (85). Оіб их свойствах см. современные
учебник* химии и соответствующие статьи в БСЭ.
18 К стр. 261. Для фтора 'Известно сюедщнеяие F20, бедаэ&здый газ с температурой
плавления —145е и F2O2 — оранжево-красные кристаллы, плавятся при температуре
—163° и превращаются э газ при -—57°. При —50° распадается.
Прш&ечания
473
Профессор Альфред Вернер
1 К стр. 267. Теория химического строения была введена в науку и разработана
А. М. Бутлеровым и приписывать честь ее открытия также Куперу и Кекуле является
ошибкой.
2 К стр. 268. О существовании позитронов во времена Л. А. Чугаева известно не
было. Они были открыты в 1932 г.
3 К стр. 274. Соединения с пятью молекулами аммиака были открыты Л. А. Чу-
гаевым в 1915 г. Электропроводность хлорпентамминхлорида Чугаева оказалась
равной 404.
Структурно- и стереохимические представления в области
неорганической химии
! К стр. 285. Встречающиеся, в этой статье Л. А. Чугаева названия происходят от
цвета солей кобальта и переносятся на весь тип комплексных солей различных
металлов, имеющих строение, аналогичное дайной соли кобальта. Названия эти выходят
в настоящее время из употребления в химической литературе. Их формулы приведены
в I томе" избранных трудов Л. А. Чугаева, стр. 634—635 (М., 1954 г.).
О неустойчивых органических соединениях
1 К стр. 333. Согласно современным представлениям, формулы а- и (З-глюкозидоа
изображаются таким образом:
СН..О-С-Н Н-С--ОСН»
н-с-он \ н-с-он \
но-с-н Хо но-с-н о
і / I /
н-с-он / н—с—он /
н_с-он н-с-он
CH2OH CHoOH
а-глюкозид р-глюкозид
[o]D-1-158,9° [a]D~34,2-
2 К стр. 334. По современным представлениям, формулы а- и р-глюкоз имеют
следующий вид:
II.,-
но—с—н
/\
/ \
н-с-он \
! \
{.НО—С-И 0 ;;
1 /
Н-С-ОН /
\ /
\/
Н—С
і
сн.он
Н—С(ОН,)
н-с-он
> но-с-н
н—с—он
н-с-он
СВ.ОН
Н-С—ОН
\
Н-С-ОН \
— НО-С-Н
і
Н -C-OH /
н-с
СН2ОН
О
474
Примечания
Открытие кислорода и теория горения
1 К стр. 373. Согласно древним китайским философским теориям, л окружающих
нас предметах существуют две стороны, противостоящие друг другу: одна из них
называется «ян», а другая — «ин». Конкретные значения «ян» и «ин» разные по разным
авторам. В данном случае .китайский ученый Мао-хоа обозначает термином «ин»
кислород.
Оксидазы, или окислительные ферменты *
1 К стр. 441. В 1899 г. Л. А. Чугаеву, который тогда работал в
бактериологической лаборатории Габричевского, было предложено написать обзорную статью по
вопросу об окислительных процессах, протекающих © организме. Эта статья под
названием «Оксидазы, или окислительные ферменты» была напечатана в 1899 г. в № 7
Русского а-рхива латологии.
2 К стр. 445. В настоящее время красящие вещества, лежащие в основе реакции
переноса кровью .кислорода, известны под названием гемов (железопорфириновых
соединений). К темам близки гистогематины или цитохромы, которые находятся в
клетках растений, животных, дрожжей аэробных бактерий и некоторых факультативных
анаэробов. На эти каталитические агенты, играющие роль в окислительных процессах,
было обращено особое внимание в течение последних 10—15 лет.
3 К стр. 452. Утверждение об отсутствии окислительных ферментов в мускулах
и в мозгу в настоящее время не считается соответствующим действительности.
4 К стр. 454. Следует отметить роль металлов (марганца, железа и др.) в переносе
кислорода; этому вопросу посвящены работы Бертрана, который в преклонном возрасте
продолжал эти исследования.
5 К стр. 457. Интересно, что Л. А. Чугаев, говоря об этом ферменте, указывает на
возможную близость между гликолитическим ферментом и дрожжевой зимазой. Этот
взгляд соответствует современному с той оговоркой, что в настоящее время
принимают, что как гликолиз, так и брожение сахара, вызываемое дрожжами, представляют
собой слоисные процессы, вызываемые комплексами ферментов.
6 К стр. 459. Приводим список современной литературы по данному вопросу.
Гюббенет Е. Р. Растение л хлорофилл. М.—-Л., Изд-во АН СССР, 1951.
Кретович В. Л. Основы биохимии растений. М., «Советская наука», 1956.
М их лин Д. М. Биологическое окисление. М, Изд-во АН СССР, 1956.
Пастер Луи. Исследования о брожениях. М., Сельхозгиз. 1937.
Дыхательные ферменты. Перевод с англ. под ред. В. А. Энгельгардта. М., ИЛ, 1952.
* Примечания к статье сделаны проф. Г. Л. Селибером.
СПИСОК ОПУБЛИКОВАННЫХ ТРУДОВ Л. А. ЧУГАЕВА
1. Uber Trimethyldiioxyglutarsaure. Ber. Dtsch. chem. Ges., 1895, Jg. 28, 2940—2942.
Студенческая работа (Совместно с Н. Д. Зелинским).
2. Об оптической деятельности эфиров ментола. Сообщение на 62-м заседании
Отделения химии Общества любителей естествознания, антропологии и этнографии
26.11.1897 г. ЖРФХО, 1897, 29, отд. 2, вып. 5, 122-123.
3. О закономерных изменениях молекулярного вращения. Сообщение на 65-м
заседании Отщеления химии Общества любителей естествознания, антропологии и
этнографии 29.Х.1897 г. ЖРФХО, 1897, 29, отд. 2, 227.
4. О действии ядов на микроорганизмы. Русский архив патологии, клинической
медицины и бактериологии (Под ред. В. В. Подвысоцкого), 1897, 4, вып. 2, 151—173.
5. К теории дезинфицирующего действия. Сообщение на 68-м заседании Отделения
химии Общества любителей естествознания, .антропологии « этнографии 11.11.1898 г.
ЖРФХО, 1898, 30, отд. 2, 18.
6. Об оптической деятельности эфиров борнеола. Сообщение на 70-м заседании
Отделения химии Общества любителей естествознания, антропологии я этнографии
5.IV.-1898 г. ЖРФХО, 1898, 30, отд. 2, вып. 3, 51.
7. Об условиях изменения молекулярного вращения. Сообщение на 72-м заседании
Отделения химии Общества любителей естествознания, антропологии и
этнографии 13.V.I898 г. ЖРФХО, 1898, 30, отд. 2, вып. 7, 188—189.
8. Молекулярное вращение и теория ассоциации жидкостей. Доклад на заседании
секции химии X съезда русских естествоиспытателей и врачей в Киеве
29.VIII.1898 г. ЖРФХО, 1898, 30, отд. 2, вып. 8, 217, 218.
9. Новая теория дезинфицирующего действия. Русский архив патологии, клинической
медицины и бактериологии (Под ред. В. В. Подвысоцкого), 1898, 5, вып. 2, 222—
234. (Критический обзор).
10. Untersuchungen uber optische Activitat. I. Mitteilung. Ber. Dtsch. chem. Ges., 1898,
Jg. 31, 360—368.
11. Untersuchungen tiber optische Activitat. II. Mitteilung. Ber. Dtsch. chem. Ges., 1898,
Jg. 31, 1775—1782.
12. Cber den Einfluss ider Aassocia'tion der FiMissigkeiten auf das optische Drehungsver-
mogen derselben. Ber. Dtsch. chem. Ges., 1898, Jg. 31, 2451—2454.
13. Новый способ получения непредельных углеводородов. 'Сообщение на заседании
Отделения химии РФХО, 4.XI.1899 г. ЖРФХО, 1899, 31, вып. 9, 959—961.
14. О переходе карвона в лимонен. Сообщение на заседании Отделения химии РФХО,
4.XI.1899 г. ЖРФХО, Л899, 31, вып. 9, 961—962.
15. О действии щавелевого эфира на 'ментол. Сообщение на заседании Отделения химии
РФХО 2.XII.1899 г. ЖРФХО, 1899, 31, вып. 9, 981, 982. (Совместно с А. М.
Щербина).
16. Оксидазы, или окислительные ферменты. Русский архив патологии, клинической
медицины и бактериологии (под ред. В. В. Подвысоцкого), 1899,7, вып. 3, 322—341.
(Критический обзор).
17. Uber eine neue Methode zur Darstellung ungesattigter Kohlenwasserstoffe. Ber. Dtsch.
chem. Ges., 1899, Jg. 32, 3332—3335.
18. О ксантогеновом методе получения непредельных углеводородов и о его
приложении к химии терпенов. Сообщение на 84-м заседании Отделения химии Общества
любителей естествознания, антропологии и этнографии 22.IV.1900 г. ЖРФХО, 1900,
32, отд. 2, вып. 5, 79.
19* О ксантогеновом методе получения непредельных углеводородов и о его
приложении к химии терпенов. Сообщение, представленное для соискания премии В. М. Мош-
нина гна 85-м заседании Отделения химии Общества любителей естествознания,
антропологии и этяапрафяи 6.V-1900 г. ЖРФХО, 1900, 32, вып. 5, 358—360.
20 О туйоне, іновом бициклическом терпене. Сообщение на заседании Отделения химии
РФХО, 4.V.1900 г. ЖРФХО, 1900, 32, вып. 5, 358—360.
21. О камфене из борнеола. Сообщение на заседании Отделения химии РФХО
4.V. 1900 г. ЖРФХО, 1900, 32, вып. 5, 360—362.
22. О фенхене из фенхилового алкоголя (предварительная заметка). Сообщение на
заседании Отделения химии РФХО, 4.V.1900 г. ЖРФХО, 1900, 32, вып. 5, 362-363.
476 Список опубликованных трудов Л. А. Чугаева
23. О новом камфене из бо.рнеола. Сообщение на 85-м заседании Отделения х.имни
Общества любителей естествознания, антропологии и этнографии 6.V.1900 г. ЖРФХО,
1900, 32, отд. 2, вып. 8, 145.
24. Четыре работы об оптической деятельности органических соединений. Сочинение,
представленное для соискания премии В. М. Мошнина, на 85-м заседании
Общества любителей естествознания, антропологии и этнографии 6.V.1900 г. ЖРФХО,
1900, 32, отд. 2, вып. 8, 145.
25. О действии ядов на микроорганизмы. Сочинение, представленное для соискания
премии В. М. Мошнина на 85-м заседании Отделения химии Общества любителей
естествознания, антропологии и этнографии 6.V.1900 г. ЖРФХО, 1900, 32, отд. 2,
вып. 8, і145.
26. О строении борнилена. Сообщение на 86-м заседании Отделения химии Общества
любителей естествознания, антропологии и этнографии 25.IX. 1900 г. ЖРФХО, 1900,
32, отд. 2, вып. 9, 161.
27. О получении оптически деятельных модификаций борнеола в чистом виде.
Сообщение на 86-м заседании Отделения химии Общества любителей естествознания,
антропологии и этнографии 25.IX.1900 г. ЖРФХО, 1900, 32, отд. 2, вып. 9, 161.
28. О новом удобном способе очистки оптически деятельных бориеолов. Сообщение на
заседании Отделения химии РФХО 5.Х.1900 г. ЖРФХО, 1900, 32, вып. 8, 654, 655
29. О борнилене. Сообщение на заседании Отделения химии РФХО 5.Х.1900 г.
ЖРФХО, 1900, 32, вып. 8, 653—654.
30. Новая реакция железа и гидроксиламина. Сообщение на заседании Отделения
химии РФХО 2.XI.1900 г. ЖРФХО, 1900, 32, вып. 9, 754.
31. О растворимости надхромовой кислоты в органических растворителях. Сообщение
на заседании Отделения химии 2.XI.1900 г. ЖРФХО, 1900, 32, вып. 9, 754, 755.
32. О растворимости надхромовой кислоты в органических растворителях. Сообщение
на 86-м заседании Отделения химии Общества любителей естествознания,
антропологии и этнографии 25.IX.1900 г. ЖР'ФХО, Л900, 32, отд. 2, вып. 9, 163.
33. О тркболюминиоценида. Предварительное сообщение на заседании Отделения химии
РФХО 7.ХИЛ900 г. ЖРФХО, 1900, 32, вып. 9, 837—839.
34. К физиологии фосфоресцирующих бактерий. СПб, 1900. 12 стр. Отд. оттиск из
¦ журн. Русский архив патологии, «клинической мр-оицины и бактериологии (Под ред.
В. В. (Подвысоцкого), 1900, 10, 352; см. также в Сб. речей и докладов,
посвященных-памяти Л. А. Чугаева. Л., НХТИ. 1924, стр. 117—121.
35. О некоторых реакциях холестерина. Русский архив патологии, клинической
медицины и бактериологии (Под ред. В. В. Подвысоцкого), 1900, 9, вып. 3, 289, 290.
¦ 36. Uber die Umwandung von Carvon in Limonen. Ber. Dtsch. chem. Ges., 1900, Jg. 33,
735—736.
37. Uber das Thujen, ein neues dicyklisches terpen. Ber. Dtsch. chem. Ges., 1900, Jg. 33,
3118—3126.
38. Аналитические заметки. Сообщение на 88-м заседании Отделения химии
Общества любителей естествознания, антропологии и этнографии 9.XI.1900 г. ЖРФХО,
1901, 33, отд. 2, вып. 19.
39. О трИ'болюминищеици-и. Сообщение на 89-м заседании Отделения химии Общества
любителей естествознания, антропологии н этнографии 9.XII.1900 г. ЖРФХО, 1901,
33, отд. 2, вып. 2, 20.
40. О переходе огг туйилами.на к ту Иону. Сообщение на заседании Отделения химии
РФХО 3.V.1901, ЖРФХО, 1901, 33, вып. 5, 371, 372.
41. О некоторых превращениях туйиламина. Сообщение на 94-м заседании Отделения
химии Общества любителей естествознания, антропологии и этнографии 3.V.1901 г.
ЖРФХО, 1901, 33, отд. 2, вып. 7, стр. 115.
42. О действии борного ангидрида на алкоголи терпенового ряда. Сообщение на
заседании Отделения химии РФХО, 13.IX.190I. ЖРФХО, 1901, 33, вып. 7, 526, 527.
43. Uber Tribolumineszenz. Ber. Dtsch. chem. Ges., 1901, Jg. 34, 1820—-1825.
44. Uberfiihrung von Thuiylamin in Thyien. Ber, Dtsch. chem. Ges., 1901, Jr. 34, 2276—
2281.
45. О различных способах получения ментена. Сообщение на 97-м заседании
Отделения химии Общества любителей естествознания, антропологии и этнографии
29.XI.1901 г. ЖРФХО, 1902, 34, отд. 2, вып. 1, 35.
46. Заметка по поводу предлагаемого бактериального населения нефти. Сообщение
на 98-м заседании Отделения химии Общества любителей естествознания,
антропологии и этнографии 28.11.1902 г. ЖРФХО, 1902, 34, отд. 2, вып. 5, 109.
47. К вопросу об оптической деятельности органических соединений. Сообщение на
заседании Отделения химии РФХО, 9.V.1902 г. ЖРФХО, 1902, 35, вып. 5, 529.
48. О ксантогенамидах терпенового ряда. Сообщение на заседании Отделения химии
РФХО, 9.V.1902. ЖРФХО, 1902, 34, вып. 5, 529-532.
49. О новом классе окрашенных соединений ксантогенового ряда. Сообщение на
заседании Отделения химии РФХО, 9.V.1902 г. ЖРФХО, 1902, 34, вып. 5, 532, 533.
Список опубликованных трудов Л. А. Чугаева
477
50. Заметка о новой цветной реакции на тиомочевины и тиоамиды. Сообщение на
заседании Отделения химии РФХО, 9.V.1902 г. ЖРФХО, 1902, 34, вып. 5, 533, 534.
51. О новом классе окрашенных соединений ксаитогенового ряда. Сообщение на 100-м
заседании Отделения химии Общества любителей естествознания, антропологии
и этнографии 2.V.1902. ЖРФХО, 1902, 34, отд. 2, вып. 6, 121.
52. О ксантогеновых амидах. Сообщение на 100-м заседании Отделения химии
Общества любителей естествознания, антропологии и этнографии 2.V.1902 г. ЖРФХО,
1902, 34, отд. 2, вып. 6, 121.
53. К вопросу об оптической деятельности органических соединений. ЖРФХО, ч. хим.,
1902, 34, вып. 6, 606—622.
54. Об 1,10-дикетоиах. Сообщение на 101-м заседании Отделения химии Общества
любителей естествознания, антропологии и этнографии 16.V.1902 г. ЖРФХО, 1902, 34,
отд. 2, вып. 7, 142.
55. Применение органо-металлических соединений магния для анализа. Сообщение на
заседании Отделения химии РФХО, 12.IX.1902 г. ЖРФХО, 1902,34, вып. 7. 652—654.
56. О нитрозохлоридах. Сообщение на заседании Отделения химии РФХО, 7.XI.1902 г.
ЖРФХО, 1902, 34, вып. 9, 851—853.
57. Удобный способ для распознавания борнеола от изоборнеола. Сообщение на
заседании Отделения химии РФХО, 7.XI.1902. ЖРФХО, 1902, 34, вып. 9, 853, 854.
58. Некоторые данные о дериватах туйона. Сообщение на заседании Отделения химии
РФХО, 7.XI.1902 г. ЖРФХО, 1902, 34, вып. 9, 854, 855.
59. Ober Imidoxanthide, eine neue Klasse geforbier organischer Verbindungen. Ber.
Dtsch. cherh. Ges., 1902, Jg. 35, 2470—2473.
60. Ober Xanthogenamide der Terpenreihe. Ber. Dtsch. chem. Ges., 1902, Jg. 35, 2473—
2483.
61. Magnesiumorganische Verbindungen als Reagens auf die Hydroxylgruppe. Ber. Dtsch.
chem. Ges., 1902, Jg. 35, 3912—3914.
62. О диагнозе гидроксила при помощи магний-органических соединений. Сообщение
на 103-м заседании Отделения хим.ип Общества любителей естествознания,
антропологии и этнографии 23.1.1903 г. ЖРФХО, 1903, 35, отд. 2, выи. 3, 30.
63. О новой константе рефракции. Сообщение .на 108-м заседании Отделения химии Об-
¦ щества любителей естествознания, антропологии и этнографии 2.1.1903 г. ЖРФХО,
1903, 35, отд. 2, вып. 3, 171.
64. К характеристике ментена, полученного по ксантогеновому методу из а-ментола
Сообщение на заседании Отделения химии РФХО, 6.11.1903 г. ЖРФХО, 1903, 35,
нып. 2, 182—184.
65. О ксантогеновой реакции. Сообщение на заседании Отделения химии РФХО,
f3.II.1903 г. ЖРФХО, 1903, 35, вып. 2, 184—185.
66. О меитенах различного происхождения. Сообщение на заседании Отделения химии
РФХО, 6.II.1903 г. ЖРФХО, 1903, 35, вып. 2, 184, 185.
67. О некоторых ороиэводных ментил-ксантогеновой кислоты и о ментенах различного
происхождения. Статья 1. ЖРФХО, 1903, 35, 1116—1179.
68. Исследования в области терпенов и камфоры. Магистерская диссертация., 1903.
69. Организм человека и животных в борьбе с бактериями, Ч. 1 и 2. Научное слово,
М., 1903, кн. 3, стр. 36—46; кн. 4, 20—37 (с табл. и рис.).
70. В. Оствальд. Катализ (перевод). Научное слово, М., 1903, кн. 5, 166
(рецензия).
71. Невидимые микробы. Научное слово, М., 1903, кн. 8, 134—136.
72. К в спросу о цвете и спектрах поглощения органических соединений. Сообщение
¦на заседании Отделения химии РФХО, 5.Н.1904 г. ЖРФХО. 1904, 36, вып. 2,
189, 190.
73 О попытке синтеза гемопиорола. Сообщение на заседании Отделения химии
РФХО, 5.II.1904 г. ЖРФХО, '1904, 36, вып. 2, 190—192. (Совместно с Н. А.
Шлезингером).
74. Возражение на замечание Ж- И. Иоцича в протоколе заседания Отделения химии
РФХО, 4.Ш.1904 г. ЖРФХО, 1904, 36, вып. 3, 340, 341.
75 Металлоам-м-иачные производные сукцинпмида. Сообщение на заседании Отделения
химии РФХО, 1.1V.1904 г. ЖРФХО, 1904, 36, вып. 4, 452, 453.
76. Заметка о происхождении нефти. Сообщение на заседании Отделения химии РФХО,
1.IV.1904 г. ЖРФХО, 1904, 36, вып. 4, 453—455.
77. Металлоаммиачные производные им-идов. Сообщение на заседании Отделения
химии РФХО 29.IV.1904 г. ЖРФХО, 1904, 36, вып. 5, 613—616.
78. По поводу заметки Вальдена в № 5 Протоколов РФХО за 1904 г. Сообщение на
заседании Отделения химии РФХО 9ЛХ.1904 г. ЖРФХО, 1904, 36, вып. 7, 925-927.
79. О трнболюминисценции. Статья 2. ЖРФХО, 1904, 36, вып. 8, 1245—1253.
80. Опыт синтеза гемопиррола. ЖРФХО, 1904, 36, вып. 8, 1258—1268.
81. К методике получения ксантогеновых соединений. Статья 3. ЖРФХО, 1904, 36,
вып. 8, 1253—1258.
31 Л. А. Чугаев, т. Ш
$78
Список опубликованных трудов Л. А. Чугаева
82. Ксантогеновая реакция и ее применение в ряду терпенов и камфоры. Статья 2.
ЖРФХО, 1904, 36, вып. 8, 988—1052.
83. Первый продукт химического синтеза в растениях. Научное слово, М., кн. 3,
144, 145.
84. Памяти проф. Е. Е. Вагнера. Научное слово, М., 1904, кн. 5, стр. 104—112.
85. А. Гол л ем ан. Учебник органической химии (перевод А. Генерозова). Научное
слово, М., 1904, кн. 7, 134, 135. (Рецензия).
86. О родственной связи между красящим веществом крови и пигментом зеленых
растений. Научное слово, М., 1904, кн. 8, 137, 140.
87. Предмет и задачи современной химии. Вступительная лекция, читанная в МВТУ
в октябре 1904 г. В кн. Сб. речей и докладов, посвященных памяти Л. А. Чугаева.
Л., НХТИ, 1924.
88. Alkohol С,оН[7ОН. Вег. Dtsch. chem. Ges., 1904, Jg. 37, S. 1036—1037. В статье
G. W a g n e г, St. M о у с h e, Fr. Z i e n k о w s k у. (Раздел Zur Kenntnis des Kamp-
fens не принадлежит перу Л. А. Чугаева).
89. Uber eine Reihe Komplexer Verbindungen des Succinim.iids. (Verlaufige Mittei-
lung). Ber. Dtsch. chem. Ges., 1904, Jg. 37, 1479—1481.
90. Uber einige Derivate des Thujens. (Auszug aus der Schrift des Verfassers: Inter-
suchungen in der Terpen —und Kampherreihe. Mosckau, 1903). Ber. Dtsch. chem.
Ges., 1904, Jg. 37, 1481 — 1486.
91. Об одном ряде комплексных соединений диметилпиррона. Сообщение на
заседании Отделения химии РФХО, 13.1.1905 г. ЖРФХО, 1905, 37, вып. 1, 146, 147.
92. О металлических соединениях а-диоксимов. ЖРФХО, 1905, 37, вып. 2, стр. 243.
93. О соединениях никельсукцинимида с аминами жирного ряда. ЖРФХО, 1905, 37,
вып. 3, 357, 358.
94. Циклические группировки в комплексных соединениях. ЖРФХО, 1905, 37, вып. 3,
358—360.
95. Новый ряд для определения конфигурации стереоизомерных оксимов. ЖРФХО,
1905, 37, вып. 360, 361.
96. Новый метод получения замещенных кобальтиаков. Сообщение на заседании
Отделения химии РФХО, 12.V1905 г. ЖРФХО, 1905, 37, вып. 5, 610—612.
97. О диоксиминокобальтиаках. Сообщение на заседании Отделения химии РФХО
15.IX.1905 г. ЖРФХО, 1905, 37, вып. 7, 956—958.
98. Искусственное получение тшнена и строение пинокамфеола. Сообщение на
заседании Отделения химии 15.IX.1905 г. ЖРФХО, 1905, 37, вып. 7, 958, 959. (Совместно
с Б. А. Еше).
99. О рефрактометрической константе г. Шенево. Тр. лаб. общ. и органич. химии
МВТУ, 1905, вып. 1, 17—19.
100. Uber Komplexe Verbindungen der ct-Dioxime. Zs. f. anorg. Chem., 1905, 46, 144—169.
101. Uber ein neues empfindliches Reagens аиГ Nickel. Ber. Dtsch. chem. Ges., 1905, Jg. 38-
2520—2522.
102. Uber Komplexe Verbindungen organischer Imide. Succinimidkupferderivate. Ber.
Dtsch. chem. Ges., 1905, Jg. 38, 2899—2914.
103. Исследования в области комплексных соединений. Докторская диссертация. М.,.
МВТУ, 1906. Тр. лаб. общ. и органич. химии МВТУ, 1906, вып. 2, 1—152.
104. Строение неорганических соединений по Вернеру. Сообщение на 119-м заседании
Отделения химии Общества любителей естествознания, антропологии и этнографии
18.XI.1905 г. ЖРФХО, 1906, 38, вып. 1, 16.
105. О кобальтиамминнитритодиметилглиоксимине. Сообщение на заседании Отделения
химии РФХО, 5.1.1906 г. ЖРФХО, 1906, 38, вып. 1, 7.
106. О комплексных соединениях а-бензоилпиридиноксима. Сообщение на заседании
Отделения химии РФХО, 5.1.1906 г. ЖРФХО, 1906, 38, вып. 1, 7—9.
107. О соотношении между химической природой аминов и способностью их к
образованию комплексных соединений. Сообщение на заседании Отделения химии РФХО,
5.1.1906 г. ЖРФХО, 1906, 38, вып. 1, 9—12.
108. Памятл Е. Е. Вагнера (младшего). Некролог ЖРФХО, 1906, 38, вып. 2, 280,
281.
109. О биуретовой реакции. Сообщение на заседании Отделения химии РФХО,
7.IX.1906 г. ЖРФХО, 1906, 38, вып. 7, 1083—1085.
ПО. Радиоэлементы и их превращения. Петроград, 1906 г. Отдельный оттиск из журн.
Практическая медицина и физиотерапия, 1906, 3, 47—142.
111. Uber Kobaltidioximine. I (IV. Mitteilung). Ber. Dtsch. chem. Ges., 1906, Jg. 39,
2692—2702.
112. Ober Komplexe Verbindungen organischer Imide: Succinimidnickel derivate (V. Mit-
teilung). Ber. Dtsch. chem. Ges., 1906, Jg. 39, 3190—3201.
113. Zur Kenntnis der Dioximine und unliche Verbindungen. Ber. Dtsch. chem. Ges., 1906,
Jg. 39, 3382—3389.
114. Исследования в области комплексных соединений. О новом ряде комплексных кис>
лот. СПб., 1907, 14 стр.
Список опубликованных трудов Л. А. Чугаева
470
115. Об искусственном получении шинена и о строении пннокамфеола. Тр. лпб. обаі-
и органич. химии МВТУ, 1907, вып. 3, 2—9; ЖРФХО, 1907, 39, 1325—1333.
(Совместно с Б. А. Еше.)
116. О кобальтиаках, не проводящих тока. Сообщение на 130-м заседании Отделения
химии Общества любителей естествознания, антропологии и этнографии 4.V.I907 г.
ЖРФХО, 1907, 39, вып. 7, 1179.
117. Исследования в области комплексных соединений. I. Комплексные соединения
органических имидов. ЖРФХО, 1907, 39, вып. 8, 1262—1323.
118. Исследования в ряду терпенов и камфоры. ЖРФХО, 1907, 39, вып. 8, 1324 —1345.
119. О некоторых производных левой дигидрокарбилксантогеновон кислоты. Тр. лаб.
орган, и общ. химии МВТУ, 1907, вып. 3, 10—16; ЖРФХО, 1907, 39, вып. 8, 1333 —
1339. (Совместно с В. П. Покровским.)
120. Об эфирном масле из «сахгыза». Тр. лаб. общ. и органич. химии МВТУ, 1907,
вып. 3, 16—20; ЖРФХО, 1907, 39, вып. 8, 1339—1343. (Совместно с Я. С. Сурень-
янцем.)
121. Памяти Г. Н. Габричевского. Бакт. іш-т Моск. Ун-та. Отчет о деятельности Ин-та
за 1906 г. М., 1906, стр. 17^27.
122. Einige Bemerkungen uber die Ringbi'ldung der Komplexverbindungen. J. prakt. Chem..
1907, Bd. 16, 88—93.
123. Uber den Einfluss der Ringbildung auf den Bestandigkeitsgrad von Komplexverbin-
dungen. (Auszug aus der Schrift: Untersuchungen uber Komplex verbindungen. M.
1906), J. prakt. Chem., 1907, 75, 153— 1G8.
124. Uber relative Bestandfgket einiger Metallaminverbindungcn (VII. Mitteilung uber
Komplexverbindungen). Ber. Dtsch. chem. Ges., 1907, Jg. 40, 173—181.
125. Uber einige komplexe Verbindungen des Oxalendiamidoxims. (VIII. Mitteilung uber
Komplexverbindungen). Ber. Dtsch. chem. Ges., 1907, Jg. 40, 181 — 185. (In Gemein-
schaft mit J. Surenjanz).
126. Uber eine neue Synthese der u-Diketone. (Verlaufige Mitteilung). Ber. Dtsch. chem
Ges., 1907, Jg. 40, 186, 187.
127. Sur une methode sensible pour la recherche du nickel en presence du cobalt. C. R
Acad. Sci., Paris, 1907, 145, 679—681. (Seance du 21 October 1907).
128. Uber Komplexe Verbindungen organischer Imide. (IV. Mitteilung). Ein Beitrag zur
Kenntnis der Biuret reaktion. Ber. Dtsch. chem. Ges., 1907, Jg. 40, 1973—1980.
129. Uber einige Komplexverbindungen des optisch-aktiven /-Propylendiamins. (X.
Mitteilung uber Komplexverbindungen). Ber. Dtsch. chem. Ges., 1907, Jg. 40, 3461—3465.
(In Gemeinschaft mit W. Sokolow).
130. Uber Kobaitidioximine. II. (IX. Mitteilung uber Komplexverbindungen). Ber. Dtsch.
chem. Ges., 1907, Jg. 40, 3498—3504. (In Gemeinschaft mit J. Tischtschenko).
131. Эволюция вещества в мертвой и живой природе. Вступительная лекаж я, читанная
в С.-Петербургском университете 11 сентября 1908 г. В кн.: Сб. 'речей и докладов,
посвященных памяти Л. А. Чугаева. Л., НХТИ, 1924.
132. Об электропроводности некоторых комплексных соединений. Сообщение па
заседании Отделения химии РФХО, 4.XII.1908 г. ЖРФХО, ч. хим., 1908, 40, вып. 9,
1781.
133. К вопросу о стереохимических условиях образования комплексных соединений.
Сообщение на- заседании Отделения химии РФХО, 4.XII.1908 г. ЖРФХО, 1908, 40,
вып. 9, 1781.
134. Uber eine Methode zur Konfigurationsbestimmung bei a-Dioximen. (XII. Mitteilung
uber Komplexverbindungen). Ber. Dtsch. chem. Ges., 1908, Jg. 41, 1678—1684. (In
Gemeinschaft mit N. Chosinsky).
135. Ober die isomeren Modifikationen des p-Tolil-dioxims und uber ihr Verhalten gege-
nuber Komplexbildung. (XIII Mitteilung uber Komplexverbindungen) Ber. Dtsch.
chem. Ges., 1908, Jg. 41, 2219—2221. (In Gemeinschaft mit L. Spiro).
136. Ober Komplexverbindungen organischer Disulfide. (XIV. Mitteilung). Ber. Dtsch.
chem. Ges., 1908, Jg. 41, 2222—2226.
137. Uber eine neue Komplexsaure. Studien uber Kobaitidioximine TII (XV. Mitteilung).
Ber. Dtsch. chem. Ges., 1508, Jg. 41, 2226—2232.
138. Einige Bemerkungen uber die Abhandlung von C. Liebermann (Ber. Dtsch. chem.
Ges., 1908, Jg. 41, 1436): Zur Theorie der Beizenfarbstoffe. Ber. Dtsch. chem. Ges.,
1908, Jg. 41, 2245, 2246.
139. О некоторых оптически-деятельных комплексных соединениях (к вопросу об
оптической суперпозиции). ЖРФХО, ч. хим., 1909, 41, вып. 2, 177. (Тр. Первого
Менделеевского съезда по общей и прикладной химии, состоявшегося с 20 по 30
декабря .1907 г.).
НЬ. Об аналогии некоторых комплексных соединений с основаниями трифенилмета-
яового ряда. ЖРФХО, ч. хим., 1909, 41, вып. 2, 177. (Тр. Первого
Менделеевского съезда по общей и прикладной химии, состоявшегося с 20 по 30 декабря
1907 г).
31*
480
Список опубликованных трудов Л. А. Чугаева
141. Демонстрация нескольких опытов с комплексными соединениями. ЖРФХО, ч. хим.,
1909, 41, вып. 2, 284, 285. (Тр. Первого Менделеевского съезда по общей и
прикладной химии, состоявшегося с 20 ло 30 декабря 1907 г.)
142. Новый ряд комплексных кислот. ЖРФХО, ч. хим., 1909, 41, вып. 2, 185—286.
(Тр. Первого Менделеевского съезда по общей и прикладной химии, состоявшегося
с 20 по 30 декабря 1907 г.).
143. Исследование в области комплексных соединений. II. О соединениях, вызывающих
биуретовую реакцию. ЖРФХО, ч. хим., 1909, 41, вып. 3, 166—184. Тр. Лаб. общ.
и органич. химии МВТУ, 1909, вып. 5, 31—48.
144. Исследование в области комплексных соединении. III. Металлические соединения
а-диоксимов. ЖРФХО, ч. хим., 1909, 41, вып. 3, 184—252.
145. Исследование в области комплексных соединений. IV. О некоторых условиях
образования комплексных соединений металлического типа. ЖРФХО, ч. хим., 1909,
41, вып. 3, 253—287.
146. К теории спинтарископа. ЖРФХО, ч. хим., 1909, 41, вып. 3, 298—301. Тр. Лаб.
общ. и органич. химии МВТУ, 1909, вып. 4, 21—24. (Совместно с В. П. Покровским).
147. Об аномальном вращении органических соединений. Сообщение на заседании
Отделения химии РФХО, 1.IV.1910 г. ЖРФХО, ч. хим., 1910, 42, вып. 4, 546. (Со-
• вместно с А. Огородниковым).
148. Об аномальной дисперсии плоскости поляризации света. Сообщение на заседании
Отделения химии РФХО, 7.V.1909 г. ЖРФХО, ч. хим., 1909, 41, вып. 5, 723,
149. О диоксиминовых кислотах. Сообщение на заседании Отделения химии РФХО,
7.V.1909 г. ЖРФХО, ч. хим., 1909, 41, вып. 5, 723. (Совместно с Сениговым).
150. Исследование в области комплексных соединений. V. О неионизированных кобальт-
идиоксим;Инах. ЖРФХО, «ч. хим., 1909, 41, вып. 8, 1332—1354. Тр. Лаб. О'рган. и
общей химик МВТУ, 1909, вып. 4, 1—21.
151. Исследование в области комплексных соединений. VI. О новом ряде
комплексных кислот. ЖРФХО, ч. хим., 1909, 41, вып. 8. 1355—1368. Тр. Лаб. орган, и
¦обшей химии МВТУ, 1909, вып. 5, 1—15. (Доложено па Первом Менделеевском
съезде).
152. О комплексных соединениях некоторых органических имидов. Сообщение на
заседании Отделения химии РФХО, 7.V.1909 г. ЖРФХО, ч. хим., 1909, 41, вып. 8,
1381. (Совместно с В. Субботиным).
153 О холестерине. Сообщение на заседании Отделения химии РФХО 5.XI.1909 г.
ЖРФХО, ч. хим., 1909, 41, вып. 9, 1591. (Совместно с В. А. Фоминым).
154. Современные задачи органической химии. Лекция, читанная 18 ноября 1909 г. в
СПб. Технологическом ин-те. (Отдельный оттиск, стр. 1—26, из 20-го тома. Известия
Технологического ин-та. СПб., 1911); Сб., посвященный памяти Л. А. Чугаева.
Л., НХТИ, 1924, стр. 153—171.
155. ОЬег Selenomercaptane unci einige Derivate desselben. (Vorlaufige Mitteilung). Ber.
Dtsch. chem. Ges., 1909, Jg. 42, 49—54.
156. Ober das a-Propylendiamin und uber einige Derivate der optisch-aktiven Propylen-
diamine. (XVI. Mitteilung uber Komplexverbindungen). Ber. Dtsch. chem. Ges., 1909,
Jg. 42, 55—58. (In Gemeinschaft mit W. Sokolow).
157. Uber anomale Rotationsdispersion. Ber. Dtsch. chem. Ges., 1908, Jg. 42, 2244—
2247. ,
158. Zur Kenntnis des Cholesterins. I. Anwendung der Xanthogen-Reaktion. Ber. Dtsch.
chem. Ges., 1909, Jg. 42, 4631—4634. (In Gemeinschaft mit A. Gastew).
159. О некоторых новейших успехах биологической химии. Сообщение на 152-м
заседании Отделения химии Общества любителей естествознания, антропологии и
этнографии 4.1.1910 г. ЖРФХО, ч. хим., 1910, 42, 722.
160. Об изомерии платиновосернистых соединений. Сообщение на заседании Отделения
химии РФХО 4ЛНЛ910 г. ЖРФХО, ч. хим., 1910, 42, вып. 2, 371. (Совместно
с В. Субботиным).
161. О некоторых производных правого антипода природного 1-ментола. ЖРФХО. 1910,
ч. хим., 42, вып. 4, 714—718.
162. О вращательной дисперсии некоторых бесцветных соединений. Сообщение на
заседании Отделения химии РФХО 22.IV.1910 г., ЖРФХО, ч. хим., 1910, 42, вып. 4,
719.
163. О дигидротуйене и дигидросабинене. Сообщение на заседании Отделения химии
РФХО 7.Х.1910 г. ЖРФХО, ч. хим., 1910, 42, вып. 7, 1330. (Совместно с В. А.
Фоминым) .
164. Памяти А. М. Зайцева и Ст. Канниццаро. ЖРФХО, ч. хим., 1910, 42, вып. 7, 1318—
1328.
165. Исследования в области комплексных соединений. VII. О некоторых комплексах из
ряда диоксиминов. ЖРФХО, 1910, ч. хим., 42, вып. 8, 1466—1487. (Совместно
с А. Постниковым, И. Кареевым, И. Тищенко, Б. Афанасьевым).
166. Комплексные соединения хрома с амино-кислотами. Сообщение на заседании
Отделения химии РФХО, 4.XI.I910 г. ЖРФХО, ч. хим., 1910, 42, вып. 8, 1490. (Совместно
с Е. Сербиным.)
Список опубликованных трудов Л. А. Чугаева
481
167. О химическом строении комплексных соединений. СПб, 1910. Отдельный оттиск
актовой речи в СПб. Ун-те. Годовой отчет СПб Ун-та, 1910 г. 155 стр.
168. Uber anomale Rotationsdispersion. II. 2s. phys. Chem., 1910, 74, H. 4, 503—512.
(In Gemeinschaft mit A. Ogorodnikoff).
169. Zur Kenntnis des Cholesterins. II. Abh.— Lieb. Ann., 1910, Bd. 375, 288—297. (In
Gemeinschaft mit W. Fomin).
170. Sur certains derives de la cholesterine. I. C. R. Acad. Sci., Paris, 1910, 150, 1435-
1437. (Seance du 30 mai 1910). (En collaboration avec W. Fomin).
171. Uber isomere Platinverbindungen organischer Sulfide. (XVIII. Mitteilung uber Kom-
plexverbindungen). Ber. Dtsch. chem. Ges., 1910, Jg. 43, 1200—1205. (In Gemeinschaft
mit W. Subbotin).
172. L'hydrogention des thuienes isomeres et du sabinene. Le Thuiane. С R. Acad. Sci.,
Paris, 1910, 151, 1058—1062. Seance du 5 dec. 1900, (En collaboration avec W. Fo-
rriin).
173. Sur des sels complexes de certains aminoacides. C. R. Acad. Sci., Paris, 1910, 151,
1361 — 1363 (Seance du 27 dec. 1910). (En collaboration avec E. Serbin).
174. ?ine Bemerkung zur Abhandlung yon Kotz und Grethe. Ober das A^-Dihydrophenol
usw. J. prakt. chem. 1910, 81, 188, 189.
175. Периодическая система химических элементов. Прил. к табл. Периодическая
система Д. И. Менделеева. СПб., «Образование», 1911, 100 стр.
176. Химические процессы. Итоги науки, т. I—II. М., 1911, стр. 277—602. 23 портрета
выдающихся химиков, 60 чертежей.
177. Вант-Гофф н основание стереохимии. Доклад на заседании Отделения химии
РФХО, 3.111.1911 г. ЖРФХО, ч. хим., 1911, 43, вып. 3, 497.
178. О борнилене. Сообщение на заседании Отделения химии РФХО 3.II.1911 г.
ЖРФХО, ч. хим., 1911, 43, вып. 1, 141. (Совместно с В. М. Бутлеровым).
179. О платиново-сернистых соединениях. Сообщение на заседании Отделения химии
РФХО 3.II.1911 г. ЖРФХО, ч. хим., 1911, 43, вып. I, 141. (Совместно с Н. И.
Беневоленским).
180. О рефракции холестерина и некоторых его производных. Сообщение на заседании-
Отделения химии РФХО 3.III.19U г. ЖРФХО, ч. хим., 1911, 43, вып. 2, 322.
(Совместно с П. Г. Коком).
181. Об утилизации нафтеновых кислот по работам покойного профессора В. В. Марков-
никова. Сообщение на заседании Отделения химии РФХО 28.1V.1911 г. ЖРФХО,
ч. хим., 1911, 43, вып. 4, 671.
182. О дигидрохолестерине. 2. Об изомерных туйопах. Сообщение на заседании
Отделения химии РФХО 12.V.19M г. ЖРФХО, ч. хим., 1911, 43, вып. 4, 678. (Совместно
с В. А. Фоминым).
ШЗ. О дитиокамфокар'боновой кислоте. Сообщение на заседании Отделения химии
РФХО, 12.V.1911 г. ЖРФХО, ч. хим., 1911, 43, вып. 678. (Совместно с Г. Пигулев-
ским).
184. Влияние растворителя іна вращательную дисперсию и на абсорбцию. Сообщение
на заседании Отделения химии РФХО I2.V.191I г. ЖРФХО, ч. хим., 1911, 43,
вып. 4, 678. (Совместно с А. Г. Огородниковым).
185. Новый тип аномальной вращательной дисперсии. Сообщение на заседании
Отделения химии РФХО, 12.V.1911 г. ЖРФХО, ч. хим., 1911, 43, вып. 4, 678.
186. Uber Rotationsdispersion. III. Farblose Verbindungen. Zeitschrift fur phys. Chem.,
1911, 76, 469—483.
187. Zur Kenntnis des cholesterins. III. Lieb. Ann., 1911, 385, 352—358. (In Gemeinschaft
mit P. Kock).
188. Sur une anomalie de la refraction moleoulaire dans la serie des dvoximes substituees.
C. R. Acad. Sci., Paris, 1911, 153, 259—261. (En collaboration avec P. Koch).
189. Sur l'acide dithiocamphocarbonique. С R. Acad. Sci., Paris, 1911, 153, 388—390.
(En collaboration avec G. Pigulewski).
190. Sur la dispersion anomale des corps colores et actifs. Ann. de Chim. Phys., 191 U
22, 137—144. (En collaboration avec A. Ogorodnikoff).
191. Uber einen neuen Typus der anomalen Rotationsdispersion. Ein Beitrag zur Kenntnis
-der optischen Superposition. (Vorlaufige Mitteilung). Ber. Dtsch. Chem. Ges., 1910,
Jg. 44, 2023—2030.
192. Об изомерных туйиловых спиртах и туйонах. Сообщение на заседании Отделения
химии РФХО, 1.IIL1912 г. ЖРФХО, ч.'хим., 1912, 44, вып. 2, 478, 479. (Совместно
с В. А. Фоминым).
193. О действии алкоголята натрия-на ментольный эфир р-камферсульфоновой кислоты.
Предварительное сообщение на заседании Отделения химии РФХО 1.III.1912 г.
ЖРФХО, ч. хим., 1912, 44, вып. 2, 479.
194 Об электропроводности платиновосернистых соединений. Сообщение на заседании
Отделения химии РФХО 26.IV.I9I2 г. ЖРФХО, ч. хим., 1912, 44, вып. 3, с. 773.
(Совместно с А. Г. Коблянским).
482 Список опубликованных трудов Л. А. Чугаева
195. О вращательной дисперсии циклических соединений с кетонной функцией.
Сообщение на заседании Отделения химии РФХО 4.X.1912 г. ЖРФХО, ч. хим., 1912,
44, вып. 7, 1560.
196. Об оптических свойствах некоторых производных пинена. Сообщение на заседании
Отделения химии РФХО 4.Х.1912 г. ЖРФХО, ч. хим., 1912, 44, вып. 7, 1560.
(Совместно с А. К- Кирпичевым).
197. Об оптически деятельных эфирах трифенилуксусной кислоты. Сообщение на
заседании Отделения химии РФХО 4.Х.1912 г. ЖРФХО, ч. хим., 1912, 44, вып. 7, 1560.
(Совместно с Г. Н. Глининым).
198. О соединениях бромистой платины с органическими сульфидами. Сообщение на
заседании Отделения химии РФХО 8.XII.1911 г. ЖРФХО, ч. хим., 1912, 44, вып. 1,
269. (Совместно с Д. Френкель).
.190. Хи.мия неорганическая. Записки по лекциям проф. Л. А. Чугаева. Сост. студенты
П. Троицмий и Н. Черможский, ч. 1. СПб., 1911.
200. Химия неорганическая. Записки по лекциям проф. Л. А, Чугаева. Сост. студ.
П. Троицкий и Н. Черможский. Ч. 1 и 2. СПб., 1911—1912.
201. Ober Bornylen. Lieb. Ann., 1912, 388, 280—293. (In Gemeinschaft mit W. Budrick).
202. Bemerkungen zur Abhandlung von Ernst Deussen «Ober eine neue methode zur
Priifung optisch-aktiver Verbindungen, zugleich ein Beitrag zur Kenntnis der anoma-
len Rotationsdispersion». J. prakt. Chem., 1912, 86, 545—550.
203. Ober isomere Tnujylalkohole und Thujene. Ber. Dtsch. chem. Ges., 1912, Jg. 45,
1293—1298. (In Gemeinschaft mit W. Fomin).
204. Sur quelques composes complexes du bromure platineux et des sulfures organiques.
С R. Acad. Sck, Paris, 1912, 154, 33-^5. Seance du 2 Janvier 1912. (En
collaboration avec D. Fraenkel).
205. Sur la dispersion rotatoire de quelques derives du camphore. Bull. soc. chim. 1912,
11, 718—722.
206. Sur quelques composes complexes du chlorure platineux avec Гатіпоасеіаі. С. R.
Acad. Sci., Paris, 1912. 155, 1021—.1025, Seance du 18 novemb. 1912. ;En collaboration
avec B. Orelkine).
207. Uber das optische Drehungsvermogen einiger aktiver Triphenyl-essigester. Ber. Dtsch.
chem. Ges., 1912, Jg. 45, 2759—2764. (In Gemeinschaft mit G. Glinin).
208. Uber Rotationsdispersion. IV. Uber den Einfluss des Losungsmitlels auf die Lichtab-
sorption und auf die Rotations dispersion gefarbter Verbindungen. Zs. phys. Chem.,
1912, 79, 471—480. (In Gemeinschaft mit A. Ogorodnikoff).
209. Периодическая система химических элементов. 1-ое изд. СПб., 1913, 258 стр.
210. О влиянии циклической связи на вращательную дисперсию. Сообщение на
заседании Отделения химии РФХО 1S.XII.1912 г. ЖРФХО, ч. хим., 1913, 4Э, вып. 1, 152.
(Совместно с А. А. Глебко).
211. Об изомерии в ряду платиновосернисгых соединений. Сообщение на заседании
Отделения химии РФХО, 7.II.I913 г. ЖРФХО, ч. хим., 1913, 45, вып. 2, 354.
(Совместно с В. Г. Хлопнным).
212. Заметка по истории химии в России (памяти Е. Е. Вагнера). Сообщение на
заседании Отделения химии РФХО 7.Ш.1913 г. ЖРФХО, ч. хим., 1913, 45, вып. 2,
377.
213. Спектры поглощения и вращательная дисперсия. Сообщение на заседании
Отделения химии РФХО 4.IV.1913 г. ЖРФХО, ч. хим., 1913, 45, вып. 3, 547.
(Совместно с А. Г. Огородниковым).
214. О влиянии оптической суперпозиции на аномальную вращательную дисперсию.
Сообщение на заседании Отделения химии РФХО 4.IV.1913 г. ЖРФХО, ч. хим.,
1913, 45, вып. 3, 648. (Совместно с В. В. Лебединским).
215. Об оптических свойствах борнилксаатогеновой кислоты. Сообщение на заседании
Отделения химии РФХО, 4.IV.1913 г. ЖРФХО, ч. хим., 1913, 45, вып. 3, 648.
216. К вопросу об оптической суперпозиции. Сообщение на заседании Отделения
химии РФХО 25.IV.1913 г. ЖРФХО, ч. хим., 1913, 45*, вып. 3, 661. (Совместно с
А. А. Глебко).
217. О диоксиминах родия. Сообщение на заседании Отделения химии РФХО
16.V.1913 г. ЖРФХО, ч. хим., 1913, 45, вып. 3, с 669. (Совместно с В. В.
Лебединским).
218. Простой аппарат для определения растворимости при различных температурах.
Сообщение на заседании Отделения химии РФХО 16.V.1913 г. ЖРФХО, ч. хим.,
1913, 45, вып. 3, 670. (Совместно с В. Г. Хлопиным).
219. Влияние температуры на вращательную дисперсию окрашенных соединений.
Сообщение на заседании Отделения химии РФХО, 16.V.1913 г. ЖРФХО, ч. хим.,
1913, 45, вып. 3, 671. (Совместно с В. Н. Пастаноговым).
220. О влиянии растворимости на вращательную дисперсию. Сообщение на заседании
Отделения химии РФХО, 16.V.19I3 г. ЖРФХО, ч. хим., 1913, 45, вып. 3, 671.
(Совместно с Г. В. Пигулевским).
Список опубликованных трудов Л. А. Чугаева 483
221. О действии хлористого хромила на метан. Сообщение на заседании Отделения
химии РФХО, 16.V.1913 г. ЖРФХО, ч. хим., 1913, 45, вып. 3, 671, 672.
(Совместно с В. А. Фоминым и В. Агрономовым).
222. Вращательная дисперсия эфирных масел. Сообщение на заседании Отделения
химии РФХО 16.V.1913 г. ЖРФХО, ч. хим., 1913, 45, вып. 3, 672. (Совместно с
Я. Гесно).
223. О действии спиртов на металлические соединения кетонов. Сообщение на
заседании Отделения химии РФХО 16.V.1913 г. ЖРФХО, ч. хим., 1913, 45, вып. 3,
674.
224. Вращательная дисперсия некоторых производных цитронеллаля. Сообщение на
заседании Отделения химии РФХО З.Х.1913 г. ЖРФХО, ч. хим., 1913, 45, вып. 7,
168Э. (Совместно с С. А. Матисеном).
225. Сообщение о результатах своего посещения 83-го съезда Британской Ассоциации,
имевшего место в Бирмингаме 10—17 сентября 1913 г. Сообщение на заседании
Отделения химии РФХО З.Х.1913 г. ЖРФХО, ч. хим., 1913 45, вып. 7, 1676—
1678.
226. О комплексных азидах хрома. Сообщение на заседании Отделения химии РФХО
З.Х.1913 г. ЖРФХО, ч. хим., 1913, 45, вып. 7, 1683—1684. (Совместно с В. М. Се-
славиным).
227. О нитропруссидах и пикратах платиновосернистых оснований. Сообщение на
заседании Отделения химии РФХО 7.XI.1913 г. ЖРФХО, ч. хим., 1913. 45, вып. 8,
1862, 1863. (Совместно с В. Г. Хлопиным).
228. О левом ментене. Сообщение на заседании Отделения химии РФХО 7.XI.1913 г.
ЖРФХО, ч. хим., 1913, 45, вып. 8, 1871, 1872. (Совместно с Б. П. Орелкиным).
229. О пимаровой кислоте. Сообщение на заседании Отделения химии РФХО
13.XII.1912 г. ЖРФХО, ч. хим., 1913, 45, вып. 1, 152. (Совместно с П. Я- Теару).
230. Об одном ряде комплексных соединений двухвалентной платины. Сообщение на
заседании Отделения химии РФХО 5.X1I.1913 г. ЖРФХО, ч. хим., 1913, 45, вып. 9,
2072, 2073. (Совместно с П. Я- Теару).
231. Краткий 'Предварительный отчет о работах по исследованию некоторых металлов
платиновой группы и их соединений. М., изд-во Об-ва содействия успехам
опытных наук и их практических применений им. X. С. Леденцова, 1913, 22 стр.
232. Zur Kenntnis der Komplexverbindungen des Rhodiums. Zs. anorg. Chemie, 1913, 83,
1—7. (In Gemeinschaft mit W. Lebedinski).
233. Uber die elektrische Leitfahigkeit einiger Platinverbindungen organischer Disulfide.
Zs. anorg. Chem. 1913, 83, 26. (In Gemeinschaft mit A. Kablianski).
234. Uber Verbindungen des Platonitrits mit organischen Dithioathern. Zs. anorg.
Chem., 1913, 82, 401—419. (In Gemeinschaft mit W. Chlopin).
235. Ober Komplexverbindungen organischer Sulfide mit vierwertigem Platin. Zs.
anorg. Chem., 1913, 82, 420—425. (In gemeinschaft mit J. Benewolensky).
236. Uber Rotationsdispersion. VI. Uber den Einfluss der Temperature auf die anomale
Rotationsdispersion gefarbter Verbindungen. Zs. phys. Chem., 1913, 85, 553—572.
(In Gemeinschaft mit W. Pastanogoff).
237. Uber Rotationsdispersion. V. Zs. phys. Chem., 1913, 85, 481—510. (In Gemeinschaft
mit A. Ogorodnikoff).
238. Sur la dispersion rotatoire de I'acide /-bornylxanthogenique Iibre. Bull. Soc. chim.,
1913, 13, 793—796.
239. Sur la dispersion rotatoire de quelques derives du fi-pinene (nopinene). Bull. Soc.
chim., 1913, 13, 796—803. (En collaboration avec A. Kirpitcheff).
240. Zm Kenntnis der optischen Superposition. Ber. Dtsch. chem. ges., 1914, Jg. 46,
2752—2762. (In Gemeinschaft mit A. Glebko).
241. Zur Kenntnis der Pimarsaure. (Vorlaufige Mitteilung) Ber. Dtsch. chem. Ges. 1913,
Jg. 46, 1769—1774. (In Gemeinschaft mit P. Tearu).
242. Вант-Гофф и судьбы стереохимии. Новые идеи в химии, СПб., 1914, вып. 1,
стр. 9—32.
243. Структурно- и стереохимические представления в области неорганической химии.
Новые "идеи в химии. СПб., 1914, вып. 1, стр. 103—154.
244. Валентность. Новые идеи в химии. СПб., 1914, вып. 3, стр. 1—87.
245. Теория Абегга и молекулярные соединения. Новые идеи в химии, СПб., 1914, вып. 3,
стр. 114—122.
246. Несколько слов о превращениях химических элементов. Новые идеи в химии, СПб.,
1914, вып. 4, стр. 159—169.
247. О неустойчивых органических соединениях. Новые идеи в химии, СПб., 1914,
вып. 6, стр. 105—160.
248. Эволюция учения о катализе. Новые идеи в химии. СПб., 1914, вьш. 8, стр. 5—37.
24.9. Об изомерии. Доклад на общем собрании РФХО, 28.ХП.1913 г. ЖРФХО, ч. хим.,
1914, 46, 1і54.
250! Памяти В. А. Бородовского. Некролог, зачитанный на заседании Отделения химии
РФХО 6.П.Ю14 г. ЖРФХО, ч. хим., 1914, 46, 155—159.
484 Список опубликованных трудов Л. А. Чугаева
251. К методике определения химических соединений. Сообщение на заседании
Отделения химии РФХО 6.H.I914 г. ЖРФХО, ч. хим., 1914, 46, вып. 1, 174.
252. О восстановительных свойствах гидросердистой кислоты. Растворение теллура и
селена. Сообщение на заседании Отделения химии РФХО, 20.11.1914 г. ЖРФХО,
ч. хим., 1914, 46, вып. 1, 185—186. (Совместно с В. Г. Хлопиным).
253. Двухкомплексное соединение третичного бутилкарбиламина rPt4{CH3)sCNS]PtCJ6.
Сообщение на заседании Отделения химии РФХО 20.11.1914 г. ЖРФХО, ч. хим.,
1914, 46, вып. 1, 186. (Совместно с П. Я- Теару).
254. Новая чувствительная реакция на железо и колориметрический метод его
определения. "Сообщение на заседании Отделения химии РФХО W.IVA914 г. ЖРФХО,
ч. хим., 1914, 46, вып. 3, 593. (Совместно с Б. П. Орелкиным).
255. Образование комплексных соединений .в неводных растворах. Сообщение на
заседании Отделения химии РФХО. 10.IV.1914 г. ЖРФХО, ч. хим., 1914, 46, вып. 3. 594.
256. О платиновых соединениях гидразина. Сообщение на заседании Отделения химии
РФХО 15.V.1914 г. ЖРФХО, ч. хим., 1914, 46, вып. 5, 632. (Совместно с М. С.
Григорьевой).
257. Об одновалентном никеле. Сообщение на заседании Отделения химии РФХО
15.V.I914 г. ЖРФХО, ч. хим., 1914, 46, вып. 3, &32, (Совместно с В. Г. Хлопиным).
258. Об электропроводности платиновых соединений моносульфидов в метиловоспирто-
вом растворе. Сообщение на заседании Отделения химии РФХО 15.V.1914 г.
ЖРФХО, ч. хим., 1914, 46, вып. 3, 632—633. (Совместно с Н. Владимировым).
259. Об электропроводности и абсорбции комплексных соединений платины с изонит-
рилами. Сообщение иа заседании Отделения химии РФХО 15.V.1914 г. ЖРФХО,
ч. хим., 1914, 46, вып. 3, 633. (Совместно с И. Е. Орловым).
260. Резюме к докладу В. Похитонова «Современное положение химической
промышленности в России». Выступление на заседании Отделения химии РФХО
1.1ЛХ.1914 г. ЖРФХО, ч. хим., 1914, 46, вып. 5, 1135, 1136.
261. Новый метод определения растворимости при различных температурах. ЖРФХО,
ч. хим., 1914, 46, вып. 8, 1659—1668. (Совместно с В. Г. Хлопиным).
262. Новая чувствительная реакция на железо и .колориметрический метод его
определения, ЖРФХО, ч. хим., 1914,. 46, вып. 9, і1874—1876, (Совместно с Б.
Орелкиным) .
263. Об углеводородобромистом алюминии Г. Г. Густавсона. Сообщение на заседании
Отделения химии РФХО 4.XII.I914 г. ЖРФХО, ч. хим., 1914, 46, вып. 8. 1738,
17Э9.
264. Профессор Альфред Вернер. Природа, 1914, № 7—8, 806—831.
265. Anomalous rotatory dispersion. A contribution to a general discussion on optical
rotatory power (held before the Faraday Society on march 27, 1914). Trans. Faraday
Soc, 1914. Читай по приглашению хим. секции Митинга Британской ассоциации
в Бирмингаме в 1913 г.
266. ОЬег Plati'nverbindungen der fsonitrile. Ber. Dtsch. chem. Ges., 1914, Jg. 47, 568—
573 (In Gemeinschaft mit P. Tearu).
267. Dber ein neues Verfahren zur Loslichkeitsbeslimmung bei hohern Temperaturen. Zs.
anorg. Chem., 1914, 86, 154—162. (In Gemeinschaft mit W. Chlopin).
268. Ober die Platinverbindungen der organischen Sulfide, welche den Salzen der ersten-
Bare von Reiset analog sind. Zs. anorg. Chem., 1914, 86, 241—256. (In Gemeinschaft
mit W. Chlopin).
269. Ober eine empfindliche Eisenreaction und uber eine Methode zur colorimetrischer»
Eisenbestimmung. Zs. anorg. Chem., 1914, 89, 401—404. (In Gemeinschaft mit
B. Orelkin),
270. Sur quelques composes du nickel monovalent. Seance du 6 juillet 1914. С R. Acad.
Sci., Paris, 1914, 159, 62—64. (En collaboration avec W. Chlopine).
271. Sur une гпё-thode de preparation des composes du platrne bivalent (par Tripropy-
Л ammonium chloroplatinite). Seance du 13 juillet 1914. C. R. Acad. Sci.t Paris, 1914».
159, 186—189.
272. The chemical constitution of dioximines. J. Chem. Soc, 1914, 105, 2187—2193.
273. Beitrage zur Kenntnis des Reaktionsvermogens der schwefligen Saure. 1. Einwirkung
von Natriumhydrosulfit auf Tellur und Selen. Ber. Dtsch. chem. Ges., ,1914, Jg. 47,
1269—1275. (In Gemeinschaft mit W. Chlopin).
274. Uber Komplexverbindungen, welche zugleich Platin und Hydrazin enthalten.
(Vorlaufige Mitteilung). Ber. Dtsch. Chem. Ges., 1914, Jg. 47, 2446—2453. (In
Gemeinschaft mit M. Grigorjewa).
275. Ufcfcr Platinverbindungen der Isonitrile, welche ein Cyanradikal enthalten. B.er.
Dtsch. chem. Ges., 1914, Jg. 47, 2643—2647. (In Gemeinschaft mit P. Tearu).
276. О тцдроксимаминовы-х соединениях платины. Сообщение на заседании Отделения
химда РФХО 8.ШШ г. ЖРФХО, ч. хим., 1915, 4.7, выв. 1, 201. (Совместно- с
И. И. Черняевым).
27Г. Новый способ цолучения солей Клеве. Сообщение на заседании Отделения химии
РФХО 8.1.1915 г. ЖРФХО, ч. хим., 191Б,; 47, вып. I, 501, 202.
Список опубликованных трудов Л. А. Чугаева
485
278. Новая реакция на соль Пейроне. Сообщение на заседании Отделения химии
РФХО 5.И.1915 г. ЖРФХО, ч. хим., 1&15, 47, вып. 1, 213.
279. О восстановительной способности гидросернистой кислоты. Действие
гидросульфита натрия на металлический селен и теллур. ЖРФХО, ч. хим. 1915, 47, вып. 2,
364—372. (Совместно с В. Г. Хлопиным).
280. Об аммиачных соединениях платонитрита. Сообщение на заседании Отделения
химии РФХО, 9.IV.I915. ЖРФХО, ч. хим., 1915, 47, вып. 3, 757, 758. (Совместна
с С. С. Кильтыновичем).
281. Электропроводность аммиачных соединений платонитрита. Сообщение на
заседании Отделения химии РФХО 9.IV.1915 г., ЖРФХО, ч. хим., 1915, 47, вып. 3, 758.
(Совместно с П. Н. Владимировым).
282. Аномальная вращательная дисперсия у соединений с одним асимметрическим
углеродом. Сообщение на заседании Отделения химии РФХО 7.V.1915 г. ЖРФХО,
ч. хим., 1915, 47, вып. 3, 774—775. (Совместно с А. А. Глебко и Г. В. Пигулев-
ским).
283. О координационном числе. Сообщение на заседании Отделения химии РФХО
7.V.1915 г. ЖРФХО, ч. хим., 1915, 47, вып. 3, 775.
284. О теории Вернера. Сообщение на заседании Отделения химии РФХО 7.V.1915 г.
ЖРФХО, ч. хим., 1915, 47, вып. 3, 775, 776.
285. О платиновых соединениях ацетонитрила. Сообщение на заседании Отделения
химии РФХО 7.V.1915 г. ЖРФХО, ч. хим., 1915, 47, вып. 3, 776. (Совместно с
В. В. Лебединским).
286. О новом ряде комплексных соединений платины. Сообщение на заседании
Отделения химии РФХО 7.V.19I5 г., ЖРФХО, ч. хим., 1915, 47, вып. 3, 776, 777.
(Совместно с М. С. Сканави-Григорьевой).
287. Действие окислителей на комплексные соединения двухвалентной платины.
Сообщение на заседании Отделения химии РФХО 7.V.1915 г. ЖРФХО, ч. хим., 1916, 4?,
вып. 3, 777, 778. (Совместно с В. Г. Хлопиным).
288. О молекулярных перегруппировках у комплексных соединений. Сообщение на
заседании Отделения химии РФХО 7.V.1915 г. ЖРФХО, ч. хим., 1915, 47, вып. 3, 778.
(Совместно с Н. К. Пшеницыным).
289. Об аквосолях двухвалентной платины. Сообщение на заседании Отделения химии
РФХО 5.XI.1915 г. ЖРФХО, ч. хим., 1915, 47, вып. 7, 1806. (Совместно с И. И.
Черняевым).
290. О мерах к развитию в России химической промышленности. Деловая Россия,
еженедельник, 1915, № 14. Отдельный оттиск, стр. 1—4.
291. Удушливые газы как средство нападения и борьба с ними. Природа, М., 1915,
? 7—8, 923-940.
292. A new method of preparing Chloro- and Bromotriamminoplatinous Haloids (Cleve's
salts). J. Chem. Soc, London, 1915, 107, 1247—12150.
293. Ober die Absorptionsspektra der Dioximine. 1. Zs. anorg. Chem., 1914, 89, 241—256.
(In Gemeinschaft mit A. Glebko).
294. Sur les complexes hydroxyammonies du platine bivalent. C. R. Acad. Sci., Paris, 1915,
161, 699, 700. (En collaboration avec W. Chlopine).
295. Sur deux series de complexes derives du platine bivalent et correspondant a l'indice
de coordination, б. С R. Acad. Sci., Paris, 1915, 161, .553, 564. (En collaboration avec
W. Lebedinski).
296. Sur la serie des sels hvdroxopentamminoplatinaques. C. R. Acad. Sci., Paris, 191JV
161, 699, 700. (En collaboration avec W. Chlopine).
297. Sur la serie de triamroimoaquosels du platine hivalent (Pt ЗЫНзЛгО)Х2. С. R. Acad.
Sci., Paris, 1915, 161, 792—794. (En collaboration avec I. Tschernjaeff).
298. Une serie nouvelle de composes du platine tetravalent pentamminochloroplatinique.
С R. Acad. Sci., Paris, 1915, 160, 840—842. (En collaboration avec A. N. "Wladi-
mirof f).
299. Химические таблицы. Календарь русской природы на 1916 г. М., Изд-во журн.
«Природа», 1916, стр. 240^-266.
300. Органические соединения в приложении к минеральному анализу. В кн.: «G6.
в честь К. А. Тимирязева». М, 1916, стр. 1—9.
301. Новая реакция на четырехокись осмия. Сообщение на заседании Отделения химии
РФХО 12.IV.1917 г. ЖРФХО, ч. хим., 1916, 48, вып. 8—9, 1956. (Совместно с
И. И. Черняевым).
302. Новые комплексные соединения родия. Сообщение на заседании Отделения химии
РФХО 27.IV.1917 г. ЖРФХО, ч. хим., 1916, 48, вып. 8—9, 1955. (Совместно с
В. В. .Лебединским).
303. Действие аммиака я аминов «а иодоплатин-иты. Сообщение на заседании
Отделения .химии РФХО 27,IV.L917 г. ЖРФХО, ч хим., 1916, 48, вып. 8—9, 1955, 1956.
(Совместно с Н. К. Пшеницыным).
304. Чувствительная реакция на платину. Сообщение на заседании Отделения химии.
РФХО 12.V.1916 г. ЖРФХО, ч. хим., 1916, 48, вып. 4, 1058.
486
Список опубликованных трудов Л. А. Чугаева
305. О трехвалентной платине. Сообщение на заседании Отделения химии РФХО
12.V.1916 г. ЖРФХО, ч. хим., 1916, 48, вып. 4, 1058. (Совместно с И. И.
Черняевым).
306. О мерах, которые необходимо принять для обеспечения рационального
использования отечественной платиновой руды в промышленном и научном отношениях
(доклад). Отчеты Комиссии по изучению естественных производительных сил
России. 1916, № 5, стр. 98, 99.
307. Совещание по вопросу об изготовлении химических реагентов в России. Природа,
1916, № 7—8, 939—941.
308. Мобилизация нашей пищевой промышленности. Биржевые ведомости (газета),
29 октября 1916 г.
309. Английская химическая литература. Природа, 1916, № 9, 1088—1094.
310. Sur une serie nouvelle cle composes platiniques analogues aux sels de Cossa. C. R.
Acad. Sci., Paris, 1916, 43—45. (En collaboration avec W. Lebedinski).
311. Памяти С. M. Танатара (некролог). Сообщение на заседании Отделения химии
РФХО, 15.11.1918 г. ЖРФХО, 1917. 49, вып. 7—9, 637—630.
312. Об одном случае химической индукции. Сообщение на заседании Отделения химии
РФХО, 15.11.1918 г. ЖРФХО, 1917, 49, вып. 7—9, 642, 643.
313. Новая характерная реакция на оомий. Сообщение на заседании Отделения химии
РФХО 15.11.1918 г. ЖРФХО, 1917, 49, вып. 7—9, 644.
314. Простой лекционный опыт для иллюстрации явлений химической индукции и
катализа. Сообщение на заседании Отделения химии РФХО 15.11.1918 г. ЖРФХО, 1917,
49, вып. 7—9, 644.
315. Памяти Б. Е. Павлова (некролог). Сообщение на заседании Отделения химии
РФХО 29.IX.1917 г. ЖРФХО, 1917, 49, вып. 7—9, 617—619.
316. Памяти Ф. М. Флав-ицкого (некролог). Сообщение на заседании Отделения химии
РФХО 23.XI.1917 г. ЖРФХО, 1917, 49, вып. 7—9, 626—631.
317. О соединениях платины с ацетонитрилом. Сообщение на заседании Отделения
химии РФХО 23.XI.1917 г. ЖРФХО, 1917, 49, вып. 7—9, 632—634. (Совместно с
В. В. Лебединским).
318. Совещание по вопросу о русской платине. Отчет комиссии по изучению
естественных производительных сил России. Петроград, 1916 г., стр. 21; 1917 стр. 14.
319. О мерах к содействию исследованиям по чистой и прикладной химии в России.
Отчет Комиссии по изучению естественных производительных сил России,
Петроград, 1917, № 8, стр. 166—172.
320. О необходимости учреждения Института для изучения платины, золота и других
благородных металлов. Отчет Комиссии по изучению естественных
производительных сил России. Петроград, 1917, № 8, стр. 173—177.
321. О необходимости издания монографии по химии и химической технологии. Бюлл.
Хим. Отд. ВТК. Петроград, 1917, № 8, стр. 284, 285.
322. История вещества в живой природе. Очерк. Знание для всех, 1917, № 10, 2—32.
26 рис.
323. Об одном способе изучения .перекиси водорода. Природа, 1917, № 1, 92, 93.
324. Получение металлического бериллия и его свойства. Природа, 1917, № 3, 390, 391.
325. О новом классе металлоорпанических соединений с пятивалентным азотом.
Природа, 1917, № 7—8, 843—845.
326. И в а н За лес с кий. Химия красящего вещества. Петроград, 1917, Рецензия.
Природа, 1917, № 7—8, стр. 865, 866.
327. Н. С. Курнаков, К. Ф. Б ел ог л азов и М. К- Шматько. Месторождения
хлористого калия Соликамской соляной толщи. Петроград, 1917, 8 стр. Рецензия.
Природа, 1917, № 7—8, 866, 867.
328. П р о ф е ее о р А. Ладенбург. История развития химии. Акад. П.И.Валь-
ден. Очерк истории химии в России. Одесса, 1917, 690 стр. Рецензия. Природа,
1917, № 11—12, 1169'—1172.
329. Новое общество, посвященное вопросам прикладной химии. Природа, 1917,№11—
12, 1168—1170.
330. Ammoniacal derivatives of platinous nitrite. J. Chem. Soc, 1916, 109, 1286—1295.
(In collaboration with S. S. Kiltinovic).
331. Наука как средство борьбы человека за существование. Речи и приветствия,
произнесенные на трех публичных собраниях Свободной Ассоциации для развития и
распространения положительных наук. Петроград, 1918, стр. 34—37.
332. Основы химии газового и противогазового дела. Петроград, 1918. С рисунками и
чертежами.
333. О солеобразующих свойствах осмиевой кислоты. Сообщение на заседании
Отделения химии РФХО 26.V.1918 г. ЖРФХО, 1917, 49, вып. 3—4, 294, 295.
334. Памяти С. С. Кильтыноэича. Сообщение на заседании Отделения химии РФХО
24.XI.1918 г. ЖРФХр, ч. хим., 1918, 50, вып. 7—9, 622—624.
335. Современные "задачи химии питательных/ веществ. Российский пищевой научио-
технич. ин-т. Программные речи. Петроград, 1918, стр. 38—52:
Список опубликованных трудов Л. А. Чугаева
487
336. Отчеты о деятельности «института по изучению плагины. Отчеты комиссии по
изучению естественных производительных сил России. Петроград, 19L8 г., стр. 11—14;
вып. 12, 47, 48.
337. 1. К 'истории фабрикации сальварсана (ареола) а России; 2. Анализ некоторых
препаратов сальварсана русского происхождения. Тр. Военно-химического К-та,
1918, 4, стр. 7.
338. Синтез чистой (100%-ной) перекиси водорода. Природа, 1918, № I, 71—73.
339. Памяти В. Е. Павлова. Природа, 1918, № 1, 81—84.
340. Азотистоводородиая кислота и ее соли (азиды). Природа, 1918, № 2—3, 183—185.
341. Интересный случай химической индукция. Природа, 1918, № 2—3, 185—187.
342. Лекционный опыт для 'иллюстрации явления химической -индукции. Природа, 1918,
№ 2-3, 187, 188.
343. П. И. Вальден. Обесценивание материи. М., 1916, 19 стр. Рецензия, Природа,
1918, N° 1, стр. 85, 86.
344. Ф. М. Флавицкий (некролог с портретом). Природа, 1918, № 2—3, 204—210.
345. Проблема утилизации спирта. Природа, 1918, № 4—6, 235—252. (Статья не окончена).
346. Sur la fonction acide du tetroxyde d'osmium. С R. Acad. Sci., Paris, 1918, 167,
162, 163,
347. Une reaction nouvelle de rosmium. C. R. Acad. Sci., Paris, 1918, 167, 235.
348. Sur unc fase nouvelle complexe de rosmium. Bull. Soc. chim., 1918, 23, 377—379.
349. Hydroxylamine platinum bases. J. Chem. Soc, 1918, 113, 884—897. (In collaboration
with I. I. Tschernjaeff).
350. Открытие кислорода и теория горения в связи с философскими учениями древнего
мира. Петроград, 1919, стр. 1—80. (РСФСР. Научно-техн. отд. ВСНХ. Науч. хим.-
техн. изд-во, серия научно-популярная, вып. 4.).
351. О назначении и задачах института гго изучению платины и других благородных
металлов. ЖРФХО, ч. хим., 1919, 51, вып. 3—5, 183—,193; Изв. Ин-та по изучению
платины и других благородных металлов, 1919, вып. 1, 1—11.
352. О гидразиновых соединениях платины. ЖРФХО, ч. хим., 1919, 51, вып. 3—5, 196—
211; Изв. Ин-та по изучению платины и других благородных металлов, 1919, вып. 1,
14—29. (Совместно с М. С. Григорьевой).
353. О лидро'ксила чиповых соединениях платины. ЖРФХО, ч. хим., 1919, 51, вып. 3—5,
211—231. Изв. Ин-та по изучению платины и других благородных металлов, 1919,
-вып. 1, 29—49. (Совместно с И. И. Черняевым).
354. Исследование над комплексными соединениями платины. ЖРФХО, ч. хим., 1920,
52, вып. 1—3, 47—63, 115—138; Изз. Ин-та по 'Изучению платины и других
благородных металлов, 1919, вып. 2, 51—89.
355. О некоторых молекулярных перегруппировках, наблюдаемых в ряду комплексных
соединений платины. III. ЖРФХО," ч. хим., 1920, 52, 47—63; Изв. Ин-та по
изучению платины и других благородных металлов, 1919, вып. 2, 51—66 (Совместно
с Н. К- Пшеницыным).
356. Новый способ получения солей хлоро- и бромоплатотриаминоівого ряда (солей
ряда Клеве). IV. ЖРФХО, ч. хим., 1920, 52, вып. 4—6, 115—119; Изв. Ин-та по
изучению платины и других благородных металлов, 1919, вып. 2, 66—70.
357. Об аммиачных соединениях платонитрата. V. ЖРФХО, ч. хим., 1920, 52, 119—135;
Изв. Ин-та по изучению платины и других благородных металлов, 1919, вып. 2,
70—86. (Совместно с С. Кильтыновичем).
358. Об изменении электропроводимости з ряду аммиачных соединений платонитрита.
VI. ЖРФХО, ч. хим., 1920, 52, 135—138; Изв. Ин-та іпо изучению платины и других
благородных металлов, 1919, вып. 2, 86—89. (Совместно с Н. Владимировым).
359. Sur un nouveau procede de preparation de certains composes complexes du platine
et de ses analogues. Bull. Soc. chim., 1919, 25, 234—237.
360. Технология платины. Горное дело, 1920, вып. 4, 127, 128.
361. Замечания к докладу В. И. Бузникова о русской канифоли. Выступление на
заседании Отделения химии РФХО 22.XII.1918 г. ЖРФХО, ч. хим., 1919, 51, вып. 3—5,
299.
362. Институт прикладной химии. Сообщение на заседании Отделения химии РФХО
7.111.1920 г. ЖРФХО, ч. хим., 1920, 512, вып. 7—9, 428.
363. О новой цветной реакции холестерина. Сообщение на заседании Отделения химии
¦ РФХО 4.V.1920 г. ЖРФХО, ч. хим., 1920, 52, вып. 5, 363.
364. О пентамминовых соединениях платины. Сообщение на заседании Отделения химии
РФХО 3.VII.1919 г. ЖРФХО, ч. хим., 1920, 52, вып. 7—9, 419—423.
365. Новая реакция для определения иридия. Сообщение на заседании Отделения хи-
• мил РФХО, 3.VII.1919 г. ЖРФХО, ч. хим., 1920, 52, вып. 7-9, 423, 424.
366 Превращения в ряду производных осмиамовой кислоты. Сообщение на заседании
Отделения химии РФХО 3.VII.1919 г. ЖРФХО, ч. хим., 1920, 52, вып. 7-9, 424,
.. 425. (Совместно с Ф. Ф. Буткевич).
367 Воспоминания' об А. Вернере. Сообщение на заседании Отделения химии РФХО
5.XI.1920 г. ЖРФХО, ч. хим., 1920, 52, вып. 7-9, 433.
488
Список опубликованных трудов Л, А. Чугаева
368. О природе химических элементов по новейшим исследованиям. Сообщение на
заседании Отделения химии РФХО 5.XI.1920 г. ЖРФХО, ч. хим., 1920, 52, вып. 7—9;
433—437.
369. Новый способ для открытия -иридия в присутствии платины и других металлов
платиновой группы. Сообщения о научно-технических работах з Республике, 1920,
вып. 3—4, № 151, 118, 119.
370. Об аципентамминовых солях платины. Сообщения о научно-технических работах
в Республике, 1920, вып. 3—4, № 152, 119—121.
371. О хлорпентаммино/вых соединениях платины. Сообщения о научно-технических
работах в Республике, 1920', вып. 3—4, № 153, 121, 122.
372. Новый способ получения гидроксопеитамминовых солей платитіы. Сообщения о
научно-технических работах в Республике, 1920, вып. 3—4, № 154, 122.
373. О новом ряде комплексных соединений четырехвалентной платины. Сообщения о
научно-технических работах в Республике, 1920, вып. 3—4. № 165. 122, 123.
374. Новая каталитическая реакция, вызываемая чернью родия. Сообщения о научно-
технических работах в Республике, 1920, вып. 3—4, № 156, 123, 124.
375. О химической природе четырехокиси осмия. Сообщения о научно-технических
работах в Республике, 1920, вып. 3—4, № 157, 124.
376. К вопросу об оптической суперпозиции. Сообщение на заседании Отделения
химии РФХО 21.IV.I92I г. ЖРФХО, ч. хим., 1921, 53, вып. 4—9, 380.
377. Нужна ли для России химия «и химическая промышленность. Природа и человек,
1921, № 2, 2—20, № 3, 25—40.
378. О пентамм'иноаых соединениях четырехвалентной платины. Доклад на заседании
секции общей химии третьего 'Менделеевского съезда по чистой и прикладной
химии 26.V.1922 г. Сообщения о научно-технических работах в Республике, 1923,
вып. 1.1, 42.
379. Российский институт прикладной х-им и и. Сообщение на заседании секции
технологии минеральных веществ третьего Менделеевского съезда по чистой и
прикладной химии 26.V.1922 г. Сообщения о научно-технических работах в Республике,
1923, вып. 11, 133.
380. О назначении и задачах Российского института прикладной химии. Изв. Российск.
ин-та прикладной химии, 1922, вып. 1, 9—18.
381. Новый способ открытия иридия в присутствии других металлов платиновой
группы. Изв. Российск. ин-та прикладной химии, 1922, вып. 1, Петроград, 69—71.
Обзор деятельности учреждений ПОНТО, 1920, вып. 1, 150—153.
382. Новый способ получения хлороплатинитов. Изв. Российск. ин-та прикладкой
химии, 1922, вып. 1, 94—96.
383. Химия брожения в на vice и в технике. Изз. Росоийск. ин-та прикладной химии,
1922, вып. 1, отд. 2, 141—166.
384. Русское Физико-химическое общество. Наука и ее работники, 1922, № 5, 14—20.
385. Природа и происхождение химических элементов в связи с новейшими
исследованиями о распаде и об изотопии. Петроград, НХТИ, 1923, 103 стр.
386. О новом ряде комплексных солей иридия, содержащих гидразин. Изв. Ин-та по-
изучению платины и других благородных металлов, 1923, вып. 4, 52—54.
387. О комплексных сульфокислотах платины. Изв. Ин-та по изучению платин-ы и
других благородных металлов, 1926, вып. 4, 44—47. (Совместно-с С. Е. Красиковым).
388. Наука и техника. Современные достижения химической промышленности. М., Изд>
во «Мир», 1923, 124 стр.
389. Uber еіпе neue Reihe von Indium Komplexsalzen, welche Hyclrazin enthalten. Ben
Dtsch. chem. Ges., 1923, Jg. 56, 2067, 2068.
390. Uber komplexe Sulfosauren des Platins. Zs. anorg. Chem., 1923, 131, 299—302. (In
Gemeinschaft mil S. Krassikow).
391. Д. И. Менделеев. Жизнь -и деятельность. Л., 1924, стр. 1—57.
392. О пентамшшовых соединениях четырехвалентной платины. I. ЖРФХО, ч. хим.,
1924, 55, вып. 1—4, 267—318. Изв. Ин-та по изучению платины и других
благородных металлов, 19'26, вып. 3—36.
393. О новом ряде ацидоамидотетрамминовых производных четырехвалентной платины..
ЖРФХО, ч. хим., 1924, 55, вып. 1—4, 319—325. Изв. Ин-та по изучению'платины и
других благородных металлов, 1926, вып. 4, 37—44.
394. Uber komplexe Verbindungen des zwelwertigen Platins mit organischen Disulfide»
und Polysulfiden. Zs. anorg. Chem., 1924, 135, 143—152. (In Gemeinschaft mit S.
Iljn). Verfasst von S. Fritzmann.
395. Uber komplexe Verbindungen .des Palladiums mit organischen Mono- und
Disulfides Zs. anorg. Chem., 1924, 135, 153—160. (In Gemeinschaft mrt Ch. Iwanoff).
Verfasst von E., Fritzmann.
396. Uber LeitfzJhigkeit einiger Verbindungen des Platins mit ThioSthern. Zs. anorg.
Chem., 1924, .135, 365—391. (In Gemeinschaft mil W. Malzschewsky). Verfasst von
E. Fritsmann.
Список опубликованных трудов Л. А. Чугаева
489
397. Uber Leitfahigkeit der PlatinmonosuUidverbindungen in methylalkoholischer Losung.
2s. anorg. Chem., 1924, 135, 392—400. (In Gemeinschaft mit N. Wladimiroff). Ver-
fasst von E. Fritzmann.
398. Ober Pentamminverbindungen des vierwertigen Platins. I. 2s. anorg. Chem., 1924,
137, 1—31.
399. Ober eine neue Reihe Acidoamidotetramminderivate des vierwertigen Platins. II. Zs.
anorg. Chem,. 1924, 137, 401—406.
400 E. Fritz man, L. Tschugajeff's uber Komplexverbindungen des Platins und
Palladiums mit organischen Sulfiden. Zs. anorg. Chem., 1924, 134, 277—282; 135, 143—
162; 153—160, 385—391, 392—400.
401. Ober eine neue Komplexe Base des Osmiums. Zs. anorg. Chem., 1925, 148, 65—68.
402. О .новом комплексном основании осмия. Изв. И-н-тз по изучению плагины и других
благородных металлов, 1926, вып. 4, 4-8—51.
403. О комплексных соединениях платины и палладия с органическими сульфидами.
Изв. Ин-та по изучению платины и других благородных металлов, 1926, вып. 4,
55—60. Работы Л. А. Чугаева и учеников. Составил Э. Фрицман.
404. Об изомерных платиновых соединениях, образуемых органическими сульфидами
(тиоэфирами). I. Изв. Ин-та по изучению платины и других благородных металлов,
1926, вып. 4, 60—73; (Совместно с В. Субботиным). Составил Э. Фрицман. Вег.
Dtsch. chem. Ges., 1910, Jg. 43, 1200.
405. О некоторых комплексных соединениях бромистой платины л органических моно-
и дисульфидов. Ст. II. (Совместно с Д. Френкель.) Изв. Института по изучению
платины и других благородных металлов. 1926, вып. 4, 74—79. Составил Э.
Фрицман. (С. R. Acad. Sci., Paris, 1912, 154, 33).
406. О соединениях, образуемых платонитритом с органическими дитиоэфирами. III.
Изв. Ин-та по изучению платины и других благородных металлов, 1926, вып. 4,
79—100. (Совместно с В. Г. Хлопиным). Составил Э. Фрицман; 2s. anorg. Chem.,
1913, 82, 401.
407. О платиновых соединениях органических сульфидов, аналогичных солям 1-го
'основания Рейзе. IV. Изв. Ин-та по изучению платины и других благородных металлов,
1926, вып. 4, 100—118. (Совместно с В. Г. Хлопиным). Составил Э. Фрицман; 2s.
anorg. Chem., 1914, 86, 241.
408. О комплексных соединениях двухвалентной плагины с органическими
дисульфидами и полисульфидами. V. Изв. Йн-та по изучению платины л других благородных
металлов, 1926, вып. 4, 118—127. (Совместно с С. И. Ильиным). Составил Э.
Фрицман; 2s. anorg. Chem., 1924, 135, 143—152.
409. О комплексных соединениях органических сульфидов с четырехвалентной платиной.
VI. Изв. Ин-та по изучению платины и других благородных металлов, 1926, вып. 4,
127—135. (Совместно іс И. Беневоленским). Составил Э. Фрицман; 2s. anorg. Chem.,
1913, 82, 420.
410. О комплексных соединениях палладия с органическими моно- и дисульфидами. VII.
Изв. Ин-та по изучению плагины и других благородных металлов, 1926, вып. 4,
136—143. (Совместно с X. Ф. Ивановым). Составил Э. Ф-рицман; Zs. anorg. Chem.,
1924, 133, 153.
411. Об электропроводности некоторых платиновых соединений органических
моносульфидов. VIII. Изв. Ин-та по изучению платины и других благородных металлов,
1926, вып. 4, 146—152. (Совместно с В. Мальчевским). Составил Э. Фрицман; 2s.
anorg. Chem., 1924, 135, 385.
412. Об электропроводности платиновых соединений моносульфидов в метиловоспирто-
всм растворе. IX. Изв. Ин-та по изучению платины и других благородных
металлов, 19'26, вып. 4, 153—158. (Совместно с Н. Владимировым). Составил Э.
Фрицман; 2s. anorg. Chem., 1924, 135, 392.
413. Электропроводность некоторых платиновых соединений органических дисульфидов.
X. Изв. Ин-та по изучению платины и других благородных металлов, 1926, вып. 4,
159 — 176. (Совместно с Л. Г. іКобляноким). Составил Э. Фрицман; 2s. anorg.
Chem., 1913, 83, 8.
414. О реакциях окисления соединений платины. I. Окисление при помощи перекиси
водорода и озона. Изв. Ин-та по изучению платины и других благородных металлов,
1926, вып. 5, 85—101. (Совместно с В. Г. Хлопиным). Составил Э. Фрицман; 2s.
anorg. Chem., 1926, 151, 253—268.
415. О платиновых соединениях гидразина и из они три лав. Изв. Ин-та по изучению
платины 'й других благородных металлов, 1926, вып. 4, 299—305. 2s. anorg. Chem..
1925, 148, 37—42. (Совместно с М. Сканави-Григорьевой и А. Позняк).
416. 2ur Kenntnis des Osmium. III. Eine ausfuhrliche Obersicht der Arbeiten von
L. Tschugajeff uber Osmium. 2s. anorg. Chem., 1928, 172, 213 (233).
.417. О некоторых комплексных соединениях хлористой платины с аминоацеталем.Изв.
Ин-та по изучению платины и других благородных металлов, 1929, вып. 7, 118—123.
(Совместно с В. П. Орелкиным).
490
Список опубликованных трудов Л. Л. Чугаева
418. О реакциях окисления комплексных соединений плагины. II. Окисление ори
помощи персульфатов и свободного кислорода. Изв. Ин-та по изучению платины и
других благородных металлов, 1929, выл. 7, 124—137. (Совместно с И. И. Черняевым)
419. О природе чстырехокисн осмия. Обзор работ Л. А. Чугаева по осмига. Изв. Ин-та
по изучению платины и других благородных металлов, 1929, вып. 7, 138—143.
Составил Э. Фрицман.
420. Комплексные производные четьгрехокиси осмия. Изв. Ин-та по изучению платины и
других благородных металлов. 1929, вып. 7, 143, 144. (Совместно с И. И.
Черняевым) .
421. О солеобразующих свойствах четырехокиси осмия. Изв. Ин-та по изучению
платины и других благородных металлов, 1929, вып. 7, 144—150.
422. О распределении четырех окиси осмия между четыреххлористым углеродом, водою
.и щелочью. Изв. Ин-та по изучению платины и других благородных металлов, 1929,
вып. 7, с. 150—156. (Совместно с А. И. Лукашук).
423. О каталитическом разложении перекиси водорода в щелочном растворе четырех-
окисью ссмия. Изв. Ин-та по изучению платины и других благородных металлов,
1929, вып. 7, 155—157. (Совместно с Я- И. Бккерман).
424. О скорости отгонки четырехокиси осмия из водных растворов. Изв. Ин-та по
изучению платины и других благородных металлов, 1929, вып. 7, 157, 158. (Совместно
с. М. Бородулиным).
425. О нозом комплексном основании осмия. Изв. Ин-та по изучению платины и других
благородных металлов, 1929, вып. 7, 159.
426. Превращение в ряду производных осмиамовой кислоты. Изв. Ин-та по изучению
платины и других благородных металлов, 1929, вып. 7, 159, 160. (Совместно с
Ф. Ф. Буткевич).
427. Новый способ открытия иридия в присутствии других металлов платиновой
группы. Изв. Ин-та по изучению платины и других благородных металлов, 1929, вып. 7,
205—207.
428. Новый способ получения хлороплатинитов. Изв. Ин-та по изучению платины и
других благородных металлов, 1929, вып. 7, 207—209.
429. Новая каталитическая реакция, вызываемая чернью родия. Изв. Ин-та по
изучению платины и других благородных металлов. 1929, вып. 7, 120.
СОДЕРЖАНИЕ
От комиссии по -изданию трудов Л. А. Чугасва 3
Г. В. Пигулевский. Жизнь и деятельность Л. А. Чугасва 5
В. В, Лебединский, Значение работ Л. А. Чугаева в развитии химии ... 11
Предмет и задачи современной химии 19
Эволюция вещества в мертвой и живой прлроде 28
Современные задачи органической химии 40
Периодическая система химических элементов 54
Валт-Гофф и судьбы стереохимии 209
Валентность 222
Профессор Альфред Верне-р . 265
Структурно- и стереохимические представления в области неорганической химии 283
О неустойчивых органических соединениях 313
Органические соединения в приложении к минеральному анализу . - . . . 344
Открытие кислорода и теория горения в связи с философскими учениями
древнего мира 350
Эволюция учения о катализе 395.
Памяти А. М. Зайцева и Ст. Канниццаро 418
Памяти В. А. Бородовского 425
Памяти С. М. Танатара 428
Памяти В. Е. Павлова 430
Памяти С. С. Кильтыновича 433
Памяти Ф. М. Флавицкого 435
Оксидазы, или окислительные ферменты 441
К физиологии фосфоресцирующих бактерий 460
Примечания 469
Список опубликованных трудов Л. А. Чугаева 475
Лев Александрович Чугаев
Избранные труды, т. Ill
Утверждено к печати
Институтом общей и неорганической химии
им. И. С. Курнакова Академии наук СССР
Редактор Д. Н. Трифонов
Технический редактор Т. В. Полякова
Корректор Л. И. Рувинская
РИСО АН СССР № 12-22В
Сдано ь набор 7/ХП 1961 г. Подписано к печати 17/IV 1962 г.
Формат 70Х1081/». Печ. л. 30,75 + 1 вкл. = 42,1 усл.-печ. л.
Уч.-изд. л. 40,9 (40,8 + I вкл.). Тираж 1600.
Тип. зак. 4123. Изд. № 721
Цена Э р. 07 к.
Издательство Академии наук СССР
Москва, Б-62, Подсосенский пер., 21
2-я типография Издательства АН СССР
Москва, Г-99, Шубинский пер., 10
ОПЕЧАТКИ И ИСПРАВЛЕНИЯ
Стр.
75
122
Л 1 А
140
162
168
170
179
182
182
185
186
250
275
278
290
299
299
305
306
308
327
329
400
402
417
418
450
450
459
462
Строка
13 сн.
3 св.
таблица,
9 графа
11 сн.
23 св.
7 сн.
18 сн.
7 сн.
8—9 сн.
27 сн.
2 сн.
10 св.
6 св.
7 св.
1 сн.
20 сн.
19 св.
16 сн.
5 св.
2 сн.
16 св.
25 сн.
22 сн.
23 сн.
4 сн.
11 сн.
15 сн.
.15 сн.
28 сн.
23 св.
Напечатано
Сг
AsCla
1
42,4
(СгНгЬ
R(OH3)
Th04
—1280
HJ/HG1
постоянной
кислорода
СЮз
Cu202Ag202
(ОН4)
(рис. 35)
С13]К
000
Co4NH3
(NH3)8
]С
СНг — С — ОН-2
= СН —
1
1
sos
азотистой
1883
1886
осадки
консервативны
J aguet
влияет
Должно быть
Cs, также
AsCl3
1,2
42,4
(СаН5)2
R(OH)3
ТЬСЦ
1280
HCl/HHr
постоянно
к водороду
СЮа
Cu20 AgaO
(0Н)4
(рис. 9 и 10)
СЦ]К
287
Co4NH3
(NH3)3
]CI
СНа—СН—СН3
|
= с —
so2
азотной
1833
1866
оксидази
констатированы
J a q u ? t
является
Чугаев Л. А., Т. III.