



































































































































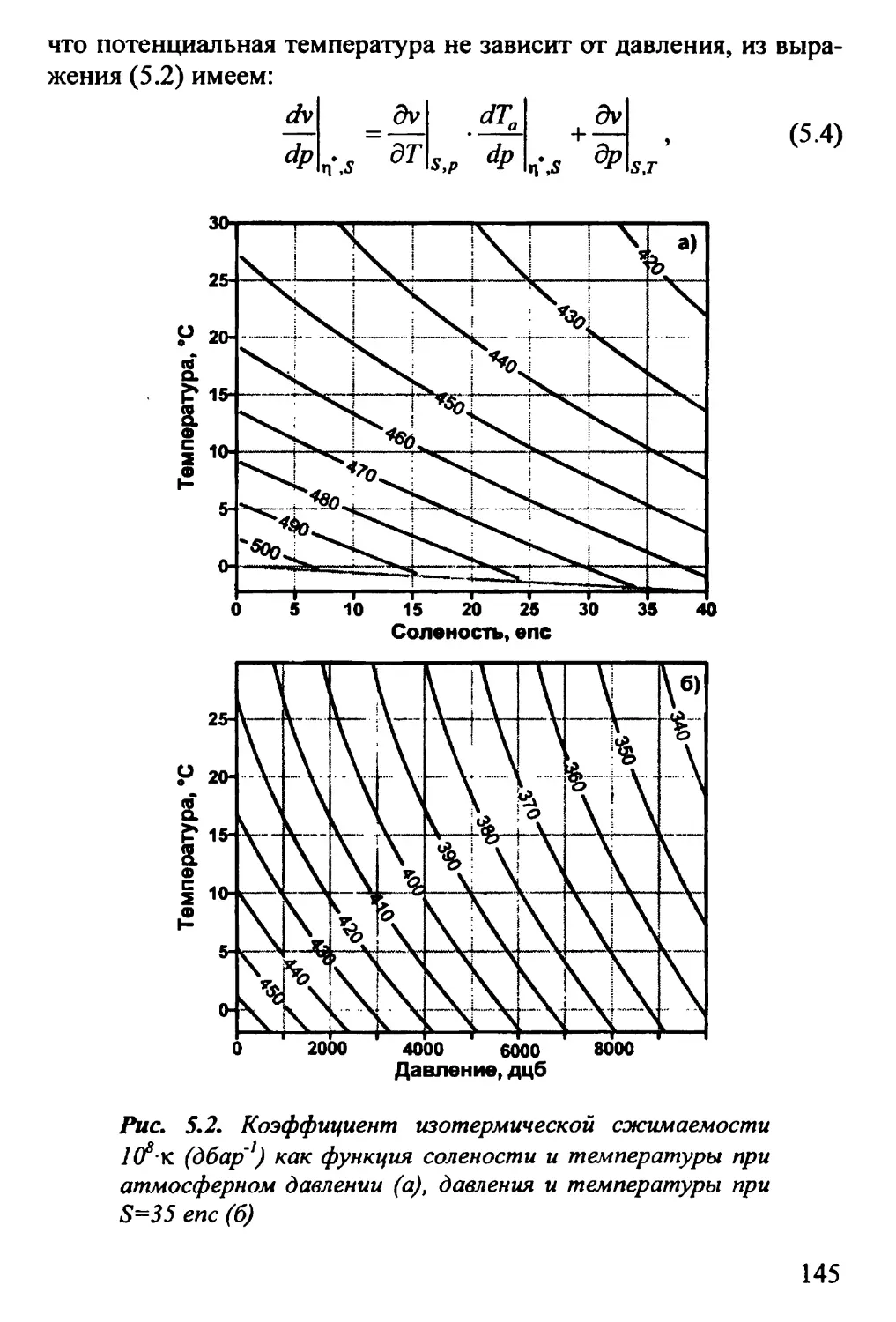

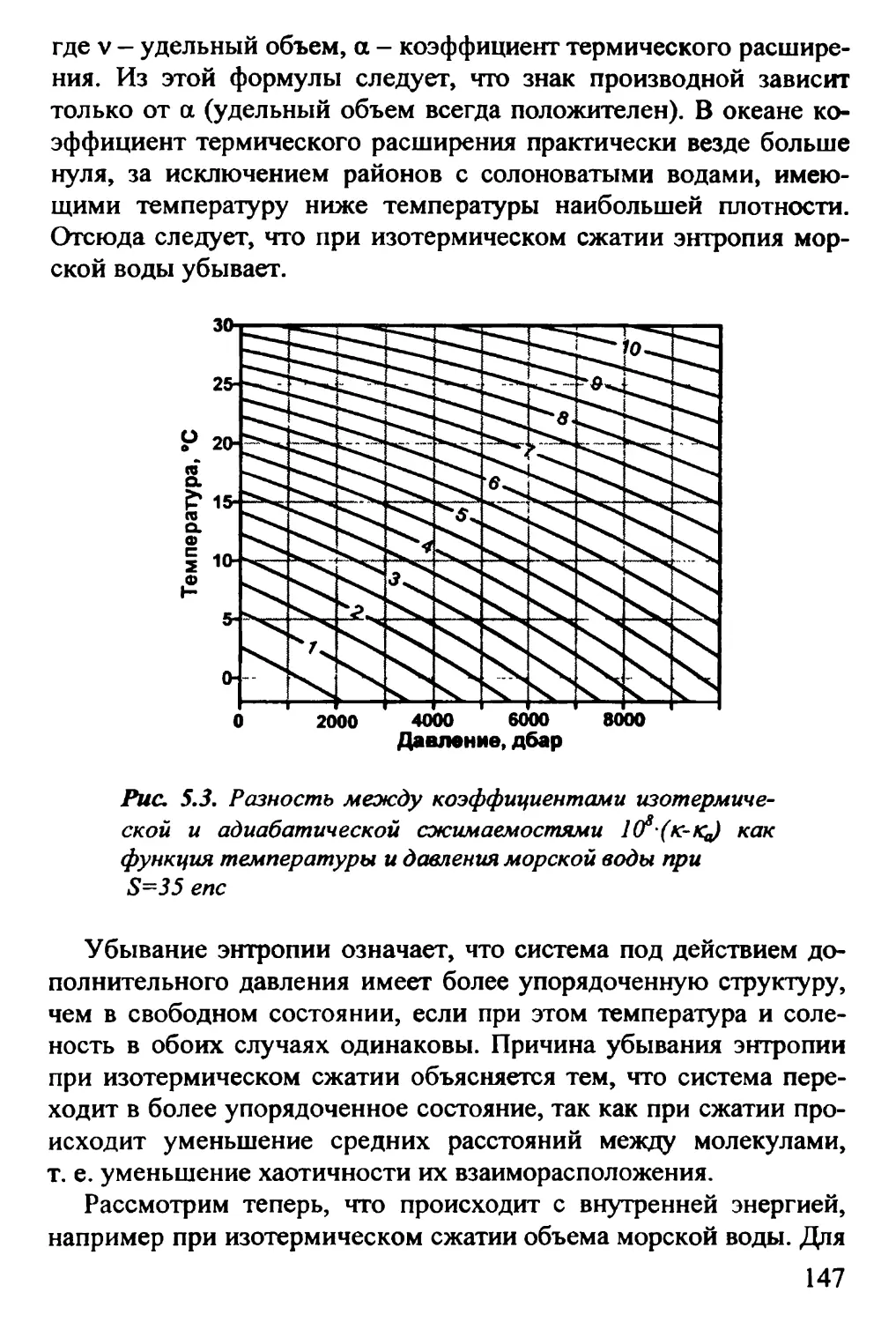





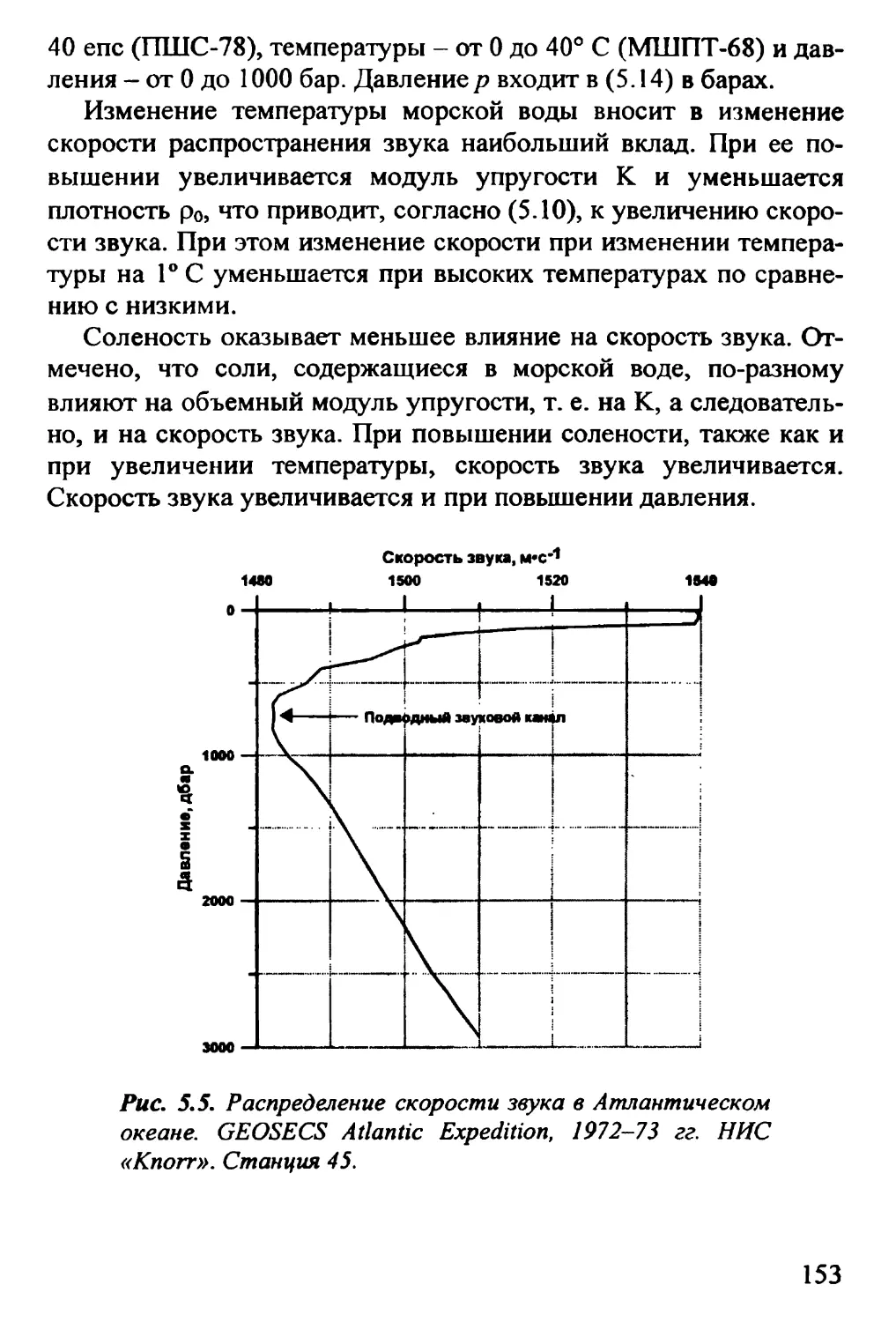






















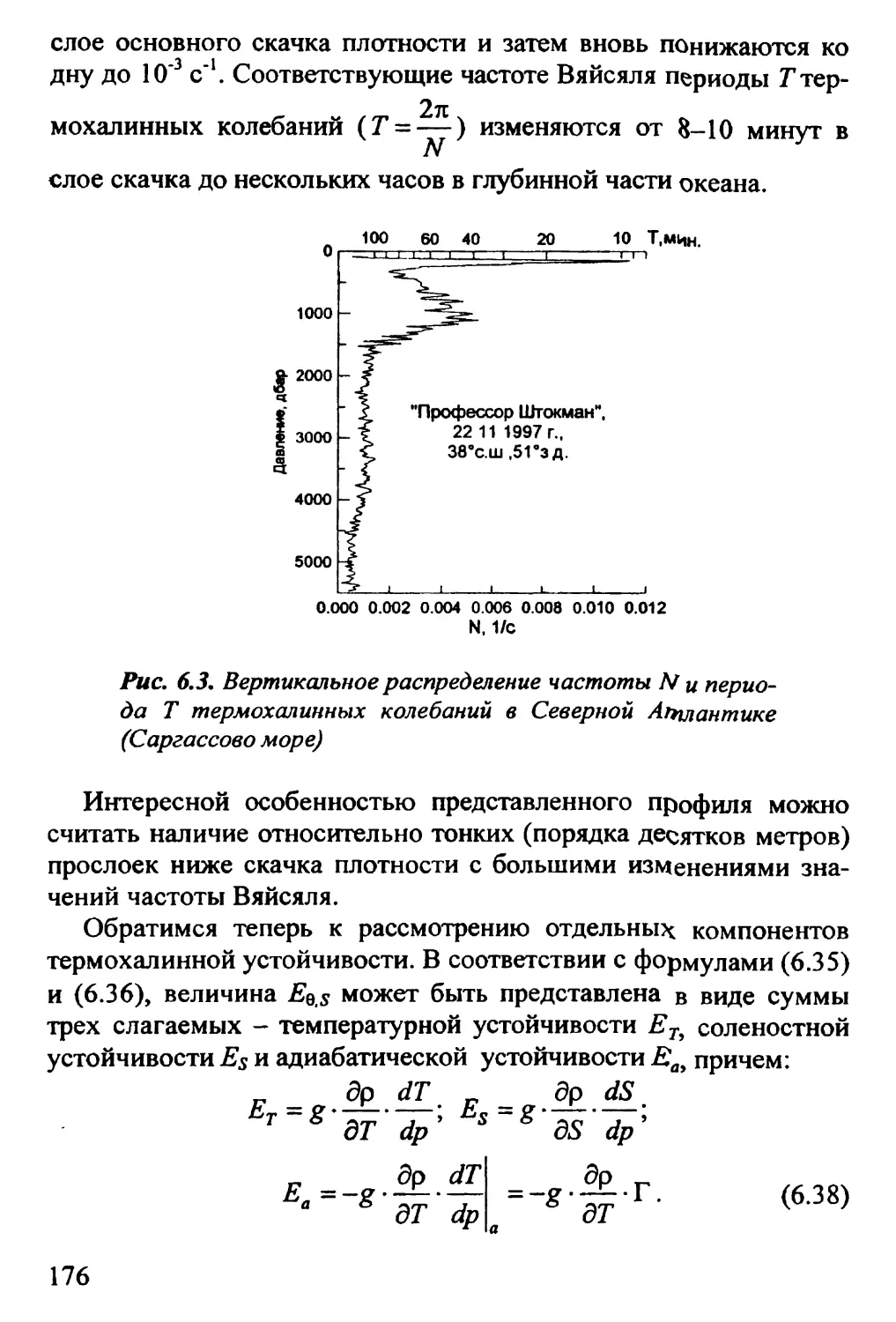






































Author: Добролюбов С.А. Архипкин В.С.
Tags: высшее образование университеты академическое обучение океанология рельеф морского дна дно океана океанография мировой океан физика и гидрология моря география биология океан моря океаны
ISBN: 5-317-01473-5
Year: 2005




МОСКОВСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ
им. М.В. ЛОМОНОСОВА
Географический факультет
Посвящается 250-летию
Московского университета
B.C. Архипкин, С.А. Добролюбов
ОКЕАНОЛОГИЯ
ФИЗИЧЕСКИЕ СВОЙСТВА МОРСКОЙ ВОДЫ
Учебное пособие
МОСКВА
2005
УДК 378(075.8):551.46
ББК 26.221я73
А87
Архипкин B.C., Добролюбов С.А.
А87 Океанология. Физические свойства морской воды:
Учебное пособие. - М.: МАКС Пресс, 2005. - 216 с: 44 ил.
ISBN 5-317-01473-5
В предлагаемом учебном пособии рассматриваются структура воды
и ее свойства, основные законы классической термодинамики и их
применение к морской воде как двухкомпонентной системе, состоящей из
чистой воды и морской соли. Приведены современные формулы
вычисления термодинамических характеристик морской воды.
Пособие предназначено для студентов - океанологов географических
факультетов университетов и гидрометеорологических институтов, а
также для научных работников, имеющих отношение к исследованию
вод океанов и морей.
УДК 378(075.8):551.46
ББК 26.221я73
Печатается по постановлению ученого совета
географического факультета МГУ им. М. В. Ломоносова
Рецензенты:
член-корреспондент НАН Украины, профессор,
д.ф.-м.н. В.А. Иванов;
профессор, д.г.н. Н.К Алексеевский
ISBN 5-317-01473-5 © Архипкин B.C., 2005
© Добролюбов С.А., 2005
СОДЕРЖАНИЕ
УСЛОВНЫЕ ОБОЗНАЧЕНИЯ 6
НЕКОТОРЫЕ ВЕЛИЧИНЫ, ИСПОЛЬЗУЕМЫЕ
В ПОСОБИИ 8
ПРЕДИСЛОВИЕ 10
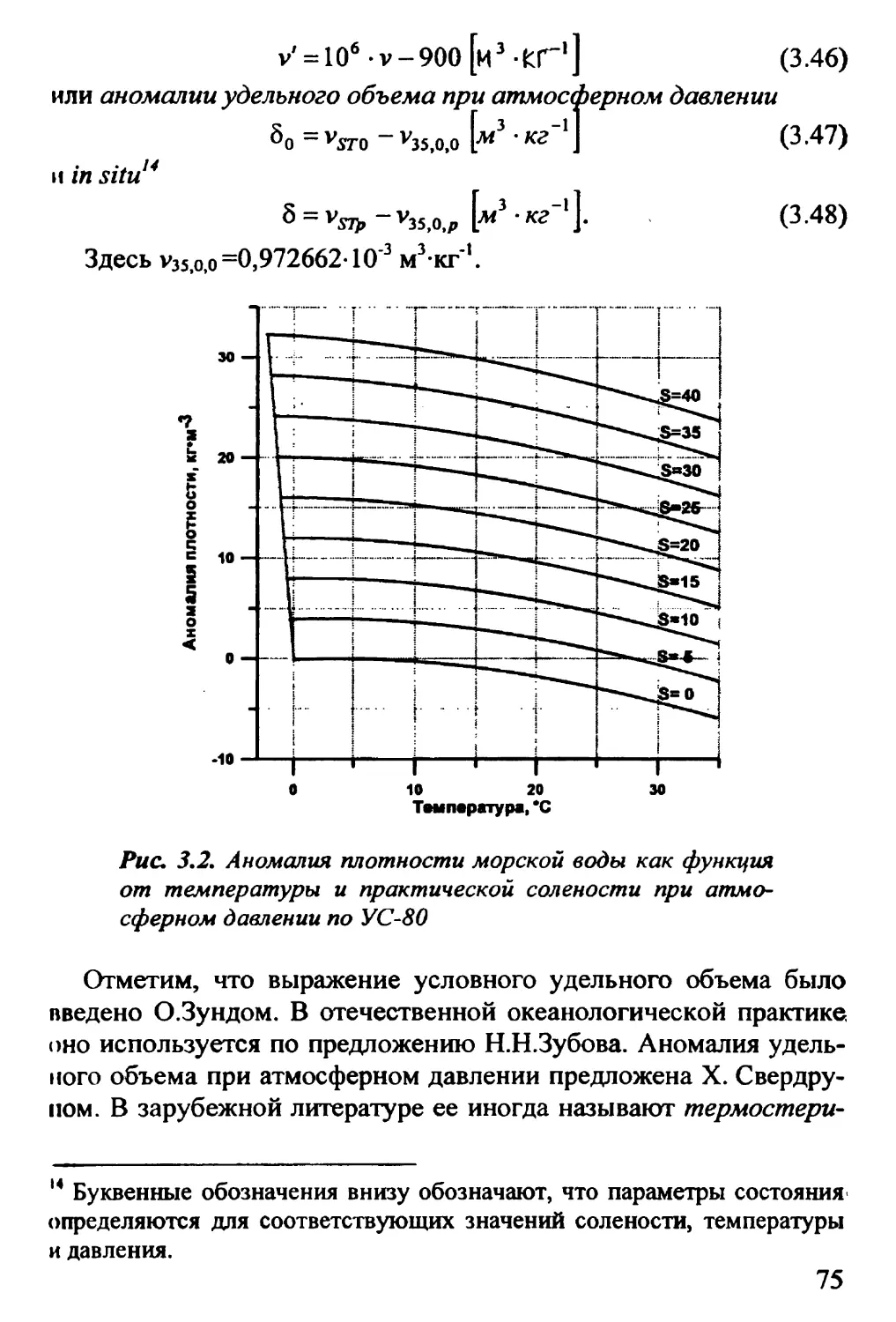
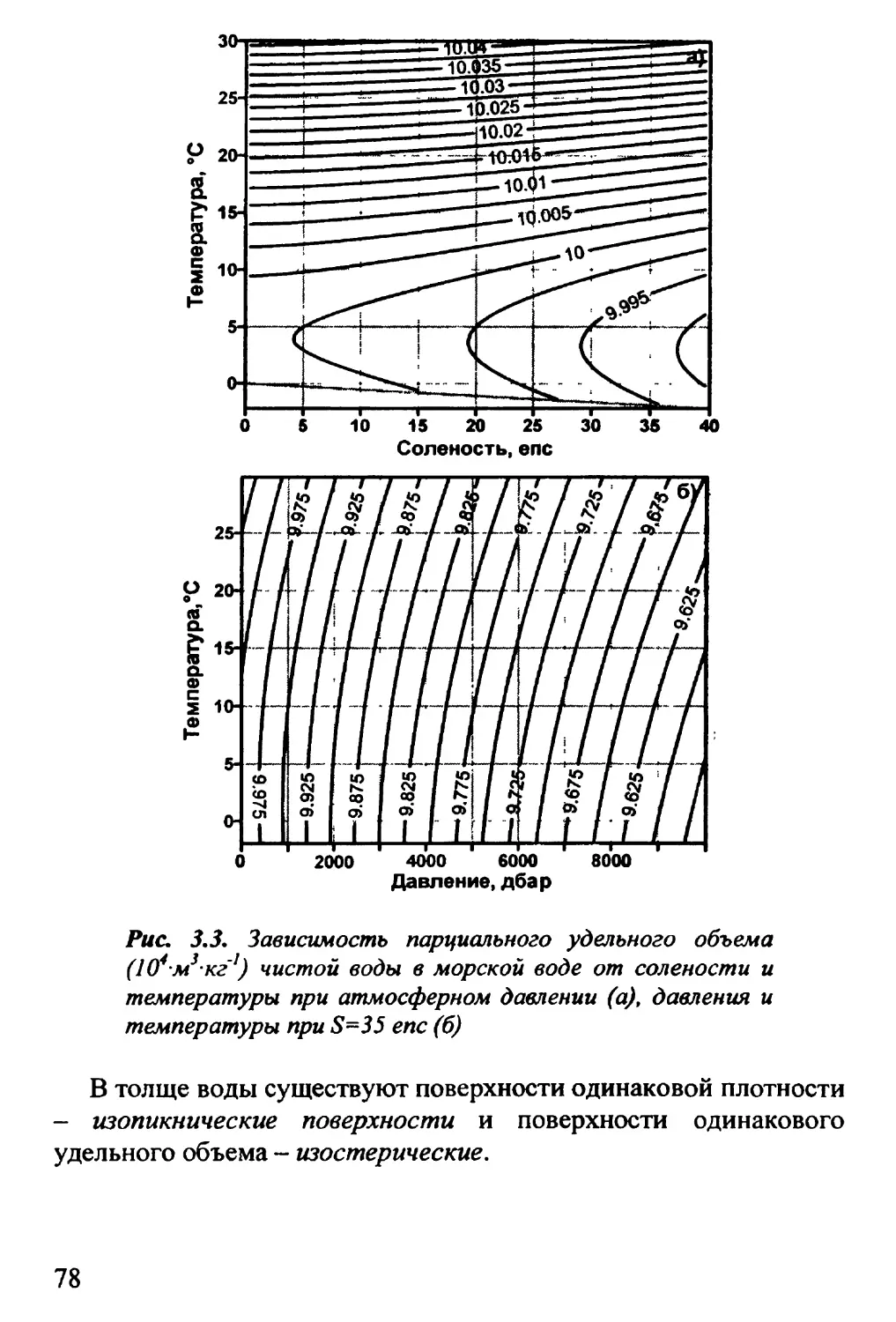
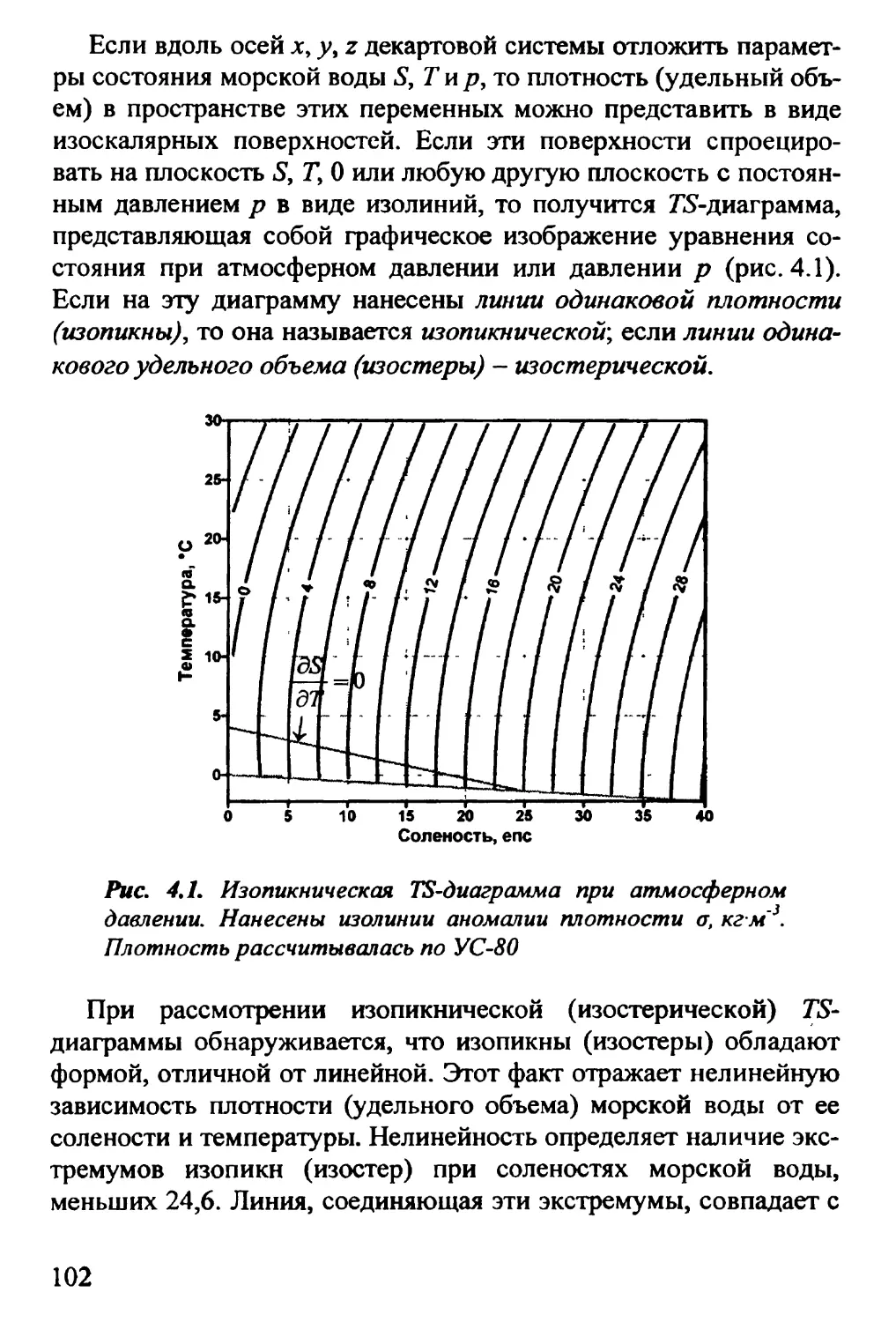
ГЛАВА 1. СТРУКТУРА ЧИСТОЙ ВОДЫ И ЕЕ СВОЙСТВА.. 12
1.1. Строение молекулы воды 12
1.2. Структура воды: наблюдения и модели 16
1.3. Аномальные физические свойства чистой воды 20
1.4. Свойства тяжелой воды 25
Задачи и вопросы для повторения 26
ГЛАВА 2. ИССЛЕДОВАНИЕ ФИЗИЧЕСКИХ СВОЙСТВ
ТЕРМОДИНАМИЧЕСКИХ СИСТЕМ МЕТОДОМ
ПОТЕНЦИАЛОВ 27
2.1. Определение термодинамических систем 27
2.2. Краткие сведения об исходных положениях и началах
термодинамики 32
2.3. Метод термодинамических потенциалов 40
2.4. Основные соотношения термодинамики для
многокомпонентных систем. Парциальные величины 45
2.5. Использование якобианов 49
Задачи и вопросы для повторения 53
ГЛАВА 3. МОРСКАЯ ВОДА КАК ДВУХКОМПОНЕНТНАЯ
ТЕРМОДИНАМИЧЕСКАЯ СИСТЕМА 54
3.1. Образование водной массы Мирового океана 54
3.2. История формирования солевого состава Мирового
океана 55
3.3. Особенности современного солевого состава Мирового
океана 57
3.4. Основные термодинамические соотношения для морской
воды 60
3.5. Параметры состояния. Температура 65
3.6. Соленость. Определение солености 69
3.7. Плотность. Удельный объем 73
3.8. Давление. Уравнение гидростатики 78
3.9. Поле силы тяжести. Ускорение силы тяжести 85
3.10. Геопотенциал. Динамическая глубина 87
3
3.11. Потенциальная энергия. Аномалия потенциальной
энергии 89
Задачи и вопросы для повторения 91
ГЛАВА 4. УРАВНЕНИЕ СОСТОЯНИЯ.
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА МОРСКОЙ ВОДЫ.. 93
4.1. Уравнение состояния морской воды. Дифференциальная
форма уравнения состояния 93
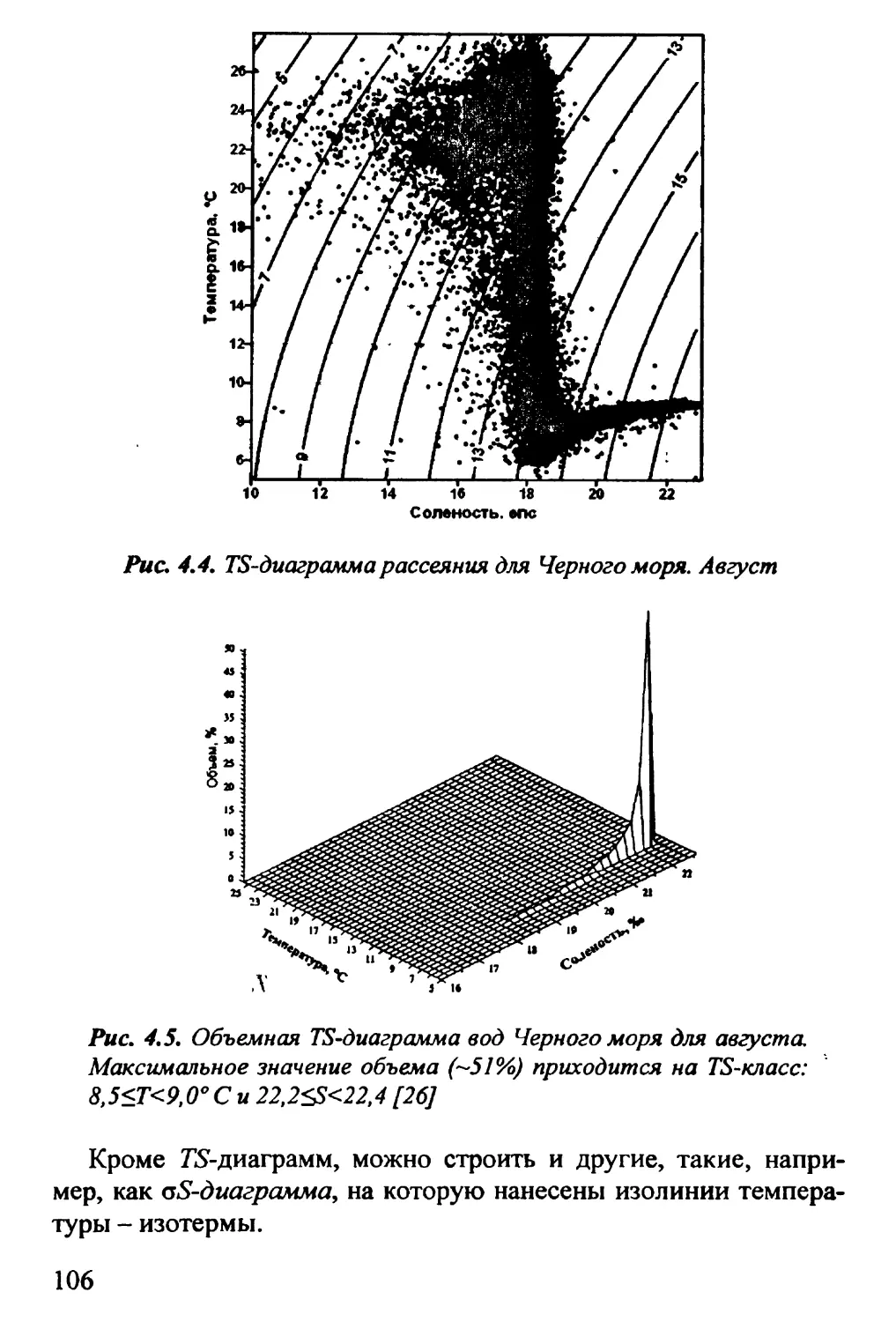
4.2. Термодинамические диаграммы. Г£-диаграмма 100
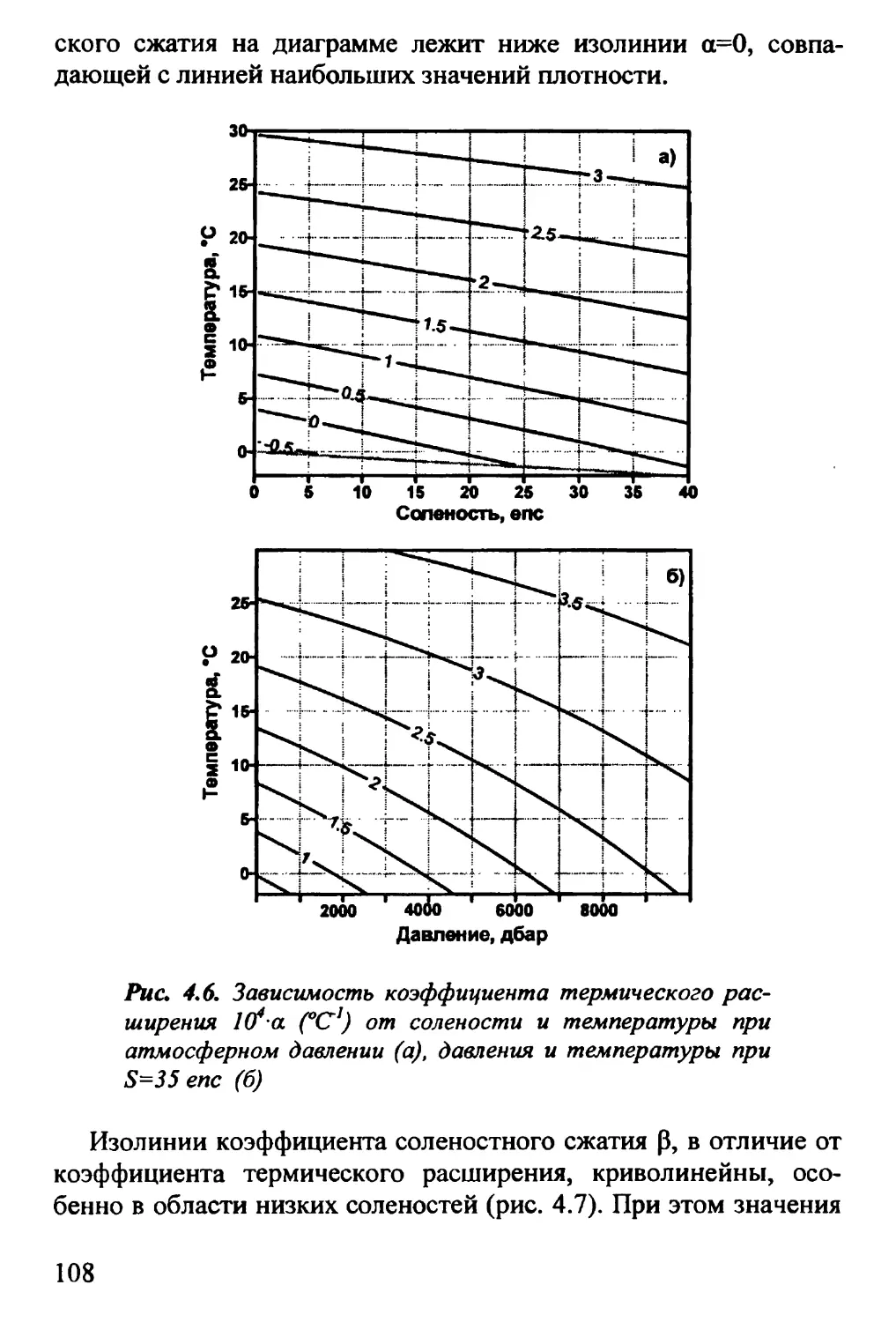
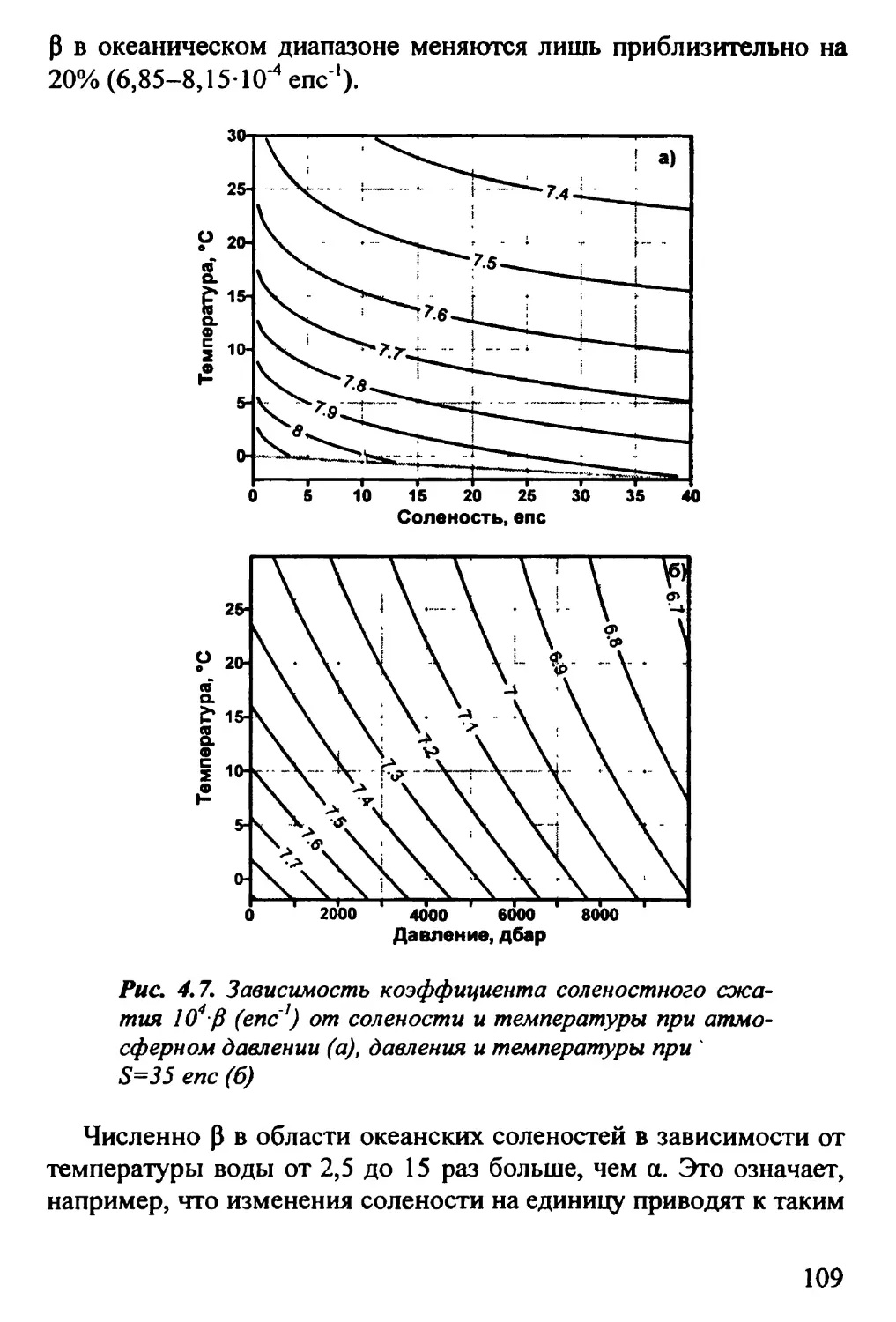
4.3. Термическое расширение. Соленостное сжатие 106
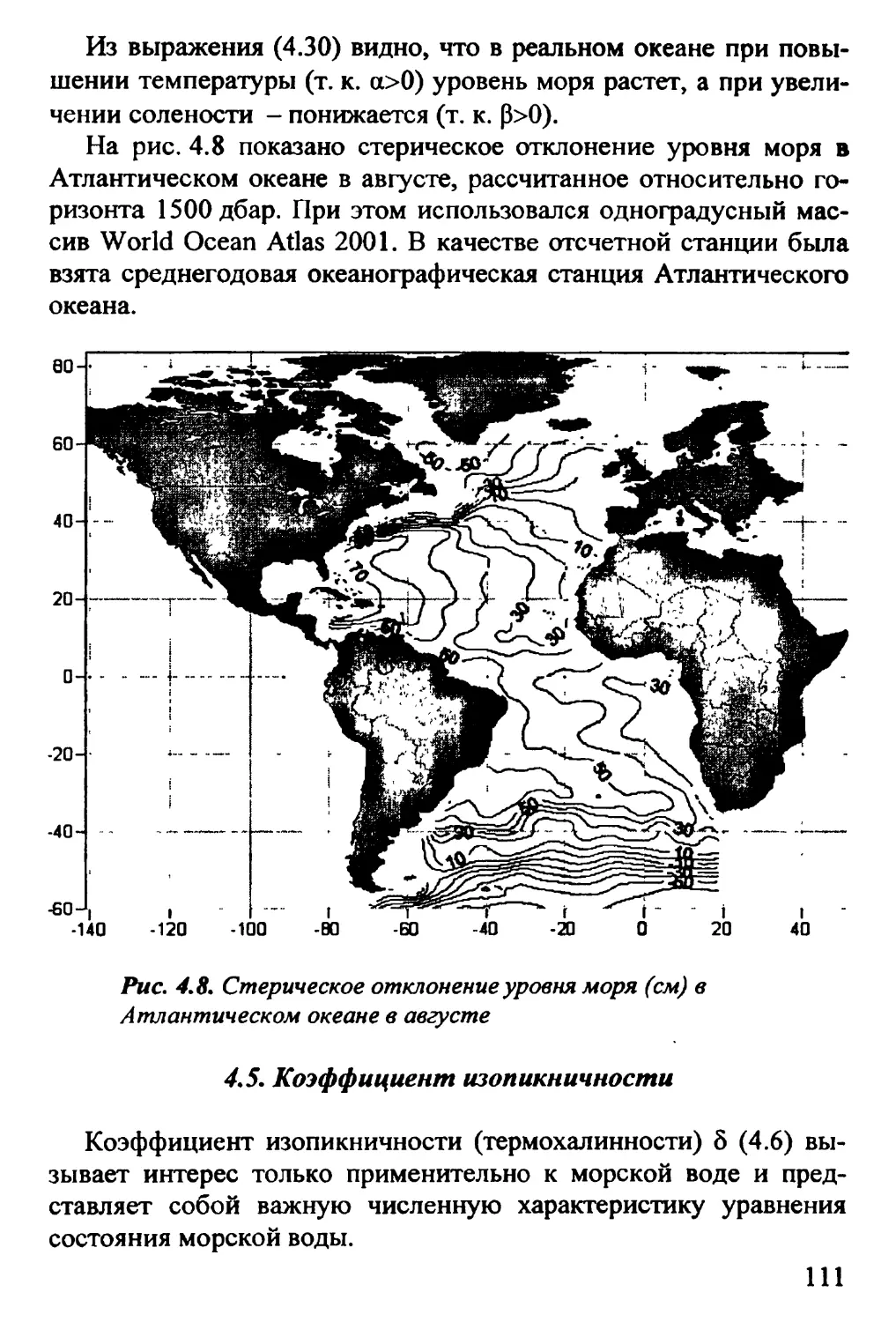
4.4. Стерические колебания уровня моря 109
4.5. Коэффициент изопикничности 110
4.6. Температура наибольшей плотности. Солоноватые и
морские воды 111
4.7. Теплоемкость морской воды 115
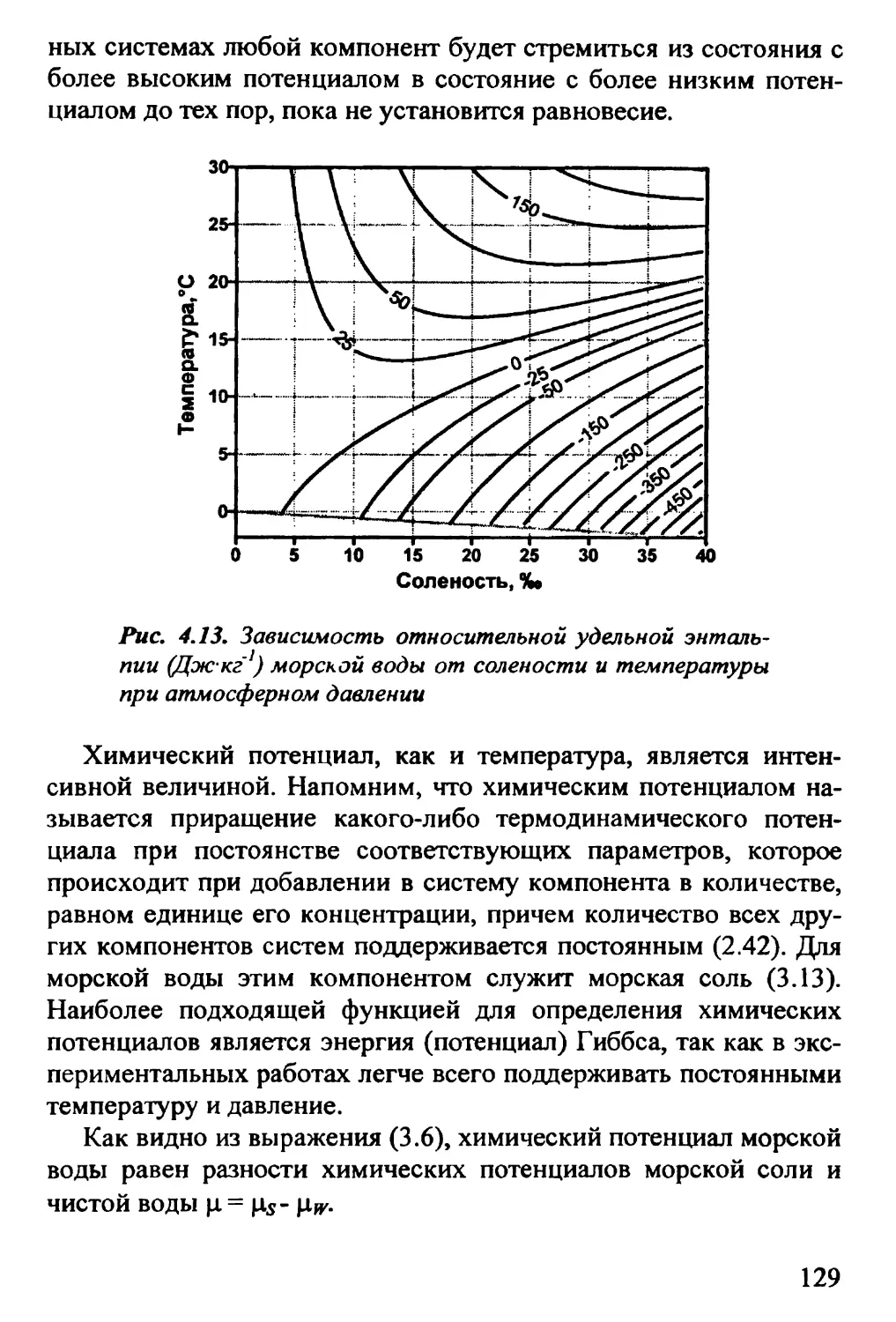
4.8. Энтропия. Энтальпия 125
4.9. Химический потенциал 127
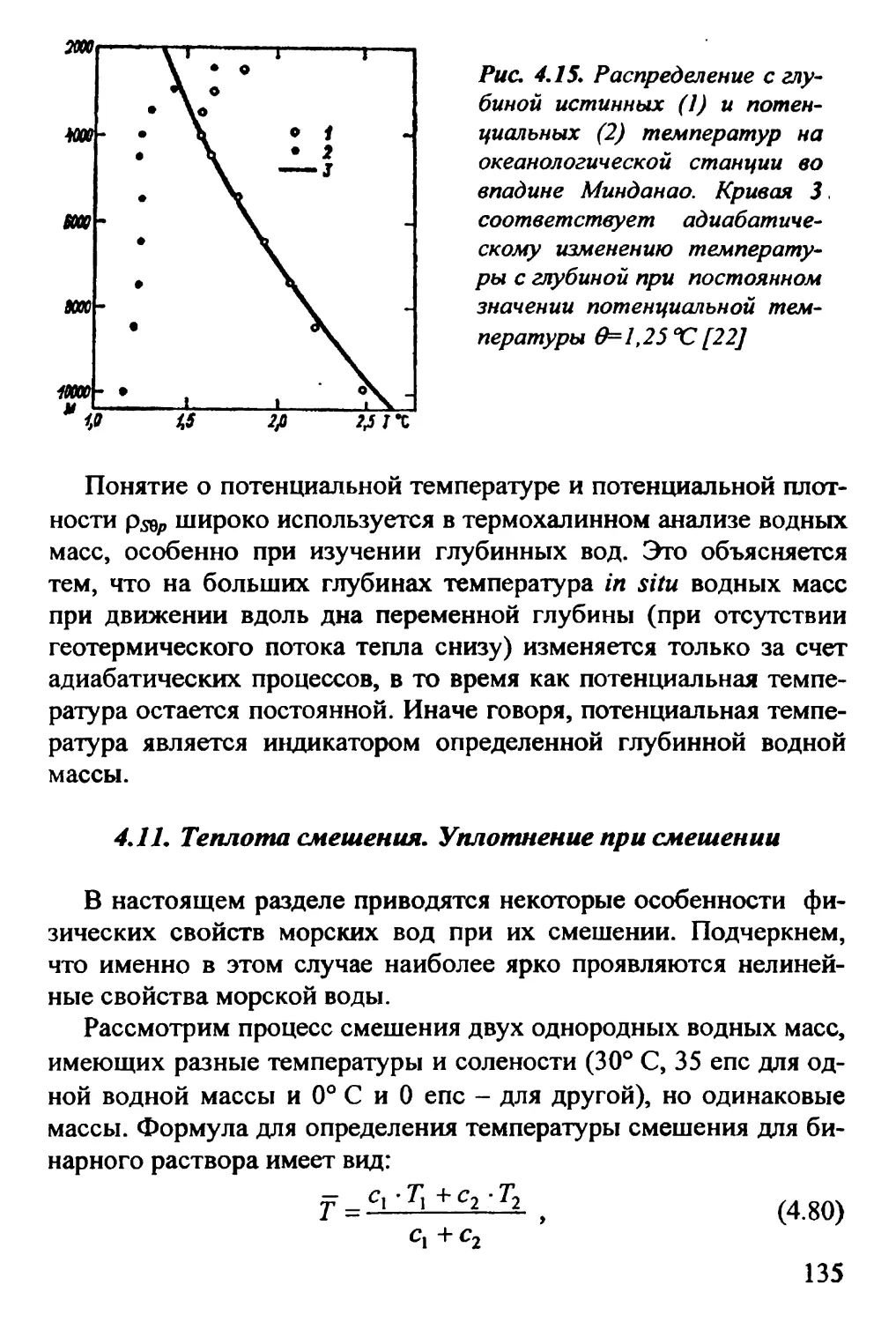
4.10. Адиабатический градиент температуры. Потенциальная
температура 129
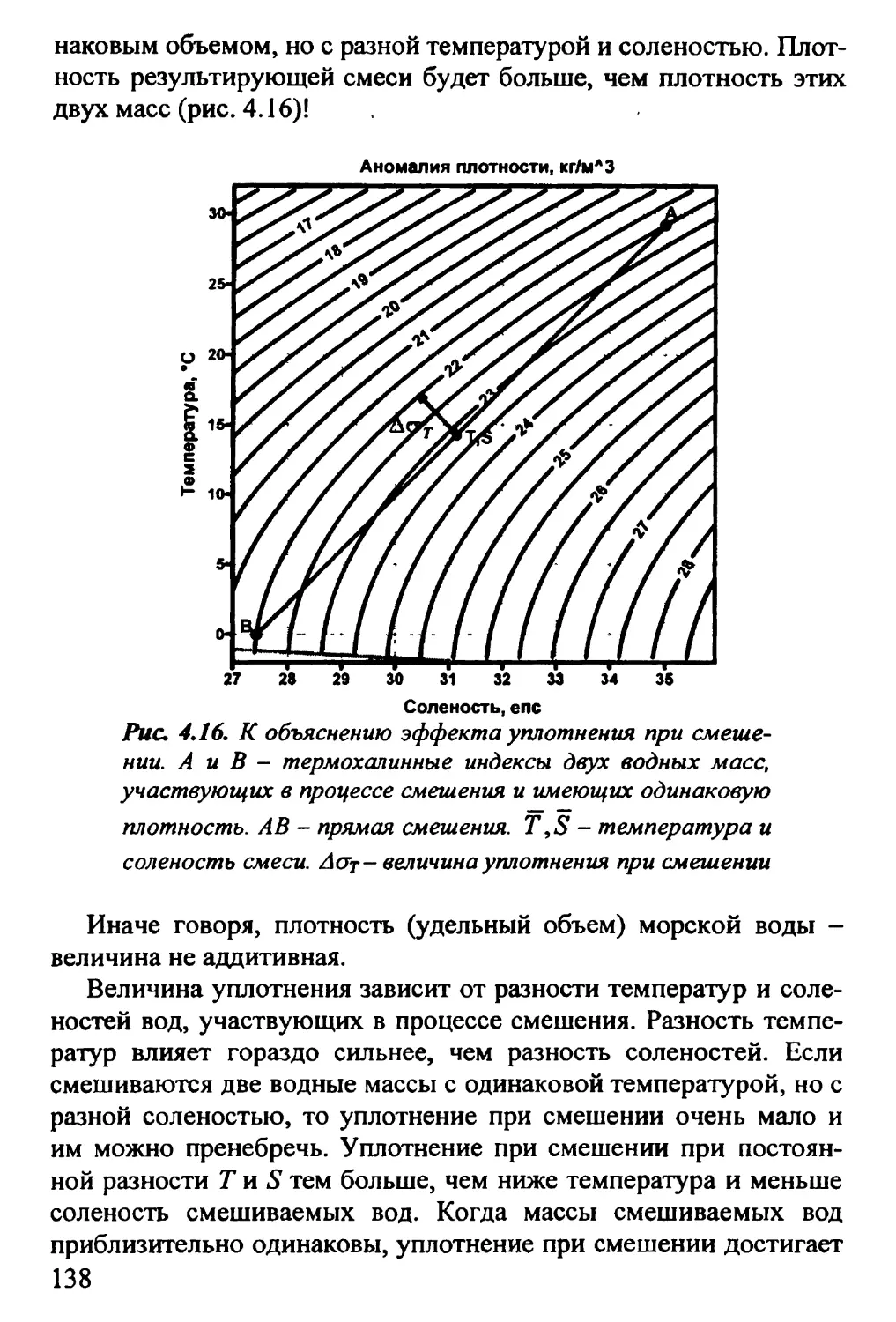
4.11. Теплота смешения. Уплотнение при смешении 134
Задачи и вопросы для повторения 140
ГЛАВА 5. МОРСКАЯ ВОДА КАК УПРУГАЯ СРЕДА 142
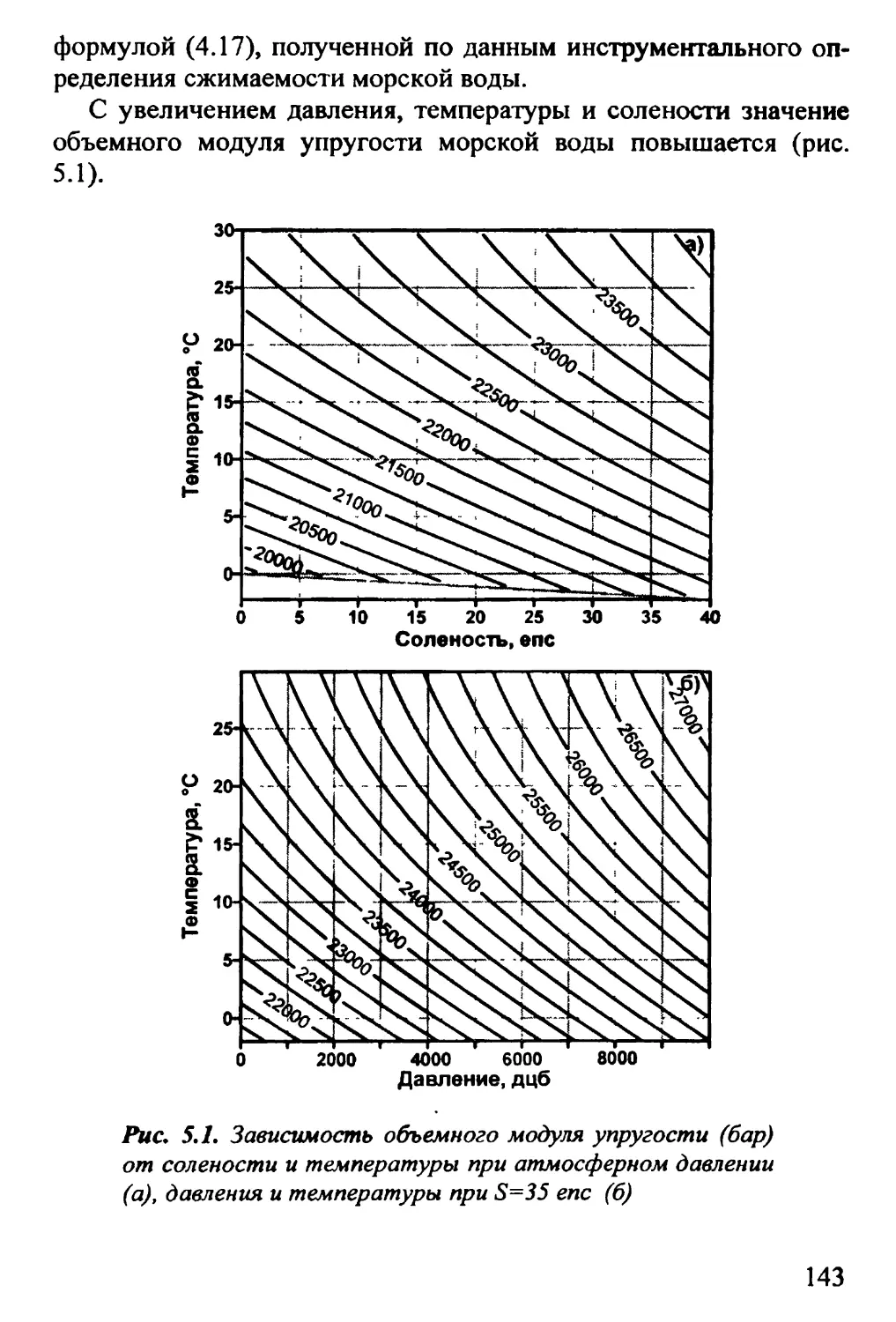
5.1. Объемный модуль упругости. Сжимаемость морской
воды 142
5.2. Скорость распространения звука 149
Задачи и вопросы для повторения 154
ГЛАВА 6. УСЛОВИЯ РАВНОВЕСИЯ МОРСКОЙ ВОДЫ .... 156
6.1. Общие условия равновесия термодинамических систем. .. 156
6.2. Фазовые равновесия и фазовые переходы. Уравнение
Клапейрона-Клаузиуса 160
6.3. Поверхностное натяжение морской воды 165
6.4. Вертикальная устойчивость. Частота Вяйсяля 171
Задачи и вопросы для повторения 180
ГЛАВА 7. КОЛЛИГАТИВНЫЕ СВОЙСТВА МОРСКОЙ
ВОДЫ 182
7.1. Температура замерзания 182
7.2. Давление насыщенного пара. Скрытая теплота испарения. 185
7.3. Осмотическое давление 188
7.4. Вязкость 192
4
Задачи и вопросы для повторения 196
ГЛАВА 8. ЭЛЕКТРОМАГНИТНЫЕ СВОЙСТВА МОРСКОЙ
ВОДЫ 197
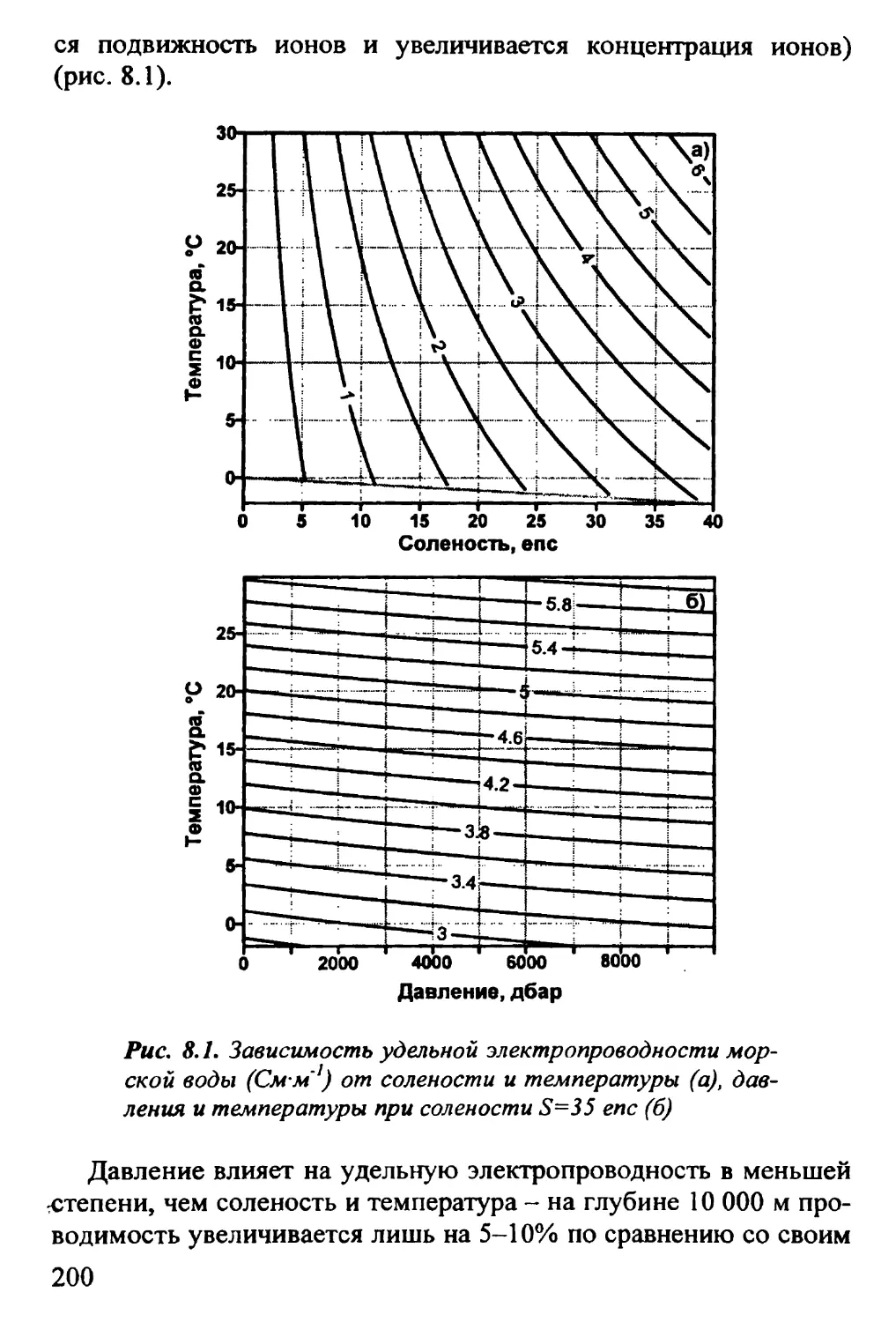
8.1. Электропроводность 198
8.2. Диэлектрическая проницаемость 202
8.3. Магнитная восприимчивость 206
Задачи и вопросы для повторения 208
СПИСОК ЛИТЕРАТУРЫ 209
УСЛОВНЫЕ ОБОЗНА ЧЕНИЯ
(в квадратных скобках приведена размерность)
Абсолютная температура - Т [°К]
Адиабатический градиент температуры -
Г рс-дбар"1 или ТС-дбар"1]
Аномалия плотности (условная плотность) - а [кг-м"3]
Аномалия потенциальной плотности - oQ [кг-м'3]
Аномалия потенциальной энергии - % [Джм~2=кг-с'2]
Аномалия удельного объема - 5 [м3-кг-1]
Вертикальная соленостная устойчивость - Es [м1 или дбар"1]
Вертикальная температурная устойчивость - Ет [м1 или дбар"1]
Вертикальная устойчивость - Е [м"1 или дбар"1]
Внутренняя энергия - U [Дж]
Географическая широта - ф [°]
Геопотенциал - Ф [м2-с"2=Дж*кг1]
Глубина - z [м]
Давление-р [Па = кг-м_|-с"2 = Нм'^10"4 дбар]
Динамическая вязкость - ц [кг-м1 с *1=9,81 Нем"2]
Динамическая глубина (высота) - D [м2-с'2]
Диэлектрическая восприимчивость - Хс [безразм.]
Диэлектрическая проницаемость - е [безразм.]
Кинематическая вязкость - v [м2-с-1]
Количество теплоты - Q [Дж]
Коэффициент адиабатической сжимаемости - ка [дбар1]
Коэффициент изотермической сжимаемости — к [дбар1]
Коэффициент соленостного сжатия - р [%о'! или епс"1]
Коэффициент термического расширения - а \°Сл или °К"1]
Коэффициент электропроводности - R [безразм.]
Магнитная постоянная - ц<> [Гнм*1]
Магнитная проницаемость - \i [безразм.]
Объем - V [м3]
Объемный модуль упругости - К [бар]
Относительная электропроводность - RT [безразм.]
Отношениетеплоемкостей cp/cv -у [безразм.]
Параметр Кориолиса -/[с*1]
Плотностное соотношение (число) -Rp [безразм.]
6
Плотность - р [кг-м ]
1 Ыотность in situ - pSrP [кгм"3]
11оверхностное натяжение - у [Нм1]
11отенциальная температура - Э [°С]
Работа-^[Дж]
Свободная энергия (энергия Гельмгольца) - F [Дж]
Скорость звука - С [мС1]
Скрытая теплота перехода (испарения, сублимации, плавления) -
X [Дж-кг"1]
Соленость - S [%о или епс]
Температура - Т [°С]
Температура замерзания - 7} [°С]
Температура максимальной плотности - Тт [°С]
Теплоемкость - С [Дж^К1]
Термический коэффициент давления (упругости) -
у[°С1 или °К'1]
Термодинамический потенциал Гиббса - G [Дж]
Удельная внутренняя энергия - 6 [Дж-кг-1]
Удельная свободная энергия - f [Дж-кг1]
Удельная теплоемкость при p=cosnt - ср [Дж-кг"1 ^К"1]
Удельная теплоемкость при постоянном объеме - су [Джкг"1оК~!]
Удельная электропроводность (электропроводимость) - у [Смм1]
Удельная энтальпия - h [Дж-кг"1]
Удельная энтропия - г|* [Дж-кг"1-""К"1]
Удельный объем - v [м^кг1]
Удельный объем in situ - vStp [м3-кг_1]
Удельный потенциал Гиббса - g* [Дж-кг1]
Универсальная газовая постоянная-R [Джмоль'^К1]
Ускорение силы тяжести - g [м-с"2=105 мГал]
Условный удельный объем - V [безразм.]
Химический потенциал - ц [Джмоль1]
Хлорность морской воды - С1 [%о]
Частота Вяйсяля-Брента - N [с1]
Электрическая постоянная - б0 [Ф-м1]
Энтальпия - Н [Дж]
Энтропия - Г| [Дж^К1]
7
НЕКОТОРЫЕ ВЕЛИЧИНЫ, ИСПОЛЬЗУЕМЫЕ В ПОСОБИИ
Давление насыщенного водяного пара над ровной поверхностью
воды или льда при Г=273° К - 610 Па
Газовая постоянная водяного пара-461,5 Дж(кгК)"1
Магнитная постоянная - 4-я-10"4 Гн-м"1
Масса Земли - 5,976-1024 кг
Молекулярный вес морской соли при 5=35 - 31,42 г-моль'1
Плотность:
воздуха при р= 1013 ГПа, 7Ч)° С - 1,292 кг-м'3
стандартной морской воды (5=35, 7=0° С,/7=0 дбар) -
1028,106 кг-м3
Площади поверхности:
Земли-5,1-1014м2
океанов и морей - 3,62-1014 м2
Постоянная Больцмана- 1,38044-10"23 Дж°К"!
Радиус Земли:
экваториальный - 6,378-106 м
полярный - 6,357-106 м
средний-6,371106 м
Скорость звука:
в воздухе при стандартных условиях - 342 м-с"1
в стандартной морской воде - 1449 м-с-1
Температура наибольшей плотности пресной воды
при атмосферном давлении - 3,982° С
Угловая скорость вращения Земли - 7,29-10"5 с"1
Удельная газовая постоянная:
сухого воздуха - 287,05 Дж-(кг-°Ку!
водяного пара - 461,51 Дж-(кг°К)*1
Удельная теплоемкость:
стандартной морской воды приp-const - 3986,533 Дж-(кг-°К)"1
стандартной морской воды при v=const - 3986,532 Дж-(кг-°К)'!
сухого воздуха при p-const - 1005 Дж^кг^К)'1
сухого воздуха при v=const- 718 Дж-(кг°К)"1
Удельная теплота:
испарения пресной воды при 7=0° С - 2,50-106 Дж-кг"1
плавления пресного льда - 0,335-106 Дж-кг'1
сублимации (возгонки) - 2,83-106 Дж-кг"1
8
Удельная электропроводность дистиллированной воды -
2-Ю-4 Смм"1
Удельный объем:
пара при 7=0° С - 206 м3кг'
пресного льда - 1,091 • 10"3 м3кг!
стандартной морской воды - 0,972662-10"3 м^кг"1
чистой воды при Т=0° С - 1,000-10"3 м3-кг1
Универсальная (молярная) газовая постоянная -
8,31441 Дж-СмольЖу1
Универсальная гравитационная постоянная - 6,672-10"11 м3-кг'1вс":
Ускорение силы тяжести на поверхности моря на широте 45° -
9,805 м-с2
Электрическая постоянная - 8,85-10"12 Фм'1
Удельная электропроводность:
стандартной морской воды - 2,9036 Смм"1
морской воды при £=35 епс, /=15° С9р=0 дбар - 4,2914 Смм"1
9
ПРЕДИСЛОВИЕ
Предлагаемое учебное пособие предназначено для студентов
университетов, обучающихся по специальности «океанология».
Раздел «Физические свойства морской воды», в соответствии с
утвержденным учебным планом, читается на втором курсе
кафедры океанологии географического факультета МГУ им.
М.В. Ломоносова. Он идет первым из всех разделов общего курса
«Океанология». В этом пособии не рассматриваются морской
лед, оптика и акустика океана, так как им посвящены отдельные
разделы общего курса.
Настоящему курсу должно предшествовать изучение
студентами общего курса физики, дифференциального и интегрального
исчислений.
На кафедре океанологии МГУ им. М.В. Ломоносова
проблемой физических свойств морской воды на протяжении многих
лет занимался профессор О.И. Мамаев. За это время им был
опубликован ряд монографий [20, 22], большое количество
статей, в которых исследовались различные аспекты термодинамики
морской воды. Многие его идеи и методические приемы
изложения этого курса взяты авторами за основу.
Начинается учебное пособие описанием структуры чистой
воды и ее свойствами. К настоящему времени единой модели воды
пока не создано, поэтому в этой главе кратко рассматриваются
основные существующие модели структуры воды.
Для исследования многих физических свойств воды
применяются два метода - термодинамический и статистический. Нами за
основу был выбран первый из них по нескольким причинам.
Во-первых, термодинамический метод не требует знания
молекулярной структуры воды. Этот момент важен в связи со
сложностью строения воды и отсутствием единой теории жидкости.
Хотя для более полного изучения статистических закономерностей
большого числа непрерывно движущихся и взаимодействующих
частиц необходимо применение статистических методов. Тем не
менее, для решения многих практических задач достаточно
применения методов термодинамики.
Во-вторых, ко второму курсу студенты-океанологи еще не
успевают прослушать многие разделы высшей математики и физики,
необходимые при изучении основ статистической физики.
10
Далее показывается, по каким причинам морскую воду можно
представить как двухкомпонентную термодинамическую
систему, состоящую из чистой воды и морской соли. Затем даются
основные термодинамические соотношения для морской воды,
описываются ее параметры.
Добавление соли еще более затрудняет построение
теоретических уравнений состояния, связывающих температуру воды, ее
соленость, плотность и давление. В настоящее время имеются
лишь эмпирические уравнения состояния. В океанографической
практике сейчас используется главным образом Международное
уравнение состояния 1980 г. В пособии рассматриваются
физические свойства морской воды, основанные на этом уравнении
состояния.
Большое внимание в пособии уделяется вопросам равновесия
морской воды, особенно частным условиям равновесия. Даются
примеры их использования.
Отдельно рассматриваются коллигативные свойства морской
воды, которые зависят от концентрации растворенных веществ в
воде и не зависят от специфических химических характеристик.
Последняя глава посвящена электромагнитным свойствам
морской воды. Упор здесь делается на электропроводность воды,
так как эта характеристика используется для определения
солености, при измерении течений электромагнитным способом и в
некоторых других областях.
Использование термодинамических принципов позволяет
расширить знания о многих процессах, протекающих в морях и
океанах, особенно в глубинных слоях. Термодинамические
методы применяются, например, в Г5-анализе водных масс, при
изучении вертикальной устойчивости вод и их сжимаемости, при
изучении взаимодействия океана и атмосферы и т. д. Однако ни
следует забывать, что большое количество процессов,
происходящих в океане, неравновесны, поэтому неправомерно их изучен
ние методами равновесной термодинамики.
Авторы рассчитывают, что эта книга будет полезна не толькой
с гудентам-океанологам, но и специалистам, занимающимся
исследованиями процессов в океане, водах суши и атмосфере.
11
ГЛАВА 1. СТРУКТУРА ЧИСТОЙ ВОДЫ И ЕЕ СВОЙСТВА
Физические свойства чистой воды и кинетика протекающих в
ней химических процессов определяются молекулярным
строением Н20: специфическим электромагнитным полем,
ориентацией соседних молекул и характером связей между ними.
Следует отметить, что свойства воды изучены недостаточно,
многие явления не имеют строгого описания. Эта ситуация
определяется сложностью построения общей теории жидкости и
аномальностью характеристик самой воды. Свойства воды
описываются в настоящее время с помощью моделей ее строения. Единой
модели, описывающей все свойства этого уникального вещества,
пока не создано. Тем не менее, существующие модели, причем
весьма разные, вполне удовлетворительно описывают основные
термодинамические свойства воды. Но прежде чем рассматривать
основные модели структуры воды, следует остановиться на
строении молекулы Н20.
1.1. Строение молекулы воды
Еще в 1780 г. Кавендиш и Лавуазье установили, что вода
состоит из кислорода и водорода. Через 25 лет Гей-Люссак и
Гумбольдт доказали, что при образовании воды взаимодействуют два
объема водорода и один объем кислорода, а в 1842 г. Дюма
получил весовое соотношение этих элементов в воде как 2:16.
В 1929 г. был открыт стабильный изотоп кислорода 180, а в
1932 г. - дейтерий 2Н, обозначаемый буквой D.
В настоящее время известны три изотопа водорода [!Н, 2Н и
3Н (тритий)] и шесть изотопов кислорода с атомной массой от 14
до 19. Стало очевидным, что естественная вода - смесь
нескольких видов с различным молекулярным весом; при этом
распространенность разных изотопов в молекуле Н20 такова, что
сколько-нибудь заметные концентрации имеют только четыре
возможные комбинации стабильных изотопов: 1Н2160 (99,73% от общей
массы), 1Н2,70 (0,04%), 1Н2180 (0,20%) и ,HD160 (0,03%).
Поскольку приготовление чистой воды с изотопным составом
!Н2|бО - очень трудная процедура, фактически все
экспериментальные исследования проводились именно с природной смесью.
12
Основное внимание мы уделим свойствам самого
распространенного типа природной воды !H2160 с молярной массой
18 грамм1, а молекулы другого изотопного состава (так
называемая «тяжелая вода») будут вкратце рассмотрены в конце этой
главы.
В соответствии с теорией молекулярных орбиталей,
электронное облако молекулы воды имеет вид неправильного тетраэдра
(рис. 1.1). При этом атом кислорода находится в центре, а два
атома водорюда - в углах. Из восьми электронов атома кислорода
первая пара находится на ближайшей к ядру сферической sx-
орбитали и прочно с ним связана, вторая пара электронов
находится на более удаленной от ядра сферической 52-орбитали и
менее прочно с ним связана, третья пара электронов располагается
уже не на сферической, а на круговой р-орбитали.
Рис. LL Электронное облако молекулы воды
Оставшиеся два электрона непарные и могут
взаимодействовать с электронами .Si-орбитали двух атомов водорода, образуя
связи О-Н. За счет объединения орбит электронов вокруг
сблизившихся протонов связь О-Н называется ковалентной.
Полагается, что при таком простом объединении электронов кислорода
и водорода угол между двумя связями О-Н был бы 90°. Однако
Молем любого вещества называется число единиц массы (например,
граммов, килограммов), равное молекулярной массе вещества. Так,
один грамм-моль воды равен 18 грамм. Один моль вещества содержит
одно и то же количество молекул 6,023-1023, названное числом Авогадро.
13
из-за сил отталкивания между ядрами водорода и добавочного
влияния электронов .у2-орбитали {эффект гибридизации)
происходит увеличение этого угла до 104,5°.
Гибридизация электронов s2 и /т-орбиталей кислорода
приводит к тому, что около его ядра образуются две области заряда.
В результате молекула воды приобретает тетраэдрическое
строение с двумя положительно заряженными областями вблизи
ядер водорода и двумя отрицательно заряженными областями
вблизи ядра кислорода.
Характерное для молекулы воды расположение ядер водорода
и кислорода и гибридизированных электронов приводит к тому,
что моменты связей О-Н и моменты ядер создают большой
дипольный момент, равный 1,86 D (D - дебай - единица дипольного
момента, она равна 3,33-10'12 кулон-метр). Остальные электроны
вклада в общий дипольный момент молекулы Н20 не дают из-за
их симметричного положения относительно ядра кислорода.
При рассмотрении параметров молекулы Н20 следует иметь в
виду, что ядра ее атомов постоянно колеблются около положения
равновесия. Поэтому расстояние между ядрами, а следовательно,
и длина связей О-Н и угол между ними, постоянно меняются
(рис. 1.2). В среднем длина связи О-Н составляет 0,96 А (А -
ангстрем, он равен 10"10 м), что несколько меньше, чем у льда
(0,99 А). При колебаниях, как длина связей, так и угол между
ними могут меняться на 5-8%.
Полная энергия молекулы воды определяется как разность
между энергией неподвижной молекулы и ее составных частей,
расположенных на бесконечном удалении друг от друга. Эта
величина при 0°К приблизительно составляет 201 МДжмоль"1.
Средняя энергия электронной связи О-Н в молекуле воды
составляет примерно 461 КДжмоль'1, изменение внутренней
энергии при испарении (100°С) - около 38 КДжмоль"1, при
плавлении льда - 5,9 КДжмоль"1, а тепловая энергия при температуре
0° С - всего 2,1 КДжмоль"1.
Рассмотрим теперь взаимодействие между молекулами воды
во всех трех фазах. Благодаря существованию значительного
дипольного момента, молекулы Н20 взаимно ориентируются
относительно друг друга, испытывая взаимное притяжение или
отталкивание. Уже при межмолекулярном расстоянии 10 А эти силы
14
практически не влияют на взаимодействие молекул. Исходя из
плотности водяного пара, среднее расстояние между молекулами
составляет около 30 А, поэтому 99% молекул остаются
разрозненными и лишь 1% составляют димеры - объединения двух
молекул н2о.
РисЛ.2. Строение молекулы воды и ее дипольный момент
Во льду молекулы Н20, напротив, связаны между собой,
Вследствие образования вокруг атома кислорода четырех
заряженных областей, каждый атом кислорода соединяется с
четырьмя другими, отстоящими от него на среднее расстояние 2,76 А.
образуя тетраэдр. При этом между неподеленной парой
электронов одного атома кислорода и связью О-Н другого возникает
притяжение, называемое водородной связью. Эта связь
сохраняется и в жидкой фазе, где ее прочность невелика - всего 21—25
КДжмоль1 или 5% от энергии связи атомов водорода и
кислорода в молекуле воды. Но при этом она оказывается гораздо
большей, нежели тепловая энергия молекул Н20 (около
2,5 КДжмоль"] при температуре 25° С) и определяет их
ориентацию и подвижность. Именно наличие водородных связей
приводит к аномальности свойств жидкой воды по сравнению с
другими соединениями - ее аналогами.
15
1.2. Структура воды: наблюдения и модели
Этот раздел излагается в соответствии с обобщением
П.А. Стунжаса [35]. Вопрос о структуре воды тесно связан с
вопросом об общих свойствах жидкости. Лаплас еще в конце
XVEI в. предположил, что все свойства жидкости определяются
межмолекулярными силами, которые практически исчезают,
когда расстояние между молекулами превышают некоторое
критическое значение. До сих пор, однако, единая теория жидкости,
как уже упоминалось, не создана. Жидкость принципиально
отличается и от газа, и от кристаллического вещества, хотя имеет
общие свойства с обоими состояниями. Имеющиеся теории берут
за основу либо газ, либо кристалл, либо базируются на
модельных представлениях о строении жидкости.
Так, в «газовой» теории жидкости Ван-дер-Ваальса
предполагалось, что в жидкости, как и в газе, молекулы расположены
хаотически и по удалению друг от друга, и по ориентации. Но
оказалось, что в обычных условиях молекулы в жидкости имеют
отчетливые наиболее и наименее вероятные расположения - так
называемый ближний порядок. В квазикристаллической модели
Френкеля предполагалось, что молекула в жидкости, как и в
кристалле, совершает большое количество колебаний вокруг
положения равновесия, обусловленного полем окружающих молекул.
Преодолев потенциальный барьер, она перемещается в новое
положение равновесия. Однако характер движения молекул в
жидкости более сложен, поэтому необходимо учитывать не только
колебания вокруг положения равновесия, но и дрейф этого
среднего положения вместе с окружающими молекулами. При этом
период колебаний (210"13 с) сравним с интервалом времени
между последовательными смещениями молекул (10"13-10"пс). Но и
при таких условиях в жидкости наблюдается ближний порядок -
среднее число соседних молекул и среднее расстояние от каждой
молекулы до ее ближайших соседей сохраняются постоянными, а
в некоторых случаях сохраняется и их взаимная ориентация. Это
сохраняющееся среднее расположение молекул и называют
структурой жидкости.
Начало современным представлениям о структуре воды было
положено в 1934 г. работой Бернала и Фаулера. В ней было пока-
16
зано, что в воде существует расположение молекул в форме
тетраэдра, которое обусловлено направленными межмолекулярными
связями. Последующими работами по дифракции рентгеновских
лучей эти представления были уточнены. Ближний порядок в
воде сохраняется до расстояний 7-8 А от выбранной молекулы,
среднее расстояние между ближайшими молекулами-соседями
составляет 2,9 А, а расстояния менее 2,5 А между молекулами
невозможны из-за их взаимного отталкивания. При этом тетраэдры
с наиболее вероятными четырьмя ближайшими соседями могут
быть «упакованы» различным образом, и остается неясным, все
ли молекулы энергетически эквивалентны или в воде существуют
локальные области с разной структурой. Более подробные детали
остаются невыясненными из-за неоднозначной интерпретации
экспериментальных данных.
Существующие модели структуры воды условно можно
разделить на три класса: однородные, двухструктурные кластерные
и двухструктурные специфических структур.
В однородных моделях предполагается, что все молекулы Н20
структурно и энергетически эквивалентны, т. е. все участки воды
однородны. В однородной модели воды Попла принимается, что
при плавлении льда не происходит разрывов водородных связей
между молекулами, а только их «растяжение и изгиб». Под
изгибом понимается отклонение атома Н от линии О-О. Такое
предположение основано на том, что изменение внутренней энергии
Н20 при плавлении льда (5,9 КДжмоль1) значительно меньше
такового при парообразовании (37,7 КДжмоль ~\ когда
происходит разрыв всех водородных связей молекулы Н20. В других
моделях однородной структуры воды предполагалось, что может
происходить частичный разрыв водородных связей, тем самым
они приближаются к моделям других классов.
Однородные модели воды наиболее обоснованы физически и
подтверждаются многими экспериментальными данными, в том
числе и рентгенограммами структур, согласно которым вода
представляет непрерывно меняющуюся связанную в тетраэдры
сеть молекул, где вместо шестиугольных колец льда
преобладающими становятся 5- и 7-членные кольца. Однако
количественные расчеты свойств воды и особенно растворов по
однородным моделям затруднены. Можно сказать, что при повышении
17
температуры в однородных моделях изменения свойств воды
происходят из-за увеличения амплитуды колебаний молекул Н20,
в результате чего уменьшается структурированность жидкости.
Другой подход применяется в двухструктурных моделях, где,
начиная с первой работы Бернала и Фаулера 1934 г.,
предполагается, что при изменении температуры одна структура воды
постепенно заменяется другой. В широко известной модели Фрэнка
и Вина предполагается, что происходит кооперирование молекул
воды, связанных водородными связями, в рои или кластеры.
Причиной этого является особенность энергии водородных
связей, когда одна связь укрепляет другую. Молекулы, не попавшие
в кластер, остаются более или менее «свободными» и плотноупа-
кованными. Благодаря тепловому движению кластеры
образуются и распадаются - «мерцают». Время жизни кластера составляет
Ю"10—10"11 с. В каждом из них содержится в зависимости от
температуры от 12 до 150 молекул воды. При комнатной
температуре в кластерах одновременно заключено около трети всех
молекул. Остальные молекулы окружают кластеры.
Кластеры находятся в динамическом равновесии со
свободными мономерными молекулами, они растут за их счет и
распадаются, вновь их образуя. При повышении температуры число
кластеров увеличивается, но размеры их уменьшаются. Хотя
такой подход получил довольно широкое распространение,
поскольку позволяет с хорошей точностью рассчитать многие
свойства воды, включая термодинамические, тем не менее, он
вызывает ряд возражений. Например, мало вероятны большие области
упорядочения структуры в воде, которые не подтверждаются
экспериментально. Для количественного описания результатов в
модель приходится вводить много дополнительных предположений.
В последующих моделях кластеров эти недостатки были
частично устранены, и наметился путь к сближению и перекрыванию
кластерных и однородных моделей.
В двух рассмотренных типах моделей допускается
значительная хаотичность пространственного расположения молекул.
В противоположность этому в моделях специфических
структур принимается существование значительной упорядоченности
молекул, объединенных в тетраэдры водородными связями. В
качестве возможных структур брались структуры газовых гидра-
18
тов, смеси кольцевых структур, смеси различных форм льда.
Чаще всего предполагалось, что в воде сохраняется частично
размытая решетка льда. К таким моделям относится и модель струк-
|уры воды, предложенная в 1946г. О.Я.Самойловым. Автор
предположил, что при плавлении льда его каркас не разрушается
полностью, а только слегка искажается тепловым движением
молекул воды. При этом некоторые молекулы воды приобретают
возможность попасть в полости структуры, имевшиеся во льду.
При таком подходе в жидкой воде уже нельзя выделить
структурно-неоднородные участки, однако молекулы воды, попавшие
в пустоты структуры, энергетически неэквивалентны молекулам,
находящимся в узлах структуры.
Полостные молекулы сохраняют часть водородных связей со
своим окружением и тем самым ослабляют прочность каркаса.
С ростом температуры и давления число полостных молекул
увеличивается. Модель Самойлова можно охарактеризовать как
пространственно-однородную, но энергетически двухструктур-
ную. Эта модель является очень наглядной, легко объясняет
многие аномалии воды, позволяет проводить расчеты некоторых ее
свойств и применима не только к чистой воде, но и к водным
растворам. В модели мало эмпирических параметров, вследствие
чего различные свойства воды оказываются непосредственно
связаны между собой, что позволяет получать одни характеристики
через другие.
Качественно модель Самойлова хорошо согласуется со
многими экспериментальными данными, например, с увеличением
плотности при плавлении льда, наиболее вероятным расстоянием
между соседними молекулами. Для количественного совпадения
тех или иных теоретических и экспериментальных величин ряд
авторов развили эту модель, несколько ее видоизменив (в
частности, принималась разная степень заполнения пустот при одной и
той же температуре). Хотя эта модель является очень
упрощенной, до температур 40-50° С она обеспечивает хорошие
результаты и наиболее часто применяется в океанологии.
Даже из приведенного очень сжатого обзора моделей
структуры воды видно, что одни и те же макросвойства воды могут быть
с определенной точностью описаны теориями, исходящими из
разных моделей структуры воды. Отсутствие единой теории и
19
приемлемость разных предположений создают ситуацию, когда
выбор той или иной модели определяется кругом
рассматриваемых свойств воды и личными интересами исследователя.
L3. Аномальные физические свойства чистой воды
Особенности структуры воды определяют особенности ее
физических свойств, имеющих «аномальный» характер по
сравнению с родственными веществами - гидридами элементов
VI группы (серы, селена, теллура) или прочими жидкостями,
имеющими сферически симметричные молекулы. Большинство
аномальных свойств воды проявляется при низких температурах
и давлениях, характерных как раз для природных условий
гидросферы Земли. При этом сами аномальные свойства сильно
зависят от температуры и давления.
Рассмотрим вначале одно из важнейших свойств -
температуру фазовых переходов.
Точки плавления водородных соединений элементов VI
группы понижаются с уменьшением молекулярного веса от Н2Те (-
60° С) к H2Se и H2S (-85° С). Экстраполируя этот ряд, можно
получить температуру плавления Н20, равную около -95° С
(рис. 1.3). Таким образом, положительная аномалия температуры
замерзания составляет почти 100° С!
Еще более значительна аномалия температуры кипения воды:
продолжая ряд свойств гидридов, можно получить
экстраполированную величину около -65° С, т. е. на 165° С ниже
существующей. Такие особенности фазовых переходов, видимо,
определяются большими дипольными моментами и наличием водородных
связей между молекулами воды: необходимостью
дополнительных затрат тепловой энергии для преодоления электромагнитных
взаимодействий молекул при плавлении или испарении.
Плотность жидкостей, как правило, при нагревании
уменьшается. Для пресной воды при повышении температуры от 0° до
4° С плотность увеличивается, а при дальнейшем нагревании
уменьшается. Это, по-видимому, объясняется перегруппировкой
молекул в данном интервале температур: происходит заполнение
пустот «рыхлой» тетраэдрической структуры. По мере
увеличения теплового движения молекул при температуре выше 4° С
20
увеличивается количество бесструктурных объемов, а плотность
начинает понижаться.
1—« Г
40 80 120
Молекулярная масса, гчиоль1
1
1*0
Рис. 1.3. Изменения температур плавления и кипения
гидридов элементов VI группы в зависимости от
молекулярной массы
Твердые тела, как правило, имеют большую плотность, чем
жидкости, получающиеся при их плавлении. Вода, наряду с
чугуном и висмутом, увеличивает свой объем при замерзании почти
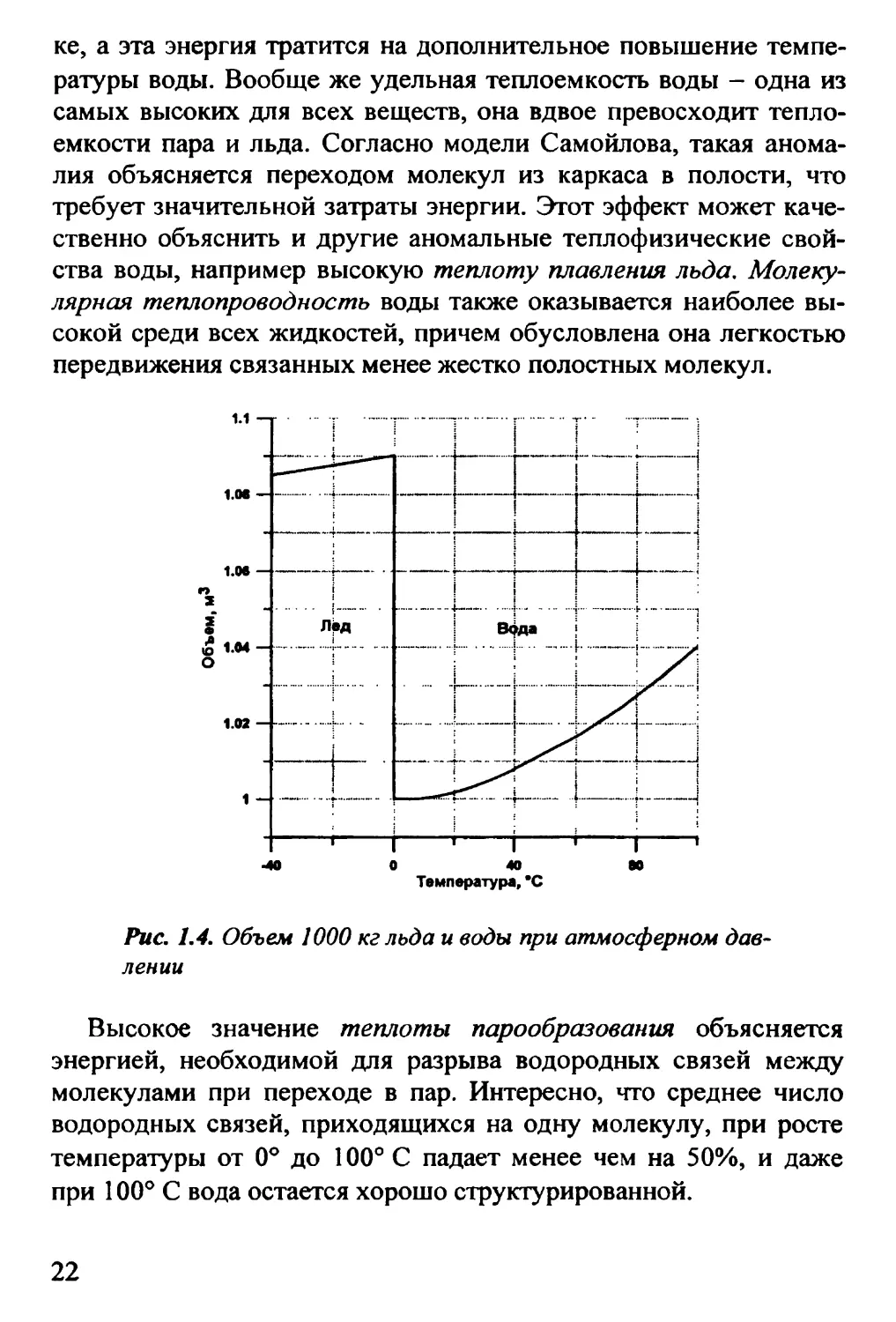
на 10%. На рис. 1.4 представлено изменение объема 1000 кг воды
в твердой и жидкой фазах при изменении температуры от -40° до
100° С. Замерзание воды, как следует из рисунка, увеличивает ее
объем вдвое больше, чем нагревание до 100° С. При этом объем
единицы массы (величина обратная плотности) у льда
увеличивается линейно с ростом температуры, а в жидкой воде эта
зависимость нелинейная и рост объема начинается только в диапазоне
температур выше 4° С.
Удельная теплоемкость воды при нагревании от 0° до 27° С, в
отличие от большинства веществ, уменьшается на 1% и лишь при
более высокой температуре начинает возрастать. Разрушение
структуры молекул Н20 при невысоких температурах
сопровождается высвобождением энергии связи в кристаллической решет-
21
ке, а эта энергия тратится на дополнительное повышение
температуры воды. Вообще же удельная теплоемкость воды - одна из
самых высоких для всех веществ, она вдвое превосходит
теплоемкости пара и льда. Согласно модели Самойлова, такая
аномалия объясняется переходом молекул из каркаса в полости, что
требует значительной затраты энергии. Этот эффект может
качественно объяснить и другие аномальные теплофизические
свойства воды, например высокую теплоту плавления льда.
Молекулярная теплопроводность воды также оказывается наиболее
высокой среди всех жидкостей, причем обусловлена она легкостью
передвижения связанных менее жестко полостных молекул.
1.1 -
1 -
1
л
»д
1
в<
—•—
да
■
-40 0 40 80
Температура, "С
Рис. 1.4. Объем 1000 кг льда и воды при атмосферном
давлении
Высокое значение теплоты парообразования объясняется
энергией, необходимой для разрыва водородных связей между
молекулами при переходе в пар. Интересно, что среднее число
водородных связей, приходящихся на одну молекулу, при росте
температуры от 0° до 100° С падает менее чем на 50%, и даже
при 100° С вода остается хорошо структурированной.
22
Производная молярного объема воды по давлению дает
коэффициент сжимаемости, который аномально низок у воды.
Возрастание давления на воду приводит к заполнению имеющихся
пустот. Если обычно в жидкостях с плотной упаковкой молекул
повышение давления упорядочивает их расположение, то в воде
повышение давления (до определенных пределов) разрушает
структуру.
В нормальных жидкостях с ростом давления уменьшается
подвижность молекул и возрастает вязкость. В воде же при
температурах ниже 20° С вязкость уменьшается с ростом давления.
Основную роль при этом играют те 10-20% молекул, которые
находятся в полостях и обеспечивают основной вклад в
кинетические свойства воды.
Вода обладает также наиболее высоким среди жидкостей (за
исключением ртути) поверхностным натяжением, это свойство
определяет особенности формирования поверхностного
микрослоя в природных водоемах и протекание многих биологических
процессов.
Несмотря на наличие у молекул Н20 дипольного момента, их
взаимная связь в твердой и жидкой фазе приводит к тому, что под
воздействием внешнего переменного электрического поля
полной переориентации диполей не происходит. Поэтому у воды
очень высок коэффициент диэлектрической проницаемости,
показывающий отношение электрической индукции к
напряженности электрического поля. Два электрических заряда в воде
взаимодействуют в 80-88 раз слабее, чем в вакууме.
С особенностями проявления аномальных свойств природной
морской воды, а также со значениями ее термодинамических
параметров мы познакомимся в последующих главах. Ряд
характеристик (гидрооптические, акустические, свойства льдов)
рассматриваются в других разделах курса «Океанология».
В заключение этого параграфа приведем с небольшими
сокращениями таблицу (табл. 1.1) описанных нами аномальных
свойств морской воды и их роли в физических и биологических
явлениях.
23
Таблица 1.1
Аномальные физические свойства жидкой воды (по [86])
Свойства
Теплоемкость
Скрытая
теплота
плавления
Скрытая
теплота
испарения
Тепловое
расширение
Поверхностное
натяжение
Растворяющая
способность
Диэлектрическая
постоянная
Теплопроводность
Сравнение с другими веще-
ствами
Наиболее высокая среди
всех твердых и жидких
веществ, за исключением NH3
Наиболее высокая из всех
веществ, за исключением
NH3
Наиболее высокая из всех
веществ
Температура максимальной
плотности уменьшается с
повышением солености; для
чистой воды она равна 4° С
Наиболее высокое из всех
жидкостей
Как правило, растворяет
большинство веществ и в
больших количествах, чем
другие жидкости
Для чистой воды наиболее
высокая из всех жидкостей
Наиболее высокая из всех
жидкостей
Роль в физических и
биологических явлениях
Уменьшает пределы
температурных колебаний; перенос
тепла течениями очень велик;
способствует сохранению
постоянной температуры тела.
Термостатирующий эффект в
точке замерзания,
обусловленный поглощением или
выделением скрытой теплоты.
Высокое значение скрытой
теплоты испарения крайне
важно для теплового и водного
переноса в атмосфере.
Для чистой и разбавленной
морской воды максимум
плотности наблюдается при более
высокой температуре, чем
температура замерзания; это
свойство играет важную роль в
регулировании распределения
температуры и в вертикальной
зональности озер.
Важно для физиологии клетки,
определяет некоторые
поверхностные явления, образование
и свойства капли.
Явно связывает между собой
физические и биологические
явления.
Имеет наибольшее значение
для поведения неорганических
растворенных веществ, так как
определяет их диссоциацию.
Основную роль играет в
процессах малого масштаба,
например в тех, которые
происходят в живых клетках; для
молекулярных процессов
оказывается гораздо важнее, чем
[ турбулентный перенос тепла.
24
L4. Свойства тяжелой воды
«Тяжелая вода» - вещество, молекулы которого состоят из
более тяжелых (имеющих большую атомную массу из-за допол-'
нительного количества нейтронов в ядре), нежели 'Ни 1бО,
изотопов кислорода и водорода.
В параграфе 1.1 уже упоминалось, что доля «тяжелой воды» в
природных условиях составляет менее 0,3%. Однако, несмотря на»
столь низкую концентрацию, относительная доля изотопов
,80/160 и D/]H в последние годы стала одним из важнейших
океанологических и гляциологических параметров, помогающих
проследить условия формирования вод и льдов.
Разное число нейтронов в ядре изотопов кислорода и водорода
приводят к различиям в физических свойствах. Для иллюстрации
этих особенностей в таблице 1.2 приводятся примеры различий в
физических свойствах изотопных разновидностей воды.
Таблица 1.2
Основные физические свойства изотопных разновидностей воды при
атмосферном давлении
Характеристика
Плотность при 20° С, кг/м3
Г Температура наибольшей плотности, °С
Температура плавления, °С
Температура кипения, °С
Н2160
997,90
3,98
0,00
100,00
D2,60
1105,10
11,24
3,81
101,42
н2[8о 1
1110,60
4,30
0,28
100,14 J
Главный механизм изменения соотношения изотопных
компонентов (фракционирования изотопов) воды — процессы
фазового перехода и прежде всего — испарение и конденсация. За счет
различия у пру гостей (давлений) паров изотопных составляющих
воды с открытой поверхности водоема в первую очередь
улетучиваются молекулы с меньшей массой. Таким образом, пар обо-,
гащается изотопами !Н и 1бО, а вода верхнего слоя - тяжелыми
изотопами ,80 и D.
При 20° С в случае равновесия пара с водой пар на 0,9%
беднее изотопом 180 и на 8,4% беднее дейтерием, при 0° С это
25
уменьшение относительной концентрации составляет
соответственно 1,2% и 11,1%.
В процессе многократного испарения и конденсации водяного
пара в атмосфере изотопное фракционирование приводит к
последовательному его обеднению изотопами тяжелой воды.
Исследования показали, что чем ниже температура воздуха и,
соответственно, упругость водяного пара, тем меньшей оказывается
концентрация 180 и дейтерия в атмосферных осадках (над
полюсами на - 3-6% ниже средних значений). Таким образом,
приповерхностный слой водоемов в высоких широтах за счет
выпадения осадков и таяния ледников оказывается также обедненным
«тяжелой водой».
Подобное фракционирование, но в меньшей степени,
происходит и при замерзании: при 0° С лед имеет более высокую
относительную концентрацию 180 по сравнению с водой - на 0,35%,
для дейтерия это обогащение достигает 2,1%.
Задачи и вопросы для повторения
1.1. Чем отличается ковалентная связь от водородной?
1.2. Чем обусловлен большой дипольный момент молекулы воды?
1.3. Какая из энергий в жидкой фазе больше: тепловая энергия
молекулы воды или энергия водородной связи?
1.4. В чем сущность эффекта гибридизации?
1.5. В каком фазовом состоянии расстояние между молекулами
больше: в водяном паре или во льду?
1.6. Что такое «тяжелая вода»?
1.7. Что понимается под фракционированием изотопов?
1.8. Какие модели структуры воды являются энергетически двух-
структурными?
1.9. Чем объясняются аномальные температуры плавления и
кипения воды по сравнению с родственными веществами - гидридами
элементов VI группы?
1.10. Как модель Самойлова объясняет очень высокую удельную
теплоемкость воды?
26
ГЛАВА 2. ИССЛЕДОВАНИЕ ФИЗИЧЕСКИХ СВОЙСТВ
ТЕРМОДИНАМИЧЕСКИХ СИСТЕМ
МЕТОДОМ ПОТЕНЦИАЛОВ
Для исследования физических свойств термодинамических
систем, находящихся в тепловом равновесии или переходящих в
равновесное состояние, используются два взаимно дополняющих
друг друга метода - статистический и термодинамический [5].
Термодинамический метод не связан с какими-либо
представлениями о молекулярном строении вещества и характере
движения образующих его частиц, т. е. имеет феноменологический
характер. Раздел физики, оперирующий этим методом, называется
термодинамикой. Термодинамика изучает макроскопические
характеристики объектов, основываясь на нескольких эксперимен-
1вльно установленных положениях - трех началах (законах)
термодинамики, обладающих весьма большой общностью.
В то же время феноменологический характер термодинамики не'
позволяет вскрыть природу исследуемых явлений. В связи с этим
параллельно с развитием термодинамики формировалась и моле-
кулярно-кинетическая теория свойств физических систем -
статистическая физика. Для более полного изучения статистических
закономерностей большого числа непрерывно движущихся и
взаимодействующих частиц необходимо применение статистических,
методов.
Тем не менее, для решения многих практических задач
достаточно применение методов термодинамики, что позволяет не прибегать
к представлениям о молекулярном строении изучаемых тел.
2.1. Определение термодинамических систем
Совокупность макроскопических объектов (тел и полей),
обменивающихся энергией в форме работы или в форме теплоты как
друг с другом, так и с внешней средой2, и находящихся в
состоянии равновесия, называется термодинамической системой.
Энергией системы называется совокупная энергия большого
числа непрерывно движущихся и взаимодействующих частиц, из
2 Под внешней средой подразумеваются внешние по отношению к
системе тела и поля.
27
которых она состоит. Полная энергия системы разделяется на
внешнюю и внутреннюю. Внешняя энергия - это часть
энергии, состоящая из потенциальной энергии системы в поле
внешних сил и энергии движения системы как целого. Внутренняя
энергия системы состоит из энергии поступательного и
вращательного движений молекул, колебательного движения атомов,
энергии молекулярного взаимодействия, внутриядерной энергии
и т. д. В термодинамике рассматривается только внутренняя энергия
системы.
Обмен энергией между системой и внешними телами
происходит двумя различными способами: с изменением внешних
параметров системы и без их изменения. Первый способ передачи
энергии называемся работой. При втором способе, названном
теплотой, при неизменности внешних параметров изменяется энтропия
системы (более подробно об энтропии речь пойдет ниже). Сам
процесс передачи энергии при втором способе называется
теплообменом. Энергия, переданная системой при первом способе,
называется/?ябо/иой W, а при втором способе - количеством теплоты Q.
Отметим, что работа W может непосредственно идти на увеличение
любого вида энергии, количество же теплоты Q непосредственно,
без предварительного преобразования в работу, может
увеличивать только внутреннюю энергию системы. Работа считается
положительной, когда она совершается системой над внешними
силами. Количество теплоты положительно, если энергия передается
системе без изменения ее внешних параметров.
При изменении п внешних параметров элементарная работа
системы равна
bW^Afda,, (2.1)
i
где At - сила, соответствующая внешнему параметру я, и
являющаяся при равновесии функцией внешних параметров и
температуры.
При квазистатическом расширении системы (увеличении
объема) под действием всестороннего равномерного давления
элементарная работа равна
bW = p dV (A = p,a = V), (2.2)
где/? - давление жидкости, dV - увеличение ее объема.
Отметим, что выражение (2.1) не является полным
дифференциалом какой-либо функции параметров состояния системы, так
28
как, согласно определению работы, в (2.1) не входит дифференциал
температуры, поэтому элементарную работу обозначают SW9 а не
dW.
Система, не обменивающаяся с внешней средой ни энергией, ни
веществом, называется изолированной (замкнутой)^ в
противном случае - открытой. Система, в которой происходит обмен
шергией с другими телами, но отсутствует обмен веществом,
называется закрытой, а в случае отсутствия обмена энергией только в
форме теплоты - адиабатически изолированной (адиабатической)
системой. В природе практически все системы - открытые, в том
числе Мировой океан или отдельные его бассейны. С большой на-
гожкой закрыто^ системой можно считать планету Земля, если из
всех видов взаимодействий на внешней границе атмосферы
рассматривать лишь обмен энергией.
Все термодинамические системы разделяются на два класса: го-
могенные и гетерогенные. В гомогенных системах свойства
изменяются непрерывно при переходе от одной точки к другой.
Гомогенными системами являются, например, смеси газов, жидкие
и твердые растворы. Если для всех равных по объему
макроскопических частей гомогенной системы состав и физические
свойства одинаковы, то такая система называется физически
однородной.
Гетерогенные системы - это системы, состоящие из нескольких
физически однородных (гомогенных) тел и, следовательно,
имеющие поверхности раздела, на которых наблюдаются разрывы
непрерывности свойств этих тел. Примерами гетерогенных систем
служат тающий лед и влажный пар.
Совокупность всех гомогенных частей гетерогенной
термодинамической системы, которые в отсутствии внешнего силового
воздействия являются физически однородными, называется фазог?.
Если система состоит из жидкости и пара, то жидкость представляет
собой одну фазу, пар - другую.
А|
} Надо помнить, что агрегатные состояния и фаза не тождественны. Если
агрегатных состояний всего четыре (твердое, жидкое, газообразное и
плазменное), то фаз - неограниченное число. Например, в жидком
состоянии в равновесии может находиться несколько фаз: морская вода и масло,
керосин и морская вода и др.
29
При исследованиях равновесия термодинамических систем
большое значение имеет понятие компонента. Компонентами
называются различные вещества, наименьшее число которых
достаточно для образования всех фаз систем. Есть и другое
определение компонента - это часть системы, содержание которой не
зависит от содержания других частей. Смесь химически не
реагирующих газов является однофазной, но многокомпонентной системой;
компонентов в смеси столько, сколько в ней различных газов.
Например, смесь газов водорода и кислорода является однофазной,
двухкомпонентной системой. Вода и лед - также однофазные
системы, но однокомпонентные, так как образующие их водород и
кислород имеют определенную зависимость друг от друга. В общем
случае, если в фазе N различных веществ, между которыми
происходит п химических реакций, то число компонентов равно N-n.
Система, состоящая из двух компонентов, называется бинарной или
двойной. Гомогенная система (твердая, жидкая или газообразная)
из двух или большего числа химически чистых веществ
представляет собой раствор; один из компонентов, масса которого
больше, называется растворителем, а остальные компоненты - рас-
творенными веществами.
Если растворяющееся вещество дробится на отдельные
молекулы (молекулярная дисперсность вещества), то образуется
молекулярный раствор. В ионных растворах происходил* распад
растворенных веществ на ионы. Кроме того, существуют коллоидные
растворы, представляющие собой взвешенные в растворителе
частицы вещества.
Состояние термодинамической системы определяется
совокупностью всех физических величин, характеризующих
макроскопические свойства системы. Эти величины называются
термодинамическими параметрами. К их числу относятся,
например, плотность, объем, упругость, концентрация, поляризо-
ванность, намагниченность и т. д. Термодинамические параметры
разделяются на внешние и внутренние.
Внешние параметры я, (i=l,...,n) - это параметры, зависящие
только от обобщенных координат внешних тел, с которыми
взаимодействует система. Примерами внешних параметров служат
объем системы (так как объем зависит, например, от формы стенок
30
сосуда), напряженность силового поля (зависимость от положения
источников поля - зарядов и токов, не входящих в систему).
Внутренними параметрами bj (/=1,...,т) называются величины,
определяемые как совокупным движением и распределением в
пространстве входящих в систему частиц, так и значениями внешних
параметров (в связи с тем, что пространственное расположение
а юмов и молекул зависит от обобщенных координат4 внешних
сил). К внутренним параметрам относятся плотность, давление,
►нергия и др.
Внутренние параметры, не зависящие от массы, относятся к
интенсивным параметрам. Таковы, например, давление,
температура, вязкость ц др. Отметим, что интенсивные параметры имеют
определенные значения в каждой точке системы. Параметры,
пропорциональные массе, называются экстенсивными (энергия,
энтропия, энтальпия и др.). Экстенсивные параметры характеризуют
систему как целое.
Параметры, не зависящие от предыстории системы и полно-
сгью определяемые ее состоянием в начальный и конечный
моменты времени, называются функциями состояния. Если параметры
системы с течением времени не изменяются, то состояние системы
инляется стационарным. Стационарное состояние системы
называется равновесным, если его неизменность во времени не
обусловлена протеканием какого-либо внешнего по отношению к
системе процесса. Например, если теплосодержание (энтальпия) водоема
остается постоянным только за счет дополнительного притока тепла
извне (из атмосферы или благодаря теплому течению через пролив),
го такой бассейн нельзя считать равновесной системой.
Всякое изменение в термодинамической системе означает, что^
н ней пропгекает термодинамический процесс. Процесс перехода
системы из неравновесного состояния в равновесное называется
релаксацией. Если все параметры системы изменяются физически
бесконечно медленно, то система все время находится в равновес-
4 Обобщенными координатами тел называются независимые
параметры, полностью определяющие положение всех точек тела по отношению к
системе отсчета.
> 31
ных состояниях5. Такой термодинамический процесс называется
равновесным (квазистатическим). Промежуток времени, в
течение которого отклонение какого-либо параметра системы
уменьшается в е раз (е~2,71) от его равновесного состояния,
называется временем релаксации.
Термодинамические процессы, протекающие при постоянном
значении какого-либо параметра состояния, называются:
• изохорными - при постоянном объеме системы;
• изобарическими - при постоянном давлении;
• изотермическими - при постоянной температуре.
Процессы, протекающие в системе без теплообмена с внешними
телами, называются адиабатическими.
2.2. Краткие сведения об исходных положениях и началах
термодинамики
В результате обобщения опытов в классической термодинамике
постулируется, что изолированная макроскопическая система с
течением времени приходит в состояние термодинамического
равновесия и никогда самопроизвольно выйти из него не может.
Это - первое (основное) исходное положение термодинамики.
В статистической физике, где явно учитывается движение частиц,
под термодинамическим равновесием подразумевается наиболее
вероятное состояние, которое чаще всего создается непрерывно
движущимися частицами и в которое с течением времени
переходит изолированная система. Отсюда следует, что классическая
термодинамика не рассматривает системы, для которых
равновесное состояние невозможно и в которых наблюдаются большие
самопроизвольные отклонения системы от равновесного
состояния. Отмечено, что самопроизвольные отклонения
термодинамической системы от равновесия при прочих равных условиях тем
меньше, чем больше частиц в системе.
Существование температуры как особой функции состояния
равновесной системы представляет второе исходное
положение термодинамики. Температура является интенсивным пара-
Физически бесконечно медленным изменением какого-либо параметра
называют его изменение со скоростью значительно меньшей, чем
средняя скорость изменения этого параметра при релаксации.
32
метром и служит мерой интенсивности теплового движения час-
шц системы.
Второе исходное положение термодинамики можно
сформулировать и по-другому. Ранее было показано, что
равновесное состояние термодинамической системы характеризуется внени
ними и внутренними параметрами, причем последние зависят oif
положения и движения частиц системы и значений внешних
параметров. Согласно же второму постулату, состояние
термодинамического равновесия определяется совокупностью внешних
параметров и температурой. Следовательно, внутренние параметры хотя и
характеризуют состояние системы, но не являются ее независим
мыми параметрами. Таким образом, все равновесные внутренние
параметры системы являются функциями внешних параметров и?>
гсмпературы (еще одно определение второго исходного
положения). Если мы выразим температуру системы через ее энергию, то
получим, что при термодинамическом равновесии все внутренние
параметры системы являются функциями внешних параметров itf
энергии.
Из второго исходного положения термодинамики следует
существование уравнений состояния системы, связывающих
температуру Г, внешние параметры aj...a„ и какой-либо
равновесный внутренний параметр 6*:
Ьк=Ьк{ах,...ап;Т). (2.3)»
Если в качестве 6* взять внутреннюю энергию U, то уравнение
U = U(a],...,an;T) (2.4)
называется калорическим уравнением состояния. С помощью
уравнения (2.4) вычисляются теплоемкости и другие аналогичные
величины. Уравнение (2.4) позволяет определить внутреннюю
энергию С/(а,,..., ал; Г) в состоянии (а,,...,ап;Т) только с
точностью до аддитивной постоянной, зависящей от выбора начального
состояния.
Если вместо bk взять сопряженную внешнему параметру а,
обобщенную силу Л(, то уравнения
А(=А((а19...ап;Т), iW,...,#i (2.5)
называются термическими уравнениями состояния. С их
помощью можно определить температуру.
33
Общее число термических и калорического уравнений состояния
системы равно числу независимых параметров,
характеризующих состояние системы. Это число обычно называется числом
степеней свободы.
Если уравнения (2.4) и (2.5) известны, то с помощью начал
термодинамики можно определить все термодинамические свойства
системы. Сами же уравнения состояния на основе термодинамики
вывести нельзя. Они находятся эмпирическим путем или
методами статистической физики.
В процессе экспериментальных и теоретических исследований в
физике и химии был открыт закон сохранения и превращения
энергии. Применительно к термодинамическим системам
первое начало термодинамики является математическим
выражением количественной стороны этого закона. Согласно первому
началу термодинамики, внутренняя энергия системы -
однозначная функция ее состояния, которая изменяется только под
влиянием внешних воздействий. Рассматриваются два типа
воздействий: связанные с изменением внешних параметров
(система совершает работу W) и обусловленные изменением
внутренних параметров или температуры (система обменивается с
внешней средой количеством теплоты Q). Из первого начала
следует, что изменение внутренней энергии системы U2-Uj при
ее переходе из первого состояния во второе под влиянием этих
воздействий равно U2'Ui=Q-fV. Для элементарного процесса
уравнение первого начала выглядит так:
5Q = dU + 6W. (2.6)
Согласно первому началу, изменение внутренней энергии при
элементарном процессе есть полный дифференциал, т. е.
конечное изменение ее не зависит от пути перехода системы из
первого состояния во второе, бесконечно близкое. Работа же W
и количество теплоты Q в отличие от U зависят от пути
перехода. То, что элементарная работа bW не является полным
дифференциалом, было доказано ранее. Доказательство же того, что
дифференциальное выражение для 8Q не есть полный
дифференциал, следует непосредственно из (2.6).
Из уравнения (2.6) видно, что работа может совершаться
или за счет изменения внутренней энергии, или за счет
сообщения количества теплоты.
34
С помощью первого начала термодинамики вычисляются,
например, такие важные свойства системы, как теплоемкость, ее
швисимость от модулей упругости, а также теплота
изотермического изменения внешних параметров.
Второе начало термодинамики более трудно для понимания, чем
первое, из-за более строгого требования к определению границ
системы. Оно, так же как и первое начало, является обобщением
данных экспериментов, и его открытие связано с анализом работы
тепловых машин сначала С. Карно, а позднее - Р. Клаузиусом и
В. Томсоном. Изучение закономерностей превращения теплоты в
работу и работы в теплоту привело к открытию у всякой
равновесной системы еще одной функции состояния - энтропии, которая, в
отличие от внутренней энергии, не изменяется у изолированной
системы при равновесных процессах и всегда возрастает при
неравновесных.
Выше уже упоминалось, что два способа передачи энергии (в
виде теплоты и работы) неравноценны. Если работа W может
непосредственно идти на увеличение любого вида энергии, то
сообщение системе количества теплоты Q, без предварительного
превращения ее в работу, приводит лишь к увеличению внутренней
жергии системы.
При этом при превращении работы в теплоту изменяется
термодинамическое состояние одного лишь теплополучающего тела,
а при преобразовании теплоты в работу наряду с охлаждением
геплоотдающего тела происходит изменение
термодинамического состояния других тел, участвующих в этом процессе. Изменение
состояния рабочего тела при незамкнутом процессе или отдача части
гепла рабочим телом другим телам и изменение их
термодинамического состояния при циклическом процессе называется
компенсацией. Опыты показывают, что без компенсации
преобразовать теплоту в работу невозможно. Работа же в теплоту
превращается полностью без всякой компенсации
Отсюда следует, что если при циклическом процессе происходит
преобразование теплоты в работу с отдачей положительного количе-
ежа тепла Q и совершением положительной работы W, то всегда
{> JV6. Если же работа W превращается в теплоту Q, то W^Q.
А Стрелка указывает на направление протекающего процесса.
35
Эти два независимых утверждения
Q^Vu W = Q (2.7)
представляют одну из формулировок второго начала
термодинамики.
Согласно второму началу, процессы в природе, при которых
превращение теплоты в работу связано с компенсацией, могут
происходить, а аналогичные процессы, не сопровождающиеся
компенсацией, не могут. Это обуславливает разделение всех
возможных в замкнутой системе процессов на обратимые и
необратимые. Процесс перехода системы из одного состояния в другое
называется обратимым, если возвращение этой системы из
конечного состояния в исходное можно осуществить без каких бы то ни
было изменений во внешней среде. Все процессы, не
удовлетворяющие этому условию, называются необратимыми.
Всякий равновесный процесс является обратимым, так как при
нем состояние системы в любое время полностью определяется
внешним параметрами и температурой, и поэтому, при изменениях
этих параметров в обратном порядке система также придет в
начальное состояние, не вызвав изменения во внешней среде.
Обратимые процессы в реальном океане крайне редки.
К ним могут бьпъ отнесены:
- медленное адиабатическое обтекание некоторым объемом воды
возвышенности на дне при сохранении потенциальной
температуры (определение будет дано позднее) и солености в начальной и
конечной точках;
- распространение волн в толще воды при отсутствии их
поглощения;
- процессы плавления и замерзания в смеси воды и льда при
273,15° К (двухфазная система при этом находится в равновесии);
- процесс нагревания воды (или ее охлаждения).
Примерами необратимых процессов могут служить процессы с
трением, теплопроводность за счет разности температур, передача
энергии посредством излучения, смешение водных масс с разной
температурой. В этих процессах при переходе системы в конечное
состояние работа может бьпъ превращена в теплоту без компенсации, а
в обратном направлении переход системы связан с переходом теплоты
в работу, что невозможно осуществить без изменения во внешней ере-
36
де. Отметим, что всякий необратимый процесс является
неравновесным.
Остановимся теперь на физическом смысле энтропии.
Согласно первому началу термодинамики, элементарная работа 50 не
яиляется полным дифференциалом, т. е. количество теплоты,
необходимое при переходе из начального состояния в конечное,
швисит от условий перехода (от пути).
Пусть этот переход может быть осуществлен по пути I или П.
Тогда количество теплоты, затраченное при переходе из первого
состояния во второе, равно
Q, = \bQ, Qa-fbQ,
I И
причем в соответствии с первым началом Qf Ф Qn. Но если
количество теплоты разделить на температуру Т, то, в соответствии с
многочисленными опытами, интегралы приведенной теплоты для
обоих путей перехода будут одинаковыми:
Jf-Jf- *»
/ •* II L
Выявленная независимость от пути означает, что величина
-У должна быть полным дифференциалом некоторой величины,
представляющей собой функцию состояния системы.
Функция состояния (термодинамический параметр) т|, полный
дифференциал которой равен
*1«^. (2-9)
называется энтропией (от греческого слова entropia - поворот,
превращение). Впервые это понятие ввел в термодинамику Р. Клау-
1иус, причем он обозначил энтропию через S. Однако, учитывая,
что в океанологии этот символ означает соленость, мы для
обозначения энтропии выбрали tj. Важно отметать, чгго значение 5Q
сопоставляется с температурой Г системы в целом.
На основе многовековых опытов установлено, что в любой
изолированной системе процессы выравнивания температуры,
давления и концентрации, а также процессы преобразования
кинетической, потенциальной, электрической, химической и
ядерной энергии во внутреннюю приводят к возрастанию энтропии
37
системы. Иначе говоря, любой спонтанный (самопроизвольный)
процесс в любой изолированной системе всегда приводит к
росту энтропии этой системы (еще одна из формулировок
второго начала)7.
Если увеличение энтропии в некоторой системе невозможно,
то в такой системе не может происходить никаких
самопроизвольных изменений. Энтропию системы можно уменьшить в
результате соответствующего обмена теплом с окружающей
средой, в то же время путем совершения работы это осуществить
невозможно. В общем случае энтропия системы может
производиться внутри системы за счет необратимых процессов или
поступать в систему в результате теплового взаимодействия с
внешними телами. Энтропия может переходить от системы к
внешним телам или наоборот, но ее количество при этом
никогда не уменьшается. Отметим, что для равновесных циклических
процессов выполняется равенство Клаузиуса
jf~0. (2.10)
Анализ процессов, происходящих в изолированных системах,
показывает, что отмеченное ранее возрастание энтропии,
сопровождающее превращение различных форм энергии во
внутреннюю, следует рассматривать как необратимую
потерю способности системы к совершению работы [36]. Так,
например, в процессе выравнивания концентрации растворенной
в воде соли могла бы совершаться работа. Однако в
изолированной системе при свободной диффузии не совершается
никакой работы, так что соответствующий потенциальный запас
работы теряется навсегда.
Более глубокий смысл энтропии основан на рассмотрении
молекулярной структуры системы. Из статистической физики
известно, что энтропия системы в данном состоянии характеризует
вероятность этого состояния:
4 = -*-I>i-lnft. (2-11)
1=1
В открытой системе при соответствующих условиях может
происходить и понижение энтропии.
38
где N - количество возможных микросостояний, к - постоянная
Ьольцмана,/7,- вероятность /-го состояния.
Вели N=ly то/7=1, а энтропия равна 0. В других случаях
П\ zp2='-.=Pir*VN9откуда
Л = *-1пЛГ. (2.12)
Возрастание энтропии в результате спонтанного процесса
соответствует переходу из состояния с меньшей вероятностью в
остояние с более высокой вероятностью. С этим связано пред-
гавление о том, что энтропия возрастает по мере увеличения
v. гепени беспорядка (хаоса) в системе.
Л. Больцман первым установил соотношение (2.12), причем
число микросостояний N он назвал термодинамической
вероятностью состояния. Эта формула выгравирована на кладбище в
Вене на его памятнике8.
Обобщая все вышесказанное, отметим, что второе начало
термодинамики определяет существование энтропии у всякой
равновесной системы и ее неубывание при любых процессах в
цитированных и адиабатических изолированных системах [5].
Подставив (2.9) в уравнение первого начала термодинамики
16) с учетом (2.1), получим основное уравнение термодинамики для
I >«щновесных процессов
T-d^^dU + J^Ardat, (2.13)
i
выраженное исключительно через параметры системы. Все
дифференциалы (энтропии, внутренней энергии, внешних
параметров), входящие в (2.13), являются полными.
Для простой системы, находящейся под всесторонним (одинако-
имм в каждой точке внешней границы) давлением р, уравнение
(2.13), с учетом (2.2), примет вид:
Tdi] = dU + p dV. (2.14)
Энтропия тела ц может быть вычислена только с точностью до
* сгоянного слагаемого:
т
П=|^ + Ло. (2.15)
о *
Отметим, что термодинамическую вероятность Больцман обозначал
буквой W, а не N
39
где интегрирование по температуре от 0°до 7° К производится
вдоль произвольного обратимого процесса, а т|0 - значение
энтропии тела при Г=0° К, которое не может бьпъ определено с
помощью первого и второго законов термодинамики.
2.3. Метод термодинамических потенциалов
Термодинамические исследования физических явлений могут
быть осуществлены при помощи двух методов: метода циклов и
метода термодинамических потенциалов. Первым возник метод
циклов или круговых процессов. Его использовали в своих
исследованиях еще Карно, Клаузиус и др. Суть этого метода
заключается в том, что для установления какой-либо закономерности
определенного явления подбирается подходящий обратимый цикл и к
этому циклу применяются уравнения первого и второго начал
термодинамики. Чаще всего используется цикл Карно. Слабостью
метода циклов является то, что всякий раз приходится подбирать
подходящий цикл, а сам его выбор произволен [5].
Сейчас при исследованиях термодинамических свойств систем
применяется, главным образом, метод термодинамических
потенциалов, предложенный Дж. Гиббсом. Основу метода
составляет определение с помощью основного уравнения термодинамики
(2.13) некоторых функций состояния в зависимости от выбора
независимых переменных. Посредством этих функций и их
производных могут быть явно выражены термодинамические
свойства системы. При этом первые производные определяют
термические свойства, а вторые - калорические.
Термодинамическими потенг^иалами называют такие функции
от соответствующих параметров, определяющих состояние
системы, с помощью которых можно вычислить полную работу (за
вычетом работы против внешнего давления) в равновесном процессе
при заданных условиях. Какая именно функция будет играть роль
термодинамического потенциала, зависит от условий, при которых
находится рассматриваемая система.
Пусть мы хотим определить полезную работу 8f^=5W-p-dV
(работу, за вычетом работы расширения). Тогда, согласно (2.1) и (2.13),
5W* =-dU + Tdr]-p-dV. (2.16)
40
Зададимся вопросом, при каких условиях левая часть уравнения
будет полным дифференциалом.
Здесь возможны несколько комбинаций, но чаще всего в
термодинамике используются четыре из них:
1. Переменные объем V и энтропия ц
При заданных V и ц из (2.16) сразу вытекает, что bif^=-dU. Это
означает, что при изохорно-адиабатических процессах работа со-
исршается за счет убыли внутренней энергии. Отсюда следует, что
при постоянных Уи т\ внутренняя энергия является
термодинамическим потенциалом U=U(W,r]).
Полный дифференциал внутренней энергии равен
dU =
dU
dn
, dU,
■ dr\ +
■dV
Сравнивая (2.17) и (2.14), получим
T =
dU
дц
Р =
ди
' dV
(2.17)
(2.18)
Продифференцируем первое выражение в (2.18) по V, а второе
нот]. Тогда
д2Ц
дцдУ
дТ
dV
d2U
дУдц
др
дц
Приравнивая полученные смешанные производные, получим:
(2.19)
дТ\ =_др_\
Отсюда следует, что изменение температуры при адиабатическом
расширении равно изменению давления при изохорном сообщении
теплоты системе с обратным знаком. Установление связей типа (2.19) -
основа метода термодинамических потенциалов.
Функция состояния U=U(V,r\) как термодинамический потенциал
мало пригодна в практических исследованиях, так как одна из
независимых переменных, энтропия т|, не может бьпъ непосредственно
измерена
2. Переменные температура Т и объем V
Преобразуем правую часть уравнения (2.16) таким образом,
чтобы в него входили дифференциалы dTv\ dV.
41
Для этого в правую часть (2.16) прибавим и из нее же вычтем
дифференциал d(Tr\) (последний раскроем)9:
-dU + Tdr\-p-dV + d(T-r\)-T dr\-\) dT.
Тогда уравнение (2.16) примет вид:
&W*=-d(U-T-r))-i\-dT-p-dV. (2.20)
При T-const и V=const
6IV*=-d(U~Tr])y (2.21)
т. е. выражение F{r, V) = U - Т • т| при заданных температуре и
объеме является термодинамическим потенциалом и называется
свободной энергией или энергией Гельмголъца.
Если 5^=0 (равновесная система находится только под
всесторонним давлением), то из (2.20) получим:
dF = -r\-dT-p-dV. (2.22)
Из (2.22) следует (вспомним определение полного
дифференциала):
3F
77=
дт
р ак1
(2.24)
(2.23)
г
d2F
Вторые производные от функции F вида позволяют
получить уравнение:
дх\\ _ др\
Из (2.24) следует, что изменение энтропии при ее изотермическом
расширении равно изменению давления при изохорном нагревании.
Свободная энергия при изотермических процессах играет роль
внутренней энергии при адиабатических процессах. Как видно из
(2.21), при изотермических процессах работа совершается
системой не за счет убыли внутренней энергии U, а за счет убыли
функции F. Величина Тг\ называется связанной энергией.
Связанная энергия - это часть внутренней энергии системы, которая не
может быть передана в форме работы в изотермическом процессе,
т. е. она является «обесцененной».
9 Подобные преобразования называются преобразованиями Лежандра.
42
3. Переменные температура Т и давление р
Для перехода к переменным Т и р из правой части выражения
\ 16) вычтем и к ней же прибавим дифференциал d(p-V) (последний
раскроем), а член Tdr\ перепишем как Т • dx\ = d(T • т|) - ц • с/Г. Тогда
6^=-€/(£/-Г-л + р -V)-T\-dT+V-dp. (2.25)
При заданных Г и р получим:
Отсюда следует, что при изотермо-изобарических процессах ра-
' <гга совершается не за счет убыли внутренней энергии С/, а за счет
1>ыли функции G.
<1>ункция состояния G(f9p) = U-T-r\ + p -V называется тер-
*их)инамическим потенциалом Гиббса или энергией Гиббса. Эта
Функция имеет важное значение, так как она определяется через лег-
<> измеряемые параметры Тир.
Если 6й^=0, то из (2.25) следует:
dG = -i\-dT+V-dp. (2.26)
Из (2.26) вытекает:
8G
1 дт
дР
(2.27)
(2.28)
Вторые производные функции G(Tp) по Тир дают уравнение
dgl дУ\
4. Переменные х\ up
Для перехода к переменным т| и р из правой части выражения
(2.16) вычтем и к ней же прибавим дифференциал d{p-V) (последний
i • скроем). Получим:
bW* =-d(U + p-V)+T-dr] + V.dp. (2.29)
11ри r\-const np^const
5W* =-d{U + p*V)=-dH{r\,p)9
т е. при изобаро-адиабатических процессах работа совершается за
v ier убыли функции H{r\9p) = U + p-V. Эта функция называется
ьшпапъпией и является термодинамическим потенциалом при
заданных х\ и/>.
11ри 5^=0 из (2.29) следует:
43
dH = Tdx\ + Vdp
Из (2.30) вытекает:
Т =
дН
дц
V =
дН_
др
(2.30)
(2.31)
Перекрестное дифференцирование (2.31) и приравнивание
вторых производных дают
дТ_
др
dV_
дц
(2.32)
Из (2.9) и (2.30) видно, что при изобарических процессах
энтальпия равна поглощенному количеству теплоты:
dH\p=T ■ dr\\p = 5$р =Cp-dT, (2.33)
где С = -=■ - теплоемкость при постоянном давлении. По этой
dT
причине энтальпию называют также тепловой функцией или
теплосодержанием.
Любой из термодинамических потенциалов можно определить
через другие. Так, зависимость внутренней энергии от свободной
энергии можно получить из определения энергии Гельмгольца
F(T,v) = U-T-t) и уравнения (2.23):
8F\
U = F-T-
дТ
(2.34)
Связь энтальпии с потенциалом Гиббса можно установить с
помощью определений потенциала Гиббса и энтальпии
G(T,p) = U-T.4 + p-V и H(j]9p) = U + p-V,
а также уравнений (2.27):
дТ
(2.35)
Уравнения (2.34) и (2.35), устанавливающие связь между
различными термодинамическими потенциалами, называются
уравнениями Гиббса-Гелъмгольца.
Отметим, что в рамках этого подхода нельзя найти явные
выражения для термодинамических потенциалов. Однако начала
термодинамики позволяют найти связи одних свойств систем с
44
другими их свойствами типа (2.19), (2.24), (2.28), (2.32). Такие
соотношения называются основными уравнениями или
соотношениями Максвелла. Другие подобные соотношения легко могут
быть получены на основании применения якобианов (см. раздел
2.5).
Таким образом, все термодинамические потенциалы
являются однозначными функциями состояния, причем их убыль при
соответствующих условиях определяет работу системы против
действующих на нее сил.
Все термодинамические потенциалы обладают аддитивными
свойствами. В случае однокомпонентной системы при
изменении числа молей N в некоторое число раз величины
термодинамических потенциалов изменяются во столько же раз.
Следовательно, аддитивная термодинамическая величина является
однородной функцией первого порядка относительно аддитивных пе-
(кгменных10. Отсюда следует, что
V S\ Щ
.. (2.36)
U = N-U\—9^-\ F = N-F\ —,Т
G = NG(p,T\ H = N-Ii(p93-\
Черта сверху означает соответствующую функцию состояния
на один моль вещества.
Заметим, что параметрыриГне делятся на N, т. к. они не
зависят от числа молей (т. е. от массы).
2.4. Основные соотношения термодинамики
для многокомпонентных систем. Парциальные величины
До этого все исходные положения и начала термодинамики
относились к термодинамическим системам, состоящим из
одного компонента. Однако в природе имеются системы с двумя или
1 "функция f(xj,.. .>xj есть однородная функция степени г относительно
пр|ументовxh..,txny если/fa Jt/,...,axn>=ar/(3c/,...>х,). Из теоремы Эйлера
об однородных функциях следует:
дхх дхг дхп
45
более компонентами, причем число молей в них или общее
число молей системы могут изменяться. Состояние таких систем
характеризуется не только внешними параметрами aJf...,an и
температурой Г, но и числом молей каждого компонента Nh.. .,Nr или
соответствующими молярными концентрациями
2Х
Рассмотрим вопрос, как изменение числа молей отражается на
энергии системы. Ведь очевидно, что, увеличивая или уменьшая
массу, мы тем самым изменяем ее энергию.
Обозначим через dNk изменение числа молей к-oro
компонента, обусловленное обменом с окружающей средой и химическими
реакциями, а через ц* - коэффициент, на который надо умножить
dNh чтобы получить энергию, и назовем его химическим
потенциалом.
Тогда основное уравнение термодинамики для систем,
состоящих из нескольких компонентов, при равновесных процессах
принимает вид:
где ц* = ял/
dNk
i-i **i
- химический потенциал k-ого компонента.
Для обратимых процессов, совершаемых гомогенной R-
компонентной системой, чей состав и масса могут изменяться, и
на которую действует только всестороннее давление,
термодинамические потенциалы внутренней энергии, свободной энергии,
потенциала Гиббса и энтальпии равны:
dU = T-dt\-p-dV + 1£\Lk-dNk ; (2.38)
R
dF^-^dT-pdV + YsVk-Mk^ (2.39)
R
dG = -r\-dT + V-dp + Y,)ik-dNk; (2.40)
А=1
46
dH = Т■ dr\ + V • dp + ^\лк ■ dNk.
(2.41)
*=i
Из уравнений (2.38)-(2.41) следует, что химический потенциал
имеет вид:
Ц* =
8U
dNt
dF
г.ц
dNk
8G
г,т
dNk
дН
P.T
dNh
(2.42)
/>.n
Химический потенциал служит количественной мерой спо-
* Юности компонента к переходу: в системе, не находящейся в
равновесии, любой компонент стремится перейти из тех участков
* истемы, где его потенциал больше, в те части системы, где по-
юнциал меньше [45]. Химический потенциал - величина
интенсивная. Она не зависит от размера системы, а зависит только от
внешних параметров, ее состава и температуры. Это следует из
а/щитивности всех термодинамических потенциалов.
Из (2.36), например, получим:
Ц = || =G(7», (2.43)
т. е. химический потенциал при изотермо-изобарических
процессах равен энергии Гиббса на один моль вещества. Сравнивая
выражения (2.36) и (2.43), имеем:
G = N-\i. (2.44)
Как видно из формулы (2.43), химический потенциал не
зависит от числа молей, а определяется только температурой и
давлением, которые в свою очередь определяются внешними
параметрами.
Для смеси веществ, состоящих из NUN2,... молей, имеем:
G{pJ;aNl9aN29..)^aG{pJ;Nl9N29..).
Дифференцируя это равенство по а и полагая а=1, получим:
<7 = 5>,
8G
/=1
3N,
=2>,-g,=2>,-ii,
(2.45)
PJ.N;
i=l
Внутренняя энергия (/является однородной функцией первого
порядка и зависит только от экстенсивных независимых
параметров г), Vk Nx, ...,Nr (2.38). Следовательно, по теореме Эйлера об
47
однородных функциях для равновесных термодинамических
систем, находящихся под действием всестороннего давления, имеем:
U =
dU
дц
■т\ +
у л,
dU_
dV
чЛ
■N,. (2.46)
y,n,Nk
Принимая во внимание уравнения (2.18) и (2.42), получим:
U^.T-p-V + YvirN,.
(2.47)
Найдя полный дифференциал (2.47) как функцию параметров
т), Т, V, р, Nj, и |1Ь и учитывая (2.38), получим важное в
термодинамике уравнение Гиббса-Дюгема:
R
При изучении термодинамических свойств растворов, таких
как морская вода, очень часто применяется понятие парциальных
молярных величин. Пусть ф является какой-нибудь экстенсивной
функцией термодинамической системы (внутренняя энергия,
энтропия, энтальпия и др.), зависящей от температуры Г, давления
р и количества молей нескольких компонентов N. Тогда ее
полный дифференциал равен:
Эф
■dT +
\р.ы ЗР
Для заданных Тир имеем:
Эф
■Ф+£
Эф
T.N
М
dN,
dq>
dN
dNt +
Эф
\T,p,Nj*l
Величина, заданная в форме
Эф
dN,
T,P.N;*i
■dN2+.
■dN,. (2.49)
(2.50)
dN,
T,p,Nj*2
:<P,-.
(2.51)
называется парциальной (молярной) функцией i-компоненты.
Отметим, что парциальные величины относятся к интенсивным
параметрам.
Подставляя (2.51) в (2.50), получим:
dy = 7prdN] +ф2 -dN2 + ... . (2.52)
48
F,cjih использовать теорему Эйлера об однородных функциях,
ю можно записать:
Ф = ЛГ1-ф,+^2-ф2+... . (2.53)
Это уравнение можно использовать для определения
зависимости любой термодинамической функции ф системы от количе-
гиа каждого компонента, из которых она состоит.
Продифференцировав уравнение (2.53), и учитывая
выражение (2.52), получим:
ЛГ1-<*Ф1+ЛГ2-Лр2+... = 0. (2.54)
Рхли принять число молей одного компонента, например N2,
л основную переменную; мы с помощью дифференцирования
I 1внения (2.54) можем получить еще одну форму уравнения
I и >бса-Дюгема:
Л^^ + ЛГ2.-^ + ... = 0. (2.55)
1 dN2 2 dN2
2.5. Использование якобианов
Если использовать восемь переменных (Г, г\9 р, V, U, Н, F> G),
о с их помощью можно получить 5108 частных производных!
1л я облегчения нахождения производных, необходимых для
шльнейших исследований, можно применить метод якобианов
11усть имеются две переменные А и В, которые можно опреде-
1Ить через другие переменные хиу:
А = А(х9у), В = В(х,у).
'Уто означает переход от координат на плоскости (ху) к (AJB)
■ оординатам. При этом функции А=А(х1у) и В=В(х1у) должны
Ы(ь непрерывны и дифференцируемы по своим координатам.
>м преобразование выражается якобианом перехода
АВЛ \A-dB
— ="7 • (2-56)
х,у \ jx-dy
ft формуле (2.56) интегралы берутся по контурам, ограничи-
ицющим достаточно малые ячейки на соответствующих коорди-
ншных плоскостях.
А
49
Якобиан можно представить как матрицу из частных произ-
водных АуВ по аргументам хиу:
А,В
,Х>У.
дА дА
дх ду
дВ дБ
дх ду
/
\
дА дВ__дА дВ\
дх ду ду дх)
(2.57)
(2.58)
Якобиан подчиняется простому правилу замены переменных:
I JAdB jx>dy JA-dB
qx -dy qu • dv qu • dv
Обозначим \AdB и ax dy как [A9B] и [xy] соответственно.
Тогда
A,B~
[xyy_
J
*,y1
u,v\
А, В
х,У
[A,B]
[x,y]
Приведем некоторые свойства якобианов, которые в
дальнейшем будут нами использоваться:
(2.59)
(2.60)
(2.61)
Щ [А,С\ш
дВ\с~[В,С]'
[а,в]=-[в,а\;
[А,А]=0;
[А,В\ dC + [В,С\ <1А + [С,А\ dB = 0;
[А,В\[С,Х]+ [В,С] [А,х] + [С,А\-[В,Х] = 0;
dC = [c,x].
Если dA = bdB + cdC, то
[Л,х]=б[Я,х]+с[С,х]. (2.62)
С учетом (2.62), соотношения (2.14), (2.22), (2.26) и (2.30)
перепишем следующим образом:
[и,х]=Т[ц,х]-р[У,Х]; (2.63)
[F,X] = -T][T,X]-P-[V,X]; (2.64)
[С,х] = -ц[т,х]+У[р,х]; (2.65)
[Н,Х] = Т[ц,Х]+У[р,Х]. (2.66)
50
Г.спи взять соотношения Максвелла (2.19), (2.24), (2.28) и
(2.32) и выразить их через якобианы, то увидим, что все они сво-
ргся к единой форме:
[Т,ц]=[р,У]. (2.67)
В качестве примера возьмем (2.19). С учетом (2.59), имеем:
дТ
dV
dp
[т,я
[у,п]
_ и
-~fov]'
Используя свойство (2.60), получим (2.67).
Соотношение (2.67) имеет важную физическую
интерпретацию: интегралы по замкнутым контурам, ограничивающим эле-
1 ||ты площади в T,i\ и /?,К-координатах, равны друг другу, т. е.
умма количества теплоты при замкнутом циклическом процессе
- i да совпадает с суммарной работой, совершаемой системой.
Ьольшинство из 5-Ю8 комбинаций производных не представ-
пет интереса и лишь небольшая часть имеет практическое зна-
1СПИС. При этом в рамках термодинамики невозможно найти
явные выражения для термодинамических потенциалов, которые
1*одят в интересующие нас частные производные, и, естествен-
10, мы не можем непосредственно их оценить. Поэтому будем
фсмиться выразить эти производные через те параметры,
которые могут быть измерены или вычислены непосредственно.
К таким параметрам относятся, во-первых, Т,Уир, во-вторых,
чующие величины (их описание приводится в главе 4):
- коэффициент термического расширения:
dV\ =I [У,р].
- коэффициент изотермической сжимаемости:
1
а=—
V дТ
(2.68)
к = —
к'ф|г~ v'[p,T]~V [Т,р]'
удельная теплоемкость при постоянном объеме:
су =Т ■
дП
= Т
WY
(2.69)
(2.70)
- удельная теплоемкость при постоянном давлении:
51
р от
- отношение теплоемкостеи:
= Т
Wp].
ЕЛ'
у = .
(2.71)
(2.72)
Таким образом, мы рассмотрели весь необходимый
математический аппарат, который будет использован в дальнейшем
следующим образом:
- записываем интересующую нас производную через якобиан;
- если в полученное выражение входят U, Н, F и G, то
исключаем их, используя (2.63)-(2.66);
- если один из якобианов содержит энтропию т|, то для ее
исключения используются (2.67), (2.70) или (2.71);
- оставляем только те якобианы, которые содержат р, Г, V, а,
к, ср, cv, у, а/к.
В качестве примера найдем изменение энтропии при
изотермическом сжатии некоторого объема воды, т. е. необходимо най-
ти производную —
др
Из (2.59) получаем
дц
дР
_ for]
М
Используя (2.67), исключаем энтропию:
др
_ [р
\pJ\
Переходим от якобианов к известным нам величинам,
используя в данном случае коэффициент термического расширения
(2.68). Откуда
дц\
др
= -V-a.
(2.73)
Анализ полученной формулы проведем на примере морской
воды в главе 4.
52
Задачи и вопросы для повторения
2.1. За счет какого вида энергии осуществляется работа при
изотермических процессах при постоянном объеме?
2 2 Для чего нужны соотношения Максвелла?
2 .V Какие параметры термодинамической системы называются
функциями состояния?
2 4. В чем отличие замкнутой системы от закрытой?
2 5. Как называется термодинамический потенциал при заданных
Ил плен ии и температуре?
2 6 Какая сила соответствует внешнему параметру объем?
2 7. Сколькими способами происходит обмен энергией между сис-
»и<1 и внешними телами?
, «S. Известно, что агрегатных состояний всего четыре. А сколько
m../истсуществовать фаз?
2.с>. Без чего нельзя преобразовать теплоту в работу?
2.10 Почему чистая вода является однофазной и однокомпонентной
• и* юмой, а смесь газов водорода и кислорода - однофазной, но двух-
- чионентной системой?
2 11. Внутренние параметры термодинамической системы зависят от
•пк у иного движения и распределения, входящих в систему частиц,
■ uu-ратуры и ... .
2.12. Какие параметры системы из перечисленных ниже зависят от
к сы: энергия, давление, температура, энтропия, вязкость? Как они
• •in ном называются?
2.13. Определите термодинамические потенциалы при независимых
«ь |«сменныхpJH и TJ7.
Ответ:
Возьмем уравнение (2.30): dH -Т dri + V-dp, из которого видно,
•| при независимых переменных р и Н термодинамическим потенциалом
«й не гея энтропия r\(pjf):
1 V
dx\- — - dH dp.
При этом
Т^дц/дН\ ' ' " dp
T = —^-r , К—^
53
Теперь возьмем уравнение (2.22): dF = —ц-dT — р-dV. Откуда
т\ 1
</Г = dT dF. Видно, что при независимых переменных Т и F
Р Р
термодинамическим потенциалом служит объем.
54
ГЛАВА 3. МОРСКАЯ ВОДА КАК ДВУХКОМПОНЕНТНАЯ
ТЕРМОДИНАМИЧЕСКАЯ СИСТЕМА
В начале этой главы рассматривается образование водной
uiccbi Мирового океана и история формирования его солевого
остава. Затем описываются особенности современного солевого
остава Мирового океана. На основании закона о постоянстве
олсвого состава и того факта, что почти все соли растворены в
юрской воде в форме ионов, морская вода представлена как
тухкомпонентная система: растворитель - вода и одно раство-
чМ1Ное вещество - морская соль. Для этой системы даются ос-
ювные термодинамические соотношения и рассматриваются ос-
юнные параметры состояния.
3.1. Образование водной массы Мирового океана
Существует несколько гипотез о происхождении водной мас-
< ы Мирового океана. Рассмотрим одну из них, предложенную
\.П. Виноградовым [8].
Исходными материалами для образования гидросферы Земли
послужили - вода в форме ледяной пыли при образовании Земли
• и газопылевого облака галактического вещества, лед в метеори-
ах, которые падали на поверхность Земли, а также облака
водорода, гидроксильных радикалов и молекул воды за пределами
^ олнечной системы.
По А.П. Виноградову, образование гидросферы обусловлено
механизмом дифференциации вещества Земли на оболочки. Этот
процесс связан с разогреванием холодного вещества Земли из-за
i равитационного сжатия и, главным образом, в результате
выделения тепла при распаде радиоактивных элементов.
При разогревании вещества мантии происходит формирование
легкоплавкой фракции силикатов и ее движение из глубин к
поверхности, сопровождающееся насыщением многими
легкоплавкими и легколетучими веществами. Основную массу их составля-
vi П20. При приближении к поверхности происходит охлаждение
и кристаллизация расплава, а также дегазация Н20, H2S, НС1, HF,
Nil* и других веществ, в результате чего он распадается на две
фазы: силикатную и водную. В тугоплавкой фазе остаются Mg,
55
Fe, Ni, Co, Cr и др. Водная фаза содержит, кроме Н20, газы,
растворяющиеся в воде (НС1, HBr, С03, HF и др.), а также частично
соединения серы. В результате на поверхность Земли поступает
ювенильный раствор, образующий водную массу первичного
океана и содержащий различные соединения.
Те газы, которые не растворяются или слабо растворяются в
воде (Не, Ar, Ne, Кг, Хе, СН4, частично С02 и др.), не
конденсировались с Н20 и остались в газовой оболочке Земли, образовав
первичную атмосферу.
Процесс разделения вещества на фазы не требует полного
расплавления Земли - механизмы выплавления и дегазации
проявлялись в виде интенсивной вулканической деятельности.
Близкое сходство состава некоторых продуктов извержения
современных вулканов с составом вод современного океана
подтверждает эту теорию. Хорошим доказательством этой гипотезы
служит также согласованность объема вод Мирового океана и массы
воды, образовавшейся в процессе дифференциации Земли на
оболочки.
Итак, А.П. Виноградов пришел к выводу, что формирование
водной массы Мирового океана происходило путем дегазации при
вулканических извержениях.
В настоящее время стал известен еще один мощный источник
поступления воды - выходы ювенильных вод или рассолов (с
соленостью до 270 г-кг"1) на дне океана.
3.2. История формирования солевого состава Мирового океана
Приспособление первичного ювенильного раствора к
изменяющимся условиям на поверхности Земли приводило к
эволюции состава вод Мирового океана, стремящейся прийти в
равновесие с этими условиями. Подобный процесс происходил во все
периоды существования океана, его суть заключается в
сохранении в воде растворимых соединений и в выпадении в осадок
компонентов, образующих труднорастворимые соединения.
Сравнение современного состава вод Мирового океана и
первичного ювенильного раствора, сделанное М.Г. Валяшко [7],
показывает, что содержание хлора и брома практически не
изменилось, а содержание остальных элементов заметно уменьшилось.
56
Ото позволяет говорить о том, что в растворе остаются
неизменными те элементы, для которых нет достаточно активных осади-
сслей, например, те же хлор и бром. Содержание тех элементов,
которые участвовали в процессах, выводящих их из раствора,
уменьшилось, причем больше всего снизилось содержание
углерода. К уменьшению содержания углерода привели в основном
две реакции. Первая - это связывание С02 в угольную кислоту с
последующим переходом главным образом в карбонат кальция,
выпадающий в осадок. Эта реакция происходит от момента
возникновения океана до настоящего времени. Вторая реакция свя-
шна с фотосинтезом - поэтому она возникла только с появлением
биосферы. Образование биосферы на Земле привело также к
уменьшению содержания азота и йода.
Выделяют три этапа формирования солевой массы Мирового
океана:
-ранний этап - при отсутствии биосферы (глубокий архей);
- этап возникновения и становления биосферы (конец архея—
палеозой);
- современный период (начиная от палеозоя).
На первом этапе развития воды Мирового океана имели
кислую реакцию, что обусловлено тем, что хлор, бром и фтор
выделялись в виде сильных кислот - НС1, НВг и HF. Кроме того,
первичные воды были насыщены по углероду и кремнезему. Связано
это с тем, что при попадании ювенильного раствора на
поверхность в условия пониженных температур и давления резко
снижалась растворимость углекислоты. Почти одновременно с
появлением гидросферы возник процесс нейтрализации первично
кислых вод - анионы первичного океана, приносимые в ювенильном
растворе, сразу же вступали во взаимодействие с катионами,
образовавшимися в процессе разрушения дна и берегов океана.
Ранний этап существовал, по-видимому, кратковременно. Воды
океана с момента своего возникновения, благодаря хорошей
растворимости солей, образующихся в них, становились солеными,
причем все анионы - это продукты дегазации мантии, а катионы
- продукты выветривания горных пород земной коры. ,
Второй этап формирования химического состава Мирового
океана начался с момента возникновения биосферы. Произошло
изменение восстановительных свойств атмосферы на окисли-
57
тельные, обусловленное выделением свободного кислорода в
процессе фотосинтеза. Это привело к образованию современной
азотно-кислородной атмосферы. Произошла перестройка
солевого состава океана, связанная с воздействием биосферы, а также с
изменившимся обменом веществ с литосферой и окисляющей
атмосферой. Увеличилось содержание углекислого газа в воде.
Постепенно стабилизировалась карбонатная система. Под
влиянием кислорода изменили свою миграционную форму азот,
железо, сера. Увеличилась подвижность кальция и магния.
После стабилизации состава атмосферы сначала
стабилизировались формы существования и миграции элементов на
поверхности Земли, затем стабилизировался и солевой состав вод
Мирового океана. Солевой состав океанических вод стал близок к
современному - преимущественно хлоридно-сульфатному.
Наступил современный период формирования солевой массы
океана. По всей видимости, стабилизация современного состава
океанических вод произошла между 1,5 и 0,5 млрд. лет назад.
Хотя на Земле непрерывно происходят процессы
взаимодействия между гидросферой, атмосферой, литосферой и биосферой,
приводящие как к поступлению солей, так и к их потери, общая
концентрация солей в Мировом океане остается практически
постоянной (В.И. Вернадский назвал концентрацию солей в
океане планетной константой).
Итак, в результате глобального процесса дифференциации
вещества Земли образовалась водная масса Мирового океана
объемом 1,34-109 км3 (что составляет около 96,5% от объема
всей воды на Земле), массой 1,38-1021 кг и с содержанием солей
481018 кг.
3.3. Особенности современного солевого состава
Мирового океана
Впервые солевой состав морской воды был определен датским
химиком Форшхиммером в 60-х гг. XIX в. Затем тщательные
исследования солевого состава были осуществлены Дитмаром,
проанализировавшим 77 проб морской воды, собранных в различных
областях Мирового океана во время плавания «Челенджера» в
58
№78-1882 гг. Позднее солевой состав исследовался такими уче-
MI кии, как Калкин, Кокс и др.
Морская вода представляет собой многокомпонентную
систему, состоящую из молекул воды, различных солей, растворенных
i i юв, органических веществ и других составляющих. В среднем
и Мировом океане на молекулы воды приходится около 96,5%
m ей массы морской воды, на растворенные соли - приблизителы
но 3,5%. Количество других компонентов пренебрежимо мало.
Несмотря на небольшой процент солей в морской воде, их
влияние на некоторые ее свойства значительно.
Исследования показали, что в морской воде растворены все
' •■-менты, встречающиеся на Земле. При этом отмечено, что
w,9% всех растворенных веществ составляют первые 20
элементе таблицы Менделеева, причем большинство солей растворе*
мы в морской воде в форме ионов (анионов и катионов).
Хлористые соединения диссоциированы полностью, только 12%
кальция, 11% магния, 2% натрия и 1% калия связаны с анионами.
Более половины сульфатов, треть бикарбонатов, от 20 до 50% фто-
ридовых соединений связаны с катионами. Незначительная часть
солей находится в морской воде в форме коллоидов и суспензий.
Таким образом, с большой уверенностью можно говорить, что в
морской воде присутствуют 10 ионов (5 анионов и 5 катионов) и
борная кислота (табл. 3.1).
Таблица 3.1
Ионный состав морской воды при содержании солей 35 гкг"1
Г Катионы
1 Название
Натрий (Na+)
Магний (Mg2+)
Кальций (Са2+)
Калий (К*)
Строниий (Sr2*)
гкг"1
10,7638
1,2970
0,4080
0,3875
0,0136
Анионы
Название
Хлоридные (СГ)
Сульфатные (S042~)
Бромидные (Вг~)
Фторидные (F~)
Борная кислота (Н3ВОз~)
Гидрокарбонатные (НС03~)
гкг"1
19,3534 !
2,7007
0,0659
0,0013
0,0265
0,1427
Других составляющих в морской воде очень мало - менее
0,01% от общего количества растворенных солей. Если из ука-
59
занных в табл. 3.1 ионов образовать соли, то состав морской воды
в форме солей был бы следующим (табл. 3.2).
Очевидно, что молекулярный вес чистой воды, состоящей из
атома кислорода и двух атомов водорода, равен 18 г-моль"1.
Эффективный же молекулярный вес суммы основных ионов,
содержащихся в морской воде с соленостью 35 г-кг"1 (табл. 3.1), равен
приблизительно 31,42 г моль"1.
Отметим, что солевой состав морских вод изменяется вблизи
устьев рек, так как основными анионами в реках являются (в
порядке убывания концентрации) НСО3, SO*" , СГ, в то время как
основными катионами - Са2+, Na+, Mg2+ и К+. Солевой состав вод
замкнутых (Каспийское море) и полузамкнутых (Балтийское,
Черное моря) водоемов также значительно отличается от
солевого состава вод открытых частей Мирового океана.
Таблица 3.2
Основные компоненты морской воды в форме солей
(на м3 морской воды с содержанием солей 35 г-кг"1 и
температурой 20° С)
Название солей
NaCl
MgCl2
MgS04
CaS04
k2so4
CaC03
КВг
SrS04
H2B03
Количество, кг
28,014
3,812
1,752
1,283
0,816
0,122
0,101
0,028
0,028
Отличительной чертой солевого состава морской воды от-
крытых частей океанов является то, что относительная доля
растворенных компонентов в ней приблизительно постоянна
(закон Дитмара или принцип Марсе). Практически во всем
Мировом океане отношения Na/Cl, K/Cl, SO4/CI и Вг/С1 являются
константами. Небольшие отклонения могут встречаться в
отношениях Mg/Cl, Sr/Cl, HCO3/CI, F/C1, а для глубинных вод и в
отношении Са/С1. В замкнутых и полузамкнутых водоемах, в устьевых
60
помах рек, а также при замерзании морской воды локальная
изменчивость этих отношений может быть значительной.
Постоянство отношений основных компонентов солевого
сошна позволяет определить общую соленость и состав морской
йоды, измерив лишь один из компонентов растворенных веществ,
м.шример, С1. Этот ион составляет 55% от общего количества и
легко определяется.
Таким образом, учитывая постоянство солевого состава и то,
•по соли растворены в морской воде в форме ионов в небольшом
Количестве, морскую воду молено представить как двухкомпо-
нентную (бинарную) систему: растворитель - вода и одно
расширенное вещество (компонент) - морская соль [58,61].
3.4. Основные термодинамические соотношения
для морской воды
Второй закон термодинамики для обратимых процессов,
совершаемых в Л-компонентной системе, находящейся под
действием только внешнего давления, выглядит следующим образом
(см. раздел 2.4):
R
dU^Td-q-p-dV + YjVi'Mi' (ЗЛ)
где U - внутренняя энергия; Т - термодинамическая
температура; г| - энтропия; р - давление; V - объем; щ - химический
потенциал *-го компонента массой Л/}.
Прежде чем применить уравнение (3.1) к термодинамической
системе - морской воде, преобразуем его для единичной массы,
введя интенсивные величины:
U
g - — _ удельная внутренняя энергия;
М
* Л
т| = —■— удельная энтропия;
М
v = удельный объем;
т. /я.
xt= _ = —■=- - массовая доля i-го компонента,
Z,mi м
i
61
где М- масса системы.
Подставляя их в (3.1), получим:
R
dz = Td^-pdv^iirdxi. (3.2)
1=1
Так как морская вода состоит в основном из чистой воды и
ионов солей, уравнение (3.2) преобразуется к виду:
п
dz = T-dx\ -pdv + \xw dxw + YaVii'dxi » (3-3)
/=i
где fiw - химический потенциал чистой воды; xw - массовая доля
чистой воды; п - количество основных ионов; ц* - химический
потенциал /-го иона, ах*- его массовая доля.
Предполагая постоянство отношений растворимых ионов,
можем написать:
*,=VS, (3.4)
где S = = —— массовая доля всех солей (так
называете +ms М
мая соленость, определение которой дается в разделе 3.6); ms -
общая масса всех солей; mw- масса чистой воды; А.,- - отношение
массы /-го иона к общей массе растворимых солей, причем
Подставляя (3.4) в (3.3), получим:
п
dz = T<h\-pdv + ]xwdxw + Y.^i\dS =
1*1 V*'5)
= Г-dj\ -pdv + \iwdxw + \xs dS9
n
где \xs = ]ГX; • \i. - химический потенциал растворимых в мор-
/=i
ской воде солей.
Введя обозначение для разности химических потенциалов:
и затем, подставив в (3.5) вместо \xs величину ц + jiw> получим:
dt = T dyf-pdv + \iw{dxw+dS) + \x dS . (3.7)
62
Выражение dxw + dS равно нулю в соответствии с условием
сохранения масс (М = mw + ms = const):
f ... \
dxw + dS = */(*„ + S) = rf
/ям
/Wo
mw+ms
mw + ms J
= d
™w + ms
M
(3.8)
= </(l) = 0.
Отсюда получаем окончательный вид основного уравнения
термодинамики для морской воды как двухкомпонентной
системы (вода и морская соль):
dz = Tdx\ -pdv + n-dS, (3.9)
де
— химический потенциал морской воды.
ч ,у
Соответствующие уравнения для других удельных термоди-
F
намических потенциалов (свободной энергии / = —, потенциа-
М
ла Гиббса g* = — и энтальпии А =—) в морской воде запишут-
М М
ся в следующем виде:
df = -T\dT-pdv + \JL dS; (3.10)
dg* =-у\ dT + vdp + \idS\ (3.11)
dh = T dx\ +vdp + \i dS . (3.12)
Из уравнений (3.9)—(3.12) можно получить выражение для
химического потенциала морской воды, выраженное через
термодинамические функции:
es
Л .v
Ж.
dS
dg
T,v
dS
Т.р
dh
dS
(3.13)
П гР
Четыре соотношения Максвелла для однокомпонентной
системы (2.19), (2.24), (2.28) и (2.32) оказываются справедливыми и
для двухкомпонентной системы; в этом случае к ним надо
приписать индекс «S» у вертикальной линии. Эти соотношения можно
дополнить еще несколькими выражениями, включающими соле-
63
ность и химический потенциал. Для этого можно воспользоваться
приемами, аналогичным описанным в разделе 2.3.
В качестве примера возьмем внутреннюю энергию е в
переменных л*, v и S, т. е. e=e(n.*,v,.S). Ее полный дифференциал
равен:
<fe =
&
an*
•<V +
v,S
де
dv
n*^
, де,
■ dv + —
dS
■dS
v,n
Сравнивая уравнения (3.14) с (3.9), получим:
Г =
де
дг\'
> Р = -
де
v,S
0V
ц =
Ц',5
dS
(3.14)
(3.15)
v,n
Дифференцируя первую формулу в (3.15) по солености, а
последнюю - по энтропии, имеем:
dS
d\i
(3.16)
Аналогично можно получить и другие соотношения Максвелла
v,S
дТ\
ds[
anas
anas
Ф
i> ^п*
1 _Ф
L дТ
>Т,р
-М
Ir.v '
U
•Р,
'.5
ф
as
Ф
dS
dv_
dS
dv_
dS
n •>■
T,v
= ф
dv
_ф
dv
r\,S
T,S
T.p Ф
= ф
T,S
n',s
(3.17)
(3.18)
(3.19)
(3.20)
(3.21)
(3.22)
(3.23)
Приведенные выше соотношения позволяют получить ряд
производных второго и более высоких порядков, выражающих
дополнительные зависимости между термодинамическими пара-
метрами.
Для двухкомпонентной морской воды обобщенное уравнение
Гиббса-Дюгема (2.48) выглядит так:
4'dT-vdp + xw-d)iw + S-d\Ls=0. (3.24)
Подставляя в (3.24) вместо \is величину ц+Щу из (3.6),
получим:
4m-dT-vdp + (xw + S)-d\iw+S-d\L = 0 . (3.25)
Учитывая, что xw + S = 1 (см. 3.8), получим соотношение
Гиббса-Дюгема в виде:
d\Lw=-r\m-dT + vdp-S-d\x. (3.26)
При изучении термодинамических свойств растворов, таких
как морская вода, очень часто применяется понятие парциальных
молярных величин (см. раздел 2.4), использующееся для
определения зависимости любой термодинамической функции системы
от количества каждого компонента, из которых она состоит.
Для удельных термодинамических функций морской воды,
выраженных обобщающей функцией ф =ф/М, вместо уравнений
(2.53), (2.54) и (2.55) имеем:
фФ=(1-5)ср^+5.ф;, (3.27)
(l-S)-d^ +5^ф; =0, (3.28)
G_S).3L+s.aL„,, (з.29)
где ф^ и фJ - парциальные (удельные) функции воды и солей в
морской воде соответственно. Из уравнения (3.29) следует, что
если при постоянной температуре и постоянном давлении
термодинамическая функция чистой воды возрастает, то эта же
функция морской соли должна уменьшаться.
Дифференцируя формулу (3.27) по солености и принимая во
внимание уравнение (3.29), получаем:
%-=Ъ-Ъ- (3.30)
65
Наконец, из формул (3.27) и (3.30) получаем уравнения для
расчета парциальных функций чистой воды и морской соли в
морской воде:
ф;=ф--5-^, (з.з1)
Ф>ср-+(1-5).^1. (3.32)
3.5. Параметры состояния. Температура
Морская вода, как всякая термодинамическая система,
характеризуется некоторыми физическими величинами,
определяющими ее макроскопические свойства. Эти величины называются
параметрами состояния. Для морской воды основными
параметрами состояния являются масса, объем, давление, температура и
соленость11. При этом только объем относится к внешним
параметрам системы, остальные - ее внутренние параметры.
Существование температуры как особой функции состояния
термодинамической системы следует из второго исходного
положения термодинамики. Температурой называется физическая
величина, характеризующая интенсивность теплового движения
молекул системы. Измерение температуры производится только
косвенным путем, основываясь на зависимости от температуры
таких физических свойств тел, которые поддаются
непосредственному измерению. Применяемые для этого тела называются
термодинамическими, а устанавливаемые с их помощью шкалы
температуры - эмпирическими. Основной недостаток
эмпирических шкал температуры состоит в их зависимости от
особенностей применяемых термометрических веществ.
В качестве исходных значений, служащих при построении
шкалы температуры для установления начала отсчета
температуры и единицы ее измерения - градуса, могут применяться темпе-
Отметим, что помимо названных параметров состояния, существуют
и другие, например внутренняя энергия, энтропия, вязкость и др.
Однако в океанологических исследованиях они используются значительно
реже. Некоторые из этих параметров будут рассмотрены в
последующих параграфах.
66
чпуры перехода химически чистых веществ из одного агрегатно-
0 состояния в другое, например, температуры плавления льда
Го) и кипения воды (Гк) при нормальном атмосферном давлении.
Температура в океанологии измеряется в градусах
стоградусной шкалы Цельсия (°С, Гс^О0 С, Гк=100° С). При этом
различают температуру in situ, обозначаемую буквой Ту и потенциальную
«смпературу Э. Под температурой in situ (с латинского языка —
6 данном месте») подразумевается температура, измеренная
термометром на определенной глубине моря. Потенциальная
температура - это температура частицы воды, адиабатически
приведенная от давления в точке измерения к заданному давлению (см.
раздел 4.10).
При некоторых расчетах применяется температура в единицах
»бсолютной (термодинамической) шкалы Кельвина (°К). Градусы
шкалы Кельвина равны градусам шкалы Цельсия (1° 0=10К), но
исчисляются они от абсолютного нуля, равного -273,15° С, т.е.
абсолютная температура равна Гк=7ч:+ 273,15.
Второе начало термодинамики устанавливает, что абсолютная
шкала температур не зависит ни от выбранного
термодинамического тела, ни от какого-либо термодинамического параметра.
В некоторых странах продолжает употребляться
температурная шкала Фаренгейта (°F, 70=32° F, Гк=212°Р). Температура в
диницах шкалы Фаренгейта связана с температурой
стоградусной шкалы следующим образом:
Г.сЛ-(Г-,-32).
При точных расчетах океанографических величин, зависящих
от температуры, важно знать, какая шкала температуры исполь-
ювалась. Большинство современных формул для расчета
физические свойств морской воды основано на Международной
практической температурной шкале 1968 г. (МПТШ-68).
В настоящее время в практике океанологических измерений
используется Международная шкала температуры 1990 г. (МШТ-
90). Чтобы использовать температуру по шкале МШТ-90 в
формулах для вычисления физических свойств морской воды, осно-
67
ванных на МПШТ-68, ее нужно пересчитать в Г6812. Формула
перерасчета температуры из МШТ-90 в МПТШ-68 имеет
следующий вид:
Га = 1,0024-Г*. (3.33)
Температура относится к интенсивным параметрам, т. е. к
параметрам, не зависящим от массы.
Температура воды в океане изменяется, в основном, от ~ -2° до
~30° С. Нижняя граница определяется температурой замерзания,
которая зависит от солености и давления. Верхняя граница
зависит от теплообмена между океаном и атмосферой. В замкнутых
морях и в мелководной прибрежной зоне температура воды
может быть и больше 30° С. Температура воды с глубиной обычно
убывает, причем чем глубже, тем медленнее (табл. 3.3). В
придонных слоях температура воды меняется в зависимости от места
от 0° до 2,5° С.
Таблица 3.3
Изменение средних температуры и солености воды с
глубиной в Мировом океане (по [12])
I Горизонт,
м
0
50
100
200
500
1000
2000
3000
! 4000
5000
Температура,
°С
17,84
16,52
14,69
11,67
7,31
4,22
2,32
1,72
1,37
1,19
Соленость,
г кг"1
34,57
34,80
34,95
34,95
34,68
34,61
34,73
34,75
34,73
34,72
Если не вносить изменения в существующие алгоритмы вычисления
физических свойств морской воды, связанные с переходом на МШТ-90,
то наиболее значимые погрешности будут при вычислениях, связанных
с соленостью, наименьшие погрешности при расчете плотности,
сжимаемости, адиабатического градиента температуры и потенциальной
температуры. После перевода температуры из МШТ-90 в МПТШ-68
ошибки всех вычислений становятся минимальными и находятся в
пределах точности ГШ1С-78 и УС-80.
68
В бассейнах осолонения, таких как, например Средиземное
морс, температура на больших глубинах имеет значительно более
высокие значения. В северных морях, где наблюдается зимняя
коннскция и формируется холодный промежуточный слой,
температура воды с глубиной в теплый период года сначала
понижайся, а затем снова возрастает. В таблице 3.4 приведены средние
шачения температуры воды в океанах.
Таблица 3.4
Средние значения температуры, солености и плотности
(при атмосферном давлении) воды океанов (по [12])
Параметры
Температу-
ра,°С
Соленость,
г кг'1
11лотность,
кгм"3
Атлантический
северная
часть
4,95
35,06
1027,69
южная
часть
3,12
34,74
1027,65
Тихий
северная
часть
3,69
34,58
1027,40
южная
часть
3,50
34,66
1027,51
Индийский
3,76
34,75
1027,56
Мировой
3,73
34,72
1027,54
Методы измерения температуры воды в Мировом океане
подразделяются на контактные и дистанционные (неконтактные).
Дистанционные (неконтактные) методы основаны на
измерении собственного теплового излучения водной поверхности
океана в инфракрасном и микроволновом диапазонах спектра
шектромагнитных волн.
Контактный метод основан на введении в толщу воды
измерительного зонда, имеющего соответствующий
термометрический элемент. В качестве такого элемента используются ртутные
термометры (обычные для измерения на поверхности и
опрокидывающиеся для измерения в толще воды), металлические
термометры сопротивления, термисторы, кварцевые резонаторы.
Принцип действия термометров сопротивления и термисторов
основан на зависимости электрического сопротивления
чувствительных элементов датчиков от температуры воды. Кварцевые
резонаторы используются в качестве термочувствительных
элементов, задающих частоту в схемах кварцевых генераторов. Вы-
69
ходная частота этих генераторов есть функция частоты
генератора и температуры воды.
Наилучшие метрологические характеристики имеют
металлические термометры сопротивления и кварцевые резонаторы.
Первые получили наиболее широкое применение в современных
СГ£>-океанографических зондах.
3.6. Соленость. Определение солености
Как уже отмечалось выше, отличительным свойством морской
воды является ее соленость (обозначается символом S). Под
истинной соленостью понимается отношение массы
растворенного твердого вещества в морской воде к ее массе. Это
определение солености было принято в начале прошлого века
Международным советом по исследованию морей.
Из-за сложности состава морской воды невозможно прямым
химическим анализом определить полное количество соли,
растворенное в пробе морской воды. Сухой остаток, получаемый
после выпаривания, очень хорошо впитывает влагу и для его
получения требуется длительное нагревание, приводящее к
разложению карбонатов и солей магния. Группа ученых, в которую
входили М. Кнудсен, К. Соренсен и С. Форх, разработали метод
обезвоживания в среде хлора, который давал высокоточные
результаты. Но этот метод слишком сложен, и на практике
соленость морской воды им никогда не измеряется.
В настоящее время соленость определяется либо по
содержанию одного из компонентов солевого состава (метод Мора-
Кнудсена), либо по электропроводности морской воды, либо по
показателю преломления. В результате возникли различные
шкалы солености, основанные на различных принципах. Единица
измерения солености зависит от способа ее определения - гкг"1
или %о (промилле) в случае метода Кнудсена и в единицах
практической солености при определении S по электропроводности13.
Отметим, что при написании практической солености знак %о
опускается. Некоторые отечественные исследователи для обозначения
практической солености пользуются аббревиатурой епс (единицы
практической солености), в англоязычной литературе эта аббревиатура выглядит
так: psu или PSS-78.
70
Метод Мора-Кнудсена основан на постоянстве солевого со-
ciaea морской воды. Вследствие возможных отклонений от
закона Дитмара определение всей массы растворенных солей по
содержанию одного из компонентов не совсем точно, но не столь
существенно по сравнению с точностью самого определения
солености. В этом методе соленость (%о) определяется по хлорно-
сти (С1) морской воды, представляющей собой сумму ионов
хлора, брома и йода и полученную стандартным аргентометрическим
методом титрования на хлор. Соотношение между соленостью и
лорностью для океанских вод выражается эмпирической
формулой:
S%о=0,030 + 1,8050С1 %о . (3.34)
Эта формула была получена в 1901 г. М. Кнудсеном. Она
справедлива для диапазона солености морских вод от 2,69 до
40,18 %о. Отметим, что для замкнутых морей (Каспийское,
Аральское), а также внутриматериковых морей (Балтийское,
Черное, Азовское) соотношения между соленостью и хлорностью
отличаются от (3.34) [34,79].
Наличие в (3.34) свободного члена приводит к тому, что
соленость оказывается величиной неаддитивной. В то же время из-
иестно, что соленость - величина консервативная,
подчиняющаяся линейному закону смешения. Для устранения этого
противоречия в 1962 г. группой экспертов при ЮНЕСКО была
предложена следующая формула {гикала Кокса):
S%o = 1,80655С1 %о . (3.35)
Для определения солености по электропроводности (см.
раздел 8.1) была разработана Шкала практической солености,
1978 г. (ШПС-78) [72]. Эта шкала основана на эмпирической
зависимости от электропроводности не природных вод, а растворов
стандартной морской воды. Первичным эталоном в этом методе
служит водный раствор хлористого калия (КС1) при температуре
15° С и давлении в одну стандартную атмосферу (101325 Па).
Практическая соленость вычисляется по следующей
эмпирической формуле:
S = а0 + axB!j.2 + a2RT + аъВ$2 + a4R^ + asRp + AS, (3.36)
71
где
я0=0,0080
a5=2,7081
64=0,0636
a,=-0,1692; я2=25,3851; я3=14,0941; a4=-7,0261;
£>o=0,0005; ^=-0,0056; 62=-0,0066; 63=-0,0375;
65=-0,0144; #=0,0162.
В выражении (3.36) RT обозначает относительную
электропроводность, равную отношению электропроводности пробы
воды к электропроводности воды с соленостью 35 при
атмосферном давлении. Обе пробы должны иметь температуру 15° С.
Относительная электропроводность RT по данным измерений
in situ рассчитывается следующим образом. Пусть y(S,Tp) -
условная электропроводность морской воды in situ (см. раздел 8.1),
у(35,15,0) - условная электропроводность морской воды при
r=15°C, практической солености 35 и атмосферном давлении
(4,2914 Смм"1). Величина
у(35,15,0)
называется коэффициентом электропроводности.
Этот коэффициент можно разложить на три части:
R = RpRTrT, (3.38)
ГПР р -У&Т,р) R _ yjSJfi) _ у(35,Г,0)
ГДе *'~ у(*,7\0)' Г~ 7(35,7,0)' Гг" у(35,15,0)-
Величины Rp и гТ также находятся по эмпирическим
формулам:
r =1, Р-{^+е2Р + е3р2)
" 1 + </,-г+</2г2 + (л3+</4:г)л' (3.39)
гт =с0 +с, -Т + с2 Т2 +с3 Т3 +с4 Т\
где е,=2,070-10'5; е2=-6,370-10"'0; е3=3,9891015; rf,=3,426-10"2;
^4,464-Ю"4; «/j-4,215-10-1; </4=-3,107-10-3; с0=0,6766097;
с,=2,00564-10-2; c2=l,104259-10J'; с3=-6,962810-7; с4=1,0031109.
Из (3.38) получим:
лт =
Т~Т~7Т • (3-40)
72
Формула (3.36) справедлива в диапазонах температуры Т
от -2° до 35° С по МПТШ-68, практической солености S от 2 до
42 и давлении от 0 до 1000 бар. Зависимость практической
солености морской воды от электропроводности и температуры пока-
iana на рис. 3.1.
Соленость в Мировом океане главным образом заключена в
пределах между 33 и 37%о. Исключение составляют приустьевые
районы, бассейны опреснения (такие как Балтийское и Черное
моря), где соленость значительно уменьшается, и бассейны осо-
юнения (Средиземное и Красное моря), где соленость превышает
*8 %о. Средняя соленость вод Мирового океана - 34,72%о.
2 3 4 5
Электропроводность, См/м
Рис. ЗА. Зависимость практической солености морской
воды от электропроводности и температуры по ШПС-78
С глубиной соленость воды в разных районах Мирового
океана изменяется по-разному. В Черном море, например,
увеличивается. В Атлантическом океане сначала растет (воды повышенной
солености), затем понижается и снова повышается. В таблице 3.4
приведены средние значения солености воды в Мировом океане.
Если бы удалось выделить из океана всю растворенную в нем
73
соль, то она покрыла бы весь земной шар слоем соли толщиной
более 40 мъесом 95 тонн на 1 м2!
5.7. Плотность. Удельный объем
В океанологии вместо параметра состояния «масса» удобнее
пользоваться удельным параметром - плотностью, а вместо
объема - удельным объемом.
Плотностью тела р называется предел отношения массы Am
элемента тела к его объему ДКпри Л V, стремящимся к нулю:
.. Am dm
р= lim = .
AV-+0&V dV
При этом масса всего тела равна:
w=JJJpdK,
V
где интегрирование осуществляется по всему объему Ктела.
В случае однородного тела его плотность р постоянна по
всему объему V и равна:
Р = -. (3.41)
Размерность плотности р в системе СИ - кгм"3.
Удельный объем v (размерность м3кг"!) есть величина,
обратная плотности:
v = p~!. (3.42)
Плотность и удельный объем морской воды являются
функциями температуры, солености и давления (рис. 3.2):
p = p{S,T,p\ v = v{S9T9p). (3.43)
Если вместо температуры Тт 5itu для определения плотности
используется потенциальная температура 0 (см. раздел 4.10), то
вычисленная плотность называется потенциальной:
рв=р(5,9,р) . (3.44)
В практических расчетах (для сокращения числа знаков и
других целей) вместо плотности иногда используется аномалия
плотности (условная плотность):
а = р-100о[хг-лГ3]; (3.45)
вместо удельного объема - условный удельный объем
74
у'^О^у-ЭДО^-КГ1] (3.46)
или аномалии удельного объема при атмосферном давлении
5o=Vsn)-v35,o,o [У-кг"1]
и in situ
14
5 = vct>-v35A;, [л^-хг"1].
Здесь v35,o,o =0,972662-10"3 м3-кт!.
5
S
2
О
30 —
20 —
0 —
IZ
~
.......
!
(
"^!"
§M0
§=35
*—^]§«5Ь^
***-**Js*15 |
"•■'■«■^•e.'in
■"-*-,
1
(3.47)
(3.48)
0 10 20 30
Температура, *C
Рис 3.2. Аномалия плотности морской воды как функция
от температуры и практической солености при
атмосферном давлении по УС-80
Отметим, что выражение условного удельного объема было
введено О.Зундом. В отечественной океанологической практике;
оно используется по предложению Н.Н.Зубова. Аномалия
удельного объема при атмосферном давлении предложена X. Свердру-
пом. В зарубежной литературе ее иногда называют термостери-
14 Буквенные обозначения внизу обозначают, что параметры состояния
определяются для соответствующих значений солености, температуры
и давления.
75
ческой аномалией. Многие океанологи умножают аномалии
удельного объема на 108, чтобы было легче с ними работать.
В различных источниках океанографических данных
приводится обычно какая-нибудь одна из указанных выше величин.
Для перевода ее в другие пользуются следующими формулами:
106
v''^)-m- (350)
50=Т—1—Ъ- 9,72662 -1(Г4; (3.51)
(а + 103)
v'= (80+0,72662) 103. (3.52)
В связи с тем, что морская вода представляет собой двухком-
понентную систему, важно знать не только ее удельный объем
(плотность), но и удельные объемы (плотности) ее компонентов:
чистой воды и соли. Для определения удельных объемов
компонентов морской воды воспользуемся результатами, полученными
в разделе 3.4.
Для удельного объема морской воды вместо формул (3.27),
(3.30), (3.31) и (3.32) имеем:
v = (l-S)-vw+Svs=vw+vs , (3.53)
^• = vs-%, (3.54)
%=v-S~, (3.55)
vs=v + (l-S)~, (3.56)
где vw ,vs - парциальные удельные объемы чистой воды и соли в
морской воде, vw, vs - удельные объемы чистой воды и морской
соли.
Разделив (3.53) на v2, получим выражение для плотности
морской воды в виде суммы плотности солей и чистой воды в
морской воде:
76
p = I = 5-^- + (1-5).^- = ps+p„ , (3.57)
V V V
где -у- = рж , ~y = ps - парциальные плотности чистой воды и
v v
соли в морской воде.
Если функция v(S,T,p) задана, то по формулам (3.55) и (3.56)
легко вычисляются парциальные удельные объемы соли и чистой
поды в морской воде (рис. 3.3 и 3.4), а затем, согласно (3.53) и
(3.57), удельный объем и плотность соли и чистой воды
соответственно.
Из формул (3.53}-(3.57) следует ряд интересных выводов. Так,
оказывается, для стандартной морской воды (5=35, Г=0° С)
плотность солей возрастает от поверхности моря до глубины 10 000
дбар на 44,6%. При этом плотность чистой воды в морской воде в
том же диапазоне давлений возрастает лишь на 3,8%, а сама
плотность морской воды in situ - на 4,2%.
Из рисунков 3.3 и 3.4 видно, что с увеличением солености
парциальный удельный объем чистой воды уменьшается,
парциальный объем соли увеличивается. С увеличением температуры
парциальный удельный объем чистой воды сначала уменьшается
примерно до температуры наибольшей плотности (около 4° С),
«атем начинает повышаться. Парциальный же удельный объем
морской соли при увеличении температуры всегда повышается.
В связи с тем, что удельный объем является параметром
морской воды, он, естественно, связан с некоторыми
термодинамическими потенциалами:
v = ^J =^| • (3.58)
дР \t,s &Wjs
Учитывая, что термодинамический потенциал Гиббса g*
определяется через легко измеряемые параметры (температура,
соленость и давление), некоторые ученые используют его для
вычисления многих параметров системы, в том числе и удельного
объема [59].
Среднее значение плотности морской воды при атмосферном
шипении в Мировом океане - 1027,54кгм"3 (табл. 3.4).
Плотность воды с глубиной практически всегда и везде повышается.
77
ou-ia
2000
4000 6000
Давление, дбар
Рис 3.3. Зависимость парциального удельного объема
(Ю4м3кг1) чистой воды в морской воде от солености и
температуры при атмосферном давлении (а), давления и
температуры при S-35 епс (б)
В толще воды существуют поверхности одинаковой плотности
- изопикнические поверхности и поверхности одинакового
удельного объема - изостерические.
78
Рис. 3.4. Зависимость парциального удельного объема
(104м*кг~1) соли в морской воде от солености и
температуры при атмосферном давлении (а), давления и
температуры при S=35 епс (б)
3.8. Давление. Уравнение гидростатики
Морская вода, как и все жидкости, характеризуется тем, что не
оказывает сопротивление сдвигу и поэтому может изменять свою
форму под воздействием сколь угодно малых сил. Для изменения
же объема морской воды требуются более значимые внешние
сипы. При этом в воде возникают упругие силы, стремящиеся урав-
79
новесить действие внешних сил. Это проявляется в том, что
отдельные части воды действуют друг на друга или на
соприкасающиеся с ними тела с силой, воздействие которой
характеризуется давлением.
Давлением называется физическая величина р, равная пределу
отношения численного значения AF„ нормальной силы,
действующей на участке поверхности тела площадью AS, к величине
этой поверхности при Д5, стремящейся к нулю:
AF dF
р= lim—"- = —*-. (3.59)
м->о AS dS
В воде, находящейся в состоянии равновесия, давление -
скаляр, абсолютно независимый от ориентации площадки AS, на
которую давление действует, иначе говоря, наблюдается
изотропность давления.
Для доказательства этого утверждения рассмотрим
треугольный двумерный элемент в неподвижной жидкости (рис. 3.5).
Поскольку движения нет, на элемент жидкости действуют только
силы тяжести и давления. Проекции этих сил на оси ОХи OY
дают равенства (с учетом, что dz=l)
px'dy-pds- since = 0, (3.60)
, dxdy
ру dx-pdscosa = mg = p-Vg = pg —.
(3.61)
i
1
p7*
t
NX
dV
1 t 1
1 s J
Dx
Рис. 3.5. К доказательству изотропности давления в воде,
находящейся в состоянии покоя
Делая подстановку dy = ds • sin а и dx = ds- cos а в (3.60)-
(3.61), получим:
80
Рх=Р'>
ds
Py=P + gP~'
При стремлении выбранного элемента жидкости стянутся в
точку, последний член в правой части второго уравнения
стремится к нулю. Тогда
Рх=Ру=Р-
Поскольку угол а произволен, давление одинаково во всех
направлениях. Аналогичное доказательство можно привести и для
грехмерного элемента воды.
На всю толщу воды Мирового океана действует атмосферное
давление, которое в большинстве формул для расчета физических
свойств морской воды непосредственно в них включено.
Поэтому, когда говорят, что такое-то свойство вычислено при давлении
равном нулю, - это означает, что расчет производился при
атмосферном давлении.
В толще воды океана, находящейся р{ • AS
в состоянии покоя под действием
только силы тяжести, давление на двух
разных горизонтах отличается на
величину, численно равную весу верти-
кального столба жидкости,
заключенного между этими горизонтами^ с - р • g • Az
площадью сечения, равной единице.
Выделим в воде цилиндр с
вертикальной осью, сечением AS и высотой Az
(рис. 3.6). Вдоль его оси, кроме сил
давления на основания, будет
действовать объемная сила тяжести -
mg = -pgAz-AS (ось z направлена
вверх), где р - плотность воды, g -
ускорение силы тяжести. Из условия
равновесия следует:
p2AS = plAS-pgAz-AS.
Откуда
Ap = Pi-P\ =~Pg-Az, (3.62)
AS
Az
p2AS
Рис. 3.6. К выводу
уравнения гидростатики
81
что и требовалось доказать. При стремлении Az -» 0 выражение
(3.62) преобразуется к виду
dp = -pgdz. (3.63)
Это выражение представляет собой широко известное
уравнение гидростатики.
В толще океана существуют поверхности равного давления -
изобарические поверхности. Расстояние между изобарическими
поверхностями, из-за различий в удельных объемах (плотностях)
воды и ускорения силы тяжести в разных частях моря, будут в
общем случае, согласно (3.63), различными на разных
вертикалях.
Кроме того, при условии гидростатического приближения
направление градиента давления совпадает с направлением силы
тяжести, поэтому как изобарические, так и изопикнические
поверхности, перпендикулярны направлению ускорения силы
тяжести и поэтому являются горизонтальными.
В Международной системе единиц СИ за единицу давления
принят паскаль (Па):
1Па=1кг-м',-с2=1Н-м"2.
Некоторые коэффициенты перерасчета одних единиц
давления в другие приводятся в таблице 3.5.
В то же время в океанологии до сих пор применяется и другая
единица давления - бар (1 бар=105 Па) или десятая доля бара -
децибар (дбар). Это объясняется тем, что изменение давления на
1 дбар соответствует изменению глубины приблизительно на 1 м;
такое соответствие позволяет иногда глубину в метрах заменять
давлением в дбарах.
Таблица 3.5
Коэффициенты перевода единиц давления
Единицы
измерения
Па
бар
атмосфера
мм. рт. ст.
Па
1
10>
101325
133,322
бар
10"
1
1,01325
0,00133
атмосфера
9,869 10*
0,9869
1
0,001316
мм. рт. ст. 1
7,5024-103
750,24
760
1
Чтобы показать это, рассмотрим давление столба воды
плотностью р и единичной площадью. Определим, какой должна быть
82
его высота Дг, чтобы давление Ар на его нижнем основании было
равным 1 дбар=104 Па. Для этой цели возьмем формулу (3.62),
выразим ее относительно высоты и подставим в нее Др=104 Па,
#=9,8 мс2, р=1027,54 кгм' (средняя плотность морской воды
при атмосферном давлении):
Az = -^- = — = 0,9931 м*\м.
gp 9,8-1027,54
Однако в ряде случаев требуется точная зависимость между
давлением и глубиной, например при проведении измерений в
придонном слое морей и океанов (океанографический зонд
измеряет давление, а эхолот по времени прохождения акустического
сигнала - глубину). Для определения этой зависимости
проинтегрируем по вертикали уравнение (3.63):
)g(z)dz = )vSJ>dp. (3.64)
О о
Учитывая приведенную ниже формулу (3.77), имеем:
g0 + -az \z = ivsrpdp, (3.65)
где vsjp ~ удельный объем океана in situ. Если z заменить
давлением р в выражении для ускорения силы тяжести, получим:
р
\VSTpdP
*= ° х - (366)
go + -ap
Если имеются наблюдения за давлением, температурой и
соленостью морской воды по глубине в океане, выражение (3.66)
может быть вычислено численно.
При отсутствии же гидрологических наблюдений
приближенную зависимость глубины от давления можно получить
следующим образом. Для этого интеграл в числителе правой части (3.66)
представим в виде суммы:
р р р
\vsTpdP = lvis,o.pdP + fop , (3.67)
83
где 5 - аномалия удельного объема относительно стандартного
океана (3.48). Второй интеграл в правой части (3.67) почти всегда
значительно меньше первого и им обычно пренебрегают. Первый
интеграл можно найти аналитически, если подставить в него
выражение для удельного объема, например, используя уравнение
состояния (4.13). Однако в результате получится очень
громоздкое выражение. Во избежание этого интеграл с помощью метода
наименьших квадратов можно аппроксимировать, например
степенным многочленом четвертого порядка:
р
\v35,o,pdP «V Р + Сг- Р2 + сз • РЪ +^4 • р4 .
(3.68)
Если использовать уравнение состояния УС-80 (4.13), то
коэффициенты в выражении (3.68) будут равны: Ci=9,72659,
с2=-2,251210"5, с3=2,27910"10, с4=1,821015, где р дано в дбар.
Подставляя (3.68) в (3.66), получим:
z =
сгр + с2р2+сгр*+с4рА
1
go+~a'P
(3.69)
где р - давление, дбар; z - глубина, м. На рис. 3.7 приведены
значения разности между давлением и глубиной, вычисленной по
формуле (3.69) как функции давления и широты.
1000 2000 3000 4000 5000 6000 7000 8000 9000 10000
Давление, дбар
Рис. 3.7. Разность между давлением [дбар] и глубиной [м]
для стандартного океана как функция давления и широты
84
В ряде случаев возникает необходимость обратной задачи -
пересчета глубины в давление. Непосредственной формулы
такого перевода нет. Для этого обращают уравнение (3.69) [1].
Обозначим зависимость глубины от давления как
z=z(p). (3.70)
Требуется решить обратную задачу - по заданной глубине
вычислить давление, т. е. решить уравнение
/p)=z(p)--z=0. (3.71)
Имеются различные методы решения алгебраических
уравнений типа (3.71). Здесь рассматривается только один -
итерационный. Уравнение (3.71) заменяют равносильным ему уравнением
Р=№- (3.72)
Затем, выбирая некоторое начальное приближение р0,
вычисляют последовательные приближения:
P»i=f(*t> "=<М,2,.... (3.73)
Существуют несколько способов приведения уравнений к
виду (3.72). Если есть возможность найти аналитическое выражение
производной ——, то можно поступить следующим образом.
dp
Разложив уравнение (3.70) в ряд Тейлора и пренебрегая членами
второго и выше порядков малости, получим:
* = Нр.)+(р-л)'^Н (3-74)
dp
Перепишем (3.74) в виде:
(<ь(ря)
А,+. = Рп + [* - *(р»)^^ 'и=0»1 А - • <3-75>
Уравнение (3.75) аналогично (3.72) и служит основой
итерационного процесса Ньютона-Рафсона. Процесс продолжается,
пока \рп+{ - рп | > 8, где е - любое малое число.
В практике океанологических исследований для измерения
давления морской воды в основном используются датчики
абсолютного давления (sealed gage). Их чувствительный элемент
монтируется в корпус так, чтобы под его мембраной был вакуум, а с
другой стороны - внешнее давление. В этом случае выходной
сигнал будет пропорционален величине внешнего давления. Эти
датчики имеют один вход для подачи измеряемого давления.
85
Кроме датчиков абсолютного давления иногда используются
датчики дифференциального давления (differential gage),
имеющие два входа для подачи давления как с одной, так и с другой
стороны мембраны, причем выходной сигнал датчика
определяется разностью этих давлений.
Затем выходные сигналы соответствующим образом
преобразуются либо в пропорциональные изменения сопротивления (ре-
зистивные датчики), либо в резонансные частоты (резонансные
датчики).
3.9. Поле силы тяжести. Ускорение силы тяжести
Как и все тела на Земле, морская вода находится под
действием поля силы тяжести. Сила тяжести Р является
результирующей силой между силой притяжения Земли fE и центробежной
силой вращения Земли /w (рис. 3.8) и равна
p = w.g = y.-^lyX + m.Co2i?£-cos(9) , (3.76)
RE
где у - гравитационная постоянная, равная 6,67-10"11 м3кг"1с'2;
Me, Re - масса и радиус Земли соответственно; т - масса тела;
ф - широта места; со - угловая скорость вращения Земли, равная
2л/86400 с.
Рис. 3.8. К определению силы тяжести и ее ускорения
Отличие силы тяжести от силы тяготения Земли невелико, так
как центробежная сила значительно меньше, чем fE. На эквато-
86
ре, например, fE почти в 300 раз больше, чем fm. Угол а между
направлениями /£ иР можно оценить, воспользовавшись
теоремой синусов
™feL А « 0,0035- cos(cp),
5ш(ф) Р ^
откуда sin(a)«0,0018зт(2ф). Отсюда видно, что в зависимости
от широты ф угол а изменяется от нуля (на экваторе и на
полюсах) до 6' (на широте 45°).
Ускорение силы тяжести g определяет силу тяжести,
действующую на единицу массы в данной точке. Согласно (3.76),
ускорение g на экваторе меньше по сравнению с полюсами (сила
притяжения Земли на экваторе меньше из-за большего радиуса
Земли на экваторе). С глубиной величина g также изменяется -
она незначительно увеличивается.
Зависимость ускорения силы тяжести g от географической
широты и глубины (рис. 3.9) можно выразить следующей
эмпирической формулой [63]:
s(<P>*)=go(<p) + «'*. (3-77)
где g0=9,780318(l+(5,278810"3+2,3610'5-sin>)sin^) - ускорение
силы тяжести на поверхности моря, м-с"2=105 мГал (миллигал);
а=2,18410"6 - средний вертикальный градиент ускорения силы
тяжести, с"2; ф - географическая широта; z - глубина, м. С
большой точностью вместо глубины можно использовать давление в
дбар.
Несмотря на изменение ускорения силы тяжести, как по
широте, так и по глубине, для многих расчетов можно принять g
постоянным и равным 9,81 мс"2.
Направление g можно определить в каждой точке отвесом.
Нормальная к линии отвеса поверхность называется изопотенци-
альной поверхностью, или поверхностью уровня. Отметим, что
через каждую точку вертикали проходит одна и только одна изо-
потенциальная поверхность. Естественно, что в любой точке изо-
потенциальной поверхности величина потенциала силы тяжести
одинакова
87
fi.84 —
0.82-
9.8 —
9.78-
1
)
f
1 1 '
20
1
: i
/
' j
1 1 ■
40
-"""~
1
!
H
1
1 j i j 1
60 80
Широта,'
Рис. 3.9. Зависимость ускорения силы тяжести g на по-
верхности моря от широты, рассчитанная по (3.77)
ЗАО. Геопотенциал. Динамическая глубина
Величина потенциала (геопотенциала) Ф определяется
работой, которую надо произвести против действия силы тяжести,
перемещая единицу массы вверх по вертикали на некоторое
расстояние от выбранной в качестве отсчетной поверхности уровня.
Элементарная работа */Ф, совершаемая над единицей массы по
вертикали на расстоянии dzb равна
d<f> = -gdz. (3.78)
Проинтегрируем уравнение (3.78) по z:
ДФ-Ф^-Ф^ =-}«*. (3.79)
h
Приняв на отсчетной поверхности геопотенциал равным нулю
(Фг = 0) и g=const, из (3.79) получим:
<& = -g(z2-z}) = -gAz. (3.80)
Очевидно, что работа Ф равна деленной на массу
потенциальной энергии (удельная потенциальная энергия) в точке, нахо-
88
дящейся на расстоянии Az от начальной изопотенциальной
поверхности. Единица измерения геопотенциала в системе СИ есть
м2-с~2 или дж-кг"1. В настоящее время в океанологии продолжают
использовать в качестве единицы измерения геопотенциала и
динамический метр: 1 дин. м=10 м2-с'2. Из (3.79) следует, что при
перемещении единичной массы по одной и той же
изопотенциальной поверхности работа не совершается.
Сравнивая (3.63) и (3.78), мы приходим к следующему
важному соотношению:
dp = p-M>, или d$> = vdp, (3.81)
связывающему между собой давление, плотность (удельный
объем) морской воды и работу по вертикали в поле силы тяжести.
В связи с тем, что при перемещении объема морской воды по
глубине совершается работа, ее количество может служить мерой
вертикального расстояния. Расстояние по вертикали, измеряемое
в море в единицах работы силы тяжести (геопотенциала),
называется динамической (геопотенциальной) глубиной или высотой^ в
зависимости от выбора отсчетной поверхности.
Динамическую глубину можно получить двумя способами,
интегрируя выражения (3.78) или (3.81):
D = A<D = -j#fe, (3.82)
ч
£> = АФ = ]уя>ф, (3.83)
Ро
где z0 и 2\ — глубины моря; р0 и р\ - давления на этих глубинах.
Единица динамической глубины (высоты) - м2с"2.
В океанографической практике для расчета динамической
глубины (высоты) используется главным образом (3.83). В связи с
тем, что измерения температуры и солености морской воды, по
которым затем рассчитывается удельный объем, проводятся на
отдельных горизонтах, выражение (3.83) вычисляется с помощью
методов численного интегрирования, например, методом
трапеций:
0 = ^^^{рм-рХ (3.84)
89
где п - количество горизонтов наблюдений, v^ - удельный
объем морской воды (м3,кг-1) на /-горизонте, /?, - давление (Па) на i-
горизонте. Если давление задается в дбар, то выражение (3.84)
умножается на переводной множитель 104.
Если интеграл в уравнении (3.83) выразить через (3.67),
получим:
D = D3Sfi_p + D* = \viSfiJp + \bdp, (3.85)
О О
где первый интеграл в правой части представляет собой
стандартную динамическую глубину (высоту) между изобарическими
поверхностями 0 и р9 а втором - аномалию динамической глубины
(высоты). Заметим, что в западных странах пользуются в
основном именно аномалией, а не динамической высотой.
3.11. Потенциальная энергия.
Аномалия потенциальной энергии
Для определения потенциальной энергии Р.Е. единичного
объема морской воды с плотностью р, находящегося на некотором
расстоянии от отсчетной поверхности, необходимо геопотенциал
Ф умножить на плотность и проинтегрировать полученное
выражение по вертикали от отсчетной поверхности до заданного
горизонта:
Р.Е.= |р-Ф-<Ь. (3.86)
Покажем, что при условии гидростатического приближения
интеграл давления по глубине также характеризует
потенциальную энергию. Для этого преобразуем его следующим способом,
используя при этом правило интегрирования по частям и
уравнение гидростатики (3.63):
]pdz = pz\2zsa - ]z-dp = p-z\[st - }z~-<k =
4
Ps-zs-Pbzb+ \pgzdz.
dz
(3.87)
90
zs 2s 2s
Учитывая, что zB(ps- pB)= zB • \dp = ]zBdp = - jzBp gdz,
*B *B *b
перепишем (3.87) следующим образом: A
*s zs
jpdz = ps{zs-zB) + jp-g{z-zB)dz. (3.88)
*в гв
Но (пренебрегая изменениями g по высоте)
*• fa-**)=**-Ф*>
где Ф — геопотенциал.
Откуда
zs 2S
]p.dz=Ps_.(<bs-<bB)+ |р.(ф_фв).&. (3.89)
Учитывая (3.86), а также тот факт, что — равно массе атмо-
g
сферы над поверхностью океана на единицу площади, уравнение
(3.89) можно интерпретировать как потенциальную энергию
столба воды относительно дна zB или геопотенциала ФЛ
состоящую из суммы двух составляющих: первая характеризует
потенциальную энергию атмосферы относительно дна океана, вторая
собственно потенциальную энергию океана. Первая
составляющая мала по сравнению со второй и ею обычно пренебрегают.
Интеграл давления можно выразить и иначе, используя
уравнение гидростатики (3.63):
']p.toJ]2±.{p.g.dz) = -^-dpJ]£?-.dp, (3.90)
*в zb * Pa ° Ps *
где v - удельный объем морской воды in situ, g - ускорение силы
тяжести, р - давление. Выразив удельный объем VsrP через сумму
удельного объема стандартного океана V35,ofP и аномалии
удельного объема 6stp, получим:
г'в Ps * Ps ° * < Ps 8
Так как интеграл давления по глубине представляет собой
потенциальную энергию столба воды относительно дна океана
(3.89), первый член в (3.91) выражает потенщалъную энергию
91
стандартного океана, а второй - аномалию потенциальной
энергии х- Размерность х-кгс"2=Джм"2
Задачи и вопросы для повторения
3.1. В чем суть гипотезы формировании водной массы Мирового
океана А.П. Виноградова?
3.2. Назовите этапы формирования солевого состава Мирового
океана.
3.3. Что понимается под законом Дитмара?
3.4. Почему морскую воду можно представить как двухкомпонент-
ную систему?
3.5. Выведите основное уравнение термодинамики для морской
воды.
дТ
3.6. Получите соотношение Максвелла —
dS
л ,р
tf
P.S
Ответ:
Возьмем независимые переменные л\ р и S. При этих параметрах
термодинамическим потенциалом является энтальпия h{r\ ,pfS) (см. разделы
2.3 и 3.4). Сравнивая ее полный дифференциал с (3.12), получим
Т =
*п
dh
. Перекрестно продифференцируем эти выра-
п,р
жения, а затем, сравнивая смешанные производные, получим искомое
выражение.
3.7. Дайте определения парциальных величин применительно к
морской воде.
3.8. Что подразумевается под температурой in situ? '
3.9. Какая шкала температуры используется в настоящее время?
3.10. Что понимается под истинной соленостью?
3.11. В чем заключается отличие определения солености методом
Мора-Кнудсена от метода электропроводности?
3.12. Что такое аномалия плотности и аномалия удельного объема
морской воды?
3.13. Как изменяются парциальные удельные объемы чистой воды и
соли в морской воде от солености?
3.14. Покажите, что гидростатическое давление в толще воды
изотропно.
92
3.15. На какую величину отличается давление на двух разных
горизонтах в воде, находящейся в состоянии равновесия в поле силы
тяжести?
3.16. Почему в океанологии до сих пор применяется единица
давления бар?
3.17. Что численно больше: глубина или давление?
3.18. Где больше ускорение силы тяжести: на экваторе или на
полюсах?
3.19. Что такое изопотенциальные поверхности (поверхности
уровня)?
3.20. В чем измеряется динамическая глубина (высота)?
3.21. Что такое аномалия динамической глубины?
93
ГЛАВА 4. УРАВНЕНИЕ СОСТОЯНИЯ.
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА МОРСКОЙ ВОДЫ
В первой главе мы уже рассмотрели сложную структуру
пресной воды. Добавление соли еще более затрудняет построение
теоретических уравнений состояния, связывающих температуру
воды, ее соленость, плотность и давление. В настоящее время
имеются лишь эмпирические уравнения состояния. В
океанографической практике сейчас используется главным образом
Международное уравнение состояния 1980 г. На его основании
рассчитаны многие физические свойства морской воды. Кроме того,
существует много различных эмпирических формул для
вычисления других ее свойств.
4.1. Уравнение состояния морской воды.
Дифференциальная форма уравнения состояния
Зависимость, связывающая между собой параметры
состояния: удельный объем (плотность), температуру, соленость и
давление, называется уравнением состояния морской воды. Из-за
сложности построения этой зависимости уравнение состояния
морской воды не может быть найдено теоретически. Поэтому
сейчас в практике пользуются только эмпирическими
формулами. Наиболее известными из них являются уравнение состояния
морской воды Кнудсена-Экмана и Международное уравнение
состояния 1980 г. (УС-80). Отметим, что эмпирические уравнения
состояния не являются уравнениями состояния в
термодинамическом смысле, так как с их помощью невозможно описать
произвольный термодинамический процесс, когда одновременно
изменяются все параметры состояния [22]. В этом состоит коренное
отличие термодинамики морской воды от термодинамики
атмосферы, где выполняется теоретическое уравнение для идеального
газа. Одновременно разрабатывались такие формы уравнения
состояния, которые удовлетворяли бы как океанографической, так и
термодинамической точкам зрения. К таким уравнениям
состояния относятся, например, уравнения Тумлирца, Тайта-Гибсона и
некоторые другие.
94
В общем виде уравнение состояния морской воды
записывается так:
или
Однако в океанологии наиболее часто применяется другая
форма:
v=v(5,r,/7). (4.1)
Уравнение (4.1) относится к термическим уравнениям
состояния. Из самого существования этого уравнения можно получить
важные следствия. Действительно, рассматривая такие изменения
состояния морской воды, при которых постоянны два из трех
параметров состояния, мы можем получить пять основных
термодинамических коэффициентов.
Термодинамическим коэффициентом термического
расширения называется величина а, характеризующая относительное
изменение объема морской воды при изобаро-изохалинном
изменении ее температуры на один градус (°С-1 или °К*1)15:
1
а = —
v
9v
дТ
или
P.S
1
а = —
Р
др
дТ
(4.2)
p>s
Термодинамическим коэффициентом соленостного сжатия
называется величина (3, характеризующая относительное
изменение объема морской воды при изобаро-изотермическом
изменении ее солености на единицу (епс1)16:
к v а?
или
Р?
(4.3)
РХ
Термодинамическим коэффициентом изотермо-изохапинной
(изотермической) сжимаемости называется величина к, харак-
15 В классической термодинамике этот коэффициент называется просто
коэффициентом расширения. Термическим коэффициентом
расширения называется другая величина:
а = -
1
dv_
дТ
где v0 - объем системы при 0° С.
16 Знак минус в формулах (4.2)-(4.4) указывает на уменьшение
удельного объема с увеличением соответствующих параметров.
95
теризующая относительное изменение объема морской воды при
изотермо-изохалинном изменении ее давления на единицу (Па-1
или дбар"1)17:
1
к = —
V
dp
T,S
1
или к = —
р
др
др
(4.4)
T,S
Термодинамическим коэффициентом давления (упругости)
называется величина у, характеризующая относительное
изменение давления морской воды при изостеро-изохалинном
изменении ее температуры на градус (°С_1 или °К"1):
.- 1 .дР\
у = -
дТ
(4.5)
v,S
Наконец, коэффициентом изопикничности (термохалинно-
сти) называется величина ("С"1 или "К"1)
6 = 1.^1
S' дТ
(4.6)
ру
Найдем полный дифференциал удельного объема морской
воды (4.1):
dS
Т,р
■dS+*\
дТ
■dT +
S.p
dp
dp.
(4.7)
StT
Затем, подставив в (4.7) термодинамические коэффициенты
а (4.2), Р (4.3) и к (4.4), получим дифференциальную форму
уравнения состояния морской воды:
— = ач/7,-р-<де-к-ф. (4.8)
v
Из (4.8) следует, что удельный объем v (плотность р) воды в
океане зависит главным образом от трех факторов:
- температуры воды Т - благодаря термическому
расширению, выраженному коэффициентом а и связанному с изменением
скорости молекул воды и, как следствие, расстояния меэюду
ними;
17 Тождественность обоих видов коэффициентов в (4.2)-(4.4) следует из
определения удельного объема как величины обратной плотности и
правил дифференцирования.
96
- солености воды S - растворенные в воде соли изменяют ее
удельный вес и приводят к соленостному сжатию, выраженному
коэффициентом ft;
- давления воды р - из-за ее сжимаемости под давлением вы-
шележащих слоев воды, определяемой коэффициентом
сжимаемости к.
Существование уравнения состояния морской воды приводит
к тому, что термодинамические коэффициенты связаны между
собой. Чтобы показать это, примем удельный объем постоянным.
(v=const) и разделим (4.7) на dT, тогда получим:
5v
dS
Т.Р
ds_
дТ
дТ
stP
dv
dp
sj
дТ
= 0.
(4.9)
S,v
ИЛИ
18
dv
dS
dS
г., дТ
dv
дТ
S.P
dv
dp
P.* , r
dp\
sj dT\
' dv
dT
S.P
S,v
= -1.
(4.10)
После подстановки термодинамических коэффициентов а
уравнение (4.10) получим соотношение, связывающее их между
собой:
а = р-8-5 + у-к р. (4.11)
Выражение (4.11) используется для более детального изучения
уравнения состояния морской воды методом термодинамических
потенциалов. В частности, его можно применить для определения
у. Иначе эту величину найти нельзя, так как морскую воду невоз-<
можно нагреть без изменения ее объема.
Рассмотрим несколько вариантов уравнения состояния
морской воды. Сначала обратимся к эмпирическому уравнению
состояния Кнудсена-Экмана, а затем - к Международному
уравнению состояния морской воды, 1980. Уравнение состояния
Кнудсена-Экмана использовалось в океанологии с начала прошлого
18 „ ^i
Строго говоря, делить на производную—|
можно только при тех
s,p
температуре, солености и давлении, где она не равна 0.
97
века и до 1981 г., когда было введено более точное
Международное уравнение.
Уравнение состояния Кнудсена-Экмана имеет вид:
vstp=vsto-(1-V--p), (4.12)
где Vsm - удельный объем при атмосферном давлении,
представляющий собой сложную эмпирическую функцию температуры и
солености; ц - средний коэффициент сжимаемости воды в
интервале давлений от 0 до р, также являющийся сложной функцией
температуры, солености и давления. Вид vS7r) и \i можно найти,
например, в работе [30].
Эмпирическая функция vsw была получена на основании
лабораторных исследований, проведенных М. Кнудсеном, К. Фор-
хом и С. Серенсеном в начале XX века. Они использовали для
своих опытов всего 24 пробы морской воды с поверхности моря,
собранные в морях Северо-Западной Европы. Основываясь на
гипотезе о постоянстве солевого состава, М. Кнудсен
распространил свои результаты на весь Мировой океан.
Эмпирическая формула среднего коэффициента сжимаемости
ц была определена В. Экманом на основании собственных
результатов и опытов Амача. Для своих опытов В. Экман
использовал всего одну пробу воды. Опыты были точными, но
неполными. В связи с этим его формула являлась основным источником
ошибок уравнения состояния Кнудсена-Экмана.
В настоящее время в океанологии применяется
Международное уравнение состояния морской воды, J980 г. (УС-80)9
основанное на шкале солености ШПС-78, шкале температуры МПТШ-68
и имеющее следующий вид:
VSTp - VST0
1-
KSTpy
(4.13)
или
Psrp = Psto тт;— • (4Л4)
l- P/KSTp
Здесь p - плотность, кг-м"3; v - удельный объем, м3кг"!; Т-
температура, °С; S - практическая соленость; р - давление, бар;
К- объемный модуль упругости, бар.
98
Плотность морской воды при атмосферном давлении
определяется по формуле:
Psto =Р„ + (*о +Ь -Т + ЬгТ2 +Ь3 Т3 +b4-T4)-S +
+ (c0+cl-T + c2-T2)-S3,2+d0-S2,
где pw - плотность пресной воды; 60=8,2449310"1; 6|=-4,089910"3;
62=7,6438-105; &з=-8,2467-10-7; 64=5,3875-10-9; с0=-5,72466-10-3;
c^l.022710-4; cf=-l,6546-10"6; ^0=4,831410Л
Плотность пресной воды pw равна:
pw = а0 + а, • Г + а2 • Т2 + а3 • Т3 + ал ■ Т4 + as ■ Ts, (4.16)
где а0=999,842594; а,=6,793952-10-2; а2=-9,095290-10-3;
а3=1,00168510^; а4=-1,120083-Ю6; o5=6,536332109.
Формула объемного модуля упругости имеет вид":
Ksr^Ksro+A-p + B-p2. (4.17)
Здесь
KSTO=Kw + {f0 + frT + f2-T2+f3T3)-S +
+ ^o+gl'T + g2T2)-S3/2,
где/о=54,6746;/,=-0,603459;/2=1,09987-1 V2\fy=-6,1670-10"5;
#,=7,944-10"2; g,=l,6483102; g2=-5,3009-10-4.
Объемный модуль упругости для пресной воды определяется
так: *
Kw=e0+el-T + e2T2+e3T3+eAT4,
где е0=19652,21; е,=148,4206; е2=-2,327105; е3= 1,360477-10'2;
е4=-5,155288-10-5.
A^Aw + (i0+i1-T + i2-T2)-S + j0-S3'2,
где io=2,2838-103; /,=-1,0981 10s; i2=-l,6078106; у0=1,91075-10Л
A„=h0+hlT + h2T2+hiT3,
где А0=3,239908; Ai=l,43713-10°; Л2=1,16092-10"4; Л3=-5,7790510'7.
В = Bw + (m0 + m, • Т + m2 • Т2 )■ S,
где m0=-9,9348-10-7; m,=2,0816-10"8; от2=9,16971010.
(4.18)
19 Следует помнить, что если вы используете давление в дбар, то для
расчета удельного объема и плотности морской воды по формулам
(4.13) и (4.14) его нужно перевести в бары, т. е. разделить на 10.
99
Bw=k0+krT + k2-T2,
где Ao=8,50935-10"5; Jfc,=-6,12293106; £2=5,2787-10-8.
Уравнение состояния УС-80 справедливо в диапазонах:
0<5<40 (ПШС-78), -2<Г<40°С (МПТШ-68), 0<р^1000 бар.
Проверочное значение: pS7>= 1059,820 для 5=40, Г=40 ир=1000 бар.
При практических расчетах полиномы типа (4.15)—(4.18)
лучше всего вычислять по схеме Горнера. Например, полином (4.16),
если использовать схему Горнера, будет выглядеть так:
Рм/ = а0-ьГ.(а1+7.(а2+Г.(а3+Г.(а4+Л5.Г)))).
Как видим, количество операций сложения и умножения не
изменилось, но операций возведения в степень в уравнении
теперь нет!
Первым, кто попытался получить уравнение состояния
морской воды, с помощью которого можно было бы описать
произвольный термодинамический процесс при одновременном
изменении всех параметров состояния, был К. Эккарт [58]. Он
показал, что уравнение состояния можно выразить уравнением Тум-
лирца:
(p + PoMv-v0) = A., (4.19)
где р - давление; v - удельный объем; р0у v0 и X - функции
температуры и солености (для морской воды). Уравнение состояния
Эккарта получено им для диапазона температур 0<Г<40° С, со-
леностей от 0 до 40 и давлений от 0 до 1000 бар. В
океанологической практике уравнение (4.19) широкого распространения не
получило.
Громоздкость уравнений состояния морской воды Кнудсена и
УС-80 иногда препятствует их непосредственному применению в
теоретических моделях, направленных на изучение океанских
течений или динамики водных масс. Учитывая это, уравнение
состояния часто аппроксимируется более простыми
зависимостями.
Линейная аппроксимация используется в основном в
аналитических моделях и может иметь различные формы, например,
такие:
р = р0[1-а(Т-Г0)] (4.20)
или
100
р = р0.[1-а.(Г-Г0)+р(5-50)], (4.21)
где ро, 7о и S0- некоторые постоянные значения плотности,
температуры и солености; а и Р - постоянные коэффициенты
термического расширения и соленостного сжатия морской воды
соответственно. В большинстве случаев принимается а=2-10"4 и
(3=8-10"4. Формулы (4.20) и (4.21) не описывают нелинейный
характер уравнения состояния морской воды, определяющий
многие ее свойства. Поэтому в океанологии часто применяются
несложные нелинейные аппроксимации, например такие:
Р~Ро="\+<*2'(т-то)+аз-(Т-Т0)2+ (4
+ [a4+as-{T-T0)l{S-S0),
где ab...,a5 - эмпирические коэффициенты, подлежащие
определению; ро, Т0, S0 - некоторые постоянные значения плотности,
температуры и солености. Коэффициенты в (4.22) можно найти с
помощью метода наименьших квадратов.
Так, в работе [11] получено упрощенное уравнение состояния
морской воды при атмосферном давлении, аналогичное (4.22).
Оно основано на УС-80 при Г0=0° С, 50-35. Уравнение имеет вид:
a=28,132-0,0734T-O,00469T2+(0,803-0,002T)(S'35), (4.23)
где a - аномалия плотности^ кг-м"3.
Наглядным методом исследования зависимости плотности
(удельного объема) морской воды от температуры, солености и
давления служат ^-диаграммы.
4.2. Термодинамические диаграммы.
TS-диаграмма
Равновесные состояния гомогенной системы и происходящие
в ней равновесные процессы можно изобразить графически на
плоскости в прямоугольных декартовых координатах, вдоль осей
которых откладываются параметры состояния системы или
однозначно связанные с ними функции состояния. Такое графическое
изображение называется термодинамической диаграммой.
В классической термодинамике наиболее часто используются
диаграммы v-/?, т]*-Ги i\-h (первый символ - величина,
откладываемая по оси абсцисс, второй - по оси ординат).
101
Если вдоль осей х, у, z декартовой системы отложить
параметры состояния морской воды S, Тир, то плотность (удельный
объем) в пространстве этих переменных можно представить в виде
изоскалярных поверхностей. Если эти поверхности
спроецировать на плоскость S, Г, О или любую другую плоскость с
постоянным давлением р в виде изолиний, то получится Г5-диаграмма,
представляющая собой графическое изображение уравнения
состояния при атмосферном давлении или давлении р (рис. 4.1).
Если на эту диаграмму нанесены линии одинаковой плотности
(изопикны), то она называется изопикнической; если линии
одинакового удельного объема (изостеры) - изостерической.
30ч
О 5 10 15 20 25 30 35 40
Соленость, епс
Рис. 4.L Изопикническая TS-диаграмма при атмосферном
давлении. Нанесены изолинии аномалии плотности а, кгм'3.
Плотность рассчитывалась по УС-80
При рассмотрении изопикнической (изостерической) TS-
диаграммы обнаруживается, что изопикны (изостеры) обладают
формой, отличной от линейной. Этот факт отражает нелинейную
зависимость плотности (удельного объема) морской воды от ее
солености и температуры. Нелинейность определяет наличие
экстремумов изопикн (изостер) при соленостях морской воды,
меньших 24,6. Линия, соединяющая эти экстремумы, совпадает с
102
- ds л as
изолинией — = 0, где — есть котангенс угла наклона изопикн
дТ дТ
(изостер) к оси абсцисс ГЯ-диаграммы (рис. 4.1). Как будет
показано ниже, на этой же линии коэффициент термического
расширения а также равен нулю. Изопикны на ГЯ-диаграмме проведены
только до температуры замерзания воды, зависящей от солености
и давления. Ниже этого значения вода в жидком виде не
содержится при атмосферном давлении в Мировом океане.
Рис. 4.2. Средние TS-кривые водных масс Черного моря для
августа (белые кружки) и февраля (черные кружки). Числа
около кружков показывают давление в дбар [26]
На ^-диаграмме может быть отражено реальное состояние
морской воды в Мировом океане. Для этого на диаграмму по
значениям температуры и солености, полученным на горизонтах
наблюдений какой-либо океанологической станции, необходимо
нанести Г^-точки, называемые термохалинными индексами.
Затем, подписав у Г^-индексов значения глубин (давления)
соответствующих горизонтов и соединив эти точки плавной кривой,
получим TS-кривую океанологической станции,
характеризующую состояние морской воды по глубине (давлению) в какой-
103
либо точке океана в конкретный момент времени (рис. 4.2). TS-
кривая в 75-координатах может рассматриваться как кривая
некоторой явной функции Т = f(s).
Интерпретацию океанологических наблюдений в виде Г5-
кривых первым предложил Б. Гелланд-Гансен в 1916 г.
Многочисленные исследования показали, что ГЯ-кривые различных
районов Мирового океана имеют значительное сходство. Это
послужило широкому использованию ГЯ-анализа для выделения
водных масс.
Отметим, что Г£-диаграмма является характеристической
диаграммой в случае процесса смешения двух водных масс,
имеющих разные температуры и солености, так как он изображается на
ней прямой линией [22]. Это свойство Г5-диаграмм является
фундаментальным.
Рассмотрим процесс смешения двух однородных водных масс
А и В. Предположим, что температура и соленость первой водной
массы равна Т\9 Su второй - Т2, S2. Тогда на Г£-диаграмме эти
водные массы будут определяться индексами А и В (рис. 4.3).
А(Т,Л)
Рис. 4.3. Прямая смешения двух водных масс
В случае аддитивности температуры и солености (см. раздел
4.11), термохалинный индекс TS водной массы Af,
образовавшийся в результате полного смешения водных масс А и В, будет
находиться на прямой АВУ называемой прямой смешения.
Температура и соленость смеси определяется формулами смешения:
104
T = Tlmx+T2m29 (4.24)
S = SX -Wj+^'/nj, (4.25)
где mu m2 - пропорции первой и второй водных масс. В
уравнениях (4.24) и (4.25) тх и гп2 выражены в долях единицы: m\+m2=l.
Часто применяется и процентное выражение долей водных масс в
смеси. Положение ГС-индекса на прямой смешения
пропорционально гп\ и гп2: чем больше доля т\9 тем ближе точка М к точке
А.
Отметим, что уравнения (4.24) и (4.25) являются
приближенными, так как не учитывается зависимость удельной
теплоемкости от температуры и солености (подробнее об этом речь пойдет
ниже), а также диссоциация морской воды как электролита.
В случае смешения трех однородных водных масс их индексы,
если они не лежат на одной прямой, образуют треугольник
смешения. ГС-индекс смеси, полученной в результате полного
смешения трех водных масс, будет находиться внутри треугольника
смешения.
Термодинамические ГС-диаграммы применяются в
океанологии для нескольких целей: во-первых, для вычисления плотности
(удельного объема) морской воды; во-вторых, для анализа ГС-
соотношений морских вод.
Иногда при исследовании распространения водных масс в
Мировом океане используются TS-диаграммы рассеяния - это ГС-
диаграммы, на которые нанесены, кроме изопикн, еще и ГС-
индексы (рис. 4.4). Они очень наглядны, однако получить по ним
хоть какую-нибудь количественную информацию нельзя.
Более информативна объемная TS-диаграмма. Она строится в
тех ситуациях, когда необходимо знать распределение объемов
вод, их теплосодержания и массы солей какого-нибудь объекта
исследования (моря, океана, района и т. д.) по ГС-классам,
заданным с определенными шагами по температуре и солености (рис.
4.5).
Если учесть, что температура воды, строго говоря, не является
аддитивной величиной, вместо нее для построения
термодинамических диаграмм можно использовать энтальпию - срТ.
105
Соленость, enc
Рис 4,4, TS-диаграмма рассеяния для Черного моря. Август
Рис. 4,5, Объемная TS-диаграмма вод Черного моря для августа.
Максимальное значение объема (-51%) приходится на TS-класс:
8,5<Т<9,0° С и 22,2<S<22,4 [26]
Кроме ГЯ-диаграмм, можно строить и другие, такие,
например, как GS-диаграмма, на которую нанесены изолинии
температуры - изотермы.
106
4.3. Термическое расширение. Соленостное сжатие
Удельный объем (плотность) морской воды, как следует из
уравнения (4.8), зависит от температуры, благодаря
термическому расширению, выраженному через коэффициент а, и
солености, из-за соленостного сжатия, определяемого коэффициентом р.
Поэтому знание термического расширения и соленостного
сжатия необходимо для исследования тех явлений, при которых
происходит изменение удельного объема. В частности, при
изучении таких процессов в океане, как распространение звука,
формирование вертикальной устойчивости вод и т. д.
Значение коэффициентов а (4.2) и Р (4.3) в зависимости от
температуры, солености и давления можно получить,
дифференцируя формулы (4.14), определяющие р,^ по УС-80, по
температуре и солености, и разделив затем результат на р^, [1]. Итак,
имеем:
Р' Psto ' лт/ ~ кзд> ' V^sTp " Pr JL
a= =2ZL1E fie а ' (426)
'STO
dPsro _ Ж^
kst> " \}^STp " PV JZ° - P' Psto ' ~c
P = —BS- dS_ (42?)
Psto *ks7> V^stp ~ P)
На рис. 4.6 и 4.7 показаны зависимости коэффициентов
термического расширения а и соленостного сжатия р от
температуры, солености и давления.
Из рис. 4.6 видно, что коэффициент а увеличивается с
возрастанием всех трех параметров, причем температура воды играет в
этом процессе наибольшую роль. При атмосферном давлении
изолинии коэффициента термического расширения практически
прямолинейны, а расстояния между изолиниями увеличиваются
с повышением температуры. Увеличение а означает, что для
изменения единицы удельного объема (плотности) морской воды
достаточно меньших изменений в температуре.
Отметим также, что на рис. 4.6а видна область, где значения
коэффициента а отрицательны, т. е. в которой плотность воды с
увеличением температуры воды увеличивается, а удельный
объем соответственно уменьшается. Аномальная область термиче-
107
ского сжатия на диаграмме лежит ниже изолинии а=0, совпа-
дающей с линией наибольших значений плотности.
Рис. 4.6. Зависимость коэффициента термического
расширения 10* а СС1) от солености и температуры при
атмосферном давлении (а), давления и температуры при
S=35 епс (б)
Изолинии коэффициента соленостного сжатия р, в отличие от
коэффициента термического расширения, криволинейны,
особенно в области низких соленостей (рис. 4.7). При этом значения
108
Р в океаническом диапазоне меняются лишь приблизительно на
гОУоСб^б-вЛЗ-Ю^епс'1).
Рис. 4.7. Зависимость коэффициента соленостного
сжатия 104ф (епс1) от солености и температуры при
атмосферном давлении (а), давления и температуры при
S=35 епс (б)
Численно р в области океанских соленостей в зависимости от
температуры воды от 2,5 до 15 раз больше, чем а. Это означает,
например, что изменения солености на единицу приводят к таким
109
же изменениям плотности морской воды, что и изменения
температуры воды на 2,5-И5° С.
Коэффициент соленостного сжатия Р уменьшается с
увеличением как солености, так температуры и давления.
4.4. Стерические колебания уровня моря
Одним из важнейших примеров использования
коэффициентов термического расширения и соленостного сжатия в практике
океанографических расчетов является оценка вкладов
температуры и солености воды в стерические колебания уровня океана, под
которыми подразумеваются колебания уровня, вызванные
изменением удельного объема морской воды. В океанологии главным
образом изучаются стерические колебания, обусловленные
сезонным ходом температуры и солености воды в Мировом океане.
Формулу для расчета стерического отклонения уровня
Azv = z-z*относительно какой-нибудь средней
океанологической станции (например, среднегодовой) можно получить, если
проинтегрировать уравнение (3.63) по вертикали от горизонта Я,
до которого проникают сезонные (межгодовые) изменения
удельного объема морской воды, до поверхности моря с учетом
постоянства ускорения силы тяжести (g=const). Тогда:
Azv=z-z* =- j\yn>-v*sip}4P = — jAv^-ф, (4.28)
& Рн % Рн
где vstp и v*stp - реальное и отсчетное значения удельного объема.
Для оценки вкладов температуры и солености в стерические
колебания разложим &vSTp = v^ - vSTp в ряд Тейлора с
точностью до членов первого порядка малости:
Avcr=— ДГ + — Д5 + ... = у(аДГ-рДГ), (4.29)
STp дТ dS ,
где AS=S-S*y ДГ=Г-7*, Ту&- соответствующие отсчетные
значения температуры и солености. Подставляя (4.29) в (4.28),
получим:
1 Ро 1 Ло
Azy = AzT +Azs =— Jv-аДГ-ф Jv • (3 - ДО • ф.
* Рн * Рн
(4.30>
ПО
Из выражения (4.30) видно, что в реальном океане при
повышении температуры (т. к. а>0) уровень моря растет, а при
увеличении солености - понижается (т. к. Р>0).
На рис. 4.8 показано стерическое отклонение уровня моря в
Атлантическом океане в августе, рассчитанное относительно
горизонта 1500дбар. При этом использовался одноградусный
массив World Ocean Atlas 2001. В качестве отсчетной станции была
взята среднегодовая океанографическая станция Атлантического
океана.
Рис. 4.8. Стерическое отклонение уровня моря (см) в
Атлантическом океане в августе
4.5. Коэффициент изопикничности
Коэффициент изопикничности (термохалинности) 8 (4.6)
вызывает интерес только применительно к морской воде и
представляет собой важную численную характеристику уравнения
состояния морской воды.
111
Рассмотрим уравнение изопикны (p=const) при постоянном
давлении:
■dT + -
P.S
dS
dS = 0.
(4.31)
р.т
Разделив (4.31) на dp/dS, получим:
dS
dS =
дТ
dT = ctg<sfdT.
(4.32)
РУ
Отсюда следует, что изопикническая (термохалинная)
производная dS/dT равна котангенсу угла наклона изопикны (изосте-
ры) (р в данной точке (S,T) к оси абсцисс, взятому с обратным
знаком:
asl
Ctg(f> =
дТ
(4.33)
pj>
Термохалинную производную можно получить как
отношение коэффициентов термического расширения и соленостного
сжатия:
-7Г = £. (4.34)
Значения термохалинной производной показаны на рис. 4.9.
Поскольку функция dS/dT характеризует отношение
термического расширения к соленостному сжатию, ее значения дают
возможность оценить влияние этих двух процессов на
формирование поля плотности.
Из рис. 4.9 видно, что соленостное сжатие в большей степени,
чем термическое расширение, влияет на плотность в области
низких соленостей (и низких температур); термическое расширение
достигает наибольшего влияния в области высоких температур (и
высоких соленостей).
4.6. Температура наибольшей плотности. Солоноватые и
морские воды
В предыдущих пунктах было обращено внимание на то, что
зависимость плотности (удельного объема) от температуры и со-
112
лености является нелинейной. Такая зависимость обуславливает
ряд нелинейных свойств морской воды. К ним относится и
наличие у воды в определенном диапазоне солености максимума
плотности. Однако важно знать не саму максимальную
плотность, а температуру, при которой она достигается.
Рис. 4.9. Изопикническая (термохалинная) производная
dS/дТ (enc0CJ) как функция солености и температуры*
при атмосферном давлении (а), давления и температуры
при S=35 епс (б)
113
Большое значение температуры наибольшей плотности Тт
объясняется ее значительным влиянием на вертикальную
устойчивость и перемешивание вод в морях и океанах. Температура
наибольшей плотности Тт определяется как температура, при
dv\
которой градиент термического расширения
дТ
равен нулю.
STp
Температура Тт чистой воды при атмосферном давлении равна
3,982° С, с увеличением давления она уменьшается. Для морской
воды температура наибольшей плотности уменьшается как с
увеличением давления, так и солености (рис. 4.10). В условиях
термодинамического равновесия экстремум плотности (удельного
объема) существует только в той области солености и давления, в
которой температура наибольшей плотности Тт больше или равна
температуре замерзания Tf (см. раздел 7.1), т. е. Тт ^ 7у [67].
• температура наибольшей плотности
температура замерзания
2 —I
а
I
Е о
Рис. 4.10. Зависимость температуры наибольшей
плотности и температуры замерзания морской воды (°С) от
солености (епс) и давления (дбар) •
114
Это обусловлено тем, что температура наибольшей плотности
уменьшается с увеличением солености и давления быстрее, чем
температура Т/°. Предельное давление существования наиболь^
шей плотности в условиях термодинамического равновесия при-*
мерно равно 270 бар, при котором Тт=Т/=-2° С. В областях, где
Тт<Т/у термодинамическое равновесие не выполняется, так как
замерзание начинается раньше достижения максимальной
плотности при охлаждении.
Точка пересечения линий температуры наибольшей
плотности и температуры замерзания (Tm—TJ) называется критике*
ской. При атмосферном давлении (т. е. на поверхности)
критическая точка наблюдается при солености 24,6 (рис. 4.10).
Морская вода с соленостью меньшей 24,6, как и пресная,
имеет температуру наибольшей плотности выше температуры
замерзания. При солености выше 24,6 соотношение температур Тт и 7}
обратное. Однако в реальных условиях температура наибольшей
плотности в этом случае не может быть достигнута, так как вода
не охлаждается ниже температуры замерзания. По предложению
Н.М. Книповича, эти два типа вод называются соответственно
солоноватыми и морскими, С глубиной соленость критической
точки уменьшается и становится равной нулю при давлении
* 270 бар [67].
Различия в соотношениях температур Тт и 7} обуславливают
различия в протекании некоторых процессов в солоноватых и
морских водоемах, в частности процесса конвективного
перемешивания. В солоноватых водах конвективное перемешивание
начинается сразу при начале охлаждения поверхностных вод и
продолжается до момента, когда температура охваченного ею слоя
достигает температуры наибольшей плотности. Затем
перемешивание до дна прекращается. В морских водах при охлаждении
моря конвективное перемешивание не прекращается вплоть до
достижения температуры замерзания, поэтому конвекция может
достичь больших глубин.
Теоретическое объяснение этому факту может дать сравнение формул.
Кельвина (4.74) и Клаузиуса-Клапейрона (6.16), связанных между собой
аналитически.
115
Температуру наибольшей плотности можно определить,
приняв коэффициент термического расширения равным нулю, так
как в него входит частная производная удельного объема
(плотности) по температуре. Тогда, с учетом (4.2) и (4.14), и проведя
некоторые упрощения, получим:
dpsro _ Р
дТ К^
Psto dKsrP t dpsro
K^ дТ дТ ;
(4.35)
Чтобы из уравнения (4.35) получить температуру наибольшей
плотности, необходимо решить полученное уравнение
относительно температуры по заданным значениям солености и
давления. Для его решения можно воспользоваться методами
численного решения алгебраических уравнений [1].
Вычислить температуру наибольшей плотности можно также
и по эмпирической формуле Д. Колдуэлла [50]:
Tm(S,p)=3,982 - 0,2229-5 - 0,02004- р -
• (l + 3,76-10"3 ■ ^-(l + 4,02 -10"4 • р\
(4.36)
где S - соленость, %>; р - давление, бары. Отметим, что Тт по
Д. Колдуэллу ниже, чем по данным УС-80, примерно на
0,1-0,2° С в диапазоне практической солености от 0 до 40 при
атмосферном давлении.
4.7. Теплоемкость морской воды
Теплоемкость относится к калорическим свойствам
термодинамических систем, так как для ее определения необходимо знать
внутреннюю энергию системы21. Под теплоемкостью
подразумевается количество теплоты, необходимое для изменения
температуры системы на один градус (1° С или 1° К), т. е.
21 Помимо теплоемкости, другой важной калорической величиной
является теплота изотермического изменения какого-либо внешнего
параметра системы (например, объема) - количество теплоты,
необходимое для увеличения этого параметра на единицу при постоянстве
температуры и внешних параметров.
116
с=
dT
(4.37)
Размерность теплоемкости в СИ: (ДжК1). Отметим, что
теплоемкость не является функцией состояния системы, так как
количество теплоты bQ, необходимое для изменения температуры
системы на dT, зависит от характера протекающего при этом
процесса. Это означает, что при нагревании одна и та же система
в зависимости от происходящего в ней процесса обладает
различными теплоемкостями. Однако теплоемкости при постоянном
давлении Ср и постоянном объеме Су являются функциями
состояния и поэтому они получили наибольшее практическое
значение.
Первое начало термодинамики позволяет найти значения
различных теплоемкостей и установить связь между ними, если
известны калорическое (2.2) и термические (2.3) уравнения
состояния системы. В случае сложных систем с несколькими
компонентами, когда внутренняя энергия зависит от внешних параметров
я,- и температуры Г, а работа может быть получена в соответствии
с (2.1), первое начало термодинамики выглядит так:
,Э£/|
* дГ
•dT + Y
k да,
da,
а,*а„Т
л п.
1-1
*=1
dU_
дТ
■dT +
(4.38)
i=i
dU
да.
+ At
<fc»+5>*-<w*-
к=\
Тогда, разделив выражение (4.38) на AT, получим:
С =
8Q _dU
dT дТ
"I °. ,=I
dU
\
да.
+ А,
dat Л
dNk
dT
(4.39)
Применим уравнение (4.39) к морской воде как двухкомгох
нентной системе, находящейся под действием только
равностороннего давления. Получим:
117
с =
дг
дТ
f
+
v,S V
дг
dv
+ Р
T,S
dv dS
+ ц .
dT dT
(4.40)
Выражение (4.40) записано в удельных величинах, где с -
удельная теплоемкость (с=С/М), определяющая количество
теплоты, необходимое для нагревания единицы массы морской воды
на один градус (Джкг'^К"1); е - удельная внутренняя энергия; v -
удельный объем; S - соленость; ц - химический потенциал
морской воды.
Приняв постоянными удельный объем и соленость, из (4.40)
получим удельную теплоемкость морской воды при постоянном
объеме и постоянной солености:
дг\
дТ
(4.41)
v,S
Удельную теплоемкость морской воды при постоянном
давлении и постоянной солености можно найти из уравнения (4.40):
СРД =
дТ
\ (дг
+ —
L 1*
>
+р
T,S j
dv\
'
дТ\
P.S
или, принимая во внимание (4.41),
cP,s ~ Cv,S +
Г*
+ Р
T,S )
' дТ\
(4.42)
(4.43)
p>s
Из уравнения (4.41) следует, что для определения cv надо знать
лишь калорическое уравнение состояния. Для определения же ср
необходимы как термические, так и калорическое уравнения
состояния системы. Чтобы избавиться в выражении для ср от
калорического уравнения (т. е. от внутренней энергии), воспользуемся
вторым началом термодинамики, с помощью которого можно
установить связь между термическими и калорическим
уравнениями состояния.
Действительно, из основного уравнения термодинамики для
морской воды (3.9) находим (раскрывая полный дифференциал
de):
118
, . _ de + p • dv - \i. ■ dS
1 - -
J_ de_
т' ST
v,S
T
(
de
dv
+ P
T.S J
, 1
dv + —
T
de
(4.44)
Сравнивая (4.44) с
1 dT
dT +
дц'
S,v
dv
■dv +
as
an'
■\i\-dS.
T,v
StT
as
•dS, (4.45)
I7>
имеем:
an'
dv
dT
S.T
_ 1
S,v
_ 1 (de
= T{dv
de_
dT
T,S
>
v,S
+ P\
J
(4.46)
(4.47)
Продифференцировав (4.46) no v, a (4.47) по Г и учитывая, что
dvdT dTdv
получим:
d_
dv
\_ de_
T' dT
"-S.
d_
dT
T
de
dv
\
Откуда
dp]
dT\
v^
ae
+ p.
гд
(4.48)
Подставляя (4.48) в (4.43), имеем:
P,S v,S qj.
v.S
dv_
dT
P,S
(4.49)
Выражение (4.49) можно вычислить, если заданы две функции
состояния р и v, что не всегда возможно. Поэтому преобразуем
(4.49) к виду, в который входит только удельный объем.
119
Для этого, умножая и деля правую часть (4.49) на —
дТ
,
поля
лучим:
cPts cv,s - Т'
dv
dT
\2
p,s)
дР
Откуда, на основании тождества'
v,S
dv_
дТ
P.S
22
dp
v,S
дТ
p,s
др
dv
= -1,
(4.50)
T,S
следует уравнение:
Cp,S Cv,S ~ * '
dv
dT
p.s J
dv_
dp
(4.51)
T,S
Таким образом, зная теперь только одно уравнение состояния
морской воды v = v(S,T,p)> можно вычислить разность
теплоемкости при постоянном давлении и постоянном объеме.
Из (4.51) легко определяется и их отношение:
у = -
P,S _
v,S
dv\
^Т\
dv
dp
\
P,Sj
2~
\t,s
-I
=
1 T a
1 V
Cp.S K
-1
. (4.52)
22
Пусть p = p(v, f). Откуда dp = —
лучаем:
120
dp
dv
dv_
dT
dp_
dT
dpi
dv\
dT
dp
-dv-
T
dp
dv
lv
dT\v
T dT\
dT. При dp = 0 no-
= 0и^ -21| .-П4 =-1.
Для морской воды отношение -^— близко к единице23.
Cv,S
Выражение (4.51) можно переписать в следующем виде:
T-va2
cPts-cv,s=—-—• (4.53)
В случае устойчивого равновесного состояния океана, т. е. ко-
гда плотность воды с глубиной увеличивается, — < 0, а к>0,
из уравнения (4.53) находим, что cpS > cvS 24. Равенство
выполняется только при а=0. Например, для пресной воды а=0 при
температуре 3,982° С и стандартном атмосферном давлении. При
этих значениях ее плотность максимальна, а а меняет знак. Для
солоноватых вод (0<5^24,б) <х=0 при температуре наибольшей
плотности. В случае морских вод (S > 24,б) равенство cpS = cvS
никогда не достигается, так как а>0 при всех температурах.
В заключение теоретического описания понятия
теплоемкости, кратко остановимся на ее связи с некоторыми
термодинамическими потенциалами.
Из определений энтропии (2.9) и теплоемкости (4.37) следует:
с = Г.^1. (4.54)
dT
Из (3.12) видно, что при изобарических процессах и
постоянной солености изменение энтальпии равно:
Откуда:
dh
p,s дт
(4.56)
23 Одним из самых точных экспериментальных способов определения у
является измерение скорости звука в морской воде. »
24 Физически это объяснимо. При постоянном давлении для нагревания,
системы на градус необходимо больше тепла, чем при постоянном
объеме, так как в первом случае часть тепла системы идет на совершение
работы, которая происходит вследствие увеличения объема.
121
и
'Pfi
дТ
рЛ
Наконец, из уравнений (4.41) и (4.46) следует:
cv,s=T-
дТ
(4.57)
(4.58)
v,S
С помощью формул (4.57) и (4.58) устанавливаются
зависимости теплоемкостей от давления и удельного объема.
Продифференцировав (4.57) по давлению при постоянной температуре и
постоянной солености, меняя затем порядок дифференцирования
и используя соотношение Максвелла (2.28), имеем:
Л • ^ г * \
дс
P.S
др
= т
T,S
д_
др
^
дТ
p,s)
дТ
з«Г
др
P.S )
= -Т
д\
дГ
(4.59)
P.S
Аналогично можно получить зависимость с„ от удельного
объема, используя соотношение взаимности (2.24):
'v,S
dv
д2Р
T,S
дГ
(4.60)
v,S
Могут быть получены и другие производные теплоемкостей,
но они не представляют практического интереса.
Первые исследования теплоемкости морской воды были
проведены в 1889 г. Туле и Шевалье. Несмотря на недостоверность
их результатов, данные, полученные ими, применялись в
океанологии более 50 лет. В 1959 г. Р. Коксом и Н. Смитом были
проделаны точные лабораторные опыты по определению
теплоемкости, на основании которых были опубликованы данные по
удельной теплоемкости ср морской воды при различных значениях
температуры, солености и атмосферном давлении. Опыты по
определению удельной теплоемкости морской воды проводились
также Д. Конорсом [54] и др.
Самые современные исследования удельной теплоемкости
растворов стандартной морской воды были проведены Ф. Милле-
ро и др. [80]. Данные этих авторов, нормированные на
температуру по МПШТ-68, приведены в новых Международных
океанографических таблицах [69].
122
Удельная теплоемкость при атмосферном давлении
рассчитывается по эмпирической формуле:
cp(S,T,0) = £а, T' + S- 2> • Г + S% • £с,. • Г, (4.61)
/=0 j=0 /=0
где а0=4,2174403; a,=-3,720283; a2=0,1412855; а3=-2,654387-10*3;
а4=2,09323610-5; &0=-7,643575; 61=0,1072763; 62=-1,38385-10"3;
с0=0,1770383; c,=-4,07718-l(r3; с2=5,148-10'5.
Коэффициенты в уравнении (4.61) приведены в соответствие с
системой СИ, МПШТ-68 и ПШС-78 [63]. Формула справедлива в
следующих диапазонах солености и температуры: 0<S<40,
0<Г<35°С. Проверочное значение: ^=3980,0513 Джкг^К"1 при
£=40, Г=35° С. Стандартное отклонение данных, полученных по
формуле (4.61), составляет 0,5 Джкг'^К"1. Зависимость ср от
солености и температуры при атмосферном давлении приведена на
рис. 4.11а.
Зависимость удельной теплоемкости ср от давления не может
быть измерена непосредственно. Ее можно получить
интегрированием уравнения (4.59):
о Ф i дТ2
Откуда следует, что:
cp(S,T,p)=cp{S,T90)-\T
dp.
p,s
(4.62)
р. я*
dzv
о
дТ2
dp (4.63)
p,s
Второй член в правой части (4.63) отражает зависимость
удельной теплоемкости от давления, выраженную через
удельный объем воды, который можно оценить по экспериментальным
данным. В работе [63] по табличным значениям этого члена с
помощью метода наименьших квадратов получена его
аппроксимация степенными полиномами. В результате уравнение (4.63)
преобразуется к виду:
123
сД5Х/>) = с,&Г,0)+р£а,Г' +р2 £б,Г'+p3YlclT' +
r=0 i-O r=0
^Sp^df +S*p£eiTi + Sp2£fiTi+goSi>2p2 + (4.64)
i-0 /=0 i=0
+ *32*|.r+*I7Sy2p3,
i=0
где ао^-4,9592-10"1; а,=1,45747-10-2; а2=-3,13885-10"4;
я3=2,0357-10-6; а4=1,7168-10"8; ^2,4931-Ю"4; ^=-1,08645-105;
62=2,8753310"7; &3=-4,0027-10-9; 64=2,2956-10п; с0=-5,422-10*8;
c,=2,6380-109; с2=-6,5637-10"п; с3=6,1361013; <Аг4,9247-1(Г3;
<*,=-1,28315-КГ*; г/2=9,802-10-7; rf3=2,5941108; ^-2,9179-Ю10;
е^ЬгЗЗЫО"4; e^l.n-lO"6; е2=3,122-10"8; /0=-2,9558-10'8;
/,=1,17054-Ю"7; /2=-2,3905-10"9; /3=1,8448-10п; йо=5,540-1010;
A,=-l,7682.10u;/i2=3,513-10-|3;*i=-l,4300-1012; р -давление,
бары.
Формула (4.64) справедлива в диапазоне: 0<5<40, 0<Г<35° С,
0<р<1000 бар. Проверочное значение: ср=3849,50 Дж-кг ^К"1 при
5=40, Г=40° С и/7= 1000 бар. Стандартное отклонение теплоемко-
стей, полученных по полиному, от теплоемкостей, вычисленных
прямым интегрированием, меньше 0,1 Дж-кг '•'ТС"1.
Вода обладает наиболее высокой теплоемкостью из
большинства веществ (кроме водорода и жидкого аммиака). Это имеет
огромное значение для формирования климатических условий на
Земле. Так, например, простые расчеты показывают, что при
охлаждении на 1°С единица объема воды нагревает на ГС почти
3200 объемов воздуха (см. задачу 4.13). Вода отдает атмосфере
огромное количество тепла, уменьшая тем самым амплитуды
колебаний температуры воздуха на большей части поверхности
Земли, особенно над самим океаном и прибрежными областями
материков, создавая здесь морской климат.
Удельная теплоемкость морской воды уменьшается с
увеличением, как солености, так и давления (рис. 4.11). В первом
случае увеличивается доля солей в морской воде, обладающих
меньшей теплоемкостью, во втором - при больших давлениях
происходит сжатие воды и, как следствие, ее дополнительное на-
124
гревание. Значит, воде уже надо дать меньше тепла, чтобы ее
нагреть на один градус.
Рис. 4.11. Зависимость удельной теплоемкости при
постоянном давлении ср (Дж-кг1-"К1 ) от солености и
температуры при атмосферном давлении (а), давления и
температуры при S=35 епс (б)
Поведение удельной теплоемкости морской воды при
постоянном давлении от температуры воды сложнее. При атмосферном
давлении в водах с соленостью большей, чем примерно 5 епс,
удельная теплоемкость сначала уменьшается, и только потом на-
125
чинает расти, причем при повышении солености начало
повышения теплоемкости смещается в область низких температур воды.
Удельная теплоемкость при постоянном давлении при соленостях
меньших 5 епс уменьшается при повышении температуры.
Как видно из рис. 4.11а, самый значительный фактор,
влияющий на изменение удельной теплоемкости, — это соленость
морской воды.
Средняя удельная теплоемкость морской воды Мирового
океана при постоянном давлении/?, рассчитанная по средним
значениям солености S и температуры Г, равна примерно
3987 Дж-кг'^К"1. Для сравнения удельная теплоемкость воздуха
при постоянном давлении равна примерно 1005 Джкг 1оК"!,
гранита - 840 Дж-кг^К"1.
4.8. Энтропия. Энтальпия
Одной из важных характеристик морской воды является
энтропия, представляющая собой функцию состояния и
характеризующая вероятностную структуру воды как термодинамической
системы, находящейся в термодинамическом равновесии.
Энтропию измерить непосредственно невозможно, поэтому она
определяется косвенно через другие параметры системы.
Для этого сначала найдем полный дифференциал удельной
энтропии в переменных 5, Тир:
, • дц
dx\ =
дТ
•л--*1'
W *
.ф+^
3S
• dS . (4.64)
TtS
Теперь заменим частные производные энтропии через другие
параметры системы, которые можно определить
экспериментально. Первую заменим через отношение между удельной
теплоемкостью при постоянном давлении (и солености) и температурой
(4.57). Изотермо-изохалинное изменение энтропии от давления
заменим следующим образом (см. раздел 2.5, формула 2.73):
дц
др
= -v • а . Последнюю производную заменим через соот-
126;
ношение Максвелла (3.18)
as
9Г
, которое можно по-
*Т,р
лучить, если использовать термодинамический потенциал Гиббса
(3.11).
В результате получим:
-~Р.АГ л- Ф
Т дТ
•dS . (4.65)
\p,S
Из выражения (4.65) видно, что для вычисления энтропии
необходимо знать удельную теплоемкость ср =cp(S,T,p),
уравнение состояния v = v(S,T9p) и градиент химического потенциала
морской воды по температуре. Если величины v и ср
рассчитываются довольно точно по эмпирическим формулам, например
(4.13) и (4.64), то градиент д]х/дТне может быть полностью
определен в рамках термодинамического равновесия [61].
В некоторых работах энтропию рассчитывают через
потенциал Гиббса [59], используя соотношение (2.27).
Ниже приводится эмпирическая формула для вычисления
относительной удельной энтропии (Дж-кг ^К"1) [80]:
ц =As-S + Bs-S3//2+CsS2, (4.66)
где S- соленость, %о;
As = 1,4218-3,1137• 10"4 • Г +4,2446-КГ6-Г2;
Bs = -2,1762 • 10"1 + 4,1426 • КГ4 • Т -1,6285 • 10^ • Т2 ;
Cs = 1,0201 • Ю-2 +1,5903 • 10~5 ■ Г - 2,3525 • 10"7 • Т2.
На рис. 4.12 приводится графическое представление
зависимости относительной удельной энтропии от солености и
температуры.
Напомним, что параметр морской воды, изменение которого
между двумя ее состояниями равно количеству теплоты,
затрачиваемому на эти изменения при постоянном давлении и
постоянной солености, называется энтальпией.
Энтальпия играет ту же роль в процессах при постоянном
давлении, какую внутренняя энергия играет в процессах при
постоянном объеме. Энтальпия, как и энтропия, является функцией
состояния.
127
25
Соленость, %о
Рис. 4.12. Относительная удельная энтропия морской
воды (Джкг1 °К) как функция солености и температуры
при атмосферном давлении
Эмпирическая формула для расчета относительной удельной
энтальпии h (Джкг1) приводится в работе [80] и имеет
следующий вид:
h^A-S + B-S^+C-S2, (4.67)
где S - соленость, %>;
Л = 3,4086-Ю'1 -6,3798- 10~2-Г +1,3877 Ю"3- Г2;
В = 7,9350-10"! +1,0760-10"! ■ Т-6,3923-КГ1 • Г2;
С = -4,7989 -10"! + 6,3787 • 10~3Г-1,1647 • 10"4Г2 + 5,717 • 10_7Г3.
Зависимость относительной удельной энтальпии от солености
и температуры показана на рис. 4.13.
Уравнения (4.66) и (4.67) не дают абсолютных значений
энтропии и энтальпии, а применимы лишь для вычисления их
разностей.
4.9. Химический потенциал
Важность химических потенциалов заключается в том, что
они определяют условия химического равновесия: в неравновес-
128
ных системах любой компонент будет стремиться из состояния с
более высоким потенциалом в состояние с более низким
потенциалом до тех пор, пока не установится равновесие.
О 5 10 15 20 25 30 35 40
Соленость, %о
Рис. 4.13. Зависимость относительной удельной
энтальпии (Джкг1) морской воды от солености и температуры
при атмосферном давлении
Химический потенциал, как и температура, является
интенсивной величиной. Напомним, что химическим потенциалом
называется приращение какого-либо термодинамического
потенциала при постоянстве соответствующих параметров, которое
происходит при добавлении в систему компонента в количестве,
равном единице его концентрации, причем количество всех
других компонентов систем поддерживается постоянным (2.42). Для
морской воды этим компонентом служит морская соль (3.13).
Наиболее подходящей функцией для определения химических
потенциалов является энергия (потенциал) Гиббса, так как в
экспериментальных работах легче всего поддерживать постоянными
температуру и давление.
Как видно из выражения (3.6), химический потенциал морской
воды равен разности химических потенциалов морской соли и
чистой воды ц = |Л$- \\w
129
Для определения химического потенциала чистой воды \iw в
морской воде (растворе) рассмотрим двухфазовую систему
вода-пар, находящуюся в условиях химического равновесия (см.
раздел 6.1), т. е. химические потенциалы обоих фаз равны друг
другу. Допустим, что насыщенный пар является совершенным
газом, подчиняющимся уравнению состояния Клапейрона:
pv = n-RT, (4.68)
где р - давление; v - удельный объем; Г- абсолютная
температура; R - универсальная газовая постоянная; п - количество молей.
Соотношение Максвелла (3.22) для совершенного газа
запишется, следовательно, в виде:
др.
дР
dv_
дп
(4.69)
Т.Р
Т,п
Подставляя в правую часть этого выражения уравнение (4.68),
получаем:
др
RT (4.70)
ir,„ Р
Проинтегрировав это выражение, получим:
\i = R • Г- In/? + const. (4.71)
После некоторых преобразований, и учитывая равенства
химических потенциалов чистого насыщенного пара и чистой воды
в растворе (морской воде), получим следующее соотношение:
цж=^+Л.7Чп%. (4.72)
Здесь \xw - химический потенциал чистой воды в растворе
(морской воде), ц% - химический потенциал чистой воды, взятой
отдельно; xw - массовая доля чистой воды в растворе.
Аналогичную формулу можно написать и для химического
потенциала солей \is:
\is=\i°s+R-T-lnx5. (4.73)
4.10. Адиабатический градиент температуры.
Потенциальная температура
Напомним, что процессы, протекающие без обмена теплом
между системой и окружающей средой, называются адиабатиче-
130
скими. Адиабатический градиент температуры определяется
как изменение температуры на единицу давления при
адиабатическом изменении давления. Он имеет большое значение в
физической океанологии, так как используется при определении
потенциальных температур, вычислении устойчивости, скорости
распространения звука и др.
Для определения адиабатического градиента температуры
воспользуемся формулой (4.65). При равновесном
адиабатическом процессе энтропия т|* и соленость S объема морской воды
остаются неизменными, т. е. ch\ = 0 и dS = 0. Тогда из уравнения
(4.65) следует формула Кельвина:
Т dv Tv ,А~,Л^
•ос, (4.74)
dp
n,s CPJS дТ Cp,S
где Т- абсолютная температура, °К; cPtS - удельная теплоемкость
при постоянном давлении и солености, Дж- кг"1 Ж"1; а -
коэффициент термического расширения, eK"1; v -удельный объем, м3-кг *.
Из формулы (4.74) видно, что знак адиабатического градиента
температуры определяется знаком производной dv/dT. При
атмосферном давлении градиент Г отрицателен только в области
температур, лежащих ниже температуры наибольшей плотности.
В связи с тем, что dv/дТ = f(S,T,p) и cpS = f(S,T9p)>
адиабатический градиент температуры также является функцией
солености, температуры и давления (рис. 4.14).
Адиабатический градиент температуры Г можно рассчитать
по формуле (4.74), воспользовавшись эмпирическим выражением
для удельной теплоемкости ср (4.64) и уравнением состояния,
например УС-80 (4.13) [24]. Для этого нужно подставить в (4.74)
выражение для частной производной удельного объема по
температуре, тогда:
dv Т др _
дТ"
T.L..
с. дТ
Т_
с„
рг
dps
^stp ' Vs-я> ~ Р)' ~~^j< Psto' Р
дК
STp
'STQ
дТ
К
дТ
STp
т
(4.75)
13Г
Вычислить адиабатический градиент температуры можно
также с помощью эмпирических формул, например формулы
X. Брайдена [48]:
ГЗТр=£агГ +(b0+brT)-{S-35) +
/«о
+ р
(4.76)
1-0
_ыо J
гдеао=3,5803105; д,=8,5258710"6; а2=-б,83б05-10"в;
а3=6,6228-10"10; 60=1,8932-10'6; £,=-4,23935-10"8; со=1,874ЫО-8;
с^-6,7795-10-10; с2=8,7330.10*12; с3=-5,448Ы014;
<й=-1,135Ы(Г10; rf1=2,775910"12; е0=-4,6206-10"13; е,=1,8676-1(Г14;
е2=-2,1687-10"16; р - давление, дбар; Г- температура, °С; S -
соленость, епс.
Формула справедлива в диапазоне практической солености -
от 30 до 40, температуры - от 2° С до 30° С и давления - от 0 до
10 000 дбар.
Из рис. 4.14 видно, что для значительной части толщи вод
океана адиабатический градиент температуры находится в
пределах от 0,1 до 0,2° С на 1000 дбар. Сравните с сухоадиабатическим
градиентом воздуха, который равен 7° С на 1000 м.
Температура in situ может быть представлена в виде суммы
так называемой потенциальной температуры 9 и адиабатической
поправки АТЛ:
7)я„Л=в + ДГв. (4.77)
Потенциальной температурой 9, по Гелланд-Гансену,
называется температура, которую вода адиабатически воспримет, если
давление сравняется с атмосферным, другими словами, если
частица будет поднята с глубины (in situ) к поверхности моря без
обмена теплом с окружающей средой [20]. Иногда под
потенциальной температурой подразумевается температура, которую
будет иметь частица воды при адиабатическом перемещении ее с
исходного уровня на некоторый другой уровень. Потенциальную
температуру можно получить из формулы (4.77), если известно
адиабатическое изменение температуры в пределах того
вертикального расстояния, которое должна пройти частица. Это
изменение находится интегрированием уравнения (4.75) при t\=consL
132
Рис. 4.14. Адиабатический градиент температуры 10* Г
ССдбар1) как функция солености и температуры при
атмосферном давлении (а), давления и температуры при
S~35 епс (б). Градиент рассчитан по формуле (4.76)
Тогда:
е = (*.Г,/Ъ,*,л)=Ъл- \r{0',S,p)dp, (4.78)
Pin situ
133
Q' = Tinsitu-jr(B,S,p)dpt
(4.79)
где p - промежуточное давление, связанное с итерационной
процедурой интегрирования.
Отметим, что интеграл в правой части уравнения (4.78),
выражающий адиабатическое изменение температуры, зависит от
температуры, которая в свою очередь меняется с изменением
давления при интегрировании, поэтому сам процесс
интегрирования оказывается итерационным [62, 63].
В качестве примера приведем процедуру вычисления
потенциальной температуры с помощью метода Рунге-Кутта
четвертого порядка. Пусть S, Г, р - значения in situ, требуется найти
потенциальную температуру Э на уровне с давлением р0. Имеем:
Др = Ро-Р>
Де^Др-Г^Г,/?); е^^-де^
Д02=Др-Г|5,,61,р + ~Др
; 82=0! +
(l-£)(M,-ft);
A9j=Ap-r|5,e2,p + -4t?|; 8j=92 +
1 +
yf2)
(Авз-ft);
ле4 =&p-r(s,o3,p+Ap); e4 =e3 +^(де4 -2Яз),
о
где qx =Дв,;
е(5,г,/7,р0)=е4.
По данным X. Свердрупа [86], вертикальное распределение
температуры во впадинах Мирового океана очень близко к
адиабатическому, и неустойчивости вод, по-видимому, практически
не существует. На рис. 4.15 изображено распределение истинных
и потенциальных температур на океанологической станции,
сделанной во впадине Минданао (глубина 10 068 м) [22].
134
Рис 4.15. Распределение с
глубиной истинных (1) и потен-
циалъных (2) температур на
океанологической станции во
впадине Минданао. Кривая J,
соответствует
адиабатическому изменению
температуры с глубиной при постоянном
значении потенциальной
температуры 9=1,25Х? [22]
Понятие о потенциальной температуре и потенциальной
плотности psQP широко используется в термохалинном анализе водных
масс, особенно при изучении глубинных вод. Это объясняется
тем, что на больших глубинах температура in situ водных масс
при движении вдоль дна переменной глубины (при отсутствии
геотермического потока тепла снизу) изменяется только за счет
адиабатических процессов, в то время как потенциальная
температура остается постоянной. Иначе говоря, потенциальная
температура является индикатором определенной глубинной водной
массы.
4.1L Теплота смешения. Уплотнение при смешении
В настоящем разделе приводятся некоторые особенности
физических свойств морских вод при их смешении. Подчеркнем,
что именно в этом случае наиболее ярко проявляются
нелинейные свойства морской воды.
Рассмотрим процесс смешения двух однородных водных масс,
имеющих разные температуры и солености (30° С, 35 епс для
одной водной массы и 0° С и 0 епс - для другой), но одинаковые
массы. Формула для определения температуры смешения для
бинарного раствора имеет вид:
Т = сгТ\+с2-Т2 ^ (480)
с, +с2
135
Я00\
где С], с2 - удельная теплоемкость первой и второй водной массы
соответственно (3999 и 4217 Джкг^К1). Подставляя в (4.80)
значения температуры и удельной теплоемкости, получим
Т =14,602° С. Если бы температура обладала свойством аддитив-
— (т + Т )
ности, то температура смеси была бы равна Г# =— —=15° С.
Разность Тщ - Т =-0,398° С представляет собой теплоту
смешения (разбавления) вод: эта величина, как видим, весьма
существенная. Таким образом, температура не обладает свойством
аддитивности [54]. Отметим, что энтальпия Л«ср- Т является свойством
аддитивным, поэтому некоторые авторы предлагают для
описания процессов смешения водных масс использовать диаграмму
«энтальпия-соленость». Что касается солености, то и она не
обладает свойством аддитивности из-за степени диссоциации
морской воды как электролита. Однако нелинейные эффекты здесь
проявляются в меньшей степени, чем для температуры.
В то же время в 75-анализе в большинстве случаях
температуру и соленость морской воды считают свойствами аддитивными.
Одно из интереснейших явлений, наблюдаемых в океане,
является уплотнение при смешении водных масс, отличающихся по
температуре и солености. Впервые на уплотнение при смешении
морских вод указал Витте. Затем это явление подробно
рассматривалось в работах Н.Н.Зубова [14,15,16] и Н.П. Фофонова [60,
61], которые показали, что уплотнение при смешении - следствие
нелинейной зависимости удельного объема (плотности) от
солености и температуры, обусловленное сложным строением воды.
Пусть смешиваются два объема морской воды Qx и Q2,
температуры, солености, массы, удельные объемы и плотности
которых равны соответственно Тх и Тъ S\ и Si, mi и m2> vx и v2, pi и p2.
Если перемешивание происходит без изменения массы и общего
количества солей, то соленость перемешивающихся объемов
должна равняться:
{тх + т2у S = тх • Sx + т2 • S2
или
S = mfSi+»>2'S2 (4gl)
/и, +т2
136
Температура смеси (если и не учитывать адиабатические
явления, изменение теплоемкости морских вод, теплоту
разбавления) может быть рассчитана аналогично:
т=тгТ,+т2-Т2 (482)
/Wj + m2
Предположим далее, что смешение происходит без изменения
объема. Тогда:
Gi=*VV„G2=m2-v2,
Q\+Qi= (m\ + тг)• v, тх • v, + т2 • v2 = (тя, + т2)- v,
откуда
-=^-v,+^V2 (483)
Для плотности получим:
щ = Gi Pi> щ = Qi Ф2> (й + Qi)'р = wi + m2>
a-pi+G2'P2=(6i+62)-p.
g=a-Pi+g2'piB (4e84)
fi+(?2
Из формул смешения (4.81)—(4.83) видно, что для солености,
температуры и удельного объема роль весов играют массы.
В формуле смешения для плотности (4.84) весами являются
объемы.
Удельный объем v и плотность р, вычисленные по формулам
(4.83) и (4.84), не равны истинным значениям удельного объема v
и плотности р, рассчитанным по Т nS, используя, например,
УС-80. Разность Др = р - р называется уплотнением при смете-
нищ а разность Av = v - v - сжатием при смешении.
Итак:
Ар = р-^уГеМ/; (4.85)
Av = v-^—'-. (4.86)
2>
Наиболее ярко уплотнение при смешении выглядит, если
смешиваются две водные массы с одинаковой плотностью и оди-
137
наковым объемом, но с разной температурой и соленостью.
Плотность результирующей смеси будет больше, чем плотность этих
двух масс (рис. 4.16)!,
Аномалия плотности, кг/мЛ3
Соленость, епс
Рис 4.16. К объяснению эффекта уплотнения при
смешении. А и В - термохалинные индексы двух водных масс,
участвующих в процессе смешения и имеющих одинаковую
плотность. АВ - прямая смешения. T,S - температура и
соленость смеси. Aoj- величина уплотнения при смешении
Иначе говоря, плотность (удельный объем) морской воды -
величина не аддитивная.
Величина уплотнения зависит от разности температур и соле-
ностей вод, участвующих в процессе смешения. Разность
температур влияет гораздо сильнее, чем разность соленостей. Если
смешиваются две водные массы с одинаковой температурой, но с
разной соленостью, то уплотнение при смешении очень мало и
им можно пренебречь. Уплотнение при смешении при
постоянной разности Т и S тем больше, чем ниже температура и меньше
соленость смешиваемых вод. Когда массы смешиваемых вод
приблизительно одинаковы, уплотнение при смешении достигает
138
наибольших величин. Уменьшение объема вод, наблюдаемое в
процессе смешения водных масс, вызывает более или менее
значительные понижения уровня моря.
Если подходить более строго, температура морской воды не
является аддитивным (консервативным) параметром. Поэтому
формулу смешения (4.82) необходимо использовать с большими
оговорками.
Вместо температуры при изучении смешения водных масс
лучше выбрать энтальпию, которая вместе с соленостью
относятся к консервативным (аддитивным) параметрам. Покажем это в
случае смешения водных масс двух при постоянном давлении.
Как уже говорилось выше, процесс смешения происходит без
изменения массы, т.е. Ат = т-(тх + т2) = 0, и с сохранением
общего количества солей - тAS = mS-(mt-Sx + т2 • S2) = 0.
Здесь т и 5- масса и соленость получившейся смеси.
Предположим также, что обмен энергией в процессе смешения
осуществляется только между двумя водными массами, но не с
окружающей средой, т. е. происходит адиабатически. Тогда
согласно основному уравнению термодинамики для морской воды
как двухкомпонентной системы (3.9) при dx\=0 и dS-09 любые
изменения в объеме системы при постоянном давлении будут
сопровождаться переносом внутренней энергии между двумя
водными массами, т. е.
Г / I S (4-87)
-p-[m-v-(ml-vx + m2-v2)\9
где s - внутренняя энергия смеси. Умножение на массу связано с
тем, что внутренняя энергия величина интенсивная, т. е.
пропорциональная массе.
Преобразуем (4.87) следующим способом:
w(s + pv)-[(ml.(81-bpv1)+m2(e2+jr?v2))] = 0/
а если вспомнить определение энтальпии (2.29) Л = 8 + р • v, то
получим:
т-АА = да-Л-(л11-А|+л12-Л2) = 0. • (4.88)
Уравнение (4.88) показывает, что сохранение энергии в
системе из двух водных масс выражается просто как сохранение
общей энтальпии.
139
Задачи и вопросы для повторения
4.1. Что называется уравнением состояния морской воды?
4.2. Какие эмпирические уравнения состояния вы знаете? Какое из
них используется в океанографической практике в настоящее время?
4.3. Что такое 7$-диаграмма? Для чего она служит?
4.4. Во сколько раз коэффициент термического расширения
численно больше коэффициента соленостного сжатия? На что это влияет?
4.5. Что такое стерическое отклонение уровня моря?
4.6. Как определяется температура наибольшей плотности?
4.7. Что такое критическая точка? Как делятся воды Мирового
океана по отношению к ней?
4.8. Может ли вода с соленостью большей 24,6 епс иметь
температуру наибольшей плотности?
4.9. Какие моря из приведенного списка относятся к солоноватым
водоемам: Балтийское, Белое, Черное, Средиземное, Азовское,
Каспийское, Красное?
4.10. Что такое удельная теплоемкость морской воды? Является ли
она функцией состояния?
4.11. Какая удельная теплоемкость морской воды больше: при
постоянном давлении или при постоянном объеме?
4.12. Как изменяется удельная теплоемкость морской воды при
увеличении температуры, солености и давления?
4.13. Сколько объемов воздуха Vх нагреет один объем морской воды
Vм соленостью 35 и температурой 25° С при охлаждении его на 1° С?
Ответ:
Из закона сохранения количества энергии следует, что
та -с* • AT = mw • с™ • AT. Перепишем это выражение в виде
V = . По условию задачи У"=\, АГ=1°С. Плотность
расрАТ
воздуха равна ра= 1,292 кгм"3, а его удельная теплоемкость при
постоянном давлении cfl=1005 Джкг'^К1. Плотность и удельная теплоемкость
морской воды при заданных температуре и солености равны
соответственно //=1023 кгм*3 и с*=4000 Дж-кг^К"1. В результате получим
^=3151.
4.14. Во сколько раз адиабатический градиент температуры морской
воды меньше сухоадиабатического градиента температуры воздуха?
4.15. Получите формулу Кельвина с помощью метода якобианов,
т. е. выясните, как будет меняться температура воды при
адиабатическом изменении давления.
140
Ответ:
дТ
dp
[р,ч ] b.n 1 T
Тогда: ——
Эр
_ r-[ft,V]
Отсюда:
л
дТ_
др
А из (2.68) следует, что ^—- = -v • а.
•rr-.pi рг.ра
р
Tv а
hi "р
4.16. Что такое уплотнение при смешении морских вод?
4.17. Докажите, что температура морской воды при адиабатическом
сжатии изменяется по экспоненте?
Ответ:
Из (4.74) следует:
dT va j
— = dp .
T CP,S
Проинтегрируем это выражение с левой части по температуре от Г0
до Г, а с правой - по давлению отр0 до р. Тогда
£.'£«.Ф.
Л"**
Отсюда:
1п(Г)-1п(Г0)= J—-ф,
т
ут
р„Ср.з
н^-г~*
p,cp-s
Взяв ехр от обеих частей уравнения, получим:
' v-a
J dp
T = T0-enCpS .
141
ГЛАВА 5. МОРСКАЯ ВОДА КАК УПРУГАЯ СРЕДА
Морская вода как любая жидкость имеет определенный объем,
но не обладает упругостью формы (отсутствие модуля сдвига) и
имеет малую сжимаемость. Ее удельный объем под давлением
1000 бар, соответствующим условиям на глубинах около 10 км,
уменьшается приблизительно всего лишь на 4% относительно
удельного объема при атмосферном давлении. Малая
сжимаемость объясняется тем, что при малых взаимных расстояниях
небольшое уменьшение промежутка между молекулами приводит к
появлению больших сил межмолекулярного отталкивания. В
результате в воде возникают внутренние упругие силы, которые
уравновешивают приложенные к ней внешние силы. При
прекращении действия внешних сил прежний объем
восстанавливается.
5.1. Объемный модуль упругости.
Сжимаемость морской воды
Как было уже показано в разделе 3.8, в толще вод Мирового
океана наблюдается равностороннее гидростатическое давление,
которое вызывает сжатие объема воды без изменения его формы.
В соответствии с законом Гука для объемной деформации,
изменение давления dp жидкости при малом изменении dV ее
объема прямо пропорционально относительной объемной
деформации:
ф> = -К~,' (5.1)
dV ^
где — - относительное уменьшение объема воды, К -
объемный модуль упругости, имеющий смысл приращения давления,
при котором относительное уменьшение объема равно единице25.
Объемный модуль упругости К входит в эмпирическое
уравнение состояния УС-80. Его зависимость от параметров
состояния (солености, температуры и давления) дается эмпирической
2 Эта величина очень большая - она соответствует нахождению объема
воды на глубине, в 25 раз большей максимальной глубины океана.
142
формулой (4.17), полученной по данным инструментального
определения сжимаемости морской воды.
С увеличением давления, температуры и солености значение
объемного модуля упругости морской воды повышается (рис.
5.1).
О 2000 4000 6000 8000
Давление, дцб
Рис. 5.1. Зависимость объемного модуля упругости (бар)
от солености и температуры при атмосферном давлении
(а), давления и температуры при 5=55 епс (б)
143
Величина, обратная К, называется коэффициентом
сжимаемости к.
Рассмотрим сжимаемость единичного объема морской воды с
температурой Т и соленостью S. Для этого найдем полную
производную удельного объема от давления:
dv _ dvl
&~as\TtP
Соленость в объеме морской воды при изменении давления не
ds Л
меняется, поэтому в дальнейшем принимаем — = 0.
dp
Если происходит изотермическое сжатие, то выражение (5.2)
имеет вид:
dS_ dv_
dp + дТ
s,P
dp dp
(5.2)
S,T
(5.3)
SJ
dv\ _ dv\
Из выражения (5.3) следует, что при изотермических
процессах барический (изотермический) градиент удельного объема in
situ отражает изменение удельного объема, вызванное только
давлением, т. е. сжимаемостью морской воды. Такие процессы
характеризуются коэффициентом изотермической сжимаемо-
1 dv
emu к =
v dp
Он, как и объемный модуль упругости К, за-
S,T
висит от давления, температуры и солености морской воды (рис.
5.2).
Если процесс сжатия объема морской воды происходит
адиабатически, то имеет место не только уменьшение удельного
объема, но и повышение температуры. Это повышение температуры
происходит вследствие уменьшения расстояния между
молекулами воды при сжатии, что увеличивает вероятность их
столкновения. Согласно кинетической теории, это и определяет
повышение температуры воды. Оно, в свою очередь, отражается в
некотором увеличении удельного объема в случае а > 0.
Для изучения адиабатического сжатия объема морской воды
представим температуру Тщ situ как сумму потенциальной
температуры 0 и адиабатической поправки Та: Т = в + Та. Учитывая,
144
что потенциальная температура не зависит от давления, из
выражения (5.2) имеем:
dv
dp
_dv
S,P dP
S,T
(5.4)
4000 6000
Давление, дцб
8000
Рис. 5.2. Коэффициент изотермической сжимаемости
J О8 к (дбар'1) как функция солености и температуры при
атмосферном давлении (а), давления и температуры при
S=35 епс (б)
145
где второй член в правой части представляет изотермический
градиент удельного объема, Г =
dT„
dp
- адиабатический гради-
Щ J
ент температуры, определяемый формулой Кельвина (4.74).
называется адиабатическим градиентом
rfv,
Величина —
dp
лД
удельного объема (адиабатической сжимаемостью) и
определяет ту часть изменения удельного объема морской воды, которая
вызвана как барическими, так и адиабатическими эффектами.
Адиабатическая сжимаемость характеризуется коэффициентом
адиабатической сжимаемости
1
*.= —
V
dp
(5.5)
r\,S
Подставляя коэффициенты изотермической (4.4) и
адиабатической сжимаемости (5.5) в (5.4), получим:
,2
кЛ = к-Га = к-
Г-vcr
(5.6)
Из (5.6) видно, что адиабатическая сжимаемость меньше
изотермической (рис. 5.3).
Для численного определения изменения объема при
адиабатическом сжатии можно воспользоваться методом якобианов:
dv\
dp
_[v>m]_ (су/Г)[у,Г]_ vk vk ( .
где v - удельный объем, к - коэффициент изотермической
сжимаемости, cpjcv - отношение удельных теплоемкостей при
постоянном давлении и постоянном объеме.
При сжатии морской воды изменяются и некоторые другие
параметры состояния, например энтропия и внутренняя энергия.
Ранее уже было выявлено, что изменение энтропии при
изотермическом сжатии некоторого объема воды равно (2.73):
дп
др
= -va,
t,s
146
где v - удельный объем, а - коэффициент термического
расширения. Из этой формулы следует, что знак производной зависит
только от а (удельный объем всегда положителен). В океане
коэффициент термического расширения практически везде больше
нуля, за исключением районов с солоноватыми водами,
имеющими температуру ниже температуры наибольшей плотности.
Отсюда следует, что при изотермическом сжатии энтропия
морской воды убывает.
О 2000 4000 6000 8000
Давление, дбар
Рис 5.3. Разность между коэффициентами
изотермической и адиабатической сжимаемостями как
функция температуры и давления морской воды при
5=55 епс
Убывание энтропии означает, что система под действием
дополнительного давления имеет более упорядоченную структуру,
чем в свободном состоянии, если при этом температура и
соленость в обоих случаях одинаковы. Причина убывания энтропии
при изотермическом сжатии объясняется тем, что система
переходит в более упорядоченное состояние, так как при сжатии
происходит уменьшение средних расстояний между молекулами,
т. е. уменьшение хаотичности их взаиморасположения.
Рассмотрим теперь, что происходит с внутренней энергией,
например при изотермическом сжатии объема морской воды. Для
147
этого опять применим метод якобианов (формулы 2.59,2.60,2.63,
2.68 и 2.69):
dU\ =[U,T] = T[r)yT]-p[v,T] =
ф|г та га (58)
¥М [т,р\
Разность /? • к - Г • а меньше нуля не только при атмосферном
давлении, но и практически во всем диапазоне давления в
реальных условиях. Следовательно 1
Эр
< 0, т. е. при изотермическом
\т
сжатии внутренняя энергия объема морской воды убывает. Это
объясняется следующим: при сжатии совершается работа, при
которой жидкость должна нагреться. Но постоянство
температуры означает, что в процессе сжатия от системы отводится тепло.
Вопрос об изменении температуры морской воды при
адиабатическом сжатии мы рассмотрели ранее в разделе 4.10.
В океанографической практике иногда используется средний
коэффициент сэюимаемости ц (в интервале от 0 до р), входящий
в уравнение состояния Кнудсена-Экмана (4.12). Дифференцируя
(4.12) по давлению и подставляя результат в (4.4), получим
соотношение между коэффициентом изотермической сжимаемости и
средним коэффициентом сжимаемости:
Ф
к = - *.. (5.9)
Отметим, что при атмосферном давлении (р=0) коэффициенты
сжимаемости равны: ksto = [iST0.
В заключение этого раздела отметим, что при полной
несжимаемости воды средний уровень Мирового океана был бы выше
приблизительно на 30 метров.
Еще одной характеристикой морской воды как упругой среды
служит скорость распространения звука.
148
5.2. Скорость распространения звука
Если в морской воде возбудить механические колебания ее
частиц (сжатия и разрежения), то, вследствие взаимодействия
между ними, эти колебания начнут распространяться в воде от
частицы к частице с некоторой скорость с. Процесс
распространения колебаний в пространстве называется волной. Частицы
жидкости, в которой распространяется волна, не переносятся
волной, они лишь совершают колебания около своих положений
равновесия. В зависимости от направления колебаний частиц по
отношению к направлению распространения волны различают
продольные и поперечные волны, В воде возможно возникновение
только продольных волн, т. е. тех волн, в которых колебания
частиц происходит вдоль направления распространения волн.
Продольные волны связаны с объемной деформацией упругой среды.
Образование поперечных волн (частицы колеблются в
направлении, поперечном распространению) в воде не происходит в связи
с тем, что они возникают только в такой среде, которая способна
сопротивляться деформации сдвига. Вода же таким свойством не
обладает.
Звуковыми волнами называются распространяющиеся в воде
слабые возмущения - колебания с малыми амплитудами.
Процесс распространения звуковых волн (скорость звука),
благодаря большой частоте колебаний, является адиабатическим,
т. е. не сопровождается обменом теплом. В связи с этим морская
вода, с точки зрения акустики, аналогична идеальному газу. В
отличие от воздуха, морская вода слабо поглощает энергию
звуковых колебаний. Кроме того, скорость звука в воде практически
не зависит от частоты колебаний, т. е. отсутствует дисперсия
волн.
Как известно из физики, скорость распространения звука в
сплошной упругой среде определяется формулой [47]:
где К = — = р0 • (ф/ф)| - адиабатический объемный модуль
упругости, ро - плотность невозмущенной среды, к,, - коэффици-
149
ент адиабатической сжимаемости. В связи с тем, что и объемный
модуль упругости К, и плотность невозмущенной морской воды
Ро зависят от ее солености, температуры и гидростатического
давления, скорость звука тоже определяется этими параметрами
состояния (рис. 5.4).
Рис. 5.4. Зависимость скорости звука морской воды (мс )
от солености и температуры при атмосферном давлении
(а), давления и температуры при S~35 епс (б). При
расчетах использовалось УС-80
150
Преобразуем формулу (5.10) таким образом, чтобы в нее
входили величины, удобные для расчетов. Для этого перепишем
производную, входящую в (5.10), следующим образом:
= _v2 -*
dv
У
= v —.
ф
м..
Сравнивая это выражение с (5.7), получим:
ф|
ф|.
Откуда
C2=v -lf (5.11)
к
где v - удельный объем, к - коэффициент изотермической
сжимаемости, у = — - отношение удельных теплоемкостей при по-
cv
стоянных давлении и объеме соответственно.
Уравнение (5.11), если использовать уравнение состояния
УС -80, можно видоизменить:
С2=-^ VsTp . , (5.12)
WsTp | г QVsTp
dp " дТ
где Г - адиабатический градиент температуры.
Формула (5.12) применяется для расчета скорости звука и
называется теоретической. Она была использована для
составления известных таблиц скорости звука Меттьюза, а также таблиц
О.И. Мамаева [25] и некоторых других.
Наряду с теоретической формулой (5.12), существуют
эмпирические формулы определения скорости звука, основанные на
современных лабораторных методах ее измерения. Наиболее
достоверными из них можно считать формулы В. Вильсона [88],
В. Дель Гроссо [56] и К. Чена-Ф. Миллеро [52].
Наиболее близкой по вычисленным значениям скорости звука
к теоретическим с использованием УС-80 является последняя.
Она имеет вид:
151
C(S,t>p) = Cw{t,p)+A(t,p)S + B(t,p)Si/2+D(t,p)S2, (5.13)
где:
Cw{^p) = ao +a\ t + a2 -t2 + a3 -t3 +a4 t* +a5 -t5 +
(b0 +6, t + b2 t2 + b3 -t3 +64 •/")•/> +
[c0+crt + c2-t2 + c3 t3 +c4t*)-p2 +
[d0+drt + d2-t2)-p3,
a0=1402,388; a,=5,03711; a2=-5,80852-10"2; аз^З.342010-4;
a4=-l,4780010"6; a5=3,1464-10-9; 60=0,153563; A,=6,8982-10-*;
62=-8,178810'6; &э=1,362Ы<Г7; Z>4=-6,11851010; co=3,1260105;
c,=-l,7107106; c2=2,597410-8; C3=-2,53351010; c4=l,04051012;
rfo=-9,772910-9; 4=3,8504-10''°; rf2=-2,3643-10-'2;
A{f,p) = aQ + al -t + a2-t2 + a3-t3 + a4-t4 +
(b0 + 6, -t + b2 -t2 + b3 -t3 + b4 -t*}p +
(co+Ct -t + c2-t2 + c3t3)p2 +
.,. (d0+dl-t + d2-t2)p3,
a0=l,389; ai=-l,26210"2; a2=7,164-105; a3=2,006-10"6; a4=-3,21-10-8;
60=9,474210'5; 6,=-1,25 80-10"5; b2=-6,№5W'*; 63=1,050710-8;
64=-2,0122-1010; co=-3,9064-10-7; c,=9)104110i'; c2=-l,60021010;
c3=7,98810'12; do=l,\00\010; </,=6,649-1012; </2=-3,3891013;
B(t,p)= a0 +a, -t + (b0 +6, i)p,
ao=-l,92210-2; a,=-4,42-105; &0=7,3637-10-5; &,=1,794510-7;
D(t,p) = a + bp,
a=l,727-103; ^-7,9836-Ю-6.
Чуть позже те же авторы предложили новую версию этой
формулы [53], отличающуюся от (5.13) только одним
поправочным членом:
Csj> = cstp ~ [(" 2,19 • Ю-4 • Г +1,4 • Ю-5 • Г2 + 0,0029)- р +
(з,47 10-7-7'-2,59 10-8Г2-4,76 10"6)-/>2+ (5.14)
2,68-Ю"9 •/>*].
Формула (5.14) справедлива в диапазоне солености от 0 до
152
40 епс (ГТШС-78), температуры - от 0 до 40° С (МШПТ-68) и
давления - от 0 до 1000 бар. Давление/? входит в (5.14) в барах.
Изменение температуры морской воды вносит в изменение
скорости распространения звука наибольший вклад. При ее
повышении увеличивается модуль упругости К и уменьшается
плотность ро, что приводит, согласно (5.10), к увеличению
скорости звука. При этом изменение скорости при изменении
температуры на Г С уменьшается при высоких температурах по
сравнению с низкими.
Соленость оказывает меньшее влияние на скорость звука.
Отмечено, что соли, содержащиеся в морской воде, по-разному
влияют на объемный модуль упругости, т. е. на К, а
следовательно, и на скорость звука. При повышении солености, также как и
при увеличении температуры, скорость звука увеличивается.
Скорость звука увеличивается и при повышении давления.
Скорость звука, м»с1
1500 1520
*
3000 —
14
(— Под»
щныйзву
совой каю
1Л
_1
Рис. 5.5. Распределение скорости звука в Атлантическом
океане. GEOSECS Atlantic Expedition, 1972-73 гг. НИС
«Кпогг». Станция 45.
153
Для океанов, где с глубиной отмечается уменьшение
температуры воды, характерно уменьшение скорости звука. Однако,
начиная с некоторой глубины, повышение гидростатического
давления перевешивает роль температуры воды и скорость звука
начинает расти. Таким образом, на некотором горизонте
формируется слой с минимальными скоростями звука - подводный
звуковой канал (рис. 5.5). В нем, благодаря рефракции, звуковые лучи,
посланные горизонтально, сосредотачиваются в слое минимума
скорости и распространяются на очень большие расстояния (до
15 00О-18 000 км).
Среднее значение скорости звука в Мировом океане
приблизительно равно 1500 м-с'1. Более подробно распределение
скорости звука в океане описано в работе [10].
Задачи и вопросы для повторения
5.1. Что такое объемный модуль упругости?
5.2. Почему адиабатическая сжимаемость меньше изотермической?
5.3. Как коэффициент изотермической сжимаемости зависит от
солености, температуры и давления морской воды?
5.4. Найдите, как меняется внутренняя энергия при адиабатическом
сжатии?
Ответ:
Применим метод якобианов - формулы 2.59, 2.60, 2.61, 2.63, 2.67,
2.69,2.70, 2.71 и 2.72. Имеем:
dU_
др
[/>>л] [ал]
Р
-Р-Н--
KI 'с
т}м_,...
р
{Т)
■м
Все параметры положительные, следовательно
> 0, т. е. при
адиабатическом сжатии внутренняя энергия возрастает. Объясняется
это тем, что при постоянстве энтропии (теплообмен с окружающей
средой отсутствует) при увеличении внешнего давления уменьшается сред-
154
нее расстояние между молекулами, увеличивается их средняя кинетиче
екая энергия, а следовательно, возрастает температура.
5.5. Какие волны называются звуковыми?
5.6. Что влияет на скорость распространения звука в морской воде?
5.7. За счет чего в океане образуется подводный звуковой канал.
155
ГЛАВА 6. УСЛОВИЯ РАВНОВЕСИЯ МОРСКОЙ ВОДЫ
Напомним, что состоянием термодинамического равновесия
системы называется такое состояние, в которое она приходит с
течением времени при определенных постоянных значениях
внешних параметров и определенной постоянной температуре
окружающих тел. В этой главе рассматриваются как общие
условия равновесия термодинамических систем, так и частные. Кроме
того, кратко описываются фазовые равновесия и фазовые
переходы. Приводятся несколько примеров равновесия применительно к
морской воде.
6.1. Общие условия равновесия термодинамических систем
Прежде чем перейти к установлению условий равновесия
термодинамических систем, необходимо сформулировать второе
начало термодинамики для неравновесных процессов.
Возьмем два близких друг к другу состояния равновесия 1 и 2
некоторой системы. Допустим, что переход системы из
состояния 1 в 2 может быть как неравновесным, так и равновесным.
Тогда:
SQq-dU + bW^ (6.1)
5Q = dU + 8W9 (6.2)
где 5g„p, бй^нр - соответственно количество сообщенной системе
теплоты и совершенная ею работа при неравновесном процессе;
bQ, 8fV- то же самое, но для равновесного процесса.
Вычитая из уравнения (6.1) уравнение (6.2), получаем для
кругового процесса:
SQHp-SQ = WHp-M. (6.3)
Эта разность не может быть равной нулю, поскольку это
означало бы, что необратимый процесс перехода системы из одного
состояния в другое можно обратить равновесно без изменения во
внешней среде. Разность (6.3) не может быть положительной, так
как это означало бы, что за циклический процесс системой была
произведена работа 5fV - 5W > О только за счет теплоты тепло-
156
источника SQHp -SQ>0 без всякой компенсации.
Следовательно, SQ„p-SQ = SfVHp-W<0. Откуда
SQ>SQHp, (6.4)
5W>5WHp. (6.5)
Учитывая уравнение (2.9), получим:
<Ь\>Цг*> (6.6)
Л2-Л, > J-zr1. (6.7)
1 1
Из уравнений (6.6) и (6.7) следует, что переход системы из
одного состояния в другое, совершаемый адиабатически равновесно
(5Q = T,dr\ = 0), нельзя осуществить адиабатически
неравновесно (8Q - 0, dr\ > 0), и наоборот. При этом в последнем случае
энтропия системы возрастает. Утверждение о возрастании
энтропии в адиабатически замкнутой системе при неравновесных
процессах - и есть второе начало термодинамики для неравновесных
процессов. Оно характеризует энтропию как меру необратимости
процессов в замкнутой системе. В связи с тем, что практически
все естественные процессы - неравновесные, энтропия в
замкнутых системах всегда возрастает. Это указывает при их
протекании на направление естественных процессов: они проходят в
направлении роста энтропии.
Основное уравнение и основное неравенство, выражающие
первое и второе начала термодинамики для систем с
переменным числом частиц, можно теперь записать в виде
Td^dU + ^Ard^-^-dN,. (6.8)
/ к
Уравнение (6.8) позволяет установить общие условия
термодинамического равновесия, а с помощью этих условий в
применении к той или иной конкретной термодинамической системе
можно найти частные для данной системы условия ее равновесия.
157
Теория термодинамического равновесия была развита Гиб-
бсом, который использовал для этого применяемый в механике
принцип виртуальных перемещений26.
При равновесном состоянии термодинамических систем все
внутренние параметры являются функциями внешних параметров
и температуры (второе исходное положение термодинамики),
поэтому если последние заданы, то для определения состояния
системы не обязательно знать внутренние параметры. В случае
неравновесного состояния внутренние параметры уже не
являются функциями только внешних параметров и температуры - для
определения состояния системы необходимы дополнительные
независимые параметры. Неравновесную систему, таким
образом, можно рассматривать как равновесную, но с большим
числом параметров и соответствующих им обобщенных сил,
стремящихся удержать систему в равновесии. Если выход системы из
состояния равновесия рассматривать как результат виртуальных
отклонений внутренних параметров от их равновесных значений,
то, используя основное неравенство термодинамики, можно
получить общие условия термодинамического равновесия.
Рассмотрим некоторые примеры для термодинамической
системы, когда работа связана только с изменением объема.
Изолированная система. В этом случае U=const> V=consty
N=const. Неравенство (6.8) принимает вид:
ch)>0,
т. е. энтропия изолированной системы при неравновесных
процессах возрастает. При прекращении этих процессов наступает
устойчивое равновесие, а энтропия системы достигает максиму-
Если состояние механической системы определяется координатами
*ь---Л> а все функциональные связи системы выражаются условиями:
Л(*,,...,*„) = 0, Л = 1,...,л,
то перемещения 5хь...,5хп, допускаемые этими связями, называются
виртуальными (возможными) перемещениями. Виртуальные
перемещения удовлетворяют условиям:
idqt
Иначе говоря, при равновесии сумма работ всех сил для любого
виртуального перемещения системы равна нулю.
158
ма. Следовательно, общим условием устойчивого равновесия
изолированной системы является максимальность ее энтропии.
Очевидно, что при этом
Лл = Л-Ло<° >
где г|0 — энтропия в равновесном состоянии.
Изотермическая система при постоянном объеме. Основное
неравенство термодинамики в независимых переменных V и Г
выглядит так (см. 2.39):
dFK-^dT-pdV^^dN,.
k=\
При T=const9 V-const9 N=const получаем:
dF<0.
Иначе говоря, в изотермической системе с постоянным
объемом энергия Гельмгольца при неравновесных процессах убывает
и имеет минимум при устойчивом равновесии. Общее условие
устойчивого равновесия изотермической системы с постоянным
объемом можно записать в виде:
AF = F-F0>0 .
Аналогично можно получить и условия термодинамического
равновесия для других условий изоляции системы:
• если x\-const9 V=const, N=const, то AU>09 £/=£/„пП;
• если r\=const,p=consti N=const> то АН>09 Н-Н^у
• если T=const, p-const, N=const9 то AG>0, G=GWm-
В связи с тем, что термодинамические потенциалы С/, Я, F, G
могут иметь несколько минимумов, а энтропия - несколько
максимумов, состояния равновесия, соответствующие абсолютным
значениями минимумов С/, Н9 F9 G и максимума г\9 называются
устойчивыми. В других случаях состояния равновесия являются
неустойчивыми (метастабилъными). Примерами метастабиль-
ных состояний равновесия служат перегретая жидкость и
пересыщенный пар.
Исходя из установленных выше общих условий равновесия
термодинамических систем, можно получить следующие частные
условия равновесия:
• условие химического равновесия: в равновесной
гетерогенной системе химические потенциалы любого компонента
159
должны быть одинаковыми для всех фаз, в которых этот
компонент содержится;
• условие теплового равновесия: температура во всех частях
равновесной системы должна быть одинаковой;
• условие механического равновесия: давление во всех
частях равновесной системы, на которую не действуют иные силы,
кроме равномерного внешнего давления, должно быть
одинаковым.
Благодаря общим условиям равновесия термодинамических
систем, внешнее воздействие, выводящее систему из состояния
равновесия, вызывает в этой системе такие процессы, которые
ослабляют это воздействие. Это положение было установлено
Ле Шателье в 1884 г. чисто интуитивно. Обосновал же его Браун
в 1887 г., поэтому оно было названо принципом Ле Шателье-
Брауна. Значение этого принципа состоит в том, что он позволяет
предсказать направление, в котором под влиянием внешнего
воздействия изменится термодинамический процесс, протекающий в
произвольной системе.
Приведем несколько примеров:
1) Сообщим смеси изо льда и воды некоторое количество
теплоты. Тогда лед начнет таять, благодаря чему не произойдет
повышение температуры смеси, которое иначе наблюдалось бы.
2) Для соли, находящейся в насыщенном растворе,
повышение температуры вызывает растворение, если последнее связано с
охлаждением, в противном случае происходит выпадение
кристаллов.
6.2. Фазовые равновесия и фазовые переходы.
Уравнение Клапейрона-Клаузиуса
Сначала рассмотрим условия равновесия изолированной
двухфазной однокомпонентной системы вода-пар, на которую
действует только всестороннее давление. Общее условие
равновесия этой системы есть:
8л = 5лЧ5т1' = 0, (6.9)
где х\ и г|" - энтропии соответственно воды и пара. С помощью
уравнения (2.38) перепишем соотношение (6.9) в виде:
160
^+£.5г-^.5лг+^+£:.5г-Т^.5*-=о.
Учитывая, что экстенсивные параметры системы подчинены
следующим уравнениям связей:
U' + U" = U = const, ЫГ + 5С/" = 0;
V +V" = V = const, 8V + 8V" = 0;
N' + N" = N = const,SN' + 6N" = 0;
предыдущее выражение можно записать в виде:
{г Г) \г г) \т9 г)
Из этого уравнения находятся следующие частные условия
фазового равновесия однокомпонентной системы:
Г = Т\ р' = р\ ц' = ц". (6.10)
Если T=const и p^const, то эти три условия можно записать в
виде одного равенства химических потенциалов вещества в
фазах:
\1'(Т,р) = 11'{Т9р). (6.11)
При равенстве же двух фаз разных веществ должны
выполняться условия
т; = т;,р'х=р'2, (6.12)
где верхний индекс означает фазу, а нижний - компонент, т. е.
равенство температур и давлений в фазах. На химические
потенциалы никаких условий в этом случае не накладывается, так как
обмена частицами между такими фазами быть не может.
Из формул (6.10) и (6.12) можно сделать общий вывод, что при
равновесии гетерогенной системы из п фаз с к компонентами
температура, давление и химические потенциалы каждого
компонента во всех фазах одинаковы.
Это позволяет определить количество фаз (состоящих из
нескольких компонентов), способных одновременно находится в
равновесии (число независимых переменных гетерогенной
системы), которые можно изменять, не нарушая ее равновесия. Эта
задача была решена Дж. Гиббсом, который установил, что в
системе из к компонент одновременно в равновесии может
находиться не больше, чем Л+2 фазы:
п<к + 2. (6.13)
161
Выражение (6.13) называется правилом фаз Гиббса. Это
правило получено для гетерогенной системы, на которую действует
только одна сила давления. Соотношение (6.13) можно записать в
виде:
f = k + 2-ny (6.14)
где / - число термодинамических степеней свободы системы
(число независимых переменных, которые могут быть
произвольно изменены без нарушения равновесия гетерогенной
системы).
При равновесии двух фаз (л=2) системы с одним компонентом
(Л=1) число степеней свободыf=l. Этот вывод можно получить и
из равенства (6.11), которое связывает температуру и давление в
фазах. Одну из них можно взять за независимую переменную,
тогда получим зависимость давления от температуры при
равновесии:
р = р(т).
На 7р-шюскости это будет соответствовать кривой равновесия
фаз. При этом точки, лежащие по обеим сторонам этой кривой,
представляют собой различные однородные состояния тела.
При равновесии трех фаз одного и того же вещества, например
лед-вода-пар, число степеней свободы f=0, т. е. равновесие в
этом случае достигается только в одной точке на 7/7-плоскости
(рис. 6.1). Температуру и давление в этой точке можно получить
из следующих уравнений:
ц'(7\р) = ц'(7>),
ц'(7>) = И7>)-
Состояние, определяемое этими значениями переменных Т и
/?, называется тройной точкой. Тройная точка для пресной воды
имеет следующие значения: Г=0,0100° С прир=509 Па. Для
морской воды температура тройной точки выше температуры
замерзания примерно на такую же величину.
При равновесном переходе вещества из одной фазы в другую,
как и при фазовом равновесии, температура, давление и
химический потенциал вещества в фазах одинаковы. В то же время
имеются такие термические и калорические величины, которые при
одних фазовых переходах терпят разрыв, при других -
непрерывны. Поэтому различают прерывные и непрерывные фазовые пе-
162
реходы. Прерывные превращения называются фазовыми
переходами первого рода. При этих переходах скачком изменяется
удельный объем (плотность) вещества (см. например рис. 1.4) и
поглощается (или выделяется) теплота. К фазовым переходам
первого рода относятся плавление, кристаллизация, кипение и
т.д.
Pw, мбар
qI I 1 I 1 I 1 1
-10 0 10 20 t°C
Рис 6*1. Диаграмма фазовых состояний. Сплошные линии
- пресная вода, пунктирные - морская вода с соленостью
35 епс
Непрерывные фазовые переходы называются фазовыми
переходами второго рода. При этих переходах скачком изменяются
теплоемкость при постоянном давлении, сжимаемость,
коэффициент теплового расширения и т. д.
Найдем уравнение, характеризующее фазовые переходы
первого рода. Для этого воспользуемся условием равенства
химических потенциалов при равновесии двух фаз (6.11).
Дифференцируя (6.11), получим:
Ф'(р,г)=ф'(р,;г)
или
163
art
дТ\
■dT+
■dp =
Эц'|
дГ\
■dT +
Щ
dpi
■dp.
Откуда:
ОТ
др\
1 Ф1
l дТ\
Ф'|
г"*1
др\
dp_
dT
Производные, входящие в это выражение найдем, используя
уравнения Гиббса-Дюгема (2.48) для одной частицы, которое
выглядит таким образом:
d\i = -r\dT + Vdp.
Имеем:
dp _ц"-г\'
л~г^г> (615)
где г\ и V- энтропия и объем одной частицы. Уравнение (6.15)
является дифференциальным уравнением кривой равновесия и
называется уравнением Клапейрона-Клаузкуса. При
изобарических процессах уравнение (6.15) можно выразить через разность
энтальпий, если воспользоваться выражением (2.33):
dp = Н"-Н'
dT~'T-(V-V')'
Чаще уравнение (6.15) записывают в виде:
dp _ А.
~dT~ T(V-V')'
где \ = Т-(у)"-г]') - скрытая теплота перехода (теплота
испарения, сублимации, плавления) на грамм (моль) вещества. При
изобарических процессах - Х = Н* - #'. Отсюда видно, что при
переходе вещества, например, воды из фазового состояния с
большей энтальпией в фазовое состояние с меньшей происходит
отдача излишков энтальпии, а при обратном переходе
необходимое количество энтальпии поступает из-за пределов
термодинамической системы. Величина X называется «скрытой» теплотой
потому, что при переходе, например, жидкости в пар это тепло от
жидкости «отнимается», но не происходит одновременного непо-
(6.16)
164
средственного нагрева окружающей воздушной среды. Нагрев
может происходить только при обратном фазовом переходе -
конденсации водяного пара, и этот процесс может происходить в
другое время и в другом месте.
Как видно из (6.16), уравнение Клапейрона-Клаузиуса
связывает между собой теплоту перехода, изменение (скачок) объема и
наклон кривой равновесия в точке перехода. Основное
применение уравнения (6.16) состоит в вычислении удельных теплот
испарения, плавления, сублимации.
Уравнение Клапейрона-Клаузиуса вида
dT = Т>{У-У)
dp А,
используется для вычисления изменения температуры фазового
перехода (например, температуры замерзания).
Ранее условия равновесия описывались нами без учета
поверхностных явлений. Теперь включим в рассмотрение эти
процессы.
6.3. Поверхностное натяжение морской воды
В связи с близостью молекул жидкости друг к другу,
межмолекулярные силы притяжения имеют значительную величину. Но
поскольку это взаимодействие быстро убывает с расстоянием, то
начиная с некоторого расстояния силами притяжения между
молекулами можно пренебречь. Это расстояние называется
радиусом молекулярного действия, а сфера с таким радиусом - сферой
молекулярного действия. Радиус молекулярного действия имеет
величину порядка нескольких диаметров молекулы.
Равнодействующая всех сил притяжения, действующих на
молекулу в пределах ее сферы молекулярного действия, в среднем
равна нулю только в том случае, если молекула находится от
поверхности жидкости на расстоянии, превышающем радиус
молекулярного действия. В противном случае на каждую молекулу,
находящуюся в поверхностном слое, толщиной равной радиусу
действия, будет действовать сила, направленная внутрь
жидкости. Это связано с тем, что та часть сферы молекулярного
действия, которая выступает за пределы жидкости, заполнена
воздухом. Эта часть сферы из-за значительно меньшей плотности газа
165
по сравнению с жидкой фазой будет менее заполнена
молекулами, чем остальная часть.
Очевидно, что величина равнодействующей силы в
поверхностном слое растет в направлении от внутренней к наружной
границе слоя. Если по каким-то причинам происходит увеличение
поверхности жидкости, то часть молекул из подповерхностного
слоя должна быть поднята к поверхностной границе жидкости из-
за сохранения массы. Такой переход молекул связан с
необходимостью совершения работы против действующих в
поверхностном слое сил. Эта работа совершается за счет кинетической
энергии молекулы и идет на увеличение ее потенциальной энергии.
Следовательно, молекулы в поверхностном слое обладают
дополнительной (свободной) потенциальной энергией, которая
входит составной частью во внутреннюю энергию жидкости.
Из-за наличия поверхностной энергии жидкость обнаруживает
стремление к сокращению своей поверхности. Поскольку
положение равновесия в поле силы тяготения соответствует
минимуму суммарной энергии (потенциальной и поверхностной),
жидкость, без воздействия на нее внешних сил, будет принимать
форму с минимальной поверхностью.
Термодинамика поверхностных явлений была развита Дж.
Гиббсом. Он принимал поверхностный слой за новую
«поверхностную фазу», отличную от объемных фаз тем, что ее толщина
крайне мала по сравнению с протяженностью в двух других
измерениях.
Площадь поверхности L фазы является наряду с объемом
новым параметром, характеризующим состояние системы.
Увеличение поверхности системы происходит при постоянстве
температуры и объема и сопровождается затратой работы, так как для
образования новой поверхности некоторые частицы из объема
должны перейти на поверхность, что, как уже говорилось,
связано с работой против сил молекулярного взаимодействия.
Обозначим у обобщенную силу, соответствующую параметру
2. Тогда элементарная работа при увеличении поверхности на dL
(при Т и V постоянных) будет равна27:
Если бы процесс увеличения поверхности протекал адиабатически, то
совершаемая над жидкостью работа была бы равна приращению
внутренней энергии. В этом случае это приращение складывалось бы не
166
bW = -dFz=-ydL. (6.17)
Термодинамическим потенциалом при постоянных
температуры и объеме служит свободная энергия (энергия Гельмгольца)
F(T,V) = U-Tt\ (см. раздел 2.3). Его дифференциал, с учетом
работы (6.17), выглядит так:
dF = -\idT-pdV + ydZ. (6.18)
Величина у, характеризующая равновесие между двумя
соприкасающимися фазами, называется поверхностным
натяжением и равна силе на единицу длины на поверхности или
изменению свободной энергии на единицу увеличения поверхности:
dFT\
Y = -
(6.19)
Эта сила направлена по касательной к поверхности жидкости.
Рассмотрим условия равновесия в системе, состоящей из двух
фаз, разделенных поверхностью раздела. В предыдущем пункте
было показано, что при пренебрежении поверхностными
явлениями, условиями равновесия двух фаз одного и того же
вещества являются выражения (6.10).
В случае равновесия двух фаз с учетом поверхностных
явлений первое и третье условия (6.10) остаются без изменения. Что
касается давлений в фазах, то, поскольку теперь на границе
учитываются силы поверхностного натяжения, равновесие между
фазами может наступить и при разных давлениях в них.
Найдем это условие механического равновесия в системе из
двух фаз: жидкость (') и пар ("), исходя из минимума свободной
энергии при T=const и V=const.
только из приращения поверхностной энергии, но и из приращения
объемной энергии. Это вызвано тем, что увеличение поверхности
сопровождается охлаждением жидкости (при переходе молекул из глубины
жидкости в поверхностный слой скорость молекул уменьшается). Для
того чтобы внутренняя энергия изменялась только за счет
поверхностной энергии, процесс увеличения поверхности жидкости нужно
проводить изотермически. В этом случае увеличение поверхности жидкости
за счет совершения работы будет сопровождаться притоком тепла
0=ТАт\=А(Тх\) из окружающей среды.
167
Дифференциал свободной энергии системы, состоящей из
жидкости, пара и поверхности раздела между ними, когда
температура и химический потенциал в фазах одинаковы, равен:
dF = -р' • dV' - pW + ydL. (6.20)
При равновесии dF = 0. Отсюда
-p'dV'-p" -dV" + y dZ = 0,
а так как V + V = V = const, то
Р' = Р' + У~, (6.21)
где —- - кривизна поверхности раздела фаз. В случае произ-
dV
dL 1 1
вольной поверхности —- = — + —, где г} и г2 - главные радиусы
dV гх г2
поверхности. Когда эта поверхность сферическая, то
dL = </(4-ят2) 2
rfF' ~ d(4/3-n-r *)~ г'
Таким образом, при равновесии сферических капель жидкости
с паром давление в каплерч и давление пара/?" связаны условием
р'-р' = ^1. (6.22)
Отсюда видно, что на поверхности раздела двух фаз сущест-
назы-
~ 2-У и (\ 1Л
вует скачок давления, равный ——. Величина у
г
вается поверхностным давлением или давлением Лапласа.
По данным исследований, в области влияния поверхностного
натяжения существует зона упорядоченной структуры толщиной
в несколько тысяч молекулярных слоев с более прочными
водородными связями [39]. Ближайший к границе молекулярный слой
ориентирован к воздушной среде атомами кислорода, а плотность
воды в этом слое может понижаться по сравнению с
нижележащими слоями на 20-25% [71]. Еще одна важная особенность
поверхностного микрослоя, оказывающая влияние на
поверхностное натяжение, - вытеснение из него ионов растворенных
веществ в подповерхностные слои, приводящее к уменьшению
солености.
168
Зависимость поверхностного натяжения от температуры
вытекает из молекулярной структуры верхнего микрослоя воды. Чем
выше температура, тем меньше поверхностное натяжение из-за
меньшей силы взаимодействия между молекулами граничного
слоя. При повышении температуры воды от 0° до 50° С
поверхностное натяжение уменьшается приблизительно на 10%. Наличие
солей в малых концентрациях (порядка 0,1 епс) понижает
поверхностное натяжение воды, а для реальных морских условий
можно отметить его незначительное повышение (менее 1% для
35 епс) по сравнению с чистой водой [39]. Наиболее точный вид
зависимости поверхностного натяжения у от температуры Т
можно найти в работе [85]. Если объединить приведенную этими
авторами формулу с зависимостью поверхностного натяжения от
концентрации ионов хлора [39] и использовать шкалу Кокса для
солености, можно получить следующее выражение для у (Н-м*1):
у(Г,5)=7,56210"2~1,392810-4-Г-3,063-10-7Т2ч-2,20910-5-5. (6.23)
Диаграмма зависимости поверхностного натяжения от
температуры и солености приведена на рисунке 6.2.
I i I г i 1 ""'" Т^^Нг» I
О 5 10 15 20 25 30 35 40
Соленость, еле
Рис. 6.2. Зависимость поверхностного натяжения (Нм~ )
от солености и температуры морской воды
Помимо обеднения поверхностного микрослоя солями,
увеличивающими поверхностное натяжение, можно отметить и другой
169
процесс - обогащение этого слоя веществами, уменьшающими
поверхностное натяжение и, следовательно, понижающими
свободную энергию поверхности. Такие вещества называются по-
верхностно-активнымщ к ним относится большинство
органических пленок на поверхности океана, обязанных своим
происхождением как природным биологическим процессам, так и
антропогенным загрязнением.
Отметим, что величина поверхностного натяжения воды -
одна из самых больших для жидкостей (табл. 6.1).
Таблица 6.1
Величины поверхностного натяжения для различных веществ
при комнатной температуре
[ Вещество
Ртуть
Вода
Бензол
Спирт
Эфир
У,Нм!
0,490
0,073
0,029
0,023
0,020
Для чистой океанской воды при температуре 20° С
характерное значение у составляет примерно 0,073 Н-м"1, естественные
биологические пленки понижают эту величину до 0,05-0,06 Н-м"1,
а пленка нефти на воде может уменьшить поверхностное
натяжение в 3-4 раза - до 0,014-0,025 Н-м"1 [31]. Свойство масла
уменьшать поверхностное натяжение иногда используется в
судоходстве для сглаживания волнения. Как показывает опыт, достаточно
сравнительно небольшого количества масла для того, чтобы
придать штормовым волнам гладкую поверхность.
Естественные пленки под действием ветра, волн, брызг
непрерывно разрушаются и время их жизни невелико. Все
возрастающее загрязнение поверхностно-активными веществами (нефтяные
углеводороды, моющие вещества-детергенты) приводит к
появлению долгоживущих пятен и значительно изменяет протекание
физико-химических и биологических процессов в океане.
Высокая величина поверхностного натяжения воды имеет
большое биологическое значение. Действительно, поверхностное
натяжение и плотность определяют высоту, на которую может
170
подняться жидкость в капиллярной системе. В некоторых
случаях поверхностное натяжение является единственной причиной
явления, называемого адсорбцией {прилипанием).
Еще одним проявлением поверхностного натяжения в море
является образование и разрушение капиллярных волн на его
поверхности.
Теперь перейдем к рассмотрению условий механического
равновесия морской воды в толще океана.
6.4. Вертикальная устойчивость. Частота Вяйсяля
Рассмотрим случай, при котором температура Г, соленость S и
плотность р морской воды, как и все другие термодинамические
параметры, зависят только от вертикальной координаты z (ось z
направлена вниз). Выясним условия, при которых морская вода
находится в состоянии механического равновесия
(термодинамическое равновесие, очевидно, в данном случае невозможно,
поскольку температура непостоянна). Пусть частица морской воды
единичного объема, имеющая плотность р, смещается по
вертикали адиабатически (т. е. с сохранением потенциальной
температуры) с уровня z0 на уровень z0+Az, где плотность окажется
равной ро=р+Др. На смещенную частицу будет действовать
архимедова сила F (сила плавучести), а соответствующее этой силе
ускорение частицы массой т (m=pl=p) составит:
£-!--,■*■ ■ <6.24,
at т р
Поведение частицы под влиянием архимедовой силы зависит
от характера изменения плотности с глубиной — плотностной
стратификации. Если плотность с глубиной уменьшается, то
Др<0, а ускорение положительно: любое смещение приводит к
еще большему удалению частицы от первоначального уровня.
При Др=0 ускорение равно нулю. Если же плотность с глубиной
увеличивается, то Др>0 и ускорение частицы будет
отрицательным. В этом случае частица будет стремиться возвратиться на
глубину z0, проскочит исходное положение и, в конечном итоге,
начнет совершать малые колебания вверх и вниз вблизи этого
уровня. Чтобы найти их частоту, необходимо преобразовать
171
формулу (6.24) таким образом, чтобы она приняла вид уравнения
.простых гармонических колебаний:
d2z _ Ар z - z,
1 —*-Т-тГ- (5-25)
Введем обозначение:
*-£•£■ **>
р Az
тогда выражение (6.26) перепишется в виде:
^- = -N>.(z-z0). ({.27)
Решение этого уравнения выглядит так:
z = y*-cos(N-/-q>), (5.28)
где А и ф - соответственно амплитуда и фаза колебаний,
определяемая начальными условиями. Очевидно, что величина IV и
представляет частоту колебаний частицы морской воды,
смещенной адиабатически по вертикали при положительной
стратификации.
В формуле (6.26) градиент плотности вычисляется по
отношению к глубине z. Вместе с тем современные океанографические
приборы определяют характеристики в толще воды как фумцию
давления. Если учесть сжимаемость морской воды, такая задела
вертикальной координаты имеет преимущества и с физической
точки зрения [21]. Для замены градиентов в выражении ((.2б)
можно воспользоваться гидростатическим приближением с
учетом направления оси z вниз (dp = gpdz)> тогда:
»2=g2-^- 0-29)
Помимо частоты колебаний N, широкое распространение
получила тесно связанная с ней величина - полная вертикальной
устойчивость Е:
N2
£ = —. (GO)
g
В соответствии с выражением (6.26), произведение gE
подставляет собой вертикальное ускорение смещенной из состояния
механического равновесия по вертикали частицы воды, деленное
172
на величину этого смещения Az. При стремлении Az к нулю
можно перейти от конечных разностей к полной производной
плотности:
Д*-*> р Az р dz
(6.31)
Домножая числитель и знаменатель в (6.31) на dp=gp-dz,
можно получить выражение для устойчивости через градиент
давления, а не глубины:
* = *•£• (6-32)
dp
Получаемая в результате расчетов по формулам (6.30) и (6.32)
размерность устойчивости, очевидно, одинакова и соответствует
м"1. Заметим, что если частота Вяйсяля может быть только
действительным числом (rf>0), то устойчивость, как и
стратификация, - более широкое понятие. Можно говорить об устойчивом
(Е>0), неустойчивом (Е<0) или безразличном (Е=0) механическом
равновесии.
Представим дифференциал плотности в выражении для
устойчивости (6.32) с учетом уравнения состояния морской воды (4.7):
dp dS dp
dp
dS dp dT dp " dp
(6.33)
где 0 - потенциальная температура, Ta - адиабатическая
поправка. Два первых члена в квадратных скобках этой формулы
представляют произведение частных производных уравнения
состояния по солености и температуре in situ на соответствующие
изменения солености и температуры при перемещении частицы с
уровня р на уровень /И-ф, последнее слагаемое - барический
градиент плотности.
Если рассмотреть однородный сжимаемый океан, в котором
вертикальные градиенты солености и потенциальной
температуры отсутствуют, то выражение (6.33) будет выглядеть
следующим образом:
E = E=g
дТ dp
dp
dp
(6.34)
173
где —- = Г - адиабатический градиент температуры, вычисляе-
dp
мый по формуле Кельвина (4.74). Величина Ер определяется по
существу адиабатической сжимаемостью воды, ее можно назвать
барической устойчивостью. Все компоненты, стоящие в
квадратных скобках последней формулы, являются функциями
температуры, солености и давления, но не зависят в общем случае от
вертикальных градиентов термохалинных характеристик. Вообще
же для характерных диапазонов изменения температуры,
солености и давления величина Ер составляет примерно (4-5)-105 м'1
[69] и может превышать все остальные слагаемые полной
устойчивости.
При этом следует помнить, что вычислять условия
стратификации слоев имеет смысл тогда, когда их характеристики
приведены к единому давлению. Действительно, в самом начале этого
параграфа при рассмотрении условий равновесия и выводе
формулы (6.1) мы пренебрегали тем, что плотность частицы воды,
перемещающейся с уровня z0 (давления р0) на уровень z (с
давлением р), может изменяться за счет адиабатической сжимаемости.
В реальных условиях механическое равновесие этой частицы
воды на новой глубине или изобарической поверхности будет
наблюдаться тогда, когда ее плотность и плотность окружающей
среды одинаковы именно на этом уровне. Значит, при
определении разности плотностей частицы и среды Ар = р0 - р в формуле
(6.29) величины в правой части должны быть приведены к одно-
му давлению. Отсюда следует, что полную производную —*- в
dp
(6.32) или — в (6.31) необходимо заменить на сумму тех компо-
dz
нентов, которые определяются изменениями потенциальной
температуры и солености по вертикали и не зависят от чисто
барических эффектов. Именно поэтому в океанологии рассчитывают
вместо полной устойчивости так называемую термохалинную
устойчивость, представляющую разность между полной и
барической устойчивостью:
174
^Q,S
•p -
p
1
p
'dp
'dp dS dp
_dS' dz+ dT'
dS dp йЮ]
dz dT dz\
(6.35)
или, используя градиенты давления:
v ,"Ф dS
dp dQ
(6.36)
IdS dp dT dp]
Впервые этот параметр с использованием градиентов
солености и температуры in situ по глубине был предложен в 1915 г.
Т. Хессельбергом и X. Свердрупом, а в 1955 г. М. Поллак
предложил выражать величину устойчивости через градиент
давления.
Если бы океан был несжимаемым, то формулы (6.33) и (6.35)
давали бы одинаковый результат. Следовательно, условие
положительной стратификации — > 0 справедливо лишь для несжи-
dp
маемого океана. Условия же равновесия для реальных условий
будут выглядеть так:
Е> Еа (Eqs^) - положительная стратификация,
Е<Еа (Eofs<0) - отрицательная стратификация,
Е = Еа (£,9.5== 0) - нейтральная (равновесная) стратификация.
В соответствии с этими условиями, частота колебаний
частицы морской воды, выведенной из положения равновесия в
сжимаемом океане, будет определяться следующим соотношением:
N2B,s=g-EQiS=g
dp dS dp dQ
dS dp dT dp
(6.37)
В океанологической литературе величину NQfS обычно
называют частотой Вяйсяля или Вяйсяля-Брента по фамилии
геофизиков, предложивших в 1925-27 гг. эту величину для анализа
колебаний в океане и атмосфере. Размерность 7V, очевидно,
соответствует с"1.
Характерный профиль вертикального распределения частоты
Вяйсяля в Северной Атлантике по данным вертикального
зондирования приведен на рис. 6.3. Как мы видим, значения частоты в
верхнем слое составляют 103 с*1, увеличиваются на порядок в
175
слое основного скачка плотности и затем вновь понижаются ко
дну до 10"3 с"1. Соответствующие частоте Вяйсяля периоды Гтер-
2л
мохалинных колебаний (Г = —) изменяются от 8-10 минут в
слое скачка до нескольких часов в глубинной части океана.
100 60 40 20 10 Т.мин.
i.i i i i 1 1 л
1000
f
2000 Ь
3000
4000 Ь
5000
"Профессор Штокман",
22 11 1997 г.,
38°с.ш,51взд.
0.000 0.002 0.004 0.006 0.008 0.010 0.012
N, 1/С
Рис. 6.3. Вертикальное распределение частоты IV и
периода Т термохалинных колебаний в Северной Атлантике
(Саргассово море)
Интересной особенностью представленного профиля можно
считать наличие относительно тонких (порядка десятков метров)
прослоек ниже скачка плотности с большими изменениями
значений частоты Вяйсяля.
Обратимся теперь к рассмотрению отдельных компонентов
термохалинной устойчивости. В соответствии с формулами (6.35)
и (6.36), величина EqS может быть представлена в виде суммы
трех слагаемых - температурной устойчивости ЕТ, соленостной
устойчивости Es и адиабатической устойчивости Еа> причем:
_ dp dT dp dS
т * дт dp' s * ds dp'
r 5p dT\
5 дт
(6.38)
176
Такое представление дает возможность оценить вклад каждого
из компонентов в формирование вертикальной стратификации
океанов.
Температура в океане в основном понижается с глубиной,
производная плотности по температуре также отрицательна,
поэтому величина Ет оказывается положительной. Производная
плотности по солености положительна, а кривая вертикального
распределения солености в океане обычно имеет несколько
экстремумов. Таким образом, кривая соленостной устойчивости для
какой-либо станции в океане также может несколько раз менять
знак. Последний компонент - адиабатическая устойчивость - на
2-3 порядка меньше двух первых в верхнем слое океана и играет
роль в сохранении положительной стратификации только на
больших глубинах, где вертикальные градиенты температуры и
солености невелики. В качестве примера на рис. 6.4 приведены
вертикальные распределения температуры, солености и
компонентов термохалинной устойчивости для той же станции в
Северной Атлантике, что и на рис. 6.3.
Между изобарами 200 и 1500 дбар на графике отчетливо
видно, как температурный компонент устойчивости компенсирует
отрицательные значения Es порядка 10 7-10"6 м'1, а в глубинном
слое с давлением более 4500 дбар положительная стратификация
на всех уровнях определяется уже прежде всего компонентом Еа,
превышающим температурную устойчивость. Вновь обращают
на себя внимание резкие скачки температурной и соленостной
устойчивости, которые практически при любом давлении
оказываются скомпенсированными.
Если объединить температурную и адиабатическую части
устойчивости, то можно говорить о влиянии на стратификацию
градиента потенциальной температуры или о «потенциальной
температурной устойчивости» Eq.
Отношение EQ к Es называется в океанологии плотностным
числом:
R =^i=_« <ю/Ф= as de/dp
р Es (3 dS/dp дТ dS/dp'
177
О 400 800Е*108м"1
1000
2000
#000
4000Н
£000
Рис. 6.4. Вертикальное распределение температуры Т,
солености S и составляющих вертикальной устойчивости
Ег, Е& ЕА в Северной Атлантике.
В формуле (6.39) вместо отношения производных плотности
по температуре и солености стоит равное ему отношение
коэффициентов термического расширения и соленостного сжатия
(действительно, коэффициенты равны соответствующим
производным, помноженным на р"1).
Плотностное число, как можно видеть по соотношению
температурной и соленостной частей устойчивости на рис. 6.4, -
очень изменчивая величина, поскольку Es на вертикальном
профиле часто меняет знак.
В практике океанографических расчетов используют как
локальное значение плотностного числа для данного давления /?,
так и среднее значение плотностного числа для слоя воды,
соответствующего главному термоклину в океане (200-1500 м).
Помимо методов оценки термохалинной устойчивости (и
частоты Вяйсяля) по вертикальному градиенту потенциальной
температуры и солености (6.36) (метод Хесселъберга-Свердрупа),
существуют и другие способы расчета этой величины.
178
Один из них был реализован Фофоновым в так называемом
методе адиабатического выравнивания. Если в формуле (6.32)
заменить дифференциал плотности на дифференциал удельного
объема v = р"1:
ф = r-rfv
(6.40)
и представить в соответствии с (3.48) удельный объем v(S,T,p) как
сумму аномалии удельного объема 5(S,T,p) и удельного объема
стандартного океана v(35,0,/?), то выражение для термохалинной
устойчивости будет выглядеть так [76]:
g
-. (6-41)
*w" v(SyTiP)2
~b(S2fQ(S29T2yp2,p)yp)-5(Sl,Q(Sl,TXipl,p)iP)
Pi-Pi
где давление p соответствует середине интервала (риРгУ
Выражение для квадрата частоты Вяйсяля, очевидно, можно получить,
домножив (6.41) на ускорение свободного падения g.
Наконец, третий метод вычисления вертикальной
устойчивости основан на представлении зависимости адиабатического
градиента плотности от скорости звука в соответствии с формулой
(5.10). Действительно, представление термохалинной
устойчивости как разности полной вертикальной устойчивости,
определяемой градиентом плотности in situ p(S,Tj))y и барической
устойчивости Ер, позволяет использовать вместо выражения (6.34)
формулу для скорости звука (5.10). Тогда:
Я„=^Г> (642)
а выражение для термохалинной устойчивости примет вид:
*•-«(*-£]• (б4з)
Расчет устойчивости в соответствии с выражением (6.43)
первым ввел в океанографическую практику М. Поллак. Хотя метод
Поллака выглядит, на первый взгляд, наиболее
предпочтительным при вычислении термохалинной устойчивости, он
подразумевает получение малой разницы двух больших величин,
определяемых по сложным эмпирическим формулам для p(S,Typ) и
179
C(S,T,p), причем эти зависимости не согласуются между собой с
точки зрения влияния давления. Сравнение всех трех схем
расчета величины квадрата частоты Вяйсяля в работе [76] показало,
что самым экономичным с точки зрения объема вычислений
остается метод Хессельберга-Свердрупа, на 20% более трудоемок
метод Фофонова, а схема Поллака требует в несколько раз
больше расчетов. Тем не менее, метод адиабатического выравнивания
(6.41) более предпочтителен с точки зрения точности вычислений
для любого вертикального расстояния между горизонтами
наблюдений. При современном уровне точности зондирующей
аппаратуры метод Фофонова позволяет добиться точности
вычисления величины N2 или EqiS с ошибкой не более 1% в главном
термоклине и не более 5% на абиссальных глубинах для
вертикального разрешения между соседними горизонтами наблюдений
(т. е. при разности p2-pi) в 20 дбар. Используя больший интервал
между горизонтами наблюдений (100-200 дбар), можно
уменьшить ошибку еще в несколько раз. Интересно отметить, что
входящее в формулы вычисления вертикальной устойчивости и
частоты Вяйсяля ускорение свободного падения g изменяется между
экватором и полюсом примерно на 0,5% и при расчетах обычно
полагается постоянным и не зависящим от широты.
Задачи и вопросы для повторения
6.1. Назовите общие условия равновесия термодинамических систем.
6.2. Какие частные условия равновесия вы знаете?
6.3. Что отражает принцип Ле Шателье-Брауна?
6.4. Назовите правило фаз Гибсса.
6.5. Что такое число термодинамических степеней свободы системы?
6.6. Сколько термодинамических степеней свободы в системе лед-
вода-пар?
6.7. Что называется тройной точкой?
6.8. Какие существуют фазовые переходы?
6.9. Какое уравнение используется для вычисления изменения
температуры фазового перехода?
6.10. Что понимается под поверхностным натяжением?
6.11. Назовите условие механического равновесия в системе из
жидкости и пара с учетом поверхностного натяжения?
6.12. Как зависит величина поверхностного натяжения от
температуры и солености воды?
180
6.13. У какой жидкости поверхностное натяжение больше: у спирта
или воды?
6.14. Что такое полная вертикальная устойчивость морской воды?
6.15. Что отражают барическая и термохалинная устойчивости?
6.16. Что такое частота Вяйсяля?
6.17. Перечислите методы вычисления вертикальной устойчивости
морских вод.
181
ГЛАВА 7. КОЛЛИГАТИВНЫЕ СВОЙСТВА МОРСКОЙ
ВОДЫ
К коллигативным свойствам растворов относят понижение
температуры их замерзания и давления насыщенного пара над
ними, повышение температуры кипения, а также явления осмоса.
С точки зрения термодинамики, каждое из этих свойств водного
раствора зависит от концентрации растворенных веществ в
воде и не зависит от их специфических химических
характеристик. Обычно это приближение справедливо для идеальных
разбавленных растворов, к которым в большинстве случаев можно
отнести и морскую воду. Поскольку все коллигативные свойства
связаны с концентрацией растворенных веществ, появляется
возможность эмпирически выразить зависимость одного свойства
через другое, более точно измеряемое.
7.1. Температура замерзания
Температура замерзания морской воды 7} имеет огромное
лимитирующее значение - ниже температуры 7} вода не
охлаждается28. Поэтому на всех Г5-диаграммах изопикны (изостеры)
должны быть проведены только до линии температуры замерзания.
Температура замерзания зависит от давления. Для
определения понижения точки замерзания чистой воды (точки плавления
льда) с увеличением давления воспользуемся уравнением Кла-
пейрона-Клаузиуса (6.16) для случая сосуществования двух фаз -
воды и льда:
dT = T-(v'-v')
dp Х§
Одновременно с охлаждением воды до температуры, при которой
твердая и жидкая фазы могут находиться в равновесии при данном
давлении, начинается рост кристаллов вокруг центров кристаллизации.
Ими могут быть взвешенные в воде твердые частицы. Хорошо
очищенную от таких частиц воду можно охладить ниже температуры
замерзания. Состояние такой переохлажденной воды является метастабилъ-
ным.
182
где А,,. =Г-(г|'г-г|') - скрытая теплота плавления чистого льда;
V и v* - удельные объемы воды и льда. Вывод уравнения
приводится в разделе 6.2, по нему строится кривая плавления на 7р-
плоскости. Слева от этой кривой лежит область твердой фазы,
справа - жидкой.
Если в уравнение Клапейрона-Клаузиуса подставить значение
абсолютной температуры Г=273,15°К, удельные объемы чистой
воды и льда ^=1,000* 1 О*3 и v"=l,091-10"3 м3-кг' и удельную
теплоту плавления ^,=0,335 106 Дж-кг"1, то получим:
— = -0,0075° Сбар1.
dp
Для определения равновесного давления в системе вода-лед
уравнение (6.16) можно проинтегрировать и, с учетом малых
изменений удельных объемов воды v' и льда v" вблизи температуры
замерзания, получить следующую зависимость [37]:
p = p0+krJi^\(ir-*y9 (7.1)
гдеро - давление при заданной температуре Т0.
Замерзание морской воды представляет собой процесс
значительно более сложный, чем замерзание чистой воды, ибо в ней
имеются растворенные вещества. При замерзании должны
возникать дополнительные связи между молекулами воды, но часть
этих связей занята ионами растворенных солей. Следовательно,
для обеспечения необходимых межмолекулярных связей и
достижения минимума потенциальной энергии вода в момент
замерзания должна выделить из себя те вещества, которые были в ней
растворены. Отсюда следует, что соленость должна влиять на
температуру замерзания.
Понижение температуры замерзания раствора определяется
формулой Вант-Гоффа:
ДГ = -:^Й..*!-, (7.2)
V NQ'
где N\ и N0 - молярная концентрация чистого растворителя
(воды) и растворенного вещества; Х-4,05 106 Дж - теплота
плавления одного моля растворителя; Г0=273,15°К -температура
плавления чистого растворителя; /2=8,314 Дж(мольК)"1 - универ-
183
сальная (молярная) газовая постоянная. В морской воде при S=35
молярная концентрация чистой воды N0 равна «54 моль-кг1, а
основных солей N\=\ моль-кг"1. Тогда Nx/N0 =0,0185. Подставив
это отношение в (7.2), получим, что АГ= -1,9° С.
В современных океанографических исследованиях при
изучении зависимости температуры замерзания морской воды от
солености и давления используется эмпирическое уравнение,
предложенное в работе [80]:
гД5,^) = -0,0575^ + 1,71052310-3^3/2-
f ' (7.3)
- 2,154996 • 10~4-S2- 7,53 10~4- р,
где р - давление в дбарах. Формула (7.2) справедлива в диапазоне
практической солености от 4 до 40 и давления от 0 до 500 дбар
(рис. 7.1).
0 5 10 15 20 25 30 35
Соленость, епс
Рис. 7.1. Зависимость температуры замерзания морской
воды (°С) от солености и давления
dTf
С помощью уравнения Клапейрона-Клаузиуса значение ——
dp
можно экстраполировать до давлений примерно 2700 дбар. Здесь,
на предельном давлении существования температуры
наибольшей плотности, температура замерзания чистой воды равна
184
«-2° С. Воды с такой температурой наблюдались под шельфовы-
ми ледниками Антарктиды [66].
7.2. Давление насыщенного пара.
Скрытая теплота испарения
Энергия взаимодействия океана и атмосферы в значительной
степени зависит от фазовых переходов между водой и паром.
Рассмотрим процесс установления равновесия между водой и
ее паром. Это равновесие наступает в тот момент, когда число
молекул, перешедших в газообразное состояние из-за испарения,
будет равно количеству молекул, перешедших в жидкую фазу29.
Это достигается при достижении вполне определенного для
данной температуры давления.
Как было уже показано в п. 6.2, условием фазового равновесия
однокомпонентнои системы вода-пар при заданных температуре
и давлении является равенство химических потенциалов
компонента в фазах, т. е.
ц'(Т,р) = 11"(Т,р).
Таким образом, должна существовать функция j{Tj?) - кривая
испарения, которая и будет соответствовать равновесному
сосуществованию этих фаз. При заданной температуре Т пар и вода
будут в равновесии, если давление пара над ровной
поверхностью воды имеет значение pw (г), называемое давлением
насыщенного пара.
Для построения кривой испарения воспользуемся уравнением
Клапейрона-Клаузиуса (6.16):
dpw _ ^
Здесь Ха - количество тепла, необходимое для испарения
жидкости единичной массы, и называется удельной скрытой
теплотой испарения (парообразование); v" и v'- удельные объемы пара
Переход молекул из пара в жидкость осуществляется в результате
того, что, двигаясь хаотически в газообразной фазе, они ударяются о
поверхность жидкости, и часть таких ударов сопровождается переходом
молекул в жидкую фазу. При этом количество этих молекул
увеличивается пропорционально росту давления.
185
и воды. Теплоту парообразования при постоянном давлении
можно трактовать также как разность энтальпий единицы массы
воды в парообразном и жидком состояниях.
В формуле (7.4) при исследовании равновесия вода-пар
вместо разности удельных объемов можно использовать значение
только удельного объема пара, поскольку характерные значения
при 0°С v,=10° м3кг"!, a v"=206 м3-кг!. Но поскольку уравнение
Клапейрона-Клаузиуса имеет универсальный характер и
применимо для других фазовых переходов (лед-пар и вода-лед), в
последнем случае разница удельных объемов составляет величину
порядка 10"5 м^кг"1 и градиент — на несколько порядков
больше, чем при переходе вода-пар.
Зная величину р° для одной из температур Т0 и
соответствующую удельную теплоту парообразования Х° , можно выразить
значения давления насыщенного пара при любой другой
температуре. Интегрируя уравнение Клапейрона-Клаузиуса и выражая
удельный объем пара v" в соответствии с уравнением состояния
идеального газа
р
получим следующую зависимость давления насыщенного пара от
температуры (R- универсальная газовая постоянная):
[fe-'-r-'k]
PM'PW R J- (7-5)
Более точные зависимости давления насыщенного пара от
температуры приводятся, например, в «Океанографических
таблицах», таблице 5.39 [30], а также в специальных
психрометрических таблицах. Аналогичным образом можно представить и
давление насыщенного пара надо льдом, в этом случае вместо
теплоты испарения Ха используют так называемую теплоту
сублимации или возгонки, первая величина при 0° С равна 2,50-106 Дж-кг"1,
а вторая несколько больше - 2,83-106 Дж-кг"1 [37]. Для сравнения
напомним, что удельная теплота плавления пресного льда А,,-,
представляющая разность энтальпий жидкой и твердой фазы,
составляет 0,335-106 Дж-кг"1.
186
До сих пор все рассуждения в этом параграфе не
рассматривали влияния солености на равновесие фаз. В формуле (6.11) при
рассмотрении равенства химических потенциалов необходимо
учесть зависимость ц- \iy(pyT,S). В соответствии с законом Рауля,
давление насыщенного пара растворителя у разбавленных
идеальных растворов пропорционально относительной доле
растворителя (чистой воды) в этом растворе и давлению пара
растворителя без примеси. Морская вода не является идеальным
раствором и парциальное давление насыщенного пара над нею
несколько меньше того, которое следует из закона Рауля. Тем не менее,
может быть построена простая эмпирическая зависимость
pw(s) = f(pw(o\s). В соответствии с работой [61], она выглядит
так (S— практическая соленость, епс):
PAS) = PW{0)4-0,00054S). (7.6)
Понижение давления насыщенного пара над морской водой по
сравнению с пресной невелико - менее 2% для океанического
диапазона солености.
Скрытая теплота испарения для всего диапазона температур
может быть получена по давлению насыщенного пара.
Зависимость Х = f(k0,pw9T) следует из уравнения (7.4). Зная величину
давления насыщенного пара для одной из температур, можно
выразить величину Х(т) для пресной воды как функцию только
температуры. Наиболее точная эмпирическая формула
приводится в работе [85]:
Х(т) = 2,50039 • 10б - 2,3683 -103 - Т - , ч
v ' (7.7)
43ЬЮ"|-!Г2-и31-1(Г2-Г3,
где Т - температура, °С; удельная теплота парообразования
выражена в Джкг"1. Если рассчитать производную dX/dS исходя из
приближенных уравнений (7.4) и (7.6) и производной для
химического потенциала d\xldS [61], то можно получить зависимость
удельной теплоты парообразования от концентрации солей. Для
морской воды при океанической солености значение X должно
отличаться в меньшую сторону от величины для пресной воды не
более чем на 3-Ю3 Джкг"1 или 0,12%, и в реальных расчетах
поправка на соленость не вводится [85]. Величина А,(г)
практически линейно уменьшается от 2,50 МДжкг'1 при 0° С до
187
2,43 МДжкг1 при 30°С (рис. 7.2),
2.52-1- - г г~ г—~
1 *«
ас
I
g 2.48
| 2.46
Б
ё
? 2.44
О 2.42
2Л-| 1 J j j 1 j i j
0 10 20 30 40
Температура, °C
Puc. 7.2. Зависимость удельной скрытой теплоты
испарения (МДжкг1) пресной воды от температуры (°С)
В заключение этого параграфа приведем также упрощенную
формулу для расчета удельной скрытой теплоты сублимации V в
Дж-кг"1 [9]:
Х'(Т) = 2,839 • 106 - 3,6 • (Т + 35)2. (7.8)
При температуре 0° С удельная теплота сублимации на 13%
выше, чем теплота испарения.
7.3. Осмотическое давление
Под осмосом (от греческого слова osmos - толчок, давление)
принято понимать явление переноса растворителя через
полупроницаемые мембраны, которые пропускают отдельные
молекулы растворителя, но препятствуют при этом прохождению
растворенных ионов. В применении к двум объемам морской воды с
разными соленостями это явление означает, что через мембрану
из раствора с меньшей соленостью в раствор с большей солено-
188
стью будет переходить больше молекул воды, чем в обратном
направлении. Равновесие при одинаковой температуре растворов
наступит только в том случае, когда химические потенциалы
чистой воды ц(р,7) по обе стороны мембраны сравняются. Этого
можно достичь, если давление в ограниченном объеме, который
имеет более высокую соленость и пополняется через мембрану,
будет выше, нежели в объеме с меньшей соленостью. Разность
давлений, при которой наступает равновесие химических
потенциалов чистой воды по обе стороны мембраны, и называется
осмотическим давлением.
Осмотическое давление в общем виде для разбавленных
растворов подчиняется закону Вант-Гоффа:
P = AcRT9 (7.9)
где Ас - разность молярных концентраций солей по обе стороны
мембраны, мольм"3; R - универсальная газовая постоянная; Т-
абсолютная температура, °К; Р - осмотическое давление, Па.
Прямые измерения осмотического давления для морской воды
весьма затруднены, поэтому обычно используются эмпирические
связи между коллигативными свойствами.
Стениус в 1904 г. эмпирическим путем установил, что
осмотическое давление в морской воде тесно связано с величиной
понижения температуры замерзания при увеличении солености.
Соотношение между осмотическим давлением Р0 (в атмосферах)
при 0° С и температурой замерзания 7} выглядит так [70]:
Р0 =-12,08-7}. (7.10)
В этой формуле осмотическое давление рассчитывается как
разность давлений между раствором с соленостью 5, имеющим
температуру замерзания 7}, и водой с нулевой соленостью, у
которой температура замерзания равна 0° С.
Если температура раствора отлична от 0° С, то осмотическое
давление Рт (Т - абсолютная температура, °К) можно выразить
следующей формулой:
Р =Р • =-Г •——— (7 И)
т ° 1,273,15) f 22,612*
Различия в величинах осмотического давления, полученных
по эмпирической формуле (7.11) и теоретическому соотношению
(7.9), не превышают 1 атмосферы [31], но эмпирическая формула
189
более удобна для практического использования. Если перейти от
давления в атмосферах к давлению в барах (1 атм=101325 Па=
1,01325 бар) и выразить величину 7}в соответствии с полиномом
(7.3), то выражение для осмотического давления Р0 (бар) как
функции температуры (°К), солености S и внешнего давления р
(бар) примет вид:
/> = Т • (2,5096 • 10"3 • S - 7,4659 • 10~5 - 53/2 +
^ (7.12)
9,4058 • 10"6 - S2 + 3,287 • КГ5 • р).
Для практической солености 5=35 и внешнего давления /7=1
атм осмотическое давление достигает 24,2 бар при температуре
278° К (5° С) и 26,7 бар при температуре 35° С. Для сравнения,
осмотическое давление кровяной плазмы человека составляет 7,7
бар, что для температуры 36,6° С соответствует 5=10,4. Величина
Р почти линейно возрастает с увеличением солености - примерно
на 0,7 бар на каждую единицу практической солености (рис. 7.3).
Несколько более сложную зависимость осмотического
давления от температуры и солености при атмосферном внешнем
давлении приводит в своей работе Ф. Миллеро [82]. Вместо
линейной зависимости величины Р от температуры Т в формуле (7.12)
автор предложил квадратичную, хотя результат при этом
изменился менее чем на 0,4 бар.
Осмотическое давление играет важнейшую роль в
физиологии, определяя перенос воды и растворенных веществ через
биологические мембраны. В частности, если пресноводную лягушку
поместить в океанскую воду, то она вскоре потеряет примерно
пятую часть своего веса из-за меньшей концентрации соли
внутри организма. С другой стороны, морские животные, попавшие в
пресную воду, набирают избыток внутриклеточной жидкости,
клетки при этом разбухают и лопаются. Это явление называется
лизисом. Автолизирующий фермент вырабатывается при нересте
у некоторых проходных рыб, что ведет к быстрому разложению
их тканей после гибели и способствует ускорению поступления в
воду органических веществ, необходимых для развития икры
[43].
190
Рис. 7.3. Зависимость осмотического давления морской
воды (бар) от солености и температуры при атмосферном
давлении (а), давления и температуры при S=35 епс (б)
По отношению к осмотическому давлению все морские
организмы можно разбить на три группы.
К первой группе принадлежат почти все морские
беспозвоночные. Осмотическое давление, температура замерзания и
химический состав их крови (внутриклеточных жидкостей) весьма
близки к тем же свойствам морской воды, в которой они обитают.
191
При переходе этих организмов из воды большей солености в воду
с меньшей соленостью они испытывают лизис.
Ко второй группе относятся акулы и скаты. Осмотическое
давление их крови также равно осмотическому давлению
окружающей воды, но выравнивание идет частично за счет солей и
частично за счет мочевины.
Третья группа объединяет организмы, у которых
осмотическое давление отличается от морской воды. Например, у морских
костистых рыб осмотическое давление несколько ниже, чем в
морской воде, а млекопитающие и некоторые рыбы являются
осмотически совершенно независимы от морской воды. У растений
осмотическое давление (тургор) всегда выше осмотического
давления окружающей воды. При этом у видов, живущих в более
соленой воде, осмотическое давление больше, чем у видов,
живущих в море с менее соленой водой.
В общем можно сказать, что чем выше развит организм, тем
более развита его осмотическая независимость от окружающей
среды.
7.4. Вязкость
Вязкость или внутреннее трение - свойство жидкостей или
газов оказывать сопротивление при перемещении одного слоя
относительно другого. Если же жидкость движется как единое
целое, то внутреннее трение не возникает. Для выяснения сущности
этого явления рассмотрим следующий элементарный опыт.
Возьмем две бесконечные плоские и параллельные друг другу
пластины, расстояние между которыми равно А. Пусть верхняя
пластина движется относительно нижней с постоянной
скоростью и. Чтобы такое движение существовало, к жидкости в
направлении движения должна быть приложена касательная сила,
уравновешивающая силу внутреннего трения. Опыт показывает,
что сила трения т, отнесенная к единице площади пластины, т. е.
касательное напряжение, прямо пропорциональна скорости и и
обратно пропорциональна расстоянию Л. Вместо отношения ulh в
общем случае одномерного движения можно взять отношение
du/dh:
192
т = ц~. (7.13)
dh
Коэффициент пропорциональности ц, который зависит от
природы жидкости, называют коэффициентом динамической
вязкости или просто вязкостью. Чем больше значение ц, тем
больше вязкость жидкости и тем больше сила, которую надо
затратить для преодоления трения между слоями для сохранения
равномерного движения пластины. Выражение (7.13) называют
законом трения Ньютона [44]. Часто вместо динамической
вязкости применяют отношение величин вязкости и плотности,
называемое кинематической вязкостью v:
v = ^. (7.14)
Р
Размерность динамической вязкости, очевидно, - кг-(м-с)"1, а
кинематической вязкости - м^с"1. Чаще при измерении ц
используется единица давления, умноженного на секунду: Н-с-м'2 или
внесистемная единица - пуаз. 1 пуаз равен 0,1 Нем"2, а
1 Kr-M"V=9,81 Нем"2.
Процесс передачи импульса от слоя с большей скоростью к
другому слою зависит от конкретных особенностей
межмолекулярных сил в жидкости. Очевидно, особенности структуры воды,
описанные в главе 1, определяют зависимость вязкости от ее
основных параметров - температуры, солености и давления. При
возрастании температуры ослабевают водородные связи, -
следовательно, переход молекул из одного слоя в другой
облегчается и значение коэффициента вязкости падает. Наличие
растворенных веществ-электролитов в целом упрочняет структуру
воды и повышает вязкость. Зависимость вязкости от давления -
одно из проявлений аномальности воды, как отмечалось в разделе
1.3. Вязкость «нормальных» жидкостей с увеличением давления
повышается, т. к. уменьшается среднее расстояние между
молекулами [39]. Напротив, значение ц в морской воде при
температуре менее 20° С понижается во всех реально встречающихся в
океане диапазонах давлений, и лишь при давлениях 1000-1500
бар вязкость начинает возрастать.
Наиболее точные эмпирические зависимости для вязкости
морской воды приведены Г. Зидлером и Н. Петерсом [85]. После
193
несложных преобразований относительных зависимостей,
представленных в этой работе, значение ц для пресной воды как
функции температуры будет выглядеть так:
ц0>г = 0,001002 • 10(22'6872-,.°978^0018277'2И7'+89>93), (7.15)
где Т- температура, °С, \i - вязкость, Нем"2. Проверочное
значение - 4,0387-Ю*4 Нем2 при Г=70° С.
В работе [85] приводится полученная Ф. Миллеро зависимость
вязкости \л от объемной хлорности морской воды СЕ которая
связана с соленостью S и плотностью р (в кг-м"3) в соответствии со
шкалой Кокса (3.35) соотношением C£(pS)/1806,55. Выражение
имеет вид:
^r=(i+^Va+^GM0,r> (716>
где^=5,18510"5Т-ь1,067510^,5=3,30010*5Т+2,59Ь10"3.
Зависимость вязкости от давления может быть представлена в
виде поправки вида [85]:
&\1 = ра\ах+а2-Т + аъ-Т2 +ра\аА + а5-Т + а6 Г2)],
где />a=p+10,13; ^=-1,8266-10"*; а2=1,381710'9; а3=-2,6362-10и;
я4=9,89651013; я5=-6,325 1014; а6=1,21151015. Давление ;> в (7.17)
задается в дбар, температура Г в °С.
Хотя формула (7.17) разработана для океанического диапазона
солености, она может применяться для солоноватых и даже
пресных вод. Ошибка этой формулы относительно наблюдаемых
значений при давлениях до 10 000 дбар составляет для пресной
воды примерно 3%, а для воды с соленостью 35 епс не более
0,4%.
На рис. 7.4а представлена зависимость коэффициента
динамической вязкости от солености при атмосферном давлении, а на
рис. 7.46 - зависимость ц от давления при 5435 епс. Наибольшее
влияние на вязкость, очевидно, оказывает температура: при ее
повышении от 0° до 30° С значение коэффициента ц падает более
чем вдвое, а увеличение солености от 0 до 35 епс повышает
вязкость менее чем на 20%. Влияние давления сказывается лишь при
температурах ниже 5° С и не превышает 4%. Для кинематической
вязкости влияние солености и давления будут еще меньше, так
194
как одновременно со значением ц аналогично изменяется и
плотность воды.
10 15 20 25 30 35 40
Соленость, епс
ЭК-
лЯг
О 9А-
У 20-
£
£* 15-
3 1°"
1
5-
'
lijii>^^
>
I
г—
_
^^
i
l 1
20
г—1
00
1 1
40
— 1 -*
1.2 ; ,
, i i
f
Г i.4i
f
Е1.6 ; 1
гЛЛ-
00
ГТ~
^t
F—1
6G
00
8С
00
]
вИ
mi
Давление, дбар
Рис. 7.4. Зависимость динамического коэффициента вяз-
кости lO3'^ (Нем2) от солености и температуры (а),
давления и температуры при солености S=35 епс (б)
В заключение отметим, что молекулярная вязкость в реальном
океане играет подчиненную роль для большинства видов
движений. Исключение составляют оценка эффектов диссипации энер-
195
гии в тепло, возникновение турбулентности, расчет параметров
мелкомасштабной конвекции, динамика взвеси и ряд других
задач. Вместо уравнения (7.13) для оценки переноса импульса
применяется его аналог, где постоянная скорость и заменяется на ос-
редненную за некоторый интервал времени соответствующую
компоненту вектора скорости течения, а коэффициент jx - на так
называемый коэффициент турбулентного обмена количеством
движения, который превосходит динамическую вязкость на
несколько порядков. Подробнее с турбулентным обменом в океане
можно познакомиться в работе О.И. Мамаева [21].
Задачи и вопросы для повторения
7.1. Что понимается под коллигативными свойствами?
7.2. Какие свойства морской воды можно отнести к коллигативным?
7.3. Как зависит температура замерзания морской воды от давления
й солености?
7.4. Как называется давление, при котором система вода-пар
находится в состоянии равновесия при определенной температуре?
7.5. Влияет ли соленость морской воды на давление насыщенного
цара?
7.6. Что такое осмос?
7.7. Как можно определить осмотическое давление в морской воде?
7.8. Что такое лизис?
7.9. Как осмотическое давление зависит от солености, температуры
и давления?
7.10. Что такое вязкость?
7.11. Какова размерность динамического коэффициента вязкости?
7.12. Как вязкость зависит от солености, температуры и давления?
196
ГЛАВА 8. ЭЛЕКТРОМАГНИТНЫЕ СВОЙСТВА
МОРСКОЙ ВОДЫ
Совершенно чистая вода очень слабо проводит электрический
ток, так как она состоит из нейтральных молекул и в ней нет
достаточного количества свободных электронов или других каких-
либо свободных заряженных частиц, которые, двигаясь под
влиянием внешнего электрического поля, создавали бы его.
Совершенно по-иному ведет себя морская вода.
Макроскопически морская вода электрически нейтральна из-за того, что
анионы и катионы растворенных в ней солей, участвуя в
тепловом движении, как и молекулы, при перемещении взаимно
компенсируются и не порождают электрического тока [17]. Однако
если приложить к морской воде внешнее электрическое поле, то
слабое упорядоченное движение свободных зарядов30 (ионов
растворенных солей) начинает преобладать над тепловым
движением - катионы перемещаются в направлении поля, анионы - в
противоположном. Возникновение электрических токов и полей
всегда сопровождается образованием магнитных полей, т. е.
формированием электромагнитного поля31.
На Земле существует квазистационарное магнитное поле,
называемое главным магнитным полем и обусловленное ее
магнетизмом как планеты. Кроме того, существуют переменные
магнитные поля {магнитные вариации), связанные с
электромагнитными процессами, протекающими в ионосфере и магнитосфере
под воздействием корпускулярного излучения Солнца.
Последние индуцируют в движущейся воде переменные электрические
Существуют два типа электрических зарядов - свободные и
связанные. Связанными зарядами называются заряды, которые входят в состав
атомов и молекул.
31 Характерная особенность электрического поля, отличающая его от
других физических полей, состоит в том, что оно действует на
электрический заряд (заряженную частицу) с силой, не зависящей от скорости
его движения. В то же время главная черта магнитного поля состоит в
том, что оно действует на движущиеся электрические заряды с силами,
пропорциональными скоростям зарядов и направленными
перпендикулярно к этим скоростям.
197
поля, называемые теллурическими, которые охватывают весь
океан.
Любые движения вод в океане (течения, ветровые волны,
приливы и т. д.) переносят свободные ионы, создавая, таким образом,
конвекционный ток. Этот ток приводит к возникновению
дополнительного электромагнитного поля в океане. Кроме этого, эти
движения воды в магнитном поле Земли индуцирует
электрические токи, плотность которых зависит от магнитного поля и
скорости движения зарядов. Индуцированные электрические токи и
поля в свою очередь создают переменные магнитные поля.
Квазистационарные электрические токи и поля в океане, вызванные
всеми видами движения морских вод в магнитном поле Земли, а
также химическими и физическими процессами в толще вод,
называют естественными токами {полями).
Полное электрическое поле в океане - сумма
квазистационарного естественного и переменного теллурического полей.
Основными параметрами, характеризующими
электромагнитные свойства морской воды, служат: диэлектрическая
проницаемость е, магнитная проницаемость \i и удельная
электропроводность у. Первые два параметра отражают способность воды
изменять свою электрическую и магнитную индукцию под
влиянием внешних электрических и магнитных полей. Удельная
электропроводность характеризует перенос электрических зарядов в
воде под действием внешнего электрического поля.
8.1. Электропроводность морской воды
По способности проводить электрический ток во внешнем
электрическом поле морскую воду можно отнести к слабо
концентрированным электролитам (проводникам второго рода).
Удельной электропроводностью (электропроводимостью) у
называют величину, обратную удельному сопротивлению
Rs ТУ г
р = , где R - сопротивление проводника (в данном случае -
столба жидкости) длиной / и площадью поперечного сечения s:
Т~~-. (8.1)
Rs
198
Размерность у - Ом^м^Смм"1 (сименс на метр)32.
Электропроводность морской воды слагается из проводимости
растворов основных электролитов, входящих в состав морской
воды, и определяется концентрацией свободных зарядов и их
подвижностью:
у = 2>лс.К+еО> (82)
где а - коэффициент активности раствора, с - концентрация
раствора, п - валентность ионов, и+ и и_ - подвижность
положительных и отрицательных заряженных ионов. Подвиэюностъю
иона называется его скорость под влиянием электрического тока
при условии, что ион движется в поле с падением потенциала,
равным одному вольту на один метр. Концентрация раствора с
*
связана с концентрацией ионов с следующей формулой: с = —,
а
где а - степень диссоциации. В числителе формулы стоит число
молекул, распавшихся на ионы, в знаменателе - число молекул
растворенного вещества.
Надо помнить, что вклад различных ионов в полную
электропроводность морской воды изменяется в зависимости от
наличия других ионов, поэтому всякое изменение относительного
состава анионов и катионов влечет за собой изменение
проводимости при постоянной солености.
Отметим, что ион К+ влияет на электропроводность сильнее,
чем ион Na+, а последний - сильнее, чем ионы Mg2+ и Са2+. Что
касается анионов, то ион СГ влияет сильнее на
электропроводимость, чем ион SOj".
Подвижность ионов морской воды зависит от ее вязкости -
чем она меньше, тем быстрее движутся ионы, т. е. увеличивается
электропроводность. Вязкость уменьшается при увеличении
температуры и, в меньшей степени, давления (рис. 7.4). Кроме того,
сжимаемость морской воды приводит к росту концентрации
ионов. Следовательно, электропроводность морской воды зависит
как от солености (увеличивается концентрация), так и от
температуры (повышается подвижность ионов) и давления (повышает-
32 Сименс - единица электрической проводимости, обратной величины
электрического сопротивления. Размерность См=Ом*1=м"2-кг !-с3#А2.
199
ся подвижность ионов и увеличивается концентрация ионов)
(рис. 8.1).
15 20
Соленость, епс
6000
Давление, дбар
Рис. 8.1. Зависимость удельной электропроводности
морской воды (Смм1) от солености и температуры (а), дав-
пения и температуры при солености S=35 епс (б)
Давление влияет на удельную электропроводность в меньшей
тстепени, чем соленость и температура - на глубине 10 000 м
проводимость увеличивается лишь на 5-10% по сравнению со своим
200
значением на поверхности (рис. 8.16). Как видно из рисунка 8.16,
удельная электропроводность с увеличением давления растет
медленнее, что связано с одновременным уменьшением
сжимаемости морской воды.
Зависимость электропроводности от давления играет важную
роль при измерениях электропроводимости in situ при помощи
океанографических CZD-зондов33.
С повышением температуры электропроводимость
увеличивается, причем эта зависимость наиболее ярко выражена при
высоких соленостях (рис. 8.1а).
Электропроводность морской воды in situ можно вычислить
по заданным значениям солености, температуры и давления, если
сначала определить относительную электропроводность RT,
обратив для этого эмпирическое уравнение (3.36), а затем применить
формулу (3.40).
Для сравнения удельной электропроводности различных
веществ ниже приведена таблица 8.1., которая свидетельствует, что
электропроводность морской воды относительно высокая.
Таблица 8.1
Удельная электропроводность различных сред
Среда
1 Известняк
Почва сухая
Почва влажная
Дистиллированная
вода
Морская вода
1 Лед
Удельная электро- 1
проводность (См-м1)
100-10*'
кИ-кг5
102-103
2-Ю"4
3-7
з-кгМ-кг7
Значительные изменения электропроводности морской воды в
зависимости от изменения ее солености позволяют применить
электрометрические методы для определения последней, что в
настоящее время широко используется в практике
экспедиционных исследований с помощью солемеров и океанографических
Аббревиатура CTD обозначает электропроводность, температуру и
глубину (в английском языке - conductivity, temperature, depth).
201
СТ£>-зондов. В океанографической аппаратуре применяется два
метода для измерения удельной электропроводности:
контактный - кондуктивный и бесконтактный — индуктивный. Первый
основан на измерении электропроводности морской воды,
протекающей в трубке между электродами. С помощью индуктивного
метода проводимость определяется по измерению
электродвижущей силы взаимоиндукции в обмотке одного из тороидальных
трансформаторов, установленных коаксиально друг к другу в
результате индуктивной связи между ними через морскую воду.
Еще одно применение удельной электропроводности —
электромагнитный метод измерения скоростей течений. Еще М. Фа-
радей высказал мысль о том, что пересечение магнитных силовых
линий земного поля потоком воды должно индуцировать в этой
воде электрический ток, тем более сильный, чем больше
электропроводность воды. Подобные условия существуют при наличии
морских течений, воды которых непрерывно пересекают силовые
линии геомагнитного поля Земли.
8.2. Диэлектрическая проницаемость
Параметром, отражающим способность морской воды
изменять свою электрическую индукцию под влиянием внешнего
электрического поля, является диэлектрическая проницаемость.
Она зависит от структуры и свойств морской воды, а также от ее
способности поляризоваться во внешнем электромагнитном поле.
Кроме того, на этот параметр влияет частота переменных
электромагнитных полей, распространяющихся в воде виде
электромагнитных волн (рис. 8.2).
Морская вода обладает аномально высокой диэлектрической
проницаемостью, что обуславливается полярностью ее молекул.
Под поляризацией понимается явление, когда в любом малом
объеме воды возникает отличный от нуля суммарный дипольный
электрический момент молекулы. Электрическим диполем
называется система из двух равных по абсолютной величине и
противоположных по знаку электрических зарядов, расстояние между
которыми мало по сравнению с расстоянием до рассматриваемых
точек поля. Выделяют эюесткий диполь - диполь, у которого
202
имеется постоянный по модулю электрический дипольный
момент. Электрическим дипольным моментом называется вектор
где q - величина заряда, /- расстояние между зарядами.
Вспомним (см. п. 1.1), что молекула воды обладает очень
большим дипольным моментом, равным 1,86 D.
О 10 20 30 40
Т#мп«ратура, *С
Рис. 8.2. Зависимость диэлектрической проницаемости
(безразм.) пресной воды от температуры (°С) для разных
частот электромагнитных волн: 1 - l(f Гц, 2 - 31(f Гц,
3-31(?Гц,4-1010Гц[37]
Количественной мерой поляризации служит вектор поляриза-
ifuu Р. Под ним понимается отношение электрического диполь-
ного момента малого объема к величине этого объема. Единица
измерения поляризации - Клм"2 (кулон на метр в квадрате)34.
Кулон - единица электрического заряда (количества электричества).
Кулон равен электрическому заряду, проходящему через поперечное
сечение проводника при постоянном токе силой один ампер за одну
секунду, т. е. К=Ас.
203
Диэлектрическая проницаемость 6 связана с вектором
поляризации Р в электрическом поле напряженностью Е следующим
образом:
(е-1) = Хе=-^ (83)
где Хс - безразмерная величина, называемая относительная
диэлектрической восприимчивостью вещества, е0 - электрическая
постоянная, равная 8,85* 10"12 Фм"1 (фарад на метр)35.
Диэлектрическая восприимчивость изотропных веществ - величина
скалярная, так что вектор поляризации совпадает по направлению с
вектором напряженности поля Е36.
В морской воде существуют электронная, дипольно-
релаксационная и ионная поляризации.
Электронная поляризация - результат смещения электронов
относительно ядра при воздействии электрического поля, при
этом формируется небольшой дипольный момент атома.
Электронная поляризация зависит от радиуса атома и температуры
воды.
В связи с асимметрией внутримолекулярных сил в воде
возникает дипольно-релаксационная поляризация. В результате центры
областей положительных и отрицательных зарядов молекулы не
совпадают друг с другом, и молекула представляет собой
жесткий диполь даже при отсутствии внешнего электрического поля.
Ориентация дипольных моментов зависит как от теплового
движения молекул, так и от присутствия внешнего электрического
поля. Тепловое движение молекул вызывает хаотичную
ориентацию дипольных моментов в пространстве, внешнее
электрическое поле ориентирует дипольные моменты в направлении поля.
Фарад - единица электрической емкости. Фарад равен емкости
конденсатора, в котором при заряде один кулон напряжение составляет
один вольт, т. е. Ф=КлВ"1= м'^кг'-с^А2.
36 Под вектором напряженности Е понимается отношение силы F,
действующей со стороны электрического поля на точечный пробный
F
заряд, к величине этого заряда - Е = —. Единица измерения напряжен-
Ч
ности - вольт на метр.
204
Таким образом, дипольно-релаксационная поляризация
определяется вектором поляризации Р и температурой воды.
Этот вид поляризации вносит основной вклад в величину
диэлектрической проницаемости. С увеличением температуры
влияние дипольно-релаксационной поляризации резко
уменьшается.
Ионная поляризация вызвана смещением ионов растворенных
в морской воде солей во внешнем электрическом поле:
положительные ионы смещаются в направлении поля, отрицательные -
против поля. Ионная поляризация зависит от зарядов ионов,
расстояния между двумя равновесными ионами и температуры.
Рассмотрим особенности диэлектрической проницаемости в
зависимости от частоты электромагнитных волн. В интервале
частот 101—108 Гц влияние дипольно-релаксационной
поляризации на диэлектрическую проницаемость наиболее значимо, при
этом оно намного больше влияния электронной поляризации. Это
приводит к тому, что океаны и моря практически непрозрачны
для радиоволн. При частотах, больших 108Гц, ориентация ди-
польных моментов молекул воды не успевает следовать за
изменением напряженности электрического поля, и поэтому вклад
^дипольно-релаксационной поляризации начинает уменьшаться,
что отражается и на уменьшении диэлектрической
проницаемости. В оптическом диапазоне частот (более 10п Гц) дипольно-
релаксационной поляризацией можно пренебречь.
Зависимость диэлектрической проницаемости морской воды
от ее температуры имеет более сложный характер, что
объясняется наличием двух факторов, вызывающих противоположные
последствия. При увеличении температуры воды уменьшается ее
молекулярная вязкость (рис. 7.4), что облегчает ориентацию ди-
польных моментов молекул воды и, соответственно, приводит к
росту е. В то же время усиливается тепловое движение молекул,
которое препятствует ориентации дипольных моментов и
уменьшает диэлектрическую проницаемость. Влияние этих двух
факторов можно проследить на рис. 8.2.
Что касается влияния солености на диэлектрическую
проницаемость, то выяснено, что с ростом концентрации раствора
электролита е сначала медленно возрастает, а затем, достигнув
некоторого уровня, начинает быстро падать, причем величина
205
максимума и скорость понижения диэлектрической
проницаемости тем больше, чем тяжелее ионы электролита. Небольшое
увеличение е в морской воде (слабоконцентрированном растворе)
связано с добавлением ионной поляризации. Однако при
больших концентрациях солей ионы электролита начинают
концентрировать вокруг себя дипольные молекулы воды, что приводит к
резкому уменьшению диэлектрической проницаемости.
8.3. Магнитная восприимчивость
По своим магнитным свойствам, которые определяются
свойствами электронов и атомов, морская вода относится к диамагне-
тикам37. Диамагнетиками называются вещества, магнитные
моменты атомов (молекул) которых в отсутствие внешнего
магнитного поля равны нулю, так как магнитные моменты всех
электронов атома (молекулы) взаимно скомпенсированы. При внесении
морской воды во внешнее магнитное поле Земли ее молекулы
приобретают наведенные магнитные моменты, т. е. формируется
собственное магнитное поле, которое накладывается на внешнее
поле. Отношение суммы наведенных магнитных моментов
макроскопически малого объема вещества к величине этого объема
называется намагниченностью J и служит количественной
характеристикой намагниченного состояния вещества.
Вектор намагниченности J зависит от магнитных свойств
среды и пропорционален вектору магнитной индукции В38:
37 Кроме диамагнетиков, существуют парамагнетики и ферромагнетики.
Парамагнетиками называются вещества, атомы (молекулы) которых в
отсутствие внешнего магнитного поля имеют отличный от нуля
магнитный момент. Ферромагнетиками называются твердые вещества,
обладающие при не слишком высоких температурах самопроизвольной
намагниченностью, которая сильно изменяется под влиянием внешних
воздействий - магнитного поля, деформации, изменения температуры.
Ферромагнетики являются сильно магнитными средами (в отличие от
диамагнетиков и парамагнетиков) - внутренне магнитное поле в них
может в сотни и тысячи раз превосходить внешнее поле.
38 Вектор магнитной индукции В служит силовой характеристикой
магнитного поля и численно равен (в СИ) отношению силы, действующей
на заряженную частицу со стороны магнитного поля, к произведению
абсолютной величины заряда и скорости частицы, если направление
206
J = X' —, (8.4)
где Цо - магнитная постоянная, равная 4-я-10'7 Гн-м"1 (генри на
метр)39; х' - безразмерная величина, характеризующая магнитные
свойства диамагнетика. Для всех диамагнетиков, в том числе и
для морской воды, х' < 0, т. е. вектор магнитной индукции
собственного магнитного поля, создаваемого диамагнетиками при его
намагничивании во внешнем магнитном поле, направлен в
сторону, противоположную внешнему.
С х' связана другая безразмерная величина - относительная
магнитная восприимчивость хт, определяемая как (в СИ):
Х„=Г^- (8.5)
1-Х
У воды х'~Ю"5, поэтому практически хт = X' •
Магнитная восприимчивость, в свою очередь, зависит от
вектора намагничивания J, создаваемого внешними магнитными
полями, и вектора магнитной напряженности Н40:
Х„=£ (8.6)
Магнитная восприимчивость хт воды очень мала и слабо
зависит от ее температуры (табл. 8.2).
С магнитной восприимчивостью хт связана магнитная
проницаемость \л среды:
Ц = 1 + Х„- (8.7)
скорости частицы таково, что эта сила максимальна. Единица
магнитной индукции - ньютон на ампер-метр - носит наименование тесла
(Тл). Тл=кгс2А"1.
39 Генри - единица индуктивности. Он равен индуктивности цепи, с
которой при силе постоянного тока в ней один ампер сцепляется
магнитный поток один вебер, т. е. Гн=ВбА"1==м2кгс"2А'2.
40 Напряженность магнитного поля Н - количественная
характеристика магнитного поля, не зависящая от магнитных свойств среды и равная
магнитной индукции, деленной на абсолютную магнитную
проницаемость, т. е. Н = B/(|i0 • \х).
207
Для воды ц<1. Магнитная проницаемость морской воды, как
видно из формулы (8.7), мало отличается от единицы, поэтому в
практике величину ц при различных частотах, температурах и
концентрациях солей принимают постоянной и равной единице.
Таблица 8.2
Зависимость магнитной восприимчивости %т от температуры
по [37]
Среда
Вода (Н20)
Тяжелая
вода (D20)
Фазовое
состояние
Жидкое
Жидкое
Твердое
Жидкое
Твердое
Температура,
°К
293,0
273,0
273,0
276,8
276,8
ХИ0б
(безразм.)
-12,97
-12,93
-12,65
-12,76
-12,54
Задачи и вопросы для повторения
8.1. Какой параметр отражает способность морской воды изменять
свою электрическую индукцию под влиянием внешнего электрического
поля?
8.2. Что понимается под удельной электропроводностью и что она
характеризует?
8.3. Какие параметры морской воды влияют на ее удельную
электропроводность и как?
8.4. Зависит ли удельная электропроводность от изменения
относительного состава ионов морской воды?
8.4. Что такое электрическая проницаемость?
8.5. Какие виды поляризации можно выделить у морской воды?
8.6. К какой группе веществ относится морская вода по своим
магнитным свойствам: диамагнетикам, парамагнетикам или
ферромагнетикам?
8.7. Что такое магнитная восприимчивость?
208
СПИСОК ЛИТЕРАТУРЫ
1. Архипкин B.C. Алгоритмы и программы на Фортране для
обработки океанологической информации. - М.: Изд-во Моск.
ун-та, 1992.-83 с.
2. Архипкин B.C., Бережной В.Ю. Стерические колебания
уровня Черного моря// Океанология. 1995. Т. 35, № 6. С. 809-816.
3. Архипкин B.C., Добролюбов С.А. Основы термодинамики
морской воды. - М.: Диалог-МГУ, 1998. - 154 с.
4. Архипкин B.C., Сарафанов А.А. Расчет нейтральных
поверхностей в Мировом океане// Вестн. Моск. ун-та. Сер. 5,
География. 2004. № 1. С. 41-46.
5. Базаров М.П. Термодинамика. - М.: Высшая школа, 1991. -
376 с.
6. Бреховских Л.М., Гончаров В.В. Введение в механику
сплошных сред. - М.: Наука, 1982. - 335 с.
7. Валяшко М.Г. Эволюция химического состава воды океана. -
М.: Наука, 1971.
8. Виноградов А.П. Введение в геохимию океана. - М.: Наука,
1967.
9. Гилл А. Динамика атмосферы и океана: В 2 т. - М.: Мир,
1986. -Т. 1. ~397 с; Т. 2.-415 с.
10. Деев М.Г. Акустика океана. - М.: Изд-во Моск. ун-та, 1992. -
78 с.
11. Добролюбов С.А., Мамаев О.И. Об уточнении упрощенного
уравнения состояния морской воды// Вестн. Моск. ун-та.
Сер. 5, География. 1987. № 1. 7 с. Деп. в ВИНИТИ от
30.09.86, № 6894.
12. Добролюбов С.А. Об определении средних термохалинных
характеристик океана// Вестн. Моск. ун-та. Сер. 5, География.
1987. №3. С. 65-71.
13. Зацепина Г.Н. Свойства и структура воды. - М.: Изд-во Моск.
ун-та, 1974.- 167 с.
14. Зубов Н.Н. Морские воды и льды. - М.: Гидрометеоиздат,
1938.-453 с.
15. Зубов Н.Н. Динамическая океанология. - М.-Л.:
Гидрометеоиздат, 1947.-430 с.
209
16. Зубов Н.Н., Сабинин К.Д. Вычисление уплотнения при
смешении морских вод. - М.: Гидрометеоиздат, 1958. - 39 с.
17. Иванов А. Введение в океанографию. - М.: Мир, 1978. -
576 с.
18. Каменкович В.М., Монин А.С. Основные положения
термогидродинамики океана// Физика океана. Т. 1. Гидрофизика
океана/ Под ред. В.М. Каменковича, А.С. Монина. - М.: Нау-
* ка, 1978. С. 85-112.
19. Кильматов Т.Р. Методы неравновесной термодинамики в фи-
. зической океанологии. - Владивосток: ДВО АН СССР,
1987.-80 с.
20. Мамаев О.И. Океанографический анализ в системе v-S-T-p. -
М.: Изд-во Моск. ун-та, 1963. - 228 с.
21. Мамаев О.И. Морская турбулентность. - М.: Изд-во Моск.
ун-та, 1970.-204 с.
22. Мамаев О.И. Термохалинный анализ вод Мирового океана. -
Л.: Гидрометеоиздат, 1987. - 296 с.
23. Мамаев О.И. О сопоставлении уравнений состояния морской
воды (Кнудсена-Экмана и Международного 1980 года)//
Океанология. 1986. Т. 26. №. 3. С. 505-513.
24. Мамаев О.И., Архипкин B.C. Таблицы для вычисления
вертикальной устойчивости вод океана// Вестн. Моск. ун-та. Сер. 5,
География. 1987. № 4. 46 с. Деп. в ВИНИТИ от 25.03.87, №
2152.
25. Мамаев О.И., Архипкин B.C., Добролюбов С.А., Суслов А.А.
Таблицы скорости звука в морской воде// Вестн. Моск. ун-та.
Сер. 5, География. 1987. № 5. 27 с. Деп. в ВИНИТИ от
03.07.87, №5514.
26. Мамаев О.И., Архипкин B.C., Тужилкин B.C. Г,5-анализ вод
Черного моря// Океанология. 1994. Т. 34. № 2. С. 178-192.
27. Матвеев А.Н. Молекулярная физика. - М.: Высшая школа,
1987.- 360 с.
28. Монин А.С. О гидротермодинамике океана// Изв. АН СССР.
ФАО. 1973. Т. IX. № 10. С. 1063-1068.
29. О введении Шкалы практической солености, 1978, и нового
Международного уравнения состояния морской воды, 1980//
Океанология. 1982. Т. 22. № 2. С. 337-343.
210
30. Океанографические таблицы. - 4-е изд. - Л.: Гидрометеоиз-
дат, 1975.-478 с.
31. Попов Н.И., Федоров К.И., Орлов В.М. Морская вода:
Справочное руководство. - М.: Наука, 1979. - 327 с.
32. Праудмэн Дж. Динамическая океанография. - М.: Изд-во
иностр. лит-ры, 1957. - 418 с.
33. Сеидов Д.Г. Синергетика океанских процессов. - Л.: Гидро-
метеоиздат, 1989. - 288 с.
34. Скороход А.И., Цыцарин А.Г. Об уравнениях состояния
каспийских вод// Тр. ГОИН. 1995. Вып. 3. С. 63-72.
35. Стунжас П.А. Структура и свойства воды и водных
растворов// Химия океана. Т. 1. Химия вод океана/ Под ред. O.K.
ь Бордовского, В.Н. Иваненкова - М.: Наука, 1979. с. 11-42.
36. Фен Дж. Машины. Энергия. Энтропия/ Пер. с англ. - М.:
Мир, 1986.-336 с.
37. Физика океана/ Под ред. Ю.П. Доронина. - Л.: Гидрометеоиз-
дат, 1978.-294 с.
38. Хефс Й. Геохимия стабильных изотопов. - М.: Мир, 1983. -
198 с.
39. Хорн Р.А. Морская химия (структура воды и химия
гидросферы). - М.: Мир, 1972. - 400 с.
40. Цурикова А.П., Цуриков В.Л. О понятии «соленость»//
Океанология. 1971. Т. 11. № 2. С. 339-342.
41. Цуриков В.Л. Новые данные о температуре замерзания
морской воды разной солености// Океанология. 1976. Т. 14. № 5.
С. 798-801.
42. Цыбанева Т.Б. О вычислении термодинамических функций
морской воды// Изв. АН СССР. ФАО. 1971. Т. 7. № 7. С. 814-
817.
43. Цыцарин Г.В. Введение в гидрохимию. - М.: Изд-во Моск.
ун-та, 1988.-104 с.
44. Шлихтинг Г. Теория пограничного слоя. - М.: Наука, 1969. -
744 с.
45. Эдсолл Дж., Гатфренд X. Биотермодинамика. - М.: Мир,
1986.-296 с.
46. Эйзенберг Д., Кауцман В. Структура и свойства воды/ Пер. с
англ. - Л.: Гидрометеоиздат, 1975. - 280 с.
211
47. Яворский Б.М., Детлаф А.А. Справочник по физике. - М.:
Наука, 1990.-624 с.
48. Bryden H.L. New polynomials for thermal expansion, adiabatic
temperatures gradient and potential temperature of sea water.
Deep-Sea Res., 1973, v. 20, pp. 401-408.
49. Brewer P.G., Bradshaw A. The effect of the non-ideal
composition of seawater on salinity and density. J. Marine Res., 1975,
v. 33, №2, pp. 157-175.
50. Caldwell D.R. The maximum density of pure and saline waters.
Deep-Sea Res., 1978, v. 25, pp. 175-181.
51. Chen C.-T., Millero F.J. Speed of sound in seawater at high
pressures. J. Acoust. Soc. Am., 1977, v. 62, pp. 1129-1135.
52. Chen C.-T., Millero F.J. The equation of state of seawater
determined from sound speeds. J. Mar. Res., 1978, v. 36, №4, pp.
657-691.
53. Chen C.-T., Millero F.J., Li K. New version of Chen-Millero's
algorithms for sound speed calculation. J. Acoustic Soc. America,
1994, v. 95, № 5, pp. 2757-2759.
54. Connors D.M. On the enthalpy of seawater. Limnol. and Ocean-
ogr., 1970, v. 15, pp. 587-594.
55. Craig H. The thermodynamics of sea water. Proc. Natl. Acad. Sci.,
1960, v. 46, № 9,pp.l221-1225.
56. Del Grosso V.A. Speed of sound in seawater. J. Acoust. Soc. Am.,
1974, v. 56, pp. 1084-1091.
57. Doherty B.T., Kester D.R. Freezing point of seawater. J. Marine
Res., 1974, v. 32, № 2, pp. 285-300.
58. Eckart С Properties of water. II. The equation of state of water
and sea water at low temperatures and pressure. Amer. J. Sci.,
1958, v. 256, № 4, pp. 225-240.
59. Feistel R. Equilibrium thermodynamics of seawater revisited.
Progr. Oceanogr., 1993, v. 31, pp. 101-179.
60. Fofonoff N.P. Energy transformation in the sea. Fisheries
Research Board of Canada/ Manuscript Report Series (Oceano-
graphic and Limnological), 1961, № 109. - 82 p.
61. Fofonoff N.P. Physical properties of sea water. In: The-Sea. Ed.
M. N. Nill. Interscience, New York, 1962, pp. 3-30.
212
62. Fofonoff N.P. Computation of potential temperatures of sea water
for an arbitrary reference pressure. Deep-Sea Res., 1977, v. 24r
pp. 489^91.
63. Fofonoff N.P., Millard R.S. Algorithms for computation of
fundamental properties of sea water. UNESCO Technical Papers in
Marine Science, 1983, v. 44. - 53 p.
64. Fofonoff N.P. Physical properties of seawater: A new salinity
scale and equation of state for seawater. J. Geophys. Res., 1985,
v. 90, № C2, pp. 3332-3342.
65. Foldvik A., Kvinge T. Conditional instability of seawater at the
freezing point. Deep-Sea Res., 1974, v. 21, pp. 169-174.
66. Fujino K., Lewis E.L., Perkin R.G. The freezing point of seawater
at pressures up to 100 bars. J. Geoph. Res., 1974, v. 79, № 12, pp.
1792-1797.
67. Gerbhart В., MoUendorf J.C. A new density relation for pure and
saline water. Deep-Sea Res., 1977, v. 24, pp. 831-848.
68. Gregg M.C. Entropy generation in the ocean by small-scale
mixing. J. Phys. Oceanography, 1984, v. 14, pp. 688-711.
69. International Oceanographic Tables. Vol. 4. Properties of Sea
Water Derived from the Equation of State. UNESCO Technical
Papers in Marine Science, 1987, v. 40. - 195 p.
70. Krummel O. Handbuch der Ozeanographie. Bd. I, Stuttgart,
J. Engelhern, 1907, 526 p.
71. Lebedev V., Aizatulin Т., Khailov K. The living ocean. M.:
Progress Publ., 1989. -328 p.
72. Lewis E.L., Perkin R.G. Salinity: Its Definition and Calculation.
J. Geoph. Res., 1978, v. 83, № CI, pp. 466-478.
73. Lewis E.L., Perkin R.G. The Practical Salinity Scale 1978:
conversion of existing data. Deep-Sea Res., 1981, v. 28a, № 4, pp.
307-328.
74. Leyendekkers J.V. The chemical potentials of seawater
components. Marine Chemistry, 1972-73, v. 1, pp. 75-88.
75. McDougall T.J. Neutral Surfaces. J. Phys. Oceanography, 1987,
v. 17, pp. 1950-1964.
76. Millard R.C., Owens W.B., Fofonoff N.P. On the calculation of
the Brunt-Vaisala frequency. Deep-Sea Res., 1990, v. 37, № 1,
pp. 167-181.
213
77. Millero F.J., Hansen L.D., Hoff E.V. The entalphy of seawater
from 0 to 30° С and from 0 to 40 %> salinity. J. Marine Res., 1973,
v. 31, №31, pp. 21-39.
78. Millero F.J., Emmet R.T. The effect of dissolved air and natural
isotopic distributions on the density of water. J. Marine Res.,
1976, v. 34, №1, pp. 15-24.
79. Millero F.J., Kremling K. The densities of Baltic Sea waters.
Deep-Sea Res., 1976, v. 23, pp. 1129-1138.
80. Millero F.J., Leung W.N. The thermodynamics of seawater at one
atmosphere. Amer. J. Sci., 1976, v. 276, pp. 1025-1077.
81. Millero F.J. The thermodynamics of sea water. Part I. The PVT
properties. Ocean Science and Engineering, 1982, v. 7, № 4,
pp. 403-460.
82. Millero F.J. The thermodynamics of sea water. Part П. Thermo-
chemical properties. Ocean Science and Engineering, 1983, v. 8,
№ l,pp. 1-40.
83. Saunders P.M., Fofonoff N.P. Conversion of pressure to depth in
the ocean. Deep-Sea Res., 1976, v. 23, pp. 109-111.
84. Saunders P.M. Practical conversion of pressure to depth. J. Phys.
Oceanogr., 1981, v. 11, pp. 573-574.
85. Siedler G., Peters N. Properties of sea water. Physical properties
(general). In: LANDOLT-BORNSTEIN, Numerical Date and
Functional Relationships in Science and Technology, New Series,
Oceanography, 1986, v. 3a, pp. 233-264.
86. Sverdrup H., Johnson M., Fleming R. The oceans. Their physics,
chemistry and general biology. N. Y.: Prentice-Hall, 1942. -
1087 p.
87. The International System of Units (Si) in oceanography.
UNESCO Technical Papers in marine Science, 1985, v. 45. -
124 p.
88. Wilson W.D. The speed of sound in sea water as the function of
temperature, pressure and salinity. J. Acoust. Soc. Am., 1960,
v. 32, pp. 641-644.
214
Учебное пособие
Архипкин Виктор Семенович,
Добролюбов Сергей Анатольевич
ОКЕАНОЛОГИЯ
ФИЗИЧЕСКИЕ СВОЙСТВА МОРСКОЙ ВОДЫ
Напечатано с готового оригинал-макета
Издательство ООО "МАКС Пресс*'
Лицензия ИДЫ 00510от01 12 99 г.
Подписано к печати 24 11 2005 г.
Формат 60x90 1/16. Усл.печ л. 13,5. Тираж 500 экз. Заказ816.
Тел. 939-3890 Тел./факс 939-3891.
119992, ГСП-2, Москва, Ленинские горы, МГУ им. MB. Ломоносова,
2-й учебный корпус, 627 к.