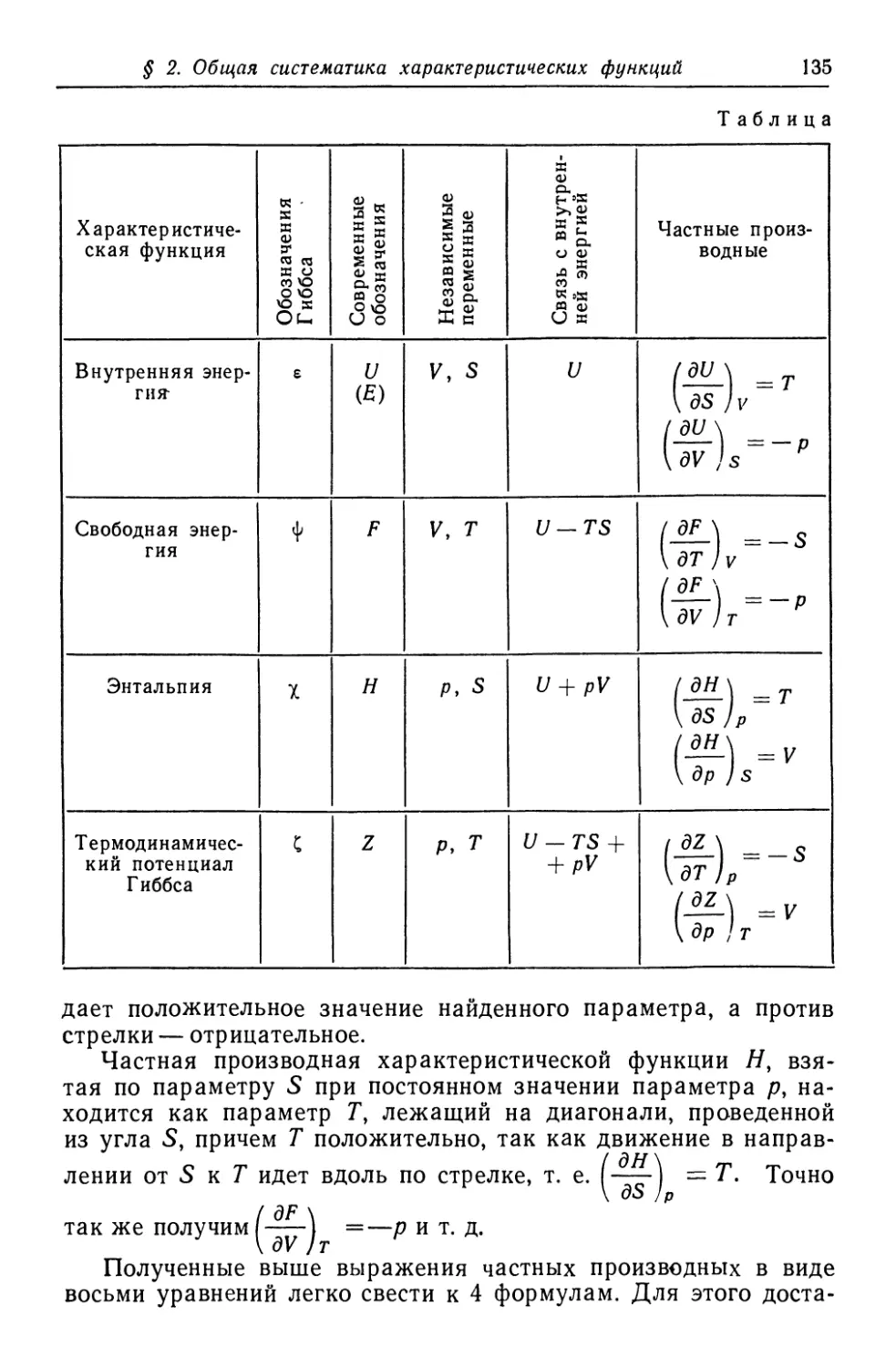
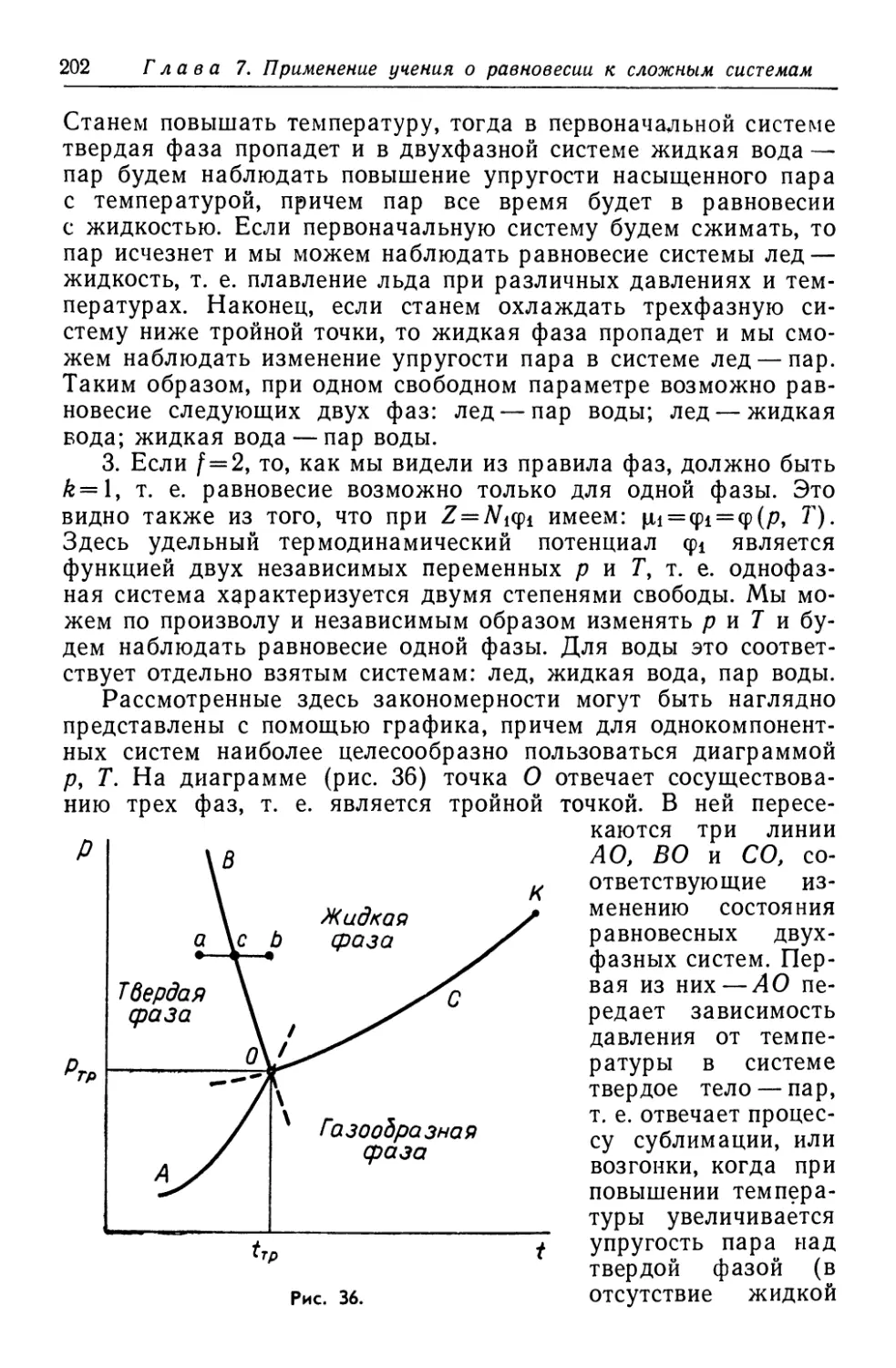
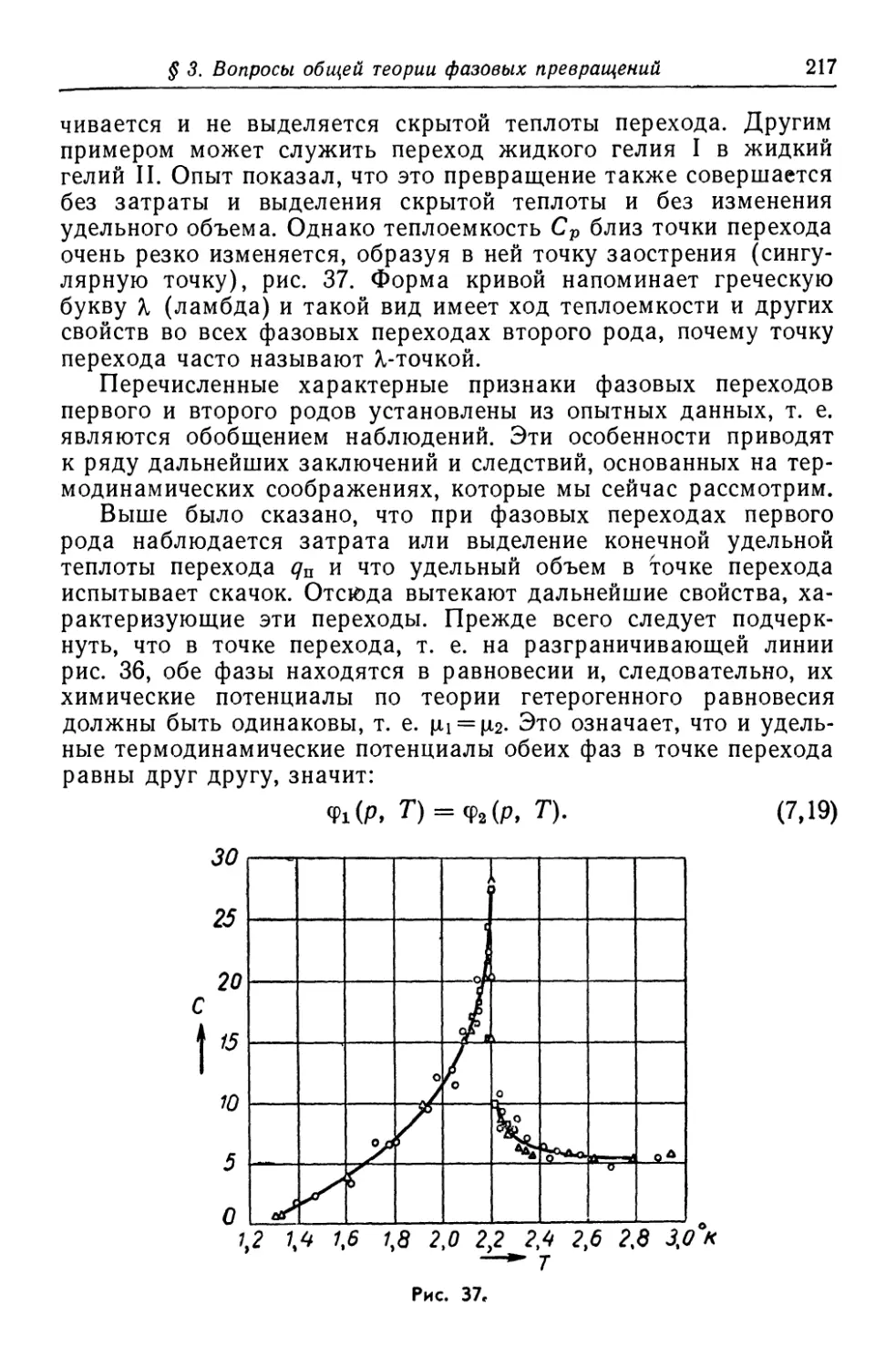
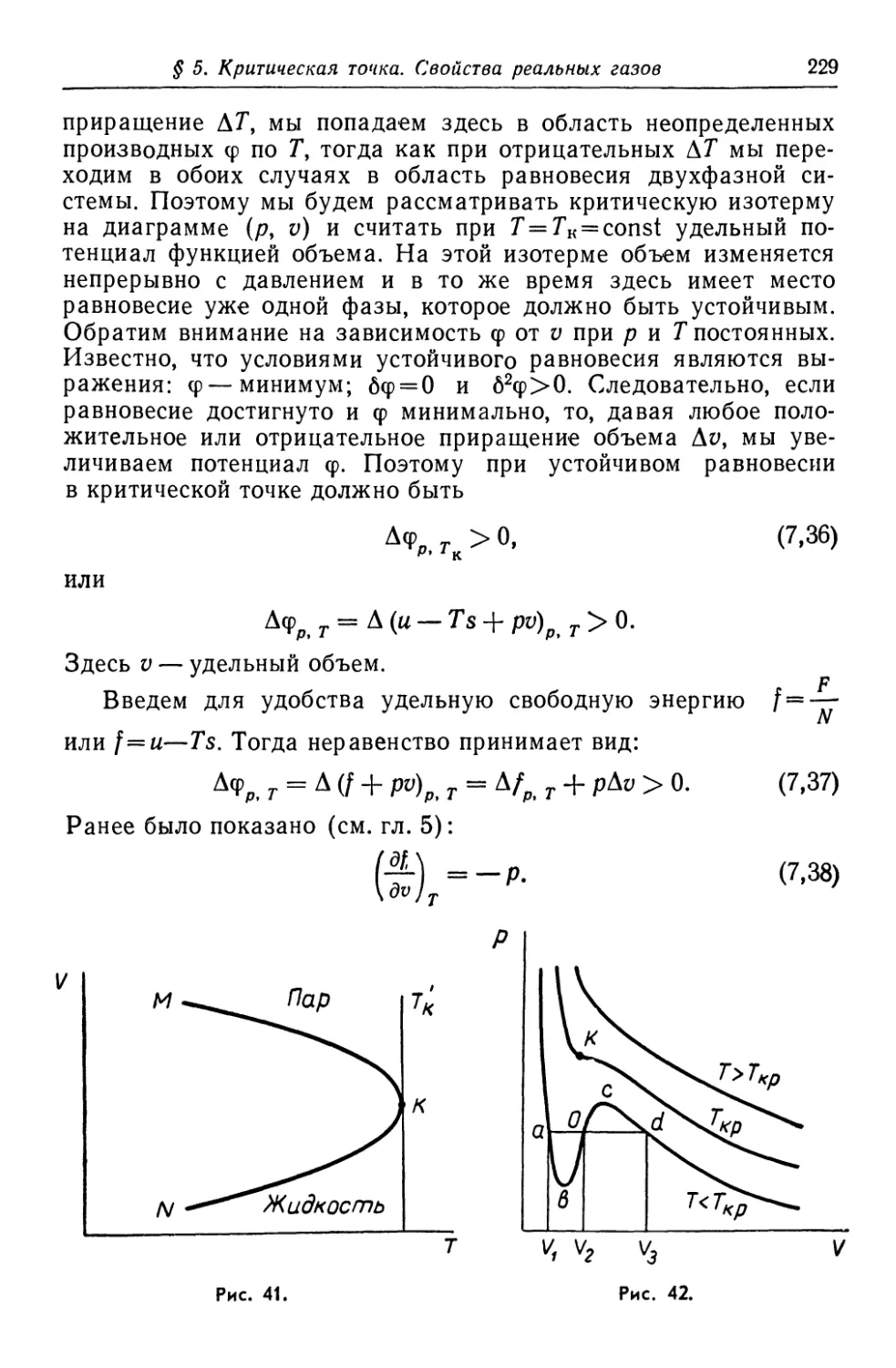
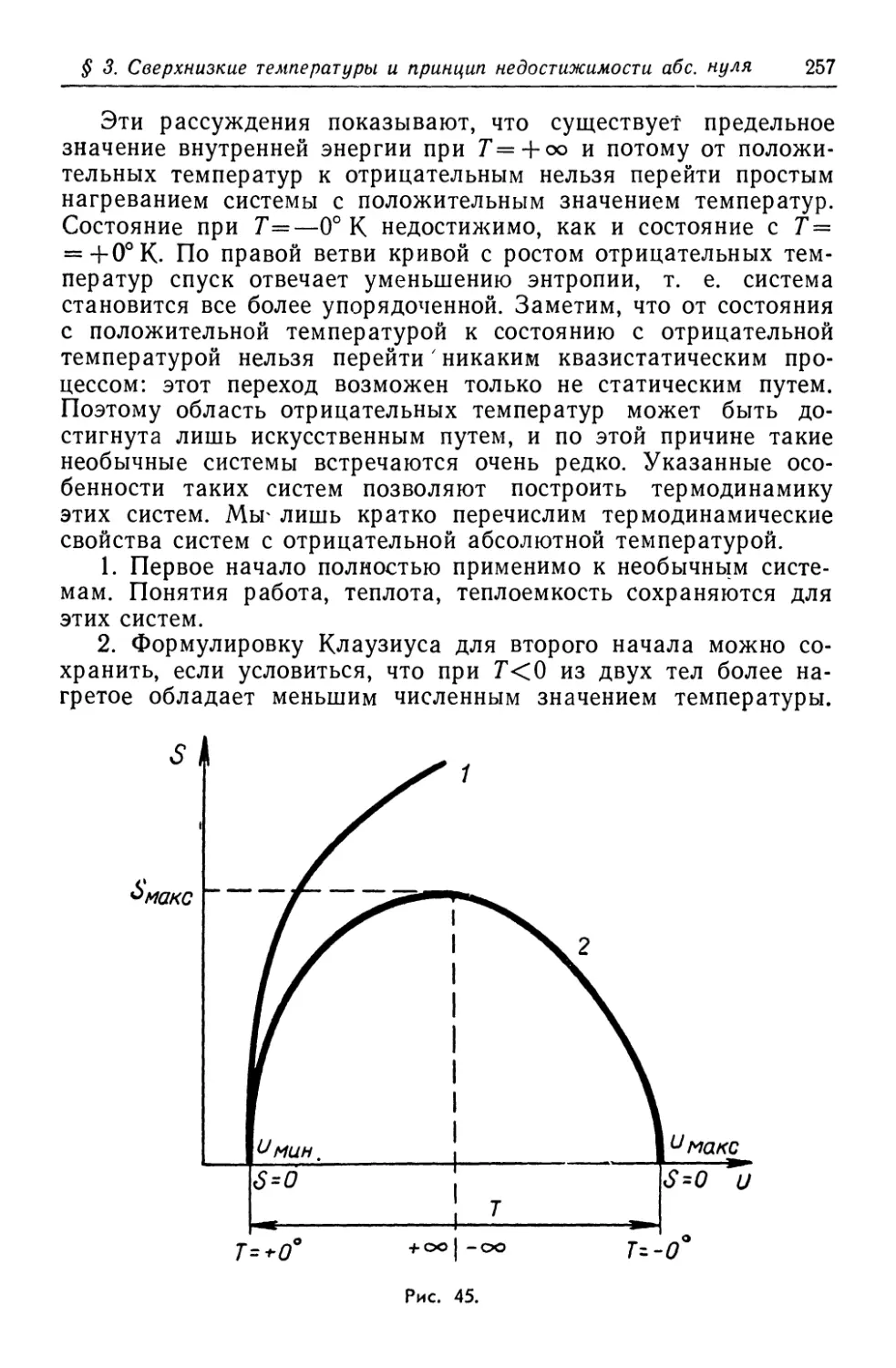
/



























































































































































































































































































Author: Радушкевич Л.В.
Tags: физика термодинамика учебное пособие общая физика издательство просвещение задачи по материалу
Year: 1971
Text
Л В. РАДУШКЕВИЧ
КУРС ТЕРМОДИНАМИКИ
Допущено Министерством
просвещения СССР в качестве
учебного пособия для студентов
физико-математических
факультетов педагогических
институтов
ИЗДАТЕЛЬСТВО «ПРОСВЕЩЕНИЕ» МОСКВА 1971
530.1
Р-15
Радушкевич Л. В.
Р-15 Курс термодинамики. Учеб. пособие для студентов
физ.-мат. фак-тов пединститутов. М., «Просвещение»,
1971.
288 с. с илл.
Книга представляет собой учебное пособие по курсу термодинамики для
студентов-физиков пединститутов.
29-71
Данный курс термодинамики представляет собой пособие
для студентов-физиков педагогических институтов и составлен
в соответствии с программой этих учебных заведений.
Построение книги основано на постепенно усложняющемся содержании:
начиная с элементарных вопросов, материал последовательно
углубляется и усложняется. Этим книга отличается по
изложению от других пособий, обычно следующих традиционно
дедуктивному изложению термодинамики. Первые главы тесно
связаны с курсом общей физики.
Хотя в программу термодинамики не входят вопросы
неравновесной термодинамики, автор счел необходимым ввести
этот материал в сжатом изложении. В настоящее время эта
область физики приобретает настолько важное значение и
настолько определилась, что целесообразно вводить ее основные
вопросы в учебные пособия.
В книге дан ряд Несложных задач, которые позволят
читателю проверить усвоение материала.
Для углубления знаний по термодинамике может быть
рекомендована следующая литература:
М. А. Леонтович. Введение в термодинамику, М., Гостех-
издат, 1950.
И. П. Базаров. Термодинамика, М., Физматгиз, 1961.
А. Зоммерфельд. Термодинамика и статистическая
физика, М., ИЛ, 1955.
Д. Тер Хаар, Г. Вер гел а нд, Элементарная
термодинамика, М., «Мир», 1968.
Ф. Морс. Теплофизика, М., «Наука», 1968.
С. Р. де Гроот. Термодинамика необратимых процессов,
М., Гостехиздат, 1956.
С. де Гроот, П. Мазур. Неравновесная термодинамика,
М., «Мир», 1964.
Кроме того,знакомство с трудами классиков можно
получить из книг;
Дж. В. Гиб б с. Термодинамические работы, М.,
Гостехиздат, 1950.
Второе начало термодинамики. Сборник работ (С. К а р н о,
В. Томсон, Р. Клаузиус, Л. Больцман, М. Смолу-
ховский),М., ГТТИ, 1934,
Глава 1
ОБЩИЕ ПОНЯТИЯ И ОПРЕДЕЛЕНИЯ
§ 1. ВВЕДЕНИЕ
Термодинамика относится к области макрофизики
и в настоящее время может бытьразделена натер мост атику
и на собственно термодинамику. Первая занимается
свойствами макроскопических систем, находящихся в так
называемом термодинамическом равновесии, и является наиболее
разработанной ветвью термодинамики. Эта область изучения
равновесных систем, т. е. систем практически не изменяющихся во
времени, аналогична разделу статики в механике, и именно
поэтому это направление часто называют термостатикой.
Термостатика дала много ценного науке: с нею связаны
коренные законы фиЗ'Ики, на ее основе выросла физическая
химия; термостатика широко применяется в многочисленных
технических расчетах (техническая термодинамика) для
проектирования большинства тепловых машин. Однако современная
техника требует изучения свойств и поведения макросистем за
конечное и очень малое время. Например, процессы,
протекающие при пуске ракеты или боевых снарядов, а также быстро
идущие процессы современной технологии с участием
катализаторов или в атомных реакторах не могут быть надежно
описаны только при помощи уравнений термостатики. В
последних время вообще не фигурирует, и потому получаемые
данные не . могут описать хода процессов, протекающих во
времени. В связи с этим за последние десятилетия быстро
развивается учение о так называемых неравновесных процессах
в макросистемах, образующее неравновесную термодинамику,
которую по существу и следует называть собственно
термодинамикой, где изучаются процессы, идущие во времени.
Заметим, что оба направления термодинамики начали
развиваться почти одновременно в виде «теории тепла» в первой
четверти XIX столетия (см. исторический очерк), но затем
термостатика получила особенно широкое развитие и приобрела
не вполне точное название термодинамики. В старых курсах
физики порознь рассматривали термостатику и учение о
теплопередаче, которое по современным взглядам относится
собственно к термодинамике. Так были построены лекции
§ 1. Введение
А. Г. Столетова, а также книга «Теории теплоты» М. Планка
и др.
В нашем курсе мы рассмотрим главным образом
термостатику, потому что она имеет наибольшее общее значение во всех
отделах физики и потому, что без изучения термостатики
невозможно освоение вопросов термодинамики неравновесных
процессов. Лишь в последней главе мы дадим изложение
важнейших частей неравновесной термодинамики.
§ 2. ЗАДАЧИ ТЕРМОДИНАМИКИ. НУЛЕВОЕ НАЧАЛО. ФЕНОМЕНОЛОГИЯ
Ранее было отмечено, что термодинамика относится к
области макрофизики. Остановимся несколько подробнее на том,
что понимают под макрофизикой. В современной физике обычно
различают прежде всего физику микрочастиц: электронов,
протонов, атомов, молекул и фотонов, называемую микрофизикой
в отличие от физики макроскопических тел (или систем),
состоящих из огромного числа микрочастиц, которая собственно
и называется макрофизикой. Заметим, что в последнем случае
речь идет о макротелах, образуемых огромным числом
микрочастиц, например ~1020—1025 частиц; к этим телам относятся
тела непосредственно видимого нами мира. Это число частиц
в макротелах столь велико, что не представляется возможным
изучать поведение отдельной частицы и потому здесь пригодны
статистические методы, которые образуют собой весьма
широкую область, называемую статистической физикой. Однако
можно описать многие свойства макротел, отвлекаясь от
подразумеваемого молекулярного строения их, а учитывая лишь
поведение системы в целом, подобно тому как это делается
во многих задачах механики, например в механике «сплошных
сред». Этим путем следует термодинамика, которая отличается
от статистической физики (или так называемой статистической
термодинамики), где рассматриваются свойства макротел,
обусловленные движениями и взаимодействием отдельных молекул
(и других микрочастиц). Обычная классическая термодинамика
подразумевает скрытое движение частиц, выражаемое
температурой. Это положение является в термодинамике столь важным,
что его иногда называют нулевым началом
термодинамики, чтобы подчеркнуть его принципиальное значение как
исходной предпосылки, и формулируют в виде аксиомы: все тела
при тепловом равновесии обладают температурой.
Поскольку молекулярное тепловое движение является
коренным свойством всех материальных тел, то нулевое начало
следует считать фундаментальным исходным положением
термодинамики. Повседневная практика показывает, что всякое
тело обладает температурой, которая экспериментально (по
Глава 1. Общие понятия и определения
крайней мере для достаточно больших участков тела) может
быть измерена.
По решению Международной комиссии (1963 г.). для
измерения температуры принята термодинамическая шкала
температуры Кельвина, в основу которой положена тройная точка
воды (см. гл. 7); для удобства практики взята близкая к ней
точка таяния льда 273,15° К, соответствующая 0°С шкалы
Цельсия. Поэтому температура в градусах Кельвина (Т)
связана с температурой в градусах Цельсия (t) соотношением.
Весьма малое (бесконечно малое) изменение температуры
в градусах шкалы Цельсия и Кельвина одно и то же, как видно
из написанного соотношения, т. е.
dT = dt.
Здесь мы пока ограничиваемся общеизвестным фактом
наличия температуры всех тел, тогда как в главе 4 будет дано
строгое термодинамическое определение этого понятия.
Подчеркнем, что нулевое начало является исходным положением
термодинамики, поскольку тепловое движение протекает во всех
телах. Движение молекул неуничтожимо, как неуничтожимо
всякое движение в природе, и это положение о молекулярном
движении является одной из основ материалистического
мировоззрения. Мы увидим, что абсолютный нуль температуры
недостижим и на этом построена так называемая теорема Нернста,
рассмотренная в главе 8.
В классической механике температура тел не учитывается,
и этим классическая механика по существу отличается от
термодинамики, основанной именно на нулевом начале. Поэтому
можно, как многие делают, считать, что механика описывает
движение и свойства тел (и систем) при абсолютном нуле
температуры.
Сказанное позволяет определить общую задачу
термодинамики как науки, в которой изучаются свойства
макроскопических тел (или систем), а также процессы в этих телах с учетом
того, что последние всегда обладают измеримой температурой.
Изучая явления в рамках классической термодинамики, как
правило, отвлекаются от характера молекулярного и атомного
строения вещества. При исследовании явлений обращают
внимание исключительно на макроскопические свойства системы,
которые оцениваются по опытным данным измерения с
макроскопическими приборами: термометрами, калориметрами,
манометрами и т. д. Поэтому классическая термодинамика
является по существу феноменологической наукой (от
слова «феномен» — явление). Она почти не пользуется ника-
§ 2. Задачи термодинамики. Нулевое начало. Феноменология 7
кими гипотезами, которые не были бы связаны с грубыми
макроскопическими явлениями.
С точки зрения современной физики классическая
феноменологическая термодинамика явно недостаточна, несмотря на ее
большое значение в описании многочисленных явлений и
общих выводов. Она недостаточна потому, что, помимо
систематики фактов и описания макропроцессов, мы стремимся еще
к объяснению этих фактов, а это без привлечения молекулярной
теории строения веществ невозможно. В термодинамике
рассматривается, например, процесс превращения тепла в работу
в тех или иных условиях, но совершенно не затрагивается
вопрос о том, каким образом этот процесс осуществляется за счет
движения и взаимодействия молекул.
Феноменологический подход к явлениям, принятый в
термодинамике, нельзя считать существенным недостатком этой
науки. Скорее эта особенность метода классической
термодинамики связана с неизбежностью применения его в тех случаях,
когда нет необходимости вдаваться в детальное изучение
скрытых микроскопических подробностей явления. Так, например,
в теории действия тепловых машин при определении их
коэффициента полезного действия нет ни возможности, ни
необходимости решать этот вопрос, рассматривая каждый раз
движение и взаимодействие молекул рабочего вещества, их
столкновения со стенками рабочего цилиндра, строение отдельных
молекул и т. п. Так как рабочее тело в машине состоит из
огромного количества молекул, то описание получилось бы столь
сложным, что мы не смогли бы решить поставленной
практически и принципиально важной задачи о к. п. д. данной
машины. С подобным феноменологическим методом мы
встречаемся также в гидростатике и гидродинамике, в механике
и т. д. В этих областях также не интересуются молекулярным
строением вещества и тем не менее правильно решают
множество вопросов течения или равновесия жидкости, равновесия
механических систем и т. д. Феноменологический подход
распространен во многих технических приложениях физики. Таким
путем рассчитывают мосты, плотины, профиль крыла самолета
и т. д. Заметим, что теоретическая физика XIX в. была
преимущественно феноменологической, хотя и привела к построению
современной атомной и молекулярной физики.
§ 3. ВНУТРЕННЯЯ ЭНЕРГИЯ
Всякое тело в определенных условиях обладает некоторым
запасом так называемой внутренней энергии, которая
состоит из кинетической и потенциальной энергии,
составляющих его частиц, например молекул. Внутренняя кинетическая
8 Г лав а 1. Общие понятая и определения
энергия обусловлена тепловым хаотическим движением
молекул, и это свойство непосредственно связано с температурой —
с увеличением интенсивности этого движения температура тела
возрастает. Внутренняя потенциальная энергия зависит от
взаимодействия молекул друг с другом; они на некоторых
расстояниях притягиваются, одна к другой, а при особенно тесном
сближении силы притяжения переходят в силы отталкивания.
Общее количество кинетической и потенциальной энергии носит
название полной внутренней энергии тела,но мы часто
будем называть его просто энергией и обозначать через U.
Очевидно, что для однородного по составу тела запас энергии тем
больше, чем больше в нем находится частиц или чем больше
взятая масса тела, так как энергия возрастает пропорционально
увеличению числа частиц. Поэтому внутренняя энергия тела
является величиной аддитивной, т. е. пропорциональной
числу частиц в теле. Если тело состоит из N однородных частиц
и его внутренняя энергия есть £/, то можно написать:
Однако следует заметить, что фактически полная
внутренняя энергия тела слагается не только из энергии образующих
его молекул, но запас ее в теле определяется еще
внутримолекулярной и внутриатомной энергией, зависящей от
взаимодействия электронов, молекул и атомов друг с другом и с ядрами
атомов. Далее, в запас внутренней энергии тела включается
также эйнштейновская энергия, определяющаяся массами
самих составляющих атом и его ядро элементарных частиц
(электронов, протонов, нейтронов); по закону Эйнштейна Е = тс2,
где т — масса частицы, с —скорость света. Отсюда видно, что
доля эйнштейновской энергии колоссально велика, тогда как
доля кинетической и потенциальной энергии молекул и атомов
составляет лишь ничтожную часть запаса полной внутренней
энергии тела. В огромном большинстве задач
рассматриваемых в термодинамике, эйнштейновская энергия остается
постоянной, за исключением тех случаев, когда рассматриваются
процессы, связанные с ядерными превращениями. Между тем
в термодинамике часто бывают важны не абсолютные значения
полной внутренней энергии тела, а лишь относительные ее
изменения, происходящие в различных процессах, поэтому
эйнштейновская энергия может рассматриваться, как некоторый
нулевой уровень энергии, от которого мы будем отсчитывать
все изменения энергии при анализе разных процессов
превращения энергии в термодинамике.
При повышении температуры, т. е. при нагревании тела,
внутренняя энергия увеличивается в простейшем случае за счет
увеличения скорости движения молекул. Во многих случаях
§ 3. Внутренняя энергия
даже при постоянной температуре запас внутренней энергии
изменяется благодаря изменению потенциальной энергии
взаимодействия молекул, как например при плавлении,
парообразовании, растворении и т. п. Отсюда мы видим, что величина U
зависит не только от числа молекул в теле, но также от
изменения условий, в которых находится тело.
В термодинамике (в термостатике) обычно не обращают
внимания на внешнюю энергию тела, обусловленную его
движением как целого, т. е. предполагаются только такие
относительно медленные движения, когда внешней кинетической
энергией тела можно пренебречь.
Рассматривая общие свойства внутренней энергии, отметим,
что в физике величины, значения которых пропорциональны
числу частиц в теле или массе тела, носят название
экстенсивных величин, в отличие от интенсивных величин,
которые тоже вообще могут зависеть от массы, но более
сложным образом или даже вовсе не зависеть от нее. Внутренняя
энергия тела, как было сказано, пропорциональна числу
частиц (или массе), и потому ее следует считать экстенсивной
величиной. Напротив, температура тела непосредственно не
связана с массой и относится поэтому к разряду интенсивных
величин. Например, внутренняя энергия иглы, накаленной в
пламени, сравнительно мала благодаря малой массе, тогда как
температура доходит до 1000° С. Наоборот, внутренняя энергия
большого стального цилиндра при комнатной температуре
сравнительно велика, а температура составляет всего 20° С. К
интенсивным величинам относится также давление, например, газа
в баллоне, поскольку оно непосредственно не связано с массой
газа и зависит от других условий. Сюда же относятся
потенциалы: гравитационный, электростатический и магнитный,
которые по своим свойствам являются интенсивными величинами.
Далее мы встретимся с рядом других экстенсивных и
интенсивных величин.
§ 4. ТЕРМОДИНАМИЧЕСКАЯ СИСТЕМА И ТЕРМОДИНАМИЧЕСКОЕ
РАВНОВЕСИЕ
Термодинамической системой называют
макроскопическое тело или совокупность таких тел, выделяемых по
какому-то признаку из окружающего их мира. Выделение
здесь подразумевает часто как настоящее выделение системы
от контактирующих или окружающих ее тел (окружения) с
помощью разделяющих перегородок или оболочек, но можно
представить себе и условное мысленное выделение одной
системы из других или же выделение так называемой подсистемы
как части большой системы. Реальными примерами термоди-
10 Г лав а 1. Общие понятия и определения
намических систем являются газ в баллоне, раствор соли в
сосуде, а также жидкая капля, кристалл, кусок сплава и т. п.
тела, ограниченные внешними поверхностями от окружающей
среды. Здесь даны простейшие примеры систем. Однако в
термодинамике изучаются свойства и поведение более сложных
систем, состоящих из сложных по химическому составу и
агрегатному состоянию разнородных тел — гетерогенные
системы. Такой системой является, например, насыщенный
раствор соли с твердыми кристаллами в закрытом сосуде и с
находящимся над ним паром. Еще более сложными системами,
с которыми приходится иметь дело, являются машины,
двигатели, холодильники, сюда же в настоящее время относят
разнообразные биологические организмы — от простейших клеток
до высших животных. Мы ограничимся здесь главным образом
наиболее простыми системами, однородными по составу,
называемыми гомогенными, но далее будут также рассмотрены
важнейшие примеры сложных систем.
В выделенной нами системе могут протекать разнообразные
физические и химические процессы, включая также и ядерные
превращения и т. п. Наиболее распространен случай, когда
в системе имеется процесс теплопроводности за счет заметного
перепада температуры на отдельных участках, а также процесс
диффузии благодаря наличию конечной разности концентраций
составных частей в разных слоях (например, в растворе).
Возможно, что в системе имеется макроскопическое течение
жидкости или газа вследствие разности давления на разных
участках системы. Если все эти процессы протекают с конечной
скоростью, то состояние системы считается неравновесным.
Однако многие опытные факты указывают, что если некоторые
внешние условия остаются неизменными во времени, то
рассматриваемая нами система постепенно переходит в состояние
некоторого равновесия, когда температура в различных слоях
системы одна и та же, когда уже не имеется различия
концентраций на участках системы, т. е. все ее части равномерно
перемешаны друг с другом, и, наконец, когда макроскопические
течения исчезли благодаря выравниванию давления в системе.
(Пока мы еще не будем точнее определять «внешние условия»,
упо'миная их только как внешнюю среду или «окружение».)
Состояние, к которому приходит система и которое описано
выше, носит название термодинамически
равновесного состояния или просто термодинамического
равновесия, причем система, как показывает опыт, может длительно
находиться в нем, пока остаются неизменными внешние
условия. Время перехода системы от исходного неравновесного
состояния к равновесному носит название времени
релаксации. Последнее для различных макросистем может изменяться
§ 4. Термодинамическая система и термодинамическое равновесие 11
в очень широких пределах от долей секунды до сроков порядка
геологических эпох.
Переход системы к состоянию термодинамического
равновесия можно проследить на простом примере. Пусть на столе
находится небольшой закрытый сосуд с водой, нагретой,
например, до 100° С. Состояние воды в сосуде сначала является
неравновесным состоянием, так как при комнатной температуре
около 20° С имеется существенный перепад температур в
данной системе. Однако вода постепенно охлаждается, причем,
кроме процессов лучеиспускания и теплопроводности, в
системе происходит еще конвекция, т. е. макроскопические
течения слоев воды благодаря разной плотности горячей и более
холодной ее частей. Спустя некоторое время можно заметить,
что температура во всех слоях воды становится одинаковой
и равной температуре окружающих тел в комнате, если эта
температура окружения сама неизменна. Наступает
термодинамическое равновесие воды. Если условие постоянства
температуры длительно соблюдается, то и термодинамическое
равновесие воды является неизменным. В этом состоянии в воде
отсутствует во всех слоях разность температуры и полностью исчезли
конвекционные токи, а также лучеиспускание. Если сосуд с
водой перенести в термостат с другой температурой,
например 5° С, то вновь возникнут теплопроводность, лучеиспускание
и конвекция, пока система вновь не перейдет к равновесному
состоянию при новой температуре.
§ 5. ПАРАМЕТРЫ СОСТОЯНИЯ. КВАЗИСТАТИЧЕСКИЕ ПРОЦЕССЫ
Данная система может обладать разными равновесными
состояниями. Например, в описанных выше опытах с водой в
первом случае равновесная температура воды была 206С, тогда
как при выдерживании в термостате равновесная температура
стала 5°С. Заметим еще,что различнее состоянии воды в первом
и втором опытах выражается в различии ее объемов или
плотностей. Следовательно, вообще каждое данное равновесное
состояние должно характеризоваться некоторыми определенными
величинами, постоянными для системы при заданном
равновесии. Эти характеристики равновесного состояния системы
называются термодинамическими параметрами
состояния, причем вполне определенными для данного равновесного
состояния. К этим параметрам относится температура тела
(системы), являющаяся важнейшей характеристикой равновесия,
а также объем, плотность, давление, концентрация и т. п. В
нашем примере с водой параметрами состояния являлись
температура и объем (плотность). Рассматривая другие
термодинамические системы, мы встречаемся с необходимостью введения
12 Глава!. Общие понятия и определения
еще других параметров состояния. Например, изучая
равновесие систем в электрическом или магнитном поле, вводят в
качестве параметров состояния такие, например, как
поляризация, намагничивание и т. п.
Необходимо отметить, что введенные здесь параметры
состояния являются макроскопическими характеристиками
состояния системы. Они всегда относятся к системе в целом, так
как подразумевается термодинамическое равновесие системы.
При отсутствии равновесия указанные параметры часто
становятся неопределенными. Так, например, в рассмотренном опыте
с охлаждением воды, пока равновесие еще не установилось,
температура не является параметром состояния, так как она
различна в разных слоях. Казалось бы, можно принять среднюю
температуру, но это было бы неправильным, так как средняя
температура в этом опыте падает с течением времени.
Следовательно, температура только в состоянии термодинамического
равновесия является определенным параметром состояния.
То же можно отметить и для других параметров, как давление,
объем и т. п. Так, например, в быстрых процессах сжатия или
расширения газа в нем распространяются волны сжатия и
расширения, и, следовательно, давление газа в разных частях
будет различным, вследствие чего в этих процессах давление газа
не является термодинамическим параметром состояния всей
системы. Только при очень медленном сжатии или расширении
можно пренебречь разницей давления в отдельных слоях, и оно
может быть принято как параметр состояния.
Термодинамическое равновесие может быть определено, если
известны параметры состояния, однако вопрос о числе
необходимых и достаточных параметров для полной характеристики
равновесия является сложным и будет рассмотрен в главе 6.
Заметим, что дЯя самых простых, однородных систем
достаточно знания трех параметров состояния. Введение небольшого
числа параметров является выгодным потому, что позволяет
геометрически изображать рабновесное состояние на так
называемых диаграммах равновесия. Для самых простых систем,
для которых параметрами состояния являются объем V,
давление р и температура Г, состояние определяется одной точкой
на трехмерной диаграмме V— р—Т. Можно еще упростить
геометрическое изображение, если состояние отмечать в плоскости
двух параметров, например, /?, V, подразумевая при этом
значение третьего параметра Т в данной точке плоскости. Тогда
получается диаграмма (рис. 1), где одно состояние показано
точкой 1 с параметрами ри У и Ти а другое —с параметрами р2,
Введение термодинамических параметров не исключает мо-
лекулярно-кинетического описания макросистем, но при этом
§ 5. Параметры состояния. Квазистатические процессы
13
состояние их должно будет определяться пространственными
координатами и скоростями всех молекул системы, и поэтому
число параметров состояния будет колоссально велико. Такой
подход к описанию состояния дается в статистической
термодинамике, тогда как в обычной феноменологической
термодинамике вводятся такие макропараметры, как температура,
давление и т. д., которые отражают собой некоторое усреднение
состояния сложной молекулярной системы. Некоторую аналогию
мы встречаем в механике, когда, отвлекаясь от движения
отдельных частей тела, рассматривают движение и координаты
только центра тяжести или центра масс системы.
Термодинамические параметры выбирают таким путем, чтобы их можно
было определить опытным путем.
Описывая состояние какой-либо термодинамической
системы, мы всегда должны иметь в виду ее «окружение», т. е. те
внешние тела, которые граничат с выделенной системой и
потому их состояние связано с данной системой. Это приводит
к необходимости во многих случаях разделять параметры на
внешние и внутренние. Первые относятся к окружению,
а вторые к выделенной системе. Примером внешних
параметров является объем. Температура, давление и концентрация
представляют собой внутренние параметры, поскольку они
зависят от внутренних свойств системы, т. е. от движения
молекул, числа их в единице объема и т. п.
Определенность параметров
состояния необходима не только для
характеристики данного состояния, нр главное,
для описания процессов. Процессом
называют изменение параметров
состояния. Однако так как параметры
состояния в каждом данном состоянии должны
быть (как сказано выше) совершенно
определенными, то, очевидно, мы должны
иметь в виду не любые процессы, а
только такие, когда каждое изменение
состояния протекает без'изменения
термодинамического равновесия. Отсюда
следует, что из всех процессов мы выделяем t
только «почти равновесные», т. е. такие,
когда из данного состояния в новое
система переходит столь медленно, что
новое состояние тоже является
равновесным. Такие равновесные (или почти
равновесные) медленные процессы
называются статическими или
квазистатическими. Изучение таких процессов оправ* Рис. 1.
-Г*
Л
I
14 Г лава 1. Общие понятия и определения
дывает само название термостатики как ветви термодинамики.
Именно поэтому время не входит в уравнения термостатики.
На диаграмме состояния (рис. 1) квазистатические процессы
изображаются линией, например АВ, где показан непрерывный
процесс с началом в точке Лис концом в точке В. Линия
состоит из непрерывного чередования бесконечно мало
отличающихся друг от друга равновесных процессов, Важное значение
в термостатике имеют круговые процессы, или циклы,
которые на диаграмме имеют вид замкнутой линии, например
CDEF на рис. 1. Известно, что циклические процессы
характерны для работы машин.
§ 6. ФУНКЦИЯ СОСТОЯНИЯ. ТЕРМИЧЕСКИЕ И КАЛОРИЧЕСКИЕ
УРАВНЕНИЯ СОСТОЯНИЯ
Свойства системы в данном равновесном состоянии
характеризуются не только непосредственно задаваемыми
параметрами состояния, но эти параметры являются функциями других
параметров. Так, например, объем газа является функцией
давления и температуры. Кроме того, во многих задачах нас
интересуют такие свойства системы, как внутренняя энергия,
энтропия и т. д. Эти величины тоже являются функциями
параметров состояния, и их мы будем называть функциями
состояния. Следовательно, функцией состояния называется
величина, зависящая от параметров состояния: она определена,
если даны эти параметры. Очевидно, функция состояния сама
может рассматриваться как параметр состояния, так как, если
вид функции известен, соответствующее уравнение можно
разрешить относительно одного из параметров и выразить его
через остальные переменные, в том числе и через функцию
состояния. Чтобы не усложнять геометрического изображения
процессов, по осям на диаграмме состояний (плоскости)
откладывают параметры состояния, а в каждой точке значение
функции состояния просто отмечают, подразумевая, что она
известна. Поэтому иногда функцию состояния называют ф у нк -
цией точки. Если независимые координаты (параметры
состояния) известны, то величин^ функции состояния может быть
определена. Уравнение, связывающее независимые внешние
и внутренние параметры состояния, называется уравнением
состояния или термическим уравнением
состояния. Очевидно, в зависимости от выбора параметров
состояния и от рода системы можно составить несколько таких
уравнений. В простейшем случае однородных систем уравнение
состояния связывает параметры /?, V и Т и в неявном виде может
быть написано как соотношение
F(p,V,T) = 0. (1,1)
§ 6. Функция состояния. Уравнения состояния 15
Отсюда видно, что любой из параметров по крайней мере
в принципе может быть выражен как функция других
параметров, т. е. из (1,1) получим:
P = fi(V,T)-> V = h(p,T); T = f3(p,V). (1,2)
Эти уравнения обычно называют собственно функциями
состояния или термическими уравнениями состояния. Следует
подчеркнуть, что уравнения состояния в конкретном виде выражения
(1,1) в термодинамике выведены быть не могут, что вполне
очевидно, так как термодинамика не рассматривает молекулярного
строения вещества. Имеются два пути вывода этих уравнений:
путь чисто экспериментальный или вывод при помощи методов
статистической термодинамики.
Для идеальных газов хорошо известно уравнение состояния
Клапейрона
PV = RT, (1,3)
которое приводится в курсах общей физики. Здесь R —
универсальная газовая постоянная для одного моля газа; ее
размерность зависит от размерности остальных величин. Известно,
что в единицах СИ ее значение равно:
R = 8,314 дж1 моль-град
при переходе к калориям имеем:
R = 1,985 кал/моль-град, или R^2 кал/моль-град.
Для реальных газов и паров хорошо оправдывается на
опыте уравнение состояния Ван-дер-Ваальса:
где а и Ь — константы для данного вещества.
Оба эти уравнения могут быть выведены при помощи
элементарной молекулярной статистики. Как показал опыт,
уравнение Ван-дер-Ваальса для паров повышенной плотности не
является достаточно точным, в связи с этим были предложены
многочисленные более точные уравнения состояния. Они
сводятся к выражению общего вида:
P^_i , В(Т) , С(Т) D (Т) . „
— - 1 -1 - h ~^- -h -— ■+-..., U,a )
где коэффициенты В (Г), С(Г), D(T) и т. д. называются
соответственно вторым, третьим, четвертым и т. д. вириальными
коэффициентами. Прибегая к методам статистической механики,
эти вириальные коэффициенты можно выразить через
потенциал сил межмолекулярного взаимодействия. Если при боль-
16 Глава 1. Общие понятия и определения
ших объемах (малых плотностях газа) можно пренебречь вири-
альными коэффициентами, то уравнение (1,3") обращается
в пределе в уравнение Клапейрша. Уравнение состояния (1,3")
было предложено Каммерлинг-Оннесом.
Приближенное уравнение состояния кристалла было
выведено Дебаем с применением модели структуры идеального
кристалла. Найдено также уравнение состояния для электронного
газа в металлах при помощи квантовой статистики. Недавно
получен ряд приближенных уравнений состояния атомарной
жидкости на основе статистической модели жидкого состояния.
Одной из важных функций состояния является внутренняя
энергия U. Величина ее зависит от температуры и других
внутренних параметров системы, например от давления. Каждой
точке диаграммы состояния (рис. 1) отвечает определенный
запас внутренней энергии тела (системы). С изменением
температуры и других параметров состояния величина U меняется,
т. е. она зависит от положения точки на диаграмме и, как
другие функции состояния, является функцией точки.
Следовательно, должно существовать уравнение общего вида
U = U(p, V, Т), (1,4)
или вообще
U = U(xl9 *a, ..., Г), (1,4')
где хи х2 и т. д.—параметры состояния. Уравнение (1,4), или
(1,4'), иногда называют калорическим уравнением
состояния, так как оно в принципе дает возможность находить
величины, которые получают с помощьк* калориметрических
измерений, как-то: теплоемкости, скрытые теплоты и т. п., о чем
сказано ниже. Так же как и термические уравнения состояния,
калорическое уравнение не может быть найдено при помощи
термодинамических методов. Исключение представляют собой
идеальные системы, для которых теплоемкость при постоянном
объеме Cv не зависит от температуры. К ним относится
идеальный газ. В этих случаях уравнение (1,4) принимает
простой вид:
Здесь и0—постоянная, не зависящая от температуры. Для
идеального газа из простых молекул сферической, формы
молекулярная теория дает, как известно, простое выражение для мо-
лярной теплоемкости Cv = — R.
Применяя методы статистической термодинамики, удалось
найти выражение для внутренней энергии реального газа,
следующего уравнению Ван-дер-Ваальса и другим уравнениям
§ 7. Термические коэффициенты и связь между ними 17
состояния. Получены также статистическим путем
калорические уравнения для кристалла и для атомарной жидкости.
Выше было отмечено, что все функции состояния, в том
числе внутренняя энергия, являются «функциями точки», т. е.
они вполне определены, если известны координаты точки или
параметры состояния на диаграмме (рис. 1). Отсюда вытекают
три важных свойства функции состояния.
1. Если w есть функция состояния, то изменение w зависит
только от начальной и конечной точек на диаграмме и не
зависит от формы «пути», т. е. не зависит от вида процесса.
2. Если w = w(x, у, z) есть функция параметров х, у, г, то
бесконечно малое изменение dw является полным
дифференциалом при бесконечно малых изменениях параметров:
dw-^-dx + ^Ldy + f-dz.
дх ду dz
где х, у, z—обобщенные параметры состояния, т. е. ими могут
быть /?, V, Т и т. д.
3. Для замкнутых процессов (циклов), т. е. таких, когда
система из начальной точки после перехода через ряд состояний
вновь возвращается в исходную точку, для функции состояния
всегда имеет место:
& dw = 0.
%)
Это получается как следствие из 1-го свойства функции
состояния; интеграл по замкнутому контуру не зависит от вида
контура.
Отметим, что и обратные заключения также верны, т. е. если
для какой-то функции мы установили указанные свойства, то
это значит, что она представляет собой функцию состояния.
Таким образом, с помощью анализа некоторых свойств удалось
открыть ряд функций состояния в термодинамике, о чем будет
сказано ниже.
§ 7. ТЕРМИЧЕСКИЕ КОЭФФИЦИЕНТЫ И СВЯЗЬ МЕЖДУ НИМИ
Некоторые тепловые и упругие свойства тел характеризуются
так называемыми термическими коэффициентами.
Так, из общего курса физики известны коэффициент объемного
расширения, термический коэффициент давления, коэффициент
сжимаемости. Мы дадим здесь общее определение этих
величин и установим связь между ними.
1. При нагревании определенной массы вещества при
постоянном внешнем давлении объем ее изменяется с температурой,
причем зависимость V от Т вообще носит сложный характер.
Для данного агрегатного состояния V является непрерывной
функцией Г, и потому изменение объема на каждый градус
2 Заказ № 2479
18 Г л а в а 1. Общие понятия и определения
повышения температуры выражается частной производной) —) ,
\дТ]р
которая может зависеть от температуры и давления.
Относительное изменение объема при нагревании на один
градус, т. е. изменение, отнесенное к объему тела при
температуре Г, называется термическим коэффициентом объемного
расширения:
а = JL/^L\ . (1,5)
V \дТ )р v '
В общем случае величина а может меняться в зависимости от Т
и р. Для идеального газа а = — , что можно легко получить из
уравнения состояния (1,3).
Если температуру выражать в градусах шкалы Цельсия,
то dt = dT и относительное изменение объема можно
представить отношением производной к объему Vo при 0°С. Тогда
*-■£■(■£„
В небольшом интервале изменения температуры возможно
допустить, что ao~const, откуда следует, что объем при
нагревании тела возрастает по линейному закону, так как тогда
отсюда после интегрирования получаем хорошо известную
формулу
V = V0(\ + at).
Для идеального газа при любом давлении
«о = ^-^- = 0,00366 1
2. Если нагревать данную массу вещества при постоянном
объеме, то давление является некоторой функцией
температуры; для определенного вещества в каком-либо агрегатном
состоянии эта функция вообще является непрерывной и мерой
изменения давления на каждый градус является частная
производная (—— )• Относительное изменение давления характери-
\ дТ ;v
зуется величиной термического коэффициента давления 0:
где р — давление при температуре Г. Вообще говоря, р зависит
от температуры. Для идеального газа р = —, как можно легко
§ 7. Термические коэффициенты и связь между ними 19
показать, применяя уравнение Клапейрона (1,3). Таким образом,
в данном случае а=|$.
Выражая температуру в градусах шкалы Цельсия, можно
отнести частную производную к давлению ро при 0°С, и тогда
Ро Ро { dt h
При малом интервале изменения температуры законно считать,
что ро = const, и в этом случае давление возрастает по
линейному закону, так как
"='* l=COnst;
после интегрирования этого выражения находим хорошо
известную формулу:
Для идеального газа ро= —=0,00366град"1 независимо
273,15 К
от температуры и объема. Следовательно, здесь cto = Po.
3. При изотермическом сжатии данной массы вещества под
действием внешнего давления происходит изменение объема:
очевидно, частная производная! [ ) характеризует собой из-
\ дР /т
менение объема при изменении давления на одну единицу
давления.
Тогда отношение этой производной к объему тела при
данном давлении является мерой сжимаемости вещества и
называется изотермическим коэффициентом сжимаемости:
. (1,7)
Так как с увеличением давления на тело объем его
уменьшается, то производная ( ) всегда отрицательна. Вводя по-
V op jt
ложительный коэффициент у» мы должны были в уравнении
(1,7) ввести знак минус. Вообще у представляет собой
величину переменную. Для идеальных газов коэффициент
сжимаемости у легко найти, так как при Т == const объем и давление
связаны законом Бойля—Мариотта:
т/ const
Отсюда
Up Jt
const __ _1_ у
20 Глава!. Общие понятия и определения
Подставляя это выражение в формулу (1,7), находим:
Следовательно, коэффициент сжимаемости есть величина,
обратная давлению газа; сжимаемость газа уменьшается с
увеличением давления. Обычно для характеристики сжимаемости
изменение объема с давлением относят к температуре 0°С.
Найдем теперь связь между термическими
коэффициентами а, р и у в общем случае. Для этого вспомним, что любой
параметр состояния х является функцией других параметров
или является функцией состояния, а значит, dx есть полный
дифференциал. Поэтому для параметров /?, V и Т мы можем
написать:
) 4Т + (Щ dV,
дТ jv \dV }т
dV = [HL\ dT + l°L) dp,
\дт)р ^ [dp jr H'
) йр + (Щ dV.
[dV )
\ dp Jv \dV /p
Из входящих сюда шести частных производных три имеют
самостоятельный смысл, тогда как остальные три являются
обратными им функциями. Подставим dp из первого уравнения
( dV \ ( др \-1 ,~
во второе. Учитывая, что [~f~ I = [w)t ' И сокращая на dT>
не равное нулю, получаем:
dV \
-1. (1,8)
i.
(8Ц . (IS.)
\др)т \дТ}\>
Подставляем в уравнение (1,8) значения частных производных
из (1,5), (1,6) и (1,7). Тогда находим:
«=Р-Т-Р- (1.9)
Это соотношение при заданном давлении связывает все три
термических коэффициента. Оно представляет интерес при
вычислении одного из коэффициентов, когда два других
определены опытным путем. Такое вычисление имеет значение в тех
случаях, когда экспериментальное определение некоторых
термических коэффициентов связано с затруднениями.
Для идеальных газов а = В = — ,и тогда из (1,9) опять
следует, что ^ = — .Для жидких тел коэффициенты сжатия очень
Р
§ 8. Теплота и работа 21
малы. Так, для воды известно, что ао = 0,000238, ро = 4,6. Отсюда
при нормальном давлении получаем уо = 0,000052, тогда как
в этом случае для газа уо= 1 - Следовательно, при увеличении
давления на одну атмосферу (при t~ const) объем воды
убывает на 0,000052 доли первоначального объема.
§ 8. ТЕПЛОТА И РАБОТА
Взгляды на сущность теплоты и применение этого термина
менялись на всем протяжении развития физики.
Первоначальное представление о теплоте было основано на допущении
существования особой невесомой «тепловой жидкости», или
«теплорода». Эти воззрения, господствовавшие особенно в XVIII в.,
подсказаны несомненно наблюдением явления
теплопроводности и выяснением понятий теплоемкости и скрытых теплот.
Если приводили в соприкосновение два тела — горячее и
холодное, то из простейшего наблюдения выравнивания
температур возникал образ текущей жидкости, которая движется в
определенном направлении. Формальная феноменологическая
теория теплопроводности, созданная Фурье в самом начале XIX
столетия, целиком основана на теории теплорода, хотя к этому
времени представление о «невесомых жидкостях» уже
становилось ненадежным. Наряду с этими взглядами в физике уже
давно пытались свести теплоту к особому виду скрытого
движения мельчайших частиц. Так, еще в 1692 г. Ньютон писал:
«Теплота есть колебание частиц друг около друга»*. В своем
«Рассуждении о причине теплоты и холода» (1744 г.)
Ломоносов убедительно для того времени доказывал, что теплота
объясняется вращательным движением молекул. Позднее Р. Клау-
зиус (в 1857 г.) выпускает сочинение под характерным
названием: «О роде движения, которое мы называем теплотой».
Из этих взглядов вытекает представление о теплоте как
энергии движения мельчайших частиц. Поэтому и теперь при
элементарном изложении физики иногда говорят, что теплота
есть вид энергии, и связывают эту энергию с молекулярным
движением. Однако в ходе развития термодинамики понятие
теплоты уточнялось, и оно к настоящему времени приобрело
вполне определенный смысл. Когда говорят о теплоте,
количестве теплоты, о затрате тепла, то во всех случаях имеют в виду
передачу энергии в определенном процессе. Передача или
отнятие энергии всегда означает определенный способ
взаимодействия тел, причем энергия распространяется в виде потока. Так,
в процессе теплопроводности распространяется поток энергии,
* См. статью «-De Natura Accidorum» для технического словаря Hariss'a;
цитир. по примечаниям акад. А Н. Крылова к «Principia» (Пр. 1915).
22 Глава!. Общие понятия и определения
имеющий определенный характер и направление (об этом см.
в гл. 9). При передаче теплоты в форме так называемого
лучеиспускания на самом деле происходит распространение потока
электромагнитного излучения, в результате поглощения эта
энергия испытывает превращение, приводящее к тому или
иному нагреванию тела, что соответствует определенному
количеству переданной телу теплоты.
В термостатике рассматриваются квазистатические
процессы, соответствующие весьма медленному теплообмену,
однако во всех случаях мы имеем дело с потоком энергии и
потому теплота есть всегда переданная в потоке энергия в
каком-либо «тепловом» процессе. Отсюда следует, что теплота
неразрывно связана с процессом передачи энергии или, как
говорят, зависит от формы пути и бессмысленно говорить о
теплоте вообще и нельзя ее приравнивать к некоторому «запасу»
энергии. Следует обратить внимание на существенное
различие понятий теплоты и внутренней энергии. В
теоретической физике переданное или полученное тепло имеет менее
принципиальное значение, чем внутренняя энергия. Возможны
разнообразные процессы, где теплообмен отсутствует
(например, во всех так называемых адиабатических процессах), и тем
не менее приходится рассматривать изменение внутренней
энергии, которая никогда не может равняться нулю. Случай перехода
теплоты является специальным, частным случаем, имеющим
специфические особенности. Применяя термин «тепловая
энергия», пытаются иногда связать теплоту с внутренней энергией
тела. Однако легко видеть, что понятие тепловой энергии
является вообще неопределенным, так как из общего запаса
внутренней энергии тела следовало бы выделить ту часть,
которая играет будто бы роль в тепловых процессах и которую
можно было бы назвать тепловой энергией. Учитывая
сложность молекулярных взаимодействий, следует признать, что
такое выделение было бы затруднительным. Вопрос о различении
понятий теплоты и внутренней энергии непосредственно связан
с привычкой словесного определения физических понятий.
Однако, как говорит Бриджмен, то, что человек обозначает
понятием, определяется наблюдением того, что он делаете этим
понятием, а не тем, что он говорит о нем. Далее, на стр. 36, мы
увидим, что коренное различие между понятиями теплоты и
внутренней энергии состоит в том, что операция дифференцирования
по отношению к теплоте имеет совершенно другой смысл, чем
по отношению ко внутренней энергии. Именно, бесконечно
малое изменение внутренней энергии dU является полным
дифференциалом функции состояния f/, тогда как бесконечно малое
dQ есть просто бесконечно малое количество теплоты,
переданной в определенном процессе. Но «если операции различны, то
§ 8. Теплота и работа 23
различны и дефинируемые (определяемые) понятия» (Бридж-
мен), и уже не требуется каких-либо словесных определений.
К понятию теплоты близко примыкает понятие работы.
Последняя представляет собой, с одной стороны, сам процесс
совершения работы, а с другой —оно указывает на количество
энергии, переданной системе (или окружению) в этом процессе.
Простейшие примеры работы в механике рассматриваются
в общем курсе физики. Элементарная (бесконечно малая)
работа силы F с компонентами Fx, Fy, Fz на бесконечно малом
пути ds с компонентами dx, dy, dz выражается, как известно,
в виде:
dW. = Fxdx + Fydy + F2dz.
В термодинамике рассматриваются другие, более сложные
процессы совершения работы; важнейшие из них мы здесь
рассмотрим.
1. Работа при изменении объема
газообразного или жидкого тела. Представим себе газ или
жидкость в оболочке, сжимающей это тело давлением р. Если дать
телу возможность расширяться, то оно будет совершать работу,
преодолевая внешнее давление р, равное его собственной
упругости (при условии очень медленного расширения). Напротив,
при сжатии будет совершаться работа внешними силами.
Найдем для этих случаев выражение элементарной работы.
Выделим на поверхности тела элементарную бесконечно малую
площадку dS (рис. 2). На нее действует внешняя сила pdS, равная
давлению изнутри тела. При незначительном расширении тела
эта элементарная площадка сместится на расстояние dh по
нормали п к площадке. Применяя обычное выражение
элементарной работы, когда направления силы и перемещения (по
нормали) совпадают, можем написать:
dW = pdS dh.
Общая элементарная работа расширения при бесконечно
малом приращении объема тела равна, очевидно, сумме работ
по всей поверхности тела:
dW = \pdSdh.
i
Но dSdh есть бесконечно малое
приращение объема тела у элемента
поверхности dS. Интеграл по всей
поверхности тела можно
рассматривать как элементарную работу рИс. 2.
24 Глава!. Общие понятия и определения
расширения при бесконечно малом приращении объема, когда р
можно считать постоянным, т. е.
(1,10)
Мь1 получили известную формулу элементарной работы
расширения, т. е. работу при увеличении объема тела. Полная
работа увеличения объема тела от V\ до V2 равна сумме
элементарных работ:
v2
W= \pdV.
Если тело совершает работу, то мы будем ее считать
положительной, напротив, работу сжатия, совершаемую внешней
силой над телом, будем считать отрицательной.
2. Работа деформации твердого тела. При
деформировании твердого тела необходимо совершать работу,
причем здесь сила не является давлением, нормальным к
поверхности, и в результате происходят, кроме сжатия или
растяжения тела, еще различного вида сдвиги. Поэтому выражение для
элементарной работы деформации должно слагаться из работы
растяжения и работы сдвига. При однородной деформации
тела с объемом V элементарная работа деформации может быть
представлена в виде:
dW = — V (sxdtx + V^tt., + a2dez + zxd^x + zyd^y + t^Tz),
где Gx, (Ту, Oz и tx, Ту, xz — составляющие нормального и
тангенциального напряжений, а е*, еу, е2 и ух, yv, yz — составляющие
величины растяжения и сдвига соответственно. Знак минус
указывает, что dW положительно, когда члены в скобках меньше
нуля. Разделив равенство на V, найдем работу на единицу
объема тела.
3. Работа изменения поверхности жидкости.
Из опытов с мыльными жидкими пленками видно, что при
увеличении поверхности пленок необходимо производить работу,
и, напротив, когда пленка сокращается, она совершает работу,
преодолевая внешнее сопротивление. Вообще, при всяком
увеличении поверхности жидкости требуется совершить работу по
преодолению сил сцепления молекул, выходящих из внутреннего
объема жидкости в ее поверхностный слой. При сокращении
поверхности система совершает работу за счет действия силы
поверхностного натяжения. Можно легко показать, что
элементарная работа в данном случае равна
dW = — adS, (1,11)
§ S. Теплота и работа 25
где а — поверхностное натяжение жидкости и dS —бесконечно
малое изменение поверхности. Знак минус указывает на то, что
положительная работа самой системы совершается при
сокращении поверхности (dS<0), так как всегда сг>0.
К этой формуле мы вернемся в дальнейшем (гл. 6).
4. Работа в электрическом поле. Известно, что
в электростатическом поле с напряженностью Е на заряд q
действует сила F = qE. Поэтому при движении заряда в поле
совершается работа, которая равна произведению заряда на
разность потенциалов двух точек поля. Следовательно,
элементарная работа при разности потенциалов dcp равна dW=qdq>.
В электростатике рассматривается изменение поля в
диэлектрике, возникающее при смещении зарядов, образующих это
поле. Работа, которая необходима для смещения зарядов при
изменении индукции D, равна на единицу объема диэлектрика:
dW=--^- (ExdDx + EydDy + E2dDz).
Здесь Ex, Ey, Ez, Dx, Dy, Dz — соответственно составляющие
вектора напряженности и индукции поля в диэлектрике.
5. Работа в магнитном поле. Элементарная работа
при изменении магнитного поля на единицу объема выражается
соотношением, аналогичным приведенному выше для
электрического поля, а именно:
dw = ~ ^ (H*dB* + HydBy + H*dB*)>
где #*, Ну, Нг и Вх, Ву, Bz—составляющие вектора
напряженности и магнитной индукции соответственно.
Здесь было рассмотрено несколько примеров вычислений
работы и из них видно, что бесконечно малая — элементарная
работа выражается в виде произведения:
dW = Akdah (1,12)
где Ak во всех случаях имеет смысл некоторой силы в общем
смысле, a da^—бесконечно малое изменение некоторого
внешнего параметра при действии этой силы. В ряде примеров
элементарная работа представлялась в виде суммы таких
произведений. Это позволяет нам в самых разнообразных случаях
выразить элементарную работу в виде:
d\V=%Akdak. (1,13)
k
В термодинамике мы иногда выделяем работу при
изменении объема и работу, не зависящую от изменения объема.
26
Глава 1. Общие понятия и определения
а также при немеханических процессах (например, в
электромагнитном поле). Условимся в дальнейшем работу,
совершенную самой системой, считать положительной, а работу,
производимую над системой внешними силами, считать отрицательной.
В термодинамике распространены случаи, когда
совершенная работа зависит от вида процесса, в котором работа
производилась, или, как принято говорить, зависит от формы пути.
Это ясно видно на примере работы при изменении объема.
На диаграмме (/?, У) элементарная работа pdV изображается
площадью бесконечно узкого прямоугольника (рис. 3); полная
работа при расширении от объема Уа до объема Уъ получается
как сумма площадей таких прямоугольников между а и Ь.
В пределе при dV—>0 полная работа равна площади
фигуры (VaabVb). Вполне очевидно, что величина этой площади
зависит не только от начального Уа и конечного Уъ, но также
от формы кривой перехода из а в Ь, т. е. от формы «пути»
между а и Ь. Следовательно, работа в этом случае определена
только, если известен ход расширения. Очевидно, в случае
замкнутого процесса (цикла) работа не равна нулю. На рис. 4
работа на пути acb равна площади фигуры (УаасЬУьУа). На
пути bda происходит сжатие тела, когда необходимо совершить
работу над телом, т. е. эта работа имеет знак обратный по
отношению к знаку в первом случае. Эта работа равна площади
фигуры (Уа^ЬУъУа)- Отсюда видно, что для всего контура
работа равна площади фигуры (acbda) и она не равна нулю.
На этом основано действие машин, которые работают
циклически и дают конечную работу.
Во многих разделах физики рассматривается работа,
которая не зависит от формы пути (в механике это пример, работы
консервативных сил). Очевидно, работа в этих случаях
является частным выражением более общего случая
термодинамики, когда работа зависит от вида пути.
Р I
Vb
Vb
Рис. 3.
Рис. 4,
§ 9. Математические методы термодинамики 27
§ 9. МАТЕМАТИЧЕСКИЕ МЕТОДЫ ТЕРМОДИНАМИКИ
В обычной термодинамике широко применяются методы
теории дифференциальных уравнений в частных производных,
что обусловлено необходимостью введения в задачи нескольких
переменных параметров состояния, которые могут быть
функционально связаны друг с другом. Эта связь между
переменными выражается дифференциальными соотношениями.
Главный результат почти всегда сводится к анализу частных
производных: к установлению их физического смысла, и особенно
к определению знака этих производных, который часто
позволяет определить собой течение и направление физического
процесса. Пример этого метода в самом простом виде нам
встречался при выводе связи между термическими коэффициентами
(§ 7). В последующих главах мы убедимся в важности
указанного метода.
Наибольшее значение в этой области получили линейные
дифференциальные уравнения в полных дифференциалах
независимых переменных, называемые формами Пфаффа,
а также дифференциальное уравнениеПфаффа.
Эти дифференциальные формы имеют общий вид:
(1Фп = X1dx1 + X2dx2 + ... + Xndxn, (1,14)
где хи *2, ..., xn являются независимыми переменными, тогда
как Хи X2i ..., Хп суть функции этих переменных.
Когда d<Pn = Oy то получаем уравнение Пфаффа:
Xxdxx + X2dx2 + . . . + Xndxn = 0. (1,15)
В простейшем случае двух независимых переменных х, у
пфаффова форма имеет вид:
d<P = М (х, y)dx + N (х, у) dy. (1,16)
Здесь М(ху у) и N(xy у)—некоторые функции независимых
переменных ху у. Этот вид часто встречается в термостатике,
где существенную роль играют два разных частных случая,
в зависимости от вида М и N.
1 т? * дМ dN
1. Если соблюдается условие = , называемое усло-
ду дх
вием Эйлера, то легко показать, что существует некоторая
функция Ф переменных х и у общего вида- 0 = F(x, y)+C, полный
дифференциал которой выражается соотношением (1,16).
Действительно, если такая функция Ф(х, у) существует, то полный
дифференциал ее есть
28 Г лава 1. Общие понятия и определения
Сравнивая (1,17) с (1,16), находим, что в этом случае
ЛЛ дФ AT дФ
дх ду
Дифференцируя М по у и N по х, получим:
дМ д2Ф dN д2Ф
ду дхду дх дудх
Так как правые части этих равенств равны друг другу, то
получаем условие Эйлера, т. е.
ду дх
Была доказана необходимость условия (1,18) для
существования функции Ф. Нетрудно видеть, что соотношение (1,18)
является также достаточным условием существования функции Ф.
Обратное заключение тоже является справедливым, т. е. если
имеется функция Ф переменных х и у, то условие (1,18)
справедливо.
2. Случай ф . Можно показать, что в этом случае
ду дх
не существует функции двух переменных х и у, для которой
уравнение (1,16) представляло бы собой полный дифференциал
какой-то функции. В самом деле, если допустим существование
функции <P = F(x, y)+C, то прежним путем получим
условие (1,18), которое противоречит принятому неравенству
частных производных. Следовательно, в этом случае йФ не есть
полный дифференциал, а представляет собой просто бесконечно
милее приращение некоторой переменной величины Ф. Однако,
как известно из теории дифференциальных уравнений, для
пфаффовой формы (1,16) двух независимых переменных всегда
существует так называемый интегрирующий множитель
|л = (х (х, у), т. е. умножением этой формы на множитель \х мы
вновь получаем полный дифференциал некоторой функции.
В самом деле, введем функцию \x=\i(x, у) и умножим
исходное уравнение (1,16) на эту величину. Тогда получаем:
с1Ф0 = рс1Ф = [хМ dx + pN dy.
Легко видеть, что йФо является полным дифференциалом
функции Ф0 = Ф0(лс, у)у т. е.
**??*-dy.
ЛФо dx+
дх ду
Сравнивая оба выражения, находим:
дФл хт бФп
—*-; (i,iV=—-2-,
дх ду
§ 9. Математические методы термодинамики 29
откуда —— = —-—, т. е. условие Эйлера соблюдается, а это
ду дх
значит, что функция Ф0 = Ф0(х, у) существует, а dФo есть ее
полный дифференциал.
Если число независимых переменных больше двух, то
пфаффова форма (1,14) может вообще и не иметь интегрирующего
множителя, т< е. нельзя в общем случае подобрать такой
функции, которая обращала бы (1,14) в полный дифференциал. Для
существования интегрирующего множителя необходимо
соблюдение некоторых условий.
Найдем условие существования интегрирующего множителя
при трех независимых переменных х, у, z, т. е. для пфаффовой
формы:
dФ = М dx + N dy + P dz. (1,19)
Пусть ц = |л(л;, у, г) есть интегрирующий множитель для
(1,19).
Тогда
dФ0 = jx dФ = [хМ dx + \*.N dy + цР dz,
где
d0Q^dx+^dy+^
дх ду дг
Следовательно,
дх ду дг
При этом должны соблюдаться условия Эйлера вида
ду дх dz ду dz дх
Выполним дифференцирование произведений в этих уравнениях,
а затем, произведя группировку, умножим их в той же
последовательности первое на Р, второе на М и третье на N. Тогда
после сокращений и помня, что [х=#=0, находим:
дх ду )^ [ду dz )^ [ dz дх)
Это выражение представляет собой условие существования
интегрирующего множителя для пфаффовой формы при трех
независимых переменных.
В термостатике нам придется встречаться с разными
случаями пфаффовых форм, а также с уравнением Пфаффа.
В дальнейшем случай, когда йФ есть полный дифференциал,
30 Г лав а 1. Общие понятия и определения
будет особенно важным, так как простым интегрированием
получаем:
2
J йФ = Ф2 — Фъ
и для замкнутого контура имеем:
' йФ = 0.
Следовательно, Ф является функцией состояния.
§ 10. КРАТКИЙ ИСТОРИЧЕСКИЙ ОЧЕРК
РАЗВИТИЯ ТЕРМОДИНАМИКИ
Можно вполне точно установить дату возникновения классической
термодинамики. В 1824 г. появился труд молодого французского инженера Сади
Карно под названием «Размышления о движущей силе огня», в котором автор
дает теорию тепловых машин и указывает пути их дальнейшего
усовершенствования. Именно такую практическую цель преследовал автор в своем
сочинении. На самом деле, однако, в труде, помимо принципиальных вопросов
теории тепловых машин, содержится первая формулировка одного из
важнейших положений термодинамики — так называемого второго начала
термодинамики. Работа Карно вначале не обратила на себя внимания, и лишь спустя
примерно 30 лет идеи автора были использованы в трудах Клаузиуса,
продолжившего учение о втором начале. Таким образом, этой работой Карно и
положено начало развития классической термодинамики. Необходимо
отметить, что учение о теплоте появляется в физике значительно раньше. Однако
с этим мы не связываем развитие именно термодинамики, так как в отдельные
эпохи развития физики теплоту рассматривали как особую невесомую
жидкость, называвшуюся теплородом. Даже в труде Карио мы встречаем
упоминание о теплороде, хотя в позднейших трудах автор уже не придает того
значения теплороду, как его предшественники.
Дальнейшим важным этапом в развитии термодинамики являются
работы 40—50-х годов XIX в., связанные с формулировкой первого начала
термодинамики, которое в науке, таким образом, появляется после второго
начала. Установление первого начала термодинамики, представляющего собой
в общем выражении закон сохранения энергии, связано с трудами трех
исследователей, подходивших с разных точек зрения к формулировке этого
основного закона природы. В 1842 г. появилась первая работа немецкого врача
и естествоиспытателя Р. Майера «Размышления о силах неживой природы»,
вначале не привлекшая внимания большинства ученых, стоявших еще на
старых позициях. Далее, в 1847 г. была издана монография молодого немецкого
врача Г. Гельмгольца (впоследствии крупнейшего физика и физиолога) под
названием «О сохранении силы». Наконец, с 1843 по 1856 г. Джеуль в Англии
проводил экспериментальные определения механического эквивалента теплоты,
представляющего собой количество механической энергии, дающее при
превращении единицу количества теплоты (выражаемой в килокалориях).
Работа Майера была в основном философски умозрительной, хотя он впервые
вычислил значение механического эквивалента теплоты по данным опытов
Реньо. В монографии Гельмгольца подчеркивается общее значение первого
начала, дается математическая формулировка и рассматривается большое
число различных его приложений в физике. Таким образом, здесь выделяется
теоретическое значение первого начала. Работы Джоуля показали постоянство
величины механического эквивалента теплоты в разнообразных процессах
превращения энергии и тем самым подвели экспериментальную базу под закон
§ 10. Краткий исторический очерк развития термодинамики 31
сохранения энергии. После исследований Джоуля и других ученых этот закон
прочно утверждается в науке. Впрочем, термин «энергия» еще не применяется
в ранних трудах. Выделение понятий «энергия» и «сила» постепенно
подготавливалось при развитии механики. Еще Лейбниц отличал живую силу
(vis viva) от других видов сил, но, хотя эта «живая сила» имеет размерность
энергии (мы называем ее теперь кинетической энергией), ранее в механике
отдельным понятием энергии не пользовались. Термин «энергия» встречается
еще у Аристотеля; позднее его применяли Кеплер, Галилей, Лейбниц, но не
связывали с ним точных представлений. Обратим внимание, что даже труд
Гельмгольца, где рассматривается сохранение энергии, носил название «О
сохранении силы». Заметим также, что о сохранении движения имелись
высказывания многих ученых еще задолго до работ Майера, Гельмгольца и Джоуля.
Так, ясно выраженная идея сохранения имеется в работах Ломоносова, у
Декарта и других. Но все эти весьма общие формулировки (скорее догадки) не
могли служить точному установлению и упрочению первого начала
термодинамики.
Последовательное развитие второго начала мы находим в ряде работ
Клаузиуса (1850—1876 гг.), где даны классические формулировки второго
начала, а также сделаны первые шаги в развитии молекулярной теории. В
работах Клаузиуса впервые вводится важнейшее понятие термодинамики —
«энтропия» и рассматриваются ее свойства. К этому же периоду — середина
XIX в. — относятся труды В. Томсона (Кельвина) и Дж. Кл. Максвелла, где
даются новые формулировки начал термодинамики, уточняется понятие
температуры, вводится абсолютная шкала температур и впервые применяются так
называемые характеристические функции термодинамики. Наконец, в работах
Л. Больцмана и особенно Дж. В. Гиббса, относящихся ко второй половине
XIX в., закладываются основы статистической термодинамики. Развитие этого
современного нам направления тесно связано с постепенным формированием
молекулярно-кинетической теории в трудах тех же ученых. Одновременно с
созданием основ классической термодинамики умножаются и растут ее
приложения в теплотехнике, в физической химии, в теории излучения, в астрофизике
и т. п. Наибольшего развития классическая термодинамика достигает в
«Термодинамических работах» Гиббса, который последовательно пользуется
термодинамическими функциями, впервые формулирует правило фаз и кладет основу'
химической термодинамики, играющей важную роль в современной физической
химии. В начале нашего века Нернст формулирует третье начало
термодинамики и создает теорию поведения систем вблизи абсолютного нуля. М. Планк
и В. Оствальд систематизируют результаты развития классической
термодинамики и рассматривают ряд важных ее приложений. Новейшее развитие
термодинамики в основном опирается на статистическую физику, хотя существует
и направление «чистой термодинамики», называемое аксиоматикой.
Задачи
1. Доказать, что выражение
dB = (х + z) dx + (y + z) dy + (x + у) dz
является полным дифференциалом.
2. Почему соотношение
dG = x2dy — уЧх
не есть полный дифференциал?
3. Доказать, что для уравнения d<t>=pdV—Vdp функция (p(F) =
—является интегрирующим множителем, приводящим уравнение к полному
дифференциалу.
Глава 2
ПЕРВОЕ НАЧАЛО ТЕРМОДИНАМИКИ
И ЕГО НЕПОСРЕДСТВЕННЫЕ ПРИМЕНЕНИЯ
§ 1. ФОРМУЛИРОВКИ ПЕРВОГО НАЧАЛА
Значение термодинамики состоит в использовании так
называемого термодинамического метода, который может быть
применен к разнообразным вопросам физики, физической химии и
техники и о котором будет сказано ниже. Здесь будут
рассмотрены наиболее важные приложения этого метода к ряду
известных вопросов общего курса физики. К ним относятся
описания свойств газов, паров и жидкостей, анализ капиллярных
явлений, теория критического состояния, принцип действия
гальванических элементов, законы излучения, теория равновесия
сложных термодинамических систем и другие приложения.
В основе термодинамики лежат два закона, или, как их
называют, начала. К ним следует добавить еще третье начало,
которое хотя является тоже принципиально важным, но имеет
более специальный характер, так как относится к поведению
систем вблизи абсолютного нуля температуры. Мы рассмотрим
прежде всего два первых начала и их непосредственные
применения, после чего перейдем к приложениям метода
термодинамики, основанного на этих началах.
Первое начало в общем виде представляет хобой закон
сохранения и превращения энергии. Этот закон, как известно,
является одним из основных законов природы и служит одним
из основных положений материалистического взгляда на
свойства окружающего мира. Многовековой опыт человечества и
неисчислимые эксперименты, доказывающие сохранение энергии
при всех ее превращениях, дают ясную реальную основу для
материалистического мировоззрения. Если энергия не творится
и не пропадает, то тем самым уже исключается идея о
всемогущем творце. Первое начало налагает строгое условие на все
процессы природы, которые при всем их разнообразии
ограничены условием сохранения энергии.
Как всякое широкое обобщение, первое начало допускает
ряд различных формулировок, общий смысл которых один и тот
же, но их используют для обсуждения разных конкретных
задач. Сначала дадим эти формулировки, а потом представим
первое начало в математической форме.
§ 1. Формулировки I начала 33
1. Все виды энергии могут взаимно превращаться в строго
равных друг другу количествах; за единицу количества энергии
(или работы) в системе единиц СИ принимается один джоуль.
В наиболее распространенном случае, а именно превращения
механической энергии в теплоту, всегда определенному
количеству механической энергии соответствует одно и то же
количество получаемой теплоты. Если механическая энергия
выражается в джоулях, а теплота — в калориях, то говорят об
эквивалентности. Многочисленные экспериментальные измерения
показали, что термический эквивалент работы,, т. е. отношение
механической энергии (или работы) к соответствующему
количеству теплоты, один и тот же, и он принимается равным
/ = 4,1868 дж/кал.
2. Невозможно построить такую периодически действующую
машину, которая давала бы полезную работу без затраты
энергии извне, т. е. даровым путем. Подобное устройство,
называемое вечным двигателем первого рода, противоречит закону
сохранения энергии и потому невозможно. Известно, что в
прошлом было сделано множество безуспешных попыток построения
такой машины, или, как ее называли (не вполне точно),
перпетуум мобиле. Неудачи с построением вечного двигателя
заставили французскую академию наук еще в 1775 г. объявить раз
навсегда, что она больше не будет рассматривать мнимых
решений этой задачи. В настоящее время в соответствии с законом
сохранения энергии мы принимаем в принципе, что вечный
двигатель первого рода невозможен. (О вечном двигателе второго
рода, который тоже невозможен, будет сказано ниже). Заметим,
что речь идет о периодически работающем двигателе, тогда как
в общем случае вечное движение возможно и вполне реально.
3. Внутренняя энергия вполне изолированной системы есть
величина постоянная.
Остановимся на третьей формулировке первого начала,
поскольку первые две достаточно хорошо известны и потому, что
эта третья связана с общим математическим выражением
закона сохранения энергии.
Пусть в некотором процессе — макросистеме сообщено
количество теплоты AQ. Часть этого тепла может пойти на
нагревание системы (тела), часть из общей теплоты идет на изменение
агрегатного состояния тел системы, например на плавление и
на парообразование. Возможно, что известная часть теплоты
может пойти на более сложные процессы, например на
перекристаллизацию, на полиморфные превращения, а также на
растворение твердых тел. Опыты показывают, что эти явления
требуют сообщения теплоты, хотя часть теплоты, выделяющейся
при этом может передаваться от таких тел к соседним телам.
34 Г л а в а 2. Первое начало и его непосредственные применения
Во всяком случае, перечисленные случаи передачи теплоты
обусловливают различные изменения внутренней энергии системы
или входящих в нее тел. Например, нагревание приводит к
увеличению кинетической энергии движения молекул тела;
изменение агрегатного состояния, например плавление твердых тел,
связано с изменением внутренней потенциальной энергии частиц
тела, обусловленной силами атомного или молекулярного
взаимодействия. В целом общая сумма полученной системой теплоты
идет на изменение внутренней энергии тела. Однако, кроме
указанных случаев, часть сообщенной телу теплоты может
превращаться во внешнюю работу, например работу увеличения объема
или изменения поверхности и т. д. Мы знаем, что при всех
перечисленных явлениях закон сохранения энергии строго
соблюдается, т. е. должен иметь место баланс полученной системой
теплоты, изменения внутренней энергии и совершенной внешней
работы. Если полученная системой теплота есть AQ, сумма
всех изменений внутренней энергии равна Д£/, а сумма всех
видов совершенной внешней работы, состоящей из слагаемых
Akkcik, то уравнение баланса энергии должно быть:
AQ = MJ + 2lAkbak. (2,1)
k
Отдельные слагаемые в этом равенстве могут иметь разные
знаки или быть равными нулю. Условимся считать теплоту п о -
ложительной, если она сообщается телу, и
отрицательной, если она отводится, т. е. отнимается от тела и передается
другим телам. Далее, если тело само совершает работу против
внешних сопротивлений, то будем считать работу
положительной, и если, напротив, работа совершается над телом, то будем
считать работу отрицательной.
Уравнение баланса энергии применяется в элементарной
физике при расчетах различных процессов в калориметрических
опытах; оно выражает баланс израсходованного и полученного
тепла; очевидно, что это «уравнение теплового баланса»
является частным случаем более общего написанного здесь
уравнения и должно рассматриваться как выражение закона
сохранения энергии. Поэтому составление подобных уравнений
баланса в элементарной физике является поучительным и
методически оправданным, так как оно позволяет обращать
внимание учащихся на закон сохранения энергии.
В дальнейшем нам часто придется йользоваться
представлением о полностью изолированной системе.
Последняя является системой, для которой всякий обмен со внешними
телами нацело устранен. Для изолированной системы, например,
невозможен теплообмен с окружающей средой, она не передает
внешним телам энергии в форме электромагнитного излучения
§ 1. Формулировки I начала 35
и не поглощает последнего извне. Полностью изолированная
система не совершает также внешней работы и внешние силы
также не производят работы над ней. Такая идеально
изолированная система представляет собой некоторую абстракцию,
однако можно различными приемами столь значительно ослабить
роль внешних воздействий, что система будет практически
изолированной и будет удовлетворять поставленным условиям. Для.
устранения обмена теплом с внешними телами можно заключить
систему в так называемую адиабатическую оболочку.
Простейшим устройством такого рода, впрочем лишь частично
устраняющим обмен теплом с внешней средой, является
калориметр. Он состоит из двух-трех сосудов, вставленных друг
в друга и снабженных общей крышкой из плохого проводника
тепла. Это схема так называемого адиабатического
калориметра. Близко напоминает адиабатическую оболочку также
обычный сосуд Дьюара или термос. Какую-либо систему можно
также считать изолированной, если рассматривать ее в течение
столь небольшого промежутка времени, за который она не
успевает обменяться теплом с внешней средой. Это обстоятельство
позволяет распространить понятие полностью изолированной
системы на многие реальные системы. Следует подчеркнуть
различие между полностью изолированной системой и
адиабатически изолированной системой. Первая из них не
может обмениваться со средой ни теплотой, ни какими-либо
другими видами энергии, ни совершать внешнюю работу, тогда
как для второй системы отсутствует только обмен теплотой,
а обмен другими видами энергии допустим и система может
совершать положительную или отрицательную работу.
Применим математическое выражение первого начала (2,1)
к полностью изолированной системе. Условия полной изоляции
дают: AQ = 0 и2 ЛьДа& = 0, поэтому уравнение (2,1) принимает
простой вид:
откуда U = const.
Следовательно, энергия вполне изолированной системы есть
величина постоянная. Это положение представляет собой
важную формулировку первого начала термодинамики. Очевидно,
первое начало налагает строгое ограничение на все процессы
в изолированной системе. Как бы разнообразны и сложны ни
были бы эти процессы, все равно внутренняя энергия такой
системы сохраняется.
Ранее было написано уравнение (2,1) как баланс конечных
величин энергии. Если в простейшем случае затрачивается
бесконечно малое количество теплоты dQ и совершается бес-
36 Г л а в а 2. Первое начало и его непосредственные применения
конечно малая работа dW, а изменение внутренней энергии
тоже бесконечно мало и равно dU, то уравнение первого начала
принимает вид:
dQ = dU + dW. (2,2)
Если учитывать возможность различных знаков величин,
входящих в это уравнение, то его можно написать в виде:
dU = dQ + dW. (2,3)
Этот вид уравнения указывает, что изменение внутренней
энергии системы (тела) происходит за счет полученной
системой теплоты и за счет совершения работы.
Обратим внимание на бесконечно малые величины,
входящие в уравнение (2,2). Ранее мы видели (гл. 1), что
сообщенная системе теплота зависит от вида процесса или от формы
пути, где эта теплота передана, т. е. запаса теплоты не
существует. Это значит, что dQ не является полным
дифференциалом какой-либо функции, а просто означает бесконечно малое
количество теплоты. Далее в § 6 главы 1 было отмечено, что
внутренняя энергия U есть функция состояния и изменение ее
не зависит от формы пути, поэтому dU есть полный
дифференциал для параметров состояния. В § 8 главы 1 мы видели, что
внешняя работа вообще зависит от формы пути, следовательно,
dW в уравнении (2,2) не является полным дифференциалом
какой-нибудь функции состояния, а тоже, как и dQ,
представляет собой бесконечно малую внешнюю работу. Таким образом,
все три величины в формуле (2,2) обладают физически и
математически существенно разными свойствами. Поэтому иногда
для dQ и dW вместо знаков дифференциалов вводят другие
обозначения, например 6Q и 6W. Здесь мы сохраним
обозначения уравнения (2,2), но будем помнить, что знаки
дифференциалов имеют иной смысл, чем знак дифференциала dU,
являющийся полным дифференциалом.
Можно строго показать, что dQ не может быть полным
дифференциалом. Ограничимся, однако, простейшим случаем, когда
производится внещняя работа по расширению системы. Тогда
dW=pdV, и уравнение (2,2) принимает вид:
dQ = dU + pdV. (2,4)
Так как мы знаем, что U к V являются функциями
состояния, то dU и dV—полные дифференциалы. Рассмотрим общий
случай независимых параметров состояния х и у. Тогда:
,7/ dU , , dU *
dUdx+dy
дх
§ L Формулировки I начала
37
Подставляя эти выражения в уравнение (2,4), после
группировки получаем:
dQ = Mdx + N dy,
где
дх
ду
f
дх
ду
(2,5)
Для того чтобы dQ было полным дифференциалом,
необходимо соблюдение условия Эйлера (стр. 27):
дМ
dN
дх
или _^L_J^ =
д дх
я ' -- я я - <2'5')
ду дх ду дх
Дифференцируя выражения (2,5) для М и N соответственно
по у и по х% находим:
дМ дЮ _х_ п д*У _±_ дУ_ др
дх ду
откуда
ду
dN
dx
dM
dxdy
d*U
dydx
dN
ду
дх
дхду
. _д*у_ , dV_ dp
дудх ду дх
дх ду ду дх
Это выражение можно представить в форме детерминанта,
представляющего собой простейший детерминант Якоби
(якобиан):
дУ_ др_
дх дх
дУ
ду
др
Можно легко показать, что в данном случае он не равен
нулю. Помня, что х и у взаимно независимы, находим
значения D для частных случаев:
= V и u = T- D = -?-
u ' дТ
= V и у = р: D=l,
= p и у = T\ D = —
дУ
дТ
38 Г лав а 2. Первое начало и его непосредственные применения
При выборе других независимых параметров состояния
можно убедиться, что всегда БФО. Значит, условие Эйлера
(2,5/) не соблюдается, и потому dQ не является полным
дифференциалом, и функции состояния Q не существует. Однако
в случае двух независимых переменных пфаффова форма имеет
интегрирующий множитель (о нем см. в главе 3).
Так как dQ не есть полный дифференциал, то
(J) dQ ф 0.
Интегрируя уравнение (2,2) по замкнутому контуру, находим:
<$> dQ = (f> dU + § dW.
Но так как первое слагаемое правой части всегда равно
нулю, то
J j dW.
<J) dQ =
Следовательно, в круговых процессах общая сумма
затраченных и отнятых теплот в каждом цикле равна работе за один
цикл, которая не равна нулю. В этом заключается смысл
применения круговых процессов в машинах.
§ 2. ПРИМЕНЕНИЕ ПЕРВОГО НАЧАЛА К ПРОЦЕССАМ
В ПРОСТЕЙШИХ СИСТЕМАХ. ЭНТАЛЬПИЯ
По отношению к окружающим телам термодинамические
системы можно разделить на закрытые и открытые
системы, смотря по тому, в какой степени они могут обмениваться
теплотой с окружающей средой. Ранее было рассмотрено
применение первого начала к полностью закрытой (изолированной)
системе. Здесь мы получим выражения первого начала в
применении к простейшим системам, способным различно
обмениваться с внешней средой; к ним относятся разные виды
открытых систем, в которых возможен обмен и теплом, и работой.
Общее уравнение первого начала для всякой системы имеет
вид:
Для анализа простейших задач из суммы элементарных работ
выделим работу, обусловленную изменением объема тела. Тогда
dQ = dU + p dV - 2 Andan. (2,6)
л
Отсюда
г* с
y2) Andan. (2,7)
§ 2. Применение I начала к процессам в простейших системах 39
В оба последних уравнения входит сумма работ, не
связанных с расширением тела.
Рассмотрим применение уравнений (2,6) и (2,7) к
простейшим процессам.
1. Изохорический процесс. Система имеет
постоянный объем V = const, например находится в жесткой оболочке
и обменивается теплотой со средой. В этом случае в ней
протекает изохорический процесс, когда1 dV=0, и уравнение (2,6)
имеет вид:
Отсюда следует, что при изохорическом процессе часть
теплоты идет на изменение внутренней энергии системы
(например, на нагревание), а другая часть идет на работу при
постоянном объеме, например на работу в электрическом или
магнитном поле. Если в системе эти последние виды работы не
совершаются, то
dQ = dU,
т. е. вся теплота идет на изменение внутренней энергии.
2. Изобарический процесс. Пусть система находится
при постоянном внешнем давлении р = const и имеет
возможность расширяться и обмениваться теплотой с окружением. Этот
процесс носит название изобарического процесса; при затрате
теплоты извне происходит увеличение внутренней энергии и
совершается внешняя работа, в том числе работа расширения
при постоянном давлении.
Пусть при передаче теплоты Q произошло увеличение объема
системы от V\ до V2 при постоянном давлении и другие виды
работы не совершаются, т. е. Ап = 0. Тогда из (2,7) находим:
Q = U2-U1+p
Сгруппируем слагаемые с одинаковыми индексами, тогда
Q = (t/2 + pl/2)_(f/1 + j0yi). (2,8)
Выражение (2,8) позволяет ввести функцию Я, имеющую
общий вид
H = U + pV. (2,80
Эта функция называется энтальпией (или тепловой
функцией). Очевидно, теплота, полученная в изобарическом
процессе, может быть выражена согласно (2,8) через
приращение энтальпии:
40 Г лава 2. Первое начало и его непосредственные применения
Энтальпия является функцией состояния, так как она
выражается согласно (2,8х) через внутреннюю энергию, которая
сама есть функция состояния, и Н явно зависит от р и V,
являющихся параметрами состояния. Следовательно, в частном
случае изобарического процесса, т. е. при условии /?=const,
величина dQ является полным дифференциалом: dQp = dH.
3. Изотермический процесс. Допустим, что система
находится в термостате, представляющим собой тело больших
размеров по сравнению с размерами выделенной системы,
обладающее большой теплоемкостью и при этом хорошо
контактирующее с данной системой. Температура искусственно
поддерживается постоянной Т= const. Процесс, протекающий
в выделенной системе, носит название изотермического
процесса. В простейших случаях внутренняя энергия зависит от
температуры и от объема тела. Поэтому полный дифференциал
функции U можно представить в виде:
где первое слагаемое означает приращение внутренней энергии,
обусловленное увеличением скорости движения молекул
(внутренняя кинетическая энергия), а второе слагаемое зависит от
изменения потенциальной энергии молекул при изменении их
взаимного расположения. В изотермическом процессе dT=0 и,
значит,
dU=(*L) dv.
Пусть внешняя работа в (2,6) ограничивается лишь работой
расширения. Тогда
{ )dV- (2>9)
Теплота идет на увеличение внутренней энергии при
возрастании объема и частью на внешнюю работу расширения.
Например, при парообразовании, происходящем в условиях
постоянной температуры, теплота расходуется на превращение
жидкости в пар, т. е. на преодоление сил сцепления молекул
жидкости и на расширение полученного пара. Выражение (2,9)
нам будет встречаться далее. Заметим, что отношение
можно рассматривать как скрытую теплоту
расширения, т. е. представить как количество тепла, необходимое при
изотермическом увеличении объема тела на единицу объема.
§ 2. Применение I начала к процессам в простейших системах 41
4. Адиабатный процесс. В этом случае система
находится в адиабатической оболочке, исключающей всякий обмен
теплотой с внешней средой, хотя, находясь в этой оболочке,
система все же может совершать внешнюю работу или над ней
может совершаться работа. В этих условиях, очевидно, dQ = 0.
Из уравнения первого начала (2,2) следует:
dU = —dW.
Это соотношение показывает, что в адиабатном процессе
система совершает работу за счет убыли внутренней энергии.
Напротив, если над системой производится работа (dW<0), то
в результате внутренняя энергия такой системы увеличивается.
Адиабатическая система является частично закрытой
системой.
§ 3. ТЕПЛОЕМКОСТЬ
Во многих процессах внешним результатом сообщения телу
теплоты является нагревание и поэтому, не разделяя
полученную теплоту на части соответственно изменению свойств тела,
вводят общее понятие теплоемкости, которая представляет
собой количество теплоты, необходимой для изменения
температуры тела на один градус. Будем называть средней
теплоемкостью в интервале температур АТ=Т2—Тх отношение:
AT '
где AQ— количество тепла, полученное в процессе повышения
температуры тела от Тх до Т2. Обычно с уменьшением
интервала температуры AT количество теплоты AQ непрерывно
убывает, поэтому, переходя к пределу, когда AT стремится к нулю,
мы получаем истинную теплоемкость при данной температуре:
Опыт показывает, что необходимая теплота всегда
пропорциональна массе тела. Поэтому для возможности сравнения
вводят, как известно, удельные величины теплоемкости, относя
необходимую теплоту к единице массы тела. Различают
удельную теплоемкость, когда необходимая теплота отнесена к 1 кг
массы
*-•£-.
и молярную теплоемкость, когда теплота отнесена к одному
молю
С= — M = ClM.
m
42 Г лав а 2. Первое начало и его непосредственные, приме нения
Мы в дальнейшем будем чаще пользоваться молярной
теплоемкостью. Эта величина зависит от особенностей структуры
отдельных молекул, и введение ее дает возможность сравнения,
так как в 1 моле любого вещества содержится одно и то же
число молекул: УУ0 = 6,023-1023 (постоянная Авогадро).
При определении истинной теплоемкости мы получили
отношение dQ к dT, которое нельзя рассматривать как производную
какой-то функции. Мы знаем, что dQ не есть полный
дифференциал и величина Q зависит от вида процесса. Поэтому также
и теплоемкость не есть функция состоякия и в различных
процессах должна быть различной. Говоря о теплоемкости при
данной температуре, мы должны иметь в виду некоторый процесс
передачи теплоты вблизи этой температуры, когда взят
бесконечно малый температурный интервал и затрачена теплота dQ.
Следовательно, так же как и Q, теплоемкость всегда зависит
от вида процесса. Так как над телом можно провести
бесчисленное множество операций или процессов, то, строго говоря,
число теплоемкостей неограниченно велико и значения С лежат
в интервале от —оо до + оо. Отрицательные теплоемкости
возможны, так как мы можем, например, затрачивать тепло
(dQ>0), одновременно сильно увеличивая объем, когда
температура тела падает (dT<0).
Как известно, наибольшее практическое значение имеют
теплоемкости в изохорическом (Су) и изобарическом (Ср)
процессах, причем всегда CP>CV, так как во втором случае тело
нагревается и, кроме того, совершает работу расширения,
соответственно чему увеличивается количество необходимой
теплоты. В справочных таблицах обычно приводятся значения Ср
по опытным данным. В элементарном изложении физики на это
обстоятельство не указывают, благодаря чему величине
теплоемкости часто неправильно придается смысл безоговорочной
константы. Легко установить, каковы теплоемкости в
рассмотренных ранее тепловых процессах.
1. В изохорическом процессе теплоемкость равна Су и
2. В изобарическом процессе теплоемкость Ср может быть
представлена выражением, если ввести энтальпию:
с dQfL = (МЛ , т. е. dHp = CpdT.
р dT \дт)р р Р
3. В изотермическом процессе теплоемкость равна
бесконечности:
§ 3. Теплоемкость 43
так как здесь при передаче системе теплоты AQ повышения
температуры не происходит: ДГ=0.
4. В адиабатическом процессе теплоемкость равна 0, так как
затраченная теплота dQ = 0 при наличии конечного изменения
температуры.
Между теплоемкостями Ср и Cv для веществ в любом
агрегатном состоянии существует зависимость, которую легко
вывести в самом общем случае. В самом деле, из выражения
первого начала следует, что:
w),dT
так как Cv = ( ) . Разделив предыдущее равенство на dT
и положив р = const, имеем:
откуда
Здесь выражение в квадратных скобках представляет собой
скрытую теплоту расширения / согласно формуле (2,9'),
поэтому
Для твердых и жидких фаз, которые очень сдабо
расширяются, при нагревании теплота расширения невелика и
изменение объема с температурой мало, а потому, как видно из
формулы (2,10"), разность между обеими теплоемкостями
незначительна. Для газов, как известно, молярные теплоемкости Ср
и Cv отличаются на R=8,313 дж (около 2 кал), т. е. разность
сравнительно велика.
Опыт показывает, что теплоемкость вообще зависит от
температуры, однако при обычных температурах эта зависимоств
выражена сравнительно слабо, благодаря чему иногда
изменениями пренебрегают, рассматривая, например, небольшие
интервалы температур, далеких от абсолютного нуля. При очень
низких температурах теплоемкость сильно изменяется с
температурой. Так, в области нескольких десятков градусов от
абсолютного нуля она возрастает пропорционально кубу
абсолютной температуры. Объяснение этой закономерности дает
квантовая статистика. Молярная теплоемкость Cv при обычных
температурах для атомарных газов постоянна и равна CV=3/2/?,
41 Г лава 2. Первое начало и его непосредственные применения
т. е. 12,471 дж1 моль-град, тогда как для газов из двухатомных
молекул она равна 5/2R, или 20,785 дж/моль-град. Эти
результаты согласуются с выводами кинетической теории газов и
связаны с числом молекулярных степеней свободы. При низких
температурах наблюдается отступление от этих значений,
которое объясняется в квантовой статистике.
Для простых веществ в твердом состоянии уже давно было
установлено эмпирическое правило Дюлонга и Пти, согласно
которому атомная (на 1 грамм-атом) теплоемкость Ср всех
таких веществ одна и та же и близка к
Ср = 6,4 кал/г-апгом- град = 26,7 дж/г-атом-град.
Общая теория теплоемкостей рассматривается в
статистической физике. В обычной термодинамике теплоемкость
является только опытной величиной.
§ 4, ИДЕАЛЬНЫЙ ГАЗ. ЭФФЕКТ ДЖОУЛЯ — ТОМСОНА
Идеальным газом называется газ, состояние которого вполне
точно определяется уравнением Клапейрона:
pV = RT.
Можно дать другое определение идеального газа, если
обратить внимание на опытный факт, впервые установленный
Джоулем и состоящий в том, что при расширении газов в пустоту
температура их изменяется столь незначительно, что можно
считать ее Почти неизменной. Опыт с расширением газа
проводился с помощью прибора, состоящего из двух сосудов,
соединенных трубкой с краном и помещенных в ящик,
наполненный хорошим термоизолятором (рис.5). В этих условиях можно
было считать, что система была практически изолирована от
внешней среды (по крайней мере на время опыта), т. е.
помещалась в адиабатической оболочке. Вначале в сосуде /
находился газ при давлении р и температуре Г, сосуд // был
практически пустым (почти полный
вакуум). При открывании крана газ
переходит из сосуда / в //, расширяясь
фактически в пустоту; при этом сна-
/—*\ ??/^ ^^ чала Ра^ота расширения равна нулю,
[ ИД * I а затем п0 меРе наполнения сосуда//
V y^v у работа сжатия вошедшего газа точно
^ ' ^—*/ компенсировалась работой
расширения газа, выходящего из первого
сосуда во второй сосуд, следовательно,
в этом процессе газ в итоге не совер-
Рис. 5. шал работы. Опыт показал, что в ре-
§ 4. Идеальный газ. Эффект Джоуля—Томсона 45
зультате температура газа оставалась почти неизменной, т. с.
изменялась очень незначительно.
Рассмотрим процесс в опыте Джоуля, применяя закон
сохранения энергии. Уравнение первого начала
AQ = Ш + ДИ7
упрощается, так как при адиабатическом процессе AQ = 0. Но
газ, расширяясь, не производил работы, т. е. AW—0.
Следовательно, в данном процессе Д£/=0, т. е. внутренняя энергия газа
постоянна: £/=const. Но мы знаем, что внутренняя энергия есть
функция параметров состояния, в данном случае функция от V
и Г; отсюда полный дифференциал есть
Опыт показал, что изменение температуры было ничтожно
мало при расширении; иными словами, допустимо принять, что
dr=0, так как f = const. Тогда из выражения дляdUполучаем:
№ dV=0.
\dV }т
Здесь dV=£0, так как газ расширился, следовательно,
Это значит, что внутренняя энергия газа не зависит от объема
(закон Джоуля). Газ, в точности следующий этому закону,
называется идеальным газом. С точки зрения молекулярной
теории молекулы такого газа, обладая кинетической Энергией
теплового движения, практически не взаимодействуют друг
с другом: разлетаясь при расширении газа друг от друга, они
не совершают работы против сил сцепления, и общая их
энергия остается неизменной, раз температура остается постоянной.
Одновременно такой газ следует уравнению Клапейрона,
который хорошо обоснован в кинетической теории газов с помощью
известного основного уравнения этой теории.
Заметим, что реальные газы строго не следуют уравнению
Клапейрона, хотя отступления очень малы при малых
плотностях и при повышенных температурах; найдено также, что для
реальных газов имеются небольшие отступления от закона
Джоуля. Обнаружено, что при сильном расширении некоторых
газов с увеличением степени расширения наблюдается сперва
растущее понижение температуры, а затем с некоторой
степени расширения начинается повышение температуры. Точка
поворота называется температурой инверсии. Для
водорода замечено только нагревание при расширении. Эти
46
Г л а в а 2. Первое начало и его непосредственные применения
данные указывают на то, что между молекулами реальных газов
действуют как силы притяжения, так и силы отталкивания.
Вследствие этого внутренняя энергия реальных газов зависит
от температуры и от объема газа. Эти явления получили
практическое использование в технике сжижения газов, и они носят
название эффекта Джоуля — Томсона. Опыты с этим
эффектом проводились пропусканием потока газа через
плотную пористую пробку (рис. 6). При заметном перепаде
давления &р = р2—Р\ можно было заметить и измерить изменение
температуры газа позади пробки. Обсуждение результатов таких
опытов будет дано в последней главе, где будет рассмотрена
термодинамика неравновесных процессов. Здесь мы пока дадим
оценку эффекта Джоуля — Томсона с помощью данных
термостатики. Величину эффекта Джоуля — Томсона можно оценить
изменением температуры АГС с изменением объема kVc при
расширении газа или отношением——
имеем при конечных АГС и AVC-
Из уравнения (2,11)
(■
дУ)
dTJv
(2,12)
Значение не равной нулю производной!——)
можно найти из
соотношения (2,10'):
откуда
\dVJT~
ср — C
— р.
Тогда из (2,12) находим:
ДГс _ _р Ср — Су
We "" Су п I dV
(2 13)
Из опыта известно, что CP>CV, а также что для газов
коэффициент теплового расширения всегда положителен, т. е.
(•$¥-) > 0. Следовательно, в последней формуле вычитаемое
дТ ) всегда больше нуля, так же
как и -£- >0. Итак, знак эф-
дТ
Г
U
Щ
л
фекта зависит от разности
Р
Су
рис.
§ 5. Применение I начала к простейшим процессам в идеальном газе 4V
тогда как АУс>0, т. е. АГс может быть больше или меньше
нуля, т. е. при расширении газа может быть его охлаждение,
но возможно и нагревание. Однако обычно указанная разность
меньше нуля. Для идеального газа, когда pV—RTy имеем
( ) =— и по известному из общего курса уравнению
Р. Майера Ср—CV=R из соотношения (2,13) следует, что
= о, т. е. эффект Джоуля — Томсона исчезает. Следова-
тельно, при расширении идеального газа в пустоту температура
его неизменна.
В главе 4 будет дана другая оценка величины эффекта
Джоуля — Томсона.
§ 5. ПРИМЕНЕНИЕ ПЕРВОГО НАЧАЛА К ПРОСТЕЙШИМ ПРОЦЕССАМ
В ИДЕАЛЬНОМ ГАЗЕ
Процессы, протекающие в идеальном газе с постоянной
теплоемкостью, называют простейшими процессами. Из них
четыре процесса являются частными случаями рассмотренных
выше для всякой системы.
1. Изохорический процесс. В этом случае V = const.
Газ не совершает никакой внешней работы. Вся полученная
теплота идет на увеличение кинетической энергии молекул,
соответственно чему возрастает внутренняя энергия газа. Будем
вести расчеты на 1 моль газа.
Тогда
dQ = dU = CvdT.
Мы получили выражение для приращения внутренней энергии
идеального газа.
Затраченная теплота равна:
На диаграмме (/?, V) изохорический процесс изображается изо-
хорой, имеющей вид прямой, параллельной оси давлений.
2. Изобарический процесс. Здесь р = const.
Уравнение первого начала напишем в виде:
dQ = dU + Р dV = С v dT + p dV = Cp dT.
Из уравнения состояния для моля газа находим при постоянном
давлении
pdV = RdT.
Подставляем это выражение в предыдущее равенство и
находим:
48
Глава 2. Первое начало и его непосредственные применения
откуда после сокращения dT получаем известную формулу
Роберта Майера:
Cp = Cv + R. (2,14)
Как известно из общего курса, постоянная R имеет смысл
работы, совершаемой молем идеального газа в изобарическом
процессе при нагревании на 1 градус. Легко видеть, что
Q = CP(T2-T1)
С молекулярно-кинетической точки зрения при
изобарическом расширении идеального газа часть тепла идет на
увеличение скорости движения молекул и некоторая часть — на
внешнюю работу молекул по преодолению внешнего давления.
Согласно (2,8) полученная теплота может быть выражена через
приращение энтальпии:
Q = H%-HV (2,14')
Изменение состояния изображается изобарой (рис. 7).
Работа на этом графике равна площади прямоугольника
высотой рис основанием (V2—^i).
3. Изотерм ический процесс: T=const. Поэтому из
dQ = CvdT + pdV
находим при dT=0:
dQ dV = dW.
Следовательно, в этом случае вся полученная теплота
полностью превращается в работу:
Q=W.
Применяя уравнение Клапейрона, исключаем р из
выражения для dQ и интегрируем полученное выражение в пределах
от V\ до V2. Тогда
1-ТГ-. (2.16)
Мы видим, что работа газа пропор-
Изобара циональна абсолютной температуре и
логарифму степени расширения —— .
Молекулярно-кинетическое
объяснение работы в изотермическом процессе
сводится к следующему. Представим
~~ \/ у себе газ в цилиндре с поршнем, который
2 может скользить без трения вдоль сте-
Рис. 7. нок. Нагрузим поршень мелкой дробью.
§ 5. Применение I начала к простейшим процессам в идеальном газе 49
Если понемногу ссыпать дробь с поршня, то газ будет
совершать работу, расширяясь и поднимая оставшийся груз (рис.8).
Эта работа совершается при ударах молекул газа о поршень.
Молекулы, подлетающие к поршню, имеют некоторую
кинетическую энергию, в результате ударов о поршень часть их энергии
передается поршню, за счет чего и совершается работа
поднятия груза. Благодаря этому кинетическая энергия отскочивших
от поршня молекул становится меньше и температура газа
должна понизиться. Для осуществления изотермического
расширения газа необходимо восполнять убыль внутренней
кинетической энергии газа подводом тепла Q, которое как раз равно
совершаемой работе, так чтобы температура оставалась
неизменной.
Теплоемкость, как при всех изотермических процессах, равна
бесконечности.
На диаграмме (/?, V) изотермический процесс изображается
изотермой, представляющей собой равностороннюю гиперболу,
уравнение которой:
pV = const.
4. Адиабатный процесс.В этом случае dQ=0, и
уравнение первого начала имеет вид:
0. (2,16)
Отсюда можно получить соотношение между р и V для
адиабатного процесса. Из уравнения Клапейрона следует при
переменных /?, V и Т:
Исключаем dT из уравнения (2,16) с помощью этого
соотношения и получаем:
— Срр dV = CVV dp, (2,17)
если учесть уравнение Р. Майера. В соотношении (2,17)
переменные легко разделяются, и тогда
где обозначено
Су
Интегрируем уравнение (2,18) в пределах от V\ до V2 и от
Pi до /?2.
Тогда
"■(■£■)*-■"*■
3 Заказ № 2479
50
Г лава 2. Первое начало и его непосредственные применения
откуда
или в общем виде
рУ% = const.
(2,19)
(2,190
Это уравнение называют уравнением Пуассона. Графически
оно изображается неравносторонней гиперболой, называемой
в данном случае адиабатой. Отношение х, называемое
коэффициентом Пуассона, всегда больше единицы. Отсюда видно,
что адиабата идет несколько круче изотермы (рис. 9).
Легко найти, что при адиабатном расширении температура
газа понижается, а при адиабатном сжатии должна повышаться.
В этом, во-первых, можно убедиться, исключая р\ и р2 из
формулы с помощью двухкратного применения уравнения
Клапейрона. Тогда
(2,20)
Так как показатель степени (х— 1) положителен, то мы видим,
что при увеличении объема температура понижается и обратно.
Адиабата идет круче изотермы потому, что в адиабатном
процессе давление газа при расширении уменьшается вследствие
увеличения объема и, кроме того, благодаря охлаждению.
Далее, из уравнения (2,16) следует соотношение
-CvdT, (2,21)
которое означает, что при адиабатном расширении внешняя
положительная работа газа совершается за счет убыли
внутренней энергии. Наоборот, при адиабатном сжатии совершенная
над газом отрицательная работа (dV<0) переходит в запас
Рис. 8.
Рис. 9.
§ 5. Применение I начала к простейшим процессам в идеальном газе 51
внутренней энергии газа. Легко видеть из (2,21), что работа
расширяющегося газа равна: W=CV(T{—Г2).
Воспользовавшись уравнением Р. Майера, находим другое выражение:
W = R Ti~T* . (2,22)
% 1
С молекулярно-кинетической точки зрения изменение
температуры при адиабатном процессе происходит вследствие того,
что когда газ совершает работу, не получая теплоты извне, то
молекулы, как мы это видели в предыдущем примере, после
ударов о поршень отскакивают от него с меньшей кинетической
энергией, что и приводит к общему понижению температуры
газа. Наоборот, при адиабатном сжатии газа он должен
нагреваться, так как молекулам сообщается некоторая добавочная
энергия за счет работы сжатия в условиях, когда отсутствует
теплоотвод во внешнюю среду.
Заметим еще, что теплоемкость в адиабатном процессе равна
нулю. В формулу для работы входит теплоемкость при
постоянном объеме, так как работа получается за счет убыли
внутренней энергии, которая выражается через теплоемкость при
постоянном объеме.
/->
Отношение теплоемкостей х = —£-, т. е. коэффициент Пуас-
Cv
сона, неоднократно измерялось на опыте для разных газов
многими методами, на которых мы здесь не останавливаемся.
Отметим, что значение х зависит от природы газа. Для
одноатомных газов х=1,67, для-двухатомных газов х близко к 1,41 и для
ряда газов, состоящих из более сложных молекул, х^ 1,33, хотя
наблюдаются и меньшие значения для некоторых газов и паров.
Теория теплоемкостей газов, основанная на кинетической
теории, дает значения х для идеальных газов, близкие к опытным
величинам.
5. Политропический процесс. Рассмотрим
изменение состояния идеального газа по уравнению, аналогичному
уравнению Пуассона, т. е.
pVn = const, (2,23)
но при этом п — произвольное, в каждом частном случае
постоянное число. Такой процесс называется политропическим.
На диаграмме (/?, V) политропический процесс в общем
случае изображается некоторой гиперболой, уравнение которой
(2,23) называется уравнением политропы. Можно легко
показать, что политропическйй процесс включает в себя все
рассмотренные ранее простейшие процессы в идеальных газах как
частные случаи этого процесса. В самом деле;
3*
52 Г лава 2. Первое начало и его непосредственные применения
1. Возводя выражение (2,23) в конечную степень — , имеем:
п
pn.V = const.
В пределе при п—*оо находим:
V = const.
Мы получили уравнение изохоры. Следовательно, при изохори-
ческом процессе п = оо.
2. Полагая в (2,23) показатель п = 0, получаем:
р = const,
что представляет собой уравнение изобары.
3. Взяв /1=1, находим из (2,23):
pV = const,
т. е. уравнение изотермы.
>"»
4. Положив п = х =—£-, находим:
C
pVx = const,
что соответствует адиабате.
Дальнейший анализ уравнения (2,23) показывает, что
политропический процесс, описываемый этим уравнением, является
процессом изменения состояния идеального газа при постоянной
теплоемкости, которая в отдельных случаях может принимать
любые значения от —оо до +оо.
Внешняя работа газа в политропическом процессе легко
может быть вычислена, если п задано. Тогда если в начальном
состоянии объем был V\ и давление ри то
pVn = ргУх = const,
Piv%
откуда р = ——.
Поэтому
v.
dV = Р;
fpdV = PlV? f
ИЛИ
w=s PiYi-PtV&ir „ftKi-^.j; r,-r, (224)
Л— 1 Л— 1 Л~- 1
Для адиабатного процесса п=к и тогда (2Э24) переходит
в прежнее выражение работы в атом процессе (2,22),
§ 6. Значение адиабатных процессов в газах 53
Выражение для затраченной теплоты в политропическом
процессе находим, применяя уравнение первого начала в
интегральном виде:
Вводя выражение для R по уравнению Р. Майера, получаем:
Q= Cvn-Cp {T2-Tl) = CK(T2-Tl),
где Сп —теплоемкость в политропическом процессе:
Cvti — C
С "
(2>25>
Отсюда видно, что всякий политропический процесс при
постоянном п характеризуется постоянной теплоемкостью, поскольку
Ср и Су постоянны. В адиабатном процессе теплоемкость
равна 0, и тогда из уравнения (2,25) следует, что
п = х =-££-.
Cv
Кстати, формулу (2,25) можно представить в виде
Г Г п — х
откуда при разных п получаются теплоемкости всех простейших
процессов. Очевидно, при 1<п<х величина Стс<0.
Уравнение политропического процесса, происходящего при
постоянной теплоемкости, относится не только к идеальному
газу, но может быть применено ко всякой другой однородной
системе.
§ 6. ЗНАЧЕНИЕ АДИАБАТНЫХ ПРОЦЕССОВ В ГАЗАХ.
РАСПРОСТРАНЕНИЕ ЗВУКА В ГАЗЕ
Мы не будем здесь касаться применений различных
рассмотренных простейших процессов в газах и остановимся только
на значении адиабатного процесса. Газы обладают плохой
теплопроводностью, благодаря чему в них теплообмен с внешней
средой всегда затруднен при отсутствии конвекции. Вследствие
этого во многих случаях изменения состояния газа процесс близко
соответствует адиабатному процессу, чему отвечает политропа
с показателем я, близким к х. Например, процессы очень
быстрых расширений и сжатий газа можно практически считать
адиабатными.
Примерами являются сжатие газовой смеси в дизелях, когда
температура резко повышается при быстром сжатии, а также
54 Г лава 2. Первое начало и его непосредственные применения
сильное охлаждение газа при расширении в машинах для
ожижения, происходящее отчасти вследствие адиабатности
процесса. В камере Вильсона при резком толчке поршня и
увеличении объема происходит сильное охлаждение насыщенного
пара вследствие того, что за короткий промежуток времени
теплообмен между паром и стенками камеры произойти не
успевает. В свободной атмосфере большое значение имеют
адиабатные процессы, например, когда теплый воздух поднимается от
земли в верхние слои атмосферы, где он быстро расширяется,
причем большие массы его не успевают обмениваться теплом
с окружающими слоями.
Наконец, распространение звуковых волн в газе
представляет собой чередование быстрых сгущений и разрежений
воздуха, где благодаря очень коротким промежуткам времени также
сильно затрудняется обмен теплом и процессы сжатия и
расширения воздушных слоев являются также адиабатными. В этом
можно убедиться, вычисляя скорость распространения звука
в газе.
Из теории колебаний известно, что скорость распространения
волн в упругой среде пропорциональна корню квадратному из
отношения коэффициента упругости е к величине плотности
среды р, т. е.
У?-
р
В газах коэффициент упругости е представляет собой
давление газа р. В гидродинамике доказывается, что скорость
распространения волн в жидкое^ или газе тем больше, чем резче
меняется давление с изменением плотности вещества, т. е.
(2,26)
Для вычисления скорости звука в воздухе Ньютон (1700 г.)
допускал, что процессы сжатия и разрежения при
распространении звука являются изотермическими процессами. Применяя
в этом случае закон Бойля — Мариотта для изотермического
процесса
pV = const,
находим:
так как плотность р обратно пропорциональна объему; здесь
К—константа. Из этой формулы
dp __ к __ £_
§ 6. Значение адиабатных процессов в газах 55
Подставляя в (2,26), находим:
и = Y~ • <2>27)
Эта формула была получена Ньютоном. Найдем скорость звука
в воздухе при нормальных условиях. Положив
р0 = 101 325 н/м\
ро= 1,293 яг/ж3,
получаем из (2,27):
Wo=l/^L = 28O м/сао.
' Ро
Опыт дает в условиях, близких к нормальным
и0 = 331,4 м/сек.
Расхождение очевидно, и это заставляет считать формулу
Ньютона неправильной. Для других газов также заметны
значительные расхождения между формулой Ньютона и опытом. В
поисках причин расхождения Лаплас в 1800 г. предположил, что
процессы сжатия и расширения газов в звуковых волнах
являются адиабатными, так как каждый слой газа не успевает
обмениваться теплом с соседними слоями вследствие частых
колебаний плотности и благодаря плохой теплопроводности.
Допущение Лапласа несколько позднее было проверено Пуассоном
(1808 г.). Предположим, что в идеальном газе распространение
звуковых волн состоит из чередующихся адиабатных процессов.
Тогда справедливо уравнение Пуассона:
рУх = const,
или
Р = К?\
Отсюда находим:
Подставляя в формулу (2,26), находим:
(2,28)
Это выражение отличается от (2,27) множителемУк. Так как
для воздуха к= 1,403, то, применяя формулу Лапласа (2,28),
находим для нормальных условий^
и0
= 1/1,403 • jAg- = 1,18-280 = 331,6 м/сек.
66 Г лава 2. Первое начало и его непосредственные применения
Этот результат находится в очень хорошем согласии с
опытом и заставляет считать, что допущение Лапласа является
справедливым. Формула (2,28) может быть использована для
нахождения коэффициента Пуассона х по скорости звука в газе.
Это один из наиболее точных способов определения х в области
обычных температур.
Применяя формулу объединенного газового закона в виде
РРо
Ро (1 + «О
можно с помощью (2,28) найти зависимость скорости звука от
температуры:
где
Л~" * Ро
§ 7. ПРИЛОЖЕНИЕ ПЕРВОГО НАЧАЛА ТЕРМОДИНАМИКИ
К ЭЛЕКТРИЧЕСКИМ И МАГНИТНЫМ ЯВЛЕНИЯМ
В своей работе «О сохранении силы» Гельмгольц
рассматривает многочисленные примеры применения первого начала
термодинамики и специально останавливается на приложениях
к электрическим и магнитным явлениям. При расчете джоулеваг
тепла в электрической цепи, в явлениях термоэлектричества,
электромагнитной индукции и в других процессах приходится
учитывать закон сохранения энергии. Когда в цепи с
электродвижущей силой Е течет ток /, то за время dx происходит
изменение внутренней энергии
dU = El dx,
часть которого представляет собой выделяющееся количество
джоулева тепла
dQ = PR dx (дж),
и часть идет на изменение запаса энергии магнитного поля,
окружающего проводник dW. Тогда согласно первому началу
термодинамики
dU = dQ + dW9
или
El dx = PR dx + dW.
§ 7. Приложение I начала к электрическим и магнитным явлениям 57
Известно, что при движении проводника с током в
магнитном поле или при движении магнита близ контура с током
совершается работа
Поэтому
Отсюда получаем:
и тогда
Величина
представляет собой э. д. с. индукционного тока, возникающего
в проводнике независимо от &. Даже при отсутствии последней
мы наблюдаем явление самоиндукции, и тогда <ft есть э. д. с.
тока самоиндукции. Знак минус в выражении для£/ следует из
закона сохранения энергии. Мы видим, что при убыли
магнитного потока (^Ф<0) э. д. с. тока индукции направлена так же,
как и э. д. с. источника. Наоборот, если магнитный поток растет
(а!Ф>0), то э. д. с. индукции направлена обратно по отношению
к э. д. с. источника тока. Это соответствует известному правилу
Ленца. Уменьшение магнитного потока, например при удалении
магнита от контура с током, вызывает возникновение
индукционного тока, магнитное поле которого препятствует движению
магнита, т. е. притягивает его. При приближении магнита в контуре
возникает индукционный ток, по направлению обратный
основному, и он тоже препятствует производимому движению, т. е.
магнитное поле этого тока отталкивает приближающийся
магнит. Энергия возникающего индукционного тока получается за
счет энергии движения магнита.
§ 8. ПРИМЕНЕНИЕ ПЕРВОГО НАЧАЛА К ХИМИЧЕСКИМ ПРОЦЕССАМ.
ЗАКОН ГЕССА
Известно, что при всякой химической реакции выделяется
или поглощается некоторое количество теплоты. Для
выяснения строения молекул в химическом соединении и природы
химической связи атомов в молекулах необходимо знать тепло-
58 Г лава 2. Первое начало и его непосредственные применения
вой эффект реакции, т. е. количество теплоты, выделяемой или
поглощаемой при получении одного моля данного химического
соединения. Реакции, которые при данных условиях их течения
идут с выделением теплоты, называются
экзотермическими, а те, которые идут с поглощением теплоты извне,
называются эндотермическими. В термохимии, изучающей
теплоты образования различных соединений, принято считать
выделенную теплоту положительной, а полученную теплоту —
отрицательной. Следовательно, в термохимии знаки для теплоты
обратны тем, которые приняты в термодинамике. Поэтому здесь
мы будет обозначать теплоту символом Q, так что Q =—Q.
Выделение или поглощение теплоты при образовании
какого-либо химического соединения вызывается изменением запаса
внутренней энергии в самих молекулах (или атомах),
зависящего от перестройки структуры электронных оболочек их
атомов. Каждому молю химически однородного вещества
соответствует определенный запас внутренней энергии, который
меняется в ходе самой химической реакции. В термохимии
приняты те же символические изображения реакций, что и
в общей химии, однако в соответствующих химических
уравнениях принято изображать не взаимодействующие массы, а
отвечающие им запасы внутренней энергии для данного
соединения и обозначать последние в квадратных скобках. По закону
сохранения энергии термохимические уравнения означают
баланс внутренней энергии до и после реакции, причем в правой
части уравнений указывается телловой эффект реакции с
соответствующим знаком. Приведем несколько примеров подобного
вида уравнений:
[Н2] + ~- [О2] = [Н2О] + 68,32 ккал (286 кдж)у
[С] + [О2] = [СО2] + 94,03 ккал (394 кдж),
2 [С] + [Н2] = [С2Н2] — 53,9 ккал (225,8 кдж),
-у [Н2] + -j- [J2] = [HJ] — 5,9 ккал (24,7 кдж).
Две первые реакции являются экзотермическими, а
остальные две эндотермическими. Коэффициенты перед скобками
означают число молей, участвующих в реакции.
Среди многочисленных химических реакций различают
реакции, идущие при постоянном объеме, и реакции при постоянном
давлении. Реакции первого типа практически протекают при
участии твердых и жидких тел, когда можно пренебречь
расширением образующегося вещества и считать V=const. Реакции,
осуществляемые для нахождения теплот горения в калориметри-
§ 8. Применение I начала к химическим процессам 59
ческой бомбе, тоже являются примером процессов этого типа.
При взаимодействии газов, которые могут свободно
расширяться, объем их меняется и возможно осуществить
превращение при постоянном давлении.
Применим уравнение первого начала термодинамики к
химическим реакциям при постоянном объеме и при постоянном
давлении. Для этого прежде всего в соответствии с переменой
знака при Q преобразуем это уравнение. Ранее мы имели:
i
С учетом, что Q = —Q, получаем:
^ (2,29)
Рассмотрим два вида химических превращений.
1. V=const. Тогда уравнение (2,29) примет вид:
Отсюда следует, что если U\> £/2, то тепловой эффект
положителен: Qv>0, что отвечает экзотермической реакции. Если
£Л<£^2, то тепловой эффект отрицателен: Qv<0, и это
соответствует эндотермической реакции. В первом случае внутренняя
энергия первоначально взятых веществ больше, чем внутренняя
энергия образовавшегося соединения, избыток ее Qv выделяется
при реакции. Обратные соотношения имеются во втором случае.
Таким образом, при постоянном объеме мы имеем вообще
2. р = const. В этом случае уравнение (2,29) имеет вид:
Отсюда
Qp = (Vi + рУг) - (U2 + PV2) = Нг - Я2.
Следовательно, в данном процессе тепловой эффект равен
изменению энтальпии системы до реакции и после реакции. Если
Я1>Я2, то QP>0y что отвечает экзотермической реакции, если
#1<#2, то Qp<0, что соответствует эндотермическому
процессу. Вообще,
60 Г лава 2. Первое начало и его непосредственные применения
Отсюда видно, что тепловой эффект реакции при р = const
равен изменению энтальпии (а не внутренней энергии, как в
первом случае).
При всех химических превращениях выполняется закон,
открытый петербургским академиком Гессом в 1840 г. Этот закон
сводится к утверждению, что теплота химической реакции не
зависит от формы пути химического превращения, а зависит
лишь от начального и конечного состояний системы, иначе:
тепловой эффект при химической реакции не зависит от того,
проведено ли данное превращение в одну или в несколько стадий.
Так, в одном из рассмотренных примеров приведена
реакция образования углекислоты при окислении углерода.
Тепловой эффект этой реакции +394 кдж/моль не зависит от того,
получена ли углекислота непосредственно из С и Ог или в
каком-либо ином химическом превращении, например при
взаимодействии СаСОз с НС1 и т. д. Во всех случаях теплота
образования СОг составляет одну и ту же величину.
Закон Гесса является непосредственным следствием первого
начала термодинамики. Действительно, применяя первое
начало к химическим реакциям, мы видели, что теплота реакции
связана с изменением внутренней энергии системы (при
постоянном объеме). Но ранее было отмечено, что внутренняя энергия
есть функция состояния и изменение ее не зависит от формы
пути. Поэтому теплота образования данного химического
соединения не зависит от того процесса, в котором произошло
образование данного химического соединения, а только от
внутренней энергии начального и конечного (после реакции) состояний
системы. При постоянном давлении теплота реакции
определяется изменением другой функции состояния — энтальпии. Это
изменение также не зависит от формы пути, а лишь от
энтальпии начального и конечного состояний. Поэтому и в данном
случае применим тот же закон Гесса о независимости теплоты
реакции от вида пути превращения.
Закон Гесса является основным положением всей
термохимии. Важное его значение состоит в том, что с его помощью
возможно рассчитать теплоту образования химического
соединения, не производя непосредственных измерений теплоты, когда
эти измерения являются сложными и неосуществимыми. В
таких случаях можно использовать данные опытов с другими
реакциями. Не затрагивая обширного опытного материала по
теплоте образования, ограничимся немногими примерами.
1. Теплота образования окиси углерода.
Теплоту сгорания углерода с образованием окиси углерода в
реакции
§ 8. Применение I начала к химическим процессам 61
непосредственно на опыте измерить не удается. Однако из
опытов известны теплота образования углекислого газа и теплота
сгорания окиси углерода по реакциям:
[С]+ [О2] = [СО2] + 394 кдж/моль,
[СО] + -i- [О2] = [СО2] + 281,5 кдж/моль.
Эти данные позволяют рассчитать теплоту образования СО
из элементов, пользуясь законом Гесса. Можно представить
себе образование СОг двумя путями:
I. С + О = СО II. С + О2 = СО2
со + о = со2
В одном случае сперва углерод окисляется до СО, а затем СО
окисляется далее до СОг; другой путь состоит в
непосредственном образовании СО2 из углерода и кислорода. Теплота
образования СО2 в процессе II известна и равна +394 кдж/моль,
известна также теплота реакции с образованием СОг во второй
стадии процесса I, она составляет 281,5 кдж/моль. Но согласно
закону Гесса теплота образования не зависит от формы пути.
Следовательно, в I и II процессах она одна и та же и равна
394 кдж/моль. Отсюда из разности находим теплоту реакции
для 1-й стадии в I процессе:
Q = 394 — 281,5 = 112,5 кдж/моль,
т. е. искомую теплоту образования СО из элементов.
2. Теплота превращения графита в алмаз. На
опыте непосредственно невозможно измерить теплоту перехода
графита в алмаз, но из калориметрических измерений известна
теплота горения обеих модификаций углерода:
[С] (графит) + [О2] = [СО2] + 394 кдж/моль,
[С] (алмаз) + [О2] = [СО2] + 395,6 кдж/моль.
Применяя закон Гесса, легко находим искомую теплоту
реакции [С] (графит) = [С] (алмаз)+"QP.
Очевидно, Qp = 394 — 395,6=— 1,6 кдж/моль.
Это ясно из сравнения двух путей перехода:
I. С (графит)—>С (алмаз) II. С (графит) + Оа —>СО2
СО2->С (алмаз)+ О2
В первой стадии процесса II выделяется теплота 394 кдж/моль,
во второй стадии этого процесса, которая является обращением
второй из написанных ранее реакций, теплота должна
поглощаться и она равна — 395,6 кдж/моль. Следовательно, по за-
62 Г лава 2. Первое начало и его непосредственные применения
кону Гесса теплота превращения графита в алмаз является
отрицательной и равной—1,6 кдж/моль. Эта величина
получается просто как алгебраическая сумма обоих уравнений
реакции.
Заметим, что наблюдаемые иногда небольшие расхождения
в термохимических расчетах с данными опыта объясняются не
отступлениями от закона Гесса, а недостаточной точностью
опыта. Закон Гесса, являясь следствием первого начала
термодинамики, т. е. закона сохранения энергии, представляет собой
точный закон.
Задачи
1. Вычислить работу, совершаемую при обратимом изотермическом
расширении I моля газа, следующего уравнению Ван-дер-Ваальса.
2. Вывести уравнение Р. Майера для идеального газа из общего
выражения (2,107) для теплоемкостей.
3. Доказать, что для идеального газа скрытая изотермическая теплота
расширения равна l—Q=RT\n ——.
4. Как изменится температура воздуха, если его адиабатически сжать от
давления в 1 атм до давления 100 атм, а начальная температура
была 27°С?
/ л f-f \
5. Доказать, что Ср =( ] , пользуясь общим выражением для
энтальпии.
6. В каких политропических процессах теплоемкость Сп является
отрицательной, когда она положительна и когда равна нулю?
Глава 3
ВТОРОЕ НАЧАЛО ТЕРМОДИНАМИКИ. ЭНТРОПИЯ И ЕЕ
СВОЙСТВА КАК ФУНКЦИИ СОСТОЯНИЯ
§ 1. ОБРАТИМЫЕ И НЕОБРАТИМЫЕ ПРОЦЕССЫ
Второе начало термодинамики, вкратце излагаемое в общем
курсе физики, как закон роста энтропии в изолированной
системе и как принцип рассеяния энергии является вполне
самостоятельным положением, которое не может быть выведено из
первого начала. В законе сохранения энергии не содержится
указания на направление процессов в изолированной системе,
тогда как из второго начала следует вывод о некоторой
направленности процессов в такой системе. Однако сначала
необходимо остановиться на разделении процессов в системе на
обратимые и необратимые процессы, так как
понимание сущности второго начала невозможно без их определения
и выяснения различия между ними.
Ранее было отмечено, что квазистатическими процессами
называются такие процессы, в которых на каждой их стадии
параметры состояния являются вполне определенными. Если
система находится в равновесии и параметры ее заданы, то
некоторое изменение последних уже выводит систему из
равновесия и ее параметры становятся переменными, различными
в отдельных частях системы (локальные параметры). Очевидно,
для того чтобы в новом, отличном от первого
состоянии-система могла быть охарактеризована новыми
макропараметрами, необходимо, чтобы изменения их были каждый раз как
можно менее заметными. Так, например, в простейшем случае,
когда происходит изменение давления и температуры тела, то
для осуществления квазистатического (равновесного) процесса
должны соблюдаться условия:
1) упругость тела должна бесконечно мало отличаться от
внешнего давления;
2) температура тела во все время изменения состояния
должна бесконечно мало отличаться от температуры источников
тепла.
Соответственно этим требованиям, если мы хотим произвести
равновесное расширение газа, мы должны последний поместить,
например, в цилиндр с поршнем, на котором находится груз,
64 Глава 3. Второе начало термодинамики. Энтропия и ее свойства
состоящий из маленьких дробинок или песчинок. Снимая
постепенно одну песчинку за другой, мы можем сколь угодно
увеличивать объем газа, так что в этом процессе допустимо считать,
что упругость газа каждый раз равна внешнему давлению. Если
необходимо нагреть жидкость, имеющую первоначальную
температуру, положим от 20 до 100° С, то мы вначале применяем
источник тепла с температурой 20,0001° С, который приводим
в соприкосновение с жидкостью; затем переходим к источнику
с температурой 20,0002° С и т. д. Каждый раз при этом жидкость
соприкасается с источником тепла, имеющим температуру, на
0,0001° С превышающую температуру жидкости. В этих
условиях допустимо считать, что процесс нагревания являлся
равновесным.
Описанные здесь равновесные процессы относятся к более
широкому классу процессов, называемых обратимыми в
отличие от необратимых процессов, примеры которых будут даны
ниже. Необходимо подчеркнуть, что это деление в
термодинамике имеет смысл лишь в применении к конечной
изолированной системе. Распространение этих понятий на неограниченную
или неизолированную систему недопустимо и может привести
к ошибкам в понимании второго начала.
Обратимыми процессами в изолированной системе
называются такие процессы, которые можно обернуть без каких-либо
изменений в свойствах этой системы. Пусть на диаграмме
состояний с координатами х, у, z такой процесс изображается
линией АВ (рис. 10) и изменение состояния происходит по
направлению А—>В. Тогда процесс, идущий по той же кривой
в направлении В—>А, называется обратным по отношению
к первому. Ясно, что в обратном процессе последовательность
состояний является в точности обратной к прямому процессу.
При заданных начальных и конечных условиях прямого процесса
обратный процесс характеризуется
обратными начальными и конечными
условиями. Обратимыми процессами
называются такие, при которых в
системе никаких изменений при
обращении процесса не происходит, т. е.
безразлично, идет ли процесс А—нВ или
В—>Л.
, Необратимые процессы представ-
X ляют собой такие изменения
состояния в изолированной системе, когда
при обращении процессов свойства
всей системы меняются. Иначе говоря,
в обратном необратимом процессе в
Риг. ю. изолированной системе протекают та-
§ 1. Обратимые и необратимые процессы 65
кие явления, которых не было в прямом процессе.
Следовательно, ход процессов А—>В и В—>-Л не безразличен для
системы в целом. Не следует думать, что по отношению к данному
телу системы необратимый процесс нельзя обернуть.
Действительно, если АВ — кривая прямого для данного тела процесса,
идущего в направлении А—*В, то принципиально всегда
возможно осуществить с выделенным телом системы в точности
обратную последовательность состояний, т. е. провести процесс
В—>А по той же кривой. Важно, что при этом в самой системе
придется совершить дополнительные изменения, которых не
было при прямом процессе. Таким образом, эти процессы
являются необратимыми именно по отношению к изолированной
системе, а не по отношению к отдельному телу системы, для
которого всегда можно осуществить прямое и обратное течение
процессов. По поводу необратимости Планк говорит, что «с
каждым необратимым процессом система делает некоторый такой
шаг вперед, следы которого ни при каких обстоятельствах не
могут быть уничтожены». Каждый необратимый процесс вносит
непоправимые изменения в систему.
К необратимым процессам относятся следующие
простейшие процессы в изолированной системе:
1. Теп л опр ово д н ость при конечной разности
температур. Если привести в соприкосновение горячее тело
с холодным, то начнется переход тепла от первого ко второму
и будет продолжаться, пока не произойдет выравнивания
температур. В начале процесса система характеризуется
следующими условиями: горячее тело имеет температуру Ти холодное
обладает температурой Г2. В конечном состоянии оба тела
имеют температуру Т и Ti>T>T2. Прямой процесс легко, как
говорят, «сам собой» протекает в системе и не требует
никаких условий,^кроме соприкосновения двух тел. Если попытаться
обернуть этот процесс, т. е. исходя из равной температуры
соприкасающихся тел Т достигнуть состояния, когда одно тело
стало бы горячим с температурой Ти а другое охладилось бы
до Гг, то можно убедиться, что в обратном процессе
потребуются дополнительные условия, которых не было в прямом, так
как сам собой обратный процесс не пойдет.
Из опыта мы знаем, что, сколько бы мы ни наблюдали двух
тел одинаковой температуры, приведенных в соприкосновение,
нам не удастся заметить, что сама по себе возникает заметная
разность температур, когда одно тело само нагреется, а другое
само охладится. Такой процесс не противоречил бы первому
началу, но мы его не наблюдаем. Таким образом, в
изолированной системе теплопроводность — необратимый процесс.
2. Движение с трением. При скольжении одного
твердого тела по поверхности другого или при взаимном скольжении
66 Г лав а 3. Второе начало термодинамики. Энтропия и ее свойства
слоев жидкости всегда возникает трение; такой процесс
является необратимым, так как в нем за счет механической
энергии движения совершается работа против сил трения. При этом
соприкасающиеся поверхности нагреваются. За счет энергии
тела, пошедшей на преодоление трения, выделилось некоторое
количество теплоты, которую необходимо сообщить
системе в обратном процессе, чтобы получить вновь механическую
энергию взаимного движения тел. И хотя полученную теплоту
вновь превратить в работу возможно, цо так как сам собой
обратный процесс не пойдет, то, чтобы его осуществить,
понадобятся специальные приспособления, т. е. дополнительные
условия, которых не било в прямом процессе, и поэтому,
следовательно, движение с трением в изолированной системе
представляет собой необратимый процесс.
3. Расширение газа при конечной разности
давлений. В опыте Джоуля и Томсона газ, первоначально
заключенный в сосуде, расширяется при открывании крана,
переходя или в «пустоту», или в пространство, где давление
значительно ниже, чем первоначальное давление газа. Такой
процесс следует считать необратимым, так как при обращении
процесса придется произвести такие манипуляции, которых не
было в прямом процессе. Газ сам собой перетекал из одного
сосуда в другой, и процесс прекратился после выравнивания
давлений. В обратном направлении процесс не пойдет, т. е. газ
сам собой не возвратится в первый сосуд с образованием
прежней разности давления. Хотя искусственно, с помощью
специальных устройств, можно обернуть этот процесс по отношению
к газу, но их применение является тем дополнительным
условием, которое приходится вводить в систему, тогда как при
прямом процессе оно отсутствовало. Следовательно, эффект
Джоуля — Томсона в изолированной системе также есть
необратимый процесс.
4. Диффузия. Этот процесс протекает в системе, где
имеется разность концентраций вещества в отдельных частях
системы. Если поверх раствора, находящегося в сосуде,
осторожно налить некоторое количество чистого растворителя, то
начнется самопроизвольный процесс диффузии,
продолжающийся до тех пор, пока не сравняются концентрации. В
начальном состоянии раствор имел концентрацию с0, тогда как в
растворителе было с = 0. В конце процесса получается некоторая
средняя общая концентрация. Этот процесс является
необратимым, так как при обращении придется применять такие
приспособления, которые отсутствовали в прямом процессе.
Действительно, самопроизвольное разделение смешавшихся веществ
не идет, хотя, как известно, искусственно можно осуществить
разделение, и это применяется во многих технологических про-
§ L Обратимые и необратимые процессы 67
цессах, например при разделении смесей, при разделении
изотопов, при выделении веществ из растворов, при перегонке и т. д.
Помимо указанных примеров, можно привести еще много
других примеров необратимых процессов. Сюда можно отнести
течение электрического тока в проводниках с сопротивлением,
распространение электромагнитных волн в поглощающей среде,
быстро текущие химические процессы (например, горение,
взрывы), а также явления радиоактивного распада. Разные
сложные процессы, представляющие собой комбинации
указанных процессов, также являются необратимыми.
К обратимым процессам относятся следующие.
1. Все чисто механические процессы, т. е. такие
механические движения, где полностью или практически
полностью отсутствует трение. К ним можно отнести, например,
незатухающие колебания маятника, движение идеальной
жидкости без трения, распространение звуковых волн без
поглощения в упругой среде, удары идеально упругих тел и многие
другие процессы. Во всех перечисленных случаях процесс легко
может протекать как в прямом, так и в обратном направлениях.
Из дифференциальных уравнений механики вполне точно
следует, что если даны начальные условия, то движение системы
однозначно определено. Если фиксировать какое-либо состояние
системы и обернуть координаты и скорости, то процесс потечет
в точности в обратном направлении, причем конечные условия
будут в точности обратными первоначальным условиям при
прямом процессе. При этом никаких изменений в окружающих
телах системы происходить не будет. Известно, например, что
при движении тела, брошенного вверх в пустоте, когда
начальная скорость равна Н-у0, то тело поднимается на высоту Л,
после чего протекает обратный процесс падения и тело падает
на землю с высоты Л, имея в конце скорость — v0. При этом
в окружающих телах никаких изменений происходить не будет.
Периодическое колебание маятника в пустоте представляет
собой процесс, где неопределенно долгое время протекают
периодически прямые и обратные процессы поднятия и опускания
маятника без всяких изменений в окружающих телах. Из этих
примеров видно, что в механике идеальных систем изучаются
только обратимые процессы, т. е. имеется симметричность
процессов прямого и обратного*.
* Эта симметрия следует из дифференциальных уравнений механики,
которые содержат вторые производные по времени, тогда как первые
производные отсутствуют. Следовательно, если f(t) есть решение такого уравнения, то
/ (—t) тоже является решением, так как при взятии второй производной
происходит повторное умножение на —1 и результат остается неизменным. Таким
же свойством обладают уравнения квантовой механики, откуда следует
обратимость по времени элементарных квантовых процессов.
68 Г лав а 3. Второе начало термодинамики. Энтропия и ее свойства
2. Незатухающие электромагнитные
колебания. В контуре, где сопротивление равно нулю, возникшие
электромагнитные колебания происходят неопределенно долгое
время и наблюдаются превращения энергии в прямых и
обратных колебаниях напряженности электрического и магнитного
полей. Очевидно, эти процессы являются обратимыми, так как
при смене фаз в окружающей среде никаких новых изменений
по мере хода процесса не происходит.
3. Распространение электромагнитных волн
в среде без погл ощени я. При поглощении энергии
электромагнитных волн происходит образование тепла. Если это
явление отсутствует, то процесс распространения волн является
обратимым, в чем легко убедиться из закона отражения волн от
гладкого почти непоглощающего зеркала. Распространение
отраженного луча непосредственно представляет собой обращение
прямого процесса.
4. Термодинамические квазистатические
процессы. При соблюдении указанных ранее условий для
квазистатических процессов, когда устранены неуравновешенное
расширение, диффузия и теплопроводность, а также отсутствует
трение, все такие равновесные термодинамические процессы
являются обратимыми. Действительно, при незначительном
изменении температуры, давления, объема, концентрации и т. д.
направление изменения не играет роли. Система в одинаковой
мере может испытывать изменения в прямом и в обратном
направлениях. Например, при соприкосновении с телом,
температура которого ничтожно отличается от температуры самого тела,
последнее получает и отдает столь малое количество тепла, что
явлением теплопроводности можно пренебречь, и потому процесс
без существенных изменений всей изолированной системы можно
вести в прямом и обратном направлениях.
Из рассмотренных примеров мы можем вывести заключение,
что все обратимые процессы являются идеальными, так как
во всех случаях при движении возникает трение, во всякой среде
происходит поглощение энергии электромагнитных волн, всегда
наблюдается явление теплопроводности и т. д. Поэтому также
и квазистатические процессы следует считать идеальными
обратимыми процессами. Кроме того, мы замечаем, что необратимые
процессы принадлежат к числу наиболее распространенных
реальных процессов окружающего нас мира. Всякий реальный
процесс в макромире необратим, и только, если можно в
отдельных случаях пренебречь трением, теплопроводностью и т. п.,
такой процесс будет близок к обратимому.
Глубокое различие между обратимыми и необратимыми
процессами, текущими* в изолированной системе, будет выяснено
в дальнейшем при анализе второго начала.
§ 2. Обратимая машина Карно
69
§ 2. ОБРАТИМАЯ МАШИНА КАРНО
Обратимая машина Карно представляет собой идеальную
схему цикла, и ее действие сводится к следующему. В цилиндре
с поршнем находится один моль идеального газа. Цилиндр и
поршень изготовлены из нетеплопроводного материала, тогда
как дно цилиндра выполнено из очень хорошего проводника
тепла. Поршень без трения может скользить вдоль стенок
цилиндра. С помощью этого устройства проведем цикл изменений
состояния газа в цилиндре, осуществляя обратимые процессы, и
найдем работу машины и коэффициент полезного действия
последней. Самыми выгодными процессами в отношении получения
работы являются изотермический и адиабатный процессы, так
как в первом из них вся сообщаемая теплота полностью
превращается в работу, а во втором работа совершается только за
счет убыли внутренней энергии газа. Пусть вначале газ имеет
температуру Ти объем и давление его У\ и р\. Поставим
цилиндр на плиту больших размеров, имеющую температуру Ти
которую условно назовем нагревателем или
источником теплоты, и постепенно, снимая нагрузку с поршня,
заставим газ обратимо расширяться при постоянной температуре
7*1 = const до объема V2. Процесс изображается изотермой (1,2)
на рис. 11. При этом теплота, полученная от нагревателя в
изотермическом процессе, равна:
Эта теплота полностью превращается в работу, величина
которой равна площади (/, Уи Уъ 2) на нашей диаграмме. После
достижения состояния 2
удалим нагреватель, приставим ко у
дну цилиндра пластину из
нетеплопроводного материала и,
уменьшая далее нагрузку на
поршень, подвергнем газ
адиабатному расширению. При
этом температура его
понизится до Г2, когда будет
достигнут объем V3. На этом
пути полученная системой
теплота равна нулю, а
совершенная газом положительная
работа равна, как было
показано ранее (стр. 50),
= CV(TX-T2).
Рис. 11.
70 Г л а в а 3. Второе начало термодинамики. Энтропия и ее свойства
На диаграмме работа изображается площадью (2, V2i VZi <?).
После этого удаляем нетеплопроводную пластину и ставим
цилиндр на поверхность тела больших размеров, имеющего
температуру Г2, равную температуре газа к этому моменту: это более
холодное тело условимся называть холодильником.
Теперь будем изотермически и обратимо сжимать газ, уменьшая
его объем от V3 до объема У4, величину которого рассчитаем
ниже. При этом газ будет отдавать холодильнику теплоту Q2,
которая как раз равна работе, совершенной над газом:
Величина работы изображается на диаграмме площадью (3, 4,
V*, Уз). После достижения объема V* удаляем холодильник и
вновь прикладываем ко дну цилиндра нетеплопроводную
пластину. Продолжаем сжимать газ, производя обратимый
адиабатный процесс. При этом температура газа будет повышаться,
и мы прекратим сжатие, когда газ вновь нагреется до
температуры 7Y Работа сжатия равна в этом случае
Она изображается на диаграмме площадью фигуры (Vu 1, 4,
V4). Если объем Vi выбран надлежащим образом, то газ,
приобретая температуру Гь примет вновь как раз первоначальный
объем Vi и перейдет в начальное состояние /, пройдя один
цикл операций.
Подсчитаем теперь работу газа за весь цикл, считая как и
ранее, работу самого газа положительной, а работу,
совершенную над газом, отрицательной:
1) работа газа при изотермическом расширении (1, 2):
2) работа газа при адиабатном расширении (2, 3)?
W^C^T.-T,);
3) работа над газом при изотермическом сжатии (3,4):
4) работа над газом-при адиабатном сжатии (4,1):
Алгебраическая сумма этих работ даст нам работу газа за
пройденный цикл. Находим:
§ 2. Обратимая машина Карно 71
Эта работа на диаграмме изображается площадью
криволинейной фигуры (1,2,3,4), ограниченной отрезками двух изотерм
и двух адиабат.
Определим теперь объем V*. Для этого воспользуемся
формулой, связывающей температуру с объемом в адиабатном
процессе (см. гл. 2, § 5). Для адиабаты (2,3) имеем:
Jj_ _
Tt ~
тогда как для адиабаты (4,1)
Отсюда следует:
Зная объемы Vu Vz и Уз, которые являются произвольными, мы
из этой пропорции определим У4 и получим адиабату,
приводящую газ к начальному состоянию, т. е. замыкающую весь цикл.
Из последней пропорции следует:
У* = У*
У* Уг #
Подставляя это выражение в формулу работы газа за цикл,
находим окончательно:
Работа W представляет собой полезную работу в нашей машине.
Отношение этой работы ко всей полученной теплоте за цикл
принято называть коэффициентом полезного
действия машины
W
В машине Карно количество теплоты Q, полученной от
нагревателя, равно:
Поэтому
т> = У-Чг-^—- = 1^-' (3,1)
72 Г лав а 3. Второе начало термодинамики. Энтропия и ее свойства
Из этого выражения вытекает прежде всего практическое
следствие, что даже для идеальной обратимой машины, где
используются самые выгодные с точки зрения получения
полезной работы изотермический и адиабатный процессы,
коэффициент полезного действия не достигает 100%. В пределе г\
обращается в единицу, когда температура холодильника равна
абсолютному нулю, но этот случай недостижим. Так как Т2ф0, то
для всех идеальных *машин Карно т]<1. Для практически
осуществимых машин коэффициент полезного действия при тех же
температурах 7\ и Т2 должен быть заведомо ниже, во-первых,
вследствие наличия необратимых процессов, при_ которых часть
полезной работы пропадает напрасно, и, во-вторых, из-за
несовершенства цикла, т. е. за счет того, что он, кроме самых
выгодных изотермических и адиабатных процессов, может
содержать и другие процессы. Таким образом, к. п. д. машины Карно
есть предел возможностей использования машины, работающей
между температурами 7\ и Т2.
Оценим теперь действие машины Карно в отношении тех
изменений, которые она вносит в систему при работе. За один
цикл некоторое количество тепла Qi было отнято у нагревателя
и другое количество тепла Q2 было передано холодильнику,
причем Q2<Qu очевидно, количество тепла Qi—Q2 перешло в
полезную работу газа за цикл. Поэтому к. п. д. машины Карно,
как и всякой другой машины, всегда может быть выражен
соотношением:
j^Qi^Sl. (3,2)
Эта формула является более общей, чем (3,1), которая выведена
только для обратимой машины Карно, для которой
и, следовательно,
J1=^=Qizl^l# (3>3)
1 1 Vl
Таким образом, результат работы машины Карно за один цикл
сводится к следующим двум изменениям:
1) переход теплоты Q2 от нагревателя к холодильнику;
2) получение полезной работы Qi—Q2, т. е. превращение
теплоты в механическую работу.
Заметим, что само работающее тело, т. е. газ, по завершении
цикла перешло в начальное состояние и, значит, внутренняя
энергия его не изменилась; поэтому из полученного от
нагревателя тепла Qi количество Qi — Q2 перешло в работу, а
остальное тепло Q2, отнятое у нагревателя, было отдано холодильнику.
Следовательно, газ в итоге не получил и не отдал теплоты, он
§ 2. Обратимая машина Карно
73
явился только посредствующим телом между нагревателем
и холодильником.
Рассмотренная машина Карно называется прямой машиной
в отличие от обратной, работающей по обратному циклу.
Отличие последней от первой состоит именно в обращении цикла
операций (но не в изменении направления хода поршня).
Рассмотрим кратко действие обратной обратимой машины Карно.
Цикл операций в ней можно разделить на следующие
обратимые процессы (рис. 12):
1) адиабатное расширение газа по адиабате (1,2) с
охлаждением газа от Ti до Т2 при изменении объема от Vi до V2\
2) изотермическое расширение по изотерме (2,3) при
постоянной температуре Т2 и с изменением объема от V2 до К3;
3) адиабатное сжатие по адиабате (3, 4), когда происходит
нагревание газа от Т2 до Т\ при уменьшении объема от У3
до У4;
4) изотермическое сжатие (4,1) при температуре Ti с
изменением объема от V* до первоначального Vi.
При изотермическом расширении газа (2,3) от холодного
тела при температуре Т2 отнимается теплота Q2, полностью
переходящая в работу; напротив, при изотермическом сжатии
по пути (4,1) теплота Qi передается к более нагретому телу
с температурой 7\ (конечно, Т\>Т2), и над газом совершается
внешняя работа. Очевидно, при работе такой машины за
цикл получается следующее:
1) работа сжатия, совершенная над газом, равная Qi — Q2
и изображаемая площадью, криволинейной фигуры (1,2,3,4),
превратилась в теплоту;
2) теплота Q2 перешла от холодного тела к более теплому.
Очевидно, количество теп-
Q, лоты Qi, переданное нагре-
7 т У тому телу, складывается из
теплоты Q2, отнятого у
холодного тела, и теплоты Q\—Q2,
полученной при работе сжатия.
Обратная машина не дает
полезной работы, так как
т4
аг т7 iq.
Рис. 12,
Рис. 13.
74 Г лав а 3. Второе начало термодинамики. Энтропия и ее свойства
совершенная за цикл работа больше работы самого газа, но сама
такая машина в принципе является полезной потому, что с ее
помощью можно отнимать у холодного тела теплоту и постепенно
вызвать значительное его охлаждение. Обратная машина Карно
является наиболее выгодной холодильной машиной. Мы не
останавливаемся на выводе ее коэффициента полезного действия,
так как очевидно, что он, как и для прямой машины, может
быть найден по формуле (3,1).
Заметим, что и в этом случае газ является посредствующим
телом и за один цикл переходит в начальное состояние; в итоге
внутренняя энергия его остается без изменения.
Представим себе, наконец, что между нагревателем и
холодильником работают две обратимые машины Карно, из которых
одна является прямой, а другая обратной (рис. 13). Очевидно,
при совместной их работе в изолированной системе не
произойдет никаких изменений. Полезную работу Qi — Q2, полученную
от первой машины, мы целиком можем израсходовать на
сжатие газа во второй машине; в результате с нагревателем и
с холодильником никаких изменений не произойдет (см. схему),
и оба количества газа останутся без изменений.
§ 3. ПОСТУЛАТ КЛАУЗИУСА. ВТОРОЕ НАЧАЛО КАК ЭМПИРИЧЕСКИЙ ЗАКОН
ДЛЯ ТЕПЛОВЫХ ПРОЦЕССОВ
Вначале второй закон может быть представлен как
обобщение простейших фактов, как некоторый эмпирический закон,
выводимый непосредственно из опыта. Эта его простейшая
формулировка основана на опытном положении, называемом
постулатом Клаузиуса: теплота не может сама собой
переходить от холодного тела к нагретому.
Этот постулат кажется вполне очевидным, так как мы
никогда не наблюдали, чтобы теплота самостоятельно переходила
от холодного тела к горячему. Повседневный опыт показывает,
что всегда сам собой идет переход теплоты от горячего тела
к холодному. Такое явление называется теплопередачей или
теплообменом. В простейшем случае контакта неподвижных
тел разной температуры этот процесс называют
теплопроводностью (об этом см. в гл. 9).
В постулате Клаузиуса существенное значение имеет
выражение «само собой», и на разъяснении его необходимо
специально остановиться, так как отсюда вытекает принципиальное
значение постулата. С подобным выражением мы встречались
ранее, рассматривая примеры необратимых процессов. Мы
подчеркивали, что в явлении теплопроводности теплота сама
собой переходит от нагретого тела к холодному, работа при
наличии трения сама собой переходит в теплоту, при диффузии про-
§ 3. Постулат Клаузиуса. II начало как эмпирический закон 75
цесс идет сам собой в направлении падения концентрации и,
наконец, газ сам собой расширяется в пустоту. Во всех этих
примерах термин «сам собой» означает, что процесс идет в системе
без участия каких-то дополнительных процессов, или, как
говорит Клаузиус, без компенсации. Иными словами,
процессы в изолированной системе, протекающие сами собой, не
требуют компенсации в виде дополнительных процессов и
могут быть единичными. Таким образом, под выражением «сам
собой» мы должны подразумевать единичность процесса в
системе. Следовательно, постулат Клаузиуса утверждает, что
процесс перехода теплоты от холодного тела к нагретому не
может быть единственным процессом в изолированной системе,
а вместе с ним должны осуществляться другие процессы. Мы
хорошо знаем, что можно за счет охлаждения какого-либо тела
получить при передаче от него теплоты нагревание другого.
К этому сводится в общем работа холодильных машин. Этот
переход тепла осуществляется не в единичном процессе,
а в сложной цепи процессов, тогда как переход теплоты от
нагретого тела к холодному протекает легко и может быть
единичным: стоит только привести в соприкосновение оба тела.
Обобщая далее известные факты, Клаузиус вводит деление
всех процессов на положительные и отрицательные.
Положительными процессами являются те, которые могут
протекать сами собой в изолированной системе, тогда как
отрицательные не могут осуществляться в единичном числе и не
могут течь сами собой.
Примерами положительных процессов являются:
1) переход теплоты от нагретого тела к холодному;
2) превращение работы в теплоту.
Примерами отрицательных процессов служат:
1) переход теплоты от холодного тела к нагретому;
2) превращение теплоты в работу.
Принимая указанное разделение процессов, мы получаем
первую и простейшую формулировку второго начала по Клау-
зиусу: положительные процессы в изолированной системе
протекают сами собой, т. е. могут быть единичными.
Отрицательные процессы в такой системе могут происходить только, когда
они сопровождаются положительными процессами, которые
компенсируют собой отрицательные процессы.
Иначе, всякий отрицательный процесс в изолированной
системе возможен только в том случае, если он компенсируется
положительным процессом. Без компенсации отрицательные
процессы в системе течь не могут. Мера компенсации в
каждом отдельном случае может быть установлена. Мы
остановимся вдесь лишь на двух примерах,
76 Г л а в а 3. Второе начало термодинамики. Энтропия и ее свойства
1. Прямая обратимая машина Карно. Пусть
такая машина работает в изолированной системе. Ранее мы
видели, что за цикл в ней происходят два процесса:
превращение теплоты в механическую работу и переход теплоты от
нагретого тела к холодному. Первый процесс является
отрицательным, и он по второму началу сопровождается и полностью
компенсируется положительным вторым процессом передачи
тепла от нагретого тела к холодному.
2. Обратная обратимая машина Карно. Если
такая машина работает в изолированной системе, то за цикл
происходят следующие процессы: отрицательный процесс перехода
теплоты от холодного тела к нагретому и компенсирующий его
положительный процесс превращения механической работы
в теплоту, что и требуется в соответствии со вторым началом.
Анализ постулата Клаузиуса приводит к общему положению
о ходе процессов в изолированной системе, которое налагает
ограничение на течение процессов, причем этот закон не может
быть выведен из первого начала, допускающего в равной мере
все процессы, согласные с законом сохранения энергии.
Формулировка второго начала, приведенная выше, недостаточна,
так как сущность этого закона остается скрытой и необходимо
дать ему более полное выражение. Постулат Клаузиуса,
очевидный как обобщение повседневного опыта, требует
теоретического обоснования.
§ 4. ОБОБЩЕНИЕ ОБРАТИМЫХ ЦИКЛОВ.
ОТКРЫТИЕ ЭНТРОПИИ КАК ФУНКЦИИ СОСТОЯНИЯ
Вернемся к циклу Карно для дальнейшего развития
полученных результатов.
Продолжаем пока рассматривать только одни обратимые
циклы. В обратимой машине Карно рабочим телом до сих пор
являлся идеальный газ, однако можно показать, что это не
является обязательным. Пользуясь вторым началом в
формулировке Клаузиуса, можно доказать принципиально важное
положение, известное как теорема Карно — Клаузиуса:
коэффициент полезного действия обратимой машины Карно не
зависит от рода посредствующего тела, совершающего работу в этой
машине. Для доказательства представим себе две обратимые
машины Карно, работающие в системе с одним и тем же
нагревателем и одним и тем же холодильником; температуры
последних соответственно равны 7\ и Т2. Пусть в первой машине
(/) работающим веществом является идеальный газ, а во
второй (2) какое-либо другое вещество. Можно разными
способами показать, что к. п. д. обеих машин не могут отличаться
друг от друга. Допустим, например, что полезные работы, да-
§ 4. Обобщение обратимых процессов 11
ваемые обеими машинами, одинаковы, но к. п. д. различаются
потому, что количества отданного и полученного тепла у них
различны. Следовательно, допустим
Q[ - $2 = Q'i—Q'v пРичем QJ > Q'r
Отсюда видно, что должно быть Qi'>Qi" и, значит, т|'<т|".
Докажем, что это невозможно. Превратим машину 1 в обратную
машину Карно; мы знаем, что от этого превращения величина
ее к. п. д. останется без изменения. В результате действия обеих
машин теперь будет получен следующий итог:
1) полезная работа машины 2 полностью будет
скомпенсирована в машине 1, где это будет работа по сжатию (согласно
нашему первому допущению);
2) так как Qi'>Qi", то нагреватель теперь получит тепла
(Q'i) больше, чем отдаст (Q/7);
3) так как Qz>Q2f\ то охладитель отдаст тепла {Qz)
больше, чем получает (Q2")-
Общий итог сводится к тому, что в системе должен
произойти переход некоторого количества теплоты от холодного
тела (охладителя) к более нагретому (нагревателю), причем
этот переход (отрицательный процесс) не компенсируется
никаким положительным процессом. Согласно второму началу
термодинамики этого не может быть и, значит, ч\' — х\". Теорему
можно доказать, исходя также из допущения одинаковых
получаемых теплот в первой и второй машинах при различных
полезных работах.
Таким образом, к. п. д..обратимой машины Карно всегда
выражается соотношением (3,1), независимо от рода
работающего вещества, что дает нам возможность
рассматривать эту формулу как общее соотношение, относящееся
ко всем веществам, а не только к идеальному газу. Общее
значение выражения (3,1) видно также из того, что при выводе
сократились множители, характеризующие свойства идеального
газа, и поэтому к. п. д. машины Карно не должен зависеть от
природы работающего вещества. Заметим, что при этом
агрегатное состояние вещества не имеет значения и цикл Карно
может быть проведен как с твердым, так и жидким телом.
Продолжая дальнейшие обобщения, можно отвлечься от
образа какой-то машины и говорить просто о циклах, подраау-
мевая под циклом замкнутый процесс, в котором вещество по
окончании цикла возвращается в начальное состояние и
происходит обмен теплом между двумя телами — нагретым и хо-
Лбдным. Обращение к циклическим процессам при решении
задач термодинамики часто оказывалось весьма полезным, так
что со временем превратилось в так называемый метод
циклов, применение которого будет рассмотрено позднее.
78 Г лав а 5. Второе начало термодинамики. Энтропия и ее свойства
Из формулы (3,1) для простейшего цикла Карно
при сравнении с формулой (3,2) следует (3,3), т. е.
отсюда получаем:
(3,4)
Величину — иногда называют приведенной теплотой,
относя ее к тому или иному телу. Заметим, что из (3,4) следует
равенство приведенных теплот нагревателя и холодильника
в машине Карно.
Можно показать, что простейший цикл Карно можно всегда
разбить на два или больше циклов промежуточными
адиабатами (рис. 14). Такое деление цикла является законным
потому, что эти промежуточные адиабаты проходятся в
противоположных направлениях и оба процесса по такой адиабате
взаимно полностью компенсируются. Разобьем цикл Карно на
два цикла с помощью адиабаты ав (см. рис. 14). Тогда
теплоты Qi и Qz разделяются на части:
Рассмотрев порознь каждый из образованных циклов, получим
опять равенство приведенных теплот согласно (3, 4):
Q\ Qr2
•* 1 •* 2
_QJ_ = J2L.
Сложив эти равенства, находим:
Qi | Q\ _ <?2 } Ql
Ti т± т2 т2
и вообще при разделении на
несколько циклов подобным же
образом:
Рис. 14. 2а ~т~ e Zj г§ • ( f '
§ 4. Обобщение обратимых процессов
79
Можно применить обратный прием, образуя общий цикл
из нескольких циклов, и тогда для составного цикла вновь
получим формулу (3,5).
Очевидно, вполне допустимо составление разных циклов
Карно, где участвуют нагреватели и холодильники с
различными температурами в разных циклах. В этом случае
составление ведется по адиабатам (рис. 15). Обобщая выражение
(3,5) на п циклов, находим при этом
(3,6)
Tf
где индекс i относится к разным циклам от 1 до /г.
Наряду с циклами, где происходит передача конечных
количеств тепла Q\ и Q2, можно представить себе так называемые
элементарные циклы, в которых в пределе передаются
бесконечно малые количества тепла dQi и dQ2 при конечной разности
температур Т\ и Г2. На диаграмме такие циклы изображаются
бесконечно узкими искривленными вытянутыми лентами с
бесконечно малыми отрезками изотерм. Для такого цикла, как
ранее,
_ dQ2
Наконец, пользуясь возможностью разбиения циклов на части
и вводя элементарные циклы Карно, можно любой обратимый
цикл разбить на большое количество узких элементарных
циклов Карно, соприкасающихся по адиабатам (рис. 16). При
этом отрезки адиабат, лежащие внутри контура, проходятся
каждый в противоположных направлениях и процессы взаимно
компенсируются. Некоторая ошибка получается вследствие
замены непрерывного контура ступенчатыми изотермами сверху
и снизу, а также нескомпенсированными отрезками адиабат,
Рис.
Рис. 16.
80 Г лав а 3, Второе начало термодинамики. Энтропия и ев свойства
прилегающих к контуру. Однако при переходе к пределу, когда
dQ(p и dQ^ стремятся к нулю, эта ошибка может быть сделана
сколь угодно малой. Каждому элементарному циклу здесь
соответствуют свои температуры Т[1) и Т£\ так как линия контура
вообще не представляет собой изотермы. На основании этих
рассуждений выражение (3,6) для конечного числа
элементарных циклов
переходит в пределе в соотношение
Следующим шагом на пути обобщений является переход
от представлений передачи тепла от нагревателей и
холодильников к более общему случаю, когда для контура на
одинаковых правах рассматриваются положительные и отрицательные
приведенные теплоты. Выражение (3,7) можно представить
в форме:
Знак минус перед вторым интегралом можно отнести к теп-
лотам dQb считая их отрицательными, и рассматривать
выражение (3,8) как алгебраическую сумму —- при переменных
dQ и Т. Так как эта сумма относится ко всему контуру, то
можно написать из (3,8):
ff (3,9)
Выражение (3,8) или (3,9) представляет собой так называемый
интеграл Клаузиуса для любого обратимого цикла. Мы
пришли, следовательно, к
заключению, что интеграл приведенных
теплот для любого обратимого
цикла для всех веществ равен
нулю.
Это положение можно
рассматривать как частную
математическую формулировку второго
начала.
Из соотношения (3,9) следует,
у что I—— не зависит от формы
Рис. 17. пути, т, е. от вида процесса.
§ 4. Обобщение обратимых процессов 8J
В самом деле, при изменении состояния от Л до В (рис. 17) по
пути АС В имеем
J ~*
ABC
тогда как при движении из А в В по пути ADB имеем
Г <<Q Г dQ
.) т ~ ) т '
DB BDA
ADB
и согласно (3,9)
АСВ BDA ACB ADB
_ dQ
Так как интеграл по замкнутому контуру от —-1- равен нулю
и вследствие того, что изменение этой величины не зависит от
формы пути, то мы приходим к выводу фундаментального зна-
* dQ
чения, а именно, что бесконечно малая величина —-1- есть пол-
Т
ный дифференциал некоторой функции параметров состояния:
dS = -f-. (3,10)
Эта функция состояния 5 называется энтропией. Из
(3,10) следует общее выражение для энтропии:
(3,11)
где константа является неопределенной постоянной
интегрирования. Отсюда следует новая математическая формулировка
второго начала: величина dS = — есть полный
дифференциал. Этим определением устанавливается существование
энтропии как функции состояния.
§ 5. ЭНТРОПИЯ И ЕЕ СВОЙСТВА. ЭНТРОПИЯ ИДЕАЛЬНОГО ГАЗА.
ПРИНЦИП КАРАТЕОДОРИ
Последовательные обобщения обратимых циклов привели
к открытию новой физической величины, называемой
энтропией, играющей важную роль в термодинамике. Физический
смысл энтропии является достаточно сложным, и он
раскрывается сначала в термодинамике и далее в статистической физике.
В физике наряду с понятиями, представляющимися для всех
4 Заказ № 2479
82 Г лав а 3. Второе начало термодинамики. Энтропия и ее свойства
простыми, благодаря тому что они опираются на повседневный
жизненный опыт, встречаются понятия, не имеющие аналогов
в повседневности, и освоение их представляет большие
трудности. К понятиям такого рода относится, например,
потенциал, а также энтропия. Вначале мы узнаем, что энтропия
представляет собой функцию параметров состояния. Если
последние даны, то принципиально энтропия всегда может быть
вычислена, причем неопределенная константа энтропии может
быть условно положена равной нулю. Так как энтропия
подобно внутренней энергии есть функция состояния, то
изменение ее зависит только от начального и конечного состояния
системы и не зависит от вида процесса. В отдельных задачах
энтропию можно рассматривать и как независимый параметр
состояния, подобно тому как объем, давление и температура
являются функциями состояния, но могут играть роль и
параметров состояния. Изменение энтропии в обратимом круговом
процессе равно нулю. Энтропия какого-либо тела или системы
вообще может возрастать или убывать, причем знак изменения
зависит от знака dQ, так как Т положительно. Очевидно, если
тело получает теплоту, то энтропия его возрастает; когда тело
отдает теплоту, то энтропия его убывает. При всех адиабатных
процессах энтропия тела остается без изменения, так как
dQ = Of и поэтому адиабатные процессы называют также изо-
энтропическими, адиабату — кривой равной энтропии или
изоэнтропой.
Энтропия обладает свойством аддитивности, т. е. энтропия
системы, состоящей из нескольких тел, равна сумме энтропии
всех этих тел. Это значит, что энтропия является величиной
экстенсивной и удельную энтропию можно отнести, например,
к одному молю. Как видно из формулы (3,10), размерность
энтропии совпадает с размерностью теплоемкости.
Неопределенная постоянная в выражении для энтропии
(3,11) может быть вычислена в отдельных случаях, если
условно задать начальное состояние. Однако во многих задачах
нас интересует не абсолютное значение энтропии, а лишь ее
изменение в результате тех или иных процессов.
Объединяя первое и второе начала термодинамики через
выражения для dQ, получаем объединенную формулу для
обоих начал термодинамики для обратимых процессов:
dQ = TdS = dU + pdV, (3,12)
или вообще:
TdS = dU + ^Akdak. (3,13)
Уравнение (3,12) представляет собой простейшую пфаффову
форму для двух независимых переменных U и V. Ранее было
§ 5. Энтропия и ее свойства. Энтропия идеального газа
83
указано (гл. 1), что она всегда имеет один интегрирующий
множитель. Величина dQ не является полным
дифференциалом, а представляет собой просто бесконечно малую величину,
но так как dQ = TdS, то, очевидно, из (3,12) получаем:
dS = — dU + — р dV, (3,12')
где dS — полный дифференциал, тогда как 5 есть функция
состояния. Отсюда следует, что величина — представляет собой
интегрирующий множитель. Далее этот результат будет
использован в общем виде (стр. 111) для доказательства
существования абсолютной температуры.
Найдем энтропию одного моля идеального газа, чтобы
показать, каким образом она может быть вычислена.
Пусть некоторое состояние моля газа изображается точкой
а на диаграмме (р, V), а состояние к нему весьма близкое —
точкой Ь (рис. 18). Найдем изменение энтропии dS при
переходе из а в Ъ. Так как изменение энтропии не зависит от
формы пути, то для простоты расчета выберем какой-нибудь
простой путь перехода. Например, сначала изменим состояние по
изохоре ас, а затем перейдем из с в Ь по изотерме. Изменение
энтропии dSac по изохоре равно:
dQa
CvdT
изменение энтропии на изотерме равно:
ня _ dQcb _ pdV _ RTdV _
р
dV
Общее изменение энтропии при переходе из а в Ь равно в итоге:
jo ло , jo cvdT , RdV
Это изменение не зависит от формы
пути перехода, так как dS есть полный
дифференциал. Величину энтропии в
данной точке мы найдем, интегрируя
последнее выражение. Тогда
о п Г dT , п
° =
(3,14)
Константа интегрирования здесь пока
остается неопределенной (см. гл. 8),
Рис. 18.
84 Глава 3. Второе начало термодинамики. Энтропия и ее свойства
Конечное приращение энтропии при переходе из состояния а
в состояние Ь, не зависящее от формы пути перехода, а лишь от
начального и конечного состояний, находим, применяя дважды
уравнение (3,14) с учетом параметров состояния. Тогда
Sa = CvlnTa + RlnVa + const,
Sb = Cv In Tb + R In Vb + const.
Отсюда
AS = S,-Sfl = Cvln^ + #ln-£L. (3,15)
1 а У а
Из формулы (3,14) видно, что энтропия газа увеличивается
с повышением температуры и при расширении газа. Применяя
уравнение Клапейрона, можно из (3,14) исключить V и
получить S как функцию р и Т:
S = Cp\nT — Rlnp + const, (3,140
так как R постоянно и
Cp = Cv + R.
Ранее мы отметили, что энтропию можно рассматривать
как независимый параметр состояния вместе с другими
параметрами. Это свойство дает возможность состояние системы,
а также процессы изображать с помощью диаграммы, где
одним из параметров является энтропия. В технических
приложениях весьма распространены так называемые
энтропийные диаграммы, где приняты параметры Т и S. Такие
диаграммы удобны потому, что с их помощью особенно легко
находить количество затраченной теплоты в том или ином
процессе. Как показывает формула (3,10),
dQ = TdS, (3,16)
поэтому на энтропийной диаграмме (TyS) бесконечно малое
количество теплоты изображается площадью бесконечно узкой
полоски, заштрихованной на рис. 19. Общее количество
затраченной теплоты в процессе на пути ab будет равно площади
криволинейной фигуры (afe&iai), так как
Sa
Для циклических процессов теплота, превращенная в
работу за один цикл, равна площади, очерченной контуром. Это
ясно из диаграммы, где на пути ABC (рис. 20) затрачивается
теплота
§ 5. Энтропия и ее свойства. Энтропия идеального газа
85
равная площади фигуры (ABCSCSA), и на CDA отнимается
теплота
изображаемая площадью фигуры (ADCScSA). Разность этих
количеств дает площадь фигуры (ABCDA), представляющую
собой теплоту, перешедшую в полезную работу за один цикл.
В системе координат (T,S) уравнение изотермы есть
Т = const и поэтому изотермический процесс изображается
прямой, параллельной оси абсцисс; уравнение адиабаты есть
S = const и, следовательно, адиабатный процесс в этой системе
графически представлен прямой, параллельной ординате.
Отсюда ясно, что цикл Карно в системе (TtS) изображается
прямоугольником со сторонами, параллельными осям
координат.
Выведем уравнения изохоры и изобары идеального газа
в координатах (Г, S). Согласно (3,14)
5 = Cv In Т + R In V + const.
Полагая V=const, имеем S — Cv\nT+const.
Из этого соотношения находим
где kx — некоторая константа. Отсюда видно, что температура
в изохорическом процессе возрастает по экспоненциальному
закону с ростом энтропии.
Для изобарического процесса р = const. Применяя
уравнение Клапейрона и формулу (3,14), находим:
R\nRT — Я In p +const,
Рис. 19.
Рис. 20.
86 Г л а в а 3. Второе начало термодинамики. Энтропия и ее свойства
ИЛИ
Отсюда
S = (Cv + R) In Т + const = Cp In T + const.
s
Мы получили опять уравнение экспоненты, однако, так как
Cp>CVt она поднимается более полого по сравнению с изохо-
рой. На рис. 21 показаны кривые рассмотренных процессов
в системе (Г, 5).
Анализ адиабатного процесса позволяет дать новую
формулировку второго начала, предложенную Каратеодори (1909)
в виде принципа адиабатической
недостижимости состояний.
Рассмотрим общее уравнение (3,12). Здесь dU есть полный
дифференциал внутренней энергии как функции переменных V
и Т. Поэтому из (3,12) следует:
fi=^ = iffi dr + lfP) +p\dV. (3,17)
Т Т [дТ )v Т [ {dv ]т J v
Для адиабатического процесса dQ = 0, следовательно,
уравнение (3,17) обращается в уравнение Пфаффа:
Отсюда следует, что dS = 0, причем S является функцией
параметров Т и V. Так как dS есть полный дифференциал, то из
равенства dS = 0 следует, что S = const, причем
S = S(T, V) = const.
Геометрически это есть уравнение адиабаты в диаграмме
(Г, V); так как постоянная может иметь разные значения, то
Изотерма
Рис. 21.
Рис. 22.
§ 5. Энтропия и ее свойства. Энтропия идеального газа 87
на графике имеем семейство адиабат, которые здесь являются
изоэнтропами (рис. 22); для каждой такой кривой энтропия
постоянна, но значения констант Su 52,..., Sn для кривых
различны. При этом очевидно, что эти адиабаты нигде одна с
другой не пересекаются, так как значения энтропии однозначны
на каждой адиабате. В пространственной диаграмме (5, V, Т)
получаем изоэнтропические поверхности F(S, Vf Г)=0, которые
между собой не связаны; это положение называется принципом
Каратеодори, состоящим в том, что в окрестности любого
термодинамического состояния системы имеются такие состояния,
которые не могут быть достигнуты адиабатическим путем. Это
видно на рис. 22. Если взять состояние а на адиабате 5Ь то из
этого состояния адиабатически нельзя перейти в состояние 6,
которое лежит на адиабате 52> так как линия аЬ не может
быть адиабатой (иначе было бы пересечение), т. е.
адиабатически из а в Ь перейти невозможно. Так как S(T, V) = const
получается интегрированием уравнения (3,17'), что допустимо
только при существовании интегрирующего множителя,
равного —, то отсюда получается определение температуры,
а также доказывается существование энтропии. Принцип
Каратеодори дает абстрактное выражение второго начала, которое
выводится формальным путем. Вывод значительно осложняется
при переходе к сложным системам, на чем мы не
останавливаемся.
§ 6. ИНТЕГРАЛ КЛАУЗИУСА ДЛЯ НЕОБРАТИМЫХ ЦИКЛОВ.
ОБЩАЯ МАТЕМАТИЧЕСКАЯ ФОРМУЛИРОВКА ВТОРОГО НАЧАЛА.
МАКСИМАЛЬНАЯ РАБОТА
Ранее было отмечено (стр. 72), что обратимый цикл Карно
является наиболее выгодным круговым процессом для
получения полезной работы. Все другие циклы, обратимые или
необратимые вообще, менее выгодны в отношении полезной работы.
Можно легко доказать, что к. п. д.
любого обратимого или
необратимого цикла при максимальной
Ti и минимальной Г2
температурах меньше к. п. д. обратимого
цикла Карно при той же
разности температур.
Рассмотрим сначала
произвольный обратимый цикл,
изображенный на рис. 23, и
покажем, что для него к. п. д. меньше,
чем к. п. д. некоторого соответ- Рис. 23.
88 Г лава 3. Второе начало термодинамики. Энтропия и ее свойства
ственного обратимого цикла Карно. В случае произвольного
цикла для контура этого цикла температура непрерывно
меняется от некоторого максимального значения 7\ до
минимальной ее величины Т2. Докажем, что к. п. д. этого обратимого
цикла меньше к. п. д. обратимого цикла Карно с
температурами 7\ и Г2. Для этого опишем цикл Карно вокруг данного
цикла так, чтобы температуры нагревателя и холодильника
в первом из них были равны максимальной и минимальной
температурам, которые встречаются на контуре данного
произвольного обратимого цикла, т. е. равны соответственно 7\ и Т2.
Разделим теперь цикл Карно и вместе с ним данный цикл
несколькими -адиабатами на ряд элементарных циклов. Пересечем наш
цикл отрезками изотерм, как указано на рисунке. Тогда весь
данный цикл будет разбит на ряд циклов Карно с разными
температурами, лежащими в интервале от Ti до Т2. К. п. д. каждого
из этих элементарных циклов равен
(О = М —У2 = j _ У2
T(i) T(i) '
Общий цикл Карно также разделен теперь на элементарные
циклы, но у всех этих последних к. п. д. один и тот же и равен
Сравнивая поочередно к. п. д. элементарных циклов на нашем
контуре с элементарными циклами, на который разбит
основной цикл Карно, мы видим, что вообще Т\>Т[1)>Т{21)>Т2 и
так как Т\ и Т%—максимальная и минимальная температуры.
Поэтому
а отсюда следует, что к. п. д. данного произвольного
обратимого цикла меньше к. п. д. обратимого цикла Карно,
построенного для тех же предельных температур.
Таким образом, из обратимых циклов самым выгодным
в смысле получения полезной работы является цикл Карно.
Вообще можно сказать, что к. п. д. любого обратимого цикла
всегда не больше и в крайнем случае равен к. п. д.
соответствующего описанного цикла Карно. Можно сказать, что в цикле
Карно теплота во всяком случае используется самым выгодным
способом при получении полезной работы.
Перейдем теперь от обратимых циклов, которые мы до сих
пор рассматривали во всех случаях, к необратимым циклам
§ 6. Интеграл Клаузиуса для необратимых циклов 89
и прежде всего сравним к. п. д. обратимого и необратимого
циклов Карно. Если при совершении цикла операций налицо
имеются необратимые процессы, то вполне очевидно, что это
обязательно приведет к уменьшению количества полезной
работы, получаемой за цикл. В самом деле, если в машине Карно
имеется трение, то часть подводимого к системе тепла пойдет
на работу против сил трения, что приведет к уменьшению
полезной работы. Если в системе происходит неуравновешенное
расширение вследствие конечного перепада давления, то это
приведет к тому, что часть энергии будет затрачена на сообщение
кинетической энергии отдельным слоям работающего вещества
(например, газа), причем эта энергия в дальнейшем перейдет
в теплоту, что приведет к уменьшению полезной работы.
Наконец, при наличии переноса теплоты путем теплопроводности
часть теплоты, отнимаемой у нагревателя, будет идти на
нагревание частей машины и затем бесполезно рассеиваться. Отсюда
ясно, что в необратимой машине Карно всегда будет
происходить потеря теплоты и, следовательно, уменьшение получаемой
полезной работы. Полезная работа будет совершаться за счет
не всей той теплоты, за счет которой она могла бы быть
произведена в обратимой машине. Сравнивая к. п. д. необратимой
машины Карно т)' с к. п. д. соответствующей обратимой
машины т), мы можем отметить, что во всех случаях
Но всегда к. п. д. представляет собой отношение полезной
теплоты Qi—Q% ко всей переданной системе теплоты Qi.
Поэтому
тогда как, если бы эта машина была обратимой, то
Т±-Тш ^
Поэтому
1 — V2 ^ М~ 1 2
Qi ^ f
или
Отсюда
f. <f-
' 1 l 2
90 Г лава 3. Второе начало термодинамики. Энтропия и ее свойства
Из последнего неравенства следует, что приведенная
теплота нагревателя в необратимой машине Карно меньше (а не
равна, как ранее) приведенной теплоты холодильника. Далее
находим
А._^1_<0. (3,18)
1 1 k 2
Опять отвлечемся от представления о машине и будем
рассматривать разные необратимые циклы. Тогда, переходя, как
прежде, к сложению нескольких циклов Карно, мы на
основании (3,18) можем написать для сложного цикла:
*1°
или в пределе для любого необратимого цикла:
b~f-^<Ot (3,19)
' 1 J ' 2
Это неравенство для всего контура цикла можно заменить
общим выражением
(3,20)
взамен прежнего выражения (3,9) для обратимых циклов.
Мы приходим таким образом к важному результату, что
в самом обшем случае
(3,21)
т. е. интеграл Клаузиуса для замкнутого контура или меньше
нуля, или равен нулю, но не может быть больше нуля. В таком
виде это утверждение представляет собой общую
математическую формулировку второго начала термодинамики.
Следствия, вытекающие из этого выражения, будут рассмотрены
ниже.
Заметим, что сопоставление к. п. д. всевозможных
обратимых и необратимых циклов с к. п. д. обратимого цикла Карно
приводит к выводу о наибольшей выгоде последнего, так что
в отношении технического использования обратимая машина
Карно была бы самой выгодной. Все реальные машины имеют
меньший к. п. д. вследствие необратимости и благодаря
отличию вида цикла от цикла Карно.
Посмотрим теперь, когда работа, совершаемая системой
при переходе из одного состояния в другое, является
максимальной. Пусть даны два состояния 1 \\ 2 простейшей системы
§ 6. Интеграл Клаузиуса для необратимых циклов 91
(рис. 24), для которых энтропия имеет значения соответственно
Si и 5г. Переведем систему из состояния / в состояние 2
необратимо по пути ABC и затем вернем ее в прежнее состояние 1
обратимо по пути CDA. Цикл является необратимым и
согласно формуле (3,20), для него интеграл меньше нуля, т. е.
в данном случае
f i!L + f-^-<o.
f + f
J г ^J т
1
(необр ) 2 (обр.)
Но для обратимого процесса при переходе из 2 в / имеем:
1
dQ_ = S о
Т 1 2'
2
Поэтому предыдущее неравенство принимает вид:
2
dQ s _s
Т 2 v
1 (необр.)
Отсюда для бесконечно малого изменения состояния
dQ ^ jo
или
TdS> dQ.
Таким образом, взамен прежнего равенства
TdS = dQ,
которое соблюдалось для обратимых процессов, теперь имеем
неравенство и, следовательно, в общем случае можно написать:
TdS>dQ, (3,22)
где верхний знак соответствует необратимым изменениям,
а нижний — обратимым. Выражение (3,22) представляет собой
полную математическую формулировку второго начала в
дифференциальном виде. Согласно уравнению
первого начала
dQ = dU + dW
находим из (3,22), что
TdS>dU + dW. (3,23)
Соотношение (3,23) является объединенной
формулой первого и второго начала
термодинамики. Рис. 24.
92 Г л а в а 3. Второе начало термодинамики. Энтропия и ее свойства
Отсюда следует:
dW^TdS — dU. (3,24)
Выражение (3,24) показывает, что при необратимом
процессе работа, совершаемая системой, всегда меньше, чем при
обратимом процессе в данных условиях. Отсюда следует,
далее, что работа при обратимых процессах является
максимальной из возможных, т. е. если из состояния / система
по заданному пути переходит в состояние 2, то при обратимом
переходе работа всегда больше, чем при необратимом по этому
же пути. Всякое вмешательство необратимости в ходе
процесса сказывается на уменьшении работы системы.
В дифференциальном выражении второго начала в форме
(3,22) или (3,23) работа dW представлена в общем виде. Если
х есть внешний параметр и У — некоторая обобщенная сила,
то в частном случае из (3,23) находим:
Т dS > dU + Y dx. (3,25)
Когда, например, совершается работа при увеличении объема,
то х= V и У=р и тогда
TdS>dU + pdV. (3,26)
Если, кроме того, совершается работа другого рода, не
связанная с увеличением объема, то в (3,26) войдут
соответствующие слагаемые и поэтому в самом общем случае
TdS>dU + ^Akdak. (3,27)
Во всех этих соотношениях обратимым процессам соответствует
нижний знак.
Задачи
1. Как вычислить полезную работу в цикле Карно, пользуясь
энтропийной диаграммой?
2. Доказать, пользуясь вторым началом, что изотерма и адиабата не
могут пересекаться более чем в одной точке.
3. Найти к. п. д. обратимой машины Карно, если температура
нагревателя 227° С, а холодильника 0° С.
4. Найти энтропию 1 моля газа, подчиняющегося уравнению Ван-дер-
Ваальса.
5. Применяя выражения полных дифференциалов энтропии, показать
справедливость формул:
[dTjv р \дт!р
6. Каково приращение энтропии 1 моля азота при нагревании от 10 до
100° С и расширении от 1 до 2f л?
7. Пользуясь приемом вычисления к. п. д. обратимой машины Карно,
доказать, что к. п. д. обратимого цикла, состоящего из двух изохор и двух
изотерм при Т\ и Тч (Г1>72), меньше к. п. д. обратимого цикла Карно при тех
же температурах.
Глава 4
РАЗЛИЧНЫЕ ОБЩИЕ ФОРМУЛИРОВКИ ВТОРОГО
НАЧАЛА И ЕГО НЕПОСРЕДСТВЕННЫЕ ПРИЛОЖЕНИЯ
§ 1. ТЕОРЕМА О РОСТЕ ЭНТРОПИИ В ИЗОЛИРОВАННОЙ СИСТЕМЕ
Второе начало термодинамики является одним из
важнейших законов природы и допускает ряд различных
формулировок. Чтобы подойти к этим формулировкам и оценить значение
этого закона, рассмотрим теорему о росте энтропии
изолированной системы, являющуюся одним из наиболее
распространенных выражений второго начала.
Рассмотрим процессы, протекающие в системе, которая
находится в идеально изолированной оболочке, исключающей
всякий обмен с внешней средой. Можно представить себе,
например, физическую лабораторию, изолированную от внешней
среды; в этой лаборатории устранена внешняя подводка
электроэнергии, выключено центральное отопление и водопровод
(и может быть устранено действие силы тяжести). В этом
помещении производятся разные физические и химические
исследования, по ходу которых требуется проводить
всевозможные процессы, такие, как, например, нагревание, сжигание
горючих материалов, растворение, различные химические
реакции, получение электроэнергии от динамо-машин или от
аккумуляторов. Кроме того, пусть в системе работают
разнообразные машины, дающие полезную энергию и вызывающие
всевозможные механические движения. Первое начало требует,
чтобы внутренняя энергия всей системы оставалась постоянной.
Рассмотрим, как изменяется со временем энтропия этой
системы, помня, что энтропия обладает аддитивностью, т. е. общая
энтропия равна сумме энтропии отдельных частей системы.
В ходе различных процессов энтропия отдельных тел системы
возрастает, у других она остается постоянной или убывает.
Допустив самые сложные процессы в нашей системе, мы,
казалось бы, ничего не в состоянии сказать об изменении
энтропии ее в таких сложных условиях. Однако мы сможем вывести
определенные суждения, если в отдельности рассмотрим все
обратимые и необратимые процессы, т. е. положим в основу
анализа общий признак: обратимость или необратимость
процесса. Тогда получается следующий результат.
94 Г л а в а 4. Различные общие формулировки II начала и его приложения
1. Обратимые процессы. Можно доказать, что любые
обратимые процессы в системе не изменяют ее энтропии, т. е.
при всех обратимых процессах в изолированной системе
энтропия ее остается постоянной.
Во всех чисто механических процессах, где отсутствует
трение, теплота не образуется, т. е. AQ = 0, и потому изменения
энтропии отдельных тел системы не происходит, а
следовательно, энтропия всей системы остается неизменной.
При любых явлениях распространения всех видов излучения
в отсутствие поглощения их в среде также не выделяется
теплота, т. е. опять AQ = 0, а значит, и от этих процессов энтропия
всей изолированной системы измениться не может.
При обратимых некруговых тепловых процессах передача
теплоты происходит всякий раз таким образом, что температура
тела на бесконечно малую величину отличается от температуры
соприкасающихся тел, иначе условие обратимости (стр. 63) не
было бы осуществлено. Следовательно, если в каком-либо
подобном процессе тело получило от источника теплоту AQ, то
AQ
энтропия источника уменьшится на и как раз на эту же
величину увеличится энтропия данного тела (пренебрегая
бесконечно малой разностью температур). Убыль энтропии точно
компенсируется прибылью ее, а отсюда следует, что энтропия
всей системы от обратимой передачи тепла измениться не
может.
Если в изолированной системе происходят обратимые
круговые процессы (циклы), например работают машины, то и в этом
случае энтропия всей системы остается без изменения. В самом
деле, за каждый цикл работающее тело возвращается в
начальное состояние, и поэтому его энтропия, являясь функцией
состояния, остается без изменения. Что касается нагревателей и
холодильников, участвующих в круговом процессе, то для них
мы на основании интеграла Клаузиуса для обратимых
процессов всегда можем написать:
Г «Wi - Г
т. е. уменьшение энтропии нагревателей равно приращению
энтропии холодильников. Особенно просто это видно из
равенства приведенных теплот для простейшей обратимой машины
Карно, где имеется нагреватель, отдающий тепло Qi, и один
холодильник, получающий тепло Q2. В этом случае, как было
показано ранее,
§ 1. Теорема о росте энтропии в изолированной системе 95
Это соотношение можно теперь рассматривать как равенство
убыли энтропии нагревателя и приращения энтропии
холодильника.
Отсюда приходим к общему выводу, что, какие бы
обратимые процессы ни происходили в изолированной системе,
энтропия ее не изменяется.
Этот вывод также формально следует из объединенного
выражения обоих начал термодинамики. Для обратимых
процессов имеем:
В изолированной системе d£/ = 0 и dW=0, следовательно,
dS = 0 или 5 = const.
2. Необратимые процессы. Докажем, что при
любом необратимом процессе в изолированной системе энтропия
последней только возрастает и никогда не может уменьшаться.
Остановимся подробнее на анализе отдельных необратимых
процессов.
1) Механические процессы с трением. При
любом механическом движении с трением за счет работы по
преодолению силы трения появляется теплота AQ>0 при
определенной температуре Т, следовательно, в системе возникает не-
которое количество энтропии ——. Значит, достаточно наличия
одного механического процесса с трением, чтобы энтропия
системы увеличилась.
2) Распространение разных видов излучения
в среде с поглощением. В процессе поглощения энергии
излучения происходит превращение ее в тепло. Среда получает
некоторое количество тепла AQ и энтропия ее увеличивается,
а следовательно, возрастает энтропия всей изолированной
системы.
3) Теплопроводность. В этом процессе происходит
необратимая передача теплоты от нагретого тела к холодному
при конечной разности температур. Рассмотрим случай, когда
соприкасаются два тела большой массы, одно имеет
температуру Ти температура другого тела Гг, причем Ti>T2. Пусть от
первого тела ко второму перешло небольшое количество тепла
AQ, и за это время температуры обоих тел изменились ничтожно
вследствие их большой массы. Тогда энтропия нагретого тела
до
уменьшилась на величину —=-, а энтропия холодного возросла
ДО тт AQ ^ AQ AQ AQ .. Л
на —=-. Но, очевидно, —-^>--г-, следовательно, — — > О,
Т2 Т2 тг т2 т±
т. е. в системе получается некоторое положительное приращение
энтропии, так как прибыль энтропии одного тела не покрыса-
96 Г лав а 4. Различные общие формулировки II начала и его приложения
ется убылью энтропии другого тела. Возможен также
необратимый процесс теплопроводности, когда два тела с
температурами Ti и Г2 (Т1>Т2) обмениваются теплом Q, в результате
чего их температуры выравниваются и спустя некоторое время
достигается общая температура Т (причем Ti>T>T2). В этом
случае энтропия нагретого тела убывает на величину
О, 0_
т тг '
а энтропия холодного тела увеличивается на
Сумма изменений энтропии системы равна:
Т тх Т2 Т Т2 Т±
Очевидно, что
т. е. и в этом случае энтропия системы возрастает.
Следовательно, любой процесс теплопроводности,
совершающийся при конечной разности температур, приводит к росту
энтропии изолированной системы.
4) Процессы неуравновешенного
расширения. При неуравновешенном расширении работа тела меньше
той, которую оно совершило бы при обратимом, т. е.
уравновешенном, расширении. Если происходит неуравновешенное
расширение, то часть энергии расходуется на сообщение
кинетической энергии движущейся массе и затем превращается
в теплоту при остановке, ударе и т. д. В результате этих
процессов выделяется теплота AQ, что приводит к росту энтропии
системы.
Рассмотрим для примера необратимое расширение
идеального газа в пустоту. Пусть один моль газа, первоначально
занимавший объем Vu расширился, переходя в пустой сосуд, до
объема V2. Мы знаем, что его температура осталась без
изменения (стр. 45). Энтропия газа до расширения равна;
Sx == Cv In Т + R In Vx + const.
После расширения энтропия имеет величину
S2 = Cv In Т + R In V2 + const.
Отсюда изменение энтропии AS составляет:
AS = S2~Sl = Rln^. (4,1)
§ 1. Теорема о росте энтропии в изолированной системе 97
Так как Vz>Vu то AS>0 и, следовательно, энтропия
системы увеличилась. Заметим, что в этом процессе AQ=0 и
увеличение энтропии происходит только за счет расширения
газа, т. е. связано с уменьшением его плотности. Этот пример
показывает, что увеличение энтропии системы может
происходить не только за счет теплообмена, но также при рассеянии
вещества.
Легко показать, что в обратимом изотермическом
расширении газа найденное приращение энтропии газа согласно (4,1)
как раз компенсировалось бы убылью энтропии другого тела, от
которого газ получал теплоту, необходимую для совершения
работы изотермического расширения
Поэтому энтропия тела, отдавшего теплоту AQ газу,
уменьшилась на величину AS" = —L'= R In—— и, очевидно, AS=AS/.
В изолированной системе от такого процесса не останется
никакого следа и энтропия ее не изменится.
5) Диффузия. Рассмотрим в виде примера диффузию
в растворе. Применяя известные представления Вант-Гоффа, мы
можем считать, что концентрация с растворенного вещества
пропорциональна так называемому осмотическому давлению р
его в растворе:
Диффузия вызвана различием концентрации или осмотического
давления в отдельных слоях. При малых концентрациях
поведение растворенного вещества аналогично свойствам идеальных
газов. Поэтому можно воспользоваться выражением (3,14х) для
энтропии моля газа
S = Ср In Т — R In р + const = — R In p + const,
если процесс диффузии протекает при постоянной температуре
Т=const. Тогда в одном слое осмотическое давление есть pi,
а в другом оно равно р2 и пусть pi>p2- Диффузия идет сама
собой в направлении падения осмотического давления. В
начальный момент энтропия равна:
51 = — R In px + const.
Спустя некоторый промежуток времени давление в слое станет
меньше, так как оно сравняется с давлением соседнего слоя и
тогда
52 = — R In p2 + const.
98 Г лав а 4. Различные общие формулировки II начала и его приложения
Отсюда
Рг
Эта величина является положительной Д5>0, так как р\>р2-
Таким образом, если в системе происходит процесс диффузии,
то общая энтропия системы возрастает. Опять мы здесь
встречаемся с необратимым процессом, когда рост энтропии
вызывается уменьшением плотности (концентрации) вещества. В
диффузионных процессах в газах также получается возрастание
энтропии и связанное с ним понижение упругости газа.
6) Необратимые круговые процессы и работа
необратимых машин. В изолированной системе могут
протекать необратимые круговые процессы, например, при
работе тепловых машин. В этих случаях мы знаем, что, какова бы
ни была форма цикла, работающее тело за один цикл
возвращается в начальное состояние и его энтропия остается
неизменной, как и при обратимом цикле. Однако для любого
необратимого цикла выполняется неравенство Клаузиуса:
или
dQ2
Это выражение показывает, что в необратимой машине убыль
энтропии нагревателей меньше приращения энтропии
холодильников. Следовательно, энтропия системы, где работает
необратимая машина, возрастает. Особенно ясно видно это на примере
необратимой машины Карно, для которой мы получили
Тг < Т2 '
ИЛИ
т. е. здесь убыль энтропии нагревателя не компенсирует собой
рост энтропии холодильника. В результате энтропия системы
увеличивается. Отсюда можно сделать общий вывод, что при
наличии круговых необратимых процессов в изолированной
системе энтропия ее возрастает.
Мы рассмотрели весьма разнообразные обратимые и
необратимые процессы в изолированной системе. Можно было бы
рассмотреть различные комбинации этих процессов, а также
более сложные изменения, среди которых встречались бы обра-
§ 1. Теорема о росте энтропии в изолированной системе 99
тимые и необратимые процессы. Анализ всех этих явлений
всегда приводит к заключению, которое мы теперь сформулируем
как теорему, доказанную выше: при всех обратимых процессах
в изолированной системе энтропия ее остается неизменной, при
всех необратимых процессах энтропия системы только
возрастает:
Ранее было отмечено, что обратимые процессы, состоящие
из непрерывной смены состояний равновесия, текут бесконечно
медленно и являются идеальными процессами. Только в
отдельных случаях можно говорить о приближенной обратимости
процесса. Реальные процессы природы, всегда протекающие с
конечной скоростью, как правило, являются необратимыми.
Какими бы ни были сложными процессы в изолированной системе,
общая направленность их такова, что суммарная энтропия
системы убывать не может. Энтропия системы не может быть
уничтожена. После завершения всех необратимых процессов
будет, наконец, достигнуто такое состояние системы, когда в ней
не удастся провести ни одного необратимого процесса и ей
будут доступны одни лишь обратимые изменения. Тогда должен
прекратиться дальнейший рост энтропии, и она достигнет
максимального значения. Поэтому второе начало термодинамики
по Клаузиусу можно сформулировать шире и сказать, что
энтропия изолированной системы стремится к максимуму.
§ 2. СЛЕДСТВИЯ ИЗ ТЕОРЕМЫ О РОСТЕ ЭНТРОПИИ И СВЯЗЬ ИХ
С РАЗЛИЧНЫМИ ФОРМУЛИРОВКАМИ ВТОРОГО НАЧАЛА
Различные выражения второго начала, которые
предлагались в ходе развития термодинамики, могут быть выведены из
теоремы о росте энтропии изолированной системы.
1. Постулат Клаузиуса. Утверждение, что теплота
не может сама собой переходить от холодного тела к
нагретому, является частной формулировкой теоремы о росте
энтропии; подобный переход приводил бы к уменьшению энтропии
системы двух тел, что, как было доказано, невозможно. Если
искусственно осуществить такой переход, то потребуется
проведение ряда дополнительных процессов, которые приведут
к росту энтропии. Обобщая это положение, мы отметим, что все
необратимые процессы могут протекать в системе в прямом
направлении сами собой, так как это не противоречит требованию
роста энтропии. Напротив, в обратном направлении эти
процессы не могут протекать в единичном числе, сами собой, так
как тогда энтропия системы должна уменьшаться, что
невозможно.
100 Глава 4. Различные общие формулировки II начала и его приложения
2. Работа тепловых машин и перпетуум
мобиле второго рода. Мы видели, что при работе
обратимых машин уменьшение энтропии нагревателей как раз
компенсируется ростом энтропии холодильников, в результате чего
энтропия системы остается постоянной. Можно сказать также, что
в таких машинах уменьшение энтропии, вызванное
превращением тепла в работу, компенсируется ростом энтропии за счет
перехода части тепла от нагретого тела к холодному.
Независимость к. п. д. обратимой машины Карно от рода вещества
согласуется с теоремой о постоянстве энтропии системы, где
протекают обратимые процессы. Если к. п. д. двух машин Карно,
работающих в одном интервале температур от 7\ до Т2 с
разными веществами, был бы различен, то, обращая одну из
машин, мы получили бы нарушение теоремы об энтропии
изолированной системы. Очевидно, нельзя построить периодически
работающую тепловую машину, которая действовала бы без
холодильника.
В своем курсе термодинамики Планк дает такую
формулировку второго начала: «Невозможно построить такую
периодически действующую машину, которая не производит ничего
другого, кроме поднятия груза и охлаждения некоторого
резервуара тепла». Далее он отмечает, что «такая машина могла бы
быть использована одновременно и как мотор и как
холодильная машина без какой бы то ни было затраты энергии и
материалов: она была бы, таким образом, самой выгодной машиной
в мире. Правда, она не была бы равноценной перпетуум мобиле,
так как производила бы работу вовсе не из ничего, а из
теплоты, заимствуемой ею из резервуара». Эта машина,
следовательно, нисколько не противоречила бы закону сохранения
энергии. Однако такая машина невозможна, так как ее
действие противоречило бы второму закону термодинамики. В этой
машине от нагревателя (резервуара тепла) отнималось бы
некоторое количество теплоты Qi и превращалось бы целиком
в работу (поднятие груза), к холодильнику же теплота бы не
переходила. В результате энтропия нагревателя непрерывно
уменьшалась бы за каждый цикл на -^- и эта убыль ничем бы
не компенсировалась. Из теоремы об энтропии изолированной
системы следует, что это невозможно.
Эти рассуждения показывают, что невозможно превращать
в работу теплоту, получаемую от тела, не производя никаких
других действий. Поэтому В. Томсон формулирует второе
начало термодинамики следующим образом: «Невозможно
получать при помощи неодушевленной материи работу от
какой-либо части материи, охлаждая ее ниже температуры
наиболее холодного из окружающих' тел». Это положение можно
§ 2. Следствия из теоремы о росте энтропии 101
выразить иначе, отметив, что «теплота наиболее холодного из
данной системы тел не может служить источником работы»
(Хвольсон). Превращение теплоты наиболее холодного тела
системы в работу привело бы к уменьшению энтропии всей
системы, что на основании рассмотренной теоремы является
невозможным. Ограничение, накладываемое теоремой об
энтропии на процессы в изолированной системе, приводит к
важному практическому следствию. Оказывается, мы не можем
использовать колоссальные запасы внутренней энергии
атмосферного воздуха, почвы, океана, применяя лишь тепловые
машины* для получения полезной работы. Действительно,
перечисленные тела являются весьма распространенными в земных
условиях и в то же время в среднем являются самыми
холодными из земных тел. Непрерывное отнятие тепла от этих тел
с превращением в работу дало бы возможность получить
колоссальные количества полезной работы, что равносильно
использованию вечного двигателя, так как запасы энергии в воздухе,
почве и в океане практически неисчерпаемы. Эта машина
нисколько не противоречила бы принципу сохранения энергии, но
все же являлась бы практически вечным двигателем. Поэтому
Оствальд называет эту воображаемую машину вечным
двигателем второго рода, в отличие от вечного двигателя первого рода,
недопустимого вследствие нарушения первого начала
термодинамики. Работа вечного двигателя второго рода приводила бы
к уменьшению энтропии системы и поэтому на основании
теоремы об энтропии изолированной системы можно сказать, что
вечный двигатель второго рода невозможен (формулировка
(В. Оствальда).
Заметим, что использование энергии океанской воды в
машинах для получения полезной работы возможно, если
использовать разницу температур отдельных слоев воды. Тогда можно
образовать цикл между этими частями, причем слои с
повышенной температурой будут служить нагревателями в машине,
а более холодные слои — холодильниками. Такая машина
допустима с точки зрения второго начала, и она была на самом деле
построена.
3. Принцип рассеяния энергии. Непрерывное
нарастание энтропии изолированной системы, вызванное
необратимыми процессами, неизбежно сопровождается рассеянием
энергии. Действительно, переход тепла от нагретого тела к
холодному приводит к выравниванию температур. В результате
течения таких процессов в системе температуры отдельных тел
сближаются, что снижает возможность получения полезной
работы в машинах. Такой процесс не противоречит закону
сохранения энергии, потому что общий запас энергии системы
остается без изменения. Однако «качество энергии», в техни-
102 Глава 4. Различные общие формулировки II начала и его приложения
ческом смысле, снижается. При достаточной близости
температур тел системы количество полезной работы, которую можно
получить в машине, становится весьма малым. Происходит,
следовательно, потеря полезной работы, что соответствует
снижению «работоспособности» системы. Одновременно с этим
большинство необратимых процессов ведет к рассеянию
энергии. При теплопроводности одно тело передает другому
некоторое количество теплоты, второе отдает теплоту третьему более
холодному и т. д. В результате энергия постепенно все
равномернее распределяется между телами системы и рассеивается.
Потенциальная энергия газа пропорциональна его давлению.
Сильно сжатый газ, расширяясь, совершает большую работу.
После расширения давление его падает и возможность
совершения работы снижается. Следовательно, расширение газа
тож^ ведет к потере работоспособности. Это становится
особенно заметным при неуравновешенном расширении, которое
является необратимым.
Соответственно указанным процессам В. Томсон
формулирует второе начало как принцип рассеяния энергии: энергия
изолированной системы постепенно обесценивается и
рассеивается.
Следует отметить, что в элементарном изложении физики,
когда рассматриваются примеры превращения энергии,
неизбежно сталкиваются с явлениями ее рассеяния, но о
принципиальном значении этих явлений обычно не говорят. Рассеяние
энергии тогда предстает нашему сознанию как досадная
частная неудача, а между тем это оказывается принципиально
важным, так как необратимость процессов ведет к рассеянию
энергии. Качание маятника обычно рассматривается как хороший
пример периодического превращения кинетической энергии
в потенциальную и обратно. Однако, демонстрируя учащимся
это явление, мы неизбежно вызываем у них представление об
исчезновении энергии, так как колебания его со временем
затухают. Неизбежно при пояснениях приходится говорить о
рассеянии энергии. В более сложных примерах превращения
энергии стараются не доходить до конца. Например, описание
превращений энергии при работе гидростанции доводят до момента
подачи энергии потребителю. Но, говоря о всех
преобразованиях энергии падающей воды, начиная с механической энергии
вращения ротора турбины и динамо-машины и далее при
передаче электроэнергии по проводам, приходится перечислять
различные «потери», и тут опять неизбежно мы сталкиваемся
с рассеянием энергии. Наконец, при использовании энергии
потребителем вновь приходится рассматривать рассеяние и
обесценение энергии. Все это проявления самостоятельного закона
природы, независимого от первого начала термодинамики и
§ 2. Следствия из теоремы о росте энтропии 103
определяемого принципом роста энтропии при необратимых
процессах.
Так как возрастание энтропии изолированной системы
связано с рассеянием и обесцениванием энергии, то иногда
энтропию рассматривают (не вполне строго) как меру
обесценивания энергии.
4. Общее изменение системы при
необратимых процессах. Возрастание энтропии системы при
необратимых процессах сопровождается не только рассеянием
энергии, но приводит также к другим изменениям свойств
системы. Необратимые процессы неуравновешенного расширения
газов ведут к более или менее равномерному распределению
давления в системе и к ее расширению. К этому же приводят
разнообразные диффузионные процессы, вызванные различием
концентрации отдельных частей системы. В результате
процессов диффузии наступает выравнивание концентраций в
отдельных частях системы. Постепенное измельчание отдельных
частиц при необратимых разрывах, раскалывании, различных
видах дробления вплоть до измельчения горных пород с
образованием песка, пыли (при эрозии почвы) и т. п. также приводит
к рассеянию вещества, так как продукты дробления легко
диффундируют, -перемешиваются и равномерно взаимно
распределяются в системе.
Таким образом, в результате всех необратимых процессов
система перерождается, или, как говорят, деградирует.
Окончательным итогом всех изменений является состояние системы,
при котором:
1) весь запас полезной работы превращен в тепло;
2) температуры всех тел системы уравнялись;
3) произошло выравнивание концентраций и давлений в
системе;
4) тела предельно измельчены и равномерно перемешаны.
В этих условиях всякий повод к дальнейшему изменению
системы оказывается устраненным и в ней возможны одни лишь
обратимые изменения. Энтропия достигает максимального
значения. Такое состояние системы иногда называют тепловой
смертью. Пока мы рассматриваем макросистемы конечных
размеров, достижение максимума энтропии и возможность
тепловой смерти для этих систем не может вызывать сомнений, так
как для них второе начало является законом, теоретически
доказанным и поэтому практически и принципиально важны'м.
Однако было бы ошибкой распространять этот вывод на всю
вселенную, как это сделал Клаузиус, и считать, что она
стремится к тепловой смерти. Мы все время говорили о конечной
изолированной системе и только для нее имеет смысл вводить
понятия обратимости и необратимости процессов и вычислять
104 Глава 4. Различные общие формулировки II начала и его приложения
значение ее энтропии, которая является суммой энтропии
составных частей. Для вселенной, которую нельзя рассматривать
как конечную изолированную систему, суждение о величине
энтропии теряет смысл и потому второе начало сформулировано
быть не может.
§ 3. НЕОБРАТИМЫЕ ПРОЦЕССЫ И НАПРАВЛЕНИЕ ВРЕМЕНИ
В МАКРОСКОПИЧЕСКОЙ СИСТЕМЕ
Описанный выше характер изменения макросистемы
согласно второму началу термодинамики ставит общую проблему
направления времени в окружающем макроскопическом мире.
Здесь мы не касаемся метрики времени, так как теория
метрических свойств времени подробно рассматривается в общей
теории относительности Эйнштейна, а остановимся на вопросе
о направлении времени в связи с феноменологическим
выражением второго начала.
Повседневные наблюдения ясно показывают нам, что
процессы окружающей природы являются необратимыми, тогда как
обратимость является той или иной идеализацией реальных
процессов. В лекциях Р. Фейнмана * приводятся примеры
необратимости в повседневной жизни. «Роняешь чашку, она
разбивается, и сколько не жди, черепки не соберутся снова и
чашка не прыгнет обратно тебе в руки». Известно, что «из елки
можно сделать палку, а из палки не сделаешь елки, в связи
с чем наш мир постоянно меняет свой характер с елочного на
палочный». Этот ход явлений представляется нам
естественный течением событий, с чем связано различение прошлого,
настоящего и будущего. «Естественное» направление хода
событий особенно остро воспринимается нами при просмотре
какой-нибудь заснятой кинопленки в «обратном направлении».
Пусть сначала на кинопленке заснято движение планеты вокруг
Солнца и мы демонстрируем такую пленку в прямом, а потом
в обратном направлениях. Мы увидим, что при обратном
показе планета вращается вокруг Солнца, правда в обратном
направлении, но ее траектория в обоих случаях представляет
собой эллипс и законы Кеплера строго соблюдаются. Это
объясняется в сущности тем, что для закона всемирного тяготения
имеет значение порядок чередования событий, а
направление времени безразлично, как для всех обратимых
процессов.
Совершенно иначе выглядит обратный запуск кинопленки,
где были засняты какие-либо необратимые процессы. Это
* Р. Фейнма^н. Характер физических законов. М., «Мир», 1968. Лекция
5, стр. 114.
§ 3. Необратимые процессы и направление времени 105
можно хорошо видеть, если на цветной кинопленке заснять,
например, опыт в приборе Джоуля — Томсона, состоящем из двух
одинаковых сообщающихся сосудов, соединенных краном (см.
стр. 44). Пусть в одном сосуде находится какой-нибудь
окрашенный газ, например пары брома бурого цвета, а из другого
газ выкачан. Снимем опыт на цветную кинопленку, начиная
с состояния, когда кран еще закрыт, ход самого опыта при
открывании 'крана и конечное состояние при открытом
неподвижном кране. Увидим, что пар потечет из первого сосуда во
второй, в результате чего в обоих сосудах будут находиться
равномерно окрашенные пары брома. Пустим теперь пленку в
обратном направлении: мы увидим, что пары будут уходить в
первый сосуд, в котором окраска начнет густеть, тогда как окраска
во втором сосуде будет бледнеть, пока весь пар не соберется
в первом сосуде, а второй станет бесцветным, после чего кран
будет закрыт. Этот ход событий представляется нереальным.
Иногда производят последовательно ускоренную съемку
распускания цветка. Бутон на экране набухает, лопается,
появляются первые лепестки, цветок полностью распускается и т. д.
Покажем кинопленку с этим эпизодом в обратном порядке и
мы увидим неестественную картину, когда распустившийся
цветок превращается в бутон, который становится все меньше и,
наконец, вовсе пропадает. Таких примеров обратного показа
кинопленки можно привести очень много *.
Рассматривая проблему направления времени, нельзя
смешивать порядок чередования событий с
направлением событий. Например, точки на прямой линии могут быть
расположены в определенном порядке, но направление здесь
отсутствует, так как сама линия не обладает каким-нибудь
направлением. Так и в чисто механических обратимых явлениях
мы различаем только порядок событий, тогда как направление
времени не имеет значения. Напротив, в необратимых
процессах приходится иметь в виду и порядок чередования, и
направление времени. Все, что соответствует росту энтропии,
рассеянию энергии и вещества, представляется нам привычным, и мы
психологически связываем с этим естественное направление
времени. «Прошлое и настоящее по-разному воспринимается
психологически»,— пишет Фейнман. Понятия прошлого,
настоящего и будущего в макроскопическом мире складываются при
действии второго начала термодинамики. Четкая
последовательность событий и привычная направленность времени при
действии второго начала приводят к очевидным аксиомам
направления времени:
* См.: Д. А. Франк-Каменецкий. Наука о времени. «Природа». 1970*
№3, стр. 11.
103 Глава 4. Различные общие формулировки II начала и его приложения
1. Время движется от прошлого к будущему.
2. Момент «теперь» есть настоящее время, отделяющее
прошлое от будущего.
3. Прошлое никогда не возвращается.
4. Мы не можем изменять прошлое, но можем изменять
будущее.
5. Мы можем иметь протоколы прошлого, но не будущего.
Направление времени есть психологический эффект,
обусловленный господством необратимых эффектов в макромире и
действием второго начала термодинамики. Временной порядок
процессов непосредственно связан с причинно-следственным
отношением, поэтому вполне основательна попытка сведения
временного порядка к причинному (она была сделана еще
Лейбницем). В нашем сознании причина и следствие идут во
времени так, что сперва действует причина, а уже за нею
выступает следствие. Это, конечно, упрощенное описание явления,
так как на самом деле причинные ряды пересекаются и
поэтому отношение между событиями сложнее. Таким образом,
второе начало затрагивает глубокие философские вопросы.
Подчеркивая общее значение второго начала как
важнейшего закона природы, нельзя не заметить глубокого
противоречия между явной необратимостью реальных процессов
макромира и идеальной обратимостью чисто механических и
элементарных квантовых явлений*. Например, механические
движения отдельных молекул газа и процессы столкновений
последних друг с другом по законам упругого удара являются
примером полной обратимости, т. е. они совершенно
симметричны по отношению ко времени и для отдельной молекулы
нельзя говорить о необратимости. Этот вопрос относится к
выяснению соотношения между вторым началом и молекулярно-
кинетической теорией. Полностью эта сложная задача
рассматривается в статистической физике и здесь может быть отражена
лишь очень кратко.
§ 4. СТАТИСТИЧЕСКИЙ СМЫСЛ ВТОРОГО НАЧАЛА ТЕРМОДИНАМИКИ
В основе молекулярно-кинетического учения лежат
статистические представления, относящиеся к массовым явлениям,
т. е. к коллективам из большого числа частиц. Поэтому с этой
точки зрения, например, газ представляет собой собрание
огромного количества молекул, причем, хотя поведение каждой
молекулы определяется законами обычной механики, поведение
* Фейнман пишет, что «во всех законах физики, обнаруженных до сих пор,
не наблюдается никакого различия между прошлым и настоящим» или
«законы обратимы, а явления — нет» (Р. Фейнман. Характер физических
законов).
§ 4. Статистический смысл II начала термодинамики 107
их в большом количестве оказывается совершенно иным. Это
объясняется тем, что начальные условия (начальные
координаты и скорости) каждой молекулы настолько затеряны среди
начальных условий множества других молекул, что они уже
никак не влияют на поведение - всей массы газа. Хаотичность
молекулярного движения приводит к тому, что в
макромасштабе проявляются новые—статистические
закономерности, которые отличны от так называемых динамических
закономерностей механики, присущих отдельным молекулам.
Следовательно, молекулярно-кинетическая теория не является
механической теорией, хотя каждая молекула строго
подчиняется законам механики. В макромасштабе происходит
усреднение всех характеристик молекулы: энергии, скорости и т. д.
Понятие температуры относится к огромному количеству
молекул макроскопического тела и неприменимо к отдельной
молекуле; также давление газа является усредненной силой
действия большого числа молекул на единицу площади. Однако
если характеристики состояния газа являются статистически
усредненными, то каждое термодинамическое состояние его
является не безусловно обязательным, а существует с той или
иной вероятностью. Последняя тем выше, т. е. состояние
тем вероятнее, чем большим числом комбинаций в
пространственном расположении молекул и в скоростях молекул оно
осуществляется. Наименее всего вероятно состояние газа, когда
скорости молекул совершенно одинаковы, так как такое
состояние реализуется всего одной комбинацией (если говорить
для простоты только о характеристике в отношении скорости).
Условно можно определить вероятность такого состояния
величиной Wo, тогда вероятность состояния w с разными скоростями
во много раз выше, чем wo, так как для разных скоростей можно
осуществить большое число комбинаций. Отношение W=w/w0
носит название термодинамической вероятности
или статистического веса состояния, причем очевидно,
что W^>\. В статистической физике доказывается в самом
общем случае (а не только для газа), что энтропия тем выше, чем
большим числом комбинаций осуществляется данное состояние.
Следовательно, существует соотношение между энтропией S и
термодинамической вероятностью W состояния. Это
соотношение было получено Больцманом, который на основании
статистических соображений показал, что энтропия прямо
пропорциональна логарифму вероятности, т. е. что
Здесь k есть отношение постоянной Клапейрона к
постоянной Авогадро, т. е. k = R/N0= 1,38-10-23 дж/°К. Эта
постоянная k называется константой Больцмана.
108 Глава 4. Различные общие формулировки II начала и его приложения
Отсюда видно, что энтропия тем выше, чем больше
вероятность состояния, а максимуму энтропии отвечает самое
вероятное состояние. В статистике формула Больцмана является
определением энтропии.
Если мы связали энтропию с вероятностью состояния, то
отсюда следует, что второе начало, строго говоря, нельзя считать
точным законом. Его следует сформулировать в виде
утверждения: весьма вероятно, что энтропия изолированной системы
возрастает, а это значит, что возможны отступления от такого
утверждения. Для макросистем эти отступления крайне
ничтожны, так как для большого числа молекул возможно
колоссально большое количество комбинаций в их скоростях и пр.
Однако для микрообъемов отступления от второго начала
могут быть существенными. Если мы, например, от большого
объема, содержащего большое количество молекул, перейдем к
таким малым объемам, где имеется всего 100—200 молекул газа
нормального давления, то плотность газа может существенно
отличаться от средней, а также давление его может отличаться
от термодинамически равновесного давления. Такие явления
отступления от второго начала называются флуктуациями,
и они были экспериментально обнаружены при изучении
броуновского движения взвешенных коллоидных частиц в малых
объемах растворов, а также в других случаях, на которых мы
не останавливаемся. Можно считать, что флуктуации опытным
путем доказаны и тем самым доказано, что отступления от
второго начала являются реальными.
Из этих рассуждений следует относительность обратимости.
В больших макросистемах, где второе начало является
практически точным законом, существуют необратимые процессы,
ведущие к максимуму энтропии. Но при переходе к достаточно
малым системам возможны процессы, когда энтропия может не
только возрастать, но и заметно убывать, т. е. необратимые
процессы могут сами собой обращаться, благодаря чему
появляется обратимость с нарушением второго начала.
В термодинамике всегда имеют дело с макросистемами и
потому не учитывают ничтожных отступлений от второго начала,
принимая его за точный закон.
Понятие энтропии, возникшее в термодинамике в настоящее
время, с успехом применяется теперь в других областях науки
и техники, совсем не связанных с термодинамикой. Так,
представление о статистическом смысле энтропии оказалось ценным
в технике сообщений и в теории информации. Последняя
охватывает весьма широкие области деятельности человека вплоть
до экономики и социологии. Природа системы в разнообразных
приложениях может быть различной; так это могут быть группы
сигналов в средствах связи и т. п. Для этих систем всегда име-
§ 4. Статистический смысл 11 начала термодинамики 109
ется некоторая неосведомленность о них в виде отсутствия
нужной информации о системе, так что последняя всегда
является несколько неопределенной. О свойствах системы можно
ставить разные вопросы до ее проверки или испытания и
получать множество возможных ответов; пусть число этих ответов
есть щ— до испытания. Если мы произведем какие-то опыты
(испытания) над системой, то число возможных ответов п —
после испытания — уменьшится, так как получена некоторая
положительная информация о свойствах системы, т. е. по>п.
Возникает вопрос о том, как наиболее рационально определить
понятие количества информации о системе. Очевидно, это
количество по смыслу должно быть положительным и
безразмерным, к тому же оно должно обладать свойством аддитивности.
Этим требованиям удовлетворяет логарифмическая функция
для отношения — э т. е. можно принять, чте количество ин-
п
формации есть р — \п^±.
при этом очевидно, что Р>0. Это соотношение сходно по виду
с формулой Больцмана для энтропии: S = klnW.
В очень сложных системах в отдельных их частях
количество информации может увеличиваться или убывать. Каждое
состояние характеризуется той или иной вероятностью, которая
представляет собой отношение w = — . Связывая количество
щ
информации с энтропией, удается показать, что средняя
величина всех отношений Р = —\nw равна:
Р = — kS,
где S — энтропия всей системы. Этим доказывается, что
энтропия и количество информации пропорциональны друг другу и
различаются только знаками. Росту энтропии отвечает
уменьшение количества информации. Так как энтропия является
выражением меры дезорганизованности в системе,
то количество информации соответствует мере ее организации.
Пусть начальное состояние системы относительно маловероятно,
что соответствует небольшой величине энтропии, тогда можно
сказать, что количество информации в системе велико. Далее
система переходит к более вероятным состояниям, и величина
энтропии растет, достигая максимума, при этом количество
информации убывает (так как S и Р отличаются по знаку).
Следовательно, росту энтропии отвечает убыль величины информации
в системе. В ней содержится все меньше и меньше «ценной
информации». Вообще, по Винеру, можно сказать, что «чем более
вероятно сообщение, тем меньше оно содержит информации» *.
* Н. В и н е р. Кибернетика и общество, ИЛ. М., 1958.
ПО Г лав а 4. Различные общие формулировки II начала и его приложения
§ 5. ПРИМЕНЕНИЕ ВТОРОГО НАЧАЛА К ПОСТРОЕНИЮ
АБСОЛЮТНОЙ ШКАЛЫ ТЕМПЕРАТУРЫ
Второе начало термодинамики может непосредственно
применяться для решения ряда вопросов термодинамического
анализа. Большое значение при этом играет так называемый метод
циклов, основанный на построении идеальных воображаемых
круговых процессов, заменяющих данный процесс.
С помощью второго начала возможно дать строгое
определение понятия температуры и построить абсолютную шкалу
температур. Эти задачи впервые были решены В. Томсоном
(Кельвином) в 1858 г.
До сих пор в наших рассуждениях мы довольствовались
элементарными представлениями о температуре и, не вдаваясь
в подробности, рассматривали ее как параметр состояния.
В элементарной физике температуру определяют как величину,
которая характеризует тепловое состояние тела, выражаемое
часто как степень нагретости, и которая обладает следующими
свойствами. Если каждое из двух тел, приведенных в
соприкосновение, не становится ни холоднее, ни горячее, то мы говорим
о равенстве температуры этих тел. Если при соприкосновении
одно тело становится теплее, то мы говорим, что его
температура была ниже температуры другого тела. При такой оценке
более целесообразно применять некоторое третье тело, которое
является пробным и имеет общее название
термометрического тела или термометра. Приводя в контакт
термометр а с телом А (рис. 25), мы выжидаем, когда наступит
тепловое равновесие в этой системе, т. е. термометр примет
температуру, одинаковую с температурой тела Л, причем
теплоемкость термометрического тела должна быть много меньше
теплоемкости тела А. Если после установления равновесия мы
перенесем термометр к телу В, приведя его в соприкосновение
с ним, то при равенстве температур тел А и В термометр не
должен ни нагреться, ни охладиться. Так мы судим о
температурах тел А и В. Установление теплового равновесия
термометра и изучаемого тела можно обнаружить, используя
какое-либо свойство вещества, зависящее от температуры.
Таким свойством может служить, например, электропроводность,
изменение цвета термометрического
тела с температурой и т. д. Однако
чаще всего, как известно, пользуются
V/ тепловым расширением. На основании
I 1 У этого свойства строится термометри-
I п ^ ческая шкала температур. Все эти
представления о температуре являются
Рис. 25. примитивными, так как ими не доказы-
а
§ 5. Применение II начала к построению абс. шкалы температуры 111
вается само существование абсолютной температуры
как параметра, который в принципе не должен зависеть от
выбора термометрического вещества и от способа измерения.
Доказательство существования абсолютной температуры
основано на применении второго начала термодинамики и
сводится к следующим положениям. Известно, что сумма dU+dW
не является полным дифференциалом, так как представляет
собой бесконечно малое количество теплоты dQ, зависящей от
формы пути. Однако известно, что для простейшей формы
Пфаффа
dQ = dU + dW
должен существовать интегрирующий множитель %, после
умножения на который последнее выражение обращается в полный
дифференциал:
Из общего выражения второго начала
следует, что таким интегрирующим множителем является Я=—,
так как dS есть полный дифференциал. Тем самым
устанавливается строгое определение абсолютной температуры Т как
интегрирующего делителя в объединенном уравнении
первого и второго начал термодинамики, т. е. доказывается
существование самой абсолютной температуры. Этот
вывод непосредственно связан с доказательством существования
энтропии как функции состояния. Однако для обратимых
процессов второе начало дает уравнение Пфаффа б/5 = 0, откуда
следует вывод о существовании S как функции состояния.
Определение абсолютной температуры из термодинамических
законов получилось формальным, но оно является вполне
строгим. Другое — статистическое — определение абсолютной
температуры дается в молекулярно-кинетической теории и
основано на так называемой статистике Гиббса, которая излагается
в статистической термодинамике. Термодинамическое и
статистическое обоснования существования абсолютной температуры
оказываются равноценными, но в термодинамике
рассматривают абсолютную температуру как феноменологический
параметр состояния для макросистем.
Для построения абсолютной шкалы температуры также
должен быть использован общий физический закон,
устанавливающий принципиальную возможность ее измерения и построения
равномерной шкалы. Последнее является также принципиально
важным, потому что при использовании различных термометри-
112 Глава 4. Различные общие формулировки II начала и его приложения
ческих веществ Шкала получается, вообще говоря,
неравномерной из-за индивидуальных особенностей теплового расширения
этих веществ и связанной с этим неравномерности расширения
в данном интервале температур. Далее необходимо отметить,
что выбор нулевой точки обычных термометров является вполне
произвольным, между тем как абсолютная шкала температур
должна иметь нулевую точку, независимую от нашего
произвола, и такая точка принципиально должна рассматриваться
как абсолютный нуль температуры.
Эти задачи непосредственно решаются с помощью второго
начала термодинамики. Пользуясь теоремой Карно — Клаузи-
уса о независимости коэффициента полезного действия
обратимой машины Карно от рода вещества, можно в принципе
осуществить измерение температуры с помощью построения
обратимых циклов.
Пусть какое-либо тело подвергается обратимому
изотермическому расширению, т. е. объем его возрастает при постоянной
температуре Ти причем о постоянстве последней мы вфобще
можем судить и без термометра. Пусть расширение (рис. 26)
происходит по изотерме ЛВ, соответствующей некоторой (пока
не установленной) температуре. Заставим тело обратимо
сжиматься по изотерме CD при другой температуре Т2 (Г2<7\).
Пересечем обе изотермы адиабатами MN и PQ. Тогда мы имеем
обратимый цикл Карно, изображаемый фигурой abed, причем
его к. п. д. не зависит от рода вещества, и на основании
предыдущего можно написать:
Qi_q2 = Tl-T%
1 Qi тг
(4,2)
В
Это соотношение прежде всего дает возможность ввести
абсолютный нуль температуры, который мы определяем как такую
температуру холодильника обратимой машины Карно, когда
коэффициент полезного действия
становится равным единице или
100%. При этой температуре
Г2 = 0 количество тепла,
переносимое к холодильнику <?2,
равно нулю, а, следовательно,
вся теплота переходит в
работу. Как будет показано из
общих соображений (теорема
Нернста) в главе 8, эта
температура недостижима. Что ка-
сается температур Тх и Г2,
* введенных в уравнение (4,2),
Рис. 26. то они должны иметь одина-
N
§ 5. Применение II начала к построению абс. шкалы температуры 113
ковые знаки, т. е. могут быть обе или положительными, или
отрицательными. Из уравнения следует, что
о т
причем при выводе (4,2) предполагаются абсолютные значения
| Qi I и | Qa | , поэтому отношение -^- всегда положительно,
т. е. абсолютная температура должна быть
знакопостоянной. Положительная шкала температур, всюду принятая
в науке, есть условность, которая означает, что более высокой
температуре соответствует более нагретое тело. Существуют
некоторые очень редкие системы (стр. 254), для которых из опыта
найдена отрицательная абсолютная температура, но мы не
останавливаемся на этих «необычных системах».
Для сравнения температуры двух тел мы мысленно образуем
между ними обратимый цикл Карно, как указано выше, и,
вычисляя к. п. д. цикла, оценим, насколько температура одного
тела выше, чем у другого. Таким путем принципиально
возможно построение идеально равномерной шкалы, независимой
от особенностей термометрического вещества. Для
доказательства покажем возможность выбора равных интервалов
температур.
Введем несколько смежных циклов Карно, т. е. таких
обратимых циклов, чтобы холодильник одного цикла служил
нагревателем для второго, и т. д. Выберем такие условия, когда
полезная работа qy равная площади цикла у всех машин Карно,
была бы одинаковой. Тогда по предыдущему:
Я ^ т\ — тг . Я _ Тъ — Тз . __Я__ _. J\zl£±. •
Qi Тг ' Q2 T2 ' Q3 T3
Но из (3,4) следует, что -^- = —— и аналогично —^- = ——
Qi T-i Q2 7*2
и т. д. Поэтому, разделив первое из написанной цепи равенств
на второе, второе на третье и т. д., мы получим:
q-Q% = (7\--Г2).Г2 # qQ3 ^ (Т2-Т3)Т3 .
Qi- <7 ^i(^2 — Т'з) Qz-Я Т2'(Т3 — Г4)
откуда следует: 7\ — Т2 = Т2 — Т3 = Т3—Г4 = ..., т. е. равные
полезные работы отвечают равным интервалам температур.
В принципе возможно построение обратимых машин Карно,
дающих равные полезные работы. Следовательно,
принципиально доказана возможность построения температурной шкалы
с равными интервалами температуры, т. е. с одинаковыми
градусами.
Если построить обратимую машину Карно между кипящей
водой при нормальном давлении и тающим льдом при тех же
5 Заказ № 2479
114 Г лав а 4. Различные общие формулировки II начала а его приложения
условиях и принять интервал температур между этими телами
за 100° С, то тогда 7\ — Г2 = 100 и к. п. д. равен:
Qi-Q« = ЮО
Q Т '
Этим устанавливается возможность построения стоградусной
абсолютной шкалы, соответствующей шкале Цельсия, и
возможность нахождения величины градуса абсолютной шкалы.
Таким образом построенная абсолютная шкала температур
обладает следующими свойствами:
1) не зависит от рода термометрического вещества согласно
теореме Карно — Клаузиуса;
2) имеет абсолютный нуль температур;
3) температуры всех обычных тел положительны;
4) шкала может быть идеально равномерной, так как
одинаковым полезным работам в циклах отвечают равные
интервалы температур;
5) шкала может быть приведена к 100-градусной шкале.
Остается выяснить, где находится на шкале Цельсия
полученный абсолютный нуль температуры. Заметим, что
предыдущие выводы действительны для любого вещества в соответствии
с теоремой Карно — Клаузиуса, а значит, они верны в том
числе и для идеального газа. Так как идеальный газ в точности
следует уравнению Клапейрона, то он имеет коэффициент
объемного расширения а= 1/273,15 град~ху а отсюда следует, что для
идеального газа, работающего в указанных обратимых циклах
Карно, абсолютный нуль должен лежать на шкале Цельсия на
273,15° ниже точки таяния льда. Последнее видно из формулы
Гей-Люссака — Шарля и определения для идеальных газов
Т = 273,15 +*.
Найдя положение абсолютного нуля для идеального газа, можем
заключить, что и для всех веществ абсолютный нуль составляет
на шкале Цельсия to=—273,15° С.
Но из всех веществ свойства водорода наиболее близки
в широком интервале температур к свойствам идеального газа.
Поэтому шкала водородного термометра ближе всех подходит
к построенной выше абсолютной шкале температур.
§ 6. ПРИЛОЖЕНИЕ ВТОРОГО НАЧАЛА К ОПИСАНИЮ ПРОЦЕССОВ
ИЗМЕНЕНИЯ АГРЕГАТНОГО СОСТОЯНИЯ.
УРАВНЕНИЕ КЛАПЕЙРОНА — КЛАУЗИУСА
Метод обратимых циклов может быть применен для
описания процессов испарения и плавления. Выясним, как зависит
скрытая теплота парообразования от температуры. Допустим,
что один моль вещества совершает обратимый цикл Карно
§ 6. Приложения II начала. Уравнение Клапейрона—Клаузиуса
115
между температурами Т и Т — dT. Рассмотрим цикл, в котором
полученное и отданное количества теплоты столь малы, что
можно принять их бесконечно малыми. Тогда на диаграмме
(рис. 27) кривблинейная фигура ABCD, изображающая цикл
Карно, может быть без большой погрешности заменена
соответствующим параллелограммом. Найдем бесконечно малую
работу dW за один цикл. В выражение к. п. д. обратимого цикла
Карно
введем обозначения для нашей задачи: Qi — Qz=
Ti — T2 = dT. Тогда формула примет вид:
dW _ dT
dQT ~~ T '
откуда
dW^dQ^ — .
(4,3)
Из рисунка видно, что dW=площадь ABCD^ площадь ABFE,
или
dW = (AE)-(ab) = dp-dV, (4,4)
где dp — приращение давления при изохорическом процессе по
изохоре (Аа). Очевидно, для этого процесса
dp = fie-
= fie-
dT.
Тогда из (4,4) следует:
\9T v
dTdV.
(4,5)
Сравнивая (4,3) и (4,5),находим:
кл.
Величина, условно принятая как
d
представляет собой количество
теплоты, затраченной при
увеличении объема тела на единицу
объема при постоянной
температуре, нам встречалась ранее и
Q
Рис. 27в
116 Глава 4. Различные общие формулировки II начала и его приложения
мы назвали ее скрытой теплотой расширения (стр. 40).Из
формулы (4,6) следует выражение для этой величины
Пусть при парообразовании на превращение в пар какой-
либо жидкости расходуется теплота при постоянной
температуре. Количество теплоты, необходимой для превращения в пар
единицы массы жидкости при постоянной температуре,
называется теплотой парообразования LK. Видимым
эффектом парообразования является расширение тела. Плотность
вещества уменьшается и, следовательно, увеличивается удельный
объем, т. е. объем единицы массы. Если удельный объем
жидкости при температуре парообразования есть vm, а удельный
объем пара равен vn при тех же условиях, то можно найти
скрытую теплоту расширения в данном процессе. Очевидно,
Сопоставляя это выражение с (4,7), находим:
В этом соотношении производная (-^-) представляет собой
скорость изменения упругости насыщенного пара с
температурой. Как показывает опыт, для данного вещества эта величина
зависит только от температуры. Поэтому предыдущее равенство
можно написать в виде:
LK = T(vn-vJ-^r. (4,9)
Эта формула носит название уравнения Клапейрона — Клау-
зиуса и находит себе многочисленные применения. Прежде всего
она позволяет весьма точно рассчитать скрытую теплоту
парообразования какой-либо жидкости при заданной температуре.
Для этого необходимо знать удельные объемы жидкости и пара
при этой температуре, а также ход изменения упругости
насыщенного пара с температурой вблизи заданных условий. Все эти
величины могут быть заранее найдены из опытов.
Помимо этого, уравнение Клапейрона — Клаузиуса дает
возможность оценить изменение скрытой теплоты парообразования
с температурой. Однако было бы неправильным по виду этого
уравнения заключить, что скрытая теплота парообразования
изменяется пропорционально абсолютной температуре. Следует
учесть, что оба. остальных множителя также зависят от темпе-
§ 6. Приложения II начала. Уравнение Клапейрона—Клаузиуса 117
ратуры, причем -— возрастает, а величина (vn — vm) резко
убывает с ростом температуры. Сильное уменьшение разности
удельных объемов с повышением температуры приводит к тому,
что LK заметно уменьшается при росте температуры, как
показывает опыт. Например, для водяного пара при 0°С удельный
объем равен 210,7 м3/кг, тогда как при 100° С он составляет
всего 1,674 м3/кг. Удельный объем vm жидкости меняется
относительно мало.
При критической температуре vn = vm и теплота
парообразования обращается в нуль. Это свойство, дало возможность
Д. И. Менделееву назвать критическую температуру
температурой абсолютного кипения.
В условиях, далеких от критического состояния, всегда
^п>^ж, и поэтому в уравнении (4,9) можно пренебречь
величиной vm. Тогда
Применяя уравнение Клапейрона для идеальных газов, находим
приближенную формулу:
i A! (4,10)
4Т dT
Вводя определение скрытой теплоты парообразования как
теплоты расширения, мы находим из общего выражения для /,
что теплота парообразования должна состоять из двух частей,
так как из (2,9) было получено:
и, следовательно, согласно формуле (4,8) имеем:
)^V + P*V' (4'П)
где До = оп— vm — приращение объема единицы массы.
Первое слагаемое
представляет собой часть тепла, затрачиваемого на изменение
внутренней энергии вещества при парообразовании. Между
молекулами жидкости действуют силы сцепления и при переходе
жидкости в пар должна быть затрачена работа преодоления этих
сил при увеличении объема тела. Соответственно внутренняя
потенциальная энергия единицы массы пара больше, чем потен-
118 Г лава 4. Различные общие формулировки II начала и его приложения
циальная энергия соответственного количества жидкости. Второе
слагаемое в формуле (4,11) представляет собой работу
расширения, затраченную на преодоление внешнего давления при
парообразовании. Таким образом, в общем виде можно написать:
Обычно большая часть тепла при парообразовании
затрачивается на преодоление внутренних сил сцепления и только
относительно меньшая часть идет на работу расширения.
Уравнение Клапейрона — Клаузиуса было выведено для
самого общего случая, и потому его можно применять и к другим
процессам перехода вещества из одного агрегатного состояния
в другое (к так называемым фазовым переходам). По аналогии
с процессом парообразования в общем виде это уравнение
можно написать как
L = T(v2-v1)^r. (4,12)
Здесь L — удельная скрытая теплота превращения; v{ и v2 —
удельные объемы начальной и образовавшейся фаз. Подробнее
эти процессы рассмотрены в главе 7.
§ 7. ВЫВОД ЗАВИСИМОСТИ ВНУТРЕННЕЙ ЭНЕРГИИ ОТ ОБЪЕМА
ИЗ ОБЩЕГО УРАВНЕНИЯ ВТОРОГО НАЧАЛА
И ОЦЕНКА ЭФФЕКТА ДЖОУЛЯ — ТОМСОНА
Ранее было отмечено, что внутренняя энергия U является
вообще функцией параметров состояния. Поэтому ее можно
рассматривать как функцию температуры и объема. Найдем
зависимость U от объема. Так как dU есть полный дифференциал, то
Уравнение первого начала примет вид, если учесть формулу
(2,9):
Подставим это выражение в уравнение второго начала:
dS = —
имеем:
§ 7. Вывод зависимости U от V из общего уравнения II начала 119
Мы знаем, что dS — приращение энтропии есть полный
дифференциал. Применяя независимые переменные V и Т, находим:
Сравнивая (4,13) и (4,14), получаем:
(*L) B1(«L) „(iL\ eiff£) +p1 • (4,15)
[dTJv T\dTJv \ду)т Т[\ду)т И\т К' '
Дифференцируя первое равенство по V, а второе по Т, имеем:
dTdV T дТдУ И дУдТ
,
Т [дУдТ
±Л 1
дТ )v j
Приравнивая правые части этих уравнений, находим после
сокращений:
(*!L) =T(*!L) -p. (4,16)
\dV }т \дТ ]v H
Формула (4,16) может быть выведена и более простым путем,
если в общее выражение скрытой теплоты расширения (2,9')
подставить ее значение (4,7), найденное в предыдущем
параграфе.
Соотношение (4,16) имеет общее значение. В частном
случае для идеальных газов, для которых внутренняя энергия не
зависит от объема, производная) ) в (4,16) должна обра-
\дУ ]т
щаться в нуль. В этом легко убедиться, применяя уравнение
состояния идеального газа. Тогда
[dTJv V '
Подставляя в (4,16), находим:
ду)т У Р
Легко видеть, что для реальных .газов, подчиняющихся
уравнению состояния Ван-дер-Ваальса, внутренняя энергия зависит от
объема. В самом деле, из уравнения состояния (1,3') находим:
-«1 £-. (4,17)
у — ь у2
Поэтому
( dp \ = R
\дТ)у V — 1
120 Г л а в а 4. Различные общие формулировки II начала и его приложения
Подставляя это выражение в формулу (4,16) и учитывая (4,17),
находим:
=.кт__ л_т (418)
)т V — Ь М V'2 V '
В формуле (4,18) производная) ] > 0,так как правая часть
\ dV It
равенства всегда положительна; отсюда следует, что при
расширении реального газа при постоянной температуре внутренняя
энергия возрастает и это связано с действием сил сцепления
между молекулами, что учитывается так называемой
аттракционной константой а уравнения Ван-дер-Ваальса.
Общее выражение для второго начала позволяет также
найти зависимость энтальпии от давления, что является важным
для оценки эффекта Джоуля — Томсона. Энтальпия как
функция состояния была введена ранее (гл. 2) и было показано, что
она может быть выражена в виде
Н = U + pV.
Проведем расчеты, аналогичные тем, которые были сделаны
для вывода зависимости внутренней энергии от объема, и най-
„ / дН \
дем выражение для частной производной
V др !т
Из объединенного уравнения обоих начал термодинамики
исключим dU при помощи дифференциала dH:
dU = dH-d (pV) = dH — pdV-Vdp.
Тогда
TdS = dH — V dp,
откуда
dS-jrdH-jrVdp. (4,19)
С другой стороны, рассматривая Н как функцию р и Г,
находим:
Вставляя это выражение в формулу (4,19), получаем:
Введем S тоже как функцию переменных р и Т и тогда:
$ 7, Вывод зависимости U от V из общего уравнения II начала 121
Сравнивая это выражение с предыдущим, получаем:
\дТ )р Т \дТ V \др )т Т [\др )т J
Дифференцируем первое равенство по р, а второе по Т, находим:
^1 =(J\±()]\
dp \дТ )р]т [др [Т \дт)р\)т '
\-Ё-(Щ 1 = (А-±\(Щ _/h
[дТ \др )т\р \дТ Т \\дР )т \)р
Левые части этих выражений должны быть одинаковыми, как
вторые смешанные производные по Т и /?, следовательно, равны-
и правые части. Однако правая часть первого уравнения равна
нулю, так как она представляет собой производные от
постоянных р и Т по нижним индексам. Поэтому
\дт т [\др )т \)Р
Выполним дифференцирование по Т выражения в больших
скобках; находим:
l_r/i"\ -V] -(dV\ -о
Окончательно получаем:
Это выражение аналогично формуле (4,16) для производной
внутренней энергии по объему.
Ранее было указано, что опыт Джоуля с быстрым
расширением газа был видоизменен Томсоном, пропускавшим сжатый
газ через плотную пробку в цилиндре (см. рис. 6). При
проталкивании газа с большим давлением pi и с температурой 7\
позади пробки создавалось пониженное давление р2 и температура
изменялась до Т2 (обычно понижалась). Этот эффект
аналогичен расширению газа в пустоту, так как газ от объема Vi
расширялся до объема V2.
В приборе Томсона с каждой стороны находятся поршни
(рис. 6). Слева над сжатым газом при постоянном давлении pi
совершается работа проталкивания его через пробку с
площадью s. Эта работа AWi может быть представлена как
произведение силы, действующей на весь поршень Fi = pis на путь
прохода через пробку, равный /ii= —, т. е. AWl = Fihi = piVi.
Справа газ, расширяясь и толкая правый поршень, сам
совершает работу ДW2, которая может быть вычислена таким же
образом, т. е. AW2 = p2V2.
122 Г лав а 4. Различные общие формулировки II начала и его приложении
Согласно общему выражению первого начала имеем:
AW.
Процесс расширения газа в опыте проводится адиабатически,
т. е. без обмена теплом со средой, следовательно, AQ = 0, и тогда
AV + AW = О,
или
— AW.
Очевидно, что изменение внутренней энергии газа при
переходе его слева направо равно разности внутренней энергии
конечного и начального состояний, т. е.
Д L/ = i/2 — Ulm
Работа AW равна алгебраической сумме работ AWi и AWz с
учетом знака или
Тогда
U 2 — Vi
откуда
tfi + PiVi = t/1 + p,V1. (4,21)
Помня общее выражение для энтальпии Я, мы на основании
(4,21) приходим к выводу, что в приборе Томсона энтальпия
постоянна, т. е.
Н = const, или dH = 0.
Рассматривая Н как функцию р и Г, можно, следовательно,
написать:
Частная производная ( ) имеет простой смысл. В самом
деле, из определения H=U + pV находим приближенно:
(Ш.) e(«L\ +рШ =limf^ =Ср
\дт)р [дТ)р^Р\дт)р ьт->о[ьт)р р
по уравнению первого начала при делении на AT для /? = const
и при переходе к пределу. Следовательно, частная производная
энтальпии по температуре при постоянном давлении, равна
теплоемкости Ср. Аналогично частная производная внутренней
энергии по температуре при постоянном объеме есть
теплоемкость Су (стр. 42).
§ 7. Вывод зависимости U от V из общего уравнения II начала 1J23
Из равенства (4,22) получаем приближенно (для конечных
AT и А/?):
~~ Ар ~" Ср[др)т
Величина £ называется коэффициентом Джоуля —
Томсона, и она характеризует собой изменение температуры
расширяющегося газа с изменением давления. Вводя сюда
производную из равенства (4,20), находим:
с
Вспомним (стр. 18), что производная Г^) »
где а — коэффициент объемного расширения. Тогда
Обычно аГ<1, т. е. коэффициент £>0. Это значит, что при
понижении давления, когда Д/?<0, температура газа понижается
(ДГ<<0). Однако возможны случаи обратного эффекта, когда
£<0, и тогда при понижении давления температура газа растет
(ДГ>0). Точке поворота или температуре
инверсии Го отвечает условие £ = 0, когда
Т ~ 1
Найдено, что для воздуха температура инверсии
сравнительно высока и составляет около 600° К и потому возможны на
практике оба случая эффекта Джоуля — Томсона. Для
водорода точка инверсии примерно равна 200° К, т. е. величина аТ
меньше единицы (а = 0,002—0,003) только при очень низких
температурах. При обычных условиях для водорода наблюдается
лишь нагревание при расширении, т. е. коэффициент £
отрицателен. Знание температуры инверсии дает возможность выяснить
условия, когда возможно сжижение газов за счет использования
эффекта Джоуля — Томсона.
§ 8. ПРИМЕНЕНИЕ ВТОРОГО НАЧАЛА В ТЕОРИИ ТЕПЛОВОГО ИЗЛУЧЕНИЯ,
ЗАКОНЫ КИРХГОФА И СТЕФАНА — БОЛЬЦМАНА
Известно, что все виды испускания света можно разделить
на два типа: люминесцентное лучеиспускание и температурное
(или тепловое) лучеиспускание. Из них первое непосредственно
не связано с температурой источника света, тогда как второй
вид излучения зависит от температуры. Состав или спектр
124 Г лав а 4. Различные общие формулировки II начала и его приложения
излучения при температурном лучеиспускании является сложным,
т. е. энергия сложным образом распределена по длинам волн.
Лучеиспускательной способностью Е светящегося тела
называется величина, характеризующая собой распределение энергии
по длинам волн и позволяющая определить мощность лучистого
потока для данной компоненты излучения. Лучеиспускательная
способность является функцией длины волны и температуры,
т. е. Е=Е (Я, Г).Мощность лучистого потока dP (Я, Т)
представляет собой часть потока лучистой энергии с 1 м2 поверхности
тела в 1 сек, приходящаяся на длины волн в интервале от X до
X + dh при температуре Г, и она равна:
dP(K T) = E(kt T)dk.
Таким образом, величина Е(к, Т) имеет смысл удельной
мощности лучистого потока. Интегральный поток, т. е.
полное количество энергии на 1 м2 поверхности излучающего
тела в 1 сек для всевозможных длин волн в спектре излучения,
равен:
оо
Р= J£(X, T)d\.
о
Очевидно, размерности введенных выше величин таковы:
[Р] = вгп/м2 и [Е] = впг/м3.
Известно, что световая энергия, падающая на поверхность тела,
может частью отражаться, а частью поглощаться этим телом.
Лучепоглощающей способностью Л (Я, Т) тела называется
отношение количества поглощенной энергии к падающей на
поверхность тела за одно и то же время. Опыт показывает, что А
является функцией длины волны и температуры.
Для температурного лучеиспускания выполняется закон
Кирхгофа (1859 г.), состоящий в том, что при условии
равновесия отношение лучеиспускательной способности к лучепогло-
щательной для данной длины волны есть величина постоянная,
независящая от природы тела и зависящая только от
температуры. Таким образом, закон Кирхгофа можно математически
представить в общей форме соотношением:
где разные индексы вверху относятся к разным телам и
<§ (К Т) —универсальная функция длины волны и температуры.
Закон Кирхгофа можно вывести из простейших
термодинамических соображений, являющихся следствием второго начала.
Представим себе два тела из различных материалов и в каждом
из них небольшую полость. Нагреем оба куска и соединим их
полости короткой трубкой (рис. 28). Если первоначально тем-
§ 8. Применение II начала в теории теплового излучения
125
пература обоих кусков была различной, то при соединении
трубкой в полостях будут происходить следующие процессы.
Внутренняя поверхность тела 1 будет испускать некоторое
количество световой энергии за единицу времени и одновременно
поглощать энергию, падающую на эту поверхность за то же
время, т. е.энергию, идущую от стенок самой полости 1 и
приходящую от полости тела 2. То же имеет место и в теле 2. Вначале
в каждой полости количества испускаемой и поглощаемой
энергии будут отличаться друг от друга, но затем согласно второму
началу само собой произойдет выравнивание температур и
наступит состояние теплового равновесия, при котором в каждом
теле количество поглощаемой за 1 сек на 1 м2 энергии будет как
раз равно количеству испускаемой энергии за то же время с той
же поверхности. Это равновесие самопроизвольно нарушиться
не может, так как в результате возникла бы разность
температур обоих тел, что противоречило бы второму началу
термодинамики.
Будем рассматривать компоненту в составе излучения с
длиной волны от X до Х+АХ. При равновесии, когда температура Т
обоих тел постоянна, количество энергии, испускаемой с 1 м2
тела / для данной компоненты, равно по определению
Ег (X, Т) АХ. Количество поглощаемой энергии в полости того
же тела легко найдем, зная количество энергии / (А,),
падающей на ту же поверхность; тогда, если А' (X, Т) есть поглоща-
тельная способность тела У, то количество поглощенной
энергии равно А'(X, Т)-1(Х). При тепловом равновесии имеем по
предыдущему условие
£'(Х, Т).ДХ = Л'(Х, 7>/(Х).
Аналогичные рассуждения для тела 2 дают нам
£"(Х, 7УДХ = уГ(Х, 7>/(Х),
Рис. 28.
126 Г лав а 4. Различные общие формулировки II начала и его приложения
где 1(1) при равновесии то же, что в первом случае. Разделив
первое равенство на второе, находим:
£'(Х, Т) = Л'(Х, Т) или Е' (X, Т) ^_ Е" (X, Т)
Е"(К, Т) Л"(Х, Т) ' Л'(Х, Т) А" (X, Т)
Свойства пары тел, взятых нами, вообще произвольны. Поэтому
последнее равенство, где в каждой части имеются величины
с одинаковыми индексами, можно обобщить на любые тела и
тогда
Е'(КТ) = Е"(КТ) = Е'"(1,Т) ^ ...
Л'(Х, Т) А" (К, Т) A'" (k, T)
Так доказывается при помощи второго начала закон
Кирхгофа (4,23), причем постоянная с? (А,, Т) может быть функцией
длины волны и температуры, которые в приведенных
рассуждениях оставались неизменными.
Физический смысл константы ё(Х, Т) в законе Кирхгофа
весьма легко выясняется, если ввести представление об
абсолютно черном теле. Известно, что абсолютно черным телом
называется тело, способное поглощать все лучи, т. е.
всевозможных длин волн. Следовательно, для абсолютно черного тела по-
глощательная способность для всех длин волн равна 100%, или
А (X, Т) = 1. Абсолютно черное тело может также испускать лучи
разных длин волн. Применяя к абсолютно черному телу закон
Кирхгофа, мы должны знаменатель отношения (4,23) положить
равным единице. Тогда
Е^- ч (Х> Т) = £(Х, Т).
1
Следовательно, константа &(Х, Т) закона Кирхгофа представляет
собой лучеиспускательную способность абсолютно черного тела
для данной длины волны при данной температуре, т. е. §
зависит от А, и от Т. Известно, что свойствами абсолютно черного
тела обладает небольшое отверстие, высверленное в стенке
полого твердого тела. Это отверстие
действительно способно поглощать все
лучи, падающие извне, так как, если его
размеры малы по сравнению с
размерами полости, узкий пучок лучей,
попавший извне, испытывает
многочисленные отражения от стенок полости,
сопровождаемые поглощением; в
результате вся энергия пучка лучей
поглощается и наружу не выходит (рис.
29). Именно поэтому небольшое отвер-
Рис. 29. стие в стенке полости представляется
§ 8. Применение II начала в теории теплового излучения 127
нам всегда черным, например зрачок глаза, открытые окна
зданий днем снаружи, щели и т. п. Абсолютно черное тело можно
заставить испускать световую энергию, если стенки полости
нагреть до температуры выше температуры окружающей среды.
Как известно, абсолютно черное тело используется в практике
фотометрии и для него установлены наиболее простые и
универсальные законы излучения.
Одним из таких законов, который может быть
термодинамически обоснован, является известный закон Стефана — Больц-
мана: количество испускаемой энергии в 1 сек с 1 м2 абсолютно
черного тела пропорционально четвертой степени абсолютной
температуры
Р = а74, (4,24)
где а — универсальная константа, равная 5,6697 • 10~8 er/jw2«°K4.
Этот закон первоначально был найден опытным путем
Стефаном, а затем получен из термодинамических соображений
Больцманом, применившим при выводе второе начало
термодинамики.
Закроем отверстие в абсолютно черном теле и рассмотрим
состояние излучения в полости при постоянной температуре.
В этих условиях можно говорить о тепловом равновесии
излучения, когда оно имеет определенный постоянный объем.
Поведение света в данном случае напоминает собой газ, содержащийся
в закрытом сосуде. Энергия этого фотонного газа является
внутренней энергией системы. Плотность энергии и есть энергия
единицы объема, которая в общем случае может быть
представлена как
и=(—\ . (4,25)
\dV )т ;
Теоретические соображения, основанные на
электромагнитной теории света, указывают на существование светового
давления, которое было измерено в опытах П. Н. Лебедева (1902 г.).
Из этих соображений следует, что давление света равно одной
трети плотности излучения, т. е.
Р = \и. (4,26)
Таким образом, к равновесию излучения в полости
применимы общие законы термодинамики, и его состояние может
характеризоваться термодинамическими параметрами состояния.
С помощью второго начала термодинамики мы получили в
общем виде выражение (4,1§) для производной внутренней
энергии по объему при постоянной температуре:
\дУ)т~ \дт)у Р'
128 Г лав а 4. Различные общие формулировки II начала и его приложения
Подставляя сюда значения и и р для фотонного газа из формул
(4,25) и (4,26), находим:
u = ±T*L-±u9
3 дТ 3
откуда
зт
Интегрируем это уравнение и получаем:
где \nk — постоянная интегрирования. Из этой формулы
получаем:
и = kT\
Поток световой энергии, выходящей за 1 сек из полости через
отверстие с площадью s столь малых размеров, что плотность
энергии остается практически неизменной, равен:
W = ucs,
где с — скорость света.
Отсюда
Р = ~Г = ис
и тогда
где a = kc есть константа. Мы вывели таким образом формулу
Стефана — Больцмана.
Следовательно, с помощью второго начала термодинамики
мы смогли вывести два основных закона температурного
лучеиспускания, имеющих важное принципиальное и практическое
значение. Механизм излучения и возникновение спектра
абсолютно черного тела в термодинамике не могут быть выяснены.
Задачи
1. Сравнить изменение энтропии 1 моля воды при испарении при 100° С
и 760 мм рт. ст. с изменением энтропии при плавлении льда при 0° С и том же
давлении (£Кип=539 кал/е\ £пл=80 кал/г).
2. Скрытая теплота парообразования воды при 100° С и давлении 1 атм
равна ^ип—ЭТЗО кал/моль. Сколько теплоты при парообразовании
расходуется в этих условиях на расширение и сколько затрачивается на изменение
агрегатного состояния?
3. Вычислить скрытую теплоту парообразования ртути в точке кипения
(/кип=357,3°С, рк = 760 мм рт. ст.). Опыты показали, что значение
производной -т|г =13,81 мм рт. ст./град.
4. Скрытая теплота плавления нафталина равна 4563 кал/моль и точка
плавления 80,1° С. Найти изменение точки плавления с давлением, если при-
раЩение объема при плавлении равно 18,7 см3/моль.
Глава 5
ТЕРМОДИНАМИЧЕСКИЕ ФУНКЦИИ И НЕКОТОРЫЕ
ИХ ПРИЛОЖЕНИЯ
§ 1. ЗАМЕЧАНИЯ О ДВУХ МЕТОДАХ, ПРИМЕНЯЕМЫХ В ТЕРМОДИНАМИКЕ.
ХАРАКТЕРИСТИЧЕСКИЕ ФУНКЦИИ
В предыдущих главах было показано, что непосредственные
применения обоих начал термодинамики дают возможность
описать важные свойства термодинамических систем. Однако
в связи с растущими многочисленными приложениями
термодинамики в науке и технике были разработаны некоторые
специальные методы, совокупность которых и составляет вообще
то, что называется термодинамическим методом исследования
макросистем. Исторически сложилось два таких пути: метод
циклов и метод характеристических (термодинамических)
функций. Первый основан на введении искусственных обратимых
циклов при анализе какого-либо вопроса. Типичным примером
является построение абсолютной шкалы температур, где, как
мы видели (гл. 4), искусственно вводится ряд последовательно
связанных циклов Карно. Таким же методом было выведено
уравнение Клапейрона — Клаузиуса и можно было бы привести
много других примеров применения этого метода. Хотя он во
всех случаях приводит к правильному решению задачи, его
нельзя считать совершенным. Метод циклов требует чисто
искусственных построений и обходных путей при решении конкретных
задач. Естественно, что с развитием термодинамики он уступил
место второму методу, основанному на применении
термодинамических функций, дающему гораздо больше возможностей и
широко распространенному в настоящее время.
Необходимость введения термодинамических функций
вытекает из простейших соображений, высказанных Гиббсом.
Объединенное выражение первого и второго начал имеет вид для
обратимых изменений:
Для простейших систем, когда работа есть dW = pdV, имеем:
TdS = dU + pdV. (5,1)
Это уравнение содержит пять величин, имеющих значение
для решения какой-либо задачи: Г, S, (/, ру V. Все они могут
130 Глава 5. Термодинамические функции и некоторые их приложения
быть параметрами состояния или могут рассматриваться как
функции состояния. Для определения состояния простейшей
системы достаточно двух независимых параметров. Остается,
таким образом, 3 параметра, входящих в уравнение (5,1), которых
мы не знаем. Следовательно, для решения определенной задачи
мы должны иметь всего 3 уравнения вместе с уравнением (5,1).
Тогда в трех уравнениях будет содержаться 5 переменных
величин, и так как 2 величины могут быть выбраны произвольно, то
получим 3 уравнения с тремя неизвестными, т. е. задача
разрешима. Откуда взять два недостающих уравнения? Одним из
них в принципе может служить уравнение состояния,
связывающее между собой параметры состояния. Вторым уравнением
является соотношение между параметрами или функциями
состояния, вытекающее из свойств рассматриваемой системы или
задачи. Это второе уравнение, приводящее к полному
разрешению данной частной проблемы, содержит новую функцию,
которая и получила название термодинамической
функции. Впрочем, необходимо заметить, что выражение
зависимости этих функций от параметров состояния в явном виде
с помощью одних термодинамических'построений не может быть
найдено; для этого необходимо привлечение молекулярных
представлений, т. е. применение статистики. В термодинамике
главную роль играют производные термодинамических функций,
дающие связь между этими функциями и известными
параметрами состояния. Ясно, что к термодинамическим функциям
вообще может относиться любая функция состояния, например
внутренняя энергия или энтропия, если мы можем их определить
как независимые функции параметров состояния.
Среди термодинамических функций, число которых вообще
неограниченно велико, выделяют так называемые
характеристические функции, которые обладают тем свойством,
что при надлежаще выбранных параметрах состояния,
например х, у, частные производные характеристической функции
ф(л:,у) по параметрам равны тому или иному параметру
состояния; благодаря этому эти производные получают наиболее
простое выражение и ясный физический смысл. Очевидно поэтому,
что число таких характеристических функций, выбранных
независимым способом, сравнительно невелико. Применение этих
характеристических функций и составляет существо второго
метода термодинамики.
Необходимость введения характеристических функций в термодинамику
была впервые осознана еще Массье в 1869 г. в работе «О
характеристических функциях жидкостей». Гиббс отмечает, что Массье показал, как все
термодинамические свойства простейшей системы могут быть выведены из
одной-единственной функции, названной характеристической функцией этой
системы. Максвелл в 1871 г. в своей «Теории тепла» отметил, что многие
выводы термодинамики упрощаются, если рассматривать внутреннюю энергию
§ 1. Замечания о двух методах, применяемых в термодинамике 131
U как функцию объема и энтропии, т. е. принять £/ = /(У, 5) и считать U
характеристической функцией. Кроме того, он же ввел другую функцию
F=U—TS, которая также весьма упрощает выводы. Гельмгольц в 1882 г.
впервые обратил внимание на то, что функция F имеет простой физический
смысл, назвал ее свободной энергией и применял ее к решению многих
вопросов термодинамики.
Наиболее полное и последовательное приложение характеристических
функций содержится в знаменитых «Термодинамических работах» Дж. Гиббса
(1873—1878), где учение об этих функциях принимает стройное и
законченное выражение. Развитию этого метода способствовали также труды П. Дю-
гема (1886—1894).
Взамен многих функций, вводимых другими
исследователями, Гиббс вводит 4 функции, которые и называет собственно
характеристическими функциями; они исчерпывающе полно
решают задачу,'если были выбраны вполне определенные
независимые параметры. Ниже будет подробно рассмотрена
систематика построения этих функций.
§ 2. ОБЩАЯ СИСТЕМАТИКА ХАРАКТЕРИСТИЧЕСКИХ ФУНКЦИЙ.
СООТНОШЕНИЯ МЕЖДУ ЧАСТНЫМИ ПРОИЗВОДНЫМИ
(УРАВНЕНИЯ МАКСВЕЛЛА)
Систематика Гиббса непосредственно следует из
обобщенного уравнения термодинамики (гл. 3):
dU = TdS-%A>dak9 (5,10
которое в частном случае одного внешнего параметра ак
имеет вид
dU = TdS — Akdak. (5,2)
Это соотношение является исходным для вывода всех
характеристических функций. Прежде всего из него следует, что
наиболее целесообразно внутреннюю энергию U рассматривать как
функцию параметров 5 и а&, так как в этом случае выражение
(5,2), представляющее собой полный дифференциал функции
состояния С/, приводит к простым значениям частных
"производных. В самом деле, полный дифференциал функции U(ak,S) есть
Сравнивая это соотношение с равенством (5,2), находим:
(Ш.) =Ги№ = -^. (5,3)
\ dS )ak \dajs k K ' '
В частности, если работа Akdak совершается по преодолению
внешнего давления, то Ah = p и а& = У и dW=pdV.
/ dU\
=
\dSjy
132 Глава 5. Термодинамические функции и некоторые их приложения
Тогда уравнения (5,3) приобретают особенно простой смысл:
rp I dU\ /с .ч
Т и =—р. (5,4)
\dv)s H K '
Следовательно, U является характеристической функцией, если
ее рассматривать как функцию параметров V и S.
Вывод остальных характеристических функций основан на
специальном преобразовании равенства (5,2). Последнее
представляет собой выражение полного дифференциала, в котором
роль независимых переменных играют 5 и а&, тогда как две
остальные величины Т w Аи являются зависимыми. Очевидно,
(5,2) можно свести к общей форме:
dU1 = Xdx+Ydy9 (5,5)
где
Х = М*, У) и К = /«(*, У).
Поставим себе задачу произвести такую замену переменных
в равенстве (5,5), чтобы в одном из слагаемых или в обоих
переменные поменялись местами (преобразование Лежандра).Тогда
получится три полных дифференциала новых функций:
Можно показать, что это преобразование выполняется путем
вычитания из dU\ дифференциалов произведения
соответствующих сопряженных переменных: d(X-x), d(Y-y) и d(Xx+Yy).
Применим преобразование Лежандра к равенству (5,2).
1. Вычтем из (5,2) дифференциал d(TS):
= -SdT-Akdak. (5,6)
Мы получим полный дифференциал функции
F = U-TS, (5,7)
которая называется свободной энергией и является
в данном случае функцией Т и ah:
Сравним это выражение с (5,6) и получим:
§ 2. Общая систематика характеристических функций 133
Если в частном случае заменить ak=V и Ak = p, то производные
в (5,8) получают простой смысл:
Следовательно, F=U—TS является характеристической
функцией.
2. Учитывая знак минус во втором слагаемом в (5,2),
вычитаем из dU дифференциал — d(Ahah). Тогда:
dU + d (А^) = d(U + Akak) = TdS- Akdak + Akdak + akdAk =
= TdS + akdAk.
Новое выражение есть полный дифференциал функции
(5,10)
называемой энтальпией, т. е.
dH = TdS + akdAk. (5,11)
Следовательно, Н является функцией 5 и Ak.
Сравнивая это выражение с (5,11), имеем:
В частном случае, как раньше:
Отсюда следует, что энтальпия является характеристической
функцией.
3. Вычтем из обеих частей уравнения (5,2) дифференциал
d{TS-Akak).
Тогда в левой части уравнения получим:
dU — d (TS — Akak) = d(U — TS + Akak)t
а в правой соответственно будем иметь:
Т dS — Akdak — TdS — S dT + Akdak+ akdAk = — SdT + akdAk,
134 Глава 5. Термодинамические функции и некоторые их приложения
т. е.
d (U — TS + Akak) = — SdT + akdAk.
Сюда входит полный дифференциал функции
Z = U-TS + Akakt
(5,14)
называемой термодинамическим потенциалом
Гиббса, причем Z является функцией Т и Аи. Очевидно, теперь
= — SdT + akdA
Имеем также
и поэтому
dH-wldT +
-Щ dAk,
dAk jT k>
' dZ\ c I dZ \
— = — i и = ak.
"T ). IdAk JT
(5,15)
(5,16)
В частности, при Ah = p и а&= V, получим:
\дТ/р \др)т
Отсюда следует, что термодинамический потенциал Гиббса
также является характеристической функцией.
Больше аналогичных преобразований исходного равенства
(5,2), представляющего собой общее выражение обоих начал
термодинамики, мы сделать не можем. Таким образом, всего
получаются 4 характеристические функции, дающие наиболее
простое выражение обеих частных производных. Мы получили
4 пары соотношений: (5,4), (5,9), (5,13) и (5,16) или более
общих выражений (5,3), (5,8), (5,12) и (5,15); они находят себе
многочисленные приложения в разных вопросах.
Результаты общего анализа сведены для удобства в
таблицу, где принято Ак = р и а^= У.
Данные этой таблицы можно для
удобства запоминания представить в
форме так называемого мнемонического
квадрата (рис. 30), смысл которого
прост и очевиден. На нем четыре
функции условно показаны по сторонам
квадрата, в вершинах которого даны
соответствующие для каждой функции
параметры состояния, в чем легко убедиться
при сравнении с таблицей. Свойства
частных производных находятся при
помощи стрелок, расположенных по диаго-
Рис. зо. нали, причем движение вдоль по стрелке
§ 2. Общая систематика характеристических функций
135
Таблица
Характеристическая функция
Внутренняя
энергия-
Свободная
энергия
Энтальпия
Термодинамический потенциал
Гиббса
яачения
са
Обоз]
Гибб
е
X
С
именные
ачения
CQ о
о \о
U о
и
(Я)
F
и
Z
висимые
1енные
Ig-
ас ё
v, s
V, Т
,.з
Р, Т
ен-
з с внутр
энергией
Связ1
ней 3
и
U — TS
U + pV
U — TS +
Частные
производные
[dS jv
(~w)s = ~P
\дТ jv
[д\/~)т~~Р
(If) =T
I dp j s
VdTJp=~S
I \ = V
[dp It
дает положительное значение найденного параметра, а против
стрелки — отрицательное.
Частная производная характеристической функции Я,
взятая по параметру S при постоянном значении параметра р,
находится как параметр Г, лежащий на диагонали, проведенной
из угла S, причем Т положительно, так как движение в направ-
лении от S к Т идет вдоль по стрелке, т. е. ( ) = Т. Точно
\ dS Jp
dF
А =—Р и т. д.
так же получим
Полученные выше выражения частных производных в виде
восьми уравнений легко свести к 4 формулам. Для этого доста-
136 Глава 5. Термодинамические функции и некоторые их приложения
точно найти вторые производные по сопряженным независимым
переменным. Тогда получим из (5,4),. (5,9), (5,13) и (5,16):
др
dSdV
dTdV
\dv)s
dV
lT
дТ
dSdp
дТдр
= i9T\ ^ /0УЛ
V ар /s \ es L
dp
dT
Окончательно находим так называемые уравнения Максвелла
(1883 г.), которые им были получены путем геометрических
построений:
(JL\ =(Jp-
\dV)T \дТ
(HL) =-IJil\
\dv)s \ds)
(JL\ =
\dV)T
(дТ\ =/^_\
\dp)s \dS)p'
(—\ =-(—)
\др)т \дТ)р
(5,17)
Найденные формулы (5,17) называются также осно вн ы ми
дифференциальными уравнениями термодинамики.
Так как для их получения применялось общее выражение
первого и второго начал (5,1), то они являются их следствием и
справедливы в такой же степени, в какой можно считать
точными основные начала термодинамики. Применение этих
формул в частных задачах будет дано ниже в этой главе, а также
позднее в главе 8 и т. п. Наряду с формулами (5,17) широко
используются и сами характеристические функции. Из четырех
выведенных функций наибольшее применение получили
свободная энергия и термодинамический потенциал, так как они
выражены как функции величин V, р и Г, непосредственно
измеряемых на опыте, тогда как в(/иЯ входит энтропия S, которая
непосредственно из опыта не может быть найдена. В связи
с наибольшим значением в приложениях величин F и Z мы
остановимся подробнее на выяснении их физического смысла, а
затем рассмотрим ряд важнейших применений.
Заметим, что термодинамической функцией в общем смысле
может быть любой параметр состояния, например объем V или
§ 2. Общая систематика характеристических функций 137
энтропия S; можно получить разными способами множество
других термодинамических функций. Но параметры состояния,
взятые в виде таких произвольных функций, содержат сложные
соотношения между частными производными и только
собственно характеристические функции устанавливают простое
соотношение между частной производной и соответствующим
параметром, выраженное в таблице 1.
Убедимся в сказанном на примере энтропии. Из общего
уравнения (5,1) для простейших систем находим:
Тогда — = ( J ; -~- = ( ) . Первое из этих равенств
может быть написано какТ=[—-) , т. е. не дает ничего но-
\ dS Jv
вого, так как уже имеется в таблице. Второе можно
представить как
н \dV )и \dSJy\dV
Отсюда видно, что один параметр р связан с произведением
двух частных производных или параметр р связан со вторым
параметром Т. Следовательно, 5 не является
характеристической функцией. Можно показать, что введение объема V как
термодинамической функции также приводит к произведению
двух частных производных, т. е. V не является
характеристической функцией. Вообще, можно ввести и другие
термодинамические функции на основе уравнения (5,1), но они приводят
к громоздким выражениям и не отвечают требованиям к
характеристическим функциям. Наконец, для сложных систем, для
которых необходимо применять общее выражение (5,Г),
появляется ряд новых характеристических функций.
§ 3. СВОБОДНАЯ ЭНЕРГИЯ. УРАВНЕНИЕ ГИББСА — ГЕЛЬМГОЛЬЦА
Физический смысл понятия свободной энергии легко
выясняется путем вычисления работы в изотермическом процессе.
Пусть система находится в термостате при Т=const, и
рассмотрим обратимый процесс перехода ее из состояния / в
состояние 2. Работа в этом случае является максимальной (3,24), и
потому в дифференциальной форме
dU.
138 Глава 5. Термодинамические функции и некоторые их приложения
Отсюда интегрированием при Т = const находим:
WT=T(Si-Sl)-(Ui-Ul).
так как S и U — функции состояния и их изменение
зависит только от начального и конечного состояний. Полученное
равенство можно представить иначе, группируя величины с
одинаковыми индексами, тогда
WT = (Ul-TSl)-(U2-TS2).
Так как согласно определению свободная энергия есть
F = U — TS,
то, очевидно,
w т ~ L 1 L 2 ~~ LSl•
Это выражение показывает, что в изотермическом обратимом
процессе работа равна убыли свободной энергии. Можно,
следовательно, представить себе свободную энергию как некоторый
запа*с энергии, составляющий, очевидно, часть внутренней
энергии системы, и тогда работа в обратимом изотермическом
процессе получается за счет этого запаса свободной энергии. Если
изотермический процесс является необратимым, то
WT<-bF,
т. е. мы получаем работы меньше, чем возможно, так как часть
ее не используется за счет потерь при необратимом процессе.
Из этих соображений следует обоснованность самого термина
свободной энергии. Так как из определения ее видно, что
U = F + TS9
то это дает повод считать, что внутренняя энергия системы
может быть разделена на две части: одна часть ее —свободная
энергия F может быть превращена в работу при обратимом
изотермическом процессе и в этом смысле является как бы
«свободной», тогда как другая, равная
G = TS,
не может быть превращена в работу в том же процессе и ее мы
называем по Гельмгольцу «связанной энергией», т. е.
[свободная] У связанная!
U = I т I »
L энергия J L энергия J
ИЛИ
U = F+G.
Определение свободной энергии как той энергии, которая
может быть использована для работы и находится как бы в
нашем распоряжении, наводит на мысль об аналогии с потенци-
§ 3. Свободная энергия. Уравнение Гиббса—Гельмгольца 139
альной энергией в механике. В самом деле, механика изучает
только обратимые процессы, причем изменение температуры там
не учитывается, т. е. всегда молчаливо предполагается, что
Г = 0. Если не рассматривать видимых движений с большими
скоростями и ограничиваться равновесными, т. е. весьма
медленными процессами, то в такой механической системе
скорость и = 0 и кинетическая энергия равна нулю. Так как обычно
в механике энергия системы разделяется на кинетическую и
потенциальную энергию, то при /С=0 мы имеем дело с
потенциальной энергией П. Применим к механической системе наше более
общее выражение
U = F + Г5.
Потенциальная энергия представляет собой в механике ту
часть энергии, за счет которой совершается работа. Так как
в механических процессах Г = 0 и они вполне обратимы, то
отсюда следует, что
т. е. свободная энергия является потенциальной энергией
системы. Таким образом, в изотермических процессах свободная
энергия F играет ту же роль, что и потенциальная энергия в
механике. Связанная энергия в механике не рассматривается, так
как она остается неизменной и при Г = 0 она условно равна
нулю.
Происхождение связанной энергии легко выясняется, если мы
обратим внимание на то, что эта часть энергии обусловлена
конечной величиной энтропии системы в данном состоянии.
Связанная энергия с молекулярной точки зрения зависит от
неупорядоченных тепловых движений частиц (молекул) системы.
Согласно второму началу энергия этих движений не может быть
превращена в работу при изотермическом обратимом процессе,
так как это соответствовало бы уменьшению энтропии. Поэтому
эта часть энергии оказывается связанной. Энергия тела,
лежащего на столе, состоит из потенциальной энергии, обусловленной
внешней силой притяжения к земле; эта энергия является
свободной. Но оно, как всякое реальное тело, состоит из молекул
и потому обладает еще энергией, зависящей от движения
молекул, от их взаимного положения, а также от энергии внутри
самих молекул. Вся эта энергия является связанной и при
падении тела на землю никак не проявляется. В изогнутой
стальной пластине, в сжатой пружине свободная энергия зависит от
деформации и может быть использована, тогда как опять-таки
энергия молекулярного движения и часть энергии
взаимодействия соседних молекул тела является связанной. Из этих
простейших примеров видно, что в обратимых изотермических
процессах в термодинамических системах свободная энергия со-
140 Глава 5. Термодинамические функции и некоторые их приложения
ответствует потенциальной энергии механики, только в
термодинамике учитывается еще роль связанной энергии.
Если система находится в изолирующей оболочке, то
согласно первому началу U = const. Тогда росту энтропии
системы отвечает по уравнению U = F + TS увеличение связанной
энергии и уменьшение свободной энергии. Когда энтропия
достигает максимума, то свободная энергия минимальна.
Практически в этих условиях вся энергия оказывается связанной.
Напротив, при температуре абсолютного нуля связанная энергия
обращалась бы в нуль и вся внутренняя энергия системы
являлась бы свободной:
Это вполне понятно, так как при абсолютном нуле
прекращаются всевозможные неупорядоченные тепловые движения и
вся внутренняя энергия могла бы быть использована. К этому
вопросу мы еще вернемся в главе 8.
Наконец, следует отметить, что свободная энергия, как и
следовало ожидать, является потенциалом внешних сил,
приложенных к системе. Из курса механики известно, что отрицательные
частные производные потенциала по координатам (параметрам
состояния) дают компоненты сил. При r = const второе
уравнение (5,8) дает нам
\dak)T kt
т. е., действительно, k-я компонента внешних сил Ah выражается
через частную производную свободной энергии по параметру а^
Отсюда, как и следовало ожидать,
т. е. опять работа равна убыли свободной энергии. В частности,
если cik=V и Ak~p> то из (5,9) ясно, что
dFT = — pdV,
следовательно, работа при расширении в изотермическом
процессе при действии внешнего давления идет за счет убыли
свободной энергии системы.
Заметим еще, что свободная энергия имеет простой смыод
лишь в изотермическом процессе, тогда как в общем случае это
есть некоторая функция состояния, т. е. она зависит от
параметров К и Г. С повышением температуры при постоянном объеме F
уменьшается и мерой убыли свободной энергии на один градус
является энтропия, как это следует из уравнения (5,9), т. е.
) *
§ 3. Свободная энергия. Уравнение Гиббса—Гельмгольца 141
Введение понятия свободной энергии облегчает многие
выводы и решение задач термодинамики. В виде примера
остановимся на выводе выражения для скрытой теплоты расширения,
служащего исходным при получении уравнения Клапейрона —
Клаузиуса (стр. 116). Ранее методом циклов мы нашли, что
скрытая теплота расширения / при постоянной температуре
выражается соотношением:
dV \ дТ
Эта формула легко выводится, если мы возьмем второе из
.уравнений Максвелла (5,17), т. е. положим
Равенство этих частных производных следует из второй
производной свободной энергии. Так как dS= ——, то для Т=const
это выражение можно подставить в уравнение (5,17) и тогда
получим:
/ dS\ = J_ дОт =
[дУ)т Т dV
откуда
dQ
dV \дТ)у*
т. е. мы получили требуемое выражение для скрытой теплоты
расширения, не прибегая к искусственному циклу.
Связь свободной и внутренней энергии позволяет установить
ряд важных закономерностей, особенно в приложениях к
физической химии. Выведенное ранее выражение
не позволяет судить об этой связи потому, что, кроме F и £/,
сюда входит еще энтропия S, которая сама является функцией
состояния, зависящей от F. Однако искомую связь между F и U
легко найти, если учесть, что из определения F как функции V
и Т следует:
Подставляя это соотношение в предыдущее, находим:
(5Л8)
142 Глава 5. Термодинамические функции и некоторые их приложения
ИЛИ
{l (6Л9)
Формула (5,18) или (5,19) носит название уравнения
Гиббса — Гельмгольца и дает искомую связь F и U.
В приложениях это уравнение часто используется в несколько
ином виде. Существует множество разнообразных процессов, где
совершается работа в отсутствие видимого увеличения объема
и при постоянной температуре. Сюда относится большой класс
явлений в растворах, а также работа в электрическом и
магнитном поле, работа гальванического элемента, механические
деформации и т. д. Для анализа этих процессов полезно
пользоваться уравнением Гиббса — Гельмгольца. Рассмотрим переход
системы от состояния / к состоянию 2. Для них мы по
уравнению (5,18) имеем:
dF±
дТ ,у
Пусть переход произошел при постоянных объеме и
температуре. Вычитая из первого равенства второе, имеем:
Но убыль свободной энергии при постоянной температуре
равна работе, т. е.
V, Т — w у
Поэтому
\^\ (5,20)
Изменение внутренней энергии при постоянном объеме
обусловливает собой поглощение или выделение тепла. Если тепло
выделяется за счет химической реакции, то принято его считать
положительным, тогда как поглощенное тепло в химической
реакции принимается отрицательным. Поэтому
Окончательно уравнение Гиббса — Гельмгольца примет вид:
Мы используем в дальнейшем это уравнение для решения
некоторых задач.
§ 4. Термодинамический потенциал Гиббса и энтальпия 143
§ 4. ТЕРМОДИНАМИЧЕСКИЙ ПОТЕНЦИАЛ ГИББСА И ЭНТАЛЬПИЯ
Рассмотрение ряда принципиальных вопросов равновесия
в сложных термодинамических системах приводит к
необходимости применения термодинамического потенциала Z как
важнейшей характеристической функции, на что было обращено
внимание особенно в классических работах Гиббса. Здесь мы
пока рассмотрим свойства и физический смысл этой функции,
тогда как приложения ее будут даны ниже.
Термодинамический потенциал Гиббса, называемый также
изобарным потенциалом, или свободной
энтальпией, выражается общим соотношением
Z = U-TS + pV,
которое было выведено ранее (стр. 133), причем было показано,
что Z является характеристической функцией * при
переменных р, Т. Тогда
Уд^/р" И lip
Мы указывали, что работа системы слагается из работы за
счет увеличения объема и какого-либо вида работы, не
связанного с увеличением объема, например работы при механическом
движении или в электрическом поле, в гальваническом элементе
и т. д. Поэтому элементарная работа может быть представлена
в виде
dW =
где dWv — элементарная работа при неизменном объеме. В
соответствии с предыдущим равенством общее уравнение обоих
начал можно представить как
dU = TdS — dWv — pdV>
откуда
dWv = — dU + TdS — pdV.
Рассмотрим теперь работу в изотермическом процессе,
осуществляемом при постоянном давлении, т. е. положим Т-= const и
р = const. Тогда написанное выше выражение для работы dWv
примет вид:
* В некоторых руководствах термин термодинамического потенциала
применяется ко всем характеристическим функциям. Поэтому
термодинамический потенциал Гиббса можно было бы назвать термодинамическим
потенциалом в узком смысле.
144 Глава 5. Термодинамические функции и некоторые их приложения
откуда
Это равенство показывает, что в описываемом процессе работа,
не связанная с изменением объема, равна убыли
термодинамического потенциала, подобно тому как при 7=const работа при
постоянном объеме равна убыли свободной энергии. Таким
образом, если изотермический процесс совершается при
постоянном давлении, то положительная работа совершается за счет
некоторого запаса энергии, называемого термодинамическим
потенциалом. Отсюда видно, что эта функция имеет сходство со
свободной энергией, представляя собой как бы часть
потенциальной энергии системы. Однако эту аналогию нельзя проводить
далеко, так как термодинамический потенциал Гиббса
непосредственно связан с энтальпией системы.
Как было показано (стр. 39), энтальпия выражается общим
соотношением
и является характеристической функцией при переменных р и S.
В изобарических процессах изменение энтальпии равно
полученной теплоте (стр. 39, гл. 2), так как
Д# = AU + p-AV,
откуда в соответствии с уравнением первого начала следует, что
АН = AQP.
Поэтому нередко энтальпию называют тепласодержа-
нием, или тепловой фу н кцие й. При изобарическом
процессе выделяющаяся теплота соответствует убыли энтальпии, и,
наоборот, при передаче теплоты при постоянном давлении она
идет на увеличение энтальпии.
Связь функций Z и Я легко найти из общих выражений:
Z = U — TS+pV9
Н = U + pV.
Отсюда следует, что
H = Z + TS. (5,22)
Этот простой результат можно представить следующим
образом. Мы видим, что энтальпия системы состоит из двух частей.
Одна из них Z может быть превращена в работу в
изотермическом процессе при постоянном давлении и потому Z правильно
называть свободной энтальпией. Другая часть
энтальпии, а именно 7\S, в работу в этих процессах превращена быть
не может и является связанной, вполне аналогично тому, как
§ 4. Термодинамический потенциал Гиббса и энтальпия 145
связанной является часть внутренней энергии в изотермическом
процессе. Следовательно,
H = Z+Gpt
где GP = TS. Эта величина зависит от энтропии и температуры
системы. Напомним, что работа за счет свободной энтальпии не
связана с изменением объема.
Выведем уравнение Гиббса — Гельмгольца для
изобарических процессов.
Заменим в выражении для энтальпии (5,22) энтропию через
частную производную термодинамического потенциала:
иначе
(^ (5,23)
Это уравнение связывает Z с Н и вполне аналогично
уравнению для связи U и F, поэтому оно называется тоже
уравнением Гиббса —Гельмгольца. В приложениях при
описании процессов при постоянном давлении формулу (5,23)
преобразуют, вводя работу Wy. При изотермическом изобарическом
переходе из состояния 1 в состояние 2 из формулы (5,23)
следует:
ZTJ i rp I C/Zo
2 = n2 -f- / I —s.
откуда
Но так как
то
Далее мы знаем, что убыль энтальпии равна количеству
выделяющейся теплоты при р = const. В химических процессах эту
146 Глава 5. Термодинамические функции и некоторые их приложения
теплоту мы считаем положительной. Поэтому так как —Д#=*
= Qv* ТО
/ ЛН7_. \
(5,24)
' Р
Мы получили уравнение Гиббса — Гельмгольца в новой
форме.
В применении к расчетам теплоты химических реакций
уравнение (5,22) можно представить в виде
Q117 f АС /£ ОК\
р === и^р — * * ^«Ь» \Э,£О)
когда химическая реакция протекает при постоянном давлении
и если учесть, что Wp=—Z. Ранее мы видели (гл. 2, стр. 59),
что тепловой эффект Qv реакции при постоянном объеме равен
убыли внутренней энергии системы. Тепловой эффект реакции
при постоянном давлении равен убыли энтальпии
Так как H=U+pV, то, очевидно,
Qp = Qv-pW. (5,26)
Отсюда следует, что если при реакции происходит
расширение, т. ^. Д V>0, jo ^)p меньше, чем Qv. Напротив, при сжатии,
когда ДУ<0, то QP>Qv. Формулы (5,25) и (5,26) используются
в термохимии для расчета теплоты образования химического
соединения, поскольку Qp не зависит от формы пути.
Важная роль термодинамического потенциала Гиббса для
описания равновесия сложных термодинамических систем будет
выяснена в главе 6.
§ 5. ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА СМЕСЕЙ ИДЕАЛЬНЫХ ГАЗОВ.
ПАРАДОКС ГИББСА
Рассмотрим смесь из нескольких идеальных газов, не
реагирующих химически друг с другом. Примером подобной смеси
является атмосферный воздух, в котором составные части
в обычных условиях ведут себя практически как идеальные
газы. Состав смеси характеризуется числом молей различных
газов; пусть смесь состоит из Ni молей одного газа, N2 молей
второго и т. д. Вообще определенное количество смеси
выражается суммарным числом молей:
При равновесии смеси температура и давление ее постоянны и
она занимает определенный объем V. Для каждой составной
§ 5. Термодинамические свойства смесей идеальных газов 147
части, взятой в отдельности, выполняется уравнение состояния
Клапейрона:
PiV = NtRT.
Давление смеси газов по закону Дальтона равно сумме
парциальных давлений составляющих газов, т. е.
Каждый газ в смеси ведет себя так, как если бы он
присутствовал бы один, зангимая весь объем смеси.
Обратим внимание на процесс образования смеси газов и
посмотрим, как изменяются свободная энергия и энтропия в
результате смешения. Сначала найдем свободную энергию одного
рода газа, считая, что газ находится в равновесии.
Свободная энергия одного моля идеального газа может быть
вычислена, если известно выражение для энтропии. Ранее
(стр. 83) было найдено, что энтропия 1 моля идеального газа
выражается формулой:
где So — константа, зависящая от природы газа; значение этой
константы мы рассматривать не будем. Подставим выражение
для S в общую формулу для свободной энергии, относя F
к одному молю:
F = U — TS = U — T (CvlnT + R\nV + So) = — RT\nV+f(T)9
где f(T)—функция только температуры, так как известно
(гл. 2), что внутренняя энергия идеального газа зависит только
от температуры, т. е.
f(T) = U — ТС v In Т — TS0. (5,27)
Итак, свободная энергия 1 моля газа как функция объема и
температуры равна:
F = — RT In V + f (T). (5,270
Найдем изменение свободной энергии в изотермическом
процессе. Пусть объем газа меняется от Vi до V2 при условии
Т = const. Тогда
Отсюда
AF = F2 — Ft = RT In -£.. (5,28)
148 Глава 5. Термодинамические функции и некоторые их приложения
В общем случае было показано, что убыль свободной энергии
при постоянной температуре равна совершенной работе.
Поэтому из (5,28) находим:
-AF = RTln-^-= Wr
Мы получили известную формулу работы газа в изотермическом
процессе. Если Vi>V2i т. е. происходит сжатие, то из формулы
(5,28) видим, что А/7>0; следовательно, при сжатии газа
свободная энергия его возрастает. Когда V\<V2, то из (5,28) видно,
что Д/7<0 и, значит, при расширении газа его свободная
энергия уменьшается.
Пусть теперь имеется система, в которой порознь находятся
порции нескольких газов, например в цилиндре, разделенном
герметическими перегородками на части, причем температура и
давления газов одни и те же: Т=const и р = const. Количество
молей газов Ni9 N2i ..., Ni и объемы Vu V2i ..., V% различны.
Можно показать, что свободная энергия всей этой
совокупности еще не смешанных газов равна сумме свободных энергий
составных частей:
^ = 2ВД, (5,29)
где Fi — свободная энергия одного моля /-го газа.
Действительно, внутренняя энергия и энтропия обладают свойством
аддитивности, если можно пренебречь взаимодействием между
телами; следовательно, если U\ и Si — внутренняя энергия и
энтропия 1 моля i-то газа, то для всей системы:
U=^NiUi и S = ]?iNiS£.
Поэтому свободная энергия всей системы равна:
где
Для 1 моля i-ro газа по формуле (5,27х) имеем:
где — объем, приходящийся на 1 моль.
Поэтому
F = - RT 2 N, In -£- + S N,ft (T). (5,30)
В рассмотренной системе газы разделены друг от друга. Если
привести их в контакт, удаляя перегородки между ними, то, как
§ 5. Термодинамические свойства смесей идеальных газов 149
известно из опыта, они будут самопроизвольно смешиваться,
причем, так как температура и давление всех газов одинаковы,
при смешении не произойдет изменения этих величин. Однако
можно показать, что процесс смешения приведет к изменению
свободной энергии за счет расширения каждого газа при
образовании смеси, так как объем до смешивания был Vu а в смеси
он равен V. Самопроизвольное смешение газов называется
диффузией, и, как было отмечено ранее, этот процесс является
необратимым. Свободную энергию F' смеси газов можно считать
величиной аддитивной, так как энергией взаимодействия газов
можно пренебречь и поэтому можно по аналогии с формулой
(5,29) принять
Тогда для смеси газов свободная энергия выразится
соотношением (5,30), в котором вместо объема Vi каждого газа следует
принять объем всей смеси У, т. е.
f = ^ ВД' = - W 2 Niln y; + 2 ШП (W)
Изменение свободной энергии, полученное в результате
смешения, равно разности энергий после смешения и до него:
AF = F — F = — RT yNiln — + RT yN{\n-^- =
^tln^-. (5,31)
Так как V>Vi, то все величины в правой части этого
выражения являются положительными, откуда следует, что
AF<0.
Итак, при смешении газов свободная энергия системы
убывает.
Найдем теперь изменение энтропии при смешении газов. До
смешения энтропия системы, состоящей из газов, разделенных
перегородками, равна сумме энтропии, так как энтропия
обладает свойством аддитивности. Поэтому, пользуясь известным
выражением для энтропии 1 моля идеального газа, имеем для
всей системы:
где /i(7") — функция температуры вида
150 Глава 5. Термодинамические функции и некоторые их приложения
После смешения энтропия, очевидно, будет равна:
так как каждый из газов будет занимать общий объем смеси V.
Это выражение показывает, что энтропия смеси идеальных
газов равна сумме энтропии отдельных газов, когда каждый из
газов занимает при той же температуре объем сМеси. В этом
состоит так называемая теорема Гиббса. Изменение энтропии от
смешения («энтропия смешения») равно:
= jR2#.lnJL. (5,32)
Так как во всяком случае V>Vi, то, очевидно,
Д5>0,
т. е. в результате смешения энтропия системы увеличивается.
Причиной рхэста энтропии является расширение каждого газа,
так как температура и давление не изменились.
Возрастание энтропии системы, где происходит
самопроизвольное смешение разных газов, т. е. диффузия, показывает, что
этот процесс является необратимым, как видно из второго
начала термодинамики. Диффузия протекает сама собой и
самопроизвольное разделение газов невозможно, так как оно
приводило бы к уменьшению энтропии системы. Вместе с ростом
энтропии при диффузии имеет место уменьшение свободной
энергии. Так как мы имели в виду процесс при постоянной
температуре, то убыль свободной энергии равна работе системы.
Следовательно, для разделения смешавшихся газов надо
затрачивать работу, которая компенсировала бы собой уменьшение
свободной энергии, причем эта работа является минимальной,
поскольку она как раз необходима для того, чтобы энтропия и
свободная энергия остались неизменными. Фактически при
разделении газовых смесей приходится затрачивать гораздо
большую работу, вследствие многих сопутствующих процессов,
осложняющих технику разделения.
Следует отметить, что, хотя мы рассмотрели смешение газов
разного рода, тем не менее в окончательные выражения для А/7
и AS не входят величины, характеризующие собой природу
(молекулярные свойства) газов. Поэтому казалось бы, что при
смешении совершенно одинаковых газов мы должны получить те
же результаты в отношении изменения свободной энергии и
энтропии. Однако такое заключение является неверным, потому
что если смешиваются, например, два различных газа, то мы
§ 5. Термодинамические свойства смесей идеальных газов 151
можем на опыте регистрировать взаимное смешение обеих
порций газов, когда молекулы одного из них распределяются среди
молекул другого; тогда согласно (5,31) имеем:
Но если первоначально две порции вполне одинаковых газов
были разделены перегородкой при Т=const, p = const, то после
удаления перегородки никакого видимого процесса мы
наблюдать не сможем, так как молекулы были совершенно
идентичными. Значит, можно 'принять, что как при наличии
перегородки, так и без нее у нас был один и тот же объем газа
Vr=V/i+V/2, а поэтому
AF = - RT (N, + N2) In -£±£- = 0.
Тот же результат получим и для энтропии. Это значит, что при
«самодиффузии» газа свободная энергия и энтропия остаются
без изменения, как и следовало ожидать из-за отсутствия
термодинамического процесса смешения. Этот вывод называется
парадоксом Гиббса. Здесь парадоксальным является то,
что даже малейшее различие в свойствах газов уже дает
конечные изменения AF и AS, тогда как казалось бы, что в пределе
при бесконечно малых различиях свойств обе величины Д/7 и AS
должны стремиться к нулю. Вопрос этот в настоящее время
имеет вполне практическое значение, так как он касается
разделения изотопов или выделения из химически однородного газа
составляющих с молекулами, обладающими тонкими
различиями (например, радиоактивных в смеси с нерадиоактивными
с разными магнитными или оптическими свойствами). Полное
разрешение парадокса Гиббса не может быть дано в
термодинамике и относится к молекулярной статистике.
Согласно квантовой теории материальных частиц
непрерывного изменения атомов, молекул и атомных весов изотопов
вообще быть не может, т. е. нельзя переходить к пределу
бесконечно малого различия свойств — эти различия всегда являются
конечными. Следовательно, если состав смеси известен, т. е.
заранее можно сказать, что смешиваются разные газы, хотя и
близкие по свойствам, и формулы (5,31) и (5,32) надо
применять, как для смеси разных газов. Планк в своей «Теории
теплоты» пишет, что «химическое различие двух газов и вообще
всяких двух веществ не может быть представлено с помощью
некоторой непрерывно изменяющейся величины; здесь можно
говорить только о прерывных соотношениях или о равенстве или
неравенстве, подобно тому как это делают при сравнении двух
152 Глава 5. Термодинамические функции и некоторые их приложения
целых чисел». Отсюда видно, что теорема Гиббса неприменима
в предельном случае идеального совпадения всех свойств
смешивающихся газов.
§ 6. СВОБОДНАЯ ЭНЕРГИЯ ПОВЕРХНОСТНОГО СЛОЯ ЖИДКОСТИ
Известно, что в тонком поверхностном слое жидкости
действует так называемая сила поверхностного натяжения,
которую принято рассматривать как силу, действующую на длине
контура, ограничивающего поверхность жидкости. Отсюда
вытекает определение поверхностного натяжения а как силы,
отнесенной к единице длины контура. Это силовое определение
свойств поверхностного слоя жидкости наглядно и потому почти
неизбежно в элементарном курсе физики при первоначальном
ознакомлении с молекулярными явлениями в жидких телах.
С ним связано представление об упругой поверхностной пленке,
которую сравнивают с пленкой резины. Однако силовое
представление все же во многих случаях является недостаточным
при описании многочисленных и разнообразных поверхностных
явлений. Более целесообразно рассмотреть энергию
поверхностного слоя и связать ее с работой. Это легко
сделать, если учесть, что всякое выделение молекулы жидкости из
общего объема в область поверхностного слоя требует затраты
работы, так как при этом приходится преодолевать силы
молекулярного сцепления. Следовательно, на увеличение поверхности
жидкости необходимо совершать работу, которая переходит
в запас энергии в поверхностном слое. Таким образом,
молекулы поверхностного слоя обладают избыточной поверхностной
энергией, которая может быть легко вычислена.
Мы определим теперь поверхностное натяжение а как
работу WT, совершаемую при увеличении поверхности жидкости
на единицу площади, при постоянной темпера-
Q туре, т. е. в общем случае:
dWT
где ds — дифференциал площади. Так,
например, если мыльная пленка натянута на
проволочной рамке с передвижной перекладиной
(рис. 31) и удерживается небольшим грузом
в равновесии, то при очень медленном
увеличении груза, обеспечивающем хороший
теплообмен (T=const), совершенная работа
растяжения для обеих сторон пленки на пути А/г
-— равна:
Рис. 31.
^ , /
/
/
/
/
§ 6. Свободная энергия поверхностного слоя жидкости 153
Здесь / — сила, действующая на длине перекладины I. Очевидно,
/ = а/.
Поэтому
AW T = а/ДЛ.
Но так как величина /А/г есть приращение площади As одной
стороны пленки, то
AWT = aAs.
В пределе при бесконечно малом растяжении имеем:
dWT = ads.
Следовательно, величина поверхностного натяжения а,
определенная через работу, совпадает с той, которая была определена
как сила на единицу длины. Это видно также из сравнения
размерностей обеих величин.
При медленном уменьшении груза пленка сокращается и
работа совершается самой системой, так что поверхностное
натяжение можно определить как работу, совершаемую силами
сцепления при уменьшении поверхности на единицу площади.
Так как мы условились считать положительной работу,
совершаемую системой, то при сокращении поверхности, когда ds<0,
мы должны положить
dWT = — ods.
Растяжение пленки приводит к увеличению запаса
поверхностной энергии, тогда как при сокращении происходит ее
уменьшение. Однако было бы неправильным отождествлять
поверхностную энергию с внутренней энергией поверхностного
слоя, как предполагал Гаусс в своих работах по капиллярности.
Для выяснения свойств поверхностной энергии с
термодинамической точки зрения необходимо обратить внимание на хорошо
установленный опытный факт, указывающий, что при
растяжении жидкостной пленки она охлаждается, а при сжатии
нагревается. Следовательно, если жидкость, поверхность которой мы
изменяем, поместить в термостат, то при изотермическом
обратимом сжатии пленки необходимо отнимать тепло от системы и
передавать термостату, а при растяжении пленки сообщать ей
некоторое количество тепла.
Изотермический процесс изменения поверхности жидкости
можно описать, применяя обычное уравнение первого начала
в виде
dUT = dQT-dWT,
где dQr — количество тепла, передаваемого от термостата или
уходящего в термостат при Т=const, а dWT — работа изотерми-
154 Глава 5. Термодинамические функции и некоторые их приложения
ческого изменения поверхности пленки. Так как dQT = TdS при
постоянной температуре, то, очевидно, из уравнения первого
начала следует:
dUT — dQT=d (UT — TS) = — dWT.
Здесь по предыдущему dWT — — ods, тогда как выражение
в скобках есть свободная энергия. Поэтому
dFT = ads,
откуда
a = -^, (5,33)
где значок Т отмечает постоянство температуры.
Выражение (5,33) показывает, что при изменении
поверхности жидкости меняется свободная энергия поверхностного
слоя, причем поверхностное натяжение представляет собой
изменение свободной энергии при изменении поверхности на
единицу площади. Так как а>0, то очевидно, что при сокращении
пленки (ds<0) свободная энергия поверхностного слоя убывает,
тогда как при растяжении происходит увеличение запаса
свободной энергии. Поэтому следует считать, что поверхностная
энергия представляет собой не всю внутреннюю энергию слоя,
а его свободную энергию, так что остальная часть внутренней
энергии, зависящая от беспорядочного теплового движения
молекул, является связанной и при изменении поверхности пленки
не используется. Этот вывод позволяет вместо (5,33)
представить поверхностное натяжение просто как отношение
a = ^f (5,34)
и считать, что
FT = as.
Следует заметить, что выделение и поглощение тепла при^из-
менении поверхности относительно мало и потому
поверхностная свободная энергия близка к внутренней энергии слоя.
Затрачиваемая теплота Qt при изменении поверхности
жидкости связана с характером зависимости поверхностного
натяжения от температуры. Для вывода этой зависимости
обозначим через qT количество тепла, отнесенное к единице
площади поверхности жидкости, т. е.
Чт— s •
Ясно, что увеличению поверхности соответствует положительное
значение теплоты Qt.
§ 6. Свободная энергия поверхностного слоя жидкости 155
Как было показано выше (стр. 133),
dF\
дТ)у
Положим, здесь F = a.s. Тогда S = — s —(объем при изменениях
dT
поверхности во всяком случае неизменен). Далее, очевидно,
QT = TS. Поэтому
4j da
Т ~ ~ 'dF9
ИЛИ
Отсюда
da ___ Ят
dT ~" T
Из этого окончательного выражения следует, что, так как при
растяжении пленки qr>0, производная отрицательна, т. е.
— <0,
dT
а это значит, что при повышении температуры поверхностное
натяжение жидкости должно уменьшаться. Это следствие
термодинамического рассмотрения поверхностных явлений
подтверждается на опыте. Различными исследователями были
предложены полуэмпирические формулы, передающие изменение
поверхностного натяжения с температурой.
Этвеш показал, что для многих жидкостей оправдывается формула
где ум — молярный объем жидкости и Гкр — критическая температура.
Близкие по характеру зависимости были установлены Ван-дер-Ваальсом и Рамзаем
и Шилдсом. Формула Этвеша показывает, что при критической температуре
поверхностное натяжение обращается в нуль; это согласуется с
представлениями о критическом состоянии, хотя более справедливо было бы считать, что
в этих условиях поверхноаное натяжение жидкости совпадает с
поверхностным натяжением ее пара. Так как vM мало зависит от температуры, то можно
приближенно принять, что а падает по линейному закону с ростом
температуры, т. е. можно положить
a ^ a — р7\ (5,35)
где аир — константы. Тогда из уравнения Гиббса—Гельмгольца
156 Глава 5. Термодинамические функции и некоторые их приложения
получаем для единицы площади
s ^ dT
Если принять приближенную формулу (5,35), то =—р и тогда
dT
U
откуда следует, что а=—, т. е. внутренняя энергия поверхностного слоя жид-
s
кости практически не зависит от температуры.
Мы не будем касаться многочисленных поверхностных
явлений в жидких телах и остановимся лишь на адсорбции из
растворов, так как это явление представляет собой хороший
пример применения термодинамического метода описания процесса
с использованием понятия свободной энергии.
§ 7. АДСОРБЦИЯ ИЗ РАСТВОРОВ И ФОРМУЛА ГИББСА
Рассмотрим поверхностные процессы в растворах при
постоянной температуре, причем будем пренебрегать
электролитической диссоциацией, что допустимо, например, для водных
растворов органических веществ — спиртов, эфиров и т. п. При
Т=const свободная энергия данной массы раствора зависит от
величины поверхности, но, кроме того, она по-прежнему может
рассматриваться как функция объема или концентрации, т. е.
FT = F(V, s),
так как объем и концентрация обратно пропорциональны друг
другу
По представлениям Вант-Гоффа, молекулы растворенного
вещества ведут себя среди молекул растворителя подобно
молекулам газа, на что указывает существование осмотического
давления, которое пропорционально концентрации
причем к сильно разбавленным растворам применимо
уравнение состояния Клапейрона
pV = nRT,
или
§ 7. Адсорбция из растворов и формула Гиббса 157
Изотермическая работа изменения состояния раствора
зависит от изменения поверхности и концентрации и поэтому
имеет вид
(5,36)
Как было показано, эта работа равнл убыли свободной
энергии системы, т. е.
dWT = —dFT. (5,37)
Дифференциал свободной энергии, являющейся функцией s
и У, есть
Сравнивая (5,36) и (5,38) и учитывая (5,37), имеем:
Первое из этих уравнений нам уже известно, второе является
аналогичным выведенному ранее в общем виде (стр. 133).
Дифференцируя (5,39) по перекрестным переменным,
находим:
dsdV
откуда
Jl) • d*F = _ (dP\
dV )s' dVds ^ \ds)
(5'40)
При этом мы предполагаем, что поверхностное натяжение
зависит от концентрации раствора, так что производная по V
вообще не равна 0, а также что осмотическое давление меняется
с изменением поверхности. Опыт показывает, что действительно
концентрация раствора влияет на его поверхностное натяжение,
причем существуют вещества, для которых с повышением
концентрации а сильно уменьшается. Сюда относятся, например,
мыла, некоторые спирты, а также органические кислоты —
валерьяновая, масляная и др. Такие вещества иногда называют
поверхностно-активными. В других случаях
повышение концентрации приводит к некоторому (обычно небольшому)
увеличению поверхностного натяжения (неорганические соли,
кислоты).
С изменением поверхностной} натяжения раствора связано
практически важное явление адсорбции. Опыт показал, что
если взять две несмешивающиеся жидкости, например воду и
158 Глава 5. Термодинамические функции и некоторые их приложения
бензин, привести их в соприкосновение, а затем добавить в
систему какое-либо вещество, понижающее поверхностное
натяжение на границе раздела, то в тонком пограничном слое
образуется избыток этого вещества; как говорят, на поверхности
происходит адсорбция. При этом, конечно, в удалении от
границы соприкосновения в каждой жидкости концентрация
вещества будет различной, она оказывается ненормально
повышенной только в пограничном слое. Схематически это
распределение концентрации можно представить на диаграмме рис. 32.
Здесь схематически изображена граница раздела ab двух
жидкостей I и II. Кроме того, по вертикальному направлению
показана концентрация примеси. В тонком слое d концентрация
ненормально повышена, тогда как в удалении от границы
концентрация примеси постоянна, хотя и различна в разных средах.
Явление адсорбции объясняется молекулярными
взаимодействиями, но может быть описано термодинамическим методом.
Термодинамика адсорбции наиболее подробно была
рассмотрена в- трудах Гиббса, нашедшего основную закономерность
для этого процесса.
Назовем величиной адсорбции Г по Гиббсу избыток
растворенного вещества, содержащегося в поверхностном слое на 1 м2,
т. е. [Г] = кг/м2. В данном растворе при равновесном состоянии
часть вещества находится в объеме У, а часть адсорбирована на
поверхности. Поэтому массу m растворенного вещества можно
представить как
m = cV + Ts, (5,41)
где s — поверхность раствора и с — концентрация. Отсюда
с=^т^- (5>42)
При отсутствии адсорбции (Г = 0) концентрация выражалась
бы просто как с = —. Если происходит адсорбция, то
концентрация зависит от s и от V. Выведем уравнение Гиббса для
адсорбции.
В формуле (5,40) производные можно представить в виде:
Но из (5,42) следует, что
m — Vs
dV
= с_ /дс\ _ Г_
~ V \dsjv~~ V *
§ 7. Адсорбция из растворов и формула Гиббса
159
Подставляя эти выражения в формулы (5,43) и переходя
к уравнению (5,40), находим:
/il\ . / М = —(1Ц . (— —
\dcjs [ V) \dc)v [ V,
откуда после сокращения
(da \
Так как для единицы массы раствора p—cRT, то
Поэтому
г / др
откуда
дс ]т
■=■1 --г^1-
RT \dcjs
(5,44)
Это выражение представляет собой так называемую
формулу Гиббса. Она дает возможность рассчитать избыток Г
вещества в поверхностном слое. Легко заметить, что знак Г
зависит от знака производной, так как остальные величины
в (5,44) всегда положительны. Если производная отрицательна:
tl
т. е. с повышением концентрации раствора поверхностное
натяжение падает, то, как это видно из (5,44), величина Г>0, т. е.
имеет место адсорбция. Напротив, при поло-
жительной производной, т. е. (—) >0,
\ocjs
величина Г<0, что соответствует десорбции,
т. е. выталкиванию вещества из
поверхностного слоя. Опыт хорошо подтверждает это
следствие, вытекающее из термодинамики;
установлено, что именно
поверхностно-активные вещества обладают способностью
накапливаться в избытке на границе
раздела, т. е. как бы уплотняться в тонком
слое. Хорошо известна также адсорбция
газов и паров на поверхности твердого
тела за счет действия молекулярных сил,
вызывающих уплотнение вещества на
поверхности.
160 Глава 5. Термодинамические функции и некоторые их приложения
§ 8. ТЕРМОДИНАМИКА ГАЛЬВАНИЧЕСКОГО ЭЛЕМЕНТА
В гальванических элементах протекает химическая реакция
и связанный с нею процесс образования электродвижущей силы.
Термодинамика не может описать детали механизма
возникновения этой электродвижущей силы, но на основании опытных
данных может вполне точно установить баланс энергии и
указать ее источник. Заметим, что термодинамические расчеты
относятся только к обратимым элементам. В простейшем элементе
Вольты имеет место явление поляризации, обусловливающее
непрерывное падение э. д. с. и приводящее в конце концов к
полному прекращению тока в цепи. Такой элемент является
необратимым. Действительно, в нем содержится раствор H2SO4
с погруженными электродами Zn и Си, причем при действии
элемента цинк переходит в раствор, а на медном электроде
наблюдается выделение водорода; для обращения процесса
необходимо через систему пропустить ток от постороннего источника
в обратном направлении и тогда должен бы происходить
переход меди в раствор с выделением водорода на цинковом
электроде. В итоге система не перейдет в прежнее состояние и,
следовательно, элемент является необратимым. Напротив,
элементы, в которых применяются деполяризаторы и
дополнительные устройства, предотвращающие явление поляризации,
способны длительно давать постоянную э. д. с, и их можно считать
практически обратимыми. К ним относится известный элемент
Даниэля, схема которого показана на рис. 33. Оба электрода
Zn и Си находятся в сосуде с пористой перегородкой, причем
в левом отделении первоначально находится раствор H2SO4,
а в правом — раствор CuSO4. При действии элемента протекают
реакции
Zn + H2SO4 — Н2 + ZnSO4,
Н2 + CuSO4 — Си + H2SO4.
Таким образом, выделяющийся
водород поступает в правое отделение,
где он вытесняет из раствора медь
и не отлагается на медном электроде.
В элементе протекает обменная
ионная реакция:
Zn + CuSO4 — Си + ZnSO4,
которая течет в обратном
направлении при пропускании тока в элемент
от постороннего источника. Отсюда
следует, что элемент Даниэля явля-
Рис. зз. ется обратимым.
§ 8. Термодинамика гальванического элемента 161
Известные элементы Вестона, Лекланше и ряд других
также обратимы.
Причиной возникновения электрического тока в элементах
является химическая реакция, тем не менее было бы
неправильным на основании этого считать, что энергия
электрического тока равна теплоте химической реакции, т. е. что в
гальванических элементах электрическая энергия как раз равна
тепловому эффекту реакции. Можно показать, что этот случай
является исключительным.
Наблюдая работу гальванического элемента, можно
заметить, что температура его меняется, причем элементы одних
типов нагреваются, а другие — охлаждаются, т. е. при действии
элемента происходит выделение или поглощение тепла.
Известно, кроме того, что э. д. с. многих элементов зависит от
температуры. Эту зависимость можно характеризовать
температурным коэффициентом, равным изменению э. д. с. при изменении
температуры на один градус. Термодинамика связывает оба
указанных опытных факта и дает точное описание баланса
энергии в обратимых элементах.
В гальваническом элементе работа тока равна
где &—электродвижущая сила и q — количество электричества.
Если вести расчеты на 1 моль превращения вещества при
химической реакции в элементе, то следует ввести число Фарадея
F = 96 487 к на 1 грамм-эквивалент и тогда, очевидно,
4 = п¥у
где п — валентность химического элемента, участвующего в
реакции. Следовательно,
W = g-nF.
Эта работа не связана, конечно, с изменением объема
системы, а затрачивается на перенос заряда в электрическом поле.
Поэтому ее можно ввести в уравнение Гиббса—Гельмгольца
(стр. 142):
где Qv — тепловой эффект химической реакции. Так как W от V
здесь не зависит, то частная производная заменена обычной
производной. Подставляя сюда выражение для работы тока,
имеем:
g.nF^Qy + T.nF-^-. (5,45)
Отсюда б=__+Г —,
162 Глава 5. Термодинамические функции и некоторые их приложения
или
S-Jk-^т^. (5,46)
nF dT V ' '
Это уравнение дает ответ на поставленный вопрос об источнике
электрической энергии в элементе. Из (5,46) следует, что лишь
в особом случае, когда э. д. с. элемента не зависит от
температуры, электрическая энергия равна тепловому эффекту
химической реакции, т. е. тогда вся теплота как раз идет на работу
тока. Действительно, в этом случае
dT
Qv
и потому S = ——, или S-nF = Qv = W.
Обычно наблюдается изменение э. д. с. с температурой и при
этом возможны два случая.
1. Когда <0, т. е. с повышением температуры э. д. с. убы-
dT
вает. Тогда из уравнения (5,46) следует, что Qv>0 и
nF '
или
В этом случае химическая реакция связана с выделением
большого количества тепла. Часть этого тепла переходит в работу
тока, а остальная часть, очевидно, выделяется при действии
элемента. Если теплоотвод затруднен, то элемент будет
нагреваться, тогда как при изотермическом процессе, который здесь
имеется в виду, эта теплота должна отводиться в окружающую
среду (термостат).
2. Когда >0, т. е. с повышением температуры э. д. с. эле-
dT
мента увеличивается; при этом возможны два частных случая:
a) Qy>0; так как правая часть равенства (5,46)
положительна, то это значит, что в данном случае
или
S-nF >QV.
Это неравенство показывает, что в элементе тепловой эффект
химической реакции невелик и вся теплота, которая должна
§ 8. Термодинамика гальванического элемента 163
была бы выделиться, переходит целиком в работу тока при
действии элемента и, кроме того, он поглощает извне теплоту,
которая тоже переходит в работу тока. Следовательно, в
изотермическом процессе источником электрической энергии в таких
элементах служит теплота реакции и теплота термостата. Если
передача теплоты из окружающей среды затруднена, то при
действии элемента он охлаждается.
б) Qv<0. В таких элементах теплота реакции поглощается
из термостата и, кроме того, часть тепла поглощается для
совершения работы тока. Следовательно, в данном случае
источником электрической энергии является запас энергии
термостата, а не энергия химической реакции.
Опыт хорошо подтверждает все перечисленные случаи
работы элементов, причем осуществление того или иного частного
случая зависит исключительно от специфичности химической
реакции в элементе данного типа.
Например, опыт показывает, что для элемента Даниеля
температурный коэффициент э. д. с. отрицателен. При температуре
15° С э. д. с. равна £ = 1,0934 в\ тепловой эффект реакции
Qv = 231,07 кдж/моль (55,189 ккал/моль). Эти данные
позволяют найти . Применяя уравнение (5,45), в котором работу
выражаем в килоджоулях, имеем при Г=288°К:
1,0934.2-96 487.10"3 =231,07 + 2-96 487.288.—,
откуда
~ = - 0,00036 в/°К.
Опыт дает близкую величину:--—= — 0,00043 в/°К. Далее от-
сюда видно, что работа тока равна S-nF = 211 кдж/моль.
Следовательно, из 231,07 кдж в электрическую энергию
превращается 1,0934-2-96487. 10~3=211 кдж, а остальные 20,07 кдж
выделяются при действии элемента, который, следовательно,
должен нагреваться, если не обеспечить хорошего теплоотвода.
Опыт подтверждает этот качественный вывод.
Задачи
1. Показать, что характеристические функции связаны между собой
соотношением: U + Z = H-\-F.
2. Проверить размерности в формулах (5, 17).
3. Найти свободную энергию газа, следующего уравнению Ван-дер-
Ваальса.
164 Глава 5. Термодинамические функции и некоторые их приложения
4. Стержень длиной / растягивается силой /, имея температуру Т. Считая,
что объем и давление стержня неизменны, написать полные дифференциалы
функций F и Z, вводя параметры состояния применительно к данной системе.
По общим формулам найги частные производные функций F и Z и
установить физический смысл этих производных.
5. Найти изменение свободной энергии и энтропии после смешения 1 моля
кислорода и 3 молей азота, взятых при равных температуре и давлении,
причем отношение первоначальных объемов составляет 2 : 3.
6. В свинцовом аккумуляторе протекает химическая реакция:
РЬ + РЬО2 + 2H2SO4 ^ 2PbSO4 + 2Н2О.
Если эту реакцию провести без аккумулятора в калориметре, то выделяется
теплота 381,4 кдж/моль (Qv =91,1 ккал/моль). Показать, что при обратимой
работе аккумулятора происходит поглощение энергии из окружающей среды
в количестве около 14,2 кдж/моль (э. д. с. аккумулятора равна 2,05 в).
7. Мыльный пузырь, имеющий начальный диаметр 2 см, раздувается то-
термически до диаметра 10 см. Каково изменение свободной энергии этой
системы? (а=23 дн/см).
Глава 6
ОБЩАЯ ТЕОРИЯ РАВНОВЕСИЯ СЛОЖНЫХ
ТЕРМОДИНАМИЧЕСКИХ СИСТЕМ
§ 1. ОБЩИЕ УСЛОВИЯ ТЕРМОДИНАМИЧЕСКОГО РАВНОВЕСИЯ
И ЕГО УСТОЙЧИВОСТИ
В этой главе будут рассмотрены термодинамические свойства
сложных систем, но перед этим необходимо более подробно
обсудить общие условия равновесия и вопрос об устойчивости
термодинамического равновесия в общем виде.
Ранее было указано, что термодинамическое равновесие
представляет собой такое состояние системы, когда в ней могут
происходить только обратимые весьма медленные процессы.
Если система находится при неизменных внешних условиях, то
она с течением времени самопроизвольно приходит к этому
равновесию, которое затем сохраняется неопределенно долгое время,
пока внешние условия неизменны. Именно по этой причине
в термодинамике (термостатике), изучающей только
равновесные состояния и процессы, не фигурирует фактор времени.
Для вывода условий равновесия рассмотрим сначала в виде
примера простую механическую систему. На рис. 34 показаны
шарики, находящиеся на поверхностях разной формы. Шарики,
лежащие на выпуклой поверхности, или на горизонтальной
плоскости, или в углублении, будут находиться в равновесии, когда
центр шара и точка опоры лежат на вертикальной прямой. Это
условие является необходимым и достаточным для равновесия,
однако оно не указывает на «качество равновесия», которое
в этих случаях является различным. Для того чтобы оценить это
качество, необходимо произвести некоторое испытание, которое
сводится к тому, что системе сообщают весьма малое, но
возможное смещение от положения равновесия и тогда по результатам
этого «виртуального» смещения судят о бывшем перед этим
равновесии. Если система в результате виртуального смещения
удаляется от положения равновесия, то последнее было
неустойчивым; если система вновь возвращается к положению рав-
У/////////////,
Рис. 34.
166-Г л а в а 6. Общая теория равновесия сложных термодинамических систем
новесия, то оно было устойчивым, и если, наконец, система
может быть в равновесии после виртуального смещения, то
равновесие было безразличным. Будем называть вариацией
переменной величины х произвольное сколь угодно малое изменение
этой величины дх. Очевидно, вариация весьма сходна с
понятием дифференциала переменной dx. Различие состоит в том,
что дифференциал соответствует действительно происходящему
изменению переменной, тогда как вариация представляет собой
любое возможное (виртуальное) изменение данной переменной.
Операция варьирования во многом аналогична
дифференцированию функции. Так, если y = f(x) есть функция х, то при
сообщении величине х вариации 8х виртуальное изменение у есть
dx
Вариация второго порядка равна о2у = —-(Щ2 и т. д.
Наиболее общим условием равновесия в механике является
положение, чтобы сумма возможных (виртуальных) работ была
равна нулю, т. е. чтобы виртуальная работа 6W была равна
нулю:
Этому условию соответствует экстремум потенциальной
энергии, которая может быть или минимальной, или максимальной.
Но равенство 6W = 0 не определяет собой качества равновесия,
поэтому необходимо произвести испытание. Если при смещении
дай от равновесия потенциальная энергия минимальна, т. е.
необходимо затрачивать положительную работу и по известному
условию для минимума 62lF>0, то равновесие было устойчивым
(шарик в углублении). Но если при смещении Ъаъ. система сама
затрачивает работу и потенциальная энергия максимальна, когда
62№<0, значит, равновесие было неустойчивым (шарик на
выпуклой поверхности). Безразличному равновесию соответствует
g2^ = o, и тогда требуется дополнительный анализ.
Условия равновесия механических систем являются частным
случаем условий равновесия более сложных по своей природе
термодинамических систем, для которых, однако можно
применить аналогичный способ рассуждений при выводе общих
условий равновесия. При заданных неизменных ^внешних условиях
термодинамическая система самопроизвольно переходит в
равновесное состояние, которое, такимобразом, соответствует
устойчивому равновесию. Неустойчивые (метастабильные) равновесия
также рассматриваются в термодинамике, но пока мы их
касаться не будем. Направление самопроизвольных процессов,
протекающих в системе при переходе к равновесию, наглядно отра-
§ 1. Общие условия термодинамического равновесия и его устойчивости 167
жается на. характере изменения термодинамических функций.
Поэтому условия равновесия лучше всего выразить через
посредство этих функций.
Все условия равновесия для различных внешних условий
выводятся из объединенного закона для первого и второго начала,
как для обратимых, так и для необратимых процессов,
представляющего собой известное нам соотношение (гл. 3):
или в более простом, но достаточно общем случае:
TdS^dU + pdV. (6,1)
Знак равенства соответствует равновесию системы, тогда как
знак неравенства отвечает неравновесному состоянию, когда
система стремится к равновесию и в ней еще не исчезли
необратимые процессы.
В связи с различными внешними условиями рассмотрим пять
важнейших частных случаев.
1. Полностью изолированная система при
неизменном объеме.
Здесь U = const и V=const. Тогда из неравенства (6,1)
следки
Т dS > 0. (6,2)
Отсюда видно, что при подходе к равновесию
dS>0
(так как Г>0), т. е. при указанных внешних условиях энтропия
системы возрастает; это представляет собой результат, нам уже
ранее известный. При достижении равновесия из (6,2) имеем:
dS = 0,
так как ТфО. Полученное равенство означает, что равновесию
соответствует максимум энтропии.
Как известно, при экстремальном значении какой-либо
функции первая производная обращается в нуль. Энтропия есть
функция параметров состояния. Поэтому, если в условиях равновесия
мы дадим небольшое виртуальное изменение любого
параметра х, то частная производная S по этому параметру должна
обращаться в нуль, а это значит, что первая-вариация S равна
нулю, потому что по определению
о,о dS <\
oS*= од:
х дх
и это выражение обращается в нуль при =0 (так как
Следовательно, в равновесном состоянии вообще
168 Г лава 6. Общая теория равновесия сложных термодинамических систем
Далее, при максимуме энтропии равновесие является
устойчивым. Максимум функции соответствует отрицательной второй
производной по соответствующей переменной. Поэтому при рав-
новесии <0, или 62S<0. Таким образом, условия равновесия
ох
при избранных внешних условиях имеют вид: 5 — максимум;
6S = 0; 62S<0.
Заметим, что необходимыми и достаточными являются два
последних условия равновесия, так как из них вытекает первое,
т. е. что при равновесии энтропия системы S максимальна.
2. Система находится в термостате при
постоянном объеме.
В этом случае T = const и V=const. Тогда из соотношения
(6,1) получаем:
или
dU — TdS^O. (6,3)
Известно, что свободная энергия выражается в общем виде
как
F = U — TS.
При r = const имеем:
dF = d(J — TdS.
Сопоставляя это выражение с соотношением (6,3), получим:
Таким образом, с течением времени, когда система
приближается к равновесию, свободная энергия убывает:
dF<0.
Следовательно, при заданных внешних условиях все
необратимые процессы в системе протекают в таком направлении, что
общая свободная энергия непрерывно уменьшается, пока,
наконец, не наступит равновесие, когда
dF = 0,
т. е. свободная энергия системы в равновесном состоянии
достигает минимума. Отсюда ясна тенденция изменения системы
в термостате при неизменном объеме: все процессы идут таким
образом, что свободная энергия стремится к минимуму,
следовательно, постепенно уменьшается полезная работа в системе.
Вновь рассматривая виртуальные изменения любых
параметров состояния, приходим к заключению, что при минимуме
свободной энеогии должно быть
oF = 0
§ 1. Общие условия термодинамического равновесия и его устойчивости 169
и, следовательно,
> О,
так как это неравенство соответствует минимуму функции F.
Отсюда условия равновесия: F — минимум; б/г = 0; 62/г>0.
Достаточно указать два последних соотношения, так как из них
вытекает первое.
3. Система находится в термостате при
постоянном внешнем давлении.
В этих условиях Т = const и р = const. Тогда из соотношения
(6,1) следует:
dU — TdS + pdV'^0. (6,4)
Известно, что термодинамический потенциал Гиббса
представляет собой функцию вида
Z = U — TS+pV.
При постоянных температуре и давлении из этого выражения
следует
dZ = dU — TdS + pdV.
Сопоставляя этот результат с формулой (6,4), находим:
dZ<0.
Отсюда следует, что если система находится в термостате при
неизменном давлении, то термодинамический потенциал или
свободная энтальпия постепенно убывает по мере приближения
к равновесию. Уменьшается, следовательно, тот запас энтальпии,
который может быть использован в качестве полезной работы
при постоянном давлении. До равновесия dZ<0, при равновесии
имеем: dZ = 0, т. е. условием равновесия является минимум
термодинамического потенциала. Аналогично прежним
рассуждениям при виртуальном изменении параметров состояния
находим: 6Z = 0; 62Z>0.
Таким образом, условия равновесия и его устойчивости для
данной системы можно сформулировать следующим образом:
Z — минимум; 6Z=0; 62Z>0.
Два последних условия являются необходимыми и
достаточными и из них вытекает первое.
4. Система занимает постоянный объем и
энтропия ее поддерживается неизменной.
В этом случае V=const и S = const. Тогда из
соотношения (6,1) следует
£Й/<0.
Следовательно, в данных условиях внутренняя энергия
системы убывает и при достижении равновесия имеет минимальное
значение, т. е.
dU = 0.
170 Г л а в а 6. Общая теория равновесия сложных термодинамических систем
Этот результат становится понятным, если учесть, что для
поддержания постоянства энтропии необходимо все время
отнимать энергию от системы и передавать эту энергию внешним
телам, иначе в необратимых процессах энтропия должна была бы
расти. По аналогии с предыдущими случаями сообщаем
виртуальные изменения параметрам системы и тогда 6f/ = 0;^ 62f/>0.
Отсюда следуют условия равновесия и его устойчивости для
системы: U — минимум; 6^ = 0; 82U>0.
5. Система находится при постоянном
внешнем давлении и энтропия ее поддерживается
неизменной.
Здесь, следовательно, р = const и 5 = const. Из соотношения
(6,1) теперь следует
dU+ pdV^O- (6,5)
Известно, что энтальпия выражается как
При заданных выше внешних условиях имеем:
Тогда выражение (6,5) примет вид:
d#<0.
Следовательно, в данном случае энтальпия или теплосодержание
системы непрерывно убывает с течением времени
dU <0
и при равновесии
dH = 0.
Таким образом, если поддерживать энтропию постоянной при
постоянном давлении, то энтальпия системы будет непрерывно
уменьшаться. При равновесии должно быть Н — минимум;
6# = 0; 62#>0.
Получаем таблицу условий равновесия:
Постоянные Условия
внешние равновесия
параметры
U, V Ь S = 0; 52 5 < 0.
V, Т Ь F = 0; 52 F > 0.
р, Т Ь Z = 0; 52 Z > 0.
V, S Ь U=z 0; 52 U > 0.
р, S Ь Н= 0; 52 Н > 0.
Заметим, что вместо V или р можно ввести любой другой
механический внешний параметр состояния и тогда получим тот
же результат, соответствующим образом определяя
термодинамические функции с помощью этого параметра.
§ 1. Общие условия термодинамического равновесия и его устойчивости 171
Необходимо иметь «в виду, что все разнообразные условия
равновесия мы получили как следствие обоих начал
термодинамики, т. е. эти условия могут быть приведены к теореме о росте
энтропии и постоянству энергии системы. Для этого достаточно
выделенную систему заключить в изолирующую оболочку вместе
с граничащими с этой системой телами и тогда для такой
большой системы вновь получается первый частный случай,
сводящийся к постоянству объема и внутренней энергии..
Формальные выводы условий равновесия полезны в том
отношении, что они дают представление об общем характере
изменения термодинамических функций при стремлении систем к
устойчивому равновесию. Устойчивость этого равновесия вполне
определяется виртуальными изменениями соответствующих
термодинамических функций. Применение вариаций является здесь
необходимым, так как оно позволяет точно установить
всесторонности термодинамического равновесия, т. е. найти
достаточные и необходимые условия его осуществления.
На простых примерах можно показать эту устойчивость
равновесия.
1. Сокращение жидкой пленки. В главе 5 было
показано, что поверхностное натяжение жидкости можно
рассматривать как свободную энергию единицы поверхности. При
равновесии жидкая пленка обладает минимальной свободной
энергией. Если растянутую пленку предоставить действию
поверхностных сил, то она самопроизвольно сокращается, т. е.
поверхность ее уменьшается, и так как поверхностное натяже-
dF
ние сг = положительно, то сокращению поверхности отвечает
ds
уменьшение свободной энергии. Равновесию при
изотермическом процессе и при V=const здесь отвечает минимум свободной
энергии в соответствии с предыдущими выводами и,
следовательно, минимум поверхности. Так можно объяснить сокращение
мыльного пузыря, выдутого на конце трубки, когда отверстие
трубки открыто. Здесь происходит уменьшение свободной
энергии с уменьшением поверхности. Иногда говорят, что системе
«выгодно» перейти в состояние с наименьшей энергией.
Шарообразная форма мыльного пузыря, а также шарообразная форма
мелких капель объясняется этим принципом. Можно показать,
что сферическая форма соответствует наименьшей поверхности
при данном объеме тела. Если действием внешней силы
сплющить капельку ртути, то после снятия внешнего воздействия
капля вновь становится сферической, так как эта форма отвечает
устойчивому равновесию.
2. Адсорбция из растворов. Было отмечено (стр. 156),
что адсорбция, т. е. аномальное накопление растворенного
вещества на границе раздела двух жидкостей, имеет место в том
172 Г лава 6. Общая теория равновесия сложных термодинамических систем
случае, когда поверхностное натяжение раствора убываете
повышением концентрации. Адсорбция происходит самопроизвольно,
и при постоянных температуре и объеме этот процесс также
объясняется переходом системы в состояние равновесия с
минимальной свободной энергией. Действительно, если поверхностное
натяжение убывает с возрастанием концентрации, то это значит,
что повышение последней ведет к уменьшению поверхностной
свободной энергии. Для системы «более выгодно» перейти в
такое состояние, когда растворенное вещество сконцентрировано
на границе раздела, так как этому состоянию соответствует
меньшая свободная энергия, чем при равномерном
распределении вещества во всем растворе.
3. Образование и рост кристалла. Правильная
геометрическая форма кристаллов, выращиваемых из раствора при
медленной постепенной кристаллизации, наводит на мысль
о действии некоторого регулярного закона роста. В одном
простейшем примере это становится наиболее наглядно, а именно
когда образуется элементный кристалл, состоящий из
однородных атомов. Каждый атом можно представить себе в виде
шарика и рост кристалла представить как упаковку шаров.
Основное условие этой упаковки состоит в том, что шары должны
быть уложены наитеснейшим образом. Именно в этом случае
энергия системы будет иметь наименьшее значение. На
расстояниях, превышающих радиусы атомов, последние
притягиваются друг к другу, тогда как при соприкосновении возникающие
значительные силы отталкивания препятствуют дальнейшему
сближению. Следовательно, касанию шаров соответствует
наиболее выгодное расстояние между центрами, когда энергия
системы минимальна. Каждый шар может
касаться не более 12 шаров одинакового
радиуса и нетрудно видеть, что такая группа
уже имеет вполне определенное правильное
строение (рис. 35). Дальнейшее
присоединение шаров идет с сохранением этой
правильности, и в результате соединения большого
числа таких шаров образуется тело с
упорядоченной структурой. Таким путем идет рост
кристалла в идеальном процессе и получается
правильная кристаллическая кубическая или
гексагональная структура, причина
образования которой заключается только в
стремлении системы к состоянию с -наименьшей
энергией при истинном равновесии. Отсюда
следует, что наблюдающиеся в реальном
кристалле дефекты упаковки соответствуют об-
Рис. 35. ластям избыточной свободной энергии.
§ 2. Условия устойчивости равновесия в применении к простым системам 173
§ 2. УСЛОВИЯ УСТОЙЧИВОСТИ РАВНОВЕСИЯ В ПРИМЕНЕНИИ
К ПРОСТЫМ СИСТЕМАМ
Выведенные ранее условия равновесия и его устойчивости
имеют общий характер. На примере простой однородной
системы можно показать, к каким конкретным требованиям они
приводят, т. е. какими свойствами должна обладать такая
система, чтобы находиться в устойчивом термодинамическом
равновесии в каком-либо выбранном случае. Рассмотрим простую
систему, находящуюся в термостате с температурой То и при
постоянном внешнем давлении ро. В § 1 было показано, что в этом
случае при равновесии необходимо, чтобы термодинамический
потенциал Гиббса Z был минимальным, что приводит к
выражениям 6Z = 0 и 62Z>0. Оба эти условия определяют собой
равновесие и его устойчивость. Для того чтобы раскрыть
написанные условия, дадим небольшое отклонение от минимума Z в
виде положительного изменения этой величины AZ>0. Если
принять условие постоянства температуры То и давления ро
в точке минимума Z, то при небольшом отклонении от
равновесия мы будем тождественно иметь:
AZ == Ш — TobS + pobV > 0, (6,6)
так как по определению Z=U—TS+рV-и потому, что надо учесть
вариации параметров 6S и 6V. При этом происходит изменение
внутренней энергии на величину Д£/.
Для нахождения второй вариации 62Z при определении
устойчивости необходимо в неравенстве (6,6) разложить Д£/ в ряд
Тэйлора, принимая во внимание по' крайней мере вторые
производные. Имеем:
dS ^ dV 2 V dS
2 _^L_ SS si/ + _^_ w2) . (6,7)
Из таблицы (стр. 135) видно, что I ) =Т и ( ) =—р.
\ dS , v \ dV /s
Подставляя эти выражения в формулу (6,7) и переходя к
неравенству (6,6), находим:
2 dW 8S W + — bV2) > 0. (6,8)
Первые два слагаемые вместе представляют собой первую
вариацию 8Z по определению изменения Z, т. е.
bZ = (Т — То) oS + (р0 — р) W.
174 Глава 6. Общая теория равновесия сложных термодинамических систем
По условию равновесия 8Z должно быть равным нулю. При
любых не равных нулю 8S и 8V это условие приводит к
требованию Т=Т0 и р = Ро> которое означает, что для равновесия
необходима неизменность температуры и давления. Эти условия
являются необходимыми. Устойчивость равновесия определяется
условием 62Z>0. Из соотношения (6,8) теперь находим:
~~ ~2~ \ dS* dSdV dV2 )
Для однородной простой системы термодинамический
потенциал является функцией двух независимых переменных. Из
курса дифференциального исчисления известно*, что экстремум
функции Y двух независимых переменных ху у зависит от
свойства дифференциала
dn dx+ 2dX dy +
2 \ дх* дхду * ду*
из которого следует, что минимум Y имеет место, когда
квадратичная форма вида
d2Y
д2У
дх2 ду2 \дх ду)
является положительной. В рассматриваемой задаче из
аналогичного соотношения (6,9), когда мы знаем, что 62Z>0,
получаем условие минимума Z в виде
d2U # d*U _ I d*U
dS* dV2 \ dSdV
которое, следовательно, является условием устойчивости
равновесия. Левая часть этого неравенства может быть представлена
в форме функционального детерминанта (якобиана), т. е. в виде
D
dVdS
dS dV dS2
Из предыдущего ясно, что
д (0U \ д (dU'
~dV~\dV~) 'W\ds'i
д I dU \ _g_ ( dU\
dS [dV ) dS [dS )
Благодаря этому якобиан принимает вид:
гд(-р)\ ( дТ\
dV Is \ dV v
(д(-р)\ (дТ\
{ dS )s [ dS )v
д(-р, Т)
(6,10)
* См.: Г. М. Фихтенгольц. Курс дифференциального и
интегрального исчисления, д. 1, М., 1947, стр. 474.
§ 2. Условия устойчивости равновесия в применении к простым системам 175
где справа представлена обычная условная форма записи
функциональных детерминатов в виде производной.
Из теории якобианов* следует их простое свойство перехода
от одних независимых переменных к другим, которое позволяет
расчленять якобиан, изображая его подобно производной
функции от функции. Поэтому якобиан (6,10) можно представить
в виде
D^ д(-Р, т) = д{р% т) д(у, т) б п
д(У, S) д(У,Т) ' д(У% S) ' V '
где осуществлен переход от переменных (V, S) к переменным
(Vf T). (В справедливости равенства (6,11) легко убедиться
путем непосредственной подстановки детерминантов и их
умножения.)
Тогда
d(V, T)
д(У, Т)
d(V, S)
А
dV )т
дТ)у
о
dV }т
дУ Js
дТ
dS
дТ_
dS
Подставляя эти выражения в формулу (6,11), находим:
\dV )т \ dS jv
) . Очевидно, что
dS Jv
V Q C
J
v = CvdT=TdSv, т.
п
V ~ теплоемкость
е.
при V=const имеем, как обычно,
I dS\ cv I дТ \ Т
\дт)у = ~ ' ИЛИ Vls"Jv = "^
постоянном объеме. Поэтому
\dV }т Cv
На основании сказанного выше детерминант D при устойчивом
равновесии должен быть положительным, т. е.
D —
дУ)т
(6,12)
* См.: Г. М. Ф и х т е н г о л ь ц. Курс дифференциального и интегрального
исчисления, т. 1, М., 1947, стр. 501.
176 Г лава 6. Общая теория равновесия сложных термодинамических систем
В этом выражении Г>0 и Cv>0. Производная (-^А свя-
\ dV )т
зана с изотермическим коэффициентом сжимаемости, который
по определению (гл. 1) есть
др )т
Следовательно, равновесие будет устойчивым, когда
1 Т
D = — — >0,
VV Cv ^
т. е. когда изотермический коэффициент сжимаемости у
является положительным. Иначе говоря, система будет находиться
в устойчивом равновесии, когда с увеличением давления объем
ее уменьшается. Если это условие не соблюдается, т. е. если
вблизи точки равновесия сжатие приводит к увеличению
объема, то равновесие неустойчиво и практически не имеет места
(гл. 7, § 5).
Наш анализ был проведен для системы, находящейся в
термостате с постоянным давлением. Можно было бы исходить
из других условий и тогда требовалось бы введение других
характеристических функций, но все равно при разложении AU
в ряд с учетом членов второго порядка получились бы
аналогичные условия устойчивости равновесия.
§ 3. ГЕТЕРОГЕННЫЕ СИСТЕМЫ. ФАЗА. КОМПОНЕНТЫ
Макроскопические системы, к изучению которых обычно
применяется термодинамический метод, являются вообще
сложными системами, т. е. они состоят из нескольких тел разного
состава в различных агрегатных состояниях, причем между
отдельными телами могут протекать разнообразные
химические реакции, процессы растворения, кристаллизации и т. п.
Такие сложные системы носят название гетерогенных
систем, и примерами их являются растворы, сплавы, смеси
газов, смеси жидкостей, кристаллов и паров и т. д. Очевидно,
гетерогенные системы близко соответствуют реальным
системам, которые никогда не могут быть совершенно однородными
по составу и другим свойствам. В самом деле, взяв даже
вполне чистую тщательно перегнанную воду, из которой
удалены все примеси, мы не можем назвать ее вполне однородной,
так как в ее состав вместе с обычной водой входит некоторое
макроскопическое количество тяжелой воды, содержащей
атомы изотопа водорода D в молекулах. Система, идеально
однородная по всем своим свойствам, называемая гомогенной,
представляет собой лишь ту или иную степень абстракции,
§ 3. Гетерогенные системы. Фаза Компоненты 177
когда мы отвлекаемся от всевозможных макроскопических
нарушений однородности и от присутствия всевозможных
примесей и загрязнений, имеющихся во всякой реальной системе.
Применение термодинамического метода является наиболее
ценным для изучения гетерогенных систем.
Равновесие в гетерогенных системах (так называемое
гетерогенное равновесие) представляется в виде условий
сосуществования отдельных частей системы при равновесии.
Эта задача была решена в работах Дж. В. Гиббса (1876 г.) и
была развита позднее Вант-Гоффом, Розебумом и другими
исследователями. В решении многочисленных вопросов
гетерогенного равновесия центральное место занимает так называемое
правило фаз Гиббса, которое является следствием условий
равновесия и которое дает возможность определить количество
фаз, могущих находиться в равновесии с отдельными частями
системы. С помощью правила фаз также можно точно
определить число независимых параметров системы.
Прежде всего рассмотрим важнейшие понятия,
применяемые в теории гетерогенного равновесия.
Фаза. Рассматривая гетерогенную систему, мы условимся
называть телом, или телесным комплексом, всякую
гомогенную механически отделимую часть такой системы.
Каждое тело, присутствующее в системе в достаточном количестве,
характеризуется различными физическими свойствами, среди
которых мы выделяем термодинамические свойства, т. е. объем,
давление, теплоемкость и т. п. Отдельные тела, обладающие
одинаковыми химическими и термодинамическими свойствами,
представляют собой одну фазу в данной сложной системе.
Отсюда следует, что фазами называются отграниченные друг от
друга видимыми макроскопическими поверхностями раздела
части системы. При этом каждая фаза в системе должна
присутствовать в таком большом количестве, чтобы нужно было
учитывать ее параметры состояния.
В настоящее время много изучаются так называемые
дисперсные системы (например, коллоидные растворы),
состоящие из дисперсионной среды и дисперсной фазы. К частицам
последней, имеющим весьма малые размеры, но состоящим из
большого числа молекул, понятие термодинамических
параметров применимо.
Понятие фазы нельзя смешивать с понятием агрегатного
состояния. Из элементарной физики известно, что существует
всего три агрегатных состояния — газообразное, жидкое и
твердое. Между тем по определению число фаз является
вообще неограниченным. Твердое агрегатное состояние химически
чистого вещества может быть в системе в виде нескольких
фаз с разными кристаллическими модификациями (например,
7 Заказ N° 2479
178 Глава 6. Общая теория равновесия сложных термодинамических систем
красный и желтый фосфор, ромбическая и моноклинная сера
и т. д.). Так же и жидкое агрегатное состояние в системе может
быть представлено в виде нескольких фаз. Приведем примеры
систем с различным числом фаз.
1. Жидкость и ее пар; в этой системе две фазы: жидкая и
паровая. Они разделены видимой границей и каждая фаза
может быть механически выделена из системы.
2. Раствор соли в воде и кристаллы этой соли; здесь две
фазы: жидкая и твердая. Граница раздела — поверхность
кристаллов.
3. Раствор этилового спирта в воде. В этой системе имеется
одна жидкая фаза.
4. Смесь воды и этилового эфира. В этой системе имеется
две жидкие фазы: насыщенный раствор воды в эфире и
насыщенный раствор эфира в воде. Обе фазы отграничены
поверхностью раздела и легко могут быть выделены из системы.
5. Ртуть и вода в одном сосуде. Здесь две жидкие фазы.
6. Ромбические кристаллы серы, соприкасающиеся с
моноклинными кристаллами серы. Эта система состоит из двух
твердых фаз.
7. Водный раствор соли вместе с кристаллами соли и льдом.
В этой системе три фазы: одна жидкая фаза и две твердые.
8. Атмосферный воздух. Здесь только одна фаза — газовая,
так как между составными частями газовой смеси нет
поверхностей раздела.
Из этих примеров видно, что число твердых или жидких
фаз в одной системе может быть неограниченно велико. Смесь
газов или паров следует рассматривать как одну фазу, так как
эта смесь всюду однородна по физическим и химическим
свойствам и не имеет поверхностей раздела между составными
частями. Так как газы могут смешиваться в любых отношениях,
то ясно, что во всякой системе газовая (или паровая) фаза
может быть только одна.
Компоненты. Гетерогенная система обычно состоит из
нескольких веществ, отличающихся между собой в химическом
отношении. Составной частью системы мы называем химически
однородное вещество, которое может быть выделено из данной
системы и может существовать в изолированном виде.
Независимые составные части системы, т. е. такие, содержание
которых в данной системе не зависит от содержания других
составных частей той же системы, называются компонентами.
Понятие компонента нельзя смешивать с понятием составной
части. Чтобы нагляднее представить смысл понятия
компонента, рассмотрим несколько примеров гетерогенных систем.
1. Система: вода — лед — водяной пар. В этой системе всего
один компонент — вода, представляющая собой одно хими-
§ 3. Гетерогенные системы. Фаза. Компоненты 179
чески однородное вещество. Было бы неправильным, исходя
из того, что вода состоит из водорода и кислорода,
рассматривать их в качестве компонентов данной системы, так как
количество водорода в воде зависит от содержания кислорода и не
является, следовательно, произвольным, т. е. независимым; то
же относится и к кислороду. Таким образом, в данной системе
имеется три фазы и один компонент (однокомпонентная
трехфазная система).
2. Смесь газов азота, кислорода и водорода при обычной
температуре. В этой системе имеется три компонента, так как
количество каждого газа в смеси независимо от количества
других газов. Это пример трехкомпонентной однофазной
системы.
3. Раствор хлористого натрия в воде. Здесь имеем два
компонента: NaCl и НгО. Хотя молекулы NaCl в воде
диссоциированы на ионы Na+ и С1~", но числа последних
взаимозависимы и, следовательно, по определению весь хлористый натрий
в системе представляет собой один компонент, т. е. данная
система является двухкомпонентной однофазной.
4. Система, состоящая из химически реагирующих веществ,
например СаСОз^СаО + СОг. Стрелки показывают, что в
системе происходит разложение и образование углекислого
кальция. Здесь имеются три составные части: окись кальция,
углекислый газ и углекислый кальций, однако по определению
в данной системе имеется два компонента, так как количество
всякой третьей составной части зависит от содержания двух
других. Следовательно, данная система является
двухкомпонентной и трехфазной, потому что в ней имеются две твердые
фазы: Са*СОз и СаО— и одни газовая СОг.
§ 4. ХАРАКТЕРИСТИЧЕСКИЕ ФУНКЦИИ ДЛЯ ГЕТЕРОГЕННЫХ СИСТЕМ.
ХИМИЧЕСКИЕ ПОТЕНЦИАЛЫ
В гетерогенной системе наряду с простейшими
термодинамическими процессами могут иметь место разнообразные
фазовые превращения, например процессы испарения, плавления,
кристаллизации и т. д., а также могут протекать различные
химические реакции между составными частями системы. Так,
в системе, представляющей собой смесь различных газов,
может происходить химическое взаимодействие между
некоторыми компонентами смеси. Если система состоит из водного
раствора какой-нибудь соли и кристаллов другого вещества,
то наряду с процессом растворения кристаллической фазы
возможно химическое взаимодействие ее с растворенным веществом.
Во всех таких процессах происходит изменение масс
компонентов, входящих в разные фазы- Поэтому термодинамический
180 Глава 6. Общая теория равновесия сложных термодинамических систем
анализ гетерогенных систем называют термодинамикой
систем с переменной массой. Очевидно, при
описании состояния гетерогенной системы недостаточно тех
параметров состояния, которыми мы до сих пор пользовались, и
необходимо дополнительно ввести параметры, которые
характеризовали бы массы различных компонентов в разных фазах
в данном состоянии. Целесообразно в качестве таких
параметров ввести концентрации составных частей в разных фазах,
определив их надлежащим образом.
Рассмотрим сперва однофазную систему, содержащую п
компонентов; примером ее может служить смесь газов или
раствор нескольких компонентов друг в друге. Пусть m', т",. ..,
т(п) — массы разных компонентов, выраженные в
килограммах, и пусть М\ №,..., Мп) — их молекулярные веса в
килограммах на моль. Тогда количество молей различных
компонентов системы равно:
#; ЛГ;...
М' М"
Эти величины пропорциональны массе и числу частиц
соответствующего компонента. Вводя значения Л№, целесообразно
определить концентрацию данного 5-го компонента как долю
числа молей его от суммарного числа всех молей 2ЛА*)
всевозможных компонентов в системе, т. е. выразить в виде:
-
Отсюда непосредственно следует, что
2 c{i) = с + с + ... + с{п) = 1. (6,14)
Это определение концентрации показывает, что если
концентрация каждого компонента выражена в относительных
величинах, то для п компонентов системы имеем (п— 1)
независимых значений концентраций, тогда как одна из них является
зависимой и может быть определена из (6, 14).
Если система состоит из п компонентов и k фаз, то
необходимо иметь концентрации каждого компонента в различных
фазах. Пользуясь относительными концентрациями согласно
(6, 13) для каждой фазы, получаем систему уравнений:
(6,15)
§ 4. Характеристические функции для гетерогенных систем 181
Здесь верхние индексы относятся по-прежнему к
компонентам, а нижние к фазам. Понятно, что в отдельных фазах
некоторые компоненты могут отсутствовать; тогда соответствующие
слагаемые в равенствах (6, 15) будут равны нулю. Выражая
концентрации через молярные доли из (6, 14), мы получаем
систему уравнений, аналогичную системе (6, 15).
Пусть даны компоненты в составе какой-либо фазы.
Очевидно, что так как массы компонентов изменяются за счет
перечисленных выше процессов, то внутренняя энергия
выделенной фазы j зависит не только от обычных параметров
состояния V и S, как ранее, но ее изменение связано с
изменением массы (или числа частиц) данных компонентов. Поэтому
в общее выражение для первого и второго начал для случая,
когда работа зависит только от изменения объема, необходимо
ввести слагаемое, зависящее от изменения числа частиц всех
компонентов данной фазы. Внутренняя энергия, как известно,
является величиной экстенсивной, т. е. обладает
аддитивностью, поэтому частное небольшое изменение ее за счет
изменения числа частиц компонентов пропорционально этому
изменению, т. е.
где |ш/г') — коэффициент пропорциональности.
Таким образом, из объединенного уравнения обоих начал
для обратимых процессов имеем:
dU = Т dS — р dV^r 2 H7°<WJ°. (6,16)
Здесь суммирование производится по всем компонентам дан*
ной /-й фазы.
Коэффициенты пропорциональности р.У>, которые здесь
введены, носят название химических потенциалов и по
определению они представляют собой изменение энергии на одну
частицу компонента при постоянных 5 и У, так как в (6, 16)
дан полный дифференциал энергии, т. е.
Так как внутренняя энергия входит в общие выражения
всех остальных характеристических функций, то последние,
очевидно, тоже зависят от изменения числа частиц компонентов
за счет различных (обратимых) процессов в данной
гетерогенной системе. Легко найти выражения полных дифференциалов
Для характеристических функций, учитывающие эти изменения.
Подставляя из (6, 16) выражение для dU, находим полные
дифференциалы функций F, N и Z.
182 Глава 6. Общая теория равновесия сложных термодинамических систем
1. Для свободной энергии F=«L/ — TS:
^ |i}° dNf\ (6,17)
2. Для энтальпии H=U+pV:
'Z4°dtf}°. (6,18)
p pZ(4
3. Для термодинамического потенциала Z=U — TS+pV:
dZ = dU — T dS + S dT + p dV + V dp =
= 5 dT + V dp + 2 jij0 diV}°. (6,19)
Все эти выражения представляют собой полные
дифференциалы соответствующих характеристических функций, и мы
видим, что в них, кроме дифференциалов известных ранее
параметров, входят еще дифференциалы.лэбусловленные переменным
числом частиц в компонентах. Из соотношений (6,16), (6,17),
(6,18) и (6,19) следует для какого-либо /-го компонента:
(6 20)
Понятие химического потенциала имеет важное значение при
анализе равновесия сложных гетерогенных систем, и на
свойствах этой величины необходимо остановиться.
Являясь величиной аддитивной, энергия пропорциональна
числу частиц системы. Все четыре характеристические функции
U, F, Н и Z представляют собой энергии разных видов, т. е. они
аддитивные величины, причем являются функциями
соответствующих параметров. Величины V и S также аддитивны.
Можно легко доказать, что все аддитивные параметры являются
однородными функциями других аддитивных параметров,
причем с первой степенью однородности. Из интересующих нас
параметров давление р и температура Т представляют собой
интенсивные величины, т. е. они не аддитивны. Все эти замечания
для простейшей системы из N частиц приводят к тому, что в
общем виде можно написать для всех характеристических
функций уравнения вида:
^, T^j = N-9i(v, T), (6,21)
-93
T).
§ 4. Характеристические функции для гетерогенных систем 183
Здесь все ф, 5 и v представляют собой удельные параметры на
одну частицу (или на один моль).
Выясним, какими свойствами обладают химические
потенциалы |аД Вернемся к соотношениям (6,21) и заметим, что фь
ф2 и фз сложным образом зависят от числа частиц. Поэтому
химические потенциалы, выраженные через соответствующие
функции £/, F и Н в равенствах (6,20), будут представлять собой
сложные функции экстенсивных величин. Мы видим, что только
термодинамический потенциал Гиббса Z является функцией
двух интенсивных параметров р и Т и потому удельный
потенциал ф4(р, Т), далее обозначаемый просто ф, не зависит от
числа частиц, т. е. является величиной интенсивной. Из (6,20) и
(6,21) имеем:
Отсюда видно, что химический потенциал вообще является
величиной интенсивной, следовательно, как бы ни были сложны
выражения фЬ ф2 и ф3, их соответствующие частные
производные являются величинами интенсивными. Этим свойством
оправдывается название химического потенциала, который
аналогичен вообще потенциалу, например, потенциалу
гравитационного, электрического и магнитного поля. Химический потенциал
связан с природой частиц и силами их взаимодействия. С
термодинамической точки зрения химический потенциал является
мерой изменения энергии в данной фазе за счет изменения
только массы определенного компонента, входящего в состав
этой фазы. Например, если в системе, состоящей из жидкости
и ее насыщенного пара, часть последнего конденсировалась, то
при данном объеме и данной температуре свободная энергия
жидкой фазы должна измениться и ее изменение на каждую
единицу массы представляет собой химический потенциал
жидкой фазы.
Мы учитывали разные компоненты лишь в одной
выделенной фазе. В гетерогенной системе в общем случае присутствует
k фаз и п компонентов, распределенных среди этих фаз.
Рассмотрим характеристические функции и их изменения для всей
сложной системы с большим числом фаз и компонентов.
Распределение концентраций различных компонентов зависит от
распределения масс (числа молей) по фазам системы.
Изменение масс компонентов в разных фазах происходит вследствие
фазовых превращений и химических реакций и благодаря этому
изменяется запас внутренней энергии, а также величин F, Н
и Z. Все четыре характеристические функции зависят не только
от введенных ранее термодинамических параметров состояния,
184 Глава 6. Общая теория равновесия сложных термодинамических систем
но также от масс отдельных компонентов в разных фазах.
Поэтому можно написать:
U = U(S, V, Nl N'u ..., N2, Nl . . . , Nin)),
F = F(V, 7\ N'u N'u ... 9 N2i N"2i . . . , Ni\
H = H(S, p, N\, N\9 . . . , N2i N2i • • • , Nkn))4
Z = Z(p, Г, N'u N'u • .., N'2, N'2, ... , Nnk).
Отсюда полные дифференциалы этих функций имеют вид:
jt/ i ^гч dU 1 \r(i)
\dV )s, nu tf dNf
dNf,
(6,23)
4Н=Ц£-\ , dS + l-Щ , dp + 2 -~^- dNf,
dZ = [^-\ , dp + (—) dT + y-^—dNf.
\ dp I t, n' V &T }p, n' fi dN^
1, ... l, ... lf 1 j
В этих формулах частные производные двух первых слагаемых
правых частей берутся при всех постоянных NjW, тогда как при
нахождении остальных производных предполагаются
постоянными два термодинамических параметра и массы всех
компонентов во всех фазах, кроме тех масс, по которым производится
дифференцирование. Суммирование производится по всем
компонентам и по всем фазам.
Частные производные в формулах (6,23) по числу частиц под
знаком суммы представляют собой по определению химические
потенциалы jx/*) в различных фазах для разных компонентов.
Они удовлетворяют условиям, аналогичным тем, которые
представлены в равенствах (6,20). Поэтому можно написать более
подробно:
\ (
a"i /sVN \OIWi /v.t.Nj...
) (6'24)
§ 4. Характеристические функции для гетерогенных систем 185
Введение понятия химического потенциала является
особенно ценным для химической термодинамики, где
рассматривается так называемое химическое равновесие при реакциях,
происходящих в системе. Далее мы обратим внимание на
применения химического потенциала в общей теории фаз.
§ 5. УСЛОВИЯ РАВНОВЕСИЯ ГЕТЕРОГЕННЫХ СИСТЕМ
Выясним, какими свойствами будет обладать гетерогенная
система, находящаяся в термодинамическом равновесии вместе
со всеми фазами и компонентами. Для определенности
рассмотрим два наиболее важных случая: когда система является
полностью изолированной от внешнего окружения и когда система
находится в термостате. Анализ этих частных случаев позволит
нам в дальнейшем вывести как следствие важное положение,
известное под названием правила фаз. Условия равновесия
мы определим путем, описанным ранее в § 1, вводя вариации
параметров.
1. Изолированная система. Допустим опять, что
система состоит из k фаз и п компонентов. На основании первого
начала мы должны считать, что, какие бы термодинамические
процессы, фазовые превращения и химические реакции не
происходили в такой изолированной системе, общий запас
внутренней энергии ее является постоянным. Полная энергия сложным
образом распределена по отдельным фазам, и из условия
U = 2 f/, = const
следует, что сумма всех вариаций энергии равна нулю:
Ъи = ЫГ1 + Ъиъ+ ... + Wk - ZWj = 0. (6,25)
Внутренняя энергия каждой фазы зависит от ее объема и
энтропии, а также от масс соответствующих компонентов. Поэтому
в соответствии с первым из уравнений (6,23) и известными
значениями частных производных по S и по V имеем:
= T.3S, - ptWt + 2-^- 8^», (6,26)
i dNf
186 Глава 6. Общая теория равновесия сложных термодинамических систем
Здесь Ти T2i ..., Tk — температуры и ри рг, ..., ph — давления,
вообще различные в разных фазах системы. Складывая все
равенства (6,26) и принимая во внимание уравнение (6,25), имеем:
W = ^ (W - pfWf) + ^ -Ц- W}0 = 0. (6,27)
/ /./ dNf
Это условие равновесия имеет место в общем случае, когда
в системе происходят фазовые превращения и химические
реакции. В отсутствие этих процессов имеем:
8#; = 8#; = ... = s#i = 8#2 = ... = *N{kn)=о,
и тогда условие равновесия принимает вид:
bU = 2 (TfiS, — pfWf) = 0. (6,28)
Исследуем два простейших случая, когда объем всей
системы является неизменным.
а) Объемы всех фаз системы постоянны. Этот
случай соответствует случаю 1 (стр. 167). Как было найдено,
при указанном условии при равновесии энтропия системы есть
величина постоянная, т. е.
S = Sx + S2 + • • • + Sk = const,
причем энтропии в отдельных фазах могут изменяться,
неизменной остается лишь их сумма. Из предыдущего равенства имеем:
8S = 8Sx + 8S2 + ... + *Sk = 0,
откуда
Заменяя в соотношении (6,28) величину dSh значением из
последнего равенства и учитывая, что при постоянных объемах
всех фаз величины 6Vj равны нулю, получаем:
W = T.8S, + T2bS2 + ... + T^bS^ - T.bS, -
или
(Гх - Tk) bSt + (Tt - Tk) 852 + (T, - Tk) 8S, + . ..
••• +(Tk_l-Tk)bSk_l=0.
Отсюда следует, что
Tl = T2= ... =-Tk = const.
§ 5. Условия равновесия гетерогенных систем 187
Таким образом, равновесию системы соответствует равенство
температуры во всех фазах и ее постоянство.
б) Энтропии во всех фазах постоянны. Тогда
X = SS2 = ... « bSk = 0.
темы мы приняли неизме!
могут меняться. Поэтому
V = Vx + V2 + ... + Vk = const
Хотя объем всей системы мы приняли неизменным, однако
объемы отдельных фаз могут меняться. Поэтому из условия
следует
W = bVx + W2 + ... + bVk = 0,
где слагаемые вообще не равны нулю. Из этого равенства
следует
Полагая в уравнении (6,28), все 6Sj равными нулю и
подставляя значение 8V& из последнего равенства, находим:
W = {pk - Pl) Wi + (Pk - Л) *Vt+ ...
••• +(Pk-Pk-l)Wk_l=0,
откуда
Pi = Рг = • • - = Pk = const.
Следовательно, в данном случае при равновесии системы
давления во всех фазах одинаковы и постоянны.
Таким образом, если гетерогенная система изолирована и
заключена в жесткую оболочку, т. е. занимает постоянный объем,
то она находится в равновесии при постоянных объемах и эн-
тропиях всех фаз, когда температуры и давления всех фаз
одинаковы, т. е.
Ti = T2= ... =Tk = const; Pi = р2 = . .. = pk = const.
2. Система в термостате. Пусть система из п
компонентов и k фаз находится в таких условиях, что за счет хоро-,
шей теплопроводности между отдельными частями системы
всегда обеспечивается быстрый теплообмен, приводящий к
выравниванию температур, и происходит отвод тепла в термостат.
Тогда для всех фаз будет соблюдаться условие:
Tt = T2= ... =Tk = T = const.
Допустим, что в этой системе происходят фазовые
превращениями имеют место химические реакции между компонентами.
В этом случае массы различных компонентов в разных фазах
188 Глава 6. Общая теория равновесия сложных термодинамических систем
будут вообще меняться. Изобразим для наглядности
распределение масс в нашей системе в виде следующей схемы:
Первая фаза: N\% N\% . . . , N[n)
Вторая фаза: N*2% N2\ . . . , N{2n)
Третья фаза: N'3, N*3, . . . , N{3n)
k-ая фаза: N'k, N*k, . . .
Итого по всем
фазам: N't N\ . . . , NW
Так как фазовые превращения представляют собой
физические процессы, то очевидно, что при таких превращениях масса
каждого компонента остается неизменной, меняется только
распределение его по фазам, т. е. все Nf, N", ..., ЛДП) постоянны,
а все Ni\ Ni", ..., Nh{n) изменяются. При химических реакциях
изменяются как массы каждого компонента, так и массы его
в различных фазах, т. е. все значения масс вообще являются
переменными. Отсюда следует, что в системе, где происходят
наряду с фазовыми превращениями также и химические
реакции, имеет место процесс изменения распределения масс по
компонентам и по фазам. Однако мы будем рассматривать так
называемое химическое равновесие в системе, считая, что
всякое уравнение химической реакции может быть представлено
в виде
А+В + С+ ... ^P + Q + R+ ...,
т. е. наряду с прямой реакцией между веществами Л, Л, С, .,.
происходит обратная реакция между продуктами Р, Q, /?,...
с образованием исходных веществ (этот характер превращения
указан противоположно направленными стрелками).
Химическое равновесие отвечает такому состоянию, когда скорости
прямого и обратного процессов одинаковы. Тогда массы
компонентов системы делаются постоянными, т. е. не меняются с
течением времени, хотя распределение гго фазам является
переменным. Следовательно, при химическом равновесии имеют место
условия для отдельных компонентов по всем фазам:
N = N[ + N2+ ... +Nk = const,
N" = N[ + N2 + ... +N'k= const, (6,29)
Nin) = N[n) + N{2n)+ ... + N[n) = const.
Рассмотрим, к чему приводят эти условия химического
равновесия для системы в двух частных случаях: р = const и
V=const при неизменной температуре r=const в обоих случаях.
§ 5. Условия равновесия гетерогенных систем 189
а) Система находится в термостате при
постоянных давлениях ри Рг, --., Ръ, для всех фаз.
Известно, что при термодинамических параметрах состояния ри Г
характеристической функцией является термодинамический
потенциал Z и согласно предыдущему (стр. 169) равновесие
системы имеет место, когда Z — минимум; 6Z = 0.
Отсюда вследствие аддитивности функции Z можно написать
8Z = IZX + 8Z2 + ... + bZk = 0. (6,30)
Здесь какое-либо /-е слагаемое выражается соотношением,
аналогичным прежним, т. е. (6,23):
dN\
Но так как все /7j = const и T=const, то 6pj = 0 и 6Г = О, и
поэтому во всех выражениях для 6Zj первые два слагаемых
обращаются в нуль. Тогда условие равновесия (6,30) можно
представить в виде:
#; +Ni + . . . +
dN[ dN\ dN[n)
dN2n)
^8МЛ) = 0, (6,31)
dNk dNk
где все производные по-прежнему представляют собой
химические потенциалы разных компонентов в различных фазах.
Уравнение (6,31) содержит в общем случае nk величин 6Nj({\
хотя в частных случаях некоторые слагаемые могут обращаться
в нуль, когда в каких-либо фазах отсутствуют некоторые
компоненты. Вообще множители при всех переменных 6Nft) не
равны нулю, так как некоторые из переменных связаны друг с
другом вследствие того, что изменение массы какого-либо
компонента в данной фазе может изменять массу его в другой фазе.
Таким образом, из nk величин 6NjM одни являются
независимыми, другие же связаны между собой. Для установления числа
независимых величин вспомним, что при равновесии должно
соблюдаться еще условие (6,29), из которого следует:
8W' = awl + ZN2 + ... + bNk = 0,
Г 1 1 = 0, (6,32)
o
N{2n) + . . . + oNin) = 0.
190 Г лава 6. Общая теория равновесия сложных термодинамических систем
Мы имеем, следовательно, п уравнений для nk неизвестных
). Поэтому у нас всего вместе с (6,31) имеется (п+1)
уравнений для nk неизвестных. Исключим зависимые переменные,
применяя для этого метод неопределенных множителей Лаг-
ранжа. С этой целью введем п пока произвольных величин \i\
\i"y ..., \in и умножим последовательно первое из уравнений
(6,32) на ц,', второе на ц," и т. д., затем сложим полученные
равенства. Находим:
или
= 0. (6,33)
Вычтем теперь это равенство из уравнения (6,31). Тогда после
перегруппировки имеем:
Так как в нашей задаче п неизвестных связаны между собой
уравнениями (6,32), т. е. п переменных являются зависимыми,
то достаточно исключить их из равенства (6,34), чтобы в нем
остались одни независимые величины. Это можно сделать,
пользуясь тем, что все \i'f ц", ..., \in представляют собой
произвольные величины. Нам необходимо из (6,34) исключить п любых
слагаемых, например все п первых слагаемых. Для этого
достаточно положить равными нулю все коэффициенты при
переменных 6Nt'9 bNt'\ ..., 6Ni<n>. Тогда находим:
S p*e-^;...,i<->e-*5L (6,35)
dN dN[n>
dN{
После этого оставшееся уравнение будет содержать только одни
независимые вариации 6Af/*). Правая часть этого уравнения
должна равняться нулю при всяких значениях независимых ва-
§ 5. Условия равновесия гетерогенных систем 191
риаций, что возможно только, когда все выражения в скобках
будут равны нулю, т. е. все \х\ \х", .., \х^ будут одновременно
равны соответствующим частным производным. В таком случае
вместе с равенством (6,35) получим:
dZk
Г
II"
г
n)
dN[
dZi
1
dN'2
dZ2
dN"2
dZ2
dN2n)
dN'k'
. dZk
dN"k '
dZk
(6,36)
Эти уравнения, являющиеся выражением условий
равновесия, показывают, что гетерогенная система, где имеют место
различные фазовые превращения и химические реакции,
находится в равновесии, когда химические потенциалы каждого
компонента во всех фазах одинаковы.
Поэтому введенные ранее множители ji', ji", ..., \xW
представляют собой химические потенциалы различных компонентов
при равновесии системы. Если вспомнить обозначения
химических потенциалов в разных фазах:
i _ dZx и п __ QZj л , dZ2 . • dZu .
гi — Г » P-i — Г » • • • Н-2 Г » Р"2 = Г » • • •
dN{ dJV{ dN2 dN{
то условия равновесия можно представить в форме:
Ь = К = • • • = Pi = !*'•
К = 1*2 = • • • = Рк = Р*
Таким образом в равновесной системе переход данного
компонента из одной фазы в другую происходит таким образом,
что химические потенциалы этого компонента во всех фазах
остаются постоянными и одинаковыми. Разность химических
потенциалов какого-нибудь i-ro компонента, положим в. первой
и второй фазах, вызываемая, например, химической реакцией,
сглаживается и стремится к нулю, когда система переходит
к состоянию химического равновесия.
192 Глава 6. Общая теория равновесия сложных термодинамических систем
б) Система находится в термостате при
постоянных объемах всех фаз. При термодинамических
параметрах V и Т характеристической функцией является
свободная энергия F системы и, как известно (стр. 169), условия
равновесия системы выражаются как F — минимум; 6F = 0.
Отсюда, принимая во внимание аддитивность свободной
энергии, имеем;
8F = 8Ft + ZF2 + ... + bFk = 0. (6,38)
Для каждой фазы может быть написано равенство, аналогичное
второму уравнению (6,23):
Здесь первые два слагаемых обращаются в нуль, так как во
всех фазах Vi = const; V2=const; ..., Vk = const и Т=const. Вводя
все dFj в равенство (6,38), получаем соотношение, аналогичное
уравнению (6,31). Поступая далее так же, как в предыдущем
случае, и имея в виду формулу (6,20) для химических
потенциалов, мы приходим к условиям равновесия системы,
выражаемым той же системой уравнений (6,37). Следовательно,
условие (6,37) является общим условием равновесия, пригодным
для обоих случаев.
Принимая во внимание равенства (6,37) и выражения (6,30)
и (6,38), мы можем услов-ия равновесия представить
объединенной формулой, применяя соотношения (6,23) и (6,20):
2t*(08tfJ° = 0f (6,39)
где все массы выражены в молях; если они выражаются в
граммах, то соответственно имеем:
ylx(08mf) = 0. (6,40)
*./
Здесь при равновесии все \х^ представляют собой химические
потенциалы компонентов.
Так как выведенные здесь условия равновесия получены при
всевозможных вариациях масс во всех фазах всех компонентов,
то эти условия относятся к так называемому истинному
термодинамическому равновесию. Полезно заметить, что в реальных
условиях возможны состояния, называемые ложными
равновесиями, при которых выведенные выше условия не соблюдаются
и тем не менее при неизменных р и Т или V и Т мы в течение
длительного времени не наблюдаем изменения равновесия
системы. Можно привести много примеров такого ложного
равновесия. Например, известно, что алюминий легко окисляется за
§ 5. Условия равновесия гетерогенных систем 193
счет кислорода воздуха при обычной температуре. Однако
обычно полного окисления каких-либо алюминиевых предметов
мы не наблюдаем, потому что на поверхности металла быстро
образуется тонкая пленка окисла, затрудняющая доступ
кислорода к неокисленному металлу под пленкой. Очевидно, такая
система может длительно находиться в равновесии, которое
является ложным, так как возможная химическая реакция в
системе заторможена образованием пленки.
Если кусок мрамора (СаСО3) ввести в раствор серной
кислоты, то вначале наблюдается бурная реакция с выделением
углекислого газа, но спустя некоторое время устанавливается
видимое равновесие между кислотой и карбонатом кальция.
Это равновесие также является ложным, потому что ионы из
раствора чрезвычайно медленно диффундируют через корку
образовавшегося продукта реакции (CaSO4) и мы не замечаем
какого-^ибо изменения системы за долгое время только
благодаря медленности диффузии. В растворе соляной кислоты корки
не образуется, и реакция идет до конца.
Стекло с течением времени кристаллизуется, т. е. переходит
из аморфного состояния в кристаллическое. Кристаллы в
данном случае находятся в ложном равновесии с аморфной частью
системы. Истинное равновесие не устанавливается вследствие
медленности процесса кристаллизации, обусловленной очень
большой вязкостью системы.
В механике часто имеют место ложные равновесия
благодаря действию трения. Например, брусок дерева может долгое.
время лежать на наклонной доске, не скользя вниз из-за
большого трения.
Выведенные выше условия равновесия характеризуют собой
всесторонность истинного равновесия. Так как
при истинном равновесии должны быть равны химические
потенциалы каждого компонента в разных фазах, то ясно, что
прибавление или убавление массы данного компонента в любую
фазу не может изменить равновесия. Если мы к системе,
состоящей из насыщенного раствора соли и кристаллов той же соли,
при равновесии добавим какое-либо количество той же соли,
то мы не изменим равновесия, так как химические потенциалы-
не изменятся.
Этот результат легко получается из написанных общих
выражений. Если, например, возьмем три фазы какого-либо п-го
компонента и допустим, что в системе установилось равновесие
первой фазы со второй и первой с третьей, тогда должно быть
и, следовательно,
a(/i) —
Г2 ~~
194 Глава 6. Общая теория равновесия сложных термодинамических систем
т. е. фаза 2 находится в равновесии с фазой 3. Если массу
одной из фаз увеличим, то равновесие не нарушится, так как
прибавленная масса по условию будет в равновесии с остальными
массами.
В приложениях теории равновесия целесообразно
пользоваться удельными значениями термодинамического потенциала
и свободной энергии, относя эти величины к единице массы,
например к единице массы данного компонента в определенной
фазе (см. ранее стр. 183). Следует иметь в виду, что Z и F для
вполне однородной фазы для данного компонента прямо
пропорциональны содержанию этого компонента, т. е.
пропорциональны массе, причем безразлично, в каких единицах выражена
эта масса. Поэтому всегда можно положить, что
где ф|° представляет собой удельный термодинамический
потенциал. Эта величина не зависит от массы, а только от
параметров состояния р и Т. Для всей /-й фазы термодинамический
потенциал равен сумме по всем компонентам:
Ранее было отмечено, что химический потенциал /-го
компонента в этой фазе равен соответствующему удельному
термодинамическому потенциалу:
и поэтому
Таким образом, условия равновесия (6,37) можно
представить в форме равенства удельных термодинамических
потенциалов каждого из компонентов во всех фазах, т. е.
Аналогичные удельные величины можно ввести и для
свободной энергии, а также для внутренней энергии и энтальпии.
§ 6. ПРАВИЛО ФАЗ. ВЫВОД
Полученные выше условия равновесия позволяют решить два
весьма важных принципиальных вопроса, связанных с общими
задачами термодинамики. Во-первых, мы можем теперь вполне
строго установить, сколько независимых параметров необхо-
§ 6. Правило фаз. Вывод 195
димо для полного определения состояния системы. Этот вопрос
мы ставили в самом начале курса, но на него не дали еще
ответа, ограничиваясь утверждением, что для простейшей системы
достаточно двух независимых параметров состояния. Во-вторых,
мы можем установить, сколько фаз в системе из п компонентов
будут находиться в равновесии. В исходной неравновесной
системе число фаз при п компонентах может быть произвольным.
Однако при равновесии системы число присутствующих в ней
фаз связано определенным соотношением с числом
компонентов. Это соотношение, называемое правилом фаз, было
открыто Гиббсом. Рассмотрим сначала вывод правила фаз, для
чего нам необходимо вернуться к результатам, полученным
в предыдущем параграфе.
Условие равновесия (6,37) представляет собой систему
уравнений, в которой в горизонтальном ряду для k фаз имеется
(k—1) уравнений, как это следует из формы записи; всего
у нас п горизонтальных рядов, следовательно, условия
равновесия гетерогенной системы выражаются n(k—1) уравнениями.
С другой стороны, параметрами состояния сложной системы
являются два каких-либо термодинамических параметра
(например, р и Т) и концентрации составных частей. В нашей
системе п компонентов, их концентрации в разных фазах связаны
уравнениями (6,15), причем в каждое уравнение входит (п—1)
независимых концентраций, так как одна из п концентраций
может быть выражена через остальные. Например, в первой
фазе имеем:
Число всех уравнений (6,15) равно числу фаз, т. е. равно k.
Таким образом, число независимых концентраций как
параметров состояния равно k(n—1). Поэтому общее число параметров
состояния гетерогенной системы равно
так как к концентрациям надо добавить еще два
термодинамических параметра (т. е., например, ри Г). Итак, мы видим, что
для k(n—1)+2 параметров у нас имеется при равновесии
n(k—1) уравнений. Следовательно, найденное выше число
параметров не равно вообще числу независимых параметров, так
как имеется n(k—1) уравнений, связывающих часть этих
параметров. Однако физический смысл имеют лишь два случая:
число уравнений равно числу параметров и число уравнений
меньше числа параметров. Во втором случае некоторые из
параметров окажутся независимыми, т. е. произвольными. Итак,
196 Глава 6. Общая теория равновесия сложных термодинамических систем
если гетерогенная система находится в равновесии, то должно
быть
n(k— 1)<А(л— 1) + 2,
откуда
£<л + 2, (6,41)
следовательно, при равновесии системы число фаз меньше или
равно числу компонентов плюс два. Это положение и носит
название правила фаз Гиббса. При строгом определении понятий
фазы и компонента правило фаз является универсальным, так
как оно термодинамически обосновано. Оно выполняется для
всевозможных систем, несмотря на происходящие в них фазовые
превращения и химические реакции, оно пригодно для веществ
в любых агрегатных состояниях. Можно также показать, что
мы придем к тому же выражению правила фаз, если в
некоторых фазах часть компонентов отсутствует. В самом деле, тогда
число уравнений (6,37) уменьшится как раз на число
отсутствующих компонентов, но настолько же уменьшится и число
концентраций, являющихся параметрами состояния. Поэтому
окончательная формула останется без изменения.
Правило фаз дает возможность легко установить, сколько
фаз может находиться между собой в равновесии при заданном
числе компонентов. Например, если система состоит из трех
компонентов, то, как видно из формулы (6,41), при п = 3 должно
быть fe^5, т. е. число фаз в равновесии не может превышать
пяти. Если в первоначально взятой системе было более пяти
фаз, то при переходе системы к равновесию часть их должна
исчезнуть, т. е. некоторые из компонентов перейдут в другие
фазы.
Пользуясь правилом фаз, можно решить вопрос о числе
независимых параметров состояния или о так называемых
степенях свободы равновесной системы. В самом деле, из
выражения (6,41) следует, что число независимых параметров при
равновесии представляет собой разность
f = n + 2-k. (6,42)
Величина / называется также числом
термодинамических степеней свободы системы по аналогии со
степенями свободы в механике. Формула (6,42) показывает, какое
число параметров состояния мы можем произвольно изменять
при равновесии, не изменяя числа фаз в системе. Очевидно,
величина / всегда является положительной, причем может быть
равной нулю, т. е. /^0. Для простейшей однородной, т. е.
однофазной системы из одного компонента, из формулы (6,42)
находим / = 2; таким образом, состояние таких простых систем, как
идеальный газ или однородная жидкость, определяется двумя
§ 6. Правило фаз. Вывод 197
независимыми параметрами в соответствии с тем, как было
принято нами в первых главах курса. Для многокомпонентных
систем число степеней свободы вообще может быть больше двух,
и для определения состояния, кроме двух термодинамических
параметров, необходимо знать концентрации компонентов.
Формула (6,42) показывает, что с увеличением числа фаз в
системе f уменьшается, т. е. число свободно варьируемых
параметров убывает, а отсюда следует, что представляется все
меньше возможностей изменения состояния системы с
сохранением равновесия и числа равновесных фаз.
Главное значение правила фаз заключается в том, что оно
дает нам рациональную основу для классификации
гетерогенных систем, находящихся в равновесии.
Сложные системы, примеры которых мы рассматривали
в этой главе, могут быть разделены на классы по числу фаз и
по числу компонентов. Так, мы различаем по числу фаз системы
однофазные, двухфазные, трехфазные и т. д.; аналогично этому
по числу компонентов мы говорим об однокомпонентных, двух-
компонеитных и т. д. системах. Правило фаз показывает, что все
эти системы обладают еще одним признаком, который может
быть положен в основу распределения систем по классам; этим
признаком является число степеней свободы системы.
Соответственно значению величины / все системы могут быть
разделены на следующие группы:
1) инвариантные системы (/ = 0);
2) моновариантные системы (/=1);
3) бивариантные системы (/ = 2);
4) тривариантные системы (/ = 3) и т. д.
Эта классификация относится только к равновесным
системам и является ценной потому, что позволяет точно указать
поведение системы при изменении состояния. Кроме того,
возникает возможность простых геометрических изображений
состояния системы, приводящих к построению так называемых
диаграмм равновесия, удобных при анализе часто весьма
сложных систем (например, сплавов, горных пород, природных
солей, силикатов и т. д.). Из формулы (6,42) следует, что
инвариантные системы обладают (в равновесии) наибольшим числом
фаз. В однокомпонентной инвариантной системе находятся
в равновесии друг с другом три фазы, в двухкомпонентной
четыре фазы, в трехкомпонентной пять и т. д. Все такие системы
не обладают степенями свободы, т. е. не могут быть изменены
без изменения числа фаз. Следовательно, инвариантные системы
существуют при вполне определенных параметрах состояния.
Если в первоначально инвариантной системе варьировать один
из параметров, то одна из фаз должна исчезнуть и система
обращается в моновариантную. Если независимо изменять два
198 Глава 6. Общая теория равновесия сложных термодинамических систем
параметра, то исчезнет две фазы и т. д. С увеличением числа
степеней свободы число фаз уменьшается.
Область применения правила фаз весьма широка. Этим
правилом приходится руководствоваться при изучении однокомпо-
нентных систем, рассматривая разные фазы одного вещества.
Далее, анализ различных сплавов и смесей также требует
применения правила фаз. Вопросы теории растворов, газовых
смесей также связаны с этим правилом. Из практически важных
вопросов можно отметить еще, например, изучение состава и
свойств нефти, представляющей собой многокомпонентную
однофазную систему, исследование которой связано с применением
правила фаз. Мы не можем рассматривать здесь всех этих
вопросов и ограничимся немногими примерами, которые будут
подробнее разобраны в следующей главе.
Задачи
1. Две очень маленькие капли жидкости одинакового радиуса г сливаются
в одну каплю. Найги изменение свободной энергии от слияния и указать,
будет ли процесс слияния капель термодинамически выгодным.
2. В соляных озерах находится водный раствор сульфата натрия и
хлористого натрия. Считая эту систему равновесной, определить при помощи
правила фаз число фаз и компонентов в системе.
3. Природная нефть представляет собой смесь многочисленных
углеводородов, растворенных друг в друге. Дать определение нефти как сложной смеси
с точки зрения учения о фазах.
4. Дана система, состоящая из кристаллов NaCl, находящихся в водном
растворе той же соли, изо льда и пара над раствором. Пользуясь правилом
фаз, доказать, что такая система может быть равновесной.
5. Сколько фаз будет в равновесии в системе, предыдущей задачи, если
изменять температуру, и какие комбинации равновесных фаз возможны?
Глава 7
ПРИМЕНЕНИЕ УЧЕНИЯ О РАВНОВЕСИИ
К СЛОЖНЫМ ТЕРМОДИНАМИЧЕСКИМ СИСТЕМАМ
§ 1. РАВНОВЕСИЕ ОДНОКОМПОНЕНТНЫ* СИСТЕМ
Пусть система состоит из нескольких фаз одного
определенного компонента. Примером подобной системы является вода
в различных граничащих друг с другом фазах. Сюда относится
система, состоящая из жидкой воды й водяного пара над ее
поверхностью, а также система изо льда с находящимся над
ним паром. Более сложной системой подобного же рода
является совокупность из жидкой воды со льдом и с паром воды.'
Ясно, что системы, о которых идет речь, состоят из одного
компонента (воды), присутствующего в сочетании различных фаз:
твердой, жидкой и газообразной. Заметим, что, как было
сказано ранее, в системе может быть несколько твердых фаз,
обладающих разной кристаллической структурой. Здесь мы
рассмотрим термодинамические свойства однокомпонентных систем,
используя правило фаз Гиббса.
Из предыдущих теоретических положений следует, что
различные фазы однокомпонентной системы будут находиться
в равновесии, когда температуры и давления в разных фазах
одинаковы, т. е. должны соблюдаться условия:
7\ = Т2 = ... = Г,
Pi = Р2 = • • • = Р-
Кроме того, необходимым условием равновесия является
требование, чтобы химические потенциалы во всех фазах были
равны друг другу, т. е. для одного компонента
1*1 = 1*2 = • • • =Pk>
где
т, N* ... \ oN2 /р, т, Nt ...
При этом термодинамический потенциал системы выражается
по-прежнему суммой (см. гл. 6, стр. 194):
Z = N1-y1 + N2.<?2 +
так что
l*i = ?i*> 1*2 = <?2; • • •
200 Глава 7. Применение учения о равновесии к сложным системам
Здесь все удельные термодинамические потенциалы ерь фг...
и т. д. являются функциями параметров состояния /?, Т.
Условия равновесия показывают нам, когда в системе,
состоящей из нескольких фаз, будет наблюдаться равновесное
состояние, но они непосредственно не дают возможность заключить,
сколько фаз может находиться между собой в равновесии. Для
выяснения этого вопроса необходимо обратиться к правилу фаз,
выводимому из условий равновесия (гл. 6). Мы видели, что
число независимо варьируемых параметров в системе,
находящейся в равновесии, выражается соотношением:
Для однокомпонентной системы здесь необходимо положить
/1=1, и тогда
/ = 3 —А.
При положительном / эта формула приводит только к трем
частным случаям:
/ = 0; тогда k = 3,
f = 1; тогда k = 2,
f = 2; тогда k = 1.
Этот результат имеет простой физический смысл,
заслуживающий, однако, подробного анализа.
1. Если /=0, то в равновесии могут находиться три фазы
данного вещества (k = 3). Большего числа фаз в равновесии
однокомпонентной системы быть не может. Так как число
степеней свободы такой системы равно нулю, то равновесие трех фаз
возможно лишь при вполне определенных, фиксированных
параметрах состояния, т. е. при определенной паре значений р и Т.
В этом еще раз легко убедиться, учитывая равенство удельных
термодинамических параметров для трех фаз одного
компонента. Действительно, при равновесии имеем:
Ф1=Ф2=ФЗ, (7,1)
причем ф1 = ф1(/?, Т)\ ф2 = ф2(/7, Г); фз = фз(р, Т). Условие (7,1)
приводит к двум независимым уравнениям, например
<Pi(p, Г) = Фз(Р, Г); ф1(р, Г) = ф8(р, Г), (7,2)
тогда как третье является их следствием. Таким образом, для
двух неизвестных р и Т мы имеем два уравнения (7,2). Решая
эти уравнения, находим вполне определенные значения
давления и температуры, отвечающие сосуществованию трех фаз
в равновесии. Эта пара значений /?Тр и Гтр соответствует
определенному состоянию трехфазной системы, называемому
тройной точкой. Например, из опыта найдено, что обычный лед,
жидкая вода и водяной пар находятся в равновесии между
§ 1. Равновесие однокомпонентных систем 201
собой при давлении /?Тр = 4,579 мм рт. ст. и температуре /тр =
= +0,0076°С. При других условиях равновесие между этими
тремя фазами не имеет места. Если первоначально
существовало равновесие между ними при указанных /?тр и /Тр, то при
нагревании этой системы растает лед, при охлаждении
пропадет жидкая вода, а при сжатии исчезнет пар. Во всяком случае,
вне указанных значений ррт и /Тр три данные фазы воды
существовать в равновесии не могут. Отсюда, кстати, следует, что
в природных условиях обычно три фазы воды не находятся
в равновесии. Опыты Таммана (1900 г.) и Бриджмена (1911 г.)
показали, что для воды возможны еще шесть различных
твердых фаз, являющихся разновидностями обычного льда. Тем не
менее по правилу фаз в равновесии между собой может быть
не более трех разных фаз, т. е. для воды имеется всего семь
тройных точек. Несколько тройных точек найдено и у других
веществ; например, у серы установлено три тройные точки,
соответствующие равновесию двух твердых фаз,— ромбической
и моноклинической и еще жидкой фазы.
2. Если /=1, то, как мы видели, число фаз данного вещества,
находящихся в равновесии, равно двум, т. е. 6 = 2. Двухфазная
система является, таким образом, унивариантной; это значит,
что в ней можно без изменения числа фаз при равновесии
произвольно изменять один параметр состояния. Этот вывод можно
получить также и из условия равенства химических
потенциалов. В самом деле, для системы из двух фаз должно быть
Н = to, или фх = фа,
иначе
<Pi(p, Л = Фв(Р, П
Из этого единственного уравнения можно найти давление как
функцию температуры
Следовательно, температура может изменяться произвольно,
тогда как давление является функцией температуры. Таким
образом, двухфазная система обладает лишь одной степенью
свободы. Можно, конечно, произвольно изменять давление, но
тогда зависимой величиной будет являться температура.
Правило фаз не указывает нам, какие две фазы удовлетворяют
условиям равновесия, т. е. для пары любых фаз данного
вещества будут соблюдаться выведенные условия равновесия и
система будет унивариантной.
Допустим, например, что система лед —жидкая вода — пар
первоначально находилась в равновесии в тройной точке.
Изменяя один из параметров состояния, мы можем идти по одному
из трех путей образования равновесной двухфазной системы.
202
Глава 7. Применение учения о равновесии к сложным системам
Станем повышать температуру, тогда в первоначальной системе
твердая фаза пропадет и в двухфазной системе жидкая вода —
пар будем наблюдать повышение упругости насыщенного пара
с температурой, причем пар все время будет в равновесии
с жидкостью. Если первоначальную систему будем сжимать, то
пар исчезнет и мы можем наблюдать равновесие системы лед —
жидкость, т. е. плавление льда при различных давлениях и
температурах. Наконец, если станем охлаждать трехфазную
систему ниже тройной точки, то жидкая фаза пропадет и мы
сможем наблюдать изменение упругости пара в системе лед — пар.
Таким образом, при одном свободном параметре возможно
равновесие следующих двух фаз: лед — пар воды; лед — жидкая
вода; жидкая вода — пар воды.
3. Если / = 2, то, как мы видели из правила фаз, должно быть
& = 1, т. е. равновесие возможно только для одной фазы. Это
видно также из того, что при Z=N^i имеем: (!ц = ф1 = ф(р, 7").
Здесь удельный термодинамический потенциал ф4 является
функцией двух независимых переменных р и Г, т. е.
однофазная система характеризуется двумя степенями свободы. Мы
можем по произволу и независимым образом изменять р и 7 и
будем наблюдать равновесие одной фазы. Для воды это
соответствует отдельно взятым системам: лед, жидкая вода, пар воды.
Рассмотренные здесь закономерности могут быть наглядно
представлены с помощью графика, причем для однокомпонент-
ных систем наиболее целесообразно пользоваться диаграммой
р, Т. На диаграмме (рис. 36) точка О отвечает
сосуществованию трех фаз, т. е. является тройной точкой. В ней
пересекаются три линии
АО, ВО и СО,
соответствующие
изменению состояния
равновесных
двухфазных систем.
Первая из них — АО
передает зависимость
давления от
температуры в системе
твердое тело — пар,
т. е. отвечает
процессу сублимации, или
возгонки, когда при
повышении
температуры увеличивается
упругость пара над
твердой фазой (в
Рис. 36. отсутствие жидкой
§ 1. Равновесие однокомпонентных систем 203
фазы). Поэтому такая кривая носит название кривой
сублимации. Известно, например, что лед может испаряться
(возгоняться), не переходя в жидкую воду. Вторая кривая — О В
соответствует равновесию в системе твердая фаза —жидкость и
передает зависимость давления от температуры в этой системе.
Равновесие твердой фазы с жидкостью возможно при точке
плавления, следовательно, кривую ОВ можно назвать кривой
плавления: с ее помощью графически передается
зависимость точки плавления от давления. Эта кривая для воды
отклоняется влево вверх от тройной точки в соответствии с хорошо
известным фактом, что с повышением давления температура
плавления льда понижается. Для большинства других веществ
кривая плавления отклоняется вправо вверх от тройной точки,
т. е. обычная температура плавления повышается с давлением.
Наконец, третья кривая — ОС соответствует фазовому
равновесию в системе жидкая фаза — пар и определяет собой
зависимость упругости насыщенного пара от температуры.
Сосуществование пара и жидкости- отвечает состоянию кипения последней,
так как жидкость кипит при той температуре, при которой над
ней имеется в равновесии насыщенный пар. Поэтому кривую ОС
можно назвать кривой кипения жидкости.
Все три кривые ОА, ОВ и ОС являются разграничивающими
линиями, делящими плоскость на диаграмме (/?, Т) на три
области, соответствующие однофазным,системам.
Очевидно, что так как для однофазных систем р и Т
являются взаимно независимыми, то точки, изображающие
состояния этих систем, не располагаются на какой-либо из трех
указанных линий, а попадают в области между ними.
Соответственно область под кривыми ОА и ОС является областью пара
(рис. 36), участок плоскости между ОВ и ОС принадлежит
жидкой фазе, а область между линиями ОА и ОВ представляет
собой область твердой фазы. Ясно, что все три линии отражают
собой процессы фазовых переходов. Так, при переходе от
твердой фазы в состоянии а к жидкой фазе в точке Ь мы пересекаем
кривую плавления, т. е. твердая фаза должна быть нагрета и
затем при заданном давлении она плавится, после чего
получается одна жидкая фаза (рис. 36).
Крайние точки кривых сублимации и плавления могут
вообще представлять собой новые тройные точки для данного
вещества, способного образовывать несколько твердых или
жидких фаз. Кривая сублимации может, кроме того, уходить в
область весьма низких температур, где прослеживание ее хода
связано только с экспериментальными трудностями. То же
следует сказать и о кривой плавления, которая может простираться
вверх в область весьма высоких давлений. Что касается
кривой кипения, то она оканчивается в конечной области давления
204 Глава 7. Применение учения о равновесии к сложным системам
и температуры, в так называемой критической точке, которая
может быть названа точкой остановки. За критической точкой
свойства газовой и жидкой фазы неразличимы. Особенности
критического состояния будут рассмотрены далее.
Опытные данные указывают, что в тройной точке кривые
фазовых превращений взаимно пересекаются, но не кончаются;
продолжения их показаны на рис. 36 пунктирными линиями. Эти
линии соответствуют так называемому метастабильному
равновесию двухфазных систем, которое представляет собой также
устойчивое равновесие, однако менее устойчивое, чем
рассмотренное выше. Таким состояниям отвечают пересыщенный пар,
переохлажденная жидкость и перегретый пар. Известно, что
небольшие внешние воздействия переводят систему из этих
относительно малоустойчивых состояний в устойчивые и она
«переходит» на соответствующую вполне устойчивую кривую
фазового превращения.
Форма разграничивающих кривых на диаграмме (р, Т)
в термодинамике в общем случае определена быть не может,
так как она зависит от особенностей структуры молекул
вещества и характера сил молекулярного взаимодействия. Поэтому
обычно пользуются эмпирическими кривыми, построенными по
опытным данным для данного вещества. Однако в некоторых
частных случаях приближенная зависимость между р и Т
может быть выведена с помощью известного уравнения
Клапейрона— Клаузиуса. Как было показано в главе 4 (стр. 118), это
уравнение (Клапейрона — Клаузиуса) для фазовых
превращений, сопровождаемых поглощением или выделением удельной
скрытой теплоты превращения L, может быть представлено
в виде общего уравнения (4, 12), откуда следует:
dp = 1 L
dT Т 02 — ^1 '
где Vi — удельный объем первоначальной фазы и v2 — удельный
объем образовавшейся фазы после затраты теплоты
превращения L. Соответственно трем видам двухфазных систем, для
которых зависимость между давлением и температурой
передается графически тремя кривыми равновесия, написанное
выше уравнение принимает одну из следующих форм:
1) для кривой сублимации:
dp = \ Lc , v
dT T vn — Утв ' V '
2) для кривой плавления:
dp _ _I_ ^пл . /эд
dT Т Ож-""тв ' V '
§ 1. Равновесие од но компонентных систем 205
3) для кривой кипения
dp = J_ LK . v
dT Т ^п — Уж ' V ;
где Lc, £Пл и LK соответственно равны удельной теплоте
сублимации, теплоте плавления и теплоте кипения
(парообразования), причем уТв, ^ж, Vn — удельные объемы твердой, жидкой и
паровой фаз. Вообще удельные скрытые теплоты зависят от
температуры, и их значения могут быть найдены для каждой
температуры из опыта, то же относится и к удельным объемам
различных фаз: они могут быть получены из опыта. Уравнение
Клапейрона — Клаузиуса оправдывается на опыте с большой
точностью, поэтому, пользуясь опытными данными, мы можем
найти для всех трех случаев фазовых переходов величины ——
dT
для каждой температуры, иными словами, мы можем получить
наклоны касательных к оси абсцисс на диаграмме (р, Т).
Отсюда по отрезкам касательных можно построить кривую
зависимости р от Т для различных двухфазных систем, т. е.
построить разграничивающие кривые ОЛ, ОВ и ОС.
Аналитические уравнения для некоторых из этих кривых могут быть
получены путем интегрирования уравнения Клапейрона —
Клаузиуса.
Рассмотрим фазовое равновесие системы жидкость — пар,
соответствующее процессу парообразования. В небольшом
интервале температур допустимо принять, что LK не зависит от 7,
т. е. можно положить LK = const. Кроме того, известно, что при
переходе жидкости в пар удельный объем сильно возрастает
и потому в формуле (с) величиной vm можно пренебречь и
положить, что vu = v. Тогда из (с) имеем:
dp _ Lk^
dT vT #
Принимая, что для пара, далекого от критического состояния,
выполняется уравнение состояния идеальных газов, исключим
из последнего равенства объем v и получаем выражение (4, 10)
главы 4:
L R
к dT
откуда
d In p LK l
dT ~~~W ~Т*Щ
Интегрирование этого уравнения дает:
£ (7,3)
206 Глава 7. Применение учения о равновесии к сложным системам
Полагая Ci = lnC, находим:
р=Се-«т. (7,4)
Это выражение дает в явном виде зависимость давления
насыщенного пара р от температуры Т и хорошо оправдывается
в широком интервале температур.
Указанные упрощения применимы также и к описанию
процесса сублимации, а следовательно, формула (7, 4) выражает
также зависимость давления пара над твердой фазой, т. е.
представляет собой аналитическую формулу для кривой сублимации
при L = LC.
Для кривой плавления упрощения в выводе недопустимы,
однако общий ход ее может быть верно оценен с помощью
того же уравнения Клапейрона — Клаузиуса. Известно, что при
плавлении твердого тела различают два частных случая:
1) удельный объем vm больше объема твердого тела vTBy
т. е. vm—vTB>0. Так как Lnjl всегда положительно, то из
уравнения (Ь) следует, что — положительно. Поэтому на диа-
аТ
Грамме (р, Т) кривая плавления отклоняется вправо от оси
давлений по мере повышения температуры, т. е. с увеличением
давления температура плавления повышается; 2) при плавлении
происходит сокращение объема или vm<vTBi как это имеет
место для воды. Тогда vm—vTB<0 и поэтому должно быть — <0.
dT
Это означает, что касательная к кривой плавления в данном
случае всюду наклонена под тупым углом к оси 7 и,
следовательно, кривая плавления отклоняется влево, приближаясь
к оси давлений (как на рис. 36). Следовательно, в этом случае
с повышением давления температура плавления понижается..
Взаимное относительное расположение кривых фазового
перехода вблизи тройной точки также может быть оценено, если
пользоваться уравнением Клапейрона — Клаузиуса. В самом
деле, при плавлении Ьил достаточно велико, тогда как
изменение объема vm—vTB сравнительно мало, т. е. в формуле (Ь) близ
тройной точки — велико, а это значит, что кривая плавления
аТ
круто поднимается вверх. При кипении LK велико, но и vn—vm
также весьма велико, так как vu^>vm. Поэтому числитель и
знаменатель в формуле (с) велики, а следовательно, —— сравни-
тельно мало, т. е. кривая кипения идет достаточно полого от
тройной точки, медленно поднимаясь вверх.
Для оценки хода кривой сублимации нужно знать величину
Lc; в свя8и с этим целесообразно использовать первое начало
§ L Равновесие однокомпонентных систем 207
в форме закона Гесса. Рассматривая фазовые превращения
аналогично химическим превращениям, мы должны иметь
в виду, что изменение внутренней энергии не зависит от формы
пути. Пусть единицу массы твердого тела мы превратили в
жидкость, затрачивая при постоянной температуре теплоту Lnjl;
затем мы полученную жидкость испарили при той же
температуре и при этом затратили теплоту LK. Очевидно, изменение
внутренней энергии в обоих процессах таково же, как если бы
мы единицу массы твердого тела превратили непосредственно
в пар при той же температуре, когда затрачивается теплота Lc.
Тогда получаем:
Lc = Lnn + LK.
Для воды при 0°С известно, что /,пл = 335 кдж/кг и LK =
=2522 кдж/кг. Поэтому Lc = 335+ 2522 кдж/кг.
Таким образом, теплота сублимации велика, причем
изменение объема (vn—vTB) так же велико, как и при
парообразовании, так как ^п^^тв- Отсюда видно согласно (а), что кривая
возгонки (сублимации) близ тройной точки идет круче, чем
кривая кипения, потому что LK<LC. Опыт подтверждает это
относительное расположение всех трех кривых фазовых
превращений.
Существование закритической области ясно следует также
из уравнения Клапейрона — Клаузиуса. В самом деле, при
критическом состоянии (точнее, непосредственно за ним) объемы
жидкости и пара совпадают, т. е. vn = vm и скрытая теплота
парообразования равна нулю, т. е. LK = 0. В таком случае ——
dT
обращается в неопределенность и, значит, нельзя провести
никакой линии, начиная с критической точки, а это указывает на
то, что последняя является действительно точкой остановки
кривой кипения.
§ 2 *. РАВНОВЕСИЕ СМЕСЕЙ ИДЕАЛЬНЫХ ГАЗОВ.
ЗАКОН ДЕЙСТВУЮЩИХ МАСС
Смесь газов с термодинамической точки зрения представляем
собой однофазную многокомпонентную систему. Если число
различных компонентов, входящих в состав смеси, есть /г, то
согласно правилу фаз число независимых параметров системы
равно
f=n + 2 — k = 2 + (n— 1),
Этот параграф может быть опущен при первом изучении курса.
208 Глава 7. Применение учения о равновесии к сложным системам
так как число фаз здесь всегда k=l. Отсюда следует, что для
простейшей смеси двух газов число независимых параметров
равно f = 3. В качестве двух термодинамических параметров
могут быть выбраны V и Т или р и Г, а третьим параметром
является концентрация одного из газов, так как концентрация
второго будет величиной зависимой, если пользоваться
относительными концентрациями. Тогда С2=1—CV
Для смеси трех газов имеем / = 4, следовательно, в данном
случае система описывается четырьмя независимыми
параметрами, из которых два по-прежнему могут быть V и Т или р и Г,
а остальные два представляют собой концентрации двух газов.
Концентрация третьего газа является зависимой величиной, так
как С3=1—Ci—Сг. Вообще, для смеси из п компонентов
необходимо знать два термодинамических параметра и (п—1)
концентраций компонентов, как видно из общего выражения для
числа / степеней свободы.
Анализ равновесия смеси идеальных газов, в которой могут
протекать химические реакции между компонентами, является
ценным примером приложения понятия химического потенциала.
Мы найдем условие химического равновесия в этой системе и
свяжем его со скоростью реакции.
В качестве независимых переменных выбираем параметры
р и 71, и число молей отдельных компонентов будем обозначать
через iVi, N2y ..., Nu ... (для удобства записи здесь пользуемся
для компонентов нижними индексами). Для выбранных
параметров р и Т характеристической функцией является
термодинамический потенциал системы Z, который связан с химическими
потенциалами компонентов соотношениями вида
Термодинамический потенциал смеси идеальных газов равен:
Z = U — TS + pV = F' + pV. (7,5)
Здесь F' является свободной энергией смешавшихся газов,
выражение для которой дано в главе 5 в виде (5,30'):
F = -RT 2 N, In !L + 2 N,ft (Т),
где V — объем смеси, a fi(T) —известная функция температуры
по (5,27). Вводим F' в формулу для Z, и тогда получим:
Z = -RT 2 Nt In -£ + 2 Njt (T) +pV. (7,6)
Исключим из этой формулы объем смеси V, применяя
уравнение состояния для каждого компонента piV=NiRT, причем по
§ 2. Равновесие смесей идеальных газов. Закон действующих масс 209
закону Дальтона должно быть р = 1,р{. Поэтому, суммируя
уравнения состояния для всех газов смеси, находим:
Полученное отсюда значение V подставим в формулу (7, 6), и
тогда имеем:
Дифференцируем это выражение по Ni при р = const, Г = const
и при Nt, N2i ... = const. После некоторых преобразований и
перегруппировки .получаем соотношение для химического
потенциала:
«*' = (w) = - RT ln 4г +х' (р> Т) • (7>7)
где для простоты введена функция %(р, Т) параметров р и Т:
Ъ(р9 Т) = RT In-£ + Ь(Т). (7,П
причем
Ъ(Т) = КГ + Ь(Т)
является известной функцией температуры для 1-го компонента.
Вводя в формулу (7,7) концентрации в виде молярных долей, т. е.
находим окончательное выражение для химического потенциала
i-то компонента:
ft = /?TlnC/ + Zi(pf T). (7,8)
Отсюда видно, что (1г- возрастает с ростом концентрации
компонента, выраженной в молярных долях.
Пусть между газами, входящими в состав смеси, протекают
химические реакции, которые, как показывает опыт, идут с
конечными скоростями и по мере образования продуктов этих
реакций в системе развиваются обратные реакции между
полученными продуктами. Спустя некоторое время устанавливается
так называемое химическое равновесие, при котором скорости
прямого и обратного процессов становятся одинаковыми ив этих
условиях число всех частиц в смеси уже больше не меняется со
временем, т. е. при химическом равновесии в системе
присутствуют постоянные количества всех веществ.
Важнейшими примерами реакций в газовой фазе, имеющими
значение для техники, являются следующие химические
реакции:
210 Глава 7. Применение учения о равновесии к сложным системам
1) диссоциация водяного пара на водород и кислород и
обратная реакция синтеза воды:
2Н2О ^ 2Н2 + О2,
2) синтез аммиака из водорода и азота:
3) реакция в генераторном газе:
Н2О + СО :£ Н2 + СО2.
Стрелки в этих символических выражениях указывают на
течение процессов в обоих направлениях. Числовые
коэффициенты перед химическими формулами означают, как известно,
количество реагирующих молей и называются стехиометрическими
коэффициентами. Очевидно, любая химическая реакция
подобного рода может быть представлена в общем виде как
v A + V*2 + • • • ^ v A + \А2 + • • • .
где все vi, v2, ...— стехиометрические коэффициенты для
веществ Аи Лг... и v/, V2X,... соответственно для веществ
A/, Л2',,.., образовавшихся из первых. Можно уравнение
реакции представить иначе, перенося все слагаемые в одну
сторону равенства, и тогда
\А[ + V2A2+ . . . — v^j—v2i42— . . . =2v/i4/ = 0.
Здесь коэффициенты v* вообще имеют разные знаки. Можно
условно положить для продуктов реакции, что все v/>0, тогда
для исходных веществ нужно принять, что все vt<0.
Рассмотрим смесь химически реагирующих газов при
условии /? = const и 7 = const. Равновесие в этой системе достигается
при условии Z — минимум.
При постоянных р и Т мы получили ранее (см. гл. 6,
формула (6,33)) общее выражение равновесия в виде:
= 0, (7,9)
где \а — химический потенциал г-го компонента, 6УУг-—вариация
числа молей его при реакции. Если V* — стехиометрический
коэффициент /-го компонента, то изменение числа молей 8Ni
можно представить в виде
где 6N — общее для всей системы изменение числа
грамм-эквивалентов во всех реакциях. Поэтому уравнение (7,9) принимает
вид
§ 2. Равновесие смесей идеальных газов. Закон действующих масс 211
или так как diV^O, то
2 (V* = 0.
Подставим сюда найденное выше выражение химического
потенциала при независимых р и Т (7,8), и тогда
2 № = ЯГ 2 Ч In С, + 2 *Л (Р. Л = 0.
Отсюда
2^('Г) . (7.10)
Правая часть этого уравнения является константой, так как она
зависит только от давления и температуры, которые были
приняты постоянными, причем все v* также постоянны. Поэтому
для удобства можно обозначить
где К(руТ) называется константой химического равновесия.
Тогда формула (7,10) принимает вид:
у, Т),
откуда, переходя от логарифмов к числам, находим:
nc;' = c;«.c?... = /c(p, п (7,12)
Это выражение называется законом действующих масс; оно
показывает, что произведение концентраций (в определенных
степенях), характеризующих реагирующие массы, является
величиной постоянной при данных р и Т. Этот закон был открыт
в 1867 г. Гульдбергом и Вааге из статистических соображений.
Здесь был дан термодинамический вывод, предложенный Гибб-
сом (1871 г.).
Если выделять продукты реакции, обозначая
соответствующие им концентрации через С/, а концентрации исходных
веществ через Си то формулу (7,12) можно представить в виде:
Cl '°2 '" =/C(p, T), (7,13)
где все v* положительны.
Постоянство произведения в формуле (7,12) впервые было
доказано при рассмотрении вероятностей столкновений молекул
различных газов в смеси. Термодинамический вывод закона
действующих масс является ценным потому, что позволяет уста-
212 Глава 7. Применение учения о равновесии к сложным системам
новить зависимость константы равновесия от температуры и
давления и показать, как эти факторы влияют на ход реакции.
Чтобы решить эти вопросы, подставим в формулу (7,11)
найденное ранее выражение (7,7х) для функции %i{p,T), тогда
получим:
\пК(р, T) = -
В формулах (7,12) и (7,13) концентрации выражены в
молярных долях; из равенства (7,14) получаем зависимость константы
равновесия от давления и температуры, положив
G = -/?Tln/?TSv, + 2v^(T)f (7,15)
где G — функция только температуры. Тогда
откуда
\пК(р, T) = lnp<-JL
К(р, Т)^р-^.е RT. (7,16)
Зависимость константы равновесия от давления дана здесь
в явном виде, зависимость от температуры найти несколько
сложнее, и мы рассмотрим ее далее. Остановимся на одном
примере приложения закона действующих масс.
Выше было дано уравнение реакции при диссоциации воды
в водяном паре (при высоких температурах). Это уравнение
имеет вид:
2Н2О ^ + 2Н2 + О2, или 2Н2 + О2 — 2Н2О = 0.
Отсюда следует: vi = 2; V2=l; V3 = —2. Следовательно,
Здесь Svi = 2+1—2=1, поэтому согласно формуле (7,16)
Kip, Г) = -1-е"^7.
Отсюда следует, что с увеличением давления константа
равновесия уменьшается и при определенном равновесном С3
концентрации водорода и кислорода С\ и С2 уменьшаются. Это
значит, что с повышением давления в равновесной смеси будет
увеличиваться относительная доля недиссоциированного
водяного пара. Следовательно, в данном случае повышение давления
сдвигает равновесие в сторону понижения степени диссоциации
воды.
§ 2. Равновесие смесей идеальных газов. Закон действующих масс 213
В важной в промышленном отношении реакции синтеза
аммиака (стр. 210) согласно закону действующих масс
повышение давления также способствует образованию большей
концентрации аммиака, как в этом легко убедиться тем же путем.
Отсюда следует, что синтез аммиака с большим выходом этого
продукта возможен лишь при очень высоких давлениях, что
практически невыгодно. Поэтому необходимо применять
катализаторы, позволяющие провести реакцию при более низких
давлениях.
Зависимость константы равновесия от температуры можно
вывести, преобразуя формулу (7,14). Для этого заметим, что
функция tyi(T) для. i-й компоненты выражается соотношением
(стр.209):
где в свою очередь fi(T) имеет значение,полученное для 1 моля
газа в главе 5 (стр. 147), формула (5,27):
/. (Т) = U (T) -TCVi In Т -TS0.
Вводя это выражение в предыдущую формулу и используя
соотношение для энтропии 1 моля газа, получаем:
где Z{ — термодинамический потенциал 1 моля /-го компонента
Подставим эти выражения в формулу (7,14), и тогда после
несложных преобразований и применения уравнения состояния
pV=IiNiRT, получаем:
Чтобы отсюда получить зависимость константы равновесия от
температуры, найдем частную производную по Т от этого
соотношения:
дТ дТ * RT RT* [^ l l * * дТ
Л 7
Как известно, —- = — S*. Поэтому
дТ
Вспомним связь термодинамического потенциала Z* и
энтальпии:
214 Глава 7. Применение учения о равновесии к сложным системам
Тогда
din л: 1 v „
дТ RT* ^ l l
Здесь 2vi#i представляет собой полную энтальпию равновесной
системы, точнее, изменение ее АН при реакции, т. е.
дТ RT* v '
Далее, нам известно, что в реакциях при р = const тепловой
эффект реакции равен убыли энтальпии
Поэтому окончательно
£М = ^-. (7,18)
Напомним, что в экзотермических реакциях, когда теплота
выделяется, величина Qp>0, тогда как в эндотермических
реакциях QP<0. Поэтому из формулы (7,18) мы приходим к
следующим выводам. В экзотермических реакциях
т. q. с повышением температуры константа равновесия убывает.
Согласно закону действующих масс (7,13) это означает, что
концентрации продуктов реакции уменьшаются. Следовательно,
нагревание препятствует протеканию экзотермических реакций,
напротив, охлаждение приводит к увеличению константы
равновесия и потому благоприятствует таким реакциям. Для
эндотермических реакций, когда QP<0, по формуле (7,18)
производная—-—>0, откуда следует, что с повышением температуры
дТ
константа К растет и возрастают концентрации продуктов
реакции согласно закону (7,13). Это значит, что нагревание
благоприятствует течению эндотермических реакций. Опытные
данные подтверждают все эти заключения, и поэтому закон
действующих масс позволяет правильно предсказывать влияние
различных условий на течение химических реакций.
§ 3. ВОПРОСЫ ОБЩЕЙ ТЕОРИИ ФАЗОВЫХ ПРЕВРАЩЕНИЙ
Фазовыми превращениями, или фазовыми переходами, мы
называем процессы, в которых происходит переход вещества из
.одной фазы в другую. Хорошо известные изменения агрегатного
состояния вещества представляют собой лишь частные случаи
§ 3. Вопросы общей теории фазовых превращений 215
фазовых превращений. К настоящему времени изучено еще
много других процессов фазовых переходов, как-то: переход из
одной кристаллической модификации в другую (например,
желтого фосфора в красный, ромбической серы в моноклиническую),
переход металлов из ферромагнитного состояния в
парамагнитное, переход металлов в сверхпроводящее состояние при очень
низких температурах, наконец, переход жидкого гелия I в
гелий II. Разделение этих многочисленных видов фазовых
превращений на отдельные классы связано с опытными данными
по изучению свойств превращающихся фаз. Положив в основу
классификации характер изменения термодинамических свойств
при фазовых переходах, т. е. внутренней энергии, теплоемкости,
удельного объема, энтропии, можно разделить все известные
превращения на два рода. Долгое время были известны лишь
переходы первого рода. Существование фазовых переходов
второго рода было установлено П. Эренфестом в 1933 г. в связи
с изучением голландскими учеными свойств жидкого гелия.
Дадим вначале краткую характеристику различных классов
фазовых переходов и затем подробнее остановимся на их
особенностях.
1. Фазовые превращения первого рода. В эту
группу превращений входят упомянутые процессы перехода
вещества из одного агрегатного состояния в другое, т. е. переход
вещества из твердой фазы в жидкую (плавление), а также
соответственно обратный процесс затвердевания жидкости, далее
превращение жидкой фазы в газообразную (испарение,
кипение) и обратный процесс конденсации пара в жидкость; к
превращениям первого рода относится процесс испарения твердого
тела (возгонка) и обратный процесс сублимации. Некоторые
превращения из одной кристаллической формы в другую
кристаллическую также принадлежат к этому классу.
Отличительные особенности фазовых превращений первого
рода сводятся к следующему:
1) при переходе затрачивается или выделяется определен-
•ное количество тепла (называемого теплотой перехода);
2) при переходе в новую фазу происходит скачок удельного
объема вещества;
3) возможно существование малоустойчивых состояний
переохлаждения, пересыщения, перегрева и т. д.
4) теплоемкость в точке перехода бесконечно велика.
Эти свойства обусловлены существованием так называемой
точки перехода (превращения), которая может быть легко
установлена на диаграмме (/?, Т). Как мы видели, части
плоскости между разграничивающими линиями на этой
диаграмме отвечают состояниям одной какой-нибудь фазы. Если
при изменении параметров состояния протекает процесс по
216 Глава 7. Применение учения о равновесии к сложным системам
линии ab (рис. 36), то вещество из твердой фазы (в точке а)
переходит в жидкую (в точке Ь). При этом пересекается кривая
плавления в точке с. Этой точке отвечает определенная
температура плавления {Тип) и определенное давление (рПл), когда
жидкая и твердая фаза находятся в равновесии и их,
следовательно, можно термодинамически сравнить. Поэтому точку с
следует называть точкой перехода. Аналогичные точки перехода
получаем и для остальных фазовых превращений.
Отмеченные признаки фазовых переходов первого рода
найдены из опытов и хорошо известны. Например, известно, что
при кипении резко скачком увеличивается удельный объем
в точке кипения и при этом затрачивается конечная теплота
перехода. Давно найдено также существование малоустойчивых
состояний вблизи точки перехода. Наконец, легко убедиться,
что в точке перехода теплоемкость Ср бесконечно велика. В
самом деле, теплоемкость является отношением Ср = (-^-) #
В точке перехода T = const и, следовательно, ДГ=0, тогда как
теплота перехода AQ = qn конечна, поэтому Ср—^оо.
2. Фазовые превращения второго рода. В эту
группу входят такие процессы, как переход металлов из
ферромагнитного состояния в парамагнитное в точке перехода,
называемой точкой Кюри; далее, переход различных металлов
в сверхпроводящее состояние, например свинца пр* ГП = 7,2°К,
олова при ГП = 3,7ГК, цинка при Гп = 0,78°К и других также при
низких температурах тоже представляет собой фазовое
превращение второго рода. К этим превращениям относится и переход
жидкого гелия I в жидкий гелий II, совершающийся при
температуре 2,2° К и некоторые превращения в кристаллических
телах. Наконец, фазовый переход в критической точке также
носит основные черты фазовых переходов второго рода.
Характерными признаками этих превращений являются
следующие особенности:
1) удельный объем вещества не испытывает скачка в точке
перехода;
2) теплота перехода отсутствует;
3) теплоемкость вещества в точке перехода меняется
скачком;
4) коэффициент теплового расширения и изотермический
коэффициент сжимаемости изменяются также скачкообразно;
5) малоустойчивых переходных состояний вблизи точки
перехода не наблюдается.
Так, когда железо при нагревании до точки Кюри теряет
свои ферромагнитные свойства, переходя в парамагнитную
фазу, то вблизи точки перехода удельный объем меняется
непрерывно, не испытывая скачка. При этом превращении не затра-
§ 3. Вопросы общей теории фазовых превращений
217
чивается и не выделяется скрытой теплоты перехода. Другим
примером может служить переход жидкого гелия I в жидкий
гелий II. Опыт показал, что это превращение также совершается
без затраты и выделения скрытой теплоты и без изменения
удельного объема. Однако теплоемкость Ср близ точки перехода
очень резко изменяется, образуя в ней точку заострения
(сингулярную точку), рис. 37. Форма кривой напоминает греческую
букву X (ламбда) и такой вид имеет ход теплоемкости и других
свойств во всех фазовых переходах второго рода, почему точку
перехода часто называют Л-точкой.
Перечисленные характерные признаки фазовых переходов
первого и второго родов установлены из опытных данных, т. е.
являются обобщением наблюдений. Эти особенности приводят
к ряду дальнейших заключений и следствий, основанных на
термодинамических соображениях, которые мы сейчас рассмотрим.
Выше было сказано, что при фазовых переходах первого
рода наблюдается затрата или выделение конечной удельной
теплоты перехода qn и что удельный объем в точке перехода
испытывает скачок. Отсюда вытекают дальнейшие свойства,
характеризующие эти переходы. Прежде всего следует
подчеркнуть, что в точке перехода, т. е. на разграничивающей линии
рис. 36, обе фазы находятся в равновесии и, следовательно, их
химические потенциалы по теории гетерогенного равновесия
должны быть одинаковы, т. е. m-i = |я2. Это означает, что и
удельные термодинамические потенциалы обеих фаз в точке перехода
равны друг другу, значит:
<Pi(P, Л = Ф2(Р, Т). (7,19)
30
25
20
15
Ю
У
<
о
J
/
1
1
'о
О
\2 1%U 1t6 1y8 2,0 2,2 2,4 2,6 2%8 3%0 к
—*~ Т
Рис. 37,
218 Глава 7. Применение учения о равновесии к сложным системам
Это равенство показывает, что при фазовом переходе
термодинамический потенциал изменяется непрерывно, несмотря на
скачок удельного объема. Однако можно легко убедиться в том,
что энтропия при этом меняется скачкообразно. Для этого
достаточно найти соотношение между теплотой перехода и
энтропией, применяя общее выражение
dQ = T dS.
В данном случае затраченная теплота Q является удельной
теплотой перехода qn и потому, вводя также удельную энтропию 5
для каждой фазы, находим для точки перехода при Т=const
при интегрировании
§s = T(s2-Sl). (7,20)
Таким образом, теплота перехода равна произведению из
температуры на приращение энтропии. Так как при фазовых
переходах первого рода теплота перехода является всегда конечной,
то равенство (7,20) показывает, что энтропия при переходе
меняется скачком, т. е. испытывает разрыв непрерывности. Если
при переходе из фазы / в фазу 2 теплота перехода
затрачивается (<7п>0), то энтропия при этом скачкообразно
увеличивается (см. рис. 38). Например, при парообразовании
энтропия пара больше энтропии жидкости, из которой пар
образовался.
Скачкообразное изменение энтропии и удельного объема
приводит далее к тому, что при фазовых переходах первого
рода внутренняя энергия (удельная) изменяется также
скачком. Общее выражение удельного термодинамического
потенциала
Ф == и — Ts -f pv
для первой и второй фаз дает нам
«Pi = "l — Ts± + pvu
Ф2 = и2 — Ts2 + pv2
в точке перехода, причем при
равновесии ф1 = ф2. Поэтому
и 1 — Tsl + pv± = u2 — Ts2 + pv2,
откуда
и2 — их = Т (s2 — Sl) — p (v2 — vj,
Tn T Нли иначе
Рис. 38. Au = qn — p- До. (7,21)
§ 3. Вопросы общей теории фазовых превращений 219
Это равенство показывает, что внутренняя энергия при
фазовом переходе изменяется скачком, что обусловлено затратой
конечной теплоты перехода и конечным изменением удельного
объема.
Заметим, что скачок энтропии и скачок удельного объема
в точке перехода приводят к скачкообразному изменению
первых частных производных термодинамического потенциала. Это
непосредственно следует из соотношений для частных
производных этой функции, приведенных в таблице главы 5, которые
для удельного потенциала принимают вид:
—№;■ -£
где s и v тоже удельные величины. Применяя (7,22) к обеим
фазам в точке перехода, имеем:
ь_„_д.__(А_А.). ,7,23,
(7,230
др )
Так как As и Av являются конечными, то из этих формул
следует, что первые производные от ф(р, Т) меняются скачком
в точке перехода.
Написанные здесь соотношения позволяют легко вывести
уравнение Клапейрона—Клаузиуса. В самом деле, при
равновесии имеем ф!(р, Г)=ф2(р, Г), причем здесь р является
функцией температуры. Дифференцируем обе части этого равенства
ф1 = ф2 по р и Т:
дТ dp dT дТ dp ' дТ #
Подставляя сюда выражения для производных (7,23) и (7,23Г),
находим:
dT v% — vi *
Наконец, вводя сюда (7,20), получаем уравнение Клапейрона —
Клаузиуса
dp _ ?п
dT T (v2 — vt)
Так как As и Av являются конечными, то первые
производные функции ф (р,'Т) меняются скачком в точке перехода. От-
220 Глава 7. Применение учения о равновесии к сложным системам
сюда далее следует, что вторые частные производные
термодинамического потенциала должны обращаться в бесконечность
в точке перехода, как это видно непосредственно из
геометрических соображений.
Но согласно равенствам (7,22) имеем:
*L = _ (*L\ • ^L = (to.\ • J!^ = (*1\ (7 24)
дТ* \дт)р9 dp* [dp )т дрдТ \дт]р' Kf )
Правые части этих трех соотношений имеют простой смысл.
Так как CD = —£ и dQD = Tdsp, отсюда \^г) = £■£. , где Ср —
р dT р р \oTjp т
удельная теплоемкость при постоянном давлении. Далее, (—) ,
\др !т
-как известно (стр. 19), связано с изотермическим
коэффициентом сжимаемости
Наконец, величина (—) входит в коэффициент объемного
\дТ)р
расширения при постоянном давлении:
1 fdv\
v \дТ)р
Таким образом, вторые частные производные от <р(р, Т)
можно представить в виде:
& C i5J* .v. (7,25)
dT* Т dp* ' дрдТ
Левые части этих равенств должны обращаться в
бесконечность из-за скачка первых производных функции ф(р, Г), и это
также подтверждается анализом правых частей равенств (7,24)
и (7,25). В самом деле, было отмечено, что в точке перехода
система обладает бесконечно большой теплоемкостью Ср, так
как теплота перехода конечна, а повышение температуры не
имеет места, т. е. Ср->оо. Далее, в точке перехода объем
меняется на конечную величину (Ди конечно), а повышения
давления нет при равновесии, т. е. у ►«>; конечное изменение
объема не связано с изменением температуры, которое здесь
отсутствует, поэтому а-^оо.
Следует обратить внимание на принципиальную
возможность существования пересыщенного пара и перегретой
жидкости при фазовых переходах первого рода. Эти состояния
являются сами по себе устойчивыми, но они все же менее устойчивы,
чем соответствующие им фазы между отрезками разграничи-
§ 3. Вопросы общей теории фазовых превращений 221
вающих линий. Здесь важна степень устойчивости,
определяемая величиной удельного термодинамического потенциала. Так,
на диаграмме (р, Т) между кривой плавления и кривой
кипения минимум термодинамического потенциала отвечает жидкой
фазе, которая обладает, следовательно, наибольшей
устойчивостью. Но точки в этой части плоскости могут соответствовать
пересыщенному пару, представляющему собой тоже
устойчивую, но относительно менее устойчивую фазу в этой области.
Фазовые переходы второго рода характеризуются
скачкообразными изменениями теплоемкости, коэффициента теплового
расширения и изотермического коэффициента сжимаемости
в точке перехода. При этих превращениях qu равно нулю и
удельный объем меняется непрерывно. Из условия равновесия
здесь также следует, что в точке перехода: (pi(p, Т)=ц>г(р, 7).
Указанные особенности фазовых переходов второго рода
приводят к следующим результатам.
Так как <7п=0 и Vi = v2 (или Да = 0), то, очевидно, согласно
формулам (7,20) и (7,21)
As = 0 и Да = 0,
т. е. в точке перехода энтропия и внутренняя энергия
изменяются непрерывно, не испытывая скачков. Поэтому на основании
(7,23) и (7,230 имеем:
дт)р [дт)р [дТ)р
др)т \др)т \др)т
Таким образом, в точке перехода не только сам удельный
термодинамический потенциал, но и его частные производные
изменяются непрерывно. Однако вследствие того, что в точке
перехода имеются скачки Ср> у и а, т. е.
то из соотношений (7,25)"и (7,26) видно скачкообразное
изменение вторых частных производных функции ф:
/ &ъ \ I д*ъ \ =A№i = L/C —С \ *Ср
V от* 1р \ дт2 )р \аг2 )р т v л pj т *
ЬЛ -(И-\ ^А(^-) =a(L)
дрдТ)р [дрдТ ) \дрдТ}р \дТ)
222 Глава 7. Применение учения о равновесии к сложным системам
Нетрудно показать, что величины ДСР, А [—) и Д( — ]
\др]т >дТ !р
связаны между собой простым соотношением. Для этого
дифференцируем уравнения (7,26) по переменным р и Г. Тогда
d7 + A
дТ2 дТдр
дрдТ dp2 y
Заменяя вторые производные их значениями из (7,27),
находим:
-MjLdT + AllL) dp = 0,
т дТ И
(7,28)
Допускаем, что оба эти уравнения совместны, и тогда после
исключения из них dp и dT получаем:
~ ' ~ = 0. (7,29)
Уравнения (7,28) и (7,29) были выведены П. С. Эренфестом.
Мы видим, что скачки теплоемкости, коэффициентов теплового
расширения и изотермического сжатия связаны между собой
соотношением (7,29). Уравнения (7,28) позволяют найти
производную — в каждой точке перехода, и отсюда по данным
ДСР, Ду и Да можно построить разграничивающую линию для
фазовых переходов второго рода. Поэтому соотношения (7,28)
являются аналогами уравнения Клапейрона—Клаузиуса,
применимого к фазовым переходам первого рода.
§ 4. ТЕРМОДИНАМИКА ПРОЦЕССА ОБРАЗОВАНИЯ ЗАРОДЫШЕЙ
НОВОЙ ФАЗЫ
Процессы образования новой фазы являются весьма
разнообразными. Очень часто новая фаза получается из старой
в виде большой массы, например при плавлении, но хорошо
известны явления, когда новая фаза сначала появляется в форме
небольших частиц, которые растут по размерам и могут
сливаться друг с другом. Так образуются капельки жидкости
в паре, кристаллики в расплаве, а также крошечные пузырьки
пара в жидкости. Для того чтобы образование новой фазы было
возможно, необходимо, чтобы исходная фаза находилась в
§ 4. Термодинамика процесса образования зародышей новой фазы 223
метастабильном состоянии, т. е. была хотя и
устойчивой, но с устойчивостью меньшей, чем это следует из условий
полного равновесия. Поэтому новая фаза образуется в
пересыщенном паре или в переохлажденном расплаве или в
перегретой жидкости. Эти состояния на диаграммах равновесия
соответствуют точкам с избыточной энергией (см. рис. 36).
Во многих практических случаях новая фаза образуется на
посторонних центрах или зародышах. В воздухе такими
зародышами служат пылинки, газовые ионы и т. д. В известной
камере Вильсона в пересыщенном паре образуются жидкие капли
на ионах, возникающих при пробеге альфа- и бета-частиц и
других частиц большой энергии. При высотных полетах за
самолетом часто видна белая полоса водяного тумана,
образующегося при конденсации из пересыщенного атмосферного пара
на частичках отработанного горючего. Наконец, если подышать
на холодное стекло, то оно запотевает, т. е. покрывается
маленькими каплями воды, образующимися на поверхностных
мелких загрязнений стекла, когда выдыхаемый пар, охлаждаясь
у поверхности, становится пересыщенным. Однако эти явления
образования новой фазы не представляют принципиального
интереса и мы их не касаемся, а обратим внимание на
образование новой фазы в отсутствие этих центров, т. е. в чистом виде.
В опытах Вильсона было обнаружено, что в камере образуется
при больших пересыщениях пара густой туман тогда, когда все
посторонние центры конденсации заранее заведомо были
удалены. Это явление носит название спонтанной
конденсации в отсутствие посторонних центров, и его теория была
разработана Фольмером (1926 г.). Согласно этой теории новая
фаза возникает в данном случае благодаря флуктуации
плотности пара, т. е. за счет случайных скоплений молекул пара.
Эти скопления или сгущения в паре то вновь образуются, то
исчезают благодаря тепловому движению молекул и в общем
случае не могут дать начало новой фазы. Для этого необходимо
соблюдение некоторых условий устойчивости таких скоплений
или очагов новой фазы, которая могла бы расти дальше и быть
сама устойчивой.
Следует обратить внимание на то, что для образования
случайного скопления молекул необходимо . совершить внешнюю
работу. Ее можно найти следующим путем. Из основного
определения термодинамического потенциала Z (гл. 5, формула
(5,14)) следует:
S + kak + kak,
где F — свободная энергия. Изменение термодинамического
потенциала есть
AZ = AF + AkAak.
224 Глава 7. Применение учения о равновесии к сложным системам
Работа AkActk является работой над скоплением молекул,
и она равна работе по созданию поверхности скопления, т. е.,
как раньше, as, где a — поверхностное натяжение, а 5 —
поверхность скопления. Последнее можно представить в виде
сферической капельки радиуса г, поэтому s = 4jtr2. Работа над
системой, как и раньше, отрицательна, поэтому
AZ = AF —
Отсюда изменение свободной энергии равно:
AF = AZ + 4-r2a.
При полном равновесии химические потенциалы старой и
новой фазы должны быть одинаковыми, но они при развитии
новой фазы не равны друг другу, т. е. AZ^O. Из определения
химического потенциала для старой фазы находим Zi = \nNy где
N — число молекул в образовавшемся скоплении; для новой
фазы соответственно имеет Z2 = (a,2^; здесь jii и |я2 — химические
потенциалы. Поэтому
№ = (^-^N + 4^.0. (7,30)
Мы рассматриваем изотермический процесс и,
следовательно, изменение свободной энергии есть совершенная в
процессе работа. Мы видим, что она состоит из двух слагаемых;
одно представляет собой работу получения скопления, тогда
как второе — работу образования поверхности. Из соотношения
(7,30) видно, что здесь возможны два частных случая:
во-первых, когда |Л2>М<1, и, во-вторых, Jjh>|12. Известно, что при
переходе к равновесию свободная энергия должна убывать,
стремясь к минимуму. Однако в первом случае изменение
свободной энергии AF является положительным, следовательно, в этом
случае равновесие не достигается. Иными словами,
образующиеся вследствие флуктуации скопления будут только
«рассасываться» и новой фазы дать не могут. Во втором случае
имеем:
AF = — (^ — ц2) N + 4тгг2-о.
Здесь изменение свободной энергии имеет вид, указывающий
на действие двух конкурирующих факторов. Первый из них,
сводящийся к сглаживанию разницы химических потенциалов
старой и новой фазы, энергетически выгоден, тогда как второй
фактор, зависящий от образования новой поверхности, является,
как известно, невыгодным. Отсюда видно, что в случае |ii>|X2
возможно образование устойчивого скопления. Если vM есть
объем молекулы в скоплении, то очевидно, число молекул в нем
равно N = —— . Тогда из (7,30) имеем:
3 vm
§ 4. Термодинамика процесса образования зародышей новой фазы
225
Представим графически зависимость AF от радиуса
скопления. В первом случае, когда ц,2>м<1, мы имеем модотонный рост
Д/7 с увеличением г (рис. 39), т. е. AF только растет, что
соответствует неустойчивости скоплений. Во втором случае, когда
имеем:
(7,31)
Теперь получаем, как легко убедиться, кривую, обладающую
максимумом (рис. 39). При очень малых радиусах преобладает
второй член, соответствующий проигрышу в работе и росту Д/\
При дальнейшем увеличении радиуса начинает сказываться
действие первого члена, дающего выигрыш, после чего AF
переходит в область отрицательных значений, когда действует почти
один первый член. Максимум функции AF соответствует
критическому радиусу гк капельки, которая еще является
неустойчивой, тогда как дальнейший рост ее может дать уже устойчивую
частицу. Положение максимума легко найти обычным путем,
полагая
dr
= 0
при r=rK. Тогда получаем:
(7,32) AF
Подставляя это значение в
формулу (7, 31), находим
величину изменения свободной
энергии для критического радиуса
скопления. После простых
преобразований имеем:
(7,33)
Работу W по образованию
поверхности, т. е. второй член в
соотношении (7, 31) легко найдем
для критического радиуса,
используя формулу (7, 32). Тогда
= 4-
(7,34)
Рис. 39.
226 Глава 7. Применение учения о равновесии к сложным системам
Сравнивая (7,33) и (7,34), находим отношение:
1
3 '
, или Д/7,
__ 1
*~~ 3
WK
Следовательно, для критического скопления одна треть
свободной энергии затрачивается на образование поверхности
капли, а две трети на объемную работу изменения состояния
системы при создании флуктуации. Для капель, радиус которых
существенно превосходит критический радиус, по мере их роста
получающиеся зародыши могут стать устойчивыми и тогда
преобладающее значение имеет выгодная объемная работа.
Как видно из формулы (7,32), радиус критического очага
новой фазы зависит от поверхностного натяжения жидкости
и от разности химических потенциалов. Последняя
соответствует степени пересыщения пара; чем пар более пересыщен,
тем разность больше и радиус критической капли делается
меньше. При высоких степенях пересыщения, как показывает
оценка, радиус первоначально уже устойчивой капли
составляет всего 5—10 А.-На этих зародышах затем идет дальнейшая
конденсация, и они растут до больших размеров.
Описанный выше механизм образования новой фазы через
флуктуации плотности относится не только к образованию
капель в пересыщенном паре, но имеет место при образовании
кристалликов в переохлажденном расплаве, а также при
образовании пузырьков пара в перегретой жидкости.
§ 5. КРИТИЧЕСКАЯ ТОЧКА. СВОЙСТВА РЕАЛЬНЫХ ГАЗОВ
Рассматривая фазовое равновесие в системе жидкость—пар,
мы отметили, что соответствующая ей разграничивающая
линия на плоскости (/?, Т) оканчивается в определенной точке К
(рис. 36), которую называют критической точкой.
Температура Тк в этой конечной точке называется критической
температурой, а давление рк при этой температуре
принято называть критическим давлением. При
температуре выше Тк вещество может быть только в газообразной фазе
и никаким давлением при Т>ТК нельзя перевести его в жидкую
фазу. Это обстоятельство, являющееся опытным фактом, может
быть наглядно объяснено с помощью молекулярных
представлений. Очевидно, при Т>ТК энергия молекул столь уже велика,
чта при столкновениях преодолеваются силы притяжения и
молекулы отталкиваются друг от друга. Пока не отнята
некоторая избыточная энергия, невозможно никаким давлением
обеспечить устойчивое действие сцепления, необходимое для
существования жидкости.
§ 5. Критическая тонка. Свойства реальных газов 227
Существование критической температуры впервые было
предсказано Д. И. Менделеевым (1861 г.), обратившим
внимание на уменьшение поверхностного натяжения жидкости
с ростом температуры и сделавшим отсюда вывод, что должна
существовать температура, при которой различие между
жидкостью и паром должно исчезнуть, он назвал ее
температурой абсолютного кипения. Позднее Эндрьюс (1869 г.)
с помощью специально сконструированного пресса изучал
изменение объема углекислого газа с давлением при различных
температурах и построил семейство изотерм, на которых было
обнаружено критическое состояние углекислоты. Критическая
температура была потом определена для многих веществ и
получены аналогичные изотермы.
Подробный анализ фазовых превращений в системе
жидкость—газ и вывод параметров Для критической точки даны
в труде Ван-дер-Ваальса «Непрерывность газообразных и
жидких состояний» (1873 г.). В основу этих исследований
положено уравнение состояния реального газа, выведенное Ван-дер-
Ваальсом из простых молекулярных соображений и пригодное
отчасти и для жидкостей. Как известно, это уравнение для
1 моля вещества имеет вид:
y-b) = RT, (7,35)
где а и Ь — константы, причем первая из них связана с
силами молекулярного притяжения, а вторая —с силами
отталкивания и является пропорциональной объему молекул.
Применимость единого уравнения к описанию поведения как жидкости,
так и газа Ван*дер-Ваальс рассматривает как непрерывность
жидкого и газообразного состояний.
Сначала рассмотрим общую термодинамическую теорию
критического состояния, а затем посмотрим, в какой мере
выводы из этой теории оправдываются для систем следующих
уравнению Ван-дер-Ваальса. Задача общей теории сводится в
данном случае к выводу существования критической точки и к
описанию свойств вещества в этой точке при условии устойчивого
равновесия системы в этом состоянии.
Прежде всего заметим, что геометрическое изображение
критического состояния меняется в зависимости от того, какой
диаграммой мы пользуемся. Так, на диаграмме состояний (р, Т)
критическая точка является точкой остановки кривой АК
(рис. 40), соответствующей равновесию двухфазной системы
жидкость — насыщенный пар. Как ясно из рисунка, пар из
состояния а может быть переведен в жидкость в точке Ь или по
изотерме ab при Т<ТК и при этом он пройдет через
разграничивающую линию АКУ но можно идти по линии acb, обойдя кри-
228
Глава 7. Применение учения о равновесии к сложным системам
тическую точку /С, и тогда переход в жидкость будет
совершаться непрерывно. Справа от критической изотермы ТКТ'К
свойства жидкости и пара неразличимы, тогда как слева
проходит линия АК двухфазной системы.
Если состояния системы изображаются на диаграмме (v, Г),
показывающей изменение удельного объема с температурой, то
получается кривая, изображенная на рис. 41, где МК
представляет изменение удельного объема насыщенного пара, а
линия NK дает изменение удельного объема жидкости с
температурой. Каждой температуре вообще отвечают два объема обеих
фаз, находящихся в равновесии. В критическом состоянии оба
объема сливаются, т. е. на кривой (у, Т) критическая точка К
есть точка поворота кривой. Справа от изотермы ТКТ'К
свойства жидкости и пара неразличимы, тогда как слева мы
опять имеем равновесную двухфазную систему.
Наконец, можно изобразить семейство изотерм на
диаграмме (р, v). Здесь при Т<ТК получаются кривые с
горизонтальными участками, которые соответствуют равновесию
двухфазной системы и которые постепенно укорачиваются по мере
повышения температуры. Для изотермы при Т=ТК на кривой
нет разрыва непрерывности, причем, как показывает опыт,
критическая точка К является точкой перегиба (рис. 42).
Легко понять, что форма кривых на трех рассмотренных
диаграммах связана с видом термодинамической поверхности,
определяемой уравнением Ф = Ф(у, р, Г)=0, представляющим
собой уравнение состояния. Изобразив семейство этих кривых
в трехмерном пространстве (р, v, Г), можно убедиться, что на
плоскости (р, Т) точка К должна быть точкой остановки, тогда
как на плоскости (у, Т) точка К является точкой поворота,
а в плоскости (р, V) она является точкой перегиба.
Для анализа свойств
критической точки мы
исходим из условий
устойчивого равновесия,
рассматривая в качестве
характеристической функции
удельный
термодинамический потенциал ф, как
при изучении фазовых
переходов. Однако с
помощью диаграмм (р, Т)
и (у, Т) мы не можем
рассматривать окрест-
f j ность точки /С, варьируя
к параметры состояния.
РИС. 4б. Давая положительное
Жидкость
§ 5. Критическая точка. Свойства реальных газов
229
приращение ДГ, мы попадаем здесь в область неопределенных
производных ф по Г, тогда как при отрицательных AT мы
переходим в обоих случаях в область равновесия двухфазной
системы. Поэтому мы будем рассматривать критическую изотерму
на диаграмме (р, v) и считать при r = rK = const удельный
потенциал функцией объема. На этой изотерме объем изменяется
непрерывно с давлением и в то же время здесь имеет место
равновесие уже одной фазы, которое должно быть устойчивым.
Обратим внимание на зависимость ф от v при р и Г постоянных.
Известно, что условиями устойчивого равновесия являются
выражения: ф—минимум; 6ф = 0 и 62ф>0. Следовательно, если
равновесие достигнуто и ф минимально, то, давая любое
положительное или отрицательное приращение объема Av, мы
увеличиваем потенциал ф. Поэтому при устойчивом равновесии
в критической точке должно быть
(7,36)
или
Здесь v — удельный объем.
Введем для удобства удельную свободную энергию / = —
или f=u—Ts. Тогда неравенство принимает вид:
Афр т = А (/ + pv)p т = Afp T + pAv> 0. (7,37)
Ранее было показано (см. гл. 5):
(i)r—P. (7,38,
р
И ^,
/V -""-'■
j
Жидкость
К
кр
Рис. 41.
230 Глава 7. Применение учения о равновесии к сложным системам
Поэтому условие равновесия (7,37) можно представить в виде
Дер Г = Д/_Ш .Ду>0. (7,39)
> т
Предположим, что в окрестности критической точки возможно
разложение функции Д/ в ряд по степеням приращения
объема Да. Тогда
(Щ (*,) +
2! \д*)РТк ' т 3!
4!
(При этом можно строго доказать, что для аналитической
функции / достаточно ограничиться производной не выше
четвертого порядка.) Подставив это выражение в формулу (7,39), по«
лучим после сокращения условие устойчивого равновесия в
критическом состоянии:
*Р-Т 21 \d*)PT } ^ 31 \)р%т
Это выражение при любых Av должно быть безусловно
положительным. Отсюда прежде всего следует, что члены с
нечетными степенями Ди должны быть равны нулю, иначе при
каком-либо Ду<0 условие (7,40) может не соблюдаться.
Следовательно,
Кроме того, обратим внимание на вторую производную в
формуле (7,40). Легко видеть из (7,38), что она может быть
представлена в виде:
Щ =(^| (7,42)
dv)T
Очевидно, для всех вообще температур устойчивое состояние
равновесия получается только, если
(d-f) <0, (7,43)
\dvJT
т. е. когда с увеличением объема при Т — const давление
падает. Напротив, условие ( —]>0 отвечает неустойчивым состоя-
§ 5. Критическая точка. Свойства реальных газов 231
ниям и не может рассматриваться в термодинамике. Таким
образом, вообще говоря, при устойчивом равновесии должно
быть
Возникает вопрос, выполняется ли условие (7,43) в критической
точке, раз оно выполняется всюду в ее окрестности? Можно
различным путем убедиться, что в критической точке, в
отличие от всех других состояний, условие (7,43) изменяется и
первая производная давления по объему обращается в нуль при
существовании устойчивого равновесия, т. е.
Во-первых, заметим, что критическая точка лежит на
разграничивающей линии диаграммы (/?, 71), где всюду для фазового
перехода первого рода изотермический коэффициент
сжимаемости равен бесконечности, т. е.
(dv\
— =оо.
\др )т
Отсюда следует выражение (7,44) для обратной производной.
Во-вторых, в критической точке при любом изменении объема^
условие (7,43) не будет соблюдаться, так как при dv<0 должно
быть d/?>0, т. е. при сжатии давление должно возрасти. Между
тем из диаграмм (/?, Т) и (v, Т) ясно, что при смещении от
точки К на первой диаграмме вверх по изотерме (dp>0) мы
попадаем в область жидкой фазы, т. е. должно иметь место
ожижение; то же должно иметь место при смещении вниз на
второй диаграмме (dv<0). Между тем на критической изотерме
не должно быть фазовых переходов. Значит, условие (7,43) не
оправдывается в критической точке при любом изменении
объема. Но одновременно условие! —) > 0 не может иметь места,
\dvjT
так как это противоречит устойчивости системы. Отсюда
следует, что в критической точке производная I —) должна обра-
\dv /т
щаться в нуль, тогда как всюду она меньше нуля.
Следовательно, в соотношении (7,40) имеем (——\ = 0; значит, первый
член равен нулю, и тогда оно примет вид:
232 Глава 7. Применение учения о равновесии к сложным системам
Это окончательное выражение для условия равновесия в
критической точке будет соблюдаться при всех Ди, если в нем все
производные положительны, в частности, когда
Таким образом, условие равновесия для критической точки
приводит к соотношениям (7,41), (7,42), (7,45), т. е.
=0;(т =0; (Л
Принимая во внимание равенство (7,38), эти соотношения
можно представить в виде:
£L = _«£- = 0, (7,46)
il = _-*£. = <>, (7,47)
^) (7,48)
Полученные значения частных производных для
критической точки позволяют вывести заключения о свойствах этой
точки на диаграмме (р, v). Будем рассматривать р как
функцию v при Т=ТК. Эта функция является, по нашему
допущению, непрерывной. Уравнение соответствующей кривой
(изотермы) мы могли бы получить из выражения (7,38), если бы
знали f=f(p, vy T)\ это значит что формула (7,38)
\dv)T ]
дает нам функцию состояния, а при Т = ТК = const — уравнение
критической изотермы. Легко видеть, что выражения (7,46),
(7.47) и (7,48) определяют собой критическую точку как точку
перегиба изотермы при Т=ТК. В самом деле, равенство (7,46)
показывает, что касательная в критической точке идет
параллельно оси v. Так как во всех соседних точках согласно (7,43)
производная отрицательна, то в критической точке —=0 до-
dv
стигает максимума. Обращение в нуль второй производной
согласно (7,47) и отрицательное значение третьей производной по
(7.48) определяют собой точку перегиба, как известно из курса
математического анализа. Этот результат согласуется с
опытными данными, показывающими, что критическая точка яв-
§ 5. Критическая точка. Свойства реальных газов 233
ляется точкой перегиба на изотерме. Для определения
критических параметров Гк, /?к и vK мы имеем систему из трех
уравнений:
f = -р; ^ = 0; ff = O. (7,49)
dv dv dv2
Таким образом, общие термодинамические соображения
устанавливают существование критической точки, определяют ее
как точку перегиба и дают возможность вычислить
критические параметры, если известно уравнение состояния. Кроме
того, было показано, что при равновесии имеет место условие
(т) <°>
\dv/T
причем знаку равенства соответствует критическая точка.
Наконец, необходимо заметить, что так как при
критическом состоянии удельные объемы пара и жидкости становятся
равными друг другу, то согласно уравнению Клапейрона —
Клаузиуса теплота парообразования в критической точке
превращается в нуль, т. е. qn = 0. Это обстоятельство приводит,
как и ранее (стр. 221), к тому, что в критической точке
энтропия не испытывает скачка, т. е. As = 0; в связи с тем что скачок
удельного объема тоже отсутствует (Аа = 0), не имеется скачка
и внутренней энергии: Ди = 0. Эти свойства критической точки
показывают, что между критическим переходом и фазовыми
переходами второго рода имеется много общего. Как мы видели,
в критической точке! —) =0, т. е. коэффициент изотермического
сжатия обращается в бесконечность. Можно из общих
соображений показать еще, что удельная теплоемкость Ср и
коэффициент объемного расширения тоже бесконечно велики в
критической точке (как для фазовых переходов первого рода).
Выясним теперь, в какой мере выполняются общие свойства
критического состояния для веществ, строго следующих
уравнению Ван-дер-Ваальса
Оно является кубическим уравнением по отношению к объ-
V2
ему. Умножим обе части уравнения на — . Тогда получим:
а
+ fv-^ = 0. (7,50)
Действительно, это уравнение третьей степени вида
х3 + ClX* + С2х + С3 = 0; (7,50')
234 Глава 7. Применение учения о равновесии к сложным системам
как известно, оно имеет вообще три корня: хи х2 и хг. При
решении физической задачи мы интересуемся только
вещественными корнями, так как v не может быть мнимым. Из теории
кубических уравнений следует, что все три корня могут быть
вещественными и разными и тогда уравнение (7,50') можно
представить как
{х — хх) (х - х2) (х — х3) = 0.
В нашей задаче это означает, что при Г=const одному какому-
либо значению р могут отвечать три разных объема. Далее,
при известных значениях коэффициентов все три
вещественных корня могут сливаться и будут равны друг другу, т. е.
Xi = Х2 = Х3 = Хк.
В этом случае кубическое уравнение можно представить в форме
(*-*к)3 = 0. (7,50")
Значит, при некоторой температуре Тк и определенном
давлении рк происходит слияние трех корней уравнения (7,50) и
получается один объем vK, соответствующий этим параметрам.
Можно показать, что это условие осуществляется в
критической точке.
Зависимость между р и v при данном Т может быть
изображена графически по уравнению Ван-дер-Ваальса, и мы получим
семейство изотерм, изображенных на рис. 42. Здесь получаются
плавные кривые, причем для какой-либо из них одному
значению р соответствуют три разные величины объема. Значения
этих объемов с повышением температуры сближаются друг
с другом и при некоторой температуре получается один объем vK.
Сравнивая эти теоретические кривые с опытными, мы не
находим горизонтальных ступенчатых участков на опытных
кривых. Это можно объяснить тем, что участок теоретической изо-
теомы abed (рис. 42) соответствует малоустойчивым и
неустойчивым состояниям, которые в принципе существовать могут,
но практически не реализуются. Участок ab отвечает перегретой
жидкости, a cd—пересыщенному пару. Отрезок be недоступен
для наблюдения вследствие неустойчивости таких состояний,
когда ( — ) >0. Остальные части теоретических изотерм соот-
\dv I т
ветствуют опытным, и поэтому можно сказать, что уравнение
Ван-дер-Ваальса правильно описывает ход изотерм при разных
температурах и, кроме того, дает сведения о ходе изотерм для
малоустойчивых состояний.
Естественно считать, что критическому состоянию
соответствуют температура Тк и давление /?к, когда все три объема vu
§ 5. Критическая точка. Свойства реальных газов 235
V2 и и3 становятся равными, т. е. когда имеет место уравнение
вида (7,50") или
(v-vK)3 = 0.
Пользуясь этим уравнением, найдем параметры в критической
точке. Выполняя возведение в третью степень, имеем:
v3 — 3v\ + Sw2K — ^ = 0.
Сравнивая коэффициенты при v в этом уравнении с
коэффициентами уравнения (7,50), находим:
При заданных а и Ь мы получили систему трех уравнений
для неизвестных рк, ^к и Тк. Решая эти уравнения, имеем:
t;K = 3&; рк = -^-; Тк= —. (7,51)
Таким образом, критические параметры просто связаны с
константами уравнения Ван-дер-Ваальса.
Согласно общей теории критическая точка является точкой
перегиба, и для нее должны оправдываться условия (7,46),
(7,47), (7,48). Для того чтобы убедиться в том, что решения
(7,51) удовлетворяют этим условиям, представим исходное
уравнение (7,35) в форме
Выполняя частное дифференцирование по v, получаем:
/др_\ = _ RT 2±. (&£\ = 2RT
/др_\
\dv)T
{p — bf v* ' \dv*)T (v — b)* &
И
(д*р\ = 6RT . 24q
Подставляя сюда значения ик, Рк и Гк из формул (7,51),
находим:
= 0; —~| =0 и —^-1 = —
Так как а>0 и fc>0, то из последнего равенства следует, что
236 Глава 7. Применение учения о равновесии к сложным системам
Полученные результаты показывают, что, действительно,
значения vKy рк и Тк по формулам отвечают критической точке,
которая является точкой перегиба.
Вычислим теперь коэффициент объемного расширения а,
коэффициент изотермического сжатия у и теплоемкость в
критической точке для веществ, следующих уравнению (7,35).
Известно, что
1 (dv\
v \дт)р
Для нахождения а вычисляем полный дифференциал dp из
уравнения (7,52) и затем полагаем dp = 0 (так как /? = const).
Имеем:
dp^-^-dT--
[V — О)" V
(7,53)
v — b (v — b)2
Отсюда
v-b
VT 2a (v~b\2
R \ v I
Полагая здесь vK = 3b и Тк = согласно (7,51), находим,
27Rb
что ак = °о, т. е. в критической точке коэффициент теплового
расширения обращается в бесконечность; это согласуется с
выводами общей теории критического состояния.
Известно далее, что
~ v [dp)
dp)/
Дифференцируя (7,52) по объему при T=const, находим:
\dv)T (v — b)2 v9 *
Подставляя обратную производную в общее выражение для у,
получаем:
fe£(7,54)
В критической точке при значениях vK и Гк, определяемых
равенствами (7,51), это выражение дает нам у = °°, т. е. в
критическом состоянии коэффициент изотермического сжатия
обращается в бесконечность, что, впрочем, следует также из анализа
производной (—] в критической точке.
^ = СР = \(Щ +p](*L) +CV,
§ 5. Критическая точка. Свойства реальных газов 237
Рассчитаем еще теплоемкость газа Ван-дер-Ваальса в
критической точке. Из выражения для первого начала
термодинамики следует (стр. 43), что
откуда
или
Для идеального газа внутренняя энергия не зависит от объема,
т. е.( —) =0,и тогда из (7,55) получается формула Р. Майера.
\ OV/T
Для газа Ван-дер-Ваальса, для которого (—] =#=0 можно
\dvjT
показать (стр. 120), что
(ди
В формуле (7,55) частная производная (—) может быть полу-
\дТ }р
чена из уравнения (7,53). Поэтому (7,55) принимает вид:
V—-Ь
R&
Подставляя сюда значения vK и Тк из (7,51), находим, что
p~LV = °°'
т. е. теплоемкость Ср в критической точке равна бесконечности.
Анализ уравнения Ван-дер-Ваальса показал нам, что оно
качественно очень хорошо описывает свойства критической точки
в соответствии с данными общей теории. Однако при
критическом состоянии обнаруживаются существенные количественные
отклонения от экспериментальных данных. Из формул (7,51)
легко получить значение так называемого критического
коэффициента /С, разделив RTK на pK-vK. Тогда
К = *^- = —=2,667.
3
Для идеального газа это отношение равно единице, так как этот
газ подчиняется уравнению Клапейрона. Мы видим, что для
238 Глава 7. Применение учения о равновесии к сложным системам
реальных газов К почти в три раза больше, чем для идеальных.
Однако опыт показал, что для большинства веществ критический
коэффициент лежит в интервале от ~3,0 до 3,8, а для воды
достигает значения 4,46. Таким образом, в критической точке имеют
место отступления от уравнения Ван-дер-Ваальса, которое все
же оказывается и в этих условиях более точным, чем уравнение
Клапейрона.
Если в уравнение (7,35) ввести относительные величины,
выражая параметры состояния отношениями v, p и Т к
соответствующим критическим параметрам, то это уравнение переходит
в выражение, содержащее только безразмерные величины.
Положим,
v p T
о = —; тг = -^-; т = —.
ок Рк Тк
Пользуясь формулами (7,51), подставим в уравнение (7,35)
введенные величины о, я и т, и тогда легко находим:
+ -!-) (3<о-1) = 8т. (7,56)
Это уравнение носит название закона соответственных
состояний, представляющего собой закон подобия, справедливый
для всех веществ, независимо от их природы.
Уравнение (7,56) обладает следующими свойствами:
1) оно не содержит констант вещества в явном виде;
2) является вполне универсальным;
3) передает ту же закономерность, как и исходное
уравнение Ван-дер-Ваальса;
4) состояния разных веществ оказываются
соответственными, если эти состояния даны для одинаковых со, я и т.
Закон соответственных состояний (7,56) хорошо
оправдывается на опыте. Аналогичный закон может быть получен для
всякого уравнения состояния содержащего три константы.
Задачи
1. Доказать, что при фазовых превращениях первого рода происходит
скачок удельной свободной энергии.
2. Доказать, что при фазовых превращениях первого рода имеет место
скачок удельной энтальпии..
3. Найти выражение для константы равновесия реакции диссоциации че-
тырехокиси азота:
Показать, как влияет давление на степень диссоциации, которая представляет
собой отношение числа диссоциированных молей к числу всех молей N2O4.
4. Найти константу равновесия реакции синтеза аммиака и показать, что
повышение давления увеличивает выход аммиака.
Глава 8
ТЕПЛОВАЯ ТЕОРЕМА НЕРНСТА
И ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА ВЕЩЕСТВ
ПРИ НИЗКИХ ТЕМПЕРАТУРАХ
§ 1. ПРИРОДА ХИМИЧЕСКОГО СРОДСТВА. ТЕПЛОВАЯ ТЕОРЕМА НЕРНСТА
Вопрос о поведении термодинамических систем вблизи
абсолютного нуля температур возник в связи с так называемой
тепловой теоремой Нернста (1906 г.). Эта теорема
вместе с примыкающими к ней теоретическими положениями
составляет содержание третьего начала термодинамики,
имеющего важное общее значение в физике и химии. Практическая
ценность этого начала состоит в том, что оно вносит
определенность в численные значения термодинамических функций.
Известно, что в выражение для энтропии входит неопределенная
постоянная интегрирования, благодаря чему нельзя
непосредственно найти абсолютную величину энтропии, хотя в
приложениях важно вычислять изменение энтропии и постоянная
интегрирования нас не интересует. Однако численное значение самой
энтропии необходимо для вычисления других
термодинамических функций, куда входит произведение TS, например в
выражение свободной энергии, и тогда неопределенность
энтропийной постоянной приводит к затруднениям при расчете.
Достаточно вычислить абсолютное значение энтропии хотя бы в
одном частном случае и тогда мы в состоянии найти энтропию
в других условиях, так как постоянную интегрирования можно
будет рассчитать, зная зависимость S от параметров состояния.
Эта задача решается с помощью третьего начала термодинамики.
Непосредственно теорема Нернста была создана им в связи
с обсуждением вопроса о химическом сродстве при низкой
температуре. Понятие химического сродства введено для
характеристики способности веществ химически реагировать друг с
другом. Уже давно было замечено, что одни вещества легко
вступают в реакцию между собой, другие реагируют плохо или
вовсе химически не взаимодействуют. Естественно возникал вопрос
о том, что считать мерой химического сродства. Так как при
химических реакциях часто наблюдается выделение теплоты, то
Томсен и Бертло высказали как общий принцип, что количество
выделяющейся теплоты реакции должно служить мерой
химического сродства, иными словами, чем больше выделяется
240 Глава 8. Тепловая теорема Нернста
теплоты, тем больше сродство между реагирующими веществами
и, значит, из всех возможных реакций данного вещества с
другими будет протекать та реакция, при которой будет выделяться
больше теплоты. Однако с принципиальной точки зрения это
утверждение следует считать ошибочным. Во-первых, в принципе
Томсена и Бертло рассматриваются только реакции
экзотермические, т. е. идущие с выделением тепла, между тем как
хорошо известны (гл. 2, стр. 58) также эндотермические
реакции, при которых теплота не выделяется, а поглощается.
Для них вообще принцип Томсена—Бертло непригоден.
Во-вторых, количество теплоты при химической реакции и само
направление последней зависят от многих условий и в первую очередь
от температуры, так что величина сродства может меняться.
Наконец, установлено, что многие реакции идут не до конца и в
системе наступает химическое равновесие. Упомянутый принцип
неприменим и для таких случаев. Таким образом, для
установления меры сродства необходимо указать условия протекания
реакции и иметь в виду переход в состояние равновесия. В
таком случае целесообразно применять общие условия
равновесия термодинамических систем.
Наиболее просто выяснить меру сродства, если система
реагирующих веществ помещена в термостат и поддерживается при
постоянной температуре Т = const. Далее условия реакции
будут определены, если система находится в твердой оболочке,
т. е. поддерживается при постоянном объеме V=const. Другой
случай соответствует условию, когда изотермическая система
находится при постоянном внешнем давлении. Если в
системе при r = const и V = const протекает химическая реакция и
затем наступает равновесие,то, как мы знаем (стр. 168), свободная
энергия системы убывает и стремится к минимуму. Пусть
свободная энергия системы до реакции есть /ч и свободная
энергия после установления равновесия есть /V, тогда ясно, что
процесс начнется и будет идти, если Fi—^2>0, причем чем больше
эта разность, т. е. чем больше убыль свободной энергии, тем
быстрее пойдет реакция. Следовательно, убыль свободной энергии,
т. е. —AFv,t, должна служить необходимым и достаточным
критерием химического сродства для изотермических реакций при
постоянном объеме; при этом имеется в виду максимальная
убыль, соответствующая максимальной работе. Как мы знаем,
максимальная работа Wv равна убыли свободной энергии
изотермического процесса. Здесь имеется в виду работа не за счет
расширения. Итак,
Wv = -bFVwT.
Поэтому можно сказать, что мерой химического сродства
является также максимальная работа.
§ 1. Природа химического сродства. Тепловая теорема Нернста 241
Аналогичный результат мы получаем для изотермических
реакций при постоянном давлении. В этих условиях
термодинамический потенциал Z стремится к минимуму, и убыль ~AZP>T,
равная максимальной работе Wp, является мерой химического
сродства.
Для процессов при постоянном объеме выполняется
уравнение Гиббса—Гельмгольца (стр. 142):
откуда следует, что
(^L) . (8,2)
Величина AF в этом уравнении и является по Вант-Гоффу
мерой химического сродства в реакциях при постоянном объеме.
Мы видим, что изменение свободной энергии AF не равно
изменению внутренней энергии Д£/ системы, а отличается от
последнего на величину, зависящую от температуры и от производной
( J . Вместо свободной энергии можно ввести
максимальную работу, так как Wy = —kFT,v> и учесть еще, чтоД£/=—QVi
где Qv — теплота реакции (черта вверху, как было отмечено
ранее, означает, что для экзотермических реакций теплоту
считаем положительной). Тогда уравнение (8,2) переходит в форму
• (8,3)
дТ
В этом выражении мерой сродства является максимальная
работа, которая не равна теплоте реакции, а зависит еще от
второго слагаемого, которое может быть как положительным, так и
отрицательным и связано с температурой.
Для изотермических реакций при постоянном давлении
нетрудно вывести соотношение, аналогичное формуле (8,3),
которое имеет вид:
(8,30
где Qp — теплота реакции при постоянном давлении.
В своих исследованиях Нернст обратил внимание на то, что
согласно многочисленным опытным данным разность
9 Заказ №2479
242 Глава 8. Тепловая теорема Нернста
всегда сравнительно невелика, особенно если реакция
протекает не в газовой фазе, а в конденсированных системах
(жидкости, твердые кристаллические тела). Тогда даже при
комнатной температуре максимальная работа Wv очень близка к
теплоте реакции QVy т. е. изменение свободной энергии практически
равно изменению внутренней энергии системы. Это означает,
что принцип Томсена—Бертло практически является
справедливым, в частности особенно для реакций с большим сродством.
Обобщая опытные данные, Нернст заметил также, что разность
в формуле (8,4) и, следовательно, разность в выражении (8,2)
(8,5)
убывает с понижением температуры не по линейному закону
зависимости от Г, а значительно быстрее. Это дало основание
допустить, что в произведении правой части (8,5) второй
множитель, т. е. производная/ ) , убывает с температурой, стре-
мясь к нулю при Т->0. Следовательно, при переходе к
абсолютному нулю разность А/7—At/ обращается в нуль за счет
обращения в нуль обоих сомножителей правой части равенства (8,5).
Таким образом, следует принять, что
о. (8,6)
дТ )v
Тогда AFV = MT при Г = 0,
или
\ /<ШМ Г = о. (8,7)
Отсюда на основании (8,6) получаем:
) =0. (8,8)
v
т-*о\ дТ jv r-o
Иначе
llmllm(^ =0. (8,9)
Равенство (8,9) представляет собой математическое
выражение третьего начала термодинамики и называется
тепловой теоремой Нернста.
Легко показать, что выражение (8,8) следует из формулы
(8,5) при допущении, что производная( ) остается не
бесконечной при абсолютном нуле. В самом деле, из (8,5) получаем:
[дТ
§ 1. Природа химического сродства. Тепловая теорема Нернста
243
Если здесь производная не обращается в бесконечность при
Г = 0, то это может быть только, когда AF = AU. В этом случае
получаем неопределенность —, которая раскрывается по
общему правилу. Дифференцируем по Т числитель и
знаменатель левой части уравнения и переходим к пределу.
Тогда
дТ
\ /dW
)у [ дТ
дТ )у
(
Отсюда ( ) =0 при Т—►(), и мы приходим к полученному
ранее равенству (8,8).
Рассматривая реакции при постоянном давлении и исходя
из уравнения (8,3'), можно показать подобным же образом, что
при Г-И)
\дТ )р~~
р[дТ )р
и что
)р
=0.
(8,90
Тщательные измерения теплоты реакции при низких
температурах, а также теоретические соображения привели Нернста
к выводу, что тепловая теорема строго оправдывается для
кристаллических тел и для жидкостей, т. е. для конденсированных
систем. Хотя вблизи Т—ИЗ все тела являются
конденсированными, все же для твердых аморфных тел при весьма низких
температурах разность (8,4) еще немного отличается от нуля,
т. е. приближается к нему медленнее, чем в других случаях.
Применение квантовой
статистики к так называемым &Uуд
вырожденным газам при
температурах, очень
близких к абсолютному нулю,
показало, что и для этих
систем, в частности для газа
из свободных электронов
при Т—И), теорема Нернста
строго выполняется. Таким
образом, несмотря на ряд
имеющихся небольших
отступлений, можно считать,
что теорема Нернста явля- Рис, 4з.
244 Глава 8. Тепловая теорема Нернста
ется законом, имеющим общее значение, а не ограничивается
применением только к некоторым системам и к химическим
реакциям.
Теорему Нернста в виде равенств (8,8) можно представить
графически, изображая ход зависимостей AF и AU от
температуры. Согласно (8,8) касательные к обеим кривым AF и AU при
Г=0 сливаются в общую касательную, идущую параллельно оси
Т (рис. 43), как это следует из геометрического смысла
производной. При этом с понижением температуры величина AU
убывает, тогда как AF при этом растет.
Тепловая теорема позволяет прийти к выводу о свойстве
энтропии при Г-*0. Именно, воспользовавшись известным
соотношением
Д/? = Д1/_ Г AS,
находим дифференцированием его по Т:
dW
dT dT dT
Отсюда, переходя к пределу при Т—>0 и учитывая равенства
(8,8), получаем:
AS = О при Т = 0.
Следовательно, при абсолютном нуле изменение энтропии
отсутствует и она остается постоянной
AS = S2 — Si=0 при Т-+0, (8,10)
или вообще
Sx = S2 = So = const. (8,100
§ 2. ПОСТУЛАТ ПЛАНКА И СВОЙСТВА ВЕЩЕСТВА
БЛИЗ АБСОЛЮТНОГО НУЛЯ
Рассматривая тепловую теорему Нернста, приводящую к
постоянству энтропии при абсолютном нуле, Планк (1912 г.)
высказал гипотезу, что константа энтропии в формуле (8,10х)
при абсолютном нуле равна нулю для всех систем, независимо
от агрегатного состояния и • различных других свойств
вещества. Следовательно,
So = 0 при Т = 0.
Это положение называют постулатом Планка, и оно
является дальнейшим обобщением теоремы Нернста. Заметим,
что этот результат может быть получен из квантовой
статистики (см. стр. 248).
§ 2. Постулат Планка и свойства вещества близ абсолютного нуля 245
Если использовать известное термодинамическое уравнение
то постулат Планка приводит при 7=0 к выводу, что
= 0.
\дТ Jv
Поэтому из общего выражения F = U—TS при So — О следует:
Itaffi =limffi =0. (8,11)
T-*o\dTJv T->o\dTJv
Этот результат показывает, что при абсолютном нуле
свободная энергия равна полной внутренйей энергии системы,
тогда как связанная энергия G=TS обращается в нуль.
Согласно постулату Планка вблизи абсолютного нуля не только
изменения AU и AF стремятся к нулю, но и сами функции U
и F сливаются вместе. Эти соображения показывают, что
с уменьшением температуры энтропия асимптотически
стремится к пределу S0 = 0. Отсюда следует важная практическая
возможность вычисления абсолютного значения энтропии. Так,
например, пусть энтропия является функцией V и Г, причем
S = f(V, T) + So,
где So — энтропия при абсолютном нуле. Из постулата Планка
следует, что 50 = 0 независимо от объема. Поэтому
5 = f(Vt T).
Если вид функции f(V, Т) известен и V и Т даны, то мы можем
вычислить абсолютное значение S. Практически вычисление
энтропии связано с нахождением неопределенных интегралов
вида
=
S(V, T)= l-^L +const, или S(p, T)= -^L+const.
Так как теперь мы знаем, что при Г=0 величина 50 = 0 во всех
случаях, то константы исключаются и интегрирование ведем
от 0 до Г, т. е. приходим к определенным интегралам
г т
Sty, T)= f£^-, или S(p, T)= j££!. (8,12)
о о
Напомним, что энтропия входит в выражения для
термодинамических функций F и 2, поэтому для их расчетов также
необходимо иметь в виду постулат Планка, т. е. полагать So = O.
246
Глава 8. Тепловая теорема Нернста
Тогда получаем:
F(V9 T) = U-'.
CvdT
Z(p, T) = #-
CpdT
T
Постулат Планка хорошо подтверждается на опыте. Так,
например, измерения теплоемкости льда при разных температурах
привели к значениям энтропии, показанным в таблице. Для
кристаллических тел в непосредственной близости Г = 0
энтропия ничтожно мала, тогда как для аморфных она имеет
значения несколько большие, но также весьма малые.
Тепловая теорема и постулат Планка приводят к общему
выводу, что при переходе к абсолютному нулю, многие свойства
вещества должны существенно
изменяться.
1. Удельные (молярные)
теплоемкости Cv и Ср обращаются
в нуль при температуре
абсолютного нуля.
В самом деле, из постулата Планка
следует, что
lim№ =0.
т-о\дТ )v
Но так как известно, что Cv
Т в°К
273
250
200
150
100
60
30
20
10
Таблица
5 в кал/°К
9,18
8,34
6,69
4,91
3,16
1,57
1,00
0,095
«0
то отсюда заключаем:
-(■
dU\
дТ jv
UmCv = 0.
г-о
Рассматривая процессы при постоянном давлении, можно
получить равенство (8, 9'), т. е.
где Н — энтальпия, - связанная с теплоемкостью Ср
соотношением:
С -(дН\
Поэтому из теоремы Нернста следует:
г-о
§ 2 Постулат Планка и свойства вещества близ абсолютного нуля 247
Оба результата могут быть также получены при рассмотрении
общих выражений (8,12) для энтропии. Оба интеграла
оказались бы расходящимися на нижнем пределе Г=0, если бы
величины Ср и Су были при этом отличными от нуля. Отсюда
видно, что в пределе при Т—>0 обе теплоемкости Cv и Ср
должны равняться нулю.
Экспериментальные исследования Нернста с сотрудниками,
а также позднейшие наблюдения многих ученых приводят к
общему выводу, что теплоемкости всех веществ в области низких
температур резко убывают с температурой. В главе 2 было
отмечено, что в области нескольких десятков градусов шкалы
Кельвина экспериментально установлен закон куба абсолютной
температуры:
С
что находится в согласии с квантовой теорией теплоемкостей
твердых тел, развитой Дебаем. В непосредственной близости
к Г=0 теплоемкость исчезающе мала; таким образом, можно
считать, что важнейшее следствие из теоремы Нернста
находит себе подтверждение на опыте.
2. Коэффициент теплового расширения при
температуре абсолютного нуля обращается
в нуль.
Из общих дифференциальных уравнений термодинамики
(5,17) находим, что
—) = (дУ\
др)т \дТ)р'
Если при Г=const вблизи абсолютного нуля изменим давление
от р до p + dpy то энтропия изменится от S=5i до S2=5xrf
] dp. Следовательно, изменение энтропии будет равно:
др /т
lim(52 — S1) = limf—)
г-о г-*о\ dp )т
AS = S2-S1==№ dp.
\ dp ]т
Переходя к пределу Т—►(), мы должны согласно формуле (8,10)
получить:
откуда
и, следовательно,
№ =0.
т-+о\дТ
248 Глава 8. Тепловая теорема Нернста
Так как а = — ( ) , то из предыдущего ясно, что
V \дТ )р
3. Термический коэффициент давления р при
температуре абсолютного нуля равен нулю.
Из общих дифференциальных уравнений термодинамики
(5,17):
tdS_\ /Jp\
\dV )т \дТ)\
Давая приращение объема dV близ температуры Г=0 и
вычисляя приращение энтропии, находим тем же путем, как
ранее, с применением уравнения (8,10), что
откуда
г-6 V dV )т °'
Ит(Щ =0.
Следовательно, коэффициент р = — (——) обращается в нуль
р \ дТ /v
при температуре абсолютного нуля.
4. Скрытая теплота перехода при
температуре абсолютного нуля равна нулю.
Это следствие может быть выведено из соотношения (7, 20)
главы 7:
Переходя к пределу при Т—^0, мы на основании формулы
(8,10) получаем:
lim qn = 0.
г-о
Можно показать, кроме того, что ряд других свойств вещества
также претерпевает изменение при абсолютном нуле, например
коэффициент поверхностного натяжения перестает зависеть от
температуры и т. п.
Теорему Нернста и постулат Планка можно обосновать
статистически, если воспользоваться законом Больцмана о связи
энтропии с вероятностью состояния (гл. 4). Следует обратить
внимание на то, что вблизи Г=0 все тела находятся в
конденсированном состоянии, по большей части в твердом агрегатном
состоянии, и хаотическое движение их молекул (атомов) уже
полностью отсутствует и заменяется колебаниями связанных
§ 2. Постулат Плапка и свойства вещества близ абсолютного нуля 249
друг с другом частиц. Эти колебания носят квантовый
характер, т. е. определяются частотами на различных квантовых
уровнях энергии. Эти уровни для макроскопических тел очень
многочисленны и лежат весьма тесно друг к другу. Вблизи
Т=0 имеется тенденция к переходу на самый низкий
квантовый уровень энергии. В пределе при Г=0 все частицы
однородного тела должны находиться на одном самом низком
уровне энергии, и так как он определяется квантовыми
условиями, то ниже его энергии быть не может. Это значит, что
состояние такой системы определяется одной лишь
комбинацией. Ранее в главе 4 мы указывали, что энтропия системы S
связана с логарифмом термодинамической вероятности WT, т. е.
S= klnWT.
Следовательно, состояние тем более вероятно, чем большим
числом комбинаций в распределении параметров частиц
(энергии) оно осуществляется. Однако при абсолютном нуле
состояние реализуется лишь одной комбинацией, когда все уровни
энергии частиц сливаются в один самый низкий уровень. Это
означает, что№т =~- = 1. Отсюда следует по формуле Больц-
мана, что \nWT=Q или что S = 0. Необходимо заметить, что
если не учитывать квантового характера энергии, то такого
вывода сделать нельзя, так как даже при самой низкой
температуре Г->0 имелись бы частицы разных энергий и состояние
определялось бы не одной, а несколькими комбинациями,
и тогда S было бы отличным от нуля.
§ 3. СВЕРХНИЗКИЕ ТЕМПЕРАТУРЫ И ПРИНЦИП НЕДОСТИЖИМОСТИ
АБСОЛЮТНОГО НУЛЯ. ОТРИЦАТЕЛЬНЫЕ АБСОЛЮТНЫЕ ТЕМПЕРАТУРЫ
В настоящее время наиболее низкие температуры, лежащие
в непосредственной близости к абсолютному нулю, достигаются
двумя методами: испарением жидкого гелия 3Не из растворов
его в 4Не и применением так называемого магнитокалори-
ческого эффекта.
Изотоп гелия 3Не содержится в гелии, извлеченном из
атмосферы в ничтожных количествах 10~4%, и потому практически
использован быть не может. Однако, после того как в атомных
реакторах стали получать в больших количествах тритий,
который после р-распада переходит в 3Не, то эксперименты с
последним дали возможность получать весьма низкие
температуры. Испарением 3Не из растворов в 4Не удается понизить
температуру до величины ~ 0,006° К и поддерживать ее в
аппаратуре в течение времени, достаточного для производства
различных опытов.
250
Глава 8. Тепловая теорема Нернста
Магнитокалорический эффект был открыт Дебаем (1926 г.)
и Джиоком (1927 г.). Сущность его состоит в том, что
намагничивание и размагничивание -некоторых парамагнитных веществ
при низких температурах при известных условиях приводит
к значительному охлаждению системы.
В этих опытах небольшое количество парамагнитной соли помещается
в специальную вакуумную рубашку, находящуюся в двойном сосуде Дьюа-
ра, причем в наружном сосуде находится жидкий водород, а во внутреннем —
жидкий гелий. В качестве парамагнитной соли в первых опытах применялся
сульфат гадолиния; в настоящее время с большим успехом применяют железо-
аммониевые или хромокалиевые квасцы. Весь прибор помещается между
полюсами сильного электромагнита, могущего давать поле в 7—10 килоэрсте-
дов. Заполняя вначале рубашку с солью жидким гелием, можно обеспечить
за счет хорошего контакта изотермические условия, при которых соль
поддерживается при температуре этого гелиевого термостата, равной 1—1,3° К.
Затем через обмотку электромагнита пропускается постоянный ток,
создающий магнитное поле в пространстве, где находится образец соли и система
выдерживается некоторое время, чтобы вновь установилось тепловое
равновесие. Находясь в сильном магнитном поле, соль намагничивается, причем ее
частицы, представляющие собой элементарные магнитики, практически все
ориентируются одинаково соответственно направлению магнитного поля. После
установления равновесия (спустя 5—10 мин) в системе создают
адиабатические условия, удаляя жидкий гелий из рубашки и осуществляя в ней путем
откачки высокий вакуум, после чего выключают магнитное поле. Соль
размагничивается в адиабатических условиях и при этом охлаждается столь
значительно, что ее температура падает до тысячных долей градуса
абсолютной шкалы. Рекордное охлаждение до 0,0012° К было впервые таким путем
достигнуто в опытах де-Гааза. Таким образом, охлаждение получается в
процессе адиабатического размагничивания предварительно сильно охлажденной
и намагниченной соли.
Для того чтобы выяснить причину охлаждения в этом
методе, рассмотрим изменение энтропии в описанном процессе,
пользуясь энтропийной диаграммой (S,T). На рис. 44 показан
ход изменения энтропии соли в отсутствие поля (Я=0) и при
постоянном магнитном поле
с напряженностью H=Hi.
Сложный ход энтропии в
отсутствие поля
объясняется тем, что при низких
температурах парамагнитные
вещества обладают
способностью проявлять некоторое
самопроизвольное
(спонтанное) намагничивание, при
котором имеет место
частичная ориентация
элементарных магнитиков,
подобно тому как это
наблюдается у ферромагнитных
Рис. 44.
тел.
§ 3. Сверхнизкие температуры и принцип недостижимости абс. нуля 251
В присутствии постоянного магнитного поля (нижняя
кривая) энтропия уменьшается, так как в образце соли
повышается степень упорядоченности структуры, а нам известно, что
энтропия является мерой неупорядоченности, связанной с
молекулярным хаосом, следовательно, упорядоченность в
структуре равноценна снижению величины энтропии. В отсутствие
поля энтропия при всех температурах выше, так как не
имеется фактора, упорядочивающего структуру, но и здесь с
Понижением Т величина S убывает. В соответствии с постулатом
Планка обе кривые проходят через начало координат.
Начальное состояние соли, находящейся в контакте с гелиевой ванной,
характеризуется точкой 1. Пр'и включении поля и после
выравнивания температуры система переходит в состояние 2 по изо-,
терме (1,2) при температуре 7Y Когда создают
адиабатические условия и выключают поле, то соль переходит в
состояние 3 по адиабате (2, 3) и температура понижается до Г2, т. е.
^2<7V Изменение энтропии, вызываемое изменением
магнитного поля от 0, на величину dH состоит в данном случае из
двух частей, во-первых, как было сказано, энтропия зависит от
намагниченности соли, т. е. от изменения ее магнитного
момента единицы объема ( ] , и, во-вторых, от изменения
\ дТ /нм
внутренней энергии, зависящей от теплоемкости Снм
намагниченной соли.
Таким образом, полное изменение энтропии равно:
dH. (8,13)
В адиабатном процессе S = const, т. е. dS=0, поэтому из
предыдущего выражения следует:
dH.
нм
Эта формула показывает, от каких причин зависит изменение
температуры. Здесь отношение > 0 при всех условиях, да-
Снм
лее из опыта известно, что ( ) < 0. Следовательно, при
\ дТ /нм
адиабатическом намагничивании, когда сШ>0, имеем dT>Q,
т. е. в этом случае происходит нагревание соли. Напротив, при
адиабахическом размагничивании, когда йЯ<0, мы имеем
dr<0, т. е. происходит охлаждение соли. В связи с тем что при
включении поля соль нагревается, необходимо было выжидать
некоторое время, чтобы восстановилась первоначальная
температура Ти прежде чем производить размагничивание.
252 Глава 8. Тепловая теорема Нернста
В целом можно сказать, что охлаждение получается
потому, что при адиабатном размагничивании полная энтропия
должна остаться без изменения, но при этом увеличивается ее
магнитная часть (второе слагаемое в формуле (8,13)) за счет
дезориентации магнитиков, а это приводит к уменьшению
другой части энтропии (первое слагаемое), т. е. понижению
температуры. Для повышения эффекта охлаждения важно, чтобы
обе кривые рисунка 44 расходились возможно сильнее в
области ~ Г К или ниже. Это заставляет применять «магнитно-
разбавленные» соли, т. е. такие, в которых ионы, обладающие
магнитными свойствами, были бы значительно удалены друг
эт друга. Такому требованию удовлетворяют, например, железо-
аммониевые квасцы Fe(NH4) (SO4)2-12НгО, где один
парамагнитный ион железа приходится на полсотни атомов, не
обладающих магнитными свойствами. Тогда даже при очень
низких температурах энергия атомов оказывается достаточной,
чтобы в стадии размагничивания преодолеть слабые силы
магнитного взаимодействия ионов железа и вызвать полное
размагничивание. Найдено, что магнитокалорический эффект
может быть весьма значительным; например, достаточно всего
1 г парамагнитной соли, чтобы охладить несколько
килограммов какого-либо вещества от Г К до сверхнизких температур
порядка 0,001° К.
Новейшее видоизменение этого метода основано на эффекте
адиабатического размагничивания атомных ядер. Первые
эксперименты по охлаждению этим методом привели к
температуре 10~5°К (1956 г.). Применение смеси 3Не с порошками
различных металлов позволило понизить температуру еще дальше.
Самая низкая температура, достигнутая этим методом, в
настоящее время оценивается (1963 г.) значением 1,2-10~~6°К *.
В связи с получением низких температур, лежащих весьма
близко к Г=0, естественно должен возникнуть вопрос вообще
о принципиальной возможности достижения абсолютного нуля
температур при дальнейшем совершенствовании
охлаждающих устройств. Анализ этой проблемы, имеющей не только
практическое, но также и глубоко принципиальное значение,
показывает, что температура абсолютного нуля недостижима;
мы можем значительно приблизиться к ней, но достигнуть ее
абсолютно точно невозможно.
Недостижимость абсолютного нуля можно показать чисто
термодинамическим путем, рассматривая в виде примера
простейший процесс охлаждения какой-либо системы. При этом
самым выгодным является процесс адиабатного обратимого
* См.: М. 3 е м а н с к и и. Температуры очень низкие и очень высокие.
М., «Мир», 1968.
§ 3. Сверхнизкие температуры и принцип недостижимости абс. нуля 253
расширения, когда система охлаждается, совершая внешнюю
работу. Энтропия является функцией объема и температуры.
Если изменяется только температура от Т\ до Г2, то изменение
энтропии равно (см. (8, 12)):
S(V, T2) — S(V, 7\) =
fx
Произведем адиабатическое расширение от объема V до V+
+ДУ, причем T2<Ti. Адиабатный процесс является изоэнтропи-
ческим, т. е. до расширения и после него энтропия остается
неизменной:
S(y, Tj^SiV + W, Т2).
Подставляя сюда S(Vt Tx) из предыдущего равенства, находим:
, 72) = S(V, T2) + J fjL
Так как CV>0, то здесь должно быть S(V+AV, 72)>S(F, Т2).
Мы хотим провести адиабатное расширение до температуры
абсолютного нуля, т. е. Г2—Я). Тогда написанное выше
уравнение примет вид:
J
AK, 0) = S(K, 0)
Отсюда следует, что должно быть S(V+AV,0)>S(Vf 0), т. е.
при абсолютном нуле мы получили: А5>0. Но этот результат
противоречит теореме Нернста (8,10), согласно которой при
Г = 0 должно быть А5 = 0. Таким образом, видно, что адиабдт-
ным расширением от 7\>0 невозможно достигнуть
абсолютного нуля. Тот же результат мы получим, если начнем процесс
при Г2>0 и будем стремиться к jTi = O. Следовательно, всегда
имеем: S(V+AV,0)=5(V,0) при Г = 0.
Легко показать, что, применяя метод адиабатического
размагничивания, также невозможно достигнуть абсолютного
нуля температуры. В самом деле, обе кривые на рис. 44
сходятся в точке Г = 0, где энтропия равна нулю согласно
постулату Планка. Переходя с верхней адиабаты на нижнюю при
включении поля и затем вновь на верхнюю адиабату при
размагничивании, мы никак не сможем прийти в точку 7=0, даже
если бы могли несколько раз чередовать намагничивание и
размагничивание. Это ясно из рис. 44. Однако если бы при
Г = 0 энтропия достигала бы отличного от нуля значения So, то
254 Глава 8. Тепловая теорема Нернста
тогда, выбрав подходящую начальную температуру, мы могли
бы сразу достигнуть точного значения нуля температуры при
размагничивании. Следовательно, в данном примере причиной
недостижимости нуля является постулат Планка о нулевом
значении энтропии 50=0 при Г = 0.
Наконец, вспомним, что для идеальной обратимой машины
Карно к. п. д. должен принимать значение 100%, когда
температура холодильника равна абсолютному нулю, т. е. Г2 = 0. Но
при этом должно быть Q2 = 0, т. е. в этом случае холодильник
вовсе не получает тепла, хотя налицо все условия для
теплопередачи, нагреватель же отдает тепло, целиком переходящее
в работу. Такая машина невозможна, так как холодильник не
может сохранять температуру абсолютного нуля, а значит,
абсолютный нуль недостижим. Все эти соображения и ряд
других, на которых мы не останавливаемся, заставили Нернста
дать общую формулировку третьего начала термодинамики:
никаким конечным процессом нельзя охладить тело до
температуры абсолютного нуля.
Теорема Нернста и принцип недостижимости абсолютного
нуля заставляют нас вновь обратиться к шкале абсолютной
температуры в связи с открытием в 1951 г. Парселлом и Паун-
дом состояний вещества с отрицательными
абсолютными температурами. Хотя этот эффект относится к так
называемым необычным системам, а именно к ядерным
спинам, тем не менее необходимо связать его с основными
положениями термодинамики.
Опыты показали, что атомные ядра обладают собственным
вращательным магнитным моментом, называемым спином.
В твердом теле в обычных условиях эти спины расположены
хаотически. В сильных внешних магнитных полях происходит
частичная ориентация спунов вдоль поля, после исчезновения
которого спины спустя некоторое время релаксации вновь
располагаются беспорядочно. Различают спин-спиновую
релаксацию за счет взаимодействия спинов друг с другом и так
называемую спин-решеточную релаксацию,
происходящую благодаря взаимодействию спинов с решеткой
кристалла. При этом первое время релаксации значительно
меньше, чем второе.
Если кристалл поместить в сильное магнитное поле, то
спины частично ориентируются вдоль поля, но с повышением
температуры эта ориентация слабеет и, наконец, при достаточно
высокой температуре (в пределе Т—^оо) ориентация исчезает,
несмотря на сильное поле. В этих условиях система будет иметь
большой, но конечный запас энергии. Допуская возможность
состояния, когда все спины повернуты против поля, мы
переходим к области отрицательных абсолютных температур.
' § 3. Сверхнизкие температуры и принцип недостижимости абс. нуля 255
Опыты Парселла и Паунда производились с очень чистыми
кристаллами фтористого лития LiF. Ход опытов был
следующим. Сначала кристалл вносился в сильное магнитное поле
(6376 эрстед), в котором происходила заметная ориентация
ядерных спинов вдоль поля. Затем кристалл быстро
переносился внутрь маленького соленоида, где напряженность поля
была 100 эрстед, а направление его сначала было такое же, как
в сильном магнитном поле. Вслед за этим поле в соленоиде
в течение короткого времени 0,2 мксек обращалось с +100 на
—100 эрстед. После этого кристалл быстро переносился
обратно в прежнее сильное магнитное поле и там измерялась
поляризация атомных ядер методом ядерного парамагнитного
резонанса. Полученные данные показали, что спины в течение
некоторого времени были направлены против поля и это
состояние соответствует состоянию с отрицательной абсолютной
температурой. Чтобы лучше понять смысл этих операций,
отметим, что для LiF время спин-решеточной релаксации
составляет около 5 мин, тогда как время спин-спиновой релаксации
равно всего 10~5 сек, т. е. много меньше первого. Пока
кристалл находился в сильном магнитном поле, спины были
ориентированы в основном вдоль поля. Перенос из сильного поля
в соленоид и обратно совершался 2—3 сек, и в этом процессе
кристалл кратковременно находился в очень слабом
магнитном поле Земли. В соленоиде сначала спины были
ориентированы вдоль поля; при обращении поля, которое длилось 1 мксек,
т. е. много больше спин-спиновой релаксации, спины успевали
изменить ориентацию на противоположную и в таком состоянии
поступали опять в сильное поле. Так как время переноса
кристалла составляло 2—3 сек и было много меньше времени
спин-решеточной релаксации, то перевернутая ориентация
держалась несколько минут, достаточных для измерения
поляризации, и это состояние соответствовало отрицательной
температуре. Спустя примерно 5 мин благодаря спин-решеточному
взаимодействию это состояние исчезало и уступало состоянию
при Г>0.
Для того чтобы убедиться, что состояние с перевернутыми
спинами отвечает отрицательной температуре, рассмотрим
новое построение шкалы абсолютной температуры.
Было показано (Рамсей, 1956 г.), что основные положения
термодинамики можно согласовать с описанными опытами и
исключить противоречия, если принять в виде условия, что
при Г<0 более нагретое тело обладает меньшим
численным значением отрицательной температуры, чем более
холодное, причем все отрицательные температуры выше
положительных. Учитывая принцип недостижимости абсолютного нуля
и принимая, что этот принцип верен как при +0° К, так и при
256 Глава 8. Тепловая теорема Вернет а
—0° К, мы находим, что эти температуры являются крайними
точками, между которыми располагаются все остальные
температуры. Поэтому шкала температуры может быть
изображена следующей схемой:
+ 0°К, +1, +2, .... + со, -со, .... -2, -1, -0°К.
Стрелкой показано направление роста температуры. Это
построение шкалы связано с зависимостью внутренней энергии от
энтропии. Ранее было показано (гл. 5), что
(dU\ =
\dS Л,...
Т.
Это соотношение можно взять за основу определения
температуры. Она показывает, что для обычных систем, для которых
Г>0, с ростом энтропии монотонно возрастает энергия. Для
удобства введем обратную производную, положив
Рассмотрим ход зависимости S от U (рис. 45), где внизу
показана шкала температуры. Для обычных систем при Г>0
согласно теореме Нернста при +0° К энтропия 5=0, а энергия
U=UMmi. Кривая 1 указывает на монотонный рост энтропии 5
с увеличением U при повышении положительной температуры,
причем этот рост не ограничен сверху, т. е. энергия при Т—*-оо
может быть неограниченной. Однако наклон кривой 1 с ростом
температуры убывает, т. е. производная уменьшается.
Поде/
этому возможно состояние, в котором при конечной величине
энергии U производная станет равной нулю, и тогда S
ди
достигает предельной максимальной величины 5Макс при Г =
= + оо. Если произвести дальнейшее нагревание, то мы
переходим в область отрицательных температур (см. рис. 45) и
тогда из формулы (8,14) видно, что производная быстро
становится отрицательной при Т =—оо и затем непрерывно убывает.
Ход зависимости S от U показан кривой 2, левая ветвь которой
соответствует положительным температурам, тогда как правая
ветвь, отвечающая убыли S, относится к отрицательным
температурам для необычных систем (ядерных спинов).
Наконец, в предельном случае Г=—0° К видим, что производная
■ обращается в —со при конечной максимальной энергии
dU
и минимальной энтропии 5 = 0.
§ 3. Сверхнизкие температуры и принцип недостижимости абс. нуля 257
Эти рассуждения показывают, что существует предельное
значение внутренней энергии при Г= + оо и потому от
положительных температур к отрицательным нельзя перейти простым
нагреванием системы с положительным значением температур.
Состояние при Т=—0° К недостижимо, как и состояние с Т =
= +0°К. По правой ветви кривой с ростом отрицательных
температур спуск отвечает уменьшению энтропии, т. е. система
становится все более упорядоченной. Заметим, что от состояния
с положительной температурой к состоянию с отрицательной
температурой нельзя перейти никаким квазистатическим
процессом: этот переход возможен только не статическим путем.
Поэтому область отрицательных температур может быть
достигнута лишь искусственным путем, и по этой причине такие
необычные системы встречаются очень редко. Указанные
особенности таких систем позволяют построить термодинамику
этих систем. Мьг лишь кратко перечислим термодинамические
свойства систем с отрицательной абсолютной температурой.
1. Первое начало полностью применимо к необычным
системам. Понятия работа, теплота, теплоемкость сохраняются для
этих систем.
2. Формулировку Клаузиуса для второго начала можно
сохранить, если условиться, что при 7<0 из двух тел более
нагретое обладает меньшим численным значением температуры.
258 Глава 8 Тепловая теорема Нернста
3. Второе начало в форме принципа существования и роста
энтропии изолированной системы остается без изменения.
4. При сообщении количества теплоты dQ>0 телу с
отрицательной температурой энтропия его не увеличивается, а
уменьшается, так как система переходит в более упорядоченное
состояние.
5. При контактировании тел с абсолютными температурами
разных знаков теплота самопроизвольно переходит от тела
с Г<0 к телу с Г>0, т. е. от более нагретого к менее
нагретому. Если оба тела имеют разные отрицательные
температуры, то теплота переходит от тела с меньшей по абсолютной
величине температурой к телу с температурой,большей по
абсолютной величине.
6. Для необычных систем возможны циклические процессы.
Цикл Карно между телами при 7\<0 и Г2<0 в обратимом
процессе возможен, когда |Г1|<|Гг| и его к. п. д. выражается,
как обычно,
но при этом г)<0. Это значит, что при работе машины Карно
приходится затрачивать работу, а не получать ее. Обратимый
цикл Карно между телами с температурами разных знаков
осуществить нельзя, так как квазистатически нельзя перейти из
области положительных температур к отрицательным
температурам.
7. Системы с Г<0 могут быть термодинамически
устойчивыми.
8. Третье начало сохраняется, если ввести положение о
различии температур +0° К и —0° К.
9. Удельная теплоемкость при Т==—0° К равна нулю, так же
как для Г=+0°К.
10. Адиабатическое размагничивание системы при Т<0
приводит к ее нагреванию, а не к охлаждению.
Поскольку необычные системы получаются в сложных
искусственных условиях и встречаются очень редко, мы можем
строить термодинамику, не придавая большого значения этим
системам.
Глава 9
ОСНОВНЫЕ ВОПРОСЫ ТЕРМОДИНАМИКИ
НЕОБРАТИМЫХ ПРОЦЕССОВ
§ 1. ОБЩИЕ ПОЛОЖЕНИЯ. ЛОКАЛЬНЫЕ ПАРАМЕТРЫ СОСТОЯНИЯ.
ПОНЯТИЕ ПОТОКА
В термодинамике необратимых процессов рассматриваются
неравновесные явления, протекающие с конечной скоростью.
Впервые в 1822 г. в своей «Аналитической теории теплоты»
Фурье рассмотрел теорию процесса теплопроводности. Вслед
за этим Ом при выводе известного закона электрического тока
использовал представления Фурье о потоке. Теория
теплопроводности детально развивалась в работах Пуассона (1834 г.).
Аналогичными методами пользовался Навье (1822 г.) при
описании вязкого течения жидкости. Позднее в 1855 г. Фик создал
теорию процесса диффузии, применяя методы Фурье и
Пуассона к частным задачам диффузии.
Современная неравновесная термодинамика ведет свое
начало с работ Онзагера (1931 г.), который вывел основные
уравнения для линейных процессов и получил первые соотношения
между так называемыми кинетическими коэффициентами.
Затем она непрерывно развивается в трудах главным образом
голландских и бельгийских ученых де-Гроота, Денбига, Мазура,
Пригожина и др. Первые успехи нового направления были
получены при описании термоэлектрических явлений, но в
настоящее время вся эта область физики представляет собой
стройную систему с многочисленными приложениями, включая
область биофизики.
Известно, что в термостатике, которую мы до сих пор
рассматривали, дается описание свойств систем в
термодинамическом равновесии и изучаются только обратимые процессы, а о
необратимых процессах выводятся самые общие заключения,
например о росте энтропии, о переходе свободной энергии
к минимуму и т. п. Интенсивное развитие метода
термодинамических функций привело к тому, что термостатика почти отошла
от описания хода процессов и свелась к описанию свойств
систем в термодинамическом равновесии. Напротив,
неравновесная термодинамика подробно анализирует течение различных
процессов в макроскопических системах во времени, являясь,
однако, феноменологической областью физики.
260 Глава 9. Основные вопросы термодинамики необратимых процессов
При изучении процессов в макроскопических системах в
неравновесной термодинамике вводится новое определение
параметров состояния. В термостатике параметры состояния всегда
относились к системе в целом. Когда в системе имеются
необратимые процессы, то термодинамические параметры приходится
относить к отдельным участкам системы, т. е. они должны
рассматриваться как локальные (местные) макроскопические
характеристики. Это значит, что в большой системе, состоящей,
как правило, из огромного числа частиц, выделяется очень
малая, но все же макроскопическая часть ее и к ней относятся
такие термодинамические параметры, как давление,
температура, концентрация и т. п. Полезно представить себе масштабы
этой «малой части» системы. Если, например, имеется объем
газа 1000 см3, в котором обычно находится примерно 1020
молекул, то для определения локальных параметров в принципе
достаточно выделить из системы объем в 0,001 см3, весьма
малый, но все же в нем содержится 1014 молекул. Это число столь
велико, что к этому объему можно применять
термодинамические понятия и состояние определять с помощью обычных
параметров. Последние относятся именно к этому выделенному
объему, составляющему ничтожную часть всего объема
системы. Применение к таким конечным участкам
(«макроточкам») различных параметров, как давление, температура,
энтропия и т. д., является законным, но их следует считать
локальными параметрами, поскольку они являются вообще
функциями пространственных координат (и времени) в отличие от
прежних параметров термостатики. Соответственно некоторые
бесконечно малые величины, такие, как объем dV и др.,
являются теперь «физически бесконечно малыми», т. е.
конечными величинами, но относительно весьма малыми.
Ранее было указано, что по отношению к окружающим телам
термодинамические системы могут быть разделены на
следующие типы систем: 1) полностью изолированная система,
которая не обменивается с внешней средой ни теплотой, ни
веществом, ни работой; 2) адиабатически изолированная система,
которая может обмениваться со средой в отношении работы и
вещества, но в которой не имеется обмена теплом; 3) открытые
системы, которые отвечают условиям обмена со средой
веществом, теплотой, работой и т. д. Этот последний тип систем —
открытые системы наиболее подробно рассматриваются в
неравновесной термодинамике, так как в них легко протекают
всевозможные необратимые процессы. Фактически все
реальные макросистемы являются в той или иной степени открытыми.
Изучая процессы обмена открытой системы с внешней
средой, обратим внимание на то, что этот обмен осуществляется
в форме потока. Так говорят о потоке вещества, потоке энер-
§ 1. Общие положения. Локальные параметры состояния 261
гии, энтропии и т. д. Поэтому понятие потока, известное из
общей теории поля, является важнейшим понятием
неравновесной термодинамики. Напомним, что поток вектора Л через
площадку 5 является скалярным произведением N=AS1 причем
вектор площади определяется положением нормали к ней.
В термодинамике поток вызывается градиентом,
характеризующим свойства данного поля. Рассмотрим простой пример
стационарного электрического потока на отрезке провода длиной /.
Если на концах последнего имеется разность потенциалов
ф2—ф1 (причем ф2<ф1), то по закону Ома сила тока равна:
где R — электрическое сопротивление участка. Так как
и направление тока мы считаем положительным, когда он
течет от высшего потенциала к низшему, то перед дробью берем
знак минус. Известно, что R = p—, где р — удельное
сопротивление и 5 — сечение проводника. Поэтому формулу Ома можно
представить в виде
JL s = _ogradcp.s,
/
где а — удельная электропроводность, тогда как <P2"~"<Pl есть
градиент потенциала gradcp, который вообще является
вектором, направленным в сторону падения потенциала, а вектор
сечения проводника $ направлен вдоль его длины. Так как из-
Ар
вестно, что/= —, то написанное выражение закона Ома
предел
ставляет собой поток электрических зарядов под действием
градиента потенциала. Далее., будут изучены наиболее
распространенные способы обмена веществом и энергией в виде
процессов диффузии, теплопроводности и их сочетания, а также
другие аналогичные процессы и понятие потока часто будет
применяться в выводах.
Многочисленные опыты подтверждают простой линейный
закон теплопроводности при небольших перепадах
температуры, открытый Фурье (1807 г.):
AQ = -x T*~Tt as.А/, (9,1)
Здесь AQ — количество теплоты, переносимой при разности
температур Г2—Tt через площадку AS за время Д^ на пути х\
к — коэффициент теплопроводности. Из опыта известно, что
теплота сама собой переносится от более нагретого слоя
262 Глава 9. Основные вопросы термодинамики необратимых процессов
с температурой 7\ к более холодному с температурой Гг (т.е. Г2<
<Ti); когда AQ>0, то в выражении закона Фурье мы ставим
минус. В этом простейшем процессе теплопроводности вдоль
прямой по оси х (одномерный случай) отношение— ^-яв-
X
ляется абсолютной величиной вектора градиента температуры,
т. е. означает падение температуры на единицу длины пути.
В общем случае потока теплоты через произвольную площадь
AS с вектором площади с нормалью п к ней при градиенте
температуры grad T выражение (9,1) принимает вид:
Если ввести вектор плотности потока теплоты
jY = -xgrad7\ (9,2)
то поток теплоты за единицу времени равен:
17 = Л-А* 0.3)
Когда происходит обмен вещества в процессе диффузии, то
в простейшем случае одномерной задачи по опытным данным
выполняется известный линейный закон Фика в виде:
Дт==_О0^р-Д5.Д/, (9,4)
где Am— масса диффундирующего вещества, переносимого в
течение промежутка времени At через площадь AS при разности
концентраций с2—й в слоях на расстоянии х друг от друга по
пути переноса, Z)o — коэффициент диффузии, который можно
считать постоянным. Опыт показывает в согласии со вторым
началом термодинамики, что диффундирующее вещество
переносится в направлении падения концентрации, т. е. всегда
Сг—й<0; поэтому положительное значение массы Am
получим, если введем перед выражением в правой части знак
минус. Обобщая закон диффузии Фика, вместо градиента как
падения концентрации на единицу пути ——— введем вектор-
X
градиент концентрации grade, и тогда имеем:
Am = — Do grad с-AS-А/,
где AS—вектор площади с нормалью п. Введем еще вектор
плотности диффузионного потока:
yD = -D0 grade. (9,5)
Тогда поток вещества за единицу времени равен:
^=Jo:^. (9,6)
§ 1. Общие положения. Локальные параметры состояния 263
По Гиббсу процесс диффузии можно рассматривать как
перенос вещества от большего химического потенциала jii к
меньшему \х2\ этот перенос равноценен переносу под действием
разности концентраций, так как химический потенциал зависит от
концентрации и можно положить:
grad[x=(—£-) grade.
\ дс Jp, т
Поэтому вместо (9,5) можно принять
(9,7)
где коэффициент диффузии D связан с Do простым
соотношением
дс
Представление потока диффундирующего вещества при
помощи градиента химических потенциалов в виде (9,7) является
ценным, потому что, как известно из главы 6, химические
потенциалы характеризуют собой изменение внутренней энергии
на одну частицу или на моль переносимого вещества.
Следовательно, химический потенциал связывает перенос массы с
изменением внутренней энергии в процессе диффузии.
Обмен веществом может происходить не только за счет
процесса диффузии, но также при регулярном течении жидкости
или газа, когда налицо поток вещества. В случае идеальной
жидкости это есть простейший гидродинамический поток
вектора скорости; для реальных жидкостей и газов необходимо
учитывать вязкость (внутреннее трение), и тогда перенос
обусловлен градиентом количества движения и в общем случае
сводится к анализу изменения тензора вязкого напряжения в
потоке; этих случаев для простоты изложения мы не
рассматриваем. Заметим еще, что в неравновесной термодинамике
вводится действие электрических и магнитных полей, а также
могут учитываться химические реакции, происходящие в системе.
Кроме того, иногда принимается во внимание механическое
движение частей системы с конечной скоростью. Здесь мы, как
раньше, рассматриваем относительно медленные движения
системы; также не будем описывать химические реакции, чтобы не
усложнять изложение.
§ 2. УРАВНЕНИЯ БАЛАНСА ЭНЕРГИИ И МАССЫ В ОТКРЫТЫХ СИСТЕМАХ
Рассматривая процессы в открытых системах, следует
заметить, что основные начала термодинамики полностью
соблюдаются для этих систем, но они должны быть сформулированы
по-новому и, в частности, необходимо иметь в виду локальные
264 Глава 9. Основные вопросы термодинамики необратимых процессов
выражения обоих начал. Обратимся прежде всего к закону
сохранения энергии. Для примера рассмотрим поток теплоты
в стержне, боковая поверхность которого изолирована от
передачи тепла извне и поток теплоты идет вдоль длины стержня;
эта задача является наиболее простой, так как тепло
распространяется вдоль только одной оси jc, т. е. мы имеем одномерную
задачу. Пусть сечение стержня есть S, и рассмотрим в нем слой
толщиной dx (рис. 46). Выведем уравнение баланса теплоты.
Снизу в слой за время dt поступает поток теплоты по закону
Фурье (см. выше):
dQx = —* — SdU
дх
так как в этом малом слое имеется градиент температуры,
который меняется вдоль слоя, то из последнего выходит другое
количество теплоты, следуя тому же закону, но с другим
градиентом, т. е.
дх дх дх
Здесь всюду даны частные производные температуры, так как Т
является функцией координаты х и времени. Разность обоих
количеств dQi и dQz дает нам изменение количества теплоты
в выделенном слое за счет его нагревания. Объем слоя есть Sdx;
если Су — удельная теплоемкость материала, а р — его плот-
йость, то масса слоя есть p-Sdx и изменение температуры равно
dT. Поэтому количество теплоты, выделившееся в слое, есть
dQ = Cvp-SdxdT.
По закону сохранения энергии и при отсутствии химических
реакций и других процессов можно составить уравнение
локального баланса энергии:
dQ = dQ± - dQ2.
Подставляя сюда написанные выше значения
для dQ, dQi HdQ2, получаем:
-н
dQ,
Рис. 46.
Cvp-SdxdT = x
дх2
dx
Разделив это выражение на Sdxdt, находим:
дТ д2Т
= x ■
дх*
или
дТ
dt
дх2
(9,8)
§ 2. Уравнения баланса энергии и массы в открытых системах .265
Где yj = — коэффициент температуропровод-
н ости.
Очевидно, что в (9,8) имеем:
дх2 дх дх дх
Обобщая поэтому процесс теплопроводности на трехмерное
пространство, мы вместо уравнения (9,8) можем написать:
-C = 7idivgrad7\ (9,9)
dt
где введено обозначение дивергенции (расходимости) вектора
градиента температуры. Уравнение (9,9) показывает, что
скорость изменения температуры пропорциональна расходимости
градиента температуры.
Мы не рассматриваем многочисленных приложений
уравнений (9,8) и (9,9), а также их конкретных решений, которые
описываются в курсах уравнений математической физики и
теплотехники. Здесь для нас важно, что уравнение (9,9) представляет
следствие уравнения локального баланса энергии.
Так как для тела с постоянной удельной теплоемкостью
u = CvT представляет собой удельную внутренюю энергию, а
согласно уравнению (9,2) мы может ввести плотность потока
теплоты /г, то уравнение (9,9) принимает вид:
P*L = -divjT (9,Ю)
или для всей системы, когда U = pu, то
-^- = _divyr (9,10')
Выражение (9, 10') показывает, что скорость изменения во
времени внутренней энергии системы равна расходимости потока
теплоты (со знаком минус); для открытой системы всегда
предполагается источник теплоты и расходимость (мощность) этого
источника дает скорость изменения внутренней энергии в
каждой макроточке согласно сохранению энергии. В том случае,
когда градиент температуры отсутствует, то Уг = 0, или
дивергенция JT равна нулю, и тогда по закону сохранения энергии из
(9, 10') получаем £/ = const.
Рассмотрим пример переноса вещества. В опыте Джоуля —
Томсона с пропусканием газа через пористую пробку,
находящуюся в цилиндрической трубке (см. гл. 2), при небольших
разностях давлений по обеим сторонам пробки допустимо
считать процесс изотермическим. Пусть в общем потоке газа
имеется небольшая примесь газа другой природы. Эта система,
266 Глава 9. Основные вопросы термодинамики необратимых процессов
состоящая из пористого слоя (например, ваты или
песка),вместе с потоком газов является открытой системой, поскольку
здесь происходит обмен вещества. При этом надо иметь в виду
два потока: регулярный, или конвективный поток,
вызываемый разностью давлений по сторонам слоя, и
диффузионный поток, вызываемый разностью концентрации
примеси, возникающий за счет диффузии в слое. Допустим, что
разность давлений поддерживается постоянной, и рассмотрим
баланс вещества в системе.
Пусть пористый слой находится в трубке с сечением S,
средняя скорость потока равна v и газы в среднем движутся вдоль
оси трубки по направлению х. Основная масса газа движется
без диффузии, тогда как примесь с концентрацией ct движется
и с регулярным потоком, и еще диффундирует вдоль х
(например, за счет различия молекулярных свойств газов).
Рассмотрим баланс вещества в элементарном слое длиной dx. Сюда
спереди по потоку поступает некоторое количество газа вместе
с регулярным потоком и за счет диффузии из предыдущих
слоев. Далее часть газа выходит из элементарного слоя
благодаря регулярному потоку и тоже вследствие диффузии в
последующие слои. Однако некоторая масса газа удерживается
в слое за счет конечной скорости процесса и образования
градиента концентрации вдоль оси х. Здесь концентрация ct
является функцией координаты х и времени /. Составим баланс
массы газа в слое dx, используя закон Фика (9,4):
1) в слой с регулярным потоком поступает за время dt
количество газа:
2) входит в слой за время dt за счет диффузии
количество газа:
dm2D
дх
3) выносится из слоя с регулярным потоком за время dt
масса газа:
дх
4) выходит из слоя благодаря диффузии за время dt масса
газа:
\ дх дх дх J
Алгебраическая сумма всех этих величин дает изменение
количества газа за время dt в элементарном слое с учетом
изменения концентрации со временем, т. е.
dmb= -^-dt-Sdx.
dt
§ 2. Уравнения баланса энергии и массы в открытых системах 267
Следовательно, уравнение локального баланса массы имеет вид:
dmx + dm2 — dm3 — dm4 = dmb.
Зная выражения всех количеств dm из предыдущего, находим
после сокращения на dt-Sdx:
^£l = ^vJ^ + DJ!£l% (9,Ц)
dt дх дх*
Этот результат можно легко обобщить на случай трехмерной
диффузии с конвективным членом, т. е. с регулярным потоком.
Тогда (9,11) переходит в соотношение:
tfgradcx + Ddvgai (9,12)
dt
Из векторного анализа известно, что если у — скаляр и х—
вектор, то
div(y-Ar) = y-Axv х + л: grad z/. (9,13)
Так как y = Ci и х = V, поэтому (9,13) принимает вид:
div (ci • v) = сх div v + v grad cv
Но в самом начале мы полагали, что v = const; поэтому dwv =з
= 0. Тогда div (ср) = v grad d. Подставляя это выражение
в формулу (9,12), находим:
—— = — div (с^) + D div grad cx.
dt
Ранее мы ввели вектор плотности диффузионного потока
в виде (9,5). Подставляя его значение в последнюю формулу,
получаем;
$L--6\v(c<o + JD). (9,14)
Это выражение представляет собой локальный баланс
примеси газа и показывает, что изменение концентрации газа за
единицу времени определяется дивергенцией регулярного и
диффузионного потоков, т. е. в системе имеются источники,
мощность которых дает скорость изменения концентрации. Если
регулярный поток отсутствует, т. е.V =0, то из (9,14) имеем:
divy^ = D div grad cr (9,14r)
Последнее выражение аналогично уравнению (9,10) для
теплопроводности. При равновесии, когда все потоки, как
регулярный, так и диффузионный, уже отсутствуют, т. е. V =0 и
grad Ci = 0, имеем:
—— = 0, или ct = const,
dt
268 Глава 9. Основные вопросы термодинамики необратимых процессов
причем в системе тогда всюду одна и та же концентрация,
неизменная во времени.
Можно вывести уравнение для однородных жидкостей и
газов, дающее баланс общей массы в виде соотношения,
аналогичного равенству (9,14).
Здесь были получены дифференциальные выражения для
баланса энергии и массы. Они могут быть легко получены из
интегральных соотношений.
Закон сохранения энергии требует, чтобы полное количество
внутренней энергии U в каком-либо произвольном объеме V
оставалось постоянным или изменялось только тогда, когда
энергия или втекала в этот объем или вытекала из него через
поверхность его й. Если введем удельную внутреннюю энергию
и на единицу массы, а через Ju обозначим поток энергии за
единицу времени через единицу поверхности, то при плотности
системы р мы по закону сохранения энергии должны получить:
Tt \pudv =\^TdV = ~\ J"dQ' (9> 15)
V V s
где вектор dQ, равный по абсолютному значению dQ, направлен
по нормали к поверхности с положительным направлением из
объема наружу. Пользуясь теоремой Гаусса о связи между
интегралом по поверхности и интегралом по объему, формулу
(9,15) можно представить в виде:
£L dV = - С judQ = - j" div judV. (9,15')
8 V
Это равенство должно соблюдаться при любом объеме V,
поэтому из (9,150 следует:
Это соотношение представляет собой локальное выражение
закона сохранения энергии; частным случаем равенства (9,15")
является соотношение (9,10') для потока теплоты.
§ 3. ТЕОРЕМА О ПРОИЗВОДСТВЕ ЭНТРОПИИ В ТЕРМОДИНАМИЧЕСКИХ
СИСТЕМАХ. ТЕРМОДИНАМИЧЕСКИЕ СИЛЫ
Второе начало термодинамики для систем, в которых
протекают необратимые процессы, выражается в виде теоремы о
производстве или о возникновении энтропии. Прежде всего заметим,
что в открытых системах изменение энтропии dS
вызывается двумя причинами: во-первых, некоторое количество
энтропии deS поступает в систему из окружающей среды и,
во-вторых, некоторое количество энтропии diS возникает (или произ-
§ 3. Теорема о производстве энтропии в термодинамических системах 269
водится) в самой системе. Поэтому для открытой системы
имеем:
dS =
d,S+соотносительно второй величины d{S второе начало
утверждает, что если не считаться с окружением, а мысленно
рассматривать систему изолированной, то
dcS > О,
т. е. внутренняя часть энтропии системы или остается
постоянной (diS = 0), или испытывает только положительное
приращение di5>0, если в системе происходят необратимые процессу.
Что% касается величины deS, то она может быть как
положительной, так и отрицательной, т. е. энтропия может извне
втекать в систему или уходить в окружающую среду и возможно,
что deS равняется нулю. Для полностью изолированной системы,
которая не обменивается со средой ни веществом, ни энергией,
имеем deS = 0, и тогда получаем обычную формулировку второго
начала в виде dS^O.
Если система может обмениваться со средой только
теплотой, то при переходе в систему теплоты dQ при температуре
перехода Г изменение поступающей энтропии deS согласно
второму началу равно:
dQ
—.
В этом случае энтропия системы возрастает.
Рассматривая открытые системы, мы должны найти
изменения deS и d{S с учетом необратимых процессов в системе. При
этом необходимо обратить внимание на поток энтропии и
считать, что последняя зависит от времени. Так как мы имеем
в виду локальные параметры, то целесообразно ввести
удельную энтропию 5 на единицу массы, помня, что энтропия
является величиной экстенсивной. Поэтому если р — плотность
вещества, то S = ps. Благодаря возможной неоднородности
системы мы должны представить S для всего объема системы V
в виде:
S= \9sdV, (9,16)
считая s и р распределенными в объеме системы. Для
открытых систем имеет место поток энтропии через поверхность Q,
ограничивающую систему. Этот поток энтропии, имеющий смысл
поступления наружной энтропии за единицу времени,
выражается интегралом, взятым по всей поверхности:
^ 1
270 Глава 9. Основные вопросы термодинамики необратимых процессов
Здесь js есть полный удельный поток энтропии для единицы
поверхности за единицу времени.
Обратив внимание на собственное изменение энтропии diS
внутри самой системы, мы должны принять, что в ней имеются
источники энтропии. В системе производится «своя» энтропия
с интенсивностью 6, которая выражает производство энтропии
на единицу объема в единицу времени. Тогда, очевидно,
скорость изменения внутренней энтропии по всему объему и для
всех источников энтропии должна быть представлена
интегралом:
J (9,18)
V
Скорость общего изменения энтропии в системе согласно
равенству (9,16) имеет вид:
V
От интеграла по поверхности в (9,17) легко перейти к
объемному интегралу, вновь применяя теорему Гаусса, и тогда:
(9,20)
v
Поэтому уравнение (9,17) принимает вид:
(9,21)
Находим баланс всех видов энтропии: наружной,
внутренней и общей, составляя алгебраическую сумму величин из
(9,18), (9,19) и (9,21), и тогда получаем уравнение баланса:
K = 0. (9,22)
Так как все эти выводы верны для произвольного объема, то
подынтегральное выражение в (9,22) равно нулю и тогда
получаем дифференциальное уравнение баланса энтропии:
^ + 6. (9,23)
В этом уравнении согласно второму началу термодинамики
для внутренней энтропии всегда должно быть:
б>0. (9,24)
Уравнение баланса энтропии (9,23) представляет собой
локальную формулировку второго начала для открытых систем,
§ 3. Теорема о производстве энтропии в термодинамических системах 271
в которой скорость изменения энтропии представлена в виде
двух слагаемых, причем первое обусловлено внешним потоком
энтропии в систему, тогда как второе слагаемое характеризует
скорость производства энтропии в самой системе благодаря
необратимым процессам, протекающим в ней.
Выведенная теорема об энтропии носит весьма общий
характер. Рассмотрим два частных случая ее применения к
процессам теплопроводности и диффузии.
Вернемся к задаче о теплопроводности в стержне и найдем
изменение энтропии в этой системе. Введем в уравнение (9,10)
дифференциал удельной внутренней энергии в виде du = Tds,
который ранее часто встречался. Тогда равенство (9,10) примет
вид:
или (9,25)
р— = div /r.
Применим к правой части свойство (9,13) из векторного
анализа, полагая здесь у= — и x=jT. Благодаря этой
подстановке уравнение (9,25) переходит в выражение:
Легко видеть, что grad (—) = — grad Г, поэтому
1 dt
Сравнивая полученный результат с формулой (9,23), видим:
— = — div -ll. — -^- grad 7\ (9,26)
dt т т* 5 v '
Следовательно, в равенстве (9,26) можно положить:
и
Js = ~- (9.27)
(9,28)
Отсюда заключаем, что равенство (9,27) дает удельный поток
энтропии из окружения в данную систему, тогда как
соотношение (9,28) выражает скорость производства энтропии в самой
системе.
272 Глава 9. Основные вопросы термодинамики необратимых процессов
Напомним, что JT = —xgradT, и представим (9,27) в виде:
JL\ (9,29)
Из этого соотношения следует, что поток энтропии в систему
из окружения тем больше, чем выше градиент температуры и
пропорционален коэффициенту к теплопроводности материала.
После замены /г его выражением в формуле (9,28) она
принимает вид:
(^)\ (9,30)
Это соотношение всегда положительно, так какх>0, скобка
также больше нуля и в соответствии со вторым началом:
е>о,
т. е. в системе производится положительная энтропия, пока
имеется градиент температуры. Когда в системе температуры
выравнялись и grad7=0, то в соответствии со вторым началом
имеем из (9,30) :
6 = 0.
Соотношениям (9,27) и (9,28) можно придать другой вид,
вводя в них так называемую термодинамическую силу
по определению Онзагера. Для процесса теплопроводности в
качестве термодинамической силы удобно принять выражение
Tj (9,31)
Тогда уравнение (9,27) примет вид с учетом определения (9,2):
Js = y.-XT.
Вводя Хт из формулы (9,31) в соотношение (9,28), получаем:
b.T=jT-XT. (9,32)
Понятие термодинамической силы оказалось ценным при
описании сложных необратимых процессов.
Перейдем теперь к описанию процесса диффузии одного
вещества в отсутствие регулярного потока в системе постоянного
объема (например, в растворе) для случая переменной
температуры и выясним, какой результат дает общее выражение
баланса энтропии (9,23).
Для систем с переменной массой в главе 6 было получено
общее выражение для дифференциала внутренней энергии вида
(6,16), которое для одного компонента можно написать как:
# 5. Теорема о производстве энтропии в термодинамических системах 273
Если объем системы постоянен и химический потенциал |л
отнесен к 1 кг/м3, то \idN = [idc. Тогда dU принимает вид:
dU = T dS + |i dc,
где с — концентрация. Перейдем к удельным величинам,
разделив это равенство на общую плотность р и отнеся результат
к единице времени. Тогда
du rp ds . dc
v dt ^ dt r dt
ds I du fx dc /n oo\
или p— = —p • (9,33)
v dt T * dt T dt v '
В первый член правой части этого равенства подставим
выражение его через (9,10), тогда как во второе слагаемое
введем — из уравнения (9,147), после чего формула (9,33) прини-
dt
мает вид:
(9,34)
г dt Т "1 Т
Используем прежнее соотношение (9,13), применяя его
дважды: к обоим членам равенства (9,34). Тогда после
перегруппировки слагаемых получаем:
p d[] [
(9,34')
Полученное выражение по смыслу и по структуре аналогично
общему виду (9,23) теоремы об энтропии. Поэтому в (9,34')
можно принять:
Л = ^гЛ—^Л>. (9.35)
6=—^-yrgradr-yDgrad^-. (9,36)
Обратим внимание на соотношение (9,36), которое выражает
собой скорость производства энтропии в самой системе.'Его
можно представить в виде:
6-71 = --fyrgradT-yDTgrad-^. (9,37)
Введем термодинамическую силу для процесса
теплопроводности
Ю Заказ № 2479
274 Глава 9. Основные вопросы термодинамики необратимых процессов
и по аналогии термодинамическую силу для процесса
диффузии
XD = -T grad-J-,
и тогда соотношение (9,37) принимает вид:
D, (9,38)
т. е. сумма произведений потоков на соответствующие
термодинамические силы является произведением скорости
образования энтропии на температуру.
Вернемся к выражению (9,36) и представим его в виде:
6 = х f
-J + D (grad ,*)« - D ±- grad p grad Г, (9,39)
заменив в (9,36) значения /г и J D согласно (9,2) и (9,7) и
подставив:
grad •£• = ± grad ц — J-j- grad Т.
Равенство (9,39) дает скорость производства энтропии при
сосуществовании процессов теплопроводности и диффузии. При
наличии градиента температуры и градиента концентрации
величина 0 не равна нулю, т. е. оба процесса вызывают
производство энтропии в системе. Согласно второму началу 0 должно
быть обязательно больше нуля, хотя это непосредственно не
видно из уравнения и мы здеСь не можем останавливаться на
прямом доказательстве. Когда в системе градиент
концентрации (или химического потенциала) равен нулю, то второй
и третий члены (9,39) исчезают и получаем
{ т
как для производства энтропии при теплопроводности в стержне
(уравнение 9,30), причем 0>О.
Если допустимо пренебречь теплопроводностью и
рассматривать изотермическую диффузию за счет градиента
концентрации, то в равенстве (9,39) первый и третий члены выпадают
и мы получаем
6 = D (grad [л)2, (9,40)
т. е. и здесь в согласии с требованием второго начала и при
D>0 всегда имеем 0>О. Очевидно, в равенстве (9,39) первый
член представляет собой вклад в энтропию процесса
теплопроводности, второй соответствует вкладу диффузии, тогда как
третий член отвечает совместному действию теплопроводности
§ 3. Теорема о производстве энтропии в термодинамических системах 275
и диффузии; этот член, определяющий собой их взаимную роль,
зависит от обоих градиентов Т и [х. Наконец, в предельном
случае, когда все градиенты исчезли (температуры и
концентрации выравнялись), получаем из (9,39), как и следовало
ожидать, 6 = 0, т. е. в соответствии со вторым началом в системе
больше нет производства энтропии.
Мы ограничились только двумя примерами приложения
теоремы о росте энтропии. Другие частные случаи дают
аналогичный результат для производства энтропии и мы на них не
останавливаемся. Оба примера дали нам повод, ввести новое
понятие термодинамической силы, которое последовательно
применяется в теории Онзагера, о чем будет сказано далее. Кроме
того, было получено равенство (9,38), допускающее широкие
обобщения при описании разнообразных неравновесных
процессов.
§ 4. ОСНОВНЫЕ ВОПРОСЫ ТЕОРИИ ОНЗАГЕРА. ПРИНЦИПЫ ЛИНЕЙНОСТИ
И ВЗАИМНОСТИ В СЛОЖНЫХ ПРОЦЕССАХ
Выше были рассмотрены исходные понятия и положения
общей теории неравновесных процессов. Наиболее
последовательно эти вопросы развиваются в теории Онзагера, имеющей
важное значение при описании наиболее сложных процессов.
В основе этой теории лежат два принципа:
1) принцип линейности;
2) принцип взаимности кинетических коэффициентов.
1. Принцип линейности. Физическая сущность этого
принципа состоит в утверждении, что вблизи состояния
термодинамического равновесия скорость необратимого процесса
прямо пропорциональна термодинамической силе. В наиболее
простой форме этот принцип сводится к тому, что скорость
процесса пропорциональна градиенту величины, определяющей
собой данный процесс.
Мы уже указывали на ряде примеров, что градиент какой-
либо физической величины служит причиной, вызывающей
течение процесса, который является следствием. Этот принцип
основан на огромном числе экспериментов как результат
многочисленных наблюдений в самых разнообразных областях
физики и химии. Мы отмечали, что из опытов по
теплопроводности следует принять, что количество переносимой теплоты
пропорционально градиенту температуры; далее, из данных по
изучению диффузии следует, что количество переносимой массы
пропорционально градиенту концентрации. Наконец, закон Ома
указывает, что электрический ток в проводнике как количество
переносимого электричества за единицу времени
пропорционален градиенту электрического потенциала. В более точной фор-
10*
276 Глава 9. Основные вопросы термодинамики необратимых процессов
мулировке этого принципа следует принимать во внимание, как
показал Онзагер, не сами градиенты температуры,
концентрации и т. д., а соответствующие термодинамические силы, про-
лорциональные градиентам. Во введении понятия
термодинамических сил имеется некоторая условность, но все же для
большинства процессов они могут быть найдены достаточно строго.
На дЬух примерах (§ 3) мы уже встречались с разными
термодинамическими силами и нашли, что:
для теплопроводности: Хт = grad T;
для диффузии: XD = — Т grad —.
В случае электрического тока в однородном металле следует
принять Хэ=—gradep.
Скорость процесса определяется как плотность потока
энергии, вещества, количества электричества и т. д. за единицу
времени, протекающих через единицу поверхности. Это понятие
было рассмотрено в § 1 и 2. В соответствии с определением
скорости процесса принцип линейности можно представить в
общем виде соотношением:
j=L-X. (9,41)
Коэффициент пропорциональности L носит название
кинетического коэффициента. Для процессов
теплопроводности и диффузии такими коэффициентами являются
соответственно коэффициент теплопроводности Ьт = к и коэффициент
диффузии LD — D\ для процесса течения электричества, как мы
видели, следует принять Ld = o, т. е. кинетический коэффициент
равен удельной электропроводности металла. Эти примеры
показывают, что кинетический коэффициент по своей физической
природе определяет собой некоторую проводимость как
величину, обратную сопротивлению и облегчающую собой течение
процесса. При равных X скорость процесса тем больше, чем
выше кинетический коэффициент L. Это легко понять,
сравнивая, например, теплопроводность меди и серебра с
теплопроводностью свинца и пр.
2. ГГр инцип взаимности кинетических
коэффициентов. Теория Онзагера приобретает исключительно
важное значение главным образом при описании сложных
процессов, состоящих из наложения более простых явлений. Так,
опыты показали, что наложение диффузии и теплопроводности
приводит к сложному явлению, которое представляется в форме
двух процессов. Первый из них, называемый эффектом
Дюфура (1873 г.), состоит в том, что при смешении двух
химически нереагирующих друг с другом газов при одинаковой
§ 4. Основные вопросы теории Онзагера 277
температуре возникает температурный градиент. В опытах Дю-
фура было замечено, что смешивание водорода с азотом при
одинаковой начальной температуре приводит к разности
температур в смеси в несколько десятков градусов. Другой процесс,
называемый термодиффузией и обнаруженный в опытах
Соре (1893 г.), заключается в том, что в бинарных смесях
жидкостей при разности температур появляется градиент
концентрации компонентов. Если однородную двойную жидкую смесь
поместить в неоднородное температурное поле, то быстро
возникает градиент температуры, сначала без разделения составных
частей, но затем появляется термодиффузионный поток,
состоящий в образовании градиента концентрации компонентов.
Очевидно, оба указанных эффекта обратны друг другу. В них
обнаруживается влияние одного процесса на другой, т. е.,
например, диффузия влияет на теплопроводность и, обратно,
последняя оказывает влияние на первую. Наблюдается также
взаимодействие трех и более процессов.
Рассмотрим случай, когда в системе протекают
одновременно два процесса, например имеется градиент концентрации
растворенного вещества и градиент температуры. В этом
процессе имеют место поток тепла и поток вещества с разными
термодинамическими силамиХ± и Х2. Оба процесса будут
налагаться друг на друга и при этом взаимно влиять один на
другой, в результате чего возникает новый эффект. Общий анализ
такого процесса был дан при выводе формулировки второго
начала (§ 3). В теории Онзагера было найдено, что скорости
процессов в данном случае выражаются в виде системы двух
уравнений:
Л = ^iA + ^i2*2, (9,42)
j2 == ^21*1 + ^22*2*
Если первый поток j\ соответствует потоку теплоты, то из
первой формулы (9,42) видно, что он зависит, во-первых, от
процесса теплопроводности с термодинамической силойХг=Хт.
и коэффициентом теплопроводности Ьц = к, как ранее,
во-вторых, на этот процесс будет влиять еще перенос вещества при
диффузии, что соответствует термодинамической силе X2=Xd,
но при этом вторая составляющая потока j\ определяется своим
кинетическим коэффициентом L12, называемым
перекрестным коэффициентом.
Второй поток j2 соответствует процессу диффузии с
термодинамической силой X2 = Xd с кинетическим коэффициентом
L22 = Dt но на этот процесс налагается еще перенос теплоты, что
отражается во втором уравнении (9,42) членом c^i с
кинетическим коэффициентом L2i. Аналогичным примером наложения
278 Глава 9. Основные вопросы термодинамики необратимых процессов
двух процессов следует считать различные термоэлектрические
явления, в которых имеется взаимное влияние электрического
тока в металле на перенос теплоты и обратно. Мы их
рассмотрим в следующем параграфе.
Когда в системе протекают одновременно три разных
связанных друг с другом процесса, то по аналогии с предыдущим
можно написать систему из трех уравнений для трех
потоков /i, j2 и у3, вводя три соответственные термодинамические
силыЛГх, Х2 и Х3, и тогда:
Jl == ^1\Х\ + L12X2 + ^13^3»
А = L21*l + ^22*2 + L^Xs, (9,43)
Уз == LSiX} -f- L32X2 -f" ^зз^з*
Здесь диагональные кинетические коэффициенты LUt L22, £33
отвечают прямым процессам, тогда как остальные Lih при
разных i и k соответствуют процессам взаимного влияния или
наложения. Примерами подобных тройных процессов являются
следующие процессы:
1) одновременные потоки энергии (теплоты) j\ и диффузии
Л и Уз двух веществ, т. е. термодиффузия в системе из двух
компонентов;
2) процесс теплопроводности в анизотропном кристалле,
когда y#i, j2 и Уз являются потоками теплоты по трем осям;
3) электрический току*1 и диффузия у2, Уз двух различных
по знаку и по подвижностям ионов в растворе.
Очевидно, число различных взаимодействующих процессов
может быть достаточно велико. Кинетические коэффициенты L^
обладают важным свойством взаимности, которое можно
представить в виде:
!** = £«; (9,44)
следовательно, кинетические коэффициенты при перекрестных
процессах равны друг другу.
Уравнения Онзагера и свойство взаимности (9,44) были
выведены с помощью методов статистической физики при
анализе затухания флуктуации в системе вблизи
термодинамического равновесия. В термодинамике неравновесных процессов
теория Онзагера рассматривается как феноменологическая
теория.
При анализе теоремы о производстве энтропии мы вывели
соотношение (9,37) для произведения скорости производства
энтропии 6 на абсолютную температуру; вводя в него, кроме
потоков, соответствующие термодинамические силы, мы
получили выражение (9,38) вида
§ 4, Основные вопросы теории Онзагера 279
Это свойство в теории Онзагера обобщается на случай
взаимодействия нескольких процессов, и оно может быть
представлено как сумма:
Формула (9,45) является общим выражением для скорости
образования энтропии и широко применяется для решения
многочисленных задач неравновесной термодинамики как общий
феноменологический закон.
§ 5. ТЕОРИЯ ТЕРМОЭЛЕКТРИЧЕСКИХ ЯВЛЕНИЙ
Уже давно было установлено три вида термоэлектрических
явлений.
1. Появление термоэлектродвижущей силы (термо-э. д. с.)
в системе, состоящей из спаев разнородных металлов, когда
спаи находятся при разных температурах (эффект Зеебека).
Это явление характеризуется величиной так называемой
дифференциальной термо-э. д. с. &у различной для разных
металлов и представляющей собой относительную разность
потенциалов для системы двух спаев на единицу изменения
температуры:
"дг""*
2. Появление в системе с электрическим током, состоящей
из разнородных металлов, помимо теплоты Джоуля—Ленца,
еще дополнительной теплоты (эффект Пельтье). Процесс
характеризуется теплотой Пельтье qn пропорциональной силе тока:
где П — коэффициент Пельтье.
3. Выделение дополнительной теплоты, кроме теплоты
Джоуля—Ленца, в цепи однородного металла, находящегося при
разных температурных условиях его участков (эффект Томсона).
Это явление определяется теплотой Томсона дт,
пропорциональной силе тока и градиенту температуры:
где т — коэффициент Томсона.
Все три явления были детально изучены Томсоном (1854 г.),
который, вводя некоторые гипотезы для их термодинамического
обоснования, получил соотношения, хорошо оправдываемые
опытом. Современная теория необратимых процессов позволила
теоретически строго связать все три явления и проверить гипо-
280 Глава 9. Основные вопросы термодинамики необратимых процессов
тезы Томсона. Мы дадим описание этих процессов при помощи
теории Онзагера, следуя при изложении де-Грооту.
Рассмотрим систему (рис. 47), состоящую из двух отрезков
металла Л, припаянных к концам отрезка металла В. Один спай
находится в большом сосуде 1 при температуре Г, а другой
в сосуде 2 также большого размера при температуре Г+ДГ,
причем два конца проводника А присоединены к пластинам
конденсатора К. При переносе заряда de затрачивается работа
yde. Из объединенного закона термодинамики следует:
TdS = dU — <?de,
где ф — электрический потенциал. Пусть некоторое количество
энергии dU в виде тепла переносится из сосуда / в сосуд 2 и
в конденсатор поступает заряд de при разности потенциалов Дф,
тогда общее изменение энтропии данной системы равно:
При этом мы принимаем температуру конденсатора
равной Г, хотя можно принять ее равной и Г+ДГ, что практически
не меняет рассуждений. В резервуаре 1 происходит убыль
энтропии dSx= , так как теплота отнимается, а в
резервуаре 2 получается увеличение энтропии dS2=l
—-Разделив dU на (Г+ДГ) и пренебрегая членами с высшими
степенями ДГ, находим:
,Q dU , dU A11 AT A de .
отсюда
dt dt T* dt T ' v ' ;
В этом уравнении величины AT и Дф пропорциональны
градиентам температуры и потенциала и вследствие малости ДГ и
Дф можно их считать самими градиентами, так как они
относятся к одной и той же
системе. Выражение (9,46)
° дает скорость производства
энтропии и согласно
общему соотношению (9,45)
для произведения 6- Т
Т+дТ
имеем:
dU AT de
Рис. 47. dt ' T dt
§ 5. Теория термоэлектрических явлений 281
Здесь следует положить \j\\ = и \Хг\ = — как поток
энергии Ji при термодинамической силе Хь тогда как | J2 | =
de
= — и |Jfa| = —^?» считая ДГ и Дф соответственно градиен-
dt
тами Т и ф. Тогда для сложного процесса наложения потока
тепла и потока электричества можно написать систему двух
уравнений Онзагера:
ji = L11X1 + L12X2,
ИЛИ
(9,47)
Здесь по смыслу Ln равен коэффициенту теплопроводности х,
a L22 является удельной электропроводностью а. В
соответствии с принципом взаимности кинетических коэффициентов
в (9,47) должно быть L12 = L21.
Рассмотрим два частных случая применения уравнений
(9,47).
1. Электрический ток в цепи отсутствует; тогда —- =0 или
J2 = 0. Тогда из второго уравнения (9,47) видно:
Дер L2i
откуда -X.==__L..
AT L22T
Мы получили выражение для термо-э. д. с. через кинетические
коэффициенты:
2. Градиент температуры равен нулю: ДГ = 0. В этом случае
уравнения (9,47) дают:
h L22
Но величина j\ по смыслу есть теплота, выделяющаяся
dU
в цепи за единицу времени, как , при силе электрического
dt
тока/2> при этом Li2 и L22 постоянны и потому
Л L22
282 Глава 9. Основные вопросы термодинамики необратимых процессов
или/1=П«Уь т. е. мы получили закон Пельтье <7/7 = П-/, причем
коэффициент Пельтье выражается как отношение кинетических
коэффициентов. Находим коэффициент L{2 перекрестного
процесса; имеем L22=а, поэтому
£12 = а.П. (9,49)
Имея в виду, что должно быть L\2 = L2u получаем сравнением
(9,48) и (9,49):
р _ Аср оП _ П
~~ "дг""~~W~~~т''
или П = £.Т. (9,50)
Эта формула называется вторым соотношением Том-
сон а, которое было получено им из других соображений и
хорошо подтверждается опытом. Оно дает связь между эффектами
Пельтье и Зеебека.
Наконец, рассмотрим непрерывную цепь разнородных
металлов Л и В, в которой выделяются теплота Пельтье и теплота
Томсона. Пренебрегая теплотой Джоуля—Ленца, можно
согласно определениям величин qn и qT написать для разных
металлов:
= П. /; qfA) = %aI grad Т = тА1\Т; ?<f > = (П + ДП) /;
= тв/ДГ.
Тогда согласно закону сохранения энергии, учитывая разные
направления токов в А и В, имеем:
П/ — (П + ДП) / + тв/ЛГ — т^/ДГ =
Сокращая на /, получаем:
ДП , Дер JI
ИЛИ - — + Тв-Тл = —=- — ,
П . ДП v , ДП
—+ — =-(? + —
откуда тБ-тл=
Этот результат, называемый первым соотношением
Томсона, хорошо подтверждается опытом, и оно связывает
эффект Томсона с остальными термоэлектрическими эффектами.
В этой главе мы ограничились лишь простейшими
примерами неравновесных процессов. Мы не могли касаться
многочисленных приложений этой области, которая быстро
развивается в настоящее время, находя много практических
применений в технике и физике.
Предметный указатель
283
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абсолютная температура 111
Абсолютная шкала температуры ПО
Абсолютно черное тело 126
Абсолютный нуль, недостижимость
112, 249, 252
Абсолютный нуль температуры 112
Авогадро постоянная 42
Аддитивность 8, 181
Аддитивные величины 8
Адиабата 50
Адиабатическая оболочка 35
Адиабатически изолированная
система 35, 260
Адиабатическое размагничивание 250,
252
Адиабатный процесс 41, 49
Адсорбция 156, 157, 171
Баланс массы (вещества) 263, 267
Больцмана константа 107
Ван-дер-Ваальса уравнение состояния
15, 227, 233
Вечный двигатель второго рода 101
— — первого рода 33
Взаимность кинетических
коэффициентов 276, 278
Внешние параметры 13
Внутренние параметры 13
Внутренняя энергия 7, 16, 33, 131
Второго начала статистический смысл
106
Второе начало термодинамики 63, 75,
81, 90, 99, 108
Газ идеальный 44, 45
Газовые смеси 146, 207
Гальванический элемент 160
Гесса закон 57, 60
Гетерогенное равновесие 177
Гиббса—Гельмгольца уравнение 137,
142, 145
Гиббса парадокс 151
Гиббса термодинамический потенциал
133, 134, 143
Джоуля — Томсона коэффициент 123
— — опыт 44, 46, 265
— — эффект 44, 46
Дифференциальные уравнения
термодинамики 136
Диффузии процесс 66, 262, 263, 272
Диффузионный поток 266
Диффузия 66, 97, 263
Закон Гесса 57, 60
Закон действующих масс 211
— Джоуля 45
— диффузии Фика 262
— Кирхгофа 123, 124
— Клапейрона 15, 44
— Ома 261
— Пельтье 279, 282
— соответственных состояний 238
— Стефана — Больцмана 123, 127
— теплопроводности Фурье 261
Зародыши новой фазы 223
Идеальный газ 44, 45
Изобара 48
Изолированная система 34, 167, 185,
260
Изотерма 49
Изотермический коэффициент
сжимаемости 19
Изохора 47
Изоэнтропа 82
Изоэнтропические процессы 82
Интеграл Клаузиуса 80, 87
Интенсивные величины 9, 183
Калорическое уравнение состояния 16
Каратеодори принцип 87
Карно — Клаузиуса теорема 76
Квазистатические процессы 13, 63,
68
Кинетический коэффициент 276
Кирхгофа закон 123, 124
Клапейрона—Клаузиуса уравнение
114, 116, 206
Клапейрона уравнение 44
Клаузиуса интеграл 80, 87
Клаузиуса постулат 74, 99
Компоненты 178
Константа Больцмана 107
Константа химического равновесия
211
Коэффициент Джоуля—Томсона 123
— кинетический 276
— критический 237
— Пельтье 279, 282
— Пуассона 51
Коэффициенты перекрестные в
теории Онзагера 277, 282
Коэффициенты стехиометрические 210
Критическая температура 117, 226
— точка 226
Круговые процессы 14
Кюри точка 216
284
Предметный указатель
Ложное равновесие 192, 193
Магнитокалорический эффект 249,250
Максимальная работа 92
Машина Карно необратимая 89
— — обратимая 69
— — обратная 73
Метастабильное состояние 222, 223
Метастабильные равновесия 166, 204
Метод циклов 77, 129
Методы термодинамики 129
Механические процессы 67
Мнемонический квадрат 134
Многокомпонентные системы 197
Направление времени 104
Нернста тепловая теорема 239, 242
Онзагера принцип взаимности 276
— — линейности 275
Онзагера уравнения 277, 278, 281
Опыт Джоуля — Томсона 44, 46, 265
Отрицательные абсолютные
температуры 254
Отрицательные процессы 75
Парадокс Гиббса 151
Параметры состояния 11, 177
— — локальные 260
Пельтье теплота 279, 282
Первое начало (формулировки) 32,
33, 35
Перекрестные коэффициенты в теории
Онзагера 277, 282
Перпетуум мобиле второго рода 100
— — первого рода 33
Поверхностно-активные вещества 157
Поверхностное натяжение 152
Политропа 51
Полностью изолированная система 34,
167, 185, 260, 269
Постулат Клаузиуса 74, 99
— Планка 244
Потенциалы химические 179, 181
Потоки 260, 261, 264, 266, 269
Правило фаз 194, 196
Принцип взаимности кинетических
коэффициентов .275, 276, 278
Лринцип Каратеодори 87
— линейности Онзагера 275
— недостижимости
абсолютного нуля 249, 252
— рассеяния энергии 101
— Томсена и Бертло 239, 242
Простейшие процессы 38, 39, 40, 41
— — в идеальном
газе 47
Процесс диффузии 66, 262, 263, 272
— политропический 51
Процессы изоэнтропические 82
— квазистатические 13, 63,
68
— круговые 14
— необратимые, обратимые 64,
65, 67, 94, 95
•— механические 67
— отрицательные 75
— положительные 75
— равновесные 64
Пуассона коэффициент 51
— уравнение 50
Работа 23
— максимальная 92
— расширения 23
Равновесие гетерогенное 177
— ложное 192, 193
— метастабильное 166
— одыокомпонентных систем
199
Равновесие термодинамическое 10
Равновесные процессы 63, 64
Распространение звука в газе 54
Рассеяния энергии принцип 101
Реакции экзотермические 58, 240
— эндотермические 58, 240
Роберта Майера уравнение 48
Сверхнизкие температуры 249
Свободная энергия 132, 137
— энтальпия 143, 144
Свойство взаимности кинетических
коэффициентов 278
Связанная энергия 138
Сжимаемости изотермический
коэффициент 19
Система в термостате 168, 187, 189
— изолированная 34, 167, 185,
260
— термодинамическая 9
Системы гетерогенные 10, 176
— гомогенные 10
— закрытые 38
— однокомпонентные 199
— открытые 38, 260, 263
— с переменной массой 180
Соотношения Томсона 282
Состояние метастабильное 222, 223
Спонтанная конденсация 223
Стефана—Больцмана^ закон 123, 127
Стехиометрические коэффициенты 210
Сублимация 202, 203
-Предметный указатель
285
Температура 5
— абсолютная 111
— инверсии 45, 123
— критическая 117, 226
Теорема Карно—Клаузиуса 76
— о росте энтропии 93
— о производстве энтропии 268
— тепловая Нернста 239, 242
Тепловая смерть 103
— теорема Нернста 239, 242
— функция 39, 144
Теплоемкость 41
Теплопроводности закон Фурье 261
«Теплород» 21
Теплосодержание 144
Теплота 21, 22
— парообразования 116
— Пельтье 279, 282
— плавления 205, 206
— приведенная 78
— расширения 40
— сублимации 205, 207
— Томсона 279, 282
Теплоты баланс 264
Термические коэффициенты 17
Термическое уравнение состояния 14
Термодинамическая сила 272, 273
— система 9
Термодинамические параметры
состояния 11
Термодинамические степени
свободы 196
Термодинамические функции 129, 130
Термодинамический потенциал Гиб-
бса 133, 134, 143
Термодинамическое равновесие 10
Термодиффузия 277
Термостат 40, 168, 169, 187
Термохимия 58
Термо-э. д. с. 279
Термоэлектрические явления 279
Томсена и Бертло принцип 239, 242
Точка критическая 226
— перехода 215
— Кюри 216
Третье начало термодинамики 239,
242
Тройная точка 200, 201
Удельные параметры 183, 194
Уравнение Ван-дер-Ваальса 15, 227,
233
—\ Гиббса—Гельмгольца 137,
142, 145
—-'Клапейрона 44
— Клапейрона—Кла>зп>са 114, 116,
206
— Пфаффа 27
— состояния 14, 16
Уравнение состояния калорическое 16
— — тер.мическое 14
Уравнения Максвелла 131, 136
— Онзагера 277, 278, 281
— Эренфеста 222
Условия равновесия гетерогенных
систем 185, 191
Условия устойчивости равновесия 173
Устойчивость равновесия 165
Фаза 176, 177
Фазовая диаграмма 202
Фазовые превращения второго рода
216
Фазовые превращения первого рода
215
Феноменология 6
Флуктуации 108
Формула Гиббса для адсорбции 159
— Р Майера 48
Формы Пфаффа 27
Функции характеристические 130, 179
Функция состояния 14
— тепловая 39, 144
Характеристические функции 130, 179
Химические- реакции 58, 191, 209
Химические потенциалы 179, 181, 183
Химическое равновесие 209
— сродство 239
Цикл необратимый 90
— обратимый 77, 80
Циклов метод в термодинамике 77,
129
Циклы 14
Экзотермические реакции 58, 240
Экстенсивные величины 9
Эндотермические реакции 58, 240
Энергия внутренняя 7, 16, 33, 131
— свободная 132, 137
— связанная 138
Энергии баланс 263, 265
Энтальпия 39, 133
Энтальпия свободная 143, 144
Энтропии баланс 270
— поток 269
Энтропийная диаграмма 84
Энтропия 81, 107, 109
— идеального газа 83
— смешения газов 150
Эффект Джоуля—Томсона 44, 46
— магнитокалорический 249, 250
Эффекты термоэлектрические 279
ОГЛАВЛЕНИЕ
Глава 1. Общие понятия и определения
§ 1. Введение 4
§ 2. Задачи термодинамики. Нулевое начало. Феноменология ... 5
§ 3. Внутренняя энергия 7
§ 4. Термодинамическая система и термодинамическое равновесие . 9
§ 5. Параметры состояния. Квазистатические процессы 11
§ 6. Функция состояния. Термические и калорические уравнения
состояния 14
§ 7. Термические коэффициенты и связь между ними 17
§ 8. Теплота и работа 21
§ 9. Математические методы термодинамики 27
§ 10. Краткий исторический очерк развития термодинамики .... 30
Задачи 31
Глава 2. Первое начало термодинамики и его непосредственные применения
§ 1. Формулировки первого начала 32
§ 2. Применение первого начала к процессам в простейших системах.
Энтальпия 38
§ 3. Теплоемкость 41
§ 4. Идеальный газ. Эффект Джоуля—Томсона 44
§ 5. Применение первого начала к простейшим процессам в
идеальном газе 47
§ 6. Значение адиабатных процессов в газах. Распространение звука
в газе 53
§ 7. Приложение первого начала термодинамики к электрическим и
магнитным явлениям 56
§ 8. Применение первого начала к химическим процессам. Закон Гесса 57
Задачи 62
Глава 3. Второе начало термодинамики. Энтропия и ее свойства
как функции состояния
§ 1. Обратимые и необратимые процессы 63
§ 2. Обратимая машина Карно 69
§ 3. Постулат Клаугиуса. Второе начало как эмпирический закон для
тепловых процессов 74
§ 4 Обобщение обратимых циклов. Открытие энтропии как
функции состояния 76
§ 5. Энтропия и ее свойства. Энтропия идеального газа. Принцип
Каратеодори 81
§ 6. Интеграл Клаузиуса для необратимых циклов. Общая
математическая формулировка второго начала. Максимальная работа . 87
Задачи , .92
Оглавление 287
Глава 4. Различные общие формулировки второго начала
и его непосредственные приложения
§ 1. Теорема о росте энтропии в изолированной системе 93
§ 2. Следствия из теоремы о росте энтропии и связь их с различными
формулировками второго начала 99
§ 3. Необратимые процессы и направление времени в
макроскопической системе 104
§ 4. Статистический смысл второго начала термодинамики .... 106
§ 5. Применение второго начала к построению абсолютной шкалы
температуры ПО
§ 6. Приложение второго начала к рписанию процессов изменения
агрегатного состояния. Уравнение Клапейрона—Клаузиуса . . .114
§ 7. Вывод зависимости внутренней энергии от объема из общего
уравнения второго начала и оценка эффекта Джоуля—Томсона 118
§ 8. Применение второго начала в теории теплового излучения.
Законы Кирхгофа и Стефана—Больцмана . 123
Задачи . . 128
Глава 5. Термодинамические функции и некоторые их приложения
§ 1. Замечания о двух методах, применяемых в термодинамике.
Характеристические функции 129
§ 2. Общая систематика характеристических функций. Соотношения
между частными производными (уравнения Максвелла) .... 131
§ 3. Свободная энергия. Уравнение Гиббса—Гельмгольца . ... 137
§ 4. Термодинамический потенциал Гиббса и энтальпия 143
§ 5. Термодинамические свойства смесей идеальных газов. Парадокс
Гиббса , . 146
§ 6. Свободная энергия поверхностного слоя жидкости 152
§ 7. Адсорбция из растворов и формула Гиббса 156
§ 8. Термодинамика гальванического элемента 160
Задачи 163
Глава 6. Общая теория равновесия сложных термодинамических систем
§ 1. Общие условия термодинамического равновесия и его
устойчивости 165
§ 2. Условия устойчивости равновесия в применении к простым
системам . 173
§ 3. Гетерогенные системы. Фаза. Компоненты 176
§ 4. Характеристические функции для гетерогенных систем.
Химические потенциалы 179
§ 5. Условия равновесия гетерогенных систем 185
§ 6. Правило фаз. Вывод 194
Задачи 198
Глава 7. Применение учения о равновесии к сложным
термодинамическим системам
§ 1. Равновесие однокомпонентных систем 199
§ 2. Равновесие смесей идеальных газов. Закон действующих масс 207
§ 3. Вопросы общей теории фазовых превращений 214
§ 4. Термодинамика процесса образования зародышей новой фазы . 222
§ 5. Критическая точка. Свойства реальных газов 226
Задачи 238
288 Оглавление
Глава 8. Тепловая теорема Нернста и термодинамические свойства веществ
при низких температурах
§ 1. Природа химического сродства Тепловая теорема Нернста . . 239
§ 2. Постулат Планка и свойства вещества близ абсолютного нуля . 244
§ 3. Сверхнизкие температуры и принцип недостижимости
абсолютного нуля. Отрицательные абсолютные температуры 249
Глава 9. Основные вопросы термодинамики необратимых процессов
§ 1. Общие положения. Локальные параметры состояния. Понятие
потока : : : 259
§ 2. Уравнения баланса энергии и массы в открытых системах . . . 263
§ 3. Теорема о производстве энтропии в термодинамических системах.
Термодинамические силы 268
§ 4. Основные вопросы теории Онзагера. Принципы линейности и
взаимности в сложных процессах 275
§ 5'. Теория термоэлектрических явлений 279
Предметный указатель 283
Леонид Викторович Радушкевич
КУРС ТЕРМОДИНАМИКИ
Редактор Т. В. Михалкевич Издательство «Просвещение» Комитета по
Художник Б. М. Мокин печати при Совете Министров РСФСР,
Художественный редактор Л. Ф. Малышева Москва, 3-й проезд Марьиной рощи, 41.
Коппект1Гг VMAfvaLecKak КвпСНЩКпЯ Ленинградская типография № 4 Главполи-
Корректор Т. М. Графовская графпрома Комитета по печати при Совете
Сдано в набор 22/Х 1970 г. Подписано к Министров СССР. Социалистическая. 14.
печати 11/III 1971 г. бОхЭО'Лв. Бумага тип.
№ 2. Печ. л. 18. Уч.-изд. л. 17,12. Тираж Заказ № 2479. Ллагт„ЛТ on *
33 000 экз (План 1971 г № 25). А07052. Пена без переплета 48 к., переплет Я к.