/




























































































































































































































Text
АКАДЕМИЯ НАУК СССР
ИНСТИТУТ ГЕОХИМИИ И АНАЛИТИЧЕСКОЙ ХИМИИ им. В. И. ВЕРНАДСКОГО
Серия: АНАЛИТИЧЕСКАЯ ХИМИЯ ЭЛЕМЕНТОВ
АНАЛИТИЧЕСКАЯ ХИМИЯ
УРАНА
ИЗДАТЕЛЬСТВО АКАДЕМИИ НАУК СССР Москва 1962
Серия: ^Аналитическая химия элементов-»
Главный редактор академик А. П. Виноградов
Редакционная коллегия:
И. П. Алимарин, А. К, Бабко, А. И. Бусев, Э. Е. Вайнштейн, А. П. Виноградов, А. Н. Ермаков, В. И. Кузнецов, П. Н. Палей, Д. И. Рябчиков, И. В, Тананаев, Ю. А. Черников
Редакторы тома «Аналитическая химия урана» Д. И. Рябчиков, М. М. Сенявин
Адрес редколлегии:
Москва В-334, Воробьевское шоссе, 47а.
Институт геохимии и аналитической химии им. В. И. Вернадского Академия наук СССР
ОТ РЕДКОЛЛЕГИИ
Институт геохимии и аналитической химии им. В. И. Вернадского АН СССР приступил к изданию серии монографий по аналитической химии отдельных элементов. Эта серия — «Аналитическая химия элементов»— составит около пятидесяти томов и рассчитана на выпуск в течение ближайших 5 лет. Потребность в подобного рода издании давно назрела. Вместе с тем у нас накопился огромный опыт многочисленных лабораторий и теперь стало возможным и необходимым его подытожить. Таким образом возникло настоящее издание — серия «Аналитическая химия элементов»,— которое осуществляется впервые. Аналитическая химия любого элемента и его различных соединений в настоящее время представляется чрезвычайно разнообразной как вследствие сложности современных объектов исследования и широты диапазона концентраций, которые бывает необходимо определить, так и вследствие разнообразия использующихся методов.
В связи с этим для монографий был разработан общий план как в смысле содержания, так и последовательности изложения материала.
В монографиях содержатся общие сведения о свойствах элемента и его соединений. Затем излагаются химические реакции, являющиеся основанием для аналитических целей. Методы как физические, физико-химические и химические излагаются применительно для количественного определения данного химического элемента, начиная с анализа сырья, далее типичных полупродуктов производства, и, наконец, конечной продукции,— металлов или сплавов, окисей, солей и других соединений и материалов. Как правило, приводятся принципы определения и, где это необходимо, дается точное описание всего процесса определения. Необходимое внимание уделяется быстрым методам анализа. Самостоятельное место занимает изложение методов определения так называемых элементов-примесей в чистых материалах.
Обращается внимание на точность и чувствительность методов в связи с общей тенденцией повышения чувствительности методов определения следов элементов-примесей.
Монографии содержат обширную литературу, доведенную до последних лет; они рассчитаны на широкий круг химиков, в первую
1
3
очередь химиков-аналитиков исследовательских институтов и заводских лабораторий различных отраслей хозяйства, а также на химиков-преподавателей и студентов химических высших учебных заведений.
К составлению монографий привлечены наши крупнейшие специалисты, имеющие собственный опыт работы в области аналитической химии того или иного химического элемента.
Отдельные тома серии «Аналитическая химия элементов» будут выходить самостоятельно, по мере их подготовки. Вышли в свет монографии, посвященные торию, таллию и урану, готовятся к печати монографии по аналитической химии плутония, рутения и молибдена.
Мы обращаемся с просьбой ко всем читателям присылать свои замечания и отзывы о монографиях.
Редколлегия
Книга «Аналитическая химия урана» составлена П. Н. Палеем.
Авторы глав: I, II — Н. И. Удальцова, III — С. Б. Саввин; IV—А. А. Немодрук, Ю. П. Новиков, Т. С. Добролюбская, С. И. Синякова, Н. И. Удальцова, Г. Н. Билимович, А. С. Сердюкова, Ю. И. Беляев, Ю. В. Яковлев; V — А. А. Немодрук, М. К. Чму-това, Н. И. Гусев; VI — Н. И. Удальцова; VII — Ю. И. Беляев, П. Н. Палей.
Глава I
ОБЩИЕ СВЕДЕНИЯ ОБ УРАНЕ
Н. И. Удальцова
До открытия трансурановых элементов (нептуния и др.) положение урана, так же как тория и протактиния (атомные номера 92, 91 и 90), в периодической системе Менделеева не вызывало сомнения; их помещали под переходными элементами шестого периода — гафнием, танталом и вольфрамом. В соответствии с тем, что у атомов Nb, Та и W идет достройка электронного уровня 5 d, принималось, что у Th, Ра и U происходит заполнение электронного уровня 6 d. Химические свойства тория, протактиния и урана в значительной степени напоминают свойства элементов переходных групп IVa (Ti, Zr, Hf), Va (V, Nb, Та) и Via (Cr, Mo, W) 1171 ]. По этой причине в большинстве довоенных учебников, а также статьях уран считали аналогом С г, Мо и W и помещали в VI подгруппу периодической системы.
После открытия Макмилланом и Абельсоном в 1940 г. нептуния (атомный номер 93) оказалось, что этот элемент по своим свойствам напоминает уран и совсем не похож на рений, стоящий в периодической таблице непосредственно выше него. Изучение химических свойств последующих элементов — плутония и других привело к выводу, что у этих элементов начинает заполняться электронный уровень 5/, и что они образуют семейство элементов, подобное семейству лантанидов.
Предположение о возможности заполнения электронами уровня 5/ у элементов, находящихся в конце периодической системы, было высказано еще Бором в 1923 г. Тщательное изучение химических свойств, спектров поглощения в водных растворах и кристаллах, магнитной восприимчивости, а также кристаллографических и спектроскопических данных элементов от актиния (атомный номер 89) до калифорния (атомный номер 98) позволило Сиборгу сделать вывод, что наиболее вероятным родоначальником семейства элементов с заполняющимся 5/ электронным уровнем является актиний и что это семейство следует называть семейством актинидов [889, 890]. Имеются и другие мнения о родоначальнике этого семейства;
5
история развития всех представлений в этой области дана подробно в обзорной статье Сиборга [227].
Как известно, название «актиниды» получило сейчас широкое распространение, и в настоящее время большинство ученых считают, что элементы, начиная с актиния, следует располагать в периодической системе Менделеева как семейство, аналогичное семейству «лантанидов» [2, 7, 50, 51, 148, 170, 221, 294]. Но все-таки электронную структуру и место этих элементов в периодической системе нельзя рассматривать как твердо установленные [227]. Сходство химических свойств актинидов, в частности Ра, Th и U, с лантанидами, с одной стороны, и элементами переходных подгрупп IVa, Va и Via, с другой стороны, говорит о двойственности химической природы актинидных элементов [147, 148]. Поскольку разность энергетических уровней таких удаленных подгрупп, как 5/ и 6d
Таблица 1
Радиохимическая характеристика изотопов урана [4]
Массовое число Тип превращения Энергия, Мэз Период полураспада
Природные изотопы
234 а 1 4,763 (74%) 4,716 (26%) 2,48-10’ лет
235 а 1 4,58 (10%) 4,47 (~3%) 4,40 ((83%) 4,20 (4%) 7,13 • 108 лет
238 а Искусствен 4,18 ные изотопы 4,49-109 лет
227 а 6,8 1,3 мин.
228 а (80%). электронный захват (20%) 6,67 9,3 мин.
229 а (20%); электронный захват (80о/0) 6,42 58 мии.
230 а 5,85 20,8 суток
231 а (0,0055%); электронный захват (>99%) 5,45 4,2 суток
232 а 5,31 (69%) 5,27 (31%) 70 лет
233 а 4,823 1,62-10’ лет
236 а 4,499 2,39-107 лет
237 Г. Y 0,245 6,75 суток
239 Г 1,21 23,54 мня.
240 Г 0,36 14±;1 час
6
сравнительно невелика, правильнее было бы говорить об уране не как о «5/-элементе», а как об элементе «области 5/—6d» 1148]. С наибольшей вероятностью электронную конфигурацию (сверх структуры радона) основного состояния нейтрального атома урана в газообразной форме можно представить как 5 / 6d 7s2 [227].
Природный уран содержит три изотопа U238, U235 и U234, относительное содержание этих изотопов равно соответственно 99,28, 0,71 и 0,006%.
Атомный вес урана, вычисленный по масс-спектрографическим и ядерным данным, равен 238,03 [227]; химическими методами найдено значение 238,07 [608]. По новой единой шкале атомных весов, принятой в 1962 г., атомный вес урана равен 238,03. Изотоп U238 — наиболее долгоживущий (период полураспада равен 4,50 • 109 лет [227]) и распространенный изотоп. U235 (актиноуран) среди природных изотопов выделяется тем, что ядро U235 способно делиться на медленных нейтронах.
Получено одиннадцать искусственных изотопов урана с массовыми числами 240, 239, 237, 236, 233, 232, 231, 230, 229, 228 и 227, важнейшим из которых является U233, получаемый как конечный продукт превращений при облучении Th232 медленными нейтронами. Ядро U233 обладает эффективным поперечным сечением деления на тепловых нейтронах, сравнимым с U235. Радиохимическая характеристика природных и искусственных изотопов урана представлена в табл. 1.
Несмотря на то, что уран обычно рассматривают как один из редких элементов, он широко распространен в природе; в земной коре его содержится значительно больше, чем таких элементов, как Cd, Bi, Hg и др.; но он находится главным образом в рассеянном состоянии. Кларк урана в земной коре, по данным А. Е. Ферсмана, равен 1 • 10~3% (вес.). Среднее содержание урана в земной коре составляет 4-10-6 г/г породы [97]. Количество урана в слое литосферы толщиной 20 км оценивают в 1,3-1014 т [97].
Основная масса урана содержится в кислых, с высоким содержанием кремния, породах.
Важнейшими из урановых минералов являются урановая смоляная руда, имеющая состав, близкий к ОзОв, и карнотит, представляющий комплексный ванадат уранила и калия; урановая смоляная руда и карнотит являются главнейшими рудообразующими минералами, но значительные их месторождения встречаются редко. Состав некоторых минералов урана представлен в табл. 2.
Более подробное описание минералов урана дано в книгах Каца и Рабиновича [97], Шубниковой [294а], а также в статье Герасимовского [45].
До второй мировой войны ввиду незначительных масштабов потребления урана переработке подвергались лишь богатые урановые руды. В послевоенные годы в связи с применением урана в военной технике и ядерной энергетике, а также в связи с выработкой
7
Таблица 2 [97]
Состав важнейших урановых минералов
Минерал Основной состав минерала Содержание урана, %
Уранинит UO2, UO3, частично 65—74
ThO2, СеО2
Карнотит K2(UO2)2(VO4)2-«H2O -50
Казолит PbO-UO3-SiO2-H2O -40
Самарскит (Y, Er, Ce, U, Са, Fe, Pb, Th)- 3,5—14
•(Nb, Ta, Ti, Sn)2O6
Браннерит (U, Ca, Fe, Y, Th),Ti5O16 40
Тюямунит CaO-2UO3-V2Os-nH2O 50—60
Цейнерит Cu(UO2)2(AsO4)2-nH2O 50—53
Отенит Ca(UO2)2(PO4)2-nH2O -50
Шрекингерит Ca3NaUO2(CO3)3SO4(OH)-9H2O 25
Уранофан CaO-2UO2-2SiO2-6H2O — 57
Фергюсонит (Y, Ce) (Fe, U) (Nb, Ta)04 0,2—8
Торбернит Cu(UO2)2(PO4)2-nH2O -50
Тухолит Содержится Th, U, С, H, О и др. Переменное
Коффинит U (SiO4)1_x(OH)4x —
запасов богатых руд возникла необходимость массовой переработки бедного сырья.
При переработке бедных урановых руд большое значение имеет их предварительное обогащение. Для отделения урана от пустой породы применяют методы механического обогащения (гравитация, флотация, магнитная сеперация, радиометрическое обогащение, использующее радиоактивные свойства урановых минералов, и др.); после механического обогащения, как правило, получаются концентраты с невысоким содержанием урана. Более богатые промышленные концентраты, содержащие до 20—60% урана, получаются при гидрометаллургических процессах переработки урановых руд, заключающихся в кислотном или карбонатном выщелачивании урана с последующим выделением урана из раствора методами осаждения, экстракции или сорбции.
Описанию технологии урана посвящена монография В. Б. Шевченко и Б. Н. Сударикова [289а].
Уран является типичным металлом; в своих соединениях, как в твердых солях, так и в растворе, он проявляет четыре степени окисления от +3 до +6, три из которых (валентности 3, 4 и 6) установлены давно и хорошо изучены; исследованиями, проведенными позднее, найдено, что уран существует и в пятивалентном состоянии.
В растворе U (III) может быть легко получен растворением солей UC13 и UBr3 или восстановлением U (IV) и U (VI); растворы U (III)
в
окрашены в интенсивно красный цвет. Трехвалентный уран в растворе очень неустойчив и подвергается окислению до U (IV) даже в отсутствие кислорода.
Растворы U (IV) имеют зеленую окраску; состояние U (IV) является довольно устойчивым, особенно в сильнокислом, холодном растворе.
Пятивалентный уран в растворе может быть получен растворением UCI5 в воде или электролитическим восстановлением U (VI) при pH 2,5—3. В растворе U (V) диспропорционирует с образованием U (IV) и U (VI):
2ио2+--4Н5о^ — иоГ + и4+4-бн2о.
Наиболее устойчивой валентностью урана является валентность + 6.
Глава II
ХИМИКО-АНАЛИТИЧЕСКАЯ ХАРАКТЕРИСТИКА УРАНА И ЕГО СОЕДИНЕНИЙ
Н. И. Удальцова
Уран представляет собой очень плотный металл, по внешнему виду напоминающий сталь. Металлический уран существует в трех кристаллических модификациях (а-, р- и у-структуры). Радиус атома урана в металлическом состоянии равен 1,421 А [799]. При высокой температуре уран может быть подвергнут ковке.
Физические свойства приведены в табл. 3.
Таблица 3
Физические свойства урана [637]
Свойства Значение
Точка плавления 1 Точка кипения [ Плотность (25°С) 1 Теплота плавления ~ ! Теплота испарения । Энтальпия (25°С) ; Энтропия (25°С) Электропроводность Теплопроводность j Поверхностная твердость (по Роквеллу) 1132 Дг 1°С 3318° С 19,04 г!см"' 4,7 ккал'моль 106,7 ккал’моль 1521,4 кал моль 11 ,99Дз0,02 кал:.град-молъ 2—-4-Ю1 (ом-см)~' 0,071 кал'см сек град 100
Уран является довольно плохим проводником электричества, слабопарамагнитен.
Металлический уран очень реакционноспособный элемент; он легко взаимодействует со всеми металлоидами, а также образует интерметаллические соединения с Hg, Sn, Си, Pb, Al, Bi, Fe, Ni, Mn, Co, Zn, Be, Ce, In, I r, P, Pt и др.
io
Полный обзор работ по химическим свойствам урана и его соединений до 1936 г. приведен в руководстве [547], а более поздних работ в [97, 145, 227].
Некоторые реакции взаимодействия урдна с металлоидами представлены в табл. 4 [227].
Таблица 4
Реакции взаимодействия урана с металлоидами
Реагирующий элемент Температура взаимодействия. ;С ।
компактный уран порошкообразный уран Продукты взаимоденствп-я
Водород 250 25 ин3
Углерод 1800—2400 800—1200 UC, U2C3, LC.
Фосфор — 600—1000 и3Р4
Азот 700 500 UN, UN1>75, UN.
Кислород 150—350 Пирофорен UO2, USO3
Фтор 25 — UF6
Хлор 500—600 150—180 UC14, UC1S, UCIe
Бром 650 210 UBr4
Под 350 260 UJS, UJ4
Вода 100 25 uo2
Фтористый водород . —• 200—400 UF4
Хлористый водород . — 250—300 UC13
Аммиак 700 400 UN1173
Окись азота .... 400—500 — USO8
Метан — 900 UC
Действие концентрированных растворов кислот на металлический уран в компактном виде дано в табл. 5 [971.
Органические кислоты (муравьиная, уксусная, масляная и др.), разбавленные или безводные, не реагируют с металлическим ураном, но в присутствии хлористого водорода протекают бурные реакции растворения урана с образованием соответствующих солей U (IV).
Ацетат урана можно получить действием уксусного ангидрида или ацетилхлорида на металл.
Растворы гидроокисей щелочных металлов слабо действуют на металлический уран, но при прибавлении к раствору щелочи перекиси водорода уран растворяется с образованием растворимых перуранатов. Растворение металлического урана в ряде кислот и других растворителях подробно описано Ларсеном [691].
Уран является довольно сильным восстановителем, в ряду напряжения уран, по-видимому, расположен близко к бериллию [97]. При действии металлического урана на растворы ряда солей
11
Таблица 5
Действие концентрированных кислот на металлический уран
Кислота Скорость реакции Продукты реакции j Примечание
HF Медленная Фторид U (IV) Образуется нерастворимая
НС1* Быстрая Переменные ко- пленка Реакция сложная
HNO3 H2SO4 Средняя Медленная личества U (III), U (IV), черный остаток Нитрат уранила Кислый сульфат С разбавленной H2SO4
Н3РО4 Медленная U (IV) Кислый фосфат не взаимодействует Реакция протекает быстро
НС104 * При рас Быстрая ** творении урана U (IV) в соляной кислоте о в горячей Н3РО4 В разбавленной НС1О4 не растворяется бразуется нерастворимый осадок
темного цвета, который, как установлено Карабашом Н 191], содержит сложные гндрнды
урана (гндроксигидрид НО—U—ОН). Для полного растворения урана в соляной кислоте добавляют окислители (Н2О2, HNO3, НС1О4 н др.).
** Мелкая стружка урана с крепкой (>60%) хлорной кислотой реагирует чрезвычайно бурно.
(Hg(NO3)2, AgNCh, CuSCh, SnCh и др.) выделяются осадки соответствующих металлов.
Важнейшими бинарными соединениями урана являются гидрид, карбиды, окислы, нитриды и галогениды.
Гидрид [97, 312] представляет собой темно-серый пирофорный порошок, обладающий большой химической активностью. Гидрид урана растворяется в НЬЮз, в концентрированной HCICh и горячей концентрированной H2SO4. Разлагается при нагревании >400° в вакууме.
В литературе [97] описаны три карбида урана UC, U2C3 и UC2, имеющие вид тугоплавких, с металлическим блеском кристаллов, которые разлагаются водой и разбавленными кислотами, особенно при нагревании, с образованием солей уранила.
Получены бориды урана UB2, UB4, UB]2 [97, 227]. Бориды UB2 и UB4 растворимы в HNO3 и HF, а также в концентрированной Н3РО4; UB12 не растворяется в горячей концентрированной НС1 и HF, но медленно растворяется в горячей концентрированной H2SO4.
Известно несколько силицидов урана состава USi, USi2, US is, U.S is. Дисилицид USi2 представляет собой светло-серый порошок, не растворимый в холодных и в горячих кислотах (НС1, HNO3, HsSCh), в царской водке, но растворимый в концентрированной HF.
12
Известны три окисла урана — двуокись UO2, закись-окись U2O8 и трехокись UO3; имеются также указания о существовании одноокиси UO [3]. Исследование системы U—О проводилось многими авторами [97, 227, 603].
UO2 — темно-коричневый порошок, обычно получаемый восстановлением высших окислов урана водородом; обладает сильноосновными свойствами. UO2 не растворяется в разбавленной и концентрированной НС1, но хорошо растворяется в НИОз с образованием уранилнитрата, а также в концентрированной H2SO4 при нагревании. .
Трехокись UO3, цвет которой меняется от красного до желтого, в зависимости от кристаллической модификации имеет амфотерные свойства; с кислотами UOs взаимодействует с образованием солей уранила, со щелочами — с образованием уранатов. 170з растворяется во всех минеральных кислотах, а также в уксусной кислоте.
ИзОв, представляющая собой черный или темно-зеленый порошок, получается при прокаливании на воздухе как UO2, так и UO3. При растворении в кислотах ПзОв дает смесь солей четырехвалентного урана и уранила. ИзОв плохо растворима в разбавленной HCI, в концентрированной НС1 при нагревании растворение идет быстрее, добавление окислителей способствует быстрому растворению ЦзОв. Закись-окись хорошо растворима в HNO3 с образованием уранилнитрата, а также полностью превращается в уранилсульфат и сульфат урана (IV) при продолжительном нагревании с концентрированной H2SO4.
В ряду UO2, ПзОв и UO3 увеличивается сродство к воде. Гидрат двуокиси урана иОг-хНгО получается при действии аммиака или щелочи на растворы солей U (IV), а также при гидролизе разбавленного раствора хлорида или ацетата урана (IV). На воздухе гидрат легко окисляется до ИОз • Н2О. После высушивания над серной кислотой гидрат имеет состав иОг-2НгО. Свежеприготовленная гидроокись урана (IV) хорошо растворима в кислотах, но при стоянии ее растворимость уменьшается.
Известен ряд гидратов ИОз : UO3H2O (или H2UO4), UO32H2O (или H4UO5), 2иОз-НгО (или H2U2O7) и др. Гидраты трехокиси урана гораздо более устойчивы, чем гидраты двуокиси. Гидраты ИОз плохо растворимы в воде.
Уран образует также перекись ПО4-2НгО, которая осаждается при добавлении перекиси водорода к слабокислому раствору соли уранила. Некоторые авторы рассматривают ПО4-2НгО как перуранат уранила (UCb^UOe [227, 985].
При добавлении щелочи к раствору уранила, содержащему перекись водорода, осаждаются перуранаты типа МгИгО^-хНгО, МгПОб-хНзО, МбПгО^-хНгО и МДЮв-хНгО, состав которых зависит от концентрации Н2О2 и щёлочи в растворе. Хорошо изучены перуранаты щелочных металлов [63, 201, 336].
13
Известны для урана тройные окислы. Наиболее хорошо изученными тройными окислами являются уранаты щелочных и щелочноземельных элементов: моно- и диуранаты состава MezUCh и MezUzCh. Уран образует и полиуранаты [731, 1001].
Нитриды урана UN, U2N3 и UN* легко окисляются на воздухе, трудно растворимы в кислотах и в растворах щелочей, но разлагаются расплавленными щелочами.
Получено шесть фторидов урана, из которых UF3, UkFj?, U2F9 и UF4 считаются нелетучими, a UFe летучим. Важнейшими из фторидов урана являются UF4 и UFe. UF4 — труднорастворимое соединение зеленого цвета получается осаждением плавиковой кислотой урана (IV) из раствора его солей или при действии газообразного фтористого водорода на ряд соединений урана [97]; описаны методы электролитического и фотолитического приготовления UF4 [324, 1050]. В химическом отношении UF4 является устойчивым, довольно неактивным соединением. В кислотах-окислителях фторид урана (IV) растворяется с образованием раствора соли уранила; быстро растворяется UF4 в горячем растворе А1(ЫОз)з или А1С 1з, а также в растворе борной кислоты, подкисленной H2SO4, НС IO4 или НС1. С фторидами других металлов UF4 образует ряд малорастворимых двойных солей (MeUFe, MezUFe, MesUF- и др.).
UFe — бесцветное, легколетучее соединение (температура кипения 56,5° [97]), получаемое, как правило, при действии элементарного фтора на соединения урана 197], является очень реакционноспособным веществом. UFe — энергично реагирует с водой с образованием UO2F2 и HF, а также с большим числом органических веществ и растворителей. Гексафторид урана взаимодействует с большинством металлов, что осложняет способы его хранения; удовлетворительным материалом для аппаратуры при работе с UFe являются медь, никель и алюминий, а также фторсодержащие полимеры (тефлон и др.). При температуре 25—100° UFe образует комплексные соединения с фторидами щелочных металлов и серебра типа 3NaF-UFe, 3KF-2UFe и др.
Известны четыре соединения урана с хлором: UCI3, UCh, UCk и UCk. Методы получения этих соединений описаны подробно в книге Каца и Рабиновича [97].
UCI3 — соединение оливково-зеленого цвета; при растворении в воде UCh дает виннокрасный, неустойчивый раствор U (III). UCI3 как в виде сухой соли, так и в растворе является сильным восстановителем.
UC14 и UCk представляют собой соли зеленого цвета. UCU очень гигроскопичен, на воздухе он притягивает влагу и легко окисляется до U (VI); с рядом хлоридов других металлов дает комплексные соединения типа NazUCk, BaUCk и др.
UCk — легколетучее вещество, очень гигроскопично; вода разлагает его по реакции:
2UCk + 2Н2О — UCI4 + UO2CI2 + 4НС1.
14
ЬтС1б образует черные или темно-зеленые кристаллы (что зависит от метода очистки и скорости роста кристаллов [97]). Гексахлорид урана летуч и исключительно неустойчив в присутствии влаги; вода бурно разлагает UCU с образованием UO2CI2.
Известны и другие галогениды урана: иВгз, UBu, Шз и UJi [97 ],
Уран образует также ряд оксигалогенидов: UO2F2, UOCI2 и др. 197].
Наиболее практически важными растворимыми солями урана являются уранилнитрат, уранилсульфат, уранилхлорид, уранилацетат, а также соли урана (IV) — сульфат и хлорид.
Таблица 6
Растворимость UO2(NO3)2-6Н2О в органических растворителях [637]
Растворитель Растворимость UO3(NO.)2, г, г растворителя Растворитель Растворимость UO2(NO3).2, г/г растворителя
Метанол 0,675 Метилпропилкетон . . 0,484
Этанол 0,615 Метил-н. бутилкетон 0,425
Пропанол 0,529 Метилизобутилкетон . . 0,428
Изопропанол .... 0,549 Метил-ги-бутилкетон . . 0,415
Бутанол 0,462 Метиламилкетон . . . 0,382
н. Пентанол .... 0,387 Ацетофенон 0,312
н. Гексанол 0,341 Нитрометан 0,140
н. Гептанол 0,310 Нитроэтан 0,051
н. Октанол 0,280 1-Нитропропан . . . . 0,011
Каприловый спирт . 0,271 Диэтиловый эфир . . . 0,491
2-Этил-1-гексанол . . 0,235 Фуран 0,003
Циклогексанол . . . 0,403 Тетрагидрофуран . . . 0,464
Ацетон 0,617 Диоксан 0,291
Дибензиловый эфир . . 0,017
Метилэтилкетсн . . . 0,547 Вода 0,540
i
Нитрат уранила иО2(МОз)г образует три гидрата, содержащих шесть, три и две молекулы воды; все гидраты хорошо растворимы в воде, а также в органических кислородсодержащих растворителях (спиртах, эфирах, кетонах и т. д.) [34 , 35 , 37, 1020]. Растворимость иО2(НОз)2-6НгО в органических растворителях при 20е дана в табл. 6. Не растворим уранилнитрат в сероуглероде и хлороформе.
Растворимость UChSCh-SHaO в воде равна 18,9 г на 100 г растворителя (при температуре 13,2°); растворимость иОг(СНзСОО)2-•2НгО равна 9,2 г на 100 г воды (при 17°).
Важнейшими труднорастворимыми солями урана являются диуранат аммония (NH^UaOz, уранаты щелочных и щелочнозе-
15
мельных элементов, фосфаты уранила типа MeUChPCh (гдеМе = Н+, К4" Na4" или др.), арсенаты AleUOsAsOi, пероксид, уранованадаты и др., растворимость которых представлена в табл. 7.
Таблица 7
Растворимость некоторых труднорастворимых соединений урана
Соединен не Произведение растворимо,ти Растворимость Литература
UO2NH4PO4 4.36-10-27 — [283]
(UO2)3(PO4)2 .... 4,73-Ю"4' — [93а]
UO2KPO4 7,76•10 24 — [283]
UO2HPO4 2,14-Ю11 — [283]
UO2SO3 2,56-1G9 — [ЮЗ]
UO2NH4AsO4 .... 1,71 - IO’ 24 — [285]
UO2KAsO4 2,52-Ю"23 — [285]
UO2NaAsO4 .... 1,35-10 22 — [285]
UF4 — МО'4 моль'л (25°) [97]
U(OH)4 (1,102=0,72)-10"’2 — [247а]
UO2(CeH5N2O2)2- -NH4C6H5N2O2 (купферонат уранила и аммония) . . (5,812=2,5) -10“10 - [101а]
UO2HAsO4 3,17-10 -11 — [285]
UO4-2H2O 7,78-10"10 *оль!л 5-10"2 г’.л (25°) Клыгин А. Е. 1954 г.
U(C2O4)2-6H2O . . . (4,3210,4)-IO"22 [56] [1054]
U(HPO4)2 3,1 • 10"23 — Моисеев И. В., 1954 г.
NH4[UO2(OH)2VO3] . 1,7-10"13 — [159]
NH4[UO2(OH)(VO3)2]. •1,5H2O 2,9- 10“14 — [159]
NH4[UO2(VO3)3]. •3,5H2O 9,6-10-’5 — [159]
Зависимость растворимости фосф ата урана (IV), определенная
И. В. Моисеевым, от кислотности среды при 20° представлена
в табл. 8.
Таблица 8
Зависимость растворимости фосфата урана (IV) от кислотности [184)
pH 4,1 I ,8 1 ,3 0,7 0,45 0,3 , 1NHC1
Растворимость, моль [л 2,1-Ю"7 2,1-10-8 4-10"® 2,6-Ю"5 6,4-10“’ 1,6-10“* б.МО'4
16
В аналитической химии находят применение также и трудно-растворимые ферроцианиды уранила. В зависимости от условий осаждения могут образовываться (L'O-2)2[Fe(C\)6], Me4(UО2)4 [Fе(СХ)б 1 з, 5(L 621 •: [Fe(СХ)в ] • Мел iFeiСХ»-? ] и др. [ 100,234 ].
Как ионы урана (IV). так и ионы уранила образуют соли, простые или комплексные с большим числом органических кислот: яблочной, молочной, тиогликолевой, 1-нитрозо-2-гидрокси-3-наф-тойной. хинальдиновой, аскорбиновой и др. Большинство этих солей трудно растворимы в воде. При прокаливании соли урана как неорганические (кроме фосфатов, арсенатов), так и органические, как показало термогравпметрическое исследование, переходят в закись-окись 1478].
Уран относится к числу элементов, весьма предр; сположенных к комплексообразованию. Известно большое число комплексных соединений как четырех-, так и шестивалентного урана. Большинство этих соединений относится к типу двойных солей, кристаллогидратов и внутрикомплексных солей. Следует отметить, что для урана малохарактерны комплексы с азотсодержащими, а также серусодержащими аддендами, хотя в литературе есть указания о существовании таких комплексных соединений; хорошо изучены соединения уранил-иона с различными аминами и мочевиной 1153].
Уран как в четырех-, так и в шестивалентном состоянии проявляет очень большую склонность к комплексообразованию с циклическими кислородсодержащими аддендами (СО2“, SO2-, С2О2" и др.).
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ УРАНА (/V)
Ион урана (IV) относится к числу комплексообразователей, характеризующихся большой величиной радиуса (г— 1,05а); в этом отношении он близок к Th и Zг. Систематические исследования, проведенные В. А. Головней и Г. Т. Болотовой, свойств некоторых
Таблица 9
Влияние температуры на комплексообразование (J (IV) [637]
Реакция комплексообразования Константа образования при p. = 2,0
10° 25° 40°
U4++C1"=UC13+ 3,3 1,21 0,91
и4+4-2СГ=иС1Г — 1,14 0,80
U4+-j-SCN-=USCN3+ 60 31 20
U4+2SCN“=U(SCN)^ 2-102 l,3-102 95
u4+4-HS0;=uscV 4- н+ 4,3 • 102 3,3-102 2,4- IO-2
,3- 0 7,4-10’ 5,7-10’
U4++2HSO4 =U(SO4)V2H +
2 Аналитическая химия урана
ацидокомплексных соединений урана (IV) (сульфатных, карбонатных и оксалатных) показывают, что в большинстве случаев уран (IV) проявляет координационное число, равное восьми [48].
Сульфатные комплексы урана (IV) изучались рядом авторов [48, 48а, 450, 583]. Считают, что в сернокислом растворе могут присутствовать комплексы USO4+, и($О4)з~, U (SO4)t~ и др. Для комплекса USO*+ рассчитана константа образования, равная 3,3-10s (при ионной силе |1 = 2 и температуре 25°) [450].
Установлено, что уран (IV) образует комплексы с хлорид-ионом [308, 450, 583, 680] типа UC1’+, UClj-1-, а также UClg- [8]. Были сделаны попытки количественно оценить константы комплексообразования этих соединений: константа образования комплекса UC1S+ равна 1,21 (при р = 2, температуре 25°); комплекса UC11+ —1,14 (р = 2, температура 25°) [450]. В бромидном растворе существует только комплекс UBrs + [310] с константой образования, равной 1,5 4-0,5 ((1=1, температура 20°).
Достаточно подробно описаны в литературе роданидные комплексы урана (IV): USC№+ и U (SCN)s+ [310, 450]; константы образования данных комплексов соответственно равны 31 и 1,3-10* (р = 2, температура 25°) [450]. Указывается также, что возможйо образование третьего комплекса U (SCN)/ [310]. Зависимость комплексообразования U (IV) с С1“, SCN" и SO1" от температуры дана в табл. 9. Из фторидных комплексов известны UFS , UOFS [8], UFS+ и UFf [450]. Указывается [450], что константа образования UFs+ равна "-10®, a Пр2+"-10& (р = 2, температура 25°).
С оксалат-ионом в нейтральной среде уран (IV) также образует комплексные соединения, подробно из которых изучен и(СгО4)1-[8, 56]. Константа нестойкости этого комплекса равна 5,7-10-2* [1054]. При подкислении раствора, содержащего ГДСгО*)!-, выпадает труднорастворимый оксалат U(CaO4)2.
В растворах, содержащих избыток НСО,- или СО*-, уран (IV) существует в виде растворимого комплекса [150, 578], состав которого точно еще не установлен.
Ион урана (IV) образует комплексы с большим числом органических соединений. Описаны комплексные соединения урана (IV) с оксикислотами (винной, лимонной, гликолевой и др.), с пирокатехином состава [U2(CeH4O2)7]6~ и [и(СвН4Оз)ОНJ4-, с салициловой кислотой, с дикетонами и др. [8]. Важными комплексами в химии урана являются: купферонат урана (IV), который не растворим в воде, но хорошо растворим в органических растворителях (эфире, хлороформе и др.), а также комплексы с реагентами арсеназо I, арсеназо II и арсеназо III [128, 216]. Константа нестойкости, комплекса урана (IV) с арсеназо I равна 6-10"17 [1038].
18
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ УРАНИЛ-ИОНА
Комплексные соединения уранила служили предметом многих исследований; было установлено, что уран (VI) может образовывать как комплексные анионы, так и катионы. Интересные сведения в этом отношении были получены изучением электрофореза водных растворов уранила [227,347]; из растворов солей уранила, содержащих НС1, КС1, NaCl, KBr, NaJ, KJ, NaClCb, NaClO*, NaNO3, NasSCh и др. уран переносится к катоду; из растворов, содержащих Избыток HaSO«, HaSeOe, Н3РО4, NaNOe-j-CHsCOONa, оксалат-, Тартрат- и цитрат-ионЫ, уран переносится к аноду. Эти данные Свидетельствуют о том, что в зависимости от типа аниона и его концентрации уранил образует комплексы анионного или катионного характера.
Уранил-ион дает с сульфат-, карбонат- и оксалат^анионами большое число комплексных соединений, исследование которых как в твердом их состоянии, так и в водных растворах физико-химическими методами указывает на следующий состав (табл. 10).
Таблица 10
Состав некоторых ацидокомплексов уранил-иона
Анион Состав комплексного соединения Литература
1 Ме4+[и0,(С0,),| [284, 346]
сся~ Me6+[(UOJ2(COs)s].nH2O [284, 346]
» Me2[UO2(CO,)2]nH2O [150, 284, 346]
[UO2(SO4)4]’~ [307, 337, 339]
SC£- Ме.4+[UO2(SO4)3] [284, 307, 321]
Mea+[UO2(SO4)2pnH2O [284, 307, 321,713]
Ме2 [(UO2)2(SO4)s]-nH2O [284]
UO2H2C2O’+ [591]
с,оГ Me4+ [UO2(C2O4)S] [284]
Me.+ [(UO2)2(C2O4)s].nH2O [58, 284]
Me^lUO^QOJJn^O [58, 284, 591]
Ме2+[(иО2)2(СЛ)з1 [284]
2
19
Подробные систематические исследования И. И. Черняева с сотрудниками [284] карбонатных, сульфатных и оксалатных комплексных соединений уранила, основанные на общих положениях координационной теории Вернера, позволили сделать вывод о существовании единых генетических рядов этих соединений; уранил-ион во всех случаях проявляет координационное число шесть. Эти ряды на примере аммонийных производных выглядят так [284]:
I. Ряд карбонатов
(NH4)4 [иО2(СОз)з]
(NH4)6[(UO2)2 (СОз)з (Н2О)2]-Н2О
(NH4)2 [иО2(СОз)2(Н2О)2]
(NH4)3 [(иО2)2(СОз)з(ОН)(Н2О)5 ]
Ш4[иО2(СОз)(ОН)(Н2О)з]
иО2СОз-Н2О.
Изучение свойств соединений этого ряда показывает, что наиболее устойчивым по отношению к гидролизу в водном растворе является анион трикарбонат-уранил [иО2(СОз)з]4-. Константа нестойкости этого аниона равна (1,7±0,6)-10~23 (р=1, температура 25°) [102].
11. Ряд оксалатов
(NH4)4[UO2(C2O4)3]
(NH4)6[(UO2)2(C2O4)5(H2O)2]-4H2O
(NH4)2[UO2(C2O4)2(H2O)2]
(NH4)2 [(UO2)2(C2O4)3 • 3H2O ]
NH4[UO2(C2O4)(OH)(H2O)3]
UO2C2O4-3H2O.
Наиболее устойчивым соединением этого ряда является диоксалат-диакво-уранил аммония (NH4)2[UO2(C2O4)2]-2НгО. Константа нестойкости аниона [UO2(C2O4)2]2- равна 8,3-10“12 (при 25°) [198], аниона [(UO2)2(C2O4)sравна ^5-10_7 (при 25°С) [58].
III. Ряд сульфатов
Met[UO2(SO4)3]
Met[(UO2)2(SO4)5(H2O)2]
Met [UO2(SO4)2(H2O)2]
Met[(UO2)2(SO4)3 . иН2О]
Me[UO2(SO4)(OH)(H2O)3]
UO2SO4-3H2O.
Наиболее легко выделяемым в твердом виде и наиболее устойчивым в водном растворе является соединение дисульфатного типа. Константа образования комплекса иО2(5О4)г~ равна 7,1 102
X
20
21
Константа
Условия (ИОН- ----------------------------
Анион Реакция комплексообразования ная сила, тем- Литература
пература, 0 С) образования нестойкости
22
(при р=2 и температуре 25°) [449]. В нейтральных растворах установлено. наличие комплексов [(UOaOH^SOd, [(UO2OH)3(804)2] и [(иОгОН)48О4]2+ с константами устойчивости, соответственно равными 13,6; 13,6 и 3,28 (С. Брусиловский, 1956 г.).
Состав и константы образования некоторых комплексов уранила даны в табл. 11.
Отсутствие комплексов с уранил-ионом доказано лишь только для перхлорат-иона [851]. Зависимость комплексообразования уранил-иона с NO3, С Г, SO2r и F" от температуры представлена в табл. 12.
Таблица 12
Влияние температуры на комплексообразование уранил-иона [637]
Реакция комплексообразования Константа устойчивости при 2,0
10° 25° 40°
ио*+ +СГ = игС1+ 0,58 0,88 1,14
ио*++ no; = uo2no3+ 0,30 0,24 0,17
UO2+ + sof = UO2SO4 63 76 96
UO2 + 4- 2SO4~ = ио2 (SOJ*- 5,8-Ю2 7,1•102 8,2-Ю2
UO2 + + HSO7 = UO2SO4 + н + 6,1 6,4 6,5
UO®+ + HF = uo2f+ 4- н+ 5,5 2,6 21
Большинство комплексов уранил-иона с неорганическими соединениями бесцветны и хорошо растворимы в воде; их существование должно учитываться как при отделении урана (экстракцией и др.), так и при его определении различными методами (спектрофотометрическими, потенциометрическими и др.).
Наибольшее значение в химии уранил-иона имеют его комплексные соединения с органическими реагентами.
Ионы лимонной, винной, яблочной и молочной кислот образуют с уранилом устойчивые даже при высоких значениях pH (8—10) комплексные соединения. В литературе описаны лимоннокислые комплексы уранила с мольными соотношениями UO1+: цитрат3"= = 1 : 1, 2 : 3 и 2 : 1; отрицательные логарифмы констант неустойчивости двух первых соединений соответственно равны рК^З.ОЗ и рК2=6,16 [503, 593].
В системе уранил — тартрат возможно существование трех комплексов с мольными соотношениями уранил : тартрат, равными 1 : 1, 2 : 1 и 3 : 1 [501].
23
Указывается, что в кислом растворе в системе уранил-ион — молочная кислота образуется комплекс с соотношением UO2+: лак-тат-ион, равным 1 : 1 [501]. Ю.И. Грызин (1958 г.), изучая систему уранил-ион — молочная кислота, нашел, что при этом образуется два комплекса [иО2(мол)] + и [иОг(мол)2] с константами неустойчивости ^=3,34-10~4 и Кг=9,45-10-1.
Установлено, что в водных растворах имеет место полимеризация цитратных, тартратных и лактатных комплексов уранила с образованием двуядерных и трехядерных комплексов [502].
С гликолевой кислотой уранил-ион образует три комплекса с соотношениями UO22+: гликолят-ион, равными 1 : 1, 1 : 2 и 1:3. Вычислены константы образования этих комплексов, равные 265±15; (9,1 ±0,6)• 103 и (1,6±0,2) • 105 [309].
В кислой среде (рН<3) уранил-ион дает окрашенный комплекс с аскорбиновой кислотой с соотношением 1 : 1 [42, 555]. В хлорнокислой среде с ионной силой р=0,1 и при температуре 20° константа нестойкости этого комплекса [иОгНА]+ равна 3,3-10[42].
Уранил-ион образует комплексы с 1,3-дикетонами (ацетилаце-тоном, бензои л ацетоном, дибензоилметаном, пиколиноилацетоном и др.). Подробно изучен бензоилацетонат уранила UO2(CH3—СО — =--СН—С—СН3)2 [59]. Соединения с дибензоилметаном и др. имеют состав подобный UO2X2 (где X — одновалентный анион 1,3-дикар-бонильного соединения) [869, 870 , 928]. Ю. П. Новиковым (1958 г.) найдено, что уранил-ион образует также с дибензоилметаном в спиртовой среде комплекс состава 1:1, константа нестойкости которого равна (1,67 ±0,1) • 10 ~4.
Важными для аналитической химии являются комплексы уранила с диэтилдитиокарбаматами (и их производными) и ксанто-генатами; эти соединения хорошо растворимы в органических растворителях.
Выделены в твердом виде труднорастворимые в воде комплексы уранила с диэтилдитиокарбаматом натрия UO2(S2NC5H,0)2 и UO2(S2NC5H10)2-NaS2NC5H,0 (Казаков В. М., 1954 г.). Спектрофотометрическим методом установлено, что в растворе присутствуют также комплексы типа UO2X-K2UO2X4 (где X — радикал диэтилдитиокарбамата) [1030]. Константа нестойкости комплексного аниона [UO2X4]2- при 25° равна приблизительно 5-10"18 [519]; константа нестойкости комплекса UO2X 3 при р=1 и 18° равна -х. 10"6 [80а]. С ксантогенатами уранил-ион образует также окрашенные комплексы, подобные диэтилдитиокарбаматным комплексам.
Окрашенные комплексы образуются при взаимодействии ура-нил-иона с салициловой [состав 1 : 2, константа диссоциации равна 1,24-10“5 [477]) и сульфосалициловой кислотами [8, 332, 606]; с л!-оксибензойной кислотой [8, 606], салициламидом [417], с мо-реллином (IUOjT : 1 мореллин) [701 ], ализарином красным S (устойчивый комплекс при pH 8,2; состава 1 : 1) [927, 993]; с морином [328], с ауринтрикарбоновой кислотой [комплекс состава 1:1, 94
константа образования комплекса равна (5,9 —0,5) • 104 при 25° ] [761 ]. .Важным соединением для химического анализа является трудно растворимый в воде, но хорошо растворимый в органических растворителях оксихинолинат уранила UO2(C9H6NO)2 • •C9H6NOH. Комплекс обладает свойствами кислоты, и поэтому считают, что его формуле правильнее писать так: Н [L'O9(C9H6NO)3] [394].
Уранил-ион образует комплексныессединения подобного же состава с производным оксихинолина [483], а также с купфероном состава NH4[UO2(C6H5N2O2)3] [373, 609].
Описаны внутрикомплексные соединения уранил-иона с шиффовыми основаниями и с некоторыми производными 2-нафтола [217], а также с производными азометина и формазила [903], с о-крезотиновой кислотой [987] и целым рядом других органических соединений. Ценными аналитическими свойствами обладают комплексы урана (VI) с реагентами арсеназо I, арсеназо II и арсеназо III [216]. По данным А. Ф. Кутейникова, константа нестойкости комплекса урана (VI) с арсеназо I равна 2,5-10-12 [1038].
СН2—N—(СН2СООН)2
Этилендиаминтетрауксусная кислота |
СН2—N—(СН2СООН)2 (комплексон II) и ее двунатриевая соль (комплексон III) образует с уранил-ионом ряд комплексов, правда, значительно менее прочных, чем с большинством других элементов, что позволяет использовать в аналитической химий урана комплексон II и III как маскирующие агенты при отделении и определении уранил-иона. Описаны следующие комплексы уранила с этилендиаминтетрауксусной кислотой, образующиеся при pH 4—6: (UO2)2y и [UO2y]2- (где у-анионный остаток комплексона II) [186, 188, 387, 403, 573, 834].
Наши исследования показали, что в более кислых растворах при pH 2,5—4 существуют комплексы состава [UO2(H2y)2l2 и [UO2(H3y)4]2"[188]. Константы нестойкости комплексов (UO2)2y и [UO2yl2" при ионной силе п—0,1 и температуре 20°С, по нашим данным, равны соответственно 1-10 17 и 4-10"12 [188]; по даннымА. Г. Козлова и Н. Н. Крота, они равны (6,7±2,8) • 10"1в, (5,2±2,4) • 10"11[104а].
СОСТОЯНИЕ ИОНОВ УРАНА В РАСТВОРЕ
В растворе уран может присутствовать в виде ионов, соответствующих четырем степеням окисления: 4-3, +4, 4-5 и 4-6.
ТРЕХВАЛЕНТНЫЙ УРАН
Водные растворы трехвалентного урана получают либо растворением его солей (UC13, UBr3), либо восстановлением растворов урана (IV) и урана (VI).
25
В растворе присутствует катион U3", который очень легко подвергается окислению до четырехвалентного урана даже в отсутствие кислорода; уран (III) в растворе медленно восстанавливает воду до свободного водорода. Комплексообразование и гидролиз трехвалентного урана изучены слабо, так как неустойчивость урана (III) затрудняет эти исследования. Но имеющиеся данные позволяют сделать предположение, что способность к комплексообразованию у урана (III) невелика. Соли U (III) в растворе гидролизованы меньше, чем соли урана (IV), но несколько сильнее, чем урана (VI), о чем свидетельствует табл. 13 [227].
Таблица 13
Значения pH 0,02 Л1 растворов ионов урана
Ион U’ + U4 + uof
pH раствора 2,4 1,3 2,9
Вычисленное методом Каснера значение первой константы гидролиза урана (III) рК —9,2 [227].
Найдено, что формальный потенциал системы U(III)/U (IV) в 1 М растворе НС1О4 при 25° равен —0,631 ±0,005 в, а в 1 М растворе НС1 равен — 0,640 ±0,005в [685]. Различие между этими значениями потенциалов приписывается образованию непрочного хлорид-ного комплекса урана (IV).
Латимер [137] дает следующую схему потенциалов урана (в вольтах) в различных степенях окисления: для кислых растворов:
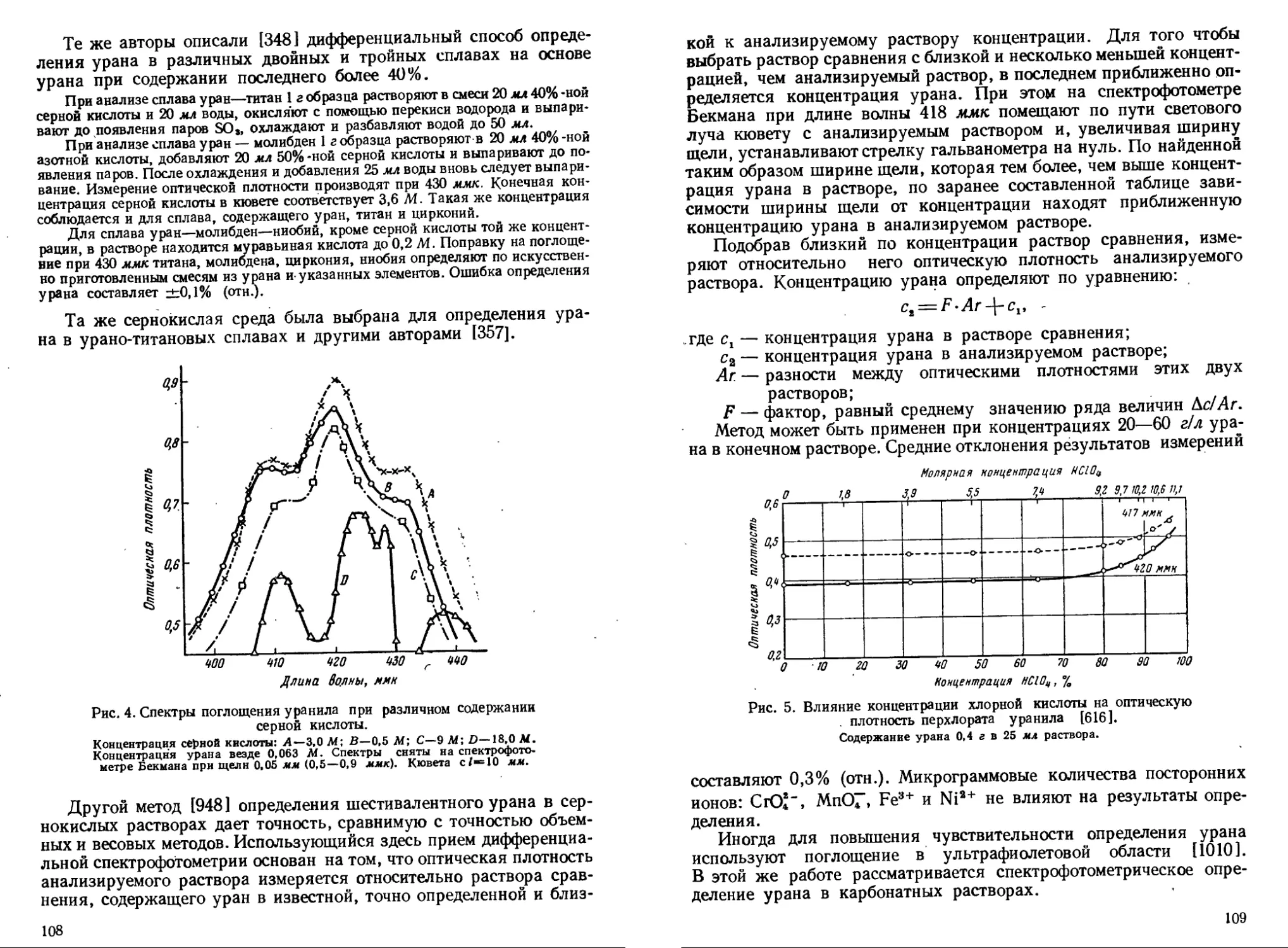
и3+ и4+ ио2+-’- иоГ,
I------0 ,354--1
для щелочных растворов:
и и (ОН), и (0Н)4 ио2 (ОН)2.
Установлено, что реакция U3+^±U4+±e обратима на ртутном и платиновом электродах [685].
ЧЕТЫРЕХВАЛЕНТНЫЙ УРАН
В кислых водных растворах четырехвалентный уран находится в виде простого иона U4+ (возможно U(H2O)n+, где п--6 или 8) [369, 6801.
Растворы солей урана (IV) имеют кислую реакцию, что указывает на их гидролиз. Установлено, что гидролиз протекает по следующей реакции:
U4+ + H2O^UOH3+ + H+[367, 585, 680, 681].
26
При глубоком гидролизе возможно образование многоядерных ионов типа U[(OH)3U]V‘ [592], а также полимеров [U(OH)JV.
Гидролиз U4* сильно зависит от температуры, что видно из табл. 14, где представлены значения константы гидролиза Ка при различной температуре [681].
Таблица 14
Значения констант гидролиза при различных температурах
(р = 0,5; U4+ + Н2О ЕОН3+ 4 НА
Температура, СС 1 0 25 43
Значение Ка 0,075 0,21 0,66
Зависимость степени гидролиза U (IV) от температуры исследовалась также и другими авторами [367, 585].
Четырехвалентный уран в растворе получают либо растворением его солей, либо восстановлением раствора шестивалентного урана. Уранил-ион можно восстановить до урана (IV) различными металлами . (Pb, Zn, Bi, Ag, Cd и др.), а также амальгамами этих элементов [8]. Восстановление урана (VI) обычно проводят в сильнокислой среде, так как при этом создаются более благоприятные условия для количественного восстановления: согласно измерениям
Таблица 15
Зависимость потенциала системы U (VI)/U (IV) от кислотности раствора [276]
Концентрация H2SO,, М 0,05 0,259 0,508 1 ,034 2,753
^U(VI)/UUV) (относительно нормального водородного электрода) 0,3359 0,375 0,3929 0,4121 0,4468
Таблица 16
Влияние pH на потенциалы некоторых металлов Е (в в) (относительно нормального водородного электрода) [3]
Среда ^Cu £Pb £Cd £Zn
НС1 pH 0 —0,202 0,072 —0,290' —0,565 — 1,225
НС1 pH 1 —0,285 0,197 —0,235 -0,515 —0,756
НС1 pH 7 4-0,1’86 — — —0,500 —
27
В. Г. Хлопина н А. ,М. Гуревич [276], окислительно-восстановительный потенциал системы U (VI)/U (IV) повышается с увеличением кислотности (табл. 15), а электродные потенциалы металлов, как правило, понижаются (табл. 16).
Потенциал металлического висмута в растворах H2SO4, как установили П. П. Палей иН.И. Гусев, зависит следующим образом от концентрации кислоты (табл. 17).
Таблица 17
Влияние концентрации кислоты на потенциал металлического висмута
Концентрация H_SO4, М 0,5 1 =.5
£Bi Bi !!! (относительно нормального водородного электрода) 0,218 0,208 0,180 0,166
Металлы, имеющие высокий отрицательный потенциал (Zn, Mg и др.), восстанавливают уран (VI) до урана (IV) и урана (III).
Bi, Cd, Си, Pb и Ag восстанавливают уран до четырехвалентного состояния. Восстановление урана серебром протекает быстро и количественно лишь в 4 Л4 растворе НС1 при нагревании (60—90°), так как присутствие большой концентрации С1~ сильно понижает потенциал системы AgCl/Ag [375].
Согласно измерениям Фурмана, потенциал системы Hg/Hg!I в растворе НС! изменяется от 0,4 в (при низкой кислотности) до 0,006 в в 9 N растворе НС1; поэтому уран (VI) количественно восстанавливается ртутью до урана (IV) в 7—9 N растворе НС] [404].
При восстановлении, в ряде случаев, частично образуется уран (III), который легко и быстро окисляется до урана (IV) кислородом воздуха. Уран (VI) можно количественно восстановить до урана (IV) электролитически на ртутном катоде [8, 260, 861], фотохимически спиртом [829], эфиром [833], или молочной кислотой [828]; в азотнокислом растворе уран (IV) можно получить восстановлением уранил-нитрата ронгалитом [57]; удобным восстановителем урана (VI) до урана (IV) является двуокись тиомочевины (NH2)2CSO2[48,48а] и гидросульфит натрия Na2S2O4[8, 184]. Кислые растворы четырехвалентного урана довольно устойчивы в темноте и на холоде [8]. Показано, что окисление урана (IV) воздухом существенно ускоряется под влиянием света, особенно прямого солнечного или ультрафиолетового [263]; при этом процесс окисления протекает следующим образом:
u4+-|-/iy —>(и4+)* (и4+)*4-о2->ио22Л
где hy — квант света;
(U4+)* — возбужденный ион урана [263].
28
Окислению урана (IV) способствует радиационное облучение (например, у-излучение); при этом процесс окисления идет даже в отсутствие кислорода [268].
Усыновлено, что окисление урана (IV) в растворе воздухом каталитически ускоряется молибденом или медью [1841.
Электрохимическое окисление урана (IV) на платиновом электроде изучалось рядом авторов [211, 366, 777].
Растворы четырехвалентного урана обладают восстановительными свойствами (нормальный окислительно-восстановительный потенциал системы U (VI),U (IV) в сернокислом растворе равен 0,407±0,003 в [276, 611; на этом основано применение урана (IV) как восстановителя в массовом анализе [361 ] и при восстановлении плутония (IV), а также плутония (VI) до плутония (III) [866].
ПЯТИВАЛЕНТНЫЙ УРАН
Исследованиями, проведенными в последнее время, установлено существование в растворе урана в пятивалентном состоянии в виде ионаИО+. Растворы пятивалентного урана в концентрации ммоль/л могут быть получены электролитическим восстановлением уранил-иона при pH 2,5—3. Например, при восстановлении урана (VI) на электроде из платинированной платины при определенных условиях (температура 1°, pH раствора 2,5—3, концентрация урана 1 -10~3 М) можно получить до 90% урана (V) [202].
В растворе уран (V) находится в равновесии с ураном (VI) и ураном (IV), так как уран (V) диспропорционирует с образованием урана (IV) и урана (VI). Считают, что наиболее вероятно диспропорционирование урана (V) в хлорнокислом растворе протекает по уравнению [6461:
ио2+ ц-н+^иоон1+
иог+ + иоон2+^=иоГ4-иоон+ иоон--> U4+ или
2UO; + 4Н3О+ — иоГ 4- U4+ -4- 6Н2О.
Константа равновесия этого уравнения равна (1,7±0 3) • 103 [778 ]. Оптимальными условиями для существования UO." является pH 2—4; при таком pH пятивалентный уран значительное время сохраняется без диспропорционирования.
Формальный потенциал системы U (V)/U (VI) в 0,1 М растворе НС1О4 при 25° равен — 0,067±0,004е; в 1 М растворе НС1О4 равен — 0,063 ± 0,004 в и в 0,5 N растворе НС1О4 — 0,066 в [685].
29
ШЕСТИВАЛЕНТНЫЙ УРАН
Шестивалентный уран в растворе присутствует в виде уранил-иона UO;~, существование которого при pH 2,5 твердо установлено [227, 303]; при более высоком pH преобладают гидролизованные ионы сложного состава. Рентгенографические исследования многочисленных кристаллических соединений урана (VI) показывают, что группа UOV является линейным радикалом, в котором связь между ураном и кислородом частично имеет ковалентный характер; в растворах UO;+, как установили последние исследования, также имеет характер линейного радикала [227].
Многие исследования свидетельствуют о неполной диссоциации солей уранила (нитрата, сульфата, ацетата и др.) в водных растворах [227]. Установлено, например, что в концентрированных азотнокислых растворах уран присутствует преимущественно в виде недис-’ социированных молекул UO2(NO3)2. Данные по электропроводности водных растворов сульфата уранила свидетельствуют о неполной диссоциации UO2SO4, константа диссоциации сульфата (в пределах концентрации урана от 5-Ю5 до 5-10~3 7И) равна 5,93-10 4 [391].
Ацетат уранила также неполностью диссоциирован и притом еще в большей степени, чем уранилсульфат. Соли уранила можно расположить в следующий ряд в порядке увеличения их степени диссоциации [227 ]: UO2(C 1О4) 2 > UO2C12 > UO2(NO3) 2 > UO2SO4 > UO2(CH3COO)2.
Коэффициенты активности уранил-иона в водном растворе уранил нитрата представлены в табл. 18.
Таблица 18
Коэффициенты активности в растворе уранилнитрата [716]
С. А. Брусиловским рассчитаны коэффициенты активности ' уранил-иона при различной ионной силе раствора, которые представлены в табл. 19.
В очень концентрированных растворах коэффициент активности уранил-иона, по-видимому, достигает громадных величин.
Поведение уранил-иона в водных растворах при pH>2,5 носит весьма сложный характер. Кислая реакция растворов ураниловых солей указывает на имеющий место гидролиз UO» . Изучению 30
Таблица 19
Зависимость коэффициентов активности уранил-иона от ионной силы раствора
Ионная сила р-ра. Коэффициент активности и0Г’ 1 Ионная j сила Р-ра, ! р- Коэффициент активности ио|+, Ионная сила Р-Ра, Коэффициент активности ио^, 1 Ионная ; сила р-ра, i Коэффициент активности ио2"", т
0,002 0,868 i 0,080 0,488 0,5 0,43 2,1 1,16
0,003 0,858 । 0,109 0,473 0,6 0,44 1 2,4 1,49
0,004 0,830 1 0,139 0,451 0,9 0,50 2,7 1,95
0,007 0,794 । 0,201 0,425 1,2 0,60 3,0 2,57
0,012 0,699 | 0,261 0,442 1,5 0,74 3,6 4,61
0,022 0,042 0,608 0,567 0,335 0,462 1,8 0,92 4,2 8,35
гидролиза уранил-иона посвящено достаточно много работ [105, 303 , 335 , 496, 586, 600, 911, 952, 953], но результаты этих работ в некоторой степени противоречивы. Считают, что существуют такие продукты гидролиза: U2OP, U3Og+ UO2(OH) + , U3O8(OH) + , U3O8(OH)2, и3о8(он); И др.
Строение многоядерных двузарядных уранкислородных ионов довольно сложное 1144]. Константы возможных гидролитических реакций уранил-иона даны в табл. 20.
С. А. Брусиловским схема гидролиза уранил-иона изучалась путем потенциометрического титрования растворов уранила щелочью. Автором было найдено, что при прибавлении щелочи, когда ОН+ , t
отношениев растворе меняется от 0 до 1 (при концентрации
урана от 0,1 до 1,10"5 моль/л), уранил-ион переходит преимущественно в UO2(OH)+. При дальнейшем прибавлении щелочи, после соот-OH"
ношения = 1, начинается процесс конденсации, и в растворе возрастает количество полимерных однозарядных ионов типа UO2(UO3)„OH +. Образование ионов типа U2O52 + и U3O* практически в этой области, как считает автор, не имеет места. При соотношении ОН"
“z2+ от 1 до 1,4—1,6 наступает область метастабильных коллоидных
растворов, где полимеры UO2(UO3)„OH+ неустойчивы и. переходят в коллоидную форму гидроокиси уранила; коллоиды постепенно ОН"
разрушаются, осаждая UO3-nH2O. При соотношении ~q2+ от 1,4—
.. 1,6 до 1,9—2 идет осаждение гидроокиси уранила; при дальнейшем
31
Таблица 20
Константы гидролиза UO*1
Реакция гидролиза Кокстантл гидролиза Литература
иог — 2Н,О_1- UO, (ОН)- — н3о- (2,0±0,4)-10~5 [303]
6,4-Ю"5 [105]
" 6,0-НН3 [227]
UO, (ОН)+ + 2Н,0 UO, (ОН), + н3о- 4,6-Ю"4 [227]
2UO2" + ЗН .0 i — иоз • L’O“ * 4 2Н3О4- 1,35-]0“6 [227]
-2-10^6 [227]
1,14-10^6 [953]
10—6f — 0'1 [600]
4,1 10“3 [303]
uof +U,O?+4-3H,O^UsOf-Ь2Н3О+ 5-109 [953]
U3of + 2Н2О^± U3O8 (ОН)+ + Н3О+ 2,8-10-* [953]
изО8 (ОН) + 4- 2Н,0 Z=± изО8 (ОН)2 + Н3О+ 3-10~7 [953]
изО8 (ОН)24- 2Н2оизО8 (ОН) ” 4- Н3О+ 4- IO'8 [953]
изО8 (ОН)” 4- 2Н2О U3O8 (ОН)2” 4- Н3О+ Ы0’п [953]
и3о8 (OH)f 4- 2Н2О^± и3о8 (ОН)3/ 4- н3о+ 4-10~12 [953]
Таблица 21
Зависимость pH начала осаждения гидроокиси уранила от концентрации UO|+ в растворе
Концентрация перхлората уранила, М 0,1 0,01 0,001 0,0001 0,00003 0,00001
pH начала осаждения гидроокиси - . 4,47-,- ... 5,2? 5,90 6,62 6,80 7,22
32
прибавлении щелочи начинается переход гидроокиси в уранаты и полиуранаты.
Автором приводится следующая зависимость значения pH начала осаждения гидроокиси от концентрации уранил-иона в растворе (табл. 21).
Значения произведений растворимости гидроокиси уранила при 20° равны:
с _ [иогон+] [ОН-J _ ч . 1S
Эр(!)— [ио2(ОН)г] 1,0-10
[UO*+HOH-]2 5-<‘>=тежг=(6±2)-10 [21а1‘
В самых последних работах [586] также обстоятельно изучен процесс гидролиза уранил-иона, исследована зависимость гидролиза от температуры и ионной силы раствора. Авторы считают, что гидролиз U02 + может быть объяснен образованием мономера UO2OH + по реакции
. иоГЧ-н2о^ио2он + 4-н -
и димера иО2-НОз+, образующегося по реакции
2иоГ + Нго^± ио2.иог + 2Н + .
Константы гидролиза
_[UO2OH+] [Н+] __ [UO2-UQ2 + ] [Н+]2
A1 [U02/] и 2 [UO2+]2
как установили авторы, следующим образом зависят от температуры и ионной силы (ц) раствора (табл. 22).
Табл ид а 22
Влияние температуры и ионной силы раствора на константы гидролиза уранил-иона
Условия (ионная сила, температура, °C) Л, к2
р = 0,347; 25° 4-10® 1,5-10®
р = 0,0347; 25° 1,5-10® 0,7-10®
р = 0,0347; 40° 8-10® 1,2-10®
3 Аналитическая химия урана
33
Глава III
КАЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ УРАНА
С. Б. С а в в и н
В предыдущей главе были описаны главные свойства ионов четырех- и шестивалентного урана: гидролиз, редоксреакции, комплексообразование и другие. В этой главе основное внимание уделяется характерным реакциям ионов урана, т. е. таким, которые благодаря своей большей или меньшей специфичности и наглядности могут быть использованы для быстрого качественного обнаружения урана в тех или иных объектах.
Для урана такими реакциями являются прежде всего цветные с неорганическими и органическими реагентами и люминесцентные. В отсутствие прочих радиоактивных элементов уран может быть быстро. определен по радиоактивности [72, 225, 635, 655]. Ультрамалые количества урана можно определить методом микрорадиографии по количеству распадов, фиксируемых специальными толстослойными фотопластинками [435, 807, 808]. Реже для обнаружения урана используют некоторые другие методы: полярографические [944], спектральные [167,442], метод нейтронного активационного анализа [724, 924]. Эти‘достаточно сложные инструментальные методы в основном применяются для количественного определения урана. Они подробно описываются в соответствующих разделах книги.
Реакции осаждения для урана в большинстве случаев мало характерны и для быстрых качественных определений не применяются. Исключение составляют некоторые микрокристаллохимические реакции, например с антраниловой кислотой [266]. Если при осаждении урана органическими соединениями осадки интенсивно окрашены, то и они могут быть использованы для обнаружения урана. В этом случае предпочитают пользоваться неводными растворителями, в которых эти осадки растворяются и далее фотометр ир уюте я по обычным схемам.
34
КАЧЕСТВЕННОЕ ОБНАРУЖЕНИЕ УРАНА ПО ФЛУОРЕСЦЕНЦИИ В ПЕРЛАХ
Почти исключительная особенность системы UO|+—NaF давать яркую желто-зеленую флуоресценцию под действием ультрафиолетового излучения делает флуоресцентный метод настолько избирательным, что в ряде случаев предварительного отделения примесей не требуется. Это особенно удобно для быстрых качественных анализов и, конечно, для количественных определений, которые подробно описываются в гл. IV. Хорошая избирательность флуоресцентных реакций сочетается с высокой чувствительностью, что делает метод полезным при анализе сбросных и природных вод и объектов с низким содержанием урана [239, 262, 364, 747, 864].
ЦВЕТНЫЕ РЕАКЦИИ УРАНА
Для обнаружения урана наиболее часто используют его цветные реакции с неорганическими или органическими реагентами.
К настоящему времени известно несколько десятков реагентов, пригодных для обнаружения урана. В то же время невозможно указать универсальный реагент, который являлся бы наилучшим при определении урана в любых объектах и в присутствии неограниченных количеств посторонних элементов. Большой ассортимент имеющихся для урана реагентов позволяет в каждом конкретном .случае выбрать наиболее удобный реагент, обеспечивающий необходимую точность и избирательность.
Цветные реакции урана по своему механизму могут быть разделены на два класса. К первому классу можно отнести реакции, основанные на хромофорном действии урана, т. е. на способности ионов U4+ и UO*+ образовывать окрашенные соединения с простейшими неорганическими или органическими реагентами. Обычно считают, что хромофорным действием обладают не сами элементы или ионы, а атомные сочетания U—О, U—S и, возможно, U—N [124 , 769]. Кроме урана, хромофорным действием обладают многие элементы: Fe, Си, Ni, Мп, Cr, V, Мо, Со, Се и др. Все они также способны образовывать окрашенные соединения с бесцветными реагентами. Напротив, Th, Zr, Hf, Al, Zn, Ca, Ba и другие, не обладающие хромофорным действием, не образуют окрашенных соединений с бесцветными реагентами. Таким образом, по крайней мере эти элементы не препятствуют цветной реакции урана, что обеспечивает некоторую повышенную избирательность цветных реакций урана этого класса.
Примерами реакций, основанных на хромофорном действии, могут служить цветные реакции урана с салициловой кислотой, KSCN, резорцином, а также возникновение окрашивания в концентрированных растворах кислот — серной, хлорной, фосфорной.
Конечными продуктами цветных реакций U4+ и UO* + могут быть самые разнообразные соединения: простые соли с неорганическими 3* 35
или органическими анионами, двойные соли, простые комплексные соединения типа (UO2-Anrt) Katm, циклические соли вида
О r/^>UO2 о
и, наконец, внутрикомплексные или хелатные соединения, причем последние являются наиболее интересными. Теория применения реагентов этого типа подробно развита в монографиях и работах Кузнецова, Кульберга и других авторов [83, 124, 131, 132, 207, 246, 264].
Ко второму классу реакций относятся цветные реакции урана с интенсивно окрашенными органическими реагентами. При этом происходит не возникновение окраски, но углубление уже имеющейся. Конечными продуктами цветных реакций этого типа являются внутренние комплексы урана с молекулой реагента. Изменение окраски вызывает любой элемент, дающий с реагентом хелатные соли, независимо от того, обладает ли он хромофорным действием или нет.
В качестве реагентов наиболее часто применяют азокрасители вида R,—N=-N—R2.
Для урана наибольшее применение нашли азокрасители, включающие остаток мышьяковой кислоты:
AsO3H2 ОН ОН AsO.H, НО
_/ I I _/ \____
N = N— и \ N = N—/
Они наиболее прочно связывают ионы урана (4- и 6-валентного), одновременно давая высококонтрастные реакции [112, 114, 117, 128]. Реагенты, включающие эту группировку, не являются специфическими только для урана. Значительное число прочих элементов также может образовывать хелатные комплексы и вызывать изменение окраски реагента. Механизм реакций этого типа подробно обсуждается в работах В. И. Кузнецова [119, 123, 124]. Им показано, что изменение окраски реагента при комплексообразовании связано прежде всего с изменением внутримолекулярного ионоид-ного состояния молекулы реагента.
Область оптимальных значений pH взаимодействия реагента с элементами различна для разных элементов. При этом замечено определенное соответствие между цветными реакциями и реакциями гидролиза [113, 115, 119]. Так, в сильнокислых средах цветные реакции с окрашенными реагентами дают лишь элементы, ионы которых значительно склонны к гидролизу: Та, Nb, Zr, Hf, U (IV), Th; в умереннокислых растворах цветную реакцию дают, кроме
36
перечисленных, ещеРе (III), Cr (III), Al; в слабокислых и нейтральных — U (VI), Fe (II), Си, 1TR, и в щелочных — Са, Sr, Mg.
Условия реакций (оптимальные pH), конечно, находятся в непосредственной связи с природой применяемых реагентов. Но для каждого реагента соотношения оптимальных значений pH реакций с элементами сохраняются всегда постоянными: легкогидролизу-ющиеся элементы взаимодействуют в более кислой среде, прочие — в менее кислой.
Таким образом, мы видим, что не безразлично, в каком виде определять уран: реакции U (IV) являются гораздо более избирательными, чем реакции UO*V
Для повышения избирательности определения как U (VI), так и U (IV) часто используют и другой прием — связывание присутствующих элементов в бесцветный прочный комплекс. Ниже будут описаны методы, где для этой цели используют комплексон III (при определении уранил-иона) и щавелевую кислоту (при определении четырехвалентного урана).
Если цветные реакции с окрашенными реагентами не всегда позволяют достигнуть высокой избирательности, то по чувствительности некоторые из них на 1—2 порядка превосходят реакции, основанные на хромофорном действии урана. Это является основной особенностью реакций второго типа.
Не всегда можно провести резкое разграничение реакций этих двух типов. Иногда имеет место смешанный механизм. В качестве примера можно привести цветные реакции с ализарином S, алюминоном, нитрозо-R-солью, морином, Н2О2 и некоторыми другими.
РЕАКЦИИ, ОСНОВАННЫЕ НА ХРОМОФОРНОМ ДЕЙСТВИИ
УРАНА
При концентрациях порядка нескольких миллиграммов на миллилитр в растворах, не содержащих окрашенных веществ, уран может быть идентифицирован по собственной окраске его простых неорганических солей. Растворы уранила желтого цвета, растворы четырехвалентного урана обладают гораздо более интенсивной зеленой окраской. Окраска солей как шестивалентного, так и четырехвалентного урана усиливается в растворах концентрированных кислот. Четырехвалентный уран рекомендуют определять в 40%-ной Н3РО4 [407], шестивалентный — в 30—50%-ной H2SO4[357, 750] или в 40—65%-ной НС1О4 [916, 938]. Используя спектрофотометр, можно определять уран и в карбонатных растворах по характерному поглощению трикарбонатного комплекса в ультрафиолете [1010].
Все эти прямые фотометрические методы, не требующие специальных реагентов, просты в выполнении и избирательны, но недостаточно чувствительны: до 0,5—10 мг/мл урана.
Много характерных реакций описано для урана с кислород-и серусодержащими реагентами.
37
В свое время широкое распространение получил метод опреде* ления урана с Н2О2 в среде Na2CO3 [79, 184, 340, 887, 893]. Качественная проба выполняется достаточно просто—добавлением к нескольким миллилитрам исследуемого раствора карбоната натрия и нескольких капель пергидроля. В присутствии уранила развивается желтоватая окраска. Чувствительность — 5—10 мкг/мл, мешают Сг, V, Mo, W, меньше Си, Fe, Ni, Мп. Избирательность реакции может быть несколько повышена, если определение проводить в среде Н2О2—NaOH—Na2CO3 при пониженной концентрации Н2О2 или в слабокислой среде при pH 4,5. Отмечают, что при этом влияние Сг, Мп, Си, Zn, Cd, Pb и Al заметно ослаблено [1012].
Оносовым и Дмитриевым предлагается следующая качественная проба на уран [179]: к 2—3 мл исследуемого раствора добавляют равный объем 10%-ного раствора комплексона III, затем раствор аммиака до ощутимого запаха и в ка честве соосадителя небольшое количество раствора бериллия. Кипятят 2—3 мин. Осадок гидроокиси урана и бериллия отфильтровывают, промывают и обрабатывают 2—3 мл 10%-ного раствора Na2CO3 или (NH4)2CO3, несколько раз пропуская раствор через фильтр. К небольшой порции фильтрата добавляют каплю 30%-ной Н2О2. Немедленное появление желтого окрашивания указывает на присутствие урана. Чувствительность определения составляет Ю мкг/мл U. Не мешают Сг, Zn, Со, Al, РЬ, Си, Се, Cd, Be, Ti, Zr, Th, VO3, MoO4, WO4.
Для обнаружения урана может быть использован роданид калия или аммония, дающий с уранилом желтое окрашивание. По данным Арланд [305], образуются комплексные ионы вида: [UO2SCNH, [UO2(SCN)2], [UO2(SCN)3I_ с константами равновесия 5,7; 5,5 и 15 соответственно. Реакция выполняется как в водных растворах, так и в смесях, содержащих ацетон, спирт или монобутиловый эфир этиленгликоля [426]. При определении в среде смешивающихся с водой органических растворителей избирательность метода увеличивается: Zr, Th, Sn, Мп, а также ацетаты, сульфаты и фосфаты определению не мешают [437]. Fe (III) предварительно восстанавливают аскорбиновой кислотой [990] или хлористым оловом [633]. Одним из достоинств метода является то обстоятельство, что окраска устойчива в широких пределах кислотности — от 0,1 до 2,0А по НС1 или HNO3, поэтому поддержание точного значения pH не обязательно. Роданидный метод является особенно удобным при определении урана на фоне больших количеств тория. При соотношении U : Th=l : 10000 торий не мешает [440]. Но чувствительность роданидного метода невысока — 20—40 мкг/мл урана.
Ион азида являющийся структурным аналогом роданида
[:S: :С: :N:]- и [:N: :N: :N:]"
также дает с ураном сходную желтую окраску (см. главу IV).
Более чувствительным является метод открытия урана с ферроцианидом калия по красно-бурой окраске [UO2]2 [Fe(CN)6], или, возможно, двойной комплексной соли n[UO2]2[Fe(CN)6 ]-m/Qx 38
x[Fe(CN)e] [254]. Определение выполняется при pH 4—6 (формиатная буферная смесь) или в слабой азотнокислой среде (3—4 капли концентрированной HNO3 на 25 мл раствора). Ванадий и железо не мешают, но многие прочие элементы, а также ацетаты, оксалаты, цитраты, фосфаты и Другие комплексообразующие вещества влияют достаточно сильно [8, 989]. По данным С. И. Синяковой и Н. С. Классовой, чувствительность реакции составляет 10—20 мкг U в 25 мл.
Для определения урана в минералах поступают следующим образом [134]: несколько миллиграммов мел ко растертой пробы помещают в небольшую фарфоровую чашку, добавляют 2—3 капли концентрированной НС1, 1 каплю HNO3 и выпаривают на водяной бане почти досуха. Прибавляют 7—10 капель воды, 1—2 капли аммиака, щепотку [NH4]2 СО3, 1 каплю сернистого аммония и фильтруют при помощи капилляра в микропробирку. Фильтрат смешивают с 3 каплями пергидроля и выпаривают на водяной бане досуха. Остаток растворяют в 1 капле воды и 3 каплях 50% -ного раствора винной кислоты. По охлаждении добавляют 1—2 капли насыщенного раствора ферроцианида калия. В присутствии урана наблюдается коричневое окрашивание.
На хромофорном действии урана также основаны методы определения с неокрашенными или слабоокрашенными органическими реагентами.
С салициловой кислотой [205, 768] и ее аналогами [184, 519] уранил дает оранжево-красное окрашивание.
Интенсивное красное окрашивание дает с ураном о-крезотиновая кислота (2-окси-З-метилбензойная). Для обнаружения урана каплю исследуемого раствора наносят на бумагу, предварительно смоченную 2%-ным раствором реагента в 80%-ном этаноле. Появление красного пятна указывает на присутствие UO*+. Чувствительность — 14 мкг U. Мешают кислоты, Fe (III), Се (IV), Th, Sn, Со, Си, Ni, оксалаты, фосфаты, сульфаты [8721.
С тиогликолятом аммония UCP+ образует желто-оранжевый комплекс, обладающий высокой прочностью. Это позволяет выполнять реакцию в присутствии многих анионов, но не карбонатов. Большинство катионов мешают. Чувствительность — 2 мкг/мл U. Определение выполняется при pH 7—11 [448, 467].
Для определения урана большой интерес представляют реагенты, дающие с ним цветную реакцию в неводной среде. Они полезны, например, для быстрого обнаружения урана непосредственно после экстрагирования нитрата уранила эфиром или другим органическим растворителем. В качестве такого реагента наиболее часто применяют дибензоилметан, дающий с UO*+ оранжево-желтое устойчивое соединение [610, 821, 1011, 1021] (см. главу IV).
8-Оксихинолин также образует с ураном растворимое в органических растворителях красно-бурое соединение [652] вида
39
Суза описывает метод микрооткрытия урана, пригодный в полевых условиях [932]: к 1 г тонкорастертой руды прибавляют 10 мл концентрированной НС! и нагревают. Прибавляют 20 мл смеси: 20 г [МН4]2СОз4- 100 мл концентрированного аммиака -у 100 мл воды, и образовавшийся осадок отфильтровывают. Каплю фильтрата помещают на фильтровальную бумагу, смоченную 5%-ным спиртовым рас твором 8-оксихинолина. Появление красно-бурого пятна указывает на присутствие урана.
Кроме перечисленных,-для открытия урана применяются и многие другие реагенты: аскорбиновая кислота [184], салициламидок-сим [351], салицилгидроксамовая кислота [372], тайрон (1,2-ди-оксибензол-3,5-дисульфонат натрия), резорцин (т-диоксифенол) [623], галловая кислота [451], хромотроповая кислота [766], диэтилдитиокарбамат натрия [8], 2-ацето-ацетилпиридин [574], R-соль и нитрозо-И-соль [831], редуктон [604], изатин-^-оксим [612], различные нафтолсульфокислоты [832], таннин [8], мореллин [827], феррон [158] и другие реагенты [8, 184, 446, 917].
Чувствительность описанных методов с использованием органических реагентов приблизительно одного порядка — около 5 мкг/мл. Избирательность невысока: помехи оказывают многие элементы и все анионы, способные связывать уран в комплекс. Некоторые свойства описанных в литературе реагентов сопоставлены в табл. 23. Рассматривая эту таблицу, следует иметь в виду, что чувствительность отдельных реакций дана лишь ориентировочно, так как в большинстве случаев данные разных авторов не сопоставимы между собой.
Обращает на себя внимание тот факт, что реакции четырехвалентного урана с неокрашенными органическими реагентами, которые по некоторым данным должны быть более чувствительными и избирательными, в литературе почти совсем не описаны.
Кроме прямых колориметрических методов, в литературе описан и ряд косвенных колориметрических методов [192, 327, 708].
ЦВЕТНЫЕ РЕАКЦИИ УРАНА С ОКРАШЕННЫМИ ОРГАНИЧЕСКИМИ РЕАГЕНТАМИ
Методы определения урана с неокрашенными реагентами в последние годы не всегда стали удовлетворять возросшим требованиям по чувствительности. Поэтому наметилась тенденция к использованию органических окрашенных реагентов, как правило, более чувствительных. Из них наибольшее применение, видимо, нашел реагент арсеназо I (или уранон), синтезированный и предложенный для определения урана в 1941 г. В. И. Кузнецовым [112]. Позднее были предложены реагенты арсеназо II [118] и арсеназо III [216], которые обладают рядом других ценных качеств.
40
41
Таблица 23 (продолжение)
Реагент Окраска и условия реакции Мешающие элементы Чувствительное! ь, мкг!мл Литератуpa
Органические реагенты
Сульфосалициловая кислота ОН Оранжево-красная, pH 4,4—4,7 Fe, V, Си, F-, РО’ и др. 10 [184]
соон
SO3H Салнцилгидроксамовая кислота ОН | Оранжево-желтая, pH 7,8—9,2 Th, Се (III), V, Pb, Zn, Al, F-, РО*~ и др. 2—5 |372]
/V- С — NOH
ОН Салициламидоксим fH Оранжево-желтая, pH 7,9-9,2 Cr, Bi, Си, Ni, Со, Мп, Fe (III), Се (IV), Th, Ti, F-, РО^ и др. 2—5 1351]
С = NOH
1н2 1 1 1
Таблица 23 (продолжение)
Реагент Окраска и условия реакции Мешающие элементы Чувствительность, мкг'мл Литер.л ура
Резорцин ОН Красно-оранжевая, pH 7-8 Мп, Со, Fe, Cr, Се, Th, F“, РО’ и др. 10—15 |623|
^/Хон о-Крезотиновая кислота СООН О”011 Красная, нейтральный раствор 80%-ного этанола Fe (III), Се (IV), _Th, Sn, Си, Ni, F", РО= и др. 5—10 [8721
ч/хн, Хромотроповая кислота ОН ОН 1 1 СП Желтая, pH 10—11 Mn,_ Fe(HI), Си, 11g, РО’ , SO* и многие другие 5—10 [8, 760]
HO3S/X/4/4SOsH 2-Нафтол-1 -сульфоновая кислота SO3H Коричневая, pH 4,0—4,5 Th, V, Zr, F~, РО’~ и др- 100-200 [832]
Гн он
\/\/
Таблица 23 (продолжение)
Реагент Окраска и условия реакции Мешающие элементы Чувствительность, мкг; мл Литература
а-Нитрозо-0- нафтол NO 1 1 1 J”0Н Красная, органические растворители, слабокислая среда Со, Си, Fe, Mo, V, Zr и многие другие Невысокая [«]
Нитрозо-И-соль NO ОИ Оранжевая, pH 4,6—6,4 Zr, Fe, Си, Mo, V, F~, PO:f и др. 2—5 [831]
8-Оксихинолин Си Красно-бурая, органические растворители или водно-спиртовый раствор, среда нейтральная Многие элементы 5—10 [8, 652, 932]
N ОН
Феррон SO,H Oj- N ОН Красновато-бурая, pH 5, уротропин Fe, Al, F“, РО’ и многие другие 10 [158]
Таблица 23 (продолжение)
Реагент Окраска и условия реакции Мешающие элементы Чувствительность, мкг/мл Литература
Тиогликолят аммония HSCH2COONH4 Диэтилдитиокарбамат натрия /Xs (CjH‘>1N-C<SNa Дибензоилметан ОНО — II 1 п /—х / >— с — с — с— < > \—/ 1 х—/ Желто-оранжевая, pH 7-11 Желто-оранжевая, водный раствор или органический растворитель, pH 6—8 Ярко-желтая, pH 6—8, водно-спиртовый рас- твор или органический растворитель Fe, Си, Zr, Al, Th, Ti и другие катионы Fe, Си, Ni, Со, F", РО’“ и многие другие V, Fe[III), Си, Mo, F", РО’~ и др. 2 1—5 2—20 [448, 467] [8] [377, 610, 631, 821, 1011, 1021]
н 2, 4-Диокси-5-хлорацетофенон СОСН, . | Желто-коричневая, реагент желтый Fe(III), F", РО’" Невысокая [356]
он
он Аскорбиновая кислота НО — С = С—ОН Желтая, pH 4,4—4,5 Сг, V, Mo, Си, Fe, Th, Al, Zn, F“, РО’” и др. 5 [184]
О = С СН—СН(ОН)СНгОН
Таблица 23 (продолжение)
Реагент Окраска и условия реакции Мешающие элементы Чувствительность, мкг/мл Л итература
Морин он о - С - он НО'/'Ч'/Ч'О - С Но/^/^ОН Красновато-коричневая, реагент лимонно-желтый, pH 4—7, водная среда или 50—60%-ный этанол Al, Fe (III), Ga, In, Se, F~, PO’~ и многие другие 2—5 [8, 331, 360, 654]
Реакции
с окрашенными органическими реагентами
Красно-фиолетовая, Ре- Си, Fe, Al, Cr, Ba, Zr, ]762, 763, 875,
агент розовый, ней- F-, POf и ДР- 2—10 994]
тральная среда
Таблица 23 (продолжение)
Реагеиг Окраска и условия реакции Мешающие элементы Чувствительность, мкг'мл Ли гература
Алюминон ОН Красная, реагент желтовато-коричневый, pH Си, Bi, Fe (II), Fe(III), Al, Be, Th, Zr, Ce(III),
Y , С—< >-ОН II ' / /\ xcoonh4 J-coonh4 II О 4,9—7,0 Fe-, PO’ и др. 2—10 1765]
4-(2-пиридилазо)-резорцин 70Н / N НО—/ \_N-~ N—/\ Красная, реагент желтый, pH 8 Многие элементы 1—2 ]815|
00
Таблица 23 (продолжение)
Реагент Окраска и условия реакции Мешающие элементы Чувствительность, мкг 1мл Литература
2-Пи ридин-1 -азо-1 -2-н афтол Ярко-красная, 80%-ный F", РО’-, Н2О2 2—10 [419, 540]
/ \ / 4 ацетон -[- пиридин
(1— N = N — / 4
N | ОН
Торон /АзО.Н, «° S°’H / /— N=N —\ / Малиново-красная, реагент оранжевый (только на U4+), pH 0,8—2 Zr, Ti, Nb, Та, F", РО*“ и некоторые другие 0,5—1 [520, 787]
д ч
SO3H
Арсен азо I _/А50,нг он он )>- N = N — Синяя, реагент розовый 1) для UO|+ pH 4,5-8 2) для U4+ pH 1—2 Th, Zr,Ti, Nb, Та, F“, POf, sof 0,2—0,5 [112, 117, 527, 529]
АА/ HO.S SO3H 9
Аналитическая химия урана
Таблица 23 (окончание)
Реагент Окраска и условия реакции Мешающие элементы Чувствительность, мкг/ мл Литература
Арсеназо II /-<3>-As0‘H= °н Г \ / Xn = N—j/X'|//'Xj j Сине-фиолетовая, реагент красный 1) для'иО|+ рН4-7 2) для U4+ 0,01—0,5 (VHCI Th, Zr, Ti, F-и-некоторые Другие 0,2-0,5 [128, 129, 160]
\ но ЛЦо н А Н03О oU3rl2
Арсепазо III /AsO3H2 НО ОН H2O3As /~ N = N -/N = N - / У Изумрудно-зеленая, реагент розовый 1)дляиО|+ pH 1,0-3,0 2) для U4+-0,01—10JV НС1 Th, Zr Fe(III), STR Th o,i. [216]
/\/\/\ п HO3S SO3H
Арсеназо I
Реагент арсеназо I (2-фениларсоновая кислота-< 1-азо-2>--1,8-диоксинафталин-З,6-дисульфокислота) пригоден для определения как U (VI), так и U (IV). С уранилом образуются внутриком-плексные соединения состава 1:1, для которых может быть предложена следующая структура:
С четырехвалентным ураном образуется комплекс состава 1 : 1 (в кислой среде при избытке металла) или 1 : 2 (в среде ближе к нейтральной). С четырехвалентным ураном реакция протекает в минеральнокислой среде; переход окраски из розовой (реагент) в синефиолетовую. С уранилом реакция протекает в слабокислой и нейтральной среде при pH 4,5—8; переход окраски из розовой в синюю или голубую. Метод определения шестивалентного урана мало избирателен : мешают Th, Zr, Ti, Al, Fe, TR и другие элементы, которые необходимо предварительно удалять [527, 529].
При определении четырехвалентного урана мешают только легко гидролизующиеся элементы: Zr, Ti, Th. Маскирующие уран анионы: фосфаты, фториды, оксалаты, ванадаты, арсенаты, присутствующие в количествах, в 5—10 раз превосходящих количество урана, подавляют реакцию и в том и в другом случае.
Открытие урана в присутствии прочих элементов может быть выполнено достаточно надежно при использовании поверочной реакции с перекисью водорода. Поверочная реакция основана на том, что при добавлении капли пергидроля к синему раствору комплекса иОг+ — арсеназо окраска моментально переходит в исходную розовую. Если синее окрашивание вызвано не ураном, a Th, TR, Al, Fe (III), Zr, Ti, Cu, Be, Cr, Ga, Pd и другими элементами, то немедленного обесцвечивания раствора не происходит [117]. Ниже приводятся методики обнаружения урана в ряде объектов,' разработанные В. И. Кузнецовым [117].
Обнаружение шестивалентного урана в растворах, не содержащих реагирующих с арсеназо I элементов, выполняется следующим образом: к 1 мл нейтрального раствора прибавляют каплю 3W НС1 и 1—2 капли 0,02—0,05%-ного водного раствора арсеназо. Розовая окраска раствора указывает на отсутствие Th, U(IV), Zr, Ti и Fe(III). Прибавляют 3—4 капли 25%-ного раствора уротропина; в присутствии урана возникает чисто голубая или фиолетовая (при избытке реагента)
50
окраска, которая моментально переходит в исходную розовую после прибавления капли Н2О2. Открываемый минимум - - 0,2 мкг L’(VI).
Четырехвалентный уран может быть уверенно открыт в растворах, не содержащих Th, Zr и Ti : к 1 мл анализируемого раствора, имеющего кислотность 0,02—0,05 N по НС1, прибавляют каплю 0,01—0,05%-ного раствора арсеназо. Возникновение синей или фиолетовой окраски, немедленно переходящей в розовую после прибавления капли Н2О2, указывает на присутствие U(1V). Открываемый минимум 1—2 мкг U. Не мешают тысячекратные количества LT(VI), V(IV), STR, Al и двухвалентных катионов.
Для обнаружения урана в рудах, минералах и других сложных объектах необходимо предварительное отделение основной массы примесей: 0,5—1 г тонко-измельченного материала нагревают с несколькими миллилитрами царской водки, слегка упаривают, смывают в стаканчик и разбавляют до 100 мл. Добавляют NaHCO3 до фиолетового окрашивания бумаги конго. Прибавляют 10—20 капель 60%-ного водного раствора SnCI4, нагревают до кипения и после коагуляции отфильтровывают. При этом отделяются фосфаты, арсенаты, а также Zr, Ti, Th, Fe(III), Nb и Та. К фильтрату добавляют 25% -ный раствор уротропина до полного покраснения конго и еще 0,5 мл избытка. Прибавляют арсеназо и раствор разливают поровну в два стаканчика. В один добавляют несколько капель Н2О2, после чего в оба стаканчика медленно прибавляют поровну насыщенный раствор Na2SiF6 или 0,5%-ный раствор NaF (для маскирования А1). Считают, что уран присутствует, если раствор без Н2О2 будет оставаться фиолетовым, в то время как раствор с Н2О2 станет почти розовым.
Образцы, содержащие 0,005% урана, дают еще ясную реакцию на уран, причем чем выше содержание алюминия, тем реакция менее отчетлива.
Для маскирования сопутствующих урану элементов может быть использован комплексон III. Определение выполняется при pH 7—8 в присутствии буферной смеси — триэтаноламина и HNO3. Многие элементы, присутствующие в ограниченных количествах, не мешают [529].
Комплекс UO2+ с арсеназо I может быть проэкстрагирован в виде его дифенилгуанидиниевой соли некоторыми растворителями, преимущественно спиртами: бутиловым, амиловым, бензиловым. Уран затем определяют непосредственно в экстракте по синему окрашиванию. Если экстрагирование проводят из растворов, содержащих комплексон III, достигается значительное повышение избирательности. Метод может быть рекомендован для качественных проб (В. И. Кузнецов, С. Б. Саввин, 1960).
Арсеназо II
Реагент арсеназо II (бифенил-4,41-диарсоновая кислота-3,З^бис [<-азо-2>-1,8-диоксинафталин-3,6-дисульфокислота]) является
близким аналогом арсеназо I [128]. Цветные реакции арсеназо II с U(VI) и U(IV) по чувствительности, возникающим окраскам, условиям реакции практически совпадают с соответствующими реакциями арсеназо I. Отличие заключается в том, что арсеназо II образует с U(VI) и U(IV) комплексы повышенной прочности. Благодаря этому определение урана с арсеназо II оказывается возможным в присутствии значительных количеств анионов: сульфатов, фосфатов, а также на фоне комплексона III и других комплексо-
4*
51
образующих веществ, применяемых для избирательного маскирования прочих элементов.
Уранил образует с арсеназо II комплексы двух составов: 1 : 1 и 1 : 2 (арсеназо II: UO*+), причем в практике анализа, когда реагент присутствует всегда в избытке, преимущественно образуется комплекс состава 1:1. Строение комплекса аналогично строению комплекса UO|+ — арсеназо I. При этом вторая функциональноаналитическая группа арсеназо II остается свободной.
Раствор чистого комплексона III существенно маскирует и уран, поэтому определение проводится в присутствии избытка нитрата кальция. При этом Fe(III), V (IV), Си и редкоземельные элементы могут быть замаскированы практически полностью. Для маскирования алюминия может быть использована сульфосалициловая кислота и для маскирования титана — винная кислота. Торий, присутствующий в 40-кратных по отношению к урану количествах, может быть практически полностью замаскирован смесью следующего состава (указываются молярные концентрации в конечном растворе): комплексон III 0,05 7И; Ca(NO3)2—0,05 М; А1С13—0,05 М; KF— 0,10 /И; сульфосалициловая кислота 0,05 М.
Таким образом, применение реагента арсеназо II позволяет повысить избирательность определения урана, но схемы обнаружения урана в основном остаются прежние.
Арсеназо III
Реагент арсен азо III (1,8-диоксинафтал ин-3,6-дисульфокисл ота-2,7<бис [<-азо-2>-фениларсоновая кислота] ) получен в 1959 г. С. Б. Саввиным [216]. Основной особенностью арсеназо III, отличающей его от ранее описанных реагентов, является способность образовывать с катионами элементов, в том числе cU(VI) hU (IV), особо прочные внутрикомплексные соединения. Благодаря этому определение элементов становится возможным в сильнокислых растворах и в присутствии больших количеств фосфатов и других комплексообразующих веществ. Состав комплекса UO^+ — арсеназо III отвечает соотношению 1:1.
Реакция с ураном весьма контрастна. Переход окраски из розовой (реагент) в изумрудно-зеленую.
Максимальное развитие окраски комплекса UO-‘! — арсеназо III наблюдается при pH 1,5—3,0. При pH 3,0, которое удобно поддерживать, применяя буферную смесь из монохлоруксусной кисло* ты и ее соли, допустимое соотношение U(VI) : Н3РО4 составляет 1 : 4000—6000. Определению UO*+ должно предшествовать возможно более полное отделение его от Th, Zr, V, Сг и STR, которые мешают. Ограниченные количества Zr, V, Сг и 2TR могут быть замаскированы комплексоном III.Влияние А1 можно полностью исключить, если определение UO®+ выполнять в 0,05 N НС1 или в при--о
сутствии сульфосалициловой кислоты. Th мешает во всех случаях.
Предварительное восстановление урана до U(IV) позволяет резко увеличить избирательность. Определение ,U(IV) удобно выполнять в 4—6 N НС1 в присутствии щавелевой кислоты для связывания Zr, Hf и Ti. Прочие элементы, кроме Th, а также фосфаты и другие анионы влияния не оказывают.
Чувствительность определения при использовании фотометров 0,01—0,02 мкг/мл урана, при визуальных определениях—0,1— 0,2 мкг/мл урана.
Реагент арсеназо III может быть использован при экстракционно-фотометрических методах для определения урана непосредственно в органической фазе. При этом из водного слоя экстрагируется комплекс UO|+—арсеназо III в виде его дифенилгуанидиниевой соли. Экстракция осуществляется из слабокислого раствора, насыщенного комплексоном III для связывания прочих элементов, и в присутствии хлорида дифенилгаунидиния. Метод позволяет собственно определение урана сочетать с одновременным отделением от большинства прочих элементов, которые удерживаются в водной фазе в виде комплексов с трилоном Б. Экстрагирование осуществляется бутиловым или амиловым спиртом. Достоинство метода — высокая чувствительность, сочетающаяся с хорошей избирательностью.
Метод достаточно прост и заключается в следующем (В. И. Кузнецов, С. Б. Саввин, 1960): авеску исследуемого вещества, предположительно содержащего 1— 100 мкг урана, разлагают способом, зависящим от его минерального состава. Упаривают, сухой остаток обрабатывают 2,0 мл 0,05 N НС1. Вводят 2,5 мл 5%-ного раствора комплексона III, 1,00мл 0,05%-ного раствора арсеназо III, 0,5 мл 20%-ного раствора хлорида дифенилгуанидиния и 5,00 мл бутилового спирта. Экстрагируют, часть верхнего слоя переносят в 10-миллиметровую кювету и фотометри-руют на спектрофотометре при 660 ммк или на фотоколориметре с красным светофильтром.
При качественных определениях окраску верхнего органического слоя сравнивают визуально с окрасками растворов стандартной серии, содержащих разные количества урана. Если уран в пробе отсутствует — окраска розовая, соответствующая чистому реагенту; при наличии урана цвет экстракта от фиолетового до чисто зеленого, в зависимости от содержания урана.
Кроме реагентов типа арсеназо, для урана описано еще ряд окрашенных реагентов.
Торон (2-фениларсоновая кислота-<-1-азо-1>-2-оксинафта-лин-3,6-дисульфокислота) предложен для определения четырехвалентного урана [520, 787]. Предварительное восстановление урана производится в Pb-редукторе. Для повышения устойчивости комплекса определение проводят в среде 80% ацетона. Чувствительность — 0,5 мкг/мл U/(IV); фосфаты, сульфаты, оксалаты должны быть полностью удалены.
Алюминон (ауринтрикарбоновая кислота) предложен для определения U(VI) [765]. Реагент — желтовато-коричневый, комплекс — красный. Окраска устойчива при pH 4,9—7,0, оптималь
53
ное pH—5,5 iO.5. Чувствительность—10 мкг/мл U. Мешают Си; Bi, Fe(II), Fe(III), Al, Be, Th, Zr, Ce(III).
Ализарин S предложен для определения U(VI) [762, 763, 875, .994]. Реакция выполняется при pH 4—6,5. Чувствительность около 10 мкг/мл U. Переход окраски из розовой (реагент) в краснофиолетовую. Мешают Си, Fe, Al, Cr, Ba, Zr и другие элементы.
В неводной среде уран может быть определен с 2-п иридии-<1- азо - 1 > - 2 - н афтол ом [419, 540]. В водных растворах реагент дает с ураном ярко-красный осадок, переходящий в органический слой при взбалтывании с о-дихлорбензоломили с хлороформом. Осаждение проводят из аммиачных растворов, содержащих комплексон III и цианиды для маскирования прочих элементов. Маскирующие уран вещества (фосфаты, Н2О2 и другие) мешают. Чувствительность—2—10 мкг урана в 10 мл органического растворителя.
В качестве возможного реагента на UOa4" предложен 4-(2- п и р и-Дилаз о)-резорцин, дающий с ураном красное окрашивание [815]. Сам реагент желтый. Реакция выполняется при pH 8, чувствительность—1 мкг/мл U.
Реже применяются другие окрашенные реагенты: родамин Б [915], родизонат натрия [760], р-диметиламинобензол-роданид 1353], рубеановодородная кислота [419], хинизарин и другие [8, 112, 114, 117, 354, 764]. По чувствительности или избирательности они не имеют преимуществ по сравнению с описанными.
Подводя итог обзору химических качественных методов определения урана, следует признать, что ни один из п р я м ы х методов обнаружения не является надежным, если его не сочетать с предварительным отделением хотя бы некоторых из присутствующих элементов. Даже при флуорометрических определениях в большинстве случаев необходимо предварительное отделение основной массы примесей.
Проблема значительно упрощается тем, что для урана в последние годы разработан ряд быстрых методов отделения. Наиболее быстрыми являются экстракционные методы,- которые к тому же могут быть совмещены с непосредственным определением урана. Для этого к раствору, содержащему уран и посторонние элементы, прибавляют реагент (например, дибензоилметан, 2-пиридин-1-азо-1-2-нафтол, арсеназо I, арсеназо III), высаливатель, комплексон III для связывания прочих элементов, дифенилгуанидин, если применяют реагенты группы арсеназо, создают необходимое pH и экстрагируют, добавляя какой-либо не смешивающийся с водой органический растворитель. В экстракте непосредственно обнаруживают уран по соответствующей окраске комплекса уранила с применяемым реагентом.
Достаточно избирательными являются методы определения четырехвалентного урана, хотя и в- этом случае присутствие тория является недопустимым. Вообще для надежного обнаружения урана можно рекомендовать одновременное, параллельное его открытие двумя-тремя методами или при использовании двух реагентов, не являющихся аналогами и работающих по разным механизмам.
Глава IV
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ УРАНА
А. ХИМИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ УРАНА
А. А. Немодрук
Химические методы количественного определения урана, к которым относятся весовые и титриметрические методы, отличаются большим разнообразием. Это разнообразие обусловлено тем, что уран принадлежит к числу элементов, способных легко проявлять различную валентность, а также обладает ярко выраженной склонностью к образованию труднорастворимых соединений и комплексов с большим числом различных реагентов. Эти свойства урана находятся в тесной связи со строением его электронной оболочки, а такж^ с легкой поляризуемостью его ионов [54, 171 ].
I. ВЕСОВЫЕ МЕТОДЫ
Все известные в настоящее время весовые методы определения урана основаны на осаждении его из растворов, в которых он находится в Четырех- и шестивалентном состоянии. В этих валентных состояниях уран обладает способностью участвовать в образований труднорастворимых соединений не только в виде катиона, но и в виде аниона.
Весовые методы по сравнению с некоторыми другими методами определения урана характеризуются большей точностью, достигающей до 0,1% (отн.) [710]. Однако наряду с этим они обладают и рядом недостатков. Одним из этих недостатков является то, что минимальное количество урана, необходимое для весового определения, всегда значительно больше, чем в случае других методов, и составляет около 5 мг. Снижение этого минимума связано со значительным уменьшением точности определения. Кроме того, минимальная концентрация урана, при которой еще возможно его весовое определение с необходимой точностью, приблизительно равна 0,05 мг/мл, что также является на много большей величиной, чем в случае применения ряда других методов. Использование микро- и ультрамикро
55
методов позволяет несколько снизить минимальное количество урана, необходимое для весового определения, но в этом отношении преимущество все же остается за такими методами, как фотометрический, полярографический, кулонометрический и др.
Вторым существенным недостатком весовых методов определения урана является, как правило, значительно большая их продолжительность.
Вследствие этого, несмотря на большое число разработанных весовых методов определения урана, они находят ограниченное практическое применение [184, 208]. Весовые методы часто применяются для определения урана в продуктах с высоким его содержанием, в различных концентратах, в металлическом уране и его сплавах. В арбитражных анализах, а также в анализах стандартных образцов весовые методы также во многих случаях часто предпочитаются другим методам.
Весовые формы
Определение урана весовыми методами заканчивается высушиванием или чаще всего прокаливанием получаемых осадков с целью достижения определенной весовой формы. Наиболее распространенной весовой формой при определении урана является закись-окись урана U3O8. Взвешиванием в виде закиси-окиси заканчивается весовое определение урана после осаждения его гидроокисью аммония, перекисью водорода, сульфидом аммония, плавиковой кислотой или фторидом аммония, а также большинством органических реагентов. Взвешивание в виде закиси-окиси удобно тем, что она негигроскопична. Кроме того, при прокаливании других окислов урана и многих его солей, независимо от его валентного состояния, конечным продуктом всегда является закись-окись урана, если температура прокаливания поддерживается в пределах 800—1050°. Прокаливание при более низких температурах может приводить к завышенным результатам определения урана вследствие более высокого содержания трехокиси урана UO3b прокаленном остатке, чем это соответствует ее содержанию в закиси-окиси урана по формуле U3O8 [374]. Для получения U3O8 иногда рекомендуется прокаливание осадков вести при постоянном токе кислорода [1026], однако такое требование является не обязательным и для получения закиси-окиси урана вполне достаточно прокаливания осадков в хороших окислительных условиях [46].
Для устранения возможных потерь при прокаливании осадков, образуемых ураном с органическими реагентами, температуру прокаливания необходимо повышать постепенно и лишь после того, как все органическое вещество сгорит, остаток прокаливать до постоянного веса при температуре 800—1050°. Более высокая температура прокаливания, чем 1050° также нежелательна, так как в этих условиях могут иметь место некоторые потери за счет изме-56
нения состава прокаленного осадка в результате его термического разложения с выделением кислорода.
В качестве весовой формы предложена также двуокись урана UO2, которая получается при нагревании и последующем охлаждении U3O8 в токе водорода [856]. Однако имеются указания,что полное превращение закиси-окиси урана в двуокись урана в этих условиях является практически невозможным [710]. Результаты определения урана весовым методом при разных режимах прокаливания осадков диураната аммония, выделенных из одинаковых аликвотных частей раствора после прокаливания при свободном доступе воздуха или в атмосфере водорода, оказались различными. Расчет показал, что в последнем случае прокаленный остаток состоял из смеси двуокиси и трехокиси урана, в которой только 65% приходилось на долю двуокиси урана.
При определении урана осаждением в виде кислого фосфата уранила UO2HPO4 или аммонийуранилфосфата UO2NH4PO4 осадки прокаливают до пирофосфата уранила (UO2)2P2O7. Однако в 1959 г. А. Е. Клыгин показал, что это соединение образуется только в интервале температур 500°—700°. При дальнейшем повышении температуры прокаливания оно переходит в U2O3P2O7, устойчивое в интервале 700—1100°. Это соединение негигроскопично, содержит 68,206% урана и может быть рекомендовано в качестве весовой формы.
Определение урана осаждением в виде аммонийуранилванадата заканчивается прокаливанием осадка до V2O5«2UO3 [376].
Ряд органических реагентов, таких как 8-оксихинолин, арсани-ловая кислота, бензолсульфиновая кислота и другие, осаждают уран с образованием осадков, непосредственно используемых в качестве весовых форм.
Многие из органических реагентов, оказавшиеся пригодными для количественного осаждения четырех- и шестивалентного урана, образуют осадки, не имеющие постоянного состава. Во всех этих случаях определение заканчивается прокаливанием полученных осадков и взвешиванием в виде U3O8.
Для прокаливания наиболее подходящими следует признать платиновые тигли. Кварцевые и фарфоровые тигли неудобны тем, что при высоких температурах (около 1000°) постепенно взаимодействуют с окислами урана, что несколько снижает точность определения.
Определение с применением неорганических реагентов
Определение осаждением гидроокисью аммония. Гидроокись аммония известна как один из первых реагентов, примененных для количественного определения урана [800]. В дальнейшем этот метод неоднократно изучался и совершенствовался [709, 710, 857].
Количественное осаждение урана с помощью гидроокиси аммония имеет место только в тех случаях, когда применяемые растворы Не содержат ионов СО““, образующих в условиях осаждения раство
римые карбонатные комплексы урана. Растворы гидроокиси аммонпя, не содержащие карбонатов, следует готовить из жидкого аммиака, хранящегося в баллонах, которым насыщают свежепрокипяченную дистиллированную воду. Применяемые растворы гидроокиси аммония также не должны содержать растворенной кремневой кислоты, присутствие которой может приводить к получению завышенных результатов.
Чтобы уменьшить растворимость образующегося осадка диураната аммония (NH4)2U2O7, осаждение следует проводить в присутствии достаточного количества солей аммония (NH4NO3 или NH4C1). Для промывания осадка применяют растворы нитрата или хлорида аммония. Предпочтение следует отдать NH4NO3 вследствие того, что при прокаливании осадков, содержащих хлориды, возможны потери за счет некоторой летучести UO2C12.
Перед осаждением к солянокислым и сернокислым растворам, содержащим уран полностью или частично в четырехвалентном состоянии, необходимо прибавлять небольшое количество азотной кислоты для окисления четырех валентного урана до шестивалентного, так как осадок U(OH)4 трудно промывается.
. Определение урана осаждением гидроокисью аммония производится по следующей методике [8]. Анализируемый раствор подкисляют азотной или соляной кислотой и кипятят для удаления СО2. Затем на каждые 100 мл раствора добавляют по 5—10 г нитрата или хлорида аммония; в случае малых количеств урана прибавляют также беззольную бумажную массу и при перемешивании постепенно добавляют разбавленный раствор гидроокиси аммония (1 : 4) до отчетливого запаха аммиака, нагревают до кипения и кипятят в течение не менее 5 мин. для переведения осадка в легкофильтрующуюся крупнозернистую форму. После окончания кипячения прибавляют еще небольшое количество раствора гидроокиси аммония, дают осадку немного отстояться и фильтруют через бумажный фильтр «белая лента». Осадок промывают 2% -ным раствором нитрата аммония, подщелоченного несколькими каплями гидроокиси аммония. Фильтр с осадком высушивают в платиновом тигле, после чего озоляют и прокаливают при температуре около 900° при свободном доступе воздуха и взвешивают в виде U3O8.
Метод применим для определения урана в отсутствие комплек-' сообразующих веществ, удерживающих его в растворе в условиях осаждения (цитраты, тартраты, фториды). Образующиеся осадки могут частично захватывать фосфаты, ванадаты, кремневую и борную кислоты, вследствие чего получаемые результаты в их присутствии могут быть завышенными.
При определении урана в присутствии элементов, гидроокиси которых частично или полностью осаждаются аммиаком, осаждение урана предложено вести после добавления комплексона III, образующего в условиях осаждения с большинством мешающих элементов прочные комплексы, остающиеся в растворе [184, 818, 820, 975]. Таким образом возможно определение урана в присутствии достаточно больших количеств Mg, Са, Sr, Ba, Си, Zn, Со, Ni, Cd, Мп, Pb, Al, Fe, Сг, La, Се, Th, Mo, W, V и других элементов [818, 820, 975]. Осаждаются вместе с ураном лишь As, Sb, Sn (IV), Nb, Ta, Ti и Be.
-58
Для устранения мешающего влияния титана его маскируют добавлением небольшого количества перекиси водорода [975].
Однако, если содержание урана в анализируемом растворе мало (меньше 0,02%), то получаемые результаты бказываются заниженными вследствие того, что в присутствии избытка комплексона III растворимость диураната аммония заметно возрастает [184].
Определение осаждением перекисью водорода. Из растворов солей уранила с pH от 0,5 до 3,5 уран количественно осаждается перекисью водорода [987]. Реакция идет в соответствии с уравнением:
UO2 (NO3)2 4- Н2О2 4- 2Н2О - • UO., • 2Н2О Д- 2HNO3.
Само собою разумеется, что уран, присутствующий в других валентных состояниях, предварительно окисляется перекисью водорода до шестивалентного, который затем уже взаимодействует с избытком перекиси водорода с образованием нерастворимого перекисного соединения.
На основании более поздних исследований предполагается, что реакция идет через промежуточную стадию с образованием надурановой кислоты H4UO8 [986, 987]:
UO2 (NOs)2 4- ЗН2О2 Н4ио8 4- 2HNO3, которая тут же взаимодействует с еще неизменившимся ионом уранила, образуя нерастворимый перуранат уранила:
H.UO, + 2UO, (NO,), — (UO2), UOS + 4HNO,.
Такое течение реакции подтверждается тем, что при действии едкого натра на выделившийся осадок образуются перуранат натрия Na4UO8 и диуранат натрия Na2U2O7.
Количественное осаждение имеет место только при определенных условиях, среди которых наибольшее значение имеют температура и pH раствора. Для’осаждения урана перекись водорода прибавляют к охлажденному раствору, после чего реакционную смесь замораживают и лишь спустя некоторое время оттаивают и при темпера-туре-[-20 отфильтровывают выпавший осадок. Чтобы получить легко-фильтрующиеся осадки, осаждение рекомендуется проводить из растворов, содержащих NH4NO3 в концентрации около 1 моля л 1741 ]. Для обеспечения полноты осаждения необходим избыток Н2О2, но добавление более чем двухкратного количества перекиси водорода нецелесообразно. В присутствии хлоридов осаждение замедляется. Сульфаты значительно затрудняют осаждение урана; если они присутствуют в количестве эквивалентном или большем, чем количество урана, то осаждение уже становится неполным. Еще большее Решающее влияние оказывают фториды, оксалаты, тартраты и дру-fyie ионы, склонные к образованию прочных комплексов с ионом Уранила. Ацетаты мешают, если они присутствуют в значительных
59
количествах. Мешающее влияние некоторых комплексообразующих веществ — ацетатов, тартратов, оксалатов и некоторых других, являющихся слабыми кислотами, может быть значительно снижено проведением осаждения при более низких значениях pH (0,5—1,0). Однако при рН<0,5 уже не происходит полного осаждения урана, так как образующийся осадок растворяется в кислотах с образованием соответствующих солей уранила и с выделением кислорода.
Определению мешает также ряд элементов, способных в тех же условиях осаждаться перекисью водорода (Th осаждается полностью, a Zr и Hf частично). Некоторые элементы, в том числе К, Fe, Al, Ti, щелочноземельные металлы и V, если они присутствуют в значительных количествах, адсорбируются образующимся осадком [4951. Мешающее влияние железа, кроме загрязнения осадков, состоит еще и в каталитическом разложении перекиси водорода. Последним свойством обладают также ионы меди. Для устранения мешающего влияния ионов железа и меди рекомендуется перед осаждением добавлять молочную, малоновую или уксусную кислоту, которые маскируют указанные элементы и сами, присутствуя в малых количествах, не оказывают заметного влияния на полноту осаждения урана. Однако применение их не всегда дает возможность из-бежать загрязнения осадка железом [81. Прибавлением перекиси водорода к предварительно охлажденному раствору удается значительно уменьшить ее разложение за счет каталитического действия ионов железа и меди.
Для определения урана осаждением перекисью водорода может быть рекомендована следующая методика [8]. pH анализируемого раствора с помощью растворов гидроокиси аммония и азотной кислоты устанавливают в пределах 2,0—2,5. В случае присутствия железа добавляют молочную кислоту. Раствор охлаждают почти до 0° и при перемешивании прибавляют холодный 30%-ный раствор перекиси водорода в двойном молярном количестве по отношению к урану. Затем с помощью бани из спирта и сухого льда замораживают и охлаждают до примерно —45°. При этой температуре выдерживают не менее 1 часа. Далее оттаивают и при температуре не выше -|-2 фильтруют через бумажный фильтр «синяя лента», который предварительно смачивают промывным раствором с pH 2,0—2,5, содержащим 3% перекиси водорода и 3% нитрата аммония. Осадок промывают этим же промывным раствором, высушивают, прокаливают и взвешивают в виде U3O8. Если осадок очень мелкий или студенистый, что обычно имеет место в случае высокого содержания железа, то его отфильтровывают через плотный стеклянный фильтр, затем вновь растворяют в разбавленной азотной кислоте и осаждают в виде диураната аммония, который отфильтровывают через бумажный фильтр, промывают, высушивают и после прокаливания до U3O8 взвешивают.
Метод позволяет определять уран в растворах, содержащих Li, Na, Mg, Са, Ni, Со, Мп, Си и Cd.
С целью повышения избирательности осаждения с помощью перекиси водорода А. Е. Клыгин (1955 г.) применил комплексон III. Им было показано, что осаждение в присутствии комплексона III, образующего с большинством мешающих примесей прочные растворимые комплексы, дает возможность избежать загрязнения
получаемых осадков. Применение комплексона III позволяет проводить определение урана в присутствии Al, Th, Zr, Hf, V и других мешающих элементов.
В соответствии с рекомендованной методикой к анализируемому раствору, объем которого не должен превышать 150 мл, прибавляют 5 мл 10% -ного раствора двузамещенной аммонийной соли этилендиаминтетрауксусной кислоты, 0,5 мл 0,1%-ного спиртового или аммиачного раствора тимолового синего и разбавленный раствор аммиака (1 : 10) до перехода розовой окраски индикатора в желтую. При этом выпадает комплексонат уранила почти белого цвета. Затем добавляют 20 мл 30%-ного раствора перекиси водорода. Через 3—7 мин. белый осадок комп-лексоната уранила превращается в светло-желтый осадок перураната уранила. Раствор охлаждают и оставляют при пониженной температуре в течение не менее 2-х час. Осадок отфильтровывают через двойной фильтр «белая лента»; первую порцию фильтрата возвращают обратно в стакан. Осадок промывают 80—120 мл раствора, в 1л которого содержится 10 мл 10%-ного раствора двузамещенной аммонийной соли этилендиаминтетрауксусной кислоты, 30 мл 30%-ного раствора перекиси водорода и 20 мл \N раствора азотной кислоты. Осадок высушивают и прокаливают при 900—1000°. Средняя квадратичная ошибка не превышает 0,2% отн.
Метод рекомендован для определения содержания урана в технической закиси-окиси урана, двуокиси урана и в других продуктах с высоким содержанием урана.
Определение осаждением фосфатами. Определение урана осаждением его фосфатами и прокаливанием полученных осадков до пирофосфата уранила не имеет никаких преимуществ перед взвешиванием в виде закиси-окиси урана [46] за исключением тех случаев, когда анализируемые растворы содержат фосфат-ионы [812]. В качестве осадителя обычно применяют первичный фосфат аммония NH4H2PO4, который по сравнению с другими солями фосфорной кислоты обеспечивает получение несколько более чистых и легче фильтрующихся осадков [809]. Произведение растворимости образующегося осадка UO2NH4PO4 равно 4,36-10-27 [283]. Применение фосфатов щелочных металлов приводит к образованию осадков, прочно удерживающих щелочные металлы, вследствие чего получаемые результаты оказываются несколько завышенными.
Взвешивание осадка UO2NH4PO4 после его высушивания не обеспечивает получения точных и воспроизводимых результатов.
Для устранения мешающего влияния ванадия и др. металлов В. И. Титов и И. И. Волков [157, 184], а также и другие исследователи [197, 748, 818, 820, 975] предложили проводить осаждение в присутствии комплексона III, удерживающего в растворе основные мешающие элементы—Fe, Al,‘Cr, Си, Ni, редкоземельные элементы и ряд других. Ванадий при кипячении раствора восстанавливается комплексоном III из пятивалентного до четырехвалентного, который затем также маскируется избытком комплексона III. Таким образом, осаждение урана фосфатами в присутствии комплексона III позволяет количественно определять уран в сложных по составу растворах. Однако этот метод нашел основное применение как способ отделения малых количеств урана от сопутствующих элементов для
61
последующего его определения объемным или фотометрическим методом, поэтому подробное изложение методики осаждения приводится в разделе «Методы отделения урана от сопутствующих элементов» (стр. 267). Для весового определения урана в богатых продуктах (концентратах, окиси, тетрафториде и т. п.) точный вариант фосфатного метода разработан А. Е. Клыгиным, Д. М. Завражновой и Н. А. Никольской (1959 г.)..
Навеску, содержащую 200—300 мг урана, разлагают смесью HNO3 и HCI, упаривают до 2—3 мл, разбавляют водой до 10—15 мл, нагревают и фильтруют. Фильтр промывают 80—90 мл горячего раствора, содержащего 5,5% NH4C1 и 1% НО. К фильтрату прибавляют 10 мл 10% -кого раствора аммонийной соли эти-лендиаминтетрауксусной кислоты. Нагревают до кипения, добавляют горячий 0,2 М раствор (NH4)2 НРО4 до появления осадка, кипятят 1 мин. и приливают еще тот же реактив до общего его объема 10 мл. Через 5 мин. вводят 0,6—0,8 мл 0,1% -ного раствора ализаринового красного S и доводят pH до 3,7—5,2 (розовая окраска) раствором аммиака (1 : 3). Спустя 5—10 мин. фильтруют, промывают 100 мл 1%-ного раствора NH4C1, содержащего 0,1% аммонийной соли этилендиаминтет-рауксусной кислоты. Осадок прокаливают при 900—1000° f час и взвешивают как U2O3P2O7.
Определению не мешают Zr, Al, Bi, Cr, Sn, Fe и др., а также F“ и тартраты; мешают Be и Ti.
Определение урана осаждением его в виде фосфата четырехвалентного урана обладает преимуществом в том отношении, что растворимость этого соединения значительно меньше растворимости фосфата уранила. Но главное преимущество данного метода, как показал И. В. Моисеев [184], заключается в том, что четырехвалентный уран количественно осаждается в виде фосфата из кислых растворов, в которых фосфаты многих элементов не осаждаются, что значительно увеличивает избирательность осаждения.
Разработанный П. А. Волковым метод осаждения урана после его предварительного восстановления до четырехвалентного состояния, вошедший в практику под названием фосфатного метода [184], применяется главным образом как метод отделения, вследствие чего подробное его описание приводится нами в разделе «Методы отделения урана от сопутствующих элементов» (стр. 270).
Определение осаждением в виде ураниламмонийванадата. В тех случаях, когда в анализируемом растворе присутствует ванадий, осаждение в виде ураниламмонийванадата удобно тем, что позволяет избежать предварительного отделения урана [159]. При добавлении к раствору соли уранила ванадата аммония и уксуснокислого аммония до полной нейтрализации минеральной кислоты уран количественно выпадает в осадок в виде ураниламмонийванадата, обладающего очень малой растворимостью, составляющей при 18° 1,7- 10~13моль/л по урану [159]. После прокаливания состав осадка соответствует формуле V2O5-2UO3 [376].
Определение осаждением сернистым аммонием. Не содержащие карбонатов растворы сернистого аммония при добавлении к растворам солей уранила количественно осаждают уран в виде сульфи-62
да уранила, представляющего собой аморфный осадок коричневого цвета, содержащий также некоторое количество диураната аммония [8841. Осадок после прокаливания взвешивают в виде закиси-окиси урана.
Цитраты, фосфаты и особенно пирофосфаты, а также другие вещества, способные удерживать уран в растворе в виде соответствующих комплексов, определению мешают. Все элементы, образующие в условиях осаждения нерастворимые сульфиды или гидроокиси, должны отсутствовать.
Вместо сернистого аммония с одинаковым успехом может быть применен полисульфид аммония, а также газообразный сероводород. В последнем случае для сохранения щелочной реакции раствора количество добавляемого раствора гидроокиси аммония соответственно увеличивают. Метод определения урана осаждением в виде сульфида не имеет преимуществ перед определением осаждением гидроокисью аммония.
Определение осаждением сероводородом в присутствии уротропина. Осаждение урана сероводородом из нейтральных растворов солей уранила, или близких к нейтральным, в присутствии уротропина (гексаметилентетрамина) [182] обладает преимуществом перед осаждением сернистым аммонием, полисульфидом аммония или сероводородом, заключающееся в том, что осаждающееся красное вещество имеет кристаллическую структуру, обладает незначительной адсорбцией и быстро фильтруется. Осадок по составу близок к формуле HSS(OH)=U=(OUO2ONH4)4, но вследствие сильной зависимости состава осадка от условий его выделения и высушивания определение заканчивают прокаливанием осадка и взвешиванием в виде закиси-окиси урана.
Для определения урана анализируемый раствор нейтрализуют раствором аммиака, нагревают до 60°, прибавляют около 2 г уротропина и в течение 15 мин. пропускают сероводород.|3атем реакционную смесь нагревают в течение 15—20мин. при продолжающемся токе сероводорода и периодическом перемешивании. После этого колбу снимают с электроплитки и, не прекращая пропускать сероводород, дают раствору остыть до комнатной температуры. По окончании охлаждения прекращают поступление сероводорода, дают осадку отстояться в течение около 15 мин., после чего его отфильтровывают и промывают 3%-ным раствором нитрата аммония, содержащим 2—3 капли раствора аммиака в 250 мл. После высушивания прокаливают и взвешивают в виде U3O8.
Присутствие щелочных и щелочноземельных металлов и магния определению не мешает.
Определение осаждением в виде йодата. При прибавлении йодата калия к кислому раствору соли четырехвалентного урана последний количественно осаждается в виде йодата [94]. Так как уран в шестивалентном состоянии в тех же условиях не осаждается, то этот метод был рекомендован для определения четырехвалентного урана в присутствии шестивалентного.
Получаемые осадки оказались непригодными в качестве весовой формы вследствие чего определение четырехвалентного урана
63
в полученных осадках заканчивалось йодометрическим титрованием [94]. Однако впоследствии [194] были найдены условия, позволяющие определять содержание урана непосредственным взвешиванием высушенного осадка.
Для определения урана (IV) 15—30 мл анализируемого раствора, содержащего 1,7—13,5 мг урана, подкисляют серной кислотой до общего ее содержания 4—5% и прибавляют горячий 10%-ный раствор йодата калия в 10%-ной серной кислоте. Если содержание урана (IV) в растворе 5 мг, то объем прибавляемого раствора йодата калия должен составлять 50% от объема анализируемого раствора; при меньших содержаниях урана (IV) объем добавляемого раствора йодата калия увеличивают до 65% от объема анализируемого раствора. Затем прибавляют 0,8 %-ный раствор йодата калия в 2%-ной H2SO4 в двойном количестве до отношению к первоначальному объему. Выпавший осадок йодата урана отфильтровывают на взвешенном фильтрующем тигле, промывают 0,4%-ным раствором йодата калия в 1%-ной серной кислоте, затем спиртом и под конец эфиром. После высушивания при ПО—120° взвешивают в виде U(JO3)4. Фактор пересчета на уран 0,2539.
Состав осадка не изменяется при нагревании до 170°. Осадки, не промытые эфиром, начинают заметно разлагаться уже при температуре около 60°.
Медь и молибден не осаждаются йодатом и определению не мешают. Алюминий мешает, если его количество более чем в 50 раз превосходит количество урана (IV). Определение в присутствии двухвалентного железа приводит к заниженным результатам, по-видимому, вследствие имеющего место частичного окисления его до Fe(III), окисляющего затем четырехвалентный уран до шестивалентного, не осаждающегося в условиях проведения реакции. В присутствии 0,5 мг Fe (II) ошибка определения достигает 10—15 %.
В несколько иных условиях возможно также осаждение ионов уранила в виде уранилиодата UO2(JO3)2 [341, 468, 469, 814, 824]. Метод количественного определения шестивалентного урана осаждением в виде йодата разработан Венугопаланом [995].
Нейтральный раствор нитрата или ацетата уранила нагревают до 60—65° и постепенно прибавляют избыток концентрированного раствора йодата натрия. По охлаждении раствора и отстаивании осадка его отфильтровывают, промывают водой, высушивают при 110° в течение 2—3 час. и взвешивают в виде UO2(JO3)2-
При определении 6—60 мг урана ошибка не превышает 0,1%. Большие количества хлоридов и нитратов определению мешают. Фосфаты, если они присутствуют в большем количестве, чем уран, также мешают определению.
Определение осаждением с помощью перйодата. Уран может быть количественно осажден [397, 3981 при добавлении перйодата калия к уксуснокислым растворам солей уранила. Осадок имеет состав, соответствующий формуле (UO2)2K2J2O10-5H2O. При нагревании до 100° осадок теряет пять молекул воды, затем разлагается с выделением кислорода и при температуре в пределах 350—400° имеет состав (UO2)2K2J2O5. Дальнейшее повышение температуры нагревания до 600° связано с полным удалением иода, причем состав осад-64
ка соответствует формуле K2U2O7 и не изменяется при дальнейшем повышении температуры прокаливания.'
Для определения урана к 10—20 мл анализируемого раствора, содержащего 50—100 мг урана в виде ацетата уранила, прибавляют насыщенный раствор перйодата калия и оставляют в течение 8—12 час. Осадок отфильтровывают через взвешенный фильтрующий тигель, высушивают, нагревают при 350—400° и взвешивают в виде (UO2)2K2J2O5 или прокаливают при температуре выше 600 °до постоянного веса и взвешивают в виде диураната калия K2U2O7.
Ошибка определения 0,5—0,9?о.
Определение осаждением в виде фторида. Фториды количественно осаждают уран в четырехвалентном состоянии [173, 275, 541] и могут применяться для определения четырехвалентного урана в присутствии шестивалентного, не образующего осадков с фторидами.
Применение в качестве осадителя плавиковой кислоты приводит к выпадению желатинообразного труднофильтрующегося осадка гидратированного тетрафторида урана. В случае применения фторида аммония или фторидов щелочных металлов осадок состоит из соответствующего двойного фторида (например NH4UF5), обладающего еще меньшей растворимостью, чем тетрафторид урана [173, 275]. Определение урана (IV) осаждением фторидами дает возможность избежать мешающего влияния даже больших количеств циркония, тантала, бора, а также железа, ванадия и других элементов, образующих в условиях осаждения растворимые фторидные комплексы [275].
Для определения урана (IV) осаждением в виде тетрафторида к анализируемому раствору в платиновой чашке добавляют значительный избыток 40—50% -ной плавиковой кислоты, хорошо перемешивают платиновой или пластмассовой палочкой, затем нагревают, переносят в пластмассовую пробирку и центрифугируют. Маточник удаляют, осадок несколько раз промывают2—4%-ным раствором плавиковой кислоты и переносят в платиновый тигель. После высушивания осадок прокаливают до закиси-окиси урана и взвешивают. Возможно также фильтрование через бумажный фильтр с применением эбонитовой или пластмассовой воронки.
В. М. Звенигородская и Л. П. Рудина [157, 184] использовали трудную растворимость тетрафторида урана для определения общего содержания урана. Предложенный ими метод основан на предварительном восстановлении шестивалентного урана до четырехвалентного солями двухвалентного железа в присутствии значительного избытка плавиковой кислоты. Так как образующиеся в результате реакции ионы трехвалентного железа связываются в прочный растворимый комплексный анион [FeF63~ ], а четырехвалентный уран выпадает в осадок в виде нерастворимого тетрафторида, то восстановление шестивалентного урана очень быстро завершается полностью. Разработанный метод, получивший название фторидного, нашел применение главным образом для отделения урана от мешающих элементов и последующего его определения другими методами. В связи с этим подробное описание метода приводится в разделе «Методы отделения».
5 Аналитическая химия урана 65
Определение осаждением с помощью едких щелочей. Весовое определение урана осаждением едкими щелочами в виде диуранатов щелочных металлов применяется крайне редко. В зависимости от количества добавленной щелочи образующиеся осадки представляют собой различного состава основные соли [46, 389], которые при дальнейшем увеличении концентрации едкой щелочи переходят в гидроокись уранила, и, наконец, более высокие концентрации едкой щелочи позволяют количественно осаждать уран в виде труднорастворимого диураната соответствующего щелочного металла. Этим путем можно определять уран в присутствии фосфатов. Необходимая концентрация едкого натра в этом случае должна быть не менее 9 V. Осадок отделяют центрифугированием, промывают водой, высушивают, прокаливают и взвешивают в виде Na2U2O7.
Элементы, образующие в растворах едкого натра труднорастворимые гидроокиси, должны отсутствовать. Получаемые результаты, как правило, несколько завышены [46, 388], что связано, по-видимому, с выпадением в осадок смеси нескольких уранатов, имеющих, как показали В. И. Парамонова и М. Д. Морачевская [190], различное содержание натрия, в среднем составляющее несколько большую величину, чем это следует для Na2U2O7.
Определение осаждением фосфорноватой кислотой. Весовое определение четырехвалентного урана может быть осуществлено при помощи фосфорноватой кислоты, осаждающей его в виде фосфорноватокислого урана (IV) UP2O6 [588, 984]. Шестивалентный уран не образует осадков с фосфорноватой кислотой в условиях осаждения и не мешает определению четырехвалентного урана [795, 796 , 858].
Для определения урана к анализируемому раствору добавляют соляную, серную или хлорную кислоту до общей кислотности в пределах 1—3 N, нагревают до 90—100° и при перемешивании добавляют избыток 5%-ного раствора фосфорноватой кислоты или ее двунатриевой соли. Осадку дают отстояться, и добавлением нескольких капель раствора фосфорноватой кислоты к отстоявшемуся раствору убеждаются в полноте осаждения. После этого нагревают на водяной бане в течение 30—60 мин., выделившийся осадок отфильтровывают, промывают 1%-ным раствором фосфорноватой кислоты, подкисленным небольшим количеством серной кислоты, высушивают, прокаливают и взвешивают в виде пирофосфата уранила (UO2)2P2O7.
Торий, цирконий, титан и другие четырехвалентные металлы определению мешают.
Определение осаждением гипофосфитом натрия. Рай и Бхатта-чарайя [159] для определения общего содержания четырех- и шестивалентного урана применили гипофосфит натрия в присутствии тиосульфата аммония.
Анализируемый раствор разбавляют водой до объема около 200 мл,прибавляют 8—10 мл концентрированной соляной кислоты, нагревают и добавляют 20ль? 20%-ного раствора гипофосфита натрия, подкисленного 2 мл концентрированной соляной кислоты. Затем нагревают до кипения, прибавляют20—25 .ил 20%-ного раствора тиосульфата аммония для восстановления U(VI) до L’(IV) и энергично размешивают в течение 2 мин. Вслед за этим начинается выделение зеленоватого осадка фосфата урана вместе с серой, образующейся за счет разложения тиосуль-66
фата аммония. Для полного выделения урана в осадок реакционную смесь выдерживают в теплом месте в течение 2—2И час. Если отстоявшийся раствор имеет зеленый цвет, то это указывает на неполное осаждение урана. В этом случае снова прибавляют оба реактива и хорошо перемешивают. Осадок отфильтровывают через бумажный фильтр, хорошо промывают 1%-ным раствором соляной кислоты и затем горячей водой до отсутствия ионов хлора в фильтрате. После высушивания прокаливают при 900° и взвешивают в виде (UO2)2P2O7.
Железо даже в больших количествах определению не мешает.
Определение осаждением пирофосфатами. Четырехвалентный уран из кислых растворов количественно осаждается ионами пиро-фосфорной кислоты [588, 984]. В тех же условиях в виде пирофосфатов осаждаются также и другие четырехвалентные металлы, в том числе торий, цирконий и титан. Шестивалентный уран образует растворимые кислые соли и остается в растворе [795, 796, 858]. Выпадающий в осадок пирофосфат четырехвалентного урана имеет кристаллическую структуру и легко отфильтровывается.
Для определения урана его предварительно восстанавливают до четырехвалентного состояния, раствор подкисляют соляной, серной или хлорной кислотой до общей кислотности в пределах 1—3 N, нагревают до 90—100° и при перемешивании прибавляют 5%-ный раствор пирофосфата натрия. Осадку дают осесть и к отстоявшемуся раствору прибавляют несколько капель раствора пирофосфата натрия для проверки полноты осаждения; в случае появления мути добавляют раствор осадителя. Реакционную смесь нагревают на кипящей водяной бане в течение 30—60 мин. При определении малых количеств урана добавляют немного бумажной массы, осадок отфильтровывают, промывает 1%-ным раствором пирофосфата натрия, содержащего 2—3% серной кислоты. После высушивания прокаливают и взвешивают в виде (UO2)2P2O7.
Определение осаждением из карбонатных растворов гексаммино-кобальтинитритом. Определение основано на количественном осаждении комплексного карбонатного аниона шестивалентного урана с помощью гексамминокобальтинитрата [776]. Метод обладает высокой селективностью.
К анализируемому раствору, содержащему 5—80 мг иона UO|+ в виде нитрата или ацетата, прибавляют раствор карбоната аммония до выпадения осадка и далее До полного его растворения. Затем добавляют концентрированный раствор гексамминокобальтинитрата до выпадения желтого осадка или, в случае малых количеств урана, до желтого окрашивания раствора. После отстаивания в течение около 10 мин. выпавший осадок отфильтровывают, хорошо промывают спиртом, высушивают под вакуумом и взвешивают.
Состав осадка соответствует формуле [Co(NH3)6]2{[UO2(CO3)3]}-•(H2O)3.(NO3)2; молекулярный вес образующегося соединения равен 950,4. Если предварительно удалены элементы, образующие нерастворимые карбонаты, то определению мешают только те элементы, которые образуют растворимые карбонатные комплексные анионы, осаждающиеся гексамминокобальтинитратом.
Определение урана (IV) по взаимодействию с селенистой кисло-той. Дешмукх и Джоши [462] предложили определять уран (IV) по количеству выделяющегося в осадок элементарного селена при обработке анализируемых растворов селенистой кислотой.
в* 67
К анализируемому раствору добавляют избыток селенистой кислогы или ее соли и концентрированную соляную кислоту до общей ее концентрации в растворе, равной 2—3 М. затем пропускают азот для вытеснения растворенного кислорода и при непрекращающемся токе азота кипятят в течение 30 мин. По охлаждении селен, выделившийся за счет восстановления селенистой кислоты четырехвалентным ураном, отфильтровывают, промывают сначала водой, затем спиртом и эфиром, высушивают при 100—110° и взвешивают. 0,1 г Se соответствует 0,6027 г U.
При определении около 25 мг урана (IV) ошибка не превышает 3%, для 400 мг она снижается до 0,2%.
Определение осаждением окисью ртути. Из растворов солей уранила уран может быть количественно осажден добавлением суспензии окиси ртути [317]. В присутствии кислородных кислот осаждение неполное. Для достижения полноты осаждения необходимо прибавление хлорида аммония. Ускоряющее действие ионов хлора состоит в том, что они связывают образующиеся в процессе реакции ионы двухвалентной ртути и тем самым ускоряют взаимодействие с окисью ртути. Осаждение проводят при кипячении, в процессе которого, по-видимому, образуется диуранат аммония и растворимый слабодиссоциированный хлорид ртути (II). Осадок промывают кипящим раствором хлорида аммония и после прокаливания взвешивают в виде U3O8. Метод позволяет определять уран (VI) в присутствии щелочных и щелочноземельных элементов с хорошими результатами .
Определение с применением органических реагентов
Определение осаждением 8-оксихинолином. Из основных и слабокислых растворов 8-оксихинолин количественно осаждает уран (VI) в виде красновато-оранжевого соединения [589]. В отсутствие избытка реагента выпадающий осадок имеет состав UO2(C9H6NO)2. При осаждении в присутствии избытка реагента состав осадка после высушивания соответствует формуле UO2(C9HeNO)2-(C9H7NO) [525, 589]. При нагревании до 105—200° его состав не изменяется. Определение содержания урана (VI) заканчивается чаще всего взвешиванием высушенного при 105—110° осадка UO2(C9H6NO)2-(C9H7NO), являющегося удобной весовой формой, или взвешиванием в виде закиси-окиси урана после предварительного прокаливания. Количественное осаждение урана (VI) имеет место при pH в пределах от 4,1 до 13,5 [8, 553]. Если осаждение проводить при pH 10—12, то определению урана не мешают умеренные количества фосфатов и тартратов, а также небольшие количества фторидов, оксалатов, лактатов и гидроксиламина [363, 436]. Правда, имеются указания [394], что при pH>11 осадок, выпадающий из растворов едкого натра, частично содержит в своем составе натрий. Это происходит вследствие того, что этот осадок представляет собой одноосновную кислоту, значительно более сильную, чем сам 8-оксихинолин, в результате чего в щелочных растворах определенная часть его находится в виде натриевой соли NaUO2(C9H6NO)3 [394].
68
ЧетВ1рехвалентный уран также достаточно полно осаждается 8-ок-сихинолином. Одновременное ураном полностью или частично осаждаются и многие другие элементы, в том числе Al, Th, Си, Zn, Mg, Fe, Bi, Cd, V, Ag, Hg, Pb, Sb, Ti, Zr, Ta, Nb, Mn , Ni и Co.
Из растворов, содержащих едкий 'натр, 8-оксихинолином осаждаются в основном те же элементы, за исключением олова, алюминия, бериллия и щелочноземельных металлов.
Для определения урана (VI) анализируемый раствор объемом около 100 мл, содержащий до 100 мг урана в виде сульфата, нитрата или хлорида уранила, нейтрализуют раствором аммиака, прибавляют 1—2 г ацетата аммония, подкисляют приблизительно 5 мл уксусной кислоты, нагревают до кипения и по каплям прибавляют 3%-ный раствор 8-оксихинолина в 3%-ной уксусной кислоте. Раствор осадителя прибавляют до прекращения образования осадка при его соприкосновении с верхним отстоявшимся слоем раствора. После охлаждения выпавший осадок отфильтровывают через фильтрующий тигель, промывают теплрй водой до бесцветного фильтрата, высушивают при 105—140° и взвешивают, “фактор пересчета равен 0,3385. В случае взвешивания в виде закиси-окиси урана осадок отфильтровывают через бумажный фильтр, промытый осадок высушивают в платиновом тигле, затем для предотвращения механических потерь при прокаливании присыпают слоем безводной щавелевой кислоты и прокаливают прн температуре около 900°. Определению урана в этих условиях не мешают щелочные и щелочноземельные металлы. *
Как указывалось выше, осаждение из щелочных растворов с pH 10—12 позволяет определять уран в присутствии умеренных количеств фосфатов и тартратов, а также ряда других комплексообразующих веществ.
Для определения урана (VI) в этих случаях анализируемый раствор нейтрализуют раствором аммиака, образующийся осадок гидроокисей металлов растворяют добавлением серной кислоты. Затем прибавляют около 10 г хлорида, нитрата или сульфата аммония и по каплям 3% -ный раствор 8-оксихинолина в 3% -ной уксусной кислоте. После этого раствор слабо подщелачивают 6 N раствором аммиака и сверх этого добавляют еще 20 мл раствора аммиака, нагревают до 60—65° и при этой температуре выдерживают в течение 30 мин. Выделившийся осадок отфильтровывают и далее поступают так же, как и при осаждении из уксуснокислого раствора.
Для повышения избирательности осаждения урана (VI) рекомендуется применение комплексона III [898, 900]. Добавление комплексона III в анализируемый раствор перед осаждением позволяет определять уран (VI) в присутствии тория и редкоземельных элементов, а также ванадия. Подробное описание соответствующих методик приводится в разделе «Методы отделения».
Определение осаждением купфероном. В солянокислых и сернокислых растворах купферон (аммонийная соль Ы-нитрозо-Ы-фенил-гидроксиламина) образует нерастворимое соединение с четырехвалентным ураном. Шестивалентный уран в тех же условиях не осаждается [345, 607]. Осаждение купфероном пригодно также и для определения общего содержания урана. В этом случае осаждение проводят в присутствии восстановителей, таких как гидроксиламин или гидросульфит натрия. Образующиеся осадки легко отфильтровываются и промываются. Однако хотя состав осадка и соответствует
69
формуле U(C6H5N2O2)4, но вследствие его зависимости от ряда условий (избыток купферона, содержание урана) непосредственное взвешивание высушенного осадка оказалось непригодным для определения урана. В связи с этим определение заканчивают прокаливанием осадка при температуре около 1000° и взвешиванием в виде закиси-окиси урана. Щелочные и щелочноземельные металлы, алюминий, цинк, хром, бериллий, марганец и никель, а также фосфаты, бораты и фторобораты определению не мешают.
Для определения урана (IV) к анализируемому раствору прибавляют серную кислоту до общего ее содержания около 8% и охлаждают в ледяной воде. Если необходимо определить общее содержание урана, то после прибавления серной кислоты добавляют еще около 5 г солянокислого гидроксиламина. К охлажденному раствору медленно при перемешивании прибавляют холодный раствор купферона с небольшим избытком, замечаемым по появлению быстро исчезающего тонкого белого осадка, отличного от хлопьевидного осадка купфероната урана. После этого размешивают в течение не менее 2 мин., прибавляют еще 5 мл раствора купферона, хорошо перемешивают и оставляют в ледяной воде в течение 5 мин. Выпавший осадок отфильтровывают через бумажный фильтр и тщательно промывают холодным промывным раствором, который готовят смешением 100 мл 7,5%-ного раствора серной кислоты и 50 мл раствора, содержащего 0,8 г купферона и 3 г солянокислого гидроксиламина. После высушивания осадка при температуре не выше 70 (при более высокой температуре влажный осадок разжижается и вскипает) его осторожно сжигают вначале при низкой температуре, затем прокаливают при 1000 й’взвешивают в виде закиси-окиси урана.
Для устранения мешающего влияния элементов, образующих в условиях осаждения урана нерастворимые купферонаты, к числу которых принадлежат Zr, Hf, Ti, Sn, Nb, Ta, Sb, Fe, Ga и V, предложено их предварительно отделять осаждением купфероном из сернокислых растворов. Для этого перед осаждением весь уран переводят в шестивалентное состояние, затем осаждают мешающие элементы, после чего восстанавливают шестивалентный уран до четырехвалентного и осаждают его купфероном. Такой прием отделения от мешающих примесей применен Тэрнером [991] для определения урана в присутствии ванадия в карнотитах, а также Ожэ [345], Холлидэй и Кеннингхэмом [607] и А. М. Дымовым и Р. С. Молчановой [69].
Вследствие необходимости полного разрушения избытка купферона после отделения мешающих элементов, чтобы избежать его мешающего влияния при восстановлении шестивалентного урана до четырех валентного, такой метод оказался достаточно трудоемким и в результате этого находил ограниченное применение. Однако С. В . Елинсону и В. А. Олезнюк [70] путем подбора подходящего восстановителя для шестивалентного урана удалось избежать необходимости разложения избытка купферона многократной обработкой серной и азотной кислотами.
В соответствии с рекомендованной ими методикой от 0,2 до 1 г анализируемого материала (в зависимости от содержания урана) разлагают азотной кислотой и после добавления 10 мл серной кислоты (1 : 1) нагревают до выделения паров SO3. Остаток разбавляют водой для понижения концентрации серной кислоты в растворе до 10% (по объему). Раствор нагревают до полного растворения солей. Не
70
отфильтровывая нерастворившегося остатка, раствор охлаждают до температуры ниже 18° и добавляют 0,1 N раствор перманганата калия до неисчезающей розовой окраски и затем малыми порциями 6% -ный водный раствор купферона до тех пор, пока избыток его не будет отмечен появлением быстро исчезающего тонкого белого осадка в отличие от хлопьевидного осадка купферонатов металлов. Осадок промывают 10%-ным раствором серной кислоты, содержащим 1,5 г купферона в 1 л. Фильтрат вместе с промывными водами разбавляют водой до объема 200 мл, добавляют 2 г гидросульфита, перемешивают и оставляют в течение 20 мин. для полного завершения восстановления шестивалентного урана до четырехвалентного. Затем повторяют осаждение купфероном. Когда осадок скоагулирует, его отфильтровывают, промывают 150 мл 4%-ного раствора серной кислоты, содержащим 1,5 г купферона в 1 л; после высушивания сжигают и прокаливают при 1000— 1050°.
При содержании урана в анализируемой руде около 50% средняя квадратичная ошибка составляет ±0,3% (отн.), а для образцов с содержанием 5—10% повышается до ±1,2%. В присутствии молибдена результаты завышены, так как молибден не отделяется от урана и при добавлении гидросульфита натрия частично осаждается в виде сульфида, который отфильтровывается вместе с купферонатом урана и при прокаливании полностью не удаляется. Подобно молибдену ведут себя медь, свинец и другие элементы группы сероводорода.
Для устранения мешающего влияния указанных элементов перед осаждением купферонатов сопутствующих металлов раствор нагревают и добавляют 10 мл 10%-ного раствора тиосульфата натрия. Выпавший осадок сульфидов отфильтровывают и из фильтрата осаждают купферонаты примесей, как описано выше.
Метод рекомендуется для прецизионных анализов руд и концентратов с содержанием более 5% урана.
Ралф и Илвинг [863] показали, что в соответствующих условиях купферон количественно осаждает также и шестивалентный уран. Хортон [609] исследовал физические и физико-химические свойства образующегося осадка купфероната уранила и нашел, что его состав соответствует формуле NH4 [UO2(CeH5N2O2)3] и не изменяется при нагревайии до 200°.
Бибер и Вечержа [373 ] и независимо от них Маджумдар и Чоудхури [7281 предложили весовой метод определения шестивалентного урана осаждением с помощью купферона. Количественное осаждение имеет место при pH в пределах 4—9. Вследствие более высоких значений pH осаждения мешающее влияние других элементов в данном случае оказалось значительно большим, чем при осаждении четырехвалентного урана. Однако теми же авторами [373, 728] было показано, что применение комплексона III позволяет устранить мешающее влияние подавляющего большинства элементов. В этих условиях полностью остаются в растворе щелочные и щелочноземельные элементы, Mg, Ag, Hg, Pb, Cu, Cd, Мп, Zn, Co, Ni, Bi, Fe, Ge, Sn, Th, La, Се и редкоземельные элементы. Определению также не мешают небольшие количества титана (IV) и циркония. Мешающее влияние алюминия, сурьмы (III), олова (IV), ниобия и тантала устраняют прибавлением винной кислоты. Присутствие
71
нитратов, хлоридов, сульфатов, хроматов, молибдатов и вольфраматов, а также ацетатов, цитратов и оксалатов на результат определения влияния не оказывает. Если осаждение производится сразу после нейтрализации раствора аммиаком, то присутствие фосфатов, арсенитов, арсенатов и ванадатов также не мешает определению. Только фториды и карбонаты, если их содержание в растворе превосходит содержание урана, затрудняют осаждение.
А. Е. Клыгин, Д. М. Завражнова и Н. С. Коляда [101 ] установили, что наиболее селективное осаждение имеет место при pH 6,7 при концентрации купферона и комплексона III, равной 1-Ю"2 М. Для уменьшения растворимости осадка рекомендуется там, где-это необходимо, вместо увеличения концентрации купферона вводить в раствор какую-либо соль аммония, в присутствии которой растворимость купфероната уранила-аммония значительно уменьшается. Осаждение проводят из нагретого до 70—80° раствора объемом около 200 мл. Оптимальное содержание урана около 250 мг. Для создания избыточной концентрации осадителя, равной 1 • 10~2 М, необходимо вводить 40 мл 2%-ного раствора купферона. Осадок отфильтровывают через 15—20 мин. Определению мешает только алюминий в том случае, если его количество в анализируемом растворе превышает 32 мг.
Ралф и Илвинг [863] показали, что купферон также количественно осаждает трехвалентный уран. Осаждение проводится из 10%-ного раствора соляной кислоты. Устойчивость образующегося при этом нерастворимого комплекса выше, чем устойчивость купфероната четырехвалентного урана.
Определение осаждением в виде оксалата. Щавелевая кислота осаждает четырехвалентный уран из солянокислых растворов в виде оксалата урана U(C2O4)2-6H2O [579]. Растворимость оксалата урана (IV) в воде найдена равной 0,05 г/л [56]. В 0,12 N растворе соляной кислоты она является минимальной и составляет 0,005 г/л, в то время как в 6 V достигает 0,5 г/л. Однако осаждение из растворов с концентрацией соляной кислоты ниже 2 N в присутствии цинка, железа (II), меди и некоторых других элементов приводит к частичному осаждению оксалатов перечисленных элементов. Вследствие этого осаждение урана (IV) в виде оксалата проводят из растворов с концентрацией соляной кислоты от 2 до 3 N. Выпавший осадок отфильтровывают через плотный бумажный фильтр только после перемешивания при комнатной температуре не менее 1 часа. Фильтрование сразу после осаждения приводит к тому, что некоторая часть осадка (0,5—1%) проходит сквозь фильтр.
Для определения рекомендуется следующая методика [8]. К раствору четырехвалентного урана добавляют щавелевую кислоту, количество которой зависит от объема анализируемого раствора, содержания в нем урана и количества других элементов. Если в растворе содержится около 1 г урана, или объем раствора превышает 250 мл, или предполагаемое содержание железа и хрома превышает 2 г, то необходимо прибавлять не менее 4 г щавелевой кислоты. После прибавления осадителя размешивают при комнатной температуре в течение 1 часа. Выпавший оса-72
док отфильтровывают через бумажный фильтр «синяя лента», промывают 10 раз 1°о-ным раствором щавелевой кислоты в 3%-ной соляной кислоте порциями по 10 м.1, высушивают и после прокаливания взвешивают в виде закиси-окиси урана .
При осаждении урана (IV) в присутствии, железа, никеля, марганца и некоторых других элементов получаемые осадки захватывают указанные элементы только в следовых количествах. Ниобий и редкоземельные элементы частично осаждаются вместе с ураном (IV), в то время как торий осаждается полностью. Определению мешают сульфаты, фосфаты и фториды.
Определение осаждением с помощью таннина. Шестивалентный уран количественно осаждается таннином из нейтральных растворов в виде темно-коричневого объемистого осадка [451,452, 460]. Для определения малых количеств урана (VI) применение таннина, по мнению Дас Гупты [452, 460], является более подходящим, чем осаждение аммиаком, которое вследствие поглощения углекислоты и образования карбонатных комплексов может давать заниженные результаты.
Для определения урана (VI) 10—20 мл раствора, содержащего 5—120 мг урана, нейтрализуют раствором аммиака, затем прибавляют уксусную кислоту до слабокислой реакции и на каждые 12 мг урана (VI) добавляют по 2 мл 2% -ного раствора таннина. Реакционную смесь, окрасившуюся в коричневый цвет, нагревают до кипения и при перемешивании постепенно прибавляют 10%-ный раствор уксуснокислого аммония, содержащего небольшое количество аммиака. Прибавление раствора уксуснокислого аммония продолжают до коагуляции осадка и полного просветления верхнего отстоявшегося слоя раствора. Осадок отфильтровывают через бумажный фильтр, промывают раствором нитрата аммония, слабо подщелоченным аммиаком, высушивают и после прокаливания взвешивают в виде закиси-окиси урана. s
В присутствии сульфатов и умеренных количеств солей щелочных металлов осаждение становится неполным [452]. Для достижения полноты осаждения в этих случаях раствор перед фильтрованием необходимо сделать слабощелочным прибавлением раствора аммиака. Уран количественно осаждается из нейтральных растворов, содержащих тартраты, а также из оксалатных растворов в присутствии небольшого избытка аммиака [880]. Количественное осаждение урана с помощью таннина происходит также из растворов карбоната аммония, содержащих пиридин [95, 96]. Та, Ti, Nb, V, Fe, Zr, Hf, Th и Al также осаждаются таннином и мешают определению 195, 882].
Определение осаждением хинальдиновой кислотой. При добавлении хинальдината натрия (натриевой соли 2-хинолинкарбоновой кислоты) к слабокислому или нейтральному раствору нитрата, хлорида или сульфата уранила уран количественно осаждается в виде основных солей хинальдиновой кислоты [837]. Состав осадка точно не установлен. По условиям осаждения и по характеру мешающих элементов осаждение хинальдиновой кислотой в основном совпадает с осаждением 8-оксихинолином.
Определение осаждением пиридином. Уран (VI) количественно осаждается пиридином в виде диурановой кислоты H2U2O7 [181,789].
73.
Применение пиридина для весового определения урана по сравнению с применением гидроокиси аммония обладает рядом преимуществ. Пиридин как более слабое основание не содержит карбонатов и не образует уранатов, вследствие чего при осаждении урана с помощью пиридина практически отсутствует опасность неполного осаждения или загрязнения осадков карбонатами щелочноземельных металлов [96, 180].
Для определения урана (VI) к слабокислому раствору прибавляют несколько капель раствора метилового красного, 5—10 г нитрата аммония и 20%-ный раствор пиридина до нейтральной реакции и сверх этого еще около 10 мл того же раствора пиридина. Нагревают до полной коагуляции осадка и в случае, если индикатор покажет кислую реакцию, добавляют раствор пиридина. Выпавший осадок отфильтровывают, промывают 3%-ным раствором нитрата аммония, содержащим небольшое количество пиридина, высушивают и после прокаливания взвешивают в виде закиси-окиси урана.
Осаждение урана пиридином позволяет определять его в присутствии щелочных и щелочноземельных элементов, магния, марганца (II), кобальта и никеля [181]. Нитраты и хлориды определению не мешают. Сульфаты, если они присутствуют в больших количествах (около 10% сульфата аммония), затрудняют осаждение. В этом случае для достижения хороших результатов рекомендуется вместо 20%-ного раствора пиридина прибавлять на каждые 10 мг урана по 3,5—4,5 мл чистого пиридина [96].
Определение осаждением уротропином. Весовое определение урана осаждением уротропином (гексаметилентетрамином) [835] аналогично определению с помощью пиридина. Являясь слабым основанием, уротропин, так же как и пиридин, не содержит карбонатов и обеспечивает большую чистоту получаемых осадков, чем это имеет место в случае применения гидроокиси аммония.
Определение осаждением изатин-^-оксимом. При добавлении к растворам солей уранила изатин-|3-оксима в присутствии ацетатного буфера уран осаждается в виде желтовато-оранжевого осадка [613, 614]. Состав осадка соответствует формуле UO2(C8H5N2O2)2, однако вследствие частичной адсорбции осадком переменных количеств осадителя прямое взвешивание его после высушивания при 105—110° не обеспечивает получения точных результатов. Вследствие этого определение заканчивают прокаливанием и взвешиванием в виде закиси-окиси урана.
Для определения урана анализируемый раствор, содержащий 8—210 мг урана в объеме 50—100 мл, нейтрализуют аммиаком, затем подкисляют уксусной кислотой, нагревают до кипения и в кипящий раствор добавляют 5—60 мл 1%-ного раствора изатин-р-оксима в 50?о-ном этиловом спирте. В горячий раствор прибавляют 5—15 мл 10%-ного раствора ацетата аммония или натрия, подкисленного уксусной кислотой по фенолфталеину, и оставляют при комнатной температуре в течение 3 час. Выпавший осадок отфильтровывают, промывают горячей водой или 0,05%-ным раствором изатин-р-оксима, высушивают и после прокаливания взвешивают. Полученные результаты несколько завышены, но ошибка определения не превышает 1 мг в расчете на U3O8.
74
Присутствие щелочных и щелочноземельных элементов, магния, марганца и цинка определению не мешает [617]. Для определения урана в присутствии больших количеств кобальта и никеля рекомендуется перед осаждением добавлять виннокислый калий 1616].
К 20—80 мл раствора, содержащего 2—100 мг урана и до 0,3 г кобальта или никеля, прибавляют 10—15 мл 10%-ного раствора ацетата натрия, подкисленного уксусной кислотой по фенолфталеину, 5—15 мл 2%-ного раствора роданида аммония, 2,5—15 мл 2'%-ного раствора виннокислого калия и 6—30 мл 1%-ного раствора изатин-р-оксима в 50%-ном этиловом спирте. После отстаивания осадка в течение 15 мин. его отфильтровывают, промывают 100 мл 0,05%-ного раствора изатин-Р-оксима, высушивают, прокаливают и взвешивают. Результаты завышены на 0,2—0,5 мг.
В дальнейшем метод весового определения урана с помощью изатин-Р-оксима улучшался главным образом за счет повышения избирательности путем подбора соответствующих маскирующих комплексообразующих веществ. Для устранения мешающего влияния серебра, свинца и меди было предложено применение тиосульфата натрия [611], а для удержания в растворе ртути (II) рекомендовано добавлять хлориды.
Определение осаждением а-нитрозо-^-нафтолом. Из растворов солей уранила с pH в пределах 4,0—9,4 а-нитрозо-Р-нафтол количественно осаждает уран в виде тонкого желто-оранжевого осадка 1657].
К анализируемому раствору прибавляют раствор аммиака до появления осадка, который снова растворяют добавлением минимального количества азотной кислоты. Затем прибавляют 1—2 капли раствора аммиака и избыток 10%-ного спиртового раствора а-нитрозо-р-нафтола из расчета 1 г реагента на 1 г урана. Выпавший осадок отфильтровывают, промывают водой, насыщенной а-нитрозо-р-нафто-лом, высушивают и после прокаливания взвешивают в виде закиси-окиси урана.
Метод позволяет определять уран в присутствии щелочных и щелочноземельных элементов, цинка и алюминия.
Определение осаждением фениларсоновой кислотой. Фениларсоновая кислота количественно осаждает четырехвалентный уран из кислых растворов [319, 848, 1013]. В этих условиях шестивалентный уран не осаждается. Одновременно с ураном (IV) осаждаются также Th, Zr, Hf, Sn (IV), Nb и Та. Церий (IV) и титан (IV) осаждаются частично.
Для определения урана (IV) с помощью фениларсоновой кислоты к раствору Добавляют гидроокись аммония или серную кислоту до pH 1—3, нагревают почти ДО кипения и добавляют избыток 2,5%-ного раствора фениларсоновой кислоты Или ее натриевой соли. Осадок отфильтровывают, промывают 0,01—0,1 N соляной Кислотой, высушивают и взвешивают в виде фениларсоната урана U(CeH5AsO3)2 или прокаливают при 1000—1050° и взвешивают в виде закиси-окиси урана.
В качестве осадителей для весового определения урана (IV) были предложены также некоторые производные фениларсоновой Кислоты, в том числе 4-(н. бутил)-фениларсоновая, З-нитро-4-оксифе-ииларсоновая,4-диметиламиноазобензол-4'-арсоновая кислоты [ 1013 ].
75
Условия определения и мешающие элементы совпадают с таковыми для фениларсоновой кислоты. Различие состоит лишь в том, что вследствие большего молекулярного веса они образуют осадки, обладающие еще меньшей растворимостью, чем фениларсонат урана.
Арсаниловая (4-аминофениларсоновая) кислота была предложена для весового определения шестивалентного урана [810].
В этом случае анализируемый раствор сильно подкисляют азотной кислотой и прибавляют горячий водный раствор арсаниловой кислоты из расчета 500 мг на каждые 100 мг урана. Затем разбавляют водой до объема 200 мл, нагревают до кипения и добавляют раствор аммиака до pH 2,5. Выпавший осадок промывают горячей водой и после высушивания прокаливают при 1000° и взвешивают в виде закиси-окиси урана.
Ошибка определения не превышает 0,5 мг урана. Если содержание сульфатов в растворе достигает 1 г и более (считая на сульфат-ион), то осаждение становится неполным. В присутствии тартратов количество добавляемой арсаниловой кислоты необходимо увеличить до 800 мг на каждые 100 мг урана. Оксалаты мешают даже в малых количествах.
Из числа других соединений, производных мышьяковой кислоты, рекомендованных для весового определения урана, следует упомянуть какодиловую кислоту [811], количественно осаждающую уран (VI) из растворов с pH 4—7. Образующийся осадок высушивают при 200° и взвешивают в виде какодилата уранила UO2(C2HeOAs)2 или прокаливают при 1000° до закиси-окиси урана.
Определение осаждением другими органическими реагентами. Число других органических реагентов, предложенных для весового определения урана, достаточно велико, поэтому подробное изложение методов определения урана с их применением здесь невозможно. Это тем более не представляется необходимым вследствие того, что подавляющее большинство их ведут себя, подобно многим из уже описанных. Так, например, изонитрозо-Ы-фенил-З-ме-тилпиразолон осаждает уран (VI) из растворов солей уранила в виде красно-оранжевого осадка [615] в тех же условиях, что и а-нитрозо-Р-нафтол. 1-Нитрозо-2-окси-3-нафтойная кислота [445] количественно осаждает уран (VI) из растворов с pH 3,4—4,5. Осадок высушивают при 120° и взвешивают в виде H2{UO2[C10 H5O(NO)COO]2) или прокаливают до закиси-окиси урана.
Этилендиамин и другие сильные органические основания осаждают уран (VI) точно так же, как и аммиак [884, 910 ], и по сравнению с ним никакими преимуществами не обладают.
Салициловая кислота [804] и ряд ее производных, в том числе салицилгидроксамовая кислота [370], 3,5-дибромсалицилальдоксим [509], дисалицилальэтилендиамин и его аналоги [804] количественно осаждают уран (VI) из слабокислых растворов и были предложены для весового определения урана (VI).
Многие карбоновые кислоты оказались пригодными в качестве реагентов для весового определения урана (IV). Из них были 76
рекомендованы бензойная [512], и-нитробензойная [804], коричная [512], себациновая[804] и фенилглицин-о-карбоновая [447] кислота.
Уран (IV) количественно осаждают и многие сульфиновые кислоты. Бензолсульфиновая кислота количественно осаждает уран (IV) в виде бензолсульфината U(C6H5SOa)4 [684]. Определению мешают Се, Fe, Th, Sn, Ti, Zr и некоторые другие элементы. Таким же образом, как бензолсульфиновая кислота, осаждают уран (IV) сс-и 0-нафтилсульфиновые кислоты [475, 684].
При добавлении к растворам солей уранила дифенилтиокар-базида [793] ди- (н.бутил)-дитиокарбамата [730] и замещенных ксан-тогенатов [729, 1030], а также при кипячении с тиозинамином (аллилтиомочевиной) [8] имеет место количественное осаждение урана (VI) в виде соответствующих соединений.
Однако методы определения урана с помощью перечисленных органических осадителей не нашли практического применения вследствие отсутствия каких-либо преимуществ по сравнению с ранее рассмотренными реагентами, а также в связи с малой доступностью многих из них.
II. ТИТРИМЕТРИЧЕСКИЕ МЕТОДЫ
А. А. Н ем од р у к
Титриметрические методы определения урана по точности в ряде случаев не уступают весовым методам, но по сравнению с ними обладают рядом преимуществ. К преимуществам титриметрических методов следует отнести возможность определения урана в широком диапазоне его содержаний, включающих также и микроколичества, выполнение анализа в значительно более короткие сроки, а также большие возможности определения урана в присутствии других элементов, мешающих при его весовом определении.
Титриметрические методы определения урана делятся на несколько групп, каждая из которых основывается на использовании определенной химической особенности урана. Очень широкое распространение имеют титриметрические методы определения урана, основанные на окислительно-восстановительных свойствах ионов уранила и урана. Несколько меньшее значение имеют методы, основанные на титровании солей урана или уранила растворами осадителей или комплексообразующих веществ. Наконец, еще меньшее значение имеют все косвенные методы, состоящие в осаждении урана при помощи осадителей, содержание которых определяют в полученных осадках тем или иным титриметрическим методом.
Методы, основанные на восстановлении урана (VI) до урана (IV) и титровании его растворами окислителей
Эти методы включают восстановление урана (VI) до урана (IV7) и последующее титрование последнего стандартными растворами окислителей.
Из всех титриметрических методов определения урана эти методы отличаются наиболее высокой точностью и имеют самое большое применение. Если требуется определить содержание урана (IV), то в данном случае определение сводится к простому титрованию раствором подходящего окислителя. Таким способом может быть определено содержание урана (IV) в закиси-окиси урана, двуокиси урана, тетрафториде и тетрахлориде урана.
Восстановление урана (VI) до урана (IV)
Для предварительного восстановления урана (VI) до урана (IV) в качестве восстановителей широкое применение нашли многие металлы в виде гранул, пластинок, стержней, стружки, спиралей, палочек и т. п., а также в виде жидких и твердых амальгам.
Из металлов наиболее часто применяются Zn, Bi, Cd, Pb, Al, Ag, Mg и Ni.
Кроме металлов для восстановления урана (VI) до урана (IV) часто применяют также соли хрома (II), титана (III), ванадия (II) и олова (II), а также такие восстановители, как гидросульфит натрия, ронгалит и двуокись мочевины. Иногда применяют электролитическое восстановление.
Недавно были предложены методы фотохимического восстановления урана (VI) до урана (IV) с помощью молочной кислоты, этилового спирта и других восстановителей [828, 829J.
Выбор того или иного восстановителя зависит от ряда условий, среди которых основное значение имеют примеси, сопровождающие уран в анализируемом растворе. С этой точки зрения применение жидких амальгам часто имеет преимущество по сравнению с восстановлением с помощью тех же металлов, взятых в виде гранул, стружки и т. п. Это преимущество состоит в том, что в случае свежеприготовленной жидкой амальгамы примеси, выделяющиеся в амальгаму, остаются в ней и меньше мешают последующему титриметрическому определению урана (IV).
При восстановлении урана (VI) многими восстановителями в зависимости от условий часть урана восстанавливается до урана (III). В этом случае для избежания завышенных результатов образовавшийся уран (III) предварительно окисляют до урана (IV) продуванием воздуха через восстановленный раствор. При этом уран (IV) в соответствующе подобранных условиях не изменяет своей валентности.
Восстановление цинком и его амальгамами
Для восстановления урана (VI) применяется как металлический' цинк [645], так и его твердые и жидкие амальгамы [458, 536, 645, 710, 772, 823, 913]. Металлический цинк полностью восстанавливает уран (VI) до урана (IV) при кипячении сернокислых или солянокислых растворов солей уранила. При этом некоторая часть урана. (VI) восстанавливается также до урана (III).
78
Восстановление амальгамированным цинком в редукторе Джонса [46] оказывается более эффективным.
Цинковый редуктор представляет собой стеклянную трубку с внутренним диаметром около 2 см и высотой до 50 см. В нижней части трубки непосредственно над краном находится пористая стеклянная или фарфоровая пластинка, на которую-помещают небольшое количество стеклянной ваты в качестве фильтрующего слоя. Редуктор наполняют амальгамированным цинком слоем высотой 35—40 см, для приготовления которого берут 180 г цинка в виде кусочков размером 10—15 лы> и промывают 100 мл 0,5%-ного раствора азотной кислоты, затем промывают 3 раза водой. После этого производят активацию поверхности кусочков цинка обработкой в течение 10 сек. 5%-ным раствором серной кислоты и снова промывают три раза водой. Промытые водой кусочки цинка в течение 10 мин. обрабатывают 100 мл 3,6%-ного раствора хлорида ртути (II), подкисленного каплей концентрированной серной кислоты. После этого раствор сливают, амальгамированный цинк промывают несколько раз 1%-ным раствором серной кислоты и заполняют им редуктор.
При проведении анализа сначала редуктор промывают три раза по 30 мл 5%-ной серной кислоты, затем пропускают через него анализируемый раствор, содержащий 5% серной кислоты, со скоростью 25—50 мл в 1 мин. После этого* редуктор промывают три раза по 30 мл 5%-ной серной кислоты и, наконец, 30 мл воды.
Для восстановления микро количеств урана пользуются редуктором с внутренним диаметром около 7 мм, заполненным амальгамированным цинком слоем высотой около 50 мм. Скорость пропускания анализируемого раствора в этом случае уменьшают до 0,5—2 мл в 1 мин. При промывании редуктора промывной раствор вливают в тот момент, когда предыдущий раствор еще покрывает столбик амальгамированного цинка слоем до 1 см.
Во время восстановления и промывания следует следить за тем, чтобы цинк в редукторе все время находился под раствором. Когда редуктор не находится в работе, он должен быть наполнен водой, чтобы избежать образования основных солей цинка, которые могут его закупорить.
При восстановлении амальгамированным цинком не мешают сульфаты, перхлораты и хлориды. Борная и борофтористоводородная кислоты также не мешают восстановлению. Нитраты и нитриты частично восстанавливаются до соединений с более низкими валентными состояниями азота, в том числе и до гидроксиламина, которые при последующем титровании растворами окислителей способны окисляться и тем самым вызывать ошибку определения урана.
В редукторе с амальгамированным цинком восстанавливаются также Fe (III), Sn (IV), Mo (VI), V (V), Ti (V), W (VI), Nb (V), Cr (HI), Eu (III), Ce (IV), Sb (V), TI (III) и некоторые другие элементы. Поэтому они не должны присутствовать в исходном анализируемом растворе или по окончании восстановления должны быть снова окислены до валентного состояния, которое не мешает титри-иетрическому определению урана. Последний прием оказывается Весьма эффективным для таких элементов, как титан (III) и хром (II), которые окисляются до титана (IV) и хрома (III) при продувании восстановленного раствора воздухом, что необходимо производить для окисления урана (III) до урана (IV). Некоторые из Металлов, восстанавливающихся цинком, как, например, церий, Ванадий и железо, могут присутствовать в исходном растворе и не
79
мешать определению, если восстановленный раствор титровать раствором соли того же самого металла с более высоким его валентным состоянием.
Металлы, которые осаждаются цинком (Cd, Pb, Ni, Си, Ag и некоторые другие), не должны присутствовать в значительных количествах.
Органические вещества, способные окисляться при титровании восстанавливаемого раствора окислителями, должны быть предварительно разрушены упариванием исходного анализируемого раствора с концентрированной серной кислотой с добавлением небольшого количества хлорной кислоты, персульфата аммония или перманганата калия.
Вещества, осаждающие уран (IV), особенно фториды, должны отсутствовать, так как образующийся в процессе восстановления уран (IV) будет осаждаться ими в редукторе. По этой же причине содержание фосфатов в количестве, превышающем количество урана, также приводит к частичному осаждению урана в редукторе. Кроме того, необходимо иметь в виду, что восстановление урана (VI) в присутствии фосфатов замедляется.
Однако имеются указания на то, что в определенных условиях присутствие фосфатов может оказаться даже полезным. Так, Шрей-нер и Бейс [883] повышают кислотность исходного раствора до 4,5 М по серной кислоте, что исключает осаждение урана (IV) в редукторе в виде фосфата. При этом уран восстанавливается только до урана (IV), вследствие чего отпадает необходимость в продувании воздуха через восстановленный раствор.
Рекомендуемая авторами методика состоит в следующем. К анализируемому раствору добавляют 20 мл концентрированной серной кислоты и 20 мл 85%-ной фосфорной кислоты. Затем разбавляют водой до объема 75—80 мл и после охлаждения пропускают раствор через редуктор со скоростью около 30 мл в 1 мин. В случае необходимости применяют отсасывание. Редуктор промывают 20 мл 3 М серной кислоты, затем 3 раза 7,5%-ным раствором серной кислоты по 40 мл и, наконец, 40 мл воды. К восстановленному раствору прибавляют 30 мл 10%-ного раствора хлорного железа и через 5—10 мин., после того как голубовато-зеленая окраска, характерная для фосфорнокислых растворов урана (IV), перейдет в чисто желтую, добавляют 15 мл 85%-ной фосфорной кислоты, 8 капель дифениламин-сульфоната натрия и титруют 0,1 N раствором бихромата калия. Метод рекомендуется для определения урана в растворах, содержащих большие количества фосфатов.
Однако Йошимура [1024] показал, что при восстановлении в присутствии больших количеств фосфорной кислоты некоторое количество урана (III) все же может образоваться.
Кроме металлического цинка и твердых цинковых амальгам часто применяют также жидкие цинковые амальгамы [7721. Основное преимущество жидких цинковых амальгам заключается в том, что помехи от никеля, меди и других металлов, осаждающихся цинком в редукторе, устраняются применением свежей порции амальгамы для каждого нового определения.
8(1
В качестве недостатков восстановления при помощи жидких цинковых амальгам следует указать на легкость образования перекиси водорода при наличии контакта между амальгамой и кислородом воздуха. Перекись водорода, попадая в восстановленный раствор, взаимодействует с окислителем, добавляемым при последующем титровании урана (IV), и тем самым искажает результат. При определении больших количеств урана эта ошибка эквивалентна около 8 мг перекиси водорода на 1 л восстановленного раствора [8]. При восстановлении микроколичеств урана ошибка холостого опыта, обусловленная образованием перекиси водорода, остается в пересчете на 1 л восстановленного раствора почти на том же уровне, что и для макроколичеств. Так как при восстановлении микроколичеств урана на воздухе ошибка холостого опыта невоспроизводима и колеблется в пределах до 50%, то для получения точных результатов восстановление рекомендуется проводить в атмосфере углекислого газа [8]. Присутствие хрома замедляет восстановление урана
Для приготовления жидкой цинковой амальгамы 50—60 г чистого гранулири ванного цинка промывают 2%-ным раствором серной кислоты, добавляют 2 кг ртути, 100 мл 0,5 М раствора серной кислоты и нагревают на водяной бане в течение 8—10 час. Затем охлаждают, тщательно промывают водой, для отделения твердого вещества пропускают через делительную воронку и хранят под слоем 0,5 М раствора серной кислоты. Твердый остаток используют в качестве добавки к навеске цинка при приготовлении следующей порции амальгамы.
Для проведения анализа к исследуемому раствору после удаления органических соединений, нитратов, нитритов и других мешающих веществ добавляют концентрированную серную кислоту до общего ее содержания в растворе 10—15 мл, охлаждают и переносят в делительную воронку емкостью около 250 мл. Затем разбавляют ледяной водой до объема 100—110 мл, добавляют 5 мл жидкой цинковой амальгамы и встряхивают в течение 5 мин. Для снятия избыточного давления воронку опрокидывают и осторожно открывают кран. Амальгаму спускают в колбу Бунзена емкостью 100—150 мл, содержащую 75 мл 1%-ной серной кислоты и соединенную с воронкой через пробку. При помощи резиновой груши в колбе создают небольшое давление, затем, осторожно открывая кран делительной воронки, да-лот выйти воздуху. После этого амальгама стекает вниз по хвосту воронки в колбу Бунзена, уровень раствора серной кислоты поднимается до нижнего края хвоста воронки, и раствор серной кислоты начинает прорываться в воронку навстречу амальгаме. После отделения амальгамы раствор серной кислоты полностью передавливают в воронку путем опускания хвоста воронки в колбу таким образом, чтобы амальгама снизу поднялась до самого крана. Восстановленный раствор из воронки переносят в коническую колбу, воронку промывают 1%-ным раствором серной кислоты, которую присоединяют к восстановленному раствору.
кДля восстановления урана (VI) жидкими амальгамами рекомендуется также применять специальные приборы [46, 189], позволяющие, в случае необходимости, проводить восстановление в атомсфере Инертного газа.
Кроме урана (VI), жидкими цинковыми амальгамами восстанавливаются также Fe (Ш), Ti (IV),V (V), W (VI), Mo (VI), Cr (III) ? некоторые другие элементы.
Для регенерации отработанной цинковой амальгамы ее промывают 20%-ным раствором серной кислоты, отделяют в делительной воронке от загрязнений и снова Медленно сливают в 20%-ный раствор серной кислоты. Затем промывают 1 N серной кислотой и снова амальгамируют, как указано выше.
«
Аналитическая химия урана
81
Восстановление висмутом и его амальгамами
Металлический висмут и его жидкие амальгамы также довольно часто применяются для восстановления урана (VI) до урана (IV) 1184, 929, 930, 1023].
И. С. Скляренко (1950 г.) показал, что при пропускании растворов солей уранила через редуктор, наполненный порошком висмута с размерами зерен около 1 мм, уран (VI) количественно восстанавливается до урана (IV).
Висмутовый редуктор перед проведением анализа промывают 500 мл воды, затем пропускают анализируемый раствор сульфата уранила и промывают 15 мл 2,5 М раствора серной кислоты. Восстановленный раствор вместе с промывным раствором титруют одним из окислителей.
Жидкие амальгамы висмута также восстанавливают уран (VI) до урана (IV). Восстановление урана (VI) при комнатной температуре не идет дальше урана (IV) 1930).
Для приготовления жидкой амальгамы висмута поступают таким же образом, как и при приготовлении цинковой амальгамы 146]. Для восстановления рекомендуется следующая методика [930, 1023].
Анализируемый раствор, кислотность которого должна составлять 6—12 А по серной кислоте, встряхивают с жидкой висмутовой амальгамой в течение 10 мин. в атмосфере углекислого газа. Отделяют восстановленный раствор и титруют его раствором бихромата калия или перманганата калия.
Восстановление также успешно может быть проведено и с применением солянокислых растворов той же нормальности, однако в этом случае для титрования восстановленного раствора перманганат калия оказывается непригодным. Кроме урана (VI), жидкие висмутовые амальгамы восстанавливают также Fe(III), Ti(IV), V(V), W (VI), Mo (VI), Sn (IV) и Си (II). Cr (Ill) висмутовыми амальгамами не восстанавливается.
Восстановление урана (VI) в висмутовом редукторе, как показали П. Н. Палей и И. С. Скляренко [184], обладает тем преимуществом, что ванадий (V) восстанавливается только до ванадия (IV), который не мешает последующему титриметрическому определению урана , (IV) раствором ванадата аммония.
Восстановление свинцом и его амальгамой
Тонкоизмельченный металлический свинец восстанавливает уран (VI) до урана (IV) [430, 661,913, 914]. Уран (III) при восстановлении свинцом не образуется. Коблик [661] рекомендует предварительно осаждать уран (VI) в виде диураната натрия.
К выделенному осадку прибавляют 25 мл разбавленной (1:1) соляной кислоты и 2 г тонкоизмельченного металлического свинца и кипятят в течение 30 мин. Восстановленный раствор отделяют от остатка свинца, который промывают разбав-«2
ленной соляной кислотой, промывной раствор присоединяют к восстановленному раствору, прибавляют 50 мл разбавленной серной кислоты (1 : 4), 1,5 г сульфата марганца в качестве катализатора и титруют при температуре 70°. Выпадающий при этом осадок сульфата свинца титрованию не мешает.
Более удобным представляется метод/ предложенный Куком с сотрудниками [430]. Для восстановления урана (VI) до урана (IV) анализируемый раствор, подкисленный соляной кислотой до общей ее концентрации 3 N, пропускают через редуктор, наполненный тонкоизмельченным металлическим свинцом.
Восстановление с помощью жидких свинцорых амальгам идет достаточно быстро и количественно [929]. В этом случае образование урана (III) также не обнаружено даже при проведении восстановления при нагревании. Для восстановления применяют солянокислые растворы. Лучшие результаты получают при кислотности, равной 4—5 N по соляной кислоте.
Для приготовления свинцовой амальгамы чистый свинец промывают соляной, кислотой, добавляют ртуть и нагревают до получения однородной жидюй массы. Затем пропускают через делительную воронку для отделения нераствсрившихся кусочков свинца.
В отличие от жидких цинковых амальгам свинцовые амальгамы не восстанавливают хрома (III) и олова (IV).
Восстановление кадмием и его амальгамами
Кадмий является более слабым вссстановителем по сравнению с цинком и восстанавливает уран (VI) до урана (IV); уран (III) образуется в совершенно незначительных количествах [982, 983], Восстановление металлическим кадмием проводят в растворах, приблизительно 3 N по серной кислоте. Редуктор наполняют кристаллами кадмия, полученного электролитическим способом [110]. Кроме урана (VI), в кадмиевом редукторе восстанавливается также Fe (III), Ti (IV), V (V), Sn (IV), Mo (VI), Nb и некоторые другие элементы. Для восстановления небольших количеств урана (VI) было рекомендовано кипятить анализируемый раствор в центрифужной пробирке с помещенной в нее заостренной кадмиевой палочкой [418].
Восстановление урана (VI) жидкими кадмиевыми амальгамами проводят точно так же, как и с помощью цинковых амальгам. В отличие от восстановления металлическим кадмием в данном случае кроме урана (IV) образуются также заметные количества урана (III) [649, 650, 773, 774]. Приготовление жидких кадмиевых амальгам аналогично приготовлению цинковых амальгам.
Восстановление серебром
Восстановление урана (VI) до урана (IV) некоторыми авторами рекомендовалось проводить в серебряных редукторах [375, 531,999]. Однако, за исключением того, что серебро можно получить вполне ** 83
Рис. 1. Серебряный редуктор с паровой рубашкой.
А— шлиф обратного холодильника; В — серебро; С— паровая рубашка; D — круглодонная колба-паровик емкостью 100 мл; Е — вода; F— стеклянная вата; G— микрогорелка.
чистым от железа, применение серебряного редуктора для восстановления урана (VI) какими-либо преимуществами перед кадмиевым редуктором не обладает. Наоборот, вследствие недостаточно высоких восстановительных свойств металлического серебра для достижения полного восстановления необходимо пропускать через редуктор горячие растворы (70—90°). Редуктор предварительно также должен быть нагретым. Это уже само по себе представляет некоторые неудобства, тем более что, как уже указывалось выше, горячие растворы урана (IV) при доступе кислорода воздуха склонны к окислению. Во избежание возможных ошибок следует применять ряд предосторожностей для предотвращения попадания кислорода воздуха в восстановленные растворы перед их охлаждением до комнатной температуры.
Для повышения восстановительных свойств серебра следует работать при более высокой кислотности, составляющей около 4 N по соляной кислоте, которая способствует повышению окислительно-восстановительного потенциала пары U (VI)/U (IV). Кроме того, высокая концентрация иона хлора способствует понижению потенциала пары Ag/AgCl.
Имеются указания, что при восстановлении в серебряном редукторе при 80° уран (VI) приблизительно на 10% может восстанавливаться до урана (III). Доказано, что растворенный кислород, содержащийся в анализируемом растворе, может восстанавливаться в серебряном редукторе до перекиси водорода [531].
р ^Кроме урана (VI), в серебряном редукторе восстанавливаются также Fe (III), Си (II), Mo (VI)и V(V); возможно также, что и некоторые другие элементы, как например W (VI), восстанавливаются до более низких валентных состояний, a Au (111) и Se (IV) — до элементар -ного состояния [46]. К числу металлов, которые не восстанавливаются в серебряном редукторе, но восстанавливаются цинком, принадлежат Ti (IV), Сг (III), Re (VII) и некоторые другие.
Серебряный редуктор готовят следующим образом: колонку диаметром около 2 еле и высотой около . 12 см наполняют приблизительно 18 г серебра.’Для приго
-84
товления этого количества серебра раствор 29 г нитрата серебра в 400 мл воды, подкисленный несколькими каплями азотной кислоты, энергично перемешивают пластинкой электролитной меди, имеющей поверхность около 10 смг, до полного осаждения серебра. Выпавшее серебро хорошо отмывают от нитрата меди и струей воды смывают в редуктор, который хранят наполненным 1 N соляной кислотой. Редуктор (рис. 1) должен иметь паровую рубашку для нагревания самого редуктора и поддержания необходимой температуры в процессе восстановления. Приготовление серебряных редукторов описано также в [8, 999].
Для восстановления урана (IV) в серебряном редукторе может быть рекомендована следующая методика [645]. К 50 мл анализируемого раствора, содержащего 100—400 мг урана, прибавляют 8 мл концентрированной соляной и 20 мл концентрированной серной кислоты. После добавления серной кислоты раствор разогревается. При температуре 70—90° его вливают в предварительно нагретый при помощи паровой рубашки редуктор и пропускают со скоростью около 20 мл в 1 мин. Восстановленный раствор принимают в колбу, содержащую 10 мл 0,5 М раствора железоаммонийных квасцов, находящуюся в ледяной бане. После восстановления редуктор промывают 150 мл горячей смеси 5 М серной и 5 М соляной кислоты в отношении 4:1. Содержание урана определяют титрованием восстановленного раствора сульфатом церия (IV) (см. стр. 93) или другим подходящим способом.
Для восстановления малых количеств урана рекомендуется пользоваться редукторами меньших размеров.
При продолжительном употреблении редуктора серебро постепенно покрывается слоем хлорида серебра. Когда этот слой достигает трех четвертей высоты столба серебра, хлорид серебра восстанавливают.
Для регенерации серебра редуктор заполняют 1 М серной кислотой и погружают в нее цинковый стержень таким образом, чтобы он касался серебра, и оставляют до тех пор, пока цвет серебра не покажет, что произошло полное восстановление. Для полного восстановления хлорида серебра требуется 10—20 час. В случае необходимости быстрой регенерации редуктора восстановление хлорида серебра рекомендуется проводить посредством раствора хлорида хрома (II) [618].
Восстановление другими металлами
Из других металлов для восстановления урана (VI) часто рекомендуется алюминий [406, 460, 556, 624, 645]. Восстановление при помощи алюминия протекает при кипячении слабокислых растворов сульфата или хлорида уранила. Алюминий применяют в виде порошка или спиралей. Для избежания окисления горячего восстановленного раствора последний охлаждают в атмосфере инертного газа. Так как уран (VI) восстанавливается при помощи алюминия до смеси урана (IV) и урана (III), для окисления последнего до урана (IV) через охлажденный раствор пропускают воздух.
Магний восстанавливает уран (VI) таким же образом и в тех же условиях, что и алюминий [645, 809].
При кипячении растворов сульфата уранила с кислотностью 3—4 V по серной кислоте медь количественно восстанавливает уран (VI) до ypanaJIV) в течение 20—45 мин. [885]. Для восстановления урана (VI) Йошимура применил металлический никель [1023]. В ртутном редукторе уран (VI) в концентрированных растворах
85
соляной кислоты восстанавливается почти количественно до урана (IV) [404]. Однако применение Al, Mg, Си, Ni и Hg для восстановления урана (VI) до урана (IV) практического применения не нашло.
Восстановление растворами хрома (II)
В. С. Сырокомскийи К. Н. Жукова [251 ] впервые показали, что уран (VI) может быть легко восстановлен до урана (IV) при добавлении небольшого избытка раствора соли хрома (II) к сернокислому раствору соли уранила. Вследствие высокой восстановительной способности хрома (II) (окислительно-восстановительный потенциал пары Сг (Ш)/Сг(11) равен—0,41 в) восстановление заканчивается в течение нескольких секунд. Избыток восстановителя удаляется простым встряхиванием восстановленного раствора в течение 2—3 мин. при доступе воздуха или при стоянии в течение 6—10 мин. Преимущество применения солей хрома (II) по сравнению с восстановлением амальгамами металлов и самими металлами состоит в том, что для восстановления солями хрома требуется очень мало времени и выполняется оно чрезвычайно просто.
Вместе с ураном (VI) восстанавливаются также Fe (III), Mo (VI), Sn (IV), Ti (IV).
Си (II), Ag (I), Pb(II), Sn (II), Bi (III), Hg(II) и Sb (III) восстанавливаются солями хрома (II) до металлического состояния.
В литературе описан ряд методов приготовления растворов солей хрома (II): электролизом [342], восстановлением цинком бихромата калия [974] или хромокалиевых квасцов [940], а также восстановлением бихромата калия цинковой амальгамой [251].
В. С. Сырокомский и К- И. Жукова [251] для приготовления раствора хрома (II) помещают в делительную воронку 25 мл раствора бихромата калия (7,5 г/л), 5 мл концентрированной соляной кислоты и около 40 мл амальгамы цинка, содержащей около 10 г цинка. Воронку встряхивают до приобретения раствором чистого синего цвета.
Применение хрома (II) для восстановления урана (VI) до урана (IV) рекомендуется многими авторами [174, 323 , 429, 568 , 966]. Иногда восстановление ведут, защищая восстанавливаемый раствор углекислым газом. Для проверки полноты восстановления урана (VI) до урана (IV) в качестве индикатора применяют сафранин, который обесцвечивается в момент полного восстановления урана (VI) [429]. Применение индикаторов для определения конца восстановления. (сафранина, нейтрального красного, и-этоксихризоидина) удобно тем, что оно дает возможность определять не только момент окончания восстановления, но и позволяет избежать больших избытков хрома (II), вводимых для гарантии полного восстановления без применения индикатора, что позволяет получать менее окрашенные растворы и тем самым облегчает установление конца титрования восстановленного раствора. Кроме того, индикатор снова приобретает первоначальную окраску, когда весь хром (II) будет
окислен до хрома (III). Восстановленные растворы после удаления избытка хрома (II) титруют ванадатом аммония [251], перманганатом калия [966] или любым другим подходящим окислителем.
Восстановление растворами олова (II)
Хлористое олово до недавнего времени не употреблялось для восстановления урана (VI) до урана (IV) для последующего титри-метрического определения в связи с трудностью удаления избытка самого восстановителя. Однако после того как Мейн [726] показал возможность устранения избытка восстановителя добавлением хлорида ртути (II), не мешающей титрованию урана (IV) при помощи окислителей, применение хлористого олова в связи с его доступностью нашло широкое применение для восстановления урана (VI) с последующим его титриметрическим определением [43, 726].
Восстановление урана (VI) до урана (IV) с помощью хлористого олова протекает количественно в растворе с кислотностью 4—6 М по соляной кислоте и более 0,5 М по фосфорной кислоте. Роль фосфорной кислоты состоит в том, что вследствие комплексообразования с ураном (IV) она сдвигает процесс в сторону образования урана (IV).
Для восстановления урана (VI) до урана (IV) может быть рекомендована следующая методика [43]. К 10 мл анализируемого раствора, содержащего 50—100 мл урана, прибавляют 6—10 мл концентрированной соляной кислоты, 1—2 мл 85%-ной фосфорной кислоты, 2 мл 5% -ного раствора хлористого олова и нагревают в течение 15 мин. при температуре около 100°. После охлаждения до комнатной температуры прибавляют 10—20 мл насыщенного раствора хлорида ртути (II) и не ранее чем через 2 мин. приливают 25 мл 0,05 N раствора железоаммонийных квасцов, 5 мл смеси (1 : 3) концентрированной серной кислоты с 85% -ной фосфорной кислотой и титруют 0,01—0,02 N раствором бихромата калия в атмосфере углекислого газа, применяя дифениламинсульфокислоту в качестве индикатора.
Преимущество применения хлористого олова состоит еще и в том, что присутствие в анализируемом раствбре до 10 мг нитрат-иона, 12,5 мг молибдата аммония, 40 мг сульфата меди, 20 мг ванадата натрия и до 20 мг хлорида титана (IV) на результат определения урана не оказывает влияния.
Восстановление растворами титана (III)
Соли титана (III) по сравнению с хлоридом олова (II) более легко восстанавливают уран (VI) до урана (IV) [344]. Вследствие этого Для восстановления урана (VI) растворами солей титана (III) нагревания не требуется. Избыток восстановителя устраняют добавлением трехокиси висмута, которая восстанавливается до металлического висмута [781]. Металлический висмут и избыток трехокиси висмута отфильтровывают и фильтрат титруют, как обычно. Для Удаления избытка восстановителя более удобным оказалось применение перхлората ртути (II) [998], так как в этом случае необходимость в фильтровании раствора перед титрованием отпадает.
87
Для определения конца восстановления часто применяют соли меди (II) [563, 998]. Восстановление выполняют в сернокислых растворах. К анализируемому раствору прибавляют небольшое количество раствора сульфата меди (II) и затем избыток раствора сульфата титана (III) до появления красного осадка металлической меди. После этого избыток титана (III) устраняют добавлением перхлората ртути (II). Одновременно металлическая медь переходит в раствор за счет восстановления ртути (II) до металлической ртути, которая последующему титрованию урана (IV) не мешает.
Электролитическое восстановление
Восстановление урана (VI) электролизом является более продолжительным, чем восстановление его вышерассмотренными восстановителями. Однако оно имеет то преимущество, что одновременно с восстановлением урана (VI) достигается отделение значительного количества примесей [8]. Кроме того, в анализируемый раствор не вводится никаких других ионов. При восстановлении урана (VI) электролитическим методом наряду с ураном (IV) образуются некоторые количества урана (III).
Неудобством электролитического восстановления является образование перекисных соединений, часто искажающих результат титрования. Для устранения этого недостатка восстановление рекомендуется проводить с применением изолированного анода (отделенного диафрагмой). Требующаяся для восстановления аппаратура в основном совпадает с аппаратурой, описанной в разделе «Электролитические методы отделения» (стр. 337).
Фотохимическое восстановление
Фотохимическое восстановление урана (VI) до урана (IV) оказалось также пригодным для последующего титриметрического определения.
В этом случае может быть рекомендована следующая методика [829]. К 0,5— 6 мл анализируемого раствора (содержащего 2,5—50 мг U) прибавляют 5 мл 1 -М раствора этанола, 5 мл 2,5 М раствора серной кислоты, разбавляют водой до объема около 25 мл и в течение 1 часа облучают светом ртутной лампы. Затем восстановленный раствор титруют ванадатом натрия, применяя дифенилбензидин в качестве индикатора.
Другой вариант фотохимического восстановления [828] предусматривает применение вместо этанола молочной кислоты в четырехкратном избытке по отношению к урану. Вместо ртутной лампы возможно также облучение солнечным светом.
Несмотря на простоту предложенного метода и полученные авторами хорошие данные этот метод, по-видимому, пока еще нельзя рекомендовать для точных определений.
88
Титрование урана (IV) растворами окислителей
Как уже указывалось выше, при восстановлении урана (VI) некоторые из восстановителей, в том числе такие, как цинк и его амальгамы, алюминий и магний, восстанавливают его до смеси урана (IV) и урана (III). Уран (III) образуется также в небольших количествах при восстановлении с помощью кадмия и его амальгам, при восстановлении в серебряном редукторе и при электролитическом восстановлении урана (VI). Так как при этом определенного постоянного соотношения между образующимися количествами урана (IV) и урана (III) достигнуть не удается, то для получения точных результатов перед титрованием уран (III) необходимо в таких случаях снова окислить до урана (IV).
Наиболее удобным для этого методом оказалось продувание воздуха через восстановленные растворы в течение 5—6 мин. или простое встряхивание раствора в течение 10 мин. При этом уран (III) полностью окисляется до урана (IV), который в холодных сернокислых растворах достаточно устойчив по отношению к кислороду воздуха. В горячих растворах кислород воздуха окисляет уран (IV) до урана (VI) уже в достаточно заметных количествах [710]. Однако окисление урана (IV) до урана (VI) [184] существенно ускоряется и на холоде, под действием света, особенно прямого солнечного и ультрафиолетового. Кроме того, присутствие в растворе молибдена и меди также ускоряет окисление урана (IV) до урана (VI). Это ускоряющее влияние молибдена и меди значительно возрастает при действии света.
Было показано, что раствор 5 мг сульфата урана (IV) в 6V серной кислоте при освещении рассеянным дневным светом за 30 мин. окисляется на 0,8%, а при освещении кварцевой лампой ЛЮМ-1 — на 32,2%. При уменьшении концентрации серной кислоты с 41V до IV скорость окисления урана (IV) возрастает в десятки раз. В присутствии урана (VI) окисление урана (IV) также ускоряется. При содержании молибдена в растворе 0,1% от количества урана в течение 5 мин. на солнечном свету без продувания воздуха окисляется около 5% урана (IV). Медь в количестве 0,01 % от содержания урана ускоряет окисление вдвое. Ванадий и титан также ускоряют окисление урана (IV) до урана (VI) [913].
В качестве окислителей для титрования урана (IV) наиболее часто применяют перманганат калия, бихромат калия, ванадат аммония и сульфат церия. Выбор окислителя зависит от требующейся точности определения, от мешающих примесей, а также от содержания урана. Конец титрования в большинстве случаев устанавливают с помощью соответствующих окислительно-восстановительных индикаторов. В случае титрования перманганатом калия установление конечной точки возможно также по появлению малиновой окраски раствора при добавлении избытка самого реагента.
89>
При определении макроколичеств урана (>10 мг) титрование любым из вышеназванных окислителей позволяет получать результаты с ошибкой до 0,1 % (отн.) и даже несколько меньшей. Однако предпочтение следует отдать бихромату калия, который с применением дифениламина в качестве индикатора дает несколько лучшие результаты. При работе с малыми количествами урана (<10 мг) ошибка определения возрастает. При определении около 100 мкг урана с использованием соответствующих бюреток и разбавленных растворов окислителей она достигает около ±2% (отн.). При титровании малых количеств (< 10 мг) лучшие результаты могут быть получены с применением растворов ванадата аммония и фенилантраниловой кислоты в качестве индикатора.
Титрование перманганатом калия
Перманганат калия, обладая высоким окислительно-восстановительным потенциалом (+1,52 в), является единственным из применяемых окислителей (кроме ванадата аммония при титровании им в -'-IOj'V H2SO4), позволяющим проводить прямое титрование растворов урана (IV) без добавления катализаторов. Титрование проводят в атмосфере углекислого газа при температуре около 80°, так как в этих условиях равновесие достигается довольно быстро и конечная точка оказывается достаточно резкой. Для установления конца титрования наиболее подходящим индикатором является ферроин [8]. Кроме того, как уже указывалось выше, конечная точка может быть определена и без применения индикатора по розовой окраске при добавлении небольшого избытка раствора перманганата калия. В этом случае получаемые результаты менее точны. Титрование проводят в растворах, содержащих около 5% серной кислоты. Для повышения точности определения рекомендуется проводить холостое титрование и вводить соответствующие поправки. 1 мл 0,1 N раствора перманганата калия соответствует 0,01191 г урана. Титр раствора перманганата калия устанавливают по щавелевой кислоте или оксалату натрия.
При титровании 0,36 г урана ошибка составляет ± 0,05% (отн.). В присутствии 10 мг железа она увеличивается до±0,35%, (отн.), а в присутствии 2 г достигает±2% (отн.). Применение в этом случае роданида аммония в качестве индикатора для определения конца титрования урана (IV) оказалось безуспешным, так как красная окраска роданида железа (III) появляется слишком медленно [8].
При титровании растворами перманганата калия хлориды в титруемом растворе должны отсутствовать. Различные варианты титрования урана (IV) растворами перманганата калия рассматриваются в работах [966, 976, 10231.
90
Титрование бихроматом калия
Ввиду хорошей сохранности растворов бихромата калия, а также благодаря тому, что он легко может быть получен в чистом виде и вследствие этого сам является стандартным веществом, его применение для определения урана является несколько более удобным, особенно для постоянной работы. Нормальный окислительно-восстановительный потенциал системы Сг (VI)/Cr (III) достаточно высок и составляет + 1,36 в [236].
Однако недостатком этого метода является то, что при непосредственном титровании растворов урана (IV) вследствие медленного протекания реакции конечная точка получается очень нечеткой. В связи с этим к раствору урана (IV) предварительно прибавляют раствор соли железа (III) для окисления урана (IV) до урана (VI) и затем уже титруют образовавшееся в эквивалентном количестве железо (II) в присутствии фосфорной кислоты, необходимой для связывания железа (III) [668]. Связывание как введенного избытка железа (III), так и железа (III), образующегося в процессе титрования, позволяет увеличить разницу между окислительно-восстановительными потенциалами систем Сг (VI)/Cr (III) и Fe (III)/Fe (II) и тем самым дает возможность проводить титрование при комнатной температуре с хорошей конечной точкой. Определение ведут в достаточно больших объемах для ослабления окраски образующихся солей С г (III).
В качестве индикатора наиболее часто применяют дифениламин-сульфокислоту или ее бариевую (натриевую) соль, дающую растворимые продукты окисления с более ярко выраженным изменением окраски, чем в случае дифениламина [877]. Имеются указания на пригодность и некоторых других индикаторов, в том числедифенил-бензидина [883].
Ошибкйопределения составляет!),1—0,2% (отн.). В присутствии нитратов определение конечной точки затрудняется и вследствие этого ошибка значительно возрастает.
Для определения урана (IV) титрованием бнхрэлштом ..алия может бы.ь рекомендована следующая методика [8]. К 300 мл анализируемого раствора, содержащего до 300 мг у рана, добавляют серную кислоту до общей ее концентрации в растворе в пределах 1,5—2N и 20 мл 4%-ного раствора хлорида железа (III). Затем добавляют 15 мл смеси (2 : 1) фосфорной и серной кислот, 8 капель 0,01 Мраство-ра дифениламинсульфоната натрия и медленно титруют 0,027V раствором бихромата калия при постоянном перемешивании до тех пор, пока чистая зеленая окраска титруемого раствора не перейдет в серо-зеленую. После этого раствор бихромата калия прибавляют очень медленно, каждый раз по одной капле, до тех •Пор, пока не появится пурпурный или фиолетово-голубой оттенок. При этом небольшое количество бихромата калия расходуется на окисление самого индикатора. Это нужно иметь в виду при титровании малых количеств урана и вносить соответствующую поправку в результат титрования. Однако, когда титр раствора бихромата устанавливают по раствору с известным содержанием урана (IV), to необходимость в такой поправке отпадает.
91
Применение бихромата калия для титриметрического определения урана (IV) описывается многими авторами [43, 563, 668, 726, 883, 966].
Пандуранга Рао, Мурти и Гопала Рао [825] исследовали оптимальные условия протекания реакции между ураном (IV) и бихроматом при прямом титровании и установили, что при повышении кислотности до 5tV по серной кислоте скорость взаимодействия настолько возрастает, что прямое титрование становится возможным. Лучшим индикатором для такого титрования оказалась N-фе-нилантраниловая кислота [253, 653, 958].
Для проведения титрования рекомендуется следующая методика [825]. К Ю—25 мл анализируемого раствора, содержащего 0,12—0,3 г урана (IV), прибавляют 50 мл 10V серной кислоты и разбавляют водой до объема около 100 мл. Прибавляют 2 капли 0,1%-ного раствора N-фенилантраниловой кислоты и титруют 0,05V раствором бихромата калия до появления розовой окраски индикатора.
Титрование сульфатом церия
Вследствие хорошей сохранности титрованных растворов сульфата церия, а также более четкой конечной точки по сравнению с титрованием бихроматом калия (зеленая окраска образующегося хрома (III) несколько затрудняет установление индикаторного перехода) сульфат церия нашел достаточно широкое применение для определения урана (IV) [375, 460, 537, 568, 726, 878, 913, 914, 976, 998, 1015, 1023].
Прямое титрование растворов урана (IV) сульфатом церия при комнатной температуре вследствие недостаточной скорости реакции затрудняется и дает размытую конечную точку. Получаемые результаты занижены на 1—2,6% (отн.) [8].
Это затруднение, как и в случае титрования бихроматом калия, устраняют добавлением избытка железа (III), которое окисляет уран (IV) до урана (VI). Выделившееся в эквивалентном количестве железо (II) титруют раствором сульфата церия (IV).
Другой вариант титрования состоит в том, что к титруемому раствору прибавляют стандартный раствор сульфата церия (IV), избыток которого затем оттитровывают обратно стандартным раствором сульфата железа (II) [536, 537].
Хорошие результаты могут быть получены и при прямом титровании растворов урана (IV), если титрование вести при температуре около 50°. Для устранения окисления урана (IV) кислородом воздуха нагревание титруемого раствора и титрование его необходимо проводить в токе инертного газа (СО2 или N2). Титрование дает хорошие результаты и в том случае, если его проводить при комнатной температуре и лишь вблизи конечной точки нагревать раствор примерно до 50°. Преимущество этого варианта состоит в том, что титрование можно проводить без применения инертного газа.
Наиболее подходящим индикатором при титровании растворами сульфата церия (IV) является ферроин (1,10-фенантролинферро-
92
сульфат) [1015]. Однако при титровании нагретых до 50° растворов в 4N соляной кислоте этот индикатор довольно сильно распадается на составные компоненты, и розовая окраска исчезает несколько раньше, чем достигается полное окисление урана (IV) до урана (VI) [375]. Для устранения этого недостатка необходимо добавлять больше индикатора. Кроме ферроина, в качестве индикаторов были рекомендованы также эриоглицин, эриозеленый и ксиленцианол FF [830], которые дают хорошие результаты при титровании растворов с кислотностью в пределах 0,5—8N. Интересным оказалось применение медьфталоцианинтетрасульфокислоты, позволяющей проводить титрование при комнатной температуре [878]. В конечной точке происходит отчетливое изменение окраски этого индикатора из бирюзово-голубой через ярко-розовую в бесцветную. Преимуществом медьфталоцианинтетрасульфокислоты по сравнению с ферроином является ее доступность и низкая стоимость, а также весьма высокая устойчивость ее растворов при хранении. Основным недостатком этого индикатора следует считать необратимость , индикаторного перехода, что, по-видимому, может являться источником дополнительных ошибок. Правда, имеются указания, что результаты титрования урана (IV) удовлетворительно совпадают с данными титрования с применением ферроина [878].
Бирнбаум и Эдмондс [375] для ускорения окисления урана (IV) сульфатом церия (IV) рекомендуют добавлять фосфорную кислоту .
При титровании урана (IV) растворами сульфата церия (IV) с применением ферроина в качестве индикатора присутствие в титруемом растворе до 5/6 азотной кислоты заметного влияния на точность определения не оказывает [11].
Присутствие до 100 мг мышьяка (III) при прямом титровании при комнатной температуре 300 мл раствора урана (IV), содержащего 5 мл фосфорной кислоты и 25 мл серной кислоты, также не мешает титрованию.
Растворы урана (IV), полученные восстановлением в цинковом редукторе, Силл и Питерсон [913] рекомендуют титровать после добавления железа (III).
Анализируемый раствор объемом около 75 мл, содержащий 6 мл концентрированной серной кислоты, охлажденный до температуры 15—20°, пропускают через цинковый редуктор. Редуктор промывают 50 мл холодной 2N серной кислоты и затем 50 мл холодной воды. Через восстановленный раствор в течение 5 мин. пропускают воздух. После этого добавляют 2 мл 20%-ного раствора железоаммонийных квасцов в 5% -ной серной кислоте, 1 мл 85% -кого раствора фосфорной кислоты, 1 мл 0,001 М раствора ферроина и титруют 0,01А раствором сульфата церия до исчезновения красной окраски индикатора. Вблизи конечной точки раствор сульфата церия добавляют порциями по 0,02 мл с промежутками в 30 сек.
Для растворов, восстановленных в свинцовом редукторе, теми же авторами [913] рекомендуется прямое титрование, которое в данном случае протекает без затруднений. Повышение скорости
93
взаимодействия урана (IV) с церием (IV), по-видимому, связано с тем, что в растворах, восстанавливаемых в свинцовом редукторе,, отсутствует серная кислота, которая образует с церием (IV) сульфа-тоцериевую кислоту и тем самым снижает концентрацию ионов Се*+ в растворе, вследствие чего окислительно-восстановительный потенциал пары Се (IV)/Ce (III) несколько снижается.
Около 80 мл анализируемого раствора, содержащего 20 мл концентрированной соляной кислоты, пропускают через свинцовый редуктор диаметром 12 мм, заполненный гранулированным свинцом слоем высотой 30 см (размерами 17— 25 мм), со скоростью 100 мл в 1 мин. Температура раствора должна быть около-25°. Редуктор промывают 80 мл 1N соляной кислоты. К восстановленному раствору добавляют 1 мл 0,001 М раствора ферроина и 1 каплю 85%-ной фосфорной кислоты и титруют 0,01 N раствором сульфата церия (IV) до заметного ослабления окраски индикатора. После этого добавляют 2 мл 85%-ной фосфорной кислоты и продолжают титрование до исчезновения красной окраски индикатора. К концу титрования раствор сульфата церия (IV) прибавляют порциями по 0,02 мл, и после добавления каждой порции дают выдержку продолжительностью 30—60 сек.
Как при титровании с применением железоаммонийных квасцов, так и при непосредственном титровании растворов урана (IV), восстановленных в свинцовом редукторе, при определении около 1 мг урана ошибка составляет ±2% (отн.), для количеств около 20 мг и более она не превышает ± 0,1% (отн.).
В случае применения медьфталоцианинтетрасульфокислоты титрование проводят по следующей методике [878]. К 5—10 мг восстановленного раствора прибавляют 10 мл 4N раствора серной кислоты, 2 капли 0,1%-ного раствора индикатора, 2,5—3 мл 85% -ной фосфорной кислоты, разбавляют водой до объема 60 мл и титруют 0,0LV раствором сульфата церия (IV) до перехода бирюзово-голубой окраски индикатора в ярко-розовую и затем до полного обесцвечивания. Для достижения большей точности рекомендуется вводить индикаторную поправку, определяемую в холостом титровании.
Дешмукх и Джоши [726] титрование сульфатом церия (IV) проводят после добавления феррицианида калия, окисляющего весь уран (IV) до урана (VI). Образующийся в этих условиях ферроцианид титруется сульфатом церия (IV), причем индикатором является уранилферроцианид, т. е. практически индикатор не добавляется.
Раствор урана (IV), полученный восстановлением с помощью алюминиевой стружки, с кислотностью 2—3 N после пропускания воздуха для окисления урана (III) до урана (IV) и последующего пропускания азота для удаления растворенного кислорода смешивают с отмеренным объемом раствора феррицианида калия. Для обеспечения полноты окисления урана (IV) до урана (VI) феррицианид калия добавляют с избытком, составляющим не менее 50% от требующегося количества. К смеси прибавляют 3N раствор едкого натра до перехода красно-коричневой окраски феррицианида урана (IV) в желтую и отмечают израсходованный объем щелочи. Выдержав в атмосфере азота в течение 15—20 мин., добавляют на 5—10 мл больше ЗА' раствора серной кислоты, чем было добавлено щелочи. Образовавшийся ферроцианид титруют раствором сульфата церия (IV) по каплям при энергичном перемешивании до перехода темно-красной окраски, обусловленной образовавшимся ферроцианидом уранила, в бледно-желтую.
Полученные данные хорошо согласуются с результатами определения урана в тех же пробах 8-оксихинолинатным методом.
94
Титрование ванадатами аммония, натрия и калия
В. С. Сырокомский и Ю. В. Клименко [252] впервые применили растворы ванадатов для титрования урана (IV).
Анализируемый раствор после восстановления в цинковом редукторе, содержащий 40—50 мл 10# серной кислоты, продувают воздухом в течение 5 мин. для окисления урана (III) до урана (IV) и титруют приблизительно 0,03# раствором ванадата аммония с применением N-фенилантраниловой кислоты [253, 653, 958] в качестве индикатора до перехода зеленой окраски раствора в фиолетовую. При определении 0,06—0,24 г урана ошибка составляет 0,06—0,6% (отн.).
Нарасинхасастри и Гопала Рао [775] несколько изменили методику титрования и к восстановленному в цинковом редукторе раствору после продувания воздуха прибавляли раствор соли железа (III) для окисления всего урана (IV) до урана (VI). Образовавшееся в эквивалентном количестве железо (II) титровали раствором ванадата натрия после предварительного добавления дифенилбензидина в качестве индикатора и 3 мл 1N щавелевой кислоты, необходимой для более резкого перехода окраски индикатора. Полученные авторами результаты хорошо совпадали с результатами, найденными при титровании тех же количеств урана (IV) растворами сульфата церия (IV).
После того как В. С. Сырокомский [249] показал, что в сернокислых растворах потенциал системы V (V)/V (IV) резко возрастает с увеличением концентрации кислоты и в 27N серной кислоте достигает 4-1,45 в, применение ванадатов для титрования урана (IV) значительно расширилось [184, 826, 828, 829].
Стандартные растворы ванадата аммония рекомендуется готовить растворением соответствующей навески в 250 мл разбавленной серной кислоты (1:1) с последующим разбавлением водой до 1 л. Для определения малых количеств урана установка титра производится по точному раствору сульфата уранила (приготовленному из чистого металла или U3O8) и восстановленному в кадмиевом или висмутовом редукторе* [184].
И. П. Алимариным |[184] было показано, что применение Af-фенилантраниловой кислоты в качестве индикатора при титровании ванадатом аммония малых количеств урана (IV) (порядка 0,1 мг) в растворах, содержащих 33% серной кислоты (по объему), позволяет получать четкие индикаторные переходы. П. А. Волков [184] нашел, что довольно четкие переходы окрасок с применением N-фенилантраниловой кислоты могут быть получены при титровании даже очень разбавленными растворами ванадата аммония. При титровании 0,0004 N раствором ванадата аммония 15—20 мл раствора урана (IV) с применением 1—2 капель 0,2%-ного раствора N-фенилантраниловой кислоты все еще наблюдается вполне четкий переход окраски индикатора.
Пандуранга Рао, Мурти и Гопала Рао [8261 нашли, что при титровании урана (IV) растворами ванадата натрия с применением N-фенилантраниловой кислоты в качестве индикатора необходи
95
мая четкость индикаторного перехода может быть достигнута даже в 4 N растворах серной кислоты, если в титруемые растворы добавлять щавелевую кислоту, катализирующую эту реакцию.
При титровании растворов урана (IV), полученных восстановлением в висмутовом редукторе, присутствие ванадия в анализируемом растворе определению не мешает, так как в висмутовом редукторе уран (VI) и ванадий (V) в 52V серной кислоте восстанавливаются только до четырехвалентного состояния [184].
Титрование растворами железа (III)
Для титрования растворов урана (IV) могут быть использованы соли железа (III) [81. Титрование необходимо проводить при повышенной температуре (около 90°). Для избежания окисления урана (IV) кислородом воздуха нагревание растворов и само титрование следует выполнять в атмосфере инертного газа. Конец титрования определяют по появлению неисчезающей красной окраски роданида железа (III). Вследствие недостаточной точности титрования (ошибка несколько более 0,3% (отн.), а также вследствие большого количества мешающих элементов (W, Mo, V, Sn, Си, фториды) соли железа (III) редко применяются для титриметрического определения урана.
Титрование‘броматом калия
Взаимодействие урана (IV) с броматом калия протекает в две стадии [8]:
1) зи1+д-вгог4-зн2о^зиоГ4-вг-4-бнт
2) ВгОГ4-5Вг-4-6Н+^ЗВг24-ЗН2О.
Первая стадия протекает очень медленно. Вторая стадия также имеет небольшую скорость. Вследствие этого прямое титрование урана (IV) броматом калия оказалось непригодным для количественного определения. Однако титрование броматом калия возможно • в том случае, если предварительно к раствору урана (IV), содержащему 2056 соляной кислоты, добавить раствор соли железа (III) и хлорид меди (II) в качестве катализатора. Образовавшееся железо (II) может быть легко оттитровано броматом калия. Титрованию мешают никель и большие количества меди. В исходном растворе урана (IV) должны отсутствовать восстанавливающие вещества, как Fe (II), V (III), V (IV) и Mo (V), которые могут быть оттитрованы вместе с образовавшимся в эквивалентном количестве железом (II). Ошибка определения составляет около 0,4% (отн.).
Вследствие недостаточной точности метода он имеет небольшое практическое применение.
96
Йодометрическое титрование
Мор [7561 показал, что уран (IV) может быть определен окислением железом (III) с последующим Йодометрическим титрованием его избытка. Однако из-за низкой точности определения, вследствие зависимости количества выделяющегося свободного иода от кислотности раствора, концентрации иодида калия и ряда других условий этот метод не находил практического применения.
После того как Десай и Мурти [4581 за счет применения тетраокиси осмия в качестве катализатора значительно повысили скорость выделения иода при взаимодействии иодида калия с избытком железа (III), точность титрования намного повысилась. Самым главным достоинством метода является возможность определения урана в присутствии больших количеств железа. Изменение кислотности титруемого^раствора в пределах 0,5—5 N на результат титрования не оказывает влияния. Присутствие фосфатов до 500 мг, считая на Р2О5, определению не мешает, однако в этом случае титрование следует проводить при кислотности не менее 2 N для предотвращения осаждения фосфата урана (IV).
В соответствии с рекомендуемой авторами методикой к анализируемому раствору, содержащему 50—100 мг урана, добавляют 2,5 мл концентрированной серной кислоты и разбавляют водой до объема 50 мл. Раствор охлаждают и пропускают через цинковый редуктор, который затем промывают 4 раза 5%-ным раствором серной кислоты порциями по 25 мл. Через восстановленный раствор продувают воздух в течение 5 мин., добавляют из бюретки определенный объем (20—25 мл) 0,05 N раствора железоаммонийных квасцов в 5% -ной серной кислоте и оставляют в течение 5 мин. для окисления всего урана (IV) до урана (VI). Затем добавляют 1 г карбоната натрия для вытеснения воздуха выделяющейся углекислотой, 10 г твердого иодида калия, 2 капли 0,25% -ного раствора четырех-окиси осмия в 5%-ной серной кислоте, закрывают колбу и размешивают до полного растворения иодида калия. Выделившийся иод титруют 0,05 N раствором тиосульфата натрия, применяя крахмал в качестве катализатора. Ошибка определения достигает 0,4% (отн.).
Возможно также окисление урана перйодатом калия с последующим восстановлением избытка перйодата добавлением иодида калия [8]. Выделившийся при этом свободный иод титруют раствором тиосульфата натрия или арсенита натрия.
Дешмукх и Джоши [460] к восстановленному с помощью металлического алюминия раствору урана (IV) добавляют избыток титрованного раствора феррицианида, раствор подщелачивают для завершения окисления урана (IV) до урана (VI), затем снова подкисляют 2V раствором серной кислоты, добавляют иодид калия и выделившийся иод титруют раствором тиосульфата натрия или арсенита натрия, применяя крахмал в качестве индикатора. При определении 25—300 мг урана ошибка определения не превышает 1 %.
Рассмотренные йодометрические методы применяются довольно редко.
7 Аналитическая химия ураиа
Методы, основанные на титровании урана (VI)j растворами восстановителей
Методы, основанные на титровании урана (VI) растворами восстановителей, обладают тем преимуществом, что позволяют исключить предварительное восстановление растворов уранила. Однако точность этих методов значительно ниже. Для титрования урана (VI) наиболее часто применяют соли хрома (II) [511, 996] и титана (III) [277, 278].
Титрование солями хрома (II)
Как ранее указывалось (стр. 86), соли хрома (II) очень легко восстанавливают уран (VI) до урана (IV). На этом основании они были рекомендованы для прямого титрования растворов уранила. Методы приготовления растворов солей хрома (II) рассмотрены в предыдущем разделе (стр. 86). Анализируемый раствор титруют в атмосфере инертного газа раствором сульфата или хлорида хрома (II) в присутствии нейтрального красного, феносафранина, метилового красного или п-этоксихризоидина в качестве индикаторов. Одновременно в идентичных условиях проводят установление титра раствора соли хрома (II) титрованием точного количества соли уранила или бихромата калия. Ого необходимо делать каждый раз, если выполняется одиночное определение, или 2—3 раза в течение рабочего дня при серийных определениях в связи с тем, что титр растворов солей хрома (II) постоянно изменяется. Растворы солей хрома (II) хранят в атмосфере инертного газа и в сосудах из темного стекла. Ошибка определения достигает 1% (отн.) [966].
Титрование солями титана (III)
Титрование солей уранила растворами титана (III) выполняется точно так же, как и титрование солями хрома (II). Разница состоит только в том, что вследствие несколько меньшей восстановительной способности титана (III) титрование проводят при температуре, близкой к 100°. В качестве индикаторов применяют новый синий Д(с), вольфрамат натрия или сафранин [277, 278]. Как в случае титрования солями хрома (II), так и в случае титрования солями титана (III) вместе с ураном (VI) титруются все другие окислители, в том числе Fe (III), V(V), Си (II), W (VI), Mo (VI).
Титрование солями хрома (II) и титана (III) применяется редко вследствие особых предосторожностей, необходимых при хранении их растворов, а также в процессе самого титрования.
11L КОМПЛЕКСОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Несмотря на то, что за последние годы методы комплексометрического титрования приобрели первостепенное значение для тит-риметрического определения многих элементов, для определения урана они нашли только очень небольшое применение. Это связано с тем, что ион уранила образует недостаточно прочные комплексы с рядом комплексонов, вследствие чего мешающее влияние других элементов оказалось очень большим. С другой стороны, титрование урана (IV), образующего прочные комплексы в достаточно кислых растворах, в которых мешающее влияние других элементов (за исключением Zr, Th, Pu, Fe и некоторых других) невелико, все же удобнее проводить оксидиметрическим методом, позволяющим с такой и даже большей точностью определять его содержание в присутствии значительно большего количества других элементов.
Для комплексометрического определения урана (VI) [518, 692] анализируемый раствор при pH 4,4—4,6 титруют раствором комплексона III, определяя конечную точку при помощи 1-(2-пиридилазо)-2-нафтола в качестве внутреннего индикатора. Для повышения растворимости окрашенного комплекса иона уранила с индикатором и повышения его устойчивости титрование проводят в смеси (1:2) воды с изопропиловым спиртом.
Анализируемый раствор, содержащий до 100 мг урана (VI) в объеме до 300 мл, подкисляют разбавленной азотной кислотой до pH 2—3, затем добавляют раствор уротропина до pH 4,4, разбавляют двойным объемом изопропилового спирта, нагревают до 80—90°, прибавляют 0,1%-ный спиртовый раствор индикатора до отчетливой розовой окраски и титруют 0,05 М раствором комплексона III до перехода розовой окраски титруемого раствора в чисто желтую.
Возможно и обратное титрование. В этом случае к титруемому раствору прибавляют избыток раствора комплексона III, нагревают до 90°, прибавляют раствор индикатора и титруют избыток комплексона III раствором хлорида или сульфата цинка до перехода желтой окраски раствора в розовую.
Ошибка определения может достигать + 0,3 мг урана. Элементы, реагирующие с комплексоном III в условиях титрования, должны отсутствовать.
Недавно П. Н. Палей и Сюй Ли-юань [187] разработали метод определения урана (IV) титрованием растворами комплексона III с применением торона [1- (2-арсонобензолазо)-2-оксинафталин- 3,6-Дисульфокислоты] в качестве индикатора [187].
Анализируемый раствор (содержащий 1—50 мг урана) восстанавливают в 1 N хлорной кислоте на ртутном катоде и в мерной колбе емкостью 50 мл разбавляют водой до метки. К 5 мл полученного раствора прибавляют 3 мл 1 N НС1О4, разбавляют водой до объема 100 мл (pH 1,0—1,8), нагревают до —30° и после прибавления 6—10 капель 0,1%-ного раствора торона титруют 0,0003—0,05 М раствором комплексона III до перехода красной окраски в оранжево-желтую.
При определении не слишком малых количеств урана (> 5 мг) ошибка не превышает 0,6% (отн.). Мешают Th, Со, Си и другие элементы, образующие с комплексоном III прочные комплексы при 7* '99
pHsc2. Присутствие нитратов, хлоридов и ацетатов на точность определения влияния не оказывает.
Разработанный метод успешно применен для определения урана (IV) в тетрафториде урана [187]. Для устранения мешающего влияния иона фтора прибавляют хлорид алюминия.
IV. МЕТОДЫ, ОСНОВАННЫЕ НА ГИДРОЛИЗЕ СОЛЕЙ УРАНИЛА
Эта группа методов обладает невысокой точностью и малой избирательностью. Все ионы, способные к гидролизу в тех же условиях, должны отсутствовать. Из числа методов, основанных на способности солей уранила к гидролизу, наиболее простым является титрование едким натром и другими щелочами. Однако такое титрование более удобно проводить потенциометрическим методом [8]. Присутствующая в растворе свободная кислота не мешает, так как получаются две вполне различные точки перегиба — одна для кислоты и другая для иона уранила.
Дешмукх и Джоши [461 ] нашли, что гидролиз солей уранила при pH 4,2—4,3 проходит количественно при нагревании до 60—70° с избытком 0,1 М раствора йодата калия и иодида калия. При этом выделяется эквивалентное количество кислоты, которое приводит к образованию соответствующего количества свободного иода. Суммарное уравнение протекающей при этом реакции представляется в следующем виде:
3U0J+ + 5J - + JO? + ЗН,О 3UO, (ОН), + 3 л,.
Анализируемый раствор нейтрализуют 0,033 IV раствором едкого натра до pH 4,2—4,3, добавляют избыток 0,1 М раствора йодата калия, 3—5 г иодида калия, 5—10 мл четыреххлористого углерода и в течение 20 мин. нагревают с обратным холодильником при 60—70°. Выделившийся иод титруют 0,1 N раствором тиосульфата натрия или арсенита натрия в боратном буфере. По одному из ранее предложенных вариантов этого метода [543] выделившийся иод отгоняют с водяным паром и поглощают раствором иодида калия, а затем титруют тиосульфатом натрия.
V. МЕТОДЫ, ОСНОВАННЫЕ НА ТИТРОВАНИИ УРАНИЛОВОЙ СОЛИ ПЕРУРАНОВОЙ КИСЛОТЫ
Метод состоит в осаждении урана (VI) перекисью водорода из слабокислых растворов в виде ураниловой соли перурановой кислоты, которую после отделения и промывания растворяют в разбавленной серной кислоте, и освобождающуюся перекись водорода титруют раствором перманганата калия. Для устранения ионов (сульфаты, фториды, оксалаты), мешающих полному осаждению урана в виде ураниловой соли перурановой кислоты, их предварительно отделяют осаждением урана едким натром в виде диураната натрия. Отрицательные ошибки, которые при этом возникают, отчасти устраняются введением эмпирического титра перманганата калия, устанавливаемого по известному количеству урана 100
(VI), подвергаемому точно такой же обработке, как и анализируемый раствор 1315, 316].
Для определения урана этим методом к 20 ял анализируемого раствора прибавляют небольшой избыток 10%-ного раствора едкого натра, не содержащего карбонатов, выпавший осадок центрифугируют в течение 5 мин. на центрифуге с 2000 оборотов в минуту, затем растворяют в минимальном объеме 10%-ной азотной кислоты, полученный раствор нейтрализуют 3%-ным раствором едкого натра, устанавливают pH в пределах 2—3, прибавляют 0,5 ял 30%-ного раствора перекиси водорода и выдерживают в течение 30 мин. После этого выделившийся осадок ураниловой соли перурановой кислоты центрифугируют, промывают 3 раза 3%-ным раствором нитрата аммония, подкисленного азотной кислотой до pH 2—3, растворяют в 20 ял разбавленной серной кислоты (1 : 2) и титруют 0,05 N раствором перманганата калия.
Метод позволяет определять уран с ошибкой 1,5—3% (отн.) при его концентрации в растворе до 1,5 мг/мл. Определению мешают, если присутствуют в количестве, равном содержанию урана, Sb (HI), Fe (III), Co, Сг (III), Th , фториды, фосфаты и ванадаты.
С. В. Блешинский и А. Г. Нечаева (1952 г.) несколько улучшили метод определения урана перманганатометрическим титрованием осадка пероксида. Они осаждают уран в слабощелочной среде в присутствии сульфата аммония. Осадок отфильтровывают и промывают 2%-ным раствором сульфата аммония до исчезновения в промывной воде перекиси водорода. Затем осадок растворяют в 25 ял разбавленной серной кислоты (1 : 3) и титруют 0,05 N раствором перманганата калия. Титр перманганата калия рекомендуется устанавливать по известной навеске соли уранила, следуя той же самой методике. При определении около 0,08 г урана в отсутствие титана ошибка определения составляет около it 0,8%.
В присутствии титана рекомендуется другой вариант, заключающийся в предварительном выделении урана (VI) в виде комплекса Ca(UO2)2(VO4)2-8H2O, соответствующего по составу минералу тюямуниту. Полученный осадок отфильтровывают, промывают нейтральным раствором нитрата аммония и растворяют в разбавленной серной кислоте. К раствору прибавляют 5 г сульфата аммония, 5 ял 5%-ного раствора алюминиевоаммонийных квасцов, затем нейтрализуют аммиаком до слабощелочной реакции по лакмусу, добавляют 1 ял 30%-ного раствора перекиси водорода, хорошо перемешивают и оставляют в течение 2-х час. Выпавший осадок отфильтровывают, промывают 2%-ным раствором сульфата аммония и далее поступают так, как описано выше.
Алкалиметрическое титрование осадка ураниловой соли перурановой кислоты [986, 987] дает худшие результаты [735].
VI. МЕТОДЫ, ОСНОВАННЫЕ НА ВОССТАНОВЛЕНИИ УРАНА (VI) ДО УРАНА (Ill) И ПОСЛЕДУЮЩЕМ ТИТРОВАНИИ ЕГО РАСТВОРАМИ ОКИСЛИТЕЛЕЙ
Г. А. Панченко [189] показал, что уран (III) в кислых растворах титруется иодом только до урана (IV). На этом основании им был предложен метод количественного определения урана. Анализируемый раствор восстанавливают амальгамой цинка в присутствии достаточного количества соляной кислоты в атмосфере инертного газа. К восстановленному раствору добавляют раствор иода в избытке, который затем оттитровывают раствором тиосульфата натрия в присутствии крахмала в качестве индикатора. При определении
101
0,25 г урана результаты занижены приблизительно на 1 %. По данным автора, ошибка определения в зависимости от количества урана может достигать 2—3%. Присутствие железа в анализируемом растворе определению не мешает, так как железо (II), образующееся после восстановления амальгамой цинка, в кислом растворе иодом не титруется.
Мейн [726] для восстановления урана (VI) до урана (III) применил хлорид олова (II). Он нашел, что хлорид олова (II) легко восстанавливает уран (VI) до урана (III) в присутствии фосфорной кислоты и железа (III) в качестве катализатора. Избыток восстановителя устраняют добавлением хлорида ртути (I) и титруют уран (III) раствором бихромата калия с применением дифениламинсуль-фоновой кислоты в качестве индикатора. Титрование и само восстановление проводят в атмосфере инертного газа. Для получения более точных результатов вводят поправку на добавленное железо и индикатор. Определению не мешают фосфаты, арсенаты, висмут и малые количества нитратов.
VII. МЕТОДЫ, ОСНОВАННЫЕ НА ТИТРОВАНИИ РАСТВОРАМИ ОСАДИТЕЛЕЙ
Титрование фосфатом
Титрование растворов солей уранила фосфатами основано на образовании нерастворимого двойного фосфата уранила типа UO2MPO4, где М — одновалентный катион. Хотя метод не отличается большой точностью, но позволяет быстро проводить определение. В качестве индикаторов применяют ферроцианид калия, салицилат натрия или 2-окси-5-метилбензоат натрия [206, 739, 954]. Титр раствора фосфата устанавливают по стандартному раствору нитрата уранила в тех же самых условиях, в которых ведут определение.
Анализируемый раствор, содержащий 50—400 мг урана, нейтрализуют аммиаком до выпадения осадка, затем добавляют такое количество уксусной кислоты, которое необходимо для растворения этого осадка, избегая большого избытка; В зависимости от содержания урана разбавляют водой до объема 50—200 мл, нагревают до 90° и осторожно титруют при интенсивном перемешивании примерно 0,1 М раствором однозамещенного фосфата калия до отсутствия положительной реакции при проведении внешней пробы с одним из вышеперечисленных индикаторов. В случае применения в качестве внешнего индикатора ферроцианида калия небольшое его количество помещают на фарфоровую пластинку и наносят каплю титруемого раствора. Если коричневая окраска не возникает, считают, что титрование закончено. Для получения надежного результата необходимо провести 2—3 титрования.
Определению мешают все катионы, образующие в условиях титрования нерастворимые фосфаты.
Кроме фосфатов, растворы солей уранила можно титровать также и другими осадителями, в частности растворами ферроцианида, применяя описанную выше внешнюю пробу с сухим порошком ферроцианида для определения конца титрования.
102
VIH. КОСВЕННЫЕ МЕТОДЫ
Большинство косвенных титриметрических методов состоит в том, что ионы урана или уранила осаждают каким-либо подходящим осадителем, выделяют образовавшийся осадок, и титриметри-ческим методом определяют в нем содержание осадителя. К числу осадителей, которые могут быть использованы для определения урана косвенными методами, следует отнести 8-оксихинолин, щавелевую кислоту, йодаты, перйодаты, салициловую кислоту, п-амино-фениларсоновую кислоту и некоторые другие. Применение этих методов иногда представляет интерес в том отношении, что для титрования связанных с ионами уранила или урана осадителей требуется значительно больше эквивалентов титранта, чем это имеет место при окси диметр ическом титровании тех же количеств урана (VI) или урана (IV). Так, например, для титрования 1 моля урана, осажденного в виде 8-оксихинолината, требуется 12 эквивалентов брома [589].
Титрование 8-оксихинолината уранила бромид-броматным раствором
Из рассматриваемой группы методов титрование 8-оксихинолината уранила бромид-броматным раствором обладает наибольшей точностью. Достоинством такого метода является еще и то, что осаждение в виде 8-оксихинолината, особенно с применением комплексона III для удержания других элементов в растворе, позволяет избавиться от мешающего влияния большого числа других элементов.
Для определения урана его предварительно осаждают в виде 8-оксихинолината уранила описанным ранее методом (см. раздел «Весовые методы определения»), отфильтрованный и промытый осадок H[UO2(C9H6NO3] растворяют в 2 N соляной кислоте, раствор разбавляют той же соляной кислотой до объема 50—75 мл, добавляют 1 г бромида калия, 2—4 капли 0,1%-ного раствора метилового красного и титруют 0,1 N раствором бромата калия до перехода красно-оранжевой окраски в желтую. Сверх этого добавляют избыток 2—3 мл раствора бромата калия, закрывают колбу и оставляют в течение нескольких минут для полного завершения выделения брома. При этом 8-оксихинолин, выделившийся при растворении 8-ок-сихинолината уранила в соляной кислоте, бромируется с образованием 8-окси-5,7-дибромхинолина; C9H7NO 2Вга—<-C9H5NOBr24- 2НВг. Затем добавляют 1 г иодида калия и выделившийся иод титруют 0,1 N раствором тиосульфата натрия.
При определении 5 мг урана ошибка определения составляет около 1% (отн.) [8].
Японские исследователи [658] модифицировали эту методику для определения микроколичеств урана (10—200 мкг).
Йодометрическое титрование осадка^ перйодата уранила
Метод основан на количественном осаждении уранила из ацетатных растворов в виде перйодата уранила с последующим растворе
103
нием его в соляной кислоте и титрованием иода, выделяющегося после добавления иодида калия [398].
К Ю—20 мл анализируемого раствора, содержащего 0,6—1,2 г урана в виде ацетата уранила, добавляют насыщенный раствор перйодата калия и оставляют в течение 8—12 час. Выпавший осадок светло-желтого цвета отфильтровывают и промывают холодной водой. Осадок по составу соответствует формуле K2(UO2)2J2On-5HaO. Его растворяют в растворе иодида калия, подкисленном соляной кислотой, и выделившийся иод титруют 0,1 N раствором тиосульфата натрия, применяя крахмал в качестве индикатора.
Ошибка определения не превышает 0,9% (отн.).
Основным недостатком метода является его большая продолжительность.
Йодометрическое титрование осадка селенита уранила
Джоши [66] разработал метод, состоящий в осаждении урана в виде селенита уранила из водно-спиртовой среды при pH 4—5; осадок после отделения растворяют в соляной кислоте и титруют селенистую кислоту раствором иода.
Вследствие малой доступности селенистой кислоты и большого числа мешающих элементов метод не нашел практического применения.
Б. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ УРАНА
I. ФОТОМЕТРИЧЕСКИЕ МЕТОДЫ
1. Колориметрические и спектрофотометрические методы определения в видимой и ультрафиолетовой областях
Ю. П. Новиков
Колориметрические и спектрофотометрические методы определения урана очень многочисленны и применяются для самых различных объектов с широким интервалом концентраций урана — от тысячных долей до десятков процентов.
Эти методы включают колориметрические определения путем визуального сравнения и работы с использованием фотоэлектрических фотометров и спектрофотометров.
Ниже употребляются следующие термины.
Пропускание:
Т = Г 7 о
где
10— интенсивность излучения, входящего в кювету;
I — интенсивность излучения, выходящего из кюветы. Оптическая плотность:
D = lg^-.
104
Пересчет от величины Т к D, которая более удобна в аналитической практике, так как прямо пропорциональна концентрации поглощающего вещества и толщине оптического слоя, можно произвести по формуле:
D=lg^.
Молярный коэффициент погашения:
где
Dx— оптическая плотность при длине волны X;
с — концентрация поглощающего вещества, моль!л\
I — толщина поглощающего слоя, см\
—соответствует оптической плотности одномолярного раствора окрашенного вещества при толщине слоя 1 см.
Определение урана по собственной окраске его солей
Степени окисления урана легко идентифицировать по спектрам поглощения. Существенные различия в светопоглощении ионов урана (IV) и уранила связаны с разным количеством электронов в 5/-облочке этих атомов. Наличие полос поглощения в видимой области объясняют взаимодействием по крайней мере двух 5/-электронов; по актинидной гипотезе [4,226] по два электрона в 5 /-оболочке имеют уран (IV), нептуний (V), плутоний (VI).
В этих валентных состояниях элементы дают характерные полосы поглощения с более высокими молярными коэффициентами поглощения. Полосы поглощения иона уранила, у которого отсутствуют электроны на указанной оболочке, имеют максимальное значение в ультрафиолетовой области спектра. Пятивалентный уран не поглощает в видимой области, так как расстояние между подуровнями в 5/-оболочке настолько велико, что полоса лежит в ультрафиолетовой области.
Эти различия в спектрах поглощения урана используются для качественного и количественного определения валентных состояний урана. Спектры поглощения соединений урана (III), урана (IV) и уранила с различными анионами изучались как в водных [4, 8, 368, 953], так и в органических растворах [4, 8, 627, 733]. Влияние различных анионов на характер спектров обусловлено комплексообразованием; как правило, для урана (IV) это влияние сказывается меньше, чем для урана (VI), что объясняют [4] трудностью воздействия внешних аддендов на поглощающие свет электроны, находящиеся во внутренней 5/-оболочке. Тем не менее и для урана (IV) необходимо учитывать влияние pH, концентрации анионов и их природу при рассмотрении его спектра.
105
Уран в водных растворах
При определении урана (IV) чаще применяют фосфорнокислые растворы, в которых спектры поглощения урана претерпевают наименьшие сдвиги в связи с изменением концентрации кислоты, чем в сернокислой и солянокислой среде (рис. 2, 3).
Рис. 2. Спектры поглощения урана (IV) и урана (VI) в фосфорной кислоте. А— уран (VI) в концентрированной Н3РО4; В— уран (IV) в концентрированной Н3РО4 С— уран (IV) в Н,РО4 (1 : 1); D— уран (IV) в Н3РО4 (1:5). Все измерения сделаны на спектрофотометре Бекмана в кювете с /=10 мм.
Метод определения урана (IV) и урана (VI) в растворах, полученных после растворения технической закиси-окиси урана в фосфорной кислоте, основан на использовании дифференциальной спектрофотометрии. Ионы урана (IV) поглощают свет в той же области спектра (410 ммк), что и уран (VI). Однако уран (IV) можно определить без помех со стороны иона уранила в более длинноволновой области спектра, при 630 ммк. Поэтому, определив концентрацию урана (IV), можно вычислить его поглощение при длине волны 410 ммк, которая выбрана для определения урана (VI). Разность между измеренной при 420 ммк суммарной оптической плотностью и вычисленной для четырехвалентного урана даст оптическую плотность, соответствующую концентрации иона уранила. Точность определения урана (VI) зависит от соотношения концентраций урана (IV) и урана (VI). Чем меньше будет это соотношение, тем точнее будет определен уран (VI). Подробности метода и пропись приведены в книге Роддена 18].
Фосфорнокислые растворы используются также и в методе Кеннинга и Диксона [407] для прямого спектрофотометрического опре-106
деления урана в присутствии ряда элементов, в том числе ванадия, хрома, редких земель в концентрациях до 2 г/л, титана до 10 г/л (в пересчете на двуокись титана) и железа как двухвалентного, так и трехвалентного до 40 г/л в пересчете на Fe?O4. Уран восстанавливают до четырехвалентного и светопоглощение измеряют в 40%-ном растворе фосфорной кислоты при 630 ммк,. Ошибка определения урана в интервале 0,5—5,0 г/л меньше 2% (отн.). Нижний предел определения урана в кювете с толщиной слоя 4 см равен 0,25 мг{мл в пересчете на закись-окись.
Рис. 3. Спектры поглощения урана (IV) в солянокислом растворе. А— уран (IV) в концентрированной НС1; В— уран (IV) в НС1 (1 : 1). Все измерения сделаны на спектрофотометре Бекмана в кювете с Z=10 мм.
Методы определения урана (VI) по собственной окраске являются менее чувствительными и менее избирательными, чем для урана (IV), так как большинство обычно сопутствующих урану элементов имеют полосы поглощения в той же области спектра, что и уран (VI). Поэтому иногда для определения шестивалентного урана его восстанавливают до урана (IV).
Один из наиболее точных фотометрических методов определения урана (VI) разработан Бейконом и Милнером [350]. Проанализировав возможные источники ошибок, авторы нашли, что определение урана в его чистых препаратах, таких как закись-окись и металлический уран, можно производить с точностью до ± 0,04% (отн.). Для этого применялась тщательная юстировка аппаратуры, термо-статирование кювет при измерениях, точное калибрование всей мерной посуды, а также строгий контроль концентрации серной кислоты. Влияние различных концентраций серной кислоты на характер спектров поглощения урана показано на рис. 4.
Оптимальными условиями для определения урана является 4 М концентрация серной кислоты; оптическая плотность измеряется при 430 ммк.
107
Те же авторы описали [348 ] дифференциальный способ определения урана в различных двойных и тройных сплавах на основе урана при содержании последнего более 40%.
При анализе сплава уран—титан 1 г образца растворяют в смеси 20 мл 40% -ной серной кислоты и 20 мл воды, окисляют с помощью перекиси водорода и выпаривают до появления паров SO8, охлаждают и разбавляют водой до 50 мл.
При анализе сплава уран — молибден 1 г образца растворяют в 20 мл 40% -ной азотной кислоты, добавляют 20 мл 50%-ной серной кислоты и выпаривают до появления паров. После охлаждения и добавления 25 мл воды вновь следует выпаривание. Измерение оптической плотности производят при 430 ммк. Конечная концентрация серной кислоты в кювете соответствует 3,6 М. Такая же концентрация соблюдается и для сплава, содержащего уран, титан и цирконий.
Для сплава уран—молибден—ниобий, кроме серной кислоты той же концентрации, в растворе находится муравьиная кислота до 0,2 М. Поправку на поглощение при 430 ммк титана, молибдена, циркония, ниобия определяют по искусственно приготовленным смесям из урана и-указанных элементов. Ошибка определения урана составляет ±0,1% (отн.).
Та же сернокислая среда была выбрана для определения урана в урано-титановых сплавах и другими авторами [357].
Рис. 4. Спектры поглощения уранила при различном содержании серной кислоты.
Концентрация серной кислоты: А—3,0 М; В—0,5 М; С—9 Af; В—18,0 М.
Концентрация урана везде 0,063 М. Спектры сняты на спектрофотометре Бекмана при хцелн 0,05 жл (0,5—0,9 ммк). Кювета с/=10 мм.
Другой метод [948] определения шестивалентного урана в сернокислых растворах дает точность, сравнимую с точностью объемных и весовых методов. Использующийся здесь прием дифференциальной спектрофотометрии основан на том, что оптическая плотность анализируемого раствора измеряется относительно раствора сравнения, содержащего уран в известной, точно определенной и близ
108
кой к анализируемому раствору концентрации. Для того чтобы выбрать раствор сравнения с близкой и несколько меньшей концентрацией, чем анализируемый раствор, в последнем приближенно определяется концентрация урана. При этом на спектрофотометре Бекмана при длине волны 418 ммк помещают по пути светового луча кювету с анализируемым раствором и, увеличивая ширину щели, устанавливают стрелку гальванометра на нуль. По найденной таким образом ширине щели, которая тем более, чем выше концентрация урана в растворе, по заранее составленной таблице зависимости ширины щели от концентрации находят приближенную концентрацию урана в анализируемом растворе.
Подобрав близкий по концентрации раствор сравнения, измеряют относительно него оптическую плотность анализируемого раствора. Концентрацию урана определяют по уравнению:
ci = F-Ar-{-c1, -
где су — концентрация урана в растворе сравнения;
е2 — концентрация урана в анализируемом растворе;
Аг. — разности между оптическими плотностями этих двух растворов;
F — фактор, равный среднему значению ряда величин Ас/Аг.
Метод может быть применен при концентрациях 20—60 г/л урана в конечном растворе. Средние отклонения результатов измерений
Рис. 5. Влияние концентрации хлорной кислоты на оптическую . плотность перхлората уранила [616].
Содержание урана 0,4 г в 25 мл раствора.
составляют 0,3% (отн.). Микрограммовые количества посторонних ионов: СгО*", МпО7» Fe3+ и Ni3+ не влияют на результаты определения.
Иногда для повышения чувствительности определения урана используют поглощение в ультрафиолетовой области [1010]. В этой же работе рассматривается спектрофотометрическое определение урана в карбонатных растворах.
109
Определение урана в хлорнокислых растворах [916, 936] удобно тем, что в широком интервале концентраций хлорной кислоты от минимальных ее количеств до 65% (7,4 Л4) оптическая плот-
Рис. 6. Спектр поглощения шестивалентного урана в карбонатом растворе. Концентрация урана в граммах.: на литр, кювета с /==10 мм.
ность при 417 и 420 ммк перхлората уранила не меняется [9161 (рис. 5). Перхлораты алюминия, железа, тория и циркония в количествах соответственно 0,1; 0,2; 1,0; 2,0 г в 25 мл раствора не мешают определению, так как не поглощают в пределах 415— 420 ммк. Влияние других ионов, таких как фториды, хлориды, карбонаты, сульфаты, существенно и они должны быть устранены (рис. 6).
Изучая влияние различных элементов на спектрофотометрическое определение урана (IV) в хлорнокислых растворах, Стил [936] установил. что Со2+, Nia+, V, Au, Сг (III) и Сг (VI) при
420 ммк сильно мешают определению. Особенно мешает Сг; уже при 0,00065% Сг (VI) и 0,006% Сг (III) ошибка определения 0,1% (отн.). Сульфаты и фосфаты завышают^ результаты определе-
Рис. 7. Спектр поглощения иО2+ в азотнокислом растворе а -f*
Концентрация UO2 =0.0398 Л4; NOg —2,40 М [368].
ПО
ния. При наличии' указанных элементов анализ урановых концентратов возможен при введении поправок на содержание примесей или после отделения урана от сопутствующих элементов.
При определении урана в азотнокислых растворах следует учитывать возможные изменения в спектре поглощения в зависимости
Рис. 8. Зависимость оптической плотности раствора UO®+ от концентрации NOS" • Концентрация UO® + равна 0,040 М [368b
от концентрации азотной кислоты (рис. 7, 8). Эти изменения связаны с комплексообразованием и ассоциацией ионов в молекулу нитрата уранила [368].
Б. Н. Гринев, И. Д. Смирнова и Т. И. Машадова (1957 г.) использовали измерение светопоглощения растворов нитрата уранила для того, чтобы по графику оптическая плотность — удельная электропроводность определять кислотность в системе UO2(NO3)2 — HNO3 — Н2О.
Уран в органических растворах
С помощью органических растворителей можно определять уран в более сложных объектах, так как экстрагирование позволяет отделять его от мешающих элементов. При экстрагировании возможно также концентрирование урана.
Часть методов определения урана в органических растворителях связана с анализом технологических экстракционных растворов.
Спектры поглощения растворов нитрата уранила в органических растворителях приведены на рис. 9, 10.
Значения молярных коэффициентов погашения растворов нитрата уранила в ацетоне, метилэтилкетоне, метилизобутилкетоне, диоксане, формамиде и других растворителях получены Б. Ежов-ской-Тшебятовской и А. Бартецким [627] (табл. 24). Ими отмечен высокий молярный коэффициент погашения, равный 29,85 при 423,2 ммк раствора нитрата уранила в формамиде.
111
Таблица 24
Значения молярных коэффициентов погашения в при различных длинах волн X (в ммк) для растворов UO2(NO3)2-6H2O в некоторых растворителях [627]
Рчс. 9. Спектры поглощения ГО2(ЬЮ?)2 • 6Н2О в воде и в органических растворителях [733].
I— метилэтилкетон; II — ацетон; III— диэтиловый эфир; IV— вода.
Пейдж, Эллиот и Рейн предложили метод [790], основанный на измерении оптической плотности комплекса урана с трибутилфосфатом в ультрафиолетовой части спектра при 250 ммк. Нитрат уранила экстрагировали из 6 М раствора нитрата натрия в 25%-ный раствор ТБФ в изооктане при невысокой кислотности (pH 3), так как азотная кислота, экстрагируемая в трибутилфосфат, сама сильно поглощает в ультрафиолете. Молярный коэффициент погашения уранил-трибутилфосфатного комплекса 7.,£0 Л(Л^=2400. Метод позволяет определять уран в пределах 0,002—5,0 мг/мл. Для данного метода предельно допустимыми количествами посторонних элементов в исходном растворе являются: по 0,5 мг/мл Al, Ba, Bi, Cd, Сг (HI), Со, Си, Fe (II), Pb, Li, Mg, Ni, Ag, Na. Zn; по 0,05д1г/ль? Be, Fe (HI), Ain (II), Hg (II); no 0,005 мг/мл Ce (III), Ru (II), Ti (III). Для анионов— по 0,5 мг/мл ацетатов, хлоридов, хроматов, перхлоратов, сульфатов; по 0,05 мг/мл фторидов, цитратов: менее
8
112
Аналитшеская химия урана
113
чем по 0,005 мг мл оксалатов, фосфатов, а также арсенатов, молибдатов, перманганатов, вольфраматов и ванадатов (в пересчете на соответствующий металл).
Рис. 10. Спектры поглощения UO2(NO,)2'6H,O в органических растворителях [627].
I— формамид; II — трибутилфосфат; III — диоксан.
К 10 мл раствора, содержащего 0,01—0,1 мг)мл урана, прибавляют 5,1 г нитрата натрия; азотной кислотой и едким натром устанавливают pH 3, используя pH-метр. Переносят раствор в делительную воронку объемом 60 мл и экстрагируют уран 10 мл 25%-ного раствора трибутилфосфата в изооктане. Оптическую плотность экстракта измеряют в кювете с Z= 10 мм при 250 ммк относительно раствора, полученного аналогичным образом, но не содержащего урана. При концентрации урана больше чем 0,1 мг!мл вводимая в раствор аликвотная часть должна быть такой, чтобы органическая фаза содержала 0,01—0,1 мг/мл. В интервале концентраций 0,002—0,01 мг/мл измерения производят в кювете с /=50 мм. Обычные отклонения при определении урана составляли меньше 1% (отн.).
Определение урана по реакциям с неорганическими реагентами
Большое количество методов описано для фотометрического определения урана с кислород- и серусодержащими реагентами. Наибольшее применение приобрели методы с использованием перекиси водорода.
Методы определения с перекисью водорода являются мало чувствительными, но относительно простыми.
Для создания оптимальных условий при определении с перекисью водорода в растворе устанавливают определенное значение pH.
114
В этих методах используют либо щелочные растворы, содержащие щелочь, аммиак, карбонат натрия или аммония, а также их различные сочетания, либо кислые растворы.
Определение урана с перекисью водорода в щелочной среде. Колориметрический метод определения в щелочных растворах изучали А. Е. Клыгин, В. К. Федорова, Н. А. Никольская (1953 г.).
Спектрофотометрический вариант этого же метода разобран в книге Роддена [81. Максимум поглощения соединения урана с перекисью водорода находится при 340 ммк; однако при этой длине волны заметнее влияние самой перекиси, что требует строгого контроля ее концентрации. Поэтому целесообразнее проводить измерения при 400—440 ммк, хотя чувствительность метода при этом снижается. Оптическая плотность при этом не меняется по крайней мере в течение 12 час.
Исследование влияния катионов показало, что наибольшие помехи при определении в щелочном растворе встречаются со стороны хрома, марганца, ванадия, железа. Спектр поглощения перекисного соединения хрома перекрывает спектр соединения урана, поэтому хром должен быть удален любым удобным способом. Мешающее влияние марганца сложно и вызывается совокупностью причин: адсорбцией урана коллоидальной перекисью марганца и каталитическим разложением перекиси.
Влияние ванадия трудно устранить такими способами, как отделение его от урана при экстрагировании и при щелочном осаждении урана. Для его устранения применяют нагревание растворов, содержащих перекись натрия и большие количества щелочи, до кипения с последующим охлаждением перед фотометр и рован и ем [8, стр. 121J.
Железо, присутствующее в больших количествах, каталитически разлагает перекись. Отделение его от урана щелочным карбонатным осаждением неудобно, так как могут произойти потери урана за счет его адсорбции на гидроокиси железа. При небольших количествах железа адсорбция урана невелика, если гидроокись железа' хорошо скоагулирована.
Определение урана с перекисью водорода в щелочной среде [8, стр. 124] производится следующим образом.
Раствор, содержащий 1—25 мг U3O8, очищенный экстрагированием эфиром, выпаривают на водяной бане. Добавляют 2 мл серной кислоты, 3 мл азотной кислоты, 3 мл хлорной кислоты и кипятят до появления паров SO3. При охлаждении стакан обмывают водой и прибавляют раствор едкого натра (1 : 1) до нейтрализации кислоты. Прибавляют около 1 г перекиси натрия и избыток раствора едкого натра (1 : 1), доведя его содержание до 10?о от конечного объема, после чего разбавляют раствор до конечного объема. Раствор фильтруют и измеряют оптическую плотность при 425 ммк. Раствором кюветы сравнения является вода.
В присутствии ванадия после нейтрализации раствора приливают 20 мл раствора едкого натра (1 : 1) и доводят объем до 100 мл. Добавляют около 0,5 г перекиси натрия и фильтруют раствор через плотный фильтр в колбу емкостью 250 мл. Колбу накрывают часовым стеклом и нагревают раствор до начала кипения. Затем еСо охлаждают и измеряют пропускаемое^ при 425 ммк по сравнению с водой.
8* 115
Определение урана с перекисью водорода в карбонатно-щелочной среде. Колориметрический метод определения урана в карбонатном растворе изучался В. М. Звенигородской [79, 80]. Метод требовал отделения урана от всех примесей, препятствующих его определению. Кроме отделения железа с помощью двухкратной обработки содой, предпринимались дополнительные операции с целью отделения меди, молибдена и кальция (аммиачное осаждение), а также ванадия (фосфатное осаждение).
А. Е. Клыгиным, В. К- Федоровой и Н. А. Никольской (1953г.) было показано, что если проводить колориметрическое определение урана в карбонатно-щелочных растворах, то количество перекиси водорода, которое следует вводить в раствор для получения окрашенного соединения, можно резко понизить. Уменьшение концентрации перекиси водорода способствует тому, что молибден и ванадий не дают окрашенных соединений с перекисью, так как их перекисные соединения в щелочной среде сильно диссоциированы.
В предыдущей работе за 1952 г. теми же авторами было установлено строение перекисного уранового соединения в карбонатном растворе. Оно отвечало составу соединения, в котором один моль урана связан с одним молем перекиси водорода. Образующееся соединение имеет гидроперекисное строение. Константа равновесия реакции UO^ -rH2O2.~z±UO2—ООН+-(-Н+ при 20°С оказалась равной
К — [UQ2-QOH+][H+]
= 1,06-10-*.
Из уравнения видно, что для уменьшения концентрации перекиси водорода, которую следует вводить в колориметрируемый раствор, необходимо понизить концентрацию ионов водорода. Это достигалось установлением в растворе pH 12,0 (1А по углекислому натрию и 0,5А по едкому калию). Количество перекиси водорода, которое необходимо вводить для колориметрирования, состоит из эквивалентного определяемому количеству урана и избыточного, необходимого для смещения, равновесия на 99% в сторону образования окрашенного соединения.
Исходя из максимального количества урана, которое возможно определять в кювете с 1=50 см при использовании сине-фиолетового фильтра, была вычислена необходимая концентрация перекиси водорода, которая для гарантии была увеличена вчетверо. Это составило 5 мл 0,02 М раствора перекиси водорода или в 90 раз меньше, чем было рекомендовано ранее Родденом с сотрудниками [8].
Ванадий до концентраций 1 мг в 100 мл раствора не мешает определению при pH 12,0. Это вытекает и из исследования А. К. Бабко и А. И. Волковой [12], установивших, что в щелочной среде (pH 7 и выше) ванадий образует с Н2О2 лишь слабоокрашенный комплекс, оптическая плотность которого уменьшается с повышением pH раствора. Мешающее влияние ванадия в карбонатном растворе в
присутствии относительно оолыпих количеств перекиси, как это показано В. М. Звенигородской, больше чем в карбонатно-щелочных растворах, где значения pH выше.
Влияние анионов на определение урана, в карбонатно-щелочном растворе изучали Виберлей и Колмэн, чьи данные приведены Родденом [8]. Они показали, что ацетаты, хлориды, фториды, нитраты, фосфаты, перхлораты и сульфаты дают лишь небольшую положительную ошибку по сравнению с раствором, не содержащим этих ионов. Наибольшие помехи оказывают хром и марганец. Малые количества меди и никеля не мешают определению урана.
Определение урана в карбонатно-щелочной среде (в рудах и т. п.) по А. Е. Клыгину, В. К- Федоровой, Н. А. Никольской (1953 г.) проводится следующим образом.
Навеску выбирают с таким расчетом, чтобы в конечном объеме 100 мл содержалось от 1,0 до 4,0 мг урана.
Рис. И. Зависимость оптической плотности растворов от концентрации урана в миллиграммах на 100 мл.
Прокаленную при 500—800° в течение 20—30 мин. навеску переносят в стакан из жаростойкого стекла емкостью в 250—300 мл, добавляют 5 мл азотной кислоты, 10 мл соляной кислоты и 10 мл серной кислоты (1 : 1). При анализе шлаков, содержащих большое количество геля кремневой кислоты, дополнительно вводят 10 мл раствора фтористого аммония. После окончания выделения окислов азота нагревание усиливают и продолжают до полного удаления избытка серной кислоты. Разложенная масса должна получаться в виде совершенно сухого порошка, легко отстающего от дна стакана. Метод разложения проб разработан В. С.Быко-вой (1952 г.). Сухой остаток смачивают небольшим количеством воды (1—2 мл) и измельчают.
К полученной кашице добавляют 50 мл 5%-ного раствора соды и около 100 мг тоцкоизмельченного животного угля. Стакан накрывают часовым стеклом, нагревают и кипятят 5—10 мин. Горячий содовый раствор фильтруют в мерные колбы емкостью 100 мл.
Осадок промывают сначала в стакане 4 раза небольшими количествами горячего 5%-ного раствора соды, затем два раза на фильтре и отбрасывают. Объем фильтрата не должен превышать 85 мл. Раствор в колбе охлаждают до комнатной температуры, добавляют сначала 5 мл 0,02 М раствора перекиси водорода, а затем 5 мл 10 А едкого калия и доводят 5% -ной содой до метки. При этом происходит растворение алюминия, если он перешел в фильтрат, а раствор становится окрашенным. Окраска развивается почти сразу и устойчива по крайней мере 12 час, если в пробах отсутствовала медь.
Полученный раствор фильтруют через сухой фильтр; прозрачный испытуемый раствор наливают в кювету с 1—50 мм и измеряют оптическую плотность на фотоколориметре ФЭК.-М-52 с применением сине-фиолетового светофильтра.
117
В качестве раствора сравнения применяют раствор, содержащий в 100 мл 5 мл 0,02 .И перекиси водорода, 5 мл 10 Л1 едкого калия и 90 мл 5%-ной соды.
Результаты анализа рассчитывают по калибровочной кривой (рис. И).
.Метод применим для продуктов, содержащих от 0,1 до 5,0% урана.
Определению урана не мешают любые количества молибдена и вольфрама и до 1 мг ванадия в навеске. Присутствие больших количеств ванадия дает некоторую положительную ошибку. Определению не мешает присутствие хрома, меди и марганца в количествах до 10 мг в навеске. При анализе продуктов с большим содержанием меди она в значительных количествах переходит в содовый раствор и вызывает быстрое обесцвечивание окраски перекисного соединения урана вследствие каталитического разложения перекиси водорода.
Точность метода при содержании урана от 0,1 до 5,0% может быть охарактеризована средней квадратичной ошибкой, вычисленной из десяти определений, которая составляет ±1,6% (отн.).
Определение урана с помощью роданида калия {натрия) или аммония. Метод определения урана с роданидом детально исследован как в водной среде [305, 633, 670, 723, 977], так и в смесях, содержащих спирт [437, 651], ацетон [459, 499] или этиленгликольмонобутиловый эфир [784], дибутиловый эфир тетраэтиленгликоля [917].
Одним из преимуществ роданидного метода является то обстоятельство,что определение возможно в широких интервалах кислотности — от 0,1 до 2,0 N по соляной или азотной кислоте.
Метод значительно уступает в чувствительности другим методам, даже перекисному, что является его недостатком. Нижний предел определения 20 мкг/мл урана.
Определение урана можно производить при использовании экстракции комплекса урана с роданидом в органические растворители. Коэффициент распределения для урана между метилэтилке-тоном и раствором, содержащим 60% нитрата аммония и 3% роданида аммония, равен 2000. Вместо метилэтилкетона можно применять амилацетат, амиловый спирт и другие кислородсодержащие растворители [184]. Таким же образом был использован дибутиловый эфир тетраэтиленгликоля при определении урана в тории [917].
Клинч и Гай [426] экстрагировали комплекс урана с роданидом в 32,5%-ный раствор трибутилфосфата в четыреххлористом углероде из раствора, содержащего комплексон III.
К анализируемому раствору прибавляют 20 мл 10%-ного раствора комплексона III, I М НО или 1 М NH4OH до pH 3,5—3.9 и 10 мл 50%-ного раствора роданида аммония. Раствор перемешивают, помещают в делительную воронку, разбавляют до 50 мл водой и прибавляют 10 мл раствора экстрагента. Экстракт фотометрируют при 350 ммк, используя в качестве раствора сравнения раствор контрольного опыта. Ряд элементов, таких как Hg, Au, Pt, мешают определению. Присутствие Ag, Си, Ni, Cd, Pb, В i, Fe(III), Cr(III), La до 2000 мг не мешает определению. Алюминий не мешает до 100 мг, ^орий и ванадий (V) — до 80 мг. Метод может быть применен для определения урана в окиси тория и в рудах.
118
С. II. Синякова и Ю. П. Новиков [184] предложили спектрофотометрический метод определения урана с предварительной экстракцией нитрата уранила для отделения от примесей. В качестве экстрагента был использован метилэтилкетон. Цветная реакция осуществлялась добавлением к экстракту насыщенного раствора роданида аммония в ацетоне.
10 м.1 раствора, содержащего уран, насыщают азотнокислым аммонием. Нитрат уранила экстрагируют двумя порциями метилэтилкетона по 2 мл Отделив органический слой, прибавляют к нему 6 мл ацетона,насыщенного роданидом аммония, несколько миллиграммов твердой аскорбиновой кислоты (для восстановления следов трехвалентного железа), доводят объем метилэтилкетоном до 10 мл и измеряют оптическую плотность в кювете с / = 10.и.и при 375 и 400 ммк. Нулевой раствор состоит из 4 мл метилэтилкетона и 6 мл ацетона, насыщенного роданидом аммония. Закон Бера соблюдается в области концентрации 0,01—0,12 мг!мл урана.
С уменьшением концентрации роданида оптическая плотность растворов уменьшается, что обусловлено диссоциацией комплекса урана с роданидом. Необходимо применять насыщенные растворы роданида.
Оптическая плотность, измеренная при 400 ммк, не изменяется .во времени до 100 мин.; при 350 и 375 ммк оптическая плотность от 10 до 40 мин. постоянна, далее медленно растет; поэтому измерения выгодно производить через 30 мин. после прибавления роданида.
Небольшие количества примесей, которые экстрагируются вместе с ураном, могут влиять на оптическую плотность. Выяснено, что трехвалентное железо экстрагируется в заметных количествах, но прибавление аскорбиновой кислоты полностью устраняет его влияние даже при отношении уран : железо, равном 1:40. Ванадий, титан и висмут заметно извлекаются и завышают величину оптической плотности, молибден ее занижает.
Для устранения влияния молибдена применяют молочную кислоту: 1 мл 40%-ной молочной кислоты на 10 мл раствора полностью удерживает 5 мг молибдена в водном слое и не мешает экстракции и определению 0,5 мг урана.
Переход ванадия в экстракт может быть устранен связыванием ванадия солями циркония. Однако применение циркония и молочной кислоты одновременно невозможно, так как выпадает объемистый осадок.
Роданидный метод в зависимости от состава руды может быть использован в нескольких вариантах.
1-й вариант применим в отсутствие Mo, V, Ti и Bin при наличии Си, Fe, Со, а также элементов, не реагирующих с роданидом; он заключается в экстрагировании нитрата уранила с последующим образованием роданидного комплекса урана в метилэтилкетоне и ацетоне. Измерение оптической плотности производится после прибавления аскорбиновой кислоты. Время анализа около 30 мин. точность 2?о (отн.).
>!'.)
2-й вариант применим в присутствии молибдена и тех примесей, что и в первом варианте, и отличается лишь тем, что до экстракции добавляют 0,5 мл 40°о-ной молочной кислоты.
3-й вариант применим в присутствии ванадия и тех же примесей, кроме молибдена, и отличается от первого тем, что до экстракции в раствор добавляют нитрат циркония (10 мг циркония).
4-й вариант применим в присутствии многих элементов: V, Mo, Т i, Bi, Си, Со и основан на двойном экстрагировании урана метилэтилкетоном. После первой экстракции с молочной кислотой уран реэкстрагируют из слоя экстракта 2%-ным раствором карбоната аммония три раза, раствор выпаривают досуха, остаток прокаливают в муфеле, растворяют в азотной кислоте и, после насыщения нитратом аммония и добавления нитрата циркония, снова извлекают уран метилэтилкетоном. Добавив раствор реагента в ацетоне, измеряют оптическую плотность. Аскорбиновая кислота в этом варианте не нужна. Время анализа 3—4 часа, точность 2% (отн.).
О возможности использования роданидного уранового комплекса для автоматического контроля в производстве урана свидетельствует работа Тейлора [967], который применил для отделения урана от примесей хроматографический метод. Определение урана проводили по измерению оптической плотности в ультрафиолете.
Определение урана с помощью ферроцианида калия. Метод определения урана по реакции с ферроцианидом калия более чувствителен, чем перекисный и роданидный. Он широко описан в литературе [8, стр. 131; 265, 466, 508, 623, 931].
При взаимодействии урана с ферроцианидом могут образоваться коллоидные растворы, подчиняющиеся закону Бера только при определенных условиях [670].
С. И. Синякова и И. С. Классова [184] нашли, что при концентрации ферроцианида 0,02 М и азотной кислоты 0,05 М оптическая плотность, измеренная через 15 мин. после прибавления реактивов на фотоэлектроколориметре с зеленым фильтром, прямо пропорциональная концентрации урана при содержании его 10—100 мкг в 25 мл.
Наличие небольших количеств железа (до 10 % от количества урана) и ванадия (до 5%) не влияет на результаты определения, точность которых 3—5% (отн.). Метод, однако, недостаточно избирателен, и поэтому почти всегда необходимо тщательное отделение урана от прочих элементов.
Ферроцианид был применен Диздаром и Обренович [466] для определения урана в органических растворах, содержащих трибутилфосфат, при реэкстрагировании урана из органической фазы в водную — в раствор ферроцианида.
Определение урана с помощью азида натрия [500]. Применение азида натрия, дающего в водном растворе аналогично
ный по структуре с ионом роданида [: S : :С : :Х :]~ ион азида [: N : : X : : X :]_, не нашло широкого распространения из-за невысокой чувствительности реакции с ураном (10—20 мкг урана в 1 .и.?), а также из-за взрывоопасности солешсопутствующих элементов — Pb, Си, Ag. Молярный коэффициент поглощения для системы U (VI) — X? при 360 льпк составляет 5300, при 375 ммк —• 4450. Однако к достоинствам метода относится то, что реагент и продх кт реакции являются очень стабильными. Определение проводится в 1—1,5°о-ной азотной кислоте [500].
Определение урана по окраске его соединений с неокрашенными и с л а б о о к р а ш е н н ы м и органическими реагентами
Взаимодействие урана с неокрашенными или слабоокрашенными органическими реагентами основано на образовании хромофорных сочетаний уран-кислород, уран-сера и т. д. Чувствительность методов, в которых применены эти реагенты, как правило, одного порядка — около 5 мкг!мл урана; избирательность относительно хорошая, но все же предварительное отделение урана от примесей всегда является необходимой операцией.
Избирательность описанных методов значительно повышается при использовании комплексонов, связывающих мешающие элементы. По данным Пржибила [820], П. П. Палея и Н. И. Удальцовой и других авторов [412, 728, 898—900, 905], при определении урана с дибензоилметаном, диэтилдитиокарбаматом, морином, салицилаль-доксимом допустимо присутствие элементов, мешающих определению в отсутствие комплексонов.
Для количественного определения урана использованы следующие реагенты: аскорбиновая кислота [8, 184, 466], салицил альдоксим [120,325], салициловая и сульфосалициловая кислоты [8,11,120, 257, 360, 519, 973], хромотроповая кислота [8, 372, 766, 876, 926, 965],‘ R-соль и нитрозо-И-соль [831], тиогликолят аммония [466, 467, 956, 1019], 8-оксихинолин [8, 256, 394, 605, 652, 862] и его производные [862], дибензоилметан [86, 295, 299, 377, 521, 522, 610, 821, 1022], теноилтрифторацетон [648], резорцин [623], таннин [451], кверцетин [669], мореллин [827] и другие [224, 351,417, 636, 934, 960].
Использование некоторых реагентов рассматривается ниже.
Определение урана с помощью аскорбиновой кислоты. Аскорбиновая кислота исследована М. П. Белой [184]. Образующийся с ураном нестойкий комплекс имеет максимум окраски лишь в случае большого избытка реактива. Окраска имеет красновато-коричневый оттенок. При измерении оптической плотности раствора, содержащего 15 мкг/мл урана, на универсальном фотометре с фиолетовым светофильтром требуется 0,4 г аскорбиновой кислоты
121
на 50 мл. В присутствии углекислых солей окраска устойчива в течение суток. Чувствительность рекции 5 мкг урана в 1 мл при pH 4,4—4,5. Регулирование pH производится по индикатору гамма-динитрофенолу, окраска которого при pH 4,5 исчезает.
Для применения этой реакции в анализе руд требуется предварительная карбонатная обработка. Следы Fe, Al, Т i, Мп, оставшиеся в растворе, определению не мешают. Влияние меди устраняют добавлением тиосульфата натрия.
Но другому методу влияние меди уничтожают путем комплексообразования с пиридином, который добавляют к раствору урана, очищенному от примесей посредством эфирной экстракции [8, стр. 147].
Комплекс урана с аскорбиновой кислотой был использован [466] для определения содержания урана в трибутилфосфатных растворах в инертном разбавителе. При встряхивании этих растворов с водным раствором, содержащим аскорбиновую кислоту, уран переходил в водную фазу, где его определяли фотометрически.
Определение урана с помощью дибензоилметана. Дибензо-илметан относится к р-дикетонам R—СО—СН2—СО—R', ко-ОН
торые в энольной форме R—С=СН—СО—R' способны к образованию комплексов с некоторыми катионами. Чувствительность этих реагентов возрастает в ряду ацетилацетон (СН3—СО— —СН2—СО—СН3) — бензоилацетон (С6Н5—СО—СН2—СО—СН3)— ди бен зои л метан (Сб Н5—СО—СН 2—СО—С6 Н 5).
Если для ацетилацетона молярный коэффициент погашения равен 580 [627], то для дибензоилметана он составляет 17810. Растворимость в воде этих реагентов уменьшается при замене алкила ароматическим ядром, и поэтому дибензоилметан в отличие от его аналогов совершенно не растворим в воде.
Как реагент для определения субмиллиграммовых количеств урана дибензоилметан изучается с 1953 г. Джой, Уилл и Блек [1022] рассмотрели его общие свойства и применили эфирную экстракцию для отделения урана от мешающих определению элементов.
Яманэ [295] предложен метод определения урана в спиртовых растворах с некоторыми р-дикетонами, в том числе с бензоилацето-ном, дибензоилметаном, изоникотилдибензоилметаном, 2-фуроил-дибензоилметаном, о-, м-, и-метоксидибензоилметаном. Исидаме и Яманэ [86] изучали и другие замещенные р-дикетоны: о, м,- п-нитрозодибензоилметан, м- и о-аминобензоилметан; соединение последнего с ураном обладает высоким молярным коэффициентом погашения, для него в спиртовых растворах j олгл/с — 30 000.
Спектр поглощения раствора комплекса урана с дибензоилметаном приведен на рис. 12.
122
Так как дибензоилметан не растворим в воде, его применение удобно при определении урана в органических растворах, а также в сочетании с экстракцией. При этом определение возможно после
экстракции урана в виде комплекса с дибензоил-метаном, а также после экстракции в виде нитрата уранила. Так, Пржибил, Елинек [821] использовали экстракцию самого комплекса урана с дибензоилмета-ном в этилацетат. Экстракция хорошо проходит в узких пределах pH, уже при pH 6 извлечение урана очень затруднено, а при pH 8 происходит выделение урана в осадок. Для отделения урана от сопутствующих мешающих
Рис. 12. Спектр поглощения раствора комплекса урана с дибензоил мета ном (1) (концентрация урана в миллиграммах на миллилитр). Поглощение реагента (2) (концентрация —0,02% [1022]).
элементов применено связывание последних раствором комплексона III. Избыточное количество комплексона тормозит извлечение урана, поэтому избыток комплексона связывают кальцием.
К 150 мл слабокислого раствора, помещенного в делительную воронку, прибавляют достаточное количество 5%-ного комплексона, заранее определенное предварительными качественными капельными реакциями в зависимости от состава раствора. Затем к раствору прибавляют 1%-ный раствор нитрата кальция, и раствор осторожно нейтрализуют аммиаком до pH 7 по универсальной индикаторной бумажке. Экстрагируют 10 мл 0,5% -ного раствора дибензоилметана и через 10 мин. экстракцию повторяют еще с J5 мл дибензоилметана. Соединенные экстракты доводят до объема 25 мл и измеряют оптическую плотность на фотоколориметре с синим фильтром. Содержание урана определяют по калибровочной кривой, построенной в пределах 0,01—0,5 мг урана в 25 мл.
Определению не мешают даже в отсутствие комплексона Cd, Zn, Mg, Ba, Ca и Sr. Комплексон связывает Cu, Fe, Ni, Co, и они не мешают определению. Мешают: Mo, W, V, Ti, Сг (III), а также карбонаты, арсенаты, фосфаты, тартраты и цитраты.
Экстракция урана в виде нитрата уранила в органические растворители для отделения его от мешающих элементов с последующим определением урана с дибензоилметаном была использована в ряде работ [ 299, 1022]. Кроме эфира как экстрагент [299] был использован этилацетат [1022]. В последнем случае цветная реакция осуществлялась в среде этилацетат—спирт.
Способы определения урана дибензоилметаном, основанные на использовании трибутилфосфата, начали разрабатываться в 1956 г. В отчете за 1958 г. Франсуа [521 ] была применена система этил-
123
ацетат—трибути.тфссфат. По другому его методу 1522] аликвотная часть экстракта, состоящего из раствора трибутилфосфата в изооктане, добавлялась в раствор дибензоилметана в ацетоне и пиридине. В качестве экстрагента был использован также триоктилфосфин-оксид [610]; последующее определение урана с дибензоилметаном проводилось в пиридинно-спиртовых растворах.
Бланке [377] применил для определения урана пиридиновые растворы из-за их буферного действия. Однако его методика длительна и предусматривает карбонатную очистку урана.
П. Н. Палеем и Ю. П. Новиковым (1956 г.) было указано на применимость реэкстракции урана из экстракта, содержащего трибу -тилфосфат и керосин, непосредственно в буферный раствор, в котором находится реагент.
По более позднему их методу (1959 г.) определение оставшихся после реэкстракции из раствора трибутилфосфата в инертных разбавителях субмиллиграммовых количеств урана проводили в аце-тонно-водных растворах. Эти растворы содержали в качестве регулятора кислотности уротропин (гексаметилентетрамин). Для фотометрирования трибутилфосфатные растворы смешивались в определенном соотношении (1:10) с раствором, который содержал ацетон, воду, уротропин и дибензоилметан. В ряде случаев вода заменялась спиртом.
Определение урана с помощью диэтилдитиокарбамата натрия. Из соединений урана с NjN-замещенными дитиокарбаминовыми кислотами наибольшее применение нашел диэтилдитиокарбамат натрия.
Взаимодействие диэтилдитиокарбамата натрия с рядом элементов, а также с ураном изучалось многими авторами: [8,282, 378, 379, 405, 457, 989], Д. П. Малюга, (1947 г., 1948 г.), И. В. Шилин (1955 г.). В. М. Казаков [184] выделил два соединения, образуемые диэтилдитиокарбаматом натрия с ураном: UO2(S2NC5H10)2— желто-оранжевого цвета, существующее при pH 2,5 — 7,0, и UO2 (S2NC5H10)2-NaS2NC5H10 — красного цвета, образующееся при pH выше 7,0. Водные растворы двойного соединения при 320 ммк показывают оптическую плотность на 75% выше, чем простого.
Из-за большого количества мешающих элементов метод определения урана с этим реагентом применялся для чистых растворов или при отделении урана от небольшого числа элементов, таких как железо и ванадий (Д. П. Малюга, Н. В. Блюер, 1948 г.). Для отделения урана в сложных объектах использовали предварительную экстракцию эфиром, методика которой приводится в книге Роддена [8, стр. 154] и экстракцию в трибутилфосфат (Ю. П. Новиков, 1953 г.).
Способность соединения урана с диэтилдитиокарбаматом экстрагироваться в органические растворители использовалась для отделения урана от элементов, не осаждающихся сероводородом. В качестве экстрагентов могут быть использованы диэтиловый эфир, 124
амилацетат, этилацетат, изоамиловый спирт, но наиболее удобным оказался хлороформ [8. стр. 151].
Экстракция в хлороформ требует регулирования pH в пределах .6,6—6,8. Из хлороформа уран реэкстрагируют в водный слой карбонатом аммония, разрушающим соединения урана с диэтилдитиокарбаматом; при этом уран отделяется от железа, меди и других элементов, остающихся в хлороформе.
Н. Агапова и И. Кривохатская (1957 г.) определили уран после отделения его от мешающих примесей экстракцией в трибутилфосфат с последующей реэкстракцией ацетатом аммония.
В цилиндры для колориметрирования отбирают пробу с содержанием урана от 5 до 100 мкг. Если проба кислая, ее нейтрализуют по лакмусу и затем прибавляют 3—5 мл 25%-ного раствора ацетата аммония. Перемешивают и прибавляют 1 мл 5%-ного водного раствора диэтилдитиокарбамата и 1—3 мл хлороформа. Тщательно встряхивают. После расслаивания пробу сравнивают со стандартными растворами урана, приготовленными в таких же условиях.
Иногда пользуются тем, что соединение урана не растворимо в четыреххлористом углероде, тогда как аналогичные соединения некоторых элементов растворимы, и проводят экстрагирование четыреххлористым углеродом, удаляя мешающие элементы [8].
Применение комплексонов при определении урана диэтилдитиокарбаматом расширяет возможности использования этого реагента.
П. Н. Палеем и Н. И. Удальцовой в 1954 г. была проведена работа по определению малых количеств урана в металлическом тории при помощи диэтилдитиокарбамата натрия; было установлено, что в присутствии комплексона III некоторые катионы, например железо, не реагируют с диэтилдитиокарбаматом и не экстрагируются вместе с ураном.
Позже Н. И. Удальцовой (1955 г.) комплексон III был применен и для анализа руд. Метод заключался в превращении большинства сопутствующих урану элементов в растворимые комплексонаты, экстракции урана из нейтрального раствора хлороформом в виде диэтилдитиокарбамата и реэкстракции его в водный раствор карбоната аммония; определение в зависимости от количества урана может быть произведено колориметрическим или иным способом.
Применение комплексона III в данном методе имеет две положительные стороны. Во-первых, как следует из предыдущей работы и данных Боде [378, 379], такие элементы, как Fe (III), Zn, Cd, Ga, Ni, Co, Pb, Мп, V , In, Nb, Sn, не связываются диэтилдитиокарбаматом и не экстрагируются в хлороформ; во-вторых, в присутствии комплексона III при создании нейтральной среды, необходимой для образования соединения урана с реагентом, не происходит выпадения гидроокисей многих металлов. Не реагируют с комплексоном III и могут выпадать в осадок только Ti, Be, Sb, отчасти — Мп. Уран в присутствии комплексона III осаждается только при pH ’8,5. Таким образом, кислые растворы, получаемые после разложе
125
ния руд, могут быть в присутствии комплексона III нейтрализованы до pH 6,6—7,0 без выделения осадков.
К прозрачному кислому раствору объемом около 50 мл, содержащему от 0,1 до 2 мг урана, полученному после разложения руды и нейтрализованному аммиаком до появления слабой мути (pH не выше 2,5—3), прибавляют комплексон III в. твердом виде из расчета 5—8 г на 1 г навески. Для руд, богатых железом, количество комплексона увеличивает до 10 г на 1 г навески. Нейтрализуют аммиаком до pH 6,6—7,0 по лакмусу и переносят в делительную воронку, приливают 5 мл 2%-ного водного раствора дпэтилдитиокарбамата натрия, встряхивают и извлекают хлороформом порциями по 5—7 мл. Экстракцию ведут до тех пор, пока слой хлороформа не перестает окрашиваться. Промывкой хлороформной вытяжки раствором карбоната аммония переводят уран в водный раствор. Карбонат аммония разрушают прокаливанием выпаренного остатка в фарфоровой чашке.После подкисления устанавливают вводном растворе при помощи ацетатного буферарН 6,6—7,0; оптическую плотность раствора измеряют на фотоколориметре с синим фильтром.
Относительная ошибка для содержания урана в образце 0,1—0,5 мг 2—3%, для 0,5—2 мг около 1%.
Определение урана с помощью сульфосалициловой кислоты. Сульфосалициловой кислоте как реагенту для определения урана посвящен ряд работ [8, 11, 257]. Окрашенный в желтый цвет комплекс образуется при pH 4,4, что достигается использованием уротропина. Наибольшая интенсивность окраски устанавливается не сразу, а через 15 мин. после приготовления реактива. Окраска устойчива в течение часа. Определению мешают Fe, Си, Мо, Мп. Хроматы оказывают влияние собственной окраской, на которую необходимо вводить поправку. Оптическая плотность соединения трехвалентного железа с сульфосалициловой кислотой примерно в 30 раз больше оптической плотности соединения урана при одинаковых концентрациях. Чувствительность реакции сульфосалициловой кислоты с ураном невелика: определение урана возможно при концентрациях выше 100 мг!л.
Использование сульфосалициловой кислоты для определения урана возможно при условии отделения его от мешающих элементов.
Определение урана с помощью тиогликолята аммония. Уран с тиогликолятом аммония образует при pH 7—11 оранжевый комплекс, который достаточно прочен, и многие анионы не разрушают его. Использование тиогликолята аммония как реагента для колориметрического определения урана описано в ряде работ [448, 467, 956].
Тиогликолят был использован и как реагент для автоматического определения урана [1019] в сбросных растворах сложного состава, содержащих 2—100 мг/л U. После смешивания с раствором нитрата уран извлекали трибутилфосфатом, разбавленным керосином, в колонке роторного типа. Вымывание урана из этого раствора в другой такой же колонке производили раствором тиогликолята. аммония; этот реагент давал с ураном прочный комплекс, оптическая плотность которого при 420 ммк не изменялась при изменении концентраций нитратов, аммиака и тиогликолята.
126
Для автоматического введения поправки на светопоглощение реактива и его соединений со следами железа и других примесей фотометрирование производили сравнением поглощения исследуемого раствора с поглощением того же раствора, переведенного в кювету сравнения с помощью газ-лифта из углекислоты, которая разрушает комплекс уранила с тиогликолятом, не изменяя прочих окрасок.
Нерастворимость тиогликолятного уранового комплекса в органической фазе была использована [466, 467] для спектрофотометрического определения урана при реэкстракции последнего из раствора трибутилфосфата в инертном разбивателе в водный раствор, содержащий тиогликолят.
Определение урана с помощью 8-оксихинолина. 8-Оксихинолин образует с ураном в нейтральном или щелочном растворе окрашенное соединение состава: UO2(C9H6NO)2CgH6NOH [394], способное экстрагироваться в хлороформ и другие растворители [8].
Наряду с ураном многие элементы, такие как Fe, Al, Zr Th Bi и другие, образуют комплексы с оксихинолином, поэтому определение урана возможно только при использовании соответствующих приемов отделения или маскировки мешающих элементов. В обзорах по применению 8-оксихинолина в аналитической химии [16, 256] приведены многочисленные методы по разделению элементов с помощью этого реагента.
В книге Роддена[8, стр. 154—155] описан ряд методик для определения урана. В одной из них, методике Смайлса и Вильсона, уран экстрагируют из раствора, не содержашего мешающих примесей, при pH 7—9, хлороформом, фильтруют и измеряют светопоглощение при 425 ммк.
Хёк [605] рекомендует проводить экстракцию уранила из хлорнокислой среды 0,1 М раствором 8-оксихинолина в хлороформе при pH 4-—9. Уран полностью извлекается за 2—3 экстракции; измерение оптической плотности осуществляется при 425 ммк. Аналогичным образом для определения урана были использованы производные оксихинолина: 5,7-дибром-8-оксихинолин и 5,7-ди-’Хлор-8-оксихинолин [862].
Применение комплексона III для связывания мешающих определению элементов расширяет возможность использования оксихино-йина [652, 898].
При определении малых количеств урана (0,01—0,1%) в висмутовых сплавах ^652] навеску образца 0,2 г растворяют в 2 мл концентрированной азотной кислоты Л упаривают до появления влажных солей. Прибавляют 1 мл концентрированной Долиной кислоты, около 25 мл воды, 2,5 г комплексона III, аммиаком и уксусной Жислотой устанавливают pH 7,0, разбавляют водой до 50 мл, прибавляют 2 мл 6%-ного спиртового раствора оксихинолина и экстрагируют в 5 мл хлороформа. 'Затем снова добавляют такое же количество оксихинолина и экстрагируют. Разбавив объединенные экстракты до 20 мл, измеряют оптическую плотность при ‘400 ммк в кювете с /—40 мм.
? П. Н. Палем и Л. А. Цветковой в 1958 г. был предложен быстрый и простой метод определения урана с оксихинолином в содовых рас
127
творах, содержащих уран до 200 мг'л, а также до 0,03 % 51g, до 0,03% А1, до 0,03% Са, до 0,03% Мо, до 0,1% V, до 0,5% К, До 6,5% Na и других элементов.
Для этого в небольшую делительную воронку помещают 0,5 мл испытуемого раствора, прибавляют несколько капель 0,5 .V азотной кислоты и слегка встряхивают, чтобы удалить образовавшиеся пузырьки углекислого газа. Пипеткой вводят 0,3 мл 5°и -ного водного раствора комплексона 111,4 мл 5% -ного раствора азотнокислого натрия и 6 мл 0,1 .И раствора оксихинолина в хлороформе. Встряхивают 30 сек., опускают нижний слой через сухой фильтр в кювету с толщиной оптического слоя 10.и и и измеряют оптическую плотность на фотоэлектроколориметре с синим фильтром. Концентрацию урана находят по калибровочной кривой.
Метод колориметрического определения урана с 8-оксихиноли-ном в растворах, содержащих железо, хром и другие мешающие
примеси, описан Н. Агаповой и И. Кривохат-ской (1957 г.). Уран определяли после предварительной экстракции в трибутилфосфат в смеси с четыреххлористым углеродом и реэкстракции в ацетат аммония.
В цилиндры для колориметрирования отбирают исследуемую пробу так, чтобы содержание урана в навеске было от 4 до 80 мкг. Раствор нейтрализуют по лакмусу. Далее прибавляют 3-5 мл буферного раствора, содержащего ацетат аммония и уксусную кислоту,с pH 5—5,5 и, после перемешивания с 1 мл
Рис. 13. Спектры поглощения:
и— раствор,! морина с концентрацией 4 10 ~'М при pH 9 на -фоне воды; б—комплексного соединения урана с морином (200 Л1ке в 25 и.г) на фоне раствора реактива пои pH 9.Толщина оптического слоя в обоих случаях 10 мм.
2,5°о-ного спиртового раствора 8-оксихинолина, 1—3 мл хлороформа.
В случае, когда содержание урана в навеске больше 80 мкг, следует брать соответственно большее количество реагента и хлороформа. Содержимое цилиндра тщательно встряхивают. После расслаивания пробу сравнивают со стандартными растворами урана, приготовленными аналогичным способом.
Определение урана с помощью морина. Сходные по строению реагенты группы пентагидрооксифлавонов: морин [328, 329, 331, 959], кверцетин [669], гематеин [934] и их производные [960] дают с ураном достаточно чувствительные реакции. Наибольшее применение в аналитической практике получил морин. Спиртовый раствор морина имеет лимонно-желтый цвет, с ураном морин образует в щелочной среде при pH 8—10 коричневое окрашивание. Спектр поглощения растворов реагента и комплекса представлен на рис. 13.
Комплексное соединение образуется во времени и довольно устойчиво, однако при pH 10 и повышенных температурах разрушается. Методом изомолярных серий найдено, что соотношение уран :
морин в комплексном соединении равно 1:1, откуда можно предположить, что комплекс имеет следующее строение:
С—-С —О
но/Хч'/ЧЬн
(А. В. Давыдов, 1959 г.)
В интервале pH 4—7, как это показали Бек и Хантос [360], образуется комплекс с соотношением НОГ: морин, равным 1:2.
П. Н. Палей и А. В. Давыдов, изучая возможность применения методики определения урана с морином Алмаши и Нади [328], показали, что уран можно определять в присутствии трехкратных количеств никеля и кобальта, тысячекратных количеств нитрат- и сульфат-ионов, десятикратных количеств фтора и фосфатов. Изучалось также влияние ванадия. В присутствии пятивалентного ванадия получаются очень заниженные данные, так как ванадий окисляет морин. Добавлением 1 мл сернистокислого натрия восстанавливают ванадий, который в восстановленном состоянии связывается комплексоном III, чем исключается окисление реагента и, таким образом, присутствие десятикратных количеств ванадия не мешает определению урана.
0,5—4 мл раствора, содержащего 25—250 мкг урана, подкисляют в 25-милли-литровой мерной колбе 0,5 мл I N соляной кислоты, затем добавляют 2 мл 25%-ного хлористого аммония (для стабилизации pH не выше 10), 2 мл 1,5% -ного раствора комплексона III, 0,6 мл 0,33%-ного спиртового раствора морина. Раствор перемешивают и оставляют стоять 5 мин., затем добавляют 1—2 мл концентрированного аммиака, доводят водой до метки и через 10 мин. фотометрируют в кювете с толщиной оптического слоя 20 л/л/на фотоэлектроколориметре с синим светофильтром или на фотометре Пульфриха со светофильтром S47. Раствором сравнения служит раствор всех реактивов-.
При определении урана в присутствии ванадия, как рекомендуют П. Н. Палей и А. В. Давыдов, после подкисления раствора урана 1 N соляной кислотой добавляют 1 мл 1 %-ного раствора сернистокислого натрия и дальше ведут определение, как описано выше.
Определение урана по окраске его соединений с окрашенными органическими реагентами Наиболее чувствительными химическими методами определения урана являются методы его определения по реакциям с окрашенными органическими реагентами. Эти реакции основаны на образовании
Аналитическая химия урана
129
циклических соединений ионов уранила и четырехвалентного урана с молекулой реагента, в результате чего изменяется внутримолекулярное состояние красителя, что приводит к изменению, чаще всего к углублению цвета красителя ( см. гл. III).
Наибольшее применение получили следующие окрашенные органические реагенты: ализарин S и его аналоги [762, 993], арсеназо I [117, 118, 231, 529, 906], торон [114, 120, 520], 1-(2-пириди-лазо)-2-нафтол [419, 540, 933] и другие соединения (1120, 128, 734, 762, 915].
Наиболее чувствительным из этих реагентов является арсеназо I. Используя спектрофотометр или фотоэлектроколориметр, можно определить от 0,2 до 10 мкг/мл урана. Реакции такого типа не всегда связаны с хромофорным действием элементов, поэтому цветную реакцию дают все те элементы, которые образуют с реагентом циклические соединения*. В связи с этим избирательность действия окрашенных органических реагентов невелика.
Условия реакции (pH среды) зависят главным образом от большей или меньшей склонности данного элемента к гидролизу, т. е. к образованию связи элемент — кислород [120]. Окраска комплекса элемента с реагентом полностью развивается при значениях pH, близких к значениям pH гидролиза. Обычно интервал pH, в котором сохраняется окраска на одном уровне, составляет 3—4 единицы pH.
Из этого следует, что уран (IV), более легко гидролизующийся, чем уранил, можно определять в более кислых средах, где будут давать реакции с указанными реагентами лишь небольшое число легкогидролизующихся элементов — цирконий, гафний, торий.
Для повышения избирательности методов с окрашенными реагентами, особенно для экспрессных определений, целесообразно применение маскирующих агентов, например, комплексонов и других соединений. Ограничение этих методов заключается в том, что комплексы урана с применявшимися реагентами недостаточно прочны, вследствие чего комплексоны в некоторой степени маскируют и уран. Кроме того, из-за небольшой прочности комплексов урана с реагентами при наличии в растворах некоторых анионов (сульфатов, фторидов, фосфатов, а в некоторых случаях ацетатов, оксалатов, цитратов) могут возникать помехи, поэтому требуется очень полно отделять указанные анионы от урана.
В этой связи большой интерес представляют реагенты арсеназо II [118] и арсеназо III [216], предложенные В. И. Кузнецовым и С. Б. Саввиным. Реагенты образуют с ураном комплексы повышенной прочности, что дает возможность определять его в присут-
* В реакциях урана с некоторыми окрашенными реагентами, например с алюминоном, наблюдается и механизм, основанный на хромофорном действии. Поэтому строгое разграничение реакций по механизмам взаимодействия элемента с реагентом в ряде случаев затруднительно.
130
ствии значительных количеств мешающих анионов и применять эффективные маскирующие агенты.
Сравнительная прочность комплексов арсеназо I, арсеназо II и арсеназо III иллюстрируется, например, данными рис. 14, на ко-
Рис. 14. Влияние фосфатов на оптическую плотность растворов комплексов урана с арсеназо I (7), арсеназо II (2) и арсеназо III (3). Условия оптимальные [216].
123456783 pH
Рис. 15. Влияние /— UOg + — арсеназо Концентрация урана — 41О 'Л4, арсеназо
pH на оптическую плотность комплексов: Г, 2— UOj4 —арсеназо II; З — UOg^—арсеназо III. 4-10 -5Af. арсеназо I — 8-10~5 М, арсеназо II— 111 — 8-10-5 М. Длины волн 620 и 600 ммк, в кювете сравнения вода.
фом показано влияние фосфатов на определение урана с указании реагентами. Из рисунка видно, что большая прочность соот-|гствующих комплексов позволяет проводить определение урана фоне больших количеств фосфатов.
131
В 1959 г. С. Б. Саввиным было изучено влияние pH среды на оптическую плотность растворов комплексных соединений уранил-иона с реагентами арсеназо I, арсеназо II и арсеназо III (см. рис. 15).
Определение урана с помощью арсеназо I. Т. Н. Кукишевой (1958 г.) был предложен экспрессный метод определения урана с помощью арсеназо I. Сущность его заключается в том, что после разложения навески к раствору, содержащему нитраты или сульфаты, добавляют комплексон III, нитрат кальция и экстрагируют уран метилэтилкетоном. Аликвотную часть экстракта растворяют в воде, пользуясь тем, что метилэтилкетон растворим в воде до 37 г на 100 г воды, добавляют уротропиновый буфер, вводят раствор арсеназо и измеряют оптическую плотность.
Добавление комплексона увеличивает избирательность экстрагирования. Если перед экстракцией в 25 мл раствора добавить 250 мг комплексона III, то определению не мешают: 500-кратное количество А1, 400-кратное количество Си, 300-кратное количество Fe и 70-кратное количество V (V) и V (IV).
Не оказывают практического влияния на определение урана присутствующие в больших количествах щелочные металлы — Mg., Са, Sr, Ba, Zn, Cd, которые в условиях колориметрирования не дают окраски с арсеназо.
Экспериментально найдено, что наличие серной кислоты до 5 а/л, фосфорной до 6 г!л, соляной до 5 г!л в условиях экстрагирования не влияет на определение урана.
Небольшие количества фторидов (до 20 мг фтора) не оказывают существенного влияния на экстрагирование урана.
Навеску анализируемого материала, содержащую предположительно 0,5— 2 мг урана, разлагают способом, обеспечивающим полное растворение урана. Раствор упаривают досуха. К сухому остатку добавляют 2,0 мл концентрированной азотной кислоты, 20 мл насыщенного раствора нитрата кальция, нагревают до кипения, и раствор, не фильтруя, помещают в мерную колбу емкостью 50 мл, доводят до метки тем же раствором нитрата кальция. 10 мл этого раствора помещают в градуированную пробирку с притертой пробкой и с ценой деления 0,1. мл. В содержимое пробирки добавляют 5 мл 5%-ного раствора комплексона III, 10 мл метилэтилкетона, закрывают пробирку пробкой и хорошо встряхивают в течение 1 мин. После расслаивания жидкостей аликвотную часть верхнего, органического слоя переносят в мерную коЛбу емкостью 50 мл, растворяют в воде, добавляют 2,0 мл 0,1%-ного раствора арсеназо I, 2,0 мл 25%-ного раствора уротропина и доводят до метки водой.
Окрашенный раствор помещают в кювету с /=30 мм и измеряют его оптическую плотность (DP на ФЭК’М с красным светофильтром или на спектрофотометре при 640 ммк. Затем в кювету добавляют 6 капель пергидроля, раствор осторожно перемешивают стеклянной палочкой и, спустя около 1 мин, снова измеряют его оптическую плотность (£)2). По разности оптических плотностей (D,—D.,) опреде' ляют содержание урана, используя калибровочный график.
Метод может быть применен для определения урана в рудах» концентратах, органических и водных растворах, а также в сила* вах урана с другими металлами, содержащих уран от сотых долей до 5%.
Продолжительность определения в одном образце 1 час (вклю
чая время, затраченное на разложение пробы), точность определения составляет около ^3% (отн.).
Определение урана с помощью арсеназо II. Арсеназо II — би-фенил-4,4'-диарсоновая кислота-3,3'-бис [< -азо-2 >-1,8-диоксинафталин-3,6-дисульфокислота представляет собой как бы удвоенный
реагент арсеназо I.
На рис. 16 приведены спектры поглощения растворов арсеназо II и комплекса арсеназо II с ураном. Переход окраски при комплексообразовании с ураном достаточно контрастен — из розовой в синюю.
В. И. Кузнецовым и С. Б. Саввиным [128] разработан метод определения урана в двух вариантах. По первому варианту уран определяется в водных растворах в присутствии умеренных количеств катионов и анионов. Помехи от анионов исключаются благодаря высокой прочности комплекса урана с арсеназо II, а помехи от катионов, дающих цветную реакцию с реагентом, исключаются добавле
нием маскирующих комплексообразующих реагентов, связывающих эти катионы и лишь не-
Рис. 16. Спектр поглощения растворов арсеназо II (1) и комплекса UOg+ с арсеназо II (.2).
Концентрацпя реагента 3,2-10~; М. Уран в избытке по отношению к реагенту. Спектр снят на спектрофотометре СФЛ, кювета с /=10 мм. В кювете сравнения вода.
значительно маскирующих уран.
Второй вариант предусматривает определение урана в присутствии больших количеств про-
чих элементов, например в рудах, технологических растворах.
Для отделения урана от прочих элементов использован экстракционный прием, связанный с применением так называемых «легкоплавких экстрагентов» [130]. Легкоплавкие экстрагенты представляют собой смесь или сплав обычного жидкого экстрагента, применяемого при решении данной задачи, с твердым инертным органическим веществом, например парафином или церезином. Использование легкоплавких экстрагентов позволяет значительно сократить время, необходимое для экстракции.
10—200 мг тонкоизмельченного образца, предположительно содержащего от 20 до 200 мкг урана, разлагают кипячением с НС1 Н2О2. Раствор упаривают Почти досуха, приливают 2 мл HNO3 (1 : 3), и раствор вместе с осадком количественно переносят в пробирку емкостью 30—40 мл, смывая стаканчик 6—7 мл насыщен
133
ного раствора нитрата кальция (300 г Са(МО3)2-411,0 в 100 мл воды). Добавляют 0,5—1,0 г комплексона III и пробирку нагревают. Вводят 0,5—1,5 г парафина. 5,0 мл циклогексанона, пробирку нагревают до плавления парафина и, энергично встряхивая содержимое пробирки, проводят экстракцию. Охлаждают. При этом твердый экстрагент удерживается на стенках пробирки. Водный слой сливают, а экстрагент, предварительно расплавив, промывают насыщенным раствором Ca(NO8)2 и реэкстрагируют двумя порциями по 5—7 мл воды. Раствор сливают через смоченный водой фильтр в мерную колбу емкостью 25 мл Нейтрализуют до нейтральной реакции по любому внешнему индикатору добавлением 25% -ного раствора уротропина и вводят 10,0 мл маскирующего и буферного раствора (9 г Ca(NO3).2 X Х4Н2О;4,3 г комплексона Ш;20 г уротропина в 250 мл воды) с pH 4,4—4.6, 1,00 мл 0,1%-ного раствора арсеназо II и до метки воду. Оптическую плотность измеряют при 620 ммк или с красным светофильтром относительно воды. Содержание урана в образце находят по градуировочной кривой, построенной на растворах с известной концентрацией урана, которые проводят через все указанные выше операции.
Определение урана с помощью арсеназо III. Реагент арсеназо III получен в 1959 г. С. Б. Саввиным [216]. Большая прочность комплексов урана (IV) и урана (VI) с арсеназо III позволяет производить определение в сильнокислой среде, а также в присутствии анионов, которые обычно маскируют цветные реакции урана (сульфаты, фосфаты, оксалаты).
Арсеназо III не является специфическим реагентом только на уран (см. главу III), но большая избирательность может быть обеспечена в сильнокислой среде при определении урана в четырехвалентном состоянии. В указанной среде вместе с ураном (IV) с арсеназо III реагируют только цирконий и торий, причем влияние циркония может быть резко уменьшено, если определение проводить в присутствии щавелевой кислоты, маскирующей его.
В. Ф. Лукьяновым, С. Б. Саввиным, И. В. Никольской [1060] разработана методика определения урана с арсеназо III. Взаимодействуя с арсеназо III, U (IV) образует изумрудно-зеленое окрашивание; при избытке реагента наблюдается смешанная фиолетовая окраска различных оттенков.
Колориметрирование дает наилучшие результаты при 2—5-кратном молярном избытке реагента. Это соответствует фиолетовым и красно-фиолетовым оттенкам раствора. Окраска образуется практически мгновенно и устойчива по крайней мере в течение двух часов.
Влияние кислотности показано на рис. 17. Из рисунка видно, что окраска комплекса U (IV) с арсеназо III образуется в сильнокислой среде и постоянна в широком интервале кислотности.
Спектры поглощения арсеназо III и его комплекса с четырехвалентным ураном представлены на рис. 18. Резкий сдвиг кривых при комплексообразовании (углубление окраски), достаточно узкий пик кривых светопоглощения комплекса, вместе с весьма высокой прочностью комплекса благоприятно сказывается на увеличении чувствительности реакции. Так, молярный коэффициент погашения для U (IV), определенный по методу насыщения при постоянной концентрации реагента, е670Л(Л(к^100000.
134
При работе на фотоколориметре ФЭК-М-1 с красным светофильтром в кювете с /=20 мм и при содержании урана 0,04 мкг/мл оптическая плотность составляет 0,030. Если общий объем раствора 5 м.г, то определяемый минимум урана равен 0,2,мкг.
Влияние анионов невелико. При соотошении U (IV) : F“ =
= 1 : 25, U (IV) : РОГ=1 : 10000 и U (IV) ; SO1"=1 : 50000 ошибка определения урана еще не превышает 5% (отн.).
Большинство катионов также не препятствует определению U (IV), даже если присутствуют в 1000—10000-кратных весовых количествах. Допустимые соотношения для суммы редких земель составляют 1 : 60. Цирконий и торий влияют достаточно сильно.
Титан не дает заметной цветной
реакции с арсеназо III, но вызывает некоторое занижение результатов определения урана. При весовом соотношении U : Т i = 1 : 9 ошибка определения урана еще не превышает 5% (отн.).
Оптическая плотность
N НО- Длина Бсг.иы, ммк
Рис. 17. Влияние кислотности на развитие окраски комплекса U (IV)—арсеназо III.
Концентрация арсеназо III— 1,29-10~5 AI; концентрация U(1V)—1,8- 10~ь М. Измерения проводились на ФЭК-М-1 с красным светофильтром, в кювете с /=30 мм.
Рис. 18. Спектры поглощения 1,4-10'’^И раствора арсеназо III с pH 2,0 (7) и комплекса U (IV) — арсеназо III с pH 1,5 (2) относительно воды. Спектрофотометр СФ-4, кювета с /—10 мм[216]
При анализе образцов, содержащих титан, необходимо иметь в виду следующее. Металлический цинк восстанавливает Ti (IV) до Ti (III), который, являясь сильным восстановителем, может разрушать реагент. Поэтому после обработки раствора цинком титан следует окислить солянокислым гидроксиламином.
Цирконий дает с арсеназо III реакцию, сходную с реакцией U (IV). Все же влияние циркония может быть резко уменьшено, если определение проводить в присутствии щавелевой кислоты, которая сильно маскирует цирконий и в значительно меньшей степени
135
уран. Это позволяет определять уран в присутствии достаточно больших количеств циркония.
Торий вызывает наибольшие помехи. Если количества урана и тория одного порядка, то возможна следующая схема анализа: до восстановления урана определяют один торий и после восстановления — сумму Th-j-U (IV), и затем по разности находят содержание урана. Если количество тория велико, то предварительное его отделение становится неизбежным (например, с применением ионного обмена).
0,1—0,5 г руды, рудного кека и др. разлагают нагреванием с НС1 4-Н.2О2. Раствор охлаждают, разбавляют 4 Л' НС1 и через фильтр переносят в мерную колбу емкостью 100 мл. Доводят объем до метки 4N НС1.
Отбирают две равные аликвотные части (по 5—10 мл) и помещают в небольшие колбочки. К обеим частям добавляют на кончике шпателя по 1,0—2,0 мг аскорбиновой кислоты и далее к одной из них 5—6 гранул цинка. Через 7—10 мин. растворы сливают в 50-миллилитровые мерные колбочки, не перенося цинк, смывая колбочки 4N НС1. В каждую из проб вводят по 2 мл раствора арсеназо III и доводят объем до метки 4JV НС1. Измеряют оптическую плотность на фотоколориметре ФЭК-М-1 с красным светофильтром в кюветах с Z— 20 мм, используя в качестве раствора сравнения невосстановленную цинком аликвотную часть.
Если в анализируемой пробе присутствует цирконий, то перед добавлением реагента в оба раствора приливают по 2 мл 4% -ного раствора щавелевой кислоты. Такое же количество щавелевой кислоты добавляют во все растворы при построении калибровочной кривой по которой находят содержание урана.
Чувствительность реакции равна 0,04 мкг)мл U. Поддающееся определению минимальное содержание урана в анализируемом объекте составляет 0,002% .
Реакция арсеназо III с уранил-ионом нашла применение в экспрессном методе определения урана, разработанном А. А. Немод-руком и А. В. Давыдовым (1959 г.). Метод основан на отделении урана от примесей экстракцией в трибутилфосфат из растворов, содержащих комплексон III, с реэкстракцией урана в раствор, содержащий арсеназо III.
1—5 мл анализируемого раствора, содержащего 5—50 мкг урана, вводят в делительную воронку и разбавляют до общего объема 25 мл 60%-ным раствором азотнокислого аммония, содержащим 0,25% комплексона III. Растворами аммиака и азотной кислоты устанавливают pH 2,5—3,0 (по универсальной индикаторной бумаге). Уран экстрагируют двумя порциями по 10 мл 20%-ного раствора трибутилфосфата в четыреххлористом углероде, к отфильтрованному экстракту прибавляют^ мл 0,06% -ного раствора арсеназо III, содержащего ацетатный буфер с pH 3, и встряхивают в течение 1 мин. После разделения слоев измеряют оптическую плотность окрашенного водного раствора на фотоколориметре ФЭК-Н-54 со светофильтром № 8 в кювете с 1= 20 мм относительно реагента.
Такие примеси, как Ni, Со, Си, Cr (IV), Cr (III), Al, Са, Zn, Na, а также хлориды, сульфаты, ацетаты не мешают определению до соотношений концентраций элемент: уран, равных нескольким тысячам. Fe (III), Th, V, Мо, фториды, не оказывают заметного действия на точность определения урана до соотношений элемент: уран, равных нескольким сотням. Церий (IV) не мешает в количествах, равных количеству урана.
136
Для определения урана в растворах, содержащих менее 1 мкг урана на 1 мл, рекомендуется несколько измененная методика, предполагающая одновременное концентрирование урана.
В этом случае берут 25—750 мл раствора, содержащего 5—50 мкг урана, добавляют 1 мл 5%-ного раствора комплексона III и такое количество сухого азотнокислого аммония, чтобы его содержание в растворе составляло 50%. Затем экстрагируют последовательно 30 и 20 мл раствора трибутилфосфата, как описано выше. К отфильтрованному экстракту добавляют 15 мл 0,006?о-ного раствора арсеназо III и после реэкстрагирования измеряют оптическую плотность окрашенного водного слоя, как описано выше. Метод позволяет определять уран в растворах, содержащих от 0,02 до 2 мкг, мл урана.
Экстракционно-фотометрический метод определения урана с арсеназо III, основанный на экстракции комплекса урана с арсеназо в присутствии дифенилгуанидина в органический растворитель, предложен В. И. Кузнецовым и С. Б. Саввиным [1036].
Дифенилгуанидин, который образует катион дифенилгуаниди-ния (C6H5NH2CNH2) + , устраняет гидрофильность ионизированных сульфогрупп, делающих весь комплекс гидрофильным ионом. Введение дифенилгуанидина дает возможность экстрагировать комплекс урана с арсеназо в органический растворитель, например бутиловый или амиловый спирт, и таким образом сочетать определение микроколичеств урана с одновременным отделением от большинства элементов.
Несколько мллиграммов исследуемого образца с содержанием урана от 1 до 50 мкг помещают в пробирку и разлагают способом, соответствующим минеральному составу объекта и полностью переводящим уран в раствор. Для тщательно растертого образца это достигается кипячением с соляной кислотой в смеси с перекисью водорода или с царской водкой. Не отфильтровывая нерастворившуюся часть, погружают пробирку в водяную баню, где упаривают досуха и обрабатывают остаток 2,0 мг 0,05 N соляной кислоты. Вводят 2,5 мг 5%-ного раствора комплексона III, 1,00 мл 0,05%-ного водного раствора арсеназо III, 0,5 мл 20%-ного раствора хлорида дифенилгуанидиния и 5,00 мл бутилового спирта. Экстрагируют, энергично встряхивают, отбирают пипеткой часть окрашенного раствора и переносят кювету с /=10лои. Оптическую плотность измеряют относительно воды при 660 ммк на спектрофотометре или на фотоколориметре с красным светофильтром.
При описанных условиях существенные помехи оказывает торий, но его влияние может быть уменьшено введением в раствор по 5 мкг KF на каждые 5 мл раствора.
Анионы, такие как хлориды или нитраты, помех не создают. Фториды при концентрации до 1 мг1мл не мешают. Сульфаты или фосфаты осаждаются дифенилгуанидинием и после насыщения ими растворителя флотируются. Небольшие их концентрации помех не вызывают, большие приводят к некоторым завышениям результатов, однако после центрифугирования или фильтрования оптическая плотность приближается к нормальному значению. При применении бутилового спирта, лучше растворяющего соли дифенилгуанидиния, влияние анионов меньше.
137
В ряде случаев, когда концентрации примесей велики, для определения урана экстракционно-фотометрическим методом требуется предварительное его отделение.
Определение урана с помощью торона. Торон (бензол-2-арсо-новая кислота-< 1-азо-1 >-2-оксинафталин-3,6-дисульфокислота) предложен В. И. Кузнецовым. В солянокислой среде реагент дает чувствительную реакцию с ураном, находящимся в четырехвалентном состоянии. А. В. Николаев и А. А. Сорокина [184] для определения урана восстанавливали его до урана (IV) иодидами. Выделившийся в результате восстановления иод удаляли из раствора продуванием азотом. Удобным для колориметрирования интервалом является 1-Ю-7—2-10~6гЛил. Нижний предел определения — 10~8г урана в 1 мл. Не мешают: уран (VI) при отношении к U (IV) до 1000 : 1, ванадий (III) до 16 : 1, Сг (III) до 6 : 1, Fe (III) до 200 : 1, Th до 1000 : 1. Влияние Al, Fe (II), Zn, Се (HI), La (III), Be, Ca ничтожно. Мешают и должны быть полностью удалены: SO1-, NO3' , F", фосфаты, оксалаты и органические вещества.
Другими авторами [520] было предложено определение четырехвалентного урана с тороном в растворах, содержащих 80% ацетона. При этом было отмечено повышение чувствительности реакции, однако методика требует тщательного предварительного отделения урана от сопутствующих элементов, для чего была применена экстракция диэтилдитиокарбаматного комплекса урана в хлороформ из раствора, содержащего этилендиаминтетрауксусную кислоту.
Определение урана с помощью реагентов группы хлорфсс-фоназо. В отличие от реагентов типа арсеназо, характеризующихся атомной группировкой — AsO3H2, реагенты типа хлорфосфоназс» содержат группировку — РО3Н2.
Введение этой группировки в молекулу повышает прочность комплекса хлорфосфоназо с ураном, что можно было предсказать, исходя из того, что фосфорная кислота, обладающая большим сходством с мышьяковой, образует более прочные соединения с ураном по сравнению с мышьяковой кислотой. Другим свойством этих реагентов является их способность взаимодействия с рядом катионов в более кислых растворах, чем это имеет место в случае других реагентов, обладающих сходным строением, но вместо — РО3Н2, содержащих группировки — AsO3H2,—СООН или —ОН.
А. А. Немодрук, Ю. П. Новиков, А. М. Лукин и И. Д. Калинина [1039] предложили реагент хлорфосфоназо— 2-(4-хлор-2-фосфоно-бензолазо)-1,8-диоксинафталин-3,6-дисульфокислоту:
РО3Н2 НО он
С1-/ Х— N —N--/Ч, z -ЗН2О
Г
138 SO3Na
и разработали метод определения урана в присутствии больших количеств катионов и анионов.
Изучение свойств этого реагента показало, что оптическая плотность растворов комплекса урана с хлорфосфоназо в отличие от арсеназо не зависит от pH растворов в'довольно широких пределах, в то время как оптическая плотность соответствующих растворов комплексов урана (VI) с арсеназо почти для всех значений pH оказывается различной [530]. Вследствие этого при определении урана (VI) с помощью хлорфосфоназо небольшие колебания в кислотности не могут влиять на точность определения. Сдвиг области, в которой оптическая плотность постоянна, в сторону большей кислотности позволяет уменьшить или полностью избежать мешающего влияния ряда комплексообразующих веществ, имеющих характер слабых кислот (тартратов, цитратов, оксалатов).
Для определения урана в растворах сложного состава с помощью хлорфосфоназо применено предварительное отделение урана экстракцией в трибутилфосфат.
К 1—5 мл анализируемого раствора прибавляют 25 мл 60%-ного нитрата аммония, содержащего 0,25% комплексона III, затем добавляют разбавленный раствор аммиака (1 : 1) до pH 2,5—3,0 (до перехода фиолетовой окраски метанилового желтого в слабо розовато-желтую) и экстрагируют уран 15 мл 20%-ного раствора трибутилфосфата в четыреххлористом углероде; органический слой фильтруют через сухой фильтр в другую воронку. Экстрагирование повторяют еще раз 10 мл 20%-ного раствора трибутилфосфата в четыреххлористом углероде. Объединенные экстракты промывают 25 мл 60%-ного раствора нитрата аммония и после отделения водной фазы реэкстрагируют уран из органической 15 мл 0,005%-ного раствора хлорфосфоназо в буферной смеси с pH 5,2 (25 г ацетата натрия и 5 мл ледяной уксусной кислоты в 1 л раствора). После отделения органической фазы оптическую плотность окрашенного водного раствора измеряют на спектрофотометре при 605дьик в кювете с толщиной слоя Ю.илс Раствором сравнения служит 0,005%-ный раствор хлорфосфоназо.
Присутствие в исходном анализируемом растворе Zr, Th, Fe, Be и Al соответственно в 150, 450 , 500, 1500 и 5000-кратном количестве по отношению к урану (VI) определению не мешает. Ошибка определения около 1,5% (отн.).
Для фотометрического определения урана был также предложен [1040] реагент хлорфосфоназо III [2,7-бис-(4-хлор-2-фосфонбен-золазо)-1,8-диоксинафталин-3,6-дисульфокислота]:
РО3Н2 Н2О,Р
I
С1-7 N = N — / ' j 7'4 - N = N- / Cl
I
HO3S Z SO3H
Сопоставление молярного коэффициента погашения раствора комплекса урана с хлорфосфоназо III, равного 73 600, с молярными коэффициентами для растворов комплексов урана с другими реаген
139
тами (табл. 25) показывает, что хлоросфоназо III и арсеназо III являются самыми чувствительными реагентами из предложенных для определения урана.
Основным преимуществом применения хлорфосфоназо III для фотометрического определения урана (VI) по сравнению с другими реагентами является незначительное, а в ряде случаев и полное отсутствие мешающего влияния маскирующих комплексообразующих веществ, таких как оксалаты, фториды и фосфаты. При определении урана (VI) с помощью хлорфосфоназо III присутствие 50-кратных количеств щавелевой кислоты, 100-кратных количеств фторидов (считая на NaF) и 5000-кратных количеств фосфатов (считая на NaH2PO4) определению не мешают.
Наиболее сильное мешающее влияние при определении урана оказывают Th, Zr, Hf, Се (IV) и Ti (IV). Однако эти элементы не препятствуют определению, если применять маскирующие комплексообразующие вещества, например NH4F для маскировки Zr и Hf, Н2О2 — для Ti (IV) или предварительно восстанавливать их до более низких валентных состояний, например Се (IV) до Се (III), Fe (III) до Fe (II).
В качестве примера использования приема маскировки приводится методика определения урана в его сплавах с цирконием 11040].
100 мг анализируемого сплава растворяют в платиновой чашке в 10 мл разбавленной соляной кислоты (1 :1) с добавлением 0,5 г NH4F или эквивалентного количества NaF (HF) при нагревании на водяной бане. Продолжительность растворения даже при грубом измельчении навески не превышает 5 мин. По окончании растворения прибавляют 20 мл 0,1 М раствора Na.,SiO3 (необходимого для связывания HF с целью предотвращения порчи стеклянной посуды), переносят в мерную колбу емкостью 250 мл, чашку споласкивают 10—15 мл разбавленной соляной кислоты и присоединяют к основному раствору. После разбавления водой до метки 1 мл полученного раствора вносят в мерную колбу емкостью 100 мл, прибавляют 5,0 мл монохлорацетатного буфера (10 г ClCH2COONa и 20 г С1СН2СООН в 1 л раствора), 2,5 мл 0,1 М раствора Na2SiO3, 12,5 мл 0,1 М раствора NH4F (или NaF), 2,00 мл 0,001 М раствора хлорфосфоназо III, разбавляют водой до метки и с помощью спектрофотометра измеряют оптическую плотность полученного раствора при 670 ммк в кювете с толщиной слоя 10 мм. В качестве раствора сравнения применяют раствор, содержащий те же количества хлорацетатного буфера, Na2SiO3, NH4F и реагента, но не содержащий урана и циркония.
Ошибка определения урана в циркониевых сплавах не превышает 2% (отн.). Продолжительность одного определения составляет около 15 мин.
Определение урана косвенным путем
К этой группе методов относится определение урана, основанное на окислении четырехвалентного урана до шестивалентного окисным железом с последующим определением двухвалентного железа, образующегося в эквивалентных количествах по отношению
140
Таблица 25
Молярные коэффициенты погашения для соединений урана
Соединение Длина волны, ммк Молярный коэффициент погашения Литература
UOf — HNO3 414,2 8,96 [627]
UO^ — HNO, 420 8,0* (Ш HNO,) [368]
UO^+ — HNO3 420 11,8* (6Л4 HNO3) [368]
uof — H2SO4 420 14,3 (ЗЛ1 H,SO4) [350]
UO^ - — н so4 420 13,5 (0,5Л1 H,S04) [350]
UO2/ — H_SO4 420 13,0 (9Л1 H2SO4) [350]
UO^—HC1O4 417 7* (2—65%-ная НС1О4) [916]
UOg —трибутилфосфат 416 9,74 [627]
U Og+—трибутилфосфат 250 2400 [790]
UOg+—ацетоуксусный 400 180 [231]
эфир
UOg+ — H2O2 400 269±2 [466]
иоГ - H2O2 Сине-фиолетовый фильтр ФЭК-М-52 446* А. Е. Клыгин, В. К- Федорова. Н. А. Никольская, 1953 г.
UO2~ — ацетил ацетон 400 580 [627]
UO2+ — аскорбиновая 400 1676^=34 [466]
кислота
UO2+ —тиогликолят 380 2019±8 [466]
UO|+ —ферроцианид 480 3806±75 [466]
UO| + —ферроцианид 525 3090* Лейфертер и др. [8, стр. 135]
UO® —ди этилдитиокарбамат 350 4560* Виберлей и др. [8, стр. 151]
380 3990* То же
UO| + — роданид 365 3031 ±26 [466]
UO^ + — роданид 350 5310 (в 32,5%-ном ТБФ в СС14) [652]
141
Таблица 25 (окончание;
Соединение Длина волны, ммк Молярный коэффициент погашения Лптератч ра
L’O* ~ — морин 475 8950* П. Н. Палей, А. В. Давыдов, 1959 г.
LTO| — кверцетин 480 12800* [669]
U(IV) — торон Фильтр Ильфорд № 605 с максимумом пропускания 535 15200* [520]
UO* —дибензоилметан 400 17810* (в С,Н5ОН) П. Н. Палей, Ю. П. Новиков, 1959 г.
СО* —бензойная кислота родамин В 555 18000* (в С6Н6) [915]
ПО|+ — 1-(2-пиридилазо)-- 2- нафтол 570 23000 [419]
С'О* +— хлорфосфоназо I 605 23100 [1039]
L’O* +—арсеназо I 595 22900 [530]
UO* ’ —о-амипобензоил-метан 410 30000 [86]
UO*+ —арсеназо III 670 около 73000 [1040]
UO* * —хлорфосфоназо III 670 73600 D [1040]
* ДЗолярныи коэффициент погашения s, =гт~7 рассчитан поданным, приведенным А [£]•»
в соответствующей работе. При этом принято, что концентрация урана в молях Суран соответствует концентрации [с] окрашенного комплекса, т. е. весь уран связан в комплекс. Строго говоря, это справедливо только для малодиссоциированных комплексов, для которых [Сурап] в выражении оуран = [cj + [суран] достаточно мало.
к урану. Для определения железа используется чувствительная реакция с а.а'-дипиридилом [327], а также с диметил глиоксимом [708].
А. И. Пономарев рекомендует применять косвенное определение урана с а.а'-дипиридилом для образцов, в которых содержание урана мало и не поддается определению по объемному фосфатному методу.
После осаждения урана (IV) в виде фосфата, по П. А. Волкову, осадок фосфатов растворяют в минимальном количестве 15%-ной серной кислоты, прибавляют 142
1 ль: раствора трехвалентного железа (0,1 мг железа в 1 .мл) и нейтрализуют карбонатом натрия до слабокислой реакции. Приоавив 0,5 мл 0,5%-ного раствора а,а'-дипиридила, перемешивают, вливают 5 мл 0,5%-ного раствора буры, разбавляют до определенного объема и через полчаса измеряют окраску соединения двухвалентного железа с дипиридилом на любом фотометре [184].
По другой схеме косвенного определения уран (IV) осаждают в виде йодата U (JO3)4, который затем растворяют и восстанавливают иодистым калием до свободного иода; иод, выделившийся в результате восстановления, извлекается хлороформом или четырех -хлористым углеродом; образовавшиеся окрашенные растворы колориметрируют [192].
К кислому раствору (4—5%-ному по серной кислоте), содержащему от 0,1 до 0,8 мг урана (IV) (получен электролизом сульфата уранила на ртутном катоде), добавляют 10%-ный раствор йодата калия в 10%-ной серной кислоте в отношении (1 :2) к объему испытуемого раствора и затем такое же количество дистиллированной воды (концентрация йодата калия в конечном объеме раствора, равном не более 15—20 мл, составляет 2,5%). Полученный осадок центрифугируют, промывают разбавленным раствором йодата калия (0,4% KJO9 в 1%-ной серной кислоте) и растворяют в серной кислоте (1 : 5); прибавляют 1 мл 1%-ного раствора иодида калия и 15 мл хлороформа. После встряхивания и отстаивания слоев оптическую плотность хлороформного раствора измеряют на фотометре Пульфриха при 496 ммк по отношению к воде.
Отрицательные значения ошибок, не превышающие, однако, 2,5% (отн.), связаны с растворимостью осадка йодата уранила. Несмотря на хорошую точность, а в случае определения урана по реакции железа с а, а'-дипиридилом большую чувствительность, косвенные методы не имеют широкого распространения главным образом потому, что прямые методы более просты в выполнении.
2. Люминесцентный метод
Т. С. Добролюбская
Оптические свойства лантанидных и актинидных элементов очень сходны. В случае лантанидных элементов они определяются переходами электронов в оболочке 4/, в случае актинидов 5/.
Надо было ожидать, что и люминесцентные свойства этих групп элементов также будут похожими. В настоящее время описана люминесценция U3+ в монокристаллах CaF2, SrF2 и BaF2[1033], Pu3+ в смешанных кристаллах трихлоридов плутония и лантана [1045] и Ams+ в смешанных кристаллах трихлоридов америция и лантана [10471.
Люминесценция актинидных элементов самовозбуждается (за счет действия а-,[3- и у-излучения), и в особенности сильно проявляется при воздействии ультрафиолетового излучения.
- У соединений, в которых уран 4- или 5-валентен, люминесценции не обнаружено. Из соединений урана (VI) люминесцируют гидратированная трехокись урана UO3-nH2O [996], при низких тем
143
пературах гексафторид урана — UFe 1195], уранат натрия — Na2U2O-[67L В особенности интенсивно люминесцируют соединения, в которых уран присутствует в виде иона UO*+.
Яркая люминесценция иона UCF+ при возбуждении ультрафиолетовым излучением была использована в аналитических целях для качественного и количественного определения урана. Качественные определения основаны на наблюдении характерной желто-зеленой флуоресценции соединений уранила [155]. В основу количественных определений положено то, что интенсивность свечения уранил-иона, находящегося в твердом [783] или жидком [1034] растворе, в широком интервале линейно зависит от концентрации элемента.
Люминесцентный метод определения урана является специфичным и самым чувствительным среди других известных в настоящее время и применяемых на практике методов. Чувствительность метода порядка 10-10aU.
Обычно этим методом пользуются при анализах проб, содержащих уран ^0,001%. Однако при наличии чувствительных фотоэлектрических фотометров для измерения интенсивности свечения границы применимости этого метода могут быть расширены до 1 % U3O8 и более [1029]. Люминесцентный метод нашел применение при определениях урана в минералах, рудах, породах, рудничных, буровых, речных и морских водах, в животных и растительных организмах, в контроле технологического процесса получения урана и при поисках урановых месторождений.
В настоящем разделе рассмотрены свойства свечения уранил-иона в связи с количественным определением урана, а также используемые в анализе приемы и аппаратура.
Свойства свечения уранил-иона в растворах
Необходимо отметить, что свойства свечения ураниловых растворов в сильной степени напоминают свечение кристаллических ураниловых солей [24, 27а, 25. 141, 142, 195, 223, 233, 782, 1005. 1041, 1046]. Это указывает на то, что ион UO^+ играет в процессах люминесценции очень важную роль.
Интенсивность свечения иона UCH+ в растворах, так же как и другие люминесцентные характеристики (спектры свечения, выход и т. д.) зависят от природы и концентрации соли и свойств растворителя [24, 142, 195, 1041].
В табл. 26 приведены результаты исследований зависимости интенсивности свечения от добавки ряда веществ к водному раствору азотнокислого уранила (концентрация 100 мкг U/мл). Измерения проводились при помощи фотометра Пульфриха при возбуждении ультрафиолетовым излучением Л 253,7 ммк (при освещении поверхности раствора сверху).
___________144
Наибольшей интенсивностью свечения обладают фосфорнокислые растворы уранила, затем растворы, содержащие однозамещенный фосфат, сульфат и фторид-ионы. Самой чувствительной люминесцентной реакцией на ион UCF+ в водных растворах в настоящее время следует считать реакцию с триметафосфатом натрия (А. В. Давыдов, Т. С. Добролюбская, А. А. Немодрук, 1959 г.).
Экспериментально было установлено, что уранил-ион с триме-тафосфат-ионом образует комплекс состава [UO2(PO3)31“, который и обусловливает люминесценцию урана (VI) в растворах фосфатов (результаты работ вышеуказанных авторов). В случае присутствия уранил-иона в других средах люминесценция, по-видимому, также обусловлена образованием комплексов определенного состава.
Интенсивность свечения водных растворов, содержащих UO*+, меньше интенсивности свечения кристаллических ураниловых солей. Она возрастает с понижением температуры, поэтому при количественных определениях урана по свечению как в жидких, так и в твердых растворах следует учитывать влияние последней; стандартный и анализируемые растворы должны иметь одинаковую температуру.
Таблица 26
Зависимость интенсивности свечения уранил-иона от состава раствора
Состав добавки Количество добавки в пересчете на анион, % Интенсивность свечения в относительных единицах
Na,CO3 5 Свечение очень слабое
NaHCO3 5 То же
NaCl 5 » »
NaJ 5 Свечения нет (раствор
окрашен в желтый
цвет)
NaF Насыщенный 22 |
раствор
NaHF8 То же 20 1 1 Яркое еве-
Na2SO4 5 30 чение
NaHSO4-H2O 5 33
NaHPO4-2H2O 5 36
Na,HPO4-7H2O 5 6 1
Na3PO4-12H2O 5 2 1 Свечение
I ослаблено
NaC2H3O2-2H,O 5 3 /
Na ,C2O4 5 Свечения иет
NasCeH5O7-2H2O 5 То же
Na2C4H4Oe-2H2O 5 > »
HSPO4 5 100 (яркое свечение)
Na3AsO3 5 Свечения нет
Ю Аналитическая химия урана
145
В ураниловых растворах рано наступает концентрационное тушение (см. стр. 148 и 152). Спектры свечения ураниловых растворов являются типичными молекулярными полосатыми спектрами. В спектрах поглощения также наблюдается структура, напоминающая структуру спектров поглощения кристаллических ураниловых солей. Послесвечение ураниловых солей, так же как и растворов, кратковременно: порядка ^10 3 сек.
Отметим, что свечение уранил-иона в водных растворах очень чувствительно к добавкам различных органических веществ, восстановителей и окислителей, включая перекись водорода. Вопрос о действии элементов-гасителей на люминесценцию урана (VI) в растворах рассматривается ниже. Вопросы же, связанные с миграцией энергии в ураниловых растворах, освещены в работах С. И. Вавилова [23а], М. Д. Галанина [1032].
Общим свойством свечения чистых ураниловых солей и их растворов является то, что интенсивность свечения пропорциональна интенсивности возбуждающего света при X-const.
Несмотря на то, что люминесценция ураниловых солей и растворов широко исследуется в течение столетия, в настоящее время механизм ее остается невыясненным. Решение этого вопроса представляло бы, безусловно, большой научный интерес.
Количественные определения урана по свечению водных растворов
В 1947 г. В. Л. Левшин с сотрудниками разработали метод количественного определения урана по свечению в сернокислых растворах. Возбуждение люминесценции проводилось длинноволновым ультрафиолетовым излучением.
Силл и Петерсон [912] повысили чувствительность этого метода, применив для возбуждения коротковолновое ультрафиолетовое излучение. Для возбуждения свечения ими применялась 18-ваттная лампа, 90% излучения которой приходится на монохроматическую линию 253,7 ммк. Наблюдения проводились визуально без применения каких-либо приборов.
Интенсивность свечения урана в растворах серной кислоты на порядок выше, чем в азотнокислых [912]. Несколько десятых миллиграмма урана можно обнаружить по свечению в сернокислом растворе (в объеме 200 мл.) Силл и Петерсон [912] отмечают, что метод определения урана по свечению в сернокислых растворах дает возможность не проводить общего химического анализа многих проб.
С увеличением концентрации серной кислоты в растворе интенсивность свечения урана (VI) растет. Определение же малых количеств урана по свечению в концентрированных растворах серной кислоты неудобно, так как последняя, обычно применяемая в анализе без специальной очистки, при освещении ультрафиолетовым 146
Рис. 19. Зависимость интенсивности свечения фосфорнокислого раствора уранила от концентрации урана (в 5%-ном растворе Н3РО4).
интенсивности свечения, кото-
светом имеет часто голубую флуоресценцию, которую объясняют присутствием органических веществ. В этих случаях необходим прогрев серной кислоты при температуре, близкой к температуре кипения, с тем чтобы разрушить органические вещества.
Силл и Петерсон показали, что кроме серной кислоты в данном случае можно использовать фосфорную и фтористоводородную кислоты, причем чувствительность определения урана даже повышается; так, можно определять 0,01 мг урана в 200 мл раствора, а в некоторых специальных случаях даже 0,001 мг U в том же объеме.
Применение фтористоводородной кислоты в химическом анализе неудобно из-за ее высокой реакционной способности. Удобнее оказалось проводить аналитические определения в растворах фосфорной кислоты: она не люминесцирует и очень слабо поглощает ультрафиолетовые излучения.
На рис. 19 представлена зависимость интенсивности свечения от концентрации урана (VI) в 5 %-ном растворе фосфорной кислоты. По оси абсцисс отложена концентрация урана в логарифмической шкале, по оси ординат — значение логарифма
рая измерялась в относительных единицах. Измерения выполнены при помощи фотометра Пульфриха (при возбуждении X 253,7 ммк, освещение сверху, рис. 20)11034]. Линейная зависимость между интенсивностью свечения и концентрацией урана в растворе сохраняется от очень малых значений до ^1.10“4 г U/мл; .при дальнейшем увеличении содержания урана кривая проходит через максимум, который соответствует--2,5• 10“4г U/мл. Снижение интенсивности свечения раствора с увеличением его концентрации называют концентрационным тушением.
Люминесценция ураниловых растворов (цвет свечения зеленый) возбуждается как коротковолновым, так и длинноволновым ультрафиолетовым излучением, однако в случае разбавленных растворов (менее 10 мкг U/мл) возбуждение лучше проводить коротковолновым ультрафиолетовым излучением А 253,7 ммк. Источником возбуждения может служить бактерицидная лампа БУВ-15 с фильтром УФС-1 ; для этой цели также удобно использовать ультрахимископ Брумберга. При возбуждении люминесценции от этого источника Можно обнаружить десятые доли микрограмма урана в 1 мл.
10*
147
Необходимо специально оговориться, что в случае количественных определений урана по свечению в водных растворах применяемые сосуды (кюветы) не должны люминесцировать при действии на них ультрафиолетового излучения.
Интенсивность свечения ураниловых растворов может быть измерена при помощи фотоэлектрического люминесцентного фотометра ЭФ-3. Для выделения коротковолнового ультрафиолетового излучения необходимо поставить фильтр УФС-1, а измерения интенсивности свечения растворов проводить в кварцевой пробирке при строго фиксированном ее положении.
Рис. 20. Схема установки для измерения интенсивности свечения растворов
1— фотометр Пульфриха; 2 — кюветодержатель; 3— источник возбуждения.
При добавлении к азотнокислому раствору уранила фосфорной кислоты наблюдается рост поглощения в коротковолновой области ультрафиолетовой части спектра, что видно из рис. 21, где кривая 1 соответствует поглощению раствора, в котором концентрация урана 1 мкг в 1 мл 5?4-ной азотной кислоты. Кривая 2 соответствует поглощению того же раствора, но после добавления к нему 5% фосфорной кислоты. Кривые измерены при помощи спектрофотометра СФ-4 только в коротковолновой области ультрафиолетовой части спектра.
Хотя добавка фосфорной кислоты к азотнокислому раствору уранила и приводит к росту поглощения, однако интенсивность люминесценции растет более значительно [1034].
Интенсивность свечения урана в фосфорнокислом растворе в определенном интервале зависит от концентрации фосфорной
148
кислоты, это можно видеть на рис. 22 (концентрация урана в мкг мл). Добавление фосфорной кислоты свыше 5° 0 не усиливает эффекта [912].
При введении большинства посторонних ионов в фосфорнокислые растворы уранила интенсивность свечения обычно падает. Силл и Петерсон подразделяли влияние примесей на свечение
урана на три группы; к первой группе они относят вещества, которые имеют малый тушащий эффект или не действуют вовсе: Li Na-. К+, NH^, Be2-, Alg2-, Са2\ Sr^, Ва2 + , Znz+, Cd2-, Hg2+, Al3\Ga3~, In3-, Bi3 + ,Pt4+, Th4+, Sn1 + , Zr4-, Ti4 + , Nb(V), Ta(V),
Рис. 21. Спектры поглощения уранил-иона:
1~ в 5%-ном растворе HNO3 2— в растворе 5%-ная HNO3+5%-ная Н3РО4.
Рис. 22. Зависимость интенсивности свечения урана (VI) от концентрации фосфорной кислоты.
AsOf, MoOf, WO! , NO3~, СЮ4 , SeO: , цитраты, ацетаты, CN-, SOf, S2or, РОГ, P2Of, РО3", ВОГ, В4ОГ, со2.
Вторая группа — очень сильные тушители: Ag+, Т1 + , Т13 + , Pbz + , Fez + , VOZ+, Hgf, Snz + , Ce3 + , редкие земли (?), станнил, J~, Вг~, СГ, SCN", SeOf, SOf, 52ОГ, Sz~, AsOr.NOf, HCOO’, H2PO2", Fe(CN)r.
Третья группа — вещества, оказывающие среднее действие: Fe3 + ,_Cuz+, Mnz+, Сг3_+, Au3+, Rh3-, Co2+, Ni2+, Ce4+, Сг2ОГ , MnO4 , Fe(CN)r, VO3 , HF, H2O2, тартраты.
Тушащее действие примесей в водных растворах сказывается при меньших содержаниях, чем в случае определения урана по сведению перлов или плавов, приготовленных на основе фтористого Натрия (см. ниже). При аналитических определениях требуется отделить уран от примесей. Для этой цели обычно применяют экстракционные методы. Результаты исследований [1034] показывают, Что если предварительно связать примеси в комплекс с комплексо
149
ном Ши провести экстракцию трибутилфосфатом (растворитель — четыреххлористый углерод, высаливатель—азотнокислый кальций), промыть раствор и реэкстрагировать уран в водную фазу, то можно получить хороший результат даже при больших соотношениях примеси (Fe3+, Сг3+, Ni2+ и др.) к урану.
Предложен метод количественного определения урана (при содержаниях 10—100 мкг U/мл) по люминесценции в 0,01 М. растворах ацетата натрия при pH 6—8 и возбуждении А 365 ммк (Н. Агапова, 3. Болотова, В. Мудрин, 1960 г,).
Количественные определения урана по свечению твердых растворов
В 1926 г. Никольс и Слеттери [783] обнаружили, что фтористый натрий, активированный ураном, при освещении ультрафиолетовым излучением очень ярко люминесцирует (желто-зеленое свечение), причем интенсивнее, чем многие ураниловые соли. Спектр
Рис. 23. Спектры свечения перлов NaF—U с различной концентрацией урана (температура среды -4-20° С).
1— 1-10-- г U/г NaF; 2— 5 10~ ’ г U,e NaF; 3— 1 • 10-1 г U/г NaF; 4— 1 -10~3 ги г NaF.
свечения такого твердого раствора подобен спектру свечения чистых ураниловых солей [921]. Спектр свечения начинается около 520 ммк и простирается в красную часть до 630 ммк, состоит он из нескольких полос. При температуре жидкого воздуха спектральные полосы расщепляются на ряд узких полос (так же как и в случае свечения ураниловых солей) [7831. На рис. 23 приведены спектры свечения плавов фтористого натрия, активированных различными
15< >
количествами урана [67]. В качестве активирующей соли был взят азотнокислый уранил.
Исследование спектров свечения показало, что вид спектра свечения не зависит от концентрации урана в цлаве. Химический состав активирующей соли также не оказывает влияния на вид спектра свечения [67] (для активации фтористого натрия употребляли азотнокислую соль уранила, диуранат натрия, фтористый уранил, тетрафторид урана и закись-окись урана; концентрация урана в плаве — 5 • 10~5г/г NaF).
Спектры поглощения плавов (перлов) NaF—U имеют полосатую структуру [921, 1042, 1048]. Перлы NaF—U возбуждаются лучше всего длинноволновым ультрафиолетовым излучением. Квантовый выход плавов NaF—U зависит от длины волны возбуждающего света. Выход остается одним и тем же, как при возбуждении светом длиной волны 335 ммк, так и 365 ммк, и быстро падает при возбуждении светом А 405 и 435 ммк [822].
Из других свойств свечения перлов NaF—U следует отметить, что послесвечение их также кратковременно [783, 1042].
В ряде работ рассматривается вопрос о составе центра свечения перла NaF—U
[921, 922, 1029, 1042, 1048, 1049]. Исследованием спектров свечения при низких температурах как перлов, так и всех тех соединений, которые могли бы образоваться в результате взаимодействия расплавленного фтористого натрия с ураном, было ' показано [67 ], что центр свечения перла NaF—U состоит из иона уранила, находящегося в окружении ионов фтора и натрия. Имеется мнение, что в случае люминесценции монокристаллов NaF—U [1031] центр свечения представляет ион Us + , изоморфно замещающий ион натрия в кристаллической решетке фтористого натрия; компенсация же избыточного заряда осуществляется путем изоморфного замещения пяти ионов фтора ионами кислорода.
На рис. 24 приведена кривая зависимости интенсивности свечения плава NaF—U от концентрации урана (измерения выполнены нами при помощи фотоэлектрического люминесцентного фотометра ЛЮФ-57, вес плава (перла) — 25 мг). Интенсивность флуоресценции изменяется линейно в зависимости от количества урана
Концентрация урана, г U/г NaF
Рис. 24. Зависимость интенсивности свечения перла NaF—U от концентрации урана (в логарифмической шкале).
!51
в перле вплоть до значения 5.10 ~5 г. Родден [81 также отмечает, что линейность по крайней мере сохраняется до 50 мкг урана. При более высоких значениях урана кривая проходит через максимум. Спад интенсивности свечения при больших количествах урана в перле обусловлен образованием нелюминесцирующих при комнатной температуре уранатов [67, 715 и др.].
Интенсивность свечения перлов NaF—U (плавов) в сильной степени зависит от присутствия посторонних элементов. Влияние примесей на свечение может быть различным:
а) посторонние примеси сами образуют люминесцирующие перлы;
б) примеси окрашивают перл;
в) примеси действуют как гасители (тушители).
Д^ногие исследователи занимались изучением влияния примесей на люминесценцию урана в перлах (плавах) фтористого натрия [8, 9, 224, 240, 492, 513, 560, 690, 786, 791, 822 и др.]. Надо, однако, отметить, что данные, принадлежащие разным авторам, иногда являются противоречивыми, что может быть объяснено тем, что при исследованиях применялись нечистые препараты солей, а также несовершенством применяемой измерительной аппаратуры.
К элементам, образующим светящиеся перлы с фтористым натрием, относят ниобий, но в отличие от свечения урана люминесценция ниобия возбуждается преимущественно коротковолновым ультрафиолетовым излучением А 253,7 ммк [224, 560, 786]. В присутствии кальция (порядка 1 %) цвет флоуресценции перлов с ураном более зеленый[224, 690], по-видимому, вследствие того, что фторид кальция активируется ураном [83].
К элементам, окрашивающим перл, относят: Fe, Со, Ni, Мп, Сг, Си, V, Pb, Bi, Sc, In, Zn, Mo, TI и редкие земли [492, 786 и др.]. Большинство из этих примесей являются также сильными гасителями свечения урана в перлах фтористого натрия.
Относительно гасящего действия посторонних элементов на интенсивность свечения плавов нами приводятся данные, взятые из работ Гримальди, Мея и др. [560]. Гасящее действие примесей на свечение урана ими определялось в 2 а флюса (состава 45,5 г Na2CO3; 45,5 г К2СО3 и 9 a NaF); измерения проводились при помощи чувствительного флуориметра. Сильными гасителями считаются те элементы, которые в количествах от 1 до 10 мкг гасят урановую флуоресценцию на 10% и более. Это — Мп, Cr, Со, Ni, La, Pt [513], Au, Pb, Ce, Pr, Nd и Hg. Средние тушители — Fe, Cu, Zn, Sn и Th; при их содержании от 10 до 50 мкг интенсивность свечения урана уменьшается на 10%. Слабые тушители — Т1 и W; присутствие их в количествах от 50 до 1000 мкг уменьшает интенсивность свечения на 10%. Остальные элементы не являются заметными гасителями. Влияние железа начинает быть заметным, когда на 1 мкг урана приходится — 1000 мкг железа [996]. Интересно отметить, что, согласно исследованиям Прайса и др. [892], нептуний и
152
плутоний в малых количествах не мешают определению урана данным методом.
В случае одновременного присутствия примесей нескольких элементов их гасящее действие, как правило, не равно сумме действия отдельных компонентов [9]; для выяснения такого влияния в каждом отдельном случае требуется проведение специального исследования. При постоянной концентрации гасителя в плаве процент гашения обычно остается постоянным даже тогда, когда содержание урана изменяется в несколько тысяч раз [8].
Высокая чувствительность свечения плавов NaF—U к присутствию большого числа примесей обязывает перед определением урана этим методом проводить трудоемкую операцию отделения его от сопутствующих элементов. Выбор метода отделения зависит от химического состава и агрегатного состояния пробы [8, 224, 265, 273, 854]. Обзор этих методов приведен в статьях Уайта [1053] и В. И. Кузнецова, С. Б. Саввина, В. А. Михайлова [1035].
В ранних работах по определению урана методом светящихся перлов отделение примесей обычно проводили аммиачно-карбонат-ным способом [261, 492, 596 и др.]. Однако даже при самом тщательном выполнении анализа по аммиачно-карбонатной схеме в урансодержащем фильтрате все же остаются значительные количества марганца и железа.
В настоящее время лучшим способом для разделения служит метод экстракции эфиром или этилацетатом [414, 596 и др. ] из азотнокислой среды в присутствии нитрата алюминия. Использование при экстракции нитрата алюминия вместо общепринятого нитрата железа в случае люминесцентного определения представляется весьма выгодным, так как небольшие количества нитрата алюминия, проэкстрагировавшиеся вместе с ураном, оказывают значительно меньшее влияние на интенсивность свечения урана в плаве фтористого натрия по сравнению с железом [561 ].
Для извлечения малых количеств урана применяли и другие экстрагенты [557]. При анализе силикатных руд, сланцев и зол для отделения урана от примесей использовалась бумажная хроматография [557].
При определении поправки на гашение интенсивности свечения урана прибегают к приему «известных добавок» [786]. Одновременно приготовляют два перла (плава), в один вводят аликвотную часть раствора анализируемой пробы, в другой — такую же аликвотную часть с добавкой известного количества урана. Измеряют интенсивность свечения обоих плавов. Разность между вторым и первым отсчетом эквивалентна значению добавки.
Прайс и др. [822] как экспрессный прием для количественного определения урана рекомендует способ разбавления, требующий предварительного разложения пробы, но позволяющий, по мнению авторов, обходиться без операции отделения урана от сопутствующих элементов. Этот прием возможен благодаря тому, что мешающее
154
влияние примесей зависит только от концентрации примеси и не зависит от отношения урана к гасителю. По мере того как концентрация примесей уменьшается, тушение постепенно исчезает. Авторы [8] предлагают брать малые аликвотные части раствора, полученного после разложения пробы, в платиновые чашки, упарить досуха, добавить фтористого натрия или смеси фтористого натрия с карбонатами натрия и калия, затем сплавить. Интенсивность свечения полученных таким путем охлажденных плавов с разным разбавлением сравнить со свечением стандартных плавов. Измерения проводят на чувствительном флуориметре. Отсутствие операции отделения урана от примесей дает возможность значительно сократить время, необходимое для проведения анализа.
Метод разбавления может быть использован при анализе растворов с соотношением гасителя к урану не более 1000 : 1 в том случае, если для измерения интенсивности свечения имеется высокочувствительный флуориметр [8]. Надо также отметить, что' при таких ничтожных концентрациях урана, с которыми приходится иметь дело при анализе по способу разбавления, особенно необходимо тщательно следить за чистотой воздуха в лабораторном помещении и предъявлять высокие требования к чистоте исходного фтористого натрия.
Для приготовления перлов (плавов), в качестве исходного вещества обычно применяют фтористый натрий, при этом плав NaF—U при освещении ультрафиолетовым излучением имеет, как было уже ранее отмечено, желто-зеленое свечение. Свечение урана интенсивно также в плавах фтористого лития, однако применение фторида лития неудобно в том отношении, что последний при высокой температуре энергичнее взаимодействует с платиной, чем расплавленный фтористый натрий. Свечение плавов LiF—U — зеленого цвета. Применение фторида калия в качестве основы для приготовления плавов представляется выгодным благодаря тому, что ниобий в этом плаве не флуоресцирует, однако свечение урана в нем менее интенсивно и, кроме того, плавы отличаются большей гигроскопичностью. Свечение урана во фториде калия—оранжевого цвета.
Ряд исследователей с целью снижения температуры образования плавов предлагают к фтористому натрию делать различные добавки [414, 539, 559, 560 и др.1. Гримальди и др. [559, 5601 применяют смесь состава: 9 ч. NaF, 45,5 ч. Na2CO3 и 45,5 ч. К2СО3. Приготовление плавов из такой смеси можно проводить при температуре ^650° (вместо ^-1000° для перлов из одного фтористого натрия). Такой флюс благодаря своей низкой температуре плавления удобен в работе. Флюс можно заготовить заранее.
Для обеспечения большей однородности флюса смесь компонентов предварительно помещают в платиновую чашку, сплавляют при температуре ^.700°, охлаждают и растирают в тонкий порошок, который хранят в банке с притертой пробкой.
Интенсивность свечения плавов (перлов), активированных ураном, сильно зависит от температуры прогрева, времени плавления,
15 \
гомогенности флюса, состава газовой атмосферы вовремя плавления и условий охлаждения плава (перла). Исследования Флетчер [513] показали, что в случае применения флюса, состоящего из фторида натрия и карбонатов натрия и калия, оптимальная температура прогрева 650—750°, время не более 25 мин. (для 1,5 г флюса). Выше этой температуры, а также с увеличением времени прогрева возрастает количество платины- (из тигля или чашки), растворенной во флюсе; из-за гасящего действия платины снижается интенсивность свечения урана. Плавление необходимо вести в окислительной атмосфере, если же его проводить в восстановительном пламени, то интенсивность свечения снижается для 1 мкг урана до 20%. При быстром охлаждении льдом интенсивность свечения уменьшается незначительно. При медленном охлаждении в окислительном пламени флуоресценция возрастает на 30% (испытания проводились в пламени горелки Меккер [8]).
Операция приготовления плава с анализируемой пробой состоит в том, что ураносодержащий остаток пробы известного веса (обычно в виде нитратов), помещают в платиновую чашку с определенным количеством фтористого натрия (или флюса) и сплавляют. Интенсивность свечения охлажденного плава сравнивают со свечением стандартных плавов, приготовленных точно при таких же условиях. Плавление проводят в печах [273] или на горелках [414, 513]. В горячем состоянии плав должен быть бесцветным, в холодном — белого цвета.
Для изготовления маленьких платиновых чашек рекомендуется применять специальный пресс [414]. При массовых анализах флюс добавляют определенной меркой, иногда из фтористого натрия предварительно при помощи пресса заготовляют таблетки [596] или гранулы [53а]. Описана специальная машина для приготовления люминесцирующих плавов [938]. Предлагался и другой способ введения флюса в виде 2%-ного раствора фтористого натрия с последующим выпариванием, но такой прием в анализе широкого распространения не получил.
В случае анализа растворов очень удобен метод «окунания» перлов [М. Ф. Коринфская, 1950 г.; 152а, 267, 1042 и др.] Из платиновой проволоки диаметром 0,5 мм, длиной 2—2,5 см, впаянной в стеклянную палочку, делают петлю диаметром -ч. 3 мм. Докрасна нагретой в пламени горелки петлей захватывают таблетку фтористого натрия и снова вносят в пламя горелки для того, чтобы фторид натрия занимал весь просвет петли. Готовый перл (в холодном состоянии) погружают в исследуемый раствор, после чего снова вносят в окислительную часть пламени. •Операцию погружения в раствор с последующим прогревом в пламени повторяют дважды. Было установлено, что перл захватывает каждый раз примерно одно и то же количество испытуемого раствора при однократном и однообразном погружении его в раствор. Интенсивность свечения исследуемого перла сравнивают со свечением стандартных перлов, приготовленных в тех же условиях.
Описан и другой способ приготовления светящихся перлов, при применении которого результаты получаются более воспроизводимые. В тигель с известным количеством фтористого натрия вводят аликвотную часть анализируемого раствора, тщательно перемешивают, упаривают досуха. Ушком платиновой проволоки, предварительно нагретым докрасна, захватывают сухой порошок и вносят в пламя горелки.
Ряд исследователей отмечает, что стандартными перлами можно пользоваться в течение недели, однако лучше всего их приготовлять одновременно с анализируемыми.
При определениях урана методом светящихся перлов (плавов) необходимо предварительно определять чистоту исходного фтори-
155
стого натрия, приготовив из него перлы без добавки урана и проверив их свечение.
Оптическая схема, обычно используемая при люминесцентных наблюдениях, включает источник возбуждения (ртутно-кварцевая лампа), фильтр, выделяющий необходимую для возбуждения спектральную область, в данном случае Л 365 ммк (для чего служит фильтр УФС-3) и прибор, воспринимающий излучение.
Поскольку главный максимум спектра излучения плава NaF—U расположен в видимой области при Z555 ммк, то их интенсивность свечения можно сравнивать визуально, спектрографически (с последующим фотометрированием), а также измерением на объективных фотоэлектрических фотометрах (флуориметрах).
Лучшими перлами для визуальных наблюдений являются те, которые содержат от 10-7 г до 10-6 г урана. При визуальных наблюдениях в качестве источника возбуждения можно применять лампу для флуоресцентного анализа витаминов Л-80 (выпускается Глав-медпромом, завод «Красногвардеец») или аналитическую лампу ЛЮМ-1 (производится заводом «Геологоразведка»). Наблюдения ведутся в затемненном помещении. Анализируемые перлы и стандарты удобно помещать в углубления (гнезда), сделанные в пластинке эбонита. Сравнение интенсивности свечения плавов при визуальных наблюдениях можно проводить при помощи различных фотометров, например отечественного фотометра ЛЮФ-51 [1042].
Спектрографическая методика применялась Хернеггер и Карлик [596] при определениях содержания урана в морской воде. Позже эта методика была принята Ланер [690]. Хернеггер и Карлик использовали светочувствительный спектрограф и фотопластинки с максимумом чувствительности в зеленой части спектра. Время экспозиции зависело от количества урана в перле. Фотографируя спектры люминесценции анализируемого и стандартных перлов и измеряя почернение фотографической пластинки микрофто-метром при Л 555 ммк (длине волны, соответствующей главному максимуму спектра излучения перла NaF—U), авторы судили о количестве урана в анализируемом перле.
Вместо перлов И. Драганич [473] предлагает готовить плавы в платиновых лодочках. В каждую лодочку помещают по 70 мг фтористого натрия, который предварительно смешивают с ураисодержащим остатком анализируемой пробы, брикетируют, после чего сплавляют. Исследуемый брикет вместе с 4 стандартами помещают в прибор для измерения. Свечение всех 5 образцов при помощи фотокамеры снимают на пленку. После проявления пленку фотометрируют. Одновременная съемка образца и стандартов исключает необходимость стабилизации напряжения источника возбуждения. Для того чтобы на пленку не действовал отраженный ультрафиолетовый свет, перед фотокамерой помещают кювету из плексигласа, наполненную 5%-ным раствором нитрита натрия. Чувствительность метода от 10“8 до 10"6 г урана, воспроизводимость, по данным автора, -±Д0% .
За последние годы как у нас, так и за границей для измерения интенсивности свечения плавов или перлов разработано несколько конструкций фотоэлектрических приборов, работающих как по 156
однолучевой, так и двухлучевой схемам. Лучшим средн отечественных приборов оказался прибор ЛЮФ-57, конструкции Государственного оптического института им. С. И. Вавилова.
На рис. 25 приведена фотография фотоэлектрического люминесцентного фотометра ЛЮФ-57 [84] (выпускается одним из заводов Московского совнархоза).
Рис. 25. Прибор ЛЮФ-57.
1— реостат; 2— стабилизатор; 3— фотометр.
Флуориметр ЛЮФ-57 построен по двухлучевой компенсационной схеме с двумя фотоэлементами. Двухлучевая схема дает возможность исключить ошибки, связанные с колебанием напряжения питающей сети. Измерения проводятся по нулевому методу путем сравнения интенсивности люминесценции образца и эталона, поочередно вводимых в одно и то же плечо прибора, которое имеет переменную измерительную диафрагму. В фотометре ЛЮФ-57 имеются две съемные кассеты, позволяющие производить измерение концентрации урана в перлах диаметром от 2 до 4 мм, плавах диаметром от 8 до 15 мм (толщиной до 2 мм). В каждой кассете установлено по 4 эталона, изготовленных из люминесцирующего стекла с различной концентрацией урана, причем яркость люминесценции одного эталона по сравнению с соседним отличается в 10 раз (при одних и тех же условиях возбуждения); этим достигается диапазон измерений концентрации урана в четыре порядка.
При помощи флуориметра ЛЮФ-57 можно определять содержание урана в перлах (или плавах) от -^-10~9г до 10 ~6 г; при этом погрешность измерений не превышает ±5% (отн.).
В литературе имеется описание американского двухлучевого флуориметра [712]. В этом приборе анализируемый образец устанавливают в одном плече прибора, стандарт — в другом. Отсчет концентрации урана в анализируемом образце ведется по шкале потенциометра в долях концентрации эталона. Прибор предназначен
157
для измерения яркости люминесценции как твердых, так и жидких растворов.
Однако большинство из описанных в литературе флуориметров [514—516, 822 и др.] построены по однолучевым схемам, приемником света люминесценции служит фотоэлектронный умножитель. При работе на таких флуориметрах требуется высокая стабилизация напряжения питания фотоумножителя и источника возбуждения.
Приложение метода в анализе проб различного состава
Руды, минералы, породы и метеориты
Изучение фотолюминесценции урановых минералов и руд показало, что не все урановые и урансодержащие минералы люми-несцируют [155, 738, 1055, 1057 и др.]. Наиболее ярко люминес-цируют фосфаты, фториды, арсенаты, карбонаты, сульфаты и сульфокарбонаты уранила. Слабо люминесцируют ванадаты и силикаты. Цвет люминесценции урановых минералов может быть желто-зеленым, голубовато-зеленым, желтым. Спектральный состав излучения можно установить с помощью карманного спектроскопа. Минералы, в состав которых входят U(IV)), а также U (VI), выступающий в качестве кислотообразующего окисла, не люминесцируют. Люминесцентная способность минералов, содержащих группу уранила, зависит от других катионов и анионов, присутствующих в минералах; так, Cu2'\ Fe2 + , Pb2 + , Bi3+, Fe3 + , Мп2Д Ag , Ni2 + , Со2+, либо полностью тушат люминесценцию уранилсодержащих минералов, либо сильно уменьшают интенсивность свечения.
Для возбуждения люминесценции урановых минералов могут применяться ультрафиолетовые источники света, как длинноволновых, так и коротковолновых лучей [22, 222], а также фиолетовая часть видимого спектра и катодные лучи. При изучении люминесценции минерала наблюдение ведут на «свежем изломе» и возбуждении светом X 300—400 ммк. Каждый люминесцирующий минерал имеет собственное положение максимумов в спектре свечения [155]. Цвет люминесценции ряда урановых минералов (желто-зеленый) очень близок по спектральному составу к свечению виллемита, однако между ними имеется и различие; так, в спектре свечения виллемита отсутствует структура полос и наблюдается длительная фосфоресценция, в то время как у урановых минералов длительная фосфоресценция отсутствует. Благодаря простоте и высокой чувствительности люминесцентный метод в комбинации с другими нашел применение при поисках урановых месторождений [155, 1058]. По наблюдению люминесценции урана, не нарушая цельности зерна и не выделяя уран, судят о распределении урансодержащих веществ на поверхности образца.
158
Урансодержащие минералы, которые сами не люминесцируют, могут быть переведены в люминесцирующее состояние путем обрызгивания их поверхности различными растворителями или путем сплавления с фтористым натрием. В. Г. Медковым и 3. М. Свердловым [156] в качестве растворителей урановых минералов (по способу обрызгивания) были испытаны серная, азотная, соляная, уксусная и фосфорная кислоты различных концентраций. Обнаружено, что во всех случаях образуются люминесцирующие (преимущественно под действием коротковолнового ультрафиолетового излучения) соединения урана, имеющие характерные полосатые спектры. Свечение сохраняется и после высыхания поверхности минерала. Для уранованадатов рекомендуют брать серную кислоту. Применяя этот прием, можно получить полуколичественную оценку (о количестве урана судят по величине люминесцирующей поверхности минерала).
Известен также и другой полуколичественный прием определения урана в минералах, основанный на том, что все без исключения урановые минералы и руды после сплавления с фтористым натрием и освещения их ультрафиолетовым излучением начинают люминесцировать (плав предварительно охлаждают, а затем уже возбуждают ультрафиолетовым излучением) и, как правило, желтым цветом. Эти плавы также имеют полосатые спектры свечения, причем вид спектра зависит от химического состава анализируемой пробы. Интенсивность свечения связана с количеством урана и наличием сопутствующих элементов. Пол у количественные определения таким способом могут проводиться для руд с содержанием урана более 5-10 4Оо. Измерения ведут по способу стандартных серий.
Для анализа пробу истирают в тонкий порошок. К 50 мг фтористого натрия добавляют 0,1 —0,3 мг порошка пробы, тщательно перемешивают, из смеси приготовляют перлы. Влияние примесей может быть учтено по способу «известных добавок». Этот прием особенно успешно может быть применен при анализе сланцев, фосфатных, высокосиликатных руд [854].
Для точных количественных определений урана люминесцентным методом требуется предварительно отделить уран от примесей. Ряд исследователй [224, 492] предлагают вскрытие основных пород проводить нагреванием с концентрированной соляной кислотой. Остаток обрабатывают смесью фтористоводородной и серной кислот для удаления SiO2- Уран осаждают совместно с гидроокисью железа; осадок гидроокисей обрабатывают карбонатом аммония, как и в обычной схеме аммиачно-карбонатного разделения. Кислые же породы сплавляют с содой, SiO2 отделяют обработкой соляной кислотой; металлы группы сероводорода осаждают сероводородом, далее анализ ведут по аммиачно-карбонатной схеме (см. стр. 283).
Разложение силикатов, содержащих цирконий, лучше проводить трехкратной обработкой смесью азотной и фтористоводородной
153
.кислот, а отделение примесей — при помощи экстракции этилацетатом или эфиром из азотнокислого раствора в присутствии нитрата алюминия, или же на целлюлозной колонке [1029]. Когда отделение примесей проводят при помощи экстракции, то осадок гидроокисей, полученный после осаждения аммиаком, растворяют в азотной кислоте, и из раствора уран экстрагируют эфиром [273]. Эфирный экстракт упаривают досуха. Остаток прогревают при температуре -^800° и обрабатывают концентрированной азотной кислотой, снова упаривают досуха и растворяют в 5%-ной азотной кислоте. Раствор переносят в градуированную колбу. Затем готовят плавы или перлы и при помощи флуориметра сравнивают их интенсивность свечения со свечением стандартных плавов.
Хернегер и Карлик [596] указывают на необходимость тщательного освобождения от серной кислоты перед операцией приготовления перлов, так как она, взаимодействуя с фторидом натрия, образует фтористоводородную кислоту, вследствие чего наблюдается изменение веса перла.
Примечание. При анализе проб, содержащих органические соединения, навеску пробы перед разложением прогревают в печи при температуре ^800°.
Для определения урана в породах, в кислых вытяжках П. А. Волковым в 1953 г. был разработан метод отделения его от сопут-ствующих элементов путем осаждения фосфата четырехвалентного урана в кислой среде с применением соосадителя — циркония. Осадок фосфатов тщательно перемешивают с известным количеством фтористого натрия. Уран определяют флуориметрическим методом. По данным автора, небольшие количества циркония, находящиеся вместе с ураном в перле, не мешают определению урана флуориметрическим методом. Этот метод был применен [143] для анализа изверженных горных пород, содержащих от 1-10-4 до 1 -10_3% урана.
Описан люминесцентный метод определения урана с предварительным выделением его на фосфате титана [157].
При определениях урана (порядка 5-10“4%) в цирконах применялась экстракция этилацетатом с использованием двузамещенного фосфата натрия для удержания циркония; высаливатель — азотнокислый аммоний. Этот метод позволяет определять уран в присутствии 100 000 кратных количеств циркония [441 ].
Люминесцентный метод был использован для определения урана в концентратах, причем получен удовлетворительный результат [1029].
При анализе метеоритов применялось полное разложение (Т. С. Добролюбская, Е. Б. Евдокимова, 1956 г.) с последующей экстракцией и люминесцентным определением по схеме, приведенной на стр. 163. Содержание урана в метеоритах оказалось равным ^-10"8а/г [839, 840, 1056].
160
Люминесцентный метод может быть использован и для определения урана в воздухе производственных помещений. Воздуходувкой или пылесосом просасывают через респиратор с ватой определенный объем воздуха. Вату помещают,в тигель и сжигают в муфеле. Остаток обрабатывают концентрированной азотной кислотой, упаривают досуха. Сухой остаток растворяют в разбавленной азотной кислоте, добавляют углекислого аммония, центрифугируют (обработку углекислым аммонием с последующим центрифугированием повторяют дважды). Раствор сливают, подкисляют азотной кислотой, измеряют объем, затем определяют уран люминесцентным методом.
Для определения содержания урана в соединениях щелочных металлов (лития, натрия и калия), которые могут быть переведены во фториды, анализируемое вещество помещают в платиновую чашку и обрабатывают фтористоводородной кислотой, перегнанной из платинового сосуда; упаривают досуха. Из полученного фторида готовят плав (или перлы), и сравнивают их интенсивность со свечением стандартных плавов (перлов), приготовленных на основе фторида того же щелочного металла, заведомо не содержащего урана.
Имеется сообщение об определении следов урана в нитрате тория люминесцентным методом [1059], причем торий связывают в комплекс с ЭДТА, уран экстрагируют 5%-ным раствором трибутилфосфата в керосине при pH 2,5, высаливатель — Ca(NO3)2. После промывания органической фазы насыщенным раствором нитрата аммония уран реэкстрагируют в водную фазу и определяют флуориметрически.
Воды (природные и производственные)
Высокая чувствительность люминесцентного метода позволяет в некоторых случаях, когда содержание примесей — гасителей небольшое, непосредственно определять уран в природных водах, внося поправку на гашение по способу добавок [557]. В две платиновые чашки берут по 10 мл раствора, в одну из них вносят известное количество урана. Растворы упаривают досуха, вносят фторид натрия или флюс (9 г NaF; 45,5 г Na2CO3; 45,5 г К2СО3) и производят плавление. После охлаждения на флуориметре измеряют интенсивности свечения плавов; сравнивают полученные значения интенсивностей со свечением стандартных плавов, вычисляют процент гашения (см. стр. 164).
В случае определения урана в водах необходимо считаться с тем обстоятельством, что уран может (при долгом стоянии раствора) частично адсорбироваться стеклом. Адсорбция понижается в присутствии бикарбонатов, карбонатов, сульфатов и ацетатов [970]. Адсорбция урана на сосудах из полиэтилена несколько меньше, чем на стенках сосуда из борсиликатного стекла.
Аналитическая химия урана 161
Для отделения урана от железа можно использовать карбонатное осаждение. Было показано, что только 0,01 мкг U теряется, когда применяют карбонат для осаждения 6,5 мкг железа в 5 мл раствора, содержащего 2,25 мкг урана [970].
Для определения содержания урана как в соленых, так и пресных водах в качестве соосадителя урана можно применять фосфат алюминия [92]. Осадок фосфатов растворяют в азотной кислоте, и уран из раствора экстрагируют этил ацетатом в присутствии нитрата алюминия. По данным Смита и Гримальди, при однократном осаждении фосфата алюминия в осадок переходит более 95% урана.
Простой и быстрый метод для определения содержания урана в природных водах (от 0,020 до 0,02 мг/л с относительной ошибкой ±15%) был предложен В. К. Марковым, В. Т. Харламовым и А. П. Медведевой [213]. Метод основан на избирательной сорбции уранилкарбонатного анионного комплекса на анионите ММГ-1, который предварительно переводят в бикарбонатную форму. После пропускания анализируемого раствора смолу растворяют при нагревании в концентрированной соляной кислоте. В этом растворе определяют содержание урана по методу «окунания» перлов.
Для концентрирования урана из морской воды В. И. Кузнецов и Т. Г. Акимова [126] разработали метод, основанный на соосажде-нии уранилроданидного комплекса с осадком роданида метилвиоле-та. Осадок отфильтровывают, озоляют. Уран в золе определяют люминесцентным методом. Соли, растворенные в морской воде, при соблюдении условий анализа, в осадок не переходят. По данным авторов, из 1 л воды можно выделить количественно 0,1 мкг урана. Для выделения урана из морских вод применялись и экстракционные методы [1017 а]. Обычное содержание урана в морских водах — от нескольких десятых до нескольких микрограммов на 1 л [92, 596, 771].
При больших содержаниях урана А. Ф. Фиолетова [267] рекомендует применять способ разбавления или добавок. Для быстрого определения урана в водах можно использовать способ «окунания», предварительно отделив примеси.
Для растворов с содержанием урана от 0,1 до 10 мкг/мл был разработан экстракционный метод отделения урана от примесей (М. Ф. Коринфская, 1958 г.). Метод заключается в экстрагировании урана из растворов --- 0,8 N по азотной кислоте, растворами ТБФ в керосине, или диизоамиловым эфиром метилфосфорной кислоты с последующим определением урана в экстрактах по способу окунания. Эталонными растворами служат органические растворы, содержащие уран.
1Г.2
Биогенные материалы Почвы
Анализ почв нами (Т. С. Добролюбская, Н. П. Муравьева, Н. И. Удальцова, 1957 г.) проводился с применением полного разложения пробы, отделения урана эфирной экстракцией; определение заканчивалось люминесцентным методом. Операции анализа были проконтролированы радиометрически введением изотопа U233 в некоторые пробы перед разложением, причем получено 100%-ное извлечение. Результаты параллельных анализов приведены в табл. 27.
Таблица 27
Данные анализов по определению урана в почвах
Ns образна Содержание урана, % № образца Содержание урана, %
1-й анализ 2-й анализ 1-й анализ 2-й анализ
1 2,9-Ю'4 3,0- 1(Г4 5 2,0-10“4 2,0-10-4
2 3,5-10~4 2,1 10~4 6 2,1-Ю"4 2,1•10~4
3 2,8-Ю-4 2,5-10-* 7 2,0-10-* 1,9-10~4
4 1,9-10~4 1,4-10~4
Ниже приведен ход анализа, который может быть использован (с небольшими изменениями) при анализах не только почв, но и различных пород.
Чистоту реактивов проверяют на отсутствие урана (сплавлением определенного реактива с шихтой и наблюдением люминесценции).
Оборудование
(для других работ не используется)
1. Фотоэлектрический люминесцентный фотометр ЛЮФ-57.
2. Муфельная печь на 700°.
3. Платиновые (d х. 10 с.и) и фарфоровые чашки (d х. 10 и 14 см).
4. Бумажные фильтры (синяя и белая лента), предварительно промытые на воронке азотной кислотой (1 : 1).
5. Платиновые мишени (d х. 1,7 см).
Ход анализа
1. Разложение пробы проводят по методике, описанной на стр. 345.
Полученный раствор выпаривают на водяной бане досуха; из этого осадка Уран экстрагируют диэтиловым эфиром на фоне 1 М раствора азотнокислого алю-Миния в приборе, описанном на стр. 293.
> 2. Экстракт выпаривают на водяной бане досуха. Остаток прокаливают в те-
чение нескольких минут при температуре х600°, охлаждают, обрабатывают концентрированной азотной кислотой, снова упаривают досуха и растворяют в 5% -ной Цветной кислоте. Раствор переносят в градуированную пробирку емкостью 5 мл, Доводят объем раствора до 5 мл. Далее уран определяют люминесцентным методом.
11* 163
3. Люминесцентное определение.
Приготовление стандартного раствора.
Точную навеску (1 г) чистого металлического урана (99,7%) растворяют в азотной кислоте в стакане емкостью 100 ,ч.г, избыток кислоты удаляют выпариванием до влажного состояния. Остаток растворяют в воде и разбавляют до 1 Получают раствор с содержанием 1 мг м.1 урана. Путем разбавления полученного раствора приготовляют стандарт с содержанием 1 мкг мл L'.
Приготовление шихты.
В платиновую чашку отвешивают 9 г NaF, 45,5 г Na.,CO3, 45,5 г 1\.,СО3, помещают ее в муфель при 650° и выдерживают до полного плавления. Охлажденный плав растирают в фарфоровой ступке до тонкого порошка и помещают в банку с притертой пробкой.
Приготовление плавов.
Платиновые мишени, предварительно очищенные кипячением в азотной кислоте (1 : 1), помещают на асбестовую пластинку, имеющую специальные углубления. Далее мишени проверяют флуориметрически на отсутствие урана. В каждую мишень помещают по 0,2 г шихты. Пластинку с мишенями вносят в муфель на 5 мин. при 650°, вынимают, охлаждают и просматривают под ртутной лампой ПРК-4, снабженной ультрафиолетовым фильтром УФС-3.
Чистые мишени обрабатывают снова азотной кислотой для удаления плава, промывают водой, сушат. В 5 мишеней микропилеткой наносят по 0,1 мл анализируемого раствора, в другие 5 мишеней — по 0,1 мл стандартного раствора урана с концентрацией 1 мкгмл и еще в 5 мишеней — по 0,1 мл анализируемого и пи 0,1 мл стандартного растворов. Мишени подсушивают в сушильном шкафу. На каждую мишень помещают 0,2 г шихты. Пластинку с мишенями вносят на 5 мин. в муфель при 650°, вынимают, охлаждают. Через 30 мил. производят измерение интенсивности свечения плавов на приборе ЛЮФ-57.
Сравнением средних значений интенсивности свечения анализируемого плава со свечением стандарта определяют кажущееся содержание урана в плаве, выраженное в микрограммах («а»). Находят поправку на гашение свечения урана примесями, частично проэкстрагировавшимися совместно с ураном; для этого сравнением средних значений интенсивностей свечения плава, содержащего 0.1 мл анализируемого раствора и 0,1 мл стандарта, со свечением стандарта определяют значение «Ь» в микрограммах урана. Значение добавки в микрограммах урана («с»), определенное из люминесцентных измерений, равняется Ь — а.
В действительности же было добавлено 0,1 мкг урана. Вычисляют процент „ 0,1 —с
гашения, равный —jy-j— -100.
Истинное содержание урана в 0,1 мл анализируемого раствора будет равно 0,1—с
а a- —Q-j— мкг урана.
В случае анализа почв поправка на гашение обычно составляла 30%. В некоторых пробах, хорошо очищенных, гашения не было.
Золы нефтей, угли, морские илы и осадочные отложения.
При анализе зол нефтей, углей и морских илов анализируемая проба, когда содержание кремнекислоты и примесей, влияющих на свечение урана, невелико, может непосредственно вводиться во фтористый натрий (или флюс) [261, 326].
Угли и морские илы предварительно озоляют; если содержание кремнекислоты большое, то ее можно удалить [566].
Анализ морских отложений на уран можно проводить путем сплавления пробы с карбонатом натрия, обработкой полученного плава азотной кислотой, удалением SiO2 и отделением урана от 16i
примесей эфирной экстракцией нитрата уранила и вторичной экстракцией бутанолом (или другими экстрагентами) или при помощи бумажной хроматографии 1566] с последующим люминесцентным определением.
Содержание урана в отложениях из Тихоокеанской глубинной скважины составляет несколько микрограммов на 1 г осадочного отложения.
Животные
Люминесцентный метод может быть использован для определения урана в разнообразном биологическом материале [513, 557, 7791. Известны два приема определения урана в биогенном материале: Гоффмана [224] и Неймана [779].
По способу Гоффмана пробу озоляют, остаток обрабатывают азотной кислотой, фосфаты удаляют, осаждая их оловом; избыток олова удаляют, действуя сероводородом. Оставшиеся примеси металлов осаждают аммиаком, предварительно связав уран в комплекс гидроксиламином. Далее анализ ведут по аммиачно-карбо-натной схеме, применяя в качестве носителя FeCl3. Уран определяют флуориметрически.
Нейман рекомендует после озоления и разложения пробы выделять уран на протеине (уран с протеином образует прочный комплекс). Тяжелые металлы, когда это необходимо, например при анализах печени, крови и селезенки, удаляют на ртутном катоде перед осаждением урана протеином. Ураново-протеиновый комплекс растворяют в соляной кислоте, белок удаляют центрифугированием, уран определяют флуориметрически.
Для извлечения урана из остатков проб после озоления могут быть использованы и экстракционные методы. В животных тканях содержатся десятые доли микрограмма урана на 1 г ткани.
II. ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ
1. Полярография у р а н а|
С. И. Синяк.ова
В последние годы широкое применение в аналитической химии получил полярографический метод анализа, обладающий рядом преимуществ по сравнению с другими методами. Для определения урана в различных материалах полярографический метод был также широко использован.
Большое значение для аналитического применения полярографического метода определения урана имеет ясное представление о механизме восстановления и окисления его в разных валентных состояниях на ртутном капельном и твердых электродах. В Настоящее время благодаря проведенным исследованиям [40,
165
211, 581, 595, 646, 664—666] значительная часть вопросов, касающихся механизма электродных процессов урана, получила однозначное решение. Некоторые же вопросы, особенно касающиеся ионного состояния продуктов промежуточных стадий электродных процессов, не выяснены еще полностью.
Механизм восстановления и окисления урана на ртутном капельном и твердых электродах
Первым исследовал поведение UO2+ и U (IV) на ртутном капельном катоде Герасименко [595]. В 0,1 V растворе КС1 им получены для ПО22+ кривые с 3 волнами, имеющими значение £1/2 =—0,15; —0,80 и 1,0 в (относительно нас. к. э.) соответственно. При этом найдено, что кислотность раствора в узких пределах не оказывает влияния на £1/2 1-й ступени, что противоречило данным Лютера и Михи [711] и Титлестада [980], которые из измерения равновесного потенциала на платиновом электроде представляли процесс восстановления UCP+ протекающим по следующему механизму:
иоГ 4-4Н + 4-2е~ и (IV) 4- 2Н2О. (1)
На этом основании Герасименко пришел к выводу, что 1-я стадия электрохимического процесса восстановления UO^+ соответствует реакции:
UO2a++e-->U(V). (2)
Это подтверждено было также построением графика 1g ~—г — Е ld 1
(где i — ток в любой точке волны, соответствующей данному потенциалу £; id—предельный диффузионный ток), для которого наклон оказался равным 0,058, что находится в хорошем согласии с теоретической величиной 0,059 для электродной реакции с участием одного электрона.
Что касается 2-й и 3-й ступени, то Герасименко считал, что они обусловлены следующими процессами:
U (V) 4-е"—U (IV) (3)
U (IV) 4-е~-+U (III). (4)
3-я стадия процесса была подтверждена им снятием полярограммы раствора U (IV), полученного фотохимическим восстановлением 13О2+ в присутствии этилового спирта. Возникавшая при этом волна соответствовала 3-й ступени на полярограмме раствора UO2+.
Наиболее интересным выводом из наблюдений Герасименко является предположение о неустойчивости и диспропорционировании в кислых растворах U (V), образующегося в 1-й стадии электрохимического восстановления UOj;+.
166
Дальнейшие исследования [40, 229, 260 , 664, 665] показали, что в растворах НС1 слабой концентрации (10_ 2 —0,2 М) 15О2Г дает две ступени с Е'/2=—0,18 в и =—0,93 в (нас. к. э.), а 3-я волна
на полярограмме обусловлена разрядом Н+ (рис. 26). Найденный
1g.——: относительно £=0,062 для 1-й сту-
ld ~~ 1
наклон графика
пени также находится в хорошем согласии с теоретической величи-
ной 0,059 для процесса с участием одного электрона.
Рис. 26. Полярограмма уранил-ионов в растворах кислот. а— в 0,1 М НС1; б— в 1 10—3 М H2SO,-j-2-10—*% тимола.
На этом основании все исследователи пришли к выводу, что продуктом 1-й стадии восстановления UO®+ на ртутном капельном катоде является UO2+, согласно следующей реакции:
ио* ,
и что реакция эта обратима.
(5)
График 1g -7-^—-.—Е, построенный по 2-й волне, состоит ld 1
из
двух отрезков, что указывает на ее сложный характер.
Полученным соотношением величин диффузионных токов 1-й и 2-й ступени, равным 1 : 2 в растворе 0,1 V КС1, 0,01 N НС1, содержащем тимол (2-10~4%), доказано, что 2-я ступень соответствует процессу с участием двух электронов. Кольтгоф и Харрис [664] подтвердили, что в кислой среде раствор сульфата урана (IV) дает
одну волну с таким же £./2, как и вторая волна UO|+-
id!ммоль этой волны равняется 73 суммарного id при восстановлении UO^ + .
Таким образом было установлено, что вторая волна восстановления UO2 является суммарной для двух процессов. Так как константа диффузионного тока U (IV) равна константе диффузионного тока 1-й стадии восстановления UO* + , то естественно было принять,
167
что при восстановлении U (IV) также участвует один электрон и продуктом реакции является U (III). Доказать это наклонов гра-
фика 1g
относительно Е не удалось, так как он оказался
по более ранним измерениям равным 0,08, т. е. процесс восстанов
ления U (IV) представлялся как необратимый. Считая, что Но-
йоны не участвуют в процессе восстановления во 2-й суммарной стадии, Кольтгоф и Харрис предложили следующий механизм для этой стадии восстановления:
иОгЧ-е- — UO2+ или UOOH+ (6)
UO2+-4~— UO+ (7)
UOOH+ 4- е~ — UOOH, (8)
причем продукты реакции (7) и (8) затем уже реагируют с ионами Н+, образуя ионы U3 + .
Анодная волна окисления U (IV) на ртутном капельном электроде не получена этими авторами даже при потенциалах +0,4 в.
Хил [581] также подтвердил выводы Кольтгофа и Харриса, что в среднекислых растворах НОг + дает три волны восстановления на ртутном капельном электроде, U (IV) — одну волну, а кислые растворы U (III) — одну волну окисления, которая по величине £,i/2 и id соответствует третьей волне на полярограмме 15Ог . Кроме того, в слабокислых растворах UOz+ Хил [581] заметил за волной Н+ еще одну большую волну, имеющую в хлоридных растворах Е1/2 ~—1,85 в, а в сульфатных Е+? ~—2,2 в. Эта волна обусловлена, по его мнению, восстановлением U (III) до металла. Из этих данных следует, что уран в твердом виде осаждается на поверхности электрода. Хотя до последнего времени считалось, что растворимость урана в ртути ничтожно мала, однако в последние годы найдено, что металлический уран с очень чистой поверхностью, полученный в отсутствие кислорода, путем разложения гидрида урана, амальгамируется, образуя массу серебряного цвета, похожую на другие амальгамы, и что амальгама с содержанием урана < 1 % жидкая и устойчива на воздухе [549]. Однако следует отметить, что ни нам, ни другим исследователям не удалось наблюдать эту волну.
Изучая систему U (IV)/U (III), Хил нашел, что волна окисления U (III) и волна восстановления U (IV) имеют совпадающие значения Eii„, равные в 1 М НС1 —0,93 вив 0,5 М H2SO4—1,10 в, а также одинаковые величины диффузионного тока. Анализ волны восстановления U (IV) по графику 1g относительно Е дал на-клон 0,064 в НС1 и 0,061 в H2SO4, что указывает на близкий к обратимому процесс с участием одного электрона. На этом основании, а также исходя из наблюденных данных о независимости Е1/, каждой стадии восстановления UO*+ от pH, Хил считает, что в элек
168
тродной реакции не участвуют ионы Н \ а следовательно, наиболее вероятные ионные формы существования U (IV) и U (III) даже в кислых растворах следующие:
для'и (IV) —«-UO2', UQH3- (9)
для U(III)^UO+ ион-. (10)
По предложенному Хилом механизму восстановления ЪТО2~ 1-я стадия соответствует реакции (5). 2-я — сумме двух следующих процессов:
UO2+ +е--^иО2 нижняя часть волны необратимая (11)
LOH3+-е ^=±UOH2+ верхняя часть волны обратимая (12) ИЛИ
UO2+4-e-z=±UO+ (13)
Как видно из представленного механизма, стадия (11) приводит к образованию твердой пленки на поверхности электрода, что обусловливает ее необратимость. Поэтому следующая стадия (12) (13) может протекать только в достаточно кислых растворах, в которых возможно превращение твердой пленки UO2 в ионы UOH3 + или UO2 + .
Стадию (11) Хил подтверждает экспериментальными данными, полученными Кольтгофом и Харрисом, которые наблюдали в почти нейтральных растворах исчезновение волны стадии (12), а также более ранними данными, из которых следует, что электрод из UO2 в растворе UO|‘*' принимает потенциал, близкий к стадии (11), что указывает на осаждение окиси или гидроокиси урана на поверхности электрода.
На том основании, что ни одна стадия восстановления UO2a+ на ртутном капельном электроде не дает значения потенциала полуволны, соответствующего величине нормального потенциала системы U (VI)/U (IV) =+0,404—(+0,407) в [276] (норм. в. э.), Хил делает вывод, что механизмы электрохимических процессов на платиновом и ртутном капельном электродах неодинаковы. Такой вывод в настоящее время вполне подтвержден доказательством образования UO2+ в 1-й стадии процесса восстановления UO^+ на ртутном капельном электроде.
Значительное количество работ посвящено изучению кинетики диспропорционирования UO2+ и влиянию на этот процесс различных факторов (pH, комплексообразования и других) [40, 260, 581, 646,. 665, 685].
Процесс диспропорционирования можно представить следующими реакциями:
2UO2+ + H+ UO:++UO+OH (14)
или
2UO2L + 2Н + ПОГ + UO2+ 4- Н2О, (15)
16»
откуда константа равновесия:
aUO; -a UO-ОН К =-----—:--------- ,
а2ОО2 а Н-
а из значения К следует, что при любых кислотностях UO* очень неустойчив в растворах и диспропорционирует, согласно уравнениям (14) или (15). Диспропорционирование UO+ ускоряется при увеличении кислотности раствора, концентрации UO2r, температуры, а также при комплексообразовании (особенно способствует этому процессу щавелевая кислота); замедляется процесс в присутствии избытка U (IV).
При подстановке значения нормального потенциала по Тит-лестаду [980] в уравнение Нернста для потенциала платинового электрода (в смеси 1 М UO2 + и U (IV)) для ЦО+ получается концентрация порядка 10"6 М. Такая же величина получена Кольт-гофом и Харрисом для равновесной концентрации из их полярографических данных.
Концентрация UO+ в смеси UO2+ и U (IV) увеличивается при освещении. Это согласуется с общеизвестным фактом об увеличении скорости редоксреакций UO2+ при освещении (эффект Беккереля). По мнению Хила, наличие чисто химической стадии в процессе восстановления UO2+ и UO+ до U (IV) в сильнокислых растворах является причиной того, что не происходит обратимое окисление U (IV) при том же потенциале.
Хил и Томас [582] установили по кривым потенциал —время, что получающийся в смеси UO2+ и U (IV) неустойчивый продукт одинаков как в случае электролитического восстановления, так и при освещении и представляет собой ион UOh.
Керн и Орлеман [260, 546] нашли, что скорость диспропорционирования UO+ пропорциональна квадрату концентраций UO+, т. е. K = [UO+]2. Эта квадратная зависимость была найдена для 10 3—10-5 М UO+ в 0,02—0,5 М растворах НСЮ4. Согласно теории Бренстедта, при увеличении ионной силы раствора скорость диспропорционирования UO+ должна расти. Влияние ионной силы на константу равновесия реакции диспропорционирования изучено Нельсоном и Краузом [778].
Приведена [788] зависимость между возрастанием тока за счет диспропорционирования, диффузионным током и константой скорости диспропорционирования. Более точные данные о кинетике диспропорционировании UO+ даны Корыта и Коутецким [676], показавшими, что константа скорости диспропорционирования UO+ в 0,5 М растворе по С10“ пропорциональна концентрации
170
[НС] и /Со = К[Н + ]=1,43-102 моль~~-л-- сек -1, а в0,5М растворе по С1" .содержащем 2-10~3?ожелатины, Ко =2,5 • 102лю.?ь-2 -л2 -сек"1.
Показано [260], что скорость реакции диспропорционирования UO+-hohob значительно больше в растворах H2SO4, чем НС1О4 (при одинаковой концентрации кислот). Скорости реакции диспропорционирования UO+ колеблются в пределах 1,2-10"® до 1,9-10 ~5 г-моль/час в зависимости от концентрации кислоты и ее природы.
А. И. Алекперов и С. И. Жданов [4а] показали, что анионы СС и NO ~ практически не влияют на скорость диспропорционирования UO^• Анионы SOP, F \ С2О42- существенно ускоряют этот процесс. По эффективности действия на процесс диспропорционирования UO^ анионы могут быть расположены в следующий ряд:
F" > С2ОГ >SOP > NO3" >Cl".
При этом ускоряющее действие анионов проявляется только в определенном интервале кислотности.
Авторы предполагают, что причина действия анионов заключается в связывании U (IV), который получается при диспропорционировании UO+, в прочные комплексы и, следовательно, выведении их из сферы реакции, что способствует ускорению диспропорционирования UO+.
По [601а] реакция диспропорционирования является реакцией 1-го порядка не только в отношении Н+-ионов, но и анионов NO-, С1_, Вг“ и J-. Поэтому автор считает, что указанные анионы сначала образуют комплексы с U (V), которые затем подвергаются диспропорционированию.
Скорость диспропорционирования UO+ (К) была также изучена в растворах НС1О4 в тяжелой воде [476]. Так как К в D2O в 1,7 раза больше, чем в Н2О, то авторы считают, что отношение констант равновесия в D2O и Н2Одля соответствующих реакций UO+H-H^' ^-UOOH+ равно примерно 3. Специфическая константа скорости реакции UO2' 7'UOOH2+— UO2 ++UOOH+ в D2O меньше, чем в Н2О.
Было также отмечено, что в сильнокислых растворах наблюдается непрерывное увеличение тока с увеличением отрицательного значения потенциала катода, как это следует из уравнения (5), ибо при этом уменьшается концентрация UO;+ у электрода, и равновесие смещается вправо. В присутствии U (IV) наблюдается уменьшение диффузионного тока с увеличением отрицательного потенциала.
Исследование поведения на ртутном капельном электроде растворов UO+, U (IV) и U (III) показало, что UO+ дает в растворе перхлората при —0,55 в (нас. к. э.) волну окисления с хорошо выраженной областью диффузионного тока. В 0,4 М растворе NaClO4
171
с pH - 3 константа диффузионного тока UO.7 равна.
К —= 1,59 мка ммоль л, ст' 1 t 3 где
id—предельный ток диффузии, мка;
с — концентрация ПОГ, ммоль/л;
т — масса ртути, вытекающая в сек.;
t — время каплеобразования, сек.:
D—коэффициент диффузии, равный 0,70• 10“5слг-сект1.
£1', в узких пределах (0,1—0,5 М) не зависит от концентрации НС1О4, если ионная сила равна 0,5. Подтверждено, что система UO|+/UOr обратима на ртутном капельном электроде и что в пределах pH 2,9—3,5 id=K. [UO+] =const [788].
Нельсон и Крауз [778] также нашли, что UO2+, UO2+ и U (IV) образуют равновесные смеси, константа равновесия которых сильно зависит от pH раствора. (В очень слабокислых растворах Д' ~ 1). По их мнению, U (IV) в кислых растворах находится в виде иона U4+ и при равновесии U4+±2H2OXUOH3+±H3O+ константа реакции Кп =0,21 ±0,02, а для реакции U4 + ±C1'XUC13 + константа Кс 0,63 (при р=0,5) и Кс=7,0 (при р—0). По Кричевскому и Хиндману [685], в растворах НС 1 >0,01 Л! ПС14дает волну с наклоном, равным 0,058, что указывает на обратимость процесса восстановления U (IV) с участием одного электрона. При более низких концентрациях НО эта закономерность нарушается; по-видимому, это можно объяснить наступающим гидролизом U(IV). Потенциал полуволны U (IV) не зависит от изменения ионной силы до ц—3. В перхлоратных растворах £-./2 более положителен, чем в хлорид-ных, что указывает на образование хлоридного комплекса UC13 + , подобно тому как UO^ + образует хлоридный комплекс UO2C1 + .
В разбавленных растворах кислот U (IV) довольно быстро гидролизуется, причем процесс необратим даже при прибавлении концентрированной кислоты; в концентрированных же кислотах U (IV) более устойчив, что известно и из других работ.
В 1 М растворах НС1 и НС 1О4 авторы получили для системы U (III)/U (IV) обратимую катодно-анодную волну с £>/,——0,885 в и —0,873 в соответственно. Обратимость электродных реакций системы U (IV)/U(III) и UO*+/UO2+ доказана также измерениями токов обмена этих реакций [260].
Как известно, потенциалы систем Np (III)/Np (IV) и Pu (111)/ /Pu (IV) также обратимы, и потенциал Pu (III)/Pu (IV) не зависит от концентрации кислоты по крайней мере при концентрации 0,3 М. Pu (IV) существует в растворах (например НС1О4) в виде гидратированного иона; Кричевский и Хиндман [685] на основании своих экспериментальных данных приходят к выводу, что U (IV) и U (III) в растворах НС1О4 и НС1 также существуют в
172
форме гидратированных ионов, а не UO2t и UO', как это принимают Кольтгоф и Харрис и другие [581, 260]. Природа второй и третьей волны UO"+ зависит не только от [Н ] в растворе, но и от природы кислоты, так как анион последней дает комплексный ион с UOV.
Действительно, было показано [8], что высота 1-й волны ЬО|+ даже при низких концентрациях H2SO4 больше, чем это соответствует одноэлектронному процессу, имеющему место в слабых растворах НС1; например, константа диффузионного тока (!</=id/c) в 0,5 N НС1 равна 4,28, а в 0,5 N H2SO4 равна 7,27 мка/ммоль/ л. Это можно было объяснить большей комплексообразующей способностью иона SO;" и восстановлением этого комплексного иона. В сильнокислых растворах 2-й волны имеет значение —0,94 в — в IV НС1 и НС1О4; —1,10 в — в 1 N H2SO4. Из этого следует, что U (IV) также образует комплекс с SO;". Авторы подтвердили это потенциометрическим методом.
В очень кислых растворах (2. N) константа диффузионного тока UO;+ в НС1 и H2SO4 имеет одинаковое значение и соответствует электродной реакции с участием двух электронов.
В связи с аномальным поведением UO;r-ионов в H2SO4 их восстановлению в сернокислых растворах посвящено значительное число работ, особенно в последние годы [88а, 202, 260, бЗОв].
С. И. Синякова [202] показала, что в весьма разбавленных растворах H2SO4 (^1 • 10-3Л4) получаются три волны ПО2+-ионов, соответствующие восстановлению UO*+ в UO+ (1-я стадия), UO+ в U (1У)(2-я стадия) и U (IV) в U (Ш)(3-я стадия). С увеличением концентрации H2SO4 количество волн уменьшается до двух, а при концентрациях H2SO4>0,5 Л1 2-я волна неограниченно растет, вследствие слияния с волной разряда Н+-ионов. Концентрация H2SO4 оказывает влияние на £i/2 и гпр UO*+-hohob. При ее увеличении Е1/2 сдвигается к более отрицательным значениям (от —0,22 до —0,25 в при концентрациях H2SO4 10"2 и 1,0 М соответственно), а предельный ток увеличивается с 2,13 до 2,50 мка.
Более отрицательное значение £1,-2 UO* + в H2SO4 по сравнению с другими кислотами объясняется большей комплексообразующей способностью SO*" по сравнению с ионами С1" и NO~. Это подтверждено также спектрофотометрическими данными [231]. Для подавления максимума применяли тимол (->- 2-10"4%). В его присутствии сумма предельных токов 2-й и3-й волн UO*T в 1-Ю"3 Л! растворе H2SO4 в 2 раза больше предельного тока 1-й волны, хотя каждый из них по величине резко отличается от величины гпр. Это может быть объяснено восстановлением в 3-й стадии гидролизованного иона U (IV). В 1 М H2SO4 восстановление UO2^ протекает с участием двух электронов, что указывает на восстановление UO22~ прямо в U (IV).
173
.Микрокулонометрическим методом установлено, что в суммарном процессе восстановления L’OV в H2SO4 средней концентрации (0,01—0,02 Л1) участвуют три электрона. Нитрат-ионы не оказывают влияния наЕ' , и ЕО2~ в 1 Л1 H2SO4, и поэтому сернокислый фон рекомендован С. И. Синяковой для определения урана в присутствии значительных концентраций нитратов.
Е. А. Каневский и Г. Р. Павловская [88а] нашли, что в растворах H2SO4 различной концентрации (от 0,02 до 4Л') Е., 1-й и 3-й волн почти постоянны, а Е. „ 2-й волны (наблюдающейся до концентрации H2SO4, равной 1 А) изменяется на 0,18 в при увеличении концентрации H2SO4 от 0,02 до 1,0 N. Показано, что Е i/3 [процесс U (V) U(IV)] связан линейной зависимостью с средней активностью H2SO4.
По данным авторов, одним из главных факторов, определяющих скорость диспропорционирования UOA в сернокислых растворах, является концентрация ионов HSO4“ и что основной процесс восстановления UOV в сернокислых растворах протекает по реакции:
uo2so4 + e-^uo2+ +sor.
Исса с сотрудниками [630 в] также указывают на образование трех волн восстановления UO22+ в 0,01 Л! H2SO4. С увеличением концентрации H2SO4 (вплоть до 0,055 М) или при добавлении Na2SO4 или сульфосалициловой кислоты 2-я и 3-я волны сливаются. В 0,25 Л! H2SO4 или при наличии комплексона III в растворе 2-я волна смещается к более положительным потенциалам и сливается с 1-й волной. В этом случае U (VI) восстанавливается, по мнению авторов, до смеси U (V) и U (IV). Предельный ток UO22+ растет с увеличением концентрации H2SO4 вплоть до 1 М, а затем при дальнейшем увеличении ее он уменьшается. Это находится в противоречии с наблюдениями нашими и других исследователей.
Предельный ток UO22+ при —0,6 в пропорционален концентрации U (VI) в присутствии 0,25—0,6 44 H2SO4, но при наличии комплексона III в растворе пропорциональность нарушается. Она лучше сохраняется при низких кислотностях, чем при высоких.
В слсбсщелочных растворах Харрис и Кольтгоф [666] обнаружили, что диффузионный ток 2-й стадии меньше теоретического, и высказали предположение, что UO2OH+ частично’гидролизуется до UO2(OH)2, который не восстанавливается на ртутном капельном катоде. Продукт же гидролиза UO22+ UO2OH+ восстанавливается вместе с UO2~ (2-я волна) до UO2+.
На ртутном капельном электроде даже при положительных потенциалах (4-0,4 в) ни одному исследователю не удалось обнаружить волны окисления U (IV). Однако такая волна была найдена с вращающимся платиновым электродом [211, 366]. Бете [366] нашел, 174
что при высоких плотностях тока скорость окисления U (IV) за-
висит от его концентрации, а при низких плотностях тока скорость процесса растет экспоненциально с увеличением потенциала анода и температуры и не зависит от концентрации U (IV); на основании этих наблюдений автор делает вывод, что при высоких плотностях тока скорость окисления определяется только скоростью диффузии ионов LT (IV) к аноду, а при низких плотностях тока скорость ли
митируется переносом электронов между ионом металла и анодом, т. е. наиболее медленной стадией ступенчатого механизма окисления U (IV). При малых анодных потенциалах скорость переноса электрона определяет, следовательно, всю скорость реакции окисления U (IV).
К. И. Розенталь и В. И. Веселовский [211] нашли, что Е>/2 окисления U (IV) до UOV в 1 N растворе НС 1О4 равен 1,05 в (норм. в. э.) (рис. 27) и что между предельным током U (IV) и концентрацией его
в растворе наблюдается линейная зависимость для интервала концентраций 1 • 10“2— —2-10“4Л1. Наличие в растворе больших количеств UO22+ не мешает определению U (IV). Th(NO3)4 катализирует процесс окисления U (IV).
Что касается механизма анодного процесса окисления U (IV), то авторы считают, что он очень сложный, так как наряду с чисто электрохимической реакцией окисления U (IV) через промежуточную стадию U (V) по элек
Рис. 27. Кривая анодного окисления U(IV) на вращающемся платиновом микроэлектроде (1-Ю-3 М UOSO4 в 1 N НС1О4).
тронному механизму протекает
еще реакция окисления U (IV) кислородом, электрохимически вы
деленном на платине, т. е. при этих потенциалах имеют место
следующие реакции:
Pt + Hso — Pt(O)„c + 2H* + 2« (16)
UO1J-:-Pi(O)a_e— иоГ+Pt. (17)
При этом стадия (17) определяет скорость процесса.
Определению урана по волне окисления U (IV) мешают NO,-, Н2О2 (и Сг3+ в концентрациях более 1 - 10-37H); в отсутствие этих веществ можно определять уран по анодной волне при концентрации до 8 • 10“s М.
Анодные волны окисления U (IV) до U (V) получены в буферных ацетатных растворах. При этом в 0,1 VI ацетате в интервале pH 2,5—4 = —0,2 в (нас. к. э.) и значение его сдвигается к более
отрицательным при pH 5,5. Однако для аналитических целей эта волна окисления U (IV) не применялась [8].
175
Полярографические методы определения урана
В результате исследования поведения UO% , UO2 и U (IV) на ртутном капельном электроде предложено много вариантов полярографического определения урана в различных природных и промышленных объектах. В основном они различаются по примененному электролиту, а также способами предварительного отделения урана от элементов-спутников.
Ион UO2? образует комплексы с различными неорганическими ионами (SO2?, SCN", [Fe(SCN)]4-, СО2? и др.) и большим числом органических соединений (см. стр. 186). Для полярографического определения урана широкое применение получили различные комплексообразующие вещества, в особенности при анализе материалов сложного состава (руды, породы, концентраты).
Минеральные кислоты и их соли в качестве фона
Харрис и Кольтгоф [664 ] рекомендуют производить для количественного определения урана полярографирование его на фоне 0,1 Л1 КС1—0,01 М НС1, в присутствии 10-4% тимола. Диффузионный ток лучше измерять при —0,5 в (отн. нас. к. э.) и вносить поправку на остаточный ток. Для устранения влияния Fe3+ авторы применяют солянокислый гидроксиламин (восстановление Fe3+ до Fe2+).
В качестве подавителя максимума авторы применяли также кофеищ впервые предложенный Карузерсом [413]. Однако этот фон может быть применен лишь в отсутствие ванадия, молибдена, меди и других ионов, имеющих близкие с ураном значения потенциалов полуволны.
Полярографическому определению урана в различных неорганических кислотах в качестве электролита посвящены также исследования С. И. Синяковой [245] и Агуаш да Сильва[302]. Измерения проводились с хлоридом, сульфатом, ацетатом и нитратом уранила, причем фоном служила обычно кислота с тем же анионом, что и взятая соль уранила. Через растворы продували азот или водород, а для подавления максимума на полярограммах прибавляли тимол. Во всех случаях найдена прямолинейная зависимость между концентрацией урана и величиной диффузионного тока [245].
Стрэбл [942] использовал в качестве фона для определения урана в присутствии большого избытка железа раствор солянокислого гидроксиламина в кислой среде. В 2 N растворе солянокислого гидроксиламина уранил-ион дает 2 волны с £V2= — 0,28 в и =—1,08 в (нас. к. э.). Соотношение высот 1-й и 2-й волны
равно 1 : 2,5, но автор не дает объяснения механизму восстановления.
176
.Между величиной диффузионного тока I-й волны и концентрацией урана в растворе существует линейная зависимость, что позволяет использовать этот фон для количественного определения урана. Большое содержание железа не мешает определению урана, так как Fe3" восстанавливается солянокислым гидроксиламином до Fe2. Ванадий сильно повышает волну урана, как, впрочем, и на фоне других электролитов.
Гелестам [594] показал влияние ВгО3 , JOF, СгОр и VO3~ на полярографическое определение урана на фоне солянокислого гидроксиламина. Было выяснено, что ВгО", JO” и СгО^" не влияют, a VO3 мешает определению. Си, Sb, As, Bi, Pb, Mo и Sn должны быть удалены, например, осаждением посредством H2S, a W можно осадить сульфатом хинина. Растворы, содержащие уран и вышеуказанные элементы, могут быть проанализированы полярографически, если отделить примеси, как уже указывалось, и если содержание VO7 и WO^” в них мало.
Автор рекомендует и другие методы отделения мешающих примесей. По одному варианту, Fe, Си, As, Bi, Pb, Sn и Mo осаждают электролитически на ртутном катоде. Но W должен при этом отсутствовать, ибо в его присутствии часть урана осаждается в процессе электролиза. Присутствующие при этом BrO3 , JC)3 и СгО.К полностью восстанавливаются. Затем VO3 осаждают купфероном и купферонат извлекают в СНС13. Вместе с V осаждаются и другие элементы.
Перед полярографированием раствор насыщают K2SO4, нагревают до кипения для удаления СНС13 и охлаждают в токе азота. Большой избыток купферона мешает при полярографировании, так как он искажает кривую силу тока — напряжение, и поэтому его необходимо удалить. Кроме того, так как концентрация SO? влияет на высоту волны урана, необходимо поддерживать ее постоянной.
По второму варианту, VO3 осаждают купфероном, после чего UCF+ восстанавливают до U (IV) посредством СгС12; U (IV) осаждают купфероном и осадок купфероната U (IV) извлекают СНС13. Экстракт выпаривают, остаток обрабатывают HNO3 и НС1О4, затем растворяют в смеси НС1О4—КС1О4 и снимают полярограмму. Все эти варианты весьма трудоемки.
Определены значения коэффициентов диффузии (D) UO^ + : в 244 растворе солянокислого гидроксиламина D=0,45-10-5 си2-сек"1; в насыщенном растворе K2SO4=0,78-10“5 саг сек"в 0,04—0,14 М НС1О4 0,73-10 5 слт-ссс1.
С. И. Синякова [229] предложила определять уран в полуна-сыщенном растворе сернокислого гидразина. Получаемая волна с Ег3=—0,20 в (отн. нас. к. э.) четкая и воспроизводима. Между величиной диффузионного тока и концентрацией урана в растворе
^2 Аналитическая химия урана 177
сохраняется линейная зависимость. Раствор сернокислого гидразина особенно удобен при определении урана в присутствии железа <и значительных количеств меди), так как сернокислый гидразин восстанавливает Fe3_r до Fe2T, а последний восстанавливается на ртутном капельном электроде при значительно более отрицательном потенциале, чем уран и поэтому не мешает его определению. Если меди содержится мало, то ее можно отделить электролизом на платиновом или ртутном электродах; можно также отделить уран от меди, переосаждая его 2—3 раза аммиаком, не содержащим СО2. При значительном содержании меди в виде сульфата она выпадает в осадок при кипячении с сернокислым гидразином в виде соединения CuSO4• (N2H5)2SO4.
Сернокислым гидразином нельзя пользоваться при определении урана в присутствии ванадия, так как волна урана при этом увеличивается и увеличение ее зависит от количества ванадия в растворе даже при малых содержаниях последнего. Поэтому при применении сернокислого гидразина в качестве фона ванадий необходимо отделять от урана.
Килнер [8] использовал фон 0,1 N HC1±2VNH2OH-HC1 для определения малых количеств урана в осадке Fe(OH)3, получающегося при очистке урана, а также в растворах после осаждения диураната аммония. Точность метода, по данным автора, ~-20?о (отн.) для 1 • 10~4 г урана в Fe(OH)3. По нашему мнению, указанная точность является сильно заниженной, так как автор работал, очевидно, не в оптимальных условиях.
Тишкоф [8] применил в качестве фона 0,17V НС1 при определении урана (VI) в тетрафториде урана. Измерялся диффузионный ток 1-й волны, для которой Е>12=—0,20 в (нас. к. э.). Точность метода ±396 (отн.).
Для полярографического определения урана в растворах, содержащих плутоний, предложен метод [740], основанный на окислении обоих элементов до шестивалентного состояния с последующим восстановлением плутония до трехвалентного посредством солянокислого гидроксиламина- (уран (VI) не восстанавливается). Уран определяют по катодной волне восстановления UO;+ до ПОД
Многие исследователи применяли в качестве фона раствор 1—2 N H2SO4. Этот фон имеет то преимущество, что 1-я волна в нем соответствует двум электронам и, следовательно, получается вдвое более высокой, чем в растворах НС1 средней концентрации.
Шил и Уотерс [8] в качестве фона применяли 1 N раствор H2SO4, содержавший 1 • 10’3% желатины. На этом фоне уранил-ион дает хорошо выраженную волну, высота которой прямо пропорциональна концентрации урана в растворе и значительно больше, чем в 1 N НС1. Точность определения урана ±2% (отн.).
Квятковский и др. [8] также применяли 1,5 N H2SO4 в качестве фона для определения урана.
178
Баленджер [8], определяя уран в атмосферной пыли, в качестве фона применил серную кислоту. После растворения пыли, собранной электрофильтром, и озоления фильтра медь отделялась в виде сульфида, а затем снималась полярограмма в 5%-ной H2SO4. Содержание урана определялось по градуировочной прямой. Для количеств урана 0,1—0,4 мг средняя сшибка составляет --8— 9 мкг.
И. Е. Старик и А. С. Старик-Смагина [244] для определения урана в количествах 8-Ю-4—2-10“5а в различных природных объектах применили для его отделения соосаждение с алюминием в виде гидроокисей с последующим отделением урана от алюминия и железа с помощью карбоната аммония. Полярографирование урана производилось на фоне А1С13 и 0,1 М НС1 после повторного соосаждения его с алюминием. Если в материале содержался ванадий, уран предварительно отделяли от него осаждением фосфатом, или ванадий осаждался купфероном. Этот метод очень трудоемкий и должен приводить к потерям некоторого количества урана, вследствие большого числа осаждений и фильтрований.
Другой метод, предложенный И. Е. Стариком и сотрудниками [243], основан на восстановлении урана до U (IV) и выделении его в виде фторида; носителем служит торий. При этом могут вместе с ураном осесть фториды кальция, свинца и редкоземельных элементов, но они не мешают при полярографировании. Метод применим для определения урана в породах и водах.
Навеску вещества, соответствующую содержанию урана 10“4 г, разлагают смесью концентрированной H2SO4 и HF и выпаривают. Нерастворимый остаток отделяют, раствор кипятят, полуторные окисли осаждают аммиаком (без СО2) и осадок отфильтровывают, а затем растворяют (в 50-миллилитровой плоскодонной колбе) в 15%-ной H2SO4 или НС1. Добавляют 20 г цинка и в течение 30 мин. раствор с цинком несколько раз перемешивают. Затем раствор отфильтровывают через воронку с ватой в платиновую чашку, в которую предварительно наливают 30 мл 30%-ной NH4F и 10 мл 40%-ной HF. Колбу и воронку с ватой промывают 25 мл 15%-ной H2SO4 или НС1 и промывные растворы присоединяют к фильтрату. Приливают в платиновую чашку 1 мл 0,5 N сульфата или хлорида тория. При этом сразу выпадает осадок фторидов тория и урана. Раствор с осадком перемешивают и оставляют на 1 час. Затем осадок отфильтровывают через эбонитовую или парафинированную стеклянную воронку и промывают его раствором плавиковой кислоты (1 : 1). Осадок смывают горячей водой в ту же платиновую чашку, в которой производилось осаждение. Прибавляют концентрированные HNO3 и H2SO4 и выпаривают раствор до появления паров SO8. Полученные сернокислые соли тория и урана растворяют в дистиллированной воде, и раствор переносят в стакан. Торий и уран (последний находится в растворе в виде UOg+) осаждают аммиаком (безСО2). Затем осадок растворяют в 10%-ной НС1, раствор переносят в бюкс, выпаривают его досуха и осадок растворяют в 5—10 мл 0,5 N НС1, 0,5 N по хлориду или сульфату тория. Раствор сливают в электролизер, продувают очищенный азот или водород (для удаления кислорода из раствора) и получают полярограмму, начиная от 0 приложенного напряжения. Снимают также полярограмму 1,2—2,4-10~4V стандартного раствора урана в 0,5 N НС1 или H2SO4 на фоне 0,5 N хлорида или сульфата тория и вычисляют содержание урана сравнением высот волн исследуемого и стандартного растворов.
12*
179
При определении урана в природных водах следует определенный объем воды, подкисленный НО, выпарить до малого объема, затем добавить FeO3 в качестве носителя, осадить железо с ураном аммиаком (без СО2), осадок растворить в кислоте и далее продолжить анализ, как указано выше.
Полярографическое исследование поведения урана в растворах фторидов [714] показало, что образующиеся соединения UO2FT и UO2F2 восстанавливаются обратимо на ртутном капельном электроде или легко и быстро диссоциируют на ион UO^, который, как известно, восстанавливается обратимо.
Ионы UO2F^_ и UO2F" восстанавливаются на ртутном капельном электроде необратимо.
В аналитических целях диспропорционирование UO+ в растворах фторида можно использовать для увеличения чувствительности определения, так как при этом регенерируются UO^y в результате чего увеличивается предельный ток.
Полярография фосфатных и полифосфатных комплексов урана
Исследовано полярографическое поведение урана в фосфорнокислых и полифосфатных растворах. Однако в истолковании полученных данных имеется ряд противоречий [577, 6306, 737а, 967а].
Одни исследователи наблюдали две волны при восстановлении UCF + в растворах Н3РО4 как высоких, так и средних концентраций [967а]. Например, в 7,5 М. Н3РО4 найдено, что Еу,=—0,12 в и Е ,=0,58 в [577, 737а]. По данным других [6306], при наличии желатины в растворе обнаруживается только одна волна, соответствующая восстановлению U (VI) в U (IV).
В отсутствие желатины на волне часто появляется максимум, который подавляется желатиной при концентрациях Н3РО4>1 44. При увеличении концентрации Н3РО4 потенциал полуволны сдвигается к более положительным значениям, а в 10 44 растворе Н3РО4 восстановление начинается при 0. Авторы [6306] показали, что характер полярограмм уранил-ионов изменяется в зависимости от концентрации Н3РО4, самого UO;+, а также от наличия желатины. Например,в 0,1 44 Н3РО4 при концентрациях UO^+ >0,5 ммоль после достижения предельного тока перед волной разряда водорода появляется минимум и .сдвигается к более отрицательным значениям с увеличением концентрации урана, но диффузионный ток, измеренный при —0,6 в, пропорционален концентрации урана в пределах 0,5—3 лшоль (в отсутствие желатины) и 0,5—4 ммоль (в ее присутствии). В 2 и 5 44 Н3РО4 получается одна волна. Е^тг зависит от концентрации UO- + и равен —0,15 в и —0,11 в соответственно. Диффузионный ток ПОД пропорционален концентрации его в 2,5 и 10 44 Н3РО4. По [6306], в растворах любой концентрации Н3РО4 диффузионный ток уменьшается от добавления желатины. Тем не менее фосфорная кислота является удобным фоном 180
для определения урана в пределах концентраций 0,5—4 ммоль. В 0,1 М Н3РО4 отношение id с равно около 2,0 для концентраций урана в пределах 0,5—2,5 ммоль. В 0,6—1 44 Н3РО4 появляется вторая волна урана, причины образования которой авторы не объясняют, игнорируя при этом возможный процесс диспропорционирования образовавшегося UOy, имеющий место в концентрированных растворах Н3РО4. Однако суммарная высота волн соответствует восстановлению U (VI) в U (IV).
Авторы считают вероятным образование комплекса [UO2-(Н3РО4)3]~ в солянокислом растворе уранила при добавлении Н3РО4. По [967а], более оптимальным фоном для анализа урана является 6 М Н3РО4, содержащая менее 0,5 44 H2SO4 и 6,0125% желатины. Автор изучил также влияние различных факторов на скорость диспропорционирования UOV в растворах Н3РО4.
В растворах триполифосфата с pH 9 UO^+ дает одноэлектронную волну восстановления с Ei/„, равным —1,07 в (нас. к. э.) [942а, 943]. С изменением pH Ей, и /пр резко меняются. Например, в незабуференных растворах с pH 3 и 9 Ei/2 равен —0,86 в и —1,10 в соответственно, а константа диффузионного тока в 0,05 44 Na5P3OI0 —0,2 44 КС1 равна 2,44 и 1,65 при pH 5 и 9 соответственно.
Природа и концентрация электролита также оказывают влия-ниенаЕ1;,и гпр. Например, в 0,04 М Na5P3O10 с pH 9 Е. .равен—0,86 <? при наличии 2,6 44 КС1 и —1,27 в в его отсутствие.
Авторы указывают также на влияние поверхностноактивных веществ (желатины, гуммиарабика, камфоры) на Еу, и znp UO^ + .
Для определения урана авторы [6306] предложили в качестве фона щелочной раствор триполифосфата. В растворе, содержащем 0,1 44 Na5P3O10 + l М KNO3+ 0,005% камфоры, соблюдается линейная зависимость между /пр и с урана в пределах концентраций 2• 101—5-10~3 44 урана.
МоО*- и WO*~ полярографически не активны в щелочном растворе. Cr3 + , Со2+, Ni2+, Мп2+ и Zn2 + не оказывают влияния, так как образуют соответствующие комплексы с триполифосфатом, которые восстанавливаются при весьма отрицательных потенциалах и поэтому не мешают определению урана. РЬ2+ и Вг{+ в малых концентрациях не мешают, ибо восстанавливаются при более положительных потенциалах, чем уран (РЬ2 при —0,65 в, Bi3+ при —0,63 в, UO-;+ при —1,05 в).
В материалах сложного состава авторы предлагают отделять уран осаждением фосфатом в присутствии комплексона III.
Рекомендована следующая методика. Кислый раствор, содержащий уран, подщелачивают аммиаком, прибавляют Зг комплексона Ш и кипятят раствор 5 мин. Прибавляют буфер, состоящий из эквимолярной смеси ацетата аммония — уксусной кислоты, 10 мг бериллия в качестве соосадителя (в виде хлорида, при малых содержаниях урана), 2 г (NH4)2HPO4 и снова кипятят 10 мин. Выдерживают раствор с осадком 30 мин. на горячей водяной бане и отфильтровывают оса док. Последний промывают раствором, содержащим 2%-ный раствор NH4NO3 и 1%-ный комп
181
лексон III, и затем растворяют в HNO3(1 : 4). Добавляют аммиак до pH 3—5, вводят 2 г безводного Na5P3OI0 и 1 мл 0,25?о-ной камфоры и доводят до 50 мл. Аликвот переносят в электролизер и снимают полярограмму. Этот метод дал удовлетворительные результаты при определении урана в минералах после их разложения (тантало-ниобаты, монациты). Навеска образца должна содержать не менее 2—3 мг урана.
По данным [981а], на фоне, содержащем 0,1 М Na5P3O104-0,l М (NH4)2CO3, UOn-ионы образуют две волны, соответствующие восстановлению U (VI) в U (V) и U (V) в U (IV). В качестве подавителя максимума добавляют «твин-80». £\,2и id каждой волны изменяются в зависимости от концентрации обоих компонентов фона. 1-я волна получается четкой, 2-я выражена плохо. На смешанном фоне с содержанием каждого компонента в концентрации 0,1 М при pH 9,6 величина тока 1-й волны, несмотря на то, что он включает кинетическую составляющую, пропорциональна концентрации урана в пределах 1-Ю-3—1-10"4 М.
На предложенном фоне можно определять уран в присутствии Со, Mn2+, Zn, Fe3 + , Си, Cd, Cr3 + , Pb, Al, ZrO2+и МоО2-- Мешает определению Ni.
Полярография карбонатных комплексов урана
Полярографическое поведение иОг+ в растворах (NH4)2CO3 изучал Стрэбл [942], который показал, что образующийся по реакции:
UO2 (NO3)2 4- 3 (NH4)2 СО3 (NH4)4 [UO2 (CO3)J + 2NH4NO3 карбонатный комплекс восстанавливается на ртутном капельном кауоде, образуя две волны (рис. 28)cEi/2=—0,83 в (/) и £i/2=—1,45 в (//) (нас. к. э.). Однако предложенный автором механизм восстановления комплекса до U (II) оказался неверным, как это показано нашими исследованиями [229], а позже Харисом и Кольт-гофом [578]. Прежде всего неизвестно ничего о существовании в растворах иона U (II), а, кроме того, U (IV) образует комплекс с (NH4)2CO3, однако он не восстанавливается на ртутном -капельном катоде (вероятно, вследствие гидролиза этих комплексов). По мнению Харриса и Кольтгофа, 1-я волна UO1+ в щелочных растворах соответствует восстановлению комплексного аниона или при меньших pH иона UO2OH + . 2-я волна вероятнее всего обусловлена восстановлением оксикомплекса UO^ до оксикомплекса U (IV).
Эти же авторы исследовали восстановление UO® + в растворах Na2CO3 и нашли, что UOj+ образует только одну волну, соответствующую процессу с участием одного электрона. В 0,1 N Na2CO3 Б-.'.=—1,1 в (нас. к. э.), а при увеличении концентрации Na2CO3 до 1 N Ei/2 сдвигается R более положительному значению, равному —0,9 в, и волна становится менее четкой. На полярограмме UOV в Na2CO3 обнаруживается еще одна небольшая волна, которая по
182
высоте приближается к 1-й по мере увеличения концентрации Na2CO3. Смещение , волны UO;- указывает на то, что в растворах Ха2СО3 ион UO;+также восстанавливается в виде комплексного аниона. Карбонатные комплексы UO;+ и U (IV) обладают большой прочностью. При добавлении KCN или Na2SO3 они не разрушаются [942].
Рис. 28. Полярограмма уранил-ионов на фоне полунасыщен-ного раствора (NH4)2CO3.
1 — 1,25»10-3 М урана; 2—7,5-Ю-1 М урана.
А. И. Стабровский [236а] показал, что образование одной или двух волн на полярограммах UCP+-hohob в карбонатных и би-карбонатных растворах связано главным образом с гидролизом комплексных карбонатных ионов U (V), так как продукты гидролиза этих ионов, а также соответствующих ионов U (IV), неспособны восстанавливаться на ртутном капельном электроде. Они могут на нем только окисляться.
1-я волна (18) восстановления ,U(VI) с Е^2 от —0,7 до—0,9 в получается во всех карбонатных растворах и, по мнению автора, отвечает реакции
[UO2 (CO3)3r- 4- е- = [UO2 (CO3)J3-, (18)
2-я волна (19) с Е^ от —1,3 до —1,7 в, отвечающая реакции
[UO2 (CO3)J5 - 4-е- =ио24-зсог, (19)
появляется только в растворах (NH4)2CO3 и бикарбонатов при условиях менее благоприятных для гидролиза комплексных карбонатных ионов U (V). Этому способствует понижение температуры и pH раствора, а также повышение концентрации U (VI), карбонат-и бикарбонат-ионов в растворе. При концентрациях урана >10“4 М на 2-й волне образуется максимум.
1S3
Предельные токи урана зависят от состава раствора и изменяются в весьма больших пределах в зависимости от концентрации карбоната и урана.
Карбонатный комплекс U (IV) окисляется на ртутном капельном электроде при напряжении от Н-0,031 до —0,094 в в зависимости от pH и ионной силы раствора. На анодных кривых также образуется максимум (в бикарбонатных растворах). Электродные процессы восстановления и окисления карбонатных комплексов урана протекают необратимо.
Несмотря на то, что Харрис и Кольтгоф не рекомендуют пользоваться растворами карбонатов в качестве электролитов при количественном определении урана, считая, что в этих растворах не соблюдается пропорциональности между величиной диффузионного тока и концентрацией урана в растворе, карбонаты нашли широкое применение в полярографии урана, в особенности в сочетании с другими комплексообразующими веществами. При попытке определить уран на фоне (NH4)2CO3 (полунасыщенный раствор) в присутствии большого избытка железа Стрэбл обнаружил, что даже малое содержание железа мешает определению урана на этом фоне, так как оно тоже образуеткомплексное соединение с (NH4)2CO3, восстанавливающееся на ртутном капельном катоде в 2 ступени, причем 1-я имеет более положительное значение Е1Д—0,47 в), чем 1-я ступень соответствующего комплекса урана (Еу2=—0,83 в) и, следовательно, определение урана в этих условиях невозможно.
Пршибил и Блажек [819] использовали способность урана восстанавливаться в виде комплексного карбонатного иона для его определения в присутствии Ni, Со, Zn, Мп, А1, Сг (III), Be, Ti, которые связывают в прочные комплексы с комплексоном III, не мешающие определению урана. При этом в растворах (NH4)2CO3 (с концентрацией более 1 VI) комплексный ион уранила [UO2(CO3)3]4-дает две волны, потенциалы которых не смещаются при добавлении комплексона III, тогда как указанные выше элементы, образуя с комплексоном III прочные комплексы, восстанавливаются при значительно более отрицательных потенциалах, чем в (NH4)2CO3. Однако определению урана в этих условиях, по указанию авторов, мешают РЬ и Си, а также Fe и V (как это было установлено нашими опытами). Поэтому указание Иса [812] о возможности применения волны иОг+ на фоне 0,2 М (NH4)2CO3, содержащем 0,02 М комплексона III, для его определения при наличии в растворе Fe (III) и Си (II) представляется нам весьма сомнительным.
Долежалу и Адаму [471 ] удалось устранить влияние железа добавлением в раствор еще одного комплексообразующего вещества— 1,2-диоксибензол-3,5-дисульфоновой кислоты (тирон). В растворе (NH4)2CO3, содержащем тирон, уранил-ион образует две волны: с Е>—0,60 в и E'.'i2=—0,91 в, т. е. ЕС.,обеих волн сдвигаются к более положительным значениям, чем в растворе одного (NH4)2CO3 184
(Ei „=—0,8 в; Ex 2=—1,4 в). С увеличением концентрации UO^ обе волны растут, однако авторы показали, что 1-я волна является кинетической, и она ограничена, по-видимому, кинетикой образования комплекса урана с тироном или кинетикой диспропорционирования UOF в иОг+ и U (IV).
Для определения урана в присутствии железа авторы рекомендуют фон 1 М. (NH4)2CO3 +1?о-ный тирон, а при наличии в растворе Pb, Bi и Си применяют фон 1 М (NH4)2CO3 +1%-ный тирон +0,02 М комплексон II 1+0,05 М KCN. На этом фоне Е^, UOg~= =—0,61 в, а Ё',-~ 0,91 в; Еу2 Fe3+=—1,21 в; F 2РЬ2+=—1,11 в и Еу, Сц-’ =—1,31 в. Следовательно, обе волны урана предшествуют волнам указанных комплексных ионов. Кроме того, авторы нашли, что на фоне 1 N NaOH и тирона уранил-ион образует одну волну с Ei/2=—1,11 в (нас. к. э.), соответствующую одноэлектронной реакции: комплекс UO^+e--^ комплекс UO+. Но на этом фоне Еуз Fe3+ очень близок к значению Еу2 UO^.
Однако все предложенные варианты определения урана на фоне (NH4)2CO3 в комбинации с другими комплексообразующими веществами не учитывают влияния V и Мо — элементов, оказывающих наибольшее влияние при полярографическом определении урана.
Хотя Власак и др. [997] указывают, что им удавалось определять малые количества урана (до 0,8%) в рудах и породах в присутствии железа на фоне Na2CO3 (после сплавления образца с Na2CO3, растворения плава и добавления Na3SO3), однако сомнительно, чтобы они получали достаточно удовлетворительные . результаты. При меньших содержаниях урана они отделяли его-от других элементов экстрагированием уранил-нитрата в эфир.
Органические кислоты и их соли в качестве фона
Полярография тартратных, комплексов урана
Хайт [8] применил солянокислый гидроксиламин в сочетании с тартратом натрия в качестве фона при полярографическом определении урана и нашел, что Мо и V не мешают при содержании эквивалентного объема 10%-ного тартрата натрия. Оказывают влияние Си и РЬ. Волна Си2+ сливается с волной урана при pH 2,7, а волна свинца — при pH 5. Для подавления максимума применялась желатина в концентрации 1,5-10~3%. Однако в рудах автор определял уран без отделения Си и РЬ, применяя электролит, состоящий из 1 N NH2OH-HC1 и 5%-ного тартрата натрия (или цитрата аммония) с pH 4—5,5. Моррисон и Хайт [8] определяли следы урана на этом фоне после отделения его от Си, Fe, Ni и Сг эфирной экстракцией.
185
0,1 М раствор битартрата натрия также дает возможность получать раздельные волны Си24-, UO^ и РЬ2“, но определению урана мешают Мо и Ti; фосфат-ионы не оказывают влияния. Для подавления максимума применялась метил-целлюлоза (0,002% в конечном объеме) [8].
Шалгоский [904] также применял тартратный фон для полярографического определения малых количеств урана. Наиболее удобным электролитом является, по его мнению, раствор 12?6-ный по Na2C4O6-2Н2О, 0,3%-ный по NaCl и 0,01%-ный по протеоз-пептону (подавитель максимума). В этом растворе Еу2 UO2+= =—0,44 в (отн. нас. к. э.) и определению урана мешает только молибден (Ец2=—0,49 в). Константа диффузионного тока урана равна 2,66 и между величиной диффузионного тока и концентрацией урана наблюдается прямолинейная зависимость в пределах концентраций урана от 10-3 до 10~4 М. Автор обнаружил, что диффузионный ток уранила уменьшается, если растворы нитрата уранила в процессе анализа выпаривают с концентрированной серной кислотой. Поэтому автор рекомендует пользоваться хлорной кислотой для удаления нитрат-ионов.
При анализе образцив'перхлоратный раствор, не содержащий молибдена, выпаривают до 0,1 мл и затем прибавляют 0,4 мл 60% -ной НС1О4. Раствор переносят в мерную пробирку емкостью 5 мл, туда же сливают промывные воды (-^2 мл). Прибавляют 0,5 мл электролита и через 2 мин. вносят 1 мл 5 М раствора H2SO4. Добавляют воду до метки и взбалтывают. Раствор сливают в электролизер, продувают азот или водород (-^5 мин.) и получают полярограмму от 0 до —0,6 в, применяя в'качестве анода слой ртути. Концентрацию урана находят по градуировочному графику. Для 100 мгк урана ошибка равна 1,5% (отн.), для 10 'мкг = ±3%',(отн.). Метод пригоден для определения урана в рудах.
Полярография цитратных комплексов урана
С. И. Синякова [229], Нойман и др. [780] исследовали полярографически систему уранил-цитрат в кислых и щелочных растворах. Количество образующихся на полярограммах волн в растворах, содержащих UO2'' и цитрат (при избытке последнего), зависит от pH. При pH 2,6—4,6 наблюдаются 3 волны (рис. 29), из которых 1-я, по мнению авторов [780], обусловлена восстановлением комплекса U (VI) до U (V), а 2-я восстановлением U (V) до U (IV). Для 3-й волны не дано объяснения; во всяком случае она не соответствует дальнейшему восстановлению урана, ибо U (IV) с цитратом в. этих же условиях не образует волны восстановления.
Цитрат аммония (5О?6-ный раствор) позволяет получать раздельные волны Cu2 + , НОЦ и РЬ2+ при pH 4—5 [8]. Уменьшение pH раствора приводит к слиянию волн UO;+ и РЬ2+, а при увеличении pH сливаются волны UCP+ и Си2+. По данным автора, Mo, Ti и V и небольшие количества Fe3+ не мешают определению урана. Наличие фосфат-ионов в растворе вызывает слияние волн 186
Pb и U0’+, так как последний образует комплекс с фосфат-ионами, Е.2 которого сдвигается к более отрицательным значениям. Для подавления максимума на этом фоне рекомендуется прибавлять метил-целлюлозу в концентрации 2-10-3?о. j
Рис. 29. Полярограмма 4,2-10 4 М раствора уранил-ионов в растворе 0,34 М лимонной кислоты 4-0,16 М цитрата аммония; pH 5.
Полярография ацетатных комплексов урана
ПОД образует комплексы в ацетатных буферных растворах; например, в растворе ацетат натрия + уксусная кислота появляется волна UO+ при 4-0,004 в (отн. в. э.), причем Е,;2 не зависит от кислотности раствора, но зависит от концентрации ацетат-иона. Реакция протекает с участием одного электрона и обратима, т. е. восстанавливается ацетатный комплексный ион уранила с образованием соответствующего комплексного иона UOo. Причем диспропорционирование UO2r в ацетатном буфере ничтожно мало и им можно пренебречь.
В ацетатном буферном растворе была получена также анодная волна окисления U (1У)-комплекса при +0,150 в (отн. в. э.). На Е^2 этой волны оказывает влияние как концентрация ацетата, так и кислотность раствора. Эта волна также соответствует обратимому процессу с участием одного электрона, что указывает на окисление ацетатного комплекса U (IV) до соответствующего комплекса UO; [979].
187
Из полярограммы (рис. 30) раствора, содержащего 1 • 10-3 М UO1+, 2-10“3 М U (IV) в 0,054 М растворе ацетата натрия _]_0,3 Л1 уксусной кислоте-у0,1 М КС1 (pH 3,6) следует, что 1-я (анодная) волна соответствует окислению U (IV) до UO^(/); 2-я (катодная) — процессу U (VI)— U (IV) (//);
Рис. 30. Полярограмма раствора 0,054 М ацетата натрия -4-0,3 М уксусной кислоты 0,1 М КС1, содержащего 1-10~3 М UOj;+ и 2-10“3 М U(IV); pH 3,6.
3-я — восстановлению UCV до U (IV) (///) и 4-я—восстановлению U(IV) до U (III) (/V), которая частично связана с волной восстановления UO7~^ U (IV).
На основании полярографических данных было установлено, что если концентрация ацетата <0,05 Л4 при 1 • 10-3 М. общей концентрации урана, то UO и UO2+ реагируют с двумя молекулами ацетата (вместо трех), a UO2’ совсем не образует комплекса. В этих условиях зависимость потенциала полуволны от значения pH опреде-вплоть до того его количества, комплексов, не более
ляется концентрацией ацетата которое обеспечивает образование с иОг+ чем би ацетатных.
Полярография комплекса урана с аскорбиновой кислотой
Аскорбиновая кислота, как указывает Шушич и др. [9511,
является хорошим фоном для ления урана. В 0,5 М. растворе 3,5—4,0 уранил-ион дает четкую волну с = — 0,36 в (нас. к. э.) (рис. 31), и диффузионный ток урана прямо пропорционален концентрации его в растворе. При более высоких значениях pH потенциал полуволны UC>2+ сдвигается к более отрицательным зна
полярографического опреде-аскорбиновой кислоты с pH
чениям, что указывает на то, что
В КИСЛЫХ растворах комплекс UO Рис. 31. Полярограмма уранил-ионов
с аскорбиновой кислотой частич- на Фоне аскоРбинов°й кислоты, но диссоциирован. По указанию авторов, Fe, V, Мо и Сг не мешают определению урана на фоне аскорбиновой кислоты, так как они восстанавливаются до более
низких валентных состояний. Ti (IV) образует комплекс, который
188
восстанавливается при более отрицательных значениях потенциала, чем комплекс с UO*-1-, и, как указывают авторы, не мешает определению урана. Си2+ дает волну восстановления на этом фоне с Ei з=—0,3 в и должна мешать, хотя авторы указывают, что небольшие количества меди не влияют на определение урана.
Шушич и др. применили этот фон для определения урана в рудах с содержанием последнего 0,01—0,1%. Однако следует отметить, что анализированные ими руды содержали мало Ti, V и Си. При 0,01—0,05%-ном содержании урана ошибка составляет -^-5—9?6 (отн.), для больших количеств точность увеличивается. Мешают определению урана также Те, Т1, РЬ и значительные количества Sb.
Полярография купферонатных комплексов урана
Элвинг с сотрудниками [490 , 491] произвели детальное полярографическое исследование системы уранил-ион — купферон .
На полярограмме этой системы в 10%-ном растворе H2SO4 они наблюдали при небольших концентрациях урана и купферона две волны, которые, по их мнению, соответствуют восстановлению U (VI) в U (V) и купферона до фенилгидразина соответственно. При возрастании концентрации купферона 1-я волна уменьшается и, наконец, совсем исчезает, а 2-я волна увеличивается и сдвигается к более отрицательным потенциалам.
Интересны также полярографические данные, полученные Руф-сом и Элвингом [863], показавшие существование купферонатного комплекса урана (III) с устойчивостью такой же или даже большей, чем купферонатный комплекс урана (IV).
Полярография комплекса урана с тираном
Исследовано также полярографическое поведение комплекса, образуемого ионами уранила с тироном (двунатриевая соль 3-5-дисульфокислоты, 1,2-диоксибензола) [955]. Показано [871], что в интервале pH 2—3,5 комплекс восстанавливается обратимо с участием одного электрона. При анализе волны установлено также, что
^=1,1, п
где р — координационное число комплексного иона U (VI); q — координационное число комплексного иона U (V); п — число электронов.
189
На этом основании авторы считают вероятным следующий электродный процесс:
Потенциал полуволны комплекса урана с тироном зависит от pH и от концентрации последнего. В табл. 28 представлена зависимость £i/2 для 1 • 10-4 Л4 раствора урана с ионной силой ц=2,55 (добавлением NaCIO4) и pH 2,2, содержащего 0,01% желатины, от концентрации тирона.
Таблица 28
Зависимость потенциала полуволны комплекса урана от концентрации тирона
Концентрация тирона, моль;л /2
0,2 —0,183
0,3 —0,195
0,4 — 0,203
Рис. 32. Полярограмма уранил-ионов на фоне 0,05 М тирона, в 0,25 М растворе NaOH.
В аммиачном растворе (0,2 N по NH4OH, 0,2 N по NH4C1 и 0,05 М по тирону) для UO1+ перед основной волной (Ех[=~ 1,13 в) получается еще маленькая волна с максимумом (Ei[2=—0,62 в). В растворе, 0,05 М по тирону и 0,25 N по NaOH, Е,/2 UO^ равен —1,24 в (рис. 32). В определенных условиях между величиной диффузионного тока комплекса и концентрацией урана наблюдается прямолинейная зависимость, что позволяет использо
190
вать тирон в качестве электролита для полярографического определения урана. Константа диффузионного тока урана в растворе, 0,05 М по тирону и с pH 3, содержащем 0,01 % желатины, имеет величину, равную 2,04.
Однако не было показано, как будет влиять наличие других элементов в растворе на определение урана в среде тирона, для которого уже известен ряд комплексов с металлами, полярографически активных и более устойчивых, чем комплекс с ураном..
Другие органические вещества в качестве фона
Как показано Иса [630а], при полярографировании урана в щелочных растворах, содержащих триэтаноламин (ТЭА), волна UC>2+ сливается с волной Fe3+ (на фоне 0,5 М NaOH+4% ТЭА).
В NH4C1+NH4OH+T9A волна урана сливается с волной Fe3+ и со 2-й волной меди (Cu+-^Cu°). Добавляя KCN, можно устранить волны Fe и Си, но при этом волна урана становится очень нечеткой.
Для совместного определения .урана и циркония рекомендован новый фон: краситель Na-соль 2-(5-хлоро-3-сульфо-2-оксибен-золазо) 5-сульфо- 1-нафтол-диокси-0-оксиазо или 0-окси-0-аминоазо (мордан синий 2R) [731а]. UO^ дает в ацетатном буферном растворе красителя с pH 5,3 четкую волну после волны самого красителя, как это получается и для алюминия [1013а]. Высота’ волны иОг+ пропорциональна концентрации урана. Константа диффузионйого тока равна 6,18. Точность метода ±6% (отн.) для 2-10-6 М растворов урана и ±3% (отн.) для 6- 10-в М. Наличие в растворе анионов 5ОГ, Cl-, NO3“ при концентрациях не более 0,1 М и РО® до 0,05 М не оказывает влияния.
Некоторые катионы (Al, Fe, Со, Pb, Ti и др.) мешают определению урана. Поэтому в их присутствии авторы рекомендуют отделять последний, извлекая его эфиром из насыщенного по NH4NO3 раствора.
Анализ проводят следующим образом: в мерную колбу емкостью 50 мл наливают 4 мл 0,25%-ного раствора красителя, 3 мл 2 М ацетата натрия, 0,5 мл 0,1%-ного тритона Х-100 (для подавления максимума) и воду до 40 мл-, pH доводят до 5,3 и добавляют водой до метки. Аликвот помещают в электролизер, продувают 20 мин. азот и получают полярограмму от 0 до —0,8 в.
Волна при —0,3 в соответствует красителю. Затем добавляют анализируемый раствор до концентрации 0,05 М и получают полярограмму. Возникает вторая волна при — 0,45 в, по высоте которой определяют уран.
Полярографическое определение урана в водных растворах в сочетании с хроматографией или экстракцией
Многие исследователи предлагают полярографические методы •определения урана, которые сочетаются с предварительным отделением его от сопутствующих элементов хроматографическим способом или экстракцией. Например, Леже [697] предложил при определении урана в рудах и продуктах их обработки отделять его от других элементов на колонке с целлюлозой, элюировать эфирным раствором с HNO3 после чего полярографировать уран в растворе 0,5 М щавелевой кислоты и 0,9 М H2SO4, содержащем 0,015% желатины. На этом фоне константа диффузионного тока UO*+ равна 2,79 мка/ммоль/л.
Этот метод позволяет определять 2,5 • 10-4% урана при 1000-кратном и более содержании других элементов (Fe, Мп, Mg, Са, А1 и др.) с точностью в среднем около±3?6 (отн.).
Арден, Бурсталь и Линстед [338] применили бумажную хроматографию и выделили уран из смеси, используя в качестве растворителя тетрагидросильван с 5 % HNO3 или тетрагидрофуран с 7% HNO3. Затем они вырезали зону металла, сжигали бумагу, остаток обрабатывали ОДА НС1 и полярографировали раствор.
Лакурт и др. [689] также отделяли уран от А1 и Fe при помощи бумажной хроматографии, используя в качестве подвижного растворителя ацетон 4-5% НС 1 в атмосфере бензола. Затем металл из бумаги извлекали кислотой и определяли полярографическим методом.
Диздар [464] применил ионообменную колонку с катионитом амберлит УК-120 для отделения урана от кадмия, затем металл селективно извлекал с колонки щавелевой кислотой, и уран определял полярографическим методом.
Другой полярографический метод определения небольших количеств урана в минеральном сырье недавно предложен Шульцек и др. [945]. Он основан на отделении урана в виде диураната аммония в присутствии комплексона III и тартрата (для маскировки остальных элементов) на колонке с силикагелем с последующим полярографическим определением урана после элюирования его раствором соляной кислоты. В качестве электролита — фона применяют раствор 0,5 М НС1О44Д,5 М НС1, содержащий тимол в концентрации 1-10"4 М (для подавления максимума). На этом фоне потенциал полуволны UCF+ равен —0,25 в (отн. нас. к. э.), и диффузионный ток прямо пропорционален концентрации урана (в интервале концентраций 4-10~5—2-Ю"4 М). Относительная ошибка для десятых долей миллиграмма урана колеблется от 1,5 до 6% и только в присутствии свинца достигает 10,5%. Олово (IV) необходимо до анализа удалить из раствора многократным выпариванием с НВг и Вг2, так как оно мешает определению урана в указанных условиях.
192
Навеску мелкоизмельченного материала, соответствующую содержанию урана не менее 0,3 мг, растворяют в смеси кислот: 15 мл Н\О3 (уд. в. 1,40) и 9 мл НС1 (уд. в. 1,19). Если образец плохо растворяется, то его сплавляют с \а2СО3 или разлагают смесью HNO3 -|- НС1 -f- HF. Затем раствор выпаривают многократно с НС1 (уд. в. 1,19) досуха, смачивают остаток 2 мл НС1 и разбавляют раствор горячей дистиллированной водой. Отфильтровывают от нерастворимого остатка, промывают последний и прибавляют в фильтрат 3 г винной кислоты и 20—30 мл 0,2 М раствора комплексона III. Раствор нагревают до кипения и по охлаждении прибавляют 25 мл аммиака, содержащего сульфат аммония. Затем раствор медленно фильтруют (со скоростью 5 мл мин) через колонку с силикагелем. Колонку промывают 100 мл воды, и адсорбированный уран вымывают 1—5 мл НО (уд. в. 1,19) в небольшой стакан. Многократно промывают силикагель водой порциями по 5 мл со скоростью 1 мл,'мин. Раствор выпаривают досуха, увлажняют остаток 0,5 мл НС1, разбавляют водой, добавляют 0,2 г аскорбиновой кислоты и нейтрализуют эквивалентным количеством твердой NaOH. Затем прибавляют 1,25 мл 70%-ной НС1О4, 0,5 мл 0,075%-ного раствора тимола, переносят раствор в мерную колбу емкостью 25 мл и доводят водой до метки. После достаточного перемешивания часть раствора (-^10 лы) помещают в электролизер, продувают азот в течение 5 мин. и получают полярограмму при анодно-катодной поляризации, начиная от 0 в (при общем напряжении 2 в). Содержание урана определяют методом добавки стандартного раствора уранил-хлорида.
Другой метод отделения урана на анионите-амберлите в сочетании с определением урана по каталитической волне NO3 описан на стр. 196.
Полярографический метод был применен для определения урана в урановых стеклах и в рудах после извлечения его эфиром. В этом случае может быть использован любой фон (КС1+НС1, H2SO4 и др.).
Метод, в котором экстракция урана сочетается с последующим полярографированием, предложен для определения его в присутствии висмута [6831. Отделение урана от висмута производят экстракцией ацетилацетоном в присутствии комплексона III. Фоном для полярографического определения урана служит 0,1 М раствор комплексона III, 2 М по ацетату натрия. В этих условиях £i/2 урана сдвигается и равен —0,47 в.
К раствору, подлежащему анализу и содержащему от 0,1 до 1 мг урана и любые количества висмута, прибавляют раствор комплексона III в таком количестве, чтобы соотношение Bi к комплексону III составляло 1 : 30. Затем доводят pH раствора до 7,5, добавляя 1 N раствор NaOH. Раствор сливают в делительную воронку, добавляют около 10 мл ацетилацетона и встряхивают воронку приблизительно 10 мин. Затем отделяют слой ацетилацетона, отфильтровывают его и доводят точно объем до 10 мл. Отбирают 2 мл раствора в мерную колбу емкостью 10 мл и выпаривают его приблизительно до 0,5 мл. По охлаждении прибавляют электролит-фон (0,1 М раствор комплексона, 2 М по ацетату натрия) до метки. Раствор сливают в электролизер, продувают через него азот (или водород) в течение 5 мин. и получают полярограмму. Концентрацию урана в растворе определяют по градуировочной Прямой, построенной по стандартным растворам сульфата уранила.
При содержании урана от 0,1 до 1 мг в присутствии 25—500 мг висмута ошибка равна в среднем ±1% (отн.). Этот метод применим также для определения урана в присутствии Си, РЬ и Zn.
13 Аналитическая химия урана
193
Определение урана по каталитическим волнам Каталитическая волна урана с нитрат-ионом
Уран катализирует, как и другие элементы (La, Се, Nd, Th), восстановление нитрат-иона на ртутном капельном катоде. Впервые это обнаружил И. Е. Старик [237], найдя при восстановлении иОо+ в виде UO2(NO3)2 в кислых растворах 2 волны, причем 2-я имела в 16 раз большую высоту, чем 1-я. Однако И. Е. Старик не объяснил замеченное им явление.
Позже было показано, что роль урана сводится к каталитическому влиянию его ионов при восстановлении NO3 -ионов, что было использовано для количественного определения очень малых количеств урана порядка 10~6—10-7 М [577, 229], а также для определения малого
-о,г -z?4 -о,в -о,8 -i,o -1,г -i,^ -1,ь
Рис. 33. Каталитическая волна NO3 -ионов в присутствии 4 -10 s АД ура-нил-ацетата рафоне 1 10~3М KNO3, 1
0.1 М КС1 и 0,01 М HC1;S= т™ '
с, в
содержания нитратов и нитритов [663, 640, 230]. Было обнаружено, что по аналогии с лантаном очень малое содержание урана в растворе вызывает появление большого тока восстановления NO3 при — 1,2 в (нас к. э.) (рис. 33), т. е. при потенциале восстановления U (V)+ 4- U (IV); хотя этот ток и не прямо пропорционален концентрации урана, но, вследствие того, что он примерно в 100 раз больше теоретического тока, позволяет определять очень малые количества урана. Что касается механизма восстановления NO3 в присутствии ионов урана, то наиболее правильным является истолкование Корыта [675] *, показавшего, что увеличение тока связано с образо-NO 3 в результате взаимодействия
ванием каталитической волны
NO3 с U (III), получающегося в 3-й стадии восстановления иО®+ -ионов.
В сочетании с экстракцией и хроматографией каталитическую волну NO3 успешно применяют для определения малых количеств урана в различных материалах. В качестве фона для определения урана по каталитической волне может служить раствор, содержащий 0,001 М KNO3, 0,1 М КС 11 и 0,01 М НС1. Содержание урана определяют по градуировочной прямой.
Следует иметь в виду, что присутствие в растворе ионов сульфата значительно понижает волну нитрата. Как показали С. И. Си
* См. также [428].
194
някова и Г. Г. Каранович [230], в присутствии 3 мг/мл сульфата (при пересчете на SO; ) величина каталитического тока иона нитрата начинает уже несколько уменьшаться, а при большей концентрации SO; — заметно уменьшается.
Кромптон и др. [8] применяют каталитический метод для определения 1 • 10-s—5-Ю-5 Л1 урана. Фоном служит раствор, 0,03 М по. NaNO3, 0,02 Л1 по HNO3, 0,005 М по NaCl и 2-10~4%-ный по метиловому красному (последний служит для подавления максимума).
Хлориды, сульфаты, перхлораты, фосфаты, оксалаты, фториды должны отсутствовать. Широкому применению этого метода мешает также влияние многих катионов, восстанавливающихся при более положительных потенциалах, чем —1,2 в, и некоторые анионы, образующие комплексы с ураном. Поэтому метод может быть применен только после предварительного отделения урана. На использовании каталитической волны нитрат-иона основан метод определения малых количеств урана в минералах, фосфатах, бокситах и других материалах [587, 672, 673].
Предварительно уран отделяют от других элементов на анионите — амберлите IRA-400 в нитратной форме. Для этого уранил-ионы связывают в анионный уранилнитратный комплекс в растворе HNO3, насыщенном NH4NO3. При этом все ионы, в том числе и фосфат-ионы, проходят через колонку и, следовательно, отделяются от урана. После промывания колонки полу насыщенным раствором NH4NO3 уран элюируют азотной кислотой (1 : 10).
Приготовление колонки с амберлитом IRA-400. Очищенную смолу заливают 0,1 N раствором HNO3 и оставляют на несколько дней. Затем ее сушат, измельчают и берут зерна диаметром 0,1—0,3 мм. Ими наполняют колонку, промывают многократно дистиллированной водой для удаления следов кислоты, пропускают 75 мл насыщенного раствора NH4NO3 со скоростью 0,25 мл'мин для перевода смолы в нитратную форму; после 2—3 анализов колонку следует заполнять новой порцией смолы.
Ход анализа. 0,5—1 г минерала растворяют в платиновом тигле в смеси HF и концентрированной HNO3 и выпаривают досуха. Эту операцию повторяют. Вскрытие минерала считают законченным, когда не остается следов SiO2. Остаток растворяют в 10 мл концентрированной HNO3 и снова упаривают досуха, а затем растворяют в 15 мл горячей 1 N HNO3; раствор фильтруют и остаток на фильтре промывают несколькими миллилитрами 1 N HNO3. Если минерал содержит значительное количество фосфат-ионов, то к фильтрату прибавляют избыток нитрата железа (III) (для связывания их в комплекс), и затем его выпаривают на водяной бане досуха. Сухой остаток растворяют при нагревании в 5 мл 1 N раствора HNO3> прибавляют 60 мл насыщенного раствора NH4NO3 и 10 мл воды, интенсивно взбалтывают, и затем раствор пропускают со скоростью 0,25 мл!мин через колонку, подготовленную, как указано выше. После этого колонку промывают 70—90 мл 50%-ного раствора NH4NO3 и элюируют уран азотной кислотой (1 : 10) в 100 миллилитровую колбу до наполнения раствора до метки тоже со скоростью 0,25 мл!мин. Содержание урана в пробе не должно превышать 0,5 мг.
Весь элюат или аликвотную часть его выпаривают досуха в кварцевой чашке. Затем осторожно нагревают ее на плитке до разложения всего нитрата аммония и еще 10 мин. нагревают на полном пламени бунзеновской горелки. Остаток растворяют в 10 мл 5 N НС1 и выпаривают на водяной бане досуха. Растворяют остаток 10 мл 0,01 # HNO3, переводят раствор в электролизер, продувают 10—15 мин.
13*
195
очищенный азот или водород и получают полярограмму, начиная от —0,5 в при общем напряжении на реохорде 2 в. Катодом служит капельный ртутный электрод, анодом—хлор-серебряный электрод. Высоту полученной волны урана сравнивают с волной стандартного раствора урана. Последнюю получают следующим образом: 1 мл стандартного раствора нитрата с содержанием 2,6 мкг урана упаривают в кварцевой чашке досуха. Остаток растворяют в 10 мл 0,01 N HNO3 помещают в электролизер, продувают азот или водород (10—15 мин.), и получают пол я рог рам* му в тех же условиях, как указано выше.
Ошибка не превышает ±3% (отн.) при определении 2—3 мкг урана в присутствии ряда элементов (Си, Fe, Pb, Со, Ni, Zn, редкоземельных и др.) при отношении урана к примесям 1 : 103—1 : 104.
Каталитическая волна урана в присутствии ванадия
С. И. Синякова и Л. А. Цветкова [233] показали, что для определения очень малых количеств урана можно применять его каталитическую волну, получающуюся на фоне молочной кислоты в присутствии любых количеств ванадия (но не менее 5-кратного по отношению к урану).
Ион иОг+ дает на фоне молочной кислоты четкую волну с Eii2=—0,20 в (отн. нас. к. э.). В присутствии ванадия в растворе высота волны увеличивается почти в 5 раз (рис. 34). При этом, если содержание ванадия не менее чем в 5 раз превышает содержание урана, между высотой волны и концентрацией урана существует прямолинейная зависимость. Ванадат аммония в растворах молочной кислоты не дает волны восстановления по крайней мере до —0,6 в (комнатная температура) или до —0,8 в (после длительного кипячения). Кипячение раствора ванадата в молочной кислоте приводит к уменьшению осцилляций на полярограмме урана.
В присутствии ванадата аммония Ei2 иОЦ в растворах молочной кислоты смещается к более положительному потенциалу. Смещение это тем больше, чем выше концентрация ванадия. Это указывает на образование соединения между UO Ц и V (V) анионного характера.
На характер волны урана оказывает влияние также концентрация молочной кислоты, особенно в присутствии ванадия. Оптимальная концентрация молочной кислоты равна 3—7%. По нашему мнению,
Рис. 34. Каталитическая волна урана на фоне молочной кислоты в присутствии ванадата аммония.
1—UOg+ на фоне 10%-ного раствора молочной кислоты; 2—то же после добавления 10-кратного количества ванадата аммония.
196
процесс каталитического влияния ванадия в растворах молочной кислоты сводится к следующему: V (V) при кипячении с молочной кислотой восстанавливается до V (IV); последний окисляет UO7— продукт восстановления уранил-ионов и, следовательно, все время вызывает регенерацию UO2+.
V(V)4~ молочная к-та —>V(IV) иоГ4-е-—UO2+
(18)
(19)
UO2+ 4-V(IV) —UO2/ + V (III) (20)
Подобный механизм был также предложен Ю. В. Морачевским и А. А. Сахаровым [162], исследовавшими процесс восстановления ионов UO2+ в присутствии ванадия (при равных приблизительно концентрациях урана и ванадия и в отсутствие молочной кислоты).
При полярографическом определении урана в растворах молочной кислоты мешают железо и медь. Эти элементы необходимо предварительно отделить.
Для определения урана в рудах и концентратах, содержащих от десятых до лей до целых процентов урана и ряд примесей (Си, Fe, V, W, Мо, Сг, Ti и др., рекомендуется [233] следующий метод анализа: навеску 0,1 г руды, предварительно прокаленной около2 час. при 500—600°, обрабатывают царской водкой, выпаривают избыток кислот и затем уран вместе с другими полуторными окислами осаждают аммиаком, не содержащим СО2. При этом медь остается в растворе. Если меди много, следует осаждение аммиаком повторить еще раз. Затем осадок, содержащий диуранат аммония и другие гидроокиси металлов, растворяют в соляной кислоте (1 : 1) и отделяют железо обработкой раствора углекислым аммонием при нагревании до 50—60°.
Полученный углекислый раствор выпаривают досуха, осторожным прокаливанием удаляют аммонийные соли. Остаток растворяют в HNO3, избыток кислоты удаляют выпариванием; к сухому остатку прибавляют 2,5 мл молочной кислоты и, если мало ванадия в руде, то некоторое количество ванадата аммония нагревают на водяной бане — 10 мин., а затем после охлаждения доводят объем раствора до 25 мл. 10 мл раствора помещают в электролизер, продувают водород или азот — 10 мин. и снимают полярограмму. Затем в исследуемый раствор прибавляют 0,5 мл стандартного раствора уранил-нитрата с содержанием 10 мг!мл урана и снова снимают полярограмму.
Содержание урана определяют по формуле:
h-с- v-100 /6U — >
где h—высота волны анализируемого раствора, мм;
Ai—высота волны после добавки стандартного раствора, мм;
с—концентрация урана в растворе за счет добавки, г,мл;
v—объем анализируемого раствора (25 мл); а— навеска, г.
Ю. В. Морачевский и А. А. Сахаров [162] предложили метод определения урана в присутствии ванадия и железа, который в одном варианте заключается в элиминировании влияния ванадия путем создания постоянной его концентрации, а в другом — применяется предварительное электролитическое восстановление урана и ванадия. Однако первый вариант требует предварительного
197
определения ванадия в исследуемом растворе до полярографиро-вания, а второй, при котором определяют уран по величине предельного тока анодно-катодной волны U (III)/U (IV), требует восстановления ванадия до четырехвалентного состояния. Этот вариант менее чувствителен и требует отсутствия элементов, восстанавливающихся при более положительных потенциалах, чем U (IV), если они находятся в растворе в значительно больших концентрациях, чем уран.
Полярографическое определение урана в неводных растворах в сочетании с экстракцией
Изучение полярографического поведения урана в различных неводных растворителях представляет значительный интерес. Можно построить схему анализа, основанную на экстракции урана каким-либо органическим растворителем с последующим полярографическим определением его непосредственно в органической фазе после добавления соответствующих веществ для повышения ионизации и проводимости, или после реэкстракции.
С. И. Синякова и В. М. Шамриков [232] исследовали полярографическое поведение ионов UO1+ в некоторых органических растворителях (трибутилфосфат, метилэтилкетон, ацетоуксусный эфир, бутиловый спирт и др.) и установили, что в этих растворах получаются растянутые пологие волны. Это находится в согласии с результатами Мак Гиллаври [1093], получившего ненормальные полярограммы в безводной уксусной кислоте для катионов, у которых £1/, положительнее, чем —0,36 в.
В смеси метилэтилкетона (25 объемн. %), этилового спирта (15 объемн. %) и воды (60 объемн. %) на фоне 0,1 М. КС1 получаются две четкие, хорошо измеримые волны уранила. £./а и id этих волн зависят от соотношения компонентов растворителя, т. е. от диэлектрической постоянной, вязкости и других факторов. Прямолинейная зависимость между диффузионным током урана и его концентрацией в растворе соблюдается только для концентраций урана 10—150 мкг/мл. При увеличении концентрации метилэтилкетона £/, UO1+ сдвигается к более отрицательным значениям (особенно для 1-й волны), что указывает на образование комплексного соединения между UOf+ и метилэтилкетоном (типа сольвата) [2311.
В растворах, содержащих бутиловый, этиловый спирт и воду при соотношении компонентов 2 : 1 : 2, на фоне 0,1 М LiC 1 также получены 2 четкие волны с £•/,=—0,22 в (нас. к. э.) и £•/,=—1,1 в (нас. к. э.). Константа диффузионного тока равна 0,25; она уменьшается при увеличении концентрации урана, поэтому для аналитических целей следует пользоваться растворами с концентрацией урана не более 0,84 ммоль!л, Величина углового коэффициента 198
(a =0,125) указывает на необратимый характер восстановления ионов UO*+ в смешанном растворителе.
В растворах, состоящих из ацетоуксусного эфира (40 объемн. %), этилового спирта (27 объемн. %) и воды (33 объемн. %) на фоне 0,1 М КС1 также получаются две четкие волны UO1+. Е./, зависит от концентрации урана и изменяется от —0,43 в (0,2 ммоль/л) до —0,34 в (1,26 ммоль/л). Это указывает на комплексообразование между UO; + и ацетоуксусным эфиром. Константа диффузионного тока урана в этом растворителе равна 0,43 и не зависит от концентрации урана, поэтому этот смешанный растворитель имеет для аналитических целей преимущество перед вышеприведенным.
Еще лучше для аналитических целей применить смешанный растворитель, состоящий из ацетоуксусного эфира (35 объемн. %), ацетона (60 объемн. %) и воды (5 объемн. %). В этом смешанном растворителе на фоне 0,1 М LiCl Еч„ UO1+ равен —0,47 в, а константа диффузионного тока равна 0,99, т. е. в 2,5 раза больше, чем при замене ацетона на этиловый спирт. Это объясняется значительно меньшей вязкостью ацетона (р.=0,3) по сравнению со спиртом (ц=1,1).
Для определения урана в рудах и других материалах уран после вскрытия переводят в уранил-нитрат, который экстрагируют бутиловым спиртом или ацетоуксусным эфиром из насыщенного по NH4NO3 раствора, а затем после разбавления спиртом и водой (или ацетоном) производят полярографическое определение урана на фоне 0,1 М КС1 или LiCl. Необходимо до снятия полярограмм тщательно удалить кислород из раствора (продувание азота 15— 20 мин.), так как в органических растворителях он более растворим, чем в воде. Концентрацию урана определяют методом добавок или по градуировочной прямой.
Для быстрого и точного определения урана в рудах и других материалах (в том числе и радиоактивных) предложен метод [505]. заключающийся в экстракции U (VI) в виде нитрата смесью три-бутилфосфата и изопропилового эфира с последующим полярографическим определением урана после разбавления раствора ледяной уксусной кислотой до концентрации урана ^50 мкг/мл. Фоном — электролитом служит 0,25 М ЫС1О4, полярограмма снимается после продувания азота ~ 10 мин. (рис. 35).
Другой вариант, предложенный этими же авторами, предусматривает реэкстракцию урана из органического слоя обработкой последнего равным объемом воды, не содержащей меди. Водный слой нагревают 3 мин. на горячей плитке *, охлаждают, подкисляют раствор азотной кислотой до концентрации 0,1 М, разбавляют до желаемого объема водой и после продувания азота в течение -^-10 мин. получают полярограмму.
* Для разрушения комплекса урана с трибутилфосфатом.
199
Метод прямого полярографирования урана в органической фазе после соответствующего разбавления удобнее и быстрее, чем с последующей реэкстракцией.
Смесь экстрагента в обоих случаях состоит из 30 объемн.% трибутилфосфата и 70 объемн.°6 изопропилового эфира; в случае экстракции урана из высокоактивных растворов выгоднее применять 5%-ный (по объему) трибутилфосфат.
Для разбавления экстракта оказались эффективными ледяная уксусная кислота, а также равные объемы этилового спирта с водой. В последнем случае хорошим электролитом оказалась 0,5—0,6 М HNO3, но концентрация HNO3 должна поддерживаться постоянной, так как она влияет на высоту волны урана.
Последний разбавитель (этиловый спирт—вода) имеет то преимущество, что при подкислении раствора HNO3 до концентрации 0,6 М на волну урана не оказывают заметного влияния ионы ртути,
J, мка
0,1 0 -о,! -0,2 -0,3 -0,4 -0,5 5,6
Рис. 35. Полярограммы урана в неводных растворах:
1— в смеси трибутилфосфата, изопропилового эфира и ледяной уксусной кислоты на фоне 0.25 М LiCl, 2—в смеси бутилового, этилового спиртов и воды на фоне б, 1 М LjCl и 1,5 -10 ~3% тимола, 3— в смеси ацетоуксусного эфира, этилового спирта и воды на фоне 0,1 М КС1.
появляющиеся в растворе при контакте его с ртутью. При этом получаются воспроизводимые волны (если контакт с ртутью не очень продолжительный) и высоты их пропорциональны концентрации урана в растворе.
При применении ледяной уксусной кислоты в качестве разбавителя следует учитывать количество воды в ней, так как она оказывает большое влияние на устойчивость среды при контакте с ртутью. При применении же безводной уксусной кислоты нельзя пользоваться насыщенным каломельным электродом, который обыч-200
но применяют при полярографических измерениях в органических растворителях в качестве электрода сравнения, соединенного через мостик с агар-агаром, насыщенным KNO3.
Другие исследователи применяли полярографический метод в сочетании с экстракцией урана 100%-ным трибутилфосфатом из растворов, слабо подкисленных HNO3 и содержащих большое количество A1(NO3)3, или из растворов 5 Л1 по HNO3 [698], и также применяли в качестве электролита 0,25 М растворы по LiC104. При этом получались в органических средах хорошие волны без максимумов [506, 972].
Чигалик и др. [422], исследуя полярографическое поведение ионов UO^+ в безводной уксусной кислоте, показали, что уранилацетат образует на фоне ацетата натрия четкую волну без максимума с Ei 9——0,32 в (отн. каломельного ацетатного электрода); при этом высота ее в пределах концентраций урана 5 • 10 5 2,4 • 10~3М линейно зависит от концентрации урана в растворе. Волна соответствует двухэлектронному процессу.
В насыщенном по LiCl растворе безводной уксусной кислоты также получаются волны урана, но при больших его концентрациях на них образуется максимум, который подавляется добавлением фуксина. Однако этот электролит менее удобен, чем ацетат натрия, так как потенциал полуволны U (VI) более положителен, и, кроме того, до образования этой волны появляется волна растворения ртути, несколько деформирующая волну урана. На фоне LiCl волна соответствует восстановлению U (VI) до U (IV) и высота ее пропорциональна концентрации урана в растворе.
Применение осциллографической и переменнотоковой полярографии для определения урана
В последние годы развиваются новые ветви полярографии — осциллографическая и переменнотоковая. Принципиальные основы первого метода изложены кратко’в [111], а второго — в [89, 386].
Оба метода имеют ряд преимуществ перед обычным полярографическим методом: обладают большей избирательностью, чувствительностью и пр. Метод осциллографической полярографии был применен для определения урана (и свинца) в минералах [53, 73]. На фоне НС1 с pH 0,7—1,7 на осциллограммах иОг+ образуются две волны (рис. 36). Количественное определение урана можно производить по 1-й или 2-й волне. При этом между
Рис. 36. Осциллограмма UO1+ -ионов на фоне 0,2 N НС1 и 0,1 N КС1.
201
током пика 1-й волны и концентрацией урана в растворе существует зависимость, приближающаяся к линейной. Количественному определению урана не мешают Fe3+, V, Мо и Си. Точность определения урана по методу добавок лежит в пределах ±7 % (отн.).
Если в качестве фона использовать 0,2—0,5 М раствор HNO3, то градуировочная кривая является прямой линией, проходящей через начало координат. На определение урана на фоне HNO3 не оказывают влияния Fe3+, Zr, Nb, Та, Ti, редкоземельные элементы, кислород и др. Уран определялся этим методом в смолке, уранините, карбуране, монаците и других минералах. Одновременно с ураном из одной навески можно определять и свинец.
Гао Цай-шэн [43а] исследовал методом осциллографической полярографии кинетику катодных и анодных процессов различных валентных состояний урана в минеральных кислотах и предложил метод определения урана в рудах непосредственно после их вскрытия.
Для определения урана методом осциллографической полярографии предложен [383а] фон 1М НС1О4—0,01 N НС1, содержащий 0,001% пептона и 0,002% фенола (для подавления максимума). На этом фоне получается линейная зависимость между величиной пика и концентрацией урана в пределах 2-10"®—4-10-3 М. Как указывают авторы, наличие в растворе Си, Pb, As, Sn, Bi в соотношениях к урану, равных 1:1, не оказывает влияния на его определение. Присутствие Sb и V в тех же соотношениях вызывает некоторое искажение волны урана. Но особенно мешает при определении урана молибден, который необходимо отделить.
Эти же авторы указывают, что при получении кривых на квадратно-волновом полярографе ионы UO22 дают в 0,1 А растворе НС1 два пика с Еп=—0,18 в и Еп ——0,86 в (относительно анода— слоя ртути). Первый пик соответствует восстановлению U (VI) в U (V), а второй —восстановлению U (V) в U (III). Первый пик выражен более четко и поэтому удобен для количественного определения урана, однако в присутствии меди солянокислый фон нельзя применять, так как этот пик сливается с пиком меди. В этом случае лучше применять в качестве фона 1 М раствор H2SO4. В нем получается пик урана при —0,34 в, соответствующий обратимому восстановлению U (VI) в U (V). Этот фон может быть применен для определения урана в присутствии свинца и меди.
Квадратно-волновым полярографом можно определять десятые доли микрограмма урана в 1 мл.
Метод квадратно-волновой полярографии был применен для определения урана в растворах, протекающих через ионообменные колонки [548].
л
Рис. 37. Смеситель в автоматической схеме анализа.
/— стеклянная трубка 8X1 см с пористой перегородкой; 2 и <?— промежуточные сосуды, в которых происходит дегазация раствора; 4— электролизер.
Полярографический метод как датчик в автоматизации контроля растворов уранового производства
В последние годы наметилась тенденция использования полярографического принципа анализа в автоматизации контроля производственных растворов; имеется описание различных типов автоматических систем, в которых осуществляется запись обычных, дифференциальных (производных) или разностных полярографических кривых, а также осциллограмм.
В качестве примера применения обычной полярографии в автоматическом контроле можно указать на непрерывный контроль щелочного раствора цианида, предложенный Юра [634], а также на автоматическую регистрацию концентраций сернистого газа в вымачивающих зерно растворах [1017].
Разностная полярография [88, 111,897] предложена в схеме для автоматической регистрации концентраций ионов кадмия и висмута при их совместном присутствии в поточном растворе [699]. Эта работа является развитием предыдущих изысканий, в которых обычная поляро
графия применялась для регистрации текущих растворов с постоянной концентрацией вещества [767] или при наличии в растворе только одного восстанавливающегося вещества[474]. Предложенный прибор состоит из двух полярографических ячеек с двумя капельными электродами, соединенных в обратном направлении. Один из них опускается в исследуемый раствор, а второй —в раствор фона. При этом измеряется разность токов, возникающих в обеих ячейках. В зависимости от подаваемого на электроды напряжения можно получить разностные полярограммы для одного или двух ионов. Разностные полярограммы имеют такой же вид, как и обычные, и высоты волн находятся в прямолинейной зависимости от концентрации ионов.
Если на оба электрода накладывать напряжение, различающееся на небольшую величину, например 50 мв, но с одинаковой скоростью, то можно получать с этим прибором разностные производные (дифференциальные) кривые, у которых точка пика соответствует потенциалам полуволн определяемых ионов.
203
Прибор, в котором происходит смешение анализируемого раствора с электролитом, удаление кислорода продуваемым азотом, а также подача раствора в ячейку для измерений, состоит из стеклянной части, приведенной на рис. 37 [699]. Разность токов в
двух ячейках регистрируется усиления. Скорость движения ка измерений не более ± 3,5 ° с
самописцем после соответствующего бумаги равна 5 дюймам в час. Ошиб-> (отн.). Авторы указывают, что этот прибор может быть применен для регистрации концентраций металлов в гальванических ваннах, а также в растворах после обработки руд и других материалов. Разумеется, что этот прибор может быть использован для автоматического контроля урановых растворов.
Для последней цели представляют интерес два прибора, недавно описанные и уже применяющиеся в производстве. Один из них, «использованием записи обычных полярографических кривых, предложен для автоматической регистрации небольших концентраций урана (10 “4—10 2 М) в радиоактивных производственных растворах [320 Ь а другая система, в которой регистрируются производные (дифференциальные) кривые— для анализа растворов с большой концентрацией урана (100—200 а/л) [365, 698]. Первая система автоматизации [320] для контроля радиоактивных растворов построена с таким расчетом, чтобы содержа-
Рис. 38. Ячейка для автоматического контроля урана.
1— капельный ртутный электрод; 2— каломельный электрод: 3— насыщенный раствор КС1; 4— смесь ртути с Hg Cl 5— полиэтиленовая трубка; 6— пористая пробка.
щаяся в производственных растворах азотная кислота в концентрации около 2 М служила электролитом. В этих растворах концентрация урана обычно менее 0,01 г/л, но при нарушении нормальных условий технологии она может достигать 10а/л. Растворы содержат так же железо, нитриты и трибутилфосфат. Автоматическая линия включает схему обычного полярографа, ансамбль, состоящий из электролитической ячейки с резервуаром для ртути, трубопроводов для подачи производственных и стандартных растворов, ловушку для ртути, трубопровод для возвращения проанализированного раствора в процесс, линию подачи гелия для вытеснения кислорода, а также самозаписывающую систему с соответствующим электронным усилением токов. Запись кривых производится через каждые 7/4 мин.
204
Полярографическая схема состоит из реохорда, сопротивление которого равно 1000 ом, приводимого в движение с помощью синхронного мотора. На реохорде используется для записей кривых урана только участок напряжений от —0,05 в до —0,25 в (отн. нас. к. э.). Чувствительность измерений токов может регулироваться по четырем шкалам: на 12,5; 50; 100 и 200 мка, а на записывающей бумаге это соответствует от 0 до 10 мв. Уменьшение осцилляций достигается, если это необходимо, включением емкостей на 2000 или 4000 миф.
Рис. 39. Схема ячейки и автоматической линии.
/— резервуар с ртутью; 2— электролитическая ячейка; 3— соединение с самописцем; 4— ловушка для ртути; 5— сигнализирующее устройство верхнего уровня ловушки; 6— то же для нижнего уровня; 7— клапан для спуска ртути; 8 и 9— подача стандартных растворов; 10— подводка гелия; 11— трубопровод для возвращения раствора в процесс; 12— соединение с потенциометром.
205
На рис. 38 показана применяющаяся в описанном приборе электролитическая ячейка, которая состоит из капельного ртутного электрода и электрода сравнения, заключенных в стальную оболочку. Контакт каломельного электрода с анализируемым раствором осуществляется через стеклянную пористую перегородку.
На рис. 39 приведен весь ансамбль прибора. Объем ячейки рассчитан на 20 мл протекающего раствора. Циркуляцию анализируемого раствора через ячейку обеспечивает воздушный аспиратор, работающий со скоростью 400 мл,'мин. Хотя весь объем подводящей к ячейке системы и отводящей от нее рассчитан приблизительно на 175 мл, тем не менее предусмотренный отрезок времени в 3 мин. для наполнения ячейки производственным раствором обеспечивает полную замену одной порции раствора новой. Ловушка для ртути предохраняет производственный раствор от попадания в него ртути. Она выполнена из стали и должна содержать инертный растворитель с уд. в. меньшим, чем у ртути и большим, чем у производственного раствора, чтобы предотвратить реакцию между ртутью и азотной кислотой. Пригодными для этой цели оказались СС14 или трихлорэтилен.
По окончании всего цикла, т. е. через 7/4 мин., на дне электролизера открывается клапан и раствор вместе с ртутью сливаются в ловушку. Здесь ртуть осаждается, а раствор возвращается по трубопроводу в процесс. В ловушке предусмотрено специальное устройство, с помощью которого ртуть, когда уровень ее достигает установленного предела, удаляется из ловушки через автоматически открывающийся клапан. Вытекание ртути выгоднее отрегулировать так, чтобы небольшое количество ее оставалось в ловушке. В этом случае радиоактивные частицы, находящиеся на границе ртуть — раствор, не попадают в отработанную ртуть.
Таблица 29
Автоматизация операций в приборе во времени
Время, мин. Выполняемая операция
0—3 Включаются клапаны, обеспечивающие подачу производственного раствора в ячейку, а также подключается линия подачи гелия и удаления воздуха
3 Клапаны все закрываются
3’/4 Включается самописец
З’/г—61/, Производится запись кривой
63/4 Самописец выключается. Раскрывается клапан ячейки для
удаления из нее ртути в ловушку и включается трубопровод для возврата раствора в процесс
7’/4 Закрывается клапан ячейки
7’/2 Начинается новый цикл
206
Автоматическое устройство предусматривает в этом приооре регистрацию концентрации урана в растворах через каждые 7Ц мин., хотя можно отрегулировать измерения через любой промежуток времени. Автоматический анализатор выполняет операции, приведенные в табл. 29, в течение 7Ц мин.
Для выполнения прибором всех указанных в табл. 29 операций служат несколько реле врехмени. Во время измерения концен
Рис. 40. Кривая автоматической регистрации концентрации урана в радиоактивных’ растворах.
трации стандартного раствора прибор переключается вручную. При этом открывается клапан сосуда с соответствующим стан
Bbico’T'a пика, мм
Рис. 41. Дифференциальные полярограммы растворов урана, записанные через разные промежутки времени.
. , 206
Содержание урана в распооре. г/л
дартным раствором урана. Цикл записи концентрации стандартного раствора осуществляется в первые 3’/4 мин. нормального цикла, так как в З’ д мин. срабатывает реле с защелкой, разъединяющее клапаны обоих сосудов, содержащих стандартные растворы, с электросистемой. Это предохраняет стандартные растворы от смешения с производственными растворами в ячейке.
207
Схема полярографического прибора автоматической записи кривых.
Рис. 42.
Для
1' . Г_. — вольтметры на 1 в; 7?i— сопротивления от 100 до2000 ом. для регулирования чувствительности; АО, — сопротивления на 500 ом для контроля напряжения; /? — потенциометр; /?-, 7?6, R-, R,— сопротивления на 1000; 10 000; 20 000; 200 000 ом соответственно; сопротивление на 10 Мом для регулирования компенсатора: С-, — электролити-
ческие конденсаторы на 4000 и 10 000 мкф соответственно; S.,5;—переключатели; 1—ячейка; 2 — самописец.
Концентрация урана в стандартных растворах должна быть такого же порядка, как и в производственных растворах. Так как определению урана в производственных растворах мешают кислород и нитриты, присутствующие обычно в них, то в схеме предусмотрена очистка раствора от этих веществ до попадания его в ячейку. Осуществляется эта очистка продуванием через раствор гелия, насыщенного парами метилового спирта (нитрит образует с метиловым спиртом летучий мети л нитрит).
На рис .40 приведена кривая, записанная для производственных растворов.
Точность определения не более ±10% (отн.), так как на диффузионный ток урана оказывают влияние находящиеся в растворе железо (III) и другие элементы, усиливая осцилляции на кривой (железо попадает в раствор главным образом вследствие коррозии оборудования, в котором ведется процесс), а также трибутилфосфат, который вызывает обрыв кривой вследствие процесса адсорбции и десорбции его на ртутной капле.
Другой автоматический прибор с использованием принципа получения дифференциальных (производных) полярографических кривых применяется для контроля производственных растворов с большой концентрацией урана (100— 200 г/л). Получаемые при
этом кривые имеют максимум (пик), появляющийся приблизительно при значениях Е,;2 данного иона (рис. 41), и они так же, как и обычные полярограммы, представляют собой изменение диффузионного тока как функцию потенциала электрода. Авторы [365] применили в этом приборе схему Левека «сопротивление — емкость» (RC), но напряжение подается в обратном направлении (от —1,0 в до 0 в) по сравнению с обычной схемой Левека, как это видно из рис. 42.
208
Рис. 43. Схема установки для автоматического контроля растворов урана с «пропорциониза-тором».
[— полярографическая ячейка; 2—капельный ртутный электрод; 3—трубка для встряхивания ртути; 4—трубка для раствора; 5—резервуар с электролитом; 6—«пропорционизатор»; 7— смеситель; 8— пористая перегородка; 9— автоматические клапаны с соленоидным приводом; 10— циркуляционный насос; 11— растворы для анализа.
Благодаря использованию такой схемы устраняются или по крайней мере сильно уменьшаются осцилляции на получаемых кривых урана, особенно в точке пика.
Прибор состоит из потенциометра (/?3), соединенного синхронно с мотором. В процессе автоматической записи потенциометр двигается обратно после достижения 60% от общего его сопротивления. Если все напряжение, подаваемое на потенциометр, равно 1 в, то напряжение изменяется от —1,0 в до —0,4 в. Сопротивления R2 и /?4 подбираются таким образом, чтобы они давали соответствующее начальное и конечное напряжение при включении двух батарей на 1,5 в. Для поддержания определенной чувствительности при записи кривой служит сопротивление При концентрации урана порядка 100—200 г/л R, должно быть равным 100 ом. Изменяя значения Rlf можно применять прибор для контроля растворов с широким интервалом концентраций урана. Оптимальные величины R, и С подбирают такими, чтобы получались кривые с заданными высотами пиков.
Самописец применяется с шкалой на 2,5 мв. Так как производственные растворы, для которых авторы сконструировали указанный автоматический прибор, содержали большую концентрацию урана, а также ванадий и молибден, которые оказывают большое влияние на определение урана, был выбран электролит, состоящий из 0,1 М H2SO4+0,l М Na2SO4+4 • 10-4% тимола. Удовлетворительные результаты получаются после разбавления этим электролитом производственного раствора в 200 и 400 раз. В этом случае расхождение результатов определения урана, полученных на автоматическом приборе и другими методами, составляло лишь ±5%. И только при большом содержании ванадия и молибдена отклонения доходили до 12%.
14 Аналитическая химия урана
209
На рис. 43 представлена та часть прибора («пропорционизатор»), в которой происходит точное разбавление раствора. Главное в ней — кран с четырьмя отростками, вращающийся синхронным мотором (1 об/мин.). Проходя через горизонтальное положение, канал крана промывается анализируемым раствором и при дальнейшем вращении отсекает строго определенный объем этого раствора; проходя через вертикальное положение, канал промывается
Рис. 44. Механическая схема, осуществляющая регулирование автоматизации.
А, В, С и D— микропереключатели; Е— синхронный мотор для потенциометра; F— регулятор скорости вращения мотора; G, Н— реле; I,J — синхронные моторы для «пропорционизатора» и «кулака»; К, L— автоматические клапаны с соленоидным приводом.
разбавляющим раствором поступающим в смеситель; при постоянном напоре разбавляющего раствора и постоянной скорости вращения крана происходит смешение анализируемого раствора и электролита в смесителе в соотношении 1 : 423 с ошибкой не более ±0,5%.
Для автоматического наполнения и удаления раствора из ячейки служит клапан, управляемый соленоидом (см. рис. 43), изготовленный из стойкого по отношению к разбавленной H2SO4 материала. Клапаны сделаны из люцита, а центральный клапан имеет диафрагму из неопрена. С помощью соленоида эта диафрагма • поднимается, позволяя раствору вытекать. Система реле регулирует подачу раствора через клапаны, а также его удаление из ячейки.
Автоматические операции выполняются циклически, причем
210
Таблица 30
Автоматизация операций в приборе в зависимости от напряжения
Время. Приложен- Выполняемая операция
У11В । ное напря- h
жение. в
0 — 1,0 Присоединяется потенциометр и самописец
1,5 —0,5 Записывается пик производной кривой урана
1,8 —0,4 Потенциометр начинает возвращаться к начальному положению. Включается «пропорционизатор», разбавленный раствор сливается в смеситель
3,0 —0,8 «Пропорционизатор» останавливается; самописец выключается; раствор сливается из ячейки
3.3 —0,9 Ячейка промывается и наполняется новым раствором из смесителя
3,6 —1,0 Потенциометр выключается на 1,5 мин.
5,1 -1,0 Цикл начинает повторяться
время каждого цикла равно 5 мин. В течение одного цикла напряжение изменяется от —1,0 в до —0,4 в, после чего потенциометр возвращается в начальное положение, т. е. к 1,0 в. Работа каждой механической части синхронизирована с изменением напряжения. На рис. 44 приведена схема механической системы прибора, а в табл. 30 приведены все операции, производимые прибором в течение одного цикла в зависимости от напряжения.
Циркуляционный насос обеспечивает подачу производственного раствора в прибор со скоростью около 4 л/мин\ это предусматривает приток свежего раствора в продолжение одного цикла.
Прибор может быть установлен для анализа любого раствора из двух или нескольких производственных точек, как это видно из рис. 43.
Калибровочные кривые авторы рекомендуют строить по известным растворам урана в статических условиях при температуре 25°.
На рис. 41 приведены производные кривые, полученные для некоторых производственных и стандартных растворов урана. Максимальная ошибка не превышает ±3,5% (отн.), если коцент-рация урана менее 1 г/л. Опыты, проведенные с автоматическим прибором, показали, что можно анализировать два раствора последовательно, так как после одного цикла происходит полная замена одного раствора в смесителе и ячейке другим. Для получения более точных результатов авторы рекомендуют производить замеры прибором при постоянной температуре как для производственных, так и для стандартных растворов. Для этого необходимо ячейку снабдить водяной рубашкой.
: Самой важной частью прибора является «пропорциональная» Схема, т. е. та часть его, которая обеспечивает разбавление. Меха-
14* 211
ническая часть прибора подобрана, по-видимому, удачно, однако фон — электролит, выбранный авторами, не позволяет с достаточной точностью производить автоматический контроль урана в производственных растворах, содержащих значительные количества ванадия и молибдена. В последнем случае можно использовать в качестве электролита некоторые комплексообразующие вещества, которые* позволят устранить влияние ванадия и молибдена.
Из приведенных примеров следует, что автоматический контроль урановых растворов с использованием полярографического принципа в качестве датчика является вполне реальным.
Амперометрические методы титрования урана
Амперометрические методы титрования урана не нашли широкого применения. Большинство этих методов основано на титровании четырехвалентного урана некоторыми окислителями. Некоторые методы амперометрического определения урана основаны на осаждении U (VI) или U (IV).
Г. А. Клейбс [99, 100] в серии работ применила амперометрический метод для изучения реакции осаждения ионов уранила ферроцианидом калия. В результате исследования была показана возможность точного определения урана в растворах KCl-f-HCl при амперометрическом его титровании ферроцианидом калия. В этом случае образуется двойной ферроцианид состава:
5(UO2)2-[Fe(CN],-3Kt[Fe(CN),].
В. Г. Сочеванов с сотрудниками [234] предлагает для амперометрического определения ионов уранила титровать его растворы ферроцианидом в 1 М растворе KNO3 с pH 3—5, используя вращающийся платиновый микроэлектрод (диаметром 0,5 мм). Конечную точку определяют по анодному току окисления ферроцианида. Титрование лучше вести при --Н),6 в (нас. к. э.) и температурах от 40 до 60°. По точности метод амперометрического титрования равноценен весовому ’даже для количеств 0,5—10 мг U.
Для амперометрического титрования ионов уранила предложена [667] jn-нитрофениларсоновая кислота, с которой UO^+ образует нерастворимое соединение состава UO2C6H4-NO2-AsO3. Так как UO’4" и реагент дают диффузионные токи, то кривая титрования будет иметь вид, наиболее удобный для определения конечной точки.
К исследуемому раствору, содержащему уран в виде UOj + , приливают 10мл ацетатного буфера и около 50 мл воды. Добавляют 2 г NaCl, 5 мл 0,1%-ного раствора желатины и разбавляют водой до 100 мл. Титрование проводят при 0,55 в. После добавления каждой порции реагента выжидают 1—2 мин.
Точность определения ^1 % (отн.).
Из амперометрических методов, основанных на редоксреак-циях, можно указать на титрование четырехвалентного урана 212
растворами трехвалентного железа. Однако, как указывают авторы 18], ошибки титрования несколько больше, чем при потенциометрическом титровании. Преимуществом по сравнению с потенциометрическим методом является то обстоятельство, что молибден не мешает определению урана.
Ю. В. Морачевский и И. А. Церковницкая [164] предложили амперометрический метод титрования урана, основанный на восстановлении его до U (IV) с последующим титрованием метаванадатом аммония.
Титрование U (IV) можно проводить при нулевом потенциале или при 0,1 в в сернокислом растворе, пользуясь 0,006 N раствором ванадата аммония.
Титрование можно также производить в растворах HCI и НС1О4 при их концентрациях в пределах 0,1—1,0 М. Концентрация урана не должна превышать 2,5 мкг/мл при титровании 0,005 N раствором метаванадата аммония. Относительная ошибка равна в среднем 1—3%. Титрование более разбавленными растворами метаванадата аммония дает мало удовлетворительные результаты.
Для определения урана в присутствии Fe, Сг, Ni, РЬ и Bi исследуемый раствор, содержащий сульфат уранила и сульфат железа (U : Fe=l : 25), восстанавливают жидкой амальгамой цинка в растворе H2SO4. Затем добавляют ^10 мг ортофенантролина (он связывает Fe2+ в прочный комплекс) и комплексон III (25— 100 г) для связывания Pb, Ni, Bi и Сг, а затем титруют метаванадатом при концентрации H2SO4, равной 0,1 N. В присутствии железа средняя ошибка титрования урана составляет 3—6% (отн.).
Другой вариант амперометрического метода титрования урана, также основанного на титровании урана (IV) ванадатом аммония и предложенного для определения урана в рудах [71а], по существу не отличается от указанного выше. Однако благодаря трудоемкому способу подготовки пробы к анализу (гидросульфитно-фосфатный или хромо-фосфатный) он не имеет преимуществ по сравнению с методом [164].
2. Потенциометрическое определение урана Н. И. Удальцова
Очень часто при объемном определении урана, основанном на окислительно-восстановительных реакциях, реже на реакциях осаждения, используют потенциометрический метод определения «Конечной точки титрования. Г*
Обычно при потенциометрическом титровании уран окисляется от четырехвалентного состояния до шестивалентного или восстанавливается от урана (VI) до урана (IV) соответствующими окислителями или восстановителями. Подробный обзор методов Потенциометрического титрования урана приведен в книге Гмелина
213
[547], а также в обзорной статье Фурмана [536]. Обзор работ до 1950 г., большей частью неопубликованных, дан в монографии Роддена [8].
Для титрования урана (IV) используют такие окислители, как КМпО4, Ce(SO4)2, К2Сг2О7, соли Fe (III), бромат калия и др.; для титрования урана (VI) восстановители Сг (II) и Ti (III).
Потенциометрическое титрование КМпО4 описано рядом авторов [8, 493, 792, 803].
Перман [803] использовал для восстановления урана (VI) цинковую амальгаму, а затем титровал потенциометрически уран(1\3 0,1 N раствором КМпО4 в 4 N растворе H2SO4 на холоде в присутствии железоаммонийных квасцов. Данным методом автор определял до 10 мкг урана в 1 мл раствора с относительной ошибкой 0,5%. При этом определению урана мешают большие количества Ti, V, Mo, Fe, W, Со, Ni, Си, Ag и Sn, которые можно предварительно отделить электролизом на ртутном катоде и экстракцией купферонатов.
Очень часто при потенциометрическом титровании урана (IV) применяют соли церия (IV) [323, 395, 536, 568, 919]. Хан и Келли [568] использовали титрование сульфатом церия (IV) при определении миллиграммовых количеств урана в присутствии железа. Восстановление до урана (IV) проводили добавлением избытка CrSO4; избыток Сг (II) удаляли продуванием через раствор воздуха или добавлением Ce(SO4)2. Мешающее влияние Fe (II) при титровании урана (IV) устраняли добавлением 1,10-фенантролина. В качестве индикаторного электрода была использована золотая проволока.
Авторы проводили определение урана по следующей методике: раствор ypana(VI), не содержащий NO~-hohob, помещают в титрационную ячейку, добавляют небольшое! количество концентрированной H2SO4, и объем раствора доводят до 4 мл. Продувают через раствор ток чистого СО2, погружают электроды и медленно приливают CrSO4 до полного восстановления урана (VI), что видно по изменению потенциала. Затем добавляют по каплям раствор Ce(SO4)2 для окисления избытка Сг (II) до Сг (III); приливают по 12,5 мл раствора 1,10-фенантролина (0,25 г мл) на каждые 0,1 мг присутствующего железа, и титруют уран (IV) сульфатом церия (IV)] до появления скачка потенциала.
Для количеств урана 0,5—2,5 мг в присутствии до 1 мг Fe средняя относительная ошибка составляет ±1,2%.
Не мешают определению урана данным методом А1, Ni и др.
Метод потенциометрического титрования урана (IV) сульфатом церия (IV) использован для определения урана в металлическом цирконии [919] и его сплаве с ураном. После разложения образца циркония уран (VI) восстанавливают в солянокислом растворе на свинцовом редукторе, добавляют раствор соли Fe (Ш) и образующееся при этом Fe (II) титруют потенциометрически стандартным раствором Ce(SO4)2, используя платиновый индикаторный электрод. Данным методом можно определять от 5 до 0,05% 214
урана в цирконии с достаточной точностью (при определении 0,05 % ошибка составляет около 8°о (отн.)).
Бане [395] определял микроколичества урана и плутония (после их восстановления цинковой амальгамой) потенциометрическим титрованием раствором церия (IV) в атмосфере азота с использованием специальных дифференциальных электродов. Титрование урана (IV) проводят в 4N растворе H2SO4, плутоний в этих условиях не титруется и не мешает определению урана; затем раствор разбавляют до 1 N по H2SO4 и проводят титрование плутония.
Как указывают авторы, миллиграммовые количества урана и плутония (1—4 мг) можно определять данным методом с точностью ±0,3 % (отн.). По-видимому, при этом все примеси, способные восстанавливаться цинковой амальгамой и титроваться сульфатом церия (IV), будут мешать определению урана.
Потенциометрическое титрование малых количеств урана (IV) раствором Ce(SO4)2 описано Хельбигом [593а]. Уран (VI) восстанавливают избытком сульфата титана (III); в качестве индикаторного электрода используют платиновую проволоку. Определение микрограммовых количеств урана проводят в объеме раствора 2—8 мкл, при этом перемешивание раствора осуществляется вибрирующим каломельным элементом (электрод сравнения). Ошибка определения миллиграммовых количеств урана составляет ±0,4% (отн.); для микрограммовых количеств ошибка повышается до 2% (отн.).
Разработан автоматический оксидиметрический метод определения микроколичеств урана (10—100 мкг) [323], в котором используется потенциометрическое титрование урана (IV) сульфатом церия (IV). При титровании применяют регистрирующий потенциометр и автоматический титратор [850]. Восстановление урана проводят раствором хрома (II).
Точность определения 10—100 мкг урана данным методом очень высокая (±1,2% (отн.)), но метод применим лишь только при определении урана в чистых растворах.
Находят применение при потенциометрическом титровании урана (IV) бихромат калия [8, 668], а также соли железа (III) 18, 562, 1007].
Вейс [1007] проводил титрование урана (IV), полученного восстановлением урана (VI) в 0,5 N растворе H2SO4 (или 0,4 N растворе HCI) в редукторе Джонса 0,1. N раствором Fe2(SO4)3 в атмосфере СО2 при температуре 70°. Как указывает автор, железо до соотношения Fe : U=100 : 1 не мешает определению Урана.
Другими авторами [562] также проводилось титрование урана(1У) раствором Fe2(SO4)3 с точностью ± 0,1 %; при этом определению урана не мешают Fe, С г, Ni, Cd, Мп, Al; сильно мешают МО3", F", РО43~, V, Mo, W, Си, Sn и Ti.
215
При определенных условиях V и Fe можно титровать в одном растворе с ураном, так как V и Fe дают определенные четкие скачки на кривой титрования [8].
Если присутствуют все три элемента вместе, то после восстановления сначала титруют (раствором К2Сг2О7, КМпО4 или др.) при температуре 80° V (II) до V (III) и U (III) до U (IV) до первого скачка; между первым и вторым скачками одновременно титруются V (III) и U (IV) до V (IV) и U (VI). Затем раствор охлаждают до комнатной температуры и Fe (II) титруют до Fe (III), что отмечается третьим скачком; после этого опять нагревают раствор до 80° и титруют V (IV) до V (V) до четвертого скачка. Уран определяют путем вычитания количества миллилитров окислителя, затраченного между третьим и четвертым скачками, из количества, израсходованного на титрование смеси V (III) и U (IV).
П. Н. Палей и И. С. Скляренко (1950 г.) использовали для титрования урана (IV) ванадат аммония, устанавливая конечную точку потенциометрически; при этом определению урана не мешает присутствие V (IV).
Симон с сотрудниками [920] разработали метод определения урана в рудах, основанный на восстановлении его цинковой амальгамой и титровании урана (IV) в среде избытка бикарбоната, цианида калия и NH4C1 стандартным раствором феррицианида калия.
Авторы предлагают следующий ход определения урана: сернокислый раствор, содержащий 0,5—50 мг урана, восстанавливают в делительной воронке 100 г цинковой амальгамы. После удаления амальгамы нейтрализуют раствор бикарбонатом натрия 'и сверх того добавляют еще 3—5 г NaHCO3 для перевода U (IV) в растворимый комплекс; затем добавляют 2—3 г сухого KCN (для маскирования Zn и других примесей) и 1 г сухого NH4C 1.Переносят раствор в титрационную ячейку и титруют уран (IV) при комнатной температуре 0,1 N раствором K3[Fe(CN)6], устанавливая конец титрования потенциометрически.
Средняя относительная ошибка определения 5—50 мг урана равна ±0,5%.
Изучение влияния примесей показало, что определению урана данным методом мешают Ti (IV), Fe (III), Al, Ag,Cd,V,Mn,Ce(IV) и большие количества PO43-. Влияние Ti устраняют продуванием воздуха через раствор после восстановления, при этом Ti(III) окисляется до Ti (IV) и не мешает титрованию урана (IV).
Железо не мешает определению урана, если его связать в комплекс винной, лимонной кислотами или F--ионом.
Определение урана в присутствии других элементов представлено в табл. 31.
Как свидетельствуют данные табл. 31, при определении урана данным методом результаты получаются удовлетворительные. Авторы применяли метод для определения урана в руде.
Для потенциометрического титрования уранил-иона обычно используют такие сильные восстановители, как соли хрома (II) [174, 511, 510, 489] и титана (III) [755].
216
Таблица 31
Определение урана в присутствии Pb, Bi, Си, As, Sn, Al, Fe, Ni, Co, Cr, W, Ti, Th, Be, Ca, Mg, Po^“
(вводится 23,84 мг V)
Введено примесей, мг Найдено U, мг Относительная ошибка, % Введено примесей, мг Найдено U, мг Относительная ошибка, %
РЬ- 5 23,84 0,00 Сг — 100 23,72 —0,50
РЬ— 50 23,60 — 1,01 W— 30 23,72 —0,50
Bi— 20 23,84 0,00 W - 50 23,60 -1,01
Bi — 100 23,60 — 1,01 Ti - 5 23,72 —0,50
Си— 10 23,84 0,00 Ti— 20 (продували воздух) 23,84 0,00
Си — 100 23,72 -0,50 Ti— 20 (продували воздух) . 24,08 + 1,01
А1 — 5 23,84 0,00 Th— 10 23,84 0,00
А1- 50 23,11 —3,06 Th —100 23,60 — 1,01
Fe— 20 Fe —20 добавлена ВИН- 23,39 —6,08 Be— 20 23,84 0,00
наякислота) Fe — 20 (добавлен 23,72 —0,50 Be— 40 23,72 -0,50
F-) 23,84 0,00 Ca— 20 23,72 —0,50
Ni— 40 23,84 0,00 Mg— 40 23,72 —0,50
' Ni — 100 23,72 —0,50 PO’~— io 23,84 0,00
1О О । 7 6 о 23,72 23,60 —0,50 —1,01 PO’“—50 4 23,60 -1,01
Эл. Щами [489] разработал метод потенциометрического титрования уранил-хлорида раствором СгС12. Стандартный раствор СгС12 приготовляли электролитически по методике, описанной ранее [511]. Индикаторным электродом служила платиновая проволока (^?=0,3 мм), впаянная в стеклянную трубку; в качестве электрода сравнения использовали насыщенный каломельный электрод. Титрование проводили в атмосфере инертного газа (СО2). Было исследовано влияние температуры, кислотности среды и комплексующих кислот (щавелевой, лимонной) на точность определения урана. Авторы рекомендуют проводить титрование уранил-хлорида раствором СгС12 в 8 М растворе НС1 при нагревании до 90—70°. Щавелевая, винная и лимонная кислоты (0,1 М) не мешают.
217’
Минчсвский с сотрудниками [754] определяли большие количества урана в присутствии V (V), Сг (VI) и железа потенциометрическим титрованием уранил-иона раствором сульфата хрома (II).
Титрование урана (VI) проводили в 5%-ном сернокислом растворе при постоянном пропускании через раствор тока СО2 и Н2 (смесь), используя платиновый и каломельный электроды. На кривой титрования получаются четкие скачки, соответствующие восстановлению Сг (VI) до Сг (III), V (V) до V (IV), Fe (III) до Fe (II) и скачок, соответствующий одновременному восстановлению U (VI) до U (IV) и V (IV) до V (III). Содержание Сг (VI), V (V) и Fe (III) можно определить по трем первым скачкам, а содержание урана рассчитывают по последнему перегибу кривой титрования за вычетом того количества миллилитров раствора CrSO4, которое требуется для восстановления V (IV) до V (III) (определяемого по второму скачку). Данный метод определения урана является не очень точным.
Для восстановления урана (VI) можно применить раствор солей титана (III), что было использовано Минчевским [755] при потенциометрическом определении малых количеств урана (VI), содержащихся в двуокиси урана.
Титрование урана (VI) раствором сульфата титана (III) проводили при температуре 70°; для получения более резкого скачка потенциала в конечной точке титрования в раствор вводили сег-нетову соль.
Для определения урана (VI) в UO2 разлагают 4—5 г двуокиси урана 10 мл концентрированной HF и 30 мл воды при нагревании; при этом образуется зеленый осадок UF4, a UO3 переходит в раствор в виде UO2F2. Приливают 100 мл воды и 10 мл полученного раствора помещают в ячейку для титрования; добавляют 10 мл воды, 3 г сегнетовой соли, включают электронагреватель, нагревают раствор до 70° и титруют уран (VI) 0,05 N раствором Т i^SOJg (в 4 N растворе HoSOJ, используя платиновый и насыщенный каломельный электроды. Для удаления растворенного кислорода; а также для перемешивания раствора пропускают непрерывно ток СО2.
Полученные авторами результаты хорошо воспроизводимы, но характеризуются ошибкой (в среднем ±3% (отн.)).
3. Кулонометрическое определение урана
Н. И. Удальцова
Кулонометрия является одним из новейших электрометрических методов, которые нашли широкое применение в аналитической химии многих элементов, в том числе и урана.
Кулонометрический метод основывается на законе электролиза Фарадея, устанавливающем связь между количеством электричества, прошедшим через раствор, и количеством вещества, подвергнувшимся электролитическому превращению под действием тока. Из закона Фарадея вытекает, что для электрохимического превра-218
щения одного грамм-эквивалента любого вещества требуется 96 484 кулона электричества.
Количественный расчет кулонометрического метода основан на уравнении:
где
G — количество вещества, претерпевшее электрохимическое превращение, г;
М — атомный или молекулярный вес вещества;
Q — число кулонов электричества, прошедшее через раствор; п — число электронов, участвующих в электродном процессе. Обязательным условием применения при расчете уравнения (1) является полное 100%-ное использование тока реакцией электрохимического превращения вещества.
Определяемое вещество может непосредственно окисляться или восстанавливаться на электроде (первичный кулонометрический анализ), или же, не претерпевая электрохимического превращения, реагировать в растворе с соединениями, возникающими под действием тока (вторичный кулонометрический анализ или, как его обычно называют, кулонометрическое титрование).
Кулонометрическое титрование получило наибольшее применение в анализе, так как оно позволяет определять большое число элементов, которые сами не способны претерпевать электрохимического превращения со 100%-ным использованием тока.
Первичный кулонометрический анализ проводят обычно при постоянном, контролируемом потенциале генераторного электрода; при этом величина потенциала должна быть такой, чтобы в растворе происходила только желаемая реакция.
Электролиз проводят до полного электролитического превращения определяемого вещества; количество электричества, затраченное на данную реакцию, определяют с помощью включенного последовательно с ячейкой кулонометра. Наиболее часто используют весовые кулонометры (серебряные или медные), отличающиеся наибольшей точностью, а также объемные (водородно-кислородные) и титрационно-химические (иодный, V (IV) и Се (III)) кулонометры. Подробные сведения об устройстве и действии кулонометров можно найти в руководствах по электрохимии [47].
Ряд авторов [678, 707, 737] предложили использовать для определения количества электричества специальные интеграторы тока по времени. Мак-Невин и Бекер [720] использовали для определения числа израсходованных кулонов метод математического расчета по кривой Igr —• t, где i — сила тока, a; t — время, сек.
Кулонометрическое титрование (вторичный кулонометрический анализ) проводят обычно при строго постоянной силе тока; цри этом измерение израсходованного количества электричества становится более точным и значительно упрощается, так как число
219
кулонов в этом случае равно произведению силы тока i (в а) на длительность электролиза t (в сек.).
Очень важным моментом кулонометрического анализа является определение конечной точки электролиза, которую можно установить при помощи обычных методов, применяемых в объемном ана-
лизе.
Колориметрические методы определения конца титрования основаны на использовании индикаторов [455, 794, 961].
При более точных определениях, особенно малых количеств веществ, конечную точку устанавливают физико-химическими ме-
Рис.' 45. Схема цепи для кулонометрического титрования.
Е— электролитическая ячейка; А, А'— индикаторные электроды; В, В'— генераторные электроды; Б—источник постоянного тока (0,1 — 0,5 в) для индикаторной цепи; Б'— источник постоянного стабильного тока; Г— гальванометр; mA— миллиамперметр; 7?i — юстирующее сопротивление; — прецизионный магазин сопротивлений для измерения стабильного тока при помощи потенциометра; Т— электрохронометр; _Н— реле для пуска приборов; Д— контакты реле.
тодами: потенциометрическими [409, 410, 433, 434, 737, 813] или амперометрическими (с использованием одного или двух поляризуемых электродов) [393, 498, 742, 859].
Для определения конечной точки применяют также метод титрования до определенного ’значения величины потенциала индикаторного электрода и др. [533, 408].
Изложению основных принципов кулонометрии и ее применению для определения ряда веществ посвящено большое число работ, многие из которых приведены в литературных обзорах [1,472,532, 841, 957, 961, 992, 1025]. Теоретический анализ процесса кулонометрического титрования дан Делахеем [456].
Предложены многочисленные схемы кулонометрических
установок и ряд автоматических приборов, позволяющих применять или стабильное напряжение [322, 703, 794], или постоянную силу тока [408, 455,702,736,798, 844, 894]. Описаны приборы, позволяющие работать одновременно при стабильном напряжении и постоянной силе тока [842, 844].
Простейшая схема установки для кулонометрического титрования представлена на рис. 45.
Величину генераторного тока трудно измерить с достаточной точностью с помощью обычных электроизмерительных приборов, поэтому ток пропускают через прецизионный магазин сопротивлений; падение напряжения на этом сопротивлении позволяет определить величину генераторного тока с большой точностью.
220
В кулонометрическом анализе предложены различные типы электролитических ячеек. Типичная ячейка для кулонометрического титрования при постоянной силе тока с внутренней генерацией титрующего агента описана Фурманом [431] (рис. 46). В‘данной ячейке генерируемый и анализируемый растворы находятся в одном и том же электролизере.
J В тех случаях, когда само определяемое вещество может подвергаться электрохимическому превращению или возможны какие-либо побочные про
цессы, генерацию реактива производят вне реак-
Рис. 46.Ячейка для кулонометрического титрования с внутренней генерацией титрующего агента.
Ег —генераторный электрод;
Е а —вспомогательный электрод: Е,, Е2— индикаторные электроды; 1— стеклянная пористая перегородка.
Рис. 47. Ячейка для кулонометрического титрования с внешней генерацией титрующего агента.
1— раствор для генерации реагента; 2— электроды из платиновой проволоки; 3— стеклянная вата; 4 — отводящая трубка; 5— механическая мешалка; 6 — сосуд с испытуемым образцом; 7—резиновые трубки.
ционного сосуда. Конструкция электролитической ячейки с внешней генерацией реагента, описанная рядом авторов [454, 455], представлена на рис. 47. Внешняя генерация реагента позволяет применять оптимальные условия и избежать влияния вторичных, побочных процессов.
Недостатком кулонометрического анализа, ограничивающим его применение, является искажение результатов из-за побочных процессов окисления или восстановления других веществ и из-за химического взаимодействия продуктов генерации не только с определяемым веществом, но и с другими, следствием чего является не 100%-ная затрата электричества на протекание основной реакции.
221
В ряде случаев этих недостатков можно избежать, правильно подбирая условия проведения электролиза и состав раствора в электролизере. Со 100%-ным использованием тока, как установлено, могут генерироваться ряд окислителей и восстановителей (Br2, J2, С1„. Fe (III), гипобромит, Се (IV), МпО4-, Си-, Fe (II), Ti (III) и др.), которые широко применяются для определения большого числа веществ как неорганических, так и органических.
Кулонометрический анализ имеет ряд существенных преимуществ по сравнению с другими аналитическими методами:
1. Кулонометрия позволяет определять очень малые количества веществ с большой точностью; можно определять вещество в количестве до 10“12 г/экв (это соответствует генераторному току порядка 10-6 а!сек:, имеющиеся приборы позволяют измерять время с точностью ± 0,01 сек. и количество электричества порядка 60 электронов в 1 сек.).
2. Метод позволяет использовать при определении ряда веществ такие нестойкие реактивы, как Cr (II), Ti (III), Си (I), Cl2, Sn (II) и др., применение которых при обычном анализе связано с большими трудностями.
3. При выполнении кулонометрического титрования нет необходимости предварительно готовить точные стандартные растворы, устанавливать их титры и измерять их объемы; стандартом в данном методе является единица количества электричества (кулон).
4. Во время кулонометрического титрования раствор не разбавляется, что особенно важно при работе с очень разбавленными растворами.
5. Кулонометрический метод имеет большие возможности для автоматизации процесса определения веществ.
Первые работы по кулонометрическому определению урана были опубликованы в 1953 г.
Карсоном [410] описан метод определения малых количеств урана, в основе которого лежит восстановление урана (VI) до урана (IV) в свинцовом редукторе и титрование урана (IV) электролитически генерируемым бромом (в присутствии соли железа (III)).
Установка кулонометрического титрования при постоянной силе тока, использованная автором в работе, схематически представлена на рис. 48. Генераторная система состоит из двух платиновых электродов, изолированных друг от друга: анод погружается в испытуемый раствор; катод, погруженный в 1 V раствор H2SO4, изолирован от анода стеклянной пористой перегородкой.
Для определения конечной точки был использован метод «производного полярографического титрования», заключающийся в потенциометрическом титровании при постоянной силе тока с применением двух поляризуемых электродов. Индикаторными электродами служили две платиновые проволоки, которые поляризовались постоянным током (1,1—1,3 лнш); в качестве источника 222
тока была использована батарея (45 в), соединенная параллельно с сопротивлением (40 Д1о.ч). Индикаторная система подробно описана в литературе [843].
Источник постоянного тока для генераторной системы, описанный автором ранее [408, 409], имеет электронную регулировку. Значение постоянной силы тока генераторной системы определялось путем измерения падения напряжения на прецизионном постоянном сопротивлений 100 cut) с помощью потенциометра.
Рис. 48. Схема установки для кулонометрического титрования урана (IV) при постоянной силе тока.
1— ячейка; 2— генераторный катод; 3— генераторный анод; 4— индикаторные платиновые электроды; 5— источник постоянного тока; 6— потенциометр; 7— часы: 8— рН-метр; 9— стеклянная пористая перегородка.
Уран (VI) предварительно восстанавливают в среде НВг на свинцовом редукторе, а затем окисляют уран (IV) в атмосфере инертного газа (СО2 или N2) при температуре 95° электролитически генерируемым бромом в присутствии соли железа (III). Данным методом определяют 0,01—7 мг урана с точностью от ±0,2% до ±10% (отн.) (для микроколичеств).
Автором исследовалось влияние примесей на кулонометрическое титрование урана (IV) бромом; было найдено, что элементы, способные восстанавливаться в свинцовом редукторе и титроваться бромом (Sb (V), As (V), Cr (VI), Л-,МоОГ, PtCls2± Sn (IV), Ti, W, оксалат-; формиат- и сульфид-ионы) мешают определению. Мешаюшее влияние оказывает также присутствие Районов.
Как указывает автор, процесс титрования может быть легко автоматизирован.
Фурман с сотрудниками [534] разработали метод кулонометрического титрования урана (IV) при постоянной силе тока (IV).
223
Для восстановления U (VI) до U (IV) была применена 20%-ная амальгама кадмия, затем U (IV) окислялся раствором Fe (III), а образующееся при этом Fe (II) титровалось электролитически генерируемым Се (IV) в атмосфере углекислого газа при комнатной температуре. Конечная точка определялась с помощью метода «производного полярографического титрования» [8431.
Се (IV) генерировался на платино-иридиевом аноде, изолированном от катода, из 3N раствора H2SO4, насыщенного сульфатом церия (III). .
1—5 мг урана можно определить данным методом с точностью ±0,3% (отн.); микроколичества урана (5—10 мкг) определяются с точностью ±5—8% (отн.). Указывается, что все элементы, восстанавливающиеся амальгамой кадмия и титрующиеся церием (IV), а именно Fe, Мо, W, Ti и др., мешают определению урана и должны быть предварительно удалены.
Преимуществом данного метода является большая точность при определении миллиграммовых количеств урана.
Возможность кулонометрического титрования уранил-иона при постоянной силе тока электролитически генерируемым титаном (III) установлена Лингейном с сотрудниками [705]. Следует отметить, что ранее некоторыми исследователями [534] делались попытки применить Ti (III) для кулонометрического определения урана, но им не удалось добиться 100%-ного использования тока при генерации Ti (III). Лингейн, используя ртутный катод и соответствующие условия (кислый раствор, pH 1, содержащий цитрат и Ti (1У)-ионы, и нагревание раствора до85°), достиг 100%-ного использования тока при получении Ti (III).
Для работы автором была использована электролитическая ячейка специальной конструкции, показанная на рис. 49. Емкость ячейки 200 мл, площадь ртутного катода равна 8 см2. Кадмиевый стержень, погруженный в 1 М раствор CdC 12, служит анодом; анодное пространство изолировано пористой стеклянной перегородкой.
Сила тока (--40 мка) при генерации Ti (III) поддерживалась постоянной автоматически с точностью ±0,01% при помощи специальной аппаратуры, включающей электронику [702]. Конец титрования устанавливали потенциометрически. Время электролиза измерялось электрочасами с точностью ±0,01 сек.
Авторы предлагают следующий ход титрования U (VI): раствор, содержащий уранил-ацетат, помещают в ячейку, разбавляют водой до 25 мл, приливают 50 мл 0,08 М раствора хлорида или сульфата титана (IV) (в 1 М растворе цитрата с pH 1); продувают через раствор чистый азот и нагревают до 85°. Вводят в ячейку определенное количество чистой сухой ртути и начинают вести электролиз при постоянном пропускании через раствор тока азота. Электролиз прекращают, когда потенциал индикаторного электрода достигает скачка, соответствующего эквивалентной точке титрования. Количество урана вычисляют по уравнению (1).
С помощью данного метода можно определить 112—20 мг U (VI) с точностью ±0,02—0,08 %(отн.).
224
из
концентрированного раствора
Рис. 49. Электролитическая ячейка для кулонометрического титрования урана (VI) при постоянной силе тока.
I— индикаторный электрод; 2— стержень из кадмия: 3 —ртутный катод; 4— солевой мостик: 5— термометр; 6— электрообогреватель; 7— ток азота; 8— магнитная мешалка.
Определению урана мешают элементы, титрующиеся титаном (III), а также F-, С2О;~ и др., что является недостатком метода.
Преимущество этого метода перед вышеописанным заключается в том, что не требуется предварительного восстановления U (VI) до U (IV).
В дальнейшем Лингейну и Кеннеди удалось добиться 100 %-ного использования тока при генерации Ti (III) на платиновом электроде при комнатной температуре H2SO4 1706, 644], что было использовано авторами для кулонометрического титрования урана (VI) при постоянной силетока [644]. Генераторным катодом служила платиновая фольга с площадью 14,9 см2; в качестве анода была использована платиновая проволока, погруженная в 1 N раствор H2SO4; анодное пространство отделено от катода стеклянной пористой перегородкой. Электролитическую генерацию Ti (III) производили из 100 мл 6—8 N раствора H2SO4, содержащего 0,6 М сульфата титана (IV), при плотности тока 3 ма/см2. Реакция восстановления U (VI) титаном (III) ускорялась добавлением соли железа (II) (в количестве 10 устанавливали амперометрически (с новых индикаторных электродов).
Для количества урана 10—200 мг точность определения очень большая (±0,03% (отн.)). Метод может быть применен для определения урана в присутствии ванадия (до соотношения U : V= 1 : 10) с точностью ±0,3% (отн.).
Буманом с сотрудниками [383] разработано кулонометрическое определение урана, основанное на электролитическом восстановлении U(VI) до U (IV) на ртутном катоде при контролируемом потенциале. Точный контроль потенциала ртутного катода осуществлялся Применением специальной аппаратуры, включающей электронные приборы. Количество электричества, израсходованное на восстановление U (VI), определялось прецизионным интегратором тока. Подробное описание аппаратуры для выполнения электролиза при контролируемом потенциале и устройства интегратора тока приведено в статье Бумана [381].
Аналитическая химия урана
мг).
Конечную точку титрования использованием двух плати-
225
Для определения конца электролиза нет необходимости в данном случае использовать индикаторную систему, так как электролиз прекращают, как только ток в цепи достигнет значения тока одного фона. Электролиз проводят в 1 М растворе H2SO4, при потенциале катода — 0,25 в (отн. электрода Ag AgCl/KC 1нас) или в цитратном растворе, 1 М по лимонной кислотен 0,1 М по A12(SO4)3 с pH 4.5. при потенциале, равном — 0,60 в. Предварительно продувают через раствор ток азота в течение 15 мин. для удаления растворенного кислорода. Электролиз в сернокислой среде следует вести 5—10 мин.; электролиз в цитратной среде протекает медленнее и длительность его увеличивают до 30—40 мин. Работать необходимо с очень чистой ртутью, продажную ртуть следует предварительно тщательно очищать.
Данным методом можно определить 76—0,0075 мг урана с большой точностью. Для количеств урана 75—7,5 мг в 1 М H2SO4 авторы получили среднее отклонение 0,03%, а для количества 0,75— 0,0075 мг в цитратном растворе — 1,3—2,6%.
Установлено, что уран можно определять таким путем в присутствии Hg (II), Си (II), Fe (III) и больших количеств NO~; мешающее влияние данных примесей устраняется предварительным проведением электролиза при более положительном потенциале. Допустимые содержания этих примесей при определении урана приведён ы в табл. 32.
Таблица 32
Допустимые содержания примесей при определении урана (381]
Примесь Hg (П) Си (П) Fe (III) Се (IV) Al (III) НС1 HNO,
Отношение примесь уран (в молях) 6,0 0,8 0,5 0,1 175,0 >170 700,0
Влияние других примесей на определение урана не изучалось. Данный метод, как указывается авторами, может быть применен для определения урана в отработанном ядерном горючем.
Феррар, Томсон и Келли [497] применили метод кулонометрии при контролируемом потенциале для определения урана в этом же. объекте. Метод основан на восстановлении урана (VI) на ртутном катоде при потенциале —0,30 в в 1 М растворе H2SO4. Применяемая авторами аппаратура для кулонометрического определения урана очень мало отличается от аппаратуры, использованной Бу-маном [381]. Количество электричества, израсходованное на восстановление урана (VI), определяется также с помощью специального прецизионного интегратора тока. Электролиз заканчивают, когда ток в ячейке достигает определенного значения (0,05 ма — ток фона).
226
Авторами было изучено влияние ряда элементов, в том числе и продуктов деления, на определение урана данным методом, и было найдено, что мешающее влияние Си (II), Fe (III), Ag, Се (IV), J2, Rh и др. можно устранить предварительным восстановлением при нулевом потенциале катода; Mo (VI) и Cr (VI) отделяют путем экстракции их насыщенным раствором се-бензоиноксима в хлороформе.
Как показывают результаты авторов, данным методом можно определить уран в количестве 5 .иг с точностью ±0,2% (отн.) в присутствии 0,3 мг Си (II); 0,5 мг Mo (VI); 0,5 мг Cr (VI) и 1 мг N i (II).
Определение урана (VI) в присутствии продуктов деления представлено в табл. 33.
Таблица 33
Определение 5 мг урана на фоне продуктов деления в 1 N растдоре H,S04
Примесь Содержание примеси, мкг Потенциал восстановления примеси в 1 jV HnSO,, в Ч исло опытов Средняя относительная ошибка, %
Тс 50 -0,5 6 0,18
Ag 500 0,0 5 0,15
Ей 500 —0,8 8 0,20
Pd 500 0,0 5 0,10
Се (IV) 200 0,0 8 0,16
J- 500 0,0 5 0,10
Rb 100 >—1 4 0,18
Те 200 —0,7 5 0,12
Rh 100 0 5 0,16
Nb 100 Не восстанав- 3 0,10
ливается
La 100 >—1 3 0,10
Cs 100 Не восста- 2 0,12
навливается
Сильно мешает определению урана F~-hoh, а также сурьма (III) в количестве более 200 мкг.
Шульц и Томсон [9081 использовали описанный выше метод для. одновременного определения урана (VI) и меди. Оба элемента сначала восстанавливают на ртутном катоде в 1 N растворе H2SO4 при —0,3 в, а затем определяют медь путем окисления ее при потенциале ±0,175 <$. Данным методом авторы определяли миллиграммовые количества U и Си с точностью ±0,1% (отн.).
Буман с сотрудниками [382] разработали высокочувствительный и очень специфичный метод определения урана в отработанном ядерном горючем, основанный на отделении урана почти от всех примесей (кроме технеция и рутения) экстракцией метилизобутил-
15*
227
кетоном [721] и на последующем кулонометрическом определении урана при контролируемом потенциале [383].
Экстракция урана метилизобутилкетоном проводилась из раствора, содержащего нитрат алюминия, NH4OH и гидроокись тетра-пропиламмония. В этих условиях вместе с ураном экстрагируются лишь Тс и Ru, которые отделяются затем от урана путем сплавления при 600° с бисульфатом натрия и НС1О4.
Отделение Тс и Ru и кулонометрическое определение проводятся в одной и той же ячейке специальной конструкции, изготовленной из боросиликатного стекла. Ячейка представляет собой трубку емкостью 50 мл, имеющую на дне U-образный отвод для соединения ртутного катода с током.
Авторы предлагают следующий ход определения урана в облученном ядерном горючем: аликвотную часть раствора (0,750 мл) помещают в экстрактор, содержащий 6 мл раствора, 2,5 М по A1(NO3)3, 1 Al по NH4OH и 0,005 М по гидроокиси тетрапромиламмония, и добавляют 3 мл метил изобутил кетона. Экстрагируют уран в течение 2 мин.Отбирают аликвотную часть органического слоя (2 мл) и помещают в электролитическую ячейку, выпаривают раствор досуха, нагревая сосуд горячим воздухом; добавляют в ячейку 0,55 г бисульфата натрия и 0,20 мл 79% -ного раствора НСЮ4. Нагревают сосуд до 600° в печи и прокаливают при этой температуре в течение 20—30 мин. После растворения плава в 2 мл воды прибавляют 0,5 мл 0,25 М раствора A12(SO4)3 в 0,25 М растворе H2SO4 и 4 мл 1,2 А1 раствора цитрата калия, имеющего pH 4,6. Затем добавляют соответствующее количество свежепе-регнанной ртути, продувают через раствор в течение 15 мин. азот, проводят электролиз при —0,25 в (отн. нас. к. э.) в течение 20 мин. для удаления легковосстанав-ливающихся примесей, и затем ведут электролитическое восстановление U (VIj до U (IV) при потенциале — 0,65 в (отн. нас. к. э.) до полного восстановления урана, что определяют по величине тока. Количество электричества, израсходованного на восстановление урана, определяют с помощью интегратора тока, собранного на блоках усилителей, которые применяют в электронных аналоговых вычислительных машинах.
Продолжительность полного анализа около 2,5 час. Данным методом можно определить очень малые количества урана (до 0,3 мкг) с большой точностью. По своей чувствительности этот метод близок к спектрофотометрическому и флуоресцентному.
Следует сказать, что в литературе опубликованы работы, в которых для кулонометрического титрования ряда элементов [Се (IV), V (V) и др.] используются электролитически генерируемые восстановители уран (IV) [909] и уран (V) [486, 805].
III. РАДИОХИМИЧЕСКИЕ МЕТОДЫ Метод изотопного разбавления
Г. Н. Б и л и м о в и ч
В настоящее время метод изотопного разбавления широко применяется в химическом [318], эмиссионном спектральном [26], а также в масс-спектральном анализах [628].
Как известно, обычные химические методы количественного анализа требуют максимально полного выделения определяемого эле
228
мента. Метод изотопного разбавления открывает в этом направлении новые возможности, позволяя при неполном выделении определяемого элемента количественно его определять. Это особенно ценно в применении к элементам, у которых отсутствуют достаточно удовлетворительные количественные методы определения, а также к трудноразделяемым элементам со сходными свойствами, где чистота и количественное выделение взаимно исключают друг друга.
В качестве индикаторов применяются как стабильные, так и радиоактивные изотопы. В случае применения стабильных индикаторов измерения проводятся на масс-спектрометре при использовании радиоактивных изотопов — на счетчике Гейгера-Мюллера.
Ниже будут рассмотрены различные варианты метода изотопного разбавления и практическое применение его для анализа урана.
Существует несколько вариантов метода изотопного разбавления, из них наиболее распространенными и перспективными являются следующие:
1. Разбавление радиоактивным изотопом (прямое изотопное разбавление)
Данный вариант метода нашел наиболее широкое распространение в практике количественного органического и неорганического анализа. Метод основан на учете изменения первоначальной удельной активности индикатора, введенного в анализируемую смесь, в результате разбавления исследуемым неактивным соединением.
Наиболее простым случаем является тот, при котором весовое количество прибавленного активного соединения настолько мало по сравнению с количеством неизвестного неактивного соединения, что им можно пренебречь. При этом извлеченное количество активного соединения прямо пропорционально количеству выделенного неактивного соединения.
В этом случае вычисление производится по формуле: д
где
— количество определяемого элемента;
Ws — выделенное количество элемента;
Л, — активность, введенная с индикатором;
А3 — активность выделенного соединения.
Если нельзя пренебречь количеством разбавляющего вещества, то к определяемому элементу с неизвестным весом W2 добавляют некоторое известное количество его радиоактивного изотопа Удельная активность которого предварительно определена и равна
1 w'f
229
Уравнение для вычисления количества определяемого элемента в этом случае будет:
/ Q \
= (2)
Так как и S, известны, то, определив S2 (удельную активность выделенного соединения), можно по формуле (2) рассчитать W 2.
л
Подсчет St = S, = легко произвести, если, например, осадить некоторую часть смеси взвесить и измерить ак-
тивность осадка.
Различие между прямой радиометрической поправкой (1) и методом изотопного разбавления (2) заключается в необходимости для последнего вычитать количество добавленного меченого вещества. В действительности, способ подсчета (по изотопному разбавлению или радиометрической поправке) зависит от удельной активности индикатора.
Метод изотопного разбавления применим и в том случае, когда неизвестное соединение активно, но имеет удельную активность, отличающуюся от активности разбавляющего вещества. Уравнение для подсчета в этом случае имеет вид:
(3)
\°2 °0/
где
= —удельная активность вводимого индикатора;
So = ^~ — удельная активность исходного соединения; Ло — активность неизвестного соединения; .
4 Д-Д
S2 = — удельная активность смеси после разбавления.
** 1 ~Г "2
Для получения препаратов требуемой чистоты применяется ряд методов, таких как осаждение, экстракция, электролиз, ионообменная хроматография, ионофорез, дистилляция, кристаллизация и др. Выделенное количество элемента определяется весовым методом, спектрофотометрическим, полярографическим или другими химическими, физико-химическими, а также масс-спектральным методами.
Точность метода изотопного разбавления зависит от ряда факторов: от радиохимической чистоты изотопа, удельной активности применяемого индикатора, соотношения количеств определяемого элемента и индикатора в исследуемых растворах,чистоты выделенных Соединений, от техники и методики радиометрических измерений [18]. При соблюдении оптимальных условий будут иметь значение лишь аналитические погрешности обычного количественного анализа, например, неточность взвешивания, колориметрического определения и т. д.
230
2. Метод изотопного разбавления
с м а с с-с п е к т р а л ь н ы м окончанием [538, 630, 1006]
Определение методом изотопного разбавления с использованием стабильных изотопов основывается на изменении изотопного состава исходного элемента при добавлении известного количества изотопически обогащенного стабильного изотопа в качестве индикатора.
Анализ состоит в добавлении к исследуемому образцу известного количества стабильного изотопа определяемого элемента, гомогенном смешении, выделении, химической очистке и определении соотношения изотопов с помощью масс-спектрометра.
Для анализа пригодны все элементы, имеющие несколько Изотопов, а также некоторые моноизотопные элементы, для которых можно приготовить второй изотоп искусственным путем, например в ядерном реакторе.
Метод является весьма чувствительным, поэтому для проведения изотопного анализа часто требуется не больше 50—100 мкг вещества. Для большей точности рекомендуется, чтобы отношение элемента в образце и в индикаторе было близко к 1. Ясно, что точность определения уменьшается не только тогда, когда достигается предел чувствительности, но и тогда, когда количество элемента в образце много больше добавленного радиоактивного индикатора.
Метод может быть применен и для определения больших количеств (^-8—11%), однако в этих случаях целесообразно использовать более простые методы анализа.
Концентрация определяемого вещества подсчитывается по формуле:
к_ Г(ailaj)M ~ (ailaj)7'1 MWS Д ~ aj L (aiiaj)s ~ (fiilaj)™ J MWT ’ где
at — относительная распространенность изотопа «/»;
а,-
~ — отношение двух изотопов;
M.WS—молекулярный вес определяемого элемента;
М. WT— молекулярный вес индикаторного изотопа.
В этом уравнении «5» относится к исходному образцу, «Г» к индикатору и «Л1» к смеси образца и индикатора.
Чувствительность метода 10-10 а, а для щелочных металлов до 10-14 г.
Точность 1—3% (отн.).
3. Разбавление активного изотопа нерадиоактивным изотопом
Как правило, разбавление нерадиоактивным изотопом применяется для определения выхода радиоизотопов при исследовании продуктов ядерных реакций, например в радиоактивационном анализе. Этот случай мы здесь подробнее не рассматриваем.
231
4. Многократное изотопное разбавление неактивным изотопом
(двойное изотопное разбавление)
Если требуется определять такие количества вещества, которые не могут быть выделены самостоятельно из сложной многокомпонентной смеси, может быть применен метод изотопного разбавления в несколько видоизмененном виде.
В этих случаях можно проведением нескольких разбавлений неактивным изотопным носителем аликвотных частей исследуемой активной смеси найти количество определяемого элемента.
Анализ проводится следующим образом: к двум равным аликвотным частям исследуемой смеси добавляют различные, но известные количества неактивного изотопа (рекомендуется 10-кратный и 20-кратный избыток по сравнению с определяемым количеством элемента), выделяют любым, но одинаковым методом части смеси (W2-Ь 1^Доб. 1) и (й^г+^доб.г) и измеряют их удельные активности Sj и S2. Расчет ведется по следующей формуле:
^добл , (5)
Аналогичные уравнения получаются, когда одинаковое количество носителя добавляется к различным количествам исследуемого радиоактивного вещества.
Основным преимуществом этого варианта метода по сравнению с предыдущим является возможность работы с микроколичествами элементов (<1 мг).
Точность определения малых содержаний элементов этим методом определяется степенью разбавления исследуемого образца неактивным изотопом, так как чрезмерное разбавление приводит (уравнение 5) к алгебраическому тождеству. Наилучшие результаты получаются при максимальном разбавлении носителем в 20 раз при условии, что IFA06.i =2Ц7дОб.2 и точности радиометрических измерений составляют 1 %.
При этом точность данного варианта может достигнуть 20%(отн.).
5. Метод производного разбавления
Данный вариант метода состоит в том, что к исследуемой смеси добавляется меченый реактив с известной удельной активностью. Метод позволяет определять чрезвычайно малые количества. Однако он еще сравнительно мало распространен и применяется лишь в биологических исследованиях [647].
6. Метод многократного радиоактивного разбавления для определения малых количеств примесей [81]
В основе метода лежит различное изменение удельной активности индикатора за счет разбавления различных количеств его радиоизотопа равными аликвотными частями исследуемого вещества. Если в раствор, содержащий «х» миллиграммов исследуемого вещества добавить лги,» миллиграммов его радиоактивного изотопа с удельной активностью So, то удельная активность смеси этого вещества станет равной
1 —J— X
При введении в такой же раствор «т2» миллиграммов радиоактивного вещества с той же удельной активностью, удельная активность смеси будет равной
2 “ + х ’
Если из обоих растворов выделить одинаковые количества анализируемого вещества «//-> мг, то, определив их активность, можно непосредственно определить количество исходного вещества.
Активность выделенного из первого раствора вещества
д. = S. • у = • у,
mi tv тл х
а из второго раствора
А — <? .11 - S0'm2 . ,,
Я2 — д2 У т2 + х У'
Решая систему двух уравнений с двумя неизвестными, получаем: г_____________mvm2 И2-ДД_____т2 (Д2 - ДД д___т2
Д1-т2 — Д2-гП1 В-А1 — А2 ’ А тл ’
Поскольку при измерении радиоактивности в определенных условиях активность пробы пропорциональна интенсивности счета пробы, то
где /1 и /2 — соответственно интенсивности счета выделенных проб из 1-го и 2-го растворов. Таким образом, для определения неизвестного количества анализируемого вещества необходимо лишь знать количество введенных радиоактивных веществ т., и т2, измерить интенсивность выделенных проб 1Х и /2.
Оптимальными условиями применимости данной формулы являются т1 = х и тя^>х.
233
Данный метод устраняет основной недостаток метода изотопного разбавления — определение удельной активности выделенного соединения, позволяя наряду с этим определять малые количества вещества порядка 10_5—10-7%.
Значительной трудностью метода является разработка в каждом отдельном случае методов выделения равных количеств исследуемого вещества.
Однако здесь не важно, какие количества будут выделены, и не мешает наличие любых примесей других веществ; важно, чтобы эти количества были одинаковыми и чтобы выделение было неполным. Точность около 10% (отн.).
При этом и в методе многократного разбавления неактивным изотопом и в методе разбавления радиоактивным изотопом очень важное значение имеет статистика радиометрических измерений.
Метод может быть применим в тех случаях, когда имеется определенная растворимость труднорастворимых соединений в некоторых средах, пропорциональность количества выделенного на катоде металла пропущенному току при электролизе в определенных условиях, создание насыщенных адсорбционных слоев на адсорбенте, образование строго заданных количеств соединений, экстрагирующихся экстрагентом в определенных условиях, и др.
Практическое применение метода изотопного разбавления к анализу урана
Уран как элемент, имеющий несколько изотопов, очень удобен для масс-спектрального анализа. Возможность получения отделенных или обогащенных стабильных изотопов с помощью магнитного сепарирования позволила использовать их в качестве индикаторов в методе изотопного разбавления.
В последнее время метод изотопного разбавления в сочетании с масс-спектральным окончанием все чаще применяется как метод анализа для определения следовых количеств элементов [630, 1006].
С помощью метода изотопного разбавления может быть решен целый ряд сложных задач как в области ядерной физики и химии {580, 847], так и в геохимии.
Тилтон и др. [629, 797] определяли микроколичества 5-10~7— —1-10~6% урана в метеоритах. Точность 1% (отн.).
На рис. 50 приведен массовый спектр, показывающий использование метода изотопного разбавления для определения следовых количеств урана.
Содержание урана в каменных метеоритах определял Патерсон. Концентрация урана составляла от 1 до6- 10~8а U/a. Поданным Тилтона, содержание урана в базальте составляет 4,98- 10-7aU/a, а в дуните - 1,6- 10-’а U/a [571].
234
Метод изотопного разбавления с масс-спектральным окончанием был применен для определения возраста минералов по соотношению pb207U238, Pb207U235 и РЬ207РЬ206[599].
Для этого к образцам минералов добавлялИ'Известные количества свинца, обогащенного изотопом РЬ208, и урана, обогащенного U235. ^Чувствительность метода анализа для урана 2-10~12а. Точность 2—3% (отн.).
Тилтон и др. [978] изучали изотопный состав и распределение свинца, урана и тория в различных геологических минералах.
Табл. 34 показывает точность метода изотопного разбавления с масс-спектральным окончанием.
Таблица 34
Содержание урана и тория в различных геологических материалах
Образец Уран Торий
частей на миллион
Циркон 2650±40 2180±20
Сфен 303±5 5375 ±75
Пертит 0,22±0,03 0,410±0,008
Каменный метеорит . . . 0,0054±0,0002 i —
Нитрат аммония .... 0,000075±0,000004 —
235
Недавно был описан также метод изотопного разбавления с масс-спектрометрическим окончанием для одновременного определения общего количества и соотношения между изотопами U234, U23e и U238 в растворах, образующихся в процессе переработки отработанных урановых блоков, обогащенных U235, с предварительным отделением от других продуктов деления и от посторонних элементов экстрагированием в 3%-ный раствор трибутилфосфата в н. гексане. Уран выделяли из органической фазы в виде UO4-2Н2О, растворяли в 12 N HNO3, раствор выпаривали на металлической нити и относительное содержание изотопов определяли на масс-спектрометре.
В качестве индикатора добавляли U233 (--50 мкг). Отношение веса урана в образце к весу добавленного урана было равно 20. В этом случае ошибка определения общего урана составляла +3,7 %, а ошибка определения главного изотопа урана ±0,13% (отн.).
Метод применим при концентрациях урана 0,001—350 мг/мл. Продолжительность одного определения 2 часа [552].
Значительный интерес представляет работа по определению урана в морской и речной воде, а также в наносных пластах [855, 1006]. Предварительно уран концентрировали экстракцией (2-этил-гек-сил)-фосфорной кислотой в СС14, реэкстрагировали его с помощью (NH4)2CO3, раствор выпаривали, растворяли в 8 V НС1 и сорбировали на анионите дауэкс-1. Выход при химических операциях контролировался с помощью U233. Азотнокислый раствор урана наносили на Та-нить накаливания, и соотношение изотопов U238/U235 измеряли на масс-спектрометре. В качестве индикатора служил обогащенный изотоп U235.
Авторами найдено среднее содержание урана в речной воде и наносных пластах 0,03—2,39-10-6 г/л, а в морской воде—3,3-10~6 °/0.
Метод изотопного разбавления был также использован для количественного спектрального определения урана в разных продуктах в диапазоне 1—0,005% U путем введения в пробу в качестве внутреннего стандарта изотопов определяемого элемента U23S или U233; или обоих вместе [40] (см. гл. VII).
в: ФИЗИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ УРАНА
1. РАДИОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ УРАНА В ПРИРОДНЫХ МАТЕРИАЛАХ
А. С. Сердюкова
Краткая характеристика радиометрических методов
Определение содержания U в природных материалах (рудахг породах и минералах) радиометрическими методами основано на использовании излучения, испускаемого U и продуктами его распада.
236
При радиометрических измерениях исключается необходимость в предварительной химической обработке пробы, так как интенсивность и качество испускаемого излучения не зависят от физического н химического состояния урановых соединений, а разработанные методики измерений позволяют исключать влияние вмещающих U соединений.
В зависимости от типа и состава исследуемого образца для определения содержания U могут быть использованы простые радиометрические методы (альфа, бета, гамма, метод микрорадиографии), комбинированные радиометрические методы или' методы спектроскопии.
В комбинированных радиометрических методах производится комплексное измерение исследуемой пробы по различным видам излучений, иногда с дополнительным применением дискриминации по энергии. Альфа, бета, гамма методы применяются в основном для определения содержания U в урановых равновесных рудах. Комбинированные радиометрические методы используются для определения содержания U в урановых неравновесных рудах, в урано-то-риевых равновесных и неравновесных рудах, для определения содержания U в горных породах.
Для определения содержания U в породах (в отдельных включениях минералов), в урановых или урано-ториевых минералах также широко применяется метод микрорадиографии.
В последние годы определение содержания U в рудах, минералах, породах, шлихах производится с помощью весьма перспективного метода у-спектроскопии.
При использовании простых — альфа (а), бета (0) или гамма (у)-методов измерение исследуемых образцов (проб) может производиться импульсным или ионизационным методами.
Из ионизационных методов практически используется только a-метод, который при соблюдении соответствующих требований является надежным методом анализа U руд с содержанием U до 0,001%.
0-Ионизационный метод из-за его малой чувствительности применяется лишь при измерении руд с высоким (порядка 5—10%) содержанием U.
Импульсный а-методс использованием а-сцинтилляционных счетчиков является основным методом анализа минералов и малых навесок проб руды.
0-Импульсный метод является очень чувствительным и применяется для измерения содержаний U в пробах в очень широком диапазоне. Существенным преимуществом его перед а- (и частично у-) ^Методом является меньшая зависимость от влияния ряда факторов, ^Которые часто бывают трудноконтролируемыми.
у-Импульсный метод со счетчиками Гейгера-Мюллера по чувствительности уступает 0-импульсному, а чувствительность у-импульс-jioro метода со сцинтилляционными счетчиками является вели
237
чиной того же порядка, что и чувствительность Р-импульсного метода.
При применении а- и у-методов необходимо, чтобы проба и эталон имели одинаковый или близкий химический состав вмещающего вещества. Следует также иметь в виду, что применение у-метода ограничивается практически неэманирующими пробами, так как результаты определения содержания U с помощью у-метода будут занижены примерно на величину коэффициента эманирования. При измерении эманирующих проб а-импульсным методом с помощью счетчиков и ^-методом результаты будут занижены примерно на 40% от величины коэффициента эманирования. При проведении измерений эманирующих проб в а-импульсных или ионизационных камерах (в настоящее время а-импульсные камеры применяются сравнительно редко) за счет выделения эманации из пробы результаты измерения будут завышены.
В связи с этим измерения проб а-, р- или у-методами следует производить только в случае низкого эманирования или практического (у-измерения) его отсутствия.
При значительном коэффициенте эманирования для определения содержания U следует пользоваться комбинированными радиометрическими методами, так как за счет эманирования в пробе будет иметь место нарушение равновесия между группой U (включая ионий) и группой Ra.
Определение содержания U в урановых равновесных рудах
Определение содержания U в урановых равновесных рудах производится с помощью простых (а-, 0-, у-) радиометрических методов. Основным методом в этом случае, как уже отмечалось выше, является импульсный р-метод [13 , 200].
При определении содержания U в руде изготовляется порошковая проба. Степень измельчения зависит от метода исследования и типа руды. Наиболее тонкое (практически до состояния пудры) измельчение требуется при a-измерениях, а в р- и у-методах допускается более грубое измельчение (от 0,05 мм при неравномерном и до 1—2 мм при равномерном оруденении), но достаточно тонкое, чтобы обеспечить равномерное распределение урановых соединений во вмещающей породе.
Определение содержания U в руде с помощью импульсных или ионизационных измерений активности пробы производится путем сравнения интенсивностей излучения или величин ионизационных токов исследуемой пробы и уранового равновесного стандартного эталона.
Для увеличения точности измерения рекомендуется, чтобы содержание U в стандартном эталоне было того же порядка, что и в исследуемой пробе. Для этого при измерениях проб с различным 238
содержанием U необходимо иметь набор урановых эталонов. Вычисление содержания U в пробе (в %) производится по формуле:
л- = Л
(1)
‘ ЭТ ' ф
где /пр и /эг — величины ионизационного тока с натуральным рассеянием от исследуемой пробы и эталона при ионизационных измерениях или соответственно интенсивность излучения от пробы и эталона, включающая в себя натуральный фон прибора, при импульсных измерениях;
/ф— величина натурального рассеяния при ионизационных измерениях или натурального фона при импульсных измерениях;
А — содержание U в эталоне, %.
При а-ионизационных измерениях проба и эталон берутся в насыщенном для а-лучей слое (порядка 40—50 л/а/сл/2) на тарелочках, одинаковой площади, помещаемых на дно ионизационной камеры.
При измерениях а-импульсным методом, кроме насыщенного' для а-лучей слоя, могут использоваться тонкие слои; при этом берут пробы и эталон одинакового веса.
В р-импульсном и ^-ионизационном методах измерения пробы и эталона производятся обычно в насыщенных для Р-лучей слоях (1—1,5 г/см2). Если исследуемой пробы или эталона недостаточно для получения слоя насыщения, измерения производятся при одинаковом весе эталона и пробы.
При проведении импульсных измерений по у-лучам следует брать одинаковый объем пробы и эталона для обеспечения идентичных геометрических условий измерения. В этом случае при различной* плотности эталона и пробы для измерений будут использованы различные веса пробы и эталона, и формула (1) для вычисления содержания U будет иметь вид:
‘ эт ф
где Рэт и РПр— вес эталона и пробы.
При всех видах измерений проба и
. циальные тарелочки, форма и размер которых зависят от типа приемника излучений.
Для увеличения чувствительности при р- и у-импульсных измерениях проб с низким содержанием U проба и эталон насыпаются в специальные тонкостенные стаканы, которые одеваются на счетчик.
Ионизационные измерения проб обычно производятся на электрометрах или электроскопах (СГ-1м, СГ-2м), а при проведении импульсных измерений применяются специальные лабораторные установки (Б; Б-2; Тобол; ПС-10000; ЛАС и др.) [235] со счетчиками типа Гейгера-Мюллера или сцинтилляционными счетчиками.
При отсутствии систематических, случайных ошибок и ошибок в работе аппаратуры относительная ошибка в определении содержа-
Р г эт
р
1 пр
(2)
эталон помещаются на спе-.
239-
ния U в пробе, определяемая флюктуациями радиоактивного распада, рассчитывается по формуле:
<3»
где Л/пр, Д/ф и Д/Эт— соответственно ошибки в определении интенсивности излучения или величины ионизационного тока пробы, фона и эталона. Для импульсных измерений
Д/ = /5 ; Д/ф = / £ ; Д/91 = /У’ , (4)
‘пр ‘ф 4ЭТ
где
/пр, /ф и /эт — время измерения пробы, фона и эталона.
Для ионизационных измерений приближенная оценка ошибки в величине ионизационного тока может быть получена по формуле:
Л/ПР = ^К^;Д/Ф=^ГЛ^; M,=k^VK. (5) ^Пр Н £-ф 1ЭТ
где К — число пар ионов, образуемых каждой частицей;
е — заряд иона:
Afnp, Л/ф и jV3t — полное число ионизирующих частиц от пробы, фона и эталона за время измерения.
Увеличивая навески пробы и эталона или увеличивая время измерения, можно добиться довольно высокой, не уступающей химическим анализам точности при определении содержания U [168].
Для характеристики описанных методов ниже приводятся некоторые краткие сведения об их точности и чувствительности (табл.35).
За пороговую чувствительность метода принята величина, равная значению утроенной флюктуации фона. Время измерения проб—10 мин. Степень дробления проб — 60—100 меш. [200].
Определение содержания урана в урановых неравновесных и в урано-ториевых рудах
Принципиально определение содержания U в урано-ториевых и неравновесных урановых рудах можно производить с помощью простых радиометрических методов, если в качестве эталона использовать руду данного месторождения, тщательно проанализированную химическими методами.
Однако такой способ является практически малопригодным из-за отсутствия данных о постоянстве коэффициента нарушения равновесия и отношения Th/U в исследуемых пробах данного месторождения, а следовательно, из-за невозможности использования типичного для данных руд эталона. Поэтому определение содержания U в неравновесных и сложных рудах производится с помощью комбинированных радиометрических методов.
240
Таблица 35
Характеристика радиометрических методов определения урана
.Метод анализа Вес пробы, г Пороговая чувствительность, % Минимальные концентрации (% U), которые могут быть измерены с точностью + ю% Примечание
1- 2. а) б) в) 3. а) *) в) а-Ионизационный метод (электрометр СГ-1 м) 0-Импульсный метод (счетчик АС-2) измерения в тарелочках измерения в стаканчиках измерение малых навесок у-Импульсный метод измерения в тарелочках со счетчиком типа Гейгера-Мюллера измерения в цилиндрических стаканах со счетчиками Гейгера-Мюллера . . . измерения с помощью сцинтилляционных счетчиков 10,0 40,0 100,0 1,0 0,5 0,1 40 200,0 100,0 0,001 0,0003 0,005 0,007 0,020 0,015 0,002 0,0002 0,010 0,004 0,0015 0,030 0,040 0,100 / 0,050 0,01 0,001 Чувствительность электрометра 13.3 делений на вольт. Диаметр тарелочки 9 см, толщина слоя пробы 0,1 см Тарелочка размером 12X3X1 см Стаканчик высотой 13 см, диаметром 3,5 см Допустимая крупность зерна 1—2 мм Тарелочка размером 12x3x1 см Стакан высотой 13 см. с внутренним диаметром 3,2 см, внешним 5,2 см
Аналитическая химия урана
241
Практически в настоящее время с этой целью применяется комбинированный р-гу-метод, впервые предложенный В. И. Барановым и Н. Н. Шашкиной в 1947 г. для анализа урано-ториевых руд и В. Л. Шашкиным для анализа урановых неравновесных руд.
В последующие годы различные варианты методик с применением р-гу-методов широко разрабатывались как в Советском Союзе, так и за рубежом [287, 487, 523].
При количественном определении содержания U в урановых неравновесных и урано-ториевых рудах с помощью р у метода производятся измерения пробы по р- и у-лучам. Результаты измерения обычно выражаются в эквивалентных единицах равновесного урана.
В результате проведенных измерений получаются две следующие системы уравнений:
А₽ ^г^а/ ДЛЯ Уран°В0Й неРавновесн°й РУДЫ (6) и
А? —U^j-yTh} для урано-ториевой руды, (7)
где Т
а, т, ban — коэффициенты, показывающие долю Р- и у-излу-чения U и Ra в равновесии с их продуктами распада от общего Р- и у-излучения равновесного U;
Р и у — урановые эквиваленты Th по Р- и у-лучам.
Ар и Ат — активность пробы, полученная при измерении по Р- и у-лучам и выраженная в эквивалентных единицах равновесного урана:
А? = (Ир Фр) Yp 1
Ay = (ny — Фу) Yy / ’
где Yp и Yy— цена одного импульса в минуту в % равновесного урана, полученная при р- и у-измерениях равновесного U эталона;
Пр и пу — скорость счета от образца и фона при р- и у-измерениях;
Фр и Фу — скорость счета фона при Р- и у-измерениях.
Формулы для определения содержания U, полученные из решения уравнений 6 и 7, имеют вид для урановой неравновесной руды:
и = (1+6и)Лр-биАу, (9)
где
би = ——l п — b
для сложной урано-ториевой руды:
U = (l 4"бть) Ар —бтьАу, (Ю)
242
где
Относительные ошибки в определении содержания U, связанные с флюктуациями радиоактивного распада, рассчитываются по формуле
для урановых неравновесных руд:
би примерно равно бть (практически полного равенства указанных коэффициентов можно добиться при использовании для у-измерений сцинтилляционных счетчиков).
Поэтому в случае нарушения равновесия в ряду U в урано-ториевой руде определение содержания U может производиться по формуле 9, если Th/U < 1, и по формуле 10, если Th/U> 1.
Сшибка в определении содержания U по формуле 9 за счет присутствия Th может быть оценена по формуле:
и'-и о 5Th-du Th
£ U Z23T-—--=== р ------- . -- .
U dTh и
(13)
Ошибка в определении содержания U, вычисляемого муле 10, будет равна:
по фор-
еи =
U' - U
дц — dTh
—K-h
(Кор-1),
(14)
U
где U' — содержание U, вычисленное по формуле;
U —истинное содержание U в пробе;
Кп\> — коэффициент равновесия в пробе.
Коэффициенты b и п, необходимые для вычисления содержания U, обычно определяются опытным путем для каждого типа установки с помощью уранового равновесного эталона и эталона из чистого U (UsOg) или чистого Ra, в которых соответственно U и Ra находятся в равновесии с продуктами своего распада.
Коэффициенты 0 и у определяются с помощью уранового равновесного эталона и эталона равновесного Th.
В литературе имеются сведения о значениях соответствующих Коэффициентов, полученных для различных типов приемников излучения [288].
16*
243
Измерения по 0- и у-лучам, так же как и в простых [3- и у-радио-метрических методах, могут производиться на любых лабораторных установках, применяемых Для 0- и у-измерений.
Для повышения чувствительности 0- и у-измерений на счетчиках типа Гейгера-Мюллера рекомендуется применять установки с параллельным включением нескольких счетчиков. При у-измерени-ях повышение чувствительности осуществляется также использованием сцинтилляционных у-счетчиков.
В радиометрической практике для проведения 0-Гу-измерений часто используют установку, предложенную И. П. Шумилиным [287]. Приемником 0- и у-излучения в ней является два ряда счетчиков типа МС-6 по четыре счетчика в каждом ряду, между которыми ставят пробу. Счетчики помещают в свинцовом домике. Пробу насыпают в тарелочку, дно которой служит экраном для поглощения 0-излучения при регистрации у-излучения.
Одновременная регистрация 0- и у-излучения производится с двумя установками типа Б или Б-2.
Для урано-ториевой руды с нарушением равновесия в ряду урана в 1955 г. Г. Р. Гольбеком была разработана методика определения содержания U с использованием порогового спектромет-рирования у-излучения при измерении со счетчиками Гейгера-Мюллера и измерения пробы по 0-лучам [49].
Формула для определения содержания U в этом случае может быть получена путем решения следующих трех исходных уравнений: MY1==(aRa4-6Th)P |
My^^Ra + ^Th)? . (15)
А'0 =си-4-Л Ra4-/\4yRa + BTh4-PByTh J
где
U, Ra Th — процентное содержание в руде соответственно U, продуктов распада Ra и Th;
a, b, alf bt — постоянные коэффициенты, определяемые опытным путем и характеризующие интенсивность общего и дискриминированного у-излучения при 1%-ном содержании Ra и Th;
Р — вес пробы, г;
С, А, В — коэффициенты, характеризующие долю регистрируемого 0-излучения соответствующих радиоактивных элементов;
и — коэффициенты, характеризующие долю у-излучения Ra и Th, регистрируемую при 0-измерениях.
В 1952 г. Витаном, а в 1956 г. Харли была предложена и проверена методика для определения содержания U и Th в равновесных урано-ториевых рудах с использованием сцинтилляционного у-спек-трометра [576, 1014].
При измерении пробы по двум каналам (методика Харли), установленным на 168 и 226 кэв и соответствующих измерениях урановых 244
и ториевых эталонов были получены два уравнения, при решении которых определяется U (и Th) в рудах.
Измерения производились в условиях одинаковой геометрии пробы и эталона. Точность определения содержания U колебалась от десятых до нескольких процентов.
В 1959 г. опубликована отечественная работа, в которой приводится описание используемого для измерений многоканального у-спектрометра и результатов определения содержания U (Ra и Th) в рудах, причем исследования проводились как на равновесных урано-ториевых и урановых рудах, так и неравновесных рудах [85].
Предел чувствительности метода с применяемым многоканальным у-спектрометром по разработанной методике измерения составлял 0,01% U.
Определение содержания урана в породах
Определение содержания U в горных породах радиометрическими методами проводится тремя способами: с помощью сцинтилляционных у-спектрометров, комбинированными радиометрическими методами и с помощью методов микрорадиографии [85, 199, 225, 576].
С помощью метода- микрорадиографии производится определение содержания U в отдельных минеральных зернах, поэтому краткое описание метода сделано в разделе, посвященном определению U в минералах.
При использовании у-спектроскопии методика исследования принципиально не отличается от исследования сложных руд. При наличии равновесия в ряду U определение содержания производится с регистрацией по двум каналам (для исключения влияния Th на результаты измерения и одновременного его определения).
В случае сдвига равновесия в ряду U для определения его содержания производится регистрация у-излучения по трем каналам. По результатам измерения составляют три исходных уравнения, при решении которых определяют содержание U.
Как и в случае комбинированных методов, результаты измерения выражают в единицах равновесного уранового эталона.
Сведений, позволяющих судить о точности этих методов, в настоящее время еще недостаточно, но, по данным работы [5761, U Может определяться с точностью до нескольких микрограммов.
При использовании комбинированных радиометрических методов с регистрацией [3- и у-излучения существенное отличие методики определения содержания U в породах от методики определения содержания U в рудах заключается в необходимости учитывать влияние р- и у-излучения К40.
В зависимости от состояния равновесия в ряду U для определения его содержания появляется необходимость в получении трех
245
(при равновесии в ряду U) или большего количества уравнений (при отсутствии равновесия в ряду U).
При использовании у, у-дискриминированных, а- и 0-измерений в описанном [225] случае определение U производилось со статистической ошибкой ±10%.
Определение содержания урана в минералах
В зависимости от типа исследуемого минерала и содержания в нем урана для определения последнего могут применяться а-, 0-импульс ные радиометрические методы, методы у-спектроскопии или метод микрорадиографии.
Простые а-, 0-методы используются при определении содержания U в урановых минералах, которое производится так же, как и в рудах с использованием порошковых проб. Особенностью методики является проведение измерений в тонких слоях.
При исследовании минералов широко используется «-импульсный метод со сцинтилляционными счетчиками.
В 0-импульСном методе чаще чем цилиндрические используются торцовые счетчики. Пробу и эталон определенного веса помещают на тарелочку, диаметр которой несколько меньше диаметра приемника излучения. При использовании цилиндрических счетчиков больших размеров пробу и эталон помещают в специальные бумажные стаканчики, которые ставят непосредственно на счетчик.
Приближенное количественное определение содержания U можно производить по 0-методу в отдельных зернах минералов, причем в качестве эталона используют зерно минерала с известным содержанием U.
При определении содержания U в урано-ториевых минералах, так же как и при измерении руд и пород, используется метод у-спек-троскопии [576]. Особенностью методики является измерение малых навесок пробы, в связи с чем для увеличения чувствительности измерений используются кристаллы с отверстием, внутрь которых помещается проба минерала и эталон.
Широкое применение для определения содержания U в минералах находит также метод микрорадиографии [199].
Определение содержания U в урановых минералах может производиться двумя способами:
1) путем сравнения числа следов а-частиц от исследуемой пробы с числом следов а-частиц от эталонного образца, приготовленного из аналогичного минерала с известным содержанием U;
2) расчетным путем по числу следов а-частиц, зарегистрированных в толстослойной эмульсии от образца, изготовленного в виде прозрачного шлифа без покровного стекла.
В первом случае сравнение числа следов от эталонного образца и пробы производится на одинаковой площади эмульсии при одинаковой экспозиции пробы и эталона.
246
Содержание U в исследуемой пробе вычисляется по формуле:
где
NnP и АЭт— число следов а-частиц от пробы и эталона.
При определении содержания U расчетным путем по числу следов производится их подсчет с остаточным пробегом в эмульсии р^2 см.
Для расчета содержания U-пользуются формулой:
Си ~3-16,6-io3 d-k » (1
где п — число следов с 1 смг поверхности образца в 1 сек;
d — плотность образца в г/см’; р
& = -£-, (R1 — пробег а-ч’астиц в воздухе, 7? — пробег в веществе);
Съ — концентрация U в г/г в исследуемом образце, изготовленном в виде тонкой пленки.
Определение содержания U в урано-ториевых минералах производится расчетным путем по числу зарегистрированных следов.
Для этого, кроме подсчета числа следов п с р>2 см, производится подсчет числа следов п1 с р 5,6 см и числа следов п2 с р 7 см.
Для вычисления содержания U используются 2 следующих исходных уравнения:
Зси4-Сть = 166.10МЛ )
си л8/л2 —0,78 где
-- П2>
Сть — концентрация Th в образце, г/г.
Для определения содержания U в урано-ториевых минералах в 1951 г. И. Кюри и Г. Фарагги был предложен метод нейтронной микрорадиографии [439].
Определение содержания U производится по числу осколков Деления U под воздействием медленных нейтронов, которые не вызывают деления ядер Th.
Расчет содержания U производится по формуле:
А 1 1 2л п
Си“ N ’ <з ’ kd (х - р) ’ F ’ где
А — масса атома U;
Л7 — число Авогадро;
ст — эффективное сечение деления;
247
,v — средний пробег осколков деления в воздухе;
о — минимальный пробег в воздухе осколка деления, который еще может быть измерен в эмульсии;
п — число актов деления на 1 см2-,
F — поток нейтронов на 1 см2;
k и а имеют те же значения, что и в формуле (17).
Существенным преимуществом метода нейтронной микрорадиографии перед обычной радиографией является отсутствие влияния состояния радиоактивного равновесия на определение концентрации урана.
II. ОПРЕДЕЛЕНИЕ УРАНА В ПОРОДАХ, РУДАХ И МИНЕРАЛАХ МЕТОДАМИ СПЕКТРАЛЬНОГО АНАЛИЗА
Ю. И. Беляев
Спектральное определение малых количеств урана в рудах представляет собой трудную аналитическую задачу, так как абсолютная чувствительность обнаружения этого элемента невелика вследствие низкой летучести его окислов и сложности спектра, обладающего большим числом линий малой интенсивности.
В ряду летучестей уран находится в конце ряда [212]. Окислы урана в процессе испарения в дуге восстанавливаются до металла, который вступает в реакцию с графитом, образуя труднолетучие карбиды.
Еще в 1926 г. Порлецца и Донатти [817] сделали попытку определения урана в антимите. Однако чувствительность, достигнутая ими, оказалась невысокой — 0,1%.
В дальнейшем Мазер [732] повысил чувствительность определения урана до 0,05%, используя прикатодную область дуги. Однако воспроизводимость результатов анализа оказалась неудовлетворительной.
Для повышения чувствительности определения урана в рудах Страсхейм [941] предложил так называемый «усиливающий агент», рекомендованный Стедманом [935]. Применение RbCl в качестве такого агента позволило Страсхейму достигнуть чувствительности 6-10_3% по линии урана 4244,4а и линии сравнения Pt 4232,5а. Однако получение надежных результатов при содержании урана <0,01 % является трудной задачей, как на это указал сам автор, так как рекомендуемая им линия находится на интенсивном непрерывном фоне.
Кроме того, Страсхейм наблюдал смещение градуировочных графиков из-за неполного соответствия химического состава эталонов и анализируемых образцов.
Предложенный недавно Н. Г. Морозовой [166] метод определения урана в рудах и продуктах их переработки позволяет достигнуть чувствительности определения 5-10"’%.
248
Н. Г. Морозовой разработано два метода: метод брикетирования при содержании урана выше 0,1 ° о и метод сжигания проб на медных электродах при содержании урана в интервале 0,005—0.1 %.
В методе брикетирования анализируемую пробу смешивают с медным порошком в отношении (1 : 9) для увеличения электропроводности и с внутренним стандартом, после чего прессуют в брикет с помощью пресса при давлении 104 кг/см2. В качестве внутреннего стандарта вводят молибден в виде молибдата аммония.
В методе сжигания проб в медных электродах проба плотно набивается в отверстие в медном диске, служащем нижним электродом дуги. Для повышения чувствительности определения урана пробы предварительно прессуют в таблетки или сплавляют в корольки. Как прессование, так и сплавление проб приводит к повышению абсолютной интенсивности линий урана. Чувствительность обнаружения урана по данным [166] составляет 5 • 10“s %.
В табл. 36 приведены условия анализа. Для повышения чувствительности обнаружения урана в известняках пробы перед прессованием разбавляют двуокисью кремния в отношении (1 : 1).
Таблица 36
Условия определения урана в рудах [166]
Параметры
при методе брикетирования
при методе сжигания проб в медных электродах
Спектрограф ...........
Аналитические линии, А Источник света .......
Сила тока, а..........
Межэлектродный промежуток, мм..............
Внутренний стандарт . .
Пластинки.............
КС-55
U II 4090,14 и Генератор дуги переменного тока 6
2,5 4%-ный раствор молибда та аммония
Спектральные, тип II,
КС-55
Mo I 4086, 025
ПС-39
8
2,5
0,5%-ный или 1%-ный р-р молибдата аммония чувствительностью 11 ед. ГОСТ
Вероятная ошибка единичного определения описываемым методом составляет ±5%.
По данным автора, непосредственное сжигание проб без прессования требует построения отдельных градуировочных графиков для руд различного состава (рис. 51). Прессование проб приводит к совпадению градуировочных графиков (рис. 52), и поэтому Н. Г. Морозова рекомендует применять описываемый метод для определения урана в любых рудах по одному градуировочному графику.
249
В табл. 37 приведен состав руд, для которых имеет место совпадение градуировочных графиков.
Таблица 37
Состав руд (в %) [166]
. № Гуды S1O, Fe2Os 1 ; А1А СаО МпО
1 71,5 3,0 6,7 0,05
2 61,9 10,0 22,6 3,6 1,1 —
3 48,4 31,7 11,8 1,6 4,4 0,6
Т. М. Морошкина, В. К. Прокофьев и М. И. Смирнова [167] повысили чувствительность определения урана в рудах до 10" 4%. Эти авторы также рекомендуют спекать пробу в королек с помощью
Рис. 51. Градуировочные графики Рис. 52. Градуировочный график для для определения урана в различных определения урана при прессовании руд рудах при непосредственном сжига- в таблетки.
НИИ проб. •— эталоны на основе руды 1; о— эталоны
на основе руды 2; X— эталоны на основе руды 3 (см. табл. 37).
специальной электропечи и использовать медные электроды, но применяют трудоемкий метод добавок [196]. Исследованные авторами [167] руды содержат 50—70% кремнезема, 3—50% окиси алюминия и небольшие примеси Mg, Са, Мп, V, Мо и Th.
Для проведения анализа используют автоколлимационный спектрограф фирмы Хильгер и дугу переменного тока. Внутренним стандартом служат вольфрам или молибден, вводимые в пробу в виде растворов вольфрамата или молибдата аммония в концентрации 0,05—0,1% (на металл). Рекомендуемые аналитические линии U II 4090, 14 А —Mo 14084,38 А и UII 4472,33 A—Mo 14484, 19 А.
250
Градуировочный график строят потрем добавкам: 0,5 • 10“5; 1,5-10"5 и 3• 10“6 г U.
Направление графика в области малых концентраций находят аналитически с помощью метода наименьших квадратов. Погрешность определений этим методом составляет ^6°6.
Повышение чувствительности определения урана при использовании пробы, спеченной в королек, авторы объясняют двумя причинами: во-первых, повышенной электропроводностью королька, благодаря чему дуга горит спокойно и не блуждает по поверхности пробы и, во-вторых, возможным частичным восстановлением окис-лов урана до металла или до кислородных соединений низших валентностей.
Точность определения урана в рудах можно повысить путем использования в качестве внутреннего стандарта изотопов урана 140, 82]. Большая близость химических и физических свойств изотопов делает их идеальными внутренними стандартами при спектральном определении элементов.
Относительная интенсивность изотопических компонентов аналитической линии элемента в минимальной степени зависит от изменения условий испарения или возбуждения атомов элемента в источнике света и определяется только относительной концентрацией изотопов в пробе. Изотопическое смещение для линий урана достигает значительной величины. Например, для линии 4244,4А оно составляет 0,23А. Изотопическая структура этой линии может быть разрешена с помощью спектрографа ИСП-51 с камерой УФ-85. Эта линия наиболее удобна для определения урана в рудах. Недостатком является наложение линии вольфрама. Однако практически вольфрам встречается редко в количестве, способном изменить относительную интенсивность линий урана.
Для проведения анализа в пробу вводят изотоп U235 в известной концентрации.
Определение содержания урана (%) в рудах ведется по формуле: pts_______________________ pts
X =-------CSSS — С238'.
/235_7235
где
у235 и psa — интенсивности изотопических компонент;
/6235 и /2*8 — интенсивность фона около этих компонент на расстоянии ДХ— Л238—X235;
с238'— содержание изотопа U238, вводимое в пробу вместе с легким изотопом U235;
с235— содержание изотопа U235.
Применение изотопов урана в качестве внутреннего стандарта Повышает точность единичного определения урана в рудах до 3—4?6 При содержании урана ниже 1% и до 2% в интервале концентраций 1—5%.
251
111. РАДИОАКТИВАЦИОННЫЙ МЕТОД ОПРЕДЕЛЕНИЯ УРАНА
Ю. В. Яковлев
Использование цепной реакции урана и возникающих в ядерных реакторах интенсивных потоков медленных нейтронов привело к созданию одного из самых чувствительных методов аналитической химии — радиоактивационного анализа. В свою очередь, радиоак-тивационный анализе успехом применяется для определения ультрамалых количеств урана, а также его изотопного состава.
Метод основан на облучении исследуемых образцов элементарными частицами (в большинстве случаев — медленными нейтронами, значительно реже — заряженными частицами или у-лучами) и образовании в результате ядерных реакций изотопов, по активности которых определяется содержание элементов в образце. Количество образующегося радиоактивного изотопа (т. е. его активность) прямо пропорционально массе (т) определяемого элемента:
At = ~ f • зиз • О 6,02 • 10as (1 — е~ М93//Г), где
At— число распадов в 1 сек. в момент конца облучения;
М — атомный вес определяемого элемента;
/ — интенсивность потока бомбардирующих частиц (число частиц на 1 см2 в 1 сек.);
оиз — изотопное сечение активации для данной реакции, см2; () — содержание стабильного изотопа в природной смеси; t — время облучения;
Т — период полураспада образующегося радиоактивного изотопа.
Чувствительность радиоактивационного анализа зависит в основном от атомного сечения активации (оак =оиз-6) и интенсивности потока бомбардирующих частиц и, в меньшей степени, от времени облучения и периода полураспада радиоактивного изотопа, по которому проводится определение. Для различных элементов величина атомного сечения активации медленными нейтронами колеблется от нескольких миллибарнов до сотен и даже тысячи барнов, т. е. от ICC27 до 10 см2. Сечения активации для заряженных частиц на 1—2 порядка меньше. Поэтому, как правило, используется активация медленными нейтронами.
Интенсивность потока зависит от типа источников нейтронов (табл. 38).
Время облучения и период полураспада входят в экспоненциальный множитель (1—e-0'693f r), стремящийся к единице при /=4—5 Т. Поэтому для радиоактивных изотопов с очень большими или очень малыми периодами полураспада чувствительность метода уменьшается, так как в первом случае необходимо длительное облучение, а во втором сильно сказывается распад радиоизотопа за период времени от конца облучения до начала измерений.
252
Минимальные количества различных элементов, которые могут быть определены методом радиоактивационного анализа при облучении потоками медленных нейтронов 2« 10”/?/ди2-се/с в течение одного месяца (для долгоживущих изотопов} или времени насыщения (для короткоживущих), лежат в пределах от 10"5 до 10“12 г.
Таблица 38
Зависимость интенсивности потока медленных нейтронов от типа источника
Нейтронные источники Интенсивность потока (п см’-сек)
Ядерный реактор ....... 1010—2-10й
Циклотрон 10 s—109
Низковольтный генератор . . . 103—106
Ra — Be (1 г Ra) 104—103
Ро —Be (1 кюри Ро) ю8—ю1
Основные этапы проведения радиоактивационного анализа заключаются в облучении исследуемого образца, химическом выделении и очистке определяемого элемента после добавления носителя (как правило, изотопного) и измерении активности выделенного препарата интересующего радиоактивного изотопа. В некоторых случаях можно избежать проведения химических операций, используя соответствующую измерительную аппаратуру, например у-сцинтилляционный спектрометр.
Для количественного определения используется эталонный метод: образец, содержащий известное количество определяемого элемента, облучается, очищается и измеряется в идентичных условиях с анализируемым образцом. При этом отношение масс определяемого элемента в образце и эталоне равно отношению соответствующих активностей.
Существенными преимуществами радиоактивационного анализа являются высокая чувствительность, которая для большинства элементов превосходит чувствительность других методов анализа и позволяет, как правило, проводить анализ без предварительного обогащения; большая специфичность, основанная на характерных свойствах радиоактивных изотопов; возможность одновременного определения ряда примесей в одной навеске образца и учета потерь при химических операциях; отсутствие поправки начистоту применяющихся реактивов, так как химические операции проводятся после облучения образца.
Хотя уран является естественно радиоактивным элементом, число а-частиц, испускаемых его долгоживущими изотопами U238 (Г =4,5-10’ лет), U235 (Т-8,8-108 лет) и U234 (Т-2,52.105 лет), содержание которых в природной смеси 99,28, 0,714 и 0,0057% соответственно, недостаточно для определения микроколичеств урана.
При облучении урана нейтронами протекают следующие ядер-ные реакции:
а) на медленных нейтронах:
U28’(n, у) U289 (Т = 23,5 мин)—> \р2зэ (Т = 2,33 дн); (1)
U:j3(/?, /)—► осколки деления; (2)
U284 (л, y)U2Si; (3)
б) на быстрых нейтронах (Е„ 1 Мэе):
U238 (n, /)—осколки деления. (4)
При облучении в ядерном реакторе вклад реакций образования U235 и деления U288 очень мал. Основную роль играют реакции (1) и (2). Определение урана можно проводить, следовательно, по U23 9, Np 239, сумме осколков деления, либо по одному из осколков— Ва140, Те182, Хе133 и т. д. В последнем случае для определения оак необходимо учесть выход данного изотопа при делении. Сечения активации этих реакций составляют ои13=2,75 барна, <уак=2,73 барна; ои23=575 барнов, оак=--4,11 барна; для Ва140 выход при делении U235—5,8% и (Так-0,238 барна [10]. Отсюда следует, что нижний предел чувствительности обнаружения урана (при следующих оптимальных условиях: f —Л013п/см2 -сек; расп/сек; вре-
мя облучения равно времени насыщения; время, прошедшее от конца облучения до начала измерения,— 1 час) составляет при определении по Ва140—1,5-10~10г; по Np239 — 1,5- 10"”г; по U289— 6-10"11 г; по сумме осколков деления —1 • 10-11 г.
Первая работа по радиоактивационному определению урана появилась в 1949 г. [494]. Для определения был использован суммарный счет осколков деления U235 в ионизационной камере. Измельченные образцы минералов (размеры зерен 150—300 мк), содержащих более 2% урана, помещали в ионизационную камеру и облучали на Ra-y-Ве-источнике нейтронов (500 мг Ra+2 кг Be). Было найдено, что число имп/мин пропорционально весовому процентному содержанию урана, плотности соединения и пробегу осколков в этом соединении.
Для счета осколков могут быть использованы не только ионизационные камеры, но и сцинтилляционные счетчики с ZnS и толстослойные фотопластинки. Было показано, что применяя сцинтилляционный счетчик и 50 мг Ra-Be-источник, можно определять уран в рудах при содержании более 10% [572]. Ирэн Кюри и Фарагги показали, что при облучении шлифов минералов на толстослойных фотопластинках в реакторе потоком \012 п/см2 • сек можно по числу треков определить 10~2% урана [439].
Сочетание счета осколков в ионизационной камере с облучением в ядерном реакторе позволило достичь очень высокой чувствительности определения урана (7-10“10 г) в воде [939]. Уран экстраги
254
ровали из объема 20—50 мл 0,5 мл дибутилортофосфорной кислоты, и экстракт упаривали на платиновом диске, помещавшемся затем в ионизационную камеру. Концентрация урана оказалась равной 5-10"6 г/л в воде Большого Соленого озера, 1,2-10"7 г/л в водопроводной воде, ЗЛО-8 г/л в дистиллированной воде.
Для определения урана по активности одного из осколков деления чаще всего используется выделение Ва140 из смеси осколков. Методика впервые была применена Смэйлсом для определения урана в минералах и породах [924]. Образцы весом --0,3 г и эталоны (смесь дунита с урановой смолкой, содержащая 10"3% U) облучали в Харуэллском реакторе потоком 1012 п/см2’сек в течение трех дней. Затем образцы и эталоны растворяли и после добавления носителя проводили химические операции выделения и очистки бария. В образцах дунита было найдено 3-10~4% U (9-Ю-7 г U), вцирконеи граните — 2-10“3%, в монаците — 0,3% U.
Фишер и Бейдон [507] определяли уран в минералах по Те132, выход которого при делении — 3,6%. После предварительного обогащения — выделения урана на 100 мг Fe(OH)3 при облучении потоком 1010 п/см2-сек — в течении 50 час. можно определить 5-10-3 г U.
Уран в каменных метеоритах определяли [484] и по Хе133, выход которого 6,5%. Следы атмосферного Хе удаляли до облучения нагреванием образца в течение нескольких часов при 400°в высоком вакууме. Облучение проводили в течение 7 дней потоком 1012 п/см2’Сек. После облучения добавляли Хе в качестве носителя, образцы сплавляли с NaOH при 900°, выделившиеся газы собирали. Хе после отделения от О2 и N2 пропусканием через нагретую до 900° бариевую печь адсорбировался на активированном угле. Активность измеряли торцовым счетчиком после десорбции.
Чувствительность определения 10“9 г U. В образцах трех хондритов найдено от 8-Ю"7 до 1,2-10-0% U.
Мальман и Леддикотт разработали метод определения урана по Np239 с чувствительностью 10"10 г/г [724]. Np239 выделяли на неизотопном носителе — лантане и измеряли на у-сцинтилляционном счетчике. При облучении потоком 1012 п/см2-сек в течение 62 час. в синтетических образцах найдено 3-10"7 г U, в почвах 5—8-10"3%, в фосфатных рудах 1,6-10-2% U.
В докладе Леддикотта и др. [694] на второй Женевской конференции упоминается о работах, связанных с определением урана по Np239 или Ва140 в металлах (2-10"2—1 • 10" 4% U), рудах (1 • 10"2— 3-10-6%), почвах (1—1,5.10"2% U), воде (1,5-10"3—1 • 10"8% U).
Короткий период полураспада U239 вызывает необходимость быстрого проведения всех операций при определении урана по этому изотопу. Применение пневматического устройства для транспортировки образца из канала реактора в лабораторию и экспрессное Химическое выделение урана из образцов алюминия и циркалоя Позволяют начать измерение активности U239 спустя 1 час после
255
конца облучения [626, 7181. Образцы облучали потоком 7-1013 п см'2-сек в течение 1 часа. При этом можно определить 10"10 г урана с точностью менее 10%. В образцах различных сортов чистого алюминия было найдено от 2-10"4 до 2,3-10~50о U.
Целая серия работ, помимо упомянутой выше [484], посвящена определению урана в каменных и железных метеоритах. И. Е. Старик и М. М. Шац [245] определяли уран после предварительного выделения эфирной экстракцией из каменных и железных метеоритов по счету осколков деления в ионизационной камере, помещенной в тепловую колонну ядерного. реактора. Среднее содержание урана в каменных метеоритах — 2,4-10" 5%, в железных — 1,9-10-6 %. Точность метода 3—4% (отн.). Холостой опыт с реактивами, применявшимися при обогащении урана, дал величину поправки —2,3-10"’%.
Рид и Туркевич [571, 839] получили совпадающие данные по определению урана по Np239 и Ва140 в каменных и железных метео-.ритах. Облучение проводили потоками 1012—1013 п/см2-сек,в течение 3—5 дней. Для определения химического выхода нептуния при выделении и очистке применяли носитель Np237 в количестве 50— 100 имп/мин. В случае железных метеоритов, как отмечают сами авторы, получены сильно заниженные результаты (-^1,5-10"8% U). При определении урана в каменных метеоритах по Ва140 активность последнего вычисляли по активности La140, выделенного из препарата бария после достижения радиоактивного равновесия. Прямое измерение активности Ва140 невозможно из-за большого влияния Ва131, образующегося из стабильного бария, присутствующего в хондритах в количестве 3—4-10"4%. Содержание урана в хондритах близко в среднем к 1,1 • 10"6%, в ахондрите в 10 раз больше.
Результаты определения урана по Ва140 в трех хондритах [597 ] (от 2,8-10"® до 9-10"’%) показали хорошее совпадение с данными [484], полученными на тех же образцах по Хе133.
Значительный интерес представляет применение радиоактива-ционного анализа для определения изотопного состава урана. Точнее говоря, радиоактивационный анализ позволяет определить лишь количество того стабильного изотопа, из которого образуется при облучении радиоактивный изотоп. Содержание же элемента находят путем расчета на основе известного содержания данного стабильного изотопа в природной смеси. Однако, например, содержание U235 в обогащенном или «выгоревшем» уране не соответствует природному соотношению, а тождественность изотопного состава урана метеоритного и земного происхождения нуждается в доказательстве. В связи с этим был проведен ряд исследований по определению изотопного состава образцов урана различного происхождения. Результаты показали, что в ряде случаев радиоактивационный анализ не уступает по точности масс-спектральному методу, а в отдельных случаях превосходит по быстроте определения.
256
Была показана возможность определения U235 по счету осколков деления сцинтилляционным счетчиком с ZnS при облучении 100-мил-лиграммовым Ra-Be источником [10021.
В работе. [551] U234 определяли по a-счету, U23?—эмиссионной спектроскопией, a U235 — радиоактивационным методом.
Несколько работ посвящено сравнительному изучению изотопного состава урана метеоритного и земного происхождения. В ранней работе [293] сравнивалась a-активность L238 и U234 и число актов деления (U235) для образцов метеоритного и земного происхождения. Отношение (U235 : U23 8)MCTPCp к (U235 : U-38)3ev оказалось равным 1,25±0,18, т. е. лежит почти в пределах ошибки опыта. При сравнении изотопного состава метеоритов и земных пород с помощью определения U2 38 по Np239 и U235 по Ва14и (с точностью 10%) было показано, что состав практически одинаков [571]. К. А. Петржак, И. Н. Семенюшкин и М. А. Бак [191] изучали изотопный состав урана, выделенного из метеоритов, и земного происхождения путем измерения в двойной ионизационной камере осколков деления U2-35 на медленных нейтронах в тепловой колонне и U235 и U238 на нейтронах деления. Результаты также показывают, что изотопный состав урана в метеоритах и на Земле одинаков (точность определения 3—496).
Сейфанг и Смэйлс [901, 902] определяли содержание U235 по Ва140 в обогащенных и обедненных этим изотопом образцах U3O8. Облучение навесок от 20 мг до 10“5 г проводили потоком 1012 г/см2 -сек в интервале времени от 5 мин. до 11 час. в зависимости от навески урана и процентного содержания U235. Было показано, что 1 мкг U235 дает достаточную активность. Достигнута точность определения ±0,44% (отн.). Результаты радиоактивационного определения (0,391 ±0,006%) не уступают масс-спектральному (0,391 ±0,007%). При концентрациях U235 более 0,1% влияние деления U238 на быстрых нейтронах при облучении потоком 1012 п/см^-сек в течение 6 час. составляет всего 0,0064%. В случае очень малых содержаний U235 необходимо проводить облучение в тепловой колонне реактора, что дает возможность определять 6-10~4% U235. Л1аклин и Ликинс [719] для устранения влияния деления U238 применили Ra-y-Be источник нейтронов (2 г Ra), испускающий нейтроны с энергией 0,69 Мэв; порог реакции деления U238 — 1 Мэв. Измерения проводили по счету осколков деления в ионизационной камере. В образце было найдено 4,5-10~4% U235.
Май и Левек [725] разработали экспрессный метод определения U235 в U3O8 без разрушения образца. Метод основан на измерении общей активности короткоживущих продуктов деления, образующихся при облучении анализируемого и стандартных образцов Весом по 5 мг потоком нейтронов 109 п/см~-сек в течение 15 сек. Образцы по пневмопочте передаются в течение 10 сек. из канала реактора в комнату для счета и измеряются одновременно на двух
Аналитическая химия урана 25/
у-сцинтилляционных счетчиках. Точность метода — 3.3'Т ютн.) дл-образцов, содержащих от 0,85 до 20% U23a.
U2SS определяли по U2’9 в растворах U233 и Ри [6261. Облучение потоком 7-Ю13 rd см--сек проводили в течение 1 часа. Влияние деления U233 и Ри уменьшалось более чем в 50 раз завертыванием образцов в кадмий при облучении. Найденное содержание U2’8 в растворах U233 лежало в пределах 0,3—6,7%. Результаты радиоактивационного и масс-спектрального анализов одного из образцов хорошо совпадают (4,6±0,3% и 4,7±0,02% соответственно). Но для двух параллельных опытов радиоактивационный анализ требует всего 2 часа, а одно масс-спектральное определение — 1 день. В плутонии активационным методом найдено 0,82 ±0,06% U2± флуориметрически — 0,78 ±0,07 %.
Радиоактивационный анализ был также применен для определения относительной концентрации частиц UO2 определенных размеров, суспензированных в натриево-калиевом сплаве при различных температурах вплоть до 800°, а также в воде и органических растворителях [296, 694]. Определение основано на активации .исследуемого порошка размером 0,08—120 мк нейтронами и измерении активности тонкого слоя суспензии у-сцинтилляционным счетчиком через щель в свинцовой защите. При этом измеряемая активность является мерой веса материала в слое.
Глава V
МЕТОДЫ ОТДЕЛЕНИЯ УРАНА ОТ СОПУТСТВУЮЩИХ ЭЛЕМЕНТОВ
ВВЕДЕНИЕ
А. А. Немодрук
Редко тот или иной метод определения урана может быть применен без предварительного отделения его от мешающих элементов.
Большинство методов отделения урана от мешающих элементов основано на выделении самого урана из анализируемого раствора, меньшая часть — на выделении примесей.
Наиболее широкое применение для отделения урана нашли методы осаждения, экстракции и хроматографические методы. Кроме того, иногда применяются и дистилляция, возгонка, электролиз, электродиализ и т. д. Для отделения малых количеств урана более подходящими являются соосаждение с носителем и экстракция, а также хроматография.
Выбор того или иного способа отделения зависит от принятого метода определения, от состава анализируемого раствора и прежде всего от характера мешающих элементов и их концентрации, от содержания урана в растворе и от требующейся точности определения. Вследствие этого невозможно указать такой из них, который обеспечивал бы лучшие результаты во всех случаях.
Широкое применение различных маскирующих комплексообразующих веществ при отделении урана от мешающих элементов значительно расширило возможности многих методов. Из комплексообразующих веществ наибольшее значение имеет комплексон III, Маскирующий в определенных условиях значительное количество Других элементов и не мешающий отделению урана.
В некоторых случаях для достижения большей полноты разделения отделение повторяют несколько раз.
Последовательное применение двух различных способов отделения часто оказывается более эффективным, чем многократное повторение отделения одним и тем же методом.
17*
Высокая избирательность при отделении урана от мешающих элементов может быть достигнута за счет использования различных валентных состояний урана. Этот прием оказался особенно успешным в методах отделения посредством осаждения. В тех случаях, когда реагент осаждает уран в одном его валентном состоянии и не взаимодействует с ним в другом, сначала переводят уран в то валентное состояние, в котором он не осаждается данным осадителем, и выделяют предварительно мешающие элементы. Затем, переведя его в соответствующее валентное состояние, осаждают этим же реагентом. При этом лишь немногие элементы, которые обладают несколькими валентными состояниями и в одном из них, как и уран, не осаждаются выбранным осадителем и осаждаются в другом, оказываются неотделенными от урана. Применение комплексона III в таких случаях, как правило, позволяет достигнуть отделения и от таких элементов.
1. ОСАЖДЕНИЕ И СООСАЖДЕНИЕ НЕОРГАНИЧЕСКИМИ И ОРГАНИЧЕСКИМИ РЕАГЕНТАМИ
А. А. Немодрук
Из методов отделения урана осаждением наибольшее распространение получили карбонатный метод, состоящий в осаждении большинства мешающих элементов при помощи карбоната аммония или карбонатов щелочных металлов, взаимодействующих с ураном (VI) с образованием растворимого карбонатного комплекса, а также осаждение урана фосфатами, перекисью водорода, купфероном, фторидами и 8-оксихинолином. Осаждение оксалатами, едкими щелочами, уротропином, пиридином и другими органическими основаниями имеет меньшее значение.
Отделение с помощью карбонатов щелочных металлов и аммония
Отделение урана, основанное на применении карбонатов щелочных металлов и аммония, является одним из наиболее простых и распространенных [8, 46, 184]. Отделение происходит вследствие осаждения не самого урана, а мешающих элементов, подавляющее большинство которых при добавлении карбонатов щелочных металлов или карбоната аммония выпадают в осадок в виде карбонатов или гидроокисей, в то время как уран образует растворимый карбонатный комплекс UO2(CO3)t— и остается в растворе.
Метод позволяет отделять уран от всех мешающих элементов, за исключением тория, а также некоторых редкоземельных элементов, в частности редких земель иттриевой группы, которые частично растворимы в карбонатных растворах. Повторение переосаждения
в этих случаях оказывается малоэффективным. Кроме того, все элементы, присутствующие в растворе в виде анионов, также не осаж-
даются и остаются в растворе вместе с ураном.
В присутствии больших количеств кальция часть урана захватывается образующимся осадком карбоната кальция, вследствие чего разделение становится неполным. Для устранения мешающего
влияния кальция уран предварительно отделяют от него осаждением аммиаком [184]. При этом одновременно с кальцием отделяются также медь, никель, цинк, кобальт и частично молибден. Если пред-
варительное осаждение аммиаком проводить в присутствии комплексона III, то на этой стадии можно достигнуть полного отделения от значительных количеств большого числа мешающих элементов (см. «Определение осаждени
ем гидроокисью аммония», стр. 57).
Заметные количества урана могут оказаться адсорбированными осадками гидроокисей алюминия и железа. Если образующиеся осадки гидроокисей этих элементов являются значительными, то после
Рис. 53. Выделение микроколичеств урана из карбонатных растворов в присутствии весомых количеств железа в зависимости от pH раствора. Содержание урана 2 10 _ 10—2 -10 7 мг/мл, железа — 0,02 мг/мл.
фильтрования их следует снова растворить и повторить отделение.
Захват урана осадком гидроокиси железа
в карбонатных раство-
рах сильно зависит от pH (рис. 53). Около 1% урана захватывается осадком уже при рН^-3. При pH 4 захват составляет 5%, затем он быстро увеличивается и при pH 5 равняется почти 50%. При дальнейшем повышении pH в результате образования ураном растворимого карбонатного комплекса количество соосажденного урана уменьшается, и при pH ^7 захват практически не имеет места. Уран, первоначально захваченный осадком гидроокиси железа, При pH >7 полностью десорбируется в присутствии достаточного избытка карбонат-ионов [242]. Однако с увеличением концентрации
карбоната щелочного металла или аммония быстро возрастает количество железа, остающегося в растворе. Так, например, из раствора объемом 50 мл, содержащего 5 мг железа и 0,25 г нитрата аммония, при добавлении 0,5%-ного карбоната аммония не осаждается 0,06% железа; при повышении концентрации карбоната аммония до 5% в растворе остается 7,4% железа.
Применение вместо карбоната аммония карбонатов щелочных металлов, особенно карбоната калия, увеличивает количество же-
261
леза, остающегося в растворе. Полное осаждение железа происходит в очень узком интервале pH. При применении карбоната аммония оно находится в пределах 7,5—7,6, и при изменении pH в любую сторону от указанного значения количество остающегося в растворе железа быстро возрастает [242].
Из карбонатов, применяемых для отделения урана, карбонат аммония применяется наиболее часто. Для удержания I г урана (VI) при обычных разведениях требуется 1,15 г карбоната аммония. Отделение с помощью карбоната аммония всегда проводится в горячих растворах. Однако при этом следует иметь в виду, что длительное нагревание может приводить к полному его разложению и улетучиванию из раствора.
Для отделения урана от железа, алюминия, титана и других элементов, осаждающихся карбонатами, рекомендуется следующая методика [8]. К раствору, содержащему уран, при перемешивании прибавляют 6 N NH4OH до появления слабой мути, которую вновь растворяют несколькими каплями 6 N азотной кислоты. Затем этот раствор вливают в стакан, содержащий 2,5% -ный раствор карбоната аммония в 3 N NHJ3H. Количество раствора рассчитывают таким образом, чтобы на 1 мг урана приходилось по 3—4 мг карбоната аммония. Колбу из-под исходного раствора споласкивают и промывную воду присоединяют к основному раствору, который затем нагревают до 90° и при перемешивании выдерживают при этой температуре в течение 4—5 мин. Прибавляют 1—4 г диатомитовой земли или какого-либо другого индифферентного носителя и фильтруют через фильтрующий стеклянный стаканчик. Осадок промывают пять раз раствором карбоната аммония, подщелоченным небольшим количеством аммиака. В полученном фильтрате определяют содержание урана любым подходящим методом.
Если анализируемый раствор содержит никель, то отделение урана вместо карбоната аммония лучше проводить при помощи карбоната натрия. В этом случае для более полного отделения гидроокисей железа, алюминия и некоторых других элементов рекомендуется вводить в раствор также перекись натрия. Щелочноземельные металлы, бериллий, марганец, кобальт, цинк и ряд других элементов отделяются с применением карбоната натрия несколько более полно, однако алюминий отделяется недостаточно хорошо. Если осадок гидроокисей и карбонатов значителен, то для более полного разделения необходимо его снова растворить в кислоте и провести повторное осаждение.
Отделение урана с помощью карбоната натрия проводится по следующей методике [8]. К раствору, полученному после разложения анализируемой рудый отделения элементов группы сероводорода, а также удаления сероводорода кипячением раствора, прибавляют 10—15 мл 3%-ного раствора перекиси водорода, разбавляют водой до 150—175 мл и прибавляют карбонат натрия до ясно щелочной реакции и сверх этого еще избыток около 3 г. Затем раствор кипятят в течение 15 мин. и отфильтровывают от осадка, который промывают 2%-ным раствором карбоната натрия. Если осадок значителен, то его снова растворяют в 10 мл разбавленной (1:1) серной кислоты и повторяют осаждение.
262
Отделение осаждением гидроокисью аммония, едкими щелочами и органическими основаниями
Отделение урана гидроокисью аммония [645, 709, 710, 800, 845, 857, 1026] часто применяется для его отделения от щелочных и щелочноземельных металлов и от элементов, образующих растворимые аммиачные комплексы (медь, никель, цинк и др.), а также от многих анионов.
Присутствие карбонатов в растворе аммиака или в анализируе мом растворе приводит к неполному отделению урана. Для устранения этого недостатка применяют растворы гидроокиси аммония, не содержащие карбонатов. Методы приготовления таких растворов описаны в разделе «Весовые методы определения» (Весовое определение осаждением гидроокисью аммония). Карбонаты, содержащиеся в анализируемом растворе, удаляют кипячением раствора после подкисления его азотной или соляной кислотой.
Неполное отделение урана происходит в присутствии ряда комплексообразующих веществ, таких как фториды, тартраты, цитраты и некоторые другие [181].
Для уменьшения растворимости выпадающего в осадок диураната аммония осаждение следует проводить в присутствии нитрата или хлорида аммония. Добавление бумажной массы также улучшает полноту отделения, особенно в случае малых количеств урана.
Эффективность* отделения урана от меди, никеля, цинка и других элементов, образующих комплексные аммиакаты, в сильной мере зависит от их количества, вследствие чего полное отделение от указанных элементов достигается только после двух- или трехкратного переосаждения. Осадки, выпадающие из растворов, содержащих фосфаты, ванадаты, бораты и силикаты, как правило, загрязняются ими, и для полного их отделения также необходимо пере-осаждение. Большинство же других элементов полностью или частично осаждаются гидроокисью аммония вместе с ураном 1709], и в их присутствии отделение урана без применения соответствующих маскирующих комплексообразующих веществ не может быть достигнуто.
Для отделения урана раствор подкисляют соляной или азотной кислотой и кипятят для удаления СО2. Если раствор содержит уран (IV), то подкисляют азотной кислотой, обеспечивающей окисление урана (IV) до урана (VI). На каждые 100 мл раствора прибавляют по 5—10 г нитрата или хлорида аммония, вносят бумажную массу и при перемешивании постепенно добавляют разбавленный раствор гидроокиси аммония (1 : 4) до появления сильного запаха аммиака, нагревают до кипения и кипятят в течение не менее 5 мин. для получения легкофильтрующегося осадка. После отстаивания осадок отфильтровывают из горячего раствора и промывают 2%-ным раствором нитрата аммония, подщелоченным небольшим количеством гидроокиси аммония.
263
Применение комплексона III значительно расширяет число элементов, от которых возможно отделение урана осаждением аммиаком. В присутствии комплексона III уран полностью отделяется от Mg, Са, Sr, Ва, Си, Zn, Со, Ni, Cd, Мп, Pb, Hg, Al, La, Се, V (IV). Сг, Th, Mo, W и некоторых других элементов [184, 820, 975].
Мешающее влияние титана рекомендуется устранять добавлением небольших количеств перекиси водорода [975]. Применение комплексона III позволяет отделять уран также и от ванадия (V), который хотя и не образует комплекса с комплексоном III, но при нагревании восстанавливается им до ванадия (IV), образующего с избытком комплексона III достаточно прочный растворимый комплекс, полностью удерживающийся в растворе.
Как уже указывалось выше, комбинирование отделения урана осаждением аммиаком в присутствии комплексона III с последующим извлечением урана из полученного осадка при помощи карбоната аммония позволяет отделять уран практически от всех элементов.
Однако осаждение аммиаком требует предосторожностей: из воздуха не должна попадать углекислота, а это связано с определенными неудобствами. С этой точки зрения применение вместо аммиака пиридина [180, 181,789], обладающего более слабыми основными свойствами и не поглощающего углекислоты из воздуха, выгоднее, так как устраняются возможные потери урана за счет неполного осаждения вследствие образования растворимых к'арбонатных комплексов урана, а также отсутствует опасность загрязнения выделяющихся осадков карбонатами щелочноземельных металлов.
Осаждение при помощи пиридина позволяет отделять уран от щелочных и щелочноземельных металлов, магния, марганца, кобальта и никеля [181 ]. По-видимому, применение комплексона III, как и в случае осаждения аммиаком, позволяет значительно расширить возможности отделения урана от большого числа элементов. Методика отделения — см. стр. 74
Отделение урана может быть осуществлено осаждением с помощью уротропина [835], являющегося (так же как и пиридин), слабым основанием, не поглощающим двуокиси углерода. Мешающие отделению элементы и чистота получаемых осадков в обоих методах как с использованием уротропина, так и пиридина, оказываются одинаковыми. Подробное описание метода см. стр. 74.
Этилендиамин [884, 910], а также другие сильные органические основания осаждают уран подобно аммиаку, и по сравнению с ним не имеют никаких преимуществ.
Состав образующегося осадка, а также количество отделяемых примесей при осаждении урана с помощью едких щелочей сильно зависит от концентрации щелочи в растворе. При небольших концентрациях едкой щелочи выпадает осадок, состоящий из основных солей различного состава. Так, например, в случае осаждения урана (VI) из растворов хлорида уранила с помощью едкого натра 264
полученные осадки соответствовали составу UO2(OH)j ,\С109 и UO2(OH), 4хС10 6. При увеличении количества добавляемого едкого натра содержание хлора в осадке уменьшается и в конце концов образующийся осадок полностью превращается в гидроокись уранила UO2(OH)2 [389].
Дальнейшее увеличение концентрации едкого натра в растворе приводит к появлению в осадке диураната натрия, содержание которого возрастает с повышением концентрации едкого натра. Состав образующихся осадков также в сильной степени зависит и от продолжительности взаимодействия с едким натром [190]. Из раствора 9 Л7 (или выше) по едкому натру выделяется осадок, полностью состоящий из диураната натрия.
Хотя с помощью едких щелочей не может быть достигнуто отделение урана от многих элементов, а также несмотря на то, что получаемые осадки содержат значительное количество самого осадителя, тем не менее осаждение едкими щелочами применяется довольно часто вследствие его эффективности при отделении урана от фосфатов. С помощью едкого натра возможно количественное выделение урана из фосфата уранила, который взаимодействует с едким натром с образованием нерастворимого диураната натрия и .растворимого трехзамещенного фосфата натрия. Образующиеся осадки диураната натрия трудно фильтруются. Отделение маточного раствора и промывание осадка осуществляются с помощью центрифугирования и декантации или фильтрованием через стеклянные фильтры или стеклянную вату. Вследствие высокой концентрации щелочи (около 9?/ и выше) применение бумажных фильтров исключается.
Если комбинировать осаждение едким натром с извлечением урана из полученного осадка при помощи карбонатов, то в этом случае его можно отделить от больших количеств различных элементов, в том числе от фосфора и ванадия. Для этого исходный раствор нейтрализуют до pH 6,5—7 едким натром и прибавляют к нему карбонат натрия до pH 10. В этих условиях осаждаются в виде гидроокисей или карбонатов многие элементы (Fe, Мп, Al, РЬ, Са), в то время как уран в виде растворимого карбонатного комплекса NaJUO2(COa)3] остается в растворе. При добавлении к этому раствору едкого натра уран полностью осаждается из раствора в виде диураната натрия.
Отделение осаждением перекисью водорода
Отделение урана осаждением перекисью водорода основано на образовании труднорастворимых перекисных соединений при добавлении перекиси водорода к слабокислым растворам солей ура-Нила. В соответствии с данными Тридо [986, 987], образующееся *Руднорастворимое соединение представляет собой ураниловую Соль перурановой кислоты, а не перекись урана, как это ранее пред-
26.5
полагалось. Реакция идет в две стадии; на первой стадии образуется перурановая кислота:
UO2 (NOs)2 4- ЗН2О2 — H4UO8 4- 2HNOS, которая тут же взаимодействует с еще не изменившейся солью уранила с образованием перураната уранила:
Н4ио8 -4 2UO2 (NO3)2 -> (UO2)2 UO8 4- 4HNO3.
Оптимальным для осаждения урана является pH в пределах 0,5—3,5. .Более низкие значения pH обеспечивают несколько лучшее отделение некоторых комплексообразующих веществ (ацетаты, тартраты, оксалаты), однако вследствие наблюдающегося при этом снижения полноты выделения урана осаждение не рекомендуется проводить при pH более низких, чем 0,5. Снижение полноты осаждения урана имеет место также при более высоких значениях pH, чем 3,5.
Отделение урана осаждением перекисью водорода применяется главным образом для выделения основной его массы из растворов при определении следов других металлов (титан, никель), так как образующиеся осадки перураната уранила обладают очень небольшой способностью адсорбировать из раствора другие элементы. Только калий, щелочноземельные металлы, железо и ванадий адсорбируются осадком в заметных количествах. Сульфаты и фториды несколько снижают полноту осаждения урана. Железо и медь затрудняют осаждение вследствие каталитического разложения перекиси водорода [741]. Для устранения мешающего влияния железа и меди рекомендуется прибавление малоновой или молочной кислот, образующих с ними достаточно прочные комплексы [8].
Методика отделения урана от других элементов с помощью перекиси водорода приводится в разделе «Весовые методы определения».
Для устранения мешающего влияния некоторых элементов (ванадий, железо, медь и др.), частично адсорбирующихся осадком, А. Е. Клыгиным предложено вести осаждение в присутствии комплексона III. В этих условиях образуются более чистые осадки, позволяющие заканчивать определение весовым методом (см. стр. 61).
Отделение осаждением фосфатами
Осаждение урана фосфатами нашло значительно большее применение для отделения урана, чем для его определения. Среди причин, ограничивающих применение осаждения урана фосфатами для его весового определения, следует указать на склонность образующихся осадков прочно удерживать щелочные металлы, что приводит к завышению результатов. Применение фосфатов для отделения урана от других элементов долгое время ограничивалось главным образом теми случаями, когда анализируемые растворы сами содер-266
жали фосфаты. Незначительные возможности отделения урана от других элементов были связаны с мешающим влиянием большого числа других элементов, образующих в условиях осаждения урана нерастворимые фосфаты. Осаждение урана'фосфатами и мешающее влияние других элементов описано в разделе «Весовые методы определения».
Однако оказалось, что мешающее влияние многих элементов можно легко устранить применением комплексона III, образующего с ними прочные растворимые комплексы и тем самым удерживающего их в растворе. Согласно исследованиям В. И. Титова и И. И. Волкова 1157], применение комплексона III при осаждении фосфата уранила позволяет отделять уран от многих элементов, в том числе таких, как Fe, Al, Сг, Си, Ni, редкоземельные элементы, V, Мои др. Разработанный указанными авторами метод отделения урана под названием «трилоно-фосфатного» метода нашел применение при определении малых содержаний урана в бедных рудах и растворах сложного состава.
Методика отделения состоит в следующем [157]. 0,2—2 г анализируемого материала (в зависимости от содержания урана) разлагают кислотами одним из обычных методов (НС1 -у Н2О2, HNO3-y НО, H2SO4-[- HNO3+ НО), нерастворив-шийся остаток отфильтровывают и промывают. К полученному фильтрату объемом ВО—100 мл добавляют 16—24г нитрата аммония и 10мл 15%-ного раствора комплексона III при исходной навеске анализируемого материала 0,2 г; в случае навески 0,5 г добавляют 15 мл раствора комплексона III и при навеске 1—2 г — 25 мл и нейтрализуют по метиловому красному 25?'о-ным раствором аммиака, прибавляя его по каплям. Так как анализируемые растворы часто бывают окрашены, то конец нейтрализации в таких случаях определяют пробой на белой фарфоровой пластинке. Затем сверх этого прибавляют еще I—2 капли раствора аммиака, нагревают до кипения и кипятят в течение 2—3 мин. Нагревание и последующее кипячение раствора способствуют образованию комплексонатов хрома (III), ванадия (IV) и некоторых других элементов. Затем раствор охлаждают до комнатной температуры, прибавляют к нему 10 мл 10%-ного раствора фосфата аммония или натрия, 5 мл раствора сернокислого титана, содержащего 5 мг TiO2 в 1 мл, играющего роль соосадителя, и нейтрализуют аммиаком по метиловому красному, добавляя его с избытком в 1—2 капли. Выделившийся осадок фосфатов титана и уранила отстаивают в течение нескольких часов и затем отфильтровывают через два вложенных один в другой фильтра «белая лента». Промывают горячим 1—2%-ным раствором нитрата аммония не менее 10—12 раз, не стремясь полностью перенести весь осадок со стакана на фильтр. Отделение урана от титана проводят по методике В. М. Звенигородской [79].Для этого промытый осадок смывают с фильтра струей горячей воды в стакан, в котором производилось осаждение, добавляют 10%-ный раствор углекислого натрия до общего его содержания в растворе, равного 5%, накрывают часовым стеклом и нагревают до кипения. По охлаждении осадок гидроокиси титана отфильтровывают через бумажный фильтр, предварительно смоченный 5% -ным раствором углекислого натрия, и промывают этим же раствором. В полученном фильтрате содержание урана определяют фотометрическим или другим подходящим методом.
Для выделения урана из пород, содержащих значительные количества фосфата кальция (фосфориты и т. п.), его также осаждают 8 виде фосфата, но осаждение проводят в слабокислой среде, чтобы предотвратить осаждение фосфата кальция. В соответствии с методикой, разработанной В. М. Звенигородской, Л. С. Василевской
267
и Т. В. Дейкиной [157], уран осаждают фосфатом в уксуснокислом растворе при pH 4,5—5,0 совместно с алюминием и железом [989]. В этих условиях кальций остается в растворе в виде кислого рас-творимого фосфата. Кроме кальция, в растворе также остаются медь, никель, кобальт, марганец и другие двухвалентные элементы, а также ванадий и молибден. Если анализируемый материал содержит церий, а также большие количества алюминия, титана и железа при низком содержании урана, то отделение урана в чистом виде этим методом уже становится невозможным. В случае руд с большим содержанием ванадия, полученный осадок фосфата уранила необходимо переосаждать.
Методика работы состоит в .следующем. От 0,2 до 3 г анализируемого материала (в зависимости от содержания урана) помещают в стакан емкостью 150—200 мл, добавляют 3—5 мл азотной кислоты (уд. в. 1,40) и 10—20 мл разбавленной серной кислоты (1 : 1), накрывают часовым стеклом, нагревают до кипения и кипятят до появления паров SO3. Сняв стакан с плитки, к горячему раствору осторожно добавляют 20—30 капель азотной кислоты (уд. в. 1,40) для окисления органических веществ и снова нагревают до появления паров SO3. Если, судя по цвету раствора (бурый, темно-желтый), органические вещества не разрушились, то добавляют азотную кислоту и повторяют нагревание. После этого нагревание продолжают еще 30—40 мин., затем снимают стекло и упаривают раствор до получения почти сухого остатка солей. По охлаждении осторожно прибавляют 15—20 мл воды, 5—10 мл концентрированной соляной кислоты и кипятят до растворения солей. Полученный раствор вместе с нерастворившимся остатком разбавляют горячей водой до 50—60 мл, нейтрализуют аммиаком до появления неисчезающей мути, которую растворяют добавлением 2—3 капель концентрированной соляной кислоты и сверх этого добавляют 20—30 капель той же кислоты. Если при нейтрализации раствора обнаружится, что осадок гидроокисей алюминия и железа очень мал, то добавляют 8—10 мл 5%-ного раствора алюминиевоаммонийных квасцов для обеспечения полного соосаждения фосфата уранила. К полученному слабокислому раствору добавляют горячей воды до 100—150 мл, 5 г хлорида аммония, 30?о-ного раствора уксусной кислоты по 5—6 мл на каждые 100 мл раствора, нагревают до кипения, добавляют 15—40 мл раствора фосфата натрия и затем отдельными порциями кристаллический ацетат натрия до отсутствия изменения красной окраски бумаги, пропитанной конго красным. Раствор хорошо перемешивают и помещают на кипящую водяную баню для коагуляции осадка фосфатов. Через 20—30 мин., когда раствор над осадком станет прозрачным, проверяют реакцию раствора смачиванием бумажки конго красного, которая при этом не должна изменять своей окраски (pH 4,5—5,0), а в противном случае добавляют еще ацетат натрия. Раствор фильтруют горячим через бумажный фильтр «белая лента», осадок промывают 7—8 раз горячим 1—2%-ным раствором нитрата аммония, содержащим 0,5% фосфата натрия, не стремясь перенести весь осадок из стакана на фильтр. Промывание осадка можно заменить переосаждением. В этом случае осадок фосфатов смывают с фильтра водой обратно в стакан, в котором производилось осаждение, и, растворив осадок добавлением нескольких миллилитров соляной кислоты, осаждают вновь, как указано выше. Переосаждение, в особенности, рекомендуется в присутствии тяжелых металлов, ванадия и молибдена. Если необходимо, то переосаждение повторяют еще раз. О присутствии тяжелых металлов (Си, Ni, Со) можно судить по цвету фильтратов. О присутствии ванадия и его количестве заключают по окраске, возникающей при добавлении капли перекиси водорода к кислому раствору. Фильтр с осадком фосфатов развертывают над стаканом, в котором производилось осаждение, и тщательно смывают осадок в стакан 50 мл 10% -ного раствора карбоната натрия. Добавляют 0,05—0,1 а животного угля (для сорбции загрязнений, коллоидной гидроокиси железа, следов тяжелых металлов и т. п.), накрывают стакан часовым стеклом и кипятят раствор до уменьшения его
268
объема до 20—25 .мл. Осадок отфильтровывают через бумажный фильтр, смоченный 5%-ным раствором карбоната натрия, и промывают его этим же раствором. В полученном растворе содержание урана определяют любым подходящим способом.
При фотометрическом определении урана карбонатный раствор вместе с осадком разбавляют в мерной колбе емкостью 100 .ч.г, отфильтровывают осадок через сухой фильтр, и аликвотную часть фильтрата берут на определение.
П. А. Волков [1841 для выделения урана из растворов сложного состава предложил метод, получивший название «фосфатного». Метод основан на осаждении урана (IV) из кислых растворов в виде труднорастворимого фосфата урана (IV) U(HPO4)2. Для обеспечения большей полноты выделения урана осаждение проводят в присутствии тория или циркония, в результате чего до 1 jf/caU можно количественно выделить вследствие соосаждения фосфата урана (IV) с фосфатами указанных элементов. Одновременно с этим происходит отделение урана от железа, марганца, ванадия и большинства других элементов. Для восстановления урана (VI) до урана (IV) применяют гидросульфит натрия Na2S2O4, ронгалит Na2H2S2O4-• 2СН2О-4Н2О, сернистый натрий или соли хрома (II). Могут быть применены также и другие восстановители.
При восстановлении гидросульфитом выделяется свободная сера. Кроме того, образуются осадки сульфидов металлов группы сероводорода. При растворении осадка фосфатов в серной кислоте они в раствор не переходят и отделяются фильтрованием. Восстановление ронгалитом ведут при нагревании до 80—90°. Соли хрома (II) легко восстанавливают уран (VI) до урана (IV) при комнатной температуре. Восстановление проводят в солянокислых или хлорнокислых растворах. Азотная кислота мешает восстановлению. Вследствие того, что в присутствии сульфатов осаждение урана (IV) замедляется, а при большом их содержании становится неполным, серную кислоту при восстановлении также не применяют. Одновременно с ураном восстанавливается железо (III) до железа (II).
Для осаждения обычно применяют двузамещенный фосфат натрия Na2HPO4- 12Н2О. Образующийся в умереннокислых растворах осадок фосфата урана имеет состав U(HPO4)2-лН2О. Состав осадка не зависит от концентрации фосфат-иона в пределах от 1-10~4 до 1,4 моль/л. Однако при постоянной кислотности (pH---2) остаточная концентрация урана, определенная по a-счету U233 в маточнике, с увеличением концентрации фосфата сначала падает, затем при концентрации фосфата 2,4-10~2 моль/л достигает минимального значения, равного 2,4-10-6 моль!л, и далее возрастает. Уменьшение полноты осаждения урана (IV) при увеличении концентрации фосфата в растворе выше 2,4-10“2 моль/л, по-видимому, связано с образованием растворимых комплексных фосфатов типа Ш(НРО4)3]2" [588, 9841, в которых уран (IV) находится в анионе. Растворимость фосфата урана (IV) в сильной мере также зависит и от кислотности раствора (см. табл. 8).
Вследствие того, что в кислых растворах растворимость фосфатов большинства сопутствующих урану элементов значительно
269
выше, чем растворимость фосфата урана (IV), уран может быть отделен от них однократным осаждением. Только цирконий, торий и частично титан и ванадий осаждаются в тех же условиях вместе с ураном.
В присутствии больших количеств свинца уран осаждается. на холоде в виде белого гидросульфита свинца, который при растворении осадка в серной кислоте разлагается с выделением сернистой кислоты, остающейся в растворе и являющейся причиной завышенных результатов при оксидиметрическом титровании урана. Для устранения этого недостатка раствор с осадком перед фильтрованием нагревают, в результате чего свинец выделяется в виде сульфида, практически не растворяющегося в разбавленной серной кислоте. При выделении урана из сульфидных руд металлы группы сероводорода необходимо предварительно отделять осаждением сероводородом или тиосульфатом натрия. Выделенные сульфиды могут частично захватывать уран, поэтому в тех случаях, когда требуется очень высокая точность, осадок сульфидов прокаливают, растворяют в хлорной кислоте, выделяют металлы на ртутном катоде, а раствор, содержащий захваченный сульфидами уран, присоединяют к основному раствору.
Как указывалось выше, при выделении малых количеств урана, что имеет место при анализе бедных руд и отвалов, необходимо в раствор вводить соли тория или циркония, образующие труднорастворимые фосфаты, соосаждающие фосфат урана. Применение циркония менее удобно, так как образующийся осадок медленнее коагулирует, и при обработке его серной кислотой уран труднее переходит в раствор.
Выделение урана фосфатным методом с применением гидросульфита в качестве восстановителя по методике П. А. Волкова [184] состоит в следующем. К кислому раствору, полученному после разложения анализируемой руды и отделения нерастворимой части, прибавляют раствор аммиака до перехода фиолетовой окраски индикатора метанилового желтого в желтую (pH ^2,3), затем добавляют 8 мл соляной кислоты (1 : 1) и разбавляют водой до 200 мл. К полученному раствору прибавляют 10 мл 10%-ного раствора двузамещенного фосфата натрия, гидросульфит натрия в количестве, равном исходной навеске руды, но не менее 0,5 г, 4—5 мл раствора хлорида тория (содержащего 20—25 мг тория), бумажную массу, хорошо перемешивают и оставляют до полной коагуляции и просветления раствора, которое наступает спустя 40—60 мин. Осадок отфильтровывают через бумажный фильтр «синяя лента» с применением отсасывания и хорошо промывают водой, содержащей 2 мл концентрированной соляной кислоты в 1 л, до полного удаления продуктов разрушения гидросульфита. Осадок на фильтре растворяют в серной кислоте (1 : 2) и промывают ею фильтр и стакан, в котором проводилось осаждение. Содержание урана в полученном сернокислом растворе определяют оксидиметрическим титрованием или любым другим подходящим способом.
Если анализируемый материал содержит менее 0,01% урана, что требует увеличения применяемой навески до 5 г, то промывание осадка фосфатов и серы в таком случае затруднено. Вместо промывания осадка повторяют осаждение. Для этого осадок фосфатов растворяют в разбавленной соляной кислоте (1 : 2), этой же кислотой промывают фильтр, раствор нейтрализуют аммиаком, затем подкисляют 6 мл соляной кислоты (1 : 1), разбавляют водой до 200 мл и повторяют осаждение с меньшим количеством реактивов (5 мл раствора фосфата натрия, 0,2 г гидросульфита, 2 мл раствора хлорида тория).
270
Вместо гидросульфита в качестве восстановителя может быть применен хлорид хрома (II). Однако в присутствии больших количеств железа расходуемое количество хлорида хрома (II) значительно возрастает, вследствие чего выделение урана.в присутствии образующихся больших количеств хрома (III) сильно затрудняется. Для устранения мешающего влияния хрома (III) его связывают в виде растворимого роданидного комплекса или уменьшают расход хрома (II) за счет предварительного восстановления железа (III) до железа (II) аскорбиновой кислотой.
Выделение урана по этому варианту фосфатного метода проводится в соответствии с методикой, разработанной К). А. Черниховым и В. Ф. Лукьяновым [184]. Анализируемый раствор разбавляют водой до 200 мл, прибавляют 3 мл концентрированной соляной кислоты, 10—15 мл 10%-ного раствора двузамещенного фосфата натрия, роданид аммония из расчета 2,5 г на 0,1 г железа, 0,5 N раствор хлорида хрома (II) до исчезновения красной окраски роданида железа и сверх этого еще 3 мл, 4—5 мл раствора хлорида тория, содержащего 20—25 мг тория, немного бумажной массы и далее поступают так же, как и в первом варианте фосфатного метода.
В. М. Бродская, Г. А. Ланской и В. Г. Сочеванов [20] показали, что при выделении урана из растворов, содержащих ванадий, выпадающий осадок фосфата урана содержит некоторое количество ванадия в результатесоосаждения с ним ванадия (III) и ванадия (IV), также образующих труднорастворимые фосфаты. Вследствие этого при последующем оксидиметрическом или фотометрическом определении урана получаются завышенные результаты. Для устранения этого недостатка указанные авторы предложили осадок фосфата урана (IV) после его промывки подкисленной водой дополнительно промывать водным раствором иода с pH 5,0—5,5 (содержащим в 1 л 8—10 мл 0,1 N раствора иода в иодистом калии) [20, 157]. Благодаря этой обработке ванадий (III) и ванадий (IV) окисляются до ванадия (V), полностью удаляющегося в процессе отмывки.
Фосфатный метод оказался также пригодным для выделения урана из горных пород, содержащих 10~3—10"4% урана. В качестве носителя в этих случаях применяют торий или цирконий [143, 184]. Промытый и прокаленный осадок сплавляют с фторидом натрия и заканчивают определение люминесцентным методом.
Отделение урана осаждением фосфорноватой кислотой [588, 984] в виде фосфорноватокислого урана (IV) UP2O6 не нашло практического применения и может рекомендоваться только для отделения урана (IV) от урана (VI).
Гипофосфит натрия в присутствии тиосульфата аммония позволяет количественно осаждать весь уран в виде гипофосфита урана (IV) вследствие восстановления урана (VI) до урана (IV) [836]. Однако в присутствии больших количеств железа, а также ряда других элементов полное отделение не имеет места.
Осаждение урана (IV) с помощью пирофосфата натрия [588, 984] позволяет отделять уран (IV) от урана (VI) и от большинства трехвалентных элементов.
271
Методики осаждения урана фосфорноватой кислотой, гипофосфитом натрия в присутствии тиосульфата, а также солями пиро-фосфорной кислоты приводятся в разделе «Весовые методы определения».
Отделение осаждением фторидами
Вследствие незначительной растворимости тетрафторида урана и в особенности двойных фторидов урана-аммония, урана-натрия или урана-калия [173, 275], а также возможности отделения урана от больших количеств циркония, ниобия, тантала, бора, железа, ванадия и других элементов, образующих растворимые фторидные комплексы [275, 991], метод отделения урана (IV) в виде фторидов нашел достаточно широкое применение. Методика осаждения урана (IV) плавиковой кислотой приводится в разделе «Весовые методы определения».
В. М. Звенигородская и Л. П. Рудина [157, 184] провели детальное изучение свойств двойного фторида четырехвалентного урана и натрия и установили, что полное осаждение урана ввидеХаЁР. имеет место даже в IV серной кислоте, в которой Fe (II), Al, V, Mo,Ti, Ni, Co, Мп, Си, В, Zr, Та и некоторые другие элементы не образуют нерастворимых фторидов. Только железо (III), частично осаждающееся в виде двойного фторида натрия, и алюминий, осаждающийся почти полностью в виде криолита, загрязняют образующийся осадок двойного фторида урана-натрия. Фторид церия также частично осаждается вместе с ураном и может быть причиной ошибок при фотометрическом определении с помощью перекиси водорода.
Осаждение в присутствии натриевых солей имеет преимущество не только в том, что образующийся двойной фторид урана-натрия обладает меньшей растворимостью по сравнению с тетрафторидом урана, но и в том, что натриевые соли парализуют растворяющее действие алюминия. Для более полного осаждения малых количеств урана иногда вводят соли кальция, осадок фторида которого является хорошим коллектором для фторида урана.
Для восстановления урана (VI) до урана (IV) применяют ряд восстановителей, в частности И. Е. Старик [184] успешно применял восстановление цинком. А. А. Гринберг, Л. Е. Никольская, Г. И. Петржак, Е. Ф. Птицын и Ф. М. Филинов [55] рекомендуют пользоваться ронгалитом NaHSO2-CH2O-2H2O. Хорошие результаты дает также восстановление в висмутовом или кадмиевом редукторе.
Образующийся частично уран (III) окисляется кислородом воздуха до урана (IV) в процессе самого осаждения.
В. М. Звенигородская и Л. П. Рудина [157, 184] в разработанном ими методе в качестве восстановителя применили двухвалентное железо. Хотя окислительно-восстановительный потенциал системы Fe3+/Fe2+ в IV серной кислоте составляет 0,67 в, что значительно выше окислительно-восстановительного потенциала системы UOI+/U4+, равного в тех же условиях 0,48 в, однако в присутствии 272
плавиковой кислоты потенциалы обеих систем сдвигаются в противоположных направлениях таким образом, что уже при концентрации плавиковой кислоты, равной IN, потенциал системы UO^/U4' значительно превосходит потенциал системы Fe:i'/Fe2*. С дальнейшим повышением концентрации плавиковой кислоты в растворе эта разница увеличивается еще больше (табл. 39). Такой сдвиг потенциалов обеих систем обусловливается связыванием ионов Fe3+ и U4+ в прочные комплексы FeFe“ и UFF- Точно такое же изменение потенциала в присутствии плавиковой кислоты имеет место и для системы VO2'/V3+ (см. табл. 39).
Таблица 39
Потенциалы систем Fe3+/Fe2+, UO|+/U4 + и VO2+/V3+ в IN серной кислоте при разных концентрациях HF, отнесенные к нормальному водородному электроду [250]
Концентрация HF, Л1ОЛ6/Л Потенциал системы, в
Fe3+/Fe2 + ио2+/и4+ vo2+/v’+
0,0 0,67 0,48 0,40
1,0 0,49 0,58 0,48
2,0 0,44 0,60 0,53
3,0 0,40 0,62 0,55
4,0 0,40 0,63 0,57
Применение железа (II) в качестве восстановителя оказалось удобным при осаждении фторида урана (IV) с последующим его оксидиметрическим титрованием или фотометрическим определением с помощью перекиси водорода. Частично осаждающееся железо (III) в случае необходимости отделяют карбонатным методом.
Для отделения урана по методике В. М. Звенигородской и Л. П. Рудиной [157, 184] анализируемый раствор подкисляют 5 мл серной кислоты (1 : 1), добавляют 2 г соли Мора, 20 мл 10% -ного раствора сернокислого натрия, 20 мл 40% -ной плавиковой кислоты и разбавляют водой до 100 мл; хорошо перемешивают и оставляют на 15 мин. для завершения восстановления урана (VI) до урана (IV). Если содержание урана в анализируемом растворе не превышает 5 мг, то для большей полноты осаждения прибавляют I—1,5 мл раствора хлористого кальция, содержащего 25 мг Са/лм. Затем добавляют бумажную массу и выдерживают в течение 1 часа при периодическом перемешивании. После этого фильтруют через парафинированную, политеновую или эбонитовую воронку с бумажным фильтром. Фильтр с осадком промывают 10—12 раз промывным раствором, который готовят растворе-' Пием 5 мл плавиковой кислоты и 4 г уксуснокислого натрия в 95 мл воды.Промытый осадок растворяют на фильтре в 25—30 мл смеси азотной и борной кислот (200 мл азотной кислоты (1 : 2), смешивают с 100 мл 4%-ного раствора борной кислоты), , .обрабатывая фильтр указанным количеством смеси повторно 3—4 раза. Затем фильтр промывают 70—75 мл воды, и полученный раствор и промывную воду быст-. ро упаривают до выделения солей. Остаток обрабатывают при нагревании 50 мл i 18 Аналитическая химия урана 273
10%-ного раствора соды, добавляют около 0,1 г животного угля, накрывают стакан часовым стеклом и кипятят до достижения объема в 20—25 мл. По охлаждении фильтруют, фильтр промывают водой, и весь раствор или часть его берут для фотометрического определения с перекисью водорода.
Е. Р. Николаева [173] для отделения урана предложила осаждение с помощью фторида аммония.
К 5—10 мл раствора добавляют 0,5 г сульфата железа (II), 0,2—0,3 г фторида аммония и разбавленную серную кислоту до pH 1,5—2,0. В течение нескольких минут нагревают до 70—80° и затем охлаждают.
Через 2—2,5 часа выпавший осадок отфильтровывают через бумажный фильтр «синяя лента», промывают 1,5%-ным раствором фторида аммония, содержащим 0,25% (по объему) серной кислоты, и растворяют в 10 мл 10 N серной кислоты, насыщенной борной кислотой. В полученном растворе уран (IV) обычно титруют ванадатом аммония.
Метод позволяет отделять 0,45—4,5 мг урана от 10 мг ванадия, 10—15 мг молибдена и до 50 мг железа (III) и определять его с ошибкой не более 1 %.
Для выделения малых количеств урана И. Е. Старик, Ф. Е. Старик и А. Н. Аполлонова [243] рекомендуют осаждать уран (IV) в виде тетрафторида > на носителе-тетрафториде тория ThF4 (см. стр. 284).
Отделение осаждением купфероном
Купферон (аммонийная соль М-нитрозо-М-фенилгидроксила-мина) из сильнокислых растворов осаждает уран (IV). Щелочные и щелочноземельные металлы, Al, Zn, Cr, Be, Мп, Ni и некоторые другие элементы, а также фосфаты, бораты, фторобораты и органические комплексообразующие вещества (оксалаты, тартраты, цитраты) при этом остаются в растворе. Так как уран (VI) из сильнокислых растворов не осаждается купфероном, предварительно его восстанавливают до урана (IV) с помощью подходящего восстановителя (гидроксиламин, гидросульфит натрия и др.). Методика осаждения приводится в разделе «Весовые методы определения».
Однако для отделения урана от других элементов с помощью купферона нашел применение иной метод, основанный на предварительном осаждении при помощи купферона всех мешающих элементов (Zr, Н f, Nb, Та, V, Fe, Ga, Sn, Sb, T i) в присутствии урана (VI), не осаждающегося купфероном из сильнокислых растворов. Затем восстанавливают уран (VI) до урана (IV) и повторяют осаждение в тех же условиях. Так как все мешающие элементы предварительно удалены, то в осадок выпадает только купферонат урана (IV) U(C6H5N2O2)4 [69, 345, 607, 996]. Вследствие того, что разработанные методы оказались довольно громоздкими и трудоемкими, они не нашли широкого применения, несмотря на большие возможности отделения.
Недавно С. В. Елинсону и В. А. Олезнюк [70, 184] путем подбора подходящего восстановителя для восстановления урана (VI) 274
до урана (IV) удалось избежать необходимости полного разложения избытка купферона многократной обработкой смесью серной и азотной кислот в фильтрате перед восстановлением урана и тем самым значительно упростить этот метод. Подробная методика, рекомендованная С. В. Елинсоном и В. А. Олезнюк [70], приводится в разделе «Весовые методы определения». Метод дает высокую точность (Н-0,3% (отн.)) при весовом окончании, но при содержании урана в анализируемом материале ниже 5% точность несколько снижается. Основным достоинством метода является возможность отделения урана от всех мешающих элементов, в том числе таких, как V, Те, Ti, Zr, Nb, Та, Р и др.
Купферон оказался пригодным также и для отделения урана осаждением его в виде купфероната уранила [863]. Количественное осаждение имеет место при pH 4—9. Вследствие более высоких значений pH осаждения мешающее влияние других элементов в этом случае значительно больше, чем при осаждении урана (IV). Для повышения избирательности осаждения рядом автором [373, 728] предложено применение комплексона III для удержания в растворе других элементов. В этом случае отделяются щелочные и щелочноземельные элементы, Mg, Ni, Со, Zn, Мп, Cd,*Си, Pb, Ag, Hg, Bi, Те, Cr, Sn, Th, La, Се и редкоземельные элементы. Прибавлением винной кислоты удерживаются в растворе Al, Sb, Sn, Nb и Та. Титан и цирконий в небольших количествах также не мешают отделению урана. Присутствие нитратов, хлоридов, сульфатов, хроматов, молибдатов, вольфраматов, а также ацетатов, оксалатов и цитратов влияния не оказывает.
А. Е. Клыгин, Д. М. Завражнова и И. С. Коляда 1101 ] исследовали мешающее влияние различных элементов и подобрали оптимальные условия осаждения. Соответствующая методика приводится в разделе «Весовые методы определения».
Отделение осаждением 8-оксихинолином
8-Оксихинолин осаждает уран (VI) из растворов с pH в пределах от 4,1 до 13,5 [8, 553]. При осаждении из растворов с pH 10— 12 уран отделяется от фосфатов, тартратов, небольших количеств фторидов, оксалатов, лактатов и гидроксиламина [436, 846]. Однако одновременно с ураном 8-оксихинолин осаждает также очень много других элементов. Осаждение урана (IV) также мало избирательно, как и осаждение урана (VI). За счет соответствующего подбора pH уран может быть отделён от ряда элементов, в частности, Из растворов, содержащих едкий натр, 8-оксихинолин не осаждает олова, алюминия, бериллия и щелочноземельных металлов. Методики осаждения урана (VI) из слабокислых и щелочных растворов Приводятся в разделе «Весовые методы определения». Однако практического значения отделение урана при помощи 8-оксихинслина
18* 275
в таком виде не получило вследствие чрезвычайно низкой селективности.
Для повышения избирательности осаждения весьма успешным оказалось применение комплексона III [898, 900], добавление которого позволяет удерживать в растворе торий, редкоземельные элементы, ванадий и ряд других элементов.
Для отделения урана (VI) от тория, редкоземельных элементов и циркония к анализируемому раствору добавляют 10 мл 10%-ного раствора комплексона III и нейтрализуют аммиаком по метиловому красному до перехода красной окраски в желтую. Затем добавляют 1,1 мл 50%-ной уксусной кислоты и 25 мл 20%-ного уксуснокислого аммония, разбавляют водой до 150—175 мл, нагревают до 70° и добавляют 5 мл 4%-ного спиртового раствора 8-оксихинолина. Если присутствует значительное количество циркония, то добавляют еще 1 мл раствора 8-оксихинолина.
После охлаждения до комнатной температуры выпавший осадок отфильтровывают, промывают 0,01%-ным водным раствором 8-оксихинолина и определяют весовым (после высушивания при 110°) или другим подходящим методом.
Описанным способом обеспечивается количественное отделение 30—40 мг урана (VI) от 100 мг тория и 30 мг редкоземельных элементов.
В случае присутствия фосфатов анализируемый раствор разбавляют водой до 125 мл, прибавляют 10 мл 10%-ного раствора комплексона III, 7,5 мл 4%-ного спиртового раствора 8-оксихинолина, нагревают до 70°, добавляют раствор аммиака до желтой окраски тимолового голубого и постепенно при перемешивании 25 мл 20%-ного раствора ацетата аммония. Дальнейший ход отделения совпадает с вышеописанным.
В этих условиях уран (VI) количественно отделяется от 100 мг Р3О5 при одновременном присутствии до 100 мг тория или 30 мг редкоземельных элементов.
Для отделения урана (VI) от ванадия к анализируемому раствору добавляют аммиак до появления осадка гидроокисей, затем 0,4—0,5 мл соляной кислоты (уд. в. 1,19), разбавляют водой до 75 мл, прибавляют 10 мл 10% -ного раствора комплексона III, нагревают до кипения и кипятят в течение 10—12 мин. для восстановления комплексоном III ванадия (V) до ванадия (IV), который затем в условиях осаждения образует с избытком комплексона III прочный растворимый комплекс и не мешает отделению урана (VI). Раствор нейтрализуют аммиаком по метиловому красному, добавляют 1,1 мл уксусной кислоты (1 : 1), 25 мл 20%-ного ацетата аммония и разбавляют водой до 150—175 мл. Далее нагревают до 70°, прибавляют 5 мл 4%-ного спиртового раствора 8-оксихинолина и поступают так же, как выше описано. Метод позволяет количественно отделять 30—40 мг урана от 100 мг V2O5 [900].
Кроме отделения от тория, циркония и редкоземельных элементов осаждение урана (VI) при помощи 8-оксихинолина из уксуснокислых растворов (pH--5,3) в присутствии комплексона III позволяет количественно отделять уран также и от Fe (III), Al, Си, Со, Ni, Zn, Cd, Pb, Bi, Мп и ряда других элементов. При проведении осаждения в аммиачно-щелочной среде (рН-^-8,4) уран (VI) может быть количественно отделен от молибдена, вольфрама и ванадия [898].
276
Отделение осаждением щавелевой кислотой
Применение щавелевой кислоты для отделения урана основано на ее способности осаждать уран (IV) из солянокислых растворов в виде труднорастворимого оксалата урана (IV) [579]. Наиболее полное осаждение урана (IV) достигается из растворов с концентрацией соляной кислоты, равной 0,12V. С целью достижения лучшего отделения урана от других элементов осаждение проводят обычно из растворов с концентрацией соляной кислоты не ниже 2N. Проведение осаждения при более низкой кислотности в присутствии цинка, железа (II), меди и некоторых других элементов приводит к частичному осаждению оксалатов этих элементов совместно с оксалатом урана (IV). Осаждение урана (IV) из растворов с кислотностью выше чем 3N становится уже неполным.
Из предварительно восстановленных растворов уран (IV) количественно может быть отделен от умеренных количеств других элементов осаждением щавелевой кислотой. Исключением являются только торий и редкоземельные элементы. Ниобий в зависимости от его содержания также может частично осаждаться вместе с ураном (IV). Полноте осаждения урана (IV) мешают сульфаты, фосфаты, фториды и некоторые органические комплексообразующие вещества (молочная кислота и т. п.). После отделения осадка содержание урана в нем определяют весовым или другим удобным методом. Методика осаждения подробно описана в разделе «Весовые методы определения».
Отделение цсаждением таннином
Таннин (глюкозный эфир танниновой кислоты) количественно осаждает уран (VI) из горячих растворов [882]. Вместо таннина с одинаковым успехом может применяться и танниновая (дигал-ловая) кислота [452, 453]. Осаждение ведут из почти нейтральных растворов в присутствии ацетата аммония, необходимого для поддержания нужного pH раствора и ускоряющего коагуляцию образующего осадка. Методика осаждения урана (VI) при помощи таннина приводится в разделе «Весовые методы определения».
Та, Nb, Ti, V, Те, Zr, Hf, Th и Al также осаждаются таннином [95, 882]. Для отделения урана от тантала, ниобия и титана эти элементы предварительно осаждают таннином из полунасыщенного хлоридом аммония слабокислого раствора, содержащего оксалат [880]. Полученный осадок отфильтровывают и затем из фильтрата осаждают уран (VI) путем дополнительного добавления таннина и подщелачивания раствора аммиаком до слабощелочной реакции.
Тартраты не мешают отделению урана осаждением таннином из нейтральных растворов [880]. В присутствии сульфатов и умеренных количеств солей щелочных металлов осаждение урана становится неполным [452]. В этом случае, так же как и в присутствии
277
оксалатов, полное осаждение достигается только в аммиачно-ще-лочной среде. Из карбонатных растворов уран осаждается таннином при добавлении пиридина [95, 882].
Осаждение таннином рекомендуется для отделения малых количеств урана вместо осаждения аммиаком [452, 453].
Отделение осаждением ферроцианидами
Ферроцианид калия очень полно осаждает ионы уранила. Вследствие малой растворимости образующегося осадка осаждение в виде ферроцианида представляет интерес для Отделения малых количеств урана. Однако большое количество других элементов также осаждается ферроцианидом калия, в результате чего отделение оказывается малоизбирательным.
Состав образующегося осадка зависит от количества ферроцианида калия и от условий осаждения (pH, температура). Наиболее полное осаждение имеет место в нейтральной или в слабокислой среде. Однако образующиеся в этих условиях чрезвычайно тонкие осадки плохо фильтруются и трудно отмываются. В. Г. Сочеванов, И. В. Шмакова и Г. А. Волкова [234] подробно исследовали осаждение урана ферроцианидом калия и нашли, что оптимальными условиями осуждения являются: pH в пределах 2—4 и температура 50— 60°. Осаждение проводят в присутствии нитрата калия (Ш). Образующийся осадок при добавлении избытка ферроцианида калия имеет состав К4 [UO2]4 [Fe(CN)6]3. Осадок отфильтровывают и промывают разбавленным раствором ферроцианида калия. Железо, марганец, цинк, медь и тяжелые металлы полностью осаждаются вместе с ураном.
Щелочноземельные металлы, а также другие легкие элементы частично или полностью осаждаются совместно с ураном. Осаждение урана ферроцианидом калия оказывается подходящим только для отделения от бериллия [881], а также от фосфатов и арсенатов [526]. В этом случае после отделения кремневой кислоты выпариванием раствора с соляной кислотой раствор нейтрализуют до слабокислой реакции, прибавляют избыток ферроцианида калия и насыщают хлоридом натрия. Осадок отфильтровывают, промывают декантацией раствором хлорида натрия, затем переносят на фильтр и тщательно промывают тем же раствором. Полученный осадок разлагают на холоде разбавленным раствором едкого калия, образовавшиеся гидроокиси промывают водой, содержащей хлорид аммония и небольшое количество аммиака, до отсутствия ионов ферроцианида в фильтрате. Затем осадок гидроокисей растворяют в соляной кислоте, нейтрализуют аммиаком до почти нейтральной реакции и прибавляют небольшой избыток карбоната аммония. После отстаивания осадок отфильтровывают и промывают водой, содержащей небольшое количество карбоната аммония. Фильтрат нагревают до разложения большей части карбоната аммония, затем подкисляют и ки-278
пятят до полного удаления углекислоты. Кипящий раствор насыщают сероводородом, осадок отфильтровывают и в фильтрате определяют уран удобным способом.
Отделение осаждением в виде ураниламмонийванадата
Осаждение урана в виде ураниламмонийванадата целесообразно применять для выделения его из богатых ванадием руд. Этот метод позволяет избежать предварительного отделения ванадия. Определение заканчивают обычно прокаливанием осадка ураниламмонийванадата и взвешиванием в виде V2O5-2UOS.Методика осаждения урана ванадатом аммония приведена в разделе «Весовые методы определения».
К. А. Ненадкевич [184] предложил выделять уран в виде ура-нованадата кальция осаждением его из уксуснокислого раствора с помощью ванадата кальция. Для отделения от ванадия уран затем осаждают в виде уранилфосфата.
Отделение осаждением при помощи сульфидов
Уран количественно осаждается сульфидами и полисульфидами, а также сероводородом из нейтральных или слабощелочных растворов. Цитраты, фосфаты, пирофосфаты и вещества, образующие с ураном растворимые комплексы, затрудняют выделение урана. Все элементы, образующие в этих условиях нерастворимые сульфиды, осаждаются вместе с ураном. Вследствие этого применение сульфидов для отделения урана путем его осаждения представляет небольшой интерес.
Метод осаждения урана сероводородом в присутствии уротропина [182] позволяет получать кристаллические осадки, значительно лучше фильтрующиеся и промывающиеся, чем осадки, получаемые осаждением в его отсутствие. Кроме того, эти осадки отличаются незначительной адсорбционной способностью, что обеспечивает хорошее отделение урана от щелочных и щелочноземельных металлов. Методика осаждением урана сероводородом в присутствии уротропина приводится в разделе «Весовые методы определения».
Сероводород может быть применен для отделения урана от других элементов осаждением их в виде сульфидов в тех условиях, в которых уран сероводородом не осаждается. Таким путем уран можно отделить от элементов группы сероводорода осаждением их из кислого раствора. Отделение урана от элементов группы мышьяка может быть достигнуто осаждением урана сульфидом аммония. Так как в присутствии карбоната аммония уран сульфидом аммония не осаждается, то осаждением из карбонатных растворов можно отделить уран от железа, алюминия, титана и ряда других элементов.
279
В виде сульфида уран не осаждается также из растворов, содержащих тартраты, и в их присутствии с помощью сульфида аммония может быть отделен от железа, меди, кобальта и многих других элементов.
Отделение осаждением в виде йодата и перйодата
Уран (IV) довольно избирательно осаждается из кислых растворов при добавлении йодата калия [94]. Улучшенный Е.С. Пржевальским, Е. Р. Николаевой и Н. И. Удальцовой [194] способ позволяет отделять уран от меди, молибдена и многих других элементов. Алюминий также отделяется полностью в том случае, если его количество не более чем в 50 раз превышает содержание урана. Получение чистых осадков йодата урана позволяет заканчивать определение непосредственным взвешиванием высушенного осадка. Однако в присутствии железа (II) полное отделение урана (IV) не достигается, что, по-видимому, связано с тем, что железо III, легко образующееся за счет окисления железа (II) кислородом воздуха, окисляет часть урана (IV) до урана (VI), неосаждающегося в условиях проведения осаждения.
Методика осаждения приводится в разделе «Весовые методы определения».
Применение йодата калия для осаждения урана (VI) [995] из ацетатных растворов обладает значительно меньшей избирательностью. Хлориды, нитраты и фосфаты, присутствующие в больших количествах, чем уран, затрудняют отделение. Мешающее влияние многих элементов в случае отделения урана (VI) осаждением в виде йодата уранила, по-видимому, можно было бы легко устранить маскированием их с помощью комплексона III.
Уран (IV) может быть отделен от многих элементов осаждением его из уксуснокислых растворов с помощью перйодата калия [397, 398]. Отделению урана при этом мешают почти те же самые элементы, что и отделению урана йодатом калия.
Методики осаждения урана (VI) при помощи йодата и перйодата калия приводятся в разделе «Весовые методы определения» (стр. 63 и 64).
Отделение осаждением с помощью арсенатов
Применение арсенатов, количественно осаждающих уран (VI) из слабокислых растворов в виде двойных арсенатов UO2NH4AsO4 или UO2KAsO4 [677], позволяет отделять его от редкоземельных элементов, алюминия, щелочноземельных элементов и от малых количеств железа. Zr, Th, Т i, Ag, Pb и ряд других элементов мешают от делению урана.
Для отделения урана (VI) осаждением мышьяковой кислотой раствор нейтрализуют аммиаком, затем добавляют столько уксусной кислоты, чтобы ее концентрация в растворе составляла 10—20%, и на каждые 100 мл раствора прибавляют
280
по 1 г хлорида аммония. Затем добавляют 35 мл 8% -ного раствора мышьяковой кислоты, нагревают до кипения и после охлаждения фильтруют. Осадок промывают разбавленным раствором хлорида аммония.
Загрязнения осадка железом можно избежать, если его предварительно восстанавливать при помощи сернистой кислоты. Для нейтрализации образующейся при этом минеральной кислоты вводят ацетат аммония. Отделение урана осаждением арсенатами не нашло практического применения вследствие большого числа мешающих элементов. Этот недостаток Н. И. Удальцова (1958 г.) сумела устранить применением комплексона III в качестве маскирующего средства.
Разработанный ею метод состоит в том, что к раствору, полученному после разложения руды (1—2 г), объем оторого должен составлять 25—30 мл, прибавляют 8—10 мг титана (в виде сульфата), необходимого в качестве соосадителя, 3—5 г комплексона III, нейтрализуют аммиаком до pH 5—7, прибавляют 5—6 мл 6% -ного раствора арсената натрия, нагревают до кипения и оставляют на 1—1,5 часа (или лучше на ночь). Выделившийся осадок отфильтровывают через фильтрующий стаканчик с пористым дном (№ 4), промывают 3—5%-ным раствором хлористого аммония, затем растворяют его в соляной кислоте (1 : 3), и содержание урана в полученном растворе определяют фотометрическим методом с помощью перекиси водорода.
Метод рекомендуется для определения урана в-бедных рудах. Для руд с содержанием урана более 0,2% осаждение проводят таким же образом, за исключением того, что в данном случае сульфат титана (IV) не добавляют. Содержание урана в растворе, полученном после растворения осадка арсената уранила, определяют иодометрически по количеству выделенного вместе с ураном арсената.
Отделение с помощью карбонатов бария и цинка
Уран может быть выделен из растворов, не содержащих солей аммония, суспензией карбоната бария [5901 в виде двойного карбоната Ba2UO2(CO3)3-6H2O или суспензией основного карбоната цинка [662]. Выделение урана при помощи нерастворимых карбонатов не обладает никакими преимуществами перед выделением щелочами и аммиаком.
Отделение в виде двойного ацетата натрия-уранила
Известная реакция для качественного открытия натрия при помощи ацетата уранила с образованием малорастворимого двойного ацетата натрия-уранила предлагалась также и для отделения урана [8]. Уран осаждают добавлением ацетата натрия до общей его концентрации в растворе, равной 2,5 моль/л; pH раствора устанавливают около 2,5. Метод обладает хорошей избирательностью, но, к сожалению, вследствие заметной растворимости осадка не пригоден для отделения малых количеств урана.
281
Отделение осаждением изатин-в-оксимом
Уран (VI) может быть отделен от щелочных и щелочноземельных металлов, магния, марганца и цинка осаждением его с помощью изатин-0-оксима из растворов, содержащих ацетатный буфер [613, 614, 617]. Для отделения урана от больших количеств кобальта и никеля добавляют виннокислый калий [616]. Мешающее влияние серебра, свинца и меди устраняют при помощи тиосульфата натрия [611]. В случае присутствия ртути полное отделение урана достигается введением хлоридов. Соответствующие методики описаны в разделе «Весовые методы определения».
Отделение осаждением а-нитрозс-^-нафтолом
Осаждение а-нитрозо-Р-нафтолом [656] - позволяет отделять уран (VI) от щелочных и щелочноземельных металлов, цинка и алюминия. А4етодика осаждения приведена в разделе «Весовые методы определения».
Отделение осаждением фениларсоновой кислотой
Уран (IV) может быть количественно отделен от урана (VI), а также от щелочных, щелочноземельных элементов, магния, большинства двухвалентных и небольших количеств некоторых трех-валетных элементов осаждением его фениларсоновой кислотой [319, 848, 1013]. Вместе с ураном (IV) полностью осаждаются Th, Zr Hf, Sn (IV), Nb и Та; Се (IV) и Ti (IV) осаждаются частично.
Кроме фениларсоновой кислоты в тех же условиях уран (IV) осаждают также и некоторые ее производные: 4-н.бутилфенилар-соновая, З-нитро-4-оксифениларсоновая и 4-диметиламиноазобен-зол-4'-арсоновая кислоты [1013]. Хотя применение их и позволяет несколько полнее осаждать уран (IV) по сравнению с фениларсоновой кислотой вследствие имеющего место эффекта утяжеления, но несколько меньшая их доступность является препятствием для их применения. С другой стороны, применение фениларсоновой кислоты и ее производных для отделения урана (IV) не обладает какими-либо преимуществами по сравнению с использованием ряда других более доступных реагентов.
Некоторые органические производные мышьяковой кислоты были также предложены для определения урана (VI), в частности, арсаниловая (4-аминофениларсоновая) [1026] и какодиловая кислоты [811]. Однако мешающее влияние других элементов в данном случае еще больше, чем при осаждении урана (IV).
Избирательность осаждения урана (VI) с помощью органических производных мышьяковой кислоты, по-видимому, может быть значительно повышена за счет применения соответствующих маскирующих комплексообразующих веществ, особенно комплексона 111-
Методики осаждения урана (IV) и урана (VI) с помощью арсо-нокислот описаны в разделе «Весовые методы определения».
282
Отделение осаждением другими реагентами
Для отделения урана предложено большое число различных соединений. Часть из них является производными уже описанных нами реагентов и по своему действию ничем не отличаются от. простейших своих аналогов. Так, например, хинальдиновая кислота [838] осаждает уран (VI) в тех же условиях и с тем же результатом, что и 8-оксихинолин; неокупферон (М-нитрозо-Ы-нафтилгидроксил-амин) позволяет отделять уран от тех же элементов, что и купферон [1009], нитрозо-Ы-фенил-З-метилпиразолон [615] осаждает ионы уранила так же, как и сс-нитрозо-Р-нафтол и т. д.
Такие реагенты, как салициловая кислота [804] и ряд ее производных [370],-ароматические и алифатические карбоновые кислоты [447, 512, 804], бензолсульфиновая кислота и ее аналоги [475, 684] были также рекомендованы для осаждения урана, однако они пока не нашли практического применения.
Некоторые из предлагавшихся в разное время реагентов кратко рассмотрены нами в разделе «Весовые методы определения». Большинство из них не обладает никакими преимуществами перед рядом более простых и доступных реагентов или являются даже менее эффективными. Так, например, щелочные цианиды [569], предложенные для осаждения.урана (VI), обладают меньшими возможностями для отделения урана (VI) от других элементов по сравнению даже с такими простыми реагентами, как. аммиак, едкие щелочи, перекись водорода или карбонаты щелочных металлов и аммония.
Отделение урана соосаждением
Когда отделяемое количество урана не обеспечивает осаждения его из раствора с образованием самостоятельной твердой фазы в связи с недостаточной его концентрацией в растворе, или если выделение имеет место, но вследствие некоторой, хотя и незначительной растворимости выделяемого соединения значительная часть его остается в растворе или удерживается в виде коллоидных частиц, то в таких случаях образующееся соединение урана выделяют из раствора с другим труднорастворимым соединением, являющимся носителем.
В качестве носителей чаще всего применяются такие соединения, Которые в дальнейшем не мешают определению или легко удаляются. Хорошими носителями для выделения следов урана являются гидроокиси многих металлов, обладающие рыхлым строением и большой поверхностью. Гидроокиси железа, алюминия, кальция, магния, олова, тория, циркония и титана были рекомендованы для со-осаждения с ними малых количеств урана [8, 19]. В качестве носителей для отделения следов урана могут применяться также перекись тория, карбонат бария, фторид кальция [8]. Соосаждение с органическими осадками также предлагалось для выделения сле-Довых количеств урана [126].
283
Удобство применения гидроокисей металлов состоит в томг что они легко выпадают из растворов при подщелачивании и увлекают с собой уран, который затем можно легко отделить обработкой раствором карбоната аммония в присутствии небольшого количества аммиака и затем определить подходящим методом.
Уран, соосажденный с гидроокисями ряда элементов или другими их соединениями, может быть определен рядом методов непосредственно без предварительного отделения от носителя (полярографическим и флуориметрическим методом, окислительно-восстановительным титрованием и др.). С этой точки зрения соосажде-ние с органическими осадками [126] представляется менее удобным, поскольку возможность определения урана без предварительного удаления носителя исключается. Кроме того, единственный метод отделения носителя от урана в данном случае состоит в высушивании и прокаливании полученного осадка, что представляется более сложной и кропотливой операцией, чем, например, отделение урана при помощи обработки осадка раствором карбоната аммония [79].
Основное значение соосаждения—выделение невесомых количеств вещества. Однако соосаждение получило значительное применение также и для улучшения полноты выделения осаждаемого элемента. При отделении урана от других элементов соосаждение применяется довольно часто. Так, например, в первой половине этого раздела изложен трилонофосфатный метод отделения урана, в котором для полноты осаждения урана вводится в раствор сернокислый титан, с фосфатом которого очень полно соосаждается фосфат уранила [157]. Л. С. Василевская и Т. В. Дейкина [157] при выделении урана из пород, содержащих значительные количества фосфата кальция, рекомендуют осаждать уран при помощи фосфатов совместно с алюминием и железом. П. А. Волков [184] для обеспечения большей полноты выделения урана (IV) в виде фосфата осаждает его совместно с фосфатом тория или циркония. Ю. А. Чернихов и В. Ф. Лукьянов [184] в разработанном ими методе также осаждают уран (IV) совместно с фосфатом тория. В. М. Звенигородская и Л. П. Рудина [157, 184] при выделении урана (IV) осаждением в виде фторида рекомендуют в тех случаях, когда отделяемое количество урана составляет менее 5 мг, осаждение проводить в присутствии кальция, фторид которого является хорошим соосадителем для фторида урана (IV). Для выделения малых количеств урана И. Е. Старик, Ф. Е. Старик и А. Н. Аполлонова [2431 осаждают уран (IV) в виде тетрафторида урана (IV) совместно с тетрафторидом тория.
Выделение урана соосаждением при помощи носителей тем полнее, чем меньше растворимость осадка носителя и соосаждаемого соединения урана и чем больше сходства в химическом поведении ионов урана и макрокомпонента. Эти закономерности характерны для изоморфного соосаждения. Распределение урана между раствором и осадком при изоморфном соосаждении находится в постоянном отношении к распределению макрокомпонента. При изоморфном 284
соосаждении уран гомогенно входит в твердую фазу осаждающегося носителя.
При адсорбционном соосаждении уран концентрируется только на поверхности образующегося осадка. Он может оказаться в этом случае также и внутри твердой фазы в результате укрупнения частиц осадка-носителя. Соосаждение урана по типу аномальной со-кристаллизации (образование неправильных смешанных кристаллов) и внутренней адсорбции (адсорбция на внутренних микротрещинах и микрокапиллярах) заметного применения не имеет.
Соосаждение применительно к радиоактивным элементам и в том числе к урану подробно рассматривается С. Е. Бреслером [19] в его монографии. Соосаждение с осадками, образуемыми органическими реагентами, менее изучено. Теоретические основы соосаждения с органическими соосадителями рассматриваются В. И. Кузнецовым в его работах [116, 121].
Как уже упоминалось, соосаждение при помощи носителей имеет большое значение для выделения малых количеств урана. Вследствие этого методы соосаждения широко применяются при определении урана в сточных и природных водах, а также для выделения его при определении в растительных и животных тканях и в материалах, содержащих уран в незначительных количествах.
Для выделения урана из сточных или природных вод его часто соосаждают на гидроокиси железа [8]. К анализируемой пробе прибавляют 2—10 мг железа (--0,005%) в виде хлорного железа и осаждают гидроокисью аммония. В полученном осадке уран определяют спектральным, рентгеноспектральным, флуоресцентным или другим подходящим методом. Соосаждение урана с гидроокисью железа позволяет выделять уран с достаточной полнотой при его содержании до 0,01 мг!л. Присутствие углекислоты до 0,01 % заметно не влияет на полноту выделения урана.
И. Е. Старик, Ф. Е. Старик и А. Н. Аполлонова [241, 2421 подробно исследовали соосаждение урана с гидроокисью железа в зависимости от ряда условий. Они установили, что при pH 5—8 уран соосаждается почти на 100% при его концентрации 10“2—10“ 5 лтгАг.
Когда введение железа по каким-либо причинам оказывается нежелательным, в этом случае уран может быть соосажден с гидроокисью алюминия, действие которой сходно с действием гидроокиси железа. Однако полнота выделения урана с гидроокисью алюминия, По-видимому, значительно меньше, чем при соосаждении с гидроокисью железа [8].
В некоторых случаях в качестве носителя рекомендуется применение гидрата окиси кальция [8, 255]. По данным Б. Тежака [255], Для соосаждения урана с гидроокисью кальция требуется не менее Чем 3000-кратный избыток кальция по отношению к урану при концентрации едкого натра в растворе около 0,5 N. Для соосаждения Урана с карбонатом бария избыток бария по отношению к урану Должен быть не менее 600-кратным.
285
В. И. Кузнецов и Т. Г. Акимова [126] для выделения урана из морской воды соосаждают его в виде роданида из солянокислой среды с осадком роданида метилового фиолетового.
К 1000 .ил морской воды прибавляют 9 мл концентрированной соляной кислоты и выдерживают в течение 2—3 час. Затем добавляют 25 .ил 40% -ного раствора роданида аммония, хорошо перемешивают и далее по каплям при перемешивании в течение 20 мин. вводят 50 .ил 2%-ного водного раствора метилового фиолетового. Через час выпавший осадок отфильтровывают через бумажный фильтр, смоченный промывным раствором, для приготовления которого к 400 мл дистиллированной воды добавляют 2—3 .ил концентрированной соляной кислоты, около 1 г роданида аммония и 2 .ил 2%-ного раствора метилового фиолетового. Осадок дважды промывают тем же промывным раствором, затем вместе с фильтром переносят в фарфоровый тигель, подсушивают, смачивают несколькими каплями серной кислоты (1 : 1) и медленно озоляют при 500—600°. В полученном остатке определяют уран люминесцентным метбдом. Описанным способом можно количественно выделять уран из морской воды при его содержании 1 • 10 ~4 мг)л.
Для определения урана в водах его рекомендуется также выделять при помощи активированного угля [108], обладающего способностью адсорбировать уран и некоторые другие катионы из водных растворов при pH 5—6.
200 мл анализируемой воды подкисляют серной кислотой до кислой реакции по метиловому оранжевому, затем нейтрализуют борно-боратной буферной смесью до pH 5, добавляют 50 мг активированного угля (ГОСТ 6217-52), перемешивают и через 5—10 мин. фильтруют через бумажный фильтр «белая лента». Уголь на фильтре промывают 2—3 раза дистиллированной водой, затем смывают его в стакан 4—7 мл воды, добавляют 10 мл 10%-ного раствора соды и через 5—10 мин. уголь отфильтровывают. В случае люминесцентного определения урана в фильтрат вводят фторид аммония, упаривают досуха и определяют содержание урана как обычно. Возможно также фотометрическое определение при помощи реагента арсеназо или другого подходящего реагента.
Рассолы, содержащие более 100 гл солей, предварительно разбавляют равным объемом дистиллированной воды. Нефтяные воды отстаивают и отделяют от нефти и мути. При неотстаивающейся мути к 1 л воды прибавляют 1 г соды и 50 мг железа в виде сульфата, хлорида или нитрата,хорошо встряхивают и после отстаивания осадка его отфильтровывают. Для удаления содержащегося в анализируемой воде сероводорода фильтрат подкисляют серной кислотой и продувают воздух до исчезновения запаха.. Затем добавляют 1—2 мл 3%-ного раствора перекиси водорода и выделяют уран при помощи активированного угля, как описано выше.
2. ЭКСТРАКЦИОННОЕ ОТДЕЛЕНИЕ УРАНА
А. А. Н ем од р у к
Экстракция в настоящее время является одним из лучших и наиболее часто применяемых методов отделения урана.
Широкое применение экстракции для отделения элементов и, в частности, для отделения урана обусловлено тем, что этот метод отличается от других малой продолжительностью, применим для выделения элементов в самом широком диапазоне их концентраций и, что особенно важно, пригоден для выделения анализируемого элемента в таких малых концентрациях, которые, как правило, значительно ниже растворимости обычных осадков. Кроме того, при-286
менение селективных экстрагентов * и различных комплексообразующих веществ дает возможность даже при одноступенчатой экстракции достигнуть такой степени очистки, которая в большинстве случаев недостижима с применением других методов и в особенности метода осаждения. К достоинствам экстракции следует отнести также отсутствие сорбционных процессов, которые характерны для методов осаждения и являются основной причиной снижения эффективности отделения этим методом. Большая специфичность экстракции позволяет осуществить даже самые трудные разделения, в том числе такие, как отделение циркония от гафния, ниобия от тантала и т. п.
Основным показателем, характеризующим процесс экстракции какого-либо вещества, является его коэффициент распределения, представляющий собой отношение концентрации этого вещества в экстракте к его концентрации в экстрагиру-
С
емом растворе: /(=—. Коэффициент распределения в большом интервале сь
концентраций остается постоянным, если только процесс экстракции не осложняется какими-либо побочными явлениями, и в большинстве случаев численно совпадает с отношением растворимости экстрагируемого вещества в обеих фазах.
Для разделения двух веществ основное значение имеет различие их коэффициентов распределения. О возможной эффективности разделения двух веществ можно судить по отношению их коэффициентов распределения, называемому коэф-
К
фициентом разделения а=-~. Чем больше это отношение отличается от 1, тем
•А 2
легче разделение. Если К—К2> то разделение невозможно.
Одновременно с высоким коэффициентом разделения необходимо, чтобы коэффициент распределения первого вещества (/(,) был не очень мал, так как иначе для его извлечения было бы необходимо многократно повторять экстракцию с применением больших объемов экстрагента.
Если первоначальное количество экстрагируемого вещества в растворе обозначить через т и объем этого раствора через о, а количество вещества, перешедшее в слой экстракта,— через т, и объем экстракта через то количество вещества, оставшееся в исходном растворе после экстракции, будет составлять т—тг Коэффициент распределения в этом случае будет:
__тх I
~~ I
т—тх v
(1)
Отсюда количество вещества, извлеченное из исходного раствора в экстракт, можно выразить с помощью уравнения:
= m (2)
Количество вещества, которое будет извлечено в результате п последовательных экстракций (тп), может быть выражено следующим уравнением:
I Kvt Л п тп = т{ ——1
(3)
При разделении смеси двух веществ, как правило, каждое из них экстрагируется независимо. Вследствие этого для их разделения необходимо, чтобы к тому моменту, когда одно из них будет извлечено практически полностью, второе, наоборот, должно практически полностью оставаться в исходном растворе.
* Вместо часто не совсем удачно применяемого термина «органический растворитель» мы предпочитаем пользоваться названием «экстрагент», более точно соответствующим существу процесса, так как в качестве экстрагентов могут применяться только те органические растворители, которые обладают ограниченной растворимостью в воде или в растворе, из которого производится экстрагирование.
Решая уравнение (3), можно рассчитать число последовательных экстракций, необходимых для извлечения определенного количества одного из компонентов смеси, а также количество второго, которое вместе с первым перейдет в экстракт. При экстракционном разделении, выполняемом в аналитических целях, часто достаточным бывает выделение 95—99,9% одного компонента, при этом количество второго компонента, экстрагирующееся вместе с первым, может составлять от нескольких сотых долей процента до одного и более процента.
Простейшими примерами экстракционного разделения являются такие, когда значительно меньшим, равным или не на много превосходящим объемом экстрагента за одну экстракцию достигается практически полное разделение двух веществ. Это возможно тогда, когда коэффициент распределения одного вещества довольно большой, а второго — очень маленький. Если коэффициент распределения одного вещества не слишком большой, а второго вещества очень мал, то полное разделение может быть достигнуто за одну экстракцию при применении значительно большего объема экстрагента, чем объем раствора, содержащего разделяемые компоненты. В этом случае вместо одной экстракции большим объемом следует применять последовательную экстракцию малыми порциями экстрагента. Этот прием позволяет достигнуть того же результата при значительно меньшем общем количестве экстрагента.
Когда коэффициенты распределения обоих веществ близки между собой, т. е. когда коэффициент разделения близок к единице, разделение таких веществ представляет значительно более трудную задачу, но все же при соблюдении определенных условий оно возможно. Полное разделение двух веществ в таких случаях достигается в результате нескольких серий последовательных экстракций. Такое разделение довольно часто применяется в промышленных масштабах, но в аналитических целях — очень редко.
Рассматриваемый закон распределения (1) и основное уравнение разделения двух компонентов (3) справедливы в условиях отсутствия протекания побочные процессов. Среди этих последних основным является изменение активной концентрации экстрагирующейся формы каждого из разделяемых компонентов, часто не совпадающей с общей концентрацией каждого из них. В случае экстракции электролитов, заметно распадающихся на ионы в водных растворахиэкстрагирующихся в виде недиссоциированных молекул, концентрация экстрагирующейся формы (не-продиссоциированной части вещества) будет меньше его общей концентрации. В случае электролитов закон распределения также соблюдается, если вместо общей концентрации вещества при расчете коэффициента распределения пользоваться концентрациями экстрагируемой формы. В случае почти полной диссоциации вещества, распадающегося с образованием двух ионов, концентрация этого вещества в экстракте линейно зависит от квадрата его общей концентрации в воде.
Особенно сильное влияние на величину коэффициента распределения и коэффициента разделения оказывает присутствие в исходном растворе других веществ, в частности, склонных к присоединению воды с образованием сольватированных молекул или ионов. Введение нейтральных солей в экстрагируемый раствор позволяет менять коэффициент распределения в очень широких пределах, вследствие чего этот прием нашел достаточно большое применение.
Однако при соблюдении постоянства всех условий закон распределения в каждом отдельном случае сохраняет свою силу. Опираясь на этот закон,' становится возможным производить расчеты оптимальных условий экстракционного раздете-ния и достаточно точно предусматривать его результаты.
Теоретические основы экстракционного отделения неорганических соединений подробно рассматриваются в ряде работ [106, 125, 172, 545, 700, 758, 7591.
Из всех элементов, по-видимому, экстракция урана исследована наиболее полно.
Способность к экстрагированию двух основных устойчивых валентных состояний урана (четырех- и шестивалентного) заметно
28 8
выделяется по сравнению с большинствохМ других элементов, что безусловно связано со склонностью ионов UO^ и U4* ксольватиро-ванию молекулами экстрагента, а также с их развитой способностью к образованию комплексов со многими органическими реагентами. Однако для отделения урана основное значение получила экстракция урана (VI).
Экстракционное отделение урана в виде молекулярных соединений с экстрагентом
Из всех известных экстракционных методов отделения урана, применяемых как в аналитических целях, так и в технологии наибольшее значение получила экстракция уранилнитрата при помощи ряда кислородсодержащих экстрагентов, таких как простые эфиры, кетоны, сложные эфиры и т. п. Преимущество экстракционного отделения урана в виде уранилнитрата состоит в том, что в данном случае вместе с ураном в виде нитратов экстрагируется очень небольшое число других элементов [125, 172], Количество элементов, экстрагирующихся вместе с ураном, непостоянно и зависит от применяемого экстрагента и условий экстракции, к которым в первую очередь следует отнести концентрацию азотной кислоты, характер применяемого высаливателя и его концентрацию, присутствие в экстрагируемом растворе анионов, способных образовывать с другими элементами экстрагирующиеся комплексы (например, хлоридов, роданидов и др.), применение маскирующих комплексообразующих веществ и т. п.
При экстрагировании из растворов нитрата уранила в органическую фазу уран переходит в виде молекулярных соединений типа UO2(NO3)2-mH2O-nS, где S — молекула экстрагента. Число молекул воды (т), входящих в комплекс, в зависимости от экстрагента и условий экстрагирования может колебаться от 0 до 2, а число молекул экстрагента в большинстве случаев находится в пределах от 2 до 4. Как видно из приведенной формулы, такой тип экстрагирования проходит по сольватационному механизму [545, 567, 622, 717, 7581. Одновременно с этим в присутствии достаточного избытка азотной кислоты образуется комплексная кислота H[UO2(NO3)3], которая легко экстрагируется по оксониевому механизму.
В случае применения в качестве экстрагентов спиртов экстрагирующееся оксониевое соединение соответствует формуле (ROH2)[UO2(NO3)31, для простых эфиров, кетонов и трибутилфосфата экстрагирующимися оксониевыми комплексами будут (R,R2OH)[UO2(NO3)3], (R^COHHUO^NO^] и [(С4Н9О)3РОН]. • [UO2(NO3)3],
Экстрагирование в виде молекулярных соединений с экстрагентом, кроме растворов нитратов, возможно также и из растворов хлоридов, иодидов, роданидов, перхлоратов и сульфатов.
19
Аналитическая химия урана
289
Экстрагирование ди этиловым эфиром
Применение диэтилового эфира для экстракционного отделения урана из растворов уранилнитрата известно давно [380, 463, 544, 601, 693, 8091.
Коэффициент распределения нитрата уранила между водой и диэтиловым эфиром по сравнению с другими экстрагентами достаточно мал и сильно зависит от абсолютной концентрации нитрата уранила (самовысаливающий эффект) (табл. 40).
Таблица 40
Распределение нитрата уранила между диэтиловым эфиром и водой при 25° [546]
Молярная концентрация нитрата уранила Коэффициент Молярная концентрация нитрата уранила Коэффициент
в водной фазе в диэтиловом эфире распределения в водной фазе В диэтиловом эфире распределения
0,348 0,00258 0,00742 0,829 0,066 0,0796
0,502 0,0097 0,0193 1,24 0,338 0,272
0,626 0,0235 0,0375 1,42 0,635 0,447
Для увеличения полноты выделения урана диэтиловым эфиром и вообще при экстрагировании любым экстрагентом в экстрагируемый раствор часто вводят различные соли в качестве высали-вателей. При выборе высаливателя руководствуются соображениями его доступности и эффективности. Само собою разумеется, высали-ватель не должен при этом сам переходить в заметных количествах в органическую фазу. При извлечении урана в виде уранилнитрата в качестве высаливателей применяют нитраты или азотную кислоту.
Повышение концентрации нитрат-иона в водном растворе сдвигает равновесие, существующее между ионом уранила и нитратными комплексами, в сторону образования последних и тем самым способствует более полному отделению урана. Одновременно высали-ватель играет и другую более общую роль — обладая большим сродством к воде, гидратируется молекулами воды, вследствие чего экстрагируемое вещество оказывается фактически растворенным в меньшем количестве воды, что равноценно соответствующему повышению его концентрации в растворе. В результате этого его коэффициент распределения между водной и органической фазами увеличивается. Кроме того, высаливатель, как правило, уменьшает содержание воды в органическом слое, а также значительно уменьшает растворимость экстрагента в водной фазе, что часто имеет существенное значение при экстракционном отделении.
Эффективность высаливающего действия зависит от природы и концентрации высаливателя. В. М. Вдовенко и Т. В. Ковалева
290
[32, 33] подробно исследовали действие нитратов различных элементов, взятых в качестве высаливателей при экстрагировании уранилнитрата диэтиловым эфиром. Они установили, что при одной и той же молярной концентрации высаливателя высаливающее действие возрастает по мере увеличения заряда и уменьшения радиуса катиона высаливателя. В. И. Кузнецов [125] высаливающее действие связывает с понижением упругости водяных паров над раствором самого высаливателя.
Применение эффективных высаливателей в достаточно высоких концентрациях позволяет значительно увеличивать коэффициенты распределения урана и обеспечивает возможность очень полного его выделения, однако при этом происходит одновременно сильное понижение специфичности экстрагирования.
При экстрагировании урана (VI) из растворов нитратов при помощи диэтилового эфира в органическую фазу уран переходит в виде молекулярных соединений типа UO2(NO3)2-mH2O-nS. Как показали В. М. Вдовенко, М. П. Ковальская и Т. В. Ковалева [38], в зависимости от условий экстрагирования ион уранила может сольватироваться двумя и четырьмя молекулами диэтилового эфира. В результате этого образуются два молекулярных соединения UO2(NO3)2-2H2O-2(C2H5)2O и UO2(NO3)2.2H2O.4(C2H5)2O. Последнее из них отличается несколько меньшей устойчивостью и выделяется из эфирного раствора при температуре около 0° и ниже. При растворении нитрата уранила UO2(NO3)2-2H2O в диэтиловом эфире выделяется 10,85 ккал/моль. При применении шестиводного нитрата уранила этим авторам удалось выделить из органической фазы при температуре 0—25° соединение UO2(NO3)2-3H2O« •(С2Н5)2О, а в случае растворения в диэтиловом эфире безводного нитрата уранила из эфирного раствора выделено соединение UO2(NO3)2-2(C2H5)2O. Таким образом, экстракция урана из водных растворов нитрата уранила при помощи диэтилового эфира состоит в сольватировании ионов уранила молекулами диэтилового эфира с образованием соответствующих молекулярных соединений, очень хорошо растворяющихся в диэтиловом эфире.
При достаточно высокой концентрации азотной кислоты 5 V) уран экстрагируется диэтиловым эфиром также и в виде оксониевого соединения (С2Н5)2О-Н[UO2(NO3)3] [28, 415, 416].
Экстрагирование урана из нитратных растворов диэтиловым эфиром не отличается высокой специфичностью. Вследствие этого определение урана в полученных экстрактах редко заканчивается взвешиванием сухого или прокаленного остатка. В этих случаях оказались более подходящими другие методы, в том числе титриметри-ческий и фотометрический. Одновременно с ураном в органическую фазу могут переходить Се (IV), Th, Pu, Zr, Hf, небольшие количества Fe (особенно в присутствии хлоридов), V, As, Мо.
В случае необходимости отделения церия (IV) его перед экстрагированием предварительно восстанавливают нитритом натрия. 19* 291
Если концентрация железа в исходном растворе велика, то оно также может в заметных количествах переходить в органический слой. Применение комплексона III в подходящих условиях (низкая кислотность) вследствие образования неэкстрагирующихся комплексов ряда мешающих элементов (Zr, Th, Fe, V, Ри) значительно повышает специфичность экстракционного отделения урана.
Ряд анионов, связывающих уран в неэкстрагирующиеся комплексы, препятствует его извлечению в органическую фазу. К их числу принадлежат фториды, фосфаты, сульфаты, ванадаты и молибдаты, а также некоторые органические комплексообразующие вещества. Мешающее влияние этих ионов может быть устранено применением в качестве высаливателей нитрата алюминия или железа, связывающих указанные ионы в прочные комплексы [184, 886].
Ион сульфата при содержании до 2 мг!мл не оказывает заметного влияния на полноту извлечения урана. В случае больших количеств его мешающее влияние может быть устранено предварительным осаждением урана (VI) аммиаком или применением в качестве высали-вателя нитрата кальция. Органические комплексообразующие вещества мешают только при экстрагировании из растворов с очень малым содержанием свободной кислоты. Повышением кислотности их влияние может быть устранено полностью. При одновременном присутствии фторидов, фосфатов и сульфатов целесообразно применять высаливатель, состоящий из смеси нитратов алюминия и кальция или железа и кальция. Хлориды уменьшают специфичность экстракционного отделения урана вследствие того, что в их присутствии некоторые элементы, как например железо (III), также экстрагируются диэтиловым эфиром в виде хлоридных комплексов.
Хотя применение диэтилового эфира для экстракции урана значительно уменьшилось вследствие применения других более эффективных экстрагентов, тем не менее им еще довольно часто пользуются [415, 416, 566, 785, 886, 1022].
Для отделения урана при помощи экстракции диэтиловым эфиром рекомендована следующая методика [1022]. К анализируемому раствору, кислотность которого по азотной кислоте предварительно устанавливают равной 1 N, на каждые 5 мл прибавляют по 10 г нитрата аммония. Вследствие этого объем анализируемого раствора увеличивается ровно в 2 раза. Затем прибавляют примерно такой же объем диэтилового эфира, встряхивают в течение 1 мин. и оставляют на 1 мин. для разделения слоев. Водный слой выпускают в стакан, а оставшийся эфирный слой снова размешивают и оставляют в течение 1 мин. Следы водной фазы отделяют и присоединяют к основной ее части, а эфирный слой помещают в чашку, содержащую около 30 мл воды. Водную фазу снова переносят в делительную воронку, и стаканчик споласкивают концентрированной азотной кислотой. На споласкивание стаканчика берут по 0,6 мл концентрированной азотной кислоты на каждые 10 мл водной фазы для возмещения уменьшения концентрации азотной кислоты вследствие частичного экстрагирования ее эфиром. Затем повторяют экстракцию, как уже описано, еще 4 раза. Объединенные эфирные экстракты упаривают до полного испарения эфира, а оставшийся водный слой нейтрализуют раствором едкого натра. В случае выпадения осадка его растворяют добавлением азот
292
ной кислоты. Затем добавляют карбонат натрия, чтобы его содержание в растворе составляло 1—5%. Раствор кипятят в >течение приблизительно 15 мин, для удаления растворенного диэтилового эфира и для коагуляции гидроокиси железа. Выпавший незначительный осадок отфильтровывают, промывают 3 раза небольшим количеством воды и затем в полученном фильтрате или его аликвотной части определяют содержание урана любым подходящим способом.
При содержании в исходном водном растворе 5 мг урана и 20-кратных количеств Fe, Мп, Cr, Mg, Cd, Си, Al, Со и Ni ошибка определения-урана фотометрическим методом при помощи дибензо-илметана не превышает 3% (отн.) [10221.
Для удаления из эфирного экстракта небольших количеств железа и алюминия П. Н. Палей и Э. И. Добкина [184] в необходимых
Рис. 54. Схема экстратора В. М. Михайлова.
Д— приемная колба с эфиром: В— экстракционная часть; С— холодильник: D— трубка с промывным раствором с двойными стенками (типа сосуда Дьюара).
Рис. 55. Прибор для экстракции нитрата уранила диэтиловым эфиром.
А — обратный холодильник; В— приемная колба с эфиром; С— пористая пластинка; D—экстрактор; Е— отверстие для введения анализируемого раствора; F— трубка для эфира, выходящего из холодильника.
293
случаях рекомендуют проводить двойную экстракцию эфиром (с применением нитрата аммония в качестве высаливателя во второй экстракции). В. М. Михайлов [184] показал, что двойная экстракция может быть отчасти заменена реэкстракцией железа и других элементов в сконструированном им приборе, в котором эфир проходит через анализируемый раствор, содержащий нитрат железа или алюминия в качестве высаливателя, затем через промывной раствор нитрата аммония (рис. 54).
Вследствие невысоких коэффициентов распределения при экстрагировании диэтиловым эфиром для достижения полного извлечения урана необходимо по крайней мере четырехкратное экстрагирование равным объемом экстрагента, что усложняет разделение и делает его достаточно трудоемким.
Для облегчения экстракционного отделения урана диэтиловым эфиром предложены специальные приборы непрерывного действия, позволяющие достаточно полно извлекать уран в течение сравнительно небольшого промежутка времени. На рис. 55 представлена схема одного из таких экстракторов.
Методика работы с применением такого экстрактора состоит в следующем [8]. 1—5 г анализируемого вещества * помещают в стакан емкостью 250 мл, добавляют 50 мл концентрированной азотной кислоты, нагревают до кипения и кипятят в течение 30 мин. По охлаждении раствор фильтруют, осадок на фильтре хорошо промывают азотной кислотой (1 : 200), фильтрат и промывную воду упаривают до 10—15 мл. В присутствии иона фосфата добавляют нитрат железа, В случае анализа раствора неизвестного состава его дважды упаривают с концентрированной азотной кислотой до 10—15 мл. Полученный раствор через отверстие Е вводят в нижнюю часть экстрактора D и стаканчик споласкивают 20—30 мл раствора нитрата аммония (700 г нитрата аммония растворяют в 1 л 10%-ной азотной кислоты). В колбу В вводят 60 мл воды и 75 мл диэтилового эфира, присоединяют к прибору и нагревают на водяной бане. Испаряющийся при этом диэтиловый эфир конденсируется в обратном холодильнике А и далее поступает в трубку F, по которой опускается вниз и через крупнопористый фильтр входит в нижнюю часть экстрактора D в виде мелких капелек, которые проходят сквозь экстрагируемый раствор и собираются над ним в виде эфирного слоя. После заполнения нижней части экстрактора эфирный слой по мере поступления диэтилового эфира из трубки F непрерывно сливается в колбу В, унося с собой некоторое количество нитрата уранила. Вследствие испарения диэтилового эфира в колбе В содержание в нем нитрата уранила непрерывно повышается, что вызывает также повышение его концентрации в водном слое, находящемся в этой же колбе. Через 30 мин. в нижнюю часть экстрактора D через отверстие Е вводят 10 мл смеси (1 : 1) насыщенного раствора нитрата аммония и концентрированной азотной кислоты для возмещения снижения их концентрации в экстрагируемом растворе вследствие перехода вместе с диэтиловым эфиром в колбу В в процессе экстракции. После этого экстракцию продолжают еще в течение 30 мин. По окончании экстрагирования колбу В отделяют от прибора, эфир удаляют упариванием на водяной бане, водный раствор переносят в стакан емкостью 250 мл, прибавляют 8 мл серной кислоты (1 : 1) и упаривают для удаления азотной кислоты. В полученном растворе после соответствующего разбавления определяют содержание урана любым подходящим методом.
Преимущество непрерывной экстракции диэтиловым эфиром состоит в том, что вследствие постоянного контакта экстрагируемого
* Если анализируемый образец содержит органические вещества, то для их разрушения навеску предварительно прокаливают.
294
раствора со свежими порциями экстрагента отделение урана в данном случае более полное, чем при пятикратной последовательной экстракции равными объемами диэтилового эфира.
Рекомендуются также и другие приборы для непрерывной экстракции диэтиловым эфиром [8, 660, 759].
Применение диэтилового эфира для выделения урана из растворов хлоридов, бромидов, иодидов, роданидов, хлоратов и сульфатов в отличие от растворов нитратов практического значения не получило главным образом вследствие меньшей избирательности.
Экстрагирование трибутилфосфатом
Трибутилфосфат (три-н • бутиловый эфир ортофосфорной кислоты) в настоящее время имеет очень широкое применение для экстракционного отделения элементов. Однако наиболее широкое применение он получил для экстракционного отделения урана.
Такое широкое применение трибутилфосфата обусловлено рядом ценных его качеств, среди которых, в первую очередь, следует назвать весьма высокие коэффициенты распределения, позволяющие в подавляющем большинстве случаев достигнуть практически полного извлечения за одну экстракцию. Так, например, при экстрагировании урана из 2 V раствора азотной кислоты, содержащего 5 мг U в 1 мл, коэффициент распределения составляет 33, а в присутствии нитрата натрия (66 г в 100 мл) он повышается до 1800 [272].
Вторым ценным качеством трибутилфосфата является то, что он не летуч в очень широком интервале температур (температура кипения+289°), и вследствие этого работа с ним совершенно безопасна.
Кроме того, трибутилфосфат обладает чрезвычайно малой растворимостью в воде (табл. 41), а также мало чувствителен к радиоактивным излучениям [52]. В химическом отношении трибутилфосфат также очень стабилен. Его гидролиз водой практически исключается. Он также устойчив по отношению к концентрированной азотной кислоте и только при ее концентрации 16 N и более имеет место заметное разложение трибутилфосфата с образованием ди- и монобутилфосфорной кислот. Трибутилфосфат устойчив к действию многих окислителей, в том числе таких сильных, как церий (IV) и др.
Т а б л и ц а 41
Растворимость трибутилфосфата в воде [313]
Температура, °C 16 17 19 22
Растворимость, г'л . . 0,420 0,410 0,397 0,380
295
Из растворов нитратов трибутилфосфат экстрагирует уран в виде молекулярного соединения U0o(N03K-2 (С4Н9О)3 РО [149, 584, 717].
Возможно также экстрагирование урана в виде H[UO2(NO3)31 по оксониевому механизму [28].
Экстракция урана (VI) из нитратных растворов при помощи трибутилфосфата рассматривается в работах [21, 93, 149, 209, 210, 314, 343, 362, 402, 465, 472, 567, 598, 638, 1003].
Кроме урана, трибутилфосфатом из нитратных растворов могут экстрагироваться также Се (IV), Zr, Hf, Th, Pu (IV), Ru (VI), Y, La и редкоземельные элементы, а также Np (IV) и Np (VI), Am (VI), Au (III), Fe (III), Sc (III), Pa (IV) и Те (III) [21, 149, 172, 362, 759, 801, 950, 1003].
Однако подбором соответствующих условий уран может быть отделен практически от всех элементов.
Так как вязкость трибутилфосфата очень велика (3,41 сантипуаза при 25°), вследствие чего разделение фаз значительно затрудняется, применяют, как правило, не сам трибутилфосфат, а его растворы в различных инертных растворителях (керосин, октан, ксилол, синтин, толуол, хлороформ, четыреххлористый углерод, дихлорэтан и др.). При этом с понижением вязкости снижается и коэффициент распределения, но одновременно увеличивается селективность.
Для повышения селективности экстракционного отделения урана, кроме выбора соответствующих условий (концентрация трибутилфосфата в инертном растворителе, концентрация высаливателя и азотной кислоты), большое значение имеет применение различных маскирующих комплексообразующих веществ, в особенности эти-лендиаминтетрауксусной кислоты [343, 427].
Предварительная обработка экстрагируемого раствора подходящими восстановителями также позволяет ряд элементов (Се (IV), Pu (IV)) переводить в более низкие валентные состояния, не экстрагирующиеся трибутилфосфатом.
Железо и висмут могут быть полностью отделены от урана промывкой органической фазы разбавленной азотной кислотой, содержащей высаливатель. Однако Th, Zr, Се (IV), Pu (IV) и Hf при этом остаются в экстракте. Для их удаления рекомендуется органическую фазу промывать несколько раз 5%-ным раствором йодата калия в 30 %-ной азотной кислоте [208], а затем 30 %-ной азотной кислотой до полного удаления йодата калия. Однако этот метод не применим для отделения урана от больших количеств тория.
Применение комплексона III при экстракционном отделении урана трибутилфосфатом оказалось особенно успешным [343, 427, 540]. Проведение экстракции в присутствии комплексона III позволяет отделять уран практически от всех элементов.
Для выделения урана из растворов нитратов экстрагированием его трибутилфосфатом может быть рекомендована следующая методика. К анализируемому 296
раствору прибавляют комплексон III до общего его содержания в растворе (в зависимости от содержания мешающих элементов) от 0,5 до 2%. Затем прибавляют приблизительно равный объем насыщенного раствора нитрата кальция, устанавливают pH раствора около 2,5 и экстрагируют небольшим объемом (не превышающим объема исходного раствора) 5?о-ного раствора Трибутилфосфата в керосине или другом инертном растворителе. Органическую фазу промывают насыщенным раствором нитрата аммония с pH 2,5. Уран из органической фазы реэкстратируют при помощи раствора карбоната аммония
Этот метод дает хорошие результаты при определении урана в рудах и породах. Он рекомендуется также для определения урана в нитрате тория [343].
Вследствие большой экстракционной способности трибутилфосфата полное извлечение урана в органическую фазу, как уже указывалось, достигается за одну экстракцию; но с другой стороны, это свойство трибутилфосфата оказывает значительное препятствие выделению урана из органической фазы. Трехкратное реэкстрагирование равным объемом воды не всегда обеспечивает полное выделение урана. Для полного извлечения урана из экстрактов, содержащих большие количества урана и азотной кислоты (вследствие эффекта самовысаливания), требуется до 9 последовательных реэкстракций равным объемом 25%-ного раствора ацетата аммония. Для реэкстрагирования урана из органической фазы, кроме ацетата аммония, рекомендуются также растворы сульфатов натрия или аммония [879]. Несколько более эффективными оказались растворы карбонатов. Однако в случае их применения имеет место обильное выделение СО2, что представляет определенное неудобство в работе.
Растворы оксалатов, не уступающие по эффективности извлечения урана карбонатным растворам, лишены этого недостатка. А. В. Давыдов, Т. С. Добролюбская и А. А. Немодрук [65] показали, что наиболее эффективными для реэкстракции урана из органической фазы являются растворы фосфатов, в том числе и фосфорной кислоты (5%-ные), позволяющие достигнуть полного извлечения урана за одну реэкстракцию равным или даже намного меньшим объемом раствора. Вследствие этого применение растворов фосфатов для реэкстракции урана оказалось особенно целесообразным в тех случаях, когда их присутствие в растворе не мешает последующему определению урана.
Для избежания трудностей, связанных с реэкстракцией урана в водную фазу, был предложен ряд методов определения урана непосредственно в получаемых экстрактах [540, 790, 505]. Так, например, Пейдж, Эллиотт и Рейн [790] экстрагируют нитрат уранила растворами трибутилфосфата в изооктане и определяют уран По собственному светопоглощению полученного экстракта при 250 ммк (см. стр. 114), обусловленному присутствием молекулярного соединения трибутилфосфата с нитратом уранила. Гилл, Ролф и Армстронг [540] экстрагируют нитрат уранила растворами трибутилфосфата в четыреххлористом углероде и определяют уран фото
297
метрическим методом непосредственно в экстракте после добавления к нему 1- (2-пиридилазо)-2-нафтола и пиридина. Указанные авторы показали, что предварительное добавление к анализируемому раствору комплексона III позволяет отделять уран от циркония, висмута, железа и многих других элементов. Однако в случае очень больших количеств тория рекомендуется его предварительно отделять осаждением в виде оксалата.
Вместо реэкстракции урана растворами карбонатов, ацетата аммония и др. было предложено использовать для этой цели растворы реагентов, пригодных для фотометрического определения урана 1467, 522).
В качестве примера может быть рекомендована следующая методика. К 5— 60 мл анализируемого раствора добавляют концентрированную азотную кислоту до общего ее содержания около 4,4 W и экстрагируют уран 10—20 мл 30%-ного раствора трибутил фосфата в четыреххлористом углероде. К аликвотной части экстракта (0,1—3 мл) в мерной колбе емкостью 25—50 мл прибавляют 3 мл 10%-ного раствора KJFe(CN)e] и pH водной фазы устанавливают в пределах 3—6. Затем водой разбавляют до метки и сверх этого прибавляют еще такой объем воды, который имела аликвотная часть экстракта. После энергичного встряхивания и разделения фаз содержание урана определяют фотометрированием водного слоя.
С таким же успехом реэкстракцию можно проводить растворами перекиси водорода, аскорбиновой кислоты, тиогликолята аммония, арсеназо I. Особенно удобным, как показали П. Н. Палей, А. В. Давыдов и А. А. Немодрук [185], оказались растворы недавно предложенного С. Б. Саввиным [216] нового реагента — арсеназо III (2,7-бис - [2 - арсонобензолазо]-1,8-диоксинафталин-3,6-дисульфокис-лота), отличающегося чрезвычайно высокой прочностью образуемого им с ураном окрашенного комплекса и обладающего одновременно высокой чувствительностью и избирательностью фотометрического определения [185].
Трибутилфосфат оказался пригодным также для экстракционного извлечения урана из растворов перхлоратов [289, 2901, хлоридов [802] и роданидов [426].
Перхлорат уранила экстрагируется в виде комплекса состава ПО2 (С1О4)2-2Н2О-2 (С4Н9О)3РО [289]; коэффициент распределения при экстракции перхлората уранила меньше, чем в соответствующих условиях для нитрата [290]. Однако полное отделение урана может быть достигнуто за одну экстракцию. Мешающее влияние других элементов при экстракционном выделении урана из растворов перхлоратов, по-видимому, примерно такое или даже меньше, чем при экстракции из растворов нитратов.
Экстракционное отделение урана в виде хлорида уранила [802] обладает меньшей избирательностью, чем экстракция нитрата уранила. Однако оно пригодно для некоторых разделений, в том числе для отделения урана от тория и особенно от протактиния 1802].
Трибутилфосфат экстрагирует уран из растворов роданида также в виде молекулярного комплекса. Экстрагирование урана
298
в виде роданидного комплекса удобно тем, что определение урана в этом случае заканчивается фотометрированием полученного экстракта.
Методика экстрагирования состоит в следующем [426]: к анализируемому раствору добавляют комплексон III в количестве, достаточном для связывания мешающих элементов, добавляют роданид аммония и при pH 3,5—3,9 экстрагируют 32,5% -ным раствором трибутил фосфата в четыреххлористом углероде.
Вместе с ураном в этих условиях экстрагируются частично Мо, Pt, Hg ,W,Sn (II), Со, Се (III) и некоторые другие элементы. Для удержания золота в водной фазе в раствор добавляют цианиды. Хром (VI) и марганец (VII) восстанавливают с помощью SO2.
•Экстрагирование [метилизобутилкетоном
Для экстрагирования урана из растворов нитратов наряду с диэтиловым эфиром и трибутилфосфатом широкое применение получил также метилизобутилкетон (гексон). Физические и химические свойства метилизобутилкетона как экстрагента подробно описаны Бредли [385]. Применение метилизобутилкетона для экстракционного отделения урана из растворов нитратов исследовано в работах [21, 392, 401, 485, 721, 722, 784 , 868]. Наибольшее применение метилизобутилкетон нашел для выделения урана из смеси продуктов деления [21, 721].
Из растворов нитратов метилизобутилкетон экстрагирует уран в виде молекулярного соединения с нитратом уранила. При достаточном содержании азотной кислоты уран в органическую фазу извлекается в виде оксониевого соединения [UO2 (NO3)3]H-•OC(GH3)(G4H9). Избирательность экстракционного отделения приблизительно такая же, как и с применением трибутилфосфата. Указывается, что в случае применения в качестве высаливателя нитрата аммония имеет место более полное отделение урана от продуктов деления, чем при применении других высаливателей. Большая селективность отделения имеет место в отсутствие свободной азотной кислоты. Увеличение кислотности экстрагируемого раствора от дефицита в 0,1 М по HNO3 (за счет частичной нейтрализации раствора нитрата алюминия, применяемого в качестве высаливателя) до ее концентрации в растворе, равной 0,1 2И, повышает коэффициент распределения осколков в 42 раза [21]. Вследствие высокой экстракционной способности метилизобутилкетона полное извлечение урана достигается в соответствующих условиях при однократной экстракции равным объемом метилизобутилкетона.
Для выделения урана из смесей сложного состава экстракцией метилизобутилкетоном может быть рекомендована следующая методика [721]. К 0,5 мл водного раствора, содержащего до 2 лег урана и с кислотностью не более 8 М по азотной кислоте, прибавляют 4 мл раствора высаливателя.
Для приготовления раствора высаливателя 105 г нитрата алюминия помещают в мерную колбу емкостью 100 мл, добавляют небольшое количество воды и нагревают до растворения. Затем прибавляют 6,75 мл 14,8 М раствора аммиака
299
и размешивают до растворения образовавшегося осадка гидроокиси алюминия. Охлаждают до 45—50°, прибавляют 1 мл 10%-ного раствора гидроокиси тетрапро-пиламмония и после растворения разбавляют водой до метки.
Вслед за прибавлением раствора высаливателя добавляют 2 мл метилизобу-тилкетона и встряхивают в течение 3 мин. Для ускорения разделения фаз центрифугируют.
Извлечение урана составляет более 99,8%. Одновременно с ураном в органическую фазу могут переходить незначительные количества Th, Pu, Np, Тс, Hg и В.
Экстрагирование этилацетатом
Экстракционное отделение урана с помощью этилацетата сущест» венно не отличается от экстракции метилизобутилкетоном.
Для отделения урана рекомендуется следующая методика [564]. В делительную воронку помещают 5 мл анализируемого раствора, содержащего до 50 мг USOB, и добавляют 6,5 мл раствора нитрата алюминия.
Для приготовления этого раствора 450 г нитрата алюминия нагревают в стакане с 25—50 мл воды до растворения и затем упаривают до тех пор, пока температура раствора не поднимется до 130° (за счет его концентрирования). Так как при охлаждении этого раствора до 60° начинается кристаллизация нитрата алюминия, то перед применением его нагревают до полного растворения твердой фазы и добавляют горячим (температура около 110°).
После добавления раствора нитрата алюминия смесь охлаждают до комнатной температуры, добавляют 20 мл этилацетата и встряхивают в течение 1 мин. После разделения фаз водный слой отделяют таким образом, чтобы иногда образующиеся хлопья на поверхности раздела фаз оставались в делительной воронке. Затем к органической фазе, оставшейся в делительной воронке, добавляют 10 мл раствора нитрата алюминия, для получения которого 100 мл раствора нитрата алюминия, применяемого для высаливания, разбавляют 73 мл воды, и добавляют 4- мл концентрированной азотной кислоты. После этого снова встряхивают в течение 1 мин. и отделяют водную фазу. Для извлечения урана из органической фазы ее промывают 15 мл воды и затем еще четыре раза по 5 мл. Водные реэкстракты объединяют и определяют содержание урана любым подходящим методом.
Адамс и Мейк [299] вместо выделения урана из органической фазы определяют его непосредственно в самом экстракте фотометрическим методом после добавления дибензоилметана. Ницел и Де Сеза [178] для определения урана прибавляют к экстрактам водно-ацетоновый раствор роданида аммония, содержащий небольшое количество хлористого олова.
Куттитта и Даниэльс [441 ] применили экстракцию урана этилацетатом из растворов нитратов для его выделения из цирконов.
Для этого плав, полученный сплавлением 0,1 г анализируемого образца с 1 г смеси буры и соды (1 : 1), растворяют в 15 мл азотной кислоты (3:7). К полученному раствору в мерной колбе емкостью 50 мл добавляют азотную кислоту до достижения ее концентрации в конечном объеме (после разбавления водой до метки), равной 7% . 5 мл этого раствора помещают в делительную воронку, прибавляют 9,5 г нитрата алюминия и нагревают на водяной бане до растворения. Затем прибавляют 50 мг фосфата натрия, для связывания циркония, снова нагревают и после охлаждения добавляют 10 мл этилацетата. Встряхивают в течение 10 мин. и отделяют водную фазу. Органический слой фильтруют для удаления следов водной фазы через сухой фильтр. Для выделения урана из экстракта его сжигают в платиновой чашке, охлаждаемой снизу водой, и затем остаток прокаливают. Определение рекомендуется заканчивать флуорометрическим методом.
300
Экстрагирование другими экстрагентами
в виде молекуля рных соединений с уран ил нитратом
К числу экстрагентов, образующих с нитратом уранила молекулярные соединения, относятся простые эфиры, спирты, кетоны, альдегиды и сложные эфиры. Поэтому кроме рассмотренных экстрагентов (диэтиловый эфир, метилизобутилкетон, этилацетат, трибутилфосфат) многие другие соединения, принадлежащие к вышеперечисленным классам органических соединений, также являются пригодными для экстрагирования урана в виде молекулярных соединений с нитратом уранила или с другими его солями. Так, например, для экстракционного отделения урана из растворов нитратов были рекомендованы дибутиловый [30, 36, 92], диизопропиловый [21] и дигексиловый [639] эфиры, которые экстрагируют уранил-нитрат, подобно диэтиловому эфиру. Некоторое отличие заключается в меньшей растворимости их в воде. Кроме того, извлечение нитрата уранила экстрагентами одного и того же класса, образующими с ним сольватные комплексы, возрастает с ростом отношения числа содержащихся в молекуле экстрагента атомов кислорода к числу атомов углерода [545, 700, 968].
Спирты вследствие большой их растворимости в воде (метиловый и этиловый спирты неограниченно смешиваются с водой) и малой экстракционной способности, которая не на много превосходит таковую для простых эфиров, не получили практического применения, за исключением амилового спирта [184] и некоторых третичных спиртов [21 ].
Кетоны, обладающие высокой экстракционной способностью, довольно часто применяются для отделения урана. Исключением является ацетон, растворяющийся в воде в любых количествах. Кроме метилизобутилкетона, для экстракционного отделения урана были рекомендованы метилэтилкетон [184, 1004] и метилциклогексанон [891, 892]. Применение первого из них несколько ограничивается вследствие его достаточно высокой растворимости в воде. Метилциклогексанон не обладает этим недостатком. Имеются указания на то, что экстракционное отделение урана из растворов нитратов с его применением имеет некоторые преимущества по сравнению с экстракцией при помощи метилизобутилкетона и трибутилфосфата [892], заключающиеся в том, что метилциклогексанон несколько дешевле и не обладает токсическим действием.
Вейдель и Баранек [892] применили метилциклогексанон для разделения урана и тория.
100—500 мг смеси нитратов обоих элементов растворяют в 20 мл 8 М раствора нитрата натрия, добавляют 20 мл метилциклогексанона и энергично встряхивают. После разделения фаз и отделения водного слоя уран реэкстрагируют насыщенным раствором карбоната натрия.
Количество выделяемого урана составляет 99,9%. В присутствии свободной азотной кислоты селективность отделения снижается.
301
Мети л эти л кетон чрезвычайно легко экстрагирует уран из растворов роданидов [184]. Коэффициент распределения между органической фазой и водным раствором, содержащим 60% нитрата аммония и 3% роданида аммония, равен 2000. В этих условиях железо также экстрагируется вместе с ураном. Механизм экстракционного извлечения в данном случае такой же, как и при экстрагировании в отсутствие роданида из растворов, содержащих нитраты. Различие заключается в том, что роданид уранила в значительно меньшей степени распадается на ионы, чем нитрат уранила. В органическую фазу уран переходит в виде молекулярного соединения роданида уранила с растворителем.
С. И. Синякова и Ю. П. Новиков [184] применили метилэтил-кетон в качестве экстрагента для выделения урана из растворов сложного состава. Из водного раствора, насыщенного нитратом аммония, уран экстрагируют двумя порциями метилэтилкетона. В отделенном органическом слое содержание урана определяют фотометрированием после добавления к нему ацетонового раствора роданида аммония (подробное описание метода см. стр. 119).
Т. Н. Кукишева (1959 г.) в разработанном ею экспрессном методе определения урана применила экстракцию урана метил-этил кетон ом с применением комплексона III для повышения избирательности экстракционного отделения.
Раствор, содержащий около 0,2 мг урана, упаривают досуха, к остатку прибавляют 2 мл концентрированной азотной кислоты, 20 мл насыщенного раствора нитрата кальция, переносят в мерную колбу емкостью 50 мл и до метки разбавляют насыщенным раствором нитрата кальция. Если образуется осадок, то его отфильтровывают. К 10 мл фильтрата добавляют 5 мл 5%-ного раствора комплексона III (полученного смещением равных объемов 10%-ного раствора комплексона III с 8%-ным раствором азотной кислоты в насыщенном растворе нитрата кальция) и экстрагируют 5 мл метилэтилкетона.
Такое отделение урана от мешающих элементов, содержащихся в урановых рудах, позволяет достаточно точно определять его в аликвотной части полученного экстракта фотометрическим методом с помощью реагента арсеназо.
Хортон и Уайт [610] для экстракционного отделения урана применили три-(н.октил)-фосфиноксид в виде 0,1 М. раствора в циклогексане.
К 0,5—8 мл азотнокислого, хлорнокислого или сернокислого раствора, содержащего 15—2500 мкг U, добавляют 0,75—1,5 мл концентрированной азотной кислоты, 2—3,5 г нитрата натрия и разбавляют водой до 10 мл. В присутствии Fe (III), Сг (VI) и V (V) их предварительно восстанавливают в хлорнокислом[или сернокислом растворе добавлением 10% -ного раствора бисульфита натрия или’2 М раствора сульфата гидроксиламина; затем прибавляют азотную кислоту и нитрат натрия и разбавляют водой до 10 мл. Тi, Th, Hf, Zr и Fe маскируют добавлением 2,5 мл 1 М раствора KF, а избыток последнего устраняют добавлением нитрата алюминия. Затем добавляют азотную кислоту, нитрат натрия и разбавляют водой до 10 мл. В каждом случае уран экстрагируют 5—10 мл 0,1 М раствора три-(н. октил)-фосфиноксида в циклогексане. После разделения слоев к аликвотной части экстракта прибавляют дибензоилметан и пиридин, растворенные в этиловом спирте, и фотометрируют образовавшуюся окраску.
302
Кроме три-(н.октил)-фосфиноксида, для экстракционного отделения урана были применены многие другие фосфорорганические соединения [396, 642], содержащие фосфиноксидную группу. Моррисон и Фрейзер [759] указывают, что уран может быть количественно экстрагирован из растворов 0,1 М по нитрат-иону, при pH около 1,0 с помощью 0,1 М раствора в керосине или четыреххлористом углероде три-(н.октил)-, три-(н.децил)-, три-(н.додецил)- или три-(3,5,5-триметилгексил)-фосфиноксида. Фосфаты и сульфаты, если их концентрация не превышает 0,5 М, на полноту экстракционного отделения не оказывают влияния. Уран может быть количественно отделен за одну экстракцию 0,05 М раствором три-(н.децил)-фосфиноксида в керосине из растворов хлоридов с кислотностью от 0,05 до 4N.
Вода не реэкстрагирует уран из органической фазы, а растворы уксусной и щавелевой кислоты реэкстрагируют весьма слабо. Только растворы фосфатов (>5 М) и плавиковой кислоты, а также концентрированные растворы сульфатов при pH 2 и растворы карбонатов хорошо реэкстрагируют уран из органической фазы.
V (V) частично экстрагируется вместе с ураном. Предварительное восстановление V (V) до V (IV) позволяет удержать его в водном растворе. Рекомендуется также отделение V (V) путем обработки, экстракта растворами восстановителей, в частности 0,25 М раствором щавелевой кислоты.
Триалкилфосфиноксиды по сравнению с трибутилфосфатом в меньшей степени экстрагируют Fe (III) и торий. В зависимости от условий вместе с ураном могут экстрагироваться Sb (III), Bi, Cd, In, Hg (II), Pt (II), Zn, Mo (VI), Sn (IV), Ti (IV), Zr и некоторые другие элементы.
Дибутиловый эфир тетраэтиленгликоля (пентаэфир) применялся главным образом для отделения урана от продуктов деления. По своей способности экстрагировать уран из растворов нитратов пентаэфир занимает промежуточное положение между диэтиловым эфиром и трибутилфосфатом. Основным недостатком пентаэфира является его нестойкость по отношению к растворам, содержащим азотную кислоту [21, 272].
Дибутиловый эфир этиленгликоля (дибутилцеллосольв) характеризуется более низкими коэффициентами распределения продуктов деления, чем метилизобутил кетон. Повышение концентрации азотной кислоты сверх 0,1 М вызывает быстрое увеличение коэффициента распределения продуктов деления, в то время как соответствующий эффект для метилизобутилкетона начинается раньше, а именно при pH 4 и ниже [21].
Так как увеличение концентрации нитрата алюминия, применяемого в качестве высаливателя, выше 1 М не вызывает заметного изменения коэффициента распределения продуктов деления, применение дибутилцеллосольва представляет интерес для извлечения Урана из растворов с большим солевым составом [21].
303
Экстрагирование в виде циклических и внутрикомплексных солей
Экстрагирование в виде циклических и внутрикомплексных солей отличается от экстрагирования в виде молекулярных соединений типа сольватов тем, что в данном случае молекула органического реагента присоединяется к иону уранила при помощи двух атомных группировок с образованием циклической или внутриком-плексной соли. Так как образующиеся при этом соединения обладают значительно большей прочностью, чем соединения сольватного типа, то оказывается достаточно очень небольших, часто почти стехиометрических количеств этих реагентов, чтобы полностью связать весь уран в циклическую или внутрикомплексную соль. Для экстрагирования образующихся при этом соединений могут применяться очень многие экстрагенты.
Чаще всего образование циклических и внутрикомплексных солей совмещают в одну операцию с экстракцией экстрагентами, содержащими в растворенном виде соответствующие органические реагенты. В тех случаях, когда применяемые реагенты представляют собою жидкости, они могут одновременно являться и экстрагентами, как например ацетилацетон.
Селективность отделения урана с применением такого вида экстракции зависит от природы применяемого органического реагента, его концентрации, pH раствора и в небольшой степени от природы экстрагента. Роль высаливателей в данном виде экстракционного отделения невелика. В целом селективность экстракции в виде циклических и внутрикомплексных солей значительно ниже, чем селективность экстракционного отделения урана в виде молекулярных соединений типа сольватов. Однако она может быть в ряде случаев значительно повышена за счет применения маскирующих комплексообразующих веществ, удерживающих мешающие элементы в водной фазе.
К числу реагентов, имеющих практическое значение для экстракционного отделения урана, относятся ацетилацетон, теноилтрифтор-ацетон, купферон, 8-оксихинолин, диэтилдитиокарбамат натрия, а-нитрозо-р-нафтол, дибензоилметан и некоторые другие.
^Экстр’акционное отделение с применением ацетилацетона
Ацетилацетон, как и другие р-дикетоны, образует с ионами уранила внутрикомплексную соль, экстрагирующуюся многими не смешивающимися с водой органическими растворителями [297, 298, 867, 870]. Кроме урана, ацетилацетон образует внутрикомплексные соли более чем с шестьюдесятью другими элементами; большинство этих комплексов также легко извлекаются из водных растворов теми же экстрагентами. Вследствие этого для удержания других элемен
304
тов в растворе их маскируют при помощи комплексона III 16831. В случае применения ацетилацетона одновременно как реагента и экстрагента достигается более полное выделение урана. Применение ацетилацетона рекомендуется для отделения урана от висмута [683]. Для удержания висмута в водном растворе прибавляют 30-кратное количество комплексона III по отношению к содержанию висмута и экстрагируют ацетилацетоном при pH 4—6.
Экстракционное отделение с применением дибензоил метана
Дибензоилметан, принадлежащий так же как и ацетилацетон к классу р-дикетонов, образует с ионами уранила внутрикомплекс-ные соли, экстрагирующиеся многими не смешивающимися с водой органическими растворителями. Условия экстракционного отделения урана с применением дибензоилметана вследствие близости его химических свойств к свойствам ацетилацетона оказываются приблизительно такими же, как и при отделении с применением ацетилацетона.
Так как дибензоилметан является не растворимым в воде твердым веществом, то его всегда применяют в виде растворов в соответствующих “ экстрагентах (хлороформ, четыреххлористый углерод, бензол, толуол, ксилол и др.). Преимущество дибензоилметана перед ацетилацетоном состоит в том, что получаемые экстракты обладают исключительно высокой интенсивностью окраски, что позволяет заканчивать анализ непосредственным фотометрированием самого экстракта. В случае больших количеств урана необходимо предварительное разбавление экстракта. Для устранения мешающего влияния других элементов в анализируемый раствор перед экстракцией добавляют комплексон III [821]. Экстракционное отделение урана в виде дибензолиметаната уранила применяется довольно часто [554, 821, 946, 1022].
Экстракционное отделение при помо щ и тено и лтрифторацетона
Теноилтрифторацетон взаимодействует с ионами уранила при более низких значениях pH, чем ацетилацетон и дибензоилметан. Это снижение pH взаимодействия связано с сильным негативирую-Щим влиянием трифторметильной группы, содержащейся в молекуле теноилтрифторацетона, и вызывающей значительное повышение 'Кислотной диссоциации енольной формы реагента.
Из всех Р-дикетонов теноилтрифторацетон получил, по-види-Мому, наибольшее применение, в особенности для разделения актинидов. Оптимальными условиями экстракционного отделения урана Являются pH раствора, равное 3 и выше,и концентрация теноилтрифторацетона в экстрагенте (бензол, толуол) в пределах от 0,1 до ^0 Аналитическая химия урана 305
0.5 А4. При более низких концентрациях теноилтрифторацетона продолжительность экстракции значительно возрастает.
Вместе с ураном в органическую фазу переходят почти все те элементы, которые извлекаются вместе с ним при экстрагировании с применением ацетилацетона. Применение комплексона III позволяет значительно повысить селективность экстракционного отделения урана (VI). Уран (IV) имеет совершенно другую экстракционную характеристику и способен экстрагироваться из более кислых растворов. Вследствие малой эффективности маскирующих комплексообразующих веществ при отделении урана (1\ ) избирательность его экстракции оказалась значительно ниже, чем для урана (VI).
Применение теноилтрифторацетона для экстракционного отделения урана исследовано в работах [272, 456].
Экстракционное отделение в виде купфероната урана (IV)
Как уже ранее указывалось (см. раздел «Отделение осаждением»), купферон является одним из наиболее эффективных реагентов для отделения урана методом осаждения. Образующийся купфе-ронат урана (IV) легко растворяется в различных органических растворителях [345]. Экстрагирование урана (IV) в виде купфероната позволяет отделять его от алюминия и некоторых других элементов группы гидроокиси аммония, а также от щелочных и щелочноземельных металлов и ряда элементов группы сульфида аммония.
Для экстракционного отделения урана с помощью купферона может быть рекомендована следующая методика [8]. К 25—30 мл анализируемого раствора, содержащего до 5 мг урана, добавляют 2,5 мл концентрированной серной кислоты и разбавляют до 50 мл серной кислотой (1 : 200). Затем прибавляют 3 мл жидкой цинковой амальгамы и энергично перемешивают в течение 5 мин. для восстановления всего U (VI) до U (IV). Восстановление при этом частично проходит и до урана (III), но он также образует купферонат, который вместе с купферонатом урана (IV) экстрагируется в органическую фазу. Восстановленный раствор экстрагируют последовательно пятью порциями по 3 мл эфирного раствора купферона.
Для приготовления этого раствора 2 г купферона растворяют в 25 мл воды, прибавляют 5 мл 6 JV серной кислоты, экстрагируют 25 мл эфира и полученный экстракт разбавляют 25 мл эфира.
После экстракционного отделения урана объединенные экстракты упаривают досуха и прокаливают для разложения органических веществ.
Так как отделение урана (IV) экстрагированием его в виде купфероната не обладает достаточной селективностью, то, как правило, проводят двойное экстракционное отделение [535, 861]. Предварительно отделяют мешающие элементы по методике, совпадающей с описанной выше, за исключением добавления цинковой амальгамы. U (VI) в водном растворе после их отделения восстанавливают на ртутном катоде до U (IV), который затем экстрагируют диэтиловым эфиром, содержащим небольшое количество купферона. Сурьма является единственным элементом, который вместе с U(VI)
303
остается в водной фазе, а затем после восстановления экстрагируется с купферонатом урана (IX ) в органическую фазу [535]. Из эфирного раствора U (IV) реэкстрагируют разбавленной азотной кислотой. Метод оказался пригодным также для выделения микроколичеств урана. Извлечение в этом случае достигает 94 [861].
Для экстракционного отделения урана были предложены также некоторые аналоги купферона. Имеются указания на несколько большую эффективность неокупферона [274l.N-бензоилфенилгидро-ксиламин при pH 3,5 также образует экстрагирующийся комплекс с ураном (VI)[480], Избирательность экстракционного отделения с применением этих реагентов практически совпадает с таковой для купферона.
Экстракционное отделение в виде 8-ок с и хинол ината уранила
8-Оксихинолин при pH 4,7—8,0 образует с ионами уранила экстрагирующиеся 8-оксихинолинаты [481, 605, 918].Вместе с ураном экстрагируются полностью или частично также Al, Си, Fe, In, Ni, Ри (IV) и Ри (VI), Th, Sn (IV), Bi, Cd, Co, Zn, V (V),Zr и некоторые другие элементы. Применение комплексона III позволяет значительно повысить селективность отделения. В этом случае-вместе с ураном (VI) экстрагируются только W (VI) и Mo (VI). П. Н. Палей и Н. А. Миркина [184] для отделения урана от меди и многих других элементов экстрагируют их из ацетатных буферных растворов, содержащих тартрат, при pH в пределах от 3,7 до 7,0 0,1 714 раствором 8-оксихинолина в хлороформе. Из экстракта уран, реэкстрагируют 3%-ным раствором карбоната аммония.
Для отделения урана (VI) от больших количеств тория с применением 8-оксихинолина рекомендуется следующая методика [424]. Канализируемому раствору, содержащему до 600 .иг тория, прибавляют 3 капли 0,1%-ного раствора бромтимолового синего, избыток (составляющий около 10%) раствора комплексона III (372,9 г комплексона III и 80 г NaOH в 1 л) и раствор аммиака до синей окраски. Затем добавляют 1 N азотную кислоту до перехода синей окраски раствора в желтую и 0,2 Л1 раствор гидроокиси аммония точно до перехода желтой окраски снова в синюю. После этого прибавляют 2 мл 10%-ного раствора 8-оксихинолина в метилизобутилкетоне, встряхивают в течение 5 мин. и центрифугируют. Отделив водную фазу, органический слой упаривают досуха и далее прокаливают для уда -ления органических веществ.
Метод рекомендуется для выделения U233 из растворов с его* содержанием до 0,01 мкг/'мл. В этом случае определение заканчивают радиометрическим методом.
Для экстракционного отделения урана вместо 8-оксихинолина были предложены также некоторые его производные, в том числе б,7-дихлор-8-оксихинолин и другие 5,7-дигалоидозамещенные 8-оксихинолина [482, 483]. Полнота отделения урана с их применением несколько повышается. Кроме того, они образуют комплексы с ионом уранила при более низких значениях pH.
20* 307
Экстракционное отделение в виде диэтилди тиокарбамата уранила
Диэтилдитиокарбамат натрия взаимодействует с ионом уранила с образованием диэтилдитиокарбамата уранила, экстрагирующегося из водных растворов хлороформом, этилацетатом, амилацетатом и другими экстрагентами [8, 224, 688].
Ю. А. Чернихов и Б. М. Добкина 1184, 281, 282] разработали метод отделения урана экстрагированием его в виде диэтилдитиокарбамата хлороформом из водных растворов с pH 6,6—6,8. Для извлечения урана из органического слоя его реэкстрагируют раствором карбоната аммония. Вместе с ураном экстрагируются также Fe, Bi, Со, Ni, Сг (VI), Ag, Sb (III), Sn (IV), Pb, Cu, Hg, TI, As, Se и Те [184, 379]. При реэкстракции насыщенным раствором карбоната аммония в водный раствор, как показали Ю. А. Чернихов и Б. М. Добкина, переходит только уран, а остальные элементы остаются в хлороформном растворе.
Е. С. Пржевальский, Е. Р. Николаева и Н. С. Климова [193] предложили следующий метод экстракционного отделения урана от 100-кратных количеств тория и алюминия: 5 мл анализируемого раствора, содержащего 0,05—0,5 мл урана, нейтрализуют аммиаком, не содержащим карбоната. Для подавления гидролиза ряда ионов добавляют 1,2 г винной кислоты, далее 5 мл 25%-ного раствора ацетата аммония, 10 мл этилацетата и 5 мл свежеприготовленного раствора диэтилдитиокарбамата натрия. После встряхивания в течение 1 мин. отделяют водный слой и повторяют экстракцию еще 3 раза. Из объединенных экстрактов уран реэкстрагируют последовательно двумя порциями по 10 мл азотной кислоты (1 : 20).
Для отделения урана от железа реэкстрагирование осуществляют при помощи насыщенного раствора карбоната аммония (двумя порциями по 10 мл). Такой прием позволяет отделять уран от 100-кратных количеств железа, меди и других элементов [184].
Е. С. Пржевальский, Е. Р. Николаева и Н. С. Климова [193] разработали простой метод отделения урана (VI) от ванадия (V), основанный на том, что из водных растворов с pH 0,4—0,5 экстрагируется только ванадий. В этих условиях уран (VI) не взаимодействует с диэтилдитиокарбаматом натрия и вследствие этого не извлекается в органическую фазу.
Для разделения урана и ванадия к анализируемому раствору с pH 0,4— 0,5 добавляют 5 мл 2% -ного раствора диэтилдитиокарбамата натрия, 10 мл амилацетата и встряхивают в течение 1 мин. После разделения фаз водный слой отделяют и повторяют экстракцию еще 3 раза. В результате весь ванадий удаляется из водного раствора.
Кроме амилацетата, этилацетата и хлороформа могут применяться многие другие экстрагенты. Для избежания потерь при экстракционном отделении урана в делительных воронках П. Н. Палей [184] рекомендует вместо легких экстрагентов пользоваться хлороформом.
Хардвик и Мортон-Смит [575] использовали экстрагирование с применением диэтилдитиокарбамата натрия для отделения ультра-308
микроколичеств (0,01—1 мкг) и микроколичеств L’233 (1 —100 мкг) от больших количеств тория.
Для выделения ультрамикроколичеств L’233 к 5 мл анализируемого раствора добавляют сухой нитрат аммония до насыщения, затем вводят 3 мл метилизобутил-кетона, перемешивают в течение 5 мин. и центрифугируют для ускорения разделения слоев. Органический слой отделяют и повторяют экстрагирование водной фазы еще двумя порциями метилизобутилкетона по 3 мл каждая. К объединенным экстрактам прибавляют 5 мл 2 Л1 раствора нитрата аммония, 1 каплю 0,1%-ного раствора метилового оранжевого (в качестве внутреннего светофильтра), перемешивают и подщелачивают концентрированным раствором аммиака. Затем добавляют 0,5 мл 20%-ного раствора диэтилдитиокарбамата натрия и 1 N HNO3 До перехода окраски водного слоя в розово-лиловую. Размешивают в течение 4—5 мин. и центрифугируют для ускорения разделения слоев. Органический слой, содержащий весь U233, отделяют, поверхность водного слоя промывают 2—3 порциями метилизобутилкетона по 0,5 мл каждая и присоединяют их к основному экстракту, который после этого упаривают досуха. Остаток растворяют в 2 лед концентрированной HNO3, которыми сначала смывают стенки стакана, затем упаривают досуха, снова стенки смывают 2 мл концентрированной HNO3 и упаривают досуха. Остаток растворяют в 4 каплях концентрированной HNO3, и полученный раствор упаривают на мишени из нержавеющей стали. В стаканчик еще раз добавляют 4 капли концентрированной HNO3 и переносят на мишень. Содержание U233 определяют после прокаливания мишени измерением а-активности.
Для определения микроколичеств U233 (1—100 мкг) органическую фазу, содержащую О233 (после ее очистки от Th реэкстракцией водным раствором диэтилдитиокарбамата натрия), разбавляют в мерной колбе емкостью 10 мл метилизо-бутилкетоном до метки и для определения U233 отбирают 0,25 мл полученного раствора.
Для повышения селективности экстракционного отделения урана П. Н. Палей и Н. И. Удальцова [184] предложили для удержания других элементов в водном растворе маскировать их при помощи комплексона III.
Рекомендуемый ход анализа состоит в следующем. Кислый раствор, полученный после разложения руды, объемом около 50 мл нейтрализуют аммиаком до появления слабой мути (pH не выше 2,5—3), прибавляют 5—8 г комплексона III на каждый грамм навески руды, а в случае руд, богатых железом, количество добавляемого комплексона III увеличивают до 10 г на 1 г навески. Затем нейтра-лизуютаммиакомдо рН6,6—7,0 по лакмусовой бумаге, переносят в делительную воронку, добавляют 5 мл 2%-ного водного раствора диэтилдитиокарбамата натрия и образовавшиеся диэтилдитиокарбаматы экстрагируют последовательно 5—7 порциями хлороформа по 5—7 мл до отсутствия окраски хлороформного слоя.Экстракты объединяют и помещают в другую делительную воронку, добавляют 2—3 мл 2%-ного раствора аммиака, 5—6 капель насыщенного раствора карбоната аммония и энергично встряхивают в течение 2—3 мин. Водный слой, содержащий уран, выпаривают досуха и прокаливают при 500—600° в течение 30 мин. Прокаленный остаток растворяют в азотной кислоте. В полученном растворе содержание урана определяют полярографическим, фотометрическим илититриметрическим методом.
Применение комплексона III для повышения избирательности экстракционного отделения урана в виде диэтилдитиокарбамата рекомендуется также другими авторами [193, 424]. Клейтон, Хардвик, Мортон-Смит и Тодд [424] применили экстракцию урана в виде Диэтилдитиокарбамата в присутствии комплексона III для выделения следовых количеств U233 из облученного тория и сопутствующих Дочерных а-излучателей.
309
Экстракционное отделение е виде а-ннтрозо-^-нафтолата уранила
II. П. Алимарин и Ю. А. Золотов [6] показали, что уран (VI) количественно экстрагируется в виде а-нитрозо-р-нафтолата из водных растворов не смешивающимися с водой органическими растворителями. Наиболее эффективными экстрагентами для извлечения ss-нитрозо-р-нафтолата уранила являются изоамиловый и н.бутиловый спирты и этилацетат. Так как в органическую фазу вместе с ураном переходит много других элементов, в том числе кобальт, медь и железо, то для повышения селективности экстракционного отделения урана в виде а-нитрозо-р-нафтолата указанные авторы применили комплексон III. В разработанных ими условиях уран может быть полностью отделен от ванадия и железа. Для отделения урана от ванадия (V) последний восстанавливают до ванадия (IV) с помощью двуокиси серы или самим комплексоном III при pH 1—2 [184]. Затем добавляют не менее чем четырехкратное по отношению к ванадию количество комплексона III, нейтрализуют аммиаком до pH в пределах 6,5—9,0 и экстрагируют несколько меньшим или равным объемом изоамилового спирта, к которому предварительно прибавляют не менее чем 100-кратный избыток сс-нитрозо-Р-нафтола <(в молярном отношении в расчете на U3O8) в виде 2%-ного раствора в этаноле. Для выделения урана из полученного экстракта его упаривают досуха и прокаливают при 900°. Определение урана может быть закончено непосредственным взвешиванием прокаленного остатка. Отделение урана от ванадия становится неполным, если содержание ванадия более чем в 3 раза превышает содержание урана.
Для отделения урана от железа последнее также связывают при помощи комплексона III. В этом случае рекомендуется прибавлять 4,5-кратный избыток комплексона III. Экстрагирование проводят при pH в пределах 7,0—7,5. Для обеспечения полноты разделения экстракт после его отделения промывают разбавленным раствором комплексона III. Метод позволяет получать хорошие результаты даже в присутствии 10-кратного количества железа.
Применение cz-нитрозо-р-нафтола обеспечивает возможность отделения урана от алюминия и цинка, не образующих сс-нитрозо-Р-на-фтолатов, а также от тория в присутствии комплексона III.
сс-Нитрозо-р-нафтол и некоторые его аналоги были успешно применены для экстракционного отделения урана от лантана и некоторых других элементов [482].
Экстракционное отделение в виде других внутрикомплексных солей
Кроме рассмотренных выше реагентов, для экстракционного отделения урана в виде его внутрикомплексных солей было предложено также много других. Так, например, Б. Н. Судариков, 310
В. А. Зайцев и Ю. Г. Пучков [248] нашли, что уран (IV) количественно экстрагируется изоамиловым спиртом в виде салицилата из водных растворов с pH в пределах от 2,5 до 5,5. Одновременно с ураном (IV) экстрагируются также торий,- частично скандий, иттрий, лантан и церий.
Салициловая кислота оказалась пригодной также для экстракционного отделения урана (VI) [606].
Дирссен [480] показал, что внутрикомплексная соль урана (IV) с N-фенилгидроксамовой кислотой экстрагируется хлороформом. В этом случае уран (1\;) может быть отделен от лантана. Торий, а также, по-видимому, другие четырехвалентные элементы, в том числе цирконий, гафний и плутоний, экстрагируются вместе с ураном (IV).
1-(2-Пиридилазо)-2-нафтол взаимодействует с ураном (VI) с образованием внутрикомплексной соли, экстрагирующейся рядом не смешивающихся с водой органических растворителей [420]. Исключительно высокая интенсивность окраски органической фазы позволяет определять очень малые количества урана непосредственным фотометрированием экстракта.
Хлороформные растворы ксантата калия экстрагируют уран (VI) из слабокислых и нейтральных водных растворов [570].
Из-за отсутствия достаточно полной характеристики экстракционного отделения урана с применением перечисленных реагентов они не имеют пока практического значения.
Другие виды экстракционного отделения урана
Экстракционное отделение в виде солей комплексных анионов урана с органическими катионами
Этот вид экстракционного отделения урана состоит в том, что экстрагент извлекает из водной фазы соль, образующуюся в результате взаимодействия комплексных анионов урана с органическими катионами.
Так, например, 8-оксихинолин образует с ураном (VI) комплекс, представляющий собой одноосновную кислоту Н[UO2 (C9H6ON)3], которая экстрагируется из растворов с pH до 8,0 [481, 605, 918]. При более высоких значениях pH указанная комплексная кислота диссоциирует с образованием комплексного аниона, который уже не обладает способностью экстрагироваться в органическую фазу. Однако при добавлении органических оснований или их солей катион основания взаимодействует с этим анионом с образованием соли, которая легко экстрагируется многими не смешивающимися с водой органическими растворителями [425]. Органическими основаниями, образующими такие соли, являются третичные амины типа диметилалкиламмония с алкилом, содержащим около 16 ато
311
мов углерода. Образовавшуюся соль экстрагируют метилизобути.т-кетоном, диизобутил кетоном, амиловым или бутиловым спиртом, эфиром, хлороформом, бензолом и многими другими экстрагентами.
Вследствие высокой прочности образующегося комплексного аниона экстракционному отделению урана этим методом карбонаты не мешают. На этом основании был предложен метод экстракционного отделения урана из карбонатных растворов, обладающий очень высокой селективностью [425].
Для отделения урана (VI) этим методом pH анализируемого раствора (содержащего до 1,0 г-моль/л Ыа2СО3)добавлением едкого натра устанавливают в пределах 11,0—12,5 и экстрагируют два раза равным объемом метилизобутилкетона, содержащим в 1 л 0,3 моля 8-оксихинолина и 0,1 моля одного из указанных выше третичных аминов. Из органического слоя уран реэкстрагируют два раза равным объемом 0,5 М раствора NaHCO3. Большая часть ванадия экстрагируется вместе с ураном.
Точно таким же образом уран может быть отделен экстрагированием его в виде солей органических оснований с такими комплексными анионами, которые образуют с ураном пирогаллол, купферон, бензоин-2-оксим [425].
Мур [757 ] предложил экстрагировать уран (VI) ксилольным раствором три-(изооктил)-амина из водных растворов, 7 N по соляной кислоте. Образующийся в этих условиях хлоридный комплекс, представляющий собой кислоту, извлекается в органическую фазу в виде соли с три-(изооктил)-амином. Реэкстракцию проводят равным объемом 0,1А7 соляной кислоты. Метод оказался пригодным для отделения урана (VI) от тория и продуктов деления. Все элементы, образующие экстрагирующиеся хлоридные комплексы, в том числе Fe, Ga, Al, In, TI и Sb, экстрагируются вместе с ураном.
В. М. Вдовенко и Л. И. Лазарев [29] исследовали экстрагирование урана (VI) при помощи анилина из растворов, содержащих уксусную кислоту. Они показали, что уран переходит в слой анилина в виде соли ацетатного комплекса урана с анилином C6H5NH2.H[UO2 (СН3СОО)3].
Так как ацетатные комплексы в указанных условиях образуют лишь немногие элементы, то указанный метод экстракционного отделения урана, по-видимому, обладает высокой селективностью.
Из сернокислых растворов уран (VI) также может экстрагироваться многими не смешивающимися с водой органическими растворителями в виде солей уранилсерной кислоты с органическими основаниями, структура которых может быть представлена формулой (R3NH)2 [UO2 (SO4)2] [610]. В качестве оснований, способных образовывать экстрагирующиеся соли уранилсерной кислоты, рекомендуются триоктиламин, дидециламин, метилдиоктиламин и тридециламин. Вместе с ураном могут, по-видимому, экстрагироваться и другие элементы, образующие сульфатные комплексы, в том числе Th, Zr, Ши Ри.
312
Экстракционное отделение урана в виде солей ал кил фосфорных кислот
Моно- и диалкилфосфорные кислоты, а также алкилпирсфосфор-ные кислоты образуют с ураном (IV) и с ураном (VI) соответствующие алкилфосфаты, экстрагирующиеся многими не смешивающимися с водой органическими растворителями [60, 146, 272, 642, 896].
Диалкилфосфорные кислоты образуют очень стойкие комплексы с ураном (VI), хорошо экстрагирующиеся н.гексаном, четыреххлористым углеродом, дибутиловым эфиром и многими другими экстрагентами. Экстракционная способность диалкилфосфатов урана настолько велика, что позволяет экстрагировать уран из очень разбавленных растворов при помощи небольшого объема экстрагента без применения высаливающих агентов. Диоктилфосфат уранила обладает большим коэффициентом распределения, чем другие диалкилфосфаты, содержащие меньше углеродных атомов в алкильном остатке. Повышение кислотности приводит к уменьшению коэффициентов распределения, однако даже в 2 М азотной кислоте при 0,1 М концентрации диалкилфосфорных кислот коэффициент распределения урана близок к 100. Если привести в равновесие 100 объемов морской воды, содержащей U233, с одним объемом 0,726 М раствора дибутилфосфорной кислоты в четыреххлористом углероде, то достаточно одной последующей промывки водного слоя еще таким же объемом четыреххлористого углерода для полного извлечения урана [939].
Моноалкилфосфорные кислоты оказались несколько менее эффективными.
Алкилпирофосфорные кислоты взаимодействуют с ураном (VI) и особенно с ураном (IV) с образованием более прочных комплексов, чем это имеет место в случае диалкилфосфорных кислот. Вследствие этого они могут применяться для извлечения урана из растворов, содержащих фосфаты [146]. Необходимая степень экстракции может быть достигнута регулированием концентрации диалкилпиро-фосфорной кислоты в экстрагенте. Реэкстракцию урана из органической фазы проводят растворами плавиковой кислоты. В связи с этим аналитическое значение диалкилпирофосфорных кислот значительно обесценивается.
Вместе с ураном при экстрагировании с применением алкилфосфорных и алкилпирофосфорных кислот в органическую фазу переходят Fe (III), Th, Zr, In, Sn (IV), Nb, Y, Ta, Mo и ряд других элементов.
3. ХРОМАТОГРАФИЧЕСКОЕ ОТДЕЛЕНИЕ УРАНА
М. К- Чмутова
Получивший широкое развитие за последние 50 лет хроматографический метод анализа ставит задачи выделения анализируемых компонентов с последующей их идентификацией и определением
313.
количеств любыми химическими, физическими и физико-химическими методами.
Основная особенность хроматографического метода состоит в многократном повторении актов адсорбции-десорбции, ионного обмена или распределения между фазами и динамическом характере этих процессов [219].
В соответствии с характером взаимодействия между раствором и адсорбентом различают три ^ида хроматографического анализа:
1) Адсорбционная хроматография (молекулярная и ионообменная).
2) Распределительная хроматография.
3) Осадочная хроматография.
В настоящей главе мы будем рассматривать отделение урана от ряда элементов методами ионообменной и распределитёльной хроматографии.
Ионообменная хроматография
Разделение смесей в случае ионообменной хроматографии основано на различной способности ионов, содержащихся в растворе, к обмену с подвижными ионами сорбента.
В последнее время в качестве сорбентов и ионообменной хроматографии применяют почти исключительно синтетические ионообменные смолы.
Их разновидностям, методам синтеза и основным свойствам посвящено большое количество работ, опубликованных в советской и иностранной литературе [5, 14, 175, 214, 215, 258, 259].-
Ионообменные смолы представляют собой нерастворимые, химически стойкие продукты полимеризации или конденсации, содержащие в своем составе ионообменные группы, способные вступать в реакции обмена с ионами, находящимися в растворе. Сорбенты этого типа делятся на два класса: 1) вступающие в реакцию обмена с катионами раствора — катиониты и 2) вступающие в реакцию обмена с анионами раствора — аниониты.
Активными группами катионитов могут быть группы — SO3H, —СООН и др. Ион водорода в этих группах подвижен и способен к обмену.
Активными группами анионитов могут быть группы —NH,, =NH, =N.
О механизме обмена ионов анионитами нет в настоящее время достаточно достоверных данных. Предполагают, что в обмен'вступают ионы гидроксила, образующиеся в процессе гидратации смолы— NH2(H3O)+OH“; =NH(H3O+)OH”; “ N(H3O)^OH~ [204]. Для хроматографических целей часто употребляют катиониты, у которых все ионы водорода активных групп замещены на какой-либо другой катион (например Na+). Переведение смолы в другую форму обычно достигается обработкой смолы солью соответствующего катиона. Точно также ОН--группа анионита может быть замещена любым другим анионом.
По степени диссоциации активных групп катиониты делятся на сильнокислотные и слабокислотные, аниониты — на сильноосновные и слабоосновные.
Сильнокислотные катиониты и сильноосновные аниониты обладают способностью к ионообменной сорбции в значительном интервале pH среды и вступают в реакции ионного обмена с любыми ионами. Это облегчает процесс поглощения ионов из раствора, но затрудняет процесс разделения вследствие малой избирательности таких сорбентов.
Обменная способность слабокислотных катионитов и слабоосновных анионитов зависит от pH среды и характера ионов в растворе.Поэтому сорбенты со сла-314
бодиссоциированными активными группами обладают избирательностью сорбции, что облегчает процесс разделения ионов [175, 259].
Важнейшей характеристикой ионообменной смолы является ее обменная емкость, определяемая числом функциональных групп, содержащих способные к обмену ионы и выражаемая числом миллиграмм-эквивалентов элементов, поглощенных граммом смолы. Полная обменная емкость определяется в статических условиях при любых, но точно известных значениях pH раствора. В динамических условиях, т. е. при пропускании раствора через слой сорбента, вводится понятие динамической обменной емкости, которая зависит не только от полной обменной емкости и структуры ионита, но и от скорости фильтрации, высоты фильтрующего слоя ионита, от концентрации раствора и пр.
При проведении анализов с помощью ионного обмена раствор фильтруется через слой ионообменной смолы, помещенный в колонку. Первой стадией в обменном цикле является поглощение, в процессе которого способные к обмену ионы ионита замещаются ионами анализируемого раствора. Следующим этапом является удаление из колонки сорбировавшихся ионов (элюция). Е. Н. Гапон [44] указывает на два возможных вида элюции:
1) элюция простая заключается в промывании сорбента раствором электролита, ионы которого вытесняют из колонки адсорбированные ионы в определенной последовательности; •
2) элюция комплексообразующая заключается в обработке колонки реагентом, переводящим ранее адсорбированные ионы в комплексные соединения, последовательность вымывания которых из колонки потоком комплексообразующего вещества зависит от их констант устойчивости.
Раствор, поступающий в колонку в целях десорбции анализируемых ионов, носит название «элюента». Раствор, вытекающий из колонки в процессе десорбции, носит название «элюата».
К настоящему времени рядом советских и зарубежных авторов разработаны теоретические основы ионообменного процесса [44 220,].
Применение ионообменных смол в процессах промышленного производства позволяет непосредственно получать высокопроцентный концентрат урана вследствие высокой селективности извлечения последнего из раствора.
Так, например, после кислотного выщелачивания концентрация урана в растворе колеблется от 0,5 до 1 г'л; концентрация же урана в элюате составляет 10—20 г!л. При этом урановый продукт не загрязняется примесями, так как содержание других элементов в элюате ничтожно [152].
В настоящем разделе мы рассмотрим отделение урана от сопутствующих элементов методами анионного и катионного обмена.
Анионный обмен
Подавляющее большинство опубликованных работ по ионообменному извлечению урана относится к области анионного обмена. Последний обладает целым рядом преимуществ, которые лежат в основе его быстрого распространения. Некоторые сильноосновные анионообменные смолы проявляют высокую избирательную способность к определенным комплексным ионам. Таким образом, имеется возможность применения анионитов при работе с концентрированными растворами электролитов, где катионный обмен не может быть осуществлен, поскольку в этих средах ионы многих металлов пере
315
ходят в отрицательно заряженные комплексы и не поглощаются катионитом. Анионный обмен позволяет широко использовать способность элементов к комплексообразованию. Методом анионного обмена часто можно разделить металлы, которые в катионном состоянии обладают сходными свойствами. Такие металлы могут образовывать отрицательно заряженные комплексы, имеющие сильно отличающиеся константы нестойкости, что может быть положено в основу их ионообменного разделения.
Сравнительное изучение комплексообразования урана и ряда других элементов bJ различных средах позволяет предсказать возможности отделения урана от сопутствующих примесей. С этой точки зрения рассмотрим поведение урана в солянокислых растворах.
Краус и Нельсон [109] разделили’все элементы на три группы в отношении их анионного обмена из солянокислых сред.
К первой группе относятся металлы, не сорбирующиеся из растворов НС1 любой концентрации. К ним относятся щелочные металлы, щелочно-земельные металлы, в значительной степени элементы левой подгруппы III группы периодической системы (от иттрия до актиния) Al (III), Ni (II), Th (IV).
Ко второй группе относятся металлы, поглощение которых растет с увеличением молярности НС1; к ним относится уран.
В третью группу входят металлы, обнаруживающие уменьшение поглощения с увеличением концентрации НС1 от 1 до 12 М. К ним относятся многие элементы, находящиеся в основном в центральной части периодической системы.
Поглощение элемента анионитом авторы характеризуют величиной коэффициента распределения D (количество вещества, поглощенного 1 кг сухой смолы/количество вещества в 1 л раствора), определяемого в статических условиях. Исследование анионообменной сорбции урана показало рост поглощения U (VI) с увеличением концентрации НС1 от D = 1 в 1 М растворе НО до D —1800 в 9 М растворе НС1. При дальнейшем увеличении концентрации НО от 9 до 12 М сорбция урана снижается. Очевидно, увеличение поглощения связано с образованием в среде НО высокой концентрации хлоридных анионных комплексов, предполагаемый'состав которых [UO2C 13]“ и [UO2C14]2~ (подробно см. в главе II).
Ю. В. Морачевский и М. Н. Гордеева [160] использовали для отделения урана от ванадия различную сорбируемость этих элементов на сильноосновных анионитах ПЭ-9 и ЭДЭ-10 из 8N соляной кислоты. Исходный раствор, 8 N по НС1, содержащий уран и ванадий, пропускали через колонку анионита в С1~-форме с последующим промыванием смолы чистым раствором 8 N НС1. При этом уран в виде хлоридного анионного комплекса сорбировался полностью, ванадий’же, не образующий сколько-нибудь устойчивых анионных комплексов в данных условиях, практически целиком переходил в элюат. Уран затем удаляли из колонки дистиллированной водой.
316
При содержании урана в пределах 0,5—12 мг и ванадия 0,25—48 мг относительная ошибка в определении каждого из них не превышала — 2%.
Шен и Реньо [292] предложили использовать солянокислые растворы для извлечения U233 из тория, облученного в ядерном реакторе. U233 образуется вследствие действия тепловых нейтронов на атомы Th232. Задача заключается в отделении урана от промежуточных элементов Th233 и Ра233.
Соотношение весовых количеств в растворе: ттгзз пя
^ = от 10-5 до 10-6.
Th Th
Исследование проводилось путем измерения коэффициентов распределения всех трех элементов между сильноосновным анионитом А300 D Филипс энд Пейн и растворами соляной кислоты различной концентрации. Для протактиния были получены следующие результаты: от 0,5 N до 4 N НС1 коэффициент распределения остается практически постоянным и равным 4. От 4 М до 8 М происходит увеличение сорбции. В 8 N НС1 коэффициент распределения равен 450. Торий независимо от кислотности практически не поглощается смолой. Сравнение этих результатов с результатами, полученными для урана [109]; показывает, что можно количественно отделить U и Ра от тория в 8N НО. U и Ра, образующие отрицательно заряженные комплексы, полностью сорбируются смолой. Th полностью проходит в элюат при промывании 8 N НС1. Ра вымывается затем 0,1 М раствором (NH4)2 SiF6 в 8 N соляной кислоте. В этих условиях коэффициент распределения урана в противоположность коэффициенту распределения протактиния практически не изменяется. Краус, Мур и Нельсон [679] для вымывания протактиния использовали смесь 9 М НО —1A4HF. После удаления Ра U вымывали 0,5 N НС1.
Описанный метод позволяет осуществить хорошее разделение трех элементов, каждый из которых извлекается с выходом более 98%.
Метод отделения урана от тория, описанный Томиком, Ладен-бауером и Поллаком [981], принципиально ничем не отличается от только что приведенного. Авторы указывают на возможность отделения урана от Mn(II), Bi(III), Cd(II), Zn(II), As(III), Ni(II), Al(III), Hg(II), Сг(П1) в солянокислых средах на анионообменной смоле Амберлит IRA-400.
Существует еще целыйж’ряд работ, в которых для отделения урана от тория на анионитах используется соляная кислота [107, 621, 686, 751 ]. Очевидно, этот метод является наиболее рациональным.
Милнер и Эдвардс [749] разработали метод определения урана в бинарных сплавах с висмутом. Процентное содержание урана изменялось от 0,001 до 10%. Адсорбция висмута сильноосновной четвертичной аммонийной смолой Деацидит-FF из раствора НС1
317
велика при низкой кислотности. Авторами найдено, что 500 мг висмута в 50 мл 10?о-ной НС1 количественно сорбируются 5 г воздушносухого анионита при скорости пропускания раствора через колонку не более 2 мл/мин. U (VI), как было сказано выше, напротив, не сорбируется при данных условиях. Следовательно, разделение этих двух элементов не представляет трудности. Ошибка определения урана составляет dz 1,6?о (отн.).
Милнером и Нанном [751 ] было осуществлено разделение урана, тория и висмута в тройном сплаве при использовании анионита Деацидит-FF и соляной кислоты.
Краус, Мур и Нельсон [679] исследовали сорбцию ряда четырехвалентных элементов на смоле Дауэкс-1 из растворов соляной кислоты (табл. 42).
Таблица 42
Сорбция четырехвалентных элементов на смоле Дауэкс-1 из растворов соляной кислоты различной нормальности
Элемент
Молярность кислоты, выше которой наблюдается значительная сорбция элемента анионитом
Sn 0,1
Се 3,5
и 6
Zr 7
Hf 8
Th 9,5
V Слабая сорбция наблюдается только
в концентрированной НС1
В эксперименте все указанные элементы были внесены на колонку в концентрированной соляной кислоте и элюированы с последовательным уменьшением концентрации НС1. Порядок элюции: ТЬ. V, Ti, Hf, Zr, U, Се, Sn.
Подобный же метод отделения урана от целого ряда примесей был предложен Броди, Фэрисом и Бьюкененом [390]. Авторы использовали для фракционного вымывания элементов с колонки Дауэкс-1 последовательно 12, 11, 10, 9, 8, 7, 6, 5 N НС1. Уран начинал двигаться по колонке только с 5 N НС1 и окончательно вымывался 1 N НС1.
Интересен метод разделения U(IV) и U(VI) [679], получающихся в эквимолярных количествах в результате диспропорционирования 0,1 М раствора UC15 в 10 А НС1. Разделение проводилось на анионите Дауэкс-1 с помощью 3 N НС1. U(VI) задерживался смолой, U(IV) проходил в элюат.
Краус, Нельсон и Мур [682] рассмотрели поведение U(VI), Mo(VI) и W(VI) в солянокислых растворах и нашли, что сорбция
318
Mo(VI) возрастает с увеличением концентрации НС1 от D = 2 в 0,5 N НС 1 доП-250 в 5 N НС 1. Далее сорбция несколько снижается с ростом концентрации соляной кислоты. Начиная с 6 М концентрации НС1, сорбция W(VI) возрастает, достигая максимума в 8 N НС1. При концентрациях НС1, меньших 6 М, происходит гидролиз W(VI). Данные по поведению LT(VI) были рассмотрены выше.
Как видно из сказанного выше, в растворах с концентрацией НС1 ниже 6 М разделение не может быть осуществлено вследствие близости значений коэффициентов распределения U(VI) и Mo(VI) и гидролиза W(VI). В достаточно концентрированных растворах разделение всех трех элементов не может быть осуществлено вследствие сходства в сорбционной способности W(VI) и Mo(VI). Но каждый из этих элементов может быть отделен от урана.
Отделение U(VI) от W(VI) проводилось на сильноосновном анионите Дауэкс-1. U(VI) и W(VI), вносили на колонку в 12 М НС1. Элюция W(VI) осуществлялась 12 М НС1. Уран затем удаляли из колонки разбавленной НО. Применяя вместо соляной кислоты смесь ее с плавиковой кислотой, можно достичь количественного разделения всех трех элементов.
Краус и Нельсон [109] изучали сорбцию U(VI) из смесей НО— HF при постоянной концентрации HF. Если концентрация НО велика, присутствие HF мало влияет на сорбцию LJ(VI). При низких концентрациях НО сорбция из смеси значительно меньше, чем из среды одной НС1 вследствие образования фторидных положительно заряженных комплексов урана. При еще более низких концентрациях НО (<ОД4 НО) поглощение урана снова увеличивается с уменьшением концентрации соляной кислоты вследствие преобладания отрицательно заряженных фторидных комплексов.
В растворах, 1 М по HF и 7—12 М по НО, коэффициенты распределения U (VI), W (VI) и Mo (VI) отличаются настолько, что элементы можно без труда разделить на анионите [682]. В ходе разделения уран элюировали из колонки анионита Дауэкс-1 0,5 М НО—IM HF. Затем вымывали W (VI) смесью 7М НО—1 М HF и Mo (VI)— 1 М НО.
В среде азотной кислоты поглощение анионитами U (VI) незначительно. При увеличении концентрации HNO3 коэффициент распределения возрастает otD = 1 в 2 /И растворе HNO3 до D=20 в 8 Л1 растворе HNO3 [109]. При дальнейшем увеличении концентрации кислоты происходит разрушение анионита. Для выделения урана из азотнокислых растворов чаще всего употребляют смесь азотной кислоты небольшой концентрации и нитрата алюминия, лития, кальция и др.
При постоянной концентрации нитрат-иона поглощение из такой смеси значительно выше, чем из среды одной HNO3.
Рассмотрим в качестве примера работу Оскендена и Формэна [787] по отделению урана от больших количеств железа и алюминия.
319
Раствор образца в смеси, 0,3 М по HNO3 и 1,6 Л1 по нитрату алюминия, пропускают через колонку, содержащую 1,5 г сильноосновной смолы Деацидит-FF, измельченной до 0,2—0,3 мм и переведенной предварительно в МО~'форму. Уран при этом целиком поглощался смолой. Затем колонку промывают 1,6 М раствором Al(NOs)s для удаления железа, 8 N НС1 для удаления алюминия и, наконец, 0,1 Л1 раствором НО для удаления урана. Метод пригоден для отделения миллиграммовых или микрограммовых количеств урана от больших количеств железа и алюминия.
Образование отрицательно заряженных комплексов тория в среде азотной кислоты, количественно сорбирующихся анионитом в условиях, когда коэффициент распределения урана достаточно низок, послужило основанием для разделения урана и тория [411].
Разделение велось на смоле Деацидит-FF, зернением 60—100 меш. Уран десорбировали 4 М HNO3, в то время как торий удерживался смолой. Th затем вымывался водой.
Кроме названных работ по отделению урана в нитратных средах, следует упомянуть метод Новака и Пекарека [169] отделения урана от ряда элементов на анионите в хлоридно-нитратных смесях.
В процессах переработки урановых руд применение серной кислоты для кислотного выщелачивания оказалось наиболее экономически выгодным. Поэтому особый интерес приобретает изучение анионообменного поведения урана в сернокислых растворах. Краус и Нельсон [13] нашли, что поглощение U (VI) уменьшается с ростом концентрации серной кислоты от £2=35000 в 0,01 М H2SO4 до D = 1 в 4 М H2SO4. В растворах сульфатов уменьшение D с увеличением концентрации происходит значительно медленнее. Так, £2=500 в 4Л4 (NH4)2SO4.
Исследователями уделялось большое внимание изучению анионообменной сорбции сульфатных комплексов урана [279, 337, 339, 947, 949[
Ряд авторов [44, 279, 949] показали, что уран сорбируется главным образом в виде [UO2(SO4)3]4“ и [UO2 (SO4)2]2~.
Суньер и Лагос [947] указали на привилегированное положение сульфатных комплексов урана в сорбционном ряду анионов, так как они могут замещать многие анионы, конкурирующие с ураном в процессе сорбции анионитом. Авторами было экспериментально показано, что возрастание концентрации сульфата вызывает увеличение сорбции урана до тех пор, пока отношение уран : сульфат не станет равным 1 : 2. Дальнейшее возрастание концентрации сульфата ведет к быстрому уменьшению сорбции урана. Кислотность раствора также влияет на степень поглощения урана, так как уменьшение pH сдвигает процесс: HSO4^H++SO42' в сторону нежелательного образования бисульфат-ионов, конкурирующих с сульфатными комплексами урана в процессе поглощения анионитом. Холлис и Мак-Артур [279] описали промышленное извлечение урана из кислой рудной пульпы при pH 1,2—1,5, концентрации урана 0,5—1 гН3О8/л и концентрации сульфата 10—20г/л. Вымывание урана осуществляли смесью 0,1 N HNO3—0,9 V
320
XH4NO3. Извлекалось более 99% растворенного урана. Конечный продукт содержал 70 ?о.
Арнфельт [340] предложила метод определения урана в растворах, содержащих до 0,001 % U и относительно большие количества Fe. V, Сг и других элементов. Метод основан на избирательной сорбции сульфатных комплексов урана из раствора с pH 2 сильноосновным анионитом Дауэкс-2 в SO*--форме. Анализируемый раствор может содержать от 0,02 до 0,4 мг/мл U. Концентрация SO*-не должна превышать 75 мг/мл. При этих условиях отмечена частичная сорбция Fe (Ш), мешающего конечному спектрофотометрическому определению урана по пероксидной методике. Вследствие этого автор рекомендует вводить в раствор, полученный после десорбции урана соляной кислотой (1 : 10), вещества, способные замаскировать железо. Тем не менее результаты получались завышенными и ошибка метода составляла -f-б,5%(отн.) Мало внимания уделено поведению ванадия, хрома и др. в условиях опыта, хотя частичная сорбция их анионитом весьма вероятна.
Фишер и Кунин [269] значительно усовершенствовали метод отделения урана от железа, ванадия и некоторых других элементов, добавляя в исследуемые растворы перед пропусканием их через анионит сернистую кислоту' для восстановления Fe (III) и V (V), благодаря чему устранялась сорбция последних анионитом.
Если анализируемый раствор содержит хлорид (>0,1 N) или нитрат (>0,01 IV), то пробу упаривают с серной кислотой до появления паров SO3 и затем разбавляют водой. Концентрация сульфат-иона в растворе должна быть по меньшей мере 2 г!л. Перед пропусканием через колонку раствор нейтрализуют щелочью до pH -~1 и обрабатывают избытком 6%-ной H2SO3. В качестве кислотно-восстановительного индикатора может быть применен метиленовый голубой. Приготовленный таким образом раствор пропускают через колонку с анионитом Абмерлит IRA-400b SO*~"форме с небольшой скоростью. Затем колонку промывают водой, уран элюируют разбавленной НС1О4. Содержание урана в полученном растворе определяют колориметрически, щелочно-пероксидным методом или объемным методом с использованием цинкового редуктора и периметрии.
Присутствие веществ, в условиях опыта являющихся катионами (Al, Са, Mg, Со, Си и др.), не мешает отделению.
При содержании урана 0,1% и более результаты всегда хорошо согласуются с результатами, полученными стандартным методом с применением купферона.
Сейм, Моррис и Фрей [893] использовали этот же метод для анализа урановых руд. Точность метода составляет ± 0,005 % при содержании окиси урана 0,01%.
Редкие земли и щелочные металлы не образуют комплексов с разбавленными минеральными кислотами, поэтому легко осуществить их отделение от урана в сульфатной среде.
Шушич [949] разработал методику отделения урана от редкоземельных и щелочных металлов. В качестве представителя редкоземельных элементов был взят Се144, щелочных металлов —Cs137.
21 Аналитическая химия урана 321
Исследуемый раствор, содержащий 2 • 10 "- г лрана, церий и цезии в 0,01 Л’ H.,S04 пропускают через колонку анионита Дауэкс-1. При этом весь Се и весь Cs оказываются в элюате. Уран вымывают затем 1 Л' Н\О3.
Этим же автором осуществлено отделение урана (2,38-10"2 г) от Zn, Мп, Ni, Cd, Со, Fe (III) и Си (по 1 • 10"2 г каждого) в 0,01 Л7 H2SO4.
После пропускания раствора через колонку смола промывается 3—4 колоночными объемами 0,01 N H2SO4. Все элементы, кроме урана, переходят при этом з элюат. Уран десорбируется затем 1 N HNO3.
Бэнкс, Томпсон и О’Лауглин [3581 отделяли редкоземельные элементы от U (VI) и Fe (Ш), так как последние мешают определению редкоземельных элементов с арсеназо.
U (VI) и Fe (III) сорбировались сильноосновной смолой Дауэкс-1 X 8 б SO;- -форме из смеси, 0,5%-ной по H2SO4 и 4%-ной по NH4SCN. U (VI) сорбировался в виде сульфатного комплекса, Fe (III) — в виде роданидного. Редкие земли элюировались 0,5?о-ной H2SO4.
Бэнержи и Хейн [355] отделяли уран (VI) от висмута при pH 1—1,5 сорбцией на смоле Дауэкс 1 X 10 в 5О;--форме из разбавленных растворов H2SO4.
Для щелочного выщелачивания низкосортных урановых руд применяется раствор карбоната натрия, который извлекает уран в виде комплексного соединения Na4 [UO2 (CO3)3j. Используя избирательную способность сильноосновных анионитов по отношению к этому соединению,Шанкар, Бхатнагар иМюрти [286] предложили промышленный метод анионообменного концентрирования урана.
Сущность метода состоит в том, что при пропускании раствора, 5?/о-ного по Na2COs, содержащего уран (порядка 0,1%) и ряд примесей в виде фосфата, ванадата, алюмината и пр., через колонку с сильноосновными анионитом Амберлит IRA-400 происходит количественная сорбция уранилкарбонатного комплекса, в то время как все примеси проходят в элюат. Уран затем извлекают I Л1 раствором NaNOa-
Авторами изучалось также поведение ванадат,-фосфат,-алюминат,-и силикат-ионов. Найдено, что в отсутствие избытка карбокатионов эти анионы поглощаются анионитом. Не следует, однако, употреблять большой избыток карбоната натрия, так как увеличение концентрации последнего от 1 до 100 г!л снижает емкость смолы по урановому комплексу от 274 мг/г до 100 мг/г (емкость выражена .waLXOs отношением ----------------- вследствие вытесняющего действия
г, сухой смолы карбонат-ионов.
Результаты применения данного метода показали, что 99,8'% адсорбированного урана может быть элюировано, причем концентрация его в элюате превышает концентрацию в исходном щелоке в среднем в 55,5 раза.
Мюрти [770] применил этот же метод для отделения урана от ванадия в аналитических целях при различных соотношениях 322
V2O5 : LT3OS от 1 : 1 до 1000 : 1. Полученные результаты показали, что даже при последнем соотношении средняя ошибка в определении урана невелика и составляет- 2%.
В. К. Марков, В. Т. Харламов и А. П. Медведева [2131 разработали быстрый и простой метод определения содержания урана в природных водах, основанный на избирательной сорбции уранилкарбонатного комплекса анионитом ММГ в бикарбонатной форме. Конечное определение урана проводили (после растворения смолы в НС1) люминесцентным методом. Уран может быть определен в концентрациях 0,2—0,02 мг/л с относительной ошибкой ^15%.
Существует ряд работ, в которых рассматривается анионообменное поведение урана в некоторых органических кислотах, а также возможности отделения урана от примесей с помощью органических кислот.
Б. П. Никольский, В. И. Парамонова и А. Ф. Вьюгина [176] разделяли уран (VI) и торий (IV), используя свойства элементов образовывать ацетатные комплексы различной прочности.
Смесь ацетатных растворов урана и тория (pH 3,3 или 4,5) обогащают на колонке со слабоосновным анионитом Вофатит-М ураном вследствие значительной сорбции UOoAc- и очень слабой сорбции ацетатного комплекса тория. Поглощенные элементы вытесняют затем 0,2 N НС1, причем в первых фракциях элюата содержится часть урана и весь торий, а в последующих фракциях — один уран.
Метод Гехта, Коркиша и др. [671] отделения урана от Fe (Ш), Th, Pb, Ca, Mg и редкоземельных элементов основан на том же принципе.
Заслуживает внимания предложенный Коркишем, Фарагом и Гехтом [587] способ обогащения урана с помощью аскорбиновой кислоты. При смешивании солянокислого раствора уранила аскорбиновой кислотой (pH смеси 4,—4,5) образуется аскорбинатный комплекс состава [UO2 (ОН)2 (СеН-Об)~ ]. Щелочные, щелочноземельные металлы, Al, Pb, As (III), Bi (III), Zn, Мп (II), Cr (III), Fe (II), Co, Ni, редкоземельные элементы образуют в этих растворах либо положительно заряженные, либо нейтральные комплексы и, следовательно, могут быть отделены от урана.
Разделение проводится по следующей методике. Навеску сухого образца, содержащую от 1 до 100 мкг U, растворяют в 5 мл 5 N НС1 при нагревании. Затем Добавляют 45 мл воды и после двухчасового стояния отфильтровывают, промывая остаток на фильтре 25 мл 0,1 N НС1.
К полученным 75 мл раствора добавляют 2 г аскорбиновой кислоты, растворенные в 25 мл воды. Добавлением аммиака pH раствора доводят до 4—5. Затем полученный раствор пропускают через колонку с анионитом в аскорбинатной форме со скоростью 1 мл мин. Колонку промывают последовательно 1%-ным раствором аскорбиновой кислоты и водой. Уран элюируют 100 мл 1 N НС1. Смола Амберлит IRA-400, иа которой проводится разделение, имеет высокую емкость в отношении ураниласкорбинатного комплекса.
Метод находит применение в материалах с содержанием урана от 0,0001 до 0,1% и выше.
21*
323
Катионный обмен
Значительно меньше места уделено в литературе катионообменному отделению урана от других элементов.
Сравнительно небольшое число работ основано на использовании зависимости сорбируемости от величины заряда иона. К ним относятся работы Дирсена [479] по разделению U (VI) и Th (IV) на катионитах Амберлит IR-100 и Вофатит KS с помощью НС1, Клемента [659] по разделению U (VI) и Fe (III) на катионите Леватит S-100 с использованием в качестве элюента 0,8 N НО и др. [632].
Следует отметить, что, основываясь на этом принципе, невозможно осуществить отделение урана от близких по свойствам элементов.
Подавляющее большинство работ по катионообменному отделению урана от примесей можно разделить на две основные группы. К первой относятся работы, в которых рассматриваются методы отделения, основанные на использовании «универсальных» сорбентов, содержащих сильнокислотные группы, способные легко вступать в реакцию с любыми катионами. Выделение в большинстве случаев сводится к поглощению всех катионов обменником с последующей избирательной десорбцией урана или примесей специально подобранным элюентом.
Вторую группу составляют работы, в основу которых положена селективная сорбция урана из раствора. Селективность сорбции обычно достигается изменением степени проницаемости катионита или подбором типа и взаимного расположения ионогенных групп.
Д. И. Рябчиков, М. М. Сенявин, 3. К- Михайлова и В. К. Сары-гина [213] предложили метод определения малых количеств урана в рудах и сбросных водах.
Анализируемый раствор, содержащий уран, алюминий и ванадий (pH 1,5— 2,0) пропускают через колонку с сильнокислотным катионитом КУ-2 в Н+-форме. Колонку промывают дистиллированной водой, затем 10%-ным раствором аммиака для избирательной десорбции алюминия и ванадия в виде алюмината и ванадата. Уран извлекают горячим 5%-ным раствором карбоната аммония в виде (КН4)4[иО2(СОз)з1.
Н. М. Собинякова, Е. А. Рубинштейн и Л. П. Макшеева [213] предложили использовать 5%-ный раствор ацетата аммония (pH 5,0—5,5) в целях избирательного извлечения урана из колонки с катионитом КУ-2 или хМСФ.
Фторид натрия, как показали В. А. Хализова и Е. П. Смирнова [213], избирательно вымывает уран из колонки с катионитом в виде фторидного комплексного соединения. Метод удобен для анализа растворов с большим содержанием железа, так как последнее не десорбируется фторидом натрия.
Принцип метода отделения урана от ванадия и молибдена, предложенный Н. Т. Воскресенской [2131, состоит в избирательном извлечении последних из колонки катионита ПФСК 5%-ным раство
324
ром перекиси водорода в виде надкислот, а урана — 5%-ной серной кислотой.
Долар и Драганич [470] в поисках селективного элюента для отделения урана от редкоземельных элементов исследовали растворы соляной, серной, щавелевой, лимонной, винной кислот, цитрата, оксалата и тартрата аммония. Наилучшие результаты были получены с щавелевой кислотой. IV раствор Н2С2О4 избирательно извлекает уран из колонки катионита Амберлит 1R-120. Европий элюируется затем 5 N НС1. Ошибка метода Д5% (отн.).
Значительный интерес представляет использование селективных катионитов, способных избирательно поглощать уран из растворов различного состава.
В. И. Кузнецов [122] рассматривает иониты как органические реагенты, у которых вследствие их высокого молекулярного веса соли стали нерастворимыми и реакции превратились в поверхностные реакции обмена. Взаимодействие солеобразующих группировок ионитов с ионами раствора ведет к образованию продуктов двух видов: 1) нормальных солей с ионным характером связи между остатком группировки ионита и ионом металла и 2) соединений с неионизованной связью.
Образованием соединений первого типа объясняют Б. П. Никольский и А. М. Трофимов [177] взаимодействие урана и тория с сульфокатионитом СБС. В основу опытов положено предположение, что избирательность поглощения зависит от концентрации водородных ионов в растворах солей урана и тория. Так, разделение этих элементов может быть осуществлено на смоле СБС из кислых растворов (0,5—1 N по НС1 или по HNO3) с избирательным поглощением тория, или из растворов с pH 3,8—5,0 с избирательным поглощением ионов уранила. Последний метод менее удобен вследствие узости интервала pH.
Детальное изучение Б. Н. Ласкориным [213] поглощения ура-нил-ионов некоторыми селективными катионитами дало основание сделать вывод о процессе комплексообразования уранил-ионов с функциональными группами смолы.
Кеннеди и Дэвис [643] установили избирательность катионитов, содержащих фосфорнокислые группы, по отношению к Th (IV), U (IV), U (VI) и Fe (Ш). Авторы предположили образование «хелатного» соединения типа:
R О OR
/ чМе^ /р/ R О OR
Избирательной сорбцией фосфорнокислым катионитом из 0,1—0,2 N растворов HNO3 уран может быть отделен отСа,Си(П), Со, но не от Fe (III), La (III), так как последние также сильно сорбируются катионитом из кислых растворов. Поэтому авторы при-
325
бегл и к использованию различий в устойчивости соединений, образуемых U (VI), La (III), Fe(III), Си (II), Со, Са с комплексоном III и с функциональными группами смолы.
На этом же принципе основана и выполненная несколько ранее работа 3. К. Михайловой (1956 г.) с той разницей, что автор использовала другой селективный катионит — Амберлит IRC-50. Как показали предварительные опыты, V, Мо, Fe, Си и др. в широком интервале pH 2—8 совершенно не сорбируются данным катионитом в присутствии комплексона III. С1“, NO3", 5О42-ионы не мешают сорбции урана из растворов, содержащих комплексон III. Фтор-ион при соотношении концентраций 10 понижает
сорбцию урана до 95,4%.
Методика определения содержания урана в рудах заключается в следующем. Навеску руды (1—3 г), содержащую целые проценты, десятые или сотые доли процента урана, разлагают соляной кислотой с добавлением Н2О2 или царской водкой с последующей обработкой нерастворимого осадка плавиковой кислотой. Избыток фтор-ионов удаляют двухкратным упариванием раствора с концентрированной HNOa. В полученный раствор (объем —50 мл) вносят навеску комплексона III (4-кратное количество по отношению к навеске руды). Устанавливают pH 5—8 и раствор пропускают через катионит Амберлит IRC-50 в Na +-форме (~10 мл смолы) со скоростью 0,78 см9'мин. Затем колонку промывают водой в течение 30 мин. Уран десорбируется 70 млЗ N H3SO4 со скоростью 1,56 см9]мин. Элюат поступает непосредственно в висмутовый редуктор, где происходит восстановление U (VI) до U (IV). Определение урана производится хроматометрическим методом. При содержании урана в навеске <2,5 мг элюат упаривают до —30 мл и восстановление урана осуществляют на кадмиевом редукторе, после чего применяют ванадатомет-рнческий метод определения. Общая продолжительность анализа 3—4 часа.
Распределительная хроматография
Разделение веществ методом распределительной хроматографии обусловлено различием коэффициентов распределе ния между двумя несмешивающимися растворителями.
Процесс разделения в общих чертах заключается в следующем.
Колонку заполняют твердым пористым веществом — «носителем», удерживающим на своей поверхности один из растворителей, так называемый «неподвижный» растворитель. Анализируемую смесь вносят в колонку, после чего начинается промывание ее чистым «подвижным» растворителем. Компоненты продвигаются вдоль колонки с различной скоростью. Наиболее медленно двигается компонент, обладающий наибольшим коэффициентом распределения К.
концентрация вещества в неподвижном растворителе, концентрация вещества в подвижном растворителе
В результате происходит образование отдельных зон чистых веществ, т. е. разделение смеси.
Теоретические положения распределительной хроматографии были разработаны Мартином и Синджем [154], Фуксом [270, 271].
В распределительной колоночной хроматографии движение зон в колонке может быть количественно охарактеризовано величиной отношения смещения максимума концентрации растворенного вещества к смещению за тот же промежуток времени свободной поверхности подвижного растворителя [154, 291].
326
Эта величина выражается уравнением:
R — А *-Ai+KAs ’ где
.4 — площадь поперечного сечения колонки;
Л t— площадь поперечного сечения подвижной жидкой фазы;
— площадь поперечного сечения неподвижной жидкой фазы;
К — коэффициент распределения.
Теория колоночной распределительной хроматографии может быть без труда распространена на случай бумажной распределительной хроматографии [203].
Так как в случае бумажной распределительной хроматографии величина R не может быть измерена, вводится новый коэффициент, представляющий отношение скорости перемещения зоны вещества к скорости движения фронта раствора:
п Ai
^-At^RAs'
и- А! Г 1 1
откуда — — 1
Значение величины R? и отношение равное отношению объемов жидких фаз, легко определить из опыта, что дает возможность вычислить R.
Два элемента могут быть разделены методом хроматографии на бумаге, если разница в значениях Rf превышает 0,1.
В качестве носителей в распределительной хроматографии могут быть использованы силикагель, крахмал, целлюлоза, фильтровальная бумага. Специфические требования к носителям сводятся к следующему [204]:
1) носители должны быть нейтральны, никакие химические и адсорбционные процессы на них по возможности не должны происходить;
2) они должны быть не растворимы в применяющихся растворителях;
3) должны обладать способностью удерживать на своей поверхности жидкую фазу, не смешивающуюся с применяемыми подвижными растворителями.
Разумеется, идеальных носителей, удовлетворяющих всем требованиям, на практике не существует.
Так, например, Блок и др. [17] указывают, что разделение смесей на фильтровальной бумаге зависит не только от распределения между растворителями, но и от адсорбции на поверхности, и от ионного обмена с полярными группами целлюлозы и с загрязнениями, присутствующими в последней.
Хроматография на бумаге
Для хроматографического разделения веществ каплю анализируемого раствора наносят на полосу фильтровальной бумаги, являющейся носителем водной фазы. После высыхания капли конец бумажной полосы помещают в подвижный растворитель,продвижение которого вдоль бумаги под действием капиллярных сил вызывает непрерывное перераспределение веществ между двумя фазами. В резуль-. тате различной скорости перемещения компонентов смеси в направлении движения растворителя происходит разделение веществ. Расположение отдельных компонентов на бумажной полосе выясняют затем опрыскиванием последней реактивами, образующими окрашенные соединения с анализируемыми веществами.
< Методы полуколичественного и количественного анализа элементов, разде-• ленных описываемым способом, включают в себя [71]:
1) метод сравнения с хроматографическими стандартами;
2) метод, основанный на существовании определенной зависимости между концентрацией хроматографируемого компонента и площадью его зоны локализации;
3) метод озоления локализованных на хроматограммах зон катионов;
327
4) метод извлечения локализованных на хроматограмме катионов с последующим определением экстрагированного вещества одним из аналитических методов.
Выбор бумаги для проведения анализа во многом определяет успешность разделения. Из отечественных сортов бумаги, применяемых в хроматографическом анализе, наилучшими являются фильтровальная бумага № 4, 5, «синяя лента ., из иностранных — бумага фирмы «Ватмана № 1, 2 и др., фирмы «Шлейхер и Шюлль».
В табл. 43, составленной на основании литературных данных, указано, от каких элементов и с помощью каких растворителей может быть отделен уран.
Высокая по сравнению с солями других элементов растворимость нитрата уранила в кислородсодержащих органических растворителях благоприятствует выделению его из смесей,
Арден, Бурсталл и Линстед [338] в одной из ранних работ получили хорошие результаты, применяя для извлечения урана методом бумажной хроматографии циклические эфиры в смеси с азотной кислотой: 2-метилтетрагидрофуран с 2,5% (по объему) HNO3 (уд. в. 1,42) и тетрагидропиран с 10% (по объему) HNO3 (уд. в. 1,42). Конечное определение урана производилось опрыскиванием бумаги ферроцианидом калия с последующим определением визуальным сравнением с хроматографическими стандартами. Метод позволяет обнаружить от 0,25 до 200 мкг урана с точностью ± 30% (отн.).
Хунт, Норф и Уэллс [620] разработали быстрый и простой метод определения урана в почвах и минералах при содержании U,O4 в последних от 0,003 до 0,0007%. В качестве растворителя использовался уксусноэтиловый эфир, содержащий 10% (по объему) HNO3 (уд. в. 1,42) и 5% (по объему) воды. Разделение занимает не более 15 мин. Методом визуального сравнения зоны, «проявленной^ ферроцианидом калия, с хроматографическими стандартами может быть обнаружено от 0,1 до 20 л/кг урана с точностью до 30 %, (отн).
Для более четкого разделения элементов иногда приходится прибегать к дополнительным приемам при хроматографировании на бумаге. Так, например, Ледерером [695, 696] было найдено при изучении значений ряда элементов в бутаноле, насыщенном различными кислотами, что предварительное насыщение фильтровальной бумаги нитратами сказывается на значении Rf для UCT% в то время как для Fe (III), Си (II), Со величины А’у практически не меняются (табл. 44).
Подавляющее большинство работ использует восходящий или нисходящий метод хроматографирования (т. е. поток растворителя движется по вертикально укрепленному листу бумаги, соответственно, снизу вверх или сверху вниз). В некоторых случаях авторы предлагают использовать метод «круговой» хроматографии, где капля анализируемого раствора наносится в центре бумажного диска, куда затем поступает и подвижный растворитель.
Этим методом Ю. В. Морачевский, М. Н. Гордеева и Т. Е. Kpyi-лова [161] отделили U от Fe (III) и V (V) с применением смеси ледяной уксусной и концентрированной азотной кислот (95 : 5)
328
Г аблг ца 4 >
Отделение урана от сопутствующих элементов методом бумажной хроматографии
Подвижный "С.С-творитель 1 1 1 Бумага ! 1 Элементы, отделившиеся т 1 урана (в скобках дань: значения R[) 1 Rt урана Ли те-rv.T' -
70% этилового эфира-4- —23% С2Н5ОН%- j -%7% концентрированной HNOS — i Fe (III) (0,14), Mg, Ca, Ni, Co. Zn, Мп (II), Al, V (V), Mo (VI), Pb, Си (II), Th i 0.84 [330]
80% изобутилметил-кетона-|-20% концентрированной HNO, Ватман № ЗММ Fe(III), Mg, Ca, Ni, Co, Al, V (V), Pb, Th, Ce, Cr(VI), Bi (III), Ti, Zr, Eu, Hg(II), Cd, Ba — [280 [
Циклогексанон, насыщенный 10%-ной HNO, № 2304в «Шлейхер и Шюлль» Fe (III), Ca, Mg, Al, Pb, Th, Се (IV) — [566!
Изобутилметилкетон, насыщенный 10%-ной HNO, № 2304в «Шлейхер и Шюлль» Fe (HI), Ca, Mg, Al. Pb, Th, Се (IV) — [566]
Метилэтилкетон, насы- — PuO22 + — [504]
щенный разбавленной HNO3
Ацетон: вода: концентрированная НС1 — 40:10:5 Ватман № 1 Fe (III) (0,984); Be (0,568); Al (0,211); Th (0,082); Zr (0,017); Ti (0,291); V (V) (0,360) 0,944 [727]
зопропиловый спирт: ацетилацетон:кои- Ватман № 1 Ti (1,00); Be (0,60) 0,83 [743]
центрированная НС1 = 6:2:2
Бутиловый спирт, насыщенный 1 N НС1 I 1 1 Ag (I) (0,0); Pb(0,0); Hg (II) (1,05); Bi (III) (0.65); Cu (II) (0,1); Cd (0,60); As (III) (0,7); Sb (III) (0,8); Sn (11) (0,95); Fe (HI) (0,12); Co (0,07); Ni(0,07); Мп (II) (0,09); Al (0,07); Cr (III) (0,07); Zn (0,76); Ca (0,03); Ba (0,0); Sr (0,0); Mg (0,11); Na (0,07); К | (0,08); Cs (0,08); Rb (0,08); TI (I) (0,0); TI (III) (1,11); j Mo (VI) (0,5); Th (0,03); i Be (0,30); In (0,33) [695]
Бутиловый спирт, насыщенный 3 N НС1 — Th (0,05—0,06) 0,19— 0,2 [8/ Ц
Бутиловый спирт, насыщенный 4 N HCI Th (0,13—0,16) 0,32— 0,33 [8/4]
329
Таблица 43 (окончание)
Подвижный растворитель Бумага Элементы, отделившиеся от урана (в скобках д^яы значения Rf) Rr ураьа i i 117% ' P -
Бутиловый спирт, насыщенный 10%-ной HNO3 Ag(l) (0.23): Pb (0.15); Bi (III) (0,27); Си (II) (0,17); Cd (0,19); Fe (III) (0,18); Co (0,17); Ni (0,17); Mn (II) (0,16); Al (0,11); Cr (III) (0,15); Zn (0,15); Ba (0,08); Sr (0,08); Cs (0,13); Rb (0,13); TI (I) (0,16); Th (0,1) 0,4 [695]
Изобутиловый спирт, насыщенный 4Л7 НС1 — Th (0,09); Pr (0,02) 9,31 [873]
Бутиловый спирт: 1 N HNO3 = 1:1 -h 0,30/6 бензоилацетона Ватман № 1 Th (0,03) 0,27 [816]
Бутиловый спирт, насыщенный 1,5АГ HNO3 Ватман № 1 * Fe (III) (0,12); Си (II) (0,1); Co (0,1) 0,88— 0,95 [696]
С2Н5ОН:4 N H2SO4 = =4:1 Ватман № 1 • Си (II) (0,41); Fe (III) (0,57) 0,72 [1008]
Ледяная уксусная кислота :концентрированная HNO3 = 95:5 —. V (V); Fe (III) — [161]
Метилэтилкетон -|--]-2,5% концентрированной HNO3; C2HSOH-|-10% 5 N НС1 Al, Cr (III), Fe (III), Ti, Zr ** [71]
s Бумага предварительно была пропитана нитратом натрия.
Катионы предварительно были осаждены на бумаге пиридином при pH 6,5 и разделены методом «двумерной хроматографии».
Таблица 44
Значения Rf для ряда элементов в зависимости от способа обработки бумаги (растворитель — бутанол, насыщенный 1,5 7VHNO3; бумага—-Ватман № 1)
Катионы ‘ Значения Rf
бумага не пропитана нитратом бумага пропитана К NO, бумага пропитана NH,NO, бумага пропитана NaNO3
! U (VI) 0,30 1 1 । 0,50 0,74 0,88—0,95
Fe (III) , 0,05 0,06 0,12 0,12
Си (II) ] 0,05 0,08 0,07 0,10
Со 0,07 1 0,07 0,07 0,10
336
в качестве подвижного растворителя. При содержании в исходном растворе от 5 до 550 мкг уран может быть отделен от 100—150-кратных количеств ванадия и железа. Относительная ошибка в определении каждого из трех элементов колеблется от 2 до 11 %.
Часто успешное разделение обеспечивается последовательным применением двух подвижных растворителей, передвигающихся по бумаге во взаимно-перпендикулярных направлениях («двумерная» хроматограмма); так, например, разделение Al, Cr, Fe, Ti, Zr и U, осажденных на бумаге пиридином при pH 6,5, Г. Д. Елисеева [71 ] осуществила путем последовательного применения метилэтилкетона, содержащего 2,5% (по объему) HNO3 (уд. в. 1,42), и этанола, содержащего 10 % (по объему) 5 N НС1.
Несмотря на ряд преимуществ метода бумажной хроматографии (возможность разделять чрезвычайно малые количества вещества, пор’ядка 1—0,1 мкг, простота проведения опыта и пр.) область применения его все же ограничена. Недостатки метода заключаются в невозможности осуществления разделения больших количеств веществ, в длительности процессов разделения, в недостаточной точности, так как определения сводятся главным образом к качественным и полуколичественным. Поэтому в анализе получил широкое распространение метод, восполняющий недостатки метода распределительной хроматографии на бумажных полосах — хроматографирование на целлюлозных колонках.
Хроматография на целлюлозных колонках
Сущность метода колоночной распределительной хроматографии была изложена выше.
Основная трудность практического применения этого метода заключается в приготовлении целлюлозной пульпы. Описываемые отдельными авторами [299, 399, 659] способы ее получения сходны между собой и в общих чертах заключаются в следующем.
Мел кона резанную фильтровальную бумагу (Ватман № 1, № 2 и др.) помещают в стакан с дистиллированной водой и перемешивают до полного превращения в однородную массу. После отсасывания пульпы на воронке Бюхнера ее помещают в кипящую 5%-ную азотную кислоту и кипятят некоторое время при постоянном перемешивании. Затем бумажную массу отсасывают на воронке Бюхнера, промывают последовательно водой, спиртом, эфиром, высушивают, измельчают и помещают в закрытый сосуд.
В отличие от хроматографии на бумажных полосах, использующей для отделения урана от примесей большое число разнообразных органических растворителей, в хроматографии на целлюлозных колонках для тех же целей используется почти исключительно диэтиловый эфир, содержащий азотную кислоту.
Бурсталл и Уэллс [399] одними из первых применили метод распределительной хроматографии на целлюлозных колонках для выделения урана из силикатных руд (содержание U3Og от 0,06 до 3,16%) и из монацита (содержание U3O8 от 0,05 до 0,48%).
331
При извлечении урана из целлюлозной колонки диэтиловым эфиром, содержащим 5°о (по объему) азотной кислоты (уд. в. 1,42) нитраты Li, Na, К, Rb, Cs, Си, Ag, Be, Mg, Ca, Sr, Ba, Ra, Zn, Cd, Al, Y, La, Ce, Pr, Nd, Sm, Eu, Ho, Er, Ga, In, TI, Ti,Hf,Ge,Sn, Pb, Nb, Ta,Cr, W, Те, Mn, Fe, Co, Ni остаются на колонке. Th, As, Mo частично экстрагируются вместе с ураном. Вымывание циркония и скандия может быть приостановлено добавлением щавелевой кислоты. Присутствие фосфорной кислоты замедляет движение урана по колонке. Поэтому для связывания фосфорной кислоты к исходному раствору добавляют трехвалентное железо. Сульфат-ионы в малых количествах не мешают проведению анализа. Присутствие галоидов нежелательно.
Анализ силикатных руд по методу Бурсталла и Уэллса [399] проводится следующим образом.
Колонку длиной 25 см, диаметром 2 см обрабатывают (СН3)2 S iC 12 для уменьшения смачиваемостилповерхности стекла, после чего заполняют целлюлозной пульпой. Навеску образца (2,5 г), взвешенную в платиновом тигле, обрабатывают 2 мл концентрированной HNO3 (уд. в. 1,42) и 5 мл HF. После выпаривания смеси досуха к остатку добавляют 10 мл концентрированной HNO3 и снова упаривают досуха. Затем остаток растворяют в 8 мл воды, подкисленной 2 мл концентрированной HNO3, и в полученный раствор вносят небольшое количество целлюлозной пульпы. Пульпу, пропитанную анализируемым раствором, переносят в верхнюю часть колонки и начинают промывать диэтиловым эфиром, содержащим 5% (по объему) концентрированной азотной кислоты. К элюату (100 мл) добавляют 50 мл воды, отгоняют растворитель, добавляют 5 мл H2SO4, 5 мл НС1О4 и упаривают до отдымления. Уран определяют либо объемным, либо колориметрическим методом.
Метод Адамса и Мека [299], разработанный для анализа ураносодержащих минералов, ничем существенно не отличается от метода Бурсталла и Уэллса, так же как и метод анализа Южноафриканских руд, описанный Леджем [66].
Работа Чирилли [423] по полярографическому, спектрофотометрическому и флуорометрическому определению урана в горных породах включает предварительное отделение от мешающих элементов на целлюлозных колонках, проводимое по методике Бурсталла и Уэллса.
Курама, Исихара и др. [135] описывают анализ урановых руд, содержащих < 0,01 % U3O8. От больших количеств сопутствующих элементов уран отделяется на целлюлозной колонке с использованием диэтилового эфира.
При проведении анализа образцов, содержащих значительные количества Th, As, Мо по методу Бурсталла и Уэллса [399] возникает ряд осложнений, так как уже было сказано выше, данные элементы частично переходят в элюат вместе с ураном. Экстрагируе-мость тория уменьшается со снижением содержания азотной кислоты в диэтиловом эфире. Исходя из этого можно, не изменяя принципа метода, отделить уран от тория, снизив содержание HNO3 в эфире до 3% по объему. Торий при этом остается в колонке и удаляется затем диэтиловым эфиром, содержащим 12,5% (по объему) HNO3 (уд. в. 1,42).
332
Описанный прием был применен Кембером [641] для разделения элементов в ураноторианите (содержание L’3OS от 13,3 до 38,4%).
Понизив концентрацию HNO3 в растворителе до 1 % (по объему), можно также достичь отделения урана от мышьяка и молибдена. Однако извлечение урана диэтиловым эфиром, содержащим столь малое количество азотной кислоты, не будет полным.
Поэтому Риан и Вильямс [865] предложили вариант метода Бурсталла и Уэллса на так называемой «совмещенной» колонке, состоящей из целлюлозы (нижний слой) и окиси алюминия (верхний слой). Дело в том, что мышьяк и молибден задерживаются окисью алюминия, а уран свободно проходит через носитель. Слой целлюлозы необходим потому, что присутствующие в образце железо, алюминий, ванадий и др. слабо удерживаются на Л12О3. Уран извлекают полностью диэтиловым эфиром, содержащим 5% (по объему) HNO3 (уд. в. 1,42). Этим методом был произведен анализ урана в образцах, содержащих от 10 до 20% мышьяка. Отклонение от данных анализа другими методами весьма незначительно при содержании U3O8 до 11,55%.
Применяя этот же метод «совмещенной» колонки для отделения урана от тория, Вильямс 11016]'рекомендует связывать мешающий цирконий фосфатами, а избыток фосфата, в свою очередь,— трехвалентным железом. Метод применим для анализа миллиграммовых количеств урана.
Опыты, в которых применялся эфир, содержащий 5% (по объему) HNO3 (уд. в. 1,42), дают хорошие результаты при концентрации урана не ниже 10"2%. При более низких концентрациях (порядка 10~3 — 10-4) выделение урана было неудовлетворительным.
Лебец и Останек [140] предложили изменить состав растворителя с таким расчетом, чтобы он содержал 20 мл петролейного эфира, 190 мл эфира и 10 мл азотной кислоты (уд. в. 1,42). При этом порцию раствора, если объем ее не превышал 10 мл, переносили непосредственно в колонку без предварительной сорбции небольшим количеством целлюлозы. Метод позволяет анализировать образцы, содержащие 10~3—10“4% урана.
Распределительная хроматография на силикагеле
По сравнению с распределительной хроматографией на целлюлозе незаслуженно мало внимания уделялось до сих пор распределительной хроматографии на силикагеле.
В настоящее время советскими учеными проделан ряд работ по распределительно-хроматографическому выделению урана на сили-кагельных колонках. В. К. Марков в своих работах отмечает, что при правильном снаряжении колонки силикагелем, смоченным не водой, как это пытались сделать Бурсталл и Уэллс, а подкисленным раствором высаливателя и применении соответствующего подвижного растворителя, можно получить полное количественное отделе
333
ние урана от сопутствующих элементов. При этом расход экстрагента значительно снижается по сравнению с разделением на целлюлозных колонках.
В. К- Марковым и сотрудниками разработаны методики изготовления стандартного химически чистого брикетированного силикагеля с заданной пористостью.
Отмеренный объем технической азотной кислоты разбавляют в 5 раз водопроводной водой. Технический силикат натрия разбавляют водопроводной водой в 4 раза и после суточного отстаивания сливают прозрачный раствор. Титрованием раствора силиката натрия раствором азотной кислоты по метиловому оранжевому устанавливают соотношение объемов, при которых происходит их нейтрализация. В керамический сосуд наливают такой объем азотной кислоты, который на 0,5— 1% больше, чем это необходимо для нейтрализации взятого объема силиката натрия. При энергичном перемешивании в сосуд медленно приливают раствор силиката натрия. Образовавшийся золь оставляют стоять в течение времени, необходимого для застудневания (1—2 суток). Гель считается созревшим, если он начал растрескиваться и отставать от стенок сосуда. Созревший гель разламывают на крупные куски и горячим 1—2%-ным раствором азотной кислоты промывают 2—3 раза в целях удаления избытка нитрата натрия. Затем гель отжимают от избыточной воды, перемешивают до состояния однородной клейкой массы и в целях его брикетирования продавливают через матрицу с заданным диаметром отверстий. Брикетированный гель проветривают в течение 30—40 мин. до тех пор, пока отдельные брикеты не перестанут прилипать друг к другу. После этого гель промывают 1—2%-ным раствором азотной кислоты, содержащим около 0,5% перекиси водорода. Перекись перестают добавлять в промывной раствор, когда гель и соприкасающийся с ним раствор перестанут окрашиваться в желтый цвет. Промывание азотной кислотой ведут до отрицательной реакции на Fe + 3 (по роданиду калия). Затем гель отмывают дистиллированной водой от избытка кислоты, переносят на стеклянные или плексигласовые листы и подсушивают при комнатной температуре до постоянного объема брикетов, после чего переносят в шкаф, где сушат сначала при 50—60 , а затем при 100—150°. Высушенный таким образом силикагель прокаливают в течение 30—60 мин. при 400— 450 в целях удаления органических веществ. Этим путем получают мелкопорпс-тый силикагель.
Для получения крупнопористого силикагеля отмытый от примесей брикетированный гель заливают 2—2,5%-ным раствором аммиака. Через 3—6 час. раствор аммиака сливают и гель отмывают дистиллированной водой доудаления запаха аммиака. Затем воду отсасывают с помощью водоструйного насоса, гель проветривают 2—3 часа на стеклянных или плексигласовых листах, сушат при 100—150е и прокаливают при 400—450° в течение 30—60 мин.
В табл. 45 дана характеристика получаемых данными способами силикагелей.
Таблица 45
Характеристика силикагелей
Силикагель Г равиме-трический УД- в., г: см- Объем пор, см , г I Преобладаю-। щий диаметр, А
Мелкопористый 0,65—0,70 0,30—0,35 -20
Крупнопористый 0,35—0,45 0,90-1,10 -100
i
334
В качестве примера распределительно-хроматографического отделения урана от примесей приведем метод анализа природных материалов (руд, минералов), предложенный В. К, Марковым и со
трудниками.
.Метод основан на том, что при распределении элементов между насыщенным водным раствором NH4NO3, подкисленным HNO3
и эфиром, численное значение коэффи-
циента распределения /\ = -° — сэфир.
для урана оказывается в десятки, сот-ни и тысячи раз меньше значений К большинства металлов.
Методика проведения анализа заключается в следующем. Навеску руды 0,5—1 г разлагают смесью азотной и плавиковой кислот. Раствор выпаривают досуха, затем выпаривание повторяют 2—3 раза с добавлением азотной кислоты, и, наконец, к сухому остатку приливают 1 мл 0,5 М по HNO., насыщенного раствора NH4NO3. В полученный раствор вносят 3 мл сухого мелкозернистого крупнопористого силикагеля (с диаметром зерен 6.5 мм). Пропитанный анализируемым раствором силикагель переносят в колонку на «очистительный слой» (рис. 56), состоящий из 6 мл силикагеля, пропитанного 2 мл 0,5 М по HNO3 насыщенного раствора нитрата аммония (т. е. неподвижным растворителем). Степки посуды, в которой проводилось разложение образца, обмывают 0,5 мл неподвижного растворителя, раствор смешивают с 1,5—2 мл силикагеля и также переносят в колонку. Слой силикагеля, пропитанный анализируемым раствором, называют «распределительным».
Уран вымывают диэтиловым эфиром, приведенным предварительно в равновесие по содержанию азотной кислоты с неподвижным растворителем. Элюат собирают в стаканчик емкостью 50—100 мл, в который наливают 5— 10 ль?воды. Кончик сливной трубки колонки
Рис. 56. Распределительно-хроматографические колонки.
а— стеклянная вата; в— очистительный слой; с— распределительный слой.
для немедленной реэкстракции урана погружают в воду. Конец экстрагирования урана определяют по отсутствию свечения в ультрафиолетовом свете перла NaF, смоченного вытекающим из колонки эфиром и прокаленного в окислительном пламени. Экстракт выпаривают
До удаления эфира, и в водном реэкстракте
определяют уран (в зависимости от его содержания) любым аналитическим методом.
Если в пробе присутствуют нитраты Th, Zr, Se, Се (IV), кислотность подвижного и неподвижного растворителей не должна превышать 0,3—0,4 моля/л, так как повышение кислотности ведет к вымыванию указанных элементов вместе с ураном. Се (IV) восстанавливают до трехвалентного, добавляя к анализируемому раствору 2 мл 6%-ной Н2О2 и упаривая раствор досуха. Присутствие вана
335
дия в пробе требует тщательной очистки эфира от перекисей. В противном случае ванадий не будет удерживаться на колонке. С 1~-ионы способствуют вымыванию Fe3+ и Sn. POI- могут задерживать экстракцию урана. Поэтому их связывают, добавляя в пробу избыток Fe(NO3)3. От мешающих анионов можно освободиться, проводя аммиачное осаждение разложенной пробы с последующим растворением осадка в минимальном количестве HNO3.
При содержании урана в рудах порядка сотых долей и целых процентов ошибка определения составляет тысячные и сотые доли процента соответственно.
На том же принципе основан распределительно-хроматографический метод очистки урана в целях использования последнего в качестве спектрально чистой основы для изготовления эталонов.
Очистку проводят на колонке с внутренним диаметром 2 см. 15 мл химически чистого крупнопористого силикагеля с диаметром зерен 0,5 мм смачивают 5 мл насыщенного раствора NH4NO3, 0,5 М по HNO3, после чего силикагель переносят в колонку.
Анализируемый раствор, содержащий 10 г урана и 1% примесей других металлов (Со, N1, Сг, Мп, Mg, Fe, Си, Al — по 0,1% каждого) упаривают до образования влажных кристаллов уранилнитрата, которые растворяют затем в 20—25 мл диэтилового эфира, 0,65 М по азотной кислоте. Полученный раствор медленно фильтруют через колонку. Затем колонку промывают 100 мл диэтилового эфира, 0.65 М по азотной кислоте. При добавлении к элюату насыщенного раствора Н2С2О4 осаждается оксалат уранила, который после отсасывания на фильтре и подсушивания под лампой переносят в платиновый тигель и прокаливают в муфеле до образования закиси-окиси.
Чистота полученных проб закиси-окиси была проверена спектральным методом.
В последнее время в литературе появились работы иностранных авторов по отделению урана от различных элементов методом распределительной хроматографии на силикагеле.
Хефнер и Хультгрен [565] разработали метод отделения урана от плутония, используя для этой цели либо дибутиловый эфир этиленгликоля, либо смесь его с керосином.
В обоих случаях растворитель содержал азотную кислоту.
Облученный уран растворяют в азотной кислоте и полученный раствор подвергают противоточной экстракции дибутиловым эфиром этиленгликоля. Органический слой, содержащий уран, плутоний и небольшое количество продуктов деления, разбавляют на Ч3 объема керосином и затем насыщенной водой смесью дибутилового эфира этиленгликоля с керосином в отношении (3 : 1) до достижения 0,35 М концентрации по азотной кислоте.
Полученный раствор пропускают через колонку, причем весь плутоний задерживается в колонке, а уран почти полностью переходит в фильтрат. Небольшое количество урана, оставшееся в колонке, извлекают 0,3—0,35 М раствором азотной кислоты в смеси дибутилового эфира этиленгликоля с керосином (3 : !)• Плутоний вымывают 0,5—1,0 М раствором азотной кислоты в органическом растворителе.
При применении в качестве подвижного растворителя дибутилового эфира этиленгликоля, 0,15—0,2 М по азотной кислоте, методика не меняется.
Приведенным способом, однако, не удается добиться полной очистки урана и плутония от продуктов деления.
336
Эти же авторы [619] описали процесс разделения урана и плутония, применяя в качестве подвижного растворителя ТБФ-30, состоящий из 30% по объему ТБФ в инертном углеводороде.
Неподвижная водная фаза, удерживаемая силикагелем, содержала 1 г Fe (II) на 1 л раствора, 0,3 .\1 по гидразиннитрату и 0,1 Л1 по Н.\О3. При пропускании анализируемого раствора через колонку плутоний переходит в трехвалентное состояние и двигается по колонке очень медленно, в то время как уран быстро переходит в элюат. Оставшийся уран элюируют раствором ТБФ-30, 0,02 Л по HNO3. Плутоний удаляют из колонки раствором HNO3.
Когда раствор содержит только малые концентрации урана, порядка нескольких граммов на литр,нет необходимости вводить в неподвижный растворитель восстанавливающий агент. Плутоний в этом случае элюируют раствором ТБФ-30, 0,2 М по HNO3.
Цветичанин [438] отделил Pu (VI), Pu (IV) иП (VI) от циркония и ниобия.
Исходный раствор плутония, урана и примесей в метилизобутилкетоне, насыщенном 2 N HNO3, пропускают через колонку силикагеля, приготовленного по способу Гордона, Мартина и Синджа [550]. В процессе промывания колонки раствором метилизобутилкетона, насыщенного 2 N HNO3, уран и плутоний проходят в элюат, a Zr95 и Nb95 нацело удерживаются в колонке.
4. ЭЛЕКТРОЛИТИЧЕСКИЕ МЕТОДЫ ОТДЕЛЕНИЯ УРАНА
Н. И. Гусев
К настоящему времени имеется значительное число работ, посвященных электролитическим методам отделения урана от сопутствующих примесей. К таким методам относятся: 1) электролиз на ртутном катоде; 2) электроосаждение урана на платиновом катоде, а также на катоде из других металлов с внешним приложением потенциала и 3) внутренний электролиз.
Электролиз на ртутном катоде
Длительной практикой выработаны оптимальные условия электролитической очистки сернокислых растворов урана на ртутном катоде [8, стр. 327]: объем электролита равен 50 мл; концентрация свободной серной кислоты — 1 N; ток 10 а (как правило 0,5 а); анод — платиновая спираль или сетка, точно контактирующая с поверхностью электролита; поверхность катода 20 см2; быстрое перемешивание поверхностного слоя ртути; температура 25—40° (достаточно охлаждение обыкновенной водопроводной водой); содержание урана =с0,25 г.
Время, необходимое для полного отделения урана от примесей, может колебаться в широких пределах в зависимости от их концентрации. Так, при силе тока 5 а и прочих оптимальных условиях требуется около 70 мин. для количественного осаждения 1 г железа в присутствии 0,25 г урана. Наличие больших количеств других осаждающихся катионов увеличивает время электролиза до 3—4
Аналитическая химия урана 33/
часов. Полноту отделения какого-либо элемента обычно проверяют, используя специфические чувствительные реакции на данный катион.
Вместо серной употребляется также хлорная кислота.
Электролиз из сернокислых сред
Катодное восстановление урана в сернокислом растворе идет следующим образом:
UOf+4Н++2е-->и*++2Н,О Катодные (1)
U4++e-—>U’+ J реакции (2)
Н2О_2г--.2Н* + */,О! 1 днне (3)
7гн,о —е-->Н+ + 7.О2 / (4)
Реакция (1) протекает одновременно с реакцией (3), а реакция (2) — с (4). Отсюда следует, что если первоначальное количество Н+-ионов менее 2 молей на 1 моль урана (VI), то выпадет осадок гидратированной окиси урана (IV).
На скорость электролитического выделения примесей оказывает влияние целый ряд факторов.
С ростом концентрации свободной H2SO4 увеличивается время, необходимое для электроосаждения Fe, Сг, Мп, Mo, Ni. Так, время, необходимое для полного восстановления железа Fe (III)-*Fe (И)-* -> Fe (0) возрастает почти в два раза с увеличением кислотности с 1 N до 5 V.
Найдено, что с повышением температуры (>25—40°) скорость выделения примесей резко падает.
Такой же эффект наблюдается с увеличением объема электролита, глубины погружения анода в раствор, с уменьшением степени очистки катодной ртути.
Отделение анодного пространства от основного объема электролита пористой перегородкой значительно уменьшает скорость отделения примесей.
Присутствие в электролите больших количеств урана (2—10 г) сильно замедляет электролитическое выделение железа и почти полностью предотвращает осаждение хрома и молибдена. Это объясняется затратой времени, необходимого для протекания реакции (1), а также действием урана как окислительно-восстановительного буфера.
Увеличение плотности тока влечет за собой увеличение скорости выделения таких, например, элементов, как железо, хром и молибден; однако ток около 10 а при прочих оптимальных условиях вызывает интенсивное газовыделение (реакции (3) и (4)) и разбрызгивание электролита.
Увеличение концентрации железа и хрома ведет к возрастанию скорости выделения этих элементов до концентрации железа 20 г/л, а хрома — 10 г/л (сила тока 3—4 а при оптимальных условиях).
338
Перемешивание поверхностного слоя ртути гораздо эффективнее, нежели перемешивание электролита.
Электролиз разбавленных сернокислых растворов обеспечивает количественное выделение Bi, Cd, Сг, Со, Ga, Ge, Au, In, Ir, Fe, Hg, Mo, Ni, Pd, Pt, Po, Re, Rh, Ag, TI, Sn и Zn. Количественно выделяясь из раствора, мышьяк, свинец, осмий и селен не переходят полностью в катодную ртуть. Сурьма, марганец и рутений осаждаются неполностью, а остальные элементы совершенно не осаждаются из кислого раствора на ртутном катоде [8, стр. 58].
В присутствии урана Сг, Си, Fe, Mo, Ni и Zn осаждаются в порядке, соответствующем возрастанию окислительно-восстановительных потенциалов, за исключением цинка, а время, необходимое для очистки урана от перечисленных катионов, приблизительно равно сумме времен электролиза каждого из этих катионов.
Мешающее влияние на процесс оказывают анионы С1“, NO ’ и РО*-
К недостаткам метода электролиза на ртутном катоде из разбавленных сернокислых растворов следует отнести то, что этот процесс не приводит к удалению таких часто сопутствующих урану элементов, как алюминий, титан и ванадий; продолжительность электролиза велика, когда^надо удалить большое количество примесей.
Необходимо отметить, что этот метод иногда используется как промежуточная стадия отделения урана от примесей. Тот факт, что сульфат U (IV) незначительно растворим в концентрированных растворах НС104, в то время как большинство других катионов обладает достаточно высокой растворимостью, был использован для осаждения и отделения электролитически восстановленного урана от небольших количеств V, Мп, А1, Мо в виде U (SO4)2 из 48 %-ного раствора по хлорной кислоте.
Метод включает следующие операции: растворение образца в царской водке, перевод его в сернокислый раствор, электролиз, осаждение U(SO4)2 добавкой двукратного объема 72% -ной НСЮ4, фильтрование на стеклянном фильтре № 4 и промывка НС1О4, растворение на фильтре водой и титрование 0,05 N КМпО4.
Ошибка метода для образцов закиси-окиси в 1 а не превышает ±0,3% (отн.) (М. А. Драгомирова, 1952 г.).
Электролиз в хлорнокислой среде
Некоторыми исследователями была опробована хлорнокислая среда при оптимальных для серной кислоты условиях [8, стр. 336]. Было найдено, что для полного выделения Fe, Си, Ni, Сг и Zn требуется несколько больше времени по сравнению с сернокислым раствором.
Этот метод был применен [674] для очистки очень малых, по-ви-Димому, количеств урана от железа, хрома, марганца (частично) с последующим осаждением урана на A1(OH)S аммиаком, экстракцией растворенного в HNOS осадка этилацетатом и флуоресцентным определением урана в 1 М серной кислоте.
22*
339
Электроосаждение урана на твердых металлических катодах с внешним приложением потенциала
Многие варианты этого метода позволяют проводить количест
венное выделение урана, но не гарантируют чистоты катодного
осадка.
Исследования в этой области проводились как с миллиграммовыми, так и с микрограммовыми количествами урана в слабокислых, нейтральных и щелочных средах.
В качестве среды использовались буферные ацетатные и карбонатные растворы [8, стр. 59]. Уран (порядка нескольких миллиграммов) выделяли на катоде в виде окисла, химическая и физическая природа которого зависела от условий осаждения. Этот осадок не пригоден для электровесового определения урана, и метод используют для изотопного анализа урана путем измерения интенсивности «-излучения. Лучшими электролитами для получения тонких, плотных и равномерных пленок являются растворы CH3COONa%-СН3СООН, (NH4)2C2O4HNaF. Осаждениеиз оксалатной среды лучше, чем из ацетатной, так как точность взвешивания пленки составляет 0,1% вместо 0,3%, а методика в первом случае несколько проще [8, стр. 348—350]. Количественное электроосаждение и воспроизводимые результаты получены для микрограммовых количеств урана при использовании раствора оксалата аммония [860].
0,033—0,13 мкг U233 выделяют с 20 мкг природного урана как носителя на '94ДсЗ% по следующей методике: к смеси, содержащей исследуемый образец и носитель, добавляют 5 мл насыщенного водного раствора (NH4)C2O4, 1 каплю 6 7И НС1, 1 каплю метилового красного и такое количество 6 М NH4OH, чтобы раствор окрасился в желтый цвет. Затем добавляют азотную кислоту (6 М) до тех пор, пока окраска индикатора не станет розовой, а также избыток 2—3 капли. Объем раствора для электролиза ~20 мл. Ячейку помещают в водяную баню (температура 80—85°), и проводят электролиз при вращении спирального платинового анода в течение 30—45 мин., используя ток 0,8—1,0 а при 9 в, время от времени добавляя азотную кислоту до розового цвета раствора. Затем добавляют 1 каплю 0,01%-ного фенолфталеина, 6 М NH4OH до розового цвета раствора и продолжают осаждение в течение 60—75 мин. при прочих прежних условиях. Промывают диск (0,15 мм толщины и 26 мм диаметра), прокаливают осадок до U3O8 и производят а-изме-рения.
Авторы считают, что оксалатная среда лучше лимонной и винной.
Осаждение микрограммовых количеств урана на никелевом диске из раствора оксалата аммония при pH 9,6 (ток 3 а) в течение часа проводится на 85±4% (цит. по 860).
А. Г. Самарцевой [218] выработаны следующие оптимальные условия выделения микроколичеств урана (на примере U233) из слабо-
кислых растворов: Кислотность...............................0,001—0,01 М по HNO,
Катодная плотность тока .... 100 ма[см2 Объем электролита...............20 мл
Комнатная температура Время...........................3 часа
340
После электролиза осадок урана промывают спиртом, прокаливают и производят измерение активности с обеих сторон платинового диска с поверхностью 1 см2 в специальной камере.
Осаждение урана [228] связано с тем, что в прикатодной области за счет повышения концентрации ОН “-ионов образуются гидратированные окислы урана, которые переносятся к катоду, восстанавливаются на нем и осаждаются в виде частично дегидратированных гидроокисей переменного состава. В более кислой среде необходимо большее подщелачивание, а следовательно, и большая плотность тока. Авторами найдено, что выделение урана не зависит от материала катода и происходит только после достижения pH осаждения гидроокиси в прикатодной области независимо от pH исходного раствора.
Имеются также данные по электролитическому выделению урана из щелочных растворов перуранатов [62, 64].
5 мл раствора уранилнитрата (2—30 мг урана) добавляют к 50 мл щелочи (КОН или NaOH), смешанной с 0,5 мл 30%-ной перекиси водорода. Раствор помещают в платиновую чашку диаметром 8 см и разводят 1 щелочью до 100 мл. Осаждение проводят при 80—100° и плотности тока 0,7 ма]см2 в течение 1,5—2 час. ;
Указанная методика применялась для выделения урана из следующей смеси: 11 мг урана, 50 мг железа, 1 г алюминия и 1 г Na2HPO4. Кислый раствор смеси был прилит к КОН (количество КОН рассчитывалось на переведение соли алюминия в алюминат с получением в растворе 5%-ного избытка щелочи). Раствор кипятили, гидроокись железа отделяли фильтрованием, и из щелочного раствора, разведенного до 100 мл водой, за 1,5 часа выделялся уран (найдено 99—101%).
Полное выделение урана происходит также из раствора, содержащего значительные количества Н2С2О4, HF, K2SO4, Na2SO4 и ацетат-иона. Не удаляются даже повторным электролизом следы ванадия. Окислители (большой избыток Н2О2 и К2СгО4) мешают электроосаждению урана, указывая на то, что процесс выделения связан с восстановлением урана на катоде. Теми же авторами изучались кинетика осаждения и состав осадка.
Похожая методика осаждения урана использовалась для отделения меди, свинца, молибдена (и перечисленных выше катионов) с целью последующего полярографического определения урана. Авторами найдено, что медь при указанных условиях осаждается на катоде, а ванадий и молибден сорбируются осадком урана. Это, естественно, приводит к завышенным результатам полярографического анализа (В. А. Заринский, Т. М. Чубукова, 1953 г.).
Внутренний электролиз
Этот метод позволяет количественно выделять уран при pH 5—5,2 из буферных растворов (объем --100 мл) Н2С4Н4О4-р Na2B4O7 и СН3СООЫа4-СН,СООН в виде UO2-2Н2О [165]. Анод — цинковый и кадмиевый стержни (диаметром 0,8 см), катод — платиновая сеточ
341
ка. Электролиз проводили при 60—80° в атмосфере СО2. Тот факт, что молибден в растворе уксуснокислого буфера осаждается на катоде, был использован для электроосаждения урана (0,2—2 мг) на молибдене как коллекторе, который вводится в раствор в количествах, несколько превышающих содержание урана. Введение молибдена позволяет выделить уран из растворов, содержащих 100—200-кратные количества железа, в течение 3 час. Добавка молибдена не обязательная, если отношение U: Fe^l : 10.
Исследования, проведенные в уксуснокислом буфере 1163], показали, что ванадий выделяется на платиновом катоде вслед за ураном, и количественное осаждение ванадия происходит лишь при малых его содержаниях. Для полного удержания ванадия (содержание U= 1,2 мг, U : Vs^l: 15) в растворе успешно использовался комплексон III. В процессе электролиза ванадий, восстановленный до V (IV), связывали в прочный комплекс с комплексоном III, и он не выделялся на катоде (найдено 1,13—1,16 мг U). Электроосаждение урана (1—2 мг) происходит на 95—98% [9] в присутствии того же комплексообразующего вещества из растворов, содержащих алюминий (U : А1;& 1 : 100), хром (U : Cr^l : 10), кобальт (U : Ох>1 : 25) и никель (U : Ni^l : 50).
Г лава VI
ОПРЕДЕЛЕНИЕ УРАНА В ПРИРОДНЫХ И ПРОМЫШЛЕННЫХ ОБЪЕКТАХ
Н. И. Удальцова
I. ОПРЕДЕЛЕНИЕ УРАНА В МИНЕРАЛАХ, РУДАХ И ГОРНЫХ ПОРОДАХ
МЕТОДЫ РАЗЛОЖЕНИЯ
Процесс извлечения и переведения урана в раствор из урановых минералов и руд является очень важной ступенью при количественном определении урана. Этот процесс осложняется тем, что руды наряду с минералами, содержащими уран, включают в себя значительно большие количества минералов других элементов.
Урановые минералы, как известно, по своему происхождению делятся на первичные и вторичные. Первичные минералы в большинстве представляют собой соединения окислов урана (IV), титана, железа, тантала, ниобия, редкоземельных элементов и др. К их числу относятся уранинит с его разновидностями, урановая смолка и др.; все первичные минералы урана хорошо растворяются в разбавленной и концентрированной HNO3, а также в НС1 и H2SO4 в присутствии окислителя.
Вторичные минералы образовались в результате изменения или разрушения первичных минералов. По составу это фосфаты, карбонаты, арсенаты, сульфаты, ванадаты, землистые окислы и др., содержащие уран (VI); наиболее распространенные из них урановые черни, отенит, уранофан, карнотит, шрекингерит, казолит, торбернит, тюямунит. Вторичные минералы урана также хорошо растворимы в минеральных кислотах.
Среди урановых руд чаще всего встречаются гидроалюмосили-катные руды, содержащие более 50% SiO2 и карбонатно-гематитомагнетитовые руды, содержащие большие количества Fe (до 70%); реже встречаются урансодержащие угли, включающие более 50% углерода, а также известняки, содержащие значительные количе
343
ства (более 40%) карбоната кальция. Уран в рудах представлен главным образом урановой смолкой, уранинитом, торбернитом, уранофаном, реже другими минералами; кроме урана, руды могут содержать Ti, Al, Fe, Мп, Mg, Ca, Na, К, Сг, P, V, W, S, Bi, Pb, Zn, Cu, Co, Ni, As, реже Zr, Nb, Та и др.
Выбор того или иного метода разложения руды или породы зависит от минералогического и химического состава образца, а также от характера распределения урана в породе. Необходимым условием быстрого и количественного извлечения урана в раствор является предварительное измельчение руды или породы; обычно руду растирают в тонкий порошок (до 150—200 меш). Выбор навески руды или породы зависит от содержания урана в образце, а также от того, какой метод определения урана будет далее применен (большей частью работают с навесками 0,2—3 г).
Если руда содержит органические вещества, то перед разложением ее следует прокалить при 500—700°; прокаливать руду при очень высокой температуре (1000—1100°) не рекомендуется, так как после такого прокаливания уран труднее извлекается в раствор. Обычно урановые руды разлагают обработкой сильными кислотами: HNO3, НС1, HBr, НС1О4, H2SO4 или же их смесями в присутствии окислителя; при этом уран в большинстве случаев количественно переходит в раствор.
Руды с высоким содержанием силикатов разлагают плавиковой кислотой, однако при этом некоторые силикаты полностью не разлагаются, например циркон.
Оставшийся после разложения руды сильными кислотами и плавиковой кислотой остаток, содержащий Zr, Th, Ti и др., может быть переведен в раствор путем сплавления со щелочами, карбонатами, перекисью натрия, бисульфатом и персульфатом, кислыми фторидами и др.
Для полного разложения почти всех урановых руд и пород может быть рекомендован следующий метод: соответствующую навеску прокаленной при 500— 600° руды обрабатывают концентрированной HNO3 или царской водкой при нагревании; неразложившийся остаток отфильтровывают, промывают; затем озоляют фильтр и прокаливают при 500—600°. Прокаленный остаток обрабатывают концентрированной HF с добавлением небольшого количества HNO3 или H2SO4; если необходимо, обработку плавиковой кислотой повторяют несколько раз до полного разложения силикатов. Так как при дальнейшем отделении и определении урана в большинстве случаев присутствие Р~-иона нежелательно, то после разложения руды плавиковой кислотой фториды переводят в сульфаты, перхлораты или нитраты выпариванием с H,SO4, НС1О4 или HNO3. Полученные соли растворяют в разбавленной HNO3; если при этом остается нерастворимый остаток, то его отфильтровывают, а затем разлагают сплавлением с бисульфатом калия.
Однако данный метод полного разложения руды используют лишь в случае очень точного определения урана. Обычно метод извлечения урана в раствор можно упростить в зависимости от химического и минералогического состава руды, а также от последующего способа определения урана.
344
Из всех многочисленных методов разложения урановых руд и пород прежде всего следует остановиться на методе разложения концентрированной НС1 в присутствии окислителей (чаще всего Н2О2) при нагревании. Как показали исследования П. А. Волкова (1953 г.), уран практически полностью извлекается из большого числа руд (известняковых, железистых, углей, ряда силикатных), а также из изверженных горных пород [143] обработкой концентрированной НС1 в присутствии Н2О2; остающийся при этом нерастворимый остаток содержит менее 1% от всего урана.
Разложение руды проводят следующим образом: к 0,5—3 г руды приливают 30—40 .мл концентрированной НС1, 2—5 .мл 30%-ного раствора Н2О2 и кипятят в течение 30—40 мин.
Хорошим методом извлечения урана из песчаников является нагревание тонкоизмельченного образца с концентрированной НС1 в запаянной стеклянной ампуле при 180—200° в течение 2 час [127].
В литературе подробно описан метод извлечения урана из руд и пород обработкой концентрированной H2SO4 или смесью H2SO4 и HNO3 [8, 70, 384, 488].
При анализе урано-танталовых и урано-ниобиевых руд их обычно разлагают смесью HNO3 и HF при нагревании [753, 943]; этот метод разложения применяют и в других случаях [687].
Для разложения руд и пород можно использовать сплавление их со щелочами, Na2O2, карбонатами и др. плавнями [143]; так, например, при определении урана в циркониевых минералах, в частности в цирконе, образец разлагают сплавлением со смесью Na2CO4 и Na2B4O7 [441].
При выполнении массовых анализов для определения урана в рудах гидротермальных и осадочных месторождений в полевых условиях удобным методом извлечения урана в раствор может оказаться обработка руды на холоде 0,1 % -ным раствором H2SO4. Исследования П. Жарова (1955 г.) показали, что при обработке разбавленной H2SO4 уран, входящий в черни, коломорфную смоляную руду, полностью переходит в раствор (к 0,1 г руды приливают 100 мл 0,1 %-ного раствора H2SO4 и оставляют стоять 12 час.). Следует отметить, однако, что этот метод разложения может быть использован при условии предварительной проверки его применимости к данному типу руды.
МЕТОДЫ ОТДЕЛЕНИЯ УРАНА ОТ ДРУГИХ ЭЛЕМЕНТОВ И МЕТОДЫ ЕГО ОПРЕДЕЛЕНИЯ
Отсутствие специфических реакций для определения урана обусловливает необходимость отделения его от сопутствующих элементов.
Для отделения урана от примесей используют методы осаждения, хроматографические, ионообменные, экстракционные, элек-
345
тролитические и др., подробно описанные выше (см. главу «Методы отделения урана»). Выбор метода отделения урана от сопутствующих элементов зависит от ряда факторов, таких как количество урана, свойства и количества присутствующих примесей, степень отделения примесей, требуемая точность определения урана и др.
Вопросу определения урана в рудах, породах и других материалах посвящен ряд литературных обзоров и монографий [183, 184, 558, 852, 853, 937, 1036].
Одним из распространенных методов отделения урана от сопутствующих примесей является осаждение фосфата четырехвалентного урана из кислых растворов. Исследования, проведенные П. А. Волковым, показали, что данный метод позволяет отделить уран от большинства элементов, кроме Th, Zr, частично Ti. Метод пригоден для отделения как больших, так и малых количеств урана (можно выделить до 1 мкг U из раствора объемом 200 мл, если в качестве соосадителя использовать соли Th или Zr).
Для определения урана в большом числе бедных руд П. А. Волковым рекомендуется следующий метод:
1—3 г руды помещают в стакан емкостью 200—250 мл, приливают 40 мл концентрированной НС 1,2—3 мл Н2О2 и 10 мл 10%-ного раствора Na2HPO4. Через 5—10 мин. раствор нагревают до кипения и кипятят в течение 40—60 мин. (конечный объем раствора должен быть не менее 15 мл). Затем приливают 50—60 мл горячей воды, 10 мл 20% -ного раствора тиосульфата натрия и кипятят в течение 10—15 мин. (при этом происходит восстановление Fe (III) до Fe (II) и выделение сульфидов Си, Pb, Bi и др.). Осадок отфильтровывают, промывают 2%-ным раствором НС1 и отбрасывают. Фильтрат охлаждают в течение 5—10 мин. и нейтрализуют аммиаком по индикатору метаниловый желтый.
После этого приливают 3,5 мл концентрированной НС1, разбавляют водой до 150 мл, добавляют 0,5—0,7 г гидросульфита натрия и 4 мл 0,5%-ного раствора ThCl4; при этом происходит осаждение фосфата урана (IV). Раствор оставляют стоять не менее часа (лучше оставлять на ночь), изредка перемешивая стеклянной палочкой для коагуляции осадка.
Осадок отфильтровывают через бумажный фильтр (или стеклянный пористый тигель № 4) под вакуумом, промывают 0,2%-ным раствором НС1 до полного отмывания от продуктов разложения гидросульфита натрия (что проверяют по обесцвечиванию 0,01 N раствора КМпО4) и растворяют на фильтре в 40—50 мл 33% -ного раствора H2SO4. К полученному раствору прибавляют 1—2 мл концентрированной Н3РО4, 1 каплю 0,5% -ного раствора дифениламинсульфоната бария, 1 каплю 0,3% -ного раствора фенилантраниловой кислоты и титруют стандартным (0,04— 0,004 V) раствором NH4VO3 до появления слабой сине-фиолетовой окраски, устойчивой в течение нескольких минут.
Для восстановления урана до четырехвалентного состояния, кроме гидросульфита натрия, могут быть использованы также ронгалит и двухвалентный хром.
При определении урана по описанному выше методу могут возникнуть осложнения только при анализе материалов, содержащих значительные количества Th, Zr, Mo, Си.
Для отделения урана от Fe, Ca, Mg, Ti и других элементов находит применение обработка раствором карбоната натрия; однако при анализе руд, содержащих большие количества кальция, осадок захватывает в значительной степени уран, вероятно, в виде мало-.346
растворимого Ca2[UO2(CO3)3]. Поэтому при анализах известняков, известковых глин и других рекомендуют отделять уран от Са осаждением урана вместе с Al, Fe и другими элементами аммиаком или пиридином. Если же в образце присутствуют большие количества фосфата, то описанный выше метод отделения от Са не применим, так как в этих условиях происходит выделение фосфата кальция. В.М.Звенигородская, Л.С. Василевская и Т. В. Лейкина предлагают при определении урана в фосфоритах отделять уран (вместе с А1, Fe) осаждением фосфатом в слабоуксуснокислой среде (pH 4,5—5); при этом Са, Си, Ni,Mn, V, Мо и другие остаются в растворе; уран затем можно определить колориметрически с Н2О2 [157].
При анализе руд и других материалов, содержащих большие количества ванадия, уран отделяют осаждением фосфата уранила или выделяют уран в виде труднорастворимого урано-ванадата кальция из уксуснокислого раствора, а затем отделяют уран от ванадия осаждением фосфата уранила (см. подробнее стр. 267). Применение комплексона III в качестве маскирующего агента при осаждении уранила в виде фосфата позволяет отделить уран не только от V, но и от Fe, Al, Cr (III), Ni, Co, редкоземельных элементов, Cr (VI) и др. Выделение двойных фторидов, например NaF-UF4 или UF4, соосаждением его с CaF2 из кислых растворов дает возможность отделить уран от Zr, Та, Ti, Мо и др., что может быть использовано при определении его в рудах, содержащих большие количества Zr, Та и др.
Хроматографические, ионообменные и экстракционные методы отделения урана от сопутствующих элементов находят в настоящее время наибольшее применение (см. соответствующие разделы). Как правило, эти методы отделения могут быть использованы при определении урана в большинстве руд и других материалов. Например, метод распределительно-хроматографического отделения, разработанный В. К. Марковым (см. раздел «Хроматографическое отделение урана»), позволяет количественно выделить уран без примесей практически из всех руд, пород и других образцов.
Для количественного определения урана после его отделения от примесей применяются химические методы (весовые [70, 753], объемные [165, 517, 1026], физико-химические методы (фотометрические [377, 421, 426, 488, 687, 1018], люминесцентные [143, 238, 300, 441, 558], полярографические [243, 672, 904, 943, 944, 951, 1052], кулонометрические, амперометрические и др.), а также физические методы (спектральные [166, 167, 442], радиометрические [72, 225, 655, 925], рентгеноспектральные, радиоактивационные и др.). Все методы количественного определения урана подробно описаны в главе IV, стр. 55.
347
2. ОПРЕДЕЛЕНИЕ УРАНА В ПРОМЫШЛЕННЫХ КОНЦЕНТРАТАХ, ПОЛУПРОДУКТАХ И ОТХОДАХ
Промышленные концентраты урановых руд, получаемые методами физического обогащения [90] (гравитационное, радиометрическое, флотационное обогащение, а также метод обогащения магнитной сепарацией и др.) по своему характеру мало отличаются от обычных руд; содержание урана в этих концентратах редко превышает 10%. Отделение и определение урана в таких концентратах можно проводить с помощью методов, описанных для руд.
Химические концентраты, главным образом диуранаты аммония, щелочных или щелочноземельных металлов, а также техническая закись-окись, получаемые в результате сложной химической переработки обогащенных руд, содержат, как правило, 30—80% урана (подробное описание схем химической переработки урановых руд приведено в литературе [4, 90, 97]).
Основными примесями, содержащимися в таких концентратах, могут быть Na, К, Mg, Са, Fe, Si, РО4~ и незначительные количества Ti, V, Al, Мо, Сг, Си и др.
Химические концентраты обычно полностью разлагаются обработкой сильными кислотами (HNO3, H2SO4, НС1О4, НС1) или их смесями при нагревании; особенных осложнений при разложении концентратов не наблюдается.
Для отделения урана от примесей могут быть использованы все описанные выше методы. Количественное определение урана в большинстве случаев проводят химическими методами: весовыми и объемными, дающими максимальную точность; реже находят применение фотометрические (после отделения примесей экстракцией) [564], а также флуорометрические методы [1027, 1028].
При определении урана в химконцентратах хорошие результаты дает разработанный А. Е. Клыгиным (1955 г.) весовой метод, заключающийся в осаждении урана в виде пероксида UO4-2H2O в присутствии комплексона III, маскирующего примеси (методика приведена выше на стр. 60).
Для определения урана в концентратах может быть использован метод, который основан на том, что после восстановления урана и отделения некоторых примесей с помощью электролиза на ртутном катоде четырехвалентный уран количественно осаждают в виде сульфата прибавлением двух объемов 72%-ного раствора НС1О4. Осадок сульфата отфильтровывают, промывают 47%-ным раствором НС1О4, растворяют в воде и титруют четырехвалентный уран раствором перманганата калия [184]. Точность метода достигает ±0,1 % (отн.).
Гидросульфитно-фосфатный метод, заключающийся в восстановлении урана гидросульфитом и осаждении фосфата четырехвалентного урана из кислых растворов, также может быть рекомендован для точного определения урана в химических концентратах.
348
Определение проводят следующим образом (И. В. Моисеев, 1950 г.): около 1,5 г тонкоизмельченного концентрата взвешивают с точностью до 0,0002 г, помещают в стакан емкостью 200—300 м.1, приливают 30 мл концентрированной НС1, 3 мл H2SO4 (1 : 1) и кипятят раствор 5—10 мин., после чего добавляют 3—4 мл пергидроля и нагревают до выделения густых паров SO3. После охлаждения приливают 50—60 мл 5%-ного раствора НС1 и кипятят 5—10 мин. Неразложившийся остаток отфильтровывают, промывают 5%-ным раствором НС1 и отбрасывают. Фильтрат собирают в мерную колбу емкостью 250 мл, охлаждают и разбавляют до метки 5%-ным раствором НС1. Для анализа берут 25 мл полученного раствора, помещают в стакан емкостью 200 мл, приливают 40—45 мл воды, добавляют 2—3 капли 0,1%-ного раствора метанилового желтого и нейтрализуют раствором NH4OH до перехода окраски индикатора от розовой до желтой. К нейтрализованному раствору прибавляют 8 мл НО (1 : 1), нагревают до кипения, приливают 4 мл 20%-ного раствора тиосульфата натрия и кипятят раствор до полной коагуляции сульфидов Си, Bi и др. Если сульфидов много, их отделяют; после охлаждения раствора до комнатной температуры приливают 10—20 мл воды, добавляют 1 г гидросульфита натрия, 10 мл 10%-ного раствора Н3РО4, немного бумажной массы, разбавляют раствор водой до 150 мл и оставляют 1—2 часа (лучше оставить на ночь). Осадок отфильтровывают, промывают до полного удаления продуктов разрушения гидросульфита и тиосульфата и растворяют в 30 мл смеси H2SO4 и Н3РО4 (20 мл 6 jVH2SO4-f- 10 мл концентрированной Н3РО4), промывая затем фильтр 6 N раствором H2SO4. К фильтрату объемом 100—120 мл приливают 15 мл 0,1 N раствора железоаммонийных квасцов, через 1—2 мин. добавляют 100—120 мл воды, 2 капли 0,5%-ного раствора дифениламинсульфоната бария или натрия и титруют 0,05 /V раствором К2Сг2О7.
Одним из важнейших технологических полупродуктов является тетрафторид урана UF4, труднорастворимое соединение (растворимость в воде при 25° равна 1 • 10-4 моль/л); для перевода его в раствор пользуются рядом способов. Часто перед растворением UF4 превращают в закись-окись прокаливанием при 800—900°. Кислоты-окислители, например дымящая НС1О4, быстро растворяют тетрафторид урана с образованием соли уранила и HF. Азотная кислота действует на UF медленно, но добавление борной кислоты усиливает растворяющее действие HNO3 (так же, как и других кислот) вследствие связывания F~-hohob по реакции:
2UF4 + 2Н3ВО3 + 6HNO, —2UO2 (NO3)2 + 2HBF4 + N2OS + 5H2O. Тетрафторид уранатакже растворяетсяврастворесульфатацерия(1У) в растворах перекисей щелочных металлов, в смеси NH4OH и Н2О2. Соли железа (III) —хлорид и фторид— реагируют с UF4 и образованием уранил-иона и комплекса железа с F'. Хорошим растворителем тетрафторида урана является горячий раствор нитрата или хлорида алюминия; в его присутствии происходит окисление урана (IV) до урана (VI) и связывание F- в комплекс с алюминием. Нагревание UF4 с Н3РО4 приводит к образованию фосфата урана (IV) и полному удалению Р“-иона.
После растворения тетрафторида урана и полного удаления Р_-иона уран может быть определен любым методом, пригодным для концентратов.
Ряд методов применим для определения урана в присутствии F~-hohob. Ч. Я. Кроль (1952 г.) разработан метод, позволяющий определять уран в UF4 с точностью ±0,2% (отн.). Метод основан
349
на разложении тетрафторида урана азотнокислым алюминием, осаждении урана перекисью водорода, растворении осадка UO4'2H2O в серной кислоте и йодометрическом титровании выделяющейся перекиси водорода.
Определение урана проводят следующим образом: UF4 (0,1500—0,1800 г) помещают в фарфоровую чашку, приливают 3—4 мл воды, 1 г азотнокислого алюминия и нагревают на водяной бане до полного разложения образца. Раствор упаривают досуха, остаток растворяют в 2 мл горячей воды. Полученный раствор переносят в центрифужную пробирку, разбавляют водой до 9 мл и добавляют несколько капель индикатора (тимолового синего); нейтрализуют аммиаком до pH 2,8— 3 и осаждают перекись урана добавлением 2—3 капель 30%-ного раствора Н2О2. Добавляют опять NH4OH до pH 2,8—3, оставляют раствор стоять около получаса, затем осадок UO4-2H2O отделяют центрифугированием, промывая его 9—10 раз 0,5%-ным раствором NH4NOS (до полного удаления избытка Н2О2). Осадок переносят в коническую колбу емкостью 250—800 мл путем взмучивания осадка с 4— 5 мл воды (операцию повторяют 3—4 раза), в колбу добавляют 80—90 мл воды, 1 г K.J и 5 лм H2SO4 (1 : 1). Центрифужную пробирку промывают H2SO4 (1 : 100) для полного удаления UO4. 2Н2О со стенок пробирки. Раствор в колбе энергично перемешивают до полного растворения осадка, приливают 1 мл насыщенного раствора молибдата аммония (катализатор реакции между Н2О2 и K.J), затем титруют выделившийся иод 0,05 N раствором тиосульфата натрия. Рекомендуется также перманганатометрическое окончание, для чего осадок перураната уранила растворяют в 10—12 мл H2SO4 (1: 4), добавляют 0,5 г мочевины и титруют 0,05 N раствором КМпО4.
Для экспрессного определения урана в его тетрафториде может быть рекомендован метод комплексонометрического титрования урана (IV). А. Е. Клыгин, Н. С. Коляда, Д. М. Завражнова и Н. Д. Никольская (1957 г.) использовали при титровании урана (IV) раствором комплексона III при pH 1,7+0,1 индикатор арсеназо I (0,1 %-ный раствор). Точность определения урана равна ±0,15% (отн.) [101а].
П. Н. Палей и Сюй Ли-юань [187] при определении урана в тетрафториде комплексонометрическим титрованием при pH 1 — 1,8 применили индикатор торон.
Авторы предлагают следующий х<5д анализа: 0,2000 г UF4 растворяют в 10 мл IN НС1О4 (или НС1), в которых предварительно растворен 1 г А1С13-6Н2О; переносят раствор в мерную колбу емкостью 50 мл и разбавляют водой до метки. Берут аликвотную часть раствора, приливают 3 мл 1 N НС1О4 (или НС1), разбавляют водой до 100 мл, добавляют 6—10 капель 0,1%-ного раствора торона и титруют уран (IV) 0,01—0,003 М раствором комплексона III до перехода окраски из розовой в оранжево-желтую. Точность определения урана данным методом составляет =±z0,2% (отн.).
Для определения урана в тетрафториде может быть использован метод титрования урана (IV) ванадатом аммония после растворения UF4 в серной кислоте (1 : 2) [11 ] или в смеси H2SO4 с борной кислотой [173].
К промышленным отходам относятся пески, остающиеся после кислотного извлечения урана из руды; гидратные кеки, получающиеся при содовой обработке раствора урана; различные шламы, а также сливные воды.
Пески представляют собой пустую породу, содержащую незначительные количества урана (0,02—0,002%) и большие количества 350
Si (более 70%), а также Ti, Al, Fe, Ca, Mn, Mg и др. Разложение песков связано с известными трудностями; обычно для их разложения используют обработку плавиковой кислотой или сплавление с NaOH, Na2CO3, Na2O2, KHF2 и др. Гидратные кеки и шламы со-держат0,02—0,002% U, большие количества Al, Fe, Ti, Сг, Мп, а также другие примеси. Кеки и шламы могут быть переведены в раствор обработкой сильными кислотами или их смесями; иногда приходится прибегать к разложению плавиковой кислотой или сплавлению.
Для определения урана в песках и кеках может быть рекомендован объемный гидросульфитно-фосфатный метод (см. описание выше). Для повышения чувствительности титрования урана (IV) раствором ванадата аммония В. Ф. Ескевич и Л. А. Комарова (1959 г.) использовали амперометрический метод определения конечной точки. Амперометрическое титрование четырехвалентного урана ванадатом аммония проводится без наложения внешней ЭДС, с использованием вращающегося платинового электрода и висмутового электрода (электрод сравнения).
При анализе отходов (песков и кеков), содержащих до 0,0025% урана, с использованием гидросульфитно-фосфатного метода отделения и амперометрического титрования урана (IV) ошибка определения урана не превышает 5% (отн.).
Для определения урана в песках, кеках, сливных водах и других материалах, содержащих тысячные доли процента урана и меньше, может быть использован метод, разработанный П. Н. Па-леем, А. В. Давыдовым, Т. С. Добролюбской и А. А. Немодруком (1959 г.). Метод заключается в отделении урана от примесей путем его экстракции трибутилфосфатом в присутствии комплексона III, последующей реэкстракции урана в водную фазу фосфорной кислотой и люминесцентном определении урана. Экстракцию урана проводят равным объемом 20 %-ного раствора трибутилфосфата в четыреххлористом углероде из азотнокислого раствора, содержащего 40% Ca (NO3)2-4H2O, при pH 2,5—3 в присутствии соответствующего количества комплексона III. Реэкстрагируют уран из органической фазы 5%-ным раствором фосфорной кислоты и измеряют интенсивность свечения полученного реэкстракта на горизонтальном фотометре Пульфриха при возбуждении ультрафиолетовым светом (А=253,7 ммк) при освещении раствора сверху. Предлагаемый авторами метод позволяет определять уран при его содержании 1 мкг/мл с точностью ±10% (отн.).
В последнее время В. С. Сахаровым разработаны экспрессные методы определения урана в различных материалах (в концентратах, производственных растворах, сбросных водах, богатых и бедных рудах, кеках, песках, отвалах и др.), заключающиеся в восстановлении урана (VI) до урана (IV) раствором двухвалентного железа В присутствии плавиковой или фосфорной кислоты, дальнейшем окислении избытка железа (II) и восстановившихся примесей сла-
351
<быми окислителями (HNO2, разбавленная HNO3, бром и др.) и титровании урана (IV) раствором ванадата аммония с использованием дифениламинсульфоната натрия как индикатора.
Автор рекомендует использовать в качестве индикатора дифе-ниламинсульфонат натрия в наполовину окисленном состоянии ([1046], что значительно повышает чувствительность титрования.
Наибольшее применение для определения урана в бедных рудах нашел ферри-фосфатно-нитритный метод, который основан на разложении навески образца фосфорной кислотой с добавлением HNO3 или Н2О2 и восстановлении урана (VI) солью Мора. Избыток соли Мора и восстановившиеся элементы с переменной валентностью (Fe, As, V, W, Мо, Sn, Си, Мп и др.) окисляют нитритом натрия, избыток которого затем разрушают мочевиной; уран (IV) титруют стандартным раствором NH4VO3.
0,2—1,0 г руды помещают в термостойкую колбу емкостью 250 мл, смачивают небольшим количеством воды (2—3 мл), приливают 15 мл Н3РО4 (уд. в. 1,6), 1 мл концентрированной HNO3 (уд. в. 1,34—1,4), тщательно перемешивают, нагревают на заранее нагретой электроплитке до кипения и кипятят в течение 6—7 мин. После этого колбу охлаждают до 70—90°.
Для легковскрываемых руд и материалов автор рекомендует применять разложение фосфорной кислотой с добавлением Н2О2, для чего навеску руды (0,25— 1 г) смачивают водой, приливают 15 мл Н3РО4 (уд. в. 1,6), 2—3 мл 26—30%-ной Н2О2. Пробу перемешивают и нагревают на плитке до прекращения разложения Н2О2 и кипятят после этого еще в течение 5—6 мин.
К охлажденной до 70—90° пробе приливают 25 мл горячего 0,1% -ного раствора мочёвины (если образец разлагали с добавлением HNO3), или 25 мл горячей воды (если разлагали с Н2О2); содержимое колбы тщательно перемешивают, приливают 2—3 мл 10%-ного раствора соли Мора, нагревают до кипения, и отфильтровывают нерастворившийся остаток через воронку с фильтром «белая лента» под вакуумом. Колбу обмывают 2 раза разбавленной (1 : 1) Н3РО4 порциями по 5—7 мл и затем один раз обмывают воронку, собирая раствор в фильтрат.
Фильтрат охлаждают до 15—20°, после чего прибавляют 5 мл 5%-ного раствора NaNO2; раствор энергично перемешивают до исчезновения коричневой окраски, появившейся при добавлении NaNO2; после этого пробу взбалтывают еще 5— 30 сек. Для разрушения избытка азотистой кислоты быстро приливают 8—10 мл 30%-ного раствора мочевины, при этом происходит бурное выделение пузырьков газа (N2,CO2).
Перед титрованием урана (IV) необходимо возможно полнее удалить пузырьки газа из раствора. К раствору приливают 0,1 мл индикатора (наполовину окисленный —0,1 М раствор дифениламинсульфоната натрия) и титруют уран (IV) 0,00252— •0,00042 Л/ раствором NH4VO3 до появления ясной фиолетовой окраски.
Точность метода при определении десятых и сотых долей процента урана лежит в пределах ±2 — 4% (отн.).
3. ОПРЕДЕЛЕНИЕ УРАНА В СПЛАВАХ МЕТОДЫ ПЕРЕВОДА СПЛАВОВ УРАНА В РАСТВОР
Способ растворения сплавов урана обусловливается главным образом природой легирующего металла, а также тем методом, который будет применен далее для определения компонентов.
Как правило, сплавы урана с различными металлами — медью, цинком, висмутом, ниобием, цирконием, молибденом и др. — до-352
вольно легко растворяются в минеральных кислотах или их смесях, а сплавы со свинцом и висмутом реагируют даже с водой [8].
Наиболее часто для растворения сплавов урана применяют азотную кислоту или царскую водку; при растворении сплавов с цирконием и ниобием требуется добавлять в HNO3 небольшие количества F"-ионов.
Для растворения большого числа сплавов можно использовать соляную кислоту с добавлением окислителя, а также хлорную и серную кислоты.
Сплавы урана с ниобием и молибденом иногда разлагают раствором едкого натра и перекиси водорода (при нагревании).
Хорошим растворителем урано-циркониевых сплавов, особенно в тех случаях, когда в дальнейшем нежелательно присутствие F", является раствор брома в этилацетате [691], при этом уран переходит в раствор в виде бромида четырехвалентного урана.
Сплав растворяют следующим образом: 10 г образца и 25 мл этилацетата помещают в колбу, снабженную обратным холодильником, и нагревают раствор до кипения. После этого нагревание прекращают и приливают 40 мл 8 М раствора брома в этилацетате порциями по 5 мл через каждые 3 мин. Для полноты разложения раствор кипятят еще 15—20 мин.
Наиболее применимые растворители для важнейших сплавов урана приведены в табл. 46.
Таблица 46 [691] Растворение сплавов урана в различных растворителях
Образец HNO3 Царская водка HNO,-+-HF HCI + окисли-тель НС1+ этил-апетат Вг2+ этил-ацетат NaOH+ Н 2О2
Металли- Раство- Раство- Раство- Р аство- Раство- Раство- Раство-
ческий и ряется ряется ряется То же ряется ряется ряется То же ряется
U-Zr Нет Нет » Нет Нет » Нет
L’-Nb » » » » » Растворяется
C’-Fe Растворяется Растворяется » Раство- р яетс я Растворяется » Нет
U—Cr Нет Нет Нет То же То же »
L’-Ru » Растворяется » Нет Нет Нет »
U- Mo » То же » Растворяется » Растворяется Растворяется
U и продукты деления1* Растворяется 2* » Нет Нет Нет
U—Si ’* Растворяется — Растворяется — — — —
U—Pu Растворяется 4* Растворяется Нет Растворяется Растворяется Растворяется Нет
1* Сплав урана, содержащий 1 — 3% Zr, Мо, Ru, Rh, Pd, Се.
2* Нужно добавлять небольшое количество F~ для растворения Zr.
3* При растворении остается осадок кремневой кислоты, которую легко растворять добавлением F-.
4* Ри медленно растворяется в HNO., поэтому предпочтительнее растворять сплав в НС1.
?3 Аналитическая химия урана
353
ОТДЕЛЕНИЕ И ОПРЕДЕЛЕНИЕ УРАНА В СПЛАВАХ
Сплавы урана с висмутом
Урано-висмутовые сплавы растворяют обычно в азотной кислоте и определяют уран одним из описанных ниже методов.
Шелтон [905] определяет уран в присутствии висмута осаждением 4°о-ным спиртовым раствором оксихинолина, маскируя висмут этилендиаминтетрауксусной кислотой (экстракционный вариант данного метода описан выше на стр. 308).
Милнер и Нанн [751 ] при определении урана в тройных сплавах на основе висмута (U—Th—Bi—сплавы) использовали ионнообменный метод отделения, сорбируя уран вместе с висмутом из 5/И раствора НС1 на анионите «деацидит FF» в солянокислой форме. После вымывания урана с колонки 0,2 М раствором НС1 его восстанавливают до урана (IV) и титруют сульфатом церия (IV) или же определяют полярографически на фоне тартрата. Метод дает вполне удовлетворительные результаты, но несколько длителен из-за необходимости удалять органические вещества, извлекаемые из анионита. Аналогичный метод использовал Милнер при определении U в бинарном сплаве с Bi [746, 749].
При анализе тройных сплавов на основе висмута, содержащих 0,2—5% урана и 0,2—5?6 неодима или празеодима, Милнер и Эдуарде [5] отделяли висмут экстракцией хлороформным раствором диэтилдитиокарбамината диэтиламмония из 1 N раствора HNO3, а затем экстрагировали уран тем же реактивом при pH 5,5—6,0 (добавляя ацетатный буфер). Количественно уран определяли фото-метрированием хлороформного раствора диэтилдитиокарбаминат-ного комплекса урана.
Керби и Кроли [652] отделяли уран от висмута, а также от небольших количеств циркония, магния и железа, содержащихся в сплаве, экстракцией оксихинолинового комплекса урана хлороформом в присутствии комплексона III как маскирующего агента.
Авторы предлагают следующий ход определения урана: 0,2 г сплава растворяют в 2 мл концентрированной HNO3 и упаривают раствор до получения влажных солей. Приливают 1 мл концентрированной НС1, 26 .ил воды, добавляют 2,5 г комплексона III и устанавливают pH 7. Затем разбавляют раствор до 50 мл, приливают 2 мл 5%-ного спиртового раствора оксихинолина и экстрагируют комплекс урана хлороформом. Экстракцию повторяют несколько раз. Экстракты объединяют, разбавляют хлороформом до 20 мл и определяют уран фотометрически при длине волны 400 ммк.
Милнер и Эдуарде [748] при анализе урано-висмутового сплава, содержащего 1—20% урана, отделяли уран осаждением в виде фосфата уранила-аммония в присутствии комплексона III; затем осадок фосфата растворяли в соляной кислоте, восстанавливали в свинцовом редукторе и титровали уран (IV) сульфатом церия (IV), используя ферроин как индикатор.
354
Урано-цинковые сплавы
Фритц с сотрудниками [528] сплав урана с цинком, содержащий до 20% урана, переводили в раствор обработкой соляной кислотой в присутствии окислителя, а затем определяли уран в присутствии объемным методом.
Определение урана прсЕОДится тгкг.м сбразсм: 0,4 г сплава растворяют в 10 лы ЕС 1 с дсбавлением перекиси водорода, разбавляют раствор водой до 2С0 мл и берут для анализа аликвотнчю часть, содержащею около 25 мг урана. Добавляют к раствору 3 A’ ЕС1 до Е0 мл, восстанавливают \ран в свинцовом редукторе, приливают 1 N раствор железа (Ill) и титруют образующееся при этом железо (II) 0,01 А’ раствором сульфата церия в присутствии феррсина как индикатора.
Сплавы урана с алюминием
Бэкон и Милнер [349] для разложения урано-алюминиевых сплавов использовали концентрированную HCI, после чего определяли уран объемным методом.
Авторы предлагают следующую методику определения урана: 2,5 г сплава, содержащего до 20% урана, растворяют в 50 мл концентрированной НО и разбавляют водой до 1С0 мл. К 20 мл этого раствора приливают 10 мл концентрированной НО, 50 мл воды и восстанавливают уран в свинцовом редукторе. К вое становленному раствору добавляют одну каплю H3FO4, 1 мл 0,001 А раствора фер-роина и титруют уран (IV) 0,02А раствором сульфата церия (IV). Точность определения урана составляет ±0,1% (отн.).
Имеются указания о возможности точного определения урана в его сплаве с алюминием рентгено-флуоресцентным методом [1043].
Сплавы урана с галлием
Хорошим, наиболее часто применяющимся растворителем сплава урана с галлием является азотная кислота.
После переведения сплава в раствор уран может быть определен весовым или объемным методом.
При весовом определении урана галлий предварительно отделяют экстракцией диэтиловым эфиром из 6 N солянокислого раствора, а в водной фазе осаждают уран аммиаком и прокаливают осадок диураната аммония до U3O8 [752].
Для объемного определения урана после разложения сплава HNO3 переводят нитраты в сульфаты обработкой H2SO4; сернокислый раствор восстанавливают в редукторе, наполненном металлическим свинцом, и титруют уран (IV) любым окислителем [744].
Галлий определяют в присутствии урана комплекеюнометриче-ским титрованием.
Сплавы урана с цирконием
Как указывалось выше, для разложения урано-циркониевых сплавов следует пользоваться азотной кислотой в присутствии фторида.
23*
355
При определении урана в этих сплавах Бэкон и Милнер [349 J использовали высокоизбирательный спектрофотометрический метод, основанный на измерении светопоглощения раствора сульфатного комплекса уранила; определение урана проводили в 7,2 N растворе серной кислоты при длине волны 430 ммк.
Данный метод дает хорошие результаты также при определении урана в его сплавах с молибденом и титаном (методика определения приведена на стр. 108).
Есть указания [745], что уран в сплаве его с цирконием можно определить осаждением таннином из водного раствора при pH 8 после отделения циркония экстрагированием хлороформом комплекса циркония с купфероном; в присутствии циркония уран может быть определен полярографическим [1051] или рентгено-флуоресцентным методом [1044].
Для определения урана в его сплаве с Zr может быть использован также метод потенциометрического титрования урана (IV) в присутствии Zr [919]; для восстановления применяют свинцовый редуктор; уран (IV) титруют раствором сульфата церия (IV). Фотометрическое определение урана в его сплаве с цирконием с реагентом хлорфосфоназо III описано на стр. 140.
Сплавы урана с бором
При определении урана в сплаве его с борол^ содержащим более 10% бора, Бэрнет и Милнер [359] растворяли сплав в HNO3, а затем осаждали уран в виде диураната аммония и прокаливали осадок до U3O8. Ошибка определения урана составлят ±0,5% (отн.).
Тройной сплав урана с плутонием и молибденом
Сплав урана с плутонием и молибденом, содержащий до 70% Ри и около 5% Мо, Филипс с сотрудниками [806] предлагают переводить в раствор следующим образом:
Около 200 мг образца нагревают в течение одного часа с 20 мл 6 N раствора НС1 и 20 мл концентрированной HNO3, затем добавляют 7 мл концентрированной НС1О4 и нагревают еще в течение часа. Если сплав не растворился полностью, то добавляют 0,1 мл концентрированной HF, продолжая нагревание до появления паров хлорной кислоты. Раствор разбавляют водой; если необходимо, то фильтруют. Молибден определяют в аликвотной части раствора в присутствии урана п плутония колориметрически с роданидом аммония.
При определении урана и плутония молибден предварительно отделяют экстрагированием хлороформом комплекса молибдена с а-бензоиноксимом. Уран и плутоний затем разделяют ионообменным методом или определяют один в присутствии другого.
356
Сплавы урана с платиной [849]
Сплавы урана с платиной, содержащие от 0,2 до 95% платины, растворяют в царской водке, после чего раствор выпаривают досуха, приливают НС1 и выпаривают снова досуха. Сухой остаток солей растворяют ъ 2 N HCI. При большом содержании платины ее отделяют от урана осаждением сероводородом; к фильтрату, содержащему уран, добавляют смесь кислот HNO3, НСЮ4 и HjSO4, кипятят раствор для удаления избытка H2S и выпаривают досуха. Затем определяют уран колориметрически с Н2О2 и NaOH при длине волны 425 ммк. При большом содержании урана (>10%) его можно определить объемным путем.
Глава VII
ОПРЕДЕЛЕНИЕ ПРИМЕСЕЙ В ЧИСТЫХ ПРЕПАРАТАХ УРАНА
/. ОПРЕДЕЛЕНИЕ МАЛЫХ КОЛИЧЕСТВ ПРИМЕСЕЙ В УРАНЕ МЕТОДАМИ СПЕКТРАЛЬНОГО АНАЛИЗА
Ю. И. Беляев
Энергии связи электронов 5/-, 6d-, 7з-оболочек, участвующих в оптических переходах атомов урана и других тяжелых элементов, имеют очень близкие значения, а сам атом урана обладает низким ионизационным потенциалом. Спектр урана, как и других трансурановых элементов, является чрезвычайно сложным. В нем вместе с линиями нейтральных атомов присутствуют линии однократноио-низированных атомов, поэтому спектр урана представляет собой сплошную сетку линий, расположенных на фоне интенсивного непрерывного спектра. В связи с этим обычные методы спектрального анализа не могут применяться для успешного определения малых количеств примесей в уране.
Фред, Нахтриб, Томпкинс [524] разработали так называемый метод «медной искры» для определения примесей в уране. Анализ по этому методу проводится путем перевода пробы в раствор, капля которого наносится на торец медного электрода. Этот электрод после упаривания пробы подвергается спектральному анализу. Чувствительность определения примесей по этому методу составляет 0,1—0,01%.
Непосредственное определение примесей в уране путем использования искры и металлических урановых электродов не приводит к желаемым результатам: чувствительность определения в 103 раз меньше требуемой [1000].
Для достижения высокой чувствительности анализа необходимо полное устранение многочисленных линий урана в спектре и ослабление интенсивного непрерывного фона.
В 1946 г. Скрибнер и Муллин [888] разработали метод определения примесей в уране, основанный на явлении фракционного испарения элементов с «носителем» в дуге постоянного тока. В качестве носителя авторы рекомендуют окись галлия, вводимую в пробу
358
в количестве 2%*. Экспозиция, режим горения дуги, форма электродов подбираются таким образом, что в спектре пробы появляются только линии определяемых элементов, а спектр урана отсутствует.
Чувствительность определения примесей в уране методом Скрибнера и Муллина составляет и• 10"4% [888, 473]. Роль носителя заключается, как это было показано позднее [271, в принудительной концентрации определяемых элементов в центре плазмы дуги в соответствии с распределением в плазме самого носителя. Это имеет своим следствием уменьшение степени рассеяния и выноса атомов элементов-примесей из зоны разряда и, следовательно, увеличение чувствительности определений. Отсутствие в спектре линий урана увеличивает возможности выбора чувствительных аналитических линий определяемых элементов.
В методе Скрибнера и Муллина процессы отделения определяемых элементов от урана путем испарения и процессы возбуждения их спектров связаны между собой, как это ясно из самого принципа метода. Это обстоятельство затрудняет раздельное регулирование и контроль этих процессов для нахождения наилучших условий анализа.
В 1947 г. С. Л. Мандельштамом с сотрудниками [40, 151 ] был разработан метод определения примесей в уране, так называемый «метод испарения», свободный от указанного недостатка. Независимо от авторов [151] А. Н. Зайдель и сотрудники 175, 76, 76а), предложили метод испарения в вакууме, характеризующийся несколько большей чувствительностью и воспроизводимостью. Однако метод испарения в вакууме не применим к анализу урана (см. ниже).
Метод испарения получил широкое распространени° в Советском Союзе. В основе метода лежит принцип полного разделения процессов испарения элементов-примесей и процессов возбуждения их спектров. Разделение этих двух процессов предоставляет широкие возможности для контролируемого выбора оптимальных условий, характеризующихся высокой чувствительностью и точностью.
Анализ по этому методу проводится в два этапа. Вначале в специальной испарительной установке (испарителе) осуществляется отделение летучих примесей от менее летучей основы и конденсация паров элементов-примесей на торец охлаждаемого медного или графитового электрода (приемника). Затем этот электрод с конденсатом используется в качестве одного из электродов в источнике света при последующем спектральном анализе. Очевидно, что применение метода испарения тем более удобно, чем больше разница в упругости паров примесей и основного вещества пробы.
На рис. 57 изображен испаритель модели ИС-3 [151]. Испаритель состоит из металлической плиты 1, графитовых щеток 3, стойки
х В роли носителя может использоваться также AgCl. См., например [443], где повторена методика Скрибнера с этим носителем.
359
4 с держателем 10. Графитовые щетки 3 охлаждаются водой, пропускаемой через держатели 2. Графитовый стаканчик с пробой 6 помещается между щетками с помощью ручки 5. Нагревание стаканчика осуществляется током от понижающего трансформатора. Температура нагревания определяется пирометром. Испарившиеся
Рис. 57. Схема испарителя.
1— металлическая плита; 2 — держатели графитовых щеток, охлаждаемые водой; 3— графитовые щетки; 4— держатель верхнего электрода; 5— ручка для отвода щеток при зажиме стаканчика с пробой; 6— стаканчик с пробой; 7— скоба; 8— винт для регулировки наклона щетки; 9— ось вращения держателя со щеткой; 10— держатель электрода, охлаждаемый водой.
примеси конденсируются на угольном или медном электродах, помещаемых в держатель 10, также охлаждаемый водой. Форма и размеры стаканчиков и электродов приведены на рис. 58.
Для количественного определения примесей, сконденсировавшихся на электроде, последний используется в качестве нижнего электрода в искровом или дуговом источнике света. Отсутствие на спектрограммах спектра урана позволяет использовать спектрографы средней дисперсии: в ультрафиолетовой области спектра— ИСП-22 или Qu-24, в видимой области — ИСП-51 с камерой f =270 мм.
Рассмотрение физико-химических свойств урана показывает, что наибольшая разность в летучестях урана и определяемых элементов имеет место для закиси-окиси урана U3O8. По данным Бильца 360
и Мюллера [374], при нагревании до 2000° закись-окись урана вследствие диссоциации переходят в UO2 и UO3 по реакции:
UsO8=UO2+2UO3.
По Бильцу и Мюллеру, на ход этой реакции существенно влияет давление кислорода. При нагревании в атмосфере воздуха скорость реакции разложения закиси-окиси очень мала и испарение летучей двуокиси урана мало заметно даже при высоких температурах (1800°). Согласно данным, приведенным в книге Каца и Рабиновича
Рис. 58. Форма и размеры графитовых стаканчиков и электродов (а, б, в, г, б—различные варианты).
[97 ], более или менее заметная упругость паров двуокиси урана имеет место лишь при 2000° (табл. 47). Поэтому испарение примесей из закиси-окиси урана можно проводить только в атмосфере воздуха при нормальном давлении, при температурах нагревания, не превышающих 1800—2000°.
С другой стороны, как было показано недавно [15], температура нагревания пробы при проведении анализа по методу испарения должна быть не ниже температуры начала освобождения примесей из решетки закиси-окиси урана. Абсолютная температура начала освобождения примесей из решетки U3O8, по данным [15], составляет 0,67 от температуры плавления основного компонента пробы, и она равна для U3O8 1470° К (=^1200°С). Эта температура начала осво-
Таблица 47
Давление паров двуокиси урана [97]
Температура, СС 1600 1800 1900 2000
Давление, Р-10-’ мм рт. ст 0,071 4,0 18 72
361
бождения примесей совпадает с температурой разрыхления кристаллических решеток, указанной Тамманом [962—964] и равной 0,57 от абсолютной температуры плавления окислов металлов.
Эксперименты, выполненные С. Л. Мандельштамом с сотрудниками [151] и Н. И. Калитеевским с сотрудниками [87], показали, что оптимальная температура нагревания пробы в виде закиси-окиси урана, характеризующаяся максимальной чувствительностью и точностью, составляет 1800—2000°С. Спектр конденсата на электроде при этих температурах практически свободен от спектра урана.
Таблица 48
Степень извлечения примесей (степень конденсации на электрод) из закиси-окиси урана при испарении в атмосфере воздуха (Продолжительность нагревания пробы 2.5 мин.; температура нагревания 1800°; навеска 50 мг [15])
Элементы-примеси и их изотопы
Степень извлечения примесей, % . . .
Bi210 Sb122 Cd1' Na21 К12 ! Вл!.. ! ХЬ' T12"* Zit'-
97 98 98 86 87 [ 5Т4 । 51 98 96
Как показали дальнейшие эксперименты с применением радиоактивных изотопов [15], степень извлечения примесей из закиси-окиси урана на электрод (степень конденсации на электрод) при этих условиях достаточно велика (табл. 48). На рис. 59 и 60 представлены зависимости степени извлечения примесей, из закиси-окиси урана от температуры нагревания пробы и продолжительности испарения [15].
Чувствительность определения методом испарения зависит лишь от абсолютной чувствительности спектроскопического определения-при возбуждении спектра примесей в источнике света. При максимальной степени извлечения примесей концентрационная чувствительность метода испарения определяется величиной навески пробы, которая может быть увеличена. Этим метод испарения существенно отличается от обычных методов спектрального анализа, основанных на непосредственном сжигании анализируемого вещества в источнике света. Однако беспредельно увеличивать вес пробы нельзя, так как степень извлечения примесей начинает уменьшаться вследствие увеличения слоя пробы, через который диффундируют определяемые примеси. Поэтому в целях увеличения чувствительности целесообразно фотографировать на одно и то же место фотопластинки спектр нескольких электродов с конденсатом. Этим самым достигается и значительное усреднение пробы. Неполная конденсация примесей на электрод приводит к значительному уменьшению чувствительности определения. Кроме того, при определении легколетучих элементов следует учитывать возможность их обратного ис-
362
парения с электрода, вследствие сильного нагревания последнего от близкорасположенного стаканчика с пробой. Этот эффект, однако. исчезает при хорошем охлаждении электрода.
Как показывает сравнение данных в табл. 49, чувствительность определения примесей методом испарения выше, чем методом Скрибнера и Муллина [138, 139].
Оценка воспроизводимости анализа методом испарения показывает, что средняя квадратичная ошибка единичного определения равна 15—2О°о [76, 76а, 1511.
Рис. 59. Зависимость степени извлече-
ния Висмута из закиси-окиси урана от температуры нагрева пробы:
Рис. 60. Зависимость степени извлече-
ния натрия из закиси-окиси урана от продолжительности нагрева пробы при
1— для случая вхождения висмута в кри-
разны.х температурах.
сталлическую решетку закиси-окиси урана: 2— висмут вне кристаллической решетки. ГОсВ— температура, по достижении которой висмут покидает кристаллическую решетку закиси-окиси урана.
При работе с искровым источником рыхлые толстые слои кон-
денсата на электроде сбиваются первыми ударами искры и возбуждаются неполно, что приводит к снижению точности и чувствительности. Поэтому метод может применяться только к анализу сравнительно чистого урана. Наличие в пробе легколетучей примеси в количестве, большем одного процента,
значительно ухудшает воспроизводимость, точность и чувствитель-
ность анализа.
Воспроизводимость анализа может быть значительно повышена введением подходящего внутреннего стандарта, что позоляет избежать систематических ошибок, связанных с ухудшением качества слоя конденсата на электроде. При определении большого количества элементов выбор внутреннего стандарта часто бывает затруднен, так как этот элемент должен обладать такой же летучестью, как и определяемые элементы. Но если удалось подобрать внутренний стандарт к одному наиболее важному элементу, то систематическая ошибка при определении остальных элементов сильно уменьшится [76]. Это связано, как уже говорилось, с качеством слоя конденсата
363
Таблица 49
Сравнение абсолютной чувствительности определения примесей методом испарения и методом фракционной дистилляции Скрибнера [76, 138]
Элемент Абсолютная чувствительность метода, мкг (1 0 ~ 9 г) (навеска 50 мг) Абсолютная , чувствительность метода Скрибнера. мкг (навеска 100 мг) Элемент Абсолютная чувствительность метода, мкг (1 0 ~ J г) (навеска 50 мг) Абсолютная чувствительность метода Скрибнера, мкг (навеска i 00 мг''
Ап 15 30 Fe 50 100
As 500 500 Ge 15 20
Ag 5 5 К 100 200
Al 250 500 Li 1 10
В 5 8 Мп 5 100
Be 5 ю Ni 50 200
Cd 10 7 Bi 2 50
Co 25 100 Pb 20 100
Cr 100 300 Sb 100 1000
Cu 10 30 Si 50 1000
на электроде. Поэтому, если почернения линий элемента сравнения остаются во всех пробах одинаковыми, то можно с большей уверенностью проводить анализ по почернениям аналитических линий 176, 138].
Определение бора [151]
Металлический уран переводят в закись-окись прокаливанием в печи при 800° в течение 2 час.
От анализируемой закиси-окиси урана квартованием отбирают пробу весом 5,5—6 г, которую прокаливают в муфельной печи при 800° в течение 30 мин. От этой пробы отбирают навеску в 5 г, в которую вводят носитель в количестве 150 мг. Носитель представляет собой смесь спектрально чистых окиси галлия и закиси-окиси урана в отношении (1 : 12) *. Смесь тщательно растирают в агатовой ступке в течение 15 мин. От приготовленной смеси пробы с носителем отбирают навеску 250 мг. Применение носителей, по данным авторов [151 ], увеличивает интенсивность линий бора. Условия испарения пробы: температура нагревания — 1800°, время установления этой температуры — 20 сек., продолжительность испарения при этой температуре — 20 сек. Наилучшая воспроизводимость анализа имеет место в случае, если электрод-приемник имеет форму, изображенную на рис. 58г. Источник спектра для анализа конденсата — дуга постоянного тока, сила тока — 10 а. Электрод с конденсатом служит анодом. Спектрограф ИСП-22, экспозиция— 6 сек. Каждую пробу снимают 3 раза. Рекомендуемая аналитическая линия в 2497,73 А.
Эталоны готовят путем введения бора в чистую закись-окись урана в виде раствора борной кислоты.
Чувствительность определения бора в уране методом испарения равна 7-10-6%. Точность анализа составляет 10—15% от определяемой концентрации. На рис. 61 приведен градуировочный график для определения бора в уране.
Метод испарения позволяет обойти трудности, связанные с от-
* Целесообразность применения носителей типа Ga2Os и AgCl в методе испарения в последнее время, однако, оспаривается [74].
364
сутствием графитовых электродов, свободных от бора. Применение различных методов очистки не обеспечивает полного удаления бора из графита, где он присутствует, по-видимому, в форме карбида.
Для разрушения карбида бора и испарения бора из графита не-
Рис. 61. Градуировочный график для определения бора в уране.
которой разложение карбида бора еще не происходит. Графитовый же электрод с конденсировавшимися на нем примесями контактирует с дугой в течение 6 сек. При сравнительно небольшой силе тока бор из массы электрода заметным образом не испаряется. Поэтому, по данным авторов [151], к чистоте графита в методе испарения можно не предъявлять высоких требований в отличие от метода Скрибнера и Муллина.
Недавно Франклин и Уилсон [1065] улучшили метод Скрибнера, увеличив навеску U3O8 до 250 мг и применив NH4 для повышения летучести бооа; чувствительность метода по бору доведена до ыо-б%.
Определение меди, никеля и железа [151]
Определение этих элементов осуществляется аналогичным образом, как и в случае определения бора. Эталоны готовят путем введения соответствующих количеств нитратов определяемых элементов в чистую закись-окись урана с последующей сушкой, про
365
каливанием при 400г и растиранием. Определение меди проводят по линиям меди 3247,5 А и 3273,96 А. Навеска пробы — 30 мг. Сила тока дуги — 5а. Достигнутая чувствительность—1 10-4%. Для определения железа используется линия 3020,6 А, никеля — 3002,49 А. Сила тока— 10 а. Чувствительность определения — 1-1О~4Оо.
Для определения ванадия вес пробы увеличивают до 2 г. Сила тока — 10 а. Анализ проводят по линии 3183,98 А. Достигнутая чувствительность— 1-10“2%.
Средняя точность анализа составляет 10—15% (потрем спектрам).
Определение кадмия, германия, индия, галлия, золота, сурьмы и свинца [871
Для определения этих элементов Н. И. Калитеевский с сотрудниками рекомендует использовать медный электрод и повысить температуру нагревания пробы до 2000°, что обеспечивает более полное испарение определяемых элементов. Испарение урана при этой температуре еще недостаточно интенсивно.
Навеска пробы составляет 50 мг *. Для повышения чувствительности определения кадмия, индия, сурьмы навеску пробы увеличивают до 200 мг. Высокая чувствительность определения этих элементов достигается при делении пробы на четыре навески по 50 мг каждая и проведении четырехкратного испарения примесей на один и тот же электрод.
Для анализа конденсата на электроде рекомендуется использовать искровой источник света — генератор ИГ-2, С=0,012 миф, Г—0,15 мгн. Рекомендуемые аналитические линии (в А):
Cd 2265,02; In 4511,32; Ge2651,17; Ga 2874,24; Au 2675,9; Sb 2311,47; Pb 4057,82.
Межэлектродный промежуток — 2 мм.
Чувствительность анализа на Cd, In, Ge, Au равна 3-10“5%, Ga, Pb — 3-10"4% и Sb— 10"3%.
Так как наиболее чувствительные линии определяемых элементов лежат в различных областях спектра, целесообразно спектр конденсата регистрировать одновременно на двух спектрографах ИСП-22 (или Qu-24) и ИСП-51 с камерой f=270 мм.
Для определения индия рекомендуются серебряные электроды, так как при использовании медных электродов на аналитическую линию индия 4511,32 А накладывается линия меди 4509,4 А. Эталонные смеси готовят путем введения в чистую основу U3O8 определяемых элементов.
В табл. 50 сведены условия определения примесей в уране методом испарения.
Определение алюминия в уране с повышенной точностью [39]
Вследствие небольшого различия в упругости паров закиси-окиси урана и окиси алюминия полного испарения последней и® закиси-окиси урана не происходит, что значительно снижает точность определения алюминия методом испарения.
* Носитель в пробу не вводят (см. по этому поводу [74]).
366
В связи с этим Е. А. Верный и В. Н. Егоров [39] разработали метод определения алюминия с повышенной точностью, использу-
ющий явления фракционного испарения с носителем в дуге постоянного тока [888]. В качестве носителя авторы рекомендуют ВаСО3, вводимый в закись-окись урана в количестве 5% в смеси с угольным порошком. Е1о данным авторов [39], угольный порошок способствует
восстановлению окислов алюминия до более летучего металла, в результате чего интенсивность линий алюминия увеличивается. ВаСО3 способствует возбуждению атомов алюминия. В конечном счете смесь угольного порошка и углекислого бария обеспечивает более стабильные условия возбуждения и испарения алюминия из закиси-окиси урана.
Рекомендуемые условия анализа: в 1 г образца закиси-окиси урана вводят 70 мг ВаСО3 и 50 мг угольного порошка. Навеску пробы 25 мг помещают в электрод специальной формы (рис. 62). Источник спектра — генератор дуги переменного тока, сила тока — 18 с. Спектрограф — ИСП-51 с камерой УФ-85А с однолинзовой системой освещения щели. Аналитические линии: А1 3944,03 А— Ва 3935,7 А. Градуировочные графики, построенные в координатах (AS, Igc), имеют прямолинейный вид в интервале концентраций 1 • 10 3— 3 10 ~ 2 % .
Относительная средняя квадратичная
Рис. 62. Форма и размер электрода для определения алюминия в уране [39].
ошибка определения при съемке трех параллельных спектров для этого интервала концентраций составляет 9—11%.
Определению алюминия в уране посвящена также работа Отдела химической службы в Спрингфильде [971], в которой используется фракционная дистилляция с носителем в дуге постоянного тока. Носителем служит фтористый натрий, вводимый в закись-окись урана в количестве 5%. Источник света-—дуга постоянного тока (12а). Аналитические линии А1 2660,4 А (в области — 2-10-3—2’10-2%) и А1 2575,1 А (5-10~4—1 -10-2 %). Данные о точности и воспроизводимости анализа в работе [971] отсутствуют.
Определение кальция (333]
Определению этого элемента в уране посвящена также работа Отдела химической службы Спрингфильда [333]. Для проведения анализа металлический уран переводят в закись-окись U3O8, которую затем прессуют в таблетки. Пробу в виде таблеток помещают в кратер медного электрода и сжигают в дуге постоянного тока; сила тока — 7 а. Спектр регистрируют на спектрографе Джарель-Эш, имеющем дисперсию 5 Х/мм в первом порядке.
367
Аналитические линии:
1 Са 3933,67 А—U 3933,04А при содержании кальция меньше, чем 410“3 % . 2. Са 3933,67 А — U 3935,4 А для содержания Са в пределах 4 10 “3?о — 10 ~2 % .
3. Са 3933,67 A — U 3932,0 А для интервала 10 “2— 2-10“2 ?о.
При содержании кальция, большем чем 2-10“2 %, образцы разбавляются чистой закисью-окисью урана.
Данные о точности и воспроизводимости анализа в работе [333] не приводятся.
Определение платины и палладия [23]
Для определения малых количеств платины и палладия в уране обычные методы спектрального анализа, описанные выше, непригодны. Для успешного анализа необходимо применение методов предварительного химического обогащения проб.
Для отделения платины и палладия от урана И. О. Буфатин, А. Н. Зайдель, Н. И. Калитеевский [23], применили методы осаждения этих элементов на меди с помощью сероводорода. Для достижения чувствительности определения платины 10“4% и палладия 3-10“5% необходимо выделить 1 мкг Pt и Pd из раствора, содержащего по крайней мере 1 г урана.
Медь вводят в исходную пробу в виде раствора сульфата из расчета 0,1 мг Си на 1 г урана. Осаждение проводят из горячих 2 N растворов серной кислоты. Полученные сульфиды растворяют в царской водке и наносят на торец угольного электрода, обработанного полистиролом.
Спектральный анализ концентрата проводят с помощью спектрографа средней дисперсии ИСП-22 или Qu-24. Источник света — конденсированная искра (генератор ИГ-2, С=0,012 мкф, L=Q),\b мгн). В качестве внутреннего стандарта авторы применяют раствор хлористого золота, вводимый в концентрат после обогащения в виде 0,01% -ного раствора в количестве 2 мкг на каждый электрод.
Рекомендуемые аналитические линии и линии сравнения: Pt I 2659,45 А— Au I 2675,95 A; Pd I 3421,24 A — Au I 3122,78 А.
В интервале концентраций 10’4—10“ 2% градуировочные графики имеют прямолинейную зависимость.
Вероятная ошибка единичного определения платины и палладия описываемым методом равна ± 20%. Чувствительность определения платины, по данным авторов [23], может быть повышена до 3-10“ 5% путем увеличения веса исходной пробы.
Определение гадолиния, европия и самария
Применение метода испарения и метода фракционной дистилляции в дуге для определения малых количеств Gd, Eu, Sm не дает положительных результатов вследствие малой разницы в лету.че-стях окислов р.з.э. и закиси-окиси урана. Непосредственное определение этих элементов в уране не обеспечивает высокой чувствительности из-за сложности спектра основного компонента пробы.
Работа Шорта и Дьютона [907] является, по-видимому, первой попыткой решения задачи спектрального определения р.з.э. в уране. 368
Таблица 50
Условия определения примесей в уране методом испарения [87, 151]
( >)фгдг.л ЯГ МЫЛ 'jJICMCI Аналитическая., линия, A Ha-веска пробы, л г Спектрограф Источник света для анализа конден< «т1 Электроды Чувствительность определения, % Темпериту н.1Г|)ев<1Ии> пробы. "С
в 2497,3 250 ИСП-22 Дуга постоянного тока, 10 а Г рафито-вые 7 -10“6 1800
Си 3247,54 3273,96 30 ИСП-22 Дуга постоянного тока, 5 а То же 1 ИГ4 1800
Fe 3020,64 250 ИСП-22 Дуга постоянного тока, 10 а » 1 10“4 1800
Ni 3002,49 250 ИСП-22 То же » 1 • 10- * 1800
Cd 2265,02 200 ИСП-22 Искра С—0,012л1кф Л=0,15 мгн Медные 3 • 10“3 2000
In 4511,32 200 ИСП-51 /=270 мм То же Серебряные 3 -10“3 2000
Ge 2651,17 200 ИСП-22 » Медные 3<10’5 2000
Ga 2874,24 50 ИСП-22 » » 3-10“4 2000
Au 2675,95 50 ИСП-22 » 3-10-5 2000
Sb 2311,47 200 ИСП-22 » » 1 10-® 2000
Pb 4057,82 50 ИСП-51 7=270 мм » » 3-Ю"4 2000
V 3183,98 2000 ИСП-22 Дуга постоянного тока, 10 а Угольные 1-10-2 1800
Таблица <51
Аналитические линии и линии сравнения лантана для определения Gd, Sm, Ей в уране {77, 78]
Спектрограф Аналитическая линия, А Линия 0 сравнения, A Абсолютная чувствительность, мкг
Qu-24 Gd 3422,47 La 3376,33 0,03
Qu-24 Gd 3358,63 La 3376,33 0,1
Qu-24 Gd 3032,85 La 3104,59 0,2
Qu-24 Eu 3907,11 La 3936,22 0,01
КС-55 Eu 3907,11 La 3936,22 0.03
КС-55 Eu 3971,99 La 4015,39 0,03
КС-55 Eu 4129,74 La 4067,39 0,03
КС-55 Sm 4467,34 La 4432,95 0,3
КС-55 Sm 4420,53 La 4432,95 0,3
24 Аналитическая химия урана
369
Авторы применили эфирную экстракцию для отделения главной массы урана, а затем из водного раствора осаждали фториды р.з.э. Сухой остаток раствора, нанесенный на торец медного электрода, сжигали в дуге постоянного тока. Метод спектроскопического анализа концентрата, разработанный этими авторами, характеризуется низкой абсолютной чувствительностью и поэтому для определения 2-10"5% Gd используется 50 г закиси-окиси урана. При выделении суммы р.з.э. авторы использовали весьма сложную методику, включающую осаждение фторидов.
Работа Хирта и Нахтриба [602] является продолжением работы Шорта и Дьютона. Эти авторы в значительной мере использовали методику обогащения Шорта и Дьютона, но для спектроскопического анализа концентрата применили метод «медной искры» [524], характеризующийся высокой абсолютной чувствительностью определения. Авторы сочли возможным уменьшить навеску анализируемой пробы до 5 г и значительно повысить концентрационную чувствительность определения р.з.э. Однако при спектроскопических определениях авторы не используют внутреннего стандарта, что не обеспечивает высокой воспроизводимости. При выделении р.з.э. из урана авторы не используют носитель, который необходим при работе с Сотыми долями микрограммов, когда необходимо считаться с возможностью значительных потерь определяемых элементов, и, следовательно, со значительными ошибками.
Метод определения Gd, Sm, Ей в уране, предложенный А. Н. Зайделем, Н. И. Калитеевским, А. Н. Разумовским, П. П. Якимовой [77, 78], свободен от указанных недостатков. Эти авторы используют лантан как носитель при отделении р.з.э. от урана и как элемент сравнения при спектральном анализе концентрата.
Отделение р.з.э. от урана и от сопутствующих элементов осуществляется по схеме, приведенной на стр. 371.
После семи экстракций в спектре проб отсутствуют чувствительные линии урана.
Для проведения спектрального анализа концентрата р.з.э. последний наносят на торец графитового электрода, пропитанного 2%-ным раствором полистирола в бензоле. Источником спектра служит дуга переменного тока или искра. При искровом возбуждении спектра используется генератор ИГ-2 с параметрами контура: L— 0,15 мгн, С=0,01 мкф. При дуговом возбуждении спектра используется генератор ПС-39, при силе тока 8—9 а. Дуговой промежуток — 2 мм. Лантан служит внутренним стандартом. Аналитические линии приведены в табл. 51.
По описанной схеме авторы определяют Gd, Sm, Ей в уране с чувствительностью 10" 5%. Ошибка анализа составляет ± 15%. Потери р.з.э. в процессе отделения их от урана не сказываются на точности анализа и не приводят к грубым ошибкам в определении содержания р.з.э., так как в процессе обогащения не изменяется отношение концентраций:
CGd fEu cSm cLa ’ cLa ’ cLa
370
Схема отделения р. з. э. от урана [78]
5 г уранилнитрата -j- загрязнения 100 мкг La 4~ ju3tfHNO,
+ эфир
------------------------------------------- :-------------------
Водная фаза: миллиграммовые количества Эфирная фаза:
U, р. з. э. и др. U
- NH.OH
Г идроокиси _______________________________
! -t-3WHNO3-|-KBrO3
Нитраты Се3+—> Се4+ —► Эфирная фаза:
. J U, Се*+
+ эфир ----------------—
Водная фаза: р. з. э., Сг, Fe, Al и др.
4- NHJDH
+
Окиси р. з. э. и Са
4-
Гидроокиси
Окиси р. 3. э.
24*
371
2. ХИМИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ ПРИМЕСЕЙ В ЧИСТЫХ ПРЕПАРАТАХ УРАНА
П. Н. Палей
Для использования металлического урана, его сплавов, солей и окислов в качестве материала ядерных реакций необходима очистка его от многих примесей до содержания их порядка 10“2—10" 5%.
Контроль примесей при таких содержаниях химическими методами требует большой затраты труда, чистейших реактивов и расходования десятков граммов материала. Поэтому усилия аналитиков за последние 15 лет были направлены на замену большинства химических методов определения примесей на спектральные, как значительно более быстрые и дешевые.
Однако ряд причин (сложность спектра самого урана, недостаточная чувствительность метода для отдельных элементов и др.) не позволяют ограничиться прямыми спектральными методами и для многих примесей требуют либо приемов комбинированного анализа, т. е. химического обогащения перед спектральным определением, либо чисто химических методов.
Из методов химического обогащения примесей заслуживают наибольшего внимания методы групповые, позволяющие отделять от урана для последующего спектрального анализа сразу большую группу элементов-примесей. К ним относятся методы соосажде-ния — например металлов платиновой группы с сульфидом меди (см. ниже); методы экстракционные — например экстракция куп-феронатов, дитизонатов, диэтилдитиокарбаминатов, некоторых ок-синатов; к сожалению, эти приемы недостаточно разработаны и мало применяются. Экстракция самого урана эфиром применяется главным образом для определения в остатке редкоземельных элементов (см. ниже).
Значительный интерес представляют приемы отделения примесей на ионитах. Так, Броди и др. [390] использовали сильноосновной анионит Дауэкс-1 (аналог нашей смолы ЭДЭ-10 П) для отделения от урана 18 примесей по следующей методике:
1 г металла или окиси растворяют в политеновом стакане в 15 мл 12 А НС1 с несколькими каплями концентрированной HNO3. Раствор пропускают через поли-теновую колонку (диаметр 10 мм, высота 200 мм) со смолой (200—400 меш), промытой предварительно концентрированной соляной кислотой. Элюируют примеси двумя объемами 12 N НС 1,затем по 1 объему 11 N, 10 N, 9 N, 8 А, 7 Д',6 N и 5 N НС1. Уран, отмеченный коричневой полосой в верху колонки, начинает вымываться только с 5N НС1;его смывают! 1VHC1, азатем элюируют остальные примеси 0,1 N НС1 (4 объема), 0,05 N НС 1 и водой (по 1объему). Сильнокислые элюаты (12— 6 N) содержат Li, Na, К, Rb, Be, В, Al, Са, Си, Fe, Ni, Cr, Mg, Мп, Ti, Pb; слабокислые — Zn и Cd. Элюаты упаривают до 1 мл и наносят аликвотные части их (по 0,1 мл) на медный или графитовый электрод —для спектрального анализа в искре.
Ниже даны краткие описания методов определения примесей, которые могут быть использованы для проверки отдельных спектральных методов или определений.
3^72
Водород
Содержание водорода в металлическом уране обычно порядка п-1О'3?о, поэтому чисто химические методы здесь невыгодны. Применяется специальная методика вакуум-плавления с «платиновой ванной» в графитовом тигле при температуре 1900°. За 10 мин. из навески 5—10 мг выделяются одновременно водород, кислород и азот. Водород в смеси газов определяют откачкой через палладиевый капилляр, нагретый до 600—700°. Аппаратура и детали методики описаны в специальном руководстве [1061] и докладе на 2-й Женевской конференции [1062].
Заслуживает внимания также спектрально-изотопный метод, или метод изотопного уравновешивания [1061, стр. 204,атакже10бЗ]. Метод основан на установлении изотопного равновесия между определенным количеством дейтерия, вводимым в кварцевый сосуд-обменник при температуре около 1000°, и водородом, содержащимся в анализируемом образце. Равновесие устанавливается за 10—20 мин., отношение H2/D2 в полученной смеси определяют спектральным методом. Чувствительность 0,1 мл в 100 г.
Бериллий
Чувствительность спектрального метода от 10~3% до 10"4% в некоторых случаях недостаточна. Для определения бериллия при содержаниях до 1-10“®% проводят обогащение на катионите КУ-2 в Н-форме по следующей методике [1064].
1,5—2 г образца растворяют в НС1 с добавкой Н2О2, раствор упаривают почти досуха, разбавляют водой до 100 мл и прибавляют 5 мл концентрированной НС1 (раствор 0,6 М по НС1). Раствор пропускают через колонку диаметром 10 мм, содержащую 15—20 а смолы, со скоростью 0,5 мл! мин. Уран и бериллий сорбируются. Промывают 50 мл воды и вымывают бериллий раствором сульфосалициловой кислоты (0,02—0,025 М раствор с pH 2,7—3,0). На смоле обычно заметно темно-фиолетовое кольцо (следы сульфосалицилата железа, вымываемые раньше, чем бериллий), спускающееся по колонке; элюат до начала вымывания этого кольца или до достижения pH 2,7 отбрасывают. Заканчивают вымывание после того, как будет вымыто 35—40 мл раствора с явной окраской уранилового комплекса; в них обычно содержится 10—20 мг U; общее количество элюата 400—500 мл. Элюат упаривают почти досуха, прибавляют 5 мл концентрированной H2SO4, отгоняют ее. Остаток прокаливают сначала при 300—400°, затем при 800—850° до U3O8, взвешивают и анализируют на бериллий обычным спектральным методом.
Бор
Определение бора — одно из наиболее важных, так как его содержание лимитируется порядком 10~5%, и в то же время одно из труднейших, так как бор широко распространен (в природе его кларк п-10'2 вес. %) в любых материалах (стекло, реактивы, угли).
Высокая чувствительность методов определения бора достигается только после отделения его от урана путем отгонки. Обычно используют летучесть метилбората, который образуется при нагревании
373
кислого раствора образца с метиловым спиртом. Для определения бора в дистиллате применяют чувствительные цветные реакции —-
с хинализарином, куркумином, диамино-хризазином.
а) Реакция с хинализарином имеет наибольшую чувствительность (до 0,1 мкг В) в крепкой (93±0,5)%-ной H2SO4. Бор,содержавшийся в дистилляте, должен быть введен в смесь кислоты и реактива вместе
с минимальным объемом воды. Поскольку упаривание кислых рас-
Рис. 63. Кварцевый прибор для отгонки мети л бората.
творов приводит к улетучиванию борной кислоты, нужно производить растворение образца и отгонку метилбората с помощью нелетучей кислоты. М. А. Драгомирова (1946 г.) применила для этого фосфорную кислоту.
Определение ведут по следующей прописи: 5 а металла или окиси (для нитрата метод не пригоден) помещают в колбу кварцевого прибора, изображенного на рис. 63, и засыпают 30 г кристаллической (орто)-фосфорной кислоты. В приемник помещают 1,0 мл 10%-ного раствора поташа и 3—4 мл воды. Колбу нагревают постепенно до полного растворения металла. Дают остыть до 40—50° и вливают через воронку 20 мл метилового спирта. Осторожным встряхиванием колбы добиваются полного смешения спирта с кислотой. Отгоняют спирт и образовавшийся борнометиловый эфир. Признаком конца отгонки спирта служит прекращение кипения жидкости при продолжающемся нагревании. Охладив колбу (погружением в стакан с холодной водой), вливают снова 20 мл метилового спирта и повторяют отгонку. Таких отгонок производят всего 10, собирая дистиллят в тот же приемник с поташем. Для контроля пол-
Стеклянный кожух холодильника (на рис. не показан)
ноты отгонки бора производят еще три отгона, собирая дистиллят в новую порцию (1 мл) 10%-ного по
таша. Реакция дистиллята должна быть щелочной по фенолфталеину. Первую и вторую группу отгонов выпаривают отдельно в небольших платиновых чашках на водяной бане досуха; остаток подсушивают над плиткой, прогревают до прекращения вспучивания и прокаливают в муфеле при 600—650° в Течение 1часа.
Остаток в чашечке растворяют в 0,5 мл воды, сливают в сухой кварцевый ци-
одевают на каучуковых трубках.
линдр, споласкивают еще 0,5 мл воды в тот же цилиндр (воду отмеривают точной пипеткой), прибавляют для нейтрализации поташа две малых капли (0,05 мл) 98,5%-ной серной кислоты, а затем точно 9,00 мл этой же кислоты, смывая ею платиновую чашку. Смешивают, дают остыть и вливают 0,5 мл раствора хинализарина; после перемешивания дают полностью развиться окраске (не менее 15 мин.)
и сравнивают с серией стандартов.
Реактивы
1. Фосфорная кислота очищается прибавлением равного объема метилового спирта и отгонкой последнего. Эту операцию повторяют 8—10 раз до тех пор, пока холостой опыт покажет содержание бора не более 0,2 мкг в 30 г кислоты.
2. Спирт метиловый очищают перегонкой с добавлением 10—15 мл 10%-ного едкого натра на 1 л спирта; отгонку ведут через кварцевый или железный холодильник в железную посуду.
374
Для осушки спирт настаивают с прокаленной окисью кальция и снова перегоняют.
3. П о т а ш, не содержащий бора и галоидов, получают разложением оксалата калия.
Оксалат калия очищают четырехкратной перекристаллизацией из горячей воды, сушат, помещают в платиновой чашке в холодный муфель (тяга, выделение СО) и постепенно поднимают температуру до 450° в течение 3—4 час.
4. Вода дистиллированная подвергается вторичной герегонке с добавлением КМпО4 и едкой щелочи.
5. Кислота серная, 98,5% -н а я по весу; допускается отступление не более чем на 0,5%. Готовят смешением обычной серной кислоты (х. ч.) и олеума.
6. Раствор хинализарина. 0,010 г хинализарина (1,2,5,8-тетра-гидрооксиантрахинон) растворяют в 100 мл серной кислоты, полученной смешением 9 объемов 98,5%-ной кислоты с 1 объемом воды. Хранить в посуде из безверного стекла, беречь от влаги.
Стандартные растворы основные
Раствор А—2,8578 г чистой борной кислоты в 1 л (0,5 мг бора в 1 мл). Раствор В — 20 мл раствора А разбавляют до 1 л (0,01 мг В в 1 мл).
Раствор С — 100 лм^раствора В разбавляют до 1 л (0,001 мг В в 1 мл).
Колориметрические стандарты
В одинаковые кварцевые цилиндрики 20 X 10 мм с плоским дном и притертой пробкой отмеривают из микробюреток растворы В или С в количествах, содержащих от 0,2 до 4 мкг В, и воду до общего объема 1,00 мл\ прибавляют в каждый цилиндр точно 9,00 мл 98,5%-ной серной кислоты, смешивают, дают остыть, прибавляют 0,5 мл хинализарина, смешивают. Окраска развивается 15 мин., устойчива в течение 2 недель при условии сохранения концентрации кислоты (беречь от влаги).
Проверка реактивов производится путем холостого опыта. Поправка на бор в реактивах не должна превышать 0,2 мкг В.
Чувствительность метода около 5-10“®% В, ошибка при содержаниях Ю-5% —около 20%.
б) Реакция с куркумином тоже весьма чувствительна, но интенсивность окрасок очень сильно зависит от условий реакции, трудно воспроизводимых. Однако в присутствии куркумина бор не улетает при упаривании солянокислых растворов, что позволяет упростить операции растворения и перегонки; поэтому куркуминовый метод широко применяется во многих вариантах. Приводим один из наиболее точных [1066, 1067].
10 г стружки, очищенной от масел, обмывают в 25 мл азотной кислоты (1 : 7), промывают водой и переносят в перегонную колбу прибора, аналогичного описанному выше. Приливают 15 мл воды и 13 мл чистой НС1 (уд. в. 1,18) малыми порциями. Когда растворение металла закончится, нагревают до начала кипения и вводят малыми порциями 3,0 мл HNO3 (уд. в. 1,42), взбалтывая колбу перед прибавлением каждой порции. Добавляют еще 0,5 мл HNQ3 и дают стоять до развития бурной реакции, после которой раствор осветляется. Прибавляют 3 мл раствора А1С13 (20 г алюминия растворяют в минимальном количестве соляной кислоты, упаривают до сиропа и разбавляют до 300 мл водой) и кипятят 0,5 мин. для удаления хлора и окислов азота. Охлаждают до комнатной температуры, вводят 2.0 мл раствора аммиака (уд. в 0,880) и 1 мл 5%-ного раствора солянокислого гидроксиламина. Охладив, прибавляют 2,0 мл индикатора (0,1%-ный водный раствор кси-
375
лен-цианола F.F.) и нейтрализуют раствором аммиака (1 : 3) порциями по 0,5 мл до бледно-зеленовато-желтой окраски. Последняя порция 0,5 мл должна дать окраску желто-зеленую (pH 1,3—1,8). Раствор переносят в цилиндр, доводят водой точно до 40 мл, переливают обратно и соединяют колбу с холодильником. Отгоняют в мерный цилиндр точно 28 мл, сразу охлаждают колбу до ^70°. Перегон отбрасывают. Под холодильник ставят платиновую чашку с 25 мл воды, 0,05 мл 0,05%-ного раствора тимолового синего и 2,0 мл щелочной смеси (3 г NaOH и 9 мл глицерина в 300 мл воды). Через воронку вливают 40 мл метилового спирта и отгоняют на водяной бане возможно полно; охладив до 60°, приливают еще 20 мл СН3ОН и отгоняют. Дистиллят выпаривают и прокаливают до выгорания угля. Охладив, растворяют в 1,0 мл воды, прибавляют 0,40 мл реактива I (0,125 г кур-кумина и 15 мл ацетона разбавлены до 25 мл водой; хранится не более недели). Нейтрализуют по каплям 5%-ной уксусной кислотой до желтой окраски, прибавляют 1 каплю избытка; выпаривают на водяной бане досуха, выдерживают на ней еще 5 мин. Охладив, прибавляют 1,0 мл ортохлорфенола (промыт 50%-ной H2SO4, высушен Na2SO4, перегнан при 170°) и, прикрыв бакелитовой крышкой, греют на масляной бане (50°) 5 мин. Прибавляют 0,200 мл реактива II (12 мл хлорфенола, 2,0 мл 60%-ной НС1О4, 3,0 мл уксусного ангидрида; беречь от света; годен лишь бесцветный), смешивают вращением, накрывают и дают стоять при 50°точно 15 мин. Сразу разбавляют 2—5 мл этилового спирта, содержащего 15% воды. Смывают еще раз спиртом в мерный цилиндр, доводя объем до 10 мл. Для осветления фильтруют или центрифугируют и измеряют светопоглощение в 4-милл и литровой микрокювете при двух длинах волн 550 (Д^ и 630 ммк (Д2). Расчет ведут по формуле:
мкг B=F(A1—1,95Л2).
Для вычисления фактора F проводят один холостой опыт и один холостой опыт с добавкой 0,2 мкг В:
Р___введенный В, мкг
(ДГ=Л) ’
где Д1 и Д2 исправлены на их значения в холостом опыте. Обычно F = 0,2.
Молярный коэффициент экстинкции образующегося розоцианина равен 180000 на грамм-атом В. Тщательной очисткой всех реактивов холостой опыт может быть доведен до 0,007 мкг В.
Точность метода авторы оценивают в ± 3% (отн.) при содержании бора 1 мкг.
в) Реакция с диаминохризазином («боран») позволяет использовать более простой метод [1068].
5 г анализируемого материала очищают 8 У HNO3, смывают водой, спиртом, сушат и переносят в прибор, приемником в котором служит точно такая же колба, как и дистилляционная. Приливают 25 мл сухого метилового спирта в дистилляционную колбу и 40 мл — в приемник. Охладив обе колбы льдом, вливают в дистилляционную колбу 2,0 мл брома; реакцию растворения регулируют охлаждением. После растворения урана в хорошо охлажденную колбу вливают еще 85 мл СН3ОН, всыпают 1,0 г мелкозерненного бериллия (для связывания брома); регулируют реакцию льдом. По окончании реакции чашку со льдом заменяют баней 85—95 и осторожно перегоняют около 60 мл. Если все реагенты хорошо высушены, то при этом перегоняется весь бор. Колбу с остатком урана удаляют, заменяют ее приемником с дистиллятом; в новый приемник вливают 45 мл воды и отгоняют 55 мл метилового спирта.
Перегон обрабатывают, как и стандарты для построения калибровочной кривой, следующим образом: в конические колбы емкостью 250 мл помещают по 15 мл взвеси Са(ОН)2 (1 г металлического кальция на 500 мл воды; хранится в кварце), 50 мл воды (или перегон) и по 0; 0,20; 1,00; 2,00 мкг В (для стандартов). Выпаривают досуха, потом просушивают при 160—170° в течение 30 мии. Закрыв пробками, охлаждают; вводят по 10,0 мл реактива (1,25 мг «борана» иа 1 мл раствора в бесцветной 95—98%-ной серной кислоте). Закрыв, взбалтывают часто в течение 30— 376
60 мин. для растворения и выделения НВ г. Измеряют светопоглощен не при 525 ммк; в кювете сравнения — стандарт «О». Прибор — Бекман DU, кювета с 1—20 мм. Чувствительность метода -х.0,1 мкг В.
г) Значительно проще методики, использующие куркуминовую новую реакцию без перегонки метилбората; однако их чувствительность-много ниже. Так, Сильвермен [1069] определяет бор в хлориде уранила (или в металле после растворения в смеси HCl-pH2O2), выпаривая слабосолянокислый раствор с куркумином и большим избытком (2,5 г на 100 мг U) щавелевой кислоты. Чувствительность метода з-ю-з%.
Росс [1070] определяет от 10 до 500 мкг В во фторидах, растворяя их в солянокислом растворе хлорида алюминия и, после прибавления этилового спирта, извлекая эти л бор ат эфиром. После перевода бора в водную фазу реакцию с куркумином проводят без выпаривания в среде 18 2VH2SO4. Чувствительность около 5-10“4% В.
Следует отметить, что при анализе некоторых сплавов возможно неполное растворение боридов в кислотах; в этих случаях [1069] применяют прокаливание нерастворимого осадка при 850° и сплавление его с содой с последующим разложением кислотами.
Углерод
Принцип метода — сожжение в кислороде и определение углекислоты по уменьшению электропроводности поглотителя — раствора Ва (ОН)2. В применении к навескам порядка 2—10 г этот принцип требует сложной аппаратуры [1071 ]. Для микронавесок [10—50 мг} нами с 1947 г. успешно применяется метод П. А. Крюкова [10721-
Схема прибора показана на рис. 64.
Рис. 64. Схема прибора для микроопределения углерода.
1 — змеевик-ячейка для измерения электропроводности; 2— главный регулировочный кран;-3 — трубочка с KJ; 4— шариковая трубка; 5— лодочка; 6— печь для сожжения навески; 7— змеевик для очистки кислорода; 8— край; 9 — трубка с окисью меди; 10 — печь.
1. Применяемый для сожжения навески металла кислород из газометра поступает в кварцевую трубку 9 с окисью меди, нагреваемую электропечью 10 до 600— 700°. Это необходимо для сжигания малых примесей СО, углеводородов и главным образом паров органических веществ, поступающих из смазок, каучуковых шлангов и т. п. Слой нагретой окиси меди должен иметь длину 10 см при диаметре 15— 20 мм. Трубка ставится вертикально, так как при горизонтальном положении легко.
377
образуется свободный канал над окисью меди, и эффективность трубки резко падает (рис. 65). Температура трубки с окисью меди обязательно контролируется термопарой с гальванометром, так как выше 700° окись меди начинает спекаться и реагировать с кварцем.
Верхний входной конец трубки со шлифом должен выступать из печи не менее чем на 10—15 см, чтобы шлиф не нагревался. Нижний выходной конец трубки изогнут под прямым углом и вынесен на 10 см в сторону с той же целью.
Рис. 65. Трубка~с окисью меди для очистки кислорода.
Рис. 66. Циркуляционный змеевик для очистки кислорода.
2. Очистка кислорода от следов СО2 производится в циркуляционном змеевиковом поглотителе (рис. 64, 7 и рис. 66), содержащем около 40 мл 40% -ной КОН. Существенно важно, чтобы соединения выходного конца трубки с окисью меди с краном (рис. 64, 8) и крана с входным концом змеевика были сделаны встык.
Рис. 67. Кварцевая трубка с лодочкой для сжигания навески.
3. Кварцевая трубка для сожжения (рис. 64, 4) имеет длину в 2,2—2,5 раза больше длины печи, т. е. 25—30 см при длине печи 10—11 см. Внутренний диаметр печи должен быть на 10 мм больше наружного диаметра трубки для того, чтобы между ними свободно помещался конец термопары, постоянно скрепленной с печью.
Печь (рис. 64, 6) должна свободно передвигаться по трубке из одного конца в другой. Внутренний диаметр трубки 9—10 мм, что позволяет легко вставлять и вынимать кварцевую лодочку шириной в 5—6 мм и длиной в 15—20 мм. Входной
:378
конец трубки изогнут штыком, чтобы его соединение со змеевиком не нагревалось излучением печи. Выходной конец трубки расширен и закрыт пришлифованным колпачком (рис. 67). Этот шлиф должен быть смазан хорошей вакуумной смазкой в минимальном количестве.
4. Для точности регулировки тока кислорода при сожжении применяется (рис. 64. 2) кран с длинной ручкой и двумя пропилами на теле крана. Эти пропи-
вид сбочу
Рис. 68, Циркуляционный змеевик для поглощения СО2 и измерения электроп роводности.
Рис. 69. Шариковая трубка с раствором иода для улавливания соединений серы.
лы длиной до 0,25 окружности делаются тонким напильником или надфелем и по* степенно сходят на нет. Этот кран тоже смазывают минимальным количеством вакуумной смазки или масла типа апьезон. Кран прочно и неподвижно монтируется на крышке термостата.
Объем выходной трубки этого крана и входной трубки поглотительного змеевика до узкого сопла должен быть минимальным (0,3—0,5 мл), без чего нельзя достичь равномерного хода пузырьков в змеевике.
5. Малый змеевик-поглотитель (рис. 68), содержащий 4 мл слабого раствора Ва (ОН)2 (0,01—0,0022V), имеет в нижней части расширение, в которое впаяны два электрода из черненой платины. Движение пузырьков газа по змеевику одновременно вызывает непрерывную циркуляцию раствора Верхняя выход
ная часть змеевика закрыта трубкой с натронной известью для защиты от СО2 воздуха. При первоначальной регулировке прибора полезно на выходе из этой трубки приключить небольшой реометр, рассчитанный на скорость газа около 10—15л<л в мин. (на схеме не показан). Змеевик-поглотитель погружают до половины верхнего шарика в водяной термостат (типа ТС-15), поддерживающий температуру 25° ztO.l.
6. В случае, если металл содержит серу, следует ставить перед змеевиком (между краном и трубкой для сожжения) шариковый поглотитель с 0,01 N раствором иода для улавливания соединений серы и трубку с твердым иодистым калием для улавливания паров иода (рис. 69).
379
7. Измерение электропроводности барита в змеевике производится по обычной схеме мостика Уитстона, в которой два плеча составляют измеряемое сопротивление и магазин сопротивлений (10—1000 ом), а другие два плеча — мостик Колъра-уша (реохорд на барабане). Питание мостика переменным током частотой 800— 10000 гц осуществляется от звукового генератора типа ЗГ-2. В диагональ схемы в качестве нуль-инструмента включают электронный осциллограф типа ЭО-4 или ЭО-5.
Процентное содержание углерода вычисляют по формуле:
0,_ Аи-4,31 • 4-6-100
/оС= —^Т000—
где
Ап —разность между начальной и конечной электропроводностью;
4,31—соотношение между удельной электропроводностью и нормальностью раствора барита (при малых концентрациях барита (0,01—0,002 Л') эта величина постоянна);
4 — объем раствора барита, мл-,
6 — V2 атомного веса С;
т— навеска, г.
После сокращения формула принимает следующий вид:
= ^10.2 и т
Определение проводят следующим образом: лодочку с навеской помещают в левую часть трубки для сожжения, но не близко к шлифу (см. схему, рис. 64); включают печь 10 с СиО. Печь для сжигания 6 отодвигают вправо до конца и включают. Соединяют все части прибора и пускают кислородсо скоростью 10—15 лл/мин. Проверяют герметичность прибора. Вся регулировка скорости газа производится краном 2 (см. схему, рис. 64). Через 20 мин. продувания всего прибора при температуре в печи с СиО 600—700° отключают змеевик-поглотитель (не прерывая тока газа) и заполняют его 4,0 мл раствора барита (0,01 или 0,02 N).
Заполнение змеевика баритом производится из микробюретки сдлинным тонко-оттянутым концом. Кончик бюретки должен при вливании раствора входить в нижнюю расширенную часть змеевика. В противном случае образуется воздушный пузырь, удалять который трудно. После заполнения змеевик вновь присоединяют к прибору и при продувании кислородом устанавливают начальную величину электропроводности раствора барита.
Измерение делается несколько раз через 3—5 мин. пока значение показаний мостика не станет постоянным.
После достижения постоянства показаний мостика печь 6 (на схеме рис. 64) надвигается на лодочку. Температура печи для сжигания поддерживается все время около 900°. Через 0,5—1 мин., в зависимости от сжигаемого металла, происходит вспышка, и через 1—2 мин. осциллограф начинает регистрировать изменение концентрации раствора Ва(ОН)2. Отсчеты по мостику Кольрауша ведутся при приведении синусоиды на экране осциллографа к прямой, что отвечает минимуму тока в диагонали. Выделение и поглощение образующейся СОг считается законченным при достижении постоянства показаний мостика в течение_ не менее 10 мин.
Чувствительность метода •— единицы микрограммов С; точность при содержании -^0,1% С около +3% (отн.).
Существенно важна полная очистка образца (стружек) от масел; она обычно осуществляется промывкой ацетоном и сушкой. Рекомендуется также непродолжительное травление азотной кислотой (1 : 1) до полного снятия оксидной пленки — до серебристо-белого цвета — с промывкой водой, ацетоном и сушкой.
380
Азот
При снятии резцом стружки с отливок урана возможно ее нагревание, а следовательно, и частичная реакция урана с азотом воздуха. Поэтому рекомендуют очищать поверхность металла травлением соляной кислотой с промыванием водой и сухим ацетоном.
При растворении металлического урана в кислотах нитриды разлагаются и превращаются в соли аммония.
Нитрид урана UN в присутствии избытка металла разлагается легко; нитрид U2N3, образующийся в избытке азота, много труднее. Для его перевода в аммиак рекомендуют [108] навеску--100 мг нагревать с 25 мл НС1 (1 : 1) и 1 мл кремнефтористоводородной кислоты в течение 30 мин., затем добавить -^-200 мг селената меди и слегка подогревать до растворения.
Нитриды некоторых примесей, например Ti, Nb, W (в особенности в сплавах), медленно реагируют с кислотами в обычных условиях; для их разложения требуется нагревание с хлорной или фосфорной кислотой.
Метод определения с применением фосфорной кислоты разработан в 1946 г. Р. Ю. Дебердеевой.
3 г металла в виде возможно тонкой и мелкой стружки растворяют в кьельда-левской колбе (емкость 250 мл) в 20 а фосфорной кислоты при осторожном нагревании. После полного растворения дают остыть до 80—90 и соединяют колбу с прибором Кьельдаля для отгонки. Через воронку с краном вливают 40—50 мл горячей воды, а затем 5 мл 30% -ной перекиси водорода и взбалтывают до полного растворения зеленого осадка и пожелтения раствора (окисление U в шестивалентный). В приемник — колбу Эрленмейера емкостью 120—150 мл наливают из микробюретки 0,01 # серную кислоту (обычно достаточно 3 мл) и 5—7 мл воды; конец холодильника погружают в жидкость. Через воронку вливают в колбу 60 мл 50% -ного раствора едкого калия, подогревают колбу горелкой и одновременно пропускают из парообразователя сильную струю пара. Отгон аммиака продолжают 15 мин., считая с момента начала конденсации паров в холодильнике.
Избыток серной кислоты оггитровывают 0,01 N едким натром из микробюретки, добавив 4 капли индикатора (50 мл спиртового 0,03% -ного раствора метилового красного смешивают с 10 мл водного 0,1%-ного раствора метиленовой сини). 1 мл 0,01 N H2SO4 эквивалентен 0,14 мг азота.
Для определения поправки на содержание азота в реактивах проводят холостой опыт с теми же количествами реактивов. После прибавления перекиси водорода прогревают раствор до кипения, кипятят 10 мин., не собирая перегона, и только потом ставят приемник с кислотой и вливают щелочь. Поправка на реактивы не должна превышать 0,2 мл 0,01 N серной кислоты.
Выщелачивание натрия из стенок прибора дает существенное увеличение поправки, потому ловушка, холодильник и приемник должны изготовляться из труднорастворимого стекла (пирекс).
Относительная ошибка при содержании азота 1-10~3% около i 10%.
При содержании азота меньше 5-10“3% точнее определять аммиак в перегоне не титрованием, а колориметрически с реактивом Несслера.
Малые количества азота, порядка 10" 4%, могут быть определены измерением pH дистиллята с помощью универсального индикатора [10731. Предложено также определение аммиака в перегоне по реак
381
ции с фенолятом и гипохлоритом натрия [ 1074 ]. Чувствительность — ью-4%.
Азот может быть определен одновременно с кислородом и водородом по методу вакуумной экстракции [10611.
Кислород
Кислород, вернее примесь окислов, в металлическом уране определяется совместно с водородом и азотом путем вакуумной экстракции [1061, 1062]; в графитовом тигле происходит его превращение в СО; после откачивания газов сжигают СО на окиси меди и конденсируют СО2 в ловушке при температуре жидкого азота.
Фтор
Для отделения фтора от урана применяют отгонку его в виде BF3; после гидролиза его щелочью определяют F- титрованием солями тория (М. А. Драгомирова, 1952 г.).
С несколько меньшей чувствительностью фтор может быть определен и спектрально с применением техники полого катода.
Натрий
Спектральный метод имеет вполне достаточную чувствительность. Для проверки может быть применен следующий химический прием (П. Н. Палей, 1946 г.).
Около 1 г металла растворяют в кварцевой или платиновой чашке в 5 мл азотной кислоты. Выпарив досуха на водяной бане, растворяют остаток в слабой HNO3 (1 : 100) и отфильтровывают кремнекислоту. Раствор выпаривают в кварцевой или платиновой чашке досуха и остаток прокаливают 2 часа при 220—250° в муфеле с пирометром (избегать перегрева, так как выше 300° образуется модификация окиси, не растворимая в уксусной кислоте). Красно-оранжевый остаток окиси выпаривают с 5 мл крепкой уксусной кислоты на водяной баие досуха, смачивают 0,2 мл крепкой уксусной кислоты, приливают 10 мл воды и, прикрыв стеклом, нагревают до полного растворения светло-желтой основной соли. Если остается небольшой красно-бурый остаток (перегретая окись), его отфильтровывают через плотный фильтр, промывают и раствор снова выпаривают досуха. Остаток растворяют 0,2 мл уксусной кислоты и 10 мл воды.
В присутствии >0,05% железа раствор принимает оранжевый оттенок — до красно-бурого. В этих случаях необходимо раствор разбавить до 150 мл горячей водой, нагреть до кипения и отфильтровать основной ацетат железа.
Фильтрат выпаривают досуха, остаток растворяют в 0,2 мл уксусной кислоты и 10 мл воды. По охлаждении прибавляют 10 мл раствора уксуснокислого цинка (40 г соли в 100 мл воды -{- 3 мл СН8СООН), перемешивают и через 4 часа фильтруют через стеклянный фильтр № 4. Частицы осадка смывают со стенок чашки фильтратом, отсасывают сильно, промывают 5 раз порциями по 2 мл спиртового раствора натрий-цинк-уранил ацетата; промывают один раз сухим эфиром, высушивают сильным отсасыванием в течение 5 мин. и взвешивают, не ставя в эксикатор.
Коэффициент пересчета с NaZn (UO2)8(C2HjOJ9-6H2O равен 0,01495.
Если навеска существенно отличается от 1 а, то количество воды, применяемой для растворения ацетата, и объем цинкового реактива должны быть изменены пропорционально навеске.
Ошибка определения 0,001 % при навеске в 1 г.
382
Магний
Для определения магния в количествах выше 5 мг применяют реакцию с эриохромчерным Т [1075]. Состав комплекса (Mg32)4+.
0,5 г металла переводят в хлорид, сульфат или нитрат уранила, нейтрализуют аммиаком до появления слабой мути, доводят объем до 25 мл, устанавливают pH~v5 добавлением 5 мл буфера (380 мл 50%-ного ацетата натрия и 10 мл концентрированной НС1 разбавляют до 1 л), нагревают до 70—90° и медленно приливают 10%-ный раствор оксихинолина в ацетоне в количестве, достаточном для осаждения урана. Коагулируют осадок кипячением в течение 3—5 мин., фильтруют промывают горячей водой. Фильтрат и промывные воды собирают в делительную воронку, экстрагируют избыток оксина и оксинаты Fe, Сг, Со, Al, Мо, Ni и (следы) U хлороформом, пять раз порциями по 10—15мл. Водный слой фильтруют от капелек СНС13; в аликвотной части раствора устанавливают pH 10,2 (буфер — 54 г NH4C1, 500 мл 25% -ного NH4OH разбавляют до 1 л), помещают в мерную колбу емкостью 50 мл, прибавляют 1 мл 0,1%-ного раствора реактива в перегнанном метаноле, разбавляют до метки водой. Измеряют светопоглощенне при 250 ммк на ФЭК с зеленым светофильтром. При содержании магния 5-10-3% и выше ошибка zt3% (отн.).
Кремний
Сотые доли процента кремния можно определить обычным методом — выделением кремнекислоты двухкратным упариванием с азотной кислотой и проверкой ее чистоты обработкой HF. Обязателен холостой опыт. Если предполагается присутствие фтора хотя бы в тысячных долях процента, в азотную кислоту добавляют борную кислоту (50 мг на 10 мл).
При содержаниях 2«10-3—1-10-1% применим колориметрический метод [1076].
1 г металла растворяют в 5 мл смеси кислот (200 мл концентрированной HNOa и 100 мл НС1 разбавляют до 1 л), охлаждают и фильтруют. Нерастворившийся остаток сжигают и сплавляют с 0,5 г Na2CO3; плав растворяют, нейтрализуют 10%-ной НС1 до кислой реакции по бромкрезоловому зеленому, нагревают для удаления СО2 и фильтруют. Фильтрат присоединяют к основному раствору и нейтрализуют NaOH до образования осадка, который растворяют прибавлением концентрированной НС1; раствор разбавляют точно до 100 мл водой. Аликвотную часть (не более 20 мл) вливают в смесь 5 мл 6,5% -ного раствора молибдата аммония и 4 мл 10%-ной НС 1; разбавляют до 30 мл, выдерживают 20—30 мин., прибавляют 2 мл 10%-ногораствора Н2С2О4, 2 мл 15%-ного раствора Na2SOs и 5 мл 25%-ной H2SO4. Выдерживают 3 мин., разбавляют водой до 49 мл, вводят 1 мл 1%-ного раствора SnCl2, перемешивают, выдерживают 5 мин. и фотометрируют в кюветах с (=20 мм с фильтром № 608. Вводят поправку на холостой опыт. Ошибка метода около 5% (отн.).
Сера
Объемное и колориметрическое определение серы производят по сероводороду, выделяющемуся при растворении металла в соляной кислоте 11078]. Для объемного определения H2S поглсщают титрованным раствором гипохлорита кальция, 1 мл которого соответствует 1 мкг S. Для колориметрии используют реакцию метиленовой сини после поглощения H2S раствором ацетата цинка; чувствительность 0,2 мкг S.
383
Хлор
Метод, основанный на окислении С1“ до С1г перманганатом, отгонке его в щелочь, переводе в хлорид и нефелометрическом определении в виде AgCl, разработан в 1946 г. Е. Б. Евдокимовой.
5 г металла (или окиси) помещают в колбу прибора (рис. 70) и засыпают 30 г кристаллической фосфорной кислоты. В приемник помещают 1 лл 10% -ного раствора поташа и 4—5 мл воды; кончик холодильника должен быть погружен в жидкость. Нагревают осторожно до полного растворения металла, дают остыть до
Рис. 70, Прибор для отгонки хлора.
70—80°, вливают 35 мл воды и тщательно перемешивают. Вливают 2,5 мл перекиси водорода (для окисления урана в шестивалентный), споласкивают воронку 15 мл воды и тщательно перемешивают до полного растворения комков. Нагревают- до разложения избытка перекиси водорода; для этого обычно достаточно отогнать в приемник 40 мл воды. Под конец отгонки во избежание толчков включают проса-сывание очищенного воздуха со скоростью 2—3 пузырька в 1 сек. Впускают через воронку 1 каплю 1%-ного раствора КМпО4, смывая ее 1—2 мл воды. Если’раствор в колбе приобретает окраску КМпО4, удаление перекиси считают законченным; если окраска перманганата исчезает, отгоняют еще 10 мл воды и снова пробуют 1 каплей 1%-ного КМпО4.
Если окраска остается, вливают через воронку 0,5 мл 5% -ного раствора KMnOs и 20 мл воды и отгоняют 20 мл раствора, с которым перегоняется практически весь хлор.
К соединенным перегонам прибавляют 1 каплю ацетона или спирта (для восстановления следов МпО^~ и перевода гипохлорита в хлорид) и выпаривают в малой платиновой чашке на водяной бане досуха. Для предохранения от поглощения хлора из воздуха при выпаривании чашку прикрывают воронкой, в которую продувают воздух, очищенный от хлора. Остаток нагревают над плиткой до прекращения вспучивания и прокаливают в муфеле при 40б—450° в течение 15—20 мин.
384
Прокаленный остаток растворяют в 1 до воды, смывают в мерную колбочку емкостью 10 мл еще 1—1,5 мл воды, прибавляют 1 мл 3 -N азотной кислоты, 5 мл 95%-ного спирта и перемешивают.
Заготовляют стандартные растворы: в мерные колбочки емкостью 10 мл вливают по 0,0; 0,1; 0,2; 0,3; 0,5; 0,8 мл стандартного раствора хлористого калия (содержат соответственно 0; 10; 20; 30; 50; 80 .мкг хлора), по 1 мл раствора К2СО3, 1 мл 3N азотной кислоты, 5 мл 95%-ного спирта и перемешивают. Затем во все стандарты и в испытуемый раствор вливают по 1 мл 0,01 /V раствора AgNCb, быстро доводят все водой до метки и перемешивают. Образовавшуюся муть сравнивают с мутью подходящего стандарта через 15—20 мин. после вливания AgNOa. Сравнение производят в щелевом нефелометре Лейтца или нефелометре НФМ.
Ошибка определения при содержании хлора 3-10~4% составляет 5-10~б% (абс.).
Определение в прокаленном остатке перегона может быть произведено при содержаниях хлора ^>20 мкг, потенциометрическим титрованием с хлорсеребряным электродом [1082].
Кальций
Метод, основанный на отделении кальция в виде сульфата, разработан в 1947 г. П. Н. Палей.
5 г металла растворяют в платиновой чашке в 10 мл азотной кислоты; по окончании бурной реакции прибавляют 20 мл 20%-ной серной кислоты и 2 мл чистой 40%-ной плавиковой кислоты, выпаривают и нагревают над плиткой до полного удаления паров SO3.
Остаток растворяют в Юлы теплой воды, прибавляют40мл 95%-ного спирта и оставляют стоять на 4—6 час. (лучше на ночь). Выпавший осадок сульфата кальция отфильтровывают через маленький плотный фильтр, промывают 2—3 раза 80% -ным спиртом (с 0,5% NazSOi) и растворяют в нескольких миллилитрах горячей 1%-ной соляной кислоты. Промывают фильтр горячей водой, собирая растворы в тот же стакан, где производилось осаждение. Подогревают до 80°, осаждают следы железа и урана аммиаком, прибавляя его до слабого запаха, отфильтровывают через маленький фильтр и промывают фильтр горячей водой.
Фильтраы упаривают до 4—5 мл, переливают в пробирку для центрифугирования, нагревают ее в кипящей воде, прибавляют 1 мл насыщенного раствора щавелевокислого аммония, оставляют в бане 10 мин. Вынув, дают остыть в течение 30 мин., центрифугируют 2 мин., отсасывают почти весь раствор капиллярной пипеткой, наливают 3 мл чистой теплой воды, взмучивая осадок, центрифугируют 2 мин., отбирают воду пипеткой. Повторяют промывание осадка еще три раза, а затем его растворяют в 4—5 мл горячей 5%-ной серной кислоты и титруют из микробюретки 0,01 N раствором КМпО4.
Ошибка составляет 0,001 % при навеске 5 г.
Титан
Титан может быть определен спектрофотометрически по реакции с тимолом в среде 85 %-ной H2SO4 после осаждения урана перекисью водорода при pH 1,4 [1077]. Метод пригоден для количеств 5—50 мкг Ti (5-10“4%), ошибка —8% (отн.).
Ванадий
Ванадий отделяют от урана купфероном и определяют в виде фосфорновольфрамбвованадиевой кислоты [1079].
5 г металла растворяют в 25лгл 50%-ной HNO3; охладив, нейтрализуют аммиаком до неисчезающего осадка, точно растворяют его, добавляя по каплям 50%-ную 25 Аналитическая химия урана о о с
HNOS, и дают еще 6 мл ее избытка. Разбавляют водой до -^100 мл в делительной воронке. Прибавляют 5 мл 5%-ного раствора купферона, перемешивают и экстрагируют дважды по 10 мл хлороформа.
Органический слой фильтруют в коническую колбу емкостью 100 мл, выпаривают досуха, прибавляют 2 мл концентрированной H2SO4, нагревают до выделения SO3. Прибавляют концентрированную HNO3 по каплям при нагревании до полного окисления органических веществ. Охладив, добавляют 20 мл воды, кипятят 5 мин.; охлаждают н разбавляют до 50 мл в мерной колбе. Отбирают аликвот, содержащий 0,2—0,4 мг V, приливают по каплям0,01 N КМпО4 до слабо розсвого цвета и еще две капли избытка. Вливают 20 мл 33% -ной фосфорной кислоты и 5 мл 8,2%-ного раствора вольфрамата натрия. Разбавляют водой до -^45 мл, кипятят 5 мин.; охладив, разбавляют точно до 50 мл и оставляют на 30 мин. Измеряют све-топоглощение с фильтрами Н503 и 601 (прибор Спеккер). Проводят холостой опыт и опыты с добавками 0,2 и 0,4 мг V; определяют из них фактор пересчета.
Метод пригоден для определения 1-10"’—4-10~1%V; ошибка ± 5% (отн.).
Хром
Хром определяют по реакции с дифенилкарбазидом [1080].
25 г образца растворяют в азотной кислоте, фильтруют, точно разбавляют до 250 мл\ аликвот 10 мл выпаривают досуха. Остаток растворяют в 20 мл воды, добавляют 1,5 мл серной кислоты (1 : 6) и 1 мл 1%-ного раствора AgNO3, доводят до кипения и вливают 5 мл 10%-ного персульфата аммония; кипятят 10 мин. Охладив, переносят в 100-миллилитровую мерную колбу, разбавляют до -х.98 мл, прибавляют 1 мл раствора дифенилкарбазида (1 г в 100 мл спирта 6 капель концентрированной H2SO4). Перемешивают и не позже чем через 5 мин. измеряют светопоглощение с фильтрами Н503 и Ильфорд 605 в кюветах (1; 2 или 4 мл). Вводят поправку на холостой опыт.
Не растворимый в HNO3 осадок сжигают вместе с фильтром (до полного выгорания угля), обрабатывают 4 мл хлорной кислоты (уд. в. 1,7) и 0,2 мл HNO3 (уд. в. 1,42) в пробирке с внутренним холодильником, нагревая на горелке 1 час. Переносят в 50-миллилитровую мерную колбу и разбавляют водой до метки. Аликвот 25 мл переносят в стакан, прибавляют 4 мл HNO3 (1 : 1), 5 мл 5%-ного бромата калия, разбавляют до 40 мл и кипятят 15 мин., не позволяя объему уменьшаться более чем до 20 мл. Прибавляют 5 мл 9,5%-ного раствора NaCl и кипятят еще 5 мин. после того как исчезнет запах брома. Охладив, разбавляют до 98 мл в 100-миллилитровой мерной колбе, прибавляют 1 мл раствора дифенилкарбазида, доводят водой до метки, смешивают и через 1 мин. измеряют светопоглощение. Расчет проводят по калибровочной кривой, построенной по стандартным растворам бихромата, содержащим 10; 20 ... 100 мкг Сг, обработанным 1,5 мл H2SO4 (1 : 6) и 1 мл дифенилкарбазида в объеме 100 мл.'
Марганец
Марганец отделяют экстракцией в виде комплекса с диэтилдитиокарбаматом и определяют после окисления до МпО4“ [1083].
25 г металла растворяют в Ю0л!лНКО3 (1 : 1), разбавляют до 250 мл. К аликвотной части (50 мл) прибавляют 15 мл 40%-ного цитрата аммония, нейтрализуют аммиаком (уд. в.=0,880) точно по лакмусу, охлаждают и устанавливают pH 8,2— 8,6 (проверка по pH-метру). Переносят в делительную воронку, прибавляют 2 мл 5%-ного диэтилдитиокарбамата натрия, перемешивают и извлекают марганец четырьмя порциями по 10 мл СНС13.
Органический слой фильтруют через сухой бумажный фильтр в 100-миллилитровую коническую колбу, отгоняют СНС13 на водяной бане, добавляют 2 мл концентрированной H2SO4 и нагревают до выделения паров SO3. Вливают концентрированную HNO3 порциями по 0,5 мл, нагревая после добавления каждой порции,
386
пока раствор не осветлится. Охладив, вливают 20 мл воды и кипятят до удаления NO2. Вводят 0,1 г перйодата калия и маленький кусочек платиновой сетки. Кипятят до полного развития окраски. Остаточный объем должен быть не меньше 8 мл. Охладив, переливают в цилиндр и разбавляют до 20 мл водой. Измеряют светопоглощение в кювете с /=20 мм (Спеккер) с фильтрами Ильфорд 604 и Н503. Обязательна поправка на холостой опыт.
Метод рассчитан на содержание марганца 1-Ю"4—4«10~3%; в пределах 1-Ю"4—2,5-10~3% результаты занижены на 10%; при 2,5-10"3% Мп ошибка около ± 10% (отн.).
Железо
Для определения железа при содержаниях п-10~2% прибавляют к 200 мл слабокислого раствора образца (навеска 2 г) раствор алюминиевых квасцов (10 мг А1) и понемногу насыщенный раствор соды до растворения выпадающего вначале осадка диураната. Нагревают до кипения, отфильтровывают гидраты алюминия и железа через малый фильтр, промывают горячей водой и растворяют осадок в 2 мл соляной кислоты; промывают водой, собирая фильтрат и промывные воды в мерную колбу емкостью50 мл. Добавляют воды до 40 мл, вводят 5 мл 25%-ного роданида аммония, доливают воды до метки и перемешивают. В другой мерной колбе к 40 мл воды, 5 мл NH4SCN и 2 мл НС1 приливают из бюретки стандартный раствор FeCl3 (0,1 мг!мл} до приблизительного совпадения окрасок; доводят до метки водой и перемешивают. Оба раствора сравнивают на приборе ФЭК или Дюбоска.
Ошибка метода при содержании 0,05% Fe равняется около 5%, Для содержаний железа 5-10“3—6-10~2% применяют реакцию с тиогликолятом аммония [1084].
1 г металла растворяют в 10 мл НС1 (3 : 7), слегка охлаждают н прибавляют 10 мл 3%-ной Н2О2; перемешивают до растворения черного осадка, кипятят 1— 2 мин.; сняв с плитки, вливают 10 мл 10%-ного сульфита натрия, снова кипятят 2 мнн. и дают остыть. Переносят в 100-миллилитровый цилиндр с притертой пробкой, вливают 5 мл реактива / (180 г лимонной кислоты в 500 мл воды нейтрализуют аммиаком по лакмусу, охлаждают, прибавляют 55 мл 75%-ной тиогликолевой кислоты и разбавляют до 1 л). Приливают по каплям аммиак (уд. в. 0,880) до оранжевой окраски. Охладив, вливают для разрушения окраски урана 25 мл реактива // (250 г карбоната аммония в порошке растворяют в холодной воде, фильтруют, прибавляют 150 мл аммиака (уд. в. 0,880) и разбавляют до 2,5 л. Разбавляют водой до 100 мл и, перемешав, отливают -^40 мл, а остаток встряхивают 10 сек.; быстро измеряют светопоглощение (Спеккер) в кюветах с 7=40 мм с фильтрами Ильфорд 605 и Н503. Вводят поправку на холостой опыт. Расчет проводят по калибровочной кривой, построенной по стандартным растворам, содержащим 0,50 ... 60 мкг Fe.
Кобальт
Метод основан на измерении окраски, образуемой кобальтом с нитрозо-R-солью непосредственно в растворе азотнокислого урана в присутствии ацетата натрия. Для облегчения сравнения окрасок при малых концентрациях кобальта прибавляют определеннее количество стандартного раствора в пробу (Д. П. Малюга, 1947 г.).
Около 1 г образца растворяют в кварцевой чашке в 5 мл азотной кислоты. Раствор выпаривают на водяной бане почти досуха, остаток смывают 10 мл воды в стакан. Прибавляют 1 мл 0,1%-ного раствора нитрозо-R-соли, 3 г твердого ацетата натрия, и раствор быстро нагревают до кипения, кипятят 1 мин. При этом выделяется объемистый осадок уксуснокислого уранила-натрня; его отфильтровывают
26*
387
через небольшой фильтр (диаметр 7 см) и промывают малыми порциями насыщенного раствора ацетата натрия (не более 5—8 .ил) так, чтобы общий объем фильтрата был не больше 15.ил; прибавляют 1 мл рабочего раствора (1Q мкг/мл Со), 0,5 .ил раствора нитрозо-И-соли и нагревают до кипения.
К фильтрату, окрашенному в оранжевый или темно-зеленый цвет комплексными соединениями кобальта (никеля и меди) с нитрозо-И-солью, прибавляют 3 мл азотной кислоты (1 : 1) или больше до изменения окраски в оранжевый или желто-вато-орайжевый цвет и нагревают до кипения. При этом комплексы никеля и меди разрушаются, а окраска кобальтового комплекса с нитрозо-R солью остается. Охладив раствор, доводят его объем дистиллированной водой до 20 мл и сравнивают окраску в колориметре с окраской стандартного раствора.
Стандартный раствор для колориметрии готовят следующим путем: к 1 мл рабочего раствора кобальтовой соли (содержание 0,01 мг Со) прибавляют 1 мл 0,1%-ного раствора нитрозо-И-соли и разбавляют водой до 10 мл. Прибавив 3 г твердого ацетата натрия, нагревают до кипения. Охладив, добавляют 2 мл азотной кислоты (1 : 1), нагревают до кипения и кипятят 1 мин. По охлаждении разбавляют до 20 мл.
Расчет содержания кобальта в навеске следует производить, исходя из следующей пропорции:
с _____hi
с ф- х h ’
откуда
с(А —АО Х - h
где
с — содержание кобальта в стандартном растворе, мг;
х — искомое содержание кобальта в навеске, мг;
h — высота столба жидкости в колориметре для стандартного раствора, мм hi — то же для пробы с добавкой, мм.
Чувствительность метода 1 • 10-б% Со.
Ошибка определения ±1*10-5% при содержании кобальта меньше чем 1-10"4% (навеска 1г).
Никель
Никель может быть отделен от урана осаждением рубеановой кислотой вместе с медью (Д. П. Малюга, 1947 г.) и определен полярографически.
Около 5 г образца растворяют в кварцевой или стеклянной чашечке в 10 .мл азотной кислоты. Удаляют избыток кислоты выпариванием на водяной бане, растворяют в 50 мл дистиллированной воды, прибавляют 20 мл 16%-ного раствора лимоннокислого натрия для связывания урана в комплекс. Затем прибавляют 5 мл 0,5%-ного спиртового раствора рубеановой кислоты, и раствор сразу нейтрализуют аммиаком до слабощелочной реакции по лакмусу.
Раствор доводят водой до 100 мл и выдерживают в течение часа при 50°, затем оставляют стоять не менее 4 час. или на ночь. Образовавшийся на дне хлопьевидный осадок рубеанатов меди, никеля (кобальта, цинка и кадмия) отфильтровывают через маленький фильтр (7 см) и промывают 1%-ным раствором хлористого аммония три раза по 5—6 мл, а затем таким же количеством воды. Фильтр с осадком слегка подсушивают и еще влажным переносят в маленькую кварцевую чашечку, смачивают 4—5 каплями серной кислоты, нагревают до озоления и прокаливают в муфеле при 500—600° для разрушения всего органического вещества.
Остаток (окиси никеля, меди и др.) смачивают 5—6 каплями соляной кислоты, выпаривают до 1—2 капель, растворяют в 2 мл воды и переносят в мерный цилиндр-388
К раствору прибавляют 1 мл 0,1 Л’ хлористого аммония, 0,25 мл 0,1 7V хлористого кальция и 3 капли 0,25%-ного агар-агара. Нейтрализуют аммиаком по лакмусу до слабощелочной реакции и доводят объем раствора в цилиндре до 5 мл.
2—3 мл раствора берут в электролизер (после прибавления ртути), прибавляют маленький кристаллик сульфита и через раствор .пропускают в течение 5 мин. водород.
Снимают полярографическую кривую; потенциал восстановления никеля 0,95 в, меди —0,35 в. При употреблении сульфита для удаления кислорода наблюдаемые потенциалы становятся более положительными. Высоту полярографической волны испытуемого раствора сравнивают с высотой волны стандартного раствора.
Стандартный раствор приготовляют так: смешивают 1,0 мл раствора меди (5 мкг/мл), 1,0 мл раствора никеля (5 мкг/мл), 2 мл 0,1 2V хлористого аммония, 0,5 мл 0,1 N хлористого кальция, 2 капли агар-агара и разбавляют до 10 мл.
Чувствительность метода для никеля и меди 0,005 мг в 10 мл или 5-10~5%. Ошибки определения — 5-10-б% ,(абс.) (при навеске в 5 г).
Для количеств никеля 5 -10-4—1 -10“2% предложен метод [1086] определения с диметилглиоксимом.
Две одинаковые навески по 1 г растворяют в HNO3, выпаривают досуха, обрабатывают 1 мл H2SO4 (1 : 1), разбавляют водой до 20мл. Прибавив 2 мл насыщенной бромной воды, дают стоять 5 мин.; вливают по каплям аммиак (1 : 1) до разрушения избытка брома и до образования неисчезающего осадка диураната. Вливают 10 мл 25%-ного раствора (МН^гСОз, 2 мл 25%-ного цитрата аммония, дают остыть. Прибавляют по каплям, перемешивая, 8 мл 25%-ного раствора NaOH; устанавливают pH 4,5 + 0,5 с помощью того же раствора NaOH. В один стакан прибавляют 2 мл 1 %-ного спиртового раствора диметил глиоксима, второй оставляют без реактива для сравнения. Оба раствора разбавляют точно до 50 мл, дают стоять 30 мин. и измеряют светопоглощение при 530 ммк в кюветах с I = 50 мм; расчет ведут по калибровочной кривой, построенной по чистому урану и количествам никеля от 5 до 100 мкг.
До 50 мкг Со (II), Си (II), Сг (III) не мешают определению, а 100 мкг приводят к ошибке +3% (отн.).
Учитывая возможную неоднородность распределения никеля в уране, химики Спригфильда [1087] рекомендуют исходить из навески 50 г, не растворимый в HNO3 остаток сплавлять с бисульфатом и из соединенных (500мл) растворов отбирать аликвот (10 мл), содержащий 1.г урана; прибавляют 5 мл 5%-ного NH2OH • •НС1 и 5 мл 25%-ного цитрата аммония, нейтрализуют аммиаком (уд. в. 0,881) по лакмусу и вводят 2 мл избытка его. Добавив 2 мл 1 %-ного раствора диметил-глиоксима в абсолютном спирте, экстрагируют никель тремя порциями СНС1з по 10 мл.
Объединенные экстракты промывают дважды по 20 мл воды и извлекают никель из хлороформа дважды по 5.и л 0,5 Al НС1. Водный слой промывают 2 мл СНС1з. Кислый раствор фильтруют через влажный фильтр в 50-миллилитровый цилиндр с притертой пробкой. Прибавляют 3 мл раствора иода (1 г KJ и 0,64 г J в 500 мл воды), 1 мл 1 %-ного диметил глиоксима, перемешивают, разбавляют до 35 мл и прибавляют 10 мл 4%-ного раствора NaOH. Перемешав, разбавляют до 50 мл и измеряют светопоглощение (Спеккер) в кювете с I = 40.мм с фильтрами Н503 и Ильфорд 602. Вводят поправку на холостой опыт.
389
Медь
Содержание меди порядка 1 • 10-4—1 • 10-3% определяют по реакции с диэтилдитиокарбаматом [1085].
0,5—1 г образца растворяют при нагревании в 3 мл азотной кислоты, разбавляют немного водой и фильтруют в делительную воронку. Прибавляют 10 мл лимонной кислоты (250 гл), нейтрализуют аммиаком до pH 9. Прибавляют 1 мл комплексона III (10 г в 250 мл) для связывания Со, Ni, Bi, Ag, и Pb, разбавляют водой до 25 мл.
По охлаждении прибавляют 1 мл 1%-ного диэтилдитиокарбэмата натрия, вливают 5 мл бутилацетата и сильно встряхивают в течение 3 мин. После отстаивания разделяют слои, органический слой промывают 2 N H2SO4, фильтруют через сухой фильтр и фотометрируют при 440 ммк..
Полярографическое определение меди после осаждения рубеа-новой кислотой см. стр. 388.
Мышьяк
Мышьяк отделяют от урана перегонкой в виде бромида и определяют по молибденовой сини (П. Н. Палей, 1947 г.).
0,5—1 г металла помещают в прибор, изображенный на рис. 70 (см. стр. 384). Приливают 5 мл азотной кислоты (уд. в. 1,35) и подогревают (очень осторожно) до полного растворения металла. Усилив нагревание и включив медленное продувание воздухом через воронку, отгоняют по возможности большое количество азотной кислоты, но не доводя до образования сухой массы в колбе. Перегон отбрасывают. Дав немного остыть, вливают через воронку 10 мл бромистоводородной кислоты, отгоняют образовавшийся бром и часть кислоты, возможно большую. Вливают еще 10 мл НВг в прибор, снова отгоняют, затем прибавляют еще 5 мл НВг и опять отгоняют.
К соединенным перегонам прибавляют 20 мл HNO3, выпаривают в фарфоровой чашке на водяной бане досуха, оберегая от пыли. Остаток в чашке должен быть небольшим и бесцветным. Остаток желтого цвета (железо) для колориметрии не пригоден — он должен быть подвергнут вторичной перегонке. Остаток смачивают 1 каплей 1,0 N H2SO4, смывают дистиллированной водой до 40—45 мл, прибавляют точно 2 мл молибденового реактива, перемешивают, прибавляют 4 капли раствора хлористого олова, доводят водой до 50 мл, тщательно перемешивают и через 15—20 мин. сравнивают в колориметре синюю окраску с окраской стандарта, приготовленного одновременно с исследуемым раствором: 1 мл образцового раствора, 45 мл воды, 2 мл реактива и 4 капли SnCh разводят до 50 мл.
Ошибка определения при навеске 1г составляет 0,001%.
Реактивы
1. Молибденовый реактив готовится смешением 1 объема 10% -ного раствора молибденокислого аммония (х. ч.) с 3 объемами серной кислоты, разбавленной (1 : 1) по объему.
2. Раствор хлористого олова готовят растворением 0,1 г металлического олова при слабом подогревании в 1 мл чистой крепкой соляной кислоты, к которой прибавляют одну каплю 5%-ного медного купороса. Раствор разбавляют водой до 10 мл\ хранится не более суток.
3. Образцовый раствор мышьяковистого натрия, содержащий 100 мкг As в 1 мл, готовят растворением 0,565 г соли (х. ч.) в 1 л воды.
390
Цирконий
Цирконий может быть определен спектрофотометрически [1088] в количествах от 1 мкг.
К раствору урана, содержащему 1 —10 мкг Zr, прибавляют 1 мл реактива <2%-ный спиртовый раствор р-(р-диметиламинофенил-азо)фенил арсоновой кислоты, содержащий 10.ил 2.V1 НС1 в 1 л). Нагревают 15 мин. при 60—75°, оставляют на ночь. Осадок отделяют, растворяют в 5 М HF и горячем 0,4 М растворе щавелевой кислоты и разбавляют до точного объема. Определяют светопоглощение при 490 ммк, сравнивая с водой.
Точность около 5% (отн.).
Молибден
Количества молибдена от 1-10“4% до 2,5-10“3% могут быть определены по реакции с толуен-3: 4-дитиолом [1089].
1 г образца растворяют в 25 мл НС1 (1 : 3), прибавляют 30%-ной Н2О2 по каплям до осветления, выпаривают досуха. К остатку прибавляют 10 мл НС1 (1 : 3), подогревают до полного растворения. Фильтруют, промывают 50 мл НС1 (1 : 20) и разбавляют до концентрацииМо меньше 30 мкг в 10 .ил. Аликвот 10 мл (или стандарт) помещают в 50-миллилитровую мерную колбу, прибавляют 20 мл НС1 (уд. в. 1,18), перемешивают, прибавляют 15 мл n-бутилового спирта.
Охладив до комнатной температуры, прибавляют 4 мл реактива *, перемешивают. Выдерживают при 20° в течение 30 мин., разбавляют до метки п-бутиловым спиртом и измеряют светопоглощение в кюветах с I = 40 мм при 680 ммк, сравнивая с водой или суммой реактивов.
Реакция с роданидом может быть тоже использована в сочетании с экстракцией [1090].
Навеску в 1 г, содержащую не более 5 -10~3% Мо, растворяют в 10 мл НС1 (1 : 1), добавляя по каплям пергидроль до полного растворения; выпаривают досуха, растворяют в воде, нейтрализуют до появления неисчезающей мути, которую растворяют добавлением минимального объема НС1 (1 : 1). Переносят в делительную воронку, вводят 2 мл НС1 (уд. в. 1,18) и разбавляют до 40 мл. Прибавляют 10 мл 5%-ного KSCN и смешивают, добавляют 2 мл 5%-ного NaNO2, затем 8лм 10%-ного раствора SnCU, перемешивают и оставляют на 2 мин. Вводят 40 мл экстрагента (2 объема диэтилового эфира и 1 объем петролейного эфира, кипящего при 100—125°; 100 мл смеси промывают смесью 10 мл 5%-ного KSCN и 10 мл 10%-ного SnCk), встряхивают сильно 1 мин. Отделяют эфирный слой, фильтруют его в кювету с Z = 40 мм фотометра (Спеккер) и сразу накрывают ее стеклом. Измеряют светопоглощение с фильтрами Н503 и Ил ьфорд 602. Вводят поправку на холостой опыт.
Рутений
Рутений в присутствии больших количеств урана может быть определен в виде рубеаната [1091].
К раствору, содержащему 10—15 мкг Ru в 25 мл, прибавляют 1 мл концентрированной НС1 и 1—2 мл 0,1 %-ного спиртового раствора рубеановодородной кислоты. Смесь нагревают 5 мин. на кипящей водяной бане, по охлаждении экстра-
* Реактив: разламывают ампулу с 1 г толуен-3 : 4-дитиола под раствором 1 %-ного NaOH (ООО мл), растирают палочкой до растворения; медленно с перемешиванием вливают 1 мл 76%-ной тиогликолевой кислоты. Хранят плотно закрытым в холодильнике.
391
гир^ют тремя порциями изоамилового спирта по 5 .ил. Экстракты собирают в мерную колбу емкостью 25 .ил и доводят до метки изоамиловым спиртом. Измеряют светопоглощение при 650 ммк.
Палладий и платину выделяют из горячих 2 N по H2SO4 растворов сероводородом при соосаждении на сульфиде меди (0,1 мг Си на 1 г U); растворенный в царской водке осадок анализируют спектрально (см. стр. 368).
Серебро
5 г образца растворяют в фарфоровой чашке, постепенно прибавляя 15 .ил концентрированной азотной кислоты. Избыток HNO3 удаляют выпариванием‘на водяной бане, и полученный нитрат растворяют в воде; отфильтровывают раствор от кремневой кислоты, хорошо промывают фильтр несколько раз водой и доводят объем раствора приблизительно до 50 мл. Раствор переносят в делительную воронку емкостью 200—250 мл, прибавляют к нему 5 мл серной кислоты (1 : 5) и 0,1г сернокислого гидроксиламина или гидразина, прибавляют из бюретки 0,2 мл 0,004%-ного раствора дитизона в СС14 и встряхивают 15—20 сек. Окрашивание слоя СС14 не в желтый цвет, а фиолетово-розовый цвет указывает на то, что в исследуемом растворе нет ни Ag, ни Hg. Фиолетово-розовый цвет появился от реакции дитизона с Си. В присутствии Ag или Hg слой СС14 окрашивается в желтый цвет. Тогда окрашенный слой СС14 отделяют от водной фазы, сливая его в другую делительную воронку. К водному раствору прибавляют 1 мл (0,008% -ного) раствора дитизона, встряхивают и снова отделяют слой СС14 во вторую воронку. Эту операцию повторяют до тех пор, пока экстракт окрасится в фиолетово-розовый цвет (в отсутствие меди раствор окрашивается при этом в зеленый цвет от избытка дитизона).
Продолжают извлечение 1 мл раствора дитизона еще 2—3 раза при более длительном встряхивании (30—40 сек.), сливая фиолетово-окрашенный слой в раствор дитизоната серебра. Полученные экстракты СС14 промывают встряхиванием в делительной воронке с S0 мл 1 %-ного раствора серной кислоты. Сливают СС14 в новую делительную воронку, затем прибавляют к СС14 3 раза подряд смесь, состоящую из 5 мл 2%-ного раствора роданистого калия и 1 мл 1%-ной серной кислоты, каждый раз сливая СС14. Образовавшийся при этом роданид серебра переходит в водный раствор.
Все растворы с роданидом промывают чистым СС14 (3—5 мл) для удаления капель окрашенного СС14, а затем водный раствор выпаривают в кварцевой чашке (диаметр 3—4 см) на водяной бане досуха.
К сухому остатку прибавляют 3 мл концентрированной серной кислоты и выпаривают раствор сначала на песчаной бане до удаления паров SO3, а затем прокаливают на электроплитке в течение 15—20 мин. (для полного разрушения роданида). После охлаждения к остатку приливают 2 мл серной кислоты (1 : 5), сливают раствор в делительную воронку, чашку споласкивают несколько раз водой (общий объем воды 18 мл). Титруют серебро 0,004%-ным раствором дитизона (свежеприготовленного разбавлением в 10 раз основного раствора) из бюретки емкостью 10 мл (лучше из коричневого стекла), прибавляя его по 0,1—02, мл непосредственно в делительную воронку и каждый раз после встряхивания отделяя желто-окрашенный слой четыреххлористого углерода от водного раствора. Титрование считают законченным, как только слой СС14 окрашивается в розоватый цвет (в присутствии следов меди) или зеленоватый (в отсутствие меди). Титр дитизона устанавливают по 1 мл стандартного раствора азотнокислого серебра (т. е. 10 мкг Ag) (С. И. Синякова, Л. А. Цветкова, 1947).
392
Кадмий
5 г образца растворяют в фарфоровой чашке в 10 мл концентрированной азотной кислоты. Чашку прикрывают часовым стеклом до тех пор, пока реакция растворения происходит бурно. Затем раствор выпаривают на водяной бане досуха, остаток растворяют в 40—50 мл воды и раствор сливают в делительную воронку емкостью 200 мл. К раствору прибавляют 12 мл 20%-ного лимоннокислого натрия, 0,2 г солянокислого гидроксиламина, нейтрализуют аммиаком до pH 7 по лакмусу или другому индикатору и прибавляют еще избыток аммиака 2—3 капли. Приливают 5 мл 0,04% -ного раствора дитизона в четыреххлористом углероде и встряхивают 1—2 мин. Окрашенному слою СС14 дают хорошо отделиться от водного и спускают в другую делительную воронку. К водному раствору снова приливают 5 мл 0,04%-ного раствора дитизона, опять встряхивают и снова сливают отстоявшийся слой СС14 во вторую делительную воронку. Эту операцию повторяют до тех пор, пока слой СС14 не перестанет изменять своей окраски. Собранные во второй делительной воронке экстракты встряхивают два раза с 5 мл воды, и водный слой отбрасывают. Затем к экстракту прибавляют 4,5 мл 0,01 N соляной кислоты и 2 мин. сильно встряхивают. При этом кадмий переходит в водный слой. Отделяют слой СС14 в другую воронку, вновь прибавляют к нему 4,5 мл 0,01 N соляной кислоты и снова встряхивают. Слой СС 14 отделяют и отбрасывают. Солянокислые растворы соединяют, прибавляют несколько капель чистого СС14 и встряхивают для удаления оставшихся в водном растворе окрашенных следов дитизонатов других металлов. Отделяют слой четыреххлористого углерода и отбрасывают. Эту операцию повторяют до тех пор, пока слой четыреххлористого углерода не перестанет окрашиваться дитизонатами. Остатки четыреххлористого углерода следует отделить очень тщательно, чтобы раствор совсем не содержал его.
Водный раствор переносят в мерную колбочку емкостью 10 мл, споласкивают делительную воронку несколькими каплями 0,01 N соляной кислоты и объем раствора доводят до метки 0,01 N соляной кислотой.
Для колориметрического определения берут 5 мл раствора. Колориметрирование производят в серии одинаковых плоскодонных пробирок емкостью 10 мл (размером примерно 1,8 X 15 см) с делениями и с притертой пробкой.
Готовят стандартную шкалу следующим образом: в первую пробирку берут 0,1 мл стандартного раствора с содержанием 0,50 мкг Cd в 1 мл, во вторую0,2 мл того же раствора и т. д. Таким образом получают шкалу с содержанием Cd в пробирках: 0,050; 0,100; 0,200 мкг и т. д. Объемы во всех пробирках со стандартами и с пробой доводят до 5 мл 0,01 N соляной кислотой, прибавляют затем по 2,0 мл 25%-ного раствора едкого натра и растворы взбалтывают. Затем во все пробирки-прибавляют по 1 мл 0,004% -ного раствора дитизона в четыреххлористом углероде, закрывают их пробкой и сильно взбалтывают 10—15 раз.
Полученную окраску пробы сравнивают со стандартной шкалой на белом фоне после взбалтывания и отстаивания слоя СС 14 в течение 5 мин. Окраски лучше сравнивать при дневном освещении. Следует также произвести контрольный опыт для определения поправки на чистоту реактивов.
Точность метода около 10% при относительном содержании'кад-мия л-10-5%. При содержании его л-10-4% точность увеличивается (С. И. Синякова, Л. А. Цветкова, 1947).
Редкоземельные элементы
Спектральный анализ выделенных из урана редких земель описан на стр. 368. Из химических методов выделения р.з.э. наиболее простым и надежным является осаждение их в виде фторидов из бикарбонатной среды (Е. К. Гольбрайх, 1950).
393.
50 г урана растворяют в стакане емкостью 800 мл в HNO3 (уд. в. 1,4), прибавляя ее небольшими порциями до полного растворения. Раствор выпаривают до небольшого объема, не доводя до кристаллизации. Охлаждают, прибавляют 200 мл воды. Вводят носители — 1 мл раствора Се (NO3)3 (1 мг СеО2) и 1 .w.z раствора Ca(NO)a (150 мг СаО).
Медленно (при энергичном перемешивании) постепенно приливают 10%-ный раствор NaOH до появления слабой постоянной мути. Добавляют порциями 60 е сухого NaHCO3. После перевода урана в уранилдикарбонат (раствор должен стать прозрачным) мешалку отмывают горячей водой. Приливают 100 мл 3%-ного раствора NaF, подогревают до 60—70°. Охлаждают и дают стоять ночь. Осадок отфильтровывают (белая лента) и промывают холодной водой, к которой прибавлен 3% -ный раствор NaF до щелочной реакции по лакмусу.
Осадок на фильтре растворяют в НС1 (1 : 1) и промывают фильтр горячей водой. Раствор упаривают досуха до отсутствия запаха НС1. Остаток растворяют в 10 мл воды, доводят до кипения и приливают несколько миллилитров аммиака (до слабого запаха).
Для отделения оставшегося урана прибавляют 100 мл 10%-ного раствора Н2С2О4; кипятят 2—3 мин. Охладив, оставляют стоять на ночь. Осадок (Са, Се, р. з. э.) отфильтровывают (фильтр «синяя лента»), промывают водой, подкисленной Н2С2О4 (20 мл 10%-ной Н,С2О4 на 500 мл) до полного отделения урана (проба с K4Fe(CN)6).
Осадок сжигают и прокаливают при 700 в течение 30 мин. Остаток переносят в стакан, растворяют в НС1 (1 : 1), раствор упаривают досуха (до удаления запаха НС1); растворяют в 10 мл воды, прибавляют 2—3 мл 0,25 N НС1 и кипятят 1 мин. Охлаждают до 70° и пропускают H2S в течение 20 мин. для выделения РЬ. Стакан накрывают часовым стеклом и дают стоять 5—10 мин. до просветления жидкости над осадком. Осадок сульфидов фильтруют (фильтр «синяя лента»), промывают 4—-5 раз сероводородной водой; фильтрат упаривают досуха. Остаток растворяют в 5—7 мл воды, доводят до кипения, прибавляют несколько капель аммиака (до слабого запаха), охлаждают. Осадок (Се, р. з. э.) отфильтровывают, промывают водой, подщелоченной аммиаком, до исчезновения реакции на кальций со щавелевой кислотой. Осадок растворяют в НС1 (1 : 1), промывают фильтр горячей водой и упаривают досуха; растворяют в 5 мл воды и повторяют аммиачное осаждение. Осадок, промыв, снова растворяют в НС1, упаривают досуха и до удаления НС1, растворяют в 3 мл воды, прибавляют 3-кратный объем 10%-ной Н2С2О4, кипятят 2 мин. и оставляют на ночь. Осадок оксалатов р. з.э. отфильтровывают через малый фильтр («синяя лента»), снимая частицы осадка со стенок кусочком влажного беззольного фильтра, сжигают и прокаливают при 800°. Взвешивают и анализируют рентгено-спектральным методом.
Чувствительность метода п-10~5%, ошибка — 15% (отн.).
Золото
1 г металла растворяют в фарфоровой чашке в 5 мл концентрированной азотной кислоты. Раствор выпаривают на водяной бане досуха, остаток растворяют в 5 мл царской водки. Раствор выпаривают на водяной бане досуха, остаток смывают в стакан с помощью 100 мл разбавленной (1 : 20) соляной кислоты.
Этот раствор в делительной воронке тщательно взбалтывают с 1 мл 0,04%-ного раствора дитизона в СС14; при этом происходит образование дитизонатов Au, Hg, Си, которые переходят в слой СС14. Затем дитизонаты в СС14 отделяют от водного раствора. Аналогичную операцию проводят еще раз. Конец извлечения определяют по появлению фиолетовой окраски, характерной для дитизоната меди. Ди-тизонат промывают 5 мл разбавленной соляной кислоты для удаления следов уранила и затем производят выпаривание СС14 на водяной бане.
Остаток от выпаривания прокаливают в муфеле при 600—700°. Прокаленный остаток растворяют в 1 мл царской водки, затем выпаривают досуха, прибавляют 1 мл концентрированной соляной кислоты и вновь выпаривают досуха. Это необходимо для полного удаления возможно присутствующего свободного хлора и азот-394
ной кислоты (которые мешают при реакции с о-толидином, давая с ним желтую окраску).
Остаток растворяют в 1 мл разбавленной соляной кислоты и к нему прибавляют 1 мл раствора о-толидина (0,1%-ный раствор о-толидина в 10%-ной соляной кислоте). При этом образуется желтое окрашивание^ которое сравнивают со стандартом в микроколориметре. Окраска возникает через 5—10 мин. и сохраняется в течение 30 мин. Колориметрировать следует через 15 мин.
Стандарты золота приготовляют путем разбавления слабой 1%-ной соляной кислотой 0,01% -ного раствора треххлористого золота до содержания 10; 5; 2 и 1 мкг Au в 1 мл стандартного раствора.
Ошибка определения около 10% при содержании золота от 1* 10"а до 1-10-5% (К. В. Троицкий, 1948).
Ртуть
Метод основан на извлечении ртути из солянокислого раствора дитизоном (серебро при этом связывается в AgCl) и разрушении полученного дитизоната ртути бромистым калием. В присутствии буферной смеси ртутный комплекс диссоциирует, и ион ртути титруется дитизоном.
2 г образца растворяют в плоскодонной с длинным горлышком колбе в возможно малом (2—3 мл) количестве концентрированной азотной кислоты, прикрыв колбу стеклом, на холоде; под конец опускают колбу в горячую воду для полного растворения металла. После окончания растворения прибавляют 5—10 мл дистиллированной воды и нейтрализуют избыток'кислоты аммиаком (1:5) до появления неисчезающей мути, которую растворяют несколькими каплями концентрированной соляной кислоты. Раствор переносят в делительную воронку, прибавляют 25 мл 0,25 N соляной кислоты, 5 мл 20%-ного раствора солянокислого гидроксиламина и 10 мл 0,004%-ного раствора дитизона в СС14. Встряхивают 1 мин., дают слою СС14 отстояться и спускают его в другую воронку, содержащую 25 мл 0,25 N НС1. Повторяют снова экстракцию с 10 мл дитизона и снова спускают слой СС14 во вторую воронку. Собранные экстракты во второй воронке встряхивают г/2 мин., затем слой СС14, содержащий все дитизонаты,спускают в третью воронку, содержащую 25 мл 0,25 N НС1, и прибавляют 5 мл 40% -ного раствора бромистого калия. Встряхивают 1/2 мин., отделяют слой СС14, содержащий дитизонат меди, и отмывают чистым СС14^5—10 мл). Отделяют последний очень тщательно и, прибавив к водному слою 10 мл буферного раствора (150 г Na2HPO4-{- 38 г К2СО3 в 1 л), титруют ртуть 0,004%-ним раствором дитизона (свежеприготовленным разбавлением основного раствора дитизона в 10 раз), прибавляя его из бюретки по 0,2 мл, непосредственно в делительную воронку и каждый раз после встряхивания отделяя оранжевый слой четыреххлористого углерода от водного раствора. Титрование считают законченным, как только слой СС14 окрашивается в ярко-розовый цвет. Титр дитизона устанавливают по стандартному раствору азотнокислой ртути (С. И. Синякова, Л. А. Цветкова, 1948).
Свинец
Микрограммовые' количества свинца отделяют от урана экстракцией дитизоната хлороформом [1092]. Экстракцию ведут в щелочной среде в присутствии цитрата, NH2OH-HC1 и KCN. Экстракт промывают аммиачным раствором цианида и свинец определяют колориметрически.
ЛИТЕРАТУРА
1. П. К. Агасян. Зав. лаб., 22, 7 (1953).
2. О. П. Ага фо шин. ЖОХ, 22, 177 (1952)
3. Г. В. Акимов, И. Л. Розенфельд. Сб.: Исследования в области электрохимического и коррозионного поведения металлов и сплавов. М.г Оборонгиз, 1950, стр. 201.
4. Актиниды (под ред. Г. Сиборга и Дж. Каца). М., ИЛ, 1955.
4а. А. И. А л е к п е р о в, С. И. Жданов. ЖНХ, 5, 1743 (I960).
5. Л. С. Александрова, Т. Б. Гапон, Н. Ф. Филатова, К- В.
Ч мутов. Труды Комиссии по аналит. химии, 6(9), 235 (1955).
6. И. П. Ал и марин, Ю. А. Золотов. ЖАХ, 12, 176 (1957).
7. В. Л. Альб а нс кий. ЖОХ, 21, 1393 (1951).
8. Аналитическая химия урана и тория (переводе англ, под ред. П. Н. Палея). М., ИЛ, 1956.
9. Л. Я. А н т р а ш е н о к, Л. Л. В о л о ш е н к о, А. Я- Крылов. Труды Радиевого ин-та АН СССР, 7, 126 (1956).
10. Атлас эффективных нейтронных сечений элементов (под ред. Ю. В. Адамчука). М., Изд-во АН СССР, 1955.
11. А. К. Бабко. Зап. Ин-та химии АН УССР, 8, вып. 2 (1946).
12. А. К- Б а б к о, А. И. В о л к о в а. ЖОХ, 22, 1108 (1952).
13. В. И. Баранов. Радиометрия. М., Изд-во АН СССР, 1956.
14. В. Бауман. Сб.: Ионный обмен (под ред. К. В. Чмутова). М., ИЛ, 1951, стр. 49.
15. Ю. И. Б е л я е в, А. Н. 3 а й д е л ь. ЖАХ, 12, 30 (1957).
16. Р. Берг. Применение оксихинолина в аналитической химии. М., ОНТИ, 1937.
17. Р. Блок, Р. Л е с т р а н ж, Г. Цвейг. Хроматография на бумаге. М.. ИЛ, 1954.
18. Г. Н. Б и л и м о в и ч, И. П. А л и м а р и н, ЖАХ, 12, 685 (1957).
19. С. Е. Бреслер. Радиоактивные элементы. М., Гостехтеоретиздат, 1957.
20. В. М. Б р о д с к а я, Г. А. Л а н с к о й, В. Г. С о ч е в а н о в. ЖАХ. 16, 185 (1961).
21. Ф. Р. Брус. Мирное использование атомной энергии. Матер. Международной конференции в Женеве, 1955, г., т. 7, М., Госхимиздат, 1958, стр. 127.
21а. С. А. Брусиловский. ДАН СССР, 120, 305 (1958).
22. Е. А. Б р у м б е р г, 3. М. Свердлов. Изв. АН СССР, сер. физ., 4, 75 (1940).
23. О. И. Б у ф а т и н, А. Н. 3 а й д е л ь, Н. И. К а л и т е е в с к и й. ЖАХ, 13, 116 (1958).
23а. С. И. В а в и л о в. Изв. АН СССР, сер. физ., 13, 18 (1949).
24. С. И. Вавилов. Соб. соч., т. 1. М., Изд-во АН СССР, 1954, стр. 242.
25. С. И. Вавилов. Соб. соч., т. 2. М., Изд-во АН СССР, 1954, стр. 261, 383.
26. Э. Е. Вайнштейн. Изотопы и излучения в химии. М., Изд-во АН
СССР, 1958, стр. 171.
396
27. Э. Е. Ва й н штейн, Ю. И. Беляев. ЖАХ, 13, 388 (1958).
27а. В. М. В д о в е н к о. Химия урана и трансурановых элементов. М., Изд-во АН СССР, 1960.
28. В. М. Вдовенко. ЖНХ, 3, 145 (1958).
29. В. М. В д о в е н к о, Л. Н. Лазарев. ЖНХ, 3, 155 (1958).
30. В. М. В довенк о, А. А. Л и п о в с к и й, М. Г. Кузьмина. ЖНХ, 2, 975 (1957).
31. В. М. Вдовенко, А. А. Л и п о в с к и й, С. А. Никитина. Тезисы доклада на Всес. конф, по химии комплексных соединений. Киев, 1959, стр. 38. *
32. В. М. В д о в е н к о, Т. В. Ковалева. Сборник работ по радиохимии. Л., Изд-во ЛГУ, 1955, стр. 44.
33. В. М. В д о в е н к о, Т. В. Ковалева. ЖНХ, 2, 1687 (1957).
34. В. М. Вдовенко, М. П. Ковальская, Э. А. Москалева, Труды Радиевого ин-та АН СССР, 8, 17 (1958).
35- В. М. Вдовенко, Т. В. Ковалева. Труды Радиевого ин-та АН СССР, 8, 22 (1958).
36. В. М. Вдовенко, И. Г. Суглобова. ЖНХ, 3, 1403 (1958).
37. В. М. Вдовенко, М. П. Ковальская, М. М. Ге р б а н ев-с к а я. Труды Радиевого ин-та АН СССР, 8, 8 (1958).
38. В. М. Вдовенко, М. П. Ковальская, Т. В. Ковалева. ЖНХ, 2, 1677 (1957).
39. Е. А. В е р н ы й, В. Н. Егоров. ЖАХ, 15, 24 (1960).
40. А. П. Виноградов. Мирное использование атомной энергии. Матер. Международной конф, в Женеве, 1955 г., т. 8, М., Металлургиздат, 1958, стр. 247.
41. С. А. Вознесенский. Труды Всес. конф, по аналит. химии, М., Изд-во АН СССР, 1, 60 (1939).
42. И. Галь. Мирное использование атомной энергии. Матер. Международной конф, в Женеве., 1955 г., т. 8. М., Металлургиздат, 1958, стр. 415.
43. Г а о С я о-с я, Л ю Ц з я-щ у, У В а н ь-с я н ь, Л и А н ь-м о. Acta sclent, natur. Univ. Petynesis, 4, 89 (1958); цит. по РЖХим, 30937 (1959).
43а. Гао Ца й-ш э н. Кандидатская диссертация. М., ГЕОХИ АН СССР (1960).
44. Е. Н. Г а п о н, Т. Б. Г а п о н. Сб.: Хроматографический метод разделения ионов (под ред. Е. Н. Гапона). М., ИЛ, 1949, стр. 9.
45. В. И. Герасимовский. Атомная энергия, 7, 47 (1959).
46. В. Ф. Г и л л е б р а н д, Г. Э. Л е н д е л ь, Г. А. Б р а й т, Д. И. 3 о ф-м а н. Практическое руководство по неорганич. анализу (перевод под ред. Ю. Ю. Лурье). М., Госхимиздат, 1957.
47. С. Г л е с с т о н. Введение в электрохимию. М., Госхимиздат, 1951.
48. В. А. Головня, Г. Т. Болотова. Тезисы доклада на Всес. конф, по химии комплексных соединений. Киев, 1959, стр. 46.
48а. В. А. Г о л о в н я, Г. Т. Болотова. ЖНХ, 6, 566 (1961).
49. Г. Р. Гольбек. В. В. Матвеева, Р. С. Ш л я п н и к о в. Мирное использование атомной энергии. Матер. Международной конф, в Женеве, 1955 г., т. 8. М., Металлургиздат, 1958, стр. 278.
50. В. И. Г о л ь д а н с к и й. Новые элементы в периодической системе Д. И. Менделеева. М., Изд-во АН СССР, 1953.
51. В. И. Г о л ь д а н с к и й. Природа, № 7, 51 (1952).
52. Б. Г о л ь д ш м и д т, П. Р е н ь о, И. Прево. Мирное использование атомной энергии. Матер. Международной конф, в Женеве, 1955 г., т. 9. Л., Госхимиздат, 1958, стр. 606.
53. Я. П. Г о х ш т е й н, В. В. Жирова. Труды V сессии Комиссии по определению абсолютного возраста геологич. формаций. М., Изд-во АН СССР, 1958, стр. 316.
-53а. В. Ф. Григорьев, В. Ф. Лукьянов, Е. П. Дудерова. ЖАХ, 15, 184 (1960).
54. А. А. Гринберг. Введение в химию комплексных соединений. Л.—М., Госхимиздат, 1951.
397
55. А. А. Г р и н б е р г, Л. Е. Н и к о л ь с к а я, Г. И. П етр ж а к, Б. Ф. Птицын, Ф. М. Филино в. ЖАХ, 12, 92 (1957).
56. А. А. Г р и н б е р г, 3. И. П е т р ж а к. Труды Радиевого ин-та АН СССР, 7, 17, 50 (1956).
57. А. А. Г р и н б е р г, Г. И. П етр ж а к, Л. Е. Никольская, Б. В. Птицын, Ф. М. Филино в. Труды Радиевого ин-та АН СССР, 8, 166 (1958).
58. А. А. Г р и н б е р г, Б. В. П т и ц ы н, Е. Н. Т е к с т е р. Труды Радиевого ин-та АН СССР, 7, 76 (1956).
59. А. А. Г р и н б е р г, А. Д. Т р о и ц к а я. Труды Радиевого ин-та АН СССР, 7, 5 (1956).
60. Р. Р. Г р и н с т е д, К. Г. Ш о у, Р. С. Л о н г. Мирное использование
атомной энергии. Матер. Международной конф, в Женеве, 1955 г., т. 8. М., Металлургиздат, 1958, стр. 90.
61. А. М. Гуревич. Труды Радиевого ин-та АН СССР, 6, 88 (1957).
62. А. М. Гуревич, М. Л. Ковальская-Ященко. Труды Радиевого ин-та АН СССР, 8, 53 (1958).
63. А. М. Гуревич, Л. Д. П р е о б р а ж е н с к а я. ЖНХ, 3, 2512 (1958).
64. А. М. Г у р е в и ч, Л. Д. Преображенская, Н. П. О с и ч е в а. Труды Радиевого ин-та АН СССР, 8, 56 (1958).
65. А. В. Д а в ы д о в, Т. С. Д о б р о л ю б с к а я, А. А. Н е м о д р у к. ЖАХ, 16, 68 (1961).
66. М. К. Джоши. ЖАХ, 11, 495 (1956).
67. Т. С. Д о б р о л ю б с к а я. ЖАХ, 11,3 (1956).
68. Т. С. Д о б р о л ю б с к а я. Труды Комиссии по аналит. химии, 8 (11),
178 (1958).
69. А. М. Дымов, Р. С. Молчанова. Зав. лаб., 7, 653 (1938).
70. С. В. Е л и и с о н, В. А. О л е з н ю к. ЖАХ, 13, 95 (1958).
71. Е. Д. Елисеева. Труды Комиссии по аналит. химии, 6 (19), 439 (1955).
71а. В. Ф. Е с к е в и ч, Л. А. Комарова, ЖАХ, 15, 84 (1960).
72. Е. И. Ж е л е з н о в а, Д. В. Т о к а р е в а. Зав. лаб., 24, 959 (1958).
73. В. В. Ж и р о в а, Я. П. Г о х ш т е й н. Труды IV сессии Комиссии по определению абсолютного возраста геологии, формаций. М., Изд-во АН СССР, 1957, стр. 241.
74. А. Н. Г а й д е л ь, Н. И. К а л и т е е в с к и й, Г. Г. К у н д, 3. Т.
Ф р а т к и н. Оптика и спектроскопия, 2, 28 (1957).
75. А. Н. Зайдель, Н. И. К а л и т е е в с к и й, Л. В. Л и п и с.
М. П. Ч а й к а. ЖАХ, 12, 17 (1957).
76. А. Н. 3 а й д е л ь, Н. И. К а л и т е е в с к и й, Л. В. Л и п и с,
М. П. Ч а й к а, Ю. И- Беляев. ЖАХ, 11, 21 (1956).
76а. А. Н. Зайдель, Н. И. К а л и т е е в с к и й, Л. В. Л и п ис, М. П. Чайка. Эмиссионный спектральный анализ атомных материалов. Л.—М., Физматгиз, 1960, стр. 348, 393.
77. А Н. 3 а й д е л ь, Н. И. К а л и т е е в с к и й, А. Н. Разумовский. Сб.: Редкоземельные элементы. М., Изд-во АН СССР, 1958, стр. 239.
78. А. Н. 3 а й д е л ь, Н. И. К а л и т е е в с к и й, А. Н. Разумовский, П. П. Я к и м о в а. Сб.: Редкоземельные элементы. М., Изд-во АН СССР, 1958, стр. 251.
79. В. М. Звенигородская. Определение малых количеств урана. М., Госгеолиздат, 1946.
80. В. М. Звенигородская,
ДЕ А. П о н е м у н с к а я,
Н. А. Т и-
х о н о в а. Матер, научн.-методич. и производ. лабораторий геол, управлений. Комитет по делам геологии при СНК СССР, № 2 (14), 1 (1944).
80а. Я- И. Зильберман, Б. И. Пищевицкий. Радиохимия, 9, 663 (1960).
81. И. Е. 3 и м а к о в, П. С. Р о ж а в с к и й. Зав. лаб., 24, 922 (1958).
82. Н. П. Иванов. Труды 10-го Всес. совещания по спектроскопии, т. 2. Изд. Львовского университета, 1958.
83, В. А. Измаильский. ДАН СССР, 26, 906, 912 (1940).
398
84. Инструкция к фотоэлектрическому люминесцентному фотометру. М., Гос-оптический институт им. С. И. Вавилова, 1956; В. А. Ф е д о р о в, С. И. Ф р е й в е р т. Сб.: Методы люминесцентного анализа. Минск, Изд-во АН БССР. I960, стр. 27.
85. С. В. И о х е л ь с о н, Е. В. Ш и т о в. Изв. АН СССР, сер. геофиз., № 1 (1959).
86. И с и д а т э, Я м а н э. J. Pharmac. Soc. Japan, 77, 386, 391, 396 (1957); цит. по РЖХим, № 1, 1028 (1958).
87. Н. И. К а л и т е е в с к и й, А. А. Л и п о в с к и й, А. Н. Ра зумов-с к и й, П. П. Якимова. ЖАХ, 13, 372 (1958).
88. Е. А. Каневский. ЖПХ, 17, 514 (1944).
88а. Е. А. Каневский, Г. Р. Павловская. ЖНХ, 5, 1738, 1964 (1960).
89. Б. Я. Каплан, Зав. лаб., 23, 771 (1957).
90. Г. Е Каплан, Б. Н. Л а с к о р и н, Б. В. Невский. Атомная энергия, 6, 113 (1959).
91. А. Г. Кара баш. ЖНХ, 3, 986 (1958).
92. С. М. К а р п а ч е в а, Л. П. X о р х о р и н а, Г. Д. Агашкина. ЖНХ, 2, 961 (1957).
93. С. М. К а р п а ч е в а, Л. П. X о р х о р и н а, А. М. Р о з е н. ЖНХ, 2, 1441 (1957).
93а. В. И. Карпов. ЖНХ, 6, 531 (1961).
94. Л. Е. Кауфман. ДАН СССР, 27, 807 (1940).
95. Л. Е. Кауфман. Зав. лаб., 9, 106 (1940).
96. Л. Е. Кауфман. Зав. лаб., 9, 228 (1940).
97. Дж. Кац, Е. Рабинович. Химия урана. М., ИЛ, 1954.
98. А. И. К и п р и а н о в, И. К- У ш е н к о. Изв. АН СССР, ОХН, 5, 492
(1950).
99. Г. А. Клейбс. ЖНХ, 1, 2177, 2198 (1956).
100. Г. А. Клейбс. ЖНХ, 3, 2621 (1958).
101. А. Е. Клыгин, Д. М. 3 а в р а ж н о в а, И. С. К о л я д а. ЖАХ,.
16, 442 (1961).
101а. А. Е. Клыгин, И. С. Коляда. ЖНХ, 4, 86 (1961).
102. А. Е. К л ы г и н, И. Д. Смирнова. ЖНХ, 4, 42 (1959).
ЮЗ. А. Е. Клыгин, И. С. Кол я да. ЖНХ, 4, 239 (1959).
104. И. М. Коган. Химия красителей. М., Госхимиздат, 1956.
104а. А. Г. Козлов. Н. И. Крот. ЖНХ, 5, 1959 (1960).
1046. И. М. К о л ь т г о ф, Е. Б. С е н д э л. Количественный анализ. М., Госхимиздат, 1941, стр. 442.
105. Н. П. К о м а р ь, 3. А. Третьяк. ЖАХ, 10, 236 (1955).
106. И. М. К о р е н м а н, Ф. Р. Ш е я н о в а. Химия и химич. техноло-
гия, 2, 151 (1959).
107. Г. Е. Кочарова, А. П. Комар, Г. А. К о р о л е в, И. Н. Маро в, Ю. А. Сурков. Изв. АН СССР, сер. физ., 23, 855 (1959).
108. Краткая энциклопедия «Атомная энергия». М., Изд-во БСЭ, 1958, стр. 456.
109. К. А. К р а у с, Ф. Нельсон. Мирное использование атомной энергии. Матер. Международной конф, в Женеве, 1955 г., т. 7. М., Госхимиздат, 1958, стр. 144.
ПО. П А. Крюков. Сб.: Современные методы исследованияфизико-химич. свойств почв, вып. 2. М.—Л., Изд-во АН СССР, 1947, стр. 61.
111. Т. А. К р ю к о в а, С. И. Синякова, Т. В. А р е ф ь е в а. Полярографический анализ. М., Госхимиздат, 1959
112. В. И. Кузнецов. ДАН СССР, 31, 895 (1941).
113. В. И. Кузнецов. ДАН СССР, 50, 233 (1945).
114. В. И. Кузнецов. ДАН СССР, 52, 39 (1946).
115. В. И. Кузнецов. ЖАХ, 2, 67 (1947).
116. В. И. Кузнецов. ЖАХ, 9, 199 (1954)
117. В. И. Кузнецов. ЖАХ, 13, 220 (1959).
118. В. И. Кузнецов. ЖАХ. 14, 7 (1959).
399
119. В. И. Кузнецов. ЖОХ, 20, 816 (1950).
120. В. И. Кузнецов. Приемы отыскания цветных реакций для неорганич. ионов. Докт. диссертация. М., ГЕОХИ АН СССР, 1948.
121. В. И. Кузнецов. Сессия АН СССР по мирному использованию атомной энергии 1—5 июня 1955 г., Заседание ОХН. М., Изд-во АН СССР, 1955.
122.
123.
124.
125.
126.
127.
128.
129.
130.
131.
132.
В. И.
В. И.
В. И.
В. И.
В. И.
В. И.
Кузнецов. Кузнецов. Кузнецов. Кузнецов. Кузнецов, Кузнецов,
Зав. лаб., 24, 1178 (1958)
Труды Комиссии по аналит. химии, 6 (9), 249 (1955). Труды Комиссии по аналит. химии, 7 (10), 174 (1956). Усп хим., 21, 175 (1952).
Усп. хим., 23, 654 (1954).
Т. Г. Акимов а, ЖАХ, 13, 79 (1958).
Г. И. Малофеева, И. В. Никольская.
В. И. Кузнецов, С. Б. Саввин. Радиохимия, 1, 589 (1959).
В. И. К У з н е ц о в, С. Б. Саввин. ЖПХ, 32, 2329 (1959).
В. И. Кузнецов, И. В. Серякова. Зав. лаб., 23, 1176 (1957).
Л. М. К у л ь б е р г. Усп. хим., 15, 605 (1946).
Л. М. К у л ь б е р г. Органические реактивы в аналит. химии. М., Гос-
химиздат, 1950.
133. Л. М. К у л ь б е р г, А. А. П о н о м а р е в, Н. И. Давыдова. ЖАХ, 9, 85 (1954).
134. Л. М. Кульберг, 3. С. Альтерзон, Р. П. Вельтман. Капельный анализ. М., Госхимиздат, 1951.
135. Курам а, И с и х а р а, Коминами, Исикава, Ито. J арап Analyst, 6, 3 (1957); цит. по РЖХим, 898 (1958).
136. А. К. Л а в р у х и н а, Ю. А. 3 о л о т о в. Трансурановые элементы. М., Изд-во АН СССР, 1958.
137. В. Латимер. Окислительные состояния элементов и их потенциалы в водных растворах. М., ИЛ, 1954.
138. Л. В. Л и п и с. Зав. лаб., 6, 736 (1958).
139. Л. В. Л и п и с. Усп. физ. наук, 68, 71 (1959).
140. Д. Л е б е ц, М. О с т а н е к. Мирное использование атомной энергии.
Матер. Международной конф, в Женеве, 1955 г., т. 8, М., 1958, стр. 340.
141. В. Л. Левшин. Изв. АН СССР, сер. физ., 2, 185, 1937.
142. В. Л. Левшин. Фотолюминесценция жидких и твердых веществ. М.,
Гостехтеоретиздат, 1951, стр. 202.
143. Л. Л. Леонова. Геохимия, № 8, 47 (1956).
144. И. И. Л и п и л и н а- ДАН СССР, 122, 238 (1958).
145. И. И. Л и п и л и н а. Уранил и его соединения. М., Изд-во АН СССР, 1959.
146. Р. С. Л о н г, Д. А. Э л л и с, Р. X. Б е й л с. Мирное использование атомной энергии. Матер. Международной конф, в Женеве, 1955 г., т. 8. М., Металлург изд ат, 1958, стр. 97.
147. Е. С. М а к а р о в. ЖНХ, 3, 1079 (1958).
148. Е. С. Макаров. Кристаллохимия простейших соединений U, Th, Рн и Np. М., Изд-во АН СССР, 1958.
149. X. А. К. М а к - К е й. Мирное использование атомной энергии. Матер. Международной конф, в Женеве, 1955 г., т. 7. М., Госхимиздат, 1958, стр. 388.
150. Л. Мак-Клейн, Е. Б а л л в и н к ел ь, Дж. Хеггинск. Химия ядерного горючего. М., Госхимиздат, 1956, стр. 59.
151. С. Л. М а н д е л ь ш т а м, Н. Н. С е м е н о в, 3. М. Т у р о в ц е в а. ЖАХ, 11, 9 (1956).
152. Г. Мервин, Т. Аптер ч, Е. Гринлиф, Е. В а н - Б л а р ко м, А. М э р ф ь ю. Мирное использование атомной энергии. Матер. Международной конф, в Женеве, 1955 г., т. 8. М., Металлургиздат, 1958, стр. 11.
152а. В. К- М а р к о в. А. В. Виноградов, С. В. Е л и н с о н, А. Е. К л ы г и н, И. В. М о и с е е в. Уран, методы его определения. М. Атомиздат, 1960.
153. В. П. М а р к о в, В. В. Ц а п к и н, И. В. Ца п кин а. Тезисы доклада на Всес. конф, по химии комплексных соединений. Киев, 1959.
400
154. А. Дж. Март и н. Со.: Хроматография (под ред. М. М. Дубинина). М., ИД. 1949, стр. 135.
155. В. Г. М ел к о в, Л. Ч. П у х а л ь с к и и. Поиски .месторождений урана. М., Гос. научно-технич. изд. литературы по геологии и охране недр, 1957, стр. 12.
155. В. Г. М е л к о в, 3. М. Свер дло в. ДАН СССР, 31, 359 (1941).
157. Методы определения радиоактивных элементов в минеральном сырье. М., Госгеолтехиздат, 1958.
158. В. А. М и х а й л о в. ЖАХ, 13, 494 (1958).
159. Ю. В. М о р а ч е в с к и й, ЖАХ, 13, 570 (1958).
160. Ю. В. М о р а ч е в с к и и, (1957).
161. Ю. В. М о р а ч е в с к и й, Зав. лаб., 24, 790 (1958).
162. Ю. В. М о р а ч е в с к и й,
163. Ю. В. М о р а ч е в с к и й, 16, 127 (1957).
164. Ю. В. М о р а ч е в с к и й, (1958).
165. Ю. В. М о р а ч е в с к и й,
по радиохимии. Л., Изд-во ЛГУ, 1955, стр. 171. 166. И. Г. Морозова. ЖАХ, 12, 185 (1957).
А. И. Беляева. Л. И. Иванова.
М. И. Гордеева. Вестн. ЛГУ, № 10, 148 М. Н. Гордеева, Т. Е. Круглова. А. А. Сахаров. ЖАХ, 13, 83, 467 (1958).
И. А. Церковницкая. Вестн. ЛГУ,
И. А. Церковницкая. ЖАХ, 13, 337
И. А. Це р ко в н и цк а я. Сборник работ
167. Т. М. Морошкина, В. К. П рокофьев, М. Н. С м и р н о в а. Зав. лаб., 23, 1324 (1957).
168. И. М. Назаров. Атомная энергия, 3, 121 (1957).
169. М. Н о в а к, В- П е к а р е к. 2-я Международная конф, по мирному использованию атомной энергии, Р/2122, Женева, 1958.
170. Р. Н а с т, Т. К р а к к а й. Усп. хим., 22, 583 (1953).
171. Б. В. Некрасов. Курс общей химии, изд. 12. М., Госхимиздат, 1955.
172. А. В. Николаев. Хим. наука и пром, 1, 548 (1956).
173. Е. Р. Николаева. Вестн. МГУ, сер. мат., мех., астроном., физ. и хим., 4, 193 (1958).
174. Е. Р. Николаева. Вестн. МГУ, № 6, 105 (1958).
175. Б. П. Никольский. Сб.: Хроматография (под ред. Б. П. Николь- ского). Л., Изд-во ЛГУ, 1956,' стр. 5.
176. Б. П. Никольский, В. И. Парамонова, А. Ф. Вьюгина. Труды Радиевого ин-та АН СССР, 8, 174 (1958).
177. Б. П. Никольский, А. М. Трофимов. Труды Радиевого ин-та АН СССР, 8, 178, 189 (1958).
178. О. Ницел, М. Д е С е з а. Мирное использование атомной энергии. Матер. Международной конф, в Женеве, 1955 г., т. 8, М., Металлургиздат, 1958, стр. 372.
179. С. П. Сносов, В. Е. Д м и т р и е в. ЖАХ, 13, 403 (1958).
180. Э. А. Остроумов. ЖПХ, 20, 9 (1938).
181. Э. А. Остроумов. Зав. лаб., 6, 16 (1937).
182. Э. А. Остроумов, Р. И. Бомштейн. Зав. лаб., 8, 558 (1939).
183. П. Н. Палей. ЖАХ, 12, 647 (1957).
184. П. Н. Палей. Мирное использование атомной энергии. Матер. Международной конф, в Женеве, 1955 г., т. 8 М., Металлургиздат, 1958, стр. 268.
185. П. Н. П а л е й, А. А. Н е м о д р у к, А. В. Д а в ы д о в. Радиохимия, 3, 81 (1961).
186. П. Н. П а л е й, Д. И. Р я б ч и к о в, 3. К- М и х а й л о в а. ЖАХ, 14, 581 (1959).
187. П. Н. П а л е й, С ю й Л и - ю а н ь. ЖАХ, 16, 51 (1961).
188. П. Н. П а л е й, Н. И. У д а л ь ц о в а. ЖНХ, 5, 2211 (I960).
189. 3. А. Панченко. ЖПХ, 8, 361 (1935).
190. В. И. Парамонова, М. Д. Морачевская. ЖНХ, 2, 1194
(1957).
2э Аналитическая химия урана
401
191. К. А. Петржак, И. Н. Семенюшкнн, М. А. Бак. Геохимия № 2, 27 (1956).
192. Е. С. П р ж е в а л ь с к и й, А. П. Гол ови на, Е. Р. Николаева. Вести. МГУ, сер. мат. мех..астроном., физ., хим., № 1, 171 (1958).
193. Е. С. Пржевальский, Е. Р. Николаева, Н. С. Климо-в а. Вести. МГУ, сер. матём.,мех., астроном., физ., хим., № 3, 217 (1958)
194. Е. С. Пржевальский, Е. Р. Николаева, Н. И. Удальцова. ЖАХ, 13, 567 (1958).
195. П. П р и и г с х е й м. Флуоресценция и фосфоресценция. М., ИЛ, 1951. стр. 392.
196. В. К- Прокофьев. Фотографические методы анализа металлов и сплавов, т. 2. М., Гостехиздат, 1951.
197. Р. П р ш и б и л. Комплексоны в- химическом анализе. М., ИЛ, 1956. К8. Б. В. П т и ц ы н, Е. Н. Т е к с т е р. ЖНХ, 4, 2248 (1959).
199. Радиография. М., ИЛ, 1952.
200. Радиометрические методы поисков и разведки урановых руд (под ред. В. В. Алексеева). М., Госгеолтехиздат, 1957.
201. А. П. Р а т и е р, А. М. Г у р е в и ч, Л. П. П о л о ж е н с к а я. ЖНХ, 2, 2316 (1957).
202. А. П. Р а т и е р, П. И. Чайкин. Сборник работ по радиохимии, М., Изд. ЛГУ, 1955, стр. 75.
203. В. В. Рачинский. Усп. хим., 19, 448 (1950).
204. В. В. Рачинский,?, Б. Г а п о н. Хроматография в биологии. М., Изд-во АН СССР, 1953.
205. Реакции и реактивы для качественного анализа неорганич. соединений. М., Госхимиздат, 1950, стр. 113.
206. Б. Р. Р е п м а н. Лабор. практика, 16, 27 (1941).
207. О. А. Реутов. Теоретические проблемы органич. химии. Изд-во МГУ, М„ 1956.
208. К. Дж. Родден. Мирное использование атомной энергии. Матер. Международной конф. вЖеневе> 1955 г., т. 8, М., Металлургиздат,1958, стр. 237.
209. А. М. Р о з е и. Атомная энергия, 2, 445 (1957).
210. А. М. Розен, Л. П. X о р х о р и н а. ЖНХ, 2, 1957 (1958).
211. К. И. Розенталь, В. И. Весе л-о в с к и й. ЖФХ, 32, 1341 (1958).
212. А. К.- Руса нов. Спектральный анализ руд и минералов. М., Госгеол-издат, 1948.
213. Д. И. Рябчиков, М. М. С е н я в и н. Мирное использование атомной энергии. Матер. Международной конф, в Женеве> 1955 г., т. 8. М., Метал-лургиздат, 1958, стр. 328.
214. Д. И. Рябчиков, М. М. С е н я в и.и, К- Ф. Филиппова. ЖАХ, 7, 135 (1952).
215. Д И. Рябчиков, М. М. Се н яви н, К. Ф. Филиппова. ЖАХ, 8, 220 (1953).
216. С. Б. Саввин. ДАН СССР, 127, 1231 (1959).
217. И. А. С а в и ч, А. К. П и к а е в, И. А. Лебедев, В. И. С п и ц ы.н ЖНХ, 1, 2736, 2742, 2746 (1956).
218. А. Г. Сама рцева. Труды Радиевого ин-та АН СССР, 7, 137 (1956).
219. Г. В. Самсонов. Хроматография. Применение в биохимии. Л., Медгиз, 1955.
220.0. Самуэльсон. Применение ионного обмена в аналитической химии. М., ИЛ, 1955.
221. Э. С. Саркисов. ЖФХ, 24, 487 (1950).
222. 3. М. Свердлов. Изв. АН СССР, сер. физ., 21, 621 (1957).
223. А. Н. С е в ч е н к о. Докторская диссертация. М., Гос. оптический институт им. С. И. Вавилова.
224. Е. Б. Сендэл. Колориметрическое определение следов металлов. М., Госхимиздат, 1949.
225. А. С Сердюкова, Ю. Т. Капитонов. ЖАХ, 13, 88 (1958).
226. Г. Т. С и б о р г. Атомная энергия, 6, 21 (1959).
402 ' "
227. Г. Т. С и б о р г, Дж. Кац. Актиниды. М.—Л., ИЛ, 1955.
228. Г. С. С и н и ц и н а, С. А. Ф а д е е в. Г. .4. Суходолов. Радиохимия, 1, 295 (1959).
229. С. И. С и н я'к о в а, Цит. по [40].
230. С. И. С и н я к о в а, Г. Г. К а р а н о в и ч. Труды Комиссии по аналит.
химии, 2 (5), 65 (1949).
231. С. И. С и н я к о в а, Н. С. К л а с с о в а. ЖНХ. 4, 2000 (1959).
232. С. И. Синякова, В. М. Ш а м р и к о в. ЖАХ (в печати).
233. С. И. Синякова, Л. А. Цветкова. Цит. по [40].
234. С о ч е в а н о в, И. В. Шмакова, Г. А. В о л к о в а. ЖНХ, 2,
2047 (1957).
235. Справочник по радиометрии. М., Госгеолиздат, 1957.
236. Справочник химии, т. 3. Л.—М., Госхимиздат, 1952, стр. 561.
236а. А. И. С т а б р о в с к и й. ЖНХ, 5, 811 (4960).
237. И. Е. С т а р и к. Отчет ЦНИГРИ, 1937.
238. И. Е. С т а р и к, Л. Я- А т р а ш е н о к. Геохимия, №’ 8," 39 (1956).
239. И. Е. С т а р и к, Ф. Е. С т а р и к, Л. Я- А т р а ш е н о к, Г. Б. К осты р е в, В. Н. К о с я к о в, А. Я. К р ы л о в, Труды Радиевого ин-та
АН СССР, 7, 114 (1956).
240. И. Е. Старик, Ф. Е. Старик, Л. Я- А т р а ш е н о к, Г. Б. Ко с-т ы р е в, В. Н.. К о с я к о в, А. Я. Крылов. Труды Радиевого ин-та АН СССР, 7, 114 (1956).
241. И. Е. С т а р и к, Ф. Е. Стар ик, А. Н. А п о л л о н о в а. ЖНХ, 3, 121 (1958).
242. И. Е. С т а р и к, Ф. Е. С т а р и к, А. Н. А п о л л о н о в а. Труды Комиссии по аналит. химии, (9) 12, 246 (1958).
243. И. Е. С т а р и к, Ф. Е. Стар ик, А. Н. А п о л л о н о в а. Труды Радиевого ин-та АН СССР, 7, 107 (1956).
244. И. Е. С т а р и к, А. С. С т а р и к - С м а г и и а. Труды Радиевого ин-та АН СССР, 5, 105 (1957).
245. И. Е. С т а р и к, М. М. Ш, а ц. Геохимия, № 2, 19 (1956).
246. Б. Н. С те п а н о в. ЖФХ, 29, 2173 (1955).
247. Б. Н. Степанов, ЖФХ, 30, 34 (1956).
247а. М. А. С т е п а н о в, И. П. Галкин. Атомная энергия,' 9, 282 (1960).
248. Б. Н. С у д а р и к о в, В. А. Зайцев, Ю. Г. Пучков. НДВШ, Химия и химич. технология, № 1, 80 (1959).
249. В. С. С ы р о к о м с к и й, Ю. В. Клименко. Ванадатометрия. М., Металлургиздат, 1950.
250. В. С. С ы р о к о м с к и й. Зав. лаб., 16, 14 (1950).
251. В. С. С ы р о к о м с к и й, К- С. Ж у к о в а. Зав. лаб., 11, 373 (1945).
252. В. С. С ы р о к о м с к и й, Ю. В. К л и м е н к о. Зав. лаб., 9, 1077
(1940).
253. В. С. С ы р о к о м с к и й, К- С. Жукова. Унифицированные методы анализа ферросплавов. Свердловск, Изд-во АН СССР, 1944.
254. И. В. Т а н а н а е в, М. И. Л е в и н а. ЖНХ, 3, 2045 (1958).
255. Б. Т е ж а к. Мирное использование атомной энергии. Матер. Международной конф, в Женеве, 1955., т. 7. М., Металлургиздат, 1958, стр. 490.
256. Е. С. Т и н о в с к а я. Зав. лаб., 17, 387 (1951).
257. 3. А. Третьяк. Диссертация. Харьковский гос. ун-т им. А. М. Горького, 1953.
258. Е. Б. Т р о с т я н с к а я, А. С. Т е в л и н а. Зав. лаб., 9, 1042 (1957).
259. Е. Б. Тростянская. Труды Комиссии по аналит. химии, 6 (9), 215 (1955).
260. Г. С. Т ю р и к о в, К- И. Р о з е н т а л ь, В. И. В е с е л о в с к и й. ЖФХ, 32, 1490 (1958). ’
261. В. А. У н к о в с к а я. ДАН СССР, 29, 379, 1940.
262. А. М. У ш а_ко в а. Укр. хим. ж., 24, 495 (1958).
263. В. Н. Ушате к и й, Ю. М. Т о л м а ч е в. Труды Радиевого ин-та АН СССР, 7, 98 (1956).
26*
403
261. Ф. Ф а й г л ь. Капельный анализ. М.. ОНТИ. 1947.
265. Фармакология и токсикология урановых соединений, т. 1 (перевод с англ.). .4., ИЛ, 1951, стр. 60.
266. Н. Г Фесенко. Зав. лаб., 10, 491 (1941).
267. А. Ф. Ф и о л е т о в а. ЖАХ, 12, 718 (1957).
268. В. Г. Ф и р с о в, Б. В. Э р ш л е р. Атомная энергия, 4, 343 (1958).
269. С. Фи шер, Р. Куни н. Мирное использование атомной энергии. Матер.
Международной конф, в Женеве, 1955 г., т. 8. М., Металлургиздат, 1958, стр. 343.
270. Н. А. Фукс. Усп. хим., 17, 45 (1948).
271. Н. А. Фукс. Сб.: Исследование в области хроматографии (под ред. М. М. Дубинина). М., Изд-во АН СССР, 1952, стр. 56.
272. Э. X а й Д. Мирное использование атомной энергии. Матер. Международной конф, в Женеве, 1955 г., т. 7. М., Госхимиздат, 1958, стр. 347.
273. М. X а с с и а л и с, Р. Му с а. Мирное использование атомной энергии. Матер. Международной конф, в Женеве, 1955 г., т. 8. М., Металлургиздат. 1958, стр. 23.
274. Е. Хеффнер, Г. Н и л ь с о н, А. X у л ь т г р е н. Мирное использование атомной энергии. Матер. Международной конф, в Женеве, 1955 г., т. 9. Л., Госхимиздат, 1958, стр. 613.
275. В. Г. X лопин, Э. К. Гер л и н г. ЖОХ, 6, 1701 (1936).
276. В. Г. X л о п и н, А. М. Г у р е в и ч. Изв. АН СССР, ОХН, 4, 271 (1943).
277. В. Г. Хлоп и н, Л. Э. К а у фм а и. Труды Радиевого ин-та АН СССР,
вып. 6, 86 (1934).
278. В. Г. Хлоп ин, Л. Э. К а у ф м а н. ЖПХ, 2, 91 (1929).
279. Р. Ф. X о л л и с, К. К- М а к-А р т у р. Мирное использование атомной
энергии. Матер. Международной конф, в Женеве, 1955 г., т. 8. М., Металлург-пздат, 1958, стр. 71.
280. Д. Ц в е т и ч а н и н, Н. Бел егиша ни н. Мирное использование атомной энергии. Матер. Между на родной конф, в Женеве, 1955 г., т. 8. М., Металлургиздат, 1958, стр. 335.
28], Ю. А. Ч е р и и х о в, Б. М. Добкина. Зав. лаб., 16, 402 (1950).
282. 1О. А. Ч е р н и хов, Б. М. Добкина. Зав. лаб., 15, 906, 1143 (1949).
283. В. Г. Ч у х л а и ц е в, С. И. Степанов. ЖНХ, 1, 478 (1956).
281. И. И. Ч е р н я е в, В. А. Головня, Г. В. Э л л е р т, Р. Н. Щелоков, В. К. Марков. 2-я Международная конф, по мирному использованию атомной энергии Р/2138, Женева, 1958.
285. В. 3. Ч у х л а н ц е в, А. К. Шаров а. ЖНХ, 1, 36 (1956).
286. Я • Ш а и к а р, Д. В. Б х а т и а г а р, Т. К. С. М у р т х и. Мирное использование атомной энергии. Матер. Международной конф, в Женеве. 1955 г., т. 8. М., Металлургиздат, 1958, стр. 83.
287. В. Л. Шашкин, И. П. Шумилин. Сб.: Вопросы геологии урана. М., Атомиздат, 1957.
288. В. Л. Шашки и, И. П. III у м и л и н, М. И. П р у т к и н а. Сб.: Вопросы геологии урана. М., Атомиздат, 1957.
289. В. Б. Ш е в ч е н к о, А. С. С о л о в к и н, И. В. Ш и л и и. ЖНХ, 3, 1955 (1958).
289а. В. Б. Шевченко, Б. Н. Судариков. Технология урана. М., Атомиздат, 1961.
290. В. Б. Шевченко, И. В. Ш и л и н, А. С. Соловки н. ЖНХ, 3, 225 (1958).
291. Ф. М. Шемякин, Э. С. М и ц е л о в с к и й, Д. В. Р о м ано в. Хроматографический анализ. М., Госхимиздат, 1955.
292. А. Ш е з н, П. Р е н ь о. Мирное использование атомной энергии. Матер. Международной конф, в Женеве, 1955 г., т. 9. М., Металлургиздат, 1958. стр. 717.
293. Л. И. Шмонин, В. В. Чердынцев, Г. Г. Танеева, Л. Л. К а ш к а р о в. Бюлл. Комиссии по определению абсолютного возраста геологии, формаций. М., Изд-во АН СССР, вып. 1, 64 (1955).
404
294. A. E. Штан дел ь. ЖОХ, 19. 981 Ц949».
294a. О. Л1. Шубникова. Минералы редких элементов и их диагностика. М., Изд-во АН СССР, 1952.
295. Я м а н э. J. Pliarm. Soc. Japan. 77, 400 (19э7); цит. по РЖХим. 21177 09о8).
296. В. М. Abraham, Н. Е. F 1 о t о w. R. D. С а г 1 s о n. Anal. Chem.
29, 1058 (1957).
297. Е. Abrahamczik. Ange*-. Chemie, 61, 89. 96 (1949).
298. Е. Abrahamczik. Mixrochemie ver. Mikrochim. Acta, 33. 213 (191-7).
299. J. A. S. Adams. W. J. M a e c k. Anal. Chem.. 26, 1635 (1954).
300. J. A d a m s, L. W h i t e. Anal. Chem., 26, 1635 (1954).
301. C. A d d i s о n, N. H о d g e. Nature, 171, 569 (1953).
302. M. T. Aguas da Silva. Techica, 28, 41 (1953),
цит. ио РЖХим,
2262 (1955).
303. S. A h r 1 a n d.
304. S. A h r 1 a n d.
305. S. A h г 1 a n dJ
306. S. Ahrland/
307. S. A h г 1 a n d,
308. S. A h r 1 a n d.
309. S. A h г 1 a n d.
310. S. A h г 1 a n d.
311. S. A h г 1 a n d,
10, 705 (1956).
Acta Chim. Scand., 3, 374 (1949).
Acta Chim. Scand., 3, 783 (1949).
Acta Chim. Scand., 3, 1067 (1949).
Acta Chim. Scand., 5, 199 (1951).
Acta Chim. Scand., 5, 1151 (1951).
Acta Chim. Scand., 5, 1271 (1951).
Acta Chim. Scand., 7, 485 (1953).
Acta Chim. Scand., 8, 137 (1954).
R. Larsson, K. Rosengren.
Acta Chim. Scand.,
312. W. Albrecht, M. Mallet. J. Electrochem Soc., 103, 404
(1956).
313. A. A 1 с о c k, S.j S. Grimley, T. V. H e a 1 y, J. Kennedy, H. A. MacKay. Trans. Faraday Soc., 52, 39 (1956).
314. A. A 1 с о c k, G. В e s t, E. H e s f о r d, H. A. M а с К a y. J. Inorg. and Nucl. Chem., 6, 328 (1958).
315. J. Alexa. Collect. Czechoslov. Chem. Comm., 23, 1187 (1958).
316. J. Alexa. Chem. listy, 51, 2254 (1957).
317. G. A 1 i b e g о f f. Liebigs Ann. Chem., 233, 143 (1886).
318. I. P. A 1 i m а г i n. G. N. В i 1 i m о v i c h, Internat. J. Appl. Rad.
Isotopes, 7, 169 (1960).
319. I. P. A 1 i m а г i n, В. I. F r i d. Mikrochemie, 23, 17 (1937).
320. C. J. A 1 k i r e m, К. К о у a m a, K. J. Hahn, С. E. M i c h e 1-
s о n. Anal. Chem., 30, 1912 (1958).
321. K. A 1 1 e n. J. Am. Chem. Soc., 80, 4133 (1958).
322. M. Allen. Anal. Chem., 22, 804 (1950).
323. K- A. Allen. Anal. Chem., 28, 1144, (1956).
324. A. A 1 1 e n, R. A n d e r s о n, E. P о w e 1 1. Цит. no Chem. Abstr., 53.
3949 d (1959).
325. G. A 1 m a s s y. Magyar Kern, folvoirat., 61, 404 (1955)- цит. no Anal. Abstr. 3, 1705 (1956).
326. G. A 1 m a s s y. Magyar tud. akad. Kem tud. oszt. Kozl., 7, 337 (1956).
327. G. A 1 m a s s у, A. К i s s. Magyar Kem. folyoirat, 64, 170 (1958).
328. G. A 1 m a s s y, Z. N a d y, J. S t г a u b. Acta chim. Acad, scient. Hung.,
7, 317 (1955).
329. G. A 1 m a s s y, Z. N a d y, Z. anal. Chem., 147, 135 (1955).
330. G. A 1 m a s s у, M. V i g v а г i. Magyar Kem. folyoirat, 61, 109, (1955).
331. G. A 1 m a s s у, C. U j e 1 v i. Magyar tud. acad. Kem. tud. oszt. kozl.
8, 47 (1956); цит. по РЖХим, 30999 (1957).
332. V. A m i г t h a 1 i n g a m, V. P a d m a n a b n a n. Anal. Chem., 31, 622 (1959).
333. Analytical Method of the spectrographie determinations of calcium in uranium metal. Chemical, services department, Springfields, Published by industrial group Headquarters, Resley, Warrington. Lancashire, 1958.
334. P. A n t i k a i n e n. Suomen kem., 31, 227 (1958); цит. по РЖХим, 27057 (1959).
405
335. T. Arden. J. Chem. Soc., 1949, 299.
336. T. A r d e n. P. M e-G lone. Nature, 166, 560 (1950).
337. T. V. Arden, M. Rowl ey. J. Chem. Soc., 1957, 1709.
338. T. V. A r d e n. F. В u r s t a 1 1, R. P. L i s t e a d. J. Chem. Soc., 1949, 311.
339. T. V. Arden, G. A. Wood. J. Chem. Soc., 1956, 1596.
340. A. L. A r n f e 1 t. Acta Chim. Scand., 9, 1484 (1955).
341. P. A r t m a n n. Z. anorg. und allgem. Chem., 79, 327 (1913).
342. A. A s m a n о v. Z. anorg. und allgem. Chem., 160, 209 (1927).
343. V. T. A t h a v a 1 e, S. В a n e r j e e, С. К- В e 1 e к a r, N. Maha de-v a n, L. M. Ma ha jan, M. N. N a d к a r n i, Das M. Sa nka r, H. D. Sha rma, A. K. Sunda ra m, M. S u n da r esa n, N. R. T h a-k о о r, M. M. T i 1 1 u, M. S. V a r d e, C h. Venkateswarlu. Second United Nations International Conference on the Peaceful Uses of Atomic Energy, Report № 1617 (1958).
344. V. Auger. Compt. rend., 155, 647 (1912).
345. V. Auger. Compt. rend., 170, 995 (1920).
346. M. В a c h e 1 e t, at all. Bull. Soc. Chim. France, № 2, 173 (1954).
347. M. M. В a c h e 1 e t, R. Cl aude, M. M. Lederer. Compt. rend., 240, 419 (1955).
348. A. Bacon, G. Milner. Atomic Energy Res. Establ., Rep. C/R 1749. (1955); цит. по РЖХим, 34710 (1957).
349. А. В а с о n, G. M i 1 n e r. Atomic Energy Res. Establ., Rep. C/R, 1813 (1956); цит. no Chem. Abstr., 50, 12745 b (1956).
350. А. В а с о n, G. W. C. Milner, Analyst, 81, 456 (1956).
351. D. Bandyopadhayay. J. Indian Chem. Soc., 33, 269 (1956).
352. C. Baes. J. Phys. Chem., 60, 878 (1956).
353. S. К. В a n e r j i, A. K- Dey. Chim. analyt., 40, 202 (1958).
354. S. К- В a n e r j i, A. K. Dey. J. prakt. Chem., 6, 62 (1958).
355. G. Banerjee, A. H. A. N e у n. Anal. Chem., 30, 1795 (1958).
356. S. К. В a n e r j i, A. K. D e y. Z. anal. Chem., 159, 293 (1958).
357. С. V. В a n к s, К- E. В u г к e, J. W. O’L a u g h 1 i n, J. A. Tom-
pson. Anal. Chem., 29, 995 (1957).
358. С. V. Banks, J. A. Tompson, J. W. O’L a u g h 1 i n. Anal. Chem.. 30, 1792 (1958).
359. G. Barnett, G. Milner. Atomic Energy Res. Establ., Report C/R 2307 (1958); цит. no Anal. Abstr., 5, 3282 (1958).
360. M. T. В e с к, E. H a n t о s. Acta Chim. Acad. Scient. Hung., 8, 233 (1955). 361. R. В e 1 c h e r, D. G i b b о n s, T. W e s t. Z. anal. Chem., 145, 293 (1955) 362. B. Bernstrom. J. Rydberg. Acta Chem. Scand., 11, 1173 (1957). 363. R. Berg. J. makromol. Chem., 115, 178 (1927).
364. P. Berthollet, A. Grimbert. Bull, inform, scient. et techn. Commissar. energie atom, № 22, 12; цит. по РЖХим, 42189 (1959).
365. H. W. В e r t r a m, M. W. Ler ner, G. F. P e t r e t i с, E. S. Ros-zkowski, C. J. Rodden. Anal. Chem., 30, 354 (1958).
366. R. Betts. Can. J. Research., 26, 441 (1948).
367. R, Betts. Can. J. Chem., 33, 1775 (1955).
368. R. H. В e t t s, R. К- M i c h e 1 s. J. Chem. Soc., Supl. Iss., 1949, 286.
369. R. В e t t s, R. Z e i g h. Can. J. Research, 28, 514 (1950).
370. A. S. В h a d u r i. Sci. and Culture, 18, 95 (1952).
371. A. S. В h a d u r i, P. R a y. Sci. and Culture, 18, 97 (1952).
372. A. S. Bhaduri, P. Ray. Z. anal. Chem., 154, 103 (1957).
373. В. В i e b e r, Z. Vecera. Chem. listy, 52, 439 (1958).
374. W. В i 1 t z, H. Mu 1 1 er. Z. anorg. und allgem. Chem., 163, 257 (1927).
375. N. Birnbaum, S. M. Edmonds. Ind. and Eng. Chem., Anal. Ed., 12, 155 (1940).
376. A. A. Blair. Proc. Amer. Philos. Soc., 52, 201 (1913).
377. P. В 1 a n q u e t. Anal. Chim. Acta, 16, 44 (1957).
378. H. В о d e. Z. anal. Chem., 143, 182 (1954).
406
379. Н. В о d е. Z. anal. Chem., 144, 165 (1955).
380. В. Boltwood. Am. J. Sci., [4]. 25, 269 (1908).
381. G. В о о m a n. Anal. Chem., 29, 213 (1957).
382. G. В о о m a n, W. Neebrook. Anal. Chem., 31, 1U (1959).
383. G. В о о m a n, W. Neebrook. J. Rein. Anal. Chem., 29, 219 (1957).
383a. E. В о о t h, E. A. T e r r y. Anal. Chim. Acta,' 22, 82 (I960).
384. M. В о r e 1 e r a. Ricerca scient., 28, 331 (1958).
385. D. Bradley. Atomic Energy Res. Establ., Repts, NCE, R 24( »8 (1859).
386. В. В г e у e r. Z. Phys. Chem., Sonderheft, Juli, 128 (1958).
387. H. В r i n t z i n g e г, С. H e s s e. Z. anorg. und allgem. Chem., 249, 113
(1942).
388. N. T. S. Brit ton. J. Chem. Soc., 127,2110 (1925).
389. N. T. S. В r i t t о n, A. E. Y о n g. J. Chem. Soc., 1932, 2467.
390. J. К. В г о d y, J. P. F a r i s, R. F. Buchanan. Anal. Chem., 30, 1909 (1958).
391. R. Brown. J. Am. Chem. Soc., 76, 1532 (1954).
392. F. R. Bruce, J. M. Fletcher, H. H. H у m a n, J. J. Katz. Progress in Nuclear Energy ser., Ill, Process Chemistry, McGraw-Hill. N. Y., 1956.
393. R. Buck, P. Farrington. Anal. Chem., 24, 1195 (1952).
394. E. P. В u 1 1 w i n к e 1, P. N о b 1 e. J. Am. Chem. Soc., 80, 2955 (1958).
395. J. Bunce. Atomic Energy Res. Establ., Repts, NC/R, 2407, 23 (1958), цит. по РЖХим, 7964 (1959).
396. L. L. Burger. J. Phys. Chem., 62, 590 (1958).
397. M. F. В u r r i e 1, G. C. Barcia. Ann. Real. Soc. espanola, fis. у quim., 47, 63 (1951).
398. M. F. В u r r i a 1, G. С. В a r c i a. Ann. Real. Soc. espanola, fis. y’quim., 50, 281 (1954).
399. F. H. В u r s t a 1 1, R. A. Wells. Analyst, 76, 396 (1951),
400. J. Byrne. Anal. Chem., 29, 1408 (1957).
401. A. Cacciari, R. De Leone, C. Fizzotti, M. Gabaglio. Energia Nucl., 3, 176 (1956).
402. A. Cacciari, R. De Leone, C. Fizzotti, M. Gabaglio. Energia Nucl., 3, 368 (1956).
403. M. Cabell. Analyst, 77, 859 (1952).
404. E. R. С a 1 e y, L. B. Rogers. J. Am. Chem. Soc., 68, 2202 (1946).
405. P. J. C a 1 1 a n, A. R. Henderson. Analyst, 54, 650 (1928).
406. E. D. C a m p b e 1 1, С. E. Griffin. Ind. and Eng. Chem , 1 661 (1909).
407. R. G. Canning, P. Dixon. Anal. Chem., 27, 877 (1955).
408. W. Carson. Anal. Chem., 22, 1565 (1950).
409. W. Carson. Anal. Chem., 23, 1019 (1951).
410. W. Carson. Anal. Chem., 25, 466 (1953).
411. D. J. Carswell J. Inorg. and Nucl. Chem., 3, 384 (1957).
412. J. A. C a r t e r, C. W. Weber. U. S. Atomic Energy Comm., Rep, TID—7516, 186 (1956); цит. no Anal. Abstr., 5, 490 (1958).
413. C. Caruthers. Ind. and Eng. Chem., Anal. Ed., 15, 70 (1943).
414. F. C e n t a n i, A. R о s s, M. D e s e s a. Anal. Chem., 28, 1651 (1956).
415. J. С e p a 1 a k, J. M a 1 у, V. M a c h a c e k. Chem. listy, 51, 2195 (1957).
416. J. С e p a 1 a k, J. M a 1 у, V. M a c h a c e k. Collect. Czechoslov. Chem. Comm., 23, 1509 (1958).
417. А. К- C h a к r a b u r t t v, D. S e n, P. R a y. J. Indian Chem. Soc.. 30, 491 (1953).
418. G. Chen. J. Jab. Clin. Med., 21. 1198 (1936).
419. K- L. Cheng. Anal. Chem., 30, 1027 (1958).
420. K. L. Cheng, R. H. В r a y. Anal. Chem., 27,782 (1955).
421. K. L. Cheng, Anal. Chem., 28, 462 (1956).
422. J. L. C i h a 1 i k, J. S i m e k, J. R u z i с к a. Collect. Czechoslov. Chem. Comm., 23, 1037 (1958).
423. V. C i r i 1 1 i. Ricerca Scient., 27, 674 (1957); цит. по РЖХим, 14181 (1958).
407
i24. R. F. Clavton, XV. H. Hardwick, M. Moreto n-S m i t h, R. Todd. Analyst, 83, 13 (1958).
425. XV. E. С 1 i f f о r d, E. P. В u i 1 w i n к e 1, L. A. MacCla i ne,
P. Noble. J. Am. Chem. Soc., 80, 2959 (1938).
426. J. Cli nch, M. J. Guy. Analyst, 82, SuO (1957).
427. J. Clinch, M. J. G u y. Analyst, 82, 850 (i957).
428. J. XV. С о 1 1 a t, J. J. L i nga ne. J. Am. Chem. Soc., 76, 4'214 (1954).
429. XV. D. Cooke, F. Hazel, X\’. M. MacNabb. Anal. Chim. Acta, 3, 656 (1949).
439. XV. D. Cooke, F. Hazel, XV. M. 51 acNabb. Anal. Chem.. 22, 654 (1950).
431. XV. Cooke, N. Furman. Anal. Chem., 22, 896 (1950).
432. XV. С о о к e, C. R e i 1 e y, N. F u г m a n. Anal. Chem., 23, 1030 (1951).
433. XV. Cooke, C. R e i 1 1 e y, N. F u г m a n. Anal. Chem., 23, 945 (1951).
434. XV. С о о к e, C. R e i 1 1 у, N. Furman. Anal. Chem., 24, 205 (1952).
435. R. С о о p e n s. Bull. Soc. franc, mineral, et cristallogr., 73, 217 (1950).
436. E. С о t t e 1 a i n. Ann. pharmac. granc., 11, 484 (1930).
437. С. E. С г о u t h a m e 1, С. E. Johnson. Anal. Chem., 24, 1780 (1952).
438. D. C v j e t i c a n i n. JENER Rept., ЛЬ 57, 10 (1958); цит. по РЖХим, 26999 (1959).
439. I. С и г i е, Н. F а г a g g i. Compt. rend., 232, 959 (1951).
440. J. E. С u г г a h, F. E. Beamish. Ind. and Eng. Chem., Anal. Ed.,
19, 609 (1947).
441. F. C u t t i t t a, G. J. Daniels. Anal. Chim. Acta, 20, 430 (1959).
442. J. C z а к о w, Z. P a d w a.n, B. S t rzy zewska. Nucleoniks, 3, 67 (1958).
443. J. Czakow, L. Pszonicki, Z. XV ale w ska. Chemia analytyczna, 3, 753 (1958).
444. N. K. D a t t, N. Gocwami.Z. ajrorg. und allgem. Chem., 298, 265 (1959).
445. S. K. D a t t a. Z. anal. Chem., 155, 241 (1957).
446. S. K. D a t t a. Z. anal. Chem., 163, 403 (1958).
447. S. K. Datta. Z. anal. Chem., 163, 349 (1958).
448. XV. H. D a v e n p о r t, P. F. Thomason. Anal. Chem., 21, 1093 (1949).
449. R. A. Day, R. M. Powers. J. Am. Chem. Soc., 76, 3895 (1954).
450. R. Day, K. Wilhite, F. Hami Iton. J. Am. Chem. Soc., 77, 3180 (1955).
451. P. N. Das C u p t a. J. Indian Chem. Soc., 6, 763 (1929).
452. P. N. Das C u p t a. J. Indian Chem. Soc., 6, 777 (1929).
453. P. N. Das Cupta. Analyst, 55, 154 (1930).
454. D. Deford, J. Pitt e, C. Johns. Anal. Chem., 23, 938 (1951).
455. D. D e f о г d, C. J о h n s, J. P i t t e. Anal. Chem., 23, 941 (1951).
456. P. Dfflahay. Anal. Chim. Acta, 6, 542 (1952).
457. M. D e 1 e p i n e. Cornpt. rend., 44, 21 (1907).
458. M. XV. Desai, T. K- S. M urty. Analyst, 83, 126 (1958).
459. M. A. De S e s a, O. A. N i e t z e 1. U. S. Atomic Energy Comm. Rep , ACCO-54, 19 (1954); нит. по РЖХим, 61840 (1956).
460. G. S. D e s h m u к h, M. K. J о s h i. Z. anal. Chem., 143, 334 (1954).
461. G. S. D e s h m u к h, M. K. Joshi. Chem. Ber., 87, 1446 (1954).
462. G. S. D e s h m u к h, M. K. Joshi. Chem. Ber., 88, 186 (1955).
463. С. XV. Dewis. Am. J. Sci., [5], 11, 201 (1926).
464. Z. I. Dizdar. Rec. trav. Inst., 2, 77 (1953).
465. Z. I. D i z d a r, O. S. G a 1, J. K- Rajnvajn. Bull. Inst. Nucl. Sci., 7, 43 (1957).
466. Z. I. Dizdar, I. D. Obrenovic. Second United Nations International Conference on the Peaceful Uses of Atomic Energy, R/471 (1958'.
467. Z. 1. Dizdar, I. D. Obrenovic. Analyst, 83, 177 (1958).
468. A. D i t t e. Researches sur 1’acide iodique, Paris, 1870.
469. A. D i t t e. Ann. Chim. Phys., [6], 21, 158 (1890).
470. D. Dolar, Z. D r a g a n i c. Rec. trav. inst. research, struct. .Mat., 2, 77 (1953).
408
471. J. Dolezal. J. Ada m. Chem. listv, 48. 32 (1954).
472. D. De Ford. Anal. Chem., 26, 135 (1954).
473. J. Dragaaic. Rec. trav. Inst, research, struct. Mat.. 1, 81 (1952); цит. no Brit. Abstr.. ser. C, 112 i!953).
474. B. Drake. Acta Chim. Scand.. 4, 554 (1950).
475. J. V. D u b s к v. E. Ora v c, A. Lange r. Chem. Obzor. 12. 41 (19371.
476. E. R. D u к e, R. C. Pi nkert on. J. Am. Chem. Soc., 73, 2361 (1951).
477. K. Dutt. N. Goswami. Z. anorg. und allgem. Chem., 298. 265 (1959).
478. C. Duval. Anal. Chim. Acta. 3, 335 (1949).
479. D. D у г s s e n. Svensk. Kern, tidskr., 7, 153 (1950).
480. D. D у г s s e n. Acta Chim. Scand., 10, 353 (1956).
481. D. Dy rssen, V. Dahlberg. Acta Chim. Scand., 7, 1186 (1953).
482. D. D v r s s e n, M. D v s s e n, E. J о h n s s о n. Acta Chim. Scand..
10, 106 (1956).
483. D. Dvrssen, M. D v г г s s e n. E. J о h n s s о n. Acta Chim. Sand.,
10, 341 (1956).
484. К. H, Ebert, H. К 6 n i g, H. W a n к e. Z. Naturforsch., 12a, 763 (1957).
485. A. R. E b e r 1 e, M. W. L с г n e r. Anal. Chem., 29, 1134 (1957).
486. К. E dwards, D. l\er n. Anal. Chem.. 28, 1811 (1956).
487. G. G. E i c h h о 1 z, J. N. H i 1 b о r n, C. McMahon. Can. J. Phvs.,
31, 613 (1953).
488. J. E 1 b e i n, M. A b о u n a g a. Anal. Chim. Acta. 19, 123 (1958).
489. H. El-Shaniy, S. El-Din Zayan. Analyst, 80, 65 (1955).
490. P. J. Elvi ng, E. С. О 1 s о n. Anal. Chem.,' 28, 338 (1956).
491. P. J. E 1 v i и g, A. F. К r i v i s. Anal. Chem., 129, 1232 (1957).
492. H. E rl enm yer, W. О p p И ger, K. St i er, M, В 1 u m in e r. Helv.
Chim. Acta, 33, 25 (1950).
493. D. Ewi ng, E. E 1 d г i d g e. J. Am. Chem., Soc., 44, 1484 (1922).
494. U, Facchini, L. Orson i. Nuovo Cimento, 6, 241 (1949).
495. T. Fairley. Chem. News, 62, 227 (1890).
496. J. F r a n c h e г r o. Compt. rend., 227, 1367 (1948).
497. L. F a г г а г, P. T h о m a s о n, M. Kelley. Anal. Chem., 30, 1511 (1958).
498. P. F a r r i ng t о n, E. S w i j t. Anal. Chem., 25, 591 (1953).
499. H. I. F e i n s t e i n. U. S. Atomic Energy Comm. Rep., ТЕ 1-555 (1955);
пит. no Anal. Abstr., 4, 3944 (1957).
500. H. I. Feinstein. Anal. Chim. Acta, 15, 288 (1956).
501. J. Feldman, J. N a v i 1 1. J. Am. Chem. Soc., 76, 2114 (1954).
502. J. F e 1 d m a n, H. H a v i 1 1, W. Neu in a n. J. Am. Chem. Soc., 76, 4726 (1954).
503. J. Feldman, W. F. Neu m an. J. Am. Chem. Soc., 73, 2312 (1951).
504. R. M. Fink, K. F. F i n k. U. S. Atomic Energy Comm., Rep. UCLA.
30, 1949: цит. no Chem. Abstr., 50, 9936 (1956).
505. D. J. Fischer, P. F. Thomason. Anal. Chem., 28, 1285 (1956).
506. D. J. Fischer. Anal. Chem. Div. Quaterly Progr. Rept. for Period Ending Dec., 26, 1951, Oak Ridge Laboratory, 02 NL. 1233, p. 9.
507. C. Fischer, J. В e v d о n. Bull. Soc. Chim. France, № 11 —12, C 102 (1953).
508. M. F i s s i e г, К. В e п а г d. Compt. rend. Soc. biol., 99, 1144 (1928).
509. J. F. F 1 a g g, N. H. Furman. Anal. Chem., 12, 529 (1940).
510. R. F 1 a t t, F. Sommer. Helv. Chim. Acta, 27, 1518 (1944).
511. R. F 1 a t t, F. Sommer. Helv. Chun. Acta, 25, 684 (1942).
512. H. R. Fleck. Analvst, 62, 378 (1937).
513. M. Fletcher. Geol. Surv. Bull., № 1006, 52 (1954).
514. M. Fletcher, J. May, J. Anderson. Geol. Surv. Bull. № 1006, 93 (1954).
515. M. Fletcher, J. May, M Slavin. Geol. Surv.Bull., № 1006, 8 (1954).
516. C. F 1 о r i d a, C. Da v e v. .1. Sment. Instrum., 30, 409 (1953).
517. M. F о d о r. Magyar tud akad. Kozp. fiz. kutato int. KozL, 5, 445 (1957); цит. по РЖХим, 60618 (1958).
409
518. M. F о <^о г. .Magyar tud. akad. Kem. tud. oszt. kozL. 9, 463 (1958).
519. R. T. Foley. R. C. Anderson. J. Am. Chem. Soc.. 71, 909 (1949).
520. J. K. Fore m a n. C. J. Rile у, T. D. Smit h. Ansalvt, 82, 88 (1957)
521. C. A. Franco i s. U, S. Atomic Energy Comm., Rep., DOW-150 (1956); цит. no Anal. Abstrs., 4, 3939 (1957).
522. C. A. Francois. Anal. Chem., 30, 50 (1958).
523. E. F г a n к 1 i n, R. Barnes. The radiometric assay of uranium and thorium ores. Atomic Energy Res. Establ., Repts. 1175 (1953).
524. M. Fred, N. N a c h t r i e b, F. T о m p к i n s. J. Opt. Soc. Am , 37, 279 (1947).
525. R. J. Frere. J. Am. Chem. Soc.. 55, 4362 (1933).
526. R. Fresenius, E. H u n t z. Z. anal. Chem., 34, 437 (1895).
527. J. S. F г i t z, E. C. Bradford. Anal. Chem., 30, 102 (1958).
528. J. S. F г i t z, M. Fulda, S. Margerum, E. Lane. Anal. Chim. Acta, 10, 513 (1954).
529. J. S. F r i t z, M. J о h n s о n-R i c h a r d. Anal. Chim. Acta, 20, 164 (1959).
530. J. S. F г i t z, M. J о h n s о n-R i c h a r d. Anal. Chim. Acta, 28, 167
-(1959).
531. C. F. F г у 1 i n g, F. V. Tooley. J. Am. Chem. Soc., 58, 826 (1936).
532. N. Furman. J. Electrochem. Soc., 101 (1954).
533. N. F u г ni a n, R. Adams. Anal. Chem., 25, 1564 (1953).
534. N. Furman, C. Brieker. Anal. Chem., 25, 482 (1953).
535. N. F u r m a n, W. В. M a s о n, J. S. P e к о 1 a. Anal. Chem., 21, 1325 (1949).
536. N. H. Furman, I. C. S c h о о n о v e r. J. Am. Chem. Soc., 53, 2561 (1931).
537. N. H. Furman, I. C. Schoonover. J. Am. Chem. Soc., 54, 1344
(1932).
538. H. G e s t, M. D. К a m e n, J. R. Reiner. Arch. Biochem., 12, 273 (1947).
539. T. G i b b, H. Evans. Science, 105, 72 (1947).
540. H. H. Gill, R. F. Rolf, G. ’W. Armstrong. Anal. Chem., 30, 1788 (1958).
541. F. G i о 1 i t t i. Gazz. Chim. Ital., 34, 166 (1904).
542. F. G i о 1 i t t i. Gazz. Chim. Ital., 35, 145 (1905).
543. B. G 1 a s in a n n. Chem. Ber., 37, 189 (1904).
544. E. G 1 e d i t s c h. Le Radium, 8, 256 (1911).
545. E. Glugckauf. Ind. chimique Beige, 23, 1215 (1958).
546. E. G 1 u e с к a u f, H. A. C. MacKay, А. К- M a t h i e s о n. Trans. Faraday Soc., 47, 437 (1951).
547. G m e 1 i n’s Handbuch der anorganischen Chemie. Uran und Isotope. Verlag Chemie. Berlin, 1936.
548. E. G о 1 d b 1 a t t, J. S. Africans. Inst. Mining. Met., 59, 369 (1959); цит. no Chem. Abstr., 53, 14815g (1959).
549. В. B. G о о d m a n, D. Shoenberg. Nature, 165, 441 (1950).
550. A. G о r d о n, A. M a r t i n, R. Synge. Biochem. J., 37, 79 (1943).
551. N. E. G о r d о n, R. A. В r i g h t s e n, H. D. С о о к, C. R. Wilson.
U. S. Atomic Energy Comm., Reports. WARD-81, 1952.
552. P. G о r i s, W. E. D u f f y, F. H. T i n g e y. Anal. Chem., 29, 1590 (1957).
553. H. Goto. Sci. Rept. Res. Inst. Tohoku Univ., 26, 391 (1937).
554. H. G о t t o. Z. Naturforsch., 38, 149 (1948).
555. Z. G r e g о r e z у k. Acta polon. pharmac., 15, 333 (1958).
556. С. E. Griffin. Engng. and Mining J., 37, 247 (1912).
557. F. Gri ma 1 di, N. Gut tag. Geol. Surv. Bull., № 1006, 111 (1954).
558. F. Grimaldi, M. I r v i n g, M. Fletcher. Geol. Surv. Bull. № 1006, 1 (1954).
559. F. G r i m a 1 d i, J. May, M. Fletcher. Geol. Surv. Circ., № 199, 200 (1952); цит. no Chem. Abstr, 47, 3180b (1957).
560. F. Grimaldv, J. Mav, M. Fletcher, J. Tictomb. Geol. Surv. Bull., №'1006, 1 (1954).
410
561. F. G г i m a ] d i, H. Levine. Geol. Surv. Bull., № 1006, 49 (1954).
562. W. G г i m e s. Nucl. Sci. Abstr., 4 , 368 (1950); цит. no Brit. Abstr., 1951, 206.
563. K. J. G u e s t, C. R. Lalonde. Can. Dept. Mines and Techn. Surveys, Alines Branch. Radioactivity Div. Topical Rept., № TK-135/56, 1956.
564. R. J. Guest, U. B. Zimmerman. Anal. Chem., 27, 931 (1955).
565. E. Haeffner, A. H u 1 t g r e n. Nucl. Sci. and Engng. 3, 471 (1958).
566. E. H a f о v e r, F. Hecht. Mikrochim. Acta, 417 (1954).
567. H. T. Hahn. J. Am. Chem. Soc., 79, 4625 (1957).
568. R. В. H a h n, M. T. Kelley. Anal. Chim. Acta, 10, 178 (1954).
569. J. Haidle n, R. Freseni us. Liebigs Ann. Chem., 43, 145 (1842).
570. D. H a 1 1. J. Am. Chem. Soc., 44, 1462 (1922).
571. H. H a m a g u c h i, G. W. Reed, A. T u г к e v i c h. Geochim. et cos-
mochim. Acta, 12, 337 (1957).
572. W. W. Happ, J. H. H о r w о о d. Science, 115, 622 ( 1952).
573. R. Hara, P. West. Anal. Chim. Acta, 12, 285 (1955).
574. T. H а г a, H. Omori. Doshisha Engng. Rev., 7, 342 (1957).
575. W. H. Hardwick, M. M о r e t о n - S m i t h. Analyst, 83, 9 (1958).
576. P. Harley. Bull. Geol. Soc. Am., 67, № 4 (1956).
577. W. E. Harris, I. M. К о I t h о f f. J. Am. Chem. Soc., 67, 1484 (1945).
578. W. E. Harris, I. M. К о 1 t h о f f. J. Am. Chem. Soc., 69, 446 (1947).
579. О. H a u s e r. Z. anal. Chem., 47, 677 (1908).
580. R. Y. Hayden, J. H. Reynol ds, M. G. Ingram. Phys. Rev.
75, 1500 (1949).
581. H. G. Heal. Nature, 157, 225 (1946).
582. H. G. Heal, J. G. N. Thomas. Trans. Faraday Soc., 45, 11 (1949).
583. H. Heal. Trans. Faraday Soc., 45, 1 (1949).
584. T. V. Healy, H. A. C. MacKay. Trans. Faraday Soc., 52, 633 (1956).
585. J. H e a r n e, A. Wh i t e. J. Chem. Soc., 1957, 2081.
586. J. Hearne, A. W h i t e. J. Chem. Soc., 1957, 2168.
587. F. Hecht, J. Korkish, R. Patzak, A. Thiard. Microchim. Acta, 1283 (1956).
588. F. Hecht, H. К r a f t - E b i n s. Z. anal. Chem., 106, 321 (1936).
589. F. Hecht, W. Reich- Rohrwig. Monatsch. 53—54, 596 (1929).
590. J. H e d v a 1 1. Z. anorg. und allgem. Chem., 146, 225 (1925).
590a. J. H e f 1 a j, E. A m i s. J. Phys. Chem., 64, 870 (1960).
591. Z. Heidt. J. Phys. Chem., 46, 624 (1942).
592. S. H e i t a n e n. Acta Chim. Scand., 10, 1531 (1956).
593. C. Heitner, M. Bob t alsky. Bull. Soc. Chim. France, 356 (1954).
593a. W. H e 1 b i g. Z. anal. Chem., 174, 169 (1960).
594. S. Hel lesta m. Svensk. kern, tidskr., 60, 253 (1948).
595. P. H era zy m e n ко. Chem. listy, 19, 172 (1925).
596. F. Hernegger, B. Karli k. Sitzber. Acad. Wiss. Wien, Math. Natur., 144, 217 (1934).
597. F. Hernegger, H. W a n к e. Z. Naturfirsch., 12a, 759 (1957).
598. E. H e s f о r d, H. A. C. MacKay. Trans. Faraday Soc., 54, 573 (1958).
599. D. С. H e s s, H. В г о w n, M. G. I n g h r a m, C. Patterson, G. Tilton. Nat. Bur. Standards, Circ., № 522, 183 (1953).
600. S. H i e t a n e n, Z. S i 1 1 e n. Acta Chim. Scand., 8, 1607 (1954).
601. W. F. Hi 1 lebrand. Geol. Survey Bull., 78, 47 (1891).
601a. Hideo Imai. Bull. Chem. Soc. Japan, 30, 873 (1957); Chem. Abstr., 13483c (1958)
602. R. H i r t, N. N a c h t r i e b. Anal. Chem., 20, 1077 (1948).
603. H. Hoekstra, S. S i e g e 1. J. Phys. Chem., 59, 136 (1955).
604. J. M. H о j m a n. Bull. Inst. Nucl. Sci., 7, 17 (1957).
605. В. H о k. Svensk. kern, tidskr., 65, 106 (1953).
606. В. H о k-B e r e s t г о m. Acta Chim. Scand., 10, 163, 341 (1956).
607. J. A. Holladay, T. R. C u n n i g. h a m. Trans. Am. Electrochem. Soc., 43, 329 (1923).
608. О. Honi gsch'i d, T. Wi ttner. Z. anorg. und allgem. Chem., 226, 289 (1936).
411
609. W. S. Horton. J. Am. Chen. Soc., 78, 897 (1955).
610. C. A. Horton, J, G White. Anal. Chem., 39, 1779 (1958).
611. V. Hovorka, Z. Holzbecker. Collect. Czechoslov. Chem. Comm.,
14, 49 (1919).
612. V. Hovorka,
613. V. Hovorka,
614. V. Hovorka, 182 (1938).
615. V- Hovorka, 70 (1939).
616. V. Hovorka,
617. V. Hovorka,
Z. H о 1 z b e c h e r. Z. anal. Chem., 130, 268 (1950).
V. D. Sy kora. Chem. listy, 32, 241 (1938).
V. S у к о r a. Collect. Czechoslov. Chem. Comm., 10,
V. S у к о r a. Collect. Czechoslov. Chem. Comm., 11,
J. V о r i s e k. Chem. listy, 34, 55 (1940).
J. V о r i s e k. Collect. Czechoslov. Chem. Comm., 11,
128 (1939).
618. E. H. Huffman. Anal. Chem., 18, 278 (1915).
619. A. Hultgren, E. Halffner. Second United Nations Conference on the Peaceful Uses of Atomic Energy, P/144 (1958).
620. E. С. H u n t, A. A. N о r t h, R. A. W e 1 1 s, Analyst, 80, 172 (1955).
621. H. Ishimori, Okuno. Bull. Chem. Soc. Japan, 29, 78 (1956).
622. E. Jackwkrth, H. Specker. Z. anal. Chem., 168, 340 (1959).
623. P. C. J a i n, G. S. Rao. Analyt. Chim. Acta, 20, 171 (1959).
624. G. J a n d e r, K- R e e h, Z. anorg. und allgem. Chem., 129, 293 (1923).
625. G. J a г a i n, J. M a i 1 1 о t. J. phys. et radiant, 19, 35 (1958).
626. R. E. J er v i s, W. D. Mac h i n t os h. Second United Nations Confe-
rence on the Peaceful Uses of Atomic Energy, Р/18Э (1953).
627. B.J ezowska-Trzebiatowska, A., Bartecki. Bull de Г Acad. Polon. des Sciences, Ser. des Sci. Chim., 6, 41 (1958).
628. M. G. Inghram. Ann. Rev. Nucl. Sci., 4, 81 (1954).
629. M. G. Inghram. J. Phys. Chem., 57, 899 (1953).
630. M. G. I n ghram. В книге J. H. Y о e, H. J. К о c h. Trace Analysis.
N. Y., 1957, p. 437.
630a. I. M. Issa, R. M. Issa. Z. anal. Chem., 174, 254 (1950).
630b. I. M. Issa, R. M. I s s a, L. A. Shalaby. Z. anaL Chem., 176,
257 (1930).
630c. I. M. Issa, R. M. Issa, L. A. S h a 1 о b y. Z. anal. Chem., 176,
250 (1930).
631. J. H. J о e, F. W i 1 1, R. A. В 1 a c k. Anal. Chem., 25, 1200 (1953).
632. G. Johansson. Svensk. kem. tidskr., 65, 79, (1953).
633. V-'S. Jovanovic h, E. F. Zukker. Bull. Jnst. Nucl. Sci., 4, 111
(1954); цит. no Anal. Abstr., 2, 72 (1955).
634. W. H. J ura, AnaL Chem., 26, 1121 (1954).
635. P. G. J ura n, J. P. M a i 1 1 о t. Phys, et Radium, 19, 35 (1958).
636. M. К an t h. а г a j u r s, K.. Neelakantam. J. Sci. and Ind. Res., HB, 78 (1952).
637. J. Katz, G. Seaborg. The Chemistry of Actinide Elements, 1957.
638. L. I. К a t z i n, D. M. S i m о n, J. R. Ferraro. J. Am. Chem. Soc.,
74, 1191 (1952).
639. L. I. К a t z i n, J. C. S u 1 I i v a n. J. Phys. Coll. Chem., 55, 346 (1931).
640. В. К e i 1 i n, J W. Ot vos. J. Am. Chem. Soc., 68, 2665 (1946).
641. W. F. Kember. Analyst, 77, 78 (1952).
642. J. Kennedy. Chem. and Ind., 1958, 950.
643. J. Kennedy, R. V. D a v i e s. Chem. and Ind., 1956, 378.
644. J. Kennedy, J. Lingan e. Anal. Chim. Acta, 18, 240 (1958).
645. E. F. Kern. J. Am. Chem. Soc., 23, 685 (1901).
646. D. M. H. К e r n, E. F. О r 1 e m a n, J. Am. Chem. Soc., 71, 2102 (1949).
647. A. S. К e s t о n, S. Udenf rien d, R. K. Cannan. J. Am. Chem-
Soc., 68, 1390 (1946).
648. S. Khopkar, A. de. Chem. and Ind., 1959, 291.
649. S. К i к u c h i. J. Chem. Soc. Japan, 43, 544 (1922).
650. S. Kikuchi. Sci. Repts. Tohoku Univ., 16, 707 (1927).
412
651. R. В. Kimball. J. E. Rein, A. E. C. Res. and Development Report I. D. O.—14380, Chemistry Ti D-4500 (1956); цит. no Anal. Abstr., 4, 3940' (1957).
652. К. К i r b y, R. Gra wll y. Anal. Chim. Acta, 19, 363 (1958).
653. A. V. Kirsanov, V. M. Tcherkasov., Bull. Soc. Chim. France, [5], 3, 817 (1936).
654. A. К i s s, G. A 1 m a s s y. Magyar Kem. foldyirat, 64, 332 (1958).
655. D. К i s s, Z. Z a m о r i. Energia es atomtechn., 11, 12 (1958).
656. T. К i у о s i, H. H i г а к u, I. T a d a s i. J. Am. Chem. Soc., 48, 895
(1926).
657. I. К i у о s i, H. H i г а к u, I. T a d a s i. J. Chem. Soc., Japan, 61, 269 (1940).
658. T. К i у о s i, T. Y о s h i о, T. C h i у e к o. J. Pharmac. Soc. Japan, 75, 724 (1955).
699. R. Ki erne nt, Z. anal. Chem., 145, 9 (1955).
660. C. A. Klienberger. Anal. Chem., 29, 1721 (1957).
661. О. К о bl i c. Chem. listy, 19, 1 (1925).
662. M. К о h n. Z. anorg. und allgem. Chem., 50, 315 (1906).
663. I. M. К о 1 t h о f f, W. E. H a r r i s, G. M a t s u у a m a. J. Am. Chem. Soc., 66, 1782 (1944).
664. I. M. К о 1 t h о f f, W. E. H a r r i s. J. Am. Chem. Soc., 67, 1484 (1945).
665. I. M. К о 1 t h о f f, W. E. H arris. J. Am. Chem. Soc., 68, 1175 (1946).
666. I. M. К о 1 t h о f f, W. E. H а г r i s. J. Am. Chem. Soc., 69, 446 (1947).
667. t. M.’ Kolthoff, R. A. Johnson. J. Electrochem. Soc., 98, 138 (1951).
668. I. M. Kolthoff, J. J. L i n g a n e. J. Am. Chem. Soc., 55, 187 (1933).
669. J. Komenda. Chem. listy, 47, 531 (1953).
670. K. S. К о p p i к a г, V. G. К о r g а о n к а г, T. К. S. M u r t h у. Anal. Chim. Acta, 20, .366 (1959).
671. J. Kor kish, A. Farag, F. Hecht. Mikrochim. Acta, 1958, 415.
672. J. Korkisch, A. Farag, F. Hecht. Z. anal. Chem., 161, 92 (1958).
673. J. Korkisch, M. Zaky, F. Hecht. Mikrochim. Acta, 1957, 485.
674. L. К о s t a. J. Repts. «I. Stefan» Inst., 2, 7—8 (1955); цит. по РЖХим,
74684 (1957).
675. I. К о г у t a. Collect. Czechoslov. Chem. Comm., 20, 667 (1955).
676. J. К о u t e с к y. Chem. listy, 48, 996, 1605 (1954).
677. J. Kraft. Compt. rend., 206, 57 (1938).
678. N. К г a m e r, R^ F i s c h e r. Anal. Chem., 26, 415 (1954).
679. К. A. К r a u s, G. E. Mo о r e, F. N e 1 so n. J. Am. Chem. Soc., 78, 2692 (1956).
680. К. К r a u s, F. N e 1 s о n. J. Am. Chem. Soc., 72, 3901 (1950).
681. K. A. Kraus, F. Nelson. J. Am. Chem. Soc., 77, 3721 (1955).
682. К- А. К r a u s, F. N e 1 s о n, G. E. M о о r e. J. Am. Chem. Soc., 77,
3979 (1955).
683. А. К r i s h e n, H. F г e i s e r. Anal. Chem., 27, 288 (1957).
684. S. К r i s h n a, H. S i n g h. J. Am. Chem. Soc., 50, 792 (1928).
685. E. S. Krittchevsky, J. C. Hindman. J. Am. Chem. Soc., 71, 2096 (1949).
686. N. S. К u n t e, D. S e n. Current Sci., 28, 94 (1958).
687. H. К u r a m a, V. 1 s h i h a r a. Bunseki Kagaku, 6, 3 (1957); цит. no Chem. Abstr, 53, 4006b (1959).
688. J. Lacoste, M. H. Earing, S. E. W i b er 1 ey. Anal. Chem., 23, 871 (1951).
689. A. Lacour t, G. Sommerlyns, J.Soete. Mikrochemie, 38, 348 (1951).
690. J. L a h n e r. Sitzber. Akad. Wiss., Wien, Math, natur. W.-Klasse, Abt. Ila, 148, 149 (1939).
691. R. Larsen. Anal. Chem., 31, 545 (1959).
692. E. L a s s n e r, R. Scharf. Z. anal. Chem., 164, 398 (1958)
693. P. Lebeau. Compt. rend., 152, 439 (1911).
413
69-1. G. \\ . Leddieotie. \\ T. Al i i 1 i n >. L. C. Bat e, J F. E m e-r y, R. E. D г u sche 1, W. А. Brooks b а п к. Second United Nations International Conference of the 'Peaceful Use< of Atomic Energy. P 927 (19581.
695. Al. E e d e г e r. Anal. Chim. Acta. 4, 629 (1950).
696. AL Lederer. Anal. Chim. Acta, 7. 458 (19521.
697. D. J. Legge. Anal. Chem.. 26. 1617 (1951).
698. R. L. L e S t r a n g e. AL W. L e г n e r, G. J. P e t r e 1 , c. 1 . S.
Atomic Energy Comm. Rep. NV 0-2047 (1954».
699. J. A. Lewi >. К. C. (herto n. Analxst, 79, 293 (195-1*.
700. W. L i b и s, AL D. S i ek ier к a. Z. Lib i; s. Roczn. chem.. 31. 129 >
(1957i.
/01. L. I. i n (i e m a n. \L. Weis о n. Spectrochim. Acta. 9. 17 «1957).
702. J. Ling a n e. Anal. Chem.. 26, 622 (1954).
703. J. Lingan e. Anal. Chim. Acta, 2, 584 (1948).
704. .1. Lingane. S. J о n e s. Anal. Chem., 22, 1169 (1956*.
705. .1. Lingan e, R. 1 w a nt о t o. Anal. Chim. Acta. 13, 465 (1955*.
706. J. Lingane, J. Kennedy. Anal. Chim. Acta. 15. i65(195(ii
707. J. Lingan e, S. Jone s. Anal, (.hem., 22, 1220 (1950).
708. F. Luce n a-Co n d e. L. Prat. Alikrochim. Acta. 1955, 799.
709. G. E. L e n d c 1 I, J. 1. H о f f m a n. (Outlines of Methods of Chemical
Analysis, John Wiley and Sons. N. Y., 1938.
710. G. E. F. I. e n d e I 1, AL В. К п о \\ 1 e s. J . Am. Chem. Soc.. 47, 2637 (1925).
711. R. Luth er, A. C. Al i c h i e. Z. Elektrochem. 14, 826 (1908).
712. F. L у n i c h. J. В a n ni g a r d n e r, Rev. Sclent. Instrnm., 26. 435 (1955).
713. W. Al acDo w ell, C. Baes, J. Phys. Chem., 62, 777 (1958).
714. D. J. Al a с E w e n. Th. D e r v i e s. Can. J. Chem., 35, 1225 (1957).
715. S. Al a c i c, Recueil trav. inst. rech. struct. Matiere. 4, 45 (1951).
716. IL A. C. Al а с К a y. Trans. Faraday Soc., 47, -136 (1951).
717. 11. A. C. Al а с К a v. Progress in Nuclear Eiiergv. series III. Process Chemistry. 1. 122 (1956).’
718. \V. D. Al а с к u n t о s h. R. E. .1 e r \ i s. Atomic Energy Can. Limited. Chalk River Project, № )81 (1957).
719. R. L. Al а с к I i n. J. H. Lvkins. J. Chem. Phvs., 19. 844 (1951).
720. W. Al a c N e v i n. B. Baker. Anal. Chem., 24, 986 (1952).
721. W. J. Al a e c k, J. L. В о о m a n. AL (.'..Elliott. J . E. R e i n. Anal. Chem., 30, 1902 (1958).
722. \V. J. Al a e c k, G. L. В о о m a n. AL C. Elliot, J. E. R e i n. Anal. Chem.. 31, 1130 (1959).
723. AL T. Al a h a d e o. D. V. В hat n aga r, T. K. S. Al и г t h y. Proc. Indian. Acad. Sci., 42, 29 (1955).
724. H. A. Al a h 1 ni a n. G. W. Led d icot I e. Anal. Chem., 27, 823 (1955).
725. S. Al a i 1 1, P. Lev eq ti e. Compt. rend, de la Conf. Inter, sur la Isotopes dans Rech. Sci. Paris. UNESCO, (ND) TC/49, 1957.
726. A. R. Al a у n. Anal. Chein., 26, 1507 (1954).
727. A. K. Al a j u m d a r, AL AL C h а к r a h a г t t v. Anal. Chim. Acta.. 17, 415 (1957).
728. A, K. At a j umd a r. J . B. C h о w d h u r v. Anal. Chim. Acta. 19, 576 (1958).
729. L. M a 1 a t e s t a. Gazz. Chim. ItaL, 69, 408 (1939).
730. L. Al a I a t e s t a. Gazz. Chim. ItaL, 69, 752 (1939).
731. J. Maly, V. Vesely. J. inorg. and Nucl. Chem., 7, 119 (1958).
731a. Al a s a j о s h i S c h i b a s h i, T a i t i г о F u i j n a g a. Kosuke
Izutsn. J. Electroanal. Chem., 1, 26 (1959/1960).
732. К. B. Al a ther. Proc. Roy. Soc. New South Wales, Part HI, 80, 187 (1946).
733. A. R. M a t h i e s о n. J. Chem. Soc., Supl. Iss., 1949. 294.
734. H. M a t s u у a m a, T. H a z а, К. К о у a ni a. J. Chem. Soc. Japan. Pure Chem. Sec.. 79. 958 (1958); цит. no Anal. Abstr., 6, 2132 (1959).
414
.35. A. Mazzucchel I i. Atti. Lincei della Reale Accad. dei (5). 15, 494 (19J4).
-3'1. D. Aleie r, R. Meyers. J. Am. Chem. Soc., 78, 2340 (1949).
/_37. L. Meites. Anal. Chem., 24, 1057 (1952).
737a. L. M e i t e s. Polarographic Techniques.— N. Y., 1955, p. 290.
738. H. Meixner. Chemie der Erde, 12, 433 (1940).
739. J. W. Mellor, H. V. T h о m p s о n. Quantitative Analysis, Charles Griffin and Co, Ltd. London, 1948.
740. C. F. Metz. Anal. Chem., 29, 1748 (1957).
741. R. J. Meyer, E. Pietsch. Gmelin’s Handbuch der anorganischen Chemie. Systemnummer 55. Uran und Isotope, Verlag Chemie G. M. В. H., Berlin, 1935, p. 104.
742. R. M e у e r s, E. S w i j t. J. Am. Chem. Soc., 70, 1047 (1948).
743. J. Micha 1. Collect. Czechoslov. Chem. Comm., 21, 1295 (1956).
744. G. Milner. Analyst, 81, 367 (1955).
745. G. Milner. Atomic Energy Res. Establ., Repts 1—9 (1953).
746, G. M i 1 n e r, G. В a r n e t t. Anal. Chim. Acta, 17, 220 (1957).
747. G. W. M i i n e r, G. А. В а г n e t t. Atomic Energy Res. Establ., Repts.
№ C/R, 2723 (1958); цит. по РЖХим, 42088 (1959).
748. G. M i 1 n e r, J. Edwards. Anal. Chim. Acta, 16, 109 (1957).
749. G. W. M i 1 n e r, J. W. E dwards. Anal. Chim. Acta, 17, 259, (1957).
750. G. Milner, J. Edwards. Anal. Chim. Acta, 18, 513 (1958).
751. G. W. M i 1 n e r, J. H. Nunn. Anal. Chim. Acta, 17, 494 (1957).
752. G. M i 1 n e r, A. Wood. Atomic Energy Res. Establ., Repts 1041 (1953); цит. no РЖХим, 9126 (1953).
753. G. Milner, A. W о о d. Brit. Abstr., 10, 433 (1952).
754. J. Minczewski, S. Kolyga. Chem. analyt., 3, 467 (1958).
755. J. Minczewski, R. Przytycka, Z. Kohma n. Chem. analyt., 3, 27 (1958).
756. C. Mohr. Ann. Chem. Pharm., 53, 105 (1858).
757. C. Moore. Anal. Chem., 30, 908 (1958).
758. G. H. Morrison, H. Freiser. Anal. Chem., 30, 632 (1958).
759. G. H. M о r r i s о n, H. F r e i s e r. Solvent Extraction in Analytical Chemistry, John Wiley and Sons. N. Y., 1957.
760. A. K. Mukh er, A. K. D e y. Chim. analyt., 39, 148 (1957).
761. A. M ц к h e r j i, A. D e y. J. Inorg. and Nucl. Chem., 6, 314 (1958).
762. А. К- M ц к h e г j i, A. K. D e y. Z. anal. Chem., 160. 98 (1958).
763. А. К. M u к h e r j i, A. K. D e y. Proc. Nat. Acad. Sci. India, 26, 242 (1957).
764. A. K- Mukher j i, A. K.Dey. Proc. Indian Sci. Cong., 44, (3), 70 (1957).
765. А. К. M u к h e r j i, A. K. D e y. Mikrochim. Acta, 1958, 736.
766. А. К. M u к h e r j i, A. K- D e y. J. Indian Chem. Soc., 35, 113 (1958).
767. О. H. Mil 1 1 er. J. Am. Chem. Soc., 69, 2992 (1947).
768. A. Muller. Z. anorg. und allgem. Chem., 109, 235 (1920).
769. A. Mftll er.. Z. prakt. Chem., 157, 89 (1940).
770. F. K. S. Murthy. Anal. Chem. Acta, 16, 25 (1957).
771. M. N a к a n i s h i. Bull. Chem. Soc. Japan, 24, 33 (1952).
772. T. N a к a z о n o. J. Chem. Soc. Japan, 42, 761 (1921).
773. K. Naotsuna. J. Chem. Soc. Japan, 43, 333 (1922).
774. K. Naotsuna. Sci. Repts. Tohoku Univ., 16, 701 (1927).
775. M. Narasinhasastri, G. Gopala Rao. Current Sci., 18, 402 (1949).
776. T. N a s c u t i u. Commun. Acad. Republ. populare Romine, 7, 51 (1957).
777. N. Neal. Trans. Faraday Soc., 45, 1 (1949).
778. F. N e 1 s о n, К. A. К r a u s. J. Am. Chem. Soc., 73, 2157 (1951).
779. W. N e u m a n; R. F 1 e m i n g, А. С a r 1 s о n, N. Cover. J. Biol. Chem., 173, 41 (1948).
780. W. F. Neuman, J. R. H a v i 1 1, I. F e 1 d m a n, J. Am. Chem. Soc., 73, 3593 (1951).
781 H D. N e w t о n, J. H u g h e s. J. Am. Chem. Soc., 37, 1711 (1915).
27 Аналитическая химия урана /лс
782. E. N i c h о 1 s, H. H о w e s. Fluorescence of the Uranyl Salts. Washington. 1919.
783. E. N ichol.s, M. K. S 1 a t t e r y. J. Opt. Soc. Am., 16, 449 (1926).
784. O. A. N i e t z e 1, M. A. De S es a. Anal. Chem., 29, 756 (1957).
785. A. N о r s t г о m, L. G. S i 1 1 e n. Svensk. kem. tidskr., 60, 227 (1948).
786. M. N ort h up, Ind. and Eng. Chem., 17, 664 (1945).
787. H. M. Ockenden, J. K. Foreman. Analyst, 82, 592 (1957).
788. E. F. О r I e m a n, M. D. H. Kern. J. Am. Chem. Soc., 75, 3058 (1953).
789. E. A. Ostroumow. Z. anal. Chem., 106, 244 (1936).
790. E. P a i g e, M. С. E 1 1 i о t, J. E. R e i n. Anal. Chem., 29, 1027, 1029 (1957).
791. J. P a p i s h, L. Hoag. Proc. Natl. Acad. Sci. U. S., 13, 726 (1927).
792. A. Pappas. Arch. math, og naturvid., 45, 83 (1942); цит. no Chem. ZbL.
2, 1494 (1942).
793. W. P a г г i. Giorn. farn. chim., 73, 207 (1924).
794. Y. Parsons, W. Seaman. Anal. Chem., 27, 210 (1955).
795. P. Pascal. Bull. Soc. Chim. France, 13, 1089 (1913).
796. P. Pascal. Compt. rend., 157, 932 (1913).
797. C. Patterson, N. Brown, G. Tilton, M.'I n ghr am. Phvs. Rev., 92, 1234 (1953).
798. H. Patton. Anal. Chem., 23, 393 (1951).
799. Z. Pauling. J. Am. Chem. Soc., 69, 542 (1947).
800. E. Pel i got. Ann. Chim. Phys., 5, 1 (1842).
801. D. F. P e p p a r d, J. P. F a r i s, P. R. Gray, G. W. Mason. J. Phys. Chem., 57, 294 (1953).
802. D. F. P e p p a r d, G. W. M a s о n, M. V. G e r g e 1. J. Inorg. and Nucl. Chem., 3, 370 (1957).
803. J. P e r m a n. Sloven Acad. Sci., J. Stefan Inst. Phys. Rept, 2, 27 (1955); цит. no Chem. Abstr., 50, 4709 h (1956).
804. P. P f e i f e r, T. Hesse, H. P f i t z n e r, W. S c h о 1 1, H. T i e 1 e г t, J. makromol. Chem., 149, 217 (1937).
805. S. P h i 1 1 i p s. D. Kern. Anal. Chim. Acta, 20, 295 (1959).
806. G. Phillips, J. Woodhead, E. Jenkins. Anal. Chim. Acta, 19, 229 (1959).
807. E. E. P i с c i о t t o. Bull. Soc. Beige geol., pale'ontol. et hydro!., 58, 75 (1949).
808. E. E. P i с c i о t t o. Bull. Soc. Beige geol., paleontol. et hydro!., 61, 215 (1952).
809. С. A. P i e r i e. Ind. and Eng. Chem., 12, 60 (1920).
810. R. P i e t s c h. Z. anal. Chem., 152, 168 (1956).
811. к. Pietsch. Z. anal. Chem., 159, 37 (1957).
812. F. Pisani. Compt. rend., 52, 72 (1861).
813. Y. P i t t s, D. D e F о r d, T. M a r t i n, E. S c h m a 1 1. Anal. Chem., 26, 628 (1954).
814. A. P 1 e i s c h 1. Schweigger’s J., 45, 1 (1825).
815. F. H. P о 1 1 a r d, P. Hanson, W. J. G e a r d. Anal. Chim. Acta, 20, 26 (1959).
816. F. H. P о 1 I a r d, F. W. M а с О m i c, J. M. E 1 b e i n. J. Chem. Soc., 1951, 470.
817. С. P о r । ezz a, A. Don at t i. Ann. Chim. Applicata, 16, 622 (1926).
818. R. P f i b i 1. Z. Anal. Chem., 138, 130 (1953).
819. R. P f i b i 1, А. В 1 azek. Collect. Czechoslov. Chem. Comm., 16, 567
(1953).
820. R. Pfibil, J.Vorlicek. Chem. listy, 46, 216 (1952).
821. R. P r i b i 1, M. J e 1 i n e k. Chem. listy, 47, 1326 (1953).
822. G. P r i c e, R. F e r r e t t i; S. S c h w a r t z. Anal? Chem., 25, 322 (1953).
823. O. S. P u r m a n. Amer. Sci., 16, 229 (1903).
824. C. F. Rammelsburg. Poggendorfs Ann., 59, 23 (1843).
825. V. P. R а о, В. V. S. R. M u r t y, G. Rao. Z. anal. Chem., 147, 99 (1955).
416
826. V. P. R а о, В. V. S. М и г t у, GF Rao. Z. anal. Chem., 147, ”161 (1955)
827. B. R. R а о, С. С. P a t e 1. Proc. Indian Acad. Sci., A43. 276 (1956).
828. G. G. R а о, V. P. R a o. Anal. Chim. Acta, 15, 97 (1956).
829. G. G. R а о, V. P. R a o, N. V e n к a t a m m a. Z. anal. Chem., 150,
•178 (1956).
830. V. P. Rao, G. G. Rao. Taianta, 1, 355 (1958).
831. M. N. Rao, R. B. S. V. Rao. Z. anal. Chem., 142, 161 (1954).
832. M. N. Rao, B. S. V. Rao. Z. anal. Chem., 159, 356 (1958).
833. V. R a o, G. R a o. Z. anal. Chem., 160, 190 (1958).
834. G. R a o, G. S о m i d e r a m m a. Z. anal. Chem., 157, 27 (1957).
835. P. R a у Z. anal.'Chem.', 86, 20 (1931).
836. H. N. R a y, N. P. В h a t t a c h a г а у у a. Analyst, 82, 164 (1957).
837. P. R а у, M. К. В о s e. Z. anal. Chem., 95, 400 (1933).
838. P. R а у, M. K. Bose. Z. anal. Chem., 95, 585 (1933).
839. Jj. W. R e e d, К- К i g о s h i, A. T u г к e v i c h. Geochim. et cosmo-
chim, Acta, 20, 122 (1960).
840. G. R e e d, A. T u г к e v i c h. Nature, 180, 594 (1957).
841. C. Reilley. Chem. Ed., 318, 543 (1954).
842. C. R e i I I e y, R. A d a m s, N. F u r m a n. Anal. Chem., 24, 1044 (1952).
843. C. R e i 1 1 e y, W. С о о к e. Anal. Chem., 23, 1223 (1951).
844. C. R e i 11 e y, W. Coo ke, N. F u r m a n. Anal. Chem., 23, 1030 (1951).
845. A. Remele. Z. anal. Chem., 4, 371 (1885).
846. R. R e r g. J. makromol. Chem., 115, 178 (1927).
847. J. H. Reynolds. Phys. Rev., 79, 789 (1950).
848. A. C. Rice, H. C. Fogg, C. J a m e s. J. Am. Chem. Soc., 48, 895 (1926).
849. M. R i c h m o.n d, J. Baldwin, E. M a i e n t h a 1, U. S. Atomic
Energy Comm., № Ps-4555, 19 (1956); цит. no Chem. Abstr., 51, 7234a (1957).
850. H. Robinson. Trans. Electrochem. Soc., 92, 445 (1947).
851. R. R о b i n s о n, C. L i m. J. Chem. Soc., 1951, 1840.
852. C. Rodden. Anal. Chem., 21, 327 (1949).
853. C. Rodden. Anal. Chem., 25, № 11 (1953).
854. C. Rodden. Anal. Chem., 25, 1598 (1953).
855. E. R о n a, L. O. G i 1 p a t r i c, L. M. J e f f r e y. Trans. Am Geophys.
Union, 37, 697 (1956).
856. H. Rose. Handbuch der analytischen Chemie. Leipzig, 1871.
857. H. Rose. Pogg. Ann., 116, 352 (1862).
858. A. R о s e n h e i m. Z. anorg. Chem., 153, 126 (1926).
859. K. R о w 1 e у, E. S w i j t. Anal. Chem., 26, 373 (1954).
860. C. L. R u 1 f s, A. K. De, P. J. E 1 v i n g. J. Electrochem. Soc., 104,
80 (1957).
861. C. L. R u I f s, A. K. D e, P. J. E 1 v i n g. Anal. Chem., 28, 1139 (1956).
862. C. L. R u I f s, A. K. D e, J. L a к r i t z, P. J. E I v i n g. Anal. Chem.
27, 1802 (1955).
863. C. L. R u 1 f s, P. J: E 1 v i n g. J. Am. Chem. Soc., 77, 5502 (1955).
864. W. A. R u n c i m a n. Nature, 175, 1082 (1955).
865. W. R у a n, A. F. W i 1 1 i a m s. Analyst, 77, 293 (1952).
866. J. Rydberg. Acta Chim. Scand., 11, 201 (1957).
867. J. Rydberg. Arkiv Kemi, № 2, 101 (1955).
868. J. Rydberg, B. Bernstrom. Acta Chim. Scand., 11, 86 (1957).
869. L. S а с с о n i, G. G i a n n о n i. J. Chem. Soc., 1954, 2368.
870. L. Sacconi, G. Giannoni. J. Chem., Soc., 1954, 2751.
871. C. J.Sambucetti, A. L. Gori. Second United Nations Interna-
tional Conference on the Peaceful Uses of Atomic Energy, 15/P/1576 (1958).
872. B. R. Sant, M. ,K. Joshi. Naturwissenschaften 44, 536 (1957).
873. B. S a r m a. Trans. Hose Res. Inst. Calcutta, 18, 105 (1949—51); цит. no Chem. Abstr., 47, 11067e (1953).
874. B. S a r m a. Sci. and Culture, 16, 165 (1950).
875. D. V. N. Sa rma, R. R а о B. S. V. Anal. Chim. Acta, 13. 142 (1955).
27* /n
417
876. В. Sarma, С. Р. S a v а г i а г. J. Sci. Ind. Research, 16, 2, 80 (1957).
877. L. A. S а г v е г, I. М. К о 1 t h о f f. J. Am. Chem. Soc., 53, 2902 (1931).
878. T. R. S a s t r i, G. R a o. Z. anal. Chem., 163, 1 (1958).
879. T. Sato. J. Inorg. and Nucl. Chem., 7, 147 (1958).
880. W. R. Sc hoe 11 er, H. W. Webb. Analyst, 58, 143.(1933).
881. W. R. S c h о e 1 1 e г, H. W. Webb. Analyst, 61, 235 (1936)
882. W. R. S с о e 1 1 e r, H. W. Webb. Analyst, 61, 585 (1936)
883. J. M. Schreyer, C. F. Baes. Anal. Chem., 25, 644 (1953).
884. R. Schwarz. Helv. Chim. Acta, 3, 330 (1920).
885. G. S c a g 1 i a r i n i, P. P r a t e s i. Ann. Chim. Applicata, 19, 85 (1929).
886. T. R. Scott. Analyst, 74, 486 (1949). .
887. T. R. Scott. Analyst, 75, 100 (1950).
888. B. S c r i b n e r, H. M u 11 i n. J. Res. Nat. Bur. Standards, 37, 379 (1946).
889. G. S e a b о r g. Nucleonics, 5, 16 (1949).
890. G. S e a b о r g. Science, 105, 349 (1947).
891. K. S e i d 1, M. Beranek. Chem. listy, 52, 337 (1958).
892. K- S e d 1, M. Beranek. Collect. Czechoslov. Chem. Comm., 24, 298 (1959).
893. H. J. Seim, R. J. M о r r i s, D. W. F г e w. Anal. Chem., 29, 443 (1957).
894. F. S e i s e y. Anal. Chem., 26, 1607 (1954).
895. I. Sekarka, J. V о r 1 i c e k. Chem. listy, 47, 512 (1953).
896. L. S e 1 m i, F. F u s s i. Chimica et industria, 40, 193 (1958).
897. G. Semer ano. Gazz. Chim. Ital., 72, 297 (1942).
898. R. N. S e n S a r m а, А. К- M a I 11 k. Anal. Chim. Acta, 12, 329 (1955).
899. R. N. Sen S a r m а, А. К- M a 1 1 i k. Sci. and Culture, 20, 135 (1954).
900. R. N. Sen S a r m а, А. К- M a 1 1 ik. Z. anal. Chem., 148, 179 (1955).
901. A. P. Seyfang. Analyst, 80, 74 (1955).
902. A. P. Seyfang, A. A. Smales. Analyst, 78, 394 (1953).
903. H. Seyhan. Ber., 87, 396 (1954).
904. И’. J. Shalgosky. Analyst, 81, 512 (1956).
905. R. A. J. Shelton. Analyst, 82,-531 (1957).
906. Sh. Shibata, T. Matsumae. Bull. Chem. Soc. Japan, 32, 279 (1959).
907. H. S h о r t, W. Dutton. Anal. Chem., 20, 1073 (1948).
908. W. Shults, P. Thomason. Anal. Chem., 31, 492 (1959).
909. W. S h u 1 t s, P. T h о m a s о n, M. Kelley. Anal. Chem., 27, 1750 (1955)
910. J. A. S i e m s s e n. Chemiker Ztg., 35, 139, 742 (1911).
911. L. S i 1 1 e n. Recueil trav. Chim., 75, 705 (1956).
912. C. Sill, H. P e t e r s о n. Anal. Chem., 19, 646 (1947).
913. C. S i 1 1, H. Peterson. Anal. Chem., 24, 1175 (1952).
914. C. W. S i 1 1, H. E. P e t e r s о n. Z. anal. Chem., 143, 306 (1954).
915. F. S i 1 v a, L. Moura. Tecnica, 33, № 285, 5 (1958).
916. L. S i 1 v e r m a n, L. M о u d y. Anal. Chem., 28, 45 (1956).
917. L. S i 1 v e r m a n, L. M о u d y. Nucleonics, 12, 60 (1954).
918. L. S i 1 v e г m a n, L. M о u d v, D. W. Hawley. Anal. Chem., 25, 1369 (1953).
919. Z. Si b verman, M. S h i d e 1 e r. Anal. Chim. Acta, 15, 181 (1956).
920. V. S i mon, E. P r i p 1 a t о v a. Chem. listy, 50, 907 (1956).
921. M. K. Slattery. J. Opt. Soc. Am., 19, 175 (1929).
922. M. K. Slattery. Proc. Nat. Acad. Sci. U. S. A., 14, 228 (1928).
923. A. Smales. Analyst, 77, 602 (1952).
924. A. A. Smales. Analyst, 77, 778 (1952).
925. A. Smith, F. Grimaldi. Geol. Surv. Bull., № 1106 (1954).
926. H. R. D. Smith. Z. anorg. Chem., 150, 163 (1936).
927. D. O. S n e 1 i v a n, P. S a d e r. J. Chem. Soc., 1957, 2916.
928. H. Sohlesinger, H. Brown, J. Katz. J. Am. Chem. Soc., 75, 2446 (1953).
929. K. Someya. Z. anorg. und allgem., Chem., 145, 168 (1925).
930. K. Someya. Z. anorg. und allgem. Chem., 152, 368 (1926).
931. J. Sonntag, L. Farad y, A. Janos i. Magyar kem. folyoirat., 61, ~ 312 (1055); цит. по РЖХим, 47302 (1956).
418
932. A. Sousa. Mfkrochemie ver. Mikrochim. Acta. 40, 319 (1953).
933. J. H. S p i n n e r, F. C. Miller. Atom. Energy Can. Ltd., Chalk River Project AECL (1959), 789; цит. no Chem. Abstr., 53, 14836g (1959).
934. K. S r i n i v a s 1 i 1 u, D. Purushottam, B. R. Rao. Z. anal. Chem., 159, 406 (1958).
935. D. T. Steadman. J. Opt. Soc. Am., 38, 1100 (1948),
936. T. W. Steele. Analyst, 83, 414 (1958).
937. T. Steele, L. Taverner. Second United Nations International Conference on the Peaceful Uses of Atomic Energy, 1958, NP/1117.
938. R. W. Stevens, W. W о о d, K. G о e t z, С. H о r r. Anal. Chem., 31, 962 (1958).
939. D. C. Stewart, W. C. Bentley. Science, 120, 50 (1954).
940. H. W. S t о п e, С. В c e s о n. Ind. and Eng. Chem., Anal. Ed., 8, 188 (1936).
941. A. Strasheim. Spectr. Acta, 4, 200 (1950).
942. R. Strubl. Collect. Czechoslov. Chem. Comm., 10, 466 (1938).
942a. P. Subba ra man, N. Yoshi, J. Gupta. Anal. Chim. Acta, 20, §9 (1959).
943. P. Subbaraman, N. Joshi, J. Gupta. Anal. Chem. Acta, 20, 190 (1959).
944. G. V. S u b b а г a у u d u, B. R. S. V. R a o. J. Sci. and Ind., Res., BC 17, 363 (1958).
945. Z. S u 1 c e k, J. Michal, J. Dolezal. Collect. Czechoslov. Chem. Comm., 24, 1815 (1959).
946. A. K. Sundaram, S. Banerjee. Anal. Chim. Acta, 8, 526 (1953).
947. A. S u n e r, E. Lagos. Second United Nations International Conference on the Peaceful Uses of Atomic Energy. P/1553 (1958).
948. C. D. S u s a n о, О. M e n t s, С. K. Talbott. AnaL Chem., 28, 1072 (1956).
949. M. V. Susie. Bull. Inst. Nucl. Sci., 7, 35 (1957).
95Q. M. V. Susie, N. J e 1 i c. Bull. Inst. Nucl. Sci., 7, 29 (1957).
951. M. S u s i с, I. G a 1, E. С u к e r. Anal. Chim. Acta, 11, 586 (1954).
952. J. Su t t о n. J. Inorg. and Nucl. Chem., 1, 68 (1955).
953. J. Sutton. J. Chem. Soc., Supl. Iss., 1949, 275.
954. F. S u t t о n, A. D. Mitchell. Volumetric Analysis, A. and J. Chur-
chill, Ltd. London, 1955.
955. E. S v a t e k, Z. R о u b a I, R. P r i b i 1. Collect. Czechoslov. Chem. Comm., 19, 674 (1954).
956. H. W. S w a n к, M. G. Mellon. Ind. and Eng. Chem., Anal. Ed., 10, 7 (1938).
957. E. Swift. AnaL Chem., 28, 1804 (1956).
958. V. S. S у г о к о m s к i, V. V. S t e p i n. J. Am. Chem. Soc., 58. 928 (1936).
959. S. Szalay, G. Al massy. Magyar tud. acad. Kem. tud. oszt. kczL, 8, 33 (1956); цит. по РЖХим, 19581 (1957).
960. P. S z a г v a s, Z. J a r a b i n, L. D e d e. Magyar Kem. folyoirat. 63, 151 (1957); цит. по РЖХим, 21095 (1958).
961. Z. Szebelledy, Z. Sdmodyi, Z. anal. Chem., 112, 313, 323, 332,
385, 391, 395 (1938).
962. G. T a m m a n n. Z. anorg. und allgem. Chem., 149, 64 (1925).
963. G. T ammann, Q. Mansuri.Z. anorg. und allgem. Chem., 126, 119 (1923).
964. G. T a m m a n n, A. S w о r i к i n. Z. anorg. und allgem. Chem., 176, 46 (1928).
965. N. A. T a n a n a j e v, G. A. P a n s c h e n к o. Z. anorg. und allgem. Chem., 150, 163 (1926).
966. J. P. T a n d о n, R. С. M e h г о t r a. Z. anaL Chem., 164, 314 (1958).
967. D. Taylor. Chem. Age, 75, 549 (1956).
967a. Те Yao Chi. Acta Chim. Sinica, 23, 79 (1957); C. A., 52, 13483e (1958)
968. С. T e m p 1 e t о n, F. H a 1 1. J. Phys. Coloid. Chem., 51, 1441 (1947).
969; J. T h a m e r. J. Am. Chem. Soc., 79, 4298 (1957).
970. L. Thatcher, F. Barken. Anal. Chem., 29, 1575 (1957)
419
971. The Spectrographic determination of aluminium in uranium metal. Cnemical Service Department. Industrial group Headquarters, Risley. Washington, Lancashire, 1958.
972. P. T h о m a s о n, L. T. С о r b i n. Anal. Chem. Div. Quarterly Progr. Rept. for Period Ending Dec. 26 (1951); Oak Ridge National Laboratory, ORNL-1233, p. 4—14.
973. P. F. Thomason, L. A. Nutting, U. Koskel a, W. M. Byer I y. U. S. Atomic Energy Comm., Rep. ORNL-1641, 15(1955j; цит. no Anal. Abstr., 4, 2183 (1957).
974. W. M. Thornton, J. F. S a d u s k. Ind. and Eng. Chem., Anal. Ed., 4, 420 (1932).
975. M. M. Till u. Current Sci., 24, 45 (1956).
976. M. M. T i 1 I u. Proc. Indian. Acad. Sci. 40A, 110 (1954).
977. M. M. T i 1 I u, D. V. Bhatnagar, T. K- S. Murthy. Proc. Indian. Acad. Sci., A42, 28 (1955); цит. no Anal. Abstr., 3, 399 (1956).
978. G. R Tilton et al. Bulk Geol. Soc. Am., 66, 1131 (1955).
979. G. H T i s h к о f f. Pharmacology and Toxicology of Uranium Compounds, New York—London, M. C. Graw. Hill Book Company, 1949, p. 125.
980. N. T i t I e s t a d. Z. phys. Chem., 72, 257 (1910).
981. E. T о m i c, J. M. La jenb ayer, M. P о I 1 a k. Z. anal. Chem., 16!
28 (1958).
981a. T oshi fumi Murat a. Nippon Kagaku Zasshi, 81, 440 (1960); C. A.-. 13981g (1960).
982. W. D. Treadwell. Helv. Chim. Acta, 5, 732 (1922).
983. W. D. Treadwell, M. L u t h y, A. R. Reiner. Helv. Chim. Acta 4, 551 (1921).
984. W. D. Treadwell. G. Schwa rzenba ch. Helv. Chim. Acta, 11, 405 (1928).
985. G. T r i d о t. Ann. Chimie, 10, 225 (1955).
986. G. T r i d о t. Ann. Univ. Paris, 24, 583 (1954).
987. G. T r i d о t. Compt. rend., 232, 1215 (1951).
988. S. T r i p a t h i, S. P г a к a s h. J. Indian Chem. Soc., 35, 119 (1958).
989. J. T sc h e r n i chow, E. Guldina. Z. anal. Chem., 96, 17 (1934).
990. H. T. Tucker. Analyst, 82, 529 (1957).
991. W. A. Turner. Am. J. Sci., 42, 109 (1916).
992. P. Tut undzic. Anal. Chim. Acta, 8, 168 (1953).
993. K- Venka teswarlu, R. B. Rao. Anal. Chim. Acta, 13, 79 (1955).
994. M. Venugopalan. Z. anal. Chem., 153, 187 (1956).
995. M. Venugopalan. Z. anal. Chem., 153, 187 (1956).
996. D. V i er, M. S c h u 1 t z, Z. В i g e 1 e i s e n. J. Opt. Soc., Am., 38, 811 (1948).
997. F. Vlasak, J. Svast a, R. Rotter, M. Bouberle. Sborn. Stat. Geol. Ustavu Ceskoslov. Rep., 16, 433 (1949).
998. J. S. W a h 1 b e r g, D. L. S к i n n e r, L. F. Rader. Anal. Chem., 29, 954 (1957).
999. G. H W a I d e n, L. P. H a m m e t t, S. M. Edmonds. J. Am. Chem.
Soc.,. 56, 350 (1934).
1000. A. Walsh. Spectr. Acta, 4, 47 (1950).
1001. C, W a m s e r, J. В e 1 1 e, E. P e n r s о h n, B. W i 1 1 i a m s о n. J. Am. Chem. Soc., 74, 1020 (1952).
1002. H. W a n к e, E. U. Monse. Z. Naturforsch., 10a, 667 (1955).
1003. J. C. W a r f. J. Am. Chem. Soc., 71, 3257 (1949).
1004. R. K, Warner. Austr. J. Appl. Sci., 4, 427 (1953).
1005. S. I. W a w i 1 о v, W. L. L e w s c h i n. Z. fur Physik, 48, 397 (1928).
1006. R. K. Web.ster. Ind. Chemist, 34, 495 (1958).
1007. G: W e i s s. Bull. Soc. Chim. France, 735 (1947).
1008. G. W e i s s, S. F a 1 1 a b, H. E r 1 e n m e у e r. Helv. Chim. Acta, 35,
1588 (1952).
420
1009. F. W e 1 c h e r. Organic Analytical Reagents, D. Van Nostrand Co. Inc. N. Y., 1947.
1010. B. W. W essli ng, M. A. De Sesa. U. S. Atomic Energy Comm., Rep. WIN-43, 12 (1956); цит. no Anal. Absir., 5, 497 (1958).
1011. T. S. West. Chem. Age, 80, 947 (1958).
1012. С. E White. Anal. Chem., 24, 1965 (1952); 32, 47R (1960).
1013. H. H. W i 1 1 a r d, H. D i e I. Advanced Quantitative Analysis, D. Van Nostrand Co, Inc., N. Y., 1943.
1013a. H. H. W i 1 1 a r d, J. A. Dea n. Anal. Chem., 22, 1264 (1950).
1014. K. W h i t h a n. Trans. Am. Geophys. Union, 33, 902 (1952).
1015. H. H. W i I 1 a r d, P. Y о u ng. J. Am. Chem. Soc., 55, 3260 (1933).
1016. A. F. Williams. Analyst, 77, 297 (1952).
1017. J. D W i 1 s о n, R. J. S m i t h. Anal. Chem., 25, 218, 334 (1953).
1017a . J. D. Wilson, R. K. W e b s t e r, G. W. С. M i I n e r, G. A. Bar-net t, A. A. Smales. Anal. Chim. Acta, 23, 505 (1960).
Ю18.чЬ. Wodkewicz. Chem. analyt., 3, 789 (1958).
1019. F. J W о о d m a n, T. G. С 1 i n t о n, W. F 1 e t c h e r, G. A. Welch. Second United Nations International Conference on the Peaceful Uses of Atomic Energy, A/conf. 15/P/1453 (1958).
1020. B. Jezowsk a-T rzebia t owska, A. В a rt ecki, M. C h m i e-lowska, H. Przywarska, T. Mi ku 1 s кi. Nukleoniks, 3, Spec. № 39 (1958).
1021. J. Yoe, F. Will. Anal. Chem., 27, 1200 (1955).
1022. J. H. Yoe, F. W i 1 1. R. A. В 1 a c k. Anal. Chem., 25, 1200 (1953).
1023. C. Y os hi mu г a. J. Chem. Soc. Japan, 75, 603 (1954).
1024. C. Yoshimura. Nippon Kagaku Zasshi, .77, 1 (1956); цит. no Chem. Abstr., 14479a (1957).
1025. C. Y r e n t e r. Anal. Chim. Acta, 8, 168 (1953).
1026. C. Zimmermann. Liebigs Ann. Chem., 232, 273 (1886).
1027. J. Zimmerman. Mining and Ind. Mag., 44, № 2, 54 (1954).
1028. J. Zimmerman, F. Rabbitts, E. Kornelson. Mines Mag., 43, № 12, 20, 44 (1953).
1029. J. Zimmerman, F. Rabbitts, E. Kornelsen. Mining and Ind. Mag., 44, 54, 1954.
1030. R. Zingaro. J. Am. Chem. Soc, 78, 3568 (1956).
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
1031. Л. M. Беляев, Г. Ф. Доброжанский, О. В. Соколов, П.П. Феофилов. Тезисы докладов 9-го Совещания по люминесценции. Киев, Изд-во АН СССР, 1960.
1032. М. Д. Галанин. Труды Физического ин-та им. П. Н. Лебедева, 12, 3 (1960).
1033. Л. Н. Галкин, П. П. Феофилов. ДАН СССР, 114, 745 (1957).
1034. А. В. Давыдов, Т.С. Добролюбская, А. А. Немодрук. ЖАХ, 16, 68 (1961).
1035. В. И. Кузнецов, С. Б. Саввин, В. А. Михайлов. Усп. хим. 29, 525 (1930).
1036. В. И. Кузнецов, С. Б. Саввин. Радиохимия, 2, 682 (1960).
1037. 3. И. К о б ы ш е в, Л. Н. Су гл обо в. Тезисы докладов 8-го Совещания по химии комплексных соединений. Киев, 1959.
1038. А. Ф. Кутейников. Бюлл. Всес. ин-та минерального сырья, № 7, 183 (1958).
1039. А. А. Н е м о д р у к, Ю. П. Новиков, А. М. Л у к и н, И. Д. Калинина. ЖАХ, 16, 292 (1951).
1040. А. А. Немодрук, Ю. П. Н о в и к о в, А. М. Л у к и н, И. Д. Калинина. ЖАХ, 16, 180 (1961).
421
1041. A. H. Севченко, Л. В. Володько. Изв. АН СССР, сер. физ.. 20, 464 (1956).
1042. 3. М. Свердлов. Оптика и спектроскопия, 3, 356 (1957).
1043. F. Bauman, Gr. Z о р a t t i. Read E. U. S. At. Energy Ccmm., TJD-7560, 105 (1958); цит. no Chem. Abstr., 53, № 21423b (1959).
1044. R. Burt on, H. С о о k, R. J а с о b s, E. V a 1 e c k e. U. S. Atomic Energy Comm., Rep. TJD-7560, 116 (1958); цит. no Chem.Abstr., 53,№21423a (1959).
1045. B. Cunningham, D. Gruen, J. Conway, R. McLaughlin. J. Chem. Phys., 24, 1275 (1956).
1046. G. D i e k e, A. Duncan. Spectroscopic Properties of Uranium Copma-unds. N. Y., 1949.
1047. D. Gruen, R. Conway, R. McLaughlin, B. Cunningham. J. Chem. Phys., 24, 115 (1956).
1048. Hay dee le Roux. Nature, 183, 1180 (1959).
1049. W. R u c i m a n. Nature, 175, 1082 (1955).
1050. D. P a t n a i k, B. S a h о o. Proc. Indian Acad. Sci., A49, №4, 200 (1959).
1051. J. P о г t e r. U. S. Atomic Energy Comm., Rep. KAPL-M-JTP-3, 17 (1958); цит. no Chem. Abstr., 53, 13886g (1959).
1052. I. Steele, L. Taverner. Progr. Nucl. Energy, Ser. 9, 1, 75 (1959).
1053. C. White. Anal. Chem., 26, 129 (1959).
1054. Ф. А. Захаров, А. И. Москвин. ЖНХ, 5, 1228 (1960).
1055. В. И. Герасимовский. Месторождения урана зарубежных стран. М„ Изд-во АН СССР, 1959, стр. 26.
1056. А. П. Виноградов. Химическая эволюция земли. М., Изд-во АН СССР, 1959, стр. 38.
1057. Р. В. Г е ц е в а, К. Т. Савельева. Руководство по определению урановых минералов. М., Госгеолтехиздат, 1956, стр. 85.
1058. Д. Я- С у р а ж с к и й. Методы поисков и разведки месторождений урана. М., Атомиздат, 1960, стр. 148.
1059. V. Т. A t h a v а 1 е, S. В а п е г j е е, G. К- В е 1 е k а г, N. М a hade v a n, L. М. М a h a j а п, М. N. N a d к а г n i, Das. М. S а п к о г,
Н. D. S h а г m а, А. К. S u n d а г m, М. S и n d а г е s а п, N. R. Tha-
к о о г, М. М. Т i 1 1 и, М. S. V а г d е, Ch. Venkateswarlu. Мат.
2-й конфер. по мирному использованию атомной энергии. Женева, 1958, доклад № 1671.
1060. В. Ф. Лукьянов, С. Б. Саввин, И. В. Никольская. ЖАХ, 15, 311 (1960).
1061. 3. М. Т у р о в ц е в а, Л. Л. Кунин. Анализ газов в металлах. М., Изд-во АН СССР, 1959.
1062. Г. М. Т у р о в ц е в а, Н. Ф. Л и т в и н о в а. 2-я Международная конф, по применению атомной энергии в мирных целях, доклад № 2205 (1958).
1063. А. Н. 3 а й д е л ь, А. А. . П е т р о в, К. И. Петров. Тезисы докладов на совещании по анализу газов в металлах (24—27. VI. 1958). М., Изд-
во АН СССР, 1958, стр. 30.
1064. Е. В. Без рогова, П. Н. П а л е й. ЖАХ, 16, 57 (1961).
1065. R. F г a n k 1 i п, A. W i 1 s о n. U. К- А. Е- A., Ind. Gr.-SCS-R- 149 (1959)*
цит. по Chem. Abstr., 54, 6 (1960).
1066. U. К. А. Е. A.—Ind. Gr.—AM/S-124 (1958).
1С67. G. S. S p i c e r, J. D. H. Strickland. Anal. Chim. Acta, 18, 3, 231 (1958).
1068. A. R. E b e г 1 e, M. W. Lerner. Anal. Chem., 32, 146 (1960).
1069. L. Si 1 v erm a n, K. Trego. Anal. Chim. Acta, 15, 439 (1956).
1070. W. J. R о s s, A. S. Meyer, J. C. White. Anal. Chem., 29, 5, 810 (1957).
1071. J. E. S t i 1 1, L. A. D u m e y, R. С. C h i г u s i d e. Analyst, № 934, 4 (1954).
1072. П. А. К p ю к о в, E. В. Рен га рте н. ЖАХ, 10, 52 (1955).
422
1073. А. И. Пономарев, А. А. А станина. Тезисы докладов на совещании по анализу газов в металлах (24—27. VI. 1958). М., Изд-во АН СССР,. 1958, стр. 46.
1074. U.K-A-E.A.-1GO-AM S-121 (1958).
1075- . Н. Н. Крот, А. П. Смирнов-Аверин, А. Г. Козлов. ЖАХ, 14, 352 (1959).
1076. Walkden, Heetfild. Atomic Energy Res. Establ., Repts, NAM-37 (1959); цит. по РЖХим, 34525 (1960).
1077. R. Fernandez-Cellini. T. В a t ueca s Rodriguez. Ann. Real. Soc. Esp. Fis. Quim., 51 (6), 409 (1955).
1078. R. Fernandez-Cellini, L. Ga sco-Sa nch ez. Ann. Real. Soc. Esp. Fis. Quim., 52, 2, 111 (1956).
1079. P. H. Scholes. Analyst, 82, 525 (1957).
1080. U. К. A. E. A.-1GO-AM/S-44 (1958).
108k Y. L a t h о u s e, F. E. H u b e r, D. L. C h a s e. Anal. Chem., 31, 9, 1606. (1959).
1082. П. А. Крюков. Гидрохим. матер., 22, 90 (1954).
1083. A. Crowther. M. Bimson, H. Neville, F. Warburton. U.K. A. E. A., Ind. Gr., SCS-R-123 (1959); цит. no Chem. Abstr., 54, 5, 4261 (1960); см. также U. К- A. E. A. IGO-AM/S-19 (1958).
1084. A. Crowther, F. Warburton, R. Dickenson, S. Dikes. U. К. A.E. A. Ind. Gr. SCS-R-108 (1959); цит. no Chem. Abstr., 54, 4261 (1960); см. также U. К. A. E. A. IGO-AM/S-49 (1958).
1085. С у д з у к и, Масами. Japan Analyst, 8,395 (1959); цит. по РЖХим, 128-34488 (1960).
1086. V. A t h a v а 1 е, L. М a h a j а п, N. Т h а к о о г, М. W а г d е. Anal. Chim. Acta, 21, 491 (1959).
1087. U. К. А. Е. A.-IGO-AM/S-42 (1958).
1088. Е. R. R us se 1 1. U. S. Atomic Energy Comm., Rep. DP-161 (1956); цит. no Anal. Abstr., 5, 443 (1958).
1089. С. O. Granger. Analyst, 83, № 992, 609 (1958).
1090. U. К. A. E. A.-IGO-AM/S-120 (1958).
1091. В. И. Ill л e и с к а я. Вести. МГУ, сер. II, химия, 2, 69 (1960).
1092. А. С г ow t h е г. U. К. А. Е. A., Ind. Gr., SCS-R-5 (1948); цит. no Chem. Abstr., 54, 4261 (1960).
1093. D. M a c G i 1 1 a v r y. Trans. Faraday Soc., 32, 1447 (1936).
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Автоматическое определение урана полярографическое 203—212 фотометрическое
с роданидом аммония 120
с тиогликолятом аммония 126
Амперометрическое титрование урана 212
метаванадатом аммония 213, 351
.и-нитрофениларсоновой кислотой 212
ферроцианидом калия 212
Весовое определение урана (см. Определение урана осаждением 56)
Восстановление урана (VI) 27, 77
алюминием 85, 89
висмутом и его амальгамами 28, 82, 272
гидроксиламином 69, 274 гидросульфитом 69, 71, 269, 274
железом (11) 270, 272, 274 кадмием, и его амальгамами 83, 89,
272
магнием 85, 89
медью 85
никелем 85
оловом (II) 87, 102
ронгалитом 269, 272
свинцом и его амальгамами 82
серебром 28, 83
сернистым натрием 269
титаном (III) 87
фотохимическими методами 88
хромом (II) 86, 269, 271
цинком и его амальгамами 89, 78, 102, 272, 305
электролитическим методом 88, 89
Гидролиз солей урана(Ш) 26 ypana(IV) 26, 27 ypana(VI) 30—33
424
Диспропорционирование урана(У) 29
Изотопное разбавление, метод определения урана 228 (см. Определение урана методом изотопного разбавления)
Изотопный состав урана, определение радиоактивационным методом 257
Изотопы урана 6
Ионообменные смолы 314
аниониты 314
сильноосновные 314
слабоосновные 314
катиониты 314, 325 сильнокислотные 314 слабокислотные 314
«Испарения» метод 360
Каталитические полярографические волны урана
в присутствии ванадия 196
с нитрат-ионом 193
Качественное определение урана 34 микрокристаллохимические реакции 34
флуоресценция в перлах 35 цветные реакции с
азидом калия 38, 41
ализарином S 37, 46, 54
алюминоном 37, 47, 54
арсеназо I 40, 48
арсеназо II 49, 51
арсеназо III 49, 52
аскорбиновой кислотой 40, 45 галловой кислотой 40 дибензоилметаном 39, 45 2,4-диокси-5-хлорацетофеноном 45 диэтилдитиокарбаматом натрия
40, 45
о-крезотиновой кислотой 39, 43 морелл ином 40
морином 37, 46
2-нафтол-1-сульфоновой кислотой 40, 43 а-нитрозо-3-нафтолом 44 нитрозо-R-солью 37, 40, 44 . 8-оксихинолином 39, 44 перекисью водорода 37, 38, 41 4-(2-пиридилазо)-резорцином 47, 54
2-пиридин-<Л-азо-1>-2-нафтолом 48, 54 редуктоном 40 резорцином 35, 40, 43 родамином Б 54 роданидом калия 35, 38, 41 «родизонатом натрия 54 рубеановодородной кислотой 54 салициламидоксимом 40, 42 салицилгидроксамовой кислотой 40, 42
салициловой кислотой 35, 39 R-солью 40 сульфосалициловой кислотой 42 тайроном (пирокатехин-3,5-ди-сульфокислота) 40 таннином 40 тиогликолятом аммония 39, 45 тороном 48, 53 ферроном 40, 44 ферроцианидом калия 38, 41 хинализарином 54 хромотроповой кислотой 40, 43 экстрационно-фотометрйческие методы с
арсеназо I 51
арсеназо III 53
Комплексные соединения урана (IV) 17 бромидные 18 оксалатные 18 роданидные 17, 18 с арсеназо I, II, III 18, 40—52 с органическими реагентами 18 фторидные 18 хлоридные 17, 18
Комплексные соединения уранила 19 ацетатные 21 диэтилдитиокарбаматные 24 карбонатные 20 купферонатные 25 лактатные 23, 24 нитратные 22 оксалатные 20 оксихинолинатные 25 роданидные 21 салицилатные 24 с арсеназо I, II, Ш 25, 40—52 с аскорбиновой кислотой 24 с ауринтрикарбоновой кислотой 24 с гликолевой кислотой 24
с 1,3-дикетонами 24 сульфатные 19, 20, 23 тартратные 23 фосфатные 22 хлоридные 22, 23 цитратные 23 этилендиамннтетраацетатные 25
Комплексонометрическое титрование урана (IV) в тетрафториде урана 99, 350
Константы гидролиза ypana(IV) 27 уранил-иона 32
Константы комплексообразования уранил-иона 20—25
Коэффициент диффузии уранил-иона 172
Коэффициенты активности уранил-иона 30, 31
Кулонометрическое определение урана в отработанном ядерном горючем 226 основы кулонометрии 219 титрование урана (IV) бромом 222 титрование урана (IV) церием (IV) 224
титрование урана (VI) титаном (III) 225, 224
титрование урана (VI) при контролируемом потенциале 226, 227
Люминесценция урана спектры 146, 150, 156 тушение 147, 152 ультрахимископ Брумберга 147
. фотометр ЛЮФ-51 фотоэлектрический фотометр ЛЮФ-57 фотоэлектрический фотометр ЭФ-3
«флуориметр») 148
156
люминесцентный 157
люминесцентный (электронный
«Медной искры» метод 358
Минералы урана 8
Молярные коэффициенты погашения
окрашенных соединений урана 140
«Носитель» в спектральном анализе 358
Окисление урана (IV) до урана (VI) кислородом воздуха 28, 89
Окислительно-восстановительные потенциалы систем ионов урана 25—29
Определение урана амперометрическими методами (см. Амперометрическое титрование урана) 212
Определение урана весовыми методами 55 и сл.
весовые формы 56 осаждение аллилтиомочевиной 77
425
арсаниловой кислотой 76
бензойной кислотой 77
бензолсульфи новой кислотой 77
ц-(н. бутил)-фениларсоновой кислотой 75
в виде ураниламмонийванадата 62
газообразным H>S 63
гексамминокобальтинитратом 67
гидроокисью аммония 57
гипофосфитом натрия 66
3,5-дибромсалицилальдоксимом 76
4-диметиламиноазобензол-4'-ар-
соновой кислотой 75
дисалицилальэтилендиимином 76
дифенилтиокарбазидом 77
едкими щелочами 66
замещенными ксантогенатами 77'
изатин-8-оксимом 74
изонитрозо-М-фенил-З-метилпиро-золоном 76
иодатами 63
какодиловой кислотой 76
коричной кислотой 77
купфероном 69
а- и 8-нафтилсульфиновыми кисло-
тами 77
,н-нитробензойной кислотой 77
1 -нитрозо-2-окси-З-нафтойной кис-
лотой 76
а-нитрозо-р-нафтолом 75
З-нитро-4-оксифеннл арсоновой кис-
лотой 75
окисью ртути 68
оксалатами 72
8-оксихинолином 68
органическими реагентами 68
перекисью водорода 59
перйодатами 64
пиридином 73
пирофосфатами 67
полисульфидом гммония 63
салицилгидроксамовой кислотой 67
салициловой кислотой 6
себациновой кислотой 77
селенистой кислотой 67
сернистым аммонием 62
сероводородом в присутствии уротро-
пина 63
таннином 73
уротропином 74
фениларсоновой кислотой 75
фенил гл ицин-о-карбоновой кислотой
77
фосфатами 61
фосфорноватистой кислотой 66
фторидами 65
хинальдиновой кислотой 73
этилендиамином 76
Определение урана в технических продуктах 343 амперометрический метод 351 гидросульфитно-фосфатный метод 346, 349
комплексонометрическое титрование
пероксидный метод 348 разложение образцов 343, 348 ферри-фосфатно-нитритный метод 352 экстракционно-люминесцентный метод 351
Определение урана в природных материалах гидросульфитно-фосфатный метод 346
метод изотопного разбавления 234, 235
полярографический метод 195, 197, 202
радиоактивационный метод 255, 257 радиометрический метод 238—248 разложение руд, пород, минералов 343
спектральный метод 248—251
ферри-фосфатно-нитритный метод 352
Определение урана в сплавах 352
уран — алюминий 355
уран — бор 356
уран — висмут 354
уран — галлий 355
уран — платина 357
уран — плутоний — молибден 356
уран — цинк 355
уран — цирконий 355
Определение урана кулонометрическими методами (см. Кулонометрическое определение урана) 218
Определение урана люминесцентными методами (см. Люминесценция урана) 143
Определение урана методами изотопного разбавления 228
в метеоритах и горных породах 234
в облученных урановых блоках 236 теория метода 229—234
Определение урана полярографическими методами 166
в ацетоуксусном эфире и ацетоне 199
в безводной уксусной кислоте 201
в бутиловом и этиловом спирте 198
в метилэтил кетоне и этиловом спирте 198
в породах 181, 195, 202
в природных водах 180
в присутствии ВгО”, J Од, СгО2~, vo;, W 177
426
в рудах 185, 186, 189, 197, 199 на фоне аскорбиновой кислоты 188, 189
карбоната аммония, комплексона III и др. 184—185 серной кислоты 173, 178 сернокислого гидразина 177 солей алюминия 179 солянокислого гидроксиламина 176, 178 солянокислого гидроксиламина и тартрата натрия 185 тартратов 185 фосфорной кислоты 180 фторидов 180 цитрата аммония 186 щавелевой кислоты 192 щелочного раствора триполифосфата натрия 181—182
Определение урана потенциометрическими методами (см. Потенциометрическое определение урана) 213
Определение урана радиоактивацион-ными методами 252 в горных породах, минералах, водах 255 в металлах 255 в метеоритах 256 по Bai4J, Те132, Хе1ээ, Nb23a, П239255 по сумме осколков деления 254 принцип метода 252
Определение урана титрованием бихроматами 91 броматом калия 96 ванадатами 95 железом (III) 96 йодометрическим 97 комплексонами 99, 351 перманганатом калия 90 перманганатом калия осадка пероксида урана 100 солями титана(Н1) 98 солями хрома(П) 98 сульфатом церия(1У) 92 ферроцианидами 102 фосфатами 102 /
Определение урана фотометрическими и спектрофотометрическими методами 105 в азотнокислых растворах 111 в карбонатных растворах 109 в сернокислых растворах 107—108 в трибутилфосфатном растворе 113 в фосфорнокислых растворах 107 в хлорнокислых растворах ПО косвенным путем с диметилглиоксимом 142 с сс.а'-дипиридилом 142 с азидом натрия 120
с арсеназо I 132
с арсеназо II 133
с арсеназо III 134
с аскорбиновой кислотой 121
с дибензоилметаном 122
с диэтилдитиокарбаматом натрия 124 с- морином 128
с 8-оксихинолином 127
с перекисью водорода в щелочных растворах 115
с перекисью водорода в карбонатнощелочных растворах 116
с роданидом калия и аммония 118, 119 с сульфосалициловой кислотой 126 с тиогликолятом аммония 126
с тороном 138
с ферроцианидом калия 120
с хлорфосфоназо III 139—140 Осциллографическая и переменнотоковая полярография урана 201—202
Отделение урана от примесей адсорбция на активированном угле 286
осаждение
ароматическими и алифатическими карбоновыми
кислотами 283
арсенатами 280
бензол сульфино вой кислотой 283 гипофосфитом натрия 271 изатин-Р-оксимом 282
нодатами и перйодатами 280 карбонатами 281, 260 купфероном 274 неокупфероном 283 а-нитрозо-Р-нафтолом 282 нитрозо-М-фенил -3-метил п ирозо-лом 283
8-оксихинолином 275 перекисью водорода 265 пирофосфатами 271
салициловой кислотой и ее производными 283
сульфидами и полисульфндами 279 таннином 277
фениларсоновой кислотой и ее производными 282
ферроцианидами 278 фосфатами 266 фторидами 272, 274, 284 хинальдиновой кислотой 283 щавелевой кислотой 277 соосаждение с
гидроокисями алюминия, железа, кальция, магния, олова, титана, тория, циркония 283 карбонатом бария 283 перекисью тория 283 органическими осадками 283
427
фосфатами алюминия, железа, тория, циркония титана 268, 284, фторидами кальция и тория 273, 274, 284
хроматографическое от
бария 329, 332
бериллия 332, 329
ванадия 318, 321, 316, 326, 328
висмута 317, 323, 329
вольфрама 318, 332
галлия 332
гафния 318, 332
германия 332
гольмия 332
европия 329, 332
железа 319, 321, 328, 331, 336
индия 329, 332
иода 332
кадмия 317, 329, 332
калия 329, 332
кальция 323, 325, 329, 332
кобальта 323, 325, 329, 332, 336
лантана 325, 332
лития 332
магния 323, 329, 332
марганца (II) 317, 323, 329, 332, 336
меди 328, 332, 326, 336
молибдена 318, 324, 329, 333
мышьяка (III) 323, 318, 329, 333
натрия 329, 332
неодима 332
никеля 317, 323, 329, 336
ниобия 332, 337
олова 318, 329, 332
плутония 329, 336
празеодима 330, 332
протактиния 317
радия 332
редкоземельных элементов 325
ртути 317, 329
рубидия 329, 332
самария 332
свинца 321, 329, 332
селена 335
серебра 329, 332
стронция 329, 332
сурьмы 329
таллия 329, 332
тантала 332
теллура 332
титана 318, 329
тория 317, 320, 323, 330, 332, 335
хрома 317, 321, 329, 336
цезия 321, 329, 332
церия 318, 321, 332, 335
цинка 317, 323, 329, 332
циркония 318, 329, 335, 337
эрбия 332
экстрагированием
амиловым спиртом 301
в виде ацетилацетоната 304
дибензоилметаната 305
диэтилдитиокарбамата 308 комплекса с арсеназо III в присутствии
дифенилгуанидина 137 купфероната 306
молекулярных соединений 289
а-нитрозо-3-нафтолата 310
8-оксихинолината 307
1-(2-пиридилазо)-нафтолата 311
салицилата 311
солей ал кил фосфорных кислот 313
солей комплексных анионов
урана с органическими катионами 311
теноилтрифторацетоната 305
N-фенилгидроксамата 311
циклических и внутрикомплексных солей 304
дибутиловым эфиром 301
дибутиловым эфиром тетраэтиленгликоля (пентаэфиром) 303
дибутиловым эфиром этиленгликоля 303
дигексиловым эфиром 301
диизопропиловым эфиром 301
диэтиловым эфиром 290
метил изобутил кетоном (гексоном), 299
метилциклогексаноном 301
метилэтилкетоном 301
трибутилфосфатом 295
Три-(н. децил)-фосфиноксидом 303.
три-(н.додецил)-фосфиноксидом 303-три-(н.октил)-фосфиноксидом 302 три (3,5,5-триметилгексил)-фосфин-
оксидом 303
этилацетатом 300, 308
электролитические методы отделения 337
Полярографическое определение урана 166
Потенциометрическое определение урана титрованием бихроматом калия 215 перманганатом калия 214 солями титана (III) 218 солями хрома (II) 217 сульфатом церия (IV) 215 феррицианидом калия 216
Приборы для экстрагирования ураиа диэтиловым эфиром 293
Примеси в уране спектральное определение 358
428
алюминия 366
бора 364
гадолиния, европия, самария
кадмия, германия, индия, галлця золота, сурьмы, свинца 3^g
кальция 367
меди, никеля, железа 365
платины и палладия 368
химическое определение 372
азота 381
бериллия 373
бора 373—376
ванадия 385
>. водорода 373
железа 387
золота 394
кадмия 393
кальция 385
кислорода 382
кобальта 387
кремния 383
магния 383
марганца 386
меди 390
молибдена 391
мышьяка 390
натрия 382
никеля 388
редкоземельных элементов 393
ртути 395
рутения 391
свинца 395
серы 383
серебра 392
титана 385
углерода 377
фтора 382
хлора 384
хрома 386
циркония 390
Произведение растворимости 16
арсенаты уранила 16
ванадаты уранила 16
гидроокись урана (IV) 16
купферронат уранила — аммония jg
оксалат урана (IV) 16
пероксид урана 16
сульфит уранила 16
фосфат урана (IV) 16
фосфат уранила 16
Радиактивационный метод определения урана (см. Определение урана радиоактивационными методами) 2$2
Радиометрическое определение урана 236
минералы 246
неравновесные и урано-ториевые руды 240
породы 245
равновесные руды 238
Разложение урановых материалов 345 концентраты, полупродукты, отводы 348
руды, минералы, горные породы 345 сплавы 352
Редуктор висмутовый 82
Джонса 79 кадмиевый 83 ртутный 85 свинцовый 82 серебряный 83 цинковый 78
Силикагель, приготовление 334
Соединения урана 12
бориды 12
гидрид 12 карбиды 12 окислы 13 перекисные соединения 13 силициды 12 уранилнитрат 15 фториды 14 хлориды 14
Состояние ионов урана в растворе 25
уран (Ш) 25
уран (IV) 26
уран (V) 29
уран (VI) 30
Спектральное определение урана в породах, рудах, минералах 248
Спектры поглощения солей урана (VI) в органических растворителях 113
Титриметрические* методы определения урана (см. Определение урана титрованием)
Ультрахимископ Брумберга 147
Уран металлический 10
’ физические свойства 10 химические свойства 11
Фотометр ЛЮФ-51 156
Фотометр ЛЮФ-57 157
Фотоэлектрический люминесцентный фотометр ЭФ-3 («электронный флуориметр») 148
Экстракционные методы отделения урана (см. Отделение урана экстрагированием) 286
Электролитические методы отделения урана 337 внутренний электролиз 341 электролиз на ртутном катоде 337 электроосаждение на твердых электродах 340
429»
ОГЛАВЛЕНИЕ
/ лава!. Общие сведения об уране....................................... 5
Глава II. Химико-аналитическая характеристика урана и его соединений ................................................................ 10
Комплексные соединения урана (IV)............................. 17
Комплексные соединения уранил-иона............................ 19
Состояние ионов урана в растворе.............................. 25
1 лава III. Качественное определение урана............................ 34
Качественное обнаружение урана по флуоресценции в перлах 35
Цветные реакции урана......................................... 35
/ л а з а IV. Количественное определение урана....................... 55
А. Химические методы определения урана......................... 55
I. Весовые методы.......................................... 55
II. Титриметрические методы................................. 77
III. Комплексометрическое титрование......................... 99
IV. Методы, основанные на гидролизе солей уранила . ' . . 100
V. Методы, основанные на титровании ураниловой соли перурановой кислоты........................................... 100
VI. Методы, основанные на восстановлении урана (VI) до урана
(III) и последующем титровании его растворами окислителей J01
VII. Методы, основанные на титровании растворами осадителей 102
VIII. Косвенные методы....................................... 103
Б. Физико-химические методы определения урана............... 104
I. Фотометрические методы................................ 104
1. Колориметрические и спектрофотометрические методы определения в видимой и ультрафиолетовой областях . . 104
2. Люминесцентный метод................................ 143
II. Электрохимические методы.............................. 165
1. Полярография урана.................................. 165
2. Потенциометрическое определение урана.............. 213
3. Кулонометрическое определение урана................. 218
III. Радиохимические методы................................ 228
Метод изотопного разбавления ......................... 228
В. Физические методы определения урана...................... 236
I. Радиометрическое определение урана в природных материалах ....................................................... 236
И. Определение урана в породах, рудах и минералах методами спектрального анализа ..................................... 248
III. Радиоактивационный метод определения урана.............. 252
430
1 л a s а V. Методы отделения храни от ссп\тств\кших элементов . .
Введение .....................................................
1. Осаждение и ссссаждение неорганическими и органическими реагентами..................................................
2. Экстракционное отделение урана...........................
3. Хроматографическое отделение урана.......................
4. Электролитические методы отделения урана ................
/ ' л а 6 а V/. Определение урана в природных и промышленных объектах ................................................................
1. Определение урана в минералах, рудах и горных породах . .
2. Определение урана в промышленных концентратах, полупродуктах и отходах ...........................................
- 3 Определение урана в сплавах . . . •.......................
/ л а в а VII. Определение примесей в чистых препаратах урана
1. Определение малых количеств примесей в уране методами спектрального анализа ...........................................
2. Химические методы определения примесей в чистых препаратах
й й g g gm й §
урана...................................................... 372
Лик ратура ... ............................................... 396
Предметный указате.и............ . ... ............... 424
Аналитическая химия урана
Утверждено к печати Институтом ^еохимии и атлетической химии им. В. И. Вернадского Академии наук СССР
Редактор М. П. Волынец
Технические редакторы Н. Д. Новичкова, О М Гиськова Корректор С. В. Мартынова
РИСО № 6-22В. Сдано в набор 4/Х 1961 г.
Подписано к печати 28/IV 1962 г. Формат 60х90‘Л» Печ. л. 27. Уч-изд. л. 28,2. Тираж 4000 экз.
Т-05805. Изд. № 130. Тип. зак. № 2426.
Цена 2 руб. П коп.
Издательство Академии наук СССР Москва, Б-62, Подсосенский пер., 21 2-я типография Академии наук СССР
Москва, Г-99, Шубинский пер. 10
ОПЕЧАТКИ И ИСПРАВЛЕНИЯ
Стр. Строка Напечатано Должно быть
10 9 сн. 2-4 2,4
10 15 сн. 3318 3818
17 1 сн. ,3-0 9,ЗЛО5
49 формула SO3H2 SO3H
138 1 сн. z^s.
SO3Na HO3S SO3H
HO OH
139 4 сн. AA
147 рис. 19 1g концентрации 1 I < концентрация
152 8 сн. Hg Hg2+
166 11 св. 1,0 в —1,0 в
186 9 св. Na2C<Oe Na2H4C4Oe
211 28 сн. от — 1,0 в до — 0,4 в от 1,0 b до 0,4 e
223 1 сн. тока (IV). тока
318 15 сн. ТЬ Th
355 4 св. в присутствии в присутствии цинка
375 22 св. 10 мм 100 мм
387 12 сн. 60 6,0
388 22 св. h hi
Аналитическая
химия урана