/





















































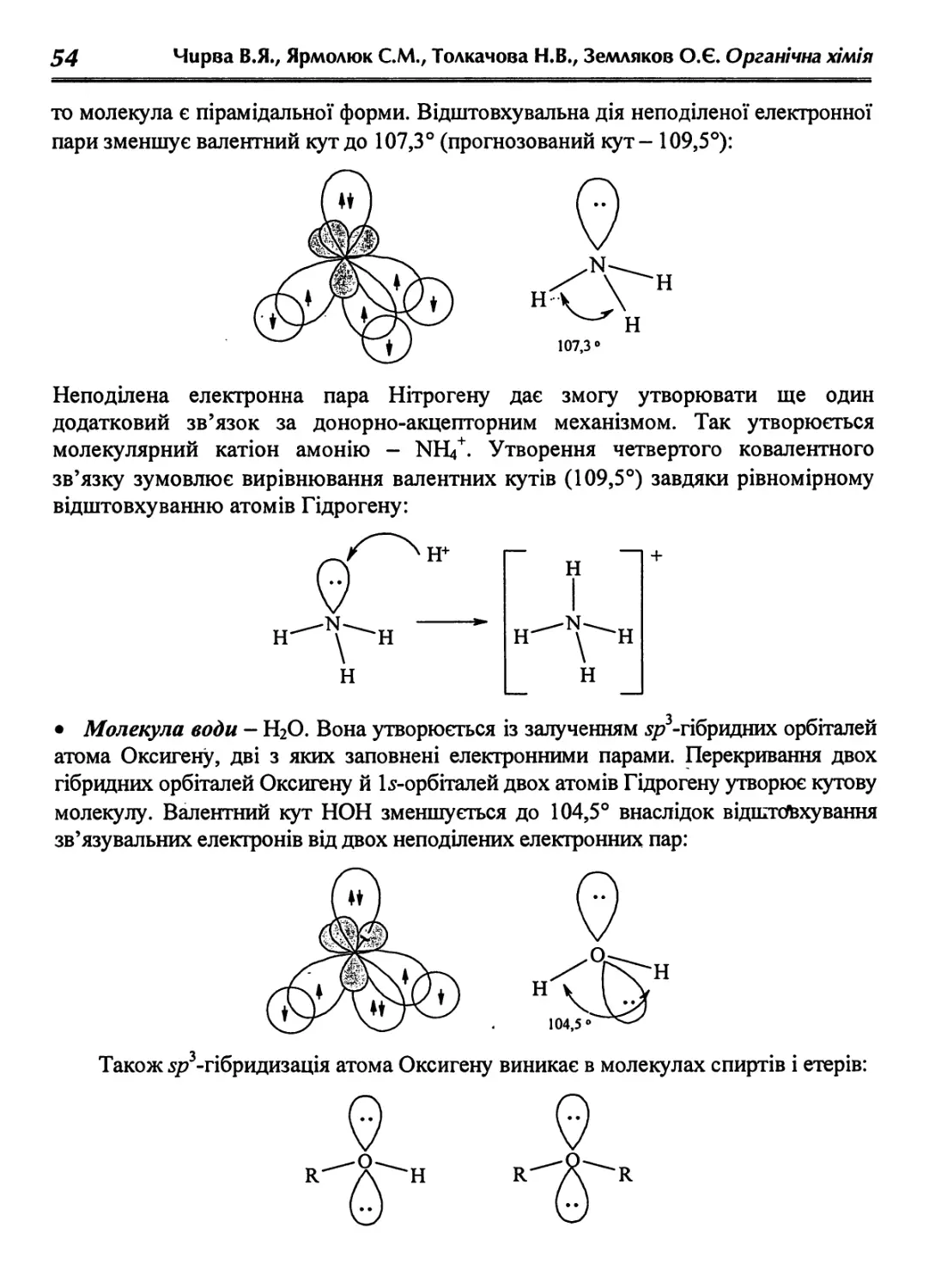













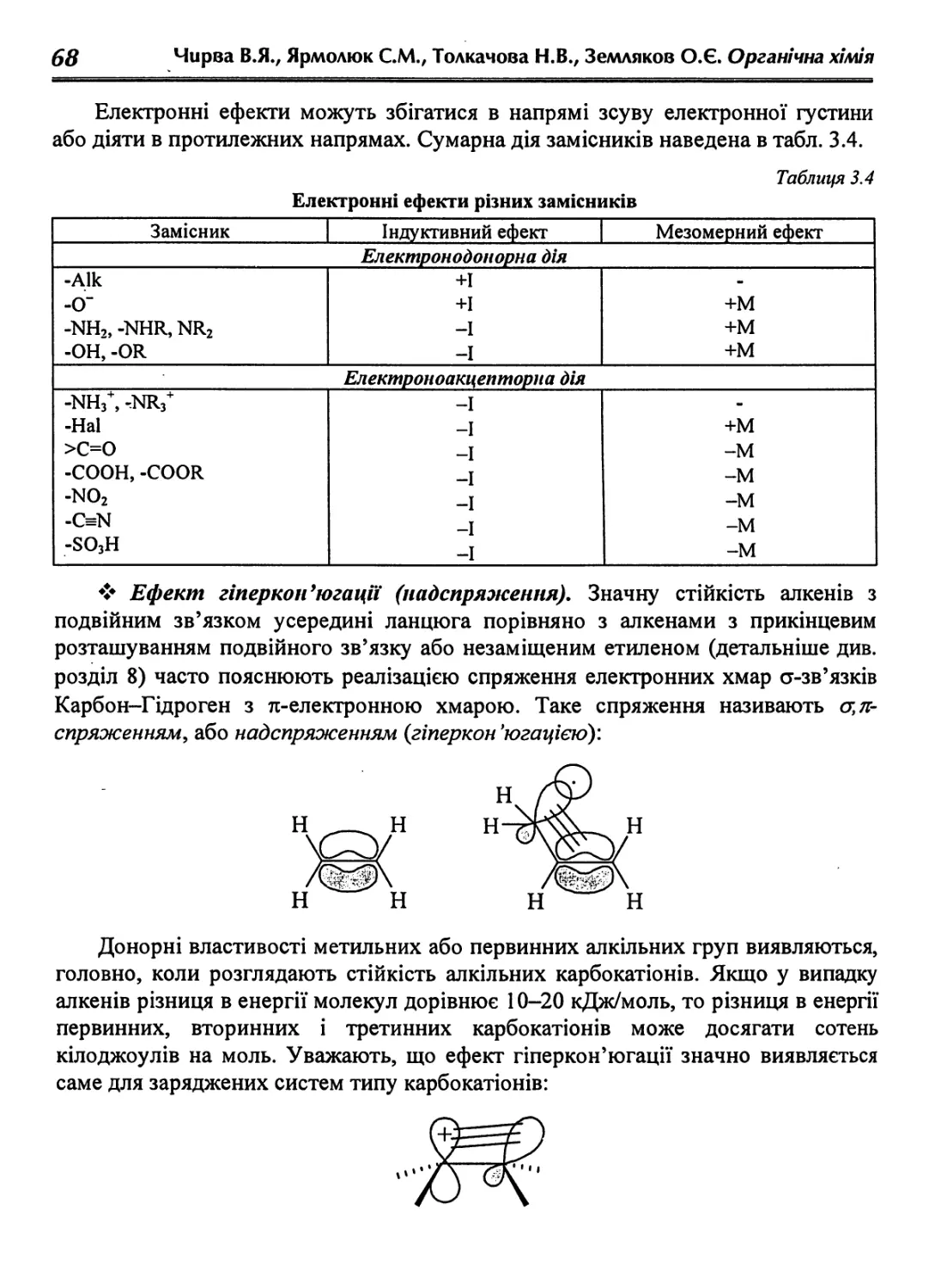




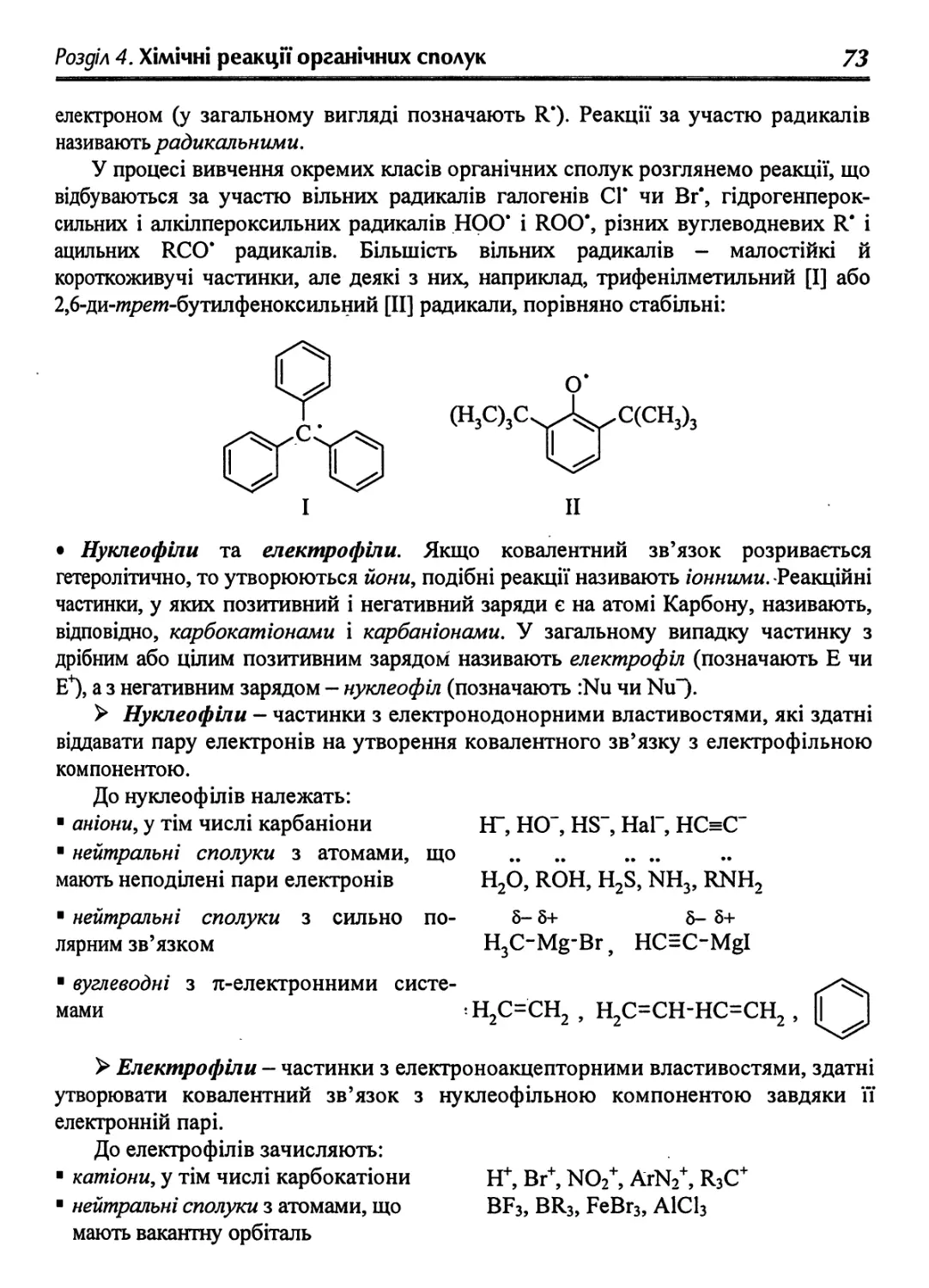





























































































































































































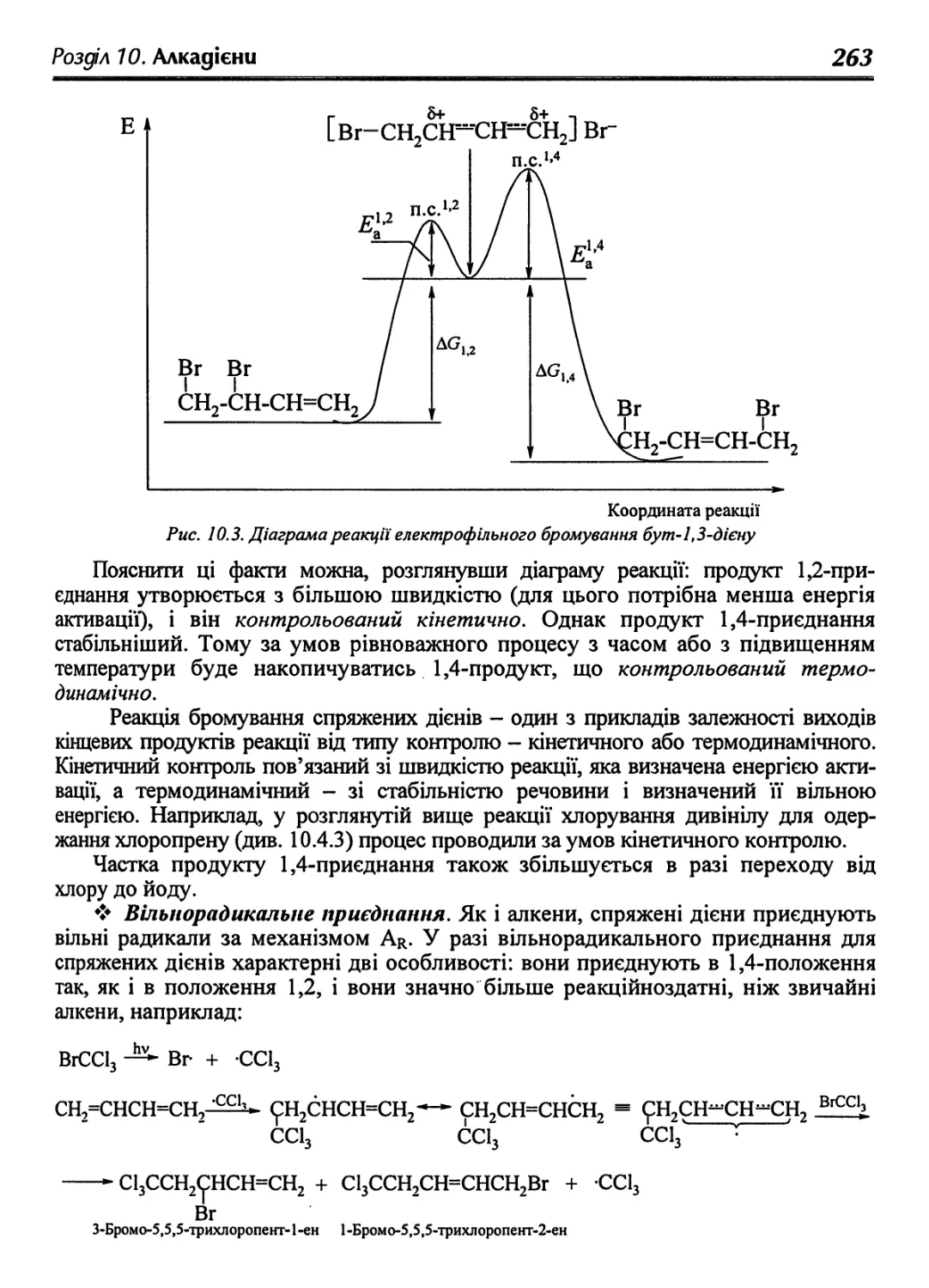





































































































































































































































































































































































































































































































































































































































































































































Text
В.Я. Чирва
С.М. Ярмолюк
Н.В. Толкачова
О.Є. Земляков
ОРГАНІЧНА
ХІМІЯ
УДК 547(075.8)
ББК Г2Я73
Ч-64
Рецензенти:
академік HAH України, д-р хім. наук, проф. М.О, Лозинсъкий
(Інститут органічної хімії HAH України);
д-р хім. наук, проф. В.П. Новиков
(Національний університет "Львівська політехніка");
д-р хім. наук, проф., член-кор. HAH України ВЛ. Хиля
(Київський національний університет ім. Т. Шевченка)
Затверджено Міністерством освіти і науки України
як підручник для студентів хімічних спеціальностей
вищих навчальних закладів
Лист № 1.4/18-Г-2726 від 16 грудня 2008 р.
Чирва В. Я., Ярмолюк С. М., Толкачова Н. В., Земляков О. Є.
Ч-64 Органічна хімія: Підручник. - Львів: БаК, 2009. - 996 с.
ISBN 966-7065-87-4.
Викладено основи сучасної органічної хімії. Розглянуто теоретичні питання
основ природи хімічного зв'язку, будову та реакційну здатність органічних молекул з
урахуванням впливу електронних факторів на перебіг хімічних реакцій. Наведено
сучасні промислові та лабораторні методи одержання основних класів органічних
сполук, їхні фізичні та хімічні властивості. Описано механізми найважливіших
хімічних реакцій. Значну увагу приділено питанням стереохімії та енантіомерії
органічних молекул. Для характеристики органічних сполук широко використано
спектральні методи, такі як УФ-, ІЧ- і ПМР-спектроскопія. Взято до уваги
рекомендації Української національної комісії з хімічної термінології і номенклатури.
Для студентів-хіміків і біохіміків, а також спеціальностей з біології, фармації
тощо та всіх, хто цікавиться органічною хімією.
УДК 547(075.8)
ББК Г2Я73
© Чирва В. Я., Ярмолюк С. М., Толкачова Н.В.,
Земляков О. Є., 2009
ISBN 966-7065-87-4 © Науково-сервісна фірма "ОТАВА", 2009
Зміст
Вступ , 19
Розділ 1. ПРЕДМЕТ ОРГАНІЧНОЇ ХІМІЇ 21
1.1. Виділення, очищення та ідентифікація органічних речовин 25
1.2. Якісний аналіз органічних речовин 29
1.3. Визначення молекулярної маси 32
Розділ 2. ТЕОРІЯ БУДОВИ ОРГАНІЧНИХ СПОЛУК 33
Розділ 3. ОСНОВИ ТЕОРІЇ ЕЛЕКТРОННОЇ БУДОВИ 42
ОРГАНІЧНИХ МОЛЕКУЛ
3.1. Основи квантово-механічної теорії будови атома 42
3.2. Основи квантово-механічної теорії хімічного зв'язку 47
3.2.1. Головні положення методу валентних зв'язків 47
3.2.2. Метод молекулярних орбіталей 55
3.2.3. Водневий 3в'явок, міжмолекулярна взаємодія 62
3.3. Взаємний вплив атомів у молекулі 66
Розділ 4. ХІМІЧНІ РЕАКЦІЇ ОРГАНІЧНИХ СПОЛУК 69
4.1. Хімічні реакції та реагенти 69
4.2. Кислотність і основність 76
Розділ 5. КЛАСИФІКАЦІЯ І НОМЕНКЛАТУРА ОРГАНІЧНИХ СПОЛУК 81
5.1. Класифікація органічних сполук 81
5.2. Ранні номенклатури органічних сполук 83
5.2.1. Тривіальна номенклатура 83
5.2.2. Раціональна номенклатура 84
5.2.3. Женевська і Льєжська номенклатури 84
5.3. Номенклатура IUPAC 85
5.5.7. Номенклатура вуглеводневих замісників 88
5.3.2. Замісникова номенклатура 90
5.3.3. Радикально-функціональна номенклатура 93
Розділ 6. ІЗОМЕРІЯ 95
6.1. Загальні положення 95
6.1.1. Хімічна структура, конфігурація, конформація органічних сполук 97
6.1.2. Зображення просторової будови молекул 98
6.1.3. Стереохімічні номенклатури 100
6.1.4. Класифікація видів ізомерії 104
6.2. Структурна ізомерія 105
4 Чирва В.Я., Ярллолкж CM., Толкачова Н.В., Земляков О.Є. Органічна хімія
6.3. Стереоізомерія . 109
6.3.1. Хіральність, енантіомери. 109
6.3.2. Головні причини хіральності молекул 110
6.3.3. Діастереомерія 115
6.3А. Рацемічні суміші й принципи їхнього розділення на енантіомери 119
6.3.5. Конформаційна ізомерія 123
6.3.6. Відносність понять хімічна структура, конфігурація і конформація 130
Розділ 7. АЛКАНИ 132
7.1. Будова і номенклатура алканів 132
7.1.1. Будова метану .' 132
7.1.2. Номенклатура алканів 133
7.1.3. Ізомерія алканів 134
7.2. Фізичні властивості 136
7.2.1. Спектроскопія алканів 137
7.3. Одержання алканів 138
7.3.1. Промислові способи отримання алканів 138
7 3.2. Лабораторні способи синтезу алканів 140
7.4. Хімічні властивості алканів 145
7 4.1. Галогенування 145
7.4.2. Нітрування 151
7.4.3. Сульфування 153
7.4.4. Каталітична дегідрогенізація алканів 154
7.4.5. Окиснення 155
7.4.6. Каталітична ізомеризація алканів 156
7.5. Крекінг алканів 156
7.6. Йонні реакції 160
7.7. Найважливіші алкани 162
7.8. Метилен 163
7.8.1. Будова метилену 163 і
7.8.2. Хімічні властивості 165
Розділ 8. АЛКЕНИ 170
8.1. Будова і номенклатура алкенів 170
8.1.1. Будова етилену 170
8.1.2. Геометрична ізомерія 172
д. 1.3. Номенклатура алкенів 173
8.2. Фізичні властивості алкенів 174
8.2.1. Спектроскопія алкенів ♦ 175
8.3. Промислові методи одержання алкенів 176 ;
8.4. Лабораторні методи одержання алкенів 178 \
8.4.1. Дегідрогалогенування алкілгалогенідів .. 178 ;
8.4.2. Дегідратація спиртів 182
8.4.3. Розщеплення амінів. Правило Гофмана 186
8.4.4. Інші лабораторні методи, що ґрунтуються нареащіях елімінування 187
8.4.5. Реакції відновлення 189
8.4.6. Реакції конденсації 190
Зміст 5
8.5. Хімічні властивості алкенів 191
8.5.1. Реакції відновлення 192
8.5.2. Реакції галогенування 195
8.5.3. Механізми реакції електрофільного приєднання 199
8.5.4. Напрям приєднання гідроген галогенідів до алкенів 200
8.5.5. Інші реакції приєднання алкенів 205
8.5.6. Реакції заміщення 210
8.5.7. Реакції окиснення 212
8.6. Найважливіші алкени 217
Розділ 9. АЛКІНИ 220
9.1. Будова і номенклатура алкінів.... 220
9.1.1. Будова ацетилену 220
9.1.2. Номенклатура алкінів • 222
9.1.3. Ізомерія алкінів 223
9.2. Фізичні властивості 223
9.2./. Спектроскопія алкінів 224
9.3. Промислові методи одержання алкінів 225
9.4. Лабораторні методи одержання алкінів 227
9.4.1. Синтез ацетилену з елементів 227
9.4.2. Синтези, що засновані на реакціях елімінування 227
9.4.3. Реакції гомологізації 229
9.5. Хімічні властивості алкінів 231
9.5.1. Гідрування 232
9.5.2. Електрофільне приєднання 234
9.5.3. Вільнорадикачьне приєднання 236
9.5.4. Нуклеофільне приєднання 236
9.5.5. Реащії заміщення 239
9.5.6. Реакції оліго- і полімеризації 243
9.5.7. Алкін-аленове перегрупування Фаворського 244
9.5.8. Реакції окисне ння 245
9.6. Окремі представники алкінів 246
Розділ 10. АЛКАДІЄНИ 248
10.1. Будова і номенклатура дієнів 248
10.1.1. Класифікація алкадієнів 248
10.1.2. Номенклатура алкадієнів 248
10.1.3. Будова бут-1,3-дієна 249
10.1.4. Будова алену 251
10.2. Фізичні властивості 252
10.2.1. Спектроскопія дієнів 252
10.3. Одержання і хімічні властивості аленів 253
10.3.1. Методи синтезу аленів 253
10.3.2. Хімічні властивості аленів 253
10.4. Одержання 1,3-ДІєнових вуглеводнів 254
10.4.1. Синтези дивінілу 255
10.4.2. Синтези ізопрену 258
S Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
10.4.3. Синтези хлоропрену 259
10.5. Реакції дієнів 260
10.5.1. Реакції 1,2- і 1,4-приєднання 260
10.5.2. Реакції "дієнового синтезу" 264
10.5.3. Реакції полімеризації 267
10.6. Каучук 272
10.6.1. Природний каучук 272
10.6.2. Синтетичний каучук 272
10.7. Терпени ациклічного ряду 274
10.8. Вітаміни, що мають полієновий ланцюг 275
Розділ 11. ЦИКЛОЛЛКАНИ 277
11.1. Моноциклічні аліфатичні вуглеводні 278
11.1.1. Номенклатура 278
11.1.2. Фізичні властивості 278
11.1.3. Будова циклопропану 279
11.1.4. Ізомерія 281
11.1.5. Конформація 282
11.1.6. Загальні методи одержання 282
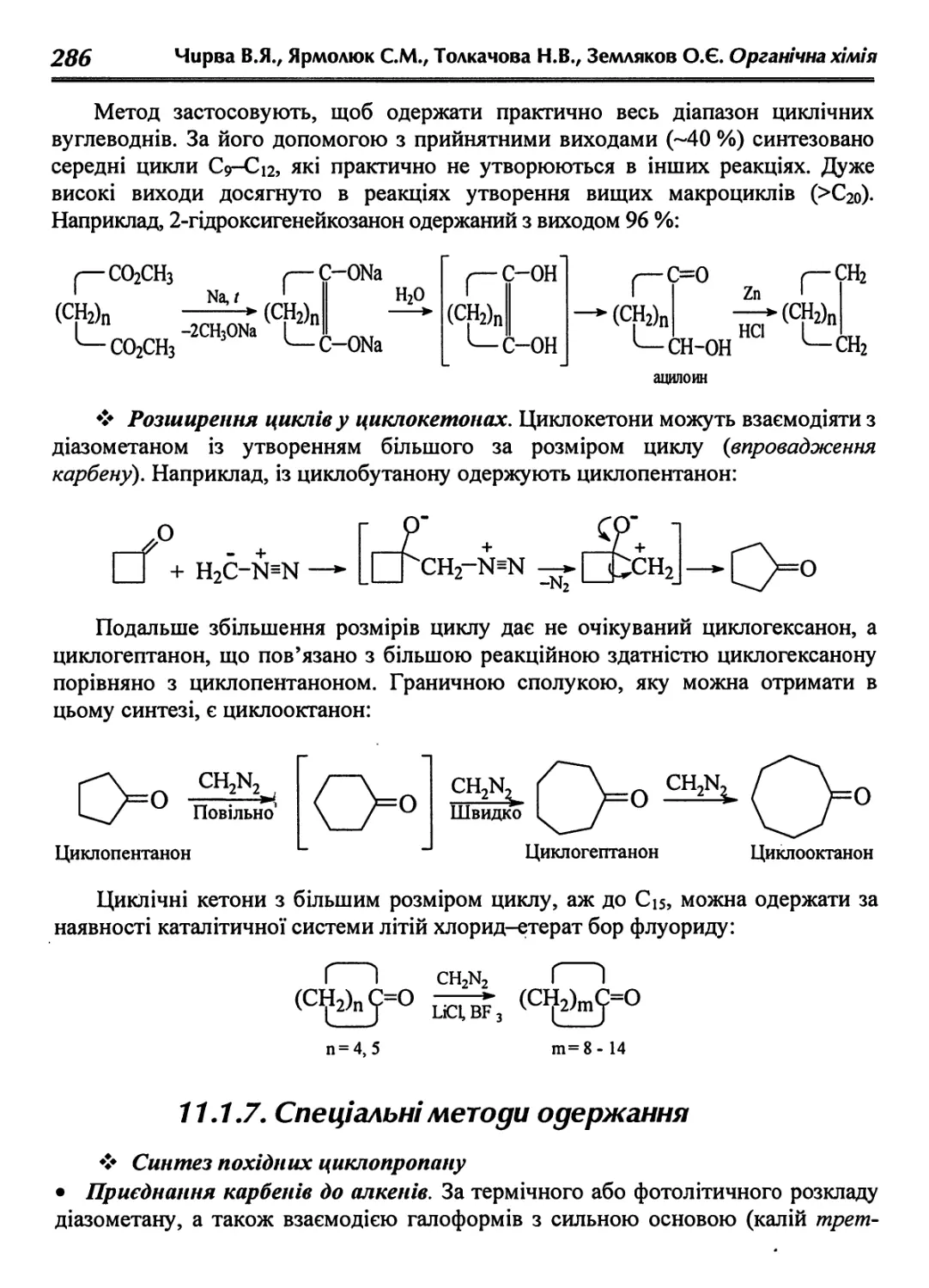
11.1.7. Спеціальні методи одержання 286
11.1.8. Хімічні властивості 290
11.1.9. Вплив просторових чинників на реакції похідних циклоалканів 294
11.2. Бі- та поліциклічні вуглеводні 298
11.2.1. Номенклатура 298
11.2.2. Декалін ЗОО
11.2.3. Адамантан 300
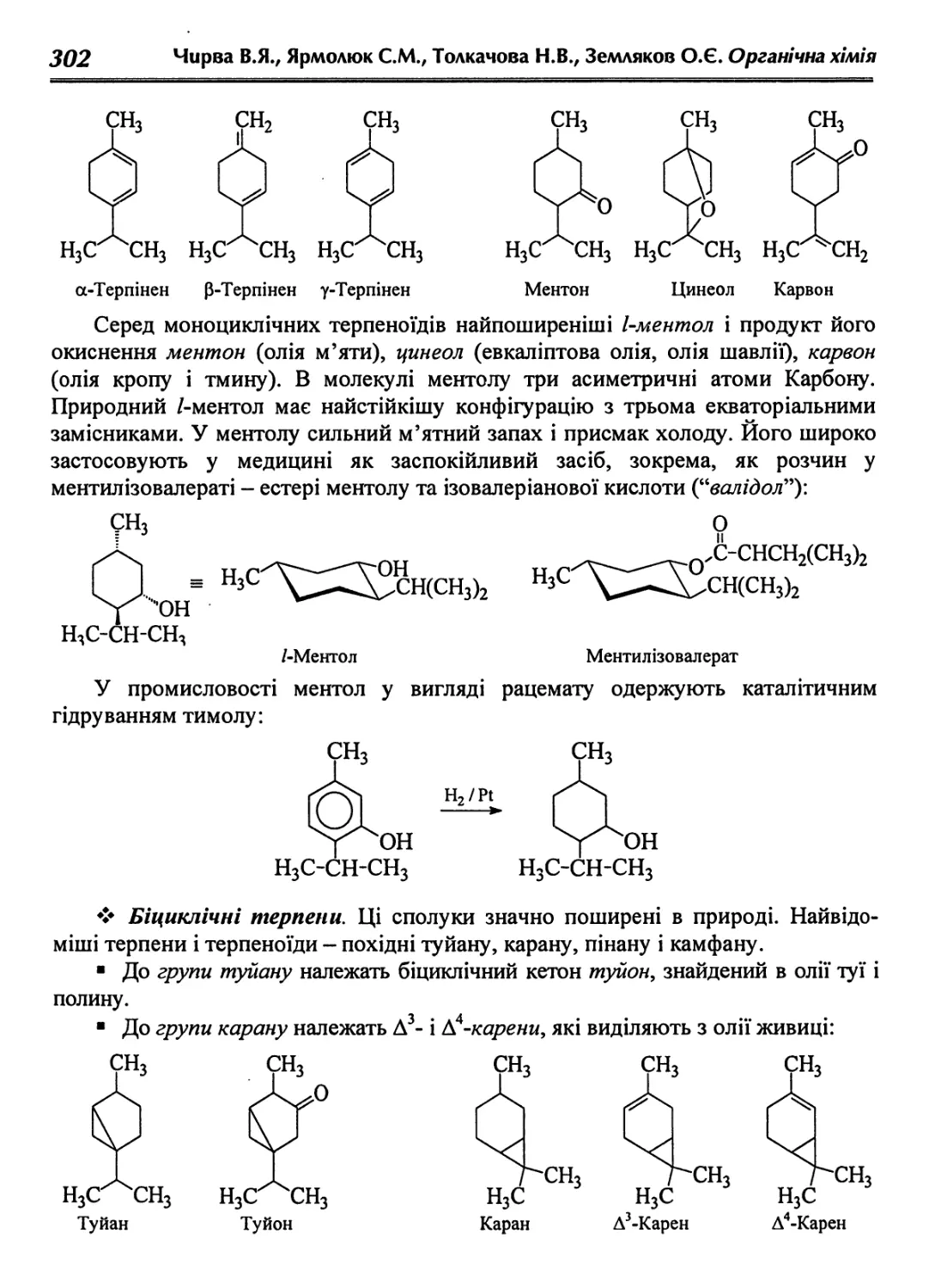
11.2.4. Моно- і біциклічні терпени 301
11.2.5. Стероїди 304
Розділ 12. ГАЛОГЕНОПОХІДНІ ВУГЛЕВОДНІВ 306
12.1. Будова і номенклатура 306
12.1.1. Класифікація галогенопохідних 306
12.1.2. Номенклатура ЮРАС. 307
12.2. Фізичні властивості 307-
12.2.1. Спектроскопія галогеноалканів 308
12.3. Синтез галогенопохідних 309
12.3.1. Одержання галогенопохідних з алканів 309
12.3.2. Синтез галогенопохідних зі спиртів 310
12.3.3. Синтез галогенопохідних з олефінів 312
12.3.4. Синтез галогенопохідних з ацетиленових вуглеводнів 313
12.3.5. Синтез дигалогенопохідних з альдегідів і кетонів 314
12.3.6. Синтез галогенопохідних з карбонових кислот 314
12.3.7. Реакції обміну 315
12.4. Хімічні властивості моногалогенопохідних 316
12.4.1. Реакції нуклеофільного заміщення 316
12.4.2. Механізм бімолекулярного нуклеофільного заміщення (SN2) 319
12.4.3. Механізм мономолекулярного нуклеофільного заміщення (SN1) 321
: м і с т 7
12.4.4. Чинники, що впливають на перебіг реакцій за механізмами S^l і SN2 323
12.5. Хімічні властивості дигалогенопохідних 327
12.6. Синтез і хімічні властивості галоформів 329
12.6.1. Синтез хлороформу 329
12.6.2. Хімічні властивості хлороформу 330
12.6.3. Іншігалоформні сполуки 331
12.7. Галогенопохідні ненасичених вуглеводнів 331
12.7.1. Методи одержання вінілхлориду 331
12.7.2. Хімічні властивості вінілхлориду 332
12.7.3. Інші хлоровані етилени 334
12.7.4. Флуорованіетилени 335
12.7.5. Алілгалогеніди 335
Розділ 13. СПИРТИ 339
13.1. Класифікація, номенклатура і будова спиртів 339
13.1.1. Класифікація спиртів 339
13.1.2. Номенклатура спиртів — 340
13.1.3. Будова спиртів 341
13.1.4. Ізомерія 342
13.2. Фізичні властивості спиртів 343
13.2.1. Спектроскопія спиртів 344
13.3. Одержання одноатомних спиртів у промисловості 344
13.4. Методи синтезу одноатомних спиртів у лабораторії 347
13.5. Хімічні властивості одноатомних спиртів 354
13.5.1. Розрив зв 'язк)> С-ОН 354
13.5.2. Розрив зв'язку О-Н 361
13.6. Окремі представники одноатомних спиртів 363
13.7. Ненасичені спирти та їхні естери 365
13.7.1. Еноли 365
13.7.2. Одержання похідних енолів 366
13.7.3. Аліловий спирт 368
13.7.4. Вищі ненасичені спирти 368
13.7.5. Ацетиленові спирти й етери 370
13.8. Гліколі 371
13.8.1 Фізичні властивості гліколів 371
13.8.2. Методи синтезу гліколів 372
13.8.3. Хімічні властивості гліколів 374
13.8.4. Застосування гліколів 379
13.9. Багатоатомні спирти 380
13.9.1. Одержання гліцеролу 380
13.9.2. Хімічні властивості гліцеролу 382
13.9.3. Застосування гліцеролу 384
13.9.4. Багатоатомні спирти •• 384
13.9.5. Жири та олії 384
Розділ 14. ЕТЕРИ 387
14.1. Класифікація і номенклатура 387
14.1.1. Класифікація етерів 387
8 Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
14.1.2. Номенклатура ЮРАС 387 \
14.2. Фізичні властивості етерів 388 \
14.2.1. Спектроскопія етерів 389 |
14.3. Одержання етерів 390 •
14.4. Хімічні властивості етерів 393
14.5. а-оксиди 397 !
14.5.1. Одержання а-оксндів ....397
14.5.2. Хімічні властивості епоксидів 398
14.6. Краун-етери 400
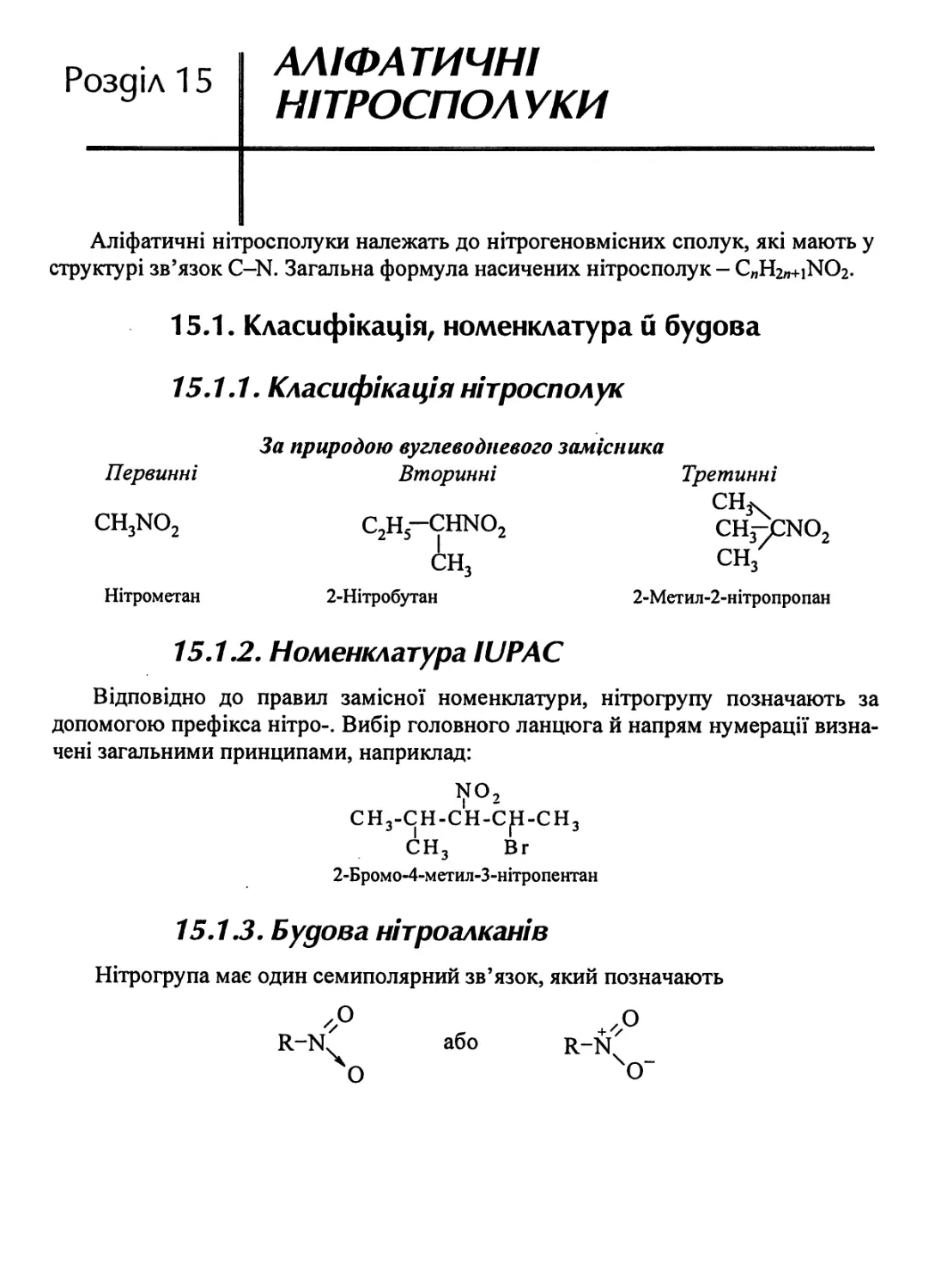
Розділ 15. АЛІФА ТИЧНІ НІТРОСПОЛУКИ 403
15.1. Класифікація, номенклатура й будова 403
15.1.1. Класифіках\ія нітросполук 403
15.1.2. Номенклатура ЮРАС 403
15.1.3. Будова нітроалканів . 403
15.2. Фізичні властивості 404
15.2.1. Спектроскопія нітроалканів 404
15.3. Одержання нітросполук 405
15.4. Хімічні властивості та застосування 407
15.4.1. Хімічні властивості нітроалканів 407
15.4.2. Застосування нітроалканів 411
Розділ 16. АЛІФАТИЧНІ АМІНИ 412
16.1. Класифікація, номенклатура й будова амінів 412
16.1.1. Класифікація амінів 412
16.1.2. Номенклатура ЮРАС 412
16.1.3. Будова амінів 413
16.1.4. Стереоізомерія амінів 414
16.2. Фізичні властивостім 415
16.2.1. Спектроскопія амінів 416
16.3. Способи одержання амінів 417
16.3.1. Реакції алкілування 417
16.3.2. Розщеплення амідів за Гофманом 420
16.3.3. Реакції відновлення 421
16.3.4. Гідроліз ізоціанатів 423
16.3.5. Розщеплення ацилазидів 424
16.3.6. Реакція Ріттера 424
16.4. Хімічні властивості амінів 425
16.4.1. Кислотно-основні реакції 425
16.4.2. Реакції амінів як нуклеофілів 426
/ 6.4.3. Окиснення амінів 428
16.4.4. Реакції амінів з нітритною кислотою 429
/ 6.4.5. Якісні реакції на первинні аміни 430
16.5. Діаміни 431
16.6. Діазометан 432
Зміст $
Розділ 17. ОКСОСПОЛУКИ 433
17.1. Номенклатура й будова 433
17.1.1. Номенклатура 433
17.1.2. Будова оксосполук 434
17.2. Фізичні властивості 436
17.2.1. Спектроскопія оксосполук 437
17.3. Способи синтезу оксосполук 437
17.3.1. Промисловий синтез 437
17.3.2. Одержання зі спиртів 439
17.3.3. Озоноліз алкенів 442
17.3.4. Синтези на основі металоорганічних сполук 443
17.3.5. Одержання з кислот та їхніх похідних 444
17.3.6. Гідратація алкінів 445
17.3.7. Оксосинтез 446
17.3.8. Синтез оксосполук на основі галогеноалканів 447
17.4. Хімічні властивості оксосполук 448
17.4.1. Реакції з О-нуклеофілами 449
/ 7.4.2. Реакції з S-нуклеофілами 451
17.4.3. Реакції з С-нуклеофілами 453
17.4.4. Реакції з галогенонуклеофілами 456
17.4.5. Реакції з N-нуклеофілами 457
17.4.6. Реакції відновлення 460
У 7.4.7. Реакції окиснення 462
17.4.8. Реакції диспропорціювання 465
17.4.9. Реакції, що відбуваються за участю атома Гідрогену в а-положенні 467
17.4.10. Реакції оліго- та полімеризації 473
17.5. Окремі представники оксосполук 474
17.6. Дикарбонільні сполуки 475
17.6.1. Класифікація й номенклатура 1UPAC 475
17.6.2. а-Діоксосполуки 475
/ 7.6.3. Р-Діоксосполуки 478
17.6.4. уДіоксосполуки 480
Розділ 18. МОНОКАРБОНОВІ КИСЛОТИ 481
18.1. Класифікація карбонових кислот 481
18.2. Номенклатура й будова монокарбонових кислот 482
18.2.1. Номенклатура 482
18.2.2. Будова карбоксильної групи 484
18.3. Фізичні властивості 486
18.3.1. Спектроскопія монокарбонових кислот 487
18.4. Одержання карбонових кислот 488
18.4.1. Промислові методи синтезу мурашиної та оцтової кислот 488
18.4.2. Способи, що ґрунтуються на окисненні 489
18.4.3. Способи, що ґрунтуються на гідролізі 491
18.4.4. Синтез карбонових кислот методом подовження ланцюга карбонових атомів 492
18.4.5. Синтез карбонових кислот та їхніх похідних без зміни довжини карбонового
ланцюга 495
10 Чирва В.Я., Ярмолюк CM., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
18.5. Солі карбонових кислот 495
18.5.1. Фізичні властивості 496
18.5.2. Способи одержання 496
18.5.3. Хімічні властивості й практичне застосування 497
18.6. Естери 499
18.6.1. Номенклатура 499
18.6.2. Способи одержання 500
18.6.3. Хімічні властивості й практичне застосування 504
18.7. Ортоестери кислот 506
18.7.1. Способи одержання 506
18.7.2. Хімічні властивості 507
18.8. Галогеноангідриди кислот 509
18.8.1. Способи одержання 509
18.8.2. Хімічні властивості 510
18.9. Ангідриди кислот 512
18.9.1. Способи одержання 513
18.9.2. Хімічні властивості 514
18.9.3. Оцтовий ангідрид 515
18.10. Аміди 516
18.10.1. Способи одержання 517
18.10.2. Хімічні властивості й застосування 519
18.11. Нітрили 521
18.11.1. Способи одержання 521
18.11.2. Хімічні властивості 522
18.12. Гідразиди й азиди кислот 525
18.12.1. Гідразиди 525
18.12.2. Ацилазиди 526
18.13. Галогенозаміщені кислоти 527
18.13.1. Способи одержання а-галогенозаміщених карбонових кислот 527
18.13.2. Способи одержання інших галогенозаміщених карбонових кислот 528
18.13.3. Хімічні властивості галогенозаміщених карбонових кислот 529
18.13.4. Окремі представники галогенозамігцених карбонових кислот 531
18Л4. Монокарбонові ненасичені кислоти 531
18.14.1. Методи синтезу 532
18.14.2. Хімічні властивості 533
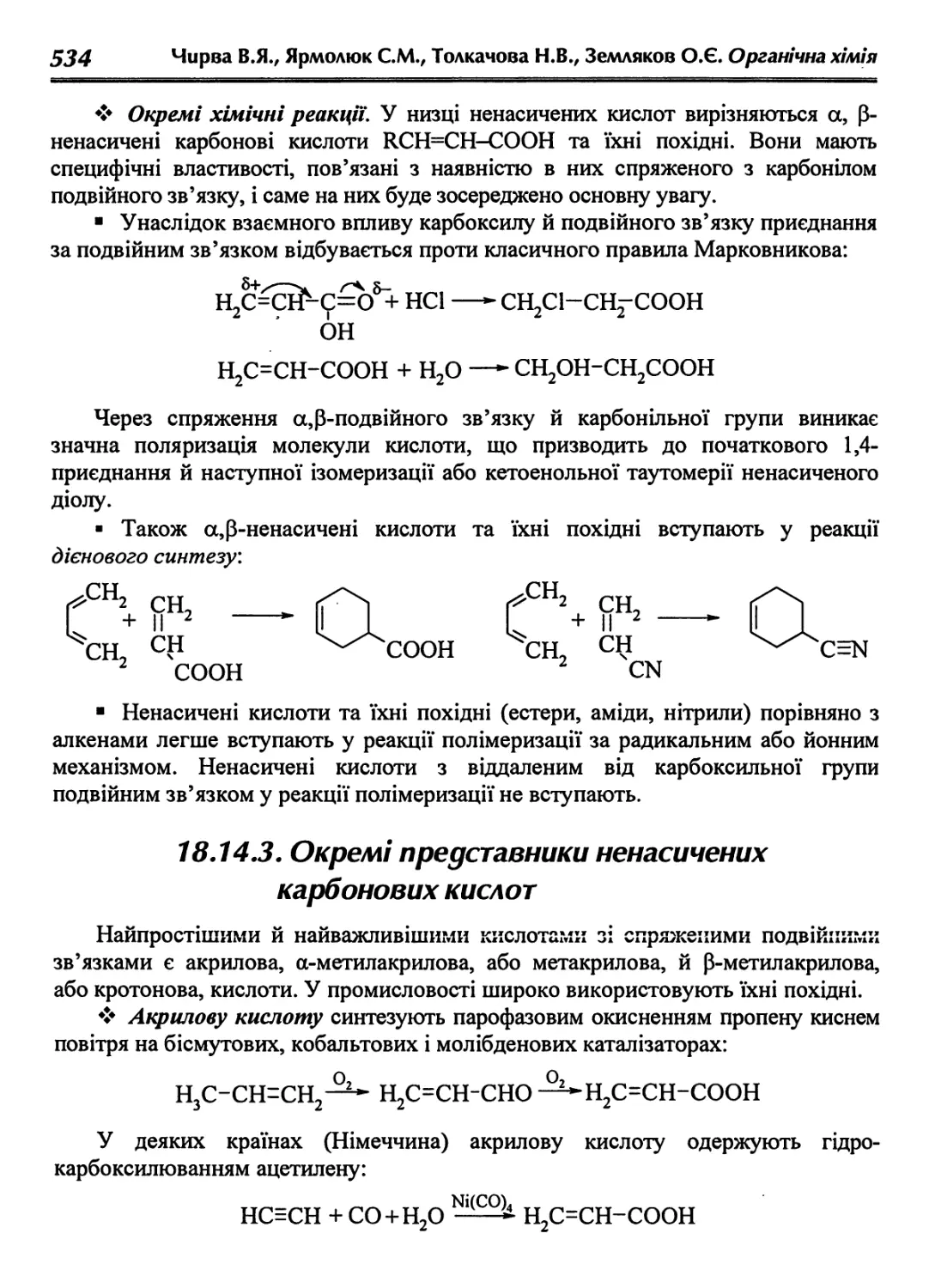
18.14.3. Окремі представники ненасичених карбонових кислот 534
Розділ 19. ДИКАРБОНОВІ КИСЛОТИ 537
19 1. Класифікація і номенклатура 537
19.1.1. Класифікація 537
19.1.2. Номенклатура 1UPАС 537
19 2. Фізичні властивості 538
193. Способи одержання 539
19.3.1. Загальні методи 539
19.3.2. Одержання щавлевої кислоти 540
19.3.3. Синтез малонової кислоти 540
19.3.4. Синтез янтарної кислоти 541
Зміст Ц
19.3.5. Одержання адипінової кислоти 541
19.4. Особливості поведінки двоосновних кислот 542
19.4.1. Щавлева кислота 542
19.4.2. Малонова кислота 545
19.4.3. Янтарна кислота 548
19.4.4. Глутарова кислота 550
19.4.5. Адипінова кислота 551
19.5. Синтези на основі малонового естеру 551
19.6. Двоосновні ненасичені кислоти 554
¡9.6.1. Будова й властивості 554
19.6.2. Методи одержання 555
19.6.3. Малеїновий ангідрид 556
Розділ 20. ПДРОКСИКИСЛОТИ 557
20.1. Класифікація і номенклатура 557
20.1.1. Класифікація 557
20.1.2. Номенклатура IVPАС 558
20.2. Фізичні властивості гідроксикислот 558
20.3. Стереоізомерія гідроксикислот 559
20.3.1. Енантіомерія 559
20.3.2. Стереоізомерія гідроксикислот з двома асиметричними атомами.
Діастереомерія 561
20.3.3. Асиметричний синтез 562
20.3.4. Вальденівське обертання 563
20.4. Синтез гідроксикислот 564
20.4.1. Синтез а-гідроксикарбонових кислот : 564
20.4.2. Синтез fi-гідроксикарбонових кислот 565
20.4.3. Синтез со-гідроксикарбонових кислот 566
20.4.4. Синтез du- і полігідроксикарбонових кислот 567
20.5. Хімічні властивості гідроксикислот 569
20.6. Окремі представники гідроксикислот 572
Розділ 21. ОКСОКИСЛОТИ 575
21.1. Класифікація та номенклатура 575
21.1.1. Класифікація 575
21.1.2. Номенклатура 1UPAC. 575
212. Синтез і властивості оксокислот 576
21.2.1. Гліоксилова кислота 576
21.2.2. Піровиноградна кислота 577
21.2.3. Ацетооцтова кислота 577
21.2.4. Левулінова кислота 578
21.3. Синтез і властивості ацетооцтового естеру 579
21.3.1. Методи синтезу 579
21.3.2. Таутомерія 581
21.3.3. Реакції, що відбуваються за кетонним типом 585
21.3.4. Реакції, що відбуваються за енольним типом 587
21.3.5. Синтези на основі ацетооцтового естеру 589
12 Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
21.3.6. Ацетооцтовий естер як амбідентний нуклеофіл 590
Розділ 22. АМІНОКИСЛОТИ 593
22.1. Класифікація, номенклатура й будова 593
22.1.1. Класифікація 593
22.7.2 Номенклатура ШРАС 596
22.1.3. Будова амінокислот 597
22.2. Фізичні властивості 597
22.2.1. Спектроскопія амінокислот 598
22.3. Способи одержання 598
22.3.1. Хімічний синтез а-амінокислот 598
22.3.2. Хімічний синтез ^-амінокислот 601
22.3.3. Хімічний синтез ^-амінокислот 601
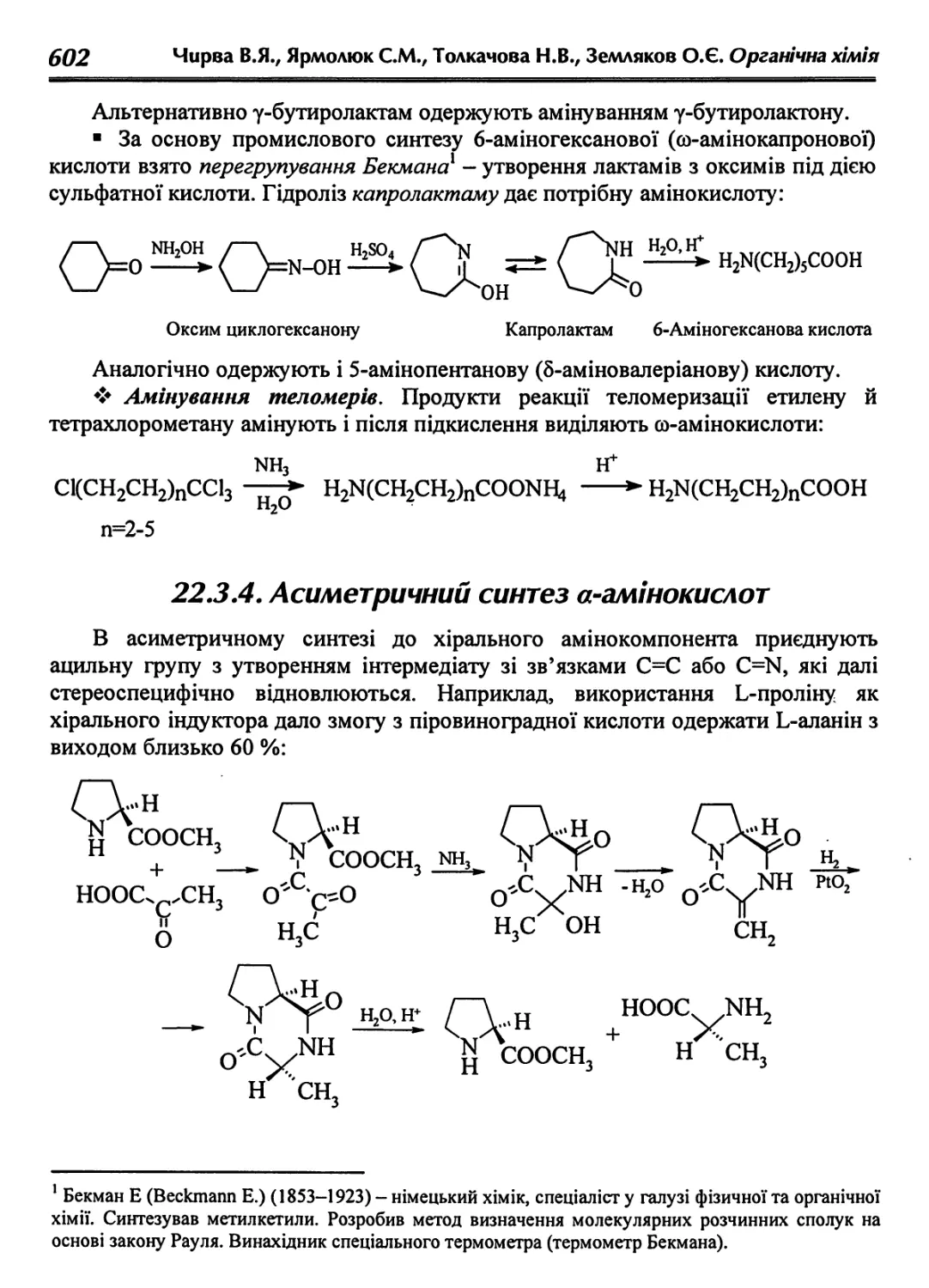
22.3.4. Асиметричний синтез а-амінокислот 602
22.3.5. Поділ рацематів на індивідуальні енантіомери 603
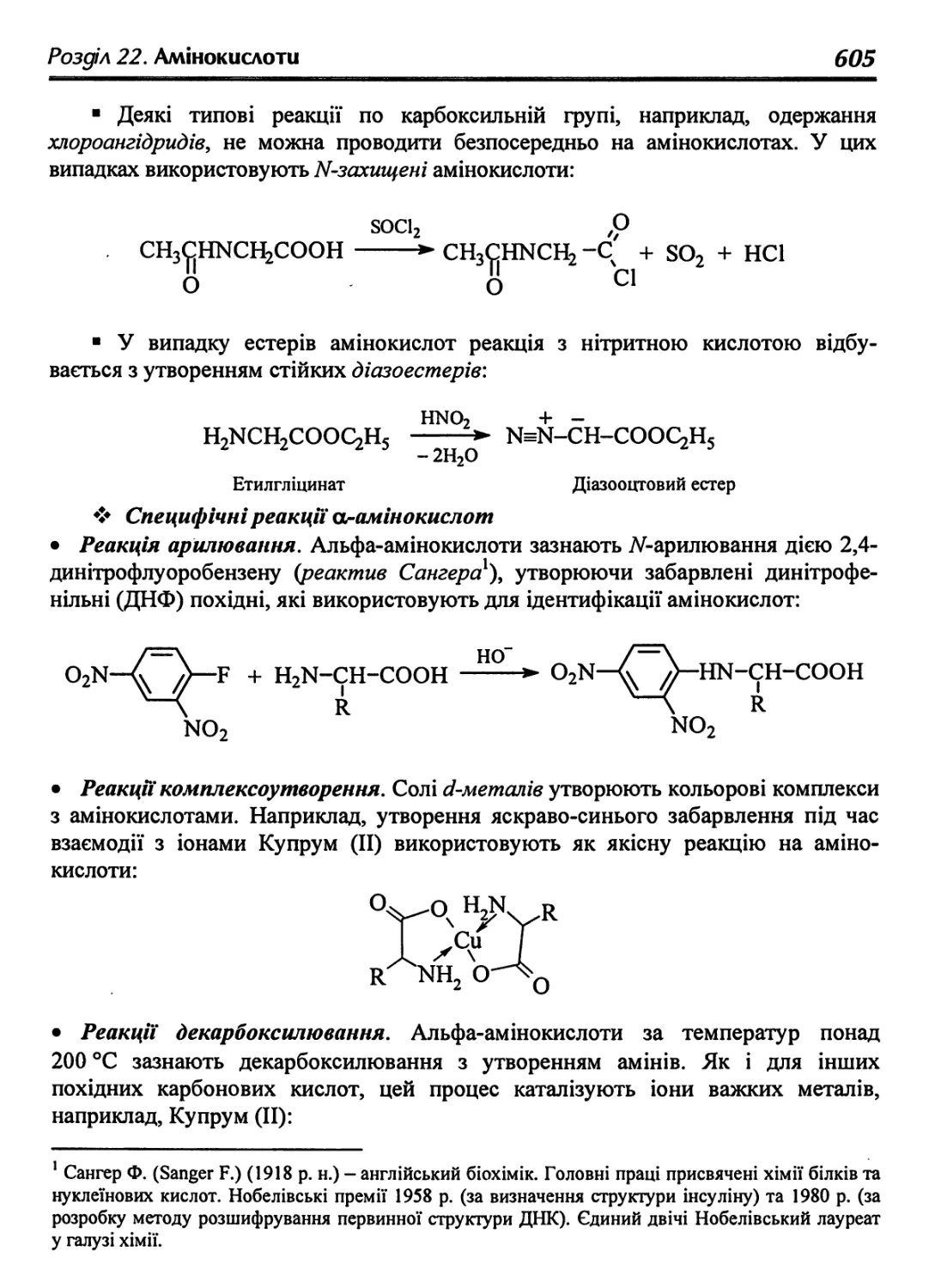
22.4. Хімічні властивості 604
22.5. Окремі представники амінокислот 608
22.6. Пептиди й пептидний синтез 608
22.6.1. Синтез пептидів 609
22.6.2. Окремі представники пептидів 6 І 2
22.7. Білки 612
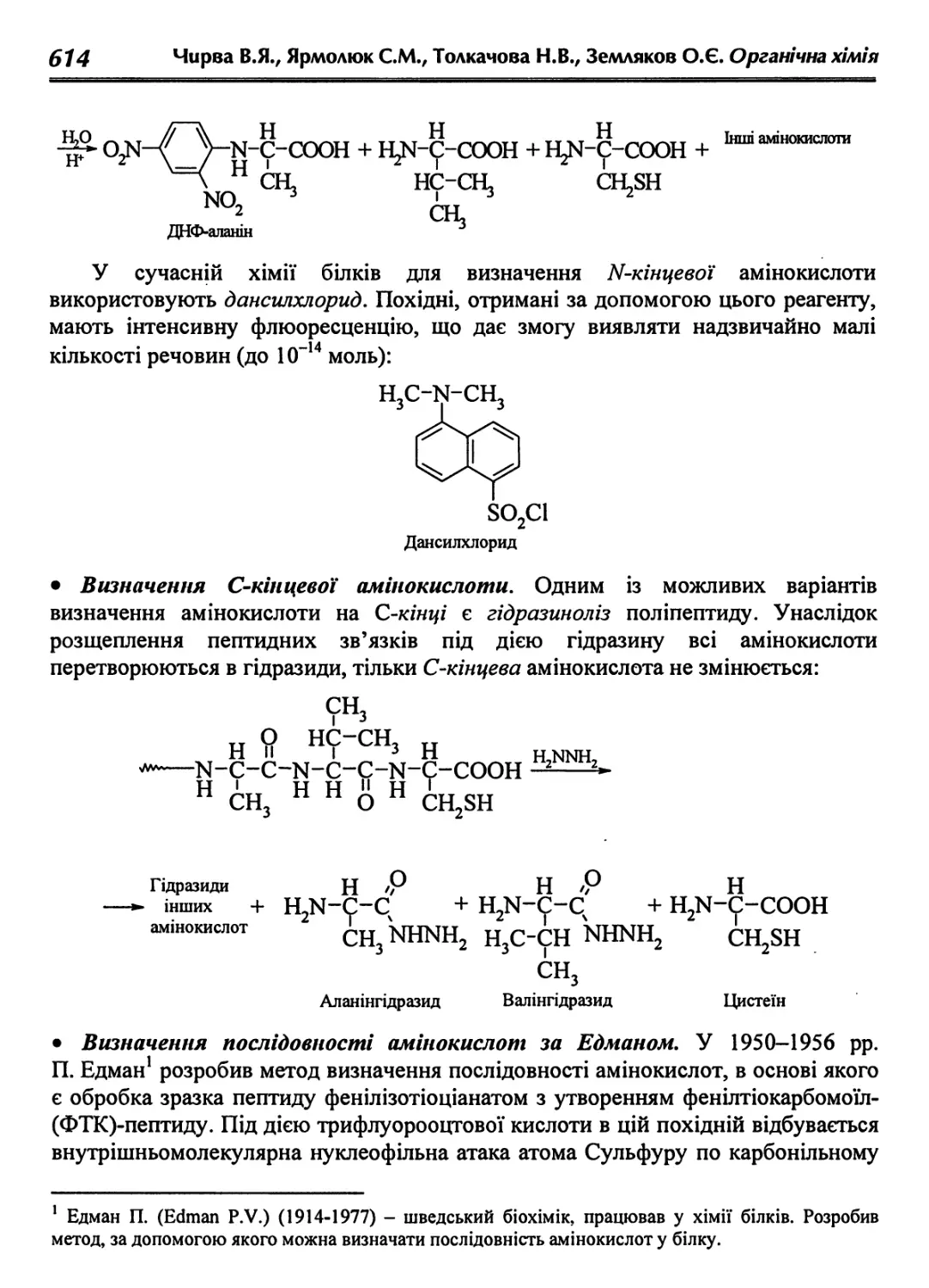
22.7.1. Первинна структура білків і принципи її визначення 613
22.7.2. Просторова будова білкових молекул 615
Розділ 23. ВУГЛЕВОДИ 619
23.1. Класифікація, будова й номенклатура моносахаридів 620
23.1.1. Класифікація моносахаридів 620
23.1.2. Структура моносахаридів 620
23.1.3. Конфігурація моносахариді 622
23.1.4. Визначення конфігурації й-глюкози й й-манози 624
23.1.5. Циклічні форми моносахаридів 626
23.1.6. Конформації моносахаридів 632
23.2. Хімічні властивості моносахаридів 633
23.2.1. Реакції відновлення 633
23.2.2. Реакції окиснення 634
23.2.3. Дія на моносахариди кислот і основ 635
23.2.4. Методи вкорочення й подовження карбонового ланцюга моносахаридів 637
23.2.5. Реакції за карбонільною групою 639
23.2.6. Реакції за участю спиртових гідроксильних груп 640
23.2.7. Реакції й похідні глікозидного гідроксилу 642
23.3. Найважливіші моносахариди 644
23.3.1. Бродіння моносахаридів 646
23.3.2. Фотосинтез вуглеводів у рослинах 648
23.4. Найважливіші олігосахариди 649
23.5. Полісахариди 651
23.5.1. Полісахариди рослинного походження 651
23.5.2. Зоополісахариди 656
Зміст із
23.5.3. Мукополісахариди 657
23.5.4. Полісахариди мікроорганізмів 658
Розділ 24, АРОМАТИЧНІ ВУГЛЕВОДНІ. БЕНЗЕН ТА ЙОГО БУДОВА 659
24.1. Будова й номенклатура аренів 659
24.1.1. Класифікаг\ія аренів 659
24.1.2. Визначення будови бензену 660
24.1.3. Сучасне пояснення будови бензену 663
24.1.4. Правила ароматичності 665
24.1.5. Номенклатура IUPAC 669
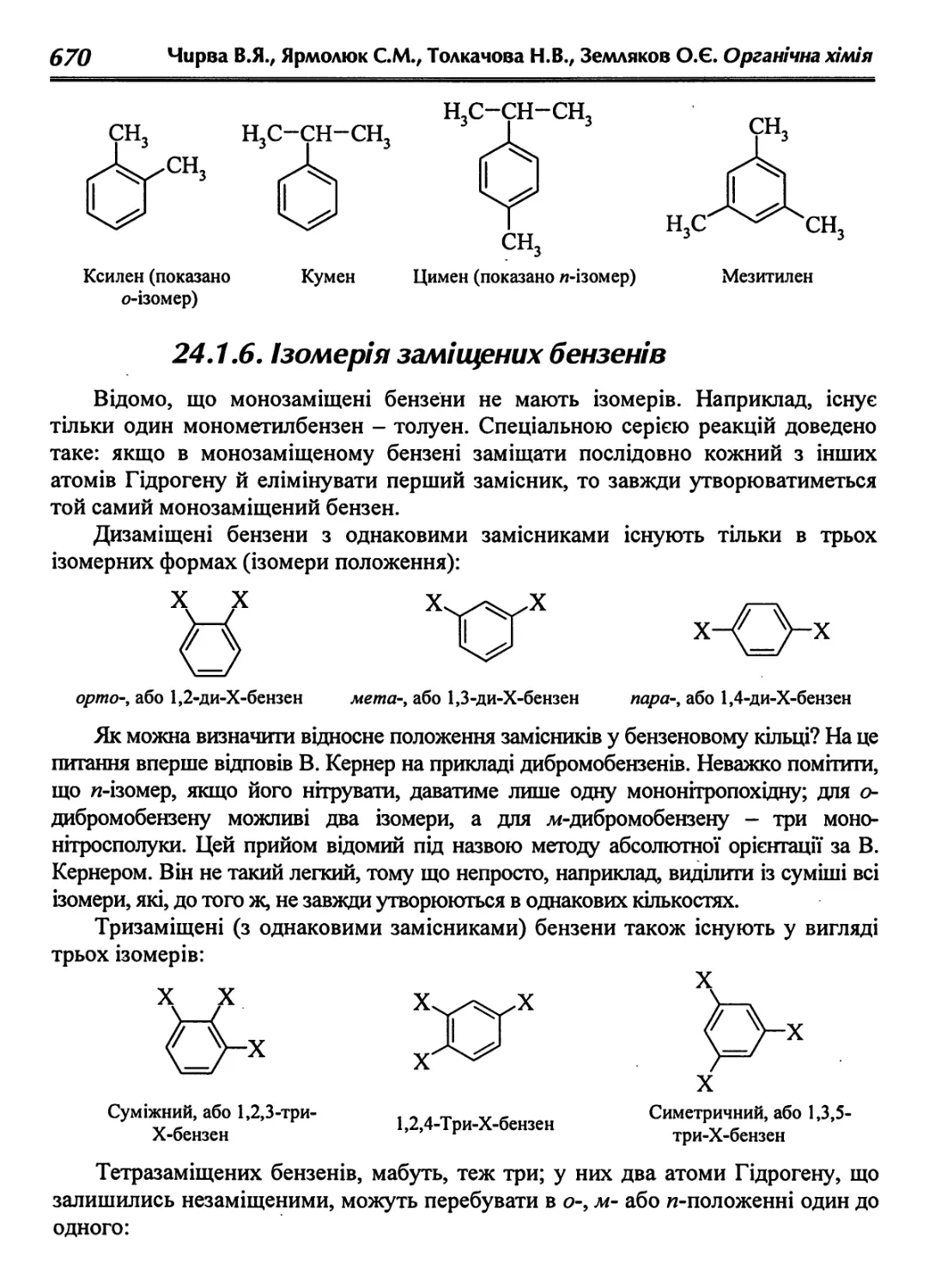
24.1.6. Ізомерія заміщених бензенів 670
24.2. Фізичні властивості 672
24.2.1. Спектроскопія аренів 672
24.3. Синтез бензену та його похідних з аліфатичних і аліциклічних сполук 673
24.3.1. Лабораторні методи 673
24.3.2. Природні джерела ароматичних вуглеводнів 675
24.3.3. Реакції гомологізації бензину 678
24.4. Перетворення бензену в аліфатичні й аліциклічні сполуки 681
24.5. Окремі представники 682
Розділ 25. ЕЛЕКТРОФІЛЬНЕ ЗАМІЩЕННЯ В АРОМ А ТИЧНИХ 683
ВУГЛЕВОДНЯХ
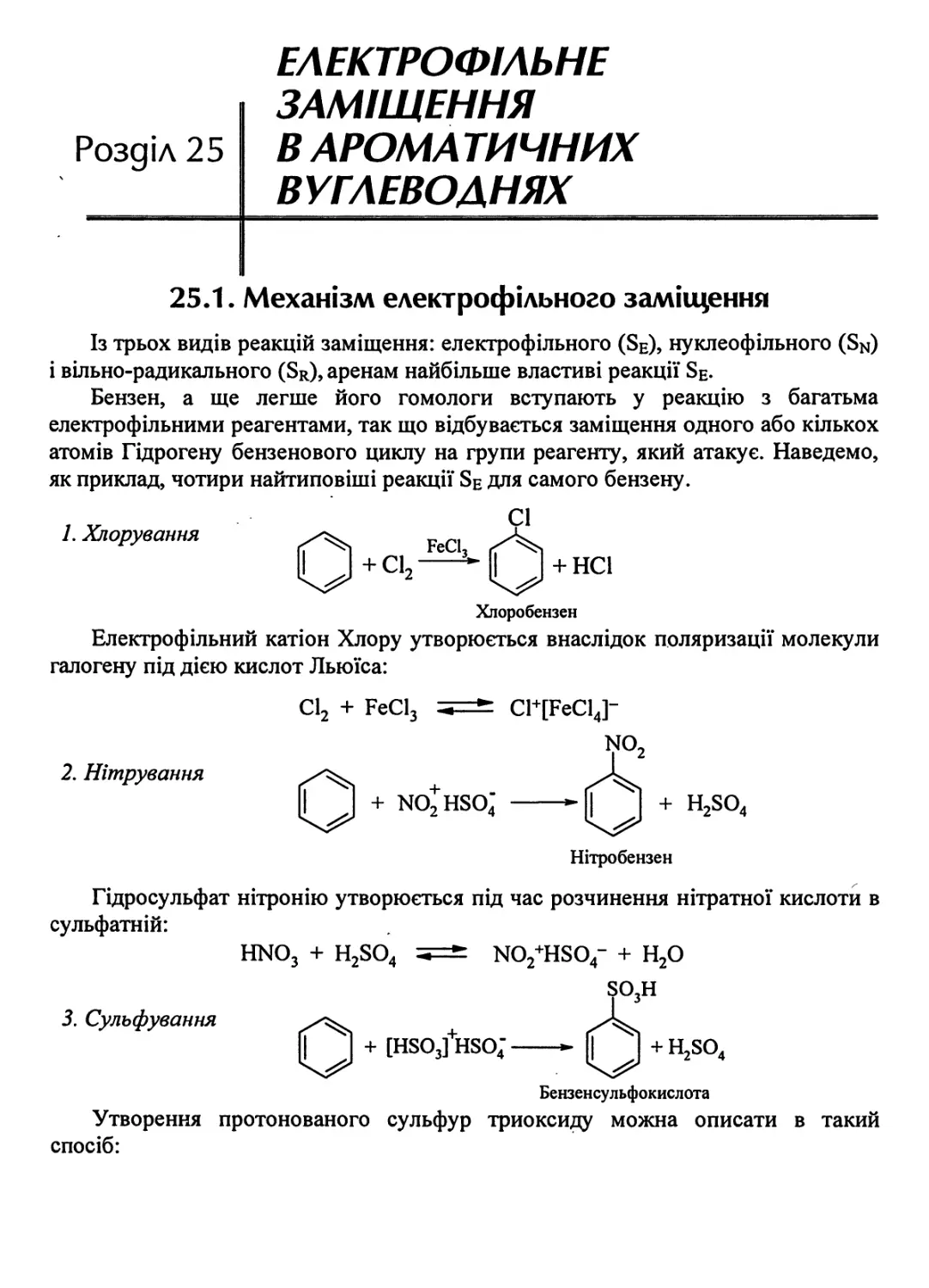
25.1. Механізм електрофільного заміщення 683
25.2. Протонування й дейтерування бензену 686
25.3. Орієнтація реакцій заміщення 687
25.3.1. Орієнтанти першого і другого роду 687
25.3.2. Причина явищ орієнтації 691
25.3.3. Гіперкон'югація й ортопараорієнтація 694
25.3.4. Метаорієнтація 695
Розділ 26. ГАЛОГЕНОПОХІДНІ АРОМАТИЧНИХ ВУГЛЕВОДНІВ 698
26.1. Класифікація й номенклатура галогеноаренів 698
26.1.1. Класифікація галогеноаренів .... 698
26.7.2. Номенклатура гапогеноаренів 698
26.2. Фізичні властивості галогеноаренів 698
26.2.1. Спектроскопія галогеноаренів 699
26.3. Одержання галогеноаренів 699
26.3.1. Галогенування в ядро 699
26.3.2. Галогенування в бічний ланцюг 703
26.3.3. Хлорометилювання 703
26.4. Хімічні властивості галогенопохідних ряду бензену 704
26.4.1. Орієнтувальний ефект галогену як замісника в реакціях електрофільного
замігцення. 704
26.4.2. Реакції електрофільного заміщення 705
26.4.3. Реакції нуклеофільного заміщення атому галогену 706
26.4.4. Реакції нуклеофільного заміщення атома галогену в бічному ланцюзі аренів 712
26.4.5. Реакції металювання 714
14
Чирва В.Я., Ярллолкж CM., Толкачова Н.В., Земляков О.Є. Органічна хімія
Розділ 27. АРЕНСУЛЬФОКИСЛОТИ ТА ЇХНІ ПОХІДНІ 715
27.1. Будова й номенклатура аренсульфокислот 715
27.2. Фізичні властивості сульфокислот 716
27.2.1. Спектроскопія аренсульфонових кислот 716
27.3. Методи синтезу аренсульфокислот 716
27.4. Реакції сульфокислот 719
Розділ 28. НІТРОСПОЛУКИ РЯДУ БЕНЗЕНУ ТА ЇХНІ ПОХІДНІ 725
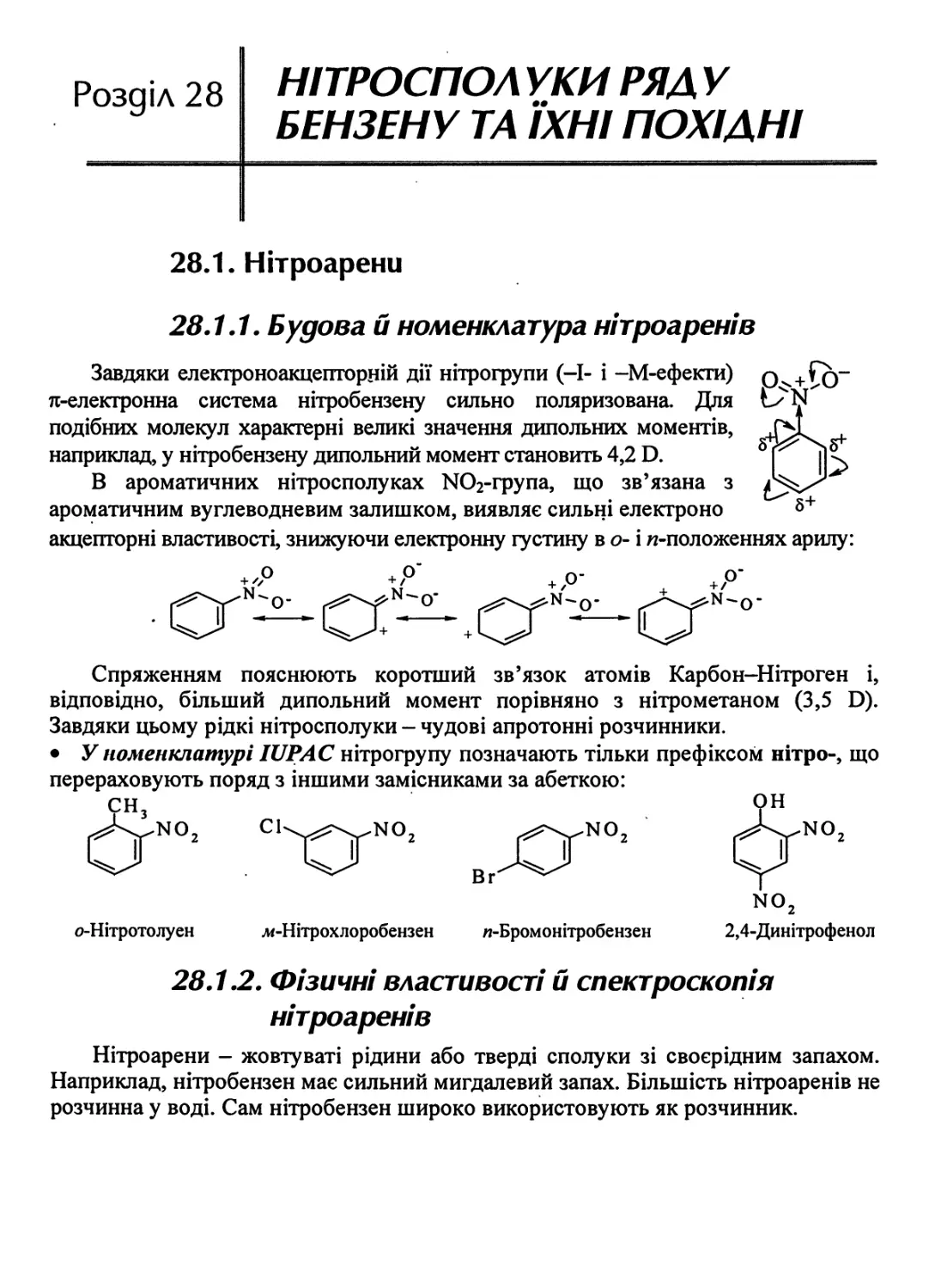
28.1. Нітроарени 725
28.1.1. Будова й номенклатура нітроаренів 725
28.1.2. Фізичні властивості й спектроскопія нітроаренів 725
28.2. Одержання нітроаренів 726
28.3. Хімічні властивості нітроаренів. 731
28.3.1. Реакції електрофільного заміщення в нітроаренах 731
28.3.2. Реакції нуклеофільного заміщення в нітроаренах 732
28.3.3. Реакції конденсацій алкілнітросполук 733
28.3.4. Реакції відновлення 733
28.4. Функції, утворені неповним відновленням нітросполук 734
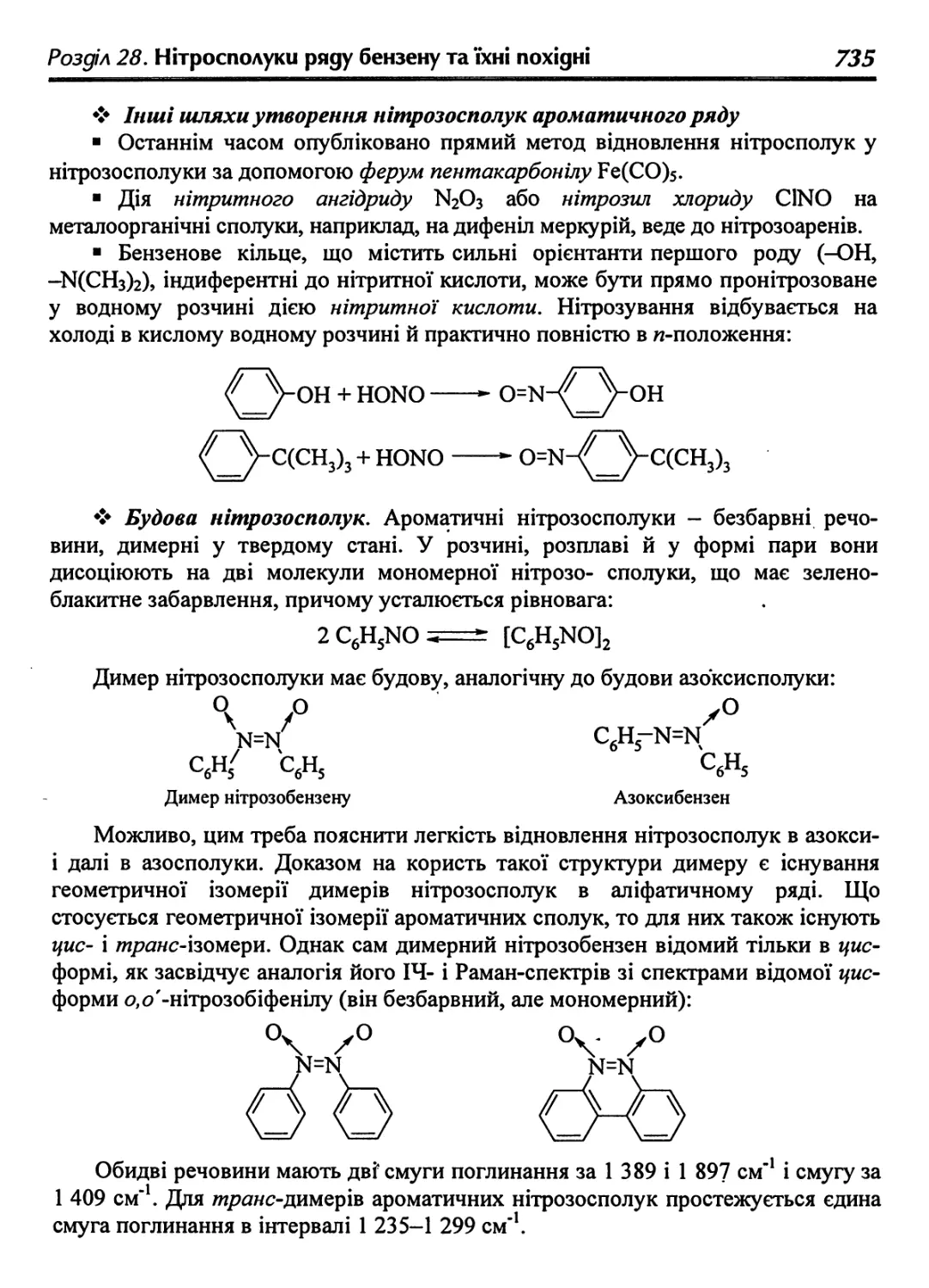
28.4.1. Нітрозобензен 734
28.4.2. Фенілгідроксиламін 737
28.4.3. Азоксибензен 738
28.4.4. Азобензен 740
28.4.5. Гідразобензен 741
Розділ 29. АРОМА ТИЧНІ АМІНИ 744
29.1. Класифікація та номенклатура ароматичних амінів 744
29.1.1. Класифікація 744
29.1.2. Номенклатура IUP АС , 744
29.2. Фізичні властивості 745
29.2.1. Спектроскопія ароматичних амінів 745
29.3. Синтез ароматичних амінів 746
29.4. Хімічні властивості ароматичних амінів 748
29.4.1. Кислотно-основні властивості ароматичних амінів 748
29.4.2. Реакції електрофільного заміщенняв бензеновому кільці амінів 750
29.4.3. Заміщення в аміногрупі 754
29.4.4. Ароматичні вторинні й третинні аміни 757
Розділ ЗО. АРОМАТИЧНІ АЗО- ТА ДІАЗОСПОЛУКИ 760
30.1. Класифікація, будова й номенклатура 760
30.2. Фізичні властивості 762
30.2.1. Спектроскопія солей діазосполук 763
30.3. Методи синтезу діазосполук 763
30.3.1. Синтез солей діазонію 763
30.3.2. Синтез солей діазопохідних 765
30.3.3. Синтез азосполук 766
30.4. Хімічні властивості 772
Зміст 15
ЗОЛІ. Реакції діазосполук без виділення азоту 772
30.4.2. Реакції діазосполук із виділенням азоту 774
Розділ 31. ГІДРОКСИПОХІДНІАРЕНІВ. ФЕНОЛИ 781
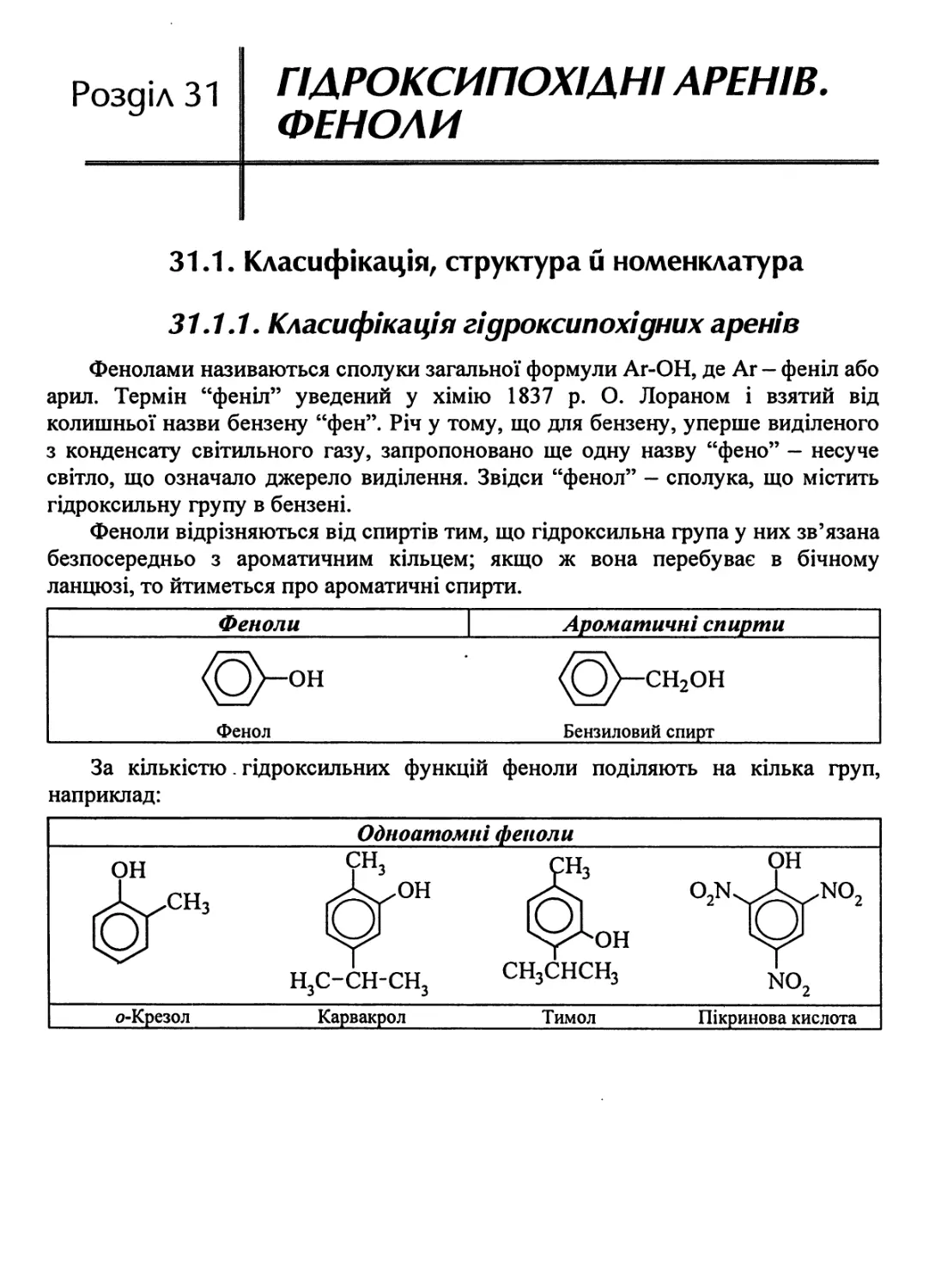
31.1. Класифікація, структура й номенклатура 781
31.1.1. Класифікація гідроксипохідних ареніе. 781
31.1.2. Будова фенолу 782
31.1.3. Номенклатура фенолів 782
31.2. Фізичні властивості 783
31.2.1. Спектроскопія фенолів 784
31.3. Промислові джерела фенолів 784
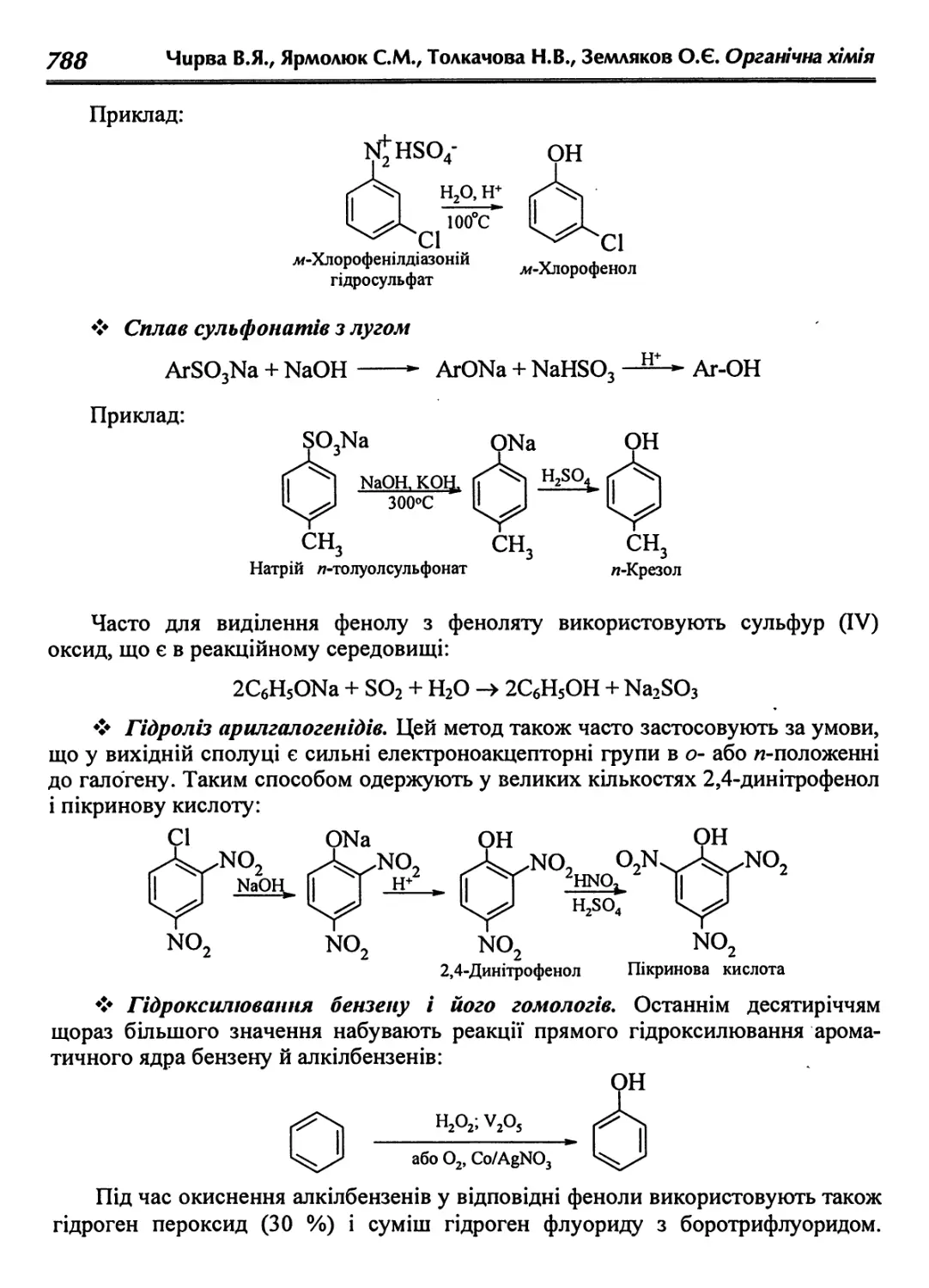
31.4. Одержання фенолів у лабораторії 787
31.5. Реакції фенолів по гідроксильній групі 790
31.5.1. Кислотність. Утворення солей 790
31.5.2. Утворення етерів 793
31.5.3. Утворення естерів 795
316. Реакції фенолів за ароматичним ядром 797
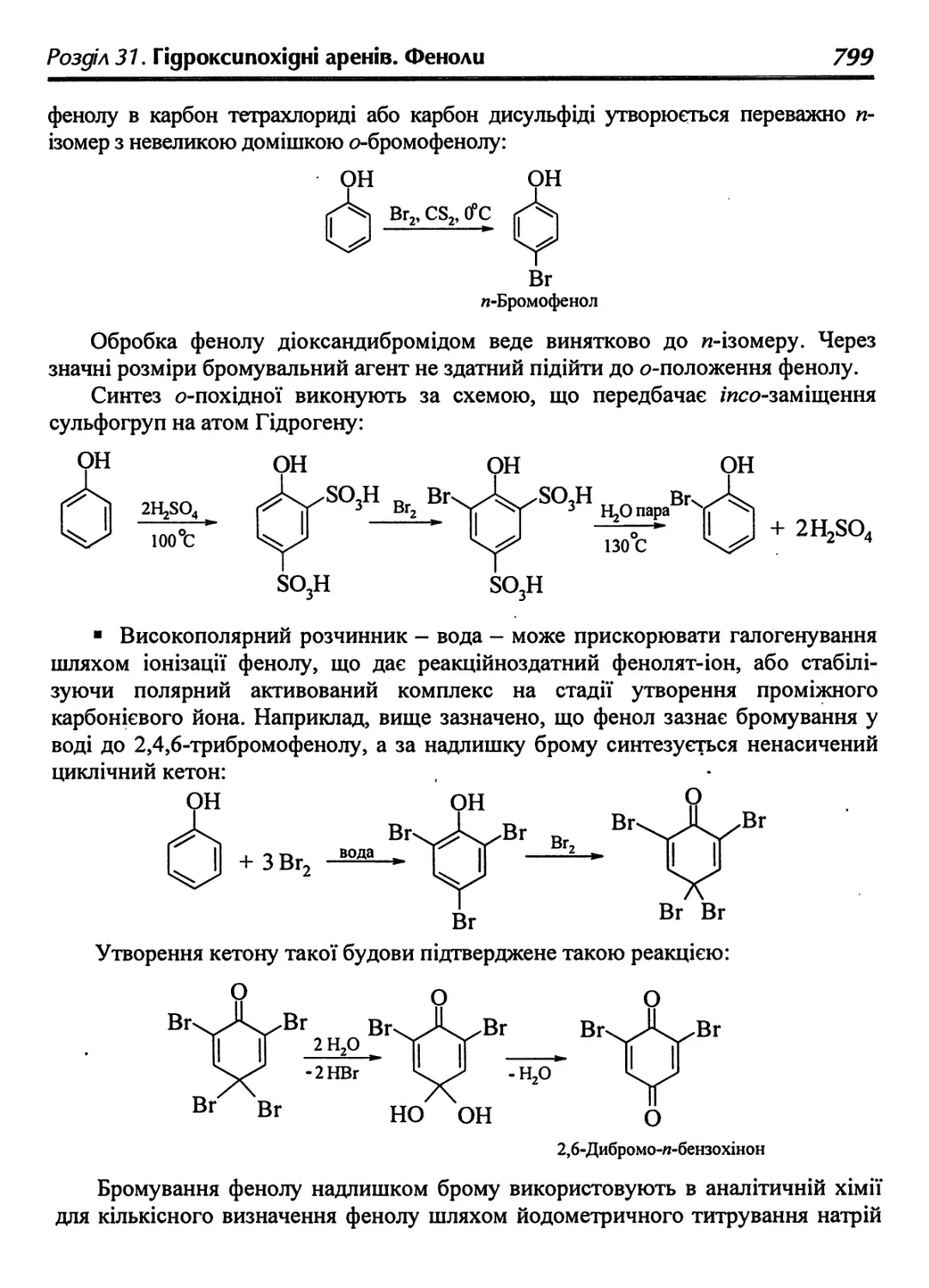
31.6.1. Галогенування 798
31.6.2. Нітрування 801
31.6.3. Сульфування 803
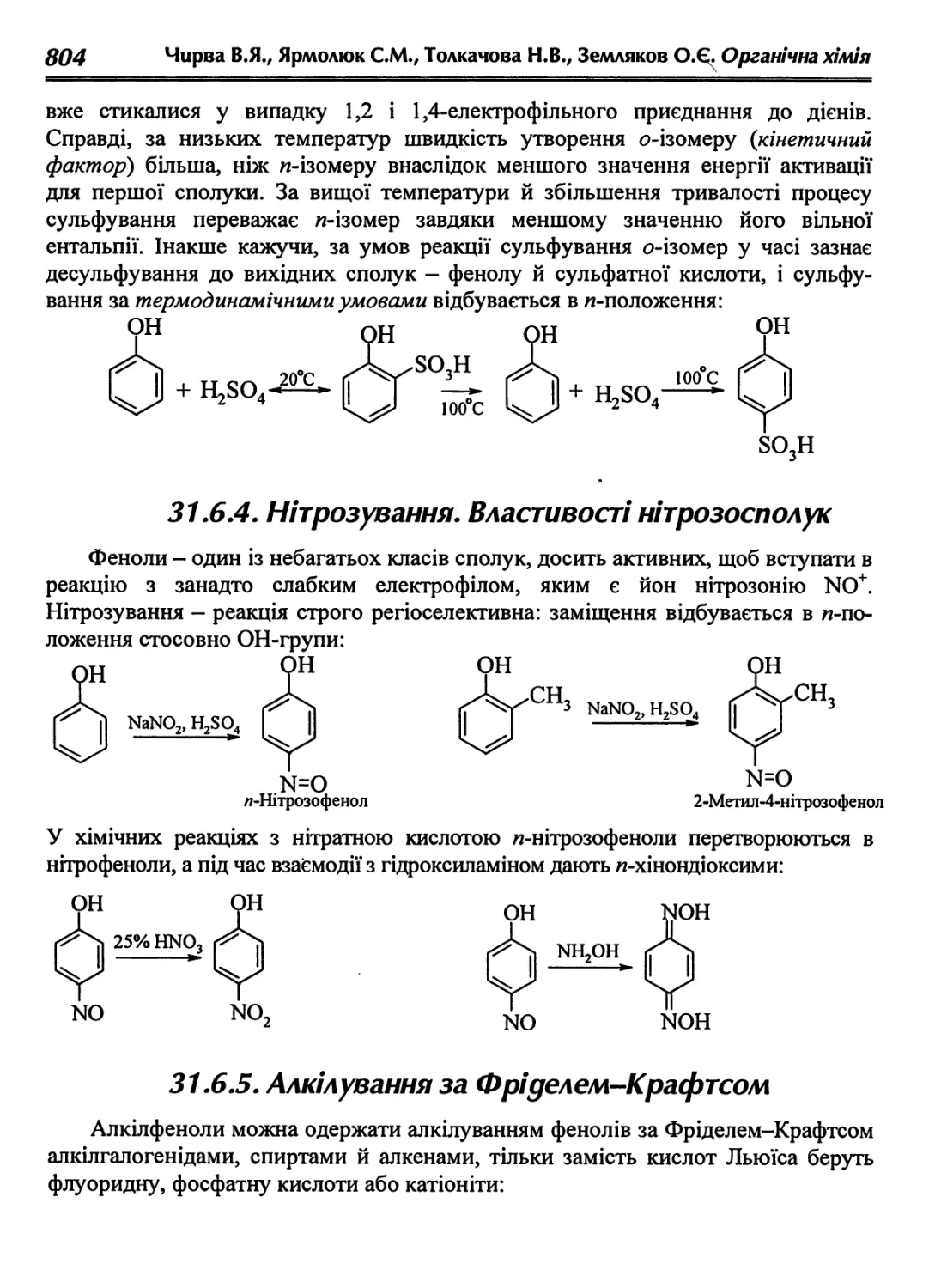
31.6.4. Нітрозування. Властивості нітрозосполук 804
31.6.5. Алкілування за Фріделем-Крафтсом 804
31.6.6. Ацилювання за Фріделем-Крафтсом 805
31.6.7. Карбоксилювання. Реакція Кольбе 806
31.6.8. Синтез фенолоальдегідів. Реакція Реймера-Тіммана 807
31.6.9. Взаємодія фенолу з карбонільними сполуками 807
31.6.10. Інші реакції фенолу. 810
31.7. Двохатомні феноли 810
31.7.1. Пірокатехол 810
31.7.2. Резорцинол 812
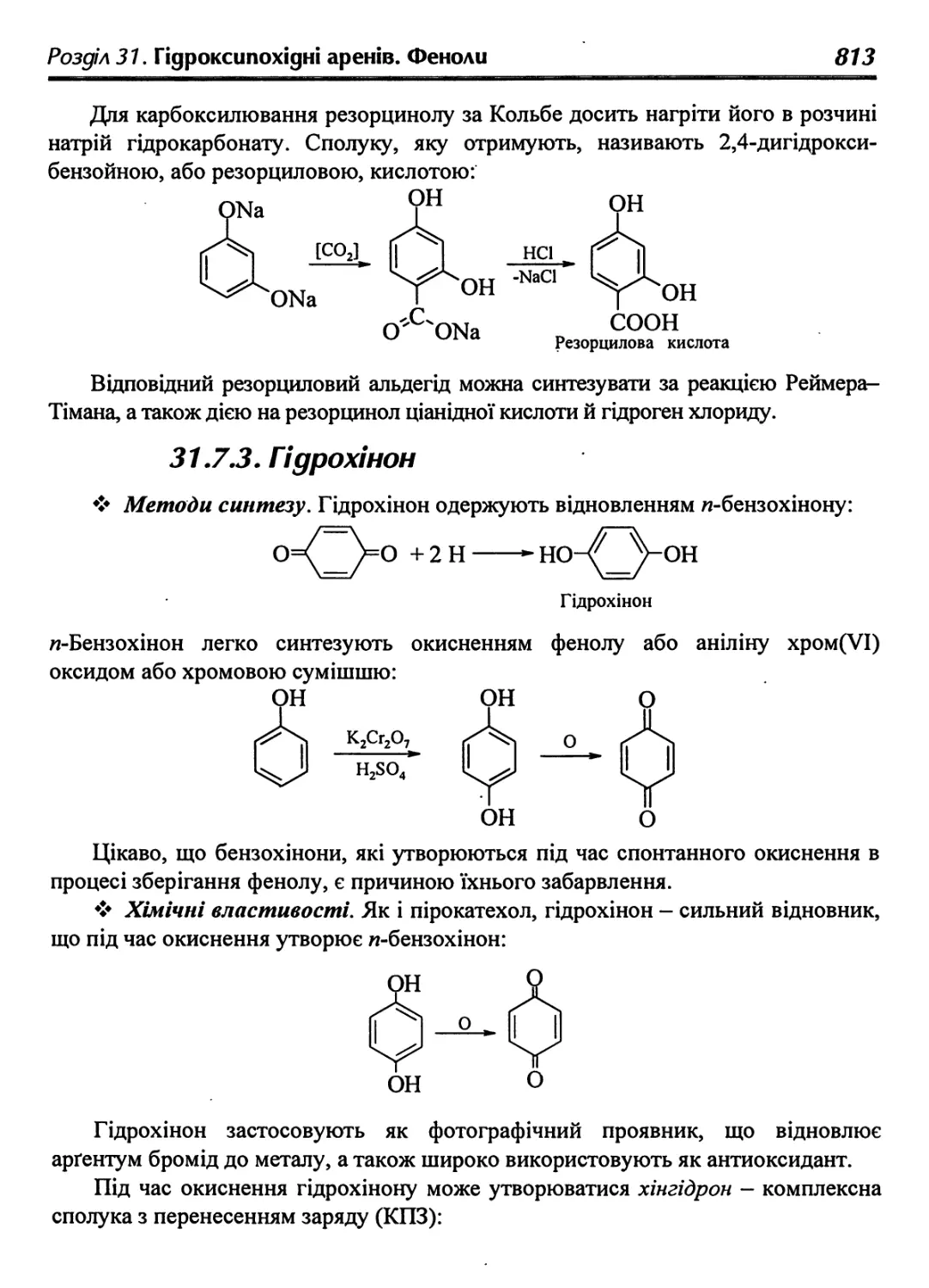
31.7.3. Гідрохінон 813
31.8. Багатоатомні феноли 814
31 9. Ароматичні спирти 815
Розділ 32. APOMA ТИЧНІОКСОСПОЛУКИ 818
32 і. Класифікація й номенклатура ароматичних оксосполук 819
32.1.1. Класифікація 819
32.1.2. Номенклатура 1UPAC 819
32 2. Фізичні властивості 820
32.2.7. Спектроскопія ароматичних оксосполук 820
32 3. Синтез ароматичних альдегідів і кетонів 821
32.3.1. Реакції перефункціоналізацїі 821
32.3.2. Уведення оксогрупи в ароматичне кільце 823
32 4. Властивості ароматичних альдегідів 825
32.4.1. Реакції окиснення 825
32.4.2. Реакції заміщення 827
32.4.3. Реакції конденсації 828
32.4.4. РеащіяКанніццаро (диспропорціювання) 831
16
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
32.5. Властивості ароматичних кетонів 832
32.5.1. Реакції конденсації • 832
32.5.2. Інші реакції арилкетонів 834
32.6. Застосування ароматичних кетонів 837
Розділ 33. АРОМАТИЧНІ КАРБОНОВІ КИСЛОТИ 838
33.1. Класифікація й номенклатура 838
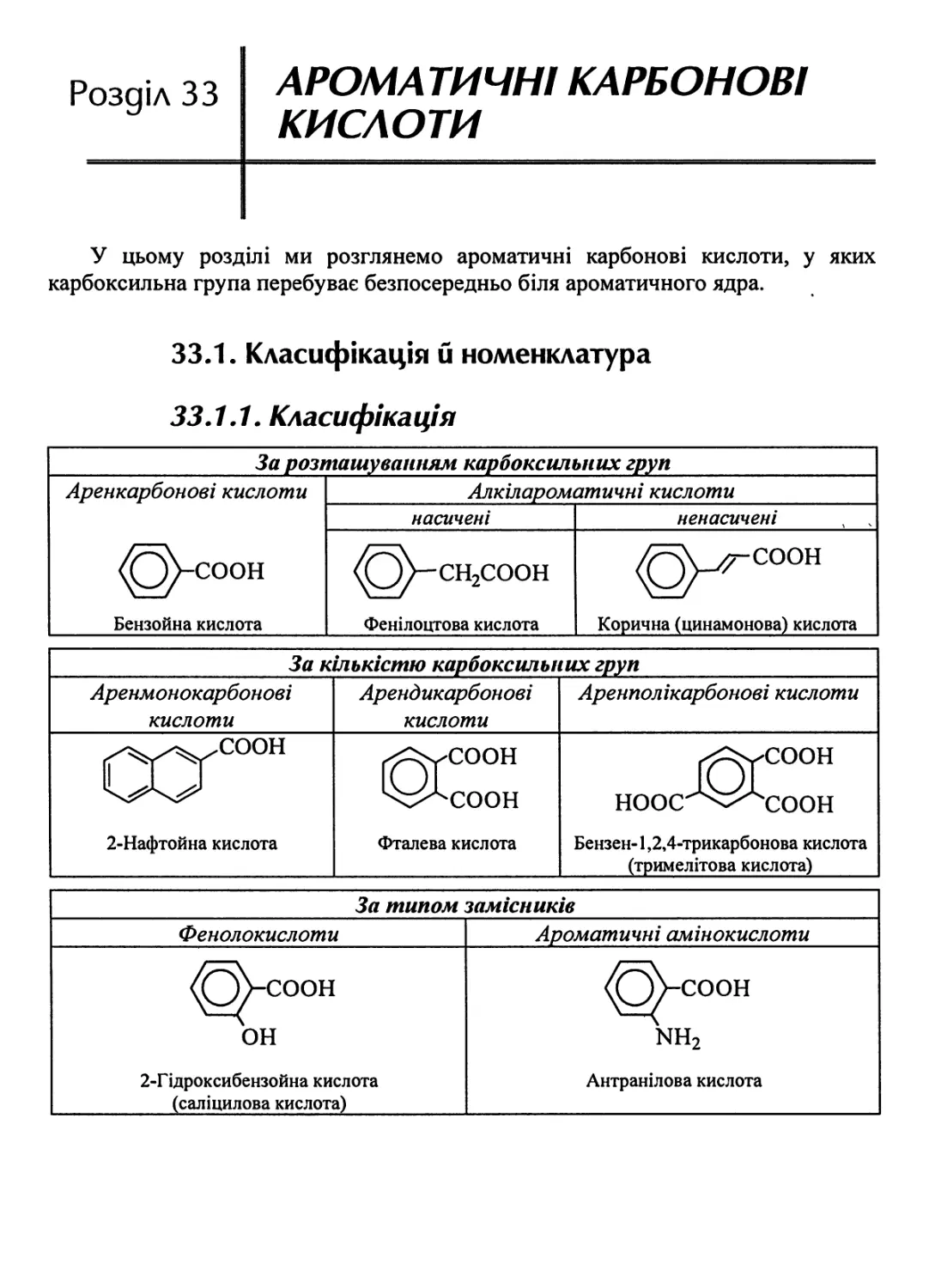
33.1.1. Класифікація . 83 8
33.1.2. Будова аренкарбонових кислот , 839
33.1.3. Номенклатура IUPАС 839
33.2. Фізичні властивості 841
33.2.1. Спектроскопія аренкарбонових кислот 841
33.3. Способи одержання аренкарбонових кислот 841
33.3.1. Реакції окиснення 841
33.3.2. Синтез за допомогою металоорганічних сполук 842
33.3.3. Ацилювання за Фріделем-Крафтсом 843
33.3.4. Гідроліз нітрилів або трихлорометиларенів 843
33.4. Бензойна кислота і її функціональні похідні 844
33.5. Ароматичні дикарбонові кислоти 847
33.5.1. Фталева кислота і її похідні 847
33.5.2. Терефталева кислота і її похідні 850
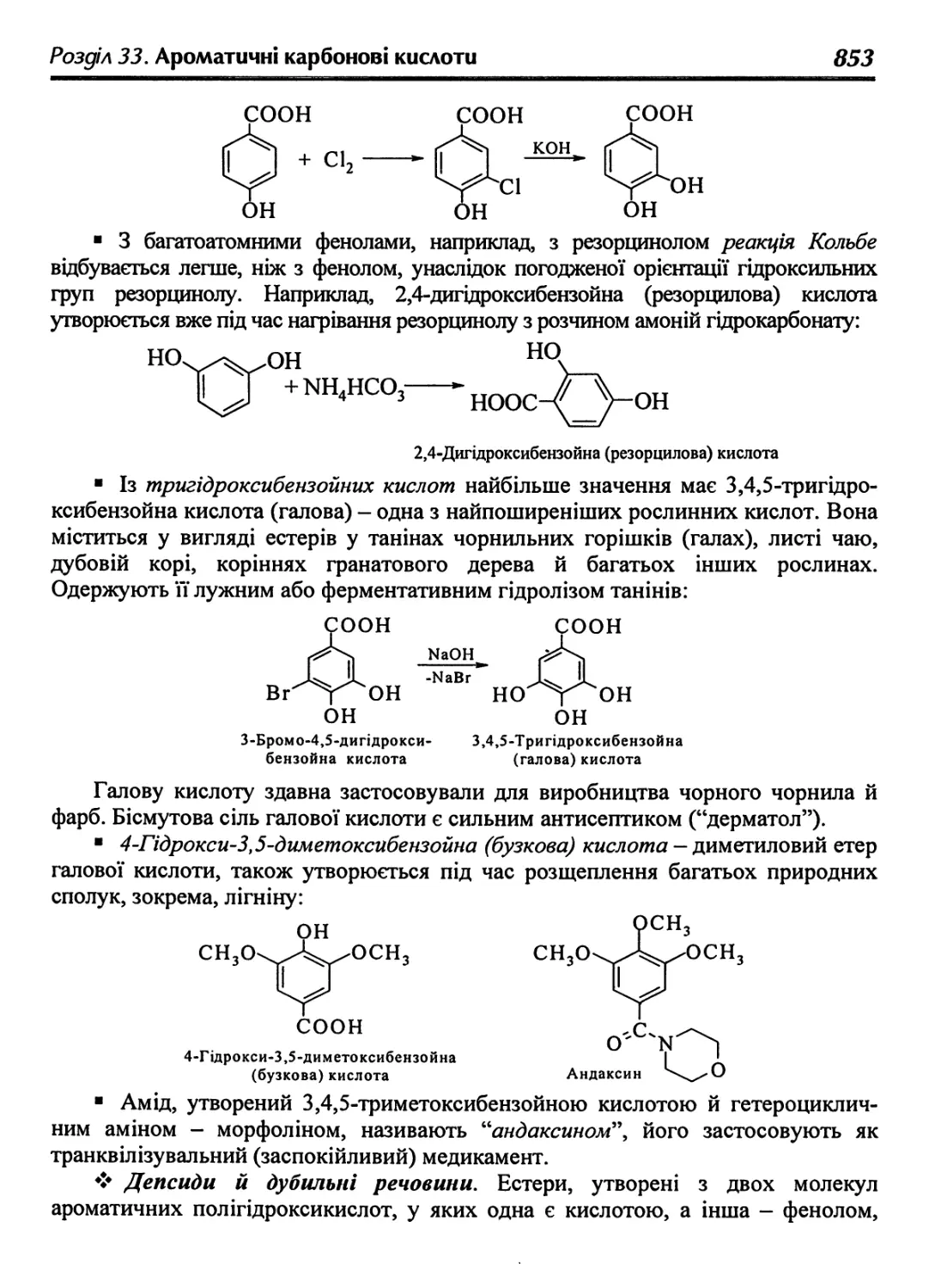
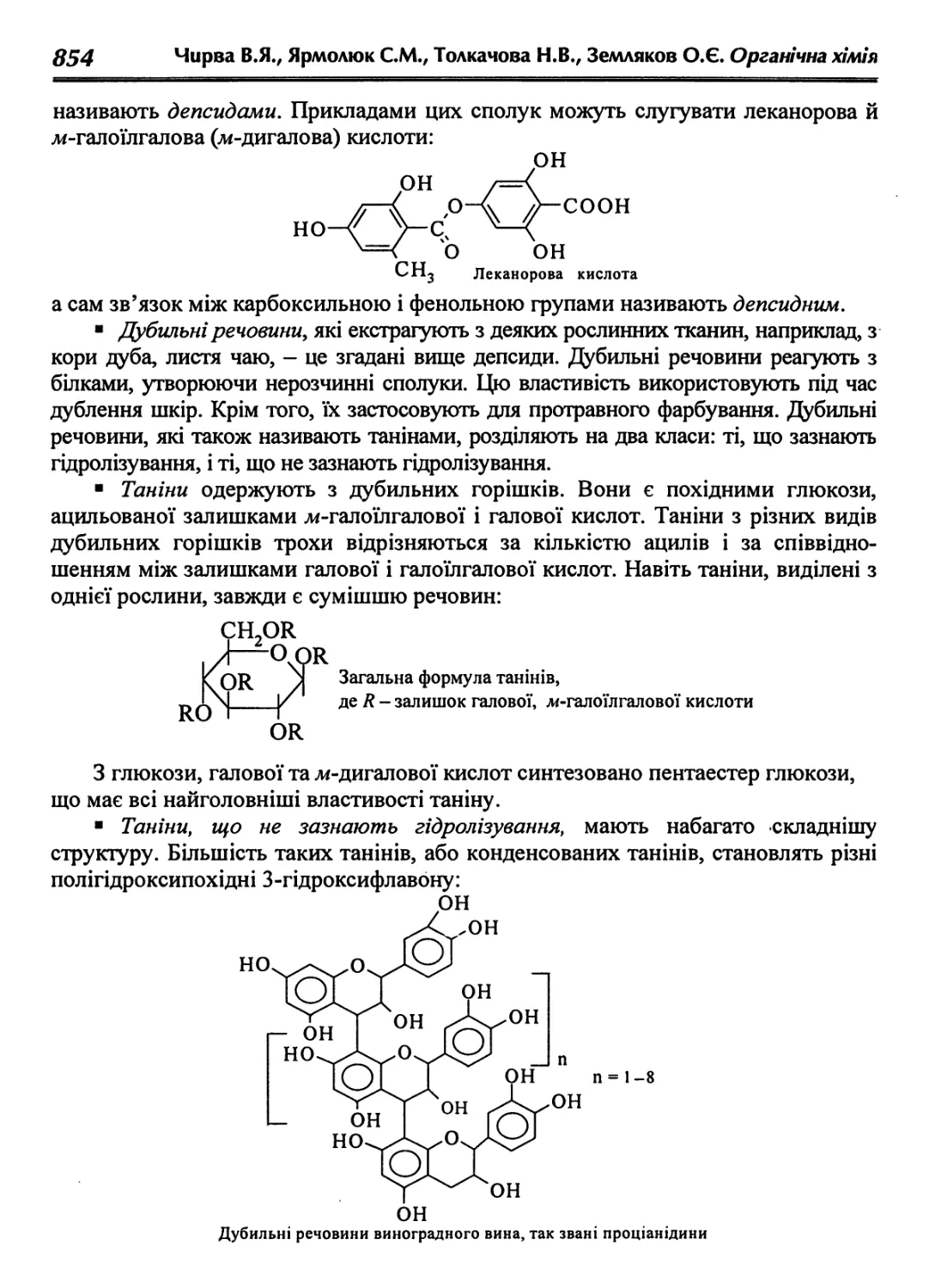
33.6. Гідроксибензойні кислоти (фенолокислоти) 850
33.7. Ароматичні амінокислоти 855
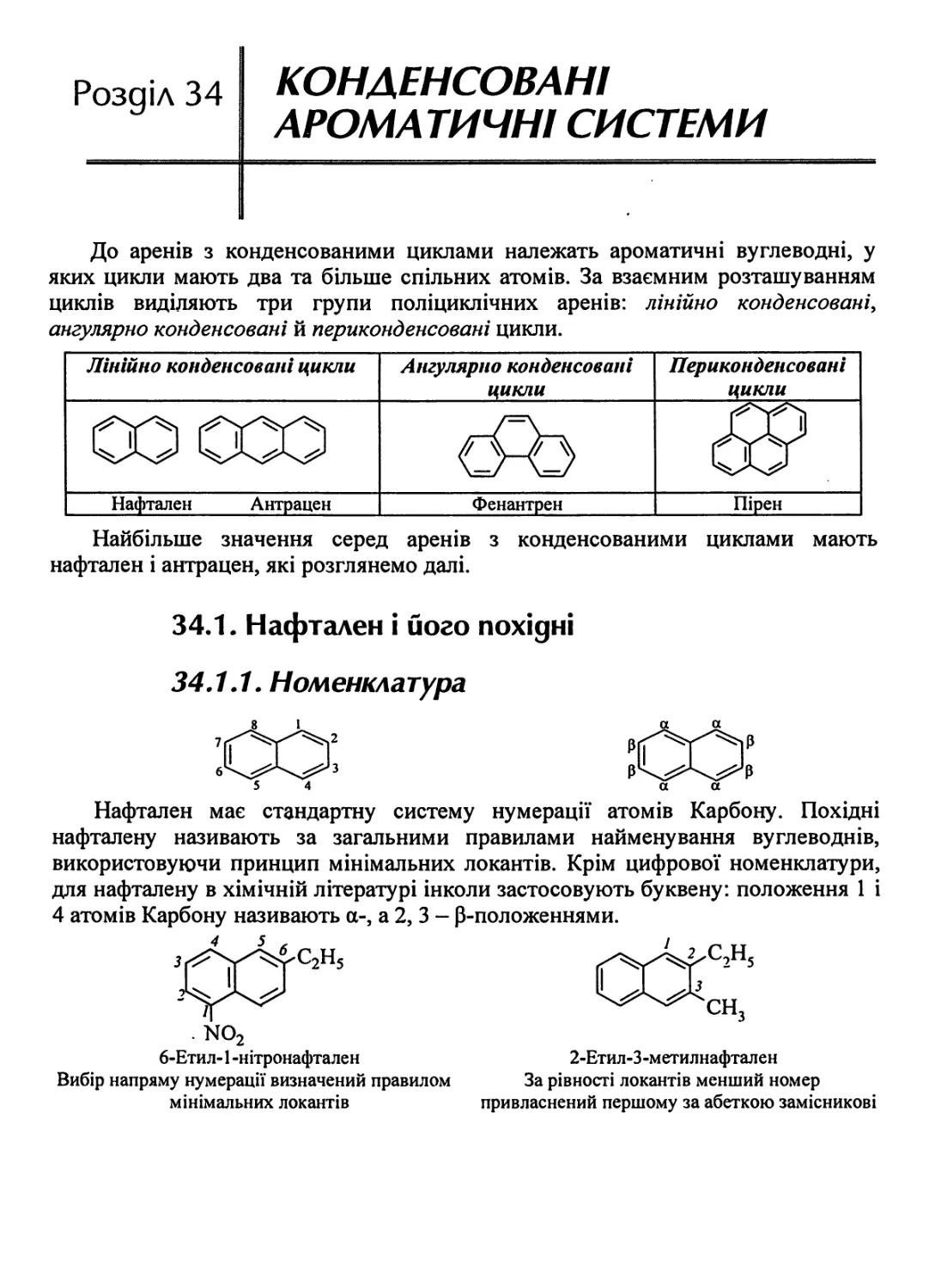
Розділ 34. КОНДЕНСОВАНІ АРОМАТИЧНІ СИСТЕМИ 857
34.1. Нафтален і його похідні 857
34.1.1. Номенклатура 857
34.1.2. Будова нафталену 858
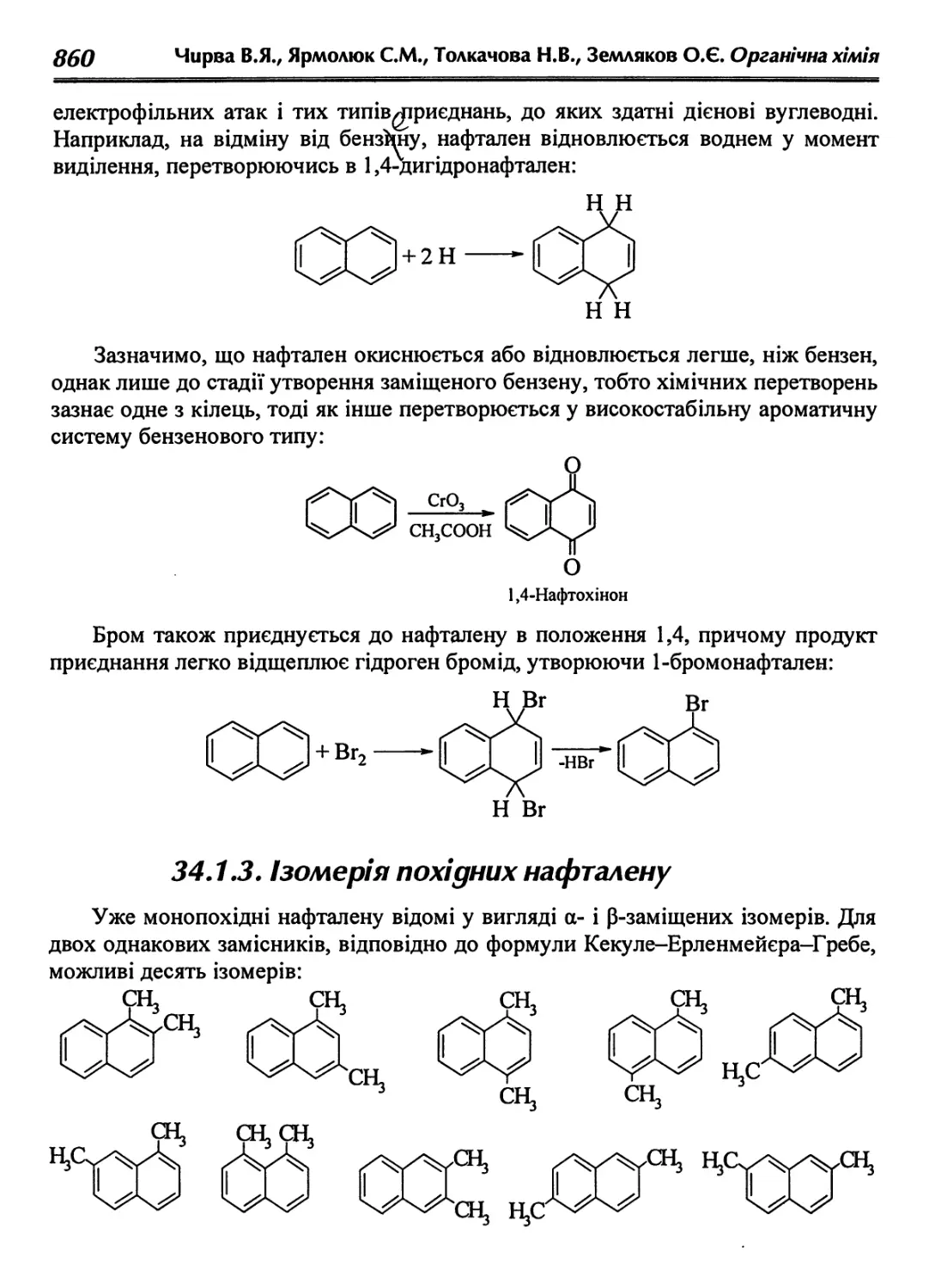
34.1.3. Ізомерія похідних нафталену 860
34.1.4. Фізичні властивості нафталену 861
34.1.5. Синтез нафталену і його похідних 861
34.1.6. Реакції електрофільного заміщення 862
34.1.7. Окиснення нафталену 866
34.1.8. Гідронафталени 867
34.1.9. Нафтоли 869
34.1.10. Орієнтація електрофільного заміщення в похідних нафталену 870
34.2. Антрацен і його похідні 872
34.2.1. Номенклатура 872
34.2.2. Будова антрацену 872
34.2.3. Фізичні властивості антрацену 873
34.2.4. Синтез антрацену 873
34.2.5. Хімічні властивості антрацену 874
34.2.6. Антрахінон і його похідні 877
Розділ 35. ПОЛЩИКЛІЧНІАРОМАТИЧНІ СИСТЕМИ 882
J ІЗОЛЬОВАНИМИ ЦИКЛАМИ
35.1.Біфеніл 882
35.1.1. Номенклатура IUPАС 882
Зміст /7
35.1.2. Будова біфенілу... 882
55.7.5. Ізомерія 883
35.1.4. Одержання біфенілу і його похідних 883
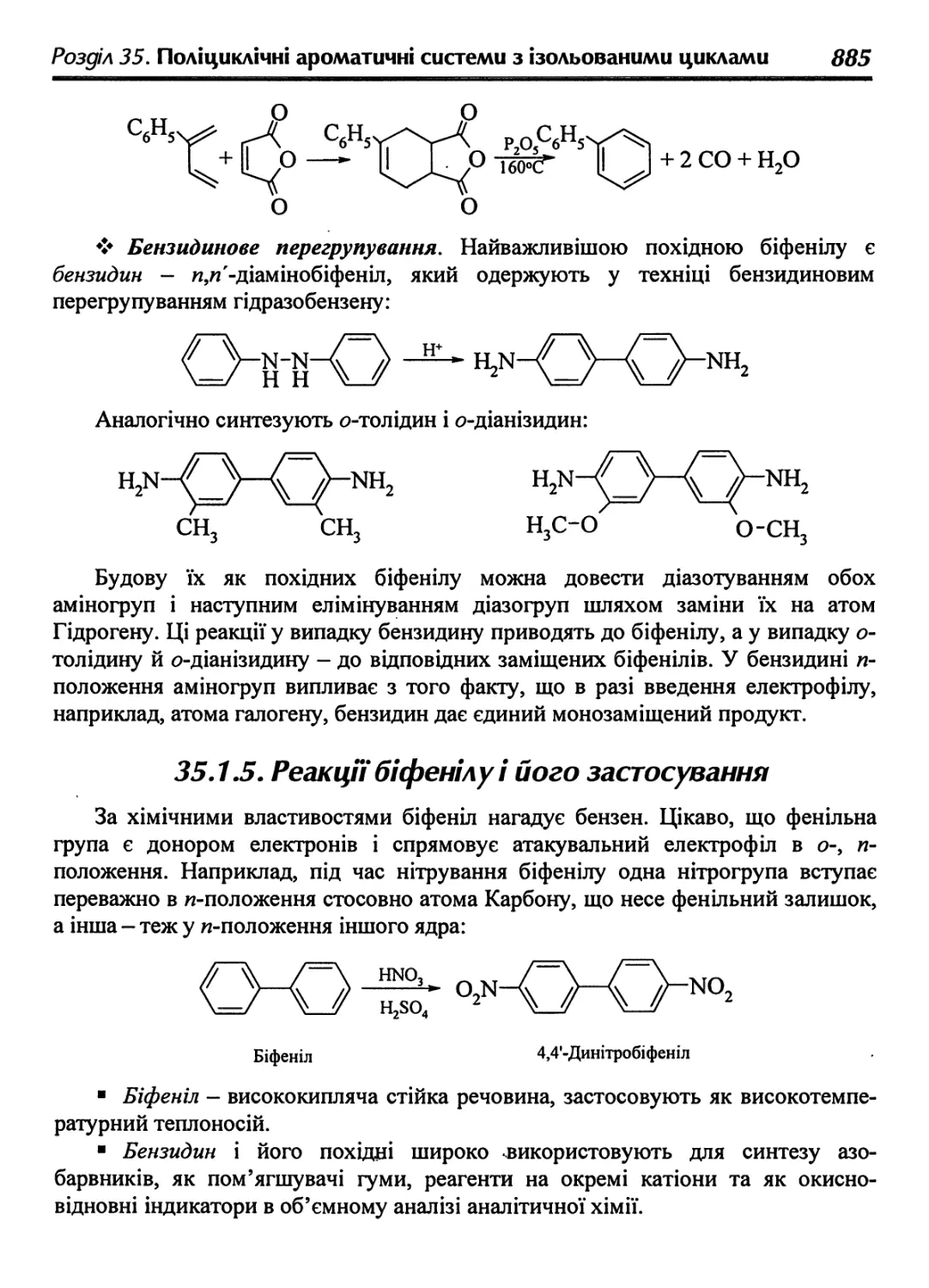
35.1.5. Реакції біфенілу і його застосування 885
35.2. Дифенілметан і його похідні 886
35.3. Трифенілметан 887
55.5.7. Синтез трифенілметану 888
35.3.2. Трифенілметиланіон 889
35.3.3. Трифенілметил-катіон 890
35.3.4. Трифенілметанові барвники • 892
35.3.5. Трифенілметилрадикал 898
35.3.6. Фталеїни, флуоресцеїн, розаміний родаміни 898
Розділ 36. ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ. П'ЯТИЧЛЕННІ 900
ЦИКЛИ З ОДНИМ ГЕТЕРОАТОМОМ
36.1. Класифікація й номенклатура гетероциклів 900
36.1.1. Класифікація 900
36.1.2. Номенклатура 901
36.2. Будова п'ятичленних гетероциклів з одним гетероатомом 902
36.3. Фізичні властивості 903
36.3.1. Спектроскопія ароматичних гетероциклів 903
36.4. Загальні способи синтезу п'ятичленних гетероциклів з одним гетероатомом... 904
36.5.Фуран . • 906
36.5.1. Синтез фурану та його похідних • 906
36.5.2. Хімічні властивості фурану ■- •.. 907
Зб.б.Тіофен 912
36.6.1. Синтез тіофену і його похідних 912
36.6.2. Хімічні властивості тіофену 913
36.7. Пірол 917
36.7.7. Синтез піролу і його похідних 918
36.7.2. Хімічні властивості піролу 919
36.7.3. Порфін і його похідні 924
Розділ 37. НІТРОГЕНОВМІСНІ КОНДЕНСОВАНІ СПОЛУКИ З ДВОМА 928
АТОМАМИ НІТРОГЕНУ. П'ЯТИЧЛЕННІ ГЕТЕРОЦИКЛИ
37.1. Індол 928
57.7.7. Фізичні властивості індолу 928
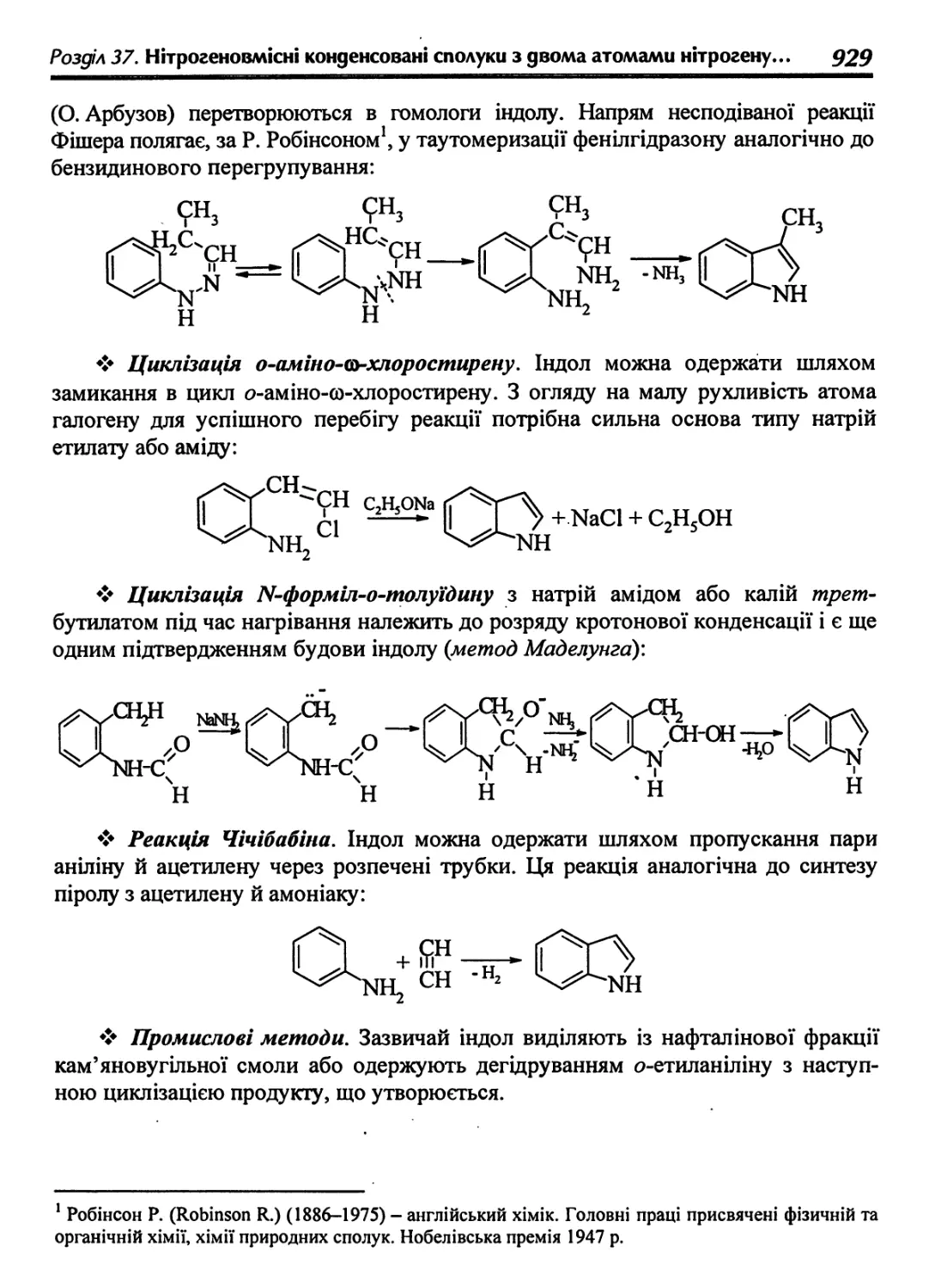
37.1.2. Методи одержання 928
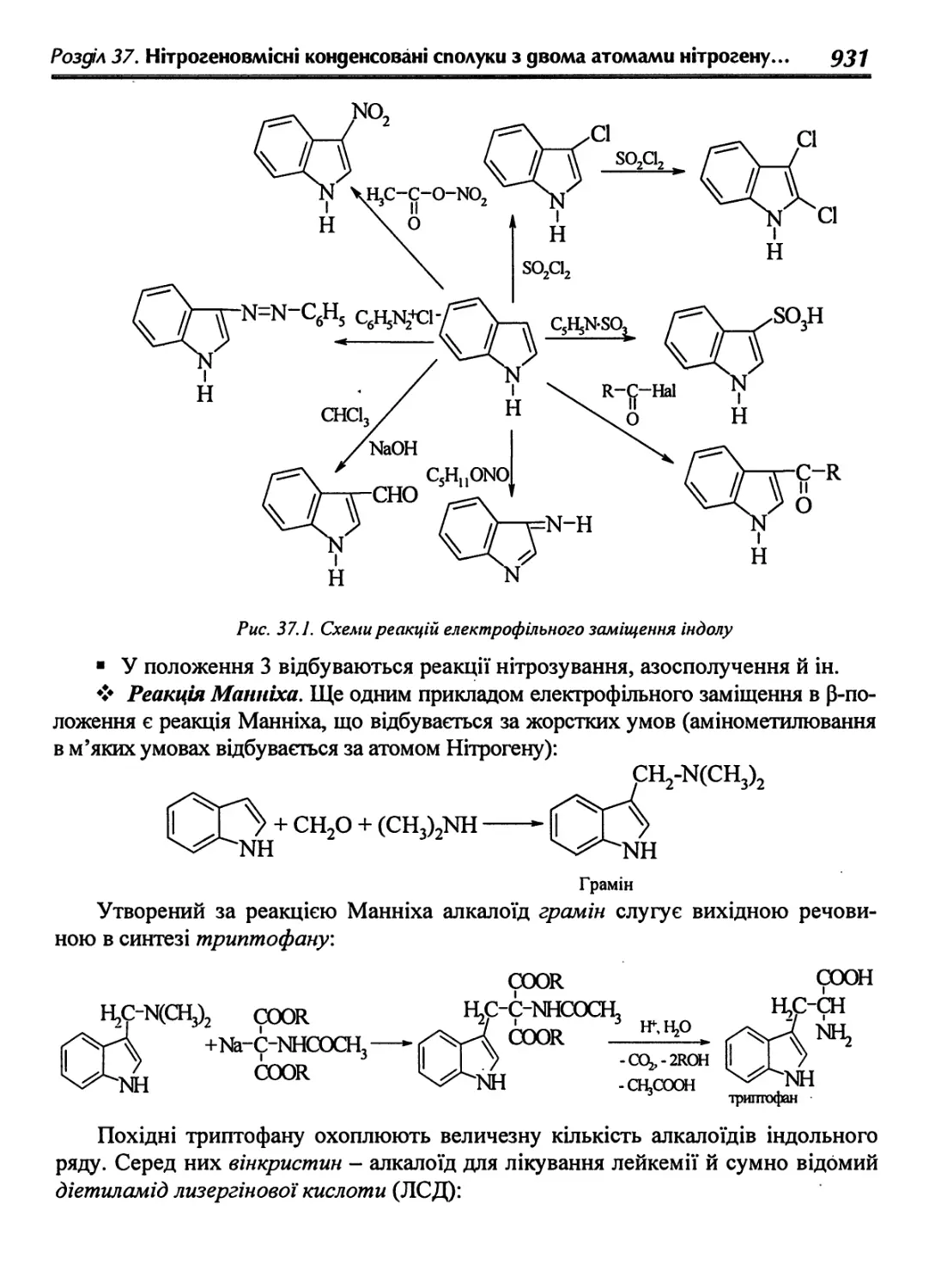
37.1.3. Хімічні властивості індолу 930
37.1.4. Застосування індолу і його похідних 932
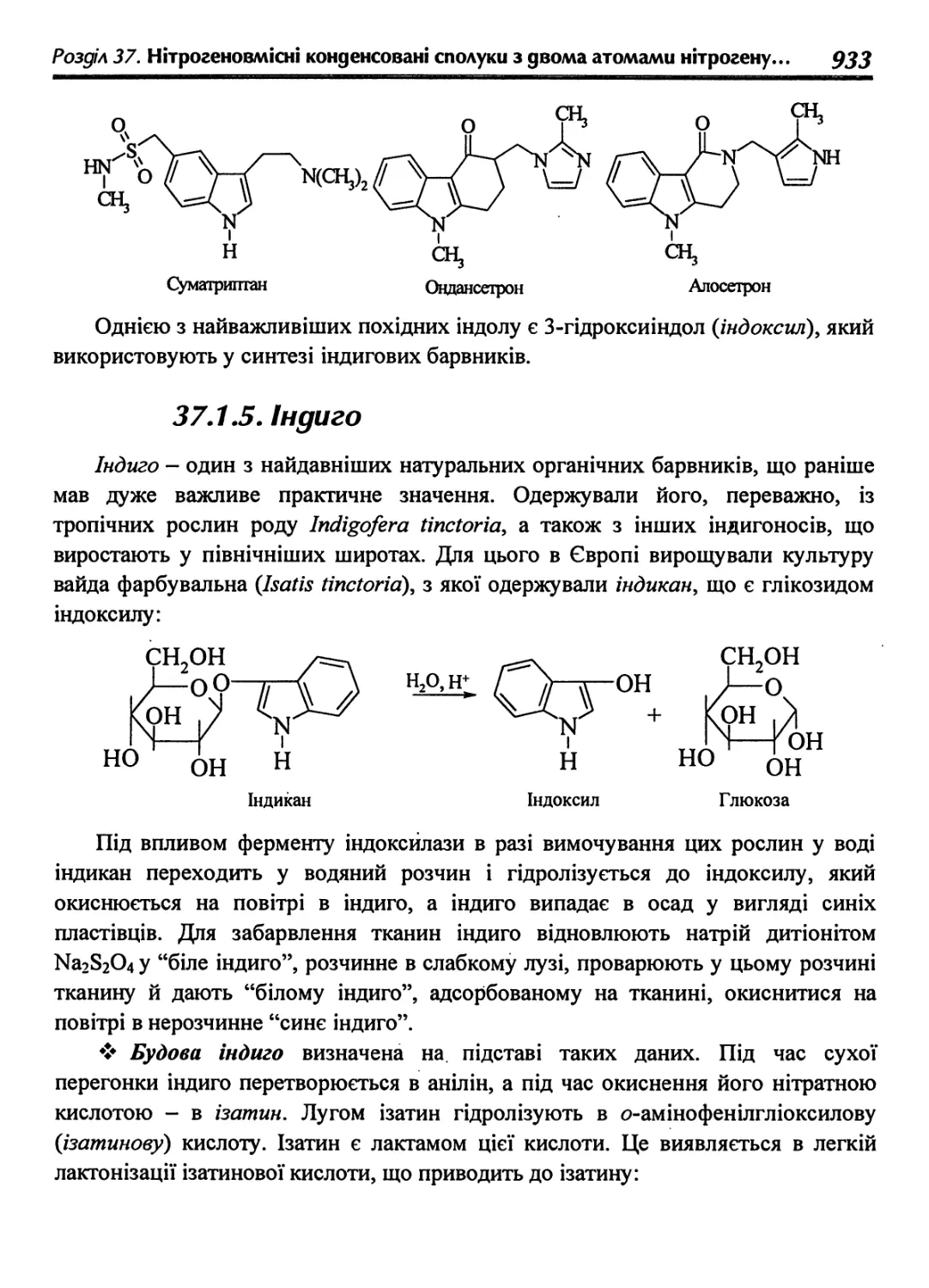
37.1.5. Індиго • 933
37.2. Карбазол 937
37.3. Піразол 938
37.3.1. Фізичні властивості піразолу 938
37.3.2. Методи одержання 938
37.3.3. Хімічні властивості 938
37.4. Імідазол 940
18
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Розділ 38. ШЕСТИЧЛЕННІ НІТРОГЕНОВМІСНІГЕТЕРОЦИКЛИ 942
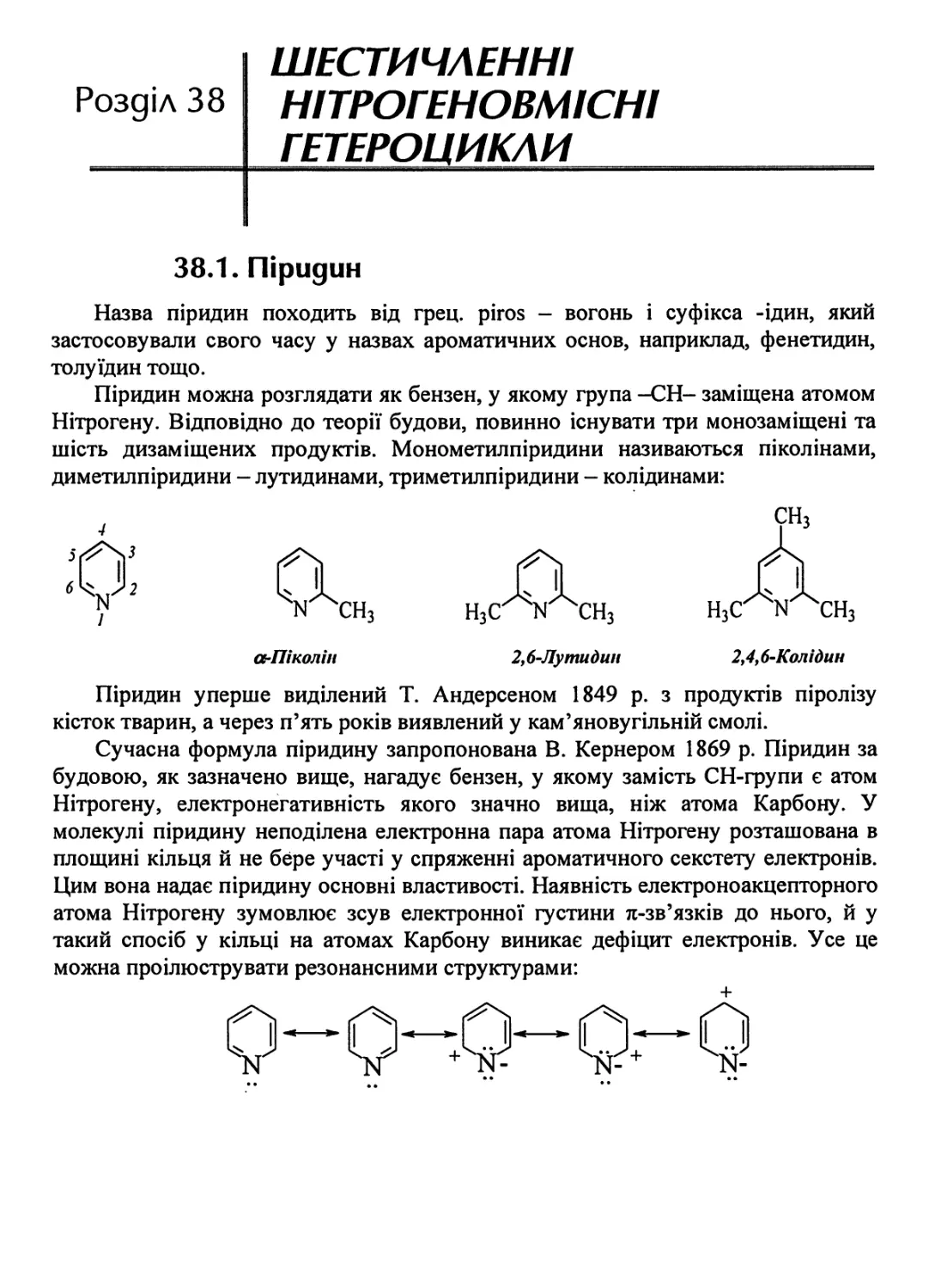
38.1. Піридин 942
38.1.1. Визначення будови піридину 943
38.1.2. Фізичні властивості піридину 944
38.1.3. Одержання піридину і його гомологів 944
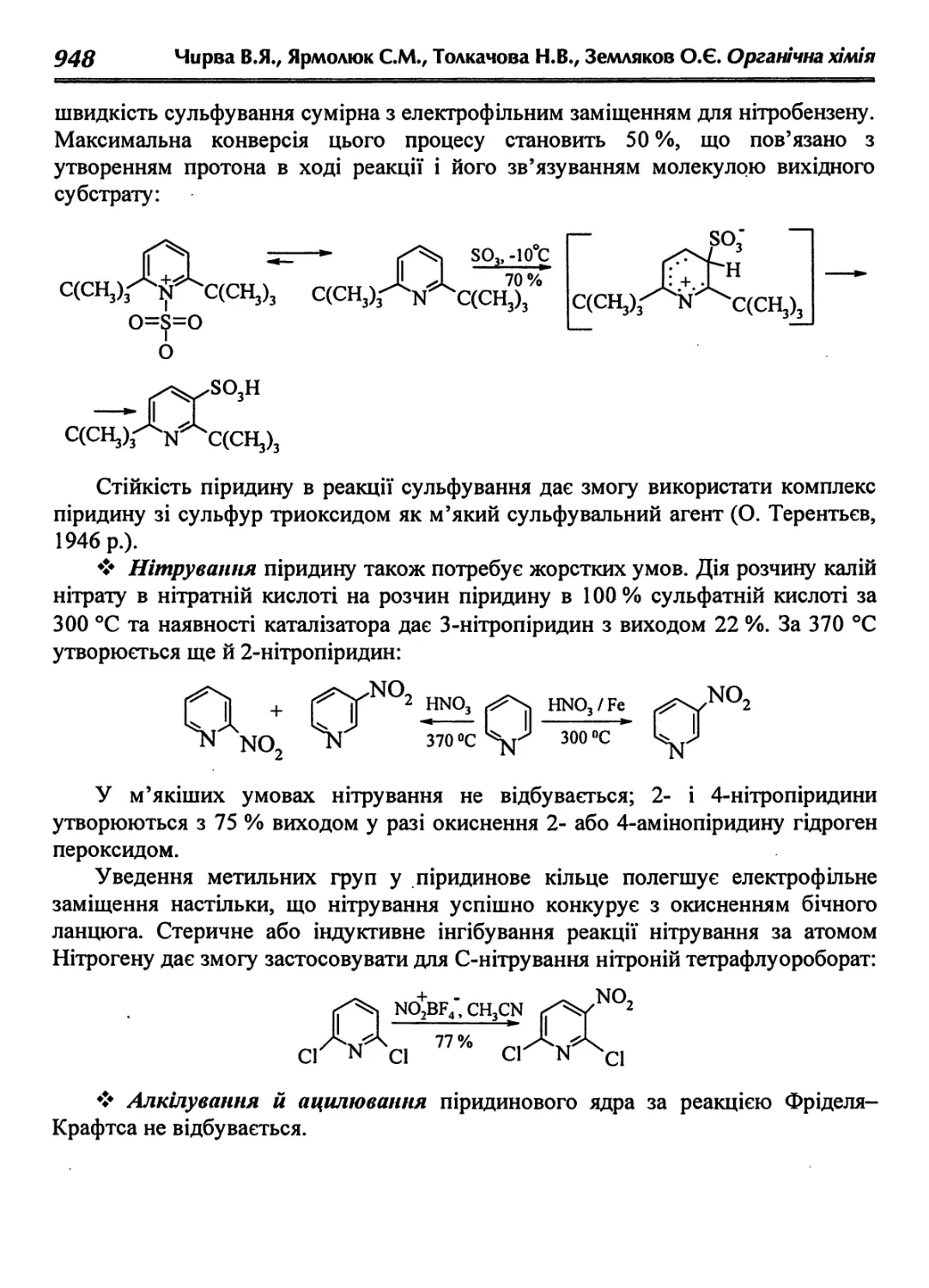
38.1.4. Реакції електрофільного заміщення 946
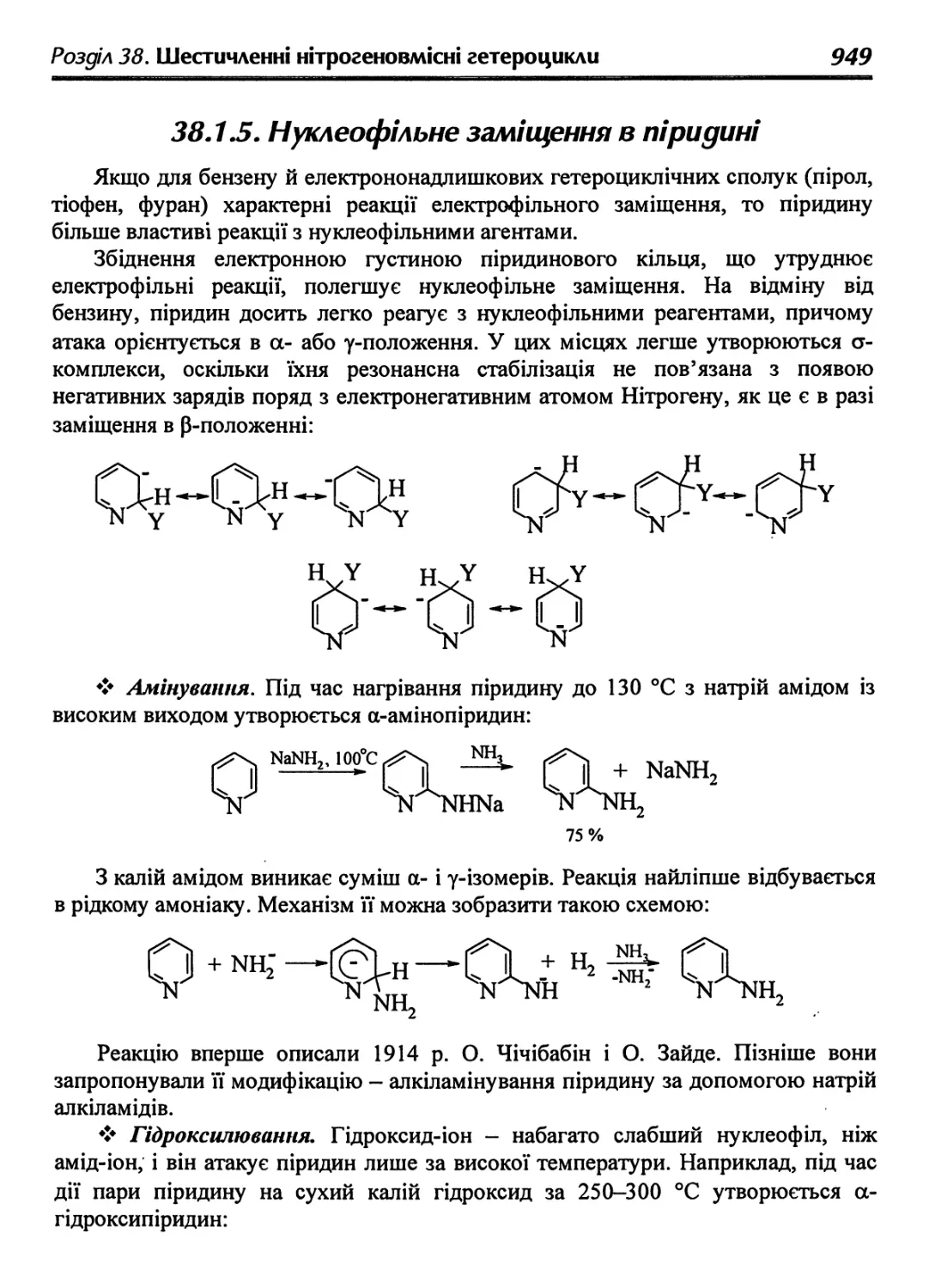
38.1.5. Нуклеофільне заміщення в піридині 949
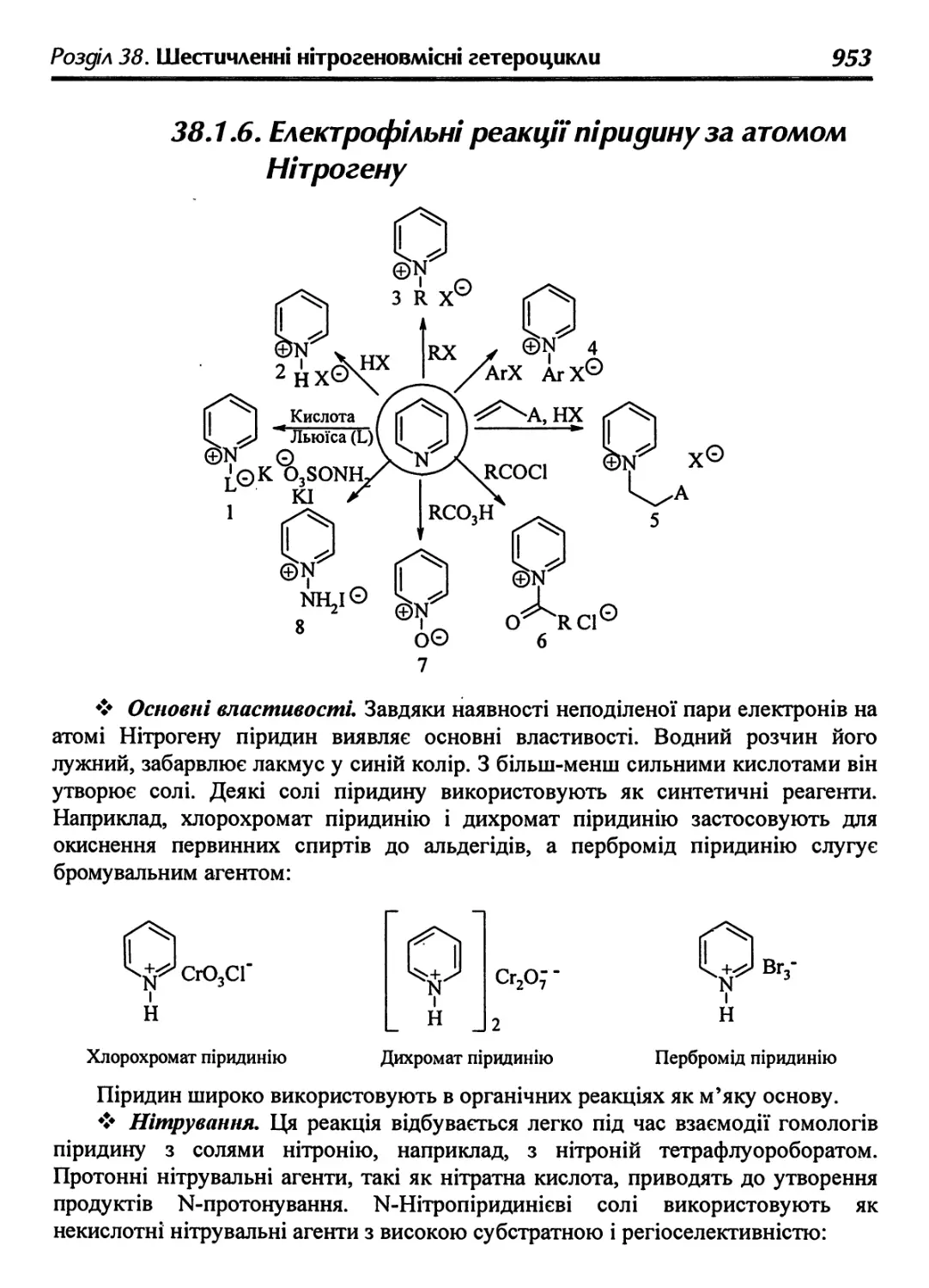
38.1.6. Електрофільні реакції піридину за атомом Нітрогену 953
38.1.7. Інші реакції піридину 956
38.1.8. Фізіологічно активні речовини з піридиновим ядром 958
38.2. Хінолін та ізохінолін 960
38.2.1. Фізичні властивості хіноліну 960
38.2.2. Одержання хіноліну і його гомологів 961
38.2.3. Хімічні властивості хіноліну 962
38.2.4. Похідні хіноліну 965
38.2.5. Ізохінолін 966
38.3. Піримідин 968
38.4. Пурин і його похідні 970
Іменний покажчик 972
Предметний покажчик 975
Список літератури 990
Вступ
Ще зовсім недавно органічна хімія посідала доволі скромне місце серед інших
хімічних дисциплін і її роль зводилася до опису властивостей низькомолекулярних
органічних сполук та їхнього виробництва. Сьогодні органічна хімія є однією з
важливих природничих наук, теоретичні дослідження та практичні результати якої
використовують в усіх сферах діяльності людини. Досягнення органічної хімії
застосовують у виробництві штучних волокон, барвників пластмас, вітамінів, ліків,
мийних засобів, під час переробки газу й нафти. Навколишній світ утворено
переважно з органічних речовин. Сюди насамперед належать білки, вуглеводи,
нуклеїнові кислоти - основні компоненти рослинних і тваринних клітин.
Дослідження їхньої будови дає потужний засіб для лікування багатьох хвороб
живих організмів.
У пропонованому підручнику збережено класичну побудову фактичного ма-
теріалу з поділом на аліфатичні й ароматичні сполуки. Вивчення алканів дає змогу
ознайомитися з реакціями радикального заміщення, а під час розгляду алкенів,
алкінів та дієнів студент вивчає основну властивість ненасичених сполук -
електрофільне приєднання і, насамкінець, на прикладі ароматичних речовин він
засвоює реакції електрофільного заміщення в ароматичному ряду.
Ми вважали за доцільне окремо подати матеріал про оксо- і нітросполуки, аміни
і карбонові кислоти аліфатичного й ароматичного рядів. У випадку ароматичних
сполук розглянуто особливості їхнього синтезу і хімічних властивостей порівняно з
аліфатичними.
На відміну від інших підручників, достатньо коротко наведено відомості з
основ теорії електронної будови органічних молекул (квантово-механічна теорія
будови атома, хімічний зв'язок тощо), оскільки ці питання обговорюють в
університетських курсах загальної хімії, а також будови молекул, квантової хімії.
З огляду на це аналогічний прийом використано у висвітленні питань,
пов'язаних зі стереохімією органічних сполук і фізичних методів.
У підручнику широко застосовано написання резонансних (канонічних)
формул, особливо у випадку гетероциклічних ароматичних сполук. Незважаючи на
певну громіздкість написання цих формул, вони допомагають студентові з'ясувати
хімічні властивості таких сполук і, що дуже важливо, напрям реакцій.
Класифікація.та особливо номенклатура величезного фактичного матеріалу
органічної хімії є складним завданням. Оскільки нинішня номенклатура в
російських підручниках далеко не досконала, то ми, висвітлюючи це питання,
20
Чирва В.Я., Ярмолкж С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
керувалися останніми рекомендаціями IUP АС та Української національної комісії з
хімічної термінології і номенклатури (УНКоХіТерм). Згідно з цими рекомендаціями
упорядковано правила IUP АС на українському мовному ґрунті. Реалізовано деякі
пропозиції щодо назв органічних сполук для уніфікації назв, особливо аренів та
їхніх похідних.
Також у підручнику висвітлено найпоширеніші лабораторні та промислові
методи одержання основних класів органічних сполук, зазначено галузі їхнього
застосування, розглянуто деякі питання їхнього впливу на довкілля.
Підручник "Органічна хімія" є базовим для подальшого засвоєння основ
біохімії, біоорганічної хімії, різних спеціальних курсів органічного профілю. Його
написано з урахуванням досвіду багаторічного викладання цієї дисципліни на
кафедрі органічної та біологічної хімії Таврійського національного університету
ім. В.І. Вернадського. Крім того, ретельно опрацьовано підручники з хімії, які
використовують в університетах США, Швейцарії, Німеччини, Англії, Франції, Росії
та інших країн.
Підручник розраховано на студентів хімічних спеціальностей класичних і
технічних університетів. Його можуть використовувати аспіранти хімічного і
біохімічного профілю.
Автори вдячні рецензентам - академіку HAH України М.О. Лозинському, член-
кор. HAH України В.П. Хилі, проф. В.П. Новікову за ретельний розгляд рукопису та
цінні зауваження, які сприяли поліпшенню якості підручника. Висловлюємо подяку
проф. М.Ю. Корнілову, доцентам кафедри органічної та біологічної хімії Таврійського
національного університету ім. В.І. Вернадського С.С. Пертелю, Д.П. Толстенку,
В.О. Кур'янову, а також доценту кафедри органічної хімії Національного універси-
тету "Львівська політехніка" В.А. Дончак. Окрема вдячність доценту кафедри культу-
ри української мови Таврійського національного університету ім. В.І. Вернадського
О.Ф. Шаталіній, а також Л.А. Єлісєєвій, В.А. Щербаковій, CO. Землякову, М.М. Марти-
няк за допомогу в підготовці рукопису.
Особливу вдячність автори висловлюють Науково-сервісній фірмі "ОТАВА"
(www.otavacheniicals.com), за фінансової підтримки якої цей підручник побачив світ.
Розділ 1
ПРЕАМЕТ
ОРГАНІЧНОЇ ХІМІЇ
Головним об'єктом вивчення хімії є хімічні сполуки та їхнє перетворення.
Спочатку органічну хімію визначали за Й. Берцеліусом1, який дав їй цю назву як
хімії сполук, що утворюються за допомогою живої природи. І хоч незабаром
віталістичні погляди були спростовані працями німецького хіміка Ф. Велера2,
термін "органічна" зберігся з тієї причини, що хімія сполук Карбону важливіша
для життя, ніж хімія будь-якого іншого елемента Періодичної системи. Достатньо
побіжного переліку головних видів органічних сполук, що є в нашому вжитку,
щоб переконатися у правомірності цієї назви: вітаміни, білки, цукри, жири,
антибіотики, полімери, пахучі речовини тощо.
Органічна хімія вивчає сполуки Карбону з іншими елементами, такими як
Гідроген, Нітроген, Оксиген, Сульфур, Фосфор і галогени. Ці елементи
називають органогенами. Ще в 30-х роках XIX ст. Л. Гмелін3 визначив органічну
хімію як хімію сполук Карбону. З огляду на це постає питання: чому з усієї сотні
відомих елементів саме Карбон посідає таке важливе місце, що його сполуки
виділяють у самостійну наукову дисципліну?
Зазначимо, що кількість відомих сьогодні сполук Карбону майже в 10-20
разів більша, ніж кількість сполук усіх інших елементів, утворених не Карбо-
ном. Однак навіть понад 10 мільйонів вивчених нині органічних сполук ніяк не
вичерпують безмежних можливостей конструювання органічних молекул. Чим
же зумовлена велика кількість цих сполук? Таких причин декілька. Надзвичайно
важливе значення в органічній хімії має відкрите 1830 р. Ю Лібіхом4 явище
ізомерії, загальне для всієї хімії, але таке, що набуло надзвичайного поширення
в органічній 'хімії. Його зміст полягає в тому, що може існувати декілька
відмінних одна від одної сполук, які мають однаковий елементний склад і
молекулярну масу, але відрізняються будовою молекули.
1 Берцеліус Й. (ВеггеІшБ ].) (1779-1848) - шведський хімік, наукові дослідження якого охоплюють
головні проблеми хімії першої половини XIX ст.
2 Велер Ф. (ЛУбЬІег Р.) (1800-1882) - німецький хімік, має праці з неорганічної та органічної хімії.
Поряд з Ю. Лібіхом виявив ізомерію солей гримучої кислоти.
3 Гмелін Л. (Отеїіп Ь.) (1788-1853) - німецький хімік, укладав довідники з експериментальних
даних, які декілька разів перевидавали.
4 Лібіх Ю. (Ьіеі^ У.) (1803-1873) - німецький хімік, засновник теорії радикалів і агрохімії.
Досліджував органічні кислоти.
22
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
В органічній хімії навіть у речовинах найпростішого складу, утворених
тільки з Карбону та Гідрогену, явище ізомерії зумовлює існування великої
кількості різних хімічних індивідуумів. Наприклад, для нонану С9Н20 існує 35
ізомерів, вуглеводню зі складом С20Н42 повинно відповідати 366 319 різних
ізомерів, а триаконтан С30Н62 утворює 4 111 846 768 вуглеводнів, які ізомерні
один одному. Будь-який з ізомерів можна синтезувати. Кількість ізомерів для
вуглеводню визначити досить просто, якщо відома кількість ізомерів для
попередника. Явище ізомерії вдало поясняють теорією хімічної будови,
розробленою у 60-х роках XIX ст. О. Бутлеровим1.
Другим чинником, що забезпечує величезну кількість органічних сполук, є
виявлене Ш. Жераром2 явище гомології. Це явище полягає в існуванні хімічно
подібних між собою рядів речовин, склад послідовних членів яких відрізняється
один від одного на групу СН2 - гомологічну різницю.
Третій чинник - це існування ізологічних рядів сполук, тобто речовин,
побудованих з однакової кількості вуглецевих атомів, що відрізняються між
собою ступенем насиченості, наприклад, етан С2Нб, етилен С2Н4, ацетилен С2Н2.
Особливою властивістю атомів Карбону є їхня здатність до утворення
ковалентних зв 'язків не лише одного з одним і атомами Гідрогену, а й з такими
атомами, як Оксиген, Нітроген, Сульфур, Фосфор, галогени та ін. Карбон,
сполучаючись з цими елементами в різних комбінаціях, утворює надзвичайно
велику кількість різноманітних органічних речовин. Ця обставина дала змогу
К. Шорлеммеру3 визначити органічну хімію як хімію вуглеводнів та їхніх
похідних.
Зрозуміло, що лише кількість органічних речовин, хоча й величезна, не
могла стати основою для виділення їх у самостійну науку. Для органічних
сполук характерна низка специфічних властивостей.
■ Відносна нестійкість. Органічні сполуки за звичайної температури є
газами, рідинами або порівняно низькоплавкими речовинами. Неорганічні
сполуки, як звичайно, на відміну від органічних, не горять. Більша частина з них
або зовсім не плавиться, або плавиться за дуже високої температури. Органічні
речовини плавляться зазвичай до 300-400 °С, причому температура плавлення є
однією з характеристик сполуки і ступеня її чистоти.
■ Складність будови. "Архітектура" деяких органічних сполук, особливо
природних, дуже складна, а їхня молекулярна маса досягає багатьох тисяч і
навіть мільйонів атомних одиниць маси (а.о.м.). Розмірність а.о.м. становить
1/12 маси атома ізотопу Карбону 12С.
Бутлеров О. (1828-1886) - російський хімік, засновник теорії будови органічних сполук. Передбачив
ізомерію багатьох сполук.
2 Жерар Ш. (Gerhardt С.) (1816-1856) - французький хімік. Працював у Ю. Лібіха, слухав лекції
Ж. Дюма. Створив теорію типів. У Ш. Жерара навчалося багато російських хіміків.
3 Шорлеммер К. (Schorlemmer К.) (1834-1892) - німецький хімік-органік, який працював у галузі
алканів, автор праць з історії хімії.
Розділ 7. Предмет органічної хімії
23
■ Тривалість перебігу реакцій. Як відомо, реакції неорганічних сполук у
водних розчинах відбуваються дуже швидко, тому що здебільшого взаємодіють
протилежно заряджені йони, що зближуються під дією електростатичного при-
тягання. Органічні сполуки, що є неіонними, взаємодіють у часі досить
повільно. Це пов'язано з тим, що не кожне зіткнення молекул приводить до
реакції: потрібне зіткнення двох активованих молекул. Підвищення температури
збільшує енергію молекул і вірогідність взаємодії зростає. Каталізатори зни-
жують енергетичний бар'єр і цим також збільшують швидкість реакції.
Більшість цих реакцій є зворотною, і необхідно стежити за зміщенням рівноваги
у потрібний бік. Перебіг реакцій у часі пов'язаний, насамперед, з переважним
ковалентним типом зв'язків в органічних молекулах.
■ Різнонапрямленість хімічних реакцій. Реакції органічних сполук відбу-
ваються часто не в одному, а в декількох напрямах і приводять до суміші
продуктів. Ця обставина має подвійне значення: з одного боку, ускладнюється
виділення з суміші та знижується вихід потрібних речовин, з іншого, - керуючи
хімічною реакцією та змінюючи швидкості окремих напрямів, можна одержати
необхідний продукт з найбільшими виходами.
■ Високий рівень організації молекул. Особливе місце органічної хімії в
системі наук зумовлене ще й тим, що вона вивчає більш високоорганізовану
матерію, ніж неорганічна хімія, і тісно пов'язана з біологією: органічні речовини
з'явилися значно пізніше, ніж неорганічні, вони є носіями життєдіяльності
рослин і тварин.
На початковому етапі розвитку науки головним джерелом одержання
органічних речовин були природні ресурси. Наведемо коротку характеристику
цих джерел.
• Природний газ і попутний нафтовий газ мають різний склад і містять від
80 до 98 % метану, до 0,5-4,0 % етану, 1,5 % пропану, які можуть бути розділені
фракційною перегонкою за низької температури. На основі природного газу
синтезують етилен, ацетилен, бутадієн, ізопрен, хлоропохідні вуглеводнів. З ме-
тану одержують сажу, водень, синтез-газ (СО + н2). Основну масу природного
газу використовують як енергоносій і в органічному синтезі.
• Нафта 'складається переважно з вуглеводнів з невеликою домішкою
сірчаних, азотистих і кисневих сполук. За складом нафти поділяють на парафі-
нові (Пенсільванія, США; Борислав, Україна), нафтенові (Баку, Азербайджан),
ароматичні (Урал, Росія) та змішаного складу. Основну масу нафти за
допомогою різних видів крекінгу переробляють на пальне: бензин, гас, мазут,
солярову оливу.
• Вугілля. Запаси кам'яного і бурого вугілля значно перевищують запаси
нафти. Вугілля використовують не тільки як енергоносій, воно є важливим
джерелом сировини для хімічної промисловості. Сьогодні існує декілька шляхів
переробки кам'яного вугілля: коксування, гідрування, неповне спалювання й
синтез кальцій карбіду.
24 Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Нагріванням вугілля у спеціальних батареях до високої температури
одержують кокс для доменного виробництва і кам'яновугільну смолу - головне
джерело ароматичних і гетероциклічних сполук. Гази коксування містять амоній,
найпростіші алкани й олефіни, а також невелику кількість ароматичних речовин.
Гідрування бурого вугілля може бути в Україні джерелом сирої нафти.
Неповне спалювання вугілля дає карбон (II) оксид, який використовують ^
разом з воднем у промисловості для виробництва різних вуглеводнів, спиртів і
альдегідів.
• Органічні речовини рослин. Продуктами життєдіяльності рослин є поши-
рені технічні матеріали: деревина, текстильні волокна (бавовна, льон, джут та
ін.) та головні продукти харчування (зернові, цукор, рослинні олії). Найважли-
віші сполуки рослинного походження - вуглеводи. Окремі хімічні виробництва
як сировину використовують рослинні відходи сільського й лісового госпо-
дарств. Сюди належать виробництво етилового (гідролізного) спирту, щавлевої і
лимонної кислот, вітаміну С та ін. Природні етерні олії, без розділення на
індивідуальні компоненти, застосовують у парфумерній промисловості..
• Органічні речовини тварин. Тут провідну роль відіграють білки. Білки є
найважливішими продуктами харчування. Тваринні волокна - вовна та шовк -
також є білковими речовини. Сюди також можна зачислити кормовий білок,
який одержують мікробіологічним шляхом із нафти.
• Органічні речовини планктону. До складу планктону належать як
рослини (фітопланктон), так і тварини (зоопланктон). Організми фітопланктону
- головні продуценти органічних речовин у морях та океанах, якими живляться
водні тварини. Окремі види зоопланктону, такі як криль, є об'єктом
промислового лову.
• Органічний синтез. Не менш різноманітними є органічні сполуки, які
одержують синтетичним шляхом. Перше місце за темпами розвитку та
впровадженням у практику посідають високомолекулярні органічні сполуки. За
технічними властивостями більшість із них переважає природні матеріали і
часто за хімічною будовою не має аналогів у природі.
Перше виробництво полімерів — виготовлення целулоїду на основі нітрату
целюлози - організовано 1872 р. у США. Сьогодні обсяг синтезу полімерів у
світі перевищив 100 млн т за рік. Пластмаси широко використовують у судно-,
автомобіле- й авіабудуванні, будівництві й сільському господарстві, легкій і
харчовій промисловості, медицині.
Особливо вражають успіхи в галузі синтетичних каучуків (СК).
Номенклатура СК налічує понад 50 тисяч найменувань. Технічний прогрес у
різних галузях промисловості і, насамперед, у шинній, спонукав до створення
СК, у яких би поєднуватися термо-, бензо- й оливостійкість, стійкість до
радіаційних випромінювань. Це завдання успішно вирішене шляхом поліме-
ризації мономерів, які містять неорганічні елементи - Бор, Фосфор, Сульфур,
Нітроген, Флуор, Силіцій та ін.
Розділ 7. Предмет органічної хімії
25
Важливого значення набули синтетичні органічні сполуки, що сприяють
підвищенню врожайності сільськогосподарських культур і продуктивності
тваринництва: гербіциди і регулятори росту рослин. Застосування високо-
ефективних речовин, які знищують шкідників рослин і тварин (інсектициди),
плісень (фунгіциди), бур'ян (гербіциди) і прискорюють ріст рослин (регулятори
росту рослин), сприяє створенню великої кількості сільськогосподарських
продуктів.
Хіміко-фармацевтична промисловість за тоннажем поступається зазначеним
вище галузям хімічної промисловості, однак її продукція становить велику цінність
для медицини. Лікарські речовини відіграють важливу роль в охороні здоров'я
людей. Незважаючи на значні успіхи, яких досягнуто у використанні
мікроорганізмів для одержання антибіотиків, і досі потрібними є синтетичні
лікарські препарати. Наприклад, систематичне застосування антибіотиків виліковує
трахому, а дієва зброя проти туберкульозу - синтетичні лікарські речовини.
Барвники є продуктами однієї з найстаріших і найважливіших галузей
органічної хімічної промисловості - анілінобарвникової. Синтетичні барвникові
речовини давно перевершили за різноманітністю, міцністю, яскравістю,
чистотою відтінків і дешевиною більшість природних барвників, їх широко
застосовують для фарбування не тільки текстильних матеріалів, а й гуми, шкіри,
деревини тощо. Номенклатура барвників складається з більш ніж 10 тис.
найменувань, а світовий обсяг виробництва досягає 1 млн т за рік.
Здатність синтетичних органічних сполук, часто безбарвних, інтенсивно
світитися різними кольорами під дією короткохвильових випромінювань (УФ-
випромінювання), що називають люмінесценцією, широко використовують на
практиці для дефектоскопії металів і виробів з різноманітних матеріалів,
з'ясування схожості насіння, для фіксування радіовипромінювань і в театральній
техніці.
1.1. Виділення, очищення та ідентифікація
органічних речовин
Кожна органічна сполука має власні фізичні та хімічні властивості, які
відображає її структурна формула. Для того, щоб визначити структурну формулу
речовини, необхідно, насамперед, виділити її з суміші в індивідуальному вигляді,
з'ясувати якісний і кількісний склад. Завершальною стадією дослідження є виведення
структурної формули на підставі її фізичних та хімічних властивостей. Якщо раніше
це питання вирішували за допомогою хімічних методів, то сьогодні провідну роль
відіграють фізичні методи, які дають змогу робити аналізи в дуже короткі терміни.
Як звичайно, найчастіше хіміку доводиться мати справу з твердими або
рідкими сумішами. Для розділення твердих сумішей використовують фільтру^
вання, кристалізацію, сублімацію тощо. Коротко розглянемо ці методи.
26 Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Фільтрування - розділення неоднорідних сумішей за допомогою пористих
фільтрів, які пропускають розчин і затримують на поверхні фільтра нерозчинені
речовини. Рушійною силою процесу фільтрування слугує різниця тисків по
обидва боки фільтра.
Кристалізація є процесом утворення кристалів з будь-якої некристалічної
(пари, розчину, розплаву) або іншої кристалічної форми. Для проведення
кристалізації суміш розчиняють у спеціально підібраному розчиннику за
максимальної температури в мінімальному об'ємі. Зі зниженням температури
розчинність більшості речовин зменшується, і вони випадають в осад. Якщо в
складі суміші є декілька речовин з різною температурою кристалізації, то
використовують дробову кристалізацію: суміш органічних речовин розчиняють
за різних температур і одержують окремі фракції, які містять певні речовини.
Сублімація - це перехід речовини з твердого стану в газоподібний без
плавлення. Зворотний процес називають десублімацією. Типовим прикладом
цього явища може бути сублімація нафталену.
Іноді органічні речовини з сумішей виділяють за допомогою екстракції
(екстрагування). Для цього суміш заливають відповідним розчинником, а саму
екстракцію проводять в апараті Сокслета1. Після завершення процесу розчинник
відганяють у перегінному апараті, так органічну речовину виділяють у чистому
вигляді.
Для розділення суміші рідин, що необмежено розчиняються одна в одній і
мають різні температури кипіння, як звичайно, застосовують перегонку або
дистиляцію. Перегонка ґрунтується на різниці температур кипіння рідин. Вона
буває простою, коли з суміші виділяється чиста речовина, або фракційною, коли
з суміші відганяється декілька фракцій.
Відомо, що речовина кипить за температури, коли тиск її пари дорівнює
атмосферному. Якщо нагрівати дві рідини, що не змішуються, то вони закиплять
за температури, коли сумарний тиск пари обох рідин зрівняється з атмосферним.
Звичайно, як іншу рідину використовують воду, тому перегонку такої суміші
можна вести за температури значно нижче 100 °С. Співвідношення перегнаних
рідин у дистиляті визначають як співвідношення добутку тиску пари кожної
речовини на її молекулярну масу.
Часто для речовин, які під час нагрівання руйнуються, застосовують
перегонку за умов високого вакууму.
Для речовин, які розкладаються за температури кипіння навіть у високому
вакуумі, використовують "молекулярну перегонку". Принцип її полягає в тому,
що в разі сильного розрідження (10"5-10"8 мм рт. ст.) з нагрітої поверхні
розплавленої речовини, що підлягає перегонці, молекули переходять у газову фазу
1 Сокслет Ф. (ЗохЬІеІ КЯ.) (1848-1926) - німецький агрохімік. Відомий працями в галузі хімії та
біохімії молока. Винайшов кілька приладів для хімічного дослідження, що названі його ім'ям. У
1886 р. запропонував пастеризувати молоко.
Розділ 1. Предмет органічної хімії
27
за температури, набагато нижчої від температури кипіння цієї сполуки. Пари
речовини потім конденсуються на холодній поверхні. Так можна очистити
речовини з порівняно великою молекулярною масою і лабільною структурою.
Для виділення й очищення органічних речовин, що важко кристалізуються або
розпадаються під час перегонки, часто використовують різні види хроматографії.
Основоположником хроматографії є російський ботанік М. Цвєт1, який застосував
цей метод для розділення компонентів хлорофілу, які мали колір, звідси й назва
хроматографія (грец. скготаїоБ - колір і &арко - пишу). Уже М. Цвєт розумів, що
метод можна використати для очищення і виділення безбарвних речовин. Згодом це
підтверджено працями англійських учених А. Мартіна2 і Р. Сінга3, які розробили
метод розділювальної хроматографії на папері й застосували його для одержання в
чистому вигляді білків, амінокислот, вуглеводів та інших сполук. У первинному
варіанті хроматографічне розділення використано на різних ступенях адсорбції
компонентів суміші, воно зводилось до багаторазового зрівноважування між
твердим адсорбентом і переміщувальним розчином розділювальної суміші.
Усі види хроматографії ґрунтуються на різній рухомості розчинених
речовин у разі проходження їх через багатофазову, а частіше - двофазову
систему. Для кожної конкретної речовини усталюється рівновага концентрацій
між фазами (у розчині та сорбувальному носії). Під час протікання розчину
через прошарок адсорбенту процес рівноваги багаторазово повторюється, так
що навіть за незначної різниці у положеннях рівноваги окремих компонентів він
приводить до помітного розділення суміші.
Залежно від типу фізико-хімічної взаємодії між активним адсорбентом і
розчиненими речовинами розрізняють такі види хроматографії: адсорбційна,
розділювальна, іонообмінна та гель-хроматографія.
Адсорбційна хроматографія ґрунтується на різній здатності речовин суміші
адсорбуватися на поверхні носія. Як адсорбенти, зазвичай, застосовують
алюміній оксид і силікагель. Значно рідше - активоване вугілля, барій сульфат,
магній силікат, поліаміди.
Здатність речовин адсорбуватися на полярному адсорбенті головно визна-
чена їхньою полярністю. За здатністю адсорбуватися речовини з різними
функціональними групами можна розташувати в такій послідовності:
ЯН < ЯОСН3 < Я-И02 < Я-К(СН3)2 < Я-СООСН3 < Я-КН2 < Я-ОН <
Я-С(ЖН2 < < Я-СООН.
1 Цвет М. (1872-1919) - російський ботанік, фізіолог та біохімік. Винахідник хроматографічного
методу. Дослідив пігменти листя рослин, отримав у чистому вигляді хлорофіли а, Ь, с.
2 Мартін A. (Martin A.J.P.) (1910-2002) - англійський біохімік. Головні праці з біохімії вітамінів та
білків. Розробив кілька методів виділення чистих речовин, насамперед розподільну хро-
матографію (разом з Р. Сінгом) та колонкову газорідинну хроматографію (разом з Т. Джеймсом).
Нобелівська премія 1952 р. (разом з Р. Сінгом).
3 Сінг P. (Synge R.L.M.) (1914-1994) - англійський біохімік. Дослідження в галузі біополімерів:
білків та полісахаридів. Розробив кілька методів очищення продуктів метаболізму білків.
28
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
За полярністю, а отже, і за здатністю елюювати, розчинники-елюенти утво-
рюють такий ряд:
Н20 > СНзОН > С2Н5ОН > СН3СОСН3> СН3СООС2Н5> С2Н5ОС2Н5> СНС13> ССІ4>
> циклогексан > н-гексан
Елюювання виконують або одним елюентом (сумішшю елюентів), або
послідовно декількома елюентами, переходячи від менш до більш полярного,
або сумішшю двох розчинників (послідовно збільшуючи концентрацію більш
полярного).
Головна вимога до адсорбенту для хроматографії - відсутність хімічної
взаємодії між адсорбентом і речовинами, що підлягають розділенню. Інша
вимога - його селективність, максимально можлива різниця до адсорбції
речовин суміші.
За основу розподільної хроматографії взято розподіл речовин між рухомою
(газ, рідина) і нерухомою (рідина, що утримується на твердому інертному носії)
фазами. Найбільшого поширення набула розділювальна хроматографія на папері
та газорідинна хроматографія.
Основою хроматографії на папері є розподіл суміші речовин, які
розділяють, між водою, адсорбованою на папері, і розчинником, насиченим
водою. За допомогою цього методу вдало виконали розділення та ідентифікацію
амінокислот і моносахаридів. Сьогодні цей варіант хроматографування втратив
актуальність для дослідників через невисоку ефективність розділення суміші та
тривалості.
Газорідинна хроматографія - це розділювальна хроматографія між ста-
ціонарною рідкою фазою, що нанесена безпосередньо на носій, і газом (зазвичай
гелій, азот або водень). Для характеристики речовин, які розділяють,
використовують "час утримання" - час від моменту введення суміші в колонку
до виходу з колонки і проходження речовини. через відповідний детектор,
наприклад, детектор, що реєструє зміни теплопровідності. Цей варіант є одним з
хроматографічних методів, який застосовують найширше, особливо з аналі-
тичною метою.
Іонообмінна хроматографія полягає в розподілі йонів речовин між рухомою
та нерухомою фазами залежно від їхньої спорідненості до йонних центрів
нерухомої фази.
За природою йонообмінника розрізняють катіонну й аніонну хроматографії.
Як елюент широко використовують воду, розчини кислот і лугів, буферні
розчини. Найчастіше йонообмінними матеріалами є катіоніти й аніоніти на
основі зшитих полімерів, що містять йоногенні функціональні групи, а також
модифіковану целюлозу.
Відмінною рисою гель-хроматографії є те, що в гелях, утворених тривимір-
ними "зшитими" макромолекулами, наявні пори певних розмірів, у які входять
менші за розміром молекули, що розділяються, і не входять більші. Тому, на
Розділ 7. Предмет органічної хімії
29
відміну від адсорбційної хроматографії, у гель-хроматографії першими крізь
колонку проходять молекули більшого розміру, а останніми — меншого.
Розділювальну колонку заповнюють зерна ліофільного або гідрофільного гелю.
Прикладами таких хроматографічних матеріалів можуть слугувати модифіковані
природні гелеутворювачі - агар, декстрини, сефадекси (зшиті декстрани) і
синтетичні сита на основі поліакриламіду або "зшитого" полістиролу.
Тонкошарова хроматографія - ефективний метод аналізу сумішей речовин
різних класів — алканів, спиртів, кислот, стероїдів, терпеноїдів тощо. Зміст
методу полягає в такому. На скляну пластинку наносять тонкий шар носія, який
може бути незакріпленим або зафіксованим за допомогою спеціальних хімічних
речовин (крохмаль, гіпс). Пробу речовини наносять у нижній частині пластинки,
яку потім поміщають у бокс з елюентом. Розчинник завдяки капілярним силам
піднімається по пластинці (висхідна хроматографія), розділяючи суміш. У
випадку речовин, які важко розділяти, вдаються до двовимірної хроматографії,
коли речовину спочатку елюють в одному напрямі, а потім елюювання
виконують у перпендикулярному напрямі.
Забарвлені сполуки спостерігають під час хроматографування безпосе-
редньо. Безбарвні речовини необхідно "виявити" - перетворити на забарвлені
сполуки. Залежно від сорбенту, закріплювача і природи речовини, яку роз-
діляють, використовують різні методи "виявлення", наприклад, вуглеводи
обвуглюють за високої температури, у тому числі й після обприскування
розчинами сульфатної кислоти, амінокислоти дають забарвлені продукти після
обробки розчином нінгідрину. За інтенсивністю забарвлення розділюваних
сполук роблять висновок про вміст їх у суміші.
Для характеристики речовин застосовують термін хроматографічна
рухомість, яку позначають як Я& - відношення довжини пробігу зони речовини
до довжини пробігу елюенту.
1.2. Якісний аналіз органічних речовин
Після одержання органічних речовин у чистому вигляді виконують якісний
елементний аналіз, щоб з'ясувати, які хімічні елементи містяться в молекулі.
Органічні речовини здебільшого не є електролітами, тому вони не здатні давати
якісні реакції на окремі хімічні елементи. Щоб виявити ці елементи, органічні
речовини заздалегідь розкладають шляхом нагрівання або дії сильних кислот чи
лугів. Цей етап називають мінералізацією. На другому етапі хімічні елементи
виявляють загальними аналітичними методами.
Визначення Карбону і Гідрогену, Наявність в органічній речовині Карбону
визначають обвуглюванням з окиснювачами, найчастіше з оксидом купруму (II),
наприклад:
С12Н22ОП + 24СиО -» 12С02 + 11Н20 + 24Си
зо
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
Вуглекислоту пропускають через баритову чи вапняну воду і спостерігають
помутніння внаслідок утворення карбонату кальцію або барію:
Гідроген визначають, додаючи до крапель води, що утворилась під час
обвуглювання органічних речовин, безводний білий порошок сульфату купруму.
Внаслідок реакції утворюється кристалогідрат сульфату купруму синього кольору:
Визначення Нітрогену. Загальноприйнятим для визначення Нітрогену є
метод Лоссеня1. За цим способом сплавляють близько 0,1 г органічної речовини
з натрієм. Нітроген, який міститься в органічній речовині, частково
перетворюється на неорганічні солі:
Якісними реакціями визначають ціанід-іон. Відповідно до класичного
варіанта розплав розчиняють у воді, додають невелику кількість солі дво-
валентного феруму. Після підкислення розчину і додавання до нього ферум (III)
хлориду утворюється синій осад берлінської лазурі або, якщо розчин дуже
розведений, з'являється синє забарвлення розчину внаслідок утворення колоїду
берлінської лазурі:
Точніше визначення Нітрогену дає реакція розчину, який містить ціанід-іон,
з реакційною сумішшю розчинів я-нітробензальдегіду, одинітробензолу і натрій
гідроксиду. За наявнрсті у сполуці Нітрогену аналізована суміш забарвлюється в
інтенсивний пурпурово-синій колір:
С02 + Ва(ОН)2 -> ВаС03>1 + Н20
CuS04 + 5Н20 -> CuS04 • 5Н20
Na+[C,N] 4 NaCN
2NaCN + FeS04 -> Na2S04 + Fe(CN)2
Fe(CN)2 + 4NaCN -> Na4[Fe(CN)6]
N02
NaCN
CN
N02
CHO
0"Na+ Na0H
+ 0,N
i2N—\CJ)/ c°2Na + NaCN + H2°
Пурпурово-синій
1 Лоссень В. (Lossen W.) (1838-1905) - німецький хімік, головні праці якого пов'язані з
дослідженням алкалоїдів, відкрив перегрупування гідроксамових кислот.
Розділ 7. Предмет органічної хімії
31
Визначення Сульфуру. Унаслідок сплавлення проби речовини, що містить
Сульфур, з натрієм за методом Лоссеня утворюється натрій сульфід, який
визначають у розчині нітропрусидною реакцією (синьо-фіолетове забарвлення)
чи реакцією із солями плюмбуму або купруму (чорний осад):
Ма + [С,8] 4 ИагЗ + С
Иа28 + РЬ(ОСОСН3)2 -> РЬ8^ + 2СН3С(0)(Жа
№28 + На2[Ре(СН)5(Ж))] -> Ка2[Ре(СН)5(Н08)]
Можна також розкладати органічні сполуки за Каріусом1. Для цього речо-
вину запаюють в ампулу з димлячою нітратною кислотою, поступово під-
вищують температуру до 300-350 °С і залишають на 3-5 год. Органічна
речовина згоряє, утворюючи воду й вуглекислий газ, а Сульфур перетворюється
на сульфатну кислоту, яку визначають за утворенням осаду барій сульфату:
НШз + [С, 8] 4 Н2804 + С02
Н2804 + ВаС12 -> ВаЗОД + 2НС1
Визначення галогенів. Цю процедуру виконують за допомогою проби
Бейльштейна. Для відкриття галогенів органічні сполуки спочатку переводять у
неорганічні, де галоген перебуває в іонному стані. Для цього використовують
мідний дріт, який змочують розчином галогеновмісної речовини і прожарюють.
Утворюються солі купруму, які забарвлюють полум'я в зелений колір, наприклад:
СНСІз + 5СиО -> 2С02 + Н20 + СиС12 + 4СиС1
Хлороформ
Визначення Фосфору. Фосфор міститься в багатьох природних органічних
сполуках, наприклад, таких як білок. Для визначення Фосфору до гідролізату
білка додають кілька крапель розчину молібденовокислого амонію і кип'ятять.
Після охолодження утворюється кристалічний осад фосфорно-молібденово-
кислого амонію лимонно-жовтого кольору:
12(НН4)2Мо04 + Н3Р04 + 21НШ3 + 6Н20 -> 2ШН4>Юз + (МН4)зР0412Мо03-6Н2ОІ
Основи кількісного аналізу органічних сполук розроблені німецьким хіміком
Ф. Преглем2. Вони полягають у такому: наважку речовини 3—5 мг спалюють за
1 Каріус Л. (СагіиБ СЬ.) (1829-1875) - німецький хімік, який розробив метод визначення
Сульфуру, галогенів та інших елементів в органічних сполуках (1860).
2 Прегль Ф. (1^1 К) (1869-1930) - німецький хімік, засновник мікроаналізу органічних сполук.
Нобелівська премія 1923 р.
32 Чирва В.Я., Ярмолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
температури 900 °С у потоці кисню, очищеного від водню, води й вуглекислого
газу. Водень від кисню очищають пропусканням газу над платиновим ката-
лізатором за 800 °С. Повне очищення від вуглекислого газу й води виконують
пропусканням через безводний магній перхлорат (ангідрон) і через азбест,
просякнутий розплавом їдкого натру (аскарит).
Після трубки спалювання з досліджуваною речовиною поставлені трубки із
вбирачами: ангідроном і аскаритом. Доважок із першого вбирача відповідає
кількості води, за якою обчислюють вміст водню в наважці речовини; доважок із
другого вбирача дає кількість вуглекислого газу, за яким визначають вміст
вуглецю в аналізованій речовині.
Галогени й Сульфур можна визначити, розклавши речовину за Каріусом,
галогени у вигляді арґентум галогеніду - ваговим способом або відтитровуючи
надлишок арґентум нітрату. Сульфур визначають у вигляді барій сульфату. Гази
після спалювання наважки пропускають над шаром металевої міді, де від-
бувається відновлення нітроген оксидів до вільного азоту. Нітроген визначають
об'ємним методом за кількістю непоглинутого газу.
1.3. Визначення молекулярної маси
Для визначення молекулярної маси сполуки часто застосовують методи
кріоскопії, що ґрунтуються на законі Рауля1. У цьому випадку визначають
температуру замерзання розчинника, а потім - розчину. Різниця цих значень Ьі
прямо пропорційна до кількості молекул речовини, розчиненої в заданій масі
розчинника. Молекулярну масу обчислюють за формулою
рм
де р - наважка речовини; Р - наважка розчинника; К - кріоскопічна стала.
Аналогічно в ебуліоскопічному методі молекулярну масу визначають через
різницю між температурами кипіння чистого розчинника й розчину.
Для високомолекулярних сполук описані вище методи не придатні. У цьому
разі користуються трьома методами: віскозиметричним, осмотичним і седимен-
таційним, які, відповідно, не придатні для речовин зі звичайною молекулярною
масою.
Сьогодні найчастіше для визначення молекулярної маси невідомої речовини
використовують мас-спектрометрію.
1 Рауль Ф. (Raoi.lt Р.) (1830-1901) - французький хімік, головний напрям його досліджень -
вивчення розчинів.
Розділ 2
ТЕОРІЯ Б УЛОВИ
ОРГАНІЧНИХ СПОЛ УК
Ще з часів винайдення вогню людина поділяла речовини на горючі й
негорючі. До першої групи належали, здебільшого, продукти рослинного і
тваринного походження, до другої - переважно мінерального. Отже, між
здатністю речовини до горіння і належністю її до живого чи неживого світу
існував певний зв'язок.
У 1807 р. Й. Берцеліус запропонував називати сполуки першої групи
органічними, а речовини, подібні до води і солей, які характерні для неживої
природи, визначив як неорганічні.
Деякі органічні речовини у більш чи менш чистому вигляді відомі людині з
давніх-давен (оцет, більшість органічних барвників). Низка органічних сполук,
наприклад, сечовина, етиловий спирт, "сірчаний ефір", одержані ще алхіміками.
Дуже багато речовин, особливо органічні кислоти (щавлева, лимонна, молочна
та ін.) та органічні основи (алкалоїди), виділено з рослин і тварин у другій
половині XVIII і перших роках XIX ст. Цей час і треба вважати початком
наукової органічної хімії.
❖ Теорія віталізму. У XVIII та першій половині XIX ст. панувало пере-
конання, що хімія живої природи принципово відмінна від хімії мертвої природи
(мінеральної хімії) і що організми виробляють свої речовини за участю
особливої життєвої сили, без якої штучно, в колбі, їх створити неможливо. То
був час панування віталізму - вчення, яке розглядає життя як особливе явище,
підпорядковане не законам Всесвіту, а впливу особливих життєвих сил.
Захисником віталізму був Г. Шталь1, засновник теорії флогістону. На його
думку, хіміки, які працювали зі звичайними речовинами, виконати їхнє
перетворення, що потребувало участі життєвих сил, звичайно, не могли.
Перші сумніви щодо слушності віталістичної теорії висловив учень
Й. Берцеліуса німецький хімік Ф. Велер, який синтезував сечовину з амоній
ціанату, беззастережно віднесеного до неорганічних речовин:
NH4CNO
NH2-C-NH,
II
О
1 Шталь Г. (Stahl G.) (1659-1734) - німецький хімік і лікар. Засновник теорії флогістону - першої
хімічної теорії, яка дала змогу покінчити з теоретичними переконаннями алхімії.
34 Чирва В.Я., Ярмолкж CM., Толкачова Н.В., Земляков О.Є. Органічна хімія
Не потрібно переоцінювати значення цієї роботи, тому що сечовина
фактично є перебудованою молекулою амоній ціанату, проте не можна й
ігнорувати важливості відкриття Ф. Велера, оскільки воно надихнуло хіміків на
синтез нових органічних речовин.
У 1845 р. А. Кольбе1, учень Ф. Велера, виконав синтез з елементів, тобто
повний синтез оцтової кислоти. Французький хімік П. Бертло2 одержав мети-
ловий і етиловий спирти, метан. Проте була думка, що таку складну речовину,
як цукор, ніколи не буде синтезовано. Однак уже 1861 р. О. Бутлеров синтезував
речовину, подібну до цукру, - метиленітан.
Одночасно з цими етапними для органічної хімії синтезами швидко зростала
загальна кількість синтезованих карбоновмісних сполук, які не трапляються в
природі. Наприклад, 1831 р. Ю. Лібіх і Е. Суберан синтезували хлороформ. Ще
раніше стали відомими етилен, етиленбромід та низка похідних бензену. У
1842 р. М. Зінін3 шляхом відновлення нітробензену одержав анілін - важливу
сировину для синтезу органічних барвників і медикаментів, а в 50-х роках
XIX ст. з аніліну одержано перші "анілінові барвники" - мовеїн (У. Перкін4) і
фуксин; після цих синтезів у середині 50-х років XIX ст. віталістична теорія
зазнала краху остаточно.
❖ Дуалістична теорія Й. Берцеліуса. Основи структурної хімії органічних
сполук заклав Й. Берцеліус, який услід за А. Лавуазьє5 поширив на органічні
об'єкти кількісний аналіз і створив для пояснення їхньої природи дуалістичну
(електрохімічну) теорію - першу наукову теорію в хімії. За Й. Берцеліусом, атом
елемента з'єднується з Оксигеном унаслідок того, що він електропозитивний, а
Оксиген електронегативний; у разі об'єднання заряди нейтралізуються.
Й. Берцеліус уважав, що його теорію можна застосовувати і до органічної хімії, з
тією різницею, що в органічних сполуках радикали в оксидах складніші, наприклад,
вуглеводневі. Тому цю теорію ще називають теорією складних радикалів.
За А. Лавуазьє радикали органічних сполук складаються з Карбону,
Гідрогену і Оксигену, до яких у випадку речовин тваринного походження
додається ще Нітроген і Фосфор.
1 Кольбе А. (Kolbe А.) (1818-1884) - німецький хімік-органік, засновник теорії радикалів. Синтезував
низку органічних кислот. Розробив електрохімічний метод одержання алканів - метод Кольбе.
2 Бертло П. (Berthelot P.) (1827-1907) - французький хімік. Один із засновників органічної хімії.
Фундаментальні праці в галузі термохімії.
3 Зінін М. (1812-1880) - російський хімік, засновник російської наукової школи. Відкрив метод
отримання ароматичних амінів {реакція Зініна). Уперше синтезував анілін та інші ароматичні
аміни, що стало базою виробництва синтетичних барвників, лікарських засобів та ін.
4 Перкін У. ст. (Perkin W. sr.) (1838-1907) - англійський хімік. Розробив промислове виробництво
барвників мовеїну, алізарину. Відкрив реакцію конденсації ароматичних альдегідів з ангідридами
карбонових кислот (реакція Перкіна).
5 Лавуазьє A. (Lavoisier А.) (1743-1794) - французький хімік, уперше з'ясував роль кисню в
процесах горіння, окиснення та дихання. Один із засновників термохімії, розробив хімічну
номенклатуру. Противник теорії "флогістону".
Розділ 2. Теорія будови органічних сполук
35
❖ Теорія радикалів. Подальшим розвитком теорії Й. Берцеліуса стала
теорія радикалів. У 1810 р. Ж. Гей-Люссак1 помітив, що група (ціанідна
група) може переходити зі сполуки у сполуку, не розділяючись на окремі атоми
Карбону і Нітрогену. Такі групи назвали радикалами.
Поступово радикали почали розглядати як незмінні складові органічних
речовин (подібні до елементів у неорганічних сполуках), які переходять у
реакціях з однієї сполуки в іншу. Деякі дослідники, особливо німецької школи
(Ф. Велер, Ю. Лібіх), натхненні відкриттям серії нових елементів, керувалися
ідеєю пошуку нових радикалів. Зокрема, вони знайшли радикали бензоїл
СбН5СО і ацетил СНзСО. До того часу стало відомо також, що речовини, які
називають нині етиловим спиртом, діетиловим етером, етилхлоридом і
етилнітритом, містять радикал етил -С2Н5. Подібним способом ідентифіковано й
інші радикали, тобто групи атомів, які залишаються незмінними у випадку
різних хімічних перетворень.
Багаторазові спроби виділити радикали у вільному стані виявлялися
невдалими або приводили до помилкових результатів. Наприклад, до відкриття
закону Авогадро2 етан, який виділявся за реакцією Вюрца3:
2СН3Вг + 2Ка -СН3-СН3 +2№Вг
вважали спочатку радикалом метилом -СН3, і лише згодом з'ясували, що його
молекулярна маса насправді вдвічі більша.
Загальноприйнятий принцип незмінності радикалів похитнули французькі
хіміки Ж. Дюма4 та його учень О. Лоран5, які відкрили реакцію металепсії. Ця
реакція полягає в такому: діючи на органічні речовини, хлор приєднується до
сполуки так, що з кожним приєднаним еквівалентом Хлору речовину покидає
один еквівалент Гідрогену у вигляді гідроген хлориду. Суперечність з теорією
Й. Берцеліуса була настільки разючою, що цю теорію відразу відкинули. Хлор -
негативно заряджений елемент - займав місце позитивно зарядженого атома
Гідрогену, і молекула не лише зберігалась, а й не змінювала хімічного характе-
ру. Виявилося можливим замінювати атом Гідрогену іншими електронега-
Гей-Люсак Ж. (Gay-Lussac J.) (1778-1850) - французький хімік та фізик, відкрив газові закони,
бор. Удосконалив методи елементного та об'ємного хімічного аналізу, технологію виробництва
сірчаної кислоти.
2 Авогадро A. (Avogadro А.) (1776-1856) - італійський фізик та хімік. Запропонував молекулярну
гіпотезу будови речовини, винайшов один з газових законів (закон Авогадро).
3 Вюрц Ш. (Wurtz Ch.) (1817-1884) - французький хімік. Навчався у Ю. Лібіха, асистент Ж. Дюма.
Синтезував аміни, феноли, етиленгліколь, молочну кислоту, провів альдольну і кротонову
конденсації.
4 Дюма Ж. (Dumas J.) (1800-1884) - французький хімік. Створив теорію радикалів. Відкрив
реакцію хлорування, виявив існування гомологічного ряду - ряду мурашиної кислоти. Запро-
понував спосіб кількісного визначення Нітрогену.
5 Лоран О. (Laurent О.) (1807-1853) - французький хімік. Вивчав продукти кам'яновугільної
смоли. Відкрив фталеву кислоту, індиго і нафтален.
36
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
тивними елементами - галогенами, Оксигеном, Сульфуром тощо. Стало оче-
видним, що постійних радикалів не існує: в одних реакціях радикали
приєднуються до речовин у незмінному вигляді, а в інших - змінюються.
❖ Теорія типів. Наступні спроби знайти щось спільне в природі органічних
сполук були пов'язані, навпаки, зі спостереженнями за частинами молекул, які
найчастіше змінюються під час хімічних реакцій, що сьогодні називають
функціональними групами. На підставі цих спостережень Ш. Жерар запро-
понував теорію типів. У спиртах і кислотах Ш. Жерар побачив аналоги води, у
хлоропохідних вуглеводнів - аналоги гідроген хлориду, в алканах - водню, в
амінах - амоніаку:
н>о СІЧо сЛ}о сн>с>
> н > н / н /
н
н х сн3. с2Но сн3соч
сі* сі* сі* сі*
н х сн3ч с2н5 ї сн3с(х
н / н ' н^н/
Н сн3 сн3 сн3
н }и н }и сн3 ^ сн3 ^
п} п} нJ сн/
Більшість прибічників теорії типів (Ш. Жерар, А. Кольбе, Ф. Кекуле1)
виходили з того, що неможливо визначити будову речовин експериментально. їх
можна тільки класифікувати залежно від того, в які реакції речовина вступає;
одну й ту ж органічну сполуку можна зачислювати до різних типів. Теоретики з
великим натягуванням класифікували величезний дослідний матеріал, а про
можливість цілеспрямованого синтезу не могло бути й мови. Подальший
розвиток хімії потребував створення нової наукової теорії.
Одним з недоліків теорії типів є прагнення вкласти всі органічні сполуки в
більш чи менш формальні схеми; перевага цієї теорії полягає в уточненні понять
про гомологічні ряди і хімічні функції, остаточно опановані органічною хімією,
її роль у розвитку науки безсумнівна, оскільки вона привела до поняття
валентності й відкрила шлях до теорії будови органічних сполук.
❖ Теорія будови органічних сполук. Зазначимо, що появі основної теорії
будови органічних сполук передувала низка досліджень. Наприклад, А. Вілья-
1 Кекуле Ф. (Кекиїе Р.А.) (1829-1896) - німецький хімік, головні праці в галузі теоретичної
органічної хімії. Синтезував антрахінон, трифенілметан.
Розділ 2. Теорія будови органічних сполук
37
мсон 1851 р. увів поняття про так звані багатоатомні радикали, тобто про
радикали, здатні замістити два і більше атомів Гідрогену. Завдяки цьому стало
можливим класифікувати окремі сполуки одразу до двох і більше типів,
наприклад, амінооцтова кислота може бути зачислена до типів води й амоніаку:
н/
Такі сполуки ми сьогодні називаємо гетерофункціональними.
Важливий крок зроблений німецьким хіміком Ф. Кекуле, який увів у хімію
тип метану:
ні
Н С
н и
н;
Ф. Кекуле в органічній хімії систематично використовував поняття
валентності, яке він поширив і на сам Карбон. Визнання чотиривалентності
Карбону невдовзі змусило Ф. Кекуле визнати також зв'язок карбонових атомів
між- собою. Щоб дотриматись сталості валентності Карбону й Оксигену,
необхідно було також погодитись з існуванням подвійного зв'язку в етилені
(С=С), в альдегідах і кетонах (С=0).
Шотландський хімік А. Купер2 запропонував сучасне зображення формул, у
яких знак елемента мав певну кількість рисочок, що відповідала його
валентності:
V
н-с-о-н н-с^и
А
Проте ні Ф. Кекуле, ні А. Купер не сприймали ідеї нерозривного зв'язку
хімічних і фізичних властивостей молекули з її будовою, що передана
формулою. Ф. Кекуле допускав опис однієї й тієї ж сполуки за допомогою
декількох різних формул залежно від того, яку сукупність реакцій цієї речовини
потрібно виразити формулою. По суті, це були так звані реакційні формули.
Головні положення теорії будови органічних сполук оприлюднені
А. Бутлеровим 1861 р. на хімічній секції З'їзду німецьких природодослідників і
лікарів у м. Шпеєрі. Теорія А. Бутлерова ґрунтувалася на атомістичному вченні
1 Вільямсон A. (Williamson А.) (1824-1904) - англійський хімік-органік, головні дослідження
присвячені етерам.
2 Купер A. (Couper A.S.) (1834-1891) - шотландський хімік, головні праці присвячені теоретичним
проблемам хімії.
38
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
М. Ломоносова1 і Д. Дальтона2. Зміст цієї теорії зводився до таких головних
положень.
1. Хімічна природа кожної органічної молекули визначена природою її
складових атомів, їхньою кількістю і будовою.
2. Хімічна будова - це певний порядок хімічних зв'язків атомів у молекулі,
взаємний вплив атомів один на одного.
3. Хімічна будова речовин визначає їхні фізичні та хімічні властивості.
4. Вивчення властивостей речовин дає змогу визначити їхню хімічну будову.
О. Бутлеров уперше запропонував такі терміни, як будова чи структура
молекули, хімічний зв'язок. Хімічною будовою він назвав певний порядок
хімічного зв'язку атомів у молекулі. Учений з'ясував, як, вивчаючи хімічні
реакції конкретної речовини, можна визначити її структуру, яка для кожного
хімічного індивідуума є адекватною. Відповідно до цієї формули можна і
синтезувати ці сполуки. Властивості певного атома в сполуці насамперед
залежать від того, з яким атомом пов'язаний атом, що нас цікавить. Приклад -
поведінка різних атомів водню в спиртах.
Теорія будови О. Бутлерова поглинула теорію радикалів, оскільки будь-яка
частина молекули, що переходить у реакції з однієї молекули в іншу, є радикалом,
проте вже не має прерогативи незмінності. Вона увібрала в себе і теорію типів,
адже наявні в молекулі неорганічні чи такі, що містять Карбон, групи, які беруть
початок від води (гідроксил -ОН), амоніаку (аміногрупа -МНЬ), вугільної кислоти
(карбоксил -СООН), насамперед визначають хімічну поведінку (функцію)
молекули і роблять її подібною за поведінкою до прототипу.
Структурна теорія будови органічних сполук дала змогу класифікувати
величезний експериментальний матеріал і накреслила шляхи цілеспрямованого
синтезу органічних речовин.
Зазначимо, що структуру речовини хімічним шляхом визначають кожного
разу індивідуально. Для цього потрібно бути впевненим в індивідуальності
речовини і знати кількісний елементний склад та молекулярну масу. Якщо
відомі склад сполуки та її молекулярна маса, то можна вивести молекулярну
формулу. Наведемо приклад виведення структурних формул для речовин зі
складом СгНбО.
Перша речовина реагує з натрієм за типом води, виділяючи один атом
Гідрогену на один атом Натрію, причому Натрій є в складі молекули продукту
реакції замість атома Гідрогену, що відійшов:
Ломоносов М. (1711-1765) - російський учений, засновник першої в Росії хімічної лабораторії,
автор принципу збереження матерії та руху. Працював у галузі фізичної хімії, мінералогії,
металургії. Займався розбудовою атомістичних уявлень.
2 Дальтон Д. факоп І.) (1766-1844) - англійський хімік і фізик, автор хімічного атомізму. Вивів
закон кратних співвідношень, увів поняття "атомна вага", уперше визначив атомну вагу декількох
елементів. Відкрив газові закони.
Розділ 2. Теорія будови органічних сполук
39
2С2Н60 + 2Иа -> Н2 + 2С2Н5ОЫа
В одержану сполуку вже не вдається ввести інший атом натрію. Тобто
можна припустити, що речовина має гідроксильну групу і, щоб позначити її в
формулі, сполуку можна записати так: С2Н5ОН. Підтвердженням цього висновку
є також те, що під час дії на вихідну речовину фосфор (III) бромідом
гідроксильна група відщеплюється від молекули як єдине ціле, переходячи до
атома Фосфору, а її заміщує атом Брому:
2С2Н5ОН + РВгз -> ЗС2Н5Вг + Н3Р03
Далі, якщо на С2Н5Вг у водно-спиртовому розчині подіяти цинковим
пилом, то виділиться газ етан С2Нб, у якому два атоми вуглецю зв'язані між
собою. Структуру молекули одержаного газу, беручи до уваги валентність
Карбону 4, а Гідрогену 1, не можна позначити інакше, ніж:
тоді речовині С2Н5Вг потрібно приписати структуру
н-^-с-вг,
н А
а для вихідної речовини буде визначена структурна формула, тобто структура
етилового спирту:
?¥
н-<р-сч)-н,
н н
Речовина, яка ізомерна етанолу, має таку ж брутто-формулу, проте не реагує
з металевим натрієм, а під час взаємодії з гідроген іодидом розкладається за
рівнянням
С2Н60 + НІ -> СН3І + СН4О
Звідси можна зробити висновок, що у вихідній речовині два атоми Карбону
не зв'язані один з одним, оскільки гідроген іодид не здатний розривати С-С-
зв'язок. У ній немає й особливого Гідрогену, який може бути заміщений на
натрій. Після розриву молекули цієї речовини в разі дії гідроген іодиду
утворюються дві молекули СН4О і СН3І. Молекулі СН3І не можна приписати
іншої структури, ніж зображена нижче, оскільки і Гідроген, і Іод одновалентні:
40 Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
і
н
н — с—І
н
Інша молекула з утворених речовин - СН4О - поводить себе в реакції не
тільки з Натрієм, а й з Фосфор (III) бромідом подібно до етанолу:
2СН,0 + 2Иа -> 2СН3ОЫа + Н2
ЗСН4О + РВгз СНзВг + Р(ОН)3
Природно припустити, що гідроген іодид розірвав зв'язок між двома
метальними групами, які з'єднані через атом Оксигену:
т г
н-(р-о-<|:-н + ні —- н-ср-і + н-о-^-н
н н н н
Справді, у разі дії одного з продуктів цієї реакції на натрієву похідну іншого
вдається синтезувати вихідну речовину, ізомерну етиловому спирту, і
підтвердити прийняту для неї структуру диметилового етеру:
г V ї ї
Н-С-І + Иа-О-С-Н Н-С-О-^-Н
Н Н Н Н
Першим наріжним каменем перевірки теорії будови органічних сполук став
синтез передбачених, але невідомих на той час /юрет-бутилового спирту й ізобути-
лену, виконаний автором створеної теорії та його учнем О. Зайцевим1. Інший учень
О. Бутлерова - В. Марковников2 - синтезував теоретично передбачену ізомасляну
кислоту і на її основі вивчив взаємний вплив атомів у молекулі.
Наступний етап у розвитку теоретичних питань пов'язують з виникненням
стереохімічних уявлень, розвинутих у працях Я. Вант-Гоффа3 і Ж. Ле Беля4. На
початку XX ст. закладено уявлення про електронну будову атомів і молекул. На
електронному рівні тлумачили природу хімічного зв'язку і реакційної здатності
органічних молекул.
1 Зайцев О. (1841-1910) - російський хімік-органік, учень О. Бутлерова. Дослідження в галузі
органічного синтезу і теорії хімічної будови.
2 Марковников В. (1837-1904) - російський хімік-органік. Дослідження в галузі теоретичної
органічної хімії, органічного синтезу і нафтохімії.
3 Вант-Гофф Я. (van't Hoff J.) (1852-1911) - голландський хімік. Один із засновників фізичної хімії
та стереохімії. Перша Нобелівська премія з хімії 1901 р.
4 Ле Бель Ж. (Le Bel J.) (1847-1930) - французький хімік. Одночасно і незалежно від Я. Вант-
Гоффа сформулював теорію просторової будови органічних сполук.
Розділ 2. Теорія будови органічних сполук
41
Створення теорії будови органічних речовин стало основою синтетичних
методів не лише в лабораторії, а й у промисловості. Виникли виробництва
синтетичних барвників, вибухових речовин і медикаментів. В органічний синтез
широко запроваджено каталізатори і високі тиски.
У галузі органічного синтезу одержано багато природних сполук (хлорофіл,
вітаміни, антибіотики, гормони). Виявлена роль нуклеїнових кислот у збере-
женні й передаванні спадковості.
Вирішення багатьох питань будови складних органічних молекул стало
ефективним завдяки залученню сучасних спектральних методів.
Розділ З
ОСНОВИ ТЕОРІЇ
ЕЛЕКТРОННОЇ Б УЛОВИ
ОРГАНІЧНИХ МОЛЕКУЛ
Поряд з традиційними завданнями (синтез, виділення, очищення і
визначення структури одержаних сполук) перед сучасним хіміком-органіком
часто постають проблеми, пов'язані з поясненням механізму перебігу реакції та
передбаченням реакційної здатності органічних сполук. Необхідною базою для
вирішення подібних завдань є знання та вміння використовувати практично
теорію електронної будови органічних молекул, яка ґрунтується на квантово-
механічній теорії будови атома і природи хімічного зв'язку. Оскільки вивчення
органічної хімії студент починає після ознайомлення з основами загальної та
квантової хімії, то коротко нагадаємо основи цих теорій.
3.1. Основи квантово-механічної теорії будови
атома
В основі квантово-механічної теорії будови атома є планетарна модель
Е. Резерфорда1 (1911), згідно з якою атом складається з ядра, яке має позитивний
заряд, та електронів, що мають негативний заряд і обертаються навколо ядра,
утворюючи електронну оболонку. Оскільки об'єкти мікросвіту не підлягають
законам класичної механіки і принципово не можливо визначити траєкторії руху
електрона, то у квантовій механіці поняття орбіти замінено поняттям "орбіталь".
Електронна орбіталь - це область простору навколо ядра, де ймовірність
перебування електрона найбільша.
Найважливішими характеристиками орбіталі є гранична поверхня і функція
радіального розподілу ймовірності перебування електрона. Гранична поверхня
орбіталі визначає її форму. Зазвичай, граничну поверхню вибирають так, щоб вона
обмежувала простір навколо ядра з імовірністю перебування електрона, що
дорівнює 90%. Функція радіального розподілу відображає ймовірність пере-
бування електрона на різній відстані від ядра. Для електронної орбіталі основного
1 Резерфорд Е. (Rutherford Е.) (1871-1937) - англійський фізик. Один із засновників ядерної
фізики. Відкрив а- і Р-промені, виконав перше штучне перетворення одного елемента в інший.
Нобелівська премія 1908 р.
Розділ 3. Основи теорії електронної будови органічних молекул
43
стану атома Гідрогену гранична поверхня і функція радіального розподілу
ймовірності мають такий вигляд:
У квантовій механіці кожній орбіталі відповідає так звана хвильова функція
ОР-функція, псі-функція). Сама хвильова функція не має фізичного сенсу, проте
квадрат її значення у будь-якій точці простору навколо ядра пропорційний до
ймовірності перебування електрона в нескінченно малому об'ємі, який містить
цю точку (сіУ).
Хвильові функції є розв'язком хвильового рівняння (Е. Шредінгер1, 1926 р.)
- основного рівняння квантової механіки:
НХР = ЕХЇ/, (3.1)
де Н - оператор повної енергії (оператор Гамільтона, гамільтоніан); Е - повна
енергія системи.
Коли гамільтоніан розкривають, то простота рівняння зникає, оскільки цей
гамільтоніан містить кінетичну частину і потенціальну енергію електрона (и):
п2 ,д2
"2т(ах2
ду2 дг2
)\|/ + и\|/ = Е\|/
(3.2)
Розв'язування рівняння Шредінгера пов'язано з колосальними складнощами.
Точно його можна розв'язати лише для систем з одним електроном, наприклад,
для атома Гідрогену. Водночас розв'язання рівняння Шредінгера навіть для
досить простих систем привело до двох цікавих наслідків.
1. Розв'язки хвильового рівняння дискретні і є набором хвильових функцій
за квантованими значеннями енергії атома, які відповідають цим функціям.
2. Хвильові функції містять числові параметри, які передають цілими числами і
які змінюються на одиницю, - квантові числа. Орбіталі атома Гідрогену можна
якісно описати за допомогою набору квантових чисел. Кожне квантове число відіграє
важливу роль, забезпечуючи квантування (дискретність) певної фізичної величини.
1 Шредінгер Е. (ЗсИгосНг^ег Е.) (1887-1961) - австрійський фізик. Один із засновників квантової
механіки. Нобелівська премія 1933 р.
44
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
■ Головне квантове число (п) визначає енергію орбіталі й середню відстань
електрона від ядра. Воно набуває значень 1, 2, 3, 4 ... оо. Відповідно до цього,
електронну оболонку атома поділяють на низку енергетичних рівнів (К, Ь, М,
що розташовані на різних відстанях від ядра. Мінімальну енергію має
перший рівень. Енергію стану, що відповідає йонізації атома (віддалення
електрона на нескінченну відстань від ядра, п = оо), прийнято за нуль.
■ Орбітальне квантове число (£) характеризує форму (симетрію)
електронної орбіталі та орбітальний імпульс електрона під час його руху. Число
і може мати значення від 0 до /7-1, якщо п фіксоване. Для атомів з багатьма
електронами і впливає також на енергію орбіталі, унаслідок чого кожен
енергетичний рівень має низку підрівнів з різними значеннями орбітального
квантового числа та орбіталями різної форми (.?,/?, сі,/...).
■ Магнітне орбітальне квантове число (т,) визначає кількість орбіталей
для кожного підрівня, набуваючи значень від —£ до +£9 і квантує проекцію
орбітального моменту електрона на одну з осей.
Підрівень 5 має лише одну орбіталь (£ = 0, т, = 0) сферичної форми, підрівень
р - три, які мають форму гантелей і напрямлені у просторі по осях прямокутної
системи координат (рх, ру, р2) (рис. 3.1).
у І У ' у Т
Рис. 3.1. Форма граничної поверхні б- і р-орбіталей. Показані математичні
знаки відповідних хвильових функцій
Розглянуті квантові числа квантують фізичні характеристики електронних
орбіталей як частин простору, де дозволено бути електронам. Природно таке: якщо
Розділ 3. Основи теорії електронної будови органічних молекул
45
електрон займає ту чи іншу орбіталь, то він автоматично набуває її квантових чисел.
До того ж, електрон має два власні квантові числа - спінове та магнітне спінове.
■ Спінове квантове число (б) квантує власний обертальний момент кількості
руху елементарної частинки. Для електрона 8=1/2. Магнітне спінове квантове
число (т8) визначає проекцію власного обертального моменту руху елементарної
частинки на одну з осей і набуває значень від -з до +б. Для електрона магнітне
спінове квантове число має два значення: -1/2 (Р-спін) і +1/2 (а-спін).
Як уже зазначено, точне розв'язання рівняння Шредінгера можливе лише
для атомів з одним електроном. Тому для опису електронних оболонок атомів з
багатьма електронами доводиться вдаватися до низки спрощень і наближень.
1. Принцип схожості на атом Гідрогену. Відповідно до цього принципу,
електронну оболонку атома з багатьма електронами можна описати набором
орбіталей і квантових чисел, які розраховані за аналогією для атома Гідрогену.
2. Принцип мінімальної енергії - електрони заповнюють орбіталі в порядку
зростання їхньої енергії. Послідовність збільшення енергії електронних орбіталей
визначена правилами Клечковськогох\
• енергія орбіталей зростає в порядку збільшення суми головного та
орбітального квантових чисел (п+£);
• за однакових значень (п+^)- енергія орбіталей збільшується зі збільшенням
головного квантового числа.
Згідно з правилами Клечковського, енергія орбіталей зростатиме в такій
послідовності:
Ь < < 2р < 3^ < Зр < 4* < ЗсІ< Ар < 5б < Ы< 5р < 6б < 4/< 5с1...
3. Принцип заборони Паулі2 - неможливе існування двох електронів з
однаковим набором квантових чисел. Звідси випливає, що електрони,
розташовані на одній орбіталі, можуть відрізнятися лише значенням спінового
квантового числа, оскільки я, і і характеризують орбіталь і автоматично
присвоюються її електронам. Оскільки спінове магнітне квантове число набуває
двох значень, то на одній орбіталі може одночасно бути лише два електрони з
різною орієнтацією спіну. Наявність в електрона спіну і його орієнтацію
позначають в електронних формулах стрілкою: а-спін (т5 = +1/2) - Т; Р-спін
(т8= -1/2) - і. У зовнішньому магнітному полі стан з Р-спіном має меншу
енергію. Якщо магнітного поля нема, то енергія станів з а- і Р-спіном однакова.
4. Правило Гунда3: заповнюючи енергетичний підрівень, електрони
1 Клечковський В. (1900-1972) - радянський агрохімік. Головні дослідження пов'язані з
запровадженням методу мічених атомів в агрохімію. Теоретичні праці з явища періодичності.
2 Паулі В. (Pauli W.) (1900-1958) - швейцарський фізик-теоретик. Один із творців квантової
механіки і теорії поля. Передбачив існування нейтрино. Нобелівська премія з фізики 1945 р.
3 Гунд Ф. (Hund F.) (1896-1997) - німецький фізик. Фундаментальні дослідження в галузі квантової
механіки, магнетизму і квантової хімії. Один із творців методу МО, увів поняття про а- і я-зв'язки.
46
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
намагаються зайняти максимальну кількість орбіталей, орієнтуючи спіни
паралельно. Наприклад, якщо електрони реалізують конфігурацію р3, то вони
заповнюють орбіталі так:
141
На підставі викладених вище принципів розглянемо будову електронних
оболонок атомів елементів малих періодів Періодичної системи. Саме ці періоди
особливо цікаві для хіміків-органіків, оскільки містять основні елементи, що
властиві живій природі (Н, С, >ї, О, Р, 8). Перший період має два елементи -
Гідроген та Гелій, у яких заповнюється перший енергетичний рівень:
К
1э
К
1э
Н: І81
Не: и2
Для складання електронної формули атома або іншої квантової частинки
числом задають головне квантове число, літерою - орбітальне квантове число, а
верхнім індексом - кількість електронів на підрівні. Оскільки ємність першого
рівня обмежена двома електронами, то наступний елемент Літій починає другий
період. В елементів другого періоду відбувається послідовне заповнення другого
енергетичного рівня, починаючи з конфігурації
конфігурацією 1.у22$22р6 (№).
(Іл) і закінчуючи
Е
І
К
2р
Е
І
К
МИ- *
4
18
Іл: 1 А*1
Ые: Ь22522/?6
Аналогічно відбувається заповнення ^ і р-підрівнів третього енергетичного
рівня в елементів третього періоду: Ыа: КХЗ^1; Аг: КЬ3.523/?6.
Далі можна було очікувати заповнення орбіталей Зб/-підрівня. Проте в
елементів четвертого періоду заповнення починається з четвертого рівня, оскільки
енергія 4у-підрівня нижча, ніж енергія Зб/-орбіталей - к: КЬМ4бх; Са: кьм4б2.
Розділ 3. Основи теорії електронної будови органічних молекул
47
Після заповнення 4^-орбіталі, відповідно до правил Клечковського, починається
заповнення З^-підрівня. Оскільки <і-підрівень уміщує десять електронів, то цей
процес поширюється на десять елементів (від Скандію до Цинку).
3.2. Основи квантово-механічної теорії
хімічного зв'язку
Хімічний зв'язок - це наслідок електростатичної взаємодії ядер та електронів,
які утворюють стійку сукупність атомів - молекулярні частинки або атомні
агрегати.
Рухомою силою утворення хімічного зв'язку є прагнення системи до
мінімуму енергії, що досягається після завершення електронної оболонки (я2 або
^р6). Залежно від способу наближення системи атомних частинок до стійкого
стану розрізняють три типи хімічного зв'язку: ковалентний, іонний і металевий.
У теорії хімічного зв'язку звичайно розглядають також сили міжмолекулярної
взаємодії (сили Ван-дер-Ваальса1), які, по суті, є фізичною взаємодією, і
водневий зв'язок, що є на межі фізичної і хімічної взаємодій.
З розвитком квантово-механічних уявлень у теорії хімічного зв'язку склалось
два методи опису ковалентного зв'язку: метод валентних зв'язків (метод ВЗ) і
метод молекулярних орбіталей (метод МО).
Згідно з методом ВЗ, атоми, що є в складі молекули, зберігають свою
індивідуальність, а хімічні зв'язки виникають унаслідок взаємодії їхніх
валентних електронів і валентних орбіталей. Метод МО розглядає молекулу як
єдине утворення, у якому кожний електрон належить частинці загалом і
рухається в полі всіх її ядер та електронів. Методи ВЗ і МО, незважаючи на
суттєві відмінності в описі молекул, добре доповнюють один одного.
3.2.1. Головні положення методу валентних зв'язків
Ковалентний зв'язок виникає завдяки утворенню спільної електронної пари,
коли електронні орбіталі атомів, що взаємодіють, перекриваються. Розрізняють два
механізми утворення спільної електронної пари - обмінний і донорно-акцепторний.
Якщо діє обмінний механізм, то кожний з атомів, які взаємодіють, надає для-
утворення спільної електронної пари неспарений електрон з валентної орбіталі.
За цим механізмом утворені, наприклад, зв'язки в молекулі метану:
Н
• С • + 4 Н • —Н : С : Н
• • •
Н
1 Ван-дер-Ваальс Я. (van der Waals J.) (1837-1923) - голландський фізик. Увів рівняння стану для
реальних газів і рідин, яке враховує міжмолекулярну взаємодію. Нобелівська премія з фізики
1910 р.
48
Чирва В.Я.Л Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
Якщо ковалентний зв'язок утворюється за донорно-акцепторним ме-
ханізмом, то один з атомів відіграє роль донора, віддаючи в загальне
користування неподілену пару електронів, розташовану на одній з його
валентних орбіталей. Інший атом - акцептор - надає для утворення зв'язку
вакантну орбіталь, приймаючи на неї електронну пару донора. За донорно-
акцепторним механізмом утворюється зв'язок >Т-Н, якщо протонується амоніак
або аміни:
Н
Н:И: + Н+
н
• За кількістю спільних електронних пар, які зв'язують атоми, розрізняють
прості, подвійні та потрійні зв'язки:
Н3С:СН3 або Н3С-СН3
Н2С::СН2 або Н2С=СН2
НС:::СН або НОСИ
• За характером перекривання орбіталей, що взаємодіють, в органічній хімії
виділяють два типи ковалентного зв'язку: а-зв'язок, якщо перекривання
орбіталей відбувається вздовж лінії зв'язку, та п-зв'язок, якщо перекривання
відбувається в площині, яка містить лінію зв'язку:
<Зе>о
❖ Характеристики зв'язку. Найважливішими фізичними характеристи-
ками хімічного зв'язку є його енергія і довжина.
> Енергія хімічного зв'язку - це кількість енергії, необхідна для розриву
зв'язку. Така ж кількість енергії виділяється, коли зв'язок утворюється.
Наприклад, енергія дисоціації молекули водню становить 435 кДж/моль. Відпо-
відно, ЕН-н = 435 кДж/моль.
> Довжина зв'язку - відстань між ядрами, що зв'язані хімічно. її
вимірюють у нано- (1 нм = 1-Ю"9 м) або пікометрах (1 пм = 1-Ю"12 м). Енергії та
довжини головних зв'язків наведено в табл. 3.1.
Н
Н:И:Н
Н
Розділ 3. Основи теорії електронної будови органічних молекул
49
Таблиця 3.1
Енергії та довжини зв'язків в органічних молекулах
Зв'язок
Енергія, кДж/моль
Довжина, пм
С-С
330-360
151-154
С=С
590-640
134
С=С
810-840
120
С-Н
402-^55
107-110
С-0
350-381
136-143
с=о
680-760
115-122
270-314
133-148
598-616
128
650-896
116
260-276
175-181
Довжина ковалентного зв'язку приблизно дорівнює сумі ковалентних радіусів
атомів:
^А-в = гА + гв. (3.3)
Значення ковалентних радіусів для кожного елемента залежать від його
стану в молекулі та найближчого оточення, наприклад, для Карбону кова-
лентний радіус залежить від стану гібридизації:
Елемент
С(5/73)
С(8р2)
С{зр)
N
О
н
г, пм
77
67
60
70
66
33-37
> Валентний кут - кут між умовними лініями, які з'єднують ядра атомів,
що хімічно зв'язані.
Наприклад, молекула води має кутову форму з валентним кутом НОН 104,5°
і довжиною зв'язків О-Н 96 пм:
Енергія, необхідна для повної дисоціації молекули, тобто для~ виконання
процесу Н20 -> 2Н + О, становить 924 кДж/моль. Отже, енергія зв'язку ОН
дорівнює 462 кДж/моль.
• Полярність ковалентного зв'язку. Якщо ковалентний зв'язок утворюють
атоми з однаковою електронегативністю, то спільна електронна пара однаково
належить партнерам. Такий зв'язок називають ковалентним неполярним. Якщо
атоми, які утворюють зв'язок, відрізняються за електронегативністю, то загальна
електронна пара зсувається до атома з більшою електронегативністю. Зв'язок,
що утворюється, називають ковалентним полярним.
Унаслідок несиметричного розподілу електронної густини двохатомні моле-
кули з ковалентним полярним зв'язком є диполями електронейтраільних частинок,
50
Чирва В.Я., Ярмолюк CM., Толкачова Н.В., Земляков О.Є. Органічна хімія
н
н
\5+ 5-
н^с—сі
н
н
центри тяжіння позитивного і негативного зарядів у яких не збігаються. У
формулі полярність ковалентного зв'язку передають декількома способами:
Н
\8+ 5-
Н^С-С1
Н
■ Кількісною характеристикою полярності є її дипольний момент, точніше -
електричний момент диполя
Ц = 8-Яе-/, (3.4)
де Яе - заряд електрона, І - довжина зв'язку.
Одиницею вимірювання дипольного моменту є кулон на метр (Кл-м (81)) або
позасистемна одиниця - дебай (1 Б = 3,34-10"30 Кл-м). Дипольний момент молекули
визначають як векторну суму дипольних моментів її зв'язків і неподілених
електронних пар. Унаслідок цього молекулярні частинки, що мають однакову
форму, але різні зв'язки, можуть значно відрізнятись полярністю, наприклад:
\ ї
Н Б
}і= 1,470 ц = 0,2Б
■ Кількісною характеристикою здатності атома відтягувати електрони, коли
утворюються хімічні зв'язки, є електронегативність (%). Електронегативність -
величина умовна і не може бути точно визначена експериментально. Сьогодні
відомо близько 20 шкал електронегативності. Найчастіше використовують
відносну шкалу електронегативності, запропоновану Л. Полінгом1.
Для типових елементів у періодах електронегативність збільшується
монотонно, наприклад: Ьі 1,0; Ве 1,5; В 2,0; С 2,5; N 3,0; О 3,5; Б 4,0. Водночас у
межах групи електронегативність змінюється немонотонно зі збільшенням
порядкового номера (вторинна періодичність), наприклад: Ьі 1,0; Иа 0,9; К 0,8;
ЯЬ 0,8; сб 0,7. Причиною виявлення вторинної періодичності є немонотонне
збільшення радіуса атома зі збільшенням кількості електронних оболонок.
1 Полінг Л. (Pauling L.) (1901-1994) - американський фізик і хімік. Головні дослідження
присвячені будові молекул і хімічного зв'язку. Один із розробників методу валентних зв'язків, у
тім числі теорії резонансу. Дослідження структури білків і нуклеїнових кислот. Нобелівська
премія з хімії 1954 р. та Нобелівська премія миру 1962 р.
Розділ 3. Основи теорії електронної будови органічних молекул
51
Електронегатнвність елемента залежить від його валентного стану і типу
гібридизації, наприклад:
Елемент
С(*/)
С(л/>)
г
2,5
2,75
3,2
3,51
3,74
• Поляризовність. Важливою характеристикою ковалентного зв'язку, яка
головно визначає його реакційну здатність, є поляризовність - здатність зв'язку
змінювати полярність (перерозподіляти електронну густину під дією
зовнішнього електростатичного поля), джерелом якого можуть бути каталізатор,
реагент, розчинник тощо. Наведений диполь частинки пов'язаний з напру-
женістю зовнішнього поля простим співвідношенням: р, = осЕ. Коефіцієнт
пропорційності а є кількісною характеристикою поляризовності.
❖ Насичуваність і напрямленість ковалентного зв'язку. Ковалентний
зв'язок має дві найважливіші властивості: насичуваність і напрямленість:
Насичуваність ковалентного зв'язку полягає в тому, що атоми здатні до утворення
максимальної кількості ковалентних зв'язків. Причиною насичуваності кова-
лентного зв'язку є обмежена кількість валентних орбіталей атома, необхідних для
утворення зв'язків як за обмінним, так і за донорно-акцепторним механізмом.
Кількісно насичуваність ковалентного зв'язку характеризують ковалент-
ністю. Ковалентність (структурна валентність) дорівнює кількості ковалентних
зв'язків, утворених атомом.
Якщо відома кількість орбіталей на валентних електронних рівнях, то можна
розрахувати максимальну теоретично можливу валентність для елементів різних
періодів. У атомів елементів першого періоду на валентному (першому) рівні є
лише одна орбіталь (1$), через те Гідроген у всіх сполуках одновалентний. Гелій,
атом якого має повністю завершений перший рівень, хімічних сполук не утворює. В
елементів другого періоду валентним є другий енергетичний рівень, який має
чотири орбіталі - 2^, 2рх, 2ру, 2рг. Тому максимальна ковалентність елементів
другого періоду, наприклад, Нітрогену, дорівнює чотирьом:
0
Н
к \ я к \ я
ук = 3; vN = 4.
Напрямленість ковалентного зв'язку зумовлена прагненням атомів утворювати
зв'язки в напрямі найбільшого перекривання орбіталей, що забезпечує
максимальний виграш енергії. Це приводить до того, що молекули, утворені за
участю ковалентних зв'язків, мають чітко визначену форму.
❖ Гібридизація. Оскільки форму більшості молекул не можна пояснити
утворенням ковалентних зв'язків зі стандартного набору атомних орбіталей, то
52
Чирва В.Я., Ярллолюк СМ., Толкачова H.B., Земляков О.Є. Органічна хімія
Л. Полінг розробив теорію гібридизації атомних орбіталей. За цією теорією процес
утворення молекулярної частинки вирівнює ковалентні зв'язки шляхом гібридизації
атомних орбіталей. Процес гібридизації можна описати як змішування хвильових
функцій атомних орбіталей з утворенням нового набору еквівалентних орбіталей.
Сам по собі процес гібридизації потребує затрат енергії, проте утворення зв'язків за
участю гібридних орбіталей енергетично вигідніше, оскільки забезпечує найповні-
ше перекривання електронних хмарок і мінімальне відштовхування електронних пар.
■ Найпростішою є Бр-гібридизація, яка виникає в разі змішування
хвильових функцій 5- і однієї/>орбіталі:
5/7
Утворені 5/7-гібридні орбіталі орієнтовані по одній осі в різні боки, тому кут
між зв'язками, утвореними за участю цих орбіталей, становить 180°. Зокрема,
.sp-гібридизація атомів Карбону виникає в молекулах органічних сполук, якщо
утворюються потрійні зв'язки (алкіни і нітрили):
180°
н—с=с—н
■ Під час гібридизації .у- і двох /?-орбіталей виникають три гібридні
орбіталі {зр2-гібридизація), орієнтовані до вершин правильного трикутника:
Валентний кут між зв'язками, що утворені за участю гібридних орбіталей
цього типу, становить 120°; ^-гібридизація атомів Карбону виникає в
молекулах органічних сполук, якщо утворюються подвійні зв'язки (алкени,
альдиміни тощо), а також ароматичні системи, наприклад, молекула бензолу:
-*vl20°
Розділ 3. Основи теорії електронної будови органічних молекул
53
■ Під час лр3-гібридизації утворюється набір із чотирьох орбіталей з
однаковою енергією, що орієнтовані під кутом 109,5° від центру до вершин
тетраедра одна щодо одної:
8р>
Зазначимо, що зр -гібридизація атомів Карбону виникає в молекулах
насичених вуглеводнів (алканів) та їхніх функціональних похідних.
Розглянемо, наприклад, будову деяких молекул, що утворені за участю ^р3-
гібридизованих орбіталей.
Молекула метану - скц. З енергетичної діаграми атома Карбону випливає,
що наявних двох неспарених електронів не достатньо для утворення кова-
лентних зв'язків за обмінним механізмом, тому молекула утворюється за участю
атомів Карбону в збудженому стані:
Е
к
4-4--
2р
Е
4
їв
к
4-4--І- *
4
їв
Рівноцінність зв'язків і тетраедрична геометрія молекули метану свідчать про
утворення зв'язків за участю .^-гібридизованих орбіталей центрального атома:
Н
Н
н
109,5«
• Молекула амоніаку - ІМНз. Атомні орбіталі Нітрогену молекули амоніаку
також перебувають в ^-гібридизованому стані. Та оскільки лише три орбіталі
утворюють зв'язки нітроген-гідроген, а четверта має неподілену електронну пару,
54
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
то молекула є пірамідальної форми. Відштовхувальна дія неподіленої електронної
пари зменшує валентний кут до 107,3° (прогнозований кут - 109,5°):
Неподілена електронна пара Нітрогену дає змогу утворювати ще один
додатковий зв'язок за донорно-акцепторним механізмом. Так утворюється
молекулярний катіон амонію - 1МН/. Утворення четвертого ковалентного
зв'язку зумовлює вирівнювання валентних кутів (109,5°) завдяки рівномірному
відштовхуванню атомів Гідрогену:
Н
• Молекула води - НгО. Вона утворюється із залученням зр3-гібридних орбіталей
атома Оксигену, дві з яких заповнені електронними парами. Перекривання двох
гібридних орбіталей Оксигену й 1^-орбіталей двох атомів Гідрогену утворює кутову
молекулу. Валентний кут НОН зменшується до 104,5° внаслідок відштсіЬхування
зв'язувальних електронів від двох неподілених електронних пар:
Також .^-гібридизація атома Оксигену виникає в молекулах спиртів і етерів:
Розділ 3. Основи теорії електронної будови органічних молекул
55
Розглянуті приклади засвідчують переваги методу ВЗ, насамперед, його
наочність і простоту опису будови молекул на якісному рівні. Метод ВЗ має і
недоліки: зокрема, він не пояснює утворення делокалізованих багатоцентрових
зв'язків.
❖ Резонансні структури. Щоб описати молекули з делокалізованими
зв'язками в методі ВЗ, потрібно використовувати спеціальний спосіб - резонанс
валентних схем. Згідно з концепцією резонансу, будову молекул з делока-
лізованими зв'язками передають не однією формулою, а накладанням декількох
валентних схем (формул), які реально не існують. Наприклад, будову молекули
нітрометану з делокалізованим трицентровим зв'язком у методі ВЗ передають
резонансом двох валентних схем (граничних структур):
н3с—щ
О
= Н,С—N " Н,С—N
3 \ 3 ^
О" о
Резонансна Граничні структури
структура
Валентні схеми, що перебувають у резонансі, мають поряд з подвійним
зв'язком нітроген-оксиген так званий семиполярний зв'язок, який можна
розглядати як комбінацію ковалентного та йонного зв'язків.
Щоб описати молекулу бензолу, у методі валентних зв'язків потрібно
вдатися до резонансу п'яти канонічних форм, найважливішими з яких є такі:
❖ Іонний зв'язок. Поняття про йонний зв'язок використовують в органічній
хімії лише для того, щоб описати подібні до солей сполуки:
О СН3
н3с-с( Н3С^ч- СІ-
О №+ НзС СНз
Натрій ацетат Тетраметиламоній хлорид
Ми детально не розглядатимемо особливості йонного зв'язку, його опис
можна знайти у відповідних розділах підручників із загальної та неорганічної хімії.
3.2.2. Метод молекулярних орбіталей
Як уже зазначено, метод МО розглядає молекулу як єдине утворення, у якому
кожний електрон належить частинці й рухається по молекулярних орбіталях у
56
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
полі всіх ядер і решти електронів молекули. Молекулярна орбіталь - це ділянка
простору в молекулі, ймовірність наявності електрона в якій суттєво відрізняється
від нуля. На відміну від атомної орбіталі (АО), молекулярна орбіталь (МО)
багатоцентрова і поширюється на декілька ядер. Електрони заповнюють
молекулярні орбіталі в порядку збільшення їхньої енергії та відповідно до
принципу заборони Паулі й правила Гунда. Хвильову функцію МО (у) будують
як лінійну комбінацію хвильових функцій АО (*рі):
Коефіцієнти Сі зумовлені нормуванням хвильової функції, згідно з яким
імовірність наявності електрона на заповненій орбіталі дорівнює одиниці:
Атомні орбіталі, придатні для складання МО, повинні мати близьку енергію,
однакову симетрію і суттєво перекриватися. У цьому разі кількість одержаних МО
дорівнює кількості вихідних АО.
Розрізняють три типи молекулярних орбіталей: зв 'язувальна, розпушувальна
(антизв'язувальна) і незв'язувальна. Зв'язувальна МО має меншу енергію, ніж
вихідні атомні орбіталі. її заповнення стабілізує молекулу. Розпушувальні
молекулярні орбіталі мають вищу енергію порівняно з вихідними АО. Заповнення
електронами розпушувальних МО дестабілізує частинку. Незв'язувальні МО
локалізовані на окремих атомах і фактично є атомними орбіталями, перенесеними
в молекулу без змін. За симетрією МО поділяють на а (симетричні відносно лінії
зв'язку) і я (антисиметричні відносно лінії зв'язку).
Щоб проілюструвати принципи методу МО, розглянемо електронну будову,
молекули Гідрогену та інших двохатомних молекулярних частинок, утворених
елементами першого періоду. Як базисний набір АО використовуватимемо і5-
орбіталі, лінійні комбінації хвильових функцій яких дадуть Ч'-функції двох
молекулярних орбіталей:
\|/ = £ Сі¥.
(3.5)
\ (1У = 1.
Ч'+ = сіЧ'„а + с2Ч',л
(3.6)
*Р- ~ Сз^^д - С4ХРі5в .
Приймемо для спрощення, ЩО Сі = с2 = с3 = с4 = 1. Тоді
*Р+ = ¥а + ¥ь
(3.7)
На рис. 3.2 зображено криві хвильових функцій *Ра і Ч'ь (штрихова лінія) і
криву молекулярної хвильової функції *Р+ (суцільна лінія). Щоб знайти значення
Розділ 3. Основи теорії електронної будови органічних молекул
57
хвильової функції, достатньо скласти значення Шг і ЧРь, що відповідають кожній
точці. Як випливає з рис. 3.2, для ділянок негативних значень координати х і
¥ь практично збігаються з кривою х¥+9 а в просторі між ядрами атомів Гідрогену
більше від нуля за будь-яких значень г. Оскільки
¥+2 = ¥а2 + ¥ь2 + Т¥ЛЧ?Ь > ^а2 + уь2, (3.8)
то ймовірність наявності електронів між ядрами, що визначена значенням Ч?+29
буде більшою, якщо електрони розташовані на орбіталі у¥+9 ніж тоді, коли
кожний з них перебував би на одній з атомних орбіталей. Відповідно, зростає
густина негативного заряду між ядрами, що спричиняє зближення ядер. Завдяки
цьому молекулярні орбіталі, які є сумою атомних орбіталей, мають меншу
енергію, ніж атомні орбіталі, які комбінуються. Заселення таких орбіталей
електронами стабілізує утворювану молекулу.
а б
Рис. 3.2. Хвильові функції і|/а, \|/ь і (а) і і|/а, і|/ь і Ч^б)
Для молекулярної орбіталі значення Ч'ь потрібно брати зі знаком мінус,
унаслідок чого для точки, рівновіддаленої від ядер атомів На і Нь, значення
дорівнює нулю, густина електронної хмари між ядрами знижується, а енергія
системи збільшується, що дестабілізує частинку.
Форма граничної поверхні одержаних МО та енергетична діаграма
молекули мають вигляд, як на рис. 3.3.
За аналогією з а-зв'язками в методі ВЗ, молекулярні орбіталі, симетричні
відносно лінії зв'язку, позначають символом а. Нижнім індексом визначають АО,
які беруть участь в утворенні МО. Праворуч угорі зазначають тип орбіталі (сгзв,
арозп або о113). Часто використають спрощені позначення: а = азв, а* = арозп. МО,
антисиметричні відносно лінії зв'язку, позначають символом я. У разі переходу
через лінію зв'язку МО цього типу змінюють математичний знак на протилежний.
58 Чирва В.Я., Ярллолюк CM., Толкачова Н.В., Земляков О.Є. Органічна хімія
0 0
Is,
Is,
'1s
Рис. 3.3. Молекулярні орбіталі та енергетична діаграма двохатомної молекули,
утвореної елементами першого періоду
Унаслідок розміщення на МО електронів отримаємо електронні формули від-
повідних молекулярних частинок. Для якісної характеристики стабільності молекул
у методі МО використовують кратність зв'язку (КЗ), яку розраховують як половину
різниці кількості зв'язувальних і розпушувальних електронів. Стійкість молекули
зростає, якщо кратність зв'язку збільшується. Частинки, для яких КЗ < 0, не
існують
З наведених у табл. 3.2 даних видно, що метод молекулярних орбіталей
описує не лише двох-, а й одноелектронні зв'язки. Поряд з цим метод МО
адекватніше відображає фізичні властивості молекул, зокрема, він пояснює
парамагнетизм молекули Оксигену, тоді як відповідно до методу ВЗ молекула
Оксигену повинна бути діамагнітною. Проте метод молекулярних орбіталей
дещо програє методу валентних схем у наочності. Незважаючи йа це, метод МО
широко застосовують у сучасній органічній хімії, особливо останнім часом у
зв'язку зі швидким розвитком обчислювальної техніки.
Таблиця 3.2
Кратність зв'язку і магнітна поведінка деяких частинок, що відповідають
елементам першого періоду
Частинка
Електронна формула
КЗ
Магнітна поведінка
н2+
(cris)1
1/2
Парамагнетик
н2
(ciis)2
1
Діамагнетик
н2-
(dls)W)1
1/2
Парамагнетик
Не2+
(tfls)W)1
1/2
Парамагнетик
Не2
(als)2(als.)2
0
Частинка нестійка
Розглянемо, як приклад демонстрації можливостей методу молекулярних
орбіталей, результати розрахунку деяких порівняно простих молекул.
На рис. 3.4 зображено енергетичну діаграму і контури МО, одержаних
під час розрахунку молекули метану розширеним методом Хюккеля1. Близькі
за формою МО дають також інші напівемпіричні методи розрахунку,
наприклад, AMI, а також неемпіричний метод розрахунку (ab initio). Однак
1 Хюккель Е. (Нііскеї Е.) (1896-1980) - німецький фізик і хімік-теоретик. Спільно з П. Дебаєм
створив теорію сильних електролітів. Розробив правила ароматичності.
Розділ 3. Основи теорії електронної будови органічних молекул
59
енергії молекулярних орбіталей, одержані різними методами, суттєво
відрізняються.
Як бачимо з рис. 3.4, розрахунок за методом МО дає дещо несподіваний
результат. Річ у тому, що замість очікуваного для .^-гібридизації атома
Карбону набору з чотирьох еквівалентних зв'язувальних МО одержана одна
сильно зв'язувальна МО з енергією -24,56 еВ, або 2 320 кДж/моль (1 еВ на
молекулу тотожний 96,452 кДж/моль), і три енергетично еквівалентні
(вироджені) зв'язувальні МО з енергією -15,52 еВ. Водночас існування в
молекулі метану двох рівнів, що відрізняються енергією, підтверджене
фотоелектронним спектром, у якому спостерігають два піки з потенціалами
йонізації приблизно 13 і 23 еВ. Цікаво, що розрахунки дають досить близькі
значення для енергії зв'язувальних орбіталей молекули. Отже, потрібно визнати
таке: незважаючи на те, що метод ВЗ дає чіткіший і правильніший опис форми
молекули, метод МО допомагає точніше визначити її енергетичні
характеристики.
Рис. 3.4. Енергетична діаграма і проекції контурів МО молекули метану
60
Чирва В.Я., Ярллолюк СМ., Толкачова H.В., Земляков О.Є. Органічна хімія
На рис. 3.5 і 3.7 показано енергетичні діаграми молекул етилену й
ацетилену, одержані розрахунком за методом АМ-1.
ВЗМО
1,438
OeV
-10,55
-11,84
-14,3
-15,79
-21,89
• -33,16
Рис. 3.5. Енергетична діаграма і контури граничних молекулярних орбіталей молекули етилену
ВЗМО
етилену
НВМО
етилену
Рис. 3.6. Взаємодія граничних орбіталей етилену з орбіталями катіона металу
під час утворення п-комплексу
Як видно з рис. 3.5, молекула етилену має складніший енергетичний спектр
порівняно з молекулою метану, що зумовлено більшою кількістю вихідних
атомних орбіталей. Проте опис особливостей електронної будови цієї та інших
молекул можна суттєво спростити, якщо розглядати не всі, а лише вищу зайняту
МО (ВЗМО) і нижчу вакантну МО (НВМО). Ці орбіталі часто називають
граничними, саме вони, передусім, визначають реакційну здатність молекулярної
частинки. Реакції, що відбуваються з приєднанням електрона до молекули етилену,
або атака на подвійний зв'язок нуклеофільного реагенту будуть спрямовані на
НВМО, тоді як атака електрофільного реагенту - на ВЗМО. Особливо яскраво це
видно в разі утворення я-комплексу етилену з катіонами перехідних металів, у
яких етилен, з одного боку, відіграє роль донора електронної пари ВЗМО, а з
іншого, - є акцептором пари <і-електронів металу завдяки НВМО (рис. 3.6).
Розділ 3. Основи теорії електронної будови органічних молекул 61
у,
Рис. 3.7. Енергетична діаграма і контури МО молекули ацетилену
(три останнірозпуіиувальні МО не показані)
Метод граничних орбіталей уперше запропонований К. Фукуї 1952 р., нині
його широко застосовують для опису і прогнозування реакційної здатності
органічних сполук.
ВЗМО молекули ацетилену мають нижчу енергію (-11,5 еВ), ніж ВЗМО
етилену (-10,55 еВ), тому можна очікувати, що до дії електрофільних реагентів
потрійний зв'язок буде стійкіший, ніж подвійний. Експериментально це
підтверджено для більшості електрофільних реагентів, хоча є винятки.
Отже, очевидно, що метод молекулярних орбіталей не лише точніше описує
енергетичні властивості молекул порівняно з методом валентних зв'язків, а й дає
змогу прогнозувати реакційну здатність молекулярної частинки.
1 Фукуї К. (Рикиі К.) (р. н. 1918) — японський хімік. Головні праці в галузі квантової хімії.
Розробив теорію граничних орбіталей. Застосував її для вивчення перехідних комплексів і
каталітичних реакцій. Нобелівська премія 1981 р.
62
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
3.23. Водневий зв'язок, міжмолекулярна взаємодія
❖ Водневий зв'язок
> Водневим зв'язком називають електростатичну взаємодію атома Гід-
рогену, зв'язаного з сильно електронегативним елементом, з іншими атомами.
Водневі зв'язки утворює атом Гідрогену, зв'язаний з атомом Флуору,
Оксигену або Нітрогену. Електронегативність інших елементів недостатня для того,
щоб утворився міцний водневий зв'язок з атомом Гідрогену. Механізм утворення
водневого зв'язку розглянемо на прикладі взаємодії гідроген флуориду. Висока
електронегативність атома Флуору приводить до того, що зв'язок гідроген-флуор у
цій молекулі сильно полярний, і спільна пара електронів зсунута до Флуору Н->Р.
Оскільки атом Гідрогену не має внутрішньої електронної оболонки, то відтягування
його валентного електрона майже повністю оголює ядро, яке є елементарною
частинкою - протоном. Тому сильно поляризований атом Гідрогену має досить
потужне електричне поле, завдяки якому він притягується до атома Флуору іншої
молекули гідроген флуориду, утворюючи водневий зв'язок:
100 пм и-'С-^Н. 155 пм
--.-.р^*1 120°
Водневий зв'язок має такі особливості.
1. Водневий зв'язок є насиченим. Атом Гідрогену утворює лише один
водневий зв'язок; його партнери можуть брати участь в утворенні декількох
водневих зв'язків.
2. Водневий зв'язок є напрямленим. Фрагмент Х-Н—У звичайно лінійний,
хоча в деяких випадках може бути і кутовим, проте в цьому разі валентний кут
не сильно відрізняється від 180°.
3. Енергія водневого зв'язку невелика (8-40 кДж/моль) і є значенням того ж
порядку, що й енергія міжмолекулярної взаємодії. Міцність водневого зв'язку
тим вища, чим більша електронегативність партнера атома Гідрогену.
Наприклад, енергія зв'язку Н— -Б становить 25—40 кДж/моль, зв'язку О - 19-
21 кДж/моль, зв'язків №--Н і в—Н- приблизно 8 кДж/моль.
4. Водневий зв'язок асиметричний: у фрагментах Х-Н—Х довжина зв'язку
Н -X більша від довжини Н-Х.
Водневий зв'язок має більшу довжину порівняно з ковалентним і меншу
енергію. Однак він суттєво впливає на фізичні властивості речовин, значно
підвищуючи їхні температури плавлення і кипіння. Наприклад, гідроген
флуорид має т. пл. -83 °С і т. кип. +20 °С, тоді як його найближчий аналог -
гідроген хлорид - плавиться при -114 °С і кипить при -85 °С Фактично завдяки
водневим зв'язкам, гідроген флуорид є полімером, який починає частково
дисоціювати лише за температури, близької до температури кипіння. Навіть у
газовій фазі гідроген флуорид існує у вигляді малих асоціатів молекул,
Розділ 3. Основи теорії електронної будови органічних молекул
63
переважно у вигляді димерів. Гідроген флуорид існує як мономерна молекула
лише за температур понад 90 °С. Дуже міцні водневі зв'язки утворює молекула
води, оточена чотирма сусідами в кристалічному стані (лід).
Тривимірна сітка водневих зв'язків, побудована з тетраедрів, існує в рідкій
воді в усьому інтервалі температур від плавлення до кипіння:
Поряд з міжмолекулярними існують і внутрішньомолекулярні водневі
зв'язки, які значно не впливають на фізичні властивості речовин:
2-Гідроксибензальдегід, т. пл. -7 °С 4-Гідроксибензальдегід, т. пл. +117 °С
Мурашина і багато інших карбонових кислот у рідкому і газоподібному
станах завдяки водневим зв'язкам утворюють циклічні димери:
163 пм 107 пм
о-~н—О
// \
н—с с—н
\ //
о—н--о
Дуже важливу роль водневі зв'язки відіграють у структурній організації
багатьох біологічно важливих макромолекул (ос-спіралі та р-структури білків і
поліпептидів, подвійна спіраль ДНК тощо).
❖ Сили міжмолекулярної взаємодії
> Силами міжмолекулярної взаємодії (вандерваальсовгши стами) нази-
вають сили електростатичного притягання диполів речовини.
Цей різновид взаємодії атомних і молекулярних частинок вирізняється
низкою таких особливостей.
1. Міжмолекулярна взаємодія є порівняно слабкою. Ефекти, спричинені
міжмолекулярною взаємодією, на один-два порядки менші від теплових ефектів
утворення ковалентних зв'язків. Наприклад, енергія зв'язку для молекули
64
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
гідрогену дорівнює 432 кДж/моль, тоді як ентальпія сублімації кристалічного
водню, пов'язана з міжмолекулярною взаємодією, становить 2,1 кДж/моль.
2. Міжмолекулярна взаємодія не є специфічною. Вандерваальсові сили діють
між найрізноманітнішими молекулами, однаковими або різними.
3. Сили Ван-дер-Ваальса мають електростатичну природу, внаслідок чого
міжмолекулярна взаємодія є ненасиченою і ненапрямленою.
За походженням диполів, що взаємодіють, розрізняють три типи сил
міжмолекулярної взаємодії.
• Орієнтацішш взаємодія - електростатичне притягання постійних диполів
речовини, орієнтованих один щодо одного протилежними полюсами:
Енергію орієнтаційної взаємодії двох однакових молекул (орієнтаційний
ефект) описує таке рівняння:
_ 2 Ц4'НА_ а
°Р "ЗК-Тт6 V
(3.9)
де ц — дипольний момент молекули; г - відстань між молекулами.
• Індукційна взаємодія - електростатичне притягання постійного і наведеного
(індукованого) диполів:
Е = -
^инд
2а//2 _
В_
б
(3.10)
де а - поляризовність молекули.
• Дисперсійна взаємодія - це електростатичне притягання миттєвих
мікродиполів речовини. Виникнення миттєвих мікродиполів спричинене ви-
падковим порушенням симетрії розподілу електронної густини в частинці, що
зумовлює виникнення і зникнення електричних полюсів. Коли виявляються сили
дисперсійної взаємодії, миттєві мікродиполі з'являються і зникають синхронно,
орієнтуючись так, щоб частинки притягались:
+ -
оо
о©
Розділ 3. Основи теорії електронної будови органічних молекул
65
-'ЛИСП
2 ЗЬсГУр _
4 .6
_D
.6
(ЗЛІ)
де Ь - стала Планка1; Уо - частота коливань молекул за температури абсолютного
нуля.
Внесок дисперсійної взаємодії в енергію міжмолекулярної взаємодії збіль-
шується зі зростанням поляризовності молекули. Наприклад, для НІ енергія
дисперсійної взаємодії (60,47 кДж/моль) становить 98,5 % енергії сил міжмо-
лекулярної взаємодії.
Дія сил Ван-дер-Ваальса зближає атомні та молекулярні частинки, що не
пов'язані хімічним зв'язком, до деякого рівноважного стану, у якому сили
притягання врівноважені силами відштовхування. У цьому разі відстань між
атомами можна записати як суму так званих вандерваальсових радіусів (табл. 3.3).
Таблиця 3.3
Вандерваальсові радіуси деяких елементів
Елемент
Вандерваальсовий
радіус, пм
Елемент
Вандерваальсовий
радіус, пм
Н
120
S
180
F
140
N
150
СІ
180
Р
190
Вг
200
С
170
І
0
220
140
Si
200
Ефективні вандерваальсові радіуси можна приписати не лише окремим
атомам, а й їхнім групам. Наприклад, ефективний вандерваальсовий радіус
метальних груп дорівнює 200 пм.
Н
Н С н
І
н
Рис. 3.8. Молекула бензолу, зображена у вигляді ковалентних і вандерваальсових радіусів атомів
Карбону і Гідрогену
З порівняння даних табл. 3.1 і 3.3 випливає, що вандерваальсові радіуси
помітно більші від ковалентних. Особливо добре це видно на рис. 3.8, де атоми в
1 Планк М. (Plank М.) (1858-1947) - німецький фізик, один із засновників квантової теорії. Головні
праці у галузі термодинаміки і теорії відносності. Винайшов закон випромінювання, названий його
ім'ям. Нобелівська премія 1918 р.
66
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
молекулі бензолу зображені як сфери, розміри яких відповідають ковалентним і
вандерваальсовим радіусам Гідрогену і Карбону.
Значення вандерваальсових радіусів дає змогу визначити конформацію
молекулярних частинок і їхнє розташування в кристалах. Енергетично
вигідними звичайно є такі конформації молекул, у яких перекривання
вандерваальсових радіусів незначне або його немає.
3-3. Взаємний вплив атомів у молекулі
❖ Індуктивний ефект. Наявність у молекулі полярного зв'язку веде до
поляризації двох-трьох найближчих карбон-карбон а-зв'язків. Такий зсув
електронної густини по ланцюгу а-зв'язків називають індуктивним (індук-
ційним) ефектом. Розрізняють позитивний індуктивний ефект (позначають +1) -
гидштовхування електронної густини від атома або групи атомів, і негативний
(позначають -І) - відтягування електронної густини до атома або групи атомів.
Ефект +1 характерний для алкільних груп і металів, -І-ефект мають електро-
ноакцепторні атоми і групи (галогени, гідроксильна група тощо). Індуктивний
ефект Гідрогену приймають за 0.
Ефект +1 алкільних груп найвиразніше виявляється під час дії на я-електронні
системи, що пов'язано з меншою електронегативністю атома Карбону в ^р3-
гібридному стані порівняно з яр2- і ^р-гібридизованими атомами Карбону
(детальніше див. розділ 9).
5Д У*-сн3 нс=с—сн3
5-
Максимальні індуктивні ефекти спостерігають для атомів, заряди яких є
цілими числами:
сн3~^ын3+ сн3*~ о-
-І-Ефект +І-Ефект
Індуктивний ефект швидко згасає по ланцюгу (5' > 8" > 8"'). Практично він
зникає на третьому-четвертому атомах від функції, яку розглядають. Індук-
тивний ефект позначають за допомогою прямої стрілки:
5,и+ 8"+ 5'+ 5-
сн3—^сн2—^ сн2—**сі
Оскільки я-електрони подвійного і потрійного зв'язків рухливіші, то їхня
поляризація відбувається сильніше. В наведеному ряді 8 < 8 < 8":
Розділ 3. Основи теорії електронної будови органічних молекул
67
8+ 5- 54- 8'- 8"+ 8"-
• Ефект поля. Різницю в реакційній здатності молекул не завжди можна
пояснити на підставі концепції індуктивного ефекту. З'ясовано, що в деяких
випадках замісник діє не по ковалентних зв'язках, а безпосередньо через
простір. Таке явище назвали ефектом поля. У наведених нижче кислотах атоми
галогену розміщені від карбоксильної групи через однакову кількість
ковалентних зв'язків, однак для іншої кислоти атоми Хлору розташовані ближче
і, відповідно, кислотність такої кислоти вища:
❖ Мезомерний ефект
> Спряження (мезомерія) - перерозподіл електронної густини, який
делокалізує я-зв'язки (молекулярна орбіталь охоплює більш ніж два атоми):
Мезомерний ефект позначають на схемах напівкруглою стрілкою, що йде
від середини зв'язку до електронегативного атома. Зсув електронних пар
унаслідок спряження називають мезомерним ефектом. Аналогічно до
індуктивного ефекту вирізняють позитивний (позначають +М) і негативний
(позначають -М) мезомерні ефекти, відповідно, — подача або відштовхування
електронної пари від атома або групи атомів. Наприклад, в амідах аміногрупа
має +М-ефект, а атом Оксигену —М-ефект:
Розрізняють ПуП-спряження (у спряженні беруть участь два і більше 71-
зв'язків) і р,я-спряження (у спряженні беруть участь я-зв'язок і р-електрони
сусіднього атома).
Н
РКа6,07
р£а5,67
5+Л Пб- 8+ 5-
с=с - с=о => с « с =-с=о
(^О -М-ефект
н3с-с/>П5+
+М-ефект
п^п-Спряження
рьп-Спряження
сн2=сн-сн=сн2
68
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Електронні ефекти можуть збігатися в напрямі зсуву електронної густини
або діяти в протилежних напрямах. Сумарна дія замісників наведена в табл. 3.4.
Таблиця 3.4
Електронні ефекти різних замісників
Замісник
Індуктивний ефект
Мезомерний ефект
Електронодонорна дія
-Аік
+1
-
-0"
+1
+м
-Ш2, -ШЯ, т2
-І
+м
-ОН, -(Ж
-І
+М
Електроноакцепторна дія
-ш3+, -т3+
-І
-
-Наї
-І
+М
>с=о
_І
-м
-СООН, -СО(Ж
-м
-N02
_І
-м
-м
-БОзН
-І
-м
❖ Ефект гіперкон'югації (надспряження). Значну стійкість алкенів з
подвійним зв'язком усередині ланцюга порівняно з алкенами з прикінцевим
розташуванням подвійного зв'язку або незаміщеним етиленом (детальніше див.
розділ 8) часто пояснюють реалізацією спряження електронних хмар ст-зв'язків
Карбон-Гідроген з я-електронною хмарою. Таке спряження називають а,п-
спряженням, або надспряженням (гіперкон 'югацією):
Донорні властивості метальних або первинних алкільних груп виявляються,
головно, коли розглядають стійкість алкільних карбокатіонів. Якщо у випадку
алкенів різниця в енергії молекул дорівнює 10-20 кДж/моль, то різниця в енергії
первинних, вторинних і третинних карбокатіонів може досягати сотень
кілоджоулів на моль. Уважають, що ефект гіперкон'югації значно виявляється
саме для заряджених систем типу карбокатіонів:
Розділ 4
ХІМІЧНІ РЕАКЦІЇ
ОРГАНІЧНИХ СПОЛ УК
4.1. Хімічні реакції та реагенти
> Хімічні реакції - це розрив старих і утворення нових хімічних зв'язків у
молекулі.
Для перебігу хімічної реакції необхідно, щоб частинки, які реагують,
зіштовхнулись. У цьому разі кінетична енергія частинок переходить у
потенціальну енергію, завдяки якій відбувається перерозподіл електронів у
реакційній системі. В певний момент часу тільки частина молекул (активовані
молекули) мають кінетичну енергію, достатню для перебігу хімічної реакції.
Мінімальна кількість енергії, необхідна для перебігу реакції, - енергія
активації (Еа). Вона потрібна частинкам, що реагують, для утворення
перехідного стану (активованого комплексу), який спонтанно перетворюється
в продукти реакції. Точніше кажучи, перехідний стан - це точка на
енергетичній діаграмі реакції, а активований комплекс - угруповання атомів,
яке відповідає перехідному стану:
Вихідні
молекули
Активовані
молекули
Активований
комплекс
Кінцеві
молекули
Чим менше Еа, тим більша швидкість реакції. Підвищення температури
реакційної суміші збільшує кількість активованих молекул і, відповідно,
підвищує швидкість реакції.
■ В екзотермічних реакціях енергія кінцевих продуктів менша від енергії
вихідних сполук. В ендотермічних реакціях енергія сполук, що утворюються,
вища від енергії речовин, які вводять у реакцію.
Не треба плутати екзотермічні й спонтанні реакції. До спонтанних належать
процеси, що відбуваються за кімнатної температури, і такі, що починаються без
Затрати додаткової енергії. Наприклад, реакція горіння метану - екзотермічна,
але не спонтанна.
Час існування активованого комплексу дуже малий - близько 10"12 с.
Визначити будову активованого комплексу безпосередньо в реакційному сере-
довищі неможливо. Зазвичай, його структуру розглядають як проміжну між
структурою вихідних і кінцевих молекул. В екзотермічних реакціях перехідний
70 Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
стан ближче за енергією до вихідних речовин, а отже, і активований комплекс
ближче за будовою до вихідних молекул. Для ендотермічних реакцій структура
активованого комплексу, навпаки, ближча до будови кінцевих речовин (рис. 4.1).
Кінцеві
молекули
Координата реакції Координата реакції
Рис. 4.1. Енергетичні діаграми екзотермічних (а) і ендотермічних (б) реакцій
■ Велика кількість реакцій відбувається через стадію утворення проміж-
ного продукту - інтермедіату (рис. 4.2, я). Час його життя значно більший, ніж
час існування активованого комплексу. Утворення багатьох інтермедіатів
зареєстроване за допомогою фізичних методів, а деякі найстійкіші проміжні
продукти вдається виділити з реакційної суміші. Якщо під час реакції можливе
утворення декількох інтермедіатів, то реакція відбувається через перехідний
стан з меншою енергією активації (рис. 4.2, б). Зазвичай, такому перехідному
стану відповідає стійкіший інтермедіат.
Е
а
Координата реакції & Координата реакції
Рис. 4.2. Енергетичні діаграми двостадійної хімічної реакції (ПС - перехідний стан) (а)
та реакції, що відбувається з утворенням двох інтермедіатів (б)
На співвідношення продуктів конкурентних реакцій впливають як швидкості
цих реакцій, так і термодинамічна стійкість кінцевих продуктів. Наприклад, у разі
конкурентної реакції В<-»А—>С (рис. 4.3), коли процес відбувається за умов
Розділ 4. Хімічні реакції органічних сполук
71
подолання обох енергетичних бар'єрів, на початковому етапі переважно
утворюватиметься сполука В, оскільки для цієї реакції менше Еа і, як наслідок,
більша швидкість реакції. Утворення продукту В контрольоване кінетично. У
випадку довготривалої реакції переважно одержуватимуть стійкіший продую С,
тобто його утворення контрольоване термодинамічно.
Координата реакції Координата реакції
Рис. 4.3. Енергетична діаграма конкурентної реакції В+-+А-+С
❖ Каталізатори. Значна частина реакцій органічної хімії відбувається за
наявності каталізаторів.
> Каталізатор - речовина, що збільшує швидкість реакції, але не
змінюється внаслідок реакції і не входить до складу її реагентів або продуктів.
Каталізатор знижує енергію активації Еа* і збільшує швидкість хімічної
реакції. Дія каталізаторів пов'язана з тим, що він спрямовує реакцію іншим
шляхом, через утворення стійкіших активованих комплексів (рис. 4.4).
Е*
\а
координата реакції
Рис. 4.4. Енергетичні діаграми некаталізованої (а) та каталізованої (б) реакцій
■ Розрізняють гомогенний каталіз - каталізатор є розчинним у реакційному
середовищі, і гетерогенний каталіз - каталізатор утворює окрему фазу відносно
реакційного середовища. Гомогенні каталізатори безпосередньо взаємодіють
72
Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
вихідною речовиною, а у випадку гетерогенних каталізаторів, зазвичай, відбу-
вається адсорбція субстрату чи реагенту на поверхні каталізатора, унаслідок
чого в їхніх структурах відбувається активація певних зв'язків. Приклади
гомогенного і гетерогенного каталізу розглянуто в розділах, присвячених
окремим класам органічних сполук.
■ Широко в органічній хімії застосовують міжфазовий каталіз. Реакцію
проводять у гетерогенній системі рідина-рідина або рідина-тверде тіло за
наявності спеціальних агентів, здатних переносити окремі компоненти
реакційної суміші через межу поділу фаз. Зазвичай як міжфазові каталізатори
використовують четвертинні амонійні солі, наприклад, бензилтриетиламоній
хлорид або тетрабутиламоній гідрогенсульфат і краун-етери (див. розділ 13).
Розглянемо принцип дії міжфазового каталізатора на прикладі реакції
нуклеофільного заміщення алкілгалогенідів. Реакцію найчастіше проводять у
системі з двох рідин, що не змішуються, - полярної і малополярної, наприклад,
вода-бензен або вода-метиленхлорид. У цьому разі у водній фазі міститься
неорганічний реагент, а в органічній фазі - субстрат і кінцевий продукт.
Четвертинна амонійна сіль здатна обмінювати аніонні компоненти, транс-
портуючи аніон нуклеофіла з водної фази в органічну, а також переносить аніон
відхідної групи з органічної фази у водну:
К4№На1- + №+n11- К4№Ии- + Иа+На1-
Вода
Бензен
Я^НаІ" + Я№ — Я4№Ш- + Я'НаІ
■ У живих організмах діють надзвичайно активні та селективні ката-
лізатори - ферменти. Наприклад, фермент уреаза збільшує швидкість реакції
гідролізу сечовини в 1014 разів.
❖ Розрив ковалентного зв'язку може відбуватися двома способами:
- гомолітично - пара електронів ковалентного зв'язку розподіляється по
одному на кожен атом
А:В -> А* + В' і
- гетеролітично - електронна пара відходить до одного більш електро-
негативного атома
А:В-+ А" + В+.
■ Перщиклічні реакції - окремий тип реакцій, у яких розрив старих і
утворення нових зв'язків відбувається узгоджено через циклічний активований
комплекс. До цього типу, зокрема, належать реакції циклоприєднання.
• Вільні радикали. Під час гомолітичного розриву утворюються високо-
реакційні малостійкі вільні радикали - частинки з неспареним валентним
Розділ 4. Хімічні реакції органічних сполук
73
електроном (у загальному вигляді позначають R"). Реакції за участю радикалів
називають радикальними.
У процесі вивчення окремих класів органічних сполук розглянемо реакції, що
відбуваються за участю вільних радикалів галогенів СГ чи Вг\ гідрогенперок-
сильних і алкілпероксильних радикалів НОО* і ROO*, різних вуглеводневих R* і
ацильних RCO* радикалів. Більшість вільних радикалів — малостійкі й
короткоживучі частинки, але деякі з них, наприклад, трифенілметильний [І] або
2,6-ди-т/?ет-бутилфеноксильний [II] радикали, порівняно стабільні:
(У
(НзС)зС^^С(СЯ^
II
• Нуклеофіли та електрофіли. Якщо ковалентний зв'язок розривається
гетеролітично, то утворюються йони, подібні реакції називають іонними. Реакційні
частинки, у яких позитивний і негативний заряди є на атомі Карбону, називають,
відповідно, карбокатіонами і карбаніонами. У загальному випадку частинку з
дрібним або цілим позитивним зарядом називають електрофіл (позначають Е чи
Е*), а з негативним зарядом - нуклеофіл (позначають :Ии чи n11").
> Нуклеофіли - частинки з електронодонорними властивостями, які здатні
віддавати пару електронів на утворення ковалентного зв'язку з електрофільною
компонентою.
До нуклеофілів належать:
■ аніони, у тім числі карбаніони НГ, НСГ, НКГ, НаГ, НСЬСГ
■ нейтральні сполуки з атомами, що
мають неподілені пари електронів Н20, ROH, Н28, МН3, ІІМН2
■ нейтральні сполуки з сильно по- 5- 5+ 5- 6+
лярним зв'язком Н3ОМ§-Вг 9 НС=С-]\^І
■ вуглеводні з л-електронними систе-
мами :Н2С=СН2 , н2с=сн-нс=сн2,
> Електрофіли - частинки з електроноакцепторними властивостями, здатні
утворювати ковалентний зв'язок з нуклеофільною компонентою завдяки її
електронній парі.
До електрофілів зачисляють:
■ катіони, у тім числі карбокатіони НҐ, Вг+, n02^ АгИ2+, RзC+
■ нейтральні сполуки з атомами, що ВБз, BRз, РеВгз, АІСЬ
мають вакантну орбіталь
74
Чирва В.Я., Ярмолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
■ нейтральні сполуки із сильно
полярним зв'язком
6+6- 6+6- 6+ 6-
R2CHBr, R2C=0, R-C=N
Cl2, Br2, h
■ нейтральні сполуки, які легко
поляризуються
❖ Головні типи органічних реакцій. Відповідно до характеру хімічних
перетворень виділяють декілька типів хімічних реакцій, зокрема:
■ реакції приєднання
(позначають А, від
англ. addition)
■ реакції заміщення
(позначають S, від
англ. substitution)
■ реакції відщеплення
(позначають Е, від
англ. elimination)
■ реакції циклоприєд-
нання
■ кислотно-основні
реакції
■ реакції ізомеризації
та перегрупування
А + В
АХ + BY —* АВ + XY
В + С
СН2=СН2 + С12 -> СН2С1-СН2С1
СНзВг + AgOH -> СНзОН + AgBr
СН3СН2ОН -> сн2=сн2 + Н20
А=В
+ -
X—Y
А—В
І І
X—Y
АН+:В ->А~ + ВН+
А=В-С ->А-В=С
A-B-C-D
А-В-С
і
D
не-. + сн -
СЩ *CN
НС1 + NH2CH3 -
Na
н2с=с=сн2 —
АВг3
СНзСН2СН2СНз *=£ Н3С-СН-СК3
СН3
СН3Шз+СГ
•нс=с-сн,
❖ Окисно-відновні реакції. В органічній хімії поширення набули окисно-
відновні (оксидаційно-відновні) процеси, у ході яких змінюється ступінь
окиснення атома Карбону або інших атомів, що є в складі молекули. Загалом
процес окиснення (оксидації) полягає в перенесенні електронів від субстрату на
реагент-окиснювач, а відновлення - навпаки, у переході електронів на субстрат
від реагенту-відновника. В органічних сполуках, звичайно, окиснення пов'язане
з відщепленням атомів Гідрогену та утворенням кратних зв'язків або заміною
зв'язків з атомом Гідрогену на зв'язки з атомом Оксигену або іншими більш
електронегативними, ніж Гідроген, атомами. Отже, відновлення зумовлене як
приєднанням атомів Гідрогену за місцем кратних зв'язків, так і заміною
гетероатомів на атоми Гідрогену.
Приклади рядів окиснення-відновлення за атомами Карбону і Нітрогену
такі:
Окиснення с
:> Н3С-СН3
-н,
Н2С=СН2
-н,
НС=СН S і Відновлення
Розділ 4. Хімічні реакції органічних сполук
75
-4 [О] -2 [О] 0 [О] +2^0 [О] +4
Окисненняс=> гн4-^—» СН3ОН ^=*: Н2С=0 Н-Сч ^=*= С02 <=3 Відновлення
[Н] [Н] [Н] он [Н]
-З -1 +1 +3
>Ш2 [О] Ш-ОН [О] N0 Ш2
Окисненняі==> 1 1 у*" - І 1 Відновлення
❖ Молекулярпість реакції визначають як кількість частинок, що необхідні
для утворення активованого комплексу. Найпоширеніші мономолекулярні і бімо-
лекулярні реакції. Прикладами мономолекулярних реакцій можуть бути реакції
нуклеофільного заміщення, що відбуваються за механізмом Бмі (див. розділ 12),
і реакції елімінування, які відбуваються за механізмом Е1 (див. розділ 8).
До бімолекулярних реакцій, зокрема, належать реакції галогенування
алкенів та реакції нуклеофільного заміщення і відщеплення, які, відповідно,
відбуваються за механізмами Бн2 і Е2.
Як приклад тримолекулярної реакції можна розглянути приєднання гідроген
галогенідів до алкенів (див. розділ 8).
❖ Поняття про механізм реакції. Багато реакцій відбувається у декілька
стадій через ланцюг перетворень. Послідовність таких перетворень, що містить
усі проміжні продукти реакції (інтермедіати), називають механізмом реакції.
Будь-який механізм органічної реакції є частково гіпотетичним. Навіть
порівняно невеликі зміни в структурі субстрату і реагенту або умов перебігу
реакції можуть принципово змінити механізм реакції. Наприклад, реакція
гідробромування алкенів може відбуватися як іонне приєднання через утворення
карбокатіону, узгоджене тримолекулярне приєднання або вільнорадикальне
приєднання (див. розділ 8). Звичайно в процесі з'ясування механізму конкретної
реакції враховують дані кінетичних досліджень, просторової будови вихідних і
кінцевих продуктів, структури інтермедіатів.
Важливу роль в описуванні механізму реакції відіграє частинка, що атакує
(радикал, нуклеофіл, електрофіл). Тип частинки часто зазначають, коли
класифікують реакції, наприклад:
■ радикальне заміщення (8я) СН4 + С12 —> СН3С1 + НС1
■ нуклеофільне заміщення (бм) снзвг + agoh —> СН3ОН + а§вг
Ос12>^ /=\
~р—\, //—^ +
Віднесення реакцій до нуклеофільних або електрофільних часто досить
умовне. Наприклад, у наведеній нижче реакції атом Карбону в алкілгалогеніді є
електрофільним, а атом Нітрогену в метиламіні, відповідно, - нуклеофільним.
Отже, цю реакцію можна розглядати як нуклеофільне заміщення галогену на
аміногрупу або як процес електрофільного заміщення за атомом Нітрогену:
76
Чирва В.Я., Ярмолюк CM., Толкачова Н.В., Земляков О.Є. Органічна хімія
5+ 5-
Н3С-С1 + ^К-СНз—*- Н3С-Н-СН3 + НСІН2К-СН3
н
4.2. Кислотність і основність
В органічній хімії найширше використовують дві концепції кислотності та
основності: теорію Бренстеда]—Лоурі2 і теорію Льюїса3.
❖ Кислотність за Бренстедом-Лоурі. За теорією Бренстеда-Лоурі
кислота спряжена з основою. Чим сильніша кислота, тим слабше спряжена з нею
основа, і навпаки:
А-Н + :В ^ А" + ВН+
Кислота Основа Основа Спряжена
кислота
> Кислотність - здатність віддавати протон. Основність - здатність
приєднувати протон.
Кількісною оцінкою кислотності може бути константа рівноваги взаємодії
кислоти й основи:
к = [А"][ВН+]
' [АН][В]
Для розчинів слабких електролітів, до яких належить більшість органічних
кислот, розчинник є основою:
А-Н + Н20 <-» А" + Н30+
У цьому разі константа рівноваги реакції має такий вигляд:
к [А-][Н30+]
' [АН][Н20] '
■ Оскільки концентрацію води вважають сталою в рівнянні константи
рівноваги, то визначають константу кислотності:
Р [АН]
1 Бренстед Й. (Bronsted J.) (1879-1947) - датський фізик та хімік. Головні дослідження в галузі
каталізу, зокрема, кислотно-основного. Розробник (1923-1929) загальної теорії кислот і основ.
2 Лоурі T. (Lowry T.) (1874-1936) - англійський хімік. Головні праці присвячені оптичній
активності органічних сполук. Незалежно від Й. Бренстеда 1928 р. розробив протеолітичну теорію
кислотно-основної рівноваги.
3 Льюїс Г. (Lewis G.) (1875-1946) - американський фізико-хімік. Дослідження в галузі хімічної
термодинаміки і будови речовини. Ввів концепцію спільної електронної пари. В 1926 р.
запропонував нову теорію кислот і основ.
Розділ 4. Хімічні реакції органічних сполук
77
Значення таких констант дуже малі, тому звичайно використовують
негативний логарифм рКа = Кй. Чим менше значення рКа, тим сильніша
кислота.
Практично всі органічні сполуки, для яких принципово можливе відщеп-
лення протона, можна зачислити до кислот. Залежно від природи атома, з яким
зв'язаний кислий атом Гідрогену, розрізняють ОН-, БН-, ЙН- і СИ-кислоти.
Приклади таких кислот і значення їхньої кислотності наведено в табл. 4.1.
Таблиця 4.1
Значення рКя кислот Бренстеда відносно води
Кислоти
Формула
Р*а
Кислоти
Формула
Р*а
ОН-кислоти
Мурашина
НСООН
3,75
Фенол
С6Н5ОН
10,0
Оцтова
СНзСООН
4,76
л-Амінофенол
Н2ТЧС6Н4ОН
10,5
Пропіонова
СН3СН2СООН
4,87
я-Нітрофенол
О^С6Н4ОН
7,1
Хлороцтова
С1СН2СООН
2,86
Пікринова
(02Ы)3С6Н2ОН
0,8
Трихлороцтова
сьссоон
0,66
Метанол
СНзОН
15,5
Трифлуороцтова
РзССООН
0,23
Етанол
СН3СН2ОН
15,9
Бензойна
С6Н5СООН
4,19
2-Метилпропан-2-ол
(СНз)зСОН
18,0
$\\-кислоти
Етантіол
СН3СН28Н
10,5
Тіофенол
С6Н58Н
6,5
ШІ-кислоти
Амоніак
33
Ацетамід
СН3СОЫН2
15,1
СН-кислоти
Метан
СН4
48
Бензен
СбН6
43
Етилен
Н2С=СН2
36
Хлороформ
СНСІз
15,7
Ацетилен
носн
25
Нітрометан
сн3>ю2
10,6
Неорганічні кислоти
Іодидна
НІ
-11
Фосфатна*
н3ро4
2,1
Бромідна
НВг
-9
Карбонатна*
Н2С03
6,4
Хлоридна
неї
-7
Вода
н2о
15,7
* Перша константа дисоціації
■ Основність водних розчинів органічних сполук, відповідно, передають
константою основності Кь, яка пов'язана з константою кислотності рівнянням
рКь = 14 - рКй, або константою кислотності спряженої кислоти ВНҐ (р£вн+).
Чим більше значення р£вн+> тим сильніша основа. Приклади основ і значення
кислотності їхніх спряжених кислот наведено в табл. 4.2.
Експериментально константи дисоціації кислот у воді можна визначити
лише до рКй - 15. В інших розчинниках діапазон визначення кислотності можна
збільшити, наприклад, у рідкому амоніаку межа визначення рКй ~ 33.
Порівнюванням значення констант кислотності однієї й тієї ж сполуки в різних
розчинниках можна виконати кореляцію цих даних. Для дуже слабких кислот,
наприклад, СН-кислот, значення р#а досить приблизні й залежать від методу
розрахунку. Оскільки для багатьох органічних сполук кислотність важко
78 Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
визначити фізичними методами, то її оцінюють через стабільність спряжених
основ (аніонів). Стабільність аніонів визначена ступенем делокалізації нега-
тивного заряду, а він, відповідно, залежить: від природи атома, можливості
стабілізації завдяки спряженню, будові радикала й інших чинників. Зокрема,
розглядаючи вплив природи атома на делокалізацію, необхідно враховувати
його електронегативність і поляризовність. Наприклад, для елементів другого
періоду (С, >І, О, Б) електронегативність зростає зі збільшенням порядкового
номера, збільшується здатність утримувати негативний заряд, тому в ряді
сполук цих елементів, що містять Гідроген, максимальна кислотність у гідроген
флуориду (СНЦ < >Шз < НгО < ТЛ7). Поляризовність у елементів головних
підгруп зростає зі збільшенням радіуса атома, оскільки заряд делокалізується по
більшій поверхні, унаслідок чого кислотність збільшується в ряді ОТ < НС1 <
НВг < Ш. Детальніше чинники, що впливають на кислотність і основність,
розглянуті в розділах, присвячених окремим класам органічних сполук.
Таблиця 4.2
Значення рА*вн+ основ Бренстеда щодо води
Основи
Формула основи
Формула спряженої кислоти
Р^вн
Амоніак
Ш3
Ш4+
9,25
Метиламін
СН3Ш2
СН3ЫН3+
10,6
Диметиламін
(СН3)2Ш
(СН3)2КН2+
10,7
Триметиламін
(СНзЬИ
(СН3)зШ+
9,8
Етил амін
СН3СН2ЫН2
СН3СН,ЫН3+
10,7
Піперидин
(СН2)5Ш
(СН2)5>Ш2+
11,1
Піридин
С5Н5Ш
с5н5ш2+
5,2
Гуанідин
(Ш2)2С=Ш
[(МН2)2С=Ш]+
13,5
Анілін
С6Н51МН2
С6Н5ЫН3+
4,6
и-Нітроанілін
о2ыс6н4ш2
о2ыс6н4ын3+
1,0
Досить суттєво на стабільність аніонів впливає сольватація. Взаємодія з
молекулами розчинника часто кардинально змінює кислотність або основність
сполуки. Відповідно, константи кислотності однієї й тієї ж сполуки в розчині та
в газовій фазі, де нема ефектів сольватації, можуть значно відрізнятись.
Наприклад, у водних розчинах анілін є слабшою основою порівняно з амо-
ніаком, а в газовій фазі - навпаки.
Ефекти сольватації багатофакторні, і їх важко оцінити кількісно. В органічній
хімії широко використовують емпіричне правило - чим менший розмір іона і чим
більший локалізований у ньому заряд, тим ліпше він сольватується.
❖ Кислотність за Льюїсом. Теорія кислотності Льюїса є загальнішою
порівняно з уявленнями теорії Бренстеда-Лоурі. За цією теорією до кислотно-
основних взаємодій належать утворення ковалентного зв'язку завдяки
електронній парі.
> Кислотність - здатність приймати електронну пару на вакантну
орбіталь. Основність - здатність віддавати електронну пару.
Розділ 4. Хімічні реакції органічних сполук
79
Na+, Ag, Си+, Си2+, Zn2+, Fe3+, Al3+
Br+, N02+, R3C+
BF3, АІСІз, FeCl3, FeBr3, ZnCl2,
S11CI4
До кислот Льюїса належать:
■ протон, який має вакантну орбіталь
■ катіони металів
■ інші катіони, зокрема, карбокатіони
■ галогеніди елементів з вакантними
орбіталями
Кислотами Льюїса є також багато електрофільних реагентів, наприклад: Вг2,
С02, S03.
Відповідно, основи Льюїса підрозділяють на:
■ п-основи Льюїса, у тому числі
аніони, включаючи й карбаніони НСГ, HS", Hal", НС=С", RCOO"
нейтральні сполуки з атомами, що ..
мають неподілені пари електронів Н20, ROH, H2S, NH3, RNH2
■ л-основи Льюїса - вуглеводні з
я-електронними системами
Н2С=СН2 , НХ=СН-НС=СН2 ,
Приклади кислотно-основних взаємодій за Льюїсом:
R-Ci: + A1CL
Основа
Льюїса
Кислота
Льюїса
+ -
R-C1-A1CL
Н 3
(С2Н5)20: + BF3 -
Основа Кислота
Льюїса Льюїса
+ -
(C2H5)20-BF3
+ Ag+
Основа Кислота
Льюїса Льюїса
. 2|4_
н2с
я-Комплекс
•Ag+
Основа
Льюїса
Кислота
Льюїса
Вг+
я-Комплекс
❖ Теорія жорстких і м'яких кислот і основ (ЖМКО). Принцип ЖМКО
останнім часом широко застосовують в органічній хімії. Його запропонував
Р.Пірсон1 1963 р. як доповнення до теорії кислот і основ Льюїса для пояснення
аномальної та зовсім незрозумілої з погляду класичних уявлень схильності певних
кислот Льюїса з'єднуватись з певними основами Льюїса. Наприклад, флуорид-іон
схильний утворювати міцні комплекси з катіонами лужноземельних металів, але не
утворює їх з катіонами важких металів, таких як Меркурій або Аргентум. Водночас
іодид-іон добре утворює комплекси саме з катіонами Меркурію та Арґентуму і
погано - з катіонами лужноземельних металів. Отже, іодид-іон є сильною основою,
якщо як кислоту використовувати катіон двовалентного Меркурію, і слабкою, якщо
як кислоту застосовувати катіон Магнію. Для пояснення безлічі подібних фактів і
запропоновано принцип ЖМКО (принцип Пірсона), який формулюють так:
1 Пірсон P. (Pearson R.G.) (1918-2003) - американський хімік, автор теорії ЖМКО. Дослідження у
галузі механізмів хімічних реакцій, теорії хімічного зв'язку та теорії функціональної щільності.
80 Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
> жорсткі основи Льюїса переважно взаємодіють з жорсткими кислотами
Льюїса і, відповідно, м'які — зм'якими кислотами.
Розподіл кислот і основ Льюїса на жорсткі й м'які виконують відповідно до
поляризовності акцепторного або донорного атома в реагенті. Якщо акцеп-
торний атом в кислоті або донорний в основі легко поляризується, то такий
реагент називають м'яким. Донорні або акцепторні атоми жорстких реагентів
мають малу поляризовність.
З високою поляризовністю пов'язані такі властивості атома: низька електро-
негативність, великі розміри, низький ступінь окиснення та здатність до деформу-
вання валентних оболонок. Навпаки, низька поляризовність притаманна атомам з
малими розмірами, високою електронегативністю, високим ступенем окиснення і
низькою здатністю до деформування валентної оболонки. Відповідно до цього,
для ізоелектронних частинок жорсткість основ у періодичній системі збіль-
шується знизу вверх і зліва направо. Жорсткість кислот зростає в групах
періодичної системи знизу вверх. Приклади реагентів, які мають жорсткість
різного ступеня, наведено в табл. 4.3.
Таблиця 43
Жорсткі та м'які кислоти й основи
Характер
кислот і основ
Кислоти
Основи
Жорсткі
Н+, и\ К+, МІ\ А13+, Ре3+, ВР3,
АІСІз, ЯСО+, С02
Н20, ОН", F", СІ", RCOO", C104",
ROH, R20, RO", RNH2
Проміжні
Ре2+, гп2+, Я3С+, ВЯ3, Си2+
N3~, Br", C6H5NH2, піридин
М'які
ЯНаІ, ЯСН2+, Си+, ВН3,
хінони, Вг+, І+, Вг2,12
RSH, RS", R2S, Г, CN", R3P,
етилен, бензен, R", H"
Застосування принципу Пірсона розглянемо на прикладі реакцій енолятів
ацетооцтового естеру в розділі 21.
Розділ 5
КЛАСИФІКАЦІЯ
І НОМЕНКЛАТУРА
ОРГАНІЧНИХ СПОЛ УК
і
5.1. Класифікація органічних сполук
Органічні речовини можна розглядати як вуглеводні та їхні похідні,
одержані шляхом уведення функціональних груп.
> Функціональна група — це атом або група атомів, які зв'язані ковалентним
зв'язком з карбоновим скелетом і визначають хімічні властивості сполуки.
Найпоширенішими є класифікації органічних сполук за будовою карбонового
ланцюга, за ступенем ненасиченості й за типом функціональної групи.
• За будовою карбонового ланцюга органічні сполуки поділяють на:
■ ациклічні (аліфатичні) - карбоновий ланцюг лінійний або розга-
лужений;
■ карбоциклічні - карбоновий ланцюг замкнений у цикл;
■ гетероциклічні — карбоновий цикл містить один або кілька гетероато-
мів (найчастіше це Нітроген, Оксиген, Сульфур2).
Ациклічні сполуки Карбоциклічні сполуки Гетероциклічні сполуки
лінійні розгалужені ^ ^
• За ступенем ненасиченості виділяють
■ насичені сполуки,
■ ненасичені сполуки.
1 В Україні правила номенклатури хімічних сполук визначає Українська національна комісія з
хімічної термінології і номенклатури (УНКоХіТерН). Рекомендуємо прочитати: Толмачова В.,
Ковтун О., Гордієнко О., Корнілов М. До впровадження сучасної хімічної термінології та
номенклатури органічних сполук // Біологія і хімія в школі. - 2005. - № 4. - С. 29-40.
2 Відповідно до Державного стандарту України ДСТУ 2439-94, для уніфікації хімічної
номенклатури введено нові назви, наближені до латинських, для таких хімічних елементів:
Гідроген Н, Карбон С, Нітроген К(, Оксиген О, Флуор Р, Силіцій Бі, Сульфур 8, Манган Мп, Ферум
Ре, Купрум Си, Арсен Аб, Арґентум Аг, Станум Бп, Стибій БЬ, Аурум Аи, Меркурій Н§, Плюмбум
РЬ, Бісмут Ві. Якщо йдеться про хімічний елемент, то назву пишуть з великої літери, у назвах
сполук - з малої. Назви відповідних простих речовин не змінено.
82
Чирва В.Я., Ярмолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Ненасичені сполуки поділяють на речовини, що містять подвійні {етиленові
сполуки), потрійні зв'язки (ацетиленові сполуки) тощо.
Серед карбо- і гетероциклічних сполук виділяють ароматичні сполуки, що
мають специфічну будову і властивості (див. розділ 24 і далі).
Насичені сполуки Ненасичені сполуки Ароматичні та гетероциклічні
ароматичні сполуки
етиленові ацетиленові
СН3СН29НСН ^
• За типом функціональної групи органічні сполуки бувають кількох класів
(табл. 5.1). Оскільки саме природа функціональної групи головно визначає
хімічні властивості певного класу сполук, то часто вуглеводневу частину
молекули позначають загальним символом -Я. Аліфатичні та ароматичні групи
позначають, відповідно, -Аік та -Аг.
• За кількістю та однорідністю функціональних груп похідні вуглеводнів
поділяють на:
■ монофункціональні - мають у складі одну функціональну групу,
наприклад, С2Н5ОН;
■ поліфункціональні - мають у складі кілька однакових функціональних груп,
наприклад, НОСН2СН2ОН;
■ гетерофункціональні - мають у складі кілька різних функціональних
груп, наприклад, НОСН2СН2Вг.
Таблиця 5.1
Класифікація органічних сполук за функціональними групами
Функціональна група
Клас сполук
Загальна формула
назва
структура
Галоген
-Р, -СІ, -Вг, -І
Галогенопохідні
Я-Наї
Гідроксил
-ОН
Спирти
Я-ОН
Феноли
Аг-ОН
Карбоніл
>С=0
Альдегіди
Я-СНО
Кетони
Карбоксил
-СООН
Карбонові кислоти
К-СООН
Ціано
-С^4
Нітрили
Аміно
-ш2
Аміни
Я-ИНг
Нітрозо
-N0
Нітрозосполуки
^N0
Нітро
-Ы02
Нітросполуки
І1-Ш2
Меркапто
-БН
Тіоли
Я-8Н
Сульфо
-БОзН
Сульфокислоти
Я-БОзН
У середині кожного класу сполуки утворюють гомологічні ряди. Приклади
таких рядів наведені у відповідних розділах.
Розділ 5. Класифікація і номенклатура органічних сполук
83
> Гомологічний ряд - група споріднених органічних сполук, подібних між
собою за будовою і властивостями, склад яких відрізняється на одну або
декілька груп сн2 (гомологічна різниця).
5.2. Ранні номенклатури органічних сполук
5.2Л. Тривіальна номенклатура
На початку розвитку органічної хімії сполуки отримували випадкові
(тривіальні) назви. Звичайно такі назви не відображали будови сполуки. Назви
цього типу часто свідчили про джерело їхнього добування, наприклад, з лимона
виділені лимонна кислота і терпен лимонен:
Назви інших речовин відображали їхні фізичні та біологічні властивості:
смак (глюкоза (від грец. glykys - солодкий), гліцерол, гліцин мають солодкий
смак); запах (кетон пахне як малина); колір (білий та червоний стрептоцид).
Н3 С~С—СН2
Лимонна кислота
Лимонен
сно
н—он
но—н
н—он
н—он
СН2ОН
СН2ОН
Н2МСН2СЧ
он
СН2ОН
Глюкоза
Гліцерол
Гліцин
Кетон малини
Білий стрептоцид
Червоний стрептоцид
Деякі назви пов'язані з іменами вчених, які відкрили ці сполуки або
запропонували для них структури (кетон Міхлера, бензен Ладенбурга):
84
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімій
2
сн3
Кетон Міхлера1 Бензен Ладенбурга:
5.2.2. Раціональна номенклатура
Пізніше виникли структурні, або систематичні, номенклатури, у яких
назва сполуки побудована за певною системою і відображає його структуру. До
таких систематичних номенклатур належать раціональна, Женевська (1892) і
Льєжська (1930).
За правилами раціональної номенклатури структуру хімічної сполуки
розглядають як продукт заміщення атомів Гідрогену в структурі сполуки -
родоначальника гомологічного ряду (або його найближчого гомолога) - на
вуглеводневі або функціональні замісники. Відповідно, назву складають шляхом
перелічення замісників (в абетковому порядку або в порядку старшинства) і
назви родоначальної сполуки гомологічного ряду.
СН3
Н3С-(р-СНз с2н—сес-сн3
сн3
Тетраметилметан Етилметилацетилен Триметилоцтова кислота
Сьогодні лише для найпростіших сполук застосовують назви, побудовані за
правилами раціональної номенклатури, зокрема, триметилоцтова кислота, вініл- і
дивінілацетилен, ди- і трифенілметан.
5.2.3. Женевська і Льєжська номенклатури
Детально розроблені правила Женевської номенклатури давали змогу
називати велику кількість класів органічних сполук. В основі є назва най-
довшого вуглеводневого ланцюга або найбільшого циклу. Перед основою
зазначали нефункціональні замісники (галогени, нітро-, нітрозо- і азидогрупи),
нітрогеновмісні функціональні групи (наприклад, аміно) і вуглеводневі заміс-
ники. Після назви головного ланцюга йшли позначення кратних зв'язків і
оксигено- або сульфуровмісних функцій. Нумерацію ланцюга визначало поло-
ження ближчого до кінця ланцюга замісника. Наведемо назви кількох сполук за
правилами Женевської номенклатури:
1 Міхлер В. (МісЬІег >У.) (1848-1889) - німецький хімік.
2 Ладенбург А. (ЬасІепЬи^ А.) (1842-1911) - німецький хімік. Головні праці присвячені вивченню
алкалоїдів і ароматичних сполук.
Розділ 5. Класифікація і номенклатура органічних сполук
85
СН3 Н3С
сн3снсн(рн-с~сн=сн2 H2NHObC00H
СІ он °
2-Хлоро-3-метилгептен-6-ол-4-он-5 6-Аміно-1-метилбензен-3-карбонова кислота
■ Льєжська номенклатура переважно ґрунтувалася на правилах Женевської
номенклатури, проте в ній головним уважають вуглеводневий ланцюг, що
містить функціональну групу, навіть якщо вона не найдовша. Функціональна
група, а не найближче розгалуження ланцюга, як і в попередній номенклатурі,
визначає початок нумерації ланцюга. Загалом ці правила багато в чому близькі
до чинної сьогодні номенклатурі ШРАС. Головний недолік Льєжської
номенклатури полягав у тому, що для однієї сполуки могло бути кілька
рівноправних назв, а це ускладнювало укладання реєстрів і предметних
покажчиків, робило неоднозначним пошук у них хімічних сполук.
5.3. Номенклатура IUPAC
На засадах правил Женевської і Льєжської номенклатур розроблено і 957 р.
опубліковано Правила номенклатури органічних сполук IUP АС *, які діють і нині.
На Міжнародних конгресах, що відбуваються кожні чотири роки, правила IUP АС
постійно доповнюють, у тому числі, номенклатурами окремих класів органічних
сполук (вуглеводів, ліпідів, стероїдів тощо). Оскільки Женевська, Льєжська
номенклатури і номенклатура ШРАС прийняті на міжнародних конгресах хіміків і
призначені для всесвітнього застосування, то ці номенклатури часто називають
міжнародними.
Правила номенклатури органічних сполук ШРАС об'єднують низку но-
мерклатур, заснованих на різних принципах. Одні з них застосовні до широкого
кола класів органічних сполук, а інші є зручними для побудови назв тільки
окремих груп органічних сполук. Перелічимо головні типи номенклатури ШРАС.
■ Замісна номенклатура - назва відображає заміщення в сполуці-основі
атомів Гідрогену вуглеводневими і функціональними замісниками, наприклад:
СН3СН2ОН Етанол N / Хлоробензен
■ Радикально-функціональна номенклатура - назва утворена з назви групи і
назви функціонального класу, наприклад:
1 IUPAC (International Union of Pure and Applied Chemistry) - Міжнародна спілка чистої
(теоретичної) і прикладної (практичної) хімії.
86
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімн
сн3с;
.0
СІ
О
Ацетилхлорид
Етилметилкетон
■ Замінна номенклатура - назва виражає заміну атома Карбону на
гетероатом або заміну атома Оксигену в гетероциклах на атоми Сульфуру,
Селену або Телуру, наприклад:
о=»о о=>о
Бензен
силабензен
Піран
тіапіран
■ Субтрактивна номенклатура охоплює назви, що виражають видалення
(від англ. subtract - віднімати) певних атомів. До них належать назви з
префіксами ангідро- (видалення молекули води), дегідро- (видалення молекули
водню), дезокси- (видалення атома Оксигену), нор- (видалення метальної групи,
від англ. по radicals) та інші, наприклад:
HOCHCH2NH-CH3 HOCHCH2NH2
Бензен
дегідробензен
чон
он он
Адреналін => норадреналін
■ Адитивна номенклатура - назва відображає приєднання (від англ. add -
додавати) атомів і молекул до сполуки-основи, наприклад:
а
нс-сн2
Стирен
стиреноксид
Нафтален => тетрагідронафтален
■ Сполучна номенклатура - назву утворюють перелічуванням назв двох
сполук, структури яких під час об'єднання в єдину молекулу втратили по
одному атому Гідрогену, наприклад:
СН3СООН
СН2СООН
Нафтален оцтова кислота => нафталеноцгова кислота
Розділ 5. Класифікація і номенклатура органічних сполук
87
■ Номенклатура гетероциклів за Ганчем1—Відманом: назви будують за
особливими правилами, що передбачають позначення гетероатома, розмірів
циклу і ненасиченості, наприклад:
Г\ н2с~сн2
N^ Триазол Оксиран
У номенклатурі IUP АС застосовують три типи назв:
■ тривіальні — випадкові назви, що часто описують якусь властивість
сполуки. Сьогодні до тривіальних зачисляють ті назви, у яких жодна частина не
має систематичного змісту. Наприклад, у назвах анілін і ванілін є суфікс -ін, але
їхні структури не містять потрійних зв'язків:
Н,СО
QbNH2 но-H-cf
4—' Анілін Х—' Н
Ванілін
■ напівтривіальні — назви, у яких тільки частина має систематичний зміст.
Наприклад, у назвах бутан або гліоксаль закінчення -ан і -аль відповідають
родовим суфіксам алканів і альдегідів, тоді як перша частина назви систе-
матичного змісту не має:
0ч р
С-С
СН3СН2СН2СН3 Бутан Н Н
Гліоксаль
■ систематичні - назви, побудовані за певними правилами зі спеціально
відібраних складів, наприклад:
СН3СН2СНСН2СН2СН3 СН3СН2СН2-С-СН3
гн О
Пентан-2-он
3-Метил гексан
У цьому розділі викладено головні правила побудови назв за замісниковою і
радикально-функціональною номенклатурами, які найширше застосовують в
органічній хімії. Деякі інші типи номенклатур розглянуто в розділах, присвя-
чених окремим класам органічних сполук, наприклад, гетероциклам.
1 Ганч А. (Наїіїксп А.) (1857-1935) - німецький хімік-органік, розробив методи одержання
піридинів, тіазолів, піролів. Є одним з фундаторів стереохімії Нітрогену. Вперше застосував
фізичні методи для визначення будови органічних сполук.
88
Чирва В.Я., Ярмолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
5.3.1. Номенклатура вуглеводневих замісників1
У замісниковій та радикально-функціональній номенклатурах широко
застосовують назви вуглеводневих замісників.
> Вуглеводневий замісник - залишок молекули вуглеводню, у якій
видалено один або кілька атомів Гідрогену.
■ Назви одновалентних вуглеводневих замісників утворюють від назв
відповідних вуглеводнів із застосуванням суфікса -ил (-іл, їл)2. Наприклад:
метан —> метил, пропан —> пропіл, пропен —> пропеніл, етин —> етиніл та ін.
■ Для деяких груп у номенклатурі ШРАС застосовують тривіальні назви.
Структури найпоширеніших вуглеводневих замісників з тривіальними назвами
наведено в табл. 5.2. Префікси ізо-, нео-, втор-, трет- застосовні тільки для
цих груп. Для нерозгалужених алкільних груп використовують префікс н~
(напр., н-бутил)3.
■ Назви складніших замісників утворюють із назв вуглеводнів за загаль-
ними правилами замісникової номенклатури. Атому з вільною валентністю
надають номер (локант) 1, наприклад:
4 3 2 1
СН3СН2СНСН2
сн3
2-Метилбутил
> Локант - цифра або літера, що позначає положення замісника в сполуці-основі.
■ Вуглеводневі замісники з двома вільними валентностями називають із
використанням суфікса -діїл, а трьома - відповідно, -триїл.
І
сн3—сн-(^н2 сн3—
Пропан-1,2-діїл Пропан-1,2,2-триїл
■ Назви двовалентних замісників, у яких обидві валентності відходять від
одного атома Карбону, утворюють додаванням до назви відповідного одно-
валентного замісника закінчення -іден, а тривалентних - -ідин. Наприклад:
СН3СН= СН2=С= С6Н5СН= (СН3)2С=
Етиліден Вініліден Бензил іден Ізопропіліден
1 Раніше вуглеводневі замісники (залишки) називали радикалами. Сьогодні ІЦРАС не рекомендує
цього терміна, щоб не плутати з вільними радикалами.
2 Після б, в, к, л, м, н, п, ф, х ("правило дев'ятки") пишуть -і, після інших приголосних -и, після
голосних -ї.
3 Префікси н-, втор- і треш- завжди пишуть курсивом і вимовляють "нормальний", "вторинний"
та "третинний".
сн2=снсн2сн2- (2^- н3с^у^
2 СІ
Бут-З-еніл 2-Хлорофеніл и-Толіл
Розділ 5. Класифікація і номенклатура органічних сполук
89
"нсе сн3с= с6н5с=
Мєтилідин Етилідин Бензилідин
■ Для двовалентних замісників, які одержані з лінійних алканів по двох
кінцевих атомах Карбону, використовують застарілі назви:
-сн2сн2сн2- - триметилен;
-сн2сн2сн2сн2- - тетраметилен тощо.
Таблиця 5.2
Тривіальні назви вуглеводневих замісників
Структура
Назва
Структура
Назва
Структура
Назва
СН3СН-
сн3
Ізопропіл*
СН3
сн,сн,с-
3 2,
сн3
трет-
Пентил
Феніл*
СНоСНСНо—
3| 2
сн3
Ізобутил*
СНз
сниссни—
3, 2
сн3
Неопентил
<0Ьсн>-
Бензил*
СН3СН2СН-
сн3
втор-
Бутил*
СН2=СН-
Вініл
^>-сн2сн2-
Фенетил
ш3
СН,С-
3|
сн3
трет-
Бутил*
СН2=СНСН2-
Аліл
<^ЬСН=СН_
Стирил
СН3СНСН2СН2-
сн3
Ізопентил
СН2=С-
2 1
сн3
Ізопропеніл
сн=снсн2
Цинаміл
сн2=
Метилен
-СН2СН2-
Етилен
-о-
Фенілен
(я-ізомер)
* Вуглеводневі замісники часто позначають скороченнями: Ме - метил, ЕХ - етил, Рг - пропіл, /*-Рг -
ізопропіл, Ви - бутил, /-Ви - ізобутил, $ес-Ви - втор-бугип, /-Ви - трет-бутил, Вп - бензил, РЬ - феніл.
90
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
5.3.2. Замісникова номенклатура
Назва органічної сполуки в замісниковій номенклатурі IUP АС принципово
може складатися з основи - систематичної назви головного вуглеводневого
ланцюга (циклу) або тривіальної назви певної стандартної сполуки, до якої
додають префікси і суфікс. Префікси позначають наявність як вуглеводневих,
так і характеристичних (функціональних) замісників, суфікс відображає наяв-
ність тільки головної характеристичної групи. Відповідно до сучасних правил
IUP АС і рекомендацій УНКоХіТерН в українській хімічній номенклатурі
локанти пишуть перед префіксами і перед суфіксами:
Вуглеводні та
функціональні замісники
Систематична назва
вуглеводневого ланцюга (циклу)
або тривіальна назва
Головна функціональна
група
Наприклад:
СІ
Замісник: хлор
СН3-СН-С=СН-СН2-СН-СН3
Основа: гепт-4-ен
0Й Головна група:-ол
Замісник: метил
/^чтт і
Основа: фенол
Замісник: хлор
11 '
іШІй Замісник: нітро
6-Метил-5-хлорогепт-4-ен-2-ол 4-Нітро-З-хлорофенол
префікс основа суфікс префікс основа
Для побудови назви за замісниковою номенклатурою ШРАС необхідно
виконати таке:
1) визначити головну групу;
2) вибрати сполуку-основу;
3) пронумерувати атоми у сполуці-основі;
4) скласти назву.
Розділ 5. Класифікація і номенклатура органічних сполук
91
• Головну групу вибирають за табл. 5.3. Чим вище в таблиці розташована функ-
ціональна група (функція), тим вона старша. У цьому разі тільки одну найстар-
шу групу позначають суфіксом, а інші розглядають як замісники і називають у
префіксах.
■ Низку функцій у замісниковій номенклатурі називають тільки в пре-
фіксах, наприклад: -Вг - бромо; -СІ - хлоро; -СЮ - хлорозил; -F - флуоро; -І -
іодо; -no2 - нітро; -NO - нітрозо; =n2 - діазо; -N3 - азидо; -OR - R-окси
(наприклад, метокси, пентилокси); -SR - R-сульфаніл (раніше R-тіо).
• Сполука-основа - найдовший вуглеводневий ланцюг (або цикл), який (у
порядку зменшення значущості) містить максимальну кількість:
а) головних (старших) груп (приклад 1);
б) кратних зв'язків (приклад 2);
в) замісників (найбільше розгалужений ланцюг) (приклад 3).
Крім того, у Правилах IUP АС перелічено сполуки, для яких дозволено
використання тривіальних назв і які можуть слугувати основою для побудови
назв їхніх похідних. Приклади таких назв наведені у відповідних розділах.
• Нумерацію головного ланцюга або циклу виконують у такий спосіб, щоб
одержати (в порядку зменшення значимості) мінімальні локанти для:
а) головних груп (приклад 1);х
б) для кратних зв'язків (приклад 4);
в) для замісників (приклад 3);
г) для першого за абеткою замісника (приклад 2).
Таблиця 5.5
Префікси і суфікси, що позначають найважливіші функціональні групи
Клас4
Формула*
Префікс
Суфікс
1
2
3
4
Карбонові кислоти
-СООН
-(С)ООН
Карбокси-
-карбонова кислота
-ова кислота
Сульфонові кислоти
-SO3H
Сульфо-
-сульфокислота
Солі
Естери
Галогеноангідриди
кислот
Аміди
Нітрили
-СООМ
-(С)ООМ
-COOR
-(C)OOR
-CO-Hal
-(С)О-НаІ
-CO-NH2
-(C)0-NH2
-C=N
-(C)-N
Карбоксилато-
R-Оксикарбоніл-
Галогенокарбоніл- {раніше
галогеноформіл-)
Карбамоїл-
Ціано-
Метал -карбоксилат
Метал -оат
R...-карбоксилат
R...-oaт
-карбонілгалогенід
-оїлгалогенід
-карбоксамід
-амід
-карбонітрил
-нітрил
Альдегіди
-сн=о
-(С)Н=0
Форміл-
Оксо-
-карбоксальдегід
-аль
92
Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Продовження табл. 5.3
1
2
3
4
Кетони
Спирти і феноли
Тіоли
Гідропероксиди
Аміни
>(С)=0
-ОН
-БН
-О-ОЯ
-мн2
Оксо-
Гідрокси-
Сульфаніл- (раніше
меркапто-)
Я-Гідроперокси-
Аміно-
-он
-ол
-тіол
-Я-гідропероксид
-амін
* Атоми Карбону в дужках належать до основи, їх зазначають у її назві, а не в суфіксі або префіксі, наприклад:
Сполука-основа: гексан
Гексанова кислота
основа суфікс
,.1
соон
Сполука-основа: циклобутан
Циклобутанкарбонова кислота
основа суфікс
• Назву складають з перелічування за абетковим порядком замісників (функ-
ціональних і вуглеводневих) з позначенням їхніх локантів і кількості (числові
префікси ди-, три-, тетра- та ін.), назви сполуки-основи і суфікса, що відповідає
головній групі (якщо вона в сполуці є), з позначенням її локанта.
З 2 1
СН3СН2СН2СНСН2СН2ОН
сн3снон
З-Пропілпентан-1,4-діол
Приклад 1
Пронумерований ланцюг коротший, але він
має дві гідроксильні групи. Початок нумерації
- за правилом мінімальних локантів для
головних груп (1,4 < 2,5).
СН2—ССН2СН(СН 3)2
І* 4
СН3С=СН2
2-Ізобутил-3-метилбутан-1,3-дієн
Приклад 2
Пронумерований ланцюг коротший, але
містить два подвійні зв'язки. Початок нумера-
ції визначає перший за абеткою замісник
(ізобутил).
СН
з
2 1
СН3СН2СН2ССН2СНСН3
\5 б 7
СН3СНСН2СН3
2,5-Диметил-4-пропілгептан-4-ол
Приклад З
Пронумерований ланцюг має більше замісни-
ків, ніж горизонтальний. Напрям нумерації
визначає перший менший локант метальних
замісників (2,5 < 3,6).
СН3
1 2 3 4 5\ 6 7
СН3СНСН=СНСНСНСН з
І
І
он он
5-Метилгепт-3-ен-2,6-діол
Приклад 4
Головні (гідроксильні) групи розташовані
симетрично відносно карбонового ланцюга,
тому напрям нумерації визначений меншим
локантом подвійного зв'язку (3). Локант
метальної групи беруть до уваги останнім,
тому в цьому разі він не впливає на вибір
нумерації.
Розділ 5. Класифікація і номенклатура органічних сполук
93
■ В абетковому порядку не беруть до уваги числові префікси ди- (ді-), три-,
тетра- й інші, втор- і трет- та префікси о-, л*-, п- у назвах сполук з
ароматичними замісниками (див. розділ 24).
■ Локанти між собою відокремлюють комами, локант від слів — дефісом,
назву основи пишуть разом. Розташування локантів у назві сполуки не є
рекомендацією Комісії з номенклатури IUP АС, а випливає з правил, що їх
регулюють національні Комісії. Тому в різних країнах для позначення положень
кратних зв'язків та інших функцій застосовують різні способи. Нижче наведено
приклади розташування локантів відповідно до традицій різних національних
мов:
2-метилпент-І-ен (укр.);
' _длгт агт атт 2-метилпентен-1; 2-метилпент-1-ен (рос);
СН2=ССН2СН2СН3 2-methylpent-1 -ene (англ., IUPAC);
Лтт 2-methyl-l-pentene (амер.);
3 2-Methylpent-l-en (нім.);
méthyle-2-pent-l-en (франц.)
5.3.3. Радикально-функціональна номенклатура
Назви сполук за радикально-функціональною номенклатурою IUP АС
складаються з назви замісника (як вуглеводневого, так і функціонального) і
назви функціонального класу (наведено в табл. 5.4). Альдегіди, карбонові
кислоти та їхні похідні, а також аміни називають за замісною номенклатурою
(див. відповідні розділи).
Таблиця 5.4
Функціональні назви, які використовують у радикально-функціональній номенклатурі
Сполука
Функціональна назва
RCO-Hal, RSOr-Hal, R-Hal
R-CN, R-NC
R,R'>C=0
Alk-OH
R-SH
R-OOH
R-O-R'
R-S-R', R-S-S-R'
RCO-N3, R-N3
Флуорид, хлорид, бромід, іодид
Ціанід, ізоціанід
Кетон
Спирт
Меркаптан, тіол
Гідропероксид
Етер або оксид
Сульфід, дисульфід
Азид
■ Якщо функціональна група є одновалентною, то сполучений з нею
замісник називають перед функціональною назвою:
CH3Ct CH3SO2-CI
СІ
Ацетилхлорид Мезилхлорид
СН3 СН2(рНСН з
СІ
emop-Бутилхлорнд
94
Чирва В.Я., Ярмолкж CM., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
■ У випадку двовалентної характеристичної групи перед функціональною
назвою зазначають обидві групи в абетковому порядку. Якщо два замісники є
однаковими, то використовують числівниковий префікс ди- (ді-):
<^^СН2-С-СН2СНСН3 сНзСН2-8-СН2СНз
О СН3
Бензилізобутилкетон Діетилсульфід
■ За традицією, в українській хімічній номенклатурі функціональні назви
спирт і етер1 пишуть окремим словом, а замісники називають у формі
прикметника:
:Н3СН2ОН СН2=СНСН2ОН СН3сн2сн2-0-СН2СН2СНз СН3-0-СН2СН2СН3
Етиловий Аліловий спирт Дипропіловий етер Метилпропіловий етер
спирт
1 На початку 90-х років XX ст. відновлено терміни, які застосовували у 20-30-х роках: етер і
естер, а також похідні терміни етеровий, естеровий, естерифікація як еквіваленти англійських
термінів ether, ester, esterification (див. відповідні розділи). Наприкінці 30-х років запроваджено
еквіваленти невдалих російських термінів "простий ефір", "складний ефір", "складноефірний",
"етерифікація". Новітня українська хімічна номенклатура позбулася застарілих термінів,
наблизившись до міжнародних стандартів.
Розділ 6
ІЗОМЕРІЯ
6.1. Загальні положення
У першій половині XIX ст. знайдено низку сполук з однаковою брутто-
формулою, але з різними фізичними і хімічними властивостями. Згідно з
теорією будови органічних речовин О. Бутлерова, властивості окремих сполук
визначені не лише кількістю і природою атомів, що є в їхньому складі, а й
хімічною будовою цих сполук. Зокрема для алкану з брутто-формулою С5Н12
теоретично можливим було існування трьох структурних ізомерів - пентану,
ізопентану та неопентану. Всі вони одержані згодом:
> Ізомери - це речовини, що мають однаковий кількісний та якісний склад,
але відрізняються будовою та, відповідно, властивостями.
У другій половині XIX ст., під час вивчення хіміками оптичних власти-
востей органічних сполук за допомогою поляриметра, виявлено ізомерні
речовини, що мали однакову хімічну будову, однакові фізичні константи, проте
відрізнялися відношенням до променя плоскополяризованого світла. Наприклад,
виділена з м'язів тварин молочна кислота СНзСН(ОН)СООН (її часто називають
м'ясо-молочною) обертала промінь плоскополяризованого світла праворуч,
молочна кислота того ж складу, одержана у процесі бродіння лактози за
допомогою деяких бактерій, - ліворуч; а молочна кислота, що утворювалась під
час молочнокислого бродіння молока, квашення капусти або синтетичним
способом, не виявляла оптичної активності.
Виявилося, що право- і лівообертальні молочні кислоти, так звані
оптичні ізомери, або антиподи, відрізняються просторовою структурою,
тобто різним розташуванням атомів у просторі. Оптично неактивна молочна
кислота - це суміш оптичних ізомерів однакової кількості, яку називають
рацемічною сумішшю, або рацематом. Існування ізомерів пояснено за
Пентан
Ізопентан
Неопентан
96
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
допомогою теорії тетраедричної будови атома Карбону (Я. Вант-Гофф, Ж. Ле
Бель, 1874 р.).
• Поляриметр. Якщо промінь звичайного світла, який є електромагнітною
хвилею з хаотично напрямленими коливаннями, пропускати через призму
Ніколя1, виготовлену з кристала ісландського шпату, то це приведе до утворення
плоскополяризованого променя світла, у якому коливання відбуваються в одній
площині. Оптично активні сполуки здатні повертати площину плоско поляри-
зованого променя світла праворуч чи ліворуч. Кут повороту можна визначити за
допомогою приладу, який називають поляриметром (рис. 6.1).
Головними частинами поляриметра є джерело світла (7) з певною довжиною
хвилі, як звичайно, 589 нм, що відповідає Б-лінії спектра натрію; поляризатор
(2) - призма Ніколя для поляризації світла; кювета (3) з розчином аналізованої
речовини; і друга призма Ніколя (4), що відіграє роль аналізатора. Під час
проходження плоскополяризованого світла через розчин оптично активної
речовини площина поляризації повертається на певний кут а, який можна
визначити, повернувши призму-аналізатор на той же кут а, щоб оптичні осі
поляризаційної призми і призми-аналізатора збігалися.
• Питоме і молекулярне обертання. На значення кута а впливає низка
чинників, у тому числі довжина хвилі світла, температура, товщина зразка,
концентрація та природа розчинника або густина у випадку індивідуальної
речовини. Тому як фізичну характеристику використовують не саме значення
кута а, а розрахункове значення питомого обертання [а], яке обчислюють за
формулою
де а - кут обертання, град.; / - товщина зразка, або довжина кювети, дм; с -
концентрація розчину, г/100 см3.
У записі питомого обертання верхній індекс відображає температуру
(звичайно 15-25 °С), а нижній - довжину хвилі світла, яке пропускають (у
випадку Б-лінії натрію ставлять індекс Б). Концентрацію (г/100 см3) і розчинник
наводять після значення питомого обертання в дужках. Наприклад, запис
а
Рис. 6.1. Принципова схема поляриметра
1 В. Ніколь (Мсої Ш.) (1768-1851) - шотландський фізик. Головні праці у галузі оптики.
Винайшов прилад (поляризатор) для отримання лінійно поляризованого світла (призма Ніколя),
який широко використовують досі.
Розділ 6. Ізомерія
97
[а]}) +2,6° (с 10; метанол) для винної кислоти означає праве питоме обертання,
виміряне для довжини хвилі 546 нм при 15 °С і концентрації 10 г в 100 см3
метанолу. Значення питомого обертання містить масову характеристику кон-
центрації, тому її не можна застосовувати, щоб порівняти речовини з різною
молекулярною масою. У таких випадках використовують поняття молекулярне
обертання [М], яке обчислюють за формулою
6.1.1. Хімічна структура, конфігурація, конформація
органічних сполук
Для опису будови органічної сполуки використовують три головні поняття.
> Хімічна структура (конституція) — порядок з'єднання атомів і типи
зв'язків у молекулі.
Для спрощеного запису структури органічних сполук хімічні зв'язки
позначають прямими лініями, маючи на увазі, що атоми Карбону містяться в
кутах, утворених цими лініями:
> Конфігурація - взаємне розташування у просторі атомів і груп атомів
молекули без урахування змін, що виникають під час обертання навколо
простих зв'язків.
Наприклад, у молекулі 1,4-диметилциклогексану метальні групи можуть
бути з одного боку від площини циклу або з обох:
> Конформація - просторове розташування атомів і груп атомів молекули
певної конфігурації, яка виникає внаслідок повертання навколо одинарних
зв'язків.
Наприклад, у молекулі транс-1,4-диметилциклогексану атоми Карбону
циклу містяться не в одній площині, а утворюють просторову структуру, яку
називають "крісло":
Н^С СН2
Н3С-НС СН-СНз або Н3С-
Н2С С Н2
Хімічна структура 1,4-диметилциклогексану
цис- і т/?а«с-Конфігурації 1,4-диметилциклогексану
98
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
сн3 н
у^ГН н)с\^л .сн,
сн3 н
Конформації крісло" для транс-1,4-диметилциклогексану
6.1.2. Зображення просторової будови молекул
Для зображення просторової будови молекул на площині використовують
низку стереохімічних і проекційних формул.
❖ Стереохімічні формули. Просторове розташування замісників в ато-
мах Карбону з ^р3- і 5р2-гібридизаціями (тетраедричний і тригональний атоми
Карбону) можна показати за допомогою клиноподібних ліній. Як звичайно,
зафарбований клин позначає розташування замісника перед площиною малюн-
ка, а незафарбований або заштрихований клин - за площиною. Звичайними
лініями показують зв'язки, що розміщені у площині малюнка:
. --^СООН
в площині ч І
\ І Н за площиною
^ Н3С ОН перед площиною
Іноді розташування замісників за площиною додатково виділяють, спря-
мовуючи на рисунку вістря клина до замісника. Часто в таких формулах
центральний тетраедричний атом Карбону взагалі не показують:
СООН СООН СООН
І^и'»Н о^н £р^Н
Н3СГ Т>Н Н3С" т>н Н3СГ т>н
Стереохімічні формули молочної кислоти
❖ Перспективні формули. Вони зручні для того, щоб показати взаємне
розташування замісників біля двох сусідніх атомів Карбону. В таких формулах
зв'язок, що з'єднує ці атоми Карбону, подовжений:
Н..Н нЧч.сн3
Перспективні формули етану та бутану
Розділ 6. Ізомерія
99
❖ Проекційні формули Фіш ера. Для зручності роботи з просторовими
формулами молекул, у складі яких є асиметричний атом Карбону, Е. Фішер1
запропонував використовувати площинні проекції (проекція Фішера).
> Асиметричний атом Карбону - атом Карбону, що має чотири різні
замісники.
Проекцію Фішера будують так:
а) асиметричний атом Карбону позначають перетином взаємно перпен-
дикулярних ліній;
б) замісники, розташовані на горизонтальній лінії, у просторі містяться
над площиною, а на вертикальній - під площиною;
в) найбільше окиснений атом Карбону розміщений зверху;
г) найдовший вуглеводневий ланцюг зображають вертикально:
Нині застосовують і модернізовані зображення проекцій Фішера, у яких
використовують клиноподібні позначення напряму зв'язків:
• Правила застосування проекцій Фішера.
1. Проекції не можна виводити з площини малюнка, оскільки енантіомер
у такому разі перетворюється у свій антипод.
2. Дозволено повертати проекцію в площині на 180°:
Наприклад, у наведеній вище проекції Фішера найбільше окиснений атом
Карбону міститься знизу. Поворот у площині на 180° приводить до правильного
вигляду проекції.
3. Заборонено повороти на 90 і 270°, оскільки в цьому випадку
енантіомер перетворюється в свій антипод.
1 Фішер Е. (Fischer Е.) (1852-1919) - німецький хімік-органік. Засновник біоорганічної хімії.
Проводив фундаментальні дослідження в хімії вуглеводів, білків, пуринових основ. Нобелівська
премія 1902 р.
соон соон
t,,CH3^.H^OH
н^бн-:::>в=:сн3
Формули молочної кислоти: стереохімічна проекція Фішера
соон соон соон соон
н-|-он - нчь-он - н-с-он - н-ф-он
CH3 CH3 CH3 CH3
.соон _
100
Чирва В.Я., Ярллолкж CM., Толкачова Н.В., Земляков О.Є. Органічна хімія
4. Парна кількість перестановок замісників не змінює конфігурації
молекули:
У наведеному прикладі, у спершу одержаній проекції, найдовший
вуглеводневий ланцюг не розташований вертикально. Дві перестановки
замісників привели до нормального вигляду проекції Фішера без зміни
конфігурації.
❖ Проекції Ньюмена1 звичайно використовують, щоб розглянути кон-
формації молекули. Такі проекції найзручніші для відтворення розташувань у
просторі замісників при двох сусідніх атомах Карбону. Щоб одержати проекції
Ньюмена, використовують проекції перспективних формул у напрямі зв'язку
між цими атомами. Близький атом Карбону зображають крапкою, далекий -
колом:
Раніше для найменування право- і лівообертальних ізомерів викорис-
товували, відповідно, позначення (+) або d (від лат. dextro - право) і (-) або / (від
лат. laevo - ліво). Відповідно, рацемат позначали (±) або di. Однак такий спосіб
найменування не мав подальшої перспективи розвитку. По-перше, знак
оптичного обертання є фізичною характеристикою речовини, що залежить від
довжини хвилі світла, концентрації, розчинника та інших чинників. Наприклад,
(-)-яблучна кислота у водних розчинах з концентрацією 8,4 % мала [а]о~2,3°, a
за концентрацій 34 і 70 %, відповідно, [a]D 0o і [a]D +3,3°. Крім того, одна й та ж
сполука в різних розчинниках може бути як і ліво-, так і правообертальною. По-
друге, і це важливіше, знак обертання не дає нам уявлення про просторову
будову сполуки. Для серії похідних з однаковою конфігурацією асиметричного
центру знаки оптичного обертання можуть бути різними:
1 Ньюмен М. (Newman M.S.) (1908-1993) - американський хімік-органік. Запропонував формулу
для зображення конформацій (1955).
Перша перестановка Друга перестановка
6.1.3. Стереохімічні номенклатури
Розділ 6. Ізомерія
101
[аЬ +2,6° (Н20) [а]0 -8,25° [а]0 -75,47° [а]578 +91,8°
Усе це зумовило розробку номенклатурних систем, що давали б змогу
позначити конфігурацію молекули. Розглянемо головні види стереохімічних
номенклатурних систем, які використовують сьогодні.
❖ ИЬ-номенклатура запропонована Е. Фішером, для позначення просто-
рових конфігурацій молекул. Згідно з номенклатурою Е. Фішера, щоб визна-
чити конфігурацію сполуки, її просторову структуру потрібно порівняти зі
структурою стандартної речовини. В 1906 р. М. Розанов1 запропонував (+)-
гліцероловий альдегід як стандарт, якому була приписана конфігурація,
позначена символом Т). Відповідно, конфігурація його дзеркального (-^-ізо-
меру позначена символом Ь.
Якщо певну сполуку одержували з гліцеролового альдегіду хімічними
реакціями, що не порушували асиметричного атома Карбону, то її конфігурація
мала теж позначення, що й у стандарту. Наприклад, послідовне окиснення
альдегідної функції і відновлення первинного гідроксилу (+)-Б-гліцеролового
альдегіду дає Б-молочну кислоту, яка є лівообертальною:
Отже, конфігурацію нової сполуки визначали шляхом її хімічних
перетворень у сполуку з відомою конфігурацією. Конфігурацію, з'ясовану цим
способом, називають відносною. З появою рентгеноструктурного аналізу для
великої кількості органічних сполук визначили абсолютну конфігурацію -
реальне розташування атомів у просторі. Зокрема, підтвердили конфігурацію Т>-
винної кислоти, яку спершу визначили відносно О-гліцеролового альдегіду, що,
відповідно, підтвердило правильну просторову будову цього стандарту,
запропонованого М. Розановим.
Тепер БЬ-номенклатуру застосовують для побудови назв вуглеводів,
гідрокси- й амінокислот. Для інших класів органічних сполук використовують
АУ-номенклатуру.
'ОН СН2ОН СН2ОН
Н
СООН
-[-он
сн.
(+)-0-Гліцероловий альдегід
(-)-Б-Молочна кислота
1 Розанов М. (ЯоБапоГГ М.А.) (1874—1951) - американський хімік українського походження.
Головні праці з теоретичної хімії, займався геометрією органічних молекул.
102
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
❖ ЛЯ-номенклатура. Р. Кан1, К. Інгольд2 і В. Прелог3 1956 р. запропонували
універсальнішу номенклатуру позначень конфігурації просторових ізомерів. Щоб
визначити конфігурації, необхідно:
1) визначити старшинство (пріоритетність) замісників, що оточують аси-
метричний атом Карбону.
■ Старшинство визначене порядковим номером елемента в Періодичній
системі. Чим більший порядковий номер елемента, тим старший атом. Нижче
наведено приклади розстановки старшинства атомів (1 - найстарший атом):
Н
НрС-ОН
®Ьсі©
®н с^о-н (і) ^ А~ (2)
■ Старшинство однакових сусідніх атомів визначають шляхом порівняння
старшинства приєднаних до них атомів. Наприклад, у структурі (3) атом
Карбону гідроксиметильної групи (один зв'язок на атом Оксигену і два - на
атоми Гідрогену) старший від атома Карбону етильного замісника (один зв'язок
на атом Карбону і два - на атоми Гідрогену), який, відповідно, є старшим від
атома Карбону метальної групи (три зв'язки на атоми Гідрогену):
®
©н © СН2ОН
@Дхн2он ол^%
нзс ®сн2сн3 с ®с
(3) НО Н (4)
■. Кратні зв'язки розглядають як відповідну кількість одинарних зв'язків
(С=0 = 2 С-О). Атом Карбону в ароматичному циклі прирівнюють до трьох С-С
зв'язків. Тому в сполуці (4) атом Карбону карбоксильної групи (три зв'язки на
атоми Оксигену) старший від атома Карбону карбонільної групи (два зв'язки на
атоми Оксигену і один - на атом Гідрогену), який, відповідно, старший від
атома Карбону гідроксиметильної групи (один зв'язок на атом Оксигену і два -
на атоми Гідрогену);
2) молекулу розміщують у просторі так, щоб асиметричний атом затуляв
замісник з найменшим номером від спостерігача:
1 Кан P. (Cahn R.) (1899-1981) - англійський хімік. Був головним редактором журналу "Journal of
the chemical Society" (Лондон).
2 Інгольд К. (Ingold К.) (1893-1970) - англійський хімік. Головні праці присвячені фізичній
органічній хімії, засновником якої він с. Розробив концепцію мезомерії. Автор фундаментальних
праць з кінетики і механізмів органічних реакцій.
3 Прелог В. (Prelog V.) (1906-1998) - швейцарський хімік. Головні праці в галузі синтезу цикланів
і антибіотиків. Нобелівська премія 1975 р.
Розділ 6. Ізомерія
103
®
сн2он
©JU'H®
HCT ^Cl©
(1)
®
СН2ОН
^ ^С1©
СІ
L.»F©
©І^^Вг®
(2)
©
f2H5
® JL...H©
©
СН2ОН
ноос
(3)
©
сн2он
©jl..H©
<4>®
або
®
сі
но
©І
Вг©
©
с2н5
© г
СН2ОН
(1)
(2)
(3)
©
ноос
®
сн,он
сно
©
(4) W
3) зі зменшенням старшинства трьох найстарших замісників за годинниковою
стрілкою ізомер позначають літерою R (rectus - правий), а якщо проти
годинникової стрілки - S (sinister - лівий).
HO
®
сн2он
-^^сі©
(1)
R-
©
СІ
©
с2н5
©I^-^Br© н3с^^сн2
(2) ®
ОН НООС
(3)
S-
©
СН2ОН
"^-^сно
(4)®
S-
Сполуку (2) за БЬ-номенклатурою однозначно назвати неможливо.
" За допомогою ЛУ-номенклатури визначають конфігурацію і для проекцій
Фішера. Після з'ясування старшинства замісників проекцію потрібно
перетворити так, щоб молодший замісник був на вертикальній лінії. У випадку
зменшення старшинства трьох замісників, що залишились, за годинниковою
стрілкою ізомер має /^-конфігурацію, у протилежному випадку - £-конфігу-
рацію:
©
H2N-
®
соон
-H®:
(роон
СН,
СН,-*)
® 3
Перша перестановка
Н
Друга перестановка
> н3с-
соон
-NH2
Н
Н3С
©
^СООН
®
Н
5-ізомер
-NH,
©
❖ цис-, транс- і Е,2-Номепклатури. Традиційно для геометричних
ізомерів розташування замісників у С=С зв'язку позначають за допомогою
префіксів цис-9 якщо однакові замісники розміщені по один бік від площини
зв'язку, і транс-, якщо - по різні.
104
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Н3С
СН2СН3
Нч СН2СН3
н3с н
с=с
/ \
н
н
і/ис-Пент-2-ен
тря«оПент-2-ен
Проте такий спосіб опису конфігурації сприйнятий лише для порівняно
простих сполук. У складніших випадках використовують ^^-номенклатуру.
Конфігурацію подвійного зв'язку за цією номенклатурою визначають так.
Спочатку з'ясовують старшинство атомів, що зв'язані з кожним із двох
атомів Карбону подвійного зв'язку. Старшинство, як і у випадку ЛУ-номенкла-
тури, визначають за порядковим номером відповідного елемента в Періодичній
таблиці. Чим більший порядковий номер елемента, тим він старший. В
однакових атомів старшинство визначають за старшинством атомів, з якими
вони зв'язані. Потім оцінюють взаємне розташування старших атомів. Якщо
вони розташовані з одного боку площини я-зв'язку, то це Z-ізомер (від нім.
Zusammen - разом), а якщо з різних - £-ізомер (від нім. Entgegen - напроти):
• Головні типи ізомерії. Розрізняють два головні типи ізомерії:
■ структурну, зумовлену відмінністю сполук між собою за хімічною будовою;
■ просторову (стереоізомерію), характерну для ізомерів з різним
розміщенням у просторі атомів, що є в складі молекули.
Відповідно, структурну ізомерію поділяють на два класи:
■ статичну ізомерію, що пов'язана зі змінами послідовності з'єднання
атомів у молекулі; для реї характерна відсутність переходу одного ізомеру в
інший за звичайних умов,
■ динамічну ізомерію (таутомерію).
> Динамічна ізомерія, або таутомерія, - це рівноважна ізомерія, що
виникає внаслідок перенесення атомів у вигляді йона (частіше - протона):
(£)-2-Хлоробут-2-ен-1-ол (2)-3-Аміно-4-метилгексен-3-ова кислота
6.1.4. Класифікація видів ізомерії
Таутомери
Кетонна й енольна форми циклогексан-1,2-діону
Розділ 6. Ізомерія
105
• Стереоізомерію також поділяють на два головні типи:
■ енантіомерію — існування сполуки в вигляді двох неідентичних прос-
торових структур, що є дзеркальними відображеннями один одного;
■ діастереомерію — різновид стереоізомерії, що пов'язана з відмінністю в
просторовій будові сполук, які не є енантіомерами.
Структурні ізомери
СН3СНСООН СН2СН2СООН
СІ СІ
2-Хлоро- і 3-хлоропропіонові кислоти
Енаптіомери
соон соон
н-|-он но-|-н
СН2ОН СН2ОН
D- і Ь-Гліцеролові кислоти
Стереоізомери
Н
СООН
,,.«СНз
сі сі-
соон
..'СНЗ
'Н
(Я)- і ^-2-Хлоропропіонові кислоти
Діастереомери
СНО СНО
Н-
НО-
-ОН
-Н
Н-
Н-
ОН
ОН
СН2ОН
СН2ОН
Ь-Треоза і О-еритроза
■ Крім того, в окрему групу виділяють конформаційну ізомерію, у якій
розглядають найбільш характеристичні конформації молекул.
Більшість із зазначених різновидів ізомерії можна далі поділяти на різні
типи. Детальніше кожний вид ізомерії розглянуто в наступних розділах.
6.2. Структурна ізомерія
❖ Статичну структурну ізомерію поділяють на три типи:
■ ізомерію вуглеводневого скелета - ізомери відрізняються порядком з'єд-
нання атомів Карбону в ациклічній або циклічній молекулі;
■ ізомерію положення функціональних груп, зумовлену різним розміщенням
функціональних груп у сполуці з одним і ти|м же вуглеводневим скелетом;
■ міжкласову ізомерію, пов'язану з відмінним порядком з'єднання атомів у
молекулі, що приводить до утворення різних класів органічних сполук.
Ізомерія вуглеводневого Ізомерія положення подвійних
скелета зв'язків
Н3 С—С~СН2
СН2=СНСН2СН3
СН3СН=СНСН3
МЬіскласова
ізомерія
Н2С-СН2
Циклобутан
СН3
2-Метилпропен Бут-1-ен Бут-2-ен
Статичні структурні ізомери стійкі за звичайних умов, хоча в спеціальних
умовах можливий перехід одного ізомеру в інший: висока температура і/або
106
Чирва В.Я., Ярллолюк CM., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
наявність каталізаторів. Наприклад, рівноважна суміш, що утворюється під час
дії на бутан алюміній броміду, містить 80 % ізобутану:
А1Вг3
СН3СН2СН2СН3 НзС-срН-СНз
сн3
❖ Таутомерія. На відміну від статичних структурних ізомерів, таутомерні
форми сполуки утворюють рівноважну суміш за нормальних умов.
Залежно від будови сполуки внесок кожного компонента в таутомерну суміш
може змінюватися в широкому діапазоні. Наприклад, циклогексанон має 0,02 %
фенольної форми, а бензоїлацетон - 99 % енольної.
Звичайна таутомерія пов'язана з перенесенням у структурі молекули
позитивно зарядженої частинки - катіонотропна таутомерія. Оскільки най-
частіше такою частинкою є протон, то таку ізомерію називають прототропною.
Рідше трапляється аніонотропна таутомерія, де відбувається міграція галогеніду,
гідроксиду, ацилоксилу та інших аніонів:
СН2-СН=СН-СН3 *—у СН2=СН-СН-СН3
СІ СІ
1-Хлоробут-2-ен, 75-85 % 3-Хлоробут-1-ен, 15-25 %
❖ Головні типи таутомерії
■ Валентна таутомерія - динамічно рівноважний перерозподіл зв'язків
між атомами без міграції замісників. Наприклад, за низьких температур
усталюється динамічний рівноважний перерозподіл зв'язків між атомами без
міграції замісників. Проте коли температура підвищується понад 20 °С, рівно-
вага повністю зсувається в бік утворення першого продукту:
оси оо
Циклооктатетраєн Біциклооктатрієн Гомотропіліден Бульвален
Для гомотропілідену обидві форми існують з однаковою ймовірністю, і
таутомерний перехід однієї в іншу відбувається дуже швидко. В трициклічному
похідному гомотропілідену - бульвалені всі 1 290 600 теоретично можливих
ізомерів існують в однаковій кількості завдяки величезній швидкості пере-
творення таутомерних форм і, як наслідок, десять атомів Карбону стають екві-
валентними.
■ Кето-єнольна таутомерія. Рівновага усталюється між у кетонною і
енольною (-єн - подвійний зв'язок, -ол - гідроксильна група) формами. Для більшості
карбонільних сполук основною є кетонна форма:
Розділ 6. Ізомерія
707
0 0 —її
н2с-с-сн2-с-сн3 н,с-с=сн-с-сня
Кето-єнольні форми циклогексанону і бензоїлацетону
■ Імінно-єнамінна таутомерія багато в чому подібна до кето-єнольної
таутомерії. До того ж, головним способом одержання імінів є конденсація
карбонільних сполук з первинними амінами. В цьому випадку рівновага зміщена
в бік утворення стійкішої імінної форми:
н3с-с-с. 4—' н3с-с=с. х—'
н н н н
Імінно-єнамінні форми іУ-пропіліденаніліну
Як окремий випадок імінно-єнамінної таутомерії можна вважати таутомери
2-амінопіридину, з яких менш стійкою формою є піридоімінна:
сі — сі
1 і
Н
Амінна і піридонімінна форми 2-амінопіридину
■ Лактим-лактамна (амідо-імідольна) таутомерія. Циклічні аміди
(лактами) перебувають у рівновазі з імідольною (імід - група -ол - група -
ОН) формою -лактимом. За звичайних умов таутомерія зсунута в бік стійкіших
лактамних похідних:
Амідна та імідольна форми капролактаму
■ Нітрозооксимна таутомерія. Первинні та вторинні нітрозоалкани не-
оборотно перетворюються у відповідні оксими:
сн-и=о —»■ с-и-он
Нітрозофеноли і нітрозоаніліни існують також у вигляді двох таутомерних
форм. Такий різновид таутомерії називають хіноксимною таутомерією. На
108
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
перевагу тієї чи іншої форми в рівноважній суміші впливають різні чинники.
Наприклад, л-нітрозофенол головно міститься в хіноксимній формі, тоді як для
о-нітрозофенолу переважною є нітрозна форма, яка зазнає стабілізації за
допомогою внутрішньомолекулярного зв'язку:
М*° ^Ч-ОН
м=о гЧ-ОН
Нітрозо- і хіноксимні форми л-нітрозофенолу і о-нітрозофенолу
■ Аци-нітро-тоутомерія. В нейтральному і лужному середовищах пер-
винні й вторинні а-нітроалкільні сполуки перебувають у рівновазі з тауто-
мерною ш/и-формою. Нітроформа значно стійкіша порівняно зі своїм тауто-
мером. Наприклад, рівноважний вміст до/и-форми для 2-нітропропану становить
близько 0,3 %:
н3с . ,р ' н3с он
сн-іч -а—^ с=н
н3с ч0 н3с ч0
Нітро- і ш/н-форми 2-нітропропану
■ Циклоланцюгова таутомерія. Низка біфункціональних сполук (у- і 8-
гідроксіальдегіди, гідрокси- й оксокислоти, вуглеводи) здатні до внутрішньо-
молекулярних реакцій і тому перебувають у рівновазі зі своїми циклічними
формами. Наприклад, у розчині 4-гідроксибутаналю у водному діоксані лише
11,4 % припадає на ациклічну форму:
Н2(^ено
н2сХ!Н2ОН
Лінійна і циклічна (лактольна) форма 4-гідроксибутаналя
Детальніше головні типи таутомерії розглянуті у відповідних розділах.
❖ Топологічна ізомерія. Останніми десятиліттями вчені одержали і вивчили
нові типи хімічних сполук, у яких частини молекул з'єднані не хімічними
зв'язками, а механічно. До таких сполук, зокрема, належать:
■ катенани - системи замкнутих один на одного циклів;
■ гротоксани - циклічні молекули, через які проходить лінійна молекула з
прикінцевими об'ємними замісниками, які перешкоджають роз'єднанню частин
такої молекули.
Розділ б. Ізомерія
109
О О
Ізомерні катенани і ротоксани
Для подібних типів сполук можуть існувати різні ізомери, у тім числі
енантіомери.
За основу пояснення існування просторових (конфігураційних) ізомерів
узято теорію Я. Вант-Гоффа і Ж. Ле Беля про тетраедричну будову атома
Карбону. Одна частина органічних молекул має хіральність, а інша - ахіральна.
> Хіральність (від грец. скеіг - рука) з погляду будови хімічних сполук -
це несумісність просторової структури молекули зі своїм дзеркальним
відображенням.
Хіральні молекули не мають площини і центра симетрії, але можуть мати
вісь симетрії. У

> Площина симетрії а проходить через молекулу так, що поділяє її на дві
частини, які є дзеркальними відображеннями одна одної.
> Вісь симетрії п-порядку С„ - вісь, яка проходить через молекулу; у разі
повертання навколо осі симетрії на кут 360°/п структура суміщається з вихідним
зображенням:
> Центр симетрії і - умовна точка, рівновіддалена від однакових атомів
або груп атомів молекули.
6.3. Стереоізомерія
63.1. Хіральність, енантіомери
Площини і вісь симетрії в молекулі дихлорометану
110
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Вг СН3 Й>-, ÇH3
СН3 Вг
Центр симетрії в молекулі Площина симетрії в молекулі
транс-13-Дибромо-тршс-2,4-диметшщиклобутану транс-13-Дибромо-і/і/с-2,4-диметалциклобутану
❖ Енантіомери. Переважна частина хіральних молекул може існувати у
вигляді двох неідентичних дзеркальних відображень - енантіомерів, або
дзеркальних ізомерів.
Енантіомери мають однакові фізико-хімічні константи, виявляють однакові
хімічні властивості, проте відрізняються за знаком оптичного обертання. Тому
термін оптичні ізомери використовують як синонім енантіомерів. Іншою
суттєвою відмінністю багатьох енантіомерів між собою є їхня різна поведінка в
біологічних системах. Наприклад, на початку розділу ми згадували про
енантіомерні право- і лівообертальні молочні кислоти, одержані в різних
біохімічних процесах. Структури і константи цих сполук такі: ^
СООН СООН
Н-)-ОН НО-|-Н
СН3 СН3
D-Молочна кислота L-Молочна кислота
т. пл. 53 °С, [ctfo -2,6° (вода) т. пл. 53 °С, [a]D +2,6° (вода)
Рацемічна молочна кислота, яка є сумішшю енантіомерів у рівних кіль-
костях, оптично не активна і має т. пл. 18 °С.
• Оптична чистота. Дослідники не завжди вивчають лише індивідуальні
енантіомери або рацемати. Часто речовина є сумішшю дзеркальних ізомерів із
вмістом одного з компонентів до 100%, але понад 50%. У таких випадках
говорять про оптичну чистоту сполуки. Визначити оптичну чистоту (%) можна
за такою формулою:
~ а суміші енантіомерів Л лп
Оптична чистота = ¿ - «100.
а чистого енантіомера
У разі розраховування оптичної чистоти вважають, що суміш складається з
чистої речовини і рацемату. Тому якщо в суміші 10% енантіомера А і 90%
енантіомера Б, то вважають, що вона складається з 20 % рацемату АБ і 80 %
чистого енантіомера Б, отже, оптична чистота суміші дорівнює 80 %.
6.3.2. Головні причини хіральності молекул
Розглянемо головні причини хіральності молекул органічних сполук.
❖ Хірольний центр. Як звичайно, хіральність пов'язана з наявністю
Розділ 6. Ізомерія
111
хірального центру. Найчастіше функції такого центру виконує асиметричний
атом Карбону. Проте хіральність виявляють чотиризаміщені похідні Силіцію,
Сульфуру, Нітрогену, Фосфору і низка інших елементів:
О а СН2СН=СН2 О
|....СН3 Iе ^.ХНз II сн3
Н3С1^ СНзСН^ СНзСНгСНг^
^-Ізомер .^-Ізомер Л-Ізомер
■ Хіральні властивості виявлено також для низки тризаміщених похідних
Сульфуру і Фосфору, які в просторі мають пірамідальну структуру.
Конфігурацію цих сполук можна розглядати як тетраедричну, якщо /?-орбіталь
неподіленої електронної пари враховувати як вершину тетраедра:
кн сн2=снсн2^
/Мзомер 5-Ізомер
■ Третинні аміни також мають пірамідальну будову, проте їхня
конфігурація не стійка, оскільки відбувається пірамідальна інверсія - швидке
перетворювання конформації. Спеціальними методами, наприклад, уведенням
замісників із гетероатомами, що мають два неспарені електрони, можна під-
вищити стійкість конфігурації та виділити індивідуальні ізомери:
Я* Я1 П
\3 ^ СН3СН20^ ^СН2СН2С02СН3
Пірамідальна інверсія амінів 5-Ізомер
❖ Вісь хіральності. У молекул без хірального центруутакож можуть бути
хіральні властивості, проте ці молекули повинні мати вісь хіральності.
■ Прикладами сполук, які мають хіральну вісь, можуть бути похідні оленів
- сполуки, що мають два подвійні зв'язки в одного атома Карбону. Оскільки два
я-зв'язки в цих сполуках розміщені у взаємно перпендикулярних площинах, і
якщо є хоча б по одному заміснику в кінцевих атомів Карбону, то сполука може
існувати в вигляді двох енантіомерів:
112
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
с=с=с
н
Вісь
^ хіральності
Н
■ Такі ж взаємно пзрпендикулярні площини існують у заміщених цикло-
алканів з екзоциклічним подвійним зв'язком і в спіросполуках - біциклічних
системах з одним спільним карбоновим атомом:
СІ
Н
\
Н
с=с=с
ноос соон
Дальній
_ н®
НООС—-^-Н Ближній
СІ
н
і н
соон
©СООН
Л-Ізомер
Дальній
©СООН
©С1-^4-Н Ближній
©Н ^
5-Ізомер
СІ
Дальній
сі®
©Н-£ тСІ
>к ©
Ближній
©Н
/Мзомер
Оскільки в такій хіральній молекулі існують чотири замісники, які
забезпечують несиметричність, то її конфігурацію можна позначати за допо-
могою /?,£-номенклатури. Для цього спочатку потрібно визначити старшість
ближніх замісників, а потім - дальніх. Якщо старшинство трьох перших за
старшинством замісників зменшується за годинниковою стрілкою, то це і?-ізо-
мер, якщо проти - .^-ізомер.
■ Низка похідних біфенілу також має вісь хіральності. Сама молекула
цього вуглеводню плоска. Взаємне відштовхування просторово зближених
замісників у положенні 2,2' спричиняє поворот навколо а-зв'язку. Якщо
додатково ввести функції в положення 6,6', то вони змушують цикли розта-
шуватися у взаємно перпендикулярних площинах. За наявності в положеннях
22' і 6,6'різних замісників молекулу можна зобразити у вигляді пари
дзеркальних ізомерів. Такий тип стереоізомерії називають атропоізомерією.
Конфігурацію атропоізомерів позначають за Л,£-цоменклатурою аналогічно до
аленових похідних:
Розділ 6. Ізомерія
113
Біфеніл
©
Вг
©вг—4н-скю
сі
©
5-Ізомер
Вісь
хіральності
©
Вг
©СІ-Д^- Вг©
СІ
©
/Мзомер
Ключовий момент реалізації атропоізомерії - наявність стерйчних
(просторових) перешкод вільному обертанню навколо о-зв'язку. Наприклад,
виділено окремі ізомери 2,2'-ди-/я/?ет-бутилбіфенілу і 1,Г-бінафтилу, у яких
об'ємні замісники у фенільних кільцях закріплюють конфігурацію молекул:
НзС СН3
(Л)-2,2'-Ди-т/?ет-бутилбіфеніл (5)-1,1 '-Бінафтил
❖ Площина хіральності. Щоб виявився цей варіант хіральності, молекула
повинна мати площинний фрагмент, звичайно, я-зв'язок або ароматичний цикл
і, з одного боку цієї площини, замісник. Типовими прикладами таких сполук є
горанс-циклоалкени, я-комплекси алкенів або аренів і циклічні похідні аренів.
■ У траяс-циклоалкенах вуглеводневий ланцюг розташований над площи-
ною подвійного зв'язку, що приводить до існування молекули у вигляді двох
енантіомерів:
(сн2)6; у і
г <с',Н!(сн2)6
Ні
Л- і 5-Ізомери транс-циклооктену
■ Аналогічно, п-комплекси алкенів і аренів з похідними металів можуть
існувати у вигляді структур з неідентичними дзеркальними відображеннями:
114
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
r- і 5-Ізомери я-комшіексу о-толуїлового альдегіду з трикарбонілом хрому
■ Циклічні поліметинові етери гідрохінону називають анса-сполуками (від
лат. ansa - ручка), оскільки через просторові перешкоди поліметиленовий
ланцюг розташовується перпендикулярно до площини фенолу. За наявності хоча
б одного замісника в ароматичному циклі можлива реалізація двох дзеркальних
конфігурацій:
U (їН2)п
г J но2с
ш/са-Сполука
Ш2С
Н02С
[2,2]-, [3,3]- і [4,4]-парациклофанкарбонові кислоти
Важливу роль у стійкості таких ізомерів відіграє розмір етерного циклу.
Наприклад, при «=8 сполука існує у вигляді двох енантіомерів, при л=9
нагрівання понад 70 °С спричиняє рацемізацію речовини, а у разі «=10 можна
вести мову лише про конформаційні ізомери, а не про конфігурацію.
■ Таке явище спостерігають і для парациклофанів - циклічних систем, у
яких кільця бензену зшиті двома олігометиленовими ланцюгами. Для [2,2]- і
[3,3]-парациклофанкарбонових кислот з етиленовими і триметиленовими
містками існують дзеркальні ізомери, а у випадку [4,4]-парациклофанкарбонової
кислоти з тетраметиленовими містками можливе обертання бензенового циклу
навколо осі. Тому оптичні антиподи не виділені.
❖ Хіральна спіраль. Низка сполук у просторі утворює елементи спіралі,
яка може бути закручена вліво або вправо. Розглянемо декілька структур з
хіральною спіраллю.
У конденсованій багатоядерній ароматичній сполуці гексагеліцені
прикінцеві ароматичні цикли не можуть розташуватись в одній площині через
взаємне відштовхування: вони розташовуються один над одним, закручуючи
структуру молекули в спіраль. Аналогічно в 4,5-диметилфенантрені метильні
групи створюють стеричні перешкоди, внаслідок яких цикли виходять з однієї
площини, утворюючи фрагмент спіралі. Цю сполуку можна розділити на
індивідуальні енантіомери, які, щоправда, легко рацемізуються:
Розділ 6. Ізомерія
115
Гексагеліцен 4,5-Диметилфенантрен
6.3.3. Аіастереомерія
Діастереомери, на відміну від енантіомерів, мають різні фізичні константи і
часто відрізняються за хімічними властивостями. їх поділяють на дві головні
групи:
■ п-діастереомери, яким властиве неоднакове розташування замісників
відносно площини симетрії я-зв'язку;
■ а-діастереомери, що мають два або більше асиметричних центрів, або
циклічні похідні особливої будови.
❖ я-Діастереомерія (геометрична ізомерія). Алкени загальної формули
RR1C=CR2R3 за умови R^R1 і R2*R3 можуть існувати у вигляді діастереомерів
(геометричних ізомерів). Потрібно виділити дві умови, необхідні для існування
геометричних ізомерів. Перша - наявність я-зв'язку, який перешкоджає
вільному обертанню і, відповідно, переходу одного ізомеру в інший. Друга -
нелінійність структури, завдяки чому замісники по-різному розташовані у
просторі.
Геометричні ізомери відрізняються за фізико-хімічними властивостями.
Наприклад, фумарова і малеїнова кислоти мають індивідуальний набір констант;
крім того, малеїнова кислота може утворювати циклічні ангідриди, а фумарова - ні:
Н СООН НООС СООН
с=с с=с
НООС н н н
Фумарова кислота, т. пл. 296,4 °С Малеїнова кислота, т. пл. 140 °С
Для позначення конфігурації я-діастереомерів простої структури
використовують цис-, /яранс-номенклатуру, а складнішої-£,2-номенклатуру.
Цей тип ізомерії стосується також інших сполук, які мають подвійний
зв'язок. Наприклад, існують геометричні ізомери відносно N=N зв'язку
арилдіазотатів:
,ОК
N=N ^ N=N
0 0 ок
транс- і #ис-Фенілдіазотат калію
116
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
■ Син-анти-ізомерія. Зв'язок С=К досить стійкий і нелінійно пов'язаний із
замісниками, що уможливлює існування я-діастереомерів для сполук з таким
угрупованням. Щоб позначити конфігурації подібних сполук, традиційно
використовують позначення син- і анти-, що аналогічні поняттям цис- і транс-
ізомери.
п
ш/лш-Конфігурація
для азометану
ii
смн-Конфігурація для оксиму
ii
N
R'NH
дити-Конфігурація
для гідразону
Потрібно враховувати, що для різних класів сполук позначення син- і анти-
стосується різних угруповань. Наприклад, для азометанів розглядають взаємне
розташування радикалів у зв'язку С=И, а в оксимів і гідразонів порівнюють
розміщення атомів Гідрогену при атомі Нітрогену.
❖ ср-Діастереомерія. Цей тип стереоізомерії звичайно виявляється для
сполук, що мають два і більше асиметричних центрів. Теоретичну кількість
стереоізомерів визначають за формулою N = 2", де п - кількість асиметричних
атомів. У складі цих просторових ізомерів є як енантіомери, так і діастереомери.
Це явище традиційно розглядають на прикладі винної кислоти-
Н02ССН(ОН)СН(ОН)С02Н, яка має два асиметричні атоми Карбону. Теоре-
тична кількість стереоізомерів для цієї сполуки становить N = 22 = 4.
Енантіомери Діастереомери
d(r)
Н
но-
соон
-ОН
-Н
d(r)
LOS)
но-
той
соон
(r,r)- або D-Винна
кислота, т. пл. 170 °С,
[oc]D+12°(H20)
Н-
L(5)
-Н
-ОН
d(r)
Н
СООН
соон
н-
L0S)
-он _ но-
соон
-н
-он но-
соон
-н
соон
(s,s)- або L-Винна кислота,
т. пл. 170 °С, [a]D-12°
(Н20)
(r,s)- або мезо-Винна кислота,
т. пл. 140 °С, [a]D 0 ° (Н20)
Розглянемо можливі просторові структури для цієї сполуки. Ліва пара кислот
є дзеркальними ізомерами. Першу з них позначають як Б-винна кислота, оскільки
обидва її асиметричні центри мають Б-конфігурацію*, або як (/?,і?)-винна кислота.
Другу, відповідно, як Ь- або (£,5)-винна кислота. Як енантіомери вони
відрізняються лише знаком обертання плоскополяризованого променя світла.
Під час вивчення конфігурації нижнього асиметричного атома Карбону структуру молекули
необхідно повернути в площині на 180°, оскільки відповідно до правил роботи з проекціями
Фішера найбільше окиснений атом Карбону повинен бути внизу.
Розділ 6. Ізомерія
117
Через наявність внутрішньої площини симетрії дві інші проекції - це
ідентичні зображення і є .мезо-формою винної кислоти. Отже, л<езо-винна кис-
лота стосовно О- або Ь-винної кислоти є а-діастереомером із власним набором
фізико-хімічних констант. Оскільки верхня і нижня частини молекули є дзер-
кальними відображеннями одна одної, то сполука оптично неактивна.
Конфігурацію діастереомерів із двома асиметричними центрами позначають
також за допомогою префіксів еритро- і трео-, якщо функціональні групи в
проекції Фішера розміщені, відповідно, з одного чи з різних боків відносно осі.
Ці позначення конфігурацій увійшли до складу низки тривіальних назв,
наприклад, моносахариди треоза і еритроза, амінокислота треонін:
-X
-X
СНО
-X
X-
еритро- і /ирео-Конфігурації
н-
н-
-ОН
-ОН
СН2ОН
О-Еритроза
но-
н-
сно
-Н
-ОН
СООН
НгИ
н
СН2ОН
Б-Треоза
Н
ОН
СН3
Ь-Треонін
Як продукт окиснення відповідного моносахариду, Ь-винну кислоту в
такому разі називають О-треаровою, ал*езо-винну - еритраровою.
❖ Діастереомерія циклічних похідних. З погляду стереохімії дизаміщені
циклоалкани багато в чому подібні до алкенів. Відсутність вільного обертання
навколо ст-зв'язків і нелінійність молекули уможливлюють існування діасте-
реомерних молекул. Для означення конфігурації таких сполук використовують
терміни ще- і транс-:
н ну н
\А-цис- і 1,4-/ираяс-Дихлорциклогексани
Особливо наголосимо, що діастереомерними можуть бути й ахіральні
молекули. Наприклад, із двох діастереомерів 1,4-транс-дихлороциклогексану і
1,4-і/мс-дихлороциклогексану, перша молекула має центр симетрії, а друга -
площину симетрії, а отже, вони ахіральні. ^
Відповідно, 1,3-/72ранс-дихлороциклогексан, який, на відміну від \,Ъ-цис-
дихлороциклогексану, не має площини симетрії, є хіральним та існує у вигляді
двох енантіомерів:
Діастереомери Енантіомери
\,Ъ-цис- і 1,3-т/>анс-Дихлороциклогексани
118
Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
■ Ще одним прикладом цис-
біциклоалкани.
Н
транс-ізомерїі можуть бути конденсовані
Н
co cd
Н Й
ще- і /ирш/с-Декапіни
■ Складніше позначити конфігурацію три- і більше заміщених циклів.
Спочатку вибирають замісник, щодо якого розглядають розташування в
просторі інших замісників. Як точки відліку беруть або старшу згідно з
правилами IUP АС групу, або замісник з меншим локантом. Позначають такий
центр літерою г:
СІ
СІ
-СІ
ОН
Н3С-<(^ СН(СН3)3
г-1 ,^мс-2,т/?анс-4 Лрихлорциклопентан 2-/и/?днс-Ізопропі л-5-і/мс-метилциклогексан-1 -ол-г
(/-ментол)
■ Для описування конфігурації похідних бі- і поліциклічних систем вико-
ристовують префікси екзо-, якщо замісники орієнтовані в зовнішню сферу
молекули, і ендо-, якщо - у внутрішню сферу.
СІ
екзо- і ендо 1-Хлоронорборнани
■ У деяких випадках назви син- і анти- використовують для позначення
конфігурації сполук, що не мають атома Нітрогену, -гіаприклад, для похідних
норборнену конфігурація анти- відображає віддалене положення в просторі
функціональної групи відносно подвійного зв'язку, а син- - наближене.
син- і анти 7-Хлорнорборнени
■ Для позначення конфігурації стероїдних сполук, похідних пер гідро-
циклопентанофенантрену, що мають тривіальні назви, використовують а,Р-
Розділ 6. Ізомерія
119
систему. В цьому випадку поліциклічну молекулу розглядають як систему, що
розміщена в площині аркуша паперу. Замісники, розташовані під площиною,
позначають символом а, а над площиною, - р.
5сс-Холестанол (дигідрохолестерол) 5Р-Холестанол (копростерин)
63.4. Рацемічні суміші й принципи їхнього
розділення на енантіомери
Енантіомери в однаковій кількості утворюють рацемічну суміш, або
рацемат. Рацемат не має оптичної активності внаслідок взаємної компенсації
оптичних властивостей дзеркальних антиподів. Звичайно рацемічна суміш
утворює окрему форму кристалів і тому має індивідуальну температуру
плавлення і розчинності. Наприклад, рацемічна винна кислота (її по-іншому
називають виноградною кислотою) оптично неактивна, має температуру
плавлення 206 °С і розчинність 20,6 г в 100 г води порівняно з 139 г/100 г води
для D- і L-винних кислот і 125 г/100 г води для мезо-вттоі кислоти.
Більшість сполук з асиметричним атомом Карбону синтезують як рацемічну
суміш. Крім того, за певних умов індивідуальні енантіомери перетворюються
(рацемізуються) у свої антиподи. Через те дослідники часто мають проблему
розділення рацематів на індивідуальні енантіомери.
Першим це завдання вирішив JI. Пастер1, який з'ясував), що натрій амонієва
сіль винної кислоти утворює дві форми кристалів, які є дзеркальними відобра-
женнями один одного. Завдяки цьому вченому вдалося механічно, за допомогою
лупи і пінцета, виділити окремі енантіомери. Оскільки енантіомери зрідка
утворюють чіткі форми кристалів, то цей метод розділення рацемічних сумішей
швидше є винятком, ніж загальним способом.
❖ Розділення рацематів у вигляді діастереометричних солей. Від
енантіомерів діастереомери відрізняються фізико-хімічними властивостями. Суміш
діастереомерів, як звичайно, можна розділити, підбираючи умови кристалізації
одного з компонентів. Отже, першим загальним методом розділення рацематів є
їхнє розділення на антиподи з попереднім перетворенням у діастереомери:
1 Пастер Л. (Pasteur L.) (1822-1895) - французький хімік і мікробіолог. Першим розділив оптичні
антиподи. Вивчав процеси бродіння. Розробив учення про штучну імунізацію, запропонував метод
знезараження нагріванням (пастеризація).
120
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Енантіомери
(Л5)-Кислота
(Л)-Амін
Діастереомери
Сіль (Л)-кислоти
і (Л)-аміну
Сіль (5)-кислоти
і (Л)-аміну
Енантіомери
(Л5)-Амін
ц (5)-Кислота]|
Діастереомери
Сіль (5)-кислоти
і (Я)-аміну
иїї Г Сіль (5)-кислоти|
1 і (5)-аміну Л
Найпростіше такий варіант розділення можна застосувати для амінів і
карбонових кислот, які легко утворюють амонійні солі. Крім того, із природних
джерел виділено достатню кількість оптично чистих карбонових кислот (0-(+)-
винна, Б-(-)-яблучна, Б-(-)-мигдальна та ін.); амінів (алкалоїди: хінін, стрихнін,
бруцин, і одержані синтетично (і?)-(+)-ментиламін, (і?)- і (5)-1-фенілетиламіни),
що дає змогу розділяти як рацемічні аміни, так і рацемічні кислоти.
Розглянемо цей спосіб розділення рацематів на реальному прикладі: СКЗ)-
З-метил-2-фенілбутанову (2-фенілізомасляну) кислоту нейтралізували (Д)-І-фе-
нілетиламіном. Одержану суміш діастереомерних (/?)(£)- і (Д)(і?)-солей
перекристалізували з водного етанолу й одержали кристалічну (Л)(і?)-амонійну
сіль, яку підкислили і виділили чисту (Л)-3-метил-2-фенілбутанову кислоту з
виходом 67 % (рис. 6.2).
рацемат
н*^—со2н
(СНзЬСН Н2Ы'
/С6Н5
сн3
Нч
с6н5*^-со| Н3Ы
(СН3)2СН
см
бН5>^—со2н
С6Н5
н»4
(СН3)2СН
(5)-
С6Щ
н^-со2н
(СН3)2СН
(Л)-
1^-со2*н3м
(СН5)2СН
сн3
н
н
неї
СбНз
н."
v
со2н3ы-
(СН3)2СН
/С6Н5
н
(ТО-
Рис. 6.2. Схема виділення (Я)-3-метил-2-фенілбутанової кислоти з рацемічної суміші
У цьому випадку, як і в багатьох інших, виділення другого енантіомера, з
більш розчинної солі, буває утрудненим. Як звичайно, ці труднощі долають,
підбираючи другий енантіомер. Наприклад, розділяючи (ОД-1-фенілетиламін,
Розділ 6. Ізомерія
121
спершу обробкою (5)-яблучною кислотою виділяють менш розчинну (Д)(5)-сіль,
а потім і сам (Д)-І-фенілетиламін. З маточного розчину виділяють суміш амінів,
збагачену (5)-енантіомером, яку нейтралізують (і?,/г)-винною кислотою,
викристалізовують відповідну сіль (5)-аміну і після розкладання амонійної солі
одержують оптично чистий (5)-1-фенілетиламін.
Цей метод можна поширити й на деякі інші класи органічних сполук.
Наприклад, щоб розділити енантіомери октан-2-олу, його спочатку дією
фталевого ангідриду перетворюють у рацемічний моноестер фталевої кислоти,
який потім розділяють за допомогою оптично активного аміну на діастереомери.
Діастереомерні солі розкладають хлоридною кислотою, а фталати обробляють
розчином лугу й одержують індивідуальні енантіомерні спирти:
с6н13х
Розділення
соон
❖ Ферментативний метод розділення рацетатів. Ще Л. Пастер довів
можливість виділення L-винної кислоти культивуванням Pénicillium glaucum у
розчині, що містив рацемічну виноградну кислоту. В процесі розвитку
мікроорганізми переважно споживали D-кислоту. У такий спосіб можна
виділити лише один з пари енантіомерів, а інший назавжди буде втрачено.
Ефективнішим є використання ферментів - біологічних каталізаторів
біохімічних процесів, що специфічно діють на субстрат певної конфігурації.
Найширше застосовують усілякі ліпази (естерази), які розщеплюють естерний
зв'язок і ацилазщ що гідролізують амідний зв'язок. Наприклад, дія ліпази на
діацетат рацемічного трянс-циклогексан-1,2-діолу руйнує естерні зв'язки лише
в (Я,і?)-ізомері, після чого розділити дві різні хімічні сполуки нескладно:
СН3 СН3
0~do fYOH f^°~d°
о-с*° ^'он + ^-Vc*0
Рацемат СН3 (Я, Я)-, 86% (5,5)-, 76% СН3
❖ Кристалізація під дією енантіомерних затравок. Іноді, додаючи в
насичений розчин рацемата затравку чистого енантіомера, вдається
викристалізувати один антипод. Наприклад, під час синтезування антибіотика
хлорамфеніколу (левоміцетин) у промисловості його синтетичний попередник
одержують у вигляді рацемічної суміші (R,R)~ і (S,S)-mpeo-2-aMmo-l-n-
нітрофенілпропан-1,3-діолу. Біологічно активний ізомер виділяють, додаючи в
пересичений розчин, частково нейтралізований соляною кислотою, затравку
(ДД)-енантіомера, що й зумовлює його кристалізацію:
а
122
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
>Ю2
ш2 ж>2
но—н о
Н—|—>Щ—с-снсі2
сн2он
ноЧ-н н-Мзн
н-|--мн2 н2к-^н
сн2он СН2ОН
Хлорамфенікол (Я, Я)
(КЯУ і (5,5)-/я/7ео-2-Аміно-1-л-нітрофенілпропан-1,3-діол
❖ Хроматографія на хіральних носіях. Сьогодні для розділення раце-
матів щораз ширше застосовують хроматографічні методи з використанням
хіральних нерухомих фаз {сорбентів). У перших експериментах як оптично
активні сорбенти використовували білки і вуглеводи, однак такі фази були
малоефективні й незручні в роботі. Тривалий час застосовували модифіковану
целюлозу, за допомогою якої розділено велику кількість рацемічних сумішей:
Сучасні дослідники у разі виділення індивідуальних енангіомерів з рацематів
віддають перевагу високоефективній рідинній хроматографії (ВЕРХ), застосовуючи
як сорбенти модифіковані силікагелі. Зазвичай, як хіральні модифікатори нерухомих
фаз використовують похідні а-амінокислот:
Хіральні сорбенти на основі целюлози
9нз
-0-^і-(СН2)з>К
сн3 н
Хіральні сорбенти на основі силікагелю для ВЕРХ
Розділ 6. Ізомерія
123
63.5. Конформаційна ізомерія
Теоретична кількість конформаційних (поворотних) ізомерів може бути
безмежна, оскільки формально поворот молекули навколо а-зв'язку, навіть на
незначний кут, створює нову конформацію. Практично розглядають лише
значущі, характерні ізомери. З-поміж таких характерних конформацій виріз-
няють енергетично найстійкіші стани - конформери.
❖ Конформації етану. Розглянемо молекулу етану в вигляді пер-
спективних формул і в проекціях Ньюмена. Можна виділити дві головні конфор-
мації:
■ затінену - з проекції Ньюмена видно, що зв'язки С-Н чітко розташовані
один над одним, торсійні кути ер = 0°, 120, 240°;
■ загальмовану — з проекції Ньюмена видно, що зв'язки С-Н рівномірно
віддалені один від одного, торсійні кути ф = 60,180, 300°:
Конформації етану:
затінена загальмована
> Торсійний кут - це двогранний кут між площинами, які проходять через
зв'язки сусідніх атомів:
Затінена конформація є найменш стабільною, оскільки має максимальну
внутрішню енергію. Підвищення внутрішньої енергії цього поворотного ізомер>
спричинене напруженням протилежних зв'язків (по-іншому,
торсійним, або пітцерівським1, напруженням), яке виникає
внаслідок взаємного відштовхування електронних хмар
зближених С-Н-зв'язків. Внутрішня енергія знижується до
мінімальної, коли молекула, повертаючись навколо С-С-
зв'язку, переходить у загальмовану конформацію з мінімаль-
ним торсійним напруженням (рис. 6.3).
Різницю енергії між загальмованою і затіненою конформаціями можна
розглядати як енергетичний бар'єр ротації, що перешкоджає молекулі
переходити в стан з максимальною внутрішньою енергією. Для молекули етану
бар'єр ротації невеликий —12 кДж/моль, його легко подолати завдяки тепловій
1 Пітцер К. (Ріггег К.) (1914-1997) - американський хімік-теоретик. Основні праці присвячені
термодинамічним властивостям молекул. Займався також дослідженнями у галузі ядерної
енергетики.
124
Чирва В.Я., Ярллолюк CM, Толкачова Н.В., Земляков О.Є. Органічна хімія
енергії. Тому за звичайних умов молекула етану вільно обертається навколо С-
С-зв'язку.
12
0 60 120 180 240 300 360 <р,град
Рис. 6.3. Залежність потенційної енергії етану від величини кута (р
❖ Конформації бутану. Молекулу бутану зручно розглядати як
дизаміщений етан. Залежно від кута повороту замісників навколо зв'язку С2-СЗ
розрізняють чотири характеристичні конформації:
■ повністю затінена (сгш-планарна) - зв'язки С-Н і С-С розташовані один
за іншим, торсійний кут ф = 0°;
■ скошена {гот або аш-клінальна) - торсійний кут ф = 60°;
■ частково затінена (аняш-клінальна) - відрізняється від повністю
затіненої конформації тим, що С-С-зв'язки це; збігаються, торсійний кут
Ф=120°;
■ загальмована (ш/яш-планарна) - торсійний кут ф = 180°:
Повністю Скошена Частково Загальмована
затінена конформація конформація затінена конформація конформація
Розділ 6. Ізомерія
125
Максимальну потенціальну енергію має сіш-планарна конформація, для якої
поряд з торсійним напруженням існує і вандервасиїьсове напруження, що
виникає внаслідок відштовхування груп, у цьому випадку метальних, які
зближені на відстань, меншу від суми їхніх вандерваальсових радіусів.
Мінімальна внутрішня енергія характерна для яняш-планарної конформації,
яка не має торсійних напружень. Бар'єр ротації для молекули бутану становить
-25 кДж/моль, унаслідок чого за кімнатної температури затінені конформації не
виникають, а 69 % молекул цієї речовини перебувають у найстабільнішій
загальмованій аниш-конформації і 31 % - у гош-конформації (рис. 6.4).
Н СН3 Н
0 60 120 180 240 300і 360 Ф, град
Рис. 6.4. Залежність потенційної енергії н-бутану від кута (р
❖ Конформації циклобутану. Якщо структура циклобутану плоска, то
виникає сильне торсійне напруження, яке виводить молекулу з площини з
відхиленням атомів Карбону на 25-30°. Оскільки бар'єр переходу в плоский
стан становить лише -6 кДж/моль, то за нормальних умов відбуваються швидкі
коливання атомів урізнобіч від площини:
Циклобутан:
перспективна формула і проекція Ньюмена вигнуті і плоска конформації
Атоми не відхиляються на значну віддаль, оскільки це збільшує від-
штовхування електронних хмарок С-С-зв'язків, якщо вони відходять від
126
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
нормального кута 109°28' в 5р3-гібридному стані, це збільшує так зване кутове
(байерівське1) напруження. Отже, реальна конформація циклобутану визначена
взаємним урівноваженням сил торсійного і байерівського напруження:
Збільшення ^Х^. Зменшення
торсійного ^ \\ кутового
напруження ^ ^Дч^>/ напруження
❖ Конформації циклопентану. Аналогічно до циклобутану, через
торсійне напруження, яке оцінюють у 10-11 кДж/моль, молекула циклопентану
не є плоскою, хоча в такій структурі внутрішній кут дорівнює 108° і практично
не відрізняється від нормального тетраедричного кута, тобто кутового
напруження нема. З просторових конформацій вирізняють конформацію
"конверт", якщо чотири атоми Карбону розташовані в площині, а один - поза
нею, і "напівкрісло" ("твіст"), якщо три атоми лежать у площині й по одному
над і під площиною:
Конформації "конверт" і "напівкрісло" для циклопентану
Різниця в енергії для цих конформерів невелика ("напівкрісло" трохи
стабільніше), через це кожний атом Карбону з великою швидкістю, по черзі,
перебуває над площиною, у площині і під площиною. Процес взаємопереходу
одного конформера в інший називають інверсією. Часто інверсію конформацій,
наприклад "конверта", розглядають як "псевдообертання":
❖ Конформації циклогексану. Для цього вуглеводню існує декілька
неплоских конформацій, які відрізняються стабільністю.
■ Найстабільніша конформація циклогексану - це "крісло". З проекції
Ньюмена видно, що всі зв'язки мають кути, близькі до валентних кутів атома
1 Байер А. (Ваеиег А.) (1835-1917) - німецький хімік-органік. Головні праці присвячені синтезу
барвників та стереохімії. В 1883 р. синтезував та визначив будову індиго. Ввів поняття про цис-
/яранс-ізомерію. Нобелівська премія 1905 р.
Зменшення
торсійного
напруження
Збільшення
кутового
напруження
Розділ 6. Ізомерія
127
Карбону в Бр -гібридизації, а отже, не має кутового напруження, нема також
торсійного і вандерваальсового напруження. В цій конформації розрізняють
аксіальні (а - перпендикулярні до площини циклу) і екваторіальні (е -
паралельні до площини циклу) замісники:
Конформація "крісло" може переходити в іншу конформацію 6Скрісло" (інверсія
циклу), у цьому разі аксіальні замісники стають екваторіальними, і навпаки.
■ Якщо один атом Карбону конформації "крісло" з положення під
площиною молекули переходить у положення над площиною, то утворюється
конформація "човен", яку в російськомовній літературі називають "ванна". Ця
конформація не має кутового напруження. Однак проекція Ньюмена засвідчує, що
в ній більшість зв'язків перебуває в затіненому положенні; у такому випадку
з'являється значне торсійне напруження. Крім того, атоми Гідрогену при верхніх
атомах Карбону (так звані флагштокові атоми) зближаються на відстань 0,183 нм
і зазнають вандерваальсового напруження. Напруження, яке виникає між
структурно віддаленими, але просторово близькими атомами, називають
трансанулярним, або прелогівським, напруженням. Загалом конформація "човен"
значно менш стійка (на 23-25 кДж/моль) порівняно з конформацією "крісло":
■ "7в/'ст"-конформація, або, по-іншому, "скривлений човен", є проміжною за
стійкістю конформацією між конформаціями "крісло" і "човен". Найнестійкіша
конформація - це "напівтвіст" або "напівкрісло", у якій чотири атоми Карбону
розташовані в одній площині, п'ятий - над площиною, а шостий - під площиною:
Інверсія конформації "крісло" для циклогексану
Проекція Ньюмена для циклогексану
Конформація "човен" для циклогексану та її проекція Ньюмена
(щоб спростити малюнок, низка атомів Гідрогену не позначена)
Конформації "твіст" і "напівтвіст" для циклогексану
128
Чирва В.Я., Ярмолкж СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
З діаграми інверсії конформації "крісло" для циклогексану видно, що
конформація "напівтвіст" є перехідною між стабільними конформаціями "крісло" і
"твіст", а конформація "човен" - між двома "твіст"-конформерами (рис. 6.5).
Е,
кДж/моль
Рис. 6.5. Діаграма інверсії конформації "крісло" для циклогексану
❖ Конформації метил циклогексану. Перебування замісників, особливо
об'ємних, в аксіальному положенні невигідне через вандерваальсове напруження
з іншими аксіальними замісниками, які розміщені через один атом Карбону. Таке
напруження називають 7,3-діаксіальним: г "
Н Н ХСХТТ
Н / н
1,3-Діаксіальне напруження в конформації "крісло" метилциклогексану
Додаткове напруження в конформації спричинює перебування замісника в
гош-конфігурації щодо С-С-зв'язку циклу. Таке напруження часто називають
гош-бутановою взаємодією. Внаслідок цього відбувається інверсія циклу;
об'ємний замісник переходить у стійке екваторіальне положення. В такому разі
говорять, що відбулося "закріплення" конформації:
гош-Бутанові взаємодії для аксіальної орієнтації конформації "крісло" метилциклогексану
Розділ 6. Ізомерія
129
Певна річ, що вирішальну роль у стійкості конформацій відіграє ефективний
об'єм замісника. Наприклад, вміст конформера з екваторіальною орієнтацією
замісника у випадку метилциклогексану становить 95 %, для ізопропілцикло-
гексану - 98 %, а трет-бутилциклогексан на 100 % перебуває в конформації з
екваторіальним положенням алкільної групи. В табл. 6.1 наведені вандервааль-
сові радіуси атомів і угруповань порівняно із вмістом екваторіального
конформера.
Таблиця 6.1
Вплив вандерваальсових радіусів замісників на вміст ^-конформера
в заміщених циклогексанах
Замісник
Н
СІ
Вг
СН3
Вандерваальсовий радіус, нм
0,120
50
0,147
57
0,175
62
0,185
66
0,20
95
Вміст е-конформера, %
❖ Конформації 1,2-диметилциклогексану. Для дизаміщених циклоалканів
діють ті ж закономірності, що обговорені раніше. Як приклад, розглянемо
конформації 1,2-диметилциклогексану. Ця сполука існує у вигляді пари цис- і
транс-діастереомерів.
Сполука і/ис-1,2-диметилциклогексан перебуває у вигляді рівноважної
суміші конформерів з одним аксіальним і одним екваторіальним замісниками
(е,а-конформер). Метальні групи цих конформерів беруть участь в 1,3-
діаксіальній взаємодії і трьох гош-бутанових взаємодіях:
СН ^ СН3 Н ^ С^
оссн; ^^-лр^сн,
е,а а,е
1,3-Діаксіальні взаємодії в і/мс-1,2-диметилциклогексані
гош-Бутанові взаємодії в і/мс-1,2-диметилциклогексані
Відповідно, транс- 1,2-диметилциклогексан може перебувати у вигляді е,е- і
я,д-конформацій. Друга практично не виникає, оскільки вона має дві метильні
групи з 1,3-діаксіальною взаємодією і чотири гош-бутанові взаємодії, тоді як у
попередньому е,е-конформері є лише одна гош-бутанова взаємодія:
130
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
1,3-Діаксіальні взаємодії в я,д-конформації транс-1,2-диметилциклогексану
гош-Бутанові взаємодії в а, а- і е,е-конформаціях трш/с-1,2-диметилциклогексану
Порівняння напружень, що виникають в \,2-транс- і 1,2-г/иоізомерах,
засвідчують велику стійкість транс-форми. Експериментально^визначена різни-
ця в енергії цих діастереомерів становить приблизно 8 кДж/моль.
Конформації великих за розміром циклів, а також деяких бі- і поліциклічних
сполук розглянуті в розділі 11.
6.3.6. Відносність понять хімічна структура,
конфігурація і конформація
Головні поняття, якими оперуємо в концепції ізомерія: хімічна структура,
конфігурація і конформація, певною мірою умовні. Визначальну роль відіграють
умови, у яких перебуває сполука, і експериментальний підхід, яким ми
визначаємо структуру сполуки. Наприклад, розглянуті в параграфі "Структурна
ізомерія" взаємоперетворення бутан <=> ізобутан і таутомерні взаємоперетво-
рення типу кетон о енол не мають принципових хімічних відмінностей.
Відмінність понять "конформація" і "конфігурація" часто пов'язана з
енергетичним бар'єром переходу одного стереоізомера або конформера в інший.
Енергія, необхідна для зміни конфігурації, зазвичай, суттєво вища, ніж для
конформаційних змін. До того ж, вчені не мають єдиного погляду щодо точного
значення енергетичного бар'єра, яке дає змогу розмежовувати ці поняття.
Наприклад, для тризаміщених фосфінів бар'єр інверсії є в межах 125—
145 кДж/моль, а для заміщених сульфоксидів - 145-190 кДж/моль. Тому ці
сполуки за кімнатної температури існують як індивідуальні енантіомери, а в разі
нагрівання вони можуть рацемізуватись:
Роздл 6. Ізомерія
131
Бар'єр інверсії 137 кДж/моль
Під час обговорення причини хіральності розглядали сполуки, наприклад,
похідні біфенілу, в яких об'ємні замісники перешкоджали конформаційним
перетворенням, що давало змогу виділити індивідуальні енантіомери. Чим
менша кількість замісників є в ортио-положеннях біфенілу, тим більший об'єм
вони повинні мати, щоб не допустити вільного обертання навколо а-зв'язку:
Бар'єр інверсії -90 кДж/моль
У цьому розділі наведено янся-сполуки з містком (СН2)9, що до 70 °С існує
як індивідуальний енантіомер, а вище переходить у-суміш.конформерів.
Іноді розділити ізомерні сполуки на індивідуальні речовини неможливо.
Наприклад, деякі таутомери малостійкі й існують у дуже маленьких кількостях.
Це стосується і конформерів. Про їхнє існування зазвичай судять зі
спектральних даних. Проте і в цих випадках умови експерименту відіграють
визначальну роль. Наприклад, відмінності в сигналах конформацій хлоро-
циклогексану з аксіальною і екваторіальною орієнтаціями зв'язків С-С1 вдається
спостерігати за допомогою ІЧ-спектроскопії за кімнатної температури, а в ПМР-
спектрах - за температури нижче -100 °С. Розділити конформери на індиві-
дуальні сполуки вдалося лише за низької температури (-150 °С).
Розділ 7
АЛКАНИ
> Апканалш називають вуглеводні з відкритим ланцюгом загальної формули
СпН2п+2>У молекулах яких атоми Карбону сполучені між собою а-зв'язками.
Інша назва сполук, які розглядаємо, - насичені вуглеводні, свідчить про їхню
хімічну особливість, тобто насиченість: усі атоми Карбону з'єднані меж собою
та атомами Гідрогену простим зв'язком. Ще одна назва цього класу вуглеводнів
- парафіни (від лат. рагит - мало і аЦіпІБ - спорідненість), відображає хімічну
інертність і має, мабуть, історичне значення. Це найпростіший клас органічних
речовин, що не має функціональних груп.
З розділу "Алкани" починають розгляд класів органічних сполук у всіх
підручниках з органічної хімії. Це пов'язано з тим, що, по-перше, алкани, або
насичені вуглеводні, мають найпростішу будову і, отже, засвоювання матеріалу
із зазначеної теми не завдасть значних труднощів. По-друге, алкани, ^чцо
містяться у нафті й природних газах, є головними промисловими джерелами
різних органічних сполук, які одержують трансформацією алканів шляхом
уведення в них різноманітних функціональних груп. По-третє, назви алканів є в
основі номенклатури органічних сполук, тобто відповідні карбонові скелети
містяться в більшості органічних сполук.
Для того, щоб відтворити картину атома Карбону, який утворює чотири
ковалентні зв'язки, потрібно розглянути його збуджений електронний стан.
Атом Карбону утворює чотири нові зовнішні орбіталі шляхом гібридизації 2^-
орбіталі та всіх трьох 2р-орбіталей. Чотири гібридні орбіталі мають однакову
енергію, і кожну з них називають 2лр3-орбіталлю (детальніше див. розділ 3).
7.1. Будова і номенклатура алканів
7.1.1. Будова метану
и 2б 2р 2р 2р
Головний і 5/Лгібридний стан атома Карбону
Структура метану
Розділ 7. Алкани
133
Яку перевагу надає така гібридизація? На відміну від негібридизованих 5- і
/7-орбіталей, 5р3-орбіталь має одну "передню" велику частку і одну "задню"
малу. Кут між гібридними орбіталями становить 109°28', що відповідає
максимальному віддаленню чотирьох орбіталей одна від одної. Така кон-
фігурація забезпечує найповніше перекриття "передньої*" частки ^р3-орбіталей
атома Карбону з іншими орбіталями, наприклад, 5-орбіталлю атомів Гідрогену в
метані, що зменшує загальну енергію молекули.
Атоми в молекулах метану та інших алканів зв'язані міцними а-зв'язками,
причому зв'язки С-С і С-Н малополярні. Тому для алканів характерні реакції з
гомолітичним розривом ковалентного зв'язку, оскільки енергія гомолітичного
розриву зв'язку значно менша, ніж енергія відповідного гетеролітичного
процесу. Зазвичай, ці реакції відбуваються під дією таких високореакційних
частинок, як вільні радикали. З огляду на повне насичення зв'язків, для алканів
характерні реакції заміщення, зокрема, радикального заміщення.
7.1.2. Номенклатура алканів
Гомологічний ряд алканів починається з метану ОНЦ. Перші чотири насичені
вуглеводні мають напівтривіальні назви: СН4 - метан, С2Нб - етан, СзНв -
пропан, С4Н10 - бутан; назви інших -утворені від відповідних грецьких
числівників із застосуванням суфікса -ан (табл. 7.1).
У хімічній літературі лінійність структури алканів часто відображають,
додаючи до назви префікс /*- (нормальний), наприклад, н-бутан. У тривіальних
назвах алканів широко використовують префікс ізо-, зокрема^у структурах
такого типу:
ц7С-СН-Я ^ = "С**3 03°бутан), -СН2СН3 (ізопентан), -СН2СН2СН3 (ізогексан).
сн3
Таблиця 7.1
Систематичні назви лінійних алканів
Формула
Назва
Формула
Назва
Формула
Назва
Формула
Назва
С5Н12
СбНм
С7Н16
С9Н20
СюН22
Пентан
Гексан
Гептан
Октан
Нонан
Декан
СцН24
СігН2б
СізН28
С14Н30
С15Н32
Ундекан
Додекан
Тридекан
Тетрадекан
Пентадекан
і т.д.
С20Н42
С21Н44
С22Н46
С23Н48
С24Н50
Ейкозан
Генейкоз
ан
Докозан
Трикозан .
Тетракоз
ан
і т.д.
СзоНбг
С40Н82
С50Н102
СбоНі22
С100Н202
Триаконтан
Тетраконтан
Пентаконтан
Гексаконтан
і т.д.
Гектан
Назви вищих алканів є комбінацією грецького числівника і суфікса -ан,
наприклад, С145Н292 - пентатетраконтагектан.
У номенклатурі ШРАС насичений вуглеводень із розгалуженою карбоновою
структурою розглядають як алкан, у якому один або декілька атомів Гідрогену
134
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
замінено на вуглеводневі замісники (назви замісників див. розділ 5). Для побу-
дови назви будь-якого алкану необхідно:
1) вибрати найдовший і найбільше розгалужений ланцюг;
2) ланцюг потрібно пронумерувати так, щоб замісники отримали міні-
мальний набір номерів (локантів) або менший номер отримав перший
за абеткою замісник;
3) назву складають з переліком за абеткою замісників та з позначенням
їхніх локантів і кількості, а потім дають назву основного ланцюга
алкану з відповідною кількістю карбонових атомів, наприклад:
СН2СН3
1 2 |3 4 5 6
СН3^НСНСН2^НСН3
сн3 сн3
3 -Ети л-2,5 -димети л гексан
Горизонтальний ланцюг складається з шести атомів
Карбону і має максимальну кількість відгалужень (три).
Ланцюг пронумерований зліва, оскільки в цьому
випадку замісники отримують менші локанти (2,3,5),
ніж у випадку нумерації справа (2,4,5).
І 2 З ^^СН3
СНз-СН^СН-СН-СН-СНз
І б\ 7
сн^сн^ сн2снз
3-Метил-4-пропіл-5-етилгептан
Якщо замісники мають однакові локанти в разі
нумерації з будь-якого кінця, то нумерацію ведуть у
такому напрямі, щоб менший локант отримав перший за
абеткою замісник (етил, а не метил).
7.1.3. Ізомерія алканів
Для алканів характерний лише один вид структурної ізомерії - ізомерія
карбонового скелета, яка починається з бутану. Кількість структурних ізомерів
швидко зростає зі збільшенням кількості карбонових атомів. Наприклад, якщо
гексан має 5 ізомерів, то декан уже 75.
Кількість ізомерів для гомолога неможливо підрахувати за якою-небудь
формулою, проте є спеціальні математичні прийоми, які дають змогу визначити
кількість ізомерів для гомолога з кількістю атомів Карбону якщо відома
кількість ізомерів для попереднього вуглеводню. У табл. 7.2 наведено кількість
ізомерів залежно від брутто-формули алкану.
Таблиця 7.2
Залежність кількості ізомерів від кількості атомів Карбону (гі) в молекулі алкану
п
Кількість ізомерів
п
Кількість ізомерів
1
1
8
18
2
1
9
35
3
1
10
75
4
2
15
4347
5
3
20
336 319
6
5
25
36 797 588
7
9
30
4 111 846 763
Розділ 7. Алкани
135
Ізомерію на прикладі бутанів уперше передбачив і вивчив О. Бутлеров.
Ізомерні пентани (їхні структури зображені на стор. 48) синтезовані його учнем
М. Львовим1. Синтез ізомерних гептанів виконаний 1929 р., а ізомерних октанів
-1946 р.
У метані та етані всі атоми Гідрогену рівноцінні (еквівалентні), проте вже в
структурі пропану розрізняють атоми Гідрогену, які входять у метальні групи і
метиленову групу. Відповідні атоми Карбону називають первинними (зв'язані з
одним атомом Карбону) і вторинними (зв'язані з двома атомами Карбону). У
складніших за будовою алканах можуть бути третинні, які мають три зв'язки С-С,
і четвертинні карбонові атоми - зв'язані з чотирма атомами Карбону:
Уже на прикладі простих алканів видно, що повні структурні формули
досить громіздкі, особливо, коли зображають складні молекули. Тому сьогодні
запропоновано використовувати стиснені структурні формули, у яких кова-
лентні зв'язки опускають, а однакові групи, зв'язані з одним атомом, записують
у дужках, зазначаючи їхню кількість, наприклад:
Нині щораз ширше використовують спрощені - скелетні формули. В них
хімічні зв'язки позначають рисками, розуміючи, що атоми Карбону містяться в
кутах, утворених цими рисками, або на їхніх кінцях. У вигляді символів
позначають лише гетероатоми, а також атоми Гідрогену, які є в складі
функціональних груп. У деяких випадках кінцеві атоми Карбону також
позначають:
Третинний атом Карбону
І (метанова група)
Первинний атом Карбону
(метальна група)
Четвертинний атом Карбону
Вторинний атом Карбону
(метиленова група)
(СН3)зССН(СНз)2
2,2,3-Триметилбутан
(СН3)2СНСН2С(СНз)з
2,2,4-Триметилпентан
2,2,3-Триметилбутан
З-Метилпропан-1 -о л
1 Львов М. (1848-1899) - російський хімік-органік. Учень О. Бутлерова. Головні праці в галузі
теорії органічної будови, зокрема, вперше одержав неопентан, і хімії ненасичених сполук.
136 Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
7.2. Фізичні властивості
За кімнатної температури перші чотири н-алкани - гази, вуглеводні С5-С17 -
рідини, далі - тверді речовини. Перші рідкі алкани мають запах бензину, а газоподібні
алкани і вищі вуглеводні не мають запаху.
Температури кипіння і плавлення алканів підвищуються зі збільшенням
кількості карбонових атомів. Процес кипіння і плавлення потребує подолання
сил міжмолекулярних взаємодій у рідині та у твердій речовині. Підвищення
температури плавлення і кипіння зі збільшенням молекулярної маси можна
пояснити зростанням міжмолекулярних сил, коли збільшуються розміри
молекул.
Ізомер з розгалуженим ланцюгом кипить за нижчої температури, ніж
нормальний, оскільки міжмолекулярні сили менші для ізоалканів, ніж для
нерозгалужених алканів. Температури кипіння підвищуються на 20-30 °С зі
збільшенням довжини ланцюга на один атом Карбону. Ця різниця зберігається не
лише для алканів, а й для деяких інших гомологічних рядів. Алкани з розга-
луженими структурами плавляться за вищої температури, ніж з нормальними. В
гомологічному ряді різниця температур плавлення у двох сусідніх алканів
повільно зменшується. Від метану до н-тетракозану вуглеводні з парною
кількістю атомів Карбону, що мають моноклінну кристалічну ґратку, плавляться^
за вищою температурою, ніж сусідні вуглеводні з непарною кількістю атомів
Карбону і ромбічними кристалами:
Цю закономірність пояснюють тим, що температура плавлення є функцією
енергії кристалічної ґратки, яка, відповідно, залежить від типу ґратки і форми
молекули. Оскільки звичайно н-алкани перебувають в антиперипланарній
конформації, то кінцеві метальні групи в ланцюзі з парною кількістю карбо-
нових атомів мають трансоїдне розташування (а), а в ланцюзі з непарною
кількістю атомів Карбону - цисоїдне (б).
Густина алканів збільшується зі збільшенням розмірів молекули, однак ця
тенденція простежується до граничного значення 0,8, оскільки всі алкани легші
від води. Це не дивно. Майже всі органічні сполуки мають густину, меншу від
густини води, оскільки вони, як і алкани, складаються головно з атомів Карбону
і Гідрогену. В загальному випадку, щоб сполука була важча від води, вона
повинна мати тяжкий атом, наприклад, атоми Брому, Хлору чи Іоду.
а
б
Розділ 7. Алкани
137
7.2Л. Спектроскопія алканів
Оскільки для алканів немає спеціальних якісних хімічних реакцій, то для
їхньої характеристики важливе значення мають спектральні методи.
• Електронна спектроскопія. В алканах під дією світла можливі лише а—>$*
електронні переходи. Відповідні їм смуги поглинання є в далекій ультра-
фіолетовій ділянці й не можуть бути виявлені за допомогою звичайних УФ-
спектрофотометрів. На цьому ґрунтується використання рідких алканів в
електронній спектроскопії як розчинників.
• ІЧ-спектроскопія. Для алканів характерні смуги валентних і деформаційних
коливань зв'язків С-Н. Як звичайно, валентні (симетричні та антисиметричні)
коливання цих зв'язків спостерігають у спектрі як комплексні смуги в ділянці
2970-2850 см"1, а смуги деформаційних коливань С-Н у метальних, мети-
ленових і метанових групах проявляються в ділянці 1470-1350 см"1.
• Мас-спектрометрія. Мас-спектри алканів мають велику кількість ліній. Пік
молекулярного йона М*-, зазвичай, має невелику інтенсивність, яка зменшується
зі збільшенням довжини ланцюга або ступеня його розгалуженості. Головний
напрям розпаду молекулярного йона пов'язаний з відщепленням алкільних
радикалів і утворенням іонів складу [СпН2п+і]+. Як приклад, наведена схема
фрагментації молекулярного йона «-гексану:
М+ш/г86
[ЩС-СНгСНіСШСНіСНз]+'
с^^-4с5н+ ^
с2н5+ с4н9+ ^
-с,н5-
-С4Н9
С2Н5
т/г29
-Сзн/ _ _ -С2Н4
С3Н7+ —СН3+
-2а а5
т/г29 т/г57 т/г43 т/г15
Іони [СпН2п+і]+ також здатні до подальшої фрагментації, утворюючи серію
йонів [СпН2п]+ і [СпН2п-і]+.
• Спектроскопія ПМР. Протони алканів, як найбільш екрановані, резонують у
сильному полі в інтервалі 0,8-1,8 м. ч. (тут і далі хімічні зсуви наведено в шкалі
8 відносно тетраметилсилану). Близькість хімічних зсувів сигналів протонів
груп СН3 (0,8-1,0 м. ч.), СН2 (1,1-1,6 м. ч.) і СН (1,4-1,8 м. ч.), а також спін-
спінове розчеплення цих сигналів спричиняє появу складних мультиплетів, які
часто важко розшифрувати:
5 0,86 5 1,33 5 1,56
НзС"""СН2~"СН2—СН2~"СН2~~СНз
Хімічні зсуви протонів у гексані (м. ч.)
138
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
7.3. Одержання алканів
У разі отримування органічних сполук важливе значення має масштаб
виробництва. У промисловому синтезі вирішальну роль відіграють вартість і
доступність вихідних матеріалів, а також екологічність та економічність
виробництва. Високий ступінь чистоти вихідних речовин потрібний не у всіх
випадках. У лабораторіях найбільшу увагу приділяють виходу продуктів реакції,
оригінальності методу, високому ступеню чистоти отримуваних сполук, а ціна
не завжди відіграє провідну роль.
7.3.1. Промислові способи отримання алканів
Алкани від метану до н-пентану, і навіть ундекану, одержують фракційною
перегонкою нафти або природного газу. Після пентану кількість ізомерів для
кожного гомолога стає наскільки великою, а різниці в температурах кипіння
настільки незначні, що індивідуальні сполуки досить важко виділити із суміші.
У промисловості останнім часом особливої ваги набувають методи розді-
лення н-алканів від розгалужених за допомогою сполук включення - клатрашів.
Ці методи ґрунтуються на різній здатності молекул, що відрізняються за формою і
розміром, розташовуватися між молекулами деяких твердих речовин, які мають
шарувату або сітчасту кристалічну структуру. Отримані у вигляді клатратів
речовини досить легко вивільняються від "хазяїна". В нафтопереробній про-
мисловості як речовини, що утворюють клатрати, найширше використовують
сечовину, тіосечовину, деякі комплексні солі металів, цеоліти та інші матеріали.
Поряд із цим прийомом застосовують розділення алканів за молекулярною масою
за допомогою молекулярних сит - модифікованих природних полімерів.
❖ Гідрогенізація бурого вугілля. Каталітична гідрогенізація вугілля
отримала назву "скраплення вугілля". Зміст методу, розробленого Ф. Бергіусом1
1913 р., полягає в тому, що тонко розмелене буре вугілля змішують із важкою
фракцією мастил, одержаною з попередньої порції вугілля, додають порошкове
залізо або каталізатори на основі оксидів і сульфідів Молібдену, Вольфраму,
Ніколу і діють воднем при 450-500 °С і тиску 200-300 атм. Одержують до 75 %
сирої нафти, з якої виділяють гази, бензин і "тяжкий" погон, який додатково
деструктивно гідрогенізують. Метод Бергіуса запроваджений у промисловість
Німеччини, де 1943 р. таким способом переробляли майже 25 млн т вугілля та
отримували понад 5 млн т бензину.
Через дефіцит нафти, який є нині в світі, цей процес привертає увагу
спеціалістів. Особливо це стосується України, яка не має великих запасів нафти,
однак багата на буре вугілля.
Бергіус Ф. (Ве^іиБ Р.) (1884-1949) - німецький хімік. Розробив метод синтезу моторного
пального з вугілля. Одержав гідролізний спирт і кормовий цукор з деревини. Нобелівська премія
1931 р. з хімії високих тисків.
Розділ 7. Алкани
139
❖ Синтез Ь карбон (II) оксиду. Ці роботи започаткував 1902 р. французький
хімік П. Сабатьє1, який, пропускаючи над тонким порошком нікелю суміш
карбон (II) оксиду і водню, одержав метан:
СО + ЗН,
250 °С ^тт тт ^
сн4 + н2о
Ф. Фішер2 і Г. Тропш3 (1922), вивчаючи цю реакцію, отримали суміш
алканів, яка на 80 % складалася з нормальних вуглеводнів. Реакція відбувається
за температури 200-300 °С, тиску 15 атм. над каталізаторами - нікелем,
кобальтом, залізом, нанесеними на алюміній оксид:
200 °С
пСО #(2п+1)Н2 СпН2п+2+ пН20
Уважають, що в основі процесу є сорбція карбон (II) оксиду на каталізаторі
з утворенням відповідних карбонілів. Коли відбувається гідрування,
утворюються карбени (детальніше див. 7.8) і метальні радикали, зв'язані з
поверхнею каталізатора. Карбени можуть приєднуватися до карбонового атома,
нарощуючи вуглеводневий ланцюг:
СО СО СО
1-І
:СН, СО СО
Каталізатор
j і
Каталізатор
•СН3 :СН2СО
1 І ї
Н3С-СН2;
:СН2 HX-CHrCTL
Каталізатор
Каталізатор
Каталізатор
Вихідну суміш карбон оксиду (II) з воднем, яку називають синтез-газом,
одержують із метану при 800-900 °С за наявності нікол оксиду, нанесеного на
алюміній оксид:
СН4 + Н20
СН4 + С02=
2СН4 + 02 =
- СО + зн2
2СО + 2Н2
2СО + 4Н,
У такий спосіб одержують "синтин", який складається з нормальних
парафінів із домішкою розгалужених та етанолу. Він послугував сировиною для
органічного синтезу і переробки в моторне пальне. Промислове виробництво
налагоджене 1936 р. в Німеччині.
1 Сабатьє П. (Sabatier P.) (1854-1941) - французький хімік. Засновник органічного каталізу.
Впровадив промислову гідрогенізацію жирів. Нобелівська премія 1912 р. за метод гідрогенізації.
2 Фішер Ф. (Fischer F.) (1877-1947) - німецький хімік.' Наукова діяльність присвячена синтезу
органічних сполук на основі карбон (II) оксиду і водню.
3 Тропш Г. (Tropsch Н.) (1889-1935) - чеський ,хімік. Спільно з Ф. Фішером вивчав каталітичний
синтез вуглеводнів.
140
Чирва В.Я., Ярмолюк СМ., Толкачова H.В., Земляков О.Є. Органічна хімія
Подібно до методу Бергіуса, він не конкурентоспроможний зі способом
одержання вуглеводів із нафти, проте останнім часом інтерес до нього також
швидко зростає.
■ Модифікацією процесу Фішера-Тропша є синтез алканів через проміжне
одержання метанолу. На основі синтез-газу одержують метанол, який перетво-
рюють у високооктанове пальне на цеолітах у псевдоскрапленому шарі
каталізатора:
СО + ЗН2=?= СН3ОН + Н20
2СН3ОН—^СН3ОСН3
СН3ОСНз1д^Г C„H2w+2 + Н20
❖ Крекінг нафти. Під час крекінгу нафти вищі алкани розщеплюються,
утворюючи суміш нижчих алканів, починаючи з метану, і дуже цінних олефінів:
^п^2п+2 *" СтГІ2т+2 + Сп.тН2(п_т)
Отже, крекінг є одним із найважливіших джерел промислового одержання
алканів. Особливо цінні для хімічної промисловості пропан, бутан, ізобутан та
ізопентан. Детальніше див. 7.5.
7.3.2. Лабораторні способи синтезу алканів
❖ Гідроліз карбідів металів. Карбіди Алюмінію і Берилію енергійно
взаємодіють із водою або розбавленими кислотами, утворюючи метан:
АІ4С3 + 12Н20 -> 4А1(ОН)3 + ЗСН4Т
❖ Синтез метану за Бертло. Сьогодні цей метод має лише історичне
значення:
CS2 + 2H2S + 8Cu -» СН4 + 4Cu2S
❖ Декарбоксилювання карбонових кислот {реакція Дюма). Ця реакція є
загальним способом одержання вуглеводнів:
RCOOH —R—H + С02
Насичені вуглеводні інколи зручніше одержувати з натрієвих солей
карбонових кислот. Під час нагрівання солей карбонових кислот із лугами, барій
гідроксидом, натрій алколятами відщеплюється вуглекислий газ і утворюється
алкан, у молекулі* якого міститься на один карбоновий атом менше, ніж у
молекулі вихідної кислоти:
Розділ 7. Алкани
141
9 t
Н3С~С* "ТГ^г СН4 + Na2C03
ONa Na0H
Це простий і зручний метод одержання метану. Реакцію можна проводити
навіть в умовах шкільної лабораторії. Для зниження температури плавлення
натрій гідроксид замінюють натронним вапном (NaOH + СаО). Однозначно ця
реакція відбувається лише з ацетатом натрію. В інших випадках утворюються
побічні продукти.
❖ Окислювальне декарбоксилювання кислот. Цим методом одержують
алкани, що мають на один атом Карбону менше, ніж вихідна кислота. З гарними
(70-80 % у перерахунку на карбонову кислоту) виходами алкани утворюються в
процесі фотохімічного декарбоксилювання первинних карбонових кислот за
наявності плюмбум тетраацетату в хлороформі. Декарбоксилювання звичайних
аліфатичних кислот має лише незначне препаративне значення через те, що
реакція відбувається за високої температури, а виходи одержують низькі,
особливо для кислот з довгим карбоновим ланцюгом.
❖ Електроліз розплаву або розчину солей карбонових кислот [реакція
Кольбе). В концентрованому водно-спиртовому розчині або розплаві солі
карбонових кислот дисоціюють:
RCOONa =ї=^ RCOO- + Na+
На аноді аніон віддає електрон, утворюючи радикал, який легко зазнає
декарбоксилювання:
9 -е- Р
R-Q —*r-q — r. + ca
о- о.
2
Унаслідок рекомбінації радикали перетворюються в алкани:
R + R-»R:R
На катоді в випадку розплавів солей утворюється лужний метал, а коли
використовують розчини, то виділяється водень і гідроксид відповідного лужного
металу. Як і синтез Вюрца, реакція Кольбе придатна для одержання вищих алканів.
❖ Відновлення галогенопохідних. Під час каталітичного гідрування за
наявності паладію галогеноалкани перетворюються у відповідні алкани:
R-X + Н2 R-H + НХ
РсІ/ВаСО,
Як альтернативні відновники використовують амальгаму натрію, комплек-
сні гідриди металів, Натрій або Цинк у спирті, Цинк у хлорній кислоті:
142
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
2п ~ тт
к-С16Н33І «-С16Н34
НС1/СН3С00Н 85о/о
Іодоалкани можна легко відновити іодидною кислотою в запаяній ампулі:
Я-І + НІ Я-Н + І2
Найдоцільніше в цій реакції застосовувати суміш гідроген іодиду з
червоним фосфором. У цьому випадку іодидна кислота, яка споживається в
процесі відновлення, легко регенерується:
2 Р + З І2 -> 2 Різ
2 Різ + 6 Н20 -> 2 Р(ОН)3 + 6 НІ
За допомогою суміші червоного фосфору та іодидної кислоти в запаяних
ампулах можна провести відновлення спиртів, альдегідів, кетонів і карбонових
кислот до алканів. У цьому разі одночасно відбуваються процеси галогенування
цих сполук та їхнє відновлення гідроген іодидом:
Я-ОН + Ш -> Я-І + н2о
Я-І + НІ -* К-Н + І2
❖ Дія води на магнійорганічні сполуки. Зручним методом відновлення
галогеноалканів є їхня взаємодія з магнієм у діетиловому етері з утворенням
магнійгалогенопохідних (реактиву Гриньяра ), які перетворюються на алкани в
разі обробки водою чи спиртом:
СпН2п+іВг + Mg -> СпН2п+іМеВг
СпН2п+1МёВг СпН2п+2 + М8(ОН)Вг
Обидві реакції відбуваються кількісно. Діетиловий етер у першій реакції є
не лише зручним розчинником, він утворює діетерат, який відіграє суттєву роль
у реакції. Реактив Гриньяра, взаємодіючи з двома електронодонорними
молекулами етеру, веде себе як кислота Льюїса. Вважають, що в цьому
комплексі зв'язок С-М^ є ковалентним, але сильно полярним:
Я-Мв-Х + 2С2Н5-Ь-С2Н5 Я-^-Х
С2Н5-0-С2Н5
❖ Гідрування алкенів і алкінів. Ця реакція найважливіша, тому що її
застосування обмежене лише доступністю алкенів, які легко можна одержати з
великої групи спиртів, синтезованих за допомогою різноманітних методів.
1 Гриньяр В. (ві^патс! V.) (1871-1935) - французький хімік-органік. Розробив простий метод
одержання і ввів у синтетичну практику магнійорганічні сполуки. Нобелівська премія 1912 р.
Розділ 7. Алкани
143
Водень за наявності платини або паладію приєднується до алкенів за місцем
подвійного зв'язку, а у випадку алкінів — потрійного зв'язку:
БІ-СЕЕСН -З*— К-СН=СН2 к-сн2-сн3
До реагентів, які звичайно використовують для некаталітичного відновлен-
ня алкенів, належить діімід МИН^Н і натрій, розчинений у гексаметил-
фосфоротріаміді.
• Каталізатори гідрування. Французький хімік Ф. Сабатьє1 запропонував як
каталізатор дешевший нікель. У випадку дрібнодисперсних або колоїдних
металів - платини або паладію - реакція гідрування відбувається за кімнатної
температури і звичайного тиску. Якщо використовувати нікель, потрібні
підвищені тиск і температура.
■ У 1927 р. американський хімік М. Реней2 суттєво поліпшив нікелевий
каталізатор, запропонувавши одержувати активніший нікелевий каталізатор
шляхом обробки нікель-алюмінієвого сплаву розчином натрій гідроксиду.
Хімічне розчинення алюмінію давало дрібнопористий порошок нікелю,
насиченого воднем. Такий каталізатор відомий як нікель Ренея, або скелетний
нікель.
■ Останнім часом як у промисловості, так і в лабораторній практиці як
каталізатор гідрування під тиском широко застосовують купрум хроміт
СиОСг2Оз (каталізатор Адкінса3).
■ Високу ефективність мають каталізатори гомогенного гідрування типу
трмс(трифенілфосфін)родій хлориду [(СбН5)зР]зЮіС1, за наявності яких
приєднання водню до алкенів відбувається в гомогенному середовищі за
кімнатної температури, оскільки такі каталізатори активують як водень, так і
ненасичену сполуку.
❖ Синтез Вюрца. Нагрівання алкілгалогенідів з металічним натрієм дає
змогу одержати алкани з подвійною кількістю карбонових атомів у скелеті.
Синтез має обмежене застосування і придатний для одержання лише симетрич-
11* їх мЛісахпв.
2СН3СН2Вг СН3СН2СН2СН3
1 Сабатьє Ф. (ЗаЬаіїег К) (1871-1935) - французький хімік, започаткував "Посібник з органічної
хімії" у 23 т. (1935-1954).
2 Реней М. (Яапеу М.) (1885-1966) - американський інженер-хімік. Працював у галузі
каталітичного гідрування рослинних олій. Винайшов кілька каталізаторів цього процесу,
насамперед, так званий нікель Ренея.
3 Адкінс Г. (АсИгіпБ Н.) (1892-1949) - американський хімік. Займався гідруванням органічних
сполук. Увів термін "гідрогеноліз" для описання деструкції молекул під дією водню.
144
Чирва В.Я., Ярмолюк СМ., Толкачова H.B., Земляков О.Є. Органічна хімія
Практично не застосовують реакцій з двома різними алкілгалогенідами, тому
що утворюється суміш алканів, яку важко розділити. Якщо брати галогенопохідні
з великою молекулярною масою, то вихід вуглеводнів буває хорошим:
Реакцію з успіхом застосовують для синтезу вищих вуглеводнів з
природних спиртів, які попередньо перетворюють у галогеноалкани.
Процес відбувається успішно для первинних галогенідів. Ліпші виходи
бувають для активніших бромо- та іодопохідних. Для третинних алкілів як
каталізатори застосовують сполуки Меркурію і Купруму (І) для підвищення
ефективності реакції.
Щодо механізму цієї реакції П. Шоригін1 висловив припущення, що в разі
дії на галогенопохідні металічного натрію спочатку утворюються натрійалкіли,
які далі реагують з галогеноалкілами, можливо, за механізмом реакцій бімоле-
кулярного нуклеофільного заміщення:
■ Альтернативний механізм для пояснення реакції Вюрца передбачає
стадію перенесення одного електрона з проміжним утворенням аніон-радикала
галогеноалкілу, який далі перетворюється в алкіл-радикал. Унаслідок димери-
зації алкіл-радикала утворюється цільовий продукт реакції:
Несиметричні алкани неможливо ефективно синтезувати за реакцією
Вюрца, що значно обмежує цей метод. До того ж, вихідні галогенопохідні під
дією проміжних натрійалкілів часто відщеплюють гідроген галогенід, пере-
творюючись в олефіни.
❖ Реакція Корі-Хауса. Ліпші результати, порівняно з реакцією Вюрца, дає
сучасний метод, запропонований Е. Корі2 і X. Хаусом, із застосуванням
літійорганічних сполук і солей Купруму. Цей метод полягає у взаємодії
літійдіалкілкупратів Я2СиЬі з галогеноалкілами. Спочатку металічний літій в
діетиловому етері взаємодіє з алкілгалогенідом, наприклад, метилхлоридом:
1 Шоригін П. (1881-1939) - російський хімік. Праці присвячені металоорганічним сполукам і
вуглеводам.
2 Корі Е. (Corey Е.) (нар. 1928) - американський хімік-органік. Дослідження в галузі стереохімії,
структурної, синтетичної та теоретичної органічної хімії. Нобелівська премія 1990 р.
2C20H4iBr + 2Na -> С4оН82 + 2NaBr
Дигідрофітилбромід Пергідролікопін (88 %)
R-X + 2Na -> R"Na+ + NaX
R~Na+ + R-X -> R-R + NaX
R-Br +Na -[
[R-BrF— R- +
R-+R-— R-R
— [R-Br] • + Na+
R- + Br"
Розділ 7. Алкани
145
СНзСІ + 2Ьі -> СН3Ьі + ЬіСІ
Далі утворюється літійдиметилкупрат:
2СН3Ьі + СиІ (СН3)2СиЬі + ІЛІ
який реагує з галогеноалкшом із утворенням насиченого вуглеводню: [
[Н3С-1/2-Си--1/2СН3]ЬІ+ +>СН3(СН2)ПСН2І -> СН3СН2(СН2)ПСН3 + ІЛІ +(Си^
Отже, можна синтезувати як симетричні, так і несиметричні алкани. У
випадку первинних галогеноалканів виходи близькі до 100%, а у випадку
третинних - 30-50 %. Зазначимо, що природа алкілу в літійдіалкілкупраті мало
впливає на вихід кінцевого продукту реакції.
Останніми роками для синтезу алканів застосовують також алкілування
магнійорганічних реагентів алкілгалогенідами. В такому варіанті вихід досягає
50 %:
СН3М§С1 + (СН3)3СС1 -> (СН3)4С
Цю реакцію можна провести дещо по-іншому:
СН3(СН2)3М8С1 -СН3(СН2)6СН3
Октан
7.4. Хімічні властивості алканів
Як уже зазначено, алкани за природою - повністю насичені сполуки. Тому
вони не вступають у реакції приєднання. Зв'язки С-С і С-Н мало поляризовані, з
огляду на це утворення йонів (на відміну від радикалів) потребує великих витрат
енергії.
Отож, для алканів характерні реакції гемолітичного розриву ковалентних
зв'язків, які відбуваються за радикальним механізмом.* Незважаючи на те, що
розщеплення сг-зв'язку між атомами Карбону потребує менше енергії
(-350 кДж/моль), ніж а-зв'язку Карбон-Гідроген (~ 420 кДж/моль), хімічні реакції
частіше відбуваються з розщепленням Гідроген, оскільки він доступніший до дії
реагентів.
7.4.1. Галогенування
❖ Хлорування метану. Незбуджений хлор не реагує з метаном. У разі
активації такого хлору ультрафіолетовим світлом або нагрівання до температури
250-400 °С він перетворює метан у хлоропохідні. Оскільки хлор є найдешевшим
галогеном, то хлорування метану застосовують у промисловому масштабі для
одержання тетрахлорометану.
146
Чирва В.Я., Ярллолюк СМ., Толкачова H.В., Зеллляков О.Є. Органічна хімія
Механізм хлорування метану є радикальним і складається з трьох головних
стадій (М. Семенов1): ініціювання (зародження ланцюга), ріст ланцюга, обрив
ланцюга (рекомбінація).
На світлі, у разі нагрівання або під дією ініціаторів радикальних реакцій,
спочатку утворюються вільні радикали Хлору. Для цього потрібно 242 кДж/моль:
Ініціювання: СІ2 —* 2СГ
Ріст ланцюга: СН4 + Cl" -> СН3 + НС1
Вільний радикал Хлору заміщує атом Гідрогену в молекулі метану. На
цьому етапі розривається зв'язок С-Н (ДЯ 423 кДж/моль) і утворюється зв'язок
Н-С1 (ДЯ -427 кДж/моль). Реакція слабко екзотермічна (ДЯ -4 кДж/моль):
СНз* + С12 -> СН3С1 + СГ
Хлорометан
Метальний радикал розщеплює молекулу Хлору, утворюючи перший
продукт заміщення - метилхлорид, і новий вільний радикал Хлору. Цей етап
також екзотермічний (ДЯ -97 кДж/моль), оскільки енергія, необхідна для
розриву зв'язку Cl—СІ (ДЯ 242 кДж/моль), компенсована енергією, що
виділяється під час утворення зв'язку С-С1 (ДЯ -339 кДж/моль).
Далі атоми Хлору послідовно заміщають інші атоми Гідрогену в молекулі
метану:
СН3С1 + СГ -
СН2С12 + СГ
СНСІз + СГ -
СН2СГ + НС1
> СНС12* + НС1
ссі3- + НС1
СН2СГ + С12 СН2С12 + СГ
Дихлорометан
СНСІ2 + С12 -> СНСІз + СГ
Трихлорометан
ССІ3 + С12 сси + СГ
Тетрахлорометан
Обрив ланцюга. В суміші завжди є вільні радикали, тому процес швидко
поширюється на щораз нові молекули. Якщо взаємодіють два радикали, то може
утворитися неактивна молекула, що спричиняє обрив ланцюга:
СНз* + СГ -> СН3С1 або СНз* + СН3* -> СН3СН3 або СГ + СГ -> С12
Енергетичні діаграми хлорування метану і н-пропану, обговорені нижче,
зображено на рис. 7.1.
Активований комплекс [С1—Н—СКЩ реакції сн4 + СГ —► СН3* +. НС1 - це
структура, У якій а-зв'язок С-Н ще не зруйнувався, а відповідний зв'язок С-С1
ще не утворився:
Н
Н
Семенов М. (1896-1986) - радянський фізик, фізико-хімік. Наукові дослідження присвячені
вченню про радикальні механізми хімічних процесів. Нобелівська премія 1956 р.
Розділ 7. Алкани
147
кДж/моль
[С1-Н-СН3]
сн3- + неї
СН4 + СІ
[С1-С1-СН3]
АЯ8
СН3С1 + СІ-
Координата реакції
СН3 СН2СН2'
+ НС1
Е,
кДж/моль
СН3СНСН3
+ НС1
Координата реакції
б
СН3СНСІСН3
Координата реакції
Рис. 7.1. Енергетичні діаграми реакцій хлорування метану (а) і пропану (б)
■ Суттєвий внесок у вивчення вільних радикалів зробив Г. Герцберг1, який зумів
записати ІЧ-спекгр метильного радикала за низької температури в твердій матриці
аргону. На підставі цих досліджень з'ясовано, що метальний радикал має плоску
будову, і максимальне відхилення зв'язків С-Н від площини не перевищує 5°:
Н
Н
ярг
-Н
Метальний радикал надзвичайно нестабільний, середній час його Існування
близько 10"3-10"2 с.
Про радикальний механізм реакції хлорування метану свідчить її галь-
мування, коли в реакційну суміш додають кисень. У цьому випадку активні
алкільні радикали перетворюються в неактивні алкілпероксидні радикали.
1 Герцберг Г. (НегеЬе^ С) (1904-1999) - канадський фізико-хімік. Народився в Німеччині,
працював у Німеччині та США, з 1935 р. в Канаді. Фундаментальні дослідження зі спектроскопії
молекул. Нобелівська премія 1971 р.
148
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Б1 + 02 іі-о-о •
Отже, кисень інгібує радикальну реакцію. Навпаки, ініціатори радикальних
реакцій прискорюють хлорування метану. Таку властивість має тетраетил-
плюмбум, що легко розпадається під час нагрівання на етильні радикали:
РЬ(С2Н5)4 —- РЬ + 4 с2н;
❖ Хлорування пропану та інших алканів. Уже реакцію хлорування
пропану через високу екзотермічність не можна провести спрямовано.
Вибірковість хлорування збільшиться, якщо проводити реакцію за температури
до 100 °С.
У молекулі пропану атоми Гідрогену нееквівалентні, тому навіть моно-
хлорування за кімнатної температури відбувається з утворенням суміші
продуктів - 1-хлоропропану і 2-хлоропропану у співвідношенні 45:55, хоча
атомів Гідрогену в метальних групах цієї сполуки втричі більше, ніж у
метиленовій групі:
сі2
СН3СН2СН3 СН3СН2СН2С1 СН3(рНСН3
СІ
Під час хлорування ізобутану реакція за атомом Гідрогену, зв'язаним з
третинним атомом Карбону відбувається також швидше, ніж для первинних:
сн3 сн3 сн3
н3с-с-н + сц-^* н3с-с-сі + н3с-с-н
сн3 сн3 сн2сі
36 % ^^^^ 64 %
З'ясовано, що співвідношення швидкостей заміщення хлором атомів
Гідрогену в первинного, вторинного, третинного атомів Карбону 1,0:3,8:5,0. На
швидкість цієї реакції впливають такі чинники. По-перше,,енергія гомолітичного
розриву зв'язку С-Н зменшується в порядку: первинний атом Карбону >
вторинний > третинний (табл. 7.3). По-друге, з діаграми (див. рис. 7.1) видно, що
енергія активації у випадку атаки радикала Хлору по атому Гідрогену у
вторинного карбонового атома менша, ніж у разі атаки атома Гідрогену в
первинного атома Карбону.
Оскільки енергію активованого комплексу можна оцінити через стійкість
відповідного інтермедіату, то для цього потрібно знати, як його будова впливає на
алкільні вільні радикали. Виявлено, що вона збільшується в порядку: первинний <
вторинний < третинний радикал. Такий ряд стійкості можна пояснити за допомогою
Розділ 7. Алкани
149
ефекту гіперкон'югації - збільшення ступеня перекривання орбіталі вільного
радикала з орбіталями С-Н-зв'язків, що розташовані поряд. Для метального
радикала такої стабілізації немає взагалі, в етильного радикала можливе
перекривання з трьома зв'язками С-Н, для ізопропільного радикала - з шістьма
зв'язками С-Н, а для /ярет-бутильного радикала - з дев'ятьма:
Гіперкон'югація в етильному, ізопропільному і /яре/л-бутильному радикалах
Таблиця 7.3
Енергія гемолітичної дисоціації зв'язків Карбон-Гідроген в алканах
Сполука
Енергія зв'язку АЯ, кДж/моль
СНз-Н
422,9
СН3СН2-Н
401,9
(СН3)2СН-Н
393,6
(СНз)зС-Н
339,8
Отже, реакції хлорування пропану та ізобутану при 25 °С відбуваються
регіоселективно.
> Регіоселективпа реакція - це реакція, під час перебігу якої хімічних змін
зазнає переважно та чи інша ділянка молекули субстрату і головно утворюється
один з декількох можливих ізомерних продуктів.
Коли реакція відбувається за високої температури (-450 °С), 1-хлоропропан
і 2-хлоропропан утворюються у співвідношенні 3:1, оскільки подача енергії в
систему внаслідок нагрівання перекриває невелику різницю в енергії розриву
зв'язків С-Н і стійкості вільних радикалів. Співвідношення продуктів реакції
відповідає ймовірності атаки по атомах Гідрогену біля первинного і вторинного
атома Карбону.
• Спеціальні методи хлорування. Алкани можна хлорувати також сульфур
діоксодихлоридом (сульфурилхлоридом) 802С12. За даними М. Караша1, засто-
сування такого хлорувального агента забезпечує значну селективність.
1 Караш М. (ЮіагаБсЬ М.) (1895-1957) - американський хімік українського походження. Займався
дослідженнями в галузі вільнорадикального приєднання та^хімії полімерів (штучного каучуку). Є
винахідником Мертіолату.
150
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Більшої вибірковості в хлоруванні алканів можна досягти в разі вико-
ристання трихлорометансульфохлориду СЬС802С1. Очевидно, реакція хлору
вання, яку ініціюють фотохімічно, відбувається за такою схемою:
За допомогою газової хроматографії визначено, що під час монохлорування н-
гексану єдиним продуктом реакції є 2-хлорогексан.
❖ Флуорування. Флуорування алканів - дуже екзотермічний процес. З одного
боку, енергія гомолітичного розщеплення молекули Флуору на радикали невелика-
155 кДж/моль, тоді як для Хлору вона дорівнює 242, для Брому - 192, а для Іоду -
150 кДж/моль. З іншого, - на стадії росту ланцюга обидві реакції екзотермічні:
З огляду на це аліфатичні вуглеводні згоряють в атмосфері флуору з
руйнуванням карбонового скелета до тетрафлуорометану і гідроген флуориду:
Напрямлене флуорування алканів без розриву зв'язків С-С проводять
кобальт (III) флуоридом або флуором в інертному газі, або флуором, що
виділяється під час електролізу.
❖ Бромування завдяки найнижчій серед галогенів екзотермічності (АЯ -
29 кДж/моль) і меншій енергії вільного радикала Брому відбувається більш
селективно і дає майже чисті продукти. Відомо, що селективність тим більша, 1
чим менша реакційна здатність агента галогенування.
Найлегше галогенування відбувається у третинного атома Карбону, потім -
вториннош/і, наприкінці, - первинного. Для Брому співвідношення швидкостей
заміщення в цих атомів 1600:82:1. Наприклад, коли бромують пропай, то
одержують ізопропілбромід з виходом 92 %, для ізобутану заміщення практично
кількісно відбувається в третинного атома Карбону, а в випадку 2,2,3-три-
метилбутану одержують (СНз)3С-СВг(СН3)2 з виходом 96 %:
С13С802С1 С13С- + 802 + СІ-
С13С-+ ІШ Я- + СНС13
Я- + С13С802С1 —- ЯСІ + С130 + 802
ОНЦ + Б- -> СН3 + НР + 130 кДж/моль
СН| + ¥2 -> СНзР + Р- + 297 кДж/моль
СпН2п+2 + Р2->СР4 + НР
СН3СН2СН3 + Вг2
Н3С-СН-СН3 + НВг
Пропан
2-Бромопропан
Розділ 7. Алкани
151
Br
Н3С-СН-СН3 + Br2 Н3С-С-СН3 + HBr
CH3 сн3
2-Метилпропан 2-Бромо-2-метилпропан (100 %)
(СН3)3С-СН(СН3)2+ Вг2-^ (СН3)3С-СВг(СН3)2
2,3,3-Триметилбутан 2-Бромо-2,3,3-триметилбутан (96 %)
❖ Йодування алканів. Йодувати насичені вуглеводні не вдається через
високу ендотермічність першої стадії росту ланцюга реакції йодування алканів
(138,2 кДж/моль). Процес неможливий навіть при 300 °С. До того ж, легко
відбувається зворотна реакція відновлення іодоалкану гідроген іодидом. Проте,
застосовуючи спеціальні агенти йодування, наприклад, wpew-бутоксиіодид
(СНз)зС-ОІ, що одержують безпосередньо в реакційній суміші з трет-
бутоксихлориду і меркурій (II) йодиду, моясливе йодування алканів з
невеликими виходами:
Hgl2 + (СНз)зСОСІ -> (СНз)зСОІ + HglCl
R-H + (СНз)зСОІ R-I + (СНз)зСОН.
7А.2. Нітрування
❖ Нітрування в рідкій фази На відміну від ароматичних сполук, алкани
окиснюються, а не нітруються, якщо на них діяти концентрованою нітратною
кислотою або сумішшю нітратної і сульфатної кислот. Нітрування алканів дією
розведеної (10-20 %) нітратної кислоти в разі нагрівання до 150 °С в запаяних
ампулах уперше виконав М. Коновалов1 (1888). Під час нітрування гексану він
одержав 2-нітрогексан:
СН3СН2СН2СН2СН2СН2 +HN03— H3C€H€H2CH2CH^:H3+ Н20
N02 63 %
Нітрування за Коноваловим розгалужених алканів переважно дає моно-
заміщені похідні в третинного атома Карбону.
❖ Нітрування у паровій фазі Цей метод набув подальшого розвитку у
працях американського хіміка Г. Хасса в так званому нітруванні у паровій фазі
(1936). Саме таким шляхом у промисловості одержують нітрометан, нітроетан і
два ізомерні нітробутани. За температури 420 °С етан, пропан і н-бутан швидко
1 Коновалов М. (1858-1906) - російський хімік-органік. Учень В. Марковникова. Головні праці в
галузі нітропохідних вуглеводнів. Професор (з 1899 р.) і ректор (1902-1904) Київського
політехнічного інституту.
152
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
вступають у реакцію нітрування. Метан за такої температури реагує повільно,
через це температуру реакції підвищують до 500 °С. В цих умовах нітрування
метану відбувається з виходом 13 %, якщо діяти на нього оксидами нітрогену
або нітратною кислотою:
СН4 + НЫОз СН3-гТО2 + Н20
Метан, що не прореагував, повертається в процес. Нітрометан застосовують у
синтезі вибухових речовин і як розчинник.
Етан реагує за двома напрямами: заміщення атома Гідрогену на нітрогрупу і
крекінг карбонового ланцюга з одночасним нітруванням.
СНГСН3 + НШ3— СН3СН2Ж)2 + СН3Ш2
Вищі гомологи ведуть себе аналогічно. Внаслідок реакції утворюються
суміші нітропродуктів, які містять сполуки з меншою кількістю атомів Карбону
порівняно з вихідним вуглеводнем. Наприклад, із пропану при 420 °С
утворюється суміш нітропродуктів, що складається з 32 % 1-нітропропану, 33 %
2-нітропропану, 28 % нітроетану і 9 % нітрометану. Загальний вихід суміші
нітропродуктів становить 21 %.
Отже, під час нітрування алканів у паровій фазі рвуться не лише С-Н, а й С-
С-зв'язки, тобто процес супроводжується крекінгом вуглеводнів.
■ Ініціатором реакції як у рідкій, так і в газовій фазах, що відбувається за
радикальним механізмом, є нітроген (IV) оксид, молекула якого має неспарений
електрон Ж)2. Він створюється за участю нітритної кислоти, яка завжди наявна
в розведеній нітратній кислоті:
о2к:он + н:о>го • о2ы:ош 2Ш2-
Н 2 . -N0, N0,
н3с-сн2-с-сн3 +. ж)2-^Г н3с-сн2-с-сн3 —і н3с-сн2-с-сн3
сн3 "нмо2 сн3 сн3
У більш концентрованій нітратній кислоті (60-70 %) джерелом радикала
•>Ю2 може бути розпад молекул нітратної кислоти:
НШ3 .М)2 + ОН
2НШ3 —Ж)з+ -Ш2+Н20
Оскільки концентрація стабільного за цих умов радикала -Ы02 досить висока,
то він швидко реагує з алкільними радикалами і тим самим унеможливлює процес
димеризації радикалів. На відміну від реакцій галогенування радикальне
нітрування не відбувається за ланцюговим механізмом, бо під час перебігу реакції
«е регенерується жоден проміжний вільний радикал.
Розділ 7. Алкани
153
На реакцію нітрування витрачається близько 40 % нітратної кислоти, а на
окиснювальні процеси - решту, унаслідок чого утворюються спирти, альдегіди,
кетони, кислоти й естери нітратної кислоти.
Тепер замість нітратної кислоти як нітрувальний агент використовують
нітроген (IV) оксид за температури понад 450 °С.
7.4.3. Сульфування
❖ Пряме сульфування. Олеум під час нагрівання повільно сульфує
парафіни, що мають метанову групу:
?03Н
СН3(рНСН2СН3 + H2S04 СНз^СН^Нз
сн3 сн3
Цю реакцію важко реалізувати в промисловості з достатньою ефективністю,
тому перспективнішою здається реакція сульфохлорування.
❖ Сульфохлорування. Під час обробки алканів УФ-світлом у газовій фазі
вони вступають у реакцію заміщення з сумішшю сульфур (IV) оксиду і хлору. В
промисловості реакцію використовують для сульфохлорування когазину.
Когазин - фракція синтину, яка складається переважно із вищих нерозгалужених
вуглеводнів і є цінним дизельним пальним.
Механізм цієї ланцюгової реакції такий:
C\2-Jï^ 2С1-
с„н2п+2+сі. - нсі + спн2п+1
О
cnH2n+1 + so2 —-cnH2nïïs-
о о °
c„H2nTls-+ci2 —^ cnH2n+Tjj-ci + сь
о о
Легко помітити, що поряд з алкансульфохлоридами повинні утворюватись
хлоропохідні, як результат ланцюгового процесу хлорування. Для пригнічення
галогенування алканів сульфохлорування проводять з великим надлишком
сульфур (IV) оксиду, який відіграє роль "перехоплювача" алкіл-радикалів.
Селективність сульфохлорування біля первинного і вторинного атомів Карбону
невелика і збігається з селективністю хлорування. Водночас атоми Гідрогену
біля третинного атома Карбону не заміщуються на сульфохлоридну групу.
Очевидно, це пов'язано з просторовими перешкодами для підходу об'ємного
реагенту до третинного атома Карбону.
754
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
■ Алкансульфохлориди - це хлороангідриди дуже сильних алкансульфо-
кислот Я-80зН. їхні натрієві солі утворюються під час обробки алкансуль-
фохлоридів водним розчином натрій гідроксиду або соди:
ЯЗОгС 1 + 2ИаОН -» ЯЗС^а + ИаСІ + Н20
Якщо алкільні групи мають відповідні розміри, то вони виявляють
властивості типової поверхнево-активної речовини (ПАР), тому їх надзвичайно
широко використовують як детергенти. Алкільна група надає цим солям
ліпофільні властивості, а йонна сульфатна група БОз" - гідрофільні. У солей з
алкільною групою, що має 8-16 атомів Карбону, ліпофільні та гідрофільні
властивості збалансовані так, що ці речовини емульгують у воді жири та оливи.
Синтетичні детергенти одержують у промисловості в величезних кількостях
сульфохлоруванням гасової фракції нафти без розділення на окремі компоненти.
Головний недолік цих ПАР, які застосовують у промисловій флотації і в побуті,
- забруднення довкілля (водоймищ, рік, озер, морів). Певні бактерії водного
середовища розкладають солі алкансульфокислот із нерозгалуженим карбо-
новим ланцюгом, але вони неефективні для деструкції алкансульфокислот із
розгалуженим ланцюгом. Для вирішення цих екологічних проблем американські
фірми вкладали величезні кошти для створення ПАР із нерозгалуженими
карбоновими ланцюгами, що сприяло вирішенню цього завдання.
❖ Сульфоокиснення. Реакцію сульфоокиснення також застосовують для
синтезу алкансульфокислот. Процес проводять при УФ-опроміненні або за
наявності радикальних ініціаторів, що сприяють утворенню алкільних радикалів:
9
К- + Б02—«- Я-802- Я-БО^ +02 " Я-§-00-
я-^-оо • + Я'Н +я-Борон
О Ажанпероксисульфокислота
Я-802ООН —- К-802О+0Н- Я8020- + Я-Н —- Я-803Н + Я-
Процес сульфоокиснення вигідніший, ніж сульфохлорування, оскільки в цьому
процесі не використовують хлору, який завдає шкоди навколишньому середовищу.
Нещодавно з'ясовано, що алкани під дією 802 при дії УФ-світла перетворюються в
алкілсульфінові кислоти, які легко і без ускладнень окиснюються в сульфокислоти.
7.4.4. Каталітична дегідрогенізація алканів
Перетворення алканів у ненасичені сполуки виконують за наявності
каталізаторів (реакція Баландіна1).
1 Баландін О. (1898-1967)-радянський хімік, головні праці присвячені органічному каталізу.
Розділ 7. Алкани
755
300-400 °С
с„н2п + н2
п"2п+2
За температури -600 °С із бутану утворюється бута-1,3-дієн, через те
реакція має важливе промислове значення для виробництва синтетичного
каучуку.
7.4.5. Окиснення
Кисень повітря і звичайні окиснювачі (КМПО4, К2СГ2О7, К2СЮ4) реагують із
парафінами лише за високих температур із розривом карбонового ланцюга й
утворенням органічних кислот. У цьому разі значна кількість алкану
окиснюється до карбон (IV) оксиду.
В обмеженому масштабі в промисловості використовують окиснення вищих
алканів у рідкій фазі при 100-150 °С із застосуванням солей Мангану як
каталізаторів. Процес передбачає стадію утворення гідропероксидів. Реакція
відбувається за умов продування потоку повітря через розплавлений парафін. У
цьому разі ланцюги карбонових атомів довільно розриваються, утворюючи
суміш насичених кислот і гідроксикислот з нормальним ланцюгом.
Передбачений такий механізм. За високої температури з вуглеводнів
виникають вільні радикали, які взаємодіють із молекулами Оксигену, утво-
рюючи алкілпероксидні радикали:
Алкілпероксидний радикал відщеплює атом Гідрогену від інших молекул
алкану, перетворюючись на відповідні гідропероксиди:
За умов реакції окиснення (висока температура і тиск) гідропероксиди
розпадаються на вільні радикали Я-О', які відщеплюють атоми Гідрогену від
молекул вуглеводнів з утворенням спиртів або розщеплюються за місцем ро-
зв'язку, і виникають альдегіди або кетони. Так розкладаються третинні
гідропероксиди:
я + о::о
я-о-о-
я-о-а+ян
я-о-о-н+я-
Гідропероксид
о
СНЗ-С-ООН
сн3
сн3-<р-о- +• он
СН3 \
сн
сн3
[3-І-ОН + я-
сн3
156
Чирва В.Я., Ярмолюк CM., Толкачова Н.В., Земляков О.Є. Органічна хімія
Вторинні гідропероксиди за низької температури перетворюються в кетони і
спирти:
Г"3 СНтС-СНз+Н-
НЧрО-ОН ' ~"
СНз СНз ^-^L (СН3)2СНОН + R-
а за високої температури вони дають альдегіди і спирти:
Н Н -сн^< СН3СНО
<р-оон СН3- <р- о + • ОН ^
снз СН3 +*R4^ (СН3)2СНОН + R-
Аналогічно ведуть себе первинні гідропероксиди.
■ Різновидом цього методу є промисловий метод одержання оцтової
кислоти з н-бутану:
СН3СН2СН2СН3 + о2 2СН3соон
50%
7.4.6. Каталітична ізомеризація алканів
Раніше зазначали, що ізомерні структури не є енергетично еквівалентними:
найбільш термодинамічно стійким є ізомер з розгалуженим ланцюгом.
Нерозгалужені алкани можна перетворити в їхні ізомери за допомогою
каталізаторів Фріделя-Крафтса, таких як алюміній галогенід, сульфатна кислота,
гідроген флуорид тощо: Каталітична ізомеризація - важливий процес нафто-
переробки. Ізомеризацію н-бутану в ізобутан (важливий продукт у реакціях
алкілування) можна провести з виходом 80 % за кімнатної температури й за
наявності алюміній броміду:
АІВг3
СН3СН2СН2СН3 5=^ НзС-^Н-СНз
СН3
7.5. Крекінг алканів
❖ Термічний крекінг. Піроліз (від грец. руг - вогонь і lysis - руйнування) -
це глибоке руйнування сполук за нагрівання до 700-1 000 °С.
Піроліз алканів, особливо, коли йдеться про нафту, відомий під назвою крекінг.
Як звичайно, термічний крекінг алканів відбувається у колоні, що нагріта до
Розділ 7. Алкани
157
температури 470-540 °С. Алкани з високою молекулярною масою перетворюються
на алкани з меншою молекулярною масою, алкени і водень. Вуглеводні, що мають
порівняно довгий ланцюг атомів Карбону, розриваються в будь-якому випадковому
місці, і осколки (алкіл-радикали) перерозподіляють атоми Гідрогену так, що
утворюється суміш еквімолекулярних кількостей алканів і олефінів, наприклад:
г СН3* ~Ь" • СН2СН2СН2СН2СН3 —*~ СН4 СН2—СНСН2СН2СН3
СН3(СН2)4СН3
СН3СН2 "Ь *СН2СН2СН2СН3 ~
СН3СН2СН2 "І" * СН2СН2СН3
* СН^ СН3 + СН2—СНСН2СН3
C3Hg + СН2=СНСН3
Незважаючи на це, є деякі закономірності в поведінці алканів. Наприклад,
чим більша молекулярна маса вуглеводнів, тим легше вони розпадаються. Якщо
підвищувати температуру крекінгу, то простежуватиметься зсув місця розриву
ланцюга до краю молекули, а з підвищенням тиску - до середини ланцюга.
Якщо проводити процес за температур піролізу, то алкани розпадаються до
ацетилену, метану і сажі.
Особливо чітко це видно на прикладі бутану:
600 °С
СН3СН2СН2СН3
600 °С
СН3СН3 + СН2=СН2 36%
СН3СН=СН2 + СН4 48%
600 °С
СН3СН2СН=СН2 + Н216%
600 °г
СН2=СН-СН=СН2 + 2Н2
11^2НСНСН + ЗН2
За низькотемпературного крекінгу із н-бутану спочатку утворюються
метальний, етильний і пропільний радикали:
боо°а
СН3СН2СН2СН3
- СН3* "Ь СН3СН2СН2*
сн3сн2- + СН3СН2-
Рекомбінація радикалів, що утворилися, дає суміш етану, пропану, бутану,
пентану і гексану.
У разі підвищування температури на перший план виходить диспро-
порціювання радикалів. В одному випадку алкіл-радикал відщеплює атом
Гідрогену від одного з алканів - вихідного чи того, що утворився внаслідок
рекомбінації і, отже, одержують новий радикал, наприклад:
К-СН2СН2 К-СН2СН2СН2СН2-К.
іі-сн2сн3 + к-сн2сн2сн2сн-я
158
Чирва В.Я., Ярллолюк СМ, Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Цей процес одержав назву реакція перенесення радикального ланцюга.
Інший тип диспропорціювання алкільних радикалів називають (і-розкладом.
Унаслідок розщеплення одержують алкен і новий радикал. Процес Р-розпаду
може тривати до тих пір, поки не буде одержано етилен і метальний радикал.
Р І а • В-Розклад Р і а
ксн2сн2снфсн2снсн2я £—- ксн24сн2сн2 + СН2=СНСН2Я
| Р-Р°зпаД
СН2=СН2 + ІІСН2- і т. д.
Більша частина алкільних радикалів утворюється не завдяки гомолітичному
розриву С-С-зв'язку в алкані, а внаслідок відщеплення від нього атома
Гідрогену за допомогою метального радикала.
За високої температури Р-розклад домінує, що спричиняє утворення
низькомолекулярних газоподібних вуглеводнів. Унаслідок термічного крекінгу
утворюється переважно етилен разом з іншими невеликими молекулами.
У 1930-1950 рр. за допомогою термічного крекінгу вдалося збільшити
бензинову фракцію внаслідок розщеплення алканів, що містилися в керосиновій
фракції (С10-С15) і фракції солярової олії (С12-С20). Проте октанове число
бензину, одержаного в умовах термічного крекінгу, не перевищує 85, що не
задовольняє вимоги двигунів внутрішнього згорання сучасного автомобіля.
Сьогодні для одержання моторного пального каталітичний крекінг повністю
витіснив крекінг термічний.
❖ Крекінг з водяною парою і гідрокрекінг. У випадку крекінгу з водяною
парою вуглеводні розбавляють парою, нагрівають до 700-900 °С і швидко
охолоджують. Цей процес набув особливої ваги для виробництва вуглеводнів,
які застосовують як реагенти, наприклад, етилен, пропілен, бутадієн, ізопрен і
циклопентадієн.
Іншим джерелом вуглеводнів з невеликою молекулярною масою є
гідрокрекінг, який проводять за наявності водню під великим тиском і за значно
нижчих температур (250-450 °С). Алкани з низькою молекулярною масою, які
одержують за умов такого крекінгу, можна розділити, очистити; їх
використовують як важливу сировину для синтезу аліфатичних сполук у
великих масштабах.
Каталітичний крекінг. У промисловому виробництві пального провідну
роль відіграє каталітичний крекінг. У цьому процесі речовини з високими
температурами кипіння, наприклад, солярову олію, вводять у контакт з
подрібненим алюмосилікатним каталізатором (Е. Гудрі1, 1934 р.) за температури
450 °С під невеликим тиском. Алюмосилікати діють як кислотні каталізатори,
Гудрі Е. (Ноисігу Е.) (1892-1962) - американський хімік і промисловець. Головні праці в галузі
каталітичного крекінгу нафти.
Розділ 7. Алкани
159
що дає змогу не лише підвищувати вихід бензину внаслідок руйнування великих
молекул до молекул меншого розміру, а й поліпшити якість бензину. Процес
відбувається через утворення карбонієвих іонів і дає, головно, розгалужені
алкани та алкени. Наприклад, синтез 2,2,4-триметилпентану (ізооктану) у
промисловості проводять взаємодією ізобутилену та ізобутану за наявності
кислого каталізатора:
<рн3 срн3 срн3 (рн3
СН^* С=СН2 +Н~ СН3 *~ СН3 (р-СгІ2СН~СН3
СН3 СН3
Ізооктан
Загальновизнаний механізм цього алкілування охоплює утворення стійкого
карбокатіону (стадія 1) та електрофільне приєднання за місцем подвійного
зв'язку (стадія 2; детальний механізм реакції див. розділ 8). На стадії (3)
карбокатіон відриває атом Гідрогену з його парою електронів (по суті, гідрид-
іон) із молекули алкану. Цей відрив гідрид-іона з наступним приєднанням його
до карбокатіону утворює алкан з вісьмома атомами Карбону, а новий
карбокатіон продовжує ланцюг реакції:
срн3 СН3
СН3-С=СН2 + Н+ — СН5-С-СН3 0)
?н3 сн3 <рн3 <рі3
СНуС=СН2 + СНГС-СН3 -СНгС-СН2-^-СН3 (2)
СН3
❖ Каталітичний риформінг. У процесі каталітичного риформінгу
величезні кількості аліфатичних вуглеводнів нафти перетворюються в ароматичні
вуглеводні, які використовують не лише як пальне вищого ґатунку, а й як вихідні
речовини для синтезу більшості ароматичних сполук. Наприклад, під час другої
світової війни потреба в толуені для виробництва тринітротолуену значно
перевищила 120-150 млн л, які щорічно одержували з кам'яновугільної смоли.
Тому розроблено спеціальні методи синтезу толуену на основі дегідрування
метилциклогексану, який видобували з нафти:
160
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
сн3 + зн2
Завдяки цьому виробництво толуену в 1944 р. зросло до 1 млрд л.
Під час каталітичного риформінгу відбувається не лише дегідрування, а й
циклізація та ізомеризація, як у синтезі толуену з н-гептану або 1,2-диме-
тилциклопентану.
Отже, каталітичні методи переробки нафти повністю витіснили старі
термічні методи крекінгу вуглеводнів. Термічні методи нині зберегли своє
значення лише тоді, коли отримують етилен з керосинової і бензинової фракцій.
❖ Октанове число. Для двигунів внутрішнього згорання потрібне пальне з
високим октановим числом. Нерозгалужені алкани, якщо стискати суміш парів
бензину з повітрям, утворюють пероксиди, які детонують передчасно і завдають
шкоди двигуну. Розгалужені вуглеводні, особливо ізооктан, не мають такого
недоліку. Для того, щоб порівняти здатність бензину до детонації, побудували
спеціальну шкалу, де гептан має октанове число 0, а ізооктан - 100. Якщо
пальне, яке досліджують, детонує як суміш 95 % ізооктану і 5 % н-гептану, то
пальному присвоюють октанове число 95. Нині якісний автомобільний бензин
має октанове число, близьке до 100.
На відміну від пального для двигунів внутрішнього згорання, пальне для
дизельних двигунів має найвищу якість, якщо містить нерозгалужені вуглеводні4.
Пальне для дизельних двигунів характеризують цетановим числом. За стандарт
взято я-цетан Сі6Н34 (цетановое число 100, а Р-метилнафтен має цетанове число 0).
Октанове число, яке характеризує якість бензину, різко збільшується, якщо
додавати невеликі кількості антидетонаторів. Найвідомішим антидетонатором
є суміш тетраетилплюмбуму з бромоетаном - так звана етилова рідина. Проте
"етильовані" бензини дуже отруйні й екологічно шкідливі. Останнім часом
знайдені металоорганічні антидетонатори, що містять Манґан, вони ефективні-
ші, ніж етилова рідина. Бензини з цією присадкою не отруйні.
Октанове число бензину різко збільшується, якщо до нього додати
ароматичні сполуки. У цьому разі воно зростає значно більше, ніж можна було '
очікувати на основі складу суміші й октанових чисел її компонентів. ;
Наприклад, и-ксилен має октанове число 100, проте в сумішах із бензинами
поводиться так, ніби має октанове число 145. Тому говорять, що и-ксилен має
"октанове число в суміші" 145.
Із наведених вище реакцій видно, що найтиповішою властивістю алканів є
здатність до радикальних реакцій. Ця обставина значно обмежує можливості
прямого введення в молекулу різних функціональних груп.
7.6. Йонні реакції
Розділ 7. Алкани
161
Ситуація різко змінилася, коли 1994 р. Дж. Ола1 провів іонні реакції з
алканами, діючи на них дуже сильними електрофільними реагентами. Він довів,
що із суперкислотами, такими як + БЬРз, молекули нижчих алканів зазнають
процесів укрупнення, тобто олігомеризації. Наприклад, із метану одержують
його гомологи. Відщеплення атома Гідрогену відбувається завдяки одно-
електронному перенесенню:
сн4—-сн4
*сн4 сн3+ + н-
н-сн3 + сн; —^н3с-сн3+ н+ і т.д.
Середні алкани, що мають нижчі потенціали йонізації, навіть під дією кислот
Льюїса утворюють карбокатіони, які зазнають ізомеризації та олігомеризації:
НСІ + А1С13 Н+[А1С14]~
СНзСНрС,Н-СН3 + Н+ СН3СН2СНСН3+ Н2
(Н
НзСтО^С-СНз ► НзС-С-СНз Н3С-СН-СН3 + Н^-С-СН^СН-СИз
\_Н + сн3 СН3
Ізобутан Ізооктан
Продукт ізомеризації Продукт димеризації
■ Алкани реагують і з іншими електрофільними реагентами:
Я-гІ + N0^""—- я-ыо2+ НРБ6
СН4 + С12 СН3С1 + НСІ
Як випливає з цих реакцій, алкани є донорами електронів. Уважають, що два
електрони а-зв'язку здатні до делокалізації між трьома центрами. У цьому разі
за участю електрофільного реагенту утворюється двохелектронний трицентро-
вий інтермедіат:
И-К + Е+ -¿+4'
1 Ола Дж. (ОІаИ С) (нар. 1927 ) - американський хімік угорського походження. Вивчав та займався
стабілізацією карбокатіонів за допомогою суперкислот. Нобелівська премія 1994 р.
162
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
7.7. Найважливіші алкани
• Метан є головною складовою частиною природного газу. У великих кількостях
він міститься у вугільних пластах і є причиною вибухів на шахтах. Крім того, метан
є головною складовою болотного газу, який утворюється на дні озер і на болотах
унаслідок анаеробного бродіння целюлози. В. промисловості та побуті метан
використовують як паливо з великою теплоємністю (-5 000 кДж/моль).
■ За високої температури (800-900 °С) метан реагує з водяною парою:
СН4 + Н90 СО + З Н, Синтез-газ
4 2 № 2
На цій реакції ґрунтується важливий шлях хімічного перетворення метану,
оскільки суміш газів, що утворюється, використовують для промислового
синтезу метанолу, він слугує джерелом водню, наприклад, для синтезу амоніаку.
Каталітичне окиснення метану за наявності амоніаку, розроблене Л. Андру-
совим, приводить до ціанідної кислоти. Ця реакція впроваджена в промисловість:
2 СН4 + 2 ЫН3 + 3 02 2 НСЫ + 6 Н20
4 3 2 Рі/Ші 2
■ За температури 1500°С метан розкладається за рівнянням:
2СН4 ^2^2 + ЗН2
Це ендотермічний процес. Він є в основі одного зі шляхів промислового
одержання ацетилену.
За часткового окиснення або термічного розкладу з метану одержують
високоякісну сажу, яку використовують як наповнювач у виробництві гуми:
СН4^оо£с + 2Н2
СН4 + 02— С + 2Н20
Важливе значення мають продукти, які одержують галогенуванням метану,
зокрема метилхлорид (органічний синтез, місцевий анестезувальний засіб) і
метилбромід (засіб для фумігації*), дихлорометан (розчинник), хлороформ
(розчинник, застосовують у синтезі фреонів), тетрахлорометан (розчинник,
застосовують у синтезі фреонів, засіб для гасіння пожеж).
Головні шляхи переробки метану відображено на рис. 7.2.
У 1950 р. Г. Ури1 виявив, що під час обробки суміші метану, води, амоніаку
і водню електричним розрядом утворюється велика кількість різних органічних
1 Ури (Юрі) Г. (Іігеу Н.) (1893-1981) - американський фізико-хімік. Відкрив дейтерій, працював над
проблемами розділення ізотопів урану, виробництва важкої води. Вивчав зародження й еволюцію
життя на Землі. Нобелівська премія 1934 р.
Розділ 7. Алкани
163
сполук, зокрема амінокислот, які є будівельними цеглинками для білків —
"матерії життя". Метан, що виникає внаслідок розкладу живих організмів, може
бути тією речовиною, з якої, зрештою, зародилося життя на Землі.
Етан можна виділити з природного газу. Його використовують для
одержання тепла й у виробництві етилену.
Бутани містяться в природному газі та нафті, а також утворюються в
процесі крекінгу нафти. їх використовують у виробництві бутенів, ізобутилену,
бутадієну.
7.8. Метилен
7.8.1. Будова метилену
В алканах два сусідні члени ряду відрізняються один від одного групою СН2
або метиленовою групою. Однак вона є не лише будівельним блоком уявної
побудови алканів. Це реальна молекула (метилен), і її хімія, і хімія споріднених
молекул - карбенів, є галуззю сучасної органічної хімії, що розвивається
найінтенсивніше.
Метилен утворюється в процесі термічного або фотохімічного розкладу
діазометану СНУ^г, а також кетену СН2=С=0 (Т. Пірсон, 1938 р.):
СН2=№=*Г—- :СН2+Н>
СН2=С=0 — :СН2 + СО
Тривалість існування метилену набагато більша, ніж метального радикала,
середній час його життя становить 0,05 с. Нормальною реакцією стабілізації
метилену в газовій фазі за високої концентрації є димеризація в етилен, а в разі
опромінення метилену ультрафіолетовим світлом утворюється поліметилен,
схожий на поліетилен.
Два незв'язані електрони в карбенах можуть бути або спареними, або
неспареними. Спарені електрони з антипаралельними спінами дають в ЕПР-спектрі
синглет, тоді як два неспарені електрони з паралельними спінами дають триплет.
Синглетний метилен несподівано виявився д
менш стійким і більш реакційноздатним, ніж од • Н-^С
ТрИПЛеТНИЙ МеТИЛеН. У ТрИПЛеТНОМу МеТИЛенІ Синглетний метилен
спостерігають .ур-гібридизацію, і два електрони ^0°
розташовані на двох негібридизованих р-орбіталях, ж • ^\
а в синглетному метилені з лр2-гібридизацією Н.С.Н Н С Н
Обидва електрони МІСТЯТЬСЯ На ОрбІталІ, ЩО має Триплетний метилен
більш ^-характер і, відповідно, меншу енергію.
N.
Водень для
синтезу амоніаку
2Н2 + С ^
Сажа (гумова
промисловість)
Трісгідроксиме-
тилнітрометан
Вибухові речовини
Силікони
ЗСН,0
СНзНОг
(СНз)28ІСІ2
8І
СО + Н,
СН3ОН
Пластмаси
Синтез-газ, водяний газ Метанол Формальдегід
Окиснення
конверсія
Піроліз
Нітрування
Піроліз
сн.
Сі,
НС=СН + Н2
Ацетилен
н-с=с-н
НСН *- н,с=сн-
-СК
Ціанова
кислота
Хлорування
Акрилонітрил
Синтетичне волокно
"нітрил", нітрильний каучук
СНЗС1
СН2С12
ссі4
НР
СНСІ3
Диметидцихлоросилан Метилхлорид Метиленхлорид Хлороформ Тетрахлорометан
Рис. 7.2. Схема промислового використання метану
СРзСІг
Фреон-12
(холодоагент)
Розділ 7. Алкани
765
У триплетному стані зменшується взаємне відштовхування електронів через
те, що вони розташовані на р-орбіталях з паралельними спінами. Тому в
незбудженому стані триплетний метилен стійкіший порівняно з синглетним.
Синглетний метилен, зіштовхуючись із навколишніми молекулами, віддає
свою енергію і перетворюється в стійкіший триплетний метилен. Він
утворюється, насамперед, у процесі фотолізу, оскільки світло, що його поглинає
діазометан або кетен у фотолізній реакції, має більшу енергію, ніж потрібно для
розриву зв'язків; метилен, що утворився, є "гарячим". Тобто молекули метилену
мають більшу енергію, ніж вони повинні були б мати за цієї ж температури.
Експериментально з'ясовано, що сам метилен зазвичай утворюється як
синглетна частинка, яка перетворюється в триплетну частинку з нижчою енергією.
Розрахунок за методом молекулярних орбіталей засвідчує, що різниця енергій
синглетного і триплетного метиленів становить 37,8-46,0 кДж/моль.
■ Одним зі способів, за яким можна одержати дихлорокарбен :ССІ2, є
реакція хлороформу з сильною основою:
СНС13+ т/?е/я-С4Н90"К+ :СС12+ КС1 + /я/?ет-С4Н9ОН
Через електроноакцепторну дію атомів Хлору хлороформ є досить сильною
СН-кислотою. Тому на першій стадії відбувається йонізація молекули
хлороформу:
СНС13 + трет-С4Н90" ^ СС13" + трею-С4Н9ОН
Наступне відщеплення хлорид-іона від аніона дає дихлорокарбен:
ССІ3" ^ :СС12+ СГ
7.8.2. Хімічні властивості
Хімічні властивості метилену залежать від умов синтезу: реагенту, з якого
утворюється метилен; довжини хвилі світла, яку використовують; проводять
реакцію в рідкій чи газовій фазі, і якщо в газовій, то за наявності якого газу.
Вплив інертного газу виявляється в зниженні енергії метилену перед тим, як
він вступить у реакцію, тобто перетворення синглетного метилену в триплетний,
або "гарячого" в "холодний". У газовій фазі це відбувається внаслідок зіткнень з
метиленом або зі збудженим діазометаном. У рідкій фазі спостерігають переважно
властивості високоенергетичного метилену, можливо тому, що він швидко реагує
з надлишковими молекулами розчинника перед тим, як втратити енергію.
Синглетні карбени, з іншого боку, аналогічно до карбокатіонів мають
нестачу електронів, але, з іншого боку, як карбоаніони, вони мають вільну
електронну пару. Проте в реакціях карбени зазвичай ведуть себе як електрофіли.
166
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
❖ Приєднання карбенів до кратних зв'язків. Простий метод роз-
пізнавання синглетних і триплетних частинок заснований на відомій реакції
приєднання карбенів за місцем подвійного зв'язку з утворенням похідних
циклопропану. Наприклад, якщо до г/нс-бут-2-ену приєднується синглетна
частинка, то синтезований циклопропан повинен бути г/нс-ізомером, оскільки
перенесення двох пар електронів повинне відбуватися або одночасно, або в
швидкій послідовності. Отже, приєднання синглетного карбену за місцем
подвійного зв'язку відбувається як електрофільне синхронне ї/ис-приєднання зі
збереженням конфігурації вихідного олефіну:
^С=С
/:СН2
• Н. .Н
Під час атаки триплетною частинкою новий ковалентний зв'язок не може
бути утворений обома неспареними електронами, оскільки, згідно з правилом
Гунда, вони мають паралельні спіни. Один із неспарених електронів карбену
може утворювати зв'язок з п-електроном подвійного зв'язку, що має
протилежний спін. У цьому разі в молекулі залишаються два неспарені
електрони з однаковим спіном, які не можуть утворювати зв'язку відразу, а
повинні чекати процесу співударяння, в якому один з електронів повинен
поміняти свій спін на протилежний. За цей час відбувається вільне обертання
фрагментів інтермедіату навколо зв'язку С-С, внаслідок чого утворюється
суміш цис- і транс- 1,2-диметилциклопропанів:
..А'" :сн<Н) *■'*>. * Д'" -*Ч. * .Д"
•с=сч ^+ к^с-Єс. =в*-^с-!£
к к І^О і ІГ Н20 \ к"
Спін-інверсія
Л Д" ~ і Лт *Ч і <Д" Я- л К"
К К" Н2С І ^Я" щС'\ чіг к V
Висловлене вище припущення про те, що синглетний метилен у газовій фазі
спочатку перетворюється в триплетний, а потім вступає в реакцію, підтверджують
одержанням як із цис-, так і трянс-бут-2-ену суміші ізомерів цис- і транс-
диметилциклопропанів. Якщо фотоліз відбувається у рідкій фазі, то синглетний
("гарячий") метилен встигає спершу прореагувати, ніж "захолоне". Про це
свідчать результати стереоспецифічності приєднання до транс- і г/мс-бут-2-енів.
Розділ 7. Алкани
167
■ Потрійний зв 'язок реагує подібно до подвійного, приєднуючи метилен:
сн3-сж;-сн3 +:сн2—► сн3-с=£—сн3
чсн2
Диметилциклопропен
Реакції вкорінення. Ймовірно, однією з найважливіших реакцій метилену є
реакція вкорінення.
-С-Н + :СН2—--С-СН3
І І
Метилен уважають найбільш неселективним реактивом органічної хімії.
Якщо він взаємодіє з я-пентаном, то одержують таку низку гомологів:
:СН2
СН3СН2СН2СН2СН3 —* СН3СН2СН2СН2СН2СН3 «-гексан, 49%
и-Пентан СН3СН2СН2СНСН3 2-Метилпентан, 34°/
\ сн3
СН3СН2(рНСН2СН3 3-Метилпентан, 17%
сн3
Аналогічно з пропану утворюються я-бутан та ізобутан:
сн =с=о
сн3сн2сн3—25^—СН3СН2СН2СН3 + СН3СНСН3
ін3
Той факт, що циклопентан взаємодіє з діазометаном при УФ-опроміненні,
даючи лише метилциклопентан, а не циклогексан, підтверджує, що метилен
реагує лише зі зв'язком С-Н і не розриває зв'язку С-С.
■ Доведено, що вкорінення метилену в алкани може відбуватися за двома
різними механізмами.
А) Пряме вкорінення:
:СН2 + -6-Н
-і,...::н
-Ь-
-сн2-н
У такий спосіб реагує синглетний метилен. Він не розрізняє зв'язки С-Н
біля первинних, вторинних і третинних атомів Карбону. Метилювання н-
пентану і пропану є яскравим підтвердженням цього.
Б) Укорінення через відщеплення - це таке приєднання, коли метилен
спочатку відриває атом Гідрогену від алкану з утворенням двох вільних алкіл-
радикалів, які потім з'єднуються між собою:
168
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
:СН2+ — (р-Н —- -(р- + -СН3^—(р-СН3
Такий перебіг реакції характерний для триплетного метилену. Наприклад, якщо
триплетний метилен реагує з пропаном, то з пропану утворюються пропільний,
ізопропільний і метальний радикали:
СН3СН2СН3 + :СН2 —- СН3СНСН3 + СН3СН2СН2 + СН3
■ Метилен як активна електрофільна частинка може приєднуватися навіть
за місцем я-зв'язку до бензену з утворенням норкарадієну і циклогептатрієну:
+ :сн2 - (^Рсн2 +
Норкарадієн Циклогептатрієн
Крім того, утворюється небагато толуену.
■ Аналогічно відбувається реакція з хлорангідридами кислот з утворенням
хлорометилкетонів:
Я-С-СІ +:сн2 Я-С-СНХІ
❖ Дихлорокарбен. На відміну від синглетного метилену, синглетний
дихлорокарбен стабільніший порівняно з триплетним ізомером. Очевидно, це
можна пояснити переходом електронної пари від атома Хлору на вакантну
орбіталь атома Карбону:
:сі-С-Сі: -—- :сі=С-Сі: -—- :С1-С=Сі:
Під час дегідрохлорування хлороформу утворюється синглетна форма
дихлорокарбену, який стереоселективно приєднується до алкенів:
н н
;С=СЧ + :СС12 — Н3С \С]/ СН,
н3с сн3
СІ
1,1-Дихлоро-г/ис-2,3-диметилциклопропан
Доказом стабільності синглетного дихлорокарбену є його інертність у
газовій фазі в реакціях укорінення. Прикладом реакцій, що відбуваються за
участю дихлорокарбену, може бути синтез діазометану з гідразину:
Розділ 7. Алкани
169
Н
кн2-кн2 + :ссі2 —* ш2уссі; сн2н2
н
Наприкінці зазначимо про неможливість перебігу димеризації карбенів
завдяки легкій добудові октету електронів:
/С:+:СЧ-^/С=С\
Причина проста. Карбени синтезуються в концентраціях, недостатньо
високих для того, щоб димеризація домінувала над реакціями вкорінення або
приєднання. ^
■ У низці реакцій утворення карбенів не доведено, хоча проміжні сполуки
ведуть себе як карбени. Такі сполуки назвали карбеноїдами. їхня наявність чітко
простежена в реакції Сіммонса1-Сміта - одному з найзручніших лабораторних
способів циклопропілування алкенів:
гп(Си) ^\
с5ніГсн=сн2 + сн2і2 с5ни—<]
Гепт-1-ен Пентилциклопропан
1 Сіммонс Г. (Simmons Н.) (1929-1997) - американський хімік. Досліджував хімічні процеси з
високореакційними інтермедіатами, реакції циклізації. В галузі теоретичної хімії вивчав
властивості кон'югованих я-електронних систем.
Розділ 8
АЛКЕНИ
8.1. Будова і номенклатура алкенів
Алкенами називають ациклічні вуглеводні загальної формули СпН2п. їх ще
називають етиленовими вуглеводнями (за родоначальником гомологічного ряду
етилену), або олефінами (від франц. olefiant, з лат. oleum - олія). Це пов'язано з
тим, що газоподібні члени етиленового ряду, з'єднуючись з хлором і бромом,
дають маслоподібні продукти.
8.1.1. Будова етилену
Найпростішим представником ряду алкенів є етилен С2Н4. Оскільки етилен
у процесі гідрування перетворюється в етан, можна очікувати аналогії в будові
цих двох сполук:
TT TT Нч ,Н
HOCH ^ ^н
Атоми Карбону з'єднані між собою ковалентним зв'язком, а до них
ковалентними зв'язками приєднано по два атоми Гідрогену. Отже, кожний атом
Карбону має шість електронів на валентному рівні замість потрібних восьми.
Умові нейтральності молекули відповідає належність ще однієї пари електронів
обом карбоновим атомам. Отже, атоми Карбону в етилені спільно володіють
двома електронними парами - тобто вони зв'язані подвійним зв'язком. Тому
подвійний зв'язок Карбон-Карбон є характерною особливістю будови алкенів.
Для реалізації такої будови атоми Карбону в етилені перебувають в sp2-
гібридному стані: три еквівалентні гібридні ^р2-орбіталі утворені комбінацією
однієї s- і двох р-орбіталей; 5р2-орбіталі лежать в одній площині й спрямовані до
кутів правильного трикутника: кут між двома орбіталями становить 120°.
Тригональне розміщення припускає максимальне віддалення гібридних орбіталей
одна від одної. Гібридизовані орбіталі беруть участь в утворенні сг-зв'язків С-С і
С-Н:
Розділ 8. Алкени
171
Н
я
Негібрндизована /?г-орбіталь, що залишилася, розміщена перпендикулярно
до площини трьох ^р2-орбіталей; вона зайнята одним електроном. Якщо р-орбі-
таль одного атома Карбону перекривається з р-орбіталлю іншого карбонового
атома, то відбувається спарювання електронів з утворенням додаткового зв'язку.
Такий зв'язок виникає завдяки латеральному (боковому) перекриванню /?-орбі-
талей, його називають п-зв'язком:
Унаслідок того, що л-зв'язок Карбон-Карбон утворюється завдяки менш
ефективному боковому перекриванню негібридизованих р-орбіталей, він менш
міцний, ніж а-зв'язок. Перекривання р-орбіталей може відбуватися лише в тому
випадку, якщо атоми Карбону і Гідрогену розташовані в одній площині. З
огляду на це етилен є плоскою молекулою:
Отже, "подвійний" зв'язок Карбон-Карбон побудований з міцного а-зв'язку
(-350 кДж/моль) і менш міцного л-зв'язку (-260 кДж/моль). Відстань між
атомами Карбону в етилені менша, ніж в етані, тобто подвійний зв'язок С=С
коротший, ніж зв'язок С-С.
7і-Електрони розташовані далі від ядер атомів і, відповідно, доступніші, ніж
електрони а-зв'язку. Крім того, я-зв'язок менш міцний, ніж а-зв'язок; тому саме
подвійний зв'язок є реакційним центром алкенів, який атакують електро-
нодефіцитні частинки (радикали й електрофіли).
0,154 нм
н
772 Чирва В.Я., Ярмолкж СМ., Толкачова Н.В., Земляков О.Є Органічна хімія
8.1.2. Геометрична ізомерія
У ряду алкенів кількість ізомерів для кожного члена, переходячи до вищих
алкенів, збільшується швидше, ніж для алканів. Річ у тому, що, окрім ізомерії
карбонового скелета, в алкенів можлива також зміна розміщення подвійного
зв'язку, яка, відповідно, може спричиняти геометричну ізомерію. Нижче на
окремих прикладах наведено різні види ізомерії.
Ізомерія карбонового
скелета
Структурна ізомерія
Ізомерія розміщення подвійних
зв 'язків
н3с-с-сн2 СН2=СНСН2СН3
СН3
2-Метилпропен
СН3СН=СНСН3
Бут-1-ен Бут-2-ен
Геометрична ізомерія
Міжкласова
ізомерія
Н2С"~~СН2
н2с-сн2
Циклобутан
н3с^ с-сн3
І
і/ис-Бут-2-ен *
Н3С\ —н
н^ и
сн,
II
транс-Бут-2-e н
Розглянемо детальніше геометричну ізомерію. Під час вивчення гомологів
етилену - бутенів, знайдено, що для них існує не три сполуки з ізомерними
карбоновими скелетами і різним розміщенням подвійного зв'язку, а чотири
алкени з формулою С4Н8: бут-1-ен, 2-метилпропен і 2 ізомери бут-2-ену.
Дві сполуки, що мають карбоновий скелет бут-2-ену, відрізняються за
температурами плавлення й іншими фізичними властивостями, тобто вони є
ізомерами. Відмінність у їхній будові пов'язана з різним просторовим розмі-
щенням метальних груп відносно площини, що проходить через подвійний
зв'язок: в одному випадку вони є по один бік (І), а в іншому - по різні (II).
Можливість існування цих ізомерів як індивідуальних речовин пов'язана з
енергією, що необхідна для повертання навколо С=С-зв'язку. Зв'язок к
утворюється через бокове перекривання р-орбіталей, частини яких лежать над і
під площиною а-зв'язків. Щоб перейти від одного з цих бут-2-енів до іншого,
необхідно фрагмент молекули повернути вздовж зв'язку Карбон-Карбон на
180°, іншими словами потрібно розірвати я-зв'язок, для чого потрібно близько
260 кДж/моль. За кімнатної температури та невеликого нагрівання, наприклад,
перегонки, теплової енергії недостатньо, щоб розірвати л-зв'язок, тому вдається
виділити два ізомерні бут-2-ени.
Оскільки ізомерні бут-2-ени мають однакову хімічну будову і відрізняються
один від одного лише розміщенням атомів у просторі, то є стереоізомерами.
Розділ 8. Алкени
173
Вони, проте, не є дзеркальними відображеннями один одного, а отже, це діасте-
реомери (точніше, я-діастереомери), або геометричні ізомери.
Отже, щоб реалізувалася стереоізомерія, потрібна наявність просторової
неоднорідності: алкени з однаковими замісниками в одного атома Карбону не
мають геометричної ізомерії:
Н\ Н\ Н\ /СН3
\сн3 н-^ х:2н5 ^сн,
Пропен (пропілен) Бут-1-ен 2-Метилпропен (ізобутилен)
■ Конфігурацію алкенів позначають префіксом цис- (від лат. на цьому боці) і
транс- (через), які відображають, що метильні групи розміщені з одного або з
різних боків від площини подвійного зв'язку. Для складніших сполук
використовують ^-систему позначення конфігурації кратного зв'язку. Якщо
два старші замісники в молекулі розташовані з одного боку площини
етиленового зв'язку, то ізомер називають 2, якщо з різних - Е (детальніше див.
розділ 6).
bk.Br НЧГВГ
п - п
сґасн3 н3Сасі
(Е)-1 -Бромо-2-хлоропропен (І)-1 -Бромо-2-хлоропропен
Явище геометричної ізомерії є загальним, воно може бути в будь-якого
класу сполук, що має подвійний зв'язок іншого типу, наприклад, >С=>1-.
8.13. Номенклатура алкенів
• Тривіальна і раціональна номенклатура. В хімічній літературі досить часто
трапляються тривіальні назви олефінів: етилен, пропілен, амілен. Іноді алкени
називають за раціональною номенклатурою як похідні етилену. Дизаміщені
етилени при одному атомі Карбону позначають префіксом несим.- (несимет-
ричний), а при різних атомах - сим.- (симетричний):
(СНз)2С=С(СНз)2 Н2С=С(СН3)2 (СН3)НС=СН(СН3)
Тетраметилетилен Несим.-диметил етилен Сим.-диметилетилен
• Номенклатура ШРАС. За основу назви алкенів беруть найдовший карбо-
новий ланцюг, що має подвійний зв'язок. Ланцюг нумерують так, щоб най-
менший номер отримав атом Карбону з подвійним зв'язком. Якщо варіанти
однакові, то напрям нумерації визначений замісниками, як розглянуто для
алканів. Назву будують аналогічно до назв алканів, замінюючи суфікс -ан на -єн, а
локант (цифра), що стоїть перед суфіксом, означає місце подвійного зв'язку:
774
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
1 2 3 4 5 6 7 8
H3C-CH=C-CHjCHjCHj^H-CH3
н3с-с-сн3 сн3
сн,
З -/я/?ет-Бутил-7-метил окт-2-ен
За основу вибраний найдовший
ланцюг, який має подвійний зв'язок.
Нумерація починається з того боку,
до якого ближче розміщений подвій-
ний зв'язок.
сн3
6 5 \4 З
сн3^нсн<рнсн2сн2сн3
СН3 СН=СН2
|4,5-Диметил-3-пропілгекс-1 -єн
За основу беруть найдовший і
найрозгалу женіш ий ланцюг,
який має подвійний зв'язок.
СН3
ІЗ 2
, 3
6 5 4 ІЗ 2 1
СН3СНСН=С-СНСН3
сн3 сн3
2,3,5-Триметилгекс-3-ен
Подвійний зв'язок розта-
шований симетрично. На-
прям нумерації визначають
мінімальні локанти заміс-
ників (якщо нумерувати
зліва, то 2,3,5 менше, ніж
2,4,5, якщо нумерувати
справа).
■ Ненасичені вуглеводневі замісники називають, додаючи до назви алкену
суфікс -ил (-іл): СН3СН=СН- пропеніл, СНз-СН=СН-СН2- бут-2-еніл (застаріла
тривіальна назва крошил). Для деяких найпростіших алкенів правилами IUP АС
залишено тривіальні назви: СН2=СН-СН2- аліл, СН2=СН- вініл і СН2=(СНз)С-
ізопропеніл.
8.2. Фізичні властивості алкенів
Фізичні властивості алкенів, по суті, такі ж, як і властивості алканів. Вони
нерозчинні у воді, але добре розчиняються в неполярних розчинниках, таких як
бензен, діетиловий етер, хлороформ або лігроїн. їхня густина менша, ніж
густина води. Алкени з розгалуженим ланцюгом киплять за нижчої температури,
ніж лінійні сполуки з тією ж кількістю карбонових атомів.
Більшість алкенів неполярна, лише деякі олефіни внаслідок певної
геометрії молекули мають невеликі дипольні моменти:
ЬК СН3 с2н5
ц = 0,350 ц = 0,37О
Зв'язок алкільної групи з атомом Карбону подвійного зв'язку мало-
полярний, і його напрям показано на приведених вище формулах: алкільна група
подає електрони до атома Карбону подвійного зв'язку.
Зокрема, і/г/с-бут-2-ен з двома метальними групами по один бік подвійного
зв'язку і двома атомами Гідрогену по інший повинен мати невеликий дипольний
момент. У т/?анс-бут-2-ені з кожного боку подвійного зв'язку в молекулі є
метальна група й атом Гідрогену. В цьому випадку дипольний момент дорівнює
нулю, оскільки індукційні ефекти метальних груп напрямлені назустріч один
Розділ 8. Алкени
175
одному. Хоча дипольні моменти не були безпосередньо виміряні, невелика
різниця в полярності відображається в вищій температурі кипіння і/ис-ізомеру:
З
цис-
транс-
цис-
транс-
цис-
транс-
1,85
0
1,35
0
0,75
0
-80
-50
-53
-6
-14
-72
60
48
110
108
188
19
іуис-Ізомер, т. кип. 4 °С транс-ізомер ц=0; т. кип. 1 °С
Оскільки і/мс-ізомер менш симетричний, то його упаковка в кристалічній
ґратці менш щільна, що зумовлює, як звичайно, нижчу температуру кипіння
порівняно з тирш/с-ізомером.
Відмінності в полярності і, відповідно, різниця в температурах плавлення і
кипіння більші для алкенів із замісниками, електронегативність яких суттєво
відрізняється від електронегативності атома Карбону. Це можна показати на
таких прикладах:
Н Н Н СІ Н Н Н Вг Н Н Н І
с=с с=с с=с с=с с=с с=с
сґ Ь сґ н в/ вг в/ н і' чі і чн
Конфігурація
т. пл. (°С)
т. кип. (°С)
Зв'язок між конфігурацією і температурами кипіння та плавлення є емпі-
ричним правилом, що має багато винятків. Водночас вимірювання дипольних
моментів часто дає змогу однозначно приписати цис- або транс-буцрву певному
ізомеру.
8.2.1. Спектроскопія алкенів
Алкени легко виявити за допомогою спектральних методів.
• В УФ-спекпгрох алкени виявляють смугу поглинання, що відповідає я—^-пе-
реходу з максимумом при 180-205 нм.
• В ІЧ-спектрах валентні коливання ус=с виявлені як смуга 1640-1680 см"1, а
деформаційні коливання 5с-н - У діапазоні 790-990 см"1. Крім того, цис- і транс-
ізомерам відповідають деформаційні коливання: 650-750 см"1 для цис- і 960-
980 см"1 для тряис-конфігурації.
• У мас-спектрах алкенів визначають характерні інтенсивні піки моле-
кулярних іонів, а також групу піків з т/г 41(СзН5), 55; 69; 83 і т.д., що
утворюються внаслідок алільного розщеплення. На жаль, подвійний зв'язок під
електронним ударом мігрує, через це мас-спектрометрію не можна використати
для визначення місця подвійного зв'язку в алкенах.
176
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
• У спектрах ПМР протони подвійного зв'язку мають хімічний зсув близько
4,5-6,5 м. ч., тобто вони значно зсунуті в слабше поле, ніж протони насичених
атомів Карбону. Приклади значень хімічних зсувів для деяких олефінів:
% На ґ^ґН
н2с=сн2 р=с [ І
СН3 Н
Н/
\ Н
.с=сч
Н Н
/с=с\
5 5,33 м. ч. 5 (м. ч.) 5,70 (а), 4,88 (Ь), 4,96 (с) 5 5,57 м.ч.
Івіи 12-18 Гц 6-12 Гц
Геометричні ізомери суттєво відрізняються за віцинальними константами
спін-спінової взаємодії, що дає змогу їх інтерпретувати.
8.3. Промислові методи одержання алкенів
У промислових масштабах алкени одержують, головно, парофазним кре-
кінгом нафти. Нижчі алкени виділяють у чистому вигляді фракційною
перегонкою, тому вони є доступною сировиною для синтезу великої кількості
важливих аліфатичних сполук, а також полімерів, наприклад, поліетилену,
поліпропілену, поліхлорвінілу.
❖ Дегідрування (ілкапів. Каталізатором цього процесу, як звичайно, є хром
(III) оксид, виготовлений спеціальним способом. Реакція відбувається за
температури 300-450 °С:
СН3СН2СН2СН3
Бутан
Сг203/Ре203
СН2=СНСН2СН3 + СН3СН=СНСН3+ н2
Бут-1-єн Бут-2-ен
■ Етилен одержують з етану за реакцією: ГНХН. НіхР0М0В| ГН^СН, + Н>
33 нитка, 800°С 2 2 2.
■ Етилен і пропілен є головними алкенами, що утворюються під час
високотемпературного крекінгу вищих алканів:
7ОО-8О0>С,
н-С6Н14
Гексан
ГСН4 + СН2=СН2 + СН3-СН=СН2 + інші продукти
Метан, 15% Етилен, 40% Пропен, 20% 25%
Серед мономерів ці алкени є лідерами. Достатньо сказати, що лише в США
(табл. 8.1) їхнє виробництво становить десятки мільйонів тонн.
Об'єм промислового виробництва найважливіших мономерів
у США в 90-ті роки XX ст.
Таблиця 8.1
Сполука
Формула
Обсяг виробництва, млн т
Етилен
СН2=СН2
17,5
Пропілен
СН3—СН==СН2
10,2
Стирен
СбЬІ5—СН=СН2
4,1
Вінілхлорид
СН2=СНС1
4,9
Бутан-1,3-дієн
сн,=сн-сн=сн2
1,6
Акрилонітрил
сн2=сн-сы
1,4
Вінілацетат
сн,=снососн3
1,3
Розділ 8. Алкени / 77
1 Циглер K. (Zeigler К.) (1998-1973) - німецький хімік. Головні праці в галузі органічного синтезу і
хімії полімерів. Нобелівська премія 1963 р. спільно з Д. Натта.
2 Натта Дж. (Natta G.) (1903-1973) - італійський хімік. Головні праці в галузі полімерів.
❖ Синтез шік-1-енів олігомеризацією нижчих гомологів. Алкени з при-
кінцевим подвійним зв'язком і парною кількістю атомів Карбону, які вико-
ристовують у промислових масштабах для синтезу детергентів, одержують
олігомеризацією етилену або інших нижчих алкенів за одним з варіантів методу
Циглера^Натта2.
Реакції полімеризації можна використати для одержання порівняно низь-
комолекулярних продуктів, якщо ріст ланцюга вдається припиняти на ранніх
стадіях. Одержані речовини називають олігомерамщ а такий процес можна
назвати олігомеризацією. Таким шляхом, зокрема, можна синтезувати вищі алк-
1-ени, а також а-розгалужені алкени, що їх застосовують як мономери для
стереоспецифічної полімеризації. Олігомеризація алкенів відбувається за наяв-
ності каталізатора триетилалюмінію:
А1(С2Н5)3 + З СН2=СН-СН3 Аі/сН^СН-С^
Пропен \ ^ /з
Реакція подібна до полімеризації Циглера-Натта (див. розділ 10), проте в
цьому випадку після акту приєднання активно відбувається передавання
ланцюга на мономер, і ріст полімерного ланцюга припиняється:
Аі/сгІ^СН-СзНзизСН^СНСНз—- А1/с^-СН^Нз^ + ЩС^-С2Я5
\ СН3 '3 * '3 сн3
2-Метилбут-2-ен
Передавання ланцюга, вочевидь, відбувається внаслідок перенесення гідрид-
іона від р-положення алкільного ланцюга до мономеру:
К'-^-СН-АІІ^ К"-((:=СН2 + К"'СН2-СН-А1К2
У цьому разі зв'язок між алкільним фрагментом і атомом Алюмінію
розривається. У кінцевого атома Карбону алкільного ланцюга виникає
подвійний зв'язок і утворюється новий зв'язок між металом і мономером.
Рушійною силою такого перерозподілу зв'язків можуть бути стеричні пере-
шкоди, створювані об'ємним і(або) Р-розгалуженим алкільним фрагментом.
178
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Так само, зокрема, одержують пент-1-ен, гекс-1-ен, 4-метилпент-1-ен, 2-ме-
тилбут-1-ен. Останню сполуку шляхом дегідрування можна перетворити в
ізопрен — мономер, який широко застосовують у виробництві каучуку.
8.4. Лабораторні методи одержання алкенів
Уведення подвійного зв'язку в молекулу, що має один простий зв'язок
Карбон-Карбон, найчастіше відбувається як елімінування атомів або груп від
сусідніх атомів Карбону:
—С—С —С=С— +
Г Г Елімінування І Г
У т
У процесі крекінгу, наприклад, відщеплюються два атоми Гідрогену:
І І
Нагрівання
—с-с — — <р=<р- + н2
н н
Реакції елімінування, розглянуті нижче, можна використовувати не лише
для синтезу простих алкенів. Вони, що важливіше, подають загальні шляхи
введення подвійного зв'язку в молекули будь-якого типу.
8.4.1. Аегідрогалогенування алкілгалогенідів
Одним з найпоширеніших і добре вивчених процесів елімінування є реакція
дегідрогсиїогенування алкілгалогенідів, що відбувається під дією основ: спир-
тових розчинів лугів, алкоголятів, амідів лужних металів або амінів (хінолін,
піридин, анілін):
І І в-
—9-9 — —*~ —9=9- + х- + НВ
Н X
В~ = основа
Наприклад: СН3СН2СН2СН2С1 -г£тЬ СН3СН2СН=СН2 + КСІ +. Н,0
і ^ / С2Н5ОН г і
1-Хлоробутан 2 * Бут-1-ен
У лабораторній практиці найчастіше алкен одержують нагріванням алкілга-
логеніду зі спиртовим розчином їдкого калі. В цьому випадку спочатку
відбувається зворотна реакція з утворенням сильнішої основи — алкоголят-аніона:
К+ + но- + С2Н5ОН в С2Н50" + К+ + н2о
Обробка вторинних і третинних галогеноалканів спиртовим розчином лугу
дає алкени з хорошим виходом:
Розділ 8. Алкени
Н3С СН3 _с2н5он,
І
2-Іодопропан
-КІ
СН3-СН—СН2
Пропен, 94%
❖ Механізми реакцій елімінування. Найчастіше елімінування в розчинах
відбувається за йонними механізмами Е1 і Е2 (Е - від англ. elimination -
елімінування, 1 - мономолекулярне, 2 - бімолекулярне).
■ Реакціям, що відбуваються за механізмом Е1, потрібний іонізуючий роз-
чинник. Стадія, що лімітує (визначає) швидкість реакції, є утворення інтерме-
діату - карбокатіону (рис. 8.1), який, відщеплюючи протон під дією основи
С2Н5О , швидко переходить у стабільний стан (олефін):
(рН^Н с2н5о
(СН3)3СС1 Is° (СН3)3С++ СГ СН3-С+ Сг^С^Сг^+СДОН
2-Метил-2-хлоропропан СН3 СН3
- 2-Метил пропен
Швидкість реакції V = k [RHal], тобто вона залежить від концентрації лише
галогенопохідного, процес мономолекулярний.
■ У реакціях відщеплення, що відбуваються за бімолекулярним механізмом
Е29 протон і галоген відриваються одночасно через активований комплекс
(рис. 8.1), у якому основа утворює зв'язок завдяки електронній парі з протоном
настільки, наскільки в якій відщеплюється галогенід-іон:
едо-4Н-(^-ш-а;--[едо-•• н-с^ш-& +вг
СНз СНз
2-Бромопропан Пропен
Швидкість реакції V- k [RHal][C2H50""], тобто залежить від концентрації як
галогенопохідного, так і основи, процес бімолекулярний.
Активність основ у реакціях, що відбуваються за механізмом Е2;
змінюється в тому ж напрямі, що і їхня сила: NH2~ > С2Н5О" > ОН" > RCOO".
(СН3)3Є
(СН3)3С-СІ / +
(СНз^ССНз-Н'-В
(СН3)2С=СН2
+ НВ
(СН3)3С-С1
[В-Н-СН2СН2-С1]-
СН3СН2С1
+в-
СН2==СН2
+ НВ + С1-
Координата реакції Координата реакції
Рис. 8.1. Енергетичні діаграми реакцій, що відбуваються за механізмом Е1 (а) і Е2 (б)
180
Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
■ Якщо є можливість відщеплення гідроген галогеніду в двох напрямах,
згідно з правилом Зайцева, переважно утворюється розгалуженіший (термо-
динамічно стійкіший) алкен (рис. 8.2). Легкість утворення алкенів зменшується
в ряду ЯгСКЖг > К2С=СНЯ > К2С=СН2 > КСН=СНЯ > КСН=СН2. У такій же
послідовності зменшується стійкість алкенів. Іншими словами, чим стійкіший
алкен, тим легше він утворюється в реакції дегідрогалогенування.
> Правило Зайцева - якщо дегідрогалогенувати галогеноалкани, то протон
відщеплюється переважно від найменш гідрогенізованого атома Карбону з
утворенням найбільш заміщеного алкену:
Координата реакції Координата реакції
Рис. 8.2. Енергетична діаграма реакції елімінування з утворенням стійкішого алкену
Вг /т|ам>ГЬнт-2-€н,41% 2-Егоксигшган,^гЛ
2-Бромопенган і^Пенг-2-ен, 14% ГЬтг-1-ен, 25% 20%
■ Як випливає з наведеного вище прикладу, реакція відбувається з певною не
лише регіоселективністю9 а й з стереоселективністю. Стереоселективність реакції
пов'язана з більшою стійкістю трш/с-ізомеру. Поряд з алкенами утворюється і етер
- продукт нуклеофільного заміщення (детальніше див. розділ 12).
Легкість дегідрогалогенування алкіл галогені дів спадає в порядку: третинні
> вторинні > первинні. У випадку первинних галогеноалканів, особливо якщо
вони не мають Р-розгалуження, домінуючим продуктом реакції буде саме етер.
Отже, головним напрямом реакції є не процес дегідрогалогенування, а
нуклеофільне заміщення:
н-С5НпВг_^^ СН3СН2СН2СН=СН2 + н-С5НпОС2Н5
1-Бромопентан -КаВг Пент-1-ен, 12% Пентилетиловий етер, 88%
Проте застосування дуже сильної основи лу?е/я-бутоксид-аніона дає алкени
з добрими виходами навіть у випадку первинних галогеноалканів з лінійним
ланцюгом:
Розділ 8. Алкени
181
"•С"Н"Вг-Д «-СІ6Н33СН=СН2 + н-СІ8Н37ОС(СН3)3
1-Бромооктадекан -КВг Октадец-1-ен, 88% т/?его-Бугилоктадециловий
етер, 12%
У конкурентних реакціях дегідрогалогенування і нуклеофільного заміщення
галогену елімінуванню сприяє висока основність реагенту і підвищена температура.
На співвідношення між продуктами елімінування суттєво впливає природа
галогену і сила основи. Наприклад, якщо дегідрогалогенувати 2-галогеногексан,
то вихід гекс-1-ену збільшується в ряду Б > СІ > Вг > І. Заміщення натрій
метилату на сильнішу основу - натрій трет-бутипат - також приводить до
збільшення виходу алкену (табл. 8.2).
Таблиця 8.2
Вміст гекс-1-ену, якщо дегідрогалогенувати 2-галогеногексан при 100 °С, %
Галоген
І
Вг
СІ
Р
СН3(Жа/СН3ОН
19
28
33
70
(СНз)зСОЫа/(СН3)3СОН
69
80
88
97
Як звичайно, реакція Е2 є стереоспецифічним аниш-елімінуванням, оскільки
аняш-копланарний активований комплекс енергетично вигідніший, ніж син-
копланарнйй, що є в малостійкій затіненій конформації:
С2Н50
ш/яш-Копланарний і син-копланарний активовані комплекси
Зазначимо, що, на відміну від процесу утворення
вільних радикалів, розрив зв'язків С-Н і С-Х відбу-
вається гетеролітично. Звідки береться енергія для Я—Оч
розриву цих зв'язків? По-перше, утворюється зв'язок між
протоном і основою. Потім унаслідок утворення 7с-
зв'язку виділяється -260 кДж/моль енергії. І, нарешті,
виділяється енергія в процесі сольватації йонів галогену.
Кожний іон-дипольний зв'язок сам по собі слабкий,
проте якщо утворюється велика кількість таких зв'язків,
то виділяється значна кількість енергії.
Можливе і термічне відщеплення (піроліз) гідрогенгалогеніду:
к-сн2сн2сі — я-сн=сн2 + неї
* Символом А позначено піроліз.
н
н~н
\
р
182
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
8.4.2. Аегідратація спиртів
Спирти перетворюються в алкени в процесі реакції дегідратації (від-
щеплення молекули води), що відбувається за наявності кислоти під час
нагрівання:
'—с-6— -с=с- + Н20
н он
У лабораторних умовах реакцію проводять, використовуючи концентровані
кислоти (Н2804, Н3Р04), що забирають воду. У цьому разі спочатку утворюється
естер мінеральної кислоти, який потім розпадається на алкен і кислоту:
СН3СН2ОН + Н2804 СН3СН20803Н + Н20
СН3СН20803Н ^^Я2С=СЯ2 + Н2804
Зниження температури реакції спричиняє міжмолекулярну дегідратацію спиртів
з утворенням етерів.
З'ясовано, що легкість дегідратації спиртів збільшується в порядку:
первинні < вторинні < третинні. Деякі третинні спирти дегідратувати дуже
легко: їх можна перегнати, лише якщо вжити заходів для ретельного захисту
спиртів від парів кислот у повітрі, які є в звичайній лабораторії. Нижче наведені
умови дегідратації спиртів залежно від їхньої будови:
СН3СН2ОН 95% н^°^ СН2=СН2
З 2 17П°Р ^ ^
Етанол игГ Етилен
-н2и
сн3сн2сн2сн2он 75^0Н^°» СН3СН=СНСН3 + СН3СН2СН=СН2
Бутан-1-ол -Н20 Буг-2-ен Бут-1-ен
(основний продукт)
60% Н2804
сн3сн2<рнсн3 іоо^> СН3СН=СНСН3 + СН3СН2СН=СН2
ОН .Н20 Бут-2-ен,80% Бут-1-ен,20%
Бутан-2-ол
?Н3 20%Н,8О4 ?Н3
нх-с-сн, нх-с=сн2
Лтт 85-90 °С 3 2
ОН .н2о
2-Метил-2-пропанол 2-Метилпропен
Реакції відщеплення води, як і відщеплення гідрогенгалогенідів, можуть
відбуватися за механізмами Е1 і Е2 залежно від будови спирту й умов реакції.
❖ Механізм реакції Е1. Реакцію дегідратації спиртів за механізмом Е1 за
наявності кислот можна описати так:
Розділ 8. Алкени
183
он н-6-н
н3с-сн-сн2сн3 + н2зо4=і=!= н3с-сн-сн2сн3+ нбо;
+ Бутан-2-ол •
н-о-н +
НзС-сн-сн2сн3^^2. н3с-сн-сн2-сн3
н3с-сн^сн-сн3+нзо; н3с-сн=сн-сн3+н2зо4
Н >^_^у^ Бут-2-ен
Зазвичай вода від спиртів відщеплюється за правилом Зайцева. Це
пояснюють тим, що реакція елімінування, яка відбувається за механізмом Е1,
проходить через утворення карбокатіонів, стійкість яких зменшується так:
третинний > вторинний > первинний > СН3, що пов'язано з компенсацією
позитивного заряду внаслідок зміщення електронної густини алкільними
групами, які мають +І-ефект.
Легкість утворення карбокатіонів зменшується в порядку: третинний >
вторинний > первинний > метил. Чим стійкіший карбокатіон і чим легше він
утворюється, тим легше відбувається дегідратація. Конкурентними процесами в
цьому випадку є реакції нуклеофільного заміщення 8>Д з утворенням етерів і
перегрупування карбокатіону.
Дуже часто дегідратація спиртів дає алкени, утворення яких не
узгоджується із запропонованим механізмом. Подвійний зв'язок з'являється в
несподіваних місцях; у деяких випадках змінюється навіть карбоновий скелет:
+
сн3сн2сн2сн2он -2-— сн3сн=снсн3 (1)
+
сн3сн2-сн-сн2он сн3сн=с-сн3 (2)
сн3 сн3
2-Метилбутан-2-ол 2-Метилбут-2-ен
9Нз н+ 9Н3 сн3
н3с-с—сн-сн3 —- н3с-с=с-сн3 +Н3С-СН-(р=СН2 (3)
сн3 он сн3 сн3
3,3-Диметилбутан-2-ол 2,3-Диметилбут-2-ен 2,3-Диметилбут-1-ен
(основний продукт)
Переважне утворення бут-2-ену в реакції (1) пояснюють тим, що «-бути-
ловий спирт утворює первинний н-бутильний карбокатіон, який перегрупо-
вується в стійкіший в/яср-бутильний карбокатіон. Стійкіший карбокатіон втра-
чає протон і дає переважно бут-2-ен:
СН3СН2СН2СН2ОН2 СН3СН2СН2СН2 + н2о
184
Чирва В.Я., Ярмолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
СН3СН2СН2СН2
СН3СН2СНСН3
СН3СН2СНСН3
СН3СН=СНСН3
СН3СН2СН2СН2ОН2
СН3СгІ2СгІ2СгІ2+
СН3СгІ2СНСН3
СН^Сг^СН-СН2
СН3СН=СНСН3
СН3СН2СН2СН2ОН + н+
Координата реакції
Рис. 8.3. Енергетична діаграма реакції дегідратації, що відбувається через утворення
стійкішого карбокатіону
Аналогічно, у реакції (2) 2-метилбутильний карбокатіон перегруповується в
карбокатіон з позитивним зарядом на третинному атомі Карбону:
СН3СгІ2СНСН2
6н,
СН3СН2ССН3
сн,
Вторинний карбокатіон, що утворюється з 3,3-диметилбутан-2-олу в реакції
(3), також перетворюється у відповідний третинний карбокатіон:
:н3
сн,-с-снсн3
сн3
сн3-с-снсн3
СН3
У кожному випадку перегрупування відбувається в напрямі утворення
стійкішого карбокатіону: вторинного з первинного, третинного з первинного або
вторинного внаслідок гідридного або алкільного зсуву - зміщується атом Гідро-
гену або алкільна група разом з парою електронів:
і
-с-с-
н +
-с-с-
н' .
1,2-Гідридний зсув
І І
-сгсг
н
Розділ 8. Алкени
185
-С-
І І
с-
+
"II"
-с-с-
V
1,2-Алкільний зсув
Якщо в процесі 1,2-зсуву гідрид-іона або алкільної групи може утворю-
ватися стійкіший карбокатіон, то відбувається таке перегрупування:
СН3СГІ2СНСН2
сн,
СНЗСН2СНСН2
+ н
сн3СгІ2І~"СгІ2
сн3
сн3і-снсн.
сн3
сн3сн2с-сн2
+ н
сн3
+ сн3
❖ Механізм реакції Е2. Якщо реакція дегідратації відбувається за меха-
нізмом Е2, то утворення карбокатіонів неможливе, і перегрупування не
простежують:
сн3сн2сн2он+н2804
Пропан-1-ол
сн3сн2сн2рн + нбо;
н
нво; + н3с-сн-сн2-о-н ==
і н
н3с-сн=СН2 + н2зо4 + н2о
Пропен
9-5
н
і
5+
но-я-о- -н-с—сн2 - 9"н
ii і тт
сн,
н
Активований комплекс
❖ Парофазна дегідратація. Значно більше значення, ніж кислотна дегід-
ратація в рідкій фазі, має парофазна дегідратація спиртів над оксидами алюмі-
нію або торію. В цьому випадку конкурентні процеси значно пригнічені, а якщо
використовувати частково отруєні каталізатори, наприклад, алюміній оксид,
піридин, то вони зовсім не відбуваються.
У промисловості для дегідратації спиртів використовують оксиди алюмінію,
торію, вольфраму, а саму реакцію проводять за температури близько 300 °С.
Вихід алкену в цьому випадку наближається до теоретичного:
300 °С
сн3сн2сн2он-
А1203
-н3с-сн=сн2
Пропен
186
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
❖ Дегідратація етерів
■ Відщеплення молекули спирту від етерів з утворенням алкенів можна
також реалізувати, як і дегідратацію спиртів, дією сильних концентрованих
кислот:
„ ~ $Нз тт H2S04,/ + _+
Н3С-С-ОС2Н5 Н3С-С-СН3 - н2с=с-сн3+ Н+
СН3 СНз СНз
2-Етокси-2-метилпропан 2-Метилпропен
Розклад етерів до алкенів тим легший, чим розгалуженіший алкіл є в складі
такого етеру (реакція типу El).
■ За аналогічним механізмом відбувається реакція і для естерів, проте в
м'якших умовах порівняно з етерами:
9Нз ,0 H2so4 9Нз
нх-с-ос н3с-с+ НХ=С-СН3 + Н
3 і 4 -СН СООН 3 1 2 і З
СН3 СН3 .HSo4- СН3 СН3
/ире/и-Бутилацетат 2-Метилпропен
8.4.3. Розщеплення амінів. Правило Гофмана1
❖ Розщеплення амінів. Зв'язок C-N в амінах менш полярний порівняно з
С-0, тому відщеплення амоніаку або амінів відбувається за гомолітичним
механізмом і в разі вищих температур, ніж дегідратація спиртів:
H3C-CH2CHi-N(CH3)2 —^ СН3СН=СН2 + NH(CH3)2
НН-Диметилпропіламін Пропен
❖ Розщеплення амонійних солей. Значно легше відбувається термічний
розклад четвертинних амонієвих основ за Гофманом:
L^2n5i\^n3;3jOn n2l^i^n2 -r п2^ т iN(^n3)3
Триметил етил амоній Етилен
гідроксид
Зазначимо, що в складі амінів є простіші алкіли, а більш високомолекулярні
залишаються в алкенах. Напрям відщеплення визначений правилом Гофмана.
> Правило Гофмана: якщо розщеплюються алкіламонійні солі, то пере-
важно утворюються менш заміщені олефіни:
1 Гофман A. (Hoffmann А.) (1818-1892) - німецький хімік-органік. Головні праці в галузі
органічних сполук, що містять Нітроген. Запропонував способи одержання амінів, зокрема, з
алкілгалогенідів (реакція Гофмана), розкладом амідів (перегрупування Гофмана).
Розділ 5. Алкени
187
У**
[С2Н5~СН-Н(СН3)3]+0Н ^Н5С—СН^СК, + Н3С-СН=:СН-СНз + М(СН3)3 + Н20
втор-Бугалтриметшіамоній Буг-1-єн, 95% Буг-2-ен, 5%
гідроксид
Як видно з рівняння реакції, амонійні солі розщеплюються протилежно до
відщеплення гідроген галогенідів від алкілгалогенідів і води від спиртів, які
звичайно відбуваються за правилом Зайцева. Таку поведінку амонійних солей
можна пояснити тим, що в Р-положенні до групи, яка відщеплюється, виникає
частково негативний заряд і вигідніше, щоб в активованому комплексі алкільні
групи не збільшували його величини:
Н6+ . Н6+
:б- :5і-
НзС-Ь^С-СН—СН2 Н3С—СН—СН-СН3 8'-> 6-
Стійкіший активований комплекс
Цю реакцію застосовують для визначення будови гетероциклічних сполук.
8.4.4. Інші лабораторні методи, що ґрунтуються на
реакціях елімінування
❖ Дегалогенуеапня віцинальних дигалогенідів. Слово "віцинальний" озна-
чає, що замісники містяться біля сусідніх атомів Карбону. Раніше для
дегалогенування використовували цинковий пил у водно-спиртовому сере-
довищі:
ii 1 і
-с-с— + —- —с=с — + гпх
і і
X X
2
Наприклад: СН3СНВг СНВгСН3"^^СН3СН-СНСН3
2,3-Дибромобуган Бут-2-ен
Проте цинк хлорид, що утворюється в реакції, спричиняє ізомеризацію
алкенів. Ефективними замінниками цинку в цій реакції є розчин натрій іодиду в
метанолі або ацетоні, солі хрому (II), літій алюмогідрид.
Дегалогенування віцинальних дигалогенідів часто обмежене тим, що їх
зазвичай одержують з алкенів. Проте іноді зручно використати перетворення
алкену в дигалогенід, щоб провести деякі реакції з іншою частиною молекули і
потім регенерувати алкен дією цинку. Таку операцію називають тимчасовий
захист подвійного зв 'язку.
188 Чирва В.Я., Ярллолюк СМ., Толкачова H.В., Земляков О.Є. Органічна хімія
❖ Реакції одночасного декарбоксилювання і дегідратації. Елімінування р-
гідроксикарбонових кислот часто використовують як регіоселективний метод
синтезу олефінів, особливо алкенів із кінцевим подвійним зв'язком, оскільки
вихідні сполуки достатньо доступні за реакцією Реформатського1:
ОН
Си, 170 С в
Хінолін
СН2СООН
1 -Гідроксициклогексил-
оцтова кислота
сн2 + со2 + н2о
Метиленциклогексан
Спорідненою реакцією є процес декарбоксилювання-елімінування похідних
Р-галогенокарбонових кислот.
❖ Окиснювальне декарбоксилювання. Олефіни можна одержати окисню-
вальним декарбоксилюванням карбонових кислот під дією плюмбум тетрааце-
тату або 1,2-дикарбонових кислот, нагріваючи ці кислоти за наявності плюмбум
тетраацетату або купрум(І) оксиду в хіноліні:
РЬ(ОСОСН3)4
СН3(СН2)3СООН >
Cu(OCOCH3)2
масляна кислота
[О]
(СН3)2С-рСНз)2
НООС соон
Тетраметиля нтарна
кислота
СН3СН2СН=СН2 + со2
Бут-1-ен
сн3\с=с/сн3
-2СО,
СНГ
-сн,
2,3-Диметилбут-2-ен
❖ ПіролЬ естерів карбонових кислот. Реакція відбувається під час нагрі-
вання естерів карбонових кислот до 500 °С. У цьому випадку виникає синхронне
г/ис-елімінування через циклічний активований комплекс:
R
СНдСН
h
R
R*
çh2-çh
о.
V.
h
с—О
R
——r ch2=ch-r:
-rcooh 2
❖ Реакція Чугаєва2. Дегідратацію спиртів до олефінів виконують терміч-
ним розпадом їхніх ксантогенатів, які одержують за такими реакціями:
1 Реформатський С. (1864—1937) - український хімік. Головні праці в галузі хімії природних
сполук і металоорганічного синтезу.
2 Чугаєв Л. (1873-1922) - російський хімік. Головні дослідження в хімії комплексних сполук.
Визначив підвищену стійкість комплексів, які утворюють п'яти- і шестичленні цикли (правило
Чугаєва), відкрив якісну реакцію на йони Ніколу (реактив Чугаєва), метод кількісного визначення
рухливих атомів Гідрогену (метод Чугуєва-Церевітінова).
Розділ 8. Алкени
189
R-CH2-CH2-OH + NaOH + CS2 ^ R-CH2-CH2-OCSSNa + H20
R-CH2-CH2-OCSSNa + CH3I ► R-CH2-CH2-OCSSCH3 + Nal
R-CH2-CH2-OCSSCH3 —^ R-CH=CH2 + COS + CH3SH
Легкість дегідратації спиртів зменшується в порядку третинний > вторин-
ний > первинний. Розклад ксантогенатів a-гліколів приводить до гомологів
ацетилену. Перевагою методу є відсутність ізомеризації карбонового скелета і
міграції подвійного зв'язку.
8 А.5. Реакції відновлення
❖ Гідрування. Залежно від умов реакції можливий перебіг двох процесів,
коли утворюються, відповідно, цис- і /иранс-ізомери. Якщо для алкінів як
відновник використовують розчини лужних металів у рідкому амоніаку (від-
новлення за Берчом\ то стереоселективно одержують яіракс-алкени (механізм
реакції див. у розділі 9).
R\ H2/Pd ^ ^ Na,Li / NH3 R\
TT JC=C. —1 RC=CR ^ ^C=C
H^ XH W \R
і/мс-Ізомер mpawc-Ізомер
Вичерпне гідрування ацетиленових вуглеводнів над платиною або паладієм
дає алкани. Неповне гідрування до алкенів можливе, якщо використовувати
каталізатор зі зменшеною активністю, наприклад, каталізатор Ліндлара -
паладій на кальцій карбонаті, модифікований солями Плюмбуму або паладій на
барій сульфаті, пасивований хіноліном. Таке відновлення веде до ^«с-ізомерів.
❖ Відновлення боранами. Останнім часом для синтезу олефінів з
гомологів ацетилену використовують діалкілгідроборати з об'ємними вугле-
водневими радикалами. Цей метод дає змогу одержувати тяранс-алкени зі
значною стереоселективністю:
RBH /Н Ь.ОН- С4Н'\ /Н ЄН, ЄН,
С4Н9С=СН 2 . /С=С\ __/С=Сч r с '
r=—С-СН
к Ж н Я
❖ Відновлення вінілгалогенідів. Іноді з препаративною метою вико-
ристовують відновлення інших похідних олефінів. Таким способом, наприклад,
відновлюють вінілгалогеніди натрієм у рідкому амоніаку:
1 Берн A. (Birch А.) (1915-1995) - австралійський хімік. Головні праці присвячені ненасиченим
сполукам і природним речовинам.
190 Чирва В.Я., Ярмолюк CM., Толкачова Н.В., Земляков О.Є. Органічна хімія
С2Н5ч /С2Н5 N. С2Н5ч ,С2Н5
с=с > С=С
' 4 VITT / 4
Н СІ NH3 н н
З-Хлорогекс-З-ен і/ис-Гекс-3-ен
8.4.6. Реакції конденсації
❖ Алкілування вінілхлоридів. Велике значення для синтезу алкенів мають
реакції конденсації, що аналогічні методу Вюрца і приводять до утворення
нового карбонового скелету:
Н3Сч /СН3 LiH С2Н5ч /С2Н5 С7Н15ВГ Н3Сч /СН3
С=С ^ С=С . С=С
Н СІ Н Li Н С7Н15
2-Хлоробут-2-ен З-Метилдец-2-ен
❖ Реакція Віттіга1. Одним із сучасних методів утворення С=С-зв'язку є
реакція Віттіга. Вона дає змогу перетворювати карбонільні сполуки в олефіни:
R-CH=0 + Н2С=Р(С6Н5)з R-CH=CH2 + 0=Р(С6Н5)3
Реагентом є алкіліденфосфоран (ріід), що утворюється в реакційній суміші
внаслідок дії на фосфонієву сіль - продукт алкілування тризаміщеного фосфіну
(зазвичай трифенілфосфіну) - сильної основи, наприклад, бутиллітію. Передба-
чуваний механізм реакції такий:
CH3Br + C4H9Li і- + — і
(С6Н5)3Р 3-+ (С6Н5)3Р-СН3 [(C6H5)3P=CH2Ä (C6H5)3P-CH2j
Вґ~ їлід
RCH=0
о
/ \
R-CH /(С6Н5)3 ^ R—СН Р(С6Я5)3
сн2 сн2
ЧС6н5)3р=о
R-CH=CH2
Перевага цього методу над іншими полягає в тому, що з'являється можли-
вість синтезу алкену з чітко визначеним розміщенням подвійного зв'язку в моле-
кулі без утворення інших ізомерних алкенів.
❖ Заміна карбонільного атома Оксигену на метиленову групу. Дією
метиленгалогеніду і магнію на оксосполуки можна одержати олефін відповідної
будови:
1 Віттіг Г. С) (1897-1987) - німецький хімік. Головні праці присвячені синтезу складних
органічних сполук. Нобелівська премія 1979 р.
Розділ 8. Алкени
191
СН2Вг2 + С=0 +2 Mg
Я'
ВгМ§СН2-С-ОЛ^г
Я1
СН2=С + (М§Вг)20
к
8.5. Хімічні властивості алкенів
Подвійний зв'язок Карбон-Карбон визначає особливості будови алкенів та
їхні властивості. Тому подвійний зв'язок можна розглядати як функціональну
групу алкенів. Унаслідок високої поляризованості і порівняно низької енергії
утворення я-зв'язку ці сполуки виявляють ненасиченість, легко вступаючи в
реакції приєднання з розривом я-зв'язку й утворюючи насичені продукти. Отже,
типовими реакціями подвійного зв'язку будуть реакції, у яких я-зв'язок
розривається і замість нього утворюються два міцні о-зв'язки. Реакції, внаслідок
яких відбувається з'єднання двох молекул з утворенням однієї молекули нової
речовини, називають реакціямий приєднання:
Як відомо, я-електрони менше беруть участь у зв'язуванні двох ядер атомів
Карбону, ніж електрони, що утворюють о-зв'язок. Завдяки цьому їх самих ядра
атомів Карбону утримують менш міцно. І, як наслідок, ці електрони особливо
доступні для реагентів із браком електронів. У багатьох реакціях подвійний зв'язок
Карбон-Карбон є донором електронів і поводиться як основа Льюїса. Він реагує зі
сполуками, яким не вистачає електронів, тобто з кислотами Льюїса. Ці кислі
реагенти, що мають вакантну орбіталь, називають електрофілами (люблять елект-
рони), тому типовими реакціями алкенів є реакції електрофільного приєднання (АЕ).
Є реагенти з браком електронів іншого типу - це вільні радикали, що мають
неспарений електрон, і карбени, у яких атом Карбону має два неспарені
електрони. Ці агенти атакують надлишкову електронну густину подвійного
зв'язку, вступаючи в реакції вільнорадикального приєднання (Аи) і реакції
циклоприєднання.
Більшість алкенів має не лише подвійний зв'язок Карбон-Карбон, а й алкільні
групи. Тому, крім реакцій приєднання, що властиві подвійному зв'язку С=С, алкени
можуть вступати в реакції вільно-радикального заміщення, яке характерне для
алканів. Ці реакції відбуваються ще легше, ніж для алканів, особливо в сс-
положення щодо подвійного зв'язку, оскільки як проміжний продукт утворюється
стабільний алільний радикал. Найважливіші реакції приєднання і заміщення
наведені нижче. Окремо розглянуто реакції окиснення, якЬ теж переважно
відбуваються за місцем подвійного зв'язку.
і і
—с=с—
+ НУ
X У
192 Чирва В.Я., Ярмолюк CM., Толкачова Н.В., Земляков О.Є. Органічна хімія
8.5.1. Реакції відновлення
❖ Реакції каталітичного гідрування. Це загальний метод перетворення
подвійного зв'язку Карбон-Карбон в одинарний С-С-зв'язок. Оскільки реакція,
зазвичай, відбувається кількісно, і об'єм витраченого водню можна виміряти, то
гідрування часто використовують з аналітичною метою.
У процесі гідрування алкенів відбувається розрив я-зв'язку (АН -260 кДж/моль) і
зв'язку Н-Н (АН ~435 кДж/моль), а також утворення двох зв'язків С-Н (АН ~
-410 кДж/моль на кожний зв'язок). Унаслідок реакції виділяється енергія (Д#.~
-125 кДж/моль):
> Теплота гідрування - кількість тепла, що виділяється під час гідрування 1 моль
ненасиченої сполуки.
Гідрування - реакція екзотермічна, проте без каталізатора вона відбувається з
незначною швидкістю навіть за підвищеної температури. Алкени також стійкі до дії
водню в момент виділення. Для того, щоб відбулося гідрування, необхідна дуже
велика енергія активації. Каталізатор її зменшує настільки, що реакція можлива за
кімнатної температури. Він, звичайно, не впливає на сумарну зміну енергії всієї
реакції, а просто знижує енергетичний бар'єр між вихідними і кінцевими сполуками.
Правда, одночасно з прискоренням реакції гідрування пропорційно
прискорюється і зворотна реакція дегідрування. Іншими словами, каталізатор
сприяє швидшому усталенню рівноваги. Тому в реакціях як гідрування, так і
дегідрування застосовують одні й ті ж каталізатори, а, використовуючи різні
умови реакції, змінні умови сорбції і десорбції вихідних або кінцевих продуктів
реакції, її вдається спрямувати в потрібний бік.
Каталізатор, зменшуючи енергію, активації, спрямовує реакцію за іншим
механізмом. Припускають, що на поверхні каталізатора відбувається не лише
руйнування л-зв'язку алкену, а й активація молекули Гідрогену.
Алкени можна гідрувати за умов гетерогенного або гомогенного каталізу:
Як гетерогенні каталізатори гідрування за кімнатної температури
використовують дрібнодисперсну платину (платинова чернь), каталізатори
Адамса1 - оксиди платини і паладію, які взаємодіючи з воднем, утворюють
насичену воднем чернь або паладій, а за температур 150-300 °С - нікель Ренея. В
R-CH=CH2+H2
Кат.
RCH2CH3
1 Адамс P. (Adams R.) (1889-1971) - американський хімік-органік. Головні дослідження пов'язані з
хімією біологічно активних сполук.
Розділ 8, Алкени
193
процесі приготування каталізаторів для надання їм структурної стійкості їх
наносять на носії, такі як активоване вугілля, кізельгур, алюміній оксид, платина.
Значний внесок у вивчення гідрування органічних сполук за наявності металів
зробив П. Сабатьє. Схематично процес реакції гідрування можна описати так:
н2с—сн,
Н
Ь^С-тр-Сг^
н н
н н
т
н
Поверхня
каталізатора
У цьому випадку на поверхні каталізатора може відбуватися синхронне цис-
приєднання водню. Наприклад, гідрування (2)-2,3-Диметилбутендіової (2,3-ди-
метилмалеїнової) кислоти над паладієм дає винятково (2&35)-2,3-диметилянтарну
кислоту у вигляді ліезо-форми:
£Н ї00**
_исоон =5= н-Ьсн,
т-
н
н3сч _ усн3
НООС СООН
нг НООС<
Н3С
т
н
-сн,
СООН
Оскільки для гідрування потрібна адсорбція молекул олефіну на ката-
лізаторі за місцем подвійного зв'язку, то алкени гідруються тим легше, чим
менше замісників у подвійного зв'язку (правило Лебедєва1),
Теплоти гідрування часто дають цінну інформацію про відносну нестійкість
ненасичених сполук, наприклад, цис-бут-2-т має теплоту гідрування 120,1 кДж/моль,
а його трада-ізомер - 115,9 кДж/моль. Це означає, що запас потенціальної енергії в
останньому ізомері на 4,2 кДж/моль менший. Іншими словами, транс-ізомер
стійкіший від і/ис-ізомеру, оскільки два об'ємні замісники в цій молекулі розташовані
по різні боки подвійного зв'язку. Тому як просторові перешкоди, так і вандєр-
ваальсові сили відштовхування між сусідніми замісниками у /я/?адо-алкенів менші:
Н~С
с^с
н .с^
Н
Н
Для циклоалкенів з малими і звичайними циклами (С3-С7) характерна тільки
ї/моконфігурація, Лише з циклооктену можливе існування стабільних сполук з
траяс-конфігурацією подвійного зв'язку. Для циклооктену я*/?ш/с-ізомер на
38 кДж/моль менш стійкий порівняно з ї/г/с-ізомером:
Лебедев С (187Ф-1934) - учень О. Фаворського. Під час Першої світової війни піролізом нафти
одержав толуен (завод у Баку). Вивчав полімеризацію бутадієну, ізобутилену. Розробив і
впровадив у промисловість метод одержання синтетичного каучуку.
794
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Н н н
цис- і /ярянс-Циклооктени
За ступенем заміщення у подвійного зв'язку і, відповідно, стійкості і
важкості гідрування алкени утворюють такий ряд:
СН2=СН2 < ЯСН=СН2 < КСН=СНК < Я2С=СН2 < Я2С=СШІ < К2С=СЯ2 .
Ця закономірність підтверджена даними відповідних теплот гідрування (табл. 8.3).
Таблиця 8.3
Теплоти гідрування деяких алкенів
Сполуки
Н2С=СН2
етилен
СН3СН2СН=СН2
бут-1-ен
СН3СН=СНСН3
ї/ис-бут-2-ен
СН3СН=СНСН3
транс-6ут-2-ен
(СН3)2С=СНСН3
2-метилбут-2-ен
-АН
(кДж/моль)
137
126,7
120,1
115,9
112
Останнім часом у лабораторії щораз частіше застосовують гомогенні каталі-
затори, зокрема, комплекси хлоридів Родію (II) і Рутенію (III) - тр«с-(трифеніл-
фосфін)родій хлорид [(С6Н5)зР]зІУіС1, тр«с-(трифенілфосфін)рутеній гідрохлорид
[(СбН5)зР]зЯиНС1 та ін. Доступніший родієвий комплекс (каталізатор Вілкін-
сона1). Відновлення у разі гомогенного каталізу відбувається за кімнатної
температури й має високу селективність: можна вибірково гідрувати алкени за
наявності алкінів, що неможливо за гетерогенного каталізу. Внаслідок того, що
алкіни ліпше порівняно з олефінами сорбуються на гетерогенних каталізаторах,
вони гідруються першими. Крім того, за гомогенного каталізу не відбувається
ізомеризації алкену, як це є в гетерогенних реакціях.
❖ Відновлення діімідом. Відоме відновлення алкенів за допомогою гідру-
вання діімідом реалізується шляхом синхронного г/мс-приєднання атомів Гідро-
гену через шестицентровий активований комплекс:
н н
/С=сч + н^мн —- /С=сС —- /С-с- + ы2
1 Вілкінсон Дж. (Wilkinson G.) (1921-1996) - англійський хімік. Дослідження в галузі
елементоорганічної хімії, зокрема, фероцену й інших сендвічевих сполук. Запропонував гомогенний
каталізатор реакцій гідрування. Нобелівська премія 1973 р.
Розділ 8. Алкени
195
Сам діімід є дуже нестійкою сполукою, її генерують безпосередньо в
реакційній суміші окисненням гідразину солями купруму (II).
❖ Відновлення дибораном. Відновлення алкенів до алканів за допомогою
диборану відбувається через тетракоординований активований комплекс:
НзС-СН=СН2-^
Пропен
СН3СН2СН3
- В(ОН)3 Пропан
Н
н3с-с—СК,
/ \
Н Н
. снзсндан,2-^^ (СН3СН2СН2)3В —
Пропілборан Трипропілборан
Завдяки наявності на атомі Бору лише секстету електронів Бор гідрид легко
утворює я-комплекси з алкенами:
Н3С-СН|СН2
ВН3
Перетворюючись у чотирицентрову проміжну сполуку, атом Бору у цьому
разі приєднується до більш гідрованого атома Карбону. Такому напряму
сприяють просторовий і електронний чинники. Атом Бору більший від атома
Гідрогену, і ймовірніша його взаємодія з найменш просторово утрудненим
атомом Карбону подвійного зв'язку. Крім того, атом Бору є електрофілом.
Утворення зв'язку В-С дещо випереджає утворення зв'язку С-Н. Фактично це
електрофільне приєднання.
н н н н
н3с—с—с—н н3с-с—с-н
н48-н н-^-н
І
н н п
Делокалізація позитивного заряду на вторинному атомі Карбону більша, ніж
на первинному, тому проміжній структурі (І) надається перевага. Зв'язок С-В
досить лабільний, тому алкілборани легко зазнають подальших перетворень.
83.2. Реакції галогенування
Алкени легко реагують з галогенами, утворюючи дигалогенопохідні. Проте
в цих реакціях іод досить малоактивний, а самі іодиди нестійкі. З іншого боку,
флуор, аналогічно до реакцій з алканами, енергійно взаємодіє з алкенами,
руйнуючи молекулу. Тому на практиці зазвичай застосовують хлорування або
бромування алкенів. Крім того, хлор реагує з алкенами на світлі з вибухом, і
196 Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
навіть реакція з бромом дуже екзотермічна. З цієї причини реагенти вводять до
реакції у вигляді розчинів.
Реакція приєднання галогенів є найліпшим методом одержання віцинальних
дигалогенідів:
СН2—СН2 Вг2
Етилен
СС14
ВгСН2СН2Вг
1,2-Дибромоетан, -100%
?н3
СН3С=СН2 + Вг2
2-Метилпропен
СС14
9Н3
СН39СН2Вг
Вг
1,2-Дибромо-2-метилпропан
Приєднання брому зазвичай використовують для виявлення подвійного
зв'язку Карбон-Карбон. Розчин брому в тетрахлорометані забарвлений у
червоний колір, а диброміди безбарвні.
Залежно від умов реакції приєднання галогенів до алкенів може відбуватися
за йонним або радикальним механізмом.
❖ Електрофільне галогенування [механізм А в). Оскільки л-електронна
хмарка виявляє електронодонорні властивості, то алкени переважно вступають у
реакції електрофільного приєднання. Спочатку під дією подвійного зв'язку
алкену відбувається поляризація зв'язку Вг-Вг, унаслідок чого виникає я-
комплекс, який швидко перетворюється в карбокатіон (а-комплекс) СН2Вг-СН2+
або містковий бромонієвий катіон:
-*Вг-Вг
Вг
)
н
с—сн
І
Вг
СН^ ~~СНл —
Вг
Вг
_І
СН2 СН2
Вг
а-Комплекс Ециклічний
відкритий карбокатіон бромонієвий іон
Утворення циклічного інтермедіату дає змогу пояснити стереоселек-
тивність приєднання аніона будь-якого галогену з утворенням продукту з
/ттранс-конфігурацією, що простежують у деяких випадках для молекул, де
неможливе вільне обертання навколо С-С-зв'язку:
в /х^Вг вг2 Вгч Н
—-і* Г Т н-с=с-н —- с=с
ссі4 к/\г
Циклогексен т/?дг//с-1,2-Дибромоциклогексан, 95% Ацетилен
Н Вг
транс-1,2-Дибромоетилен
У випадку перебігу реакції через карбокатіон такої стереоселективності не
може бути. Циклічний бромонієвий іон стабільніший, ніж відкритий 2-бромоетил-
катіон, тому що в циклічному бромонієвому йоні всі атоми мають по октету
Розділ 5, Алкени
197
електронів у зовнішньому електронному шарі, тоді як атом Карбону 2-бромоетил-
катіону - лише шість електронів. Механізм аняш-приєднання також підтверджено
на прикладах реакції брому з лінійними алкенами. У випадку бромування г/мобут-2-
ену утворюється суміш енантіомерів, аз транс-бут-г-ену-ліеда^З-дибромобутан:
н сн,
н н
>=<
сн,
Вг,
Н3С
і/ис-Бут-2-ен
Вг+
Вг Н
н^Н-СН' .
Н3С Вг
Вг-
-Вг
(1Л,2Л>2,3-Дибромобутан
Н Вг
НіС*И"н -
сн3
сн
Вг-
Вг
сн,
з
Вг
СН,
Н3С
н
>=<
н сн,
Вг,
трвнс-Бут-2-ен
нх.
(15',25)-2,3-Дибромобутан
Вг Н
\__^сн3
н,с"Г\ =
сн
н
Вг
з
Вг
Вг
(1Л,25)-2,3-Дибромобутан
Н3С Вг
Н-Н'н -
сн3
сн,
Вг-
Вг-
Вг
СН,
СН,
(15,2Л)-2,3-Дибромобутан
■ Циклічні галогеноонієві йони вважають звичайними проміжними сполу-
ками за електрофільного приєднання брому до алкенів. Оскільки схильність
атома Хлору до утворення такого йона значно менша, то є приклади приєднання
хлору і через утворення відкритого карбокатіону. На користь існування відкри-
того карбокатіону свідчать перегрупування і депротонування карбокатіонів, які
відбуваються в таких реакціях:
(СН3),С=СН, + с1
2 _с1г*(СН3)2С-СН2с1-^гН2С=С-СН2с1
СН3
2-Метил-3-хлоропропен
2-Метилпропен
■ Приєднання галогенів, зокрема брому, до сиікенаренів також відбувається
через відкритий карбокатіон, оскільки він стабілізований спряженням з
ароматичною системою арену:
Вг,
Стирен
СН-СНгВг
Вг
^~у~ СН-СН2-Вг
Дибромостирен, -100%
198
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Як видно з наведених вище прикладів, реакції галогенування відбуваються у
дві стадії, перша з яких є лімітувальною. Двостадійність процесу підтверджена
фіксацією карбокатіонів за допомогою спектральних методів і утворенням
іонних сполук із суперкислотами.
■ На користь двостадійного механізму з утворенням проміжного карбо-
катіону свідчить той факт, що під час галогенування за наявності інших
нуклеофільних агентів (змішане приєднання) ці агенти також реагують з
проміжним карбокатіоном:
сі"
СН2=СН2-^
Етилен
ВгСН2СН2-
ВгСН2СН2с1
1 -Бромо-2-хлороетан
^ЬРН ВгСН2СН2ОСН3 1-Бромо-2-метоксиетан
ОН
Н20
ВгСН2СН2ОН
ВгСН2СН2ОН2
2-Бромоетанол
4.
-н
ВгСН2СН2ОН
■ Збільшення ступеня заміщення в подвійного зв'язку збільшує швидкість
реакції галогенування (табл. 8.4), що пов'язано як зі збільшенням електронної
густини на подвійному зв'язку завдяки +І-ефектам алкільних груп, так і зі
стабілізацією позитивно заряджених інтермедіатів:
Н2 Н3С-— СН=СН2 Н3С-^- СН=СН — СН3
+1 +1 +1
Таблиця 8.4
Відносна реакційна здатність алкенів стосовно реакції бромування
(розчинник - метанол)
Н2С:
Сполука
Н2С=СН2
СН3СН2СН=СН2
і/г/с-СН3СН2СН=СНСН3
(СНз)2С=С(СН3)2
Відносна
швидкість
реакції
1
103
105
107
❖ Радикальне галогенування. Під час радикального приєднання брому до
алкенів (механізм на стадії ініціювання реакції молекула галогену розпа-
дається на атоми:
2Вг
Далі вільні радикали гомолітично розривають я-зв'язок, приєднуючись до
одного з двох атомів Карбону подвійного зв'язку. Реакція переважно відбу-
вається через стабільніший радикал. Наприклад, вторинний алкільний радикал
(1) стійкіший, ніж первинний (2). Подальша взаємодія з молекулою галогену дає
продукт приєднання, і генерується новий радикал:
Розділ 8. Алкени
199
Вг
Пропен
СН3СНСН2Вг —2-+ СН3СНВгСН2Вг + Вп
Н3С СН—СН^ —Ч ( ) 1,2-Дибромопропан
"Х^ СН3СНВгСН2
(2)
Активність галогенів у реакціях радикального приєднання до алкенів
знижується в ряду СЬ > Вг2 > І2. Особливо інертний у цих реакціях іод через
його низьку активність в атомарному стані.
83.3. Механізми реакції електрофільного приєднання1
❖ Механізм АЕ* Перш ніж ознайомитись з іншими реакціями приєднання до
алкенів, варто розглянути механізм, який виникає в більшості таких реакцій.
Уважають, що приєднання кислого реагенту Ж передбачає стадію утворення
карбокатіону і відбувається у дві стадії:
II ||,
—С=С— +Н:г —£-С— + 2 (1)
Н
Н:г = НС1,НВг, Ш,Н2804
ii - ii.
—С-С— + г: —С~С— £к:і;вг",і;н8о;,н2о (2)
н н г
Стадія (1) полягає в перенесенні протона до алкену з утворенням
карбокатіону; по суті, це перехід протона від однієї основи до іншої:
: :ОҐ С:С—
г :Н
Перша стадія відбувається повільно і її швидкість значно або повністю
визначає швидкість приєднання. Оскільки протон - типовий електрофільний
реагент, то і реакцію називають електрофільним приєднанням.
Далі карбокатіон може приєднувати негативний іон з утворенням галогеніду,
гідросульфату, спирту тощо. У цьому разі електронодефіцитний атом Карбону
одержує пару електронів. Друга стадія, здебільшого, - це йонна реакція, і вона
відбувається практично миттєво:
(1) СН3СН=СН2 + НС1 СН3--СН-СН3 + СІ"
Пропен
+
9і
(2) СН3СН-СН3+ СІ" ~СН3-СН-СН3
2-Хлоропропан
1 Твердження, що та чи інша реакція є електрофільною або нуклеофільною, стосується реагенту,
тобто речовини, яка простіша за структурою.
200
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
(1) СН3СН=СН2 + Н:0803Н -СН3-£н-СН3 + "0803Н
пропен ОБОзН
(2) СН3СН-СН3 + "0803Н СН3-СН-СН3
Ізопропілсульфат
(1) СН3СН=СН2 + Н:ОН2 =ї= Н3С-СН-СН3
Пропен
(2) СН3С№-СН3 + :ОН2 =5==^ СН3~-СН-СН3
+ОН2
(3) СН3СН-СН3 СН3-СН-СН3+ Н+
+(^Н2 ^Н Пропан-2-ол
❖ Інші механізми реакцій приєднання. Для приєднання біполярних
молекул Х6^5*" можливий перебіг реакції через циклічний катіон:
\. ^ Х:У \І +/ Ч А / у ЧІ '
/с=с\ —* /С-сч —/>Х—-/С-сч
у
У розглянутих раніше процесах приєднання молекул типу НЕ такий механізм не
може виникати, оскільки атом Гідрогену не має вільної електронної пари,
завдяки якій відбувається циклізація.
■ Третій можливий механізм приєднання АеЗ охоплює погоджену взаємо-
дію алкену і двох молекул реагенту. Реакція відбувається через шестицентровий
активований комплекс:
8.5А. Напрям приєднання гідроген галогенідів
до алкенів
❖ Правило Марковпикова. Взаємодія алкенів з гідроген галогенідами
залежить від будови ненасиченого вуглеводню і сили кислоти. Реакційна
здатність зменшується в ряду НІ > НВг > НС1 > НБ.
У випадку несиметричних алкенів приєднання гідроген галогенідів відбу-
вається згідно з правилом Марковпикова (1869).
Розділ 8. Алкени
207
> Правило Марковникова у старому формулюванні таке: у реакціях приєднан-
ня до алкенів атом Гідрогену приєднується до найбільш гідрогенізованого атома
Карбону подвійного зв'язку.
Правило Марковникова можна пояснити поляризацією алкену під дією
електронодонорних ефектів замісників (+І-ефект і ефект гіперкон'югації):
6+ 5-
СНз^О=СН2 + Н-Вг
Н
СН3 СН3
Вг
Нижче наведено двостадійний механізм приєднання гідроген броміду до
алкенів, який також пояснює правило Марковникова. З двох проміжних карбо-
катіонів стійкішим є вторинний, тому що він стабілізований двома алкільними
групами, які мають +І-ефект:
Г^-НзС—СН—СН3
2-Бромопропан
СН3СН—СН2
Пропен
СН3(рНСН3
• СН3СН2
►сн,
Реакція переважно відбувається через стійкіший перехідний стан (рис 8.4),
для якого, відповідно, енергія активації буде меншою.
і н3с
-сн^сн2
Н-^Вг
+ Вг
\ /\/3^2
V Ч / \\
' У сн3снсн,+ вг- Vх-
_СН3СН2СН2Вг
сн3сн=сн2 х—
— нх-сн-сн,
3 і 3
+ НВг
Вг
Координата реакції
Рис. 8.4. Енергетична діаграма реакції гідрогенохлорування протну
> Сучасне тлумачення правила Марковникова - електрофільне приєднання
до подвійного зв'язку Карбон-Карбон відбувається через стадію утворення
найстійкішого карбокатіону, наприклад:
+ІСН,
н,
СН3С—СН2
2-Метилпропен
НС1
+1
СН3СН-
+1
с
сн3
Н3С-^С—сн3
сі-
сн3
сн,
—- СН3(рСН3
СІ
2-Метил-2-хлоропропан
202
Чирва В.Я., Ярмолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
• Стійкість карбокатіонів. Карбокатіон - це плоска структура з ^-гібрид-
ним електронодефіцитним атомом Карбону. Вакантна /7-орбіталь перпендику-
лярна до площини молекули. Алкільні замісники, що мають позитивний
індуктивний ефект, компенсують позитивний заряд на атомі Карбону і цим
збільшують стійкість карбокатіону. Найстійкіші третинні алкільні карбокатіони,
найменша стабільність у метального карбокатіону:
Збільшення стабільності
< + ■
н3с—>-с—сн3 н3с—*-сн-«—сн3 н3с—сн сн3
+1 і +1 +1 +1 +1
+ІСН3
Поряд з індуктивним ефектом для пояснення стабілізації карбокатіону
використовують ефект гіперкон'югації- спряження електронних хмар а-зв'язків
С-Н із вакантною орбіталлю:
Стабілізація карбокатіону через гіперкон'югацію
Ще більшу стійкість, ніж алкільні третинні карбокатіони, мають карбокатіони,
стабілізовані я-електронними системами, що розміщені поряд, наприклад, алільний
або бензильний карбокатіони. Внаслідок спряження позитивний заряд у таких
карбокатіонах делокалізується по системі атомів Карбону:
+ 1+
н2с=сн-сн2 сн2-сн=сн2 = [сн2—сн-сн2]+
+м
+м
Легкість утворення карбокатіонів змінюється в такому ж порядку, як і їхня
стійкість: третинний > вторинний > первинний. Це дає змогу розташувати
алкени за реакційною здатністю щодо приєднання кислих реагентів типу КЕ в
такому порядку: (СН3)2С=СН2 > СН3СН=СНСН3 > СН3СН2СН=СН2 >
>СН3СН=СН2 > СН2=СН2 > СН2=СНС1.
• Приєднання до алкенів, що мають електроноакцепторну групу. Сучасне
тлумачення правила Марковникова пояснкЬє утворення продукту приєднання
гідрогенгалогеніду до подвійного зв'язку Карбон-Карбон, поряд з яким
розміщена електроноакцепторна група. Класичне правило в цьому випадку не
Розділ 8. Алкени
203
спрацьовує. Наприклад, у наведеній нижче реакції приєднання відбувається проти
класичного правила Марковникова. Вторинний карбокатіон (1) менш стабільний
порівняно з первинним карбокатіоном (2), оскільки в першому випадку діють
сильні -І і -М-ефекти карбоксильної групи, які дестабілізують карбокатіон (1),
тому реакція йде через утворення стабільнішого карбокатіону (2):
НООССН=СН2
Акрилова кислота
неї
-І, -м +
ноос—сн-сн3
(1)
-і, -м +
НООС^СНз— сн2
(2)
сі-
НООССН2СН2С1
Аналогічно відбувається
трифлуоропропену:
8^ 8+
СК-
З-Хлоропропіонова кислота
приєднання гідроген галогеніду до 3,3,3-
СН=СН2 + НС1
— СР3~СН2СН2С1
1,1,1 -Трифлуоро-З-хлоропропан
Оскільки реакції електрофільного приєднання відбуваються через стадію
утворення карбокатіонів, то в деяких випадках можливі перегрупування,
характерні для цих частинок. Зазвичай перегрупування йде в стійкіший
карбокатіон:
СН7
СН3
нх-с-сн=сн9
сн3
3,3-Диметилбут-1 -єн
НСІ^
-78 °С
гг
н3с-с-сн-сн3
сн3
і
сн,
І
нх-с-сн-сн,
3+| з
сн3
сн,
сі- І 3
—- н,с-с-сн-сн,
І І 3
н3с СІ
2,2-ДиметилгЗ-хлоробутан, 37%
СН,
І 3
н,с-с-сн-сн,
сі-
I І
СІ сн,
2,3-Диметил-2-хлоробутан, 61%
■ Якщо під час приєднання гідроген галогеніду до алкену виникає
хіральний центр, то утворюється рацемічна суміш в еквімольному співвідно-
шенні:
н3с-сн=сн-сн3 + НВг
Буг-2-ен
сн3
Н-С-Вг
с2н5
сн,
І 3
Вг-С-Н
с2н5
(5)-2-Бромобутан, 50 % (Я)-2-Бромобутан, 50 %
204
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Такий результат пояснюють утворенням карбокатіону, у якому прохі-
ральний атом Карбону перебуває в .^-гібридному стані, а сам інтермедіат має
плоску структуру. Тому атака карбокатіону аніоном однаково ймовірна з обох
боків:
■ Сучасні дослідження засвідчують, що в багатьох випадках приєднання
відбувається не через карбокатіон, а утворюється інтермедіат, що несе
частковий позитивний заряд на найбільш заміщеному атомі Карбону, де він
максимально компенсується. Сама ж реакція відбувається як тримолекулярне
анти-приєднання (механізм АеЗ):
❖ Пероксидний ефект Карата. Зазначимо, що правило Марковникова
виконується лише за йонного приєднання гідроген галогенідів і повної відсутності
кисню й інших окиснювачів у вихідних реагентах і розчинниках. За радикального
приєднання гідроген броміду відбувається зворотне за напрямом приєднання.
Першим цю реакцію вивчив М. Караш, який виявив вирішальну роль у зміні
напряму реакції незначної кількості пероксидів, що наявні в багатьох розчинниках
і вихідних алкенах. Звідси й назва - пероксидний ефект Карата.
Причина зміни механізму реакції полягає в дії пероксидів (на схемі задля
простоти показаний гідроген пероксид) на гідроген бромід з утворенням вільних
радикалів Брому. Далі процес відбувається через стійкіший вторинний
алкільний радикал. Під час перебігу реакції постійно генеруються вільні
радикали, тому для ініціювання реакції за механізмом Ац достатньо слідів
пероксидів:
2 5 (5)-2-Бромобутан
Вг-Н
Н202 + 2НВг
2Вг-+ 2Н20
— СН2СНСН2Вг
x- СН2СНВгСН2
НВг
СН2СН2СН2Вг + Вг
н3с-сн=сн2—
1-Бромопропан
Пропен
/
Розділ 8. Алкени
205
За низьких температур (-80 °С) радикальна реакція гідроген броміду з
алкенами відбувається через циклічний радикал і як ян/гш-приєднання.
Пероксидний ефект властивий лише гідроген броміду. Ні гідроген хлорид,
ні гідроген іодид не здатні приєднуватися таким способом. У випадку НС1
процес не відбувається через міцність зв'язку Хлор-Гідроген, у якому
електронна густина сильно зсунута до атома Хлору. Для цієї сполуки характерне
гетеролітичне, а не гомолітичне розщеплення зв'язку. В випадку НІ вільний
радикал Іоду легко генерується, однак процес унеможливлений через високу
енергію активації приєднання атомарного Іоду до подвійного зв'язку.
8.5.5. Інші реакції приєднання алкенів
❖ Приєднання сульфатної кислоти (сульфування). Алкени реагують на
холоді з концентрованою сульфатною кислотою, унаслідок чого утворюються
кислі алкілсульфати загальної формули Я-ОБОзН. Ці речовини виникають
шляхом приєднання протона до одного з атомів Карбону, що зв'язаний
подвійним зв'язком, а гідросульфат-іон - до іншого. Важливо наголосити, що
атом Карбону утворює зв'язок з атомом Оксигену, а не атомом Сульфуру:
II ? II?
—с=с— + н-о-|-он —- — ер—ер—о—^—он
о но
Якщо розчин кислого алкілсульфату в сульфатній кислоті розвести водою і
нагріти, то утвориться спирт, який має ту ж алкільну групу, що і вихідний
кислий алкілсульфат. Кислі алкілсульфати розкладає вода з утворенням спиртів і
сульфатної кислоти, тому її приєднання до алкенів використовують для
одержання спиртів:
98% Н2804 Н,0
СН2=СН2 ^СН3СН20803Н —СН3СН2ОН +Н2804
Етилен Етанол
СН3СН=СН2 8°%Н25?т,СНСН3 СНз^НСНз + Н2804
Пропен ОБОзН ОН
Пропан-2-ол
СНз 63%Н,80, н,0
сн3с=сн2 МСН3ССН3 сн3ссн3 + н2о
ОБСШ ОН
2-Метилпропен ^о^3і
2-Метилпропан-2-ол
Це хороший промисловий метод синтезу спиртів, оскільки алкени порівняно
легко одержують під час крекінгу нафти. Приєднання сульфатної кислоти
відбувається за правилом Марковникова, і через те первинні спирти, за винятком
206
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
етилового, не можна отримати цим методом. Властивість алкенів розчинятися в
сульфатній кислоті використовують також для їхнього очищення від алканів і
галогеноалканів, які в цій кислоті не розчинні.
❖ Приєднання води (гідратація). Реакція гідратації алкенів відбувається за
наявності мінеральних кислот, частіше сульфатної:
н2о
н+
і і
-с-с-
но н
Механізм
галЬгенідів:
8+ 05-
СН3—С^=СН2
сн3
2-Метилпропен
реакції аналогічний до механізму приєднання гідроген
:0
Н
СНз-С]-СНз
сн3
Карбокатіон
н
СН3 п + СН3
СНз-С-6^ СН3-С-ОН
сн, н сн,
Оксонієвий катіон
2-Метилпропан-2-ол
Лімітувальною стадією реакції є утворення карбокатіону. Ця стадія зворотна, як
і весь процес гідратації. Важливо, що взаємодія карбокатіону з надлишком води є
швидшою, ніж оборотна реакція регенерації алкену. Поряд із взаємодією
карбокатіону з водою відбувається його конкурентна реакція з аніоном суль-
фатної кислоти:
СН2=СН2 +
Н-
■ СН3СН2
Етилсульфат, що утворився, у разі нагрівання гідролізується до етилового
спирту.
Під час гідратації несиметричних алкенів реакція відбувається за правилом
Марковникова, а гідратація розгалужених алкенів часто супроводжується
перегрупуванням. Наприклад, із 2,2-диметилбут-1-ену під час гідратації утво-
рюється головно 2,3-диметилбутан-2-ол. На початку вторинний карбокатіон, що
виникає під час протонування алкену, перегруповується в стабільніший
третинний карбокатіон (алкільний зсув), який і атакує молекула води:
СН,
СН3
н3с-с-сн=сн2 + н+-
сн3
3,3-Диметилбут-1 -єн
СН3
— н,с-с—сн-сня
3,1 з
/ \ з
н н
■ н,с-с-сн-сн,—-н,с-
(сн
СН3
-с-сн-сн,
сн,
Н20
Н20
-Н30+
СН3
н3с-с—сн-сн2
онсн3
2,3-Диметил бутан-2-ол
Розділ 8. Алкени
207
■ Останнім часом розроблено ефективний промисловий метод гідратації за
наявності менш агресивної й менш нуклеофільної фосфатноі^кислоти в суміші з
вольфрамовою кислотою, що нанесена на силікагель (тиск 100 атм., температура
300-350 °С). У цьому випадку карбокатіон, що утворюється, реагує лише з
водою з утворенням спирту.
Реакцію гідратації можна також провести і в газовій фазі (підвищені
температура і тиск). Як каталізатор застосовують алюміній оксид, цинк хлорид
та ін. Оскільки таке приєднання відбувається за правилом Марковникова, то
одержують ті ж спирти, що і в попередній реакції. Така безпосередня гідратація
- найпростіший і найдешевший із двох способів. Гідратацію широко засто-
совують у промисловості для синтезу трет-бутипового спирту з ізобутилену і
/ирет-амілового (2-метилбутан-2-ола) - із триметил етил єну:
ртт ^тт сни СН7
гпз н2о,н+ Упз і 3 н2о,н+ тт^тт і ^тт
сн3с=сн2 -2— сн39сн3 н3с-с=сн-сн3 2 > сн3сн2— с-сн3
он он
2-Метилпропен 2-Метилпропан-2-ол 2-Метилбут-2-ен 2-Метилбутан-2-ол
❖ Приєднання меркурій ацетату {оксимеркурування). Обробка алкенів
меркурій ацетатом у воді, спиртах або карбонових кислотах із наступним
відновленням натрій борогідридом дає, відповідно, спирти, етери й естери.
Детальніше метод розглянуто в розділі 13:
(СН,СОО)2Н8
Н3С-СН=СК, _ ' »
3 ^ -сн3соо-
^ (Ж
НЕ5*
ососн3
У»
ОСОСНз К=Н.А1к,А1кСО
❖ Одержання галогеногідринів (гіпогалогенування). Галогеногідрини —
сполуки з віцинальним розташуванням гідроксильної групи й атома галогену:
—С=С— + X, +Н20 -—- —І- С—
II * X ОН •деХ = с1-Вг
Етилен реагує з бромом за наявності води з утворенням етилен-
бромогідрину. Аналогічно поводяться й інші алкени. Під дією катіона Брому
утворюється проміжний карбокатіон, який реагує з молекулою води:
Вг2 + Н20 НО"Вг+ + НВг
(СН3)2С=СН2 (СН3)2С-СН2Вг (СН3)2С-СН2Вг
2-Метилпропен ОН
1 -Бромо-2-метилпропан-2-ол
208
Чирва В.Я., Ярмолюк CM., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
❖ Реакція ПрінсаК Алкени приєднують формальдегід у кислому середо-
вищі за таким механізмом. Спочатку формальдегід зазнає протонування:
НСНО + Н+— СН2ОН
Катіон, що утворився, атакує олефін за місцем подвійного зв'язку, утворюючи
новий карбокатіон, який за наявності води і кислих каталізаторів (h2so4, Н3РО4,
BF3, ZnCb) перетворюється в 1,3-діол:
СН3-СН=СН2 + СН2ОН —- СН3-СН-СН2СН2ОН ^ CH3ÇHCH2CH2OH
Пропен ОН
Бутан-1,3-діол
Вагомість цієї реакції полягає в тому, що такий діол легко перетворюється в
бутан- 1,3-дієн СН2=СН-СН=СН2.
В іншому випадку, у безводному середовищі за температури близько 200 °С
з 2-метилпропену утворюється ненасичений спирт - 3-метилбут-3-ен-1-ол, який
можна перетворити в ізопрен:
н3с-с=сн2 + СН20 -jjgw н2с=с-сн2сн2он
сн3 3 сн3
❖ Реакція гідроформілювання алкенів {реакція Penne1). Одним із найефек-
тивніших методів уведення в олефіни карбонільної групи є синтез альдегідів за
наявності комплексів перехідних металів у потоці водню:
CH3-CH=CH2 + СО + Н2 10o_i8(fc?îoa-300aTM. СН3СН2СН2СНО
Пропен Бутаналь
Уважають, що каталізатором слугує гідроген тетракарбонілкобальтат
Н[Со(СО)4], що утворюється за цих умов. Сьогодні це промисловий метод
одержання альдегідів.
Заміна каталізатора на комплекс пентакарбонілкобальтату з три феніл-
фосфіном на порядок знижує тиск, проте в цьому разі альдегіди відновлюються
до спиртів:
сн3-сн=сн2 + со + н2 (Сб^25ат!іСО'5 СН3СН2СН2СН2ОН
Пропен Бутан-1 -ол
1 Прінс Г. (Prins H.) (1899-1958) - голландський хімік-органік. Відкрив реакції приєднання
формальдегіду до алкенів і галоформів або тетрагалогенометанів до полігалогеноолефінів, що
названі його іменем.
2 Реппе В. (Reppe W.) (1892-1969) - німецький хімік. Головні праці присвячені хімії ацетилену. Відкрив
низку реакцій, що названі його ім'ям.
Розділ 8. Алкени
209
❖ Димеризація й алкілування. В розділі 7 обговорено реакції димернзації
алкенів і конденсації алкенів з алканами, що відбуваються за наявності
кислотних каталізаторів. Реакції проходять через утворення карбокатіонів:
сн3 <рн3 + <рн3 <рн3 сн3 сн3
СН3С=СН2 + СН3С=СН2 -^1*СН3С)СН=ССН3 + СН3С[СН2ОСН2
СН3 СН3
2-Метилпропен 2,4,4-Триметилпент-2-ен 2,4,4-Триметиллент-1 -єн
СН3С=СН2+ НССН3 -2—► СН3СНСН2С|СН3
сн3 сн3
2-Метилпропен 2-Метилпропан 2,2,4-Триметилпентан
❖ Гідроборування-окиснення - один із найефективніших методів одержання
спиртів (детальніше вони розглянуті в розділі 13):
н вн2 н он
З СН3-СН=СН2 — (СН3СН2СН2)3В З СН3СН2СН2ОН + н3во3
Пропен Пропан-1-ол
❖ Вільнорадикальне приєднання тетрагалогенометанів. За умов утворення
вільних радикалів тетрагалогенометани приєднуються до подвійного зв'язку
алкенів:
і к , Пероксидиабо^ І І
^ ^ Лї освітлення У у
У X
Наприклад: сбНІЗСН=СН2 + ВгСС13 КА . СбН^НСН-гССІз
Вг
Окт-1-ен 3-Бромо-1,1,1-трихлорононан
С6Н13СН=СН2 + СВг4 —^ с6и13^псн2свг3
Вг
Окт-1 -єн ], 1,1,3-Тетрабромононан, 88%
Ця реакція є в основі одержання Р-гідроксикарбонових кислот.
❖ Приєднання карбенів. У попередньому розділі детально розглянуто
синтез карбенів і їхні реакції з алкенами з утворенням похідних циклопропану:
І І сн,ы2 І І
-с=с- -с-с-
сн2
210 Чирва В.Я., Ярмолкж С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
З-Хлоропропен
(алілхлорид)
Хлорування пропілену в алільне положення {аномальне хлорування) за
високої температури вперше спостерігав М. Львов 1883 р.
Висока температура потрібна для перебігу реакції заміщення з низькомоле-
кулярними лінійними алкенами, тоді як у разі сполук із розгалуженим ланцюгом
процес може відбуватися і за кімнатної температури:
НзС СН3 С12 Н3С СНг-СІ
с=с —*• с=с
н3с сн3 -НС1 н3с сн3
2,3-Диметилбут-2-ен 2-Метил-3-хлорометилбут-2-ен
Під час високотемпературного хлорування радикал Хлору може приєдну-
ватися за місцем подвійного зв'язку з утворенням первинного і вторинного
алкільних радикалів, а також вільнорадикального заміщення атома Гідрогену в
метальній групі. Другий процес переважає, оскільки алільний радикал стійкі-
ший, а хімічна реакція, як відомо, відбувається через перехідний стан з меншою
енергією активації, якому і відповідає стійкіший інтермедіат:
Низька
температура
т 9
СН3СН—СН2"
Пропен
сі,
ссь
>400 °С
СН3СН-СН2
Іонна реакція
С1СН2СН=СН2
Вільнорадикальна реакція
Наприклад: СН3-СН=СН2 + СН^Г^)^ Н3С~СН-СН2 + Ц]*
Пропен ^? СН2
Метилциклопропан
8.5.6. Реакції заміщення
❖ Алільне галогенування. Оскільки алкільні групи в олефінах мають струк-
туру алканів, то вони повинні вступати в реакції заміщення:
II II
Н—С—С=С- +Х2 Х-<р-ОС— х=сі,вг
Якщо необхідно виконати галогенування алкільної частини молекули алкену,
то реакцію проводять за умов вільнорадикального заміщення (механізм 5д):
1,2-Дихлоропропан
Розділ 8. Алкени
211
СН3СН—•СН2
Пропен
СІ-
9 •
сн3сн-сн2 -x*
сн3сн-сн2 ^*
-неї
СН2СН=СН2
сі,
С1СН2СН=СН2 + сі-
Алілхлорид
Стабільність проміжного алільного радикала СН2=СН-СН2- пояснюють
р,я-спряженням електронів подвійного зв'язку з /7-орбіталлю неспареного
електрона, що делокалізує електронну густину і вирівнює зв'язки:
н2с=сн-сн2
сн2-^н=сн2
= [СН-—СН-СНЛ"
Підтвердженням високої стабільності алільного радикала може бути
зменшення енергії розриву зв'язку С-Н в алільному стані порівняно зі зв'язком
С-Н у насиченому вуглеводні - відповідно, 364 і 402 кДж/моль.
Якщо розглядати структуру алільного радикала методом МО, то можна
побачити, що перша зв'язувальна орбіталь Ч7! охоплює всі три атоми Карбону.
Друга МО 0¥2) - незв'язувальна. Вона має одну вузлову площину і демонструє
делокалізацію вільного електрона По кінцевих атомах Карбону. Третя
розпушувальна МО (ЧРз) має дві вузлові площини:
-71*,
НІ-*.
Рис. 8.5. Енергетичні рівні й обриси АО і МО алільного радикалу
■ Бромування в алільне положення можна виконати в м'яких умовах дією Лґ-
бромосукциніміду, який створює постійну, але невелику концентрацію брому:
о
НВг
Вг2+ |
снга
+ І >І-Вг
СН~Ч
о
М-Бромосукцинімід
н2с=сн-сн2н ^ Н2С=СН-СН2
Пропен
енг-с:
Вг,
- Н^СН-СН^г +Вг-
З-Бромопропен (алілбромід)
212
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Низька концентрація брому унеможливлює реакцію електрофільного
приєднання галогену до подвійного зв'язку алкену. Застосований вперше 1942 р.
А. Волем і К. Циглером, цей варіант радикального бромування значно поши-
рився в органічному синтезі:
Р
II + Г М-Вг —-І У + І >ин
о
Циклогексен И-Аромосукцинімід 3-Бромоциклогексен
❖ Окиснювальний амоноліз (амоксидування). Відносну легкість утворення
алільного радикала використовують у промисловості в окиснювальному амонолізі
пропілену для одержання акрилонітрилу:
500 °С
СН2=СН-СН3 + 1ЧН3 + 3/2 о2 сн2=сн-си + з Н20
2 3 3 2 Ві203, Мп02 2 2
❖ Високотемпературне хлорування етилену. Заміщення атома Гідрогену
на атом Хлору в етилені за короткочасного нагрівання (-2 с) реакційної суміші
до високої температури набуло важливого промислового значення. Цілком
можливо, що спочатку в реакції утворюється 1,2-дихлороетан, який унаслідок
елімінування гідроген хлориду перетворюється у вінілхлорид:
Н2С=СН2^ГН^С=СНС1+НС1
Етилен Вінілхлорид, 38%
Важливе препаративне значення має реакція заміщення алкенів, розроблена
Хеком 1968 р. Вона дає змогу за допомогою комплексу паладію виконати з
хорошими виходами синтез регіо- і стереоселективних продуктів:
К\ /Н „ (С2Н5)3Н Р(1-комплекс К\ /Н
С=С + ЯХ 4 2 5П ^ С=С
н чн -(с2н5)3ш+ „' ^
де Я - арил, гетарил, вініл.
Ця реакція заміщення поширена Соночаширою на алкіни:
„ Рсі-комплекс, Си(І), ІІЛ^ „ .
н-с=с-к' + я"х ,КзШ+ я-сеес-к'
8.5.7. Реакціїокиснення
На відміну від алканів, алкени легко окиснюються, насамперед, за місцем
подвійного зв'язку. Глибина окиснення залежить від типу окиснювача й умов
перебігу реакції. ^
Розділ 5. Алкени
213
❖ Окиснення до епоксидів. Окнснення алкенів 25 % гідроген пероксидом в
оцтовій кислоті дає епоксиди (а-оксиди). Реакція відбувається через утворення
надоцтової кислоти:
СН5СООН + Н202 СН3СОООН + Н20
Надоцтова
кислота
М. Прилєжаєв1 довів, що для одержання а-оксидів можна використовувати
готові надкислоти. Припускають, що реакція відбувається через циклічний
активований комплекс:
К2С=СК2 к^с и^С" ~с&2
/°ч —- а —- °
но н
+
0—£ д : ІІ О
б 5 о—с-с6н5 но-с-ан
6х А5
Раніше для епоксидування алкенів переважно використовували л*-хлоро-
пероксибензойну кислоту, яку тепер у лабораторних синтезах замінила магнієва
сіль монопероксифталевої кислоти:
О
и
НзСч ,СН, ^.С-ООН 0ч ХН, ^/СООН
.с=с. + г її ► 3 чс—сґ 3 +
Н ЧСН3 ^^с-0"і/2МЕ^ К ЧСНз ^^СОО-1/2МЄ^
2-Метилбут-2-ен О Триметилоксиран, 98 %
■ Олефіни можна окиснювати до епоксидів також за допомогою полі-
мерного матеріалу - йонообмінної смоли, що має карбоксильні групи й обробле-
на гідроген пероксидом. Відпрацьована смола легко регенерується.
■ У промисловості етиленоксид (оксиран) одержують окисненням етилену
повітрям за наявності срібного каталізатора:
н2с=сн2-^Г Н2С—сн2
А8 о
❖ Окиснення до гліколів. Холодний розбавлений розчин калій перманганату
(реакція Вагнера2, 1888 р.) перетворює олефіни в гліколі:
1 Прилєжаєв М. (1877-1944) - радянський хімік-органік. Головні праці присвячені сполукам, що
містять атом Оксигену.
2 Вагнер Є. (1849-1903) - російський хімік-органік. Дослідження в галузі органічного синтезу і
хімії терпенів.
214
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Н2С=СН2+ Мп04—Ї^^МпОз -^СН2ОН-СН2ОН + Мп02
Етилен СН2 О Етиленгліколь
■ Реакція Вагнера сьогодні має незначне синтетичне значення через її
неселективність, проте може бути використана як якісна проба на подвійний
зв'язок (знебарвлення перманганату).
Вивчення механізму окиснення алкенів з застосуванням калій перманганату, з
міченим ізотопом 180, засвідчило, що джерелом обох атомів Оксигену в гліколі є
перманганат-аніон. Реакції стереоселективні і є г/г/с-приєднанням гідроксильних
груп до подвійного зв'язку:
^ /°НЗ КМп04 "зС',. ..-СН3
Н20
но^^он
1,2-Дим етил цикл опентен цис-1,2-Д им етил циклопентан-1,2-діол, 45%
■ У сучасних методах для гідроксилювання алкенів частіше застосовують
осмій (VIII) оксид {реакція Кріге, 1936 р.). Перевага над реакцією Вагнера
полягає у тому, що як розчинники можна використовувати органічні сполуки -
етиловий етер, бензен, хлороформ. Реакцію прискорює піридин. Як і в попе-
редньому випадку, утворюються і/мс-віцинальні гліколі. Ця реакція потребує
еквімольної кількості досить дорогого реагенту. Тому замість реакції Кріге
використовують реакцію Майлса, де потрібні каталітичні кількості осмій оксиду
спільно з гідроген пероксидом:
О-Об' ОН
Циклогексен О */мс-Циклогексан-1,2-діол, 58%
■ Алкени зазнають аииш-1,2-гідроксилювання під дією 30% гідроген
пероксиду в оцтовій кислоті за наявності каталітичних кількостей сульфатної
кислоти:
,ОН
" <3
СН3СООН, Н2804 ^
ор
Циклогексен /гг/7дгяс-Циклогексан-1,2-діол, 53%
■ В органічному синтезі щораз частіше застосовують окиснення олефінів у
неводному середовищі плюмбум тетраацетатом, що дає діацетильні похідні
гліколів, які можна деацетилювати до діолів:
Розділ 8. Алкени
215
РЬ(ОЛс)4 9СОСН3
КСН=СНЯ' —-—± ЯСН-СНК'
ОСОСН3
❖ Окиснення жорсткими окиснювачами. Дією концентрованих розчинів
сильних окиснювачів (КМ11О4, НИОз, СЮз, К2СГ2О7) молекула алкену
розщеплюється за місцем подвійного зв'язку, утворюючи кислоти і кетони.
Кінцева метиленова група окиснюється до вуглекислого газу:
СЮ3
няс-с=сн-сн, *~ нх-с=о + о=с-сн3
З , 3 3 , ,3
СН3 СН3 ОН
2-Метилбут-2-ен Ацетон Оцтова кислота
н^с=с'СНз н3с-с=о + о=с-сн3
нзС СНз СН3 СН3
2,3-Диметилбут-2-ен Ацетон Ацетон
Для синтетичних потреб цю реакцію використовують зрідка через низькі
виходи цільових продуктів. Проте цього недоліку можна позбутись, якщо
реакцію окиснення калій перманганатом проводити в бензені за наявності краун-
етеру або додаючи рутеній (IV) оксид. Є реагенти, що дають змогу припинити
окиснення альдегідів до кислот, наприклад, хромілхлорид СЮ2СІ2.
Ці реакції до появи спектральних методів використовували для визначення
будови алкенів. За допомогою аналізування структури продуктів окиснення
можна визначити як кількість карбонових атомів в олефіні, так і місце
подвійного зв'язку, а також ступінь його заміщення, наприклад:
Сг03 .
сНзСЕ^сн^с^о + о=с-сн3 СН3СН2СН2СН|=СНСН3
ОН ОН
Масляна кислота Оцтова кислота Гекс-2-ен
КМп04
_ _ СНаСН^СНчСН^СН)
ї
он
Масляна кислота Пент-1-ен
КМп04
СН^С^СН^-С^О + С02 СН3СН2СН2СН|=Сіх2
■ Реакція Лем'є1. Діючи на олефіни періодною кислотою НЮ4 і ката-
літичною кількістю калій перманганату, Р. Лем'є провів таку реакцію розщеп-
лення подвійного зв'язку: \
1 Лем'є Р. (Ьетіеих Я.) (1920-2000) - канадський хімік. Дослідження в галузі теоретичної і
синтетичної хімії вуглеводів.
216
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
NaI04 R'—С— ОН ^ R'—С=0
кмп04 КІ_г_он R,-o=o
1 к
❖ Озоноліз. Аналогічних результатів можна досягнути, окиснюючи алкени
озоном за Гаррієсом1. Озон приєднується за місцем подвійного зв'язку, утворюючи
нестійкі, вибухові озоніди, які відразу розкладають водою до альдегідів або кетонів.
Гідроген пероксид, що утворюється в цьому разі, окиснює альдегіди до карбонових
кислот. Якщо ж розклад озонідів проводити водою в кислому середовищі за
наявності цинкового пилу, то окиснення альдегідів не відбувається:
СН3 сн3 сн3
І І Спонтанно і
н3с-с=сн-сн3 + о3 —- н3с-с—сн-сн3 н3с-с-о-сн-сн3
/ 2-Метилбут-2-ен 0 О О О
О Мольозонід 0зоніД
н2о гп Н+
—- н3с-с-сн3 + сн3сно + н2о2 н3с-с-сн3 + сн3сно + Н20
о о
Ацетон Ацетальдегід
Під час обробки озоніду натрій борогідридом ЫаВНЦ кінцеві продукти
виділяються у вигляді суміші відповідних спиртів:
03 ЫаВН4
СН2С12
НО(СН2)6ОН
Циклогексен О °С Гексан-1,6-діол, 63%
Реакція озонування успішно застосована К. Гаррієсом для визначення
будови каучуку, унаслідок якої одержано єдиний продукт реакції - левуліновий
альдегід, що схематично виглядає так:
~снгс=сн-с%снгс=сн-сн2сн^(^=сн-сн^
СН3 СН3 СН3
0-0 ' 0-0 о-о
-СН^Сг /СН-СНзСН^С^ /СН-СН^СН^-С^ /СН-СН2- 32
сн3° сн3° сн3°
Гаррієс К. (Harries С.) (1866-1923) - німецький хімік. Головний об'єкт дослідження - реакції
ненасичених сполук, у тому числі каучуку.
Розділ 8. Алкени
217
сн3 Левуліновий альдегід
❖ Каталітичне окиснення етилену (Вакер-процес). Ця реакція відкрита
1957 р. німецьким хіміком Хафнером, який тоді працював у приватній фірмі
"Wacker Chemie", звідки й виникла назва цього процесу. До реакцій окиснення
належить м'яке перетворення етилену в ацетальдегід:
PdCl2+H2C=CH2— U>d...]fH2 2.1£Г Cl-Pd-CH2-CH2OH — Pd +CH3CHO
Купрум (І) хлорид, що утворюється, знову окиснюється до купрум (II) хлориду.
Сьогодні ця реакція є головним джерелом одержання ацетальдегіду в промисловості.
Наприкінці зазначимо, що важливі для алкенів реакції полімеризації
детально розглянуті у розділі 10 , а також у курсі "Високомолекулярні сполуки".
8.6. Найважливіші алкени
Найширше використовують перші представники гомологічного ряду алкенів -
етилен і пропен (пропілен). Промислове застосування цих сполук відображено на
рис. 8.6 і 8.7.
НаС=СН2 + 02^g^ СН3СНО
Можливий механізм реакції такий:
Купрум (II) хлорид відіграє роль окиснювача паладію:
2CuCl2 + Pd -> PdCl2 + 2CuCl
н,с=сн-сн=сн.
Дивініл
Синтетичні
каучуки
"—сн-сн—
^- Полістирен
синтетичні
каучуки
- Н3С-СН2ОН
Етиловий спирт
Дегідрування
Н3С—СНО
Оцтовий альдегід
Окиснення
(С2Н,)П-
Поліетилен
Полімеризація
СН^СІ-ССН^)!!—ССІ3-
ССІ,
Синтетичне волокно
й інші матеріали
СН3СООН
Оцтова кислота
[О]
Н3С-СНО
Оцтовий альдегід
—СН2|
СбН,
1 Полімеризація
Етилбензен
Дегідру-
1 рування
с^н^сн^сн^
Стирен
СІ,
[СиСІг], [РсіСІг]
сн^он-сн^сі
НОСІ
неї
2 \ / 2
о
Етиленоксид
Окиснення (каталізатор А§)
СН2СІ—СН^СІ
Дихлороетан
Дегідрогало-
генування ^
Н^С^СНСІ
Вінілхлорид
Полімеризація
-сименсі
Jn
Полівінілхлорид
Рис. 8.6. Схема промислового використання етилену
СНЗСОСНЗ
Ацетон
СН20Н~СН(ОН)СН20Н
Гліцерол
|2Н0Н
СН2СІ-СНОН-СН2СІ
-Н2С=СН-СН20Н
Аліловий спирт
Н2О
Окиснення
дегідрування
Полімери
Н3С—СН
Ізопропіловий спирт
ОН-СН3 Н2С=СН—СН
Акрилонітрил
Н,0
КИз; О2
[Кат.]
Н2С~СН—СН,С1
Епіхлорогідрин
Епоксидні смоли
Алілхлорид
СІ,
Спирти
4500С
Н2С=СН-СНз
нх=сн-сно --^
'Акролеїн Окиснення
СО + Н,
Полімери
Поліме-
ризація
СН
Поліпропілен
-сн^-сн-
Jn
Алкілування
СНз СНз
[Кат.]
С^Н^ СН СН3
кумен
Н3С-С СН-СНз
СНз
"Триптан"
[Н]
Н3С—сн^-сн^-сно
Н3С-СН-СНО с.НзОН СН3СОСН3
СН,
Фенол
Ацетон
Рис. 8.7. Схема промислового застосування пропілену
Розділ 9
АЛКІНИ
Значення хімії ацетилену швидко зростає через дешеві способи одержання
алкінів за допомогою крекінгу нафти. Це привертає увагу до них як до сировини
для перетворення в безліч цікавих продуктів з широким застосуванням.
Загальна формула алкінів ОД^. Як видно з формули, вони мають менше
атомів Гідрогену, ніж алкени, і виявляють ще більший ступінь ненасиченості. Не-
зважаючи на однакову загальну формулу з дієнами, алкіни і дієни мають різні функ-
ціональні групи і, відповідно, мають різні властивості, тому їх розглядають окремо.
Найпростіший представник ряду алкінів - ацетилен С2Н2. Той же підхід, що
раніше використаний для опису молекули етилену, приводить до структури, у
якій атоми Карбону мають три спільні електронні пари, тобто зв'язані
потрійним зв'язком. Отже, потрійний зв'язок Карбон-Карбон є характерною
особливістю будови алкінів і, по суті, їхньою функціональною групою:
Квантово-механічний підхід дав таке уявлення про потрійний зв'язок
Карбон-Карбон. Для утворення зв'язків з двома іншими атомами Карбон
використовує дві еквівалентні ^р-гібридні орбіталі, утворені з однієї s- і однієї р-
орбіталі. Ці орбіталі розташовані на прямій лінії, що проходить через ядра
атомів Карбону. Кут між двома орбіталями становить 180°. За такого лінійного
розташування гібридні орбіталі максимально віддалені одна від одної:
9.1. Будова і номенклатура алкінів
9.1.1. Будова ацетилену
н:с;;с:н
або
Н-С=с-Н
лр-Гібридизація
Розділ 9. Алкіни
221
Якщо розташувати два атоми Карбону і два атоми Гідрогену ацетилену так,
щоб орбіталі максимально перекривались, то ми одержимо таку структуру:
Ацетилен — лінійна молекула, у якій всі чотири атоми розташовані на одній
прямій. Зв'язки Карбон-Гідроген і Карбон-Карбон симетричні відносно лінії,
що з'єднує ядра, а отже, є о-зв'язками.
Негібридизовані 2рг і 2/у-орбіталі кожного з карбонових атомів взаємно
перпендикулярні і перпендикулярні до осі гібридних орбіталей. їхнє бокове
(латеральне) перекривання утворює два я-зв'язки. Якщо одну хмарку гс-зв'язку
зобразити над і під лінією, що з'єднує ядра, то інша хмарка л-зв'язку буде
попереду і позаду цієї лінії. Проте між ними існує перекривання, таке, що два
зв'язки зливаються, утворюючи одну циліндричну я-електронну хмарку навколо
лінії, яка з'єднує ядра:
Отже, "потрійний" зв'язок побудований з міцного о-зв'язку і двох менш
міцних ті-зв'язків. Його енергія становить -815 кДж/моль. Він міцніший, ніж
зв'язок С=С в етилені (-610 кДж/моль) або простий зв'язок Карбон-Карбон в
етані (-350 кДж/моль), а тому коротший, ніж будь-який з них. Варто зазначити,
що енергія потрійного зв'язку в алкінах не є адитивним значенням трьох
одинарних о-зв'язків (350-3 = 1050 кДж/моль) або одного а- і двох тс-зв'язків
(350+2-260 = 870 кДж/моль).
Квантово-механічне уявлення про будову ацетилену підтверджене експери-
ментально. Методами дифракції електронів, дифракції рентгенівських променів,
а також спектральними методами доведено, що ацетилен є лінійною молекулою:
0,120 нм 0,106 нм
Н—С=С—H
4s^-180o-^
Довжина зв'язку Карбон-Карбон в ацетилені становить 0,120 нм порівняно
з 0,134 нм в етилені і 0,154 нм в етані. Будова потрійного зв'язку, як і
подвійного, підтверджена, хоча і шляхом одержання негативних результатів,
даними про кількість ізомерів. Як видно з моделей, лінійне розташування
зв'язків унеможливлює геометричну ізомерію. Справді, геометричні ізомери,
зумовлені наявністю потрійного зв'язку, невідомі.
В ацетилені довжина зв'язку С-Н становить 0,106 нм, тобто менше, ніж в
етилені (0,109 нм). Унаслідок збільшення ^-характеру .ур-орбіталі, ніж у .sp2-op6i-
222
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
sp3 -q
зр*
талі, лр-гібридизований атом Карбону утворює коротші зв'язки, ніж ^-гібри-
дизований. Довжина зв'язку С-Н відповідає ковалентним радіусам. Вона
становить для ^-орбіталі 0,060, для sp2 - 0,067 і sp3 - 0,072 нм. З іншого боку, я-МО
ацетилену компактніші, ніж у етилену. Ця особливість будови ацетилену
повинна дещо ускладнювати електрофільні реакції приєднання.
Іншим активним центром у молекулі ацетилену є атом
Y Гідрогену потрійного зв'язку, який виявляє кислі властивості
^^4-^ й може брати участь у реакціях заміщення. Кислі властивості
атома Гідрогену пояснюють тим, що в разі переходу від
етану й етилену до ацетилену змінюється тип гібридизації, і,
відповідно, a-зв'язок С-Н утворюється перекриванням орбі-
талей sp3-s, sp2-s і sp-s. В цьому ряду гібридних орбіталей
(ГТЧ атома Карбону збільшується внесок 5-орбіталі, максимум
sp 9)—7—електронної густини якої є ближче до центра атома Карбону.
Тому в С-Н-зв'язку ацетилену максимум електронної густи-
ни зсунутий до атома Карбону НС^С^П5*. Іншими словами,
зв'язок С-Н поляризований, атом Гідрогену має частковий
позитивний заряд і здатний відриватись у вигляді протона. Проте ацетилен -
дуже слабка кислота, значно слабша, ніж вода (рКа = 25 і 15,7, відповідно).
Енергія дисоціації зв'язку С-Н в алкінах —505 кДж/моль, що більше, ніж в
алкенах (443 кДж/моль). У цьому випадку лр-гібридизація, яка утруднює
гомолітичний розрив зв'язку С-Н з утворенням радикалів, полегшує
гетеролітичний розрив цього зв'язку з утворенням іонів.
Н—С=С:Н —► Н—С=С* + Н* — гомолітичний розрив
Н—С=С:Н —> Н—С=С:~+ КҐ — гетеролітичний розрив
9.1.2. Номенклатура алкінів
Алкіни, як алкани й алкени, утворюють гомологічний ряд з тією ж
гомологічною різницею СН2.
• Тривіальна номенклатура. Сьогодні з тривіальних назв алкінів вико-
ристовують лише ацетилен. Назви його найближчих гомологів: алілен
(СН3С=СН), кротонілен (СДІб), валерієн (С5Н8), застаріли і рідко трапляються в
хімічній літературі.
• Раціональна номенклатура. Згідно з принципами цієї номенклатури,
алкіни розглядають як ацетилен, у якого один або два атоми Гідрогену заміщені
на алкільні групи. В назві спочатку зазначають замісники, а потім основу:
метилацетилен СН3С=СН, диметилацетилен СН3С=ССН3. Подібні назви також
застаріли і їх використовують обмежено.
• Номенклатура ТОР АС. За основу структури вибирають найдовший
вуглеводневий ланцюг, який має потрійний зв'язок. Ланцюг нумерують так, щоб
Розділ 9. Алкіни
223
мінімальний номер одержав атом Карбону з потрійним зв'язком. Назву будують
аналогічно до назв алканів, замінюючи суфікс -ан на -ин (-ін). Відповідно до
сучасних правил ШРАС і рекомендацій УНКоХіТерН, в українській хімічній
номенклатурі локант -ин (-ін) зазначають перед префіксами і перед суфіксами.
У сполуках, що мають подвійний і потрійний зв'язки, за основу вибирають
ланцюг з обома зв'язками, який нумерують так, щоб мінімальні номери
одержали обидва кратні зв'язки. Якщо вони розташовані симетрично (однаковий
набір локантів), то менший номер присвоюють подвійному зв'язку. В назві
спочатку зазначають подвійний, а потім - потрійний зв'язок:
СН3
1 \2 3 4 5
СН3СНОССНСН2СН3
\б 7
СН3СНСН3
5-Етил -2,6-диметилгепт-1 -ин
За основу вибраний найдов-
ший і найрозгалуженіший лан-
цюг з потрійним зв'язком.
Нумерація починається з
того боку, до якого найближ-
че розташований потрійний
зв'язок.
б 5
сн,с=
сн
сн3
ЗІ 2 1
-с-ос
сн
сн3 сн3
р,3,5-Триметилгекс-4-ен-1 -ін
і
Нумерація ланцюга почи-
нається з атома Карбону з
потрійним зв'язком, оскіль-
ки в цьому випадку кратні
зв'язки одержують менші
локанти (1,4), ніж тоді,
коли нумерувати зліва -
(2,5).
СН2СН2СН3
/ 2 З 4\ 5 6 7
СН3С=СНСОССН3
снз СН2СНЗ
4-Етил-2-метил-4-пропілгепт-2-ен-5-ін
Якщо кратні зв'язки розташовані
симетрично, то ланцюг нумерують
з того боку, до якого ближче
розташований подвійний зв'язок.
9.1.3. Ізомерія алкінів
Структурна ізомерія починається з С4, проте ізомери С4Н6 відрізняються лише
розташуванням потрійного зв'язку, а ізомерія карбонового скелета починається з
С5. Виникає і міжкласова ізомерія, зокрема, з дієнами. За кількістю ізомерів алкіни
посідають проміжне місце між алканами й олефінами:
Ізомерія карбонового Ізомерія положення Міжкласова ізомерія
скелета
НС=ССНСН, нс=ссн2сн2сн3 СН3С=ССН2СН3 сн2=снсн=снсн3
І 3
сн3
З-Метилбут-1-ин Пент-1-ин Пент-2-ин Пентан-1,3-дієн
9.2. Фізичні властивості
У гомологічному ряду ацетилену за нормальних умов газами є ацетилен,
метилацетилен, етилацетилен (бут-1-ин). Від диметилацетилену (бут-2-ина) і до
гептадецинів алкіни є рідинами, а з октадец-1-ину - твердими речовинами.
224
Чирва В.Я., Ярмолюк CM., Толкачова Н.В., Земляков О.Є. Органічна хімія
Густини, температури кипіння і плавлення алкінів вищі, ніж у відповідних
алкенів і алканів.
Оскільки алкіни - малополярні сполуки, то їхні фізичні властивості подібні
до властивостей алканів і алкенів. Проте лр-гібридизований атом Карбону
значно більш електронегативний, ніж карбонові атоми у стані sp3- і sp2-
гібридизацій (відповідно, 3,1; 2,5 і 2,8). Тому несиметричні гомологи ацетилену
мають відмінні від нуля дипольні моменти:
СН3СН2-СН=СН2 СН3СН2-С=СН (СН3)зС-С=СН СНз-ОС-СНз
Бут-1-ен Бут-1-ин 3,3-Диметилбут-1-ин Бут-2-ин
H=0,30D ц=0,8(Ш H=0,65D ja=0D
Алкіни погано розчинні у воді, проте ліпше, ніж алкани й алкени, і добре
розчиняються в органічних розчинниках з низькою полярністю: лігроїні,
бензині, етері, тетрахлорометані. Густина їх менша від густини води.
Температура кипіння алкінів підвищується зі збільшенням кількості атомів
Карбону; розгалуження ланцюга впливає так само, як і у випадку алканів і
алкенів. Алкіни мають температури кипіння, які дуже близькі до температур
кипіння алканів і алкенів, у яких такий же карбоновий скелет. Проте наявність
потрійного зв'язку підвищує температуру кипіння. Наприклад, бут-1-ин кипить
при 8,5 °С, а бут-2-ин - при 27 °С, тоді як обидва бутани і всі бутени за
звичайних умов - гази. Температури кипіння алкінів з кінцевим потрійним
зв'язком нижчі для алкінів, у яких кратний зв'язок ближчий до середини
ланцюга. Ця різниця температур дає змогу розділити ізомери фракційною
перегонкою.
9.2.1. Спектроскопія алкінів
• УФ-спектроскопія. В УФ-області монозаміщені алкіни мають смугу погли-
нання À,max 185 нм, а дизаміщені - X тах 178 і 196 нм, що відповідає переходу
я—»я*.
• ІЧ-спектроскопія. В ІЧ-спеклрах валентним коливанням vc=c відповідають
смуги поглинання 2 150-2 250 см"1, а валентні коливання v=c_h виявлені вузькою
лінією 3 310-3 200 см"1.
• Mac-спектроскопія. Мас-спектри алкінів мають інтенсивні піки з miz 27 і 39,
що дає змогу відрізнити ацетилени від ізомерних сполук.
• ПМР-спектроскопія. Сигнали протонів у .ур-гібридного атома Карбону суттєво
зміщені в сильне поле порівняно з протонами в атома Карбону подвійного зв'язку
(4,5-6,5 м.ч.), що пов'язано з екранувальним ефектом індукованого магнітного
поля потрійного зв'язку. На хімічний зсув протона H-C^CR сильно впливає
Розділ 9. Алкіни
225
природа радикала R, і його значення змінюється в діапазоні 1,7-3,1 м.ч.,
наприклад:
Hb3C-feC-Ha НОСН2-С=С-На <(^)_С=С-На
Ô (м.ч.) 1,80 (а), 1,75 (b) Ô 2,33 м.ч. (a) Ô 3,05 м.ч. (а)
9.3. Промислові методи одержання алкінів
Найпростіший представник класу алкінів - ацетилен, що є одним з най-
важливіших для промисловості сполук. Багато напрямів синтетичного вико-
ристання ацетилену стали можливими внаслідок робіт, виконаних В. Реппе в
Німеччині до і під час Другої світової війни в фірмі "Фарбеніндустрі". Його роботи,
метою яких була заміна нафти, якої в Німеччині практично немає, як сировини для
синтезу органічних речовин, доступнішим вугіллям, спричинили революцію в
промисловій хімії ацетилену. Зазначимо, що основи хімії ацетилену і його похідних
закладені працями О. Фаворського1 наприкінці XIX і на початку XX ст.
Ацетилен у великих кількостях можна одержувати низкою способів.
❖ Карбідний метод. Метод синтезу ацетилену з кальцій карбіду, запро-
понований Ф. Велером 1862 p., набув промислового значення тільки після роз-
робки технології виробництва самого кальцій карбіду, який одержують шляхом
випалюванням вапняку з наступним спіканням з коксом при 2 000 °С.
Синтезований кальцій карбід гідролізують до ацетилену:
С + СаО ^£ СаС2 НС^СН
2 -Са(ОН)2
Головна кількість ацетилену, яку використовують для газового зварювання,
сьогодні одержують цим способом. Головним недоліком карбідного методу є
його велика енергоємність.
Кальцій карбід — це сірувато-коричнева маса, забруднена домішками
сульфідів і фосфідів, які під дією води утворюють сірководень і фосфін. Вони й
зумовлюють неприємний запах технічного ацетилену.
Одержання ацетилену гідролізом кальцій карбіду дає великі об'єми
карбідного шлаку, який після попередньої обробки можна використовувати у
будівництві, оскільки він містить переважно гашене вапно.
Із магній карбіду дією води одержують пропін і частково ален:
Mg2C3 + 4Н20 -> СН3-С=СН + 2Mg(OH)2
❖ Піроли етилену і метану. В цьому методі метан або етилен миттєво
1 Фаворський О. (1860-1945)-російський, радянський хімік-органік. Головні дослідження в галузі
хімії ненасичених сполук. Один з основоположників хімії ацетилену.
226
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
нагрівають (менше 0,1 с) до 1 500 °С і потім різко охолоджують, щоб уникнути
розкладу ацетилену до вуглецю і водню:
2СН4==£ НС=СН + ЗН2
1200 °С
н2с=сн2=^нс=сн + н2
Розрахунки умов утворення ацетилену з етилену засвідчують, що рівновага
реакції настає за температури понад 1 200 °С. Проте за цих температур
термодинамічно найстійкішою є система 2С + Н2, тому ацетилен прагне швидко
розкластись на вуглець і водень. З огляду на це вміст ацетилену тим більший,
чим менший час нагрівання середовища, де міститься утворений газ. Тому
ацетилен потрібно різко охолодити після того, як він вийде з реакційної зони, і
хоча він термодинамічно нестійкий, але вже не розкладається з помітною
швидкістю завдяки високій енергії активації реакції розкладу. Оскільки реакція
метану відбувається з розширенням об'єму, то знижений тиск також сприяє
утворенню ацетилену.
Одержана газова суміш реально містить 12-15 % ацетилену.
❖ Окиснювольний піроли метану. Він відрізняється від методу,
розглянутого вище, тим, що до метану додають кисень:
6 СН4 + 02 ^£ 2 НС=СН + 2 СО + 10 Н2
У цьому процесі частина метану витрачається на підтримку високої
температури процесу реакції, а решта зазнає піролізу. Вихід ацетилену -15 %.
Ацетилен виділяють із суміші газів, користуючись його більшою розчинністю в
диметилформаміді (тиск-10 атм) порівняно з іншими вуглеводнями. Цей процес
економічно вигідний ще й завдяки використанню побічних продуктів - карбон
(II) оксиду і водню.
❖ ПіролЬ вуглеводнів. Як сировину використовують газоподібні насичені
вуглеводні або рідкі нафтові фракції - прямогонні бензини, гас. Це один з
головних методів одержання ацетилену. У процесі піролізу рідких вуглеводнів
при 1 200-1 500 °С ацетилен утворюється легше, ніж під час крекінгу нафти.
Зокрема, запропоновано спосіб одержання ацетилену із гасу за допомогою
замикання вольтових дуг під шаром рідини.
Ацетилен відкритий Г. Деві1 у світильному газі 1836 р., де він наявний у
незначній кількості й утворюється, очевидно, також унаслідок крекінгу вуглеводнів.
❖ Одержання ацетилену із "синтез-газу". Як засвідчили останні дослід-
ження, ацетилен можна одержувати в промисловості з карбон (II) оксиду і
водню ("синтез-газ") у м'яких умовах за наявності каталізаторів на основі
похідних графіту:
1 Деві Г. (Davy Н.) (1778-1829) - англійський фізик і хімік. Наукові праці присвячені електрохімії,
засновником якої він був.
Розділ 9. Алкіни
227
ЗН2 + 2СО 100°с > НС=СН + 2Н20
¿ C9FeCl3 + КС10Н8 L
9.4. Лабораторні методи одержання алкінів
9.4.1. Синтез ацетилену з елементів
П. Бертло, пропускаючи вольтову дугу між вугільними електродами в
атмосфері водню, одержав ацетилен з виходом близько 4 % (ендотермічний
процес, АЯ -230 кДж/моль):
2С + Н2 НС=СН
Однак термодинамічний розрахунок засвідчує, що рівновага зміщена
праворуч лише за дуже високої температури (понад 3 000 °С). Цей метод не має
будь-якого практичного значення, проте важливий історично.
9.4.2. Синтези, що засновані на реакціях елімінування
Потрійний зв'язок Карбон-Карбон утворюється так, як і подвійний:
унаслідок відщеплення атомів або груп від двох сусідніх атомів Карбону:
А В А В
_ І І І_І _ _ _
f"-^"" -cd —с"с— -ав —С=С —
С D
Групи, що відщеплюються, і використовувані реагенти ті ж самі, що і в синтезі
алкенів.
❖ Дегідрогалогенування віцинальних дигалогеноалканів і галогеноалкенів.
Ця реакція має особливе значення, оскільки віцинальні дигалогеніди можна легко
одержати з відповідних алкенів, приєднавши галоген. По суті, цей метод є
перетворенням подвійного зв'язку на потрійний у декілька стадій:
нн н н н хтхттт
I І Х7 II КОН І NaNH2 v оі о
С=С^- —С-С— —С=С С=С—, ДеХ = С1, Вг
II 11 1
11 XX X
Наприклад: ^ у-СН-Сг^ К°Н> $ \—С=СН
' ' СН3ОН \—/
Вг Вг 3 4 '
1,2-Дибромостирен Фенілацетилен, 66%
Перша стадія - це важливий метод одержання ненасичених галогенідів. Такі
сполуки з галогеном при подвійному зв'язку називають вінілгалогенідами, вони
228
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
інертні. Отже, в м'яких умовах процес дегідрогалогенування припиняється на
стадії вінілгалогеніду; а в жорстких, у разі використання сильнішої основи,
утворюється алкін:
СН3СН=СН2 + Вг2 СН3(^Н2™™ СНзСВг=СН2^^сСН3С-СН
Пропен Ві» Зг 2-Бромопропен Пропін
1,2-Дибромопропан
За помірних температур (-80 °С) реакцію можна припинити на стадії
одержання вінілгалогеніду, а підвищення температури реакції до 200 °С дає
безпосередньо алкіни. Варто пам'ятати, що застосування дегідрогалогенувального
агента у спиртовому розчині може спричинити міграцію термінального (кінцевого)
потрійного зв'язку до середини ланцюга. Щоб уникнути цього ускладнення,
рекомендують як основу застосовувати суспензію натрій аміду в мінеральній оливі.
❖ Дегідрогалогенування гемінальних дигалогеноалканів. Взаємодія альде-
гідів і кетонів з галогенідами фосфору дає галогенопохідні вуглеводнів з двома
атомами галогену біля одного карбонового атома - гемінальних дигалогенідів,:
Якщо обробляти їх спиртовим розчином їдкого калі, або, ще ліпше, натрій
амідом, то ці сполуки відщеплюють дві молекули гідроген галогеніду й
утворюють ацетилен або його гомологи:
БОСІ, ИаНН,
СН3СН2СНО-^ СН3СН2СНС12 сн3с=сн
Пропаналь 2 1,2-Дихлоропропан. 2ИН3' Пропін
РСЦ КОН
н3с-с-сн2сн3 сн3ссі2сн2сн3^н3с-сес-сн3
О Бутанон 2,2-Дихлоробутан "2Н20 Бут-2-ин
Реакція відбувається за правилом Зайцева. У разі застосування натрій
гідроксиду або калій гідроксиду цю реакцію варто проводити за якомога нижчої
температури, інакше можуть виникнути перегрупування з міграцією кратного
зв'язку в середину ланцюга.
❖ Дегалогенування тетрагалогенідів. Сфера застосування цієї реакції
дещо обмежена, оскільки тетрагалогеніди, власне, одержують з алкінів. Як і у
випадку подвійного зв'язку, потрійний зв'язок можна тимчасово захистити
перетворенням алкіну в насичений тетрагалогенід з наступним відновленням
потрійного зв'язку під дією цинку:
? т"
—с=с— + 2Х2 —► — +2гп —- н-с=с-н + 2 гпх2
X X
❖ Синтез алкінів за Кольбе. Цікавим способом одержання деяких
вуглеводнів - гомологів ацетилену, є електроліз лужних солей ненасичених
Розділ 9. Алкіни
229
дикарбоновнх кислот. Цей спосіб — спеціальний випадок методу Кольбе, що
розглянутий у розділі 7:
-ООС<р=ССОО- Я'СееСК" + 2 с02
Якщо, наприклад, піддати електролізу розплав гідрату калієвої солі
фумарової або малеїнової кислоти, то на катоді утвориться водень, а на аноді -
ацетилен і карбон (IV) оксид:
2Н20 +2е~* Н2 + 20Н"
СНСООК к™!*^ 2к + 2н20 ~2кон + Н2
;г Сі
СНСООК дтт™_-^~ СН-СН + 2с02
;нсоо- _2е_
Калій малеїнат
СНСОО Ацетилен
9 АЗ. Реакціїгомологізації
До такого типу реакцій належать процеси, у яких алкіни одержують зі
сполук, що вже мають потрійний зв'язок. Звичайно, вихідною сполукою є
ацетилен, продукт промислового виробництва.
❖ Взаємодія натрій ацетшіенідів з алкілгалогенідами. Під дією натрію в
рідкому амоніаку або безпосередньо натрій аміду з ацетилену утворюються
натрій ацетиленіди. В цих сполуках різко підвищується нуклеофільність атомів
Карбону порівняно з ацетиленом, і вони легко зазнають алкілування. Отже,
можна послідовно нарощувати карбоновий ланцюг спочатку в одного атома
Карбону з потрійним зв'язком, а потім - в іншого. Поряд з натрієвими
похідними аналогічно можна використати й літійорганічні сполуки, які також є
дуже сильними основами:
5+ 5-
№МН2 , СНХНХНХН.Вг
НС=СН -г-гті НС=С Иа+ 3 2 2 2 » НС=С-СНХН,СН7СН,
. -Ш3 -ИаВг 2 2 2 3
Ацетилен Гекс-1-ин, 89%
№Ш2
н3с-с=сн
Пропін
-Н3С-С=С"Ка+ —, 8+5-
•н,с-с=с У+_1
СН3СН2Вг
н3с-с=с-сн2сн3
Пент-2-ин
-с4н10 з
Ацетиленід-іон атакує електрофільний атом Карбону, заміщуючи йон
галогену в алкані, внаслідок чого утворюється новий алкін:
230
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Ацетиленіди натрію та літію є досить сильними основами -сильнішими, ніж
гідроксид-аніон. З'ясовано, що в процесі синтезу алкенів з акілгалогенідів
основний гідроксид-іон може відщеплювати йон Гідрогену, тобто зумовлює
елімінування (див. розділ 8). Не дивно, що ще більш основний ацетиленід-іон
також дає продукт елімінування:
* І II
-Ст-С -С=С- + X- + НОееСН
І і...
^с^сн
Отже, ацетиленід-іон може реагувати з алкілгалогенідами за двома
напрямами, атакуючи атом Карбону (реакція заміщення) або діючи на атом
Гідрогену {реакція елімінування). Виявлено, що реакційна здатність алкіл-
галогенідів у реакції елімінування зменшується в такому порядку: третинний >
вторинний > первинний. Якщо заміщення відбувається за механізмом Бм2, то
активність змінюється у зворотному порядку: первинний > вторинний >
третинний. Якщо заміщення й елімінування є конкурентними реакціями, то
кількість продуктів елімінування зростає в порядку: первинний < вторинний <'
третинний алкілгалогенід. Багато третинних галогенідів, які швидше вступають
у реакцію елімінування і повільніше - в реакцію заміщення, за цих умов дають
винятково алкени:
Тенденція до елімінування зростає
ДХ: первинний, вторинний, третинний
Тенденція до заміщення зростає
Однозначно реакції гомологізації відбуваються лише з первинними
галогеноалкілами, що не мають розгалужень у а-карбонового атома відносно
галогену; а-розгалужені первинні алкілгалогеніди, а також вторинні й третинні
галогеноалкіли за цих умов переважно перетворюються в алкени:
СН3(СН2)3С=С-СН^СН-СН3 +
СН3(СН2)ГС=С" ЬІ+ + ВгСН— СН-СН3 2-Метилнон-4.ін,32%СН3
СН3 СН3(СН2)3С=СН + (СН3)2С=СН2
1-Бромо-2-метилпропан , Гекс-1-ин 2-Метилпропен,
68%
СН3(СН2)3С=С-СН(СН3)2 +
2-Метилокт-З-ин, 6%
СН3(СН2)3-С=С Ьі+ + СН3СНВгСН3
2-Бромопропан СН3(СН2)3С=СН + СН3СН=СН2
Гекс"-1-ин Пропсн
85%
Розділ 9. Алкіни
231
❖ Реакція Йоцича1. Використання реактиву Гриньяра дає змогу синте-
зувати гомологи ацетилену через проміжний комплекс Йоцича:
НС-СН + СНзї^І -> ПС=С*-МІ+\ + СН4Т
Комплекс Йоцича
НС=С-^І + К-І -> НС=СГЇІ + Ї^І2
Комплекс Йоцича має ті ж властивості, що і реактив Гриньяра, однак меншу
реакційну здатність і, відповідно, більшу стійкість. Його широко застосовують у
синтезі алкінів, каротиноїдів і стероїдних гормонів.
9.5. Хімічні властивості алкінів
Як хімія алкенів переважно є хімією подвійного зв'язку Карбон-Карбон, так
і хімія алкінів - власне хімія потрійного зв'язку Карбон-Карбон. Алкіни, як і
алкени, вступають у реакції електрофільного приєднання, оскільки вони мають
порівняно доступні для атаки я-електрони.
Потрійний зв'язок, порівняно з подвійним, більш реакційно здатний до
реагентів, що є донорами електронів, тобто нуклеофілів. Наприклад, ацетиленіди,
алкоголяти, меркаптиди вступають у реакції нуклеофільного приєднання, які не
властиві алкенам. Відомо, що чим більша частка ^-електронів у гібридних
орбіталях, тим більша електронегативність відповідного атома Карбону. З іншого
боку, завдяки лінійній будові молекули ацетилену ядра атомів Карбону
доступніші для нуклеофільних реагентів.
Оскільки атом Карбону в ацетилені утримує електронну пару зв'язку С-Н
значно міцніше, ніж в алкенах і алканах, то електронна густина зсунута до атома
Карбону, і через те атом Гідрогену здатний до протонізації. Отже, ацетилен
поводить себе як СН-кислота.
Наскільки ж сильною кислотою є ацетилен? Порівняємо його з двома
відомими протонними сполуками - водою та амоніаком. Металічний натрій
реагує з амоніаком з утворенням натрій аміду ЫаЫНг, який є сіллю слабкої
кислоти Н-ЇЧНг:
ЫН3 + Ыа -> Ыа'МЪ" + Н2.
Якщо додавати ацетилен до аміду натрію, що розчинений у діетиловому етері,
то виділяється амоніак і утворюється натрій ацетиленід:
Н-С=С-Н + Ка"*Ш2' -> Н-ОД + НС=С"Ыа+
Сильніша Сильніша Слабша Слабша
кислота основа кислота основа
1 Йоцич Ж. (1870-1914) - сербський хімік. Учень О. Фаворського. Головні праці присвячені
синтезу та ізомеризації ацетиленових вуглеводнів.
232
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Отже, слабшу кислоту (Н-МН2) з її солі витісняє сильніша кислота (НС=С-Н).
Іншими словами, сильніша основа (ИНг-) відщеплює протон від слабшої основи
(НОСГ).
Якщо додавати воду до натрій ацетиленіду, то утворюється натрій гідроксид
і ацетилен. Слабшу кислоту (НС=С-Н) з її солі витісняє сильніша кислота (Н-
ОН).
Н-ОН + НС=СШ+ -> НС=СН + Ка+ОН"
Сильніша Сильніша Слабша Слабша
кислота основа кислота основа
Отже, ацетилен - сильніша кислота, ніж амоніак і слабша, ніж вода. Таку ж
кислотність виявляють інші алкіни, що мають атом Гідрогену в потрійного
зв'язку.
Описаний метод порівняння кислотності ацетилену, амоніаку і води є
загальним, його використовують для визначення кислотності дуже слабких
кислот. Одна сполука буде сильнішою кислотою, ніж інша, якщо вона здатна
витісняти цю сполуку з її солі:
А-Н + В"М+ -» В-Н + АЧУҐ
Сильніша Слабша
кислота кислота
Експериментальні дані свідчать, що ацетилен, як кислота, на 11 порядків
сильніший від етилену і на 9 порядків слабший від води (табл. 9.1).
Таблиця 9.1
Кислотність деяких органічних і неорганічних сполук
Кислота
СНзСООН
Н20
С2Н5ОН
НОСН
Ш3
Н2С=СН2
Н3С-СН3
рК.
4,76
15,7
15,9
25
33
36
48
Отже, для ацетилену характерні три типи реакцій: електрофільне і
нуклеофільне приєднання, а також реакції заміщення, зумовлені кислотністю
гідрогенного атома, що зв'язаний з атомом Карбону потрійним зв'язком. Окрім
того, алкінам, як і іншим ненасиченим вуглеводням, притаманні реакції
відновлення та окиснення.
9.5.1. Гідрування
Гідрування алкінів до алканів проводять за наявності тих же каталізаторів .
(платина, паладій, нікель), що й гідрування алкенів. Помічено, що алкіни
подібно до алкенів не реагують з воднем у момент його виділення.
Реакції відбуваються за наявності каталізаторів, залежно від природи яких
утворюються цис- або /ярянс-алкени чи алкани:
Розділ 9. Алкіни
233
Наприклад:
/я/?ш/с-Гекс-3-ен, -90% Гекс-З-ин і/^-Гекс-З-ен, -98%
2Н
СН^С=ССН^ 7 * СН3СН2СН2СН3
Бут-2-ин Бутан
Реакція гідрування потрійного зв'язку алкінів відбувається повільніше, ніж
подвійного алкенів аналогічної будови. Однак селективне гідрування
ацетиленових вуглеводнів у суміші з алкенами можливе, насамперед, тому, що
алкіни легше адсорбуються на поверхні каталізаторів і не допускають на неї
молекул алкенів. Ця обставина дає змогу селективно гідрувати ацетилен в
етилен.
■ Механізм відновлення алкінів за методом Берна до т/шнс-алкенів такий.
Перехід електрона від атома Натрію спричиняє розщеплення потрійного зв'язку
з утворенням аніон-радикала, який зазнає протонування амоніаком до вінільного
радикала:
Я-СЕЄ-ІІ +На— [КОС*]^ ,С=Сч*-=* ,С=С.
2 н к н
Такий /нрш/с-радикал стійкіший, ніж г/нс-ізомер. Далі вінільний радикал
відриває від атома Натрію електрон, перетворюючись у вініл-аніон, а
протонування вініл-аніона дає трянс-алкен:
н її * н я 2 н я
Неактивовані алкени за методом Берча далі до алканів не відновлюються.
■ Дією літій алюмогідриду дизаміщені алкіни можна відновити до транс-
алкенів:
Т ЛІН ^^Я^^2\
СНзСН^С^С-СН^СНз І^Зі ,С=СЧ
Гекс-З-ин Н СИ^СНз
/и/нжс-Гекс-З-ен, 96%
234
Чирва В.Я., Ярмолкж С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
9.5.2. Електрофільне приєднання
❖ Галогенування. Реакція відбувається за тим же механізмом АЕ> як і в
алкенів, однак повільніше, ніж для олефінів. Цю реакцію каталізують світло або
кислоти Льюїса БеСІз, БЬСІз тощо. На першій стадії утворюється я-комплекс:
Н3С-С=СН +Вг2^=^Н3С-СїСН
5^ 8+
Вг-Вг
Друга стадія реакції утворення місткового бромонієвого катіона є лімітувальною:
НХ-СЇСН =^ Ш>С=СН + Вг
3 т v/
Вг-Вг Вг
На третій стадії відбувається атака аніона Брому до бромонієвого йона з
протилежного боку, що забезпечує тираис-приєднання:
^ Вг Н
НХ-С=СН+Вг ^ чг=г/
V/ Н3С Вг
Вг 3
траисЛ ,2-Дибромопропен
Далі трянс-дигалогеноалкени зазнають галогенування важче, через що їх
можна легко виділити:
Н3СЧ Вг ВГз ВгВг
н3с-с=с-сн3+вг2— ,с=с — н3с-<р-<р-сн3
Бут-2-ин -20 3 20 °С Вг Вг
/Ьрш/с-2,3-Дибромобут-2-ен, 67% 2,2,3,3-Тетрабромобутан, 95%
Сповільнення реакції електрофільного приєднання до алкінів порівняно з
алкенами пояснюють більшою стійкістю і, відповідно, меншою нуклеофільністю
я-орбіталей алкіну, а також меншою стійкістю вінілкатіону, який утворюється,
що дає вищу енергію активації:
Я-С^С-Я + Е+ К/С=С-Я
Е зр
Алкени в цьому випадку утворюють стійкіший карбокатіон унаслідок приєднан-
ня електрофіла:
/С-С. + Е к с_с
Розділ 9. Алкіни
235
■ Різниця реакційної здатності алкенів і алкінів достатня для того, щоб
селективно приєднати бром до активнішого подвійного зв'язку:
Н2С=СН-СНГСЕСН + Вг2 СН2Вг-СНВг-СНгС=СН
Пент-І-ен-4-ин 4,5-Дибромопент-1-ин
Проте у випадку спряження подвійного і потрійного зв'язків приєднання
галогенів відбувається за місцем потрійного зв'язку більш термодинамічно
стійкого спряженого дієну:
Я-С=С-СН=:СН2 + Вг2 —- ІІ-СВг=СВг-СН=:СН2
■ Ацетилен згоряє в потоці хлору з вибухом, тому процес хлорування
проводять у розчиннику. За наявності активованого вугілля еквімольні кількості
реагентів дають продукт монохлорування:
нс=сн + сі/Т°Глл>я С1СН=СНС1
Ацетилен 1,2-Дихлороетен, 90%
❖ Приєднання гідроген галогенідів. Дві молекули гідроген галогенідів
приєднуються до потрійного зв'язку згідно з правилом Марковникова. В м'яких
умовах можливе виділення проміжного продукту:
X Н X
—с=с ^* -с=с—^—<у—с^-
де X = Вг, СІ, І.
СІ
Наприклад: СН3С=СН СН3С=СН2 СН3ССН3
СІ СІ
2-Хлоропропен, 56% 2,2-Дихлоропропан, 44%
Пропін
// у_с=с„. _їїа_ // >-с=СН2
\_/ сн.соон \=/ і
Фенілацетилен (І-Хлоровініл)бензен
❖ Гідроборування алкінів. Ця реакція відбувається стереоселективно як
і/ис-приєднання:
я-с=с-к ——-
с=.с
1_ н ВИ'
* с=с
Н ВЯ',
Одержані борани після обробки відповідними реагентами можуть давати цис-
алкени або оксосполуки:
236
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
с=с
н н
н2о
щсоон~/ С^
на
с=с
н он
2 II
о
9.5.3. Вільно радикальне приєднання
❖ Галогенування. Галогени до алкінів можуть приєднуватися і за
вільнорадикальним механізмом:
ц Н3Сч Вг Н3С Н
н3с-с=сн + Вг2 ^* с=с + с=с
Пропін Вг Н Вт Вг
транс- і і/мс-1,2-Дибромопропен
У цьому випадку стереоселективності нема.
Вільний радикал галогену приєднується з розривом одного я-зв'язку. У
цьому разі утворюються два ізомерні алкенільні радикали, у яких неспарений
електрон перебуває на $р2-гібридній орбіталі. Подальша взаємодія з молекулою
брому дає суміш геометричних ізомерів:
Ізомерні алкенільні радикали
❖ Гідробромування. За наявності пероксидів гідроген бромід приєднується
за радикальним механізмом, що пов'язано з пероксидним ефектом Караша:
С4Н—С=СН 4- НВг К0(Ж » С4Н9СН=СНВг
Гекс-1 -ин 1 -Бромогекс-1 -єн, 74%
9.5А. Нуклеофільне приєднання
Як уже зазначено, на відміну від алкенів, алкіни можуть вступати в реакції
нуклеофільного приєднання (Ан). Приєднання до ацетилену таких нуклеофіль-
них реагентів, як вода, спирти, аніони органічних кислот, відбувається за
наявності солей Купруму (І) або Меркурію (II). Каталізатори відіграють клю-
чову роль в утворенні я-комплексу за потрійним зв'язком алкіну.
❖ Приєднання гідроген хлориду. За наявності каталізаторів приєднання
гідроген хлориду відбувається як нуклеофільна атака хлорид-аніону тс-ком-
плексу алкіну і катіона металу:
Розділ 9. Алкіни
237
Н
І
С
III
с
І
н
+ Си+С1"
н
¿5+
■ . ІН—
С5+
сі- і
Си+
сі-н+
Н Си
С і сі-
с^н+
СІ н
н н
И + Си+С1"
С1 н
За наявності солей двовалентного Меркурію з ацетилену і гідроген хлориду
вперше в промисловості налагоджено виробництво вінілхлориду:
150-300 °С
НСНСН + НС1
Ацетилен
Hg2"
Н2С=СНС1
Вінілхлорид
❖ Приєднання води {реакція Кучерова1). Алкіни гідратуються важче
порівняно з алкенами, проте, за наявності солей Меркурію (II) цей процес
значно полегшений. На першій стадії йон Меркурію утворює я-комплекс, який
зазнає нуклеофільної атаки молекулою води. Утворення я-комплексу сприяє
збільшенню електрофільності ацетиленового атома Карбону. Далі меркурований
вініловий спирт зазнає ізомеризування з демеркуруванням до оцтового
альдегіду: v
2+
HOCH
Hg*
нсХ*
сн
л-Комплекс
Hg-CH=CH-0-H-
—-н
Hg*
• [сн2=о-о-н] —*
сн3сн=о
Гомологи ацетилену в цій реакції дають кетони:
СН3(СН2)3-С=СН + Н20 СН3(СН2)ГС-СН3
Гекс-1-ин О
Гексан-2-он, 80%
■ Структуру з ОН-групою біля подвійного зв'язку називають енолом.
Здебільшого, енольна форма нестійка і перегруповується в оксосполуку. Тут
виникає кето-єнольна таутомерія - динамічна рівновага між цими двома
структурами, однак зазвичай вона дуже посунута в бік кетонної форми. Отже,
якщо гідратувати ацетилен, то як проміжна сполука утворюється вініловий
спирт, який швидко перетворюється на ацетальдегід.
Такі кето-єнольні перегрупування відбуваються особливо легко внаслідок
полярності зв'язку О-Н. Протон легко відщеплюється від атома Оксигену, але у
випадку оборотного приєднання він може приєднуватись як до атома Оксигену,
так і до атома Карбону. Якщо він приєднується до атома Карбону, то повторне
відщеплення протона ускладнене, тобто енергія гетеролітичного розщеплення
Кучеров М. (1850-1911) - російський хімік. Головні праці присвячені органічному синтезу.
238
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
зв'язку С-Н більша, ніж зв'язку О-Н. Отже, оксосполука буде переважати в
рівноважній суміші. Ця реакція слугує ще одним прикладом перенесення
протона від слабкої основи до сильнішої:
І І
—с=с-он
Сильніша кислота Слабша кислота
слабша основа сильніша основа
❖ Реакції віншування
• Приєднання спиртів. Процеси віншування почав досліджувати 1887 р.
О. Фаворський, який відкрив реакцію вінілування спиртів алкінами за наявності
порошкоподібного їдкого калі й тиску в декілька атмосфер:
кон
нс=сн + с2н5он -~ н2с=сн-ос2н5
Ацетилен Етанол Вінілетиловий етер
(надлишок)
Про механізм реакції можна сказати таке: оскільки спирт як кислота значно
сильніший від ацетилену (рЯа, відповідно, — 15,9 і 25), то під дією основи зі спирту
утворюється етилат-аніон,- який, як сильний нуклеофіл, атакує ацетилен.
Карбаніон, що утворився, відриває протон від етилового спирту і так далі:
кон „ тт ^_
С2Н5ОН =р= с2н5о
НС=СН + С2Н50" —^ сн=снос2н5
сн=снос2н5+с2н5он —- н2с=сн-ос2н5 + С2Н50"
Швидкість вінілування зменшується в разі переходу від первинних спиртів
до третинних.
Приєднання ще однієї молекули спирту утворює ацеталь, тому вінілові
етери синтезують за надлишку ацетилену:
ô~0 >^N.. с2н5он
н2с-сн-^ос2н5 c^5j^ сн3сн(ос2н5)2
1,1-Діетоксиетан
■ Спирти приєднуються до ацетилену також за наявності солей Меркурію і
кислих агентів:
нс=сн iw? сн2=сн-ос2н5
■ Гідролізом вінілетилового етеру О. Фаворський і М. Шостаковський1
одержали оцтовий альдегід:
1 Шостаковський М. (1905-1983) - радянський хімік-органік. Головні дослідження в галузі хімії
ацетилену та елементоорганічних сполук.
\5-
I 5-
-с—О
+ н+
¥ і
-<р—с=о
Розділ 9. Алкіни
239
ьі2с=сн-ос2н5-^НзС-сно + С2Н5ОН
Ацетальдегід
Цю реакцію взято за основу екологічно чистого виробництва ацетальдегіду
в Німеччині і Радянському Союзі замість реакції Кучерова, де застосовують солі
меркурію і сульфатну кислоту.
• Синтез вінілацетату та акрилонітрилу. Приєднання до ацетилену оцтової
та ціанідної кислот дає, відповідно, вінілацетат і акрилонітрил:
О
НС=СН + СН3СООН * сн2=сно-с-сн3
Н3Р04/С Вінілацетат
НС=СН + HCN CH2=CH-CN
CuCl + NH4C1 Акрилонітрил
Сьогодні такий метод одержання акрилонітрилу не має промислового
значення, оскільки розроблено ефективніший метод його синтезу - неповне
спалювання пропілену в потоці амоніаку.
• Інші реакції віншування. Як виявив В. Реппе, нуклеофільне приєднання до
алкінів за наявності гідроксидів або алкоголятів лужних металів характерне не
лише для спиртів, а й для фенолів, тіолів, амінів, амідів. Процес відбувається за
підвищеної температури (150-180 °С) і тиску (7-35 атм):
r—sh rnh2
2-+ H^CH-NHR
h- НС=СН —
Вініламін
Я о
R-C-NH, II
2HX=CH-NH-C-R
Н2С=СН-8Я
Вініловий тіоетер
Н2С=СН~"0 С^Н^ —
Вінілфеніловий етер Вініламід
Одержані ненасичені сполуки використовують як мономери в реакціях поліме-
ризації та кополімеризації.
9.5.5. Реакції заміщення
❖ Реакції металування. Завдяки кислій природі атома Гідрогену в
потрійного зв'язку ацетилен легко утворює металоорганічні сполуки під дією
сильних основ або металоорганічних реагентів, наприклад, реактиву Гриньяра:
НС=СН ^ нСЕЕС*а+ Ж+Ыа-СЕСг4а+
НС^СН ^ НС^С Ьі+ ^ +ЬГС-С Ьі+
240
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
НСЕЕСН ^ НСЕСМвІ ^ ІМ^СМеІ
Кислі ацетилени, тобто ті, що мають атом Гідрогену в потрійного зв'язку,
реагують з деякими йонами важких металів, головно, іонами Арґентуму і
Купруму, даючи практично нерозчинні ацетиленіди. Наприклад, утворення
осаду, що є комплексом арґентум ацетиленід-арґентум нітрат, якщо додавати
алкін до розчину Арґентум нітрату в спирті, є якісною реакцією на групу =СН:
Я-С=СН + 2AgNOз -* Я-ОС^^ИОз)! + Нг\Ю3
Цю реакцію можна використати, щоб відрізнити алкіни з потрійним
зв'язком на кінці ланцюга від алкінів з потрійним зв'язком в іншому положенні,
які не здатні вступати в реакцію.
Якщо ацетилен або його гомологи з кінцевим потрійним зв'язком
взаємодіють з амоніачним розчином купрум (І) оксиду, то випадають осади
нерозчинних ацетиленідів, забарвлені від вишнево- (СиС=ССи) до жовто-бурого
(ІІС=ССи) кольорів:
НС=СН + 2[Си(НН3)2]+ОН- -> СиС=ССи| + 4Ш3 + 2Н20
ЯС=СН + [Си(НН3)2]+ОН" -> КС=ССи| + ЖН3 + Н20
Арґентум ацетиленід у сухому вигляді досить чутливий до ударів і вибухає без
виділення газоподібних продуктів. Купрум ацетиленід стійкіший, і його можна
застосовувати в лабораторних синтезах без особливого ризику. Обидві сполуки стійкі
в нейтральних водних розчинах і зазнають гідролізу розбавленими кислотами.
У металоорганічних сполуках ацетилену є різні типи зв'язку Карбон-метал.
Наприклад, у НС=СПЧа+ зв'язок іонний, а в НС^С5-!^54! - ковалентний
полярний. Такі ацетиленіди легко розкладаються водою.
£_о=с_си В ацетиленідах Арґентуму і Купруму зв'язок кова-
| лентний малополярний, що дає змогу їм існувати у воді.
ІІ-С=С-Си Через сильні міжмолекулярні донорно-акцепторні зв'язки
| (я-електрони потрійного зв'язку відіграють роль донорів
^"~С=С-Си електронної густини, а вакантні орбіталі металу -
акцепторів) утворюються псевдополімерні структури.
Ацетиленіди легко вступають у реакції нуклеофільного заміщення з
галогенопохідними (див. детальніше 9.4.3) або приєднання з альдегідами,
кетонами, карбон (IV) оксидом, утворюючи ненасичені спирти і кислоти.
Окиснення купрум моноалкілацетиленідів використовують для одержання
ди- і полієнових сполук (див. далі 9.5.6).
■ Реактив Йоцича досить зручний для синтезу не лише гомологів
ацетилену, а й спиртів і органічних кислот, що мають потрійний зв'язок.
Принципово ці реакції є перекладанням загальних реакцій з реактивом Гриньяра
на реакції з алкінієвим нуклеофілом, наприклад:
Розділ 9. Алкіни
241
g g і ÇH3 ÇH3
HCHC-MgI+CH,CCH, —- hcsc-ç-ch,—hc=c-ç-oh
V oMgi3-M^(0H) CH3
Метоп 2-Метилбут-3-ин-2-ол
ô- 5+ b+r\ * H, О
HC=OMgI + C=0 —" HC=OCOOMgI 2 > нс=с-соон
" 11 c " Mgl(OH) Пропіолова кислота
❖ Реакція Фаворського. О. Фаворський, у лабораторії якого працював Ж.
Йоцич, пішов шляхом спрощення цих синтезів і запропонував проводити
подібні реакції в абсолютному диетиловому етері й за наявності сухого
порошкоподібного їдкого калі:
R'
j^C=p + НС^СН Шїі^ RCC^CH
R^ ^ I
OH
Напевно, в цьому синтезі під час взаємодії з безводним їдким калі
утворюється ацетиленід-аніон, який реагує з оксосполуками (мурашиний
альдегід і його гомологи, кетони) за механізмом нуклеофільного приєднання
(детальніше див. розділ 17). Так одержують первинні, вторинні і третинні спирти
з етинільними радикалами:
НС=СН + НСГ — НС=СГ + Н20
НС=СГ+ СН20 —^ НС=С~СН2ОН
Формальдегід Пропаргіловий спирт
нс=сг + h3c-çQ5"— нс=с-с~сн3 нс=с-сн-сн3
А ^ . H Бут-З-ин-2-ол
Ацетальдегід J
ÇH3
НСНС" + H3C-Çf-CH3 ^§gr нс=с-с-он
о сн3
Ацетон 2-Метилбут-З -ин-2-o л
❖ Реакція Penne. За наявності купрум (І) ацетиленіду або хлориду, що стабі-
лізовані сполуками Вісмуту, ацетилен приєднується до альдегідів:
H
r>C=0 + HCWH 2^ RCC^CH
ОН
242
Чирва В.Я., Ярмолюк СМ., Толкачова H.В., Земляков О.Є. Органічна хімія
2 ^>С=0 + НС^СН
H
6н
он
Особливо важливе приєднання ацетилену до формальдегіду, що веде до
утворення пропаргілового спирту СН=С-СН2ОН або бутин-1,4-діолу:
НОСН + 2НСНО НОСН2С=ССН2ОН
Бутин-1,4-діол
Бутин-1,4-діол є вихідною речовиною для промислового синтезу тетрагідро-
фурану і бутадієну (Німеччина).
❖ Галогенування. До реакцій заміщення атома Гідрогену в ацетилені
належить взаємодія ацетилену з натрій гіпогалогенітом ЫаНаЮ у лужному
середовищі:
Вг7 + ЫаОН
-NaBr,
-Н20
Вг-С=СН
Бромоацетилен
-NaBr,
-Н20
Вг-С=ОВг
Дибромоацетилен
Ці сполуки нестійкі й самозаймаються на повітрі.
❖ Карбонілювання алкінів {В. Penne). До цієї групи реакцій належить
карбонілювання ацетилену карбон (II) оксидом і сполуками, що мають рухливий
атом Гідрогену (такі як вода, спирт, первинні й вторинні аміни) за наявності
Нікол тетракарбонілу під тиском з утворенням акрилової кислоти або її
похідних.
НС=СН + СО
Ni(CO)4
H2Q
ROH
R—NH,
- гі^СН-СООН
Акрилова кислота
- Н2С=СН-СО(Ж
Естери акрилової кислоти
•н2с=сн-сошіі
Аміди акрилової кислоти
Уважають, що я-комплекс ацетилену з Нікол тетракарбонілом атакує нукле-
офільна молекула карбон (II) оксиду з утворенням інтермедіату -
циклопропенону, що розкладається під дією нуклеофілів.
н
н
8+
с
І
H
+ Ni(CO)4-
Ô=C
^ 'Jt*"Ni(CO)3 -
Ґ*- Ç8+ v '3-ЩСО)3
0=
нх
H
н
С=С, +0
с:
X
H
Розділ 9. Алкіни
243
9.5.6. Реакції оліго- і полімеризації
На відміну від алкенів, алкіни неохоче вступають у реакції ланцюгової
полімеризації, що пов'язано з інертністю їхнього я-зв'язку. Проте вони легко
вступають у різні реакції олігомеризації.
❖ Ди- і тримеризація ацетилену. Вперше димеризацію ацетилену виконав
Дж. Ньюленд1 у 1925 р. за наявності хлоридів Купруму (І) й амонію. Реакція
відбувається як нуклеофільне приєднання НОСГ за місцем потрійного зв'язку:
НС=СН нс=с-сн=сн2
Вінілацетилен
Процес може тривати і далі з утворенням дивініл ацетилену Н2С=СН-С=С-СН=СН2.
Вінілацетилен - важливий промисловий продукт. На його основі одержують
низку важливих продуктів органічного синтезу, зокрема, різні мономери:
неї
нс=с-сн=сн2
Н2С=С-СН=СН2
СІ
Хлоропрен
нЕ*н+ Н3С-С-СН=СН2
о 0
Вшілметилкетон
и_
-н2с=сн-сн=сн2
Бутадієн
Особливо цікавий мономер хлоропрен. Полімеризуючи його, У. Карозерс2 і
Дж. Ньюленд 1934 р. одержали бензостійкий американський синтетичний
каучук - неопрен.
❖ Циклоолігомеризація ацетилену. М. Зелінський3 і Б. Казанський4, нагрі-
ваючи ацетилен за наявності активованого вугілля, синтезували бензин:
450 °С
знс=сн ^£
вугілля
1 Ньюленд Дж. (>4іеи\УІап(і ].) (1878-1936) - американський хімік-органік. Дослідження у галузі
хімії ацетилену. Спільно з У. Карозерсом розробив метод одержання вінілацетилену і на його
основі синтетичного каучуку хлоропрену (неопрену).
2 Карозерс У. (Сагойїеге W.) (1896-1937) - американський хімік. Головні праці в галузі хімії
полімерів. Брав участь у розробці виробництва першого в США синтетичного каучуку неопрену і
поліамідного волокна найлону.
3 Зелінський М. (1861-1953) - російський, радянський хімік-органік. Головні дослідження
присвячені органічному синтезу, хімії гетероциклів, органічному каталізу, хімії амінокислот і білків.
4 Казанський Б. (1891-1973) - радянський хімік-органік. Головні дослідження присвячені нафтохімії
та органічному каталізу.
244
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
Цю ж реакцію "мокрим" методом при 60 °С і тиску 15 атм за наявності
Нікол дикарбонілбіс(трифенілфосфіну) пізніше виконав В. Релпе. Він же провів
тетрамеризацію ацетилену над Нікол (II) ціанідом до циклооктатетраєну.
60°С,р \
4НСЕСН——-
Циклооктатетраєн, 80-90%
Завдяки цій реакції циклооктатетраєн став доступною речовиною, яку широ-
ко застосовують в органічному синтезі.
❖ Окиснювольна конденсація ацетилену. Гомологи купрум ацетиленіду під
дією кисню повітря можуть димеризуватись. Залежно від умов реакції одержують
діацетилен (реакція_Глазера!-Залкінда2\ або вінілацетилен (реакція Штрауса):
2ііосн Си2°І2 > гяосси ЯОС-ОСгІ
Н20,МН4С1
2ІЮеСН Си2СІ2 > 2ЯС=ССи -^2^ КС=С-СН=СгЖ
СН3СООН
❖ Окиснювольна полімеризація. Полімеризацією ацетилену за наявності
кисню і каталізатора одержують карбін - полімерну сполуку, що складається
практично з чистого Карбону (нова алотропна форма Карбону):
пНОСН Н-[ОС]п-Н
[Си+] 1 -ш
Реально цей полімерний ланцюг є полікумуленом, де всі атоми Карбону
перебувають взр-гібридизації: •,С=С=С=С=С".
9.5.7. Алкін-аленове перегрупування Фаворського
Алкіни з потрійним зв'язком, що віддалений від кінця ланцюга, під час
нагрівання з металічним натрієм перетворюються в алкіни з термінальним
положенням потрійного зв'язку:
Ка-' Н20
К-С=С-СН3 * Я-СН2-СЕС-Ка Я-СН-СЕСН
Найліпші виходи спостерігають, якщо використовувати як основу калій 3-
амінопропіламід:
1 Глазер K. (Glaser К.) (1841-1935) - німецький хімік. Головний напрям досліджень - хімія
ацетилену, зокрема, вперше одержав фенілацетилен.
2 Ю.Залкінд (1875-1948) - радянський хімік, головні праці присвячені каталітичному гідруванню
ацетиленових сполук. Синтезував низку сполук, близьких до вітаміну А.
Розділ 9. Алкіни
245
NH2(CH2)3NH-K+ Н20
С2Н5-СЕС-С2Н5 *~ НзССС^з СЕСН
Гекс-З-ин Гекс-1-ин, 96%
У разі нагрівання ацетиленідів з крайнім положенням потрійного зв'язку зі
спиртовим лугом, натрій амідом або алкоголятами лужних металів відбувається
переміщення потрійного зв'язку до центру молекули. Реакція відбувається через
утворення аленіву які можна виділити, якщо проводити процес за низької
температури:
R-CH2-OCH К°" ^с"50" R"CH=C=CH2 -R-OC-CH3
(сн3)2сн-с=сн 150о2с5* (СН3)2С=С=СН2
З-Метилбут-1 -ин 3-Метилбутан-1,2-дієн
Механізм реакції, вочевидь, полягає в тому, що спочатку алкоксид-іон
відриває протон від групи СН2, сусідньої з потрійним зв'язком. Далі пара
електронів, що звільнилася, взаємодіє з я-системою потрійного зв'язку, утво-
рюючи систему кумульованих я-зв'язків. Нарешті, за допомогою основи відри-
вається протон від аленового угруповання, і відбувається перерозподіл
електронної густини, що спричиняє переміщення потрійного зв'язку:
\ г< ... C2HsOH ^
RCTC^CH RCH=C=CH і'» RCH=C=CH2
С,Н,07 H^
-г1'5V
CS, ^ C2H5OH
яс^с=сн2 яс== сен, ' ' » кс=с— сн3
2 -с,н,он 2 -с2н5о- 3
с2н5о-^н
■ За наявності купрум (І) галогенідів ацетилен-аленове перегрупування
спокійно відбувається для ацетиленових спиртів і галогенідів:
\ Си2На12 ^
^С-С=СН = ^С=С=СН-Х X = Наї, ОН
X
Си2СІ2
н3с-сн-с=сн н3с-нс=с=сн-сі
СІ 1 -Хлоро-1,2-бутадієн
3-Хлоро-1-бутин
9.5.8. Реакціїокиснення
❖ Окиснення сильними реагентами. Алкіни, подібно до алкенів, зазнають
розщеплення сильними окиснювачами з розривом я- і сг-зв'язків і утворенням
карбонових кислот:
246
Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
К-С=С-К' ^тг4 ЯСООН + НСЮСЯ1
рНЮ
Змінюванням рН середовища процес можна припинити на стадії утворення
дикетонів:
КМпО,
о о
❖ Окиснення киснем. Якщо окиснювати алкіни киснем без каталізатора, то
реакція йде в а-положення відносно атома Карбону з потрійним зв'язком, і
утворюються гідропероксиди:
НСЕС-Сіуі + 02 ► НСНС-СНзООН
Пропін Пропаргілгідропероксид
9.6. Окремі представники алкінів
❖ Ацетилен. Легкий газ, що легко скраплюється. Ацетилен використо-
вують у зварюванні. Для цього його розчиняють під тиском в ацетоні й
зберігають у спеціальних балонах.
Ацетилен - одна з найважливіших для промисловості сполук. Його річне
виробництво перевищило 5 млн т. Він слугує сировиною для промислового
синтезу важливих органічних сполук, які, зокрема, використовують для вироб-
ництва пластмас і синтетичного каучуку. Головні галузі його застосування
показані на рис. 9.1.
• Природні ацетиленові сполуки. Потрійний зв'язок зрідка трапляється в
природних молекулах. Перша природна поліацетиленова сполука відкрита 1935 р.
Далі ця група сильно розширилася. Наприклад, поліацетилени знайдені в багатьох
рослинах (особливо багатими виявились складноцвіті), а також у великій
кількості базидіоміцетів. Зокрема, з різноманітних видів кареопсису вдалося
виділити низку поліацетиленових вуглеводнів з 13 карбоновими атомами:
СН3-С=С-С=С-С=С~С=С—С=С-СН=СН2
СН3-СН=СН-С=С-С=С~С=С-С=С~СН=СН2
сн3-сн=сн~с=с-с=с~сн=сн-сн=сн-сн=сн2.
Ще одним прикладом природних сполук з потрійним зв'язком можуть бути
рослинні ізомерні токсини цикутоксин і енантотоксин, що виділені із
зонтичних - цикути (Cicuta viroson L.) і лабазника (Filipéndula vulgaris L.).
OH OH
Цикутоксин Енантотоксин
Синтетичний каучук
Синтетичне волокно
СНСІ2—СНСІ2
Тетрахлороетан
-Н2С=сн-ск-
Акрилонітрил
Си*
СІ,
СНС1=СНСІ
СІ,
н.
НОСН2—СНЗ-*— Н2С-СН-
Етиловий спирт
СН^ОН-СНОН-СНгОН-
Гліцерол
СН,0Н-С=С-СН20Н-
' Бутин-1,4-діол
НОСН2-СН2-СН2-СН2ОН
Бутан-1,4-діол
Н,С=СН-СН=СН2
" Дивініл
1
Синтетичний каучук
НОНО
НСНО
^НС=С-СН20Н-^
Пропаргіловий спирт
Н2С=СН-СС1=СН2
Хлоропрен
неї
н,с=сн-с=сн
Вінілацетилен
Си^СІг
С4Н5ОН
нс=сн|_н20н,с-сно-
-1 1 г" ug^* з
Ацетапьдепд
КОН
н,с=сн-ос,Н9
Вінілбутиловий етер
неї
СН3СООН Си*
Н* Н^С-
'снсі
Вінілхлорид
Н2С=СН-ОСОСНз
Вінілацетат
. (С4Н5С1)П
Поліхлоропрен (СК)
(СбН,20)п (С^н'Ог)!! (С,ЩС\)п
Полімер етеру Полівінілацетат Поліхлоровініл
(бальзам Шостаковського)
Рис. 9.1. Схема промислового використання ацетилен
Н3С-СН2ОН
Етиловий спирт
НзС-СООН
Оцтова кислота
Н3С-СООС2Н5
Етилацетат
• СН3СНОН-СН2-СНО
Альдоль
СН3СНОНСН2СН2ОН
Бутан-1,3-діол
Н,С=СН-СН=СН2
Дивініл
Синтетичний каучук
Розділ 10
АЛКААІЄНИ
10.1. Будова і номенклатура дієнів
Дієни - сполуки з відкритим ланцюгом, які містять два подвійні Карбон-
Карбонові зв'язки. Загальна формула дієнів аналогічна до брутто-формули
алкінів - С„Н2,,.2. Родоначальник гомологічного ряду Н2С=С=СН2 - ален
(пропадієн).
Якщо подвійні зв'язки розділені одним або декількома атомами Карбону, то
такі сполуки не мають будь-яких нових властивостей порівняно з алкенами, які
описані вище. Наприклад, пент-1,4-дієн і гекс-1,5-дієн мають два рівноцінні
подвійні зв'язки. Вони приєднують хлор, гідроген хлорид, водень за наявності
каталізаторів з такою ж швидкістю, як і пропілен. До того ж, два подвійні
зв'язки реагують незалежно один від одного.
Властивості дієнів з іншим взаємним положенням подвійних зв'язків
відрізняються від властивостей простих алкенів. Саме такі відмінності розгля-
немо в цьому розділі.
10.1.1. Класифікація алкадієнів
Дієни поділяють на три головні групи залежно від розміщення подвійних
зв'язків:
■ кумульовані (алени) - два подвійні зв'язки є в одного атома Карбону,
наприклад: Н2С=С=СН2 - ален;
■ спряжені - подвійні зв'язки розділяє одинарний зв'язок: Н2С=СН-СН=СН2
- дивініл;
■ ізольовані - подвійні зв'язки, розділені одним або декількома атомами
Карбону, наприклад: Н2С=СН-СН2-СН=СН2 - пент-1,4-дієн.
10.1.2. Номенклатура алкадієнів
• Тривіальна номенклатура. Для найпростіших дієнів збереглися тривіальні
назви, наприклад: СН2=С=СН2 - ален, СН3-СН=С=СН2 - метилален, СН2=СН-
СН=СН2 - дивініл, СН2=С(СН3)-СН=СН2- ізопрен.
Розділ 10. Алкадієни
249
• Номенклатура ШРАС. За основу структури вибирають найдовший вугле-
водневий ланцюг, який має обидва подвійні зв'язки. Ланцюг нумерують так,
щоб мінімальні номери одержували обидва атоми Карбону з подвійним
зв'язком. Назву будують аналогічно до назви алканів, замінюючи суфікс -ан на -
дісн. Локанти подвійних зв'язків зазначають перед префіксами і перед
суфіксами. *
2 1
СНзСН2СН2С=СН2
\3 4 5
сн3-с=снсн3
З-Метил-2-пропілпент-1,3-дієн
За основу вибрано найдовший ланцюг, що має обидва
подвійні зв'язки. Нумерація на рисунку забезпечує
мінімальні локанти двом подвійним зв'язкам.
10.1.3. Будова буг-І^-дієна
У молекулі бут-1,3-дієну довжина одинарного С-С-зв'язку 0,146 нм. Це
менше, ніж в алканів або алкенів:
Н2ОСН-СН=СН2
0,146 нм
СН3—СН2—СН2—СН3
0,154 нм
СН3-СН=СН-СН3
0,152 нм
Н ^jkj Н
я-зв'язок
Таке зменшення довжини раніше пояснювали частковим перекриванням я-орбі-
талей двох подвійних зв'язків з неповним усередненням зв'язків у молекулі (я,я-
спряження).
З сучасного погляду в бутадієні зв'язок а-С-С я-зв'язок
утворюється перекриванням 5р2-^р2-орбіталей, а не
5р3-^р3-орбіталей, як в алканах або 5р3-^р2-орбіталей,
як в алкенах. Раніше (див. розділ 9) зазначено, що зі
збільшенням внеску ^-орбіталі в гібридну орбіталь
максимум електронної густини наближається до
ядра атома, тому і зв'язок, побудований з "коротких"
орбіталей, буде коротший.
Під час розгляду структури бутадієну методом МО можна побачити, що
чотири я-електрони розташовуються на двох зв'язувальних МО, а дві
розпушувальні МО є вакантними. Перша МО ОРі) делокалізована по всіх
чотирьох атомах Карбону. Вища заповнена МО (ВЗМО, Ч^) має вузлову
площину між С2- і СЗ-атомами. Третя МО 0Р3) є нижчою вакантною МО
(НВМО) і має дві вузлові площини, а четверта МО (Ч^) - три вузлові площини.
ВЗМО і НВМО мають домінантне значення для реалізації хімічних властивостей
молекули дієну. Фактично ВЗМО має електронодонорні властивості й визначає
взаємодію з електронодефіцитними реагентами, а НВМО, відповідно, -
електроноакцепторні властивості і взаємодію з нуклеофільними реагентами
(рис. 10.1).
250
Чирва В.Я., Ярмолюк CM., Толкачова Н.В., Земляков О.Є. Органічна хімія
*\ *г '•'з *4
Рис. /0./. Обриси AO і MO молекули бут-1,3-дієну
Розрахунки за методом MO засвідчують наявність невеликої делокалізації
я-електронів. Порядок зв'язків С1-С2 і СЗ-С4 -1,89, а зв'язку С2-СЗ -1,47. У '
дивінілі довжина зв'язків С1-С2 і СЗ-С4 більша, ніж подвійного зв'язку в
алкенах (0,134 нм) і становить 0,137 нм.
Спряження подвійних зв'язків збільшує стабільність реальної молекули
бутадієну порівняно з ідеалізованою, у якій немає взаємодії між подвійними
зв'язками. Раніше з'ясовано, що теплоти гідрування алкенів схожої будови-
практично однакові й дорівнюють 113-125 кДж/моль. Для сполук з більшою
кількістю подвійних зв'язків можна було б очікувати, що теплота гідрування
буде сумою теплот гідрування індивідуальних подвійних зв'язків.
Таблиця 10.1
Теплоти гідрування дієнів
Дієн
АЯ, кДж/моль
Дієн
АЯ, кДж/моль
Бут-1,3-дієн
239,4
2-Метилбут-1,3-дієн (ізопрен)
224,7
Пент-1,3-дієн
226,8
2,3-Диметилбут-1,3-дієн
225,4
Пент-1,4-дієн
255,4
Ален
299,5
Гекс-1,5-дієн
254,1
На підставі аналізу даних табл. 10.1 видно, що для неспряжених дієнів ця
адитивність справді простежується. Зменшення теплоти гідрування спряжених
дієнів свідчить про їхню стійкість. Виграш в енергії, що одержують унаслідок
спряження, називають енергією спряження (резонансу), її прирівнюють до
зниження енергії молекули внаслідок делокалізації електронів.
Теорія резонансу описує спряження подвійних зв'язків у молекулі,
наприклад бут-1,3-дієну, набором таких резонансних структур:
снгсн=сн-сн2^- н2с=сн-сн=сн2— сн2-сн=сн-сн2
Навіть неповна делокалізація я-електронів зумовлює специфічний перебіг
низки реакцій. Наочним виявом взаємодії я-орбіталей у дивінілі є відсутність за
звичайних умов вільного обертання навколо С-С-зв'язку. Подвійні зв'язки
розташовані в "s-трансоїдній" конформації відносно простого зв'язку (s - single).
Розділ 10. Алкадієни
251
Бар'єр обертання (інверсії) невеликий і, наприклад, у реакції Дільса^Альдера2
бутадієн реагує в "^-цисоїдній" формі:
СН2
не
8-12 кДж/моль
Н2С^.
сн
ii
сн.
Н2С
сн
,сн
ь-транс-Буї-1,3-дієн б-цис-Бут-1,3-дієн
Про підвищену стійкість спряжених дієнів свідчить і те, що у випадках,
коли це можливо, вони є основними продуктами в реакціях елімінування.
Існування спряження в молекулі бута- е
дієну випливає з УФ-спектра. Довгохвильо-
ва смуга поглинання для цього дієну є на
217 нм і зумовлена електронним переходом
з 7і2- на яз*-рівень. Для порівняння, етилен
поглинає світло на 187 нм. Річ у тому, що
спряження в молекулі бутадієну зменшує
відстань між вищою зайнятою молеку-
лярною орбіталлю Я2 і нижчою вільною
молекулярною орбіталлю Яз*, а це, відпо-
відно, зменшує енергію збудження. Справді,
для сполук з декількома спряженими зв'яз-
ками різниця енергії між граничними орбіталями настільки мала, що молекули
поглинають світло у видимій ділянці, тому полієни є забарвленими сполуками
(рис. 10.2).
р я*
я*4
я*3 НВМО
-Н-яі взмо
/?-Орбіталь Етилен
Бутадієн
Рис. 10.2. Енергетичні рівні ізольованої
р-орбіталі і молекулярних
орбіталей етилену і бут-І,3-дієну
10.1.4. Будова алену
У молекулі алену СН2=С=СН2 два атоми Карбону (перший і третій) є у стані
^р2-, а один - у стані лр-гібридизації. Завдяки цьому виникає таке розташування
л>зв'язків:
1 Дільс О. (Diels О.) (1876-1954) - німецький хімік-органік. Дослідження в галузі органічного
синтезу, зокрема стероїдів і дієнів. Нобелівська премія 1950 р. (спільно з К. Альдером).
2 Альдер К. (Alder К.) (1902-1956) - німецький хімік-органік. Учень О. Дільса. Головні праці в
галузі дієнового синтезу.
252
Чирва В.Я., Ярмолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Система я-зв'язків лінійна, обидва я-зв'язки і кінцеві замісники розташовані
на взаємно перпендикулярних площинах. У випадку несиметричного заміщення
алени існують як енантіомери. Це явище передбачене Я. Вант-Гоффом. Згодом
припущення вченого підтверджене синтезом оптично активних аленів як
рацематів і наступним розділенням їх на оптичні антиподи:
СНз
н3с^сх —і- щс С=С< + Н3С /С=С=С(
г с=сн ^ с хн Нзсг хн
^ сн3 НзС сн3
3,4,4-Триметилпент-1-ин-3-ол (Я)- і (5)-3,4,4-триметил-1 -хлоропент-1,2-дієн
Особливість таких енантіомерів - у їхніх молекулах нема хірального центру,
але є осі хіральності.
10.2. Фізичні властивості
За нормальних умов перші представники алкадієнів (Сз і С4) - гази, інші -
рідини або тверді речовини.
10.2.1. Спектроскопія дієнів
• УФ-спекпгроскопія. Спряжені дієни поглинають УФ-світло у більш
довгохвильовій частині спектра (215-245 нм), ніж алкени. Збільшення кількості
спряжених зв'язків зсуває смугу поглинання світла у видиму ділянку, а
речовини з п'ятьма спряженими подвійними зв'язками забарвлюються в жовтий
колір.
Для аленів характерне поглинання в ділянці 170-185 нм.
• ІЧ-спектроскопія. В ІЧ-спектрах спряжених полієнів з'являються додаткові
смуги поглинання в ділянці 1 600-1 650 см"1. Наприклад, дієни мають дві смуги,
які відповідають валентним коливанням Ус=с 1 600 см"1, 1 650 см"1.
• ПМР-спектроскопія. Сигнали протонів у атомів Карбону з подвійним
зв'язком у спектрах !Н-ЯМР дієнів є в інтервалі 4,5-6,5 м.ч., наприклад:
Н Н НЬ На „ нЬч ,на
С=С=С С=С=С г-р/ іл <\ ЇЇ
н Н Нь сі нн н
5 4,55 м.ч.
8 5,76 (а), 5,05 м.ч. (Ь)
5 5,11 (а), 5,20 м.ч. (Ь)
6 6,42 м.ч.
Розділ 10. Алкадієни
253
10.3. Одержання і хімічні властивості аленів
10.3.1. Методи синтезу аленів
■ Найпростішим вуглеводнем цього типу є ален СН2=С=СН2, який у лабо-
раторії раніше одержували перетворенням гліцеролу в 1,2,3-трибромопропан,
який спочатку піддавали дегідробромуванню, а потім дебромуванню:
ВгСН2-СНВг-СН2Вг + СНзСЖа -+ СН2=СВг-СН2Вг + СН3ОН + №Вг
СН2=СВг-СН2Вг + Хп -+ гпВг2 + СН2=С=СН2
Аналогічно ален можна одержати із пропену:
Н2С~СпН_снз 500»с* Н2С=СН-СН2С1 сс2і4 * СН2С1СНС1СН2С1 сдод
СІ
—- н,с=с-сн,сі-^»н,с=с=сн,
2 2 -2пС12 2 длен 2
■ Алени можна синтезувати із алкінів перегрупуванням Фаворського (див.
розділ 9).
НзС>н-сесн к-2^н£н П^с=с=сщ
Нзсх 170 -с нзСх 2
2-Метилбут-1 -ин 2-Метилбут-1,2-дієн
■ Сьогодні алени виділяють з ален-метгиіацетиленової фракції, що утво-
рюється під час піролізу бензинів і яку іноді використовують для зварювання
металів замість ацетилену.
10.3.2. Хімічні властивості аленів
Алени помітно не відрізняються за реакціями від олефінів, за винятком
декількох специфічних реакцій. Електрофільне і радикальне приєднання до
аленів відбувається легше, ніж до олефінів через наявність двох сусідніх зв'язків
С=С Вони легше гідруються до алканів, швидко взаємодіють з гідроген
галогенідами та окиснюються до карбонільних сполук.
❖ Гідратація. Під час перегонки водного розчину алену за наявності
концентрованої сульфатної кислоти з водяною парою дієн приєднує воду і
перетворюється в ацетон:
н2с=с=сн2+н2о —
н2с=с-сн3
он
СН0ССН7
3ІІ 3
о
254
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Реакція гідратації алену відбувається за електрофільним механізмом. Як
проміжний продукт утворюється вініловий спирт (енол). О. Ельтеков1 з'ясував, що
вінілові спирти нестійкі - вони швидко зазнають прототропного перегрупування
(міграція протона), перетворюючись в оксосполуки.
> Правило Ельтекова і Ерленмеєра2 - сполуки, що мають гідроксильну
групу біля подвійного зв'язку або дві гідроксильні групи в одного атома
Карбону, нестійкі й перетворюються в карбонільні сполуки.
Інші алени поводяться аналогічно. Наприклад, у разі гідратації димети-
лалену (СНз)2С=С=СН2 головним продуктом реакції є З-метилбутан-2-он
(СН3)2СНСОСН3.
❖ Перегрупування ФавОрського. Під час нагрівання алену з натрієм в етері
він перегруповується в метил ацетилен:
СН2==С==СН2 сн^~с=сн
❖ Циклізація. Алени відрізняються від олефінів здатністю до олігомеризації в
похідні циклобутану за нагрівання понад 130 °С У цьому разі ален утворює димери,
тримери і тетрамери, як описано в працях академіка С. Лебедєва:
н2с=с=сн2
130 °С і # 2 200 °С і г *
500 °С Суміш
л , . ,к олігомерів
1,3-Диметиленциклобутан 1,2-Диметиленциклобута
Димеризація аленів є прикладом реакції циклоприєднання. Най характерні-
шою рисою подібних процесів є утворення стійкої циклічної системи без
виділення побічних продуктів, і кінцева сполука є сумою інгредієнтів двох
реагентів, тобто аддуктом.
Алени широко застосовують в органічному синтезі для одержання
простагландинів, феромонів, вітамінів, лікарських препаратів. Вони виявлені серед
метаболітів нижчих грибів, вищих рослин, а також продуктів життєдіяльності
морських гідробіонтів і комах. З'ясовано, що можна синтезувати кумулени -
сполуки, що мають декілька кумульованих подвійних зв'язків: К2С=С=С=С=С=СК2..
10.4. Одержання 1,3-дієнових вуглеводнів
Для 1,3-дієнових вуглеводнів, що слугують мономерами у виробництві
синтетичного каучуку, розроблено багато спеціальних способів одержання.
1 Ельтеков О. (1846-1894) - російський хімік-органік. Професор Харківського і Київського
університетів. Праці в галузі органічного синтезу.
Ерленмеєр Р. (Егіептеуег Я.) (1825-1909) - німецький хімік-органік. Фундаментальні
дослідження в структурній органічній хімії. Першим висунув гіпотезу подвійного зв'язку.
Запропонував правильні формули етилену, ацетилену, нафталену.
Розділ 10. Алкадієни
255
Особливо це стосується двох вуглеводнів зі спряженими подвійними зв'язками:
бут-1,3-дієну (дивінілу) і 2-метилбут-1,3-дієну (ізопрену).
10А. 1. Синтези дивінілу
❖ Дегідратація бутан-1,3-діолу. Сполуку, що одержують альдольною
конденсацією двох молекул ацетальдегіду, відновлюють у Р-гліколь, який потім
за наявності алюміній оксиду каталітично дегідратується в бут-1,3-дієн:
2Н АІ О /
сн3<рнсн2сно —^— сн3(рнсн2сн2он сн2=снсн=сн2
он он
3-Пдроксибутаналь Бутан-1,3-ДІол Бут-1,3-дієн
❖ Метод Лебедєва. Промисловий синтез 1,3-бутадієну, який застосовували
в колишньому СРСР з 1932 р., зрештою зводиться до попереднього. За цим
методом пари етилового спирту пропускають при 400-500 °С над каталізатором,
який складається з оксидів магнію і цинку. Цей каталізатор має здатність
дегідрувати спирти і, будучи лужним, може зумовлювати альдольну
конденсацію. Наступні процеси гідрування і дегідратації пришвидшують тим же
каталізатором. Отже, різнобічна дія каталізатора Лебедєва дає змогу за один
прохід парів етанолу над каталізатором перетворити його в бутадієн з виходом
-20 %:
С2Н2ОН СН3С<„ +Н2 2СН3СНО^і5^ СН3<^НСН2СНО
Ацетальдегід ОН
З-Гідроксибутаналь
(р-гідроксимасляний альдегід)
— н3с-сн=сн-сно н3с-сн=сн-сн2он
2 Кротоновий альдегід Кротиловий спирт 2
н3с-сн-сн2сно-^-н3с-сн-сн2сн2он н2с=сн-сн=сн2
0Н °Н Бутан-1,3-діол 2 Бут-1,3-дієн
Сумарне рівняння процесу: 2С2Н5ОН —> С4Н6 + 2Н20 + Н2
Механізм реакцій детально вивчений Ю. Горіним1. Модифікацією методу
Лебедєва є одержання дивінілу з ацетальдегіду за наявності складного ката-
лізатора, що має одночасно дегідрувальні й дегідратувальні властивості (оксиди
Магнію, Феруму, Титану, Цинку на казеїні).
Сьогодні цей метод не застосовують, оскільки для одержання бутадієну є
дешевша сировина з нафти.
1 Горін Ю. (1907-1987) - радянський хімік, автор досліджень зі створення синтетичного каучуку.
256
Чирва В.Я., Ярмолюк CM., Толкачова H.В., Зеллляков О.Є. Органічна хімія
❖ Реакції елімінування дигалогеноалканів, що супроводжуються утво-
ренням спряжених дієнів, вирізняються високою регіоселективністю:
СН2С1СН2СН2СН2С1 ^^Н2С=СН~СН=СН2
1,4-Дихлоробутан Бут-1,3-дієн
Серед продуктів реакції завжди переважає спряжений, стабільніший дієн.
Альтернативний 4-метилгекс-1,4-дієн утворюється в мізерній кількості: .
сн3 сн3
н2с=сн-снгс-сн2сн3 -^gi н2с=сн-сн=с-сн2сн3
Вг
4-Бромо-4-метилгекс-1 -єн 4-Метилгекс-1,3-дієн, 78%
❖ Синтези на основі ацетилену. У Німеччині внаслідок нестачі харчової
та нафтогазової сировини змушені були розвивати способи одержання дивінілу
на основі ацетилену.
• Метод Penne ґрунтується на конденсації ацетилену з формальдегідом за
наявності купрум-бісмутового каталізатора, потім - гідруванні одержаного
бутин-1,4-діолу в бутан-1,4-діол, частковій його дегідратації з фосфорною
кислотою в тетрагідрофуран і остаточній дегідратації в бутадієн з загальним
виходом близько 90 %:
2СН20 +НС=СН HO-CHjC=C-CHfOH НОСН2СН2СН2СН2ОН^^
^ ^ н q3 *• СН2=СНСН==СН2
Сьогодні цей метод переважно використовують для синтезу бутан-1,4-діолу.
• Синтез па основі ацетальдегіду. В другому способі одержання бутадієну,
що розроблений у Німеччині, для одержання ацетальдегіду використана реакція
Кучерова:
НСЕСН н"^2+' СН3СНО ==>н2с=сн-сн=сн2
• Синтез на основі вінілацетилену. За основу третього методу взято
селективне відновлення вінілацетилену, синтезованого димеризацією ацетилену:
2НС=СН^!£^
❖ Синтези на основі бутану і бутену: Сьогодні дієни зі спряженими
подвійними зв'язками в промисловості одержують з бутану - головного
компонента супровідних газів у видобутку нафти, або частіше з бутан-бутенової
Розділ 10. Алкадієни
257
фракції нафтогазової сировини, що утворюється під час крекінгу, пропускаючи
їх за високої температури над промотованим хромоапюмінієвим каталізатором:
CHзCH2CH2CHз^^^ СН2=СН-СН=СН2
■ У двостадійному процесі бутан спочатку, за температури 650 °С,
дегідрують у бутени. Потім їх відділяють від бутану, який не прореагував,
розбавляють перегрітою парою і дегідрують за 550 °С до бутадієну. Вихід
кінцевого продукту становить 33-40 %.
■ В одностадійному процесі бутан дегідрують у бутадієн на нерухомому
каталізаторі у вакуумі з контактуванням до 10 хв. Вихід у цьому випадку не
перевищує 14 %.
■ Реакція дегідрування ендотермічна, тому процес ефективніший, якщо
замість звичайного використовують окиснювальне дегідрування. Бутени в суміші
з водяною парою і гарячим повітрям дегідрують за 400-500 °С у реакторах з
нерухомим шаром каталізатора на основі оксидів Феруму:
Н^СН-СН^Нз + 1/2 02 Н2С=СН-СН=СН2+ Н20
Процес має високий вихід (60-75 %) і менші енергетичні витрати порівняно
з методами, що описані вище.
■ Найекономічніше одержання бутадієну з фракції с4 через піроліз рідких і
газоподібних нафтопродуктів. Хоча вихід кінцевої сполуки не перевищує 3-5 %
від сировини і 12-18 % щодо етилену, для якого цей метод розроблений, проте
за кордоном цим способом виробляють понад 80 % бут-1,3-дієну.
■ Промислового використання набуває димеризація пропену над каталіза-
тором Циглера до 2-метилпентену-1, що під час нагрівання перетворюється в
ізопрен і метан:
H2C—-CH ► Н3С""СН^"СН^"С—"СН^ CHj-СН~С—СН2
СН3 СН3 4 CHj
❖ Реакція Прінса. Серед інших методів треба згадати конденсацію
пропілену з формальдегідом над кислотним каталізатором. Спочатку одержують
4-метил-1,3-діоксан, який далі втрачає формальдегід і воду, що веде до
утворення бутадієну:
№ +
НрО —HjC-OH
/.кат.
ЩСН=СК —- RC—( ОН —*- RC—( О СНзКЖБ^Са.
н-оу ас-он -щр
н
258
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
❖ Окиснення вінілкупратів. Один із сучасних лабораторних методів
синтезу дієнів полягає в окисненні вінілкупратів, що утворюються як проміжні
сполуки з вінільних літійорганічних сполук і солей одновалентного Купруму.
Вінілкупрати швидко окиснюються до дієнів:
Кч /Вг СДУл Кч /ЬІСи2Вг2,02 с=Е К
С—С » Г=Г — » / Ч /
/и Ч -78 °С А К .зо °С р С=С
н н н н к а Ч
н н
10.4.2. Синтези ізопрену
❖ Синтези на основі пентану. Ізопрен - гомолог бут-1,3-дієну - у
промисловості сьогодні одержують ізомеризацією пентанової фракції продуктів
переробки нафти (В. Іпатьєв1, 1897 р.). Після ізомеризації ізопентанову фракцію
нагрівають до 600 °С в кип'ячому шарі алюмохромового каталізатора і пе-
ретворюють в ізопентени. Потім ізопентени розводять перегрітою водяною парою
і дегідрують у стаціонарному шарі хромкальційнікельфосфатного каталізатора за
550-650 °С. Вихід ізопрену становить 32-34 %:
А1СЦ 6П0°Г 550-650°С
ЩСНру^СЦ—^н3с-сн-сн2сНз ^ > н^с-с^чн, -^щс^с-т-с^
Шпж СНз ЛДСВД СНз Кат. Сг^
Ізопенган 2-Шилбут-2-ен Ьопрен
■ Пентан-ізопентанова суміш утворюється також під дією кислот Льюїса
(АІОз, А1Вг3, ВР3) і протонних кислот на фракцію газів крекінгу нафти або
природного газу:
н2с=сн2+ сн3сн2сн3-^р сн3(сн2)3сн3 + н3с-сн-сн2сн3
СН3
Цей процес досить важливий з екологічного погляду, оскільки дає змогу
утилізувати супровідні нафтові гази і гази крекінгу.
■ Найекономічнішим способом виробництва ізопрену є його виділення в
процесі піролізу рідких нафтопродуктів с5, який використовують для вироб-
ництва етилену. І хоча вихід за етиленом становить лише 2-5 %, проте у світі
гаким способом виробляють понад 70 % ізопрену.
❖ Реакція Прінса. Конденсацією ізобутилену з формальдегідом у про-
мисловості за реакцією Прінса одержують 4,4-диметил-1,3-діоксан, який
дегідратують в ізопрен:
1 Іпатьєв В. (1867-1952) - російський хімік-органік. Головні дослідження в галузі каталітичної
органічної хімії.
Розділ 10. Алкадієни
259
н2с=с(сн3)2 <(~У"3 Са-(Р^Р0- н2с=с-сн=сн2+н2о + сн2о
1 5 1 Н+, 220 °С \ / Утт 220 °С 2 ■ 2 2 2
снз СН3
Як вихідну сировину використовують фракцію вуглеводнів с4 з вмістом
ізобутилену не менше 40-50 %.
❖ Синтез на основі реакції Фаворського. Ізопрен можна синтезувати з
ненасиченого спирту 2-метилбут-3-ин-2-олу (так званий клей Назарова1),
одержаного за реакцією Фаворського, який електрохімічно відновлюють до
диметилвінілкарбінолу (2-метилбут-3-ен-2-ол). Дегідратація диметилвінілкарбі-
нолу дає ізопрен (у сучасній технології цей метод не застосовують):
(Сг^зОО +НСЖИ (СНз)2(^-С^СН ^^С-СН^Сг^ Щ=<рСгКБ2
2-К^ггилбуг-3-ин-2-ол 2-^fc^илDyг-3-eн-2чш
10АЗ. Синтези хлоропрену
Близьке до бутадієну та ізопрену за принципами синтетичних схем
виробництво ще одного дуже важливого мономеру - хлоропрену (2-хлоробут-
1,3-дієну).
❖ Синтези на основі вінілацетилену. Одержання цього ключового
продукту для багатьох мономерів розробили 1932 р. Дж. Ньюленд і У. Карозерс.
Приєднання гідроген хлориду відбувається вибірково за місцем потрійного
зв'язку. Синтез на обох стадіях характеризують високі виходи:
нс=сн + нс=сн 2н*654» нсес-сн=сн2-^ н2с=с-сь^сн2
Вінілацетилен сі Хлоропрен
❖ Синтези на основі дивінілу. Сучаснішим уважають метод одержання
хлоропрену з бутадієну:
^с-сн-сн^о^
а а
3,4-Дклоробуг- 1-ен
Си+ МаШ
—^щ:-ш-сн=сн,—«►н2с=с-сн=снг
а а " а
д Ьомгризація 3,4-ДкгюробуН-ен
1,4-Дкпоробут-2-ен
1 Назаров І. (1906-1957) - радянський хімік. Наукові дослідження присвячені хімії ацетилену.
260
Чирва В.Я., Ярллолюк СМ., Толкачова H.В., Земляков О.Є. Органічна хімія
10.5. Реакції дієнів
Як відомо, реакції дієнів з ізольованими зв'язками не відрізняються від
реакцій олефінів: приєднання відбувається за двома подвійними зв'язками, а за
браком реагенту - по одному. Наведемо лише особливі властивості метиленової
групи в дивінілметані. Раніше зазначено про підвищену реакційну здатність
атомів Гідрогену метальної і метиленових груп, що примикають до подвійного
зв'язку. У сполуках із системою зв'язків -СН=СН-СН2-СН=СН- рухливість
атомів Гідрогену метиленової групи також висока. Такі дієни здатні до легкого
окиснення з утворенням гідропероксидів:
RCH=CH-<pH-CH=CHR
ООН
10.5.1. Реакції 1/2- і 1,4-приєднання
Інші властивості має бут-1,3-дієн, який називають просто бутадієном. К
ньому два подвійні зв'язки перебувають у спряженому стані. Це означає, що за
участю спряженого дієну в реакції приєднання обидва центри ненасиченності,
як звичайно, функціонують як єдине ціле, а не як ізольовані подвійні зв'язки.
Якщо пент-1,4-дієн, що має ізольовані подвійні зв'язки, реагує з браком брому
за йонним механізмом (АЕ), то утворюється 4,5-дибромопент-1-ен, який і
прогнозували. Приєднання великої кількості брому дає 1,2,4,5-тетрабромопентан:
СН2=СНСН2СН=СН2
Вг,
* СН^СНСН^СН—CHw-
І ! 2
Вг Вг
Вг,
сн^снсн^снсн^
І і 21 І 2
Вг Вг
Вг Вг
У реакції бутадієну з одним еквівалентом брому за тих же умов утворюється
не лише очікуваний продукт - 3,4-дибромобут-1 -єн, а також і 1,4-дибромобут-2-
ен. Дією 1 моль гідроген броміду одержано не тільки З-бромобут-1-ен, а й 1-
бромобут-2-ен. Відновлення воднем у момент виділення, наприклад, дією
натрієм в амоніаку або амальгамою натрію в водному спирті, дає як бут-1-ен, так
і бут-2-ен. Подальше гідрування алкенів, що утворилися за цих умов, не
відбувається:
вг ^ (рн2<|:нсн=сн2 + <^н2сн=сн(|н2
Вг Вг Вг Вг
СН2=СНСН=СН2
НВг
(j^HCB^CHj + (];н2сн=снс]:н2
Н Вг Н Вг
ÇH2ÇHCH=CH2 + ÇH2CH=CHCH2
н н н h
Розділ 10. Алкадієни
261
Дослідження взаємодії серії спряжених дієнів з багатьма реагентами
засвідчило, що така поведінка характерна для всіх спряжених дієнів - реагент
може приєднуватися не лише до сусідніх карбонових атомів (1,2-приєднання,
або пряме приєднання), а також до двох кінців спряженої системи (1,4-
приєднання, або спряжене приєднання). Здебільшого, продукт 1,4-приєднання є
головним продуктом реакції.
❖ Гідрогалогенування. Відомо, що електрофільне приєднання відбувається
через найстійкіший карбокатіон. Застосуємо цей принцип і у випадку при-
єднання гідроген хлориду, наприклад, до гекс-1,3-дієну, яке дає 4-хлорогекс-2-
ен і 2-хлорогекс-З-ен:
СН3СН=СНСН=СНСН3 ^ СН3СНСгіСН=СНСН3 + СН3СНСН=СНСНСН3
Н СІ Н СІ
4-Хлорогекс-2-ен 2-Хлорогекс-З-ен
Спочатку приєднання протона до подвійного зв'язку приводить до
утворення стійкого алільного карбокатіону (1), у якому позитивний заряд
делокалізований завдяки спряженню з подвійним зв'язком. Алільний карбо-
катіон стійкіший, ніж найстійкіший третинний алкільний карбокатіон. Оскільки
реакція відбувається через найстійкішу проміжну сполуку, то йон (2) у реакції
практично не бере участі:
СН3СН=СНСН=СНСН3 + Н+
^СН3СНСНСН=СНСН3—СН3СНСН=СНСНСН3
н о) й
^сн3£нснсн=снсн3
Н Р)
На другій стадії хлорид аніон атакує два атоми Карбону, що несуть
позитивний заряд, і утворюються продукти 1,2- і 1,4-приєднання:
СН3(рНСН^СН^СНСН3 СН^Н^НСНКЖНз + сн3снсн=сн([:нсн3
н + н сі н сі
Відносні кількості продуктів 1,2- і 1,4-приєднання залежать від умов
перебігу реакції - температури, тривалості процесу, природи розчинника.
Продукт 1,4-приєднання термодинамічно стійкіший, тоді як 1,2-приєднання
відбувається з більшою швидкістю.
Наприклад, якщо гідробромування бут-1,3-дієну проводити при -80 °С, то
зворотна реакція дегідробромування не відбудеться. За цих умов реакція
контрольована кінетично, а головним продуктом є З-бромобут-1-ен (81 %). Якщо ж
температуру реакції підвищити до 20 °С, то поряд з прямою реакцією
262
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
відбуватиметься процес дегідробромування, і в реакційній суміші буде
накопичуватись 1-бромобут-2-ен, тому що, згідно з правилом Лебедева, як більш
заміщений алкен, він термодинамічно стійкіший. Вихід цього продукту при -80 °С
збільшується з 19 до 58 %. А якщо проводити реакцію за температури 40 °С, то
вміст 1-бромобут-2-ену досягне 85 %. Отже, за підвищеної температури йдеться про
реакцію, що контрольована термодинамічно.
❖ Електрофільне галогенування. Із наведених вище даних випливає, що
електрофільне приєднання до дієнів починається з утворення л-комплексу по
одному з подвійних зв'язків, який швидко перетворюється в стабілізований
резонансом аліл-катіон, наприклад:
щ^т-ш-а^ ь^с^сн-сн^сНз^г Вгсн^^н-а^--- Вгсн^^-щ
За низької температури (-80 °С) бромід-аніон не встигає відійти від
карбокатіону, який тільки що утворився, і приєднується до С2 атома, даючи 3,4-
дибромобут-1 -єн.
Ця сполука синтезується швидше від іншої, її називають продуктом реакції,
що контрольована кінетично:
Н2С=СН-СН=СН2 + Вг2 —- ВгСН2-СН-СН=СН2—- СН2Вг-СНВг-СН=СН2
Вг"
У разі підвищення температури до 40 °С збільшується не лише швидкість
1,2-приєднання, а й іонізація продукту реакції з утворенням того ж резонансно
стабілізованого алілкатіону і броміданіону, який здатний приєднуватися до
кінцевого атома Карбону, утворюючи продукт 1,4-приєднання:
Сг^СН-СН^СНз-^г сн^сн-сн^Сг^--- ^-сн^сн-сь^-^ о^-сн^сн-^
Вг Вг Вг Вг Вг Вг
Іонізація продукту 1,4-приєднання відбувається важче, ніж 1,2-аддукту,
тобто ецергія Г,2-диброміду вища, ніж 1,4-аддукту (рис. 10.3). Це пояснюють
тим, що подвійний зв'язок усередині ланцюга стійкіший, ніж кінцевий.
Експериментально з'ясовано, що 1,2-приєднання відбувається за низьких
температур, а 1,4-приєднання - за вищих. Наприклад, у процесі бромування
бутадієну при -80 °С одержують 80% продукту 1,2-приєднання, а при +40 °С,
навпаки, - стільки ж продукту 1,4-приєднання. Довготривале нагрівання дає такий
же склад суміші. Отже, за умов рівноваги переважає стійкіший 1,4-продукт, а за
знижених температур, коли рівноваги ще не досягнуто, - 1,2-продукт, який швидко'
утворюється.
Розділ 10. Алкадієни
263
Е
6+ 6+
~сн2сн--=--сн=--сн2] Вг
І п.сЛ4
Вг Вг
СН2-СН-СН==СН2
;г Вг
:н2-сн=сн-сн2
Координата реакції
Рис. 10.3. Діаграма реакції електрофільного бромування бут-1,3-дієну
Пояснити ці факти можна, розглянувши діаграму реакції: продукт 1,2-при-
єднання утворюється з більшою швидкістю (для цього потрібна менша енергія
активації), і він контрольований кінетично. Однак продукт 1,4-приєднання
стабільніший. Тому за умов рівноважного процесу з часом або з підвищенням
температури буде накопичуватись 1,4-продукт, що контрольований термо-
динамічно.
Реакція бромування спряжених дієнів - один з прикладів залежності виходів
кінцевих продуктів реакції від типу контролю - кінетичного або термодинамічного.
Кінетичний контроль пов'язаний зі швидкістю реакції, яка визначена енергією акти-
вації, а термодинамічний - зі стабільністю речовини і визначений її вільною
енергією. Наприклад, у розглянутій вище реакції хлорування дивінілу для одер-
жання хлоропрену (див. 10.4.3) процес проводили за умов кінетичного контролю.
Частка продукту 1,4-приєднання також збільшується в разі переходу від
хлору до йоду.
❖ Вільнорадикальне приєднання. Як і алкени, спряжені дієни приєднують
вільні радикали за механізмом АЛ. У разі вільнорадикального приєднання для
спряжених дієнів характерні дві особливості: вони приєднують в 1,4-положення
так, як і в положення 1,2, і вони значно більше реакційноздатні, ніж звичайні
алкени, наприклад:
ВгССІз Вг + ССІ3
сн2=снсн=сн2-^- срн2снсн=сн2— сн2сн=снсн2 - дн2сн-сн-сн2
СС13 СС13 СС13' • '
ВіССІ,
264
Чирва В.Я., Ярллолюк CM,, Толкачова Н.В., Земляков О.Є. Органічна хімія
У цьому виг^адку цьому переважно утворюється продукт 1,4-приєднання,
оскільки алільний радикал існує протягом більшого часу порівняно з
алілкатіоном до зіткнення з СВгСЬ, тобто кінетичний фактор не такий суттєвий
порівняно з електрофільним приєднанням.
Клас реакцій приєднання в 1,4-положення відомий як дієновий синтез, або
реакції Дільса-Альдера. В цих реакціях спряжені дієни за порівняно м'яких умов
і з хорошими виходами взаємодіють зі сполуками, що мають реакційно здатний
подвійний зв'язок (дієнофілами).
Процес відбувається за схемою синхронного [4+2]-циклоприєднання.
Необхідною умовою для перебігу реакції є перебування дієну в .у-г/нс-конформації.
Можливо, що комплекс з перенесенням заряду (КПЗ) є інтермедіатом, у якому дієн
виконує функцію донора електронів, а алкен - акцептора. Утворення продукту
реакції пов'язане з перерозподілом електронів, у цьому разі з двох я-зв'язків
утворюються два нові а-зв'язки:
З іншого погляду, реакції дієнового синтезу не супроводжуються
утворенням проміжних сполук, а сама взаємодія відбувається як узгоджений
процес із формуванням активованого комплексу, у якому один я- і два а-зв'язки
утворюються синхронно з розривом трьох я-зв'язків:
Тривалий час нічого не було відомо про механізм реакцій дієнівого синтезу і
їх навіть називали реакціями без механізму. І лише 1965 р. Р. Вудворд1 і Р. Хоф-
ман" на підставі власних досліджень пояснили цей феномен, відомий сьогодні як
правило збереження орбітальної симетрії Вудворда-Хофмана. За цим прави-
1 Вудворд P. (Woodward R.) (1917-1979) - американський хіміка Фундаментальні праці з
органічного синтезу, у тому числі таких складних природних сполук, як хінін, холестерол,
стрихнін, хлорофіл, вітамін В12. Нобелівська премія 1965 р.
Хофман P. (Hoffmann R.) (1937 p. н.) - американський хімік. Головні дослідження присвячені
механізмам хімічних реакцій, зокрема, реакцій циклоприєднання. Нобелівська премія 1981 р.
10.5.2. Реакції "дієнового синтез/'
КПЗ
Розділ 10. Алкадієни
265
лом, можливість легкого перебігу узгоджених реакцій визначена правилами
орбітальної симетрії: відбуваються лише ті реакції, у яких симетрія граничних
орбіталей - ВЗМО дієну і НСМО алкену або, навпаки, НСМО дієну і ВЗМО
алкену - збігається. Симетрія граничних орбіталей визначає стереохімію
одержаних сполук.
У дієнієвому синтезі максимальне перекривання орбіталей відбувається за
термічної активації системи. У цьому разі л-електрони молекули етилену
переходять на нижчу вільну, а дієну - на вищу заповнену молекулярну орбіталь,
або навпаки:
Правило орбітальної орієнтації Вудворда-Хофмана застосовують для
реакцій циклізації спряжених дієнів, які детально розглянуті в розділі 11.
• Найпростішою реакцією цього типу є синтез циклогексену на основі
бутадієну та етилену. Реакція відбувається важко за підвищеної температури
-200 °С і високого тиску (200-400 атм), вихід продукту реакції в цьому разі
незначний, оскільки молекула дієнофілу не активована:
• Як дієнофіли застосовують різні сполуки, що мають не лише зв'язки С=С, а й
зв'язки С=С або N=14. Добрі результати дають дієнофіли, у яких подвійний
зв'язок активований електроноакцепторними замісниками. До класичних
дієнофілів належать малеїновий ангідрид, акролеїн, акрилонітрил, естери
ацетилендикарбонової кислоти, 1,4-хінони, кротоновий альдегід тощо. В цих
випадках реакції відбуваються кількісно навіть за кімнатної температури або
слабкого нагрівання.
Як дієни звичайно використовують бут-1,3-дієн і його похідні, заміщені
різними групами (метил, етил, хлор, феніл):
ВЗМО
ВЗМО
'о
266
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
н.
+
Кімн.
н ^с.
Малеїновий ангідрид
темп.
+ ([ н
'єн
Акролеїн
100 ^'С
о
Ангідрид циклогекс-4-ен-
1,2-дикарбоновоі' кислоти
,.--^^сно
Циклогекс-З-енкарбальдегід,
100%
^СООСНз
СООСНЗ
Диметилацетилен-
дикарбоксилат
Диметил-3,6 -
дигідрофталат
Диметил-1,4,5,8,9,10-гексагідро-
нафталін-9,10-дикарбоксилат
Бензохінон
Тетрагідронафтохінон
Октагідроантрахінон
■ У дієновому синтезі існує син-{цис)"приєднання, причому в аддукті обидва
синтони зберігають вихідну конфігурацію. Справді, у синтезах з малеїновою і
фумаровою кислотами утворюються діастереомери естерів циклогекс-4-ен-1,2-ди-
карбонових кислот:
+
\
ОООСН3
ДимгтипмаїнЕіг
^^ООССНз
СООШз ОООСН3
^ ізо-с ^
СН^СНзООС
димеггалфумараг
ОООСН5
• Синтез листкових сполук. Використання в синтезі Дільса-Альдера.
циклопентадієну або циклогексадієну як дієнових компонентів дає місткові
циклічні сполуки:
Розділ 10. Алкадієни
267
• Димеризація дієнів. 1,3-Бутадієн димеризується у вінілциклогексен за 180 °С
(С. Лебедєв):
Це типова реакція циклоприєднання, де одна молекула в з-цисоїдній
конформації, відіграючи роль дієну, реагує у положеннях 1 і 4, а друга, в
трансоїдній формі, слугує дієнофілом і реагує у положеннях 1 і 2.
■ У реакції димеризації можуть вступати молекули й інших дієнів.
Наприклад, димеризацією ізопрену можна одержати терпен лимонен:
10.5.3. Реакції полімеризації
Алкени і дієни є вихідною сировиною для одержання полімерів. Це одна з
найважливіших галузей їхнього застосування. Значення і масштаби світового
виробництва полімерів безперервно зростають. З огляду на це реакції
полімеризації заслуговують на окремий розгляд.
> Полімери - високомолекулярні сполуки, які складаються з простих ланок,
що повторюються - мономерів.
Полімери мають такі фізико-хімічні, гігієнічні й інші властивості, яких
можуть і не мати традиційні природні матеріали. Сьогодні хімія дає змогу, у
деяких випадках, одержувати полімери із заданими властивостями. Цим
визначене наперед їхнє місце в житті людини і науково-технічному прогресі.
268
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Полімери, які одержують на основі олефінів, називають поліолефінсти.
Залежно від механізму реакції розрізняють радикальну, катіонну, аніонну і
координаційну полімеризації. Всі ці реакції є ланцюговими, проте, на відміну ьщ
класичних ланцюгових процесів, вони дають полімерні молекули. Такий
результат пов'язаний з тим, що під час перебігу реакції активний центр не
передається на іншу молекулу, як це звичайно відбувається в ланцюгових
реакціях, а залишається на тій же частинці, яка внаслідок приєднання нових
молекул мономеру постійно збільшує свій розмір.
❖ Радикальну полімеризацію застосовують найчастіше і вивчена вона
найдетальніше. Як мономери використовують етилен, пропілен, тетра-
флуороетилен, трифлуороетилен, стирен, вінілхлорид, вінілацетат, метил акри-
лат, метилметакрилат, акрилонітрил та ін.
■ Процес полімеризації починається з ініціювання реакції. Для цього вико-
ристовують речовини, що здатні в певних умовах (нагрівання, опромінення тощо)
утворювати радикальні частки - ініціатори. До них належать пероксиди.
азосполуки:
С6Н-(f °С-С6Н5 2С6Н5СОО 2С6Н5- + 2С02
О—О
Бензоїлпероксид
Взаємодія радикала з першою молекулою мономеру належить до стадії
ініціювання:
С6Н5- + Н2С=СН — С6Н5-СНГСН,
Х Х де X = Hai, Alk, CN, СООСН3 і т.д.
■ Далі відбувається ріст ланцюга:
с6нгсн2-сн +пн2с=сн -с6н5-<снг<рн)гснг9н
XX хх
■ Обрив ланцюга настає внаслідок рекомбінації, диспропорціювання або
передавання ланцюга на іншу сполуку:
рекомбінація
2С6Н5+СН2-ОІттгСН2СН - С^СН^ЬД^
XX XXX
диспропорціювання
2С6н5Ч-щ-онЧіга^^н — С6Н5Н~сн^сріЧтгС^ан^х + сдЧ-а^срн-тпСН^снх
XX X х
передавання ланцюга
СДЧСН^ОТ+тСБ^ +R'H—-C^+CHjCH-hC^CH^ + R'
XX "х
Розділ 10. Алкадієни
269
Методом радикальної полімеризації етилену за наявності пероксидів у разі
підвищеної температури й тиску одержують поліетилен високого тиску.
Радикальною полімеризацією похідних етилену, що мають замісник у
подвійному зв'язку, отримують низку полімерів з цінними властивостями, які
широко застосовують. Такі полімери належать до атактичних. У них замісник
X розташований хаотично щодо площини, у якій може бути зигзагоподібний
ланцюг полімеру. Вони мають малий ступінь кристалічності, і внаслідок
знижених міжмолекулярних взаємодій такі полімери мають невелику міцність і
високу пластичність:
X Н X Н Н X X Н Н X
Структура атактичного полімеру
❖ Катіонна полімеризація відбувається за електрофільним механізмом
приєднання. Вона легко виникає за наявності в мономерах електронодонорних
замісників. Ініціаторами слугують протонні кислоти і кислоти Льюїса,
наприклад: Н¥, Н2804, Н3Р04, ВБз, А1С13, ТіСЦ та ін.
■ Ініціювання ланцюга починається з утворення карбокатіону:
Н,С
(СН3)2С=СН2+Н+
■ Потім відбувається ріст ланцюга:
СН,
СН
СН,
(рн3
*с=сн2 + н3с-с+ 3 —- н3с-с^сн2-^снгс+
н3с
СН,
І 3
,С-СНз
СН,
СН,
СН,
СН,
СН,
■ Обрив ланцюга може трапитися внаслідок приєднання до карбокатіонного
центру нуклеофіла або через відщеплення протона з утворенням термінального
(кінцевого) подвійного зв'язку.
❖ Аніонної полімеризації зазнають мономери, що мають електроноакцеп-
торні замісники, такі як акрилонітрил, естери акрилової і метакрилової кислот,
дієни тощо. За здатністю до аніонної полімеризації ненасичені сполуки
розташовані в такій послідовності: акрилонітрил > метакрилонітрил > метил-
метакрилат > бут-1,3-дієн.
■ Для ініціювання реакції полімеризації застосовують основи, досить сильні
для того, щоб генерувати карбаніон після приєднання до подвійного зв'язку.
Ініціатори аніонної полімеризації іноді в розчинах здатні дисоціювати з
утворенням аніона, який приєднується до атома Карбону молекули мономеру,
що несе частковий позитивний заряд:
270
Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
КЖІ2 К+ + МН2~ N112" + н2с=Сн —- н,м-сн СН" к+
€Н Н^-СН^і
У
Ініціаторами можуть бути, наприклад, аміди лужних металів, їхні гідриди,
металоорганічні сполуки, лужні метали тощо. Приєднання настає без
попередньої дисоціації ініціювального реагенту.
■ Ріст ланцюга відбувається аналогічно до катіонної полімеризації через
впровадження нових молекул мономеру в іонну пару, що утворена карбаніонним
центром і протиіоном:
У У У У Макрокарбаніон у
■ Обрив ланцюга може настати внаслідок реакції перенесення реакційного
центру на молекулу розчинника або приєднання протона:
Н2К(СН2СНУ),2+УС^ -Н2К(СН2СНУ)Л+УСН2СН2У + Ш2" + К+
У
Методами аніонної полімеризації часто можна одержати так звані
стереорегулярні полімери ізотактичної будови, у яких замісники розташовані
по один бік ланцюга, що перебуває в зигзагоподібній конформації, і
синдіотактичної будови. В цьому випадку замісники розташовані поперемінно
то по один, то по інший бік полімерного ланцюга. Такі стереорегулярні полімери
мають цінніші фізико-хімічні властивості порівняно з нерегулярними атак точ-
ними полімерами. Зокрема, у них більша густина і вони більш кристалічні.
Таким методом сьогодні одержують поліетилен низького тиску, поліпропілен,
полістирен, поліакрилонітрил тощо:
НН. XX. Н
Структура синдіотактичного полімеру
❖ Координаційна полімеризація. Найефективнішим методом синтезу
стереорегулярних полімерів є так звана стереоспецифічна, або координаційна,
полімеризація з використанням каталізаторів Циглера-Натта. Такі каталізатори
утворюються в разі змішування металоорганічних сполук металів І-ПІ груп
Періодичної системи (наприклад, триетилалюмінію) і сполук сі-металів,
наприклад, титан (IV) хлориду. Полімеризація за методом Циглера-Натта дає.
полімери, що мають високий ступінь стереорегулярності і, відповідно, хороші
механічні властивості.
Розділ 10. Адкадієни
271
Каталізатори Циглера-Натта поділяють на гомогенні, або розчинні, — на них
утворюються синдіотактичні полімери, і гетерогенні, або нерозчинні, - за
їхньою допомогою одержують ізотактичні полімери, у яких замісники роз-
ташовані по один бік від площини ланцюга:
Механізм такої полімеризації складний і ще не визначений до кінця. Якщо
змішувати компоненти каталітичної системи, то відбувається алкілування і
відновлення похідних <і-металу металоорганічною сполукою:
Активним центром у разі ізотактичної полімеризації є алкілований і
оточений хлоридними лігандами атом Титану зі ступенем окиснення +3, що
міститься на поверхні кристала ТіСЬ. Мономер попередньо координується в
активному центрі завдяки утворенню я-зв'язків з атомом Титану. Така
координація спричинює поляризацію подвійного зв'язку і збільшення електро-
фільності найбільше заміщеного ^р2-гібридного атома Карбону.
Алкілування цього атома вуглеводневим радикалом при атомі Титану
супроводжується розривом зв'язку С=С і зв'язку Ті-С та утворенням нового
зв'язку між найменш заміщеним атомом Карбону мономеру і атомом Титану.
Внаслідок такого алкілування полімерний ланцюг пересувається в нове
положення біля атома Титану і збільшується на один мономерний залишок. Далі
процес полімеризації відбувається за схемою, що наведена вище. Обрив ланцюга
може виникати через передавання активного центру на відповідний регулятор
росту ланцюга, наприклад, молекулу Гідрогену:
ХНХНХНХНХН
Структура ізотактичного полімеру
ТІС14 + А1(С2Н5)3
:с2нз)з С2Н5ТІС13 + (С2Н5)2А1С1
СІ + 2ТІС14 2С2Н5А1С12 + 2ТіС13 + С2Н4 + С2Н6
С2Н5А1С12 + ТіС13 —- С2Н5ТіС12 + А1С13
2(С2Н5)2А1С1 + 2ТіС14
Кристалічна
ґратка ТіС13
пН2С=СН2
8- 5+
СНзС^СС^СНз^СНзСН^іСІз
^^^^
СН3СН2(СН2СН2),,СН2СН3 + НТіС12
272
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
За вивчення металоорганічних каталізаторів, розробку і впровадження
процесів координаційної хімії -К. Циглер і Д. Натта отримали Нобелівську
премію 1963 р.
10-6. Каучук
10.6.1. Природний каучук
■ Каучук - це високомолекулярний ненасичений вуглеводень складу (C5Hg)n.
Основну кількість каучуку свого часу одержували з молочного соку гевеї
бразильської {Hevea brasiliensis), де його вміст досягає 50 %. Молекулярна маса
цього полімеру коливається в межах 150-500 тис. Да.
-СНз^СНСНзСНз^СНСНзСНз^СНСНз-
СН3 СН3 СН3
Методом рентгеноструктурного аналізу визначено г/мс-будову ізопреноїдно-
го ланцюга, при якому метиленові групи розташовані по один бік від подвійного
зв'язку:
СН3 СН3
■ Гутаперча має втричі коротші ланцюги і /я^анс-конфігурацію подвійних
зв'язків.
■ Подвійні зв'язки в молекулі каучуку відіграють важливу роль, оскільки
зумовлюють реакційну здатність атома Гідрогену в алільному положенні; їхня
наявність дає змогу проводити вулканізацію за наявності сірки. Ці поперечні
зв'язки надають вулканізованому каучуку еластичності й міцності, а також
позбавляють липучості, яку має натуральний каучук:
СН3 СН3 СН3 СН3
-сн2оснсн2сн2с=снсн2- -снс=снсн2снс=снсн2-
- СН2С=СНСН2СН2С=СНСН2- -снс=снсн2снс=снсн -
сн3 сн3 Ьщ сн3
10.62. Синтетичний каучук
Найважливішою з практичного погляду реакцією 1,3-дієнів, безсумнівно, є
полімеризація, яка може відбуватися за радикальним, аніонним, катіонним і
Розділ 10. Алкадієни
273
аніонно-радикальним механізмами. Залежно від умов перебігу реакції і типу
полімеризації дієни можуть реагувати у положеннях 1,2- чи 1,4- або двома
способами одночасно. Окрім бутадієну, як мономери широко використовують
ізопрен, хлоропрен і суміші бутадієну з різноманітними алкенами, такими як
стирен, акрилонітрил, ізобутилен і пропілен. У підсумку одержують синтетичні
каучуки. Розглянемо найважливіші з них.
• Бутадієнові каучуки вперше одержані 1931 р. С. Лебедєвим за допомогою
аніонної полімеризації бутадієну. Вони були відомі в СРСР під назвою СК-
каучуки, а в Німеччині - Буна. В методі Лебедева ініціатором полімеризації є
металічний натрій, на поверхні якого відбувається адсорбція і полімеризація
бут-1,3-дієну. Малий вміст у них ланок 1,4-приєднання (30%) знижує їхню
еластичність порівняно з природними каучуками:
гЩ<ЖН=Щ -О^ОМЇтЧ^(<^
СНКН,
■ За умов реакції Циглера-Натта вдається синтезувати ^нс-1,4-полібутадієн
(стереорегулярний бутадієновий каучук), який не має перелічених вище
недоліків і за властивостями подібний до натурального каучуку (в СРСР
виробництво такого каучуку організоване 1956 р.):
Нч Н
V /С—С^ /СН7 / ч2
хсн2 сн2 2;с=сч ^
2 н н
■ Співполімер бутадієну (три частини) зі стиреном (одна частина) за
радикальної полімеризації дає каучук СКС {бутадієн-стиреновий каучук), який
використовують для виготовлення шин:
-сн2-сн=сн-сн2сн2-сн=сн-сн2сн2-сн-сн2-сн=сн-сн2-
сбн5
■ Співполімер бутадієну з акрилонітрилом за тих же умов перетворюється в
бутадієн-нітрильний каучук, стійкий до олив, бензинів і до стирання.
■ Бутилкаучук — співполімер бутадієну й ізобутилену — одержують за
допомогою низькотемпературної йонної полімеризації за наявності бор (III)
флуориду. Має високу хімічну і термічну стійкість, газонепроникність, є
хорошим ізолятором для дротів і кабелів:
СН3 СН3 СН3
лСН2=9 + /иСН2=СНСН=СН2—■—сн2£сн2сн=снсн2сн29-
сн3 сн3 сн,
274 Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
■ Хлоропрен (2-хлоробут-1,3-дієн) - легко полімеризується в хлоропреновий
каучук, який вирізняється бензостійкостю.
• Ізопреновий каучук. Застосування каталізаторів Циглера-Натта дало змогу в
промислових масштабах синтезувати стереорегулярний поліізопрен, який не
уступає за властивостями природному каучуку. В СРСР перший такий каучук
(під маркою СКИ-3) випущено 1963 р.
10.7. Терпени ациклічного ряду
Клас біологічно активних сполук терпенів вирізняється великою різнома-
нітністю хімічних структур, значним поширенням у природі та різноманітною
фізіологічною дією. До терпенів, зокрема, належать природні глікозиди (сапоніни),
рослинні пігменти, продукти смоли хвойних дерев, компоненти ефірних олійл
У природі трапляються ациклічні терпени і циклічні терпени з різною
кількістю кілець. Поряд з терпенами чисто вуглеводневої природи відомі й їхні
оксигенові похідні, які називають терпеноїдами. Об'єднує ці сполуки те, що
карбоновий скелет більшості з них можна розглядати як продукт об'єднання
декількох молекул ізопрену, тому терпени по-іншому називають ізопреноїдами.
Л. Ружичка1 сформулював "ізопренове правило", за яким формальна конденсація
ізопрену в терпени відбувається за принципом "голова" до "хвоста":
Мірцен Лімонен
За кількістю ізопренових ланок терпени поділяють на монотерпени (дві
ізопренові ланки — Сю), сесквітерпени (три ізопренові ланки - с15), дитерпени
(чотири ізопренові ланки - С20), тритерпени (шість ізопренових ланок - с30),
тетратерпени (вісім ізопренових ланок - с40) і поліізопрени (каучук і гутаперча
-[с5]„).
❖ Ациклічні терпени. Велика кількість ациклічних ізопреноїдів міститься в
рослинах і має приємний запах ^ефірні олі'ґ). Прикладами таких сполук можуть
слугувати мірцен (є в складі олій хмелю і благородного лавру), гераніол (олії
троянди і герані), цитраль (олія лимона й інших цитрусових), ліналоол (оліі^
коріандру, лаванди і винограду):
1 Ружичка Л. (Яи2ібка Ь.) (1887-1976) - швейцарський хімік, спеціаліст у галузі циклічних сполук
і терпенів. Нобелівська премія 1939 р.
Розділ 10. Алкадієни
275
Мірцен Гераніол Цитраль Ліналоол
10.8. Вітаміни, що мають полієновий ланцюг
Деякі вітаміни, які належать до групи жиророзчинних вітамінів, мають у
структурі полієнові фрагменти. До них, зокрема, належать вітаміни А, К і Q.
❖ Вітамін Aj (ретинол) міститься в печінці морських тварин і риб, а
також утворюється в організмі людини внаслідок окиснювального розщеплення
каротину, який є в овочах і фруктах (морква, червоний перець). Вітамін А
необхідний для нормального зору, оскільки його альдегідна форма (1\-цис~
ретиналь) є в складі родопсину - світлочутливого білка зорових клітин:
11ч(кс-ретііналь
❖ Вітаміни К - вітаміни коагуляції, необхідні для нормального згортання
крові. До них, зокрема, належать менахінони (вітаміни Кг), що їх в організмі
людини утворює мікрофлора кишківника, а у тварин - печінка. Ациклічний
фрагмент цього вітаміну - поліізопреновий ланцюг.
Вітамін К2 («=4-9) Убіхінони (Зб-ю(л=6-Ю)
276
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
❖ Вітаміни {? (убіхінони). Вітаміни групи (2 поширені в живих об'єктах'
(убіхінон - від лат. повсюдний). В основі структури цих сполук є метил-
диметокси-и-бензохінон, до якого приєднано від шести до десяти ізопреноїдних
ланок. Кількість ізопреноїдних ланок відображена в назві убіхінону цифровим
індексом. Головна біохімічна функція вітаміну - участь у процесах дихання,
зокрема, в окисно-відновних реакціях, у яких убіхінони є посередниками транс-
портування електронів.
Розділ 11
ЦИКЛОАЛКЛНИ
Вуглеводні ланцюги можуть замикатись, утворюючи циклічні похідні.
Неароматичні циклічні вуглеводні об'єднують у клас сиііциклічних сполук. За
кількістю циклів циклоалкани поділяють на moho-, бі- та поліциклічні сполуки.
З групи бі- та поліциклічних вуглеводнів виділяють:
■ спірани - сполуки, у яких для двох сусідніх циклів існує лише один
спільний атом;
■ конденсовані циклоалкани — системи циклів, у яких для двох сусідніх
кілець існують два спільні атоми;
■ місткові вуглеводні - циклічні сполуки, у яких два кільця з'єднані
"містком" з одного або більше атомів;
■ каркасні (поліедричні) вуглеводні - поліциклічні сполуки об'ємної будови,
у яких кожний цикл зв'язаний із сусідніми двома й більше спільними атомами.
Спірани
Спіро[2.4]гептан
Моноциклічні аліфатичні вуглеводні
-СН2 , і
І 2 або Q
н2с-
н2с—сн2
Циклобутан
Бі- та поліциклічні вуглеводні
Конденсовані Місткові вуглеводні
циклоалкани
РО 00
Біцикло[4.4.0]декан
(декалін)
Біцикло[2.2.1] гептан
(норборнан)
Каркасні
вуглеводні
Адамантан
Найглибше вивчені й мають важливе практичне значення моноциклоалкани,
яким у цьому розділі приділено основну увагу.
278
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
11.1. Моноциклічні аліфатичні вуглеводні
Моноциклоалкани мають загальну формулу ОДЬ,, відповідно, вони структурно
ізомерні з алкенами. Родоначальник гомологічного ряду - СзНб, циклопропан.
На відміну від класів вуглеводнів, розглянутих раніше, перший представник
моноциклоалканів - циклопропан - не є типовим представником цього класу
органічних сполук. Крім того, і для інших представників моноциклічних
аліфатичних вуглеводнів є суттєві відмінності у властивостях і способах
одержання, що залежать від розміру циклу. Тому моноциклоалкани поділяють
помсті (С3 і С4), звичайні (С5-С7), середні (Св-Сц) і великі (> С12) цикли, які ще
називають макроциклами.
11.1.1. Номенклатура
Назву циклоалканів будують із назви алкану з відповідною кількістю
карбонових атомів, додаючи префікс цикло-. У разі називання заміщених
циклоалканів за основу беруть циклічну систему. Цикл нумерують так, щоб
замісники отримали найменші номери (правило "мінімальних локантів"). Якщо
варіанти однакові, то мінімальний номер присвоюють першому за абеткою
заміснику. Якщо в циклі є кратні зв'язки, то використовують правила
номенклатури для ненасичених сполук (див. розділ 8).
3-Метилциклопентен
Нумерація
починається від
подвійного зв'язку.
НзС-СНН^СНз
СН3І-УчсНз
3-Ізопропіл-1,1 -диметилциклопентан
Замісники повинні одержати мінімаль-
ні локанти (1,1,3 < 1,3,3).
СН3
сн3
1 -1зопропіл-2-метил циклогептан
Мінімальний локант присвоєний
першому за алфавітом заміснику.
11.1.2. Фізичні властивості
Моноциклоалкани мають дещо вищу температуру плавлення і температуру
кипіння порівняно з відповідними ациклічними сполуками. Менша конформа-
ційна рухливість циклічних вуглеводнів полегшує їхнє укладання в кристали.
Водночас, на відміну від алканів, фізичні властивості циклоалканів змінюються
не плавно. Наприклад, циклододекан (С12Н24) має вищу температуру плавлення
порівняно з циклоундеканом (СцН22) і циклотридеканом (Сі3Н2б).
• ІЧ-спектроскопія. В ІЧ-спектрах циклоалканів нема смуг поглинання з
частотою -1 380 см"1, що характерні для деформаційних коливань зв'язків С-Н
Розділ 11. Циклоалкани
279
метальних груп. Метиленові групи мають смуги поглинання в ділянці 2 850-2 920 і
1 460 см"1, що належать, відповідно, до валентних і деформаційних коливань.
Особливі властивості циклопропану підтверджені існуванням валентних
коливань зв'язків С-Н v 3 050 см"1, які, зазвичай, властиві алкенам.
• Мас-спектрометрія. Для циклоалканів характерне утворення молекуляр-
ного йона М+. У процесі фрагментації, як звичайно, губиться молекула етилену і
утворюється пік [М -с2н4]+. Подальша фрагментація складна і малоінфор-
мативна.
• ПМР-спектроскопія. У ПМР-спектрах циклоалканів сигнали протонів
метиленової групи є в інтервалі 1,4-1,8 м.ч. Яскравий виняток — циклопропан, у
якого протони мають хімічний зсув 0,22 м.ч. Також треба зазначити про
випадкову еквівалентність протонів в адамантані - різні за хімічною природою
протони мають однаковий хімічний зсув. Приклади хімічних зсувів протонів
різних циклоалканів такі:
d o o o db о* А
нь
5 1,96 5 1,51 м.ч. 8 1,44 м.ч. 5 1.54 м.ч. 5 1,4 м.ч. 6 1,78 м.ч. (a, b 5 1,51 (а), 2,1 м.ч.
11.1.3. Будова циклопропану
Унаслідок визначення теплот згорання циклоалканів з циклами різних
розмірів з'ясовано, що з розрахунку на одну метиленову групу найбільша
теплота згорання є в циклопропану, а потім - у циклобутану. Інші циклоалкани
мають близькі значення теплот згорання з розрахунку на групу СН2. Щоправда,
у циклогексану це значення найменше.
Таблиця 11.1
Теплота згорання й енергія напруженості циклоалканів (СН2)„
п
3
4
5
6
7
8
•АН / С#2, кДж/моль
697
686
664
659
662
663
Енергія напруженості / СН2*,
38
27
5
0
3
4
кДж/моль
Загальна енергія напруженості **,
114
108
25
0
21
32
кДж/моль
* Різниця між теплотами згорання циклоалканів і теплотою згорання найменш напруженого
циклоалкану з розрахунку на групу СНг.
** Енергія напруження з розрахунку на групу СНг, помножена на кількість груп СНг.
Значні теплоти згорання в циклопропану і циклобутану порівняно з іншими
циклоалканами цілком природно пояснити більшою внутрішньою енергією.
280
Чирва В.Я., Ярмолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Чинники, що збільшують внутрішню енергію молекули, називають напруженням.
Розрахунки, наведені в табл. 11.1, свідчать про велику енергію напруження в
молекулах циклопропану і циклобутану. Вище (див. розділ 6) обговорено, що в
малих циклах реалізується кутове і торсійне напруження, які пояснюють взаємним
відштовхуванням, відповідно, коли зменшується кут між електронними хмарками
одного атома Карбону, і зближуються в просторі електронні хмарки двох сусідніх
атомів Карбону.
У правильному трикутнику внутрішній кут дорівнює 60°. У молекулі
циклопропану такий кут між С-С-зв'язками неможливий для атомів Карбону в
5р3-гібридизації з валентним кутом 109°28\ Утворення С-С-зв'язків відбу-
вається по кривій поза прямою лінією, що з'єднує центри атомів. Тому в цьому
разі не виникає максимального перекривання атомних орбіталей. Такі а-подібні
зв'язки, які не мають кругової симетрії, називають т-зв'язками, або '''банановими
зв 'язками".
Реальний кут між С-С-зв'язками становить -106°, а кут між двома С-Н-зв'яз-
ками одного атома Карбону 116-118°. Проте навіть таке відхилення від
значення тетраедричного кута зумовлює сильне кутове напруження. Додатковим
чинником, що збільшує внутрішню енергію молекули, є перебування зв'язків С-Н в
затіненому стані, що спричиняє значне торсійне напруження. Менш ефективне
перекривання атомних орбіталей у разі утворення С-С-зв'язку зменшує його
міцність. Для циклопропану характерні реакції приєднання, що супрово-
джуються розривом С-С-зв'язку.
Оскільки утворення С-С-зв'язків неможливе шляхом перекривання
звичайних 5р3-гібридних орбіталей, то в них збільшується внесок р-орбіталей, а в
^р3-гібридних орбіталях, що беруть участь в утворенні С-Н-зв'язків, відповідно,
збільшується внесок ^-орбіталі. Такий ефект спричиняє зсув електронної густини
до атома Карбону, бо максимум електронної густини 5-орбіталі є ближче до
центру атома порівняно з р- або лр3-орбіталями. Атом Гідрогену набуває частково
позитивного заряду, тобто виявляє кислі властивості.
Циклопропан є сильнішою кислотою порівняно з алканами або звичайними
циклоалканами, проте значно слабшою, ніж ацетилен (рКй = 25). Оскільки
кислотність для більшості вуглеводнів - розрахункова величина, то залежно від
методу розрахунку константи кислотності можуть суттєво відрізнятися. Наприклад,
"Бананові" зв'язки в циклопропані
Проекція Ньюмена для циклопропану
Розділ 11. Циклоалкани
281
за однією шкалою для циклогексану рКл =51, метану pKâ = 48, циклопропану рКа =
46, а за іншою - для циклогексану рКа = 45, метану рКъ = 40, циклопропану рКа = 39.
11.1.4. Ізомерія
Для циклоалканів характерна структурна ізомерія, а також міжкігасова
ізомерія з алкенами. Серед структурних ізомерів можна розглядати окремо
ізомерію розмірів циклу, ізомерію положення замісників у циклі й структурну
ізомерію ланцюгів замісників.
За наявності двох замісників у циклі можлива реалізація діастереомерії
(геометричної ізомерії), до того ж, транс-форми таких ациклічних .сполук
можуть існувати у вигляді двох енантіомерів (дзеркальні ізомери).
Ізомерія розмірів
циклу
Ізомерія карбонового скелета
Ізомерія положення замісникі Ізомерія замісників
Н<С
А
5^2
Т9Н
2П5
СН2СН2СН3
Циклогептан
сн,
1,2-Диетилциклопропан
,с,н5
І^сл
Метилциклогексан
-2П5
1,1 -Диетилциклопропан
МЬіскласова ізомерія
Н3С
1 -Метил-2-пропілциклопропан
СН-СНз
сн3
Н3С
1-Ізопропіл-2-
метилциклопропан
О
Циклогексан
Н2С=СНСН2СН2СН2СНз
Гекс-1-ен
Стереоізомерія
Діастереомерія (геометрична ізомерія) Енантіомерія (дзеркальна ізомерія)
• Н,
Н5С2
H
с2н5
Н5С2,
H
H
с2н5
н5с2,
H
H
C2H5
H
с2н5
H
1,2-
Диетилциклопропан
транс-\,2-
Диетилциклопропан
Н5С2
(S,.S)- і (R,R)-\,2-Диетилциклопропан
282
Чирва В.Я., Ярллолюк CM,, Толкачова Н.В., Зеллляков О.Є. Органічна хімія
11.1.5. Конформація
На стійкість конформацій циклічних вуглеводнів впливають кутове, торсійне
і вандерваальсове напруження. В розділі 6 ми розглянули головні конформації
циклобутану, циклопентану і циклогексану. В цьому розділі опишемо
конформації деяких циклоалканів з великою кількістю карбонових атомів. Для
них досить складно виконувати конформаційний аналіз, бо кількість конформерів
різко зростає і виникають труднощі з урахуванням усіх чинників, що впливають
на конформації. Тому зазвичай визначають найстійкіші конформери.
• Циклогептан. Для цієї молекули виділяють чотири енергетично вигідні
конформації, з яких найстійкіша - "твіст-крісло". Проте бар'єр переходу між
конформерами невеликий (~11 кДж/моль):
"Крісло" "7в/с/и-крісло" "Гв/ст-ванна" "Ванна"
• Циклооктан. Питання про найстійкішу конформацію цієї сполуки досі без
відповіді. В кристалах похідні циклооктану переважно перебувають у конфор-
мації "ванна-крісло" і менше - у конформації "корона". В газовій фазі
циклооктан існує як суміш конформерів. Цей висновок підтверджують і деякі
розрахункові дані, за якими три основні конформери тотожні за енергією:
"Ванна-крісло" "Сідло" "Корона"
• Циклодекан. Уже для циклононану розрахунок конформацій надзвичайно
складний, однак для циклодекану на підставі його подібності до стійкої
структури алмаза доведено реалізацію енергетично вигідної конформації
"ванна-крісло-ванна" (рис. 11.1).
Рис. 11.1. Конформація "ванна-крісло-ванна" циклодекану та її подібність
до кристалічної ґратки алмаза
11.1.6. Загальні методи одержання
В основі більшості загальних методів є замикання циклу в реакціях а,со-бі-
функціональних сполук. Практично завжди в цих реакціях утворюються похідні
Розділ 11. Циклоалкани
283
циклоалканів. Щоб одержати власне циклоалкани, застосовують процеси
дефункціоналізації.
❖ Внутрішьомолекулярна реакція Вюрца. Цикл замикається дією металів
на дигалогеніди з атомами галогену на кінцях вуглеводневого ланцюга:
< СН2Вг , СН2
¥2)11
СН2Вг ^—сн2
Використання класичного реагенту реакції Вюрца - натрію — малоефектив-
не, навіть для одержання циклопропану. Добрі виходи циклопропану і
циклобутану є, якщо застосовувати в цій реакції цинк або магній. Циклобутан і
циклопентан утворюються з високими виходами і під дією амальгами літію.
Середні і макроцикли за цих умов одержують з низькими виходами або вони не
утворюються:
н^с/СН2Вг ^ д Н2С-СН2Вг
"СН2Вг '—* Н2С-СН2Вг
1,3-Дибромопропан Циклопропан 1,3-Дибромобутан Циклобутан
❖ Реакція Перкіна1. Цикл утворюється внаслідок взаємодії натрій малоно-
вого естеру з а,о>дигалогеналканами. Детальніше реакції малонового естеру
обгово-рено в розділі 19. Наступне омилення естеру дикарбонової кислоти і
декарбоксилювання дає циклоалканкарбонові кислоти. За допомогою цієї реакції
одержано цикли, що мали від трьох до шести атомів Карбону:
с^-вг соусд, адодЧ/^^ -2едон ЧД^ -со2 V ^
1,3-Дцбромопрогандаептшонаг Цчююбуганкарбонова
кислота
В. Перкін молодший реалізував також альтернативну схему синтезу
похідних циклоалканів. Дією на малоновий естер 1,3-дибромопропану за
наявності натрій етилату одержують тетраестер, який через динатрієве похідне
зазнає циклізації за наявності галогенів. Подальший стандартний шлях при-
водить до циклопентан-1,2-дикарбонової кислоти:
1 Перкін В. мол. (Регкіп W. ]г.) (1869-1929) - англійський хімік, працював у галузі циклоалканів і
природних сполук.
284
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
<СН2Вг ^СООС2Н5 2С2Н50На ^СЬ^СЩСООС^Нз^ 2С2Н50Ыа
+ 21^0^ ►
СН2Вг ТОООД -2С2н5он, ^СНгСЩСООедЬ-гс2н5он
-2ЫаВг
/СНг^сооедь н с/сн2с(соос2н5)2 н2о і {> Л^СООН
СН2СЫа(СООС2Н5)2 "2МаВг 2 ХСН2С(СООС2Н5)2 -4С2н5он -2С0,\^С00Н
❖ Декарбоксилювшшя солей дикарбонових кислот {метод Ружички).
Нагрівання барієвих або кальцієвих солей дикарбонових кислот дає циклічні
кетони, що мають на один атом Карбону менше, ніж вихідна сполука. Наступне
відновлення кетогрупи до метиленової (детальніше див. розділ 17) дає
циклоалкани:
О
с; ( СН2 ( СН,
[Н]
4^ ^^^^
г
(СН2)п.і
с 4—сч 4—сн2
о о 2
Метод застосовують для синтезу п'яти-семичленних циклоалканів, наприклад:
О
счха
СчгО" -СаО,-С02 \ -СаО,-С02
Кальцій адипінат Циклопентанон, 45 % Кальдій суберонат Циклогептанон, 35 %
Використання в цій реакції торієвих солей дає змогу одержати циклогептанон
з виходом -50 %, циклооктанон —20 %. Виходи циклічних кетонів Сд-Сп дуже
низькі (~1 %). Більші за розміром цикли утворюються дещо ліпше: вихід
циклопентадеканону - 5 %, циклогептадеканону - 8 %.
❖ Циклізація динітрилів за Торпомх-Циглером. Димеризація а,со-динітрилів
за наявності основ дає циклічні імінонітрили, які потім зазнають гідролізу до
кетокарбонових кислот і декарбоксилювання:
,—СН2С=Ы ,—СН2 /—СН2 /—СН2
^ -^^)п СН-С^^+(СН2)п СН-СООН-^(СН2)п сн2
1—СЙ2СМ4 1—С=Ша 1—0=0 2 1—0=0
Повільним додаванням розчину динітрилу до конденсувального реагенту ми
забезпечуємо перебіг реакції за умов сильного розведення, що, відповідно,
1 Торп Дж. (Тпогре т.) (1872-1940) - англійський хімік, що працював у галузі синтезу ди- і
полікарбонових кислот.
Розділ 11. Циклоалкани
285
перешкоджає побічним процесам. Цим методом з дуже високими виходами
одержують циклічні кетони Су-Сз і С14-С32. Вихід сполук С9-С11 дуже малий (1-
2 %):
Ж:-(СН2)17-СН
NaN(CHз)C6H5
(С3Н7)20
(СН2)
16
Н20,Н+ і
(СН2)16
СН,
С=Ша
-с=о
Циклооктадеканон, 80 %
❖ Естерна конденсація Дікманах - це варіант конденсації Кляйзена2
(детальніше див. розділ 21). За наявності одного еквівалента натрію або натрій
алкоголяту естери а,со-дикарбонових кислот циклізуються в кетоестер, який
після гідролізу легко зазнає декарбоксилювання до циклічного кетону:
О
и
^-С-ООД
(СН)П _№
С02СНз
он
он
І
^-с-оед ^-с-осд
тх -*(сшп
Чсн Ч,
оо.сд
сн
ґ
-(СЩ,
-сдон ^
СН -00,
ООуСД
Найліпші результати зафіксовано в синтезах п'яти-семичленних циклів.
Помірні виходи (24-48 %) одержані для циклічних кетонів Сц-Сів:
ґ^С02С2Н5
к^С02С2Н5
Диетиладипінат
N3
а'
В>0, Н+, /
-С2Н5ОН,
С02С2Н5 -со2
Циклопентанон
❖ Ацилоїнова конденсація естерів дикарбонових кислот. Естери а,ес>-
дикарбонових кислот у разі дії надлишку натрію (4 екв.) зазнають циклізації за
іншим механізмом, утворюючи а-гідроксикетон (ацилоїн). Реакція відбувається
через натрієву похідну ендіолу. Відновленням ацилоїну за Клемменсеном3 (див.
розділ 17) одержують потрібні циклоалкани.
1 Дікман В. (Оіескліап W.) (1869-1925) - німецький хімік, який досліджував етерну конденсацію
диетерів.
2 Кляйзен Л. (СІаІБеп Ь.) (1851-1930) - німецький хімік. Головні праці присвячені органічному
синтезу, ацилюванню карбонільних сполук і таутомерії.
3 Клемменсен Е. (СІеттепБеп Е.С.) (1876-1941) - американський хімік данського походження.
Працював у фармацевтичній промисловості.
286
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органична хімія
Метод застосовують, щоб одержати практично весь діапазон циклічних
вуглеводнів. За його допомогою з прийнятними виходами (~40 %) синтезовано
середні цикли с9-С12, які практично не утворюються в інших реакціях. Дуже
високі виходи досягнуто в реакціях утворення вищих макроциклів (>Сго).
Наприклад, 2-гідроксигенейкозанон одержаний з виходом 96 %:
(— СО2СН3
г
C-ONa
(CH2)n ^ (СНЛ..
І т -2CH3ONa І т\\
^—СОгСНз ^—г~'
Н,0
C-ONa
г
(CH2)n
с-он
с-он
f— с=о
(СН2)П
^сн-он
ацилоин
Zn Г
сн2
сн2
❖ Розширення циклів у циклокетонах. Циклокетони можуть взаємодіяти з
діазометаном із утворенням більшого за розміром циклу {впровадження
карбену). Наприклад, із циклобутанону одержують циклопентанон:
.0
H2C~№N
CH2-№N
-N2
?сн2]^[^о
Подальше збільшення розмірів циклу дає не очікуваний циклогексанон, а
циклогептанон, що пов'язано з більшою реакційною здатністю циклогексанону
порівняно з циклопентаноном. Граничною сполукою, яку можна отримати в
цьому синтезі, є циклооктанон:
CH2N2
Повільно
Циклопентанон
О»
сн
Циклогептанон Циклооктанон
Циклічні кетони з більшим розміром циклу, аж до с15, можна одержати за
наявності каталітичної системи літій хлорид-етерат бор флуориду:
cum
п = 4,5
т=8- 14
11.1.7. Спеціальні методи одержання
❖ Синтез похідних циклопропану
• Приєднання карбенів до алкенів. За термічного або фотолітичного розкладу
діазометану, а також взаємодією галоформів з сильною основою (калій трет-
Розділ 11. Циклоалкани
287
бутилатом) утворюються електронодефіцитні частинки — карбени (детальніше
див. розділ 7). Карбени здатні приєднуватись до подвійного зв'язку алкенів,
утворюючи похідні циклопропану:
СН^2 * :СН2 + N2 СН3СН=СН2 + :СН2 * Н3С-
Пропен
V
Метилциклопропан
У випадку менш активних дигалогенокарбенів приєднання до алкенів
відбувається стереоспецифічно. Наприклад, із цис- і транс-2-бутену
утворюються похідні циклопропану з цис- і т/?анс-розташуванням метальних
груп:
Г-С4Н90К
СНВгз > :СВг2
3 -ґ-С4Н90Н, 2
-КВг
Ц /Н :СВг2
С=С —-
Н3С сн3
Бромоформ Дибромокарбен цис-Бут-
2-ен
і/ш>1,1-Дибромо- /и/?ш/с-Бут-2-ен трансЛ,\-
2,3-диметил- Дибромо-2,3-
циклопропан диметилциклопропан
• Реакція алкенів з іодидом іо дом етил цинку. Для зручного синтезу карбену,
який важко отримувати, запропоновано використовувати реакцію метиле-
ніодиду з цинк-купрумною парою (реакція Сіммонса-Сміта, див. також стор.
94). Дія такого реагенту на олефіни дає тричленні цикли. Згодом доведено, що
карбен у цій реакції не утворюється, а активною частинкою слугує іодид
іодометилцинку (карбеноїд), у цій реакції атом Карбону є як електрофільний
центр:
сн2і2
Тп-Си
Циклогексен
Біцикло[4.1.0]гептан
• Дегідрогалогенуванпя у-галогенокетонів і у-галогенонітрилів. За наявності в
галогеновмісній сполуці рухливого атома Гідрогену в у-положенні відщеплення
гідроген галогеніду супроводжується утворенням тричленного циклу, а не
алкену:
н3с-о-сн-сн2
кон
НіС—С—Т 7
-квг,-н2о її \у
8-А
мо-сн-сн, -
V
5-Бромопентан-2-он Метилциклопропілкетон 4-Хлоробутиро-
нітрил
Циклопропанкарбо-
нітрил
288
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
❖ Синтез похідних циклобутану
• [2+2]-Циклоприєднання. До цієї групи реакцій належать процеси утворення
циклу з молекул двох ненасичених спЬлук, що відбуваються через перерозподіл
електронної густини двох я-систем (також див. розділ 10). Найвідомішими є реакції
термічної або фотохімічної димеризації аленів. Наприклад, унаслідок нагрівання
ален зазнає циклізації, що веде до утворення 1,2-диметиленциклобутану як
основного продукту і мінорної кількості 1,3-диметиленциклобутану:
Н2С—С~СН2~
150 °С
н9с-7с—СНо НоС-гС—СНо
н9с--С—СН2 Н20~"С—СН2
ЕГ2
сн2 Н2С=^
Міжмолекулярна циклизація успішно відбувається і для різних похідних
етилену й ацетилену. Наведемо приклади таких реакцій:
Н2С=С=СН2 + Н2С=СН-СК
Н,С=ї
Ален
Акрилонітрил
0=ї
н2с=с=о + НС=С-ОС2Н5
Кетен
3-Метиленциклобутанкарбонітрил
90% Н2$р4 0=ї І
-ОС2Н5 °°с І 1=0
Циклобутан-1,3-діон
• Циклоізомеризація. До цього типу реакцій належить утворення циклів
унаслідок зміни кратності зв'язків. Зазвичай, такі процеси відбуваються за
досить сильного нагрівання або опромінення ультрафіолетовим світлом:
НС—С=СН <>с
НС-ОСН
Гекс-1,5-діїн
гсн2
сн2
<сн3
^н
сн.
^Н
3,4-Диметиленцикло- цис-Ъ,4-Диметил-
бутен циклобутен
(£,2)-Гекс-2,4-
дієн
транс-Ъ ,4- Диметил-
циклобутен
Наприклад, (£,2)-гекс-2,4-дієн, якщо його нагрівати, зазнає циклізації в
похідну з г/ис-розташуванням метильних груп, а під впливом фотоізомерізації
утворюється транс-ізомер. Для утворення нового зв'язку необхідно, щоб знак
орбіталей збігся відповідно до правила орбітальної орієнтації Вудворда-Хофмана
(див. розділ 10), що і є причиною одержання в цій реакції різних просторових
ізомерів. За термічної циклоізомеризації\р-електрони дієнової системи містяться
на вищій зайнятій молекулярній орбіталі, і, щоб утворились а-зв'язки, необхідні
повороти за С-С-зв'язками в одному напрямі (конротаторне обертання). У разі
Розділ 11. Циклоалкани
289
фотоізомеризації молекула збуджується, і електрони займають нижчу вільну
молекулярну орбіталь, тому, щоб цикл замкнувся, поворот навколо С-С-зв'язків
відбувається у протилежних напрямах (дисротаторне обертання):
СН,
нГ
н
СНз " ~Н ~ иН СНз СН,
Конротатсрне ВЗМО НВМО Дисротаторне
обертання обертання
❖ Синтез похідних циклопентану. Циклопентан з добрими виходами
одержують деякими загальними методами. Зі спеціальних методів можна
навести приклад міжмолекулярної естерної конденсації діетилоксалату з
діетилглутаратом (детальніше про цю реакцію див. у розділі 21):
СООС2Н5 СООС2Н5
о* ос2н5 н2с ~ ' 25
V + 2сн2
0*иОС2Н5 Н2С -2С2н5он
СООС2Н5 СООС2Н5
❖ Синтез похідних циклогексану
• Гідрування бензену і його гомологів. Каталітичним гідруванням бензену
одержують циклогексан. Аналогічно, з гомологів бензену можна одержати
гомологи циклогексану:
СН3
н2/мі Г ґ^чї н2/иі
200 °с 200 °с
Бензен Циклогексан Толуен Метилциклогексан
• Дієновий синтез. Взаємодія дієнів з алкенами або алкінами відбувається за
механізмом [4+2]-циклоприєднання і дає похідні циклогексену (детальніше див.
розділ 10). Реакція 1,3-бутадієну з етиленом відбувається за жорстких умов з
невеликим виходом, а заміщені етилени добре реагують навіть за помірного
нагрівання. Сам дивініл димеризується за температури 150 °С:
сн, сн2'
ii 1
Бут-1,3-ДІєн 4-Вінілциклогексен
290
Чирва В.Я., Ярмолюк СМ., Толкачова H.В., Земляков О.Є. Органічна хімія
❖ Синтез похідних циклогептану. Одержання цнклогептатрієну за
реакцією розширення циклу між бензеном і карбеном, який генерується з
діазометану, має особливе значення. Як інтермедіат утворюється біцикло-
гептадієн, що перегруповується в циклогептатрієн:
❖ Синтез похідних цикло октану. Зручними методами одержання
похідних циклооктану є реакції циклоолігомеризації. Зокрема, ацетилен
тетрамеризується над нікелевим каталізатором за тиску 2 МПа і температури
60 °С {реакція Penne), а дивініл за наявності комплексів Ніколу може
утворювати ряд циклічних сполук, у тому числі циклоокт-1,5-дієн:
4 НСЬСН
Ni(CN)2
і
н2с=^> Ґ [Ni] і—^
Циклооктатетраєн
Циклоокт-1,5-дієн
11.1.8. Хімічні властивості
❖ Реакції малих циклів. У циклопропану напружений цикл. Для нього
характерні реакції, що супроводжуються розривом циклу. Поведінка
циклопропану в хімічних процесах нагадує поведінку етилену. Проте повної
збіжності хімічних властивостей цих сполук немає. Наприклад, циклопропанове
кільце стійке до реакцій окиснення. Для циклобутану, як менш напруженого
циклу, мало характерні процеси з розривом С-С-зв'язків, тому, окрім процесу
гідрування, в реакціях він поводиться як типовий алкан.
• Гідрування. Циклопропан легко зазнає каталітичного гідрування, утворюючи
молекулу пропану. На необхідну для реакції температуру впливає природа
каталізатора. Наприклад, на платинових каталізаторах гідрування відбувається за
50-80 °С, а якщо використовувати нікелевий каталізатор, то потрібне нагрівання до
120 °С. Для гідрування циклобутану до бутану потрібна вища температура:
AH2/Pt і—і H2/Pt
іо^с снзсн2сн3 І і 12(М50;С сн3сн2сн3
• Галогенування. Вільнорадикальне хлорування циклопропану за м'яких умов
(температура до 100 °С) відбувається як звичайний процес заміщення. Вищі
температури або наявність каталізаторів (кислот Льюїса) спричиняють
розщеплення циклу й утворення 1,3-дихлоропропану:
Розділ 11. Циклоалкани
291
С12, Ьу Г\
<100 °С 1^
С12> Ьу
>100 °С
С1СН2СН2СН2С1
Хлороциклопропан
1,3-Дихлоропропан
• Гідр о галогенування. Дія водних розчинів гідроген галогенідів на циклопро-
пан спричиняє розрив С-С-зв'язків. На відміну від алкенів, циклопропан не
реагує з сухими гідроген галогенідами:
Для заміщених похідних циклопропану напрям приєднання гідроген
галогенідів формально відбувається за правилом Марковникова - атом
Гідрогену приєднується до найбільш гідрогенізованого атома Карбону:
2-Бромобутан
• Гідратація. Аналогічно до алкенів, циклопропан утворює ациклічні спирти,
якщо його витримувати в концентрованій сульфатній кислоті:
❖ Реакції звичайних циклів. Представники цієї групи циклоалканів за
хімічною поведінкою близькі до насичених вуглеводнів. Для них характерні
реакції вільнорадикального галогенування, нітрування, сульфохлорування. Вони
стійкі до дії окиснювачів і окиснюються лише за досить жорстких умов.
Каталітичне гідрування відбувається за високих температур, за яких можливий
піроліз С-С-зв'язків. Фактично можна говорити про відновлення продуктів
крекінгу циклоалканів. Нижче наведено типові реакції цих циклічних вугле-
воднів.
• Каталітичне гідрування:
1-Хлоропропан
Пропан-1-ол
• Галогенування:
Хлороциклогексан
292
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
• Нітрування. В промисловості циклогексан нітрують у паровій фазі до
нітроциклогексану, який потім відновлюють до оксиму циклогексанону -
вихідного продукту в синтезі капролактаму (також див. розділ 22):
О
150 °С \ / Р.і \ /
Нітроциклогексан Оксим циклогексанону
• Окиснення циклогексану. В промисловості циклогексан окиснюють киснем
над каталізаторами на основі кобальту або мангану до циклогексанолу,
циклогексанону й адипінової кислоти. Окиснення циклогексанону концентро-
ваною нітратною кислотою також розриває С-С-зв'язки з утворенням
адипінової кислоти:
ОІ°1 АЛ ™т 1° 1 АЛ_^ 1°' [0] = 02/Со(Мп),Г;
_^0Н--(^=0--НООС(СН2)4СООН ^о^оЦ;іоо.200»с
Циклогексанол Циклогексанон Адипінова кислота
Аналогічно окисненням циклопентану можна одержати глутарову кислоту
НООС(СН2)3СООН.
• Дегідрування циклогексану. Нагрівання циклогексану і його похідних з
паладієвим, платиновим або нікелевим каталізаторами дає ароматичні
вуглеводні. Практично за тих же умов відбувається і каталітичне гідрування
аренів. Реакція зсувається вправо внаслідок сорбції водню на каталізаторах, і,
навпаки, вліво за наявності надлишку водню. Ця реакція відома як зворотний
каталіз Зелінського.
?й(Рі,N0 г^С| Г ] разімо
>200 °С І^-^І + ЗН2 к у >200 °С
НЗС-СН-СНЗ н30сн-СНЗ
Циклогексан Бензен Ментан я-Цимен
• Незворотпий каталіз Зелінського. Ненасичені похідні циклогексану - цикло-
гексен і циклогекс-1,3-дієн - за наявності платинового каталізатора диспро-
порціонують на циклогексан і бензен:
О^ОО зО^?і 1+211
Циклогексен Циклогекс-1,3-ДІєн
❖ Реакції звуження і розширення циклу. В цю групу реакцій об'єднують
хімічні процеси, що супроводжуються зміною розмірів циклу. Одну з таких
Розділ 11. Циклоалкани
293
реакцій - розширення циклів у циклокетонах дією діазометану - ми вже
розглянули в загальних методах одержання циклічних сполук.
Практично всі ці процеси відбуваються через проміжний карбокатіон. Якщо
позитивний заряд міститься на атомі Карбону циклу, то зазвичай виникає звуження
циклу, а якщо він локалізований на атомі Карбону поряд з циклом, то цикл
розширюється. Оскільки зменшення розмірів циклу супроводжується збільшенням
внутрішньої енергії молекули, то зазвичай процес звуження циклу відбувається між
тотожними за напруженням циклічними системами: циклобутан —» циклопропан
або циклогексан -> циклопентан. Нижче наведено приклади таких реакцій:
Іодоциклогексан Циклогексан Метилциклопентан
н2с-он СН2
* ♦ о
Циклобутилметан Метиленциклобут Циклопент
Циклопропілметанол Бромрметилциклопропан Бромометилциклобутан
• Ізомеризація циклів. Циклоалкани, аналогічно до насичених вуглеводнів за
наявності кислот Льюїса, утворюють рівноважну суміш сполук. Наприклад, за
наявності алюміній хлориду за кімнатної температури усталюється рівновага в
суміші, що складається із 88 % циклогексану і 12 % метилциклопентану. Реакція
також відбувається через утворення і перегрупування карбокатіонів:
ІіОАІСІз НАІСІз"
1^/-СН2 5=* ІУ~СН2 \^_у+ НАІСІз «
• Перегрупування Дем'янова1. Ізомерні первинні аміни з різними розмірами циклів
(наприклад, метилциклопропіламін і циклобутиламін), взаємодіючи з нітритною
1 Дем'янов М. (1861-1938) - російський, радянський хімік-органік. Дослідження в галузі
аліциклічних сполук.
А1С13^
294
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
кислотою, утворюють суміш одних і тих же спиртів - циююпропілметанолу і
циклобутанолу (детальніше про реакції амінів з нітритною кислотою див. розділ 16).
Отже, можна вважати, що в одному випадку розмір циклу збільшується, а в іншому -
зменшується. Вивчення механізму цієї реакції засвідчило, що вона відбувається через
карбокатіони, які легко зазнають ізомеризування один в інший, взаємодіючи з водою,
та дають суміш ізомерних спиртів. Реакція не має препаративного значення, тому що
разом зі спиртами одержують велику кількість побічних продуктів:
Звуження циклу описано лише для амінів, у складі яких є циклобутанові та
циклогексанові кільця.
11.1.9. Вплив просторових чинників на реакції
похідних циклоалканів
Функціональні замісники в циклогексані можуть бути розташовані в
аксіальному та екваторіальному положеннях. На їхню реакційну здатність
впливають різні просторові чинники. Щоб спростити оцінку просторових чинників,
необхідно запобігти інверсії шестичленного циклу. Це можна досягнути, додавши в
цикл об'ємний замісник, наприклад, трет-бутильну групу, яка розташовується
чітко екваторіально. Другим стабільним конформером є трянс-декалін. Наявність
другого циклу в цій сполуці також сприяє закріпленню конформації. Розглянемо
декілька прикладів впливу просторових чинників на реакційну здатність
аліциклічних похідних.
❖ Кислотність. Із двох ізомерних 4-трет-бутилциклогексанкарбонових
кислот трянс-ізомер (екваторіальна карбоксильна група) має більшу кислотність
порівняно з */мс-похідною (аксіальна карбоксильна група). Цей факт можна
пояснити тим, що відповідний карбоксил-аніон просторово доступніший у разі
екваторіальної орієнтації. Отже, він легше сольватується і ліпше стабілізується:
/?Ка 7,79 /?Ка 8,23
транс- і і/ис-4-т/?е/и-Бутилциклогексанкарбонові кислоти
Розділ 11. Циклоалкани
295
❖ Окиснення. Хромовий ангідрид окиснює 4-тре/и-бутилциклогексанол з
екваторіальною орієнтацією гідроксильної групи до відповідного кетону в 3,23
раза повільніше, ніж його г/нс-ізомер:
Розщеплення проміжного естеру хроматної кислоти є лімітувальною
стадією реакції. Оскільки перебування об'ємного естеру в аксіальному
положенні невигідне, то швидке елімінування знімає напруження в молекулі.
❖ Відновлення. Атака гідрид-аніона в процесі відновлення похідного
циклогексанону може відбуватися з двох боків. Використання як відновника
літій три-втор-бутоксиалюмогідриду зумовлює утворення переважно цис-4-
/и/?ет-бутилциклогексанолу, оскільки великий об'єм реагенту перешкоджає
аксіальній атаці. Менший за розміром літій алюмогідрид перевалено відновлює
кетон до /иранс-ізомеру:
ІЛАІН4 ЬіА1[ОСН(СН3)СН2СНз]зН
Літій алюмогідрид Літій три-в/ио/?-бутоксиалюмогідрид
❖ Омилення. Омилення т/?аяс-етил-4-тре^-бутилциклогексанкарбокси-
лату 1 відбувається в 19,8 раза швидше, ніж його і/ис-ізомеру 2. Із вихідних
сполук продукт з екваторіальною орієнтацією естерної функції на -5 кДж/моль
стійкіший, ніж конформер з аксіальною етоксикарбонільною групою (рис 11.2).
В активованому комплексі (див. механізм омилення в розділі 18) відбувається
зміна гібридизації атома Карбону з Бр2 в ^р3. До того ж, збільшується об'єм
296
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
замісника, що за аксіальної конфігурації збільшує вандерваальсове напруження
і, відповідно, збільшує енергію активованого комплексу. Як відомо, реакція
переважно відбувається через менший за енергією перехідний стан:
0*_.ОС2Н5
рн
со;
(Н3С)з<
НО"
(НзС)з'
оХос2н5
(НзС)з<^/ ^/
~5 кДж/моль
1
Координата реакції
Рис. ¡1.2. Енергетична діаграма омилення
транс- і цис-етил-4-трет-
бутилциклогексанкарбоксилату
Координата реакції
Рис. 11.3. Енергетична діаграма сольволізу
транс- і цис-1 -трет-бутил-4-
тозилоксициклогексану
❖ Нуклеофільне заміщення за механамом .Улгі. Щоб порівняти швидкості
реакцій нуклеофільного заміщення у двох стереоізомерів, необхідно, щоб реакції
відбувалися через один активований комплекс. У випадку механізму 8ц1 реакція
сольволізу тозилатів відбувається через один і той же карбокатіон (рис. 11.3):
(Н3С)3(
ОТз (Н3С)зС^С^У в
С2Н5ОН
(Н3С)3<
З
Тз—НОьсНз
О ^
Розділ 11. Циклоалкани
297
Відомо, що енергія активації визначена різницею між енергією перехідного
стану і внутрішньою енергією вихідної сполуки. Очевидно, що легше буде
реагувати напруженіша сполука з аксіальною конфігурацією. Справді, цис-
ізомер 2 реагує в 4 рази швидше, ніж трш/оаналог 1.
❖ Нукпеофільне заміщення за механізмом 5^2. Дня перебігу реакції за цим
механізмом необхідно, щоб група, яка відходить, і нуклеофіл, що атакує з тилу,
перебували на одній прямій. За аксіального розташування групи, яка відходить,
активований комплекс реалізується легко, а за екваторіального положення цієї
групи атаці нуклеофілу "в тил" стеричні перешкоди створює сам цикл. У цьому
випадку для перебігу реакції потрібна зміна конформації циклу, що спричинить
зростання внутрішньої енергії, і, відповідно, зменшить швидкість процесу. В
експерименті г/«с-1-шрет-бутил-4-тозилоксициклогексан 2 в 19 разів швидше реагує
з натрій тіофенолятом порівняно з шранс-похідною 1:
❖ Елімінування за механізмом Е2. За цим механізмом, щоб утворився
активований комплекс, групи, які відходять, повинні перебувати на одній
гоющині й мати протилежну орієнтацію. Наприклад, і/мс-1-метил-2-то-
зилоксициклогексан 1 під дією сильної основи утворює суміш 1-метилцикло-
гексену 2 (59 %) і 3-метилциклогексену 3 (41 %), а відповідний транс-ізомер 4
дає лише 3-метилциклогексен 3.
Щоб виникла необхідна для перебігу реакції конфігурація, сполука 1
переходить у конформацію з аксіальним розташуванням тозилоксигрупи.
Основа може атакувати два сусідні аксіальні атоми Гідрогену. Відщеплення
відбувається переважно від більш заміщеного атома Карбону. В випадку транс-
ізомеру 4 за аксіальної конфігурації тозилату лише один сусідній атом Гідрогену
має аксіальне розташування:
298
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
11.2. Бі- та поліциклічні вуглеводні
Класифікацію бі- та поліциклічних сполук ми навели на початку розділу.
Для цих вуглеводнів характерне велике різноманіття в будові і, відповідно, у
властивостях, тому ми розглянемо лише найтиповіших представників цього
класу аліциклів.
11.2.1. Номенклатура
❖ Спіровуглеводні. В цих сполуках один спільний (вузловий, або спіро)
атом. Назва сполук цього класу починається з префікса спіро-, далі в квадратних
дужках арабськими цифрами через крапку зазначають кількість атомів Карбону
по різні боки від вузлового атома, починаючи з меншого за розміром циклу.
Після квадратної дужки записують назва алкану, що відповідає загальній
кількості атомів Карбону. Нумерацію починають у меншому циклі від
найближчого до вузла атома, закінчують вузловим атомом і продовжують по
великому циклу. Для похідних спіранів, з урахуванням особливостей нумерації,
застосовують усі інші принципи побудови назв за замісниковою номенкла-
турою, зокрема, надання мінімальних локантів кратним зв'язкам або замісникам:
Спіро[2.3]гексан 4-Метилспіро[2.3]гексан Спіро[3.4]октан [3.4]-Спіро-1-октен
❖ Місткові вуглеводні. Біциклічні системи, як конденсовані, так і місткові,
мають два вузлові атоми Карбону. їхню назву утворюють з назви вуглеводню,
який має таку ж кількість карбонових атомів, що і циклічна система, додаючи
префікс біцикло-. Після префікса в квадратних дужках арабськими цифрами,
розділяючи їх крапками, зазначають кількість атомів Карбону по різні боки від
вузлових атомів (починаючи з великого циклу) і кількість атомів Карбону в
Розділ 11. Циклоалкани
299
містку (для конденсованих систем 0). Нумерацію починають від одного з
вузлових атомів, ведуть по периметру циклу в напрямі великого кільця і
закінчують атомами містка:
□О
CO <D ©
Біцикло[3.2.0]гептан Біцикло[5.4.0]ундекан Біцикло[3.1.1]гептан Біцикло[3.2.2]нонан
Для бі- та поліциклічних систем, які можна розглядати як продукти повного
відновлення відповідних конденсованих ароматичних систем, можливе
використання назв, які утворюють від тривіальних назв аренів додаванням
префікса пергідро-:
Пергідронафталін (декалін)
Пергідроантрацен
Пергідрофлуорен
❖ Поліциклічні системи. В основі назв поліциклічних вуглеводнів є назва
відповідного вуглеводню, до якого додають префікси трицикло-, тетрацикло- і
т.д. Для цього класу сполук числовий префікс позначає кількість зв'язків, які
умовно потрібно розірвати в поліциклані, щоб утворилася ациклічна сполука
(показано знаком ~).
Між префіксом і основою в квадратних дужках наводять спеціальні цифрові
позначення. Щоб їх правильно визначити, спочатку вибирають головний цикл і
головний місток, які мають максимальну кількість атомів Карбону. Першими
трьома цифрами за правилами місткових вуглеводнів позначають головну систему.
Наступні цифри означають кількість карбонових атомів у містках, що залишились, а
надрядкові цифри відображають, які атоми Карбону вони з'єднують:
о 1 2 ,
1
2
Трициклононан Трицикло[4.2.1.02,5]нонан
4
Трицикл одекан Трицикло[3.3.1.13,7]декан
Для великої групи каркасних вуглеводнів є тривіальні назви, що включені
до номенклатури IUP АС:
Кубан
Квадрициклан
Адамантан
Баскетан
Твістан
300
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
11.2.2. Лекалін
Типовим представником конденсованих біциклічних систем є декалін
(пергідронафталін, біцикло[4.4.0]декан). Ця сполука існує у вигляді двох
стереоізомерів цис- і транс-декаліну, що відрізняються орієнтацією атомів
Гідрогену біля вузлових атомів Карбону. Найстійкіші конформації цис- і транс-
декаліну - це два з'єднані циклогексанові "крісла". Однак г/г/с-декалін
приблизно на 11 кДж/моль менш стійкий порівняно з транс-ізомером через
реалізацію гош-бутанових напружень (на приведеному нижче рисунку виділені
жирними лініями) і екзо-орієнтації атомів Гідрогену, що зближені на відстань
0,2 нм з аксіальними атомами Гідрогену другого циклу:
Н
Н
Н
Н
Н
CD
н
н
н
н
/ираяс-Декалін і/ис-Декалін
Типовий спосіб одержання цієї сполуки полягає у вичерпному
каталітичному гідруванні нафталену; у цьому разі утворюється суміш ізомерів:
н2/т
300 °с
11.2.3. Адамантан
Трициклічний каркасний вуглеводень трицикло[3.3.1.13,7]декан відомий як
адамантан, оскільки його структура відповідає кристалічній ґратці алмаза
(adamant). Молекула адамантану дуже стійка, тому що вона вільна від
напруження і її можна розглядати як сполучення трьох циклогексанових
конформацій "крісло".
Адамантан має легкий камфорний запах. Власне специфічний запах деяких
фракцій нафти і спонукав дослідників їх розділити, що й дало змогу одержати
цю сполуку. Адамантан має досить високу для вуглеводнів температуру
плавлення (270 °С), проте він легко сублімує за невисоких температур, що
пов'язано зі значною компактністю його структури і, відповідно, слабкими
міжмолекулярними взаємодіями у кристалах.
Основну кількість адамантану одержують з нафти. Синтетично його можна
одержувати з димеру циклопентадієну.. Гідрують та ізомеризують ендо-
дициклопентадієн за наявності кислот Льюїса. Найліпші результати досягнуто
для каталітичної системи гідроген бромід-бор флуорид:
Розділ 11. Циклоалкани
301
со
A1CU
Циклопентадієн ewóo-Дициклопентадієн
ю
(HBr-BF3) P^J
Адамантан
Адамантан досить легко за наявності кислот Льюїса зазнає бромування до 1-
бромоадамантану, який, відповідно, взаємодіючи з мурашиною кислотою, дає
іншу важливу похідну - адамантан-1-карбонову кислоту. Адамантан стійкий до
окиснення, проте дія такого сильного окиснювача, як озон, перетворює його в
адамант- 1-ол:
Вг2/АВг3
-НВг
1 -Бромоадамантан Адамантан-1 -карбонова кислота
Адамантан-1-ол
Поєднання великої гідрофобності адамантильного фрагмента з мінімальним
для вуглеводнів ефективним об'ємом надає похідним адамантану особливих
фізико-хімічних властивостей, які використано у створенні лікарських засобів.
Адамантильні радикали є в складі таких ліків, як ремантадин (противірусний
засіб), мемантин (препарат для лікування хвороби Паркінсона) і глудантан
(противірусний препарат для лікування кон'юнктивіту і засіб для лікування
хвороби Паркінсона):
СН-СНз н с
NH9
Ремантадин
соон
но^—^А.
он
Глудантан
11.2.4. Moho- і біциклічні терпени
❖ Мопоциклічні терпени. В природі найпоширеніші моно- і біциклічні
терпени; трициклічні структури трапляються дуже зрідка. Природні моноциклічні
терпени належать до дієнових похідних вуглеводню ментану і відрізняються
взаємним розташуванням подвійних зв'язків. Найвідоміші моноциклічні терпени:
лимонен (/-лимонен - компонент скипидару, лимонної і м'ятної олій, а його rf-ізомер
є в складі олії тмину) та а-, р- і у-терпінени (одержані з олії живиці - продукту
перегонки смол хвойних дерев). Також а- і у-терпінени трапляються в ефірних оліях
коріандру, кропу, майорану:
302
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
СН,
Н3С" "СН3
а-Терпінен
н3с сн3 Н3С СН3
Р-Терпінен у-Терпінен
сн3
СН3
л
Н3С СН3
НзС^ХНз
Ментон
Цинеол
СН,
Н3С^СН2
Карвон
Серед моноциклічних терпеноїдів найпоширеніші 1-ментол і продукт його
окиснення ментон (олія м'яти), цинеол (евкаліптова олія, олія шавлії), карвон
(олія кропу і тмину). В молекулі ментолу три асиметричні атоми Карбону.
Природний /-ментол має найстійкішу конфігурацію з трьома екваторіальними
замісниками. У ментолу сильний м'ятний запах і присмак холоду. Його широко
застосовують у медицині як заспокійливий засіб, зокрема, як розчин у
ментилізовалераті - естері ментолу та ізовалеріанової кислоти ^валідол"):
сн3
О
п
І,.,он3 НзС'\^23сН(СНз)2
Н,С-СН-СЩ
НзС V Д^СН(СН3)2
/-Ментол
У промисловості ментол у
гідруванням тимолу:
СН3
Ментилізовалерат
вигляді рацемату одержують каталітичним
Н2/РІ
чОН
н3с-СН-сн3
❖ Біциклічні терпени. Ці сполуки значно поширені в природі. Найвідо-
міші терпени і терпеноїди - похідні туйану, карану, пінану і камфану.
■ До групи туйану належать біциклічний кетон туйон, знайдений в олії туї і
полину.
■ До групи карану належать А3- і -карени, які виділяють з олії живиці:
СН3 СН3 СН3 СН3 9Нз
1 *0
Н3С" "СН3
Туйан
Н3С" ХН3
Туйон
ГСН3
ГСНз
Розділ 11. Циклоалкани
303
■ Найвідоміші похідні пінану - а- і ^-пінени - часто трапляються в ефірних
оліях. Зокрема, а-пінен є головним компонентом скипидару (-60 %):
-СН3 ШС^СНз Н.С^СНз
н,с
сн,
сн,
Пінан
а-Пінен
р-Пінен
■ Головними представниками групи камфану є спирт борнеол і продукт
його окиснення камфора. Обидві ці сполуки належать до біцикло[2.2.1]-систем,
шестичленне кільце яких перебуває в конформації "ванна". Замісників у цьому
циклі розрізняють за просторовою орієнтацією: екзо - напрямлені в бік містка,
та ендо - орієнтовані в бік від містка:
Н3ССН3
Конформація камфану
Н
Я І^^У н
екзо- і ендо-Заміщені біцикло[2.2.1]гептани
Природний борнеол має ендогідроксильну групу. З природних джерел
виділені обидва його дзеркальні антиподи. Головними компонентами ефірної
олії валеріани є (-)-борнеол і його естер ізовалеріанової кислоти. У складі смоли
хвойних дерев як головний компонент є борнілацетат:
Н3С
СН,
ОН
0-С-СН2СН(СН3):
Камфан (-)-Борнеол Борнілізовалерат Борнілацетат
Камфора має специфічний запах і гірко-пекучий смак. Зокрема, <і-камфора
знайдена в складі деревини камфорового дерева, що росте в Південно-Східній
Азії. Вона також є в складі олій камфорної шавлії, камфорного лавру, скипидару
хвойних дерев. Камфору широко застосовують у медицині як препарат, що
підсилює серцеву діяльність і стимулює дихання. Фізіологічні властивості має
як чистий енантіомер, так і рацемічна суміш:
СН3 НзС^СНз
^/-Камфора
304 Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
.О-СН НСЮчХ
>ГСН3 + НзР^СІСНз
а. нсг
Ь.[0]
Я3
:чсн3
:н3
о*
+ Нзр-с|сн3
11.2.5. Стероїди
Стероїди — велика група біологічно активних сполук, що об'єднані
наявністю скелета пергідроциклопентанофенантрену (естрону). До цього класу
сполук належать статеві гормони і гормони надниркової залози, серцеві
глікозиди, вітаміни, стерини (ліпіди, які не зазнають омилення), жовчні кислоти
й інші природні сполуки.
Конформаційно стійкою формою пергідроциклопентанофенантрену є
трянс-сполучення шестичленних циклів у вигляді конформацій "крісло" і
п'ятичленного циклу в конформації "конверт":
Естран
З похідних естрану найчастіше трапляються два види ізомерів - а- і Р-сте-
роїди, що відрізняються розташуванням замісників відносно площини циклічної
системи (детальніше див. розділ 6). Для 5ос-ізомерів стероїдів виникає транс-
з'єднання двох кілець декалінової системи, а для 5р-похідних, відповідно, - цис-
з'єднання.
Головну кількість камфори як лікарського засобу одерясують синтетично.
Вихідним продуктом є сс-пінен, який ізомеризують сумішшю мурашиної та
сульфатної кислот до форміату. Естер омилюють і одержаний спирт окиснюють
до кетону. Продукт є рацемічною сумішшю:
Розділ 11. Циклоалкани
305
ОН
5а-Стероїд (андростенол) 5р-Стероїд (холева кислота)
■ Типовим представником стероїдів є холестерол - компонент ліпідних
мембран. Його особливо багато в клітинах головного мозку (10% ваги) і в
жовчних каменях (до 90 %):
Холестерол Естрадіол Тестостерон
■ До стероїдних гормонів належить один з найактивніших — жіночий
статевий гормон естрадіол, який широко застосовують у медичній практиці. Ця
речовина також знайдена в рослинах (кокосова пальма, верба). Чоловічий
статевий гормон тестостерон не лише впливає на ендокринну систему, а й є
анаболічним гормоном.
Розділ 12
гллогенопохілні
вуглевоанів
Усю різноманітність органічних сполук хіміки пов'язують з вуглеводнями
та їхніми похідними, під якими розуміють сполуки, що мають інші атоми, ніж
атоми Карбону і Гідрогену. Такі атоми називають гетероатомами.
Вуглеводні, у яких один або декілька гідрогенних атомів заміщені атомами
галогенів, називають галогенопохідними. Ця група охоплює різні типи галогено-
похідних, зокрема moho-, ди-, три- і полігалогеноалкани, а також галогено-
похідні ненасичених вуглеводнів.
12.1. Будова і номенклатура
12.1.1. Класифікація галогенопохідних
Галогенопохідні вуглеводнів класифікують за декількома принципами:
■ за природою атома галогену поділяють на флуоро-, хлоро-, бромо- та
іодопохідні:
CH3F СН3С1 СН3Вг СН3І
Флуорометан, або Хлорометан, або Бромометан, або Іодометан, або
метилфлуорид метилхлорид метил бром ід метиліодид
■ за кількістю атомів галогенів розрізняють moho-, ди-, три- і
полігалогеновуглеводні:
СН3С1 СН2СЬ СНСІз СС14
Хлорометан, або Дихлорометан, або Трихлорометан, або Тетрахлорометан
метилхлорид метиленхлорид хлороформ
■ за природою заміщеного атома Карбону бувають первинні, вторинні й
третинні галогенопохідні:
СНз^НСНз-Вг CH3CH2CjH-Br
СН3 СН3
СН3
СНз-С-Вг
СН3
І-Бромо-2-метилпропан, або 2-Бромобутан, або 2-Бромо-2-метилпропан, або
ізобутилбромід emop-бутилбромід mpe/w-бутилбромід
Розділ 12. Галогенопохідні вуглеводнів
307
■ за природою заміщеного атома Карбону поділяють на алкілгалогеніди,
вінілгалогеніди й ацетиленілгалогеніди:
СН3СН2СН2СН2СЬ
1-Хлоробутан, або
бутилхлорид
Н2С=СНСЬ
Хлороетилен, або
вінілхлорид
носсь
Хлороацетилен, або
етинілхлорид
12.1.2. Номенклатура ШРАС
Зомісникова номенклатура. В замісникові номенклатурі галогени
розглядають як замісники, їх називають лише в префіксі. Вибір головного
ланцюга і порядок нумерації виконують за тими ж правилами, що й для
вуглеводнів:
/ 2 3 4 5 6
СНзСНСНоСНСНоСН*
З, 2, 2 3
СІ СН3
4-Метил-2-хлоротексан
Нумерація — відповідно до прин-
ципу мінімальних локантів для
замісників.
5 4 3 2 1
сн3снсн=ссн3
І І
СІ СН3
2-Метил-4-хлоропент-2-ен
Менший локант присвоюють
атомові Карбону подвійного
зв'язку.
1 -Бромо-3-хлороциклогексан
Початок нумерації з першого
за абеткою замісника.
• Радикально-функціональна номенклатура. В радикально-функціональній
номенклатурі назва сполуки складається з назви вуглеводневого радикала і
назви галогеніду, які записують без розподілення. Приклади назв
галогенопохідних за радикально-функціональною номенклатурою наведені в
параграфі "Класифікація галогенопохідних".
12.2. Фізичні властивості
Уведення галогену в алкан підвищує густину сполуки. Серед них
іодопохідні мають найбільшу густину. Тому рідкий метиленіодид СН2І2
застосовують у мінералогії для визначення густини мінералів.
З огляду на більшу молекулярну масу температура кипіння алкілгалогенідів
вища, ніж температура кипіння алканів з тією ж кількістю карбонових атомів.
Для заданої алкільної групи вона підвищується зі збільшенням атомної маси
галогенів, так що флуориди мають найнижчі, а іодиди - найвищі температури
кипіння. Загалом для галогеновуглеводнів температури кипіння і плавлення
порівняно невисокі через слабкі міжмолекулярні взаємодії диполь-дипольного
типу. Наприклад, температура кипіння етилхлориду настільки низька, що шар
рідини, випаровуючись на шкірі та забираючи тепло, заморожує тканину,
роблячи її нечутливою до болю. Тому етилхлорид застосовують як місцевий
анестетик у випадку нескладних травм.
308 Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Галогенопохідні вуглеводнів мають полярний ковалентний зв'язок Карбон-
галоген і, на відміну від розглянутих алканів, вирізняються помітним дипольним
моментом, що порівнюваний за значенням з дипольними моментами спиртів
(табл. 12.1).
Таблиця 12.1
Дипольні моменти галогеноалканів і деяких органічних сполук
Сполука
Дипольний момент, Т>
Сполука
Дипольний момент, Т>
н3с-сн3
0
СН3СН2Р
1,95
Н3С-МН2
1,32
СН3СН2С1
2,02
НзС-ОН
1,69
СН3СН2Вг
1,95
Н3С-Р
1,81
СН3СН2І
1,82
Н3С-С1
1,86
СН2Р2
1,91
Н3С-Вг
1,78
СН2С12
1,63
Н3С-І
1,59
СН2Вг2
1,48
СН3СН2ОН
1,69
СНСІз
1,12
СН3СН2ОСН2СН3
1,16
СР2С12
1,18
Н3С-С-СН3
2,85
С6Н5С1
1,70
0
Н2С=СНС1
1,44
Хоча галогеноалкани - полярні сполуки, вони нерозчинні у воді, імовірно,
тому, що не здатні утворювати водневі зв'язки. Вони розчинні в більшості
органічних розчинників, мають трохи солодкуватий запах, розчиняють жири.
Полігалогеновуглеводні добре знежирюють поверхні металів, одягу. Цікаві
перфлуориди алканів загальної формули Сп¥2п+2- Незважаючи на різке
збільшення молекулярної маси порівняно з алканами, їхні температури кипіння
мало відрізняються. Це свідчить про те, що між атомами Флуору в органічних
флуоропохідних виявляються надзвичайно малі вандерваальсові сили. Завдяки
цьому вироби з полімерних перфлуоровуглеводнів не змочує вода, а
високомолекулярний матеріал перфлуорополіетилен (тефлон) має високу
хімічну інертність і низьке стирання, що зумовлює його застосування, зокрема, у
виготовленні підшипників.
12.2.1. Спектроскопія галогеноалканів
• УФ-спектроскопія. В УФ-спектрах галогеноалкани здатні до %—^-пере-
ходів, мають смуги поглинання з ХтйХ для хлоропохідних - 173 нм, а для
бромоалканів - 210 нм.
• ІЧ-спектроскопія. В ІЧ-спектрах смуги валентних коливань уС-наі середньої
інтенсивності спостерігають у діапазоні для Хлору 850-795 см"1, Брому - 680-
500, Іоду - 500-200 см"1. Для флуоропохідних вуглеводнів характерні сильні
валентні коливання 1 100-1 000 см"1.
Розділ 12. Галогенопохідні вуглеводнів
309
• Мас-спектрометрія. В мас-спектрах хлоро- і бромопохідних вуглеводнів
легко знайти молекулярний іон, оскільки він виявляється як два сигнали М+ і
(М+2)+ зі співвідношенням інтенсивності 3:1 для хлоровмісних і, відповідно, 1:1
для бромовмісних сполук. Таку форму сигналів пояснюють тим, що атоми
Хлору мають два основні ізотопи 35С1 (-75,5 %) і 37С1 (-24,5 %), а атоми Брому -
79Вг (-50,5 %) і 81Вг (-49,5 %).
• Спектроскопія ПМР. В ПМР-спектрах алкілгалогенідів збільшення
електронегативності галогену в ряду від Іоду до Флуору зміщує сигнал протонів у
слабше поле, наприклад, НзС-І 8 2,16 м.ч., НзС-Вг 8 2,68 м.ч., Н3С-СІ 8 3,05 м.ч.,
НзС-її 8 4,26 м.ч. Перехід від фрагмента СН3-С1 до -СНт-СІ і >СН-С1 зміщує
сигнали протонів від 3,05 до 3,44 м.ч. і 4,1 м.ч., відповідно.
12.3. Синтез галогенопохідних
12.3.1. Одержання галогенопохідних з алканів
• Фпуорошікапи. Після 1940 р. хімія флуоропохідних почала швидко розви-
ватися. Флуорування вільним флуором спричиняє повну деструкцію молекули
вуглеводню через високу екзотермічність реакції і, як наслідок, неспроможність
керувати процесом:
С„Н2„+2 + (Зп+1)¥2 -> пС¥4 + (2и+2)НР
У разі нестачі потрібної кількості Флуору Карбон виділяється у вигляді
сажі. Через сильне розведення Флуору інертним газом з'являється можливість
вести реакцію заміщення, однак ліпше для цього використовувати спеціальні
прийоми флуорування вуглеводнів.
■ Флуорування дією кобальт трифлуориду. Навперемінне пропускання
алканів і Флуору над каталізатором дає змогу виконувати флуорування і
регенерувати кобальт трифлуорид із кобальт дифлуориду, що одержують у ході
реакції
Я-Н + 2Со¥3 20(>-300.оск-Р + СоР2+ НБ СоР2 + Р2 СоР3
Таким шляхом можна синтезувати перфлуоровані вуглеводні з дуже
високим виходом. Через великі витрати Флуору цей метод є досить дорогим.
• Хлоро- і бромоалкани. Прямі реакції галогенування, за винятком метану і ще
деяких алканів, не мають практичного застосування. Детальніше про методи
синтезу хлоро- і бромопохідних з алканів див. у розділі 7.
• Іодоалкани не можна одержати реакцією прямого йодування.
310 Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
123.2. Синтез галогенопохідних зі спиртів
Оскільки пряме галогенування алканів не дає змоги вибірково ввести один атом
галогену, а призводить до суміші продуктів з різним ступенем заміщення, то
широкого використання набув метод синтезу моногалогенопохідних зі спиртів.
❖ Реакція з гідроген галогенідами. Спирти легко реагують з гідроген
галогенідами, утворюючи алкілгалогеніди і воду. Цей метод має як промислове,
так і лабораторне значення, його використовують для синтезу хлоро-, бромо- та
іодоалканів. Швидкість реакції залежить від будови спиртів і природи гідроген
галогенідів. Реакційна здатність спиртів зменшується в ряду: третинні спирти >
вторинні спирти > первинні спирти, а також у ряду: гідроген іодйд > гідроген
бромід> гідроген хлорид. Зазначимо, що з гідроген іодидом і гідроген бромідом
реакції відбуваються легко, з гідроген хлоридом - важче, з гідроген флуоридом
реакція не відбувається. Тому монофлуороалкани одержують заміщенням атома
Брому або Іоду флуорид-аніоном за реакцією обміну (див. далі 12.3.7).
Зазвичай сухий газоподібний гідроген галогенід пропускають через спирт.
Рідше, наприклад, для третинних спиртів, процес проводять з концентрованими
розчинами галогеноводневих кислот. Гідроген бромід інколи отримують у
реакційній суміші дією сульфатної кислоти на натрій бромід.
Гідроген хлорид реагує з первинними спиртами, як звичайно, за наявності
цинк хлориду, а трет-бутиловий спирт перетворюється в хлорид у разі
струшування його з концентрованою хлоридною кислотою вже за кімнатної
температури. Нижче наведені приклади подібних реакцій:
о" °Н "ІГ Су ВГ СНзСН2СН2СН2ОН^4 сн3сн2сн2сн2вг
Циклогексанол Бромоциклогексан Бутан-1-ол "Н20 1-Бромобутан
сн, сн.
НС1 (газ) „„ „„ Г З „СІ „„ І 3
СН3СН2СН2ОН СН3 СН2СН2С1 СН—С-ОН -ТГ5- сн—с-сі
сн, 2 сн.
Пропан-1-о л 1-Хлоропропан
З ^"3
2-Метилпропан-2-ол 2-Метил-2-хлоропропан
• Перегрупування. Особливістю цих реакцій є можливість перегрупувань, які
особливо легко відбуваються у випадку розташування гідроксильної групи
поряд з третинними або четвертинними атомами Карбону:
І|І (рНз 1^ <^Нз СНз СНз
ш3-с-с-сн3 сн3-^с-сн3 сНз-с-шрн-1^ СНГС-СН2СН3
НО Н Н СІ СНз СІ
З-Метилбутан-2-ол 2-Метил-2-хлоробутан 2,2-Диметилпропан-1-ол 2-Метил-2-хлоробутан
Розділ 12. Галогенопохідні вуглеводнів
311
Існування перегрупувань свідчить про утворення проміжних карбокатіонів,
а це означає, що реакція відбувається за механізмом вці. Процес починається з
протонування гідроксильної групи спирту. Лімітувальна стадія реакції -
дисоціація - дає карбокатіон, який швидко реагує з нуклеофілом:
Я~ОН + НХ^=^Я-іЗН2 + Х
+ Повільно
Я-ОНз я+ + Н20
Швидко
я++х~^—- я-х
Якщо структура сполуки забезпечує моясливість утворення стійкішого
карбокатіону, то відбуваються 1,2-гідридні або 1,2-алкільні зсуви:
нанз№ н ш, нш, н СНз нсн,
н3с-9-С-сн3 — НзС-СГ9-СНз н3с-С-С-сн3— н3с-С-С-сн3 -*НзОС-С-СНз
но н н-о н "Н2° 1 н) пфидшйзсув н на
^ Вторинний кзрбокагіон Третинний карбокатіон
Нз9 № н3с + н3с
НзС-9-о^ —- н3с-С-СК, -2» НзС-9-а^ —*. НзС-с-о^-оі, —- ^09-0^-01,
н3с он н3с сьн НзСО
Н Первинний карбокатіон Третинний карбокатіон
■ Лінійні первинні спирти не перегруповуються тому, що вони реагують за
8м2-механізмом:
х-+ЯОН2 [Х-- -Я— ОН2] Я-Х + Н20
❖ Дія галогенідів фосфору і сульфуру. Галогеніди фосфору вважають
одними з найефективніших галогенувальних агентів. Проте в цьому випадку
можливі побічні реакції утворення естерів фосфористої кислоти.
• Використання тіонілхлориду зручне тим, що всі побічні продукти - гази,
які легко вилучити нагріванням:
Я-ОН + С1-80-С1 -> Я-0-80С1 + НС1|
Я-0-80С1 -> Я-С1 + БОгТ-
У реакцію вступають первинні, вторинні, а також ацетиленові спирти. Третинні
спирти в цьому випадку зазнають дегідратації. Метод не зумовлює ізомеризації,
його успішно використовують для спиртів, лабільних до дії галогенокислот.
■ Алкілхлориди одержують також взаємодією спиртів з фосфор (III)
хлоридом або фосфор (V) хлоридом:
ЗЯ-ОН + РСІз -> ЗЯ-С1 + Р(ОН)3
Я-ОН + РС15 -> Я-С1 + РОСЬ + НС1
(1)
(2)
(3)
312 Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
• Бромоолтни можна одержати дією на спирти фосфор (III) броміду чи суміші
фосфору з бромом:
2Р + ЗВг2-> 2РВг3
оон ^ 0-вг
Циклогексанол Бромоциклогексан, 70%
• Іодоолкани синтезують аналогічно обробкою спиртів іодидом фосфору (III),
що утворюється під час внесення йоду до суспензії червоного фосфору в спирті.
■ Метиліодид кількісно одержують взаємодією диметилсульфату з водним
розчином калій іодиду:
(СНзО^Ог + 2КІ -> 2СН3І + К2804
• Флуоропохідні алканів можна одержати обробкою спиртів сульфур
тетрафлуоридом:
Я-ОН + 8Р4 -> Я-Р + БСЬТ + ИБТ
12.3.3. Синтез галогенопохідних з олефінів
Детальніше перераховані нижче реакції розглянуто в розділі 8.
❖ Синтез моногшіогенопохідних реакцією приєднання гідроген галогенідів
до алкенів. Взаємодія алкенів з гідроген галогенідами залежить як від будови
ненасиченої сполуки, так і від кислотності Н-Наї. Зміна реакційної здатності
гідроген галогенідів порівняно з алкенами зменшується в ряду: Ш > НВг > НСІ >
НК Розрізняють два головні механізми приєднання гідроген галогенідів до алкенів.
• За електрофільного приєднання гідроген галогенідів спочатку утворюється
я-комплекс, який швидко перетворюється в карбокатіон (о-комплекс), а той
приєднує нуклеофільну частинку НаҐ, перетворюючись у галогеноалкан:
НН НН НН^НН
с=с + н-сі — с=рс — н-с-с —- н-с-с-сі
Н- Н НІН н н н н
н+ сі-
Етилен ті-Комплекс а-Комплекс Хлороетан
Приєднання гідроген галогенідів до несиметричних алкенів відбувається за
правилом Марковникова:
СН3СН2СН=СН2 — СН3СН2СНСН3 СН3-ОСН-СН3 —^~ сн3- с-сн2сн3
І І
Буг-1-єн 2-Іодобуган 2-Метилбут-1-ен 2-Іодо-2-метилбуган
Розділ 12. Галогенопохідні вуглеводнів
313
• Вільнорадикальне приєднання. За наявності пероксидів гідроген бромід
приєднується до алкенів за механізмом Ая. Приєднання відбувається проти
правила Марковникова:
СН3 СН—СН2
Пропен
Нема
пероксидів^ СН3-СНВГ-СН3 За правилом Марковникова
2-Бромопропан
^ СНї~ СН^СН0Вг Проти правила Марковникова
Занавності ^ її
пероксидів 1-Бромопропан
У реакцію радикального приєднання не вступають гідроген хлориди і
флуориди через високу енергію гомолітичного розщеплення зв'язку гідроген-
галоген, а енергія атомарного Іоду недостатня для розриву я-зв'язку алкену.
❖ Алільне галогенування. Високотемпературне галогенування алкенів або дія
на них АГ-бромосукциніміду дає заміщення в ос-положення щодо подвійного зв'язку
(алільне заміщення). Реакція заміщення в алільне положення відбувається за
радикальним механізмом через утворення стабільного алільного радикала:
О + сі-* 3£ Сгв,+
ттр' ^ -на цр ^ * *
2-Мгтилпропен 2-Метил-З-хлоропропен Циклопентен ^Бромосукцинімід З-Бромоциклопентен, 63%
❖ Синтез віцинальних дигалогенопохідних реакцією приєднання галогенів.
Наведемо декілька прикладів одержання віцинальних дигалогенопохідних:
Н3С СНз в Н3С СНз ^ ^гі
С=С —і. Н3С-9-9-СН3 Г ] ґ Т
Н3С сн3 ¿r¿r Ч>-С1
2,3-Диметилбут-2-ен 2,3-Дибромо-2,3-диметилбуган Циклогексен 1,2-Дихлороциклогексен
❖ Синтез 1У3-дигалогенопохідних реакцією приєднання галогенів.
Простим способом одержання триметиленброміду ВгСН2СН2СН2Вг є приєднан-
ня гідроген броміду до алілброміду за низької температури:
СН2=СНСН2-Вг + НВг Вг-СН2СН2СН2-Вг
12.3.4. Синтез галогенопохідних з ацетиленових
вуглеводнів
У розділі 9 детально розглянуто приєднання гідроген галогенідів і галогенів
до потрійного зв'язку, що дає, залежно від реагентів і умов перебігу реакції,
різні галогенопохідні, наприклад:
314
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
■ гемінальні дигалогеноалкани
Н3С-сес-СН3 2НС1> сн3сн2-с-сн3
Бут-2-ин "„ С1
2,2-Дихлоробутан
■ галогеноалкени
HBr, hv НВг с4н0—с=сн.
с4н-сес-Вг—— с4н-сесн 4 9 ' 2
4 9 -60 °С 10 °С Вг
1-Бромогекс-1-ен Гекс-1-ин 2-Бромогекс-1-ен
■ віцинальні дигалогеноалкени
Cl,,hv С2Н5ч ,С1
с2н5-сесн 2 » с=с
сГ h
Бут-1-ин 1,2-Дихлоробут-1-ен, 90%
■ тетрагалогеноалкани
2Br2 ВгВг
h3c-cec-œ3 — h3c-ç-ç-ch3
Бут-2-ин Вг Вг
2,2,3,3-Тетрабромобутан
1233. Синтез дигалогенопохідних з альдегідів і кетонів
Взаємодією альдегідів і кетонів з галогенідами фосфору і сульфуру (РСІ5,
РВгз, SOCk, SF4) одержують відповідні гемінальні дигалогеніди:
НзЧ SF4 Н3С О SOci2 Cl
с=о—- сС н,с-с *- н3с-сн
н3с -so^h3c f 3 н -so> Cl
Ацетон 2,2-Дифлуоропропан Ацетальдегід 1,1-Дихлороетан
12.3.6. Синтез галогенопохідних з карбонових кислот
Декарбоксилювання аргентумних солей карбонових кислот за наявності
галогенів дає алкілгалогеніди (реакція Бородіна1-Хунсдіккерсг):
CF3COOAg + І2 A CF3I + Agi + С02
Аргентум трифлуороацетат Іодотрифлуорометан
1 Бородін О. (1833-1887) - російський хімік і композитор. Дослідження галузі органічного
синтезу, у тому числі одержання галогенопохідних карбонових кислот і конденсації альдегідів.
2 Хунсдіккер Г. (НипБсііескег Н.) (1904-1981) - німецький хімік. Розвинув реакцію Бородіна в
практичний метод (1939), розробив метод одержання макроциклічних кетонів з со-гало-
геноалкілацетооцтових естерів {реакція Хунсдіккера).
Розділ 12. Галогенопохідні вуглеводнів
315
Простіший варіант цієї реакції припускає дію галогену на розчин кислоти в
тетрахлорометані за наявності меркурій оксиду:
І>С0ОН^5Г~ І>-Вг
Циклопропанкарбонова кислота Бромоциклопропан, 46 %
12.3.7. Реакції обміну
Флуоро- та іодопгіхідні, які важко одержати іншими методами, можна
синтезувати з доступних хлоро- і бромоалканів реакціями обміну.
❖ Монофлуоропохідні звичайно одержують за механізмом нуклеофільного
заміщення, наприклад, дією арґентум флуориду на іодопохідні вуглеводнів:
Я-І + А£Р Я-Б + AgЦ
■ Альтернативно в реакціях обміну використовують також інші флуориди
металів, що належать до кислот Льюїса:
—С-С1 + Мїїп * +С[МРпСіГ ^ —С-¥ + МР(П-1)С1
¿4 I / ^ I
Наприклад: ;' . 2СН3С1 + Н%2¥2 -> 2СН3Р + Н§2С12.
■ Реакційну здатність галогенід-іона можна збільшити, якщо проводити
реакцію за наявності краун-етерів (детальніше див. розділ 14), які міцно зв'язують
протиіон. Отже, суттєво збільшується нуклеофільність галогенід-аніона:
О О^ 25 °С
СН3(СН2)7Вг + + £ ^)
1-Бромооктан О О ^6^6
18-Краун-6
*- СН3(СН2)7Р + СН3(СН2)5СН=СН2 + [ К+ 1 вг
о о
1-Флуорооктан, 92% Окт-1-ен, 8% '^Оч^
❖ Ди- і поліфлуороалкапи. Для обміну на Флуор двох і більше атомів
галогену в одного атома Карбону застосовують хлоропохідні алканів і стибій
трифлуорид:
316
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
CCl4 + SbF3 —- CCl2F2 + SbCl3 SbCl3 + 3HF — SbFj, + ЗНСІ
Дифлуородихлорометан
Дифлуородихлорометан (фреон-12І) далі використовують для одержання
тетрафлуороетилену, який легко полімеризується до політетрафлуорретилену
(тефлон, флуоропласт-4).
❖ Моноіодопохідні найліпше одержувати за реакцією обміну дією натрій
іодиду в ацетоновому розчині на відповідні алкілхлориди і броміди, оскільки
натрій хлорид і бромід в ацетоні не розчиняються (реакція Фінкельштейна2):
' R-Cl + Nal -> R-I + NaCU 'або R-Br + Nal -> R-I + NaBr|.
12.4. Хімічні властивості моногалогенопохідних
12.4.1. Реакції нуклеофільного заміщення
Хімічні властивості галогеноалканів головно визначені полярністю зв'язку
Карбон-галоген, яка зменшується в ряду: R-I > R-Br > R-Cl > R-F (табл. 12.2).
Найлегше заміщується (найрухоміший) атом Іоду, що пов'язано з його більшою
здатністю поляризуватися, проте алкіл іодиди^порівняно дорогі й тому можуть бути
використані лише в лабораторних синтезах. Хлориди і броміди мало відрізняються
за реакційною здатністю, і через те в промислових синтезах надають перевагу
доступнішим і дешевим алкілхлоридам. Флуоропохідні алканів у цьому випадку .не
придатні, оскільки атом Флуору досить інертний через малу здатність
поляризуватися. Властивість зв'язків до поляризації залежить від розмірів атома
галогену: чим далі від ядра перебувають електрони зовнішнього енергетичного
рівня, тим більша їхня рухливість, яку можна відобразити таким рядом: C-F < С-С1
< C-Br < С-І. З іншого боку, на швидкість реакції заміщення атома галогену сильно
впливає будова вуглеводневого радикала.
Заміщення відбувається під дією частинок, що мають надлишкову електронну
густину, — нуклеофілів. Загальні відомості про нуклеофіли розглянуті у розділі 4.
Таблиця 12.2
Деякі характеристики зв'язку С-НаІ у галогеноалканах
Зв'язок
Енергія,
Дипольний момент,
Довжина,
Ковалентний радіус
кДж/моль
D
нм
галогену, нм
C-F
443
1,81
0,141
0,060
С-С1
328
1,83
0,176
0,099
С-Вг
279
1,79
0,191
0,114
С-І
240
1,60
0,210
0,136
1 Фреони (холодоагенти) позначають номерами, у яких остання цифра означає кількість атомів Флуору,
а передостання - кількість атомів Гідрогену +1, наприклад: СС12Р2 фреон-12, СНІ^СІ фреон-22.
2 Фінкельштейн Г. (Ріпке&еіп Н.) (1885-1938) - німецький хімік. Досліджував властивості
галогенопохідних.
Розділ 12. Галогенопохідні вуглеводнів
317
Галогеноалкани вступають у численні реакції нуклеофільного заміщення
(табл. 12.3). Тому в багатьох синтезах вони - ключові сполуки. Реакції, у яких
нуклефілом є розчинник, називають реакціями сольволізу. Такі реакції значно
поширені в органічній хімії і відіграють важливу роль. До них належать реакції
гідролізу (нуклеофіл-вода), амонолізу (нуклеофіл-рідкий амоніак), алкоголізу
(нуклеофіл-спирт) та ін. Крім того, реакції гідролізу - одні з найважливіших
біохімічних процесів.
Таблиця 12.3
Реакції нуклеофільного заміщення в галогеноалканах
Схема реакції
Продукт реакції
Я:Х + :ОН Я:ОН + :Х
Спирти
Я:Х + Н20->Я:ОН + НХ
Спирти
ЯгХ+ЮЯ'-^ЯЮЯ' + іХ
Етери {реакція Вільямсона)
Я:Х + :С=СЯ' -> Я:С=СЯ' + :Х
Алкіни
Я:Х + Ыа+Ы Я:Я' + ЫаХ
Алкани (синтез Вюрца)
Я:Х+:СМ->Я:СЫ + :Х
Нітрили {реакція Кольбе)
Я:Х +:ОСОЯ' -> Я:ОСОЯ' + :Х
Естери
Я:Х + :Ш3 -» Я:1ЧН2 + Н:Х
Аміни (реакція Гофмана)
Я:Х + ЯгШЯ' + Н:Х
Вторинні аміни
Я:Х + :Ш2' -> Я:ЫЯ2'+ Н:Х
Третинні аміни
Я:Х+:ЫЯ3->К4:Н+Х-
Четвертинні солі амонію
Я:Х + :Р(С6Н5)3 -^(СбВДзГЭГ
Солі фосфонію
Я:Х+:8Н-»Я:8Н + :Х
Тіоли (меркаптани)
^Х+^Я'-^Я^Я' + гХ
Тіоетери (сульфіди)
Я:Х+:^->Я:^ + :Х
Азиди
Я:Х+:8СЫ->Я:8СЫ + :Х
Тіоціанати
Я:Х+[СН(С02С2Н5)2Г -* Я:СН(С02С2Н5)2 + :Х
Алкілмалонові естери
Я:Х +[СН3СОСНС02С2Н5]" -> СН3СОСНЯС02С2Н5 + :Х
Ацетооцтові естери
• Реакції заміщення атома галогену на різні нуклеофільні групи широко
використовують в органічних синтезах. Розглянемо найважливіші з них.
■ Реакція обміну одного галогену на інший має препаративне значення як
метод одержання флуоро- та іодопохідних, які важко синтезувати іншими
шляхами (детальніше див. "Синтез галогеноалканів").
■ Гідроліз алкілгалогенідів як препаративний метод синтезу спиртів має
обмежене значення, оскільки спирти, як звичайно, доступніші, ніж алкілгалогеніди.
Гідроліз первинних алкілгалогенідів часто використовують для синтезу спиртів,
мічених 180, які застосовують у вивченні механізмів реакцій:
І1-На1+ Н18ОН=Я18ОН +ННа1
■ Синтез етерів за реакцією Вільямсона відбувається через заміщення
галогену на алкоксидну групу. Реакцію проводять за наявності однойменних
318 Чирва В.Я., Ярмолюк CM., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
спиртів, оскільки вони, як донори електронів, пригнічують елімінування гідроген
галогеніду:
н-С4Н9Вг + С2Н5(Жа с7^н* н-С4Н9ОС2Н5 + NaBr
1-Бромобутан Натрій етилат Бутилетиловий етер, 80%
■ Синтез тіолів і тіоетерів. Найзручнішим реагентом для одержання
тіолів є натрій гідросульфід:
и-С4Н9Вг+ НаБН Щ0ІСЩ^ я^ДОН + ШВг
1-Бромобутан Н28 Бутан-1-тіол
Несиметричні тіоетери одержують так само, як і етери дією натрієвих
похідних тіолів:
я-С4Н9Вг + С2Н58Иа нр/с^оЬ н"С4Н98С2Н5 + ИаВг
1-Бромобутан Натрій етантіолят 1-Етилтіобутан
Симетричні тіоетери зручно одержувати алкілуванням натрій сульфіду:
2СНХН2Вг + Иа.Б (СН3СН2)28 + 2ИаВг
3 2 ^ Н20/СН3ОН
Бромоетан Діетилсульфід, 65%
■ Синтез нітрилів та ізонітрилів. Ціанід аніон ("СгМ:) налеясить до
амбідентних іонів - сполук, що мають два реакційні центри, тому нуклеофільним
центром може бути як атом Карбону, так і атом Нітрогену. Взаємодія
алкілгалогенідів з ціанідами металів першої групи дуже важлива з препаративного
погляду. Ціаніди лужних металів зазнають алкілування алкілгалогенідами,
утворюючи нітрили, тоді коли аргентум ціаніди дають ізонітрили:
«-С..Н„СН,Вг + КСИ - н-СпН9ХНСК + КВг
^ ^ (СН) 80 11 23
1-бромододекан 3 2 тридеканнітрил, 90%
ІІСН2Вг + А§СК -Я-СН^№С + А§Вг
■ Синтез нітроалканів і нітроестерів. Нітрит-аніон (:М02~) також є
амбідентним іоном. Первинні іодиди і броміди алкілів у неполярних
розчинниках реагують з нітритами лише за атомом Нітрогену незалежно від
природи металу (реакція Майєра1): ф
СН3(СН2)7Вг (^ . СН3(СН2)7Ш2
1-Бромооктан 1-Нітрооктан, 78%
Майєр В. (Meyer V.) (1848-1897) - німецький хімік. Працював у Німеччині і Швейцарії. Головні
праці в галузі органічної хімії, зокрема, визначив будову саліцилової кислоти, відкрив тіофен,
оксими.
Розділ 12. Галогенопохідні вуглеводнів
319
У випадку вторинних і третинних алкілгалогенідів, а також алкілування
нітритів у полярних розчинниках, окрім нітропохідних, завжди утворюються ще
й алкілові естери нітритної кислоти:
НзСч AgN02 НзСч нзСч
НС—І HC-N02 + HC-ONO
Н3С Н3С Н3С
2-Іодопропан 2-Нітропропан, 30% 2-Нітрозооксипропан, 27%
NaN02
СН3(СН2)7Вг——^CH3(CH2)7N02 + CH3(CH2)7ONO
1-Бромооктан * 3'2 1-Нітрооктан 1-Нітрозооксиоктан
Реакції нуклеофільного заміщення відбуваються лише в рідкій фазі,
зазвичай, у розчинах. У газовій фазі вони відбуватися не можуть, тому що
дисоціація, наприклад, зв'язку С-С1 потребує витрати понад 920 кДж/моль. У
рідкій фазі витрати енергії на дисоціацію значно нижчі (~ 250 кДж/моль)
завдяки виділенню теплоти сольватації йонів.
Швидкість заміщення атомів галогенів у галогеноалканах досить різна і
значно залежить від будови радикалів, з якими зв'язаний атом галогену, а також
від природи галогену, нуклеофілу і розчинника. Висновок про вплив будови
радикала на швидкість нуклеофільного заміщення можна зробити, порівнюючи
відносні швидкості гідролізу метилброміду і mpem-бутилброміду за наявності
гідроксильних іонів. Для пояснення різної кінетики цих реакцій, а також низки
інших спостережень припустили, що реакція нуклеофільного заміщення може
відбуватися за двома різними механізмами (К. Інгольд, Е. Хьюз1, 30-ті роки
XX ст.). Розглянемо ці механізми.
12.4.2. Механізм бімолекулярного нуклеофільного
заміщення (SN2)
Розглянемо першу реакцію:
СН3Вг + ОН" -> СН3ОН + ВГ
Метилбромід Метанол
Уважають, що в цій реакції гідроксид-аніон атакує позитивно заряджений атом
Карбону в "тил" відхідної групи - бромід-аніона. Електронна пара атома Оксигену
взаємодіє з електроноакцепторним атомом Карбону, і утворюється активований
комплекс.
Згідно з теорією молекулярних орбіталей, перехідний стан виникає як
наслідок перекривання ВЗМО нуклеофіла і НВМО молекули, що реагує. В
перехідному стані орбіталі нуклеофіла і групи, що відходить, перекриваються з
1 Хьюз Е. (Hughes Е.) (1906-1963) - англійський хімік. Учень K. Інгольда. Дослідження в галузі
механізмів органічних реакцій.
320 Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
протилежними частками однієї й тієї ж р-орбіталі атома Карбону, що є у вигляді
5р2-гібридизації. Орбіталь р розташована перпендикулярно до площини, у якій
розміщені С-Н-зв'язки. Негативний заряд розподілений між атомами Брому й
Оксигену. Такий стан можливий лише на максимумі кривої потенціальної
енергії, і в жодному разі його не молена трактувати як проміжну сполуку -
інтермедіат. Якщо відбувається перехід із втратою енергії до хімічної сполуки,
то одна з груп повинна відійти, оскільки атом Карбону не може мати більше
чотирьох замісників і, відповідно, утримувати більше восьми електронів. У
процесі утворення активованого комплексу зв'язок Карбон-Оксиген
посилюється, а зв'язок Карбон-галоген послаблюється. Зазначимо, що розрив
старого зв'язку й утворення нового відбуваються одночасно (енергетична
діаграма реакції показана на рис. 12.1). Загалом, механізм цієї реакції можна
уявити так:
/-^АоЧ 5- І /
НО- ^С—Вг -+ [НО- • С; . Вг]- ^ НО -С., + Вг
Отже, реакція відбувається як одностадійний синхронний процес.
Оскільки в реакції беруть участь два реагенти, то реакція бімолекулярна.
Справді, з'ясовано, що швидкість гідролізу метилброміду залежить від
концентрації обох реагентів відповідно до такого рівняння:
І
Г=А;-[СНзВгИОгГ],
де к - стала хімічної реакції.
Е
[но-
1
••C--.Br]-
/
но- + СН3-Вг/
\^ ин3-ин + вг
Координата реакції
Рис. 12.1. Енергетична діаграма 5^2 реакції
• Стереохімічні аспекти реакції. У випадку перебігу реакції з оптично
активною сполукою відбувається обертання конфігурації асиметричного атома
Карбону:
Розділ 12. Галогенопохідні вуглеводнів
321
НО" Л-Вг
СН3
^6Н13
НО—С -Вг
н сн3 _
/С6Н13
НО-С,, + Вг
v сн3
Н 3
(Л)-2-Бромооктан (5)-Октан-2-ол
Власне, щоб пояснити обертання конфігурації в Бм2 реакціях, уперше висунуто
ідею про атаку з "тилу". У міру того як гідроксильна група наближається до атома
Карбону, три зв'язки починають розходитися, доки не досягнуть в активованому
комплексі плоского розташування; потім, з відходом атома Брому, вони знову
набувають тетраедричного розташування, яке протилежне до вихідного. Тому
процес образно називають "вивертанням парасольки".
12.4.3. Механізм мономолекулярного
нуклеофільного заміщення (Бы1)
Розглянемо реакцію між /ире/я-бутилбромідом і гідроксид-аніоном:
«рНз СН3
сн3-с-сн3 +он-—-сн3-с-сн3 + Вг
Вг 6н
2-Бромо-2-метилпропан 2-Метилпропан-2-ол
За загальними уявленнями реакція відбувається як двостадійний процес.
Перша стадія - зворотна - дисоціація галогеноалкілу на йони:
Повільно +
(СН3)3СВг . (СН3)3С + вг
Дисоціація ковалентного зв'язку є енергетично невигідним процесом. її
полегшує те, що одержані карбокатіон і бромід-аніон стабілізуються завдяки
сольватації молекулами розчинника. Карбокатіон далі швидко реагує з
молекулами реагенту (або розчинника), що можуть давати пару електронів для
заповнення вакантної орбіталі атома Карбону:
(СНз)зС + ОН ^°(СН3)3С-ОН
або
(СН3)3С + :6:Н^ [(СН3)3С:рн] « (СН3С)3СОН + Н+
Н н
Карбокатіон - реальний інтермедіат, тому він має меншу внутрішню
енергію порівняно з перехідним станом, через який утворюється (див.
енергетичну діаграму реакції на рис. 12.2).
322 Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Е
(СН3)3С + Вг
+
8+ 5-
(СН3)3С Вг
(СН3)3С ОН
5+ 8-
(СН3)3СВг + -ОН
(СН3)3СОН + Вг
Координата реакції
Рис. 12.2. Енергетична діаграма Бці реакції
> Швидкість багатостадійної реакції визначена швидкістю найповшьнішої
(лімітувальної) стадії.
Лімітувальна стадія (у цьому випадку - утворення карбокатіону) не залежить
від концентрації лугу. Тому це мономолекулярний процес, а механізм реакції
позначають як 8>Д. На практиці з'ясовано, що в такому випадку швидкість реакції
справді залеясить лише від концентрації галогеноалкани і не залежить вЦ
концентрації лугу:
• Стереохімічні аспекти реакції. Під час реакції з оптично активним
алкілгалогенідом за умов реалізації механізму Бці відбувається часткова
рацемізація - утворюються два енантіомерні спирти:
Реакція відбувається» через утворення карбокатіону, що має плоску^
структуру. Відповідно, нуклеофіл може його атакувати з двох боків. Часткову, а
не повну рацемізацію пояснюють екрануванням фронтального боку йоном
галогену, що відходить.
У=к-[КВг]
(5і)-Октан-2-0 л
(Д)-Октан-2-ол
Розділ 12. Галогенопохідні вуглеводнів
323
12.4.4. Чинники, що впливають на перебіг реакцій
за механізмами SN1 і SN2
Низка чинників сприяє перебігу реакції або за механізмом SnI, або за
механізмом Sn2. Зокрема, до таких чинників належать будова галогенопохідної,
природа відхідної групи, нуклеофільність реагенту і розчинник.
• Вплив структури галогенопохідної
■ На ліміту вальній стадії реакції, що відбувається за механізмом .SnI,
утворюється карбокатіон. За цим механізмом відбуваються реакції зі сполуками,
які дають стабільні карбокатіони. Здатність галогенопохідних реагувати за
механізмом SN1 зменшується в ряду CH2=CHCH2-Hal > Alk3C-Hal > Alk2CH-Hal,
оскільки, відповідно, зменшується стійкість карбокатіонів (Alk = алкіл):
СН3 СН3
г<+ + ^ \ І
сн2=снсн2 —^СН2СН=СН2 > СН3—(J+ > сн3-^сн
сн3
У випадку алільного карбокатіону висока стійкість пов'язана з
делокалізацією позитивного заряду внаслідок його взаємодії з я-електронною
системою; інші алкільні карбокатіони стабілізовані електронодонорною дією
алкільних груп (+І-ефект).
■ За механізмом на лімітувальній стадії нуклеофіл атакує позитивно
заряджений атом Карбону. Значення цього заряду зумовлює легкість перебігу
реакції. Здатність галогенопохідних реагувати за механізмом Sn2 зростає в ряду
Alk2CH-Hal > AlkCH2-Hal > CH3-Hal. Тут максимальний позитивний заряд має
атом Карбону в метальній групі, який не компенсується внаслідок +І-ефекту
алкільних груп:
СН,ч8и+ 5_ 54 6_ 5+ 5_
J>CH-HBr СН3 СН^ -*Вг СН3-НВг (8" < 5' < 8)
СН3^
Наявність поряд зі зв'язком С-Hal електроноакцепторних груп збільшує
позитивний заряд на атомі Карбону і, відповідно, збільшує швидкість реакції за
механізмом Sn2. Наприклад, бромооцтова кислота Вг«—СН2—»COOH за будовою
- сполука з вторинним атомом Карбону, тому вона реагує з нуклеофілами
швидше, ніж метилбромід з первинним атомом Карбону.
Одночасно з електронним чинником діє і стеричний (просторовий). У
третинних алкілгалогенідах нуклеофіл не може підійти на відстань,
необхідну для взаємодії з позитивно зарядженим атомом Карбону, через
екранувальну дію трьох алкільних груп, які повністю закривають задню
324
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
напівсферу молекули. Тому третинні алкілгалогеніди реагують лише за
механізмом Бні:
Н,
н
н
н
-Вг
но-=
\ /лЛ
•с—с\
Стеричні перешкоди, які виникають, коли /ирет-бутилбромід і неопентилбромід
заміщуються за механізмом 8М2
Навіть у випадку неопентилгалогенідів екранування метальними групами
електрофільного атома Карбону не дає змоги реалізуватися механізму 8н2.
Вплив стеричних чинників на швидкість реакції заміщення алкілбромідів в
ацетоні за температури 25 °С вивчений за допомогою ізотопів атомів Брому
(табл. 12.4).
Я-Вг + Вг*" -> Я-Вг* + ВГ
Таблиця 12.4
Відносна швидкість реакції заміщення для різних алкілбромідів
СН3-
СНзСНз-
СНз СН2СН2—
(СН3)2СН-
(СНз)зС-
Відносна
швидкість
100
1,31
0,81
0,015
0,004 ^
• Вплив природи групи, що відходить. У випадку перебігу процесу за
механізмом Бні природа галогену впливає на його швидкість. Енергія зв'язку
С-Наї зменшується в ряду Б > СІ > Вг > І. Оскільки лімітувальна стадія - це
розрив зв'язку Карбон-галоген, то найменша реакційна здатність буде у флуоро-
похідних.
Загалом група, що відходить, повинна мати малу основність, тому аніон іу
який відповідає найсильнішій галогенідній кислоті НІ, є в цьому ряду
найслабшою основою і, відповідно, відходить найліпше. Наприклад, відносні
швидкості реакцій заміщення за механізмом 8н2 для різних алкілгалогенідів
співвідносяться так:
Я-Б : Я-С1: ІІ-Вг : Я-І = 1 : 70 : 7000 : 20000.
Тому неможливо провести зворотну реакцію - заміщення гідроксильної
групи в спиртах на галоген за нейтральних умов (див. "Синтез галогено-
похідних"). Гідроксид-аніон є сильною основою і, відповідно, поганою групою,
що відходить. Проте за наявності кислот заміщення в спиртах відбувається
легко, тому що в цьому випадку гідроксильна група зазнає протонування, а.
відхідна група - молекула води - слабка основа:
Розділ 12. Галогенопохідні вуглеводнів
325
R-OH + Ві R-Br + HO-
R-OH + ЬҐ R-бн R-бн + Br- * R-Br + Н20
Н Н 2
Найліпшими групами, що відходять, є алкіл- і арилсульфонат-аніони, що
мають дуже низьку основність. В органічній хімії широко використовують
естери метансульфокислоти (мезгтати) і и-толуенсульфокислоти (тозилати).
Для скорочення замість структур сульфонатних груп часто застосовують
відповідні буквені позначення Ms і Ts (Tos):
НзС-S-OH з MsOH H3C^Q)— S-OH = TsOH
Ö — О
За легкістю заміщення групи, що відходять, можна розташувати так:
MsO" > TsO" > Г > ВГ > Н20+ > СГ > F" .
• Вплив розчинника
■ Механізм SmI переважно виникає в полярних протонних розчинниках
(вода, мурашина кислота, етанол тощо). Біполярні молекули цих розчинників
сприяють дисоціації, сольватуючи йони, що утворилися:
■ Механізм .5^2 зазвичай відбувається в середньополярних апротонних
розчинниках (тетрагідрофуран, діоксан, ацетон). Уважають, що молекули таких
розчинників завдяки електронним парам атома Оксигену сольватують іон
металу і збільшують нуклеофільність реагенту.
Полярні протонні розчинники, утворюючи водневі зв'язки, значніше
стабілізують аніон нуклеофіла, ніж активований комплекс, у якому негативний
заряд делокалізований.
■ Різке збільшення швидкості реакції заміщення простежується в разі
використання апротонних біполярних розчинників, які, з одного боку, мають
високу діелектричну сталу і великий дипольний момент, а з іншого, - не
утворюють водневих зв'язків. Типовими представниками цієї групи розчинників
є ацетонітрил, диметилсульфоксид (ДМСО), диметилформамід (ДМФА),
гексаметилфосфотриамід (ГМФТА):
326 Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
н3с +
Н3С
0
и
ЬҐС^(СНз)2
(СНз^ -Р-0
снз-ои
(СН3)гН
Ацетонітрил ДМСО (ц 3,960, є 48) ДМФА (ц 3,820, є 38) ГМФТА (ц 4,300, є ЗО)
(ц 3,520, є 36)
Вплив полярності розчинника на швидкість реакції заміщення можна
оцінити за даними, що наведені в табл. 12.5 і 12.6.
Таблиця 12.5
Відносна швидкість реакції заміщення в розчинниках різної полярності
СН3І + СГ -> СН3С1 + Г
Розчинник
СН3ОН
НС(ЖН2
НСОШСНз
СНзССЩСНз)2
1,7
3,7
3,83
3,81
Відносна швидкість
1
12,5
1,210ь
7,4106
Таблиця 12.6
Вплив полярності розчинника на відносну швидкість реакції заміщення
(СНз)зС-СІ +С2Н5ОН -* СНз)3С-ОС2Н5 + НС1
Розчинник
Н20
20 % С2Н5ОН
40 % С2Н5ОН
С2Н5ОН
є
80,4
24
Відносна швидкість
100
14
0,1
0,01
• Вплив нуклеофільності реагенту. Чим вища нуклеофільність реагенту, тим
імовірніший механізм 8м2 через зменшення енергії активації, тоді як за механізму
8ц1 нуклеофільність не має значення. Висока концентрація нуклеофільного
реагенту також сприяє 8н2-реакції, оскільки він бере участь у лімітувальній стадії.
Нуклеофіл атакує атом Карбону, що має позитивний заряд. Тому чим більший
заряд на частинці, яка атакує, тим сильніші нуклеофільні властивості. Аніони -
сильніші нуклеофіли порівняно з нейтральними молекулами: Б2" > НБ" > Н28.
Наявність у реакційного центру частинки, що атакує, електронодонорнп^
замісників збільшує нуклеофільність, а електроноакцепторні замісники, відповідно,
зменшують її. В ряду, наведеному нижче, нуклеофільні властивості зменшуються:
К> > Н3С-С^
Для атомів, що є в одному періоді Періодичної системи елементів,
нуклеофільність збільшується зі зменшенням порядкового номера, оскільки зі
збільшенням порядкового номера зростає заряд ядра, і електронні пари
притягуються сильніше:
:І\ГН3 > Н20: > НБ: і, відповідно, СН3~" > Ш2" > НСГ > Г
Для атомів, що є в одній групі Періодичної системи елементів, нуклеофіль-
ність збільшується зі збільшенням порядкового номера: Г > Вг" > СГ > Г".
Розділ 12. Галогенопохідні вуглеводнів
327
Нуклеофільність головно визначена легкістю поляризації електронних пар, тому
зі збільшенням радіуса йона електрони зовнішнього шару менше утримувані
зарядом ядра і легше зазнають поляризування.
Проте наведена вище зміна нуклеофільності для галогенід-аніонів властива
протонним розчинникам. Можливість сольватування аніонів сильно впливає на
їхні нуклеофільні властивості:
Н. Н
Х0 І
н^оч і н'° н
*ҐЖ> и-о
р—Н^^н
/ Н \
н І о-н
•
В аніонах більшого розміру, наприклад Іоду, негативний заряд
стабілізований його делокалізацією по більшій поверхні йона. В аніонах
меншого розміру, наприклад Хлору, стабілізація досягнута можливістю
утворення водневих зв'язків з молекулами розчинника. Отже, у реакціях
нуклеофільного заміщення аніон перед атакою по електрофільному центру
повинен "втратити" сольватну оболонку, на що потрібні витрати енергії.
У полярних апротонних розчинниках аніони не сольватовані, тому в розчині
ДМФА нуклеофільність галогенід-іонів зменшується в ряду СГ > Вг~ > Г.
Загалом за нуклефільністю реагенти можна розділити на декілька груп:
■ дуже сильні нуклеофіли - Г, НБ", "ЗСИ (відносна активність > 105);
■ сильні нуклеофіли - Вг", НО~, ІЮ~", Х>Т, N3" (відносна активність -104);
■ помірні нуклеофіли - СГ, ЯСОО", >Ш3 (відносна активність ~103);
■ слабкі нуклеофіли - Н2О, ІЮН (відносна активність 1);
■ дуже слабкі нуклеофіли - ЯСООН (відносна активність ~1(Г2).
Зазначимо, що сильні нуклеофіли одночасно є сильними основами, і багато
реакцій нуклеофільного заміщення супроводжуються процесами відщеплення. В
деяких випадках продукт реакції елімінування є основним. Детальніше про
реакції відщеплення див. у параграфі "Одержання алкенів" розділу 8.
12.5. Хімічні властивості дигалогенопохідних
Два атоми галогену вуглеводневого ланцюга можуть бути розташовані по-
різному. Найцікавішими варіантами є їхнє розташування біля одного атома
Карбону {гемінальні дигалогеніди), біля двох сусідніх атомів (вщинальні
дигалогеніди) і біля кінцевих атомів Карбону. Спеціальні методи одержання
різних дигалогенопохідних розглянуті в параграфі "Синтез галогенопохідних".
328
Чирва В.Я., Ярмолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Крім того, можна використати методи синтезу моногалогенопохідних, якщо як
синтони взяти біфункціональні сполуки. Наприклад:
ЗНОСНзС^СНзСНзОН +2РВг3 — ВіСНзСг^СН^Вг +2Н3Р03
Бутан- 1,4-діол 1,4-Дибромобутан
За реакційною здатністю дигалогенопохідні насиченого ряду цілком подібні
на алкілгалогеніди. Обидва галогени в цих сполуках можна заміщувати на різні
нуклеофільні групи ("ОН, "БН, ~ТЧН2, ХЫ) і одержувати таким способом різні
дизаміщені похідні вуглеводнів, наприклад:
ВгСН2СН2Вг + 2І\ГН3 -> Н2Ы-СН2СН2->Щ2 + 2НВг
1,2-Дибромоетан Етан-1,2-діамін
СН3СНС1-СН2С1 + 2А80СОСН3 -* СН3СН(ОСОСНз)СН2(ОСОСН3) + 2АвС1
1,2-Дихлоропропан Ді-О-ацетил-пропан-1,2-діол
❖ Гідроліз. Якщо обидва атоми галогену є біля одного атома Карбону, то
гідроліз дає альдегіди і кетони:
сн3сн2снсі2^ сн3сн2снС^^ сн3сн2-<°
1,1-Дихлоропропан ^1 Пропаналь Н
НХ-ССІг-СН
н2о
3^ ~~ж2 ~ХАз -неї
ш
НЗС-9-СНЗ
он
ттгч нх-с-сн,
-н,о 3 и 3
О
А2^
2,2-Дихлоропропан Ацетон
❖ Елімінування. Дія лугів залежно від температури реакції й концентрації
лугу спричиняє відщеплення однієї або двох молекул гідроген галогеніду.
■ 3 віцинальних дигалогенопохідних спочатку одержують вінільні похідні,
а потім ацетиленові вуглеводні:
СН2Вг-СН2Вг НХ=СНВг НС=СН /
2 2 -КВг 2 .КВг /
1,2-Дибромоетан -Н20 Вінілбромід -Н20 Ацетилен
■ Якщо використовувати цинковий пил, то з віцинальних дигалогенідів
утворюються алкени:
СН2Вг-СН2Вг СН2=СН2
1,2-Дибромоетан " п г2 Етилен
■ У дигалогенопохідних, що мають атоми галогену в положенні 1,3- і,
особливо, 1,4- чи 1,5-, виявляється прагнення до утворення циклічних сполук
(подібні синтези розглянуто в розділі 11):
сн—сн2 2п сн2сн2вг ннсн ^
сн—СН2 "2пВг2 СН2СН2Вг ^ 3 ^
Циклобутан 1,4-Дибромобутан М-Метилпіролідин
Розділ 12. Галогенопохідні вуглеводнів
329
12.6. Синтез і хімічні властивості галоформів
Серед галогенопохідних, в яких три атоми галогену знаходяться у одного
атома Карбону, важливими є найпростіші члени цього ряду - тригалогенопохідні
метану СНХз, які також називають галоформами. Серед них найбільше значення
має хлороформ СНСІз.
Хлороформ - важка рідина, що мало розчинна у воді і добре розчинюється в
спирті й етері. Він широко застосовується в якості розчинника і вихідної
речовини в різних органічних синтезах. Раніше хлороформ широко засто-
совувався як дихальний загальнонаркотичний засіб. Проте через небажану
побічну дію на серце і печінку він повністю замінений діетиловим етером.
12.6.1. Синтез хлороформу
❖ Галоформна реакція. Під час взаємодії хлору в лужному розчині з
етиловим або ізопропіловим спиртами, з ацетальдегідом або ацетоном утворюється
хлороформ. Спочатку під дією окиснювача спирти перетворюються, відповідно, до
оцтового альдегіду й ацетону. Далі рухливі а-гідрогенні атоми метальних груп
сполук заміщаються на атоми Хлору. В трихлорацетальдегіді {хлоралі) і
трихлорацетоні два сусідні атоми мають значні позитивні заряди. Тому зв'язок між
ними неміцний і легко розщеплюється дією лугу, унаслідок чого утворюються
хлороформ і* солі мурашиної та оцтової кислот, відповідно. Останню реакцію
називають галоформним розщепленням. Цим шляхом хлороформ уперше
одержаний 1831 р. Ю. Лібіхом:
[О] ,0 ЗС12 &>5ьО кон ,?
СН3СН2ОН ► Н3С-С. т *> сі3с-С' ► С13СН + Н-С.
* Н -ЗНС1 Н 3 ОК
хлораль
[О] ЗС12 ^ кон О
н3сснсн3 *н3с-с-сн3 * сі3с-с-сн3 ^сі3сн +н3с-с.
он о ЗНС1 о
Для синтезу цієї сполуки також застосовують хлорне вапно Са(ОС1)2 в Са(ОН)г:
2Н3ССОСН3 + ЗСа(ОС1)2 -> 2С13ССОСН3 + ЗСа(ОН)2
2С13ССОСН3 + Са(ОН)2 -> (Н3ССОО)2Са + 2СНС13
У галоформну реакцію вступають усі речовини, що мають угруповання
СН3С(0)С- або СН3СН(ОНЬ
❖ Хлорування метану. Сьогодні хлороформ одержують у значних
кількостях прямим хлоруванням метану:
СН4 + ЗС12 -> СНС13 + ЗНС1
330 Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
❖ Синтез із тетрахлорометану. Після того, як тетрахлорометан став
легкодоступним продуктом, розроблено спосіб його часткового відновлення
залізом у сульфатній кислоті до хлороформу:
СС14 + Н2 -> СНСІз + HCl
12.6.2. Хімічні властивості хлороформу
❖ Окиснення хлороформу. Електроноакцепторна дія трьох атомів Хлору
приводить до відтягування електронної густини від атома Гідрогену, зв'язок С-Н
поляризується. На вологому повітрі відбувається повільне окиснення
хлороформу з утворенням отруйного газу фосгену, який значно впливає на
органи дихання:
СІзСН + О -> [СІзСОН] -* СОСІ2 + HCl
Побічні токсичні явища під час хлороформного наркозу зумовлені саме
наявністю фосгену і подібних до нього продуктів розпаду. Для вивільнення від
фосгену до медичного хлороформу додають 1 % спирту; спирт зв'язує фосген,
/творюючи естер карбонатної кислоти:
СОСЬ + 2С2Н5ОН -> ОС(ОС2Н5)2 + 2НС1
❖ Синтез ортоформіатів. Дія на безводний хлороформ алкоголятів
чатрію дає естери ортомурашиної кислоти, яка не існує у вільному стані:
НССІз + 3NaOC2H5 -* НС(ОС2Н5)3 + 3NaCl
Під час реакції з водним лугом замість нестійкої ортомурашиної кислоти
/творюється звичайна її "мета"-форма:
Р
СНС13 +ЗКОН [НС(ОН)3] —-н-с
он
❖ Синтез хлоропікрину. Коли хлороформ взаємодіє з нітратною кислотою,
го утворюється хлоропікрин - нітрохлороформ, що має інсектицидну і
шьозогінну дію:
CHCU-^^CCL—N0,
3 -Н20 3 • 2
1 Хлоропікрин
Раніше хлоропікрин використовували як бойову отруйну речовину.
❖ Одержання дихлорокарбенів. У разі дії на хлороформ лугів як проміжна
полука утворюється нестійкий дихлорокарбен, який використовують у синтезах
сучасної органічної хімії:
СНС13 + кон :СС12 + КС1 + Н2°
Розділ 12. Галогенопохідні вуглеводнів
331
Дихлорокарбен можна розглядати як галогенопохідну найпростішого карбену
- метилену (детальніше див. розділ 7). Прикладом реакції, що відбувається за
участю дихлорокарбену, може бути реакція одержання діазометану:
Н
■ | 9НР1
ХСІ^И^-Ш, н^-і^-ссі,1^ СН2К2
н
12.6.3. Інші галоформні сполуки
• Флуороформ можна одержати, наприклад, із бромоформу нагріванням зі
стибій флуоридом. Він менш активний як хімічно, так і фізіологічно.
• Бромоформ одержують аналогічно до хлороформу. Його застосовують як
терапевтичний засіб у разі коклюшу, причому в цьому разі для стабілізації його
змішують з 4 % спирту.
• Іодоформ. Іодоформ утворює жовті кристали, що мають сильний специфічним
запах. У лабораторії одержують тими ж методами, що і хлороформ. У
промисловості іодоформ синтезують електрохімічно, піддаючи електролізу
розбавлений розчин калій іодиду в спирті або ацетоні. На катоді виділяються калій і
вода, утворюючи їдке калі, а на аноді - іод. Обидві речовини реагують зі спиртом
або ацетоном, що міститься в електролітичній рідині, виділяючи іодоформ.
Іодоформну реакцію використовують як аналітичний метод для відкриття в
молекулі груп СН3С(0)- і СН3СН(ОН)-.
У хімічних реакціях він поводиться подібно до хлороформу. .
Іодоформ застосовують як антисептик, оскільки він перешкоджає заг)юєнню
ран. Сам іодоформ не має бактерицидних властивостей, проте коли він
контактує з органічними речовинами, то постійно виділяється іод, який і має
антисептичну дію.
12.7. Галогенопохідні ненасичених вуглеводнів
Дня галогенопохідних ненасичених вуглеводнів найцікавішим з погляду
хімії є наявність атома галогену або біля карбонового атома за місцем кратного
зв'язку, або в сс-положенні до кратного зв'язку. Наприклад, заміщенням у
пропені атомів Гідрогену атомами Хлору можна одержати такі сполуки:
С1СН=СН-СН3 Н2С=СС1-СН3 СН2=СН-СН2С1
1-Хлоропропен 2-Хлоропропен З-Хлоропропен (алілхлорид)
12.7.1. Методи одержання вінілхлориду
Найпростіша галогенопохідна етилену - вінілгалогенід - має специфічні
хімічні властивості. З практичного погляду дуже важливий вінілхлорид
332 Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
СН2=СНС1, який застосовують у величезних кількостях для полімеризації в
полівінілхлорид (ПВХ).
Синтез на основі ацетилену. Найдавніший метод одержання полягає в
приєднанні гідроген хлориду до ацетилену. Спосіб застосовували в країнах, що
не мають своєї нафти, для яких етилен, що є продуктом нафтопереробки,
недоступний:
СН^СН + НС1 f^hn СН2=СНС1
150-220 °С 2
Синтези на основі етилену. У разі отримання вінілхлориду з етилену
етилен перетворюють у дихлороетан, який дегідрохлорують за 400-550 °С:
С1СН2-СН2С1 СН2=СНСІ
2 2 _нс1 2
■ За сучасною технологією вінілхлорид одержують високотемпературним
хлоруванням етилену за наявності кисню. Реакція відбувається за радикальним
механізмом:
250-350 °С
н2с=сн2+ С12 н^снсі + неї
Комбінований метод. Можна поєднувати обидва методи, використовуючи
для гідрохлорування ацетилену гідроген хлорид, що утворюється в процесі
крекінгу дихлороетану.
У сучасних технологіях гідроген хлорид, що виділяється, окиснюють до
хлору:
4НС, + °2^Г 2С12 + 2Н20
Це дає змогу знову використовувати регенерований хлор для синтезу
дихлороетану з етилену.
Синтези на основі дихлороетану. В лабораторії вінілхлорид синтезують
дегідрохлоруванням дихлороетану спиртовим розчином їдкого калі за 60-70 °С:
С1СН2СН2С1 + КОН -* Н2С=СНС1 + KCl + Н20
12.7.2. Хімічні властивості вінілхлориду
Сполуки, що містять галоген, пов'язаний з ^-гібридизованим атомом
Карбону, різко відрізняються за хімічною поведінкою від сполук з галогеном
біля насиченого атома Карбону.
Атом Хлору у вінілхлориді СН2=СНС1, вініленхлориді СН2=СС12 і
дихлороацетилені CHCl= =СНС1, а також атоми Брому та Іоду у відповідних
положеннях зрідка вступають у звичайні для алкілгалогенідів реакції
нуклеофільного обміну. Такі сполуки не реагують з водою, водними розчинами
Розділ 12. Галогенопохідні вуглеводнів
333
лугів, амоніаком. Проте за наявності сильних нуклеофілів деякі реакції можна
провести. Наприклад, вінілхлорид взаємодіє з натрій етилатом, утворюючи
вініл етиловий етер:
Н^СН-СІ + ИаОС2Н5 Н2С=СН-ОС2Н5+ ЫаС1
Уважають, що дія сильної основи зумовлює реакцію елімінування гідроген
хлориду, а потім ацетилен приєднує етоксильну групу:
С9Н,0-Иа+ 5- 5+
н2с=сн-сі ,уаС1 > НС=СН НС=СН + С2Н50 Н н2с=сн-ос2н5
- С^ОН
Такий механізм одержання вінілових етерів цілком правдоподібний,
оскільки дія сильної основи - аміду натрію - на вінілхлорид зумовлює
дегідрогалогенування:
Н2С=СН-С1 нс=сн
Вінілхлорид Ацетилен
■ Іноді атом галогену у вінілгалогенідах називають "мертвим", маючи на
увазі його низьку реакційну здатність. Проте сучасна хімія металоорганічних
сполук робить деяку поправку. Наприклад, вінілгалогеніди реагують з магнієм у
тетрагідрофурані (ТГФ). В хімічній літературі цей синтез відомий як реакція
Нормана:
Н2С=СН-С1 ^Н2С=СНМёС1
Така інертність зв'язку С-С1 пов'язана зі слабким мезомерним ефектом, що
діє в напрямі, протилежному до звичайної поляризації зв'язку С5+->С15"~, і, як
наслідок, відбувається зменшення негативного заряду на атомі Хлору і
позитивного на атомі Карбону:
Н
СН2ТС^" = СНГС=С1:- -НХ=СН-Сі: и СНГС^
<3> " " V.1-
Символіка кривих стрілок, що позначають зсув електронних пар, дає змогу
порівняти розподіл електронної густини у вінілхлориді та насичених
хлоропохідних типу хлороетану, де немає взаємодії електронних пар атома
Хлору з електронами я-зв'язку. Часткове (слабке) надходження вільної
електронної пари атома Хлору на атом Карбону (+М-ефект) не може повністю
нейтралізувати позитивного заряду цього атома, створюваного потужним
відтягуванням електронної густини атомом Хлору (пряма стрілка С-+С1). Проте
наслідком спряження є часткова двозв'язність атома Хлору з атомом Карбону і
334
Чирва В.Я., Ярмолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
зменшення довжини зв'язку С-С1 до 0,169 нм порівняно з алкілгалогенідами
(0,179 нм в етилхлориді), а також частковий зсув електронної густини я-зв'язку
до крайнього карбонового атома. Все це зменшує дипольний момент молекули
загалом (для вінілхлориду він дорівнює 1,44В, а для етилхлориду —2Э).
• Електрофільне приєднання гідроген хлориду до вінілхлориду відбувається
за таким рівнянням, проте вже з певними труднощами:
СН2=СН-С1 + НС1 [СН3СНС1 ] + сі-—- СН3СНС12
І + .СІ
1-Х— сн2-с-н
н
Атака протона Р-карбонового атома однозначно не доводить наявності
часткового негативного заряду на ньому. Річ у тому, що карбокатіон, який
утворюється в цьому випадку завдяки +М-ефекту атома Хлору, значно
стійкіший, ніж його ізомерний проміжний катіон, який утворився б, якби
приєднання відбувалося у зворотному порядку. Отже, енергія активації першої
реакції нижча, ніж останньої. Окрім того, під час електрофільної атаки лише в
цьому випадку можливий спряжений активований комплекс:
х -н -н2с=сн-х
12.7.3. Інші хлоровані етилени
• Вініліденхлорид СН2=СС12, як і вінілхлорид, застосовують для полімеризації.
Його одержують відщепленням гідроген хлориду від трихлороетану, а той,
відповідно, утворюється приєднанням хлору до вінілхлориду:
СН2=СНС1 + С12 СН2СЬСНС12 СН2=СС12
-Н20
Відщеплення гідроген галогенідів від несиметричних алкілгалогенідів
відбувається відповідно до правила Зайцева, яке для цього випадку можна
сформулювати так: протон відщеплюється від атома Карбону, що зв'язаний з
найбільшою кількістю атомів галогену.
• Симетричний дихлороетилен - ізомер вініліденхлориду - синтезується
через приєднання еквімолярної кількості хлору до ацетилену:
СН=СН + С12 -> СНС1=СНС1
• Трихлороетилен СНС1=СС12 одержують відщепленням гідроген хлориду від
тетрахлороетану, який синтезують приєднанням 2 моль хлору до ацетилену:
Розділ 12. Галогенопохідні вуглеводнів
335
НС=СН + 2С12—-снсі2-снсі2 С12СН-СНС12 ~± СНС1=СС12
-Н20
Використовують для знежирення металів, оскільки він не горить і не
спричинює корозії.
• Тетрахлороетилен СС12=СС12 можна синтезувати відщепленням 1 моль
хлору від гексахлороетану, який є побічним продуктом реакції хлорування
етилену й ацетилену, дією цинку або нагріванням з алюміній хлоридом:
СС1ГСС13 ——- СС12=СС12
3 3 -гпсі2 2 2
Використовують головно у хімчистці одягу. Трихлороетилен і тетрахло-
роетилен не можуть полімеризуватися.
12.7.4. Флуоровані етилени
З флуоропохідних етилену найважливіше значення мають тетрафлуоро-
етилен (СР2=СР2) і трифлуорохлороетилен (СС1Р=СР2), які полімеризуються,
даючи винятково стійкі полімери.
• Тетрафлуороетилен одержують, діючи на хлороформ рідким гідроген
флуоридом або стибій (III) флуоридом. Спочатку синтезований дифлуорохлоро-
метан після відщеплення гідроген хлориду перетворюється в проміжний
дифлуорокарбен, який рекомбінують у тетрафлуороетилен:
— 2 НСІ
2СНС1Р2 [2СР2] -СР2=СР2
гЯ2С=Сїг -> [-СР2СР2-]„
Тетрафлуороетилен є винятково важливим вінільним мономером, на основі
якого одержують політетрафлуороетилени (флуоропласти, або тефлони) -
надзвичайно стійкі до агресивних середовищ полімери.
Досить багато написано про вплив флуорохлороалканів на озоновий шар
Землі. Проблема виникає через інертність цих сполук, наприклад, фреону, доки
вони не досягнуть стратосфери. Там під впливом короткохвильового УФ-світла
може відбуватися їхній фотоліз з утворенням атомарного Хлору, який каталізує
розклад озону. Проте сьогодні неможливо сказати, чи дія цього ефекту значна
порівняно з варіаціями озонового шару, наприклад, зі зміною широти і
наявністю озонових дірок над Антарктикою.
12.7.5. Алілгалогеніди
• Синтез алілгалогенідів. Алілгалогеніди в лабораторії синтезують з
алілового спирту, обробляючи його фосфор (III, V) галогенідами:
336
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
H^CHCRjOH + РВг3—- Н2С=СНСН2Вг + Р(ОН)3
Алілбромід, 60%
Алілхлорид також одержують з алілового спирту, однак трошки інакше:
н2с=снсн2он+нсі —-н^сн-сь^сі
У промисловості його синтезують, обробляючи пропілен хлором
(співвідношення пропілен - хлор 5:1) у разі високої температури за методом
Львова. За цих умов заміщення виникає, головно, в метальній групі. Реакція
відбувається за радикальним механізмом через найстійкіший алільний радикал
(детальніше див. розділ 8):
❖ Властивості алілгалогепідів
• Реакції нуклеофільного заміщення. Алілхлорид, як і його аналоги та
гомологи, легко вступає в реакції нуклеофільного заміщення як за механізмом
Бці, так і за механізмом 8ы2.
■ У випадку бімолекулярного заміщення тг-електронна система через
спряження стабілізує активований комплекс:
■ За мономолекулярного нуклеофільного заміщення алільний катіон, що
утворюється, має підвищену стійкість завдяки делокалізації позитивного заряду
внаслідок взаємодії з я-електронами подвійного зв'язку:
Для таких сполук характерна висока реакційна здатність у реакціях
заміщення навіть за досить низьких температур, що пояснюють як порівняно
неміцним зв'язком Карбон-галоген, так і легкістю утворення алільного катіона.
Наприклад, відносна швидкість заміщення алілхлориду в реакції Фінкельштайна
Алілхлорид, 70%
S00 °С
Н3С-СН=:СН2 + С12^Н2С=СН-СН2С1
¥ н
Розділ 12. Галогенопохідні вуглеводнів
337
в 33 рази вища, ніж етилхлориду. З іншого боку, під дією навіть 5 % водного
розчину лугу за невисоких температур відбувається заміщення алільного атома
Хлору на гідроксильну групу. Легкість гідролізу і леткість алілхлориду дає
змогу використовувати його як сльозогінний газ.
• Ллільні перегрупування. Експерименти заміщення атома галогену в
алілгалогенідах або в його несиметричних гомологах, що мічені 14С,
засвідчують, що в цьому випадку зазвичай простежується перегрупування,
внаслідок якого група, що заміщає галоген, зв'язується не з тим атомом, де був
галоген, а з третім атомом системи:
Н^-СН-Сг^СІ +КСЫ — СЫ~СН2-СН=СН2+КС1
■ У тих випадках, коли відбувається мономолекулярна реакція заміщення,
утворюється алільний карбокатіон, який завдяки спряженню має два центри
реакції, що реагують з нуклеофілами. Тому й одержують суміш продуктів.
Наявність перегрупування потрібно враховувати, плануючи синтез, наприклад,
гомологів алілового спирту:
н3с-сн=сн-сн2сі н?с-сн=сн-сн:— няс-сн-сн=сн7
ОН
н3с-сн=сн-сн2он+н3с-сн-сн=сн2
Бут-2-ен-1-ол Бут-З-ен-2-ол
(кротиловий спирт)
■ Проте алільне перегрупування може відбуватися повністю й у тих
випадках, коли реакція бімолекулярна. В цьому разі аніону легше атакувати
крайній карбоновий атом з його 8+-зарядом, ніж зв'язаний з галогеном атом
Карбону, який екранує негативний заряд галогену. Аніон унаслідок здатності до
поляризації 7г-зв'язку, атакуючи крайній атом Карбону з 8+-зарядом, зміщує його
електрони до атома Карбону, що несе галоген, і виштовхує останній у вигляді
аніона:
[=снч:н2-с
я її
Ы^С:+СН=СНЧ:Н2-С1 :Ы^С-СН-СН=СН2 + С1-
Система працює як єдине ціле зі спряженими зв'язками С=С і С-С1 (о,л-
спряження). Формально таку атаку можна описати як 1,4-приєднання, таке ж, як
і приєднання до я,7і-спряжених систем. Також, як і у випадку 1,4-приєднання до
я,л>спряжених систем, тут розриваються зв'язки 1,2 і 3,4, а я-зв'язок
усталюється в положенні 2,3.
338 Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
• Комплекси с металами. Під час взаємодії алілгалогенідів і алілових спиртів
з солями перехідних металів утворюються я-комплекси:
СН2 СН СН2 ХО
сн2 сн2х сн2 сі
Коли ці комплекси окиснюються, то зв'язок ліганд-метал руйнується і
утворюються олефіни, альдегіди і кетони. Алільні комплекси перехідних металів
- це високоактивні й селективні каталізатори.
Розділ 13
СПИРТИ
Спиртами називають сполуки, які мають одну або декілька гідроксильних
груп біля атомів Карбону, що перебувають у стані 5р3-гібридизації. Гідроксильна
група "ОН є функціональною групою і визначає властивості, характерні для
цього класу сполук. Кількість гідроксильних груп визначає атомність спиртів.
Будова вуглеводневого замісника впливає на швидкість, з якою спирти
вступають у реакції, а іноді й на характер реакції.
13.1. Класифікація, номенклатура і будова спиртів
13.1.1. Класифікація спиртів
За природою вуглеводневого замісника
Насичені спирти Ненасичені спирти Ароматичні спирти
СНзСНСНї
3| 3
он
Пропан-2-ол
Ізопропіловий спирт
Одноатомні спирти
СН2=СНСН2ОН
Пропен-2-ол
Аліловий спирт
он
Бензиловий спирт
За кількістю гідроксильних груп
Двохатомні спирти Три- і багатоатомні спирти
СНо—СН—СНо
II І
он он он
Етиленгліколь Гчіцерол (гліцерин)
НОСН2СН2ОН
СН3СН2ОН
Етанол
Етиловий спирт
За природою атома Карбону з гідроксильною групою
Вторинні спирти
Первинні спирти
СН3СНСН2ОН
3|
сн3
2-Метилпропан-1 -ол
Ізобутиловий спирт
СН3СН2С|НОН
СН3
Бутан-2-ол
втор-Бутиловий спирт
Третинні спирти
СН3
СНз-С-ОН
І
СН3
2-Метилпропан-2-ол
тре т-Бутиловий спирт
340
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
13.1.2. Номенклатура спиртів
• Тривіальна номенклатура. Для деяких простих спиртів іноді використо-
вують тривіальні назви: СН3ОН - деревний спирт, С2Н5ОН - винний спирт,
С5НііОН - аміловий спирт, СібН33ОН - цетиловий спирт.
• Раціональна номенклатура. В цій застарілій номенклатурі як основу
використовують родоначальника гомологічного ряду - спирт СН3ОН, який
називають карбінолом. Складніші спирти розглядають як продукти заміщення
одного або декількох атомів Гідрогену в метальному заміснику цього спирту,
наприклад:
СН2СН3 Н3С н
сн3сн2срон
СН2СНЗ
Триетилкарбінол
СНзСНзСНС^ОН
Н
в/иор-Бутилкарбінол
Сьогодні цю номенклатуру не рекомендують застосовувати.
❖ Номенклатура ШРАС
• Замісна номенклатура. Гідроксильну групу в замісній номенклатурі позна-
чають суфіксом -ол, якщо вона є головною групою, та префіксом гідрокси-,
якщо ця функціональна група розміщена в боковому ланцюзі або в сполуці, де є
старша група.
■ Гліколі та багатоатомні спирти. За наявності двох і більше гідроксильних
груп за основу вибирають найдовший ланцюг, що має максимальну кількість
гідроксилів, а в назві цих сполук перед суфіксом -ол додають числовий префікс ді-,
три- і т. д.:
Вг^Ч^СНз
3
4-Бромо-2-метилциклогексанол
Нумерація від гідроксильної групи (старша
функціональна група) в напрямі найближчого
замісника
2 1
СН3СН-СНСН2ОН
1 |з
СІ СН2ОН
2-( 1 -Хлороетил)пропан-1,3-ДІол
За основу вибирають ланцюг з максимальною
кількістю гідроксильних груп
ноч
У-сн2он
З-Гідроксиметилциклопентанол
За основу беруть п'ятичленний цикл. Гідро-
ксильну групу в боковому ланцюзі називають
за допомогою префікса гідрокси-
4 3 2 1
НОСН2СН2-С-СН3
2 2 II 3
0
4-Гідроксибутан-2-он
Старша функціональна група - кетон, вона і
визначає нумерацію сполуки
Розділ 13. Спирти
341
■ Недопустимою є назва ізопропанол та подібні, оскільки не існує вугле-
водню ізопропан. Варто називати цю сполуку пропан-2-ол або за радикально-
функціональною номенклатурою - ізопропіловий спирт.
• Радикально-функціональна номенклатура. Велика кількість традиційних назв
спиртів побудована за принципами радикально-функціональної номенклатури (у
деяких посібниках такі назви неправильно зачисляють до так званої спиртової
номенклатури). Назви спиртів складаються з назви радикала і назви функції -
спирт. Приклади таких назв наведено курсивом у параграфі "Класифікація".
У цій номенклатурі положення замісника в алкільній групі спирту можна
позначати не лише цифрою, а й буквою грецького алфавіту. Грецька буква со
засвідчує наявність замісника, що пов'язаний з найвіддаленішим атомом
Карбону щодо функціональної групи:
у Р а р а
ВгСН2СН2СН2ОН СН3ОСН2СН2ОН
у-Бромопропіловий спирт р-Метоксиетиловий спирт
■ Двохатомні спирти. Назви цих сполук складають із назв двовалентних
вуглеводневих замісників, додаючи назву функції - гліколь. Наприклад,
НО(СН2)гОН - етиленгліколь, НО(СН2)40Н - тетраметиленгліколь тощо.
• Тривіальна номенклатура. Низку тривіальних назв спиртів рекомендовано
ШРАС як родоначальні назви, наприклад: етиленгліколь, гліцерин, пінакон,
ментол, пентаеритрит. Українська національна комісія з хімічної термінології і
номенклатури рекомендує використовувати назви гліцерол, пінакол, пента-
еритрол.
Н3С СН3
С Н3 ^~"^ї' ^
но он
снзснснз
Пінакол (пінакон) Ментол Пентаеритрол (пентаеритрит)
13.1.3. Будова спиртів
У насичених спиртах зв'язки Н-О і О-С розташовані один щодо іншого під
кутом -106°. Зв'язок Н-О поляризований через більшу, ніж у атома Гідрогену,
електронегативність атома Оксигену. Алкільні замісники, що мають +І-ефект,
подають електронну густину на атом Оксигену, частково компенсуючи його
електроноакцепторні властивості. Тому в спиртах зв'язок Н-О поляризований
менше, ніж у воді, у якій атом Гідрогену не має індукційного ефекту. Отже,
спирти мають слабші кислотні властивості порівняно з водою (відповідно, рКг ~17
і 15,7), проте значно сильніші, ніж раніше вивчений ацетилен (рКй -25):
но-сн,-с-снгон
2 І 2
снгон
342 Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
К-^О-Н Н-О-Н КгОН К-^О" + № к-ОН Н-О" + №
Слабша Сильніша
кислота основа
По-іншому кислотні властивості спиртів і води можна оцінити, порівнюючи
основність спряжених основ. В алкоголятах +І-ефект вуглеводневого замісника
збільшує негативний заряд на атомі Оксигену, тоді як атом Гідрогену в гідроксид-
аніоні ніяк не впливає на нього. Алкоголят-аніони є сильнішими основами
порівняно з гіроксид-аніоном, а отже, спирти, як спряжені кислоти, відповідно,
слабші за кислотними властивостями, ніж вода.
13.1.4. Ізомерія
Очевидно, що для метану й етану, у яких усі атоми Гідрогену рівноцінні,
заміщенням одного атома Гідрогену на гідроксил можна одержати лише
єдиний спирт: це метиловий СН3ОН і етиловий СН3СН2ОН, відповідно.
Пропан має вже дві можливості - заміщення на гідроксил одного з атомів
Гідрогену метильних груп і одного з атомів Гідрогену метиленової групи. І
справді, існує два пропілові спирти: первинний, у якому гідроксил зв'язаний з
первинним атомом Карбону (пропіловий спирт, або пропан-1-ол), і вторинний
- з гідроксилом біля вторинного атома Карбону (ізопропіловий спирт, або
пропан-2-ол):
СН3СН2СН2ОН СНзСН(ОН)СНз
Первинний спирт (пропан-1 -ол) Вторинний спирт (пропан-2-ол)
Отже, ізомерія спиртів, як і взагалі ізомерія заміщених вуглеводнів, є
двоякою - ізомерія вуглеводневого скелета, вже відома нам за алканами, та
ізомерія положення гідроксильної функції в цьому скелеті. Справді, для
четвертого члена гомологічного ряду алканів - бутану - спирти походять від
двох різних вуглеводневих ланцюгів: від н-бутану та ізобутану:
Структурна ізомерія
Ізомерія карбонового Ізомерія положення Міжкласова ізомерія
ланцюга функції
СН3СНСН2ОН СНзСН2С|НОН
СНз СН3СН2СН2СН2ОН СН3СН2ОСН2СН3
2-Метилпропан-1-ол Бутан-2-ол Бутан-1-ол Етоксіетан
ізобутиловий спирт втор-бутиловий спирт н-бутиловий спирт діетиловий етер
Розділ 13. Спирти
343
Для спиртів можливі і всі основні види стереоізомерії:
Стереоізомерія
<т-Діастереомерія
СН3
НО+Н
н-|-он
СН3
ҐЛ>Бутан-2-ол (£>Бутан-2-ол ері/тро-Бутан-2,3-діол
т/?ео-Бутан-2,3-діол
13.2. Фізичні властивості спиртів
У разі порівняння температури кипіння спиртів подібної структури видно,
що, переходячи від одного члена гомологічного ряду до наступного,
температура кипіння підвищується приблизно на 20 °С. Якщо ланцюги
розгалужені, то це, як і у вуглеводнях, підвищує температуру плавлення
(особливо сильно для третинних спиртів, у яких "розгалужений" атом Карбону
безпосередньо примикає до функціональної групи) і знижує температуру
кипіння. Порівняно з вуглеводнями, спирти киплять за значно вищої
температури.
Щоб пояснити аномалії в температурах кипіння, використано поняття
водневого зв 'язку. Можна вважати, що в спиртах атом Гідрогену гідроксильної
групи слугує містком між двома електронегативними атомами Оксигену; до того
ж, з одним із них він зв'язаний ковалентним зв'язком, а з іншим -
електростатичними силами притягання. Енергія водневого зв'язку в спиртах
близько 20 кДж/моль (для більшості ковалентних зв'язків вона дорівнює 210—
420 кДж/моль):
Молекули, утримувані разом водневими зв'язками, називають асоційованими;
їхні аномально високі температури кипіння зумовлені додатковою енергією, що
необхідна для руйнування водневих зв'язків. Детальніше про водневий зв'язок
див. розділ 3.
Суттєва відмінність спиртів від вуглеводнів полягає в тому, що нижчі
спирти змішуються з водою в будь-якому співвідношенні. Через наявність ОН-
групи молекули спирту утримувані разом такими ж міжмолекулярними силами
взаємодії, які існують у воді. Внаслідок цього можливе змішування двох типів
молекул, до того ж, енергія, потрібна для відривання молекули води або спирту
одна від одної, виникає внаслідок утворення аналогічних зв'язків між
молекулами води і спирту. Проте це справджується лише для нижчих спиртів, у
яких ОН-група становить значну частину молекули. Довгий аліфатичний ланцюг
\5-8+ К\8-8+ К\5-5+ К\5-5+
•О-Н-О-Н О-Н О-Н-
344 Чирва В.Я., Ярмолюк CM., Толкачова H.В., Зеллляков О.Є. Органічна хімія
з невеликою ОН-групою значно подібний до алканів, і фізичні властивості таких
сполук відображають це. Зменшення розчинності у воді зі збільшенням кількості
атомів Карбону відбувається повільно: перші три первинні спирти необмежено
змішуються з водою; розчинність w-бутилового спирту становить 8 г на 100 г
води, н-пентилового -2 г, н-гексилового - 1 г, а для вищих спиртів вона ще
менша.
Відповідно до дипольних моментів ((і = 1,6-1,8 D), спирти - полярні
речовини, які мають слабкі електронодонорні або нуклеофільні властивості,
зумовлені наявністю неподіленої пари електронів атома Оксигену.
13.2.1. Спектроскопія спиртів
• УФ-спектроскопія. Спирти практично не поглинають в УФ-діапазоні. Слабка
смуга з А,тах 180-185 нм відповідає я—>о*-переходу електрона неподіленої пари
атома Оксигену.
• ІЧ-спектроскопія. В ІЧ-спектрі спиртів простежуються сильні валентні
коливання v0h близько 3635-3615 і 3600-3200 см"1, відповідно, для сильно
розбавлених, де нема водневого зв'язку, і концентрованих з водневими
зв'язками. Окрім того, деформаційні коливання 8он виявляються близько 410-
1250 см"1, а валентні коливання vC-o - близько 1150-1050 см"1 залежно від
будови спиртів.
• Mac-спектрометрія. Для спиртів, починаючи з бутилового, характерна мала
інтенсивність піка молекулярного йона. Вона зменшується паралельно зі
зростанням молекулярної маси спиртів, а також у разі переходу від первинного
спирту до вторинного. Для третинних спиртів піка молекулярного йона
практично нема. Для первинних і вторинних спиртів основна фрагментація
починається з відщеплення молекули води. У випадку третинних спиртів
спочатку електронний удар відщеплює найдовший вуглеводневий радикал,
утворюючи осколковий іон з гідроксильною групою.
• Спектроскопія ПМР. У ПМР-спектрах сигнал протона гідроксилу
виявляється в діапазоні від 1,0 до 5,5 м.ч. залежно від концентрації і природи
розчинника.
13.3. Одержання одноатомних спиртів
у промисловості
Вимоги до промислових синтезів інші, ніж до лабораторних. Зокрема, вели-
котоннажні виробництва економічніше виконувати безперервним способом з
багаторазовою рециркуляцією великих мас речовин, які реагують. Тому у
виробництві перевагу надають газофазовим процесам.
Для одержання спиртів у промисловості найширше використовують два
головні методи: гідратація алкенів, що виділяються під час крекінгу нафти, і
Розділ 13. Спирти
345
ферментативний гідроліз вуглеводів. Окрім цих двох методів, є і деякі інші, що
мають обмежене застосування.
❖ Гідратація алкенів. Відомо, що алкени, які мають до п'яти атомів
Карбону, можна виділити із суміші, одержаної у процесі крекінгу нафти. Ці
алкени легко перетворюються в спирти внаслідок безпосереднього приєднання
води або сульфатної кислоти з наступним гідролізом алкілсульфатів, що
утворилися (детальніше див. розділ 8):
„. 9Н= <?">
СН3С=СН2 + Н20==: СН3ССН3-£г* сн3(рсн3
2-Метилпропен 2 2-Метилпропан-1-ол
СН2=СН2 + Н2804 СН3СН20803Н СН3СН2ОН
Етилен Етанол
СН3СН=СН2 + Н2804 -СН3(|НСН3 СН3С|НСН3
пР°пен 0803Н ОН
Пропан-2-ол
н бо н о
СН3СН2СН=СН2 ^ СН3СН2С|!НСН3 ~ *"СН3СН2С|НСН3
Бут-і-ен ОБОзН ОН
Бутан-2-ол
Таким способом можна синтезувати лише ті спирти, які утворюються за
правилом Марковникова: наприклад, ізопропіловий, але не пропіловий; втор-
бутиловий, але не н-бутиловий, тре/и-бутиловий, але не ізобутиловий. Цими
методами молена одержати лише один первинний спирт - етиловий. Окрім того,
цей метод не має стереоспецифічності, а в процесі гідратації можливі пере-
групування. Зазначені проблеми можна обійти двостадійним синтезом спиртів
через оксиран, що дає врешті-решт антигідратацію (див. далі).
У промисловості гідратація алкенів, каталізована кислотами, є в основі
виробництва етанолу з етилену і пропан-2-олу з пропену:
Н,Р04/8Ю2
СН2=СН2 + Н20 3оо°с,70а^. СН3СН2ОН
Етилен Етанол, 95%
СН3СН=СН2 250°С,440атм* СН3СНОНСН3
Пропен Пропан-2-ол, 95%
Цей метод має досить обмежену сферу застосування, оскільки гідратація
алкенів часто супроводжується ізомеризацією вуглеводневого скелета внаслідок
346
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
перегрупування карбокатіонів, що суттєво звужує синтетичні можливості цього, на
перший погляд, простого способу одержання вторинних і третинних спиртів. У
лабораторії застосовують інший метод, що ґрунтується на реакції гідрокси-
меркурування—демеркурування алкенів. Детальніше про нього йтиметься далі.
❖ Ферментативний гідроліз сировини, що містить вуглеводи (виноград,
ягоди, пшениця, картопля тощо), має величезне практичне значення, особливо
для одержання етилового спирту:
С6Н1206 -> 2С2Н5ОН + 2С02
Основну його масу використовують для виробництва алкогольних напоїв.
Звідси і назва "винний спирт". У разі застосування крохмалю як вихідної
речовини разом з етиловим спиртом виділяють сивушні масла, які є сумішшю
пентилових спиртів, а також пропілового та ізобутилового спиртів, що мають
токсичну дію.
Для промислових цілей використовують етанол, одержаний гідролізом і
бродінням деревини, відходів целюлозно-паперової промисловості {гідролізний
спирт).
■ Цікавою є реакція Вейцмана^ - ферментативний гідроліз вуглеводнів дією
бактерій Clostridium acetobutylicum, унаслідок чого утворюється суміш н-
бутилового спирту (60 %), етилового спирту (10 %) і ацетону СН3СОСН3 (ЗО %).
❖ Гідроліз алкілгалогенідів. Реакція не має суттєвого значення, оскільки
власне галогенопохідні алканів частіше синтезують зі спиртів. Однак у
промисловості хлоруванням суміші н-пентану й ізопентану з наступним гідро-
лізом галогеноалканів одержують суміш п'яти ізомерних спиртів, яку застосо-
вують як розчинник. Із цієї суміші перегонкою одержують малодоступний
чистий пентан-1-ол.
❖ Оксосинтез. Внаслідок нагрівання суміші карбон (II) оксиду і водню над
каталізаторами можна одержати різні спирти, склад яких залежить як від умов
перебігу реакції, так і співвідношення речовин, що реагують, наприклад:
со +2Н2 тоо^гооатм^НзОН
Метанол
6 CO +12Н2 й -СН.СНХН.ОН + СНХНСН, + 4Н20
2 400 °С, 200 атм. 3 2 2 3j
ОН
Пропан-1-ол Пропан-2-ол
■ Карбонілювання спиртів дає змогу подовжити вуглеводневий ланцюг:
СН3ОН + CO +2Н2—- СН3СН2ОН + Н20
Метанол Етанол
1 Вейцман X. (Weizman H.A.) (1874-1952) - ізраїльський біохімік. Головні праці присвячені
біохімії вуглеводів. Перший президент Ізраїлю.
Розділ 13. Спирти
347
• Гідроформілювання алкенів. Приєднання карбон (II) оксиду і водню до
алкенів за наявності каталізатора дає альдегіди і кетони, які можна відновити до
спиртів:
СН3 СН3 СЯ,
Ш3С<^<р=€Н2 сц%^23«с' СНзССН2СНСН2СНО — ОТзСС^ЖНгСНрН
СН3 СН3 СН3 СН3 СН3 СН3
2,4,4-Триметилпент-1 -єн 3,5,5-Триметалгексаналь 3,5,5-Триметилгексан-1 -ол
Оксосинтез, відкритий у США (О. Ройлен1, 1938 р.), який спочатку активно
розвивали в Німеччині, нині набуває щораз більшого значення в хімічній
промисловості. Наприклад, таким способом одержують н-бутиловий спирт з
пропілену і н-пропіловий спирт з етилену:
со н
НХ-СН=СН, п ' '» СНзСН2СН2СН2ОН
3 2 Со,(СО)о ,
Пропен Бутан-1-ол
н2с=сн2 Со; » ■ сн3сн2сн2он
Етилен 24 '* Пропан-1-ол
❖ Альфол-процес. Головним конкурентом попереднього методу є альфол-
процес - спосіб одержання н-алканолів олігомеризацією етилену за наявності
каталізатора на основі титан хлориду і триетилалюмінію за Циглером (детальні-
ше див. розділ 8) з наступним окисненням продуктів олігомеризації. Зокрема,
цим методом синтезують первинні спирти С\?-С\%:
едснр^сн^^Озі^^
альфол
❖ Окиснення алканів. Унаслідок окиснення вищих алканів молекулярним
киснем переважно одержують вторинні спирти Сі2-С2о> які використовують для
синтезу поверхнево-активних речовин. Реакція каталізована солями або
комплексами перехідних металів: Кобальту, Купруму, Феруму, Мангану і
відбувається через стадію розкладу гідропероксидів (детальніше див. розділ 7).
13.4. Методи синтезу одноатомних спиртів
у лабораторії
❖ Гідроліз алкілгалогенідів. Зазвичай спирти одержують гідролізом хлоро-
алканів, нагріваючи їх з водою або водним розчином лугів. У першому випадку
1 Ройлен О. (Яоеіеп О.) (1897-1993) - німецький хімік. Працював разом із Ф. Фішером та
Г. Тропшем. Розробив метод промислового синтезу альдегідів (оксосинтез).
348
Чирва В.Я., Ярллолюк CM., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
реакція зворотна, а в другому - часто супроводжується елімінуванням гідроген
галогенідів, наприклад:
НзС-Ш-С^С^ НзС-С=СН-СНз
СН^ CHj CHj
З-Метил- 1-хлоробутан 3-Метилбуган-1-ол З-Метилбуг-1-ен 2-Метилбут-2-ен
Щоб уникнути побічних процесів, спочатку з хлороалканів ліпше синте- ^
зувати естери, які потім омилюють до спиртів:
НХ~СН-СНХН9С1 + CH3COONa————Г HX-CH-CHXH-OCOO-L+NaCl
3 І 2 2 З ДМФА, 100 °С 3 і 2 2 З
сн3 сн3
З-Метил-1 -хлоробутан З-Метилбутилацетат
Н3С-СН-СН2СН2ОСОСН3 -|^Н3С-СН-СН2СН2ОН + CH3COONa
сн3 2 сн3
3-Метилбутан-1 -ол
Для ліпшої гомогенізації реакційної суміші до неї додають деяку кількість
розчинника, що змішується з водою, наприклад, діоксану.
❖ Гідроборування окиснення алкенів. Алкени реагують з дибораном
(ВН3)2, утворюючи спочатку алкілборани, які, окиснюючись, перетворюються у
спирти:
(ВН3)2 + СН2=СН2 СН3СН2ВН22СН2=СН2. (СН3СН2)3В ЗСН3СН2ОН + В(ОН)3
Реакцію проводять у тетрагідрофурані. Диборан одержують реакцією між
двома комерційними реагентами: натрій борогідридом і борофлуоридом,
переважно in situ (у реакційній суміші за наявності алкена) або відновлюють Бор
(III) хлорид воднем:
3NaBH4 + BF3 —2(ВН3)2 + 3NaF
Алкілборани не виділяють, а обробляють їх у тій же реакційній суміші
лужним розчином гідроген пероксиду. Як видно з наведених реакцій, вони
відбуваються проти класичного правила Марковникова і без перегрупувань:
СН3СН2СН=СН2 н^о^он- СН3СН2СН2СН2ОН
Бут-1-ен Бутан-1-ол, 93%
СН3 СН3
CH3(f=СН2 fg^. СН3^НСН2ОН сн3с=снсн3 ^^.СН.СНСНСН,
сн3 сн3 2 2 он
2-Метилпропен 2-Метилпропан-1-ол, 98% Метилбут-2-ен З-Метилбутан-2-ол, 98%
Розділ 13. Спирти
349
Зазначимо, що в реакції бере участь не диборан, а його мономер, який
утворюється в розчині:
(ВН3)2= 2ВН3
Поряд з дибораном в органічному синтезі застосовують комплекс борану в
тетрагідрофурані:
-вн,
■ Механізм реакції гідроборування можна описати як типове електрофільне
приєднання бор гідриду до подвійного зв'язку, у якому електрофілом є атом
Бору. Із сучасного погляду цю реакцію розглядають як процес, що відбувається
через чотирщентровий активований комплекс:
СН3СН—СН2~
вн3
СН3СН2СН2
вн,
СН3СН^СН2
Н - ]=НН
н
Очевидно, реакція гідроборування алкенів починається з електрофільної
атаки атома Бору. В я-комплексі, що утворився, на атомі Бору збільшується
негативний заряд з тенденцією до утворення вторинного карбокатіону. Проте
карбокатіон не утворюється, оскільки атом Бору з набутим негативним зарядом
легко втрачає атом Гідрогену у вигляді гідрид-іона, синхронно утворюючи
продукт і/ис-приєднання.
Реакція окиснення алкілборанів відбувається так. На першій стадії гідро-
пероксид-аніон атакує електронодефіцитний атом Бору:
ІІ-В + ООН
І
я-в-оон
І
Інтермедіат, що утворився, зазнає перегрупування завдяки міграції алкільної
групи з електронами до атома Оксигену за схемою, що подібна до перегрупу-
вання карбокатіонів:
я-в-о он — я-в-оіі + он"
Взаємодія з гідропероксидом у лужному середовищі відбувається швидко і з
виділенням тепла:
ІЦВСЖ + 200Н — В(ОЯ)3 + 20Н"
350 Чирва В.Я., Ярллолкж СМ., Толкачова H.B., Земляков О.Є. Органічна хімія
Утворений естер борної кислоти легко розпадається в умовах реакції,
вивільняючи спирт:
Щоб уникнути подальшого окиснення продуктів реакції до альдегідів і
кислот, процес проводять в атмосфері азоту за наявності борної кислоти
(О. Башкіров1), яка утворює зі спиртами стійкі до окиснення естери борної
кислоти B(OR)3, які потім легко гідролізують лугами. Таким способом у
промисловості, зокрема, одержують цетиловий спирт СібНззОН.
Реакція гідроборування проста і зручна, виходи дуже високі. її можна
використати, щоб синтезувати сполуки, які важко одержати з алкенів будь-яким
іншим способом. Для ациклічних, моно- і дизаміщених алкенів гідроборування-
окиснення відкриває унікальну можливість синтезу первинних спиртів з
сумарним виходом 80-95 %.
❖ Алкілборування карбон (II) оксиду. Подальшого розвитку метод
одержання спиртів з алкілборанів набув у працях Г. Брауна2 і М. Рашке, які
запропонували карбон (II) оксид як акцептор алкілборану. Реакція відбувається
за температури 100-125 °С. У проміжному комплексі виникає послідовна
міграція алкільних груп з атома Бору на атом Карбону:
Цим методом, залежно від умов перебігу реакції, можна одержати з високим
виходом первинні, вторинні та третинні спирти.
❖ Гідроксшіеркурування-демеркурування алкенів дає змогу синтезувати
спирти і не супроводжується перегрупуваннями. Напрям реакції відповідає
правилу Марковникова; вона відбувається за м'яких умов, а виходи
наближаються до теоретичних-:
1 Башкіров А. (1903-1982) - хімік-технолог, член-кореспондент Академії наук СРСР, працював у
галузі нафтосинтезу, хімії та технології палива. Досліджував хімію і технологію винних спиртів.
2 Браун Г. (Brown Н.С.) (1912-2004) - американський хімік, започаткував праці з хімії бору;
розробив кількісні методи вивчення стеричного напруження молекул; Нобелівська премія 1979 р.
(разом з Г. Віттігом).
B(OR)3 + ОН"+ ЗН20—- 3ROH + В(ОН)4
RCH=CH2 + Hg(OCOCH3):
з>2 -|V R-CH-CH2HgOCOCH3
ОН
Розділ 13. Спирти
351
Механізм цієї реакції можна описати так. Спочатку відбувається дисоціація
меркурій (II) ацетату з утворенням йона СНзСООН^*+. Ацетоксимеркурат-катіон
реагує з подвійним зв'язком алкену подібно до протона. Далі карбокатіон
взаємодіє з водою, утворюючи сіль алкілмеркурату:
Н§(ОСОСН3)2= СН3СОСГ+ Н^ОСОСНз
+ ■ \ / І /
Ь^ОСОСНз + /С=сч ^ сн3соо^-с-с+
І / н2о II
сн~соон§-с-с+ ^ снхоонг-с-с-
3 ' + ' он
Демеркурування утворених меркурійованих спиртів відбувається кількісно,
якщо обробляти їх натрій борогідридом, наприклад:
ИаВН4
І І
сн3соон8-с-с-он ——^ н-с-с-он
|| н2и
СН3(СН2)3СН=СН2^^ ^ сн3(сн2)з-сн-сн3
он
Гекс-1-ен Гексан-2-ол, 96%
Унаслідок заміни води на спирт або карбонову кислоту можна одержати етери
або естери. В лабораторії цей метод повністю витіснив реакцію гідратації алкенів.
❖ Відновлення естерів і карбонових кислот дає первинні спирти.
■ Каталітичне гідрування естерів зазвичай проводять над платиновими
каталізаторами, нікелем Ренея або купрумхромітовим каталізатором:
СНз(СН2)12-С-ОС2Н5 сио^о^ СНз^Н^зОН + С^ОН
О 250°С,220атм. Тетрадекан-1-ол, 98%
Етилтетрадеканоат
■ У лабораторних умовах як відновник частіше використовують літій
алюмогідрид:
сн3сн2сн=сн-соос2н5 !2Ь'А^ СН3СН2СН=СН-СН2ОН
Етилпентен-2-оат ' 3 Пент-2-ен-1-ол, 91 %
/ \ 1) ЬіА1Н4 / \
^-сн2соон ^—¡4- <^Ьсн2сн2он
Циклогексилоцтова 2-Циклогексилетан-І-ол, 97%
кислота
■ Великі кількості спиртів з нерозгалуженим вуглеводневим ланцюгом і
парною кількістю атомів Карбону раніше одержували шляхом відновлення
352 Чирва В.Я., Ярмолюк CM, Толкачова Н.В., Зеллляков О.Є. Органічна хімія
натрієм в етиловому або бутиловому спирті естерів органічних кислот чи жирів
за методом Буво1—Блана2:
СНз(СН2)10-С-ОС2Н5 №» СН3(СН2)1ІОН + С2Н5ОН
Етилдодеканоат ° Додекан-1-ол, 75%
❖ Оксосполуки можна відновлювати воднем до спиртів за наявності
каталізаторів, таких як нікель Ренея або платина, а також літій алюмогідридом
або натрій борогідридом. У цьому разі з альдегідів одержують первинні спирти,
а з кетонів - вторинні:
а^^аю-^он3(сн2)6он ^^-с-с^снз щх^^-^щ
Гепганаль Гептан-1-ол, 86% О ОН
Нонан-5ч>н Нжш-5-ол, 89%
Зазначимо, що натрій борогідрид, на відміну від літій алюмогідриду, не
відновлює карбоксильної та естерної груп, що дає змогу відновлювати
карбонільну групу за їхньої наявності:
н3с-с-сн2сн2соос2н5 н3с-сн-сн2сн2соос2н5
о 3 он
Етил-2-оксопентаноат Етил-2-гідроксипентаноат
Алкіл- і арилзаміщені борогідриди поряд з селективністю відновлення
забезпечують і стереоселективність.
❖ Синтези за допомогою реактиву Гриньяра. Реактиви Гриньяра легко
взаємодіють з карбонільними сполуками. Формальдегід у цьому разі утворює
первинний спирт, решта альдегідів - вторинні, а кетони - третинні спирти:
8- ОМЄІ
р 5- 5+ І
Н-С6+ + CH3MgI - н-с-н СН3СН2ОН
H Лтт Первинний спирт
8-
о
8- 8+ І Н20
он .
Вторинний спирт
R-C5. + CHjMgI R-C-H R-CH-CH,
Н СН, О»
" т
СН
R-C£ + CH3MgI — R-(f-R _7SS; R-C-R
З Третинний спирт
1 Буво Л. (Bouveault L.) (1864-1909) - французький хімік-органік. Головна діяльність - органічний
синтез.
2 Блан Г. (Blanc G.) (1872-1927) - французький хімік-органік. Дослідження в галузі хімії терпенів і
органічного синтезу.
Розділ 13. Спирти
353
Взаємодією реактиву Гриньяра з естерами одержують третинні спирти, за
винятком естерів мурашиної кислоти, яка дає вторинні спирти:
Н^ф ™~ Н3С-С-СН3 —-г НзС^^^С-С-СНз^ Н^С-С-СНз
ОСА ОСД .ш ' 05" СНз ^ Д СНз
„ ^ 2-Мяилпропан-2-<ш
етилацетат , ч
(третинний спирт)
Кетон, що утворився, активніший, ніж естер, тому він насамперед реагує з
реактивом Гриньяра.
❖ Одержання спиртів із оксиранів
■ Органічні сс-оксиди (оксирани або епоксиди) також вступають у реакції з
алкілмагнійгалогенідамщ утворюючи первинні спирти:
СН2—СН2 + СН3М£ СН3СІ^СН2ОМ^ СИзСН^^ОИ + Ug{OЩ
Пропан-1-ол, 60%
Етиленоксид
■ Епоксиди, взаємодіючи з літій алюмогідридом, перетворюються в
спирти. Реакція полягає в нуклеофільній атаці гідрид-аніона атома Карбону з
найменшим ступенем заміщення (менш екранованим) та утворенням вторинного
або третинного спирту:
с6н5сн—рн2 С6Н5СН-СН3
^ 3 он
Стиролоксид 1-Фенілетан-1-ол
Оскільки ос-оксиди, зазвичай, одержують з олефінів, то такий двостадійний
процес можна розглядати як альтернативу реакції гідратації алкенів. На відміну
від реакції гідратації, реакції відновлення епоксидів відбувається регіо- і
стереоселективно. В системах, для яких неможливе вільне обертання навколо а-
зв'язків, гідроксильна група і атом Гідрогену мають яняш-конфігурацію, звідси і
назва цього процесу — антигідратація:
Циклогексен Циклогексанол
❖ Взаємодія первинних амінів з нітритною кислотою дає спирти:
С„Н2„+,МН2 + НОНО -> СН2„+ІОН + И2Т + Н20
Реакцію практично не застосовують у синтетичній хімії, оскільки вона
супроводжується утворенням великої кількості побічних продуктів.
354 * Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
❖ Взаємодія галогеноалканів із калій супероксидом - один із надсучасних
методів синтезу спиртів:
к-х + ко2 18$Г' ^он
25 С 35-75%
Заміщення атома галогену на гідроксил у вторинних асиметричних атомів
Карбону супроводжується повним обертанням конфігурації.
13.5. Хімічні властивості одноатомних спиртів
З огляду на те, що спирти виявляють кислотно-основні властивості, їхні
реакції можна розділити на такі, що відбуваються з розривом зв'язку С-ОН, і
такі, що супроводжуються розривом зв'язку СО-Н.
13.5.1. Розрив зв'язку С-ОН
❖ Заміщення гідроксильної групи на галоген. Є велика кількість реакцій
заміщення гідроксильної групи на галоген. Найвідоміші з них - взаємодія
спиртів з гідроген галогенідними кислотами, а також галогенідами Фосфору і
Сульфуру. Залежно від будови вихідного спирту реакція заміщення може
відбуватися за механізмом або 8м2.
• Взаємодія спиртів з гідроген галогенідами. На вихід реакції, окрім умов
перебігу, значно впливають природа спирту і кислотність гідроген галогеніду,
реакційна здатність яких зменшується в ряду НІ > НВг > НСІ » НІ7, а в ряду
спиртів швидкість заміщення ОН-групи різко зменшується, якщо переходити від
третинного до первинного. Наприклад, третинний спирт реагує з гідроген
галогенідами, за винятком гідроген флуориду, уже на холоді. Первинні й
вторинні спирти перетворюються в галогеноалкани, коли їх нагрівати з
сумішшю галогенідної і сульфатної кислот протягом декількох годин:
(СН3)3СОН ЩШ. (СН3)зСС1 сн3сн2сн2сн2он ш\^сн?°ї СН3СН2СН2СН2Вг
2-Метил- _но 2-Метил-2-хлоро- Бутан-1-ол -Н 0 1-Бромобутан
пропан-2-ол 2 пропан 2 95%
94%
Іноді гідроген галогенідні кислоти одержують з їхніх натрієвих і калієвих
голей дією концентрованої сульфатної кислоти.
■ Зазначимо, що хлорид-іон є досить слабким нуклеофілом через його
високу сольватацію у водних середовищах. Щоб збільшити швидкість реакції,
додають цинк хлорид, який полегшує заміщення ОН-групи на хлорид-іон:
КСН2ОН +гпС12=ї= ЯСН2-6^ — ІІСН2С1 + йі(ОН)Сі;
2ліС12
Розділ 13. Спирти
355
За механізмом 8к2 реагують метанол і більшість первинних спиртів, що мають
просторово не утруднену гідроксильну групу. Протонування спиртів сприяє тому,
що гідроксильна група добре відходить:
Вг"+ К-СН2ОН2-^ Я-СН2Вг
У реакціях реакційна здатність первинних спиртів Я-СНгОН нижча, ніж
метанолу. Це пов'язано зі збільшенням кількості просторових перешкод для
атаки протонованого спирту галогенід-іоном.
■ Третинні й частково вторинні спирти реагують за механізмом Бці,
оскільки протонований спирт легко і швидко вивільняє молекулу води, утво-
рюючи карбокатіон. Його подальша стабілізація визначена атакою сильнішого
нуклеофіла, ніж вода, - галогенід-аніона:
^ ™+ Швидко ^ + „ л
Я3СОН + Н К3С + н2о
К3СЧ СІ Я3СС1 Я3С++ Н2О^^К3СОН + Н+
■ Проте варто враховувати, що карбокатіон, який утворився із вторинних
спиртів, може перетворюватися в третинний унаслідок 1,2-гідридного або
апкільного зсуву, наприклад:
СН3СН-СН-СН, СН3СН-СН-СН3 3= СНзСН-С=СН3
II II І
ОН СН3 +0^0^ сн3
З-Метилбутан-2-ол ^
—*■ сн3сн2-с^сн3 — сн3сн2-с-сн3
сн3 сн3
2-Бромо-2-метилбуган
(СН3)3С-СН-СН3 -д^ (СН3)3С-СН-СН3 —- (СН3)2С-СН-СН3
І І
ОН +ОН2
3,3-Диметилбутан-2-ол
—* (СН3)2СІСН-СН3 — (СН3)2С-СН-СН3
І II
СН3 ВгСН3
2-Бромо-2,3-диметилбутан
3;2^-^п-
<5У
356 Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Остання стадія ускладнена побічною реакцією Е1 - відщепленням протона з
утворенням алкену:
(СН3)2С-СН-СН3=^ (СН3)2С=С(СН3)2
сн,
-НВг
2,3-Диметилбут-2-ен
■ Деякі просторово утруднені первинні спирти, наприклад, неопентиловий
спирт, можуть реагувати за механізмом 8ц1. Первинний карбокатіон, що утворився,
швидко перегруповується в третинний унаслідок 1,2-метильного зсуву:
<РНз
•• неї
Н3С-С-СН2ОН5=:
сн3
2,2-Диметилпропан-1 -ол
?н3
-9-е
сн,
н3с-с-сн2он
1 н
: Н3С-С-СН2
сн3/
+
•н3с-(р-сн2сн3-
сн,
СІ"
сн,
н3с-<р-сн2сн3
2-Метил-2-хлоробутан
Вторинні спирти можуть реагувати як за механізмом Бні, так і за 8ц2.
Механізм реакції визначений концентрацією спирту, кислоти, температурою
перебігу реакції і природою розчинника.
• Проба Лукаса. Чи є спирт первинним, вторинним або третинним, можна
визначити за допомогою проби Лукаса, яка ґрунтується на різній реакційній
здатності трьох класів спиртів стосовно гідроген галогенідів. Третинні спирти
реагують з реактивом Лукаса (суміш концентрованої НС1 з безводним гпСЬ)
відразу, про що свідчить миттєве помутніння реакційної суміші, вторинні -
протягом 5 хв, а первинні спирти помітно не реагують за кімнатної температури.
Третинні спирти легко утворюють карбокатіони, вторинні - повільніше, а первинні
- зовсім не утворюють. Оскільки спирти розчиняються в концентрованій хлоридній
кислоті за наявності цинк хлориду, а утворені з них галогеніди - ні, то, відповідно,
простежується помутніння. Винятком є первинні аліловий і бензиловий спирти, які
утворюють стійкі карбокатіони і тому дають позитивну реакцію.
• Взаємодія спиртів з галогенідами Фосфору і Сульфуру. Порівняно з
гідроген галогенідами найзручнішими реагентами для одержання галоген-
ноалканів зі спиртів є галогеніди Фосфору і Сульфуру та гідроген галогеніди
деяких неорганічних кислот, наприклад: 80СЬ, РСЬ, РС15, РОСЬ, СОС12:
БІ-ОН + РС15 -> Я-С1 + РОСЬ + НС1
З ІІ-ОН + РВгз -> 3 Я-Вг + Н3РО3
6 СНзОН + 2Р + ЗІ2->6 СНзІ + Н3РО3 (Р + З І2 -> 2РІ3)
Розділ 13. Спирти
357
■ Для реакцій з фосфор тригалогенідами найімовірніший такий механізм
реакції. Спочатку утворюється триалкілфосфіт і, якщо реакція відбувається за
наявності основ, ця сполука може бути кінцевим продуктом реакції:
ЗІІ-ОН + РВг3 (ЯО)3Р + ЗНВг
Якщо гідроген бромід не нейтралізувати, то проміжна сполука три алкіл-
фосфіт легко зазнає протонування, і алкільні групи перетворюються в галогено-
алкани:
(ЯО)3Р + ЗНВг -3 ІІ-Вг + Н3Р03
■ Реакції спиртів із фосфор пентагалогенідами зазвичай не супроводжуються
перегрупуваннями. Це приводить до зміни конфігурації асиметричного атома
Карбону, що зв'язаний з гідроксильною групою:
/СНз
НС-ОН + РС1— ОТ н.>С-гСХ-РС13 СІ-С-н + РОС13 + СІ"
с2к{ ^ с? >с2н5
(Л)-Буган-2-ол (5)-2-Хлоробуган
■ У реакціях спиртів з тіонілхлоридом спочатку утворюється хлоросульфіт-
ний естер:
ЯОН + 80СІ2 -> ЯОЗОСІ + НС1
Далі реакція може відбуватися за двома механізмами. У нуклеофільних
розчинниках, таких як діоксан, розчинник бере участь у реакції, і в кінцевому
продукті зберігається вихідна конфігурація:
О \) + Я-О-Б-СІ — (/ \?-Я+802+ СІ" -Я-СІ + с/ \)
О ^ /
У випадку, якщо розчинник не бере участі в реакції, атака хлорид-аніона
молекули хлоросульфітного естеру відбувається з тилу і супроводжується
обертанням конфігурації продукту реакції:
СІЧК-гОг-в-СІ -Я-С1 + 802+ СГ
її 2
о ,
❖ Застосування п-толуенсульфохлоридів для заміщення гідроксигруп.
Відомо, що спирти взаємодіють з я-толуенсульфохлоридом (ТзСІ) за наявності
піридину, утворюючи алкіл-л-толуенсульфонати (тозилати):
я-сн2он + НзС-/-\-^-сі а-сн2о-^-^~^-сн3+нсі
О О
358
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Оскільки я-толуенсульфат-іон є дуже доброю відхідною групою, то він
може бути легко заміщений без перегрупувань у реакціях з нуклеофілами,
зокрема, з галогенід-іонами.
❖ Дегідратація спиртів за допомогою таких кислот, як сульфатна,
ортофосфатна й оксалатна, приводить до алкенів:
і і н+
-—
н ОН
Як зазначено вище, найлегше дегідратування відбувається у третинних
спиртах, потім - у вторинних і, нарешті, у первинних. Процес дегідратації
спиртів підлягає правилу Зайцева, згідно з яким атом Гідрогену відщеплюється
від найменш гідрогенізованого атома Карбону, що міститься в Р-положенні до
ОН-групи, наприклад:
Н3С-9Н-9Н-СН3 —"-Н3С-СН=С-СН3
ОН СН3 " 2 СН3
З-метилбутан-2-ол 2-метилбут-2-ен
Дегідратація має два етапи. Спочатку відбувається протонування ОН-групи,
а потім - елімінування молекули води за механізмом Е2, якщо це первинні
спирти, або за механізмом Е1, якщо спирти третинні.
Вторинні спирти, залежно від умов, реакції можуть зазнавати дегідра-
тування за Е2- або Е1-механізмом, наприклад, за механізмом Е1 відбувається
дегідратування /яре/я-бутилового спирту:
СН. , СН, Снт
Г Н Т ">+ ПппІпї.нп ' Швидко
сн3с-ОН СН3С-0Н2 По™ь"? СН39+ - СНз-С=СН2
СН3 СН3 " 2 СН3 "Н+ інз
2-Метилпропан-2-ол 2-Метилпропен
Третинні спирти дегідратувати наскільки легко, що можлива вибіркова
дегідратація діолу, що має первинну і третинну гідроксильні групи:
(сн^с-ссн^сьірн40 % "2^4 (СН3)2С=СН(СН2)2СН2ОН
ОН ^0("
с \а „^і. і с „• 5-Метилгекс-4-ен-1-ол
5-Метилгексан-1,5-дюл
Дегідратацію третинних спиртів можна проводити в 20-50 % сульфатній
кислоті за 85-100 °С. Вторинні спирти дегідратують за жорсткіших умов: 85 %
фосфатна кислота, нагрівання до 160 °С або 60-70 % сульфатна кислота за
температури 90-100 °С:
СН3СН2СН2СНОНСН3 62%ц££і СН3СН2СН=СНСН3 + СН3СН2СН2СН=СН2
Пентан-2-ол Пент-2-ен,80% Пент-1-ен, 20%
Розділ 13. Спирти
359
■ Утворення алкену визначене стабільністю проміжного карбокатіону і
термодинамічною стабільністю розгалуженого алкену. Наприклад, для ізоамілового
спирту, згідно з правилом Зайцева, повинен утворюватись лише З-метилбут-1-ен, а
реально утворюються три алкени:
щ-^ш^снрії ^(^-ш^^бі^ ^ш3чрі«^а^-^ сНз^рі^імн,
СН,
З-Мгтилбутан- 1-ол
сн,
СНз
СНз
З-Метилбуг-1-ен
Первинний карбокатіон, що утворюється спочатку, найменш стабільний, тому
він через 1,2-гідридний зсув переходюь у стабільніший вторинний карбокатіон:
сн3-сн-сн-сн2—► сн3- сн-сн-сн3
сн,
сн,
Н,С-С=СНСН3
сн
сн,
^-СН-СН-
сн,
2-Метилбут-2-ен
[=СН2
З-Метилбут-1-ен
Відповідно, вторинний карбокатіон легко перетворюється в третинний, який
є найстабільнішим:
СН3-О-СН-СН3
СН,
СН3 (р СН2 СН3
сн,
СН2—СН2СНЗ
сн,
2-Метилбут-1-ен
сн3-с=сн-сн3
сн
З 2-Метилбут-2-ен
Головним продуктом реакції буде 2-метилбут-2-ен як найрозгапуженіший алкен.
Зазначимо,, що ізоаміловий спирт належить до первинних спиртів, однак його
дегідратація відбувається за механізмом Е1 тому, що механізм Е2 не можливий
через стеричні утруднення.
■ Первинні спирти зазнають дегідратування в концентрованій сульфатній
кислоті в інтервалі температур 170-190 °С:
СН3СН2СН2ОН і8о °С ^ СНзСН==СН2
Пропан-1-ол
Пропен
Для них реалізується механізм відщеплення Е2. У реакцію вступає не сам
спирт, а алкілсульфат, а роль нуклеофіла відіграє гідросульфат-аніон або вода.
СН3СН2СН2ОН + Н2804
9
сн3сн2сн2о-£-он + Н20
о
СН3СН2СН20-^-ОН 170-і8ос СН3СН=СН2 + Н2804 + Н804
О
360
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Цікаво, що в разі перебігу реакції за низької температури її можна
припинити на стадії утворення алкілсульфату:
СН3СН2ОН + Н2804=
Етанол
сн,сн,6^Н
Н
803Н"=г= Н20+СН3СН20803Н
Етилсульфат
■ Для дегідратації спиртів у промисловості замість сульфатної кислоти як
дегідратувальний агент зручніше використовувати алюміній оксид. Гетерогенну
каталітичну дегідратацію можна провести для первинних, вторинних і
третинних спиртів.
Поряд із сульфатною і фосфатною кислотами та алюміній оксидом для
дегідратації спиртів також застосовують оксалатну кислоту, бензенсульфо-
кислоту, цинк хлорид і торій (IV) оксид. У разі нагрівання вторинних спиртів з
торій (IV) оксидом одержують алкени з термінальним (кінцевим) подвійним
зв'язком. Крім алкенів, залежно від умов реакції (температура і концентрація
кислоти), спирти можуть перетворюватися в етери, про які йтиметься у
наступному розділі.
❖ Синтез естерів сульфокислот. Спирти реагують із сульфохлоридами,
утворюючи естери:
едбн + СІ-Б-Я Піриди" С2Н50-!5-ІІ +НС1
о о
Частіше застосовують хлорангідриди толуенсульфокислоти, метансуль-
фокислоти і трифлуорометансульфокислоти:
НзС~Ч //^°гСХ СН3802С1 СР3802С1
я-Толуенсульфохлорид Метансульфохлорид Трифлуорометансульфохлорид
(тозилхлорид, ТбСІ) (мезилхлорид) (трифлілхлорид)
Естери сульфокислот - зручні сполуки для різних нуклеофільних реакцій,
оскільки сульфонатна група легко, часто за кімнатної температури, піддається
заміщенню; особливо це стосується "трифлатів" К-О-802СР3.
сн3сн2-(рн-сн2снз1ї^г сн.снгсрн-сн.сн,сн^-сн-сн.сн,
ОН ОТб Вг
Пентан-З-ол З-Бромопентан
Реакції відбуваються стереоспецифічно з обертанням конфігурації:
Розділ 13. Спирти
361
л Хй цис-1 -Метил-4-метил-
/иш//с-4-Метилциклогексанол п ,
сульфанілциклогексан
❖ Синтез амінів зі спиртів. Амоніак або аміни алкілують спиртами, якщо
нагрівати реагенти в кислому середовищі:
СН3ОН + ИН3 + НХ==СН3ОН2Х"+ Ш3^^СН3КН2-НХ
Залежно від співвідношення реагентів одержують первинні, вторинні й
третинні аміни, а також четвертинні амонієві солі. Використаний як каталізатор
алюміній оксид за 300 °С дає ті ж самі результати.
13.5.2. Розрив зв'язку О-Н
❖ Реакції спиртів як кислот. Як відомо, силу кислоти характеризують її
здатністю відщеплювати протон. Для спиртів вона визначена різницею між
електронегативностями атомів Оксигену і Гідрогену, а також характером і
кількістю замісників біля атома Карбону, що має гідроксильну групу. Наявність
алкільних замісників з позитивним індукційним ефектом (+І-ефект) зменшує
кислотність спиртів. Справді, кислотність спиртів зменшується в ряду СН3ОН >
первинний > вторинний > третинний.
Якщо вводити електроноакцепторні замісники, то кислотність спиртів
збільшується, наприклад, спирт (СР3)зСОН однаковий за кислотністю з
карбоновими кислотами.
■ Спирти, як слабкі кислоти, реагують з лужними, лужноземельними металами,
алюмінієм, галієм, талієм, утворюючи алкоголяти з іонним або ковалентним
зв'язком, і можуть бути сильними основами та ефективними нуклеофілами:
2СН3СН2ОН +2Ш 2СН3СН2(Жа + Н2
3(СН3)3СОН + АІ [(СН3)3СО]3АІ + Н2
■ Алкоголяти можна одержати також, діючи на спирти натрій або калій
гідридами, натрій або калій амідами, реактивом Гриньяра:
СН3СН2ОН + КаМНгСН3СН2ОКа + №ї3
СНзОН + СН3Ь%І -> СНзСад + СН4
Останню реакцію застосовують для кількісного визначення в органічних
сполуках рухливих атомів Гідрогену.
362
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Спирти за кислотністю значно поступаються воді, тому навіть наявність
сильних лугів не зсуває рівноваги в бік утворення алкоголятів:
СН3ОН + ЫаОН « _^ СН3ОЫа + Н20
Проте цією реакцією іноді користуються у промисловості, щоб одержати
алкоголяти найпростіших спиртів. Під час процесу в реакційну суміш додають
бензен, що дає змогу позбуватися води у вигляді азеотропної суміші.
Серед алкоголятів треба назвати алюміній ізопропілат (/-РЮ)зА1 і трет-
бутилат (Г-ВиО)зАІ, які слугують реагентами для окиснення за Оппенауером1 і
відновлення за Верлейєм-Мейєрвейном2-Пондорфом3.
❖ Окиснення або каталітичне дегідрування спиртів. Окиснення спиртів
дає карбонільні сполуки. Первинні спирти в цьому разі перетворюються в
альдегіди, які далі можна перевести в карбонові кислоти. Вторинні спирти
окиснюються до кетонів. Третинні спирти за звичайних умов не окиснюються.
Окиснення первинних і вторинних спиртів до альдегідів або кетонів
проводять за допомогою таких реагентів: КМпОд, К2СГ2О7, СЮз, МпОг, А§гО,
Ag2COз тощо. З калій біхроматом реакція відбувається за рівнянням:
ЗН3С-С-СН3 + К2Сг207 + 4Н2804 — ЗН3С-С-СН3+ Сг2(804)з+ к2804+7Н20
О
ОН
Пропан-2-ол А^0"
Визначено такий механізм реакції:
<ґНз ЇЇ
Н-С-О-Сг-ОН + Н20
СН3 О
СН, О СН3 О
і 3 и6+ . і 3 ІЦ+
Н20' + НхСгО-Сг-ОН -Н30 + С=0 + :СгОН
2 її 3 і її
сн3 о сн3 о
Тоді як окиснення вторинних спиртів припиняється на стадії одержання
кетонів, первинні спирти за цих умов перетворюються в альдегіди, які через
гідратну форму окиснюються до карбонових кислот:
1 Оппенауер Р. (Оррепаиег Я.У.) (1910-1969) - австрійський хімік-органік, є автором кількох
іменних реакцій.
2 Мейєрвейн Г. (Меепуеіп Н.Ь.) (1879-1965) - німецький хімік-органік. Досліджував хімію
карбонієвих іонів. Автор кількох іменних реакцій, зокрема, "відновлення Мейєрвейна-
Понндорфа-Верлея", "перегрупування Вагнера-Мейервейна" та ін.
3 Понндорф В. (Рсппсіог^ АТУ.) (1894-1948) - німецький хімік-органік. Головні праці присвячені
вивченню механізмів реакцій.
Розділ 13. Спирти
363
>° н,о ?Н_ К,Сг,0,/Н^ <р
к_сн2онЬ^+к-сС ^ к-с-он^^к-с;
чн н он
Альдегід Гідрат альдегіду Карбонова кислота
Якщо потрібно припинити реакцію на стадії альдегіду, то процес проводять
у безводному метиленхлориді. У цьому випадку неможливе утворення гідрат
альдегіду, і, отже, карбонова кислота не синтезується. Окиснення спиртів калій
біхроматом супроводжується заміною жовтого забарвлення розчину хрому (Сг6+)
на зелене (Сг3+) і може слугувати контролем за перебігом реакції.
Третинні спирти за звичайних умов не окиснюються, проте в кислому
середовищі вони можуть зазнавати дегідратування до алкенів, які потім
окиснюються з деструкцією вуглеводневого ланцюга:
СН- СН,
н3с-с-он-^н3с--с=сн2 нзС-с-сн3 + С02
СНз О
2-Метилпропан-2-ол 2-Метилпропен Ацетон
• Каталітичне окиснення. Останнім часом первинні спирти почали окиснювати
до альдегідів киснем повітря з кількісним виходом над змішаним каталізатором:
ясн2он + о Ре^о!з^с К"СН0 + Н2°
• Каталітичне дегідрування. Дегідрування первинних і вторинних спиртів
проводять, пропускаючи їх над мідним дротом або мідно-срібним каталізатором
за 400-500 °С.
• Йодоформна реакція. Наявність у спирті структурного фрагмента СН3-СН-
ОН можна визначити за допомогою йодоформної реакції. Для цього спирт
обробляють йодом і натрій гідроксидом, які у суміші утворюють натрій
гіпоіодит №01; спирти, що мають згаданий структурний фрагмент, дають
жовтий осад СНІз.
13.6. Окремі представники одноатомних спиртів
■ У промисловості метиловий спирт одержують із синтез-газу, вико-
ристовуючи високий тиск і контактний каталізатор гпО/Сг203:
СО + 2Н2ІЙІ^СН3ОН
200 атм.
Використання як каталізатора Си/2п/А1 дає змогу знизити температуру
процесу до 250 °С і тиск до 50 атм. та зробити реакцію енергетично виграшною
порівняно з попередньою.
364
Чирва В.Я., Ярмолюк CM., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Метанол широко використовують у промисловості для метилювання
аніліну, виробництва диметилсульфоксиду і формаліну та як розчинник для
лаків. Зазначимо, що вже невеликі кількості метанолу, якщо вони потрапляють у
середину людського організму, спричинюють важкі отруєння. Летальна доза
метанолу для людини становить 25 см3.
■ Етиловий спирт у промисловості одержують адсорбцією етилену
концентрованою сульфатною кислотою за температури 55-80 °С. Спочатку
виникає диетилсульфат, який потім гідролізують до етанолу:
н,с=сн, /
r^cr, + h2S04 —*~ ch3ch2OS03h — * (c2h50)2S02
ch3ch2oso3h нр0 » c2h5oh + h2S04
3 z 3 70-100C 25 L 4
(c2h50)2S02 c2h5oh + h2so4
Значну кількість етанолу також одержують гідратацією етилену за наявності
фосфатної кислоти:
н^сн, + н2о СН3СН2ОН
Промислове значення має відновлення ацетальдегіду:
Ni
сн3сно + н2 —► СН3СН2ОН
Спиртове бродіння цукрів широко використовують у харчовій про-
мисловості для виготовлення алкогольних напоїв. Етанол є в складі пива, вина,
горілки й інших спиртних виробів. На противагу метанолу, етиловий спирт у
невеликих кількостях збуджує організм, а у великих - спричинює його отруєння.
З водою він утворює азеотропну суміш, яка складається з 96 % спирту і 4 %
води. Тому звичайною перегонкою 100% ("абсолютний") спирт одержати
неможливо. Для одержання чистого спирту воду, що в ньому міститься,
зв'язують хімічно, наприклад, додаванням перед перегонкою кальцій оксиду. Як
звичайно, етанол використовують у вигляді 96 % розчину. Його застосовують у
виробництві діетилового етеру, етилацетату й ацетальдегіду.
■ н-Пропіловий спирт утворюється в процесі спиртового бродіння
вуглеводів і під час гідроформілування етилену.
■ Ізопропіловий спирт одержують гідратацією пропілену. Пропілові спирти
використовують як замінники етилового спирту і для одержання ацетону.
■ Бутиловий спирт у великих кількостях отримують із суміші, що
утворюється під час бродіння цукру під дією Bacterium acetobutylicum, де його
вміст становить 60 %. Окрім того, я-бутиловий спирт у промисловості
Розділ 13. Спирти
365
одержують гідроформілуванням пропілену. Застосовують у виробництві бутил-
ацетату, гербіцидів, а також як розчинник у виробництві лаків і фарб.
■ втор-Бутиловий спирт одержують гідратацією бутилену.
■ Ізобутиловий спирт отримують з водяного газу за наявності солей
кобальту. Його застосовують для виготовлення фруктових ефірів або есенцій.
■ трет-Бутпловий спирт одержують гідратацією ізобутилену, що утво-
рюється під час крекінгу нафти. Застосовують як алкілувальний агент і
розчинник.
Спирти з довгими ланцюгами трапляються у восках рослин, знайдені в
комахах і деяких тваринах. Вихідною сировиною для їхнього одержання
слугують природні жири або олії, які в разі омилення дають вищі жирні кислоти.
Вищі жирні кислоти перетворюють у спирти:
4Н(СиО/Сг203)
СНГ(СН9)п-СООН LJ— СН3-(СН2)п-СН2ОН
32 -Н20 з * /.
На їхній основі готують синтетичні мила. Для цього спирти естерифікують
концентрованою сульфатною чи хлоросульфоновою кислотою. Натрієві солі
алкілсульфатів розчинні у воді й мають властивості мил. Перевагою таких мил
перед традиційними є розчинність їхніх кальцієвих солей у воді.
Сьогодні для синтезу вищих первинних спиртів віддають перевагу реакції
Циглера:
3/2 О, Щ}
Аікснр-сн^педіз —і Аі[о-(а^а^(зд3--^ с2щснГ-сн2)п'-ои
Алкоголят алюмінію
13.7. Ненасичені спирти та їхні естери
13.7.1. Еноли
Відомо, що олефіни не можуть мати гідроксил біля атома Карбону в ^р2-
гібридному стані, тому такі структури нестійкі та ізомеризуються в оксосполуки
за правилом Ельтекова-Ерленмейєра:
>с=<г — *г-С-
он но
Для структур, що мають гідроксил біля ненасиченого атома Карбону, не
зв'язаний з електроноакцепторними групами (>С=0, ->Ю2 й інші), правило
Ельтекова-Ерленмейєра завжди спрацьовує, тому вініловий спирт і його
гомологи не існують, а спроби їх одержати завершуються перегрупуванням в
ацетальдегід або, відповідно, у його гомологи.
366
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Сьогодні відомо багато сполук, правда, зазвичай складніших або з
декільками атомами Оксигену, які стійкі й можуть бути виділені не лише в
карбонільній формі, а й у вигляді ненасиченого спирту - енолу, наприклад:
Ш>СНСООС2Н5 =^ К<^СН2СООС2Н5
ОН О
Ізомерію між карбонільною сполукою і ненасиченим спиртом-енолом, що
утворився внаслідок переміщення одного атома Гідрогену від атома Карбону до
Оксигену, називають таутомерією, або десмотропією, а рідинні суміші
таутомерних форм, у яких обидва ізомери перебувають у рівновазі, -
алелотропними. Детальніше про таутомерію див. розділ 5.
У випадку вінілового спирту перегрупування спричинює мезомерний ефект,
що йде до кінця:
6 г)Ч *П?\ н
О'ПТТ ТТ *~\ І -г т л
■сн^с —"Н2С-СЧ ) —- щс-с.
ЮН О Н+
Унаслідок мезомерного ефекту атом Гідрогену гідроксильної групи
відщеплюється у вигляді протона й атакує заряд &~ на другому ненасиченому
атомі Карбону.
13.7.2. Одержання похідних єно лі в
На відміну від енолів, етери й естери вінілового спирту не лише існують, їх
широко використовують у промисловості як мономери. Зрозуміло, що їх
одерясують непрямим шляхом.
❖ Синтез вінілових естерів
■ Вінілові естери, як зазначено, одержують у промисловості через при-
єднання до ацетилену карбонових кислот за наявності солей Меркурію (II),
Кадмію або Цинку:
ЯСООН + НС^СН ЫСООСН=СН2
Сюди треба додати дію оцтового ангідриду на кетони за наявності
каталітичних кількостей сульфатної кислоти:
СН3ССН3 + СНзСОССНз-111- СН2=СОССН3 + СН3СООН,
Зц 3 Зц Ц 3 2 і н з з
О 0 0 Н3С о
а також дію кетену на ацетальдегід:
СН3СНО + СН2=С=0 СН3СООСН=СН2
Розділ 13. Спирти
367
Із вінілових естерів особливо важливий вінілацетат, що полімеризується за
радикальним механізмом у полівінілацетат. Полівінілацетат використовують для
одержання емульсійних фарб і лаків, прозорих пластмас, у виробництві
триплексу для склеювання шарів силікатного скла і для синтезу полівінілового
спирту:
-сн^н-сн^н—ch2-ch-ch2-<jh-
сн3с00 сн3с00 н ОН ОН
Полівініловий спирт застосовують у виробництві синтетичної крові, плазмо-
замінників, емульгаторів важливих полімерних матеріалів, таких як волокно
вінол. У практиці використовують полівінілові спирти після їхнього ацилю-
вання, наприклад, бутиральдегідом (полівінілбутираль r = С3Н7):
-ch^hch^hc^c^hch^h- + rcho -c^chch^hch^hch^h-
он он он он о-срн-о о-сн-о
r r
де R = Н, алкіл, фурфурил
■ Вінілові етери одержали О. Фаворский, М. Шостаковський і В. Реппе
незалежно один від одного через взаємодію ацетилену зі спиртами:
НС=СН + R-OHJg^ ch2=ch-or
90-98%
Вінілові етери можна синтезувати також взаємодією вінілацетату з відпо-
відними спиртами. їхньою особливістю є гідроліз, який легко відбувається в
кислому середовищі:
roch=ch2 + Н20 -je— roh + сн3сно
Головне призначення вінілових етерів - з'єднувальна основа багатошарових
плівок, виробництво клеїв, лаків, емалей. Вінілові етери легко полімеризувати
переважно ініціюванням реакції кислотами Льюїса, наприклад, ферум (III)
хлоридом. Одержують рідкі в'язкі полімери, які застосовують як загусники.
Полімеризацією вінілбутилового етеру одержують бальзам Шостаковського:
FeCl7^h2=<p^^ -СНГСН-СН2- СН-СН2-СН -
or or or or or or
Вінілові етери утворюють і тверді співполімери, проте внаслідок гомолі-
тичної полімеризації, ініційованої пероксидами.
368 Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
1 Кляйзен Л. (СІаІБеп Ь.) (1851-1930) - німецький хімік; головні праці присвячені органічному
синтезу, ацилюванню карбонільних сполук, таутомерії.
Під час нагрівання алкілвінілові етери перегруповуються за Кяяйзеном1 із
перетворенням в альдегіди внаслідок сильного М-ефекту /?,7г-спряженої системи:
(^=сн^дсн3 —- сн3сн2сно
Метоксіетен Пропаналь
13.73. Аліловий спирт
■ Донедавна аліловий спирт одержували гідролізом алілхлориду, що
синтезували хлоруванням пропілену в разі високої температури за Львовим:
СН2=СНСН3^^ СН2=СНСН2С1 5%№0Н> СН2=СНСН2ОН
Ще раніше аліловий спирт виробляли, нагріваючи гліцерол з оксалатною
кислотою. Естер, що утворювався після гідролізу, втрачав фрагменти оксалатної
кислоти у вигляді вуглекислого газу:
(рн2<рі^н2 + нос-сон —-с;н2с;нсн2он :—■ сн2=снсн2он
он он он оо 9 9
о=с—с=о
Сьогодні в промисловості аліловий спирт одержують ізомеризацією
пропіленоксиду.
■ 3 алілового спирту одержують гліцерол, акролеїн, акрилову кислоту.
Естери алілового спирту використовують у виробництві пластмас.
Будова алілового спирту зумовлена, з одного боку, його ненасиченістю, що
полягає, наприклад, у здатності цієї сполуки приєднувати два атоми галогену або
Гідрогену. З іншого боку, аліловий спирт можна окиснити до ненасиченого
альдегіду - акролеїну, і далі - до ненасиченої кислоти (акрилової кислоти), що
свідчить про наявність первинної спиртової групи. Каталітичне відновлення
алілового спирту воднем за наявності платини або нікелю дає нормальний
пропіловий спирт.
13.7.4. Вищі ненасичені спирти
Окремі вищі ненасичені спирти, що мають один або два подвійні зв'язки,
знайдені в етерних оліях рослин. Вони вирізняються сильним приємним запахом і є
головними складовими цих олій. За будовою вони належать до терпенів (див.
розділ 10) або легко можуть бути в них перетворені.
Серед етерних олій найбільше поширені такі представники.
Розділ 13. Спирти
369
■ Цитронелол (СН3)2С=СНСН2СН2СН(СНз)СН2СН20Н міститься до 50 % у
трояндовій, геранієвій і цитронелоловій оліях. У рослинах він часто трапляється
в суміші з ізомером 3,7-диметилокт-7-ен-1-олом. Таку суміш називають
родинолом. Найбільшу цінність має лівообертальний цитронелол, запах якого
ніжніший, ніж правообертального. Виділяють його з етерних олій або
синтезують відновленням цитралю - головного компонента етерної олії лимона
-3,7-диметилокта-2,6-дієналю, або селективним гідруванням гераніолу.
■ Гераніол існує у вигляді суміші транс-ізомерів а- і Р-форм:
СН2=С(СН2)3-С-СН3 СН3-С=СН-(СН2)3-С-СН3
СНз НС-СН2ОН СНз НС-СН2ОН
а-Форма р-Форма
Його виділяють з етерних олій багатьох рослин і синтезують ізомеризацією
ліналоолу - (СНз)2С=СНСН2СН2С(СНз)(ОН)СН=СН2 або гідруванням цитралю.
■ Нерол - і/ис-ізомер гераніолу - також існує у вигляді а- і Р-форм. Його
виділяють із трояндової і бергамотової олій. Обидві сполуки використовують в
парфумерних композиціях, для ароматизації мил і мийних засобів.
■ Ліналоол має запах конвалій, його використовують у парфумерній
промисловості як сам по собі, так і у вигляді естеру (ліналілацетат). Він є в
лавандовій, бергамотовій і коріандровій оліях. Синтезують з гераніолу алільним
перегрупуванням, а також із 6-метилгепт-5-ен-2-ону (Л. Ружичка):
он
(СН3)2С=СН-СН2СН2-£-СН3 + НС=СН -^(СНз^СНСНзСНХС^СН-^
о
сн,
(СНз^СНСН^Н^СН^Н,
Ліналоол СН3
■ Завдяки стійкому запаху, що нагадує запах конвалій, у парфумерії має
важливе значення фарнезол - спирт з трьома подвійними зв'язками, що
міститься в липоврму цвіті:
(СН3)2С=СН-СН2СН2С(СН3)=СН-СН2СН2С(СН3)=СН-СН2ОН
■ У вигляді естеру в складі молекули хлорофілу є ненасичений аліфатичний
спирт фітол:
сн3(рнсн2сн2сн2(рнсн2сн2сн2(^нсн2сн2сн2с=снсн2он
СН3 СН3 СН3 СН3
Якщо озонувати і розщеплювати цей спирт, то утворюється гліколевий
альдегід і кетон Сі8Н360, який виявився ідентичним кетону, одержаному з
гексагідрофарнезолу.
370
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
■ А. Бутенант, вивчаючи пахучі речовини, що їх виділяють самки
метеликів для приманки самців {статеві атрактанти), виділив із самок
шовкопряда ненасичений спирт бамбікол:
¥ ¥ ¥
СН3СН2СН2С=С~С=С(СН2)8СН2ОН
H
Самці відчувають його запах у надмалих концентраціях на величезних
відстанях від самок.
13.7.5. Ацетиленові спирти й етери
■ Ці речовини не набули важливого значення і вивчені мало. Найвідоміший
із них - пропаргіловий спирт НС=С~СН2ОН, який переважно одержують за
методом Реппе:
НС^СН + СН20 НС=ССН2ОН
Він має звичайну спиртову групу, і якщо її замістити, то пропаргіловий
спирт здатний до алільного перегрупування. Ацетиленовий атом Гідрогену
пропалгілового спирту, як і в ацетилені, може бути заміщений на метали,
зокрема, Арґентум і Купрум, або зазнавати гідроксиметилювання за Реппе до
бутиндіолу. Цей спирт у промисловості застосовують для одержання алілового
спирту, а також як розчинник поліамідів, ацетату целюлози й інших полімерів.
■ Алкілацетиленові етери вперше одержані А. Щукіною з вінілових етерів:
_ _ _ ^тт_ C2H5ONa тт^
CH2=CHOR + Вг2 CH2ÇHOR 2-J— HC^COR
Br Br
За В. Якобсом, їх одержують, діючи цинковим пилом у спирті на ацеталі
дибромоацетальдегіду з наступним відщепленням гідроген броміду від алкіл-
бромовінілового етеру:
Br2CHCH(OR)2c^i^5fï BrCH=CHOR + BrZnOR
C2H5ONa
BrCH=CHOR J-J— HC=COR
Ці речовини застосовують y дієновому синтезі, оскільки вони легко зазнають
конденсації з дієнами.
■ Спирти з подвійними і потрійними зв'язками вперше одержані учнем
А. Фаворського І. Назаровим конденсацією вінілацетилену з альдегідами і
кетонами за наявності порошкоподібного калій гідроксиду:
Бутенандт А. (Віиепапск А.) (1903-1996) - німецький хімік. Головні праці присвячені хімії
статевих гормонів. Нобелівська премія 1939 р.
Розділ 13. Спирти
371
(сн3)2с=о + н<х:сн=сн2 (сн3)2с|см:сн=сн2
он
2-Метилгекс-5-ен-3-ин-2-ол
(диметилвінілетинілкарбінол)
Диметилвінілетинілкарбінол легко зазнає полімеризування у прозору склу-
вату масу, яку застосовують як клей (карбінольний клей Назарова).
13.8. Гліколі
Досі ми розглядали лише властивості одноатомних спиртів, хоча, зазвичай,
акцентували на тому, як ці властивості змінюються за наявності інших замісників.
Тепер проаналізуємо найважливіші класи поліфункціональних сполук.
Функціональні групи цих класів настільки впливають на властивості один одного,
що виникають певні властивості, які не характерні для жодної з цих груп окремо,
а лише для комбінації. Першими розглянемо двохатомні спирти, або гліколі.
Зазначимо, що наявність двох гідроксильних груп біля одного атома
Карбону неможлива, оскільки такі сполуки внаслідок дегідратації перетво-
рюються в альдегіди або кетони:
І 0Н І „о | ОН, ||
—С-С-0Я—гг7г — С-Сч —С-С-С 777Г —С-С-С—
і н -н>° І чн і он" 2 1 6 і
Проте їхні похідні — ацеталі й ацилалі є реальними речовинами:
о-о. 3
СН-СН(ОС2Н5)2 Н3С-СН °
Ацеталь Ацилаль СН3
У звичайному стані сполуки стійкі, якщо дві й більше гідроксильних груп
розміщені біля різних атомів. До найважливіших багатоатомних спиртів
належать гліколі (дві гідроксильні групи), гліцероли (три гідроксили), інозити
(шість гідроксилів) і моносахариди з різною кількістю гідроксильних груп.
Моносахариди, окрім гідроксилів, мають ще альдегідну або кетонну групу.
Розглянемо спочатку двохатомні спирти.
Зокрема, 1,2-гліколі мають солодкий смак, звідси й назва цього класу
речовин. Цікаво, що зі збільшенням кількості гідроксильних груп солодкий смак
посилюється.
13.8.1. Фізичні властивості гліколів
Як і очікували з розгляду структури гліколів, що мають більше одного
центра для утворення водневого зв'язку, для них характерні високі температури
372
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
кипіння. Вже для найпростішого члена ряду - етиленгліколю температура
кипіння становить 197 °С. Нижчі гліколі змішуються з водою, і навіть гліколі,
що мають сім атомів Карбону, виявляють помітну розчинність у воді. Гліколі -
в'язкі рідини більшої густини, ніж одноатомні спирти.
Етиленгліколь застосовують як компонент антифризу через високу
температуру кипіння, низьку температуру замерзання і високу розчинність у
воді. Суміш 66,7 % етиленгліколю й 33,3 % води, яка є основою антифризу,
замерзає за -75 °С.
13.8.2. Методи синтезу гліколів
❖ Синтез Вюрца. Гліколі відкрив Ш. Вюрц дією арґентум ацетату на
віцинальні дигалогенопохідні з наступним омиленням утворених гліколевих
естерів оцтової кислоти:
íH>Br + 2CH3COOAg — ?Н2°СОСНз _2на їН2°Н + 2СН3СООН
СН2Вг 3 - 2AgBr сн2ососн3 СН2ОН 3
Цей спосіб одержання гліколів найширше застосовували донедавна. Правда,
тепер замість арґентум ацетату використовують дешевший, однак повністю
придатний калій ацетат. Можлива також пряма заміна галогенів гідроксильними
групами дією слабких основ, наприклад, розчину соди, та цей процес дає погані
виходи внаслідок побічних реакцій.
❖ Гідроксилювання алкенів
ОН
_і і кмпо4 _¿==(J;_ rco2qh _¿_¿_ н?о,н; ¿_¿_
но он н*° " " V" но 1
Приклад:
СН2=СН2 ^qoc8" QH2—£Н2 -LJ— (^Н2—Cj!H2
О ОН ОН
Різні способи гідроксилювання алкенів детально розглянуті вище.
Нагадаємо лише, що окиснення алкенів розбавленим розчином калій
перманганату {реакція Вагнера) дає г/г/с-1,2-гліколі, так само, як і окиснення
гідроген пероксидом за наявності осмій (VIII) оксиду {реагент Майлса).
Гідратація гомологів оксиду етилену в кислому середовищі відбувається за
механізмом Sn2, унаслідок чого епоксидне кільце розщеплюється, утворюючи
транс-ізомер. Зазначимо, що для одержання вихідних синтонів - епоксидів,
окрім традиційних методів - реакції Прилежаева або окиснення алкенів над
срібним каталізатором - використовують галогеногідрини:
Розділ 13. Спирти
373
сн2он-сн2сі -ЙЙ*- н2с—сн2
❖ Гідроліз галогеногідринів
X он
И^СОз
НО ОН
Приклад:
СН2-СН2 — с^н2
СІ ОН
2 ГП2 Н,0
(рн2 сн2
ОН ОН
❖ Бімолекулярне відновлення карбонільних сполук. Класичним методом
одержання симетричних 1,2-діолів є відновлювальна димеризація кетонів. Для
цього як відновники застосовують магній і цинк. На першій стадії дві молекули
кетонів приєднують по одному електрону. Такі аніоноподібні частинки
називають кетгілами. Вони зазнають димеризації, потім утворюють алкоголят,
який розщеплюють водою до пінаколу. Тому реакцію називають пінаколовим
відновленням:
Н,
СН,
СН,С +ССН,
3ІІ II 3
о о
Н3С
сн
<рн3
з9* * 9й 131
ч я
н3с сн.
.сн3с-ссн
ЧЯ
м8
3 що ^
3-і^2СНз"С~С~СН'
І І
но он
❖ Синтез 1,3-діолів за реакцією Прінса. Формальдегід у кислому сере-
довищі приєднується до алкенів, утворюючи циклічний ацеталь і 1,3-діол:
(СН3)2С=СН2 + НСНО н2'Я°с Я + (снз)г(г"СН,СН'0Н
2-Метилпропен
н3сч
Нзс/ О-/" ОН
4,4-Диметил-1,3-діоксан 3-Метилбутан-1,3-діол
❖ Відновлення естерів двохосновних кислот. 1,3-Діоли і сполуки з відда-
ленішими один від одного гідроксилами одержують відновленням естерів вищих
двохосновних кислот за методом Буво-Блана або літій алюмогідридом. Дикар-
бонільні сполуки добре відновлює натрій борогідрид, наприклад:
С2Н5ООС(СН2)пСООС2Н5-^^ НОСН2(СН2)пСН2ОН
СНз^(СН2)зССНз СН3^Н(СН2)3СНСН3
оо 25 он он
Гептан-2,6-діон Гептан-2,6-діол
374
Чирва В.Я., Ярмолюк CM., Толкачова H.В., Зеллляков О.Є. Органічна хімія
❖ Гідруванням бутин-1,4-діолу у промисловості одержують бутан-1,4-діол
- важливий продукт, який є проміжною сполукою у виробництві бутадієну й
далі синтетичного каучуку. Вихідний бутин-1,4-діол легко синтезують з
ацетилену і формальдегіду за реакцією Реппе.
13.83. Хімічні властивості гліколів
Як і одноатомні спирти, гліколі можуть мати первинні, вторинні й третинні
гідроксили і все, що сказано про властивості одноатомних спиртів, стосується
також гліколів.
❖ Дегідратація гліколів. Аналогічно до спиртів, діоли здатні де гідра-
туватись залежно від їхньої будови й умов перебігу реакцій.
• Внутрішньомолекулярна дегідратація до спряжених дієнів. Цей тип
реакції легко відбувається для гліколів з гідроксильними групами біля
третинних і вторинних атомів Карбону.
(СНЛС-^СНз), .ОЩк. н^-С-СН,
он он сн3сн3
2,3-Диметилбутан-2,3-діол 2,3-Диметилбут-1,3-дієн, 62%
Ефективніше дегідратація відбувається на алюміній оксиді з виходом від 70
до 85 %.
Якщо нагрівати етиленгліколь з цинк хлоридом, то відбувається де гідра-
тація з утворенням ацетальдегіду: р
СН2ОН-СН2ОН [СН2=СНОН] —- СН3СНО
• Внутрішньомолекулярна дегідратація, що супроводжується піна коліно-
вам перегрупуванням. Під дією мінеральних кислот 2,3-диметилбутан-2,3-діол
[пінакол) перегруповується і перетворюється в метил-иарет-бутилкетон
(пінаколін):
Нз^ с:н3 срн3
СНз"~ СН3 СНз^ —С| —СН3
но он о сн3
Пінакол Пінаколін
Уважають, що пінаколінове перегрупування має дві важливі стадії.
Спочатку відбувається відщеплення води від протонованого гліколю з
утворенням карбокатіону (1), а потім - його перегрупування з 1,2-метильною
міграцією, унаслідок чого одержують протонований кетон (2):
Розділ 13. Спирти
375
Я-С-С-Я === Я-С-^-Я - Н20 + К-<р-<р-ІІ (1)
но он но он, но я
+ ^
Я-^-С-Я К-р- р- Я =3= ЯН^-Ср-К + Н+ (2)
НО Я НО+ Я о я
Під час 1,2-міграції до електронодефіцитного атома Карбону метильна
група не буває повністю "вільною", вона доти не відривається від
"материнського" атома Карбону, доки не утворить новий зв'язок з електроно-
дефіцитним центром (3):
НО
К8+
5+/' \6+ -|
-?-9-
НО
т
ї Т (3)
но+
• Дегідратація з утворенням циклічних етерів і епоксидів. Етиленгліколь і
ряд 1,2-гліколів здатні в разі нагрівання з концентрованою сульфатною
кислотою або 80 % фосфатною кислотою перетворюватись в 1,4-діоксани:
сн2он-сн2он ^ УСІІіСІІ%
ХСН2СН£ Діоксан, 55%
Зокрема, 1,4- і 1,5-діоли зазнають циклізування за наявності каталітичних
кількостей мінеральних кислот у тетрагідрофуран і тетрагідропіран. Ці реакції
вперше реалізовані О. Фаворським:
но^сн2)4-он2605Р8^4ат* ГП
О Тетрагідрофуран (ТГФ), 98%
Н0-(СН2)5-0Н 51%н^°і (^|
О Тетрагідропіран, 100%
Останнім часом тетрагідрофуран переважно одержують гідруванням фурану
за 80-140 °С, а каталізаторами слугують нікель і осмій або паладій.
• Міжмолекулярна дегідратація дає змогу одержати лінійні олігомерні етери.
Як звичайно, одержують суміш етерів:
2НОСН2-СН2ОН—^ НОСН2СН2ОСН2СН2ОН
Діетиленгліколь
З НОСН2-СН2ОН —- НОСН2СН2ОСН2СН2ОСН2СН2ОН
Триетиленгліколь
376
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
У промисловості ді- і триетиленгліколь одержують як побічний продукт у
синтезі етиленгліколю з оксиду етилену.
❖ Кислотні властивості гліколів. Завдяки існуванню другої гідроксильної
групи, яка виявляє -І-ефект, кислотність гліколів, особливо a-гліколів, вища
порівняно з одноатомними спиртами. Тому гліколі здатні реагувати не лише з
лугами, а й з гідроксидами важких металів, утворюючи комплексні сполуки. У
цих молекулах, окрім ковалентних зв'язків, існують донорно-акцепторні:
сн2он носн2 снь-ск ^-.он:^
І 2 + Cu(OH)2 + і 2 ^ І 2 ^с< Т
СН2ОН НОСН2 CH2~0\„ °^н2
❖ Утворення етерів і естерів. Гліколі зі спиртами за наявності міне-
ральних кислот утворюють повні й неповні етери. їх можна також синтезувати
дією галогеноалканів на гліколяти:
СН2ОН ^тт_т н+ СгірСД CHLONa 2NaBr CFLOQIL
І + ОДОН І І * +2СДОГ І ^* Н
СН2ОН ""2° СН2ОН CI^ONa СЦОСД
Еталцелозоль Діетиловий етер ети
Аналогічно до спиртів гліколі або гліколяти реагують з органічними
одноосновними кислотами або їхніми похідними, утворюючи естери:
CH2ONa н+ СН.ОСОСН,
І 2 + 2СН3СОС1 І 2 3
CH2ONa ~2NaCl СН2ОСОСН3
Діацетат етиленгліколю
З двоосновними кислотами етиленгліколь утворює складні лінійні полімери:
п НОСН2СН2ОН + п НООС(СН2),СООН -[СН2СН20С0(СН2)ХС00]-
Поліестер
■ 3 мінеральними оксигеновмісними кислотами гліколі утворюють естери
по одній або двох гідроксильних групах, наприклад, моно- і динітрогліколі:
HOCH2-CH2ON02 і 02NOCH2-CH2ON02.
■ Під дією лужних каталізаторів гліколі взаємодіють з ацетиленом,
утворюючи вінілові та дивінілові етери:
НОСН2СН2ОН+НСЕСН—Н2С=СНОСН2СН2ОН + н2с=сн-осн2сн2о-сн=сн2;
з акрилонітрилом, утворюючи моно- і біс-ціанетилові етери:
НОШ2О^ОН+Н2С=СН-Ш—- НООН^^
з епоксидами за 140-180 °С, утворюючи ди-, три- і полігліколі:
Розділ 13. Спирти
377
А
НОСН2СН2ОН+ н2с-сн2—- НОСН2СН2ОСН2СН2ОН щс СН2.
о о
—-HO-[CH2CH20]jH—- 3- НО-[СН2СН20]п+-зН
❖ Утворення галогеногідринів. Під дією гідрогенгалогенідних кислот
гліколі легко перетворюються в галогеногідрини. Наприклад, якщо нагрівати
пропіленгліколь з хлоридною кислотою, то виникає суміш хлоргідринів:
НОСН2СН(ОН)СН3 + HCl С1СН2СН(ОН)СН3 + НОСН2СН2С1СН3 + Н20
Зазначимо, що заміщення другої гідроксильної групи відбувається значно
важче. Для перетворення хлоргідринів у дигалогенопохідні реакційну суміш
необхідно обробити фосфор (V) хлоридом РСЬ або тіонілхлоридом SOCl2.
❖ Окиснення гліколів. Реакції окиснення гліколів можна розділити на дві
групи:
а) реакції окиснення зі збереженням карбонового ланцюга;
б) реакції окиснення ^wc-гліколів із розщепленням карбонового ланцюга.
■ Дією на етиленгліколь нітратної кислоти, залежно від її концентрації,
одержують гліоксаль або гліоксилову кислоту:
HNO,
НОСН2-СН2ОН —І
ОНС-СНО
Розв.
Гліоксаль
HN03
онс-соон
Гліоксилова кислота
■ Окиснення етиленгліколю гідроген пероксидом за наявності солей Феруму
(II) приводить до гліколевої кислоти НОСН2СООН, а окиснення калій
перманганатом - до оксалатної кислоти НООС-СООН.
■ Другий тип реакцій спричиняє розщеплення Карбон-Карбонового зв'язку.
Наприклад, обробляючи періодатною кислотою сполуки, що мають дві або
більше гідроксильних груп, розміщених біля сусідніх атомів Карбону (а-глікалі)9
окиснюються з розщепленням зв'язку Карбон-Карбон. Аналогічно відбувається
реакція і для відповідних а-гідроксикарбонільних і а-дикарбонільних сполук:
К^Н^НК1 + НІ04 ЯСНО +Я'СНО + НЮ3
НО ОН
К-С^-К/ +■■ НІ04 ЯСООН + Я'СООН
О О
Я-СрН-С-К1 + НІ04 ЯСНО + Я'СООН
но о
378 Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
ясн-сн-снк' + 2НЮ4—- ЯСНО + НСООН + Я'СНО
он он он
Жр-^НЯ' + НІ04 Я2СО + Я'СНО
но он
я<рнсн2<рня' + ніо4
он он
Ця реакція особливо важлива для визначення структури цукрів. Окиснення
періодатною кислотою супроводжується утворенням іодатної кислоти (НІОз),
яку якісно можна впізнати за білим осадом, який утворюється в разі додавання
арґентум нітрату. Оскільки реакція зазвичай кількісна, то можна одержати цінну
інформацію про структуру вихідних речовин.
Метод окиснення 1,2-гліколів періодатною кислотою, розроблений Л. Мала-
праде 1928 р., вдало доповнений їхнім окисненням плюмбум (IV) ацетатом, яке
запропонував 1931 р. Р. Кріге1. Якщо перша реакція відбувається лише у водних
середовищах, то другу можна проводити в бензині, оцтовій кислоті тощо.
Обидві реакції відбуваються через циклічні естери, утворені гліколем з
окиснювачем. Цим пояснюють той факт, що і/ис-гліколі руйнуються значно
швидше, ніж транс-ізомери. Пропонують такий механізм окиснення гліколів:
Я
Я
Н-<р-ОН + р^_с_сн\ -снзсоон Н-С-ОРЬ(ОСОСН3)
Н-С-ОН 1 и 3 +сн3соон, н-г-он
Т>і \ / Швидко І
Я'
я*
-сн3соон,
Повільно
н~с=0 и_Л_п^ ,0~с-сн,
+ рь(ососн3)2 ^2г0 н с^р(
н-с=о н-с-о о-с-СН3
Я1 Я1 о
Кінетичні вимірювання засвідчують, що утворення циклу є найповільнішою
стадією реакції.
❖ Одержання циклічних а це талів. Альдегіди в кислому середовищі
ацилюють 1,2-гліколі, утворюючи циклічні ацеталі:
Кріге Р. (Сі^ее Я.) (1902-1975) - німецький хімік. Автор механізму озонолізу. Досліджував
циклічні реакції та механізми перегрупувань. Отримав ті ж самі результати, що й Р. Вудворд та
А. Гофман (Нобелівська премія 1981 р.), та не встиг їх завчасно надрукувати. Є автором так
званого перегрупування Кріге.
Розділ 13. Спирти
379
сн2он н "н сяцУ 3
?н2он + ч » ?нгск
Якщо обробляти ацеталі кислотами, то можна регенерувати вихідні
речовини. У лужних умовах ацеталі досить стійкі.
1,3-Гліколі здатні реагувати подібно, даючи шестичленні циклічні ацеталі.
Необхідною умовою для перебігу реакцій ацилювання є наявність гідроксильних
груп у цисоїдній конфігурації.
13.8.4. Застосування гліколів
Із представників гліколів на найбільшу увагу заслуговує сам етиленгліколь,
а також низка сполук, що синтезовані на його основі.
■ Етиленгліколь - в'язка безбарвна рідина, солодка на смак, що сильно
знижує температуру замерзання води. Він досить гігроскопічний, тому його
застосовують для виготовлення друкарських фарб.
Одержувати етиленгліколь можна шляхом гідролізу 1,2-дибромоетану за
допомогою розведеного розчину калій гідроксиду, проте вихід цільового продукту
не перевищує 50 %, оскільки паралельно відбувається реакція дегідробромування
з виділенням вінілброміду. Тому ліпше 1,2-дибромоетан перевести спочатку в
ацетат, а потім виконати гідроліз ацетату до етиленгліколю:
^Н2Вг +2СН3С00К> ^Н2ОСОСНз НС1вСН3ОН ^Н20Н
СН2Вг "2КВг сн2ососн3 -2СНзсоон сн2он
Етилендіацетат 85 %
У промисловості етиленгліколь одержують з оксирану в разі нагрівання з водою:
Н2С ;СН2 + Н20 8А0°С » СН,ОН—СН2ОН
\ / 2 1 20атм 2 2
О
Нітратний естер етиленгліколю - динітроетиленгліколь - сильна вибухова
речовина, яка замінює нітрогліцерол.
Поліетери етиленгліколю широко застосовують у різних галузях промисловості
як плівкоутворювальні речовини для лаків і фарб, у виробництві пластмас і
особливо синтетичних волокон, наприклад, лавсану.
■ Серед похідних етиленгліколю одним з найважливіших є етиленхлорогідрин,
який використовують для різноманітних синтезів. На його основі готують новокаїн,
індиго і гірчичний газ (іприт) (С1СН2СН2)28. Назва іприт пов'язана з тим, що 1917 р.
німці застосували цей отруйний газ проти французів біля міста Іпр.
380 Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
З амоніаком і амінами хлоргідрин утворює аміноспирт етаноламін (коламін),
що є в складі гліколіпідів:
НОСН2СН2С1 + 2Ш3 -> НОСН2СН2КН2 + Т^СІ
Колам ін
Дією сильних лугів етиленхлорогідрин перетворюється в етиленоксид.
■ Діетиленгліколь - рідина, яку застосовують як розчинник і робочу рідину
гальмівних гідравлічних механізмів.
■ Діоксан - рідина, що змішується з водою у будь-яких співвідношеннях.
Зазвичай його одержують взаємодією етиленгліколю з дихлороетаном. Переважно
застосовують як розчинник, зокрема, для етерів целюлози, олій і жирів.
■ Бутандіол-1,4 - вихідна речовина для одержання бутадієну в країнах з
нестачею нафти, для синтезу тетрагідрофурану.
■ Тетрагідрофуран застосовують як апротонний розчинник природних і
синтетичних смол, естерів целюлози й ацетилену, для одержання бутадієну.
13.9. Багатоатомні спирти
З багатоатомних спиртів найпростішим і найважливішим представником є
гліцерол (пропантріол-1,2,3), відкритий 1779 р. відомим шведським хіміком
К. Шеєле1.
13.9.1. Одержання гліцеролу
❖ Гідроліз жирів. Гліцерол уперше отриманий гідролізом жирів, які є
естерами гліцеролу і вищих гомологів оцтової кислоти та їхніх олефінових
ізологів. У разі гідролізу жирів перегрітою парою гліцерол залишається в водному
розчині, який відділяють від шару розплавлених органічних кислот.
У процесі лужного гідролізу жирів одержують натрієві солі вищих кислот -
мила. Звідси процес гідролізу естерів під дією лугів одержав назву омилення.
Сьогодні цей метод є в основі промислового виробництва гліцеролу для
косметики і фармацевтики.
❖ Синтез на основі пропілену.
■ Перший синтез гліцеролу виконали Ш. Фрідель2 і М. Сільва 1872 р. за
такою схемою:
1 Шеєле К. (Scheele К) (1742-1786) - шведський хімік. Першим отримав низку органічних та
неорганічних сполук, у тому числі хлор, гліцерин, синільну кислоту, довів, що повітря є складним
утворенням.
2 Фрідель Ш. (Friedel С.) (1832-1899) - французький хімік. Виявив пінаколінове перегрупування й
алкілування ароматичних речовин спільно з Дж. Крафтсом.
Розділ 13. Спирти
381
сн2=снсн3 + сі2 - СНСНСН3 сн2-сн-сн2 сн2-сн-сн2
СІ СІ -неї сі сі сі он он он
■ Сьогодні гліцерол одержують з пропілену, що виділяється у процесі
крекінгу нафти:
сн3сн=сн2 тг^ сн2сн=с^ ^» сн2с№=сн2-^- а^-сн-сНг а^-сн-а^
а
он он а он он он он
|-^сн2-с^ирн2-^--- а^-сн-сНг
а она он он он
Недоліком цього методу є великий об'єм стічних вод, що містять мінеральні
солі й органічні речовини, які потребують дорогої утилізації, і непродуктивні
витрати хлору.
■ Спосіб без застосування хлору ґрунтується на окисненні пропілену до
акролеїну з наступним його відновленням до алілового спирту і гідрокси-
люванням цього спирту до гліцеролу:
*»» н т он онон
❖ Синтез на основі пропаргілового спирту. Внаслідок конденсації ацетилену
з формальдегідом утворюється пропаргіловий спирт, який селективним гідруванням
перетворюють в аліловий спирт. Потім за методом, що описаний у попередній
схемі, одержують гліцерол:
Деяка кількість гліцеролу утворюється під час бродіння цукрів за певних умов.
Дуже перспективний технічний синтез гліцеролу розробили Я. Береш і
Л.Якубович. Відновленням найпростішого ненасиченого альдегіду - акролеїну,
одержаного за умов кротонової конденсації за Оппенауером, синтезують аліловий
спирт, який перетворюють у гліцерол:
НСНО + СН3СНО—А НО(СН2)2СНО СН2=СНСНО"-^^-СН2=СНСН2ОН
СН2=СНСН2ОН + Н202 НОСН2С|НСН2ОН
он
Розроблені промислові способи одержання гліцеролу дали змогу зменшити
витрати харчових жирів для технічних потреб.
382
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
13.9.2. Хімічні властивості гліцеролу
❖ Кислотні властивості. Хімічні властивості гліцеролу зумовлені
взаємним впливом трьох гідроксильних груп; його кислотність вища, ніж
кислотність одно- і двохатомних спиртів. З цієї причини він утворює розчинні у
воді сполуки - гліцерати - не лише з лужними металами, а й з оксидами
металів, наприклад, кальцій оксидом:
❖ Естернфікація. Гліцерол, взаємодіючи з одноосновними органічними
кислотами, дає повні або неповні естери. З двоосновними карбоновими
кислотами утворюються поліестерні волокна і смоли, які застосовують для
лакофарбових або плівкоутворювальних композицій. У разі взаємодії гліцеролу
з неорганічними оксигеновмісними кислотами він утворює повні й неповні
естери, наприклад:
Повний нітратний естер гліцеролу — гліцеролтринітрат, який неправильно
називають нітрогліцеролом - вибухова речовина, яку застосовують у
виробництві для виготовлення динаміту. Після відкриття А. Нобелем1 динаміту
(суміш гліцеролтринітрату і кізельгуру) побудовано заводи для його
виробництва. Широке застосування динаміту в військовій справі та будівництві
давало величезні прибутки, тому А. Нобель зробив заповіт, згідно з яким
щорічно спеціальна комісія нагороджує учених за видатні роботи в галузі хімії,
фізики, медицини та літератури, а також у боротьбі за мир. Зазначимо, що сам
гліцеролтринітрат легко вибухає з виділенням газоподібних продуктів, а самого
оксигену достатньо для перебігу реакції, що дає змогу використовувати його без
доступу повітряГ
Розчин 1 % спирту під назвою нітрогліцерол - лікарський засіб.
❖ Реакції заміщення.
■ Дією хлоридної кислоти на гліцерол одержують суміш монохлоргідринів,
яка має більше а-монохлоргідрину СН2ОН-СНОН-СН2С1 і менше Р-ізомеру
Нобель А. (Nobel A.B.) (1833-1896) - шведський хімік та інженер, винахідник багатьох
вибухових речовин, насамперед динаміту. Засновник Нобелівської премії.
СН2ОН-СНОН-СН2ОН
HN03/H2S04
іо°с
CH2ON02-CHON02-CH2ON02 + З Н20
<pH2ON02
(pHON02-
CH2ON02
З С02 + 2,5 Н20 + 1,5 N2 + 0,25 02
Розділ 13. Спирти
383
СН2ОН-СНСІ-СН2ОН. Якщо обробляти лугом обидва ізомери, то утворюється
один і той же гліцидний спирт:
СЯ,СНСН,ОН
У
О Гліцидний спирт
За жорсткіших умов гліцерол з хлоридною кислотою утворює два
дихлорогліцероли: СН2С1-СНС1-СН2ОН і СН2С1-СНОН-СН2С1. Після обробки
їх лугом отримують епіхлоргідрин гліцеролу:
(^Н?СНСН,С1
О Епіхлорогідрин гліцеролу
Аналогічні результати одержують у разі взаємодії гліцеролу з бромідною
кислотою.
■ У реакції з гідроген годидом усі три гідроксили заміщуються на атоми
Іоду, проте утворений 1,2,3-триіодопропан далі відновлюється до 2-іодопропану:
СН2ОН-СНОН-СН2ОН шщ5 СН2І-СНІ-СН2І СН3-СШ-СН3
■ Дією хлоридів і бромідів Фосфору в гліцеролі на галоген заміщуються всі
три гідроксили:
СН2ОН-СНОН-СН2ОН + РВг3 СН2Вг-СНВг-СН2Вг,
а якщо гліцерол взаємодіє з іодом і фосфором, то утворюється 1,2,3-
триіодопропан, який далі розпадається до аліліодиду:
СН2ОН-СНОН-СН2ОН СН2І-СШ-СН2І ^ СН2=СН-СН2І
<г Дегідратація гліцеролу водовідділювальними реагентами в разі
нагрівання дає акролеїн:
сь^он-снон-с^он А^о > СН2ОН-СН=СНОН снрн-с^-сно-^^сНз^сн-сно
❖ Окиснення гліцеролу залежно від умов реакції й типу окиснювача дає
великий набір продуктів:
С^ОН-ОТО№(^ОН Жа^ОН-СНОН-СНО + ОН^НуО^ОН + СН2ОН-СНОН-СООН-^
Гліцероловий альдегід Дигідрошіацетон Гліцеролова кислота
— НООС-СНОН-СООН -Н. НОООС-СООН-^ НООС-СООН
II
Тартронова кислота О
Мезоксалева кислота Оксалатна кислота
384
Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
13.93. Застосування гліцеролу
Гліцерол широко використовують у харчовій і лікеро-горілчаній про-
мисловості, виробництві косметики і лікарських препаратів. Синтетичний
гліцерол застосовують у текстильній промисловості й поліграфії. Як вихідну
речовину його застосовують у виробництві вибухових речовин, антифризу,
гліфталевих смол і лаків.
13.9.4. Багатоатомні спирти
■ Еритришол (бутанЛ,2,3,4-тетраол) трапляється у вільному стані й у
вигляді естерів у водоростях і деяких плісенях. Синтетичний еритритол
одержують з бутадієну:
О О
II II
(рН=СН2 ВГ2> уНСН2Вг сн^сооАр (^НСНрССНз Вг2 ^ ВгСНСН2ОССН3
СН=СН2 СНСН2Вг СНСН2О^СН3 ВгСНСН2О^СН3
сн3сооа6 СН3СО(^НСН2ОССН3 _4Н£ гН-СН-СН-СН2
сн3^оснсн2оссн3 £н2 £н ін ін
■ Пентаеритритол одержують синтетичним способом взаємодією форм-
альдегіду з водним розчином ацетальдегіду в лужному середовищі:
СН3СНО + 4НСНО^с?2 С(СН2ОН)4 + НСООН
13.9.5. Жири та олії
Усі жири й олії - гліцериди, естери трьохатомного спирту гліцеролу і вищих
карбонових кислот, як звичайно, з парною кількістю атомів Карбону (від 12 і до
22). Найпоширеніші в жирах кислоти з 16 і 18 атомами Карбону. Трапляються
також карбонові кислоти від 4 до 10 атомів:
О О О
Н2С-0-С-* Н2С-0-С-(СН2)16СН3 Н2С-0-С-(СН2)14СН3
І 4.0 | Д) І „0
не—о-с' не—о-сґ не— о-с'
І К І Ч(СН2)16СН3 І Ч(СН2)7СН=СН(СН2)7СН3
Н2С-0-С-Г Н2С-0-С-(СН2)16СН3 Н2С-0-С-(СН2)16СН3
0 0 о
Загальна формула Тристеарат Змішаний тригліцерид
тригліцеридів
Розділ 13. Спирти
У складі жирів є як насичені кислоти, наприклад, пальмітинова С15Н31СООН
і стеаринова С17Н35СООН, так і ненасичені карбонові кислоти: олеїнова
С8Н17СН=СН(СН2)7СООН, лінолева СзНцССН^НСНгМСНг^СООН, ліноленова
С2Н5(СН=СНСН2)з(СН2)бСООН.
Залежно від консистенції жири поділяють на тверді (яловичий, баранячий і
свинячий) та рідкі, які називають оліями (соняшникова, лляна, бавовняна,
оливкова і кокосова). Як звичайно, тверді жири — це речовини тваринного
походження, а олії виділяють з рослин. Тверді жири містять зазвичай насичені
кислоти, а олії - ненасичені. Зазначимо, що консистенція жирів не завжди
відповідає характеру складових карбонових кислот. Наприклад, кокосова олія
містить переважно насичені кислоти.
Жири та олії людина використовує головно як харчові продукти. Вони є
необхідною і досить цінною складовою частиною їжі. З жирами організм
одержує значно більшу кількість енергії, ніж з такою ж кількістю протеїнів і
вуглеводів. Завдяки цьому жири в організмі слугують резервним джерелом
енергії. У шлунку під дією ферменту ліпази вони розкладаються на гліцерол і
органічні кислоти. Стінки кишківника всмоктують продукти гідролізу, з них в
організмі синтезуються нові жири. Додатково гліцерол одержують унаслідок
розщеплення вуглеводів.
Наголосимо, що окремі ненасичені кислоти синтезують лише рослини, тому
вони є незамінними компонентами їжі. Рослини необхідні для людини і тварин
як вихідний матеріал у синтезі простагландинів, нестача яких спричиняє
сповільнення росту, ураження шкіри, порушення функціонування нирок і
репродуктивних органів.
Додатковим джерелом жирів можуть бути рослинні олії, які в промисловості
за допомогою гідрогенізації переводять у тверду консистенцію. Синтетичні
тверді жири використовують у виробництві харчового продукту маргарину. Для
поліпшення смакових і поживних властивостей до нього додають яйця, молоко,
солі, а для отдушки - діацетил СН3СОСОСН3 і ацетоїн СН3СОСН(ОН)СНз.
Значну кількість рослинних олій використовують для виготовлення фарб і
лаків. Оскільки олії мають гліцериди кислот з двома і більше подвійними
зв'язками, то, окиснюючись, вони утворюють прозорі плівки внаслідок
руйнування кратних зв'язків. Такі олії одержали назву висихальних. Проте
висихання фарб - це не лише випаровування розчинника (скипидару), а й
хімічний процес: оліфу готують, проварюючи, наприклад, лляну олію з
невеликою кількістю оксидів мангану або плюмбуму. Під час цього
відбуваються двоякі процеси. Ізольовані подвійні зв'язки лінолевої і ліноленової
кислот стають спряженими, а продукти, що утворились, окиснюються за схемою
дієнів:
9 о
—с=с-с=с— +о2—- -с—с=с—с-
386
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
Далі пероксиди розпадаються, утворюючи вільні радикали:
і і
— 9—0=0—9— >
О- О'
які спричиняють полімеризацію за спряженими й ізольованими подвійними
зв'язками. Наприклад, тунгова олія, яка є основою знаменитого китайського
лаку, містить гліцерид елеостеаринової кислоти, що має три спряжені подвійні
зв'язки; вона полімеризується під впливом повітря швидше, ніж лляна олія.
Розділ 14
ЕТЕРИ
Етерами називають такі похідні спиртів, у яких атом Гідрогену гідроксиль-
ної групи заміщений на алкільний замісник.
14.1. Класифікація і номенклатура
14.1.1. Класифікація етерів
Ациклічні є тер и
Симетричні Змішані, або
НзС-О-СНз
Метоксиметйн
диметиловий
етер
несиметричні
-ОСН3
Циклічні етери
Епоксиди, Циклічні етери
або а-оксиди
О
Н9С—снсн*
\ /
о
/—\
о о
Метоксициклогексан 1>2.Епоксипропан Тетрагідрофуран і)4.Діоксан
метилциклогексшовии пропіленоксид Р шр ФУР ' Д
етер
14.1.2. Номенклатура 11)?АС
• Замісна номенклатура. За замісною номенклатурою в етерах типу ЯОЯ'
один вуглеводневий замісник розглядають як вуглеводень-основу, а інший - як
Я-оксизамісник. Назву таких замісників утворюють із назв вуглеводневих
замісників із додаванням закінчення -окси. Для алкоксигруп, що містять від
одного до чотирьох карбонових атомів, використовують скорочені назви:
СН3(СН2)40- СН2=СНСН2 СН30- СН3СНО- СНзСН2СНО-
О-
СН3
СН,
Пентилокси- Алілокси- Метокси-
Ізопропокси-
втор-Бутокси-
388
Чирва В.Я., Ярмолюк CM., Толкачова Н.В., Земляков О.Є. Органічна хімія
1 2 3
1 2
І 2
СН3СН2ОСН2СН2СН3
СНзСН2СН20СН=СН2
СН3СН2ОСН2СН2С1
Етоксипропан
Пропоксіетен
1 -Етокси-2-хлороетан
За основу беруть довший
За основу беруть ланцюг з
За основу беруть ланцюг з більшою
ланцюг.
подвійним зв'язком.
кількістю замісників. Початок ну-
мерації — від першого за алфавітом
замісника.
Етилпропіловий етер
Вінілпропіловий етер
Етил-2-хлороетиловий етер
■ Атом Оксигену, приєднаний до двох сусідніх атомів Карбону, позначають
префіксом -епокси, звідси назва подібних сполук "епоксиди". Циклічні етери
звичайно називають як гетероциклічні сполуки.
• Радикально-фунаціональна номенклатура. Назва симетричних етерів
охоплює назву вуглеводневого замісника у формі прикметника з додаванням
числівникового префікса ди- і назви функції етер. Для несиметричних етерів
назви вуглеводневих замісників записують за абеткою.
■ Епоксиди в радикально-функціональній номенклатурі називають, вико-
ристовуючи назви двовалентних вуглеводневих замісників і функції -оксид.
Приклади таких назв наведені вище курсивом.
14.2. Фізичні властивості етерів
^Загалом просторова структура молекул етерів подібна до
Д10° молекулярної будови спиртів і води. Кут між зв'язками С-О-С не
q д ^ Д°рівнює 180°, отже, дипольні моменти двох зв'язків С-0 не
компенсують один одного. Внаслідок цього етери мають невеликий
дипольний момент. Його наявність суттєво не впливає на температури кипіння
етерів, які близькі до температур кипіння алканів з такою ж молекулярною
масою й значно нижчі, ніж температури кипіння ізомерних спиртів. Порівняйте,
наприклад, температури кипіння н-гептану (98 °С) метил-н-пентилового етеру
(100 °С) і н-гексилового спирту (157 °С). У етерах, на відміну від спиртів, немає
міжмолекулярної асоціації завдяки утворенню водневих зв'язків.
Лише два перші представники етерів - диметиловий і етилметиловий - за
нормальних умов перебувають у газоподібному стані, інші всі - рідини або
тверді речовини. Питома густина етерів менша від питомої густини води.
Розчинність етерів і спиртів у воді приблизно однакова. Наприклад, діетиловий
етер і н-бутиловий спирт розчиняються у воді в кількості 8 г на 100 г води.
Розчинність нижчих спиртів у воді пояснюють утворенням водневих зв'язків між
молекулами спирту й води. Розчинність етерів у воді можна пояснити аналогічно:
R-C^-H-Ox
R Н
Розділ 14. Етери
389
Зокрема, 1,4-діоксан містить два етерні атоми Оксигену на чотири атоми
Карбону, тому він утворює вдвічі більшу кількість водневих зв'язків з водою й
може змішуватися з нею в будь-яких співвідношеннях. Необмежено змішується
з водою і тетрагідрофуран, оскільки завдяки циклічній структурі молекули
неподілена електронна пара Оксисену доступніша для атаки молекулою води
порівняно з діетиловим етером.
Розчинність етерів у протонних кислотах зумовлена головними властивостя-
ми атома Оксигену. Як і у випадку атома Нітрогену в амоніаку, атом Оксисену
виявляє основні властивості внаслідок наявності в його електронній оболонці
двох вакантних електронних пар (в атома азоту одна така пара електронів). Ці
електрони не можуть брати участі у формуванні нових зв'язків, оскільки в
молекулі етеру атом Оксигену вже має вісім електронів (октет). Тому атом
Оксигену не може прийняти електрони, проте здатний односторонньо надати
одну пару електронів у спільне володіння для утворення зв'язку, наприклад, із
протоном:
(С2Н5)20 +Н+^— [(С2Н5)2ОН]+
14.2.1. Спектроскопія етерів
• УФ-спектроскопія. Етерний зв'язок не дає характерних смуг коливання у
видимій УФ-ділянці спектра (А^ах 180-190 нм).
• ІЧ-спектроскопія. В ІЧ-спектрах спостерігають дуже сильне поглинання
валентних асиметричних коливань vc.0 1 150-1 050 см"1.
• Мас-спектрометрія. У мас-спектрах етерів інтенсивність молекулярного
йона здебільшого мала. Фрагментація під дією електронного удару починається
з видалення електрона з неподіленої електронної пари атома Оксигену. Потім
відбувається фрагментація з утворенням радикалів -ОСН3 (m/z 31), -ОС2Н5 (m/z
45), -ОС3Н7 (m/z 59) тощо. Подальше розщеплення спричиняє розрив а-С-С-
зв'язку. Зазначимо, що радикали відщеплюються перевалено від найбільш
заміщених атомів Карбону.
• Спектроскопія ПМР. Сигнали протонів у ряді -ОСН3, -ОСН2СН3 й -
ОСН(СНз)2 у ПМР-спектрах зсуваються у бік слабшого поля й виявляються в
діапазоні 3,4-3,6 м.ч. Значення хімічних зсувів протонів у типових циклічних
етерах такі:
5 2,54 м.ч.
5 3,63 (а), 1,79 (Ь)м.ч.
5 3,59 м.ч.
390
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
14.3. Одержання етерів
Деякі симетричні етери з невеликими алкільними групами використовують
як розчинники, їх синтезують у промисловості. Найважливішим з них є
діетиловий етер, відомий з часів алхімії як сірчаний. Раніше його застосовували
як наркотичну речовину і як розчинник для екстракції, а також для синтезу
реактиву Гриньяра. Крім діетилового етеру, у промисловості синтезують
діізопропіловий і ди-н-бутиловий етери.
❖ Міжмолекулярна дегідратація спиртів. Утворення етерів, а не алкенів
у процесі дегідратації спиртів досягають вибором відповідних умов реакцій.
Наприклад, етилен одержують нагріванням етилового спирту з концентрованою
сульфатною кислотою за 180 °С; діетиловий етер утворюється в разі нагрівання
цієї суміші до 140 °С, причому в реакційну суміш увесь час додають спирт, щоб
він був у надлишку.
Утворення етеру під час дегідратації є прикладом реакції нуклеофільного
замігцення, у якій протонований спирт діє як субстрат, а друга молекула спирту - як
нуклеофіл. Теоретично реакція може відбуватися за механізмом як Бці, так і 8н2
залежно від того, елімінує протонований спирт молекулу води раніше чи одночасно
з атакою другої молекули спирту. Вторинні й третинні спирти реагують за
механізмом вмі (1). Однак варто пам'ятати, що за цих умов третинні спирти
перетворюються в алкени. Первинні спирти реагують за механізмом (2):
^ ЯОЯ + Н+
Я-О-Я ^ ЬЮІІ + НзО
Наприклад, з н-бутилового спирту утворюється ди-н-бутиловий етер, тобто
реакція відбувається без перегрупування й, отже, переважно без утворення
проміжних карбокатіонів. На відміну від інших вторинних і третинних спиртів
ізопропіловий спирт за умов кислотного каталізу дає непоганий вихід
діізопропілового етеру:
2(СНз)2СНОН-^^(СНз)2СН-0-СН(СН3)2
Діізопропіловий етер
У реакції міжмолекулярної дегідратації спиртів зазвичай одержують
симетричні етери, оскільки у випадку використання двох різних спиртів утвориться
суміш трьох етерів: И-О-ІІ, К-О-Я' й И'-О-Я'. Крім того, дегідратацію головно
застосовують для первинних нижчих спиртів, оскільки вищі спирти за цих умов,
як звичайно, перетворюються в алкени. Вторинні й третинні спирти, замість
очікуваних етерів, також переважно дають алкени.
яон2-Г -н2°
+
(2)^ ЯОН2 + ІЮН
Розділ 14. Етери
391
Однак, змінюючи умови синтезу й співвідношення реагентів, таки можна
цілеспрямовано одержувати змішані етери. Відомо, що первинні спирти
залежно від молекулярної маси здатні в інтервалі температур від кімнатної до
100 °С утворювати з надлишком сульфатної кислоти моноалкілсульфати:
чС2Н5ОН + HOS02OH -> C2H5OS02OH + н2о
Якщо далі температуру реакції підвищити до 140 °С і додавати надлишок
іншого найпростішого первинного спирту, то вдається синтезувати змішаний
етер:
C2H5OS02OH + СН3ОН « С2Н5ОСН3 + H2S04
■ Крім того, з алілового спирту в суміші з яким-небудь іншим спиртом за
наявності тетрахлороплатинової (II) кислоти селективно утворюються алкіла-
лілові етери.
Н PtCl
СН2=СН-СН2ОН + R-OH CH2=CH-CH2OR
■ Під час пропускання етанолу над алюміній оксидом (дегідратувальний
каталізатор), нагрітий до 375 °С, утворюються вода й етилен, однак за 250 °С
дегідратація дає етер:
2 С2Н^ОН 250 °сГ ^2^5^^2^5
❖ Синтез Вільямсона. Незважаючи на те, що реакція виявлена ще 1852 p.,
вона і досі є головним методом одержання етерів. У лабораторії синтез
Вільямсона сьогодні особливо важливий через його багатосторонність: його
можна використати для синтезу як змішаних, так і симетричних етерів. У синтезі
Вільямсона алкілгалогенід реагує з натрій алкоголятам за такою схемою:
R-X + Na+OR"-^ R-0-R^eX=Cl,Br,I.
Реакція є нуклеофільним заміщенням іона галогену на алкоксид-аніон,
наприклад:
(j:h3 ^Н3
CH^r + NaO^CHj СНзО^СНз
СН3 СН3
Треба пам'ятати, що під час синтезу змішаних етерів можливі дві
комбінації, причому одна з них майже завжди ліпша. Наприклад, для трет-
бутилетилового етеру можливі два варіанти синтезу:
392
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
9нз
СН3СН2Вг + КаО(|:СНз
сн3
СНзСН2ОИа + ВгС(СН3)3
Який же варіант вибрати? Як завжди, варто враховувати можливість реакції
елімінування. Вона повинна бути найімовірніша внаслідок сильної основності
алкоголят-іона. Тому третинний алкілгалогенід, що дає лише або головно
продукт елімінування, не застосовують. Варто використати іншу комбінацію. Як
відомо, натрій повільно реагує з третя-бутиловим спиртом. Однак ця
незручність сповна компенсована властивістю первинних галогеноалканів
вступати в реакцію заміщення, а не елімінування:
У випадку (1) є заміщення, а у випадку (2) - елімінування. Останнього
процесу можна уникнути, якщо третинні галогеноалкани етерифікувати у
слабкоосновних умовах, наприклад, у піридині або з використанням меркурій
перхлорату. Під час планування синтезу етеру за Вільямсоном треба пам'ятати,
що тенденція галогеноалканів вступати в реакцію дегідрогалогенування
зменшується в такому порядку: третинний > вторинний > первинний гало-
геноалкан.
За умов застосовування алкілтозилатів синтез симетричних і змішаних
етерів відбувається досить чітко і без ускладнень:
Я-0"Ка++ Н3С-С6НГ8020К' Я-О-К' + СН3С6Н480; №+
❖ Синтез на основі алкенів. Спирти здатні приєднуватися до олефінів. Як
каталізатори використовують бор (III) флуорид або неорганічні кислоти. Таким
способом у промисловості синтезують діізопропіловий етер, приєднуючи
пропан-2-ол до прбпену:
СН3СН2Вг + -09СН3
сн3
СН3С=СН2 + С2Н5ОН + сі- (2)
(СН3)2СНОН + СН3СН=СН2
Мі
(СН3)2СНОСН(СН3)2
Метод також придатний для синтезу в промисловості несиметричних етерів:
Розділ 14. Етери
393
Н2С=СН-СН3+ СН3СН2ОН н3с-СН-СН3
о-с2н5
Етилізопропіловий етер
❖ Алкоксимеркурування алкенів. Дей спосіб дає змогу одержувати зміша-
ні етери з високими виходами:
СН3(СН2)5-СН=СН2 2.2£^^н сн3(сн2)5-сн-сн3
Окт-1-ен ' ОС2Н5
2-Етоксиоктан, 92%
Реакція, очевидно, починається з атаки катіоном "І^ОСОСНз подвійного
зв'язку алкену, що веде до утворення інтермедіату - меркурієвого катіона, який
потім розкривається внаслідок нуклеофільної атаки спирту з тилу найбільше
заміщеного атома Карбону:
К-СН=СН2 + Н§ОСОСН3
я-сн—СН,
С^ОН р_Гтт_Гтт
снхоо- *авн4 Кі"„ 3
ОС2Н5
Для синтезу етерів, що мають вторинний або третинний алкіл, варто
використовувати меркурій (І) трифлуороацетат.
сн3(сн2)3сн=сн2 + (сн3)3сон Нб2(^СРз)>2 СН3(СН2)3СНСН3
Гекс-1-ен 4 ОС(СН3)3
2-/и/?е/и-Бутоксигексан
Цей найпоширеніший спосіб одержання етерів нагадує гідратацію алкенів за
правилом Марковникова шляхом гідроксимеркурування-демеркурування, а
відмінність полягає в тому, що реакція відбувається в спирті, а не у воді.
❖ Метилювання спиртів. Алкілметилові етери одеряеують взаємодією
спиртів з діазометаном за наявності кислот Льюїса - ВБз або АІСІз, наприклад:
ІІ-ОН ^2ї Я-О-АІСІз Я-О-АІСІз ^ Я-ОСН3
н 2 сн3
14.4. Хімічні властивості етерів
Етери - нейтральні й малоактивні сполуки, тому їх часто використовують у
різноманітних органічних реакціях як розчинники. Оскільки вони здебільшого
не реагують з натрієм, то цей метал застосовують для висушування етерів. На
394
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
них не діють розведені мінеральні кислоти, луги. Етерів не розщеплюють
металоорганічні сполуки, гідриди й аміди лужних металів.
Деякі хімічні властивості цих сполук пов'язані з наявністю вакантної
електронної пари біля атома Оксигену, що надає етерам основні властивості, а
також з наявністю полярних зв'язків С-О, розрив одного з яких спричиняє
розщеплення етерів.
❖ Утворення солей оксонію. Незважаючи на те, що етери є слабкими
основами й слабкими нуклеофілами, вони здатні взаємодіяти з сухим гідроген
хлоридом і утворювати солі діалкілгідроксонію:
(С2Н5)20 + HCl -> (С2Н5)2ОН+СГ
Утворена сполука як сіль слабкої основи легко гідролізується при розбавленні
водою:
(С2н5)2ОНҐСҐ + Н20 -+ (С2Н5)20 + HCl
Про основність етерів свідчить їхня розчинність у концентрованій сульфатній
кислоті з утворенням за низької температури кристалічних оксонієвих солей:
с2н5ос2н5 + h2so4 —- [с2н5ос2н^]+ hso;
Цю реакцію застосовують, щоб відділити етери від алканів і галогеноалканів.
■ У 1928 р. X. Мейєрвейн відкрив третинні оксонієві солі, які можна
одержати з етерів унаслідок такої реакції:
(СН3)20 +CH3F + BF3 — ^3/6-СНз
У цій реакції галогеніди бору відщеплюють атом галогену від галогеноалкану і
зв'язують його в міцний аніон з наступним утворенням третинної оксонієвої солі.
Триалкілоксонієві сполуки з комплексними аніонами - тверді, цілком стабільні,
солеподібні. Під час спроби замінити складний аніон BF4" у цих солях на аніони
звичайної кислоти або на ОНГ групу оксонієві солі розпадаються, утворюючи етер і
галогеноалкани. Тому триалкілоксонієві солі є сильними алкілувальними засобами,
значно ефективнішими порівняно з галогеноалканами і діалкілсульфатами.
Діетиловий етер використовують як розчинник у реакції Гриньяра, оскільки
він здатний сольватувати й, отже, розчиняти реагент Гриньяра. Він діє як основа
на атом Магнію:
С2Н5^0^С2Н5
C6h5Br + Mg + 2С2Н5ОС2Н5 С6Н— Mg—Вг
С2Н5—Ö^c2h5
Розділ 14. Етери
395
Діетиловий етер у цій реакції можна замінити на тетрагідрофуран.
До речі, реактиви Гриньяра можна також одержувати з високим виходом у
бензені за наявності триетиламіну як основи. Для цього потрібно лише один
моль основи на один моль галогеноалкану.
■ Як основи Льюїса, етери здатні утворювати комплекси з галогенами, у
яких етер відіграє роль донора електронів, а галоген - акцептора. Наприклад,
розчин іоду в діетиловому етері має коричневий колір, на відміну від
фіолетового кольору в інертних розчинах. Такі комплекси одержали назву
комплексів із перенесенням заряду (КПЗ):
❖ Розщеплення етерів. Етери в разі нагрівання до 140 °С з концентро-
ваними кислотами (Н2804, НВг й, особливо, НІ) здатні розщеплюватися. Цю
реакцію відкрив О. Бутлеров 1861 р. на прикладі 2-етоксипропанової кислоти:
Під впливом іодидної кислоти етер спочатку перетворюється в діалкіл-
гідроксоній іодид. Це збільшує полярність зв'язків С-О і полегшує гетеро-
літичне розщеплення одного з них, завдяки чому утворюється гарна відхідна
група у вигляді молекули спирту, функцію нуклеофіла виконує іодид-іон:
У випадку розщеплення метил- і етилалкілових етерів дія нуклеофіла
спрямована на просторово доступніший алкіл. На цій особливості ґрунтується
метод Цейзеля1 — кількісне визначення метокси- й етоксигруп в органічних
сполуках. Зазначимо таке, якщо один з алкілів третинний, то розщеплення
відбувається особливо легко. Реакція протонованого етеру з іоном галогену, як і
відповідна реакція протонованого спирту, може відбуватися як за 8м 1-, так і 8н2-
механізмами залежно від будови етеру. Треба обґрунтовано сподіватися, що
первинна алкільна група має тенденцію до 8к2-, тоді як третинна - до 8м1-
заміщення.
Н3С-СН-СООН + НІ Н3ОСН-СООН
ОС2Н5 ОН
+
СН3ОСН2СН3 + ні — н3с-о-сн2сн3 — СН3І + С2Н5ОН
1 Цейзель С. ^єібєі Б.) (1854—1933) - австрійський хімік. Займався фармацевтичною хімією,
досліджував алкалоїди.
396
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
Н
sN1: (1) r-o-r' повільно> r++hqr,
(2) r++x" 4I*M™rx
s*T2: r-ó-r'+x" гх- r..o-R4 —- rx+hor'
i i
h h
❖ Реакції з атомом Гідрогену в оь-попоженні. Наявність в етерах атома
Оксигену позначається на хімічній поведінці атомів Гідрогену, особливо тих, що
перебувають в a-положенні. Таку регіоселективність пояснюють стабільністю
радикала r-ch-ó-r, де неспарений електрон 2р-орбіталі атома Карбону
перекривається з неподіленою парою 2р-електронів атома Оксигену. Най-
ефективніше й вибірково відбуваються вільнорадикальні реакції хлорування
етерів. Наприклад, під час обробки діетилового етеру розрахованою кількістю
хлору на світлі одержують а-монохлоридні похідні етерів:
СН3СН2ОСН2СН3 + С12 ^ СН3СНОСН2СН3 + НС1
СІ
Діетиловий етер - .
v Етил-1-хлороетиловии етер
Швидкість реакцій а-хлорозаміщених етерів на багато порядків вища
порівняно з реакціями відповідних галогеноалканів. Зокрема, а-хлоропохідні
етерів надзвичайно легко вступають у реакції нуклеофільного заміщення,
оскільки як проміжна сполука виникає стійкий карбокатіон (механізм SnI). Ця
стійкість відображена такими резонансними структурами:
СН3СН-0-СН2СН3 СН3СН=ОСН2СН3
Подібні реакції широко використовують в органічному синтезі:
KCN CHXOONa
н3с-сн-осн2сн3 -— сн3снсюсн2сн3—^ н3с-сн-осн2сн3
cn І Nal ОСОСН3
СН3СНІОСН2СН3
Примітно, що, змінюючи умови перебігу реакції, її можна спрямувати на
дегідрогалогенування з одержанням вінілових етерів:
СН3СНС10СН2СН3 §^ СН2=СН-ОСН2СН3
Етил-1-хлороетиловий етер Вінілетиловий етер
• Реакції аутоокиснення. Етери схильні до реакцій аутоокиснення на повітрі за
радикальним механізмом навіть без опромінення, що пояснюють стабільністю
Розділ 14. Етери
397
вільного радикала, який утворюється. Стабілізація відбувається через делокалізацію
неспареного електрона Карбону з електронною парою сусіднього атома Оксигену:
Зародження ланцюга
Х'+ СН3СН2ОСН2СН3—» НХ + СН3СНОСН2СН3
Подовження ланцюга
СН3СН0СН2СН3 + о2—- н3с-сн-осн2сн3
о-о-
н3с-сн-осн2сн3 + сн3сн2осн2сн3 — н3с-сн-осн2сн3 + СН3СНОСН2СН3
о-о- ООН
Особливо легко аутоокиснення зазнають етери, що містять атом Гідрогену
біля третинного атома Карбону. Гідропероксиди, що спонтанно утворюються в
разі довготривалого зберігання етерів, винятково вибухонебезпечні. Оскільки
вони менш леткі порівняно з вихідними етерами, то не відганяються разом з
етерами, а накопичуються в реакційній суміші. З цієї причини етери не можна
відганяти до сухого, бо може статися вибух. Гідропероксиди потрібно ретельно
вилучати з етеру за допомогою відновників - солей Феруму (II) або Стануму (II).
Наявність пероксидів виявляють обробкою проби етеру водним розчином
калій іодиду. Поява характерного коричневого забарвлення, а за наявності
крохмалю - синього кольору свідчать про наявність гідропероксидів.
14.5. а-оксиди
З циклічних етерів найцікавіші властивості мають тричленні цикли, які
називають а-оксидамщ або епоксидами.
14.5.1. Одержання а-оксидів
З органічних оксидів практичне значення мають етиленоксид і пропіле-
ноксид. Для їхнього синтезу є два головні методи: дегідрогалогенування гало-
геногідринів і окиснення алкенів.
❖ Одержання а-оксидів з галогеногідринів виконують за наявності сильних
основ:
С1СН2СН2ОН ^сі^ 0*2~>СН2
о'
2-Хлороетанол Епоксиетан
Вихідний етиленхлорогідрин одержують з етилену й гіпохлоритної кислоти:
СН2=СН2 (о^+^Ь^о) ^^2^~^^2^^
398
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
По суті, реакція дегідрогалогенування - це внутрішньомолекулярний синтез
етерів за Вільямсоном, де оксид-аніон, що утворюється під дією основи, відіграє
роль нуклеофільного агента, а атом галогену - відхідної групи:
Н(\
ртт ОН'
^ -Н.0
"(О
СНоСН-5
СІ
'¿1
н
■ сг
ш2 сн2
Необхідною умовою цієї реакції є трянс-розташування гідроксилу й атома
Хлору, що дає змогу атомові Оксигену атакувати атом Карбону з тилу.
❖ Окиснення алкенів уважають найперспективнішим методом синтезу
циклічних етерів етилену й пропілену. Як окиснювач застосовують молеку-
лярний кисень або надкислоти (реакція Прилєжаєва, 1909 р.).
Окиснення алкенів надкислотами відбувається стереоспецифічно: із транс-
алкенів одержують трянс-епоксиди, із г/ис-алкенів - г/ис-епоксиди:
н3сх
н
н3с
А
і
н
с
II
О:
Н
Чис-Бут-2-ен
н3сч
н
і?
А
:0
і
Н
с
II
О:
Н СН3
транс-Буг-2-єн
н н й
і/ис-2,3 -Епоксибутан
он
НчД/СН3+а_с_0Н
н о
н3с
т/?ш/с-2,3-Епоксибутан
14.5.2. Хімічні властивості епоксидів
Епоксидне кільце має форму правильного трикутника, тому валентний кут
С-О-С значно деформований. З цієї причини в епоксидах існує не повне, а лише
часткове перекривання атомних орбіталей Карбону й Оксигену. Тому епоксидне
кільце нестійке і легко розкривається. Подібного деформування валентних кутів
немає в циклічних етерах з більшою кількістю атомів Карбону, наприклад, у
таких сполуках, як тетрагідрофуран і тетрагідропіран. Тому тетрагідрофурац і
тетрагідропіран значно менш реакційноздатні порівняно з епоксидами.
Для органічних епоксидів характерні реакції приєднання з розщепленням
кільця за механізмами 8ц1 та 8ц2. Загальна схема показана на прикладі
етиленоксиду. Протон зі сполуки НХ приєднується до атома Оксигену, а
нуклеофільна складова молекули НХ - до електрофільного атома Карбону:
Розділ 14. Етери
399
8-0,
НОСН2СН2Х
За кислотного каталізу розщеплення циклу відбувається за механізмом як
8ц1, так і 8к2, а за лужного - тільки за в^-механізмом.
■ Загальна схема розщеплення епоксидного кільця за кислотного каталізу
виглядає так:
о
+ н-
но
т/?я//с-Приєднання
но
т/?ш/с-Приєднання
но X
і/мс-При єднання
Визначити 8н1 механізм реакції допомагає стереохімічна спрямованість
синтезу. Відсутність стереоспецифічності свідчить про утворення проміжного
карбокатіону. Зазначимо, що за наявності в оксирановому циклі замісників
реалізація механізму 8м1 відбувається через найстійкіший карбокатіон.
СНз
•НОСНрС-СНз
сод
2-Етокси-2-метилпропан-1 -ол
яр.
\ /
о
с, + н+-
СНз
,СНз
0+ СНз
Н
+ '*~^ с нон
^ ОН СНз 'Н
1,1-Диметилепоксиетан
Інакше кажучи, нуклеофільна атака за Бмі механізмом спрямована на найбільш
заміщений атом Карбону.
■ Механізм лужного каталізу має такий вигляд:
СН30
/СН3
\
О
СН,
СН3ОСН2-
СН3
-ср-сн
:ОГ
СНзОН^
3 -сн3о-
СН3
сн,осн-с-сн
1,1 -Диметилепоксиетан
он
2-Метил-1 -метоксипропан-2-ол
Метоксид-аніон атакує менш заміщений атом Карбону, оскільки він має
більший позитивний заряд порівняно з незаміщеним. Річ у тому, що заряд на
сусідньому атомі Карбону делокалізований за допомогою +І-ефекту двох
метильних груп, які можуть слугувати, крім того, стеричним блокуванням
підходу метоксид-аніона. Тому приєднання нуклеофіла відбуватиметься лише з
протилежного боку щодо атома Оксигену за механізмом 8м2.
400
Чирва В.Я., Ярмолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
■ Етиленоксид - один з найважливіших синтонів в органічній хімії. З нього
можна одержувати етанол, етиленгліколь, гідроксипропаннітрил, оцтовий альде-
гід, етаноламін, діоксан тощо.
Останніми роками в лабораторній практиці й техніці важливого значення
набули краун-етери (від англ. crown - корона). Вони є макрогетероциклічними
сполуками, що містять у циклі 12 і більше атомів, з яких не менше чотирьох -
гетероатоми, зв'язані між собою метиленовими містками. У тривіальних назвах
загальна кількість атомів у циклі й кількість гетероатомів позначають цифрами,
які ставлять перед і після слова краун, наприклад:
Найпростіші краун-етери одержують, конденсуючи дигалогеніди з
діаніонами діолів за наявності лугів:
Вихід краун-етерів суттєво залежить від катіона основи. Він відіграє
вирішальну роль у розмірі циклу, що визначений координаційним числом
катіонів. Координаційне число калію стосовно оксигеновмісних лігандів
дорівнює шести, тому катіон калію найефективніший у разі синтезу 18-краун-6-
етерів. Координаційне число катіона літію дорівнює чотирьом, тому він
найефективніший у випадку синтезу краун-етерів з тією ж кількістю гетеро-
атомів. Вплив катіона на розмір циклу, що утворюється, одержав назву
матричного ефекту, що зумовлено відповідністю розмірів атомних радіусів
катіонів і внутрішніх порожнин макроциклів.
Якщо один або кілька атомів Оксигену замінені атомами Нітрогену або
Сульфуру, то відповідні сполуки називають азакраун- (А) або тіакраун-етерами
(Б). Краун-етери, конденсовані з бензеновим і циклогексановим кільцями,
назвали бензокраун- (В) і циклогексанокраун-етерами (Г):
14.6. Краун-етери
ГЛ
1_/
12-Краун-4
Розділ 14. Етери
401
гл
С °~)
ш ж
^-о
\_7
о
о
в
Як відомо, солі неорганічних кислот погано розчинні в органічних
розчинниках, і це веде до утворення гетерогенних систем під час реакцій, що
негативно позначається на ефективності різних реакцій. Розчинність сполуки,
катіон якої потрапляє в порожнину краун-етерів, різко зростає. Це дає змогу
розчиняти солі лужних і лужноземельних металів у малополярних розчинниках.
Аніон у такому випадку слабко сольватований, що підвищує його нуклеофіль-
ність та основність. Ефективність цього процесу, тобто утворення комплексів
краун-етерів з катіонами, залежить від відповідності діаметра катіона розмірові
порожнини кільця, а також координаційного числа катіона металу. Збільшення
за допомогою краун-етерів розчинності неорганічних сполук і, отже, створення
гомогенного середовища дає змогу реалізувати в органічній хімії реакції,
неможливі за інших умов:
Н
/С<н ™*><, 2 Гу.
>=х 18-Краун-6 Z \у //
Стильбен
КМп04
Циклогексен
18-Краун-6
у бензені
СООН
> " \\ //
у бензені Бензойна кислота,
100%
(
НООС-(СН2)4-СООН
Адипінова кислота, 100%
КМпОд
сн3(сн2)5сн2он СН3-(СН2)5-С00Н
У бензені
Гептан-Іол
Гептанова кислота, 70%
СН3(СН2)4СН2ВГіз^* СН3(СН2)4СН2І
і г> в ацетоні л , щппп,
1-Бромогексан 1-Іодогексан, 100%
Перенесення неорганічних іонів з однієї фази в іншу назвали міжфазовим
каталізом, а краун-етери - каталізаторами міжфазового перенесення.
402
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
Краун-етери широко використовують у поділі катіонів екстракцією. В
аналітичній хімії їх застосовують як іоноселективні електроди. На їхній основі
створюють ефективні лікарські препарати, які дають змогу виводити з організму
радіоактивні ізотопи.
Розчинення за допомогою краун-етерів нерозчинних за звичайних умов
неорганічних солей застосовують у боротьбі з закупореннями під час буріння
свердловин.
За допомогою оптично активних краун-етерів можна розділяти рацемічні
суміші на антиподи. В органічному синтезі застосовують макроциклічні
поліаміноетери - криптандщ що утворюють з катіонами металів сполуки
включення - криптати.
Розділ 15
аліфатичні
нітросполуки
Аліфатичні нітросполуки належать до нітрогеновмісних сполук, які мають у
структурі зв'язок С-И. Загальна формула насичених нітросполук — С„Н2,,+іЖ)2.
15.1. Класифікація, номенклатура й будова
15.1.1. Класифікація нітросполук
За природою вуглеводневого замісника
Первинні Вторинні Третинні
СНз\
2
CH3N02 C2H5-CHN02 СНА*Ю.
СН3 СН3
Нітрометан 2-Нітробутан 2-Метил-2-нітропропан
15.1.2. Номенклатура IUPAC
Відповідно до правил замісної номенклатури, нітрогрупу позначають за
допомогою префікса нітро-. Вибір головного ланцюга й напрям нумерації визна-
чені загальними принципами, наприклад:
СН3-СН-СН-С]Н-СН3
СН3 Вг
2-Бромо-4-метил-3-нітропентан
15.1.3. Будова нітроалканів
Нітрогрупа має один семиполярний зв'язок, який позначають
*° +*°
r-К або r-n
Ч0 о"
404 Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Завдяки неподіленій парі електронів семиполярного атома Оксигену та двох
л-електронів зв'язку N=0 утворюється л-електронна система з вирівняними
зв'язками N-0:
Я-й' або Я-Ж.
£5г 6-ше
Обидва атоми Оксигену в нітрогрупі рівноцінні, і довжини зв'язків між
атомами Нітрогену й Оксигену однакові, причому вони коротші (0,122 нм) і
міцніші від одинарного №-0-зв'язку через часткову двозв'язаність. Атом
Нітрогену є чотиривалентним і несе позитивний заряд. Угруповання С-ІМОг
плоске, з низьким енергетичним бар'єром обертання навколо зв'язку С->І.
Завдяки наявності двох сильно електронегативних атомів Оксигену
нітрогрупа виявляє значні електроноакцепторні властивості (-1-, -М-ефекти).
15.2. Фізичні властивості
Нітросполуки жирного ряду - рідини або кристалічні речовини, мало-
розчинні у воді, але добре розчинні в багатьох органічних речовинах. Багато
нітросполук під час нагрівання розпадаються, причому цей процес може
супроводжуватись вибухом. їхні водні розчини мають нейтральну реакцію.
Нітросполуки мають значні дипольні моменти |i = 3,5-3,9D, що зумовлено
елеКТрОННОЮ буДОВОЮ НІтрОГруПИ. ВІДПОВІДНО, ЦЄ ПРИЗВОДИТЬ ДО аНОМЯ' -ліО
високої температури кипіння нітросполук. \; '
15.2.1. Спектроскопія нітроалканів
• УФ-спектроскопія. В УФ-спектрі нітрогрупа поглинає в ділянках 270-285
(я—>л*-перехід) і 200-210 нм (л—>л*-перехід).
• ІЧ-спектроскопія. В ІЧ-спектрах нітросполук є дві дуже інтенсивні смуги
поглинання в ділянках 1 320-1 360 і 1 550-1 570 см"1, які належать симетричним
і асиметричним валентним коливанням зв'язків ONO. Середні за інтенсивністю
валентні коливання vc-n спостерігають за 830-870 см"1.
• Mac-спектрометрія. У мас-спектрах аліфатичних нітросполук піка моле-
кулярного йона нема. Осколкові йони відповідають фрагментам алкільного
ланцюга. У спектрі є сигнали йонів NO+ й Ж>2+.
• Спектроскопія ПМР. Нітрогрупа сильно зміщує сигнали протонів, що
перебувають щодо неї в а-положенні, у слабке поле ( 4,3-4,6 м.ч.).
Розділ 15. Аліфатичні нітросполуки
405
15.3. Одержання нітросполук
❖ Нітрування алканів виконують за рідко- або парофазовим способом.
У першому випадку алкани нітрують розведеною нітратною кислотою
{реакція Коновалова):
сн3сн2сн2снз120^50:с-р н3с-снсн2сн3+Н20
1 2-Нпробутан
■ У другому випадку нітрування проводять концентрованою нітратною
кислотою в інтервалі температур 350-500 °С:
СН3СН2СНз Ш°3> Н3С-СН-СН3+ СН3СН2СН2Ш2+СН3СН2Ж)2 + СН3Ж)2
Ш2
2-Нітропропан 1-Нітропропан Нітроетан Нітрометан
(40%) (25%) (10%) (25%)
Загальний вихід продуктів нітрування коливається в межах 35-45 %. Процес
нітрування в газовій фазі відбувається за радикальним механізмом і тому
приводить до суміші нітросполук з різною довжиною ланцюга:
Я-Н —*~ Я- + Н- або Я—Я1—Я- + Я'-
Я + НОК02—- я-ж>2+ • ОН
Я-Н + - ОН—я- + Н20
❖ Нуклеофільне заміщення під дією арґентум нітриту. В 1872 р.
В. Майєр розробив загальний метод одержання нітроалканів, що полягає у
взаємодії первинних, вторинних алкілбромідів або алкіліодидів (алкілхлориди
не реагують) з арґентум нітритом в етері або без розчинника:
С2Н5Вг+ А§Ш2 С2Н5Ш2 + А§Вг
Побічним продуктом реакції є складний ефір нітритної кислоти - етилнітрит
С2Н5~0-К=0. Він утворюється внаслідок амбідентності нітрит-іона:
['0->ї=0 -—- 0=№-0~1
що забезпечує можливість атаки двома нуклеофільними центрами - атомами
Нітрогену й Оксигену.
За допомогою полярних апротонних розчинників, таких як диметилформ-
амід НСОИ(СНз)2 або диметилсульфоксид (СНз^БО, можна виконати нітру-
вання за допомогою нітритів лужних металів, наприклад, ІОЮ2 або №N02
406 Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
(Н. Корнблюм1, 1955 р). Утворення нітроалканів відбувається, якщо використо-
вувати первинні та вторинні галогеноалкани:
СН3(СН2)5-СНІ-СН3 + КаШ2—СН3(СН2)5-СН-СН3 + СН3(СН)5-СН-СН3
58% 30%
Використання арґентум нітриту значно підвищує вихід цільового продукту:
СН3(СН2)6СН2І + АвШ2— СН3(СН2)6СН2Ш2 + СН3(СН2)6СН2ОШ
83% 11%
Як видно з наведених реакцій, у випадку вторинних галогеноалканів зростає
вихід етерів нітритної кислоти. їх можна легко відокремити, оскільки, з одного
боку, вони мають значно нижчі температури кипіння, а з іншого, - на відміну від
нітроалканів легко гідролізуються водою до спиртів.
Третинні галогеноалкани перетворити в нітроалкани таким шляхом не вдається,
оскільки в цьому разі переважно виникають естери нітритної кислоти та алкени:
СН~ СН,
сн3
СН3-С-Вг + КаШ, —|
сНз-^-ош сн3-(|:-он
■9-
сн,
сн3 сн3
трет-Бутилоьш етер
нітритної кислоти
СН3-(р==СН2
сн3
Ізобутилен
❖ Окиснення амінів. Окремі первинні й третинні нітросполуки одержують
через окиснення відповідних амінів водним розчином калій перманганату або
іншими окиснювачами, наприклад, трифлуоронадоцговою кислотою (СР3СОООН)
або гідроген пероксидом:
СН3СН2СН2Ш2 СН3СН2СН2Н02 + Н20
Залежно від умов реакції утворюються проміжні продукти, які можуть бути
виділені як кінцеві сполуки, наприклад, алкілгідроксиламіни ІІ-МНОН* оксими
Я-СКНЧОН і нітрозоалкани Я-№Ю.
❖ Декарбоксилювання нітрооцтової кислоти. Ця реакція, запропонована
А. Кольбе, є одним з найпоширеніших у лабораторії методів одержання
нітрометану. Вихідну нітрооцтову кислоту, відповідно, одержують із хлороцто-
вої. Нітрогрупа - сильна електроноакцепторна функція, тому нітроацетатний
1 Корнблюм Н. (КогпЬІшп N0 (1914-1993) - американський хімік, вивчав реакції, що відбуваються за
радикальним механізмом.
Розділ 15. Аліфатичні нітросполуки
407
аніон легко зазнає декарбокснлювання в іон СНгІЧОг, який потім утворює
нітрометан з виходом 35-38 %:
С1СН2СО(Жа СН——"СН2Ш2+С02 +Ш+-І^СН3Ш2+№ОН
О" ЧОИа
Для вищих гомологів оцтової кислоти така реакція непридатна через украй
низькі виходи цільового продукту.
15.4. Хімічні властивості та застосування
15.4.1. Хімічні властивості нітроалканів
Головні хімічні властивості нітросполук визначені реакційною здатністю
групи NO2 і підвищеною активністю атомів Гідрогену, що перебувають в а-
положенні до цієї групи.
❖ Відновлення нітросполук. Якщо використовувати різні відновники, такі
як водень, Fe + HCl, Zn + КОН, літій алюмогідрид, нітросполуки можна
перетворити в аміни з набором проміжних нітрозопохідних і гідроксил амінів:
h2/Pt або
RN02 z.h2oC1> RN=0 ~ RNHOH-^- RNH2
Нітрозосполука ^ Алкілгідроксиламін Амін
Перебіг процесів у кислому середовищі унеможливлює виділення проміжних
продуктів. Тому нітроалкани можна частково відновити до нітрозосполук та
оксимів лише в лужному середовищі або електрохімічним шляхом:
сн3-(рн-сн3 C2H5QH+N»a сн3-<рн-сн3^ СН3-£-СН3
N02 NO NOH
2-Нітрозопропан Оксим ацетону
Зазначимо, що аліфатичні аміни зазвичай одержують доступнішим шляхом
- алкілуванням амоніаку. Навпаки, ароматичні аміни одержують саме від-
новленням нітросполук.
❖ Аци-нітротсіутомерія нітросполук. Підвищена активність атомів
Гідрогену в а-положенні щодо нітрогрупи (С-Н-кислота) зумовлена а,я-спря-
женням зв'язків С-Н з подвійним зв'язком і дозволяє первинним і вторинним
нітросполукам існувати у двох таутомерних формах:
rc-nT -у— rch=nn
н о- он
Алканнітронова
кислота
408 Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
Така таутомерія відбувається лише в лужному середовищі, її називають аци-
нітротаутомерією:
ясн2<° ^
ЯСНІ^Т^ ЯСН=]<
н+ /ОН
Ка+=^ ІІСН=]<
но- \0
Аніон, що утворюється під дією лугу, стабілізується завдяки мезомерії:
+<0 +/0"
я-сн->ґ ІІ-СН=:Й
0. 0_
Із двох таутомерних форм н/троформа набагато стабільніша й тому
переважає в рівноважній суміші Наприклад, частка ш/и-форми для нітрометану
дорівнює 1-Ю*7, для нітропропану - 3-Ю"3. Нітронові кислоти у вільному стані,
як звичайно, нестійкі, тому їх одержують обережним підкисленням їхніх солей.
Нітронові кислоти проводять електричний струм і дають червоне забарвлення з
ферум (III) хлоридом.
Лі/м-форма нітрогрупи виявляє сильні кислотні властивості й утворює солі з
лугами. Завдяки цьому нітрометан легко розчиняється в водному розчцні натрій
гідроксиду. Процес утворення солей відбувається в часі, на відміну від іонної
нейтралізації типових кислот, що відбувається миттєво. Первинні й вторинні
нітросполуки називають псевдокислотами, оскільки під впливом лугів вони дають
солі ізомерних нітросполукам кислот. Таку назву псевдокислоти одержали тому, що
у водних розчинах дисоціації зв'язку ИО-Н не спостерігають, незваж^^~ї на
здатність ш/м-форми до солеутворення. Крім того, якщо лужні розчини нітросполук
обробляти мінеральною кислотою, то спочатку відбувається утворення аци-форми
нітросполуки, яка потім легко зазнає ізомеризування у звичайну нейтральну форму.
Для псевдокислот характерна нейтральність.
Наявність ш^м-нітроформи доводять її швидкою взаємодією з діазометаном:
СН3СН=ШОН СН3СН=ШОСН3
Метилнітронат
Подібні естери можна синтезувати з натрієвих солей і галогенувальних
агентів. Під час нагрівання естери розпадаються на оксими й альдегіди:
СН3СН=>ЮОСН3--^— СН3СН=ИОН + Н2С=0
Лі/н-нітросполуки - це досить сильні кислоти, які реагують не тільки з лугами, а
й з натрій карбонатом. З іншого боку, нітросполуки містять в а-положенні щодо
нітрогрупи рухливі атоми Гідрогену, тому їх можна схарактеризувати як СН-кислоти.
Третинні нітросполуки до таутомерії не здатні, оскільки нема атомів
Гідрогену в а-положенні щодо нітрогрупи. _
Розділ 15. Аліфатичні нітросполуки
409
❖ Гідроліз пітрогрупи
■ В. Майєр довів, що первинні нітросполуки взаємодіють з концентрованою
сульфатною кислотою під час нагрівання з перетворенням у карбонові кислоти й
гідроксиламін:
h2s(V ru ç//
сн3сн2к. +н2о8^_904£о сн3
о
"NHOH
Ацетогідроксамова кислота
Передбачуваний механізм реакції такий:
СН3СООН + NH2OH
R-CHjNO^ R-CH=N-OH
ht
+ oh + /
R-ch=n; — r-ch-n.
oh
oh
oh
+ h.0
— R-C^N-OH—
• R-C=N-OH *=t R-C-NHOH -
I
oh
h*
Пароксамова кислота
R-COOH + NHjOH
Таким способом у промисловості з 1-нітропропану одержують гідроксиламін.
У минулому нітроалкани були сировиною для синтезу окремих карбонових кислот.
■ Дією 50 % сульфатної кислоти солі нітронових кислот за кімнатної темпе-
ратури гідролізують до альдегідів і кетонів з виходом 85 % через стадію утворення
нітронових кислот (реакція Нефа1, 1894 р.):
2RCH=N
/°Na 2НС1
чо
_^2RCH=ljJ-oh-
он
-н+
2R(j:hn=o
oh
2RÇ=<^Ia.^r
R ^о
+/Он -н+ Рн
2RC=r< — 2RÇN=0-
r ч°н k
•2RCHO + N20 + H20
2RÇ=0 + N20 + H20
R
Є кілька модифікацій перетворення нітросполук в альдегіди й кетони, що
дають змогу одержати оксосполуки з високими виходами. Серед них треба
назвати метод Нефа, що полягає в обробці нітросполук титан (III) хлоридом.
Якщо у випадку первинних нітросполук реакцію проводити не у воді, а в
метанолі, то вдається синтезувати ацеталі альдегідів.
❖ Елімінування нітритної кислоти. Аліфатичні сполуки, що містять
рухливий атом Гідрогену в Р-положенні до нітрогрупи, дією лугів легко
елімінують її у вигляді НП^Ю2 з утворенням олефінів. Аналогічно відбувається
розпад нітроалканів за температур понад 450 °С.
1 Неф Д. (Nef J.W.) (1862-1915) - американський хімік. Головні праці з органічного синтезу.
Запропонував існування карбенів.
410
Чирва В.Я,, Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
❖ Реакція з нітритною кислотою. Первинні й вторинні нітропохідні по-
різному реагують з нітритною кислотою.
■ Зокрема, первинні нітросполуки утворюють нітролові кислоти, здатні до
таутомерії:
ЯСН2Ш2 + НОЖ) К(рш2 —- Я<рю2 Я(^02
N=0 ШН " 2 КСЖа
Сіль червоного кольору
■ Вторинні нітроалкани утворюють псевдонітроли синього або зеленого
кольору:
/СНИ02 + НСЖО— /СЇТО2
Реакції з нітритною кислотою використовують для визначення типу нітро-
алканів, оскільки розчини й розплави нітрозопохідних вторинних нітроалканів
забарвлені в інтенсивно синій колір, а лужні розчини нітролових кислот - у
темно-червоний.
■ Третинні нітросполуки з-нітритною кислотою не реагують.
❖ Конденсація з альІЇегідами^й кетонами. Аніони, що генеруються з
первинних і вторинних нітроалканів, поводяться як нуклеофільні реагенти й
легко вступають у реакцію з альдегідами й окремими кетонами за типом
альдольної конденсації: ^
он
СН3(СН2)7СНО + СН3Ш2 с'н^оН» СН3(СН2)7СНСН2М02
1-Нітродекан-2-ол, 80%
Відповідно, відновлення Р-нітроалканолів - зручний метод синтезу 1,2-
аміноспиртів:
носн2сн2ш2 носн2сн2ш3+нзо; НОСН2СН2КН2 ^
Для ароматичних альдегідів процес супроводжується дегідратацією {реакція
Генрі):
СН3СН2Ш2 ->^СНз-СН-Ш2 с*н*сно, Сбн5.сн.сН-Ш2 С6Н5-СН=С-Ш2
О" сн3 "Нг° СН3
2-Нітро-1 -фенілпропен
■ Зазначимо, що альдольну конденсацію для первинних нітроалканів можна
спрямувати залежно від співвідношення реагентів за шляхом утворення нітродіолів:
Розділ 15. Аліфатичні нітросполуки
411
СН2ОН
псн2ш2+нсно —- шжо2м^-кс^о2
сн2он сн2он
■У випадку альдольної конденсації нітрометану можна використати всі
три атоми Гідрогену метильної групи:
у срн2он
З НСНО + н-(р-ш2 —- НОСНгСрК02
Н СН2ОН
2-Гідроксиметил-2-нітропропан-1,3-діол
■ Третинні нітроалкани з альдегідами й кетонами не реагують, оскільки не
мають рухливого атома Гідрогену в ос-положенні щодо нітрогрупи.
❖ Реакція Манніха1. Первинні й вторинні нітросполуки вступають у
реакцію амінометилювання за Манніхом:
к-сн2-мо2 + сн2о + ш(с2н5)2 ^£ я-(рі-сн2н(с2н5)2
N0,
❖ Реакції заміщення атома Гідрогену в а-положенні. Аліфатичні нітро-
сполуки, що містять а-гідрогенний атом, легко зазнають алкіл^раі-шя й ацилю-
вання, утворюючи, як звичайно, О-похідні. Однак взаємодія літієвих солей ,
первинних нітросполук з галогеноалканами, ангідридами, галогеноангідридам^і^
карбонових кислот приводить до продуктів С-алкілування або С-ацилювання,
наприклад:
Я-СН2Ж)2 ^я-сн-ж)2и+ ^^сн-жх
Відомі приклади внутрішньомолекулярного С-алкілування:
Вг-СН2СН2СН2Ж)2ч у И02
15.4.2. Застосування нітроалканів
Нітропарафіни використовують як розчинники, для синтезу окремих альдегідів
і кислот, у реактивній техніці, як вулканізатори гуми. Найважливішою галуззю
застосування нітросполук є вибухові речовини, що пов'язано з їхньою високою
термодинамічною нестійкістю. Наприклад, теплота розкладання нітрометану
становить 282 кДж/моль:
3|ЙзШ2 ІШ2 + С02 + 3)$Н2 Д# = -282кДж/моль.
1 Манніх К (МаппісЬ С.) (1877-1937) - німецький хімік. Уперше синтезував аміноспирти й аміно-
кетони.
Розділ 16
ааіфа тичні аміни
Продукти заміщення атомів Гідрогену в амоніаку на вуглеводневі замісники
називають амінами. Вперше їх отримав Ш. Вюрц. Аміни - це сполуки, що мають
високу біологічну активність. До них належать вітаміни групи В, амінокислоти,
алкалоїди, адреналін та ін.
16.1. Класифікація, номенклатура й будова амінів
16.1.1. Класифікація амінів
За ступенем заміщення^
Первинні аміни Вторинні аміни Третинні аміни Четвертинні
амонієві солі
CH3-N-CH2CH2CH3 СН3
CH3CH2-NH2 CH3-NH-CH2C jlh І +
Н3 3 H3C-N±CH3 СГ
Етанамін ЛГ-Метилетанамін N,N-Діт е тил-1-пропан амін £л
етиламін N-метилетиламін ИДчІ-лимсгтлпротламш З
етилазан ЛГ-етил^-метилазан К^-дішетм-К-гфопишан Тетраметиламоній хлорид
16.1.2. Номенклатура IUP АС
• У залисній номенклатурі аміногрупу як головну позначають суфіксом -
амін. Для вторинних і третинних амінів один карбоновий ланцюг вибирають за
основу, а інші (простіші) називають у префіксі як вуглеводневі замісники із
зазначенням їхнього знаходження біля атома Нітрогену за допомогою буквеного
локанта N.
Якщо аміногрупа є замісником, то використовують префікси аміно-, R-аміно-
або КДІ'-аміно-:
Розділ 16. Аліфатичні аміни
413
6 5 4 3 2 1
СН3СН2СН2СНСН2СН3
N42
З -Гексанам ін
Нумерація ланцюга почи-
нається з того краю, до якого
ближче аміногрупа.
сн
т
н,
3МСНСН2СН3
/СН,
ЬІ,ЬІ-Ц,ш,іетш-2-бутанамІн
За основу беруть найдовший
вуглеводневий ланцюг.
5 4 3 2 1
СН3СН2С|НСН2СН20Н
1МН2
З-Аміно-1-пентанол
Головна група - гідроксил.
Нумерація ланцюга починаєть-
ся з головної групи.
• Альтернативно назви первинних амінів будують із назви вуглеводневого
замісника й слова амін.
■ У несиметричних вторинних і третинних амінах простіші вуглеводневі
замісники розглядають як замісники біля атома Нітрогену. Приклади таких назв
наведені курсивом у параграфі "Класифікація амінів".
■ У назвах симетричних вторинних і третинних амінів до назви вугле-
водневого замісника додають числові префікси ди-(ді-) і три-, наприклад:
СгНзІМНг - етиламін, (С2Н5)2>Ш - діетиламін, (Сг^^И - триетиламін.
• За правилами ШРЛС 1993 р. родоначальний гідрид №їз називають азан,
відповідно, назви первинних амінів ІІ-МІг складають з назви вуглеводневого
замісника і назви родоначального гідриду. ^^Х^
16.1.3. Будова амінів
Спектроскопічні дані свідчать, що молекула амоніаку пірамідальна, причо-
му атом Нітрогену перебуває у вершині піраміди, а атоми Гідрогену - у кутах
трикутної основи.
У разі утворення амоніаку атом Нітрогену перебуває в 5р3-гібридному стані.
Три лр3-гібридні орбіталі перекриваються з 5-орбіталями атомів Гідрогену,
утворюючи ст-зв'язки, напрямлені до кутів тетраедра, а місце четвертого зв'язку
займає неподілена пара електронів Нітрогену.
Оскільки аміни є похідними амоніаку, в яких один або більше атомів
Гідрогену заміщені на алкільні групи, то можна припустити, що аміни також
пірамідальні. Це доведено методом дифракції електронів: згідно з отриманими
даними, молекула триетиламіну пірамідальна, і валентні кути С-И-С (108°)
дуже близькі до валентних кутів Н-М-Н (107°) амоніаку.
414 Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
16.1.4. Стереоізомерія амінів
Досі під час вивчення органічної хімії значну увагу приділяли питанням
просторового розташування атомів і груп, пов'язаних з атомом Карбону, тобто
стереохімії Карбону. Розглянемо тепер стереохімію сполук з атомом Нітрогену.
З аналізу моделей можна переконатися, що молекулу, у якій атом Нітрогену
несе різні групи, не можна сумістити з її дзеркальним відображенням, отже, вона
асиметрична, тому такий амін, як, наприклад, И, ЛГ-метилетилпропіламін, теоре-
тично повинен існувати у двох енантіомерних формах (І) і (II), кожна з яких має
виявляти оптичну активність:
^с3н7 с3н7^'\^с2н5
сн3 1 псн3
Спектральні дослідження засвідчують не тільки те, що амоніак і найпростіші
аміни є пірамідальними молекулами, а й те, що енергетичний бар'єр для
перетворення однієї пірамідальної конфігурації (III) в іншу (IV) становить усього-
25,2 кДж/моль. Навіть за кімнатної температури енергія зіткнень є достатньою для
подолання цього бар'єра, тому відбувається швидке перетворення між пірамі-
дальними конфігураціями - швидка інверсія атома Нітрогену:
сд \"сн3
9 ..хн,
с2ик ,сн,
ш н rv
У плоскому активованому комплексі атом Нітрогену має .^-гібридизацію з
неподіленою електронною парою, що перебуває на 2/?2-орбіталі.
Усі спроби розділити нециклічні аміни на антиподи виявилися безуспішними.
Розглянемо четвертинні амонієві солі - сполуки, у яких чотири алкільні групи
пов'язані з атомом Нітрогену. У цьому випадку чотири 5р3-орбіталі використані для
утворення зв'язків. Сіль четвертинного амонію, у якої при атомі Нітрогену є чотири
різні групи, повинна існувати у вигляді енантіомерів, здатних виявляти оптичну
активність, оскільки інверсія атома Нітрогену в цих сполуках неможлива. Таку сіль
можна розділити на оптично активні форми. Іодид алілбензилметилфеніламоній
справді існує у вигляді двох енантіомерних форм, поділ яких виконав 1899 р.
англійський хімік В. Поуп1:
1 В. Поуп (Sir Pope W.J.) (1870-1939) - видатний англійський хімік, професор Кембриджського
університету, президент IUPAC (1922-1925), провадив дослідження в галузі стереохімії.
Розділ 16. Аліфатичні аміни
415
сн2=снсн2
6і *5
/
ХН2СбН5
сн2=снсн2
сн
з
На відміну від четвертинних тетразаміщених амонієвих солей, інші солі
амонію швидко зазнають рацемізування через депротонування й утворення
нестійкого аміну. Внаслідок приєднання до нього протона утворюється раце-
мічна суміш солей амонію:
Дуже^
швидко
я.
н+
Стійка
конфігурація
Стійка
конфігурація
16.2. Фізичні властивості
Нижчі аміни є газоподібними, середні - рідкими, а вищі - твердими речови-
нами. Запах нижчих амінів подібний до запаху амоніаку, середні мають
неприємний запах гнилої риби.
Подібно амоніаку, аміни - полярні сполуки й, за винятком третинних амінів,
можуть утворювати міжмолекулярні водневі зв'язки. Тому аміни киплять за
вищих температур, ніж неполярні сполуки тієї ж молекулярної маси, проте їхні
температури кипіння нижчі, ніж температури кипіння спиртів або карбонових
кислот, наприклад:
СНзСН2СН2СНз СН3СН2СН2>Ш2 СН3СН2СН2ОН СН3СН2СООН
т. кип. 0 °С
т. кип. 49 °С
т. кип. 97 °С
т. кип. 141 °С
Причина цього полягає в утворенні спиртами й кислотами міцнішого
порівняно з амінами водневого зв'язку, що відображено в енергії водневого
зв'язку і його довжині.
Первинні й вторинні аміни утворюють водневі зв'язки як електронодо-
норних, так і протонодонорних компонентів, а третинні можуть бути тільки
електронодонорними:
ъг-н-
З огляду на це асоціати молекул первинних і вторинних амінів плавляться і
киплять за вищої температури, ніж третинні аміни з такою ж молекулярною
масою. Оскільки третинні аміни не містять гідрогенних атомів при Нітрогені, то
416
Чирва В.Я., Ярллолюк СМ., Толкачова H.B., Земляков О.Є. Органічна хімія
вони не утворюють міжмолекулярних водневих зв язків, тому їхні температури
кипіння нижчі, ніж первинних і вторинних амінів, і ближчі до температур
кипіння неполярних сполук, наприклад:
сн •
H3C-N-CH3
т. кип. +3 С
сн3
H3C-C-CH3
н
т. кип. -12 °С
Аміни всіх трьох типів здатні утворювати водневі зв'язки з водою, тому
нижчі аміни досить добре розчинні у воді, причому межа розчинності проходить
біля Сб-атома. Аміни розчинні в малополярних розчинниках, подібних до етеру,
спирту, бензену тощо.
Розходження в розчинності амінів і їхніх солей можна використати для
виявлення амінів та їхнього відділення від сполук, що не є основами. Органічна
сполука, нерозчинна у воді, проте розчинна в холодній розбавленій хлоридній
кислоті, повинна мати помітні основні властивості, що маюкд напевно означає,
що вона є аміном: Ч
RNH2
R2NH
R3N _
н+
он-
RNH3
R2NH2
LR3lfe
Нерозчинні у воді Розчинні у воді
Амін можна відокремити від сполук, що не є основами, розчинивши його в
кислоті; після поділу амін можна регенерувати, змінюючи рН водного розчину з
кислого до лужного.
16.2.1. Спектроскопія амінів
• УФ-спектроскопія. В УФ-спектрах амінів виявляються смуги поглинання
за 190-210 нм (л—»о*-перехід електрона з неподіленої пари електронів атома
Нітрогену).
• ІЧ-спектроскопія. За смугами поглинання в ІЧ-спектрі можна розрізнити
первинні, вторинні й третинні аміни.
■ У первинних амінах спостерігають смугу поглинання валентних коливань
Vnh 3 400-3 500 см"1 і сильнішу смугу деформаційного коливання за 1 640—
1 560 см"1.
■ У вторинних амінах смугу поглинання валентного коливання спостері-
гають у тій же ділянці, що й у первинних амінів (vN_H 3 300-3 350 см"1), однак
смуга деформаційного коливання виявляється за 1 490-1 580 см"1.
■ Оскільки біля атома Нітрогену нема протонів, то у третинних амінів
зазначених смуг поглинання немає.
Розділ 16. Аліфатичні аміни
417
Крім того, для первинних і вторинних амінів є смуга поглинання середньої
інтенсивності за 1 030-1 230 см"1 завдяки валентним коливанням С-М-зв'язку. У
третинних амінах у цій ділянці спостерігають дві смуги поглинання.
• Мас-спектр ометрія. У мас-спектрах первинних амінів найбільшу інтенсив-
ність має іон, що містить Нітроген - КСН2-МН2+. У спектрах вторинних і
третинних амінів переважають фрагменти алкільних радикалів.
• Спектроскопія ПМР. У ПМР-спектрах хімічний зсув протонів аміногрупи
перебуває в інтервалі 1,1-1,8 м.ч. для первинних амінів і 1,2-2,1 м.ч. для
вторинних амінів.
16.3. Способи одержання амінів
16.3.1. Реакції алкілування
❖ Алкілування амоніаку й амінів (реакція Гофмана, 1849). Алкілування
амоніаку або амінів галогеноалканами, діалкілсульфатами або третинними оксо-,
нієвими солями є одним з найстаріших методів синтезу аліфатичних первинних,
вторинних і третинних амінів, а також солей тетраалкіламонію. Реакція відбу-
вається через стадію приєднання електрофільного атома Карбону алкллгалогеніду
до вакантної пари електронів Нітрогену, після чого відщеплюється гідроген
галогеніду, що зв'язується з надлишковим амоніаком або аміном:
м Лзі"-^ ішн2+мну
М я2іч[н2Г-^- Я2Ш + Ш4І
У цій реакції атом Карбону, пов'язаний з атомом галогену, зазнає
нуклеофільної атаки атомом Нітрогену амоніаку чи амінів, або, що те ж саме,
атом Нітрогену є місцем електрофільної атаки атомом Карбону алкільного
замісника.
Оскільки під час алкілування виникають проміжні речовини високої
полярності, то використання таких розчинників, як метанол, етанол, диметил-
формамід сприятиме збільшенню швидкості реакції.
Залежно від співвідношення реагентів та умов перебігу реакції Гофмана
утворюється суміш первинного, вторинного й третинного амінів і четвертинної
амонієвої солі з переважанням низько- або високоалкілованих продуктів. Для
одержання первинних і вторинних амінів беруть надлишок амоніаку або первин-
ного аміну. Надлишок у реакційній суміші галогеноалкану спричиняє синтез
третинного аміну в суміші з четвертинною амонієвою сіллю, що розкладається під
час перегонки з натрій гідроксидом, перетворюючись теж у третинний амін. Якщо
418
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
використати один еквівалент алкілувального агента в реакції з вторинними
амінами, то можна одержати з високим виходом третинні аміни.
Реакційна алкілувальна здатність зменшується в ряду М > ЯВг » ЯСІ »
>ЖР. Якщо додавати в реакційну суміш іодид-іон, то це ініціює обмін галогену
на іод і полегшує реакцію алкілування.
Реакційна здатність знижується, якщо збільшується ступінь заміщення в
аміні або алкілгалогеніді. Ця обставина дає змогу припинити реакцію на ранній
стадії. Зростання ступеня розгалуження в галогеноалкані збільшує ймовірність
реакції дегідрогалогенування під дією аміну. На практиці значення цього методу
обмежене тим, що здебільшого не можна припинити реакцію на конкретній
стадії, а поділ суміші часто є досить складним завданням, що видно з наведеного
нижче прийому. 4—.
■ Поділ суміші амінів. Четвертинні амонієві солі можна кількісно відокремити,
обробивши суміш лугом. Під час перегонки з водяною парою четвертинна амонієва
сіль залишиться в реакційній суміші. Подальший поділ відігнаних амінів пов'язаний
з відомими труднощами й не завжди може бути виконаний одним і тим сами^
способом. Метод, що виправдав себе в багатьох випадках, полягає в обробці суміші
амінів бензен- або л-толуенсульфохлоридом. Ці реагенти не взаємодіють із
третинними амінами у водно-лужному розчині; у разі ж дії на первинні й вторинні
аміни вони утворюють аміди бензенсульфокислоти або, відповідно, л-то-
луенсульфокислоти:
С„Н2п+1НН2 + С1802С6Н5-^С„Н2^,НН802С6Н5 + КСІ + Н20
(СИН21Н.1)2МН + С1802СбН5-^ (С„Н2й+1)2М802СбН5 + КСІ + Н20
Моноалкіламіди бензенсульфокислоти, на відміну від діалкіламідів, здатні
утворювати водорозчинні лужні солі: C6H5S02NNaC/lH2/,+l. Завдяки цьому їх
можна відокремити, а потім відщепити залишок сульфокислоти, нагріваючи з
концентрованою хлоридною або сульфатною кислотою.
С6Н5802МНС„Н2л+1 + Н20 -И— СбН5803Н + С„Н2л+1КН2
Вторинні аміни реагують з бензенсульфохлоридом у водному розчині лугу,
утворюючи Л^-дизаміщений сульфамід, який не розчинний у водному лузі,
оскільки не має кислого атома Гідрогену при Нітрогені. Різне поводження амінів у
реакції з бензенсульфохлоридом називають тестом Гінзберга.
■ За механізмом реакції алкілування амінів належать до реакцій заміщення
8н2. Кінетика відповідає реакції другого порядку, причому відносні швидкості
реакції різних іодидоалканів, згідно з даними Меншуткіна1, такі:
Меншуткін М. (1842-1907) - російський хімік, працював у галузі кінетики органічних реакцій,
особливо реакцій етерифікації спиртів та омилення естерів. Винайшов вплив розчинника на швидкість
хімічних реакцій. Автор першої книги з історії хімії, а також підручника з аналітичної хімії.
Розділ 16. Аліфатичні аміни
419
СНз (100); С2Н5 (8,8); я-С3Н7 (1,7); н-С7Н15 (0,9); /зо-СзН7 (0,18).
❖ Алкілування амоніаку й амінів спиртами. У промисловості амоніак або
аміни алкілують спиртами над оксидами Алюмінію, Силіцію, Титану,
алюмосилікатами, фосфатами металів в інтервалі температур 300-500 °С:
NH3 + СН3ОН CH3NH2 (CH3)2NH (CH3)3N
Очевидно, це найвигідніший метод одержання нижчих амінів.
Moho-, ди- і триметиламін у промисловості одержують з метанолу й
амоніаку. Вихідні продукти пропускають над нагрітим алюміній оксидом або
купрум хромітом СиСЮ2 за 300 °С
Інакше алкіламіни промислово синтезують шляхом нагрівання амоніаку з
метиловим або етиловим спиртом і невеликою кількістю сульфатної кислоти:
СН3ОН + H2S04 -> CH3OSO3H + Н20
NH3 + 2CH3OSO3H -> (CH3)2NH + 2H2S04
Як проміжний продукт утворюється метилсульфатна кислота, яка безпе-
рервно відновлюється. Внаслідок дегідратувальної дії сульфатної кислоти її
іноді заміняють на хлоридну. Суміш амінів розділяють перегонкою на ефектив-
них колонах.
■ Взаємодією спиртів з аміаком цинк хлориду одержував аміни Г. Мерц:
C„H2ffHOH + NH3 C„H2„+1NH2 + 2Н20
Загалом алкілування амінів спиртами відбувається значно гірше, ніж
галогеноалканами.
❖ Синтез Габрієля1. Варіант методу алкілування, що дає змогу одержувати
чисті первинні аміни, запропонований 1887 р. німецьким хіміком 3. Габрієлем.
Метод полягає в алкілуванні галогеноалканами "захищеної" форми амоніаку.
Вихідний фталімід готують, обробляючи фталеву кислоту або її ангідрид
амоніаком за 300-350 °С. Утворений імід має кислі властивості через електроно-
акцепторні властивості карбонільних груп, тому у водно-спиртових розчинах
лугів він повністю перетворюється на аніон:
1 Габрієль'З. (Gabriel S.) (1851-1924) - німецький хімік, учень А. Гофмана і Р. Бунзена. Головні
праці присвячені синтезу нітрогеновмісних гетероциклічних сполук. Відкрив спірани
чотиривалентного Нітрогену. Синтезував ізохінолін.
420
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Фталімідний аніон - досить сильний нуклеофіл і легко заміщає галоген у
галогеноалканах, утворюючи ІЧ-алкілфталіміди. Теоретично реакція може
відбуватися за атомом як Нітрогену, так і Оксигену, та оскільки Нітроген більш
нуклеофільний порівняно з Оксигеном, то процес відбувається переважно по
Нітрогену. Подальше алкілування неможливе, бо немає протонів. ІУ-алкіл-
фталіміди в разі лужного або кислотного гідролізу перетворюються в первинні
аміни і фталеву кислоту:
"с:ы"к++ сн3сн2сн2сн,вг—А 1~^:н-сн,сн,сн,сн,+КВЙ^
^ о^соо- + н2^н2сн2сн2сн3
Ефективнішим реагентом для зняття фталоїльного захисту з атома Нітро-
гену є гідразин:
я
О II
О
Отже, синтез Габрієля можна трактувати як один із найліпших методів
синтезу первинних амінів з первинних і вторинних алкілгалогенідів. Третинні
алкілгалогеніди для синтезу Габрієля непридатні, оскільки в лужному
середовищі вони перетворюються в алкени.
16.3.2. Розщеплення амідів за Гофманом
Труднощі, пов'язані з одержанням чистих амінів, зокрема первинних,
повністю усуває відкрита Гофманом реакція, яку він запропонував 1882 р.
Унаслідок дії брому (хлору, іоду) в лужному середовищі на ацетамід
утворюється ІУ-бромоацетамід:
СН3СОШ2 + Вг2 — СН3СО№ЛЗг,
який під дією натрій гідроксиду розщеплюється до метиламіну:
СН3СО№1Вг + КОН -> КВг + С02 + СН3№.
З'ясовано, що реакція відбувається через метилізоціанат. У випадку
відщеплення гідроген броміду утворюється проміжна сполука зі зниженою
Розділ 16. Аліфатичні аміни
421
електронною щільністю на атомі Нітрогену, що зумовлює міграцію метальної
групи з її парою електронів від Карбону до Нітрогену:
СН3:С:>Л:Вг:|
Ш СН3:С:^СН3:К::С::0
Ізоціанати, взаємодіючи з водою, перетворюються в нестійкі карбамінові
кислоти, які втрачають Карбон (IV) оксид, утворюючи амін:
я_к=с=о + Н20
Я-Ш-СООИ ^ Я-ЫН2
Синтез амінів за допомогою перегрупування Гофмана веде до вкорочення
ланцюга на один атом Карбону. Зазначимо, що перегрупування Гофмана
придатне для одержання оптично активних амінів. Відповідно до внутрішньо-
молекулярності реакції, простежується збереження конфігурації, якщо вико-
ристовувати оптично активні аміди:
СНз СН,
СН3СН2
СГ
Вг2, 2ИаОН
СН3СН2
(/?)-Бутанамін-2
(Л)-2-Метилбутирамід
Оскільки аміди кислот легкодоступні, то їхній розпад за Гофманом має
препаративне значення для синтезу первинних амінів.
16.3.3. Реакції відновлення
❖ Відновлення амідів. Аміди можна перетворити в аміни, не вкорочуючи
карбонового ланцюга, шляхом їхнього відновлення літій алюмогідридом або
дибораном в етері (тетрагідрофурані) з дуже високим виходом, наприклад:
к-с,-сі
о
я-ссжн,
ІЛАІН.
4 Я-СН21МН2
Первинний амін
II
о
Я-СН^НК'
■ын
Вторинний амін
Третинний амін
сн^сн^-мссн^
о
о
вн,
СН3(СН2)4СН2Ы(СН3)2
422 Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Таким способом одержують первинні, вторинні й третинні аміни.
❖ Відновлення нітрилів за допомогою літій алюмогідриду в етері або
каталітичного гідрування дає з високим виходом первинні аміни з домішкою
вторинних:
н,
Я-СМ + Н
n1
2 100-130°С
я-сн=ш
К-СН2Ш2
Я-СН=ЫН + Я-СН2ЫН2 —г
Я-СН=Ж:Н2ІІ
н,
(ЯСН2)2МН
Щоб під час перебігу відновлення унеможливити утворення вторинних
амінів, гідрування нітрилів проводять в оцтовому ангідриді: первинний амін, що
утворився, негайно ацилюється й тому далі не зазнає змін. Цей метод
застосовують, головно, для виробництва вищих первинних і вторинних амінів
нормальної будови.
Оскільки первинні нітрили легко одержують із відповідних галогено-
алканів, то інколи метод має препаративне значення, як, наприклад, у випадку
синтезу діамінів.
❖ Відновлення оксшіів і гідразонів. Оксосполуки можна легко перетворити
в нітрогеновмісні функціональні похідні (оксим, гідразон), а ті потім відновити в
первинні аміни каталітичним гідруванням або літій алюмогідридом:
ЯСЯ' + Н,1МОН
-ЯСЯ'
н2/№
СГ с2н5он
1МОН
ЯСНЫ' + Н,0
❖ Відновне амінування. Багато альдегідів і кетонів перетворюється в аміни
в разі їхньої обробки амоніаком, гідроксиламіном, гідразином за одночасного
відновлення воднем. Як видно зі схеми, як проміжні продукти утворюються
малостабільні іміни, які легко можна відновити до амінів:
Я—С—Н
II
О
я—с—я
II
о
Я— СН=Ш
Я—с—Я
II
ш
н,
я— сн— ш2
Первинний амін
я—сн—^
мн2
Первинний амін
Відновне амінування кетонів дає первинні аміни, що мають втор-алкільні
групи. Подібні аміни синтезувати шляхом амонолізу втср-алкілгалогенідів
важко, бо втяр-алкілгалогеніди схильні до реакцій елімінування.
Розділ 16. Аліфатичні аміни
423
■ Етилювання первинних і вторинних амінів відбувається під час нагрівання
з формальдегідом за наявності мурашиної кислоти (реакція Ешвайлера-Юіарка1):
К-гт2 + 2СН20 + 2НСООН -> К->ї(СНз)2 + 2С02 + 2Н20
Це реакція призначена винятково для синтезу третинних амінів.
■ Формальдегід, взаємодіючи з амоній хлоридом, утворює гідрохлорид
метиламіну:
і оо°с 4- -
2 СН20 + Ш4С1 СН3Ш3 СІ + нсоон
50%
❖ Відновлення нітросполук до амінів можливе за допомогою різних
відновників:
спн2п+1ио2 + ЗН2 -> СпН2п+1ї<Н2 + 2Н20
Цей метод, запропонований М. Зініним для нітробензену, не набув такого
значення в хімії аліфатичних сполук, оскільки ці сполуки менш доступні, ніж
ароматичні нітропохідні.
❖ Гідрування ізонітрилів дає вторинні аміни:
І1-№ЕС Я-ШСНз
16.3.4. Гідроліз ізоціанагіз
❖ Синтез Вюрца. Як уже зазначено, уперше аліфатичні аміни відкрив
Ш. Вюрц 1848 р. унаслідок гідролізу естерів ізоціанової кислоти (ізоціанатів):
R-NH-C=0 + Н20
ос2н5
R-NH2 + C02+C2H5OH
Самі ізоціанати одержують у разі взаємодії галогеноалканів з арґенгум ціанатом.
❖ Реакція Лоссеня. Аміни одержують унаслідок дії на гідроксамову
кислоту або її ацильні похідні дегідратувальними агентами, такими як фосфор
(V) оксид, оцтовий ангідрид, тіонілхлорид. Процес відбувається через стадію
утворення ізоціанату:
он
О -r'o-
RCN
О J
RN=C=0
н,о
RNH2 + C02,
де R = алкіл, В! = ацил.
Виходи амінів за реакції Лоссеня становлять 60-80 %.
1 Кларк Г. (Clarke Н.Т.) (1887-1972) - англійський хімік та біохімік, займався синтезом органічних
сполук, дослідженням структури біосполук, наприклад, вітаміну В( та пеніциліну. Є автором
кількох підручників з органічного синтезу.
424
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
16.3.5. Розщеплення ацилазидів
❖ Реакція Курціуса1. Під час розщеплювання азидів кислот СЛН2,,+іС(Жз за
Курціусом відбувається їхнє внутрішньомолекулярне перегрупування. У разі
нагрівання ацилазидів у спирті спочатку настає втрата молекули Нітрогену; далі
нестійкий інтермедіат зазнає перегрупування в естер ізоціанової кислоти, який
миттєво приєднує молекулу спирту і перетворюється в так званий уретан -
естер алкілкарбамінової кислоти. Уретани омиляються до амінів у разі
нагрівання з кислотами або лугами:
С^^О^^^^Н^^О-К:] -СпИ2^СО \
С„Н2„+1ЫНСООС2Н5 СпН2^Ш2 + С02 + С2Н5ОН
\
Уретан
Вихідні ацилазиди одержують взаємодією хлорангідридів карбонових
кислот з натрій азидом:
ЯСОСІ + — К-С-Н=№=Й
О
Азиди кислот також можна одержати дією нітритної кислоти на гідразиди
карбонових кислот:
о
я-с^-ын,
О N0
о
❖ Реакція Шмідта. В 1923 р. К. Шмідт розробив метод безпосереднього
одностадійного перетворення карбонових кислот в аміни. За наявності сульфатної
кислоти карбонові кислоти, взаємодіючи з натрій азидом, перетворюються не в
ацилазиди, а в спряжені сполуки, що легко відщеплюють молекулу Нітрогену,
перегруповуючись в ізоціанати, які вода розкладає до амінів: /
Я-СООН + НЫ3 -і£ Я-С-МН-гЬы Я-СО-ЙН ^ Я-Ы=С=0 Ш Я-Щ, + С02
О
16.3.6. Реакція Ріттера
Цей метод досить зручний для синтезу первинних амінів, що мають
третинні алкіли. Процес відбувається через утворення ізонітрилу:
ЯзСОН^ЯзС та кзс-ЙЕС-Я'_^ ЯзС-Ш-С^ Я3С-Ш2
1 Курціус Т. (СштіиБ ТИ.) (1857-1928) - німецький хімік. Відкрив діазооцтовий етер, гідразин.
Розробив метод синтезу пептидів.
Розділ 16. Аліфатичні аміни
425
16.4. Хімічні властивості амінів
16А. 1. Кислотно-основні реакції
❖ ИИ-кислоти. Первинні й вторинні аміни є дуже слабкими кислотами. З
металоорганічними сполуками або лужними металами вони повільно утворюють
солеподібні сполуки, які легко гідролізує вода:
я2>щ ^ Я2їГьі + -^ я2>щ + ЬіОН
■ Рухливість атомів Гідрогену в амінах можна підтвердити за допомогою
реакції Чугаєва—Церевитиновах-Терентьєва:
Я->Щ2 + СН3МвІ -> Б^Н^І + СКЦ
Я2№1 + СНзї^І -> ^NN^1 + СН4
❖ Основність амінів. Основні властивості амінів виражені достатньо
сильно. Аміни, як основи, утворюють солі навіть з такими слабкими кислотами,
як карбонатна, ціанідна й сульфідна; з водою вони дають алкіламоній
гідроксиди:
СН3>Ш2 + НОН 5=2: СН3ЙН3 + ОН",
які виявляють лужну реакцію з індикаторами.
Основність аліфатичних амінів вища, ніж амоніаку завдяки позитивному
індукційному ефекту алкільних груп (+І-ефект), які підвищують електронну
густину на атомі Нітрогену. З огляду на це метиламін, як основа, сильніший, ніж
амоніак:
СН3— -СН3 > СН3— СН3 > СН3>ІН2 > КН3
І
сн3
Ця закономірність сили основності справджується для неводних розчинів або в
газовій фазі. Вирішальну роль у водних розчинах відіграє не електронний, а
просторовий чинник, тобто доступність атома Нітрогену для протона, тому
третинні аміни за силою основності поступаються вторинним. Розгалуженість
алкільних груп в амінах також знижує їхню основність. Наприклад, діізопропіламін,
як основа, слабкіший, ніж пропіламін. Вільне обертання алкільних залишків
навколо одинарного зв'язку також позначається на силі основи. Наприклад,
піролідин, де атом Нітрогену доступніший для протона, сильніший від діетиламіну:
1 Церевітінов Ф.В. (1874—1947) - радянський учений, професор московських ВНЗ. Головні праці у
галузі хімії та технології харчових продуктів. Досліджував хімічний склад плодів та овочів.
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
;н2-сн2\
:н2-сн/
,ЫН > (СН3СН2)2МН
Електронодонорні алкільні групи виявляють здатність делокалізувати
позитивний заряд у заміщеному йоні амонію й цим забезпечують стабілізацію
йона, неможливу для незаміщеного йона амонію. Отже, заміщений іон амонію
зазнає стабілізації завдяки подачі електронів точно так само, як карбокатіон.
Відповідно до іншого погляду, алкільна група зміщує електронну густину в бік
атома Нітрогену, внаслідок чого четверта пара електронів стає доступнішою для
узагальнення з протоном.
■ Четвертинні амонієві солі можуть замінювати свій аніон на гідроксильну
групу. Цього досягають, зокрема, за допомогою реакції обміну з вологим
арґентум оксидом, що дає відповідні гідроксиди:
які є сильнішими основами, ніж водні розчини амоніаку й амінів. Наприклад,
тетраметиламоній гідроксид (СН3)4М+ОН" за основними властивостями майже не
поступається їдким лугам. У водних розчинах вони повністю дисоційовані.
Під час кип'ятіння такі амоній гідроксиди поступово розпадаються на третинні
аміни, спирти або алкени:
Реакція має важливе значення для з'ясування структури азотистих гетеро-
циклів, наприклад, піридину, і відома в літературі як розщеплення за Гофманом.
Як і амоніак, аміни утворюють комплексні катіони з низкою катіонів
важких металів (А%+, Си2+, Со3+ та ін.).
■ Для ідентифікації амінів часто одержують пікрати, які зазвичай легко
кристалізуються й мають чітку температуру плавлення.
Солі амінів утворюють подвійні сполуки з багатьма солями металів,
наприклад, сполуки з аурум (III) хлоридом 113ЫН[АиС14] і платинум {IV)
хлоридом (ЮШз)+2(Рі:С1б)~, які часто застосовують для характеристики амінів.
❖ Алкілування амінів. Як нам уже відомо, первинні, вторинні й третинні
аміни взаємодіють з галогеноалканами та спиртами. Реакція відбувається як
бімолекулярне заміщення, тобто за механізмом 8н2:
2 (СН3)4Ы+С1 + Ag20 + Н20
2(сн3)4ы+он +2 АйСІ,
(с3н7)4н+оьг -> (с3н7)3к + сн3сн2сн2он сн3-сн=сн2 + н20
16.4.2. Реакції амінів як нуклеофілів
к-кн2 + сн3г
І / \
н н н
ішн2—СН3І
+
Розділ 16. Аліфатичні аміни
427
Унаслідок обробки їх водним розчином лугу одержують аміни у вільному стані:
+
Я-КН^-СНзІ + ИаОН-
Я-МН-СН3 + КаІ + Н20
❖ Ацшіюеання амінів. Первинні й вторинні аміни, що мають атом
Гідрогену при атомі Нітрогену, зазнають ацилювання тими ж реагентами, що й
амоніак, тобто галогеноангідридами, ангідридами кислот й арилсульфохлори-
дами, утворюючи аміди:
СН3СС1 + 2(СН3)2КН-
О
СН3СС1 + 2С2Н5КН2
О
(СНз)2КССН3 + (СНз)2КН2СГ
о
М^-Диметилацетамід
с2н5ш рсн3 + с2н5ын3с7
о
ЛГ-Етилацетамід
Кислота, що утворюється, зв'язує еквівалентну кількість аміну, який не
прореагував. Цей метод неекономний, якщо амін важко синтезувати або він є
дорогим реагентом. Реакція відбувається як нуклеофільне приєднання-
відщеплення по карбонільній групі:
СН,СН,гШ, + н,с-сї_. ■
сі
н с?~
-сн,сн,—Ж—с-сн, -—■
н (І
сі
сн3сн2-ш-с;
сн,
Первинні аміни реагують із фосгеном - дихлорангідридом карбонатної кислоти;
карбомоїлхлорид, що утворюється, далі перетворюється в ізоціанат. Усього на
реакцію витрачається три молі аміну, з яких два - на нейтралізацію гідроген хлориду:
н 9
К-М: + С1-С-С1 —
Н
Нч О
І і II
-М-І— Г".
я-ы^с—СІя-ы=с=о
Ізоціанат
Карбомоїл-
хлорид
Ізоціанати, в основі яких є кумульовані подвійні зв'язки, легко реагують з
водою, спиртами й амінами. У всіх випадках атаки зазнає електрофільний атом
Карбону ізоціанатної групи:
0
►я->ж,+со,
-5+5-5
я-и=с=о
Н20
я-ш-с-он —
оя
я-ш-с=о
Уретан
2 я-ш-с^-шя
о
Заміщена сечовина
Амін
428
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
Третинні аміни взаємодіють із хлорангідридами кислот, даючи четвертинні
солі, які самі стають ацилювальними агентами.
16.4.3. Окиснення амінів
Хід окиснення залеясить як від типу аміну, так і від умов окиснення.
• Первинні аміни дією органічних надкислот або кислоти Каро1 (h2S05)
перетворюються в гідроксиламіни, оксими, нітрозо- і нітроалкани: —"
^ roi
,CH-NH2 ;c=N-OH
R R
R^ roi R,
,ch-nh2 -u. ch-no
r r
R^ roi R,
,ch-nh2 ;ch-no2
R r
Дією сильнішого окиснювача - трифторонадоцтової кислоти - первинні
аміни окиснюються відразу до нітросполук. Цей метод має практичне значення
для синтезу нітросполук з третинним алкільним замісником.
• Вторинні аліфатичні аміни за тих же умов дають діалкілгідроксиламіни:
R> -a R N-он /
R R'
З препаративною метою окиснення первинних і вторинних амінів недоцільне
• Третинні аміни в разі окиснення пероксидом Гідрогену кількісно
перетворюються в оксиди третинних амінів, у яких атоми Нітрогену й Оксигену
зв'язані семиполярним зв'язком:
R3N + Н202 -> R3N+-CT + Н20
Якщо нагрівати амін оксид, який має рухливий атом Гідрогену в р-поло-
женні, до 150 °С, то він розкладається на алкен і похідну гідроксиламіну:
¥ 97 ч
—(^-ch^-r - /с^н, + r,noh
R
1 Каро Н. (Caro N.) (1871-1935) - німецький хімік. Праці з каталітичного окиснення аміаку,
газифікації торфу. Розробив ціанамідний метод фіксації азоту.
Розділ 16. Аліфатичні аміни
429
Однією з переваг цього методу синтезу є те, що він дає змогу одержувати
алкени з чітко фіксованим подвійним зв'язком.
16.4.4. Реакції амінів з нітритною кислотою
Для з'ясування ступеня заміщення аміну можна використати різне реагуван-
ня первинних, вторинних і третинних амінів на дію нітритної кислоти.
• Первинні аліфатичні аміни реагують з нітритною кислотою, утворюючи
солі діазонію; та оскільки аліфатичні солі діазонію дуже нестійкі, вони легко
розкладаються, утворюючи складні суміші органічних продуктів, серед яких
переважають спирти. У цьому разі виділяється азот:
RNH2 + HONO RNHNO -—- RN=NOH
НП " + ho
RN=N СІ" N2 + СІ" + R+ ROH + HCl
Передбачають такий механізм реакції. Якщо до водного розчину амін
гідрохлориду додавати натрію нітрит, то утворюється нітритна кислота. Вона
перебуває у рівновазі з нітроген (III) оксидом, який взаємодіє з первинним аміном.
За звичайних умов він перегруповується в діазогідрат. У разі взаємодії з гідроген
хлоридом виникає нестійка діазосполука, що розпадається до карбокатіону:
NaN02 + HCl— HN02 + NaCl 2HN02^ N203 + H20
+
RCH^-NHj +0=N-0-N=0^ RCH2CH2-NH2~ N~ ff-NO^
H+ + dr
RCH^N-NO^ RC^CH^N-OH ^ RCH2CH2N^N — RCH2CH2 + N2
H
Подальший напрям реакції залежить від природи розчинників. Кінцевими
продуктами можуть бути спирти, етери та ізомерні алкани й алкени, що
утворилися з карбокатіону внаслідок гідридних зсувів, наприклад:
CH3CH2CH2NH2 ІШ- [CH3CH2CH2N2J [СН3СН2СН2]
— СН3СН2СН2ОН + СН3СН(ОН)СН3 + сн3сн=сн2 + ^c-cr,
сн2
Ця реакція має лише обмежене синтетичне значення. Однак той факт, що
азот виділяється кількісно, важливий в аналізі амінокислот і білків.
• Вторинні аміни під час взаємодії з нітритною кислотою перетворюються у
нітрозаміни:
R2NH + HONO -» R2N-NO + Н20
430
Чирва В.Я., Ярмолюк С.М., Толкачова H.B., Земляков О.Є. Органічна хімія
Нітрозаміни - нейтральні речовини білого або злегка жовтуватого кольору,
розчини яких, як і всіх інших нітрозосполук, забарвлені в зеленкувато-блакитні
кольори. Вони нерозчинні в розведених водних мінеральних кислотах. Якщо
нагрівати їх з концентрованими мінеральними кислотами, то можна одержати
вторинні аміни:
(Q,H2,í+1)2N-NO + НС1 -> NOCÍ + (CH^NH
Доказом будови нітрозамінів може бути їхнє відновлення у двозаміщений
несиметричний гідразин:
R2N-NO -> R2N-NH2.
Неорганічні нітрити, які протягом тривалого часу використовували як
консерванти в м'ясній промисловості, виявилися мутагенами. Очевидно, що в
організмі нітрити взаємодіють з гетероциклами нуклеїнових кислот, спричиняючи
мутацію генів.
• Третинні аміни з нітритною кислотою за звичайних умов не реагують.
7 6 А.5. Якісні реакції на первинні аміни
❖ Ізонітрильна реакція. Первинні аміни під час нагрівання з хлороформом у
спиртовому розчині лугу утворюють карбіламіни, які частіше називають
ізонітрилами:
RNH2 + СНСІз + 3NaOH -» 3NaCl + 3H20 + RNC
Ізонітрили, на відміну від нітрилів, отруйні й огидно пахнуть, тому їхнє
утворення — чутлива якісна реакція на первинні аміни.
❖ Проба на гірчичну олію. Первинний амін взаємодіє з сірковуглецем із
утворенням алкілдитіокарбамінової кислоти:
RNH2 + CS2 R-rj ~^~SH
Якщо до розчину цієї сполуки додавати солі важких металів (HgCl2, AgN03
й ін.), то утворюватиметься відповідна алкілдитіокарбамінова сіль, яка під час
кип'ятіння розчину розпадається на сульфід металу й гірчичну олію - естер
ізороданідної кислоти:
2RNH-CSSAg 2R-NOS + Ag2S + H2S
Гірчична олія
Гірчичні олії мають різкий запах, що нагадує запах гірчиці або часнику, за
цією ознакою їх можна легко виявити.
Розділ 16. Аліфатичні аміни
431
16.5. Діаміни
Діаміни - це сполуки, що містять дві аміногрупи. Назви цих сполук можна
побудувати за правилами замісної номенклатури ШРАС. Також використо-
вують назви, що складаються з назв двовалентних вуглеводневих замісників і
суфікса -діамін. Наприклад:
НзИ-СНг-СНг-ИНг Н2К-СННСН2)4-СНг->ІН2
Етандіамін-1,2 Гександіамін-1,6
етилендіамін гексаметилендіамін
■ Діаміни можна отримати всіма тими ж способами, що й моноаміни:
алкілуванням амоніаку а,со-дигалогеноалканами, відновленням динітрилів або
амідів двоосновних кислот та ін.
■ Етилендіамін використовують як ліганд для одержання комплексних
сполук.
■ Широкого застосування набув гексаметилендіамін, який синтезують з
адипінової кислоти. Спочатку її амонієву сіль сухою перегонкою перетворюють
у діамід. Підвищуючи температуру реакційної суміші, з діаміду через де гідра-
тацію синтезують динітрил адипінової кислоти, який каталітично гідрують до
гексаметилендіаміну:
НООС-(СН2)4-СООН-?І^Ш4ООС-(СН2)4--СООШ4
—Ш2СО-(СН2)4-СОМН2 т.н-с-(сн2)4-с^н ^Ш2-(СН2)6-Ш2
^ Гексаметилендіамін
Старіший метод одержання гексаметилендіаміну такий:
СН^СН—СН=СН2 + С12— СІ—-СН—СН=СН—СН2С1 —
Дивініл 1,4-Дихлор-2-бутен
мес-сн2сн=снсн2-сек ш2-(сн2)-кн2
■ 3 гексаметилендіаміну й адипінової кислоти шляхом реакції поліконден-
сації одержують синтетичне волокно найлон:
«ноос-ссн^-соон + пщ-(сн2)6-ш2 --со-ссн^-аьш-(ау^м-
найлон
■ Продукти метаболізму аргініну й лізину - путресцин і кадаверин - є отру-
тами. Вони спричинюють неякісність м'яса і багато в чому визначають трупний
запах:
Н2>ЇСН2СН2СН2СН2КН2 Н2НСН2СН2СН2СН2СН2Ш2
Путресцин Кадаверин
432
Чирва В.Я., Ярмолюк СМ., Толкачова H.B., Земляков О.Є. Органічна хімія
16.6. Діазометан
❖ Одержання діазометану. Діазометан виявив Г. Пехманн1 1894 р., діючи
лугом на нітрозометилуретан. Намагаючись одержати ізонітрил H2N-N+=C з
хлороформу й гідразину, Г. Штаудінгер2 синтезував діазометан:
H2N-NH2 + СНСІ3 + 3NaOH CH2=tf=N"
Зазвичай для синтезу діазометану, який став важливим реагентом,
використовують одну з таких реакцій:
HNO, V 2NaOH
CH3NHCNH2 ^ CH31}JCNH2 CH2N2 + Na^C^ + NH3 + H20
О NO
Метилсечовина ЛШітрозометилсечовина
HNO,
CH3NH<j:OC2H5 1 CH3^ -COC2H5 ™ CH2N2 + Na2C03 + NH3 + H20
О ON О
Метилуретан W-Нітрозометилуретан
❖ Реакції діазометану. Найважливіша реакція аліфатичних діазосполук -
взаємодія з рухливим атомом Гідрогену:
CH2N^N: + Н+ CH3ft=N СН3 + N2
СН3 + ROH CH3OR + Н+
Так уводять метальну групу в молекули органічних кислот або фенолів:х
RCOOH + CH2N2 RCOOCH3 + N2
С6Н5ОН + CH2N2 С6Н5ОСН3 + N2
Метилювання діазометаном - надзвичайно вдалий спосіб перетворення
кислот у метилові естери, чим користуються в разі експрес-аналізів сумішей
кислот.
Спирти можна метилювати діазометаном, попередньо перетворивши їх в
ансольвокислоти, що на практиці виконують додаванням до спиртів невеликої
кількості цинк хлориду (II):
ROH + ZnCL [ROZnCl2]-H+^Ä ROCH3 + N2 + ZnCl2
1 Пехманн Г. (Респтапп Н.) (1850-1902) - німецький хімік. Головні праці присвячені препаративному
органічному синтезу.
2 Штаудингер Г. (Біаисііг^ег С) (1881-1965) - німецький хімік. Один із засновників хімії полімерів.
Розділ 17
ОКСОСПОЛУКИ
Функціональною групою оксосполук є карбонільна група, або оксогрупа
>С=0. Залежно від замісників, пов'язаних з оксогрупою, ці сполуки поділяють
на альдегіди й кетони. В альдегідах з карбонільною групою зв'язані вугле-
водневий замісник і атом Гідрогену (у найпростішому випадку два атоми
Гідрогену), тоді як у кетонах карбонільний атом Карбону зв'язаний з двома
вуглеводневими замісниками:
ьі-с=о я-с-я
і м
н о
Альдегіди Кетони
Назва альдегіди походить від скорочення латинських слів а! (соЬої) (ІеЬусІ
(п^епаШт) - алкоголь, позбавлений Гідрогену. Назву кетони дав Л. Гмелін
1848 р. Вона походить від назви найпростішого представника цього класу -
ацетону, якій одержували з кальцій ацетату (лат. асеШт - оцет).
17.1. Номенклатура й будова
17.1.1. Номенклатура
❖ Альдегіди
• Раціональна номенклатура. У літературі трапляються застарілі назви альде-
гідів, побудовані за правилами раціональної номенклатури. За основу назви
беруть оцтовий альдегід, а складніші сполуки розглядають як продукти
заміщення атомів Гідрогену в його метальній групі на вуглеводневі замісники,
наприклад:
н3с сн2 сн с н3с-с-с'
сн3 н сн3 н
Метилетилоцтовий альдегід Триметилоцтовий альдегід
• Замісна номенклатура ШРАС. Як головну групу альдегідну функцію
називають за допомогою суфіксів -аль або -карбальдегід. Для позначення
434
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
альдегідної групи в бічному ланцюзі або за наявності старшої групи засто-
совують префікс форміл-:
5 4 3 2 1
сн3сн=снснсн=о
і
СН3
2-Метилпент-3-еналь
За основу взято найдовший
вуглеводневий ланцюг із голов-
ною (альдегідною) групою. Ну-
мерація від головної функції.
о"сн=о
Циклогекс-2-енкарбальдегід
Основа - циклогексен. Поча-
ток нумерації від карбоніль-
ної функції.
НООССН^НСООН
сн=о
2-Формілянтарна кислота
Головна функція - кар-
боксильна група. Основа -
бурштинова кислота.
• Дозволено використовувати папівтривіольні назви альдегідів, утворені від
тривіальних назв карбонових кислот, наприклад:
НСНО - формальдегід, СН3СНО - ацетальдегід, СН2=СНСНО - акрилальдегід.
❖ Кетони
• Замісна номенклатура. У замісний номенклатурі карбонільну групу в
кетонах позначають суфіксом -он або префіксом -оксо.
• Збережена тривіальна назва СН3СОСН3 ацетон.
• Радикально-функціональна номенклатура. Назва кетонів складається з
перерахування за абеткою вуглеводневих замісників (для симетричних кетонів
використовують числовий префікс ди-) і назви функції кетон. Приклади таких
назв наведені далі курсивом:
н3с-с-снсн2сн3
О СН3
3-Метилпентан-2-он
бтор'Бутилметіиікетон
Основа - пентан. Нумерація
починається з того краю, до
якого ближче головна функція.
5 4 3 2 1
НОСН2СН2ССН=СН2
2 2II 2
о
5-Гідроксипент-1 -ен-3-он
Вініл-2-гідроксиетилкет он
Головна функція (кетон) роз-
ташована в середині ланцюга,
тому нумерація йде від подвій-
ного зв'язку.
3 2
сн
о
4-( 1 -Оксоетил)циклогексанон
Основа - циклогексан. Кето-
група в бічному ланцюзі
позначена префіксом -оксо.
17.1.2. Будова оксосполук
Доказом наявності в оксосполуці карбонільної групи може бути реакція
заміщення оксигенового атома карбонілу під дією фосфор (V) хлориду на два
атоми Хлору з утворенням гем-дихлороалкану:
Х=0 + РСЬ
/СІ
хсі +РОС13
Те, що в цьому разі будова карбонового ланцюга не змінюється, свідчить
синтез тієї ж оксосполуки під час гідролізу дихлоралкану:
Розділ 17. Оксосполуки
435
х:\ н2о
^=0 + 2 неї
Саме за цією реакцією гідролізом метиленіодиду СНгЬ О. Бутлеров
синтезував найпростішу з оксосполук - формальдегід СНг=0.
Розглянемо будову карбонільної групи. Атом Карбону карбонільної групи
має три ^-гібридизовані орбіталі, які зв'язують його з трьома іншими атомами
о-зв'язками. Оскільки ці зв'язки використовують 5р2-орбіталі, то вони
розташовані в одній площині під кутом 120° один до одного. Орбіталь р2 атома
Карбону, що залишається, перекривається з /?г-орбіталлю атома Оксигену,
утворюючи я-зв'язок; отже, атоми Карбону й Оксигену з'єднані подвійним
зв'язком:
я^-Ч200
К ^20°
Подвійний зв'язок карбонільної групи складається, як і у випадку алкенів, з
я- і сг-зв'язків, проте, на відміну від алкенів, електрони подвійного зв'язку С=0
з'єднують атоми, що сильно відрізняються електронегативністю, і тому
електронна густина розподілена нерівномірно: зокрема, рухлива я-хмара сильно
зсунута у бік більш електронегативного атома Оксигену. Про це, зокрема,
свідчать суттєві дипольні моменти альдегідів (ц -2,5 ц) і кетонів (р, -2,7 Б):
Нерівномірний розподіл електронної густини в карбонільній групі можна
також зобразити відповідно до теорії резонансу за допомогою двох канонічних
.структур:
\
С=0
\ + -
-/С-0
Унаслідок полярності зв'язку С=0 атом Карбону отримує ефективний
позитивний заряд, його називають електрофільним центром. Тому він взаємодіє
з нуклеофілами, а атом Оксигену карбонільної групи - з електрофілами.
Неподілені пари електронів атома Оксигену карбонільної групи надають їй
властивості слабкої основи Льюїса. Справді, альдегіди й кетони зазнають
протонування кислотами, унаслідок чого утворюється оксонієвий катіон:
кЛҐ( + н+
к*
он
Я-С-Я'
:0Н -
436 Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
Взаємодія з кислотою збільшує поляризацію карбонільної групи, підвищує
електрофільність карбонільного атома Карбону, що є причиною кислотного
каталізу реакцій нуклеофільного приєднання.
Особливості електронної будови оксосполук слугують причиною СН-
кислотності атомів Гідрогену біля а-карбонового атома відносно карбонільної
групи.
Як альдегіди, так і кетони мають карбонільну групу С=0, і їх часто
розглядають разом як карбонільні сполуки. Цілком природно, що більшість
властивостей альдегідів і кетонів подібні. Однак по сусідству з карбонільною
групою в альдегідах перебуває атом Гідрогену, а поряд із карбонільною групою
в кетонах - два вуглеводневі замісники. Це розходження в структурі зумовлює
відмінності у хімічних властивостях:
- альдегіди легко зазнають окиснення, тоді як кетони за цих умов
залишаються незмінними;
- альдегіди зазвичай активніші, ніж кетони, в реакціях нуклеофільного
приєднання - характерних реакціях карбонільних сполук.
Останню особливість пояснюють тим, що в альдегідах реакційний центр -
атом Карбону карбонільної групи - просторово не утруднений і має більший
частковий позитивний заряд, оскільки він зв'язаний тільки з одним, порівняно з
кетонами, вуглеводневим замісником, що виявляє позитивний індукційний
ефект. Треба, однак, зазначити, що самі вуглеводневі замісники можуть мати
атоми або групи акцепторної та донорної природи. Тому вони можуть, особливо
в ос-положенні щодо атома Карбону карбонілу, відповідно, збільшувати або
зменшувати позитивний заряд на атомі Карбону С=0-групи.
17.2. Фізичні властивості
Наявність карбонільної групи робить молекули альдегідів і кетонів полярними,
тому вони мають вищі температури кипіння, ніж неполярні сполуки з такою ж
молекулярною масою. Самі по собі вони не здатні утворювати міжмолекулярних
водневих зв!язків, оскільки містять атоми Гідрогену, зв'язані тільки з атомом
Карбону. Внаслідок цього їхня температура кипіння нижча, ніж у відповідних
спиртів і карбонових кислот. Для прикладу можна порівняти бутиральдегід (76 °С) і
етилметилкетон (80 °С) з я-пентаном (36 °С) і діетиловим етером (35 °С), з одного
боку, і н-бутиловим спиртом (118 °С) та пропіоновою кислотою (141 °С), з іншого.
Формальдегід - газ із гострим запахом, розчинний у воді. Нерідко його
використовують у вигляді 40 % водного розчину, який називають формаліном.
Нижчі альдегіди - рідини з різким неприємним запахом. Вищі гомологи мають
запах квітів і фруктів.
Нижчі альдегіди й кетони помітно розчинні у воді, імовірно, внаслідок
утворення водневих зв'язків між молекулами речовини й розчинника.
Розділ 17. Оксосполуки
437
Розчинність різко зменшується для членів гомологічного ряду, починаючи з С5-
атома, оскільки подовження вуглеводневого замісника збільшує гідрофобність
сполук, що спричиняє, відповідно, зменшення розчинності у воді.
Зазвичай розгалуження ланцюга спричинює зниження температури кипіння
й питомої густини.
17.2.1. Спектроскопія оксосполук
• УФ-спектроскопія. В УФ-спектрах альдегіди й кетони мають інтенсивну
смугу поглинання за 185-195 нм унаслідок л—>я*-переходу й слабку - за 270-
290 нм, що відповідає переходу я—>7с*.
• ІЧ-спектроскопія. В ІЧ-спектрах альдегідів зазвичай наявні смуги погли-
нання, зумовлені валентними коливаннями С=0- і С-Н-зв'язків: vc=o 1 725-
1 685 см"1; Vc-h 2 820-2 720 см"1. ІЧ-спектри кетонів мають смуги поглинання
1 710-1 720 см"1, що належать валентним коливанням С=0-групи.
• Мас-спектрометрія. У мас-спектрах альдегідів у разі електронного удару, як
звичайно, відбувається розрив С-С-зв'язку в Р-положенні щодо карбонільної групи:
RCH2CH2CH2CHO — RCH2CH2CH2CH(f — R-CH=CH2 + С2Н60
З молекулярного йона альдегідів утворюються осколкові йони з масою 18 й 28.
Кетони порівняно з альдегідами дають інтенсивніший пік молекулярного
йона, який розщеплюється за Р-С-С-зв'язком. Утворюються два інтенсивні піки
йонів R+ й RCO+, що виникають у разі розриву а-С-С-зв'язків.
• Спектроскопія ПМР. У ПМР-спектрах є характерний сигнал протона
альдегідної групи при 8,5-11,0 м.ч. Приклади хімічних зсувів атомів Гідрогену
деяких карбонільних сполук наведені нижче:
О Н3С-С-СН3 Ґ"\-п
8 9,72 (а), 2,17 (СН3) м.ч. 5 2,07 м.ч. 5 2,06 (а), 2,02 (Ь) м.ч.
17.3. Способи синтезу оксосполук
17.3.1. Промисловий синтез
Альдегіди й кетони одержують низкою загальних методів. Однак перші два
представники гомологічного ряду альдегідів і ацетон синтезують специфічними
способами. До багатотоннажних промислових продуктів належать формальдегід,
ацетальдегід і ацетон.
438
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
❖ Формальдегід сьогодні у промисловості одержують окисним дегідру-
ванням метанолу над срібним та оксидним ферум-молібденовим каталізаторами
за наявності кисню повітря:
сн3он-^сн2о + н2
СН3ОН + 02 Н20 + СН20
Газоподібний формальдегід, що утворюється, поглинають врдою. Отри-
маний у такий спосіб 40 % розчин містить деяку кількість метанолу.
Зазначимо таке: якщо перша реакція відбувається з виділенням тепла, то
друга - це ендотермічний процес. Багатотоннажне виробництво формальдегіду в
проточних апаратах безперервної дії з частково контрольованим окисненням дає
змогу зменшити ендотермічність процесу.
■ Пряме окиснення метану за 400-600 °С із залученням нітроген (IV)
оксиду не може конкурувати з попередніми методами через низькі виходи
цільового продукту, а також окиснення формальдегіду до мурашиної кислоти:
N0^ + СН4 — СН3 + Ш02
СН3- + 02 — н3оо-о-
н3с-о-о-+ СН4— СН3ООН + СН3-
сн3оон — сн3о • + он—- сн2о + Н20
н3с-о-о-+сн3о—- сн3оон + сн2о
Формальдегід — газ, тому його зручніше використовувати у вигляді водного
розчину (формаліну), твердого полімеру - параформальдегіду (СН20)п або оліго-
меру - триоксану (СН20)3.
❖ Лцетальдегід щораз частіше в промисловості воліють одержувати за
екологічно безпечним методом Фаворського й Шостаковського:
нснсн+с2н5он— Н2С=сн-о-с2н5^^ н3с-с^ + с2н5он
Вінілетиловий етер Н
замість традиційної гідратації ацетилену за реакцією Кучерова:
Н^04 'р
НСЕСН+НаО^НзС-С
Обидва методи застосовують у країнах, які мають скруту з вуглеводневою
сировиною.
■ Сьогодні у промисловості віддають перевагу рідкофазовому каталі-
тичному окисненню етилену киснем. Вихід ацетальдегіду - близько 98 %:
Розділ 17. Оксосполуки
439
СиС12/Рс1С12
О
н2с=сн2 + 02
н
Детально про цей метод описано в розділі, що присвячений хімічним
властивостям алкенів.
Головну кількість синтезованого ацетальдегіду використовують для
одержання оцтової кислоти.
■ Найчастіше ацетальдегід зберігають у формі його тримеру - паральдегіду:
❖ Ацетон нині у промисловості одержують разом із фенолом куменовим
(кумоловим) методом (див. розділ 31).
Ацетон також одержують під час бродіння крохмалю за Вейцманом.
Зі структурного споріднення альдегідів з первинними спиртами й кетонів із
вторинними випливає можливість одержання як спиртів з оксосполук, так і
оксосполук із цих груп спиртів шляхом каталітичного дегідрування або
окиснення.
❖ Каталітичне дегідрування спиртів у промисловості виконують, про-
пускаючи пари спиртів над порошком міді, срібла, нікелю, платини, не додаючи
окисника:
Цим методом досягають чудових виходів, його з успіхом застосовують у
виробництві, головно, формальдегіду й альдегідів складу С5 і вище. Реакція,
очевидно, полягає в дегідруванні спиртів з наступним згорянням водню, і тепла,
що виділяється, досить для підтримки потрібної температури реакції.
❖ Окиснення в розчині. Окиснення можна виконувати також "мокрим"
методом, використовуючи такий реагент, як хроматна кислота в 90-95 %
оцтовій кислоті:
Н3С О СН3
17.3.2. Одержання зі спиртів
ясн2он
ясно + н2
яснрн-^ ЯСНО + Н20
440
Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Під час синтезування альдегідів шляхом окиснення спиртів продукт реакції
варто негайно видаляти зі сфери реакції, щоб не допустити його подальшого
окиснення, тим більше, що нижчі альдегіди переганяють за нижчої температури,
ніж відповідні спирти. Незважаючи на те, що самі по собі альдегіди порівняно
повільно окиснюються, їхні гідрати легко й швидко перетворюються в кислоти:
[О]
Повільно
я-соон
^ К-СН(ОН)2^оІІ-СООН
Оскільки кетони стійкіші в реакціях окиснення, ніж альдегіди, то окиснення
хроматною кислотою стає загальновживаним методом одержання кетонів з
вторинних спиртів, наприклад:
ОН
СН^СНз СЬІ^СНз V-/ 112804
4-Етилциклогексанол 4-Етилциклогексанон, 90% циклооктанол циклооктанон
Давно відому реакцію окиснення спиртів хроматною кислотою детально
досліджував Ф. Вестхаймер1 на прикладі ізопропілового спирту. За помірної
концентрації кислоти швидкість реакції пропорційна до концентрації аніона
НСЮ4" який естерифікує спирт. Кислий естер спирту й хроматної кислоти за
участі води як акцептора протона зазнає внутрішнього окиснєння-відновлєнрш,
унаслідок яких утворюється карбонільна сполука:
НО^гО- + Н+ +(СН3)2рОН =5=^ (СН3)290^гОН + Н20
о ?н н о
НргчКСНз^СуО-^г-ОН Н30+ + (СН3)2СО + (рЮН
^—-н о о-
Те, що ланцюг електронних переміщень завершується на атомі Хрому,
означає, що атом Хрому, одержавши пару електронів, зі ступеня окиснення +6
переходить у нестійкий стан зі ступенем окиснення +4, а потім Сг+4
диспропорціонує в Сг+6 й Сг+3. Утворення естерів хроматної кислоти під дією
спиртів доведене їхньою екстракцією органічним розчинником.
■ Досить зручне в лабораторній практиці застосування манган (IV) оксиду:
1 Вестхаймер Ф. (ШезШеітег Р.Н.) (1912-2007) - американський хімік. Головні праці присвячені
дослідженню механізмів реакцій.
Розділ 17. Оксосполуки
441
нс=с-сн=ссн2он с-^-.нс=с-снг^-сно
сн3 сн3
2-Метилпент-2-ен-4-ин-1-ол 2-Метшшент-2-ен-4-иналь, 72 %
❖ Окиснення комплексами хромового ангідриду. Застосування реагентів,
розчинних в органічних розчинниках, дає змогу вести селективне окиснення.
Одним з ефективних окисників є комплекс піридину, хром (VI) оксиду і гідроген
хлориду - піридиній хлорохромат:
н-С9Н19СН2ОН + С5Нз>ШСгОзСГ ^'сГ*—- н-С9Н19-СНО
Декан- 1-ол Деканаль,92%
■ Для окиснення речовин, що містять чутливі до кислот групи, придатний
розчин комплексу хром (VI) оксиду з піридином. Особливо зручний цей метод
окиснення у випадку насичених та ненасичених спиртів. Як приклади можна
навести синтези таких альдегідів:
сн3(сн2)4-сн2он С^;"55ГЬ СН3(СН2)4-СНО
Гептан-1-ол Гептаналь, 93%
СНзСНгСНгСНзСН^СЕС-СНрН ^С^; СН3СН2СН2СН2СН2-С=С-СНО
Окт-2-ин-1-ол Окт-2-иналь, 84%
❖ Окиснення за Оппенауером. Надзвичайно вживаним у лабораторній
практиці є окиснення вторинних спиртів за Оппенауером надлишком ацетону за
наявності алюміній алкоголяту, який є слабкоосновним каталізатором і приско-
рює усталення рівноваги в реакції. Для синтезу кетонів рівновагу зміщають
праворуч, застосовуючи надлишок ацетону:
. [(СН3)2СНО]3А1
ОН О 6 ОН
ШрНЯ' + СН3^СН3^^ ^ Яр' + СН3(:НСНз
Роль алюміній алкоголяту полягає в координаційному зв'язуванні кетону.
Гідридне переміщення, що визначає швидкість реакції, відбувається в межах
координаційної сполуки, що утворюється, внаслідок чого настає рівновага:
?гч >і( н =^ н ;аі( н
сн^ н 3 н
442
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Гідридне переміщення доведене застосуванням міченого дейтерієм вихідно-
го вторинного спирту. У цьому разі від угруповання >СБОН до ацетону перехо-
дить дейтерій, і синтезується дейтерований вторинний спирт:
СНХОСН,
ОН
Оскільки окиснення за Оппенауером специфічне для спиртових груп і не
зачіпає подвійних зв'язків, то метод широко використовують для окиснення
ненасичених спиртів.
Для окиснення первинних спиртів реакція Оппенауера має обмежене засто-
сування, тому що за умов реакції можлива альдольна конденсація альдегідів, які
утворюються в процесі синтезу.
■ Ту ж рівноважну систему використовують для реакції, зворотної окиснен-
ню вторинних спиртів за Оппенауером, - для відновлення кетонів у вторинні
спирти за Мейєрвейном-Понндорфом-Верлеєм. У цьому випадку в надлишку
беруть ізопропіловий спирт, а нагрівання проводять так, щоб з рівноважної
суміші відганявся весь ацетон, що утворюється.
❖ Окиснення диметилсульф оксид ом. Останніми роками в лабораторіях як
окисник спиртів застосовують суміш диметилсульфоксиду й оцтового ангідриду
або безводної фосфатної кислоти. Карбонільні сполуки синтезуються з виходом
близько 80 %:
<р
Я-СН2ОН + (СН3)280 Я-С^ + (СН3)28 + Н20
н
■ Як з'ясував Н. Корнблюм, первинні спирти перетворюються з виходом
60-85 % в альдегіди під час обробки диметилсульфоксидом и-толуен-
сульфонатів (тознлатів) відповідних спиртів:
ІІ-СН2ОН + ТбСІ —- Я-СН2ОТ8 (СНз)г^ Я-СНО Тб = -80 —/~\-СН3
17.3.3. Озоноліз алкенів
Детально реакція розглянута в розділі 8. Цей метод прийнятий для си-
метричних дизаміщених алкенів, і це дає змогу одержувати тільки один альдегід:
О 7 'р
НХ-СН=СН-СН, —
3 3 сн2сі2 сн3соон 3 хн
Бут-2-ен Ацетальдегід
У випадку тетразаміщених алкенів озоноліз приводить до кетонів:
Розділ 17. Оксосполуки
443
н,с сн3 л не
Н3С^ ^СНзСН2с,2сн3соон НзС/С-°
2,3-Диметилбуг-2-ен Ацетон
Сьогодні з озонолізом конкурує оксидна деструкція алкенів під дією
періодату натрію ЫаЮ4 і тетраоксиду осмію ОбО^
17.3.4. Синтези на основі металоорганічних сполук
■ Під дією на магнійорганічні сполуки естерів мурашиної кислоти за
низької температури утворюються альдегіди:
Ю^Бг + НСООС2Н5 -> ЯСНО + Вг№^ОС2Н5
■ У разі взаємодії хлорангідридів гомологів мурашиної кислоти з реакти-
вом Гриньяра одержують кетони:
Ю^Вг + Я'С\° -ЯСОЯ' + ВгІУ^СІ
Реактиви Гриньяра також приєднуються за потрійним зв'язком групи -С=Ы,
даючи солі кетимінів. Під час їхнього розкладу водою з досить високими
виходами одержують кетони:
Я-С^Ы + Я'Мё1 — ^С=ЫМё1+ ^с=0 + Мё(ОН)1
я к
Для реакції необхідно брати еквівалентні кількості вихідних речовин, тому
що надлишок реактиву Гриньяра приводить до спиртів.
❖ Кадмійорганічні сполуки. Як засвідчили дослідження Г. Гільмана1 й
Дж. Нельсона (1936), ліпшим методом перетворення хлорангідридів кислот у
кетони є використання кадмійорганічних сполук. Для цього спочатку шляхом
взаємодії реактиву Гриньяра з сухим кадмій хлоридом одержують відповідні
кадмійорганічні сполуки Я-СсіС1, які, реагуючи з ацилхлоридами, дають кетони з
високим виходом:
Я-С(1С1 + СН3СОС1 інсГ я~£~снз
О
Під час взаємодії двох еквівалентів реактиву Гриньяра з одним еквівалентом
кадмій хлориду утворюється діалкілкадмій, що також легко реагує з ацилхлоридом:
1 Гільман Г.(Оі1тап Н.) (1893-1986) - американський хімік. Головні праці присвячені метало-
органічній хімії. Є автором "реагенту Гільмана".
444
Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
2Ю*^І + Сс1С12—К2СА + М£С\
Я2Сс1 +2СН3СОС1-^К-С~СН3+ СсіС12
О
Лише кадмійорганічні сполуки, які мають первинну алкільну групу, досить
стійкі, щоб їх можна було використати для синтезу кетонів.
Порівняно низька реакційна здатність кадмійорганічних сполук не тільки дає
змогу синтезувати кетони, а й сприяє широкому застосуванню цього методу, оскільки
кадмійорганічні сполуки не реагують з багатьма функціональними групами, з якими
реагують реактиви Гриньяра, наприклад, -N02, -СН >СО, -СООЯ. Тому наявність
цих груп у молекулі хлорангідридів не позначається на синтезі кетонів:
СНзОССНгСНзСШ + КСН^СНСН^]^ СН3ОССН2СН2С(СЬ^)2ОТ(СН3)2
О О О
Метил-7-метил-4-оксооктаноат(у-кетоестер)
❖ Літійдіалкілкупрати. Кетони з кількісними виходами синтезують
взаємодією хлорангідридів кислот з літійдіалкілкупратами:
Я'2СиЬі + 2 Я-С-СІ —-2 Я-С-Я1 + СиСІ + ЬіСІ
6 6
173.5. Одержання з кислот та їхніх похідних
❖ Піроліз карбонових кислот. Для синтезу кетонів пару кислоти про-
пускають за температури понад 300 °С над каталізатором - манган (II) або торій
(IV) оксидом:
^О 400-450 °С
гяс^он^^* ІІССЖ+С02 + Н20
У випадку двох різних кислот утворюється суміш кетонів, у тому числі й
кетон ЯСОЯ:
ЯСООН + Я'СООН —і^ЯСОЯ' + ЯСОЯ + Я'СОЯ' + С02 + Н20
МпО А *
Якщо одна з узятих кислот мурашина, то головним продуктом реакції буде
альдегід:
ЯСООН + НСООН -> ЯСНО + со2 + н2о
■ Цей метод розвився зі старого методу Піріа1 - сухої перегонки кальцієвої
солі кислоти (або суміші кислот). Свого часу він був єдиним промисловим
методом одержання ацетону:
1 Піріа Р (Рігіа Я.) (1814-1865) - італійський хімік. Головні праці в галузі синтетичної органічної
хімії. Вчитель С. Канніццаро.
Розділ 17. Оксосполуки
445
(СН3СОО)2Са -» СН3СОСН3 + СаСОз
Ця реакція висвітлює й роль оксидів металів як каталізаторів описаного вище
способу одержання кетонів.
❖ Відновлення хлорангідридів кислот
• Каталітичне відновлення за Розенмундом1-Зайцевим хлорангідридів кислот
(каталізатор — паладій, осаджений на ВаЕЮ^ воднем:
ЯСОС1 + Н2 ЯСНО + НС1
Успіх методу залежить від різниці швидкості синтезу альдегіду й швидкості
його відновлення до спирту. Тому К. Розенмунд запропонував до реакційної
суміші як каталітичну отруту додавати сірку, що сильно сповільнює відновлення
альдегідів. Самі кислоти відновлюються погано.
• Відновлення комплексними гідридами металів. Одним з ліпших методів
одержання різних альдегідів є відновлення хлорангідридів кислот літій три-
шрет-бутоксиалюмогідридом за -78 °С. Специфічний відновник і низька
температура унеможливлюють подальше відновлення альдегіду до спирту:
•» 2)Н20,Н+ •»
❖ Відновлення нітрилів. Нітрили можна відновити в альдегіди з виходами
близько 90 %. Реакцію виконують дією на субстрат одного еквівалента літій
алюмогідриду в тетрагідрофурані. На першій стадії відбувається відновлення
потрійного зв'язку до подвійного:
Я-С^ + ЬіАІЩ ЯСН=1чГ-Ьі + АІНз
Далі, під дією розведеної кислоти, цей проміжний продукт перетворюється в
альдегід:
Я-СН=М-Ьі + Н20 -> Я-СН=>Щ + ІЛОН
Я-СН=НН + Н20 Я-СНО + Ї<Н4+
Використання замість літій алюмогідриду дезактивованого відновника - літій
триетоксиалюмогідриду - унеможливлює подальше відновлення альдегідів:
СНз(СН2)4-СИ 1)УА1(0С?Ч3 СН3(СН2)4-СНО
^ 3 ,2 4 2)Н20 + Н+ V 24
Гексанітрил 2 Гексаналь
173.6. Гідратація алкінів
❖ Реакція Кучерова. Ацетилен і його гомологи під дією води за наявності
каталізатора перетворюються в оксосполуки. У цьому разі тільки ацетилен дає
альдегід, а саме - ацетальдегід.
1 Розенмунд К. (ЯоБептипсі КЖ) (1884-1965) - німецький хімік, учень О.Дільса. Є автором
кількох іменних реакцій.
446
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Усі гомологи ацетилену утворюють кетони за правилом Марковникова:
НОСЯ + Н20 СН3С^я
Має сенс говорити про синтез кетонів з алкінів, якщо молекули алкінів
симетричні:
СН3СН2СН2СН2С=ССН2СН2СН2СН3 СНзСНзСНзС^СН^С-С^СН^СН,
Дец-5-ін Декан-5-он,80% ^
В іншому випадку одержують суміш кетонів. Метод також має препа-
ративне значення, якщо потрійний зв'язок є на кінці карбонового ланцюга. У
цьому випадку утворюється метил-1-гідроксициклогексилкетон, наприклад:
ночуснсн но 9
нео,н2804 і^^^СНз
н2о 1^
1 -Етинілциклогексанол Метил-1 -гідроксицик-
логексилкетон, 84%
❖ Реакція гідроборувания-окиснення. Оскільки гідратація алкінів дає
кетони, то, щоб одержати на основі алкінів альдегіди, застосовують реакцію
гідроборування-окиснення:
ВН Н2°2
ЗС5НПС=СН—і. [ОД.^НЧИ-ІзВ-^* 3[С5Нп-СН=СН-ОН] —ЗОД.-СНг-СНО
Гепт-1-ін . Геїгганаль ^
17.3.7. Оксосинтез
У 1938 р. В. Реппе відкрив важливу реакцію синтезу альдегідів з олефінів і
суміші карбон (II) оксиду з воднем (водяний газ). Реакція відбувається за
наявності кобальту або нікелю як каталізатора за 200 атм. і температури, трохи
вищої від 100 °С (за вищої температури синтезуються спирти). Очевидно, що
найпростіший альдегід, який можна отримати таким способом, - це пропіоновий:
СН2=СН2 + СО + Н2 СН3СН2СНО
З гомологів етилену виникає суміш альдегідів з нормальним і розгалуженим
ланцюгами.
Реакцію з етиленом і пропіленом проводять у газовій фазі, а зі складнішими
(С4-С20) - У рідкій. Як видно з наведеної вище реакції, під час оксосинтезу
утворюються альдегіди, що містять на один карбоновий атом більше, ніж алкен.
Передбачають такий механізм реакції:
Розділ 17. Оксосполуки
447
2Со + 8СО —- Со2(СО)8 Co2(CO)g + Н2—►2НСо(СО)4
R-CH=CH2 + НСо(СО)4—- RCH2CH2Co(CO)4
R-CH2CH2Co(CO)4 + CO —- R-CH2CH2CO-Co(CO)4
R-CH2CH2CO-Co(CO)4+ HCo(CO)4-^RCH2CH2CHO +2Co(CO)4
Нижче наведемо, як приклади, низку таких реакцій:
СО + н, ?Нз
СН3(СН2)3СН=СН2 -2 СН3(СН2)5СНО + СН3(СН2)3-СН-СНО
Гекс-1-ен Гептаналь, 32% 2-Метилгексаналь, 32%
со + н
сн3СООСН2СН=СН2 -2 СН3СООСН2СН2СН2СНО
Алілацетат 4-Оксобутилацетат, 70%
' СО + Н2 |^^| СНО
Циклопентен Циклопентанкарбальдегід, 65%
17.3.8. Синтез оксосполукна основі галогеноалканів
❖ Гідроліз гемінальних дигалогенопохідних приводить до альдегідів, якщо
галогени перебувають біля кінцевого атома Карбону; якщо ж вони є у середині
ланцюга, то виникають кетони. Метод зручний за доступності гемінальних
похідних:
ЯСННа12 + Н20 -+ ЯСНО + 2ННа1
ЯСНа12Я' + Н20 -> ЯСОЯ' + 2ННа1
❖ Карбонілювання алкілгалогепідів. Перетворення галогеноалканів в
альдегіди з подовженням карбонового ланцюга на один атом досягають
обробкою їх натрій тетракарбонілферитом за наявності трифенілфосфіну:
я-сі + ка2[Те(со)4] я-сорє(со)3р(с6н5)3 СНзС0°" я-с^°
н
Зазначимо, що є ще низка інших реакцій одержання оксосполук, про які вже
йшлося раніше. Серед них:
а) ізомеризація етиленоксиду;
б) розщеплення а-гліколевих угруповань за допомогою йодної кислоти та
плюмбум (IV) ацетату;
в) пінаколінове перегрупування а-гліколів;
г) синтез на основі нітроалканів тощо.
448
Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
17.4. Хімічні властивості оксосполук
Реакції альдегідів і кетонів визначені наявністю карбонільної групи. Ця
група, по-перше, є місцем нуклеофільної атаки й, по-друге, збільшує кислотність
атомів Гідрогену, пов'язаних з а-карбоновим атомом. Обидва ефекти цілком
узгоджуються з будовою карбонільної групи, і, по суті, зумовлені здатністю
атома Оксигену приймати негативний заряд.
Якого типу реагенти будуть атакувати таку групу? Оскільки найважливіша
стадія в цих реакціях - це утворення зв'язку з електронодефіцитним (кислим)
карбонільним атомом Карбону, то карбонільна група найбільше схильна до
взаємодії з електрононадлишковими нуклеофільними реагентами, тобто
основами. Тому типовими реакціями альдегідів і кетонів є реакції нуклеофіль-
ного приєднання (механізм Ац).
В активованому комплексі атом Оксигену починає отримувати електрони й
негативний заряд. Саме тенденція атома Оксигену отримувати електрони,
точніше - його здатність нести негативний заряд, і є справжньою причиною
реакційної здатності карбонільної групи щодо нуклеофілів:
Ш Ми
к^Я^г к~ 9~ он
Я' У- Я'
Спочатку нуклеофільний агент атакує атом Карбону карбонільної групи,
змінюючи його гібридизацію з ^р2 на ^р3 і переводячи в тетраедричний інтермедіат.
Нуклеофільний агент може бути нейтральною молекулою, що має неподілену
електронну пару - вода, спирти, аміни, похідні амінів, тіоли; або аніоном - С>Г,
Н80з~, гідрид-іон, карбаніон. Як звичайно, приєднання нуклеофілів за
карбонільною групою є зворотним процесом. Виняток - відновлення карбонільної
групи до спиртів, а також реакції оксосполук з реактивом Гриньяра.
Зазвичай альдегіди легше вступають у реакції нуклеофільного приєднання,
ніж кетони. Ця різниця в реакційній здатності узгоджується з характером
проміжного стану реакції й, очевидно, передбачає спільну дію електронних і
просторових чинників. Кетони мають другу алкільну групу, а альдегіди - атом
Гідрогену. Алкільна група подає електрони й цим дестабілізує активований
комплекс унаслідок посилення негативного заряду на атомі Оксигену.
За наявності кислоти протон зв'язується з карбонільним атомом Оксигену.
Це попереднє протонування знижує енергію активації для нуклеофільної атаки,
оскільки дає змогу атому Оксигену одержати пару тг-електронів, не отримуючи
негативного заряду. Отже, приєднання до альдегідів і кетонів може бути
каталізоване кислотами:
Розділ 17. Оксосполуки
449
,/С=0
К'
с=он-
Я' он
я-с-он
Я'
Нижче наведено головні реакції оксосполук з їхнім детальним аналізом.
17.4.1. Реакції з О-нуклеофілами
❖ Гідратація оксосполук. Серед численних нуклеофілів, які беруть участь
у реакціях з оксосполуками, вода є найслабшим 0-нуклеофілом. Тому вона може
реагувати тільки з тими альдегідами, які несуть на карбонільному атомі Карбону
значний позитивний заряд. Серед них формальдегід, ацетальдегід, гекса-
флуороацетон та ін. Продукти гідратації таких сполук - геик-діоли - можуть бути
навіть виділені в індивідуальному стані, наприклад:
-Р
ссі3-с' + нр
н
сі3с-сн(он)2 сРз-с-СТз+нр —^С-ОрН)^^
Хлоральгідрат ^
Хлораль
Однак вони є скоріше винятком: звичайні гідрати розпадаються на вихідні
сполуки, й виділити їх не вдається.
Гідратація альдегідів і кетонів каталізована як кислотами, так і основами.
Розглянемо ці механізми на прикладі ацетальдегіду.
У кислому середовищі відбувається швидке протонування карбонільної
групи:
н3с-с^ +нв=:
н
/ОН
н3с-с' -
н
н3с-сх
ОН
н
В"
На другій повільній стадії, що визначає швидкість усього процесу,
приєднується молекула води:
,ОН~
н3с-сх
+ /ОН
Н
В" + Н,0
Повільно
н3с-с^
о+
в- СН3СН(ОН)2
-нв
У лужному середовищі реакція починається з атаки атома Карбону карбо-
нільної групи гідроксид-аніоном:
450
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зелдляков О.Є. Органічна хімія
н
Н3С\ /°" н2о
Сч
/ \ -НО-
НО Н
СН3СН(0Н)2
❖ Приєднання спиртів. Утворення ацеталів і кеталів. Під час взаємодії
альдегідів зі спиртами утворюються два типи похідних - напівацеталі й
ацеталі:
иг, „*° снзон Н3СХ ,0-СН3 сн3он,н* Н3С 0-СН3
3 н н^ он -н>° к ^о-сн3
Напівацеталь
(1 -метоксиетанол)
Диметилацеталь
ацетальдегіду
(1,1 -диметоксиетан)
Утворення напівацеталів може відбуватися як у кислому, так і в лужному
середовищі.
■ У лужному середовищі спочатку утворюється метоксид-аніон:
СН3ОН + ОН"
СН30"+Н20,
який атакує електронодефіцитний атом Карбону карбонільної групи (повільна
стадія)
го „ . сг
5-к/ - Повільно тт ^ > л л„
н3с-счч^_рсн3 « н3с-с-о-сн3
н н
з подальшим утворенням напівацеталю:
н3сч р
н,с ,он
с; + н,о ^ша?° 3 с
н о-сн,
н чо-сн,
■ У кислому середовищі спочатку відбувається активація карбонільної
групи альдегіду:
н3сч
н
.с=о +н+
Нзсч+ Нзс\ +
/С-он— ":с=он
Н ¥ґ
а потім метоксилювання інтермедіату з наступним викидом протона:
Розділ 17. Оксосполуки
457
н3сч+>^—н3с о-сн3 н3с о-сн3
с-он + носн3^= )с(н в )с( + н+
н н хон н он
■ На противагу утворенню напівацеталів реакція утворення ацеталів
каталізована тільки кислотами:
^зС\ /ОН Повільно Н,С\ /ОН, Повільно
іґ ХОСНз і/Ч-сн,
+
НзС-СНОСНз— Н£<В=6-СН3
сн3он Н-О-СНз
" НзС-СНо-СБз т^Г СНзСЩОСН^
Природно, щсГацеталі, які утворюються лише в кислому середовищі, стійкі
до дії основ. Цією обставиною користуються для тимчасового захисту
карбонільної групи в різних синтезах, що відбуваються тільки в лужному
середовищі, оскільки в кислотному регенеруються вихідні альдегіди:
СН3СН(ОСН3)2 + Н20 СН3С^н + 2СН3ОН
Зазначимо, що реакція утворення напівацеталів та ацеталів досить чутлива
до просторових чинників. Наприклад, кеталі важко синтезуються в разі
безпосередньої взаємодії кетонів зі спиртами, мабуть, за винятком промислового
виробництва 2,2-диметоксипропану:
НХ-С-СН, + 2СН3ОН =±: НзС^с(осн3)2
3 II 3 3 -27 °С и Г *
О
Тому кеталі одержують за методом Кляйзена-Арбузова1 з кетону й орто-
форміатів:
ОС2Н5
Н3С-С-СН3 + НС(ОС2Н5)3 н3с-с-сн3 + НСООС2Н5
о ос2н5
Кеталі мають ті ж властивості, що й ацеталі.
17.4.2. Реакції з Б-нуклеофілами
❖ Приєднання тіолів. Із сірчистими аналогами спиртів - тіолами -
альдегіди й кетони утворюють тіоацеталі й тіокеталі. їхні виходи зазвичай вищі,
ніж відповідних ацеталів і кеталів:
1 0. Арбузов (1877-1968) - академік АН СРСР (1942), засновник наукової школи фосфоро-
органіків, дослідник у галузі таутомерії, хімічної технології, історії хімії.
452
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
ОН
Азот
ВР3-С2Н5ОС2Н5 ^ С{*2
Н2СО + Н8СН2СН28Н —— Н,С СН~
2 2 2 СН,С1, 2 \ / 2
Б—СН2
1,3-Дитіан(85%)
Ця реакція відкрила простий і доступний шлях перетворення карбонільної групи
кетонів у СНг-групу:
К\ вмс2н5),0 ^—СН, о /м і і
2>=0 +Н8СН2СН28Н * ' 7' 0Х І 2 НгШі > ^СНАКІЗ
рГ 2 2 CH2C12,250CR2/\S_¿H2 СН3СН2ОН 2
Обидва процеси - утворення тіокеталів і їхнє "знесульфурення" - відбу-
ваються у м'яких умовах.
❖ Синтез гідросульфітних похідних. Взаємодія надлишку 40 % розчину
натрій гідросульфіту з альдегідами і кетонами (особливо метилалкілкетонами)
відбувається внаслідок нуклеофільної атаки гідросульфіт-іона карбонільного
атома Карбону:
\/ . . _
Н- " 9" 303Н=-С- 803
О 8°зН О" ОН
Зазначимо, що приєднання до електрофільного атома Карбону відбувається
через атом Сульфуру, який більше нуклеофільний порівняно з атомом Оксигену.
Продукти приєднання - гідросульфітні похідні - є солями сс-гідроксіалкан-
сульфонових кислот, які не розчиняються в органічних розчинниках. Подібно до
інших реакцій карбонільного приєднання, ця реакція також зворотна. Додавання
кислоти або основи руйнує гідросульфітну сіль і приводить до регенерування
вихідної карбонільної сполуки:
, \ / н> 802 + Н20 + Иа+
-С- 803Иа С + Ш^О"^^.
ОН О 8032" + Н20 + №+
Завдяки специфічним фізичним властивостям, легкості утворення й
розпаду гідросульфітні похідні використовують для виділення й очищення
карбонільних сполук.
У якісному органічному аналізі для розрізнення альдегідів від кетонів
гідросульфітній пробі віддають перевагу перед реакцією срібного дзеркала, тим
більше, що метилалкілкетони можна легко додатково виявити іодоформною
реакцією.
Розділ 17. Оксосполуки
453
17.4.3. Реакції з С-нуклеофілами
❖ Утворення ціангідринів. Ціанідна кислота приєднується до альдегідів і
просторово неутруднених кетонів, утворюючи гел*-гідроксиціаніди, які нази-
вають ціангідринами, або а-гідроксинітрилами:
Реакцію каталізують основи (В-), оскільки вони дають змогу одержати
активний аніон СИ~ зі слабкодисоційованої ціанідної кислоти:
Н-С^І + В" ВН + ОТ швидко
0 9"
я-с-н з^ я-^-н
Повільно
от
Я-^-Н + ВН ^ Я-ср-Н + В Швидко
Реакція на всіх стадіях зворотна. Вихід ціангідрину зумовлений просто-
ровою будовою карбонільної групи й досягає 80-100 %.
Унаслідок високої токсичності НСИ ціангідрини одержують, додаючи міне-
ральну кислоту в суміш карбонільної сполуки з водним розчином натрій ціаніду.
Ціангідрин, що утворюється, шляхом гідролізу можна перетворити в а-
гідроксикислоту або ненасичену кислоту, наприклад:
Н ЯО ^ 2н О
СНзСНО + ИаСК СН3С-СК -„^СНзСНСООН
ОН ОН
Ціангідрин ацетальдегіду Молочна кислота
СН3ССН3 + КаСК—^
О
9нз
СНз^СИ
ОН J
н2о
н280' ^112
9Н3
СН,=С-СООН
Ацетонціангідрин Метакрилова кислота
Як випливає з особливостей розподілу електронної густини в карбонільній
групі, альдегіди легко утворюють ціангідрини, кетони - важче. Крім того,
унаслідок просторових утруднень кетони із розгалуженими алкілами взагалі є
практично нереакційноздатними.
■ Зручніший метод одержання ціангідринів з оксосполук - дія калій ціаніду
на гідросульфітну похідну альдегіду. Як приклад можна навести синтез мигда-
левої кислоти:
454
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
о-
О /=\ ?Н
cf ч-ИзГОО, -6 у-С-БО^а №СН
ХН 4-У н 3 -N^0,
н н
/=\ « Н20вНС1 І
^ />-С-С=К— у-С-СООН +ЫН4С1
— он 4=7 он
Мигдалева кислота, 67 %
❖ Приєднання металоорганічних сполук до карбонільної групи. У разі
приєднання реактиву Гриньяра до альдегідів і кетонів відбувається синтезування
вторинних і третинних спиртів. Лише формальдегід утворює первинний спирт:
Н
С4Н<,МеВг + НСН0 — Н-С-0МёВг СНзССН^СНрН
С4Н9 Пентан-1 -ол, 92 %
(СН3)2СИУ^г +СН3СН0—-НзС-СН^Ш(СНз)2 Н3С-СН-СН-СН3
0М§Вг СН3 ОН
3-Метилбуган-2-ол, 54 %
С4Н9МЕВг +Н3С-С-СНз — ЭД-С-СНз ► ОД-С-СНз
О OMgBr °Н
2-Метилгексан-2-ол, 90 %
Одержання вторинних і третинних спиртів через реактив Гриньяра
супроводжується побічними процесами, такими як енолізація:
?МеІ нон» ^
* (СНз)2СН-С-СН(СНз)2 •5—► (СНз^СН-С-СНССНз^
(СНз)2ОІ-С-СН(СНз)2^МЕ.
о
СНз ОН
ом^
—г (СНз^СН-С^СНз),1^ (СНз^СН-С-СЩСНз),
' * о
Цих ускладнень вдається уникнути в разі застосування літійорганічних
сполук. Завдяки їхній високій реакційній здатності процес можливо проводити
за низьких температур.
■ Продуктом реакцій нерідко є рацемічна суміш двох енантіомерів. Коли
один з реагентів хіральний і в реакції утворюється новий хіральний центр, то два
продукти є діастеомерами й утворюються вони не в рівних кількостях:
Розділ 17. Оксосполуки
455
Н
СН 2)Н20/Н+
+
(1К, 25)-1,2-Димєтил-
циклопентанол, 90%
(/£ 25)-1,2-Диметил-
циклопентанол, 10%
❖ Реакція Віттіга є типовою реакцією нуклеофільного приєднання по
карбонільній групі оксосполук і дає змогу заміщати атом Оксигену на
метиленову ланку й так із альдегідів і кетонів синтезувати алкени. Реактив
Віттіга готують за такою схемою:
ЗС6Н5Ї^Вг
РС13
ЗК^СІВг
:(С6Н5)3Р
[(С6Н5)3Р-СНІ12]СГ
(С6Н5)3Р=СК2— (С6Н5)3Р-СК2
-с2н5он
Отриманий реактив є біполярним іоном, що легко вступає в реакцію з
оксосполуками:
^>0+ :с-р(сл)^ — ЦснгЦ %
к Я С-РГСЬП Лч™сбн5)з 5 / \
я я як
Наприклад: —
(СбН5)3Р + СН3Вг [(С6Н5)зР-СН3]Вг- -їш (С6Н5)3Р-СН2
(С6Н5)3Р-СН2 + н3с-с-сн3 (СбН5)3Р=0 +н3с-с=сн2
о сн3
2-Метилпропен
❖ Синтез ацетиленових спиртів. Взаємодія ацетилену з оксосполуками
належить до типових нуклеофільних реакцій. Оскільки ацетилен як кислота
нагадує ціанідну кислоту, то для посилення нуклеофільності застосовують той
же прийом: дію сильних основ, таких як порошкоподібній калій гідроксид або
натрій амід. Таким шляхом одержують ацетиленові спирти й гліколі:
*аНН2 ъ~ - ^
і) 2 ;с=о кі
И-С=СН КСЕСНа К-С=С-СС 2
^з(ж) 2)Н30+ І К
ОН
456
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
НСЕЕСН^И^ НаСЕЕСИа к
і)2 >=о .
2/
№і3 (ж)
С2Н2
2) Н,0+
220 °С;
-С2Н2
1)2 _>=0
2№С=СН
2/
2) Н30+
к он он
•2 ^с—с=сн
к он
17.4.4. Реакції з галогенонуклеофілами
❖ Галогенування карбонільної групи. Альдегіди й кетони реагують за
механізмом реакції Ам з галогенонуклеофілами. Як реагенти застосовують
фосфор (V) галогеніди:
я-с^ + РС15 ==:
Н
.о-рсі^
н
я-^-орсі4
н
сг
.орсі.
я-сн-сі
я-с-сі -а* я-снсі,
-РОСІ,, І
-сі- Н
Аналогічно відбувається реакція й для кетонів:
я-с-я + РС15 —- я2СС12 + РОС13
О
У поєднанні з відщепленням за механізмом Е2 вона зручна для одержання
галогеноалкенів, аленів та ацетиленів:
я'-с-с. ,с=с^ +НС1
н
сі я-
СІ
Заміщення Оксигену на два атоми Флуору можна виконати за допомогою
сульфур (IV) флуориду:
я-с-я + брл —► ясрд + БОХ
и
О
4і 2
Розділ 17. Оксосполуки
457
17.4.5. Реакції з N-нуклеофілами
Як ЛГ-нуклеофіли можуть бути молекули амоніаку, гідразину, гідроксиламіну,
арилгідразинів, семикарбазидів і тіосемикарбазидів. Реакції з N-нуклеофілами відбу-
ваються у дві стадії. Перша - нуклеофільне приєднання за механізмом AN, друга -
відщеплення. Розглянемо ці процеси на прикладі реакції оксосполук з амоніаком.
❖ Реакції з амоніаком. На першій стадії відбувається повільне приєднання
нуклеофільного атома Нітрогену до атома Карбону карбонільної групи, а потім -
перенесення протона з атома Нітрогену на атом Карбону з утворенням
альдегідамоніаку. Проміжний аддукт - альдегідамоніак зазвичай малостійкий і
легко піддається дегідратації до альднмінів:
С<6Ь
R-< + NH3 ^= R-CH-NH3 ^= R-CH-NH, R-CH=NH
Vх Н у 3 І 3 І 2 -н2о
V / О ОН ' . .
Альдимін, або
Альдегідамоніак основа Шиффа
Альдиміни за умов реакції зазнають тримеризування в циклічні альде-
гідамоніаки:
сн3
ну ун
,-СН
Н3С ^NH ^СН3
Вулкацит
■ Своєрідною реакцією формальдегіду з амоніаком є одержання гексаме-
тилентетрааміну, або уротропіну:
6 СН20 + 4 NH3 - І <
Уротропін
Зазначимо, що будова уротропіну з'ясована за допомогою рентгено-
структурного аналізу. Це було перше практичне використання цього методу.
Уротропін застосовують у медицині окремо і з кальцій хлоридом (кальцекс).
З нього одержують вибухову речовину - циклопіт, або гексоген:
уо2
С6Н12М4 + ЗНЖ)3—- І І + зсн2о + ш3
0^ N0;,
Циклоніт
458
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
Амоніак і формальдегід, що вивільняються, знову переводять в уротропін.
■ Кетони реагують з амоніаком інакше й значно повільніше:
5+О мн, і 2
н3с-с=о + н3с-с-сн3—- н3с-с=сн-с-сн3—- н3с-с-снгс-сн3
СН, О СН <Об- сн3 8
х3
4-Ам іно-4-метил пентан-2-он
■ За надлишку амоніаку кетони за наявності водню під тиском можна
каталітично перетворити в первинні аміни. Цю реакцію називають відновним
амінуванням кетонів:
/С=0 +N11,-
К 3
к>-кн2 -^К/С=кн
кат., / К г
Якщо амоніаку не вистачає, то виникає суміш вторинних і третинних амінів.
Багато речовин, у яких є аміногрупи, взаємодіють з оксосполуками, утво-
рюючи похідні, які мають фрагмент >С=№-. Зрозуміло, що подібні нуклеофільні
реакції каталізовані кислотами. Максимум швидкості визначатимуть два прямо
протилежні процеси. Перша реакція
(СН3)2С=0 + Н+ ^ (сн3)==с6н
сприятиме перебігу реакції нуклеофільного приєднання. З іншого боку, прото-
нування неподіленої пари електронів атома Нітрогену унеможливлює
нуклеофільне приєднання. Тому процес проводять у слабкокислому середовищі
(рН 3-5), що дає змогу протонувати оксосполуки й водночас зберігати достатню
концентрацію сполуки з непротонованою аміногрупою. Загалом рН залежить від
основності аміносполуки: чим вона нижча, тим концентрованішу кислоту треба
застосовувати.
❖ Приєднання похідних амоніаку
• Реакції з гідроксиламіном. Продукти взаємодії альдегідів і кетонів - оксими
- альдо- і кетоксими:
Я-СГ + Ш2ОН — я-сн-шон — Я-СН=ШН
14 Атт -н2о
п иН Альдоксим
я-с-я + №ШН — я-с-шон — с=шн
и гч. IV Кетоксим
Під дією фосфор (V) хлориду альдоксими перетворюються в нітрили:
Розділ 17. Оксосполуки
459
Н 2
■ Оксими можна відновити до первинних амінів:
>=Ж>Н ^СН-ИН, , де 11' = Аік, Н
II 1?.
■ Цікаво, що оксими можуть існувати у вигляді двох стереоізомерів, які
позначають префіксами син- і анти-\
с с
II II
но он
Назву син- приписують ізомеру, у якому ОН-група є на одному боці зі
старшим замісником, і навпаки.
• Реакції з гідразином. Під час взаємодії гідразину з альдегідами й кетонами
реагують обидві аміногрупи з утворенням азинів (альдазинів, кетазинів), і тільки
за великого надлишку гідразину одержують гідразони:
2Н3С-СчЧ + ш2імн2 —- сн3сн=кьк=сн-сн3
О Альдазин
2н3с-с=о + ш2мн2—-н3с-с=к-н=с-сн3
сн3 сн3 сн3
Кетазин
я-сно + н2к-ш2 —- ксн=к-кн2
Гідразон
• Реакції з заміщеними гідразинами. Для одержання з альдегідів і кетонів
кристалічних похідних з високою й чіткою температурою плавлення вико-
ристовують фенілгідразин, семикарбазид, тіосемикарбазид й особливо 2,4-
динітрофенілгідразин:
*ч_ . Рх
,с=о+н2к-ш-с6н5— ;с=№-*тс6н5
^ Фенілпдразон
/С=о +мн2шсош2—- хс=к-кн-сокн2
Семикарбазон
)с=о +ш2ш-с-ш2 — /с=к-ш-^-кн2
Тіосемикарбазон
460
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімій
ОД 0,И
^ >=\ ^
2
2,4-Динітрофенілгідразон
Усі ці продукти конденсації є кристалічними речовинами, їх застосовують
для ідентифікації альдегідів і кетонів.
17.4.6. Реакції відновлення
Альдегіди відновлюються в первинні спирти, а кетони — у вторинні. Для
відновлення використовують каталітичне гідрування (платина, скелетний
нікель), або такі агенти, як літій алюмогідрид. Відновлення кетонів амальгамою
натрію детально висвітлене в розділі 13.
❖ Каталітичне гідрування. Ці реакції застосовують для синтезу низки
спиртів, які менш доступні, ніж відповідні карбонільні сполуки, особливо ті, які
можна одержати альдольною або кротоновою конденсаціями:
СН3СН=СНСНО СН3СН2СН2СН2ОН
❖ Відновлення альдегідів і кетонів воднем у момент виділення веде до
одержання, відповідно, первинних і вторинних спиртів, наприклад:
снхоон
н-С6Н13СНО 3 0 » н-С6Н13СН2ОН
6 13 Ре, 100 С 6 13 2
78%
^с5н11сосн3№ч"С2Н5°" ^С5НпСНОНСН3
65%
Зазначимо: якщо відновлення альдегідів за участю металів (порошок заліза
й оцтова кислота; цинковий пил і спиртовий луг) відбувається нормально, то
кетони, поряд з нормальним відновленням, можуть утягуватися в бімолекулярне
відновлення до пінаколів. Такого типу реакції мають препаративне значення.
❖ Відновлення комплексними гідридами металів. Натрій борогідрид
ИаВНЦ не відновлює подвійні карбон-карбонові зв'язки, навіть якщо вони
спряжені з карбонільними групами, тому його використовують для відновлення
ненасичених карбонільних сполук у ненасичені спирти. Крім того, натрій
борогідрид забезпечує вибіркове відновлення карбонільної групи за наявності
естерної, нітрильної, амідної, лактонної й оксиранової груп.
Як вже зазначено, альдегіди й кетони зазнають відновлення літій алюмо-
гідридом у спирти легко і з кількісними виходами, але без перерахованих вище
функцій:
Розділ 17. Оксосполуки
461
Н
411,00 + ЬіА1Н4 (Я2СНО)4А1Ьі 4К>С0Н + ЬЮН + А1(ОН)3
Нуклеофілом у цьому разі слугує атом Гідрогену, що переноситься разом з
парою електронів у вигляді гідрид-іона від металу на карбонільний атом
Карбону:
Н
^С=0 + Н :ХА1Н3" ► -^-0А1Н3 3^-^ (-Є-0)4А1"
(-9-0)4
Реакція каталізована кислотами Льюїса: похідними алюмінію або, можливо,
іоном літію:
Х=0 + а1- Х=0"Ці-
Отже, відновлення оксосполук гідридами металів, де вирішальною стадією є
приєднання гідрид-іона до електронодефіцитного атома Карбону карбонільної
групи, варто зачислити до нуклеофільних реакцій приєднання.
❖ Відновлення за Мейєрвейном-Понндорфом-Верлеєм. До реакцій
нуклеофільного відновлення кетонів до спиртів належить метод, розроблений у
1925-1926 рр. X. Мейєрвейном, А. Верлеєм і В. Понндорфом. Цей метод полягає
в селективному відновленні оксосполук за допомогою алюміній триізопро-
пілату, що слугує донором гідрид-аніона:
R R
>0 + А1[0СН(СН3)2]3 = )с-0-А1[ОСН(СН3)2]3=
RN^5LcH3 s RN /СН3 неї R4
Г г Г 4- Г ^ Г-ПТ4
Rf і у cil r'\ TTCHL -сіаі[ОСН(СН3)2]2 R"frun
о c° О О ^
/Äi ;aiv
(СНз)2НСО хОСН(СНз)2 (СНз)2НСО хОСН(СН3)2
Гідрид-іон як чинник, що визначає механізм реакції, ще буде фігурувати в
реакціях диспропорціювання за С. Канніццаро1 і В. Тищенком2.
❖ Відновлення до метиленової групи
• Реакція Вольфа-Кіжнера3. Глибше відновлення оксосполук можна виконати
за Вольфом-Кіжнером через гідразони:
1 Канніццаро С. (Саппіггаго 8) (1826-1910) - італійський хімік, один із засновників атомно-
молекулярної теорії. Розмежував поняття "атом", "еквівалент" і "молекула".
2 Тищенко В. (1861-1941) - російський хімік. Працював у лабораторіях О. Бутлерова і Д. Мен-
делєєва. Вивчав склад нафти. Цікавився лісохімією, історією хімії. Розробив рецептуру хімічного
скла, вивчав кольські апатити.
3 Кіжнер М. (1867-1935) - російський хімік. Головні праці присвячені нітрогеновмісним сполукам.
462
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
ясно + НзИ-ад -> ксн=>^Н2,
які під час нагрівання у висококнплячому розчиннику - діетиленгліколі -
перетворюються в алкани:
ксн==н-нн2 ІІСНз + N2
Використання як розчинника сильнополярного диметилсульфоксиду дає
змогу розкладати гідразони за кімнатної температури.
Механізм розкладу гідразонів має такий вигляд:
к-сн=г>ККН2 — [я-сн^НЙН— к-сн-^ин] я-сн^ш
Угруповання -СН2-И=>Щ нестійкі й розкладаються з виділенням молекули
азоту.
Реакцію успішно застосовують для аліфатичних, ароматичних, аліциклічних
і гетероциклічних сполук.
• Відновлення за Клемменсеном1. У випадку оксосполук, чутливих до лугів,
застосовують відновлення за Е. Клемменсеном:
к-^-я' + 4Н ясн^' + 2Н20
О
Як відновник використовують амальгаму цинку в хлоридній кислоті. За цих
умов відновлюються аліфатичні, ароматичні та алкілароматичні карбонільні
сполуки.
Зазначимо, що спирт не є проміжним продуктом відновлення, оскільки за
умов реакції Клемменсена відновлення спиртів не відбувається.
17.4.7. Реакції окиснення
З усіх органічних сполук, які сьогодні вивчені, альдегіди окиснюються
найлегше. Вони перетворюються в карбонові кислоти навіть під дією таких
слабких окиснювачів, як іон Арґентуму або Купруму у вигляді фелінгової
рідини, не кажучи вже про такі класичні реагенти, як калій біхромат аро
перманганат. Останніми роками хіміки віддають перевагу реагенту Джонса.
Реакція відбувається швидко і з високим виходом:
сщсна-сС ——^сн3(сн2)ГСС'
3^ ^2/4 \ Аацетон,0°С 34 ~'4 он
Гесаналь Гексанова кислота, 89 %
1 Клемменсен Е. (Сіеттепзеп Е.СЬ.) (1876-1941) - американський хімік данського походження.
Отримав ступінь доктора з хімії за відкриття так званого відновлення за Клемменсеном. Емігрував
у США, працював у фармацевтичній промисловості.
Розділ 17. Оксосполуки
463
❖ Окиснення альдегідів. Відповідно до методики окиснення за Толленсом ,
для запобігання осадженню нерозчинного у воді арґентум оксиду додають
амоніак, тому реактив Толленса містить комплексно зв'язаний іон Арґентуму
[Аё(МН3)2]+:
Я-СНО + 2^(ННз)2]+ + ЗОН" -> ЯСОСГ + 2Аё| + 4ЫН3 + 2Н20
Безбарвний розчин Срібне дзеркало
Реакція одержала назву реакції срібного дзеркала з огляду на те, що
металеве срібло осаджується у вигляді щільного дзеркального шару.
Надзвичайну легкість, з якою окиснюються альдегіди, використовують для
ідентифікації цих сполук і особливо для того, щоб відрізнити їх від кетонів.
Метод становить синтетичний інтерес у тих випадках, коли альдегіди більше
доступні для синтезу ненасичених кислот з ненасичених альдегідів:
ЯСН=СН-СНО -> ЯСН=СН-СООН
Легкість окиснення альдегідів пов'язана з порівняно невеликими
енергетичними витратами на гомоліз зв'язку Карбон-Гідроген (368 кДж/моль).
Ацильний замісник, що утворюється, легко зазнає окиснення навіть на повітрі:
Я-СНО Я-ОО + Н
Я-С=0 + о2 — Я-СООО
Я-СООО • + Я-СНО — Я-СОООН + ЯСО
2 Я-СОООН —- 2 Я-СООН + 02
■ Інший реагент, рідину Фелінга2, готують, змішуючи водний розчину
купрум (II) сульфату і лужний розчин сегнетової солі ЫаООС-СНОН-СНОН-
СООН. Темно-синій розчин, що утворюється, є комплексом Купруму. Під час
взаємодії з альдегідами йони Купрум (II) відновлюються до Купруму (І), які
випадають у вигляді червоного осаду купрум (І) оксиду:
ЯСНО +2Си2++ ОН"+ Н20 —- ЯСОО"+ Си20( + 4Н+
❖ Окиснення кетонів потребує розриву С-С-зв'язку й тому відбувається
лише в досить жорстких умовах. Винятком є галоформна реакція, у яку
вступають метилалкілкетони:
1 Толленс Б. (Tollens В.) (1841-1918) - німецький хімік і біохімік. Розробив кількісний метод
одержання фурфуролу з пентоз. Вивчав обмін вуглеводів у рослинних організмах.
2 Фелінг Г. (Fehling Н.) (1812-1885) - німецький хімік. Головний напрям наукових досліджень -
аналіз та ідентифікація органічних сполук. Вивчав багатоосновні органічні кислоти.
464
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
с2н5ссн3 + зої- —- с2н5соо- + СНІ3
О Галоформна реакція
г*ы СН3
сн3>:=сн(||:сНз сні3 + ^с=снсоок
б СН3
4-Метилпент-З-ен-2-он Калій 4-метилпент-З-еноат
Галоформна реакція є якісною пробою на фрагмент -СО-СНз, оскільки в
разі йодування утворюється йодоформ - речовина жовтого кольору.
■ Окиснення кетонів жорсткими окисниками, наприклад, лужним розчи-
ном КМпОд або гарячою нітратною кислотою, відбувається з розривом С-С-
зв'язків. Кетони розщеплюються відповідно до правша Попова1 з будь-якого
боку карбонільної групи, що приводить до утворення суміші чотирьох кислот:
Розрив по С2-С3
і
КМп04
СНоСНоСНоССНоСН-»
і3^ а і2^х 1 А2^А Аз н+
О
СН3СН2СН2СООН + СН3СООН
Розрив по С3-С4
СН3СН2СООН + СН3СН2СООН
Ця реакція особливо важлива для симетричних циклічних кетонів, які дають
дикарбонові кислоти.
■ На відміну від деструкції кетонів під дією таких окисників, як калій
перманганат або нітратна кислота, окиснення симетричних кетонів пероксидами
кислот дає естери:
сгхооон О0
снхн2сосн2сн3—2 - сн3снга
23 <ЗД 3 2 хосн2сн3
Етилпропіонат, 78 %
Для цієї реакції запропонований такий механізм:
г:ОН О
Сі її
Я-С-Я + СРХОООН ===== К-С-ОтО-С-СР, -
О Я (
бн 1
я-с-сж + СР3СОО"—- я-с-оя + СР3СООН
о
Ця реакція, відкрита А. Байєром і В. Віллігером 1899 р., набула особливого
значення для циклічних симетричних кетонів, з яких утворюється лише одна
сполука:
Попов О. (1840-1881)-російський хімік. Учень О. Бутлерова. Головні праці в галузі структурної
органічної хімії.
Розділ 17. Оксосполуки
465
СРзС000^
є-Капролактон, 90 %
Несиметричні кетони утворюють суміш двох естерів.
17.4.8. Реакції диспропорціювання
❖ Реакція Канніццаро. Ця реакція, відкрита італійським хіміком
С. Канніццаро 1853 р., характерна для альдегідів, що не мають атомів Гідрогену
біля а-атома Карбону. Механізм її виглядає так: на першій стадії утворюється
комплекс з одного моля натрій гідроксиду й двох молів альдегіду. Гідроксид-
аніон комплексу атакує атом Карбону однієї з карбонільних груп, зумовлюючи
збільшення електронної густини на атомі Карбону, чим полегшує перехід
гідридного йона до іншої карбонільної групи. Водночас відбувається зсув
електронів і утворення кислоти та алкоголят-аніона. Оскільки алкоголят-аніон є
сильнішою основою, ніж карбоксилат-аніон, то в підсумку одержують спирт і
сіль кислоти:
ОН 0№
я-с +яонр~№+—я-с + яснрн
о о
Механізм реакції підтверджено виділенням 1951 р. описаних комплексів.
Такий тип збалансованого відновлення-окиснення-відновлення одержав назву
реакції диспропорціювання.
Реакція С. Канніццаро відбувається за наявності концентрованих розчинів
лугів за кімнатної температури й не супроводжується альдольною конденсацією,
її можна поєднувати з альдольною конденсацією, як це є в синтезі
пентаеритриту. У цьому випадку карбонільним компонентом є формальдегід:
тт г_г*° са(он) -/^^сщо ?г''°ІЕі£
нзс % сн2 % — носн-9
сн2он
СН2ОН-С—С(СН2ОН)4 + нсоон
сн2он н
II
о -,
он
н
І
II
о
466
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Остання стадія реакції свідчить про те, що саме мурашиний альдегід
окиснюється до кислоти. Це пояснюють тим, що на атомі Карбону
формальдегіду позитивніший заряд порівняно з іншими оксосполуками,
оскільки формальдегід - єдина в цьому ряді сполука, в якій нема алкільного
замісника, що компенсує позитивний заряд на карбонільному атомі Карбону. З
огляду на це він легше порівняно з іншими оксосполуками віддає для
відновлення гідрид-іон:
н-< + он- —- ~íp HCt
н н' to- он
Здатність формальдегіду давати гідрид-іон зумовлює його використання як
відновника деяких альдегідів.
❖ Реакція Тищенка. Альдегіди, на відміну від кетонів, під дією низки
алкоксидів, наприклад, (ЯО)зАІ й (RO)4Ti, у неводному середовищі зазнають
диспропорціювання: одна молекула альдегіду відновлюється завдяки окисненню
іншої, і в підсумку утворюється естер:
2 СН3СНО^^)з СН3СООСН2СН3
Реакція В. Тищенка відбувається з гідридним переміщенням Гідрогену
альдегідної групи. Механізм цієї реакції має такий вигляд:
¥ / 9нз \ н , / єн, \ Л=о
r-c=o + ai b-c-CHj ==: r-c-ó-a"i4o-9-ch3
\ сн3 /3 + " \ сн3 /з
н R0./ 9н3 \ / 9н3
—- r-C-0-CJ02Alf0-9-CH3 — r-CH20-C-r+А1 0"9-сн3|
+ ^ \ сн3 k о \ сн3
+Хн)
Взаємодія атома Алюмінію з атомом Оксигену альдегіду значно збільшує
електрофільність атома Карбону карбонільної групи, стимулює гідридне
перенесення й полегшує саму конденсацію.
Недавно з'ясовано, що гіпофосфатна кислота є ефективним каталізатором
естерної конденсації. Наприклад, параформальдегід під час нагрівання з
гіпофосфатною кислотою в автоклаві утворює метилформіат:
2СН20 d—г НСООСН3
2 250 °С 3
Розділ 17. Оксосполуки
467
17.4.9. Реакції, що відбуваються за участю атома
Гідрогену в сс-положенні
❖ Лльдольна й кротонова конденсації. Терміном "конденсація" позна-
чають реакції, у яких приєднання реагентів супроводжується відщепленням
простих молекул, наприклад, води, спирту тощо. Однак, як доведено нижче, така
назва не завжди коректна, оскільки деякі реакції, визначені терміном "конден-
сація", формально такими не є.
Величезна кількість реакцій альдегідів і кетонів пов'язана з конденсаціями,
що ведуть до утворення простих або подвійних карбон-карбонових зв'язків.
Обов'язковою умовою перебігу реакції є наявність в а-положенні порівняно
карбонільної групи атомів Гідрогену. Це так звані С—Н-кислоти. Рухливість цих
атомів пов'язана з наявністю карбонільної групи, що виявляє негативний
індукційний та мезомерний ефекти. Такі електроноакцепторні фрагменти, як -СИ,
-ССІз, -N0, -N02, можуть зумовлювати в а-положенні аналогічну рухливість
атомів Гідрогену.
Під дією каталітичних кількостей мінеральної кислоти або лугу альдегіди,
що містять атоми Гідрогену в а-положенні стосовно карбонільної групи,
перетворюються в р-гідроксіальдегіди, інакше названі альдолями (від слів
альдегід й алкоголь), звідки утворена назва цієї реакції.
■ Конденсація, каталізована основами, починається з утворення енолят-
аніона альдегіду:
,о
н3с-сн-с^
+ он
Швидкої
.0
.0"
н3с-сн-с^
н
н3с-сн=сч
Енолят-аніон
н
Далі карбаніон, що виник, атакує карбонільну групу другої неіонізованої
молекули альдегіду:
5-
Н
сн3сн-с; + сн3сн2-с
сн3сн2сн-сн-с
6-сн, н
Приєднання нуклеофіла до карбонільної групи неіонізованої молекули
альдегіду визначає швидкість усього процесу.
На третій стадії відбувається протонування алкоксид-іона водою й утворюється
кінцевий продукт альдоль з регенерацією каталізатора гідроксид-іона:
468
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
/О но /°
сн3сн2сн-сн-< сн3сн2сщон)-сн-сС
о_ сн3 н " сн3 н
Реакція відбувається за 0 °С протягом 5 год з виходом З-гідрокси-2-
метилпентаналю близько 60%. У жорсткіших умовах (100 °С, 3 год) альдолі
зазнають дегідратації, утворюючи сс,р-ненасичені альдегіди. Вихід становить
близько 90 %.
■ Під час альдольно-кротонової конденсації в кислому середовищі на
першому етапі відбувається повільне утворення енолу:
О +ОН ОН
II н+ м 1
Н: СН9-С-Н Н: СН2-С~Н === Н2С=С-Н
Друга молекула альдегіду після його протонування атакує енол, утворюючи
альдоль:
+.ОН ,р
сн3сн(он)-снгс^н^ сн3сн(он)сн2-сСн
Дією кислот припинити реакцію на стадії альдольної конденсації
неможливо, оскільки вже за кімнатної температури відбувається дегідратація
альдолів до а,Р-ненасичених альдегідів.
У випадку ацетальдегіду такий ненасичений альдегід СНзСН=СН-СНО
називають кротоновим, і, відповідно, ця стадія реакції одержала назву
кротонової конденсації. Цей процес відбувається легко через наявність
рухливого сс-гідрогенного атома і завдяки утворенню системи спряжених
подвійних зв'язків:
я-сн=с-с=о
І І
■ Цікаво зазначити, що в кротоновому альдегіді вплив карбонільної ^рупи
досить ефективно передається через вінільну ланку - метальна група цієї
сполуки активна настільки, наскільки метальна група ацетальдегіду. Таке явище
в органічній хімії називають вінілогією. Тому кротоновий альдегід, що спочатку
утворюється з оцтового альдегіду під дією сильних основ, здатний вступати в
наступну конденсацію. Реакція вперше виконана О. Вороніним і Ж. Вюрцом за
наявності калій ацетату, карбонату й сульфіту:
псн3сно сн3(сн=сн|-сно
Розділ 17. Оксосполуки
469
• Перехресна альдольно-кротонова конденсація відбувається у випадку двох
різних альдегідів з утворенням складної суміші. Практично альдольну
конденсацію є сенс проводити, якщо один з альдегідів не має а-гідрогенних
атомів, тоді він відіграватиме роль тільки карбонільного компонента. Такими є
ароматичні й гетероциклічні альдегіди, формальдегід, триметилоцтовий альдегід
і його аналоги, наприклад:
(СНз)3С-С^°+ Н,С-<° — (ОУзС-СрН-С^СЯЮ ~ (СНз)3С-СН=СН-СНО
^ ^ ОН 4,4-Диметилгаент-2-еналь
2,2-Диметгилпропаналь Ацетальдегід
сн,он
ЗН-С^.НзС-С^^НОС^-СНО
н н снрн
Формальдегід Ацетальдегід 2Д-Дигідроксиметилпропаналь,82%
• Кетони значно важче вступають у реакцію альдольної конденсації: потрібні
нагрівання й сильніші основи:
?\ Ва(ОН)2 9Н н+ Н3СХ
Н3С-С-СН3 ==^н3с-с--снгс-сНз— /С=с-с-сн3
СН3 О Нзс О
4-Гідрокси-4-метилпентан-2-он 4-Метилпент-3-ен-2-он
(мезитилоксид)
Застосування головних каталізаторів веде до незначного виходу продукту
альдольної конденсації. Це пов'язане з низькою швидкістю атаки аніоном "СН2-
СО-СНз карбонільного атома Карбону другої молекули ацетону. Однак якщо
реакцію проводити з використанням кислого каталізатора, то на другій стадії
відбувається процес дегідратації, і рівновага процесу зсувається вправо з
повною витратою ацетону. Мезитилоксид, що утворився, за наявності гідроген
хлориду утворює форон, а під дією концентрованої сульфатної кислоти -
мезитилен:
НзС-С=СН-С-СНз+Н3С-С-СНз ^ Н3С-С=СН-С-С№С-СНз
сн3 о о сн3 о сн3
Мезитилоксид, або 2,6-Диметилгептан-2,5-дієн-4-он
ізопропіліденацетон (форон)
470
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
Н2804 конц.
Н3С^^г\^СН3
нх-с=сн-с-сн=с-сн,
3 І II І 3 -н,о
сн3 о сн3 у
сн3
Мезитилен
• Перехресна конденсація альдегідів з кетонами. У разі такого типу конден-
сації варто враховувати, що позитивний заряд на атомі Карбону карбонільної
групи альдегіду завжди вищий, ніж у кетонів, тому карбоніл альдегіду буде
взаємодіяти з метиленовим фрагментом кетону:
*°
сн3сн7— с + нх-с-сн, —► н,с-снгсн-снгс-сн,
З 2 \ 3 її З 3 2| 2 Ц З
н 5 он о
4-Пдроксигексан-2-он
З огляду на заряд на карбонільному атомі у випадку перехресної реакції як з
альдегідами, так і кетонами як карбонільний компонент варто брати формальдегід.
❖ Реакції заміщення в а-положенні. Поляризація я-зв'язку карбонільної
групи й наявність на карбоновому атомі карбонілу часткового позитивного
заряду позначається на поведінці Гідрогену біля а-карбонових атомів альдегідів
і кетонів. Природно, що атоми Гідрогену, які перебувають під впливом
сусіднього 5+-заряду, утримуються менш міцно, ніж у вуглеводнях, і легше
видаляються з молекули у вигляді протонів: /
У наведеній вище системі зв'язків Н-С-С=0 я- і а-зв'язки спряжені, і
система діє як єдине ціле: розрив зв'язку С-Н веде до розриву тг-зв'язку
карбонільної групи й локалізації я-пари електронів біля атома Оксигену, до
якого приєднується протон:
н-сД
СНІ—'
Н+ Енольна форма
Для значного зміщення рівноваги в оксосполуках необхідна наявність груп,
що відтягують електрони (наприклад, С=0). Швидкість багатьох реакцій
оксосполук із заміщенням атома Гідрогену в а-ланці дорівнює швидкості
енолізації й не залежить від концентрації реагенту. Це підтверджено тим, що
хлорування і йодування відбуваються з тією ж швидкістю, що й бромування.
Розділ 17. Оксосполуки
471
• Галогенування карбонільних сполук у кислому середовищі дає монога-
логенопохідні. Досить імовірний такий механізм реакції. На першій стадії під
впливом кислоти повільно утворюється енол:
Я-СН^^О:
Н
н+
к-снгс=он я-с^-с-он
н н
н2о
"н3о+
я-сн=с-он
н
Потім відбувається швидке електрофільне приєднання галогену до подвійного
зв'язку енолу:
я-сн=снон + сі, = я-сн-сн-он я-снсі-с^
сі сі -НС1 н
■ Галогенування в лужному середовищі відбувається за таким механізмом:
у'О х Повільно
Я-СН-С: +Т)Н «
н
я-сн-с
я-сн=с
н
н
а2 ,р
~=г к-сн-с; + сі
Швидко ^ Н
Ця реакція є вичерпною й у випадку ацетону та ацетальдегіду закінчується
галоформним розщепленням. Такого ж розщеплення зазнають усі метил-
алкілкетони:
І2,№ОН /р наон
Я-С-СН,-2 - К-С'-^^-
О СІ,
Я-С-СІз
ОН
КСООИа + СНІ,
• Дейтерообмін у кетонах у важкій воді також відбувається через енол або
енолят-аніон і теж є вичерпним:
Н _ со
Н3С-С-С-Н +ОН —*Н3С-9=СН2~Н}С-С-СН2 ^ НзС-С-С^О
О н
и
О
О
Н3С-£-СН3 Л ЩС-^СЩ ^= Н3С-$-СЦр
0 ОН п
О
• Рацемізація оптично активних альдегідів за асиметричним а-карбоновим
атомом також відбувається через еноли (кисле середовище) або енолят-аніони
(лужне середовище), наприклад:
472
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
н сн, сн3 н ¥
НзС Н чн н Н3С Н Н CHj
❖ Окиснення кетонів. Подібно через еноли відбувається окиснення
кетонів хроматною кислотою й селен (IV) оксидом до дикетонів:
?Н
0=Сг-ОН 0Н
'о "О-Сі-ОН
С\> -н+ н/С с\>—- R-^-g-r
+ Н2СЮ3
Селен (IV) оксид під час розчинення у воді перетворюється в селенисту
кислоту (НО)г8е=0, яка бере участь у реакції окиснення {реакція Раті):
HO^Se-OH CJau ^ Ґ—*
HW/R' ► uJ^_r/R' ► R-C-C-R' + Se
R-^ СфН -НР Н^С С^ -\
R
❖ Метилювання за Нефом. Оскільки кетони, на відміну від альдегідів,
стійкі до дії лугів, їх можна метилювати метиліодидом по a-ланці за наявності
калій гідроксиду або натрій аміду:
RCOCH2CH3 + КІ + Н20
R-C-CH3 + СН3І + КОН C^J RCOCH(CH3)2 + КІ + н2о
O RCOC(CH3)3 + КІ + Н20
Однак за жорстких умов (>350 °С) твердий луг розщеплює кетон, утворюючи
солі й насичені вуглеводні:
Сп^+іСОСНз + NaOH Cn^+iCOONa + СН,
❖ Утворення оксимів під дією нітритпої кислоти. Реакційноздатною
формою карбонільної сполуки й у цьому випадку є енол:
?Н
RCCH2CH3=R-C=CH-CH3+HON=0 — R-C-CH-СНз R-fi-fí-CH,
O O NO О NOH
Нітрозокетон Монооксим кетону
Розділ 17. Оксосполуки
473
■ Усі оксими (гідроксиіміни) легко зазнають гідролізу, утворюючи дикетони:
СН3С-ССН3 + Н20 СН3С-ССН3 + NH2OH
О NOH О О
■ Якщо на диметилдикетон, що утворився, або його монооксим подіяти
гідроксиламіном, то одержимо диметилгліоксим:
СН3С-ССН3 +NH2OH СН3С-ССН3 ,
О NOH HON NOH
Диметилгліоксим
відомий як реактив Чугаєва (якісна реакція на йон Ніколу).
■ 3 іншого боку, монооксими відновлюють цинком в оцтовій кислоті до
амінокетонів, які використовують у синтезі піролів:
R-C-C-R' —-—- R-C-CH-R'
и и снхоон и і
О NOH О NH2
17.4.10. Реакції оліго- та полімеризації
• Формальдегід у газоподібному стані спонтанно зазнає полімеризування в
тример — триоксиметилен, або триоксан:
Н HLC^ ^CHL
о \
Триоксан
У разі зберігання 40 % водного розчину формальдегіду (формаліну)
відбувається мимовільне випадання полімерів іншого роду - лінійних
високомолекулярних сполук:
иСН20 + Н20 -» HOCH2(OCH2)w+i-OH(w~20-200).
Ці полімери називають поліоксиметиленом, або параформом. Реально вони
відрізняються кінцевими групами. Коли полімеризації досягають за допомогою
сульфатної кислоти, то кінцеві гідроксильні групи естерифіковані сульфатною
кислотою:
Р V
HO-f-0-CH2(OCH2)nO-S-OH
о о
Якщо у формальдегіді наявний метанол, як це буває в товарному формаліні,
то на кінцях полімеру можуть бути метоксильні групи - СНзОСН2(ОСН2)лОСНз.
474 Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Під дією водного розчину лугу параформ зазнає деполімеризування,
утворюючи формальдегід.
■ Порівняно недавно запропоновано метод полімеризації сухого форм-
альдегіду в поліформсиїьдегід більшої молекулярної маси. Умовою полімери-
зації, яку виконують сьогодні в промислових масштабах, є висока чистота
формальдегіду. Поліформальдегід - один з найдешевших полімерів з цінними
властивостями, його застосовують для виготовлення деталей машин.
• Ацетальдегід. Якщо до ацетальдегіду додати краплю сульфатної кислоти, то
відбудеться екзотермічна реакція циклоолігомеризації з утворенням паральде-
гіду. Його утворення відбувається за температури нижче 0 °С. За наявності
сухого НС1 оцтовий альдегід перетворюється в метальдегід:
Обидва олігомери легко зазнають деполімеризування до ацетальдегіду під час
нагрівання з сульфатною кислотою.
■ Формальдегід. Головну масу формальдегіду одержують під час де гідру-
вання метанолу.
Використовують у виробництві фенолоформальдегідних і карбамідних
смол, для синтезу ізопрену через 4,4-диметилдіоксан, виробництва гліцеролу,
нітро- й аміноспиртів. Його широко застосовують для дезінфекції зерно- й
овочесховищ, парників, теплиць, протравлення насіння. Таблетки уротропіну,
параформу або триоксиметилену використовують як пальне (сухий спирт).
■ Ацетальдегід СН3СНО одержують, головно, за допомогою вакер-проце-
су. Це вихідна речовина для виробництва оцтової кислоти, оцтового ангідріду,
етилового й бутилового спиртів, альдолю, ацеталю, етилацетату, хлоралю.
■ Ацетон сьогодні одержують за допомогою куменового (кумолового)
методу, а також вакер-процесу:
СН3СНО
Ізч
13
Паральдегід
Метальдегід
17.5- Окремі представники оксосполук
Н3С-СН=СН2 + 1/2 02
Р<іС12/СиС12
Н3С-С-СН3
ПОЪ, 10-14 атм.
О
94%
Як розчинник, ацетон застосовують у великих кількостях у лакофарбовій
промисловості, у виробництві ацетатного шовку, кіноплівки, бездимного
пороху. Він слугує вихідним реагентом у виробництві органічного скла.
Розділ 17. Оксосполуки
475
17.6. Дикарбонільні сполуки
Діальдегіди й дикетони мають деякі властивості, відмінні від властивостей
монокарбонільних сполук.
17.6.1. Класифікація й номенклатура ШРАС
Дикарбонільні сполуки відрізняються між собою. Характер відмінностей
залежить від природи карбонільних груп і взаємного розташування обох
функціональних груп.
За природою карбонільних груп
Діальдегіди Кетпоальдегіди Дикетпрни
Н3С-С-СНО Н3С-С-С-СН3
ОНС-СНг-СНО II II II
О 0 0
Пропандіаль Оксопропаналь Бутан-2,3-ДІон
малональдегід (біацетил)
За взаємним розташуванням функціональних груп
1,2- або а- 1,3- або /?- 1,4- або у-дикарбононіль-
дикарбонільні дикарбонільні ні сполуки
сполуки сполуки
ОНС-СНО 3 II 2 II 3 3 II 2 2 II 3
0 0 0 0
Гліоксаль / Пентан-2,4-діон Гекса-2,5-діон
(ацетилацетон) (ацетонілацетон)
Крім назв, побудованих за загальними правилами замісної номенклатури,
використовують напівтривіальні назви діальдегідів, утворені з тривіальних назв
дикарбонових кислот, наприклад: СН2(СНО)2 - малональдегід, ОНС-(СН2)з-
СНО - глутаральдегід тощо.
Збереглася тривіальна назва гліоксаль.
17.6.2. ОгАіоксосполуки
Найтиповішими представниками цього класу речовин є гліоксаль, пропа-
нональ (метилгліоксаль або піровиноградний альдегід) і біацетил.
❖ Синтезують а-діоксосполуки з оксосполук шляхом їхнього окиснення
селен (IV) оксидбм:
476
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
СН3СНО + 8е02 ОНС-СНО + Н20 + Бе
СН,ССН, + БеО, СН,ССНО + Н,0 + Бе
О 6
СНз^СН^Нз + 8е02 СНТ<^-СН3 + Н20 + Бе
О 0 0
■ Гліоксаль також одержують окисненням етиленгліколю повітрям над
мідним каталізатором за температури 250-300 °С. Він легко зазнає полімери-
зування, утворюючи безбарвний продукт, який легко розпадається до мономеру
під час перегонки з фосфор (V) оксидом.
Інакше гліоксаль можна одержати з 1,1,2,2-тетрахлоретану СІ2СН-СНСІ2,
який у разі обробки концентрованою сульфатною кислотою перетворюється в
кристалічний сульфат. Кристалічний сульфат під час гідролізу розпадається з
утворенням діальдегіду.
■ Нітрозування кетонів є загальною реакцією й відбувається або внаслідок
безпосередньої обробки кетонів нітритною кислотою на холоді в кислому
середовищі, або за допомогою реакції кетонів з естерами нітритної кислоти за
наявності натрій етилату:
СН3ССН2СН3 + НШ2 СН3-Ч^-СН3
О О ЫОН
СНз-^-^-СНз-^ СНз^-^-СНз+КНрН
О ШН О О
❖ Хімічні властивості. Альфа-діоксосполуки виявляють вищу реакційну
здатність порівняно з монопохідними, особливо в разі взаємодії з нуклеофіль-
ними реагентами, такими як амоніак, гідразин, гідроксиламін, ціанідна кислота,
натрій гідросульфіт.
■ Гліоксаль має типові властивості альдегідів. У реакції може брати участь
як одна, так і дві карбонільні групи:
^Ш > онс-сн=шн +но№=сн-сн=мш
ОНС~СНО _ Монооксим гліоксалю Діоксим гліоксалю
сукнин, онс-сн^^-с^ +С6Н5ШК=СН-СН=К-КНСбН5
Фенілгідразон гліоксалю Озазон гліоксалю
Однак простежуються й деякі специфічні реакції, зумовлені наявністю двох
сусідніх оксогруп. Про них ітиметься нижче.
■ Зв'язок між оксогрупами легко розривають дією гідроген пероксиду:
СН3СОСОСН3 + Н202 -> 2СН3СООН
■ 1,2-Дикарбонільні сполуки в лужному розчині піддаються внутрішньо-
молекулярній реакціїКанніццаро:
Розділ 17. Оксосполуки
477
н,о
ОНС-СНО -^г НОСН2СООН
Гліколева кислота
СН3СОСНО СН3СН(ОН)СООН
Молочна кислота
■ 1,2-Дикетони конденсуються за наявності розбавлених лугів за
альдольно-кротоновою схемою з утворенням циклічних сполук:
0 0 ОН О
II II І II
СН3С-ССН3 СН3СН-ССН3
СН3С-ССН3 Сн2с-ссн3
00 °о о
2,5-Диметилбензохінон-1,4
■ 1,2-Дикарбонільні сполуки легко зазнають конденсації з первинними 1,2-
діамінами, утворюючи похідні піразину:
н3с-с=о+ щк-сщ _НзС^]
Н3С-С=0 Н^-СНг
а в реакції з амоніаком й альдегідами перетворюються в імідазоли:
к_с=0 Ш3 К-С=ЫЧ
я-с=о Шз к-с=к
■ Біацетш одержують гідратацією вінілацетилену з наступним окисненням або
взаємодією метилетилкетону з нітритною кислотою з наступним гідролізом. Він має
властивості кетонів, причому в реакцію може вступати одна або дві кетогрупи:
нзС-с!-£-сн3+ ин2он —^с-^—с-сн3+н3с-р—^-сн3
0 ° ион о жшжш
Монооксим Диметилгліоксим
Діоксим біацетилу дає з солями Ніколу хелатну сполуку:
Т № V
•н-о
Комплекс Ніколу з реактивом Чугаєва
478
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Ця реакція, відкрита Л. Чугаєвим, слугує для якісного й кількісного визна-
чення йонів Ніколу.
Сам біацетил є в складі коров'ячого масла й надає йому специфічного
запаху, тому його додають до маргарину як смаковий додаток.
17.6.3. р-Аіоксосполуки
❖ Для синтезу Р-діоксосполук найважливішою є конденсація кетону з
естером карбонової кислоти під дією основ, зокрема, таких як натрій амід або
етилат. У випадку естеру мурашиної кислоти синтезується Р-альдегідокетон, а за
інших гомологічних естерів - р-дикетони:
ГН:СН2-С-СН3 СН.-С-СНз — СН2=С~СН3
1 ^ 5 -яон 0) 6"
" СНз-(рОС2Н5 + СН2-^-СН3-^ СНз-^СН^СНз + СзНзО"
^ ^ Ацетил ацетон
Н-СОС2Н5 + СН2-£-СН3 — Б-ССН^СН, + С2Н50"
о о бо
Формілацетон
❖ Хімічні властивості
■ Бета-дикарбонільні сполуки схильні до помітної кето-єнольної тауто-
мерії, внаслідок чого вони можуть давати похідні за гідроксильною та
карбонільною групами. Вони одночасно можуть бути як СН-, так і ОН-
кислотами:
Н
І он" он"
—с-с-с— =5= -с-сн-с— -с-сн=с- -с-сн=с-
іі і и и м м і и •
ОНО 00 о он оо-
Кетоформа Енольна форма
Положення рівноваги визначене, насамперед, стабільністю таутомерів:
наявність в 1,3-діоксосполуках спряженої системи подвійних зв'язків в єнольній
формі й утворення водневого зв'язку робить її стабільною:
Н3Сч ^СНч ,СЩ
н3сч ,сн2Ч ,сн3 у с
ї ї в ¿ о
о о V
23,6% 76а%
Розділ 17. Оксосполуки
479
■ Спряжені 1,3-дикарбонільні сполуки можуть зазнавати алкілування
галогеноалканами за атомом Карбону або за атомом Оксигену і поводяться як
амбідентний субстрат:
Напрям реакції у бік С- або О-алкілування енолу залежить від природи групи, що
відходить, і вуглеводневого замісника в галогеноалкані. Якщо група, яка відходить,
належить до жорстких основ Льюїса, то переважає заміщення під дією жорсткого
оксигенового центру. У тих випадках, коли використовують м'яку основу (іодид,
бромід), переважає заміщення під дією м'якого карбонового центру єноляту.
■ Ацетилацетон одержують взаємодією ацетону з оцтовим ангідридом за
наявності бор (III) флуориду або з етилацетатом за наявності натрій етилату,
амідів натрію або літію.
Ацетилацетон утворює міцні внутрішньокомплексні єноляти дво- і
тривалентних металів. У них карбонільний атом Оксигену дає пару електронів
металу алкоголяту єнольної форми дикетону. Отже, атом двовалентного металу
спряжений з чотирма оксигеновими атомами (зайняті чотири координаційні
місця навколо металу) — з двома єнольними й двома кетонними.
Координаційне число тривалентного металу дорівнює 6, а число кетонних і
єнольних атомів Оксигену дорівнює, відповідно, 3. Такі внутрішньокомплексні
сполуки ацетил ацетону з іонами Купрум (П), Хром (III), Ферум (Ш), Кобальт (Ш)
дуже міцні, нерозчинні у воді і є забарвленими кристалічними речовинами. Колір їх
відмінний від кольору відповідного катіона. Вони добре розчинні в органічних
розчинниках. Деякі дикетони цього типу використовують для екстракції катіонів
металів, наприклад, урану органічними розчинниками з розведених водних розчинів:
НХ
II
О
R-X
ОН
Комплекс двовалентного металу
Комплекс тривалентного металу
Застосовують ß-дикетони для синтезу шестичленних гетероциклів.
480
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
17.6.4. у-Аіоксосполуки
Гамма-діальдегіди можна одержати відновленням дихлорангідридів відпо-
відних кислот за Розенмундом-Зайцевим:
СЬ^-СН2СН2СН2у~с1 + 2 Н2-^— Н-^СНзСНзСН^-Н
О О О О
Гамма-дикетони одержують дією хлорацетону на натрійацетооцтовий естер,
виконуючи "кетонне розщеплення" отриманого ацетонілацетооцтового естеру
або діючи на натрійацетооцтовий естер іодом і піддаючи "кислотному"
розщепленню діацетилянтарний естер:
Н3С-С~СН2с1 + Н3С-С=СН-СООС,Н5—-Н3С~С-СН-С^Г 9ШЕ2^>
О (Жа О СН?С-СН,"
2 м З
/СООН , 0
н3с-с-сн -аз- н3с-с-сн2-сн2-с-сн3
О ин2<ігч'нз О О \
о \
НзС-с=сн-с; о
(Жа 0сл н.С-С-СН-СООС.Н, р-рн (ршб.)
+і2 о ^НзС-с-сн-соосл"^ ~нзс £ ^(і сн>
У ^ м 25 о о
НзС-с^сн-с, о
Серед у-діоксосполук широкого застосування в лабораторному синтезі,
зокрема для одержання гетероциклів, набув сукцинальдегід ОНС(СН2)2СНО.
Для цього ж використовують найпростішого представника у-Дикетонів -
ацетонілацетон СНзСО(СН2)2СОСНз.
Розділ 18
МОНОКАРБОНОВІ
КИСЛОТИ
Серед органічних сполук, що мають помітну кислотність, найважливішими є
карбонові кислоти.
Карбоновими кислотами називають похідні вуглеводнів, які містять кар-
боксильну групу СООН. Назву "карбоксильна" утворено поєднанням назв
карбонільна (С=0) і гідроксильна (ОН).
18.1. Класифікація карбонових кислот
Залежно від кількості карбоксильних груп кислоти поділяють на moho-, ди-,
три- й полікарбонові (більше трьох карбоксильних груп). їх ще називають одно-,
дво- або триосновними кислотами, оскільки їхня молекула може зв'язувати певну
кількість еквівалентів основи:
Одноосновні
(монокарбонові)
СН3СН2СООН
Пропанова кислота
пропіонова кислота
За основністю
Двохосновні
(дикарбонові)
НООССН2СН2СООН
Бутандіова кислота
бурштинова кислота
Трьохосновні
(трикарбонові)
І0ГОН
ноос^^соон
Бензен-1,2,4-трикарбонова кислота
тримелітова кислота
Залежно від природи вуглеводневого замісника, з яким зв'язана
карбоксильна група, кислоти поділяють на аліфатичні (насичені й ненасичені),
аліциклічні й ароматичні.
Насичені
СН3(СН2)2СООН
Бутанова кислота
масляна кислота
За природою вуглеводневого замісника
Ненасичені Аліциклічні
Н2С=СНСООН
Пропенова кислота
акрилова кислота
СООН
Ароматичні
-СООН
Циклопропанкарбонова
кислота
<0ь<
Бензойна кислота
482
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Велика група карбонових кислот у бічному ланцюзі може мати додаткові
функції, що надають їм специфічних властивостей:
За природою додаткової функціональної групи
Галогенозаміщені Гідроксикислоти Оксокислоти Амінокислоти
карбонові кислоти
С1СН2СООН НОСН2СООН ОСНСООН Н2КСН2СООН
Хлороцтова кислота Гліколева кислота Гліоксилова Гліцин
кислота
Двохосновні й гетерофункціональні карбонові кислоти розглянемо окремо,
цей розділ присвячений лише монокарбоновим кислотам.
18.2. Номенклатура й будова монокарбонових
кислот
18.2.1. Номенклатура
• Раціональна номенклатура. За цією застарілою номенклатурою кислоту
формально розглядають як продукт заміщення одного або декількох гідрогенних
атомів в оцтовій кислоті на вуглеводневі замісники, наприклад: (СзНз^СН-
СООН - діетилоцтова кислота, (СНз)3С-СООН - триметилоцтова кислота.
• Замісна номенклатура ШРАС. Карбоксильну групу карбонових кислот, атом
Карбону якої є в складі вуглеводневого ланцюга, позначають суфіксом -ова
кислота. Для циклічних сполук, у яких цикл безпосередньо зв'язаний з
карбоксильною функцією, використовують суфікс -карбонова кислота. Для
позначення групи -СООН, що перебуває в бічному ланцюзі, застосовують
префікс карбокси-:
6 5 4 3 2 1
. СООН
2/СН2СООН
сн3сн2сн=снснсоон
■
1
СІ
^н2
( У-соон
2-Хлорогекс-З-енова кислота
1 -Аміноциклопро-
панкарбонова кислота
2-Карбоксиметилцикло-
гексанкарбонова кислота
Нумерація від головної функції.
Основа
- циклопропан.
Основа - циклогексан. Карбоксиль-
Початок нумерації від кар-
ну групу в бічному ланцюзі
боксильної функції.
називають за допомогою префікса
карбокси-.
■ Назву ацильних замісників ЯСО- - структур, що утворені відщепленням
від карбоксильної групи гідроксилу, утворюють із назв карбонових кислот
шляхом заміни суфіксів -ова кислота й карбонова кислота на -оїл і -карбоніл.
Розділ 18. Монокарбонові кислоти
483
■ Назву кислотних залишків ІІСОО— структур, отриманих відщепленням
атома Гідрогену від карбоксильної групи, одержують із назв карбонових кислот
шляхом заміни суфіксів -ова кислота й -карбонова кислота на -оат і -кар-
боксилат, наприклад:
СН3(СН2)4СО- Гексаноїл
СН3(СН2)4СОО- Гексаноат
Циклогексанкарбоніл
^ ^—СОО— Циклогексанкарбоксилат
Для низки карбонових кислот, але не їхніх похідних, IUP АС рекомендує
використовувати тривіальні назви. їхні кислотні залишки й ацильні замісники
наведені в табл. 18.1.
Таблиця 18.1
Тривіальні назви насичених аліфатичних монокарбонових кислот,
їхніх кислотних залишків й ацильних замісників, дозволених правилами IUP АС
Структура
Назви кислот
Тривіальні назви
систематичні
тривіальні
кислотних
залишків
ацильних
замісників
НСООН
СНзСООН
СН3СН2СООН
СН3(СН2)2СООН
(СН3)2СНСООН
СН3(СН2)14СООН
СНз(СН2)16СООН
Метанова
Етанова
Пропанова
Бутанова
2-Метилпропанова
Гексадеканова
Октадеканова
Мурашина
Оцтова
Пропіонова
Масляна
Ізомасляна
Пальмітинова
Стеаринова
Форміат
Ацетат
Пропіонат
Бутират
Ізобутират
Пальмітат
Стеарат
Форміл
Ацетил
Пропіоніл
Бутирил
Ізобутирил
Пальмітоїл
Стеароїл
■ Тривіальну назву оцтова кислота можна використати для побудови назв
відповідних похідних, наприклад, нітрооцтова кислота.
■ Для низки кислотних залишків та ацильних замісників застосовують
назви, утворені від англійських назв відповідних карбонових кислот: форміл і
форміат від formic acid, ацетил і ацетат від acetic acid, бутирил і бутират від
butyric acid.
■ У літературі трапляється застарілий спосіб побудови назви похідних
карбонових кислот. Розгалужені й заміщені карбонові кислоти називають,
використовуючи як основу тривіальні назви кислот і позначаючи положення
замісників грецькими буквами а, р, у, 8 і т.д.; буквою а позначають атом
Карбону, безпосередньо зв'язаний з карбоксильною групою, наприклад:
СН3
СН3СН2СНСООН
а-Метилмасляна кислота
СН2СН2СНСООН
СІ
у-Хлоро-а-метилмасляна кислота
484
Чирва В.Я., Ярмолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
18.2.2. Будова карбоксильної групи
Хоча карбоксил і є групою, яка структурно складається з гідроксилу й
карбонілу, її властивості різко відрізняються від уже відомих нам властивостей
гідроксилу спиртів і карбонілу оксосполук. Карбоксил - носій кислотних
властивостей, і дисоціацію карбонових кислот у воді описують рівнянням
ясоон + н2о ясоо- + н3о+
Кислотні властивості карбонових кислот зумовлені електроноакцепторним
впливом двох атомів Оксигену. Крім того, карбоксилат-іон, який утворюється
відповідно до теорії резонансу, є гібридом двох структур, що роблять однаковий
внесок у кінцеву структуру. Атом Карбону зв'язаний з кожним із атомів
Оксигену одним "полуторним" зв'язком. Негативний заряд рівномірно
розподілений (делокалізований) між двома атомами Оксигену. У спиртів такого
явища немає, тому для них слабкіше виражені кислотні властивості.
Те, що карбоксилат-аніон справді є резонансним гібридом, підтверджене
значеннями довжин зв'язків. Наприклад, у мурашиній кислоті наявні один
подвійний та один простий зв'язок Карбон-Оксиген. Тому ці зв'язки повинні
мати різну довжину. Натрій форміат, якщо він справді є резонансним гібридом,
повинен містити два еквівалентні зв'язки Карбон-Оксиген. Можна очікувати,
що вони матимуть однакову довжину, проміжну між довжинами подвійного й
простого зв'язків. Дані рентгеноструктурного аналізу й дифракції електронів
засвідчують, що ці припущення правильні. У мурашиній кислоті один зв'язок
Карбон-Оксиген має довжину 0,136 нм (простий зв'язок), а інший - 0,123 нм
(подвійний зв'язок), тоді як у натрій форміаті є два еквівалентні зв'язки С-0
довжиною 0,126 нм. Тому в теорії резонансу карбоксильну групу відтворюють
набором резонансних структур:
о-н он ^он
Карбоксильний атом Карбону зв'язаний з трьома іншими атомами с-зв'яз-
ками. Оскільки ці зв'язки використовують 5р2-орбиталі, то вони перебувають
один щодо одного під кутом 120°. Орбіталь р атома Карбону, що залишається,
перекривається однаково добре з /?-орбіталями обох атомів Оксигену,
утворюючи гібридні зв'язки. Отже, електрони зв'язані не з одним або двома
ядрами, а з трьома одночасно (один атома Карбону і два атома Оксигену). Саме
така участь електронів в утворенні більш ніж одного зв'язку, або, іншими
словами, делокалізація електронної хмари дає змогу відобразити аніон як
резонансний гібрид двох граничних структур:
Розділ 18. Монокарбонові кислоти
485
Карбоксилат-аніон
Цікаво, що тенденція вирівнювання довжин зв'язків Карбон-Оксиген про-
стежується навіть біля карбоксильної групи. Зокрема, довжина С=0-зв'язку в
альдегідах і кетонах дорівнює 0,121 нм, а в карбонових кислотах - 0,127 нм,
довжина С-О-зв'язку в спиртах становить 0,143 нм, тоді як у кислотах -
0,139 нм.
Константа дисоціації є характеристикою кислотності карбонових кислот:
_ [ЯСОО-] [Н3ОЧ __.
к>- [ясоон] рК*~ 1§к° .
Більшість карбонових кислот має значення рКа близько 4,8. Електроно-
акцепторні замісники, відтягуючи на себе електрони, стабілізують карбоксилат-
аніон і цим підсилюють кислотність та знижують рКа, наприклад:
Кислота С13С-СООН С12СН-СООН С1СН2-СООН СН3-СООН
/>Ка0,7 />Ка1,48 />Ка2,85 рКа4,75
Однак індукційний ефект швидко згасає в міру того, як електроно-
акцепторний замісник віддаляється від карбоксильної групи:
СН3СН2СНС1СООН СН3СНС1СН2СООН СН2С1СН2СН2СООН СН3СН2СН2СООН
а-Хлоробуганова кислота Р-Хлоробуганова кислота у-Хлоробуганова кислота Бутанова кислота
/^2,86 рК.4,52 ^4,82
Електронодонорні замісники, навпаки, знижують силу кислот:
СН3СН2СООН СН3СООН в НСООН
Пропіонова кислота Оцтова кислота Мурашина кислота
рКа4,86 рКа4,76 рКаЗ,37
Порівняно з сильними мінеральними кислотами, карбонові кислоти є
слабкими, та все ж значення їхніх констант дисоціації набагато вищі, ніж води,
не кажучи вже про спирти.
І ще одна особливість карбоксильної групи в органічних кислотах.
Карбонільна група в карбоксилі інертна. Вона не здатна вступати в жодні
реакції, що характерні для карбонільної групи альдегідів і кетонів, окрім реакцій
з магнійорганічними сполуками.
Карбонові кислоти, реагуючи з сильними кислотами, поводяться як слабкі
основи. Основність кислот.порівняна з основністю альдегідів і кетонів.та
виявляється в сильнокислому середовищі (рН < 3). У цьому разі можливе
протонування як карбонільного, так і гідроксильного атома Оксигену:
486
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
+
сн3-с
\
+ Н2804
яр
сн3-сч.. •
он
.он
сн,-с
,ОН
+ Н804
он
он
Я-С^ + Н2804
ОН
+ НБ04
он
н
Наявність карбоксильної групи позначається також на рухливості атомів
Гідрогену в а-положенні (СН-кислотність). Електронні пари обох атомів
Оксигену сприяють прояву кислотами основних і нуклеофільних властивостей, а
наявність на атомі Карбону карбоксилу невеликого позитивного заряду дає
змогу проводити нуклеофільні атаки й реакції заміщення в а-положення.
Нарешті, електроноакцепторні замісники, особливо в а-положенні, підсилюють
лабільність карбоксильної групи й можуть спричинювати декарбоксилювання:
Перші три представники кислот - леткі рідини з характерним гострим
подразнювальним запахом. Більшість представників від С4 до С9 - маслянисті
рідини з неприємним запахом. Вищі жирні кислоти без запаху, оскільки вони
мають низьку леткість.
Кислоти Сі_з змішуються з водою в будь-якому співвідношенні. Далі зі
збільшенням молекулярної маси кислот їхня розчинність зменшується. ВІД Сю і
далі - це тверді речовини з незначною розчинністю у воді. Цікаво, що
розчинність карбонових кислот у воді трохи вища, ніж у спиртів, оскільки з
водою кислоти утворюють міцніші водневі зв'язки.
Температури кипіння кислот підвищуються зі збільшенням молекулярної
маси, причому нормальні кислоти киплять вище від кислот з /зо-структурою,
наприклад, н-масляна кислота має температуру кипіння 163,5 °С, а /зо-масляна-
154,4 °С. Температури кипіння карбонових кислот вищі, ніж спиртів, що мають
ту ж молекулярну масу; це пояснюють більшою полярністю карбонових кислот і
міцнішими водневими зв'язками, які вони утворюють у димерах.
• Що стосується температур плавлення, то кислоти з парною кількістю
карбонових атомів плавляться за вищої температури, ніж сусідні гомологи з
непарною кількістю атомів. Це пояснюють тим, що в кислотах з парною кількістю
н
18.3. Фізичні властивості
Розділ 18. Монокарбонові кислоти
487
атомів Карбону метальна й карбоксильна групи перебувають по різні боки осі
молекули, а з непарною кількістю - по один бік. Завдяки симетричній будові
молекули кислоти з парною кількістю карбонових атомів сильніше взаємодіють між
собою в кристалічній ґратці, і її важче зруйнувати в разі нагрівання:
/сщ ^ /Сн2 Ш? СН^ /СН^СООН
Н3С СН2 сщ СООН Н3С СНз сн2
Гептанова кислота, Октанова кислота,
т. пл.-105°С т. пл. 16,2 °С
/СН./СН^СН^СН,
н3с сн2 сщ сщ соон
Нонанова кислота,
т. пл. 12,5 °С
Наявність рухливого атома Гідрогену в кислоті й надлишкового негативного
заряду на атомі Оксигену карбонілу засвідчує, що саме ці атоми відповідальні за
утворення асоціатів між кислотами типу димер або олігомер:
ї ї ї //0-Н-Оч
НЧС С .С~СН~
0"%Н о^он о^он о-н
З'ясовано, що за нормальних умов оцтова кислота існує на 68 % у вигляді
димеру, 15% - олігомеру й 17% - мономеру. Природно, що підвищення
температури веде до збільшення кількості мономерної форми кислоти.
18.3.1. Спектроскопія монокарбонових кислот
• УФ-спектроскопія. В УФ-спектрах насичених кислот спостерігають слабкий
пік поглинання в ділянці 195-210 нм, зв'язаний з и—►7с*-переходом електронів
карбонільної групи.
• ІЧ-спекпгроскопія. В ІЧ-спектрах для насичених кислот є сильна смуга
поглинання карбонільної групи Vc=o 1 750-1 765 см"1, широка voh 3 550 см"1 і
середня смуга 80н 1 400-1 200 см"1. У випадку наявності димерних структур
спостерігають додаткову смугу поглинання при 1 710-1 720 см"1.
• Mac-спектрометрія. У мас-спектрах органічних кислот найінтенсивніший
пік в ацил-катіона:
RCH2COOH ^ [RCH2COf + ОН
Ще один інтенсивний пік для нерозгалужених карбонових кислот є при miz
60, що відповідає оцтовій кислоті.
488 Чирва В.Я., Ярмолкж С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
• Спектроскопія ПМР. У ПМР-спектрах протон карбоксильної групи вияв-
ляється у вигляді широкого сигналу в слабкому полі при 10-13 м.ч.
18.4. Одержання карбонових кислот
18.4.1. Промислові методи синтезу мурашиної
та оцтової кислот
❖ Мурашина кислота. Нижчі члени ряду зазвичай одержують особливими
методами. Наприклад, мурашину кислоту синтезують у великих масштабах
взаємодією карбон (II) оксиду з водним розчином натрій гідроксиду за високих
температури й тиску:
СО + ИаОН НСОСЖа —^НСООН
❖ Оцтова кислота, безумовно, найважливіша з усіх карбонових кислот,
утворюється під час окиснення ацетальдегіду киснем повітря. Сам ацетальдегід
раніше синтезували гідратацією ацетилену або дегідруванням етанолу:
нс^н ^^4с^сно0-^сн^соон
сн,сн,он
_Си_
]
3^хх2^х 250_30()оС
Продуктом окиснення оцтового альдегіду киснем у промисловості є оцтовий
ангідрид, який потім гідролізують до оцтової кислоти:
*° о
2СН3СНО-^ Н>С"% ^2ЩС-С:
2 н,с-с' он
3 чо
■ Метод, який широко використовують для синтезу оцтової кислоти
сьогодні, полягає в каталітичній оксидній деструкції бутану:
Со(ОСОСН3)2 .
сн3сн2сн2сНз+о2і80ОС 15аш- 2СН3СООН
50 %
■ Нещодавно розроблено ефективніший метод отримання цієї кислоти шля-
хом карбонілювання метанолу над родієвим каталізатором:
СН,ОН + СО —№* » СН,СООН
3 180'С,30атм 31(Ю%
Розділ 18. Монокарбонові кислоти
489
У лабораторії в разі потреби оцтову кислоту синтезують із метану дією
концентрованої сульфатної кислоти, що відіграє роль окиснювача за наявності
паладієвого каталізатора:
Pd(II)
2СН4 + 4H2S04 - * СН3СООН + 4S02 + 6Н20
ІоО u
Значні об'єми оцтової кислоти виробляють також у вигляді розведеного
водного розчину, що називають оцтом. У цьому випадку оцтову кислоту
одержують окисненням етилового спирту киснем повітря, а каталізаторами є
ферменти бактерій Acetobacter.
18.4.2. Способи, що ґрунтуються на окисненні
❖ Окиснення алкапів.
■ Під час розгляду реакцій алканів зазначено, що в разі дії кисню на ці
сполуки за 100 °С та наявності солей Мангану як каталізатора (беруть зазвичай
КМПО4, що відновлюється) завдяки розриву ланцюга внаслідок окиснення різних
метиленових ланок виникає суміш кислот. Справді, у промисловості з бензинової
фракції С5_8 цим способом синтезують суміш мурашиної, оцтової й пропіонової
кислот, які далі розділяють на індивідуальні речовини. Із фракції алканів Сю-20
таким способом одержують синтетичні жирні кислоти, потрібні у виробництві
мил та інших мийних засобів.
❖ Окиснення алкенів і алкінів
■ Алкени і алкіни можна окиснювати до карбонових кислот за допомогою
калій перманганату або суміші калій перманганату і натрій періодату. В
останньому випадку необхідні каталітичні кількості перманганату, оскільки
надлишок періодату знову окиснює відновлену сіль Мангану. Застосування
каталітичної кількості солей Мангану веде до селективності реакції. Метод
використовують для з'ясування будови алкенів та алкінів і в препаративних
цілях.
На відміну від окиснення алкенів калій перманганатом або біхроматом, що
дає низький вихід карбонових кислот, розщеплення алкенів сумішшю калій
перманганату й натрій періодату відбувається кількісно:
снзсс^-с^с-сс^соон ^7о°с сщсн^соон + нсоцсн^соон
н H ' 100 % 100 0/0
■ Досить ефективним є окиснення калій перманганатом за умов між фа-
зового каталізу за наявності солей тетраалкіламонію:
ouchjxhkxl ^№)£?гсн (СН ) соон
34 2/7 2 Вода-бензен, 28 °С 3V Vl
91 %
490
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
■ Пряме окисне розщеплення алкенів до карбонових кислот можна вико-
нувати за допомогою озонолізу з наступним окисненням альдегідів:
Яч _ /Я "\ /'
-оч ,р Со
о
н ^о-о
кч ,Оч Л Н20
н о—о н
,0 .о
н,о,
—- Я-С + К-Сч -2£Х. Я-СООН + к-соон
н н
❖ Для окиснення первинних спиртів до відповідних кислот вико-
ристовують хроматну кислоту. Оскільки в процесі реакції альдегід може
взаємодіяти зі спиртом і давати напівацеталь із наступним перетворенням у естер:
[О] [О]
О
К-СН2ОН — Я-СНО —* Я-СООН
[О]
Я-СНО + Я-СН,ОН з=±= ІІ-^НОСН^ Я-С-ОСН,!*.
он
то процес проводять у дві стадії з проміжним видаленням із реакційної суміші
леткішого порівняно зі спиртом альдегіду.
❖ Окиснення оксосполук
• Альдегіди окиснюють амоніачним розчином арґентум оксиду, комплексними
сполуками Купрум (II) у лужному середовищі, наприклад, фелінговою рідиною і
сильними окиснювачами в кислоти з тією ж кількістю карбонових атомів:
• Кетони окиснюють за О. Поповим сильними окиснювачами (КМПО4) до
суміші карбонових кислот. Окиснення відбувається по обидва боки карбонільної
групи:
СН3СН2СН2СН2- С-СН2СН3 +[0] СН3СН2СН2СООН +
+ СН3(СН2)3СООН + СН3СООН + СН3СН2СООН
Окиснення кетонів частіше застосовують для аліциклічних сполук. Зокрема,
окисненням циклогексанону в промисловості одержують адипінову кислоту.
■ Обмежене застосування має реакція Байєра-Віллігера для окиснення
симетричних кетонів. Синтезовані естери перетворюються в кислоти лужним
омиленням:
Розділ 18. Монокарбонові кислоти
491
СР3СОООН №ОН
СН3СІ^С-СІ^СНз-^^ сн3сн2-с-осн2сн3 СН3СН2СООН
о 2 о 78 0/о
■ Метилалкілкетони кількісно перетворюються в карбонові кислоти внаслідок
галоформної реакції:
Я-СОСНз + З С12 НаОН» Я-СОСС13 +3 КаСІ
Я-СОСС13 + Н20 Л-СООН + СНС13
18.4.3. Способи, що ґрунтуються на гідролізі
х ?
Будь-які сполуки типу К_С^^ та К-^-У
де X, У, 7. - атоми галогену, Оксигену або Нітрогену (в останніх двох випадках
зв'язані з атомами Гідрогену або будь-якими групами атомів), під час гідролізу
утворюють кислоти ЯСООН:
Я-ССЦ + зн2о Я-С^ + З НСІ (1)
ОН
Я-С^ + 2Н20 -Ни к-С^°+ КН4+ (2)
Нітрил ОН
/ОСН3 и+ .о
Я-С-ОСНз + 2Н20 -Н-— К-С<Птт + ЗСН3ОН (3)
чосн3 он
Ортоестер
Л> (4)
К-С\^|+ Н20 *" я~^^он
Хлорангідрид
У перших трьох реакціях варто було б очікувати, що внаслідок гідролізу
утвориться сполука ЯС(ОН)з, так звана оршокислотпа. Однак навіть два
гідроксили біля одного атома Карбону стійкі лише в особливих випадках, а три -
ніколи. Унаслідок відщеплення води з ортосполуки утворюється карбонова
кислота:
/ОН
Я-С-ОН -ЯСООН + Н20
^ОН 2
Реакції (3) і (4) не становлять препаративного інтересу. Дещо важливим є
синтез карбонових кислот з доступних тригалогеноалканів (1). Найбільше
практичне значення має синтез карбонових кислот через нітрили (2).
492
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
❖ Гідроліз нітрилів. Аліфатичні нітрили одержують обробкою алкілгало-
генідів натрій ціанідом. У диметилсульфоксиді за кімнатної температури реакція
відбувається швидко і з виділенням тепла. Алкілнітрил, що утворюється,
гідролізують до кислоти водним розчином лугу або кислоти.
ЮС + С№ Я-О^ + X-
^^ï^RCooн + NH4+
^он^-КСОО- + №
RCN + H20:
Реакція алкілгалогенідів з ціанід-іонами відбувається як нуклеофільне замі-
щення атома галогену на нітрильну групу. Оскільки HCN - дуже слабка кислота, то
ціанід-аніон є сильною основою; завдяки цьому, як і треба було сподіватися, він
може відщеплювати протон, зумовлюючи елімінування поряд із заміщенням.
Справді, для третинних алкілгалогенідів елімінування - головна реакція; навіть для
вторинних алкілгалогенідів вихід продуктів заміщення невисокий. У цьому випадку
ми знову стикаємося з тим фактом, що реакція нуклеофільного заміщення
синтетично важлива тільки в разі використання первинних галогенідів:
CHj-^-Br + CN -CH3q=CH2 Елімінування
СН3 СН3
Третинний алкілгалогенід
СНзСН2СН2СН2Вг + CN" -> CH3CH2CH2CH2CN заміщення
Первинний алкілалогенід
■ Альдегіди й кетони можна перетворити в ціангідрини, які зазнають
гідролізу з утворенням а-гідроксикислот:
R4 ОН r
С=0 + HCN ^ R-C-C=N -7^?- /f-СООН
R І' " 4 ROH
Ці гідроксикислоти під час кип'ятіння відщеплюють молекулу води,
утворюючи ненасичені кислоти.
18.4.4. Синтез карбонових кислот методом
подовження ланцюга карбонових атомів
❖ Реакціїкарбонілювання
• Карбонілювання лугів і алкоголятів. Нагрівання лугів і алкоголятів спиртів
із карбон (II) оксидом за підвищеного тиску дає солі карбонових кислот:
NaOH + CO HCOONa
RONa + CO RCOONa
Розділ 18. Монокарбонові кислоти
493
Промислове значення має лише перша реакція, за якою сьогодні виробляють
усю мурашину кислоту.
• Карбонілювання ненасичених сполук та спиртів. Важливе промислове
значення одержав синтез кислот, починаючи з пропіонової, дією на олефіни
карбон (II) оксиду і води за наявності Нікол або Кобальт тетракарбонілу як
каталізаторів:
Н3С-СН-СООН
ы-сн=сн2 + СО
ИІ(СО)4
200-500°С,
15 атм.
я-сн-сн2
ї
о
У 1958 р. В, Хааф й Ю. Кох відкрили спосіб синтезу кислот дією карбон (II)
оксиду на олефіни за наявності концентрованої сульфатної кислоти. Зміст
реакції полягає в алкілуванні карбон (II) оксиду карбокатіоном, що утворюється
під час протонування алкену:
ЯСН=СН2 + Н+ ЯСНСН3
ЯСНСН3 + СО + НОН -ЯСНСН3 + Н+
У цих реакціях замість карбон (II) оксиду можна використовувати
мурашину кислоту, яка з сульфатною кислотою утворює карбон (II) оксид,
сульфатну кислоту можна замінити флуоросульфоновою або бор (III) флуорид
гідратом ЕҐ [НОВРз]".
■ Карбонілювання ацетилену за цих умов дає акрилову кислоту, а моно-
заміщених похідних ацетилену - а-заміщені акрилові кислоти:
НС=СН + СО + н2о СН2=СН-СООН
ЯС=СН + СО + Н20 СЙ^-СООН
Дизаміщені ацетилени дають суміші ізомерних кислот:
Ю-НСОК2 К^С^СН-К2
СООН СООН
■ Одноатомні спирти в процесі карбонілювання утворюють монокарбонові
кислоти, наприклад:
СНзОН + СО™^СНіСООН
494
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
❖ Синтез за допомогою металоорганічних сполук. Під час синтезу
карбонових кислот за допомогою метилмагнійброміду газоподібний вугле-
кислий газ пропускають в етерний розчин реактиву Гриньяра або виливають на
подрібнений сухий лід (твердий СО2). В останньому випадку сухий лід слугує не
лише реагентом, а й охолоджувальним агентом.
Реактив Гриньяра приєднується до подвійного зв'язку С=0 карбон (IV)
оксиду точно так само, як і в реакції з альдегідами й кетонами. У підсумку
синтезується магній карбоксилат, який розкладають сильною кислотою, як
звичайно, хлоридною:
ЯСООГ^Х -ї^— ЯСООН + Мї2+ +х-
Реактив Гриньяра можна використати в синтезі карбонових кислот, виходячи із
третинних спиртів.
Ме 9^3 т Н+ 9^3
снз-ср-он-^ сНз-^чл — снГ$^с\ сНз-^-соомвсі —- сн3"9ч:оон
сн3 сн3 сн3 сн3 сн3
тримегилоцгова
кислота
■ Набагато ефективнішим виявилося використання літійорганічних сполук:
,0
я-п + со2—- я-с' ш- Я-СООН + Іл
ОЬі
❖ Алкіліденфосфорани також реагують із карбон (IV) оксидом. Після
відщеплення трифенілфосфіноксиду під дією гідроксид-аніона з високими
виходами утворюються карбонові кислоти:
К к Р(С6Н5)3 Яч ,0
>=р(сбн5)3 ^ ^-соо- -8^ ,СН< + 0=Р(СбН5)з
Я я * он
❖ Синтез гомологів кислот Ь карбонових кислот. Спочатку карбонову
кислоту відомими методами перетворюють у галогеноангідрид, з якого
обробкою діазометаном синтезують діазокетон. Діазокетон під впливом
арґентум (І) оксиду перегруповується в кетен, який, взаємодіючи з водою, дає
наступний гомолог вихідної кислоти:
к_С00н^Я-С-с1^Я"-С-СН^=Н ^я-сн=с=о^ я-о^соон
II II 2
О О
Розділ 18. Монокарбонові кислоти
495
Виходи в цьому методі становлять 50-80 %. Стадія перетворення діазоке-
тонів у кетони має самостійне значення.
18.4.5. Синтез карбонових кислот та їхніх похідних
без зміни довжини карбонового ланцюга
❖ Реакція Вільгеродпга дає змогу перетворювати кетони дією на них
водного розчину полісульфіду амонію (NH^S,, (л=2-6) в аміди. Після відкриття
цей метод поширився на альдегіди, тіокетони, олефіни, ацетилени, спирти тощо:
R-CHO ^ RCONH2
RCH=CH-CH3T55^RCH2CH2CONH2
R2C(OH)CH3^^ R2CHCONH2
■ У лабораторній практиці широко використовують модифікацію Кіндлера
- одержання тіоамідів шляхом нагрівання кетонів із сумішшю сірки й амоніаку
або аміну (зазвичай морфоліну) з наступним гідролізом до кислот:
(Щ(^п(^сщтол^ снзссн^с^^ одод^аюн+н^+Шз
Я№Уггт/ггп
V/
❖ Перегрупування Фаворського. Важливе синтетичне значення має пере-
групування а-хлорокетонів під дією лугу в циклічні кетони з наступним
розкриттям циклу до карбонових кислот:
R-^HHj^-C^R
R-^№hCH-R
HjO(OH-)
R-CHfCH-R
s?
❖ Гідроліз жирів. Найважливіше джерело вищих аліфатичних карбонових
кислот - це тваринні й рослинні жири. За допомогою гідролізу жирів можна
одержати кислоти з нерозгалуженим ланцюгом і парною кількістю атомів
Карбону (від С6 до Сі8).
18.5. Солі карбонових кислот
Хоча карбонові кислоти зазвичай набагато слабкіші від мінеральних, вони
незрівнянно сильніші кислоти, ніж ті органічні сполуки, які ми дотепер
розглядали (спирти, ацетилен); вони - сильніші кислоти, ніж вода. Тому водні
496
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
розчини лугів легко переводять карбонові кислоти в солі, а водні мінеральні
кислоти легко перетворюють солі знову у карбонові кислоти:
ясоон =г=^ ЯСОО" + Н20
❖ Номенклатура. Назва солей карбонових кислот складається з систе-
матичної або тривіальної назви кислотного залишку (суфікс -оат або -ат) і назви
катіона, наприклад:
СН3(СН2)4СООК Калій гексаноат СН3СООКН4 Амоній ацетат
18.5.1. Фізичні властивості
Солі карбонових кислот, як і всі солі, - це кристалічні нелеткі речовини, що
складаються з позитивно й негативно заряджених іонів. їхні властивості цілком
відповідають тому, що можна було б очікувати для солей неорганічних кислот.
Сильні електростатичні сили, що втримують іони в кристалічній ґратці, можуть
бути зруйновані лише шляхом нагрівання до високої температури або в разі
використання сильнополярних розчинників. Температура, необхідна для
плавлення, виявляється настільки високою, що ще до того, як її можна досягти,
розриваються С-С-зв'язки, і молекула руйнується.
Амонійні солі й солі лужних металів (Калію, Натрію) карбонових кислот
розчинні у воді. Більшість солей важких металів (Феруму, Арґентуму, Купруму)
у воді нерозчинна.
Отже, за винятком кислот, що містять чотири і менше атомів Карбону і
розчинні у воді, карбонові кислоти і їхні солі лужних металів мають прямо
протилежні характеристики розчинності. Подібне розходження можна вико-
ристати для розпізнання кислот і їхнього розділення: нерозчинна у воді органічна
сполука, яка розчиняється у водному розбавленому розчині натрій гідроксиду,
може бути кислотою або однією з невеликої кількості інших органічних сполук,
кисліших ніж вода:
ЯСООН + МаОН -> КСООИа + Н20
Нерозчинна у воді Розчинна у воді
18.5.2. Способи одержання
■ Карбонові кислоти витісняють із солей слабшу карбонатну кислоту. Цю
реакцію використовують як якісну пробу на карбоксильну групу:
ЯСООН + МаНСОз — КСООИа + Н20 + С02|
■ Це ж виявляється й у взаємодії з алкоголятами спиртів і ацетиленідами:
Я-СООН + СгНзОИа -> Я-СООИа + С2Н5ОН
Розділ 18. Монокарбонові кислоти
497
Я-СООН + Иа+С=СН -> ІІ-СО(Жа + НС=СН
■ Аналогічно до звичайних кислот, карбонові кислоти реагують з активними
металами, оксидами, амоніаком, металоорганічними сполуками й гідридами
металів, які є сильними основами:
МНз
І
1
-я-соон
(ІІ-ССЮ)22п + Н2
(К-СОО)2ї^ + Н20
к-сосжн4
сНзме* Я-СООЇ^! + СН4
-*^К-СО(Жа + Н2
18.53. Хімічні властивості й практичне застосування
❖ Декарбоксилювання. Натрієві солі насичених карбонових кислот під час
нагрівання з натронним вапном утворюють насичені вуглеводні:
СНзСОСЖа ^ЦЗІ СН4 + ^СОг
■ Як зазначено вище, солі двовалентних металів і карбонових кислот під
час нагрівання дають кетони, а у випадку змішаних солей за участю мурашиної
кислоти — альдегіди:
н3с-сч н3с-с
^Са СН3-^СНз °Ха СН3СНО
НзС-с; О Н-<
О О
Надалі цю реакцію почали проводити, пропускаючи пари кислот над
каталізаторами - оксидами Мангану, Цирконію й Торію за 380-400 °С:
2ЬІСООН + С02 + н2о
О
Цей спосіб застосовують у промисловості, оскільки вихід кетонів становить
близько 80 %. Самі кислоти досить стійкі в разі нагрівання, а за 600-700 °С
утворюють кетени:
,0 600 °с
НХ-С' в ^=0, « СН2=С=0 + Н20
3 он ОН
498
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
❖ Реакція Кольбе (1849). Електроліз розплаву натрієвих солей відбувається
через декарбоксилювання за радикальним механізмом і дає змогу одержувати
насичені вуглеводні (детальніше див. розділ 7):
Електроліз
СН3СО(Жа СН3СН3 + Иа + С02
Анод Катод Анод
Це був перший в історії синтез алканів. У подальшому розвитку реакції
Кольбе вона виявилася корисною для синтезу діестерів дикарбонових кислот:
2СН3ООС(СН2)пСОО" "2е~» СН3ООС(СН2)2пСООСН3 + 2С02,
75%
а також в електролізі суміші солей карбонової кислоти й моноестеру
дикарбонової кислоти:
КСОО-+СН3ООС(СН2)пСОО" К-К + Я(СН2)пСООСН3 +СН3ООС(СН2)2пСООСН3
Поділ цих продуктів з використанням перегонки не дуже складний.
❖ Реакція Бородіна-Хунсдиккера. Очевидно, проміжне утворення ради-
калів відбувається й під час взаємодії брому з арґентум карбоксилатами, що дає
змогу синтезувати бромоалкани:
<р <р
я-с' + Вг2 —- Я~с' Я-СООЧ- Вг'
0Аё -АбВг 0Вг
Я-СОО* — Я* + С02 Я" + Вг-— Я-Вг
Модифікований метод полягає в нагріванні суміші карбонової кислоти, мерку-
рій (II) оксиду і брому:
НеО, Вг,
СН3(СН2)15СН2СООН С-^ТС СН3(СН2)15СН2Вг + НёВг2+ со2+н2о
Стеаринова кислота 1-Бромогептадекан, 93 %
Реакція відбувається через проміжне утворення солі.
■ Однією з модифікацій декарбоксилювання карбонових кислот є їхнє
нагрівання з плюмбум (IV) ацетатом і літій хлоридом (реакція Кочі, 1965 р.).
Вона особливо ефективна для синтезу вторинних алкілгалогенідів:
СООН СІ
□ рь(ОСГСН3)4ліо СІ + СНзС00ЬІ +СН3СООН +РЬ(ОСОСН3)2
Циклобутанкарбонова Хлороциклобутан, 100%
Розділ 18. Монокарбонові кислоти
499
❖ Мила. Натрієві солі вищих жирних кислот (наприклад, стеаринової
С17Н35СООН), які одержують дією лугу на рослинні й тваринні жири (цей
процес відбувається в миловарінні), називають милами. Звідси реакції гідролізу
естерів лугами одержали назву омилення.
Мила, які одержують з рослинних і тваринних жирів, після прання
потрапляють у стічні води, де зазнають біодеградації. До 1937 р. усі мийні засоби
були милами. Після того в побуті з'явилися синтетичні детергенти, які, на
відміну від звичайного мила, добре працювали у твердій воді. Річ у тому, що йони
Магнію й Кальцію позбавляли звичайні мила піноутворювальної здатності
внаслідок утворенню нерозчинних у воді солей. Тому прання в такій воді
неефективне. Синтетичні детергенти не мали цього недоліку. Перші синтетичні
детергенти були октилбензенсульфонатами:
Подібно до мил вони мають довгий неполярний вуглеводневий хвіст і полярну
групу на кінці молекули. Незабаром з'ясували, що детергенти з розгалуженим
хвостом не підлягають руйнуванню мікроорганізмами, що негативно впливало на
навколишнє середовище. Після інтенсивних досліджень синтезовано детергенти з
лінійними ланцюжками, які легко зазнавали деградації за допомогою бактерій.
❖ Сикативи. Манганові й деякі інші солі вищих жирних кислот засто-
совують як ініціатори окисних процесів і полімеризації (наприклад, "висихання"
оліф), їх називають сикативи.
Назва естеру складається з назви вуглеводневого залишку спиртового
компонента й систематичної або тривіальної назви кислотного залишку (суфікси
-оат/-ат або -карбоксилат). Естерну групу в бічному ланцюзі або за наявності
старшої групи позначають префіксом К-оксикарбоніл-:
803 Ыа+, де Я - розгалужений алкільний залишок
18.6. Естери
18.6.1. Номенклатура
СН3СООС2Н5
СН3(СН2)4СООСНз
Етилацетат
Метилгексаноат
СН.СНСН
Ізопропіл-2-метилбутаноат
2-Етоксикарбонілциклогексанкарбонова кислота
500
Чирва В.Я., Ярмолкж С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
18.6.2. Способи одержання
Для одержання естерів використовують три головні підходи, а саме: реакцію
спиртів з карбоновими кислотами, яку каталізують найчастіше неорганічні
кислоти; попереднє перетворення кислот у їхні активні похідні; нарешті,
активування спиртів щодо нуклеофільної атаки карбоксильних груп. В
останньому випадку найчастіше спирт активують перетворенням його в галогенід
або з використанням інших сполук, таких як діазометан, діалкілсульфати або
триалкілоксонієві флуороборати.
❖ Естерифікація кислот спиртами за Фіш ер ом
ЯСООН + Я'ОН =г= ЯСООК1 + Н20
Метод Е. Фішера застосовують для одержання естерів метилового, етилового
та інших нижчих спиртів. Реакцію каталізують іони гідрогену мінеральних
кислот. Це так званий специфічний кислотний каталіз. Сильна кислота, зазвичай
сульфатна або хлоридна, протонуючи атом Оксигену, збільшує електрофільність
атома Карбону карбонільної групи й тим самим полегшує приєднання такого
слабкого нуклеофіла, як спирт:
ОН Повільно І І
онн
Н+
н-6-н
к-9-ок' ^°к-с-ок'^°к-с:-ок^№
он он о
Досліди з органічною кислотою за допомогою міченого 180 засвідчують, що
вода Н2180 утворюється з гідроксилу карбонової кислоти й протона спирту.
Реакційна здатність до естерифікації спиртів знижується в такій послі-
довності: СН3ОН > первинні > вторинні > третинні, а ряд активності кислот має
такий вигляд:
НСООН > СНзСООН > ІІ-СНг-СООН > Я2СНСООН > ЯзССООН.
Отже, швидкість естерифікації максимальна у випадку первинних спиртів.
Вихід естеру значно знижується для вторинних спиртів. Естери третинних
спиртів і карбонових кислот прямою естерифікацією одержати не можна,
оскільки за умов реакції спирти легко зазнають дегідратування, перетворюючись
в алкени. Тому естери вторинних і третинних спиртів варто одержувати
ацилюванням цих спиртів ангідридами кислот та ацилгалогенідами за наявності
піридину або третинних амінів для нейтралізації кислот, що виділяються.
Розділ 18. Монокарбонові кислоти
501
Особливістю реакції естерифікації є її зворотність. Для зсуву рівноваги в бік
утворення естеру необхідно брати надлишок дешевшого реагенту (частіше
спирту) або видаляти з реакційної суміші воду, зазвичай у вигляді азеотропу з
бензеном, або, якщо це моясливо, відганяти естер.
• Реакції переестерифікації. Нові естери можна отримати шляхом взаємодії
доступних естерів з надлишком інших спиртів (алкоголіз) за умов реакції
переестерифікації. Для цього суміш естеру й надлишок спирту нагрівають за
наявності кислотних або основних каталізаторів: -
х>-С^° + р2-гш ~Н+» р-г^° + С2Н5ОН
к с^о-с2н5+ к он "— к %-к2
Прикладом алкоголізу естерів, каталізованих основами, може бути синтез
тре/и-бутилтерефталату, неможливий за умов кислотного каталізу:
СООСН3 СООС(СН3)
І| + 2(СНз)3СОН(С2!^^ Г || + 2СН3ОН
Аз'з
СООСН, СООС(СН3)3
*3 - *3'3
Переестерифікацію можна виконати й дією на естер іншої кислоти:
Я-С^° + ІГСООН К"-Сґ° + ясоон
О-Я О-Я'
Нарешті, можна міняти обидва реагенти:
Н,С-ССГ° + НзС-СНгС^° = НзОСНг-С^° + сн^с^
^ хо-с2н5 ^ ^ 0-СН3 3 ^ хо-с2н5 О-СНз
Усі ці реакції рівноважні, тому, щоб змістити рівновагу в потрібний бік,
необхідно використати відповідні прийоми. Синтетичні можливості цього методу
обмежені доступністю вихідних реагентів.
❖ Ацшіювання спиртів. Карбонові кислоти - порівняно малоефективні
ацилювальні агенти. Для підвищення реакційної здатності їх переводять у
похідні з більшою ацилювальною активністю. Нижче наведено ряд похідних
карбонових кислот:
/О .О //О /р
л-с? < я-< • < < &со^° < к~с^
хнн2 хоя' он сі
Ацшіювання спиртів похідними карбонових кислот може відбуватися як за
наявності неорганічних кислот:
502
Чирва В.Я., Ярллолюк CM., Толкачова H.В., Зеллляков О.Є. Органічна хімія
R-Cf + Н+ = R-cC°Hà R-g-X R-C-OR-,
чх Nx Д 6
H R'
так і під дією основ:
г
І
\
9'
R'-C( + RO" = R-Ç-OR' = R-Ç-OR' + X'
X X О
Для синтезу естерів найчастіше використовують ангідриди й галогено-
ангідриди кислот.
• Взаємодія ангідридів кислот зі спиртами
R-C^0
р ^О + Л'ОН ^СОСЖЧ^СООН
За винятком оцтового ангідриду, інші симетричні ангідриди кислот зрідка
використовують для ацилювання спиртів, оскільки вихід естерів, перерахований
на кислоту, не перевищує 50 %. Для синтезу естеру зазвичай застосовують
змішані ангідриди кислот, особливо карбонатної, причому атака спирту як
нуклеофіла переважно відбувається по карбонільній групі сильнішої кислоти:
сісоосл, /Я Я\ я «он ^
К"Сч (сн/н К~С\ /С-ОС2Н5 -ь^я-с-сж + С02+ С2Н5ОН
он 2 о о
Змішаний ангідрид оцтової й мурашиної кислот застосовують для швидкого
формілювання спиртів. Форміати також одержують із суміші мурашиної
кислоти й оцтового ангідриду.
• Взаємодія галогеноангідридів кислот зі спиртами. З усіх галогено-
ангідридів кислот найчастіше використовують бромо- і хлороангідриди:
ЯСОСІ + Я'ОН ЯСООК1 + НС1 '
Ця реакція відбувається дуже швидко й без каталізатора. У реакційну суміш
для зв'язування гідроген хлориду, що виділяється, вносять третинний амін
(піридин). Часто замість спирту, особливо третинного, застосовують його
алкоголят:
Я-С' + КОС(СН3)3 —-К-С-ОС(СНА+КС1
сі о
Розділ 18. Монокарбонові кислоти
503
❖ Приєднання карбонових кислот до алкенів
ЯСООН + СН2=С\ 3 ЯСООС-СНз
СНз сн3
Так одержують естери третинних спиртів. Реакція відбувається через проміжне
утворення карбокатіону:
Н3С.
Л>СН~ + Н+—- (СНЛ,С+
Н3С ^
Алкілування карбоксилат-аніонів алкілгалогенідами. Обмеження цієї
реакції зумовлене низькими виходами естерів через сильне сольватування
карбоксилат-аніона протонними розчинниками, що є причиною його низької
реакційної здатності. Використання умов міжфазового каталізу тетраалкіламо-
нієвими солями дало змогу уникнути цього недоліку завдяки полегшенню
перенесення карбоксилат-аніонів у апротонний розчинник. Як каталізатор
можна використати краун-етери:
СН3СН2СОО№ + СН3(СН2)3С1 ' СН3СН2СОО(СН2)3СН3
2^ 0£ Бутилпропюнат, 87 %
(СН3)3ССООК + ВгСН2С6Н5 (СН3)3ССООСН2С6Н5
Бензилтриметил ацетат, 95 %
❖ Алкілування солей кислот діалкілсульфатами. Для синтезу метилових
й етилових естерів використовують солі карбонових кислот, які піддають
обробці диметил- або діетилсульфатом:
О
ЯСООКа +С2Н50-|з-ОС2Н5 - Я-СООС2Н5+ С2Н50803На
6
Такі реакції відбуваються за механізмом 8к2, тому для їхньої реалізації
використовують полярні розчинники апротонного типу, наприклад: дим етил-
формамід, ацетон, диметилсульфоксид тощо.
❖ Метилювання кислот діазометаном. Метод особливо ефективний у
разі ідентифікації жирних кислот за допомогою газорідинної хроматографії:
Я-СООН +н2с=й=й — КСОО'СН3— №И — КСООСН3+И2
❖ Реакція Тищенко детально розглянута у розділі 17, присвяченому
хімічним властивостям оксосполук.
504
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
❖ Синтез Ь нітрилів. Зручним і легкодоступним методом синтезу естерів :
є алкоголіз нітрилів за наявності кислот.
+ОН о
я-с=к+н+ —- я-с=>щ — я-с=:ын -—- я-с=кн2 -£г Я-С-СЖ' + ш4+
о
❖ Окиснення за Байєром-Віллігером. Як уже зазначено, окиснення кето-
нів надкислотами дає естери.
Я-С-Я' ср^^°? Ч/\ —- Я-С-О^+СРзСООН
^ О Н-0 О—ОСОСБз 0
❖ Синтез Гриньяра. Аліфатичні й ароматичні естери легко синтезують на
основі реактиву Гриньяра.
?\ я
с2н5м§вг +С1-С-0-сн3 —~с2н— с-о-сн3+ М§С1Вг
18.6.3. Хімічні властивості й практичне застосування
❖ Гідроліз естерів. Реакція естерифікації зворотна, і тому йон Гідрогену
каталізує гідроліз естеру. Роль протона можна уявити в такий спосіб:
О ОН ОН ОН
н _ н+ _ і ^ ^ І
я-с-о-я' я-с-оя' = я-с-оя я-с-оя'
+ + '
і н-о-н
== Я-С—О. Я-С-ОН + Я'ОН
ІіГхн -№
Як видно з наведених рівнянь, цей механізм зворотний до механізму
естерифікації за Фішером. Із експерименту з міченими атомами випливає, що за
кислотного гідролізу естеру атом Оксигену з молекули води з'являється в
молекулі карбонової кислоти:
О
я-с-оя/ +н218о я-с-он + Я'ОН
II ^
о
Гідроліз більшості естерів відбувається за таким механізмом. Винятком є
гідроліз естерів третинних спиртів з утворенням третинного карбокатіону:
Розділ 18. Монокарбонові кислоти
505
0 т' ?Н ^ .ОН +/* „р .„ Л
Я-ОО-С-Я' + Н+ Я-С-ЮтС-Я = я-сч + с-я
но-с-я
я + ~ V 'о \, чя
Гідроліз естерів можна проводити в лужному середовищі (омилення) :
0
ясоя + он-
я-^-оя
. он .
ясоон+-оя —- ясоо- + НОЯ'
Для цього застосовують водний або водно-спиртовий розчин лугів, а омилення
просторово утруднених естерів виконують за наявності краун-етерів.
Порівняно з кислотним гідролізом омилення естерів відбувається незво-
ротно. На ньому ґрунтується миловарне виробництво. Ще в античні часи люди
вміли одержувати мило. Тваринні жири, у складі яких є тристеарат гліцеролу,
нагрівали з деревним попелом, що містив поташ:
СН2ОСОСІ7Н35 СН,ОН
срнососІ3н35 КгС7Н2°. <рнон + ЗС17Н35СООК
сн2осос17Нз5 сцрн
Гліцерол Калій стеарат
Калій стеарат є основою рідкого мила, а натрій стеарат - твердого.
❖ Синтез Гриньяра. Естери як вихідні речовини можуть взаємодіяти з
реактивом Гриньяра у двостадійному синтезі, утворюючи третинні спирти:
КаЮК' + БП^І Я-С-БГ Я-С-БГ я-с-я» + MgI(OH)
О ОМ§І он
Естери мурашиної кислоти в цьому випадку слугують основою для одер-
жання симетричних вторинних спиртів:
*°
Н-с' + 2СН3СН2СН2М£Вг-* СН3СН2СН2СН(ОН)СН2СН2СН3
ос2н5
Етилформіаг Гептан-4-ол
❖ Відновлення естерів алюмо- або борогідридами лужних металів є
важливим синтетичним методом і дає первинні спирти:
ІлВН
сн3(сн2)14соос2н5 4» СН3(СН2)14СН2ОН + С2Н5ОН
95 %
506
Чирва В.Я., Ярллолкж C.MV Толкачова Н.В., Земляков О.Є. Органічна хімія
Останніми роками як відновник найліпше зарекомендував себе алюміній
діізобутилгідрид:
Він має знижену порівняно з літій алюмогідридом відновну активність і тому є
селективнішим. Відновлення гідридами металів витіснило старий метод
відновлення естерів за Буво-Бланом натрієм у спирті.
❖ Естери нижчих карбонових кислот і найпростіших спиртів - рідини з
сильним фруктовим запахом. їх застосовують для ароматизації напоїв. Багато
естерів (етилацетат, бутилацетат, ізопентилацетат) широко використовують у
техніці як розчинники, особливо для лаків.
❖ Реакції еістерів з амоніаком та амінами розглянуті у відповідному
розділі.
Алкільні похідні неіснуючих ортоформ карбонових кислот ЯС(ОН)з
називають ортоестерами. За номенклатурою IUP АС їх називають як похідні
відповідних ориюкарбонових кислот або як алкоксипохідні насичених
вуглеводнів:
❖ Алкоголіз трихлоралканів. Нагрівання 1,1,1-трихлоралканів Я-ССЬ, що
не мають при а-карбоновому атомі Гідрогену, з алкоголятом натрію дає
ортоестери:
18-7. Ортоестери кислот
18.7.1. Способи одержання
RCCl3 + 3NaOR
/ORf
RC-OR' + 3NaCl
\)R'
Із хлороформу таким способом одержують ортомурашиний естер:
9R
НССІ3 + 3NaOR' H4:-OR + 3 NaCl
ÖR
Якщо біля а-карбонового атома є атом Гідрогену, то під дією алкоголяту
відбувається процес відщеплення гідроген хлориду:
RCH2CClз + ИаО^ -> RCH=CCl2 + ИаСІ + R'OH
Розділ 18. Монокарбонові кислоти
507
❖ Синтез на основі нітрилів. Дія на нітрили кислот гідроген хлориду й
надлишку спирту спочатку дає гідрохлорідімідат, що далі зазнає алкоголізу,
унаслідок чого утворюється ортоестер з однаковими алкоксигрупами:
КС=И — ЯС^ИН *,0НШЯ? ЯС=>Ш.НС1 *-1Е5їі-К~С(ОК,)з + ИН4С1
СІ ОЯ'
Естери ортомурашиної кислоти можна синтезувати таким методом із
гідроген ціаніду, проте одержання з хлороформу зручніше.
❖ Синтез на основі кеталів. Для одержання ортоестерів іноді викорис-
товують інші сполуки, які мають кратні зв'язки. Наприклад, каталітичне
приєднання спиртів до кеталів дає ортоестери:
1^0=0(0^)2 + БЮН -^Б^СН-С^ЬОз
❖ Переестерифікацією ортоестерів успішно синтезують складніші за
будовою ортоестери:
К.С(ОК2)3^ к'-с-сж2 ^ к'-б-ок2^
оя2 °к2
о
(ж
сж3
= Я-С-ОК2=іН К'С(СЖ3)з + 2К2ОН
2 (Ж2
Дня переестерифікації беруть спирти з більшою молекулярною масою, ніж в
ортоестері, щоб відігнати з реакційної суміші спирт, що виділяється в процесі
синтезу.
18.7.2. Хімічні властивості
❖ Гідроліз ортоестерів. Ортоестери стійкі до дії лугів і легко зазнають
гідролізу в кислому середовищі по стадіях. Спочатку утворюються естери, які
потім дають кислоти й спирти:
• К-ф-(Ж + Н20 КСОСЖ + 2Я'0Н
(Ж
КСООЯ' + Н20 -Е— ЯСООН + Я'ОН
508
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
❖ Синтез ацетилів. У препаративній хімії ортоестери мурашиної кислоти
застосовують для одержання ацеталів і особливо кеталів. Кеталі найлегше
синтезувати саме таким шляхом (Л. Кляйзен):
сн3 ос2н5 НзС ос н
0=С + НС-ОС2Н5 —" + НСООС2Н5
СН3 ОС2Н5 НзС ос2н5
❖ Синтез тіоортоестерів. Своєрідна переестерифікація відбувається під
час взаємодії ортоестерів з тіолами:
КС(ОК!)3+ЗК28Н=^ КС(аК2)з+ЗК'ОН
❖ Естерифікація кислот. У деяких випадках ортоестери використовують
для естерифікації карбонових кислот:
к-с' + нс(ОС2н5)3—— я-с; + с2н5он+н-<
чон ос2н5 ос2н5
Реакція відбувається з високим виходом і, що особливо важливо, не потребує
участі каталізатора. Тому її можна розглядати як зручний метод одержання
естерів кислотолабільних карбонових кислот.
❖ Відновлення. Під дією розрахованої кількості літій алюмогідриду
ортоестери відновлюються до альдегідів або їхніх ацеталів:
КС(ОСН3)3 ЯСН(ОСН3)2 + СН3ОН
❖ Конденсація. Ортоестери легко зазнають конденсування зі сполуками, що
містять реакційноздатні метиленові групи, утворюючи алкоксиметиленові похідні:
9с2н5 срооя
Н<р-ОС2Н5 + СН2 С2Н5ОСН=С(СООК)2 + 2С2Н5ОН
ОС2Н5 сооя
❖ Синтез ацеталів на основі реактиву Гриньяра. Ортоформіати
взаємодіють з реактивом Гриньяра, утворюючи ацеталі, які, як відомо, легко
розщеплюються до альдегідів:
НС(ОІІ)з + ІЛ^І ^ Я'СЩОК^ —^ ІІ'-СНО
У літературі цю реакцію називають реакцією Бодру-Чічібабіна1, її вико-
ристовують для одержання аліфатичних і ароматичних альдегідів.
1 Чічібабін О. (1871-1945)
сполук.
- російський хімік. Головні праці присвячені хімії гетероциклічних
Розділ 18. Монокарбонові кислоти
509
18.8. Галогеноангідриди кислот
❖ Номенклатура. Назва галогеноангідридів загальної формули RCO-Hal
складається з назви ацильного залишку й назви галогеніду. Якщо в сполуці є
старша група, то використовують префікс галогенокарбоніл- (раніше застосо-
вували префікс галогеноформіл-).
,0 ,0 ^^СОСІ
СН3(СН2)4-С СН3СН2СН2-С f Ь
СІ Вг /^-^СООСНз
Гексаноїлхлорид Бутирилбромід
хлорокарбонілциклогексанкарбоксилат
18.8.1. Способи одержання
❖ Хлороангідриди. Найважливішими сполуками цього класу є хлоро-
ангідриди. їх одержують із кислот за однією з таких реакцій:
RCOOH + PCl5 ►RCOCl + РОСІ3 + HCl
RCOOH + SOCl2 RCOCl + S02 + HCl
2 RCOONa + S02Cl2 RCOCl + Na^
RCOOH + COCl2—^RCOCl + HCl + C02
(1)
(2)
(3)
(4)
Для реакції з тіонілхлоридом запропоновано такий механізм нуклеофільного
заміщення:
<Х
О СІ О СІ | а +
п'8=0~^ ^с-^о-—к^8=о—к-с-о-?=о ^^9"а
сн а ^0СХ о н а о-н а о
Для лабораторного синтезу ацетилхлориду користуються головно реакціями
(1) і (2); у техніці його одержують за реакціями (3) і (4). У промисловому
виробництві ацилхлоридів вигідніше використовувати солі карбонових кислот.
Особливо м'якими умовами вирізняється синтез із карбонових кислот і
трифенілфосфіну в тетрахлорометані:
Я-С ' + Р(С6Н5)3 + СС14 *• R—С^ + (СбН5)3Р=0 + СНС13
ОН СІ
❖ Бромоангідриди готують зазвичай дією фосфор (III) броміду на кислоти:
510 Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
О 5+5- § +
Л + Вг,Р-Вг—- Я-^С^-р-РВг,—►Я-С-Вг+НОРвг,
2 І ^-»Т 2 и 2
Я ОН Вг н о
Окремо є реакція синтезу галогеноангідридів з кетену й гідрогенгалогенідів:
9
н2с=с=о + НВг Н3С-С-Вг
❖ Іодоангідриди малостійкі, тому їх застосовують зрідка. Одержують дією
фосфор (III) іодиду на кислоти або переведенням ацилхлоридів в ациліодиди за
реакцією
О 1
❖ Флуороангідриди синтезують обміном на атом Флуору атома галогену в
хлороангідридах, діючи на них калій гідрофлуоридом КНР2.
Нарешті зазначимо, що найліпшими лабораторними методами синтезу
бромідів, іодидів і флуоридів є дія на хлороангідриди карбонових кислот сухих
НВг, Ш та Ш\
18.8.2. Хімічні властивості
Галогеноангідриди карбонових кислот - сильні електрофільні реагенти, що
зумовлено наявністю часткового позитивного заряду на атомі Карбону карбонільної
групи. Внаслідок електроноакцепторних властивостей атомів галогену (-І-ефект) й
Оксигену густина електронів на карбонільному атомі Карбону різко зменшується. З
огляду на це галогеноангідриди -сильніші електрофільні реагенти, ніж відповідні
карбонові кислоти. Завдяки збільшенню радіуса атомів у ряді від Флуору до Іоду
електрофільність ацилгалогенідів зростає в такій послідовності:
Я-СОР < Я-СОСІ < Я-СОВг < Кг-СОІ,
однак практичне значення мають хлороангідриди, рідше через вартість -
бромоангідриди.
У цих речовин різко виражені ацилювальні властивості. У катіоні до
позитивно зарядженого атома Карбону частково відтягується вільна пара
електронів атома Оксигену, унаслідок чого позитивний заряд розподіляється
між атомами Карбону й Оксигену. Реальну середню структуру ацил-катіона
можна виразити за допомогою двох резонансних формул:
Я-С^О Я-С^)+
Розділ 18. Монокарбонові кислоти
511
❖ Гідроліз. Ацилгалогеніди легко гідролізувати водою, ще легше - лугами:
я-с-сі н
о
ясоон + неї
-^Ш-ЯСООКа + ШС1 + Н20
Це типова реакція нуклеофільного заміщення за атомом Карбону:
О
К-С-С1 +Н,0
Ч +
я—с—ОН,
- С1 -
ЧС1
0 + -
я-с-он2+сі
к-с{ + неї
ОІГ
Реакція незворотна, оскільки аніон хлору - слабший нуклеофіл, ніж гідроксид-
аніон.
❖ Реакції нуклеофільного заміщення. Ацилгалогеніди легко взаємодіють з
нуклеофілами, утворюючи різні сполуки:
я-с.
Естери
К'ОН
ОЯ
(0
Я~С. Я'СОСЖа
,0
Я-С,
О
Ангідриди
Р
я-с,
СІ
Ыа8Н
Р
я-с.
ин2
Аміди
Р
я-с.
Азиди^З
Р
я-сч
8Н
тіокислоти
Алкоголіз ацилгалогенідів спиртами дає естери за механізмом, подібним до
гідролізу. Хлоридну кислоту, яка виділяється, нейтралізують піридином.
Під час взаємодії ацилгалогенідів з амоніаком, первинними й вторинними
амінами з високими виходами (85-95 %) утворюються незаміщені або ІУ-замі-
щені аміди:
(СН^СНС?°+ 2Ш3 (СН3)2СН-^° + >*Н4С1
❖ Відновлення. Залежно від умов реакції ацилхлориди відновлюються до
альдегідів (реакція Розенмунда1-Зайцева) або первинних спиртів. У першому
випадку гідрування проводять над паладієм на барій сульфаті, дезактивованому
хіноліном або сіркою.
1 Розенмунд К. (КоБептипсі КЖ) (1884-1965) - німецький хімік, учень О. Дільса. Головні праці
присвячені органічному синтезу.
512
Чирва В.Я.*, Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Сучаснішим методом синтезу альдегідів з ацилгалогенідів є застосування як
відновника літій три-тре/я-бутоксіалюмогідриду, що, на відміну від літій
алюмогідриду, відновлює ацилгалогеніди до оксосполуки, а не спирту:
СН ҐГН Ї-С^° ЬІА1Н[ОС(СН3)3]з <>0
СН^СН^ сч - СН3(СН2)ГС
СІ "78 С Н
Октаналь, 60%
Сам відновник одержують взаємодією літій алюмогідриду з трет-бути-
ловим спиртом:
ІЛА1Н4 + 3(СН3)3СОН ЬіА1Н[ОС(СН3)3]3 + ЗН2
❖ Синтези з металоорганічними сполуками. З еквівалентних кількостей
ацилгалогеніду й реактиву Гриньяра одержують кетони, а як побічний продукт -
третинні спирти:
—Я-С-Я' + МеСІХ
чсг
н
О
к-^-к —- Я-С-ОМеХ -^КЯ^ОН+М8(ОН)Х
СІ І'
■ Для синтезу кетонів доцільніше застосовувати кадмій- або меркурійорга-
нічні сполуки:
СІ о
(80-100%)
■ Після відкриття діалкілкупратів хіміки надають перевагу синтезам з їхнім
використанням:
(СН3)3С-С^ + [(СН3]2Си]ІЛ —*(СНз)зС-С-СН3
СІ 95 о/о
18.9. Ангідриди кислот
Формально ці сполуки розглядають як продукти, що утворилися внаслідок
відщеплення води від двох молекул одноосновних карбонових кислот або однієї
молекули води від двоосновної карбонової кислоти:
Розділ 18. Монокарбонові кислоти
513
ясоо_[н
ясо [он
-н2о ясС~ сн^-соон -н^ сн2-с^о
ЯС^ СНз-СООН СНз-С^
❖ Номенклатура. Назви симетричних ангідридів монокарбонових кислот
загальної формули (ЯСО)20 утворюють із назв відповідних кислот, замінюючи
слово кислота на ангідрид. Назви змішаних ангідридів загальної формули
ЯСО-О-ОСЯ' будують з перерахованих за абеткою назв карбонових кислот і
слова ангідрид.
(СН3СО)20 (СН3СН2СО)20
н-сС
НзС-С^
3 ^О
Оцтовий ангідрид, Пропіоновий ангідрид Мурашинооцтовий
або ацетангідрид ангідрид
18..9К1. Способи одержання
❖ Реакції дегідратації. За наявності таких сильних засобів, що забирають
воду, як фосфор (V) оксид, карбонові кислоти легко зазнають дегідратування,
перетворюючись в ангідриди:
2СН3СН2СООН (СН3СН2СО)20
Цей метод особливо зручний для одержання ангідридів сильних кислот:
СБзСООН Р'°5» (СР3СО)20 + НР03
Трифлуорооцтовий ангідрид, 80%
■ Вищі монокарбонові кислоти під час нагрівання з оцтовим ангідридом
перетворюються в ангідриди. Спочатку утворюється змішаний ангідрид, який за
вищих температур зазнає диспропорціювання.
✓ О
/>о н,с-сС
Я-С^ + (СН3СО)20 ► 3 + СН3СООН
он я-<
о
2 Н3С-С^ Н3С-С'С° Я-С^
514
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
■ Ангідриди можна одержати, діючи на карбонові кислоти тіонілхлоридом
або фосгеном:
CHjCOOH
-неї, so, 3
(СН3СО)20 + Н20
\
❖ Ацилювання солей карбонових кислот. Ангідриди можна синтезувати
ацилюванням солей кислот хлороангідридами:
БІ^-СІ + КаС^Я' -Ир-О-ра' + ИаСІ
О О О О
У цьому разі Я і Я' можуть бути однаковими або різними. Зокрема, так
одержують змішані ангідриди трифлуорооцтової кислоти
Ко~°_($СРз'
які є найсильнішими ацилювальними засобами. За їхньою допомогою вводять
ацил ІІСО- в різні органічні сполуки.
■ На реакції солей кислот з їхніми хлороангідридами ґрунтується спосіб
одержання оцтового ангідриду, який раніше застосовували в промисловості.
Хлороангідрид не виділяли, а одержували безпосередньо в реакційному
середовищі дією сульфурилхлориду або фосгену на натрій ацетат. Друга
молекула натрій ацетату дає з ацетилхлоридом, що утворився, оцтовий ангідрид.
■ Самі карбонові кислоти теж реагують з ацилгалогенідами. Процес
проводять за наявності піридину:
RCOOH + R'COCl + C5H5N
R-CO-O-OCR' + C5H5NHCI
Ангідрид
❖ Реакції з кетенами. Карбонові кислоти взаємодіють із кетенами, утво-
рюючи ангідриди:
лО/ 8+ 5-
о-н
«О
cHjCHj-c;
о
нас=сч
он
'Р
сНзС^-с:
р
н3с-сч
Сам кетен відкритий 1905 р. Г. Штаудінгером.
18.9.2. Хімічні властивості
Ангідриди карбонових кислот, подібно до галогеноангідридів, є активними
електрофільними реагентами. Звичайно, частковий позитивний заряд на карбо-
Розділ 18. Монокарбонові кислоти
515
нільному атомі Карбону менший, ніж у галогеноангідридах, проте більший
порівняно з кислотами. Ефект +М атома Оксигену в ангідридах діє на два
карбонільні атоми Карбону, тоді як у кислоті — лише на один:
❖ Гідроліз. Ангідриди карбонових кислот легко гідролізувати водою, а ще
легше-лугами:
р£ 2СН3СООН
(СН3СО)20 —
2СН3СО(Жа
Ці реакції мають сенс тільки тоді, коли ангідрид доступніший, ніж сама кислота.
❖ Реакції ацилювання. У реакціях зі спиртами ангідриди утворюють естери:
3 /О + яон -т— НзС-^-ф-я —- СН3СОСЖ+СН3СООН
НзС_С^0 О Н
н3с-с=о
■ Третинні спирти в таких реакціях потребують активування кислотами
Льюїса:
(СН3)3СОН + (СН3СО)20 ^ (СН3)3СОСОСН3 + СН3СООН
Приклади ацилювання ангідридами різних сполук такі:
СН3(
Естери
сн3сосж-
К0Н СНзССЯН
Тіокислоти
снхомн. —^
(СН3СО)20
^ СНХОБІІ
Аміди Тіоестери
■ Трохи незвичайно виглядає реакція ацилювання третинних амінів:
(СН3СО)20 + БЦИ —- НзС-С-^ + СН3СО(Ж
О
18.9.3. Оцтовий ангідрид
Оцтовий ангідрид - рідина, що має неприємний різкий запах. Цей продукт
широко застосовують у техніці, у різноманітних процесах ацилювання
516
Чирва В.Я., Ярмолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімій
(целюлози, фенолів, ароматичних амінів). Йому надають перевагу перед хлоро-
ангідридом, що має занадто низьку температуру кипіння й украй агресивний, а
тому менш зручний у заводських умовах.
■ Сучасним способом одержання оцтового ангідриду, що набуває щораз
важливішого значення, є дія кетену на "крижану" оцтову кислоту: \^
СН3СООН + СН2=С=0 (СН3СО)20
95%
Сам кетен одержують на основі оцтової кислоти:
уР (С2Н50)3Р=0
нх-с' і н2с=с=о + н2о
Як бачимо з цієї реакції, кетен сам є ацилювальним агентом. Справді, його
можна використовувати замість оцтового ангідриду або ацетилхлориду.
Незручність застосування полягає в тому, що він газоподібний, швидко зазнає
димеризування, тому його важко зберігати.
■ Іншим промисловим методом одержання оцтового ангідриду є окиснення
ацетальдегіду киснем за наявності кобальт і купрум ацетату за реакцією Хехста:
О, 'Р СН3СНО _ _
сн3сно н3с-сХоонЖ^ (СН3СО)20 + н2о
Разом з оцтовим ангідридом одержують оцтову кислоту.
Невигідність застосування оцтового ангідриду як ацилювального агента
полягає в тому, що половина його молекули не ацилює, а перетворюється в
кислоту.
18.10. Аміди
Амідна група СОМІ12 є важливою складовою частиною багатьох біологічно
активних сполук, тому знання способів одержання, властивостей і реакцій амідів є
важливим для подальшого розвитку хімії пептидів і білків. Крім біологічних аспектів,
аміди становлять інтерес для фундаментальних хімічних досліджень, оскільки
спряження між неподіленою електронною парою атома Нітрогену й л-електронами
карбонільної групи відображене в характерних фізичних і хімічних властивостях.
❖ Номенклатура. Назви амідів загальної формули ЯСО-КНг утворюють із
систематичних назв карбонових кислот, замінюючи суфікси -ова кислота й
-карбонова кислота на суфікси -амід і -карбоксамід, або з тривіальних назв
кислот, додаючи до основи закінчення -амід. Аміди, отримані з первинних і
вторинних амінів, називають якіУ-заміщені аміди:
Розділ 18. Монокарбонові кислоти
517
О о О О
сн3(сн2)5-с; ҐЛ-С* СНз-С н-с
N11 ^ кн2 МНС2Н5 К(СН3)2
Гептанамід Циклопентанкарбоксамід ІУ-Етилацетамід ІУ,ІУ-Диметилформам ід
18.10.1. Способи одержання
❖ Ацилювання амоніаку й амінів. Вище наведено реакцію ацилювання
амоніаку ангідридом кислоти, а ще вище - аналогічну реакцію з хлороангідри-
дом. Продуктом ацилювання в обох випадках є амід кислоти з загальною
формулою КСОІЧНг.
Під час ацилювання амінів утворюються //-алкіл- і Л/;М-діалкіламіди:
ЯСОСІ + 2СН3>ІН2 ЯСОШСНз + СН31\ІН3С1
ЯСОСІ + 2(СН3)2№І КС(Ж(СН3)2 + (СН3)2ИН2С1
■ Реакція ацилювання амінів може відбуватися за механізмом 5^7. У цьому
разі настає дисоціація ацилгалогеніду з наступною швидкою атакою ацилка-
тіона, що утворюється, амоніаком чи аміном:
Повільно ^ +^ ЯТГЩ
Швидко^ ІТ
я-с-х бі-со ^^к-с-нк'я" + нх
о о
Цей механізм реалізується тільки для декількох реагентів, у яких стабільність
аніона X" надзвичайно висока, як у випадку ацилтетрафлуороборату.
■ Здебільшого реакція відбувається за механізмом А^-Е - тобто приєднання-
відщеплення:
Я-С—СІ ►Я-С£С1—►іі-с-К-Н + СГ—^-£-N^,+N^1
н ^ін,
Цю реакцію широко застосовують у промисловості й лабораторіях. Процес
проводять за кімнатної температури або з охолодженням. Аміди утворюються з
високими виходами. Для нейтралізації кислоти, що виділяється, беруть надлишок
амінів або амоніаку. Як звичайно, як ацилювальні агенти використовують
доступні бромо- і хлороангідриди, хоча для одержання формамідів ліпшим
реагентом виявився формілфлуорид.
■ Ацилювання ангідридами карбонових кислот амоніаку та амінів відбу-
вається аналогічно до ацилювання ацилгалогенідами хоча й з меншою швидкістю.
518
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
Цей підхід застосовують у синтезі амідів тоді, коли галогеноангідриди реагують
занадто бурхливо. Як звичайно, використовують симетричні ангідриди. Як
каталізатор застосовують піридин або мінеральні кислоти, однак процес може
бути й автокаталітичним, оскільки одним із продуктів реакції є кислота.
■ Кетени легко реагують з амоніаком та амінами, утворюючи аміди:
2СН2=С=0 + №І3 -> (СНзССОгИН
КГС=С=0 +КНЯ2— іі2сн-<Х
Надзвичайно високу активність карбонільної групи кетенів пояснюють тим,
що я-зв'язки в них розташовані у взаємно перпендикулярних площинах,
унаслідок чого частково позитивний заряд на карбонільному атомі Карбону не
компенсований сусіднім я-зв'язком. З цієї причини він, як ацилювальний агент,
у багато разів перевершує ангідриди й галогеноангідриди карбонових кислот.
❖ Амоноліз естерів. Ацилювати амоніак можна навіть таким слабким агентом,
як естери. Щодо естерів цю реакцію називають амонолізом:
ЯСООС2Н5 + Щ, ЯС(ЖН2 + С2Н5ОН
❖ Піроліз амонійних солей карбонових кислот. Під час взаємодії кислоти
з амоніаком спочатку утворюється амонієва сіль:
сн3сн2сн2-соон + мн3^- сн3сн2сн2-соо~ш4+
Карбоксилат-аніон має низьку реакційну здатність у реакціях нуклеофіль-
ного заміщення, тому аміди із солей у розчинах не утворюються.
При нагріванні (суха перегонка) амонійні солі карбонових кислот розпа-
даються до амідів з виділенням води:
185 РС
СН3СН2СН2-СООТч[Н4+ —- сн3сн2сн2-ссжн2+н2о
85 %
Реакція відбувається з тієї причини, що сіль утворена слабкою кислотою й
слабкою основою:
а-с-о"кн4+5=і яс-он + ш3
О О
Рівноваги реакції достатньо для того, щоб відбулося нуклеофільне
приєднання молекули амоніаку з наступною дегідратацією:
(Я ?• с?- + о
Я-С-ОН +:>Ш3=г=Е Я-С-ОН =г= К-<р-^ОН2 « К-С-Ш2+Н20
^ у +>ш3 ш2
Розділ 18. Монокарбонові кислоти
519
Це важлива реакція, яку використовують для промислового одержання
амідів. На жаль, вона не є загальною й не відбувається для просторово
утруднених аліфатичних кислот.
❖ Гідроліз нітрилів. Нітрили є доступним синтоном, з якого легко
одержують аміди. Пряма гідратація нітрилів приводить до первинних амідів:
Я-С^И +Н20 ЯС(ЖН2
Процес треба проводити обереясно, оскільки аміди часто зазнають подаль-
шого гідролізу до відповідних карбонових кислот та амоніаку. Найліпших
результатів досягають у разі використання водного розчину натрій гідроксиду й
гідроген пероксиду.
Гідратація з алкілюванням нітрилів веде до вторинних амідів:
я-с=м+яон -Н1- я-д-ічия'
або 0
алкен
Як бачимо, у кислому середовищі зі спирту або алкену утворюється
карбокатіон, що реагує з нітрилом, утворюючи нітрилієву сіль, яка зазнає
гідролізу до вторинного аміду:
. + 1ГС=Н + н2о
Я'ОН + Н+ — Я* - Я'-С^Я' Я'-С-ШЯ"
- н2и 11
О
❖ Реакція переамідування. Первинні аміди молена одержувати за методом
Шербульє, нагріваючи карбонові кислоти з сечовиною (діамідом карбонатної
кислоти):
ЯСООН + Н2КС0І\ІН2 ЯСОИН2 + С02 + Ш3
18.10.2. Хімічні властивості й застосування
Щоб чітко зрозуміти хімічні властивості амідів, спочатку розглянемо
електронну будову амідної групи. її описують такими резонансними структу-
рами:
*° '°~
Ш2 Ш2 КН2
Через зсув Ятелектронної густини до атома Оксигену основність амідів
значно нижча, ніж амінів, і протонування амідів відбувається за карбонілом, а не
за атомом Нітрогену.
З цієї ж причини аміди мають підвищену ИН-кислотність.
520
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
❖ Розщеплення за Гофманом. Аміди легко зазнають розщеплення за
Гофманом до амінів, втрачаючи один атом Карбону:
СпН2п+1^ч
гО° ИаОН гн •
мн2 Вг> п 2п+,С
ЫНВг
#-Бромоамід
СпН2п+іС\
Нітрен
.0
N
СпН2п+1ЖЮ
н,о
Ізоціанат
—[с„н2п+І ш-соон]
Карбамінова кислота
с„н2п+1нн2+со2
Амін
Як зазначено вище, спочатку відбувається бромування за атомом Нітрогену,
каталізоване основами. Під впливом гідроксид-аніона ІУ-бромоамід далі утворює
нітрен. Нітрен зазнає стабілізування завдяки перегрупуванню в ізоціанат.
Ізоціанат, відповідно, під час взаємодії з водою утворює нестійку карбамінову
кислоту, яка перетворюється в амін.
❖ Відновлення. У разі прямого відновлення амідів кислот за допомогою
літій алюмогідриду в тетрагідрофурані можна одержати аміни з тою самою
кількістю карбонових атомів з високим виходом:
ед^сомк.
ІлА1Н4
С„Н2ігИСН2М12
■ Інший напрям відновлення - розщеплення зв'язку С-^ і утворення аміну
й альдегіду. У цьому випадку як відновник рекомендують застосовувати літій
триетоксиалюмінійгідрид:
(СН3)3С-С-К(СН3)2 + ЬІА1(ОС2Н5)3Н -(СН3)3С-СХ + Ш(СН3)2
О Н
❖ Гідроліз. Під час нагрівання з водними розчинами кислот або лугів аміди
гідролізують до карбонових кислот з високим виходом. Обидва процеси
незворотні й потребують жорсткіших умов, ніж у випадку естерів.
❖ Реакція з нітритною кислотою. Первинні аміди під час обробки
нітритною кислотою або нітрозилхлоридом зазнають дезамінування і,
відщеплюючи азот, перетворюються у карбонові кислоти:
о 2 2 о
❖ Формамід. Найпростіший амід - формамід - рідина, що має високу
діелектричну проникність. Його одержують, діючи на амоніак карбон(ІІ)
оксидом за наявності каталітичних кількостей натрій алкоголяту:
С0 + Ш,-^ НСОШз
Розділ 18. Монокарбонові кислоти
521
К(СН3)2
Дгшетилформамід (ДМФА) одержують взаємодією мурашиної кислоти з
диметиламіном або карбонілюванням диметиламіну:
,0
НСООН + Н1\Г(СН,)2 —н-с'
і 1 -Н20
/ 'Р
СО + Ш(СН3)2 —н-с;
Н(СН3)2
■ Формамід і його діалкільна похідна - М,И-диметилформамід НСОЫ(СНз)г
- є важливими промисловими розчинниками. їх застосовують у виробництві
синтетичних волокон. Незважаючи на те, що ці сполуки мають високу
полярність, вони належать до апротонних розчинників.
18.1.1. Нітрили
❖ Номенклатура
■ За замісною номенклатурою нітрили загальної формули Я-С=Ы нази-
вають за допомогою суфіксів -нітрил і -карбонітрил або префікса ціано-.
■ За радикально-функціональною номенклатурою назва нітрилів складається
з назви вуглеводневого замісника й назви функції ціанід. Приклади таких назв
виділені курсивом:
СН3(СН2)4С^ СНзОИ ^^-С^ ІЧ=С(СН2)зСООСН3
Гексаннітрил Ацетонітрил Циклогексанкарбонітрил Метил-4-ціанобутират
пентилціанід метилціанід циклогексилціанід
18.11.1. Способи одержання
❖ Нуклеофільне заміщення на ціаногрупу. Нітрили карбонових кислот
одержують алкілюванням первинними й вторинними галогеноалкілами солей
ціанідної кислоти за механізмом 8к2:
ЯСІ + КаСИ -> ЯСИ + ^СІ
Оскільки ціанід-аніон є амбідентним іоном, то як побічні продукти
утворюються ізонітрили:
Я-Х + С=№ 1
Нітрил
Я-№С"
Ізонітрил
522
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
Оптимальні умови для високих виходів алкілціанідів виникають під час про-
ведення реакції в біполярних розчинниках типу диметилсульфоксиду (ДМСО):
СНзССН^СНзСІ +КаСИ СН3(СН2)8СН2СК
Ундеканнітрил, 94 %
Ще ефективніше застосування краун-етерів, таких, наприклад, як [18]-
краун-6. У цьому випадку вихід алкілнітрилів досягає кількісного навіть для
вторинних алкілгалогенідів.
Третинні галогеніди не вдається перевести в нітрили, тому що ціанід-іон,
відіграючи роль основи, зумовлює реакцію елімінування алкілгалогеніду,
унаслідок чого утворюється алкен.
Замість алкілгалогенідів можна застосувати алкілсульфати або алкілові
естери и-толуенсульфокислоти (алкілтозилати).
❖ Дегідратація амідів. Інший шлях одержання нітрилів - збезводнювання
амідів над фосфор (V) оксидом:
2 КСОКН2-^* 2ЯС=Н +2НР03+Н20
Цей метод вдало доповнює попередній, оскільки він не обмежений будовою
вуглеводневого замісника.
❖ Дегідратація оксимів альдегідів за допомогою тіонілхлориду, оцтового
й, особливо, трифлуорооцтового ангідридів, також дає нітрили:
БОСІ,
я-сн=н-он *А» к-с=к
ДМФА
70-90 %
Можна замість оксимів використати альдегіди, якщо їх нагрівати з
гідрохлоридом гідроксиламіну й натрій ацетатом у мурашиній кислоті.
❖ Приєднання ціанідної кислоти. Промислово важливі нітрили одержують
через приєднання ціанідної кислоти до сполук із кратними зв'язками, такими як
етилен та ацетилен:
Я-СН(ОН)-СМ
а-Пдроксинітрил
Я-СН=Ш
ЯСНО н2с=сн2>
Пропанонітрил
СН3СН2СК
НС-СН>СТ н„с=сн-см
КСН(Ш2)СК - -
а-Амінонітрил Акрилонітрил
18.11.2. Хімічні властивості
Реакційна здатність нітрилів зумовлена наявністю ціаногрупи -С=И. Атоми
Нітрогену й Карбону перебувають у $р-гібридному стані й утворюють між собою
потрійний зв'язок, електронна густина якого зміщена до атома Нітрогену:
ІІ-С=К-—-Я-С=>1:
Розділ 18. Монокарбонові кислоти
523
Відповідно до такого розподілу електронної густини, нітрили вступають у
реакції з електрофілами за атомом Нітрогену й нуклеофілами за атомом Карбону.
Крім того, ціанідна група зумовлює рухливість атомів Гідрогену біля а-карбонового
атома, що дає змогу проводити з їхньою участю реакції конденсації.
❖ Гідроліз. Найважливішою реакцією нітрилів є їхній гідроліз, що відбу-
вається у дві стадії. Спочатку до зв'язку приєднується одна молекула води,
й утворюється амід, який можна виділити в індивідуальному стані. Подальший
гідроліз веде до карбонової кислоти:
Н+або ОН- V Н+або 0№
кс=и + н2о—- ясмн2—- КСО(ЖН4
Розглянемо механізми гідролізу нітрилів до амідів.
■ Гідроліз нітрилів за наявності лугів або амрніаку має такі стадії. Спочатку
відбувається приєднання гідроксид-іона до нітрильної групи:
?н
Потім - протонування продукту приєднання:
ОН ОН
І _ Н20 І
Я-С=Ы я-с=м-н + ОН
і, нарешті, - таутомерне перетворення в амід:
2
■ У кислому середовищі спочатку зазнає протонування атом Нітрогену
молекули нітрилу:
К-С=№ + Н+=^=: я-с^й-н-—- я-с^и-н,
а потім відбувається нуклеофільне приєднання молекули води:
Я-С=М-Н ^Я-С=ЙНї=К-С=к
О ^
н+чн;
он
і, на завершення, - депротонування:
•^•т -г —- Т^» У-Ч \ТТТ І ТТ^
я-с=ш2 я-с-кн2+Нл
ОН О
524
Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
❖ Синтез іміноестерів та їхній гідроліз. У безводному середовищі нітрили
за наявності гідроген галогеніду реагують зі спиртами, утворюючи іміноестери:
с2н5он.
яс^и + неї —- яс;=ш ^^і^=ш
сі ос2н5
Імінохлориди Іміноестери,
або імідати
Іміноестери й тіоіміноестери в кислому середовищі зазнають гідролізу до
естерів і їхніх сульфурних аналогів:
н2о/н+ ,р ^NH н2о/н+ ,р .
\ о°с 3 R~< + NH4 R-C^ 0оС а R-C4 + NH4
XOR' 0 С NOR' SR' 0 C SR'
❖ Синтез тіоамідів. Приєднання гідроген сульфіду до нітрилів дає змогу
синтезувати тіоаміди:
❖ Синтез імідів. Під час нагрівання нітрилів з карбоновими кислотами
утворюються іміди:
R-C=N + R'-Cv
ОН
9н 9
А А
R N R'
9 9
А А
R N R'
н
❖ Синтез кетонів. Нітрили приєднують реактиви Гриньяра, утворюючи
солі імінів. Після кислотного гідролізу вони дають кетони:
н о/н"*"
R-C=N + R'MgX R-C=N-MgX——- R~ft~R'
R' О
Це один із найстаріших і надійних методів синтезу несиметричних кетонів.
❖ Відновлення. Нітрили в разі відновлення перетворюються в первинні
аміни. Процес гідрування можна вести за наявності платини або паладію за
невисокого тиску й температури 70-80 °С. Як відновники можна використо-
вувати комплексні гідриди металів ЬіАІКЦ TaNaBH4:
RCNIÄ rch2NH2
Реакція відбувається через альдиміни RCH=NH. Гідролізом цих проміжних
сполук можна одержати альдегіди.
Розділ 18. Монокарбонові кислоти 525
18.12. Гідразиди й азиди кислот
18.12.1. Гідразиди
❖ Номенклатура, Назви ацилгідразидів будують на підставі систематичної
або тривіальної назви карбонової кислоти, використовуючи суфікс -огідразид,
наприклад:
НзС-сГ НзС-(СН2)^С, 1/"^^
Ацетогідразид Бутирогідразид Циклопентанкарбогідразид
❖ Способи одержання. Оскільки гідразини більше нуклеофільні, ніж
аміни, то їх можна одержувати ацилюванням гідразинів навіть такими слабкими
агентами, як естери:
/.О
к-с^ + н,к-ш, — к-с-кнш,+к'он
ОК' 2 2 о
Ацилювання гідразинів кетеном відбувається за м'яких умов з кількісним
виходом.
❖ Хімічні властивості. За хімічними властивостями гідразиди карбонових
кислот нагадують аміди, проте в гідразидах є Р-атом Нітрогену, неподілена
електронна пара якого, на відміну від а-атома Нітрогену, не бере участі в
спряженні з карбонільною групою:
З огляду на це гідразиди виявляють як основні, так і кислотні властивості,
тобто є амфотерними сполуками.
• Гідроліз. Під час обробки кислотами гідразиди розщеплюються за меха-
нізмом, подібним до механізму розщеплення амідів:
К-С-МШН, К-СООН ч-МИгШ,
О
• Алкілювання гідразидів за нейтральних умов відбувається по більш основному
Р-атому Нітрогену:
НзС-С-ННМНг + КСІ НзС-С-МНМНК
О 'о
526
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
• Конденсація з оксосполуками, З цієї ж причини утворення ацилгідразонів,
які використовують для ідентифікації оксосполук, відбувається легко:
R-C-NH-NH2 + КіКзСО R-C-NH-N=CR^R2
О О
• Нітрозування простих гідразидів є однією з найважливіших реакцій замі-
щення для цього класу сполук і веде, залежно від умов реакції, до утворення
ацилазидів або амідів:
R-C-NHNH2
6
R-C-NHNH-NO=f=^ R-C-NH-N=NOH
,0
к-с.
N3
Ацилазвд
Амід
18.12.2. Ацилазиди
Найімовірніша електронна структура азидів має такий вигляд:
к-с-м=к=м
II
О
❖ Номенклатура. Назви азидів карбонових кислот будують із назви
ацильних замісників з додаванням суфікса -азид, наприклад:
Н,С-С. СН3СН2С.
N3
Ацетилазид
Пропіонілазид
❖ Способи одержання. Крім методу, згаданого щодо гідразидів, ацилазиди
синтезують ацилюванням галогеноангідридами кислот солей азидної кислоти:
R-C-C1 + КаКз Я-С-Кз + КаСІ
6 О
❖ Хімічні властивості
• Гідроліз азидів дає карбонову й азидну кислоти:
яр,
о
ясоон + нк.
Уперше азидну кислоту отримано саме таким способом.
• Перегрупування Курціуса є однією з найважливіших реакцій азидів карбо-
нових кислот і синтетично подібна до перегрупування Гофмана:
Розділ 18. Монокарбонові кислоти
527
.О
R-C>
"N—N=N "N2
RtC4..
Нітрен
R-N=C=0
Ізоціанат
ЩО
R-NH-C4
OHJ
Карбамінова кислота
-RNHyfCCX
Перегрупування Курціуса каталізоване сильними кислотами, а перетворення
оптично активних азидів відбувається зі збереженням конфігурації.
18.13. Галогенозаміїцені кислоти
18.13.1. Способи одержання а-галогенозаміщених
карбонових кислот
❖ Пряме хлорування карбонових кислот під час нагрівання й опромінення
не відрізняється вибірковістю й приводить до суміші • а,р,у-ізомерів. Воно,
мабуть, ефективне тільки у випадку оцтової кислоти. Наприклад, хлорування
оцтової кислоти за наявності слідів іоду, червоного фосфору або в разі
опромінення сонячним світлом приводить послідовно до одержання moho-, ди- і
трихлороцтових кислот:
СН3СООН С1СН2СООН^^ CHCl2COOH^^CCl3COOH
Шляхом дозування хлору всі три кислоти вдається одержати з високим
виходом і в досить чистому вигляді.
Уважають, що іод слугує переносником хлору. У цьому разі утворюється
трихлорид Іоду, який є ефективним хлорувальним агентом:
І2 + ЗС12 -> 2ісіз
ІСЬ + 2СН3СООН -> ICI + 2СН2С1СООН
ICI + Ch -> ІСЬ
❖ Бромування карбонових кислот, на відміну від хлорування, дає тільки а-
ізомери кислот:
СН3СН2СН2СН2СН2СООН BhJ0ç3* СНзСП^СНзСІ^СН-СООН
Вг
Гексанова кислота 2-Бромогексанова кислота
У цьому випадку виявляється рухливість атомів Гідрогену в а-положенні,
зумовлена карбоксильною групою, хоча вона не така сильна, як в альдегідах і
кетонах.
■ Під час бромування набагато зручніше застосовувати реакцію Гелля-
Фольгарда—Зелінського. Для цього на кислоту діють бромом за наявності малої
528
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
кількості фосфору. Фосфор (III) бромід, що утворюється, переводить деяку
кількість кислоти в бромоангідрид:
2Р + ЗВг2 2РВг3 О
ЗК-СН2СООН + РВг3 -ЗЯСНг-С^
Вг
На відміну від самої карбонової кислоти, бромоангідрид здатний у кислому
середовищі перетворюватися в енол, який реагує з бромом:
, ^ОН ПН
я-снгсС + н+— я-сн2-сС я-сн=сґ
Вг Вг Вг
/ОН
Вг~Вг + Я-СН=С. — Я-СН-С-Вг
Яг -НВг Т II
Ьг Вг О
Бромований бромоангідрид ІІ-СНВг-СОВг реагує з надлишком кислоти:
ІІСНВгСОВг + ЯСН2СООН ЯСНВгСООН + ЯСН2СОВг
❖ Йодування. Альфа-іодозаміщені кислоти не можна одержувати прямим
йодуванням. їх синтезують із бромозаміщених кислот шляхом реакції обміну:
Я-СНВг-СООН + Ю -> Я-СНІСООН
❖ Під час флуорування оцтової кислоти за умов електролізу утворюється
флуороангідрид трифлуорооцтової кислоти, який легко зазнає гідролізу до
відповідної кислоти:
СН3СООН р^^Г,СР,-С-Р СР3СООН
і ЬЛЄКТрОЛ13 3 || _|^р •>
о
❖ Окиснення хлоралю ССІ3-СНО концентрованою нітратною кислотою є
зручним способом одержання трихлороцтової кислоти.
18.13.2. Способи одержання інших
галогенозаміщених карбонових кислот
❖ Бета-галогенозаміщені карбонові кислоти, як звичайно, одержують
шляхом приєднання гідроген галогенідів до ненасичених кислот:
СН2=СН-СООН + НСІ -> С1СН2-СН2-СООН
Акрилова кислота 3-Хлоропропанова кислота
■ Галогенозаміщені кислоти іноді синтезують з гідроксикислот:
Розділ 18. Монокарбонові кислоти
529
носн2—сн2соон -£^і- сісн2—сн2соон
❖ Омега-галогенозаміщені карбонові кислоти. Кислоти, що несуть атом
галогену на найбільшій відстані щодо функціональної групи (со-ізомери),
одержують за допомогою реакції теломеризації, що відбувається за наявності
передавача ланцюга (телогену), наприклад, ССЦ або СВг4, з додаванням
пероксидних ініціаторів радикальних реакцій:
иН^О^ + СС14 СІССНгСН^ССІз-^- сіссь^сн^-соон
со-Хлоралканова кислота
18.13.3. Хімічні властивості галогенозаміщених
карбонових кислот
❖ Кислотність. За ступенем заміщення в ланцюзі карбонової кислоти
гідрогенних атомів на атоми галогенів константа дисоціації кислоти збільшується.
Особливо різко вона збільшується в разі заміщення атомів Гідрогену в а-положенні.
Вплив різних атомів галогенів на їхню силу зменшується в ряді: її > СІ > Вг > І. Він
також залежить від кількості й положення атомів галогенів. Це чітко видно з
наведених нижче даних:
Кислота
Ка
Кислота
Ка
РСН2СООН
2,1-10*
С1СН2СООН
1,4-Ю"3
Р2СНСООН
5,7-10"2
С12СНСООН
3,32-10"2
РзССООН
5,9-10-1
сьссоон
2,0-10"1
❖ Реакції нуклеофільного заміщення. Галогенозаміщені кислоти, особ-
ливо а-заміщені, легко вступають у різні нуклеофільні реакції. Як нуклеофіли
можна використати основи, спирти, аміни, тіоли тощо. Часто такі реакції
відбуваються за механізмом 8н2, проте зі збереженням конфігурації. Справді, у
разі гідролізу (5)-2-бромопропанової кислоти в слабколужному середовищі
утворюється (£)-2-гідроксипропанова кислота.
Уважають, що карбоксилат-іон, який утворився, атакує хіральний центр з
обертанням конфігурації й синхронним відходом бромід-аніона. Далі а-лактон,
що утворився, під впливом ОНГ-аніона легко й швидко розщеплюється з новим
обертанням конфігурації. Отже, незважаючи на те, що реакція відбувається за
механізмом 8н2, подвійне обертання при хіральному центрі дає змогу зберігати
конфігурацію:
530
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Н0\ //° /О я . . (Р^-сн - <° СООН
с _о-с: повене, ^y^gu о-с; -HU «
А -№ ^С-^Бг -Br Cfc Швидко ..С-ОН „Л
HdH> н «і н^ ни
кислота
Загалом наявність у р-положенні функцій з неподіленими електронними
парами, таких як -ОН, -OR, -СООН, -SR, -Hal, -COOR, -NH2, впливає на
просторовий напрям реакції нуклеофільного заміщення. Вони унеможливлюють
атаку нуклеофіла з тилу, залишаючи тільки що наведену реалізацію процесу.
• Заміна атома галогену на нітрогрупу. Взаємодію естерів а-галогено-'
заміщених кислот з калій нітритом використовують для синтезу естерів а-нітро-
заміщених карбонових кислот:
NaNO,
R-CH-COOC2H5 -j^p R"9H-COOC2H5
CI " 3 N02
■ Аналогічно з калієвих солей а-галогенозаміщених карбонових кислот
одержують солі ос-нітрозаміщених карбонових кислот, які внаслідок термічного
розпаду дають нітроалкани:
R-(pH-COOK ^-R-CH-COO"K+ t§^R-CH2N02
CI N02
❖ Де карб оке плювання. Наявність в а-положенні карбонової кислоти атома
галогену з його -І-ефектом різко послаблює міцність С-С-зв'язку, що підвищує
лабільність цих сполук і виражене в їхній здатності зазнавати декарбоксилю-
вання під час нагрівання.
❖ Дегідрогалогенування.
■ Під час кип'ятіння водні розчини Р-галогенозаміщених кислот
перетворюються в ненасичені кислоти. Реакція відбувається через проміжні Р-
гідроксисполуки:
R-^H-CH.-COOH-112^ R-CH-CH2-COOH R-CH=CH"COOH
СІ ОН
■ Гамма-галогенозаміщені кислоти під час нагрівання спочатку утворюють
відповідні гідроксикислоти, які відразу переходять у лактони (циклічні
внутрішні естери):
НХ-СН-СНХНХООН —jj пЛ^Мгл
ОН Н3С о О
Розділ 18. Монокарбонові кислоти 531
18.13.4. Окремі представники галогенозаміщених
карбонових кислот
■ Хлоромурашина кислота СІСООН сама по собі не існує, проте відомі її
похідні, наприклад, естери. їх одержують дією фосгену на спирти:
СН3ОН + С0СІ2 СІСООСН3
Оскільки фосген є дихлороангідридом карбонатної кислоти, то отриманий естер
також можна розглядати як похідну цієї кислоти.
■ Хлороцтові кислоти
У промисловості монохлороцтову кислоту одержують двома методами:
а) нагріванням трихлоретилену з 70 % сульфатною кислотою:
С1НС=СС1, > ССЬІ,—ССШ50зН—^С1СН-С^
б) окисненням 2-хлороетанолу нітратною кислотою:
СН^СІ-СНзОН > СН3СІ-СООН
Дихлороцтову кислоту синтезують із хлоральгідрату дією кальцій карбонату
за наявності натрій ціаніду:
2С1зС~СН(0Н)2+ 2СаС0з ^^^(СІ2СН-С00)2Са + СаСІ^ч- 200^+ Н^О
Трихлороцтову кислоту в промисловості одержують окисленням хлор-
альгідрату концентрованою нітратною кислотою:
[01
С1зС~СН(ОН)з сцс-соон
18.14. Монокарбонові ненасичені кислоти
❖ Номенклатура. Для назви монокарбонових ненасичених кислот
користуються загальними правилами номенклатури IUP АС. Як і у випадку
насичених монокарбонових кислот, рекомендовано використання тривіальних
назв для великої групи ненасичених карбонових кислот (табл. 18.2), проте не їхніх
похідних.
Виняток - тривіальна назва акрилова кислота, яку можна використати для
побудови назв відповідних похідних, наприклад, СІ2С=СНС00Н 3,3-
дихлороакрилова кислота.
532
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Таблиця 18.2
Тривіальні назви ненасичених монокарбонових кислот,
їхніх кислотних і ацильних залишків, рекомендовані правилами номенклатури ШРАС
Структура
Назви кислот
Тривіальні назви
систематичні
тривіальні
кислотних
залишків
ацильних
залишків
СН2=СНСООН
НС=ССООН
Пропенова
Пропінова
Акрилова
Пропіолова
Акрилат
Пропіолат
Акрилоїл
Пропіолоїл
соон
н2с=с'
СН3
2-Метилпропенова
Метакрилова
Метакрилат
Метакрилоїл
Н17С8ч ,(СН2)7СООН
с=с
н' н
і/мс-Октадец-9-енова
Олеїнова
Олеат
Олеоїл
18.14.1. Методи синтезу
• Окиснення а&ненасичених альдегідів. Вихідні альдегіди досить доступні
завдяки кротоновій конденсації. їх перетворюють у кислоти шляхом обробки
м'якими реагентами, які не зачіпають подвійного зв'язку:
*°
Я-СН=СН-СЧ + А§(Ш3)20Н -А§ + КСН=СН-СОО'Ш+
н
2 СН=СН-С^° + 02 Р^І. 2 СН^СН-С^0
Н ОН
Акролеїн Акрилова кислота
В останньому випадку вихідний акролеїн синтезують з пропілену:
/О
нх=сн-сн,+ о2 » н9с=сн-сС
2 3 1 270 °С 2 хц
• Синтез із а- і $-галогенозаміщених карбонових кислот. Особливо легко
зазнають дегідрогалогенування Р-галогенозаміщені карбонові кислоти:
СН2Вг~СНГСООН кон> н2>°н С=СН-СООК -^тг- нх=сн-соон
^ ^ / * н2о ^
• Дегідратація $-гідроксикарбонових кислот. Бета-гідроксикислоти легко
зазнають дегідратування навіть у разі обережного нагрівання:
Розділ 18. Монокарбонові кислоти
533
сн2он-сн2соон н2с=сн-соон
• ГідролЬ нітрилів. Ненасичені нітрили, синтезовані на основі ацетилену,
можуть перетворюватися шляхом гідролізу в кислому або лужному середовищі
у відповідні карбонові кислоти:
НС=СН +НСК нх=сн-ск
ш4сі ^
H2C=CH-CN -^-Н2С=СН~СООН + Ш4+
• Синтез Кневенагеля1 (1896). Конденсація карбонільних сполук з малоновою
кислотою або її естерами за наявності основ дає а,|5-ненасичені кислоти або їхні
естери:
Я-СНО + СН2(СООС2Н5^ "^о^"' КСН=С(СООС2Н5)2
я-сно + сн2(соон)2 т"оГос" ^"СН=СН-СООН
18.14.2. Хімічні властивості
❖ Кислотність. Ненасичені кислоти за аналогією з насиченими утво-
рюють усі звичайні похідні кислот - ангідриди, аміди, нітрили, естери,
галогеноангідриди тощо. З іншого боку, завдяки наявності кратного зв'язку вони
вступають у реакції приєднання, окиснення й полімеризації.
Зазначимо, що а,Р-ненасичені кислоти сильніші, ніж насичені:
СН3СН2СООН Н2С=СН-СООН НОС-СООН
Пропіонова кислота, рКа 4,87 Акрилова кислота, рКа 4,26 Пропіолова кислота, рКа 1,84
Причиною підвищення кислотності є дія -І-ефекту подвійного чи потрій-
ного зв'язку. Аніон, що утворюється під час дисоціації, додатково стабілізується
завдяки негативному індукційному ефекту:
н2с=сн-с; ^н,с=сн-с<^+ н+
2 он 2 чо-
У випадку віддалення кратного зв'язку від карбоксильної групи сила
кислоти приблизно однакова порівняно з її насиченим аналогом.
1 Кневенагель Е. (Кпоеуепа§е1 Е.) (1865-1921) - німецький хімік. Головні дослідження в галузі
органічного синтезу.
534
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
❖ Окремі хімічні реакції. У низці ненасичених кислот вирізняються а, Р-
ненасичені карбонові кислоти ЯСН=СН-СООН та їхні похідні. Вони мають
специфічні властивості, пов'язані з наявністю в них спряженого з карбонілом
подвійного зв'язку, і саме на них буде зосереджено основну увагу.
■ Унаслідок взаємного впливу карбоксилу й подвійного зв'язку приєднання
за подвійним зв'язком відбувається проти класичного правила Марковникова:
Через спряження а,р-подвійного зв'язку й карбонільної групи виникає
значна поляризація молекули кислоти, що призводить до початкового 1,4-
приєднання й наступної ізомеризації або кетоенольної таутомерії ненасиченого
діолу.
■ Також а,р-ненасичені кислоти та їхні похідні вступають у реакції
дієнового синтезу:
■ Ненасичені кислоти та їхні похідні (естери, аміди, нітрили) порівняно з
алкенами легше вступають у реакції полімеризації за радикальним або йонним
механізмом. Ненасичені кислоти з віддаленим від карбоксильної групи
подвійним зв'язком у реакції полімеризації не вступають.
18.14.3. Окремі представники ненасичених
карбонових кислот
Найпростішими й найважливішими кислотами зі спряженими подвійними
зв'язками є акрилова, а-метилакрилова, або метакрилова, й Р-метилакрилова,
або кротонова, кислоти. У промисловості широко використовують їхні похідні.
❖ Акрилову кислоту синтезують парофазовим окисненням пропену киснем
повітря на бісмутових, кобальтових і молібденових каталізаторах:
У деяких країнах (Німеччина) акрилову кислоту одержують гідро-
карбоксилюванням ацетилену:
ЩС^^-С=СҐ+ неї —- сн2сі-сн^-соон
ОН
н2с=сн-соон + н2о — сн2он-сн2соон
н3с-сн=сн2
н2с=сн-сно
н2с=сн-соон
НС=СН+СО+Н20
Ы1(СО)4
н2с=сн-соон
Розділ 18. Монокарбонові кислоти
535
або взаємодією етиленхлорогідрину з калій ціанідом із наступним гідролізом
етиленціангідрину:
носн2-сн2сі +неи-— носн2-сн2си ^ос0, н2с=сн-соон + Ш4+
❖ Акрилонітрил у промисловості одержують за такими реакціями:
НС^СН + НСМ ^нс^^сн^снск
СН2=СНСН3 + 3/202 + ИН3 СН2=СНСК + ЗН20
Остання реакція - окиснення суміші пропілену й амоніаку недостатньою
кількістю повітря над каталізатором - найекономічніша й найперспективніша.
Процес проводять за 400-485 °С над бісмут молібдатом протягом 2-4 с. З
реакційної суміші, крім акрилонітрилу (80 %) одержують товарні ацетонітрил і
ціанідну кислоту.
■ Акрилонітрил легко приєднує аміни. Такий процес називають ціанети-
люванням:
я-мн2+н2с=сн-с=н —^ якн-сн^ь^с^
■ Важливу роль відіграють полімери акрилонітрилу, які застосовують для
одержання хімічного волокна (орлон, нітрон) - найліпшого замінника вовни, а
також кополімери акрилонітрилу з бутадієном, які дають бензостійкі синтетичні
каучуки ("буна №\ або пербунан).
Полімери акрилонітрилу, як і всі "вінілові естери", мають структуру типу
^Н-СН^-^Н-СН^Н-
CN CN CN
-сн2-
CN
❖ Метилакрилат. В техніці широко використовують метиловий естер
акрилової кислоти - метилакрилат, який одержують за методом Penne:
4HC=CH+4CH3OH+Ni(CO)4+2HCl « 4^0=0^0000^+NiCl2+H20
або алкоголізом акрилонітрилу:
H,SOd
2H2C=CH-CN+2H20+2CH3OH—2H2C=CHCOOCH3+(NH4)2S04
❖ Метакрилову кислоту звичайно синтезують з ацетону й ціанової
кислоти:
?Н HSO
CH3ÇCH3 + HCN — СН3ССН3-7^СН2=ССООН + Н20 + NH4HS04
О CN 2 СН,
536
Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Практично в суміш ацетону з сульфатною кислотою додають метанол і
одержують відразу метилметакрилат.
Метакрилову кислоту та її естери в промисловості одержують також
окисненням ізобутилену:
Н2С=С(СН3)2-^ н2с=с-сно -^н2с=с-соон
СН3 СН3
■ Широке практичне застосування мають полімери метилових естерів
акрилової й метакрилової кислот, що утворюють прозорі безбарвні пластичні
маси (поліметилакрилат і поліметилметакрилат) - "органічне склії\ що
розм'якшується приблизно за 100 °С і здатне формуватись:
9н3 9н3 9н3
- сн-сн2-сн-снгсн-сн2- -сн2- с-сн2- с- сн2-с-
соосн3 соосн3 соосн3 соосн3 соосн3 соосн3
Поліметилакрилат Поліметилметакрилат
❖ Кротонову кислоту можна одержати конденсацією ацетальдегіду з
малоновою кислотою в піридині (Г. Кневенагель):
£оон (роон
сн3сно + <^н2 —- сн3сн=с;; сн3сн=снсоон
соон соон
Кротонова кислота відома у двох формах. Одна з них - стійка (тверда) - є
транс-ізомером, а друга - нестійка (рідина) - ізокротонова кислота - г/ис-ізомер.
За наявності слідів брому, іоду або під впливом сонячного світла лабільна цис-
форма швидко й кількісно переходить у стабільну транс-форму.
Розділ 19
АИКАРБОНОВІ
КИСЛОТИ
19.1. Класифікація і номенклатура
19.1.1. Класифікація
Насичені
НООССН2СН2СООН
Бутандіова кислота,
бурштинова кислота
Ненасичені
НООС Н
чс=с
Н СООН
/иракс-Бутендіова кислота,
фумарова кислота
Ароматичні
СООН
СООН
1,2-Бензендикарбонова кислота,
фталева кислота
19.1.2. Номенклатура ШРАС
Номенклатура двоосновних карбонових (дикарбонових) кислот і їхніх
похідних аналогічна до номенклатури одноосновних карбонових кислот.
Рекомендовано використання тривіальних назв для низки дикарбонових кислот,
але не їхніх похідних, і відповідних кислотних та ацильних залишків (табл. 19.1).
Допускають побудову назв похідних дикарбонових кислот шляхом заміни атомів
Гідрогену у вуглеводневому ланцюзі для малонової, бурштинової, малеїнової й
фумарової кислот. Наприклад, хлоробурштинова кислота.
Таблиця 19.1
Тривіальні назви дикарбонових кислот, їхніх кислотних
залишків та ацильних замісників
Структура
Назви кислот
Тривіальні назви
систематичні
тривіальні
кислотних
залишків
ацильних
замісників
1
2
3
4
5
Насичені аліфатичні дикарбонові кислоти
НООС-СООН
НООС-СН2-СООН
НООС(СН2)2СООН
НООС(СН2)3СООН
Етандіова
Пропандіова
Бутандіова
Пентандіова
Щавлева
Малонова
Бурштинова
Глутарова
Оксалат
Мал он а т
Сукцинат
Глутарат
Оксаліл
Мало ніл
Сукциніл
Глутарил
Дикарбонові кислоти - тверді речовини. Нижчі члени ряду помітно розчинні у
воді й лише незначно розчиняються в органічних розчинниках. Межа розчинності
є при С7-8. Ці властивості здаються цілком природними, оскільки полярні
карбоксильні групи становлять значну частину молекули кислоти.
Цікаво, що кислоти з непарною кількістю атомів Карбону плавляться за
нижчої температури, ніж із парною кількістю. На противагу одноосновним
кислотам температура плавлення двоосновних кислот знижується зі збільшенням
молекулярної маси, принаймні, у низці кислот із парною кількістю карбонових
атомів. Дикарбонові кислоти дисоціюють поетапно, утворюючи моно- і діаніони:
Перша константа дисоціації для двоосновних кислот, особливо щавлевої й
малонової, більша, ніж для відповідних одноосновних кислот. Відмінність у
значеннях константи за першим і другим ступенями дисоціації пояснюють тим,
що неіонізована карбоксильна група виявляє електроноакцепторну дію щодо
йонізованої карбоксильної групи й тим стабілізує карбоксилат-іон, що
утворюється:
(
Стабілізація
19.2. Фізичні властивості
Назви низки ацильних замісників і кислотних залишків утворені від
англійських назв відповідних кислот, наприклад, щавлева - oxalic, бурштинова -
succinic
í/Mcr-Бутендіова Малеїнова Mалеam Малеоїл
/и/7ог//сг-Бутендіова Фумарова Фумарат Фумароїл
Гександіова Адипінова Адгтінат Адипініл
Продовження табл. 19.1
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
538
Розділ 19. Дикарбонові кислоти
539
Крім цього ефекту, в моноаніонах важливу роль відіграє внутрішньо-
молекулярний водневий зв'язок, наприклад:
19.3. Способи одержання
Методи синтезу двоосновних кислот аналогічні до методів синтезу моно-
карбонових кислот. Однак зазначимо, що для окремих представників, особливо
для перших членів гомологічного ряду, є свої специфічні методи, які ми
розглянемо детальніше.
19.3.1. Загальні методи
❖ Окиснення циклічних кетонів є звичайним способом синтезу вищих
двоосновних кислот, наприклад:
HNO,
Г >0 ї НООС(СН2)3СООН
Циклогіентанон Глутарова кислота
QO НООС(СН2)4СООН
Адипінова кислота
Циклогексанон
HNO,
О НООС(СН2)10СООН
Додекандіова кислота
Циклододеканон
❖ Синтез на основі продуктів теломеризації. Вищі дикарбонові кислоти
можна одержувати з тетрахлороалканів С1-(СН2СН2)п-СС1з:
н,о,
С1(СН2СН2)ПСС13 к^С(СН2СН2)пСС13-н^ НООС(СН2СН2)пСООН
С1(СН2СН2)ПСС13-^^ С1(СН2СН2)пСООН HO(CH2CH2)nCOONa
НООССН2(СН2СН2)^СООН
За першою реакцією одержують кислоти з парною кількістю атомів Карбону, а
за другою - з непарною. Необхідні вихідні тетрахлороалкани синтезують з етилену й
карбон тетрахлориду за реакцією теломеризації за О. Несмеяновим1.
1 Несмеянов О. (1899-1980) - радянський хімік. Головні інтереси пов'язані з металоорганічними
сполуками.
540
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
❖ Реакція Кольбе. Кислоти з парною кількістю карбонових атомів можна
отримати також електролізом солей моноестерів нижчих двоосновних кислот:
гедоосссн^соо- —- 2 едск^сн^го
19.3.2. Одержання щавлевої кислоти
Щавлеву кислоту - перший член у ряді двоосновних кислот - синтезують
окисненням багатьох органічних речовин, таких як спирти, вуглеводи, гліколі,
сумішшю нітратної й сульфатної кислот за наявності ванадій (V) оксиду, або
окисненням етилену й ацетилену нітратною кислотою над паладій (II) хлоридом,
або окисненням пропілену нітроген (IV) оксидом. Порівняно донедавна її
одержували з деревини сплавленням з лугом або дією суміпіі ШОН і КОН у
струмі кисню повітря.
■ Сьогодні у промисловості її одержують піролізом натрій або калій
форміату:
0=(ГШа О. СК /ОН
Н >збо*с 0=?Ша „ сасі2д "С^ч
н І 2 і / с
V 0=СОЫа п^С^0 /' 4
0=СОЫа ° и О ОН
або з карбон (IV) оксиду і водню:
С02 + Н2 -> НООС-СООН
■ Доречно згадати перший синтетичний шлях одержання щавлевої кислоти
гідролізом її нітрилу - диціану (Ф. Велер, 1824 р.):
№С-С-М + 2Н20 НООС-СООН
■ Далі треба зазначити про синтез щавлевої кислоти із сухого карбон (IV)
оксиду і лужних металів, який проводять, пропускаючи газ за 360 °С над натрієм
або калієм:
2 С02 + 2 № (СОО№)2
19.3.3. Синтез малонової кислоти
■ Мсиїонову кислоту зазвичай одержують через її мононітрил - ціанооцтову
кислоту:
С1СН2СООЫа + ЖСЫ МНХН2СОО№ ^£ НООС-СНгСООН + Ш4+
■ Уперше малонову кислоту отримано окисним розщепленням яблучної
кислоти, з якої як проміжний продукт утворюється оксалілоцтова кислота:
Розділ 19. Дикарбонові кислоти
541
нооссн2снсоон -^ноосснхсоон-^* НООССН2СООН+С02
он о ,
Яблучна кислота Оксалілоцтова
кислота
19.3.4. Синтез янтарної кислоти
■ Янтарну кислоту можна синтезувати з етилену:
СН2=СН2 + Вг2 -> ВгСНг-СН2Вг
ВгСНгСН2Вг ^С-О^-СТ^С^К НООССН2СН2СООН + 2Ш4+
Янтарна кислота
■ Для одержання янтарної кислоти, як звичайно, використовують
бактеріальне бродіння яблучнокислого кальцію, причому утворюється янтарна
кислота високої чистоти.
■ Значна кількість янтарної кислоти синтезується під час спиртового
бродіння, де вона утворюється, ймовірно, із глутамінової кислоти:
НООССН2СН2СНСООН
кн2
■ Янтарну кислоту можна отримати також гідруванням бутендіових кислот
(фумарової або малеїнової):
НООССН=СНСООН —^ НООССН2СН2СООН
■ Інші способи. Відомий синтез янтарної кислоти через натріймалоновий
естер. Вона є побічним продуктом виробництва адипінової кислоти. Янтарна
кислота виділяється із суміші кислот, що утворюються під час окиснення
вуглеводнів С4-С10. її можна отримати окисненням фурфуролу гідроген
пероксидом.
Відоме виробництво янтарної кислоти з відходів янтарю (бурштину).
Остання назва послужила причиною того, що в деяких випадках її називають
бурштиновою. Ми віддаємо перевагу назві "янтарна". Саме в продуктах
перегонки бурштину цю кислоту вперше виявив Г. Агрікола1 1555 р.
19.3.5. Одержання адипінової кислоти
Адипінова кислота є в соці цукрового буряка. її моясна одержати синтезом
на основі малонового естеру, використовуючи 1,2-дибромоетан:
1 Агрікола Г., справжнє прізвище Бауер (Agrícola (Bauer) G.) (1494—1555) — німецький учений-
дослідник, працював у галузі геології, гірської справи та металургії. Розробив основи хімічної
оцінки і переробки свинцевих, мідних та срібних руд.
542
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
вісні-сі^г + гкгісщсос^^
НООС. /СООН
>н-снгсн,-сн —^нгос-ссн^-соон
ноос' соон -2С0
Адипінова кислота
У промисловості адипінову кислоту одержують каталітичним або хімічним
окисненням (розбавлена нітратна кислота) циклогексанону.
19.4. Особливості поведінки двоосновних кислот
Природно, що все, викладене щодо властивостей карбоксильної групи
одноосновних кислот, стосується також двоосновних кислот. У цьому розділі
розглянемо тільки специфічні особливості поводження двоосновних кислот
різного типу.
Вплив взаємного розташування карбоксильних груп виявляється насамперед
у різному реагуванні дикарбонових кислот на нагрівання.
19.4.1. Щавлева кислота
Щавлева кислота не утворює ангідриду. Вона дає два ряди похідних з однієї
або обох карбоксильних груп. їй властива низка специфічних реакцій.
❖ Естерифіксіція. Щавлева кислота - одна з найсильніших карбонових
кислот, що значно перевершує за силою своїх гомологів, тому вона утворює
діетиловий естер шляхом звичайного нагрівання щавлевої кислоти й етанолу з
одночасною відгонкою води:
НООС-СООН + 2С2Н5ОН -> С2Н5ООС-СООС2Н + 2Н20
Кислий каталізатор у цій реакції не потрібний, оскільки сама кислота є досить
сильною.
■ 3 етиленгліколем щавлева кислота, на відмін}' від інших двоосновних
кислот, що дають полімери, утворює етиленоксалат або 1,4-діоксан-2,3-діон:
(^ООН + (рн2он
СООН СН2ОН
1,4-Діоксан-2,3-діон
❖ Щавлева кислота під дією сульфатної кислоти розпадається з
утворенням карбон (IV) оксиду, карбон (II) оксиду і води:
НООС-СООН С02 + СО + Н20
Розділ 19. Дикарбонові кислоти
543
У разі нагрівання до 150 °С вона розпадається в такий спосіб:
НООС-СООН 150 °С> НСООН + С02
❖ Окиснення. Калій перманганат у кислому розчині легко окиснює
щавлеву кислоту до карбон (IV) оксиду і води. Цю реакцію застосовують в
об'ємному аналізі для визначення титру розчинів перманганату:
5(СООН)2 + 2Mn04~ + 3H2S04 -> S042" + 2MnS04 + 10СО2 + 8Н20
❖ Відновлення. У разі відновлення цинком у сульфатній кислоті щавлева
кислота перетворюється в гліколеву, а з магнієм у сульфатній кислоті
відновлення припиняється на гліоксиловій кислоті:
СООН
І
СООН
Zn/H,S04
-—*• НОСН2СООН-™іколева кислота
^^£2*ОНС—СООН -гліоксилова кислота
Ці ж сполуки можна отримати й за допомогою електрохімічного віднов-
лення.
❖ Дихлор о ангідрид, або оксалілхлориду одержують дією на безводну
щавлеву кислоту фосфор (V) хлориду:
НООС-СООН +2РС15—-С1-С-С-С1+ 2РОС13+2НС1
О О
Необхідну для синтезу безводну кислоту одержують з гідрату щавлевої
кислоти С2Н204-2Н20 шляхом азеотропної відгонки води з тетрахлорометаном.
• Ацилювання алканів. Схильність оксалілхлориду до гомолітичного розриву
С-С-зв'язку через великий позитивний заряд на карбонільних атомах
використовують для одержання хлороангідридів монокарбонових кислот з
алканів за радикальним механізмом:
Я-Н + С1СОСОС1 Я-СОСІ + НС1+СО
• Реакція із сечовиною. Важливою похідною щавлевої кислоти є її уреїд -
парабанова кислота (оксалілсечовина), яку одержують із сечовини й
оксалілхлориду:
Н
/НН2 С1С=0
о=с 2 + ыт и —- о=< Т
>Ш2 С1С=0 N-^0
н
Парабанова кислота
544
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Під дією лугу вона розщеплюється, утворюючи оксалурову кислоту:
н,ж:—ш-с-соон
6 6
Оксалурова кислота
Електролітичне відновлення парабанової кислоти можна виконати так, що
утвориться гідантоїн:
О О
^—' Гідантоїн
0 0 о
З цих сполук синтезують лікарські препарати, гербіциди, згущувачі пластмас
і стабілізатори полівінілхлориду.
• Синтез хлороангідридів. Оксалілхлорид застосовують для одержання з
карбонових кислот їхніх хлороангідридів:
Я-СООН + СІСОСОСІ-
9і
я-с-о-с-с-сі
І II II
О" о о
н+ — К-С-С1+С02+С0+НС1
II г-
о
❖ Оксалати. Цікаві властивості мають різні солі щавлевої кислоти. Середні
оксалати луясних металів легкорозчинні у воді, кислі ж, навпаки, важкорозчинні.
Майже зовсім нерозчинні солі лужноземельних металів, і цю властивість,
зокрема, кальцій оксалату, використовують в аналітичній хімії для кількісного
визначення Кальцію або щавлевої кислоти. Оксалати важких металів також
нерозчинні у воді, але багато з них легко розчиняється в оксалатах лужних
металів. Це пояснюють тим, що оксалати лужних і важких металів у разі
змішування утворюють комплексні солі — "оксало-солі", наприклад:
Ре2(С204)3 + ЗК2С204 + 6Н20 ■
Сг2(С204)3 + ЗК2С204 + 6Н20
мю2о4 + К2С204+6Н20
-2К3[Ре(С204)3]-6Н20
-2К3[Сг(С204)3]-6Н20
К2[№(С204)2]-6Н20
На розчиненні оксалатів важких металів в оксалатах лужних металів
ґрунтується розчинювальна дія "кисличної солі" на іржу.
❖ Застосування. Щавлеву кислоту використовують як відбілювач соломи,
горіхової й цінної деревини, для виготовлення барвників, просочення тканин у
друкуванні на ситці, очищення урану, в реакціях дегідратації й поліконденсації,
для виробництва фенолоформальдегідних смол.
Розділ 19. Дикарбонові кислоти
545
19.4.2. Малонова кислота
Малонова кислота також має особливості, що різко відрізняють її від інших
членів ряду, таких як янтарна, глутарова тощо.
❖ Декар бокс плювання. Під час нагрівання малонова кислота легко зазнає
декарбоксилювання, перетворюючись на оцтову кислоту:
НООССН2СООН С02 + СН3СООН
Цю властивість зберігають і С-заміщені гомологи малонової кислоти, тобто
алкілмалонові кислоти.
❖ Конденсація. Атоми Гідрогену метиленової ланки в малоновій кислоті,
перебуваючи в а-положенні щодо двох карбоксилів, мають набагато більшу
здатність відщеплювати протон порівняно з одноосновними карбоновими
кислотами. Зокрема, малонова кислота особливо легко вступає в конденсацію з
оксосполуками й, залежно від співвідношення реагентів, із молекулою альдегіду
реагує одна або дві молекули кислоти:
СООН НООС /СООН
RCHO + 2СН2< —^ ^СН-СН-СН + 2Н20
СООН НООС ^ СООН
Спочатку під впливом основного каталізатора - піридину або третинного аміну
- із малонової кислоти утворюється аніон, що зазнає конденсування з оксосполукою.
Алкіліденмалонова кислота, що утворилася, під час обережного нагрівання зазнає
декарбоксилювання, перетворюючись в а,Р-ненасичену кислоту:
но ,0
ги
yh—"
но'%
- о о
НО-С_ R ,R НО-С RWR
СН+ с — сн-с
. О»- О -
0
но-с r r ,р
С=С -^г с=сн-с.
но-с к'^я он
о
У такий спосіб з ацетону можна отримати метакрилову кислоту
СН2=С(СН3)СООН, а із кротонового альдегіду СН3СН=СН-СНО - сорбінову
кислоту СН3-СН=СН-СН=СН-СООН.
Реакція конденсації малонової кислоти з оксосполуками із одержанням а,р-
ненасичених кислот відома як реакція Кневенагеля.
❖ Галогенування. Малонова кислота легко зазнає галогенування в а-
положення.
❖ Дегідратація. У разі збезводнювання малонової кислоти фосфор (V)
оксидом одержують недооксид Карбону:
СН2(СООН)20=С=С=С=0
546
Чирва В.Я., Ярмолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
У цій реакції також виявляється підвищена рухливість а-гідрогенних атомів.
Недооксид Карбону належить до класу кетенів і поводиться подібно до
своєрідного ангідриду малонової кислоти:
0=С=С=С=0
ноос-сн2-соон
с2н5оос-сн2-соос2н5
Звичайного ангідриду малонова кислота не утворює.
❖ Циклічні ацилали Малонова кислота реагує з ацетоном в оцтовому
ангідриді, утворюючи циклічний ацилаль ацетону, так звану кислоту Мельдрума:
О 0
,&0Н ^снз-'Й) /-о СН,
н2< ип + о=£ — Н2СЧ /С^
2 с-он "сн3 2 чс-о СНз
\\ -її
о о
❖ Барбітурова кислота. Значний інтерес становить уреїд малонової
кислоти - барбітурова кислота. Вона та її С-алкілпохідні можуть бути
синтезовані багатьма способами. Зазвичай для цього застосовують конденсацію
малонової кислоти з сечовиною за наявності фосфор (V) хлороксиду або
оцтового ангідриду, або малонового естеру й сечовини за наявності натрій
етил ату:
0 н Р
0=сС + ,сн2^-* о=сч ,сн2
Ш, С2Н50-С N-0, 2
2 2 5 о н о
Барбітурова кислота
■ Барбітурова кислота поводить себе як сильна кислота. Кислотні власти-
вості зумовлені кето-енольною і лактам-лактимною таутомерією:
ы^ыи" нк ,ын нк^н
он о 6
її метиленова група зберігає реакційну здатність метиленової ланки малонової
кислоти. Тому, діючи димлячою нітратною кислотою, можна замістити в ній атом
Гідрогену на ЫСЬ-групу. С-Нітробарбітурову кислоту, що утворюється, можна
відновити до амінобарбітурової кислоти (урамілу), а ту за допомогою калій ціанату
перетворити в уреїдобарбітурову або псевдосечову кислоту:
Розділ 19. Дикарбонові кислоти
547
9г9 Й^9 НР Н Р
н о н ь н ь н ь 5
Ураміл Псевдрсечова кислота
Усі ці сполуки утворюються як проміжні продукти під
час синтезів рибофлавіну, піримідину й сечової кислоти, ^
мононатрієва сіль якої є складовою частиною каменів N
сечового міхура. Висохлі екскременти птахів (гуано) містять ^^-^
до 25 % сечової кислоти й можуть слугувати джерелом її
одержання. Сечову кислоту використовують для синтезу
гтА**А\жч, Сечова кислота
кофеїну.
■ Досить цікава ізонітрозобарбітурова, або віолурова кислота, яку
одержують із барбітурової кислоти шляхом її нітрозування або з алоксану й
гідроксиламіну. Віолурова кислота вирізняється яскравим забарвленням солей:
відомі, наприклад, синя й червона калієві солі, червона й темно-жовта літієві солі
тощо:
н Р
о=сч ,с=жш
н ь
Віолурова кислота
■ Свого часу важливе практичне значення мали С-діалкілбарбітурові
кислоти - барбітурати, багато з яких пригнічують нервову систему й виявилися
свого часу придатними як снодійні засоби (група вероналу). Це насамперед сам
веронал — діетилбарбітурова кислота (Е. Фішер), діал, люмінал тощо:
н Р н Р н Р
о-г^"С^С2Н5 л „^"^ .СН,СН=СН2 л ^"Сч
о-сч ис. 2 5 о=сч ,с 2 2 о=сч ,с
N-0, С,Н5 N-0, СН,СН=СН, N-0. С,Н
н ь н о ^ 2 н Ь 2 ■
Веронал Діал Люмінал
Однак їхнє вживання в значних дозах або в ^ ^ ^
поєднанні з алкоголем спричинювало важкі отруєння і 3
призводило до смертельного результату. 0=с' С
Для наркозу часто застосовують евіпан {гексенал) - N-0^ сн
натрієву сіль іУ-метил-С-метил-С-циклогексенілбарбі- №+ О 3
турової кислоти: Бвіпан (гексенал)
5
548
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
19.4.3. Янтарна кислота
❖ Дегідратація. У разі тривалого нагрівання або, ліпше, збезводнювання
оцтовим ангідридом чи тіонілхлоридом янтарна кислота, на відміну від нижчих
гомологів, легко перетворюється в циклічний ангідрид - сукцинангідрид, що має
властивості ангідридів монокарбонових кислот:
р
сн7соон , Н2?"°ь
СН2СООН -н2о ' с-
о
• Сукцинімід. У разі нагрівання янтарної кислоти або її ангідриду з амоніаком
одержують імід {сукцинімід):
Срь-
<^Н2СООН ньу НХ-С' в+
СН2СООН -2Н20 нс-сч'
¿05-
Сукцинімід
У сукциніміді основні властивості атому Нітрогену пригнічені більше, ніж в
амідах. Навпаки, атом Гідрогену іміногрупи легко зазнає протонування, що
зумовлене акцепторним впливом двох сусідніх карбонільних груп. Наприклад,
під дією алкоголятів луясних металів відбувається заміщення атома Гідрогену
іміногрупи на метал з одержанням калій- або натрійсукциніміду, з якого
синтезують ІУ-бромосукцинімід:
,0 .0 +0
СНг-СС ма0Н СНг-СС . + вг2 сн^с
сн2-с^0 2 снгс^0 сн2-с^0
Бромосукцинімід
■На рухливості імідного атома Гідрогену ґрунтується одержання
бромосукциніміду, що широко використовують для бромування за Волем-
Циглером. За його допомогою можна заміщати атом Гідрогену на атом Брому в
алільному положенні пропілену. Річ у тому, що бромосукцинімід завжди містить
незначну домішку молекулярного брому, який під час опромінення або
нагрівання перетворюється в атомарний та ініціює галогенування в алільне
положення алкену:
Вг2-^2Вг-
Н2С=СН-СН3+ Вг' Н^СН-СН^ +■ НВг
Розділ 19. Дикарбонові кислоти
549
Гідроген бромід, що виділяється, реагує з бромосукцинімідом, утворюючи
молекулярний бром, який бере участь в утворенні алілброміду з вивільненням
атомарного Брому — продовжувача ланцюгової реакції:
ї**2 С>Вг + НВг С>Н + Вг2
СН—СНг-С^
О О
Вг2 + Н2С=СН-СН2 ►Н2С=СН-СН2Вг + Вг
■ Сукцинімід під час перегонки з цинковим пилом переходить у пірол, а під
час нагрівання з фосфор (V) сульфідом - у тіофен:
Н Н
нс*с нс*са
н н
Пірол Тіофен
■ Сукцинангідрид, як і сукцинілхлорид, - хлороангідрид янтарної кислоти
С1СО(СН2)2СОС1, можна відновити до у-бутиролактону:
Р
н2с-сч н2с-с;
о о
у-Бутаролактон
■ Янтарний ангідрид застосовують для одержання багатьох барвників,
вітамінів групи В, інсектицидів.
❖ Естерифікація. Прямою естерифікацією кислоти можна отримати її
повні естери:
СН2СООН н2зо4 СН2СООС2Н5
І 2 + 2С2Н5ОН І
сн2соон -2Н*° сн2соос2н5
а взаємодією ангідриду зі спиртом - кислі естери:
<,0
^ с> + с2н5он ай-?
н2соос2н5
сн^сс- сн2соон
■ 3 етиленгліколем янтарна кислота утворює поліестер:
«НООС(СН2)2СООН .+ иНО(СН2)2ОН 4* [-(0)С(СН2)2С(0)0(СН2)20-]„
550
Чирва В.Я., Ярмолюк CM., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
❖ Конденсація. Хоча метиленові ланки в повному естері янтарної кислоти
не мають такої рухливості, як у малоновій кислоті, проте її достатньо, щоб
провести в основних умовах конденсацію з оксосполуками (конденсація
Штоббе):
нг хСООС2Н5 Н3(Х /СООС2Н5
т ' /С=0 + CPL - /С=С
нзс чсн2соос2н5 нзС с^сосед
Цей метод застосовують в галузі ароматичних кетонів для введення залишку
естеру пропіонової кислоти.
19.4.4. Глутарова кислота
❖ Дегідратація. Глутарова кислота під час нагрівання у вакуумі також
утворює циклічний ангідрид. Він виникає завдяки нуклеофільній атаці
гідроксилу на атом Карбону іншої карбоксильної групи:
Зазначимо, що такий хід реакції відповідає правилу Блана. Згідно з ним,
дикарбонові кислоти, карбоксильні групи яких роз'єднують дві або три метиленові
групи, під час нагрівання дають ангідриди. Якщо кількість таких груп дорівнює 4
або 5, то кислоти в цих умовах перетворюються в циклічні кетони.
Вищі карбонові кислоти також дають кетони, однак для цього треба нагрівати
їхні кальцієві солі:
/у
О
/CH-CHj-C
Н2С ^Са СаС03 + ЛЛ=0
ХСНГСНГС ^
О
Пімеліноат кальцію
❖ Циклізація за Дікманом. Етилглутарат можна перетворити у п'ятичленну
циклічну діоксодикарбонову кислоту під дією натрію в спирті внаслідок естерної
конденсації (детальніше див. розділ 11):
о=сос2н5 сн2соос2н о=/>°°С2Н5
0=СОС,Не У 2 -2С2н5он І Т
25 сн2соос2н5 о=с—6нсоос2н5
Розділ 19. Дикарбонові кислоти
551
19.4.5. Адипінова кислота
Адипінову кислоту в промислових масштабах витрачають для синтезу
синтетичного волокна найлону-66 (дакрону). Найлон - надзвичайно міцне й
еластичне волокно, що нагадує за властивостями натуральний шовк, його
виготовляють з поліаміду, який одержують сплавленням гексаметилендіаміну й
адипінової кислоти:
иНООС(СН2)4СООН + «H2N(CH2)6NH2
-> [-OC(CH2)4CONH(CH2)6NH-]w + wH20
Гексамешилендіамін одержують у такий спосіб. Спочатку з адипінової
кислоти синтезують її діамонійну сіль. Потім нагріванням її переводять у діамід,
з якого дегідратацією одержують динітрил. Каталітичне гідрування динітрилу
дає гексаметилендіамін:
2NH, -2Н20
НООС-(СН2)4СООН —І- NH4OOC-(CH2)4COONH4—■
2Н О Н
NH2OC(CH2)4CONH2"-^NEC-(CH2)rCHN H2N-(CH2)g-NH2
Гексаметилендіамін
В іншому промисловому способі одержання гексаметилендіаміну вихідною
речовиною є дивініл:
2HCN
н2с=сн-сн=сн2+сі2—- сісн2сн=сн-сн2сі
N=C-CH2CH=CH-CHrC~NH2N-(CH2)g-NH2
19-5. Синтези на основі малонового естеру
Рухливість атомів Гідрогену метиленової ланки особливо зручно спосте-
рігати в естерах малонової кислоти, де нема рухливіших кислотних атомів
Гідрогену карбоксильних груп, які впливають на реальну картину кислотності
Гідрогенів метиленової групи.
Діетиловий естер малонової кислоти одержують із натрій ціанідацетату:
С1СН2СООКа+ + КаШ—КЕС-СН2СОО"Иа+ 2С^Ша, °^СЧЗ^<£°
Натрій ціанідацетат С2Н50 ^^2^5
Малоновий естер
Алкілмалоновий естер є слабшою СН-кислотою, ніж малоновий. Тому для
його йонізації потрібна сильніша основа, наприклад, калій трет-бутилат:
552
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
^COOC^
R—СН
^СООС^з -(снз)зСОН
(СН3)3сок -/СООС2Н5 R-x> Rx /СООС2Н5
СООС.Н, R' СООСД
к+
'2і А5
2А15
Під дією на діетиловий естер малонової кислоти, який називають просто
"малоновим естером", металевого натрію або натрій алкоголяту атом Гідрогену
може заміщатися на Натрій, утворюючи натріймалоновий естер, який як
нуклеофіл широко застосовують у різних синтезах:
о=с;
N
■ або -
,сп ■—- \сн Ich
о-сч
оед
/ОСА
о-сч
o-cN
Na+
Натріймалоновий естер як типовий нуклеофіл взаємодіє з алкілгалогенідами
або іншими галогенопохідними з досить рухливим атомом галогену за
механізмом 8к2, унаслідок чого відбувається введення за метиленовою групою
алкільного замісника.
Так можна одержувати гомологи й різноманітні інші заміщені малонових
естерів, а після їхнього гідролізу - кислоти (синтез Конрада1). Декарбоксилю-
ванням у разі обережного нагрівання можна видалити один з "малонових"
карбоксилів і отримати гомологи або інші заміщені похідні оцтової кислоти. Отже,
синтез М. Конрада є важливим методом одержання одно- і двоосновних кислот.
Найвищі виходи алкіл- і діалкіл малонових естерів досягають у реакціях з
первинними алкілгалогенідами, нижчі - із вторинними. Третинні алкілгалогеніди
непридатні для цього, оскільки за умов синтезу вони перетворюються в алкени.
Нижче наведено окремі приклади синтезів на основі натріймалонового естеру.
❖ Синтез гомологів оцтової кислоти. Алкілюванням галогеноалканом
мононатрієвого естеру одержують алкілмалоновий естер, який після омилення й
декарбоксилювання перетворюється на гомолог оцтової кислоти:
RCl + Na+
900С2Н5
СООС2Н5
-NaQ
90ос2н5
RCJH
СООС2Н5
2Н20
-2С2Н5ОН
9ООН
R£H -
СООН
-со,
RCH2COOH
За допомогою такого прийому можна ввести в алкілмалонову кислоту ще
один алкіл і, в остаточному підсумку, синтезувати діалкілоцтову кислоту:
1 Конрад М. (Conrad М.) (1848-1920) - німецький хімік. Спільно з К. Бішопом розробив загальний
метод синтезу на основі пропандіового естеру.
Розділ 19. Дикарбонові кислоти
553
rch:cooc2h5 {стсок ^сооед R_CI
сооед -слон ^соос2н5
Rs .соос2н5 но- R. СООН , R4
R^cooc2h5 -едон R"c^cooh "^*r-ch-cooh
❖ Синтез янтарної кислоти
с2н5о
/CCH2C1 + Na+
СООСД,
СООС2Н5
-NaCl
<\ /COOCjHj
/ССНг^Н -//^НООССНзСНзСООН
CjHjO СООС2Н5
Зазначимо, що в процесі цього синтезу утворюється триосновна карбонова
кислота, яка зазнає декарбоксилювання до двоосновної.
Альтернативно янтарну кислоту можна одержати димеризацією малонового
естеру:
. с2н5оос соос2н5
2[(C2H5OOC)2CH]Na++12 — СН -СН
С2Н5ООС СООС2Н5 -4V2Hs°h
-^(НООС)2СНСН(СООН)2 ^^-ноосссн^соон
❖ Синтез глутарової й інших двоосновних кислот
+ СООС2Н5 С2Н5ООС СООС2Н5
Cl(CH2)„Cl + 2Na(jH- ► (*Н(СН2)/н
СООС2Н5 С2Н5ООС СООС2Н5
ноос соон
СН(СН2)„СН ^¿0* НООССН2(СН2)„СН2СООН
ноос соон
Цікаво, що за допомогою цієї реакції можна одержати не тільки дво-, а й
чотириосновні карбонові кислоти. Успіх цього синтезу залежить від доступності
дигалогеноалканів такого типу.
❖ Синтез оксокислот. Нуклеофільне приєднання натріймалонового естеру
до вінілкетонів, що мають карбонільну групу біля подвійного зв'язку, веде до
утворення оксокарбонових кислот:
554 Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
(С2Н5ОСО)2СН-Na++H2C=CH-£-R— (C2H5OCO)2CH-CHrCH=:C-R
О ONa
НООС^ ,
— /СН-CHrCHr-C-R —HOOC-CH,CH,CHrC-R
НООС о 2 О
❖ Синтез гідроксикислот. Застосування оксиду етилену дає змогу одержу-
вати на основі малонового естеру у-гідроксикислоти:
(С2Н5ОСО)2СН2 + H2C-CH2-^(C2H5OCO)2CH-CH2CH2OH
О
ОН"
(НООС^СН-С^СНгОН НООССН2СН2СН2ОН
J1
❖ Синтез двоосновних кислот. Використання акрилонітрилу в малоновому
синтезі відкриває шлях до одержання янтарної кислоти та її гомологів:
(С2Н5ОСО)2СН2+H2C=CH-CN °снова» (C2H5OCO)2CH-(CH2)jCN
❖ Синтез циклоолканів. Дією на <х,со-дигалогеноалкани натріймалонового
естеру У. Перкіну вдалося синтезувати циклоалкани - від циклопропану й до
циклогептану:
скус СООС2Н5 CHjX соос2н5 ^ сопе и
({н2)п.2 ф ^^(сн^сСсоос2н5
СЩХ СООС2Н5 СООС2Н5 .roh v_y соос2н5
19.6. Двоосновні ненасичені кислоти
19.6.1. Будова й властивості
Найпростіші ненасичені двоосновні кислоти - t/ис-бутендіова (малеїнова) й
/иранс-бутендіова {фумарова) — мають однакову структурну формулу, але різні
просторові конфігурації:
^'С00Н НООС. .COOH
.С-С. г=с
HOOC Н н' хн
Фумарова кислота Малеїнова кислота
(транс-ізомер) (і/ис-ізомер)
Фумарова кислота вперше виділена з гриба рутка (Fumaria officinalis L.), звідки
й походить її назва. Малеїнову кислоту в природних джерелах не виявлено.
Розділ 19. Дикарбонові кислоти
555
Ці кислоти різко відрізняються за фізичними і деякими хімічними
властивостями. Фумарова кислота, на відміну від малеїнової, погано розчинна у
воді, на чому ґрунтується їхня індивідуалізація за допомогою кристалізації.
Теплоти згоряння цих кислот неоднакові: 1340 кДж/моль для фумарової й
1370 кДж/моль для малеїнової. Це дало змогу припустити траяс-розташування
карбоксильних груп для фумарової кислоти і г/нс-конфігурацію для малеїнової, що
підтверджене першими константами дисоціації двоосновних кислот. Перша
константа і/ис-сполуки повинна бути значно більшою, ніж юряис-кислоти,
оскільки електроноакцепторні групи СООН зближені. Справді, Кі для малеїнової
кислоти дорівнює 1,17*10"2, а для фумарової - 9,3-10"4, а ось друга константа
дисоціації для малеїнової кислоти повинна бути вже меншою, оскільки протон
притягується ще карбоксилат-аніоном, який утворився. Це підтверджено
експериментально: К2 і/ис-ізомеру становить 2,6-10"3, а транс-ізомеру - 2,9-10"5.
Прямим доказом г/ис-розташування карбоксильних груп у малеїновій кислоті
є легкість утворення нею ангідриду під дією фосфор (V) оксиду:
Фумарова кислота ангідриду не утворює.
Цікаво зазначити, що на прикладі цих кислот 1877 р. Й. Вісліценус1 уперше
виявив передбачену Я. Вант-Гофом цис-транс-ізомерію сполук із С=С-зв'язком.
19.6.2. Методи одержання
■ Обидві кислоти у вигляді суміші стереоізомерів отримують дегідратацією
яблучної кислоти за умов нагрівання:
■ Під дією спиртового розчину лугу на хлороянгпарну кислоту відщеп-
люється гідроген хлорид, і утворюється малеїнова кислота:
О
Малеїновий ангідрид
ноос-сн2снон-соон-^-ноос-сн=сн~соон
І-Т5аСЇГ
-с2н5он
ЫаООС-СН=СН--СО(Жа
1 Вісліценус Й. (ШізІісепиБ І.) (1835-1902) - німецький хімік-органік. Головні праці в галузі
ізомерії.
556
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
■ Для цис- і транс-ізомерів кислот характерні взаємні переходи в разі
нагрівання до 150 °С. В обох випадках відбувається розрив я-зв'язку з
утворенням бірадикала, в якому існує вільне обертання навколо сг-зв'язку:
ноосч Я ноосч Н Нч СООН нч СООН
А А А А
н соон н соон н соон н соон
19.6.3. Малеїновий ангідрид
❖ Синтез. Головний спосіб одержання малеїнового ангідриду - парофазове
окиснення бензену на ванадій-молібденовому каталізаторі:
Р
|І ]| (ґ О + 2С02 + 2Н20
Крім того, його можна одержати окисненням н-бутану або бутиленів на
ванадій-фосфорному каталізаторі.
❖ Хімічні властивості. Малеїновий ангідрид гідратацією переводять у
яблучну кислоту:
е
О + 2Н20 ноос-сн2снон-соон
Малеїновий ангідрид - ефективний дієнофіл у реакціях дієнового синтезу
(реакція Дільса-Альдера). Практичне значення він має як вихідна речовина для
одержання ненасичених поліестерів, алкідних смол, пестицидів, ПАР, присадок
для мастил.
Розділ 20
ПАРОКСИКИСАОТИ
Гідроксишслошами називають карбонові кислоти, що мають додатково одну
або кілька гідроксильних груп.
20.1. Класифікація і номенклатура
20.1.1. Класифікація
За кількістю карбоксильних груп (основністю)
Монокарбонові
гідроксикислоти
НОСН2СНСООН
2|
он
Дигідроксипропанова
кислота
(гліцеринова, або
гліцеролова кислота)
Дикарбонові
гідроксикислоти
НООССН^СНСООН
он
2« Гідроксибутандіова
кислота
(яблучна кислота)
Трикарбонові
гідроксикислоти
ОН
ноос-НзС-с-соон
СІ^СООН
З-Гідрокси-З-карбоксипентандіова
кислота
(цитринова, або лимонна
кислота)
За кількістю гідроксильних груп
Моногідроксикарбонові Дигідроксикарбонові Тригідроксикарбонові
кислоти кислоти кислоти
Н3ОСН-СООН
ОН
НООС^Н-С^НСООН
ОН ОН
НОСН2СН-СНСООН
І І
он он
2-Гідроксипропанова
кислота
(молочна кислота)
2,3-Дигідроксибутандіова
кислота
(винна кислота)
2,3,4-Тригідроксибутанова
кислота
558
Чирва В.Я., Ярллолюк CM., Толкачова Н.В., Земляков О.Є. Органічна хімія
За положенням гідроксильних груп відносно карбоксильної функції
а-Гідроксикислоти ß-Гідроксикислоти ю-Гідроксикислоти
СН3СН2СНСООН
ОН
сн3снсн2соон
он
2-Гідроксибутанова кислота З-Гідроксибутанова кислота
(а-гідроксимасляна кислота) (ß-гідроксимасляна кислота)
носн2сн2сн2соон
4-Гідроксибутанова кислота
(со-гідроксимасляна кислота)
20.1.2. Номенклатура IUP АС
Номенклатура гідроксикарбонових кислот і їхніх похідних аналогічна до
номенклатури одноосновних карбонових кислот. Гідроксильну групу в цих
сполуках розглядають як замісник.
Для деяких гідроксикислот використовують тривіальні назви (приклади
наведені курсивом вище; назви, рекомендовані 1993 р. IUP АС, виділені жирним
шрифтом), від яких утворюють відповідні назви ацильних залишків. Наприклад,
гліколева кислота (гліколоїл), молочна кислота (лактоїл - від англ. lactic acid),
гліцеролова кислота (гліцероїл), винна кислота (тартароїл - від англ. tartaric acid),
цитринова кислота (цитроїл - від англ. citric acid).
Розташування гідроксильних груп щодо карбоксильної функції можна
зазначати за допомогою букв грецького алфавіту (буквені локанти). Приклади
таких назв наведені в дужках вище.
20.2. Фізичні властивості гідроксикислот
Гідроксикислоти - це рідкі або кристалічні речовини, добре розчинні у воді
завдяки додатковій можливості утворення водневого зв'язку порівняно з
карбоновими кислотами або спиртами. З огляду на це вони мають вищі
температури кипіння й плавлення, ніж незаміщені карбонові кислоти. Для а-
гідроксикислот характерна оптична ізомерія завдяки наявності хірального атома
Карбону.
Гідроксильна група впливає також на дисоціацію сусідньої з нею
карбоксильної групи. Для а-гідроксикарбонових кислот зазначено зростання
їхньої кислотності. Наприклад, константи дисоціації гліколевої (гідроксиоцто-
вої) й оцтової кислот за 25 °С становлять 1,52-10"2 і 1,76-10"5, відповідно. Це
збільшення зумовлене двома чинниками: по-перше, негативний індуктивний
ефект гідроксильної групи знижує рКа приблизно на 0,45; по-друге, відбувається
взаємодія карбоксилат-іона з гідроксильною групою при сусідньому карбоно-
вому атомі через внутрішньомолекулярний водневий зв'язок. З'ясовано, що
Розділ 20. Гідроксикислоти
559
останній ефект залежить від взаємного розташування гідроксильної й карбо-
ксильної груп.
с-с
/ \ -
Доведено, що чим далі перебуває гідроксильна група від карбоксильної, тим
менше вона впливає на силу кислоти.
20.3. Стереоізомерія гідроксикислот
20.3.1. Енантіомерія
Головні принципи стереоізомерії розглянуті в розділі 6. Низка природних
гідроксикислот, наприклад, молочна, яблучна, мигдалева, мають у структурі
асиметричний атом Карбону й, отже, можуть існувати у вигляді двох
енантіомерних форм. В енантіомерних гідроксикислотах є однаковий набір
фізико-хімічних властивостей, відрізняються вони тільки знаком обертання
плоскополяризованого світла й поведінкою у біохімічних реакціях.
Рацемати, що є сумішшю однакових кількостей енантіомерів, оптично не
активні. їхні кристали, що містять два різні за просторовою будовою типи
молекул, відрізняються за упакуванням від утворених індивідуальними енантіо-
мерами. Тому рацемат плавиться за іншої температури (вищої або нижчої), ніж
оптично чисті форми, його розчинність інша:
СООН
Н—|—ОН
СН3
Б-Молочна кислота
т.пл. 53 °С,[аЬ-2,60 (Н20)
СООН і
н—|—он І
сн2соон І
О-Яблучна кислота
т. пл. 99-100 °С, [а]0+5,7°
(ацетон)
СООН
но-
-н
сн3
Ь-Молочна кислота
т. пл. 53 °С, [ссЬ +2,6° (Н20;
СООН
но—|—н
сн2соон
Ь-Яблучна кислота
т. пл. 99-100 °С, [а]в -5,7°
(ацетон)
СООН
но—с— н
і
СН3
В,Ь-Молочна кислота
т. пл. 17 °С, [а]в 0° (Н20)
СООН
но—с—н
і
СН2СООН
О.Ь-Яблучна кислота
т. пл. 129-130 °С, [а]в 0°
(ацетон)
560
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
СООН
Н—]—ОН
с6н5
О-Мигдалева кислота
т. пл. 133 °С, [аЬ-154°
(с2;Н2ПРО)
СООН
но—|—н
с6н5
Ь-Мигдалева кислота
т. пл. 133 °С, [а]0+154о
(с 2; Н2ПРО)
СООН
І
но—с— н
І
с6н5
Э.Ь-Мигдалева кислота
т.пл. 118,5 °С, [а]о0° (Н20)
Перетворення одного енантіомера в інший називають рацемізсщією.
Найлегше зазнають рацемізування сполуки, що мають асиметричний атом
Карбону в а-положенні до карбонілу, карбоксилу й загалом до електрофільних
замісників, за умови, що асиметричний атом Карбону має атом Гідрогену.
Пояснюють це легкістю протонування цього атома Гідрогену. Тому, зокрема, а-
гідроксикислоти рацемізувати порівняно легко під час кип'ятіння з лугами:
но-
соо
нхг^он-^0
сн,-
сЗ^
он
СНз\ /О-
но о
н,ок
-но-
Н3С
СОО"
Дсон
н
Для позначення конфігурації асиметричного центру гідроксикислот традиційно
використовують В,Ь-номенклатуру. У сучасних виданнях її витісняє однозначніша
К,8-номенклатура.
Стандартом у визначенні відносної конфігурації молекул є
гліцериновий альдегід. Важливо було з'ясувати співвідношення конфігурації
цієї речовини з іншими опорними речовинами: (+) і (-)-яблучною НООС-
СНОН-СН2-СООН та (+) і (-)-винною НООС-СНОН-СНОН-СООН кислотами.
Для цього за допомогою ціангідринного синтезу (+)-гліцериновий альдегід
перетворюють у суміш нітрилів (-)-винної кислоти й так званої мезо-винної
кислоти, неактивної внаслідок симетрії молекули. Після гідролізу нітрилів та
окиснення СНгОН-групи до карбоксилу утворюються ці кислоти:
СООН
но—н „
н—он
СООН
Ь-(-)-Винна кислота
н2о,НО-|-Н нсы
н+
Н-ИЭН
СН2ОН
СНО
н4юн
НСЫ
9м
Н~ОН
"Н—ОН
СН2ОН
н,о,
н+
СН2ОН
СООН
Н—ОН
Н—ОН
СООН
л/взо-Винна кислота
Так усталюються відносні конфігурації 0-(+)-гліцеринового альдегіду й Ь-(-)-
винної кислоти. Зокрема, Б-(+)-винна кислота міститься у виноградному соці й
"винному камені" у вигляді кислої калієвої солі, що випадає під час
витримування вина у дубових бочках. Оскільки в разі відновлення Б-(+)-винної
Розділ 20. Гідроксикислоти
561
кислоти за допомогою гідроген іодиду утворюється 0-(+)-яблучна кислота, то
так усталюється й відносна конфігурація асиметричного центру цієї кислоти:
ні
СООН
—ОН
но—
СООН
0-(+)-Винна кислота
СООН
—он ^
СООН
СООН
но-
соон
0-(+)-Яблучна кислота
Конфігурацію інших сполук визначають подібним способом стосовно однієї
з трьох зазначених опорних сполук.
У 1952 р. французьким ученим для рубідієвої солі обох оптично активних
винних кислот вдалося шляхом рентгеноструктурного дослідження визначити
їхню абсолютну конфігурацію й, отже, абсолютні конфігурації всіх речовин,
відносні конфігурації яких стосовно винних кислот були відомі, у тому числі й
конфігурації гліцеринових альдегідів.
20.3.2. Стереоізомерія гідроксикислот з двома
асиметричними атомами. Ліастереомерія
При двох асиметричних атомах кількість стереоізомерів досягає N = 22 = 4.
Розглянемо, наприклад, сполуку з двома асиметричними атомами — хлорояблучну
кислоту - й випишемо всі можливі її конфігурації:
Енантіомери
СООН СООН
но—н н—он
сі—н н—СІ
СООН СООН
(1) ' (2)
Енантіомери
СООН
Н—ОН
—Н
СООН
(3)
СІ
СООН
но—н
н—СІ
СООН
(4)
Діастереомери
Речовини (1) і (2), а також (3) і (4) - енантіомери (оптичні антиподи).
Водночас пари сполук (2) і (3), як і (1) та (3), (1) та (4) або (2) та (4), є
стереоізомерами, проте не енантіомерами, отже, вони один щодо одного є
діастереомерами.
На відміну від двох енантіомерів, два діастереомери аж ніяк не тотожні за
властивостями і мають різні фізико-хімічні характеристики. Це зрозуміло,
оскільки в діастереомерах функціональні групи не можуть бути розташовані так,
щоб усі замісники в обох сполуках виявились на однаковій відстані один від
одного. А якщо відстані різні, то і взаємний вплив буде різним, звідси й
відмінність у константах.
562
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
Важливий окремий випадок - тотожність замісників біля обох асиметрич-
них карбонових атомів, який уперше Л. Пастер спостерігав на прикладі винних
кислот:
Енсснтіомери
0(/?)СООН
н-
но-
-ОН
-Н
Діастереомери
ЬфСООН В(Л)СООН
нЧ-он
НО
н-
або О-винна кислота,
т.пл. 170 °С,[аЬ+12°
(Н2ПРО)
-н
-ОН
н-
ь©соон
(5,5)- або Ь-винна кислота,
т. пл. 170 °С, [осЬ-12°
(Н20)
но-
соон
-н
-он = но-
м^соон
н
соон
(Я 5)- або мезо-винна кислота,
т. пл. 140 °С, [<х]0 0 ° (Н20)
Як бачимо, (/г,5)-винна кислота або інакше мезо-ьттг. кислота має
внутрішню площину симетрії, тому її дзеркальні відображення тотожні. Ізомерні
Б- і Ь-винні кислоти енантіомерні одна щодо одної і мають трео-конфігурацію.
Діастереомерна їм л*езовинна кислота має е/?мга/?с^конфігурацію. Отже, для
винної кислоти існує три стереоізомери (два трео- і один - еритро-):
но-
ОН
но-
ОН
-ОН
-ОН
но-
но-
трео-Конфігурація ери/иро-Конфігурація
Рацемічну суміш Б- та Ь-винних кислот {виноградну кислоту) одержують
тривалим нагріванням Б- або Ь-винної кислоти.
203.3. Асиметричний синтез
Синтез у лабораторії майже завжди дає рацемічну суміш, тому постає
питання: звідки в живій природі виникли чисті енантіомерні форми? Щоб
відповісти на це питання, треба згадати, що всі каталізатори живої природи -
ферменти, за допомогою яких відбувається більшість біохімічних реакцій, -
складні асиметричні молекули. Тому проміжні реакційні комплекси молекули з
каталізатором є діастереомерами. Саме цим пояснюють той факт, що дія
ферментів специфічна, і вони синтезують лише один з двох можливих
енантіомерів.
Порівняно з природою, успіхи хіміків з асиметричного синтезу скромніші.
Зокрема, Н. Марквальд і К. Маккензі, нагріваючи кислу сіль етилметилмало-
нової кислоти з алкалоїдом бруцином (чистий природний енантіомер) до
відщеплення карбоксилу, одержали карбонову кислоту з асиметричним
карбоновим атомом. У продукті реакції трохи переважав енантіомер 2-
метилбутанової кислоти з лівим обертанням:
Розділ 20. Гідроксикислоти
563
С2Н5\Х:ООН
СНз^СООНбруцин
-со,
.C2H5Nc/H
СНз^ СООНбруцин
HCl.
CHf^COOH
+ НСІбруцин
Як же вперше в живій або неживій природі з'явилися окремі енантіомери?
Відповіді на це питання дають досліди з так званого асиметричного синтезу. У
природі є два чинники, здатні впливати на асиметричний синтез. По-перше,
поляризовані промені світла. Відомо, що, відбиваючись від поверхонь,
наприклад, води, світло частково зазнає поляризації, а в разі певного кута
падіння - повністю. О. Кун і Ю. Браун, розкладаючи поляризованим світлом
етил-2-бромопропіонат, виконали асиметричний синтез з переважанням одного
ізомеру. Л. Тенн і Т. Акерман опромінювали поляризованим світлом суміш
гідроген пероксиду й естеру фумарової кислоти та одержали дигідроксибу-
тандіову кислоту з невеликим переважанням D-енантіомера:
Нч /СООН <г°он 1оон
Jj' _jO]_ н+он ноЧ-н
но-
і-і
соон
н-
он
соон
По-друге, у природі є енантіомерні форми кристалів, наприклад кварцу.
К. Шваб дегідрував втор-бутиловий спирт у метилетилкетон так, що в залишку
переважав один з енантіомерів бутан-2-олу, використавши кристали однієї з
модифікацій кварцу з нанесеним на них нікелем. А. Терентьєв і Е. Клабуновський
нанесли луг на кристали кварцу і виконали ціанетилювання метилциклогексанону
на цьому каталізаторі. Виникла оптично активна суміш із невеликим пере-
важанням правообертального нітрилу.
203.4. Вальденівське обертання
П. Вальден1 (1896) виявив оптичну інверсію в таких циклах перетворень
так зване вальденівське обертання:
СООН
Н—|—СІ
СН2СООН
D-Хлороянтарна
кислота
AgOH
он
СН2СООН
_н
Стабілізований
карбокатіон
СООН
н—|—ОН
1.РСЦ
СН2СООН
D-Яблучна
кислота
2. Н20
СООН
СІ—:А;-ОН
Н ~"
СН2СООН
С1-
соон
-1-н
AgOH
сн2соон
L-Хлороянтарна
кислота
СООН
но-|-н
сн2соон
L-Яблучна
кислота
1 Вальден П. (Waiden Р.) (1863-1957) - латвійський вчений. Наукові праці стосуються стереохімії,
історії хімії.
564
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімій
Зміст явища полягає в тому, що заміщення гідроксильної групи на галоген
відбувається за механізмом 8Н2 з обертанням конфігурації. Заміна галогену на
гідроксил проходить за механізмом Бці, однак рацемізації не відбувається, а
простежується збереження конфігурації через внутрішньомолекулярну стабілі-
зацію карбокатіону карбоксилат-аніоном.
20.4. Синтез гідроксикислот
20 А. 1 • Синтез а-гідроксикарбонових кислот
❖ Гідроліз ціангідринів. Метод, який широко застосовують для синтезу а-
гідроксикарбонових кислот, полягає в перетворенні альдегідів, кетонів та
епоксидів у відповідний ціангідрин з наступним кислотним або лужним гідро-
лізом ціаногрупи:
ЩС-С(° + НСИ -HзC-CH"CN^^HзC--CH-COOH
н он 3 ОН
❖ Синтез на основі літійорганічних сполук. Реакція літієвої похідної ІУ.Л/"-
діізопропілформаміду з кетонами дає корисний альтернативний підхід до
синтезу а-гідроксикислот, що особливо важливо у тих випадках, коли утворення
ціангідрину утруднене:
О ОН ОН
ЬІС^[СН(СН3)2]2 К'К2С°> КіК2С-^[СН(СН3)2]2 ЯЧ^С-СООИ
О
❖ ГідролЬ а-галогенозаміщених карбонових кислот. Після впровадження
в органічну хімію реакції Гелля^Фольгарда2—Зелінського а-галогенозаміщені
кислоти стали зручною вихідною сполукою для синтезу а-гідроксикислот. Щоб
прискорити реакцію, можна використовувати вологий арґентум оксид або калій
гідроксид:
СН3СНС1СООН + Н20 5= СН3СНОН-СООН + НСІ
❖ Синтез на основі амінокислот. У деяких випадках а-гідроксикислоти
одержують із доступних природних а-амінокислот:
1 Гель К. (von Hell CM.) (1849-1926) - німецький хімік, працював у галузі органічного синтезу. У
1889 р. синтезував вуглеводень гексаконтан С6оН|22-
2 Фольгард Я. (Volhard J.) (1834-1910) - німецький хімік, автор багатьох методів органічного
синтезу та кількісного елементного аналізу. Синтезував саркозин, креатин та тіофен.
Розділ 20. Гідроксикислоти
565
н3с-сн-соон "^^н3с-сн--соон
>ІН2 -нр ОН
❖ Бродіння вуглеводів відбувається під впливом молочнокислих бактерій.
Наприклад, із глюкози одержують рацемічну молочну (гідроксипропанову)
кислоту із виходом 90 %:
• с6н12о6 — 2Н3С-СН-СООН
он
Інші реакції, такі як селективне окиснення первинної спиртової групи в
гліколях до карбоксильної, відновлення оксогрупи в а-альдегідо- або кето-
кислотах до гідроксильної, застосовують зрідка.
20.4.2. Синтез р-гідроксикарбонових кислот
❖ Реакція Реформатського. Найчастіше р-гідроксикислоти синтезують за
реакцією О. Реформатського. Дією цинку на суміш естеру а-галогенозаміщеної
кислоти й оксосполуки одержують цинковий алкоголят Р-гідроксикислоти, яку
гідролізують до потрібної гідроксипохідної:
Я-СНО+ ВгСН2СООС2Н5 Я-СН-СНзСОСК^Нз ^0ї^ Я-СН(ОН)СІ^ССЮС2Н5
ОгпВг
Ця реакція аналогічна до реакції Гриньяра. Цинк використовують замість
магнію тому, що цинкорганічні сполуки менш реакційноздатні й інертні до
естерної групи, проте взаємодіють з альдегідами або кетонами. Замість
карбонільних сполук у цьому синтезі можна використати нітрили. Часто на
кінцевій стадії синтезу відбувається відщеплення води, що дає естери а,Р-
ненасичених кислот.
❖ Синтез на основі оксиду етилену: Також р-гідроксикислоти можна
отримати шляхом взаємодії оксиду етилену і його гомологів з ціанідною
кислотою, наприклад:
Н20 + Н2804 ^О +
н2с—СН2+ НСИ-^носн2-сн2сн —ОПоП » н2с-снгс^ + Ш4-Н804
^ 90 С ОН ОН
р-Гідроксипропіонова кислота
❖ Гідратація а$-пенасичених карбонових кислот, що відбувається всу-
переч класичному правилу В. Марковникова, приводить до Р-гідроксикарбо-
нових кислот:
566
Чирва В.Я., Ярмолкж СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
б+/—ч. Со Л>
н2с=сн^с^ + н2о — сн2он-снгсч
он он
❖ Окиснення ольдолів. Альдолі є зручним джерелом для одержання
різноманітних Р-гідроксикислот як з лінійними, так і розгалуженими ланцюгами.
,о
СНзСНОН-СН^С; + 2 [Ае(Ш3)2]+0Н- Н3С-СН-СН2С00Ш4 +2£% + ЗЖ3 + Нр
Н ОН
20.4.3. Синтез со-гідроксикарбонових кислот
❖ Гідроліз лактонів. Загальний метод одержання у-, 5- і ш-гідроксикислот
полягає в окисненні циклічних кетонів за Байєром-Віллігером до лактонів з
наступним розкриттям циклів лугами й подальшим обережним підкисленням.
Якщо як окиснювач використовувати лужний розчин гідроген пероксиду, то це
приведе відразу до солі гідроксикислоти.
Циклобутанони й циклопентанони розширюють цикл, даючи у- і 8-лактони,
відповідно. Самі ж гідроксикислоти одержують у разі дуже обережного
підкислення, тому що вони часто мимовільно зазнають дегідратування й знову
перетворюються в лактони.
Гідроксикислоти з віддаленішими одна від одної функціями значно ста віль-
ніші, тому їх можна одержувати зі значно меншими труднощами з лактонів із
розміром циклу понад сім:
о2
Чсн2)/
снрщсн^соон
• Вихідні лактони в окремих випадках можна отримати відновленням
циклічних ангідридів двоосновних кислот:
сн2-с*° ^ сн2-ф2
сн2-с;ч сн2-с;
о о
або нагріванням Р,у-ненасичених кислот із розведеними мінеральними кислотами:
неї СН^СН,
н,с=сн-сн,соон —- І о
сн^
Розділ 20. Гідроксикислоти
567
❖ Гідроліз продуктів реакції теломеризації. Кислоти з віддаленим кінцевим
положенням гідроксилу можна одержувати послідовним кислотним, а потім
лужним гідролізом тетрахлороалканів - продуктів теломеризації етилену з
тетрахлорометаном (О. Несмеянов):
С1(СН2СН2),СС13-|^ С1(СН2СН2)„СООН *^НО(СН2СН2),,СООКа
20.4А. Синтез ди- і полігідроксикарбонозих кислот
Для синтезу дигідроксикарбонових кислот використовують окиснення
ненасичених кислот гідроген пероксидом (безпосередньо або із залученням
пероксикислот) з одночасним розкриттям епоксидного циклу. Цей метод особливо
ефективний у випадку кислот, що мають одночасно подвійний і потрійний зв'язки,
оскільки потрійний зв'язок порівняно стійкіший до дії пероксикислот.
Аналогічне перетворення можна виконати й іншими способами:
- окисненням алкенових кислот у юрет-бутиловому спирті дією вищих
оксидів металів, таких як Осмій(УІІІ), Ванадій(У), Хром(УІ), Манган(УІІ);
- приєднанням галогену до алкенової кислоти й обробкою сполуки, що
утворилася, водним розчином натрій гідроксиду або вологим арґентум оксидом.
♦> Синтез дигідроксибутандіових (винних) кислот. Найліпше вивченими
представниками серед дигідроксидикарбонових кислот є винні кислоти, які можна
отримати як рацемічну суміш або у вигляді оптично неактивної л*езо-форми.
■ Рацемат одержують окисненням фумарової кислоти калій перманганатом.
Гідроліз ціангідрину, отриманого з гліоксалю, також приводить до рацемічної
суміші, з якої можна виділити оптично активні кислоти за допомогою стандартних
методик:
СООН СООН
ноос-
онс-сно
КМп04
-СООН
HCN
он
•NC-CH-
ОН
І
-сн-
CN-
но-
ОН но-
+
СООН
ОН
СООН
■ Кислоту л<езо-винну одержують окисненням малеїнової кислоти або
фенолу калій перманганатом або фумарової кислоти за допомогою натрій
хлорату й осмій (VIII) оксиду у воді:
або
(
СООН
СООН
СООН
СООН
КМпО.
NaClO
СООН
он
•он
AgOH
соон
Вг
-В г
Os04, Н20
СООН
мезо-Вітнл кислота
СООН
568
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Реакція вологого арґентум оксиду з 2,3-дибромоянтарною кислотою також
дає л*ез0-винну кислоту.
Велику кількість Б-винної кислоти у вигляді кальцій тартрату (так зване
виннокисле вапно) одержують переробкою відходів виноробної промисловості.
❖ Синтез полігідрокситрбонових кислот
• Окиснення ді- й полієнових кислот. У загальному випадку кислоти, що
містять більше двох спиртових груп, можна одержати гідроксилюванням
ненасичених кислот із більш ніж одним подвійним зв'язком, наприклад:
СН3(СН2)4СН=СНСН2СН=СН(СН2)7СООН
Октадекан-9,12-дієнова кислота
-Н* СНзСС^СН-СН-СНз-СН-СН-СС^^ООН
он он он он
9,10,12,13-Тетрагщроксиоктадеканова кислота
• Окиснення моносахаридів. Однак найбагатшим джерелом полігідрокси-
кислот є вуглеводи. М'яке окиснення цукрів галогенами в лужному середовищі
перетворює альдегідну групу в карбоксильну. Наприклад, із глюкози синтезують
глюконову кислоту. Ці кислоти в загальному вигляді називають альдоновгши.
Виділяють їх, як звичайно, у вигляді лактонів:
Н-
но-
Н-
Н-
СООН
-ОН
-Н
-ОН
-ОН
СН2ОН
О-Глюконова кислота
сн2он
О-Глюконо-1,4-лактон
(у-лактон О-глюконової кислоти)
■ Окисненням відповідно захищених похідних моносахаридів можна
одержати уронові кислоти:
СН2ОН СООН
НО
ОСН,
ОСН,
н2о
он
О-Глюкуронова кислота
■ Другий шлях синтезу таких кислот полягає в утворенні ціангідринної похідної
моносахариду за методом Кіліані з наступним гідролізом нітрилу, що утворився.
Розділ 20. Гідроксикислоти
569
20.5. Хімічні властивості гідроксикислот
❖ Загальні властивості. Гідроксикислоти поводяться і як спирти, і як
кислоти. За значного віддалення цих функцій одна від одної в багатьох реакціях
вони не пов'язані між собою. У разі звичайних умов реакцій карбоксильну групу
можна перетворити в естерну, амідну, нітрильну групи або хлороангідрид.
Модифікацію гідроксильної групи часто виконують після тимчасового
захисту карбоксильної групи. Наприклад, галогенозаміщені карбонові кислоти
зазвичай одержують з естерів гідроксикислот. В інших випадках попередня
естерифікація не потрібна. Наприклад, ацилювання гідроксильної групи можна
безпосередньо проводити на кислоті.
У низці реакцій карбоксильна й гідроксильна групи реагують одночасно.
Справді, під час дії металевого натрію участь у реакції беруть обидві
функціональні групи. Вони синхронно реагують із фосфор(У) хлоридом. Усе
сказане можна проілюструвати такою схемою:
ІІ-СН-С^
он осн,
сн,к,
N8 <>0
СЖа (Жа
<Р ИаОН <Р НВг <Р
к-сн-с/ -^к-сн-с( — к-сн-<
ОН ОИа Нг° ОН ОН 2 Вг ОН
уО 2РС15
к.-сн-с(
(сн3со)20
сі
СІ
■ 2РОС13
■ 2нс1
-сн3соон
іі-сн-
ОСОСН3 ОН
Прикладом використання тимчасового захисту може слугувати синтез а-
метоксипропанової кислоти. Подібно до звичайних карбонових кислот, молочна
кислота здатна утворювати зі спиртами естери:
СН3СНСООН + С2Н5ОН
ОН
н+
СН3СНСООС2Н5 + Н20
он
Атом Гідрогену гідроксильної групи дією алкоголяту може бути заміщений
на Натрій:
СН3СНСООС2Н5 + С2Н5ОИа
СНз^НСООСг^ + С2Н5ОН
ОИа
Синтезований алкоголят, що утворився, далі алкілують:
570
Чирва В.Я., Ярмолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
СНз^НСООС^з + СН3І СН3СНСООС2Н5 + Nal
ONa ОСН3
Під час гідролізу цього продукту утворюється етер гідроксикислоти:
СНз^НСООСзНзї^^СНз^НСООН + С2Н5ОН
ОСН3 ОСН3
❖ Реакції дегідратації. До перетворень, специфічних для гідроксикислот,
належить їхня дегідратація. Зазвичай вона відбувається під час нагрівання;
залежно від взаємного розташування функціональних груп, утворюються
продукти різної будови.
• У разі нагрівання до 150 °С а-гідроксикислоти зазнають дегідратування,
утворюючи лактиди:
h3cvííoh но *о н3с^о^о
о^он но"с\Нз о^оАсн3
У кислому середовищі під час кип'ятіння відбувається зворотна реакція.
Конкурентною реакцією є розпад а-гідроксикислот до альдегідів і мура-
шиної кислоти:
R-CH-COOH —* RCHO + НСООН
І
ОН
Для збільшення виходу альдегіду ліпше використовувати ацетати кислот
або їхні естери.
Нагріванням карбонових кислот із третинною гідроксильною групою в а-
положенні до карбоксильної одержують кетони й а,Р-ненасичені кислоти:
(рООН R2
R1^— C-R2 —R!CH=C—COOH + R'CHj- C~R2
OH O
Зі збільшенням довжини карбонових ланцюгів підвищується вихід
ненасичених кислот.
• Бета-гідроксикислоти під час нагрівання утворюють а,р-ненасичені кислоти:
НОСН.-СН7СООН ' » нх=сн-соон
2 2 -Н20 2 ї
• Гамма- і дельна-гідроксикислоти в разі нагрівання й навіть за звичайних
Розділ 20. Гідроксикислоти
571
умов спонтанно дегідратують, утворюючи циклічні внутрішні естери - лактони.
Вивчення рівноваги цих гідроксикислот з відповідними лактонами засвідчило,
що частка лактону в рівноважній суміші досить велика.
❖ Лактони. На підставі номенклатури ШРАС лактони називають за
тривіальною назвою кислоти, використовуючи суфікс -олактон і зазначенням
локантів атомів Карбону, що замкнуті в лактонний цикл, наприклад, Б-глюконо-
1,4-лактон. Назви лактонів аліфатичних гідроксикислот утворюють від відпо-
відного алкану, додаючи суфікс -олід, наприклад: бутанолід, 5-пентанолід.
■ Під час дегідратації Р-гідроксикислот через малу стійкість чотиричлен-
ного циклу лактони не утворюються. Однак іншим шляхом деякі ^-лактони
можна отримати, наприклад, з формальдегіду й кетену:
■ Як уже зазначено, дуже легко синтезувати у- і 8-лактони. Наприклад, із
кумаринової кислоти спонтанно утворюється шестичленний лактон кумарин, що
зумовлює запах свіжоскошеної трави:
■ Полігідроксикислоти легко утворюють у- і 8-лактони, як це показано на
прикладі Б-глюконової кислоти.
■ Відомо лише декілька є-лактонів, які утворюються тільки тоді, коли
геометричні чинники сприяють утворенню циклу.
■ Синтез макроциклічних лактонів з со-гідроксикислот досить утруднений.
Вони утворюються під час нагрівання з сильно розбавлених розчинів кислот; ш-
лактони з дев'ятьма й десятьма атомами Карбону (нонанолід і деканолід) у
малих кількостях трапляються в молоці й молочних продуктах і частково
зумовлюють запах вершкового масла поряд з біацетилом та іншими речовинами,
їх також використовують для ароматизації маргарину.
Відомі лактони зі ще більшими циклами, хоча со-гідроксикислоти й не
циклізуються спонтанно в такі лактони. Наприклад, лактон тібетолід широко
застосовують як духмяну речовину з запахом мускусу і використовують як
стабілізатор запаху парфумів. Тібетолід синтезують циклізацією 15-гідрокси-
пентадеканової кислоти - продукту конденсації моноетилового естеру
адипінової кислоти з 11-ацетоксиундекановою кислотою.
❖ Одержання й властивості бутиролактону. З лактонів найбільше
значення мають у-лактони і, насамперед, бутиролактон. Його вперше отримав
О. Зайцев шляхом відновлення ангідриду янтарної кислоти:
0=С=СН2
о=с-сн2
Кумаринова кислота
Кумарин
572
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
■ Глибше відновлення бутиролактону приводить до тетрагідрофураиу.
Реакція відновлення ангідриду до тетрагідрофураиу однозначно доводить
будову бутиролактону:
Сьогодні бутиролактон у промисловості одержують каталітичним
окисненням бутан-1,4-діолу або шляхом його дегідрування над мідним
каталізатором:
Си, 200 С
СН2ОН СІ^ОН °
у-Бутиролактон
(бутан-4-олід)
■ Наведена далі схема ілюструє використання бутиролактону в різних
органічних синтезах:
СН3(СН2)2СООН
Карбонові кислоти
>ТС(СН2)2СООН
Глутаронітрил
Піролідон
ИН3
р
о
ИаОН
нх
СН3Ш2
НО(СН2)3СОСЖа
Натрій 4-гідроксибутаноат
Х(СН2)3СООН
у-Галогенобутанова
кислота0
С^СНз
#-Метилпіролідон
З реактивом Гриньяра лактони взаємодіють з утворенням гідроксикетонів:
0. о +ям§сі-
+ішйсі —- 0<оМ§С1 —- ^СН2СН2СН2ОН
о
20.6. Окремі представники гідроксикислот
• Гліколева кислота НОСНг-СООН є найпростішою з гідроксикислот. Вона
міститься в незрілому винограді й цукровій тростині.
Будова гліколевої кислоти зрозуміла з її поведінки щодо етиленгліколю й
щавлевої кислоти:
Розділ 20. Гідроксикислоти
573
HO-CHrCHrOH-I5L-HOCH2COOHJ2L-HOOC-COOH
її одержують шляхом гідролізу хлорооцтової кислоти. Застосовують для
протравлення під час фарбування вовни й дублення шкір.
• Молочну кислоту відкрив 1740 p. К. Шеєлє у формі рацемату. Зокрема, (5)-
(+)-2-гідроксипропанова кислота (м'ясомолочна) міститься в м'язовій тканині
людей і тварин; (і?)-{-)-2-гідроксипропанова кислота виявлена в кислому молоці.
Альфа-гідроксипропанову кислоту відновлюють гідроген іодидом у
пропіонову, що доводить будову її скелета, і окиснюють в кетокислоту -
піровиноградну, з чого можна зробити висновок, що спиртова група вторинна:
СН3СН2СООН — Н3С-С-СООН Н3С-С-СООН
он о
Пропіонова кислота 2-Пдроксипропанова кислота Піровиноградна кислота
У промисловості рацемічну 2-гідроксипропанову кислоту одержують
молочнокислим бродінням мальтози, лактози або глюкози.
• Винна кислота у вільному стані й у формі кислої калієвої солі міститься у
винограді й спричинює помутніння вина.
Будова винних кислот визначена перетворенням їх у дихлороянтарну
кислоту шляхом заміни гідроксилів на атом Хлору за допомогою фосфор(У)
хлориду й відновленням гідроген іодидом спочатку в яблучну, а потім у янтарну
кислоти:
1 РС1
ноос-сн-снсоон —-^ноос-сн-сн-соон
он он 2Нг° ¿1 ¿1
Винна кислота 2,3-Дихлоробутандіова, або
а,Р~дихлороянтарна кислота
і і
НІ НІ
НІ
HOOCCRr^HCOOH —- НООС-СН2СН2-СООН
он
Яблучна кислота 2,3-Дихлороянтарна кислота
Винну кислоту можна отримати гідролізом 2,3-дибромобутандіової кислоти
або ж гідроксилюванням фумарової кислоти. Застосовують, головно, у вигляді
солей (тартратів) під час виробництва прохолоджувальних напоїв та випікання
хліба. Сегнетова сіль KNaC4Hi06 -4Н20 дала назву низці речовин, які широко
використовують у п'єзоелектричних пристроях, конденсаторах, електрооптич-
них системах тощо, - сегнетоелектрики.
• Цитринова (лимонна) кислота виділена 1784 p. К. Шеєлє з лимонного соку,
де її вміст досягає 10 %. Вона має три карбоксили й один третинний гідроксил. її
574
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
відновлення дає трикарбалілову кислоту. У разі нагрівання із сульфатною
кислотою утворюється мурашина й ацетондикарбонова кислоти; остання
розпадається під час нагрівання на карбон(ІУ) оксид і ацетон:
Н,СООН СНХООН СН2СООН
сн2
Ацетондикарбонова кислота Цитринова Трикарбалілова кислота
нсоон +• С=о но-с-соон -и* енсоон
:н2соон сн2соон сн2соон
2С02 + СН3СОСН3^
Утворення оксогрупи з відщепленням мурашиної кислоти, як уже зазначено,
типове для а-гідроксикислот, що несуть третинний гідроксил. Це означає, що
центральний атом Карбону, який став карбонільним, ніс і карбоксильну, і
гідроксильну групи.
Будову лимонної кислоти також підтверджено її синтезом з 1,3-дихлоро-
ацетону:
С1СН-С-СН2С1 + 2K.CN ►МС-СН2-С-СН2-СК -НЗ!»
о рн
^снз-си -^г* ноос-сн2-с -СН2-€ООН+Ш+
соон
Розділ 21
оксокислоти
Оксокислотами називають органічні сполуки, що мають у молекулі одно-
часно і карбонільну, і карбоксильну групи.
21.1. Класифікація та номенклатура
21.1.1. Класифікація
ОгАльдегідокислоти
ОСН-СООН
2-Оксооцтова кислота
гліоксилова кислота
сс-Кетокислоти
Н3С-С-СООН
О
2-Оксопропанова кислота
піровиноградна кислота
Альдегідокислоти
^-Альдегідокислоти
ОСН-СН2-СООН
З-Оксопропанова кислота
(формілоцтова кислота, або
малональдегідова кислота)
Кетокислоти
^-Кетокислоти
нх-с-снхоон.
-> II *•
о
З-Оксобутанова кислота
ацетооцтова кислота
^-Альдегідокислоти
ОСН-СН2СН2-СООН
4-Оксобутанова кислота
(формілпропіонова кислота, або
сукцинальдегідова кислота)
^-Кетокислоти
сн3-^ч:н2сн2-<:оон
о
4-Оксопентанова кислота
левуліноеа кислота
21.1.2. Номенклатура IUPAC
За замісною номенклатурою наявність карбонільної групи як замісника
позначають префіксом -оксо, наприклад: СН5СН2СН2СОСН2СООН - 3-оксо-
гексанова кислота.
Для низки представників оксокислот традиційно використовують тривіальні
назви. Приклади таких назв виділені курсивом у таблиці вище; рекомендовані
1993 p. IUPAC назви виділені жирним шрифтом. Також тривіальні назви
використовують для утворених із них ацильних замісників, наприклад, гліоксилоїл,
пірувоїл (від англ. pyruvic acid), ацетоацетил (від англ. acetoacetic acid).
576
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Якщо альдегідна група альдегідокислот перебуває в головному ланцюзі, то її
можна позначати префіксом форміл-. Для альдегідокислот, що є похідними
дикарбонових кислот, можна скласти назви на підставі тривіальних назв
дикарбонових кислот за допомогою суфікса -альдегідова кислота. Приклади таких
назв наведено в дужках у таблиці вище.
21.2. Синтез і властивості оксокислот
21.2.1. Гліоксилова кислота
Гомологічний ряд оксокислот починається з найпростішого представника
гліоксшової кислоти ОСН-СООН. Серед природних джерел вона міститься в
незрілих плодах.
❖ Методи одержання. Гліоксилову кислоту одержують гідролізом
дихлорооцтової кислоти:
сі2сн-соон H2Ö,/> ОСН-СООН
■ Відновлення щавлевої кислоти воднем у момент виділення або
електрохімічне відновлення також приводять до гліоксилової кислоти:
СООН Mg/H,S04
І w 2 4> ОСН-СООН
СООН
■ Також гліоксилову кислоту синтезують окисненням гліколевої кислоти, а
також етиленгліколю, наприклад, нітратною кислотою:
НОСНгСН2ОН-t^HOCH^OOH -t^OHC-COOH
Етиленгліколь Гліколева кислота Гліоксилова кислота
❖ Хімічні властивості. Внаслідок негативного І-ефекту карбоксильної
групи гліоксилова кислота утворює стійкий гідрат: НООС-СН(ОН)2. Як альдегід
вона взаємодіє з гідросульфітом, амоніачним розчином арґентум оксиду,
ціанідною кислотою, гідроксиламіном, а як кислота дає солі, естери тощо.
Оскільки в гліоксиловій кислоті нема атомів Гідрогену в а-положенні щодо
карбоксильної групи то це дає змогу їй вступати в реакцію Канніццаро:
2СН0 _кон_ COOK + СН2ОН
СООН COOK СООН
Етандіова
кислота
2-Гідроксиетанова
кислота
Розділ 21. Оксокислоти
577
21.2.2. Піровиноградна кислота
Найпростішою а-кетокислотою є 2-оксопропанова, або піровиноградна,
кислота - найважливіша проміжна речовина в метаболізмі вуглеводів і білків.
❖ Методи одержання. Уперше її синтезував Й. Берцеліус дегідратацією
виноградної кислоти, й саме за цією реакцією її синтезують тепер:
нооссрнс^нсоон —
но он " :
Виноградна кислота
(рн^нсоон-т^сн^-соон
он он 2 он
СНз^СООН
Піровиноградну кислоту, як і інші а-кетокислоти, можна отримати й через
нітрил:
СН3СОС1 + КСИ -=Ш- СН3(^ -04 ^^нСНз^ -СООН + NH4+
Також а-кетокислоти зручно одержувати окисненням а-гідроксикислот або
гідролізом а,а-дихлорозаміщених карбонових кислот.
❖ Хімічні властивості. Як оксосполука, вона дає оксим і гідразон, а як
кислота - звичайні похідні карбоксильної групи. Солі піровиноградної кислоти
називають піруватами.
Взаємний вплив функціональних груп позначається на хімічній поведінці
піровиноградної кислоти. Вона значно сильніша від оцтової кислоти. Відновлює
амоніачний розчин арґентум оксиду, окиснюючись у цьому разі до оцтової
кислоти й карбон (IV) оксиду. Здатна зазнавати декарбоксилювання під дією
теплої розведеної сульфатної кислоти й декарбонілювання під час нагрівання з
концентрованою сульфатною кислотою:
Н3С-С-СООН
О
Po3b.h2so4> СНздН0'+
Конц. H,S04
г—*—* СН3СООН + CO
У процесі спиртового бродіння піровиноградна кислота, як побічний
продукт, під дією ензиму піруватдекарбоксилази здатна відщеплювати карбон
(IV) оксид уже за кімнатної температури і перетворюватись в ацетальдегід.
21.2.3. Ацетооцтова кислота
Гомологічний ряд Р-кетокислот починається з ацетооцтової, або 3-оксобу-
танової, кислоти.
578
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
❖ Методи одержання. Ацетооцтова кислота трапляється в сечі хворих на
цукровий діабет, її можна отримати обережним омиленням її естерів або
гідратацією дикетену:
Н
н2с=сч ,с=о + н2о СН3СОСН2СООН
о
Дикетен Ацетооцтова кислота
❖ Хімічні властивості. Ацетооцтова кислота дуже нестійка й легко зазнає
декарбоксилювання до ацетону:
Нзсу^г^° -со2> Н3СЧ^СН2 Н3С-С-СН3
сО (То он 0
чн
Навпаки, етиловий естер 3-оксобутанової кислоти або, коротше, ацетооцтовий
естер стійкий і відіграє важливу роль в органічному синтезі. Його застосовують для
одержання різних кислот, кетонів та інших сполук.
21.2.4. Левулінова кислота
❖ Будову левулінової (4-оксопентанової) кислоти доведено за допомогою
синтезу через натрієву похідну естерів Р-кетонокислот:
н3с-с=сн-соос2н5 + сісн2-соос2н5 —г нх-с-сн-соос2н5
3 Т 2 5 2 2 5 . МаСІ 3 и | 2 5.2СНОН
ОЖ О СН^СООС2Н5 2 5
—- НзС-с-сн-соон н3с-с-сн2-снгсоон
о сн^-соон 2 о
❖ Одержання. Левулінову кислоту синтезують зазвичай дією на вуглеводи
- фруктозу, глюкозу, сахарозу й крохмаль - розведених неорганічних кислот.
Походження назви цієї кислоти випливає зі старої назви фруктози, яку з огляду
на лівий кут обертання називали левулозою, на відміну від глюкози - декстрози,
що має правий кут обертання (від лат. Іаечо - ліво й (Лехїго - право).
❖ Хімічні властивості. Левулінова кислота дає два ряди похідних - за
карбоксилом і кетогрупою. Як кислота, вона утворює солі, естери, хлороангідри-
ди, а як кетосполука - реагує з гідразином, гідроксиламіном, ціанідною кислотою.
Під час нагрівання левулінова кислота перетворюється в суміш ізомерних
лактонів:
Розділ 21. Оксокислоти
579
СН3
н2с'%
нх^.он
н& -ОН
■ Н20
У лабораторній практиці левулінову кислоту використовують для розщеплен-
ня оксимів і гідразонів кетонів та їхнього кількісного визначення.
21.3. Синтез і властивості ацетооцтового естеру
213.1. Методи синтезу
❖ Есшерифікація дикетену. Ацетооцтовий естер (етил-3-оксобутаноат)
одержують дією спиртів на дикетен, що спонтанно утворюється під час
зберігання кетену:
СН
' 5с=о + с,н5он —► н,с-с-сн—соос,не
н2с=с;
о
о
-2і "5
❖ Естерна конденсація Кляйзена. Головний метод одержання
ацетооцтового естеру полягає в автоконденсації етилацетату під дією натрій
етилату. Механізм його утворення такий. Під впливом натрій етилату етилацетат
утворює карбаніон: с
н2с-сч
н
С2Н5(Жа
ос2н;С2Н5°н
н2с-сч
ос2н5
N3
Як сильний нуклеофіл, аніон атакує іншу молекулу етилацетату за карбо-
нільним атомом:
5-
н3с-с-ос2н5 + н2с-с
ос2н5
н3с-(р-н2с<
0С2Н5 ОС2Н5
580
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Цей аддукт завдяки напівацетальному естерному залишку й рухливому атомі
Гідрогену, що перебуває в а-положенні щодо етоксикарбонільної групи,
відщеплює молекулу етанолу, утворюючи амбідентний аніон, будову якого можна
відобразити такими резонансними структурами:
н,с-с-сн-с-ос,н-— н3с-с=сн-с-ос2н5
З її П 2 5 Зі. її 25
0 0 0 0
на
0 н
1 і -с,н5он
н3с-с-нс-с-ос2н5 2 5 '
ос2н5 о
—► Н3С-С-СН2СООС2Н5
о
Ацетооцтовий естер
Натрієву похідну аніону називають натрійацетооцтовим естером. Значний
внесок у вивчення синтезу ацетооцтового естеру зробив Л. Кляйзен, чиїм ім'ям і
названо цю реакцію.
■ Як і треба було сподіватися, карбонільним компонентом у разі естерної
конденсації можуть бути будь-які естери як одно-, так і двоосновних кислот:
СНзСНзСНзСООед + СНзСОСОД Сг^СНзСНзС-СНзСОІХзНз + СДОН
О
С,Н50№
с2н5о-с-с-ос2н5 + н3с-с-ос2н5^—- с2н5о-с-с-сн2- с-ос2н5
о о о о о о
■ Зазначимо, що найсильнішим ацилювальним агентом естерної конденса-
ції є диетилоксалат.
Незважаючи на те, що за ацилювальною здатністю вони утворюють такий
ряд:
•<р Ф5- Ф5" Ф5"
6+0-0^+ > Н-С>н > СН3-*С,5+
С2^9У \9.с2я5 \!ос2н5 Сіос2н5 5
у реакції естерної конденсації варто використовувати похідні тільки одного
естеру. У випадку двох різних естерів утворюється суміш чотирьох різних
естерів Р-оксокислот, розділення яких є досить складним завданням.
■ Крім того, у разі естерної конденсації двох різних двоосновних кислот,
згідно з реакцією Дікмана, відбувається реакція ацилювання з утворенням
циклічних сполук:
Розділ 21. Оксокислоти
581
2Н5 н7с-соос,н,
2 \ 2 5
соос2н5
+ сн2
ос2н5 н2с-соос2н5
С2Н5СОЫа
соос2н5
■ Естери двоосновних кислот з довгим ланцюгом карбонових атомів здатні
самоконденсувсипися. Під час циклізацій, як завжди, легко синтезуються лише
п'яти- і шестичленні цикли:
Усі ці приклади свідчать, що конденсація пов'язана з наявністю в естері, що
є другим компонентом реакції, рухливого атома Гідрогену в ос-положенні.
Власне кажучи, реакція є ацилюванням одним естером іншого за а-карбоновим
атомом.
■ Конденсація з карбонільними сполуками. З естерами можуть конденсу-
ватись будь-які сполуки, що містять рухливий атом Гідрогену, тобто який
перебуває в а-положенні щодо карбонільної групи, наприклад, ацетон. Ацетон
дає похідні 1,3-дикетонів:
Отже, метод естерної конденсації - один із потужних синтетичних методів
побудови скелетів органічних молекул.
У хімії оксокислот, особливо (З-оксокислот та їхніх функціональних
похідних, величезну роль відіграє кето-енольна таутомерія^ то розглянемо
питання таутомерії саме на їхньому прикладі.
Незважаючи на те, що етил-3-оксобутаноат синтезований А. Гейтером ще
1863 р., потрібно було 50 років для з'ясування будови й особливостей його
поведінки. Річ у тому, що ацетооцтовий естер - це рівноважна суміш кетонної й
енольної форм, яка є класичним прикладом таутомерної системи. Метиленова
ланка в естері настільки активована карбонільною й естерною групами, що
енольна форма тут значно стійкіша, ніж у кетонах, де вона становить,
наприклад, в ацетоні усього 2,5-10"4 %. Особливо великий внесок в
ацидифікацію атомів Гідрогену метиленової групи робить кетофункція, яка за
дією на п'ять-вісім порядків перевищує естерну групу.
гедоос-соос^Нз+СН3СОСН3
N3
Іа
213.2. Таутомерія
582
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Припускали, що в естері існує рухлива рівновага між двома таутомерними
формами:
-я -р
н3с-с-сн-с. ^ н,с-с=сн-с
сн-с zzr. н3с-с
ö н, ос2н5 ін_і ос2н5
Двоїста природа ацетооцтового естеру спочатку виявлена в якісних
реакціях. Наприклад, він дає всі позитивні проби на карбонільну групу: утворює
гідросульфітну похідну, гідроксинітрил, гідразон, оксим (Франкланд1 і Дуппа,
1865 р.). З іншого боку, він реагує з ферум (III) хлоридом подібно до фенолів; з
одним еквівалентом натрію утворює натрієву похідну, виділяючи водень і,
нарешті, з купрум(ІІ) ацетатом дає сіль Си(СбН90з)2.
У 1911 р. Л. Кнорр2 провів блискучий експеримент із розділення кетонної й
енольної форм естеру. Він з'ясував, що за низьких температур рівновага між
таутомерними формами практично заморожена. З охолодженого ацетооцтового
естеру Л. Кнорр виділив кристалічну речовину з температурою плавлення -39 °С,
що є його кетоформою і не реагує з ферум (III) хлоридом, купрум (II) ацетатом.
Із маточника за -78 °С вдалося виділити сполуку, яка виявилася чистим енолом,
що й підтверджено якісними реакціями на еноли.
Взаємне перетворення таутомерних форм помітно каталізоване кислотами й
основами, навіть такими, як скло. Кислотно-основний каталіз - один із
численних прикладів прискорення реакцій під впливом іонів Гідрогену й
гідроксилу. Причину цього явища можна зрозуміти з таких схем.
■ Каталіз лугами відбувається в такий спосіб:
сн3-с^нчюос2н5--— СН3-С=СНСООС2Н5 СНГС=СНС00С2Н5
Зворотне перетворення виглядає так:
СНгС=СНС00С2Н5 СНгС=СНСООС2Н5 -j^*CH3-C-CHCOOC2H5
9_.он Woh " он
■ Каталіз кислотами відбувається за таким механізмом:
1 Франкланд Е. (Ргапкіапсі Е.) (1825-1899) - англійський хімік. Синтезував органічні похідні
металів. Впровадив термін "металоорганічні сполуки". Розробив методи синтезу цинк алкілів і
їхнього використання в органічному синтезі. Вивчав ацетооцтовий естер.
2 Кнорр Л. (Кпогг Ь.) (1859-1921) - німецький хімік. Головні праці присвячені кето-енольній
таутомерії. Зробив вагомий внесок у вивчення структури алкалоїдів.
Розділ 27. Оксокислоти
583
СН,-С-СНСООС,Нс
сн3-±с-снсоос2н:
І
2П5 _Н+
ch3(j:=chcooc2h5
Зворотне перетворення:
+
снг р=снсоос2н5-
Того ж року К. Мейєр одержав чисту енольну форму ацетооцтового естеру
шляхом перегонки суміші в глибокому вакуумі у кварцовому посуді. Остання
обставина досить важлива, оскільки звичайне скло має достатню основність для
прискорення усталення рівноваги. Несподівано виявилося, що енол має більшу
леткість порівнянні з кетонною формою. Цю аномалію пояснюють наявністю в
енолі внутрішньомолекулярного водневого зв'язку, що унеможливлює
асоціацію з міжмолекулярних водневих зв'язків:
Справді, в ІЧ-спектрі ацетооцтового естеру виявляють смуги поглинання,
характерні для карбонільних груп кетонів - 1 715 см"1, і їхніх естерів - 1 740 см"1,
а також спряженого етиленового зв'язку - 1 630 см'1 і карбонільної групи,
з'єднаної внутрішньомолекулярним водневим зв'язком, - 1 650 см'1. У ПМР-
спектрах видно сигнали протонів метиленової групи (6,15-6,35 м.ч.), а також
сигнали вінільного протона (5,5 м.ч.) і протона гідроксильної групи.
На підставі порівняння показників заломлення рівноважної суміші (п™ =
1,4232), чистого енолу (п™ = 1,4480) і чистої кетоформи (я])0 = 1,4225) Л. Кнорр
дійшов висновку, що вміст енолу в суміші невеликий, що експериментально
підтвердив К. Мейєр. К. Мейєр не без підстави вважав, що в рівноважній суміші
реакція знебарвлення брому енолом відбуватиметься швидше, ніж зсув рівноваги.
З огляду на цю особливість, він обробив ацетооцтовий естер надлишком брому.
Залишок брому, який швидко не прореагував, зв'язав ß-нафтолом. У разі
/;
не
\
,с—о
н
С=0"
1 Мейєр К. (Meyer К.Н.) (1883-1952) - німецький хімік. Відкрив декілька реакцій азосполучення.
Організував виробництво формаліну з оксиду (II) карбону і фенолу з хлоробензену.
584
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
підкислення й додавання натрій іодиду а-бромокетон зазнає відновлення гідроген
іодидом. Титрування іоду, що виділяється, дає змогу розрахувати вміст енолу:
Вг
н3с-с=сн-соос2н5
ОН
Вг,
Н,С-С-СНВг-СООС,Н5
З п 2 5
О
НХ-С-СНВг-СООС,Н<+ 2 НІ"
3 II 1 ъ
н3с-с-сн2соос2н5+ НВг +12
о
Згідно з експериментальними даними, вміст енолу в ацетооцтовому естері не
перевищує 8 %. У розчинах рівновага може зміщуватися в той або інший бік. У
полярних розчинниках полярна кето-форма буде ліпше зазнавати сольватування
порівняно з енолом, тому рівновага зміщується у бік кетосполуки. У неполярних
розчинниках рівновага, навпаки, зміщується у бік збільшення концентрації енолу
(табл. 21.1). Рівновага також значно залежить і від концентрації розчину.
Таблиця 21.1
Вміст енольної форми в ацетооцтовому естері в різних розчинниках
Розчинник
Вода
Метанол
Етанол
Ацетон
Бензен
Діетиловий етер
Гексан
Вміст енолу, %
0,4
6,9
12,0
7,3
16,2
27,1
46,4
Ацетооцтовий естер виявляє кето-енольну (прототропну) таутомерію,
аналогічну до діетилового естеру малонової кислоти й 1,3-дикетонів.
Найбільший вміст енолу й висока кислотність характерні для 1,3-дикетонів
(80,4 % і рКа = 8,8), що виявляється у відтягуванні електронної густини
карбонільною групою — СНг-С—СН..
II4)
О'
Позитивний індукційний ефект метальної групи незначний. Менша
рухливість (кислотність) атомів Гідрогену метиленової ланки в ацетооцтовому
естері й діетилмалонаті пов'язана із сильним позитивним мезомерним ефектом
етоксигрупи:
-сн2-с^6с2н5
(У
Це підтверджено вмістом енолів і показниками рКа, які становлять для
ацетооцтового естеру 8 % і 10,7, а для діетилмалонату, відповідно, 0,77-10"2 % і
13,3. Порівняно слабка С-Н кислотність потребує використання сильних основ
типу С2Н50№.
Розділ 21. Оксокислоти
585
Хоча енольна форма енергетично менш вигідна порівняно з кетонною, проте
система спряжених кратних зв'язків і наявність водневого зв'язку приводять до
побудови циклічної структури енолу з певним виграшем енергії. Дня
натрійацетооцгового естеру виграш енергії ще значніший, оскільки катіон натрію
зв'язується координаційно з двома атомами Оксигену, на яких зосереджена висока
електронна густина:
н3с^с^с ^ос2н5
21.3.3. Реакції, що відбуваються за кетонним типом
Унаслідок кето-енольної таутомерії ацетооцтовий естер виявляє властивості
як енолу, так і кетону й утворює С-похідні кетонної форми й О-похідні енольної.
Однак зазначимо, що взаємні переходи в разі таутомерії відбуваються легко,
оскільки енергія активації таких переходів досить незначна (8 кДж/моль), тому
часто немає сенсу говорити, у якій формі реагує естер. Наприклад, утворення
ціангідрину, з одного боку, можна тлумачити як реакцію нуклеофільного
приєднання до Карбону карбонільної групи:
сн3-с—сн2соос2н5 є*0]- сн3-с—сн2соос2н5,
2 он
а з іншого, - як нуклеофільне приєднання до ненасиченої енольної сполуки:
СЫ
і а.. н2о
СЫ
І
сн3-с-сн-соос2н5+ск—-н3с-с-сн-соос2н5-^сн3-с-сн2-соос2н5
он он он
Проте умовно можна виділити реакції, що відбуваються за кетонним чи
енольним таутомером.
❖ Дія фенілгідразину приводить до фенілгідразону, що відщеплює
молекулу спирту й перетворюється в метилфенілпіразолон:
" 19™Р* +СДОН
а^сооед 2 6 4
СНзСНМ-ІМНОД
5\
О
3-Метил-№(|)сніл-5-піразолон
586
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Метилювання 3-метил-1-феніл-5-піразолону приводить до антипірину -
відомого жарознижувального засобу:
НзС\*Н сил H3SrN;
рн3
к-с6н5 і>-с6н5
он" ^
о о
Антипірин
З нього синтезують широко вживані жарознижувальні препарати з
антиневралгічною дією - амідопірин (2,3-диметил-4-диметиламіно-1-фенілпіра-
золон-5) і анальгін (2,3-диметил-4-(Н-метил-Н-натрій сульфометиламіно)-1-
фенілпіразолон-5).
рн3 рн3
(Н3С)2Л ^зЗСН^А^
3 2 о сн3 о
Амідопірин Анальгін
■ Аналогічно відбувається взаємодія й з іншими похідними гідразину:
Піразолони О
сас=о
13?"~ +NH,NHR
CHjCOOCjHs 2
CH3(p=N-NHR
CH2COOC2H5
У цих випадках звичайна реакція кетону ускладнена другою фазою -
циклізацією в Л/'-алкілпіразолон.
❖ Реакція з гідрокс тіаміном. Реакція ацетооцтового естеру з гідрокси-
ламіном приводить до оксиму, який подібно до гідразону перетворюється в
циклічну сполуку:
СН3 £СН2СООС2Н5^^ СН3^СН2СООС2Н5 СН^ -€|Н2
О N04 £=0
Метилізоксазолон
❖ Із карбамідом ацетооцтовий естер дає шестичленний гетероцикл:
9Н2 Н2К -С2Н5ОН >—N
СООС2Н5 НО
Метилдигідроксипіримідин
Розділ 21. Оксокислоти
587
❖ Приєднання натрій гідросульфіту приводить до типового кристалічного
гідросульфітного похідного:
9н
СНгССН2СООС2Н5 + ЫаН803—- СНг(|:СН2СООС2Н5
О 803Ыа
21.3.4. Реакції, ир відбуваються за енольним типом
❖ Бромування. Швидкість, з якою енол зазнає бромування, цілком
зрозуміла: електрофільний атом Брому швидко атакує подвійний карбон-
карбоновий зв'язок, даючи фенолу легко перетворитися в кетон унаслідок
відщеплення протона:
_ Дуже швидко
сн3с-сн2соос2н5сн3с=снсоос2н5+ Вг2 _Вг »
6 он
Вг
—-н3с-£-снсоос2н5=^= сн3£-9НС00С2н5
+0Н
н+ О Вг
❖ Ацилювання хлороангідридами кислот у піридині приводить до естерів
енолів:
СНгС=СНСООС2Н5 + СН3СС1 -СН3<^=СНСООС2Н5 + неї
он О ососн3
❖ Одержання енолятів. Під дією на ацетооцтовий естер натрій або калій
метилату або етилату з огляду на те, що кислотність енолу значно вища від
кислотності спиртів, легко утворюються відповідні еноляти:
н3с^х<сн ос2н5
сн3-9ГсоосЛ^- £ ~;1
Така структура еноляту однозначно підтверджена його ІЧ-спектром, де є
тільки одна смуга поглинання спряженої системи аніона, і нема смуг для оксо- й
енольної груп.
■ Взаємодія з солями сі-металів приводить до комплексних енолятів,
наприклад:
588
Чирва В.Я., Ярмолюк CM., Толкачова Н.В., Земляков О.Є. Органічна хімія
°W° (СНзСОО^Си РеС1з / 'Fe^, ,Щ
A " ^gçHcc^H,— „уц>£
У хелатах атом металу ковалентно зв'язаний з атомом Оксигену енолу й
координаційно - з карбонільним атомом Оксигену етоксикарбонільної групи.
❖ Дія фосфор (V) хлориду приводить до заміщення ОН-групи на галоген:
СН3С=СНСООС2Н5 + РС15 СН3(р=СНСООС2Н5
ОН СІ
❖ Реакція Міхаєля\ відкрита 1887 p., є приєднанням сполук з реакційно-
здатною метиленовою групою, що діє як донор електронів, до речовин зі
спряженими кратними зв'язками (дієнофілами). Реакція відбувається за
наявності основ. Як С-Н-кислоти можуть діяти діетилмалоновий і ацетооцтовий
естери, Р-дикетони. Дієнофілами можуть бути <х,Р-ненасичені альдегіди й
кетони, естери, нітрили й аміди. Реакція починається з утворення карбаніону, що
виникає під дією основи:
CH3-CO-ÇH-COOC2H5 .^н'он" Н3С-СО-СН~СООС2Н5
H
Далі карбаніон атакує атом Карбону подвійного зв'язку з найменшою
електронною густиною. Аніон, що утворився, знову зазнає стабілізування
протоном з реакційного середовища:
снгсо-нс-соос2н5 + сн3-сн=сн-соос2н5
но сн3-со-нс-соос2н5
-он-
СН3~СН-СНгСООС2Н5
Реакція Міхаеля - одна з найважливіших реакцій в органічному синтезі для
одержання поліфункціональних сполук.
• Реакція з нітратною кислотою має такий механізм. На першій стадії
відбувається нітрозування алкілацетооцтового естеру:
№Ш2 + СН3СООН НШ2 + СН3СОО№
Н-0-№0+Н+=*= Н-0+-№Ю
і
Н
1 Міхаєль А. (Michael А.) (1853-1942) - американський хімік. Головні праці присвячені хімії сполук,
що мають реакційноздатні метиленові групи. Синтезував арилглікозиди. Має дві іменні реакції.
Розділ 21. Оксокислоти
589
н3с-с=с-с-ос2н5 + н-о+^=о—-Н3С-С-СК-СООС2Н5+Н20 + Н+(Н30)
ОН О N=0
Нітрозоестер, що утворився під час кислотного гідролізі, легко зазнає
декарбоксилювання до монооксиму:
СНгС—С—Я
3 II II
О N04
який у кислому середовищі перетворюється в дикетон: ^Г^[—^—^
о о
21.3.5. Синтези на основі ацетооцтового естеру
Під час взаємодії натрій ацетооцтового естеру з первинними або вторин-
ними (однак не третинними) галогеноалканами відбувається С-алкілування за
механізмом із утворенням алкілацетооцтового естеру. У випадку повторення
подібної послідовності операцій отримують діалкілпохідні.
• У разі кепгонного розщеплення розведеними розчинами лугів, розведеною
сульфатною або фосфатною кислотою з алкіл- і діалкілпохідних як основні
продукти реакції утворюються відповідні кислоти, які зазнають декарбокси-
лювання з утворенням кетонів. Декарбоксилювання дизаміщеної ацетооцтової
кислоти відбувається ще легше, ніж подібних аналогів малонової кислоти.
Процес може відбуватися навіть до підкислення суміші після омилення естерів.
Синтез кетонів за допомогою ацетооцтового естеру приводить до похідних
ацетону, у яких один або два атоми Гідрогену при одному атомі Карбону
заміщені на алкільні групи:
СН3ССН2СООС2Н5?2?Жа [СН3СОСНСООС2Н5]-№+ -М
41
О
сн
^-ССООС2Н5
С2Н,ОЫа
- сн^-срісоос^
о я
Алкілацстооцтовий естер
Я'Х
[СН3СОСЯСООС2Н5]^а+
но-
о а
но-1
сн3с-ссоо-
н2о, н+
О Я
Діалкілацетооцтовий естер
СН3С-^(
соон
■ со,
О к
СНХСНЯЯ"
3ІІ
о
Дизаміщений ацетон
сн.
СНз^НСОО-
о я
І Н2°'н*
(^НСООН
о я
сн7ссн0гч
3ІІ 2
о
Монозаміщений ацетон
590 Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
• Кислотне розщеплення алкіл- і діалкілпохідних естеру концентрованими
розчинами лугів відбувається як зворотна конденсація за Кляйзеном. Як
основний продукт реакції утворюються лінійні або розгалужені монокарбонові
кислоти:
сн3со-сн-соос2н5 сн3сос-соос2н5
ИаОН (конц)
№ОН (конц)
СН3СО(Жа + ІІСН2СООНа СН3СООКа + ІІ-СН-СООШ + С2Н5ОН
+ С2Н5ОН Я
Отже, у разі відповідного підбору реагентів цим методом можна одержати
чимало різноманітних кетонів і кислот.
21.3.6. Ацетооцтовий естер як амбідентний
нуклеофіл
Еноляти цієї сполуки є типовим прикладом так званих амбідентних
реагентів, у цьому випадку - амбідентних нуклеофілів. Такі нуклеЬфіли мають
два зв'язані між собою завдяки мезомерному ефекту реакційні центри. Наявність
спряження приводить до того, що такі системи реагують як єдине ціле (як одна
функціональна група), однак завдяки наявності двох нуклеофільних центрів їхня
взаємодія з електрофілами не є однозначною.
Двоїсту реакційну здатність подібних реагентів можна зрозуміти,
розглядаючи відповідні резонансні структури, у цьому випадку граничні
структури енолятів етил-3-оксобутаноату:
н3с-с=сн-с-ос2н5 — н3с-с-сн=с-ос2н5 —- н3с-с-сн-с-ос2н5 =
0"~ + О О+О" 0+6
м м м
= н3с-с-сн-с-ос2н5
о"" "'"■■о
м
У них надлишкова електронна густина і негативний заряд локалізовані на
атомах Карбону й Оксигену. Отже, атака електрофіла може бути спрямована на
атом як Карбону, так і Оксигену.
Логічно припустити, що внаслідок взаємодії амбідентного нуклеофіла з різними
електрофілами буде утворюватись суміш продуктів приблизно одного складу,
причому співвідношення продуктів визначатиме відносна реакційна здатність
Розділ 21. Оксокислоти
591
нуклеофільних центрів. На практиці, однак, співвідношення між продуктами
взаємодії варіює в широких межах і залежить як від природи реагентів (електрофіла й
катіона металу в еноляті), так і від умов, головно, від природи розчинника.
Це явище можна пояснити за допомогою підходу, що ґрунтується на так
званій теорії твердих і м'яких кислот та основ (див. розділ 4). Чим жорсткішим
є електрофільний реагент, тим сильніша тенденція до утворення зв'язку з
атомом Оксигену в еноляті як із жорсткішим порівняно з атомом Карбону
нуклеофільним центром.
Жорсткість електрофільного реагенту зростає зі збільшенням електронега-
тивності відхідної групи, зв'язаної з елекгрофільним центром, - електроноакцепторна
група, що здатна ефективно відтягувати електронну густину від електрофільного
центру, збільшує на ньому позитивний заряд і зменшує його здатність до поляризації.
З огляду на це стає зрозумілим порядок зміни вибірковості алкілування в низці
однотипних реагентів Я-Х, що мають різні за електронегативністю відхідні групи:
О О
І-<Вг<С1-<р-< Н3С-^-ОГ< РзОБ-СГ
О О
Збільшення електронегативності відхідної групи X-
Якщо алкілування натрій еноляту ацетооцтового естеру в діетиловому етері
проводити алкіліодидами, то реакція відбувається переважно по м'якому центру
- атому Карбону:
СН3
Н3С-СТСН7(р--ОС2Н5 Н3С-С-СН-С^ОС2Н5 (97о/о)э
0'хт +'° О О
а в разі використання алкілтрифлуорометансульфонатів за тих же умов
селективно зазнає алкілування жорсткий атом Оксигену:
СНзОБОХР,
Н3С-С-СН-(р-0С2Н5 Н3С-С=СН-£-0С2Н5 (100%).
" " 0СНз О
Очевидно, що вплинути на жорсткість електрофільного реагенту можна,
змінюючи природу замісника Я, наприклад, уводячи в нього електроноакцепторні
групи. Наприклад, алкілування галогенометиловими етерами (И'ОСН2На1) відбу-
вається практично винятково за атомом Оксигену, навіть якщо використовують
іодометилові етери. Алільний замісник, що містить подвійний зв'язок у спря-
женому положенні щодо електрофільного центру, легко зазнає поляризування,
тому алілування відбувається переважно за атомом Карбону, навіть у тих
випадках, коли замісник є групою з високою електронегативністю.
592
Чирва В.Я., Ярмфыок СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Зазначимо, що ацилювання енолятів ацетооцтового естеру повинно відбу-
ватися за атомом Оксигену, оскільки карбонільний атом Карбону в ацилю-
вальних реагентах, таких як ангідриди й галогеноангідриди карбонових кислот, є
жорсткою кислотою Льюїса. Насправді у разі використання енолятів лужних і
лужноземельних металів у таких розчинниках, як вуглеводні, бензен і етери,
відбувається переважно С-ацилювання, причому схильність до С-ацилювання
залежить від природи металу й збільшується в ряді
Сб+ < Яг/ < К+ < №+ < Ьі+ < М2+,
де М2"1" - катіон лужноземельного металу.
Цей ефект пояснюють екрануванням атома Оксигену в енолят-аніоні,
пов'язаному електронною парою з катіоном металу. Очевидно, що ступінь
асоціювання йонної пари й, відповідно, ступінь блокування атома Оксигену як
жорсткої основи Льюїса залежить від жорсткості катіона металу, що
збільшується від цезію до літію, й максимальна для катіонів лужноземельних
металів, здатних утворювати з атомом Оксигену ковалентний зв'язок.,
"Звичайні" розчинники не спроможні сольватувати катіони металів або
енолят-аніони й практично не впливають на ступінь асоціювання йонних пар.
Водночас використання полярних апротонних розчинників, таких як
диметилформамід, диметилсульфоксид, які можуть специфічно сольватувати
катіони металів, взаємодіючи з ними за типом жорстка кислота-жорстка основа,
зменшує асоціацію йонів і збільшує доступність атома Оксигену в еноляті. Це
приводить до зростання частки О-ацилювання або О-алкілування. Наприклад, у
разі алкілування натрієвого еноляту ацетооцтового естеру діетилсульфатом у
діоксані переважно утворюється продукт С-алкілування, тоді як у
високополярному гексаметилфосфотриаміді переважає О-алкілування:
О О
(снлбо, м и
^^н3с-с-сн-с-ос2н5
^2П5 (85%)
н3с-с-сн-с-ос2н5
н3с-с=сн-?-ос2н5
ОС2Н5 (83%)
Збільшення ступеня дисоціації йонних пар спостерігають і в гідрокси-
ловмісних розчинниках (вода, спирти тощо), які здатні специфічно сольватувати
енолят-аніони завдяки утворенню водневих зв'язків з атомом Оксигену еноляту.
Незважаючи на збільшення відстані між атомом Оксигену і катіоном металу або
навіть на повну дисоціацію, взаємодія з електрофілами відбувається за атомом
Карбону, оскільки за цих умов атом Оксигену блокований молекулами
розчинника, що утворюють з ним досить міцні зв'язки.
Розділ 22
АМІНОКИСЛОТИ
До амінокислот належать сполуки, що мають у складі карбоксильну й
амінну групи.
22.1. Класифікація, номенклатура й будова
22.1.1. Класифікація
За взаємним розташуванням аміно- і карбоксильної груп
а-Амінокислоти ^-Амінокислоти ^-Амінокислоти
Ш2-СН-СООН
^н №12СН2СН2СООН КН2(СН2)5СООН
2-Амінопропанова кислота З-Амінопропанова кислота 6-Аміногексанова кислота
а-аланін $-аланін (й-амінокапронова кислота
За природою бічного ланцюга
Аліфатичні амінокислоти Ароматичні амінокислоти Гетероі^иклічні амінокислоти
н2к-сн-соон н2*і-сн-соон н^-сн-соон
НзсінСНз <0^-СН2 Т^Н,
2-Аміно-З-метилбутанова 2-Аміно-3-фенілпропанова
кислота
кислота
валін , .
фенілаланін
N
Гістидин
За кислотно-основними властивостями
Нейтральні амінокислоти Кислі амінокислоти Основні амінокислоти
НН2-СН-СООН КН2-СН-СООН
ш,снгсоон ^сн.соон !снЛнн2
Амінооцтова кислота 2-Амінопентандіова кислота 2,6-Діаміногексанова кислота
гліцин глутамінова кислота лізин
594
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
■ 3 біохімічного погляду виділяють 20 основних протеїногенних або, інакше,
тих, що генетично кодовані, амінокислот (табл. 22.1). Інші амінокислоти
зачисляють до незвичайних (рідкісних) амінокислот. Серед них багато сполук
можна розглядати як продукти хімічної модифікації (метилювання, гідрокси-
лювання, декарбоксилювання й дезамінування тощо) основних амінокислот,
наприклад:
Н2М-СН-СОО
НООС-С-СН,
II 2
сн2
у-Метиленглутамінова
кислота
н2м-сн-соон
(Н3С)2С8Н
Р-Тіовалін
но-
I Н2И-СН-СС НгИ-СН-СО НО
| І
СН7 Н2К-(СН2)3
І
3,5-Диіодотирозин
соо
Орнітин
Н2К(СН2)3СООН НзС-ИНСНгС
ООН
у-Аміномасляна
кислота
4-Гідроксипролін
н-,с-кн-сн-соо
Нзсснснз
#-Метил валін
Саркозин
■ Виділяють також групу незамінних амінокислот, які не синтезуються в
організмі людини й повинні надходити з їжею (назви таких амінокислот виділені
жирним шрифтом у табл. 22.1).
Тривіальні назви протеїногенних амінокислот
Таблиця 22.1
Назва
Структура
Умовне
позначення
1
2
3
Аланін
н2к-сн-соон
сн3
Аіа
Аргінін
Н2Н-СН-СООН
™ |
С-КН(СН2)3 ч
Н2К
Аспарагін
Н2М-СН-СООН
"с-сн2
н2м
Розділ 22. Амінокислоти
Продовження табл. 22.1
—n 1
595
Аспарагінова
кислота
Валін
Гістидин
Гліцин
Глутамін
Глутамінова
кислота
Ізолейцин
Лейцин
Лізин
Метіонін
Пролін
Asp
Val
His
Gly
Gin
Glu
He
Leu
Lys
Met
Pro
596
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Закінчення табл. 22.1
1
2
3
Серин
Бег
НгИ-СН-СООН
^ |
но-сн2
Тирозин
н2ы-сн-соон
Туг
но~С5~сн2
Треонін
Н2Ы-СН-СООН
1
Тпг
НО-СНСНз
Триптофан
н2ы-сн-соон
1 1
Тгр
Фенілаланін
РЬе
НгЫ-СН-СООН
1
0"снг
Цистеїн
н2м-сн-соон
1
Н8-СН2
22.1.2. Номенклатура ШРАС
■ За замісниковою номенклатурою амінокислоти розглядають як заміщені
карбонові кислоти. Приклади таких назв наведені вище.
■ Для великої групи а-амінокислот природного походження прийняті
тривіальні назви, наведені в табл. 22.1.
■ Назву ацильних замісників природних а-амінокислот утворюють заміною
суфікса -ін (-ин) на суфікс -іл, (-ил), наприклад: аланін => аланіл, лізин лізил.
Винятком є цистеїніл, аспарагініл, глутамініл, триптофіл.
■ Для моноацильних замісників дикарбонових амінокислот використовують
назви з суфіксами -ил, а для діацильних замісників - суфікси -оїл, наприклад:
НООССН2СН(ЫН2)СО- -ОССН2СН(КН2)СООН -ОССН2СН(Ш2)СО-
а-Аспартил р-Аспартил Аспартоїл
Розділ 22. Амінокислоти
597
22.1.3. Будова амінокислот
Амінокислоти мають групи як з кислотними, так і з основними власти-
востями, тобто їх можна розглядати як амфотерні органічні сполуки. У
кристалічному вигляді й у розчинах амінокислоти перебувають у вигляді
біполярного йона - цвітер-іона (внутрішня сіль), що утворюється внаслідок
приєднання протона карбоксильної групи до аміногрупи:
Р + /Р
гИНг-СНЯ-С МНз-СНЬІ-С7
ОН о
Альфа-амінокислоти, за винятком гліцину, мають асиметричний атом
Карбону й можуть перебувати у двох енантіомерних формах. Природні
амінокислоти, які виділяють із білків тваринного й рослинного походження,
головно належать до Ь-ряду. Як стандарт для амінокислот використовують
природний Ь-серин. Наприклад, конфігурацію природного треоніну позначають
Ь, оскільки конфігурація його верхнього тетраедра збігається з конфігурацією
Ь-серину. Його діастереомер позначають як Ь-алотреонін:
СООН
СООН
Н,ґ>Г-
-Н
Н2№
н-
СН2ОН
Ь-Серин
-н
-он
Н2№
но-
СООН
-н
-н
сн3
Ь-Треонін
сн3
Ь-Алотреонін
22.2. Фізичні властивості
Амінокислоти - безбарвні кристалічні речовини. Через цвітер-іонну природу
вони мають високу температуру плавлення.
Водні розчини амінокислот через наявність іонів є електролітами. Низка
природних амінокислот у бічному ланцюзі має карбоксильні або амінофункції,
завдяки яким вони виявляють, відповідно, кислі або основні властивості
(приклади див. у класифікації).
В електролітичному середовищі молекули амінокислот будуть мігрувати до
катода або анода. За певного значення рН середовища, яке називають
ізоелектричною точкою, амінокислота не буде рухатися ні до катода, ні до анода.
На значення ізоелектричної точки вирішальний вплив має бічний замісник.
Оскільки кожна амінокислота має свій бічний ланцюг, то, відповідно, і значення
ізоелектричної точки для кожної амінокислоти своє, наприклад, для гліцину - 5,97,
глутамінової кислоти - 3,22, лізину - 9,74.
598
Чирва В.Я., Ярмолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
22.2.1. Спектроскопія амінокислот
• УФ-спекмроскопія. Основні амінокислота, окрім ароматичних і гетероаро-
матичних, не поглинають світла в УФ-ділянці. Для ароматичних амінокислот
спостерігають смуги поглинання, типові для їхніх ароматичних залишків.
Наприклад, тирозин має інтенсивні смуги поглинання з 245 і 280 нм.
• ІЧ-спектроскопія. Завдяки біполярній природі амінокислот в ІЧ-спектрах
замість коливань, характерних для аміногрупи (3 300-3 500 см"1), виявляється
смуга поглинання Нз^-групи - Ум.н 3 070 см"1. Також нема сигналів вільної
карбоксильної групи, зате простежується типове для солей карбонових кислот
сильне поглинання Ус-о 1 560-1 600 см"1.
Унаслідок хімічного синтезу зазвичай утворюються рацемічні суміші
амінокислот. Чисті енантіомери можна одержати гідролізом білків або за
допомогою біосинтезу. Певне препаративне значення мають методи аси-
метричного синтезу й поділ рацематів на індивідуальні енантіомери.
22.3.1. Хімічний синтез а-амінокислот
❖ Амінування галогенозаміщених карбонових кислот. Дією надлишку
амоніаку на а-галогенозаміщені карбонові кислоти, які легко одержують за реак-
цією Гелля-Фольгарда-Зелінського (див. розділ 18), утворюються амінокислоти:
Для запобігання можливому процесу діалкілування амоніак можна замінити
на уротропін (гексаметилентетраамін) з наступним гідролізом.
■ Зручніше заміщення галогену на амінофункцію в галогенозаміщених
карбонових кислотах виконувати методом Гийрієля за допомогою калій
фталіміду. Фталімідну похідну розщеплюють дією гідразину, одержуючи
гліцин:
22.3. Способи одержання
ш3
С1-СН2СООН
^ Н2Н-СН2СООН
С1-СН2СООН (У
її М-СН2СООН
мн2ш2
+Н2К-СН2СООН
❖ Синтез на основі ацетооцтового естеру. Послідовне алкілування й
амінування субстрату дає змогу синтезувати а-амінокислоти:
❖ Синтез на основі натріймалонового естеру, Натрійзаміщений малоно-
вий естер амійують за допомогою хлораміну. Подальше розщеплення естеру дає
потрібну амінокислоту:
Бромомалоновий естер можна використати як галогенозаміщений
карбоновий компонент у взаємодії з калій фталімідом. Синтезовану ІУ-фта-
лоїльну похідну далі алкілують і обробляють гідразином з виділенням а-аміно-
кислоти:
❖ Синтези на основі ціанідоцтового естеру. Похідні ціанідоцтового
естеру можна перегрупувати до «-амінокислот.
• Перегрупування Курціуса. Ціанідоцтовий естер традиційним способом
переводять у відповідний ацилазид, який перегруповується в стоксикарбо-
ніламін, а той легко зазнає гідролізу до відповідної амінокислоти:
• Перегрупування Гофмана. Нітрильну функцію в заміщеній ціанідоцтовій
кислоті спочатку частково гідролізують до аміду, а потім розщеплюють за
Гофманом до аміну:
Розділ 22. Амінокислоти
599
600
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
CN ноно- CN „«w™»^ CONH, Rr NH,
r-ch r-ch ІМ2А> R_cH 2^ r-CH 2
соос2н5 соон соон Na0H соон
❖ Синтез Штрекера1. Приєднання ціанідної кислоти до альдимінів, що
утворюються з альдегідів та амоніаку (див. про карбонільні сполуки), приводить
до амінонітрилів, гідроліз яких дає змогу синтезувати цільові амінокислоти:
NH3 HCN Н20,Н+ №
R-CH=0 —R-CH=NH R-CH-CN 1 ■ R-OCOOH
Н
На практиці на карбонільні сполуки діють сумішшю амоній хлориду і натрій
ціаніду, які в реакційному середовищі утворюють необхідні реагенти:
NH4C1 + NaCN 5=^ NaCl + NH4CN
NH4CN 5=^ NH3 + HCN
❖ Азлактоновий синтез. Гіпурова кислота (N-бензоїлгліцин) під дією
оцтового ангідриду зазнає циклізування в азлактон, активна метиленова група
якого легко конденсується з ароматичними альдегідами. Продукт конденсації
піддають відновленню й наступному гідролізу:
Н_ .с6н5
Щс"~% ^2>Є VC6"5 ArCH=C V с<н'
СООН -HP £_0' ^_о/
О Азлактон q О
_ а ™-^N~G~C6H5 И HN-C-C6H5 н2о,н+ NH2
СООН ArCH.-C-COOH АгСН^С-СООН
❖ Алкілування основ Шиффа2. Етиловий естер гліцину легко утворює з
бензальдегідом основи Шиффа. Під дією такої сильної основи, як калій трет-
бутилат, утворюється карбаніон, який легко реагує з різними алкілгалогенідами.
Виходи а-амінокислот досягають 90 %. Гідроліз продукту реакції приводить до
а-амінокислоти:
1 Штрекер А. (Strecker A.F.L.) (1822-1871) - німецький хімік, учень Ю. Лібіха. Займався аналізом,
з'ясуванням структури та синтезом різних органічних сполук, насамперед, азотовмісних. Є одним
із засновників металоорганічної хімії.
2 Шифф Г. (Schiff Н.) (1834-1915) - італійський хімік. Головні праці стосуються різних галузей
органічної хімії. Вперше синтезував тіонілхлорид, запропонував якісну реакцію на альдегіди з
фуксин сульфатною кислотою.
Розділ 22. Амінокислоти
601
С~^-(Р + Н2С-СООС2Н5 /ГЛ^ СООС2Н5 (сн3)3сокг
ч=/ н ш,
\=/ н
н-с-соос2н5
КНаї
2і х5
н
к-с-соос,н
N
н
н,о, н+
>ш2
я-сн
соон
22.3.2. Хімічний синтез ^-амінокислот
❖ Синтез ^-аланіну. Приєднання амоніаку за подвійним зв'язком акри-
лової кислоти дає р-аланін. Напрям приєднання визначає відтягування
електронної густини я-зв'язку карбоксильної групою.
5+ /Ч5- н2М-Н
СН2=СН-СООН ——»-Ш2СН2СН2СООН
❖ Синтез на основі пропандіової кислоти. Реакція Родіонова1. Конденса-
цією альдегідів з малоновою кислотою за наявності амоніаку одержують
амінодикарбонову кислоту, яка під час нагрівання зазнає декарбоксилювання до
Р-амінокислоти:
СООН
ЯСНО + Н£
СООН
СООН
ш:н=с
соон
ш н соон , н
Я-С-СН —К-С-СНгСООН
м^соон 2 N1^
22.3.3. Хімічний синтез ^-амінокислот
❖ Гідроліз лактамів. Лактами - внутрішньомолекулярні циклічні аміди -
зазнають гідролізу до со-амінокарбонових кислот. Наприклад, у-аміномасляну
кислоту одержують гідролізом у-бутиролактаму, який, відповідно, синтезують
неповним відновленням сукциніміду:
0^Х0^д.0^н2н(снЛсоон
н н
Сукцинімід у-Бутиролактам у-Аміномасляна кислота
1 Родіонов В. (1878-1954) - радянський хімік-органік. Праці в галузі хімії барвників, алкалоїдів,
фармацевтичних препаратів.
602
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Альтернативно у-бутиролактам одержують амінуванням у-бутиролактону.
■ За основу промислового синтезу 6-аміногексанової (о>амінокапронової)
кислоти взято перегрупування Бекмана1 - утворення лактамів з оксимів під дією
сульфатної кислоти. Гідроліз капролактаму дає потрібну амінокислоту:
о
NH2OH /-Д H2S04
О ► ( V=N-OH ►
Оксим циклогексанону
Н НА^ H2N(CH2)5COOH
ОН "О
Капролактам 6-Аміногексанова кислота
Аналогічно одержують і 5-амінопентанову (8-аміновалеріанову) кислоту.
❖ Амінування теломерів. Продукти реакції теломеризації етилену й
тетрахлорометану амінують і після підкислення виділяють ©-амінокислоти:
С1(СН2СН2)ПСС13
п=2-5
NH3
>
Н,0
н+
H2N(CH2CH2)nCOONI^
H2N(CH2CH2)nCOOH
22.3.4. Асиметричний синтез а-амінокислот
В асиметричному синтезі до хірального амінокомпонента приєднують
ацильну групу з утворенням інтермедіату зі зв'язками С=С або C=N, які далі
стереоспецифічно відновлюються. Наприклад, використання L-проліну як
хірального індуктора дало змогу з піровиноградної кислоти одержати L-аланін з
виходом близько 60 %:
д,.н
£} соосн3
+
ноос. хн3
о
Ос?
¥ СООСН, nh3
-С —
0'V°
о"схш -що o"cy
сн.
NH рю2
н3с он
o"cxNH
н сн,
J хт V
N
н
СООСН,
НООС NH2
н "сн,
Бекман Е (Beckmann Е.) (1853-1923) - німецький хімік, спеціаліст у галузі фізичної та органічної
хімії. Синтезував метилкетили. Розробив метод визначення молекулярних розчинних сполук на
основі закону Рауля. Винахідник спеціального термометра (термометр Бекмана).
Розділ 22. Амінокислоти
603
• Синтез Штрекера. Основу Шиффа, яку одержують дією оптично активно-
го аміну на етилгліоксилат, перетворюють взаємодією з ферум карбонілом у
комплекс. Синтезовані діастереомери розділяють. Далі комплекс стереоспе-
цифічно алкілують. Після розкладу інтермедіату вихід чистих оптичних ізомерів
досягає 95 %:
СбїІ5\ о=сн-соос,н< СбЬІ5\ ге2(со)9 СбН5\ ЯВг
Н3С Й Н3СУЙ • Н3С й \І(С0^
С6Н5Ч *\ а.Н2/Рсі І\
—* ЯҐЧС-СООС2Н5 » С6Н5СН2СН3 + Н2№-С-СООС2Н5
Н3С н І Н ь.н2о,но ^
Ре(СО)зВг
22.3.5. Поділ рацематів на індивідуальні
енантіомери
❖ Кристалізація діастереомериих солей. Принципи цього методу викла-
дено в розділі 6. Рацемічні амінокислоти, зазвичай у вигляді ІУ-ацильних
похідних, переводять у солі дією оптично активного аміну (бруцин, ефедрин,
один з енантіомерів 1-фенілетиламіну тощо). Підбирають умови кристалізації
однієї з діастереомерних солей, і після одержання оптично чистої солі її
розкладають до енантіомерної амінокислоти.
❖ Ферментативні методи полягають у тому, що одну з енантіомерних
амінокислот або її похідну в рацематі модифікують дією специфічних
ферментів, тоді як друга амінокислота залишається без змін. Далі дві різні
хімічні речовини розділяють традиційними методами.
• Використання ацилаз. Ацилази - ферменти, що розщеплюють амідний
зв'язок. Такий фермент з нирок свині діє лише на Ь-амінокислоти. Тому обробка
ацилазою, наприклад, И-ацетил-ОЬ-валіну дасть суміш Ь-валіну і И-ацетил-й-
валіну9 яку легко розділити на оптично чисті сполуки:
н^-соон ЄЄ* НзС-^-соон ^ ^Л-соон + ^Ьсоон
СН(СНз)2 0НСН(СН3)2 (НАСН (НзОзСН^
• Використання оксидаз. Під дією цих ферментів амінокислоти окиснюються
до а-кетокислот, наприклад, аміноксидази зміїної отрути діють на Ь-аміно-
кислоти, а відповідні ферменти з нирок свині — на їхні антиподи:
Н21\ Ь-аміноксидаза Г СООН 1 Н,0 СООН
кч-соон ~^н ~|к-с=ин ] ГїїнТ к-с=о
604
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
СООН
я-с=о
Н20
-МН,
СООН
Я-С^Н
Н N
й-Аміноксидаза 2 \
- н СООН
•2Н
22.4. Хімічні властивості
❖ Кислотно-основні властивості амінокислот. Амінокислоти мають
властивості амфотерних сполук. Залежно від рН середовища вони перебувають у
вигляді карбоксилат-аніона, цвітер-іона або амонієвого катіона:
НзИ-СН-СООН
Я
рН~1
Амонієвий катіон
-Н+
н3й-сн-соо~
рН~7
Цвітер-іон
-н+
*=£ НгИ-СН-СОО"
А
рН~11
Карбоксилат-аніон
Навіть нейтральні амінокислоти мають дві константи дисоціації, наприклад,
для гліцину рКі =2,4, а рК2 = 9,8. Перша константа відповідає дисоціації
карбоксильної групи, а друга - депротонуванню амонієвого катіона, тобто в
цьому випадку амінокислота поводиться як ИН-кислота:
Н31\Г-СН2-СООН
ї=£ н3м-сн2-соо
+н+
+н+
рк2
н2к-сн2-соо~
❖ Загальні властивості амінокислот. Амінокислоти значно поєднують у
собі властивості амінів і карбонових кислот (див. розділи 16 і 18). Як приклади,
нижче наведені деякі типові реакції гліцину:
+ неї
сГ н3ж:н2соон <*
Пдрохлорид гліцину
_^Н3СОС1
сн3<^нж:н2соон
О Л^-Ацетилгліцин
НОСН2СООН <+-
Гліколева кислота
НШ9
С6Н5СН=Ж:Н2СООН
ІУ-Бензиліденгліцин
С6Н5СНО
ИаОН
Н2ИСН2СООН
Н2НСН2СООНа
Гліцинат натрію
СН,ОН
7^ Н2НСН2СООСН3
н Метилгліцинат
ЬіА1Н4
► Н2НСН2СН2ОН
2-Аміноетанол
Розділ 22. Амінокислоти
605
■ Деякі типові реакції по карбоксильній групі, наприклад, одержання
хлороангідридів, не можна проводити безпосередньо на амінокислотах. У цих
випадках використовують И-захищені амінокислоти:
ЬОС\2 ,?
СН3<^НМСН2СООН ► СН3СНМСН2-С + 802 + НС1
О О
СІ
■ У випадку естерів амінокислот реакція з нітритною кислотою відбу-
вається з утворенням стійких діазоестерів:
HNO, + -
H2NCH2COOC2H5 ^ N=N-CH-COOC2H5
- 2н20
Етилгліцинат Діазооцтовий естер
❖ Специфічні реакції а-амінокислот
• Реакція арилювання. Альфа-амінокислоти зазнають iV-арилювання дією 2,4-
динітрофлуоробензену {реактив Сангера\ утворюючи забарвлені динітрофе-
нільні (ДНФ) похідні, які використовують для ідентифікації амінокислот:
HO""
02N—<ч h—F + H2N-CH-COOH ► 02N—^ //-HN-CH-COOH
I^q-HN-Cp
і
Я
Ж)2 N02
• Реакції комплексоутворення. Солі ^металів утворюють кольорові комплекси
з амінокислотами. Наприклад, утворення яскраво-синього забарвлення під час
взаємодії з іонами Купрум (II) використовують як якісну реакцію на аміно-
кислоти:
• Реакції де карооке плювання. Альфа-амінокислоти за температур понад
200 °С зазнають декарбоксилювання з утворенням амінів. Як і для інших
похідних карбонових кислот, цей процес каталізують іони важких металів,
наприклад, Купрум (II):
1 Сангер Ф. (Sanger F.) (1918 p. н.) - англійський біохімік. Головні праці присвячені хімії білків та
нуклеїнових кислот. Нобелівські премії 1958 р. (за визначення структури інсуліну) та 1980 р. (за
розробку методу розшифрування первинної структури ДНК). Єдиний двічі Нобелівський лауреат
у галузі хімії.
606
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
я-снсоон —іі-сн2їга2
І -со2
>щ2
Ця реакція належить до найважливіших біохімічних процесів. Унаслідок
декарбоксилювання, каталізованого ферментами, з амінокислот утворюються
незвичайні амінокислоти й так звані біогенні аміни. Наприклад, декарбокси-
люванням аспарагінової й глутамінової кислот отримують р-аланін і у-аміно-
масляну кислоту, а з гістидину й 5-гідрокситриптофану - гормоноподібні сполуки
гістамін і серотонін:
ІШ-СН-СООН декарбоксилаза Н^-СН-СООИ декарбоксилаза
| —->Н2Н(СН2)2СООН І —^НДООТ^ССЮН
СН2СООН _с°2 (СН2)2СООН ~с°2
Аспарагінова кислота Р-Аланін Глутамінова кислота у-Аміномасляна кислота
НгИ-СН-СООН
І тт декарбоксилаза
І х>—СН2 — ► к х>— (СН2)2>ІН2
Гістидин Гістамін
Н2К-СН-СООН
Атт декарбоксилаза НО^ / (СН2)2МН2
5-Гідрокситриптофан Серотонін
• Реакції оксидного дезамінування. Під дією сильних окиснювачів а-аміно-
кислоти зазнають далі розщеплення, виділяючи амоніак і вуглекислий газ.
Отриманий як проміжний продукт, альдегід далі окиснюється до карбонової
кислоти:
[О] [О]
Н2М-СН-СООН хттт > я-сн=о ► Я-СООН
^ І -ИНз, -с02
Я
• Реакція з нінгідрином. Під час взаємодії амінокислот з нінгідрином утворюється
пурпурово забарвлений продукт конденсації, що дає змогу використовувати цю
властивість як якісну реакцію на амінокислоти. За допомогою нінгідрину виявляють
амінокислоти за різних варіантів хроматографування, зокрема в амінокислотних
аналізаторах, де суміші амінокислот розділяють на йонообмінних смолах.
НО
Розділ 22. Амінокислоти
607
о.
Нінгідрин
он+ш2снксоон
+ Н20 + СОг + ЯСНО
о о
Уважають, що спочатку відбувається одночасне оксидне дезамінування
амінокислоти й відновлювальне амінування нінгідрину. Потім амін зазнає
конденсування з іншою молекулою нінгідрину, утворюючи барвник:
ш,снксоон
О—'- *-
чно о
❖ Реакції циклізації
■ Естери а-амінокислот під час нагрівання зазнають димеризування,
утворюючи циклічні аміди — дикетопіперазини:
н д ог"гК
2 Н,М-С-СООС,Н, » 1 І
н
■ У разі нагрівання Р-амінокислоти відщеплюють амоніак і перетворю-
ються в а,Р-ненасичені карбонові кислоти:
ЫН2СН2СН2СООН
• Н,0
*- СН2=СН-СООН
■ Під дією специфічного конденсувального реагенту - карбодііміду - Р-амі-
нокислоти можуть утворити просторово утруднений р-лактам:
608
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
н2кснасн2соон + с6ніГ№с=^с6нп — я~Л=о + сд^-с-и-сд.
Карбодіімід д О
■ На відміну від Р-амінокислот, у- і 5-амінокислоти легко утворюють п'яти-
і шестичленні циклічні аміди - у- і 8-лактами:
Н2К(СН2)3СООН (^Х0 Н2К(СН2)4СООН £\
н й °
22.5. Окремі представники амінокислот
■ Гліцин застосовують у медицині як заспокійливий (седативний) препарат.
Він поліпшує роботу головного мозку.
■ Гамма-аміномасляна кислота (ГАМК) - амінокислота, що забезпечує
нервове гальмування. Утворюється в організмі внаслідок біохімічного
декарбоксилювання глутамінової кислоти. Механізм дії ГАМК пов'язаний з
дією на специфічні рецептори нервових клітин. Сама ГАМК є лікарським
засобом, що поліпшує роботу головного мозку й активує пам'ять.
■ Ь-Глутамінова кислота має солодкий смак, а її натрієва сіль - натрій
глутамат - надає розчинам смаку вареного м'яса, завдяки чому її широко
використовують як харчосмакову домішку. Головний спосіб одержання Ь-глу-
тамінової кислоти - гідроліз клейковини пшениці. Світові обсяги її виробництва
перевищили 200 тис. т за рік.
В організмі людини Ь-глутамінова кислота бере участь у передаванні
нервових збуджень. її використовують для лікування деяких нервових
захворювань.
■ Ь-Метіонін належить до незамінних амінокислот. Як лікарський засіб
його застосовують для нормалізації ліпідного обміну.
Його метальну похідну — метилметіонін ^ И-СН-СООН
{вітамін Ц) - використовують для лікування |
виразок шлунка й дванадцятипалої кишки. У СГ(Н3С)28(СН2)2
промисловості його одержують алкілуванням
метіоніну метилхлоридом.
22.6. Пептиди й пептидний синтез
Продукти поліконденсації амінокислот з молекулярною масою менше
5000 Да називають пептидами. Амінокислоти, з'єднуючись одна з одною
пептидним зв'язком, утворюють пептидні ланцюги. У таких ланцюгах виді-
Розділ 22. Амінокислоти
609
ляють амінокислоту, у якої є вільна аміногрупа (так званий И-кінець), і
амінокислоту з вільною карбоксильною групою (відповідно, С-кінець).
❖ Номенклатура пептидів. Під час побудови назв коротких пептидів за
основу беруть С-кінцеву амінокислоту, а інші амінокислоти розглядають як
ацильні замісники, назви яких будують, починаючи з И-кінця, наприклад:
❖ Будова пептидної групи. Пептидний зв'язок практично плоский,
відхилення від площини не перевищує 5-10°. Зв'язки 1М-Н і С=0 зазвичай
перебувають у транс-положенні один щодо одного. Через р,ті-спряження
електронної пари атома Нітрогену з карбонільною групою нема вільного
обертання навколо зв'язку С-Ы, що обмежує конформаційну рухливість
пептидного ланцюга. Довжина пептидного зв'язку - 0,132 нм, що менше, ніж у
звичайного зв'язку С-Ы:
Біологічно активні пептиди виділяють з природних джерел або синтезують.
Хімічний синтез пептидів має низку особливостей.
По-перше, карбоксильна група, взаємодіючи з амінами, не утворює амідного
зв'язку й, отже, карбоксильну групу необхідно активувати.
По-друге, для того, щоб утворити пептидний зв'язок між двома різними
амінокислотами, потрібно унеможливити хімічну взаємодію однакових
амінокислот між собою. Для цього вводять захисні групи - проводять таку
хімічну реакцію з карбоксильною або аміногрупою, щоб отримана сполука була
стійкою в реакціях утворення пептидного зв'язку й легко розщеплювалася за
умов, що не діють на пептидний зв'язок.
По-третє, реакції необхідно проводити в м'яких умовах, за яких не
відбувається рацемізації амінокислот або пептидів.
Принципово в пептидному синтезі можна виділити чотири етапи.
Перший етап. Уведення захисних груп в амінокислоти, у тому числі по
аміногрупі (И-захист) і карбоксильній групі (С-захист).
^-кінець ^тт С-кінець
СН3 п п О п СН28Н
Аланіл-валіл-цистеїн
22.6.1. Синтез пептидів
610
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
Другий етап. Активування карбоксильної групи И-захищеної амінокислоти.
Третій етап. Конденсація амінокислот з утворенням пептиду. Низка
сучасних методів дає змогу об'єднати активування з конденсацією.
Четвертий етап. Повне або вибіркове видалення захисних груп, що,
відповідно, дає вільний або частково захищений пептид.
Розберемо, як приклад, синтез дипептиду Ь-аланіл-Ь-глутамінової кислоти.
• Уведення захисних груп
Спочатку потрібно захистити аміногрупу .в Ь-аланіні. Ацетильні або
бензоїльні похідні, які широко використовують у класичній органічній хімії,
практично не застосовують у синтезі пептидів, оскільки амідні зв'язки
розщеплюються в жорстких умовах кислотного або лужного гідролізу за високої
температури.
Для захисту аміногрупи зручно використовувати естери карбонатної кислоти,
наприклад, бензилоксикарбонільний захист. Цю групу легко вводять в
амінокислоти дією бензилоксикарбонілхлориду (хлороангідриду бензилового
естеру карбонатної кислоти) за наявності лугу. Захист стійкий у широкому
діапазоні рН: від лужного до середньокислого середовища. Звичайний м'який
спосіб видалення бензилоксикарбонільного захисту полягає в гідрогенолізі над
паладієвими каталізаторами, у цьому разі утворюються толуен і вуглекислий газ:
Н2М-С-СООН + /~УсНгО-<* ^Л-СНгО-С^-С-СООН
Аланін Бензилоксикарбонілхлорид ІУ-Бензилоксикарбонілаланін
■ Для захисту карбоксильної групи в хімії пептидів дуже зрідка вико-
ристовують метилові або етилові естери, оскільки вони розщеплюються за умов,
які сприяють рацемізації. Найпоширеніший С-захист - це утворення бензилових
естерів. Головною їхньою перевагою над іншими способами захисту є
можливість м'якого розщеплення за умов каталітичного гідрогенолізу.
Амінокислоти зазвичай естерифікують бензиловим спиртом за наявності
кислотних каталізаторів. Наприклад, під час кип'ятіння суміші бензилового
спирту й глутамінової кислоти в бензені за наявності и-толуенсульфокислоти з
високим виходом одержують відповідний дибензиловий естер:
Ш2СНСООН +^усН20Н ННХНСООСНг-^
сн2соон \=/ 2 (снлсоосн,-^
Глутамінова кислота Бензиловий спирт Дибензилглутамат
• Далі активують карбоксильну групу в похідній аланіну. Є безліч способів
активування карбоксилу. Розглянемо метод активованих естерів, у якому
Розділ 22. Амінокислоти
611
карбоксильну групу переводять у естер, що має електроноакцепторні групи.
Порівняно зі звичайними естерами в таких естерах збільшений частковий
позитивний заряд на карбонільному атомі Карбону, що полегшує атаку
нуклеофільної аміногрупи. Прикладом такого активованого естеру може бути ДО-
гідроксисукцинімідний естер. Найефективніший метод його одержання - вико-
ристання як конденсувального реагенту дициклогексилкарбодііміду, що зв'язує
воду, перетворюючись у заміщену сечовину:
9 н .с-,
£^снго-с-м-с-соон + но-н ^ + <^-к=с=н-^)-
^ Дициклогексилкарбодіімід
3 Р 2 Дициклогексилсечовина
• На третьому етапі проводять конденсацію двох амінокислот:
0 Н Р % Н
{ Уснго-с-и-с-а /С~СН, + Н^-С^СООСН-гС' х)
снз-о-с-м-с-с; /^н, + н^-с-соосн,-^; ™*
^ н сн3 0 *уя, (і^-соосчРО
х==/ н сн3 Ц-^-сооснД/ >_ + но-мС
, \=а-ь + ш А^с'Сн;
(СН^-СООСК,-^ о
• На завершення каталітичним гідрогенолізом видаляють захисні групи:
нЇЇ н
г-х о и о „ г-^ н2н-с-с-м-с-соон
(^сн.-о-^н-^с-кТсоосн.-О Ь!И сн3 н(ін2)2-соон
(сн^соосн^) + з + СОі
612
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
22.6.2. Окремі представники пептидів
■ Аспаршам (метиловий естер L-аспартил-Ь-фенілаланіну) - дієтичний
замінник цукру, солодший від сахарози в 200 разів.
н° н
H2N-C-C-N—С-СООСН3
✓СН~ KLC>,
НООС 2 2 ||
■ Глутпатіон (у-Ь-глутаміл-Ь-цистеїніл-гліцин) - трипептид, бере участь в
окисно-відновних реакціях, що відбуваються у клітинах, а також виконує
функцію захисту цистеїновмісних білків.
?оон Я Н9
н2к-с-(сн2)—с-и-с-с-ічг-снхоон
2 н ні н 2
СН2БН
■ Тимоген (глутамініл-триптофан) - імуностимулювальний. лікарський
засіб.
н9 н
H2N-C-C-N—9-СООН
■ Тироліберин (піро-Ь-глутаміл-Ь-гістидил-Ь-пролінамід) - гормоноподібна
сполука, яку виділяє гіпоталамус; контролює продукування гіпофізом гормону
тиреотропіну. Піроглутамінова кислота є продуктом внутрішньомолекулярного
амідування.
1 )-C-N—C-C-N /
O^N Hu А
HN^N
22.7. Білки
Білки - природні полімери, що складаються з залишків а-амінокислот,
з'єднаних за допомогою амідного (пептидного) зв'язку. Молекулярна маса білків
може досягати декількох мільйонів Да.
Розділ 22. Амінокислоти
613
В організації білкових молекул виділяють чотири рівні: первинну, вторинну,
третинну й четвертинну структури.
22.7Л. Первинна структура білків і принципи
її визначення
Первинна структура - це послідовність з'єднання амінокислот між собою:
Саме хімічна природа амінокислот і порядок їхнього з'єднання визначають
просторову структуру білка й, отже, його властивості. Кожен білок має
індивідуальну первинну структуру. Комбінуванням порівняно невеликої
кількості амінокислот можна отримати величезну розмаїтість білкових структур.
Це нагадує те, як за допомогою невеликої кількості букв можна написати
нескінченну кількість книг. Недарма амінокислоти називають "хімічною мовою
природи".
❖ Принципи визначення будови первинної структури білка. Ключовими
етапами у визначенні структури білка є з'ясування амінокислотного складу,
визначення N- і С-кінцевих амінокислот, а також визначення послідовності
амінокислот.
• Зуясування амінокислотного складу. Зразки білка в запаяних ампулах
гідролізують 5,7 н HCl за 110 °С. Продукти гідролізу розділяють за допомогою
хроматографії на йонообмінних смолах. Амінокислоти детектують за допомо-
гою реакції з нінгідрином. Кожна амінокислота має свій час утримування на
хроматографічній колонці, а за інтенсивністю фарбування оцінюють кількість
амінокислоти.
• Визначення N-кінцевої амінокислоти. Зразок поліпептиду обробляють 2,4-
динітрофлуоробензеном, що реагує з амінокислотою із вільною амінофункцією.
Продукт реакції піддають гідролізу. Діетиловим етером екстрагують динітро-
феніл-(ДНФ)-похідну й проводять хроматографічний аналіз, порівнюючи з
відомими зразками ДНФ-амінокислот:
N02
614
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
"і*1 о^нО-м-с-ссон + н^-озон + н^-9-ооон + ^ аміно~
^ Н СНз НС-СНз СН^Н
Ш2 СН,
ДНФ-аланш
У сучасній хімії білків для визначення И-кінцевої амінокислоти
використовують дансилхлорид. Похідні, отримані за допомогою цього реагенту,
мають інтенсивну флюоресценцію, що дає змогу виявляти надзвичайно малі
кількості речовин (до 10~14 моль):
н3(ж-сн3
802С1
Дансилхлорид
• Визначення С-кінцевої амінокислоти. Одним із можливих варіантів
визначення амінокислоти на С-кінці є гідразиноліз поліпептиду. Унаслідок
розщеплення пептидних зв'язків під дією гідразину всі амінокислоти
перетворюються в гідразиди, тільки С-кінцева амінокислота не змінюється:
сн3
Н2№Ш2
Н 9 Н? СНз Н
-и-с-с-ічі-с-с-к-с-соон
и І тт тт II тт Т
п СН3 п п О П СН28Н
Гідразиди Н /,° Н /Р Н
— інших + н2ї<-с-с + н2к-с-с + н2ы-с-соон
амінокислот СН3ННШ2 нзС-СН ННМН2 СН.БН
сн3
Аланінгідразид Валінгідразид Цистеїн
• Визначення послідовності амінокислот за Едманом. У 1950-1956 рр.
П. Едман1 розробив метод визначення послідовності амінокислот, в основі якого
є обробка зразка пептиду фенілізотіоціанатом з утворенням фенілтіокарбомоїл-
(ФТК)-пептиду. Під дією трифлуорооцтової кислоти в цій похідній відбувається
внутрішньомолекулярна нуклеофільна атака атома Сульфуру по карбонільному
1 Едман П. (Есітап Р.У.) (1914-1977) - шведський біохімік, працював у хімії білків. Розробив
метод, за допомогою якого можна визначати послідовність амінокислот у білку.
Розділ 22. Амінокислоти
615
атому Карбону, що приводить до відщеплення И-кінцевої амінокислоти у вигляді
заміщеного 2-анілінотіазолінону-5:
Н и
Я'
і
Фенілізотіоціанат
к-с-с-ы-с-с
' НН Д
1
о
но-
Н Я
л-с-с-м-с-с-
ФТК-пептид
Н+
н ?~н+** .
ы-с-с-^с-с-
с
гін
о
о-*
\=/ Н Б^О
2-Анілінотіазолінон-5
Я'
+ нІі-с-с—*
з и II
п о
Подібні похідні важко аналізувати, тому їх піддають короткочасній обробці
1 н НС1, унаслідок чого вони зазнають ізомеризування в стійкі похідні 3-феніл-
2-тіогідантоїну (ФТГ-похідні):
Н,
2-Анілінотіазолінон-5
•Н20
ФТК-кислота
ФТГ-похідна
Хроматографічними методами шляхом порівняння з відомими зразками
ФТГ-амінокислот визначають, яка амінокислота відщепилася від пептиду.
Пептид, що залишився, піддають другому циклу розщеплення за Едманом для
визначення другої амінокислоти й т.д.
22.7.2. Просторова будова білкових молекул
❖ Вторинна структура - це просторово впорядковані ділянки пептидних
ланцюгів, стабілізовані водневими зв'язками. Вирізняють а- і ^-структури.
• Спіраль а. Пептидний ланцюг здатний спонтанно закручуватися в спіраль,
яку скріпляють водневі зв'язки. Бічні замісники амінокислотних залишків у
цьому разі опиняються на периферії спіралі. Найпоширеніша а-спіраль, у якій
616
Чирва В.Я., Ярмолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
амідний атом Гідрогену утворює водневий зв'язок з атомом Оксигену четверто-
го від нього амінокислотного залишку, так званий (5-И)-зв'язок:
а-Спіраль
На стійкість спіралі впливає природа амінокислот. Стабілізують спіраль
нейтральні амінокислоти, наприклад, аланін, валін, лейцин, метіонін, тоді як
заряджені амінокислоти, наприклад, лізин, глутамінова кислота й амінокислоти,
бічні ланцюги яких створюють просторові перешкоди (ізолейцин, треонін),
ускладнюють утворення спіралі. Прикладом білка, пептидний ланцюг якого має
ос-структуру, може бути а-кератин (волосся, вовна).
• Структура р ^складчастий аркуш") - з'єднаний водневими зв'язками
асоціат витягнутих, зиґзаґоподібних ланцюгів. Бічні ланцюги розташовуються
над і під площиною "складчастого аркуша". Стабільні Р-структури побудовані з
амінокислот із невеликим об'ємом замісника, наприклад, гліцин й аланін.
Наприклад, білок натурального шовку - фіброїн, побудований із білкових
ланцюгів з Р-структурою, має велику кількість цих амінокислот:
Водневі зв'язки в Р-структурі "Складчастий аркуш"
Розділ 22. Амінокислоти
617
❖ Третинна структура
• Глобулярні білки. Більшість розчинних у воді білків у просторі існують у
вигляді глобули (неправильної сфери або еліпсоїда). Зазвичай, у білковому
ланцюзі таких білків ділянки вторинної структури сполучені з неструктуро-
ваними фрагментами ланцюга.
Різні амінокислоти, у тому числі віддалені одна від одної в амінокислотному
ланцюзі, можуть зв'язуватися за допомогою внутршшьомолекулярних водневих
зв'язків притяганням протилежно заряджених функцій бічного ланцюга, а поряд
розташовані ділянки ланцюга можуть, навпаки, відштовхуватися одна від одної
через наявність однойменно заряджених груп.
Більшу жорсткість третинній структурі білка надають дисульфідні містки,
утворені димеризацією тіольних функцій залишків цистеїну:
Дисульфідний місток
Величезну роль у формуванні просторової структури відіграє вода:
гідрофобні групи прагнуть максимально взаємодіяти одна з одною й уникнути
контактів з водою, тоді як полярні групи виходять на поверхню глобули.
Сукупність подібних взаємодій формує третинну структуру білка. Для
більшості білків третинна структура є повною просторовою структурою, що
забезпечує їхні специфічні біологічні властивості:
Третинна структура цитохрому с
618
Чирва В.Я., Ярмолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
• Фібрилярні білки. До цієї групи білків належать нерозчинні у воді білкові
молекули, що мають ниткоподібну (фібрилярну) будову. Прикладом такої
структури може слугувати білок волосся і вовни - <х-кератин. Три а-спіральні
ланцюги кератину закручені один на одного, утворюючи протофібрилу.
Комплекс протофібрил утворює мікрофібрилу, з яких і складається волосина:
❖ Четвертинна структура. Багато білкових структур є комплексом
білкових молекул з третинною структурою, що зв'язані між собою головно
водневими зв'язками, гідрофобними й електростатичними взаємодіями. У
деяких випадках компоненти з'єднані дисульфідними зв'язками. Фактично всі
білки з молекулярною масою понад 50 кДа - це олігомери.
Біологічна активність властива саме білковому комплексу, а не окремим
субодиницям. Наприклад, в еритроцитах є білок гемоглобін, який переносить
молекули кисню в організмі. Він складається з двох а- і двох Р-субодиниць, які
мають 141 й 146 амінокислот, відповідно. Кожна субодиниця містить
порфіринову структуру - гем, який забезпечує фіксування однієї молекули
кисню:
а-Спіраль
Протофібрила
Мікрофібрила
Гемоглобін
Розділ 23
ВУГЛЕВОЛИ
Вуглеводи (цукри) - це полігідроксикарбонільні сполуки та їхні похідні.
Більшість із них має брутто-формулу Ся(Н20)л. Перші відомі цукри - глюкоза й
фруктоза - мали саме такий склад, що дало змогу німецькому хімікові
К. Шмідту 1844 р. дати іншу назву - вуглеводи. Пізніше з'ясували, що багато з
них не відповідає формулі С/;(Н20)„, однак назва "вуглеводи" міцно вкоренилася
в хімічній літературі.
Серед численних речовин довкілля вуглеводи та їхні похідні відіграють
винятково важливу роль у промисловості й повсякденному житті людини,
забезпечуючи їжею, одягом і різними виробами з деревини. Вуглеводи у вигляді
різних похідних є в складі клітин будь-якого живого організму, відіграючи роль
конструкційного матеріалу, субстратів і регуляторів біохімічних процесів, а
також постачальника енергії. Взаємодіючи з нуклеїновими кислотами, ліпідами
й білками, вуглеводи утворюють складні високомолекулярні комплекси, які є
основою субклітинних структур - основ живої матерії. Вуглеводи є початковими
продуктами фотосинтезу, тобто першими органічними речовинами у кругообігу
вуглецю в природі, і слугують сполучною ланкою між мінеральними й
органічними речовинами.
Величезне практичне значення вуглеводів привернуло до них увагу людини з
давніх часів. Обробка деревини, виготовлення паперу й бавовняних тканин,
хлібопечення й бродіння - усі ці процеси, відомі з глибокої давнини,
безпосередньо пов'язані з переробкою вуглеводовмісної сировини. Тростиновий
цукор був, очевидно, першою органічною речовиною, яку людина отримала в
хімічно чистому вигляді. Услід за тростиновим цукром 1792 р. виділено фруктозу,
а 1802 р. Т. Ловіц1 одержав кристалічну глюкозу. В 1811 р. К. Кірхгоф2 обробкою
крохмалю хлоридною кислотою провів перший хімічний гідроліз полісахариду, а
через три роки - перше ферментативне розщеплення крохмалю під впливом
ензимів слини. Нарешті 1861 р. О. Бутлеров дією вапняної води на водний розори
формальдегіду одержав суміш цукрів, яку назвав метиленітаном. Так проведено
перший історичний синтез у хімії вуглеводів.
Ловіц Т. (1757-1804) - російський хімік. Головні дослідження в галузі адсорбції і кристалохімії.
2 Кірхгоф К. (1764-1833) - російський хімік. Головні дослідження в галузі каталізу.
620
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Вуглеводи поділяють на три великі групи:
■ моносахариди — найпростіші полігідроксикарбонільні сполуки, що не
піддаються гідролізу;
■ олігосахариди - проміжний клас вуглеводів, що містять від двох до
десяти моносахаридних залишків;
■ полісахариди - природні полімери, що складаються з моносахаридних
залишків і здатні гідролізуватись.
• Моносахариди поділяють на:
■ альдози - мають у складі альдегідну групу;
■ кетози — мають у складі кетогрупу.
• За кількістю карбонових атомів у ланцюзі моносахариди розділяють на:
■ тріози - три атоми Карбону;
■ тетрози - чотири атоми Карбону;
■ пентози - п'ять атомів Карбону;
■ гексози - шість атомів Карбону;
■ вищі цукри - більше шести атомів Карбону.
• За функціональними групами розрізняють:
■ нейтральні цукри — мають тільки гідроксильні й карбонільні функції;
■ кислі цукри - мають карбоксильну або іншу кислотну функцію;
■ аміномоносахариди — мають аміногрупу;
■ дезоксимоносахариди - замість гідроксильної групи мають атом Гідрогену.
23.1.2. Структура моносахаридів
Центральне місце в хімії моносахаридів посідають альдогексози (І),
кетогексози (II) й альдопентози (III):
23.1. Класифікація, будова й номенклатура,
моносахаридів
23.1.1. Класифікація моносахаридів
іСНО
2СНОН
зСНОН
4СНОН
5СНОН
6СН20Н
іСЬШН
І 2
2С=0
І
іСНО
2СНОН
зСНОН
4СНОН
5СН2ОН
II
III
Розділ 23. Вуглеводи
621
Оскільки розмаїтість моносахаридів пов'язана, насамперед, зі стереохіміч-
ними відмінностями, а структури звичайних, найвідоміших і найпоширеніших
моносахаридів відрізняються кількістю карбонових атомів і відносним положен-
ням функціональних груп, то методи визначення хімічної будови різних
моносахаридів досить близькі. Розглянемо їх на прикладі двох найпоширеніших
представниках моносахаридів - глюкози й фруктози. Ці роботи виконані
наприкінці XIX ст. Б. Толленсом, Е. Фішером і Г. Кіліані.
До початку цих досліджень було відомо, що глюкоза має склад СбНі2Об. Під
час ацилювання оцтовим ангідридом вона утворює пентаацетат, а це означає, що в
моносахариді є п'ять гідроксильних груп.
Глюкоза відновлює аміачний розчин арґентум оксиду й фелінгову рідину. її
легко окиснює бром. Ці реакції ведуть до утворення глюконової кислоти (IV) з
тією ж кількістю гідроксильних груп, що й у глюкозі. Наявність альдегідної
групи в моносахариді також підтверджена відновленням глюкози до шести-
атомного спирту сорбітолу (V).
Наявність у глюкозі нерозгалуженого ланцюга з шести атомів Карбону
підтверджена відновленням моносахариду іодидною кислотою до 2-іодогексану
(VI) і глюконової кислоти до гексанової (VII). Цей же висновок можна зробити
на підставі синтезу ціангідрину (VIII) з глюкози і його відновленням після
омилення (IX) до гептанової кислоти (X). Все викладене можна проілюструвати
наведеною нижче схемою:
СН2ОАс
СНОАс
СНОАс
СНОАс
СНОАс
СН2ОАс
СН=0 СООН
СНОН СНОН
Ас.0 СНОН на/нё СНОН вг2> СНОН
СН2ОН
СНОН
ні
СНОН
СНОН
сн2он
СНОН
СНОН
сн2он
нсы
сн3
СНІ
сн2
сн2
сн2
сн3
VI
СНОН
СНОН
СНОН "
СНОН
снон
сн2он
VIII
снон
снон
сн2он
IV
соон
снон
снон
снон~
9нон
снон
сн2он
IX
СН3(СН2)4СООН
VII
НІ
СН3(СН2)5СООН
X
Будова кетогексози - фруктози - запропонована на підставі таких реакцій.
Аналогічно до глюкози, фруктоза утворює пентаацетат, а її відновлення
приводить до суміші двох шестиатомних спиртів, один із яких - сорбітол (V),
що свідчить про структурну подібність до глюкози. §
622
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
На відміну від глюкози, фруктозу не окиснює бром, а жорстка дія, наприклад,
нітратною кислотою розщеплює карбоновий ланцюг і дає гліколеву та винну
кислоти. Отже, у фруктозі, на відміну від глюкози, існує кетогрупа. її локалізація
випливає з подальшої серії реакцій. У разі дії на фруктозу (її) ціанідної кислоти
утворюється ціангідрин (XI), омилення якого дає гідроксикислоту (ХП). Під час її
відновлення отримано 2-метилкапронову кислоту (XIII). Звідси зрозуміло, що
кетогрупа міститься біля другого карбонового атома фруктози:
1 СН2ОН
2 С=0
3 СНОН
« СНОН
5 СНОН
6 СН2ОН
СН2
І п
НСЫ
.2ОН
^ОН
у-см
фнон
СНОН '
СНОН
сн2он
XI
сн2
і2ОН
г-ОН
УЧЮОН
СНОН
СНОН
СНОН
сн2он
XII
НІ
сн3
сн3сн2сн2сн2-снсоон
XIII
23.1.3. Конфігурація моносахаридів
З наведених вище структурних формул моносахаридів видно, що вони
мають значну кількість асиметричних атомів Карбону. Кількість стереоізомерів
для альдогексоз при чотирьох асиметричних атомах дорівнює 24 = 16, а для
альдопентоз і кетогексоз (три асиметричні атоми) вона зменшується до восьми.
Для позначення конфігурації стереоізомерних моносахаридів зрідка
використовують ІІ^-систему. Замість цього традиційно застосовують набагато
раніше введену В,Ь-номенклатуру.
Родоначальником генетичного ряду Б-альдоз є правообертальний Б-глі-
цериновий альдегід:
СНО
Н+ОН
СН2ОН
(Я)- або О-гліцериновий альдегід
Від цієї тріози синтетичним шляхом можна перейти до тетроз. За основу
синтезу Кіліані (детальніше див. 23.2) взято приєднання ціанідної кислоти до
альдегідної групи гліцеринового альдегіду. У цьому разі поряд зі збільшенням
карбонового ланцюга на один атом виникає новий асиметричний центр і, отже,
утворюється пара діастереомерів. Специфічне відновлення амальгамою Натрію
нітрилів до альдегідів завершує синтез:
нсы * [н 1 *
я-сн=о > їі-сн-си >- я-сн-сн=о
І І
он он
Цей метод дає змогу з одного моносахариду одержати два нові з більшим на
один карбоновий атом ланцюгом, що відрізняються конфігурацією верхнього
Розділ 23. Вуглеводи
623
тетраедра. Такі діастереомерні пари називають епімерами. Пари епімерів
розділяють зазвичай кристалізацією їхніх похідних, наприклад, фенілгідразонів.
Перехід від Б-тріози до Б-тетрози і так далі утворює ряд О-альдоз.
У всіх цих моносахаридів однакова конфігурація нижнього асиметричного
центра, а розходження у взаємній конфігурації позначають тривіальними назвами,
наприклад:
но-
ОН
-ОН
ОН
ОН
НО-
но-
-он
-ОН
но-
но-
ОН
ОН
трео-
рибо-
гстакто-
Огже, назва альдози складається з позначення конфігурації нижнього
тетраедра, тривіальної основи, що позначає конфігурацію всіх асиметричних
центрів, і суфікса -оза.
Ряд Б-альдоз
сно
н+он
снрн
І>Глщериновий альдегід
/
сно
н+он
н—он
н+он
а^он
І>Рибоза
/
\
сно сно
н-4-он но-І-н
н+он н+онно
н+он н+он н
сно
н+он
н+он
сь^он
ЕМЕригроза
сно
но+н
н—он
н+он
Е)-Арабіноза
/ \
сно сно
н+он но+н
н но—н
сно
но+н
н
он
снрн
ЕКГреоза
н+он н+он н+он н+он но+н но
н
сно
н+он
но—н
н+он
снрн
ІЖсилоза
/ \
сно сно
он но+н
сно
он н+он н+он н+он н+он н+он
но—н
но—н
н+он
снрн
О-Ліксоза
/ \
сно сно
н+он но+н
н+он н+он но+н но+н
-н но-
-н но+н
снрн снрн сщм снрн снрн снрн снрн снрн
В-Алоза І>Альтроза І>Глюкоза І>Маноза І>Гупоза І>Ідоза І>Галакгоза ІУГалоза
z ¿ ¿ ¿
D-Псикоза D-Фруктоза D-Сорбоза D-Тагатоза
D-Ксилулоза
L-Еритрулоза
D-Еритрулоза
D-Рибулоза
624 Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Аналогічно, на підставі Ь-гліцеринового альдегіду, будують також Ь-ряд
альдоз.
З використанням як вихідної сполуки дигідроксіацетону можна скласти ряд
В-кетоз:
23.1.4. Визначення конфігураціїО-глюкози
й О'Манози
До 60-х років XIX ст. виділено значну кількість моносахаридів, і постало
питання про визначення їхньої конфігурації. Ці дослідження виконав Е. Фішер
на підставі доказу конфігурації В-глюкози й В-фруктози. Для цього шляхом
окиснення або відновлення він "зрівнював кінці" моносахаридів, перетворюючи
їх у двоосновні карбонові кислоти або поліоли. Унаслідок цього для окремих
молекул з'являлася площина симетрії, що призводило до втрати молекулою
оптичної активності. У разі такого прийому не зачіпалися асиметричні атоми
молекул. Це давало змогу вилучити окремі конфігурації для моносахаридів.
Простежимо ці прийоми на прикладі визначення стереохімічних формул В-
глюкози й В-манози.
Розділ 23. Вуглеводи
625
З іншого боку, у разі окиснення арабінози нітратною кислотою отримано
оптично активну арабінарову кислоту (XIV). Для неї, а отже, і для арабінози
можливі конфігурації арабіно- (XV) або ліксо- (XVI), а вилучені кето- (XVII) і
рибо- (XVIII) як оптично неактивні завдяки наявності площини симетрії:
СООН
но-
-ОН
ОН
СООН
XIV
НО-
ОН
он
но-
но-
соон
он
-не-
он
СООН
он
ен-
-он
СООН
XV
XVI
XVII
он
СООН
XVIII
За допомогою ціангідринового синтезу з арабінози отримано глюконову й
манонову кислоти. Отже, конфігурації атомів С4, СЗ і С2 в арабінозі збігаються
з конфігурацією атомів С5, С4 і СЗ у манозі й глюкозі. З огляду на це глюкозі й
манозі не можуть відповідати сіло-, альтро-, гуло-, ідо-конфігурації, які утво-
рюються з ксилози й рибози.
Під час окиснення Б-глюкози й Б-манози виникають оптично активні
глюкарова (XIX) і манарова (XX) кислоти:
СООН
ОН
НО-
СООН
НО-
НО-
ОН
ОН
СООН
XIX
СООН
ОН
-он
-он
СООН
XX
но-
но-
-он
СООН
XXI
-он
но-
но-
-он
но-
но-
но-
-он
XXII
XXIII
У випадку окиснення галактози утворюється оптично неактивна галактарова
кислота (XXI). З цієї причини для Б-глюкози й О-манози вилучена галакто-
конфігурація (XXII). Як випливає з генетичного ряду, наведеного вище,
галактоза й талоза відрізняються стереохімією атома С2. Отже, тало-
конфігурація (XXIII) також не може відповідати О-глюкозі й Б-манозі. Для них
залишаються тільки дві можливі структури:
СНО
ОН
НО-
СНО
но-
но-
ОН
-ОН
ОН
-ОН
СН2ОН
СН2ОН
Вибір між ними зроблений за допомогою ціангідринового синтезу на основі
глюкози й манози з наступним окисненням до гептарових кислот. Одержання з
626
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
глюкози оптично неактивної дикарбонової кислоти (XXVI) дає змогу приписати
для Б-глюкози будову XXIV, а для манози - XXV:
СООН
ОН
ОН
-не-
[О]
СООН
он
он
но-
-он
-он
СООН
XXVI
СООН
он
СООН
сно
он
-он
-он
но-
но-
но-
сн2он
СООН
он
но-
но-
[О] НО-
' но-
-он
-он
СООН
-он
-он
сн2он
О-Глюкоза(ХХІУ)
сно
СООН
но-
-он
І2ІНО-
-он
-он
-он
сн2он
СООН
но-
но-
-он
-он
сн2он
но-
но-
но-
-он
-он
сн2он
О-Маноза (XXV)
-он
-он
СООН
СООН
но-
[О] НО-
~^но-
-он
-он
сн2он
-он
-он
СООН
З огляду на той факт, що фруктоза дає однаковий озазон з Б-глюкозою й Б-ма-
нозою, Е. Фішер запропонував для неї таку структуру:
СН2ОН
СЮ
но-
-ОН
-ОН
СН2ОН
Незабаром аналогічно визначили відносні конфігурації інших пентоз
гексоз.
23.1.5. Циклічні форми моносахаридів
Фішерівські проекції з альдегідними й кетонними групами, так звані
ланцюгові, або лінійні, структури, добре застосовувати для опису конфігурації
моносахаридів. Проте окремі властивості цих сполук не можна пояснити за
допомогою фішерівських формул. Передусім карбонільна група не дає деяких
альдегідних реакцій, наприклад, з фуксинсульфітною кислотою, не утворює
гідросульфітної похідної. Повні ацетати цукрів зовсім не виявляють ніяких
альдегідних властивостей. Крім того, один з гідроксилів у моносахаридах має
особливі властивості. Він легко під час нагрівання з 3 % розчином гідроген
Розділ 23. Вуглеводи
627
хлориду в метанолі утворює ізомерну суміш монометоксильних похідних.
Зазвичай у таких м'яких умовах спирти не утворюють етерів.
Свіжоприготовлені розчини моносахаридів мутаротують, тобто змінюють
значення оптичного обертання. Наприклад, перекристалізований з води зразок
D-глюкози у випадку розчинення у воді має первісне питоме обертання [а]о
+111°, a водний розчин глюкози, кристалізованої з піридину, - [а]о +19,2°. Під
час відстоювання розчинів питоме обертання змінюється і в обох випадках
досягає [cc]d +52,5°. На підставі ланцюгових формул моносахаридів ці феномени
пояснити неможливо.
Для пояснення виявлених фактів А. Колі ще 1870 р. запропонував щодо
моносахаридів циклічну структуру з тричленним оксидним кільцем:
носн2-снон-снон-снон-сн-снон
О
Після появи теорії напруги А. Байєра Б. Толенс виділив п'ятичленний
оксидний цикл:
НОСНгСН-СНОН-СНОН-СНОН
2^ о >
Напівацетальна циклічна форма добре пояснює особливості властивостей і
поведінки моносахаридів. Зазначимо, що в цьому випадку з'являється новий
асиметричний атом (у формулі Толенса він позначений зірочкою), гідроксильна
група при якому може перебувати над і під площиною кільця. Такі діастерео-
мерні пари називають а- і $-аномерами. Напівацетальний гідроксил, що виник,
відповідно, одержав назву аномерного, або глікозидного.
❖ Експериментальне визначення розмірів циклу моносахаридів. Вислов-
лене Б. Толленсом припущення про наявність у моносахаридах п'ятичленного
оксидного кільця потребувало експериментального підтвердження. Для вирі-
шення цього питання У. Хеуорс1 вирішив досліджувати метилглікозиди
моносахаридів, у яких глікозидний гідроксил блокований, що унеможливлює
наявність рівноваги між циклічною (кільчастою) й ланцюговою формами.
Під час метилювання а-метилглюкозиду (XXVIII) отримано метилтетра-О-
метилглюкозид (XXIX), кислотний гідроліз якого привів до тетра-О-метил-
глюкози (XXX). У разі її окиснення нітратною кислотою, всупереч очікуванням,
отримано триметоксиглутарову (XXXI), а не диметоксиянтарну кислоту. Отже,
цикл є не п'яти-, а шестичленним:
1 Хеуорс В. (Налуогйі W.) (1883-1950) - англійський хімік-органік. Головні дослідження при-
свячені вуглеводам, а також вітаміну С. Нобелівська премія 1937 р.
628
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
но-
-ОМе
ОН
-ОН
З
(МеО)2802 —
ЫаОН * МеО"
сн2он
Ме = СН, XXVIII
-ОМе
-ОМе
-ОМе
Н20>
н+
СНО
ОМе
МеО-
[О]
СООН
ОМе
МеО-
С^ОМе
XXIX
-ОМе
-ОН
СН2ОМе
ххх
-ОМе
СООН
XXXI
Інше підтвердження циклічної структури глікозидів отримане Хадсоном і
Джексоном за результатами періодатного окиснення. Під час дії на а-
метилглюкозид періодатної кислоти, що, як відомо, розщеплює тільки а-
глікольні угруповання, відбувається поглинання 2 моль окиснювача з утво-
ренням 1 моль мурашиної кислоти. Неважко переконатися в тому, що такі
результати можна отримати тільки у випадку піранозного циклу:
но-
-ОСН,
-ОН '
оса
2НІ04
СНО
-он
j
нсоон +
я.
но
і
з
сн2он
сн2он
Нижче наведено дані щодо очікуваного складу продуктів окиснення й
витрати окиснювача для всіх циклічних структур, можливих для а-метил-
глюкозиду:
осн3о
но-
ОН
:ОН
СН2ОН
1 моль СН20
2 моль НСООН
3 моль НЮ4
-ОСН3
-он
ОН
:0Н
СН20Н
но-
-ОСН3
-ОН
0
j
но-
:0Н
СН20Н
-ОСН.
-ОН
ОН
Jo ноЕЕ
ОСН,
он
о
1 моль СН20 1 моль СН20
1 моль НСООН НСООН
2 моль НЮ4 не утворюється
2 моль НЮ4
СН20Н
СН20
не утворюється
1 моль НСООН
2 моль НІ04
ОН
ОН
сн2-
СН20
не утворюється
2 моль НСООН
3 моль НЮ4
Наявність піранозного циклу доведена на прикладі інших моносахаридів. У
дослідах У. Хеуорс виявив і моносахариди з п'ятичленними циклами. Моносаха-
риди в п'ятичленній циклічній формі називають фуранозами, а в шестичленній -
піранозами.
❖ Аномери. Як зазначено, кожен моносахарид може існувати у двох
діастереомерних формах, що пов'язане з наявністю додаткового асиметричного
Розділ 23. Вуглеводи
629
атома Карбону, який з'являється внаслідок замикання піранозного або фура-
нозного циклу. Такі стереоізомерні моносахариди назвали аномерами.
Аномер позначають як а-, якщо в проекції Фішера конфігурація його
аномерного гідроксилу збігається з конфігурацією нижнього асиметричного
центра, в іншому випадку - Р:
СН2ОН
СН2ОН
СН2ОН
СН2ОН
Р-О-Глкжопіраноза
а-О-Глюкофураноза
Конфігурацію при СІ в аномерних альдозах з'ясував експериментальним
способом Я. Безекен. Він визначив, що електропровідність водних розчинів
борної кислоти різко зростає в разі додавання а-г/ис-гліколів:
І
-с-он
І
-с-
I
—с-он
но
+
но
х /он но-с—
/в + но-с-
I
-с-о^о-с-
с-о'%-с-
н+
а-Б-Глюкоза
Б-Глюкоза
Цей факт послугував основою для
експериментального визначення конфі-
гурації аномерів. Якщо додати свіжо-
приготовлений розчин а-Б-глюкози до
водного розчину борної кислоти, то це
різко збільшить електропровідність роз-
чину, після чого електропровідність посту-
пово зменшуватиметься, поки не досягне
сталого значення. Додавання розчину Р-Б-
глюкози зумовлює поступове збільшення
електропровідності до досягнення того ж
сталого значення, що й у випадку а-Б-
глюкози (рис. 23.1).
Із цих результатів випливає, що а-Б-глюкопіраноза повинна мати цис-, а Р-
Б-глюкопіраноза — тярянс-конфігурацію при С2- і С7-атомах.
❖ Проекція Хеуорса. Порівняно з проекційними формулами Фішера, ліпше
уявлення про конфігурації циклічних форм моносахаридів можна одержати за
допомогою перспективних формул Хеуорса:
Н—І—І—І—І
2 4 6 8 10 г,час
Рис. 23.1. Зміна електропровідності
борної кислоти за наявності
а- і р-й-глюкози
630
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Цикл у цій проекції розташовують перпендикулярно до площини паперу
так, щоб атом Оксигену був максимально віддалений від спостерігача, а зв'язки
Н-С-ОН спрямовані паралельно до площини паперу. Часто для спрощення
структури, зв'язки С-Н не зображають.
Побудову проекцій Хеуорса можна зобразити як зближення гідроксильних
груп, що реагують з карбонільним атомом Карбону і перебувають біля у- і 5-
карбонових атомів. У цьому разі для замикання циклу необхідний оберт на 120°
по зв'язках СЗ-С4 і С4-С5, відповідно:
Для гексапіраноз спрощений перехід від проекцій Фішера до проекції
Хеуорса можна виконати в такий спосіб. Гідроксиметильну групу моносахаридів
Б-ряду розташовують над площиною малюнка, а Ь-ряду - під площиною.
Гідроксильні групи моносахаридів О-ряду, які в проекції Фішера розташовані
ліворуч, спрямовують униз, а розташовані ліворуч - угору, наприклад:
КНЧ-ОН
СНоОН
СН^ОН р-О-Глюкопіраноза
Кетогексози, як і альдогексози, здебільшого існують у шестичленній
(піранозній) формі, причому замикання циклу відбувається за участю кетогрупи й
гідроксильної групи при атомі Карбону Сб:
Розділ 23. Вуглеводи
631
но
осн2он
он
но
гсн2он
а -О-Фруктопіраноза
р-О-Фруктопіраноза
У випадку альдопентоз кільчасто-ланцюгова таутомерія веде до замикання
циклу за участю альдегідної групи й гідроксилу при атомі Карбону С5, що
приводить до утворення піраноз. На відміну від альдогексоз, альдопентози частіше
існують у фуранозній формі. Особливо це стосується пентоз у полісахаридах:
❖ Мутаротація. Причиною мутаротації моносахаридів є кільчасто-
ланцюгова таутомерія, що виникає у водних розчинах. Згодом усталюється
рівновага між лінійною й циклічними піранозними й фуранозними формами
моносахаридів. І хоча вміст лінійної форми невеликий (зазвичай близько 0,1 %),
однак завдяки зсуву рівноваги моносахариди дають деякі якісні реакції на
карбонільну групу:
ОН
а-О-Ксилопіраноза
ОН он
Р -О-Рибофураноза
но
ч-он
-(-он
сн2он
сно
_он ^
сн2он
но4-
он
О-Глюкоза
он
р-О-Глюкопіраноза
он
р-О-Глюкофураноза
632 Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
23.1.6. Конформації моносахаридів
Більшість моносахаридів, що перебувають у формі піранозних циклів,
насправді мають неплоску будову. Для них найстабільнішими е конформації
"крісла" (позначають С від англ. chair - крісло). З огляду на наявність атома
Оксигену, що зумовлює асиметрію в конформації "крісла", для моносахаридів
існують дві форми, які називають *С4- й 4Сі-конформаціями (верхній індекс
означає, який атом перебуває вище площини, а нижній — відповідно, нижче
площини):
P-D-Глюкопіраноза
Вибір між двома конформаціями залежить від просторового розташування
замісників: конформація з найменшою кількістю аксіальних замісників є
найстійкішою. У цьому разі чим більший об'єм замісника в аксіальному
положенні, тим менш стійка конформація. Особливо це стосується гідрокси-
метильної групи СН2ОН у гексопіранозах, просторове розташування якої є
вирішальним чинником стійкості конформації.
■ Як і в молекулі циклогексану, особливо сильний негативний вплив на
стійкість конформацій моносахаридів має 1,3-діаксістьне розташування
замісників. Наприклад, в а-рибопіранозі 1,3-діаксіальна взаємодія виникає в
обох конформаціях:
■ Крім того, на стійкість конформації моносахаридів впливає "А -ефект",
пов'язаний з наявністю в циклі атома Оксигену. Якщо гідроксильна група біля
С2 розташована аксіально, а замісник у СІ - екваторіальний, то оксигенові
атоми гідроксилів і Оксиген циклу виявляються просторово зближеними, що
веде до сильного взаємного відштовхування й, отже, до лабільності конформації.
Це легко продемонструвати на прикладі Р-Б-манопіранози за допомогою
проекцій Ньюмена:
Розділ 23. Вуглеводи
633
2
он
■ Через наявність атома Оксигену в циклі моносахаридів виникає ще один
чинник нестійкості - "аномерний ефект". Наприклад, у рівноважній суміші а- і
Р-метил-О-глюкопіранозидів несподівано переважає сс-аномер, що перебуває в
аксіальному положенні. Це явище пояснюють взаємодією диполя С7-Х і
сумарного диполя вакантних електронних пар Оксигену циклу. Відштовхування
між цими диполями сильніше для екваторіального аномеру, коли диполі
паралельні, і слабше для аксіального аномеру:
Є кількісні критерії розрахунку нестійкості конформацій, які задовільно
узгоджуються з експериментальними даними. Очевидно, відсутність чинників
нестійкості для Р-О-глюкози визначає її поширення в природі.
Подібно до всіх карбонільних сполук моносахариди здатні до відновлення в
поліоли:
23.2. Хімічні властивості моносахаридів
23.2Л. Реакції відновлення
—он
но—
—он
—он
сн2он
сно
сн2он
—І-он
И но
—І-он
—Нон"
сн2он
О-Глюкоза
О-Сорбіт
Під час відновлення альдоз утворюється тільки один поліол, а відновлення
кетоз дає два стереоізомерні спирти. Наприклад, із Б-фруктози одержують Б-
сорбіт і Б-маніт:
634
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
СН2ОН
С = 0
СН2ОН
но-
ОН -
ОН
сн2он
но-
•он
•он '
■он
сн2он
но-
но-
сн2он
-он
-он
сн2он
Відновлення
О-Фруктоза
моносахаридів
виконують за
О-Сорбіт О-Маніт
у лабораторії найчастіше
допомогою натрій борогідриду ИаВНі або амальгами Натрію. Гідрування в
промисловості проводять над платиновими, паладієвими, скелетними нікеле-
вими каталізаторами за кімнатної температури й атмосферному тиску.
23.2.2. Реакції окиснення
❖ Окиснення альдегідної групи. Найлегше в моносахаридах окиснюється
альдегідна група, унаслідок чого утворюються альдонові кислоти. Найліпшим
окиснювачем для препаративних цілей є бром. У промисловості, щоб одержати
кальцій глюконат, проводять електролітичне окиснення глюкози бромом за
наявності кальцій карбонату. Бром, що виділяється з кальцій броміду на аноді,
окиснює глюкозу до альдонової кислоти, яка під час взаємодії з кальцій
карбонатом дає глюконат.
Альдонові кислоти, що утворюються внаслідок окиснення, легко перетво-
рюються в лактони, у рівноважній суміші яких переважає у-форма:
СН^ОН
НО^О
ОН
у-Лактон
соон
он
но-
-он
-он
сн2он
О-Глюконова кислота
сн2он
но^ (
он
8-Лактон
❖ Окиснення первинної спиртової групи. У разі каталітичного окиснення
найлегше окиснюються первинні спиртові групи. У цьому випадку синтезуються
урокові кислоти. Для перебігу реакції моносахарид перетворюють у глікозид або
яку-небудь іншу похідну, щоб захистити аномерний центр:
снрн
О^1—-/о
сурі
СООН
кон >
о4*—/6
но ^—г осн3 но^—г сен,
он он
а-Метил-О-глюкопіранозид а-Метил-О-глюкопіранозидуронова кислота
Розділ 23. Вуглеводи
635
❖ Окиснення а-глікольних угруповань. Для реакції використовують
періодатну (IV) кислоту й Плюмбум (TV) ацетат. Такі реакції широко застосовують
у хімії вуглеводів для визначення будови moho-, оліго- і полісахаридів. За
допомогою реакції розщеплюють a-гліколі, що веде до утворення двох альдегідів:
R-CHOH-CHOH-Rf +НЮ4-
RCHO +Н20 + R'CHO + НІ03
Отже, на окиснення а-глікольного угруповання витрачається 1 моль
періодатної кислоти.
Сполуки, що мають тріольне угруповання, розпадаються з утворенням
молекули діальдегіду і мурашиної кислоти. З первинної спиртової групи
утворюється формальдегід. Реакція відбувається кількісно, тому на її основі
розроблено низку аналітичних методів для визначення структури вуглеводів.
❖ Одночасне окиснення альдегідної і первинної гідроксильної груп. Під
час окиснення моносахаридів нітратною кислотою утворюються дикарбонові
гідроксикислоти — альдарові кислоти:
СНО СООН
но-
ОН
HNO, но-
-он
ОН
сн2он
D-Глкжоза
■он
■он
-он
СООН
D-Глюкарова кислота
(цукрова кислота)
23.2.3. Лі я на моносахариди кислот і основ
Під дією кислот і основ моносахариди зазнають численних перегрупувань.
Першою стадією цього процесу є утворення енолу:
но-
Ч СНО
Н+ОН
Н+ОН
R
он
н
R
СНО н+
снон
R
он с-он
он = н+он
R
■ Енол трансформується до вихідної альдози, її епімеру по 0.2-атому, й
кетози. Ця реакція відома як реакція Добрі де Брюїна1 й еон Екенштейна (1895):
1 Лобрі де Брюїн К. (Lobry van Troostenburg de Bruyn C.A.) (1857-1904) - нідерландський хімік,
досліджував ізомерні форми та алкалоїди. Синтезував гідроксиламін та гідразин.
636
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
сн2он
+ОН =
снон^
с-он ^
—гон
її
сно
он
он
сно
но-
-он
■ Відщеплення від енолу молекули води в кислому або лужному сере-
довищі веде до утворення З-дезокси-2-кетоальдози:
снФн
снон
н+
СїіФн
-он
сн^
он
сно
с-он
сн
сно
с=о
■ У кислому середовищі З-дезокси-2-кетоальдози перетворюються в
похідні фурану - 5-гідроксиметилфурфурол:
НОНр
XX,
о
1
СНО
5-Гідроксиметилфурфурол за подальшого впливу хлоридної кислоти
розпадається до левулінової кислоти СН3СОСН2СН2СООН.
Перетворення моносахаридів у похідні фурану має важливе практичне
значення. У промисловості для одержання фурфуролу переробляють різні
відходи сільськогосподарського виробництва, що мають великий вміст
пентозанів - полісахаридів, що складаються з пентоз. Для цього сировину
нагрівають з кислотами, що спочатку зумовлює гідроліз, а потім перетворення
пентоз у фурфурол.
■ У лужному середовищі 3-дезоксигексозулози через низку перегрупувань
перетворюються в сахаринові кислоти:
Розділ 23. Вуглеводи
637
соон
Hjc-T-OH
-он
-он
сн2он
а-Глюкосахаринова кислота
Сахаринові кислоти застосовують у синтезі розгалужених моносахаридів.
23.2.4. Методи вкорочення й подовження
карбонового ланцюга моносахаридів
Для визначення будови моносахаридів або в синтетичних цілях часто
застосовують вкорочення або подовження ланцюга моносахаридів.
❖ Метод Руффа1 полягає в дії гідроген пероксиду на сіль альдонової
кислоти за наявності йонів Феруму (III). Вважають, що реакція окиснення
відбувається через стадію утворення 2-кетоальдонової кислоти:
СООН
ОН
НО-
COOH
І
с=о
сно
но-
-он
-он
сн2он
D-Глюконова
кислота
Fe3+
но-
-ОН -сог
-ОН
сн2он
2-OKCO-D-apa6mo-
гексонова кислота
-он
-он
сн2он
D-Арабіноза
❖ Розпад за Волєм полягає в дії на оксими моносахаридів оцтового
ангідриду з наступною обробкою отриманого нітрилу амоніачним розчином
арґентум гідроксиду:
но-
CH=NOH
ОН -
(СН3со)2о СН,ОСО-
ОН
-ОН
скри
Оксим D-глюкози
C=N
-ОСОСН3
- AgOH
-ОСОСН3 NH,
-ОСОСН,
СНО
но-
CHpCOCHj
-он
-он
СН2ОН
D-Арабіноза
❖ Розщеплення за Веєрманом. Третій метод деградації моносахаридів
полягає в розщепленні аміду альдонової кислоти під дією гіпохлоритної кислоти
за умов реакції Гофмана:
1 Руф О. (Ruff О.) (1871-1939) - німецький хімік, учень Е. Фішера. Автор кількох праць з хімії
вуглеводів. Потім займався неорганічної хімією, насамперед сполуками галогенів. Є автором
кількох підручників.
638 Чирва В.Я., Ярллолюк СМ,, Толкачова Н.В., Зеллляков О.Є. Органічна хімія
В-Глюконолактон О-Глюконамід, 80 % В-Арабіноза, 49 %
❖ Синтез Кіліані. Ціангідринний синтез є класичним методом подовження
ланцюга альдоз. Е. Фішер з'ясував, що в цьому разі утворюються два
енантіомерні нітрили, які після гідролізу перетворюються в альдонові кислоти, а
тц легко переходять у у-лактони. На цій стадії відбувається поділ епімерів на
індивідуальні сполуки завдяки різній розчинності. їхнє відновлення амальгамою
Натрію дає альдози:
L-Глюконова кислота L-Глюкоцолактон L-глюкоза
L-Арабіноза
L-Манова кислота L-Манонолактон L-Маноза
❖ Нітрометановий метод. За основу нітрометанового методу взято
конденсацію первинних і вторинних нітроалканів з альдегідами й гідроліз солей
аци-форм нітросполук мінеральними кислотами. Отримані в реакції епімери
порівняно просто розділяють за допомогою кристалізації:
Розділ 23. Вуглеводи
639
23.2.5. Реакцїі за карбонільною групою
❖ Реакції з гідроксиламіном. Під час взаємодії моносахаридів з гідро-
ксиламіном утворюються добре розчинні у воді й спирті оксими. Більшість
оксимів мутаротує у розчинах, що підтверджує наявність таутомерних пере-
творень між відкритою й циклічною формами оксимів:
нс=шн сн2он
Он
ынон
он
сн2он
снрн
З огляду на це оксими не використовують для ідентифікації моносахаридів.
❖ Реакції з гідразинами. На первинному періоді розвиток хімії вуглеводів
гальмували труднощі одержання кристалічних похідних вуглеводів, оскільки
вони внаслідок високої гідрофільності мають тенденцію до утворення сиропів,
що не зазнають кристалізації. Цю перешкоду подолав Е. Фішер, увівши 1884 р.
у синтетичну практику фенілгідразин, що реагує з багатьма карбонільними
сполуками, утворюючи малорозчинні гідразони, які добре кристалізуються:
СНО
ОН
НО-
НС=І\[-МН-С6Н5
но-
-ОН +
-ОН
СН2ОН
О-Глюкоза
-он
-он
-он
сн2он
Гідразон Э-глюкози
Під час взаємодії моносахаридів з надлишком фенілгідразину утворюються
озазони. Спочатку синтезується гідразон. Потім під дією фенілгідразину
відбувається окиснення гідроксильної групи в а-положенні до кетону, який
також вступає в реакцію з фенілгідразином:
СНО
ОН
но-
но-
сн=к-ынсбн5
С=к-ш-с6н5
-ОН
-ОН
ЗСбН5МНг\ІН2
с6н5і>м2>
сн2он
О-Глюкоза
-ОН
ОН
СН2ОН
Озазон О-глюкози
Оскільки в молекулі моносахариду в цій реакції зникає один асиметричний
центр, то епімери альдоз, а також 2-кетози й 2-аміноцукри, що мають однакову
640
Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
конфігурацію інших карбонових атомів, дають один і той же озазон. Ці сполуки
відіграли важливу роль у розвитку хімії вуглеводів, їх широко використовували
в з'ясуванні будови й стереохімії моносахаридів.
23.2.6. Реакції за участю спиртових
гідроксильних груп
❖ Естери моносахаридів. У хімії вуглеводів найбільшого поширення
набули ацетати й бензоати. Ці естери використовують як тимчасовий захист
гідроксильних груп, а також для виділення й розділення моносахаридів. Естерні
групи легко відщеплюються в разі омилення натрій метилатом або амоніаком,
що приводить до регенерування вихідних речовин.
Ацилювання зазвичай проводять дією оцтового ангідриду в піридині на
холоді. За цих умов утворюються повні ацетати зі збереженням конфігурації в СІ:
СН2ОН СН2ОСОСН3
0 (СН3СО)20 ^ 0
CH3OCON f ОСОСН3
ОСОСН3
На відміну від ацетатів, бензоїльну групу можна ввести селективно. Крім
того, вона має меншу схильність до міграції й стійкіша за умов гідролізу.
Бензоїлювання виконують як звичайно дією бензоїлхлориду в піридині або за
наявності лугу за Шоттен}-Бауманном2. Ця реакція відбувається менш
енергійно, ніж ацилювання, що використовують для вибіркового введення
захисту за первинною спиртовою групою або глікозидним гідроксилом.
Наприклад, у випадку одержання 1,6-ди-С?-бензоїл-В-маніту або 1-О-бензоїл-
4,6-(9-бензиліден-В-глюкози:
но-
но-
сн2о-с
с6н5
-он
ОН ,0
с6н5-сн
с6н5
снр-с
1,6-Ди-0-бензоїл-О-маніт 1 -О-Бензоїл-4,6-0-бензиліден-О-глюкоза
1 Шоттен K (Schotten С.) (1853-1910) - німецький хімік, учень А. Гофмана. Працював у
Берлінському університеті.
2 Бауманн Е. (Baumann Е) (1846-1896) - німецький хімік. Автор досліджень з фізіологічної та
харчової хімії.
Розділ 23. Вуглеводи
641
❖ Сульфати. З огляду на те, що сульфати полісахаридів значно поширені в
природі й відіграють важливу біологічну роль, сульфати моносахаридів цікаві як
модельні фрагменти природних біополімерів. їх зазвичай одержують дією
піридинсульфотриоксиду в піридині за кімнатної температури. Естерифікування
зазнає передусім первинна спиртова група:
H2OS03H
ОН
Сульфоестери легко зазнають розщеплення кислотами.
❖ Етери моносахаридів
■ Найбільший інтерес серед етерів становлять метилові, тому що частково
метильовані моносахариди значно поширені в природі. Зокрема, вони є в складі
полісахаридів, серцевих глікозидів й антибіотиків. Метилювання виконують
дією на моносахариди метиліодиду за наявності арґентум оксиду (метод
Пурді1-Ірвіна2) або диметилсульфатом
гідроксиду (метод Хеуорса):
(СНзО)2802 за наявності натрій
СН2ОН
НО
осн,
Ag2o
сн
он
сн2осн3
,oN—Г осн3
осн,
На відміну від естерів метилові етери є дуже стійкими сполуками, що
відіграло вирішальну роль у визначенні будови оліго- і полісахаридів.
■ Тритилові (трифенілметилові) етери мають важливе значення в синте-
тичній хімії вуглеводів, оскільки тритилюванню піддаються переважно первинні
спиртові групи. їх одержують дією на моносахариди трифенілхлорометану
(ТгСІ) у піридині:
сн2он
НО41 ГОСН3
он
ТгСІ»
C}H5N
СН2ОТг
НО41 ГОСНз
он
Тг = -С(С6Н5)3
1 Пурді Т. Ригсііе ТИ. (1843-1916) - британський хімік, засновник наукової школи вуглеводнів
університету Сан Андре (Шотландія).
2 Ірвін Дж. (Sir Irvine J.C.) (1877-1952) - I
британський хімік, учень Т. Пурді, автор досліджень з
хімії вуглеводів. З 1921 до 1952 рр. - ректор університету Сан Андре (Шотландія).
642
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Вибірковість пояснюють, очевидно, значним об'ємом тритильної групи.
Вона стійка до дії основ і легко відщеплюється в кислому середовищі.
23.2.7. Реакції й похідні глікозидного гідроксилу
Напівацетальна, або, інакше, глікозидна, гідроксильна група відрізняється
поведінкою від звичайних гідроксилів. Вона легко зазнає нуклеофільного
заміщення на галоген або алкоксильну групу.
❖ Глікозилгологеніди. Типовим способом одержання глікозилгалогенідів є
дія безводних гідроген галогенідів на ацетати моносахаридів:
СН,ОСОСН, СН2ОСОСН3
ососнЛ
СНзОСО^ ГОСОСНз сн3осо
3 ОСОСН3 ОСОСН3
У глікозилгалогенідах атом галогену легко зазнає заміщення під дією різних
нуклеофілів та елімінування під дією основ:
-o.cn -он
—осі
, -1
ОСОК осоя'
-от У8см — оок
осок ~°\ осок
-о 8К ^ —А "О ОАг
у осок /
( ■ н2о/ \^осоа "^сок,
_о —оосоя
юн ^
осок осоя-
❖ Глікозиди. Найбільшим класом сполук, утворених завдяки напівацеталь-
ному гідроксилу, є глікозиди.
Під час кип'ятіння розчинів моносахаридів зі спиртами за наявності кислот
відбувається заміна напівацетального (глікозидного) гідроксилу на алкоксильні
групи з утворенням змішаного ацеталю, який називають глікозидом:
ОСОЯ'
Розділ 23. Вуглеводи
643
Залежно від природи гетероатома, що з'єднує моносахарид з карбоновим
атомом, розрізняють О-глікозиди, .У-глікозиди, ІУ-глікозиди, С-глікозиди тощо:
Х=09 8, N,0
У рослинному світі найпоширеніші (9-глікозиди. Як невуглеводна (агліконна)
складова є залишки фенолів, флавоноїдів, терпеноїдів, стероїдів тощо. Серед
моносахаридів найчастіше трапляються О-галактоза, Б-глюкоза, Ь-арабіноза, Ь-
рамноза й О-глюкуронова кислота. Нижче наведені приклади окремих глікозидів:
ОН ОН
Арбутин (брусниця) Коніферин (спаржа)
644
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
р-Глюкозид р-ситостерину
(олії вищих рослин)
он
он
конваллятоксин (конвалярія)
Глікозидні зв'язки розщеплюють у кислому середовищі або за допомогою
ензимів. Фермент емульсин з кісточок мигдалю розщеплює тільки Р-глюкозидний
зв'язок, а фермент мальтоза із дріжджів діє тільки на а-глюкозидний зв'язок.
У нуклеозидах, що належать до ^глікозидів, гетероциклічний аглікон
зв'язаний з О-рибозою або 2-дезокси-В-рибозою через атом Нітрогену.
Агліконами є тимін, цитозин, урацил, аденін і гуанін. Нижче наведені структури
окремих нуклеозидів:
О хти О
НО ОН
Аденозин
но он
Гуанозин
но он
Уридин
но он
Цитозин
но он
Риботимідин
233. Найважливіші моносахариди
Формально до моносахаридів належать О- та Ь-гліцериновий альдегід і
дигідроксіацетон. їхні естери з фосфатною кислотою відіграють важливу роль у
розщепленні вуглеводів в організмі. Однак найбільше поширені в природі
пентози й гексози.
❖ Пентози
■ Ь-Арабіноза й В-ксилоза - структурні одиниці пентозанів, звідки їх
одержують кислотним гідролізом. Відновленням ксилози готують ксиліт -
замінник цукру для діабетиків.
■ В-Рибоза є в складі нуклеїнових кислот.
❖ Альдо- і кетогексози. З 16 стереоізомерних альдогексоз у природі
знайдені Б-глюкоза, О-маноза, Б-галактоза й О-талоза.
Розділ 23. Вуглеводи
645
■ й-Глюкоза (виноградний цукор, декстроза) міститься у винограді й
фруктах, є структурною ланкою сахарози, клітковини, крохмалю, глікогену.
■ І>- і Ь-Галактоза трапляються в рослинних і тваринних біополімерах,
містяться в лактозі. У вигляді уронової кислоти є структурною ланкою
пектинових речовин і полісахаридів бактерій.
■ В-Маноза у вільному стані знайдена в цедрі апельсинів; у структурному
фрагменті полісахаридів - мананів, звідки її й одержують.
■ И-Фруктоза (фруктовий цукор, левулоза) міститься в дозрілих фруктах,
камеді, вона'є структурним фрагментом сахарози й полісахариду інуліну.
❖ Дезоксицукри - моносахариди, у молекулі яких одна або кілька гідро-
ксильних груп замінені на атоми Гідрогену. Вони є структурними фрагментами
нуклеїнових кислот, полісахаридів, серцевих глікозидів, антибіотиків.
■ 2-Дезокси-В-рибоза міститься в ДНК.
■ Ь-Рамноза (6-дезокси-Ь-маноза) є структурним фрагментом рослинних
глікозидів.
■ Ь-Фукоза (6-дезокси-Ь-галактоза) є в складі олігосахаридів молока й
різних глікопротеїнів.
❖ Аміноцукри формально одержують заміною гідроксилу на аміногрупу.
■ И-Ацетил-В-глюкозамін (2-ацетамідо-2-дезокси-В-глюкоза) — структур-
ний фрагмент глікопротеїнів, гіалуронової кислоти, олігосахаридів молока,
полісахаридів бактерій. Із залишків ІУ-ацетилглюкозаміну побудований полімер
хітин, з якого кислотним розщепленням виділяють О-глюкозамін.
■ В-Галактозамін - структурний фрагмент глікопротеїнів, деяких бакте-
ріальних полісахаридів, хондроїтиносульфату (склоподібне тіло).
❖ Аскорбінова кислота (вітамін С) перебуває в тісному зв'язку з моно-
сахаридами, міститься у великій кількості в шипшині, свіжих фруктах. Добова
потреба людини становить близько ЗО мг. Нестача вітаміну С спричинює цингу
й підвищену інфекційну сприйнятливість. Аскорбінова кислота - перший віта-
мін, отриманий у чистому вигляді й перший синтетичний вітамін. %
З хімічного погляду вітамін С є похідною моносахариду - у-лактон 2-оксо-Ь-
гулонової кислоти:
СООН
N=0
СООН
.ион
НО—і
Нон
НО
н-
но-
н
-он
•н
н
но-
—он
—он
—н
НО он
СН2ОН
СН2ОН
Таутомерні форми 2-оксо-Ь-гулонової кислоти
Аскорбінова кислота
646
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Сьогодні аскорбінову кислоту у промисловості одержують з глюкози, яку
перетворюють у О-сорбітол шляхом гідрогенізації над купрум-хромовим
каталізатором. Далі сорбітол піддають селективному окисненню за допомогою
АсеіоЬасіег хуііпит до Ь-сорбози:
но-
но-
сн2он
но-
н,
но-
но-
сн2он
-он
но-
сили
с=о
АсеЮЬаМег НО"
"ОН хуііпит —
но-
сно
сн2он
97%
-он
сн2он
70%
Для окиснення гідроксиметильної групи необхідно захистити інші спиртові
групи, що досягають перетворенням Ь-сорбози в її діацетонід (дикеталь). Після
окиснення захист знімають кислотним гідролізом, 2-оксо-Ь-гулонову кислоту
енолізують з одночасною внутрішньомолекулярною циклізацією:
О.
Н(
)Н
сн
НзС^^СН,
н3с, ,сн3
£ОСН3 ^0^0
КМпО,
нон2сч—*сщоп
он
р-Ь-Сорбофураноза
Н+
9\л
скрп
н+
соон Нг°
Діацетонід
2-оксо-Ь-гулонової
соон
сю
но-
но-
н+
-он
sCs ДІаЦвТОНІД и р' "^ц і-ииді-ілулит
Н3С СН, Ь-сорбофуранози "з^ СН3 кислоти (97%)
но—
І—он
о
СНрН
2-Оксо-Ь-гулонова
кислота (82 %)
он он
Аскорбінова кислота
23.3.1. Бродіння моносахаридів
Багато моносахаридів, а також дисахариди мальтоза, сахароза й лактоза
після гідролізу здатні вступати в анаеробне (без доступу кисню) розщеплення
під впливом ферментного препарату з дріжджів (зимаза). Це набір ферментів,
багато з яких міститься в клітинах тварин і рослин. Характер продуктів бродіння
залежить від типу мікроорганізмів, типу субстрату, рН середовища, наявності
кисню.
Розділ 23. Вуглеводи
647
❖ Спиртове бродіння - це розщеплення не самих моносахаридів, а естерів
моноз і фосфатної кислоти, які утворюються у водних розчинах за участю
аденозинтрифосфату (АТФ), що естерифікує гідроксильну групу С6 глюкози,
перетворюючись в аденозиндифосфат (АДФ). Процес каталізує фермент
гпюкокіназа. Під впливом ферменту ізомерази 6-фосфо-глюкоза трансформується
в 6-фосфо-фруктозу, а та ще раз фосфатується, перетворюючись в 1,6-дифосфат
фруктози. Отриманий продукт під впливом ферменту альдолази розщеплюється
на фосфат дигідроксіацетону. й 3-фосфат гліцеринового альдегіду, які здатні до
взаємного перетворення за наявності ізомерази:
ОН
НС-
н-
НО-
Н-
н-
-ОН^
Н О
-ОНІ
ОН
НС
атф
Гпюкокіназа
ПС n
н-|-он І
но+н о
сн2он
Н-г-ОН ) І30мераза
н-І-—^
но-
но-
н-
н-
сн2он
АТФ ^
Фосфофруктокіназа
но
но
сн2оро3н2
сн2оро3н2
СН,ОРО,Н
сн2оро3н2
^0
нн-ону
н-1 J
Т-2
с=о
3і *2
СН20Р03Н2
но-
-н
н-
-он
н-
-он
сн2оро3н2^
Альдолаза
сн2оро3н2
с=о
сн2он
н-сю
н-|-он
сн20Р03н2
З-Фосфат гліцеринового альдегіду за участю ферменту дегідрогенази,
глутатіону (ГБН) - донора меркаптогрупи, і нікотинамідаденіннуклеотиду
(НАД), що є переносником гідрогену, перетворюється через кілька стадій у
піровиноградну кислоту:
Н-С=0 дегідрогеназа ГЗ-С-ОН над _
Н-С-ОН -над-н, Н-С-ОН -над-ь^
ге. ,р
С
Н-С-ОН -ген
КОЛЮ .с
НзГО4 ^ 3 с
Н-С-ОН
енрю^
адф; но .о ^. . но .о но .о
фосфогліцераткіназа £ Фосфогліцеромутаза £ енолаза ^ адф>
^атф " и-Л-ои н-9-ою3Н2 '"^ ^-оюд "АТФ
СНрН СН,
н-с-он
но р
с
С=0
СНз
Піровиноградна
кислота
648
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Під час спиртового бродіння піровиноградна кислота під впливом ферменту
піруватдекарбоксилази дає оцтовий альдегід, який відновлюється ферментом
алкогольдегідрогеназою до етанолу:
Ши Алкагольдегідрогеназа
ТТіг*л>ж>люЛт6пшт,пл*гі П ТТ А ТТ ТТ
труеапгЬекарбоксипаэа Л НАД^ ^ СН3СН2ОН
q о -со2 3 чо -над 3 2
❖ Оцтовокисле бродіння. Якщо бродіння відбувається за наявності кисню,
то як основний продукт виділяють оцтову кислоту. В атмосфері повітря етанол
окиснюється за наявності альдегіддегідрогенази, яку виділяють оцтовокислі
бактерії:
СН3СН2ОН -—-——- СН3СООН+Н202
3 L Алкогольоксидаза
Таким способом одержують харчову оцтову кислоту. Цей процес відбувається
під час скисання молодого вина в разі порушення технології бродіння.
❖ Молочнокисле бродіння. Під час ферментативного бродіння за участю
молочнокислих бактерій родів Lactobacillus і Leuconostoc піровиноградна
кислота відновлюється до молочної за допомогою НАДН2:
' ^ НАД Н ' ^
н3с-с-с; Т?"2 • сн,снон-сч'
3 о он ™ 3 он
Цю реакцію взято за основу виробництва самої молочної кислоти, а також
хліба, кисломолочних продуктів, силосування кормів у сільському господарстві.
Квашення плодів й овочів відбувається за участі гетероферментативних
молочнокислих бактерій. У цьому випадку утворюються молочна й оцтова
кислоти, етиловий спирт.
На закінчення зазначимо, що відомі й інші види бродіння, наприклад,
пропіоновокисле, маслянокисле, ацетон-бутанольне тощо.
233.2. Фотосинтез вуглеводів у рослинах
Весь складний органічний матеріал зелених рослин синтезований з карбон
(IV) оксиду й води завдяки сонячній енергії, у перетворенні якої безпосередньо
бере участь хлорофіл:
бсо2+ бн2о і Ьу» 602+ С6Н1206
Дихання
Організми тварин не здатні до такого синтезу і тому цілком залежать від
органічного матеріалу, який одержують з їжею.
Розділ 23. Вуглеводи
649
За допомогою міченого карбон (IV) оксиду 14СОг з'ясовано, що першою
стійкою похідною асимільованого вуглекислого газу є фосфогліцеринова кислота
Н20зРОСН2СН(ОН)14СООН, що утворюється внаслідок взаємодії карбон (IV)
оксиду з дифосфатом рибулози. Фосфатний естер гліцеринової кислоти під дією
світла відновлюється у фосфат гліцериновий альдегід, а той вступає в альдольну
конденсацію зі своїм ізомером - фосфатом дигідроксіацетону й стереоспецифічно,
як і в більшості ензиматичних реакцій, перетворюється в дифосфат фруктози.
Водночас утворюється й дифосфат рибулози, необхідний для нової асиміляції
карбон (IV) оксиду:
СН2ОР03Н2
сн2ОР03н2
с--о
н—он
н+он
сн2оро3н2
н20+ С02
но-(-н
С
но о
+
но: о
н-|-он
-1/20,
н-с=о
н-|-он
сн2ОР03н2
сн2оро3н2
сн,оро,н,
і 2 3 2
с=о
сн2он
1
н-с=о
н-|-он
сн2оро3н2
сн2оро3н2
с=о
но+н
н—он
н+он
сн2оро3н2
23.4. Найважливіші олігосахариди
Олігосахариди утворюються завдяки О-зв'язку між глікозидним гідрокси-
лом однієї молекули моносахариду й будь-яким гідроксилом іншої молекули. У
тому випадку, коли в утворенні цього зв'язку беруть участь обидва глікозидні
гідроксили, синтезуються дисахариди, які є невідновлювальними. До них
належать трегсиїоза й сахароза.
Якщо ж один з моносахаридних залишків дисахариду має вільний
напівацетальний (глікозидний) гідроксил, то такі дисахариди називають від-
новлювальними, оскільки в цьому випадку можливе окиснення олігосахариду до
карбонової кислоти. Найпоширенішими відновлювальними дисахаридами є мальтоза,
целобіоза й лактоза. Коротко розглянемо методи визначення будови цих дисахаридів.
❖ Мальтозу С12Н12О11 одержують з виходом близько 80% у разі дії на
крохмаль ферменту амілази. Дисахарид відновлює реактив Толенса, окиснюючись до
мальтобіонової кислоти. Також мальтоза реагує з фенілгідразином, даючи озазон
650
Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
Сі2Н2о09(=Н-МН-СбН5)2. Оскільки мальтозу а-глюкозидаза (мальтаза) розщеплює до
глюкози, то мальтоза є а-глюкозилглюкозою. Треба з'ясувати, яка з відновлювальних
гідроксильних груп моносахариду бере участь в утворенні глікозидного зв'язку в
мальтозі та які розміри циклів обох ланок Б-глюкози. Відповідь на це питання дає
вичерпне метилювання й наступний гідроліз перметильованої мальтобіонової
кислоти. Унаслідок цих процедур утворюються 2,3,5,6-тетра-0-метил-0-глюконова
кислота й 2,3,4,6-тетра-0-метил-В-глюкоза. Ці продукти засвідчують, що мальтоза є
4-0-(а-В-глюкопіранозил)-В-глюкопіранозою:
Н0СН2\ НОСН,
НО-^ тлп ^пм тлп-^І^ С
но^он но^^ссш
Мальтоза Мальтобіонова кислота СООН
-осн,
ЩОЄНг, о ,г ___ СН3ОСН2 „ ОЦУ
+
-он
-сен,
Метилчзкта-О-метил-В-мальтобіоноаг 2,3,4,6-тетра-О- 2Д5,6-тетра-0-Метил-
Метил-Г>глкжоза Г>глкжонова кислота
❖ Целобіозу С12Н22О11 можна отримати в разі часткового гідролізу
целюлози. Целобіоза є відновлювальним дисахаридом, утворює озазон.
Послідовність тих же реакцій окиснення, метилювання й гідролізу, що й для
мальтози, засвідчує, що целобіоза має два піранозні кільця, й глюкозидний
зв'язок існує з ОН-групою С4. Відмінність від мальтози полягає в тому, що
глюкозидний зв'язок між моносахаридами зазнає розщеплення емульсином з
гіркого мигдалю, здатного гідролізувати лише Р-глюкозидні зв'язки. Отже,
целобіоза є 4-0-(Р-В-глюкопіранозил)-Б-глюкопіранозою:
РН
ОНЮН
❖ Лактоза С12Н22О11 є в складі жіночого й коров'ячого молока. У
промисловості її одержують як побічний продукт виробництва сиру. Лактоза -
цукор, який відновлює, тому вона утворює озазон. Оскільки лактобіонова кислота,
що утворюється під час окиснення лактози бромом, розпадається в разі гідролізу
на галактозу й глюконову кислоту, то фрагментом відновлювального дисахариду є
глюкоза. Метилювання й наступний кислотний гідроліз засвідчують піранозні
цикли моносахаридів й 1-»4-зв'язок між ними. Оскільки лактозу розщеплює
Розділ 23. Вуглеводи
651
фермент Р-галактозидаза, лактоза є 4-0-(Р-Б-галактопіранозил)-В-глюкопірано-
зою:
З невідновлювальних дисахаридів розглянемо будову сахарози.
❖ Сахароза С12Н22О11 не відновлює реактиву Толленса, не утворює озазону, не
зазнає окиснення бромом і, на відміну від розглянутих дисахаридів, не мутаротує в
розчинах. Отже, у сахарозі обидва глікозидні гідроксили беруть участь в утворенні
зв'язку між моносахаридами. Під час кислотного гідролізу сахарози утворюються
фруктоза й глюкоза. Висока лабільність сахарози до кислоти засвідчує, що один з
моносахаридів у дисахариді перебуває у фуранозній формі. У разі метилювання
сахарози й наступного гідролізу одержують 2,3,4,6-тетра-0-метил-Б-глюкозу й
1,3,4,6-тетра-0-метил-Б-фруктозу. На підставі різних методів, у тому числі
рентгеноструктурного, дійшли висновку, що сахароза є Р-фруктофуранозидом й а-
Б-глюкопіранозидом. Тому її можна назвати як а-Б-глюкопіранозил-( 1 —>2)-Р-0-
фруктофуранозид, або р-Б-фруктофуранозил-(2->1)-а-Б-глюкопіранозид.
Сахароза - важливий харчовий продукт, відомий уже кілька сторіч.
Спочатку її добували із соку цукрової тростини. Сьогодні половину сахарози
добувають із цукрового буряка. Хоча про наявність сахарози в коренеплодах
відомо давно, її промислове виробництво почали тільки під час британської
блокади в період наполеонівських війн, коли завезення тростини з Центральної
Америки стало неможливим.
Під впливом інвертази, що міститься в організмі бджіл, відбувається
інвертування сахарози з нектару до суміші Б-глюкози й Б-фруктози. Цей
інвертований бджолами цукор є медом.
Головними полісахаридами цієї групи є целюлоза, геміцелюлози, пектинові
речовини, крохмаль, фруктозани й біополімери водоростей.
❖ Целюлоза надзвичайно поширена в рослинному світі. її вміст становить
від 40-50 % у деревині до 98 % у ваті бавовнику. У промисловості целюлозу
НОСН
СН,ОН
ОН
23.5. Полісахариди
23.5.1. Полісахариди рослинного походження
652
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
виділяють хімічною обробкою деревини, за ДОПОМОГОЮ ЯКОЇ ВІДДІЛЯЮТЬ ЛІГНІН,
геміцелюлози й інші супутні речовини. Для цього проводять екстракцію
сировини сумішшю кальцій гідросульфіту й сульфітної кислоти (сульфітний
метод), їдким натром (натронний метод) або натрій гідроксидом із натрій
сульфідом (сульфідний метод) під тиском за підвищеної температури.
Будова целюлози визначена за допомогою часткового й повного кислотного
гідролізу, метилювання й окиснення періодатною кислотою. Повний кислотний
гідроліз целюлози дає тільки глюкозу, а гідроліз її перметилату приводить до
2,3,6-три-0-метил-Б-глюкози з невеликою домішкою 2,3,4,6-тетра-О-метил-О-
глюкози. Отже, результати метилювання свідчать про лінійну будову целюлози з
1-»4-зв'язками між моносахаридними ланками. На основі спектральних даних
визначено Р-конфігурацію глікозидних центрів. Кількість моносахаридних
залишків може досягати 10 000:
Наведена структура підтверджена одержанням під час часткового й
ферментативного гідролізу целобіози та її олігомерів, а також періодатним
окисненням, за якого на кожен моль моносахариду витрачається 1 моль
окиснювача. Лінійні макромолекули целюлози, розташовуючись паралельними
пучками, утворюють завдяки водневим зв'язкам дуже міцні тривимірні
структури. У деревині ці целюлозні пучки оточені лігніном, що дає структури,
які можна зрівняти з армованим бетоном.
Ферментів, що розщеплюють Р-глікозидні зв'язки, в тракті людини, на
відміну від тварин, немає, тому людина не здатна перетравити целюлозу, проте
целюлоза є необхідною речовиною нормального харчування.
■ Целюлоза та її похідні мають колосальне практичне значення. Під час її
обробки сумішшю концентрованих нітратної й сульфатної кислот утворюється
повністю нітрована целюлоза, з якої виготовляють порох. Целюлоза, що містить від
двох до трьох нітрогруп на залишок глюкози, є піроксиліном - необхідною
сировиною у виробництві целулоїду й колодію. З колодію раніше готували нітролаки й
фотоплівки. Однак через займистість та отруйність сьогодні їх знято з виробництва.
■ Під час дії на целюлозу суміші оцтового ангідриду й оцтової кислоти за
наявності каталітичних кількостей сульфатної кислоти синтезують ацетат
целюлози, який потім піддають частковому омиленню. У цьому разі відбувається
часткова деградація макромолекули. Такий ацетат целюлози застосовують
замість нітрату у виробництві безпечної кіно- і фотоплівки. Шляхом про-
давлювання ацетонового розчину ацетату целюлози через фільєри з наступним
випарюванням розчинника одержують ацетатний шовк.
Розділ 23. Вуглеводи
653
■ Унаслідок обробки целюлози сірковуглецем у лузі вона перетворюється в
ксантогенат целюлози:
Продавлюванням її розчину через фільєри в підкислені розчини
регенерують вихідну целюлозу. Так одержують віскозне волокно для вироб-
ництва текстильних тканин і шинного корду. Якщо продавлювати розчин через
довгі вузькі щілини, то отримають целофан.
Також целюлоза слугує сировиною для виробництва паперу. Після
відбілювання її змішують з наповнювачем (гіпс або важкий шпат) і проклеюють.
На такому папері чорнило не розпливається. Фільтрувальний папір складається з
чистої целюлози.
Величезне значення має целюлоза у виробництві етилового (гідролізного)
спирту: його одержують гідролізом целюлози до глюкози, яку далі піддають
бродінню.
❖ Геміцелюлози залежно від моносахаридного складу головного ланцюга
поділяють на ксилани, глюкоманани й галактани.
■ Ксилани є головною частиною геміцелюлоз покритонасінних рослин.
Найбільше вивченим представником цієї групи є 0-ацетил-(4-0-меггші-
глюкуроно)-ксилан. 4-0-Метил-0-глюкуронова кислота приєднана до
гідроксилу С2-ксилози коясного десятого залишку пентози. Головний лінійний
ланцюг ксилану має Р(1—>4)-зв'язки. Ацетильні залишки локалізовані переважно
в СЗ-ксилози:
■ Полісахариди зернових культур - це арабіноксилани, у яких арабіно-
фуранозні залишки приєднані до залишків ксилози основного ланцюга 1->3-
зв'язками.
■ Глюкоманани голонасінних рослин мають лінійну структуру із Р(1—>4)-
зв'язками з нез'ясованою регулярністю чергування залишків глюкози й манози.
ОН
ОН
654
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
-—і СООСН3
■ Галактани хвойних складаються з мономерів галактози із Р(1—>3)- і
(1—>6)-зв'язками. На кінцях полісахаридів є арабофуранозні й арабопіранозні
фрагменти.
❖ Пектиновими речовинами називають
поліуроніди, наявні у всіх рослинах й у низці
водоростей. Головною будівельною ланкою є
О-галактуронова кислота, часто естерифіко-
вана метанолом.
Зв'язки 1->4 між моносахаридними
залишками визначені періодатним окиснен-
ням із доокисненням, унаслідок чого була
виділена О-винна кислота:
Важливою у практичному сенсі властивістю пектинових речовин є здатність
їхніх розчинів до утворення міцних гелів або киселів. Звичайним промисловим
джерелом є яблука, цитрусові та відходи цукрової промисловості. Головне
застосування пектини мають у фармацевтичній і харчовій промисловостях.
О —
---тСООН
Н0А
—і соон
о—-
НІ04 Н-СН>ЧС-Д
о 0 6
соон
н+ но-
Вг,
Г СООН
ii м і
о о 0 —
соон
-он + соон
соон
❖ Крохмаль є головним резервним полісахаридом насіння злаків -
пшениці, рису, кукурудзи. Він також міститься в бульбах картоплі. Зазвичай
крохмаль має близько 20 % розчинної у воді фракції амілози й 80 %
амілопектину.
■ Амілоза є лінійним полісахаридом з ос(1->4)-зв'язками між залишками
глюкопіранози. Ці дані отримані на підставі метилювання, періодатного
окиснення, часткового й ферментативного розщеплення амілози. Віскозиметричні
вимірювання дають значення ступеня полімеризації амілози близько 1 000
залишків глюкози. Амілоза утворює інтенсивне синє забарвлення з іодом.
Припускають, що її ланцюги закручені у вигляді спіралі, внутрішні розміри якої
дають змогу розміститися молекулі іоду:
Розділ 23. Вуглеводи
655
Просторова будова амілози й амілопектину
■ Амілопектин, на відміну від амілози, має високорозгалужену структуру.
Залишки D-глюкози в основному ланцюзі з'єднані а(1—»4)-зв'язками. Під час
метилювання поряд з 2,3,4,6-тетра-Ометил- і 2,3,6-три-0-метил-Б-глюкозою
виділяють 2,3-ди-(9-метил-0-глюкозу, що свідчить про наявність розгалуження
за участю Сб-атома глюкози в амілопектині. Як і амілоза, амілопектин утворює
іодний комплекс, що має червоний колір. Причому існує лінійна залежність між
ступенем розгалуження полісахариду й довжиною хвилі поглинання у видимій
ділянці спектра.
Крохмаль широке застосовують у харчовій і текстильній промисловостях, у
виробництві паперу, клеїв тощо.
❖ Фруктани побудовані із залишків D-фруктози й
відіграють аналогічну до крохмалю роль харчового резерву
рослин. Прикладом може слугувати інулін, що міститься в
бульбах і коріннях складноцвітих (топінамбур, артишок,
кульбаба). За результатами метилювання виявлено, що залишки
D-фруктози пов'язані між собою 2-» 1-зв'язками, а самі
моносахариди перебувають у фуранозній формі.
Відсутність відновлювальних властивостей в інуліні й
наявність у гідролізаті невеликої кількості D-глюкози
пояснюють тим, що кінець відновлювальної молекули
замінений глікозид-глікозидним угрупованням типу сахарози.
Ступінь полімеризації становить близько 35.
інул 1Н
❖ 3 полісахаридів водоростей частіше трапляються агар, карагінін та
альгінова кислота.
■ Агар одержують у промисловості екстракцією червоних водоростей і
використовують як гелеутворювальний агент. Головний компонент агару -
агароза - побудована з D-галактози й 3,6-ангідро-Ь-галактози:
656 Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
На підставі метилювання з'ясовано, що залишки D-галактопіранози мають
замісників у положенні 3, а 3,6-ангідро-Ь-галактози — у положенні 4. Вміст
залишків сульфатної кислоти невеликий, і вони, очевидно, належать Сб-атому
D-галактози. Будова агарози запропонована з урахуванням ферментативного й
часткового гідролізу.
■ У карагінанах, побудованих аналогічно до агарози, сульфатна група
перебуває в С2 і Сб-положеннях галактози.
■ Головним компонентом бурих водоростей є альгінова кислота —
поліуронід, що складається з D-мануронової й L-гулуронової кислот:
НООСНО СООН ноосно
ОН
Альгінову кислоту одержують з бурих водоростей, головно ламінарії.
Використовують як гелеутворювальний агент і емульгатор.
■ Камеді - це полісахариди, що виділяються у вигляді в'язких розчинів, які
утворюють склоподібну масу в разі ушкодження кори багатьох рослин. їхнє
утворення пов'язують з патологічним станом - механічними або інфекційними
ушкодженнями.
Складаються камеді з кислих і нейтральних біополімерів. Кислі полісахариди
містять D-глюкуронову кислоту, D-ксилозу й D-фукозу, а нейтральні - D-га-
лактозу й L-арабінозу.
■ Полісахариди слизів є лінійними галактомананами з ß(l—>4)-зв'язками.
Деякі представники мають розгалуження в положенні 3. Слизи є продуктами
нормального метаболізму рослин і слугують або харчовим резервом, або
речовинами, що утримують воду, особливо в тканинах сукулентів.
Камеді й слизи широко застосовують у медицині, харчовій промисловості,
виробництві паперу, текстильних виробів, у разі одержання емульгаторів і клеїв.
23.5.2. Зоополісахариди
■ Глікоген - це резервний полісахарид, загальний для всіх тваринних
організмів. Він побудований винятково із залишків D-глюкози. Методами
метилювання, періодатного окиснення, часткового кислотного гідролізу й
Розділ 23. Вуглеводи
657
ферментативного розщеплення доведено, що він є найближчим аналогом
амілопектину. Відмінність від амілопектину полягає в більшій розгалуженості й
тіснішій "упаковці" макромолекули.
■ Хітин — головний компонент зовнішнього кістяка ракоподібних і комах.
У разі повного кислотного гідролізу хітину за жорстких умов утворюються
еквімолярні кількості глюкозаміну (2-аміно-2-дезокси-0-глюкози) й оцтової
кислоти. З аналізу продуктів часткового гідролізу видно, що хітин є однорідним
лінійний полімером ІУ-ацетилглюкозаміну, у якому моносахаридні залишки в
піранозній формі сполучені між собою Р(1—>4)-зв'язками:
За хімічною будовою, фізико-хімічними властивостями і біологічною роллю
хітин подібний до целюлози.
23.5.3. Мукополісахариди
■ Гісиїуронова кислота - полісахарид, близький за структурою до хітину, його
виділяють зі склоподібного тіла ока, синовіальної рідини суглобів. У складі
гіалуронової кислоти є Б-глюкуронова кислота й ІУ-ацетилглюкозамін. На підставі
ферментного розщеплення макромолекули мікробіальною гіалуронідазою і
наступного аналізу продуктів розпаду за допомогою періодатного окиснення й
метилювання для гіалуронової кислоти запропоновано таку будову:
О
Розчини гіалуронової кислоти завдяки в'язкості виконують у суглобах
організму функцію мастильного матеріалу, а також регулюють розподіл води в
організмі й забезпечують селективну проникність тканин.
■ Гепарин - мукополісахарид з молекулярною масою 10-20 тис. Да, має
блокову будову. Виділяють два блоки, у складі яких є глюкозамін і, відповідно,
а-Ь-ідуронова й а-Б-глюкуронова кислоти. Сильнокислості гепарину надають
залишки сульфатної кислоти по аміно- і Сб-гідроксигрупах глюкозаміну й
гідроксилу Сг-уронових кислот:
658
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
Гепарин має специфічну властивість збільшувати час коагуляції крові, його
застосовують у медицині для запобігання утворенню тромбів після деяких видів
хірургічних операцій та інфарктів міокарда. Одержують з печінки великої рогатої
худоби.
■ Досить близькі до гепарину за будовою хондроїтиносульфати — сульфа-
товані мукополісахариди. Вони є в складі сполучної тканини тварин. Цукрові
ланцюги побудовані з залишків, що чергуються, 4-0-заміщеної P-D-глюкуронової
кислоти й 3-0-заміщеного Н-ацетил-р-О-галактозаміну, сульфатованого в
положення 4 (хондроїтиносульфат А) або в положення 6 (хондроїтиносульфат С).
Ступінь полімеризації дорівнює 150 дисахаридним одиницям:
Хондроїтиносульфат А Хондроїтиносульфат С
Хондроїтиносульфат В, або Р-гепарин, відрізняється від хондроїтиносуль-
фатів А і С тим, що замість О-глюкуронової кислоти має її епімер за С5 - Ь
ідуронову кислоту.
233.4. Полісахариди мікроорганізмів
■ Декстрани - це глюкани з а(1—»6)-зв'язками, продуковані бактеріями
Leuconostoc і Streptococcus. Вони мають антигенні властивості. Декстрани у великих
кількостях виробляють у промисловості як замінники плазми крові, а також як
вихідні речовини в разі одержання молекулярних сит - сефадексів, які широко
застосовують у лабораторній практиці для поділу речовин за молекулярною масою.
■ Мурамін - полісахарид клітинних стінок бактерій. Структурно нагадує
хітин, у якому один із залишків глюкозаміну утворює естер з D-молочною
кислотою (А/-ацетилмурамова кислота):
Розділ 24
АРОМАТИЧНІ ВУГАЕВОАНІ.
БЕНЗЕН ТА ЙОГО БУАОВА
Термін "ароматичні сполуки" (пізніше з'явилася назва "арени") почали
застосовувати на початку XIX ст. для бензену та його похідних, в основі яких є
вуглеводні із бензеновими або конденсованими бензеновими кільцями (нафта-
лен, антрацен та ін.).
Першими відомими хімікам були речовини, що гарно пахнуть, виділені з
ароматичних бальзамів (толуен з толуанського бальзаму) або інших запашних
екзотичних продуктів (бензойна кислота з росного ладану). Найпростіший
вуглеводень ароматичного ряду - бензен - виділений 1825 p. М. Фарадеєм з
конденсату світильного газу. Він виявився тотожним речовині, отриманій
Е. Мітчерліхом1 1834 р. унаслідок декарбоксилювання бензойної кислоти за
наявності кальцій оксиду. Елементний склад сполуки цієї речовини, яку
Ю. Лібіх назвав бензеном, Е. Мітчерліх визначив як С6Н6 за густиною пари. В
1845 р. А. Гофман виділив бензен як продукт коксохімічного виробництва, й
відтоді бензен є сировиною промислового органічного синтезу.
24.1. Будова й номенклатура аренів
24.1.1. Класифікація аренів
Карбоциклічні ароматичні сполуки
Гетероциклічні ароматичні сполуки
@^ со
Толуен Нафтален
q g 0
Фуран Тіофен Піридин
Моноциклічні
Поліциклічні-арени
арени
арени з ізольованими
арени з конденсованими
циклами
циклами
@-сн2сн3
@@
ООО
Етилбензен
Біфеніл
Антрацен
Мітчерліх Е. (МкБСпегІісп Е.) (1794-1863) - німецький хімік. Головні праці стосуються загальної
хімії. Виконав аналіз молочної кислоти, одержав бензен з бензойної кислоти, синтезував нітро-
бензен (нітрування за Мітчерліхом), азобензен, бензенсульфонову кислоту, бензофенон.
660 Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
24.1.2. Визначення будови бензену
У головних рисах будова бензену визначена А. Кекуле вже 1865 р., тобто
через чотири роки після того, як О. Бутлеров розробив теорію хімічної будови
органічних сполук. Він запропонував для бензену структурну формулу, що
відповідає 1,3,5-циклогексатрієну. Незважаючи на це, у будові бензену і його
похідних залишалася загадка, до якої хіміки поверталися протягом усієї
подальшої історії і яку з'ясували порівняно нещодавно. Відомі А. Кекуле факти,
що послугували основою запропонованої ним для бензену "формули Кекуле",
були такими.
Бензен і його гомологи мають формулу С„Н2и-б, яка у моноциклічній
структурі бензену повинна позначати наявність трьох подвійних зв'язків. Однак
ці подвійні зв'язки дивно інертні: вони стійкі до дії окиснювачів і не вступають
у звичайні реакції електрофільного приєднання, що характерні для алкенів,
алкадієнів і алкінів.
У бензені рівноцінні всі атоми Карбону, як і всі атоми Гідрогену, що
сумісно тільки з припущенням про циклічну структуру сполуки. Отже,
заміщення будь-якого атома Гідрогену бензену приведе до одного й того ж
монозаміщеного продукту. З іншого боку, існують завжди три ізомерні між
собою двозаміщені похідні бензену: орто- (о-), мета- (м-) і пара- (я-). Формула
А. Кекуле задовольняє всі ці факти, за винятком кількості о-заміщених.бензенів,
що знав і сам учений:
(а) (б)
о-Дихлоробензен л*-Дихлоробензен я-Дихлоробензен
Справді, якщо ґрунтуватися на знанні хімії аліциклічного ряду, то формули
(а) і (б) повинні відповідати двом різним ізомерним речовинам. Насправді таких
двох о-ізомерів не існує. Пояснити властивості бензену (відсутність структурних
двозаміщених о-ізомерів) спробував Ф. Тіле1, який 1889 р. запропонував теорію
"парціальних валентностей". Відповідно до цієї теорії, в органічних сполуках з
подвійними зв'язками в області простих зв'язків існує часткова двозв'язаність
завдяки залишковим "парціальним валентностям":
1 Тіле Ф. (Thiele F.K.J. ) (1865-1918) - німецький хімік. Головний напрям наукових досліджень -
розробка теорії родинності ненасичених органічних сполук.
Розділ 24. Ароматичні вуглеводні. Бензен та його будова
661
А. Кекуле ж запропонував спеціальну гіпотезу про швидку "осциляцію" -
міграцію подвійних зв'язків у бензені:
Багато вчених, намагаючись пояснити властивості бензену особливостями
будови, висували свої гіпотези. Наприклад, Г. Армстронг1, А. Байєр припустили,
що в молекулі бензену четверті валентності всіх шести атомів Карбону
спрямовані до центра й насичують одна одну. Крім того, оригінальні структури
бензену запропоновані Д. Дюаром2 і А. Ладенбургом:
Структура Армстронга й Байєра Структура Дюара Структура Ладенбурга
Зазначимо, що структури Дюара й Ладенбурга є реальними й отримані згодом
у фотохімічних реакціях. Усі вони мають неплоску будову і є малостійкими, легко
переходять одна в іншу і в бензен під час нагрівання й опромінення.
Наступний розвиток хімії зумовив появу численних підтверджень структури
А. Кекуле. Зокрема, А. Байєр з'ясував, що терефталеву кислоту можна відновити
воднем у момент виділення в циклогексадієндикарбонову, а потім у цикло-
гександикарбонову кислоту. Так уперше були пов'язані ряди бензену й цикло-
гексану й доведено існування у бензені шестичленного циклу:
СООН СООН СООН
СООН СООН СООН
На початку XX ст. П. Сабатьє, використовуючи відкритий ним метод
гідрування ненасичених сполук воднем над нікелем, довів, що ця ж залежність
існує для самого .бензену й циклогексану:
У 1912 р. М. Зелінський виконав зворотну реакцію - ароматизацію
циклогексану в бензен над платиновим або паладієвим каталізатором:
1 Армстронг Г. (Armstrong Н.) (1848-1937) - англійський хімік, запропонував сучасну номенкла-
туру органічних сполук. Висунув хіноїдну теорію кольору.
2 Дюар Дж. (Dewar J.) (1842-1923) - англійський фізик і хімік, уперше отримав рідкий водень.
Працював у галузі органічної хімії.
662
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
У 1904 р. К. Гаррієс проозонував бензен, причому взаємодія відбувалася
аналогічно до озонування олефінів. Аналіз продуктів реакції засвідчує, що всі
шість карбонових атомів бензену за валентним станом олефінові: озонід бензену
після гідролізу перетворюється на гліоксаль і гідроген пероксид:
Г*-Г" 0 О тт тт
^-?оР ?"-^зЧс-с+зн2о2
сьс.с.0^н о' Ъ
Озонування о-ксилену дає гліоксаль, метилгліоксаль та діацетил у моле-
кулярному співвідношенні 3:2:1, що підтверджує рівноцінність усіх С-С-зв'язків
у циклі о-ксилену до озонування, і отже, відсутність жорсткого фіксування
подвійних зв'язків:
снз зо3 о^с'с-0с9сн32і^Нс-сД + 2н,стсно
и0?г с-н 3-зн^о Ъ 6.
н н о
СН, /О £Нз
гРо г-С-0 "!НА О' О 6' Ъ
На світлі молекули бензену приєднують по шість атомів Хлору,
перетворюючись у суміш геометричних ізомерів гексахлороциклогексану, який
використовували як інсектицид "гексахлоран", або "гаммексан":
с. с
\\ //+6С1—~С1" ^
СІ СІ
У разі УФ-опромінення бензен аналогічно до дієнів приєднує малеїновий
ангідрид:
Розділ 24. Ароматичні вуглеводні. Бензен та його будова
663
.0
he
he
Незважаючи на таке, здавалося б, блискуче узгодження хімічної формули з
експериментальними фактами, багатьох видатних хіміків не задовольняла
формула Кекуле, яка ґрунтувалася на тому, що бензен і всі ароматичні сполуки у
реакціях приєднання незрівнянно інертніші, ніж олефіни. Можливі лише деякі
реакції приєднання (хлорування, озонування, гідрування), а багато реагентів, які
легко приєднуються до олефінів, заміщують атоми Гідрогену в бензені.
Більша інертність бензену порівняно з олефінами випливає з такого
енергетичного зіставлення:
|^j)+H2 ^^[^ + 120кДж/моль (ґ^| +ЗН2-^[^
+ 208 кДж/моль
Під час гідрування бензену як циклогексатрієну повинно б виділятися 120-3
= 360 кДж/моль, тобто на 152 кДж/моль більше, ніж насправді. На це значення
бензен енергетично бідніший і, отже, стійкіший, ніж гіпотетичний циклогексан-
1,3,5-трієн, циклоолефін з фіксованими подвійними зв'язками.
24.1.3. Сучасне пояснення будови бензену
Атоми Карбону в бензені перебувають у стані 5/?2-гібридизації. Кожний атом
утворює два аС-с і один Сс-н-зв'язок. Шість негібридизованих р-орбіталей
перпендикулярні до площини циклу й паралельні одна до одної. Бічне перекри-
вання цих р-орбіталей дає єдину я-електронну хмару у вигляді двох торів над і під
площиною циклу. Шість р-електронів делокалізовані по шести карбонових
атомах. Довжина С-С-зв'язків у бензені 0,139 нм, що менше, ніж в алканах
(0,154 нм), однак більше порівняно зі зв'язком С=С Ь олефінах (0,134 нм).
Утворення єдиної я-електронної хмари дає значний виграш
в енергії порівняно з локалізованою системою (-150 кДж/моль).
Тому для ароматичних сполук характерні реакції заміщення, у
яких зберігається ароматичність системи, а не реакції
приєднання. Оскільки ароматичні сполуки мають надлишок
електронної густини (я-електронна хмара), то вони головно
реагують з електронодефіцитними частинками - електрофілами
й вільними радикалами. Нуклеофільне заміщення відбувається
з великими труднощами за жорстких умов.
Щоб ліпше зрозуміти причини більшої стійкості бензену порівняно з
іншими ненасиченими системами, корисно розглянути його структуру з позиції
664 Чирва В.Я., Ярллолюк CM., Толкачова Н.В., Земляков О.Є. Органічна хімія
теорії молекулярних орбіталей. Шість />орбіталей атомів Карбону кільця
комбінуються, утворюючи шість молекулярних орбіталей - три зв'язувальні
\|/2 і v|/3) і три розпушувальні (\|/4, v|/5 і \|/б). Однак, на відміну від нециклічних
тріенів, де всі молекулярні орбіталі відрізняються за енергією, для бензену
характерна наявність шести молекулярних орбіталей, з яких дві зв'язувальні й
дві розпушувальні молекулярні орбіталі мають попарно однаковий енерге-
тичний рівень. У цьому разі зв'язувальні орбіталі заповнені, а розпушувальні -
порожні (рис. 24.1,24.2).
Рис. 24.1. Енергетична діаграма молекули
бензену рис 24.2. Обриси АТі МОмолекули бензену
Зв'язувальна орбіталь охоплює всі шість карбонових атомів, а однакові за
енергією орбіталі \|/2 і \|/з відрізняються за симетрією й мають по одній вузловій
площині.
Для оцінки стійкості плоских повністю спряжених циклічних 71-систем за
відносною енергією молекулярних орбіталей зручно користуватися графічним
методом (за Фростом). Систему подають правильним багатокутником і вписують
його в коло так, щоб одна з вершин розташувалася в точці перетину вертикально
проведеного діаметра з нижньою частиною кола. Точки, у яких вершини
багатокутника торкаються кола, вважають рівнями енергії молекулярних орбіта-
лей, а вертикальний діаметр - координатою енергії, на якій горизонтальний
діаметр позначає незв'язувальний рівень.
Е Е
а б
Рис. 24.3.
Розділ 24. Ароматичні вуглеводні. Бензен та його будова
665
Використаємо цей підхід для оцінки міцності бензену й циклобутадієну -
систем плоских і спряжених. Як видно з рис. 24.3, а, у випадку бензену всі шість
я-електронів розташовані попарно на трьох зв'язувальних орбіталях. Система
стійка. У випадку ж циклобутадієну (див. рис. 24.3, б) із чотирьох я-електронів
на зв'язувальній орбіталі можуть бути розташовані тільки два електрони, два ж
інші повинні бути на незв'язувальних орбіталях, правильніше (за правилом
Гунда), по одному на обох незв'язувальних.
Отже, циклобутадієн повинен бути бірадикалом, тобто досить нестійким, що
й спостерігають на практиці. Таку оцінку можна використати й для заряджених
систем.
24.1.4. Правила ароматичності
Стійкість сполуки до дії окиснювачів або інших електрофільних агентів
звичайно залежить від реакційної здатності цих агентів та особливостей будови
вихідної сполуки. З огляду на це тривалий час поняття ароматичності
формулювали не завжди досить однозначно. В 1931 р. на підставі уявлень
квантової механіки Е. Хюккель дав визначення, що дає змогу зачислювати ту чи
іншу систему до ароматичної.
До ароматичних належать моноциклічні сполуки, що мають такі ознаки.
1. Циклічність структури.
2. Компланарність структури (плоска молекула).
3. Наявність (4л + 2) сполучених (делокалізованих) я-електронів, якщо п = 0,
1,2 і т.д.
Якщо хоча б один пункт правил не дотриманий, то сполука не є арома-
тичною. Наприклад:
0
Бензен
Система плоска, циклічна, містить шість спряжених п-
електронів (л = 1) => ароматична сполука
О
Циклооктатетраєн
Система циклічна, містить вісім спряжених я-електронів,
тобто не дотримане правило Хюккеля => неароматична
сполука
О
Циклогептатрієн .
Система циклічна, містить 6 я-електронів, однак вони не
повністю спряжені (немає повної делокалізації по семи
карбонових атомах) => неароматична сполука
Більш ніж через десятиліття гіпотезу Е. Хюккеля підтвердили: отримано
стійкі катіони шропілію та аніон циклопентадієнілію, які мають властивості
ароматичних сполук.
666
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
■ Наприклад, аніон, що утворюється під час нагрівання циклопентадієну з
натрієм або калієм у ксилені, ароматичний, оскільки в аніоні два неподілені
електрони не локалізовані біля одного атома Карбону, а утворюють із я-
електронами подвійних зв'язків ароматичний секстет. Енергія резонансу
циклопентадієнілій-аніона дорівнює 167,4 кДж/моль:
н н |_ н
або «0
К + н,
сн
Аніон
циклопентадієнілію
Система циклічна, плоска, має шість л-електронів (п = 1), які
делокалізовані по п'яти карбонових атомах => ароматична сполука
■ У 1951 р. доведено здатність циклопентадієнілій-аніона утворювати дуже
стійкі комплекси з йонами перехідних металів. Одним з таких комплексів є
фероцен, структура якого з'ясована за допомогою рентгеноструктурного аналізу.
Фероцен є "сандвічем", у якого йон Ре2+ розміщений між двома розташованими в
паралельних площинах циклопентадієнілій-аніонами. Зв'язок між атомом Феруму
і циклопентадієнілій-аніонами містить дванадцять я-електронів обох кілець, і атом
Феруму набуває електронної конфігурації криптону:
30-
4з,
Ре2+(1^22^2^6 3524^23^)
Шість вакантних орбіталей атома Феруму (Ре2+) беруть участь в утворенні
шести зв'язків за донорно-акцепторним типом. Фероцен не розкладається в разі
нагрівання до 470 °С, стійкий до кислот, основ, має хімічні властивості,
характерні для бензену.
Крім атома Феруму, структури типу "сандвіча" (металоцени) з циклопента-
дієнілій-аніоном утворюють також інші метали.
■ Правило Хюккеля прогнозує ароматичність катіона циклогептатрієнілію
(катіона тропілію), що підтверджено 1954 р. В. Дерінгом1:
1 Дерінг В. (Ооег^ W.v.E.) (нар. 1917) - американський хімік, професор Гарвардського
університету. Займався структурами органічних речовин і механізмами органічних реакцій.
Досліджував хінін, працював у хімії карбенів.
Розділ 24. Ароматичні вуглеводні. Бензен та його будова
667
=\ м
Н
+ Вг,
=\ м
Вг
^^-Набо
Катіон тропілію
Вг
Катіон тропілію
Система циклічна, плоска, має шість я-електронів (п = 1), які
через наявність позитивного заряду делокалізовані по семи
карбонових атомах => ароматична сполука
Солеподібність бромоциклогептатрієну (розчинний у воді й не розчинний в
органічних розчинниках), уперше виявлену в 1891 р., В. Дерінг пояснив
легкістю утворення катіона тропілію, що є ароматичним.
■ Циклопропен не є ароматичною сполукою, тому що подвійний зв'язок у
цій сполуці локалізований. Проте циклопропен досить легко відщеплює гідрид-
іон, утворюючи стійкий катіон циклопропенілію (п = 0; ІУ= 4-0 + 2 = 2):
Р\ або /^К
НС=СН
Катіон циклопропенілію
Система циклічна, плоска, містить два тс-електрони
(л = 0), які через позитивний заряд делокалізовані по
трьох карбонових атомах => ароматична сполука
■ Циклооктатетраєн має неплоску конфігурацію і є типовою ненасиченою
сполукою з підвищеною реакційною здатністю. Однак діаніон циклоокта-
тетраєну із плоскою конфігурацією й десятьма я-електронами (п = 2\И= 4-2 +
+ 2=10) задовольняє вимоги правила Хюккеля та є ароматичним (Д. Курсанов1):
Діаніон циклооктатетраєну
2К+
Система циклічна, плоска, має десять я-електронів
(я = 2), які делокалізовані по восьми карбонових атомах =>
ароматична сполука
Діаніон циклооктатетраєну
■ Існування ароматичної системи настіль-
ки вигідніше
подвійних:
Н
ягідніше порівняно із системою спряжених [К Л н+
ійних зв'язків, що той же циклооктатетраєн ^ у-І/
1 Курсанов Д. (1899-1983) - радянський хімік-органік. Досліджував механізми реакцій та
реакційну здатність органічних сполук. Разом із М. Вольпіним займався вивченням ароматичних
небензенових сполук.
668 Чирва В.Я., Ярмолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
легко зазнає протонування, утворюючи ароматичну сполуку катіон гомотропілію, у
якому сім атомів Карбону перебувають в одній площині і їх зв'язує єдина я-
електронна хмара, а восьмий атом Карбону розміщений над площиною, і його
електрони не беруть участі в делокалізації я-системи.
Правилу Хюккеля повинні також підлягати системи з десятьма (п = 2; N =
=4-2 + 2 = 10), чотирнадцятьма (п = 3; ІУ= 4-3 + 2 = 14), вісімнадцятьма (п = 4; Лґ=
= 4-4 + 2 = 18) і так далі електронами.
• Приклади подібних систем можна знайти серед моноциклічних полієнів із
суцільно спряженими подвійними зв'язками, які називають ануленами (від лат.
аппиіш - кільце). Щоб позначити кількість атомів Карбону й одночасно кількість я-
електронів у молекулі анулену, перед словом "анулен" ставлять у квадратних
дужках відповідну цифру. Наприклад, бензен може бути названий [6]-анулен.
Відповідно до правила Хюккеля, властивості ароматичних систем повинні мати
[2]-, [6]-, [10]-, [14]-, [18]- і так далі анулени. Зокрема, [2]-Анулен - це етилен, який
не можна розглядати як циклічну систему, [6]-анулен, як уже зазначено, - бензен,
загальна характеристика властивостей якого наведена вище. Натомість, [10]-анулен
виявився досить реакційно здатним - легко зазнавав окиснення, бурхливо реагував з
бромом, тобто не проявляв ароматичних властивостей. Це пов'язане з тим, що через
відштовхування атомів Гідрогену всередині циклу система перестає бути плоскою,
тобто порушується одне з положень правил Хюккеля:
Система циклічна, має десять спряжених ті-електронів
(л = 2), проте два атоми Гідрогену, що перебувають у
середині циклу, зближені й через вандерваальсове
відштовхування роблять цикл неплоским => неароматична
сполука
Правила Хюккеля, відверто кажучи, застосовують для моноциклічних
систем, однак з деяким наближенням їх можна застосувати й для поліциклічних
вуглеводнів із замкнутою системою спряження.
Ароматичними є нафтален, азулен (десять я-електронів), антрацен,
фенантрен (чотирнадцять я-електронів), хризен, пірен, тетрацен (вісімнадцять я-
електронів), бензпірен, дибензантрацен (двадцять два я-електрони). Останні два
арени є сильними канцерогенними речовинами, тобто спричинюють рак. Вони
містяться в кам'яновугільній смолі, тютюновому димі, можуть утворюватися під
час розкладання природних продуктів, у пригарі, що утворився внаслідок
готування їжі (смаження). Канцерогенні властивості має й бензен, що
спричинило обмеження на його застосування в хімічних лабораторіях.
З появою спектроскопії ЯМР знайдені критерії ароматичності: сигнали
протонів бензену є за 7,37 м.ч., тоді як сигнали протонів етилену виявляють за
5,3 м.ч. З'ясовано, що причиною сильного дезекранування протонів в аро-
матичних сполуках є наявність кільцевих струмів, які виникають у сполуках із
замкнутою спряженою я-електронною системою.
Розділ 24. Ароматичні вуглеводні. Бензен та його будова
669
24.1.5. Номенклатура IUP АС
За правилами IUP АС 1993 р. тривіальні назви бензен, толуен, стирен
можна и використати для побудови назв відповідних похідних, які утворені
заміщенням атомів Гідрогену тільки в ароматичному кільці:
Толуен Стирен 1,2-Дихлоробензен Бензен Бензенсульфокислота
CH2CH2CH2CH3
СН2СН2СН2СН3
^І^^^СНоСНоСН-}
^^СН2СН3
Ту^СНгСНгСНз
СН2СН3
1 *Бутил-3-етил-2-пропілбензен
4-Бутил-1 -етил-2-пропілбензен
Така нумерація забезпечує мінімальний набір
Якщо локанти однакові, то перший номер буде
локантів.
привласнений першому за алфавітом замісникові.
Для позначення положення двох замісників у бензеновому кільці можна
використати введені В. Кернером позначення: орто- (р-) - замісники перебувають
поряд, мета (лі-) - замісники перебувають через один атом Карбону, пара- (я-) -
замісники перебувають навпроти один одного, або цифрові локанти. У разі
цифрового позначення положення груп менший номер одержує замісник у сполуці-
основі.
У випадку наявності в сполуці декількох однакових метальних або вінільних
замісників як основу для побудови назви похідних арену використовують бензен, а
не толуен або стирен:
сн3
нс=сн2
(У,
о-Етилтолуен, або 2-етилтолуен
Вг
Правильно: 4-бромо-1,2-дивінілбензен
Неправильно: 4-бромо-2-вінілстирен
■ Дозволено використання тривіальних назв таких аренів, однак вони не
можуть слугувати основами для назв їхніх похідних:
670
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
н3с-сн-сн3
нх-сн-сн,
З і з
Ксилен (показано Кумен Цимен (показано л-ізомер) Мезитилен
о-ізомер)
24.1.6. Ізомерія заміщених бензенів
Відомо, що монозаміщені бензени не мають ізомерів. Наприклад, існує
тільки один монометилбензен - толуен. Спеціальною серією реакцій доведено
таке: якщо в монозаміщеному бензені заміщати послідовно кожний з інших
атомів Гідрогену й елімінувати перший замісник, то завжди утворюватиметься
той самий монозаміщений бензен.
Дизаміщені бензени з однаковими замісниками існують тільки в трьох
ізомерних формах (ізомери положення):
* * ХУТ" х-> v
орто-, або 1,2-ди-Х-бензен мета-, або 1,3-ди-Х-бензен пара-, або 1,4-ди-Х-бензен
Як можна визначити відносне положення замісників у бензеновому кільці? На це
питання вперше відповів В. Кернер на прикладі дибромобензенів. Неважко помітити,
що «-ізомер, якщо його нітрувати, даватиме лише одну мононітропохідну; для о-
дибромобензену можливі два ізомери, а для лі-дибромобензену - три моно-
нітросполуки. Цей прийом відомий під назвою методу абсолютної орієнтації за В.
Кернером. Він не такий легкий, тому що непросто, наприклад, виділити із суміші всі
ізомери, які, до того ж, не завжди утворюються в однакових кількостях.
Тризаміщені (з однаковими замісниками) бензени також існують у вигляді
трьох ізомерів:
X
X
Суміжний, або 1,2,3-три- п 4Т у - Симетричний, або 1,3,5-
V ^ ' * ' р 1,2,4-Три-Х-бензен
Х-бензен *
три-Х-бензен
Тетразаміщених бензенів, мабуть, теж три; у них два атоми Гідрогену, що
залишились незаміщеними, можуть перебувати в о-, м- або л-положенні один до
одного:
Розділ 24. Ароматичні вуглеводні. Бензен та його будова
671
X
X
X
X
X
X' ^ "X
1,2,3,5-Тетра-Х-бензен
X
X
X
1,2,3,4-Тетра-Х-бензен
1,2,4,5-Тетра-Х-бензен
Нарешті, пентазаміщений бензен (знову-таки з однаковими замісниками)
буває тільки один.
Набагато більше комбінацій утворюється у разі використання різних
замісників. Наприклад, у випадку трьох різних замісників можливі десять
ізомерів положення.
Інший шлях з'ясувати положення замісників у бензені, особливо для о-по-
хідних, - це здатність утворювати циклічні похідні. Наприклад, фталева кислота
(я-бензендикарбонова кислота) - єдина серед своїх ізомерів утворює циклічний
ангідрид:
о-Фенілендіамін - єдиний серед своїх ізомерів утворює гетероцикли з низкою
діальдегідів і дикетонів:
Тепер уже немає потреби визначати положення замісників яким-небудь
первинним способом, оскільки відомі положення замісників у безлічі похідних, до
яких можна трансформувати будь-яку нову речовину через низку відповідних
реакцій. Наприклад, знаючи, що у фталевій кислоті карбоксили перебувають у о-
положенні один до одного, можна визначити відповідність положень у фталевій
кислоті й оксилені, окиснюючи оксилен дихроматною кислотою у фталеву кислоту:
або, діючи бромом на арґентум фталат і заміщуючи карбоксили на атоми Брому,
одержати о-дибромобензен:
+ 2н20
Вг
Вг
+ 2 К% + 2 с02
672
Чирва В.Я., Ярмолкж СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
24.2. Фізичні властивості
Ароматичні вуглеводні за звичайних умов - рідини, інколи тверді речовини,
мають сильний специфічний запах. Ізомери з декількома алкільними
замісниками киплять за вищої температури, ніж з одним, що дорівнює за масою
сумі замісників, оізомери киплять за вищої температури, ніж и-ізомери. Ізомери
з ізобудовою замісників киплять за нижчої температури, ніж з нормальним
ланцюгом замісника. Ізомери з симетричною будовою мають вищі температури
плавлення. З дизаміщених л-ізомер плавиться за найвищої температури.
24.2.1. Спектроскопія арені в
• УФ-спектроскопія. Ароматичні вуглеводні мають характерні смуги погли-
нання в ділянці 170-210 і 230-270 нм. Наприклад, в УФ-спектрі бензену є
інтенсивні смуги з Хтах 183 і 204 нм, менш інтенсивна група смуг з А,тах 256 нм.
• ІЧ-спектроскопія. В ІЧ-спекграх ароматичних вуглеводнів наявні смуги
поглинання середньої інтенсивності валентних коливань Ус=с У ділянці 1 580—
1 600 і 1 450-1 500 см"1. Крім того, до характеристичних зачисляють смуги
поглинання деформаційних коливань зв'язків С-Н за 675-890 см"1.
• ПМР-спектроскопія. Зсув сигналів протонів ароматичних сполук у слабше
поле (5,6-8,2 м.ч.) порівняно з сигналами протонів біля подвійного зв'язку (4,5-
6,5 м.ч.) є характерною ознакою ароматичності сполук:
5 7,27 м.ч. 8 5,57 м.ч. 5 9,28 м.ч. 5 5,69 м.ч.
у тетрагідрофурані) (у 802) (у тетрагідрофурані)
8 м.ч. 6,6 (а), 8,6 (Ь),
5,2 (с), -0,6 (сі)
Природа замісника в монозаміщених бензенах впливає на хімічні зсуви
протонів в о-, м- і «-положеннях, наприклад:
НЬ Н;
нь н
НЬ На
НЬ На
5 м.ч. 7,07 (а, Ь, с) 5 м.ч. 7,30 (а), 7,25 (Ь), 5 м.ч. 7,45 (а), 7,19 (Ь), 5 м.ч. 7,67 (а), 7,06 (Ь),
7,18 (с) 7,23 (с) 7,27 (с)
Положення замісників дизаміщених бензенів у спектрах ПМР можна оцінити за
кількістю сигналів і характером їхнього розщеплення. Константа спін-спінової
Розділ 24. Ароматичні вуглеводні. Бензен та його будова
673
взаємодії для срто-протонів - 6,0-9,5 Гц, для м- - 1,2-3,1 Гц, а для п- — 0,2-1,5 Гц.
Приклади хімічних зсувів протонів в ізомерних бромохлоробензенах:
5 м.ч. 7,54 (а), 7,03 (Ь), 7,16 (с), 5 м.ч. 7,51 (а), 7,35 (Ь), 7,07 (с), 7,40 (d) 5 м.ч. 7,14 (а), 7,36 (Ь)
Із синтетичних методів одержання аренів промислове значення мають
ароматизація парафінів і дегідрогенізація циклогексану. Інші реакції є лише
пізнавальними, зокрема, підтверджують-наведені висновки про будову бензену.
Лише в деяких випадках, наприклад, для мезитилену, трифенілбензену, гекса-
фенілбензену такі синтези мають препаративне значення і їх використовують у
лабораторії.
❖ Циклоолігомеризація ацетилену. Пропускаючи через ацетилен електричні
розряди, П. Бертло одержав невелику кількість бензену. М. Зелінський,
пропускаючи ацетилен над активованим деревним вугіллям, який він уперше
запропонував застосовувати для заповнення протигазів, синтезував з гарним
виходом суміш ароматичних вуглеводнів, що містила бензен:
■ Цю ж реакцію можна виконати "мокрим методом" під дією каталізатора -
нікол дикарбоніл-5/с-трифенілфосфін [(СбН5)зР]2№(СО)2, який отримують із три-
фенілфосфіну й нікол тетракарбонілу. Той же каталізатор тримеризує монозаміщені
ацетилени в 1,3,5-заміщені бензени й ізомерні їм 1,3,4-похідні бензену.
■ Каталізатори Циглера [Al(C2Hs)3 + ТіСЦ], що зумовлюють полімери-
зацію етилену та його гомологів, тримеризують як moho-, так і дизаміщені
ацетилени, відповідно, в 1,3,5-тризаміщені й гексазаміщені бензени.
Дифенілацетилен перетворюється в гексафенілбензен під дією багатьох
каталізаторів, зокрема, хром (III) хлориду в момент його відновлення реактивом
Гріньяра.
7,37 (d)
24.3. Синтез бензену та його похідних
з аліфатичних і аліциклічних сполук
24.3.1. Лабораторні методи
674
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
❖ Циклоконденсація кетонів. Ацетон під дією концентрованої сульфатної
кислоти утворює мезитилен (1,3,5-триметилбензен). Реакція є прикладом
циклічної кротонової конденсації трьох молекул кетону:
О ^ ЇМ +ЗН2°
СН3
о—СН3 _ц,
СНз п
н3сХхо н3сХхсн3 НзС
Подібно поводяться всі кетони типу ЯСОСНз. Наприклад, з ацетофенону
СбН5СОСНз можна одержати 1,3,5-трифенілбензен.
Цей тип реакцій циклізації загалом досить поширений, а за наявності у
вихідній сполуці альдегідної групи, спряженої з реакційноздатною метиленовою
групою, відбувається спонтанно з кількісним виходом. Зокрема, реакції, у яких
повинен синтезуватися напівальдегід малонової кислоти (НООС-СНг-СНО),
насправді приводять до кротонової конденсації напівальдегіду в бензен-1,3,5-
трикарбонову (тримезинову) кислоту:
итг v0 н2с'ссюн соон
ноос-сн2 2т /
оч о''ан ^ ноос-{ Ч
с-сн2 ч=<
н соон соон
❖ Ароматизація парафінів. Реакцію відкрили М. Зелінський, Б. Казанський,
А. Плате1. З'ясовано, що під час пропускання н-гептану, «-гексану й інших
парафінів з ланцюгом не менше шести карбонових атомів над платиною за 300 °С,
утворюються, відповідно, бензен, толуен або їхні гомологи, і виділяється водень:
Виявлено, що того ж ефекту можна домогтися, застосовуючи як каталізатор
хром (III) оксид, висаджений на алюміній оксиді (Б. Молдавський), за вищої
температури:.
Сг203/А12-
СН3СН2СН2СН2СН2СН2СН3 -^к^ К У-сн, + 4 Н
500 С \—/
1 Плате А.Ф. (1906-1984) - радянський хімік, професор Московського державного університету,
автор багатьох праць у галузі нафтохімії. Був першим головою оргкомітету Всесоюзних хімічних
олімпіад.
Розділ 24. Ароматичні вуглеводні. Бензен та його будова
675
Ці реакції широко використовують для ароматизації нафтових фракцій.
❖ Ароматизація циклопарафінів. Як зазначено в розділі 11, пропускаючи
пару циклогексану (або його гомолога) над платиною або паладієм за 300 °С,
можна кількісно перетворити його в бензен або гомологи бензену (М.
Зелінський, 1912 р.). Пізніше виявлено, що подібну дегідрогенізацію похідних
циклогексанів можна виконати за 400 °С з сіркою або селеном (500-600 °С),
причому атоми Гідрогену відходять у вигляді гідроген сульфіду або гідроген
селеніду.
■ Реакція "незворотного каталізу" Зелінського. Циклогексен, циклогексадієн і
їхні гомологи під дією платини або паладію диспропорціонують у бензен і
циклогексан (або гомологи) вже за кімнатної температури:
24.3.2. Природні джерела ароматичних
вуглеводнів
Ароматичні вуглеводні є вихідними продуктами для одержання найрізно-
манітніших необхідних людині речовин: фізіологічно активних, смакових,
запашних, вибухових, полімерів, барвників тощо.
❖ Кам'яне вугілля. Найважливішим джерелом одержання ароматичних
сполук є вугілля. Оскільки саме вугілля в історії розвитку цивілізації було одним
з перших освоєних природних ресурсів, то головною галуззю органічної хімії,
промислового органічного синтезу в XIX ст., у період становлення органічної
хімії, була хімія ароматичних сполук. Переорієнтація досліджень і техноло-
гічних процесів органічного синтезу на насичені й ненасичені вуглеводні стала
необхідною й можливою завдяки доступності нафтогазової сировини як
економічнішої й зручнішої для переробки.
Під час одержання з кам'яного вугілля коксу, одного з головних компонентів
металургійного циклу, одночасно утворюється кам'яновугільна смола й коксовий
газ. Коксування проводять на коксохімічних заводах, нагріваючи кам'яне вугілля
без доступу повітря в коксових печах за 1 000-1 100 °С
• Кам'яновугільна смола є складною сумішшю ароматичних сполук (понад
300 компонентів), з якої фракційною перегонкою виділяють понад 160
індивідуальних продуктів. Переробку кам'яного вугілля провадять у такий
спосіб (рис. 24.4).
676
Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
Вугілля
Кокс
(78 %)
Кам'яновугільна смола
(3%)
Коксовий газ
(16%)
Легкі олії
до 100°С
Середні
олії
Важкі
олії
230-270°С
(8-10%)
Антраценова
олія
Пек
(залишок)
(50-60 %)
(0,2 % від маси
смоли)
100-230°С
(10-12%)
270-360°С
(18-26%)
Рис. 24.4. Схема переробки кам 'яного вугілля
• 3 легкої олії за допомогою вторинної фракційної перегонки одержують
бензен, толуен, ксилени, піридин. Із середньої олії фракційною перегонкою й
кристалізацією виділяють нафтален, фенол, хінолін. У важкій олії містяться
нафтален, фенол, хінолін, крезоли, біфеніл, аценафтен, флуорен та ін.
Антраценова олія поряд з компонентами важкої олії містить карбазол, антрацен,
фенантрен та інші ароматичні вуглеводні. Залишки антраценової олії
застосовують для просочення шпал. Феноли і крезоли із фракцій зазвичай
виділяють обробкою лугами, а піридин, хінолін, карбазол та їхні похідні -
розведеною сульфатною кислотою.
• Коксовий газ розділяють на легку олію, феноли, піридинові основи й амоніак
пропусканням, відповідно, через поглинальні олії, луги й кислоти. Легкі олії
коксового газу є головним джерелом кам'яновугільного бензену. Вони містять
також толуен, ксилени, нафтален. Амоніак далі переводять у амоній сульфат, який
використовують як добриво або перетворюють шляхом окиснення в нітратну
кислоту. Зворотний коксовий газ, що складається переважно з водню й метану (52 і
32%, відповідно), використовують як висококалорійне паливо (обігрів коксових
печей) або як джерело водню (під час синтезу амоніаку) і метанолу.
❖ Нафта. З нафти виділяють більше половини всіх ароматичних сполук.
Деякі види нафти (Майкоп, Західна Україна, о. Борнео) у помітній кількості містять
бензен і його гомологи. З відповідних фракцій можна ізолювати ароматичні
вуглеводні, користуючись селективними розчинниками, наприклад, рідким сульфур
(IV) оксидом або фурфуролом, адсорбцією на силікагелі, за допомогою якої
відділяють ароматичні вуглеводні, що легко зазнають сорбування.
Зазвичай у разі прямої переробки нафти виділяють такі фракції:
■ газова фракція - містить алкани нормальної й розгалуженої будови (Сі-С5);
■ бензин - містить вуглеводні С6-Сю різної будови: нормальні й
розгалужені алкани, циклоалкани, алкілбензени. Використовують як пальне у
двигунах внутрішнього згоряння, розчинники;
■ гас - містить вуглеводні Сц-Сіг, використовують як авіаційне й
реактивне пальне;
Розділ 24. Ароматичні вуглеводні. Бензен та його будова
677
■ солярова олія (газойль) - містить вуглеводні Сіз-Сп, використовують як
дизельне пальне та сировину для подальшої переробки в бензин за допомогою
крекінгу;
■ мазут - залишок після прямої перегонки нафти (близько 50 %),
використовують як котельне паливо, для одержання мастил і для крекінгу;
■ мастила - вуглеводні Сгв-Сзв* отримують вакуумною перегонкою мазуту.
По фракціях одержують моторні, реактивні, трансмісійні, циліндрові й інші олії;
■ гудрон - залишок після вакуумної перегонки мазуту, використовують як
будівельний і дорожній бітум.
❖ Переробка нафти. Крекінг. Значне зростання автомобільного транспорту
потребувало збільшення виробництва бензину, і проста перегонка нафти не
давала змоги вирішити цю проблему. Тому сьогодні застосовують вторинну
переробку нафти. Найчастіше для цього використовують крекінг, який
проводять нагріванням нафти до температури 450 °С. Залежно від умов крекінгу
утворюються різні продукти. Є кілька видів крекінгу:
■ термічний крекінг мазуту або солярової олії за невеликого тиску й
температури 350-550 °С дає суміш алкенів, яка потребує подальшої переробки
шляхом риформінгу;
■ високотемпературний крекінг низького тиску за 550-650 °С солярової
олії утворює легші вуглеводні, які далі переробляють у моторне пальне;
обробкою мазуту й гудрону за температури 650-750 °С одержують газ, що
містить до 50 % алкенів (етилен, пропілен, бутилен) і ароматичні вуглеводні. Ці
продукти зазвичай використовують як сировину в органічному синтезі;
■ каталітичний крекінг відбувається за нижчих температур і тиску, ніж
термічний, і тільки в газовій фазі, як каталізатори використовують алюмо-
силікати. Порівняно з термічним крекінгом збільшується вихід бензину і його
якість, попутно виділяється сірка. В цьому процесі для переробки вико-
ристовують солярову олію;
■ риформінг - процес каталітичного крекінгу з одночасним каталітичним
облагороджуванням низькооктанових бензинів; виконують з використанням
різних каталізаторів. Значного поширення набули два варіанти риформінгу:
гідроформінг (гідрокрекінг) і платформінг;
■ гідроформінг виконують воднем за тиску й температур 350-450 °С,
каталізаторами є оксиди й сульфіди Ніколу та Молібдену на алюмосилікатах.
Застосування водню забезпечує гідрування високомолекулярних і сірчистих
сполук з наступним їхнім розпадом. Зазвичай використовують низькосортний
бензин і важкі фракції нафтоперегонки. Одержують високооктановий бензин з
виходом 70 % у перерахунку на нафту;
■ платформінг - каталітичний крекінг, що відбувається за допомогою
паладію й платини на алюміній оксиді, алюмосилікатах. У цьому разі
низькосортний бензин стає високооктановим завдяки процесам ізомеризації,
дегідроциклізації й ароматизації. /
678 Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
❖ Походження нафти. Сьогодні загальновизнаним уважають органічне
походження нафти. Головною вихідною речовиною нафти є планктони.
Головними чинниками нафтоутворення вважають тривалий вплив підвищених
температур і відновне середовище. Джерелом водню може слугувати вода, яка під
час взаємодії з магмою землі розпадається до водню і кисню. Відновлення
органічних сполук до алканів - найінертніших органічних сполук, стійких до
впливу кислот, основ, кисню, - пояснює склад нафти. Нафта накопичується в
порожнинах і пастках осадових басейнів. Викачана на поверхню, вона містить
воду, солі й попутний нафтовий газ; газ спалюють у факелах. Утилізація
нафтового газу є важливим економічним завданням комплексного й раціо-
нального використання природних ресурсів.
24.3.3. Реакції гомологізаціїбензену
❖ Реакція Вюрца-Фіттіга1. Гомологи бензену можна синтезувати, діючи
натрієм на суміш алкілгалогеніду й галогенобензену, наприклад:
С6НзВг + СДОг + 2 Иа ОД-ОД + 2 ШВг
Однак ця реакція лише частково відбувається в потрібному напрямі. Поряд
з цільовими продуктами утворюються діарил (біфеніл СбН5-С6Н5) і продукти
взаємодії двох замісників С2Н5, тобто алкан, алкен, діалкіл (СгНб, С2Н4, С4Н10).
❖ Реакція Вюрца-Гріньяра. Вона дає змогу цілеспрямованіше вводити
алкільний замісник в ароматичне кільце, хоча в цьому випадку можна вводити
лише первинні алкіли:
❖ Реакція Корі-Хауса. Досить перспективним методом синтезу алкілбен-
зенів є реакція Корі-Хауса:
АгВг + А1к2СиІЛ ТГФ'°^ Аг-АІк + ІЛВг + СиВг
Взаємодія, по суті, аналогічна реакції Вюрца, однак відбувається за м'яких
умов, з високою вибірковістю й не супроводжується утворенням побічних
продуктів.
1 Фіттіг В. (Рі«і§ W.R.) (1835-1910) - німецький хімік, займався органічним синтезом та
дослідженням механізмів реакцій. Відкрив та синтезував лактони, вперше отримав фенатрен,
запропонував структуру бензохінону. Є автором кількох іменних реакцій (реакція Фіттіга, Реакція
Вюрца-Фіттіга тощо).
Розділ 24. Ароматичні вуглеводні. Бензен та його будова
679
Н
❖ Реакція Фріделях-Крафтса2-Густавсонаг. Набагато частіше в синте-
тичній практиці використовують алкілування бензену алкілгалогенідами або
олефінами. Безводний алюміній хлорид слугує каталізатором. Реакція відбу-
вається вже за кімнатної температури:
А1С1,
С6Н6 + С1СН3 - С6Н5-СН3 + НС1
Аісі3
с6н6 + сісн2сн3—- с6н5сн2сн3 + неї
С6Н6 + СН2— СН2 С^Н5СН2СН3
А1С1
СбНб + СН2=СН(СН3)2 3* С6Н5С(СН3)3
Механізм цієї реакції можна описати такими стадіями:
(1) RC1 + А1С13 ===== А1С14" + R+
(2) Г + С6Н6=^С6Н5(
+ Л
(3) C6H5' =C6H5-R + H+
Н
У цьому випадку електрофілом слугує алкільний карбокатіон. Роль
алюміній хлориду полягає в генеруванні карбонієвого йона: він відщеплює аніон
галогену від алкілгалогеніду. Не дивно, що інші кислоти Льюїса можуть
реагувати подібним способом, тому їх можна використати замість алюміній
хлориду:
О :С1: :F: :F:
R:X: + Fe:Ci:== R++ Cl: Fe:Cl:- R:X: + A1:F: ===== R++ F'-AkFr
:C1: :C'l: " :F:" ":*£:"
Як випливає з наведеного, можна очікувати, що карбокатіони, які
виникають іншим способом, наприклад, у разі дії кислоти на спирт, також
атакуватимуть бензенове ядро:
\ / + I
ROH + H+=^ ROH2+ R+ + H20 С=Сч + Н+ ~С"С~
1 Фрідель Ш. (Friedel С.) (1832-1899) - французький хімік. Виявив пінаколінове перегрупування й
алкілування ароматичних речовин спільно з Дж. Крафтсом.
2 Крафтс Дж. (Crafts J.) (1839-1917) - американський хімік, працював у Р. Бунзена, Ш. Вюрца і
Ш. Фріделя.
3 Густавсон Г. (1843-1908) - російський хімік. Відкрив нестійкі комплексні сполуки галогенідів
алюмінію з вуглеводнями, що мають каталітичні властивості.
680 Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Це припущення правильне: спирти й алкени за наявності кислот здатні
алкілувати ароматичне ядро, тому ці реакції можна трактувати як варіант реакції
Фріделя-Крафтса:
ед + ССНз^СОН-1^ С6Н5-С(СН3)3+Н20
С6Н6 + (СН3)2С=СН2^- С6Н5-С(СН3)з
Реакцію алкілування аренів спиртами детально розробив І. Цукерваник1.
Промислові процеси подібного типу, наприклад, одержання етилбензену й
кумену, розробили в СРСР М. Долін і Ю. Мамедалієв2. З огляду на цей механізм
можна сподіватись, що алкілування повинне супроводжуватися перегрупуван-
ням, яке характерне для карбонієвих іонів. Справді, алкілбензени, що мають
алкільні групи, які перегрупувалися, не лише утворюються в реакціях
алкілування, а й іноді є єдиними продуктами. У кожному випадку відбувається
перегрупування менш стійкого первинного карбокатіону в стійкіший вторинний
або третинний карбокатіон:
А1Г1 ^г^з
СбН6 + 2 СН3СНСН2С1 С6Н5ССН3
СН3 СН3
ель + снхнхнхі-
А1СК
^ С6Н5СН2СН2СН3 + С6НХН(СН3)2
31-55% 65-69%
А1С13
С^Н^ + СНз(СН2)3С1 С^Н^(СН2)зСНз + С^Н^СНСЖ^СНз
34% 66% снз
■ Альтернативний механізм реакції алкілування аренів. Є докази на користь
існування іншого механізму алкілування за Фріделем-Крафтеом. У ньому
електрофілом є не карбокатіон, а алкілгалогенід, сильно поляризований
комплексоутворенням з кислотою Льюїса, і алкільна група в одну стадію
переміщається від галогену до ароматичного ядра:
91
сьАі-асі-к:а+ с6Нб-
С!
, Н
я-аісі;
■ф5/Н+А1С14-
1 Цукерваник І. (1901-1968) - узбецький хімік-органік, професор академії наук Узбекистану.
Автор багатьох досліджень у галузі алкілування та ацетилювання ароматичних сполук.
2 Мамедалієв Ю. (1905-1961) - азербайджанський хімік, президент АН Азербайджану. Головні
праці присвячені хімії та переробці нафти і природного газу. Розробив промисловий метод
хлорування метану на стаціонарних каталізаторах, налагодив виробництво дибромоетану. Запро-
вадив промислове виробництво алкілбензенів шляхом алкілування бензену ненасиченими
вуглеводнями.
Розділ 24. Ароматичні вуглеводні. Бензен та його будова
681
Цей двоїстий механізм не є чимось винятковим і укладається у звичайну
схему нуклеофільного заміщення в аліфатичному ряді, до якого, якщо
розглядати алкілгалогенід як електрофіл, а ароматичну сполуку як нуклеофіл,
можна зачислити цю реакцію. Крім того, первинні алкілгалогеніди й
метилгалогенід реагують за іншим механізмом, як і треба було сподіватися з
їхньої будови.
Реакцію Фріделя-Крафтса широко використовують у лабораторії й про-
мисловості. Серйозним її недоліком є утворення побічних продуктів -
поліалкілзаміщених бензену.
❖ Ацшіювання за Фріделем-Крафтсом. Гомологи бензену можна синте-
зувати й шляхом ацилювання за Фріделем-Крафтсом з наступним відновленням
за Клеменсеном. Ацилювання відбувається лише до стадії моноацилбензену й не
супроводжується ізомеризацією:
Відновлення алкіл-ароматичних кетонів є надійним методом одержання
гомологів бензену шляхом відновлення оксогрупи до метиленової за Клеммен-
сеном або за реакцією Вольфа-Кіяснера.
❖ Окиснення бензену. Яким би міцним не був бензен, однак енергійні
впливи руйнують його ароматичну систему. До таких процесів, насамперед,
належить розщеплення киснем повітря над ванадій (V) оксидом, що приводить
до малеїнового ангідриду. Процес використовують у промисловості:
В організмі кроликів бензен окиснюється в муконову кислоту й у такий
спосіб виводиться з організму. Під час цієї реакції зберігається карбоновий
кістяк і два подвійні зв'язки бензену:
❖ Відновлення бензену за Кіжнером. Відновлення бензену за допомогою
гідроген іодиду відбувається з ізомеризацією й приводить до металциклопентану:
24.4. Перетворення бензену в аліфатичні
й аліциклічні сполуки
Р
О
682 Чирва В.Я., Ярмолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
СН3
❖ Ізомеризація бензену. Під час обробки бензену ультразвуком К. Цейх-
мейстер1 одержав ацетилен, ідентифікований ним у вигляді купрум ацетиленіду;
діючи УФ-світлом, він ізомеризував бензен у фульвен:
У 1968 р. опроміненням бензену в рідкому стані УФ-світлом отримано
валентний ізомер бензену - дюарівський бензен (біцикло-[2,2,0]-гексадієн-2,5)
поряд з двома іншими структурними ізомерами бензену - бензваленом і фульвеном:
■ Бензен виробляють переважно каталітичним риформінгом з нафти.
Широко застосовують у синтезі циклогексану, аніліну, фенолу, лікарських
препаратів, вибухових речовин, барвників тощо.
■ Толуен застосовують у виробництві вибухових речовин, барвників,
парфумерної й харчової промисловості.
■ Ксилени використовують як розчинники, для підвищення октанового
числа бензину; у виробництві лавсану.
■ Етилбензен одержують алкілуванням етиленом бензену з наступною
переробкою в стирен.
■ Кумен одержують алкілуванням бензену пропаном за наявності
фосфатної кислоти:
Фульвен
Бензвален
24.5. Окремі представники
н3с-сн-сн3
Його застосовують як вихідну сировину у виробництві фенолу й ацетону.
■ Стирен - мономер у виробництві пластмас і каучуків.
1 Цейхмейстер Л. ^есптеІБІег Ь.) (1889-1972) - угорський хімік, працював у лабораторіях
Угорщини, Данії та США. Досліджував природні органічні речовини, такі як каротиноїди,
вуглеводи, вітаміни, займався органічним синтезом.
Розділ 25
ЕЛЕКТРОФІЛЬНЕ
ЗАМІЩЕННЯ
В АРОМАТИЧНИХ
ВУГАЕВОАНЯХ
25.1. Механізм електрофільного заміщення
Із трьох видів реакцій заміщення: електрофільного (Se), нуклеофільного (Sn)
і вільно-радикального (Sr), аренам найбільше властиві реакції Se.
Бензен, а ще легше його гомологи вступають у реакцію з багатьма
електрофільними реагентами, так що відбувається заміщення одного або кількох
атомів Гідрогену бензенового циклу на групи реагенту, який атакує. Наведемо,
як приклад, чотири найтиповіші реакції Se для самого бензену.
1. Хлорування ^ 2
{J+ci2—^(Гр+нсі
Хлоробензен
Електрофільний катіон Хлору утворюється внаслідок поляризації молекули
галогену під дією кислот Льюїса:
С12 + FeCl3 Cl+[FeCl4]-
2. Нітрування
+ N02HS04" І + H2S04
Нітробензен
Гідросульфат нітронію утворюється під час розчинення нітратної кислоти в
сульфатній:
НШ3 + Н2804 Ш2+Н804- + Н20
3. Сульфування ^ ^
+ [Н803]+Н8б; - Ц І + Н2804
Бензенсульфокислота
Утворення протонованого сульфур триоксиду можна описати в такий
спосіб:
684
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хіллія
2 Н2ЗГ>4-; - [Н3804]+Н80"4^^= [Н803]+Н80"4 + Н20
Протонований
сульфур триоксид
4. Алкілування за Фріделем-Крафтсом
Толуен
Електрофілом є атом Карбону метилхлориду, поляризований під дією
кислоти Льюїса:
СН3С1 + АІСЬ -> СНз^СІ^АІСЬ
Усі ці реакції будуть детально розглянуті у відповідних розділах. Наразі для нас
важливо зазначити, що такого типу реакції заміщення, які легко відбуваються за
кімнатної температури або незначного нагрівання, є найхарактернішою властивістю
ароматичних сполук. Це різко відрізняє їх від насичених сполук, для яких також
характерні реакції заміщення, проте вони зазвичай відбуваються за радикальним
механізмом. Що стосується олефінів, то під впливом тих же реагентів вони вступають
за звичайних умов в інші реакції - електрофільного приєднання за подвійним
зв'язком або полімеризації, так що можливість реакції заміщення залишається
нереалізованою.
Цілком імовірно, що першою фазою реакції електрофільного заміщення є
утворення я-комплексу (Дж. Дюар), у якому електрофільна частинка - у наших
прикладах С1+, >Ю2+, ЗОзИҐ, СН3+ - зв'язується з молекулою бензену завдяки
всім його л-електронам (як побачимо далі, іноді можуть утворюватися й міцні л-
комплекси внаслідок перекривання вакантних орбіталей електрофіла з л>
електронною системою бензенового кільця).
Координата реакції
Рис. 25.1. Енергетична діаграма реакції електрофільного заміщення бензену
Розділ 25. Електофільне заміщення в ароматичних вуглеводнях
685
З енергетичного погляду (рис. 25.1) утворення нестійкого я-комплексу 1
відбувається через перехідний стан ПСі легко і з малою енергією активації Е&\.
Як звичайно, стадія утворення л-комплексу минає швидко й не лімітує
швидкості всього процесу. Потім електрофільна частинка витягує з арома-
тичного секстету пару електронів я-зв'язку, причому інші чотири я-електрони
утворюють два спряжені подвійні зв'язки (структура в):
ОД н ож н
+ NO.
або, у разі використання формули Кекуле,
Ж)2
02N Н
Зазвичай, ці я-електрони під дією карбокатіону делокалізовані по всіх
п'ятьох карбонових атомах, що залишилися в другому валентному стані, а
атакований атом Карбону стає тетраедричним (5/?3-стан). Тому замість формули
(в) часто пишуть формулу (г) з позитивним зарядом, розосередженим по всіх
п'ятьох карбонових атомах. Структуру цієї проміясної сполуки, яку за аналогією
називають о-комплексом, моясна описати як резонансний гібрид граничних
структур (в'). Надалі ми будемо користуватися формулами як типу (в), так і
типу (г), не роблячи між ними різниці.
Далі від тетраедричного карбонового атома, що утворився, відділяється
протон, і електронна пара, яка вивільнилась, вливається до складу ароматичного
секстету л-електронів (д), що знову утворився. Видалення протона з л-
комплексу енергетично вигідніше, ніж приєднання нуклеофіла (як це є в
реакціях електрофільного приєднання для алкенів), тому що в цьому випадку
відбувається регенерування ароматичної системи, що дає виграш в енергії
(енергія ароматизації близько 150 кДж/моль).
Як видно з рис. 25.1, реакція може відбуватися через кілька перехідних станів.
Перший невисокий пік - перехідний стан ПСі, ЩО веде до малостійкого гс-
комплексу (перший мінімум). Другий пік - перехідний стан ПС2, через який реакція
відбувається від я- до а-комплексу. З енергетичного профілю реакції випливає, що
утворення нестійкого я-комплексу через перехідний стан відбувається легко і з
малою енергією активації Ел\. Для утворення а-комплексу через перехідний стан
686
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
ПСг необхідна енергія активації яку одержують завдяки руйнуванню а-ком-
плексу й енергії делокалізації позитивного заряду в карбокатіоні. Отже, утворення
с-комплексу не потребує значної витрати енергії ззовні. Ще раз варто нагадати, що
о-комплекс, як доведено нижче, - реальна проміжна сполука (інтермедіат\ а не
проміжний стан молекули (активований комплекс).
Загалом о-комплекс переважно малостійкий порівняно з ароматичним
секстетом і легко перетворюється в бензенову структуру шляхом викидання
протона й утворення о-комплексу з енергіями активації £аз І ^а4, відповідно.
Зазначимо, що існують випадки, коли електрофільний реагент заміщує не
атом Гідрогену в ароматичній системі, а вже наявний замісник, наприклад, -
ЗОзН на -N02 або на гідроген. Такі реакції називають шсозаміщенням:
Примітно, що шсо-заміщення характерніше для нуклеофільних реакцій.
25.2. Проточування й дейтерування бензену
Найпростішою і тому повчальною реакцією електрофільного заміщення в
бензені й узагалі в ароматичному ряді є протонування й дейтерування. Бензен
розчинний у міцних безводних кислотах, а в дейтерокислотах обмінює один за
одним усі свої атоми Гідрогену на Дейтерій. З гідроген галогенідом за наявності
алюміній галогеніду (тобто з кислотою НА1СЦ) бензен і його гомологи
утворюють тверді за низької температури комплекси. Аналогічні комплекси
утворюються з НВР4 (наприклад, комплекс толуену, т. пл. -65 °С). Як засвідчує
детальне спектроскопічне дослідження цих речовин, вони мають таку будову:
Протонований бензен Протонований толуен
В. Ола синтезував зазначений комплекс іншим шляхом:
ги
+ ^^%г
Бромування в алільне
щодо СН3-групи положення
Причому продукт реакції виявився ідентичним сполуці прямого про-
тонування толуену. З такого типу структурами, постульованими К. Інгольдом як
Розділ 25. Електофільне заміщення в ароматичних вуглеводнях
687
проміжні л-комгоіекси в разі електрофільннх реакцій в ароматичний циклах, ми
вже стикалися.
Д. Макор зі співавт. безпосередньо визначили основність низки арома-
тичних вуглеводнів, у тому числі бензену і його гомологів, вимірюючи їхній
розподіл між гексаном і безводним гідроген флуоридом (табл. 25.1). Константа
основності Кь у цьому випадку
[ArH^][F]xf±28
[ArH] f
де у квадратних дужках зазначені концентрації відповідних іонів або молекули; f -
коефіцієнт активності йонів (f+ і f-) і молекули.
Таблиця 25.1
Константи основності ароматичних вуглеводнів
Кь=і
Сполука
lgKb
Сподука
lgKb
Бензен
-9,4
лі-Ксилен
-3,2
Толуен
-6,3
Мезитилен
-0,4
и-Ксилен
-5,7
Гексаметилбензен
+1,4
Як видно з табл. 25.1, бензен - дуже слабка основа, що захоплює протон
зазначеним вище механізмом. Якщо ароматичні вуглеводні алкілувати, то їхня
сила як основ підвищується на багато порядків.
25.3. Орієнтація реакцій заміщення
25.3.1. Орієнтанти першого і другого роду
Якщо в бензеновому ядрі наявний який-небудь замісник, то він впливає на
характер наступних реакцій заміщення так, що сприяє або пасивує заміщення.
Місце введення нового замісника визначене природою вже наявного замісника,
тому що поява першого замісника порушує рівномірність розподілу я-елек-
тронів, зумовлюючи на атомах Карбону часткові ефективні заряди.
Реакції електрофільного заміщення в ряді бензену підпорядковані таким
правилам, що визначені емпірично А. Големаном1.
1.Місце введення електрофільного замісника визначене головно характером
уже наявних одного або декількох замісників у бензеновому ядрі.
2. Замісники (орієнтанти) поділяють на дві групи.
1 Големан А. (Ноііетап А.Р.) (1859-1953) - професор університету Гронінгенц (Нідерланди), автор
одного з найвідоміших підручників з неорганічної хімії. Досліджував також механізми реакцій.
688 Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
• Замісники першого роду - електронодонорні, або о, и-орієнтанти, що ске-
ровують новий замісник в о- або «-положення стосовно себе (тут і нижче
орієнтанти наведені в порядку зменшення орієнтувальної здатності):
-О",-NR2, -NHR,-NH2,-ОН,-OR,-NH-CO-R-OCOR,-СН3 (-Alk),
алкеніли, -І, -Вг, -СІ, -F.
Всі замісники першого роду за характером орієнтувальної дії поділяють на
такі, що активують або слабко дезактивують бензенове ядро:
■ активувальні орієнтанти
сильні
О: > —NR2 > —NHR >
-NH0
середні -ОН > —OR >
0-C-R:
О
NHCOCH,
слабкі -Аік $ С6Н5, де Аік = -СН3, -С2Н5 і т.д.
слабко дезактивувальні: -І > -Б > -СІ > -Вг
с Найпростішими за дією є алкільні групи. Виявляючи позитивний
індукційний ефект, вони полегшують утворення а-комплексу. Однак
зазначимо, що об'ємні алкільні групи перешкоджають о-заміщенню.
До орієнтантів першого роду належать також атоми або групи
атомів, у яких атом з неподіленою (або неподіленими) парою електронів
О—R безпосередньо пов'язаний з ароматичним ядром. Зазвичай
подібні замісники мають позитивний мезомерний (+М) і
негативний індуктивний (-1) ефекти.
Для алкоксильної, гідроксильної й амінної груп
позитивний мезомерний ефект суттєво перевершує
негативний індуктивний:(+М > -І). З цієї причини зазначені
групи виявляють сильні електронодонорні властивості та є
ефективними о-, л-орієнтантами. Найсильніший о-, п-
орієнтувальний вплив на бензенове ядро має негативно
заряджений атом Оксигену у фенолятах (+М- і -І-ефекти).
Це особливо наочно ілюструють граничні структури для
фенолят-аніона:
0 0 0
Атомам галогенів, подібно до розглянутих вище оксигено- і нітрогеновмісних
груп, властиві позитивний мезомерний і негативний індуктивний ефекти. Однак, на
відміну від оксигеновмісних, для них характерна перевага індуктивного ефекту над
мезомерним (+М < -І). Унаслідок цього атоми галогенів утруднюють вхід
Розділ 25. Електофільне заміщення в ароматичних вуглеводнях
689
електрофільної частинки в пов'язане з ними ароматичне ядро, оскільки загалом
збіднюють його електронами:
Водночас вони є о-, и-орієнтантами, оскільки здатні завдяки позитивному
мезомерному ефекту брати участь у делокалізації позитивного заряду в а-комплексі,
що утворюється під час о- або «-атаки, і так знижують енергію його утворення.
Отже, галогенозаміщені бензену повинні вступати в реакції електрофільного
заміщення за жорсткіших умов, ніж сам бензен або бензени, що містять в
ароматичному ядрі розглянуті вище замісники, і давати в цьому разі продукти о-
і «-заміщення. Відповідно до значень +М- і -І-ефектів галогени за силою
орієнтувальної дії розташовані так: І > Вг > СІ > Б.
• Замісники другого роду електроноакцепторні, або л*-орієнтанти, вони
скеровують замісник ^улі-положення:
їх поділяють на сильно дезактивувальні (-NO2, -[ЫЯз]+, -CF3, -ССІ3) і
помірковано дезактивувальні (-C=N, -SO3H, -СООН, -СНО, -COR).
Якщо замісники першого роду полегшують (прискорюють) вступ електро-
фільного замісника, то замісники другого роду - утруднюють (сповільнюють).
Нижче наведено схеми, що ілюструють перші два правила орієнтації:
-NH2
0
сі2
FeCl,
HNO.
СН3СОСІ
АІСІ3
СІ
о=ссн3
сі
HN03 (У'
а
сі
N02
+ СІ
N02
690 Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
У спрощеному вигляді реакції електрофільного заміщення для монопо-
хідних бензену можуть давати такі продукти:
■ а) замісник електронодонорний, реагент електрофільний:
N0.
N0.
0-Нітротолуен, л-Нітротолуен, л*-Нітротолуен,
62% 33 % 5 %
б) замісник електроноакцепторний, реагент електрофільний:
803Н
л/-Нітробензенсульфокислота
За наявності двох замісників різного типу місце вступу наступного
електрофільного замісника визначає замісник першого роду, тому що він
активує бензенове ядро до електрофільної атаки, наприклад:
СООН
Якщо ж обидва є орієнтантами одного роду, то місце вступу визначає
активніший. Зокрема, у наведених нижче прикладах метальна група є
сильнішим орієнтантом порівняно з Хлором, а гідроксильна група - порівняно з
метальним замісником:
Розділ 25. Електофільне заміщення в ароматичних вуглеводнях 697
Якщо орієнтанти суттєво не відрізняються за здатністю орієнтувати, то
одержують усі ізомери, що властиві як одним, так і іншим орієнтантам.
Орієнтація може бути узгодженою, якщо обидва наявні замісники
орієнтують в одне й те ж положення, або неузгодженою, коли вони орієнтують у
різні положення. Приклади узгодженої орієнтації:
СІ
СІ
Приклади неузгодженої орієнтації:
25.3.2. Причина явищ орієнтації
Орієнтант змінює вільну енергію перехідного стану порівняно з
незаміщеним бензеном. Зазначимо, що про вплив замісника на перехідний стан
можна судити згідно з постулатом Дж. Хеммонда1 за його впливом на о-ком-
плекс, оскільки а-комплекс близький до перехідного стану за енергією (див.
рис. 25.1), а отже, і за структурою. Орієнтанти першого роду знижують Ей,
особливо в разі введення нового електрофільного замісника в о- і «-положення.
У підсумку відповідно до рівняння для константи швидкості реакції різко
збільшується константа швидкості реакції електрофіла
1 Хеммонд Дж. (Hammond G.S.) (1921-2005) - американський хімік, викладач. Автор досліджень
механізмів вільнорадикальних та йонних реакцій. Займався фотохімією. С розробником так
званого постулату Хеммонда.
692
Чирва В.Я., Ярмолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
К = —е *т
А
з заміщеним бензеном порівняно з незаміщеним. Зниження Ел відбувається
завдяки подачі електронів орієнтантом першого роду до атома Карбону, з яким
цей орієнтант з'єднаний і на якому за перевагою в перехідному стані виникає
позитивний заряд. Інакше кажучи, вирішальним чинником, що визначає напрям
реакції електрофільного заміщення, вважають вплив замісника на стійкість а-
комплексу або на його здатність делокалізувати позитивний заряд.
Наведемо приклад пояснення дії орієнтувального замісника в електро-
фільній реакції, використовуючи резонансні структури. У випадку о- і «-за-
міщення о-комплекс стабілізують структури І-ІУ, тоді як л*-ізомер - тільки
структури І—III. Особливий внесок у стабілізацію о-комплексу робить структура
IV, у якій позитивний заряд додатково локалізується на гетероатомі:
II III iv
і II III IV
Механізм процесу аналогічний до того, що розглянутий на прикладі бензену.
Електрофільний реагент, атакуючи бензенове кільце, витягує з ароматичного
секстету пару електронів, за допомогою якої утворює ковалентний зв'язок з
атакованим атомом Карбону. У цьому випадку одночасно він зберігає в
активованому й у а-комплексі зв'язок також з атомом Гідрогену. Атакований атом
Карбону стає в а-комплексі тетраедричним, а чотири л-електрони, що залишились,
утворюють два спряжені зв'язки. Внаслідок відтягування пари електронів, що
утворила зв'язок з атакувальною групою, на сусідньому з атакованим атомі
Карбону виникає позитивний заряд, що, з одного боку, зазнає стабілізування
Розділс25. Електофільне заміщення в ароматичних вуглеводнях
693
делокалізацією завдяки взаємодії зі спряженою я-електронною системою, а з
іншого, - може бути частково нейтралізований (компенсований), якщо він
перебуває на атомі Карбону, що несе електронодонорний орієнтант, тобто за умови,
що атаці піддається атом Карбону в о- або «-положенні щодо атома Карбону, що
несе орієнтант.
Механізм компенсації позитивного заряду різний залежно від характеру о-,
и-орієнтанта. Якщо орієнтант першого роду несе на ключовому атомі вільну
(неподілену) пару електронів (аміно-, гідроксильна, метоксильна групи, галоген
тощо), то він передає цю пару в спільне володіння зв'язаному з ним атому
Карбону бензенового кільця, утворюючи з цим атомом подвійний зв'язок і
переходячи внаслідок цього в позитивно заряджений амонієвий, оксонієвий
тощо йон. Така система енергетично набагато бідніша, ніж система позитивного
карбонієвого йона. Тому вільна енергія перехідного стану набагато нижча, ніж
тоді, коли нема орієнтувальної групи.
Якщо о-, «-орієнтант, наприклад алкіл, не має вільної пари електронів, то він
частково компенсує позитивний заряд, що виник на зв'язаному з ним атомі
Карбону, завдяки індукційному ефекту (+1), властивому алкільним групам (треба
пам'ятати, що поняття +І-ефекту виникає лише в разі порівняння ефекту, що
його надає замісник, з ефектом, що його надає стандарт — атом Гідрогену).
Порівняємо, наприклад, бензен і толуен:
Під час атаки нітроній-катіоном позитивний заряд, що виник у «-положенні
до місця атаки, ліпше нейтралізує сусідня СН3-група (толуен), ніж атом
Гідрогену (бензен), тому що електронна пара зв'язку НзС:С зміщена до
бензенового атома Карбону більше, ніж електронна пара зв'язку Н:С:
СН3 9Нз 9нз СН3
іР+чо,..— — ф — ф + н*
Н И02 N02
У випадку електрофільної атаки в лі-положєння компенсації заряду завдяки
орієнтанта не відбувається:
694
Чирва В.Я., Ярллолкж CM., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
N02 N02 NQ2
Тому, наприклад, нітрування толуену в л*-положення мало б відбуватися
приблизно з тією ж швидкістю, що й незаміщеного бензену. Насправді
орієнтанти першого роду, які різко прискорюють процес електрофільного
заміщення в о- і «-положення, все-таки трохи прискорюють і заміщення в м-по-
ложення. Наприклад, толуен нітрується в о і «-положення приблизно в 40 разів
швидше, ніж бензен, а в лі-положення - в 2-3 рази швидше. Це передавання
впливу на л*-положення пояснюють зазвичай індуктивним ефектом (+І-ефектом),
що позначають прямою стрілкою, яка символізує зсув електронної пари о-зв'яз-
ку порівняно зі стандартом:
Оскільки І-ефекти швидко згасають у ланцюзі атомів, то вони дуже слабкі у
м- та «-положеннях порівняно з ефектом спряження, тому збільшення швидкості
заміщення в л*-положення невелике.
2533. Гіперкон'югація й ортопараорієнтація
Усі алкільні замісники в багато разів збільшують швидкість електрофільного
заміщення в бензеновому ядрі. Однак І. Бекер1 і У. Натан зазначили про
несподівану послідовність алкілів у випадку їхнього розташування в ряд за
впливом, зокрема, на швидкість хлорування й бромування (табл. 25.2).
Ця послідовність прямо протилежна до ряду збільшення +І-ефекту тих же
алкілів. Якщо порівняти дипольні моменти алкілбензенів, то виявиться, що толуен у
цьому ряді має найменший дипольний момент, а Аире/и-бутилбензен - найбільший,
тобто етил (у молекулі, яка не реагує) передає ядру менший негативний заряд, ніж
/ире/и-бутильна група. Це узгоджується з поняттям індуктивного +І-ефекту як
ефекту подавання ковалентної пари електронів зв'язку атомом X атомові У у
комбінації Х-У порівняно з Н-У. Зрозуміло, що чим більше алкільних груп несе
ключовий (пов'язаний з бензеном) атом Карбону, тим більший його +І-ефект.
1 Бекер Дж. (Baker J.W.) (1898-1967) - англійській хімік, працював разом з Т. Торпом та
K. Інгольдом. Досліджував механізми органічних реакцій. Разом з У. Натаном є автором ефекту
Бекера-Натана.
Розділ 25. Електофільне заміщення в ароматичних вуглеводнях
695
Таблиця 25.2
Відносні швидкості галогенування аренів за 24 °С у 15% водній оцтовій кислоті
Алкілбензени
Швидкість
реакції
0
сщсн3)2
0
С(СН3)3
0
Хлорування
Бромування
0,29
100
110
94
76
51
44
32
27
Таблиця 25.3
Дипольні моменти алкілбензенів
Алкілбензени
СНз-*С—"*СбН5
сн3
СН3
н-с^сбн5
сн3
СН3~-*в СН^~**С6Н5
снгс6н5
Дипольний
момент, В
0,7
0,65
0,58
0,37
Описуваний парадокс, що одержав назву ефекту Натана-Бекера, пояснили
більшою здатністю до поляризації Н-С-зв'язку порівняно з С-С-зв'язком і
можливістю завдяки цьому більшого подавання електронів алкільної групи вже
завдяки +М-ефекту, оскільки ключовий атом зв'язаний з більшою кількістю
атомів Гідрогену (гіперкон 'югація):
Поняття гіперкон'югації не обмежене тільки що розглянутою галуззю або
явищами взаємодії алкілів з ароматичним ядром, а має також загальне значення.
Це частковий, але найважливіший випадок о,я-спряження, що стосується вже не
тільки взаємодії систем Н-С=С- або Н-С-СбН5, а й систем з негідрогенними
атомами.
25.3.4. Метаорієнтація
Замісникам другого роду, або ж-орієнтантам, властиві, як звичайно,
одночасно -М- і -І-ефекти або тільки -І-ефект. Перші мають кратні зв'язки:
4? -§-он -с -с
о" о он я
696 Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Інші замісники діють тільки завдяки наявності часткового або повного
позитивного заряду на атомі, що пов'язаний з бензеновим кільцем:
+ +5 +5
-Н(СН3)3 -СБз -СС13
Негативний індукційний ефект найсильніше виявляється в оположеннях і
найменше - у найбільш віддалених «-положеннях. Водночас негативний мезо-
мерний ефект виявляється однаково як в о-, так і «-положеннях.
В обох випадках ця дія завдяки відтягуванню електронів закінчується
утворенням позитивного заряду на тому атомі Карбону бензенового кільця, що
несе л*-орієнтант. Тому відповідний активований комплекс і а-комплекс менш
вигідні, ніж аналогічні комплекси для незаміщеного бензену:
N0+
$(СН3)3 $(СН3)з
Під час атаки ж у л*-положенні настільки невигідної комбінації не виникає, і
ця атака виявляється єдино можливою. Оскільки відбувається загальне відтя-
гування електронів з бензенового кільця, зокрема з лі-положєнь (-І-ефект), то у
^и-положенні атака відбувається важче, ніж у незаміщеному бензені, і бензенове
кільце, з лі-орієнтантом, виявляється загалом пасивнішим до електрофільних
заміщень:
$(СН3)3 ^(СН3)3 ЖСН3)3 ^(СН3)3
Коли відбувається електрофільна атака похідної бензену, що містить
орієнтант з неподіленими парами електронів, наприклад:
н Он ОТ
-< -с, -Н
О: О: О:
то поряд з уведенням електрофільного замісника переважно в л<-положення,
заміщення спрямовується значно й у о-положення. Тому в цих прикладах під час
нітрування утворюються, крім м-, також о-ізомери:
Розділ 25. Електофільне заміщення в ароматичних вуглеводнях
697
N0,
^02
СООН
-™2
Ж)2
Очевидно, це можна пояснити первинним зв'язуванням атакувального
електрофіла парами р-електронів або я-електронами орієнтанта з наступним
переміщенням електрофіла в найближче о-положення:
З викладеного випливає, що бензенове ядро може збагачуватися та збіднюватися
електронами. Отже, саме воно може як подавати електрони, так і забирати їх. Така
"двоїстість" бензенового ядра є однією з його найважливіших властивостей і дасть
змогу надалі пояснити його поведінку в багатьох хімічних реакціях.
Крім дії орієнтувального замісника, на співвідношення продуктів реакції
впливають, і в деяких випадках суттєво, природа, активність і концентрація
каталізаторів, характер розчинника, температура й стеричні чинники.
За допомогою сучасних методів дослідження з'ясовано, що розглянуті
реакції можуть відбуватися за всіма можливими напрямами так, що не йдеться
про винятковий синтез тих або інших ізомерів, а лише про переважний напрям
реакції заміщення. Наприклад, виявлено, що зі значним підвищенням
температури орієнтувальний вплив замісників поступово втрачається, тому що
швидкості всіх трьох процесів (о-, м- і «-заміщення) вирівнюються.
Дуже цікаве, *оча ще й мало вивчене питання про співвідношення між о і «-
ізомерами. Тут варто враховувати декілька чинників. По-перше, у бензеновому
кільці оположень два, а «-положення лише одне. По-друге, в оположенні можуть
простежуватися просторові утруднення. Зокрема, під час нітрування толуену
утворюється 58% о-ізомеру, а під час нітрування за тих же умов треш-
бутилбензену - 16 % о і 73 % «-ізомеру. По-третє, індукційному впливу особливо
піддаються оположення. Отже, якщо замісник індукційно дезактивує бензенове
кільце, то він виявляється найбільше в 0-положенні. По-четверте, ефект спряження
за нез'ясованими причинами найбільше виявляється в «-положенні.
З розглянутого вище матеріалу бачимо, що найбільший вплив на напрям
реакцій електрофільного заміщення в ароматичних сполуках має ефект
спряження. Водночас спряження може виявлятись тільки у випадку, якщо групи,
які можуть брати участь у спряженні, компланарні. Якщо ж осі хмар /?- або я-
електронів замісника й я-електронів бензенового кільця не паралельні, то
спряження послаблюється, і замісник втрачає свій вплив на бензенове кільце. Ця
особливість ефекту спряження підтверджена багатьма прикладами.
г+Ж)П
N0,
Розділ 26
ГАЛОГЕНОПОХІАНІ
АРОМАТИЧНИХ
ВУГАЕВОАНІВ
26.1. Класифікація й номенклатура галогеноаренів
26.1.1. Класифікація галогеноаренів
Арени
з одновалентними
галогенами
Арени з
багатовалентним
и галогенами
Алкіларени з галогенами
в бічному ланцюзі
<©ЬВг
Бромобензен
(фенілбромід)
Іодобензен
@)-СН2Вг @-СНВг2
Бензилбромід Бензиліденбромід
26.1.2. Номенклатура галогеноаренів
• Замісна номенклатура. У замісній номенклатурі галогени розглядають як
замісники і називають лише як префікси. У цьому разі дотримуються всіх
правил номенклатури вуглеводнів.
• Радикально-функціональна номенклатура. У радикально-функціональній
номенклатурі назва сполуки складається з назви вуглеводневого радикала й
назви галогеніду, які записують разом. Приклади назв галогенопохідних за
радикально-функціональною номенклатурою наведені курсивом вище.
26.2. Фізичні властивості галогеноаренів
Моногалогенобензени є порівняно висококиплячими рідинами зі специ-
фічним "ароматичним" запахом. Температури кипіння залежать від дипольного
моменту молекули, тому, наприклад, одихлоробензен, що має найбільш
полярну молекулу порівняно з м- і «-ізомерами, кипить за найвищої
температури. Ароматичні сполуки з галогеном у бічному ланцюзі в а-положенні
спричинюють сльозовиділення. Температура кипіння природно підвищується в
ряді від флуоробензену до іодобензену. Зазвичай, и-галогенозаміщені похідні
Розділ 26. Галогенопохідні ароматичних вуглеводнів
699
бензену мають вищу температуру плавлення, ніж відповідні о- і л*-ізомери, що
пов'язано зі щільнішим упакуванням у кристалічну ґратку «-ізомеру.
Наявність негативного індукційного й мезомерного ефектів, що діють у
прямо протилежних напрямах, вкорочує зв'язок С-Наї і зменшує дипольний
момент галогеноаренів порівняно з галогеноалканами й робить їх подібними на
галогеноалкени (табл. 26.1).
26.2.1. Спектроскопія галогеноаренів
• УФ-спектроскопія. В УФ-спектрах моногалогенобензенів наявні інтенсивні
смуги з А,тах 204-210 і 254-264 нм.
• ІЧ-спектроскопія. Галогенопохідні ароматичних вуглеводнів поряд зі
смугами поглинання, що властиві аренам, мають характерні смуги поглинання
середньої інтенсивності валентних коливань Ус-сі~1 100 см"1 та інтенсивні смуги
поглинання валентних коливань \с-? в ділянці 1 100-1 250 см"1.
• ПМР-спектроскопія. Приклади впливу атомів галогену на хімічні зсуви
сигналів протонів ароматичних сполук наведені в розділі 24.
26.3. Одержання галогеноаренів
26.3.1. Галогенування в ядро
❖ Пряме галогенування аренів. Як відомо, реакційна здатність галогенів під час
взаємодії з алканами змінюється в ряді Рг > СЬ > Вгг > І2. Ця ж закономірність
зберігається й у разі їхньої взаємодії з аренами. Пряме галогенування в ядро
застосовують зазвичай у випадку хлору й, рідше, брому. Наприклад, атоми
Гідрогену в бензені і його гомологах легко зазнають заміщення на Хлор або Бром за
наявності каталізаторів, так званих переносників галогену (РеС13, А1С13 та інші
галогеніди), які звичайно готують безпосередньо в реакційній суміші. Загальна
властивість цих галогенідів полягає в здатності утворювати з аніоном галогену
більш-менш міцні аніони типу: РеСЦ", АІСЦ", РеВгЛ За силою дії переносники
брому розташовують у такий ряд:
РеВгз > А1Вг3> 2пВг2 > ІВг3 > 8ЬВг3 > СиВг2 > 82Вг2 > РВг3.
Під дією такого каталізатора молекула галогену зазнає поляризування через
захоплення одного з двох атомів молекули у вигляді аніона "переносником
галогену". У цьому разі другий атом одержує частковий позитивний заряд і стає
електрофільним реагентом:
С!2 + РеС13 СІ-
СІ
-СІ-Ре-СІ
і
сі .
1-5
700
Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Загалом механізм хлорування полягає у взаємодії комплексу каталізатора й
галогену з бензеном:
СІ СІ
тг-Комплеке о- комплекс
Стадією, яка лімітує заміщення, є утворення о-комплексу. Те, що реакція
відбувається через утворення галогеном комплексів з кислотами Льюїса,
підтверджено за допомогою ізотопу хлору. Внаслідок взаємодії мітка виявилася
не тільки в хлоробензені, а й у алюміній хлориді.
Отже, хлорування й бромування в ядро бензену й толуену описують такі
підсумкові рівняння (вихід хлоро-і бромобензену - 80-90 %):
СІ
|| Л +СЇ[¥еС14] -(і "] + НРеС14
3 +С,+[РеСІ4]"
Уперше хлоробензен отримав 1865 р. М. Соколов1.
Процес галогенування (хлорування й бромування) супроводжується
регенеруванням каталізатора. Із цієї причини його потрібно всього близько 1 %
від маси субстрату. Іноді як каталізатор застосовують іод. У цьому випадку
галогенувальний агент утворює з іодом міжгалогенну сполуку, яка генерує
іодид-катіон. Під час взаємодії з молекулою Хлору іодид-катіон дає міжгало-
генну сполуку й катіон Хлору:
С12+ І2 =2: 2ІС1
ІС1=^І++СҐ
І++С12 ІС1 + С1+
Хлор-катіон, що утворився, вступає в реакцію електрофільного заміщення.
Реакція Зандмейера2. Галогенування ароматичних вуглеводнів за наявності
кислот Льюїса зрідка дає моногалогенопохідні. Тому, щоб синтезувати
1 Соколов М. (1826-1877) - російський хімік, стажувався у Ю. Лібіха і Ш. Жерара. Видавав
перший російський хімічний журнал спільно з А. Енгельгардтом.
2 Зандмейєр Т. (Бапсітеуег Т.) (1854-1922) - швейцарський хімік. Піонер створення хімії
барвників.
Розділ 26. Галогенопохідні ароматичних вуглеводнів
701
моногалогенопохідні, застосовують солі діазонію. Для цього їх нагрівають у
водному розчині за наявності купрум (І) хлориду або броміду. Реакція
відбувається за рівнянням
СбН^СІ^2- С6Н5С1 + Ы2 або С6Н5К2Вг^^ С6Н5Вг + И2
Роль купрум (І) галогенідів полягає в тому, що вони утворюють із сіллю
діазонію продукти приєднання, які під час нагрівання розпадаються, утворюючи
галогенопохідні. Ці реакції відкриті Т. Зандмейєром і мають важливе препа-
ративне значення.
Не менше значення має реакція Л. Гаттермана1: розпадання солей діазонію
за наявності міді й гідроген галогенідів. Обидві реакції детально розглянуті в
розділі ЗО.
Уведення більш ніж одного замісника в ядро відбувається важко внаслідок
дезактивувального впливу вже наявного атома галогену.
❖ Використання інших галогену вальних реагентів. Найактивнішими
галогенувальними агентами вважають перхлорати Брому й Хлору:
Аё"сю;+ сі2 —- сі+сю;+ AgCl
Якщо застосовувати міжгалогенні сполуки, то електрофіл утворюється з
галогену, електронегативність якого менша:
С16^-Вг+=СГ+Вг+ С16^І6+=С1+І+
Для гіпогалогенітів утворення електрофілів спостерігають тільки за
наявності кислот:
НОС1 + Н+ =^ Н20-С1 = Н20 + СҐ
Передбачають, що електрофіл існує як сильнополярний комплекс, тому що
немає прямого доказу існування катіонів СҐабо Вг+.
Щоб уникнути утворення дигалогеноаренів, використовують надлишок
субстрату, і процес ведуть за кімнатної температури.
■ У промисловості хлоробензен синтезують у газовій фазі за методом
Рашіга2:
СІ
Онеї + 02 1
-5Г- ф + н>°
1 Гаттерман Л. (Gattermann L.) (1860-1920) - німецький хімік, автор практикуму з органічної хімії.
2 Рашіг Ф. (Raschig F.) (1863-1928) - німецький хімік та політик. Працював разом з Р. Бунзеном у
Берлінському університеті, потім - у фірмі BASF. Є автором промислових методів одержання
кількох речовин: гідроксиламіну (процес Рашіга) та фенолу (процес Рашіга-Хукера).
702
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
❖ Одержання флуороаренів. У випадку флуору пряме галогенування засто-
совують зрідка й тільки в струмі інертного газу через сильний тепловий ефект. У
цьому разі частіше відбувається не процес електрофільного заміщення, а реакція
приєднання з утворенням перфлуороциклоалканів і далі флуороалканів, які можна
дегідрофлуорувати, щоб одержати суміш поліфлуоробензенів.
У деяких випадках як флуорувальний агент зручно використовувати
флуориди металів з їхньою вищою валентністю:
0'+a6f>^- 0"f+ ag
Як звичайно, монофлуоробензен одержують через діазосполуки за методом
Шимана1 (див. розділ ЗО) або реакцією обміну атома Хлору на Флуор.
❖ Одержання іодоаренів. Трохи окремо стоять іодопохідні аренів. У
випадку сильних о-, и-оріентантів (-NH2, -ОН-групи) реакція відбувається легко
без застосування каталізатора, як під час хлорування або бромування.
Іодування слабко активованих ароматичних вуглеводнів іодом проводять за
наявності окиснювачів. їхнє застосування мотивують необхідністю зв'язування
сильного відновника - гідроген іодиду, який здатен відновлювати іодобензен до
бензену:
2Ш + HN03 І2+Н20 + HN02
Однак гідроген іодид за звичайних умов не відновлює іодоарену. Очевидно,
річ у тому, що внаслідок окиснення з молекули іоду або іодид-аніона утво-
рюється активна іодувальна частинка -1+.
Гарні результати синтезу ароматичних іодосполук дає взаємодія калій
іодиду з солями діазонію, які одержують з первинних ароматичних амінів і
нітритної кислоти в розчині мінеральної кислоти:
hno, v1
c6h5nh2-^ сда.сі-^- c6h5i+n2 + KC1
Сіль фенілдіазонію
❖ Одержання полігалогеноаренів. Дія надлишку галогену на арен за
100 °С дає полігалогенопохідні:
1 Шиман Г. (8спіетапп С) (1899-1967)-німецькийхімік. Головні праці в галузі флуороорганічних
ароматичних сполук.
Розділ 26. Галогенопохідні ароматичних вуглеводнів
703
Галогенування алкілбензенів відбувається легше, ніж бензену. Наприклад,
толуен зазнає бромування діоксандибромідом винятково в «-положення, що
пояснюють стеричним чинником, тоді як бензен у цій реакції є індиферентним.
Просторовий чинник особливо виявляється під час галогенування гомологів
бензену з об'ємними алкільними групами. Узгоджена орієнтація алкілів в арені
сприятливо позначається на швидкості галогенування й виході продуктів
реакції.
26.3.2. Галогенування в бічний ланцюг
Метальна група толуену під час електрофільного хлорування з "пере-
носником" залишається незачепленою. Якщо ж хлорувати або бромувати толуен
та інші гомологи на світлі, а ще ліпше за підвищеної температури або в парі,
коли ретельно забезпечено відсутність феруму й інших "переносників", то
галогенування відбувається за законами ланцюгових реакцій і тільки в алкільну
групу. Толуен у цьому разі послідовно перетворюється в бензилхлорид,
бензиліденхлорид і трихлорометилбензен, а етилбензен - в а-хлороетилбензен і
а,а-дихлороетилбензен.
Алкілбензени легко зазнають галогенування саме в а-положення бічного
ланцюга, а у віддаленіші від бензенового ядра положення - сутужніше, майже
так само, як у насичених вуглеводнях. Це відбувається тому, що вільні радикали
типу бензильного СбН4-СН2-або фенілетильного С6Н5-СНСНз, які є інтерме-
діатами в ланцюговій реакції, стійкіші і, отже, утворюються з меншою енергією
активування, а, відповідно, і з більшою швидкістю, ніж ізомерні їм радикали,
наприклад, СбНг-СН2СН2-:
С12 ^ »2 СІ • (ініціювання ланцюга)
С6Н5СН3 + сі- —*~ с6н5сн2- + НС1
С6Н5СН2 + С12 —*~ С6Н5СН2С1 + СІ •
Таким же способом утворюються бензиліденхлорид і трихлорометилбензен:
С6Н5СН3 + 2 С12 —- С6Н5СНС12 + 2 НС1
еденз + з сі2 —~ с6НзСсі3 + з неї
Іодування в бічний ланцюг не відбувається. Іодовані й флуоровані в ланцюг
продукти одержують шляхом обміну відповідних хлоридів на арґентум флуорид
в етері й натрій іодид в ацетоні.
26.3.3. Хлорометилювання
Реакцією електрофільного заміщення в ароматичні вуглеводні можна ввести
групу СН2С1 (хлорометилювання). Для цього на ароматичний вуглеводень діють
704
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
формальдегідом у вигляді формаліну або параформу за наявності цинк хлориду,
пропускаючи одночасно гідроген хлорид (Г. Блан):
СНгО + Н+ +СН2ОН -—- СН2=Ь-Н
АгН + 'С^ОН - АгСН2ОН ^ (Г^рСН2С1
Аналогічно виконують бромометилювання, іодометилювання й флуороме-
тилювання аренів.
■ Цікаву реакцію обміну атома Гідрогену на Хлор під час взаємодії толуену
з тетрахлорометаном (225-235 °С) описав Ю. Ольдекоп1:
С6Н5СН3 + СС14 С6Н5СН2С1 + СНС13
26.4. Хімічні властивості галогенопохідних ряду
бензену
26.4.1. Орієнтувальний ефект галогену
як замісника в реакціях електрофільного
заміщення
Атоми галогенів - це найслабші о-, и-орієнтанти, і, на відміну від всіх інших
орієнтантів цього роду, вони не прискорюють, а сповільнюють реакції
електрофільного заміщення. Це пояснюють протилежним знаком індукційного
ефекту й ефекту спряження. Атоми галогенів виявляють щодо ядра -І-ефект
(відтягування електронів з ядра порівняно зі стандартним Н-С-зв'язком) і
слабкий ефект спряження (здатність у момент атаки в о- і «-положення подати
пари електронів і нейтралізувати в такий спосіб позитивний заряд, що виникає в
циклі). Це виражають такою схемою:
Пряма стрілка означає, що бензенове ядро хлоробензену збіднене електронами
порівняно з бензеном. Однак у випадку елекгрофільної атаки в о або «-положення
все-таки виникає активований комплекс, звичайний для о, «-орієнтації. Правда,
рівень вільної енергії цього активованого комплексу вищий, ніж у випадку
1 Ольдекоп Ю. (1918-1992) - білоруський хімік, професор. Є автором багатьох праць у галузі
металоорганічних сполук, досліджував поліхлорорганічних сполук, а також органічних та
елементоорганічних пероксидів.
Розділ 26. Галогенопохідні ароматичних вуглеводнів
705
незаміщеного бензену, оскільки компенсація позитивного заряду завдяки неспареним
електронам атома галогену менш повна, ніж для будь-якого іншого звичайного
орієнтанта першого роду. Не компенсується повністю навіть те відтягування
електронів, що відбувалося завдяки сильному індуктивному ефекту атома Хлору:
Як відомо, атоми галогенів належать до орієнтантів першого роду (о-,«-
орієнтанти), однак, на відміну від інших орієнтантів першого роду, вони
утруднюють перебіг реакцій електрофільного заміщення:
З галогеноаренів найбільшу схильність до реакцій електрофільного
заміщення виявляють флуоро- та іодобензени. Менший дезактивувальний ефект
атомів Флуору й Іоду пояснюють найбільшим позитивним ефектом спряження
внаслідок /?,7і-спряження між 2р-електронами атома Флуору й я-електронами
бензенового кільця, а атом Іоду має найнезначніший індукційний ефект
порівняно з іншими галогенами.
Орієнтувальну дію атомів галогенів пояснюють тим, що вони стабілізують
а-комплекси, які відповідають о- і «-заміщенням, а перевагу «-ізомерів у
продуктах заміщення - дезактивувальним впливом атомів галогенів на о-
положення завдяки -І-ефекту:
К02
26.4.2. Реакції електрофільного заміщення
ж>2
СІ
6'
н
706
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
с\ СІ СІ
нх хн нх нх
Як зазначено, усі чотири галогени — o-, л-орієнтанти, що зменшують, однак,
швидкість електрофільного заміщення в бензеновому ядрі; +М-ефект атома
Флуору більший, ніж інших галогенів. Швидкості нітрування флуоро-, хлоро- і
бромобензену співвідносяться як 5:1,1:1 (К. Інгольд). Проте у випадку
конкурувальної орієнтації між атомами Хлору і Флуору, за даними
М. Ворожцова (мол.), місце входження нітрогрупи визначає атом Хлору.
Наприклад, 3,5-дифлуоро-2,6-дихлоробензен зазнає нітрування в оположення
щодо атомів Хлору і в л*-положення щодо Флуору.
26.43. Реакціїнуклеофільного заміщення атому
галогену
Атоми галогену, безпосередньо пов'язані з атомами Карбону бензенового
циклу, "нерухомі" і мало здатні вступати в реакції нуклеофільного заміщення.
Найбільше здатний до цього типу реакцій атом Іоду, найменше - Флуору.
Однак зазначимо, що реакції нуклеофільного заміщення атома галогену, за
винятком Флуору, в арилгалогенідах каталізовані металевою міддю (Ф. Ульман1),
іноді купрум (І) та купрум (П) оксидами. Нижче наведені приклади таких реакцій:
C6H5Cl + NaOH-^— C6H5OH + NaCl
С6Н5І + CH3COONa -^U* СН3СООС6Н5 + Nal
С6Н5С1 + NaCN C6H5CN + NaCl
C6H5C1 + NIV^*— C6H5NH2 + NH4C1.
Роль каталізатора остаточно не з'ясована, однак припускають, що він
зменшує електронну густину на атомі Карбону, який несе галоген. Уважають,
що реакція відбувається через купруморганічні сполуки. Найбільший
практичний інтерес серед такого роду сполук має синтез ароматичних нітрилів з
галогеноаренів — реакція Розенмунда—Брауна:
ff\ CuCN Лш^ fYCH>
0-Бромотолуен о-Метилбензонітрил
1 Ульман Ф. (ІЛІтапп Р.) (1875-1939) - швейцарський хімік. Головні праці присвячені синтезу
біфенілу та акридину. Редактор 12-томної "Енциклопедії технічної хімії*".
Розділ 26. Галогенопохідні ароматичних вуглеводнів
707
Без міді хлоробензен також можна прогідролізуватн лугом, однак для цього і:
потрібні дуже жорсткі умови, наприклад, тривала дія 20-40 % лугу за 300 °С і
300 атм. Порівняно нещодавно з'ясовано, що пара хлоробензену реагує з парою
води за атмосферного тиску й 500 °С за наявності силікагелю, обробленого
сіллю Купрум (II) (метод Ф. Рашіга). Цей спосіб став основою одного з
промислових методів синтезу фенолу:
с6н5сі + н2о Уц™* СбН5он+неї
Причина інертності атома галогену, зв'язаного з ароматичним кільцем, та ж,
що й вінілгалогеніду. Вона зумовлена меншою полярністю й більшою міцністю
зв'язку С-Наї в ароматичних сполуках і вінільних галогенідах порівняно з
галогеноалканами. Атоми галогену, маючи вільні пари електронів, взаємодіють
як з я-зв'язком вінільної групи, так і з л-системою бензенового ядра. Цей
мезомерний ефект (+М-ефект) дуже незначний, тому що здатність атома Хлору
подавати електронну густину неподілених пар /7-електронів мала. Навпаки,
індукційний ефект атома Хлору, тобто відтягування електронів до атома Хлору
як до електронегативного атома, завжди дуже великий, і він наводить
позитивний заряд на атом Карбону, що зв'язаний з цим атомом Хлору, і далі -
на атоми Карбону в о- і «-положеннях щодо галогену арену або на р-атом
Карбону у вінілхлориді. Однак ці часткові позитивні заряди в о-, «- або,
відповідно, Р-положеннях виявляються меншими, ніж вони були б, якби не
мезомерний ефект атома Хлору, що має зворотний напрям і зменшує згадані 5+-
заряди. У правильності цього твердження можна переконатися, зіставивши
дипольні моменти низки галогеноалканів, галогеноалкенів і галогеноаренів
(табл. 26.1).
Таблиця 26.1
Дипольні моменти галогенопохідних
Галогенопохі
дна
Дипольний
момент, Б
Галогенопо
хідна
Дипольний
момент, Т>
С2Н5С1
2,0
С2Н5Вг
2,09
СН2=СНС1
1,44
СН2=СНВг
1,41
С6Н5С1
1,56
С6Н5Вг
1,54
З наведених значень дипольних моментів бачимо, що у вінілгалогенідів і
галогенобензенів вони значно нижчі, ніж у відповідних насичених галогенідів.
Як уже зазначено, атом Карбону, зв'язаний з атомом галогену в арома-
тичних і вінільних галогенідах, має менший позитивний заряд і тому менш
сприятливий до нуклеофільних атак. Проте варто збільшити цей заряд, увівши в
о- або «-положення електроноакцепторну групу (-N02,)» як пов'язаний з
галогеном атом Карбону стає здатним сприймати нуклеофільні атаки і, як
наслідок, атом галогену стає рухливим і легко вступає в обмін з групами ОН",
МН2", СИГ, ЯСОО".
708 Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Реакції нуклеофільного заміщення атома галогену в аренах відбуваються за
двома механізмами:
-відщеплення-приєднання (ариновий механізм), характерний для
неактивованих сполук;
-приєднання-відщеплення, що властивий активованим сполукам.
❖ Реакції відщеплення—приєднання. Що стосується галогенобензенів, які
не мають електроновідтягувальних груп, то часто, якщо не завжди, заміщення
атома Галогену на нуклеофіли відбувається не прямо, а через попереднє
відщеплення гідроген галогеніду з утворенням нестійкого, досить реакційно-
здатного бензину або дегідробензену, що має потрійний зв'язок у циклі й здатний
по ньому приєднувати воду, амоніак та інші нуклеофільні молекули. Такий
висновок зроблено на підставі того, що під дією лугу на и-хлоротолуен
утворюються два ізомери - п- і л*-крезоли:
СІ ОН Н
Остаточно цей механізм доведено шляхом одержання аніліну дією амоніаку
на хлоробензен, у якому атом Хлору був зв'язаний з радіоактивним ізотопом
Карбону 14С:
У 50 % молекул аніліну аміногрупа виявилася зв'язаною з радіоактивним
атомом Карбону, а в 50 % - із сусіднім з ним карбоновим атомом. Таке непряме
заміщення називають кінезаміщенням.
■ Дегідробензен - нестійка проміжна сполука, у якій, крім а- і я-зв'язків, є
ще один додатковий зв'язок завдяки слабкому (бічному) перекриванню ^р2-
орбіталей, що розташований у площині молекули перпендикулярно до
електронної хмари я-зв'язків. Однак він досить неміцний, оскільки її яр2-
орбіталі не паралельні:
Н
Слабке перекривання
^-орбіталей
❖ Реакції приєднання-відщеплення в галогенонітробензенах. Як з'ясо-
вано, нуклеофільне заміщення атома галогену в арилгалогенідах стає можливим
Розділ 26. Галогенопохідні ароматичних вуглеводнів
709
за м'яких умов та наявності електроноакцепторних замісників, таких як -N02,
-СООН, -С=Н в о-, «-положеннях до атома галогену. Справді, одержання 2,4-
динітрофенолу з 2,4-динітрохлоробензену відбувається за 100 °С і з 5 % натрій
гідроксидом за атмосферного тиску. Такі галогеноарени % називають акти-
вованими. Доведено, що реакція відбувається за двостадійним механізмом, що
передбачає утворення проміжного продукту А під час приєднання нуклеофіла до
субстрату:
N0,
+ НО
- Повільно
N0,
Швидко
N0,
N0.
Розглянемо механізм нуклеофільного заміщення з використанням резо-
нансних структур. На першій, найповільнішій стадії нуклеофільний реагент
приєднується до атома Карбону, що несе галоген, з утворенням а-комплексу.
Електроноакцепторні замісники в о- і «-положеннях до атома галогену
збільшують стійкість а-комплексу й цим прискорюють реакцію заміщення:
СІ
і
"ОН
Повільно
Січ он
.о-^ю
т
-0-№0
+
На другій стадії відбувається швидке відщеплення аніона Хлору й
утворення кінцевого продукту реакції:
Ск ОН
Реакція є бімолекулярною, двостадійною, а її швидкість залежить від
концентрації субстрату й нуклеофіла:
У = А:[АгНа1] [НО"].
Водночас групи -N02, -СООН і -С=Н що перебувають у лі-положєнні
стосовно атома галогену, сповільнюють реакцію заміщення, оскільки ці
замісники дестабілізують проміжні о-комплекси.
710 Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Одним з головних доказів реалізації подібного механізму реакції нуклеофіль-
ного заміщення в активованих арилгалогенідах є одержання стабільних аналогів
проміжної сполуки. Хоча проміжної сполуки заміщення атома галогену в
активованих арилгалогенідах ще не виділено, однак у реакціях нуклеофільного
заміщення алкіларилових етерів це вдалося зробити. Хімічними й спектральними
методами доведено, що такі проміжні сполуки є аніонними а-комгаїексами, тобто
вони утворюються внаслідок формування нового а-зв'язку. Атом Карбону, який
атакують, стає .ур3-гібридним. Ці комплекси одержали назву комплексів
Мейзенгеймера1, який уперше синтезував їх 1902 р.:
Кислота
N0,
Н3СО .ОС2Н5
N0,
ос2н5
К
N0,
СН3ОК
N0,
2,4,6-Тринітроанізол
Комплекс Мейзенгеймера
1,3,5-Тринітро-2-етоксибензен
Утворення аніонних о-комплексів стає можливим за наявності електроно-
акцепторних замісників, які стабілізують аніон за механізмом спряження.
Будову аніонного комплексу можна відобразити набором граничних (резо-
нансних) структур:
N0,
С1Ч ОН
N0,
N0,
С1Ч ОН?^
N0.
Замісники в лі-положенні до атома галогену в арилгалогенідах мало
впливають на їхню реакційну здатність, оскільки не беруть участі в ефективній
делокалізації негативного заряду аніонного о-комплексу завдяки спряженню.
Цікаво, що заміщення атома Флуору на нуклеофіли в и-нітрофлуоробензені
відбувається значно легше, ніж атома Хлору в л-нітрохлоробензені. Тому
нітрофлуоробензени, особливо 2,4-динітрофлуоробензен, широко використо-
вують для введення 2,4-динітрофенільного залишку в кінцеві аміногрупи білків.
❖ Синтез пікринової кислоти. Чи є можливе на практиці нуклеофільне
заміщення в арилгалогенідах за м'яких умов? Це питання стає актуальним у разі
Мейзенгеймер Я. (МеІБепЬеітег ].) (1876-1934) - німецький хімік. Дослідив механізм
"перегрупування Бекмана", а також відкрив та вивчив структуру так званих комплексів
Мейзенгеймера.
Розділ 26. Галогенопохідні ароматичних вуглеводнів
711
організації промислового виробництва хімічного продукту, одержання якого
передбачає реакцію подібного типу. Розглянемо цю ситуацію на прикладі
синтезу пікринової кислоти, яка має широке практичне застосування як барвник,
вибухова речовина, аналітичний реагент. Можливі дві схеми її одержання з
доступної сировини - бензену:
1)
сі,
РеСЦ
СІ №ОН
(водн.)^
300-340°С
НЫ03 (конц.)
Н2804 (конц.)
0-Нітрофенол
N0
ОН
2
+
О >г
2 я-Нітрофенол
НЫОз
(конц.)
.БОзН
О N
НШ3(конц.} 2"
Н2804 (конц.)
2)
БОзН
Фенол-2,4-дисульфокислота
)Н
N0.
N0,
N0.
ЫаОН (водн.)
70-100 °С
N0.
N0.
2,4,6-Тринітрофенол
(пікринова кислота)
(конц.)
2,4-Динітрохлоробензе
2,4-Динітрофенол
За першою схемою спочатку одержують фенол з хлоробензену в апаратах
високого тиску — автоклавах — за високих температур. Далі фенол нітрують у дві
стадії - спочатку розведеною нітратною кислотою до суміші нітрофенолів, потім
концентрованою НЖ)з або сульфують до фенол-2,4-дисульфокислоти з
наступним нітруванням концентрованою нітратною кислотою.
Обидва варіанти першої схеми мають недоліки, пов'язані з частковим
осмоленням під час нітрування фенолу й, відповідно, зниженим виходом
нітрофенолів або з необхідністю сульфування фенолу, що ускладнює утилізацію
стічних вод.
Друга схема економічніша. Оскільки нітрування хлоробензену нітрувальною
сумішшю виконують за температури його кипіння, то можна не побоюватися
смолоутворення. Ключовою стадією в другій схемі є нуклеофільне заміщення атома
галогену в 2,4-динітрохлоробензені, що, на відміну від аналогічної реакції самого
хлоробензену за першою схемою, відбувається за м'яких умов.
❖ Діоксин. На закінчення зазначимо, що необхідний суворий контроль на
всіх стадіях виробництва різних хлороаренів, тому що вони за певних умов
712
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
перетворюються на вкрай токсичні речовини діоксини, небезпечні тим, що,
розчиняючись у воді, вони потрапляють в організм людини й тварин,
послаблюють їхній імунітет і призводять до незворотних генетичних змін.
Нижче наведені приклади утворення діоксинів у природі.
Трихлороетилен, який широко застосовують у хімчистці, під час нагрівання
перетворюється в дихлороацетилен, що легко зазнає тримеризування в
гексахлоробензен з наступною димеризацією до діоксину:
сі сі сі
Зазначимо також про надзвичайно токсичний і дуже стійкий за природних
умов 2,3,7,8-тетрахлородибензо-и-діоксин (ТХДД), що як домішка утворюється
під час синтезування гербіциду 2,4,5-Т. За даними Книги рекордів Гіннеса,
2,3,7,8 - ТХДД є найотрутнішою речовиною в світі:
2,4,5-Т
Доведено, що діоксини утворюються й під час згоряння полівінілхлориду.
Тому спалювання відходів, які містять цей матеріал, призводить до небезпечних
для людини й тварин наслідків.
26.4.4. Реакції нуклеофільного заміщення атома
галогену в бічному ланцюзі арені в
Зовсім інша ситуація з реакційною здатністю атомів галогенів, що містяться
в бічному ланцюзі. Тоді як віддаленіші атоми галогену бічного ланцюга мають
нормальну рухливість, що відповідає рухливості атома галогену в насичених
сполуках, галоген у а-положенні щодо ароматичного ядра активніший.
Бензилхлорид - сльозогінна сполука; роздратування слизових оболонок носа й
ока - наслідок високої реакційної здатності сполуки. Ще більшу активність має
бензиліодид - "поліцейський сльозогінний газ".
Розділ 26. Галогенопохідні ароматичних вуглеводнів
713
Бензилгалогенідн зазнають гідролізування і взагалі вступають в обмін з
нуклеофілами в спиртовому розчині за мономолекулярною реакцією Бці. Це
свідчить про те, що вони спочатку дисоціюють (повільна стадія реакції, що
визначає перший порядок усього процесу), утворюючи галогенід-аніон й
бензилкатіон, бензилкатіон потім зв'язується дуже швидко з нуклеофілом.
Оскільки концентрація нуклеофіла ніяк не впливає на швидкість електролітичної
дисоціації бензилгалогеніду, то швидкість реакції пропорційна тільки до
концентрації бензилгалогеніду (реакція першого порядку):
С6Н5СН2С1 —- с6н5сн2+ + сі-
едсн^ + нр—- одснрн+н*
Такі реакції успішно відбуваються в добре сольватувальних середовищах з
високою діелектричною проникністю.
Легкість електролітичної дисоціації бензилгалогенідів порівняно з алкілга-
логенідами пояснюють більшою стабільністю (нижчим енергетичним рівнем)
бензильного катіона порівняно з алкільними катіонами.
Бензилхлорид широко використовують для бензилювання — уведення
бензильної групи в різні органічні сполуки.
❖ Гідроліз бензиліденхлориду й трихлорометилбензену. Як завжди,
скупчення двох і більше атомів Хлору біля одного атома Карбону робить сполуку
стабільнішої^ і менш сприятливою до нуклеофільних атак. Однак бензиліденхло-
рид, хоча й під час нагрівання, зазнає гідролізування в бензальдегід:
С6Н5СНС12С6Н5СНО
Трихлорометилбензен за високої температури та наявності сульфатної
кислоти зазнає гідролізування (1 моль Н2О) у бензоїлхлорид - хлороангідрид
бензойної кислоти:
С6Н5СС13 + Н2б С6Н5СОС1 + 2 НС1
Останню реакцію використовують у промисловості.
За надлишку води трихлорометилбензен зазнає гідролізування в бензойну
кислоту:
С6Н5СС13 + 2 Н20 С6Н5СООН + З НС1
❖ Одержання трифлуорометилбензепу. Дією ЗЬБз трихлорометилбензен
легко перетворюється в трифлуорометилбензен:
С6Н5СС13 + 8ЬР3 С6Н5СР3 + 8ЬС13
Ця сполука винятково стійка, зовсім не зазнає гідролізування й не схильна
вступати в реакції обміну. Цікаво, що групи -ССЬ і, особливо, -СР3 - сильні м-
орієнтанти, що пасивують бензенове ядро до електрофільних атак.
714
Чирва В.Я., Ярмолкж СМ., Толкачова H.В., Земляков О.Є. Органічна хімія
26.4.5. Реакції металювання
Ароматичні галогенопохідні, окрім флуоропохідних, вступають у взаємодію
з літієм, натрієм і магнієм. Іодобензен і бромобензен в етерному розчині
реагують з магнієм не так активно, як алкілгалогеніди, хоча й утворюють
галогенофенілмагній. Так само реагують і гомологи галогенобензенів. Із
хлоробензеном подібна реакція відбувається за вищої температури (-150 °С),
тому його можна перетворити в фенілмагній хлорид в автоклаві без етеру
(П. Шоригін) або в тетрагідрофурані за звичайних умов. У лабораторії, зазвичай,
для синтезу Гріньяра ліпше застосовувати броміди й іодиди. У такий же спосіб в
етері або у вуглеводневому розчині дією металевого літію одержують
феніллітій, який використовують для тих же реакцій, що й галогенофенілмагній.
Обидві реакції відбуваються через стадію утворення вільного радикала:
ОДВг + Mg ОД- + MgBr C^MgBr
ОДВг + Li C6bV + LiBr —^ СвЩл
Це зрозуміло з того факту, що як побічний продукт утворюється досить
багато біфенілу:
2 С6Н5 С6Н5-С6Н5
❖ Реакція Вюрца. Реакцію галогенобензенів з натрієм можна затримати на
стадії утворення натрійорганічних сполук:
C6H5Br + Na NaBr + С6Н5-^— C6H5Na
однак фенілнатрій реагує з бромобензеном:
С^Вг + C6H5Na С6Н5-С6Н5 + NaBr
так що синтез можна відразу вести як реакцію Вюрца:
2 CgHjBr Na * С6Н5-С6Н5
❖ Реакція Вюрца-Фіпгпгіга. Можна ґрунтуватися на двох різних арил-
галогенідах або брати арилгалогенід і алкілгалогенід. В останньому випадку ця
реакція відома як реакція Вюрца-Фіттіга:
СуН^Вг + С2Н5Вг + 2 Na C^-Cft + 2 NaBr
❖ Реакція Ульмана. Арилгалогеніди, реагуючи між собою за наявності
міді, теж утворюють діарил:
2 С6Н5І + 2 Си С6Н5-С6Н5 + 2 СиІ
Розділ 27
аренсульфокисаоти
та їхні похіані
27Л. Будова й номенклатура аренсульфокислот
Загальна формула ароматичних сульфокислот АгЗОзН. Атом Сульфуру у
цих сполуках має тетраедричну будову. У сульфонат-аніоні, як і в карбоксилат-
аніоні, всі зв'язки Б-О мають однакову довжину, а негативний заряд рівномірно
розподілений по трьох атомах Оксигену:
Аг"
О
Аг-5-0
9
^гон
Ъ і о J
Сульфокислота Сульфонат-аніон
Не треба плутати сульфокислоти, у яких атом Карбону безпосередньо
пов'язаний з атомом Сульфуру, з ариловими естерами сульфатної кислоти, у
яких атом Карбону забезпечує зв'язок через атом Оксигену:
О
АЮ^-ОН
О
Сульфат (естер)
Назви аренсульфокислот утворюють додаванням до назви ароматичного
вуглеводню суфікса -сульфонова кислота, або скорочено -сульфокислота. За
наявності в сполуці старшої групи функцію сульфонової кислоти позначають
префіксом сульфо-:
<0«іо,н
БО,Н
но3&
СООН
Бензенсульфонова кислота, л-Толуенсульфонова кислота,
або бензенсульфокислота або л-толуенсульфокислота
ж-Сульфобензойна кислота
Сульфокислоти ароматичного ряду та їхні похідні відіграють винятково
важливу роль як проміжні продукти в синтезі барвників, а також вихідних
сполук у виробництві фенолів, ароматичних амінів, карбонових кислот. На
основі арилсульфокислот синтезують сульфамідні препарати.
716
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
27.2. Фізичні властивості сульфокислот
Як можна було очікувати на підставі розгляду структури, сульфокислоти
мають фізичні властивості, характерні для сильнополярних сполук. Загалом
вони ліпше розчинні у воді, ніж органічні сполуки будь-яких інших класів.
Справді, сульфогрупи дуже часто вводять у великі молекули, наприклад,
барвників або лікарських препаратів для того, щоб зробити їх розчинними у
воді. Оскільки сульфокислоти є сильними кислотами й за силою близькі до
сульфатної кислоти, то вони повністю йонізовані у водних розчинах:
ArS03H + H20 - ArS03- + H30+
Вони розчинні також у деяких інших полярних розчинниках. Сульфокислоти
мало леткі й, як звичайно, під час нагрівання розкладаються нижче температури
кипіння.
27.2.1. Спектроскопія аренсульфонозих кислот
• ІЧ-спектроскопія. Безводні ароматичні сульфонові кислоти поряд* зі
смугами поглинання, що властиві аренам, мають інтенсивні характеристичні
смуги поглинання валентних коливань vs=o 1 350-1 340 см"1 (асиметричні) і
1 165—1 150 см"1 (симетричні). Для сульфонат-аніона ці смуги зміщуються в
ділянку 1 250-1 150 і 1 100-1 000 см"1, відповідно. .
• ПМР-спектроскопія. Сигнал протона сульфогрупи зміщений у слабке поле й
має хімічний зсув (11-12 м.ч.)
27.3. Методи синтезу аренсульфокислот
❖ Пряме сульфування аренів. Ароматичні сульфокислоти майже завжди
одержують прямим сульфуванням аренів димлячою сульфатною кислотою за
температури 40-50 °С У промисловості застосовують вищу температуру. Для
проведення процесу на холоді потрібен великий надлишок кислоти, оскільки
вода, яка утворюється в реакції, розводить сульфатну кислоту, а та за
концентрації 65% перестає сульфувати. Це утруднення можна подолати, якщо
вести процес під* час нагрівання приблизно до 170-180 °С, тому що за цієї
температури вода відганяється, і концентрація кислоти не змінюється. Про
завершення реакції свідчить повне розчинення у воді проби, взятої з реакційної
суміші, оскільки аренсульфокислота, на відміну від арену, розчинна у воді:
нз50< , <T\SOH
170-180 °С \ / bU3n
Бензенсульфокислота
Отримані внаслідок сульфування ароматичні кислоти відокремлюють від
надлишку сульфатної кислоти за допомогою солей Барію, які, на відміну від
Розділ 27. Аренсульфокислоти та їхні похідні
777
барій сульфату, розчинні у воді. Для виділення аренсульфонових кислот можна
також наситити розведений водою сульфатний розчин натрій хлоридом,
унаслідок чого натрієві солі аренсульфокислот виділяються в кристалічному
стані, а неорганічні сполуки залишаються в розчині.
■ Шляхом енергійного сульфування бензену димлячою сульфатною
кислотою під час нагрівання можна одержати ди- і трисульфокислоти, однак
увести цим шляхом у бензенове кільце більше трьох сульфогруп не вдається.
Солі деяких металів, а саме - арґентум сульфат і меркурій сульфат,
прискорюють процес.
Під час сульфування до дисульфокислот водночас одержують м- і «-ізомери
(перший ізомер в більшій кількості); о-бензендисульфокислота не утворюється,
очевидно, з огляду на просторові утруднення, створювані двома поряд
розташованими сульфогрупами.
Енергійне тривале нагрівання лі-бензенсульфокислоти із димлячою сульфат-
ною кислотою зумовлює її часткове перегрупування в «-сполуку; з іншого боку,
за тих же умов и-дисульфокислота частково перегруповується в л*-сполуку, так
що з часом під час нагрівання досягається стан рівноваги:
О ^ 0-8°зН в Н038НО-803Н
Л#-Бензендисульфокислота и-Беюендисульфокислота
■ Додаванням ртуті, або ліпше меркурій (II) сульфат, можна значно
змінити виходи ізомерів, що утворюються. Це взагалі стосується сульфування
похідних бензену й пов'язане з тим, що в разі дії солей Меркурію на арен
утворюються меркурійорганічні сполуки, у яких потім атом Меркурію зазнає
заміщення сульфогрупою (шсо-заміщення).
■ Дисульфокислота фенолу (4-гідрокси-л*-бензендисульфокислота) відіграє
винятково важливу роль у синтезі пікринової кислоти. Річ у тому, що пряме
нітрування фенолу його обвуглює. Наявність же сульфогрупи (орієнтант другого
роду) надає стійкість бензеновому кільцю до дії димлячої нітратної кислоти. Цю
обставину використовують у промисловості для синтезу пікринової кислоти
(шсо-заміщення):
■ Гомологи бензену, особливо з об'ємними алкільними замісниками, легше
зазнають сульфування порівняно з бензеном і переважно в «-положення:
718
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
СНгСЗ + Нг8^^5^1^ СН^^-803Н + Н20
Поряд з цим утворюється невелика кількість оізомеру.
■ Як сульфувальні агенти найчастіше використовують, як уже зазначено,
концентровану сульфатну кислоту, а також сульфур (VI) оксид в інертному газі
(азот) або його розчин у сульфатній кислоті (олеум), піридинсульфотриоксид
СбНбМ-ЗОз, діоксансульфотриоксид, сульфурилхлорид БОгСЬ.
❖ Сульфування олеумом. Сульфування олеумом відбувається під час
слабкого нагрівання, а в багатьох випадках потрібне навіть охолодження:
О ^ О-50*» снїО ^ сО°=н
Бензенсульфокислота л-Толуенсульфокислота
Побічними продуктами реакції сульфування олеумом за надлишку бензену
можуть бути сульфони:
С6Н5802ОН + 803+С6Н6^= С6НГ80ГС6Н5+ Н2804
Причина цієї побічної реакції полягає в тому, що за жорсткіших умов роль
сульфувального агента відіграє аренсульфокислота:
Аг-802ОН + Аг-Н Аг-80гАг + Н20
Цікаво, що під час обробки сульфонів розведеною сульфатною кислотою
шсо-реакції не відбувається.
❖ Механізм сульфування. Вважають, що сульфувальним агентом є гідро-
сульфонієвий катіон або протонований сульфур оксид Н080г+:
2 Н2804 =5=- Н20+802ОН +Н80;
Н20+802ОН==^ Н20 + 802ОН
Реакція сульфування починається з утворення о-комплексу, відщіплення
протона від якого є стадією, що лімітує швидкість сульфування:
+ Швидко
+ 802ОН
^ 4§0зн
сг-Комплекс
802ОН
Це відрізняє механізм сульфування від механізму інших реакцій заміщення
в ароматичному ряді (див. розділ 25).
Справді, кінетичні дослідження засвідчили, що сульфування дейтеробензену
відбувається в п'ять-вісім разів повільніше, ніж звичайного бензену (ізотопний
Розділ 27. Аренсульфокислоти та їхні похідні
719
ефект). Отже, лімітувальною стадією сульфування є не утворення о-комплексу, а
відщеплення від нього протона або дейтерію.
Привертає увагу той факт, що реакції сульфування зворотні. Це підтверджене
збільшенням виходу сульфокислоти під час відгонки води у вигляді азеотропу або
її зв'язування. Недотримання цих умов призводить до прискорення зворотної
реакції, яка відбувається через утворення того ж о-комплексу внаслідок
протонування атома Карбону бензенового кільця, що несе замісник, тобто, по суті,
маємо справу з /«созаміщенням сульфогрупи на протон.
Оборотність реакції сульфування неможлива у разі використання сульфатного
ангідриду як у вільному вигляді, так і у вигляді розчинів у моногідраті (олеум),
піридині й діоксані.
Зворотною реакцією іноді користуються для синтезу деяких важкодоступних
0-ізомерів. Наприклад, під час хлорування /ярет-бутилбензену утворюється
винятково «-ізомер. Тому, для одержання о-ізомеру трети-бутилбензен спочатку
сульфують в «-положення, а потім хлорують .(^-орієнтувальний ефект сульфо-
групи) і, нарешті, виконують десульфування перегрітою водяною парою:
803Н Б03Н
❖ Сульфохлорування. Якщо необхідно одержати хлороангідрид сульфо-
кислоти, то групу ЗОгСІ можна ввести в одну стадію шляхом обробки
ароматичної сполуки надлишком хлоросульфонової кислоти 0803Н:
Аг-Н + СІБОзН -> Аг-БОзН + НС1
Аг-803Н + СІБОзН -> Аг-803С1 + Н2804
Хлоросульфонова кислота є активним сульфувальним агентом, тому вона
реагує з бензеном за м'якших умов (0 °С), ніж олеум.
27.4. Реакції сульфокислот
❖ Кислотність. Утворення солей. Солі сильнокислих сполук легко утво-
рюються під час обробки їх навіть слабкими основами. Тому сульфокислоти
зазвичай зручніше виділяти у вигляді солей, і часто їх використовують саме в
цій формі:
Аг803Н + Н20 - Аг803- + Н30+
Повна дисоціація
за кислотним типом
720
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
^2^803Н + ШОН ^^803Ш + Н20
Бензенсульфокислота Натрій бензенсульфонат
^^803Н + КаНС03 СН3-^^803Ка + С02 + Н20
Сульфокислоти легко утворюють солі з лужними й лужноземельними
металами, які не зазнають гідролізування, з огляду на що в такому вигляді їх
легко виділяти з реакційної суміші.
❖ Хлороангідриди сульфокислот і їхні властивості. Хлороангідриди
сульфонових кислот можна одержати традиційним методом — дією фосфор (V)
хлориду на кислоту або її сіль:
О О
АгДгОН + РС15 Аг^гС1 + Р0С1з+ НС1
СНз<^3^80з>Іа + ?СІ5 " СН^^-802С1 + NaCl + РОС13
я-Толуенсульфохлорид
З іншого боку, ароматичні вуглеводні зручніше перетворювати в
сульфохлориди аренів обробкою їх надлишком хлоросульфонової кислоти:
+ 2 С1803Н У-802С1 + НС1 + Н2804
н3с-сч 4=7 н3с-сч 4=7
о о
Хлороангідриди аренсульфокислот моясна легко очистити перегонкою у
вакуумі або перекристалізацією й далі використати для одержання як самих
кислот, так і їхніх похідних.
Сульфохлориди - важливі проміжні сполуки у синтезі інших функціональних
похідних, які не можна одержати прямо із сульфокислот. Вони реагують зі
спиртами або фенолами, даючи естери, а з амоніаком - аміди:
АіЗО^І + ІЮН В°ДНИЙЛуг. Аі803И
Алкіловий естер
сульфокислоти
АгБ02С1 +Аг'ОНПіриди" АгБОзАг'
Ариловий естер
сульфокислоти
Аг802С1 + 2 ИН3 АгвОИНг +Ш4С1
Аренсульфонамід
Розділ 27. Аренсульфокислоти та їхні похідні
721
Серед аренсульфохлоридів треба назвати и-толуенсульфохлорид (тозилхло-
рид\ який часто застосовують для естерифікації гідроксильних сполук
(тозилування). В естерах толуенсульфокислот під час омилення розщеплюється
зв'язок С-О, а під час відновлення алюмогідридом літію часто утворюються
сполуки, які не мають гідроксильних груп:
N^SS- CH3-C6H4-S03Na + RR'CHOH
CH3-C6H4-S02OCHRR' 3 6 4 3
LiAlt^ CH3-C6H4-S03H + RCH2R'
❖ Естери арепсульфокислот. Важливе практичне значення мають алкілові
естери аренсульфокислот, зокрема, метиловий естер и-толуенсульфокислоти. Під
час дії їх на аміни, а також на феноли або спирти в лужному розчині відбувається
алкілування функціональних груп цих сполук (нуклеофільне заміщення в
аліфатичному ряді). Отже, естери сульфокислот, подібно до діалкілсульфатів, є
сильними алкілувальними агентами, тому їх широко використовують в органічній
хімії:
CH3-C6H4-S03CH3 + 2 RNH2 CH3NHR + CH3-C6H4-S07'H3NR
CH3-C6H4-S03CH3 + C6H5OK
CH3-C6H4-S03K + C6H5OCH3
❖ Сульфонаміди. Аміди сульфокислот мають вираженіші кислотні
властивості порівняно з аліфатичними амідами й розчиняються в лугах,
утворюючи нейтральні речовини. Під час хлорування амідів сульфокислот
відбувається заміщення одного або двох атомів Гідрогену сульфонамідної групи
на Хлор, і синтезування так званих хлорамінів, які зазнають гідролізу,
утворюючи хлоридну кислоту і. вихідний сульфонамід, завдяки чому мають
сильну дезинфікувальну дію:
0=S=0 o=s=o
NCl2
o=s=o
o=s=o
NCL
ШСІ А^42 N1^01
Хлорамін Б Дихлорамін Б Хлорамін Т Дихлорамін Т
Аренсульфонаміди - основа для синтезу сульфамідних препаратів, таких як:
Дисульфан
H2N
S02NH-
S02NH2,
722
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Норсульфазол ^~~^3~8^Н та ІН'
❖ Сахарин. Серед сульфонамідних похідних особливе місце посідає
сахарин. Ця речовина, що приблизно в 500 разів солодша, ніж цукор, є похідною
о-сульфобензойної кислоти. її відкрив І. Ремзен1 1879 р. Вихідною сполукою для
одержання сахарину є толуен, що під час сульфування дає суміш п- і о-сульфо-
кислот. Необхідний о-ізомер легко виділяється, тому що він є рідиною, на
відміну від «-ізомеру. Потім його перетворюють у хлороангідрид, який також
можна отримати безпосередньо з толуену під дією хлоросульфонової кислоти,
далі - в амід і нарешті окиснюють перманганатом калію. Під час випарювання
розчину осульфонамідобензойна кислота, яка утворилася, зазнає де гідрату-
вання, перетворюючись в імід о-сульфобензойної кислоти, або сахарин:
Натрієва сіль сахарину, яка чудово кристалізується,
>Л^а надходить у продаж під назвою кристалоза:
§0 * Десульфування. Під час нагрівання водних розчинів
2 ароматичних сульфокислот до 100-175 °С утворюється суль
фатна кислота й ароматичний вуглеводень. Ця реакція десульфування зворотна
до реакції сульфування, за допомогою якої отримано вихідну сульфокислоту:
АгН + Н2804=^ Аг803Н + Н20
Вуглеводень, Сульфокислота,
леткий нелетка
З використанням звичайних принципів зсуву рівноваги молена підібрати умови,
які спрямовуватимуть реакцію в потрібний бік. Для сульфування використовують
великий надлишок концентрованої або димлячої сульфатної кислоти; високу
концентрацію сульфувального агента й малу концентрацію води або її видалення за
допомогою сульфур (VI) оксиду. Всі ці чинники зміщують рівновагу в бік
утворення сульфокислоти.
Для десульфування застосовують розведену кислоту й часто через реакційну
суміш пропускають перегріту пару - мала концентрація кислоти (надлишок води) і
видалення леткого вуглеводню перегонкою з парою зміщують рівновагу у бік
десульфування.
1 Ремзен І. (Ііетзеп І.) (1846-1927) - американський хімік. Засновник хімічної освіти в США.
Розділ 27. Аренсульфокислоти та їхні похідні
723
❖ Відновлення аренсульфокислот. Обробка сульфонілхлоридів цинковим
пилом дає сульфінові кислоти:
я-Толуенсульфохлорид л-Толуенсульфінова кислота
Сульфінати є амбідентними нуклеофілами й у реакціях з галогеноалканами
реагують по атому Сульфуру, утворюючи сульфони:
:6-£бГ№+ °=Г°
Алкіл-л-толілсульфон
❖ Відновлення сульфогрупи до тіофенолу засвідчує, що в сульфокислотах
атом Сульфуру безпосередньо зв'язаний з бензеновим кільцем:
С6Н5803Н ^ С6Н58Н+Н20
❖ Реакції сульфокислот із заміщенням сульфогрупи. Сульфогрупа бензе-
нового кільця, на відміну від більшості замісників, у спряження з я-електронами
ароматичного кільця не вступає внаслідок тетраедричної будови. Як відомо, реакція
сульфування - процес зворотний, тому варто очікувати, що в разі дії досить
потужних електрофільних агентів можливе її шсо-заміщення. Крім того,
сульфогрупа, як і інші електроноакцепторні замісники в ароматичному ядрі, здатна
нуклеофільно заміщуватись. Отже, ароматичні сульфокислоти можна використати
як вихідні речовини для одержання сполук з іншими функціями. Тому вони
відіграють в ароматичному ряді таку ж роль, як і галогеноалкани в аліфатичному, і
їхнє препаративне, а також промислове значення пов'язане, головно, з можливістю
використання як проміжних продуктів для органічного синтезу.
■ Найважливішою з таких реакцій є реакція "лужного плавуп (А. Кекуле),
за допомогою якої дотепер одержують фенол:
Аг803Ка + ИаОН АгіЖа + ИаН803
АгОШ + НС1 АгОН + ШСІ
■ Аналогічно відбувається реакція з ціанідами, яка не має важливого
препаративного значення (М. Кучеров):
СдеОзШ + №СМ С6Н5СН + Иа^Оз
724
Чирва В.Я., Ярмолкж С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
❖ Заміщення в ароматичне ядро. Відомо, що сульфогрупа пасивує
бензенове кільце в реакціях електрофільного заміщення, орієнтуючи в жорстких
умовах електрофільну групу у л*-положення:
Водночас під час сульфування галогенобензену синтезують, майже
винятково, «-ізомер:
Розділ 28
НІТРОСПОЛ УКИ РЯА У
БЕНЗЕНУ ТА ЇХНІ ПОХІАНІ
28.1. Нітроарени
28.1.1. Будова й номенклатура нітроаренів
Завдяки електроноакцепторяій дії нітрогрупи (-1- і -М-ефекти) ск+£о~
я-електронна система нітробензену сильно поляризована. Для
подібних молекул характерні великі значення дипольних моментів, .+
наприклад, у нітробензену дипольний момент становить 4,2 Т>. ^
В ароматичних нітросполуках М02-група, що зв'язана з
ароматичним вуглеводневим залишком, виявляє сильні електроно §+
акцепторні властивості, знижуючи електронну густину в о-1 «-положеннях арилу:
♦я*
Спряженням пояснюють коротший зв'язок атомів Карбон-Нітроген і,
відповідно, більший дипольний момент порівняно з нітрометаном (3,5 О).
Завдяки цьому рідкі нітросполуки - чудові апротонні розчинники.
• У номенклатурі ШРАС нітрогрупу позначають тільки префіксом нітро-, що
перераховують поряд з іншими замісниками за абеткою:
)Н
о-Нітротолуен ж-Нітрохлоробензен л-Бромонітробензен 2,4-Динітрофенол
28.1.2. Фізичні властивості й спектроскопія
нітроаренів
Нітроарени - жовтуваті рідини або тверді сполуки зі своєрідним запахом.
Наприклад, нітробензен має сильний мигдалевий запах. Більшість нітроаренів не
розчинна у воді. Сам нітробензен широко використовують як розчинник.
726
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
• УФ-спектроскопія. В УФ-спектрах мононітробензенів є інтенсивні смуги з
Хтах 252 і 280 нм, що відповідають я—>я*-переходам, а також менш інтенсивна
смуга з Хтак 330 нм, пов'язана з п—>я*-переходом у
нітрогрупі.
• ІЧ-спектроскопія. Нітрогрупа ароматичних спо-
лук має інтенсивні смуги поглинання валентних
коливань Ум_о 1 560-1 500 см"1 (асиметричні) і 1 355-
5 м.ч. 8,22 (а), 7,53 (Ь), 7,65 (с) 1 ш Ш-1 (симетричні).
• ПМР-спектроскопія. Нітрогрупа зсуває сигнали ароматичних протонів у
слабке поле. Особливо сильний вплив цієї групи на оположення.
28.2. Одержання нітроаренів
Завдяки легкості одержання ароматичні нітросполуки мають більше
промислове й препаративне значення, ніж нітросполуки аліфатичного ряду.
❖ Пряме нітрування. Звичайним нітрувальним реагентом є нітрувальна
суміш, тобто розчин концентрованої нітратної кислоти (60-100%) у концентро-
ваній сульфатній кислоті (температура процесу - 45-50 °С). Електрофільним
нітрувальним агентом є нітроній-катіон Ж)г+:
НОШ2 + Н2804 = N0^04+ Н20
У такому розчині нітратна кислота поводиться як основа. Сама нітратна
кислота нітрує набагато повільніше, а оскільки вона - сильний окиснювач, то
переважають реакції окиснення. Наявна в суміші сульфатна кислота не сульфує
ароматичного ядра, оскільки швидкість нітрування приблизно в 1000 разів
більша, ніж швидкість сульфування.
Процес нітрування аренів починається з утворення я-комплексу, що
повільно перетворюється в о-комплекс:
я-Комплекс ^-Комплекс я-Комплекс
Швидкий і незворотний розпад о-комплексу приводить до того, що в разі
видалення води з реакційної суміші вихід продуктів нітрування аренів не
змінюється. Тому нітратну кислоту застосовують у мінімальному надлишку (5-
10 %), домагаючись практично повного перетворення арену. Вихід нітробензену
-85 %.
■ Нітрувальні реагенти. Оскільки йдеться про взаємодію ароматичної
сполуки з електрофільною часткою >Ю2+? то нітрувальними агентами є сполуки
загальної формули Т<Ю2-Х9 де X- аніон, і чим легше він відходить від молекули,
Розділ 28. Нітросполуки ряду бензену та їхні похідні
727
тим ефективніший процес нітрування. Нижче наведено ряд нітрувальних агентів
за зменшенням активності:
Nö|;bf; > N02ClCf4 > N02Cl"> NOlpNO; > N02ONO"> N02OCOCH; > NO+OH "> N02OC2H5"
(N205) (N204)
Нітрувальний агент - ацетилнітрат, який широко застосовують у лабораторній
практиці, особливо часто використовують для нітрування ацидофобних сполук;
готують його, додаючи оцтовий ангідрид до димлячої нітратної кислоти за -50 °С.
Найефективніший нітрувальний агент - нітроній тетрафлуороборат, який
одержують дією на димлячу нітратну кислоту безводного гідроген флуориду й
боротрифлуориду в нітрометані:
(СН3СО)20 + HON02^^N02OCOCH3 + СН3СООН
HN03 + HF • 2 BF3 - NO+2BF;+ H20 • BF3
Постає запитання: чому саму нітратну кислоту не використовують як
нітрувальний агент, як у випадку сульфатної кислоти, де активним електрофілом
є протонований сульфур триоксид HSCV? Річ у тому, що процес само-
протонування нітратної кислоти відбувається не настільки легко, і рівновага
зміщена в лівий бік:
HON02 + HN03 ^ H2ON02N03"^ H20 + NO+2 + N03
Стабільність катіона нітронію зумовлена частковою взаємодією неподілених
електронних пар атома Оксигену з вакантною р-орбіталлю атома Нітрогену:
0=N=Ö — Ö=N=0
sjP- sp sp2
Нітрування аренів нітрувальною сумішшю має низку недоліків - високу
витрату кислот (500 кг нітратної кислоти й 800 кг сульфатної кислоти для
синтезу 1 000 кг нітробензену); необхідність розділення ізомерів, що утво-
рилися, та великий об'єм стічних вод.
Значно економічнішим є метод, апробований у промисловості Японії, в якому
нітрування ведуть нітроген (IV) оксидом у суміші з озоном або киснем за наявності
ферум (III) хлориду. Метод відрізняється високою регіоселективністю, м'якими
умовами реакції (0 °С, нейтральне середовище), відсутністю побічних продуктів:
сн3
l^J) FeCl3
100%
728
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
■ Ди- і тринітробензен. Зазначимо, що найуживанішим і найдешевшим
методом є нітрування за допомогою нітрувальної суміші, який дає змогу
вводити в арени декілька нітрогруп. Вихід л*-динітробензену досягає 80%:
Ю,-
6+
N0
N0;
100°С
N0.
+ Ж)2
N0.
80%
45%
Тринітробензен (ідеальна бризантна вибухова речовина) утворюється за
жорстких умов і з поганим виходом: унаслідок п'ятиденного нагрівання м-
динітробензену з великим надлишком димлячих нітратної й сульфатної кислот
вихід тринітробензену становить лише 45 %. Незважаючи на зусилля хіміків
багатьох країн, провести цю реакцію з прийнятним виходом не вдалося. Тому в
лабораторних синтезах замість нітрувальної суміші застосовують нітроній
тетрафлуороборат, що дає змогу знизити температуру реакції до кімнатної й
домогтися прийнятних виходів тринітробензену.
❖ Нітрування алкілбензенів. Нітрування толуену, як і всіх алкілбензенів,
на всіх стадіях відбувається легше, ніж бензену. На схемі показані всі стадії його
нітрування:
СНЯ СН3
\Ж)2 02И^ЦЛЖ)2
СН,
N0,
N0,
N0,
Серед гомологів бензену толуен зазнає нітрування найлегше завдяки участі
метальної групи в о-7і-спряженні (гіперкон'югації) і мінімальної стеричної
утрудненості для реагенту.
Таким шляхом отримують 2,4,6-тринітротолуен (тол, або тротил) -
бризантну вибухову речовину, що слугує для заповнення снарядів. На відміну
від 1,3,5-тринітробензену, тротил одержують з високим виходом.
Накопичення алкільних груп в арені, що не створюють просторових
утруднень і узгоджено орієнтують замісники, приводить до активування
ароматичного ядра. Наприклад, мезитилен (1,3,5-триметилбензен) нітрують
навіть ацетилнітратом з гарним виходом.
■ Вибуховими речовинами називають хімічні сполуки, здатні до швидкої
хімічної реакції, що супроводжується вивільненням великої кількості енергії,
Розділ 28. Нітросполуки ряду бензену та їхні похідні
729
виділенням газів, передаванням енергії за допомогою тепло- або масообміну чи
ударної хвилі (детонація). До вибухових речовин належать, наприклад, тротил,
пікринова кислота, гексоген, амоній нітрат.
За вибуховими властивостями їх поділяють на ініціювальні речовини -
плюмбум азид РЬ(Ыз)г і гримуча ртуть Н§((ЖС)г, що легко вибухають під час
нагрівання або удару, бризантні - тротил, тетрил, октоген, що детонують під час
вибуху ініціатора, і порох, що згоряє з високою швидкістю, але без детонації.
■ Нітрування л*-/я/7ет-бутилтолуену дає 3-т^е/и-бутил-2,4,6-тринітрото-
луен - штучний мускус, що має запах, подібний до запаху природного мускусу.
Мускуси застосовують у парфумерії як запашні речовини й фіксатори запаху.
Формули низку штучних мускусів наведені нижче:
❖ Нітрування інших ароматичних сполук
■ Галогеноарени з огляду на двоїсту поведінку галогенів зазнають
нітрування в жорсткіших умовах, ніж бензен. Уведення другої нітрогрупи
потребує застосування 8 % олеуму.
■ Феноли легко нітрувати нітратною кислотою в оцтовій кислоті за
кімнатної температури до онітрофенолу. Спроба ввести другу нітрогрупу у
фенол за допомогою нітрувальної суміші веде до деструкції молекули. Для
цього зручно використати динітроген (IV) оксид:
Нітрування цієї сполуки нітрувальною сумішшю дає пікринову кислоту.
■ Нітрування аніліну розглянуте в розділі 29.
■ Нітрування похідних бензену з електроноакцепторними замісниками
вимагає досить жорстких умов - димлячої нітратної кислоти з концентрованою
сульфатною кислотою.
Амбровий мускус
Ш2
Кетонний мускус
ио2
Ксиленовий мускус
Толуеновий мускус
ж>2
730 Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
'2
18,5%
Підвищення температури реакції до 100 °С дає змогу одержати 3,5-ди-
нітробензойну кислоту з виходом 60 %.
■ Такі м'які л*-орієнтанти, як альдо- і кетогрупи, спрямовують електрофільне
заміщення в л*-положення, хоча в цьому разі утворюються й інші ізомери.
Особливо це видно на прикладах нітрування бензальдегіду й ацетофенону:
Наведені результати засвідчують, що правила орієнтації відображають
тільки основний напрям реакцій електрофільного заміщення.
❖ Нітрування за Коноваловим. Під дією на толу єн розведеної (10-14 %)
нітратної кислоти за високої температури (100-150 °С) електрофільне заміщення
в ядро пригнічується, тому гомолітичне нітрування толуену відбувається за
ланцюговим механізмом у метильну групу:
нш3+нш2^—»» 2ш2+н2о с6Н5-сн3+ ш2 —-сдан,+ НШ2
ОД-СН^+ НОШ2 — ОДОН^ + ОН ЗД-СНз + ОН ОДОН^ + Р^О
В а-положення за цих умов відбувається нітрування і гомологів бензену з
довшим бічним ланцюгом, що пояснюють більшою стійкістю проміжного
радикала порівняно з іншими радикалами, у яких атом, що несе неспарений
електрон, віддалений від ядра.
■ Також а-нітротолуен можна одержати, діючи бензилхлоридом на натрій
нітрит:
с^снд+№ш2 —- с6Н5<:н2ш2+шсі
Зокрема, а-нітротолуен - одна з декількох нітросполук, для якої можна
Розділ 28. Нітросполуки ряду бензену та їхні похідні
731
ізолювати лабільну сильнокислу ш/и-форму, таутомерну з а-нітротолуеном:
СдаНгШг+ЫаОН
С6Н5СН=Й
О
О
Ка-&С6Н5СН=К
О
^ с6н5снгмч
он о
❖ Окиснення нітросполук. Інший загальний метод одержання нітроаренів
полягає в окисненні ароматичних амінів трифлуоронадоцтовою кислотою, яку
готують безпосередньо в реакційній суміші, змішуючи ангідрид трифлуороцто-
вої кислоти з 90 % розчином гідроген пероксиду:
(СР3СО)20 + Н2Оз
40°С
N0,
86%
Галузь застосування цього методу обмежена, тому що самі ароматичні аміни
одержують через відновлення нітроаренів.
28.3. Хімічні властивості нітроаренів
28.3.1. Реакції електрофільного заміщення
в нітроаренах
Реакції нітрування, сульфування й галогенування в ядро відбуваються
сутужніше, ніж для бензину, за участю ж-положення стосовно нітрогрупи,
наприклад:
Однак така регіонапрямленість не завжди дотримана. Особливо чітко це
виявляється у випадку нітроаренів, що мають у л*-положенні орієнтант першого
роду. Наприклад, під час нітрування ^-нітроанізолу утворюється суміш речовин,
у яких наступна нітрогрупа вступила в о-положення до уже наявної нітрогрупи:
Аналогічний результат отриманий під час нітрування л-бромотолуену:
732
Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
Вг Вг Вг
Уважають, що такий хід реакції зумовлений координацією атакувального
електрофіла >Юг+ з я-електронами гетероатома замісника, його називають орто-
ефектом:
283.2. Реакції нуклеофільного заміщення
в нітроаренах
Нуклеофільне заміщення атома Гідрогену в о і «-положеннях щодо
нітрогрупи стає можливим з огляду на брак у цих положеннях електронної
густини. Однак виконати типові реакції нуклеофільного заміщення, наприклад,
амінування за Чічібабіним, дією аміду натрію практично не вдається через
відновлення гідрид-іоном нітрогрупи й подальшого осмолення в лужному
середовищі продуктів реакції. Проте в о- або л-динітробензенах одна з нітрогруп
легко піддається шсо-заміщенню на нуклеофіл:
и2м 4=/ ^и2 и2м \=/ У=ОН, тмн2, осн3
Після заміщення однієї нітрогрупи друга втрачає здатність до заміщення,
оскільки вона більше не перебуває під сильним електроноакцепторним впливом
замісника. Поряд з шсо-заміщенням нітрогрупи можливе нуклеофільне
заміщення атома Гідрогену в активованому арені, що містить кілька потужних
замісників, які відтягують електрони. У цьому випадку повинно бути забез-
печене ефективне зв'язування гідрид-іона, що виділяється (наприклад, взаємо-
дією його з розчинником):
Розділ 28. Нітросполуки ряду бензену та їхні похідні
733
28.3.3. Реакції конденсації* алкілнітросполук
Рухливість атомів Гідрогену в а-положенні алкілбензенів значно збільшується,
якщо в о- або «-положеннях перебуває нітрогрупа. Відомо, що толуен не
конденсується з бензальдегідом, зате 2,4-динітротолуен легко реагує з ним:
Електронодефіцитність аренів за наявності двох, а ще ліпше - трьох,
нітрогруп уможливлює здатність цих сполук взаємодіяти з деякими нуклео-
філами або ароматичними сполуками, що мають електронодонорні замісники. У
цьому випадку утворюється молекулярна комплексна сполука донорно-
акцепторного характеру з перенесенням заряду. У табл. 28.1 наведено дані про
деякі такі комплекси.
Таблиця 28.1
Фізико-хімічні властивості комплексів з переносом заряду на основі нітроаренів
Акцептор
Донор
Темп, пл., °С
Кольори комплексу
1,3-Динітробензен
Анілін
41
Червоний
N. Л^Циметил-л-толуїдин
41
Чорний
1,3,5-Тринітробензен
Анілін
123-124
Помаранчевий
И, МД иметил-л-толуїдин
106-108
Темно-фіолетовий
28.3.4. Реакції відновлення
Мононітросполуки, які промисловість випускає у величезних масштабах,
застосовують головно для подальшого відновлення в аміни. Ця реакція слугує
також доказом того, що атом Нітрогену нітрогрупи з'єднаний з атомом Карбону
безпосередньо, а не через атом Оксигену.
Електрохімічне відновлення нітросполук залежно від рН середовища й
густини струму дає серію продуктів, які містять нові для нас функціональні
групи, що містять атом Нітрогену.
734
Чирва В.Я., Ярмолюк CM., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Кожний з цих продуктів можна отримати відновленням нітросполуки за
допомогою відповідного реагенту. Кінцевим продуктом серії відновлень
нітробензену у кислому й лужному середовищах є анілін:
Fe(CO)5
C6H5-N=0
Нітрозобензен
C6H5N02
Нітробензен
NaOH
СН,ОН
C6H5~N=N-CAH
О Азоксибензен
Zn
•"5 S~ C6H5-N=N-C6HS
NaOH
Zn в CHjCOOH
C^-NH-OH
Фенілгідроксиламін
Zn
HC1
Азобензен
Zn/NaOH
C6H5-NH-NH-C6H5
Гідразобензен
Zn/HCl
C6H5NH2
Анілін
28.4. Функції, утворені неповним відновленням
нітросполук
28.4.1. Нітрозобензен
❖ Методи одержання нітрозобензену. Відновлення нітробензену в кислому
середовищі важко затримати на стадії утворення нітрозобензену. Останню сполуку
легше одержати окисненням наступного продукту відновлення - фенілгідро-
ксиламіну (Е. Бамбергер1). Це і є звичайний шлях синтезу нітрозобензену:
3C6H5NHOH + H2Cr207
6Н^
fc
З C6H5-N=0 + 2Сг3+ + 7Н20
Імовірно, це ліпший спосіб одержання ароматичних нітрозосполук, які не
вдається виділити іншим методом як проміжні продукти відновлення нітросполук.
Під час обробки нітробензену алкілмагнійбромідом алкільний замісник як
нуклеофіл вступає в о- і «-положення до нітрогрупи, яка в цьому разі
відновлюється до нітрозогрупи:
0"ч+ О'Г^Вг"
+ RMgBr-
04+^OMgBr
ї я
- MgBrCl, ЩО
1 Бамбергер Е. (Bamberger Е.) (1857-1932) - швейцарський хімік. Головні праці присвячені
вивченню ароматичних і нітрогеновмісних сполук.
Розділ 28. Нітросполуки ряду бензену та їхні похідні
735
❖ Інші шляхи утворення нітрозосполук ароматичного ряду
■ Останнім часом опубліковано прямий метод відновлення нітросполук у
нітрозосполуки за допомогою ферум пентакарбонілу Fe(CO)s.
■ Дія нітратного ангідриду N2O3 або нітрозил хлориду C1NO на
металоорганічні сполуки, наприклад, на дифеніл меркурій, веде до нітрозоаренів.
■ Бензенове кільце, що містить сильні орієнтанти першого роду (-ОН,
-Ы(СНз)2), індиферентні до нітритної кислоти, може бути прямо пронітрозоване
у водному розчині дією нітритної кислоти. Нітрозування відбувається на
холоді в кислому водному розчині й практично повністю в и-положення:
^^>-ОН + HONO 0=N^j^-OH
^^С(СН3)3 + HONO 0=NH^^C(CH3)3
❖ Будова нітрозосполук. Ароматичні нітрозосполуки - безбарвні речо-
вини, димерні у твердому стані. У розчині, розплаві й у формі пари вони
дисоціюють на дві молекули мономерної нітрозо- сполуки, що має зелено-
блакитне забарвлення, причому усталюється рівновага:
2C6H5NO^=^ [C6H5NO]2
Димер нітрозосполуки має будову, аналогічну до будови азоксисполуки:
°ч Р /°
n=n; c6h5-n=n
сбн/ с6н5 сбн5
Димер нітрозобензену Азоксибензен
Можливо, цим треба пояснити легкість відновлення нітрозосполук в азокси-
і далі в азосполуки. Доказом на користь такої структури димеру є існування
геометричної ізомерії димерів нітрозосполук в аліфатичному ряді. Що
стосується геометричної ізомерії ароматичних сполук, то для них також існують
цис- і транс-ізомери. Однак сам димерний нітрозобензен відомий тільки в цис-
формі, як засвідчує аналогія його 14- і Раман-спектрів зі спектрами відомої цис-
форми о.о'-нітрозобіфенілу (він безбарвний, але мономерний):
,0
Обидві речовини мають дв? смуги поглинання за 1 389 і 1 897 см 1 і смугу за
1 409 см"1. Для транс-димерів ароматичних нітрозосполук простежується єдина
смуга поглинання в інтервалі 1 235-1 299 см"1.
736
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
■ Нітрозофенол - особливий випадок нітрозосполуки. У його розчинах
спостерігають таутомерію нітрозофенольної та хіноноксимної форм:
ОН=:Н-0-Н=
Положення таутомерної рівноваги залежить від розчинника, який
застосовують.
■ Нітрозодиметиланілін також відрізняється від звичайних нітрозосполук.
Унаслідок узгодженої взаємодії електронодонорної диметиламіногрупи й
електроноакцепторної нітрозогрупи рівновага помітно зміщена у бік хіноїдної
форми:
0=К"СЗ^(СНз)2 =ї=^"°"н={3)=:^(СНз)2
О^И Це виявляється за збільшеним порівняно з нітробензеном і
^ Р диметиланіліном дипольним моментом, батохромним зсувом смуги
у поглинання видимого світла (від жовтого до зеленого) і нездатності
^ утворювати димер.
■ Іншу взаємодію двох нітрозогруп спостерігають в оди-
нітрозобензені, який реально є гетероциклом бензфуразану:
❖ Хімічні властивості нітрозосполук
• Реакції конденсації. Нітрозосполуки здатні до численних реакцій конденсації.
Вони реагують, виділяючи воду, зі сполуками, що мають угруповання НгН-Х, і зі
сполуками, що мають реакційноздатну СН2-групу. Приклади:
С6Н5^=0 + ш2-с6н5 — С6Н5-№К-С6Н5
Азобензен
С-СН3 р~снз
СбН5-К=0 +ЩС — с6н5-к=<
,с-сн, ,с-ск.
о о
Пентан-2,4-діон 3-Фенілімінопентан-2,4-діон
(ацетилацетон)
Р Р |
., н
2-Феніліміноізатин
• Реакції циклізації. Друга група реакцій - приєднання ненасичених сполук до
нітрозогрупи з утворенням чотиричленного циклу:
Розділ 28. Нітросполуки ряду бензену та їхні похідні
737
с н
(сбн5)2с=сн2 ^(сбн5)2с-сн2 с6н5-и=о ^ 6 5Ч^-с-
с6н5-к=о гн/*~° н2ом-с6н5 н,с-н
• Дієновий синтез. Нарешті, з дієнами відбувається синтез Дільса-Альдера, у
якому нітрозогрупа діє як дієнофіл (Ю. Арбузов):
28.4.2. Фенілгідроксиламін
❖ Синтез феніпгідроксшіоміну. Нітрозобензен, як і інші ароматичні
нітрозосполуки, має високий оксидний потенціал, тому він легко відновлюється
до Н-фенілгідроксиламіну. Його можна також одержати відновленням нітро-
бензену за допомогою суспензії алюміній хлориду у водному розчині або
цинковим пилом у слабкокислому середовищі:
с6н5імо2++н2о —""є* » с6н5шон + гп(он)2
68%
З водного розчину фенілгідроксиламін висолюють натрій хлоридом. Це
безбарвна кристалічна речовина із запахом, що дратує слизові оболонки носа.
Електролізом нітро- і нітрозосполук в оцтовому ангідриді за наявності
натрій перхлорату можна отримати діацетильні похідні арилгідроксиламіну,
лужний гідроліз яких безпосередньо веде до арилгідроксиламінів:
+4е-
Аг-1М02
ar^n-ococh3-^Ar-NHOH
NaC104 і 3
Ar-NO -j V снз
+2e- О
❖ Утворення озоксибензену. Фенілгідроксиламін легко зазнає окиснення й
темніє на повітрі, а за енергійнішої дії перетворюється в нітробензен. З
нітрозобензеном у лужному середовищі він утворює азоксибензен:
C6H5NHOH + 0=N-C6H5 C6H5-N=N-C6H5
І
О
738
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
Цим пояснюють появу азоксисполуки як первинного продукту в разі
відновлення нітробензену в лужному середовищі й неможливість виділення
арилгідроксиламіну й нітрозобензену в лужному середовищі:
~Н>0
І- О" ОН 4)8- Чзно-
аг 6^ |
О
Правильність запропонованого механізму підтверджена реакцією л-то-
лілгідроксиламіну з нітрозобензеном. Отримана азоксисполука має атом
Оксигену біля атома Нітрогену, зв'язаного з фенілом, а не з толілом:
*-сн3-сбн4шон + с6н5к=о »-сн3-сбн4-№^-сбн5
о
❖ Ізомеризація в п-амінофенол. Фенілгідроксиламін - дуже слабка основа.
Його унікальною властивістю є перегрупування (ізомеризація) під час
нагрівання з кислотою в сильнішу основу - и-амінофенол:
/~Л-і^нон НО—
4=/ — \=/ _Ш2
и-Амінофенол - відомий фотопроявник. Проявниками слугують і його
похідні:
№ЮН3 Ш2
ОН
Амідол
Промисловий процес одержання и-амінофенолу виконують в одній ванні,
відновлюючи нітробензен до феніл гідроксил аміну з наступним перетворенням
його в цільовий продукт.
28 АЗ. Азоксибензен
❖ Синтез азоксибензену. Азоксибензен, отриманий уперше М. Зініним,
можна синтезувати декількома способами.
■ Відновлення нітробензену натрій метилатом або розчином лугу в метанолі:
,о
4 С6Н5->Ю2 + 3 СН3ОКа З НСООШ + З Н20 + 2 №И
С6Н/ с6н5
Розділ 28. Нітросполуки ряду бензену та їхні похідні
739
■ Відновлення нітросполук натрій борогідридом у диметилсульфоксиді або
арсен (III) оксидом:
с6н5мо2 + А82о3-^Г с6н5-н=н-с6н5
н2о, 80 с
Окиснення азобензену гідроген пероксидом:
о-
О-ч /=\ -^з ^~V-* /=n
N4J
\=/
о
Раніше вважали, що азоксисполуки мають циклічну будову:
с6н-н-к-с6н5
о
Однак А. Анжелі1 з'ясував, що несиметричні азоксибензени існують у
вигляді двох ізомерів, що принципово неможливо у разі циклічної структури:
Аг-^И-Аг' Аг-КИчІ-Аг'
\ \
О О
Унаслідок окиснення ароматичних азосполук одержують обидва ізомери.
■ Те ж відбувається й під час реакції взаємодії арилгідроксиламінів з
нітрозоаренами:
Ar-NHOH+0=N-Ar-
Агч Ar*
N-N
но b"
ArN Ar"
N-N
"6 OH
-HjO
Ar-N=N-Ar' Ar-N=N-Ar'
I + I
О О
♦♦♦ Перегрупування Валаха : Нагрівання азоксисполук у кислому середо-
вищі веде до їхнього перегрупування в и-гідроксіазосполуки:
с6н5-ір-с6н5^ нон(^^^(3
1 Анжелі А. (Angeli А.) (1864-1931) - італійський хімік. Головні праці присвячені вивченню
продуктів неповного відновлення нітробензену.
2 Валах О. (Wallach О.) (1847-1931) - німецький хімік. Головні праці присвячені хімії аліцик-
лічних сполук і терпеноїдів. Нобелівська премія1910 р.
740
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
За М. Шемякіним1, азоксибензен передає атом Оксигену в «-положення обох
бензенових кілець, що доводять дослідом з азоксибензеном, який містить один
атом Нітрогену 15Ы. Відновлення отриманого и-гідроксіазобензену дає суміш
аніліну й и-амінофенолу з еквівалентним розподілом між ними ізотопу
Нітрогену. Цей результат свідчить про утворення симетрично побудованого
інтермедіату:
г п \^Ьк=к-4^он
о
—- +
❖ Відновлення до озобензену. Азоксибензен легко відновлюють порошком
заліза або дією триалкілфосфату до азобензену:
слі^=н-ан< —слш=н-ан<
о 5 і 6 5 0т_|- 6 5 6 5
Азобензен
З іншого боку, азобензен легко зазнає окиснення до азоксибензену
пероксикислотами або гідроген пероксидом й легко — відновлення до гідразо-
бензену:
65 ^ 6 5 Н202 6 5 6 5 02або№ОС1 6 5 6 5
28.4.4. Азобензен
❖ Ізомерія азобензену. У випадку опромінення УФ-світлом стійкий транс-
ізомер азобензену перетворюється у малостійкий геометричний г/мс-ізомер. Це
приклад г/нс-/?у?ш/с-ізомерії, що для випадків геометричної ізомерії за участю
атома Нітрогену зазвичай позначають як сіш-яняш-ізомерію; аниш-азобензен
стабільніший від смн-ізомеру приблизно на 46,2 кДж/моль:
^"С6Н5
транс- або шши-азобензен цис- або син-азобензен
1 Шемякін М. (1908-1970) - радянський хімік. Головні праці присвячені біоорганічній хімії, одним із
засновників якої він був.
Розділ 28. Нітросполуки ряду бензену та їхні похідні
741
Сш/-азобензен - неплоска сполука, оскільки в іншому випадку бензенові
кільця накладалися б одне на одного. Питання про ізомерію обох сполук
вирішене за допомогою рентгеноструктурних досліджень.
❖ Синтез азобензену
■ Азобензен синтезують прямим відновленням нітробензену натрій
станітом і багатьма іншими відновниками, наприклад, цинком у лужному
середовищі, амальгамою Натрію, літій алюмогідридом у діетиловим етері:
2 С6Н5-Ш2 + 4 Ш28п02 С6Н5-К=>І-С6Н5 + 4 Ш^пОз
6 5 2 або ЬіА1Н4 6 5 6 5
■ Окиснення гідразобензену нітробензеном дає азобензен (див. також 28.4.5):
С6Н5-Ш->Щ-СбН5 + С6Н5Ж)2 -* СбН5-К=Н-С6Н5 + С6Н5Ж> + н2о
■ Крім того, ароматичні азосполуки можна отримати за допомогою реакції
азосполучення (див. розділ ЗО).
■ Несиметричні азосполуки можна одержати конденсацією нітрозобензену
з ароматичними амінами:
с6н5ш + к-с6н4ш2 -— с6нг^-с6н4-іі
❖ Азобензен - основа ще слабша, ніж фенілгідроксиламін. Він розчинний у
концентрованій сульфатній кислоті, проте випадає в осад під дією води
внаслідок гідролізу солі, що утворилася. Така сіль у середовищі концентрованої
Н28С>4 легко забирає в безлічі типів органічних сполук гідрид-аніон і
перетворюється у гідразобензен, який перегруповується в бензидин.
Азобензен - це легкоплавкі, розчинні в органічних розчинниках яскраво
помаранчеві кристали. Однак фарбування ними далеке за інтенсивністю від
фарбування азосполуками. Крім того, він не здатний закріплюватися на волокні.
Азосполуки застосовують для синтезу лікарських препаратів і барвників. Це
канцерогенні сполуки. Під час кип'ятіння з розчином станум (II) хлориду вони
кількісно відновлюються в первинні аміни.
28.4.5. Гідразобензен
❖ Синтез гідразобензену. Шляхом відновлення азобензену цинковим
пилом у лузі одержують безбарвний кристалічний гідразобензен (надлишок
відновника, нагрівання):
С^^К-С^-^-- С6Н5-КН-НН-С6Н5,
який поступово під дією кисню повітря перетворюється знову в азобензен,
забарвлений у помаранчево-червоні кольори.
742
Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
❖ Окиснетія до азобензену. Для препаративних цілей використовують
окиснення гідразобензену бромом у лузі:
с6нгкшн-сбн5КаОН+в»2 с6н5н=н-с6н5
❖ Бензидинові перегрупування. Чудовою властивістю гідразобензену є
перегрупування в бензидин («, «'-діамінобіфеніл), що кількісно відбувається під
час кип'ятіння у водних розчинах сильних кислот:
Його відкрив М. Зінін 1845 р. і назвав бензидиновнм перегрупуванням.
Бензидинове перегрупування вивчало багато дослідників. З'ясовано, що
поряд з «.«'-перегрупуванням відбувається також <?,«-перегрупування і утво-
рюється, щоправда, у меншій кількості, о, «'-діамінобіфеніл (ЗО %):
ИН2
Якщо одне з «-положень у бензенових ядрах зайняте карбоксильною або
сульфогрупою, то вони зазнають елімінування в процесі утворення бензидину.
❖ Семідинове перегрупування. У випадку наявності в одному з «-положень
замісника, що не відходить, відбувається семідинове перегрупування, яке
призводить до утворення первинно-вторинного діаміну ряду бензену - семідину,
що є «-заміщеною похідною «-амінобіфеніламіну:
■ Перегрупування відбувається інтрамолекулярно, тобто всередині однієї
молекули. Це підтверджене тим, що із суміші двох різних гідразосполук
синтезуються тільки два симетричні бензидини, наприклад:
Розділ 28. Нітросполуки ряду бензену та їхні похідні
743
Продукту міжмолекулярної взаємодії не виникає зовсім.
■ За найімовірнішим механізмом цієї реакції передбачене протонування
обох атомів Нітрогену:
Такий стан із двома повними позитивними зарядами на сусідніх атомах
Нітрогену нестійкий. Оскільки кожний атом Нітрогену набуває тетраедричної
конфігурації, то протилежні {пара-) кінці бензенових ядер зближаються один з
одним. Для гомолітичного розриву зв'язку N-14 потрібно по одному
додатковому електрону на кожний атом Нітрогену. Ці електрони надходять від
«-карбонових атомів, які, відповідно, стають вільнорадикальними. Бензенові
ядра з'єднуються своїми «-карбоновими вільнорадикальними атомами в систему
біфенілу:
Утворення нового карбон-карбонового зв'язку й розрив зв'язку між двома
атомами Нітрогену відбувається синхронно.
Зазначимо, що бензидин і його похідні є канцерогенами й спричинюють рак
сечового міхура, тому їхнє виробництво скорочують.
■ Під дією на азобензен сульфатної кислоти й донора гідрид-іона,
наприклад, первинного або вторинного спирту, також відбувається бензидинове
перегрупування. Спочатку утворюється гідразобензен, що далі перегруповується
так, як описано вище:
+
Розділ 29
APOMA ТИЧНІ АМІНИ
29.1. Класифікація та номенклатура ароматичних
амінів
29.1.1. Класифікація
Ароматичні аміни аналогічно до амінів аліфатичного ряду класифікують за
ступенем заміщення біля атома Нітрогену:
Первинні
аміни
Вторинні
аміни
Третинні аміни
Четвертинні солі
амонію
н3с-ш
H3C-N-C2H5
N(CH3)3 СГ
Анілін
іУ-Метиланілін N-Мтил-ІУ-метиланілін Триметиланіліній хлорид
29.1.2. Номенклатура IUP АС
За правилами 1993 р. родоначальний гідрид NH3 називають азан, відповідно,
назви первинних амінів R-NH2 складають з назви вуглеводневого замісника і
назви родоначального гідриду. Зберігають і попередні варіанти назв амінів:
позначення амінофункції як головної групи суфіксом -амін або додавання назви
вуглеводневого замісника до назви функції амін:
NH,
нафт-1-ілазан
або
нафтален-1-амін
або
нафт-1-іламін
■ Для несиметричних вторинних і. третинних амінів за основу вибирають
назву ароматичного аміну, а інші (простіші) вуглеводневі замісники називають у
Розділ 29. Ароматичні аміни
745
префіксі із зазначенням їхньої наявності біля атома Нітрогену. Приклади таких
назв наведені вище.
■ У назвах симетричних вторинних і третинних амінів до назви вугле-
водневих замісників додають числові префікси ди- і три-, наприклад: (СбН5)2МН
- дифеніламін, (СбН5)зИ - трифеніламін.
■ Допустимо як родоначальні використовувати тривіальні назви анілін і
бензидин. Тривіальну назву толуїдин застосовують тільки для незаміщеної
сполуки:
Вг
Анілін о-Бромоанілін Бензидин Толуїдин (показано п-
ізомер)
На базі цих тривіальних назв будують назви відповідних замісників,
причому назву толуїдіно- використовують тільки для незаміщеної структури:
Аніліно л-Нітроанілінобензойна Бензидино Толуїдино (показано
кислота я-ізомер)
29.2. Фізичні властивості
• Ароматичні аміни - рідкі або кристалічні безбарвні речовини зі специфічним
запахом. З огляду на легкість окиснення вони часто жовтуватого забарвлення.
Зазвичай «-ізомери мають вищу температуру плавлення порівняно з о- і м-
ізомерами.
29.2.1. Спектроскопія ароматичних амінів
• Уф-спектроскопія. Максимуми поглинання ароматичних амінів зміщені в
довгохвильову ділянку порівняно з ароматичними вуглеводнями. Наприклад, в
УФ-спектрі аніліну є дві інтенсивні смуги з А,^ 230 і 280 нм, а бензен має смугу
зА,тах 183 і 204 нм.
• ІЧ-спектроскопія. В ІЧ-спектрах первинних ароматичних амінів є смуги
поглинання амінофункції: слабкої інтенсивності - валентні коливання Уц-н в
ділянці -3 500 (асиметричні) і ~3 400 см"1 (симетричні), а також інтенсивні смуги
деформаційних коливань 5м-н 1 640-1 560 см'1. Для вторинних амінів ці сигнали
746
Чирва В.Я., Ярмолюк CM., Толкачова Н.В., Земляков О.Є. Органічна хімія
зміщені в ділянки 3 350-3 310 (vN_H) і 1 580-1 490 cm'1 (5n-h). Смуги поглинання
коливань vc-n в ділянках 1 360-1 250 і 1 280-1 180 см"1.
• ПМР-спектроскопія. Хімічний зсув сигналів протонів аміногрупи в
ароматичних сполуках зміщений у слабше поле (3,3-4,0 м.ч. для первинних і
3,1-3,8 м.ч. для вторинних амінів) порівняно з відповідними сигналами протонів
аліфатичних амінів (1,1-1,8 і 1,2-2,1 м.ч.). Вплив аміногруп на хімічні зсуви
сигналів ароматичних протонів можна описати такими прикладами:
Нс^О)^
НЬ На
НЬ На
H2NH^~NH2
На
5 м.ч. 6,52 (а), 7,02 (Ь), 6,62 (с)
5 м.ч. 7,7 (а), 7,5 (Ь), 7,5 (с)
5 м.ч. 6,37 (а)
29.3. Синтез ароматичних амінів
Найпростіший ароматичний амін - анілін СбН5МН2. Уперше його отримано
сухою перегонкою індиго К. Фріцше1 1840 p., звідки й походить назва (від ісп.
anil - індиго). Сліди аніліну знайдені також у кам'яновугільній смолі.
❖ Реакція Зініна та її варіанти. Промислового значення анілін набув
після одержання його М. Зініним 1842 р. реакцією відновлення нітробензену
гідроген сульфідом:
С6Н5>Ю2 + З H2S C6H5NH2 + 2 Н20 + З S
Донедавна головним способом промислового синтезу аніліну й низки інших
ароматичних амінів був варіант реакції Зініна - відновлення нітросполук чавунними
ошурками за наявності невеликої кількості хлоридної кислоти (А. Бешам2):
4 C6H5N02 + 9 Fe + 4 Н20 4 C6H5NH2 + 3 Fe304
■ Зі збільшенням виробництва аніліну на перший план у промисловості
виходить каталітичне відновлення нітросполук:
Кат
C6H5N02 + З Н2-^ C6H5NH2 + 2 Н20
Найчастіше як каталізатор використовують мідь у парофазовому методі або
нікель Ренея в рідкофазовому відновленні.
■ У лабораторії улюбленим відновником є металеве олово з міцною
хлоридною кислотою:
1 Фріцше К.Ю. (1808-1871) - російський хімік і ботанік, академік Петербурзької АН. Виділив
антрацен і фенантрен.
2 Бешам A. (Bechamp А.) (1816-1908) - французький хімік-органік. Головні праці присвячені
ароматичним нітросполукам.
Розділ 29. Ароматичні аміни
747
2 С^Ж^ + 3 Бп + 14 НС1 - 2 С6Н5Ш2- НС1 + 3 8пС14 + 4 Н20
Відновленням відповідних нітросполук одержують гомологи аніліну: о-, м- і п-
толуїдини СНз-СбНЦ-МНг, а також заміщені аніліни: о, м-, «-хлороаніліни СІ-СбНр-
МН2, о- і «-анізидини СНзСМ^НН^Нг, о і «-фенетидини С2Н5(>^бН4-->М2 тощо.
Відновлення лі-динітробензену чавунними ошурками або каталітично
збудженим воднем дає л/-фенілендіамін:
■ У разі відновлення сульфідами лужних металів вдається виконати
часткове відновлення (як звичайно, у цьому разі найлегше відновлюється нітро-
група, що перебуває в «-положенні до іншого замісника):
а з лі-динітробензену утворюється лг-нітроанілін.
❖ Амінування галогеноаренів. У деяких країнах Західної Європи про-
мислове значення іуає спосіб синтезу амінів з галогенопохідних аренів:
с, Ш3,Си20 /~\_
\ / ^! 200 °С, 65 атм\ /^П2
У цьому випадку одержують відповідні первинні аміни. Реакція відбувається за
нуклеофільним механізмом або через утворення проміжного дегідробензену. За
наявності в о- або «-положенні сильних електроноакцепторних угруповань
нуклеофільне заміщення атома галогену на аміногрупу значно полегшене.
❖ Інші способи одержання ароматичних амінів мають допоміжне
значення.
■ Відновлення (наприклад, за допомогою олова й хлоридної кислоти)
кожного з проміжних продуктів відновлення нітросполук, зокрема, азосполук.
■ Дія азидної кислоти на ароматичні вуглеводні:
С6Нб + №N=N=14" С^ИН, + И2
■ Дія натрій аміду за жорстких умов на галогенобензени. Перебіг реакції з
галогенобензеном, у якому мічений атом Карбону зв'язаний з галогеном,
засвідчив, що аміногрупа приєднується лише на 50 % до цього атома Карбону, а
на 50 % - до окарбонового атома. Це пояснюють попередгіІМ відщепленням
гідроген галогеніду сильноосновним агентом, яким є амід натрію - ИаІМНг, і
748
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
утворенням дегідробензену, а потім уже відбувається приєднання амоніаку до
потрійного зв'язку:
-НВг ^ ^"МН2
■ Відновне амінування ароматичних альдегідів приводить до бензиламіну і
його похідних. Відновне амінування кетонів дає аміни, що мають вторинні
алкільні групи: • • *
4=7 о 4=7 ш2
■ Розщеплення похідних карбонових кислот. За лабораторних умов
ароматичні карбонові кислоти можна перетворити в аміни тим же шляхом, що й
карбонові кислоти жирного ряду, тобто через їхні аміди {розщеплення за
Гофманом) або азиди {розщеплення за Курціусом):
Аг-СО-ЮТ. °Н
2 Вг2
,0
Аг-СОКНВг-^Аг-С'
-н2о
—* Аг-ЫСО^ [Аг-ИН-СООН]— АгШ2 + С02
Ізоціанат
+ — Р н ОН
Аг-СО-Ы=Н=Ы^г [Аг-СО-Ічі:] — АгЫССЯ^
• АгЫНСООС2Н5— Аг-Ш2 + С02 + С2Н5ОН
29.4. Хімічні властивості ароматичних амінів
29.4.1. Кислотно-основні властивості ароматичних
амінів
Анілін у водних розчинах - основа набагато слабша, ніж амоніак і
аліфатичні аміни. Справді, він погано розчинний у воді й не забарвлює лакмусу і
фенолфталеїну; не утворює солей з такими слабкими кислотами, як карбонатна,
ціанідна або сульфідна, проте із сильними кислотами дає тверді солі, які у
водному розчині сильно гідролізовані:
Розділ 29. Ароматичні аміни
749
н ¥+ -
с6н5-к +нсі нгн-н сі
н 6 5 н
Ослаблення основних властивостей тривалентного атома Нітрогену в аніліні
зумовлене мезомерним ефектом аміногрупи, або, інакше кажучи, спряженням
пари електронів атома Нітрогену з я-електронами ароматичного циклу (р,я-спря-
ження). Це означає, що орбіталь р-електронів атома Нітрогену (неподілена пара)
перекривається з орбіталями я-елеклронів бензенового циклу. Така взаємодія
можлива, очевидно, лише в тому випадку, коли осі /^-електронних орбіталей
атома Нітрогену приблизно паралельні до осей я-електронних орбіталей
карбонового циклу. Саме тому бензенове кільце в, аніліні збагачене електронами
й дуже легко вступає в реакції електрофільного заміщення. Справді, виведення
аміногрупи з, умовно кажучи, площини циклу миттєво збільшує її основні
властивості й зменшує дипольний момент молекули. Таке порушення
компланарності можливе за наявності двох о-замісників. їхній вплив видно,
наприклад, з порівняння дипольних моментів аніліну й 2,3,5,6-тетра-
метиламінобензену (амінодурол):
н3с^^сн3
амінодурол
р=1,53 0 р=1,39Б
Метальні групи, що загалом надають дипольний момент ароматичному
ядру, у цьому випадку розташовані симетрично, так що їхній власний сумарний
вплив на дипольний момент дорівнює нулю. На жаль, не можна порівнювати
основні властивості аміногрупи в обох сполуках, тому що в разі о-розташування
метилів поля їхніх атомів Гідрогену відштовхують атакувальний протон, що
послаблює основні властивості й приховує ефект, який нас цікавить. Різниця
дипольних моментів двох ароматичних амінів зумовлена тільки наявністю
мезомерного ефекту у випадку аніліну і його відсутністю в амінодуролі.
Основність анілінів значно залежить від природи й положення замісників в
ароматичному ядрі. Електроноакцепторні замісники послабляють її, а електроно-
донорні - збільшують. Наприклад, основність «-заміщених анілінів зменшується зі
збільшенням електроноакцепторних властивостей замісників у такому ряді:
амінофенол > анізидин > толуїдин > анілін > бромоанілін > нітроанілін.
На основності заміщених анілінів позначається місце положення замісника.
Замісники з індукційним ефектом мають найбільший ефект, перебуваючи в о-
положенні й найменший - в «-положенні. Замісники з мезомерним ефектом
найефективніші в о- і «-положеннях стосовно аміногрупи.
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
29.4.2. Реакціїелектрофільного заміщення
в бензеновому кільці амінів
❖ Галогенування. Взаємодія аміногрупи з бензеновим ядром особливо сильно
виявляється в момент електрофільної атаки в о і «-положення, наприклад, під час
галогенування. У цих випадках а-комплекси мають таку будову:
Здатність амінного атома Нітрогену легко перетворюватися в позитивно
заряджений амонійний і брати на себе в проміясному о-комплексі позитивний
заряд, що виникає на зв'язаному з ним атомі Карбону циклу в момецт атаки
електрофіла на о- і «-карбонових атомах, і є причиною зниження рівня енергії
перехідного стану й великої швидкості перебігу електрофільних заміщень. Отже,
слабкі основні властивості ароматичної аміногрупи та її сильна о-орієнтувальна
дія мають ту саму причину.
Активувальна дія аміногрупи в аніліні та його гомологах настільки сильна,
що прямою взаємодією йоду за кімнатної температури анілін перетворюється на
2,4,6-триіодоанілін. Ще енергійніше він реагує з бромом і.хлором, утворюючи
2,4,6-тригалогеноаніліни.
Галогенування ароматичних амінів відбувається настільки легко, що не
потрібно каталізаторів (кислот Льюїса), а припинити реакцію на стадії моно-
галогенування вдається, лише знизивши о,«-орієнтувальний ефект аміногрупи.
❖ Тимчасові захисти аміногрупи. Цікаво, що під час хлорування аніліну
може відбуватися окиснення аміногрупи. Для запобігання цьому процесу
використовують тимчасовий захист аміногрупи:
Об'ємність ацетамідного залишку сприяє селективному галогенуванню
аніліну в «-положення.
❖ Сульфування. Під час оброблення аніліну концентрованою сульфатною
кислотою він, як і аміни аліфатичного ряду, утворює гідросульфат. Амонійний
атом Нітрогену спрямовує атаку електрофіла в л*-положення, що дає метанілову
кислоту:
І 2 г -і і .
Н
Н
№1
-н+
Розділ 29. Ароматичні аміни '
751
ЫН3+НБО; НН3+Н80"4
24% Г і)
Н,0
Олеум X - Н2804
^^803Н ^ ^03Н
Метанілова кислота
Якщо ж сіль аніліну "запікати", тобто нагрівати доти, доки проба не перестане
давати анілін у разі нейтралізації розчином натрій гідроксиду, то через проміжний
продукт - фенілсульфамінову кислоту - утворюється сульфанілова кислота:
Ш3 Н804 НЫ-803Н
2—4 1^ .н20
180"С _ Ц Н+
Фенілсульфамінова
кислота
Н 802ОН Н 802ОН 802ОН
Сульфанілова
кислота
Під час запікання аніліну з сульфатною кислотою за температури нижче
100 °С здебільшого утворюється о-ізомер - ортанілова кислота. Очевидно, такий
результат можна пояснити оборотністю сульфування, більшою швидкістю
утворення ортанілової кислоти (кінетичний контроль) і більшою стійкістю
сульфанілової кислоти (термодинамічний контроль). Обидві кислоти існують у
вигляді бетаїну, або цвітер-іон:
йн3
Важливо зазначити, що ацильовані аміни через стеричні чинники зазнають
сульфування тільки в «-положення.
❖ Сульфамідні препарати. Величезне практичне значення мають аміди
сульфанілової кислоти, або сульфонаміди, яким притаманні ефективні лікарські
властивості. Уперше антибактеріальні властивості сульфонамідів виявлені на
прикладі червоного стрептоциду:
752
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
II .1 4=7 ихт_УЛ
Червоний стрептоцид Білий стрептоцид
Подальші дослідження засвідчили, що ефективний антимікробний препарат
- білий стрептоцид, або просто стрептоцид (п-сульфонстід)9 що є в основі всіх
сульфамідних препаратів.^ Сучасний промисловий метод одержання п-сульфо-
наміду ґрунтується на хлоросульфуванні ацетаніліду хлоросульфоновою
кислотою:
ны-с:-сНз НЫ-£-СНз Н№-С-СН3
л (СН3СО)20 НСКО^ НН3^ Н20(Н+)
СН3СООН -н2° "нс1 -сн3соон
802С1 802Ш2
Численні модифікації (понад 6 000) структури сульфонамідів спрямовані на
створення похідних типу
Сульфамідні препарати відіграли величезну роль під час Другої світової
війни, зберігши життя сотням тисяч хворих і поранених, тому що ефективних
засобів проти стрептококових, пневмококових, стафілококових, гонококових,
менінгококових мікроорганізмів, деяких вірусів для лікування ран, гангрени в
той час практично не було.
Основний механізм дії сульфамідних препаратів - генерування в організмі
незаміщеного сульфонаміду (Я', ІГ, Я=Н) і його конкуренція з я-амінобензойною
кислотою за взаємодію з певними ферментами в мікробній клітці. Унаслідок
зв'язування цих ферментів сульфонамідом припиняється ріст мікроорганізмів. На
жаль, інтенсивне застосування будь-якого одного сульфонаміду призводить до
появи стійких штамів мікроорганізмів, що значно знижує ефективність препарату.
Приклади деяких сульфонамідів, які широко використовують, наведено нижче:
сн3
( У-802Ш-С-СН3 Н,Ы-< ^БО^нХД
Н2К" \_/ —2*— з \_/ —2*— -сн
Альбуцид (сульфацил) Сульфадимезин
Розділ 29. Ароматичні аміни
753
/—\ N—п
^ \=/ 2 N ОМе норсульфазол
Сульфадиметоксин
СООН Фталазол
❖ Нітрування. Нітрувати безпосередньо анілін не можна, тому що реакція
занадто енергійна, а нітроген оксиди, що виділяються внаслідок окиснення
аніліну, діазотують неокиснений анілін. Тому анілін попередньо ацетилюють
або формілюють, а похідну, що утворилася, нітрують, одержуючи суміш о- і п-
нітроацетанілідів (або о- і и-нітроформанілідів). Видаляючи захисну ацильну
групу, одержують суміш о і и-нітроанілінів:
>Ш^О" ХН3
і»
Ж>2 + Н2804
Ш-С-СН3 + Н20 - 02N-^^>-NH2 + СН3СООН
О
^КН-СОСНз /унн2
І і + н2о —- У \ ■+сн3соон
Під час дії на ацетанілід нітрувальної суміші реакція відбувається
переважно в «-положення, а оброблення ацетаніліду нітратною кислотою в
оцтовому ангідриді дає о-ізомер.
Для одержання л*-нітроаніліну використовують л*-динітробензен, який
вибірково відновлюють сульфідами або гідросульфідами лужних металів (вони,
754 Чирва В.Я., Ярллолюк CM., Толкачова Н.В., Земляков О.Є. Органічна хімія
як відомо, здатні відновлювати тільки одну нітрогрупу з двох наявних у
молекулі).
❖ Форміпювання. Третинні аміни можна формілювати в «-положення
сумішшю диметилформаміду й фосфорхлорооксиду:
K2N-O +HCON(CH3)2|^k*NR2-<Q^CHO
80-85 %
Реакцію запропонував А. Вільсмейєр 1927 р.
29.4.3. Заміщення з аміногрупі
❖ Аніліди. У багатьох синтезах аміногрупа повинна бути блокована, щоб
уникнути по ній будь-яких реакцій. Ми вже розглядали реакцію ацилювання
аніліну. Ароматичні аміни можна ацилювати хлороангідридами й ангідридами
кислот або сильним нагріванням амінів з кислотами аж до відщеплення води.
Для формілювання придатний тільки останній метод:
C6H5NH2 + RC0C1 —- C6H5NHC0R + HC1
R R
,C-0-Q +C6H5NH2 —- C6H5-NH-CO-R + RCOOH
O O
RCOOH + C6H5NH2—- RCOÖNH3-C6H5-^- C6H5NH-CO-R + H20
Сьогодні, однак, є ефективніші ацилювальні агенти. Вони дають змогу
вводити і знімати захист за дуже м'яких умов. Це важливо, якщо в молекулі є
інші реакційні центри.
Ацильовані аміни нейтральні й не розчиняються ні у водних розчинах лугів,
ні в кислотах, однак під час тривалого нагрівання з кислотами або лугами
зазнають гідролізування до амінів і кислот:
C^-NH-CO-R + Н20 C6H5NH2 + RCOOH
■ У безводному середовищі атом Гідрогену, зв'язаний з атомом Нітрогену
ацильованих амінів, може бути заміщений лужним металом. Натрієві або калієві
солі, наприклад, C6H5N(Na)COCH3, під дією води повністю розкладаються на
вихідні сполуки, а під дією алкілувальних засобів утворюють ацильні похідні
вторинних алкілариламінів:
C6H5vXTXOCTL ^ тт 9
6 5 N 3 PHP
• + СН3І Y СН3 + NaI
сн3
Розділ 29. Ароматичні аміни
755
■ Деякі анілідн застосовують як лікарські седативні та жарознижувальні
засоби. Це сам ацетанілід або антифебрин, М-ацетилфенетидин С2Н5О-С6Н4-
NH-CO-СНз або фенацетин - жарознижувальний засіб і анальгетик.
• Похідні карбамінових кислот. З інших найпростіших ацильних похідних
ароматичних амінів заслуговують на увагу похідні карбонатної кислоти. Самі
карбамінові кислоти Ar-NH-COOH нестійкі. Натомість естери карбамінових
кислот — уретани, а також їхні аміди — прості й змішані похідні сечовини -
стабільні й добре відомі.
Для одержання уретанів зазвичай діють арилізоціанатом, наприклад,
фенілізоціанатом, на спирти:
C6H5-N=C=0 + HOC2H5 C6H5NHCOOC2H5
Фенілуретан (етиловий естер
фенілкарбамінової кислоти)
Ароматичні похідні сечовини утворюються з амінів і фосгену або з амінів і
арилізоціанатів:
СОС12 + 2 C6H5NH2 CO(C6H5NH)2
C6H5N=C=0 + H2NC6H4CH3 C6H5-NH-CO-NH-C6H4-CH3
■ Деякі фенільовані похідні сечовини мають солодкий смак. До таких
належить похідна сечовини й фенетидину C2H5O-C6H4-NH-CO--NH2, яку
називають дульцин і часто застосовують як замінник цукру. Ця сполука в 200
разів солодша від цукру.
❖ N-Алкіланіліни. Під час метилювання аніліну шляхом його нагрівання з
метйліодидом утворюється суміш моно- і диметиланіліну з четвертинною
амонієвою сіллю.
Під час нагрівання анілін сульфату з метанолом під тиском відбувається
заміщення одного або двох атомів Гідрогену аміногрупи на метальний замісник
і утворюються iV-метил- і Л/;Л/-диметиланілін (перевага того чи іншого продукту
залежить від умов реакції). Цього ж можна досягти, пропускаючи пари аніліну
та метанолу над дегідратувальним каталізатором, яким є алюміній оксид:
C6H5NH2 + СН3ОН^- C6H5NHCH3 + H20
C6H5NH2 + 2 СН3ОН^- C6H5N(CH3)2 + 2H20
iV-Метил- і А/;Л/-диметиланілін виробляють у промислових масштабах,
оскільки моноалкіламіни застосовують у деякихГкраїнах як антидетонатори
моторного пального. їх же, особливо діалкіламіни, використовують у
виробництві синтетичних азо- і трифенілметанових барвників.
Монометиланілін і диметиланілін не можна розділити фракційною перегонкою,
тому що вони киплять за одної й тої ж температури. Однак їхній поділ можна
756
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
виконати за допомогою оцтового ангідриду або «-толуенсульфохлориду (ТбСІ),
оскільки лише вторинна основа - монометиланілін - здатний утворювати ацетильну
або тозильну похідну.
Для синтезу лише М-метиланіліну аміногрупу попередньо захищають шля-
хом ацилювання оцтовим ангідридом або ацетилхлоридом, а потім проводять
реакцію метилювання:
О
с.н^н.+снхосі—-г сдан-с-сн,
Ацетанілід
—- с н їч—с-сн Н*'Н2° сйн5шсн3
^6П5^ ^ '-"з -снхоон 6 5 3
СН3
Л^-Метилацетанілід
• Монометиланілін під час нагрівання до 330 °С унаслідок переміщення
метальної групи частково перегруповується в «-толуїдин:
Вторинні ароматичні аміни під дією нітритної кислоти дають ТУ-нітро-
зоаміни:
С6Н5ШСН3 + НОШ С6Н5-Ы(КО)СН3 + Н20
ІУ-Нітрозоаміносполуки під дією спиртового розчину НС1 повністю перегру-
повуються в «-нітрозопохідні:
• Диметиланілін дуже вразливий у «-положенні. Завдяки цьому під дією
різних реагентів він легко вступає в реакції заміщення. Наприклад, під час
оброблення М.ЛГ-диметиланіліну нітритною кислотою одержують «-нітрозо-М,ІУ-
диметиланілін, що має зелене забарвлення:
<^^Ы(СН3)2+ ЖЖО ONнf~^N(CHз)2 + Н20
■ Завдяки мезомерному ефекту нітрозогрупа є сильним електроноакцепто-
ром і відтягує електронну густину від атомів Карбону в о- та «-положеннях.
Унаслідок цього «-карбоновий атом, що несе диметиламіногрупу, стає вразливим
Розділ 29. Ароматичні аміни
757
до нуклеофільних атак. Тому нітро-М,ІУ-диметиланілін під час кип'ятіння з лугом
відщеплює диметиламін, перетворюючись у нітрозофенол:
о=^ Ук(сн3)2-ЫаОН
ю^=о<он
■ Під час нітрування ДІУ-диметиланіліну поряд із заміщенням у ядро
відбувається заміщення на нітрогрупу одного метилу, і одержують тетрил -
вибухівку з сильною бризантною дією:
Н3С СН3
N
N
+ш2н8о; —- 11*1 +
Продукти окиснення
метильної групи
N0.
■ Деякі хлороангідриди кислот, наприклад, фосген, також порівняно легко
вступають у реакцію з ІЧМ-диметиланіліном, заміщуючи атом Гідрогену в п-
положенні до диметиламіногрупи. Продуктом цієї реакції є кетон Міхлера, що
має значення для синтезу барвників:
С0С12 + 2^-М(СН3)2— (СН3)2Ы-^^-^-^-Н(СН
з)2
29.4.4. Ароматичні вторинні й третинні аміни
❖ Дифеніламін одержують нагріванням за 200 °С еквімолярних кількостей
аніліну та його солі:
СбН5МН2- НС1 + СбН5Ш2 - С6Н5-№Ї-С6Н5 +ЫН4С1
Очевидно, синтез дифеніламіну відбувається як нуклеофільне заміщення
аміногрупи в амонійній формі на феніламінну:
<Онцсг+н2кчО>— СУшДн,7^ 0"ш_0
Можливість нуклеофільного заміщення амонійної групи в ароматичному
ядрі підтверджена експериментально:
Бензен-1,3,5-тріол
(флороглюцин)
758
Чирва В.Я., Ярмолкж С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
Дифеніламін є антиоксидантом. Для захисту пластмас від окисного руйну-
вання використовують низку аналогічно побудованих ароматичних вторинних
амінів, наприклад:
Ю О-гО-Ю
М-Фенілнафтил-2-амін N.N'-Дифеніл-я-фенілевдіамін
Сам дифеніламін застосовують як стабілізатор піроксилінових порохів
(зв'язування нітроген оксидів).
З мінеральними кислотами дифеніламін утворює солі, які, однак, зазнають
гідролізування вже під дією води. Отже, основні властивості в цієї сполуки
виражені дуже слабко, що пов'язано зі спряженням неподіленої електронної
пари атома Нітрогену з я-електронами двох бензенових ядер. З окисниками,
зокрема, з нітритною кислотою, дає синє забарвлення в сульфатній кислоті. Ця
кольорова реакція зумовлена утворенням імонієвих солей дифенілбензидину, її
використовують як якісну реакцію на нітрат-іон:
+
hso;
❖ Трифеніламін. Фенілюванням натрієвої похідної дифеніламіну за
Ульманом іодобензеном за наявності порошку міді можна одержати третинний
амін - трифеніламін:
С Н
6 V Na++ еді-^ (C6H5)3N + Nal
с6н/
На відміну від інших третинних аміні, він не зазнає метилювання
метиліодидом і не утворює четвертинних амонієвих солей. Трифеніламін
індиферентний навіть до концентрованої сульфатної кислоти. З такою сильною
кислотою, як перхлоратна, він усе-таки утворює сіль.
У трифеніламіні досягається настільки повне перекривання /?-орбіталі пари
електронів атома Нітрогену з усіма я-електронами бензенових ядер, що атом
Нітрогену практично не виявляє основних властивостей.
❖ Азатриптицен. Молекула трифеніламіну плоска, інакше неможливе
було б р,я-спряження. У випадку порушення площинної структури основність
атома Нітрогену зростає. М. Віттігу вдалося, забираючи гідроген хлорид від 9-
(о-хлорофеніл)-9,10-дигідроакридину за допомогою натрій аміду в рідкому
амоніаку, замкнути просторовий гетероцикл в азатриптицен. Азатриптицен є
структурою трифеніламіну, у якому група НС зв'язує три о-карбонові атоми
фенільних залишків і виводить їх з площини:
Розділ 29. Ароматичні аміни
759
Азатриптицен
Азатрйптицен є сильною основою, і він утворює з метиліодидом іодоме-
тилат. У цій сполуці /7-електронні орбіталі атома Нітрогену вже не можуть
вступати в спряження з орбіталями я-електронів ароматичних кілець унаслідок
взаємної перпендикулярності їхніх осей. Отже, розосередження електронів по
фенільних кільцях неможливе, і можна сказати з упевненістю, що о- і и-
орієнтувальної дії амінного атома Нітрогену в азатриптицені зовсім нема.
Розділ ЗО
АРОМАТИЧНІ АЗО-
ТА ЛІАЗОСПОАУКИ
30.1. Класифікація, будова й номенклатура
Солі діазонію
| Діазопохідні
Азосполуки
Фенілдіазоній хлорид
Натрій фенілдіазотат
Азобензен
• Солі діазонію. Реакція первинних ароматичних амінів з нітритною кислотою
приводить до утворення важливого класу сполук, відомих як солі діазонію. Ці
солі мають загальну формулу АгЫ2+ЛГ, де Х' - будь-який із численних аніонів,
таких як СГ, ВГ, И03", Н804".
Арилдіазонієві солі - іонопобудовані сполуки, і їхні водні розчини - електроліти.
Це правильно, якщо вони мають ненуклеофільні або слабконуклеофільні аніони
(ВР4", СГ, НБОО; якщо ж в аніонів достатня нуклеофільність (С>Г, ОСОСНз",
І"), то зв'язок у таких солях ковалентний Аг-Ы=Ы-ОСОСНз. Цікаво, що атоми
Нітрогену в іонопобудованих солях не фіксовані, й існує рівновага, що доведено
методом ЯМР 15Ы:
15+ + 15 .
Аг-Ы=ЫХ =5= Аг-Ы=ЫХ
Солі арилдіазоніїв, на відміну від солей алкілдіазоніїв, досить стабільні.
Стабілізувальна дія арильного радикала зумовлена спряженням я-електронів
ароматичного кільця з діазогрупою. Діазогрупа - це дуже сильний електроно-
акцепторний замісник, тому вона відтягує на себе електронну густину з
ароматичного кільця. Внаслідок цього на діазогрупі зменшується позитивний
заряд, і збільшується кратність її зв'язку (а водночас і її міцність) з
ароматичним ядром. Для найпростішого випадку - катіона фенілдіазонію -
це можна відтворити за допомогою граничних структур:
+
Катіони арилдіазоніїв - плоскі системи з лінійно розташованими в площині
ароматичного кільця атомами Нітрогену. Рентгеноструктурні та спектральні дані
Розділ ЗО. Ароматичні азо- та діазосполуки
761
свідчать про те, що зв'язок між атомами Нітрогену в катіоні діазонію - потрійний, і
що позитивний заряд у ньому зосереджений переважно на діазогрупі. Частково
позитивний заряд локалізований на крайньому атомі Нітрогену, який можна
трактувати як електрофільний центр у реакціях азосполук.
Наприклад, аніон у бензендіазоній хлориді розташований з одного боку
площини від діазогрупи і трохи ближче до кінцевого атома Нітрогену, ніж до
атома Нітрогену, зв'язаного з бензеновим ядром.
■ Назви солей діазонію утворюють шляхом додавання закінчення - діазоній
до назви вуглеводневого замісника вихідної ароматичної сполуки із зазначенням
назви аніона, наприклад:
Сі" СН;^^^ Вг" 02К-^^-№ЫВР^
Фенілдіазоній хлорид л-Толілдіазоній бромід
и-Нітрофенілдіазоній
борофлуорид
• Діазопохідні. Будову діазонієвих солей, багато в чому аналогічних до
амонієвих солей, визначив через 12 років після їхнього відкриття
К. Бломстранд1. Справді, ці речовини можна розглядати як онієві сполуки
молекули Нітрогену, зіставляючи з амонієвими, оксонієвими, сульфонієвими й
іншими сполуками, що є похідними атомів Нітрогену, Оксигену, Сульфуру.
Діазонієві катіони утворюють з аніонами слабких кислот (С>Г, ЗОзЬГ, ОН")
ковалентно побудовані діазосполуки:
Аг-№К:+ ОН" -Аг-И^-ОН
Аг-№№+ С14" Аг-№=1*-С*І
Аг-М=>І:+Н807 -Аг-№Н-803Н
Найвідомішими сполуками цієї групи є арилдіазотати загальної формули
Аг-^=К-0"М+. Вони можуть існувати у вигляді двох ізомерів: син- і анти-.
Перший з них менш стійкий через взаємне відштовхування атома Оксигену та
атома Гідрогену в о-положенні ароматичного ядра й легко зазнає ізомерйзування
в сполуку з яниш-конфігурацією:
Ша
// \\
Натрій сш/-фенілдіазотат Натрій ш/лш-фенілдіазотат
• Азосполуки. Вони містять у структурі азогрупу -К=1Ч-, зв'язану з двома артіль-
ними залишками. Ароматичні азосполуки, на відміну від аліфатичних аналогів, є
1 Бломстранд К^ВІоіши-апсі СЖ) (1826-1897) - шведський хімік. Головні праці в галузі неорга-
нічної хімії.
762
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
стійкими речовинами. Вони мають геометричну ізомерію (я-діастереомерія). У цис-
(син-)ізомері ароматичні кільця через просторові утруднення не можуть перебувати
в одній площині. У стійкішому транс-{анти~) ізомері максимально реалізоване
спряження. Причому якщо в конформері 1 р-електрони атомів Нітрогену не
спряжені з я-електронами ароматичних систем, то для конформера 2 таке
спряження є:
0
сш/-Азобензен
-К
яилш-Азобензен
■ Назви азосполук будують із назви відповідного вуглеводню, додаючи
префікс азо-. Для похідних азобензену використовують напівтривіальні назви, у
яких у префіксі зазначають положення й назву замісника. Несиметричні
азосполуки називають на підставі більшого за розміром вуглеводню, вико-
ристовуючи префікс арилазо-.
За правилами IUP АС 1993 р. групу -N=N- називають діазен. Відповідно,
азосполуки можна називати як дизаміщені діазени:
Азобензен,
або
дифенілдіазен
и-Гідроксіазобензен,
або
я-гідроксифенілфенілдіазен
2-Фенілазонафтален,
або
нафт-2-ілфенілдіазен
30.2. Фізичні властивості
Солі діазонію добре розчинні у воді, гірше - у спирті, нерозчинні в етері,
вуглеводнях. Сухі солі діазонію є кристалічними речовинами, багато з них
вибухають. Наприклад, фенілдіазоній хлорид вибухає не тільки під час тертя, а й
навіть у разі легкого натискання. Через їхню нестійкість ці сполуки зрідка
виділяють в індивідуальному стані, зазвичай використовують свіжоприготовлені
розчини. Солі з комплексними аніонами менш розчинні й стійкіші. Найміцніші
солі типу АгН2+Вр4", які моясна зберігати місяцями.
Уведення в о- і «-положення арену електроноакцепторних замісників
збільшує стійкість солей діазонію, а введення електронодонорних замісників,
навпаки, знижує їхню стабільність.
Розділ ЗО. Ароматичні азо- та діазосполуки 763
1 Грісс П. (Grieß Р.) (1829-1888) - німецький хімік, працював у Великій Британії. Синтезував
низку барвників.
Азосполуки - стабільні кристалічні речовини, більшість із них забарвлені.
Для використання їх я барвників у структуру вводять полярні угруповання, які,
зокрема, поліпшують розчинність сполук у воді.
30.2Л. Спектроскопія солей діазосполук
• Електронна спектроскопія. Азосполуки поглинають світло як в
ультрафіолетовій, так і видимій ділянках спектра. Сам азобензен має інтенсивну
смугу поглинання з Хтгх 318 нм в УФ-ділянці, пов'язану з я—»я*-переходом, і
малоінтенсивну смугу поглинання з А,тах 440 нм у видимій ділянці, пов'язану з
я—>я*-переходом. Уведення елекгронодонорних груп зсуває смугу поглинання,
пов'язану з я—>я*-переходом.
• ІЧ-спектроскопія.. Характеристичною в ІЧ-спектрах солей діазонію є
інтенсивна смуга поглинання vNsN+2 280-2 240 см"1.
Валентні коливання Vn=n азосполук виявляються в інтервалі 1 440-1 410 см"1
(транс-ізомери) і 1 510 см"1 (і/ис-ізомери).
30.3. Методи синтезу діазосполук
30.3.1. Синтез солей діазонію
❖ Реакція діазотування. Солі арендіазоніїв утворюються під час взаємодії
первинних ароматичних амінів з нітритною кислотою. Цю реакцію відкрив
1857 р. студент П. Грісс1 у лабораторії А. Кольбе. Вона набула важливого
промислового значення для одержання різноманітного набору барвників, а в
лабораторії - для синтезу різних класів ароматичних сполук.
Нітритна кислота - нестійка сполука, тому її генерують за наявності аміну
взаємодією натрій нітриту з мінеральною кислотою, зазвичай хлоридною або
сульфатною. Сумарне рівняння діазотування таке:
ArNH2 + NaN02 + 2 НХ -^j- ArN2X + NaX + 2 H20
Первинний
ароматичний амін
Реакцію проводять з охолодженням, оскільки за кімнатної температури
ароматичні аміни реагують з нітритною кислотою аналогічно до аліфатичних,
унаслідок чого утворюються відповідні гідроксисполуки з виділенням азоту.
Кінетичні дослідження К. Інгольда засвідчили, що насправді реакція відбувається
не за наведеним вище наочним рівнянням, а в такий спосіб:
764
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
+ С Н N14
н-о-м=о + н+ —* н2о-н=о ■ сбн5мн-к=о+ н3о+ 3=
Нітрозамін
=сбн5н=к-он^ с6н5-к=нсі~
Фенілдіазогідрат Фенілдіазоній
хлорид
Таке тлумачення процесу випливає з того, що діазотування - реакція
третього порядку, швидкість якої пропорційна до добутку концентрацій вільного
аміну, нітритної кислоти й іона Гідрогену.
За іншими припущеннями вважають, що протонована нітритна кислота в
розведених розчинах перетворюється в нітроген (III) оксид або похідні нітрозилу:
Н-0-№0 + НОШ ^=*=: 0=И-0-М=0 + Н30+
Н
Н-0-М=0 + Вг ^=5: Вг-И=0 + Н20
Н
Особливо зручними для діазотування є хлорид і бромід нітрозилу.
До найактивніших діазотувальних агентів належить катіон нітрозонію
"^N=0, що утворюється в сильнокислому середовищі.
Значення рН не повинне перевищувати 0,5-1,5, оскільки в іншому випадку
синтезований арендіазоній буде взаємодіяти з вихідним ароматичним аміном з
утворенням триазенів і аміноазосполук:
Аг-№И +ЬО*-Аг - Аг-№№Ш-Аг
триазен
2
амшоазосполука
Тому реакцію діазотування ведуть саме за таких умов. Стадією, що лімітує
діазотування, є утворення амонієвої солі
С6Н5№І2 + 0=К-0-№0 С6Н5Ш2Ш +N0^ ,
яка швидко перетворюється в нітрозамін. Далі нітрозамін зазнає ізомеризування
в діазогідрат, а той у кислому середовищі перетворюється в сіль діазонію, як
уже зазначено.
Наголосимо, що ариламіни - слабкі основи, і їхні солі у водних розчинах
дисоційовані:
Аг>Ш2 + НС15=±: АгМН3С1===: Аг>Ш3 + Сі"
Розділ ЗО. Ароматичні азо- та діазосполуки
765
Завдяки цьому концентрація вільної основи достатня для перебігу реакції
діазотування. Солі арилдіазоніїв із простими аніонами, як уже відомо, порівняно
малостійкі, і їх не виділяють із водного розчину, а безпосередньо далі
перетворюють. Однак із водного розчину їх можна виділити, додаючи відпо-
відну сіль, унаслідок чого випадають подвійні стійкі солі, наприклад:
C6H5-N sNHgCb" (C6H5N ^N)2SnCl6-
C6H5-N =NBF4~ (C6H5N =N)2Si62"
C6H5-N sNSbOT C6H5-N =NSbCl6"
C6H5-N =NZnCl3" QH5-N s-NFeClT
На практиці під час реакції діазотування необхідно витримувати
температуру не вище 0-5 °С, оскільки за вищої температури відбувається
розклад солей діазонію. Закінчення реакції діазотування визначають за
наявністю надлишку нітратної кислоти за допомогою іодкрохмального папірця:
2КІ + Крохмаль + 2 KN02 + 2 HCl—-
—-2 KI + Крохмаль + 2 KN02 + 2 HCl
Синє забарвлення
❖ Метод Бамбергера. Крім головного шляху синтезу діазонієвих солей -
діазотування амінів, солі діазонію також можна одержати з нітрозосполук,
діючи на них гідроксиламіном або нітроген (II) оксидом:
C6H5-N=0 + NH2OH C6H5-N=NOH + Н20
C6H-N=0 +2 NO ~ C6H5-N=NN03"
❖ Синтез із арилсульфінілімідів. Нещодавно описано новий шлях синтезу
ароматичних діазонієвих солей дією на арилсульфініліміди нітрозил перхлоратом:
Ar-N=S=0 + NO+C10"4 -Ar-N=N СК^ + S02
303.2. Синтез солей діазопохідних
Особливо прискіпливо вивчали взаємодію катіонів арилдіазонію з гідро-
ксильним аніоном. Першою досить повільною реакцією є утворення діазогід-
рату, що має властивості протонних кислот:
Ar-N=N: + 0:Н Ar-N=N-0~H (1)
тому швидко реагує з лугом як кислота, утворюючи солі діазотатів:
Ar-N=N-OH+0 ' Ar-N=N-0"+ Н2 (2)
766
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Завдяки швидкому перебігу реакції (2) під час зливання еквівалентних
кількостей діазонієвої солі й лугу лише половина солі діазонію встигає
перетворюватися в діазогідрат і далі швидко в діазотат-аніон, а половина
залишається у формі діазоній-катіона:
2 Аг-№1М: + 2 ОН" -Аг-ыЬ|:+Аг-№Н-0~+ Н20
Діазотат-аніон амбідентний і може реагувати за атомом як Оксигену, так і
Нітрогену. Це підтверджене результатами метилювання фенілдіазотатів Натрію
й Арґентуму метиліодидом:
М=^а С6Н5М-№0
сн3
с6н5к=ы-о-сн3
Методами потенціометрії й УФ-спектроскопії доведено, що в середовищах,
близьких до нейтральних, діазогідрат у чистому вигляді не існує. Навіть одразу
ж після одержання він є таутомерною сумішшю нітрозаміну із син- і анти-
ізомерами діазогідрату, які переходять один в одного через стадію утворення
нітрозаміну:
/Ж
№ЇМЧ =ї=^С6Н5Ш-Н=0 №И
С6Н5Х он свк{
сш/-Форма ^-Нітрозамін <ш«*Форііа
діазогідрату діазогідрату
3033. Синтез азосполук
❖ Перегрупування діазоаміносполук. Під час нагрівання з солями амінів
діазоаміносполуки або триазени (одержання див. нижче) перегруповуються в
аміноазосполуки. Наприклад, діазоамінобензен під час нагрівання з розчином
аніліній хлориду перетворюється на и-аміноазобензен:
На відміну від бензидинового й інших внутрішньомолекулярних перегру-
пувань, перетворення діазоамінобензену в аміноазобензен - міжмолекулярне
перегрупування. Воно відбувається з ацидолізом діазоаміносполуки до катіона
діазонію та аміну, які й вступають далі в реакцію азосполучення:
Розділ ЗО. Ароматичні азо- та діазосполуки
767
сбн5-№к-ічгн<:6н5+ сбн5кн3С1 —-сбн5-й=к сі"+ 2 с6н5їш2
я-Аміноазобензен, що містить як одну з функціональних груп уже відому нам
азогрупу, - найпростіший азобарвник.
❖ Реакція азосполучення. Під дією солей арилдіазоніїв на ароматичні спо-
луки, що містять дуже сильні орієнтанти першого роду (-№ї2, -N№1, -№12, -ОН),
відбувається реакція азосполучення і утворюються гідроксіазо- й аміно
азосполуки:
Аг-И=Й — АіНЙ=її + ^у"ОН Аг-№К-^^0Н
АНН* — АіН*=Й+^^К(СН3)2— Ar-N=N-(^)-N(CHз)2
Азосполучення - новий приклад електрофільного заміщення в бензеновому
ядрі (фенолу або аміну). Швидкість реакції азосполучення залежить як від
електрофільних властивостей катіона діазонію, так і електронодонорних
властивостей азоскладової. Катіон діазонію є слабким електрофільним агентом,
тому що позитивний заряд на атомі Нітрогену делокалізований завдяки
спряженню з ареном. Тому він вступає в реакцію зі сполуками, що мають сильні
електронодонорні замісники. Спочатку формується я-комплекс фенолу або
аніліну з електрофілом (катіоном діазонію), що переходить у а-комплекс, з якого
видаляється протон, і утворюється продукт заміщення - відповідна азосполука:
—- Аг^И-^"^-!^
а-комплекс
Аг-И=М-^^-ОН
а-комплекс
З'ясовано, однак, що спочатку катіон арендіазонію в реакції азосполучення з
амінами атакує атом Нітрогену як місце найбільшої електронної густини,
768
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
утворюючи триазени. Проте ця реакція зворотна, і триазени в кислому
середовищі розпадаються на вихідні сполуки. Повільніша реакція азосполучення
дає азосполуку як кінцевий продукт термодинамічно контрольованого процесу.
Таку ж ситуацію спостерігають у реакції з фенолят-іонами. Спочатку
швидко утворюється діазоетер, що у воді дисоціює на вихідні речовини, і тільки
потім відбувається повільна незворотна взаємодія катіона арилдіазонію з
амбідентним фенолят-іоном.
Заміщення відбувається переважно в «-положення, і лише якщо воно
зайняте — у о-положення. Такий напрям зазвичай пояснюють тим, що в
перехідному стані й кінцевому продукті реакції виникає довший, ніж у випадку
0-положення, ланцюг спряжених 7г-зв'язків. У цьому разі рівень вільної енергії в
перехідному стані нижчий, і, як наслідок, швидкість реакції більша.
Діазосполуки, що вступають в азосполучення, називають першим компо-
нентом, або діазоскладовою, а ароматичні фенол або амін - другим компо-
нентом, або азоскладовою. Чим сильніші орієнтанти другого роду перебувають в
о- і «-положеннях у діазоскладовій, тим швидше й енергійніше відбувається
реакція азосполучення. Тому активовані солі діазонію на основі 2,4-динітро-
аніліну або 2,4,6-тринітроаніліну вступають у взаємодію навіть із такими
слабкими азоскладовими, як поліметилбензени:
02К^Ь№К + ^ЬСН3 - О2Кч0нК=М-<Р^СНз
К02 СН3 К02 СН3
Реакція азосполучення може також відбуватися з неароматичними
азоскладовими, якщо вони містять рухливі метиленові атоми Гідрогену, подібно
до малонового й ацетооцтового естерів або циклопентадієну. По суті, реакція
відбувається між катіоном арилдіазонію й аніоном сполуки. Тому потрібне
лужне середовище для відривання протона від метиленової групи:
<СООС2Н5 СООС2Н5
АгНМ^ + СН - Аг-№^СН
СООС2Н5 СООС2Н5
О" СНз 0ч
" Аг—N-N-01
о^ос2н5 соос2н5
Аг-№К + ^ Аг-№№-^|
Реакції азосполучення з речовинами, що містять активну метиленову групу,
ускладнені наступним переміщенням другого атома Гідрогену цієї групи до
Розділ ЗО. Ароматичні азо- та діазосполуки
769
Нітрогену, що є сусіднім з ароматичним ядром, і перетворенням азосполуки в
арилгідразон. У випадку малонового естеру виникає арилгідразон діетилового
естеру мезоксалевої кислоти, а у випадку циклопентадієну - арилгідразон
циклопентадієнону:
СООС2Н5 СООС2Н5
Аі-№И-СН Аг-Н-№С
СООС2Н5 н СООС2Н5
У випадку ацетооцтового естеру в лужному середовищі відбувається
розщеплення за кислотним типом. Азопохідна ацетооцтового естеру (у цьому
прикладі заміщена на II) у лужному середовищі зазнає кислотного розщеплення
й перетворюється в арилгідразон а-кетонокислоти, відновленням якого можна
одержати а-амінокислоту (метод Феофілактова1):
V4, ^
Ar-№N-C-R1^CHзCOONa+Aг-N-N=C, Аг-М^ + ^-СНЯ-СООН
Л н соон
О' оод
Що стосується азосполучення з ароматичними азоскладовими, то на
практиці для цього застосовують лише феноли й аміни. Феноли вступають у
реакцію в слабколужному середовищі, оскільки у сильнолужному сіль діазонію
переходить у діазотат. Для амінів рекомендують користуватися слабкокислим
середовищем, тому що в сильнокислому практично весь амін перебуватиме у
вигляді амонієвої солі. Остання умова пов'язана з тим, що в нейтральному або
слабколужному середовищах ароматичні аміни випадають в осад. Крім того,
реакція відбувається за аміногрупою з утворенням триазенів (див. далі)/
Активність орієнтантів першого роду в цих азоскладових, як завжди, спадає
в такій послідовності:
Аг-О" > Аг-№гІ2 > Аг-ОН > Аг-Ш3+
В останньому випадку орієнтантом також є група МНг, яка в малій
концентрації утворюється внаслідок гідролізу солі:
Аг-№із+Н20^=^ Аг^Н2+Н30+
• Азобарвники. Реакцію азосполучення широко застосовують у техніці для
приготування безлічі марок азобарвників, що дають найбільшу розмаїтість серед
усіх типів синтетичних барвників.
1 Феофілактов В.В. (1891-1956) - російський радянський хімік, був завідувачем кафедри
органічної хімії МГУ, автор багатьох праць з синтезу біологічно активних речовин (ауксини,
гербіциди тощо). Розробив метод синтезу а-амінокислот (метод Феофілактова).
770
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Практично всі азобарвники, які застосовують, мають зазвичай дві та більше
азогрупи. Для введення декількох азогруп і одержання б/с-, трис- і тетракис-
азобарвників є різні способи. Обмежимося окремими прикладами синтезу
відомих азобарвників:
н°з§нСЗ^і"к + <^3~к(сНз)2_~~ ноз8_^З^м:=м^^Зк"і^(сНз)2
Метиловий оранжевий
У кислому середовищі за певного значення рН цей барвник переходить у катіон,
що супроводжується різким поглибленням забарвлення, яке називають індикаторним
переходом. Це використовують у методі кислотно-основного титрування:
НОз8-^^№1Ч-^^К(СН^ Н038Н^У-№^
Н
+ 2 НЖ)2 + 2 НС1—-
л/-Феніленддамін
кн2
/=\
2
МН2
ч
Один з барвників везувину
НОзБ
>щ2 н2и
Н038 БОзН
Найпростіший барвник типу
Феноли вступають у реакції азосполучення в слабколужному середовищі:
,0Н ^ ^Ша+
а - ос
СООН ^ СОО"Ка+
неї
>І02 ш2 Ш2
О^ COONa
Алізариновий
жовтий
Розділ ЗО. Ароматичні азо- та діазосполуки
771
У кислому середовищі азобарвники приєднують протон за азогрупою, що
супроводжується перебудовою зв'язків у бензеновому циклі, який несе аміно-
(гідрокси-)групу, і зміною кольору (індикаторні властивості):
<0>~^=н-<^^-н(сНз)2+н+—- СЗ^-ыЦ^
Масляний жовтий 3
Отримана сіль азобарвника має хіноїдну, а не бензоїдну структуру.
Приєднання протона відбулося не до диметиламіногрупи, а до атома Нітрогену
азогрупи внаслідок реакції з перенесенням реакційного центра, проте амонійним
усе-таки став атом Нітрогену диметиламіногрупи.
Наявність кольору азобарвників пов'язана з мезомерією їхньої структури,
що виражене в наведеному вище прикладі двома граничними структурами:
Внесок першої структури більший, проте, як видно з напряму атаки прото-
ном, друга теж дається взнаки.
З наявністю спряження пов'язане зближення головного енергетичного рівня
азобарвника і його збудженого рівня. Тому він поглинає світло в більш
довгохвильовій ділянці спектра не тільки порівняно з бензеном, а й з азобензеном.
Цей батохромний зсув зумовлений наявністю в «-положенні орієнтанта першого
роду, здатного нести позитивний або негативний заряд (у цьому випадку
позитивний). Такий замісник називають ауксохромом. Він "працює", передаючи
головні властивості (у цьому випадку негативного заряду) віддаленому атому
Нітрогену, і батохромно зміщує максимум поглинання лише за наявності спряженої
системи, що виникає в разі площинного розташування молекули. Тільки за цієї
умови виявляється спряження (мезомерія) і його ефекти.
Наприклад, за А. Кіпріановим1, під час переходу від барвника А до барвника
Б спостерігають гіпсохромний зсув (зсув максимуму поглинання в коротко-
хвильову ділянку спектра) унаслідок того, що о-метильний замісник спонукає
диметиламіногрупу вивернутися й так частково вийти із системи:
А Б
Якщо, однак, діалкіламіногрупу зв'язати в цикл, що міцно втримує її в майже
плоскому положенні, то, навпаки, зсув поглинання порівняно з А буде
батохромним:
1 Кіпріанов А. (1896-1972) — український хімік. Головні праці присвячені хімії барвників.
772
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
Для барвників існують загальні правила. Як відомо, поява забарвлення
виникає під час поглинання речовиною світла. Енергія поглинутого світла
витрачається на збудження а- або я-електронів. Тому, змінюючи будову
молекули барвника, можна змінювати характер та інтенсивність поглинання.
Промені світла, що не поглинулись, відбиваються від поверхні, їх і сприймає
око, внаслідок чого поверхня забарвлюється в ті кольори, які відбилися від неї:
нм
400
500 600
700
Поглинуте
світло
фіолетовий, синій,
зелений, жовтий, жовтогарячий,
червоний
Видиме світло
жовтий,
червоний, фіолетовий, синій,
зелений
Для появи забарвлення необхідна наявність у сполуках довгої системи
спряжених кратних зв'язків (хромофорів). Для того, щоб барвник зв'язувався з
волокном, потрібно, щоб він мав полярні групи, здатні взаємодіяти з полярними
групами волокна (пряме фарбування), або "розчинявся" у матеріалі волокна
(дисперсійні барвники) чи зв'язувався з іншими речовинами, нанесеними на тканину
(протравне фарбування). Азобарвники часто одержують безпосередньо на волокні
(льодяне фарбування): спочатку обробляють тканину азоскладовою, а потім
наносять малюнок холодним розчином солі діазонію.
У сучасній промисловості азосполуки становлять понад половину барвників
за номенклатурою й близько чверті за тоннажем.
30.4. Хімічні властивості
Розрізняють два типи хімічних перетворень солей арилдіазоніїв і арилдіазо-
сполук: без виділення та з виділенням азоту.
30.4.1. Реакції діазосполук без виділення азоту
❖ Відновлення. Під час відновлення діазосполук солями сульфітної кислоти
(гідросульфітом) арилдіазосульфонат, що утворюється спочатку, відновлюється в
арилгідразинсульфонат, який зазнає гідролізування в кислому середовищі до солі
фенілгідразину:
О О
Ь 5 6 5 ~Н2804 НН \ф 2
Фенілдіазосульфонат Фенілгідразинсульфонат
Розділ ЗО. Ароматичні азо- та діазосполуки
773
С6Н5-Ш-Ш3Н804 +ШН804
Фенілгідразоній сульфат
Реакцію розробив Е. Фішер 1875 р.
Цим шляхом фенілгідразин одержують у промисловості. Аналогічно синте-
зують 2,4-динітрофенілгідразин, який використовують для ідентифікації альдегідів і
кетонів.
■ Якщо в катіоні арилдіазонію, крім діазогрупи, немає інших фрагментів,
що відновлюються (наприклад, нітрогрупи), то відповідний арилгідразин молена
одержувати, використовуючи як відновники станум (II) хлорид і концентровану
хлоридну кислоту (метод В. Мейєра, 1883 р.) або цинк в оцтовій кислоті:
❖ Триазени. Первинні й вторинні ароматичні аміни в нейтральних або
слабкокислих розчинах спочатку вступають в азосполучення за атомом
Нітрогену, утворюючи діазоаміносполуки:
Аї-^кк + йн^Аг'*^^2 Аг-^Н-КН-Аг' + Н+
За менших значень рН діазоаміносполуки зазнають діазоаміно-аміноазо-
перегрупування, про яке йшлося у 30.3.3.
Триазенам властива тринітрогенна таутомерія:
Аг-М^-НН-Аг' =5=- Аг-Ш^Н-Аг'
Рівновага зміщена у бік тієї сполуки, у якій протон зв'язаний з більш
основним атомом Нітрогену.
■ Діазоаміносполуки утворюються також під дією арилмагнійгалогеніду на
арилазиди:
•ґ А'
Аг-№№Н +Аг'Ї^Вг ^Аг^И-И -^-Аг-№Н-Нч
М§Вг н
Цією реакцією можна отримати й аліфатичні триазени.
■ Якщо в середовищах, близьких до нейтрального, у реакцію з сіллю
діазонію вводити не первинні ароматичні аміни, а арилгідроксиламіни, то
утворюватимуться відповідні гідрокситриазени, що є комплексонами, їх
застосовують в аналітичній хімії для визначення катіонів деяких металів:
774
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
+ - .. Си(ОСОСН,)2 '• .О
Аг-№МС1 + Ну-Аг' - Ат-^^-^-Ат1 -± ' си
он "НС1 он Успирті 9Х \
Аг*—/У*—Аг
>Г
■ Під дією сильних кислот діазоаміносполуки розщеплюються на сіль
діазонію й первинний амін:
Аг-№:іМ-1ЧН-Аг'+ НС1 ►Аг-№ЕМСҐ+ Аг'-т,
У цьому разі, якщо триазен несиметричний, то розпад відбувається так, що
відщеплюється амін, який є сильнішою основою, а фрагмент, що залишився,
утворює відповідну сіль:
^Ь№к-ш-^0>-осНз с6н5-ш2- неї+сн3о-с6н4-ш2
30.4.2. Реакції діазосполук із виділенням азоту
❖ Заміщення на галоген
• Реакція Зандмейєра-Гагптермана. Реакцію заміщення діазогрупи на атом
Хлору або Брому виконують, змішуючи свіжоприготовлений розчин солі
діазонію із купрум (І) хлоридом або бромідом за кімнатної або іноді за
підвищеної температури; у цьому разі виділяється азот, і через кілька годин
арилхлорид або арилбромід можна виділити з реакційної суміші. Для одержання
арилхлоридів вихідний амін треба діазотувати у хлоридній кислоті, а
розкладання солей діазонію вести за наявності купрум (І) хлориду - С112СІ2, а у
випадку бромідів - відповідно, у бромідній кислоті:
70 %
Ця реакція з використанням купрум (І) галогенідів відома як реакція
Зандмейєра:
Розділ ЗО. Ароматичні азо- та діазосполуки
775
Аг^Х-^^АтХ + Нг
Іноді застосовують модифікацію цієї реакції, відому як реакцію
Гаттермана, у якій замість купрум (І) галогенідів використовують порошок
міді й гідроген галогенід. Роль міді в цих реакціях не зовсім з'ясована; можливо,
купрум(І)-катіон відновлює діазонієві солі й сприяє утворенню арильного
радикала, що далі за наявності великої кількості йонів Купруму(ІІ) пере-
творюється в продукт заміщення:
На користь цього механізму свідчить утворення побічного продукту -
дифенілу завдяки рекомбінації фенільних радикалів:
сбн;+сбн;—~сбн5-с6н5,
а також інгібувальний вплив кисню на цю реакцію.
Виходи арилхлоридів і арилбромідів із солей діазонію без каталізаторів не
перевищують 20 %.
• Утворення аршііодидів. Заміщення діазогрупи на атом Іоду не потребує
застосування купрум (І) галогеніду або порошку міді; реакція відбувається за
простого змішування солі діазонію з калій іодидом:
25°С
АгМ2Х + КІ-=^ АгІ + Л2 + КХ
Препаративно це одна з найпростіших реакцій діазосполук:
с А-іЬгсґї^ с6н5-і
78%
• Реакція Шимана. Заміщення діазогрупи на атом Флуору виконують трохи
інакше: до розчину солі діазонію додають тетрафлуороборидну кислоту (НВР4).
Осаджується діазоній борофлуорид АгН2ВР4, який відфільтровують, проми-
вають і висушують. Діазоній борофлуориди - досить стійкі сполуки. Під час
нагрівання сухий діазоній борофлуорид розпадається, утворюючи арилфлуорид,
борофлуорид з виділенням азоту:
Аг1Ч2Х-^ АгИ2ВР4-^ АгР + ВР3+^
Виходи аренфлуоридів значно залежать від природи замісника в бензе-
новому кільці. Електронодонорні замісники підвищують вихід кінцевих
продуктів реакції, електроноакцепторні - навпаки, знижують:
776
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
н3с н3с
69%
ГУ-і
соон соон
9%
Вихід реакції також сильно залежить від розчинності солі діазонію.
Наприклад, малорозчинна у воді сіль гексафлуорофосфату РБб" дає вихід тієї ж
о-флуоробензойної кислоти 61 %.
Флуороборати можна використати для синтезу нітропохідних аренів:
Флуороборати легко карбоксилювати карбон (II) оксидом:
СН3СОО)2Р
0,5 ч, 2^С
+ . (СН3СОО)2Р<і 9 9 100°с
Аг-И2ВР4+ СО + СН3СО(Жа—-—-^-Аг-С-ОС-СН3
(АгСО)20 + (СН3СО)20
Сумарний вихід ангідридів становить майже 100 %, і цей метод можна
розглядати як спосіб заміни аміногрупи на карбоксил.
Синтез арилгалогенідів із солей діазонію має низку переваг порівняно з іншими
методами. Арилфлуориди й ариліодиди взагалі не можна одержати прямим
галогенуванням аренів. Арилхлориди й арилброміди можна синтезувати прямим
галогенуванням, проте в тих випадках, коли утворюється суміш о і и-ізомеров, її
буває важко розділити на індивідуальні речовини через близькість температур
кипіння. Сировиною для одержання солей діазонію є, в остаточному підсумку,
мононітросполуки, які зазвичай можна легко одержати у чистому вигляді.
❖ Заміщення на нітрильну групу. Синтез карбонових кислот. Діазогрупа
легко зазнає заміщення на ціаногрупу під час взаємодії солі діазонію з купрум (І)
ціанідом:
Аг^Х-д^АгСК + К2
Для запобігання втратам у вигляді ціанідної кислоти розчин солі діазонію
перед внесенням купрум ціаніду нейтралізують натрій карбонатом. Сьогодні
замість ціанідної кислоти застосовують комплексний іон Купруму:
Розділ ЗО. Ароматичні азо- та діазосполуки
777
С|Г"СНз к[си(сыц ґ^]ГСНз
Н20,5°С
70%
Гідроліз нітрилів дає карбонові кислоти. Отже, синтез нітрилів із солей
діазонію є прекрасним способом перетворення нітросполук у карбонові кислоти:
сн3
сн3
сн3
сн3
сн3
|^ СиСї^ |і
Н20 [Г^Ч
Л неї
\ \ неї 11
ш2
К2С1
но'%
Такий шлях синтезу ароматичних карбонових кислот зазвичай універсальні-
ший, ніж карбоксилювання реактиву Гриньяра або окиснення бічних ланцюгів.
Ми щойно переконалися, що чисті бромопохідні, необхідні для одержання
реактиву Гріньяра, найчастіше синтезують через солі діазонію. Як звичайно,
легше ввести в молекулу нітрогрупу, ніж алкільний ланцюг; крім того, алкіл не
можна перетворити в карбоксильну групу в тому випадку, коли молекула
містить інші групи, чутливі до окиснення.
❖ Заміщення на гідроксил. Синтез фенолів. Солі діазонію реагують із
водою, перетворюючись у феноли:
АгЫ2С1 + Н20
АгОН + Ы2 + НС1
Ця реакція відбувається навіть у крижаному розчині солей діазонію, і вона є
причиною, через яку солі діазонію негайно використовують після їхнього
приготування. За підвищених температур та наявності 40—50% сульфатної
кислоти цей процес може стати головною реакцією солей діазонію, і вихід
фенолів становитиме 50-95%. Це типова реакція нуклеофільного заміщення в
ароматичному ядрі, що відбувається за мономолекулярним механізмом:
+ Повільно . ,
Аг-№Ы ~Аг++К,
Аг^Н^^^Аг-ОН+Н^
❖ Заміщення діазогрупи на атом Гідрогену. Солі діазонію можна
відновлювати, використовуючи чимало відновлювальних агентів. Універсаль-
ним відновником є гіпофосфітна кислота Н3РО2, а також її солі, формальдегід
(параформ), мурашина кислота, етиловий спирт. Сіль діазонію просто зали-
шають на добу за 0-5 °С біля 30-35 % гіпофосфітної кислоти; у цьому разі
виділяється азот, а гіпофосфітна кислота окиснюється у фосфітну:
АгЫ2Х + Н3Р02 + Н20 АгН + Ы2 + Н3Р03 + НХ
778
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Особливо елегантний метод перебігу цієї реакції з використанням гіпо-
фосфітної кислоти як агента під час діазотування: амін розчиняють у
гіпофосфітній кислоті й додають натрій нітрит; сіль діазосполуки реагує в міру її
утворення. Реакція відновлення відбувається за радикальним механізмом, про
що свідчить синтез діарилів. За допомогою цього перетворення можна видаляти
аміно- або нітрогрупи з ароматичного кільця. Останніми роками замість
гіпофосфітної кислоти застосовують натрійборогідрид.
❖ Заміна діазогрупи на алкоксил. Процес проводять нагріванням
діазосполуки зі спиртами:
Ar-N=N + ROH ArOR + N2 + H+
Реакція має обмежене значення, тому що супроводжується заміщенням
діазогрупи на атом Гідрогену:
ArN2X + СДОН АгН + N2 + СН3СНО + НХ
Зі збільшенням розміру алкільної групи спирту вихід арену збільшується, а
етеру фенолу - зменшується.
❖ Реакція Несмеянова. Як відомо, галогеніди арилдіазонію утворюють
стійкі солі з галогенідами Меркурію, Стануму, Стибію, Вісмуту й інших
перехідних металів. У 1928 р. О. Несмеянов виявив, що дія металів-відновників
на солі арилдіазоніїв з комплексними аніонами може слугувати методом синтезу
арильованих важких металів. Як відновники найчастіше застосовують порошко-
подібну мідь, залізо й цинк:
C6H5N2ClHgCl2 + 2Cu -» C6H5HgCl + N2 + 2CuCl
2C6H5N2Cl.HgCl2 + 6Cu -> (C6H5)2Hg + 6CuCl + 2N2
2C6H5N2BF4 + 2T1 -> (C6H5)2T1 + 2N2 + Tl + 2BF3
(C6H5N2Cl)2SnCl4 + Zn -> (C6H5)2SnCl2 + 2N2 + ZnCl2
2C6H5N2CbSbCl3 + Zn -» (C6H5)2SbCl3 + 2N2 + SbCl3 + ZnCl2
3C6H5N2BF4 + 2Bi -»(C6H5)3Bi + 3N2 + 3BF3 + BiF3
Ця група реакцій одержала назву "діазометоду" Несмеянова-Бахмана.
❖ Реакція Гомберга1. Під час дії діазонієвих солей на надлишок арома-
тичної сполуки, що не здатна до азосполучення, ароматичний радикал
діазосполуки зв'язується С-С-зв'язком з ароматичним ядром другого реагенту,
заміщаючи в ньому атом Гідрогену:
1 Гомберг М. (ОотЬе^ М.) (1866-1947) - американський хімік українського походження. Заснов-
ник хімії радикалів. Президент американського хімічного товариства.
Розділ 30. Ароматичні азо- та діазосполуки
779
Аг-І^И СН3СОО- + (^У~Х Аг"^3^Х + К2+ СНзС00Н
40-60%
У цьому випадку можуть синтезуватися о, п- і л*-заміщені біфеніли:
N0.
Цікаво, що наявність електроноакцепторної групи сприяє утворенню
біарилів, а електронодонорних замісників - одержанню азосполук. Метод
Гомберга-Бахмана (1924) тривалий час був єдиним способом синтезу біарилів.
❖ Реакція Мейєрвейна. Ефективними акцепторами радикалів, що утворю-
ються в процесі розпаду арилдіазоній хлоридів, виявилися сполуки з кратними
карбон-карбоновими зв'язками.
Як каталізатори, що зумовлюють розпад діазонієвих солей, найчастіше
використовують купрум (І) та титан (ПІ) хлориди, а як субстрати, що зазнають
арилюванню, - спряжені дієни або алкени, що мають при подвійному зв'язку
нітрильну або карбонільну групу. Реакцію зазвичай проводять у водно-метанольному
або водно-діоксановому середовищі, що забезпечує гомогенність розчину.
Перетворення відбувається за тією ж схемою, що й реакція Зандмейєра, з
тією тільки різницею, що арильний радикал, який утворюється, реагує спочатку
не з сіллю Купруму (II), а з ненасиченою сполукою, наприклад:
АгЙ2С1+СиС1— [АгК2+СиС12 — Аг+СиС12 АіО^^+СиСу — АіО^ОіД
Оскільки реакція відбувається як приєднання арильного радикала до
ненасиченої системи, то її зазвичай називають арилюванням. Нижче наведені
окремі приклади цієї реакції:
СиСІ
Н^ШЧГССН.+и^^
«-ш2с6н4сн=сн-сн=сн2
Діоксан 60о/о
780 Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Діоксан - вода
Метанол - вода
Розділ 31
ПАРОКСИПОХІАНІАРЕНІВ.
ФЕНОЛИ
31.1. Класифікація, структура й номенклатура
31.1.1. Класифікація гідрокси похідних арені в
Фенолами називаються сполуки загальної формули Аг-ОН, де Аг - феніл або
арил. Термін "феніл" уведений у хімію 1837 р. О. Лораном і взятий від
колишньої назви бензену "фен". Річ у тому, що для бензену, уперше виділеного
з конденсату світильного газу, запропоновано ще одну назву "фено" - несуче
світло, що означало джерело виділення. Звідси "фенол" - сполука, що містить
гідроксильну групу в бензені.
Феноли відрізняються від спиртів тим, що гідроксильна група у них зв'язана
безпосередньо з ароматичним кільцем; якщо ж вона перебуває в бічному
ланцюзі, то йтиметься про ароматичні спирти.
Феноли
Ароматичні спирти
0-он
@>-сн2он
Фенол
Бензиловий спирт
За кількістю. гідроксильних функцій феноли поділяють на кілька груп,
наприклад:
Одноатомні феноли
ОН <ґНз СНз он
9"
н3с-сн-сн3 сн3снсн3 НОг
о-Крезол Карвакрол Тимол Пікринова кислота
782
Чирва В.Я., Ярмолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Двохатомні феноли
Трьохатомні феноли
ÖH
Гідрохінон
он
Пірокатехол Резорцинол Гідрохінон
(раніше (раніше
пірокатехін) резорцин)'
Бензен-1,2,3-тріол
(раніше пірогалол)
Бензен-1,3,5-тріол
(раніше флороглюцин)
31.1.2. Будова фенолу
У молекулі фенолу неподілена електронна пара атома Оксигену перебуває в
спряженні з я-електронною хмарою ароматичного ядра. Таке відтягування
електронної густини від атома Оксигену у бік кільця компенсоване більшим
зсувом електронної густини від атома Гідрогену до атома Оксигену. Тому фенол
є сильнішою кислотою, ніж вода (рКа -10,4). Інакше вищу кислотність фенолу
порівняно зі спиртами можна пояснити меншою основністю спряженого з ним
фенолят-аніона порівняно з гідроксид-іоном. Це зменшення основності пов'я-
зане зі зменшенням ефективного негативного заряду на атомі Оксигену через
-М-ефект фенільної групи:
Атом Оксигену в гідроксильній групі має -І-ефект. З іншого боку,
неподілена пара електронів атома Оксигену вступає в /?,л-спряження з я-елек-
тронною хмарою бензенового кільця (+М-ефект, що перевищує -І-ефект). У
підсумку відбувається подача електронної густини в ароматичне кільце й, отже,
полегшується перебіг реакцій електрофільного заміщення. Електронна щільність в
ароматичному кільці збільшується в о- і и-положеннях, які й атакує електрофіл:
ОН
6
—
31.1.3. Номенклатура фенолів
За правилами IUP АС 1993 р. низку тривіальних назв одноатомних фенолів
(фенол, нафтол, фенантрол, антрол) використовують як основу для побудови
назв їхніх похідних, утворених заміщенням будь-яких атомів Гідрогену:
Розділ 31. Пдроксипохідні аренів. Феноли
783
1-Нафтол і його ізомери 2-Фенантрол і його ізомери 9-Антрол і його ізомери
Для іншої групи одно- і двохатомних фенолів збережені тривіальні назви,
які також можна використовувати як родоначальні для створення назв похідних
за гідроксильними групами. Структури й тривіальні назви таких фенолів
наведені вище.
Раніше дозволені тривіальні назви ксиленол, пірогалол і флороглюцин за
новими правилами рекомендовано замінювати за правилами замісної номенклатури.
Інші феноли в замісній номенклатурі розглядають як похідні вуглеводнів з
використанням суфікса -ол. За наявності старшої групи фенольну функцію
позначають префіксом гідрокси-:
Вг^гмн°2
соон
0
4
2-Бромо-6-нітрофенол
З-Хлоробензен-1,2-діол
он
я-Гідроксибензойна
кислота
Основа - фенол. Напрям
нумерації за рівності локан-
тів визначає перший за алфа-
вітом замісник.
Тривіальна назва пірокатехол не може
бути використана як основа, тому як
основу беруть назву вуглеводню -
бензену. Нумерація за принципом
мінімальних локантів головних груп.
Основа - бензойна кисло-
та
ОН
ОН
4-Ам і но-2-м етил-1 -нафтол
2-Метилнафтален-1,4-діол
Основа - 1-нафтол. Початок нумерації - від
-головної групи.
У номенклатурі ШРАС немає тривіальної назви
для такого двохатомного фенолу, тому як основу
використовують назву вуглеводню - нафталену.
31.2. Фізичні властивості
Найпростіші феноли є рідинами або низькоплавкими твердими речовинами.
Через утворення між собою водневих зв'язків вони зазвичай мають високі
температури кипіння. Сам фенол помітно розчинний у воді (9 г на 100 г води),
784
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
імовірно через утворення водневих зв'язків з водою; більшість інших фенолів
практично нерозчинні у воді. Феноли - полярні речовини з дипольним
моментом 1,5-1,6 D,. що зумовлено взаємодією гідроксильної групи з я-
електронами бензенового кільця.
Феноли - безбарвні речовини, якщо тільки вони не містять яких-небудь груп,
що спричиняють появу забарвлення. Однак феноли подібно до ароматичних
амінів легко окиснюються, і багато з них, якщо тільки їх спеціально не очищати,
забарвлені завдяки наявності домішок продуктів окиснення.
Феноли - отруйні речовини, і у випадку потрапляння на шкіру спри-
чинюють опіки; вони є антисептиками.
31.2.1. Спектроскопія фенолів
• УФ-спектроскопія. Максимуми поглинання в УФ-ділянці спектра фенолу
Хтах 210 і 270 нм зміщені у бік довших хвиль (батохромний зсув) порівняно з
бензеном через взаємодію неподілених пар електронів атома Оксигену з я-елек-
тронами арену.
• ІЧ-спектроскопія. В ІЧ-спектрах фенолів характерне поглинання Voh в ділянці
З 600 см'1 для сильно розбавлених розчинів. Інтенсивна смуга Veo фенолів
простежена близько 1 200 см"1, на відміну від спиртів з veo 1 150-1 050 см'1.
• ПМР-спекпіроскопія. У ПМР-спектрах сигнал протона гідроксилу виявляється в
досить широкому діапазоні (4,0-12,0 м.ч.) залежно від природи й концентрації
розчинника, температури й наявності внутрішньо- або міжмолекулярного водневого
зв'язку:
Більшу частину фенолів отримують у промисловості тими ж методами, що й у
лабораторії. Однак є спеціальні методи синтезу цих сполук у промисловості, у тому
числі найважливішого - фенолу. Фенол посідає одне з перших місць за масштабами
промислового виробництва серед синтетичних ароматичних сполук. Головну
кількість фенолу використовують у виробництві фенолоформальдегідних полімерів.
❖ Виділення з кам'яновугільної смоли. Деяку кількість фенолу, а також
крезолів (метилфенолів) виділяють із кам'яновугільної смоли, що утворюється
під час коксування вугілля. Отриману смолу шляхом хімічної обробки й
фракційної перегонки розділяють на низку основних фракцій. Феноли з
а
8 м. ч. 6,68 (а), 7,15 (Ь), 6,82 (с)
31.3, Промислові джерела фенолів
Розділ 31. Пдроксипохідні аренів. Феноли
785
відповідних фракцій виділяють за допомогою обробки їх 12-15 % лугом, з яким
вони утворюють водорозчинні феноляти.
❖ Лужне сплавлення арилсульфонатів. В основі методу є реакція
сплавлення натрій бензенсульфонату з лугом. Процес зазвичай ведуть за
температури 320—350 °С із натрій гідроксидом, хоча ліпші виходи дає дорожчий
калій гідроксид. Вихід фенолу становить близько 60-80 % від узятого для
сульфування бензену. Реакція супроводжується побічними окиснювальними
процесами: поряд з фенолом утворюється резорцинол, бензен-1,3,5-тріол
(флороглюцин) і п,п -дигідроксибіфеніл:
п,п -Дигідроксибіфеніл
У випадку алкілбензенів відбувається часткове окиснення алкільних груп.
Одержання фенолу варто зачислити до реакції нуклеофільного заміщення, у
якій гідроксид-іон відіграє роль нуклеофіла, а сульфіт-іон - групи, що відходить:
На користь такого механізму (приєднання-відщеплення) свідчить результат
одержання фенолу з фенілсульфонату, у якому ОН-група зв'язана тільки з
атомом Карбону 14С, де раніше була сульфогрупа. Цей факт унеможливлює
перебіг реакції через дегідробензен.
❖ Гідроліз хлоробензену. Відповідно до цього методу, хлоробензен уводять
у реакцію з 8 % водним розчином натрій гідроксиду за температури 350 °С.
Процес ведуть в автоклаві за наявності солей Купруму або розчин лугу
пропускають разом із хлоробензеном через систему мідних трубок довжиною
понад 1 км за 300 °С і тиску 250 атм. Подібно до синтезу аніліну із
хлоробензену, ця реакція є реакцією нуклеофільного заміщення, проведеною в
умовах, які зазвичай не використовують у лабораторії:
0*f^ (>ONa -И- <q-OH
Хлоробензен Натрій фенолят Фенол
На підставі дослідів з 1-хлоробензеном 14С з'ясовано, що реакція відбу-
вається через ариновий механізм, тобто відщеплення-приєднання.
Уведення в о- і «-положення електроноакцепторних груп дає змогу
збільшити швидкість заміни атома галогену на гідроксил у реакціях без
каталізатора.
786 Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Методи одержання фенолу шляхом лужного сплавлення бензенсульфокисло-
ти й гідролізу хлоробензену втратили значення й становлять тільки історичний
інтерес.
❖ Куменовий (кумольний) метод. Метод синтезу фенолу, значення якого
останнім часом щоразу зростає, - це синтез на основі кумену (стара назва
кумол). Під час його обробки киснем повітря виникає гідропероксид кумену, що
під дією водного розчину кислоти розпадається на фенол і ацетон:
Метод розроблений у спецтаборі ГУЛАГ 1942 р. латиським хіміком
Р. Удрісом1 під керівництвом П. Сергєєва2. В зарубіжній літературі цей метод
відомий як метод Хока.
Перетворення гідропероксиду кумену у фенол, мабуть, передбачає стадію
перегрупування, оскільки фенільна група зв'язана з атомом Карбону в пероксиді
й атомом Оксигену у фенолі. Ми вже стикалися з 1,2-зсувами, спрямованими до
електронодефіцитних атомів Карбону й Нітрогену. Вивчення реакції засвідчило,
що перегрупування гідропероксиду кумену передбачає стадію 1,2-зсуву до
електронодефіцитного атома Оксигену. Вірогідно, що реакція відбувається через
такі стадії:
Н
нх-с-сн
ООН
нх-с-сн
ОН
Кумен
Фенол
Ацетон
+
(і)
сн?с-о-он +н+
сн3 і
СНз"С-0-ОН2
сн3
Гідропероксид кумену
+
+
(2)
(3)
1 Удріс Р. (1899-1949) - радянський хімік-органік.
2 Сергеев П. (1885-1957) - радянський хімік-органік.
Розділ 31. Гідроксипохідні аренів. Феноли 787
Пероксид І під дією кислоти перетворюється в протонований пероксид
(стадія 1). Він втрачає молекулу води, утворюючи інтермедіат, у якому атом
Оксигену несе лише шість електронів (стадія 2); 1,2-зсув фенільної групи від
атома Карбону до електронодефіцитного атома Оксигену дає карбонієвий іон II
(стадія 3), що реагує з водою, утворюючи гідроксисполуку III (стадія 4).
Гідроксисполука III є напівацеталем, що розпадається в кислому середовищі
(стадія 5), утворюючи фенол та ацетон.
Передбачають, що стадії 2 і 3 відбуваються одночасно, так що мігрувальна
фенільна група полегшує видалення молекули води. Як і раніше, перегрупування
молена розглядати як особливий випадок електрофільної атаки ароматичного кільця,
причому реагентом у цьому випадку буде електронодефіцитний атом Оксигену.
Кожна стадія реакції є вже відомим хімічним процесом: протонування
гідроксисполуки з наступною йонізацією та утворенням електронодефіцитної
частки; 1,2-зсув до електронодефіцитного атома; реакція карбонієвого йона з
водою з утворенням гідроксисполуки; розклад напівацеталю. Одержання фенолу
з кумену - одна з найчудовіших технологій нашого часу. Вона вирізняється тим,
що практично нема відходів, малими енерговитратами й простим апаратурним
оформленням.
31 .4. Одержання фенолів у лабораторії
У лабораторії феноли звичайно одержують одним з наведених нижче
методів.
❖ ГідролЬ солей діазопію. Дія на первинні аміни нітритної кислоти
приводить до утворення солей діазонію, які під час стояння або, ліпше, за
незначного нагрівання в кислому розчині розпадаються до фенолів:
АгЫ2 + Н20 АгОН + Ы2 + Н+
788 Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Приклад:
ї£Н804-
100 С
ж-Хлорофеншд.азон.й „.х^ро,^
гідросульфат г г
❖ Сплав сульфонатів з лугом
А
Приклад:
АгБОзЫа + ИаОН АгОЫа + ЫаН803 Аг-ОН
БО-^а
^1 №ОН.КОН, |
Л 300°С 1
V
сн3
т
сн3
Натрій л-толуолсульфонат л-Крезол
Часто для виділення фенолу з феноляту використовують сульфур (IV)
оксид, що є в реакційному середовищі:
2СбН5(Жа + БОг + Н20 -> 2С6Н5ОН + Ш-ДОз
❖ Гідроліз арилгалогенідів. Цей метод також часто застосовують за умови,
що у вихідній сполуці є сильні електроноакцепторні групи в о- або «-положенні
до галогену. Таким способом одержують у великих кількостях 2,4-динітрофенол
і пікринову кислоту:
Жа ОН ОН
ИаОЬі.
N0
і. 2нш, 2 її іі 2
Н2804 к^>
К02 N0;,
2,4-Динітрофенол Пікринова кислота
❖ Гідроксилювання бензену і його гомологів. Останнім десятиріччям
щораз більшого значення набувають реакції прямого гідроксилювання арома-
тичного ядра бензену й алкілбензенів:
)Н
\
Н202;У205
або 02, Со/АеШ3
Під час окиснення алкілбензенів у відповідні феноли використовують також
гідроген пероксид (ЗО %) і суміш гідроген флуориду з боротрифлуоридом.
Розділ 31. Гідроксипохідні аренів. Феноли
789
Початком, що діє, у цьому випадку є тетрафлуороборат протонованої форми
гідроген пероксиду:
н2о2+нр + вр3—►но-о, вр4
н
Електрофільність реакції підтверджена переважним утворенням о- і п-
ізомерів під час окиснення моноалкілбензенів:
О •■»■•:>■■ І5г£%^ 0-°"
Зі збільшенням об'єму алкільної групи зменшується зміст о- і збільшується
відсоток «-заміщених, що, загалом кажучи, характерно й для інших реакцій
електрофільного заміщення за участю алкілбензенів. У разі збільшення кількості
алкільних груп гідроксилювання гомологів бензену полегшене. Наприклад,
моногідроксилювання мезитилену виконують трифлуоронадоцтовою кислотою
за зниженої температури:
СР3СОООН
❖ Синтез фенолів за допомогою реактиву Гриньяра. Під час взаємодії
триалкілового естеру борної кислоти з реактивом Гриньяра утворюється
фенілдіалкіловий естер, що зазнає гідролізування в кислому середовищі до
фенілборної кислоти. Під дією на неї гідроген пероксиду з виходом 70 %
синтезується фенол:
-6 8+ /ОСЛ -80°С
одмбвг.снзсн.о-в^^.^^ ед-всосд),
2Н,0/Н+ Н,0,
"* -2сЯон ' САВСО^-^СДОН + НзВОз
❖ Виділення з е тер них олій. Деякі феноли та їхні етери ізолюють з
етерних олій різних рослин. Нижче наведені деякі з них:
790 Чирва В.Я., Ярмолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
он он осн3
^Ц^осн3 ^Л^ОСНз
v р
СН^С№СН2 С№СН-СН3 СН=СН-СН3
Евгенол Ізоевгенол Анетол
(гвоздикова олія) (олія мускатного горіха) (олія анісу)
он он сн, Р'ЯР2
СНО нзс СН^-СН=СН2
Ванілін Тимол Сафрол
(олія бобів ванілі) (олія чабрецю й м'яти) (сасафрасова олія)
31.5. Реакції фенолів по гідроксильній групі
Як феноли, так і спирти містять ОН-групу й тому певною мірою подібні один
на одного. Наприклад, їх можна перетворити в етери й естери. Однак за більшістю
властивостей і методами одержання ці типи сполук відрізняються між собою
настільки сильно, що цілком обґрунтовано їх розділяють на два окремі класи.
Гідроксильна група фенолу забезпечує прояв не тільки кислотних
властивостей і, відповідно, можливості перебігу реакцій за цією функцією, а й
активує ароматичне кільце в реакціях електрофільного заміщення в о- і «-
положення. У цих реакціях також дуже важливу роль відіграє кислотність
фенолу, тому що йонізація фенолу приводить до появи аніона СбН50". З огляду
на постійний негативний заряд Оксисен у феноляті є сильнішим донором
електронів, ніж ОН-група в фенолі.
31.5.1. Кислотність. Утворення солей
Реакції гідроксильної групи фенолів мають особливості порівняно з іншими
гідроксиловмісними сполуками. Вони - досить кислі сполуки, і щодо цього
помітно відрізняються від спиртів, які є навіть менш кислими сполуками, ніж
вода:
Сполука С2Н5ОН Н20 С6Н5ОН Н2С03 СН3СООН
рКа 15,9 15,7 10,0 7,0 4,8
Вищу кислотність фенолів порівняно зі спиртами легко побачити з
резонансних структур, у яких чітко простежена стабілізація фенокси-аніонів
завдяки спряженню неподілених електронів атома Оксигену з я-електронною
системою ароматичного ядра:
Розділ 31. Пдроксипохідні аренів. Феноли
791
-/, +Л£ефекти
На відміну від фенолів, у спиртах такої стабілізації алкокси-аніона немає,
тому його можна зобразити єдиною структурою:
С2Н50:Н + Н20
Н30+ С2Н56:
Як випливає з показника кислотності фенолів, під дією водних розчинів лугів
вони перетворюються в солі, а далі водні мінеральні кислоти перетворюють солі
знову у вільні феноли. Феноли і їхні солі протилежні за розчинністю: солі
розчинні у воді й нерозчинні в органічних розчинниках:
АЮН + ОН- АгО- +
Фенол, нерозчинний
у воді;сильніша
кислота, ніж вода
Н20
Фенолят-іон (сіль), Слабша кислота,
розчинний у воді ніж фенол
Більшість фенолів має значення Ка близько 10"10, тобто вони - значно
слабші, ніж карбонові кислоти (Ка~10'5). Тому феноли, на відміну від
карбонових кислот, не розчиняються у водному розчині гідрокарбонату, що
зумовлено їхньою низькою кислотністю. Феноли зручно виділяти із солей,
діючи на них карбонатною кислотою:
со2 + н2о-
:Н2С03 + АЮЫа
АгОН + ЫаНСО,
Сильніша
кислота
Розчинни
й у воді
Слабша кислота,
нерозчинна у воді
Електронодонорні замісники в ядрі фенолів зменшують їхню кислотність, тому
що утруднюють делокалізацію негативного заряду феноксид-іона й цим зменшують
його стійкість. Навпаки, електроноакцепторні замісники підвищують кислотність
фенолів. Особливо сильно позначається введення таких замісників у «-положення.
Наприклад, кислотність «-нітрофенолу збільшується майже на три порядки
порівняно з фенолом, що пов'язано з ефективною резонансною стабілізацією
фенокси-аніона:
Н30+|
Зазначимо, що внесок останньої резонансної структури в стабільність
фенокси-аніона найвагоміший.
792 Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Наявність у молекулі фенолів декількох електроноакцепторних замісників
різко збільшує відтягування електронної густини від атома Гідрогену
гідроксильної групи. Зокрема, 2,4,6-тринітрофенол (пікринова кислота) за
кислотністю наближається до мінеральних кислот. Замісники в о-положенні
часто впливають на кислотні властивості фенолів через стеричний чинник.
❖ Поділ гідроксиловмісних сполук. Кислотність фенолів і розчинність
їхніх солей у воді можна використати як під час аналізу, так і для їхнього
розділення. Нерозчинна у воді сполука, що добре розчиняється у водному
розчині лугу, але не розчиняється у водному розчині гідрокарбонату, повинна
бути сильнішою кислотою, ніж вода, але менш сильною кислотою, ніж
карбонатна кислота; більша частина сполук, кислотність яких є в цьому
інтервалі, належить до фенолів. Феноли можна відокремити від сполук, що не
мають кислих властивостей, завдяки їхній розчинності в основах: метод
відділення від карбонових кислот ґрунтується на нерозчинності фенолів у
розчині гідрокарбонату. Це використовують для поділу сумішей спиртів,
фенолів і карбонових кислот за такою схемою:
Не реагує
Я"ОН Не реагує
.О
ОН
Аг-ОН -
NaHC03
Н20
Реагує
R-ОН, Аг-ОН
NaOH
R-OH
sONa
Реагує
ArONa
Наведений прийом не застосовують, якщо хоча б одна вихідна сполука
розчинна у воді.
❖ Комплекс фенолу з іоном Ферум (III). Феноли здатні утворювати
феноляти не тільки з лужними металами, а й із солями важких металів.
Наприклад, солі Феруму(ІІІ), що їх використовують як реактив на енольний
гідроксил, як це продемонстровано на прикладі кето-енольної таутомерії
ацетооцтового естеру, з фенолом, дають комплексну сіль темно-фіолетового
кольору. Більшість інших фенолів також дає забарвлені солі під час взаємодії з
розчинами ферум (III) хлориду:
с6н5о
С6Н5ОН
2С6Н5ОН+ FeCl3
СбН50
ОСбН5
ОС6Н5
Розділ 31. Гідроксипохідні аренів. Феноли
793
31.5.2. Утворення етерів
❖ Синтез Вільямсона. Феноли перетворюються в етери під час взаємодії з
первинними алкілгалогенідами в лужному розчині; метилові етери можна
одержати реакцією з диметилсульфатом. У лужному розчині фенол існує у
вигляді фенолят-іона, що діє як нуклеофільний реагент, атакуючи галогенід (або
сульфат). Наявність лугу необхідна для утворення фенолят-іона, тому що сам
фенол не здатний брати участь у цій реакції. Річ у тому, що значне спряження
неподіленої електронної пари атома Оксигену з я-електронами бензенового
кільця сильно знижує основність і нуклеофільність фенолу порівняно зі
спиртами:
АЮН АгО—АгОЯ + Х-
О
С6Н5Ша++СН3— 0-8-ОМе ™- С6Н5ОСН3 + СН30803Ыа
0 96%
Зазначимо, що використання в останній реакції другої метальної групи
потребує жорсткіших умов.
Приклади:
^^--ОН + С2Н5І нагріваній ^^^ОС2Н5
Етоксибензен
(фенетол)
н3с-<0-он+ВгСн2-^-ко2^^н3с-^осн2-Оько2
я-Крезол л-Нітробензилбромід я-Нітробензил-л-толіловий етер
Д°2 осн '
°Н+ (СН3)2804 ^¿¡3 |ГД і Ка(СН3)804
о-Нітроанізол
(метил-о-нітрофеніловий етер)
О^ОН + С1СН2СООН ^^ОьОСН.СООЫа ^^ОСН/ЛОН
Феноксіоцгова кислота
Найліпші результати О-алкілування досягають у полярних апротонних
розчинниках, які добре сольватують катіони лужних металів. Якщо ж
розчинники сольватують за допомогою водневих зв'язків атома Оксигену
феноксиду, то відбувається реакція С-алкілування:
794 Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
+ СНзССН^СН-Зг
ацетон
60°С
ОСН2(СН2)3СНз
N0,
80%
^>-ОЫа + Н2С=СН-СН2С1 ■
^Оюсн^сн-сн,
100%
СУОСН2СН=СН2+ О~0Н
49% ^\
СН^СН^СЬ^
■ Деякі етери можна синтезувати взаємодією натрій алкоголятів з
арилгалогенідами, якщо арилгалогеніди особливо активовані, наприклад:
ОСН,
+ ЫаОСН,
2,4-Динітрохлоробензен
ш2
2,4-Динітроанізол
❖ Алкілування діазометапом. Алкілування фенолів за атомом Оксигену
можливе за допомогою діазометану. Цей метод можна використати як якісну
реакцію на кислотні гідроксильні групи. У ході реакції зникає жовте
забарвлення розчину діазометану й виділяється азот:
С6Н5ОН+СН2-№*ї
С6Н5ОСН3+М2
❖. Алкілування епоксидами. Взаємодія фенолів з епоксидами, наприклад,
етиленоксидом, дає фенілові етери поліетиленгліколів:
о-н + н2с—7сн2
/°\
о
осн2сн2он
пН2С СН2 у-»
<осн2-сн2^он
У випадку, якщо Я = октил, синтезовані поліетери використовують як
неіоногенні детергенти типу ОП-7 або ОП-10. Цифра означає кількість
етиленоксидних ланок.
Розділ 31. Гідроксипохідні аренів. Феноли
795
❖ Властивості етерів фенолів. Хоча алкоксигрупи є о-, я-орієнтантами у
реакціях електрофільного заміщення в ароматичному ряді, вони значно менш
активні, ніж ОН-група. Внаслідок цього етери, як звичайно, не вступають у ті
реакції, які потребують підвищеної активності субстрату: реакції азосполучення,
реакцію Кольбе тощо. Подібне розходження в реакційній здатності пояснюють
тим, що, на відміну від фенолу, етер не може йонізуватися, утворюючи
надзвичайно активний фенолят-іон.
■ Унаслідок зниженої реакційної здатності кільця ароматичні етери менш
чутливі до окиснення, ніж феноли:
сн3онГ>сн3^н|- сн3о-^соон
^ з \==/ нагрівання з \ /
■ Розщеплення метилових етерів концентрованою іодидною кислотою є в
основі важливого аналітичного методу - методу Цейзеля, кількісного визна-
чення в органічних сполуках метокси- або етоксигруп. Для цього алкоксіарен
нагрівають із концентрованою іодидною кислотою до 100 °С. Іодометан або
іодоетан, що виникає під час розщеплення етерів, відганяють у спиртовий
розчин арґентум нітрату, а утворений унаслідок гідролізу іодоалкану арґентум
іодид визначають гравіметрично.
■ За наявності кислот Льюїса етери фенолу під час нагрівання здатні до
ізомеризації (М. Курсанов1):
Пи л_П и
с2н5
ос.нс—^ к у-он+сль—<' у-он
31.5.3. Утворення естерів
Феноли зазвичай перетворюють у естери дією хлороангідридів кислот або їхніх
ангідридів. У випадку застосування як ацилювальних агентів хлороангідридів
ароматичних кислот, що мають значно меншу реакційну здатність порівняно з
аліфатичними, естери одержують за методом Шоттена-Баумана, тобто за наявності
лугів або піридину.
Приклади:
Фенол Фенілбензоат
1 Курсанов М. (1874-1921) - російський хімік, учень Марковнікова. Головні наукові праці в галузі
хімії аліциклічних сполук, ментолу та його похідних. Розробив екстракційний метод одержання
скипидару та каніфолі.
796 Чирва В.Я., Ярмолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
сн3
я-Нітрофенол
он
л—\ rr CHr^yso,o
+ CH^j^SO^l^™- 3 w 2
я-Нітрофенілацетат
/=
\ //
о-Бромофенол
Вг
о-Бромофеніл-я-толуенсульфонат
Через низьку нуклеофільність фенолів вони не вступають у реакцію
естерифікації з карбоновими кислотами.
Широко використовувана взаємодія аліфатичних спиртів з органічними
кислотами за наявності сульфатної кислоти з утворенням естерів для фенолів
відбувається вкрай повільно. Це пов'язано зі зменшенням електронної густини
на протонованому фенольному атомі Оксигену.
❖ Властивості естерів фенолу
• Перегрупування Фріса1. Під час нагрівання естерів фенолів з алюміній
хлоридом відбувається міграція ацильної групи з атома Оксигену ОН-групи
фенолу в о- або «-положення кільця з утворенням кетонів:
С2Н5СОС1
О A1CL
CS,
Фенол Фенілпропіонат Етил-о-гідроксифеніл-
кетон,
летить з парою
Етил-я-гідроксифеніл-
кетон,
не летить з парою
Сумарний вихід продуктів реакції - 70-90%.
Міграція ацильної групи під час перегрупування Фріса в о- або «-положення
залежить головно від температури, природи розчинника й каталізатора. Зазначимо,
що за низьких температур утворюються «-ізомери, а за високих - о-ізомери:
)Н
А1С1,
25°С
сосн
з
A1CL
165 С
о=с-сн3
я-Ізомер
о-Ізомер
1 Фріс К. (1875-1962) - німецький хімік. Головні праці присвячені вивченню біциклічних гете-
роциклів. Відкрив метод синтезу фенолокетонів (перегрупування Фріса).
Розділ 31. Пдроксипохідні аренів. Феноли
797
■ Відомо, що алкілування й ацилювання фенолів у ядро за Фріделем-
Крафтсом відбувається повільно й має низькі виходи. Річ у тому, що за наявності
кислот Льюїса феноли утворюють солі типу С6Н5ОАІСІ2. Тому ароматичні о- і п-
гідроксикетони одержують за допомогою перегрупування Фріса.
Те, що причиною повільного перебігу реакції є утворення зазначеної солі,
свідчить той факт, що ацилювання етерів фенолів за Фріделем-Крафтсом
відбувається легко, регіоселективно й зависокими виходами:
RO-/~\ + R'-C° RO-^V-C-R'
\=/ сі °°с \=/ о
85-95%
• Перегрупування Кляйзена. Алілфеніловий етер під час нагрівання за
наявності калій карбонату кількісно перетворюється в оалілфенол:
<Г>о-сн2-сн=сн2^- (>он
\=/ 2 2 200 °С л \
снгсн=сн2
❖ Аралові естери фосфатних кислот. Під час обробки фенолів фосфор
оксохлоридом і фосфор (III) хлоридом утворюються арилові естери фосфорних
кислот:
РОС13
—^0=Р(ОАг)3
Аг-ОН
Р(ОАг)3
❖ Галогенування. Заміщення гідроксильної групи фенолів на атом
галогену відбувається тільки під дією фосфор (V) галогенідів за дуже жорстких
умов. Це пояснюють тим, що атом Оксигену у фенолах, по суті, двозв'язаний з
ароматичною системою.
❖ Відновлення гідроксильної групи. Під час перегонки фенолів із цинко-
вим пилом гідроксильна група заміщується на атом Гідрогену. Цим методом
користуються для визначення структури складних фенолів:
С6Н5ОН + Ъъ С6Н6 + гпО
31.6. Реакції фенолів за ароматичним ядром
Гідроксигрупа фенолу й атом Оксигену феноляту є дуже сильними
орієнтантами заміщення в о-, и-положення в електрофільних реакціях арома-
тичного ряду. Етерна група фенолу - менш потужний активатор порівняно зі
згаданими сполуками.
798 Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Механізм активування пов'язаний з утворенням як проміжних сполук,
найімовірніше, оксонієвих іонів (типу І і II), у яких кожний з атомів, за винятком
атомів Гідрогену, має повний октет електронів; вони утворюються набагато
швидше, ніж карбонієвий іон із бензену:
ОН ОН О 9
НУ 111 н ¥іу
Атака фенолят-іона дає ще стійкіший інтермедіат - ненасичений кетон (типу
III і IV).
У випадку фенолів, як і амінів, необхідно вживати спеціальних заходів для
того, щоб запобігти полізаміщенню й окисненню. Крім того, феноли піддаються ще
низці інших реакцій, які також охоплюють стадію електрофільного заміщення й
можливі лише завдяки надзвичайно високій активності ароматичного кільця.
31.6.1. Галогенування
Висока реакційна здатність фенолів приводить до того, що навіть під час
обробки їх бромною водою без каталізатора відбувається заміщення всіх атомів
Гідрогену в о- і «-положеннях щодо ОН-групи, і навіть можливе заміщення
інших груп, наприклад:
Вг Вг
Фенол 2,4,6-Трибромофенол о-Крезол 2,4-Дибромо-6-метилфенол
+ ЗНВг + Н2804
803Н Вг
л-Гідроксибензен- 2,4,6-Трибромофенол
сульфокислота
Реакція одержання 2,4,6-трибромофенолу настільки чутлива, що дає змогу
виявити фенол у концентрації 10"5М у водному розчині.
■ Якщо галогенування проводити з дотриманням еквімолярних співвідношень
реагентів у розчиннику низької полярності або взагалі без розчинника, то реакцію
можна спрямувати у бік моногалогенування. Наприклад, під час бррмування
Розділ 31. Гідроксипохідні аренів. Феноли
799
фенолу в карбон тетрахлорнді або карбон дисульфіді утворюється переважно п-
ізомер з невеликою домішкою обромофенолу:
)Н ОН
Вг2, С82, 0°С
Вг
/ї-Бромофенол
Обробка фенолу діоксандибромідом веде винятково до «-ізомеру. Через
значні розміри бромувальний агент не здатний підійти до о-положення фенолу.
Синтез о-похідної виконують за схемою, що передбачає шсо-заміщення
сульфогруп на атом Гідрогену:
2Ц2$Ол
100 °с
80,Н
80^Н Віч
3 Н^пара 4
130 С
+ 2Н2804
803Н
803Н
■ Високополярний розчинник - вода - може прискорювати галогенування
шляхом іонізації фенолу, що дає реакційноздатний фенолят-іон, або стабілі-
зуючи полярний активований комплекс на стадії утворення проміжного
карбонієвого йона. Наприклад, вище зазначено, що фенол зазнає бромування у
воді до 2,4,6-трибромофенолу, а за надлишку брому синтезується ненасичений
циклічний кетон:
ОН ОН 9
Вг Вг Вг
Утворення кетону такої будови підтверджене такою реакцією:
О О
Вг НО ОН О
2,6-Дибромо-я-бензохінон
Бромування фенолу надлишком брому використовують в аналітичній хімії
для кількісного визначення фенолу шляхом йодометричного титрування натрій
800 Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
тіосульфатом іоду, що утворюється після додавання до реакційної суміші калій
іодиду:
О ОН
І2 + 2 Иа^Оз 2ШІ + Иа^Оз
■ Для введення іоду у феноли процес необхідно проводити за наявності
окиснювачів типу гідроген пероксиду або нітритної кислоти.
■ Флуоропохідні найпростіше синтезувати через солі діазонію за реакцією
Піймана.
❖ Застосування галоген о фенолів. Деякі галогенофеноли застосовують як
антисептики. Масляними розчинами пентахлорофенолу просочують деревину для
запобігання її руйнуванню грибками й термітами. Натрієву сіль пентахлорофенолу
використовують для обробки промислових вод для попередження росту слизових
грибів і водоростей. Вісмутову сіль трибромофенолу застосовують як антисептик
під назвою ксероформ.
Галогенофеноли є також проміжними продуктами під час одержання
багатоатомних фенолів.
Гербіцидами називають речовини, використовувані для боротьби з
бур'янам. Вони здатні або повністю знищувати всю рослинність (суцільна дія),
або вибірково - окремі класи рослин, наприклад, тільки дводольні чи однодольні
рослини або певні родини рослин (наприклад, хрестоцвіті, складноцвіті тощо).
Часто та ж сама речовина, залежно від концентрації й норм витрат, може діяти
як гербіцид суцільної або вибіркової дії. У кожній групі розрізняють гербіциди
контактної дії (вражають листя й стебла в разі нанесення на них препарату),
системні гербіциди (здатні переміщатися по судинній системі рослин) і
гербіциди кореневої дії, що вражають насіння, яке проростає, або корінь рослин.
Як кореневі гербіциди суцільної дії використовують хлоропікрин,
карбондисульфід, дихлоропропан, метилбромід, арилдиметилсечовину й ін.
Прикладом контактних гербіцидів суцільної дії є нафтові й кам'яновугільні
масла, що мають багато ароматичних вуглеводнів, пентахлорофенол, натрій
пентахлорофенолят, натрій хлорат та ін. Гербіциди суцільної дії використовують
для знищення рослинності на залізничному полотні, спортивних спорудженнях,
доріжках парків.
Дводольні бур'яни, що засмічують посіви злаків, які належать до
однодольних рослин, часто знищують 2,4-дихлорофеноксиоцтовою і 2-метил-4-
хлорофеноксиоцтовою кислотами, як у вільному вигляді, так і у вигляді солей
або естерів. Однодольні бур'яни в посівах просапних культур знищують натрій
Розділ 31. Гідроксипохідні аренів. Феноли
801
трихлороацетатом, натрій дихлоропропіонатом, 3-хлорофенілізопропілкарбама-
том (хлор-ІФК) і ін.
Гербіцид, що широко застосовують, 2,4-Д (2,4-дихлорофеноксиоцтова
кислота) одержують із натрій 2,4-дихлорофеноляту й натрієвої солі хлороцтової
кислоти: "~
9Н
~ СІ
+ С1СН2СООКа - С1^^-ОСН2СООМа
Гербіцид 2,4-Д
Гербіциди дають змогу механізувати таку трудомістку роботу, як
просапування посівів, є засобом підвищення врожайності сільськогосподарських
культур.
31.6.2. Нітрування
❖ Синтез нітрофенолів. Для одержання мононітрофенолів доводиться
нітрувати феноли на холоді розведеною нітратною кислотою (20 %), найліпше -
змішуванням водного розчину селітри з сульфатною кислотою, щоб уникнути
наявності нітроген оксидів. Утворюється суміш о- і «-нітрофенолів:
он он
Розв. НЖ)3 ^Л^->Ю2
'О +
N0.
о-Нітрофенол л-Нітрофенол
з якої сннітрофенол видаляють відгонкою з водяною парою. Леткість о-нітро-
фенолу зумовлена утворенням міцного внутрішньомолекулярного водневого
зв'язку:
-н.
«-Ізомер виділяють за допомогою кристалізації.
л*-Ізомер доводиться синтезувати окружним шляхом, наприклад, з лі-нітро-
аніліну через лі-нітрофенілдіазоній.
2,4-Динітрофенол найпростіше одержувати за допомогою гідролізу 2,4-ди-
нітрохлоробензену.
Пікринову кислоту синтезують у промисловому масштабі, обробляючи
міцною нітрувальною сумішшю 2,4-фенолдисульфокислоту, яку одержують
802 Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
сульфуванням фенолу, без виділення її із сульфураційної маси. У цьому разі
нітрування зазнає вільне шосте положення, а сульфогрупи заміщаються на
нітрогрупи - явище, яке спостерігають й у деяких інщих випадках. Наявність у
фенолі сульфогруп захищає його від окиснення нітроген оксидами. Пікринову
кислоту можна одержати й прямо з бензену, якщо нітрувати його за наявності
солі Меркурію, яка каталізує окиснення проміжного лі-динітробензену, до 2,4-
динітрофенолу. Сіль Меркурію далі перетворюється в пікринову кислоту.
❖ Властивості нітрофенолів. Уведення нітрогрупи в о- і л-положення
значно підвищує константу дисоціації фенолу. Узгоджену дію на кільце обох
орієнтантів (ОН і N02), що перебувають в о- і и-положеннях один до одного,
можна описати такою схемою:
(^л^ 0"%
Л//\Г\ і +
н^о
N
и
О
Менший ефект має л*-нітрогрупа. 2,4-Динітрофенол у шість разів сильніший
від оцтової кислоти, а 2,4,6-тринітрофенол наближається за силою до
мінеральних кислот (табл. 31.1).
Таблиця 31.1
Кислотність нітрофенолів
Сполука
ка
Сполука
Ка
Фенол
о-Нітрофенол
я-Нітрофенол
1,3-Ю"10
5,9-10"8
7,08-Ю'9
лі-Нітрофенол
2,4-Динїтрофенол
2,4,6-Тринітрофенол
4,8-10*
1-Ю"4
1,610і
Цікавою є поведінка нітросполук у лужному середовищі. Луг зв'язує протон
і сприяє зсуву електронів у тенденції, позначеній стрілками. Тому натрій
нітрофеноляти, по суті, не мають будови фенолятів: заряд аніона зосереджений у
них на нітрогрупі, а не на атомі Оксигену колишньої гідроксильної групи, і ядро
має хіноїдне розташування подвійних зв'язків:
Н
6 Х==/ о
У твердому вигляді червоний
Жовтий
Таке переміщення зв'язків веде до різкої зміни ділянки поглинання світла й
зумовлює властивості нітрофенолів (а також динітрофенолів) як індикаторів
концентрації водневих іонів.
Незначно перехід протона від гідроксилу до нітрогрупи, що супроводжується
хіноїдним переміщенням зв'язків, відбувається й у самих нітрофенолах, особливо
Розділ 31. Гідроксипохідні аренів. Феноли
803
в розчинниках, здатних, подібно до води, зв'язувати протон, тобто йдеться про
таутомерію:
оНітрофенол досить різко відрізняється від свого «-ізомеру й від самого
фенолу тим, що він не асоційований у вуглеводневих розчинах; «-нітрофенол, як
і всі гідроксильні сполуки, асоційований завдяки водневим зв'язкам між
гідроксилом однієї молекули й нітрогрупою іншої:
У о-нітрофенолі водневий зв'язок відбувається всередині молекули. Така
особлива будова о-нітрофенолу зумовлює і його ліпшу розчинність, низьку
температуру плавлення, леткість із водяною парою, оскільки молекули хімічно
не зв'язані одна з одною, а також забарвлення.
Пікринова кислота є сильною. З безліччю навіть слабких основ (таких як
алкалоїди) вона легко утворює кристалічні солі, які широко використовують для
виділення, характеристики й аналізу основ. Цим справа, однак, не обмежена, тому
що і з конденсованими ароматичними вуглеводнями (нафтален, антрацен)
пікринова кислота також утворює кристалічні "молекулярні" сполуки (Ю. Фріцше)
у співвідношенні 1:1. Ця властивість уже не пов'язана з кислотністю пікринової
кислоти, оскільки подібні речовини утворюють усі тринітросполуки, наприклад,
тринітробензен з нафталеном. У таких молекулярних сполуках одна молекула є
донором електронів, інша - акцептором.
Раніше пікринову кислоту застосовували як жовтий барвник. Як усі
ароматичні тринітросполуки, її уперше під час російсько-японської війни 1904 р.
використали як бризантну вибухову речовину під назвою меленіт, медіт,
шимоза. Згодом пікринову кислоту витиснув тротил, тому що її кислотні
властивості створюють низку незручностей - корозія металу, утворення солей,
крайня чутливість до удару. Як вибухову речовину інколи застосовують сіль
пікринової кислоти - амоній пікрат.
Напрям реакції сульфування цілком залежить від температури проведення
процесу. Наприклад, за кімнатної температури утворюється о-ізомер, а за
температури понад 100 °С - «-ізомер. Під час нагрівання о-ізомер повністю
перегруповується в «-ізомер. З подібним ходом і умовами перебігу реакції ми
31.6.3. Сульфування
804 Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
вже стикалися у випадку 1,2 і 1,4-електрофільного приєднання до дієнів.
Справді, за низьких температур швидкість утворення оізомеру (кінетичний
фактор) більша, ніж «-ізомеру внаслідок меншого значення енергії активації
для першої сполуки. За вищої температури й збільшення тривалості процесу
сульфування переважає «-ізомер завдяки меншому значенню його вільної
ентальпії. Інакше кажучи, за умов реакції сульфування о-ізомер у часі зазнає
десульфування до вихідних сполук - фенолу й сульфатної кислоти, і сульфу-
вання за термодинамічними умовами відбувається в «-положення:
І) + Н2804-
20°С
803Н
100°С
І] + Н2804
100 с
803Н
31.6.4. Нітрозування. Властивості нітрозосполук
Феноли - один із небагатьох класів сполук, досить активних, щоб вступати в
реакцію з занадто слабким електрофілом, яким є йон нітрозонію N0*.
Нітрозування - реакція строго регіоселективна: заміщення відбувається в «-по-
ложення стосовно ОН-групи:
ОН
ИаШ2, Н2504
он
^СН3 ШШ2,Н2804 ^С"з
N=0
2-Метил-4-нітрозофенол
У хімічних реакціях з нітратною кислотою «-нітрозофеноли перетворюються в
нітрофеноли, а під час взаємодії з гідроксиламіном дають «-хінондіоксими:
N=0
А7-НІТрОЗОфеНОЛ
25%Ш03
Ш2ОН
N0.
N0
31.6.5. Алкілування за Фріделем-Крафтсом
Алкілфеноли можна одержати алкілуванням фенолів за Фріделем-Крафтсом
алкілгалогенідами, спиртами й алкенами, тільки замість кислот Льюїса беруть
флуоридну, фосфатну кислоти або катіоніти:
Розділ 31. Гідроксипохідні аренів. Феноли
805
А Л СН3 г > СН3
но 4=/ + сі-ср-сНз-1^-* но-^У^-сНз
фенол СН3 СН3
п-трет-бутлфеноп
он он
(СН3)2СН^^СН(СН3)2
| + СН3СН(ОН)СН3 Г/
СН(СН3)2
2,4,6-Триізопропілфенол
он он
!і + ф™, ^ <™^г^
2,6-Ди-т/?ет-бутил-4-метилфенол
«-Крезол (іонол)
Як звичайно, така реакція дає суміші о- і и-алкілфенолів, у яких за тривалого
впливу високої температури відбуваються процеси ізомеризації алкільних груп,
їхня міграція, дезалкілування й диспропорціювання. Хоча все-таки є приклади
вибіркового алкілування:
)Н ОН
0-/яре/я-бутилфенол, 99%
У промисловості алкілування ведуть за наявності алюміній феноляту для
одержання антиоксидантів:
ОН ОН
(СНз)зС^^.С(СН3)з
| + (СН3)2С=СН2
юо°с
2,6-ди-/и/?е/и-бутилфенол, 75%
Алкілфеноли також використовують під час синтезі поверхнево-активних
речовин і барвників.
31.6.6. Ацилювання за Фріделем-Крафтсом
Ароматичні кетони можна синтезувати з незадовільним виходом прямим
анулюванням фенолів, тому їх частіше одержують у дві стадії за допомогою
перегрупування Фріса (див. 31.5). У деяких випадках використовують модифікацію
806
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
реакції Фріделя-Крафтса - комплекс карбонової кислоти й боротрифлуориду.
Ацильну групу вводять винятково в «-положення бензенового кільця:
он он
+ CH3(CH2)4COOHBF3-
ОН
ОН
0=С(СН2)4СН3
2,4-Дигідроксифенілгексан-1-он, 95%
■ Для одержання ароматичних фенолокетонів також застосовують метод
Геша. Вихідними речовинами слугують багатоатомні феноли:
Н,С-С=Ш-НС1 НОС=0
31.6.7. Карбоксилювання. Реакція Кольбе
Нагрівання натрій феноляту з карбон (IV) оксидом приводить до заміщення
атома Гідрогену в кільці на карбоксильну групу. Ця реакція відома як реакція
Кольбе. її найважливіше застосування - перетворення натрій феноляту в о
гідроксибензойну кислоту, яку називають саліциловою:
ОИа о 9Н
^.COONa ^^СООН
¿5+ 120-140 °С, 4-7 атм.
6
Натрій саліцилат Саліцилова кислота
Хоча в цьому разі утворюється також деяка кількість и-гідроксибензойної
кислоти, ці два ізомери легко розділити перегонкою з парою, причому леткішим
буде о-ізомер.
Здається ймовірним, що первісна атака карбон (IV) оксидом спрямована на
атом Оксигену феноляту, а не в ароматичне кільце. У будь-якому разі кінцевий
продукт утворюється внаслідок електрофільної атаки електронодефіцитного
атома Карбону бензенового кільця.
З використанням як вихідної сполуки калій феноляту вибірково одержують
гс-гідроксибензойну кислоту:
ОК ОН
5 атм, 220 °С
COOK
Розділ 31. Гідроксипохідні аренів. Феноли
807
31.6.8. Синтез фенолоальдегідів. Реакція
Реймерал-Тіммана2
Обробка фенолу надлишком хлороформу й водним лугом дає змогу ввести
альдегідну групу в ароматичне кільце, як звичайно, в о-положення до ОН-групи:
NaOH (воду,.)
СНС13, 70°С
(Унс,]
о" он
^А^СНО н+, ^Д^СНО
Спочатку утворюється заміщений бензиліденхлорид, що зазнає гідролізування,
оскільки реакцію проводять у лужному середовищі.
Реакція Реймера-Тіммана передбачає стадію електрофільного заміщення в
дуже активному кільці фенолят-іона. Електрофільним реагентом є дихлорометилен
:ССЬ, який генерують із хлороформу дією лугу. Хоча дихлорометилен електрично
нейтральний, однак він містить атом Карбону, що несе лише секстет електронів і
тому є сильним електрофілом:
НО- + СНС13 -Н20 + :ССі7 -С1"+:СС12
+ :СС1
СНС12
Ми вже розглядали дихлорометилен як частинку, що приєднується до
подвійного карбон-карбонового зв'язку. У тих реакціях так само, як і в
розглянутій, припускали, що дихлорометилен утворюється з хлороформу під
дією сильної основи. Правильність припущення про існування цієї частинки
підтверджена насамперед тим фактом, що дихлорометилен дуже добре пояснює
характер продуктів деяких реакцій: зокрема, він приєднується до алкенів точно
так само, як у випадку реагенту, що безумовно є метиленом (:СН2).
Утворення дихлорометилену в наведених вище реакціях підтверджене
численними даними, отриманими переважно в працях Дж. Хайна.
31.6.9. Взаємодія фенолу з карбонільними
сполуками
❖ Фенолоформольдегідні смоли. Найважливіша реакція - гідроксимети-
лювання фенолів формальдегідом - відбувається за наявності як кислот, так і лугів.
1 Реймер К. (Reimer K.L.) (1856-1921) - німецький хімік. Головні наукові праці стосуються орга-
нічної хімії. Працював у фірмі "Ванілін".
2 Тімман Ф. (Tiemann F.) (1848-1899) - німецький хімік-органік. Головні праці присвячені хімії
терпенів.
808
Чирва В.Я., Ярмолюк CM., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Каталітична дія кислот полягає в протонуванні формальдегіду з утворенням
карбокатіону, здатного заміщати атоми Гідрогену в о- або «-положенні фенолу, і
веде до синтезу фенолоспиртів:
Н.5+ҐЧ5- , Н + Н\ +
,С=0 + Н+ /С-ОН С=ОН
Н Н Н
ОН
2 І J +2СН2ОН -т
сн2он
сн2он
Каталітична дія лугу полягає в утворенні фенолят-аніона, який, маючи
високу нуклеофільну здатність, атакує електронодефіцитний атом Карбону
формальдегіду:
ОН
+ ОН"
СН2ОН
СН2ОН
Під час нагрівання надлишку фенолу з формаліном за наявності сульфатної
кислоти відбувається бурхлива реакція і утворюється розчинний у спиртах,
ацетоні, естерах і розчинах лугів полімер лінійної будови - "новолак".
За лужної конденсації фенолу з надлишком формаліну спочатку утворюється
легкоплавкий, порівняно низькомолекулярний полімер "резол", подібно до
новолаку, розчинний в органічних розчинниках. Обидва продукти належать до так
званих термореактивних полімерів: під час нагрівання відбувається подальша
конденсація вільних гідроксиметиленових груп з утворенням метиленових
містків, і полімер набуває сітчастої структури. Отриманий "резитол" нерозчинний
в органічних розчинниках, проте зберігає деяку пластичність. Під час нагрівання
до 150 °С конденсація відбувається далі, і одержують хімічно дуже стійкий,
неплавкий і нерозчинний полімер "резит", який можна нагрівати до температури
300 °С. Такі три стадії процесу конденсації поєднані назвою "бакелітизація" за
іменем американського хіміка Л. Бакеланда1.
Більшу частина фенолу й крезолів використовують для одержання
фенолоформальдегідних полімерів. Хімічний зміст процесів, що відбувається,
відображає така можлива схема:
1 Бакеланд Л. (Ваекеїаікі Ь.Н.) (1863-1944) - американський хімік. Головні праці присвячені хімії і
технології полімерів. Винахідник фотографічного паперу.
Розділ 31. Гідроксипохідні аренів. Феноли
809
НО
^Ц/СНзОН рн он
НО-^УснрН СНрН
н\ ?н
снг^у-он
он он
'С^^Ц.СНз^к/СНз-
и у
сн,
/=1 '
он
СНз
Отже, відбувається поступове "зшивання" метиленовими групами щораз
більшої кількості молекул фенолу в хаотично побудовані макромолекули резолу,
резитолу й, нарешті, резиту. Хімічну стійкість резиту пояснюють не стільки тим,
що значні кількості активних о- і «-положень фенолу заміщені метиленовими
групами, скільки тим, що внаслідок повної нерозчинності бакеліту реагенти
можуть діяти тільки на поверхні полімеру.
Як звичайно, резол перед наступною стадією конденсації змішують із
наповнювачем (мінеральним - типу азбесту, органічним - типу деревини,
лігніну або целюлози) або просочують ним деревину, волокнисті матеріали й
потім піддають подальшій бакелітизації.
❖ Конденсацією фенолу з ацетоном одержують "бісфенол А", який
широко використовують для синтезу поліконденсаційних полімерів, наприклад,
полікарбонату, епоксидних смол тощо:
ОН
СНЧ
-с-он
І
СН,
с=о
-но
О~он
но-
сн,
І 3
с
І
сн
ОН
Бісфенол А
Застосування ацетону для синтезу бісфенолу А, а потім і полімерів на його
основі, дало змогу свого часу утилізувати ацетон, який одержують у порів-
няльних кількостях з фенолом за куменовим методом.
810 Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
31.6.10. Інші реакції фенолу
❖ Реакції відновлення. Відновленням фенолу в промисловості одержують
циклогексанол, який, відповідно, слугує вихідною сполукою для синтезу
адипінової кислоти. На її основі виробляють капрон (перлону) і найлон (аніду).
❖ Реакції окиснення. З огляду на те, що фенол легко зазнає окиснення,
його використовують у синтезі и-бензохінону шляхом окиснення калій
біхроматом у кислому середовищі:
ОН
К2Сг207>
Н2804
О
Заміна калій біхромату на гідроген пероксид дає змогу синтезувати піро-
катехол:
О
)Н ОН
^ ^6гон
31.7. Двохатомні феноли
Ізомерні двохатомні феноли пірокатехол, резорцинол і гідрохінон - добре
розчинні у воді речовини, тверді, позбавлені запаху.
31.7.1. Пірокатехол
❖ Методи синтезу. Спочатку пірокатехол виділили як продукт декарбокси-
лювання під час нагрівання 3,4-дигідроксибензойної (протокатехової) кислоти, що
поширена в рослинах:
Протокатехова кислота Пірокатехол
Крім того, пірокатехол можна одержати сухою перегонкою лігніну,
гідролізом о-дихлоробензену або о-хлорофенолу перегрітою парою, а також
лужним сплавленням о-гідроксибензенсульфокислоти. Синтез пірокатехолу
дією гідроген пероксиду на фенол описано вище.
У промисловості пірокатехол виробляють шляхом деметилювання його
монометилового етеру - гваяколу, нагріваючи з алюміній хлоридом або із
саліциловим альдегідом за такою схемою:
Розділ 31. Гідроксипохідні аренів. Феноли
811
,сно _ он
Н202,№ОН
нсоон ^
ОН ^ ОН
❖ Хімічні властивості. Пірокатехол - сильний відновник, і, окиснюючись
гетеролітично, наприклад, за наявності йонів Арґентуму, він перетворюється в о-
бензохінон:
сс-"- а>
о-Бензохінон
Пірокатехол використовують у виробництві адреналіну і як відновник у
фотографії.
❖ Похідні пірокатехолу
• Гваякол. Із етерів двохатомних фенолів назвемо монометиловий етер
пірокатехолу — гваякол, що трапляється в дьогті, особливо буковому, і який
застосовують у медицині. Сьогодні його одержують із о-метоксіаніліну (о-ані-
зидину):
ОСН3 ОСН3+ пснз
НН2 неї + №n0, їД^^Н С1" |Ру0Н
Анізидин Гваякол
Гваякол споріднений із важливими запашними речовинами, тому його
застосовують у парфумерії. У фармацевтичній промисловості є синтоном для
виробництва фтивазиду та папаверину.
Уведенням у гваякол тим або іншим способом альдегідної групи в л-по-
ложення до гідроксилу одержують ванілін. Таке формілювання раніше
виконували дією метилформаніліду за наявності фосфор оксохлориду:
ІА С„>< 1 1 +с6н5кнсн3
"^ОСНз сбн5' н онс^^осн
• Ванілін - запашна речовина ванілі. У сучасній промисловості його
одержують окисненням лігніну. Використовують в харчовій, парфумерній і
фармацевтичній промисловостях.
• Вератрол (диметиловий етер пірокатехолу) синтезують за реакцією
812
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Жа ?снз
А^ОСН3
+ 2 С6Н5802ОСН3 [[ Т ' + 2С6Н5803Иа
З1.7.2. Резорцинол
❖ Методи одержання. Резорцинол одержують у техніці, сплавляючи з
лугом натрій л*-бензендисульфонат:
803Ш ^.ОН
+ 2КаОН -|| } + 1Ш£Ог
803№ ОН
Резорцинол
Цікаво, що о- і и-бензендисульфокислоти, а також о і и-хлоробензенсульфо-
кислоти під час сплавляння з лугом за жорстких умов (понад 300 °С) теж
утворюють резорцинол. Можливо, що за цих умов відбувається відщеплення
ИаН80з з утворенням похідної дегідробензену, а потім - приєднання води або
лугу:
80^а ^^/ЗОЛа ^ 0>Іа
+ №ОН
80,На^иС0№-й53;(Я0№
-з1"" ~а"" ~ "ОИа
❖ Хімічні властивості. Резорцинол стійкіший до окиснення, ніж його
ізомери. Кислотні властивості виражені сильніше, ніж у фенолу. Уже в момент
виділення водню (амальгама Натрію й вода) він відновлюється в
дигідрорезорцинол, що перебуває в таутомерній рівновазі з 1,3-циклогександіоном:
НО^^ОН НО^^ОН О^х^О
+2Н— и ху
И02 Резорцинол ще легше, ніж фенол, сприймає
ііО^^ґОН різноманітні електрофільні атаки, оскільки обидві його
ДХТГІ гідроксильні групи мають узгоджену орієнтацію. Тому
2 2 резорцинол легко зазнає галогенування, сульфування,
Стифнінова кислота нітрування, нітрозування, вступає в реакції азоспо лучення.
Одне з головних його застосувань - синтез азобарвників, у якому він слугує
азоскладовою:
Під час вичерпного нітрування резорцинолу виникає тринітрорезорцинол,
або стифнінова кислота, яка багато в чому нагадує пікринову кислоту.
Розділ 31. Гідроксипохідні аренів. Феноли
813
Для карбоксилювання резорцинолу за Кольбе досить нагріти його в розчині
натрій гідрокарбонату. Сполуку, яку отримують, називають 2,4-дигідрокси-
бензойною, або резорциновою, кислотою:
>н он
[со2] і неї
0'-С~ОКа „ соон
Резорцинова кислота
Відповідний резорциловий альдегід можна синтезувати за реакцією Реймера-
Тімана, а також дією на резорцинол ціанідної кислоти й гідроген хлориду.
31.7.3. Гідрохінон
❖ Методи синтезу. Гідрохінон одержують відновленням и-бензохінону:
0=^^)=0 + 2Н -НО^^МЭН
Гідрохінон
и-Бензохінон легко синтезують окисненням фенолу або аніліну хром(УІ)
оксидом або хромовою сумішшю:
к2Сг2о7 о
Н2804
ОН О
Цікаво, що бензохінони, які утворюються під час спонтанного окиснення в
процесі зберігання фенолу, є причиною їхнього забарвлення.
❖ Хімічні властивості. Як і пірокатехол, гідрохінон - сильний відновник,
що під час окиснення утворює и-бензохінон:
Гідрохінон застосовують як фотографічний проявник, що відновлює
арґентум бромід до металу, а також широко використовують як антиоксидант.
Під час окиснення гідрохінону може утворюватися хінгідрон — комплексна
сполука з перенесенням заряду (КПЗ):
814
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
о—н • • • О
Хінгідрон використовують як електрод для визначення рН розчинів.
31,8. Багатоатомні феноли
❖ Пірогалол. Бензен-1,2,3-тріол також називають пірогалолом, тому що
його отримують піролізом (декарбоксилюванням) галової (3,4,5-тригідрокси-
бензойної) кислоти. Саме таким шляхом його уперше отримав шведський хімік
К. Шеєле 1786 р.:
НО НО
н°""^3~соон —" н°~^3 +с°2
НО НО
Галова кислота Пірогалол
Галову кислоту виділяють із продуктів гідролізу дубильних речовин типу
таніну.
Пірогалол у лужних розчинах легко зазнає окиснення навіть повітрям, тому
такі розчини використовують для поглинання кисню. У фотографії пірогалол
застососують я проявник, а також під час синтезування адреналіну й
приготування барвників.
❖ Флороглюцин. Симетричний ізомер пірогалолу - бензен-1,3,5-тріол, або
флороглюцин, у вигляді похідних значно поширений у рослинному світі. Багато
з подібних сполук одержують через флороглюциновий альдегід, відповідно,
синтезований із флороглюцину за Реймером-Тіманом або дією ціанідної
кислоти й гідроген хлориду.
Сьогодні флороглюцин одержують гідролізом симетричного бензен-1,3,5-
триаміну, який готують відновленням 1,3,5-тринітробензену:
ОН
[Н].
НгО,/
II \ +змн4+
о2>ґ ^ Ж)2 Н2^^^"Ш2 НО'^^ЮН
Флороглюцин
За властивостями флороглюцин подібний до резорцинолу. Він часто реагує
в десмотропній карбонільній формі, і схильність вступати в реакції, властиві
Розділ 31. Гідроксипохідні аренів. Феноли
815
кетону, у нього виражена ще сильніше, ніж у резорцинолу. Однак виділити
десмотропні форми флороглюцину у вільному стані не вдається. Як і фенол,
флороглюцин реагує, наприклад, з діазометаном і фенілізоціанатом; у цьому разі
утворюються опохідні:
осошсд
С6Н5№ЮСО
6а а5
3 С6Н5ИСО
осошс6н5 но
осн,
з сн2кг2
он н3со
осн,
У формі кетону він вступає в реакцію з гідроксиламіном, утворюючи
триоксим:
О ШН
О
з Ш2ОН
О НОК
ОН
Під час алкілування флороглюцину метиліодидом утворюються С-метильні
похідні, і кінцевим продуктом реакції є С-гексаметилфлороглюцин, який,
будучи Р-трикетоном, легко розпадається на диметилмалонову кислоту й
тетраметилацетон. Таке С-алкілування найлегше зрозуміти, якщо припустити,
що в цьому випадку флороглюцин вступає в реакцію у вигляді реакційно здатної
метиленової сполуки:
о - 9
± —у ^—і н II и 1 І і-
СН3І, ИаОН " 3
О
о
НзСИСІ
н3с сн3
З КОН (СНз)гСН СН(СН3)2Тетраметилкетон
~ ноосч соон
Диметилмалонова
СН кислота
н3с
Флороглюцин застосовують в аналітичній хімії для визначення пентоз.
Метод ґрунтується на тому, що фурфурол, який утворюється з пентоз під час
кип'ятіння їх з хлоридною кислотою, дає з флороглюцином нерозчинний осад.
Крім того, флороглюцин застосовують для відкриття речовин деревини або
лігніну, з якими він за наявності хлоридної кислоти дає вишнево-червоне
забарвлення.
31.9. Ароматичні спирти
❖ Бензиловий спирт. Найпростіший ароматичний первинний спирт -
бензиловий С6Н5СН2ОН - можна отримати відновленням бензальдегіду, гідро-
лізом бензилхлориду:
816
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
С6Н5СН=0 С6Н5СН2ОН
сбн5сн2сі + н2о КаНС°3 сбн5сн2он + неї
з
або реакцією Гриньяра:
С6Н5!У^Вг + СН20 С6Н5СН2С^Вг С6Н5СН2ОН
Доречне застосування реакції Канніццаро:
сбн5 с\ + Н"СЧ с6н5сн2он + н-с'
н н ок
Загальним методом, який застосовують у лабораторії для синтезу
бензилових спиртів, є відновлення естерів бензойної кислоти натрієм у спирті:
с6н5-с-ос2н5 ^ С6Н5СН2ОН + С2Н5ОН
о
■ Бензиловий спирт за функціональною поведінкою цілком подібний до
аліфатичних первинних спиртів. Досить суттєвою його рисою є легка віддача а-
гідрогенного атома з парою електронів (гідрид-аніона); сам він цьому разі зазнає
окиснення до бензальдегіду:
С^-С^ІОН + А+ С6Н5СНО + АН + Н+
[ні""
де А+ - акцептор гідрид-аніона, наприклад, (С6Н5)3С+.
Крім того, так само як від бензилхлориду відщеплюють йон Хлору, так і з
бензилового спирту легко видаляють кислотами гідроксил, залишаючи відносно
стійкий бензил-катіон, що вступає в подальші перетворення.
■ Бензилацетат має свіжий квітковий запах і є в складі етерних олій
жасмину, туберози й гіацинта, чим зумовлене його застосування в парфумерії.
❖ Бета-фенілетиловий спирт (ФЕС), гідроксил якого більше віддалений
від фенільного ядра, цілком подібний до аліфатичних спиртів. Готують його в
промисловості з бензену й оксирану (оксиду етилену) за варіантом реакції
Фріделя-Крафтса:
С6Нб + ^Г^Щі С6Н5СН2СН2ОН
О
У лабораторії його одержують через реактив Гриньяра:
о
С6Н5М^г ч-Н^-СН, — С6іуСН2СН20}^г-^Сби5СМ2СН2ОЯ+ М^ОН)Вг
Розділ 31. Гідроксипохідні аренів. Феноли
817
Зазначимо, що спирти з ОН-групою в Р-положенні не можна синтезувати
омиленням відповідних галагенопохідних, оскільки спирт, що утворюється,
легко зазнає дегідратації:
6 5 2 2 -КаС1,Н20 6 5 2
Стирен
■ ФЕС застосовують у парфумерії як стабілізатор запаху за назвою штучна
трояндова олія.
❖ Вторинні й третинні ароматичні спирти одержують реакцією
Гриньяра - взаємодією фенілмагнійброміду з альдегідами й кетонами, а
вторинні, крім того, відновленням арилкетонів.
Ароматичні спирти як у вільному вигляді, так і у вигляді естерів містяться в
етерних оліях багатьох рослин, їх застосовують у парфумерній і лакофарбовій
промисловостях.
Розділ 32
АРОМАТИЧНІ
ОКСОСПОЛУКИ
У цьому розділі головну увагу приділено альдегідам і кетонам, карбонільна
група яких безпосередньо зв'язана з ароматичним ядром, оскільки саме вони мають
специфічні властивості й їх синтезують особливими методами. Типовим
представником є бензальдегід. Альдегіди, подібні до фенілоцгового СбН5СН2СНО
або які мають віддалену від бензенового ядра альдегідну групу, за способами
синтезу й реакціями цілком подібні до аліфатичних альдегідів, тому їх ми не
розглядатимемо. Ненасичені альдегіди, такі, наприклад, як коричний (цинамоновий)
СбН5-СН=СН-СНО, також суттєво не відрізняються від аліфатичних альдегідів зі
спряженою системою л>зв'язків типу кротонового альдегіду.
Бензальдегід - сполука, давно відома як олія гіркого мигдалю. Його
виділяють під час гідролізу глікозиду амігдалину, що міститься в гіркому
мигдалі, а також у кісточках вишень, абрикосів тощо. Глікозид розпадається під
час гідролізу на дисахарид генціобіозу й ціангідрин бензальдегіду, що легко
регенерує сам альдегід і ціанідну кислоту:
>н+
но
н
он
сбн5-с+нш
н
Розділ 32. Ароматичні оксосполуки
819
32.1. Класифікація й номенклатура ароматичних
оксосполук
32.1.1. Класифікація
Альдегіди
Кетони
аліфатично-
ароматичні
ароматичні
®— ОСТ0
Бензальдегід Нафтален-2-карбальдегід
(Р-нафтальдегід)
Ацетофенон
метилфенілкетон
Бензофенон
дифенілкетон
32.1.2. Номенклатура IIIРАС
❖ Ароматичні альдегіди. У номенклатуру ШРАС уведено два способи
побудови назв ароматичних альдегідів. За першим назву дають на підставі
ароматичного вуглеводню з додаванням суфікса -карбальдегід. Наприклад, нафта-
лен-2-карбальдегід. За другим назву альдегіду утворюють з основи тривіальної
назви карбонової кислоти, до якої додають суфікс -альдегід. Наприклад, бензойна
кислота => бензальдегід, нафтойна кислота => нафтальдегід.
Для позначення альдегідної групи в бічному ланцюзі або за наявності
старшої групи застосовують префікс форміл-.
Широко використовують історично сформовані тривіальні назви альдегідів,
утворені від відповідних карбонових кислот, наприклад: коричний (цинамо-
новий) альдегід, саліциловий альдегід, ганусовий альдегід тощо.
❖ Ароматичні й аліфатично-ароматичні кетони. Аліфатично-арома-
тичні кетони можна називати за правилами замісної номенклатури як заміщені
аліфатичні кетони:
о=сн-^^-соон ^^-С-СН2СН2СН3
О
4-Формілбензойна кислота 1-Фенілбутан-1-он (замісна номенклатура),
Головна функція - карбоксильна група. пропілфенілкетон (радикально-
Основа - бензойна кислота. функціональна
номенклатура)
Для групи кетонів дозволено використання тривіальних назв, однак вони не
можуть слугувати основою для побудови назв похідних:
820 Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Ацетофен Пропіофено Бензофен Бензил Халкон
он н он
■ За радикально-функціональною номенклатурою назва кетонів складається
з перерахування за абеткою назв вуглеводневих замісників (для симетричних
кетонів використовують числовий префікс ди-) і слова кетон. Приклади таких
назв наведені курсивом вище.
32.2. Фізичні властивості
Ароматичні оксосполуки - висококиплячі рідини або кристалічні речовини.
Багато з них має приємний запах. Оскільки карбонільна група сильно поляри-
зована, то для ароматичних сполук характерна наявність значних дипольних
моментів - 2,7-3,3 В.
32.2.1. Спектроскопія ароматичних оксосполук
• Уф-спекпіроскопія. Максимуми смуг поглинання ароматичних оксосполук
зміщені в довгохвильову ділянку (батохромний зсув) порівняно з аренами.
Наприклад, в уф-спектрі бензальдегіду є інтенсивні смуги з А,тах 244 і -280
(я—>7і*-перехід) і 328 нм (и—>тс*-перехід). Для ацетофенону ці смуги мають
максимум поглинання за 240, 278 і 319 нм, відповідно.
• Іч-спектроскопія. В іч-спектрах ароматичних альдегідів є інтенсивні смуги
поглинання валентних коливань ус=0 в інтервалі 1 700 см"1. За наявності
внутрішньомолекулярного водневого зв'язку ця смуга зміщається, наприклад, у
саліцилового альдегіду до 1 666 см"1.
• Валентні коливання ус=0 змінюються в разі переходу від алкіл-ароматичних
кетонів до чисто ароматичних з 1 690 до 1 665 см"1.
• ПМР-спектроскопія. Сигнал протона альдегідної групи перебуває в слабко-
му полі ~10 м.ч. Хімічні зсуви протонів типових ароматичних оксосполук
наведені нижче:
Нс—^-СН=0 Нс-^^-С-СНз
Нь На Нь на
5 м.ч. 7,83 (а), 7,49 (Ь), 7,56 (с)
6 м.ч. 7,89 (а), 7,41 (Ь), 7,56 (с)
Розділ 32. Ароматичні оксосполуки
821
32.3. Синтез ароматичних альдегідів і кетонів
32.3.1. Реакціїперефункціоналізації
До цієї групи методів одержання оксосполук ароматичного ряду належать
реакції, у яких відбувається перетворення різних функцій, що перебувають у
бічному ланцюзі ароматичного ядра, у карбонільну функцію. Граничним
випадком можна вважати перетворення алкільної групи в оксогрупу.
❖ Гідроліз а,а-дигалогенопохідних. Синтез бензальдегіду в промисловості
донедавна зводився до гідролізу бензиліденхлориду:
едснс^-^- сдено + 2 неї
Для одержання бензиліденхлориду толуен хлорують за опромінення, потім з
продуктів реакції відділяють фракцію, що відповідає бензиліденхлориду, гідро-
лізують водяною парою за 100 °С та наявності порошкового заліза. Вихід
бензальдегіду —90 %.
У лабораторних умовах бензиліденхлорид гідролізують за наявності калій
карбонату в атмосфері С02.
❖ Синтези на основі бензилхлориду. Із фракції, що відповідає бензилхло-
риду, можна одержати альдегід, діючи на неї, наприклад, водним розчином
плюмбум (II) або купрум (II) нітрату чи калій хромату в гексаметилфосфотриаміді
за наявності краун-етерів або йонообмінних смол, що містять хроматну кислоту:
2С6Н5СН2С1 + РЬ(Ш3)2 -> 2С6Н5СНО + РЬС12 + ЫО + Н02 + Н20
■ Важливим загальним методом перетворення групи -СН2С1 в альдегідну
слугує метод Соммле\ що полягає в кип'ятінні бензилхлориду з водним
розчином уротропіну:
С*Н<СН,С1
C6H5-CH2-NH2 + 6 СН20 + 2 NH3 + NH4C1
СН20 Н+ + C6H5CH2-NH2
C6H5CH2NH2—C6H5CH2-N=CH2 C6H5CH2NH=CH2 1—-
QH,CH,-NH-CH, + QH,-CH=NH, —^ C6H5-CHO + NH4+
н2о
Ароматичні альдегіди одержують з виходом 50-80%. Вихідні бензилгало-
геніди можна отримати в чистому вигляді під час дії на толуен N-бромо- або АГ-хло-
росукциніміду. -
1 Соммле М. (Боттеїеі М.) (1877-1952) - французький хімік. Головні праці присвячені ароматич-
ним вуглеводням.
822
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
❖ Окиснення гомологів бензену. Можливе й пряме окиснення толуену в
бензальдегід і загалом метальних груп, зв'язаних з ароматичним ядром, в
альдегідні. У лабораторних умовах це можна виконати за допомогою реакції
Етара дією хроміл хлориду:
З С6Н5СН3 + 4 СЮ2С12 З С6Н5СНО + 4 Сг(ОН)С12 + Н20
Цю реакцію можна використати для окиснення метальної групи в аліфатичних
сполуках. У промисловості окиснення виконують хром (VI) оксидом у розчині
оцтового ангідриду або манган пероксидом із сульфатною кислотою за дотримання
деяких застережень. Подальшому окисненню альдегідів до карбонових кислот
перешкоджає утворення ацилалю, стійкого до дії окисників:
сЮз + (сн3со)2р ^усн(0С0СН) н2зо4 , /тусш
\=( 3 Н2804 \=( 3/2 Н20 + С2Н5ОН \=<
Вг Вг Вг
Останніми роками застосовують пряме окиснення гомологів бензену киснем
над ванадій (V) оксидом або за наявності солей Манґану (II):
о <Р
сбн5-сн3^сбн5-сч + н2о
25 н
СбН5СН2СН, °* 130°С. С6Н5-С-СН3
Мп(ОСОСН3)2 0
❖ Окиснення ароматичних спиртів. Серед цих методів певне значення
має окиснення ароматичних спиртів:
с6н5сн2он -2- с6н5-сч +н20
н 2
сбн5-сн-сн3 с.Нз-с;-сн3 + н2о
он о
❖ Відновлення хпороангідридів кислот. Реакція Розенмунда. Цей метод
набуває щораз ширшого застосування. Наприклад, на паладієвих і нікелевих
каталізаторах, отруєних сіркою, щоб відновлення не відбувалося до спиртів,
хлороангідриди ароматичних кислот перетворюються в альдегіди:
С6Н5-С-С1 -23— СбН5-СНО
6
Розділ 32. Ароматичні оксосполуки
823
32.3.2. Уведення оксогрупи в ароматичне кільце
Не менш важливі синтетичні методи, що полягають у введенні альдегідної
групи на місце атома Гідрогену ароматичного ядра.
❖ Метод Гаттермшш-Коха. Під дією на бензен або загалом на арома-
тичний вуглеводень карбон (II) оксиду і гідроген хлориду за наявності алюміній
і купрум (І) хлоридів утворюються ароматичні альдегіди:
Си,СІ,
СО + НС1
А1С13
сі-с(
н
СбН6 + [С1-СН0] —'* с6н5сно + неї
Для введення альдегідної групи у феноли та їхні етери використовують
варіант реакції Гатгермана, у якому замість карбон (II) оксиду застосовують
ціанідну кислоту, а замість алюміній хлориду - цинк хлорид:
Н
НСИ + НСІ ► с=лн
снз°^Э + %н^сн3о-{^сн^н сн3о^О-сно
❖ Формілування за Фріделем-Крафтсом. Ароматичні альдегіди одержують
також дією на арени несиметричного дихлорометилового етеру за наявності кислот
Льюїса:
Аг-Н + С12СНОСН3 Аг-СНСЮСНз -^2- Аг-С^
Н
❖ Реакція Вільсмейєра1. Ароматичні феноли й аміни формілують диметил-
формамідом за наявності фосфор оксохлориду:
РОСІ,
Аг-Н + (СН3)2К-СНО - Аг-СНО + (СН3)21МН
❖ Реакція Реймера-Тіммана. У розділі 31 детально розглянуто метод
уведення формільної групи в молекулу фенолу:
)Н ОН
^А^СНО
|| І + СНС13 + ЗКаОН И + ЗЫаС1 + 2Н20
❖ Реакція Гриньяра. Зрозуміло, альдегідну групу можна ввести в бензе-
нове ядро за допомогою реакції Гриньяра. Як електрофільний компонент у
цьому разі можна використати форміати або ортоформіати:
1 Вільсмейєр А. (УіЬтеіег А.) (1894—1962) - німецький хімік, досліджував механізми реакцій.
Відкрив реакцію, в якій бере участь імінієвий іон, так званий реагент Вільсмейєра.
824
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
НСОСЖ^ р тт ртт/^
C6H5MgBr— и ок
^^адНмж Селено
Н
❖ Реакція Фріделя-Крафтса. Кетони з карбонілом біля ароматичного
ядра зручно синтезувати реакцією Фріделя-Крафтса. Ацилювальними реагента-
ми слугують хлороангідриди та ангідриди карбонових кислот:
а1г1
С6Н6 + ЯСОСІ С6Н5С(Ж + неї
А1С1
С6Н6 + КСОООСЯ ^ С6Н5СОЯ + КСООН Я = А1к,Аг
Алюміній хлорид у цій реакції доводиться брати в еквівалентній хлоро-
ангідриду або ангідриду кількості, оскільки він повністю зв'язується в комплекс
і є зв'язаний з кетоном після його утворення.
С1 -і+ На підставі УФ-спектрів, термохімічних досліджень
Й.-С І 1 д™ ПР° молекулярну масу припускають таку
- структуру обговорюваного комплексу.
[А1С14] Природно, що зв'язок з атомом алюмінію під-
силює позитивний заряд на карбонільному атомі й
ц-С' і робить його сприятливішим до еле^сгрофільної атаки.
СІ _| Ароматичне ядро, пасивоване наявністю м-
орієнтанта, не вступає в цю реакцію.
Тому реакцією Фріделя-Крафтса не вдається ввести в бензенове ядро другу
ацильну групу. Таким шляхом не можна ацилювати також нітробензен,
бензенсульфокислоту тощо.
До реакцій ацилювання належить і синтез кетону Міхлера, що застосовують
у виробництві барвників. У цьому випадку ацилювальним агентом є дихлоро-
ангідрид карбонатної кислоти (фосген):
2(сн3)2х^О>+сосі2^Ь (сн3)2к^0^^^ужсн3)2
❖ Реакція Геша. У випадку більш нуклеофільного, ніж бензен, арома-
тичного ядра (феноли, їхні етери) для синтезу кетонів застосовують реакцію
Геша-ція нітрилу й гідроген хлориду (варіантреакціїГаттермана):
О. /СІ
р СІ
❖ Реакція Гриньяра й інші реакції з металоорганічними сполуками.
Ароматичні нітрили - зручні синтони для одержання кетонів взаємодією з
реактивом Гриньяра:
Розділ 32. Ароматичні оксосполуки
825
К = А1к, Аг
С=КМ§Вг
• £ + ВгМ§ОН + >Щ3
■ Кетони одержують часто з високими виходами в разі взаємодії
хлороангідридів ароматичних карбонових кислот з алкілцинковими або
алкілмагнієвими солями:
■ У лабораторії кетони іноді синтезують з кадмійорганічних сполук, які
одержують взаємодією кадмій хлориду з реактивом Гриньяра:
2СН31\^Вг + СсіСЬ Сс1(СНз)2 + 2К^ВгС1
Діалкіл- або діарилкадмій, що утворився, взаємодіє з хлороангідридами
кислот з утворенням кетонів:
❖ Піроліз солей карбонових кислот. Серед інших методів треба назвати
одержання оксосполук шляхом сухої перегонки суміші кальцієвих солей
ароматичної й аліфатичної кислот. Утворення змішаних алкіл-ароматичних
кетонів під час пропускання суміші парів ароматичної й аліфатичної карбонових
кислот над нагрітим приблизно до 450 °С торій оксидом можна розглядати як
видозміну цього методу, оскільки як проміжні продукти в цій реакції
утворюються торієві солі застосовуваних кислот.
Бензальдегід та інші альдегіди, у яких альдегідна група зв'язана з ароматичним
ядром, багато в чому подібні на аліфатичні альдегіди. Наприклад, вони вступають у
реакції нуклеофільного приєднання, альдольної і кротонової конденсації,
відновлюються в первинні спирти, окиснюються в карбонові кислоти.
Карбонільна група належить до л*-орієнтантів середньої сили, дезактивує
ароматичне ядро й направляє замісника, який вступає в реакцію, головно в м-
положення.
Проте взаємний вплив альдегідної групи й ароматичного кільця надає їм
низки специфічних властивостей.
С6Н5СОС1 + СН31^С1
С6Н5СОСН3 + М8С12
- Сс1(СН3)2+2С6Н5.С-С1
О
2С6Н5-С-СН3 + СсіС12
32.4. Властивості ароматичних альдегідів
32.4.1. Реакції окиснення
Для ароматичних альдегідів, як і для аліфатичних, є два типи реакцій
окиснення, що приводять до утворення відповідних кислот: гомолітичне
(ланцюгове) і гетеролітичне (іонне).
826 Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
О
о-он
с6н5-с=о+о2—-сбн5-с,
О Ланка ланцюга
о-он о-он ц
с6нгсч + с6н5сно—-с6н5-сх +С6НГС=0
О О
О-ОН ОН
^б^Г^ + С6Н5СНО *~ 2 С6Н5-СХ Кінцеві продукти
О О
Легкість окиснення альдегідів пояснюють тим, що інтермедіатом на першій
стадії, яка визначає швидкість реакції, є бензоїл-радикал Аг-С=0, стійкіший
порівняно з аліфатичним Я-С=0. Легкість його утворення з бензальдегіду
підтверджена тим, що бензальдегід легко зазнає хлорування за умов радикальної
реакції, даючи бензоїлхлорид, тоді як для аліфатичних альдегідів галогенування
зазнає вуглеводневий ланцюг:
С12 >2С1- С6Н5СН0 +С1 -С6НГС=0 + НС1
О
с,н,—с=о + сі2 —- ед-сГ + С1"
Такий спосіб окиснення ароматичних альдегідів киснем повітря не має
препаративного значення. Замість нього застосовують окиснення надкислотами
або гідроген пероксидом.
❖ Гетеролітичне окиснення. Прикладом іншого типу окиснення може
слугувати гетеролітичне окиснення альдегідів калій перманганатом. Цю реакцію
каталізують іони Гідрогену. Механізм досліджено із застосуванням міченого
калій перманганату КМп1804 і міченого атомом Дейтерію бензальдегіду
СбНбСОО. Виявилося, що швидкості окиснення звичайного й дейтерованого
бензальдегідів досить різні: дейтерований окиснюється в сім разів повільніше
(ізотопний ефект). Це означає, що у стадії, яка визначає швидкість реакції,
беруть участь С-О або, відповідно, С-Н-фрагменти. Оскільки отримана
і
❖ Гемолітичне окиснення. Ароматичні альдегіди зазнають окиснення
киснем повітря протягом декількох годин до карбонових кислот, що не
характерно для аліфатичних альдегідів. Солі перехідних металів, наприклад,
Феруму, Мангану прискорюють процес радикально-ланцюгового окиснення:
Ініціювання
н
СбН5-Сч + Бе3+ ► Бе2* + Н+ +СбН5-С=0
Розділ 32. Ароматичні оксосполуки
827
бензойна кислота містить 50% атомів Оксигену-18 з перманганату, то зрозуміло,
що атоми Оксигену для окиснення бензальдегіду надходять не з води, а з
перманганату. На підставі цих даних моясна зробити висновок про такий
механізм реакцій гетеролітичного окиснення:
Р +ОН
с6н5-с+н+—-с6н5-с
н н
+РН он он
СбН5-С _ С6Н5-£7Н -С6Н5-СХ + НМп'«03
1Wo3 'Ч^з
НМп03 + НМп04 + Н20 2 Н2Мп04
❖ Фотоліз. Під час фотолізу бензальдегіду утворюється вільний радикал
бензоїл СбНг-С=0, стійкий за —200 °С, а за -140 °С лише повільно (хвилини)
приєднує бром, перетворюючись у бензоїлбромід СбН5СОВг. Радикал бензоїл -
помаранчево-червона речовина, дає сигнал ЕПР, приєднується за подвійними
зв'язками.
32.4.2. Реакції заміщення
Низка реакцій заміщення альдегідного атома Оксигену також відбувається
аналогічно до реакцій аліфатичних альдегідів. Сюди належать реакції з гідрази-
ном, семікарбазидом, арилгідразинами.
❖ Реакція з гідроксиламіном приводить до двох стереоізомерних оксимів
бензальдегіду, що відрізняються не тільки за фізичними константами, а й за
реакційною здатністю. Під дією оцтового ангідриду й інших водовідбиральних
засобів яняш-ізомер відщеплює воду й утворює бензонітрил, а сгш-ізомер зазнає
ацетилювання за гідроксильною групою:
СНЛСН3СО)20 ^ 1.нс1 [ґ4] (СН3СО)20 /=\
Цу^-с^" Цчрн Чий ІЯс-н ІУ-0*1
Н Ж-ОН НО"^ +2СН3СООН
а-, або син- або анти- 5
+ СН3СООН бензальдоксим беюальдоксим
(т.пл. 39°С) (т.пл. 130°С)
Зазначимо, що син-аниш-ізомерія, з якою ми вже стикалися на прикладі
азобензену, цілком подібна до і/ис-иірш/с-ізомерії з карбоновим л>зв'язком. Роль
другого замісника при атомі Нітрогену відіграє вакантна пара електронів;
/ирш/с-елімінування (у цьому випадку відщеплення Н і ОН із /яранс-положення)
є загальним правилом.
828
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
❖ Основи Шиффа1. Новою для ароматичних альдегідів порівняно з
аліфатичними альдегідами є реакція конденсації з ароматичними амінами, що
приводить до важливого класу азометанів або анілів:
С6Н5СНО + Н2№С6Н5 С6Н5СН=М-С6Н5
❖ Реакція з амоніаком. З амоніаком ароматичні альдегіди також реагують
по-іншому, ніж альдегіди аліфатичного ряду, й дають не альдегідамоніаки, а
5/с(ариліденаміно)метиларени:
с6н5, РА
зс6н5сно + 2ш3—- нС"%н н
с6н5
Одна з головних особливостей ароматичних альдегідів від аліфатичних
полягає в тому, що для них не відома реакція утворення тримерів.
32.4.3. Реакції конденсації
❖ Бензоїнова конденсація. Для ароматичних альдегідів неможлива ні
альдольна, ні кротонова конденсації, оскільки для цих реакцій необхідна
наявність сусідньої з альдегідною групою метиленової ланки з рухливими
атомами Гідрогену. Проте бензальдегід і загалом ароматичні альдегіди здатні до
іншої - бензоїнової конденсації, що відбувається під дією калій ціаніду. Подібні
реакції в аліфатичному ряді відбуваються тільки під впливом ензимів:
СА-^П+/-СЛ — с6н5-с-с Бензоїн
О О ОН О
Роль ціанід-іона полягає в первинному приєднанні до альдегіду (за типом
приєднання ціанідної кислоти) і відтягуванні електронів зв'язку від атома
Гідрогену альдегідної групи аж до його протонізації й надання йому здатності
переміщатися до атома Оксигену, що має негативний заряд, після чого
утворюється Карбон-карбоновий зв'язок, відбуваються повторна міграція
протона й викид ціанід-іона:
с6н5-9н ^с6н-9-
о он
1 Шифф Г. (Schiff Н.) (1834-^1915) - італійський хімік. Головні праці стосуються різних галузей
органічної хімії. Вперше синтезував тіонілхлорид, запропонував якісну реакцію на альдегіди з
фуксинсульфатною кислотою. Спільно з С. Канніццаро заснував журнал "Газетта хіміка Італіана".
Розділ 32. Ароматичні оксосполуки
829
ф 9
С6Н5-С:<^С-С6Н5^С6Н
ОН Н
ОНО:-
СИН
<&он 9 9Н
^сдм-с-ед, ^с6н5-с-с-с6н5
Ч^н _с>г н
Розглянемо деякі факти, що підтверджують запропонований механізм
реакції. Міграція протона відбувається внутрішньомолекулярно, оскільки
перебіг реакції у важкій воді не приводить до включення дейтеромітки в
бензоїн. Така міграція не характерна для аліфатичних альдегідів. У випадку
ароматичних сполук проміжний аніон зазнає стабілізування за участю
бензенового кільця. Це припущення підтверджує той факт, що ароматичні
альдегіди із сильними електроноакцепторними й донорними замісниками в
бензоїнову конденсацію не вступають. Реакцію відкрили Ю. Лібіх і Ф. Велер
1834 р., а її механізм з'ясував А. Лепуорт1 1903 р.
Будова бензоїну випливає з його окиснення в 1,2-дикетон - бензил.
Під дією лугу бензил зазнає внутрішньомолекулярного перегрупування в
бензилову кислоту:
Уважають, що це перегрупування ініційоване атакою гідроксил-аніона
карбонільної групи з утворенням аніона, що перегруповується завдяки 1,2-
арильному зсуву.
❖ Конденсація Кляйзена-Шмідта. Ароматичні альдегіди здатні вступати
в реакцію конденсації з аліфатичними альдегідами за наявності водних лугів з
утворенням а,Р-ненасичених альдегідів. Процес відбувається за тих же умов, що
й кротонова конденсація. Тому реакцію Кляйзена-Шмідта варто трактувати як
окремий випадок альдольно-кротонової конденсації:
,с-сн
)' ОН
чтт [О]
Бензилова кислота
1 Лепуорт А. (Lapworth А.І.) (1872-1941) - шотландський хімік, професор Манчестерського
університету. Був одним з засновників фізичної органічної хімії. Має іменну реакцію.
830 Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
С6Н5СНО + СН3СНО —- с6н5-сн-сн2сно С6Н5-СН=СН-СНО
ОН Коричний альдегід
Дуже легко за наявності розведених лугів відбувається кротонова
конденсація бензальдегіду з ацетоном та іншими аліфатичними кетонами.
Залежно від співвідношення реагентів одержують бензальацетон або дибне-
зальацетон. Обидва ненасичені кетони - жовті кристалічні речовини:
С6Н5СНО + СН3ССН3 С6Н5-СН=СН-ССН3
О О
2 С6Н5СНО + СН3ССН3 С6Н5-СН=СН-С-СН=СН-С6Н5
о о
❖ Конденсація Пер кін а. Взаємодія альдегіду, ангідриду кислоти та її
натрієвої солі, що приводить до утворення ненасичених кислот типу коричної
(цинамонової), є різновидом альдольно-кротонової конденсації. У цьому процесі
натрій ацетат відіграє роль основи, і реакція починається як нуклеофільна атака
аніон ангідриду (І) на карбонільний атом бензальдегіду:
гн-г° СН-С3 СНгС°
С"!Ср + СН,СОШа [ р ВОЄН. Ц ' Р И
СН-С -сн3соон СЩС, С6Н5-9:СН2-Ск-сн3соона
0 Na+ 0
Р Р
СНз< CHÍ< НО
► Н О _ Н 0-^
ед-c-CHjq -н2о сбн5-с=сн-<
он о о
С6Н5-СН=СН-СООН + СН3СООН
Корична кислота
■ Окремий випадок реакції Перкіна - синтез кумарину, що має запах
свіжоскошеної трави:
ХНО
^^он w ^-о^^о
Кумарин
■ Корична (цинамонова) кислота у вільному вигляді або переважно у
вигляді естерів трапляється в етерних оліях і смолах, у перуанському й
толуанському бальзамах, а також у листях Erythroxylon Coca. Завдяки наявності
подвійного зв'язку корична кислота існує у вигляді двох геометричних ізомерів:
Розділ 32. Ароматичні оксосполуки
831
н>^с6н5 н>^с6н5
н соон ноос н
і/ноКорична кислота /я/?а//с-Корична кислота
Корична кислота потрібна для промисловості. її бензиловий, метиловий і
етиловий естери застосовують як запашні речовини; також її використовують для
синтезу ß-бромостирену СбН5-СН=СН~Вг (запах гіацинту) і фенілацетальдегіду, що
відіграє важливу роль у парфумерії. Є кілька зручних способів її синтезу.
■ У промисловості коричну кислоту отримують з бензиліденацетону
СбН5СН=СН-СО-СН3, окиснюючи його гіпохлоритною кислотою.
■ Етиловий естер коричної кислоти одержують за реакцією Кляйзена
шляхом конденсації бензальдегіду з етилацетатом за наявності натрій
алкоголяту:
С6Н5СНО + СН3СООС2Н5 -^тг" С6Н5СН=СН-СООС2Н5
32.4Л. Реакція Канніццаро (диспропорціювання)
Відкрита С. Канніццаро 1853 р. окисно-відновна реакція ароматичних
альдегідів полягає в тому, що альдегіди, які не мають атома Гідрогену при атомі
Карбону в а-положенні, зазнають диспропорціювання під час обробки водно-
спиртовим лугом з утворенням однакових кількостей первинного спирту й
карбонової кислоти:
2С6Н5СН0 |^-с6н5сн2он + с6н5соон
2 ' 85% 85%
Механізм реакції Канніццаро з'ясували через 100 років після її відкриття,
він полягає в такому. На першій стадії відбувається приєднання гідроксид-іона
до карбонільної групи альдегіду:
О ОН
Аг-С? +:ОН" - Аг-С:Н
н • Д
На наступній стадії гідрид-іон переміщається на атом карбонільної групи
другої молекули альдегіду:
4M'
832
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
Потім відбувається швидке перенесення протона від карбонової кислоти до
аніона:
Аг-Сч + -ОСН2С6Н5 з=е С6Н5СН2ОН + Аг-СОСГ
Як відомо, у реакції Канніццаро окиснюється та молекула альдегіду, що
була атакована гідроксид-аніоном. Отже, якщо, крім бензальдегіду, взяти
дешевий формальдегід, то обробка суміші лугом дасть бензиловий спирт і
мурашину кислоту. Це дуже вигідно, тому що відбувається повне відновлення
бензальдегіду, і він не окиснюється до бензойної кислоти як у класичному
варіанті реакції Канніццаро. Такий спосіб відновлення назвали перехресною
реакцією Канніццаро його розробив 1936 р. радянський хімік В. Родіонов.
32.5. Властивості ароматичних кетонів
Алкіл-ароматичні й, особливо, чисто ароматичні кетони мають порівняно з
альдегідами низку специфічних властивостей, тому ми вважали за доречне
розглянути їх окремо. Наприклад, активність карбонільної групи суто ароматичних
кетонів порівняно невисока: воца-не^ вступає в реакції ціангідринового синтезу, не
дає гідросульфітних похідних, а в реакціях заміщення реагує значно повільніше.
У реакціях електрофільного заміщення електрофіл в арилкетонах спрямо-
ваний переважно в л*-положення до карбонільної групи.
Типовим представником алкіл-ароматичних кетонів може слугувати
ацетофенон (метилфенілкетон). Це рідина із запахом квітів черемшини, яку
застосовують у парфумерії. Ацетофенон здатний до реакцій заміщення
карбонільного атома Оксигену. Як і всі кетони, він вступає в кротонову
конденсацію й щодо цього нагадує ацетон.
32.5.1. Реакції конденсації
❖ Кротонова конденсація. Продуктами кротонової конденсації ацетофе-
нону є дипіон і трифенілбензен:
2 сл-й-сн, ^=>- С(„г9=сн-<;-сЛ
ЗС6Н5 я СН3
с6н5ш2-нсі
о сбн5^<^сбн5
❖ Конденсація Кляйзена-Шмідта. Зазначимо, що легкість перебіг
конденсації (кімнатна температура) алкілароматичних кетонів з ароматичними
Розділ 32. Ароматичні оксосполуки
833
альдегідами дала змогу застосовувати як конденсувальний агент основний оксид
Алюмінію:
// wh,
Ці реакції засвідчують рухливість атомів Гідрогену метальних груп.
❖ Конденсація з функціональними похідними кислот в 1,3-дикетони
• Естерна конденсація
Р
Н С І?г~С
КСО(Ж' + На+ 3 с=0 - СК, + КОШ
С6Н5' СбНГ< ^
О
• Конденсація з ангідридами кислот
О ,0
сн3-с ВР сбн5-с;
С6Н5СОСН, + О СН2+СН3СООН
сн3-сч сн3-сч
о о
❖ Реакція Манніха1 полягає в амінометилюванні сполук з рухливим атомом
Гідрогену під дією формальдегіду й вторинних амінів з утворенням так званих
основ Манніха:
С6Н5СОСН3 + СН20 + (СН3)2Ш + НС1 ~С6Н5СОСН2СН2ЙН(СН3)2С1- + Н2С
Можливий механізм:
сн2=о+н+=* 6^он^о^2"== сн2-6н2 ^ с^мц
+
С6Н5-С-СН3= с6н5-с=сн2 од-с-сн^н^^ од-с-сн^н^
6 он Он о
Основи Манніха - проміжні продукти одержання ненасичених кетонів і
альдегідів.
1 Манніх К. (Mannich C.U.F.) (1877-1947) - німецький хімік. Займався фармацевтичною хімією в
Берлінському університеті. Досліджував похідні піперидину, папаверин, а також глікозиди
наперстянки (Digitalis). Є автором іменної реакції. Вперше синтезував аміноспирти й амінокетони.
834
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
32.5.2. Інші реакції арилкетонів
❖ Галогенування ацетофенону за метильною групою. Галогенування
відбувається надзвичайно легко й саме в бічний ланцюг навіть за наявності
таких каталізаторів, як залізо. Такий напрям заміщення зумовлений взаємодією
галогенувального агента з енольною формою карбонільної сполуки:
С6Н5ССН3 + С12 С6Н5ССН2С1 + НС1
О О
Хлороацетофенон
■ Хлороацетофенон, що містить досить рухливий атом галогену, дуже сильно
дратує слизові оболонки. У минулому це бойова сльозогінна речовина. Його можна
отримати також реакцією Фріделя-Крафтса з бензену й хлороангідриду моно-
хлороцтової кислоти СІ-СНг-СО-СІ, що доводить його будову.
❖ Відновлення ацетофенону. Аналогічно до ацетону ацетофенон легко
перетворюється в пінакол під впливом амальгами Алюмінію:
2С6Н5СОСН3^ °бЩ >-сССбН5
6 5 3 тт х Т ^ГН
П^ ОН ОНииз
■ За умов пінаколінового перегрупування фенільний залишок легше
порівняно з метальним зазнає 1,2-гідридного зсуву:
/^6**5
❖ Розщеплення кетонів. Для суто ароматичних кетонів характерне
розщеплення концентрованим лугом за високої температури:
СДООС^ + ЫаОН С^СООЫа +
Реакція, очевидно, відбувається через стадію приєднання гідроксид-аніона до
карбонільного атома й наступного розщеплення С-С-зв'язку, що супроводжується
утворенням феніл-аніона. Про те, що це так, свідчить розщеплення феніл-и-
толілкетону. Утворення феніл-аніона енергетично вигідніше, ніж толіл-аніона,
оскільки метальна група подає електрони в кільце, і тому головним продуктом
реакції буде сіль толу'іїювої кислоти:
Розділ 32. Ароматичні оксосполуки
835
он- ?н
«сн3-сбн4-с-с6н5 + он"= «н3с-с6н4-|-с6н5
о о-
п сн3-с6н4-соон + с6н; иСН3-СбН?+СбН5СООН
»сн3-с6н4соо-+ сбн6^ »сн3-с6н5 + сбн5соо^
>^ V
87% 13%
❖ Оксими кетонів і перегрупування Бекмана1. Більшість кетонів реагує з
гідроксиламіном і похідними гідразину за звичайною схемою:
Аг2С=0 + ИНгОН -> Аг2С=Ж)Н
Кетоксим
З усіх нітрогенних похідних кетонів найцікавіші оксими. Несиметрично
побудовані кетони ЯСОЯ', як чисто ароматичні, так і алкілароматичні,
утворюють з гідроксиламіном по два ізомерні оксими, що відрізняються
фізичними константами й поведінкою в бекманівському перегрупуванні.
Дослідники цієї ізомерії дійшли висновку, що це сии-ан/ии-ізомерія,
аналогічна до цис-транс-ізомерїі олефінів та їхніх похідних:
син-о-Толілфенілкетоксим дн/тш-о-Толілфенілкетоксим
Зазначимо, що сіш-формою вважають ізомер, який має менший вугле-
водневий залишок у і/ш>положенні до ОН-групи. Для оксимів ароматичних
кетонів зручніше використати Е,2-номенклатуру9 де 2 - конфігурація, що
відповідає і/нс-положенню старших груп, а Е - транс-потюженто. Сьогодні син-
або аняш-конфігурацію ізомеру досить легко з'ясувати за його дипольним
моментом, якщо хоча б один замісник кетону має дипольний момент, напрям
якого відомий.
■ Важливе значення має бекманівське перегрупування оксимів, що відбу-
вається під впливом концентрованих мінеральних кислот, кислот Льюїса або
1 Бекман Е. (Beckmann Е.) (1853-1923) - німецький хімік, спеціаліст у галузі фізичної та
органічної хімії. Синтезував металкетили. Розробив метод визначення молекулярних розчинних
сполук на підставі закону Рауля. Винахідник спеціального термометра (термометр Бекмана).
836
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
фосфор (V) оксиду. Напрям перегрупування Бекмана залежить від конфігурації
оксиму. Два ізомерні оксими дають у цьому разі два ізомерні аміди:
С6Н5-£-С6Н4ОСН3-^ 0=С-С6Н4ОСН3
И-ОН ИН-С6Н5
ш/ти-л-Метоксифеніл- л-Метоксйбензанілід
фенілкетоксим
с6н5-с-сбн4осн3-^ сбн5-с=о
но-й МН-СбН4ОСН3
сю/-/і-Метоксифеніл- #-л-Метоксифенілбензамід
фенілкетоксим
■ Передбачуваний механізм перегрупування Бекмана такий. Під дією
кислоти оксимний гідроксил зазнає протонування і перетворюється в групу, що
легко відходить. Синхронно з відщепленням молекули води до атома Нітрогену,
що отримує частковий позитивний заряд, з ш/тгш-положення стосовно
гідроксилу мігрує карбаніонний фрагмент з парою електронів. Карбокатіон, що
утворюється, захоплює із середовища молекулу води:
НОН
РЧГ* .н2о * НО. Ж' О*
Н+ 2 3 4
Відповідно до правила Ельтекова-Ерленмейєра, проміжна сполука "З" з
гідроксилом біля атома Карбону при подвійному зв'язку нестійка й
перегруповується в амід "4".
Промислового застосування перегрупування Бекмана набуло в процесі
виробництва капролактаму й з нього високомолекулярної сполуки - капрону
або "найлону-6". Вихідною сировиною слугує бензен, з якого куменовим
методом одержують фенол. Гідрування фенолу й наступне окиснення приводить
до циклогексанону, який під час взаємодії з гідроксиламіном перетворюється в
оксим, а той унаслідок перегрупування Бекмана дає капролактам, що зазнає
полімеризування під час нагрівання в капрон:
о Г 1ш,он н+ / \ Г \ '
о
Каталізатор
О
•••КН-(СН2)5-СО-МН-(СН2)5-СО-МН-(СН2)5-СО-
Найлон-б
Розділ 32. Ароматичні оксосполуки
837
❖ Утворення металкетшіів. Своєрідна реакція приєднання натрію або
калію відбувається під час їхньої дії на ароматичні кетони. У цьому випадку
утворюються забарвлені парамагнітні (наявність неспарених електронів)
металкетили з тривалентним атомом Карбону:
(С6Н5)2СО + Иа - (С6Н5)2-С-бКа+
Металкетили легко окиснюються в кетони.
Порівняно висока стійкість аніонорадикалів, мабуть, пов'язана зі стабілі-
зувальним впливом на вільнорадикальний центр двох бензенових ядер і
негативно зарядженого атома Оксигену.
32.6. Застосування ароматичних кетонів
■ Бензофенон переводять ближнім ультрафіолетовим випромінюванням у
збуджений стан, тому його використовують у фотохімічних процесах як
індуктор.
■ Деякі похідні бензофенону також збуджуються під дією світла, віддаючи
енергію у вигляді випромінювання - флуоресціюють.
■ 2,2'-Дигідрокси-4,4'-диметоксибензофенон з назвою "увінол" застосовують
для захисту тканин і пластмас від руйнівної дії світла, тому що він "бере удар на
себе", поглинаючи короткохвильове ультрафіолетове випромінювання, яке має
велику енергію, і потім віддає отриману енергію вже у вигляді нешкідливого,
довгохвильового флуоресцентного випромінювання.
■ Гідроксикетони застосовують для захисту Р ОН
пластмас від руйнівної дії ультрафіолетової й корот-
кохвильової видимої частини спектра. Як приклад,
наведемо один із них - ціаносорб: ^ОК
К=Сі8Н37 або СІ2Н25.
Ціаносорб
Розділ 33
АРОМАТИЧНІ КАРБОНОВІ
КИСАОТИ
У цьому розділі ми розглянемо ароматичні карбонові кислоти, у яких
карбоксильна група перебуває безпосередньо біля ароматичного ядра.
33.1. Класифікація й номенклатура
33.1.1. Класифікація
За розташуванням карбоксильних груп
Аренкарбонові кислоти
<^>-соон
Бензойна кислота
Алкілароматичні кислоти
насичені
ненасичені , ч
^^-сн2соон
Фенілоцтова кислота
^р^-соон
Корична (цинамонова) кислота
За кількістю карбоксильних груп
Аренмонокарбонові
кислоти
Арендикарбонові
кислоти
Аренполікарбонові кислоти
СХУ
2-Нафтойна кислота
^еоон
Фталева кислота
^соон
ноос^^хоон
Бензен-1,2,4-трикарбонова кислота
(тримелітова кислота)
За типом замісників
Фенолокислоти
Ароматичні амінокислоти
<Ц>-соон
он
2-Гідроксибензойна кислота
(саліцилова кислота)
^-соон
Антранілова кислота
Розділ 33. Ароматичні карбонові кислоти
839
33.1.2. Будова аренкарбонових кислот
Константи дисоціації ароматичних карбонових кислот зазвичай вищі, ніж у
гомологів оцтової кислоти, проте нижчі, ніж у мурашиної. Це пов'язано з
наявністю я, ;г-спряження бензенового ядра й карбонільної групи, яке впливає на
йонізацііо О-Н-зв'язку:
б"
Природа й положення замісників в ароматичному ядрі впливають на силу
кислоти. Загалом електронодонорні замісники зменшують кислотність, а
електроноакцепторні, навпаки, збільшують.
У цьому разі в карбоксилатному аніоні нема прямого спряження атомів
Оксигену з замісником у бензеновому ядрі. Відомо, що кислота має тим більшу
силу, чим стабільніший аніон. У випадку розміщення замісника в о-положенні
щодо карбоксильної групи відбувається аномальне збільшення сили кислоти, що
поясняють утворенням внутрішньомолекулярного водневого зв'язку, який
сприяє йонізації кислоти. Цей ефект найбільше характерний для о-гідрокси-
карбонових кислот:
ОН саліцилова кислота
33.13. Номенклатура ШРАС
У замісній номенклатурі в назвах циклічних сполук, безпосередньо пов'язаних
з карбоксильною групою, використовують суфікс -карбонова кислота:
HOOG
СООН
Нафтален-2,6-дикарбонова кислота
За правилами IUP АС 1993 р. низку тривіальних назв ароматичних
карбонових кислот використовують як основу для побудови назв їхніх похідних,
які утворені заміщенням будь-яких атомів Гідрогену. Для іншої групи кислот
збережені тривіальні назви, які можна застосовувати також як родоначальні для
створення назв похідних за атомом Гідрогену гідроксильної групи.
840 Чирва В.Я., Ярмолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
У табл. 33.1 наведено тривіальні назви ароматичних карбонових кислот і
утворених з них назв кислотних (Аг-СОО-) і ацильних (Аг-СО-) залишків.
Таблиця 33.1
Тривіальні назви аренкарбонових кислот, їхніх кислотних залишків
і ацильних замісників
Структура
Назви кислот
Тривіальні назви
систематичні
тривіальні
кислотних
залишків
ацильних
замісників
Допустиме необмежене заміщення атомів Гідрогену
Бензенкарбонова
Бензойна
Бензоат
Бензоїл
ОСУ00"
Нафталін-2-
карбонова
2-Нафтойна
2-Нафтат
2-Нафтоїл
ос;
Бензен-1,2-
дикарбонова
Фталева
Фталат
Фталоіл
О-соон
ноосу
Бензен-1,3-
дикарбонова
Ізофталева
Ізофталат
Ізофталоїл.
ноос-<^)-соон
Бензен-1,4-
дикарбонова
Терефталева
Терефталат
Терефталоїл
Допустиме заміщення атома Гідрогену карбоксильної групи
^^соон
транс-Ъ-
Фенілпропенова
Корична
(цинамонова)
Цинамат
Цинамоїл
но соон
Гідроксидифеніл
етанова
Бензилова
Бензилат
Бензилоїл
^-соон
мн2
о-Аміно-
бензенкарбонова
Антранілова
Антранілат
Антранілоїл
Назви функціональних похідних аренкарбонових кислот утворюють за
загальними правилами, які розглянуті в номенклатурі карбонових кислот
аліфатичного ряду. У хімічній літературі також часто трапляються історично
сформовані тривіальні назви кислот.
Розділ 33. Ароматичні карбонові кислоти
841
33.2. Фізичні властивості
Ароматичні карбонові кислоти - безбарвні кристалічні речовини з темпера-
турами плавлення понад 100 °С. Водночас «-заміщені аренкарбонові кислоти
мають значно вищі температури плавлення порівняно з о- і л*-похідними.
Ароматичні кислоти зазвичай погано розчинні у воді й добре розчинні в
полярних органічних розчинниках.
• Уф-спектроскопія. Максимуми смуг поглинання аренкарбонових кислот
зміщені в довгохвильову ділянку (батохромний зсув) порівняно з аренами.
Наприклад, в уф-спектрі бензойної кислоти є інтенсивні смуги з ^тах 230 і
270 нм (я—>я*-перехід).
• Іч-спектроскопія. Іч-спекгри ароматичних карбонових кислот мають
інтенсивну смугу поглинання валентних коливань ус=0 1 720 см"1 (для мономерів)
і 1 690 см"1 (для димерів). Для порівняння: відповідні коливання в аліфатичних
карбонових кислотах спостерігають в інтервалах 1 750-1 765 і 1 710-1 720 см"1.
• ПМР-спектроскопія. Сигнал протона карбоксильної групи перебуває в
слабкому полі -10-13 м.ч. Хімічні зсуви ароматичних протонів бензойної
кислоти наведені нижче:
33.3. Способи одержання аренкарбонових кислот
33.3.1. Реакціїокиснення
❖ Окиснення олкіларенів. Ароматичні карбонові кислоти одержують
унаслідок досить глибокого окиснення органічних сполук. Наприклад, алкіл-
бензени за наявності каталізаторів кобальт нафтенату, кобальт або манґан
бензоату, промотованих солями Брому, незалежно від довжини бічного ланцюга
зазнають окиснення киснем повітря до бензойної кислоти:
33.2.1. Спектроскопія аренкарбонових кислот
а
8 м.ч. 8,12 (а), 7,43 (Ь), 7,51 (с)
С6Н5СН3
Нафтенат кобальту
140° С
'С6Н5СООН
У лабораторії як окиснювачі використовують калій перманганат, калій
біхромат, нітратну кислоту:
842
Чирва В.Я., Ярллолюк CM., Толкачова Н.В., Земляков О.Є. Органічна хімія
КМп04
СбН5СН2СН3 С6Н5СООН
Таким шляхом з о-9 м- і л-ксиленів одержують фталеву, ізофталеву й
терефталеву кислоти, відповідно, наприклад:
02, кобальт нафтенат
150 С
соон
Терефталева кислота
❖ Окиснення ароматичних карбонільних сполук. Ароматичні альдегіди
легко перетворюються в кислоти під дією кисню повітря за радикальним
механізмом або хімічними окиснювачами за йонним механізмом:
2 С6Н5СНО + 02 -+ 2 С6Н5СООН
Арилметилкетони перетворюються в кислоти під час окиснення натрій
гіпохлоритом {галоформна реакція):
C6H5-C-CH3i!!^C6H5COONa + СНС13 + NaOH
❖ Окиснення нафталену. Фталеву кислоту зазвичай одержують у
промисловості окисненням нафталену киснем повітря над ванадій (V) оксидом
за 380-450 °С:
33.3.2. Синтез за допомогою металоорганічних
сполук
Ароматичні одноосновні або монокарбонові кислоти можна отримати
карбоксилюванням магній- або літійорганічних сполук аналогічно до синтезів в
аліфатичному ряді. Зокрема, зберігають значення для синтезу ароматичних кислот
реактиви Гриньяра. Особливо цей метод зручний для одержання мічених сполук:
АгГ^Вг + 14С02 Ar-14COOMgBr Аг-14СООН + М%ВхС\
Кислоти з карбоксилом в ароматичному ядрі утворюються також під час
карбоксилювання натрій- і літійарилів, які синтезують взаємодією арилгало-
генідів з металами або металюванням ароматичних сполук, наприклад:
Розділ 33. Ароматичні карбонові кислоти
843
С6Н5Вг + 2 Li C6H5Li + LiBr C6H5Li + C02 C6H5COOLi
C6H6 + C2H5Na C6H5Na + C2H<^ C6H5Na + C02 C6H5COONa
3333. Лцилювання за Фріделем-Крафтсом
Специфічним для ароматичного ряду є синтез карбонових кислот за
реакцією Фріделя-Крафтса; якщо як ацилювальний агент використати фосген
або хлорокарбоновий естер, то в ароматичне ядро ввійде хлороформільна або
алкілкарбоксилатна група:
Ar-H + С1-СО-С1 -^ь- Ar-CO-СІ АгСООН
Аг-Н + С1-СООС2Н5-^- Аг-СООС2Н5-^- АгСООН
У першій реакції синтез часто припиняють на стадії галогеноангідриду,
оскільки бензоїїгхлорид є однією з найважливіших похідних бензойної кислоти.
33.3.4. Гідроліз нітрилів або трихлорометиларенів
У випадку використання гідролізу нітрилів вихідні продукти зазвичай
одержують методами, що не мають аналогів для аліфатичного ряду. Наприклад,
потенційний карбоксил у вигляді ціаногрупи можна ввести на місце сульфо-
групи сплавленням солей сульфокислот із натрій ціанідом, а на місце аміно-
групи — через діазосполуки:
C6H5-S03Na + NaCN C6H5-CN + Na2S03
2 C6H5-N2S04H"+ Cvb(CN)r^* 2 C6H5CN + 2 N2 + Cu2S04 + H2S04
Цікавий синтез ароматичних moho- і полікарбонових кислот виконують
через метил- і поліметилбензени, відповідно:
СбН5СН3 + ЗС12-^ C6H5CCl3±|g- [С6Н5С(ОН)3] — с6н5соон
Безпосередній гідроліз трихлорометилбензену СбН5ССЬ відбувається
погано, тому цю речовину попередньо перетворюють у бензоїлхлорид шляхом
нагрівання з бензойною кислотою, після чого піддають гідролізу:
С6Н5СС13 + С6Н5СООН 2 С6Н5СОС1 + НС1
С6Н5СОС1 + Н20 -> С6Н5СООН + НС1
Що стосується кислот з карбоксилом у бічному ланцюзі, то їх можна
синтезувати звичайними методами, що застосовують в аліфатичному ряді й в
особливих випадках - деякими специфічними методами.
844 Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
33.4. Бензойна кислота і її функціональні похідні
Бензойна кислота - одна з найдавніше відомих органічних кислот. Ще на
початку XVII ст. її виділили під час сублімації бензойної смоли або росного
ладану, звідки й походить назва. Детально цю кислоту дослідили Ю. Лібіх і
Ф. Велер, які працею про "бензойний радикал" започаткували хімію похідних
бензойної кислоти.
Сьогодні бензойну кислоту широко застосовують у промисловому
виробництві барвників, наприклад, для синтезу "синього анілінового" й низки
антрахінонових барвників. Бензойна кислота має антисептичні властивості, тому
її використовують у вигляді натрієвої солі для консервування харчових
продуктів. Значне застосування мають також різні похідні бензойної кислоти.
Схарактеризуємо їх детальніше.
• Солі одержують шляхом нейтралізації кислоти натрій гідроксидом, амо-
ніаком. Вони мають, як і сама кислота, високу бактерицидну й бактеріостатичну
активність. Завдяки цим властивостям, а також нетоксичності їх застосовують як
консерванти у харчовій промисловості й антисептики у медицині. Амоній
бензоат використовують як інгібітор корозії і стабілізатор у виробництві
латексів і клеїв.
• Естери синтезують нагріванням кислот зі спиртами за наявності сульфатної
кислоти. Якщо замісників у о-положенні немає, то естерифікація карбоксильної
групи відбувається так само легко, як і у випадку аліфатичних кислот. Якщо
одне з оположень зайняте, то швидкість естерифікації зменшується, а у
випадку, коли обидва о-положення зайняті, реакція через просторові перешкоди
не відбувається зовсім.
Естери о-заміщених кислот можна отримати реакцією солей Арґентуму із
галогеноалканами. Через причину, що зазначена вище, вони важко піддаються
омиленню, зате легко й кількісно розщеплюються за наявності краун-етерів.
Гідроліз естерів ароматичних кислот відбувається як у кислому, так і в лужному
середовищі. Під час гідролізу в лужному середовищі електроноакцепторні
замісники збільшують швидкість гідролізу, а електронодонорні - сповільнюють
реакцію.
Естери бензойної кислоти від метилового до ізоамілового - запашні
речовини. Тому вони є в складі фруктових есенцій і мил. Бензилбензоат -
фіксатор запаху в парфумерії, розчинник запашних речовин і репелент від молі.
• Аміди. Заміщений амід бензойної кислоти - гіпурова кислота
СбН5СОННСН2СООН - продукт життєдіяльності тварин.
• Хлороангідриди. Серед похідних бензойної кислоти найбільше значення має
бензоїлхлорид, насамперед, як ацилювальний агент. За реакційною здатністю він
поступається аліфатичним аналогам завдяки зсуву електронної густини з
бензенового кільця на атом Карбону оксогрупи:
Розділ 33. Ароматичні карбонові кислоти 845
СІ
Значення 8+ на атомі Карбону зменшується, а отже, зменшується і його
реакційна активність у синтезах з нуклеофілами.
■ Бензоїлхлорид у промисловості одерясують гідролізом трихлоро-
метилбензену надлишком оцтової кислоти за наявності 3-4 % сульфатної
кислоти:
н so
СбН5СС13 + СН3СООН г'І' С6Н5СОС1 + СН3СОС1 + HCl
100 С 95%
Як побічний продукт одержують ацетилхлорид, який також застосовують у
промисловості.
Також його синтезують з трихлорометилбензену та бензойної кислоти:
С6Н5СС1, + С6Н5СООН - 2С6Н5СОС1 + НС1
Дg
Крім того, бензоїлхлорид можна одержати хлоруванням бензальдегіду на
холоді за реакцією, яка неможлива для аліфатичного ряду:
,о ,о
С6Н5-С + С12—^ СЛІгС + HCl
Н СІ
Останніми роками в промисловості віддають перевагу взаємодії бензойної
кислоти з фосгеном:
с6н5соон + сосі2—с6н5с' + неї + со2
СІ
■ У лабораторії бензоїлхлорид синтезують дією фосфор (V) хлориду або
тіоніл хлориду на бензойну кислоту:
с^нгсоон +■ РС15 С6Н5СОС1 + РОС13 + НС1
■ За допомогою бензоїлхлориду легко бензоїлюють спирти, феноли, аміни,
а також уводять бензоїльний залишок в ароматичні сполуки за реакцією
Фріделя-Крафтса тощо. Бензоїлхлорид трохи пасивніший, ніж хлороангідриди
аліфатичних кислот, зокрема, він значно повільніше зазнає гідролізування. Цю
обставину використовують у реакції Шоттенах—Баумана, що полягає в
проведенні бензоїлювання у водно-лужному середовищі. Наприклад, у розчин
1 Шоттен K. (Schotten С.) (1853-1910) - німецький хімік, учень А. Гофмана. Працював у Бер-
лінському університеті.
04$ /
846
Чирва В.Я., Ярмолюк С.М, Толкачова Н.В., Зеллляков О.Є. Органічна хімія
фенолу в розведеному лузі поступово вводять бензоїлхлорид (швидкість
бензоїлювання натрій феноляту значно перевищує швидкість гідролізу хлоро-
ангідриду):
АгСОСІ + Аг'ОИа АгСООАг'
У випадку хлороангідридів аліфатичних кислот гідроліз лугом відбувається
набагато швидше, ніж процес ацилювання.
Бензоїлювання фенолів, спиртів і амінів проводять також у безводних
розчинах за наявності піридину або третинних амінів:
с6н5сосі + с2н5он Піридин» С6Н5СООС2Н5
Етилбензоат
С6Н5СОС1 + 2С2Н5Ш2 - C6H5CONHC2H5 + С2Н5гШ2 НС1
Л^-Етилбензамід
С6Н5СОС1 + 2Ш3 С6Н5СОШ2+МН4С1
Уперше метод бензоїлювання застосований К. Шоттеном для ацилювання
амінів (1884), а потім Бауманом на спиртах (1886).
■ Бензоїлхлорид використовують для одержання пероксиду бензоїлу й
надбензойної кислоти (гідропероксид бензоїлу). Реакція бензоїлхлориду з натрій
пероксидом приводить до утворення пероксиду бензоїлу:
сі р-р
СбН5-Сч + НаА ► С6Н5-СЧ ,С-С6Н5 + 2 КаСІ,
О 0 0
Пероксид бензоїлу
який є поширеним ініціатором багатьох радикальних реакцій, особливо реакцій
полімеризації, що мають величезне промислове значення.
■ Надбензойну кислоту одержують взаємодією бензоїлхлориду з гідроген
пероксидом у лужному середовищі:
С6Н5СОС1 + Н202 + 2 ИаОН С6Н5СОО(Жа + №С1 + 2 Н20,
або дією натрій метилату на пероксид бензоїлу:
0-0
С6Н5-СЧ ,С-С6Н5 + СН30№ С6Н5СОООИа + С6Н5СООСН3
О О
Надбензойна кислота стійкіша, ніж гідропероксиди аліфатичних кислот, її
можна отримати в кристалічній формі. Однак найчастіше її застосовують у момент
утворення й не виділяють в індивідуальному вигляді. Надбензойну кислоту
використовують, головно, для синтезу а-оксидів і гліколів з ненасичених сполук
(М. Прилежаєв). Вона зручніша, ніж аліфатичні рідкі надкислоти:
Розділ 33. Ароматичні карбонові кислоти
847
р-он яч /Я1
R-CH=CH-R, +С6Н-СЧ НС-СН + С6Н5СООН
О о
■ м-Хлоронадбензойну кислоту використовують для окиснення ариламінів і
арилгідроксиламінів у відповідні нітросполуки.
На закінчення зазначимо, що карбоксильна група бензойної кислоти є
орієнтантом другого роду, тому спрямовує електрофіли в ^-положення.
33.5. Ароматичні дикарбонові кислоти
33.5.1. Фталева кислота і її похідні
Найважливіша фталева кислота, яку, як уже зазначено, у промисловості
синтезують окисненням нафталену або о-ксилену. З огляду на о-розміщення
функціональних груп фталева кислота дає низку циклічних похідних і щодо
цього нагадує малеїнову або янтарну кислоти.
• Фталевий ангідрид легко утворюється під час нагрівання фталевої кислоти
або дією на неї зневоднювальних агентів:
асоон
соон
р + Н20
Він є безбарвною кристалічною речовиною з температурою плавлення 130—
131 °С. Під час звичайного нагрівання зі спиртами фталевий ангідрид приєднує
одну молекулу спирту, утворюючи кислі, або неповні, естери фталевої кислоти,
які можна далі перетворити в повні естери звичайною естерифікацією за
наявності мінеральних кислот:
♦ с2н5он— Гїстонаі^»^С00СЛ
к^-соос2н5 ^сооед
Моноетилфталат Діетилфталат
Моноестери фталевої кислоти використовують для поділу спиртів на оптичні
ізомери.
Фталевий ангідрид легко вступає в реакцію Фріделя—Крафтса, наприклад, з
бензеном, утворюючи о-бензоїлбензойну кислоту, яку за жорсткіших умов
можна перетворити в антрахінон:
848 Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
О
О+І
Під час нагрівання за наявності сульфатної кислоти або цинк хлориду
фталевий ангідрид конденсується з фенолами:
О
о
Фенолфталеїн
■ Значна частина фталевого ангідриду витрачається на синтез бутилового й
октилового естерів фталевої кислоти, які застосовують як розчинники з високою
температурою кипіння й пластифікатори таких полімерів, як нітроцелюлоза,
полівінілхлорид, синтетичні каучуки. Крім того, диметил-, діетил- і дибутил-
фталати - засоби, що відлякують комарів і мух {репеленти).
Головна ж кількість фталевого ангідриду витрачається на синтез гліфта-
левих смол, які використовують у виробництві лаків і плівок:
О -
О
н
\
С с<хщь-сцр-\
\0 ш /„
9 о
^у^с-о^-сн-снзо-
о
о
II
-с
9/
с=о
і
о
. С-ОСНг-СН-СЬ^О-
О
• Фталамінова кислота і фталімід. У разі дії амоніаку на фталевий ангідрид
утворюються два продукти: фталамінова кислота й фталімід (імід фталевої
кислоти). Дія водного амоніаку на холоді приводить до фталамінової кислоти, а
фталімід утворюється під час пропускання газоподібного амоніаку над фталевим
ангідридом:
Розділ 33. Ароматичні карбонові кислоти
849
NH Фталімід
Фталамінова кислота
Фталамінову кислоту синтезують також під час неповного гідролізу фталіміду,
який, відповідно, утворюється в разі нагрівання' фталамінової кислоти. З цієї
кислоти за умов перегрупування Гофмана готують технічно важливий продукт -
антранілову кислоту.
■ Фталімід застосовують у синтезі Габріеля1 для введення первинної
аміногрупи на місце галогену. У цьому разі починають з каілій фталіміду, який
реагує з алкілгалогенідами, унаслідок чого утворюється заміщений фталімід під
час гідролізу заміщений фталімід дає відповідний амін і фталеву кислоту:
Особливо важливий цей метод для отримання аліфатичних амінів, що
містять у ланцюзі інші замісники.
Калій фталімід одержують дією на фталімід сильних основ, наприклад,
алкоголятів калію або спиртових розчинів лугів. Фталімід має слабкокислі
властивості, що пов'язано з наявністю двох електроноакцепторних груп безпо-
середньо біля атома Нітрогену: завдяки р,я-спряженню між електронною парою
атома Нітрогену й двома я-зв'язками карбонільних груп. Унаслідок цього зв'язок И-Н
значно поляризований, і атом Гідрогену під дією лугу зазнає заміщення на метал.
Раніше іУ-заміщений фталімід перетворювали в первинний амін лужним
гідролізом. Однак виявилося, що реакція відбувається набагато легше й за
м'якших умов під час обробки гідразином:
NK + C1CH2R
CCncHiR
о
р
о
о
,NCH2R
NH2NH2
R-CH2NH2 +
NH
.NH
О
Фталілгідразид
1 Габріель 3. (Gabriel S.) (1851-1924) - німецький хімік, учень А. Гофмана і Р. Бунзена. Головні праці
присвячені синтезу ніїсрогеновмісних гетероциклічних сполук. Відкрив спірани чотиривалентного
Нітрогену. Синтезував ізохінолін. ІмекіГаГреакція.
850
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Похідну фталілгідразиду - люмінол (4-амінофталілгідразид) Qx q
- використовують у криміналістиці для виявлення слідів крові. r^^TJs^
■ Ще сильніші порівняно з фталімідом кислотні власти- (І I^NH
вості має, наприклад, сахарин (імід о-сульфобензойної кисло- ^ч/^^
ти), у якого до атома Нітрогену приєднаний потужний акцептор Сахарин О
електронів - група S02:
33.5.2. Терефталева кислота і її похідні
Калієва сіль фталевої кислоти за 400 °С та наявності цинк або кадмій
фталату зазнає ізомеризування в сіль терефталевої кислоти. Терефталеву
кислоту синтезують у великих кількостях окисненням и-ксилену. Також її
одержують карбоксилюванням калієвої солі бензойної кислоти за 340 °С під
тиском 300 атм. за наявності калій гідрокарбонату з виходом 60%:
ЮОК COOK
со2
КНС03
COOK
■ Поліетилентерефтпалат одержують у промисловому масштабі шляхом
переестерифікації диметилтерефталату етиленгліколем:
О. /=ч р
о_...
Використання диметилтерефталату замість самої кислоти зумовлене тією
обставиною, що кислоту, на відміну від естеру, важко очистити в промисловості.
Текстильні волокна на основі поліетилентерефталату в Росії називають
лавсаном, у Великій Британії - териленом, у США - дакроном.
■ М,ЛГ-Диметил-ІЧЛГ-динітрозотерефталамід застосовують для одержання
пінопластів і губчастої гуми. Під час нагрівання він розкладається з виділенням
азоту:
НС/Ь ^ \_/ V А^іл -м^ -з—- \_/ ——з
:n-c-^Vc-nC„ h3cooc-fy-coocH3
II \=/ II NCH. -N2 3 \=/ 3
о о 3
33.6. Гідроксибензойні кислоти (фенолокислоти)
❖ Синтез гідроксикислот. Фенолокислоти можна отримати введенням
карбоксилів у фенол або гідроксигрупи в бензойну кислоту.
Розділ 33. Ароматичні карбонові кислоти
851
• о-Гідроксибензойну (саліцилову) кислоту одержують у промисловості
реакцією Кольбе — нагріванням до 130 °С сухого натрій феноляту під тиском 5
атм. у середовищі газоподібного карбон (IV) оксиду:
N3
О Иа
4>
н
с=о
о-
а
СООЫа
Саліцилову кислоту можна також одержати окисненням саліцилового
альдегіду або дією на натрій фенолят тетрахлорометаном і лугом.
• м-Гідроксибензойну кислоту, яку використовують для виробництва барвни-
ків, одержують лужним плавленням л*-сульфобензойної кислоти:
^ООС^^^^СШа КаООС>ч^^(Жа
• п-Гідроксибензойну кислоту синтезують за методом Кольбе, застосовуючи
калій гідроксид.
• п-Метоксибензойну (ганусову) кислоту одерясують окисненням анетолу:
СН
Анетол
сн-сн-сн,
[О]
СН.
соон
Ганусова кислота
❖ Властивості гідроксикислот
■ Саліцилова кислота значно сильніша від бензойної та
своїх ізомерів. Такий оефект гідроксигрупи пояснюють
утворенням водневого зв'язку між функціональними групами,
що приводить до збільшення позитивного заряду на карбо-
ксильному атомі Карбону й посилення протонізації атома
Гідроген^' карбоксильної групи.
■ Саліцилова кислота утворює за участю обох функціональних груп два
ряди похідних. Ацилхлориди й ангідриди кислот ацилюють її за гідрокси-
групою:
^ч^соон
II ] +(сн3со)2о-
СООН
он
сн +сн3соон
Аспірин 0
Аспірин застосовують у медицині як жарознижувальний та анальгетичний
препарат.
852
Чирва В.Я., Ярмолкж СМ., Толкачова Н.В., Земляков О-Є. Органічна хімія
■ Відповідно* хлороангідрид саліцилової кислоти утворює з фенолами або
спиртами естери іншого типу, наприклад:
асоон
+ РОС13 + С6Н5ОН-
он
он
Салол
Салол є антисептиком, що рекомендують для дезинфекції в разі шлунково-
кишкових захворювань.
Сама саліцилова кислота - сильний дезинфікувальний засіб. її натрієву сіль
застосовують як ліки від суглобового ревматизму. У великих кількостях її
використовують у промисловості для виробництва азобарвників.
■ Метиловий естер саліцилової кислоти, який одержують за схемою
^^соон
он
он
соосн
з »
застосовують для лікування ревматизму. Метилсаліцилат трапляється у вигляді
глікозиду в різних рослинах, його використовують у медицині.
■ Гідрування саліцилової кислоти над платиною або відновлення її натрієм
в ізоаміловому спирті дає пімелінову кислоту; у цьому разі утворюється
проміжна тетрагідросаліцилова кислота, що зазнає гідролізування за типом
кислотного розщеплення ацетооцтового естеру:
гГ^СООН,и ^/СООН ^ХООН
IX ШкСї вСі — ноосчсн^-соон
ОН ^^ОН \^^о Пімелінова кислота
Пімелінову кислоту використовують у синтезі поліамідів, поліуретанів і під
час виробництва пластифікаторів.
❖ Полігідроксибензойні кислоти досить поширені в природі. Вони
містяться в рослинах у вигляді глікозидів або інших похідних. Приклади таких
кислот та їхні систематичні й історичні назви наведені нижче:
СООН
3,4-Дигідрсксибензойнг
(протокатехова)
кислота
осн.
СООН
4-Гідрокси-З-
метоксибензойна
(ванілінова) кислота
осн,
он
(7°СНі
СООН
З-Гідрокси-4-
соон
3,4-Диметоксибензойна
метоксибен-зойна (вератрова) кислота
(ізованілінова) кислота
Протокатехову кислоту синтезують з я-гідроксибензойної кислоти:
Розділ 33. Ароматичні карбонові кислоти
853
соон соон соон
кон^
он он
■ 3 багатоатомними фенолами, наприклад, з резорцинолом реакція Кольбе
відбувається легше, ніж з фенолом, унаслідок погодженої орієнтації гідроксильних
груп резорцинолу. Наприклад, 2,4-дигідроксибензойна (резорцилова) кислота
утворюється вже під час нагрівання резорцинолу з розчином амоній гідрокарбонату:
Н<Х^Х>Н НОх
+ш<Нсо,—-ноосЧГуон
2,4-Дигідроксибензойна (резорцилова) кислота
■ Із тригідроксибензойних кислот найбільше значення має 3,4,5-тригідро-
ксибензойна кислота (галова) - одна з найпоширеніших рослинних кислот. Вона
міститься у вигляді естерів у танінах чорнильних горішків (галах), листі чаю,
дубовій корі, коріннях гранатового дерева й багатьох інших рослинах.
Одержують її лужним або ферментативним гідролізом танінів:
ЮОН СООН
ИаОН
-ИаВг
^ОН но^^г^он
ОН ОН
3-Бромо-4,5-дигідрокси- 3,4,5-Тригідроксибензойна
бензойна кислота (галова)кислота
Галову кислоту здавна застосовували для виробництва чорного чорнила й
фарб. Вісмутова сіль галової кислоти є сильним антисептиком ("дерматол").
■ 4-Гідрокси-3,5-диметоксибензойна (бузкова) кислота — диметиловий етер
галової кислоти, також утворюється під час розщеплення багатьох природних
сполук, зокрема, лігніну:
ОН ?СНз
СН30\^Д^ОСНз СН3О^Л-/ОСН3
4-Гідрокси-3,5-диметоксибензойна І '
(бузкова) кислота Андаксин
■ Амід, утворений 3,4,5-триметоксибензойною кислотою й гетероциклич-
ним аміном - морфоліном, називають "андаксином", його застосовують як
транквілізувальний (заспокійливий) медикамент.
❖ Депсиди й дубильні речовини. Естери, утворені з двох молекул
ароматичних полігідроксикислот, у яких одна є кислотою, а інша - фенолом,
854
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
називають депсидами. Прикладами цих сполук можуть слугувати леканорова й
л*-галоїлгалова (м-дигалова) кислоти:
ОН
ОН /=
о-
СООН
но-<' х)-сч
о он
СН3 Леканорова кислота
а сам зв'язок між карбоксильною і фенольною групами називають депсидним.
■ Дубильні речовини, які екстрагують з деяких рослинних тканин, наприклад, з
кори дуба, листя чаю, - це згадані вище депсиди. Дубильні речовини реагують з
білками, утворюючи нерозчинні сполуки. Цю властивість використовують під час
дублення шкір. Крім того, їх застосовують для протравного фарбування. Дубильні
речовини, які також називають танінами, розділяють на два класи: ті, що зазнають
гідролізування, і ті, що не зазнають гідролізування.
■ Таніни одержують з дубильних горішків. Вони є похідними глюкози,
ацильованої залишками л*-галоїлгалової і галової кислот. Таніни з різних видів
дубильних горішків трохи відрізняються за кількістю ацилів і за співвідно-
шенням між залишками галової і галоїлгалової кислот. Навіть таніни, виділені з
однієї рослини, завжди є сумішшю речовин:
Загальна формула танінів,
де Я - залишок галової, лі-галоїлгалової кислоти
З глюкози, галової та л*-дигалової кислот синтезовано пентаестер глюкози,
що має всі найголовніші властивості таніну.
■ Таніни, що не зазнають гідролізування, мають набагато складнішу
структуру. Більшість таких танінів, або конденсованих танінів, становлять різні
полігідроксипохідні 3-гідроксифлавону:
.ОН
.ОН
[О]
1-8
Дубильні речовини виноградного вина, так звані проціанідини
Розділ 33. Ароматичні карбонові кислоти
855
33.7. Ароматичні амінокислоти
Амінобензойні кислоти можна отримати каталітичним відновленням над
платиною або залізом у хлоридній кислоті відповідних нітробензойних кислот.
У техніці так синтезують п- і лі-амінобензойні кислоти:
02N-C6H4-COOH
H2N-C6H4-COOH
о-Ізомер {антранілову кислоту) одержують із фталевого ангідриду через
фталімід за допомогою перегрупування Гофмана:
NH,
о 6
соон
а..
C^-N-Br
ó "
-Br"
СООН
о
^усоон ^соон
■ Антранілова кислота є проміжним продуктом у разі синтезу індиго,
також її використовують як діазоскладову для одержання низки азобарвників і
запашних речовин.
Метиловий естер антранілової кислоти є в складі етерної олії квітів жасмину
разом з бензилацетатом і невеликою кількістю індолу. Він має запах суниці, тому
його застосовують у парфумерії. Цікаво, що саме наявністю метілантранілату
пояснюють характерний аромат, властивий винам з винограду американського
походження Vitis Labrusca (Ізабелла, Лідія тощо).
■ п-Амінобензойну кислоту (ПАБК) застосовують для синтезу таких
анестезувальних засобів, як анестезин, новокаїн і пантокаїн:
кн-ссиуз-сНз
соосн2сн3
Новокаїн
OCHz-CHrN(C2H5)2- НС1
Анестезин
O' OCHjCH¿N(C2H5)2HCi
Пантокаїн
и-Амінобензойна кислота - фактор росту багатьох мікроорганізмів, а також
вітаміноподібна речовина. Дія сульфамідних препаратів полягає саме в тому, ще
вони підміняють необхідну для життя бактерій и-амінобензойну кислоту
856
Чирва В.Я., Ярллолюк СМ., Толкачова H.В., Земляков О.Є. Органічна хімія
(вітамін Ні). Достатня концентрація цих речовин у крові призводить до загибелі
бактерій.
м-Амінобензойну кислоту застосовують у виробництві кубових барвників і
азобарвників, кольорових компонентів для фотопаперу.
■ п-Аміносаліцилова кислота (ПАСК) - важливий антитуберкульозний
препарат, що діє за тим самим принципом, що й и-амінобензойна кислота:
НО
S03 + H2SOé {Г
S03H
Fe +HC1
100°C
NH9
I 2
SO3H
кон
250°С
соон
Розділ 34
КОНЛЕНСОВЛНІ
АРОМАТИЧНІ СИСТЕМИ
До аренів з конденсованими циклами належать ароматичні вуглеводні, у
яких цикли мають два та більше спільних атомів. За взаємним розташуванням
циклів виділяють три групи поліциклічних аренів: лінійно конденсовані,
ангулярно конденсовані й периконденсовані цикли.
Лінійно конденсовані цикли
Ангулярно конденсовані
цикли
Периконденсовані
цикли
00 осо
¿2?
Нафтален Антрацен
Фенантрен
Пірен
Найбільше значення серед аренів з конденсованими циклами мають
нафтален і антрацен, які розглянемо далі.
34.1. Нафтален і його похідні
34.1.1. Номенклатура
5
Нафтален має стандартну систему нумерації атомів Карбону. Похідні
нафталену називають за загальними правилами найменування вуглеводнів,
використовуючи принцип мінімальних локантів. Крім цифрової номенклатури,
для нафталену в хімічній літературі інколи застосовують буквену: положення 1 і
4 атомів Карбону називають а-, а 2, 3 - Р-положеннями.
4 '^с2н5 ^^Ц/С2н5
6-Етил-1 -нітронафтален 2-Етил-З-метилнафтален
Вибір напряму нумерації визначений правилом За рівності локантів менший номер
мінімальних локантів привласнений першому за абеткою замісникові
858
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
34.1.2. Будова нафталену
Нафтален виділений англійським хіміком А. Гарденом1 1819 р. з кам'яновугіль-
ної смоли. Його вміст становить 6 %, тому дотепер це головне джерело нафталену.
Додатково нафтален одержують зі смол, які залишаються після піролізу нафти і
бензинових фракцій. Елементний склад нафталену визначений російським хіміком
О. Воскресенським2. У 1868 р. Р. Ерленмейєр запропонував використовувати
бензенову формулу Кекуле для нафталену. На підставі цієї пропозиції він
запропонував для нафталену таку структуру:
Через два роки К. Гребе підтвердив цю будову хімічним синтезом.
Докази на користь цієї формули нафталену такі: нафтален - явно
ароматична сполука, тому що молекулярній формулі СюНв повинні відповідати
за наявності одного циклу шість подвійних зв'язків, а за наявності двох циклів -
п'ять, однак звичайних реакцій на подвійні зв'язки нема.
Будова нафталену доведена окисненням його до фталевої кислоти, точніше
- фталевого ангідриду. На підставі цього факту доведено наявність у нафталені
бензенового кільця й о-положення атомів Карбону іншого циклу. Відомо, що
нітрогрупа стабілізує ароматичне кільце, з яким вона зв'язана. Тому в
нітронафталені окиснюється незаміщене кільце з одержанням нітрофталевого
ангідриду. Якщо нітрогрупу відновити до амінної, то заміщене кільце стає
лабільним, і окиснення а-амінонафталену дає фталевий ангідрид:
Гарден A. (Harden А.) (1865-1940) - англійський біохімік, дослідник у галузі ферментів
спиртового бродіння, першовідкривач коферментів. Нобелівська премія 1929 р.
2 Воскресенський О. (1809-1880) - російський хімік-органік, викладач, першовідкривач хінону та
теоброміну, досліджував вітчизняні горючі копалини.
3 Гребе K. (Graebe С.) (1841-1927) - німецький хімік. Працював у Кенігсберзькому та Женевсь-
кому університетах. Уперше ввів поняття opmo-, мета-, лярд-замісників у бензеновому кільці.
Разом з K. Ліберманом синтезував алізарин.
Розділ 34. Конденсовані ароматичні системи
859
Звідси ясно, що молекула нафталену складається із двох бензенових ядер,
що мають два спільних сусідніх атомів Карбону.
Нафтален уважають ароматичною сполукою, оскільки його властивості
нагадують властивості бензену. До нафталену й антрацену можна застосувати
правила ароматичності, якщо розглядати розподіл я-електронної хмари в умовно
моноциклічних системах (10 я-електронів, л=2). Відповідно до його молекулярної
формули CioHg, можна було б припустити наявність . високого ступеня
ненасиченості. Однак нафтален стійкий, хоча й у меншому ступені порівняно з
бензеном, до реакцій приєднання, що характерні для ненасичених сполук. Замість
цього дня нього є типовими реакції електрофільного заміщення.
У бензені ароматична шістка л-електронів розподілена ідеально рівномірно
стосовно шести атомів Карбону. У нафталені вирівнювання настільки неможли-
ве, і він унаслідок цього менш ароматичний і більш "ненасичений", ніж бензен.
Це випливає з меншого зниження енергії нафталену порівняно з розрахованою
енергією ненасиченого вуглеводню, що відповідає структурі, побудованій за
формулою Ерленмейєра з фіксованими подвійними зв'язками (енергія резо-
нансу). Це значення для нафталену становить 255 кДж/моль, тобто помітно
менше від подвоєного значення (2-150 кДж/моль) для бензену.
Справді, якщо використати теорію резонансу, то структура нафталену
повинна бути проміжною між трьома граничними структурами Ерленмейєра
(структури а, б і в нижче). Уважний аналіз цих резонансних структур засвідчує
нерівноцінність зв'язків у молекулі нафталену:
Нерівноцінність подвійних зв'язків, менша симетричність молекули нафталену,
а отже, і ароматичність порівняно з бензеном випливає з рентгено- і електроно-
графічних досліджень, які засвідчують, що довжина зв'язків між атомами С1-С2 і
С2-С3 різна (відповідно, 0,136 і 0,143 нм). Унаслідок цього в молекулі нафталену
електронна густина в кожному з кілець нагадує дієнову систему, тому для
зображення нафталену ліпше використати формулу Кекуле—Ерленмейєра-Гребе,
ніж вписувати кола в шестикутниках, як у випадку бензену. Однак у цьому разі
варто пам'ятати, що нафтален є ароматичною, а не полієновою сполукою.
Завдяки особливостям будови нафталену а-комплекси, що утворилися
шляхом приєднання електрофіла в Р-положення, є менш стабілізованими
порівняно з а-приєднанням, що, відповідно, спричиняє переважний напрям
електрофільних реакцій в а-положення.
Особливості структур а, б і в, де одне з ядер бензенове, а інше - дієнове,
відображені в особливій активності атомів Карбону в а-положеннях до
a
в
860 Чирва В.Я., Ярллолкж СМ, Толкачова Н.В., Зеллляков О.Є. Органічна хімія
електрофільних атак і тих типів^риєднань, до яких здатні дієнові вуглеводні.
Наприклад, на відміну від бензину, нафтален відновлюється воднем у момент
виділення, перетворюючись в 1,4-дигідронафтален:
Н Н
Н Н
Зазначимо, що нафтален окиснюється або відновлюється легше, ніж бензен,
однак лише до стадії утворення заміщеного бензену, тобто хімічних перетворень
зазнає одне з кілець, тоді як інше перетворюється у високостабільну ароматичну
систему бензенового типу:
О
1,4-Нафтохінон
Бром також приєднується до нафталену в положення 1,4, причому продукт
приєднання легко відщеплює гідроген бромід, утворюючи 1-бромонафтален:
Н Вг Вг
Н Вг
34.1.3. Ізомерія похідних нафталену
Уже монопохідні нафталену відомі у вигляді а- і Р-заміщених ізомерів. Для
двох однакових замісників, відповідно до формули Кекуле-Ерленмейєра-Гребе,
можливі десять ізомерів:
Розділ 34. Конденсовані ароматичні системи
861
Положення 1,2- або 2,3- називають сряю-розташуваннями, проте у випадку
нафталену це "позначення не однозначне; розташування 1,3- називають мета-, 1,8- -
пери-, 2,6- - амфі-. Якщо в молекулі є два різні замісники, то кількість ізомерів
збільшується до 14. Всі наведені формули нафталену підтверджені на практиці.
Нафтален - кристалічна речовина з температурою плавлення 80 °С, зазнає
сублімування, летка вже за кімнатної температури, переганяється з водяною парою.
Метилнафталени мають нижчі температури плавлення, ніж сам нафтален.
• УФ-спектроскопія. В УФ-спектрі нафталену є інтенсивні смуги з Хтгх 167,
190,220, 286 і 312 нм.
• ІЧ-спекпіроскопія. Смуги поглинання зв'язків С-Н і скелетних коливань
нафталену й антрацену, який детально розглянуто нижче, мають частоти,
близькі до бензену.
• ПМР-спектроскопія. Хімічні зсуви сигналів ароматичних протонів нафталену
наведені нижче:
34.1.5. Синтез нафталену і його похідних
❖ Циклоолігомеризація ацетилену. Під час пропускання ацетилену над
деревним вугіллям за 400 °С поряд з бензеном утворюється нафтален (М. Зелінсь-
кий, Б. Казанський):
Як різновид реакції М. Зелінського й Б. Казанського, можна розглядати
синтез нафталену з бензену й ацетилену, у якому суміш реагентів пропускають
через розпечені залізні трубки:
CHECH ^"N^
❖ Дегідр о циклізація. Під час витримування над платиною за 300 °С гомо-
логів бензену з бічним ланцюгом із чотирьох і більше атомів Карбону
утворюються нафтален і його гомологи:
34.1.4. Фізичні властивості нафталену
8 м. ч. 7,81 (а), 7,46 (Ь)
862
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
■іг^СЩ зоо»с Ц
СНз
+ ЗН,
❖ Дієновий синтез. Різні варіанти взаємодії дієнів з дієнофілами приво-
дять до нафталену та його похідних:
+гпо
о 6п он
❖ Гідродеметилювання гомологів нафталену. Сьогодні нерідко вико-
ристовують продукти каталітичного риформінгу нафти, у яких у значних
кількостях міститься метилнафтален. Під час обробки метилнафталену воднем
за наявності каталізаторів відбувається гідродеметилювання:
СН,
+ СН4
❖ Внутрішньомолекулярна конденсація фенілвінілоцтової кислоти.
Найдавніший шлях, що послугував підтвердженням формули Кекуле-Ерлен-
мейєра-Гребе, - замикання фенілвінілоцтової кислоти в 1-нафтол:
1-Нафтол
❖ Гомологи нафталену можна отримати з нафталену алкілуванням або
ацилюванням за Фріделем-Крафтсом з наступним відновленням кетонів за
Клемменсеном в алкілнафталени.
34.1.6. Реакції електрофільного заміщення
Як відомо, енергія стабілізації двох конденсованих ароматичних ядер
бензену менша, ніж двох ізольованих бензенових ядер. Звідси можна зробити
висновок, що всі реакції нафталену, за яких одне з бензенових ядер втрачає
ароматичність, а інше зберігає, будуть супроводжуватися меншою втратою
енергії, ніж у випадку бензену. Це, мабуть, і є причиною підвищеної порівняно з
бензеном реакційної здатності нафталену.
Розділ 34. Конденсовані ароматичні системи
863
У разі заміщення в нафталені можливе утворення двох регіоізомерних
продуктів під час атаки електрофіла в а- або Р-положення. Атоми Гідрогену в а-
положенні мають вищу реакційну здатність. Це можна пояснити, розглянувши
можливість делокалізації позитивного заряду у відповідних с-комплексах.
■ Електрофільна атака за ^положенням:
І II III IV V
Помітний внесок у делокалізацію позитивного заряду можуть робити тільки
граничні структури І і II, у яких ароматична система порушена лише частково
(зберігається ароматичний секстет електронів в одному із шестичленних циклів),
а не структури ИІ-У, у яких вона порушена повністю.
■ Електрофільна атака за ^-положенням:
VI VII VIII IX X
У цьому випадку помітний внесок у делокалізацію позитивного заряду в о-
комплексі може зробити тільки гранична структура VI, тому що в інших
структурах VII-X повністю порушена ароматична система.
Отже, оскільки під час а-атаки електрофіла кількість граничних структур,
що роблять внесок у делокалізацію позитивного заряду, більша, ніж під час Р-
атаки, то така орієнтація електрофільного заміщення ліпша.
❖ Галогенування. Хлорування й бромування нафталену відбувається
настільки легко, що не потребує каталізатора - кислоти Льюїса. Спочатку за
кімнатної температури відбувається 1,4-приєднання; наступне підвищення
температури до 50-70 °С веде до утворення продукту а-заміщення:
Н Вг
За наявності каталізаторів реакція відбувається через а-комплекс, і теж
утворюється а-бромонафтален з невеликою домішкою Р-ізомеру, який у разі
864
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
потреби можна синтезувати шляхом підвищення температури реакції до 500-
600 °С. За цих умов змінюється механізм реакції з електрофільного на ради-
кальний, і утворюється суміш а- і Р-ізомерів.
Незважаючи на те, що атоми галогенів у нафталені схожі на бензенові, тобто
вони мало рухливі, усе-таки бромопохідні можна використати для приготування
реактиву Гриньяра.
❖ Нітрування нафталену відбувається легко, й нітрогрупа вводиться
практично в а-положення, а вміст Р-нітронафталену не перевищує 5%. Для
введення ще однієї нітрогрупи потрібні жорсткіші умови, причому друга нітрогрупа
вступає в інше ядро теж в а-положення, і синтезується 1,8-динітронафтален з
домішкою 1,5-ізомеру. Це свідчить про деяку автономію обох ядер, оскільки
нітрогрупа пасивує до електрофільних атак більше те ядро, у яке вона вступила.
Відновленням а-нітронафталену М. Зінін уперше одержав а-нафтиламін.
Зазначимо, що Р-нітронафтален можна одержати лише окружним шляхом,
наприклад, окисненням Р-нафтиламіну або через Р-нафтилдіазонієві солі за
Гаттерманом.
Загалом нітропохідні нафталену використовують для одержання нафти-
ламінів, які, відповідно, необхідні для синтезу барвників.
❖ Сульфування. Реакція сульфування найкорисніша, оскільки сульфо-
нафталени є вихідними сполуками в синтезі різних барвників, тим більше, що а- і Р-
ізомери можна легко одержувати, змінюючи температуру процесу.
Наприклад, сульфування нафталену концентрованою сульфатною кислотою
за 80 °С відбувається майже повністю як а-заміщення. Швидкість утворення за
цієї температури альтернативної 2-сульфокислоти дуже мала, тобто є кіне-
тичний контроль. Водночас сульфування за 160 °С приводить до продукту, що
містить 80 % 2-сульфокислоти і лише 20 % 1-ізомеру. Той факт, що за цих умов
діє термодинамічний контроль, підтверджений таким спостереженням. Під час
нагрівання чистої нафтален-1- або нафтален-2-сульфокислоти в концентрованій
Н2804 також утворюється рівноважна суміш: 80 % 2-сульфокислоти й 20 % 1-
сульфокислоти:
Зворотна реакція десульфування полегшена завдяки малій термодинамічній
стабільності а-ізомеру, тому що в ньому існує відштовхування між сульфо-
Розділ 34. Конденсовані ароматичні системи
865
групою і атомом Гідрогену біля Сз-атома. Крім того, атом Карбону в а-
положенні більш нуклеофільннй порівняно з таким атомом в Р-положенні
завдяки особливостям будови нафталену:
$о3н
Десульфування
+ Н2804
У подібній ацидолітичній реакції може вціліти тільки сульфогрупа, що
потрапила в Р-положення. Отже, обидва ізомери - а- і Р-нафталенсульфокислоти
- доступні й слугують вихідною речовиною для синтезів у нафталеновому ряді.
За надлишку концентрованої сульфатної кислоти а-нафталенсульфокислота
зазнає сульфування далі, утворюючи дві ізомерні дисульфокислоти - 1,5- і 1,6-; Р-
нафталенсульфокислота також дає два ізомери: 2,6- і 2,7-нафталендисульфо-
кислоти. Утворення 1,8-дисульфокислоти неможливе зі стеричних причин:
503Н
$03Н
+ Н2804
+ Н2804
$03Н
Н038
803Н
Н038
803Н
Нафталенсульфокислоти є вихідними речовинами в синтезі нафтолів і
нафтиламінів, які застосовують для одержання барвників.
❖ Лцшіювання й алкілування за Фріделем-Крафтсом
■ Нафтален найліпше ацилювати ацетилхлоридом за наявності алюміній
хлориду. Орієнтація заміщення визначена природою розчинника:
О^-СН,
сн3сосі
А1СЦ
Вплив нітробензену пояснюють тим, що він утворює з реагентом об'ємний
комплекс СН5СОС1-А1С1з-СбН5>Ю2, який може атакувати тільки просторово
доступне Р-положення субстрату, тоді як менш об'ємний комплекс
СНзСОС1-А1С1з-С82 здатний ацилювати ос-положення. Отже, ацетилювання дає
866 Чирва В.Я., Ярмолюк CM, Толкачова Н.В., Зеллляков O.G. Органічна хімія
змогу одержувати порівняно важкодоступні Р-ізомери. Наприклад, обробка р-
ацетилнафталену гіпохлоритом - ліпший метод одержання Р-нафтойної кислоти.
■ Алкілування нафталену за Фріделем-Крафтсом використовують мало,
оскільки внаслідок високої реакційної здатності нафталену відбувається різні
побічні реакції й поліалкілування. Тому алкілнафталени ліпше одержувати аци-
люванням з наступним відновленням кетонів або циклізацією гомологів бензену.
34.1.7. Окиснення нафталену
Під час обробки нафталену киснем повітря за наявності ванадій (V) оксиду
руйнується одне кільце і утворюється фталевий ангідрид. Оскільки нафтален
виділяється головно з кам'яновугільної смоли, а потреба у фталевому ангідриді
досить велика, то цей синтез широко використовують у промисловості.
❖ Нафтохінони. Окиснення нафталену й деяких його похідних реагентами
на основі Хрому (VI) у кислому середовищі веде до порушення ароматичності
одного з кілець, проте іншим способом, і приводить до одержання дикетопо-
хідних, відомих за назвою хінонів:
О
2-Метилнафтален 2-Метил-1,4-нафтохінон, 70%
Схильність нафталену до утворення хінонів не завпеди дає змогу синте-
зувати нафталенкарбонові кислоти шляхом окиснення метальної групи в
бічному ланцюзі, тобто методом, який використовують для одержання
бензойних кислот. Нафтойні кислоти можна синтезувати окисненням гомологів
нафталену натрій біхроматом у водному розчині.
Під час окиснення хромовою кислотою нафтален-1,4-, 1,2- і 2,6-діолів або
відповідних амінонафтолів синтезують три нафтохінони:
1,4-Нафтохінон 1,2-На.фтохінон 2,6-Нафтохінон
(я-нафтохінон) (о-нафтохінон) (ом$/-нафтохінон)
■ Похідними «-нафтохінону є низка природних і синтетичних барвників. З
синтетичних барвників назвемо лише аналогічний до хіназарину 5,8-дигідрокси-
1,4-нафтохінон (нафтазарин), який одержують дією сульфатної кислоти й сірки
на 1,5-динітронафтален:
Розділ 34. Конденсовані ароматичні системи
867
❖ Вітаміни групи К. Похідними «-нафтохінону є вітаміни групи К:
філохінон (вітамін Кі) і фарнохінон (вітамін К2). Вітамін Кі міститься в зелених
частинах рослин, а вітамін К2 - у бактеріях і рибі. Обидва вітаміни побудовані
аналогічно до 2-метил-1,4-нафтохінону, про що можна судити за подібністю
спектрів поглинання. У всіх трьох сполуках хромофором є структура 1,4-
нафтохінону. Будова вітамінів Кі і К2, визначена Л. Фізером, така:
О О
Н
О \ /з О \ /з
Вітамін Кі Вітамін К2
Вітаміни Кі і К2 мають кровоспинну дію, яка властива синтетичному 2-метил-
1,4-нафтохінону, виробництво якого провадять за методом, що розроблений
О. Корольовим:
НС"СН2 /уШ
НО
СНз
О
34.1.8. Пдронафталени
На відміну від бензину, нафтален через нерівномірність розподілу
електронної густини можна легко відновлювати хімічними реагентами.
Наприклад, гідрування нафталену амальгамою Натрію у водному спирті дає
головно 1,4-дигідронафтален, а під дією натрію в ізопентиловому спирті
утворюється 1,2,3,4-тетрагідронафтален - тетралін:
1,4-Дигідронафтален
У промисловості тетралін одержують шляхом каталітичного гідрування
нафталену:
868
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Він є ароматичним, легко зазнає сульфування та нітрування. Під дією
концентрованої Н2804 утворюється суміш 5- і 6-тетралінсульфокислот, а 5- і 6-
мононітропохідні виділяють під час нітрування тетраліну на холоді; за 35-40 °С
вдається ввести два замісники в положення 5 і 6. Хлорування на холоді веде до
утворення 5-хлоротетраліну, а за температури 200 °С синтезується 2-хлоротетралін.
Це свідчить про те, що в останньому випадку галогенування відбувається за
радикальним механізмом.
Тетралін легко окиснюється киснем за радикальним механізмом в алільне
положення щодо подвійного зв'язку до тетрсиїону й далі до о-карбокси-
бензоїлмурашиної (фталонової) кислоти:
О О
^^с-соон
ч
о,
70°С
КМп04
соон
Фталонова кислота
Тетралін застосовують як розчинник жирів та лаків, а в суміші з етанолом -
як моторне пальне.
❖ Декалін. Під час вичерпного гідрування дуже чистого нафталену воднем
над нікелем (П. Сабатьє) одержують декагідронафтален, або декалін, відомий у
вигляді двох ізомерів:
н н
н
^нс-Декалін /ярянс-Декалін
Гідрування р-нафтолу дає вже чотири стереоізомерні декалоли, одна пара
яких має каркас цис-, а інша — тиранс-декалолів:
Н Н
Н
Н
но
н
он н
і/ис-Декалоли
транс- Декалоли
Розділ 34. Конденсовані ароматичні системи
869
Окиснення декалолів у кетони - декалони - знову приводить до двох ізомерів, і
з двох перших декалолів утворюється один |3-^мс-декалон, а із двох інших - його Р-
транс-ізомер. Відновлення декалонів веде, відповідно, до цис- і транс-декалінів.
Під час окиснення */ис-декаліну утворюються дві кислоти - г^с-р-2-карбоксицикло-
гексилпропіонова й г/ис-циклогексилен-1,2-діоцтова; окиснення гарянс-декалону
приводить до утворення транс-ізомерів тих же кислот.
34.1.9. Нафтоли
❖ Синтез нафтолів. Нафтоли, подібно до фенолів, можна синтезувати з
відповідних сульфокислот сплавленням з лугом:
,І -| розв. Н2804ґ
зоо°с II
Натрій 2-нафтолят
Крім того, нафтоли одержують кислотним гідролізом нафтиламінів. Ця
реакція, яку не вдається виконати в ряді бензену, ефективніша, ніж гідроліз
відповідних солей діазонію:
)Н
+ Ш3
1-Нафтол, 95%
❖ Перефункціоналізація 2-иафтолу. Подібно до заміщених бензену, а-
заміщені нафталени найчастіше одержують за допомогою послідовності реакцій,
у яких вихідними речовинами слугують нітросполуки. Однак р-заміщені
нафталени не можна синтезувати з нітросполук, оскільки нітрування в Р-по-
ложення виконати не вдається. Шлях синтезу нафтіл-2-аміну, а з нього - дуже
реакційноздатних солей діазонію відбувається через стадію одержання 2-наф-
толу, який отримують із 2-нафталенсульфокислоти; далі його перетворюють у
нафтиламін, нагріваючи під тиском з амоніаком і амоній сульфітом за реакцією
Бухерера1, що, за рідкісним винятком, не можна виконати в ряді бензену.
Виходи реакції близькі до кількісних, а характер та положення замісників у
нафтеновому циклі мало впливають на хід реакції:
Ш3,15(Л:,5агм.
^о3н ^^"он
1 Бухерер Г. (ВисЬегег Н.ТЬ.) (1869-1949) - німецький хімік. Професор університетів у Дрездені,
Берліні та Мюнхені. Є автором кількох іменних реакцій (карбазольний синтез Бухерера, реакція
Бухерера, реакція Бухерера-Берга).
870
Чирва В.Я., Ярллолюк CM, Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Реакція амінування зворотна. Наприклад, нафтіл-2-амін під час кип'ятіння з
лугом перетворюється в 2-нафтол.
❖ Барвники. Для нафтолів характерні звичайні реакції фенолів. Реакція
азосполучення з солями діазонію особливо важлива у виробництві барвників:
Конго червоний - кислотно-основний індикатор: синій у кислому середо-
вищі й червоний в основному.
34.1.10. Орієнтація електрофільного заміщення
в похідних нафталену
Раніше ми обговорили синтез різних монозаміщених похідних нафталену і
їхню доступність. Розглянемо тепер, як уже наявний замісник впливає на напрям
електрофільного заміщення в монозаміщених нафталенах.
Орієнтація заміщення в ряді нафталену складніша, ніж для бензену: вхідна
група може вступати або в кільце, що вже несе замісник, або в інше кільце.
Усього в нафталені є сім різних положень, які можуть зазнавати атакування.
Напрям заміщення можна пояснити з погляду структурної теорії й уявлень про
електрофільне заміщення в ароматичному ряді.
Активувальна група орієнтує заміщення в те кільце, у якому вона перебуває.
Якщо група, що активує, перебуває в положенні 1, то наступний замісник
спрямовується переважно в положення 4. Якщо ж група, що активує, перебуває
в положенні 2, то наступне заміщення відбуватиметься по атому С-1.
Дезактивувальна група спрямовує подальше заміщення у вільне кільце: в а-
положення під час нітрування, галогенування або а-, Р-положення залежно від
температури реакції під час сульфування, наприклад:
Розділ 34. Конденсовані ароматичні системи
871
ЫаОН,0-10°С
НЫ03 ^
^ л 20 °С
№Ы-С6Н,
ы=к-с6н5
N0,
Орієнтацію в цьому ряді можна пояснити на підставі оцінки відносної
стійкості а-комплексів, які виникають під час приєднання електрофіла до
нафталену. Очевидно, що найстійкішими будуть карбокатіони, під час
утворення яких ароматична система не руйнується повністю, а зберігається у
вигляді секстету 7і-електронів в одному з кілець/Сполуки, у яких зберігається,
ароматичний секстет - це структури, де позитивний заряд переважно перебуває
в кільці, що зазнає заміщення. Оскільки саме в
цьому місці зосереджений заряд, то найлегше
зазнає атакування те кільце, у якому розміщення
позитивного заряду енергетично найвигідніше: це
кільце, що має електронодонорний замісник, або
кільце, що не містить електроноакцепторної групи:
Електронодонорна група в положенні 1 найліпше компенсуватиме позитивний
заряд, якщо атака відбувається в положення 4 або 2. Це правильно незалежно від
того, завдяки якому ефекту надходять електрони:
+0Н
Н У
+0Н
Н У
У всіх випадках кільце, що не має замісників, залишається ароматичним.
Електронодонорна група в положенні 2 може найліпше брати участь у делокалізації
позитивного заряду, якщо атака відбуватиметься в положення 1 або 3:
872 Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
У
н
II
Насправді електрофільне заміщення відбуватиметься тільки в положення 1,
оскільки в цьому випадку зберігається ароматичний секстет у кільці, що не
зазнало атакування електрофілом, як у структурі І.
Антрацен, як і нафтален, має стандартну систему нумерації атомів Карбону,
з використанням якої будують назви його похідних:
Уперше антрацен виділений 1832 р. французькими хіміками Ж. Дюма й
О. Лораном з важкої фракції кам'яновугільної смоли, звідки його одерлсують і
сьогодні шляхом кристалізації з наступною сублімацією. Будова антрацену
з'ясована Г. Армстронгом - автором сучасної номенклатури органічних сполук
- на підставі результатів гідрування.
Молекулярна формула антрацену Сі4Ню за наявності трьох ароматичних
кілець повинна охоплювати сім подвійних зв'язків. З огляду на нерівномірний
розподіл електронної густини в молекулі антрацен легко окиснюється до дикетону
- антрахінону, що зберігає кількість атомів Карбону антрацену і його циклічну
структуру, тому що відновленням антрахінону можна знову отримати антрацен.
Будову антрахінону визначають за способом синтезу з о-бензоїлбензойної
кислоти, що замикається під дією фосфор (V) оксиду в антрахінон:
34.2. Антрацен і його похідні
34.2Л. Номенклатура
34.2.2. Будова антрацену
Г н -їй
С12Н8(СО)2 =
0-Бензоїлбензойна кислота
Розділ 34. Конденсовані ароматичні системи
873
Отже, для самого антрацену ми отримуємо структуру конденсованого
трициклічного вуглеводню - бензнафталену. Ця формула має такі резонансні
структури:
І II III IV
і підтверджена у синтезах як антрацену, так і продуктів його окиснення або
відновлення.
До антрацену також можна застосувати правила ароматичності, якщо
розглядати розподіл л-електронної хмари за умовно моноциклічною системою
(14 л-електронів, п=3). Як і в нафталені, в антрацені не всі зв'язки однакові, й
вони мають меншу енергію делокалізації порівняно з бензеном. Про це ж
свідчить той факт, що енергія резонансу в молекулі антрацену - 352 кДж/моль,
що на 104кДж/моль менше порівняно з трьома молекулами бензену (152 х
*3 = 456 кДж/моль). Звідси можна зробити висновок, що антрацен термодина-
мічно менш стійкий, ніж нафтален, не кажучи вже про бензен.
34.2.3. Фізичні властивості антрацену
Антрацен - безбарвна кристалічна речовина з
температурою плавлення 217 °С, сублімується. Для його
розчинів спостерігають фіолетову флуоресценцію.
• УФ-спекшроскопія. В УФ-спектрі антрацену є
інтенсивні смуги з А,тах 186,221,256 і 375 нм.
• ПМР-спектроскопія. Як приклад, наведені хімічні
зсуви сигналів протонів антрацену:
м.ч. 8,31 (а), 7,91
(Ь),7,39(с)
34.2.4. Синтез антрацену
❖ Дієновий синтез. З використанням и-бензохінону як дієнофілу за
методом Дільса-Альдера одержують трициклічну систему антрацену:
О О
Декагідроантрацен
❖ Реакція Фріделя-Крафтса. Взаємне алкілування двох молекул бензил-
хлориду дає 9,10-дигідроантрацен, що легко зазнає дегідрування або окиснення
в антрацен:
874 Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
СН,С1
сісн
і.
9,10-Дигідроантрацен
■ Ацилювання бензену за Фріделем-Крафтсом фталевим ангідридом
приводить до о-бензоїлбензойної кислоти, яка замикається під дією фосфор (V)
оксиду в антрахінон:
9 о
о+н І—Ч II II
р
о
о-Бензоїлбензойна
кислота
О
Антрахінон
❖ Реакція Вюрца-Фіттіга. Використання о-бромобензилброміду за умов
реакції Вюрца-Фіттіга також приводить до 9,10-дигідроантрацену, що далі
перетворюється в антрацен:
Н. Н
Вг ВгСН2
І + 4Иа +
СН2Вг
Вг
н н
❖ За Р. Левіною1 можна одержати антрацен і його похідні в разі нагрівання
о-бензилтетрагідробензойної кислоти з фосфор (V) оксидом і сіркою:
.і
о;Счон
Р205
о
34.2.5. Хімічні властивості антрацену
❖ Реакції антрацену як дієну. З даних рентгеноструктурного аналізу
бачимо, що електронна густина в антрацені розподілена ще нерівномірніше, ніж
1 Левіна Р. (1900-1970) - радянський хімік. Головні праці стосуються хімії вуглеводнів. Запро-
понувала синтез циклопропанових похідних, відкрила ацетилен-дієнове перегрупування, а також
ароматизацію аддуктів дієнового синтезу.
Розділ 34. Конденсовані ароматичні системи
875
у нафталені; менша енергія резонансу свідчить про те, що антрацен (порівняно з
нафталеном або, тим більше, з бензеном) термодинамічно нестійкий і виявляє в
різних реакціях ненасиченість.
Найактивнішими атомами Карбону в антрацені є атоми 9 і 10, які ще
називають л/езо-карбоновими атомами. Вони відрізняються за хімічною
поведінкою від ароматичних бензенових атомів ще більше, ніж а-атоми
нафталену, і наближаються до крайніх атомів Карбону дієнових спряжених
систем. Приєднання по атомах 9 і 10 не пов'язане з великою втратою резонансу.
• Дієновий синтез. Антрацен легко приєднує малеїновий ангідрид. Аддукт
зазначеної будови вже за слабкого нагрівання розпадається по утворених о-
зв'язках, регенеруючи вихідні продукти, чим користуються для очищення
антрацену і його кількісного визначення:
Під час взаємодії антрацену з дегідробензеном за схемою дієнового синтезу
утворюється триптицен Віттіга:
• Димеризація. Під час опромінення ультрафіолетом антрацен зазнає димеризу-
вання в важкорозчинний діантрацен, що в темряві спонтанно деполімеризується в
мономер:
• Окислення. Кисень у разі УФ-опромінення також приєднується до антрацену
в положення 9 і 10, утворюючи ендопероксид, що вибухає під час нагріванні:
• Мсталювання. Подібно до дієнових вуглеводнів антрацен приєднує лужні
метали. Наприклад, з натрієм утворюється 9,10-динатрій 9,10-дигідроантрацен -
речовина, що окиснюється не так легко:
876 Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є Органічна хімія
+ 2Ш
Ш Н
❖ Реакції електрофільного заміщення. Цікаво, що під час приєднання
реагентів у л*езоположення утворюється нова молекула з двома незалежними
бензеновими кільцями з мінімальною втратою енергії резонансу. У випадку реакції
електрофільного заміщення втрата енергії резонансу компенсована регенеруванням
ароматичності внаслідок руйнування о-комплексу й може бути перекрита завдяки
виграшу енергії спряження нового замісника з бензеновим кільцем.
• Галогенування. З хлором і бромом спочатку також відбувається приєднання
до атомів 9 і 10 і лише потім - відщеплення гідроген галогеніду:
Вг Н Вг
Вг Н
• Нітрування. Концентрована нітратна кислота окиснює антрацен в
антрахінон, а розведена нітратна кислота, розчинена в оцтовій, або нітроген (IV)
оксид нітрують його в положення 9:
+м2о4
• Сульфування. Концентрована сульфатна кислота легко сульфує антрацен в а-
положення (кінетичний чинник). Аналогічно до нітрування спочатку утворюються
похідні за атомами Карбону в положеннях 9 і 10 антрацену, що потім легко
зазнають шсозаміщення на атоми Гідрогену.
Розведена сульфатна кислота під час нагрівання дає Р-похідну. Сульфогрупа
в антрацені мало впливає на реакційну здатність інших кілець, тому він легко
утворює дисульфокислоти:
$03Н
Н038
ЯОзН Н038
803Н
Незважаючи на різні реакції приєднання й заміщення, антрацен витрачається
майже винятково для окиснення його в антрахінон, що широко використовують у
хімії барвників.
Розділ 34. Конденсовані ароматичні системи
877
34.2.6. Антрахінон і його похідні
Назва антрахінону походить від грец. ántrax - вугілля й кіпа — кора хінного
дерева. Незважаючи на те, що в молекулі антрахінону є два фрагменти а,Р-не-
насиченого кетону, за хімічними властивостями він скоріше нагадує
бензофенон. Його інертність у реакціях з дієнами пов'язана з неможливістю
залучення для синтезу л-зв'язків С=0-атомів, оскільки вони відповідальні за
забезпечення ароматичності кілець А і С. Руйнування ароматичності не тільки
не дало б виграшу енергії, а й призвело б до її втрати. Подібність антрахінону з
бензофеноном виявляється й у поведінці під час сплавлення з лугами, що
приводить до бензойної кислоти:
О
_.COONa
а| в II с l+2NaOH
Незважаючи на те, що весь антрацен переробляють на антрахінон,
антрахінону систематично не вистачає, і його одержують синтетичним шляхом
за реакцією Фріделя-Крафтса між фталевим ангідридом і бензеном, а також за
допомогою дієнового синтезу між 1,4-нафтохіноном і бутадієном і, нарешті,
димеризацією старену СбН5~СН=СН2.
■ Антрахінон відновлюють цинковим пилом у лужному середовищі в
антрагідрохінон, що здатний легко зазнавати окиснення на повітрі з утворенням
гідроген пероксиду. Цей процес використовують для одержання концентро-
ваного гідроген пероксиду:
+ Н202
■ Обробка антрахінону оловом у середовищі хлоридної й оцтової кислот
приводить до селективного відновлення однієї карбонільної групи й утворення
антрону:
О
Антрон не утворює оксиму й дає фіолетове забарвлення із ферум (III)
хлоридом. Проте, на відміну від фенолу, він легко відновлюється до 9,10-ди-
878 Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
гідроантрацену. На підставі цих даних і даних ІЧ-спектрів уважають, що
антрону властива кето-енольна таутомерія:
О ОН
Кетоформа термодинамічно вигідніша, ніж гідроксиформа. За групою СН2
антрон конденсується з альдегідами й кетонами, внаслідок чого утворюються
похідні бензантрону.
❖ Функціоналізація антрахінону. Однією з промислових операцій є
сульфування антрахінону. Під час нагрівання подвійного надлишку антрахінону
з димлячою сульфатною кислотою (20-40 % олеум) відбувається реакція
моносульфування. У цьому разі сульфогрупа вступає в Р-положення одного з
ядер. У випадку додавання в реакційну масу солей меркурію (II) сульфогрупа
спрямовується в а-положення. Всі ці сульфокислоти застосовують для синтезу
барвників.
Під час нагрівання а(Р)-антрахінонсульфокислот з лугом відбувається
заміщення сульфогруп на гідроксильні, і утворюються а(р)-гідроксіантрахінони.
Нагрівання сульфокислот з амоніаком і борною кислотою до 200 °С приводить
до утворення аміноантрахінонів, які використовують у синтезі так званих
індантренових барвників:
❖ Барвники на основі антрахінону. Найвідомішим і важливим
антрахіноновим барвником є алізарин. Будова його доведена 1868 р. К. Гребе й
К. Ліберманом1 на підставі таких фактів. У молекулі антрахінону є дві
1 Ліберман К. (ЬіеЬегтапп С.ТЬ.) (1842-1914) - німецький хімік, учень А. Байєра. Головні праці
присвячені синтезу фарбників. Разом з К. Гребе і Н. Каро розробив і впровадив у виробництво
алізарин. З'ясував природу протравного фарбування.
Розділ 34. Конденсовані ароматичні системи
879
карбонільні й дві гідроксильні групи. Під час перегонки з цинковим пилом він
перетворюється в антрацен. У разі окиснення алізарин дає фталеву кислоту.
Отже, обидва гідроксили перебувають в одному бензеновому ядрі. Тому для
алізарину можливі чотири гіпотетичні структури:
О О О ОН О
І II III IV
Вибір на користь формул (І) і (II) зроблений на засадах того, що алізарин
можна синтезувати із фталевого ангідриду й пірокатехолу:
HO ОН Q ОН О
О О
З одержуваних цим шляхом двох ізомерних продуктів один подібний до
природного алізарину, а інший не має властивості протравного барвника, тобто
не дає лаків з іонами Алюмінію, Хрому, що дало змогу приписати алізарину
структуру (І). З кінця 50-х років XX ст. алізарин втратив значення як барвник
для текстильних матеріалів через складну технологію фарбування. Його
використовують лише у виробництві художніх лаків і в поліграфії.
Із тривалентними металами алізарин дає внутрішньокомплексні забарвлені
нерозчинні сполуки, так звані лаки.
На утворенні лаків ґрунтується спосіб протравного фарбування: спочатку
на тканину наносять протраву - сіль металу, а потім - барвник. Залежно від
катіона металу алізарин забарвлює тканину в різні кольори: з алюмінієм
утворюється кумачевий, з хромом - фіолетово-чорний, з ферумом - коричневий
лак тощо.
/=\ ,о
880 Чирва В.Я., Ярллолюк CM., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
S03Na
+ NaOH + 0,
O O
Цей метод розроблений К. Ліберманом, К. Гребе й Н. Каро (1889).
Як протравні барвники, крім алізарину, використовують інші похідні
гідроксіантрахінонів.
■ До появи синтетичного алізарину (1875) головним його джерелом були
коріння марени фарбувальної {Rubia tinctorum), яку вирощували з давніх часів у
південних регіонах Європи й Азії, у тому числі й у Криму. Після появи
економічніших синтетичних методів ця галузь сільського господарства
збереглася тільки в окремих місцях Азії, де барвник використовують головно
для фарбування килимів. У марені фарбувальній алізарин міститься у вигляді
глікозиду такої будови:
ОН
т он
Руберитринова кислота
Під час вимочування коренів глікозид під впливом ферментів або
сульфатної кислоти розпадається на моносахариди й алізарин.
■ 3 Р-аміноантрахінону сплавленням його з лугом і наступним окисненням
одержують кубовий барвник індантрен синій:
О
о
Індантрен синій
■ Деякі барвники одержують із бензантрону, який синтезують під час
нагрівання антрахінону і гліцеролу з порошком заліза та сульфатною кислотою.
У цьому випадку спочатку гліцерол дегідратує до акролеїну з наступною його
конденсацією з антрахіноном:
■ Найчастіше використовуваний шлях синтезу алізарину полягає в одно-
часній дії на антрахінон-Р-сульфокислоту або 2-хлороантрахінон лугу, або
окиснювача (натрій нітрату чи хлорату):
О О ОН
Розділ 34. Конденсовані ароматичні системи
881
+ Н2С=СН-СНО
-2Н20
Під час сплавлення бензантрону з натрій гідроксидом за 225 °С відбувається
окисна конденсація його у віолантрон - темно-синій кубовий поліциклоке-
тоновий барвник, що належить до індантренового ряду:
Na2S204
О
Бензантрон
Віолантрон
NaO
ONa
"Куб"
■ Після попереднього хлорування або бромування бензантрону й сплав-
лення його з калій гідроксидом синтезується ізомер віолантрону - фіолетовий
кубовий барвник — ізовіолантрон {фіолетовий):
Ізовіолантрон
У цих барвниках усі цикли, що не містять кетонної групи, ароматичні.
Відновленням натрій гідросульфітом поліциклокетонові барвники переводять у
розчинні в лузі феноляте - "куби", у яких виварюють тканину, що адсорбує
"куб". На повітрі "куб" знову окиснюється в поліциклокетон і дає дуже міцне
фарбування. Подібне фарбування називають кубовим.
Індантренові й поліциклокетонові кубові барвники часто поєднують під
загальною назвою індантренових. їх уважають одними з найцінніших барвників,
які перевершують за якістю азобарвники.
Розділ 35
поащикаічніарома тичні
системи з ізольованими
циклами
Із вуглеводнів з ізольованими ароматичними ядрами найбільший інтерес
становлять біфеніл, дифенілметан і трифенілметан.
35.1. Біфеніл
35.1.1. Номенклатура IUP АС
Молекула біфенілу має спеціальну нумерацію - локанти
1 і Г привласнені атомам Карбону, що з'єднані а-зв'язком.
Похідні біфенілу називають за загальними правилами з
5' б 65 огляду на те, що цифри без штрихів привласнюють циклу з
меншим локантом.
Н3С3 2 / *'
З-Метил-4-етилбіфеніл
З-Метил-З'-нітробіфеніл
Цифри без штрихів привласнені циклу з
меншим локантом.
За однаковості локантів цифри без штрихів
привласнені циклу з першим за алфавітом
замісником.
35.1.2. Будова біфенілу
Молекула біфенілу лежить в одній площині. За нормальних умов немає
обертання навколо о-зв'язку, тому що я-електронні системи обох циклів
перебувають у спряженні.
Біфеніл - типовий ароматичний вуглеводень, у якому обидва ядра більш
незалежні один від одного, ніж можна було очікувати з огляду на його плоску
структуру й зменшену порівняно з нормою відстань між зв'язаними простим
зв'язком атомами Карбону двох бензенових ядер (0,148 нм замість 0,154 нм). Ця
відстань ясно виражає відтінок додаткового зв'язування, тобто перекривання л-
електронних хмар двох ядер:
Розділ 35. Поліциклічні ароматичні системи з ізольованими циклами 883
35.1.3. Ізомерія
За звичайної температури біфеніл має планарну структуру. Однак за високої
температури вона порушена внаслідок обертання бензенових кілець навколо а-
зв'язку. Зазначимо, що таке обертання неможливе у випадку наявності замісників у
бензенових кільцях у положеннях 2 і 6. На прикладі заміщених біфенілів з'ясовано,
що для них можуть існувати оптично активні ізомери - дзеркальні антиподи, навіть
у тому випадку, коли сполука не містить асиметричних атомів Карбону. Наприклад,
молекула 2,2'-динітро-6,6'-дикарбоксибіфенілу не має площини симетрії й може
бути зображена у вигляді двох несумісних у просторі структур, які й виділено в
чистому вигляді.
Ця ізомерія пов'язана з тим, що арильні залишки містяться в різних площинах,
унаслідок чого неможливий поворот навколо осі С-С, що зв'язує два ядра. Йому
заважає вандерваальсове відштовхування груп, що перебувають у оположеннях
аренів. У підсумку молекула не має площини симетрії, і її дзеркальне відбиття не
тотожне оригіналу:
Такий різновид ізомерії назвали атропоізомерією (від грец. тропос -
поворот, а - заперечення). Вона характерна для сполук у деяких випадках з
двома і навіть одним об'ємним замісником, що зменшує можливість обертання
бензенових кілець.
35.1.4. Одержання біфенілуі його похідних
❖ Метод Б ер під о. Біфеніл можна одержати, пропускаючи пари бензену
через розпечену до 800 °С залізну трубку:
2 С6]\ — С6Н5-С6Н5
❖ Реакція Вюрца-Фіттіга. Одержання біфенілу в цій реакції підтверджує
його будову:
2С6Н5Вг + 2Ш^+ С6Н5-С6Н5 + 2ШВг
❖ Реакція Гриньяра. Біфеніл синтезується як побічний продукт у синтезі
884
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
реактиву Гриньяра з галогенобензену й магнію за реакцією, подібною до реакції
Вюрца-Фіттіга:
2 С6Н5Вг + Мё ОД-ед + МёВг2
Високі виходи біфенілу отримують у синтезі з реактивом Гриньяра за
наявності купрум (II) хлориду:
2СбН5МёВг С6Н5-С6Н5
Певне значення мають способи синтезу сполук ряду біфенілу, що ґрун-
туються на побудові одного з двох бензенових кілець:
О=0 + л-СН3С6Н4М8Вг — «-НзС-ОД-^ Н3С^О^О) + 2Н2
ОН
❖ Реакція Гомберга. Біфеніл і його різноманітні похідні можна одержати,
діючи сіллю діазонію за наявності натрій ацетату на бензен і заміщені бензени:
Х-С6Н;№Ы СН3Чо_ + С6Н5-У -Х-С6Н4-С6Н4-У + Ы2 + СН3СООН
Оскільки реакція відбувається гомолітично, то місце вступу радикала,
одержаного з діазосполуки, у бензенове кільце не залежить від характеру
орієнтанта. Тому зазвичай одержують суміш о-, п- і л*-заміщених біфенілів з
переважанням «-ізомеру.
❖ Реакція Гаттермана. Біфеніл з домішкою терфенілу С6Н5-СбН4-СбН5 і
кватерфенілу СбНз-СбНЦ-СбЩ-СбНз - звичайний продукт гомолітичного розпаду
солей діазонію. Його одержують шляхом взаємодії двох фенільних вільних
радикалів. Терфеніл і кватерфеніл також утворюються внаслідок реакції
Гомберга між біфенілом і солями діазонію.
❖ Реакція Ульмана. Металеву мідь з успіхом застосовують як каталізатор
для синтезу біфенілу з іодобензену за реакцією Ульмана:
2С6Н5І С6Н5-С6Н5
❖ Метод Корі-Хауса, який успішно використовують для алканів, засто-
совують і в цьому випадку:
(СбН5)2СиІл С6Н5-С6Н5
❖ Р. Левіна запропонувала метод синтезу ароматичних вуглеводнів, що
приводить до біфенілу і його похідних, він ґрунтується на дієновій конденсації
2-арилбутадієну з малеїновим ангідридом:
Розділ 35. Поліциклічні ароматичні системи з ізольованими циклами
885
^ Т| 1 + 2 СО + Н20
❖ Бензидинове перегрупування. Найважливішою похідною біфенілу є
бензидин - и,я'-діамінобіфеніл, який одержують у техніці бензидиновим
перегрупуванням гідразобензену:
о-н-н-о ^оон>
Аналогічно синтезують о-толідин і о-діанізидин:
н3с-о о-сн3
Будову їх як похідних біфенілу моясна довести діазотуванням обох
аміногруп і наступним елімінуванням діазогруп шляхом заміни їх на атом
Гідрогену. Ці реакції у випадку бензидину приводять до біфенілу, а у випадку о-
толідину й о-діанізидину - до відповідних заміщених біфенілів. У бензидині л-
положення аміногруп випливає з того факту, що в разі введення електрофілу,
наприклад, атома галогену, бензидин дає єдиний монозаміщений продукт.
35.1.5. Реакції біфенілу і його застосування
За хімічними властивостями біфеніл нагадує бензен. Цікаво, що фенільна
група є донором електронів і спрямовує атакувальний електрофіл в о-, п-
положення. Наприклад, під час нітрування біфенілу одна нітрогрупа вступає
переважно в «-положення стосовно атома Карбону, що несе фенільний залишок,
а інша - теж у «-положення іншого ядра:
Біфеніл 4,4'-Динітробіфеніл
■ Біфеніл - висококипляча стійка речовина, застосовують як високотемпе-
ратурний теплоносій.
■ Бензидин і його похідні широко використовують для синтезу азо-
барвників, як пом'ягшувачі гуми, реагенти на окремі катіони та як окисно-
відновні індикатори в об'ємному аналізі аналітичної хімії.
886
Чирва В.Я., Ярмолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
35.2. Дифенілметан і його похідні
Для дифенілметану характерна відсутність безпосереднього сполучення
бензенових кілець, як це є у випадку біфенілу. Тому він більше нагадує бензен.
❖ Одержання дифенілметану. Для синтезу дифенілметану і його
похідних можна використати два варіанти реакції Фріделя-Крафтса:
ІС^ + СЩС^-^^ С6Н5-СН2-С6Н5 + 2 нс1
С6Н6 + С6Н5СН2с1 С6Н5-СН2-С6Н5 + нс1
Деякі розглянуті нами сполуки, такі як бензофенон СбН5СОСбН5 і продукт
його відновлення - вторинний спирт бензгідрол СНг-СНОН-СбН5, так званий
кетон Міхлера, одержуваний дією фосгену на Л^М-диметиланілін, є похідними
дифенілметану.
❖ Хімічні властивості дифенілметану. Метиленова ланка дифенілметану
легко зазнає окиснення, утворюючи бензофенон:
с6н5сн2с6н5-^ с6н5-^-с6н5
о
■ Під час пропускання пари дифенілметану через нагріту до червоного
кольору трубку утворюється 0,0 -дифеніленметан - флуорен:
П'ятичленне кільце флуорену виявляє подібність до циклопентадієну й
індену, що відображене, насамперед, у легкій протонізації атомів Гідрогену
метиленової групи в реакціях з '■ високоосновними реагентами, наприклад,
метилмагнійіодидом СНзМ§І і в реакціях конденсації з альдегідами:
❖ Дифенілметан і його похідні застосовують переважно в синтезі барв-
ників і парфумерній промисловості.
Розділ 35. Поліциклічні ароматичні системи з ізольованими циклами
887
35.3. Трифенілметан
Властивості бензенових ядер у трифенілметані такі ж, як у звичайних
алкілбензенах. Особливості їхньої хімічної поведінки виявляються у
властивостях С-Н-зв'язку аліфатичної ("метанової") частини молекули. Легкість
гетеро- або гомолітичного розриву цього зв'язку залежить, насамперед, від
можливості делокалізації позитивного або негативного заряду, що виникає, у
випадку гетеролітичного розриву або неспареного електрона у випадку
гомолітичного розриву. У трифенілметановій системі можливість такої делока-
лізації винятково велика.
Розглянемо здатність фенільованих метанів до дисоціації С-Н-зв'язку з
відщепленням протона (СН-кислотність). Сила СН-кислот, як і звичайних
протонних кислот, визначена стійкістю, а отже, і легкістю утворення відпо-
відних аніонів (у розглянутому випадку - карбаніонів). Стійкість і легкість
їхнього утворення, відповідно, визначені можливістю делокалізації в них
негативного заряду. Кожне бензенове ядро, пов'язане з бензильним атомом
Карбону, бере участь у делокалізації негативного заряду, який виникає на ньому,
що можна зобразити за допомогою граничних (резонансних) структур:
Оскільки зі збільшенням кількості граничних структур зростає здатність до
делокалізації, то трифенілметил-аніони повинні мати особливо високу стійкість.
З огляду на це можна очікувати, що СН-кислотність фенілметанів збіль-
шуватиметься зі збільшенням кількості фенільних кілець, які можуть брати
участь у делокалізації заряду на центральному атомі Карбону, тобто зростати в
такому ряді:
СН4 < С6Н5СН3 < (С6Н5)2СН2 < (С6Н5)3СН.
888
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Значення рКа цих вуглеводнів, визначені спеціальними методами, під-
тверджують це припущення. Дифенілметан (рКа 33) за кислотністю приблизно
дорівнює амоніаку, а трифенілметан (рКа 31,5) - юрею-бутиловому спирту;
трифенілметан як кислота більш ніж у 1010 разів сильніший, ніж метан.
35.3.1. Синтез трифенілметану
❖ Реакції Фріделя-Крафтса. Трифенілметан і його похідні найпростіше
синтезувати з хлороформу й бензену тією ж реакцією, що й дифенілметан:
з + СНСІз (С6Н5)3СН+з неї
❖ Реакція Гриньяра. Крім синтезу за Фріделем-Крафтсом, сполуки три-
фенілметанового ряду можна отримувати за допомогою реактиву Гриньяра:
(С6Н5)2СО + С6Н5МёВг—-(С6Н5)3С-ОМ§Вг^(С6Н5)3СОН + МвВЮН
С6Н5СООС2Н5 + 2 С6Н5М§Вг (С6Н5)
С-ОМ£Вг + С2Н5ОГ^Вг
н2о
(С6Н5)3С-ОН + MgBЮH
❖ Реакції конденсації карбонільних сполук. У промисловості багато
сполук цього ряду одержують конденсацією ароматичних оксосполук з
вуглеводнями -за жорстких умов (концентрована сульфатна кислота) або за
м'якших умов з ароматичними сполуками, які містять сильні о-, и-орієнтанти,
що є активаторами:
н яо
СбН5СНО + 2 С6Н«р^ (С6Н5)3СН + Н20
/СбН4К(СН3)2
С6Н5СНО + 2 С6Н5-К(СН3)2-^-С6Н5-СН + н2о
С6Н^(СН3)2
од)^-сбн4-с-сд-к(от3)2 + с6н5-к(снз)2^. [(от3)2кед]3с-он
о
❖ Реакції конденсації. Застосовують конденсацію ароматичних амінів із
трихлорометилбензеном:
СІ
М(СН3)2—СС13 + £^м(Снз)2— Ы(СНз)2-Ч^-4-^^-Ы(СН3)2
Розділ 35. Поліциклічні ароматичні системи з ізольованими циклами
889
353.2. Трифенілметиланіон
Центральний атом Карбону в трифенілметиланіоні
перебуває в лр2-гібридизації, однак ця структура не є
плоскою через вандерваальсове відштовхування атомів
Гідрогену в оположеннях фенільних груп, що приводить до
повертання бензенових кілець на 30-40° ("повітряний
гвинт"). Аналогічну будову мають трифенілметильний
катіон і радикал трифенілметилію. Трифенілметил-аніон
Забарвлений у вишневі кольори трифенілметилнатрій зазвичай одержують
відновленням трифенілхлорометану натрій амальгамою:
(С6Н5)3СС1 п > (СНЛСЫа
V о 5/3 В атмосфері азоту 4 6і ^/З^1^"
Трифенілметилнатрій - винятково реакційноздатна речовина, що швидко
розкладається водою або спиртом:
(С6Н5)3СЫа + Н20 (С6Н5)3СН + ШОН
З карбон (IV) оксидом і оксосполуками він реагує подібно до реактиву
Гриньяра:
СА + сбн5
СбН5-С N3 + С02 С6Н5-С-СООКа
С6Н5 С6Н5
Кисень повітря окиснює трифенілметилнатрій у пероксид трифенілметилу:
2 (С6Н5)3СКа + 2 02 [(СбН5)3-СО)]2 + №202
Визначальний вплив делокалізації негативного заряду на стійкість
відповідного карбаніону підтверджений на прикладі трис-л-нітрофенілметану:
натрієва похідна утворюється вже під час обробки його спиртовим розчином
натрій гідроксиду, який на трифенілметан не діє. Отриманий аніон має синє
забарвлення й стійкий до дії вологи й кисню повітря:
(«-Ш2С6Н4)3СН (и-Ш2СбН4)3С Ка-
У цьому випадку в делокалізації негативного заряду в аніоні беруть участь
три бензенові ядра й три нітрогрупи. Для кожного з и-нітрофенільних залишків в
аніоні можливі п'ять граничних структур:
890 Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Аг "О' \=/ 'ЧАг
Звичайно, розосередження заряду по атомах Карбону відіграє деяку роль і в
тринітрофенілметил-аніоні, де заряд більше зосереджений уже на атомах
Оксигену нітрогруп.
353.3. Трифенілметил-катіон
Інший різновид гетеролітичного розщеплення бензильного СН-зв'язку -
відривання гідрид-іона з утворенням відповідних карбокатіонів бензильного типу:
С6Н5СН2-Н — С6Н5СН2+ (С6Н5)2СН-Н — (С6Н5)2СН+
(С6Н5)3С-Н — (С6Н5)3С+
Оскільки бензенові ядра здатні стабілізувати як позитивний, так і
негативний заряди, то фенільовані метани за гідридною рухливістю атома
Гідрогену в аліфатичній частини утворять такий самий ряд, що й за протонною
рухливістю, тобто
СН4 < С6Н5СН3 < (С6Н5)2СН2 < (С6Н5)3СН.
Однак експериментально зрівняти легкість відривання гідрид-іона, як
звичайно, буває важко, оскільки для цього процесу здебільшого використовують
досить активні кислоти Льюїса. Порівняльні оцінки легко можна зробити
шляхом зіставлення рухливості атома галогену (зазвичай Хлору) за умов Бні-
реакцій, оскільки в цьому випадку, як і в разі відщеплення гідрид-іона, стадією,
що визначає швидкість перетворення, є утворення відповідного карбокатіону:
Аг-СЯ2-С1 -> АгСЯ2+ + СГ; де Я = Н або Я = Аг
Справді, виявилося, що в зазначених умовах найбільшу рухливість атом
Хлору має в трифенілхлорометані, а найменшу - у бензилхлориді. Швидкість
реакції зменшується в ряді (СбН5)3С-С1 > (СбН5)2СН-С1 > СбН5СН2-С1.
Реакційна здатність атома Хлору в трифенілхлорометані нагадує реакційну
здатність у хлороангідридах карбонових кислот, а в бензилхлориді - реакційну
здатність в алілхлориді. У табл. 35.1 наведені дані про відносні швидкості
сольволізу хлоридів у мурашиній кислоті за 25 °С:
Роздл 35. Поліциклічні ароматичні системи з ізольованими циклами 891
К-СІ + н-с
Р
с-н + неї
он
Я-0
Таблиця 35.1
Відносні швидкості сольволізу хлоропохідних
Відносні
Я
Відносні
швидкості
швидкості
Н2С=СН-СН2-
0,04
(С6Н5)2СН-
300
СбН5—СНг—
0,08
(С6Н5)3С-
3-Ю6
(СН3)3С-
1
Порівняльна стійкість трифенілметильного (тритильного) катіона підтер-
джена також багатьма іншими експериментальними даними. Прикладом може
слугувати легкість утворення його солей з ненуклеофільними аніонами, розчини
яких у полярних апротонних розчинниках типу Л^Д-диметилформаміду (ДМФА)
електропровідні. Вони мають іонну будову і характерно забарвлені в жовті
кольори:
(с6н5)3сон(СНзСО)г0 ^ (С6Н5)3С ріу ■
Про це ж свідчить здатність трифенілхлорометану до дисоціації на
трифенілметил-катіон і хлорид-аніон у розчині рідкого сульфур (IV) оксиду:
802 (рідкий) .
рь3с-сі = рь3с++ сі
❖ Тритильний захист. Висока реакційна здатність атома Хлору та
великий об'єм вуглеводневого залишку в трифенілхлорометані, а також легкість,
з якою можна генерувати трифенілметильний катіон, зумовили можливість
використання цього хлориду для захисту первинних гідроксильних груп у
молекулах сполук, які модифікують. Трифенілхлорометан, реагуючи зі спиртами
за наявності піридину, здатний утворювати відповідні етери. Однак просторові
утруднення, спричинювані об'ємною трифенілметильною групою, приводять до
того, що в цю реакцію вступають тільки спирти з найменш екранованою
гідроксильною групою, тобто первинні спирти. Отже, виявилося можливим
одержувати трифенілметилові етери спиртів тільки за первинними гідроксиль-
ними групами за наявності в молекулі вторинних або третинних, тобто
регіоспецифічно. Це дає змогу модифікувати ці спирти (наприклад, окиснювати,
алкілувати), зберігаючи незачепленим первинний гідроксил:
892
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
а-сн(он)сн,он ТгС1 » к-сн(он)сн2отг-^асосн2отг
2 Піридин; 25°С 1 рН 12
Тг=С(СбН5)з
Трифенілметилові етери зазвичай називають тритиловими. Вони стійкі в
нейтральному й лужному середовищах, тобто за умов, у яких виконують
модифікацію, однак легко зазнають розщеплення протонними кислотами з
регенеруванням первинної спиртової групи, що є наслідком легкості утворення
трифенілметильного катіона:
я-с-а^отг-^
О
К-С-СН^О-Тг
о н
— Я-С-С^ОН* [(С6Н5)3С]+-^ (ОД)3СОН
о -н
353.4. Трифенілметанові барвники
Стійкість трифенілметильного катіона можна збільшити введенням у
бензенові кільця електронодонорних груп (наприклад, аміно-, алкіл- і
діалкіламіно-, гідроксильної, алкоксильної). Подальше збільшення стійкості
карбокатіону приводить до ситуації, коли він стає стійким навіть у водному
розчині, тобто рівновага реакції
Я++Н20=ї=^ ЯОН + Н+
зсувається ліворуч. Подібні тритильні катіони не тільки стійкі, а й забарвлені.
Прикладом може слугувати інтенсивно забарвлений у фіолетовий колір /ир/с-(4-ди-
метиламінофеніл)метильний катіон. Його хлорид застосовують як барвник -
кристалічний фіолетовий. У кристалічному фіолетовому позитивний заряд
розосереджений між трьома атомами Нітрогену й дев'ятьма атомами Карбону
бензенових ядер. Участь одного з трьох л-диметиламінофенільних замісників у
делокалізації позитивного заряду можна відобразити за допомогою таких
граничних структур:
• (сн3)2к^З-сСА— (сн3)2к-{3=с;Аг
\—' Аг 4—' Аг
(СН3)2К^}=С^(СН3)2КЦЗ=С^СН3)2К^>=<
+ Аг Аг Аг
Усі три фенілметанові барвники, що містять амінні або заміщені амінні
групи в бензеновому кільці, отримують забарвлення в кислому середовищі, що,
Аг
Розділ 35. Поліциклічні ароматичні системи з ізольованими циклами
893
як з'ясовано вище на прикладі кристалічного фіолетового, сприяє виникненню
структури з протяжним ланцюгом спряження, так званої хіноїдної структури.
Унаслідок делокалізації заряду катіона й аніона між двома й більше
атомами, зв'язаними системою 7г,7с-спряження подвійних зв'язків, рівень енергії
першого збудженого стану молекули завжди зменшується. Тому такі молекули
збудасувані квантами меншої енергії, тобто випромінюванням більшої довжини
хвилі порівняно з УФ-випромінюванням, що збуджує безбарвний трифеніл-
хлорометан. Якщо безбарвний трифенілхлорометан дисоційований на йони,
наприклад, у розчині 802, то його катіон зазнає збудження фіолетовими
квантами, поглинає їх і тому має додаткове до фіолетового жовте забарвлення. У
разі переходу до катіона кристалічного фіолетового, що має різко знижений
енергетичний рівень у збудженому стані, довжина хвилі збуджених і тому
поглинених квантів різко переміщається в ділянку жовтого спектра, виникає
фіолетове забарвлення. Подібна ситуація з усіма трифенілметиловими катіонами
й аніонами, і всі вони мають інтенсивне забарвлення. Однак як барвники
застосовують лише сполуки зі стійкими катіонами.
❖ Головна група трифенілметанових барвників - це амінопохідні три-
фенілхлорометану. Всі вони побудовані за одним із трьох типів:
Я = Н, Аік, СН2С6Н5, С6Н5
■ Барвники першого типу не мають практичного значення.
■ До другого типу належать важливі барвники: малахітовий зелений
(Я=СНз), діамантовий зелений (]1=С2Н5), а також один з найпростіших за
структурою, але не застосовуваний на практиці фіолетовий Дебнера1 (К=Н).
Звичайні методи синтезу барвників цього типу проілюструємо на прикладі
фіолетового Дебнера й малахітового зеленого:
Фіолетовий Дебнера
1 Дебнер О. (ИбЬпег О.) (1850-1907) - німецький хімік, досліджував реакції конденсації. Є автором
іменних реакцій (реакція Дебнера, реакція Дебнера-Міллера).
894
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
н>4 /г^°Хм <СН=,..,НЛ ^ ,сн
н,с' ITS ^ сн,-н'°н,с' снз
Лейкооснова малахітового зеленого
Карбінольна основа Малахітовий зелений
Нагрівання амінів із трихлорометилбензеном приводить безпосередньо до
утворення катіона барвника, як у випадку фіолетового Дебнера. Під час
конденсації амінів з бензальдегідом утворюється амінопохідна трифенілметану -
безбарвна речовина, яку називають лейкоосновою (від грец. leucos - білий), яка
легко окиснюється вже повітрям або плюмбум (IV) оксидом у карбінол - основу
барвника. Під дією навіть розведеної хлоридної кислоти вона віддає гідроксил і
утворює катіон барвника.
■ До барвників третього типу належать парафуксин і фуксин. Фуксин
відрізняється від парафуксину тим, що в одному з бензенових кілець в о-
положенні щодо аміногрупи є метальна група.
Першим представником трифенілметанових барвників був фуксин або
розамін, отриманий окисненням аніліну, що містить домішку о- і я-толуїдинів,
арсен (V) оксидом:
NH2 NH,
. СН, As,05>HCl
Під час окиснення тим же реагентом або нітро-
бензеном суміші аніліну з ^-толуїдином отримано п—л ^ /=\
парафуксин: \ / 0(А /Ґ
Сі"
Новий "фуксиновий процес" полягає в нагріванні аніліну з формаліном і
окиснювачем. Передбачають таку послідовність реакцій:
Розділ 35. Поліциклічні ароматичні системи з ізольованими циклами
895
Шляхом алкілування парафуксину метиліодидом одержують барвник
метиловий фіолетовий (дуже подібний за кольором на кристалічний
фіолетовий) - суміш метильованих за атомом Нітрогену яу?мс-я-амінофеніл-
хлорометанів з переважанням пентаметильної похідної, поряд з цим отримують
також і гексаметильну похідну {кристалічний фіолетовий). Метиловий фіоле-
товий загальновідомий, тому що з нього роблять фіолетове чорнило.
Кристалічний фіолетовий у чистому вигляді одержують конденсацією біс-п-
диметиламіно-бензофенону (кетон Міхлера) з І^ІУ-диметиланіліном і наступним
підкисненням:
Як бачимо з наведеного вище, аміногрупи трифенілметанового барвника-
катіона відіграють вирішальну роль у стійкості катіона та його світлопоглинанні;
такі групи називають ауксохромами. Обидва ефекти є наслідком розосередження
заряду з метанового атома Карбону на атоми Нітрогену, однак ці ефекти діють
непаралельно. Зі збільшенням у трифенілметильному катіоні кількості ауксохромів
у ядрі від одного до двох і далі до трьох стійкість катіона зростає, а швидкість
знебарвлення основою зменшується. Це відбувається внаслідок зняття щораз
більшої частини заряду з метанової частини молекули, і з огляду на це щораз
більшого зростання енергії активації взаємодії сполуки з гідроксильним іоном. Що
стосується світлопоглинання, то зі збільшенням кількості ауксохромів від одного до
двох жовтий або жовтогарячий колір, який доповнює колір, що поглинає -
фіолетовий, переходить у зелений (поглинається пурпурно-червоний), а в разі
введення трьох ауксохромів - у фіолетовий (відбувається поглинання жовтого або
жовтогарячого), тобто батохромний ефект максимальний у випадку двох, а не трьох
ауксохромів. Це відбувається тому, що рівень енергії першого збудженого стану
досить близький до незбудженого, якщо заряд делокалізований лінійно, а не
розгалужено, як у випадку трьох ауксохромних груп.
896
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Ауксохромні групи діють завдяки наявності в атомів Нітрогену неподілених
пар електронів. Якщо ці електронні пари "зв'язати", приєднуючи до них іони
Гідрогену дією кислоти або перетворенням у четвертинну амонієву сіль під дією
метиліодиду, то хімічна й оптична дія ауксохромів припиниться. Тому, додаючи
до розчину кристалічного фіолетового концентровану хлоридну кислоту, можна
спостерігати поступовий перехід від фіолетового кольору до зеленого, точно
подібного на колір малахітового зеленого (вимикається один ауксохром), потім
до помаранчево-жовтого (вимикаються два ауксохроми) і, нарешті, до слабко
забарвленого жовтого розчину.
Будову парафуксину визначили Е. і О. Фішери1 шляхом діазотування
первинних амінофункцій його лейкооснови й подальшого елімінування діазо-
груп, унаслідок чого отримано трифенілметан:
(Н2Ы-С6Н4)3СН + ЗНС1 -(С1НзИ-С6Н4)зСН ™°2 ' (С1ЙЕЫ-С6Н4)3СН
СНзСН2°? (С6Н5)3СН+ЗСН3СНО+ И2 +ЗНС1
Діазотування парафуксин-катіона й заміна діазогрупи на гідроксили кип'я-
тінням з водою приводить до утворення аурину:
(С1>М^сбН4)3СС1 + З Н20—-— (НО-С6Н4)3СС1 —
о n2, -3 нсл
ОН аурин
З фуксину цим шляхом синтезують розолову кислоту, а з фіолетового
Дебнера - бензаурин:
ОН ^
Розолова кислота Бензаурин
Усі три речовини - барвники трифенілметанового ряду з гідроксилами як
ауксохромами. Як бачимо з формул, гідрокситрифенілметанові барвники є
похідними фуксону, який можна одержати, відщеплюючи воду нагріванням від
гідрокситрифенілкарбінолу:
1 Фішер О. (Fischer О.) (1852-1932) - німецький хімік. Головні праці присвячені синтетичним
фарбникам. Одержав малахітовий зелений. Разом з Е. Фішером визначив структуру розаніліну.
Вперше синтезував фенілгідразин.
Розділ 35. Поліциклічні ароматичні системи з ізольованими циклами 897
У лужному розчині ці барвники різко поглиблюють кольори, особливо
бензаурин (батохромний зсув ділянки поглинання); жовтий аурин і розолова
кислота в лужному середовищі стають червоними, а бензаурин навіть фіолето-
вим. Усі три речовини застосовують як індикатори:
X
Формули відображають розподіл зарядів в аніоні бензаурину (Х=Н) і в
аурині (Х=ОН). До речі, в аурині, що містить три ауксохроми, делокалізація
заряду розгалужена, і поглиблення кольору менше.
Бензенові ядра повинні впливати на стійкість трифенілметильного радикала так
само, як і на трифенілметильні аніони і катіони, що розглянуто вище. У цьому
випадку легкість розриву зв'язку, утвореного центральним атомом Карбону з
"нефенільним" замісником, зумовлена частково й іншими причинами. Річ у тому,
що в трифенілметані, трифенілхлорометані, трифенілкарбінолі тощо центральний
атом Карбону перебуває в ^р3-гібридному стані й, відповідно до цього, має
тетраедричну конфігурацію. З огляду на це фенільні ядра розташовані не в одній
площині й не спряжені. У разі переходу до трифенілметилкатіону (гетеролітичний
розрив) або радикала (гомолітичний розрив) центральний атом Карбону зазнає
регібридизації до зр2-гибрідного стану. Внаслідок цього структура стає плоскою, і
взаємодія (сполучення) між трьома фенільними ядрами посилюється. Це частково
компенсує енергетичні витрати, пов'язані з розглянутою дисоціацією, і в такий
спосіб полегшує її.
Трифенілметанові барвники використовують для забарвлення різних
тканин, шкір, для готування чорнила й паст, як індикатори.
898
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
35.3.5. Трифенілметилрадикал
Трифенілметильний радикал можна генерувати з відповідного хлориду дією
цинку, міді або срібла, які в цьому випадку є донорами електронів:
(с6н5)3с-С1
(с6н5)3с +а8С1
Цей радикал досить стійкий у розведених розчинах (етер, бензен) і зазнає
димеризування лише частково. Тривалий час цьому димеру приписували
структуру гексафенілетилену, однак виявилося, що насправді під час димери-
зації виникає зв'язок між центральним атомом Карбону одного радикала й п-
положенням одного з фенільних ядер іншого:
2РЬ3с
рь3с-срь3
/=\ /СбН5
2(с6н5)3с +'^=/=сч
с6н5
(с6н5)3с /=\ с/с6н5
н \=/ \
с6н5
Очевидно, у розглянутому випадку один трифенілметильний радикал атакує
найменш просторово утруднене місце іншого, причому, природно, одне з тих
місць, що бере участь у делокалізації неспареного електрона.
35.3.6. Фталеїни, флуоресцеїн, роз аміни
й родаміни
■ Фенол і фталевий ангідрид під час нагрівання з сульфатною кислотою
утворюють фенолфталеїн. У хімії ця сполука відома як індикатор, у медицині - за
назвою пурген - як проносне. Під дією лугу безбарвний фенолфталеїн (І) утворює
червоно-фіолетовий аніон (II) з таким же характером розподілу аніонного заряду
між двома бензеновими ядрами, як у бензаурині. Надлишок лугу, як завжди,
знебарвлює трифенілметановий барвник, перетворюючи його в карбінол (III):
и ш
■ Відомі й інші індикатори - фталеїни. Під час сплавлення фталевого
ангідриду з резорцинолом за наявності цинк хлориду відбувається подібна
Розділ 35. Поліциклічні ароматичні системи з ізольованими циклами
899
конденсація, ускладнена додатковим виділенням молекули води з двох
фенольних гідроксилів і утворенням гетероциклічного кільця. У лужному
середовищі з розмиканням лактонного циклу утворюється аніон флуоресцеїн,
який інтенсивно флуоресціює жовто-зеленими кольорами:
■ Бромування флуоресцеїну приводить до утворення барвника еозину -
тетрабромофлуоресцеїну (всі чотири оположення стосовно гідроксилів заміщені на
атоми Брому).
■ Конденсація фталевого ангідриду з л*-діалкіламінофенолами дає барвни-
ки, що мають практичне значення -родаміни:
ит.д.
■ Позбавлені карбоксилу родаміни називають розамінами. їх одержують
конденсацією трихлорометилбензену з лі-диалкіламінофенолами:
Розамін
Розділ 36
ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ.
П'ЯТИЧЛЕННІ ЦИКЛИ З
РАНИМ ГЕТЕРОАТОМОМ
Гетероциклічними називають сполуки із замкнутим ланцюгом, молекула яких
поряд із атомами Карбону містить щонайменше один некарбоновий - гетероатом.
Отже, уже вивчені нами сполуки оксид етилену, янтарний і фталевий ангідриди,
бутиролактон, гліколід, паральдегід, діоксан, фталімід і так далі, формально
належать до гетероциклів. Однак водночас ці речовини функціонально так тісно
пов'язані з "базовими" аліфатичними або ароматичними сполуками, що їх зручніше
було б розглянути у відповідних розділах.
З усіляких гетероциклічних сполук найважливішими є ароматичні
гетероцикли. Саме їм і приділимо головну увагу. Паралельно ми розглянемо й
відповідні гідровані системи, що належать до ароматичних гетероциклів, як
циклогексен і циклогексан - до бензену.
Аналогічно до карбоциклічних сполук п'яти- і шестичленні цикли найстійкіші.
Теоретично будь-який атом, здатний утворювати мінімум два ковалентні зв'язки,
може брати участь в утворенні кільця. Найліпше вивченими й поширеними є
циклічні молекули, що містять атоми Оксигену, Сульфуру та Нітрогену.
36.1. Класифікація й номенклатура гетероциклів
36.1.1. Класифікація
Гетероциклічні сполуки можна класифікувати за декількома ознаками,
наприклад:
за ароматичністю
за природою гетероатома
неароматичні
ароматичні
нітрогеновмісні
сульфуровмісні
о
N
Н
Оксолан
Оксол
Азол
Тіол
тетрагідрофуран
фуран
пірол
тіофен
Розділ 36. Гетероциклічні сполуки. П'ятичленні цикли з одним гетероатомом 907
за розміром циклу
за кількістю циклів
тричленні
шестичленні
моноциклічні
біциклічні
N
Н
О
1 н
Пурин
Азирин
Азин
піридин
1,2-Діазин
піримідин
Найважливішою ознакою для класифікації гетероциклів слугує кількість
атомів у циклі. Ми обмежимося вивченням п'яти- і шестичленних циклів.
36.1.2. Номенклатура
Для багатьох гетероциклів усталилися тривіальні назви, не уніфіковані ні за
принципом побудови, ні за закінченнями. Найпоширеніші тривіальні назви
гетероциклів дозволені до використання правилами IUP АС.
Для заміщених гетероциклів назви будують так само, як для заміщених
ароматичних вуглеводнів. Положення замісників зазначають цифрою відповідно до
нумерації, прийнятої для кожного гетероциклу. Крім того, положення замісника в
гетероциклі в найпростіших випадках позначають грецькими літерами а, а', ß, ß' і
так далі, починаючи з атома Карбону, найближчого до гетероатома.
• Система Ганча-Відмана. Комісія ШРАС увела нову номенклатуру гетеро-
циклів, згідно з якою назву моноциклічних гетеросистем будують із префіксів, що
позначають гетероатом: для Оксигену -окса, для Сульфуру -тіа, для Нітрогену -аза
тощо, опускаючи для милозвучності букву "а", де це необхідно, і кореня, що
позначає розмір циклу й ступінь насиченості (табл. 36.1). За наявності декількох
гетероатомів використовують числові префікси ди-, три- тощо.
Таблиця 36.1
Основи назв гетероциклів за Ганчем-Відманом
Кількість
членів у циклі
Цикли, що містять Нітроген
Цикли, що не містять Нітрогену
ненасичені*
насичені
ненасичені *
насичені
3
-ирин
-иридин
-ирен
-иран
4
-ет
-етидин
-ет
-етан
5
-ол
-олідин
-ол
-олан
6
-ин
-инан
-ин
-ан
7
-епін
-епан
-епін
-епан
8
-один
-окан
-один
-окан
9
-онін
-онан
-онін
-онан
10
-ецин
-екан
-ецин
-екан
* Максимальна кількість некумульованих подвійних зв'язків.
902
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Наведені нижче приклади пояснюють ці правила:
^7
о
іО
N
Н
^7
N
Н
о
N
Оксиран Азиридин Діазирин
0
1,2-Діазол 1,2-Оксазол 1,3-Тіазол
о
N
Н
Азол
N
Н
Азан
н
Азолідин
0
Оксол
О О г°і
V Vм V
Оксолан
о
1,2-Діазин 1,4-Діоксан 1,3-Діоксан
36.2. Будова п'ятичленних гетероциклів з одним
гетероатомом
Найпростіші п'ятичленні гетероциклічні сполуки ароматичного характеру -
фу ран, тіофен і пірол, а також їхні гідрогенізовані похідні:
Фуран
н
9
Тіофен
н
о
N
н
Пірол
9
2,3- і 2,5-дигідропіроли, або Тетрагідрофуран,
піроліни Або фуранідин
2,3-дигідрофуран 2,3-дигідротіофен
Тетрагідротіофен,
Або тіофан
9
Н
Тетрагідропірол,
Або піролідин
Ароматичність п'ятичленних гетероциклів з двома л-зв'язками пояснюють
тим, що одна вільна пара електронів атома Оксигену, Сульфуру або Нітрогену
бере участь у делокалізації й утворенні ароматичного секстету. Отже, вико-
нується правило ароматичності: 6 я-електронів (и=1) делокалізовані на п'ятьох
атомах. Делокалізацію електронів можна проілюструвати за допомогою п'яти
резонансних структур:
Пірол
Такий спосіб зображення, незважаючи на громіздкість, добре ілюструє
делокалізацію я-зв'язків і пари ^-електронів гетероатома й пояснює підвищену
схильність до електрофільного заміщення, ослаблену нуклеофільність гетеро-
атома, посилену кислотність Гідрогену при гетероатомі.
Розділ 36. Гетероциклічні сполуки. П'ятичленні цикли з одним гетероатомом
903
У молекулах цих гетероциклів шість р-електронів утворюють, як і в бензені,
загальну електронну хмару. Унаслідок спряження кільце стає плоским, а прості
зв'язки коротшають, як це видно з рентгенографічного визначення міжатомних
відстаней у молекулах фурану, тіофену, піролу. Тому поряд з граничними
структурами їх можна зобразити, подібно до бензену, формулами:
^ ^ ?
Н
Фуран Тіофен Пірол
Наслідком утворення ароматичного секстету електронів, з урахуванням
неподіленої пари електронів гетероатома, є, з одного боку, прояв відповідних
гетероциклом властивостей, характерних для ароматичних сполук, з іншого, -
втрата гетероатомом льюїсівської основності. Наприклад, атом Нітрогену піролу
зовсім не має основних властивостей.
36.3. Фізичні властивості
Фуран, тіофен і пірол, а також найпростіші їхні похідні - безбарвні рідини,
практично розчинні в воді. Температури їхнього кипіння значно вищі, ніж у
відповідних їм за кількістю атомів Карбону сполуках аліфатичного ряду (маємо
на увазі етери, сульфіди й аміни), а дипольні моменти - нижчі.
36.3.1. Спектроскопія ароматичних гетероциклів
• Уф-спектроскопія. Фуран і тіофен мають інтенсивну смугу поглинання з
А^ах 205 і 231 нм, відповідно. Для УФ-спектра піролу характерна наявність
ІНТеНСИВНИХ СМуГ 3 А,п,ах 210 і 240 нм.
• ІЧ-спектроскопія. В ІЧ-спектрі піролу простежується інтенсивна смуга
поглинання зв'язку И-Н в ділянці 3 500 см"1. Валентні коливання зв'язків С-О-С
із частотою близько 600 см'1 малоінтенсивні. Відповідні коливання зв'язку С-55-С
також малоінтенсивні. Смуги поглинання, що відповідають валентним коливан-
ням зв'язку С-Н, для всіх трьох п'ятичленних гетероаренів мають смугу
поглинання близько 3 100 см"1, а деформаційні коливання цього зв'язку
розташовані в інтервалі 700-935 см"1 для тіофену, 710-770 - для піролу і 725-
990 см"1 для фурану.
• ПМР-спектроскопія. З п'ятичленних гетероциклів ароматичні властивості
найчіткіше виражені в тіофену, тому й сигнали його ароматичних протонів
мають менше розходження в хімічних зсувах. І навпаки, для фурану, що більше
904 Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
нагадує дієн, хімічні зсуви протонів у а- і 0-положеннях максимально
відрізняються.
Сигнал протона біля атома Нітрогену в молекулі піролу зміщений у слабше
поле (-8 м.ч.) порівняно із сигналами протонів ароматичних амінів (3-4 м.ч.),
що підтверджує його кислість:
НЬ НЬ НЬ
О на хс н
7 \\
Н
5 м. ч. 7,58 (а), 6,30 (Ь) 5 м. ч. 7,19 (а), 7,04 (Ь) б м. ч. 6,62 (а), 6,05 (Ь)
36.4. Загальні способи синтезу п'ятичленних
гетероциклів з одним гетероатомом
Однотипність структур п'ятичленних гетероциклів з одним гетероатомом
визначає існування деяких загальних методів їхнього синтезу. Однак є й
численні спеціальні методи, які розглянуті у відповідних розділах.
❖ Циклізація 1,4-дикардоцільних сполук. Найважливішим загальним
способом одержання гетероциклів з одним гетероатомом із 1,4-діоксосполук є
дія на них водовідбиральних засобів (Р2О5, СН3СОС1), що приводить до фурану і
його гомологів. Ця реакція відома як синтез Пааля-Кнорра. У випадку тіофену і
його гомологів діють фосфор (V) сульфідом, а для синтезу піролу і його
гомологів - амоніаком.
■ Під час одержання фурану нуклеофільний атом Оксигену однієї з карбо-
нільних груп атакує електрофільний атом Карбону іншої карбонільної групи:
/СН2СН2 рц^
н-сч ,с-н н-с^ с-н
0 0 0 он
СНГСН2 /СН-СН2 /г-л
° он о он
Обмеження синтезу пов'язані лише з доступністю 1,4-діоксосполук. Сама
циклоконденсація проходить з кількісними виходами.
» Аналогічно відбувається реакція і у випадку тіофену, з тією лише
різницею, що попередньо одна з карбонільних груп під дією фосфор
пентасульфіду перетворюється в тіонову:
Розділ 36. Гетероциклічні сполуки. П'ятичленні цикли з одним гетероатомом
905
сн2-с^2 .снГсн2
о о \§
Н Б ОН
Виходи значно зростають у разі використання замість фосфор пентасульфі-
ду реактиву Лоуссана1:
Н3С-О^У;Р=8
н3с-о^^р=о
Застосування фосфор (III) сульфіду Р28з, що має відновлювальні власти-
вості, дає змогу використовувати похідні карбонових кислот:
нзс-сн-сн2 _ра^ НзСч|П ^-сн, О
Н3С'^ /Я^ОЫа ирА^ КаО'Яч /Я^ОИа
0 0 "з*- ь 0 0
■ Проміжним продуктом аналогічного синтезу піролу є імін:
/СН2-СН2 сн2-сн2
Н Чч. ^.С-Н Н С<> ^С-Н -
н н
,сн2-сн н /Сн2-сн
н
•н2о >Г
Цей метод дає змогу одержати з високими виходами не лише пірол, а й ІУ-
алкілпіроли, для чого амоніак необхідно замінити на первинні аміни.
❖ Реакція Юр'єва. В 1936 р. Ю. Юр'єв2 відкрив реакцію взаємопере-
творення п'ятичленних гетероциклів, ароматичних і гідрованих, над дегідра-
тувальним каталізатором (алюміній оксид) за -350-400 °С у струмені гідроген
сульфіду, амоніаку або водної пари, відповідно. Однак з високим виходом
відбуваються тільки перетворення фурану й фуранідину. Тому пірол у
промисловості одержують із фурану й амоніаку:
1 Лоуссан С.-О. (ЬахуеБЗОп Б.-О.) (1926-1988) - шведський хімік, працював у галузі органічного
синтезу, насамперед тіокетонів. Є автором реактиву Лоуссана.
2 Юр'єв Ю. (1896-1965) - радянський хімік-органік. Головні праці присвячені хімії гетероциклів.
906
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
У \^н3 н2о^ ^
'О =^ О О =^ О
Н Н
❖ Ф. Первеєв і його співробітники 1948 р. розробили загальний метод
синтезу гетероциклів з одним гетероатомом, наведений тут на прикладі синтезу
похідних тіофену. Замість гідроген сульфіду можна застосувати гідроген селенід
або амоніак:
о ?н ,3Н /К'
❖ Д. Шульте розробив синтез п'ятичленних гетероциклів із одним гетеро-
атомом з діацетилену:
Б н
36.5. Фу ран
36.5.1. Синтез фуранута його похідних
❖ Фуран уперше синтезований сухою перегонкою барієвої солі піросли-
зової кислоти. Його також можна легко виділити під час термічного декарбокси-
лювання самої пірослизової кислоти:
^соон -^ф+с*
❖ Синтез на основі фурфуролу. Кислотний гідроліз пентозовмісних
полісахаридів; які добувають із доступної природної сировини (лушпиння
насіння, овес, кукурудзяні качани, цукрова тростина), з високим виходом
приводить до утворення фуран-2-карбальдегіду (фурфуролу) за такою
схемою:
Розділ 36. Гетероциклічні сполуки. П'ятичленні цикли з одним гетероатомом
907
СНО
снон
снон-
сно
с-он
сн
снон 'Нг° снон
сн2он сн2он
СНО
с=о
сн, —
1 2 -2Н.
снон 2
сн2он
СНО
Фурфурол можна перетворити у фуран двома різними методами:
о 2 ч0 соон о
о-
О
СООН
-со
-о
О
Декарбонілювання фурфуролу використовують як промисловий метод
синтезу фу рану:
400 »С і ,
7хіОІСт2(
О ^СНО -со
❖ Циклізація мезитилоксиду. Під дією концентрованої сульфатної
кислоти в оцтовому ангідриді мезитилоксид перетворюється в шестичленний
оксигено- і сульфуровмісний цикл, який зазнає десульфування кальцій оксидом
у хіноліні:
(СН3СО)20
н3с о СНз
сн
/СТ^СН.
НзС О н
з -н*
СН,
2 +н* нс- оНСН2
сн3
СаО
250°С
| +Н2504
сн,
сн,
сн,
*з~ п-
С—БО
БОзН
❖ Внутрішньомолекулярна дегідратація 1,4-діолів. Тетрагідрофуран
також одержують дегідратацією бутан-1,4-діолу:
НО-СН2-СН2-СН2-СН2-ОН кзатор*
36.5.2. Хімічні властивості фурану
❖ Дієновий синтез. Фуран має проміжні властивості між властивостями
ароматичної сполуки й звичайного дієну. Здатність до реакцій приєднання для
908
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
фурану характерна більше, ніж для інших гетероциклів. Це пов'язане з тим, що
неподілена електронна пара атома Оксигену в фурановому циклі порівняно з
атомами Нітрогену і Сульфуру найменше схильна до утворення ароматичної
системи. Показовим щодо цього є його здатність вступати в дієновий синтез із
такими дієнофілами, як малеїновий ангідрид або диметиловий естер ацетилен-
дикарбонової кислоти:
О О
О О
Ще одним прикладом поведінки фурану як 1,3-дієну слугує реакція з
бромом у метанолі за наявності натрій ацетату:
НзС~?+ О +в^Вг —^А"^1 Х\ .
Н СН3О^О^Вг СН3О^О^ОСН3
❖ Окиснення. Фу ран можна окиснити до малеїнового ангідриду:
Глибше окиснення фурану на повітрі супроводжується полімеризацією.
❖ Ацидофобність. Фуран значно легше, ніж тіофен, осмолюється за
наявності кислот. Такий ефект дії кислот полягає в тому, що спочатку відбу-
вається протонізація атома Оксигену фу ранового циклу:
У цьому разі здатність атома Оксигену надавати електронну густину
неподіленої електронної пари різко знижується, ароматичність фурану
практично зникає, й відбувається полімеризація утвореної активної дієнової
системи. Цю властивість називають ацидофобністю - "острахом кислоти".
За наявності у фурановому ядрі електроноакцепторних замісників стійкість
таких похідних фурану щодо кислот підвищується.
❖ Реакції електрофільного заміщення. Незважаючи на невисоку стабіль-
ність, фуран, як ароматична сполука, здатний до реакцій електрофільного
заміщення, а саме - сульфування, нітрування, ацилювання за Фріделем-
Крафтсом, галогенування, хоча умови їхнього перебігу значно відрізняються від
умов перебігу відповідних перетворень у випадку бензену.
о
СООСН3
С ^.СООСНз
0+ї — ^
с соосн,
СООСН, ^чл^л3
Розділ 36. Гетероциклічні сполуки. П'ятичленні цикли з одним гетероатомом
909
У реакціях електрофільного заміщення переважно утворюються сс-похідні,
чому сприяє більша термодинамічна стійкість проміжного а-комплексу
порівняно з а-комплексом, що утворюється в разі р-заміщення. У першому
випадку делокалізація позитивного заряду в комплексі відбувається між трьома
граничними структурами, а в другому - між двома:
Н Н
ф—ф
+
■ Фуран через високу ацидофобність зазнає сульфування за методом
Терентьева піридинсульфотриоксидом:
У випадку використання концентрованої сульфатної кислоти відбувається катіонна
полімеризація фурану. Дія розведеної кислоти приводить до розкриття циклу:
/\ в г\ НзС-С-СНз-СН.-С-СНз
НзС^О^СНз ^сЛЛа? О О
■ Нітрування фурану з цієї ж причини виконують ацетилнітратом за
наявності піридину:
О сн,соош?[ /П\
Ж>2
■ Галогенування. Безпосередня взаємодія фурану з бромом може призвести до
окиснення й осмолення продукту внаслідок виділення гідроген броміду. Тому для
бромування застосовують комплекси піридину або діоксану з бромом. У всіх
випадках першими заміщення зазнають атоми Гідрогену в а-положеннях. Фуран в
омположення хлорують за -40° С.
■ Ацильні замісники вводять у фуран або його похідні за реакцією Фріделя-
Крафтса, використовуючи, однак, замість алюміній хлориду м'якші каталізатори,
такі як станум (IV) хлорид:
910
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
■ Як уже зазначено, сполуки, що містять електроноакцепторні замісники, такі
як СНО, СООН, N02, стійкіші порівняно з фураном і не так зазнають осмолення в
електрофільних реакціях. Тому зручніше нітрувати або галогенувати відповідні
похідні, наприклад, пірослизову кислоту, а потім видалити замісник, що стабілізує
фурановий цикл. У наведеному прикладі це карбоксильна група.
Дія меркуріум (II) хлориду і натрій ацетату дає змогу меркурувати фуран:
Металювання фурану. В гексані н-бутиллітій металює фуран в а-положення^
тоді як надлишок реагенту за підвищеної температури дає 2,5-дилітійфуран:
/г\ + НВиЬі А~\ + СН3СН2СН2СН3
❖ Похідні фурану. З похідних фурану найважливішою сполукою є альдегід -
фурфурол. У промислових масштабах його одержують як побічний продукт під час
кислотного гідролізу деревини й пентозовмісних відходів сільського господарства,
головно, кукурудзяних качанів, соняшникового лушпиння, соломи тощо.
Фурфурол забезпечує всі типові реакції альдегідів (рис. 36.1). Він приєднує
гідросульфіт-іон, окиснюється в пірослизову кислоту, відновлюється у фурфу-
риловий спирт, зазнає реакції Канніццаро, утворює фероїн, аналогічний до бензоїну,
реагує з амоніаком з утворенням фурфураміду й дає основу Шиффа з аніліном.
■ Фурфурол застосовують як селективний розчинник для очищення
нафтових фракцій, у виробництві пластмас, для одержання фумарової кислоти, а
також багатьох сполук з фурановим циклом, зокрема, лікарських препаратів.
■ Багато похідних 5-нітрофурфуролу є сильними антисептиками (фурацилін,
фурагенін, фурадонін, фуразонол, фуразолідон), їх широко застосовують у
медицині. Суттєву роль у їхньому відкритті й виробництві в СРСР відіграв
С. Гілер1:
і о (
Фурацилін
02И О СН=Н-№і-СО-Ш2
1 Гілер С. (1915-1975) - латвійський хімік. Головні праці присвячені синтезу фізіологічно
активних сполук на основі фурану. Одержав низку аналогів нуклеотидів та нуклеїнових кислот.
Синтезував і впровадив у виробництво серію лікарських препаратів (фурацилін, фуразолін, ПАСК
та ін.).
Розділ 36. Гетероциклічні сполуки. П'ятичленні цикли з одним гетероатомом
911
СґО>0 tvAl
Малеїновий ангідрид
^-С№СН СОС№СН-^
Дифурфуриліденацетон
І Фурфурол +NaOH
СУ
п, СН=СНСОСН3
^ Фурфуральацетон
о
€№СН€ООН
Фурилакрилова
кислота
О-С0-?Н4^
ОН
Фуроїн
(СН3СО)20
» ^Q^CH=N-C6H5
Фурфурал ьанілін
С^УсЩОН
Тетрагідрофурфуриловий спирт
н2; [Ni]
Q, СН2ОН
Фурфуриловий спирт
[Ni]
^-соон
Пірослизова кислота
Фурфурамід
NH3 [А120,]
-сн2
хн2
Тетрагідрофуран
Рис. 36.1. Схема перетворення фурфуролу
>4Н
Пірол
■ Тетрагідрофуран широко використовують у промисловості для одержання
дивінілу, бутан-1,4-діолу і 1,4-дихлоробутану. З 1,4-дихлоробутану через нітрил
адипінової кислоти синтезують 1,6-гександіамін (гексаметилендіамін) і адипінову
кислоту, що слугують вихідними продуктами для одержання поліамідних волокон:
неї
Cl-CCrL^OH
4-Хлоробутан-1 -ол
H,N—(СН2)4—NH2
Гексаметилендіамін
н^сн-сн^сн.
Дивініл
H2c-qH2
[Кат.] H2C^q^CH2
Тетрагідрофуран
НС1
снсн^-сі
KCN
;[№]
нс-(сн2)^см
Нітрил адипінової
кислоти
1,4-Дихлоробутан
н2о,н+ |нсі ,
1 Н20, Н+
НО-(СН2)^ОН \
Буган-1,4-діол НООС-(СН2)гСООН
Адипінова кислота
912 Чирва В.Я,, Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
36,6. Тіофен
Тіофен уперше виявлений А. Байєром 1879 р. у технічному бензені, виділе-
ному з кам'яновугільної смоли. Через три роки вчений отримав тіофен у
чистому вигляді. Труднощі його виділення з бензену полягали в подібності їхніх
хімічних властивостей і досить близьких температур кипіння. Вміст тіофену в
"кам'яновугільному" бензені становить 0,5%.
Інтерес до хімії тіофену посилився останніми роками з огляду на те, що
окремі його похідні використовують для лікування анафілактичного шоку, а
деякі речовини мають протизапальну дію.
36.6.1. Синтез тіофену і його похідних
❖ Циклоароматизація, У промисловості тіофен одержують із бутану або
бутилену й сірки. Для цього попередньо нагріті до 600 °С реагенти змішують у
реакційній трубі за 650 °С, час контакту - 0,07 с Вихідний газ швидко охоло-
джують. Очевидно, на першій стадії відбувається дегідрування бутану сіркою з
наступним приєднанням гідроген сульфіду, що утворився, до ненасичених
структур:
СН3СН2СН2СН3 + 4 S ^3 + 3 H2S
S
Спосіб застосовують для вуглеводнів, що містять не більше п'яти атомів
Карбону, для інших вуглеводнів за цих умов відбувається крекінг.
❖ Циклізація диацетиленів. Останнім часом знайдено шлях синтезу похідних
тіофену через діацетилени, які взаємодіють з аніонами SFT за м'яких умов:
С2Н5— С=С~С=С-С2Н5 сноН; с o/^c^V ід
20°С 25 С2Н5
65%
Очевидно, процеси біосинтезу тіофенів пов'язані з цією реакцією, оскільки
тіофени утворюються в тих частинах рослин, де сконцентровані поліацетилени.
❖ Циклізація янтарної кислоти. Тіофен можна отримати нагріванням
солей янтарної кислоти із фосфор (III) сульфідом, що в цьому випадку не тільки
слугує для введення атома Сульфуру, а й відіграє роль відновника:
^9-9^ +ps л
НООС COOH + P2S3 "4S>
❖ Синтез на основі ацетилену. Ацетилен реагує із сірководнем за 400-
450 °С над алюміній оксидом, утворюючи тіофен (О. Чічібабін):
ffÍH ALO, /7—Д
СН СН II \
+ H2S S
Розділ 36. Гетероциклічні сполуки. П'ятичленні цикли з одним гетероатомом
913
❖ Циклізація алкенів. Похідні тіофену утворюються під час нагрівання
деяких олефінів із сіркою. Наприклад, зі стильбену синтезують тіонесаль -
тетрафенілтіофен:
❖ Циклізація у-алкінонів. У разі дії гідроген сульфіду і гідроген хлориду в
спиртовому розчині на у-алкінони вони замикаються в похідні тіофену (вихід до
70%). Цю реакцію можна розглядати як комбінацію циклізації 1,4-дикарбо-
нільних сполук і діацетиленів:
Багато похідних і гомологів тіофену зручно одержувати з самого тіофену,
оскільки це досить міцна водночас реакційноздатна сполука.
Тіофен більше від інших п'ятичленних гетероциклів з одним гетероатомом
подібний на бензен. Це зумовлено декількома причинами. Значно більший, ніж у
атомів Оксигену й Нітрогену, атомний радіус атома Сульфуру дає змогу циклу
замкнутися з меншим напруженням валентних кутів. Тіонієвий атом Сульфуру
стабільніший, ніж оксонієвий, і отже, у тіофені електронна густина легше
зміщується від атома Сульфуру й делокалізується по кільцю:
Можливо, відіграє роль і здатність атома Сульфуру як елемента третього
періоду використати вакантні <і-орбіталі для переміщення електронної густини.
Зазначимо, щоя-зв'язки тіофену не здатні до реакцій приєднання й, на
відміну від фурану, він не вступає в реакції з дієнофілами. Тіофен не окиснюють
пероксиди й взагалі які-небудь окисники в сульфоксид або сульфон. Атом
Сульфуру в тіофені інертний і не зазнає алкілування метиліодидом, тільки
третинні оксонієві сполуки типу ЯзО+Вр4 алкілують тіофен за атомом Сульфуру
з утворенням тіонієвих солей:
❖ Реакції електрофільного заміщення. Оскільки тіофен належить до л-
надлишкових гетероароматичних систем, то він має яскраво виражені
36.6.2. Хімічні властивості тіофену
914
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
ароматичні властивості в реакціях електрофільного заміщення (галогенування,
нітрування, формілювання тощо), набагато активніші, ніж бензену (у деяких
випадках у 100 000 разів). Реакції відбуваються звичайно, а іноді винятково, в а-
положення.
■ Нітрування. На відміну від фурану й піролу, тіофен практично не має
ацидофобних властивостей, хоча під час його нітрування нітратною кислотою
замість реакції електрофільного заміщення відбувається окиснення з руйнуван-
ням молекули. Тому тіофен нітрують готовим ацетилнітратом або синтезують
безпосередньо в реакційній суміші з безводної нітратної кислоти й оцтового
ангідриду:
Поряд з переваленим утворенням 2-нітросполуки одерлсують 3-ізомер з
виходом 10 %.
■ Сульфування. На відміну від бензину, тіофен сульфують концентрованою
сульфатною кислотою на холоді або за незначного нагрівання:
Цю реакцію застосовують для очищення кам'яновугільного бензену від
тіофену, оскільки тіофенсульфокислота добре розчинна в водному шарі
сульфатної кислоти.
Використання комплексу піридин-сульфотриоксид є зручнішим методом
сульфування тіофену:
80%
■ Галогенування. Тіофен галогенують хлором і бромом за низької темпера-
тури без каталізатора. У цьому разі моясуть утворюватися як моно-, так і
полігалогенотіофени:
Одержання а-хлоропохідної моясливе у випадку обробки тіофену сильфу-
рил хлоридом.
■ Реакція Фріделя-Крафтса. Алкілування й ацилювання тіофену відбу-
вається тільки в а-положення, якщо використати як каталізатор бор (III)
флуорид або станум (IV) хлорид:
Розділ 36. Гетероциклічні сполуки. П'ятичленні цикли з одним гетероатомом
915
O + C1C0R -f\
Бензен за цих умов не реагує.
У реакціях ацилювання майже завжди утворюються а-заміщені ізомери,
однак, якщо обидва а-положення зайняті, легко відбувається Р-заміщення.
❖ Реакція Манніха. Тіофен легко зазнає конденсування з альдегідами й,
зокрема, вступає в реакцію Манніха:
/г\ + сн2о + нт2—-
❖ Реакція Гриньяра. Зокрема, а-бромотіофен утворює магнійорганічні
сполуки нормальним способом, що можна використати для синтезу похідних
тіофену, наприклад:
но 6 5
❖ Амінотіофен. Під час відновлення нітротіофену виникає амінотіофен,
що так само, як і амінофуран, дуже легко зазнає окиснення нітритною кислотою
й не може бути продіазотований. Однак діазотування амінотіофенів, що мають у
ядрі електроноакцепторні замісники, відбувається нормально.
❖ Меркурування. Характерною для тіофену й фурану реакцією є їхня
взаємодія з меркурій (II) ацетатом уже на холоді. Причому залежно від умов
процесу можуть синтезуватися моно- й полімеркуровані сполуки. Меркурій-
похідні під час обробки хлоридною кислотою регенерують тіофен. Цю реакцію,
поряд із сульфуванням, застосовують для очищення тіофену від бензену,
меркурування якого відбувається лише під час нагрівання, і для визначення
вмісту тіофену в бензені:
Г\ + (CH3COO)2Hg ~f~\^ + СН3СООН
4S^ 4s^HgOCOCH3 3
|нсі
S-^Hgci \s
Реакція, що становить особливий історичний інтерес і дала змогу виявити
тіофен у кам'яновугільному бензені, полягає в конденсації тіофену з ізатином у
концентрованій сульфатній кислоті, унаслідок якої утворюється суміш геомет-
ричних ізомерів індофеніну, що має глибоке синє забарвлення:
916
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
і
Н
Н2804
Н
Індофенін
Металювання тіофену. Аналогічно до фурану тіофен зазнає металювання
в положення 2. Синтезовані сполуки використовують для одержання гомологів
Реакції приєднання. В окремих випадках тіофен поводиться як дієн і може
вступати в реакцію з активними дієнофілами за високого тиску:
Реакція десульфування тіофену. Нікель Ренея є ефективним реагентом для
видалення шкідливих сульфуровмісних сполук із бензинів:
Гі Д Нікель / ч
❖ Тіофан. Під час гідрування тіофену натрієм в амоніаку або спирті поряд
з Д2- і Д3-дигідротіофенами одержують тетрагідротіофен (тіофан).
Під дією водню, адсорбованого на нікелі, відбувається відновлювальна
десульфуризація заміщених тіофену з утворенням аліфатичних сполук. Ця
реакція - один зі шляхів синтезу сполук різних класів, наприклад, карбонових
кислот, вищих спиртів, етерів, аміноспиртів, кетокислот, з похідних тіофену.
Тіофан і його гомологи містяться разом з аліфатичними тіоетерами й
меркаптанами в сірчистих нафтах. На відміну від тіофену, у тіофановому кільці
електрони атома Сульфуру неподілені - не беруть участі в утворенні
ароматичного секстету електронів. З огляду на це тіофан, як звичайні тіоетери,
тіофену:
Розділ 36. Гетероциклічні сполуки. П'ятичленні цикли з одним гетероатомом
917
легко зазнає окиснення й може бути перетворений у сульфоксид і сульфон;
метиліодид алкілує тіофен з утворенням солей сульфонію:
СН3 О О О
Сульфоксид Сульфон
❖ Біотин. Найважливіший природний продукт, у молекулі якого є
тіофанове кільце, - біотин (вітамін Н). Його синтезує мікрофлора кишківника, з
огляду на що авітаміноз простежується вкрай зрідка й пов'язаний з уживанням
сирих яєць, білок яких з'єднується з біотином:
О
Біотин
Багато похідних тіофену - лікарські препарати, наприклад, антигельмінтний
препарат комбантрин, модифіковані антибіотики цефалотин, цефалофідин.
Окремі сполуки застосовують як мономери для виробництва електропровідних
полімерів.
36-7. Пірол
Пірол уперше виявлений у кістковому маслі - продукті сухої перегонки
кісток і в невеликій кількості в кам'яновугільній смолі (Ф. Рунге1, 1834 р.). У
чистому вигляді отриманий Т. Андерсоном2 1858 р. під час сухої перегонки
рогів і ратиць. Будову піролу визначив А. Байєр 1870 р.
Слово "пірол" походить від грец. - червоний. Це пов'язане з тим, що пірол
надає яскравого червоного забарвлення сосновим стружкам, змоченим у
концентрованій хлоридній кислоті.
Пірольний і гідрований пірольний цикли, як структурні одиниці, є в складі
багатьох біогенних сполук - амінокислот (пролін, гідроксипролін, триптофан),
алкалоїдів, гемоглобіну, низки коферментів окиснення, хлорофілу, пігментів
жовчі, вітаміну ВІ2у деяких антибіотиків тощо.
1 Рунге Ф. (Runge F.F.) (1795-1867) - німецький хімік. Відкрив хінін, кофеїн, фенол, анілін,
атропін і його здатність розширювати зіниці. Засновник паперової хроматографії.
2 Андерсон Т. (Anderson Т.) (1819-1874) - англійський хімік. Вивчав склад дьогтю із кам*яного
вугілля. Виділив піколін та лутидин. Визначив структуру піролу.
918
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
36.7.1. Синтез пі ролу і його похідних
❖ Циклізація 1,4-дикарбонільних сполук. Дією амоніаку на 1,4-діоксо-
сполукн синтезують пірол. Реакцію відкрив Л. Кнорр1 1884 р., а згодом детально
вивчив К. Пааль. Спосіб аналогічний до синтезу фурану й тіофену та пояснює
споріднення всіх трьох гетероциклів:
н2с сн2 m не сн +nhv /п\
00 ноон У
н
❖ Піроли солей слизової кислоти. Звичайний спосіб синтезу піролу,
подібно до синтезу фуранкарбонової (пірослизової) кислоти, полягає в піролізі
амонієвої солі слизової кислоти:
HO
-COONH,
^ NH + 2C02 + 4H20 + NH3
У разі застосування замість амоніаку первинних амінів одержують Ы-
заміщені піроли.
❖ Під час перегонки сукциніміду з цинковим пилом утворюється пірол:
й н
❖ Циклоконденсація ацетилену. Під час пропускання ацетилену й
амоніаку через нагріті скляні трубки утворюється пірол:
2НС=СН + Ш3 30^00>с ^
і
н
Заміна амоніаку на первинні аміни веде до М-заміщених піролів.
❖ Циклізація бутиндіолу. У випадку взаємодії ацетилену з формальде-
гідом і амоніаком під тиском відбувається амінування утвореного ацетиленового
діолу із замиканням циклу:
1 Кнорр Л. (Кпогг Ь.) (1859-1921) - німецький хімік. Головні праці присвячені кето-енольній
таутомерії. Зробив вагомий внесок у будову алкалоїдів.
Розділ 36. Гетероциклічні сполуки. П'ятичленні цикли з одним гетероатомом
979
нс=сн + сн2о 300 °с> носнГс=с-сн2он А1^3+ т»ю ^ + 2Н20
н
❖ Циклізація похідних бутадієну. Нещодавно А. Павда й В. Норман
опублікували синтез піролу шляхом приєднання амінів до активованих
бутадієнів. Для процесу необхідне активування дієну для нуклеофільного
приєднання шляхом уведення акцепторних груп у положення 2 і 3:
І
❖ Реакція розширення циклу. З виходом 90 % пірол можна отримати
розширенням азирідинового циклу. Процес каталізують карбонілами металів:
N МсХСО)^ -~ ■
сбн5 Ч— ат^Чі соосн,
соосн3 н
❖ Конденсація кетоксимів з ацетиленом за наявності лугів приводить до
синтезу гомологів піролу:
СНЛГ Я''
X + Йн -най >1
І І
он н
36.7.2. Хімічні властивості піролу
❖ Відновлення. Під час відновлення піролу воднем у момент виділення в
лужному або нейтральному середовищі утворюється 2,5-дигідропірол, а під час
каталітичного гідрування 2,5-дигідропіролу або вихідного піролу -піролідин:
кий
н н н
Пірол 2,5-Дигідропірол Піролідин
■ Піролідин можна одержати також сухою перегонкою гідрохлориду -
путресцину - одного з представників "трупних отрут":
н2с-сн2
н,с сн, —- С) + >*Н4С1 + неї
сгн3н кн3сі й
920 Чирва В.Я«, Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
❖ Реакції піролу як дієну. Пірол стосовно водню в момент виділення
поводиться як типовий дієн. Аналогічно він реагує з вільними радикалами
трифенілметилу:
О + 2 (СбН5)3С - Г-ГХГГГ н >
Н Н
Схожість зі спряженими дієнами пірол виявляє і в реакції окиснення калій
перманганатом у лужному середовищі:
н н
Імід малеїнової кислоти
■ Інших реакцій дієну, на відміну від фурану, пірол не виявляє, і з
малеїновим ангідридом він реагує інакше, ніж звичайні дієни:
р
-с
.0
о н о
❖ Кислотні властивості піролу. Атом Нітрогену піролу, як уже
зазначено, повністю позбавлений основних властивостей, іміногрупа має слабкі
кислі властивості, приблизно такі, як у фенолу. Аналогію з фенолом далі
проілюструємо низкою хімічних реакцій. Зникнення основності й збільшення
кислотності ИН-групи відбувається завдяки спряженню неподіленої пари
електронів атома Нітрогену з я-електронами циклу.
■ За допомогою низки реакцій можна одержати И-металеві похідні піролу,
які широко застосовують у синтезах:
^ Ц^-М# + СН4
Замість металів можна брати безводний калій гідроксид, алкоксиди металів
типу С2Н5ОК.
■ Під час алкілування й ацилювання цих металевих похідних (у цей час
віддають перевагу пірилмагнійгалогенідам) ниясче О °С утворюються ІУ-алкіл- і,
відповідно, ІУ-ацилпіроли:
Розділ 36. Гетероциклічні сполуки. П'ятичленні цикли з одним гетероатомом
921
Алкілування й ацилювання у разі нагрівання дають а-алкіл- і а-ацилпіроли.
Ці ж сполуки можна одержати нагріванням іУ-алкіл- і Л/-ацилпіролів:
сн3сосі ^—. у^"\
N СОСН3 '-месн %4 ~м&іг N снз
Н ЩІ н
Для ацилювання замість хлороангідридів можна застосовувати естери.
Наприклад, з естеру мурашиної кислоти за низької температури синтезують №
формілпірол, за високої - а-піролкарбальдегід:
уГ\ нсоос2н5 Л нсоос2н$ Л
\^СНО ' > <0°с * N
н м%\ сно
Дія на пірилмагнійгалогенід хлорокарбонатного естеру приводить до
утворення естеру а-піролкарбонової кислоти:
О +сі-с-ос2н5 ^
V І ЧЛсооса
МеІ 0 н
❖ Ацидофобність піролу зумовлена тим, що в кислих середовищах катіон,
який утворюється під час протонування, як електрофіл атакує наступну
молекулу піролу; новий катіон знову атакує непротоновану молекулу й т.д. в
остаточному підсумку такий процес приводить до полімеризації піролу:
н н Лн Лн
н V ,
Н Jn
Н Н
Реакції електрофільного заміщення. Пірол — ароматична сполука. Подібно
до фурану і тіофену, він належить до електрононадлишкової сполуки. Як
зазначено вище, електрофільне заміщення відбувається, як звичайно, у поло-
ження 2 пірольного кільця:
922
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
н н н н н
З наведеної схеми бачимо, що у випадку електрофільного заміщення в а-
положення в делокалізації заряду карбокатіону, що утворюється, беруть участь
три резонансні структури, тоді як у разі заміщення в р-положення - тільки дві.
Об'ємні або електроноакцепторні групи біля атома Нітрогену піролу орієнтують
електрофільне заміщення в положення 3:
N Ч>Ґ ° чу Ч>Г и
С(СН3)3 С(СН3)3 !502С6Н5 802С6Н5
■ Бромування й годування піролу приводить до тетрабромо- і тетраіодо-
піролу (антисептик "йодол"). Хлорування сульфурилхлоридом є найпростішим
шляхом, що веде до а-хлоропіролу.
■ Концентрована сульфатна кислота приводить до полімеризації піролу,
сульфування виконують за методом Терентьєва піридинсульфотриоксидом
(метод сульфування "ацидофобних" сполук):
Н Н
■ Оскільки пірол "ацидофобний", то його нітрування виконують нітратною
кислотою в оцтовому ангідриді за -10 °С.
■ Активні алкілгалогеніди (алкіл- і бензилгалогеніди) за наявності слабких
основ алкілують пірол у положення 2 і 5, а метиліодид ще й у положення 3 і 4.
■ Ацилювання піролу оцтовим ангідридом за 100 °С приводить до суміші
2-ацетил- і 2,5-діацетилпіролів.
❖ Реакція Реймера-Тіммана. И-Металеві похідні піролу за низкою реак-
цій нагадують феноляти лужних металів. Справді, відповідні альдегіди можна
синтезувати характерною для фенолів реакцією Реймера-Тіммана:
о
•х/ + СЫСК + 2 ^ОН + З N301 + Н20
N з ^СНО
N3
N
Н
Як побічний продукт утворюється 3-хлоропіридин.
Розділ 36. Гетероциклічні сполуки. П'ятичленні цикли з одним гетероатомом
923
❖ Реакція Геша. Хоча пірол чутливий до кислот (за їхньої наявності він
зазнає полімеризування), все ж альдегіди й кетони похідних піролу можна
одержати, використовуючи реакцію Геша, яку застосовують до фенолів:
й н >Щ,с1 н о
Я = Н, Аік А
Ці реакції відображають особливу доступність а-положень піролу для
електрофільних атак. Тут є аналогія з реакційною здатністю о-положень
фенолів. Якщо обидва а-положення заміщені, то більша частина цих і наступних
реакцій відбувається в Р-положення.
❖ Реакція азосполучення. Пірол може слугувати азоскладовою в реакціях
азосполучення в слабкокислому або нейтральному середовищах. Це ще одна
риса, що зближає пірол з фенолом:
н н
Цікаво, що в лужних розчинах реакція приводить до утворення біс-
азопохідної:
ф ЩИІ»і-СбнгМ^^=н-сЛ
' Н
н и
❖ Конденсація піролу з формальдегідом. Надзвичайно важливі реакції
конденсації піролу з формальдегідом або мурашиною кислотою. У лужному
середовищі формальдегід гідроксиметилює пірол аналогічно до фенолу, а в
кислому - реакція відбувається далі: 2 моля піролу зв'язуються метиленовим
містком за а-положенням, утворюючи дипірилметан — проміжну сполуку в
процесах синтезу порфінів і порфіринів:
Чг^СНЮН он- чм н+ N СН2 N
Н Н Н Н
❖ Застосування піролу і його похідних. Пірол головно використовують
для одержання піролідину.
■ 3 похідних піролідину дуже важливе значення має піролідон - лактам у-
аміномасляної кислоти, який під впливом лугу вступає в реакцію поетапної
полімеризації з розщепленням амідного зв'язку й утворенням поліаміду:
924 Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
о
-ш-(сн2)гс-
'І - Найлон-4
н
■ Мономер И-вінілпіролідону легко зазнає полімеризування за радикальним
механізмом до полівінілпіролідону, який використовують як замінник плазми
крові й клей:
Г~Л + нс=сн —- Г~\ Г"Л
н сн=сн2 (-сн-сн2)п
36.7.3. Порфін і його похідні
Під час нагрівання а-піролкарбапьдегіду з мурашиною
НС^^т^^СН кислотою з виходом в один відсоток утворюється нова
И Ц ^кГ винятково важлива гетероциклічна структура, що є в основі
|| хлорофілу, деяких вітамінів і гема крові - порфін.
Порфін - міцна ароматична сполука, здатна зазнавати
сульфування, нітрування, ацилювання. Його я-електрони
делокалізовані, і реально молекула має симетричну структуру.
Порфін Молекула порфіну є 16-членним кільцем плоскої будови й
належить до небензоїдних ароматичних систем. Кількість дело
калізованих електронів становить ЗО: (11x2) = 22 електрони одинадцяти я-
зв'язків і 8 електронів чотирьох неподілених пар атомів Нітрогену), що
відповідає правилу Хюккеля: 4и + 2 при п = 7.
Порфіни з вуглеводневими замісниками в пірольних і піроліденових циклах
називають порфіринами.
❖ Гемоглобін - головний компонент еритроцитів крові. Він виконує
транспортування кисню від легенів до тканин і карбонатної кислоти від тканин у
легені. Гемоглобін - це білок глобін, зв'язаний з похідною порфіну, що містить у
центрі молекули атом двовалентного Феруму. Цю гетероциклічну сполуку
називають гем. Зв'язок глобіну з гемом, очевидно, координаційний - атом
Нітрогену гістидину, однієї з амінокислот білка глобіну, зв'язаний з атомом
Феруму молекули гема. Гем, координаційно зв'язаний з глобіном, утворює
гемоглобін, здатний утримувати газоподібний кисень і з кров'ю розносити його
по всіх клітинах організму.
Комплекс гемоглобіну з молекулою Оксигену - оксигемоглобін, цікавий
тим, що йон Феруму (II) під час такого комплексоутворення не окиснюється, а
переходить з високоспінового стану в низькоспіновий, зменшуючи свій радіус.
Ліпофільна молекула Оксигену виявляється в гідрофобній кишені.
Розділ 36. Гетероциклічні сполуки. П'ятичленні цикли з одним гетероатомом
925
Механізм отруєння чадним га-
зом, амоніаком або синильною
кислотою полягає в тому, що вони
утворюють міцніші комплекси з
гемоглобіном, ніж молекула Оксиге-
ну, унаслідок чого порушується
транспортна функція гемоглобіну в
процесі дихання й настає кисневе
голодування тканин.
Під дією хлоридної кислоти гем
відділяється від глобіну. У цьому разі
на повітрі Ферум гема окиснюється до
ступеня +3, і утворюється гемін, який
н2с=сн снЗБІ/^
Центральна частина
окисненого гемоглобіна
за складом відрізняється від гема
наявністю аніона Хлору. Кристали геміну мають яскраво-червоне забарвлення. У
випадку його деструкції гідроген іодидом утворюється суміш гомологів піролу
(група А) і Р-пірилпропіонових кислот (група Б). Підсумком структурних
досліджень гемоглобіну був синтез глобіну, виконаний 1930 р. Г. Фішером1.
■ Жовтогарячий барвник жовчі - білірубін - є продуктом розпаду гема:
Підвищення його концентрації в крові спричинює жовтяницю.
❖ Хлорофіл. Зелені частини рослин містять розчинний у спирті пігмент
хлорофіл, завдяки якому за допомогою світлової енергії рослини асимілюють
вуглекислий газ із атмосфери й воду з навколишнього середовища,
перетворюючи їх у кисень і вуглеводи. В остаточному підсумку всі життєві
процеси на Землі пов'язані з процесом включення атмосферного карбон (IV)
оксиду у вуглеводи. Цей процес, який називають фотосинтезом, потребує
великих енергетичних витрат, і джерелом енергії для нього слугує сонячне
світло. На першій стадії процесу відбувається поглинання фотона пігментами,
причому в багатоклітинних рослинах ключову роль на цій стадії відіграє
хлорофіл. Енергія фотонів згодом перетворюється в хімічну енергію, що
витрачається для зв'язування атомів Карбону, включених у молекули карбон
НООС НООС
Білірубін
1 Фішер Г. (Fischer Н.) (1881-1945) - німецький хімік. Головні праці присвячені хімії піролу.
Синтезував порфірин (1927), визначив будову хлорофілів А і Б. Нобелівський лауреат 1930 р.
926
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
(IV) оксиду, що, зрештою, супроводжується вивільненням кисню. Отже, як
побічний продукт у цьому процесі виділяється кисень, що забезпечує еволюцію
всіх аеробних організмів, у тому числі й людини. Як довів М. Цвєт1, адсорбуючи
хлорофіл на твердому носії, його можна розділити на дві індивідуальні речовини
- хлорофіли А і Б, що відрізняються забарвленням. Функціональним аналізом
підтверджено, що хлорофіл Б відрізняється від хлорофілу А наявністю
альдегідної групи замість однієї з метальних груп. Обидва хлорофіли - естери.
Під час їхнього омилення вивільняється спирт фітол С20Н39ОН.
Під дією мінеральних кислот хлорофіли відщеплюють координаційно
зв'язаний іон Магнію, й одночасно відбувається гідролізування естерного
зв'язку з відщепленням фітолу. У підсумку одержують феофорбіди А і Б.
Обробка хлорофілів іодидною кислотою приводить до дивного на перший
погляд результату - утворюється така сама суміш похідних піролу групи А і Б,
що й у разі аналогічної обробки геміну.
Хлорофіл А Хлорофіл Б
Будова хлорофілів визначена внаслідок тривалих досліджень (Л. Мархлевсь-
кий2, Г. Фішер, Р. Вільштеттер3).
Повний синтез хлорофілу виконав 1960 р. Р. Вудворд.
❖ Вітамін Вї2. Вітамін В\2 (кобаламін, ціанкобаламін, оксикобаламін) є
похідною диметилбензімідазолілкобамілдиціаніду досить складної структури. У
разі нестачі вітаміну Вп порушується кровотворення, що призводить до
злоякісної анемії та змін у нервовій тканині. Одержують його біотехнологічним
шляхом із гриба БігеріотусеБ ^ібєш.
1 Цвєт М. (1872-1919) - російський фізіолог і біохімік. Засновник хроматографії.
2 Мархлевський Л. (МагспІелУБкі Ь.Р.) (1869-1946) - польський хімік. Виявив спорідненість
хлорофілу та гемоглобіну. Працював у Львівському університеті.
3 Вільштеттер Р. (\УПкШег Я.) (1872-1942) - німецький хімік. Визначив будову кокаїну. Виділив
кристалічний хлорофіл. Нобелівська премія 1915 р.
Розділ 36. Гетероциклічні сполуки. П'ятичленні цикли з одним гетероатомом 927
Структура вітаміну В12 має багато загальних рис зі
структурами гема й хлорофілів. В основі планарної
частини його молекули перебуває ядро корину, що за
будовою близький до порфіну.
У складі молекули вітаміну В12 є Кобальт - елемент,
що досить зрідка трапляється в ароматичних сполуках. Іон
Со2+ координований у центрі коринового ядра аналогічно
до йонів ¥е2+ і Mg2+, відповідно, в гемі й хлорофілі.
Будову вітаміну В12 за допомогою рентгеноструктурного методу визначила
Д. Ходжкін11956 р., а повний синтез виконав Р. Вудворд 1972 р.
1 Ходжкін Д. (Hodgkin D.M.C.) (1910-1994) - англійський хімік. Головні праці присвячені
рентгеноструктурному аналізу складних біологічних систем. Нобелівська премія 1964 р.
Розділ 37
НІТРОГЕНОВМІСНІ
КОНАЕНСОВАНІСПОЛ УКИ З
ЛВОМЛ АТОМАМИ НІТРОГЕНУ.
П'ЯТИЧАЕННІ ГЕТЕРОЦИКАИ
37.1. Індол
індол є прикладом конденсованої гетероциклічної системи, у якій
сполучаються бензенове кільце й фрагмент піролу. Якщо вважати цю систему
умовно моноциклічною, то до неї можна застосувати правило Хкжкеля.
Делокалізовано десять електронів (« = 2), із яких вісім я-електронів і два р-
електрони атома Нітрогену. Стандартна нумерація атомів у молекулі індолу така:
• н
37.1.1. Фізичні властивості індолу
Індол - безбарвна кристалічна речовина. Він має приємний запах у малих
дозах і досить неприємний у великій концентрації. Не розчинний у воді.
• УФ-спектроскопія. В УФ-спектрі індолу є три інтенсивні смуги з А^ах 226,
282 і 290 нм.
• ІЧ-спектроскопія. ІЧ-спектри індолу і карбазолу, який розглянуто далі,
мають смуги поглинання, що характерні для аренів і піролу.
• Мас-спектрометрія. Для індолу характерний інтенсивний пік молекулярного
йона, а також пік, менший на 27 а.о.м., що відповідає відщепленню ціанідної
кислоти.
37.1.2. Методи одержання
Уперше виділений під час перегонки з цинковим пилом гідроксііндолу -
продукту розпаду природного барвника індиго (див. 37.1.5).
❖ Циклізація фенілгідразону. Перший синтез індолу і його похідних
виконаний Е. Фішером у 1886 р. Вихідними сполуками є різні фенілгідразони, які
під час нагрівання до 180 °С із сульфатною кислотою або купрум (І) хлоридом
Розділ 37. Нітрогеновмісні конденсовані сполуки з двома атомами нітрогену... 929
(О.Арбузов) перетворюються в гомологи індолу. Напрям несподіваної реакції
Фішера полягає, за Р. Робінсоном1, у таутомеризації фенілгідразону аналогічно до
бензидинового перегрупування:
9нз 9н3 ?н3 СНз
н н 2
❖ Циклізація о-аміно-ау-хлоростирену. Індол можна одержати шляхом
замикання в цикл о-аміно-со-хлоростирену. З огляду на малу рухливість атома
галогену для успішного перебігу реакції потрібна сильна основа типу натрій
етилату або аміду:
ссмн^^'Оа-с, + с2ніо„
❖ Циклізація №форміл-о-толуїдину з натрій амідом або калій трет-
бутилатом під час нагрівання належить до розряду кротонової конденсації і є ще
одним підтвердженням будови індолу (метод Маделунга):
н н н *н н
❖ Реакція Чічібабіна. Індол можна одержати шляхом пропускання пари
аніліну й ацетилену через розпечені трубки. Ця реакція аналогічна до синтезу
піролу з ацетилену й амоніаку:
❖ Промислові методи. Зазвичай індол виділяють із нафталінової фракції
кам'яновугільної смоли або одержують дегідруванням оетиланіліну з наступ-
ною циклізацією продукту, що утворюється.
1 Робінсон P. (Robinson R.) (1886-1975) - англійський хімік. Головні праці присвячені фізичній та
органічній хімії, хімії природних сполук. Нобелівська премія 1947 р.
930
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
37.1.3. Хімічні властивості індолу
Реакції індолу і його похідних подібні до реакцій піролу. Індол, аналогічно
до піролу, має дуже слабку основність й водночас слабкі кислотні, властивості.
Він виявляє явно виражену ароматичність характер і схильний до реакцій
електрофільного заміщення. Як і треба очікувати, індол зазнає осмолення
кислотами (ацидофобність).
❖ Кислотні властивості індолу. Атом Гідрогену, що зв'язаний з атомом
Нітрогену індолу, може легко бути заміщений на метали (Калій, Натрій). Однак
зазвичай використовують реакцію з реактивом Гриньяра:
(ґ^]3+СНзМ8Вг "ОО^ +СН4
З індолілмагнійгалогенідом можна виконати ті ж реакції, що й з
пірилмагнійгалогенідом, проте відмінністю цих реакцій є вступ замісників за
підвищеної температури не в а-, а в Р-положення індольного ядра, і тільки якщо
це положення зайняте, замісник надходить в а-положення. Пояснюють це тим,
що в індолі одна з електронних пар пірольного ядра належить також
бензеновому кільцю й делокалізована в ньому сильніше, ніж у пірольному.
Отже, у піролі й індолі лабільна пара електронів зв'язку Нітроген-Магній
зумовлює різний ефект концентрації електронів:
М§1 або
О
05-со-а?
ту
М%\ Mgl ЩІ
❖ Реакції електрофільного заміщення. Схеми типових реакцій електро-
фільного заміщення індолу зображені на рис. 37.1.
■ Галогенувати індол через його ацидофобність моясна тільки м'якими
реагентами, наприклад, за допомогою сульфурилхлориду, діоксандиброміду.
■ Нітрують індол бензоїлнітратом за наявності натрій етилату в етері.
■ Під час сульфування індолу комплексом піридину з сульфур (VI) оксидом
одержують індол-3-сульфокислоту.
■ Алкілування за Фріделем-Крафтсом ведуть за наявності станум (IV) хло-
риду активними алкілгалогенідами (бензилгалогенід, алілгалогенід). Ацилюван-
ня в оцтовій кислоті відбувається в положення 3; в оцтовому ангідриді
утворюється 1,3-діацетиліндол.
Розділ 37. Нітрогеновмісні конденсовані сполуки з двома атомами нітрогену... 937
3 II 2
т'т . / І \ р-Г_ттаі N
с-я
Рис. 37.1. Схеми реакцій електрофільного заміщення індолу
■ У положення 3 відбуваються реакції нітрозування, азосполучення й ін.
❖ Реакція Манпіха. Ще одним прикладом електрофільного заміщення в Р-по-
ложення є реакція Манніха, що відбувається за жорстких умов (амінометилювання
в м'яких умовах відбувається за атомом Нітрогену):
£Н2-Н(СН3)2
СН20 + (СН3)2КН
Грамін
Утворений за реакцією Манніха алкалоїд грамін слугує вихідною речови-
ною в синтезі триптофану:
н^-кссн^ сооя
+Ыа-С-ЫНСОСН3
ССХЖ
сооя
^р-с-шсосНз
ссхж _
-со^-жон
-СН3СООН
триптофан
Похідні триптофану охоплюють величезну кількість алкалоїдів індольного
ряду. Серед них вінкристин - алкалоїд для лікування лейкемії й сумно відомий
діетиламід лизергінової кислоти (ЛСД):
932 Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
ОН
СШ(С2Н5)2
<ы-сн3
ОАс
Н
НО
С02Ме
Діетиламід
лізергінової кислоти
Вінкристин
Під час гниття білків із триптофану утворюються триптамін і Р-метиліндол
{скатол). Скатол - це речовина, що спричиняє запах фекальних випорожнень. У
мексиканських грибах родини РзіїосуЬе міститься похідна 4-гідроксііндолу -
псилоцибін, що належить до так званих психоміметичних речовин, тобто
галюциногенів:
37.1.4. Застосування індолу і його похідних
Індол міститься в етерній олії жасмину, білої акації й цитрусових, поряд з
іншими оліями його застосовують у парфумерії.
З похідних індолу важливий триптофан (Р-індолілаланін) - незамінна
амінокислота, складова частина білка; Р-індолілоцтова {гетероауксин) і р-
індолілмасляна кислоти, які застосовують як стимулятори росту й коренеутворення
в рослин; індиго - у минулому природний, нині синтетичний барвник.
Структура індолу - фрагмент ваясливих природних сполук, таких як
серотонін, мелатонін і буфотемін. Серотонін (5-гідрокситриптамін) - гормон,
що регулює кров'яний тиск і потік крові через нирки й пов'язаний з нормальною
діяльністю мозку. Порушення його концентрації веде до шизофренії.
Структурно подібний гормон мелатонін бере участь у контролі зміни
денного й нічного ритмів фізіологічних функцій.
Вивчення й класифікація серотонінових рецепторів дали змогу змоделювати
й синтезувати високоселективні лікарські препарати, такі як суматриптан для
лікування мігрені, ондансетрон для подолання нудоти й блювання, спричи-
нюваних протираковою хіміо- і радіотерапією, і алосетрон для лікування
запальних процесів у кишківнику:
о. .ОН
Триптамін
Скатол
Псилоцибін
Розділ 37. Нітрогеновмісні конденсовані сполуки з двома атомами нітрогену... 933
Суматриптан Ондансетрон Алосетрон
Однією з найважливіших похідних індолу є 3-гідроксиіндол (індоксил), який
використовують у синтезі індигових барвників.
37.1.5. Індиго
Індиго - один з найдавніших натуральних органічних барвників, що раніше
мав дуже важливе практичне значення. Одержували його, переважно, із
тропічних рослин роду índigo/era tinctoria, а також з інших індигоносів, що
виростають у північніших широтах. Для цього в Європі вирощували культуру
вайда фарбувальна (Isatis tinctoria), з якої одержували індикан, що є глікозидом
індоксилу:
Індикан Індоксил Глюкоза
Під впливом ферменту індоксйлази в разі вимочування цих рослин у воді
індикан переходить у водяний розчин і гідролізується до індоксилу, який
окиснюється на повітрі в індиго, а індиго випадає в осад у вигляді синіх
пластівців. Для забарвлення тканин індиго відновлюють натрій дитіонітом
На28204 у "біле індиго", розчинне в слабкому лузі, проварюють у цьому розчині
тканину й дають "білому індиго", адсорбованому на тканині, окиснитися на
повітрі в нерозчинне "синє індиго".
❖ Будова індиго визначена на підставі таких даних. Під час сухої
перегонки індиго перетворюється в анілін, а під час окиснення його нітратною
кислотою - в ізатин. Лугом ізатин гідролізують в о-амінофенілгліоксилову
(ізатинову) кислоту. Ізатин є лактамом цієї кислоти. Це виявляється в легкій
лактонізації ізатинової кислоти, що приводить до ізатину:
934
Чирва В.Я., Ярмолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
О
II
ру СООН ^н2о (|^ГС
р
КН2 +н^он- Ч^КН~°
Ізатинова кислота Ізатин
Відношення ізатину до індолу й інших його оксопохідних визначене такими
реакціями:
Ізатин Індоксил
,р рн н
с^н2=!:ІҐ^СРсн СХ
Індол
Р он
Оксопохідним індолу (точніше індоліну) властива таутомерія, відображена
на наведених вище схемах; ізатину - лактим-лактамна, індоксилу - кето-
енольна. Індикан є глікозидом саме енольної форми індоксилу.
Індоксил у лужному розчині окиснюється на повітрі в індиго, склад якого -
СібНю^Ог; дві молекули індоксилу СвНуЖ) кількісно утворюють одну
молекулу індиго, втрачаючи у вигляді води чотири атоми Гідрогену. Одна
молекула індиго окиснюється нітратною кислотою у дві молекули ізатину
С8Н5Ж>2, втрачаючи подвійний зв'язок і приєднуючи два атоми Оксигену.
Звідси, як і з деяких інших міркувань, А. Байєр визначив формулу індиго, що на
підставі нових даних, невідомих А. Байєру, має транс-форму:
р 0
с=о + о=с ]| І -— У А ,с-с.
6 Ізатин О Індиго
Р
Ізатин
ас нм-^ч.
Індоксил О Індоксил
Розділ 37. Нітрогеновмісні конденсовані сполуки з двома атомами нітрогену... 935
Зазначимо, що /яраяс-розташуванню індоксильних залишків сприяють
водневі зв'язки між карбонільним атомом Оксигену й атомом Гідрогену,
приєднаним до атома Нітрогену:
❖ Методи синтезу. Після декількох синтезів індиго, виконаних А. Байє-
ром і К. Гейманом, розроблено два способи, що ввійшли в промислову практику
й дали змогу виробляти індиго так дешево, що культивування індигоносних
рослин виявилося збитковим.
■ Перший технічний метод синтезу індиго, запропонований 1890 р.
К. Гейманом, полягав у нагріванні феніламінооцтової кислоти (Лґ-фенілгліцину)
з лугом до 300 °С. Проте внаслідок термічного розкладу вихід індоксилу був
низьким. У 1898 p. К. Пфлегер з'ясував, що метод Геймана можна значно
поліпшити, якщо лужне плавлення робити за наявності натрій аміду, що дає
змогу знизити температуру процесу до 180 °С:
ОН
'•О
Кетонна форма
синього кольору
Лейкооснова
білого кольору
а
NH2 сГ
ONa
0=с
СН, юо°с
Зазначимо, TV-фенілгліцин ліпше одержувати за такою схемою:
Н
а
■ Другий спосіб К. Геймана на основі антранілової кислоти також відбу-
вається за порівняно низької температури:
936
Чирва В.Я., Ярллолюк CM., Толкачова Н.В., Земляков О.Є. Органічна хімія
9
C^ONa
Ж,
CICHCOONa
NaOH
О
II
с
"ONa a)NaOH,200°C
XT.CTL б)н+
N V2
9
с
рн-соон
nh
Н СОСЖа
Обидва способи приводять до індоксилу, який потім окиснюють в індиго.
■ Нещодавно розроблений безперервний спосіб одержання індиго. З аніліну
й оксиду етилену синтезують Р-гідроксіетиленанілін, лужна обробка якого дає
індоксил:
NH,
HjÇ-CHj
О
NaOH
300°С
NH-CHjCH2OH
Р-Пдроксіетиленанілін
І
H
О
❖ Властивості індиго. Індиго не дає реакцій, що характерні для карбонільної
групи. Воно легко зазнає сульфування й бромування. За різних умов чотири атоми
або групи можуть бути введені в молекулу в положення 5,5'і 7,7'. Продукти
галогенування індиго дають забарвлення з ліпшою стійкістю до світла й прання.
Для текстильного друкування й фарбування набув значення індигозоль, який
синтезують з індиго білого й хлоросульфонової кислоти в піридині з наступним
підлу жненням:
1)С1803Н
2)№ОН
^ їх л
ОБС^а
Індигозоль зазнає гідролізування розбавленими кислотами з утворенням
індиго білого й, отже, барвник переходить на бавовняне волокно прямо з
водного розчину. На закінчення його окиснюють у кислому середовищі й так
фіксують барвник на волокні. Оскільки під час одержання кубового розчину не
використовують луги, то цим методом можна також фарбувати вовну й шовк.
❖ Старовинний пурпур - дорогоцінна в стародавні часи (відома за 1800
років до н.е.) червонясто-фіолетова фарба, яку отримували з молюсків, за
дослідженням П. Фрідлендера1, виявилася 6,6'-дибромоіндиго.
❖ Тіоіндигоїдні барвники. Поряд з класичним індиго сьогодні застосовують
тпіоіндигоїдні барвники, що мають принаймні один сульфуровмісний гетеро-
циклічний залишок. їх використовують як кубові барвники й пігменти.
Найпростішим з тіоіндигоїдних барвників є 2,2'-5/с-тіонафтеніндиго, що утворює
забарвлення червоних кольорів із синюватим відтінком:
1 Фрідлендер П. (Ргіесіїапсіег Р.) (1857-1923) - німецький хімік. Головні праці присвячені барвни-
кам. Отримав пурпур.
Розділ 37. Нітрогеновллісні конденсовані сполуки з двома атомами нітрогену... 937
а:
О
Тіоіндигоїдні барвники застосовують для друку по бавовні, льону, віскозі,
для кубового забарвлення вовни, хутра, а також як пігменти в поліграфії. За
стійкістю й спектром забарвлення вони перевершують індигоїдні барвники.
У кам'яновугільному дьогті у фракції, яку називають антраценовою олією,
поряд з антраценом і фенантреном перебуває нітрогеновмісний гетероцикл -
дибензопірол, або карбазол, який можна відділити від нейтральних вуглеводнів
завдяки характерним для нього слабкокислим властивостям, наприклад,
здатністю утворювати калієві солі під час обробки смоли калій гідроксидом.
Карбазол - це безбарвні кристали, нерозчинні у воді. Будову карбазолу
доводять утворенням його під час нагрівання солі о,о'-діамінобіфенілу:
Карбазол можна одержати також, пропускаючи дифеніламін через розпечені
трубки:
Ця речовина більше схожа на дифеніламін, ніж на похідні піролу, що й
зрозуміло, оскільки виграш енергії в разі ароматичної делокалізації електронів
по бензенових ядрах значно вищий, ніж по пірольному ядру.
Ароматичність карбазолу зумовлена наявністю 14-тс-електронної системи за
участю неподіленої електронної пари атома Нітрогену. Для нього характерні
властивості ЫН-кислот - утворює солі з лужними металами й взаємодіє з
реактивом Гриньяра. Практично не ацидофобний. Для карбазолу характерні
електрофільні реакції заміщення, головно, у положення 1 і 3. Він має обмежене
застосування у виробництві сульфурних барвників і термопластиків на основі ІУ-
вінілкарбазолу, який одержують з ацетилену й карбазолу під дією лугу.
37.2. Карбазол
неї,
8 >Ш 1
Дибензопірол (карбазол)
СПМНз Ш3СІ
938
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
«о
37.3. Піразол
з Піразол і його похідні не знайдені в природі. Ароматичну
систему цієї сполуки утворюють три я-електрони атомів
N Карбону, один я-електрон атома Нітрогену й неподілена пара
^ електронів іншого атома Нітрогену.
37.3.1. Фізичні властивості піразолу
Піразол - безбарвна кристалічна речовина.
• ІЧ-спекпгроскопія. ІЧ-спектри піразолу й імідазолу, про який ітиметься далі,
мають смуги поглинання, аналогічні до смуг поглинання піролу.
• ПМР-спектроскопія. На рисунку показано віднесення сигналів протонів і
піразолу.
37.3.2. Методи одержання
❖ Взаємодія алкінів або алкенів з діазометаном приводить, відповідно, до
піразолу й 4,5-дигідропіразолу:
СН N СН: N
N Н N ^ Н
❖ Циклізація гідразопів. Дією гідразину або заміщеного гідразину на
ненасичені альдегіди синтезують піразоліни, які моясна окиснити в піразоли:
К R
❖ Циклізація $-дикетонів. У разі взаємодії 1,3-дикетонів з гідразином
Л. Кнорр одержав 3,5-дизаміщені гомологи піразолу:
д Я'
н
3733. Хімічні властивості
За хімічними властивостями піразол належить до основ, оскільки один з
атомів Нітрогену зберігає неподілену електронну пару. Із сильними кислотами
він дає солі. На відміну від піролу, піразол не виявляє ацидофобних власти-
востей.
Розділ 37. Нітрогеновмісні конденсовані сполуки з двома атомами нітрогену... 939
Електрофільне заміщення відбувається в положення 4. Піразол зазнає
сульфування олеумом, хлорування й бромування, нітрування й меркурування.
Він дуже стійкий до дії не тільки кислот, а й окисників. Під час окиснення бічні
ланцюги можна перетворити в карбоксильні групи.
З іншого боку, піразол - слабка NH-кислота, що дає металеві похідні.
❖ Піразолон і його похідні. Найважливішим представником піразолу є
піразолон - складова частина деяких ліків. Найпростішу похідну піразолону
синтезував Л. Кнорр з ацетооцтового естеру й феніл гідразину:
ЦСуСН^^О NH2NH-CtH, нг_Сн: ^п ^ Н3С^уЮ
О ОС2Н5 ÖC2H-CA°H N-N
¥н 2 5 сбн5
С Н
6 5 3-Метил-1-фенілпіразол-5-он
■ 3 цієї сполуки синтезують антипірин, з якого, відповідно, у кілька стадій
одержують знеболювальний і жарознижувальний препарат амідопірин
(пірамідон):
СН, СН, o=N СН,
/З уЗ w \ / з
HCl, Zn
чсн,
СН3 СН3 o=N С
Г-^ СН3І Г=і HNO, )=(
І І І
С6Н5 С6Н5 с6н5
сн
h2n сщ НзС-н 3 сн3
2 W ' сиР W
жсн
С6Н5 С6Н5
—\ ин^ — \
Антипірин • Пірамідон
■ Ефективніший лікарський препарат - анальгін - одержують за такою
схемою:
HjN сНз NaOS02CH2NH_CH3 NaOSO,-CH,-N
с,н,
CA с6н5 4"5
940
Чирва В.Я., Ярмолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
4/7-Ц1
37А. Імідазол
з Імідазол у вигляді різних похідних поширений у тваринному й
...*2 рослинному світі та відіграє важливу біологічну роль. Імідазол-
¥ структурний фрагмент молекул пурину, гістаміну, гістидину,
4 Н пілокарпіну, уреїдів, вітаміну Ві2. Ароматична система імідазолу
побудована аналогічно до системи піразолу:
❖ Синтез імідазолу. Імідазол і його похідні одержують з гліоксалю,
амоніаку й формальдегіду:
сно кн3 ^ гн
сно + сн2о О
NHз Ш
■ Альтернативно його можна синтезувати взаємодією а-дикетонів з
амоніаком і формальдегідом у льодяній оцтовій кислоті.
■ У промисловості імідазол і його гомологи одержують взаємодією етилен-
діаміну зі спиртами, альдегідами, кислотами або естерами при 400 °С за
наявності платини, висадженої на алюміній оксиді:
снгнн2 но >
н
❖ Хімічні властивості. Імідазол - сильніша основа, ніж ізомерний до
нього піразол; утворює із сильними кислотами солі. Водночас він має
властивості кислоти завдяки зв'язку N-11:
^ + СН3М§І V +СН4
■ Імідазоли здатні до всіх головних типів реакцій електрофільного
заміщення, які відбуваються в положення 4; нуклеофільні реакції спрямовані на
Сг-атом.
❖ Похідні імідазолу. Важливою природною похідною СООН
імідазолу є гістидин - одна з незамінних амінокислот, \ -^СН-СН
необхідний компонент їжі людини. Гістидин, або Ь-амі- №І2
ноімідазоліл-2-пропіонова кислота, є в структурі активних Гістидин
центрів багатьох ферментів:
■ Під час ферментативного розщеплення гістидин перетворюється в
гістамін:
^сн2сн2-ш2
Гістамін
Розділ 37. Нітрогеновмісні конденсовані сполуки з двома атомами нітрогену... 94/
Гістамін має сильну фізіологічну дію: знижує кров'яний тиск і розширює
капіляри, активізує гладку мускулатуру. Дії гістаміну приписують деякі
алергійні нездужання. Є багато антигістамінних препаратів, що знімають
гістамінний ефект. До них, зокрема, належить димедрол:
■ До похідних імідазолу належать такі лікарські препарати, як нафтизин,
дибазол і клофелін:
(С6Н5)2СН-0-СН2~СН2-Н(СНз)2
■ Імідазол є в основі алкалоїду пілокарпіну, що підвищує тонус гладкої
мускулатури. Його синтез виконали М. і В. Преображенські:
Пілокарпін
Нафтизин
Дибазол
Клофелін
Розділ 38
ШЕСТИЧЛЕННІ
НІТРОГЕНОВМІСНІ
ГЕТЕРОЦИКЛИ
38.1. Піридин
Назва піридин походить від грец. piros - вогонь і суфікса -ідин, який
застосовували свого часу у назвах ароматичних основ, наприклад, фенетидин,
толуїдин тощо.
Піридин можна розглядати як бензен, у якому група -СН- заміщена атомом
Нітрогену. Відповідно до теорії будови, повинно існувати три монозаміщені та
шість дизаміщених продуктів. Монометилпіридини називаються піколінами,
диметилпіридини - лутидинами, триметилпіридини - колідинами:
а-Піколін 2,6-Лутидші 2,4,6-Колідин
Піридин уперше виділений Т. Андерсеном 1849 р. з продуктів піролізу
кісток тварин, а через п'ять років виявлений у кам'яновугільній смолі.
Сучасна формула піридину запропонована В. Кернером 1869 р. Піридин за
будовою, як зазначено вище, нагадує бензен, у якому замість СН-групи є атом
Нітрогену, електронегативність якого значно вища, ніж атома Карбону. У
молекулі піридину неподілена електронна пара атома Нітрогену розташована в
площині кільця й не бере участі у спряженні ароматичного секстету електронів.
Цим вона надає піридину основні властивості. Наявність електроноакцепторного
атома Нітрогену зумовлює зсув електронної густини я-зв'язків до нього, й у
такий спосіб у кільці на атомах Карбону виникає дефіцит електронів. Усе це
можна проілюструвати резонансними структурами:
+
Розділ 38. Шестичленні нітрогеновллісні гетероцикли
943
38.1.1. Визначення будови піридину
Будова піридину тісно пов'язана з піперидином. Справді, під час обробки
піридину воднем у момент виділення або над каталізатором він перетворюється
в піперидин:
О
Na, С2Н5ОН
N
Н
Піперидин
Будову піперидину доводять його зустрічним синтезом шляхом нагрівання
дигідрохлориду пентаметилендіаміну (кадаверину):
№Ш А
+ в3- ^ + №і4С1 + НСІ
ш3сі
н
Крім того, будову піперидину доводять його деструкцією через вичерпне
метилювання за Гофманом:
СН£_Г ^| AgOH ^ ґ > д Г П СН*І
NH ^N(CH3)2I ^N(CH3)2 НО ^n3h™
i II III IV
її О (РіІ + (CH3)3N-
.W «і ho.(CHj)j1.J ch;-H.O
V VI VII
- сн2=сн-сн=сн-сн3
VIII
Деструкція піперидину (І) за Гофманом полягає спочатку в його метилюванні
до четвертинної амонієвої солі (II) з наступним переведенням її в основу (III).
Піроліз четвертинної основи веде до розкриття циклу з утворенням ненасиченого
аміну (IV). Повторення цих прийомів до сполуки (IV) приводить до
триметиламіну (VI) і 1,4-пентадієну (VII), який спонтанно перетворюється в
стійкіший спряжений дієн (VIII). Цей метод зазвичай застосовують для з'ясування
послідовності С-С-зв'язків у гетероциклах. У цьому випадку це нерозгалужений
ланцюг з п'яти атомів Карбону. Подібним способом молена визначити будову й
гомологів піридину.
944
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
38.1.2. Фізичні властивості піридину
Піридин - безбарвна рідина, що має сильний неприємний запах. Змішується з
водою у всіх співвідношеннях. Зсув електронної густини від атомів Карбону до атома
Нітрогену зумовлює утворення в піридині значного дипольного моменту (2,2 Б).
• ПМР-спекмроскопія. Сигнали ароматичних прото-
(м.ч. 8,50 (а), 7,06 (Ь), 7,46 (с) нів піридину виявляються у слабкому полі.
До 1950 р. кам'яновугільна смола була, по суті, єдиним промисловим
джерелом піридину, хоча його вміст становить лише 0,1 %. Зі смоли його
виділяють у вигляді солі за допомогою розведеної хлоридної кислоти. Для
одержання у вільному стані сіль обробляють лугом.
Кількості піридину і його гомологів, що виділяється з кам'яновугільної смоли й
частково із продуктів сухої перегонки деревини й торфу, були недостатніми, щоб
задовольнити потреби хімічної та фармацевтичної промисловості. Тому розроблено
низку реакцій для синтезу як самого піридину, так і його гомологів.
❖ Циклоконденсація ацетилену й ціанідної кислоти. Під час про-
пускання пари ціанідної кислоти й ацетилену через розпечені трубки У. Рамзай1
1877 р. синтезував піридин:
Через низький вихід цільового продукту метод має тільки історичне
значення.
❖ Синтез Дільса-Альдера передбачає застосування як дієнофіли алкілнітрилів;
3,6-дигідропохідна, що утворюється, легко зазнає окиснювальної ароматизації:
❖ Реакція Байєра. Під час конденсації акролеїну з амоніаком утворюється
Р-піколін. Ця реакція лягла в основу гіпотези Байєра про виникнення піридину і
його гомологів у кістковій олії з гліцеролу жирів і нітрогенних основ:
1 Рамзай В. (Ramsay W.) (1852-1916) - англійський хімік і фізик. Відкрив аргон (разом із Дж. Релеєм),
неон, ксенон і криптон (разом із М. Траверсом), виділив гелій. Нобелівська премія 1904 р.
• Уф-спектроскопія. Піридин має характерні смуги
поглинання з А™* 195 і 251 нм, пов'язані з я—>я*-
ПЄРЄХОДОМ, І МеНШ ІНТеНСИВНу СМугу 3 Атоах 270 нм
(п—>я*-перехід).
38.1.3. Одержання піридину і його гомологів
2НС=СН + H-C^N
Розділ 38. Шестичленні нітрогеновмісні гетероцикли
945
2Н,С=СТ-СНО + МН3 — (УСЩ
Акролеїн N
❖ Синтез Чічібабіна. З використанням насичених амоніаком оцтового і
мурашиного альдегідів О. Чічібабін синтезував піридин і його гомологи:
сн3соош4
4 СНтСНО + ІЧН, -гг^ І , .
250 °с ^^СН3
5-Етил-2-метилпіридин
2 СН3-СНО + NH3 +НСНО ^ Q + ЗН20 + Н:
Останніми роками піридин з виходом у 60-70 % одержують газофазовою
високотемпературною обробкою кротонового альдегіду, формальдегіду, водяної
пари, повітря й амоніаку на алюмосилікатному каталізаторі.
❖ Реакція Penne. Ацетилен і амоніак за наявності складного нікелевого або
кобальтового каталізатора перетворюються в гомологи піридину:
4HC=CH+NH3
С2Н5>^
СН3
2-Метил-5-етилпіридин, що утворився, дегідруванням переводять у 2-метил-
5-вінілпіридин, який широко використовують як мономер у виробництві
синтетичних каучуків і пластичних мас.
❖ Циклізація 1,5-дикетонів. У разі циклоконденсації 8-дикарбонільних
сполук з амоніаком утворюються похідні 1,4-дигідропіридинів, які під час
окиснення ароматизуються в гомологи піридинів:
NH3. \\ ÎI [о]
я о о & -н2о к у я к- ^
н
Потреба окиснення на завершальному етапі відпадає в разі використання
замість амоніаку гідроксиламіну, оскільки в цьому випадку 1,4-елімінування в
молекулі приводить до гомологів піридину.
Вихідні сполуки можна легко отримати приєднанням кетонів за Міхаелєм до
а,Р-ненасичених кетонів або озонолізом циклопентенів.
❖ Синтез Ганна полягає в конденсації ацетооцтового естеру з альдегідами
й амоніаком:
946 Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
2СНз-С-СН2-С-ОС2Н
V нзс-сч, с,н,оос^>^.соос,н
т ґ\г*< тт ГІ _ ^ •> Ті ІТ ^
О Шз НзС'^Г^СНз
н
Діетші-2,4,6-триметил-1,4-дигідро-
піридин-3,5-дикарбоксилат
У разі потреби естерні й алкільні групи можна легко вилучити:
СНз СН3 СН,
С2Н5ООСук^СООС2Н5 нр, н+ НООСууСООН д ^ X кмпо,
НзЛЛсН, -ВД0Н НзС^СНз -^НзсЛЛсН,
ґ^ґлґлтт 2,4,6-Триметилпіридин- сии-Колідт
ССХ-/Н 3,5-дикарбонова кислота
НООС^^ГХООН"ЗС°2
38.1.4. Реакціїелектрофільного заміщення
Піридин набагато індиферентніший щодо електрофільних атак, ніж бензен і
навіть нітробензен. Це пояснюють тим, що атом Нітрогену виявляє негативні
індукційний (-1) і мезомерний (-М) ефекти, які зумовлюють певну елекро-
нодефіцитність кільця. Вона особливо виявляється в а- і уположеннях. Крім
того, атом Нітрогену піридину взаємодіє з електрофілами в кислому середовищі,
унаслідок чого на атомі Нітрогену виникає позитивний заряд, що ще більше
дезактивує цикл щодо електрофільних реагентів. Тому о-комплекси в <х-
положенні, утворення яких у такому випадку потребує утворення другого
позитивного заряду, малоймовірні:
н~ и^н —Іп>н
х 7 х 7 х
X - X X
Трохи більшу стійкість мають сг-комплекси в Р-положенні, резонансна
стабілізувальна дія яких не пов'язана з виникненням другого позитивного
заряду на атомі Нітрогену:
і і
X X
Розділ 38. Шестичленні нітрогеновмісні гетероцикли
947
❖ Галогенування. Галогени за низьких температур приєднуються до
піридину з утворенням М-галогенідів, які під час нагрівання до 200 °С перетво-
рюються в р-галогенопіридини:
+ СІ,
І. СІ J
сг
9'
СІ
+ неї
Також 3-хлоропіридин одержують хлоруванням піридину при 100 °С за
наявності алюміній хлориду; 3-бромопіридин синтезують із високим виходом
дією на піридин брому в олеумі. Процес відбувається, очевидно, через
утворення піридиній-1-сульфонату як реакційноздатної проміжної сполуки,
оскільки бромування не виникає в 95 % сульфатній кислоті:
N
С12? А1С13,100°С
33%
о
N
Вг2, олеум
86%
СҐ
N
Найефективніший спосіб одержання 2-бромо- і 2-хлоропіридину полягає у
взаємодії піридину з галогенами при 0-5 °С за наявності паладій (II) хлориду.
За 300-400 °С без каталізатора заміщення настає тільки в Р-положення
(положення 3 і 5). За 500 °С бромування відбувається дуже легко з утворенням
продуктів заміщення в а-положеннях (положення 2 і 6):
„Вг Вг^^^Вг
ВГ^Ґ^Вг ^ Вг
❖ Сульфування. Під час нагрівання піридину протягом 24 год із
концентрованою сульфатною кислотою до 220-230 °С за наявності меркурій (II)
сульфату утворюється піридин-3-сульфокислота:
+ НОБОзН
конц. Н2504, Н§504> 220°С
70%
803Н
+
Н20
Підвищення температури реакції до 360 °С приводить до синтезу у-ізомеру.
Це свідчить про термодинамічний контроль процесу сульфування. Роль солі
^804 зводиться до перешкоджання протонуванню атома нітрогену.
Сульфування піридину без меркурієвого каталізатора відбувається за 300 °С
і дає низькі виходи піридин-3-сульфокислоти. Сульфування 2,6-ди-трет-
бутилпіридину добре ілюструє справжню реакційну здатність піридинового
циклу в реакціях електрофільного заміщення. Об'ємні замісники ефективно
перешкоджають приєднанню сульфур триоксиду по атому Нітрогену піридину, і
948 Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
швидкість сульфування сумірна з електрофільним заміщенням для нітробензену.
Максимальна конверсія цього процесу становить 50 %, що пов'язано з
утворенням протона в ході реакції і його зв'язуванням молекулою вихідного
субстрату:
С(СН3)з^^С(СНз)3 С(СНз)з^к^С(СН7з)з%
0=^=0
о
н
С(СНз)3^ ^С(СН3)3
-XX
С(СНз)з^^с(СНз)3
Стійкість піридину в реакції сульфування дає змогу використати комплекс
піридину зі сульфур триоксидом як м'який сульфувальний агент (О. Терентьєв,
1946 р.).
❖ Нітрування піридину також потребує жорстких умов. Дія розчину калій
нітрату в нітратній кислоті на розчин піридину в 100% сульфатній кислоті за
300 °С та наявності каталізатора дає 3-нітропіридин з виходом 22 %. За 370 °С
утворюється ще й 2-нітропіридин:
Н02 N 370 °С 300 °С
У м'якіших умовах нітрування не відбувається; 2- і 4-нітропіридини
утворюються з 75 % виходом у разі окиснення 2- або 4-амінопіридину гідроген
пероксидом.
Уведення метальних груп у піридинове кільце полегшує електрофільне
заміщення настільки, що нітрування успішно конкурує з окисненням бічного
ланцюга. Стеричне або індуктивне інгібування реакції нітрування за атомом
Нітрогену дає змогу застосовувати для С-нітрування нітроній тетрафлуороборат:
аЛЛ, 77% сіЛАс,
❖ Алкілування й ацилювання піридинового ядра за реакцією Фріделя-
Крафтса не відбувається.
Розділ 38. Шестичленні нітрогеновмісні гетероцикли
949
38.1.5. Нуклеофільне заміщення в піридині
Якщо для бензену й електрононадлишкових гетероциклічних сполук (пірол,
тіофен, фуран) характерні реакції електрофільного заміщення, то піридину
більше властиві реакції з нуклеофільними агентами.
Збіднення електронною густиною піридинового кільця, що утруднює
електрофільні реакції, полегшує нуклеофільне заміщення. На відміну від
бензину, піридин досить легко реагує з нуклеофільними реагентами, причому
атака орієнтується в а- або у-положення. У цих місцях легше утворюються а-
комплекси, оскільки їхня резонансна стабілізація не пов'язана з появою
негативних зарядів поряд з електронегативним атомом Нітрогену, як це є в разі
заміщення в Р-положенні:
❖ Амінування. Під час нагрівання піридину до 130 °С з натрій амідом із
високим виходом утворюється а-амінопіридин:
75%
З калій амідом виникає суміш а- і у-ізомерів. Реакція найліпше відбувається
в рідкому амоніаку. Механізм її можна зобразити такою схемою:
Реакцію вперше описали 1914 р. О. Чічібабін і О. Зайде. Пізніше вони
запропонували її модифікацію - алкіламінування піридину за допомогою натрій
алкіламідів.
❖ Гідроксшіювання. Гідроксид-іон - набагато слабший нуклеофіл, ніж
амід-іон, і він атакує піридин лише за високої температури. Наприклад, під час
дії пари піридину на сухий калій гідроксид за 250-300 °С утворюється а-
гідроксипіридин:
950 Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
ОК
н2о ^
x + КОН
^ОН
Цей синтез аналогічний до реакції нітробензену з концентрованим лугом,
що дає о-нітрофенол.
. Розглянемо детальніше амінопіридини і гідроксипіридини.
❖ Амінопіридини можна одержати такими методами: з амідів піридин-
карбонових кислот за реакцією Гофмана, з нітропохідних піридину відновлен-
ням (3-амінопіридин), реакцією Чічібабіна (2-амінопіридин). Зазвичай 3-амінопі-
ридин одержують з 3-бромопіридину дією амоніаку за 140 °С та наявності
купрум (II) сульфату.
Усі три ізомерні амінопіридини існують в іміноформі, оскільки а- і у-
ізомери схильні до таутомерії:
Зазначимо, що 2- і 4-амінопіридини є сильнішими основами, ніж анілін або
піридин. Зокрема, 3-амінопіридин утворює дигідрохлорид, тоді як інші
амінопіридини утворюють тільки моногідрохлориди. Він суттєво відрізняється від
2- та 4-ізомерів і за іншими властивостями. Амінопіридини з аміногрупою в 0-поло-
женні виявляють властивості ароматичних амінів. Вони можуть бути діазотовані, а
діазонієва група заміщена галогеном, нітрильною та іншими групами. Такі
діазонієві солі вступають у реакції азосполучення з фенолами й амінами.
Якщо аміногрупа перебуває в 2- або 4-положенні, то спроба діазотування
зазвичай веде до утворення піридонів. У концентрованій хлоридній або
флуоридній кислоті аміногрупа під час дії натрій нітриту дає відповідну хлоро-
або флуоропохідну.
■ 2-Амінопіридин можна перетворити в натрій діазотат дією натрій
амілнітриту за наявності алкоголяту:
Н
+
. і
Н
+ С5НіГСК№=0
ЫаОС2Н5
-С2Н5ОН
+ С5НиОН
і2(Жа
Підкислення концентрованого розчину діазотату оцтовою кислотою дає 2-
гідроксипіридин, а за наявності калій іодиду - 2-іодопіридин. Діазотований
Розділ 38. Шестичленні нітрогеновллісні гетероцикли
951
таким способом 2-амінопіридин може вступати в реакцію сполучення з
фенолами.
■ 2-Амінопіридин легко зазнає хлорування або бромування за кімнатної
температури з утворенням 5-галогено- і 3,5-дигалогено-2-амінопіридинів:
■ 2-Амінопіридин зазнає сульфування за нижчої температури, ніж піридин
(145 °С). У цьому разі утворюється з 70-80 % виходом 2-аміно-5-піриди-
носульфокислота:
■ 2-Амінопіридин реагує з нітрувальною сумішшю за низької температури з
утворенням нітроаміну, що за 40 °С перегруповується в 5-нітро-2-амінопіридин:
■ Серед похідних 2-амінопіридину назвемо сульфамідний препарат
сульфідин, який деякий час широко застосовували в медицині:
❖ Гідроксипіридшш. Гідроксильна група в піридиновому ядрі поводиться
по-різному залежно від положення. У положенні 3 вона виявляє властивості
фенольного гідроксилу й дає реакції із ферум (III) хлоридом; 2- і 4-
гідроксипохідні піридину здатні до таутомерії.
За нормальних умов а- і уізомери існують практично тільки в кетонній
формі й відомі як піридони. Гідроксиформи можна виявити в значних кількостях
тільки в дуже розведених розчинах у неполярних розчинниках або газовій фазі.
Поляризовані форми ліпші в разі сольватації; 3-гідроксипіридин існує в рівно-
вазі зі цвітер-іонною формою, причому положення рівноваги залежить від
природи розчинника:
Н
Н
Н
Н
н
952 Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Під час обробки діазометаном 2- і 4-гідроксипіридини утворюють
метоксипохідні. З іншого боку, якщо їх нагріти з метиліодидом, то утвориться
И-метилпіридон:
❖ С-Алкілування й арилювання піридину. Реакції піридину з алкіл- і
ариллітієвими похідними відбуваються у дві стадії: спочатку літійорганічні
сполуки приєднуються з утворенням ЛГ-літієвих солей дигідропіридинів, які
потім перетворюються в заміщені ароматичні піридинові структури. Атака
літійорганічними реагентами відбувається в а-положення:
[Ґ^ІпВиІл, 100°С
пВиА>Г 87 %
и СА
❖ Нуклеофільне заміщення груп, що добре відходять. Атоми галогенів, а
також у деяких випадках нітро- і метоксигрупи, що перебувають в а- і у-
положеннях, але не в Р-положенні піридинового циклу, порівняно легко
зазнають заміщення багатьма нуклеофілами. Заміщення відбувається за
механізмом приєднання-відщеплення. У реакціях нуклеофільного заміщення у-
галогенопіридини більше реакційноздатні, ніж відповідні а-ізомери. Флуоро-
похідні найактивніші серед усіх галогенопіридинів:
Ьі
Розділ 38. Шестичленні нітрогеновмісні гетероцикли
953
38.1.6. Електрофільні реакції піридину за атомом
Нітрогену
©у
З Я X
®У хнх
Кислота
II Л Льюїса (Ь)
©>ї ©
1
КІ
о
©к
о©
❖ Основні властивості. Завдяки наявності неподіленої пари електронів на
атомі Нітрогену піридин виявляє основні властивості. Водний розчин його
лужний, забарвлює лакмус у синій колір. З більш-менш сильними кислотами він
утворює солі. Деякі солі піридину використовують як синтетичні реагенти.
Наприклад, хлорохромат піридинію і дихромат піридинію застосовують для
окиснення первинних спиртів до альдегідів, а пербромід піридинію слугує
бромувальним агентом:
^Сг03СГ
і
Н
9
н
Сг207"
Н
Вг,-
Хлорохромат піридинію Дихромат піридинію Пербромід піридинію
Піридин широко використовують в органічних реакціях як м'яку основу.
❖ Нітрування. Ця реакція відбувається легко під час взаємодії гомологів
піридину з солями нітронію, наприклад, з нітроній тетрафлуороборатом.
Протонні нітрувальні агенти, такі як нітратна кислота, приводять до утворення
продуктів Ы-протонування. М-Нітропіридинієві солі використовують як
некислотні нітрувальні агенти з високою субстратною і регіоселективністю:
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
^т х тт ^ ^т ^ Н,С N сщ N0,
Н3С N
сн3
Н3С N
N0,
СН3 "з-
Використання нітроген (V) оксиду як нітрувального агента піридину дає
високі виходи (70 %).
❖ Амінування. Ця реакція за атомом Нітрогену відбувається в разі
використання солей гідроксиламіно-о-сульфокислоти:
Н2Ш803Н, К^СОз, 90°С
N
68%
N
ш
N
ш2 г
Л^-Амінопіридиній іодид
❖ Сульфування. Реакція піридину із сульфур триоксидом приводить до
утворення кристалічного цвітер-іонного піридиній-1-сульфонату. Ця сполука
зазнає гідролізування гарячою водою з утворенням сульфатної кислоти й
піридину:
N
БОз, СН2С12,20°С
90%
Н20
Ч^і Нагрівання І* і + Н2804
0=8=0
І
О"
Піридиній-1 -сульфонат
Піридиносульфотриоксид використовують як сульфувальний агент ацидо-
фобних сполук.
❖ Алкілування. Під час дії на піридин галогеноалканів за порівняно м'яких
умов відбувається реакція М-алкілування з утворенням іУ-алкілованих піридинів,
здатних під час нагрівання перетворюватися в 2- або 4-алкілпіридин:
І] + СН3І-
сн7
г-а
і
н
сн,
г +
сн,
І- Н J
-ын41
сн,
сн,
Розділ 38. Шестичленні нітрогеновмісні гетероцикли
955
❖ Ацилювання. Галогеноангідридн карбонових і арилсульфонових кислот
швидко реагують із піридинами з утворенням 1-ацил- і 1-арил-сульфоніл-
піридинієвих солей:
о
І + *-?ГСІ І* )
N о тСГ
с=о
О + ад^а —- (ТУ
N СІ
0=^=0
с6н5
Ці сполуки використовують для синтезу естерів і сульфонатів із відповідних
спиртів, амідів і сульфамідів з амінів.
❖ Окисиепня. Аналогічно до звичайних третинних амінів піридин легко
реагує з надкислотами з утворенням И-оксиду піридину:
ч Н202, СН3СООН, 65°С
95 % v
О"
Зокрема, №оксиди піридину відрізняються від самого піридину більшою
схильністю до реакцій електрофільного заміщення, що пов'язане з мезомерним
електронодонорним впливом атома Оксигену ^оксидного фрагмента в
структурах Ь-сІ. Підтвердженням цього є невеликий дипольний момент И-оксиду
піридину, що, відповідно, свідчить про суттєвий внесок у структуру ІМ-оксиду
піридину канонічних форм, у яких атом Оксигену нейтральний, а піридинове
кільце заряджене негативно:
NN NN N
о- о- О О О
а а* Ь с * сі
Отже, К-оксидна група піридину полегшує перебіг реакцій електрофільного
заміщення в а- і у-положення. Наприклад, нітрування з виходом, близьким до
кількісного, відбувається в у-положення:
-5
Сі
її
о
3 100 °С
956
Чирва В.Я., Ярмолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
Атоми Оксигену потім видаляють відновленням М-піридиноксиду трифеніл-
фосфіном:
ио2
89%
Шляхом відновлення у-нітропіридину легко одержати у-амінопіридин.
Чудовою властивістю И-оксиду піридину є його здатність приймати
нуклеофільні атаки. Це пояснюють тим, що поза електрофільною атакою Ы-оксид
піридину близький до структури а, і її атом Нітрогену завдяки позитивному заряду
ще більше здатний відтягувати електрони, ніж атом Нітрогену в піридині. Це й
виявляється в нуклеофільних атаках в а- і у-положення:
38.1.7. Інші реакції піридину
❖ Окислення. Серед інших властивостей піридину треба назвати його
стійкість до окиснювачів, гомологи ж поводяться аналогічно до гомологів
бензену, тобто окиснюються калій перманганатом до кислот або киснем над
ванадій (V) оксидом до альдегідів і кислот:
СН0 о, г^уСНз кмпо^ О^СООН
-"2
Піридин-3-карбальдегід Нікотинова
кислота
❖ ОзонолЬ піридину відбувається сутужніше, ніж бензену. Під час взаємодії
3,4-диметилпіридину з озоном утворюється гліоксаль, метилгліоксаль і діацетил:
:н3
!І онс-сно + н,с-со-сно + сн.сососн,
❖ Відновлення. Піридин, на відміну від бензену, відновлюється натрієм в
етанолі:
Розділ 38. Шестичленні нітрогеновмісні гетероцикли
957
j^j] C2H5OH + Na jf^jj CjH5OH + Na (f^| C2HsOH + Na
н н н
Піперидин
■ Бензен під час каталітичного гідрування (180 °С) дає циклогексан, тоді як
піридин - первинний аміламін:
l|~ C5HHNH2
■ Бензен відновлюється іодидною кислотою за 280 °С до метилцикло-
пентану, піридин за подібних умов дає н-пентан і амоній іодид:
❖ Реакції піколінів. На особливу увагу заслуговує реакційна здатність
метальних груп в а- і у-положеннях піколінів. В а-піколіні метальна група має
досить сильні СН-кислотні властивості, й атом Гідрогену в цій групі здатний
зазнавати заміщення літієм під дією бутиллітію. Піколіллітій, що утворився,
може реагувати з формальдегідом, утворюючи спирт:
C4H10Li
сн3—* к^сн^ґ снг° ~~~ CJ-c^oyi V-ch^oh
Н20
а
Порівняно висока СН-кислотність метальних груп в а- і у-піколінах
приводить до того, що ці сполуки, подібно до ацетооцтового естеру, здатні
конденсуватися з альдегідами й нітрозосполуками:
^N^CHrCH(OH)R "Н2° ^^^CH^CH-R
<Ок:н3 + ок-<Оь-к(сНз)2 ^VcH=NЧ^-N(CHз)2
Метильна група в Р-положенні цієї властивості не має.
❖ Піперидин подібний до аліфатичних амінів. Він є значно сильнішою
основою, ніж піридин. Основність його така ж, як і діетиламіну.
Подібно до атомів Гідрогену в аміногрупі аліфатичних вторинних амінів,
атом Гідрогену в піперидині здатний зазнавати заміщення різними залишками
(алкіли, ацили, нітрозогрупа тощо). Піперидин може бути дегідрований у
піридин нагріванням із сульфатною кислотою або за допомогою нікелевих або
паладієвих каталізаторів. Досить стійкий до таких окисників, як калій
перманганат у кислому середовищі, хром (VI) оксид, нітратна кислота.
958 Чирва В.Я., Ярмолкж С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
■ Продукти окиснення й відновлення похідних піридину належать до
природних біологічно активних речовин з широким спектром дії на організм
людини. Наприклад, важливою похідною піперидину є знеболювальна лікарська
речовина промедол, отримана в СРСР І. Назаровим. У промисловості його
синтезують за схемою:
(СН3)2СОН-С=С-СН=СН2
2-Метилгексен-5-ин-3-ол-2
+Н20
•н^с-сесмжн,
сн,
н2с=с-со-сн=сн-сн3
СНз
9
н^сн3 сНз-НС'^СН.
СЛІЛ.І
Н2С^ХН-СН3 н2о
сн,
о
5 с2н—со с6н5
Н,С*^>\ (сн3сн2со)^о Н3С^ГХ.
. сн3
сн3
промедол
38.1.8. Фізіологічно активні речовини
з піридиновим ядром
■ Нікотинова кислота належить до вітамінів і є 3-піридинкарбоновою
кислотою. Вітамінну активність має також амід нікотинової кислоти. Його
нестача в організмі призводить до захворювання на пелагру. Добова потреба
людини становить 0,05 мг. Нікотинамід, або вітамін РР, синтезують із
нікотинової кислоти, яку одержують окисненням нікотину. Нікотин добувають з
махорки. Нікотинамід є в складі деяких необхідних для життя ферментів, які
відіграють роль каталізатора гідрогенізаціі-дегідрогенізації відповідно до схеми
Н Н
+ 2Н
и
N
і
АяГ
-2Н
О^СОШ.
N
і
+ Н+Ап-
Розділ 38. Шестичленні нітрогеновмісні гетероцикли
959
■ Діетнламід нікотинової кислоти - кордіамін - є лікарським препаратом,
що стимулює центральну нервову систему, збуджує дихання і тонізує серцево-
судинну діяльність.
■ Похідні ізонікотинової кислоти - ізонікотингідразид і фтивазид -
застосовують як протитуберкульозні препарати:
/=\ Р
^-У HN~NH2
N
/=\ Р Н
HN-N
-он
ОСН,
Ізонікотингідразид
Фтивазид
Високотоксичний алкалоїд - нікотин - впливає на вегетативну нервову
систему. Це наркотик, що має ефект звикання. Смертельна доза для людини -
близько 40 мг. Широко використовують для боротьби зі шкідливими комахами.
Ізомерний до нікотину анабазин добувають із середньоазіатської рослини Anabasis
aphylla. Подібно до нікотину, дуже отруйний і має високу інсектицидну дію:
Нікотин
Будову анабазину доведено окисненням його до нікотинової кислоти й
дегідруванням до а-,Р-дипіридину:
а-,Р-Дипіридин
■ До сполук, що містять гідрований піридиновий цикл, належить коніїн (2-
пропілпіперидин). Він міститься в болиголові (Сопіит тасиїаШт). Дуже отруйний.
У 399 р. до н.е. водну витяжку цієї рослини був змушений випити філософ Сократ.
■ Вітаміни групи В6 представлені трьома похідними 2-метил-З-
гідроксипіридину — піридоксалем, піридоксолом, або піридоксином, і піридокса-
міном, що виділені з рису 1939 р.:
сно *
НОуЛуС^ОН
Піридоксаль
CRjOH
HO^W^CHjOH
НО^Ч^СНгОН
N
Н3С
Піридоксол
(піридоксин)
Піридоксамін
960 Чирва В.Я., Ярллолкж CM., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Вони є в структурі ферментів, що регулюють білковий обмін і каталізують,
зокрема, реакції переамінування амінокислот та їхнє декарбоксилювання. У
випадку нестачі цих вітамінів у їжі затримується ріст дітей, виникають шлун-
ково-кишкові розлади, втрата апетиту тощо.
38.2. Хінолін та ізохінолін
Піридинове кільце може бути сконденсоване з бензеновим двома спосо-
бами. Вони відображені в хіноліні й ізохіноліні:
^ ЇМ 3 4
Хінолін уперше виділений поряд з ізохіноліном Ф. Рунге з кам'яновугільної
смоли (1834). Незабаром після цього його отримали під час піролітичного
розкладу цинхонаміну - алкалоїду, спорідненого з хініном. Назва "хінолін"
походить від слова "хінін" (хіна - іспанський варіант місцевої південно-
американської назви кори дерева Cinchona^ що містить хінін). Велика кількість
похідних хіноліну виділена з деяких сортів нафти. Цикл хіноліну - основа
частини молекул великої групи алкалоїдів, серед яких є хінін.
Доказ структури хіноліну як 2,3-бензопіридину, або, що те ж саме, 1-
азанафталену, зводиться до окиснення його в хінолінову кислоту, яку можна
декарбоксилювати в піридин:
о-Положення замісників випливає з легкості утворення ангідриду. Будова
хіноліну також підтверджена низкою його синтезів, із яких найважливішою є
реакція Скраупа1.
Хінолін та ізохінолін - безбарвні рідини з сильним неприємним запахом,
погано розчинні у воді.
8
1
Хінолін
Ізохінолін
38.2.1. Фізичні властивості хіноліну
1 Скрауп 3. (Бкгаир Z.Щ (1850-1919) - австрійський хімік. Головні праці присвячені хімії алкалоїдів
групи хініну, вуглеводів та білків. Запропонував метод синтезу хіноліну (реакція Скраупа).
Розділ 38. Шестичленні нітрогеновллісні гетероцикли
961
• Уф-спектроскопія. В уф-спектрі хіноліну є смуги поглинання з А,тах 228, 270
і 315 нм. Для ізохіноліну характерна наявність смуг поглинання з л.гаах 218, 265 і
313 нм.
• ПМР-спекмроскопія. Всі атоми
Гідрогену в молекулі хіноліну
нееквівалентні, тому в його спектрі
спостерігають сім сигналів арома-
тичних протонів: ^ "На м.ч.8,81 (а), 7,26 (Ь),
8,00 (с), 7,68 ((і),
4,43 (е), 7,61 (і),
8,05(8)
38.2.2. Одержання хіноліну і його гомологів
❖ Синтез Комба. Конденсація 1,3-дикарбонільних сполук із ариламінами з
високими виходами приводить до утворення ароматичних похідних хінолінів:
❖ Реакція Скраупа полягає в тому, що ароматичний амін з вільним о-
положенням нагрівають із гліцеролом, сульфатною кислотою й ароматичною
нітросполукою або ванадієвою кислотою як окисником. Сульфатна кислота
перетворює гліцерол в акролеїн, що зазнає конденсування з аніліном, унаслідок
чого утворюється Р-анілінопропіоновий альдегід. Цей альдегід втрачає воду й
дає дигідрохінолін, що його потім окиснює до хіноліну нітробензен:
9Н
Ш2 СН2
КНСН2
Виходи хінолінових основ можна підвищити, застосовуючи різні окиснювачі
(ванадієву кислоту) або каталізатори дегідратації (алюміній, торій оксиди).
Конденсацією акролеїну з гомологами аніліну або його похідних, такими, як
галогено-, нітрозаміщені, а також амінокарбонові кислоти, аміносульфокислоти
962
Чирва В.Я., Ярллолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
й амінофеноли, одержують найрізноманітніші похідні хіноліну із замісниками в
бензеновому кільці.
Синтез Скраупа найпридатніший для одержання незаміщених за гетероциклом
хінолінів.
❖ Синтез Дебнера-Міллера1. Ароматичний амін нагрівають із двома
молями альдегіду й концентрованою хлоридною кислотою. У цьому разі, цілком
імовірно, спочатку дві молекули альдегіду вступають в альдольну конденсацію з
утворенням Р-гідроксіальдегіду, який потім зазнає дегідратування до а,Р-
ненасиченого альдегіду. Аналогічно до синтезу Скраупа, а,р-ненасичений
альдегід утворює дигідрохінолінову сполуку, яка під дією азометану,
перетворюється в похідну хіноліну:
ОНС. .Я
4- тг
сн
2 СЬІД
N ХН2Я
н
сн2к
Цей метод, по суті, є різновидом реакції Скраупа.
❖ Синтез Фрідлендера. В цьому способі о-амінобензофенон конденсують
з альдегідами або кетонами, що мають поряд з карбонільною групою
метиленову ланку; реакцію каталізують як кислотами, так і основами, що дає
хіноліни:
кон, с2н5он, о°с
71%
N С6Н5
с6н5
СН,
^с
СН^
Кип'ятіння
конц. Н2504, СН3СООН
ш2 о' чсн3
88%
Паралельно із синтезом хіноліну відбувається альдольно-кротонова конденсація
альдегіду й утворення основ Шиффа, що загалом знижує цінність реакції.
38.2.3. Хімічні властивості хіноліну
Для хімічних властивостей хіноліну характерні електрофільні реакції, що
відбуваються за участю бензенового кільця, й нуклеофільні реакції, спрямовані
на піридиновий цикл. Крім того, хінолін виявляє хімічні властивості, властиві
третинним амінам. Подібно до піридину він утворює солі з сильними
мінеральними кислотами (І), четвертинні амонієві солі (II), Аґ-оксид (III):
1 Міллер В. (Міііег (1848-1899) - німецький хімік-органік.
Розділ 38. Шестичленні нітрогеновллісні гетероцикли
963
О
III
Галогеніди 1-алкілхінолінію (II) під час нагрівання в запаяній ампулі до
300 °С піддаються реакції Ладенбурга з міграцією алкільного замісника
переважно в положення 2, 4 і 8.
❖ Реакції електрофільного заміщення. Електрофільні реагенти спочатку
утворюють з хіноліном амонієві солі, а потім атакують бензенове кільце в
положення 5 і 8. Наприклад, нітрат хіноліну в сульфатній кислоті через 1 год за
кімнатної температури перетворюється в суміш 5- і 8-нітрохінолінів:
■ Нітрування хіноліну сумішшю нітратної і сульфатної кислот приводить
головно до двох продуктів - 5,7- і 6,8-динітрохінолінів, тоді як нітрування під
дією цирконій (IV) нітрату дає 7-нітрохінолін. Використання як нітрувального
реагенту ацетилнітрату (нітратна кислота в оцтовому ангідриді) забезпечує
перебіг нітрування хіноліну за механізмом приєднання-відщеплення і приводить
до утворення з високими виходами 3-нітрохіноліну.
Сульфування хіноліну приводить до різних продуктів залежно від темпе-
ратури реакції:
5
30 % олеум
90°С
ЗО % олеум
170>С
8
30 % олеум
300<€
Н2804
300°С
ч 3
300°С
Н2804
80,Н
Сплавлення 8-сульфохіноліну з лугом приводить до 8-гідроксихіноліну, або
океану.
964
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
■ Процес галогенування хіноліну може відбуватися за різними меха-
нізмами. Наприклад, обробка бромом у сульфатній кислоті за наявності А§2804
дає 5- і 8-монозаміщені похідні в співвідношенні 1:1. Дія комплексу Вг2 АІСЬ
приводить лише до 3-бромохіноліну, обробка хіноліну бромом у піридині дає
змогу синтезувати 3-бромохінолін за таким механізмом:
■ 3 галогеноалканами хінолін, подібно до піридину, утворює солі И-
алкілхінолінію.
❖ Реакції нуклеофільного заміщення. Амінування хіноліну. Реакція калій
аміду з хіноліном приводить до утворення дигідроаддуктів унаслідок атаки амід-
іонів головно у положення 2 і в меншому ступені за атомом С4-хіноліну. їхнє
окиснення дає, відповідно, 2- і 4-амінохіноліни:
■ Гідроксилювання хіноліну. Реакцію проводять прямою взаємодією хіноліну
з калій гідроксидом:
2-Хінолон
Розділ 38. Шестичленні нітрогеновллісні гетероцикли
965
■ Заміщення атома галогену. Для реакцій цього типу діє загальний
принцип - реакційна здатність атомів галогену в бензеновому кільці хіноліну
така ж, як у галогенобензенів. Водночас для атомів галогену в положеннях 2 і 4
хіноліну характерні ті ж реакції з нуклеофільними агентами, що й для а- і у-
галогенопіридинів.
38.2.4. Похідні хіноліну
Низку заміщених хіноліну використовують у синтезі лікарських препаратів і
барвників.
❖ 8-Гідроксихінолін (рксин) одержують за реак-
цією Скраупа або під час сплавлення 8-сульфохіноліну з
лугом. Його широко застосовують як реагент в
аналітичній хімії. Просторова близькість гідроксильної
групи й неподіленої пари електронів атома Нітрогену
зумовлює виникнення внутрішньомолекулярного водне-
вого зв'язку, що виявляється в низькій температурі
плавлення (75-76 °С) і леткості з водяною парою:
З низкою металів 8-гідроксихінолін утворює міцні й нерозчинні у воді
координаційні сполуки - "хелати". Цю властивість 8-гідроксихінолину вико-
ристовують у кількісному аналізі для визначення Магнію:
8-Пдроксихінолін (оксин)
2НаОН /=\^ Mg.a _/=
❖ Хінін. Хіноліновий цикл існує у важливій природній сполуці - алкалоїді
хінін, виділеному 1820 р. з кори хінного дерева. Його солі надзвичайно гіркі на
смак. Розчини мають сильну синю флуоресценцію. Будова підтверджена
численними перетвореннями й повним синтезом Р. Вудвордом у 1944 р.
Молекула хініну містить дві різні гетероциклічні системи: хінолінове й
хінуклідинове ядра. Вона має чотири асиметричні атоми Карбону, що дуже
ускладнило синтез хініну:
н3с-о
носн—сн ?^сн2
N
-сн=сн.
Хінін має токсичну дію на нижчі організми, особливо ті, що спричинюють
малярію.
966
Чирва В.Я., Ярллолкж С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
■ Через відсутність природного хініну в Радянському Союзі в 30-х роках
XX ст. отримано синтетичні антимапярійні препарати примахін і плазмохін:
СН30
(СН2)3М(С2Н5)2
І
Н3ОСН-(СН2)3МН2
Примахін
н3с-сн
2'3
Плазмохін
■ На основі 8-гідроксихіноліну синтезований 8-гідрокси-7-іодо-5-хлоро-
хінолін (ентеросептол)^ який завдяки кількісному зв'язуванню йонів Кобальту
ефективно застосовують у боротьбі з кишковими захворюваннями.
❖ Ціанінові барвники. Похідні хіноліну використані як перші фотографічні
сенсибілізатори: ціаніновий барвник етиловий червоний дав змогу зробити
фотографію чутливою не тільки до блакитного, а й до зелених кольорів. Нижче
наведено схему його синтезу:
ох+н хо - ссхло
11 "Ц А+*1
СН N
С,Н
2**5
сн
г
2**5
с2н5 с2н5
Етиловий червоний
У 1904 р. з появою пінаціанолу фотоплівка стала чутливою до червоних кольорів:
х3
Пінаціанол
Згодом отримано й досліджено тисячі сенсибілізувальних барвників, а
хінолінові барвники замінені на інші, ефективніші.
38.2.5. Ізохінолін
Ізохінолін - ізомер хіноліну. Під час окиснення він утворює фталеву кислоту
й 3,4-піридиндикарбонову кислоту, що ізомерна хіноліновій кислоті, чим
доведено наявність і відносне положення бензенового й піридинового ядер:
КМп04 р^С00Н + НООС
3,4-Піридиндикарбонова
кислота
Розділ 38. Шестичленні нітрогеновмісні гетероцикли
967
■ Синтез за Біииіеромх-Напиральським. У класичному варіанті синтезу
Бішлера-Напиральського амід, отриманий реакцією 2-арилетиламіну із
хлороангідридом або ангідридом карбонової кислоти, зазнає циклізування з
втратою молекули води до 3,4-дигідроізохіноліну, який легко дегідрує під дією
паладієвого каталізатора, сульфуру або дифенілсульфиду. Як конденсувальні
агенти зазвичай використовують фосфор (V) оксид, фосфорилхлорид і фосфор
(V) хлорид:
+
.СІ
Н3С-С^0
95%
№1
V
СН,
Р205,
82%
■ Синтез Пікте2-Шпенглера. Арилетиламіни легко реагують із альдегідами,
внаслідок чого утворюються іміни з високими виходами. їхня циклізація,
каталізована кислотами, приводить до утворення 1,2,3,4-тетрагідроізохінолінів:
сн3о
N^100% Ч^^'
сн,о
ш2100%
неї, 100°С,
80%
Звичайні прийоми дегідрування дають змогу легко перетворити похідні
тетрагідроізохіноліну в повністю ароматичні сполуки.
Хімічні властивості ізохіноліну подібні на властивості хіноліну. Сульфу-
вання ізохіноліну приводить до 5-сульфонової кислоти. Амінування ізохіноліну
калій амідом дає 1-амінопохідну, а гідроксилювання калій гідроксидом - 1-
ізохінолін (ізокарбостирил). Нуклеофільне заміщення атома галогену в положенні
4 ізохіноліну відбувається аналогічно до галогену в положеннях 2 і 4 хіноліну.
■ Кістяк ізохіноліну, зазвичай гідрованого, є в структурі низки важливих і
складно побудованих алкалоїдів - морфіну, папаверину, алкалоїдів кураре.
1 Бішлер А. (ВібсЬієг А.) (1865-1957) - швейцарський хімік російського походження. Займався
органічним синтезом. Разом з учнем Б. Напиральським (В. №ріега1$кі) розробив синтез Бішлера-
Напиральського.
2 Пікте А. (Рісіеі А.) (1857-1937)- швейцарський хімік. Наукові праці присвячені гетероциклічним
сполукам, алкалоїдам та вуглеводам. Виконав синтез піюбіну, алкалоїдів опіуму, а також маль-
този, мелібіози, лактози і рафінози.
968
Чирва В.Я., Ярллолюк СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
38.3. Піриллідин
Дуже важливим гетероциклом є піримідин. Ароматичні
властивості в цій сполуці поєднуються з підвищеною основ-
ністю завдяки двом атомам Нітрогену з неподіленими елект-
ронними парами:
❖ Синтез піримідину. Піримідин і його гомологи можна синтезувати з
барбітурової кислоти (уреїд малонової кислоти, або малонілсечовина) через
стадію 2,4,6-трихлоропіримідину:
,с-он н2н
н,с + с=о
с-он
її
Сам піримідин синтезують конденсацією 3-аміноакролеїну із сечовиною або
каталітичним відновленням 2-хлоро-, 2,4-дихлоро- чи 2,4,6-трихлоропохідних
піримідину за наявності основ.
❖ Хімічні властивості. До електрофільного заміщення піримідин здатний
ще менше, ніж піридин. Реакція спрямована тільки на положення 5. Взаємодія з
електрофілами може також відбуватися по атому Нітрогену. Піримідини, що
містять ОН-, ЫН2- та інші замісники, які його активують, зазнають нітрування,
нітрозування і вступають в азосполучення в положення 5; замісники, як
звичайно, перебувають у положеннях 2, 4 або 6 та орієнтують атаку електрофіла
в о- і «-положення щодо себе.
У метилпіримідинах метальні групи в положеннях 2, 4 і 6 вступають з
альдегідами в реакції кротонової конденсації.
Зазначимо, що 2-, 4- і 6-хлоропіримідини мають (подібно до 2-, 4- і 6-хлоро-1-
нітробензенів) реакційноздатний атом галогену, що не тільки зазнає заміщення
внаслідок "звичайних" нуклеофільних атак, а й забезпечує досить високу здатність
хлоропіримідинів у реакціях Фріделя-Крафтса з ароматичними вуглеводнями.
Гідроксипохідні піримідину мають сильніші кислотні властивості, ніж
феноли.
Бічні алкільні групи гомологів піримідину можна перетворити в кар-
боксильні групи, не зачіпаючи піримідинового циклу, дуже стійкого до
окиснення.
Розділ 38. Шестичленні нітрогеновмісні гетероцикли
969
Нуклеофільне заміщення в пірнміднні відбувається в електронодефіцитні
парні положення. Ці реакції часто супроводжуються розкриттям циклу з
наступною рециклізацією.
❖ Похідні піримідину. Три важливі похідні піримідину є в складі
природних нуклеозидів, нуклеотидів і, відповідно, нуклеїнових кислот, що
містяться в ядрі клітин і можуть бути отримані з них шляхом гідролізу:
1Н ї
НзС>ґ% нзс>г>н
н
N ОН N
Урацил Тимін
ш2
Цитозин
■ До піримідинових похідних належать ліпші з сульфамідних препаратів -
супьфазин, метилсульфазин, сульфадимезин:
сн,
— о *Н[
Сульфадимезин *
■ Вітамін В/, або тіамін, теж містить піримідиновий цикл:
носн2-сн2
Н3С 8
Вітамін В (тіамін)
Відсутність вітаміну Ві у їжі спричинює нервове захворювання - поліневрит
із наступним виснаженням, діареєю, недоумкуватістю, дерматитом. Потреба в
цьому вітаміні пов'язана з тим, що вітамін В| є в структурі ферменту
кокарбоксилази.
970
Чирва В.Я., Ярллолкж СМ., Толкачова Н.В., Земляков О.Є. Органічна хімія
38.4. Пурин і його похідні
1 ^6 Біциклічний гетероцикл, що є конденсованою системою
ї^::::^у^^7 піримідину й імідазолу, називають пурином.
2 х- ^ Пурин - електронодефіцитна гетероциклічна ароматична
У '♦И^8 система, тому для неї не характерні електрофільні реакції. їх
^ можна виконати тільки із замісниками, що активують і
п спрямовують реакції в положення 8. Зате пурин і його похідні
легко вступають у реакції нуклеофільного заміщення в
положення 6.
■ Похідні, що містять пуринову систему, поширені в природі й відіграють
важливу роль у біологічних процесах. Це насамперед аденін і гуанін, що містяться
як аглікони (невуглеводна компонента, що зв'язана глікозидним центром із
цукровим залишком) у нуклеїнових кислотах, нуклеозидах, нуклеотидах:
ОН
Гуанін спричиняє блиск луски риби, його застосовують для виробництва
штучних перлів.
■ До пуринів належать алкалоїди теобромін (какао), кофеїн (чай," кава) і
теофілін (чай):
Н3С,
О
і N
сн3 н
Теофілін
Кофеїн - важливий лікарський засіб, що збуджує нервову систему,
стимулює роботу серця й прискорює пульс. Теобромін і теофілін - діуретики,
вони розширюють судини, тому їх застосовують для лікування гіпертонії.
■ До ключових сполук пурину належить сечова кислота:
Розділ 38. Шестичленні нітрогеновллісні гетероцикли
971
Сечова кислота - продукт нітрогенного обміну в організмі людини й тварин,
її мононатрієва сіль - складова частина каменів сечового міхура. Висохлі
екскременти птахів (гуано) містять до 25% сечової кислоти і слугують джерелом
її одержання.
Синтезують сечову кислоту шляхом конденсації урамілу (амінобарбітурової
кислоти) з ізоціанатами або калій ціанатом:
ОН ОН ?н
„сЛАон н* „оЛ.Аон но^н^он
Амінобарбітурова кислота Псевдосечова кислота Сечова кислота
Сечова кислота - синтбн для одержання алантоїну, парабанової кислоти і
кофеїну.
Пурин є в основі сипденофілу, який одержав світову популярність під
торговою назвою віагра для лікування імпотенції:
Силденофіл
Ім енний
Авогадро А. 35*
Агрікола Г. 541
Адамс Р. 192
Адкінс Г. 143
Акерман Т. 563
Альдер К. 251,264,266, 556,73:
873,944
АндерсонТ.903, 942
Анджелі А. 739
Андрусов Л. 162
Арбузов 0.451, 737,929
Армстронг Г. 661. 872
Байер А. 126,464,490, 504,566,
627,661,878,912,917,934,935,
944
Бакеланд Л. 808
Баландін 0.154
Бамбергер Е. 734, 765
Бауманн Е. 640,795, 845, 846
Бахман В. 778,779
Башкіров 0.350
Безекен Я. 629
Бейльштейн Ф. 31
Бекер І. 694, 695
Бекман Е. 602. 710, 835, 836
Бергс Г. 869
Бергіус Ф. 138, 139
Береш Я. 381
Бертло П. 34, 140,227, 673,883
Берцеліус Й. 21,33-35, 577
Берч А. 189,233
Бешам А. 746
Бішлер А. 967
Бішоп К. 550
Блан Г. 352,373, 506, 550, 704
Бломстранд К. 761
Бодру Ф. 508
Бородін 0.314,498
Браун Г. 350
Браун Дж. 706
Браун Ю. 563
Бренстед Й. 76,77,78
Буво Л. 352,373, 506
* Жирним виділений номер сторінки,
на якій наведена біографічна довідка
покажчик
Бунзен Р.419,679,701
Бутенандт А. 369
Бутлеров 0.22,34,37,38,40,95,
134,135,395,435,461,464,619,
660
\ Бухерер Г. 869
Вагнер Є. 213,214,362,372
Валлах О. 739
Вальден П. 563
Ван-дер-Ваальс Я. 47,64,65
ван Екенштейн А. 635
Вант-Гофф Я. 40,96, 109,252,
555
Веєрман Р. 637
Вейцман Х.346,439
Велер Ф. 21.33-35,225, 540,
829,844
Верлей А. 362,442,461
Вестхаймер Ф. 440
Відман О. 87
Вілкінсон Дж. 194
Віллігер В. 464,490,504, 566
Вільгеродт К. 495
Вільсмейєр А. 823
Вільштеттер Р. 926
Вільямсон А. 37,317,391,392,
398,793
Вісліценус Й. 555
Віттіг Г. 190,350,455, 758, 875
Воль А.212,548,637
Вольпін М. 667
Вольф Л. 461,681
Ворожцов М. мол. 706
Воронін О. 468
Воскресенський О. 858
Вудворд Р. 264, 565,288, 378,
926,927, 965
Вюрц Ж.468
Вюрц Ш.35, 141, 143,144, 190,
283,317,372,412,423, 678,679,
714, 874, 883. 884
Габрієль 3.419,420, 598,849
Ганч А. 87,945
Гарден А. 858
Гаррієс К. 216, 662
Гаттерман Л. 701, 774, 775,823,
824, 884
Гей-Люссак Ж. 35
Гейман К. 935
Гейтер А. 581
Гелль К. 527,564,598
Генрі Л.410
Герцберг Г. 147
Геш К. 806, 824, 922
Гілер С. 910
Гільман Г. 443
Гінзберг 0.418
Глазер К. 244
Гмелін Л. 21,433
Големан А. 687
Гомберг М. 778,779,884
Горін Ю. 255
Гофман А. 186,317,378,417,
419-421, 519,526,599,637,640,
659, 748, 849, 855,943,950
Гребе К. 858,859,860,862,878.
880
Гриньяр В. 148,231,240,241,
352,353, 390,394,395,443,444,
448,454,494, 504, 505, 508,512,
524, 565, 572, 673, 678, 714,777,
789, 816, 817, 823-825,842,883,
884,888, 889,915,930,937
Грісс П. 763
Гудрі Е. 158
Гунд Ф. 45, 56,665
Густавсон Г. 679
Дальтон Д. 38
Дебай П. 58
Дебнер О. 893, 894,896,962
Деві Г. 226
Дем'янов М. 293
Дерінг В.666,667
Джеймс Т. 26
Джексон Е. 628
Джонс Е. 462
Дікман В. 285, 550, 580
Дільс 0.251,264,266,445,556,
737, 873, 944
Долін М. 680
Дуппа Д. 582
Дюар Дж. 661, 684
Дюма Ж.22,35, 140,872
Едман П. 614,615
Ельтеков 0.254, 365, 836
Іменний покажчик
973
Енгельгардт А. 700
Ерленмеєр Р. 254,365,836, 858.
859,860,862
Етар А. 822
Ешвайлер В. 423
Жерар Ш. 22,36,700
Зайде 0.949
Зайцев 0.40,180,183,228,358,
359,445,480,511,571
Залкінд Ю. 244
ЗанцмейєрТ.700,701,774
Зелінський М. 243,292, 527, 564,
598,661,673-675, 861
Зінін М. 34,423,738,742,746,
864
Інгольд ІС 102,319,686,694,706,
763
Іпатьєв В. 258
Ірвін Дж. 641
Йоцич Ж.231,241
Казанський Б. 243,674,861
Кан Р. 102
Канніццаро С. 444,461,465.476,
576,600,816,831.832,910
Караш М. 149,204,236
Каріус Л.31,32
Каро Н. 428,878,880
Карозерс У. 243,259
Кекуле А. 723
Кекуле Ф.36, 37,660,661,663,
685,858,859,860, 862
Кернер В. 665, 670,942
Кіжнер М.461,681
Кіліані Г. 568,621,622,637
Кіндлер К.495
Кіпріанов А. 771
Кірхгоф К. 619
Клабуновський Є. 563
Кларк Г.423
Клемменсен Е. 285,462,681
Клечковський В. 45,46
Кляйзен Л. 285,366,451,508,
579,580,795,829,831,832
Кневенагель Е. 533,536,545
Кнорр Л. 582,583,904,918,938,
939
Колі А. 627
Кольбе А.34,36, 141,228,317,
406,497,498,540,763, 806,813,
850,853
Комб А. 961
Коновалов М. 151,405,730
Конрад М. 552
Корі Е. 144,678, 884
Корнблюм Н. 406,442
Корольов О. 867
Кох Ю. 493,823
Кочі Дж. 498
Крафтс Дж. 156,380,679,680,
681,684,797,806,805,816,823,
824,834. 843. 845,847, 862,865,
866,873,874, 877,886, 888,908,
909,914,930,946
Кріге Р.214,378
Кун О. 563
Купер А. 37
Курсанов Д. 667
Курсанов М. 795
Курціус Т. 424,526,527,748
Кучеров М. 237,239,256,438,
445,723
Лавуазьє А. 34
Ладенбург А. 83,84,661,963
Ле Бель Ж.40,96,109
Лебедєв С. 193,254,255,267,273
Левіна Р. 874,884
Лем'є Р. 215
Лепуорг А.829
Ліберман К. 858,878,880
Лібіх ІО. 21,22,34,35,329,600,
659,700,829,844
Лінцлар Г. 189
Лобрі де Брюїн К. 635
ЛовіцТ. 619
Ломоносов М. 38
Лоран О. 35,781,870
Лоссень В. 30,31,423
Лоурі Т. 76,78
Лоуссан С.-0.905
Лукас Г.356
Львов М. 135,210,336,368
Льюїс Г. 74,78-80,140,191,234,
258,269,290,300. ЗОЇ, 315,367,
393,395,435,461,479, 515,592,
679,680, 683,684,700,750,795,
797,804, 823, 863, 890,953
Маделунг В. 929
Майєр В. 318.405,409,773
Мейєр К. 583
Майлс Н. 214,372
Маккензі К. 562
Макор Д.687
Малапраде Л. 378
Мамедалієв Ю. 680
Манніх К. 411,833,915,931
Марквальд Н. 562
Марковников В. 40,151,200-203,
205-207,235,291,312,313,345,
348,350,393,446,534,565,795
Маргін А. 27
Мархлевський Л. 926
Мейєрвейн Г. 362,394,442.461,
779
Мейзенгеймер Я. 710
Мсльдрум А. 546
Менделєєв Д. 461
Меншуткін М. 416
Мерц Г.419
Міллер В. 893,962
Мітчерліх Е. 659
Міхаєль А. 588,945
Міхлер В. 83,84,757,824,886
Молдавський Б. 674
Назаров і. 259,370,371,958
Напиральський Б. 967
Натан у. 694,695
НаттаДж. 177,270-274
Нельсон Дж. 443
Неф Д. 409,472
Нєсмєянов 0.539,567,778
Ніколь В. 96
Нобель А.382
Норман В. 919
Норман Г. 333
Ньюленд Дж. 243,259
Ньюмен М. 100,123,125,127,
280,632
Ола Дж. 160,686
Ольдекоп Ю. 704
Оппенауер Р. 362,381,441,442
Пааль К. 904,918
Павда А. 919
Первеєв Ф. 906
Пастер Л.119,121,562
Паулі В. 45,56
974
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
Перкін В. 34, 554, 830
Перкін В. мол. 283
Пехманн Г. 432
ПіктеА. 967
ПіріаР.444
Пірсон Р. 79, 80
ПірсонТ. 164
Пітцер К. 123
Планк М. 65
ПлатеА. 584
ПолінгЛ. 50,51
Понндорф В. 362,442,461
Попов 0.464,490
Поуп В. 414
Прегль Ф. 31
ПрелогВ. 102
Преображенський В. 941
Преображенський М. 941
Прилєжаєв М. 213,372,398, 846
ПрінсГ.208,257,259,373
Пурді Т. 641
ПфлегерК. 935
Райлі Г. 472
Рамзай У. 944
РаульФ.32,602
Рашіг Ф. 701,707
Рашке М. 350
Резерфорд Е. 42
Реймер К. 807, 813, 814, 823,922
Релей Дж. 944
Ремзен А. 722
Реней М. 143,192,351,352,746,
916
Реіше В. 208,225,239,242,244,
256,290,367,370,373,446,535,
945
Реформатський С. 188, 565
РітгерДж. 424
Робінсон Р. 929
Родіонов В. 601. 832
Ройлен 0.347
Розанов М. 101
Розенмунд К. 445,480.511,706,
822
РужичкаЛ. 274,284,369
РунгеФ.917,960
Руфф 0.937
СабатьєП. 139, 193,661,868
СабатьєФ. 143
Сангер Ф. 605
Семенов М. 146
Сергєєв П. 786
Сільва М. 380
Сіммонс Г. 169,287
СінгР. 27
Скрауп 3.960,961,962
СмітР. 169,287
Соколов М. 700
Сокслет Ф. 26
Сократ959
Соммле М. 821
Соночашира К. 212
Суберан Е. 34
ТеннЛ. 563
Тереетьєв 0.425,563,909.922.
948
' Тищенко В. 461,466, 503
Тіле Ф. 660
Тімман 1.807,813,814,823,917
Толленс Б. 462,463, 621,627,
649,651
ТорпДж. 274,694
Траверс М. 944
Тропш Г. 139, 140,347
Удріс Р.786
УльманФ.706,714,758.884
Ури (Юрі) Г. 162
Фаворський 0.193,225,231,
238,241,244,253,254,259,367,
370,375,438,495
Фарадей М. 659
ФелінгГ. 463
Феофілакгов В. 769
ФізерЛ. 867
Фінкельштейн Г. 316,336
Фітгіг Р. 678,714. 874,883, 884
ФішерГ. 925,926
ФішерЕ. 99,100,101,102,116,
117,500.504,621,626, 629,630,
631,637,639,773,896,928,929
Фішер 0.896
ФішерФ. 139, 140.347
ФольгардЯ. 527,564,598
Франкланд Е. 582
Фрідель Ш. 156,380,67^81,
684,797,804,805,816,823,824,
834,843,845,847,862,865,864,
873,874,877,886,888,908,909,
914,930,948
Фрідлендер П. 936,962
Фріс К. 796
ФріцшеЮ.746.803
Фрост А. 664
ФукуїК.61
Хааф В. 493
ХадсонК. 628
ХайнДж. 807
ХассГ. 151
ХаусХ. 144,682,884
ХафнерК.217
ХекР.212
Хеммонд Дж. 691
Хеуорс В. 627,628-630,641
Хехст516
Ходжкін Д. 927
ХокХ. 786
ХофманР. 264,265,288
Хунсдіккер Г. 314,498
Хьюз Е. 319
Хюккель Е. 58,665-^67,924,928
ЦвєтМ.27,926
ЦейзельС.395,795
Цейхмейстер К. 682
ЦеревітіновФ. 188,425
Циглер К. 177,212,258,270-274,
284,347,365. 548,673
Цукерваник 1.680
Чічібабін 0.508,732,912,929,
945,949,950
ЧугаєвЛ. 188,425,473,477
ШвабК. 563
ШеєлеК. 380,573,814
Шемякін М. 740
Шербульє519
Шиман Г. 702,775
Шифф Г. 457,600,602,828,910,
962
ШмідтК.414,619,829,832
Шоригін П. 144, 714
Шорлеммер К. 22
Шостаковський М. 238,247,367,
438
Ш(УітенК.640,795, 845,846
ШпенглерТ.967
Шредінгер Е. 43
ШтальГ.ЗЗ
Штаудингер Г. 432, 514
ШтоббеГ. 550
Штраус Ф. 244
ШтрекерА. 600,602
ШультеД. 906
ЩукінаА. 370
Юр'єв Ю. 905
Якобс В. 370
Якубович Л. 381
Предметний покажчик
а-Спіраль 615
р-Аланін 601,606
а-Діастереомерія 116, 276, 343, 561, 868
N-Фенілгліцин 935
Агар 655
Агароза 655
Аглікон 643
Адамантан 277, 299
Аденін 970
Адреналін 86
Азатриптицен 758
Азиди 317, 773
Азини 459
Азлактон 600
Азоксисполуки 734, 737, 931
Азосполуки 734,736,739,761,764,766,923
Азометіни - див. Іміни
Акролеїн 265, 381, 383, 532, 944, 961
Аланін 602,610
Алени 253
Алізарин 878
Алкани 132
Алкени 170
Алкіни 220
- природні сполуки 246
Алкогольдегідрогеназа 647
Алосетрон 932
Алотреонін 597
Альбуцид 752
Альдегід
-гліцероловий 101, 383, 560, 622,647
-коричний (цинамоновий) 818, 830
- кротоновий 468, 545
-левуліновий 216
-саліциловий 810, 823
-толуїловий 114
Альдегіддегідрогеназа 648
Альдолаза 647
Альдоль 467, 567
Амігдалин 818
Аміди 239, 420, 495, 511, 515, 522, 525, 599, 637,
748,750,752,835,846,855,967
Амідол 738
Амідопірин (пірамідон) 586,939
Амілаза 649
Амілоза 654
Амілопектин 655
Аміни 412, 744
Амінодурол 749
Амінокислоти 593, 855
- незамінні 594
- незвичайні (рідкісні) 594
- протеїногенні 593
Аміноксидази 603
Анабазин 959
Анальгін 586, 939
Ангідриди карбонових кислот 265, 366, 488, 501,
511,548,550,555, 566,571,640,662,671,681,737,
750,754,796,824, 830,833,847,851,855,858,874,
879,884,898,908,920,930
Андаксин 853
Андростенол 305
Анетол 790, 851
Анестезин 855
Анізидини 811
Аніліди 745, 750, 752, 754
Анілін 87, 734, 736, 748, 893, 935
Аномери 627
Анса-сполуки 114
Антидетонатор 160
Антиоксиданти 758, 805, 813
Антипірин 585, 939
Антифриз 371
Антрагідрохінон 877
Антрацен 872
Антрон 877
Анулени 668
Арабіноза 625, 637
Арабіноксилани 653
Арбутин 642
Ароматичність 663, 665, 859, 873, 902, 924, 928,
942
Аспартам 612
Аспірин 851
Атрактанти статеві 369
Атропоізомерія 112, 883
Аурин 896
Ацеталі (кеталі) 238,371,378,450,507, 642,646
Ацетальдегід 217, 237, 255, 329, 364, 374, 379,
384, 438, 442, 445, 450, 453, 459, 466, 474, 488,
516, 536, 564, 577, 647, 946
Ацетофенон 730, 748, 822, 825, 832, 842
Ацидофобність 908, 921
Ацилази 603
Ацилазиди 424, 511, 525, 599, 748
976
Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Земляков О.Є. Органічна хімія
Ацилалі 371, 546
Ацилгідразиди 424, 524, 599, 614
Ацетилацетон 475, 479
Бакелітизація 808
Бамбікол 370
Барвники 769, 870, 878, 891, 899, 936, 966
Баскетан 299
Бензвален 682
Бензальацетон 830
Бензальдегіди ЗО, 63, 410, 454, 600, 713, 730, 733,
748,815,818,821,826,842,845,894
Бензантрон 880
Бензаурин 896
Бензен та його гомологи 659
Бензидин 742, 745, 885
Бензил 829
Бензин 138
Бензин (дегідробензен) 708, 747
Бензоїлацетон 107
Бензоїн 828
Бензофенон 834, 886
Біацетил 475
Білірубін 925
Білки 612
-глобулярні 617
-фібрилярні618
Бінафтил 113
Бісфенол А 809
Біфеніли 112, 678, 714, 779, 822, 937
Борнеол 303
N-Бромосукцинімід 211, 313, 548
Будова
- алену 251
-амінів 413
- амінокислот 597
- антрацену 872
- арилдіазоніїв 760
- ацетилену 220
- бензену 663
-біфенілу 882
-бут-1,3-дієну 249
- етерів 388
-етилену 170
- карбонових кислот 483, 839
-метану 132
- нафталену 858
- нітрозосполук 739
- нітросполук 403, 725
- оксосполук 434
- пептидної групи 609
- піридину 942
- піролу 902
-спиртів 341
-фенолу 782
-хімічна 38
- циклопропану 279
Бульвален 107
Валін 603
Ванілін 87, 790,811
Вератрол 811
Веронал 547
Вінілацетат 176
Вініліденхлорид 334
Вінілогія 467
Вінкристин 931
Віолантрон 881
Вісь симетрії 109
- хіральності 111
Вітаміни
-Аі (ретинол) 275
-В, (тіамін) 969
-Вб (піридоксаль, піридоксин, піридоксамін) 959
- Ві2 (кобаламін, ціанкобаламін, оксикобаламін)
926
т С (аскорбінова кислота) 645
-Н (біотин) 917
- Кі (філохінон) 867
- Кг (менахінони) 276, 867
- РР (нікотинамід) 958
- Р (убіхінони) 276
- и (метилметіонін) 608
Волокно віскозне 653
Вугілля 23, 138, 675
Вуглеводи 619
Вулканізація 272
Вулкацит 457
Галактани 653
Галогеноангідриди кислот 168, 424, 427, 443,
479, 494, 501, 509, 517, 526, 543, 549, 569, 587,
605, 610, 640, 713, 754, 756, 796, 822. 824, 843,
851,915, 920, 955, 967
Галогеногідрини 207,373,377,382,397,534
Галогенопохідні вуглеводнів 306, 698
Галоформи 329, 363, 463
Гваякол 811
Гексагеліцен 114
Гексаметилфосфотриамід 325
Гексахлоран 662
Гем 618, 924
Геміцелюлози 653
Гемоглобін 618, 924
Предметний покажчик
977
Генціобіоза 818
Гепарин 657
Гераніол 275, 369
Гербіциди 25, 800
Гетероциклічні сполуки 900, 928, 942
Гіалуронідаза 657
Гібридизація 49, 51, 66. 98, 132, 164, 170, 202,
220, 231, 236, 244, 249, 251, 271, 280, 295, 320,
332, 339, 365, 413, 435, 448, 484, 522, 663, 708,
726, 889, 897
Гідантоїни 544, 615
Гідразини 168, 195, 420, 430, 432, 459, 476, 525,
585,598,614,639,649,772,849,938
Гідразони 116, 459, 461, 476, 525, 639, 769,
928, 938
Гідразосполуки 734, 740, 885
Гідроксамові кислоти 409,423
Гідроксикислоти 557
Гщроксиламіни 407, 428, 458, 472, 476, 586, 639,
734,737,765,773,804,815,827,835,954
Гідропероксиди 155, 246, 786
Гідрохінон 813
Гістамін 606, 940
Гістидин 606, 940
Глікоген 656
Глікозиди 942
Гліколі - див. Спирти
Гліоксаль 87,377,475,567,662,671,940,956
Гліцерол (гліцерин) 83, 368, 380, 505, 961
Гліцеролтринітрат (нітрогліцерол) 382
Гліцин 83, 604, 608
Гліциризин 642
Глобін 924
Глудантан 301
Глутатіон612
Глюкоза 83, 624, 633, 638
Глюкозамін 645, 657
Глюкокіназа 646
Глюкоманани 653
Головні типи реакцій 74
Гомологія 22
Гомотропіліден 106
Гомотропілію катіон 668
Грамін 931
Група
- функціональна 81
-захисна 187, 228, 420, 451, 569, 609, 634, 640,
646, 750, 753, 891
Гуанін 970
Гутаперча 272
Дансилхлорид 614
Дегідробензен 86
Дегідрогеназа 647
Декалін 118, 277, 299, 868
Декалоли 868
Декстрани 658
Депсиди 853
Детергенти 154, 499, 794
Дибазол 941
Дибензальацетон 930
Дикетен 578
Дикетопіперазини 607
Димедрол 941
Диметилсульфоксид 325
Диметилформамід 325, 520, 754, 823
Дипіон832
Дипіридин 959
Дисульфан 721
Дифенілметан 886
Діазометан 164, 168, 286, 290, 331, 393, 408,
432, 494, 503, 793, 815, 938, 952
Діазосполуки 760
Діал 547
Діанізидин 885
Діантрацен 875
Діацетил 385, 662, 956
Діацетилен 906
Дієни 248
Дієнофіли 265, 556, 588, 737, 908, 916
Діоксини 711
Довжина зв'язку 48, 171, 221, 249, 316, 334,
404, 484, 609, 663, 859, 882
Дульцин 755
Евгенол 790
Евіпан 547
Електронегативність 50
Електрофіли 73, 79, 161, 165, 191, 195, 199,
229, 234, 237, 271, 287, 324, 351, 398, 417, 427,
435, 452, 466, 500, 510, 514, 522, 587, 590, 679,
683, 699, 729, 732, 750, 761, 767, 782, 804, 807,
823, 832, 847, 859, 863, 885, 904, 921, 946, 968
Емульсин 644, 650
Енантіомерія
-аленів 111, 252
Енергія зв'язку 48, 62, 146, 149, 171, 192, 211,
221,316,319, 343, 463, 663
Еноли 365,467, 470, 581, 587, 635
Еозин 899
Епімери 622, 635, 639
Епоксиди (а-оксиди, оксирани) 213, 353, 372,
376, 379, 387, 397, 565, 794, 816, 846, 906, 936
Еритроза 105, 117
975 Чирва В.Я., Ярллолкж CM., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Еритритол 384
Естер 499
- ацетооцтовий 480,579,598,768,939,945
- діазооцтовий 605
- малоновий 283,533,541,546,551,559,768
- ціанідоцтовий 599
Естери сульфокислот 503, 720
Естрадіол 305
Естран 304
Етер 387
-діетиловий 174
-тротиловий 641, 891
Ефект
- аномерний 633
- гіперкон'югації (надспряження) 68, 149, 201,
694, 728
- ізотопний 718, 826
- індуктивний (індукційний) 66, 68, 174, 183, 198,
201,323,341,361,375,399,404,425,436,467,485,
510,530,533,558,576,584,688,693,699,704,725,
749,782,802,946
- матричний 400
- мезомерний (спряження) 70, 203, 249, 333, 337,
366,404,467, 514,584,590,609,688,695,699,704,
725,749,756,762,771,782,802,839,946
- орто-732
- пероксидний 204, 236
- поля 67
-Л2- 632
Жири-див. Олія
Замісники вуглеводневі 88, 174
Захист тимчасовий - див. Група захисна
Зв'язок
- водневий 47, 62, 325, 343, 388, 415, 436, 486,
539, 558, 587, 592, 615, 652, 783. 793, 803, 839,
851,935
-депсидний 854
- іонний 55, 241, 270, 361, 592, 891
- ковалентність 51
- пептидний 609, 612
- семиполярний 55, 403, 428
- хімічний 47
-а-48, 57, 66, 68, 112, 117, 123, 131, 145, 155,
161, 170, 202, 222, 245, 249,'264, 288, 353, 413,
435, 470, 556, 882
-тс- 48, 67, 104, 111, 113, 115, 167, 170, 198,
221, 236, 243, 245, 251, 264, 313, 435, 470, 518,
708, 849, 902,913
-т- 282
Зсув
- алкільний (арильний, гідридний) 184, 206,
311, 355, 374, 441, 465, 786, 829, 834
-батохромний 771
- гіпсохромний 771
Ізатин 736,915, 933
Ізовіолантрон 881
Ізоевгенол 790
Ізомераза 646
Ізомерія 21, 95
-алканів 134
-алкенів 172
- алкінів 223
- бензену 670
- геометрична (л-діастереомерія) 115, 172, 175,
193,281,536,545,740,761,766,827,831,835
- конформаційна - см. Конформація
- нафталену 860
- оптична (дзеркальна, енантіомерія) 95, 110,
252, 281, 343, 414, 559, 597, 883
- спиртів 342
- структурна 39; 95, 104, 134, 172, 223, 281,
342, 672, 860
-топологічна 108
- циклоалканів 281
Ізонікотингідразид 959
Ізонітрили 318, 423, 43.0, 521
Ізотіоціанати 615, 815
Ізоціанати 423, 427, 755, 971
Ізохінолін 960, 966
Імідазоли 477, 940
Іміди 524, 548, 601, 847, 855, 918, 920
Іміни (азометіни, основи Шиффа) 107, 116,
284, 422, 457, 522, 600, 602, 737, 828, 905, 920,
950, 957, 962, 967
Іміноестери 522
Інвертаза 651
Індиго 933
Індигозоль 936
Індикан 933
Індоксил 933
Індол 928
Індофенін 915
Ініціатори 268
Інсектициди 25, 662
Інтермедіат 70, 75, 148, 161, 166, 179, 196, 203,
210, 240, 264, 290, 320, 349, 393, 424, 448, 602,
686, 703, 740, 787, 798, 826
Інулін 655
Іонол 805
Предметний покажчик
979
Іприт 379
Йодоформ - див. Галоформи
Йон амбідентний 318, 521, 580, 764
Кадаверин 431, 943
Камеді 656
Камфан 303
Камфора 303
Капролактам 107, 602, 836
Карагінан 656
Каран 302
Карени 302
Карбазол 937
Карбаніон 73, 79 238, 270, 448, 467, 579, 588,
600, 837, 887, 889
Карбен - див. Метилен
Карбеноїди 169, 287
Карбін 244
Карбодііміди 607, 611
Карбокатіон 68, 73, 75, 79, 159, 161, 165, 179,
183, 196, 201. 234, 261, 269, 293, 311, 321, 323,
334, 346, 349, 351, 355, 358, 374, 390, 394. 399.
426, 429, 493, 503, 519, 563, 679, 685, 693, 787,
798, 807, 836, 890, 892, 909, 922
Карбонільні сполуки (оксосполуки) 433,818
Карбонові кислоти 481, 838
— ароматичні 661, 671, 713, 730
— галогенозаміщені 528, 533, 551, 555, 564,
572, 576, 598
— двохосновні 537, 905
— ненасичені 531, 565, 568, 570, 607
Карвон 302
Каротин 275
Каталізатори 71
-гетерогенні 71, 143, 192, 194, 271, 360
-гомогенні 71, 143, 192, 194, 271
-міжфазові 72, 401, 489, 503
Катенани 108
Каучук
-природний 216, 272
— синтетичний 24, 273, 535
Квадрициклан 299
Кверцитрин 642
Кератин 614
Кеталі - див. Ацеталі
Кетени 164,288,366,494,497,514,545, 571
Кетон малини 83
Кислота
— у-аміномасляна 606, 608, 923
— СН- 77, 231, 478, 485, 551, 887, 957
NH- 77, 425, 519, 849, 920, 930, 940
N-ацетилмурамова 658
ОН- 77, 478
8Н- 77
адипінова 401, 431, 539, 541,551
акрилова 239, 528, 532, 534
альгінова 656
альдарова 639
альдонова 568, 634
амінобарбітурова 971
антранілова 855, 935
арабінарова 625
аспарагінова 606
ацетондикарбонова 574
ацетооцтова 577
барбітурова 546, 968
бензилова 829
бензойна 401, 730, 831, 834, 841 -
бузкова 853
винна 116, 119,560,573,654
виноградна 119, 562, 577
віолурова 547
галактарова 625
галова 814, 853
ганусова 851
гіалуронова 657
гіпурова 600
гліколева 377, 476, 572, 576
гліоксилова 377, 576
гліцеролова 105, 383
глутамінова 606, 608, 610
глутарова 539, 550, 553
глюкарова 625, 635
глюконова 568, 625, 634, 637
глюкуронова 568, 657
гулуронова 656
ідуронова 657
ізатинова 933
карбамінова 421, 424, 430, 520, 755
корична(цинамонова) 830
• кротонова 536
■ кумаринова 555
■ левулінова 578, 636
■ леканорова 854
• лимонна (цитринова) 83, 573
• лінолева 385
• ліноленова 385
• малеїнова 115, 541, 554, 567
• малонова 533, 536, 540, 545, 601, 968
- мальтобіонова 649
• манарова 625
980 Чирва В.Я., Ярмолюк С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
- манонова 625, 638
- мануронова 656
- мезоксалева 383
- метакрилова 453, 535, 545
- метанілова 750
-мигдальна 120, 454
- молочна 101,110,453,476,559,573,648,658
-муконова 681
- нікотинова 956, 958
- оксалурова 543
- олеїнова 385
- ортанілова 751
- оцтова 156, 213, 239, 329, 372, 473, 487, 516,
527, 545, 558, 577, 648, 845, 877
- пальмітинова 385
- парабанова 543
-пікринова710, 717
- пімелінова 852
- піровиноградна 573, 577, 602, 647
- пірослизова 906
-пропіолова 241
- пропіонова 573
- протокатехова 810, 852
- псевдосечова 546, 971
-резорцилова 813, 853
- розолова 896
-руберитринова 880
- саліцилова 806, 850
- сахаринова 636
- сечова 547, 970
-слизова 918
- сорбінова 545
- сульфамінова 751
- сульфанілова 751
-стифнінова 812
- стеаринова 385
-тартронова 383
-терефталева 661, 842, 850
- толуїлова 834
- трикарбалілова 573
- тримезинова 674, 680
- триметилоцтова 494
- уронова 568, 634
- фталамінова 848
-фталева 671,842, 847
- фталонова 868
- фумарова 115, 541, 554, 563, 567
- хінолінова 560
-хлоросульфонова 720, 752, 936, 955
- холева 305
-щавлева (оксалатна) 377,383,540,542,577,576
-яблучна 120, 540, 555, 559, 563, 573
-янтарна (бурштинова) 541.548.553.573,912
Кислоти сульфінові 723
- сульфонові - див. Сульфокислоти
Кислотність 67, 76, 222, 231, 280, 294, 341,
361, 375, 382, 448, 485, 519, 528, 533, 558, 584,
587, 604, 719, 782, 790, 802, 839, 851. 887, 902,
904, 920, 930, 957
Клатрати 138
Клофелін 941
Когазин 153
Коламін (етаноламін) 379
Колідини 942, 946
Комплекс
-активований 69, 75, 146, 179, 181, 187, 194, 200,
213,234,295,319,321,334,336,349,414,448.465,
684,696,704,799,824
- з йонами металів 79, 194,208,212,231,290,347,
376,401,426,441,463,477,499,544,587, 605,666,
773,776,778,792,879,924
- з перенесенням заряду (КПЗ) 264,395,733,813
- а- 196, 312, 349, 685, 689, 691, 696, 700, 705,
710, 718, 726, 750, 767, 859, 863, 871, 896, 909,
946, 949
- ті- 54, 113, 142, 195, 234, 236, 240, 262, 311 338,
349,394,602,684,700,726,768,909,914,930,948
Конваллятоксин 642
Коніїн 959
Коніферин 642
Константа
- кислотності 76
- спряженої кислоти 77
- основності 77
Контроль реакції кінетичний 71, 262, 750, 803,
864, 876
-термодинамічний 71,262,750,768,804,864,947
Конфігурація 97, 421
- обертання 320, 360
Конформація 66, 97, 100, 105, 111, 114, 123, 136,
181,250,267,270,282,294,297,300,303,609,632
Кополімеризація (співполімеризація) 239
Корин 927
Кофеїн 970
Краун-етери 315, 400, 500
Крезоли 798, 805
Крекінг 23, 140, 152, 156, 163, 176, 178, 205, 220,
225,257,291,332,344,365,381,677,912
Криптанди 402
Крохмаль 654
Ксилани 653
Ксилени 662. 671,842
Кубан 299
Предметний покажчик
981
Кумарин 571, 830
Кумен 682, 786
Кут валентний 48, 52, 62, 126, 133, 170, 220,
280, 341, 388, 398, 413,435, 484, 913
-торсійний 123
Лактами 601,607, 923
Лактиди 570
Лактоза 650
Лактони 465,530,549,566,568,570,578,634,637
Лігроїн 174
Лимонен 83, 267, 301
Ліналоол 275, 369
ЛСД (діетиламід лизергінової кислоти) 931
Лутидини 942, 954
Люмінал 547
Люмінол 849
Мальтаза 644, 649
Мальтоза 649
Маніт 633
Маноза 624, 638
Масла сивушні 346
Мезилати 325, 360
Мезитилен 469, 674
Мезитилоксид 469, 907
Мемантин 301
Ментол 302, 341
Ментон 302
Металоорганічні сполуки 142, 144, 229, 239,
258, 333, 352, 365, 443, 477, 493. 497, 504, 508,
512, 564, 572, 678, 706, 714, 734, 773, 778, 816,
823, 836, 842, 875, 883, 886, 888, 910, 915, 920,
930, 940, 952, 957
Метальдегід 474
Метилени (карбени) 163, 286, 290, 330, 807
Метіонін 608
Метод
— валентних зв'язків 47
-молекулярних орбіталей 55,249,265,289,664
Метол 738
Механізм реакції АЕ алкенів 196,199,312,334
алкінів 233
дієнів 260
— АЕ3 алкенів 200, 204
— Ам карбонільних сполук 241, 448
— Ак алкенів 198, 204
дієнів 263
— Е1 і Е2 галогенопохідних 179
спиртів 182, 355, 358
— Е2 спиртів 185,358
- SE бензену 679, 683, 718, 726
- 8лалканів 145
алкенів 210
- SN1 галогенопохідних 321, 336, 713
спиртів 311,355, 390
- SN2 галогенопохідних 230; 319, 336
спиртів 312, 355, 390
Мила 498, 504
Мірцен 275
Молекулярність реакцій 75
Момент дипольний 50, 174, 175, 226, 308, 316,
326, 334, 404, 435, 707, 749, 784
Мономер 176, 177
Мурамін 658
Мускуси 729
Мутаротація 627, 631, 639
Надкислоти-див. Пероксиди
Найлон 431,551,836, 924
Напівацеталі 450
Напруження
- вандерваальсове (прелогівське) 125, 127
- 1,3-діаксіальне 128 в
- кутове (байерівське) 126
- торсійне (протилежних зв'язків, пітцерівське)
123,127
Нафта 23, 132, 138, 140, 154, 159, 163, 176,
205, 220, 225, 255, 300, 332, 344, 365, 380, 675,
800, 856, 916, 960
Нафтазарин 866
Нафтален 857
Нафтизин 941
Нафтоли 862, 869, 871
Неопрен 243
Нерол 369
Нікотин 959
Нітрили 214, 239, 318, 337, 376, 422, 431, 443,
445, 458, 493, 503, 514, 518, 521, 532, 534, 540,
551, 554, 572, 574, ^77, 622, 706, 723, 776, 780,
806, 824, 827, 843, 923, 944
Нітроамін 951
Нітрозаміни 429, 756, 763
Нітрозосполуки 407, 428, 734, 741, 756, 765, 804,
957
Нітролові кислоти 410
Нітросполуки 403, 725
Новокаїн 855
Новолак 808
Номенклатура алканів 133
-алкенів 173
- алкінів 222
982 Чирва В.Я., Ярллолюк CM, Толкачова H.В., Зеллляков О.Є. Органічна хімія
-амінів 412, 431, 744
- амінокислот 596
- антрацену 872
- бензену 669
- галогенопохідних 307, 698
- гетероциклічних сполук 901
- гідроксикислот 558
-діазосполук 761
-дієнів 248
- екзо-, ендо- 118, 303
- естерів 499
- етерів 387
- Женевська 84
- карбонільних сполук 433,475, 819
- карбонових кислот 482, 495, 531, 537, 839
- Льєжська 85
- нафталену 857
- нітросполук 403, 725
- оксокислот 579
- раціональна 84, 173, 222, 340, 433, 482
- син-,анти- 116, 118, 459, 740, 761, 827, 835
- спиртів 340
- сульфокислот 715
-тривіальна 83, 173, 222, 248, 340, 475, 483
- фенолів 782
- циклоалканів 278, 298
—цис-, транс- 103,118,172,300,740,762,868
-DL- 101, 116, 597, 622
-EZ- 103, 173, 835
- ШРАС 85, 135, 173, 222,248, 298, 307, 340, 387,
412,431,433,477,482,499,506,509,512,516,521,
524,531,537,558,571,575,669,698,715,725,744,
761,782,819,839,857,872,882,901
-RS- 86, 96, 100
Норадреналін 86
Норборнан 277
Норкарадієн 167
Норсульфазол 722, 753
Нуклеозиди 644
Нуклеофіли 60, 72, 75, 198, 196, 229, 236, 240,
249, 269, 297, 311, 316, 322, 325, 337, 354, 358,
390, 394, 398, 410, 420, 426, 435, 448, 500, 511,
525, 529, 552, 579, 590, 611, 642, 681, 685, 708,
723, 732, 760, 785, 796, 845, 904, 949, 952
Обертання
- молекулярне 97
- питоме 96
Одержання
- аленів 245, 253, 328
-алканів 138, 192, 209, 232, 239, 290,497
-алкенів 154, 176, 232, 297, 358, 455
-алкінів 162, 225, 328
- амінів 239, 361, 407, 411, 417, 431, 458, 519,
524, 526, 551, 605, 706, 733, 738, 742, 746, 849,
869,915, 931,949, 958, 964
- амінокислот 598, 769, 855
- антрацену 873
- бензену 243, 292, 661, 673, 679, 682, 684, 686,
777, 797
- галогенопохідних 145, 195, 209, 233, 236,
242, 240, 290, 309, 328, 354, 368, 383, 434, 456,
498, 662, 683, 699, 749, 774, 843, 860, 863, 876,
911
- гетероциклічних сполук 671, 904, 912, 91 î
928, 935, 938, 940, 944, 961, 968
- гідроксикислот 453,492,529,533,554,562,648
-діазосполук 605, 763, 869, 950
-дієнів 154, 208, 243, 254, 374, 911
- естерів 239, 347, 366, 376, 432, 466, 490, 499,
511,515, 530, 569, 610, 640, 706, 795, 831, 845,
851,921
- етерів 238, 317, 333, 367, 370, 374, 390, 432,
569, 641, 778, 793, 801, 908, 952
- карбонільних сполук 208, 215, 235, 237, 245,
253, 292, 295, 328, 346, 362, 374, 377, 409, 437,
475, 497, 508, 511, 520, 522, 524, 551, 570, 577,
589, 634, 681, 713, 754, 796, 805, 807, 812, 816,
821, 833, 865, 909, 915, 921, 931, 956
- карбонових кислот 156, 215, 241, 245, 292,
362, 377, 407, 462, 465, 476, 488, 504, 511, 515,
520, 532, 539, 545, 552, 555, 572, 577, 590, 601,
606, 648, 713, 777, 795, 806, 841, 889, 946, 956
-нафталену 861
- нітросполук 151, 292, 318, 405, 428, 530, 687,
726, 752, 801, 864, 876, 885, 909, 914, 930, 948,
951,954,963
- оксокислот 553, 576
-спиртів 205,205,213,216,241,291,295,317,337.
344,363,370,372,429,454,460,465,504,512,633,
647,807,810,815,834,911,915,923,957
- сульфокислот 153, 683, 716, 731, 750, 803, 864,
876,878,909,914,922,930,947,951,963
- фенолів 706, 723, 738, 757, 777, 784, 795, 853,
862, 877
- циклоалканів 165, 168, 209, 264, 288, 328,
411,534, 550, 554, 580, 661,681
Озазони 476, 639, 649
Оксазолони 586
N-Оксиди 955, 962
Оксими 116, 292, 407, 422, 428, 458, 472, 476*
522, 589, 602, 637, 639, 804, 815, 827, 835, 919
Предметний покажчик
983
Оксин 963, 965
Оксокислоти 579
Олігомер 177
Олігосахариди 649
Оліфа 385
Олія (жири) 365, 380, 384, 495, 498
- гірчична 430
-ефірна 24,274,301,368,789,816,830,855,932
-кам'яновугільна 676, 937
-солярова 158, 677
Ондансетрон 932
Орбіталь електронна 42
Ортоестери кислот 330, 451, 506, 823
Основи 229, 238, 240, 244, 425, 759, 764, 953
Основність 76, 176, 324, 342. 392, 394, 401,
425, 458, 485, 519, 583, 686, 749, 758, 783, 793,
903, 920, 930, 957, 968
Пантокаїн 855
Паральдегід 439, 474
Параформ (параформальдегід) 438,473
Парафуксин 894
Парациклофани 114
Пектини 654
Пентаеритрол 341, 384
Пептиди 608
Перехідний стан 69, 201, 210, 296, 321, 685,
691, 750, 768
Пероксиди 268, 385, 464, 504, 731, 789, 846,
875, 889, 955
Піколіни 942, 944, 957
Пілокарпін 941
Пінакол 341,374
Пінаколін 374
Пінан 303
Пінаціанол 966
Пінени 303
Піперидин 943, 957
Піразини 477
Піразол 938
Піразолони 585, 939
Піранози 628
Піридони 952
Піридин 942
Піримідини 586, 968
Пірогалол 814
Пірокатехол 810, 879
Піроксилін 652
Піроли 549, 905,917
Піролідин 919
Піролідони 572, 923
Піроліз 158, 181, 188, 225, 253, 257, 291, 444,
518, 540,814, 825, 858,918,912
Піруватдекарбоксилаза 647
Плазмохін 966
Площина симетрії 109
-хіральності 113
Полівінілацетат 367
Полівінілбутираль 367
Полівініловий спирт 367
Полівінілпіролідон 324
Полі естери 549
Поліетилен 269
Поліетилентерефталат 850
Полімери 24, 176, 244, 267, 367, 376, 431, 473,
535, 551,808, 836, 848, 850, 923
Поліметилакрилат 535
Поліметилметакрилат 535
Полісахариди 651
Поляризовність зв'язку 51
Поляриметр 95
Полярність зв'язку 49
Порфін 924
Примахін 966
Природний газ 23, 132, 138, 161, 163, 258
Пролін 602
Промедол 958
Псилоцибін 932
Пурин 970
Пурпур старовинний 936
Путресцин 431, 919
Радикали 72, 75, 133, 137, 139, 141, 144, 152,
157, 164, 167, 171, 181, 191, 198, 204, 209, 222,
233, 236/263, 268, 313, 336, 344, 386, 389, 396,
417, 497, 557, 663. 665, 703, 715, 730, 743, 760,
775, 778, 837, 884, 889, 897, 919
Рацемат 95, 119, 322, 402, 415, 454, 471, 559,
567, 603
Реакції азосполучення амінів 764, 767, 770,
773, 870
— бензену 768
—діазосполук 764,767,770,773,870,923,931
— фенолів 767, 770
-алкілування амінів 417, 426, 755, 943
— бензену 679, 682, 684, 816, 886, 888
— галогенопохідних 190, 229,328
— карбонільних сполук 472, 478
— нафталену 865
— нітросполук 411
— фенолів 804, 811
— амідування карбонових кислот 519
984
Чирва В.Я., Ярллолюк CM., Толкачова H.В., Земляков О.Є. Органічна хімія
— амінів з нітритною кислотою 353,429,756,763
— амінокислот з нінгідрином 606
— з нітритною кислотою 564, 604
— амінолізу естерів 518, 525
— амінометилювання карбонільних сполук 411
— нітросполук 411
— амінування бензену 747
— амоксидування алкенів 212
— арилювання алкенів 779
— амінокислот 605, 613
— бензену 884
— аутоокиснення етерів 396
— ацилювання алканів 543
— амінів 427, 515, 551, 750, 752, 846, 967
— амінокислот 604
— бензену 681, 689, 797, 824, 843, 847, 874
— гідроксикислот 569
— естерів 921
— карбонових кислот 513, 753, 806
— нафталену 865
— спиртів 501, 511, 515, 549, 846
— фенолів 805
— бродіння вуглеводів 346, 364, 565, 646
-відновлення (гідрування) алкенів 142,192,194
—-алкінів 142, 189, 232
— амінокислот 604
--бензену 289, 661, 663, 666815
— вуглеводів 633, 645
— галогенопохідних 141, 189, 330
— гетероциклічних сполук 919, 943, 956
—діазосполук 772, 777
— естерів 351, 505, 816
— етерів 973
карбонільних сполук 295, 351, 373, 460,
681,815, 834
— карбонових кислот 351, 365, 543, 852
— нафталену 300, 859, 867
— нітросполук 407, 423, 733, 737, 747, 855
— сульфокислот 723
— фенолів 797,810, 862
— відновного амінування амінів 422
карбонільних сполук 422, 458, 748
— віншування (приєднання) алкінів 238, 376,
438, 522, 534, 924
-вкорінення алканів 166
— вкорочення й подовження карбонового
ланцюга вуглеводів 622, 636
— галогенометилювання бензену 703
-галогенування алканів 145, 309, 329
— алкенів 195, 210, 313, 332, 336, 548
— алкінів 233, 236, 242, 334
— амінокислот 605
— антрацену 876
— бензену 662, 683, 689, 695, 699
— вуглеводів 642
— дієнів 260, 262, 551
— карбонільних сполук 314, 470, 845
— карбонових кислот 509,527,544,833,845,853
— нафталену 860, 863
— фенолів 798
— циклоалканів 290
— галоформні 329, 363
— галоформного розщеплення карбонільних
сполук 329, 463,471,491, 842
— гетероциклізації алкенів 938
— алкінів 912, 918, 929, 938, 944
— дієнів 919
— карбонільних сполук 904,913,918,938,944
— карбонових кислот 940
— гідратації аленів 253
— алкенів 206, 345, 364
— алкінів 237,438, 445
— циклоалканів 291
— гідроборування алкінів 235,446
— гідроборування-окиснення алкенів 209,348
— гідрогалогенування алкенів 199, 312
— алкінів 236,259, 332
—дієнів 261
— циклоалканів 291
— гідролізу амінів 814, 869
— вуглеводів 627, 649
— галогенопохідних 317, 328, 337, 346, 347, 380,
447,567,713,788,815,843,845,853
— діазосполук 293, 787, 811, 896
— - (омилення) естерів 295, 372, 379, 384, 490,
504, 570
— нітросполук 409
— гідроксилювання бензену 788
— гідроформілювання алкенів 208, 347, 446
— гідрування циклоалканів 290
— гіпогалогенування алкенів 207
— гомологізації алкінів 212,229
— дегалогенування галогенопохідних 187, 228,
253, 283, 328, 335
-дегідратаціїгідроксикислот 532, 555, 570
— естерів 186
— етерів 186
— карбонових кислот 513,548,550,555,578,847
--спиртів 182, 185, 255, 259, 358, 374, 390,
834, 907
— дегщрогалогенування галогенопохідних 178,
227,253,256,259,287,328,330,332,334
Предметний покажчик
985
— дегідроциклізації бензену 861
-дегідрування алканів 154,176,257,674,912
— бензену 887
— каталітичного спиртів 362, 438
— циклоалканів 292, 661, 675
-дезамінування амінокислот 606
-дейтерування бензену 686
-декарбоксилювання амінокислот 605
— і дегідратації гідроксикислот 188
--карбонових кислот 140, 188, 228, 284. 314,
406, 444, 497, 540, 542, 545, 551, 577, 589^ 671,
810,814, 906, 946
-декарбонілювання карбонільних сполук 907
— десульфування сульфокислот 722, 865
— димеризації алкенів 209, 258
— алкінів 244
— диспропорціювання карбонільних сполук
465, 476, 576,816, 829, 831
— дієнового синтезу (циклоприєднання) 264,
289, 534, 662, 737, 862, 873, 884, 944
гетероциклічних сполук 907, 916
— екзотермічні 69
— елекгрофільного заміщення аренкарбонових
кислот 730
ареноксосполук 730
аренсульфокислот 724
ариламінів 749, 757
арилгалогенідів 704, 711, 724
гетероциклічних сполук 908, 913, 921,
930, 946, 951,953, 963
нітроаренів нітросполук 731
-елімінування галогенопохідних 230,492,816
— ендотермічні 69
— естерифікації амінокислот 604
— вуглеводів 640
— гідроксикислот 569
—карбонових кислот 432,508,542,549,852
— спиртів 500, 610, 850, 852
— фенолів 795, 845, 851
— етерифікації вуглеводів 641, 649
— гідроксикислот 569
--фенолів 432, 793, 801
-заміщення алкенів 212
— алкінів 212, 240, 370
— вуглеводів 639
— гідроксикислот 528, 569
— діазосполук 700, 742, 869, 884
— спиртів 309, 336, 354, 361, 377, 391, 419
— звуження циклу циклоалканів 292
— ізомеризації аленів 254
— алканів 156, 161
— алкінів 244, 253
— бензену 681
— іпсо-заміщення сульфокислот 686, 723, 785,
788,812, 851,869, 878
— фенолів 717
— карбоксилювання карбонових кислот 850
— фенолів 806, 813, 850, 853, 856
— карбонілювання алкенів 493
— алкінів 239,493
— галогенопохідних 447
— спиртів 346, 488,493
— кислотно-основні амінів 425, 748
амінокислот 604
— комплексоутворення амінокислот 605
— комплексування спиртів 376
— фенолів 792
— конденсації алкінів з оксосполуками 241,
370, 455,918
— амінів 671, 736, 741, 828, 833, 915, 931, 940,
961, 967
— амінокислот 604, 611
— антрацену 875
— галогенопохідних 143, 873, 898
— естерів 287,285,289,478,550,579,833,945
— - карбонільних сполук 190, 384, 410, 467,
476, 478, 533, 536, 545, 550, 571, 581, 604, 638,
671, 674, 733, 807, 828, 832, 886, 888, 915, 923,
931,945, 957, 961,967
— нітросполук 410, 733
— фенолів 807, 848, 898
— крекінгу алканів 156, 225, 257
— металювання антрацену 875
— метилювання алкінів 240
— вуглеводів 627
— діазосполук 767
— спиртів 393,432
— нейтралізації карбонових кислот 496
— нітрилювання алкенів 522
— нітрозування бензену 735
— карбонільних сполук 472, 476
— фенолів 735, 804
— нітросполук з нітритною кислотою 410
— нітрування алканів 151,405
— антрацену 876
— бензену 683, 689, 726
— нафталену 864
— фенолів 711,729, 801,812
— циклоалканів 292
— нуклеофільного заміщення ариламінів 757
галогенопохідних 316,347,354,372,391,405,
419,426,492,503,506,521,678,706,712,714,730,
747,754,758,793,849,883,920
986
Чирва В.Я., Ярмолкж CM, Толкачова H.В., Зеллляков О.Є. Органічна хімія
гетероциклічних сполук 949, 964
нітроаренів 732
— приєднання естерів 443, 823, 888
карбонільних сполук 241,370,378,449,522,
535,585,622,816,827,888, 915,938,957
— обміну галогенопохідних 315
-окиснення алканів 155, 162,438, 488
— - алкенів 212, 372, 398. 438, 442, 474, 489,
532, 534, 846
— алкінів 245
— амінів 406,428, 731
— антрацену 875
— бензену 556, 662, 681, 795, 822
—вуглеводів 568,627,634,639,645,649,654
— галогенопохідних 330
--гетероциклічних сполук 908, 917, 923, 946,
955, 960, 966
карбонільних сполук 462, 471, 479, 488,
490, 504, 516, 532, 539, 807, 825, 842
— карбонових кислот 543
— нафталену 842, 858, 860, 866
— - спиртів 295, 362, 377, 439, 490, 531, 572,
576, 816, 822
— фенолів 567,810,813, 877
— циклоалканів 292
— оксокислот 905
— оксимеркурування алкенів 207, 350, 393
— олігомеризації алканів 161
— алкенів 177 •
— алкінів 243
— карбонільних сполук 473
— перегрупування амінів 756
— вуглеводів 635
— естерів 796
— етерів 367, 795
— переестерифікації естерів 500, 850
— перициклічні 72, 264
-піролізу галогенопохідних 181
— естерів 188
— полімеризації алкінів 244
—- дієнів 267
— карбонільних сполук 473
— приєднання карбенів до алкенів 165, 168,
209, 286
алкінів 165
бензену 168
— карбонових кислот до алкенів 503
— нітрозосполук до алкенів 736
— протонування бензену 676
-розширення циклів гетероциклічних сполук 919
циклоалканів 286, 292
-розщеплення амінів 186, 943
- етерів 395, 795
- карбонільних сполук 834
- сольволізу галогенопохідних 890
- спиртів як кислот 361
- спонтанні 69
- сульфоокиснення алканів 154
- сульфохлорування алканів 153
- бензену 718
- сульфування алканів 153
- алкенів 205
- антрацену 876
- бензену 683, 689,716
- нафталену 864
- фенолів 717, 803
- теломеризації алкенів 209, 528
-утворення солей оксонію 394
сульфокислот 719
фенолів 790
- формілювання бензену 823
- фенолів 807,811
-хімічні 69
- хлорування етерів 396
- сульфокислот 720
- циклізації алкенів 913
- амінокислот 607
- циклоізомеризації дієнів 288
- циклоолігомеризації алкінів 243, 288, 673,
712, 861,912
- циклоприєднання аленів 254, 288
- ціанетилювання амінів 535
Реакція ізонітрильна амінів 430
Регіоселективність 149, 153, 180, 188, 194, 233,
256, 352, 369, 381, 391, 396, 441, 461, 489, 506,
565, 591, 640, 645, 727, 750, 797, 804, 877, 953
Регулятори росту рослин 25
Резит 808
Резитол 808
Резол 808
Резорцинол 806, 812, 853, 898
Ремантадин 301
Різниця гомологічна 22
Родаміни 899
Розаміни 899
Ротоксани 108
Салол 820
Сафрол 790
Сахарин 722, 850
Сахароза 651
Семикарбазони 459
Предметний покажчик
987
Семідин 742
Серин 597
Серотонін 606
Сечовини (карбаміди) 427, 432, 519, 543, 546,
586,611,755, 968
Сефадекс 658
Сикативи 499
Силденофіл 969
Сили міжмолекулярної взаємодії (вандерва-
альсові) 63,125,129,193,308,648,883,889
Синтин 139
Скатол 932
"Складчастий аркуш" 616
Слиз 656
Смола
- гліфталева 848
-йонообмінна211, 606, 613, 821
- кам'яновугільна 23, 159, 668, 675, 746, 784, 858,
866,872,912,917,929,937,942,944,960
- фенолоформальдегідна 807
Сорбіт 633, 645
Сорбоза 645
Спектроскопія 175
-алканів 137
-алкенів 175
- алкінів 224
-амінів 416, 745
- амінокислот 598
- антрацену 873
- бензену 668, 672
- галогенопохідних 308, 699
- гетероциклічних сполук 903,928,938,954,960
-діазосполук 763
-дієнів 252
- етерів 389
- карбонільних сполук 437, 820
- карбонових кислот 487, 841
-нафталену 861
- нітросполук 404, 726
- спиртів 344
- сульфокислот 716
- фенолів 784
- циклоалканів 278
Спирти 339
-аліловий 368, 381
-бензиловий 610, 713, 815, 831
- пропаргіловий 370, 381
Спіраль хіральності 114
Спірани 112, 277, 298
Стереоізомерія амінів 414
- гідроксикислот 559»
Стереоселективність 168, 180, 189, 196, 212,
214, 235, 287, 352
Стероїди 118, 304
Стильбен 401
Стирен 86, 176,816
Стрептоциди 83, 722, 751
Структура
-білка 613
- резонансна (гранична) 55, 250, 262, 435, 484,
510, 519, 552, 580, 589, 685, 688, 692, 696, 705,
709, 725, 760, 782, 791, 859, 862, 873, 882, 887,
889, 891, 897, 902, 909, 913, 921, 930, 942, 946,
949, 951,955
- хімічна (конституція) 97
Сульфадимезин 752, 969
Сульфадиметоксин 753
Сульфідин 951
Сульфініліміди 765
Сульфонаміди (сульфаміди) 720, 750
Сульфокислоти 715
Сульфоксиди 917
Сульфони718, 722,917
Суматриптан 932
Таніни 854
Таутомерія 106, 237, 366, 407, 478, 546, 581,
631, 639, 645, 736, 766, 773, 802, 812, 815, 878,
929, 934, 950, 968, 971 .
Твістан299
Теломеризація 209, 529, 539, 567, 602
Теобромін 970
Теорія
- будови органічних сполук 36
- віталізму 33
-дуалістична 34
- жорстких і м'яких кислот і основ 79, 591
- квантово-механічна 42
- парціальних валентностей 660
-радикалів 35
-типів 36
Теофілін 970
Теплота гідрування 192, 249, 663
Терпінени 301
Терпени 374, 301
Тестостерон 305
Тетрагідрофуран 256,333,349,375,380,396,398,
421,445,520, 572,714, 900,905,911
Тетралін 867
Тетралон 868
Тетрил 757
Тефлон 316, 335
988 Чирва В.Я., Ярмолкж С.М., Толкачова Н.В., Зеллляков О.Є. Органічна хімія
Тимоген 612
Тимол 302, 790
Тимін 969
Тироліберин 612
Тіоаміди 524
Тіоіміноестери 523
Тіоацеталі (тіокеталі) 451
Тіоестери 515
Тіоетери 239, 297, 318, 952
Тіокислоти 511, 515
Тіоли 318, 508
Тіонесаль913
Тіоортоестери 508
Тіофан 916
Тіофени 549, 904,912
Тіофеноли 723
Тозилати 296,325,357,360,390,442,522,721,796
Толідин 885
Толуен 159, 689, 695, 700, 703, 718, 727, 730,
822, 841
Толуїдини 745, 756, 884, 929
Точка ізоелектрична 597
Треоза 105, 117
Треонін 117,597
Триазени 764, 766, 773
Триптамін 932
Триптицен 875
Триптофан 931
Трифенілметан 887, 919
Трифлати 360
Тропілію катіон 666
Тротил (тол) 728
Туйан 302
Туйон 302
Ураміл 546
Урацил 969,
Уретани 424, 427, 432, 755
Уротропін 457, 821
Фарнезол 369
Фенацетин 754
Феноли 781
Фенолфталеїн 848, 898
Ферменти 72, 121, 644
Фероцен 666
Фіброїн 616
Фізичні властивості алканів 136
— алкенів 174
— алкінів 223
— амінів 415, 745
— амінокислот 597
— антрацену 873
— бензену 672
— галогенопохідних 307, 698
—гетероциклічних сполук 903,928,938,944,960
— гідроксикислот 558
— діазосполук 762
— дієнів 252
— етерів 388
— карбонільних сполук 436, 820
— карбонових кислот 486, 495, 538, 841
— нафталену861
— нітросполук 404, 725
— спиртів 343,371
— сульфокислот 716
— фенолів 783
— циклоалканів 278
Фітол 369, 926
Флороглюцин 806, 814
Флуорен 886
Флуоресцеїн 899
Формалін 438, 914
Формальдегід 208, 242, 259, 352, 373,381, 386,
410, 413, 425, 437, 440, 456, 459, 467, 469, 473,
571, 704, 807, 833, 915, 918, 923, 931, 940, 957,
967
Формули
— перспективні 98
— проекційні 99
— стереохімічні 98
Форон 469
Фотосинтез вуглеводів 648
Фреон 316
Фруктани 655
Фруктоза 626, 633
Фталазол 753
Фталеїни 898
Фталімід 419, 598, 848, 855
Фтивазид 959
Фуксин 894
Фуксон 896
Фульвен 682
Фунгіциди 25
Фурани 904
Фуранідини - див. Тетрагідрофуран
Фуранози 628
Фурацилін 910
Фурфуроли 636, 906, 910
Хінгідрон 813
Хінін 965
Предметний покажчик
989
Хінолін 960
Хінолон 964
Хінони 265, 477, 799, 810, 813, 847, 860, 862,
866, 872, 877
Хіральність 109
Хітин 657
Хлораль 329,449, 528
Хлораміни 721
Хлорамфенікол (левоміцетин) 121
Хлороацетофенон 834
Хлоропрен 243, 259, 274
Хлороформ 174, 306, 329, 432, 506, 807, 823,
888, 922
Хлоропікрин 330
Хлорофіли 925
Холестанол 119
Холестерол 305
Хондроїтиносульфат 658
Хроматографія 27, 122
Хромофори 772
Цвітер-іон 599, 606, 953
Целобіоза 652
Целофан 652
Целюлоза 122, 651
Центр симетрії 109
- хіральності 110, 203
Циклоалкани 277
Циклоніт 457
Цинеол 302
Цитохром с 617
Цитозин 969
Цитраль 275, 369
Цитронелол 368
Ціангідрини 453, 492, 564, 567, 622, 637, 818
Ціаносорб 837
Число
- квантове 43
-октанове 160
-цетанове 160
Чистота оптична 110
Шовк ацетатний 652
Список літератури
1. Белобородое В.Л., Зурабян С.Э., Лузин АЛ. и др. Органическая химия. - М.: Дрофа,
2002.-Т. 1.-С. 639.
2. Вацуро КВ., Мищенко ГЛ. Именные реакции в органической химии. - М.: Химия,
1976. -528 с.
3. Волков В.А., Вонский КВ., Кузнецова Г.И. Выдающиеся химики мира. - М.: Высшая
школа, 1991. - 656 с. /
4. Гауптман КЗ., Греффе Ю., РеманеХ. Органическая химия. - М.: Химия, 1979. - 832 с.
5. Гордон А., Форд Р. Спутник химика. - М.: Мир, 1976. - 541 с.
6. Ким A.M. Органическая химия. - Новосибирск: Сиб. ун-тское изд-во, 2002. - 971 с.
7. Ластухін Ю.О., Воронов С.А. Органічна хімія. - Львів: Центр Європи, 2000. - 864 с.
8. Мищенко ГЛ., Baijypo КВ. Синтетические методы органической химии. - М.:
Химия, 1982.-440 с.
9. Нейланд О.Я. Органическая химия. - М.: Высшая школа, 1990. - 751 с.
10. Несмеянов А.Н., Несмеянов H.A. Начала органической химии. - М.: Химия. 1969-
1970.-Т. 1.-663 с; Т.2.-824 с.
11. Обушок М.Д., Біла Є.Е. Органічна хімія. - Львів: Видавничий центр ЛНУ
ім. І. Франка, 2004. - 233 с.
12. Общая органическая химия. - М.: Химия, 1981. - Т. 1. - 736 с; ТЛО. - 704 с; Т. 11. -
736 с.
13. Овчинников Ю.А. Биоорганическая химия. - М.: Просвещение, 1987. - 815 с.
14. Пaccent Б.В. Основные процессы химического синтеза биологически активных
веществ. - М.: Изд. дом ГЭОТАР-МЕД, 2002. - 370 с.
15. Петров A.A., Балъян Х.В., Трощенко А.Т. Органическая химия. - СПб.: Иван Федо-
ров, 2002. - 622 с.
16. Реутов O.A., Курц АЛ., Бутин К.П. Органическая химия. - Ч. 1. - М.: Изд-во МГУ,
1999. - 555 с; Ч. 2. - М.: Изд-во Моск. ун-та, 1999. - 623 с; Ч. 3. - М.: БИНОМ.
Лаборатория знаний, 2004.
17. Терней А. Современная органическая химия. - М.: Мир, 1981. - Т. 1. - 678 с; Т.2. - 651 с.
18. Травень В.Ф. Органическая химия. - М.: ИКЦ "Академкнига", 2004. - Т. 1.-727 с,
Т. 2.-582 с.
19. Химия углеводов / Кочетков Н.К., Бочков А.Ф., Дмитриев Б.А. и др. - М.: Химия,
1967.-672 с.
20. Шабаров Ю.С. Органическая химия. -М.: Химия, 2000. - 847 с.
21. Buddrus J. Grunlagen der organischen Chemie, I. Auflage, Walter de Gruyter, Berlin;
New York, 2003.-863 s.
Список літератури
991
22. Carey F.F., Sundberg R.J. Advanced Organic Chemistry: 4th Ed. - N. - Y., Boston,
Doptrecht, London, Moscow: Kluwer Academic / Plenum Publisher, Part A, 2000. - 823 p.;
PartB, 2000.-965 p.
23. Hart H., Craine L.K, Hart DJ. Organische Chemie. Zweite Auflage. - Wiley-VCH, 2000.
-7165 s.
24. Morrison R.T., BoydRM Organic Chemistry: 6th Ed. - Prentice Hall (NJ), 1992. - 1278 p.
25. Seyhan N.E. Organic Chemistry. - Lexington (MA), Toronto: D.C. Heath and Company,
1984.- 1163 p.
26. Solomons G.T.W. Organic Chemistry: 6th Ed. - N.-Y., Chichester, Brislane, Toronto,
Singapore: John Wiley & Sons, Inc., 1998. - 1218 p.
27. Smith M. B., March J.f March's Advances organic chemistry S. Edition Wiley-intercience
Publication. - New York; Chichester; Weinheim; Brisbane; Singapore; Toronto, 2001. -
2083 p.
28. Streiwieser A., Heathcock C.H., Kosower KM. Introduction to Organic Chemistry: 4th Ed.
- Upper Saddle River (NJ): Prentice Hall, 1998. - 1256 p.
Науково-сервісна фірма "ОТАВА" спеціалізується в галузі інноваційних
досліджень для хімічних, біотехнологічних, фармацевтичних та агрохімічних
компаній, а також на постачанні лабораторного обладнання.
Головні напрями діяльності НСФ "ОТАВА":
• наукові дослідження на контрактних засадах для вітчизняних і зарубіжних
хімічних, біотехнологічних, фармацевтичних та агрохімічних компаній;
• продаж лабораторного обладнання західного та вітчизняного виробництва, як
нового, так і уживаного;
• синтез на замовлення хімічних реагентів у форматі 1-50 кг;
• консультації з питань оформлення наукових контрактів з іноземними партнерами;
• допомога в написанні наукових і науково-популярних статей, що рекламують
продукцію замовника.
Контрактні наукові дослідження. Співробітники НСФ "ОТАВА" мають
величезний досвід роботи з іноземними фірмами та науковими установами в галузі
контрактних досліджень. Науковцями фірми в рамках дворічного наукового контракту з
Fluka Chemie GmbH (Швейцарія) розроблено флуоресцентні барвники для фарбування
білків в електрофоретичних гелях. Спільно з Chromeon GmbH (Німеччина) запатентовано
новий метод флуоресцентного мічення біологічних молекул і полімерних частинок (патент
US2003175988).
Лабораторне обладнання. Важливим напрямом комерційної діяльності фірми є
продаж нового лабораторного обладнання таких провідних світових виробників, як ЇКА
Werke, Bandelin, Sanyo, Fisher Scientific, Metrohm, Linseis, Carl Zeiss, Millipore, Kern, Sartorius,
BioSan, Leika, Hanna Instruments, WTW, Perkin Elmer, Agilent, Shimadzu, Bio-Rad та ін.
Окрім того, ОТАВА імпортує обладнання, яке використовували в медичних і наукових
закладах Європи й Америки. Таке обладнання, якщо потрібно, ремонтуть у спеціалізованих
майстернях країн-експортерів, воно має необхідні сертифікати відповідності.
Синтез органічних речовин у кг-форматі. Органічний синтез - один із
найпотужніших напрямів діяльності фірми "ОТАВА". Досвідчені хіміки розглянуть
пропозиції щодо синтезу будь-яких речовин, і комерційно доступних, і таких, яких
нема в каталогах.
Наукові консультації. Співробітники НСФ "ОТАВА" мають багаторічний
досвід проведення експериментальних досліджень у галузі органічної і біоорганічної
хімії, біології та спектральних досліджень. Фірма може бути консультантом або
виконавцем під час проведення наукових досліджень. За результатами цих робіт наші
науковці можуть підготувати рекламні статті до фахових наукових чи науково-
популярних журналів, що рекламують продукцію фірми-продавця або виробника.
ОТАВА - колектив професіоналів, які здатні виконати замовлення будь-якого
ступеня складності. Співпрацюючи з нами, Ви отримуєте якість, реальні ціни та
максимально короткі терміни виконання замовлення!
а/с 88, Київ-187,03187
тел./факс: +38 (044) 522 24 58
e-mail: info@otava.com.ua
http://vsrww.otava.com.ua
Інформаційний ресурс Інтернет-портал Labprice.ua - ефективна
рекламна площа для українських дистриб'юторів реактивів, лабораторного
обладнання і послуг. Портал забезпечує онлайновий доступ до інформації
про необхідні товари та послуги для широкої аудиторії користувачів
мережі Інтернет, зокрема, науковців, медиків, працівників лабораторій.
На Labprice.ua можна знайти все необхідне для лабораторії,
ознайомитися зі спеціалізованими статтями, новинами і прес-релізами,
безкоштовно розмістити оголошення, отримати повний комплекс послуг із
рекламування власної продукції для лабораторій. На порталі подано
максимально можливий перелік підприємств та організацій України, що
пропонують продукцію і послуги для лабораторій.
Labprice.ua пропонує велику кількість корисних послуг, що
знадобляться працівникам лабораторій. На порталі можна ознайомитись із
цікавою науковою інформацією, отримати безкоштовну юридичну
допомогу, обрати спеціальну пропозицію товарів, скористатися однією з
найбільших добірок веб-посилань.
Labprice.ua є найліпшим довідником цін на реактиви, обладнання та
послуги на лабораторному ринку України. Він призначений для
працівників наукових, медичних і виробничих лабораторій, які не хочуть
витрачати зайвого часу на пошук необхідної продукції. Для зручності
відвідувачів портал містить її детальний опис і систему порівняння цін.
На Labprice.ua продукцію та послуги можуть рекламувати як великі
підприємства й організації, так і представники малого приватного бізнесу.
За інформацією звертайтесь: .
www.labprice.ua
а/с 485, м. Київ, 03150, Україна
Тел.:+38 (044) 221 55 31
E-mail: info@labprice.ua
UKRAINICA BIOORGANICA ACTA
журнал біоорганічної і біологічної хімії
вул. Академіка Заболотного, 150
м. Київ, 03143, Україна
Тел./факс: +38 (044) 522 24 58
E-mail: bioorganica@ukr.net
http://www.bioorganica.org.ua
Наукове видання "Ukrainica Bioorganica Acta" презентує праці з усіх галузей
біоорганічної і біологічної хімії. Проблематика журналу охоплює питання синтезу
й вивчення властивостей біологічно активних органічних сполук, дослідження
природних біорегуляторів, білків, нуклеїнових кислот, ліпідів, вуглеводів.
Розглядають як природні сполуки, так і їхні синтетичні структурні аналоги,
значну увагу приділяють проблемам біокон'югатів.
Видання призначене для наукових співробітників, викладачів ВНЗ, фахівців у
галузі біоорганічної, органічної, біологічної і медичної хімії, біохімії, біології,
фармакології і біотехнології.
Журнал внесено до переліків наукових фахових видань України, у яких
можуть публікувати основні результати дисертаційних робіт (галузі - "хімічні
науки", "біологічні науки"; постанови президії ВАК України від 08.06.2005 р.
№2-05/5, і №4-05/5, 30.06.2005 р. № 1-05/6), його надсилають до наукових
бібліотек, науково-дослідних інститутів і установ України.
"Ukrainica Bioorganica Acta" внесено до переліків наукових видань, які
реферують у Chemical Abstracts Service (www.cas.org), електронну версію видання
www.bioorganica.org.ua зареєстровано в Директорії журналів відкритого доступу
(Directory of Open Access Journals, www.doaj.org).
У виданні публікують повідомлення про діяльність відомих учених,
інформацію про наукові з'їзди, конгреси і конференції з хімії, а також інші
інформаційні та рекламні матеріали.
Журнал виходить двічі в рік.
Мови видання: українська, англійська. к .
ISSN: 1814-9758 (друкована версія), 1814-9766 (електронна версія). /, 77
Навчальне видання
Чирва В. Я., Ярмолюк С. М., Толкачова Н. В., Земляков О. Є.
ОРГАНІЧНА ХІМІЯ
Підручник
Затверджено
Міністерством освіти і науки України
Підп. до друку 12.04.2009. Формат 70x100/16.
Папір друк. Офсет друк. Ум. друк. арк. 80,3.
Наклад 500 прим.
Видавництво "БаК"
вул. Чайковського, 17
а/с 9009, Львів 79011
тел. (032)-261-00-12
факс (032)-261-10-81
e-mail: office@bak.lviv.ua
Internet: http://www.bak.lviv.ua
Чирва В. Я., Ярмолюк С. М., Толкачова Н. В., Земляков О. Є.
Органічна хімія: Підручник. - Львів: БаК, 2009. - 996 с.
Ч-64
ISBN 966-7065-87-4.
Викладено основи сучасної органічної хімії. Розглянуто теоретичні питання
основ природи хімічного зв'язку, будову та реакційну здатність органічних молекул з
урахуванням впливу електронних факторів на перебіг хімічних реакцій. Наведено
сучасні промислові та лабораторні методи одержання основних класів органічних
сполук, їхні фізичні та хімічні властивості. Описано механізми найважливіших
хімічних реакцій. Значну увагу приділено питанням стереохімії та енантіомерії
органічних молекул. Для характеристики органічних сполук широко використано
спектральні методи, такі як УФ-, ІЧ- і ПМР-спектроскопія. Взято до уваги
рекомендації Української національної комісії з хімічної термінології і номенклатури.
Для студентів-хіміків і біохіміків, а також спеціальностей з біології, фармації
тощо та всіх, хто цікавиться органічною хімією.
УДК 547(075.8)
ББК Г2Я73
Доктор хімічних наук, професор,
завідуючий кафедри органічної та
біологічної хімії, декан хімічного
факультету Таврійського національного
університету ім. В.І. Вернадського. Автор
400 наукових публікацій, двох монографій.
Доктор хімічних наук, професор, завідувач
відділу комбінаторної хімії Інституту
молекулярної біології і генетики HAH
України. Автор понад 280 наукових праць.
Ярмолюк Сергій Миколайович
Чирва Василь Якович
Толкачова Наталя Василівна
Кандидат хімічних наук, доцент
кафедри органічної та біологічної хімії
Таврійського національного університету
ім. В.І. Вернадського. Автор 50 наукових
робіт.
Земляков Олександр Євгенович
доктор хімічних наук, професор, професор
кафедри органічної та біологічної хімії
Таврійського національного університету
ім. В.І. Вернадського. Автор 150 наукових і
навчально-методичних робіт.
www.otava.com.ua
www.bioorganica.org.ua
www.labprice.ua