/

























































































































Author: Сумм Б.Д.
Tags: химия коллоидная химия физико-химия дисперсных систем физико-химическая механика
ISBN: 978-5-7695-4041-7
Year: 2007




Text
Высшее профессиональное образование
Б. Д. Сумм
ОСНОВЫ коллоидной химии
2еиздайие
Естественные науки
ВЫСШЕЕ ПРОФЕССИОНАЛЬНОЕ ОБРАЗОВАНИЕ
Б. Д. СУММ
ОСНОВЫ коллоидной химии
Допущено
Учебно-методическим объединением по классическому университетскому образованию в качестве учебного пособил для студентов, обучающихся по специальности 020101.65 (011000) «Химия» и направлению 020100.62 (510500) «Химия»
2-е издание, стереотипное
ACADEMA
Москва Издательский центр «Академия» 2007
УДК 541.182(075.8)
ББК 24.6я73
С897
Ре цензе нты:
зав. кафедрой коллоидной химии Московской государственной академии тонкой химической технологии им. М. В. Ломоносова, д-р хим паук, профессор И. А Туторскищ
зав. кафедрой физической химии Московского государстве иного института стали и сплавов, д-р хим. наук, профессор М. В. Астахов
Сумм Б.Д.
С897 Основы коллоидной химии : учеб, пособие для студ. высш, учеб, заведений /БД Сумм — 2-е изд., стер. — М. : Издательский центр «Академия», 2007. — 240 с.
ISBN 978-5-7695-4041-7
Представлено современное состояние коллоидной химии, ее прикладное значение применительно к новым материалам и технологиям с учетом наиболее крупных научных достижений за последние 20—25 лет Подробно изложены основы коллоидной химии: поверхностные и капиллярные явления, термодинамика поверхностей раздела фаз, адсорбция поверхностно-активных веществ, мицеллярные системы, электроповсрхностные явления, устойчивость и молекулярно-кинетические свойства дисперсных систем, ультрадисперсныс системы («наносистемы»). Рассмотрены главные процессы коллоидной химии: коагуляция, коалесценция, седиментация, образование коллоидных структур.
Для студентов высших учебных заведений, обучающихся по химическим. биологическим, медицинским, геологическим специальностям.
УДК 541.182(075.8)
ББК 24.6я73
Оригинал-макет данного издания является собственностью Издательского центра «Академия», и его воспроизведение любым способом без согласия правообладателя запрещается
© Сумм Б.Д., 2006
© Образовательно-издательский центр «Академия», 2006 ISBN 978-5-7695-4041-7 © Оформление. Издательский центр «Академия», 2006
ПРЕДИСЛОВИЕ
Коллоидная химия возникла в середине XIX в. и, развиваясь очень быстро и успешно, сформировалась как самостоятельная научная дисциплина в начале XX в. В период с 1925 по 1932 г. четыре Нобелевские премии были присуждены за открытия в области коллоидной химии
Современная коллоидная химия — это наука «на стыке» химии, физики и биолоши. Особое междисциплинарное положение коллоидной химии подчеркивается тем, что в зарубежной литературе часто используют название «коллоидная наука» (Colloid Science). Коллоидная химия давно вошла в число обязательных общеобразовательных химических дисциплин для многих специальностей (химия, химическая технология, биология, медицина, геология, охрдна среды и др.). Преподавание коллоидной химии в высшей школе в нашей стране имеет богатые традиции и опирается на прекрасные учебники и пособия, подготовленные выдающимися учеными и преподавателями.
Создание данного учебного пособия мотивировано главным образом тем, что за последние 15 — 20 лет в коллоидной химии возникли и развились новые крупные направления. Среди них — коллоидная химия нанодисперсных систем (с дисперсными частицами размером несколько нанометров), коллоидная химия белковых систем, химические реакции в коллоидных микрореакторах (мицеллярных системах и микроэмульсиях), самоорганизованные коллоидные структуры. Кроме того, совершенно новые перспективы открылись в применении коллоидной химии в биологии, медицине, в области зашиты окружающей среды, создании принципиально новых материалов и технологий для микроэлектроники, фармакологии, полиграфии, биотехнологии, биоинформатики и многих других отраслей. Большие успехи достигнуты в развитии методов исследования коллоидных систем. Значительно обогатились физические и химические методы получения дисперсных частиц и дисперсных систем. Стало возможным получение дисперсных частиц нужного состава, размера и формы, тонкое регулирование их свойств.
Теоретические методы коллоидной химии достигли высокого уровня, особенно в области термодинамики мицеллярных систем, теории образования дисперсных систем, теории тонких пленок.
Успешно используются методы неравновесной термодинамики, фрактальной геометрии, теории самоорганизации и некоторых других современных научных направлений.
Новые идеи и методы коллоидной химии, наиболее принципиальные результаты должны быть представлены в учебной литературе. Поэтому главная цель данного учебного пособия — отразить современное состояние коллоидной химии, ее связи с достижениями других наук (физики, биологии, геологии, медицины, науки о материалах) и новые приложения коллоидной химии в высоких технологиях. в области охраны окружающей среды, создании материалов с уникальными свойствами и некоторых других направлениях.
Предлагаемое учебное пособие включает основополагающие разделы коллоидной химии, входящие в учебные программы классических и технологических университетов: поверхностные и капиллярные явления, адсорбция поверхностно-активных веществ, мицеллярные системы, электроповсрхностные явления, устойчивость дисперсных систем, образование коллоидных структур, молекулярно-кинетические свойства дисперсных систем. Впервые включены несколько направлений коллоидной химии, которые сложились в последние годы: методы получения дисперсных частиц. строение и свойства (особенно размерные эффекты) дисперсных систем, химическое модифицирование твердых поверхностей, новые поверхностно-активные вещества, влияние смесей поверхностно-активных веществ, в частности при их взаимодействии с полимерами и белками. Выбор именно этих направлении носит субъективный характер и, конечно, не дает всей картины современного состояния коллоидной химии. Тем не менее в рамках учебного пособия даны необходимые сведения об основных направлениях, методах и практических приложениях современной коллоидной химии
Учебное пособие создано на основе многолетнего опыта преподавательской и методической работы кафедры коллоидной химии химического факультета Московского государственною университета им. М В Ломоносова, педагогических принципов, сформулированных академиком Петром Александровичем Ребиндером, который руководил кафедрой 30 лет (1942—1972). Вместе с тем учитывалось и мнение студентов об изложении разных разделов в лекционном курсе коллоидной химии
Учебное пособие адресовано в первую очередь студентам химических и химико-технологических специальностей. Вместе с тем основные принципы коллоидной химии, ее методы и практические приложения необходимо знать студентам многих других специальностей В Московском государственном университете им. М. В. Ломоносова коллоидная химия преподается на биологическом и геологическом факультетах, факультете почвоведения,
факультете наук о материалах. Опьп чтения лекционных курсов но коллоидной химии на химическом факультете и других факультетах МГУ им. М. В. Ломоносова также был использован при написании данного учебного пособия. Особо следует отметить, что при определении новых направлении коллоидной химии, включенных в учебное пособие, очень полезным было сотрудничество с преподавателями и научными сотрудниками факультета наук о материалах, поскольку свойства дисперсных и особенно нанодис-псрсных частиц играют ключевую роль в создании функциональных материалов для микроэлектроники, а также многих других современных высоких технологий. Большую помошь оказало обсуждение различных вопросов с преподавателями кафедр коллоидной химии Санкт-Петербургского юсударственного университета, Московской государственной академии тонкой химической технологии им. М В. Ломоносова, Российского химико-технологического универси тета им. Д. И. Менделеева, Сан кт-П с гербу р-гского государственного технологического института, с преподавателями других кафедр химического (факультета МГУ им. М. В. Ломоносова. Очень полезным был анализ современных проблем преподавания коллоидной химии в высшей школе, который был проведен в 2001 г. на сессии Научного совета по коллоидной химии и физико-хим ическои механике Российской академии наук
Автор глубоко признателен доценту кафедры коллоидной химии МГУ им. М. В. Ломоносова Нине Ивановне Ивановой за огромную помощь в подготовке этого учебного пособия.
ГЛАВА 1
ВВЕДЕНИЕ В КОЛЛОИДНУЮ ХИМИЮ
Объекты и задачи коллоидной химии. Коллоидная химия изучает свойства разнообразных систем (неорганических, органических, полимерных, белковых), в которых хотя бы одно из веществ находится в виде частиц размером примерно от 1 нм (несколько молекулярных размеров) до 10 мкм. Частицы таких размеров называют дисперсными (лат. dispergo — рассеивать, распылять).
Дисперсные частицы могут находиться в любом агрегатном состоянии: твердом (микрокристаллы, волокна), жидком (капли в туманах и эмульсиях), газообразном (пузыри в пенах). Существуют и более сложные по строению дисперсные частицы. Их внутренняя структура существенно отличается от твердого тела или жидкости. Примером таких необычных структур, изучаемых в коллоидной химии, могуг служить мицеллы, находящиеся в воде или органической жидкости, — наноразмерные агрегаты из нескольких десятков молекул органических (поверхностно-активных) веществ. Мицеллы играют важную роль во многих биологических процессах (ферментативный катализ, перенос жиров в организмах и др.).
Дисперсные частицы по размерам занимают промежуточное положение между молекулами (атомами, ионами) и макроскопическими объектами (фазами). Частицы таких размеров широко распространены в природе и участвуют во многих технологических процессах. Приведем несколько примеров дисперсных частиц:
Дисперсные частицы Размер частиц, нм
Коллоидные металлы (золото, платина, серебро)..................................... 3 — 50
Дым.......................................... 30-40
Вирусы....................................... 10 — 20
Капли в микроэмульсиях....................... 5 — 20
Эритроциты крови ............................(5— 10) • 10
Взвеси в воде................................ 10—100
Выделение в особую группу дисперсных частиц вызвано несколькими обстоятельствами. Прежде всего многие физические и химические свойства дисперсных частиц значительно отличаются от аналогичных свойств крупных (макроскопических) объектов для одного и того же вещества К числу таких свойств относятся
прочность, теплоемкость, температура плавления, маипппыс и электрические характеристики, реакционная способность. Эти различия называю! размерными (или масштабными) эффектами. Они выражены тем сильнее, чем меньше размер дисперсных частиц, и поэтому особенно характерны для частиц нанометровых размеров (наночастиц). Особые свойства наночастиц (в том числе и наличие у них квантовых свойств) открывают принципиально новые практические приложения химии, физики, биологии Поэтому изучение дисперсных частиц (методов получения, структуры, физических и химических свойств) относится к наиболее актуальным и перспективным задачам ряда научных дисциплин. Размах и темпы исследований по развитию нанотехнологии весьма наглядно иллюстрируются объемом их финансирования. В США только бюджетные ассигнования на эти цели составили* 270 млн долл, в 2000 г., 422 млн долл — в 2001 г. и 604 млн долл, в 2002 г В коллоидной химии всестороннее изучение дисперсных частиц всегда было одним из фундаментальных направлении.
Другое фундаментальное направление коллоидной химии — исследование дисперсных систем. Поясним это важное понятие.
Дисперсные системы обычно состоят из двух фаз: дисперсной фазы, ее образуют дисперсные частицы, и дисперсионной среды — сплошной (непрерывной) фазы, в которой распределены дисперсные частицы. Дисперсионная среда, как и дисперсная фаза, может находиться в любом агрегатном состоянии: твердом, жидком или газообразном.
Таким образом, дисперсные системы — это микрогетерогеппыс системы, в которых одна из фаз представлена дисперсными частицами, а другая — дисперсионной средой. Термин «микрогсте-рогенность» подчеркивает малые размеры дисперсных частиц. Положение о микрогетеро те иной природе коллоидных растворов и других дисперсных систем имеет фундаментальное значение За его открытие и экспериментальное обоснование австрийский ученый Р. Зигмонди стал лауреатом Нобелевской премии по химии 1925 г.
Дисперсные системы чрезвычайно разнообразны как по химическому составу частиц и среды, гак и по их агрегатному состоянию (см. подразд. 2.2).
На поведение и свойства дисперсных систем оказывают влияние в основном две группы факторов:
1) свойства (химический состав, строение, размер) дисперсных частиц;
2) взаимодействие дисперсных частиц с окружающей их дисперсионной средой.
* Нанотехнология в ближайшем десятилетии. Прогноз направлений исследований; пер с англ. — М.: Мир, 2002
Основополагающее значение имеют физико-химические явления, происходящие на поверхности контакта дисперсных частиц с дисперсионном средой. В коллоидной химии важнейшую роль играют поверхностные явления — смачивание, адгезия, адсорбция, электрические эффекты. Принцип ключевой роли поверхностных явлении в коллоидной химии был выдвинут, обоснован и развит в трудах академика П.А. Ребиндсра.
Фундаментальное значение поверхностных явлений обусловлено особенностью дисперсных систем. Из-за малого размера дисперсных частиц их общая поверхность очень велика. Количественной характеристикой здесь служит удельная поверхность (QJ — отношение общей плошали (Qz;) поверхности всех дисперсных частиц к их суммарной массе (///):
Qj — Qn/m.
(1.1)
Для монодисперсных частиц (т.е. при одинаковом размере (d) всех частиц дисперсной фазы), которые имеют форму шара или куба, удельная поверхность обратно пропорциональна размеру частиц:
Ц/ = 6/(р(/г7),
(1.2)
где pd — плотность вещества дисперсной фазы.
Отсюда следует, что для частиц размером 1 мкм удельная поверхность составляет ~ 101 м2/г, а для частиц нанометрового диапазона 10 м /г.
Таким образом, в дисперсных системах удельная поверхность дисперсной фазы очень велика. Это определяет значительный вклад поверхностных явлений в свойства различных дисперсных систем. Соответственно изучение поверхностных явлений и развитие методов их регулирования традиционно составляет еще одно фундаментальное направление коллоидной химии. Исследование поверхностных явлений, как и дисперсных частиц, представляет комплексную задачу с привлечением ряда научных направлений (химия и физика поверхности, капиллярные явления, адсорбция, электрохимия и др.).
Одно из наиболее важных следствии большой поверхности дисперсной фазы заключается в том, что по этой причине большинство дисперсных систем сильно неравновесны, т.е. они сильно отклоняются от состояния термодинамического равновесия данной двухфазной системы. Это положение имеет фундаментальный характер и его необходимо обосновать и разъяснить.
Термодинамика гетерогенных систем (Дж. Гиббс) устанавливает, что поверхность раздела обладает избыточной свободной энергией по сравнению с энергиями внутренних объемов граничащих фаз Этот избыток называю! удельной свободной поверхностной лнер-
гией (о, Дж/м2). Соответственно каждая единица массы дисперсном фазы обладает избыточном свободном энергией (энергией Гельмгольца) ДГизб = Этот избыток определяет термодинамическую неус тойчивость дисперсных систем. Он стимулирует самопроизвольное протекание таких процессов, коюрые ведут к уменьшению избытка энергии. Наиболее общими являются процессы уменьшения удельной поверхности, что в соответствии с уравнением (1.2) означает укрупнение дисперсных частиц — увеличение их размера.
Конкретные механизмы укрупнения дисперсных частиц могут быть разными, например прямое столкновение капель в эмульсиях или аэрозолях с их последующим слиянием (гак называемая коалесценция). Укрупнение частиц постепенно приводит к тому, что они становятся слишком крупными и утрачивают те физические и химические особенности, которые присуши именно дисперсным частицам. Поэтому проблема устойчивости дисперсных систем, т.е. сохранения размеров частиц в течение длительного времени, является центральной в коллоидной химии. От ее решения зависят «жизнь и смерть» дисперсных систем. Поэтому разработка методов зашиты от самопроизвольного укрупнения (агрегации частиц, слияния капель и пузырьков) дисперсных частиц входит в число основных направлении и задач коллоидной химии.
Интересно в связи с этим отметить, что М. Фарадей, впервые получивший в 1857 г. высокодисперсные частицы золота, взвешенные в воде (так называемый коллоидный раствор золота), одновременно разработал два метода их защиты. Они оказались настолько эффективны, что приготовленные М.Фарадеем коллоидные растворы сохранились до сих пор и демонстрируются в одном из английских музеев.
Неравновесное состояние дисперсных систем помимо их термодинамической неустойчивости имеет еще одно важное следствие. С позиций неравновесной термодинамики эволюция систем, далеких от равновесия, может протекать по биологическому типу — с образованием упорядоченных диссипативных структур (лауреат Нобелевской премии по химии 1977 г. И. Р. Пригожин). Такая самоорганизация может происходить внутри дисперсионной среды. Примером может служить упоминавшееся выше образование мицелл в растворах поверхностно-активных веществ (ПАВ). Процессы самоорганизации могут протекать и на поверхностях раздела фаз. Самоорганизация органических веществ (особенно белков) на поверхности воды с образованием определенных упорядоченных структур имеет исключительно большое значение для формирования биологических обьектов. Это направление, изучающее эволюцию дисперсных систем на основе положений неравновесной термодинамики, является пограничным между коллоидной химией и биологией.
Изучение дисперсных структур (процессов их образования, физико-химических и механических свойств, методов регулирования этих свойств) также представляет фундаментальное направление коллоидной химии. Основополагающую роль здесь играют взаимодействия дисперсных частиц при их непосредственном контакте друг с другом или через тонкую прослойку дисперсионной среды. Такие структуры по своим свойствам существенно отличаются от обычных твердых материалов (металлов, полимеров, стекол и т.п.). Один из примеров — после разрушения определенного типа дисперсных структур (коагуляционных структур) происходит постепенное частичное (или полное) восстановление исходной структуры и се механической прочности. Этот эффект называют тиксотропией, он играет важную роль в залечивании студнеобразных тканей и органов в организмах.
Методы коллоидной химии. Охарактеризуем методы исследовании, применяемые в коллоидной химии. Пограничное положение коллоидной химии между химией, физикой и биологией во многом определяет и се методологию как в области теории, так и при проведении экспериментов.
Химические теории обычно преобладают в области высокодисперсных систем, поверхностных слоев жидкостей и твердых тел и нанесенных на них адсорбционных слоев различных веществ (в первую очередь — поверхностно-активных веществ). Например, для расчета электронной структуры наночастиц (кластеров) и ее влияния на их каталитические свойства успешно применяют методы квантовой механики. Другой пример — константу адсорбционного равновесия на поверхности жидкости или твердого тела рассчитывают на основе фундаментального закона химических реакции — закона действующих масс. Кинетика агрегации дисперсных частиц при их столкновениях вследствие броуновского движения в дисперсионной среде (кинетика коагуляции) построена на аналогии этого процесса с бимолекулярной реакцией. Теоретические представления электрохимии о строении двойного электрического слоя лежат в основе теории электроповерхност-ных и электрокинетических явлений (электрофорез, электроосмос). Они использованы также в теории устойчивости дисперсных систем.
Химические реакции лежат в основе многих методов получения высокодисперсных части, в том числе наночастиц. Особенно часто используют химическое восстановление металлов. Контроль температуры, pH растворов и их концентрации позволяет достаточно гибко регулировать размеры получаемых частиц. Наряду с химическим восстановлением в ходе обменных реакций в растворах для синтеза дисперсных частиц успешно применяют фотохимическое и радиационно-химическое восстановление металлов.
Химические методы синтеза твердых дисперсных частиц были успешно развиты еще на начальном этапе становления коллоидной химии — во второй половине XIX в. В это время были получены дисперсные частицы самых разнообразных веществ. Важнейший обобщающий вывод этих исследований — принцип универсальности дисперсного состояния, т е. в виде дисперсных частиц может существовать (при определенных условиях) любое вещество. Этот принцип установил русский химик П. П.Веймарн в 1905- 1910 гг.
Важный химический аспект коллоидной химии связан с поверхностно-активными веществами, которые играют роль наиболее эффективных и гибких регуляторов поверхностных свойств дисперсных систем. В создание этого, исключительно плодотворного направления коллоидной химии основополагающий вклад внесли работы академика 11. А. Ребиндера и лауреата Нобелевской премии но химии И.Ленгмюра
Химический состав поверхностно-активных веществ и строение их молекул определяют такие важные для коллоидной химии процессы, как формирование адсорбционных слоев на поверхности дисперсных частиц, образование мицелл в растворах, стабилизация пен, эмульсий и других дисперсных систем. Современная коллоидная химия обладает широким ассортиментом поверхностно-активных веществ, что позволяет достаточно точно, адресно проводить их подбор применительно к конкретным задачам и условиям. За последние 15 —20 лет эти возможности еще более расширились благодаря применению смесей поверхностно-активных веществ. Кроме того, сейчас синтезированы принципиально новые вещества; широко применяют полимерные и белковые поверхностно-активные вещества.
Необходимо отмстить еще один химический аспект коллоидной химии, связанный, однако, не с изучением дисперсных систем, а с их применением. Некоторые типы дисперсных систем (мицеллярные растворы, микроэмульсии) оказались чрезвычайно перспективны, как микрореакторы. Приведем пример. Важным материалом для микроэлектроники является порошок из нанодиспсрсных частиц полупроводника CdS. Существенно, чтобы эти частицы имели одинаковый размер. Для решения этой задачи используют две микроэмульсии — одна из них содержи! микрокапли нужного размера водного раствора Cd(NO3)2 в среде органической жидкости, другая — микрокапли водного раствора Na2S в той же органической жидкости. При сливании и перемешивании этих микроэмульсий происходит попарное слияние (коалесценция) капель и в них происходит обменная реакция с образованием CdS. Размер частиц определяется стерическим условием — диаметром слившейся капли (размером микрорс-актора).
Физические теории и методы также очень широко и плодотворно используются в коллоидной химии. Благодаря им современная коллоидная химия представляет количественную науку. Назовем несколько наиболее крупных физических концепций коллоидной химии: броуновское движение и диффузия дисперсных частиц (А. Эйнштейн, М.Смолуховский): термодинамика поверхностных явлений (Дж. Гиббс, Я. Ван-дер-Ваальс, А. И. Русанов), теория капиллярных явлении (Т. Юнг, П.Лаплас); гидродинамика дисперсных систем (А. Эйнштейн); седиментационно-диффузионное равновесие дисперсных систем (Ж. Перрен — Нобелевская премия по физике 1926 г.); образование дисперсных частиц при фазовых переходах первого рода (Дж. Гиббс, М.Фоль-мер); теория устойчивости дисперсных систем (Б. В. Дерягин, Л. Д. Ландау, Э.Фервей, Т.Овербек — теория ДЛФО).
Физические методы занимают доминирующее место в изучении различных свойств дисперсных систем. Остановимся на одной из наиболее важных задач — измерение размеров дисперсных частиц (так называемый дисперсионный анализ). Размер сравнительно крупных частиц и капель наиболее просто определяют по скорости их осаждения (седиментации) или всплывания в жидкой дисперсионной среде, т.е. расчет размера проводят на основе законов Архимеда и Ньютона (для силы вязкого трения в жидкости). Использование центробежной силы вместо силы тяжести позволяет применять этот принцип и для высокодисперсных частиц, в том числе и паноразмерных. За создание ультрацентрифуги для дисперсионного анализа коллоидных растворов шведский ученый Т.Сведбсрг получил Нобелевскую премию по химии 1926 г.
Для частиц размером примерно несколько десятков нанометров используют оптические свойства дисперсных систем, прежде всего способность коллоидных растворов рассеивать свет (в отличие от истинных растворов — молекулярных и ионных).
Для прямого измерения размера наночастиц применяют электронную микроскопию.
Структуру дисперсных частиц и тонких пленок изучают соответствующими физическими методами: рентгеноструктурный анализ, дифракция электронов, эллипсометрия. Внутреннюю динамику нежестких дисперсных частиц (мицелл) исследуют с использованием резонансных методов.
Большая группа методов коллоидной химии связана с измерением рсоло! ических свойств дисперсных структур и частиц, а также вязкости эмульсий, суспензий, паст и других дисперсных систс?»« с жидкой дисперсионной средой. Современные приборы для этих целей весьма разнообразны, они позволяют определять достаточно точно эти механические и гидродинамические характеристики.
Наряду с химическими и физическими методами в последнее время стали применять, хотя и в меньшей мере, некоторые био
физические и биохимические методы, в особенности при изучении различных явлений в белковых и полимерных коллоидных системах (гелях, адсорбционных слоях, пленках Ленгмюра— Блоджетт).
Современные направления коллоидной химии. Назовем несколько новых направлений коллоидной химии, которые сформировались за последние 15 — 20 лет.
1. Самоорганизация — самопроизвольное образование высокоорганизованных диссипативных структур.
2. Строение и свойства нанодисперсных частиц (размерные эффекты).
3. Дисперсные системы с ультранизким поверхностным натяжением (вплоть до 10 3 мДж/м2) на границе дисперсной фазы и диснерсион нон сре. iы (м икроэмул ьси и).
4. Коллоидные системы в условиях частичной и полной невесомости.
5. Применение методов компьютерного моделирования коллоидно-химических процессов.
Ь. Синтез и применение новых поверхностно-активных веществ, в том числе и высокомолекулярных и белковых.
7. Применение методов коллоидной химии в медицине (например. создание заменителей крови, проблема образования тромбов), в области охраны среды и биотехнологии.
Становление и развитие коллоидной химии. Рождение коллоидной химии как самостоятельной области знания относят к 60-м тг. XIX в. Начало коллоидной химии положено работами английского ученого М Фарадея. В 1857 г. М. Фарадей синтезировал коллоидные растворы (золи) золота — взвесь дисперсных частиц золота в воде размером от нескольких единиц до десятков нанометров. М.Фарадей разработал эффективные методы, препятствующие слипанию (агрегации) дисперсных частиц, и полученные им коллоидные растворы золота сохранились до нашего времени.
Другое важное открытие, ставшее стартовой точкой коллоидной химии, сделал в 1861 г. английский химик Т. 1рэм, который обнаружил, что некоторые вещества (например, оксид алюминия, кремниевая, оловянная и титановая кислоты, желатина, агар-агар) при выпаривании водного раствора не кристаллизуются, а образуют студенистые осадки (гели). Кроме тою, их диффузия в растворе происходит крайне медленно по сравнению с электролитами — солями, кислотами и основаниями. Некоторые из этих веществ обладают клеющим действием. Группу некристаллизу-ющихся и медленно диффундирующих веществ Т. Грэм назвал «коллоиды» (фсч. kolla — клеи). Отсюда позднее возникло и название науки, изучающей такие вещества, — коллоидная химия.
К началу XX в. было открыто много веществ с типичными коллоидными свойствами, разработаны методы получения, очистки
и стабилизации разнообразных коллоидных систем (неорганических, органических и белковых веществ), созданы методы их изучения, в том числе и методы измерения размера дисперсных частиц.
На основе этих результатов в 1906— 1910 гг. профессор Горного института в Санкт-Петербурге П П.Веймарн выдвинул фундаментальный принцип универсальности коллоидного (дисперсного) состояния вещества: «Коллоидное состояние не является обособленным, обусловленным какими-либо особенностями состава вещества. При определенных условиях каждое вещество может быть в коллоидном состоянии».
Прямым следствием принципа универсальности стало введение понятия о дисперсном состоянии вещества и в связи с этим осознание важнейшей роли поверхносгных явлений. Именно с этого времени коллоидная химия становится самостоятельной дисциплиной со своими объектами, методами и практическими приложениями.
Первая треть XX в. — период исключительно плодотворного и быстрого развития коллоидной химии. Пограничный характер коллоидных систем — на границе химии и физики, между миром молекул и макроскопическими телами, новизна и оригинальноеть этой области науки привлекают внимание крупнейших ученых того времени. А. Эйнштейн разработал теорию броуновского движения и диффузии дисперсных частиц, а также теорию течения дисперсных систем. М. Смолуховскии создал кинетическую теорию процесса слипания (коагуляции) дисперсных частиц в коллоидных растворах.
В этот период за работы в области коллоидной химии были присуждены четыре Нобелевские премии. Лауреатами стали:
австрийский химик Р.Зигмонди (1925 г.) — «за установление гетерогенной природы коллоидных растворов и за разработанные в этой связи методы, имеющие фундаментальное значение в современной коллоидной химии»;
французский физик Ж. Перрен (1926 г.) — «за работу по дискретной природе материи и в особенности за открытие седиментационно-диффузионного равновесия»;
шведский ученый Т.Сведберг (1926 г.) — «за работы в области дисперсных систем» (прежде всего за создание ультрацентрифуги для определения размеров высокодиспсрсных частиц и макромолекул);
американский физикохимик И.Ленгмюр (1932 г.) — «за открытия и исследования в области химии поверхностных явлений».
К этому списку следует добавить Нобелевскую премию шведского биохимика А Тиселиуса (1948 г.) — «за исследование электрофореза и адсорбционного анализа, особенно за открытие гетерогенной природы сывороточных белков».
Наряду с перечисленными работами, отмеченными Нобелевскими премиями, в коллоидной химии в первой трети XX в. было сделано еще несколько очень крупных открытий. В 1913 г. английский химик Дж. Мак-Бен обнаружил замечательное явление: молекулы и ионы поверхностно-активных веществ в водных растворах и органических растворителях при определенных условиях образуют крупные агрегаты (из нескольких десятков молекул) с упорядоченной самоорганизованной структурой. Эти агрегаты называют мицеллами (см. гл. 8). Они обладают целым комплексом уникальных физико-химических свойств, благодаря чему мицеллы играют чрезвычайно важную роль в коллоидной химии, химии полимеров, во многих биологических процессах, в медицине.
Другое крупное открытие этого периода сделал в 1928 г. советский ученый П. А. Рсбиндер. Он установил, что адсорбция поверхностно-активных веществ приводит к существенному понижению прочности кристаллов и других твердых тел. Это открытие (эффект Ребиндера) сыграло в дальнейшем очень важную роль в создании новых технологий обработки и получения различных материалов, в бурении горных пород и ряде других процессов. Вместе с тем это открытие стало впоследствии основой нового научного направления — физико-химической механики.
Еще одно (фундаментальное достижение этого периода — в 1935 г. Б. В. Дерягин открыл особое свойство тонких жидких пленок, которое он назвал расклинивающим давлением (см. гл. 4). Расклинивающее давление оказывает сильное влияние на свойства многих дисперсных систем (пен, эмульсий, золей) и в первую очередь — на их устойчивость.
В 30-х гг. XX в. коллоидная химия сформировалась в крупную научную дисциплину. Наряду с перечисленными выше фундаментальными открытиями и теориями коллоидная химия достигла больших успехов в прикладных областях. Это обусловлено двумя факторами. Во-первых, дисперсные системы чрезвычайно широко используются в многочисленных технологических процессах: в металлургии, при добыче нефти, в строительной промышленности и др. Во-вторых, коллоидная химия представляет научную основу для регулирования разнообразных свойств дисперсных систем и происходящих в них процессов. Ключевую роль в таком регулировании играют поверхностно-активные вещества. Принцип регулирующей роли поверхностно-активных веществ в коллоидно-химических процессах был убедительно обоснован в работах П.А. Ребиндера и его учеников. Ими были разработаны коллоидно-химические основы многих важных технологий: флотационного обогащения руд, бурения горных пород, приготовления красок, получения высококачественных строительных материалов (цементов и бетонов) и др.
Следующий период развития коллоидной химии относился ко второй половине XX в. Он характеризуется глубоким изучением различных дисперсных систем и формированием нескольких новых научных направлений, пограничных между коллоидной химией и некоторыми другими дисциплинами. Преимущественное внимание имели следующие направления:
• термодинамика поверхностных явлении и дисперсных систем;
• поверхностно-активные вещества: растворы, адсорбция; смеси поверхностно-активных веществ;
• электроповерхпостные явления;
• физико-химическая механика;
• свойства тонких пленок;
• коллоидная химия полимеров и белков.
В этот период получила развитие прикладная коллоидная химия (работы П.А. Ребиндсра, И. В. Петрянова-Соколова, Ф.Д. Овчаренко, М.П Воларовича, В Н. Еременко и др.). Полученные результаты эффективно используются в методах и технологиях флотации, полиграфии, нефтедобычи, в производстве строительных и конструкционных материалов, во многих других традиционных и новых технологиях.
Таким образом, для этого периода характерно освоение и развитие идей, которые сформировались в коллоидной химии еще в первой половине XX в. Значительное место по-прежнему занимают проблемы, близкие к разным разделам физики, механики, гидродинамики. Химические направления связаны преимущественно с поверхностно-активными веществами (синтез, очистка, адсорбция, мицеллообразование, применение в различных промышленных процессах).
Тем разительнее перемены, которые произошли в современной коллоидной химии и в тенденциях ее развития в последнее время. Самое важное достижение последних 1Э —20 лет заключается в решительном повороте в сторону фундаментальных проблем химического характера. Центральное место начинают занимать химические реакции и явления в различных коллоидных системах: микроэмульсиях и мицеллах, тонких жидких пленках и адсорбционных слоях, пенах и гелях. Новые идеи в этой области основаны на том, что высокодиспсрсные коллоидные частицы (капли микроэмульсии, мицеллы ПАВ, тонкие пленки) могут выполнять функции микрореакгоров. По ряду причин проведение процессов в коллоидных микрореакторах имеет существенные преимущества по сравнению с протеканием тех же реакций в .макрофазах.
Такое использование коллоидных систем известно давно Один из наиболее ярких примеров — эмульсионная полимеризация. Принципиальная особенность исследовании последнего периода
заключается в том, что «центр тяжести» смещается в сторону у ль-традиспсрсных систем, т.с. наносистем.
Перекресток «нанохимия — коллоидная химия» оказывается весьма плодотворным и перспективным. В качестве иллюстрации ограничимся только одним типом коллоидных микрорсакторов— микроэмульсиями. Эти системы были открыты еще в 1943 г., но интенсивное изучение началось только в конце 1970-х гг. в связи с применением их для повышения нефтеотдачи
Микроэмульсии используют для проведения биохимических, электрохимических и каталитических реакции, процессов полимеризации. Поскольку содержание дисперсной фазы в микроэмульсиях может быть весьма большим, они позволяют проводить реакции органических веществ в водной среде. Среди технических приложении отметим, наряду с нефтедобычей, жидкостную экстракцию. очистку почв oi нефтяных загрязнений, моющее действие, импрсгнированис тканей, а также использование микро-эмульсий в фармакологии и парфюмерии. Другой хорошо известный пример применения коллоидных систем как микрорсакторов — мицеллярный катализ в водных и неводных средах.
Выше говорилось о существенно большей роли химических аспектов в развитии коллоидной химии в конце XX в. Среди конкретных процессов и явлений, которые весьма успешно изучались, можно назвать: строение дисперсных частиц (особенно уль-традисперсных) и поверхностных слоев жидкостей и твердых тел, получение адсорбционных слоев определенного состава и структуры. включая модифицирование поверхностей разной природы; конформационные изменения макромолекул на межфазных поверхностях; совместное действие двух и нескольких поверхностно-активных веществ (синергизм и антагонизм); биологические и меди 1 in некие аспекты.
Существенный прогресс в теории лих процессов и явлении был достигнут благодаря сочетанию классических методов коллоидной химии со сравнительно новыми подходами: методами термодинамики необратимых процессов, синергетики, фрактальной геометрии и др. Весьма эффективно стали внедрять методы компьютерного моделирования. В экспериментальных исследованиях стали широко применять методы высокого разрешения, спектроскопию ядерно!о магнитного резонанса и электронною парамагнитного резонанса, атомную силовую микроскопию, прецизионные методы измерения поверхностного натяжения (до 10_> мДж/м2) и сил контактного взаимодействия между дисперсными частицами, фотон-корреляционную спектроскопию (измерение размеров коллоидных частиц), малоугловое отражение нейтронов.
Таким образом, наиболее крупные достижения коллоидной химии за последние 10 —20 лет заключаю 1ся в существенном сдвиге исследовании в сторону химических реакций и явлении в колло
идных микрореакторах и получении прямой информации о строении мицелл, микрокапель, адсорбционных и поверхностных слоев, строении и свойствах наночастиц.
Имеются веские основания полагать, что в ближайшие 10— 20 лет положение коллоидной химии среди других научных дисциплин может существенно измениться. Исходно коллоидная химия занимала промежуточное положение между химией и физикой. Такая ситуация обусловлена природой коллоидного состояния вещества — между молекулярным и фазовым состоянием. Вместе с тем коллоидные системы обладают ярко выраженной тенденцией к образованию самоорганизованных структур. Наиболее известные примеры — периодические коллоиды (кольца Ли-зеганга, тактоиды), адсорбционные слои поверхностно-активных веществ, поверхностные слои жидкости (см. гл. 10). Эта принципиальная особенность коллоидной химии сближает ее с биологией, особенно с биофизикой. Поэтому вполне реальны крупные успехи коллоидной химии на стыке с биологическими дисциплинами.
Современная коллоидная химия развивается очень быстро. В 1997 г. в США был опубликован учебник «Химия поверхности. Коллоидная химия», написанный группой ведущих американских ученых. Уже через несколько лет новые результаты стимулировали подготовку существенно расширенного второго издания этого учебника гораздо большего объема*. За последние 10—15 лет появилось также много монографий по различным направлениям коллоидной химии; некоторые из них приведены в списке литературы.
Контрольные вопросы
1 Укажите основные причины выделения дисперсных частиц в особую группу.
2. Приведите определение понятия «дисперсная система*. Из каких фаз состояг дисперсные системы?
3. Назовите две группы факторов, определяющих основные свойства дисперсных систем
4. Назовите важнейшие дня коллоидной химии поверхностные явления.
5. Выведите уравнение (11), которое связывает удельную поверхность монодисперсной суспензии с размером дисперсной частицы. Рассчитайте удельную поверхность частиц золота для двух случаев: 1) d = 1 нм; 2) d = 100 нм. Плотность золота 19.32 г/см3
6 Объясните термодинамическую неустойчивость дисперсных систем.
7. Перечислите основные направления развития современной коллоидной химии.
* Handbook of Surface and Colloid Chemistry / Ed. K. S. Birdi. — 2nd cd — N Y.. CRC Press, 2003. - 765 p
ГЛАВА 2
ДИСПЕРСНЫЕ СИСТЕМЫ
Дисперсные системы — это гетерогенные системы, в которых одна из фаз представлена частицами небольшою размера — примерно от 1 нм до 10 мкм. Дисперсные системы распространены в природе и широко используются в промышленности. Приведем несколько примеров дисперсных систем:
эмульсии — капли одной жидкости распределены в среде другой жидкости;
аэрозоли — твердые частицы или капли взвешены в воздухе (дым и туман);
коллоидные растворы (золи) — твердые частицы распределены в жидкости.
Частицы, размер которых отвечает интервалу 1 нм—10 мкм, назы ва ют дисперсными.
По агрегатному состоянию дисперсные частицы могут быть твердыми, жидкими или газообразными; вместе с тем во многих случаях они имеют более сложное строение.
Совокупность (ансамбль) дисперсных частиц называют дисперсной фазой. Дисперсные частицы находятся в сплошной фазе — дисперсионной среде. Дисперсионные среды могут быть газообразными, жидкими и твердыми.
2.1. Количественные характеристики дисперсных систем
Для количественного описания дисперсных систем используют ряд характеристик.
I. Средний (г/ ), минимальный (t/mjn) и максимальный (б/тач) размер дисперсных частиц.
2. Концентрация частиц (v, м" ), равная числу дисперсных частиц (nd) в единице объема дисперсионной среды (И):
V = П(]/ V.
(2.1)
3. Удельная поверхность дисперсной фазы (Qf/, м2/кг) — отношение суммарной плошали поверхности (Г2„) всех дисперсных част иц к их суммарной массе (т):
Lid=an/m. (2.2)
Для дисперсных частиц сферической или кубической формы, имеющих одинаковый размер (<7), удельная поверхность дисперсной фазы равна удельной поверхности одной частицы:
Q, = 6/W), (2.3)
где pj — плотность вещества дисперсной фазы.
Важнейшее свойство всех дисперсных систем — очень большая удельная поверхность. В соответствии с уравнением (2.3) она изменяется обратно пропорционально размеру дисперсных частиц Удельная поверхность наноразмерных частиц (d - 1 — 10 нм) составляет 102— К)3 м2/г (рис 2.1).
4. Дисперсность (D) — отношение суммарной площади поверхности дисперсных частиц (Qw) к суммарному объему (И„) дисперсной фазы:
Я = ЯЛ/И„. (2.4)
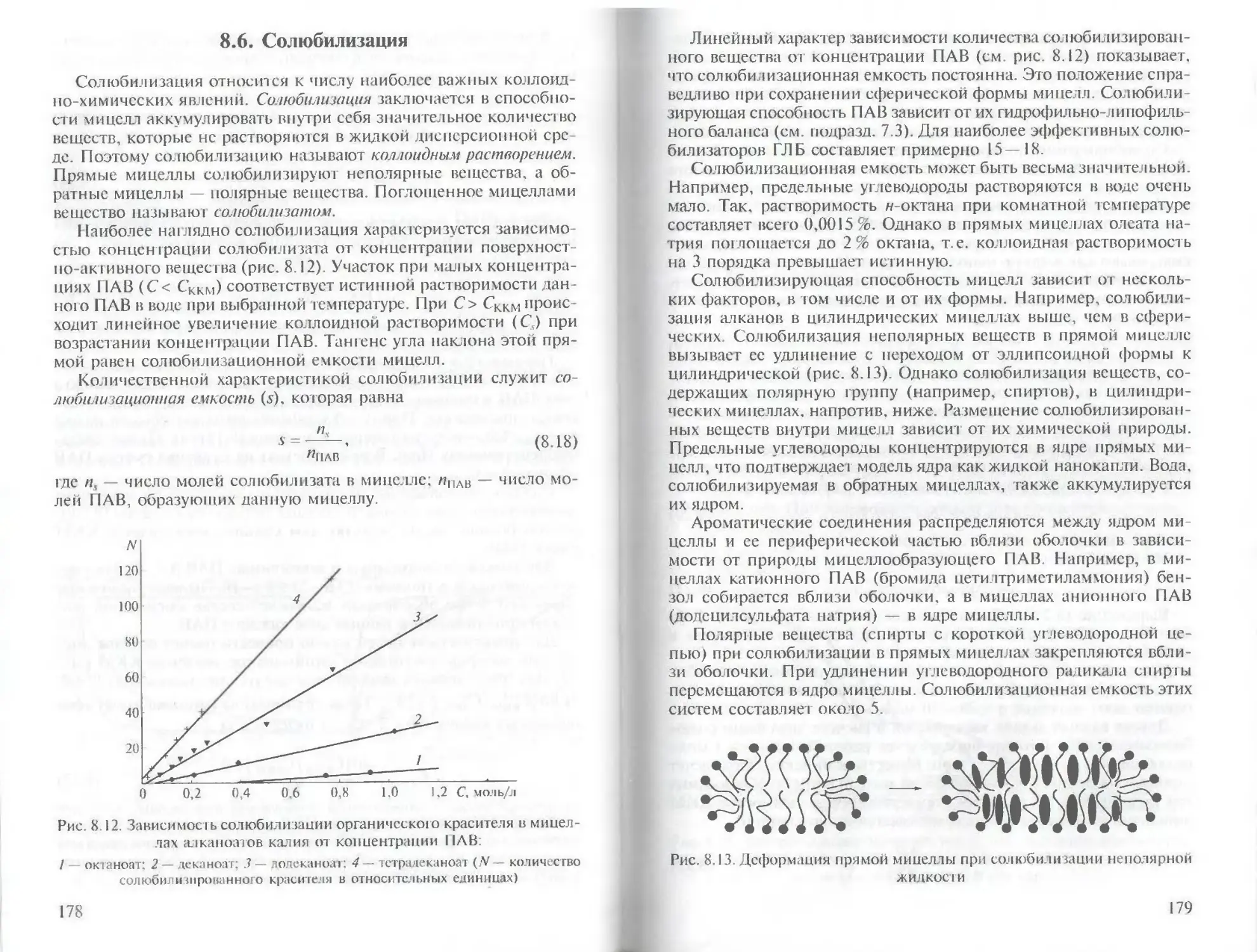
Дисперсность сферических или кубических частиц одинакового размера (J) равна
D = b/d. (2.5)
5. Поверхностное натяжение (о, Дж/м2) на границе дисперсных частиц с дисперсионной средой — основной термодинамический параметр, характеризующий свойства поверхности раздела фаз: для жидкостей численно равно удельной свободной поверхностной энергии (см. подразд. 4 1).
В зависимости от химической природы дисперсной фазы и дисперсионной среды поверхностное натяжение может изменяться в очень широких пределах — примерно от 10 Здо К)2 мДж/м2.
6. Время жизни дисперсной системы — количественная характеристика важнейшего общего свойства дисперсных систем — их термодинамической неустойчивости. Причина неустойчивости —
Рис. 2.1 Зависимость удельной поверхности (QJ от размера частиц (J):
I — нанодисперсные (ультрадиспсрсные) системы: II — высокодисперсные системы II! — щубодиспсрсиые системы
большая суммарная поверхность дисперсных частиц и, как следствие, избыток энергии Гельмгольца (Л/) по сравнению с компактной макроскопической фазой того же вещества. Этот избыток равен oQ„. Избыточная энергия стимулирует процессы, которые ведут к укрупнению, или агрегации, дисперсных частиц. В результате происходит «гибель» дисперсной системы (подробнее см. подразд. 10.1). Количественной мерой продолжительности существования дисперсных систем с жидкой или 1азовой дисперсном фазой служит время жизни (т) одной дисперсной частицы — капли в эмульсиях, тонкой жидкой пленки в пенах и газовою пузырька в газовых эмульсиях
Другая характеристика продолжительности жизни дисперсных систем — период полураспада (т ) Он определяется временем, за которое начальная (в момент образования дисперсной системы) концентрация частиц (v()) уменьшается в два раза. Эту характеристику используют преимущественно для систем с твердой дисперсной фазой (коллоидных растворов) и эмульсии.
2.2. Классификация дисперсных систем
Дисперсные системы очень разнообразны, их классификация проводится по нескольким признакам (табл. 2.1).
Рассмотрим классификацию, представленную в табл. 2.1, подробнее.
Классификация по размеру частиц дисперсной фазы. В зависимости от среднего размера (d ) дисперсных частиц принято выделять три I руины дисперсных систем:
1) ультрадисперсные системы, или наносистсмы, d = 1 — 10 нм;
2) высокодисперсные системы, d = 10 нм—1 мкм;
3) грубодисперспые системы, d = 1 — 100 мкм.
Ультрадисперсные системы соответствуют предельно возможной степени дисперсности и соответственно предельно большой удельной поверхности дисперсной фазы. Образующие их ультра-дисперсные частицы (наночастицы) могут содержать всего несколько сотен атомов или молекул. Для наночастпц характерна сильно выраженная зависимость ряда физических и химических свойств от размера (размерные, или масштабные, эффекты; см. подразд 3 4).
Для высокодисперсных частиц характерно интенсивное тепловое (броуновское) движение. Поэтому высокодисперсные частицы не оседают в поле силы тяжести. Другая важная особенность высокодисперсных систем — очень большая удельная поверхность дисперсной фазы. В соответствии с уравнением (2.3) для веществ плотностью 2 000 — 3 000 кг/м3 она составляет 2 000 — 3 000 м2/кг.
Т абл и па 2.1
Классификация дисперсных систем*
Признак классификации Название системы
Размер частиц дисперсной фазы: 1 — 10 нм 10 нм—1 мкм 1 — 100 мкм Ульградисперсная В ысо кол и с п срс ная Г рубодисперсная
Фракционный состав частиц дисперсной фазы: частицы одинакового размера частицы разного размера Мон од и с п с рс н ая П од идисперс ная
Концентрация част иц дисперсной фазы: малая большая Свободнодисперсная и пт связиодисперсная Связнодисперсная ил и свобод 1 юдисперсная
Характер взаимодействия дисперсных частице дисперсионной средой: слабое сильное Лиофобная Лиофильная
Характер распределения фаз: сплошное сетка тонких прослоек Континуальная Би конти нуальная
* Классификация дисперсных систем по агрегатному состоянию дисперсной фазы и дисперсионной среды приведена в табл. 2.2
В грубодисперсных системах с частицами размером более 10 мкм под действием силы тяжести дисперсная фаза сравнительно быстро оседает, если ее плотность (р^) больше плотности дисперсионной среды (р0). Напротив, более лет кие частицы (р^< р0) всплывают.
Таким образом, граница между высокодисперсными и грубо-диснсрсными системами определяется действием силы тяжести на дисперсные частицы. Количественно эта граница определяется капиллярной длиной:
a = [2o/(Apg)]’ \ (2.6)
где Др = (prf - ро) — разность плотностей дисперсной фазы и дисперсионной среды; g— ускорение силы тяжести.
Для высокодисперсных систем выпо шлется неравенство: d> а. Величину а2 называют капиллярной постоянной жидкости В космических условиях ускорение силы тяжести значительно меньше,
чем на Земле. Поэтому граница между этими группами смешается в сторону более крупных частиц.
Классификация по фракционному составу частиц дисперсной фазы. В зависимости от однородности размеров дисперсных частиц выделяют две группы:
1) монодисперспыс системы с частицами одинакового размера;
2) полидисперсные системы, в которых размер частиц изменяется от минимального (t/min) до максимального (dmn). Средний размер d изменяется в интервале dmin < d < Jmax; полидисперсные системы характеризуются распределением частиц но размерам, для установления которого проводят седиментационный анализ (см. подразд. 3.6).
Классификация по концентрации частиц дисперсной фазы. В зависимости от концентрации частиц выделяют:
1) свободнодисперсные системы;
2) связнодиспсрсные системы.
В свободподиспсрсных системах частицы находятся на больших расстояниях L » d друг от друга: L v_,/3. Поэтому дисперсные частицы не взаимодействуют друг с другом и представляют собой самостоятельные кинетические единицы. Такие системы по молекулярно-кинетическим свойствам сходны с обычными растворами.
В связнодисперсных системах большинство частиц контактируют друз с друзом и образуют различные типы коллоидных структур Механические и реологические свойства связнодисперсных систем резко отличаются от свойств свободнодисперсных систем (см. гл. 10).
При делении дисперсных систем на свободнодиспсрсные и связнодисперсные необходимо учитывать следующее важное обстоятельство Если дисперсные частицы сильно вытянуты (т.е. имеют анизодиаметричную форму), то переход от свободнодисперсной к связнодиспсрсной системе (например, переход от золя к гелю) происходит при весьма низкой концентрации частиц.
Образование связной сетки дисперсных част иц приводит к резкому изменению большинства свойств дисперсной системы (см гл. 10). Концентрацию частиц, при которой образуется связная сетка, называют порогом перколяции.
Классификация по характеру взаимодействия дисперсной фазы с дисперсионной средой. Интенсивность взаимодействия дисперсных частиц с дисперсионной средой количественно характеризуют- удельной свободной поверхностной энергией на границе дисперсная фаза—дисперсионная среда. В зависимости от характера этого взаимодействия различают две группы дисперсных систем:
1) лиофильные;
2) лиофобные.
Лиофильные системы отличаются сильным взаимодействием. Такая ситуация имеет место при сходной химической природе дисперсной фазы и дисперсионной среды, однако при отсутствии химических реакций между ними и условии большой взаимной растворимости. В лиофильных системах межфазная поверхностная энергия мала — ниже 10-2 мДж/м2. Примером лиофильных систем могут служить мицеллярные растворы поверхностно-активных веществ (см. гл. 8) и микроэмульсии.
Лиофобные системы соответствуют слабому (молекулярному) взаимодействию дисперсной фазы и дисперсионной среды, поэтому удельная свободная поверхностная энергия границы раздела фаз достаточно велика и составляет 101— 102 мДж/м2. Примеры лиофобных систем — частицы малорастворимых солей и оксидов (Agl, V2O5 и т.п.) в воде, капли воды в органической жидкости (нефти) и др.
Классификация по характеру распределения фаз (дисперсных частиц и дисперсионной среды). Наиболее распространенный вид дисперсных систем характеризуется тем, что дисперсионная среда представляет сплошную (континуальную) макроскопическую фазу — газ, жидкость или твердое тело.
В ряде случаев имееч место существенно иное распределение. Дисперсионная среда состоит из сообщающихся друг с другом тонких прослоек (каналов), размеры которых малы и соответству-
Таблица 2.2
Классификация дисперсных систем по агрегатному состоянию дисперсной фазы и дисперсионной среды
Агрегатное состояние Название системы
дисперсионной среды дисперсной фазы
Газ Газ Жидкость Жидкость Жидкость Твердая фаза Твердая фаза Твердая фаза Твердая фаза Жидкость Твердая фа )а Жидкость Газ Твердая (раза Жидкость Газ Аэрозоль (дым) А эрозол ь (тума 11) Золи (коллоидные растворы) d< 0.1 мкм. Суспензии d > 1 мкм. Гели — би конги нуальные системы Эмульсии Газовые эмульсии (свободнодисперсные системы). Пены (связнодисперсные системы) Ком।юзи гныс материajiы Пористые среды Пористые среды и материалы. Мембраны
ют размерам дисперсных частиц. В таких системах обе фазы по существу являются дисперсными и их можно рассматривать как смесь.
Подобные системы называют биконтинуальнылиц поскольку в них дисперсная фаза является не дискретной (как в обычных дисперсных системах), а континуальной: большинство частиц этой фазы находятся в контакте друг с другом. Пример биконтинуаль-ной системы — пористая среда с частицами и порами дисперсных размеров.
В биконтинуальных системах важную роль играет трехмерная сетка, образуемая элементами дисперсионной среды. Механические и гидродинамические свойства таких сеточных структур изучает одно из современных направлений математики и физики — теория перколяции.
Классификация по агрегатному состоянию дисперсной фазы и дисперсионной среды. Эта классификация (табл. 2.2) используется наиболее часто.
2.3. Получение, стабилизация и очистка дисперсных систем
Получение дисперсных систем связано в первую очередь с получением дисперсных частиц. Методы получения дисперсных частиц рассмотрены в гл. 3. Кроме того, нужно решить следующие задачи:
I) распределить дисперсные частицы в дисперсионной среде до необходимой концентрации;
2) стабилизировать дисперсную систему, чтобы сохранить ее структуру и свойства в течение достаточно длительного времени;
3) провести очистку дисперсной системы от различных примесей.
Эти задачи решают в зависимости от специфики (типа) той или иной дисперсной системы.
Получение дисперсных систем. Эмульсии. Поскольку эмульсии — грубодисперсные системы, их обычно получают диспергациоп-ным методом (см. подразд. 3.3). Жидкости, которые должны образовать эмульсию, интенсивно перемешивают или подвергают воздействию механических вибраций или ультразвука. Чтобы получить капли одинакового размера (т.е. монодисперсную систему), проводят гомогенизацию. Этот процесс заключается в продавливании жидкости дисперсной фазы в дисперсионную среду через небольшие отверстия требуемого диаметра под большим давлением. Такой прием используют, например, при обработке молока. В результате гомогенизации средний размер капель жира уменьшается примерно от 1—3 до 0,1 —0,2 мкм.
Эмульсии получают также конденсационными методами (обычно — заменой растворителя; см. подразд. 3.3).
Самостоятельную задачу представляет получение высококонцентрированных эмульсий. К ним относят эмульсии с концентрацией дисперсной фазы более 74 об. %, вплоть до 99 об. %. Капли дисперсной фазы в таких эмульсиях, имеющие форму многогранных призм, разделены тонкими пленками жидкой дисперсионной среды.
Концентрированные эмульсии могут обладать механическими свойствами твердых тел — прочностью и упругостью.
Специфика приготовления концентрированных эмульсий заключается в том, что дисперсная фаза вводится в дисперсионную жидкую среду небольшими порциями при интенсивном перемешивании.
Пены. Как и эмульсии, иены — грубодиснсрсные системы. Поэтому во многих технологических процессах пены получают теми же диснергационными методами, которые применяют для получения газовых пузырьков.
Конденсационные методы получения пен основаны на пересыщении раствора газа в данной жидкости при соответствующем изменении температуры или давления. Используют также химические реакции с выделением газа. В качестве примера приведем реакцию, лежащую в основе приготовления пены в огнетушителях:
NaHCO3 + HCI -> NaCI + Н2О + СО,Т
Еще один конденсационный метод получения пен основан на использовании микробиологических процессов.
Коллоидные растворы Получают коллоидные растворы (золи) различными конденсационными методами. Для получения высокодисперсных золей необходимо обеспечить выполнение следующего условия: скорость образования твердых частиц должна во много раз превышать скорость их роста. Чтобы выполнить эго условие, при получении дисперсных частиц с помощью химических реакций часто используют такой способ: концентрированный раствор одного компонента вливают в небольшом количестве в сильно разбавленный раствор другого компонента при очень и н те н с и в н о м г г е р е м е ш и ва н и и.
Гели. Приведенные выше системы являются свободнодиспср-сными. Получение св? игодисперсных систем имеет определенную специфику. Рассмотрим в качестве примера получение гелей. Обычно их получают из коллоидных растворов (золей). При определенных условиях дисперсные частицы слипаются друг с другом — происходит процесс коагуляции (см. подразд. 10.2).
Если частицы имеют анизодиамстричсскую форму (стержни, эллипсоиды), то они соединяются преимущественно своими кон
нами и образуюг пространственную структуру (сетку'), в ячейках которой находится жидкая дисперсионная среда (см. подразд. 10.4). Процесс превращения золей в гели называют золь—гель-переход. Он имеет важное значение в нанотехнологии. Таким образом, гели, как и концентрированные эмульсии, иногда могут быть биконти-нуальными дисперсными системами.
Свойства гелей весьма эффективно регулируют, изменяя концентрацию дисперсной фазы и форму дисперсных частиц. Еще один важный фактор — температура: ее повышение затрудняет образование контактов между дисперсными частицами и поэтому прочность гелей снижается (см. гл. 10).
Стабилизация дисперсных систем. Необходимость стабилизации дисперсных систем возникает из-за их термодинамической неустойчивости, которая в свою очередь обусловлена большой суммарной поверхностью дисперсных частиц. Цель стабилизации заключается прежде всего в сохранении размеров дисперсных частиц и предотвращении непосредственных контактов между н и м и.
Основные методы стабилизации, применяемые в коллоидной химии, базируются на следующих принципах:
1) создание одноименно заряженных дисперсных частиц, что вызывает их электростатическое отталкивание (ем. гл. 9, 10); этот метод применяют главным образом для обеспечения устойчивости аэрозолей и коллоидных растворов;
2) создание на поверхности дисперсных частиц тонких защитных слоев (оболочек), которые не разрушаются при сближении частиц и препятствуют их контакту и последующему укрупнению; такую защиту (структурно-механический барьер) обеспечивают адсорбционные слои поверхностно-активных веществ, в особенности — высокомолекулярных; этот метод применяют преимущественно для стабилизации эмульсий и пен (см. гл. 6, 7).
Обеспечение устойчивости дисперсных систем представляет одну из основных проблем коллоидной химии, она будет подробно рассмотрена в гл. 10.
Очистка дисперсных систем. Чаше всего необходимо очистить дисперсные системы (после их получения) от низкомолекулярных примесей, преимущественно электролитов. Для этого используют такой метод, как диализ — фильтрация через полупроницаемую мембрану. Низкомолекулярные вещества свободно проходят через поры мембраны, а дисперсные частицы задерживаются. Для диализа часто используют мембраны, приготовленные из раствора нитрата целлюлозы (нитратцеллюлозные пленки).
Наиболее эффективным способом очистки дисперсных систем является мембранное разделение. Частицы дисперсной фазы могут отличаться размерами и физико-химической природой, поэтому для их разделения применяют разные мембранные методы.
Последние отличаются типом используемых мембран и движущей силой, т.е. условиями процесса разделения.
Для разделения наиболее грубых частиц, взвешенных в растворе, имеющих размер 0.1 — 10 мкм (например, бактерии, коллоидные частицы, капли в эмульсиях), применяют микрофильтрацию. Используют пористые мембраны (размер пор 0,05—10 мкм). а движущей силой является небольшое давление (до 200 кПа), приложенное к очищаемому раствору.
Дисперсии более «мелких» частиц (размер 1—500 нм) очищают методом ультрафильтрации. Здесь также используют пористые мембраны (размер пор 1 — 100 нм), а движущей силой служит давление, но более высокое (100 кПа — 1 МПа). Типичные объекты ультрафильтрации — растворенные в воде молекулы полимеров (белков, крахмала, ферментов), а также фармацевтических объектов.
Общим для этих процессов является необходимое для разделения давление, поэтому эту группу мембранных процессов, важнейшую по разнообразию областей применения, называют баро-мембранными.
Еще одним из баромембранных процессов является обратный осмос, используемый сейчас в особо крупных масштабах для обессоливания морской и солоноватой воды. Растворенные в воде ионы окружены гидратной «шубой», поэтому представляют собой диспергированные частицы размером 0,2 —2,0 нм. Осмотическое давление над такими растворами составляет несколько десятков бар (например, для морской воды 2,5 МПа). Однако если приложить к раствору давление, большее осмотического, то через мембрану пойдет поток пресной, не содержащей соли воды. Обратноосмотические мембраны имеют наименьшие поры, хотя некоторые исследователи считают, что обратный осмос воды может осуществляться в непористых мембранах.
Обессоливание воды (или удаление электролитов) может осуществляться и под действием другой движущей силы — градиента электрического поля. Такой процесс называют электродиализом.
Многие мембранные процессы нс укладываются в подобную классификацию. Процессы диализа протекают под действием градиента концентрации (без прилагаемого внешнего давления). В зависимости от характера разделяемых сред и природы материала мембраны речь может идти о пористых и нспористых мембранах. Явление диализа лежит в основе работы почки, а гемодиализ — мембранный метод очистки крови пациентов с почечной недостаточностью.
Существуют мембранные методы, объектами разделения которых являются уже не коллоидные частицы, а молекулярные объекты — индивидуальные атомы, молекулы газов и органических веществ (газоразделенис, первапорация, нанофильтрация). Для их
реализации применяют, главным образом, напористые мембраны.
Следует отмстить, что и сами мембраны (точнее их селективные слои) являются дисперсными системами. Дчя пористых мембран решающим фактором является узкое распределение пор по размерам. Напористые полимерные мембраны нс содержат открытых пор, позволяющих селективно разделять частицы определенного размера. Однако скорость массопереноса через них определяется наноструктурой свободного объема в полимере, который может быть представлен сферическими или цилиндрическими замкнутыми порами (микрополостями) с характерным размером в пределах 0,3 —2,0 нм.
2.4. Движение частиц в дисперсных системах
В свободнодисперсных системах с газовой и жилкой дисперсионной средой (аэрозоли, коллоидные растворы, газовые эмульсии, мицеллярные растворы поверхностно-активных веществ) дисперсные частицы не фиксированы в каких-либо позициях. Напротив, они перемешаются; кроме того, частицы могут вращаться и совершать колебания с разной амплитудой. Подвижность дисперсных частиц, особенно высоко- и ультрадисперсных, представляет собой фундаментальное свойство свободнодисперсных систем.
Движение дисперсных частиц обусловлено различными факторами и зависит главным образом от их размера. Высокодисперсные частицы благодаря своим малым размерам активно участвуют в броуновском движении.
Броуновское движение дисперсных частиц рассматривают как проявление молекулярно-кинетических свойств дисперсных систем.
Другое молекулярно-кинетическое свойство — диффузия дисперсных частиц — процесс переноса частиц вследствие различия их концентрации в разных областях дисперсной системы. Диффузия приводит к постепенному выравниванию концентрации частиц. В соответствии со вторым началом термодинамики диффузия сопровождается увеличением энтропии дисперсной системы.
Крупные дисперсные частицы (твердые частицы, капли, газовые пузыри) в броуновском движении практически не участвуют. Соответственно грубодисперсные системы не обладают молекулярно-кинетическими свойствами. Именно на этом отличии базируется классификация дисперсных систем на высокодисперсные и грубодиспсрсныс (см. подразд. 2.2).
Основной причиной движения крупных дисперсных частиц служит различие плотностей дисперсной фазы (р/) и дисперсионной среды (р0). Если плотность дисперсной фазы больше (pf/> р0). частицы постепенно оседают под действием силы тяжести Этот процесс называют седиментацией. Более легкие дисперсные частицы (при условии р^< ро), напротив, постепенно всплывают вверх. Этот процесс называют обратной седиментацией.
Перемещение дисперсных частиц происходит и под действием других внешних сил. В коллоидной химии важную роль играет движение заряженных частиц в электрическом поле. Такое движение называют электрофорезом (см. подразд. 9.6).
Отдельную группу составляют перемещения дисперсных частиц, которые происходят вместе с движущейся дисперсионной средой. Такие потоки называют двухфазными. Они существенно отличаются от однофазных потоков газа или жидкости. Например, присутствие в жидкости даже небольшого количества дисперсных частиц приводит к возрастанию вязкости дисперсной системы по сравнению с вязкостью дисперсионной среды (см. гл. 10).
Рассмотрим теоретические закономерности разных форм движения дисперсных частиц.
Седиментация. Это процесс осаждения дисперсных частиц в дисперсионной среде (обычно — в жидкости) под действием силы тяжести. Важнейшая характеристика седиментации — скорость (г) оседания индивидуальной дисперсной частицы.
Седиментация твердых частиц. Проведем расчет скорости оседания твердой сферической частицы радиусом г.
Движущая сила (/) создается разностью плотностей Др = pf/-p0 дисперсной фазы (pj и дисперсионной среды (р0):
где g — ускорение силы тяжести, равное 9,81 м/с2.
При движении частицы в вязкой жидкости возникает противоположно направленная сила вязкого трения (£.,), ее определяет один из основных законов i идродинамики — уравнение Спюкса.
Для равномерно движущегося шара уравнение Стокса имеет вид
= блттч], (2.7)
где г| — вязкость жидкости, Па-с; v — скорость частицы; г — радиус частицы.
Уравнение Стокса (2.7) выполняется при достаточно медленном движении частицы Необходимое условие — встречное течение жидкости, облекающее частицу, должно быть ламинарным.
Условие ламинарности определяет неравенство: число Рейнольдса Re < 1. Безразмерный критерий ламинарности (число Рейнольдса) определяется уравнением
Rc=
П
(2-8)
где d — размер частиц.
При больших скоростях седиментации Re » I речение становится турбулентным (вихревым) и уравнение Стокса для расчета силы вязкого трения уже неприменимо.
При равномерном движении частицы с постоянной скоростью v движущая сила / и сила трения f , взаимно уравновешены:/ = fv. Из этого условия находят скорость седиментации частицы радиусом г:
2 Apffl*2
9 1]
(2.9)
Таким образом, скорость седиментации резко растет с увеличением размера частиц (пропорционально г2). Поэтому крупные частицы оседают гораздо быстрее мелких.
Седиментация капель. Оседание капель происходит несколько сложнее, чем твердых частиц. Причина заключается в изменении формы движущихся капель. Капли при большой скорости сужаются в передней и расширяются в задней части, приобретая пулеобразную форму. Соответственно в уравнение Стокса (2.7) нужно вводить поправочные коэффициенты, зависящие от скорости оседания. Другой осложняющий процесс — при оседании внутри капли реализуются сложные вихревые потоки. В результате возникает дополнительная сила сопротивления, зависящая от вязкости (ц,/) жидкости, образующей каплю. С учетом вязкости общая сила вязкого сопротивления при оседании капли определяется уравнением Ада мара— Рыбчинского\
% +П
(2.10)
При оседании твердых частиц (п^>> Г|) уравнение (2.10) принимает вил уравнения Стокса (2.7).
При всплывании пузырьков газа в пенах и газовых эмульсиях, напротив, r|rf« 1] и сила вязкого трения равна
/. = 4лгиг|. (2.11)
Броуновское движение и диффузия дисперсных частиц. Броуновское движение — это хаотическое движение дисперсных частиц в
дисперсионной среде. Главная особенность броуновского движения заключается в том, что частица перемешается хаотически. Эти скачки совершаются с большой частотой в произвольном направлении, т.е. независимо от того, каким был предшествующий скачок. Кроме того, скачки имеют разную длину. Направление движения частицы может изменяться до К)20 раз в течение 1 с (рис. 2.2).
Задача заключается в расчете расстояния, на которое переместится дисперсная частица размером d за время /. Из-за хаотичности скачков нельзя прогнозировать место (точку), где окажется частица за это время. Поэтому данный вопрос должен ставиться иначе: каким будет наиболее вероятное (среднеквадратичное) расстояние Т от исходной точки через время /. Такая постановка соответствует статистической природе броуновского движения
Перемещение дисперсной частицы при броуновском движении определяется уравнением Эйнштейна:
P=2Dr, (2.12)
где D — коэффициент диффузии дисперсных частиц. м2/с.
Коэффициент диффузии равен массе вещества, перенесенного вследствие диффузии через единицу площади поперечного сечения стационарного диффузионного потока за единицу времени пол действием единичного градиента концентрации (gradC = -1 г/см4).
Коэффициент диффузии сферической дисперсной частицы радиусом /' определяется уравнением Эйнштейна:
= (2J3)
бтпу*
где кн — постоянная Больцмана, равная 1,38 • 10-23 Дж/К; Т — температура, К; q — вязкость дисперсионной среды, Па-с.
Уравнение Эйнштейна получено на основе анализа сил, действующих на дисперсную частицу при ее броуновском движении.
Движущая сила создается тепловыми колебаниями молекул жидкости, окружающей частицу. Средняя энергия этих колебаний в расчете на одну степень свободы равна ’ЛА'/Д. Силу сопротивления создает вязкое трение жидкости, которое определяется уравнением Стокса (2.7).
Рис. 2.2. Схема броуновского движения дисперсной частицы
В соответствии с уравнением (2.13) коэффициент диффузии дисперсных частиц изменяется обратно пропорционально их размеру d- 2г. Поэтому броуновское движение и диффузия присуши именно высокодисперсным и ультрадисперсным частицам.
Коэффициент диффузии резко растет с увеличением температуры. Зависимость D = f(T) определяется возрастанием энергии тепловых колебаний квТ. Вместе с гем вязкость жидкостей с увеличением температуры уменьшается по экспоненциальному закону — пропорционально ехр|-(7л/(АйГ)], где ИА — энергия активации вязкого течения.
Диффузия дисперсных частиц. Диффузия молекул, атомов, ионов, дисперсных частиц — один из основных процессов, происходящих в газах, жидкостях и твердых телах.
Диффузией называю! самопроизвольный процесс выравнивания концентрации какого-либо вещества в газе, жидкости или твердом теле. Диффундирующее вещество может находиться в виде отдельных атомов, молекул и ионов, а также в виде дисперсных частиц. Диффузия дисперсных частиц происходит под влиянием броуновского движения и ведет к увеличению энтропии системы.
Основной закон диффузии (закон Фика) определяет количество вещества dm, которое переноси гея через площадь Q (площадь поперечного сечения диффузионного потока) за время dr под действием градиента концентрации gradC = -dC/dx (х — направление (ось координат), в котором происходит диффузия):
dm = -D (dC/dx) Q dr.
(2.14)
Для расчета диффузии дисперсных частиц в дисперсионной среде (жидкости) следует пользоваться концентрацией частиц (v, м“3). Соответственно уравнение (2.14) принимает вид
dm = -D — \idr. dx J
(2.15)
Количество вещества
dm
Qdr’
которое переносится диффузией в единицу времени через единицу площади, называю! удельным диффузионным потоком. После подстановки значения id в уравнение (2.15) получаем
(2.16)
Уравнение (2.16) лает макроскопическое (феноменологическое) определение коэффициента диффузии (D) дисперсных частиц: коэффициент диффузии равен массе дисперсной фазы вещества,
которое продиффундировало через единицу плошали в единицу времени при градиенте концентрации dv/dx = 1 м-4.
Физический смысл коэффициента диффузии дисперсных частиц раскрывает приведенное выше уравнение Эйнштейна (2.13). Важнейшее следствие этого уравнения — коэффициент диффузии обратно пропорционален радиусу частицы г. Поэтому диффузия происходит достаточно интенсивно только в ультрадисперсных и высокодисперсных системах. Коэффициент диффузии дисперсных частиц размером 1 — 100 нм в воде при комнатной температуре изменяется примерно от 4 10-14 до 4 • 10 10 м2/с. По уравнению (2.12) при известном коэффициенте диффузии можно рассчитать среднеквадратичное перемещение дисперсной частицы за выбранный интервал времени /. Например, для частиц размером 100 нм коэффициент диффузии в воде равен 4- 10“14 м2/с, соответственно за период / = 1 ч среднеквадратичное перемещение / составит около 10 мкм.
Диффузия дисперсных частиц играет в коллоидной химии очень важную роль. Одно из наиболее важных следствий диффузии — в высокодисперсных системах устанавливается динамическое равновесие между оседанием (седиментацией) частиц и их диффузией в противоположном направлении. Это равновесие обеспечивает длительную седиментационную устойчивость высокодисперсных систем (например, коллоидных растворов) в поле силы тяжести.
Седиментационно-диффузионное равновесие дисперсных систем. Рассмотрим возникновение седимептационно-диффузионного равновесия в монодисиерсной системе, например в суспензии, содержащей твердые сферические частицы радиусом г плотностью р^. Пусть эта суспензия получена перемешиванием до достижения одинаковой концентрации частиц v0 во всем объеме дисперсионной жидкой среды.
Далее перемешивание прекращают и начинается процесс седиментации. Оседание нарушает равномерное распределение частиц. В нижней части дисперсной системы концентрация частиц v„ становится больше исходной (v„ > v0). В верхней части системы (выше средней линии) концентрация частиц vB понижается по сравнению с исходной (vB < vfl).
Неравномерное распределение частиц приводит, таким образом, к изменению концентрации частиц вдоль вертикальной оси х. Разность концентраций Av = v„ - vb будет увеличиваться по мерс седиментации. Под действием разности концентраций возникает диффузионный поток (/j), направленный снизу вверх, т.е. противоположно седиментации. Величина этого потока определяется уравнением (2.16); в данном случае х— вертикальная координата. По мерс седиментации градиент концентрации растет, соответственно увеличивается и диффузионный поток.
Седиментационный поток рассчитывают по формуле ix = vv, где и — скорость седиментации частиц данного размера; v — кон-нентрация частиц. Когда встречные потоки (диффузионный и сегментационный) сравняются: z’j= в дисперсной системе насупит динамическое равновесие между седиментацией и диффузней. Оно заключается в том. что на каждом уровне (т.е. на данном расстоянии х от дна сосуда) устанавливается стационарная концентрация частиц, которая со временем не меняется.
Распределение концентрации частиц вдоль вертикальной оси х в высокодиснерсной системе можно получить из условия: id = is. После соответствующих подстановок получаем
ln^- = jKr3(prf -р0)^х/(А>7). (2.17)
Таким образом, благодаря диффузии не происходит осаждения всей дисперсной фазы. Основная ее часть остается в дисперсионной среде, т.е. дисперсная система в целом может сохраняться («жить») в течение длительного времени.
Яркий пример седиментационно-диффузионного равновесия представляют коллоидные растворы золота с частицами размером около К) нм, приготовленные М.Фарадеем в 1857 г. С тех пор прошло почти 150 лет, но эти дисперсные системы сохранились: несмотря на очень большую плотность золота, дисперсные частицы не выпали в осадок.
Размер частиц имеет ключевое значение для седиментационной устойчивости дисперсных систем. Приведем такой пример. Частицы коллоидного серебра диаметром менее 0,1 мкм распределяются в объеме золя практически равномерно; крупные частицы (d > 10 мкм) переходят в осадок; частицы промежуточных размеров (0,1 < d< 1 мкм) находятся в седиментационно-диффу-знонном равновесии и распределяются но высоте дисперсной системы (золя) в соответствии с логарифмическим соотношением (2.17).
Седиментационно-диффузионное равновесие дисперсных систем представляет интерес еще в одном аспекте. Уравнение (2.17) позволяет по экспериментально установленной зависимости концентрации частиц от высоты х рассчитать константу Больцмана и число Авогадро NA- R/kB(R — универсальная газовая постоянная). Такие опыты были проведены французским физиком Ж. Перреном на суспензиях гуммигута в 1908 г.
Число Авогадро, определенное методом Перрена, оказалось очень близко к известному значению. Этот результат — весьма убедительное подтверждение атомно-молекулярной теории. В тот период эта проблема была очень актуальна и работа Ж.Перрена была отмечена Нобелевской премией по физике (1926).
2.5. Течение дисперсных систем
Свободнодисперсные системы с жилкой и газовой дисперсионной средой (золи, суспензии, эмульсии, пены, аэрозоли) характеризуются большой подвижностью.
Под действием внешних воздействий: гидростатического давления, электрического напряжения — возникает течение жидкости или газа, которое увлекает находящиеся в них дисперсные частицы Таким образом происходит перемещение всей дисперсной системы. Основная проблема заключается в учете влияния размера, формы и концентрации дисперсных частиц па скорость течения.
В свободнодисперсных системах основную роль играют 1идро-динамические эффекты, возникающие при взаимодействии частиц с жидкостью или газом. При большой концентрации v частицы начинают контактировать между собой. Возникающая структура значительно уменьшает текучесть дисперсионной среды и течение может полностью прекратиться (см. гл. 10).
Рассмотрим следующую задачу: по трубке длиной / достаточно большого диаметра d - 2г (несколько сантиметров или более) под действием перепада давления на ее концах (Др) течет коллоидный раствор (золь) — взвесь твердых частиц в жидкости. Для упрощения будем рассматривать монодиспсрсную систему, т с все дисперсные частицы имеют одинаковый размер. Примем также, что частицы имеют сферическую форму, а концентрация частиц v мала, поэтому они находятся далеко дру| от друга и не контакт и-руют между собой. При отсутствии дисперсных частиц, т е. при течении одноротной жидкости вязкостью т|, средняя скорость ламинарного течения (г) определяется законом Пуазе иля:
При таком же перепаде давления Др течение дисперсной системы (коллоидного раствора) происходит с меньшей скоростью. А.Эйнштейн объяснил это icm, что присутствие дисперсных частиц приводит к возрастанию вязкости дисперсной системы (г|*) по сравнению с вязкостью жидкости р, т.е. q*> г|. Теория Эйнштейна определяе т вязкость дисперсной системы следующим уравнением:
т|* = п(1 + щр), (2.19)
где <р — объемная доля дисперсной фазы; а — безразмерный коэффициент, который зависит от формы дисперсных частиц.
Для сферических частиц коэффициент а равен 2,5. Для анизо-диаметрических частиц (эллипсоидов, стержней и т.д.) коэффи-36
iinciiT а имеет более высокое значение. Чем больше вытянутость частиц, тем больше коэффициент а.
В соответствии с уравнением Эйнштейна вязкость коллоидной системы зависит от общего количества дисперсных частиц (от объемной доли дисперсной фазы), но не зависит от размера части.
Уравнение Эйнштейна (2.19) можно преобразовать к форме
= аф. (2.20)
1
Отношение (ц* -П)/л характеризует относительное увеличение вязкости дисперсионной среды при введении в нес дисперсной фазы; оно растет линейно при увеличении объемной доли дисперсной фазы (р.
Уравнение (2.20) позволяет но экспериментальной зависимости rj* = /((р) определить коэффициент а, зависящий от формы частиц. Э ти данные используют в свою очередь для оценки размера дисперсных частиц и макромолекул.
Контрольные вопросы
1. Дайте определение понятий «дисперсная фаза», «дисперсионная среда».
2. Дайте определение понятий «концентрация частиц», «удельная поверхность дисперсной фазы».
3. Назовите основные признаки и свойства, по которым проводят классификацию дисперсных систем.
4 Охарактеризуйте границу между высокодисперсными и грубодисперсны ми системами.
э В чем заключается главное отличие между свободнодисперсными и свя знодисперсн ы м и с исте мам и?
6. В чем главное отличие лиофильных и лиофобных дисперсных систем?
7. Назовите основные классы дисперсных систем в зависимости от агрегатного состояния дисперсной фазы и дисперсионной среды.
8 Назовите основные принципы получения и стабилизации дисперсных систем.
9 Охарактеризуйте процесс седиментации Напишите уравнение ско-pociH оседания дисперсной частицы, укажите условия его выполнения.
10. Рассчитайте скорость оседания дисперсных частиц мела (плотность 2,7 г/см3) размером 10 мкм в воде (плотность 1 г/см3, вязкость 0,8 МПа с). Найдите соответствующее число Рейнольдса.
II. Рассчитайте по уравнению Эйнштейна коэффициент диффузии тля дисперсной части пы радиусом 0.1 мкм в воде (вязкость 0,8 МПа с) при температуре 300 К.
12. Охарактеризуйте седиментационно-диффузионное равновесие дисперсной системы
ГЛАВА 3
ДИСПЕРСНЫЕ ЧАСТИЦЫ
Физические и химические свойства дисперсных частиц существенно отличаются от свойств макроскопических образцов данного вещества. Эти отличия выражены тем сильнее, чем меньше размер дисперсных частиц. Изучение методов получения, структуры, физических и химических свойств дисперсных частиц — одна из наиболее актуальных задач коллоидной химии.
3.1. Классификация дисперсных частиц
Дисперсные частицы очень разнообразны по размеру, форме, строению и химическому составу. Классификация дисперсных частиц проводится по нескольким признакам (табл. 3.1).
Поясним приведенную в табл. 3.1 классификацию.
Классификация дисперсных частиц по размерам. Классификация частиц проводится в основном по тем же принципам, что и классификация дисперсных систем.
Самыми малыми дисперсными частицами являются наночастицы. Наночастицы содержат несколько сотен (или менее) атомов. Они имеют сложную внутреннюю структуру. Важная особенность напочастип по сравнению с более крупными частицами заключается в том, что из-за предельно малых размеров у них отсутствует различие между поверхностными и объемными свойствами К числу наиболее известных наночастиц относятся фуллерены — разветвленные образования, состоящие из атомов углерода. Характерный признак ультрадисперсных частиц (напочастиц) — зависимость их свойств от размера частиц, т с. наличие размерных (масштабных) эффектов.
Высокодисперсные частицы благодаря своим малым размерам способны участвовать в броуновском движении. Грубодисперсные частицы вследствие отличия их плотности от плотности дисперсионной среды быстро оседают или всплывают, что приводит к расслоению фаз и «гибели» дисперсной системы.
Классификация дисперсных частиц по форме. Форма дисперсных частиц очень разнообразна. На многие свойства дисперсных частиц и образуемых ими дисперсных систем сильно влияет соотношение между их размерами: длиной (/), шириной (Л) и тол-
Классификация дисперсных частиц
1 — — — 1 Признак классификации Название дисперсных частиц
Ра «мер частиц: 1 —10 нм 10 нм—1 мкм 1 — 100 мкм Ул ьтрадисперсные Высокодисперсные Г рубодисперсные
Форма частиц. длина, ширина и толщина частиц примерно одинаковы и составляют от 1 нм до 10 мкм Объемные (трехмерные)
толщина частиц составляет от 1 нм до 10 мкм, длина и ширина — более 100 мкм Поверхностные (двухмерные): например, тонкие пленки и слои
диаметр поперечного сечения составляет от 1 нм до 10 мкм Линейные (одномерные); тонкие нити, волокна
сферическая, кубическая Симметричные
эллипсоидная, призмагическая Анизодиаметричные
( троение частиц: твердые частицы; жидкие капли и пленки; газовые пузырьки пленки Фазовые
мицеллы в растворах поверхностно-активных веществ; кластеры Псевдофазовыс
Химический состав частиц Неорганические Органические Полимерные Биополимерные
шиной (d). По этому признаку частицы классифицируют на три । руппы.
I. Объемные (трехмерные) частицы: все три размера (Л, /, d) примерно одинаковы и находятся в дисперсном (от I нм до 10 мкм) интервале.
2. Поверхностные (двухмерные) частицы: дисперсному интерва-iy отвечает только один размер (толщина d)y а два других размера (длина / и ширина Л) значительно больше и могут иметь макроскопические значения.
3. Линейные (одномерные) частицы: очень гонкие нити, волокна: интервалу дисперсных размеров отвечает поперечный размер.
Классификация дисперсных частиц по строению. В зависимос ти от строения дисперсные частицы классифицирую! на фазовые и пссвдофазовые.
Фазовые частицы. Такие частицы представляют собой малые объемы макроскопической фазы вещества. Они могут находиться в трех агрегатных состояниях: в виде твердых частиц (микрокристаллов), небольших жидких капель или тонких пленок, газовых пузырей. Основные особенности дисперсных фазовых частиц определяются их большой удельной поверхностью
Пссвдофазовые (коллоидные) частицы. Большая группа дисперсных частиц имеет сложное и своеобразное строение, которое нельзя отнести к одному из трех агрегатных состояний. Дисперсные частицы с такой особой структурой называют псевдофазовы-ми, или коллоидными.
Яркий пример псевдофазовых дисперсных частиц — агрегаты, состоящие из нескольких десятков молекул поверхностно-активных веществ; такие агрегаты называют ми цел.лами (см. гл. 8). Другой важный класс псевдофазовых частиц — агрегаты из небольшого числа наночастиц
Классификация дисперсных частиц по химическому составу. Эта классификация, на наш взгляд, вполне очевидна (см. табл. 3.1) и не требует дополнительных пояснений.
3.2. Получение дисперсных частиц
Дисперсные частицы — это основные «строительные блоки» дисперсных систем. Подобно тому как в неорганической и органической химии одна из главных задач заключается в синтезе определенных веществ, фундаментальная и общая проблема коллоидной химии состоит в разработке методов получения дисперсных частин с необходимым набором химических и физических свойств (состав, агрегатное состояние, размер, форма, структура, поверхностные свойства). Поскольку дисперсные системы очень широко используются в разных областях промышленности, получение дисперсных частиц имеет большое прикладное значение.
Для получения дисперсных частиц, как правило, решают две принципиальные задачи.
1 Разработка методов, обеспечивающих получение дисперсных частиц нужного размера и формы. Дисперсные частицы чрезвычайно разнообразны по химическому составу, а требуемые размеры могут изменяться в очень широком интервале (от нескольких нанометров до десятков микрон). Поэтому в коллоидной химии разработан большой набор физических и химических методов получения дисперсных частиц, которые позволяют достаточно гибко регулировать их размер и форму.
2. Стабилизация дисперсных частиц, т.е. сохранение их размеров в течение достаточно длительного времени. Стабилизация относится к числу основных проблем коллоидной химии вообще. <Ь\ и да ментальное свойство большинства дисперсных систем — их термодинамическая неустойчивость, которая возникает из-за большою избытка свободной поверхностной энергии (см. гл. 4, 10). По ному проблема стабилизации особенно остро стоит для высо-кодисперсных частиц и наночастиц, у которых удельная поверх-пость очень велика. Без надчежашеи стабилизации неизбежно бу-iyi протекать процессы укрупнения дисперсных частиц, ведущие к «гибели» дисперсной системы. Современная коллоидная химия располагает несколькими эффективными методами, обеспечивающими надежную стабилизацию дисперсных систем с сохранением их свойств в течение очень длительного времени. Эти методы широко применяют на практике — для сохранения лекарственных препаратов, нишевых продуктов и в ряде других отраслей.
Методы получения дисперсных частиц принято разделять на те большие группы: диспергационные и конденсационные.
Диспергационные методы. Эти методы заключаются в измельчении крупных (макроскопических) образцов данного вещества ю частиц дисперсных размеров. При диспергировании химический состав и arpeiатное состояние вещества обычно не меняемся, все изменения затрагивают только размер частиц, а также их форму.
Диспергационные методы используют в основном для получения грубодисперсных частиц — от 1 мкм и выше. С помощью специальных приборов удается снизить границу размеров примерно до 0,1 мкм, но в промышленных процессах диспергирования обычно получают более крупные частицы.
Процессы диспергирования твердых тел, жидкостей и газов имеют существенные различия. Поэтому рассмотрим их отдельно дня каждого агрегатного состояния вещества.
Диспергирование твердых материалов. Для диспергирования твердых тел используют механические (дробление, истирание и т.п.), шектричсские (распыление в электрическом поле) методы, взрывы. )т процессы широко применяют в промышленности: в строительстве при производстве цементов и бетонов, в пищевой промышленности для помола зерна и других продуктов, в энергетике (измельчение угля), в производстве красок, наполнителей и т.д. Объемы этих производств весьма велики. Например, мировое производство порошков превышает 1 млрд т в год. Поэтому затраты на промышленные процессы диспергирования огромны. Совершенствование методов диспергирования может приносить весьма большие экономические результаты. Один из эффективных путей заключается в использовании поверхностных явлений (смачива
ния, адсорбции, элсктрокапиллярности) для снижения прочности и пластичности материалов.
Диспергирование играет большую роль во многих природных процессах, например при выветривании и эрозии горных пород. Еше один важный аспект — продукты измельчения, особенно высокодисперсныс фракции помолов, часто служат причиной сильных загрязнений воздушного бассейна, воды и почв. Поэтому диспергирование (техническое и природное) может наносить значительный ущерб окружающей среде; в связи с этим требуются специальные методы защиты природы.
Диспергирование твердых материалов проводят несколькими способами. Самый распространенный способ — механическое измельчение. Оно используется во многих технологических процессах. Для этой цели разработаны соответствующие установки (мельницы, дробильные аппараты и т.п.). В них измельчаемый материал подвергается сильным механическим нагрузкам (растяжению, сжатию, уларам). Нагрузки создают высокий уровень напряжений, под действием которых массивные образцы разрушаются на мелкие частицы. Процесс измельчения идет достаточно эффективно в случае хрупких материалов (многие минералы, керамика, стекло). Пластичные материалы (металлы) диспергировать гораздо труднее. Механические нагрузки вызывают вначале большую пластическую деформацию и только после этого происходит их разрушение. Поэтому механическое диспергирование пластичных материалов до достаточно мелких частиц требует очень больших энергетических затрат.
В лабораторных условиях диспергирование обычно проводят в шаровых мельницах. Измельчаемый материал помещают в цилиндрическую камеру, где находятся шары из материала с высокой твердостью (сталь, агат, фарфор). При вращении камеры шары наносят многократные удары по исходному материалу и постепенно измельчают его на все более мелкие частицы. Форма и размер отдельных частиц при таком способе помола могут сильно различаться.
Более эффективны вибрационные мельницы, в которых измельчаемый материал подвергается действию периодических механических колебаний, а также струйные и коллоидные мельницы, в которых дополнительное измельчение достигается благодаря взаимным ударам и истиранию дисперсных частиц.
Кроме механического диспергирования используют несколько более сложных вариантов. Ультразвуковой метод с частотой колебаний порядка 20 тыс. Гц применяют для диспергирования твер-тых матсригглов, помещенных в жидкость. Ультразвук создает в небольших локальных объемах жидкост и резкие чередования сжатия и расширения, что приводит к разрушению материалов и их диспергированию. Ультразвуковой метод удобен для получения
uicncpcHbix частиц металлов в органических жидкостях (органо-юлен), а также коллоидных растворов серы, гипса, гидрофильных полимеров в воде и т.д.
I ще один вариант — электрогидравлическос диспергирование, которое основано на действии разряда высокого напряжения в ни электрической жидкой среде. При электрическом методе диспергирования используют вольтову дугу между электродами из шепергируемого материала (металла), которые помешены в воду. Высокая температура дуги приводит к распылению металла в виде очень мелких частиц.
К числу диспергационных методов относится коллоидно-химический процесс, называемый пептизацией, или дезагрегацией. В этом случае исходным материалом служат связподисперсныс структуры — осадки (коагуляты), образующиеся при агрегировании и седиментации твердых частиц в коллоидных растворах (см. гл. 10).
Пептизацией называют распад структуры на образующие ее частицы при промывании осадка достаточным количеством жидкости или под действием специальных веществ (пептизаторов).
Диспергирование жидкостей. Для диспергирования жидкос тей и получения мелких капель в аэрозолях и эмульсиях используют преимущественно механические методы: встряхивание; быстрое перемешивание, сопровождаемое кавитационными разрывами; воздействие ультразвука. Применяют также распыление при течении жидкости через тонкие отверстия и при быстром движении струи. Неустойчивость струи с последующим дроблением на отдельные капли возникает при достаточно большой скорости (г), когда кинетическая энергия будет достаточно велика для преодоления сил сцепления (когезии) между молекулами жидкости. Количественно эти силы характеризуются поверхностным натяжением жидкости (о) на Гранине с соседней фазой, например газом. Условие разрыва (диспергирования) количественно определяет (безразмерный критерий — число Вебера:
We = p/^V/a, (3. 1)
iде р/— плотность жидкости; г — радиус струи.
Диспергирование происходит при достаточно больших значениях числа Вебера. Процесс диспергирования жидкости очень сложен и может определяться разными механизмами. Известно шесть механизмов этого процесса и каждому из них отвечает свое кри-гическое число Вебера.
Процессы диспергирования жидкостей имеют большое прикладное значение в энергетике — для обеспечения эффективного сжигания жидкого топлива; в медицине; при приготовлении различных эмульсионных систем (в частности, для дезинфекции).
Самопроизвольное диспергирование. При определенных устовиях макроскопическая фаза (твердая или жидкая) может распадаться на частицы или капли дисперсных размеров без совершения какой-либо внешней работы — механической, электрической и т п. Такой процесс называют самопроизвольным диспергированием (см. подразд. 4.4). Он наблюдается только для лиофильных систем, характерное свойство которых заключается в том, что межфазное натяжение на границе дисперсных частиц с дисперсионной средой достаточно мало.
Примером самопроизвольно образующихся дисперсных систем могут служить так называемые критические эмульсии. Они состоят из двух ограниченно смешивающихся жидкостей, например анилина и воды. При повышении температуры межфазное натяжение на границе анилин—вода уменьшается и вблизи критическом температуры оно становится очень малым (порядка 0,01 мДж/м2 и менее; см. подразд. 4.1). В этих условиях одна из жидкостей распадается на микрокапли размером несколько десятков нанометров и в результате происходит самопроизвольное образование эмульсии. Микрокапли имеют короткое время «жизни», т.е. непрерывно происходят два процесса: образование капель и их распад. Такое динамическое равновесие очень чувствительно к изменениям температуры, и критические эмульсии достаточно устойчивы только в очень узком интервале температуры.
Самопроизвольное диспергирование может происходить и с твердыми материалами, особенно материалами, имеющими слоистую структуру. Характерные примеры — «распускание» некоторых минералов (бентонитовой глины) в воде, графита — в жидком натрии.
Самопроизвольное диспергирование облегчается, если в систему вводят поверхностно-активные вещества, которые существенно уменьшают межфазное натяжение на границе частиц дисперсной фазы с дисперсионной средой (см гл. 6). Этог прием широко используют для получения эмульсий разного типа, например капель органической жидкости (ароматических или предельных углеводородов) в водной дисперсионной среде
Диспергирование газов. Для получения газовых пузырьков в жидкости применяют несколько вариантов диспергирования:
I) барботирование — прохождение газовой струи через жидкость с достаточно болы пой скоростью, при которой струя становится неусгоичивой и начинает дробиться на отдельные небольшие пузырьки;
2) одновременное течение жидкости и газа через устройства, которые смешивают эти потоки; при этом формируются газовые пузырьки, в качестве диспергирующих устройств используют пористые перегородки, узкие трубки, мембраны, сопла и т.п.
Радиус пузырька (гД. образующегося при прохождении газа через отверстие радиусом г0 со сравнительно небольшой скорос-н.ю, определяется уравнением
гЛ= [3r0O//(2Apg)|b ,
гас о — поверхностное натяжение жидкости; Др = (pz- р ) — ра шость плотностей жидкости (ру) и газа (р ); g — ускорение сво-Ьо.-пюго падения.
Гаким образом, размеры пузырьков в этих условиях не зависят от объемной скорости подачи газа (t>x, м3/с). Напротив, при больших скоростях газа пузырьки получаются тем крупнее, чем больше объемная скорость.
\ rh = 13^/(2ягЛ)1'/2,
где vb — скорое!ь всплывания пузырька, которая зависит от вязкости жидкости.
Конденсационные методы. Эти методы позволяют получать вы-сокодиспсрсные и ультрадисперсные частицы, поэтому их широко используют в нанотехнологиях. Рассмотрим физические и химические конденсационные методы.
Физические конденсационные методы. Основной принцип получения дисперсных частиц с использованием методов этой группы включается в выделении частиц новой фазы из пара (при кон-!.енсации) или из жидкости (при кристаллизации) Образование дисперсных частиц происходит в результате фазового перехода первого рода. Необходимое условие физической конденсации со-сгоит в отклонении исходной гомогенной системы (пара или жидкости) от состояния термодинамического равновесия, что достигается соответствующим изменением температуры и ш давления. Например, изменение температуры и давления используют для получения аэрозолей. Так, в системе, содержащей насыщенные пары воды, при понижении температуры образуется туман В камере Вильсона образование тумана происходит при адиабатическом расширении воздуха, насыщенного парами воды, что вызывает переохлаждение системы и образование капелек волы. Аналогичные процессы конденсации имеют место и в том случае, когда воздух содержит насыщенные нары таких веществ, как оксид фосфора(У), оксид цинка, сера, мышьяк и др. В этом случае при понижении температуры происходит образование твердых частиц и образуется дым.
Для получения металлических наночастин широко применяют двухстадииные физические методы. Первая стадия заключается в диспергировании металла до атомных размеров с образованием пара, вторая сталия состоит в последующей конденсации этих паров и образовании наночастиц. Существует несколько вариантов этой методики.
Метод молекулярных пучков. Исходное вещество помещаю! в вакуумную камеру с узким выходным отверстием (диафрагмой). После нагревания до достаточно высокой температуры вещество испаряется. Проходя через диафрагму, испарившиеся частицы образуют молекулярный пучок. Его направляют на подложку, на поверхности которой происходит конденсация пара с образованием дисперсных частиц или тонкого покрытия толщиной около 10 нм.
Аэрозольный метод. Металл испаряется в разреженной атмосфере инертного газа. При понижении температуры пары конденсируются и образуются дисперсные металлические частицы размером от 1—3 до 100 нм. Аэрозольный метод используют для получения наночастиц металлов (железо, кобальт, никель, медь, серебро, золото, алюминий) и их соединений (оксиды, нитриды, сульфиды).
Распылительная сушка. Этот метод является комби пированным. На первой стадии раствор данного вещества (например, соли) диспергируют на мелкие капли в потоке нагретого газа (воздуха). При умеренных температурах газа происходит испарение растворителя и продуктом процесса является порошок из дисперсных частиц соли. При достаточно высоких температурах наряду с испарением растворителя может произойти термическое разложение соли и исходным продуктом будет оксидный порошок.
К р и о х и м и ч е с к и й с и н г е з. Основная особенное! ь этого метода заключается в том, что сначала металл испаряют в потоке инертного газа (аргона или ксенона) при интенсивном нагреве, катодном распылении с помощью электроразрыва или другим способом. Далее происходит конденсация паров металла на поверхности подложки (субстрата) при низких и сверхнизких температурах в большом избытке (в тысячи раз) инертного газа. В результате на подложке образуются наночастицы. Очень низкие температуры в сочетании с сильным разбавлением препятствуют диффузии наночастиц по поверхности субстрата, поэтому они сохраняют свои малые размеры.
Плазменный метод. В инертной атмосфере (или с некоторой примесью водорода) создают электрическую (волыову) дугу. Анодом служит испаряемый материал. В струе пара, исходящей из анода, создают очень высокую температуру (до 7 000 К). За пределами дуги темпера гура газа гораздо ниже. В результате достигается очень высокое пересыщение металлического пара, что приводит к его конденсации в виде наночастиц.
3 о л ь — г с л ь - м с т о д. Это т метод применяют для выделения высокодисперсных твердых частиц или наночастиц из коллоидного раствора (золя). При определенных условиях дисперсные частицы слипаются друг с другом и образуют пространственную струк-
।spy — гель. При быстром высушивании геля получают порошок и । высокодисперсных частиц.
Весьма эффективен способ сверхкритической сушки. Он за-। помается в нагревании влажного геля в закрытом аппарате, так чтобы давление и температура превысили критические значения температуры и давления жидкости, которая находится в норах геля. В результате происходит превращение влажного геля в высо-копористый аэрогель с очень малыми (до 2 нм) порами. Таким образом, сверхкритическая сушка гелей позволяет получать газовые нанодисперсные частицы (норы).
Метод замены раствор ите л я. Этот метод широко используют для получения твердых частиц коллоидного раствора (юля). Метод заключается в том, что раствор данного вещества при постоянном перемешивании приливают к жидкости, в которой это вещество практически нерастворимо. Возникающее пересыщение приводит к образованию дисперсных частиц. Их размер регулируют пересыщением: чем оно больше, тем мельче частицы. Методом замены растворителя получают гидрозоли серы, фосфора, канифоли и других веществ, которые хорошо растворимы в органических жидкостях, но практически нерастворимы в воде.
Органические вещества, применяемые в методе замены раствори геля, должны обладать неограниченной растворимостью в воде и достаточно высоким давлением паров при комнатной температуре. По этим причинам обычно используют ацетон, а также спирты: этиловый, изопропиловый и др.
Метод замены растворителя используют для решения ряда современных задач нанотехнологии. Например, с его помощью в 1986 г. впервые было получено соединение, обладающее высоко-।емпературнои сверхпроводимостью.
Химические конденсационные методы. Химические методы заключаются в осуществлении реакций, приводящих к образованию дисперсных частиц определенного состава и размера.
Химические реакции с получением нерастворимых веществ. Этот метод широко применяют для получения коллоидных растворов (золей). Впервые его применил М. Фарадей (1857) для синтеза наночастиц коллоидного золота размером 5 — 20 нм Принцип метода заключается в том, что пересыщение, необходимое для образования зародыша новой фазы в рас-|ворс, может быть достигнуто в результате протекания химической реакции с образованием нерастворимою соединения. Это могут быть реакции окисления—восстановления, обмена, гидролиза. В качестве примера можно привести реакции образования юля золота:
НАиС14+ 2К2СО3 + Н2О = Au(OH)3i + 2СО2Т + 4KCI
2Au(OH)3 + К2СО3 = 2КАиО2 + ЗН2О + СО2?
2KAuO2 + ЗНСОН + K2CO3 = 2Aui + ЗНСООК + KFICOj + Н2О
Другие примеры — получение коллоидного раствора берлинской лазури и образование золя гидроксида железа:
4FeCl3 + 3K4|Fe(CN)J = Fe4[Fe(CN)6|3 + 12KCI
FeCl3 + 3H2O Fe(OH):4 + 3HC1
Размер получаемых частиц зависит от условий образования дисперсных систем, а именно — от соотношения скоростей одновременно протекающих процессов образования и роста зародышей новой фазы. Для получения высокодиспсрсных частиц необходимо преобладание скорости образования зародышей новой фазы над скоростью их роста. Эти условия могут быть реализованы, если концентрированный раствор одного из компонентов добавляют к разбавленному раствору другого компонента при сильном перемешивании. Таким образом, влияя на скорость образования и роста зародышей новой фазы, можно целенаправленно изменять дисперсность получаемой системы.
При малой растворимости соединений, получаемых в ходе реакции, могут быть достигнуты большие пересыщения и низкие скорости роста частиц, что приводит к образованию высокодиспсрсных систем. Примером влияния соотношения скорости образования и роста зародышей новой фазы на дисперсность образующегося золя может служить получение монодисперсного золя серы (метод Ла Мера). Так, при медленном разложении Na2S2O3 (в присутствии кислоты) концентрация растворенной серы постепенно увеличивается, достигая значения, при котором начинается образование новой фазы. Вследствие этого концентрация серы в растворе уменьшается и образование новых зародышей прекращается. При дальнейшем разложении тиосульфата вновь образующаяся сера идет на рост зародышей. Поскольку скорость роста зародышей велика и практически постоянна, критическая концентрация, необходимая для возникновения новых зародышей, не достигается и образование новых зародышей уже не происходит. Таким образом, первоначально сформировавшиеся зародыши новой фазы равномерно увеличиваются в размерах, приводя к образованию практически монодисперсного золя.
Г и д р о т е р м а л ь н ы и синто з. Это наиболее эффекли вн ый современный метод получения неорганических дисперсных частиц. Он заключается в проведении химических реакций в водных растворах при давлении р > I атм и температуре выше 100°C. В гидротермальных условиях, т.е. при повышенных значениях давления и температуры, в воде растворяется большинство неорганических соединений, в том числе силикаты, сульфиды, фосфаты, оксиды. Наряду с водой растворителем могут быть водные растворы кислот, оснований и солей.
Принцип гидротермального синтеза заключается в нагревании । меси исходных компонентов (А и В) до температуры, нсобходи-н»и л 1Я их растворения. После этого они реагируют друг с другом t образованием нового соединения С: А -ь В С. Гидротермаль-iii.ni синтез используюг для получения оксидов, сульфатов, ферритов и дру! их неорганических вешсств. Размеры дисперсных части могут изменяться в очень широких пределах, вплоть до нано-уровня Варьирование температуры и давления гидротермального рас । вора, pH и некоторых других факторов позволяет получать час!ины требуемой дисперсности (т.с. заданного размера).
Для получения органических дисперсных частиц используют 1НИЛОГИЧНЫЙ метод, ио вместо воды применяют органические рас творители при повышенных значениях температуры и давления (по сравнению с точкой кипения при атмосферном давлении). В случае органических веществ этот метод называют сольвотермальным синтезом.
Возможности гидротермального синтеза существенно расширяем создание микроволновых колебаний. Комбинированный гид-|кнсрмалыю-микроволновои метод обеспечивает высокую скорость naipeea, скорость реакции можно повысить на несколько порядков. Благодаря этим преимуществам гидротермально-микроволновой синтез применяют для получения дисперсных частиц керамических оксидов (в том числе используемых при получении мац-риалов с высокотемпературной сверхпроводимостью) и феррита.
Другой комбинированный вариант представляет гидротермально-электрохимический синтез, при котором происходит электро-III) металла в растворе щелочи. Этот метод весьма эффективен для получения тонких пленок солеи.
Важное направление гидротермального синтеза — получение нанотрубок. Например, метод был использован для получения нанотрубок диоксида титана диаметром около 8 нм длиной 100 нм и । порошков рутила и анатаза в растворе NaOH при температуре НО С.
Синтез дисперсных частиц в микрореакторах. Чля многих технологических процессов (например, в микроэлектронике) важно, чтобы дисперсные частицы имели узкое распределение по размерам, т.е. в пределе были бы моиодисперсными. Для решения этой задачи химический синтез проводят в ограниченном объеме — в условиях микрореактора.
Микрорсакторы, размер которых составляет несколько нано-метров, называют нанореакторами. Основные типы применяемых п.пюрсакторов представляю! дисперсные системы.
• м и кроэ м ул ьс ин;
• мицеллярные системы;
• высокопористые тела (например, цеолиты).
Поясним принцип получения дисперсных частиц в нанореакторах на примере микроэмульсиопной системы.
Микроэмулъсии — это жидкие прозрачные (или слегка опалесцирующие) тонкодисперсные термодинамически устойчивые системы. Микроэмульсии (капельные) бывают прямые и обратные. В прямых эмульсиях дисперсная фаза представлена микрокаплями «масла», в обратных — микрокаплями воды. Размер капель воды в зависимости от условий получения микроэмульсии и природы стабилизатора, в качестве которого используют различные ПАВ, может изменяться в широких пределах — от 1 —3 до 100 им Микрокаплю в данном случае можно рассматривать как микрореактор, в котором образуется новая фаза. Размер образующейся частицы ограничен размером микрокапли, а форма частицы повторяет форму капли. Таким способом были получены не только сферические, но и нитевидные наночасти цы металлов, оксидов металлов, труднорастворимых солей.
Метод получения ультрадисперсных частиц в мицеллярных системах называют также темплатным синтезом (англ, temple — шаблон, форма). Метод имеет следующие достоинства. В зависимости от концентрации раствора поверхностно-активного вещества мицеллы имеют разную форму: сферическую (при низких концентрациях), цилиндрическую (при высоких концентрациях). Подробно эти формы описаны в гл. 8. Благодаря этому темнлат-ный синтез позволяет получать частицы разной мерности: трехмерные частицы в сферических мицеллах, одномерные частицы (нановолокна). Другое достоинство метода — после завершения синтеза достаточно просто очистить частицы от «формы» — от поверхностно-активных веществ.
Синтез наночастиц в нанопорах также применяют достаточно широко благодаря возможности получения частиц очень малых размеров и узкому распределению пор по размерам. С помощью этого метода синтезированы, например, наночастицы золота размером около 2 нм в порах цеолита.
3.3. Строение дисперсных частиц и поверхностных слоев жидкостей и твердых тел
Дисперсные частицы. Строение дисперсных частиц очень разнообразно. Оно зависи । от многих физических и химических факторов. К ним относятся агрегатное состояние частиц, их химический состав, размер и форма частиц, температура, условия получения и др.
Дисперсные частицы в зависимости от их строения можно классифицировать на две большие группы: фазовые и псевдофазовые (или коллоидные) частицы (см. подразд. 3.1).
Фазовые частицы представляют собой малые объемы макро-»• омической фазы вещества. Поэтому в основном они имеют та-। нс же строение, как и макрофаза. Фазовые частицы существуют в р.ниых агрегатных состояниях: твердые частицы в аэрозолях и коллоидных растворах, капли в эмульсиях, газовые пузырьки в пенах, гонкие пленки жидкости, поры в пористых материалах.
( ।роение псевдофазовых частиц не имеет макроскопических ini.i югов. Оно присуще только данному типу дисперсных частиц. Характерные примеры псевдофазовых частиц — прямые и обратные мицеллы поверхностно-активных веществ (см. гл. 8), гигант-• кис кластеры металлов. Рассмотрим строение фазовых и нссвдо-Ф новых частиц более подробно.
Фазовые частицы. Строение фазовых дисперсных частиц во мно-н>м определяется их агрегатным состоянием. Поэтому целесообразно рассмотреть отдельно строение газовых, жидких и твердых частиц.
Газовая дисперсная фаз а. Газы не имеют структуры. По-• юму газовые пузырьки в пенах и газовых эмульсиях, поры и каналы в пористых средах и мембранах идентичны по строению макроскопической газовой фазе во всем интервале их размеров, вп лоть до наноуровня.
Жидкая дисперсная ф а за. Достаточно крупные капли в •мульсиях и аэрозолях, а также достаточно толстые жидкие пленки в эмульсиях, пенах и в зазорах между твердыми поверхностями имеют такую же структуру, как макроскопические объемы жидкости. Особенности структуры характерны для капель и пленок, таметр или толщина которых составляет менее 0,1 мкм. Эти изменения обусловлены главным образом следующими причинами:
I) в объектах малого размера происходит перекрывание поверхностных слоев, что приводит к возникновению расклинивающего пиления (см. нодразд. 4.3);
2) адгезия жидкостей к подложке (см. подразд. 5.3) приводи! к формированию тонкого слоя, который имеет более упорядоченную структуру, чем объемная жидкость; такой слои называют по-сраничнылц он играет очень важную роль в аэродинамике и гидро-щнамике.
Некоторые свойства жидкости в пограничном слое отличаются •и аналогичных свойств макроскопической жидкой фазы. Особенно сильно различается вязкость. Например, пленки смазочных масел толщиной в сотни нанометров имеют вязкость в десятки раз большую, чем толстые слои. Эти особенности вызваны тем, чю гонкие жидкие слои являются структурированными жидко-i1ями. В этом отношении они имеют некоторое сходство с жидкими кристаллами.
Твердая дисперсная фаза. Структурные различия меж-iy твердой дисперсной фазой и макроскопической фазой одною
и того же вещества наиболее значительны для твердых дисперсных частиц.
Структура твердых дисперсных частиц зависит от нескольких факторов: их химической природы, условий получения и размера (d) частиц. Отметим наиболее важные особенности. Сравнительно крупные частицы (d > 0,1 мкм) существуют как в аморфном, так и в кристаллическом состоянии. Возможно также смешанное строение- частично аморфное, частично кристаллическое. Аморфное состояние характерно, например, для частиц в свежеприготовленных коллоидных растворах диоксид:! титана, диоксида кремния, сульфида мышьяка, пептаоксида ванадия. Стечением времени аморфное состояние постепенно переходит в кристаллическое. Длительность периода, в течение которого частицы остаются аморфными, в зависимости от химического состава и температуры различается очень сильно. Например, при комнатной температуре частицы золота в коллоидных растворах становятся кристаллическими уже через несколько минут после приготовления золя. Для частиц золя кремниевой кислоты этот период очень продолжителен — около двух лет.
Строение улырадисперсных твердых частиц (наночастиц) весьма значительно отличается от строения макроскопических образцов того же вещества. Следует подчеркнуть, что за последние 10 — 15 лет в изучении строения ультрадиспсрсных частиц (наночастиц) достигнуты огромные успехи Это стало возможным благодаря применению экспериментальных методов с очень высокой разрешающей способностью: электронная микроскопия, сканирующая зондовая микроскопия, в том числе сканирующая туннельная и атомно-силовая, дифракционные методы (рентгенография, дифракция электронов), фотоэлектронная и рентгенофлюоресиснт-ная микроскопия.
Вместе с тем благодаря применению компьютеров сейчас можно рассчитывать структуры димеров, тримеров и нанодисперсных частиц методами квантовой химии.
Основная особенность строения улырадисперсных частиц заключается в том, что они часто имеют другую кристаллографическую структуру, чем макроскопические кристаллы тою же вещества. Теоретически показано, что для частиц, состоящих из 150— 300 атомов, наиболее стабильной является структура икосаэдра (симметрия 5-го порядка) вместо более простых кристаллических решеток, присущих макроскопическим фазам (кубической или гексагональной). Наименьший икосаэдр состоит из 13 атомов: 12 атомов на равных расстояниях от центрального атома. Такая структура определяется тем, что для наночастиц действует принцип максимального координационного числа, а нс условие минимума свободной энергии. Такие особые кристаллические структуры обнаружены экспериментально в металлических наночастицах.
Этот вывод подтвержден экспериментальными исследованиями структуры металлических частиц размером d < 10 нм. Например, с помощью электронного микроскопа были изучены нано-ч.н тины золота, платины и палладия, осажденные на углеродную пленку. Для частиц размером 4—15 нм преобладают декаэдриче-» не и икосаэдричсскис структуры Для более крупных частиц (15 — <1 нм) характерна структура гранецентрированной кубической решетки, свойственная массивным кристаллам золота Икосаэд-рическую структуру имеют также фуллерены из 60 атомов углерода Наночастицы платины и палладия могут иметь октаэдрическую । рнсталличсскую решетку.
При переходе от макроскопического образна к наночастицам p.i тмером менее 5 нм и кластерам отмечают также сокращения межатомных расстояний (до 15 %). Общая физическая причина изменения структуры состоит в том, что число атомов на поверхно-( hi частицы составляет большую долю от общего числа атомов, образующих данную частицу.
Пссвдофазовые частицы. Размеры псевдофазовых частиц, как правило, очень малы, т.е. пссвдофазовые частицы относятся к группе v 1ырадисперсных частиц (наночастиц). К числу псевдофазовых дисперсных частиц относится один из главных объектов коллоидной химии — мицеллы в растворах поверхностно-активных веществ. 11одробно их строение описано в гл. 8. Здесь отметим следующее.
Мицеллы представляют собой самоорганизованную структуру in нескольких десятков молекул или ионов ПАВ. В сферической мицелле в водном растворе молекулы или ионы ПАВ (например, ю (сцилсульфата натрия CL H25SO Na) располагаются вдоль ра-шуса мицеллы, а на поверхности мицеллы, обращенной к водной дисперсионной среде, находятся полярные группы. Таким образом, структуру псевдофазовых частиц этого типа нельзя счи-ыгь жидкой или твердой. Наиболее близкий аналог — жидкокри-с ылличсскос состояние. Другой пример относится к строению так и । «ывасмых «гигантских кластеров» палладия. Металлическое <ядро» 1икпх кластеров содержит 570 ± 30 атомов палладия. Оно имеет форму сферической частицы диаметром около 2,5 нм. Расстояния между отдельными атомами палладия в кластере соответствуют пкосаэдричсской структуре. Выше отмечалось, что именно этот нт структуры ультрадиспсрсных частиц предсказан теорией. Ре-1ИЫЮ гигантские кластеры имеют более сложную структуру. Они представляют набор примерно одинаковых по составу и строению еще более мелких частиц, каждая из которых близка по структуре к идеализированной частице.
Поверхностные слои жидкостей и твердых тел. В коллоидной химии определяющую роль играют поверхностные явления. Поэтому с । роение поверхностных слоев жидкостей и твердых тел имеет очень Н.1ЖНОС значение. Поверхностные слои очень тонки, их толщина
обычно имеет молекулярный размер. Из-за предельно малой толщины поверхностных слоев жидкостей и твердых тел для изучения их структуры необходимы методы высокого разрешения.
Поверхностные слои жидкостей и твердых тел по физическим и химическим свойствам значительно отличаются от объемных (макроскопических) фаз того же вещества.
Важные области химии, в которых большую роль играют особые свойства поверхности: гетерогенный катализ, электрохимия, физико-химическая механика.
Основная причина особых свойств поверхностных слоев жидкостей и твердых тел заключается в том, что они имеют иную структуру, чем внутренний объем этих фаз (для одного и того же вещества). Это положение можно считать следствием принципа Эренфеста о различиях физических законов для трехмерных (объемных) и двухмерных (плоских) объектов.
Рассмотрим вначале особенности строения поверхностного слоя твердого тела (кристалла) на границе с газом. Для каждого вещества характерен свой тип кристаллической решетки, для которого координационное число (Z() и расстояния между атомами (ионами) соответствуют минимальной энергии межатомных взаимодействий. Координационное число на поверхности кристалла (Zs) меньше Zv. Кроме того, атом на поверхности взаимодействует не только с атомами своей фазы (Zs—число этих связей), но и с атомами граничащей фазы В случае газа из-за ею малой плотности вклад этих взаимодействий мал. В результате атом на поверхности кристалла находится в другом энергетическом состоянии, чем во внутреннем обьеме. Некомпенсированные связи с ближайшими соседями делают уровень энергии каждою атома на поверхности более высоким, чем внутри фазы. В соответствии с принципом Ле Шателье поверхностный слой должен перестроиться таким образом, чтобы уменьшить произошедшие отклонения от исходного равновесною состояния. Возможны различные механизмы такой перестройки.
1. Релаксационный механизм. Этот механизм характерен для ионных кристаллов. Поверхностный (наружный) монослой, сохраняя структуру исходною кристалла, перемешается вглубь перпендикулярно поверхности кристалла. В результате вблизи поверхности происходит сжатие вещества и уменьшение расстояния между соседними атомными плоскостями.
Релаксационный механизм можно наблюдать экспериментально методом дифракции электронов. Возможность такого механизма подтверждена расчетами методом динамического моделирования (например, для кристаллов хлорида натрия). Такой сжатый слой очень тонок —1—2 атомных размера. На большей глубине кристаллическая решетка остается такой же, как во всем внутреннем объеме.
2 Перестройка кристаллической решетки поверхностного слоя Этот mi хапмзм присущ металлам и ковалентным кристаллам. Он пришли! к изменению взаимного расположения атомов и образованию решетки с другой симметрией. Этот механизм также измени-ci роение твердого тела, но только в очень тонкой зоне — нс ни тес нескольких монослоев. Поверхность металлов персстраива-IKH, как правило, незначительно. У полупроводников из-за направленных ковалентных связей происходит более существенная нерестройка поверхностной решетки или нескольких монослоев.
3. Образование наноразмерных дефектов кристаллической структуры (ступеней, уступов, вакансий).
Указанные механизмы перестройки поверхностного слоя от-но- я гея к однокомпонентным системам (чистым веществам). Для тух и многокомпонентных систем возможны значительные из-mi пения состава поверхности — из-за адсорбции компонентов с 1Ш <ким поверхностным натяжением (см. гл. 4). Например, в сплаве медь—никель поверхность сильно обогащена медью.
Структура поверхностного слоя жидкости также отличается от re объемной структуры. Несимметричные молекулы обычно принимают на поверхности жидкость—газ одинаковую ориентацию. Например, молекулы алканов, начиная с гексана, располагаются in риендикулярно поверхности жидкости. При такой ориентации достигается максимально плотная упаковка молекул в поверхно-i гном монослое Другое отличие — в поверхностном слое многих жидкостей (ртуть, галлий, спирты и алканы с достаточно длинным углеводородным радикалом) методами высокого разрешения обнаружена упорядоченная структура, состоящая из двумерных кристаллов.
В поверхностном слое жидкости на границе с газом непрерывно происходят несколько физико-химических процессов. Прежде шею это испарение. Для воды при комнатной температуре скорость испарения составляет примерно 0,02 моль/(см2 с). При этом • |Ч*днестатистическос время пребывания одной молекулы воды на поверхности с воздухом составляет около 10~7с. С такой же скоро-»гыо происходит конденсация водяных паров, благодаря чему поверхностный слой волы остается в равновесии с газовой фазой. Поверхностный слой жидкости ynaciBvei в массообмене нс толь-io с граничащей фазой (газом), но и с внутренним объемом этой ।идкости.
Другая важная особенность жидких поверхностей заключается и юм, что в атомном масштабе они не являются гладкими. На поверхности жидкости происходят периодические колебания, которые называют капиллярными волнами. Скорость их распространения (г) определяется уравнением Кельвина
v2 = 2ho//(pA), (3.2)
где G/ — поверхностное натяжение жидкости; р/ — плотность жидкости; X — длина капиллярной волны
3.4. Физические и химические свойства дисперсных частиц
Замечательная особенность высокодисперсных и ультрадиспер-сных частиц заключается в том, что их свойсгва зависят не только от химического состава образующего их вещества, но и от размера частиц. Зависимость свойств от размера частиц называют размерным или масштабным, эффектом.
Размерные эффекты, наблюдаемые в дисперсных системах, можно разделить на две большие группы.
1. Эффекты, связанные с кривизной поверхности жидкой или газовой дисперсной частицы (см. гл. 5):
а) зависимость поверхностного натяжения жидкости от радиуса капли или газового пузырька в жидкости;
б) зависимость давления насыщенного пара (рг) от радиуса и знака кривизны (выпуклая или вогнутая) поверхности жидкости на границе с газом;
в) зависимость капиллярного давления (рс) от радиуса поверхности жидкости.
2. Изменения физических и химических свойств, обусловленные малыми размерами дисперсных частиц; от размера частиц, в частности, зависят:
а) кристаллическая структура и степень симметрии кристаллической решетки (см. подразд. 3.3);
б) термодинамические параметры: теплоемкость, температура плавления (кристаллизации), температура Дебая;
в) механические свойства: прочность и пластичность;
г) магнитные и электрические свойства;
д) химические свойства, например каталитическая активность.
Важная особенность размерных эффектов в коллоидной химии заключается в следующем. Интервал размеров (d) частиц, в котором обнаруживаются эти эффекты, не является универсальным, т.е. одним и тем же для разных свойств. Напротив, для каждого конкретного свойства (физического или химического) характерен свой интервал, где размерные эффекты значительны. Например, капиллярные свойства проявляются достаточно ярко для капель, пузырьков и пленок при условии d < а (а — капиллярная длина; см. подразд. 2.2).
Размерные эффекты особенно значительны для ультрадиспер-сных систем, т.е. в интервале наноразмеров. В этом одна из причин большого значения наносистем в современной науке и высоких технологиях.
Iругая важная черта — некоторые размерные эффекты в нано-ч.н типах носят квантовый характер.
Рассмотрим несколько размерных эффектов, характерных для ni.li окодиснерсных частиц.
Гермодинамические свойства. Понижение температуры плавлении При уменьшении размеров твердых дисперсных частиц наблюдается постепенное снижение температуры плавления (Гпл) различных веществ. Для металлических частиц этот размерный •ффект проявляется достаточно сильно в интервале размеров d < 50 нм. Например, для частиц золота (рис. 3.1) разность температур ДГ = Гпп 0- становится заметной при d < 20 нм (Тпл0, /и,,/—температура плавления макроскопического и высокодиспер-1:11010 образца соответственно). В интервале d < 5 нм понижение температуры плавления составляет сотни градусов; при d = 2 нм имеем ДГ= 1 000 град. Значительное понижение температуры плавания обнаружено также для свинца, висмута, олова, индия.
Таким образом, размер наночастиц можно рассматривать как • пособразный аналог температуры Поэтому при использовании правила фаз для высокодисперсных систем нужно вводить дополнительную переменную величину (степень свободы).
Понижение температуры плавления высокодисперсных частиц 1Ы1ЛЯДНО объясняет следующая упрощенная модель процесса плавления. Предполагается, что при подводе теплоты кристалл начинает плавиться, когда амплитуда (60) колебаний атомов в кристаллической решетке будет соизмерима с межатомным расстоянием (Ь). Температура плавления макроскопических кристаллов соответствует в пой схеме условию 60~ Ь. Для атомов па поверхности кристалла амплитуда колебаний §(1> So. Уменьшение размеров частиц приводит к увеличению относительной доли поверхностных атомов в данной частице. При d= 3 нм эта доля дост игает уже 50 %. В результате условие > b достигается при более низкой температуре плавления.
Для теоретического описания размерных эффектов при плавлении используют следующие уравнения:
-----= ехр----------------
lL 3/? d/d{}-[
(3.3)
^пл.О / d{} — I
nn.d ^пл.О
2-Vo 1 1
3/? d/d. 1 I d/d, -1
(3.4)
(3-5)
»не 5пл>о, S^d — энтропия плавления соответственно макроскопического и высокодисперсного образца исследуемого вещества,
Рис. 3.1. Зависимость температуры плавления наподисперсных частиц золота от их размера (d); пунктиром показано изменение температуры плавления макроскопическою образца золота
Дж/(мольК); /7„лЛЬ — энтальпия плавления соответственно макроскопического и высокодисперсного образна, Дж/моль; R — универсальная газовая постоянная, 2J0 — минимальный размер частицы, при котором отсутствуют структурные различия между твердым телом и жидкостью; при размере J() различия энтальпии и энтропии макроскопической фазы и дисперсной частицы равны нулю.
Рассмотрим сравнительно большой интервал, когда d/d^» 1, т е. d> 5— 10 нм. При этом условии имеем
25„.i,.o 1 ~ о dp
3R d/d(}-\~ 3R ~d
Тогда при разложении экспоненты в уравнении (3.5) в ряд достаточно учесть только первые два члена ряда:
^п.ч.О __
^пл.о^пл.о 3Rd
(3.6)
Уравнение (3 6) имеет глубокий физический смысл. Оно показывает, что изменение термодинамического параметра (в данном случае — энтальпии плавления) происходит обратно пропорционально размеру дисперсной частицы d: ЛИ - l/d.
Такой характер размерных эффектов обусловлен тем, что величина \/d Q/У, т.е. представляет отношение площади поверхности частицы Q к ее объему К; таким образом, термодинамический параметр определяется дисперсностью частиц (см. уравнение (2.5)). Поэтому чем меньше размер тем больше доля поверхностных атомов в данной дисперсной частице. Этот принцип изменения термодинамических свойств дисперсных частиц (обратно пропорционально их размеру d или радиусу г) является в коллоидной химии достаточно общим. Пример такой связи дают закон Томсона (Кельвина) для давления насыщенного пара над искривленной поверхностью жидкости (см. подразд. 5.4) и уравнение Гиббса—Оствальда (см. гл. 5).
Характер зависимости температуры плавления дисперсных частиц от их размера существенно связан с агрегатным состоянием окружающей частицу дисперсионной среды. Приведенный выше эффект понижения температуры плавления при уменьшении размера частиц наблюдается для аэрозольных дисперсных систем, когда дисперсионной средой является газ. Во многих технологических процессах дисперсные частицы распределены в тонких порах другой твердой фазы. В таких системах наблюдаются как положи-1ельные, так и отрицательные размерные эффекты. Например, наночасишы индия в порах внутри железа плавятся при пониженной температуре (7]ul|j/ < Т^о). Внутри алюминиевой матрицы имеет место противоположный по знаку эффект — частицы индия плавятся при более высоких температурах, чем макроскопические образцы (Тпл^ > Г1и1Л)).
Переохлаждение капель. Размер дисперсных частиц оказывает сильное влияние не только на процесс плавления, но и на процесс кристаллизации. Высокодисперсные капли разных жидкостей могут длительное время сохраняться в жидком состоянии при сильном переохлаждении.
Для жидких металлов максимальное переохлаждение (Д7^тох) капель диаметром 2—100 мкм, например, составляет (град):
Ртуть 77 Золото 230
Галлий 76 Медь 236
Олово 118 Марганец 308
Свинец 80 Никель 309
Серебро 227 Платина 370
Значительное переохлаждение наблюдается и для капель дру-гих веществ, например воды, органических жидкостей и расплавов солей (град):
Вода.......................................... 39
Хлороформ .................................... 49
Бензол........................................ 70
Тстрахлормстан................................ 50
Октан......................................... 30
Декан ........................................ 29
Гексадекан.................................... 14
Фторид лития................................ 232
Фторид натрия ............................... 2X1
Хлорид натрия................................ 168
Бромид калия................................. 168
Бромид цезия ................................ 161
Теплоемкость. Влияние размера металлических наночастиц на 1сплоемкость проявляется особенно резко в области очень низких температур. Например, для кластеров палладия температурная зависимость теплоемкости отклоняется от соответствующей зави-
си мости с = f(7~) для макроскопических образцов. Эти различия растут по мере уменьшения числа атомов палладия в кластере. Таким образом, уменьшение размеров дисперсных частиц влияет на их тепловые свойства аналогично уменьшению температуры.
Механические свойства. Основное механическое свойство твердых тел — их прочность. Количественной характеристикой прочности материалов служит предельное напряжение (рсч Н/м2), при котором происходит разрыв образца (стержня) при одноосном растяжении. Предельное напряжение определяется уравнением
(3.7)
где/. — растягивающая сила, вызывающая разрыв; Q — плошадь поперечного сечения образца.
Для образцов с достаточно большим поперечным сечением (диаметр d > 0,1 мм) прочность зависит только от химической природы вещества Однако для гонких образцов, диаметр которых соответствует размеру дисперсных частиц, показан явный размерный эффект. Он заключается в том, что предельное напряжение увеличивается по мере уменьшения диаметра (J) стержней, волокон, частин и т.д. Этот масштабный эффект иллюстрирует зависимость прочности (рс) стеклянных нитей от их диаметра (J):
мкм............... 22,0 16,0 12,5 8,0 2,5
рс, Н/м2.............. 220 1070 1460 2 070 5 600
Приведенные данные показываю!, что прочность тонких нитей резко возрастает по мере уменьшения их диаметра. Размерную зависимость рс = fld) в этой области описывает уравнение
Рс = A-.min + PAA <3-8)
где р — константа, которая зависит от химической природы материала, Н/м.
Таким образом, как и для термодинамических свойств, имеет место обратно пропорциональная связь между свойством дисперсной час I и цы (нити) и ее размером.
Стекло но своей структуре аморфно, поэтому дисперсность нитей характеризуется их диаметром. Поликристаллическис материалы (металлы, сплавы, минералы) представляют собой конгломерат мелких кристаллов («зерен»), разделенных гонкими амор-физированными прослойками — границами зерен. Дисперсность таких материалов поэтому характеризуется нс только размером поперечного сечения, но и размером зерен. С уменьшением размера зерен прочность поликристаллов растет. Особенно сильное увеличение прочности наблюдается в интервале малых размеров зерен. Поэтому для производства материалов с высокой прочностью используют методы получения мелкозернистой структуры: внутренний диаметр зерен составляет от одного до нескольких
и пюмстров. Прочность нанотрубок примерно в 10 раз выше, чем и ни, хотя их пластичность в 6 раз меньше.
Основная физическая причина повышения прочности при yMi пыиснии диаметра образца и размера зерна заключается в сле-i\кинем Предельное напряжение зависит не только от химиче-। коп природы вешества, но и от рахчичных дефектов структуры. II шболее сильное влияние на механические свойства оказывают |.п называемые «линейные» дефекты — краевые и винтовые дис-!Окании. Вероятность нахождения в образце «опасною» дефекта, iHHCiciвенного за ею разрушение, тем ниже, чем меньше диа-мчр образца. Среднее расстояние между дислокация ми в кристаллах i оч гавляет примерно 100 нм Поэтому рост прочности характерен । I I образцов меньших размеров. Это положение легло в основу по (учения высокопрочных композитных материалов. Их основой । гужат очень тонкие («нитевидные») волокна, обладающие повышенной н роч н ос I ью.
Другое замечательное механическое свойство наноразмерных мвразцов некоторых веществ заключается в их большой пластичности. Установлено, что композитные медно-ниобиевые и медно-чромовые проволоки диаметром 15 — 20 нм при очень сильном охлаждении (в жидком гелии) деформируются пластично — пре-к* ibiioe удлинение достигает 10 %. Массивные образцы такою же состава в этих условиях очень хрупки. Другой пример высокой прочности наноразмерных образцов — углеродные нанотрубки.
Магнитные свойства. Для магнитных свойств размерный эффект проявляется в значительном понижении точки Кюри (Тс) — гемпературы перехода от ферромагнитного состояния к парамагнитному. Для наночастиц железа, кобальта, никеля размером менее 10 нм точка Кюри находится на сотни градусов ниже, чем для макроскопических образцов. Измерение намагниченности таких малых частиц сопряжено с большим разбросом данных (как и при непосредственном измерении температуры плавления). Поэтому пя определения точки Кюри наночастиц используют мессбауэровскую спектроскопию
Магнитные фазовые переходы происходят и в более крупных (2Q—50 нм) дисперсных частицах. Однако для осуществления таких переходов нужна дополнительная (по сравнению с наноча-i гицами) энергия. Источником этой энергии служат внутренние напряжения и дефекты, которые возникают при получении таких частиц с помощью топохимических реакций.
Mai нигные размерные эффекты проявляются очень ярко у кла-tлеров палладия. Макроскопические образцы палладия обладают парамагнетизмом и их магнитная восприимчивость почти не зависит от температуры вплоть до температуры жидкого гелия. Магнитная восприимчивое! ь гигантских кластеров палладия в несколько раз меньше, чем макроскопического металла, но они остаются
парамагнитными. При значительном уменьшении размеров кластера (до десятков атомов палладия) они становятся диамагнитными. Размер дисперсных частиц влияет также на коэрцитивное ногте (//f, А/м). Эта величина характеризует размагничивающее поле, при котором остаточная намагниченность образна становится равной нулю. Коэрцитивное ноле представляет важную характеристику ферромагнитных материалов. При Нс< 100 А/м материалы считаются магнитомягкими, при Нс > 100 А/м — магнитожесткими.
Коэрцитивное поле нанокластеров (d < 4 нм) железа почти нулевое. Такие низкие значения обусловлены тепловыми колебаниями. Для малых частиц их энергия достаточна, чтобы разрушить упорядоченную ориентацию магнитных доменов и перевести кристалл в парамагнитное состояние. При комнатной температуре для железг! значение коэрцитивного поля максимально для кристаллов размером 20—25 нм. Поэтому нанокристалл и чес кие ферромагнетики можно использовать для получения запоминающих устройств с большой памятью.
Еше одно важное магнитное свойство высокодисперспых частиц — низкая магнитная анизотропия. В поликристаллических материалах минимальная магнитная анизотропия феррома! нети ков достигается при размерах зерен 10 — 20 нм. Потери при перемагничивании таких наноматсриалов малы.
Очень перспективно использование нанодисперсных намагниченных частиц диаметром около 10 нм для приготовления феррома! нитных жидкостей — коллоидных растворов, в которых дисперсной фазой являются наномагнитные частицы, а дисперсионной средой — жидкость, например вода или керосин. При наложении внешнего магнитного поля наночастицы начинают двигаться и включают в движение окружающую жидкость. Перспектива промышленного использования этого эффекта весьма велика (например, для охлаждения мощных трансформаторов в электротехнике и для магнитною обогащения руд).
Каталитические свойства. Высокодисперсные и особенно на-нодисперсные твердые частицы металлов и оксидов металлов имеют высокую каталитическую активность, что позволяет проводить различные химические реакции при сравнительно невысоких температурах и давлениях. Приведем несколько примеров, иллюстрирующих каталитические свойства высокодиспсрсных частиц.
1. Наночастицы золота размером 3 — 5 нм обладают высокоспецифической каталитической активностью. Ее появление сопряжено с переходом кристаллической структуры золота от гране центрированной кубической в более крупных частицах к икосаэдри-ческой структуре наночастиц Важнейшие характеристики этих нанокатализаторов (активность, избирательность, темпера!ура)
ишисят от материала подложки, на которую их наносят. Кроме uno, очень сильно влияют даже следы влаги. Наноразмсрныс частим золота эффективно катализируют окисление монооксида y i тсрода при низких (до -70 °C) температурах. Вместе с тем они обладают очень высокой избирательностью при восстановлении оксидов азота при комнатной температуре, если частицы золота । i.iнесены на поверхность оксида алюминия.
2. Высокодиспсрсный катализатор на основе пестехиометри-чсского оксида церия значительно снижает температуру восстановления оксида cepbi(IV) под действием монооксида углерода. Кроме того, этот катализатор обладает более высокой стойкостью, чем обычные катализаторы на основе оксида церия, по отношению к отравлению участвующими в реакции потоками водяною пара и диоксида углерода.
3. Повышенную каталитическую активность имеют нс только уш.традиспсрсныс частицы, но и тонкие слои катализатора, если их толщина составляет несколько нанометров. Такая высокая ак-гннность обнаружена, например, для нанослосв MoSi2 в реакции । идродссульфуризации. Максимальная каталитическая активность достигается при высокой степени упорядоченности наноструктуры кристаллического MoSi2. Избирательность этого катализатора регулируется числом монослоев MoSi2. Каталитическую избира-ц*льность можно также регулировать изменением структуры на-i «счастии MoSi2 в одном (по толщине слоя) или двух измерениях, что открывает весьма перспективное направление в гетерогенном катализе органических реакций.
Высокой каталитической активностью обладают предельно малые наночастицы — кластеры. Например, гигантские кластеры палладия, состоящие из нескольких сотен (около 600) атомов, проявляют высокую избирательную каталитическую активность во многих органических реакциях (окисление кислородом п нитробензолом, карбонилирование, гидрирование, ацетилирование карбонильных соединений, изомеризация олефинов и др.). При этом эффективный катализ идет в сравнительно «мягких» условиях — при температурах и давлениях, гораздо меньших, чем при использовании обычных промышленных катали-пн оров.
Высокая каталитическая активность металлических кластеров связана с их строением. Как правило, их поверхность имеет достаточно сложную структуру; на ней есть и очень острые выступы, и плоские участки. Локальные положительные заряды в кластере распределяются по-разному. В результате на поверхности кластерного катиона существуют сильные льюисовские центры (в вершинах полиэдрической структуры), более слабые льюисовские центры (на ребрах), положительно заряженные атомы металла с наименьшей электрофильностью (на гранях).
Биологические свойства. Высокая химическая активность нано-частиц позволяет использовать их в ряде биологических и медицинских процессов. Одно из актуальных направлений — защита от некоторых видов биологического оружия. Например, термостои кие возбудители сибирской язвы эффективно уничтожаются н воздухе при комнатной температуре распылением наночастиц оксида магния. Этот эффект тем сильнее, чем меньше размер наночастиц.
3.5. Дисперсное состояние вещества
Анализ строения дисперсных частиц, их физических и химических свойств показывает, что высокодиспсрсные и особенно ультрадисперспые частицы (наночастины) принципиально отличаются от более крупных частим и гем более от макроскопических фаз одних и тех же веществ. Поэтому для характеристики таких малых объектов в коллоидной химии используют представление о дисперсном состоянии вещества.
Понятие об универсальности дисперсного состояния веществ было введено в начале XX в. (П. П. Веймарн) и относился к числу фундаментальных принципов коллоидной химии. Исходно этоз принцип опирался на два положения:
1) любое вещество может находиться в дисперсном состоянии, т.е. в виде дисперсных частиц (твердых, жидких, газообразных);
2) дисперсные частицы имеют очень большую удельную поверхность, поэтому свойства дисперсных систем определяются в основном поверхностными явлениями на границе раздела дисперсной фазы с дисперсионной средой.
С современной точки зрения дисперсное состояние вещества характеризуется еще несколькими важными особенностями, отличающими его от макроскопических образцов того же вещества; перечислим наиболее принципиальные из них.
1 Сложная внутренняя организация ультрадиспсрсных частиц, которая существенно отличается от структуры крупных частиц (в том числе и по типу структуры кристаллической решетки и по степени ее симметрии). Особая структура присуща и пссвдофазо-вым наночастицам (кластерам и мицеллам поверхностно-активных веществ).
2. Сильная зависимое! ь различных свойств от размера частиц. К числу этих свойств относятся физические (тепловые, механические, электрические, магнитные) и химические (каталитическая активность, растворимость) свойства. В ряде случаев свойства наночастиц яатяются квантовыми, т.е. наночастицы можно рассматривать как своеобразные макромолекулы с квантовыми свойствами.
3. Наночастицы состоят из небольшого числа (К)2—I О4) атомов. поэтому неизбежны значительные флуктуации и большой । гппсгический разброс различных свойств.
I. Для описания процессов и реакций, происходящих с участи-• м нточастиц, наряду с масштабом «размер» очень важен и мас-III i6 «время». Для одних процессов он измеряется фемтосекундами (10 15 с); для других (например, для химической кинетики) он может быть гораздо продолжительнее (от 10 2 до 106 с).
5. Поверхность твердых высокодисперсных частиц обычно имеет сложную структуру, которая весьма далека от идеальной ।палкой плоскости. Эта поверхность имеет сложный рельеф с и* |рыми выступами и впадинами (также наноразмерными). Свой-i та таких поверхностей описывает фрактальная геометрия.
На свойства жидких и газовых высокодисперсных частиц (капе ль и пузырьков) сильное влияние оказывает друюи геометрический фактор — очень большая кривизна поверхности, обратно пропорциональная радиусу кривизны. Одно из следствий большой кривизны — очень высокое капиллярное давление.
Таким образом, дисперсное состояние вещества хараклсризу-г.тся геометрическими, физическими и химическими особенно-। 1ями по сравнению с обычным aipciaTHbiM состоянием — твер-и IM, жидким или газообразным.
3.6. Измерение размеров дисперсных частиц
Дисперсные системы очень разнообразны по физическим, химическим и биологическим свойствам В зависимости от типа дисперсной системы наиболее ярко могул проявляться оптические, > 1сктрические, магнитные и другие явления. Вместе с тем не-сколько характеристик являются общими для всех дисперсных систем. К ним относятся в первую очередь размер дисперсных ча-• гпц и концентрация частиц.
Интервал размеров дисперсных частиц очень велик — примерно от I нм до 10 мкм, поэтому для их измерения применяют раз-1ичные методы. Выбор метода определяется главным образом в иютвстствии с классификацией дисперсных частиц но их размерам (см. подразд. 3.1).
Рассмотрим основные методы, применяемые в коллоидной химии для измерения размеров дисперсных частиц.
Седиментационный анализ. Для измерения размеров 1рубодис-псрсных частиц (более 1 мкм) широко используют седиментационный анализ. Метод основан на зависимости скорости оседания (v) hi размера частиц. В соответствии с уравнением (2.9) по результант измерения скорости оседания можно рассчитать радиус (/•) сферической частицы:
9п
----v
J/2
=
(3.9)
где К — константа седиментации:
рдрг, ее значение определяется свойствами дисперсионной среды и дисперсной фазы (вязкость, плотность), а также ускорением силы тяжести.
Уравнение (3.9) применимо для определения размеров частиц одинакового размера, т.е. для монодиснерсных систем.
Для полидиспсрсных систем (суспензий), содержащих частицы разных размеров, измеряют зависимость веса (Р) осадка на чаше весов, помешенной в сосуд с суспензией, от времени оседания (/). В монодисперсной суспензии все частицы оседают с одинаковой скоростью v = H/ts (Н — высота верхнего слоя; rv — время окончания оседания, которое на кривой оседания соответствует началу плато).
В полидисперсной суспензии частицы оседают с разной скоростью. Поэтому сначала заканчивается седиментация самых крупных частиц радиусом rindV Время их оседания (7min) определяется по моменту отклонения кривой накопления от начального линейного участка (рис 3.2). Время /тах (начало плато) соответствует полному оседанию всех частиц. Отсюда находят скорости оседания самых крупных частиц радиусом и самых мелких частиц радиусом rmin.
Далее по уравнению (3.9) рассчитывают максимальный (rmax) и минимальный (rmin) радиусы. Промежуточные rmin < г< г|11ах размеры находят фафическим дифференцированием кривой накопления для нахождения производных dP/dr в разные моменты времени. Затем используют уравнение материального баланса
dP dr ’
(3.10)
где Q — вес частиц, которые полностью осели к моменту времени t после начала седимешации.
Произведение z(dP/d/) даст вес частиц размером г < г,, осевших ко времени t.
Отношение Q/Ртлх (Ртак — общий вес осадка) дает интегральную кривую распределения частиц но размерам Q/Pmax =f(r). Дифференцирование интегральной кривой даст дифференциальное распределение частиц по размерам (рис. 3.3). При наличии одного максимума на дифференциальной кривой дисперсные системы
Гис 3 2. Зависимость веса (Р) осев-। их дисперсных частиц от времени (/) (кривая накопления)
Рис. 3.3. Кривые распределения дисперсных частиц по размерам на основе данных седиментационного анализа суспензий:
/— интегральная кривая (?/Р||1йх =/(г); 2 — дифференциальная кривая Af/dr
к шссифи пируют как моподисперсные; если таких пиков два, их пазывают бимодалыiыми.
Седиментационный анализ применим для дисперсных частиц и интервале размеров примерно от 0,1 до 20 — 30 мкм. Более крупные частицы оседают слишком быелро и для них не выполняется условие ламинарности Re < 1. Размер частиц менее 0,1 мкм не удается установить этим методом из-за наступления ссдимента-нионно-диффузионного равновесия, связанного с броуновским шижением и диффузией коллоидных частиц (см подразд. 2.5).
Измерение размеров дисперсных часгиц в центрифугах. Для определения размеров дисперсных частиц используют центрифуги, t осуд с дисперсной системой (суспензией) помещают в центрифугу, чтобы заменить ускорение силы тяжести g центробежным ускорением g* = оку (со — угловая частота вращения центрифуги; г — расстояние от частицы до оси вращения). В современных уль-грацентрифугах ускорение g* достигает очень больших значений (вплоть до 10б£). При таких больших ускорениях седиментационное равновесие резко смещается в сторону частиц малых размеров. Использование ультранентрифу! позволяет определять размеры наподиспсрсных частиц и макромолекул, в том числе и макромо-юкул белков. Значение этого метода для химии, биологии и медицины очень велико, его создание было отмечено Нобелевской премией (Т.Сведберг, 1926).
Метод рассеяния света. Коллоидные растворы (золи), размер шспсрсных частиц (d) которых меньше длины волны падающего < вета (X), обладают способностью рассеивать свет. Интенсивность света (/), рассеянного дисперсными частицами, определяется законом Рэлея:
I 24jr3vr2f
7“ = —p— 4.w ' (3.11)
/0 Л ynd + 2ло
где /0 — интенсивность света, падающего на кювету с коллоидным раствором; v — концентрация частиц; V — объем дисперсной частицы; nd, д0 — показатель преломления дисперсионной среды и вещества дисперсной фазы соответственно.
Закон Рэлея (3.11) справедлив для сферических частиц, не обладающих электропроводностью. Поэтому для измерения размера частиц металлических золей приходится вводить определенные поправки.
В монодисперсной системе концентрация частиц v (м-3) связана с массовой концентрацией С (кг/м3) вещества дисперсной фазы в дисперсионной среде соотношением С = vKpj (pj — плотность дисперсной фазы). Уравнение Рэлея (3.11) преобразуется к следующему виду:
/ _ 24л3 С Л и2 - n02
/0 л4 prf [«(/ + 2ло?
(3.12)
При постоянной концентрации С дисперсной фазы интенсивность светорассеяния пропорциональна объему дисперсной частицы И, следовательно, светорассеяние растеч пропорционально г3. Поэтому измерение светорассеяния (нефелометрия} — очень чувствительный метод определения размеров дисперсных частиц.
Другое применение метода нефелометрии к дисперсным системам заключается в определении концентрации частиц v. Для этого необходимо знать объем дисперсных частиц данного коллоидного раствора. Обычно для измерения концентрации частиц используют сравнительный метод, т.е. сопоставляют светорассеяние изучаемого коллоидного раствора (/) и стандартного золя (/(|) с известной концентрацией частиц.
Фотон-корреляционная спектроскопия. На измерении интенсивности рассеянною света в коллоидных системах основан широко используемый в настоящее время метод динамического светорассеяния, или фотон-корреляционной спектроскопии. Метод используют для измерения размеров коллоидных частиц от 10-9 до 10-6 м. Лазерный луч рассеивается на частицах дисперсной фазы. Поскольку частицы в таких системах участвуют в броуновском движении, наблюдается уширение спектра рассеянной световой волны Полуширина (Г) рассеянного спектра описывается выражением
где D — коэффициент диффузии частиц; g — значение волнового вектора исходного излучения.
Для монолисперсной системы, состоящей из сферических не н ыимодействующих частиц, коэффициент диффузии связан с hi |родинамичсским радиусом частиц (7?) уравнением Стокса— >и и штейна
нс kR — константа Больцмана; р — вязкость растворителя; Т — к-мнература.
Существуют два взаимодополняющих метода, позволяющих определить величину Г путем анализа светового потока: анализ частотного состава фототока и метод автокорреляционной функции путем счета фотонов.
Для полидиснсрсной системы используют два способа обра-(нпки экспериментальной автокорреляционной кривой: метод кумулянтов и метод регуляризации. Первый метод позволяет най-III средний радиус частиц и ширину распределения. Второй ме-к»д, основанный на аппроксимации экспериментальной кривой суммой экспонент, позволяет найти вклад разных фракций в ин-к’нсивность рассеянного света и распределеште частиц по размерам. Сравнение результатов, полученных этими двумя методами, позволяет судить о правомерности описания совокупности размеров частиц в реальной системе с усредненным радиусом
Таким образом, несомненным достоинством метода фотон-коррелянионной спектроскопии является возможность определения радиуса частиц дисперсной фазы непосредственно в диспер-I ной системе.
Контрольные вопросы
I. Охарактеризуйте две основные группы методов получения дисперсных частиц.
2. Укажите основные способы диспергирования твердых материалов.
3. В чем заключается процесс самопроизвольного диспергирования?
4. Приведите примеры физических и химических конденсационных методов получения дисперсных частиц.
5. Объясните принципы синтеза дисперсных частиц в микрореакторах. Назовите основные типы коллоидных микрореакторов
6 В чем заключается главная особенность строения ультрадиспер-< пых частиц9 Охарактеризуйте принцип максимального координационною числа?
7. Приведите примеры размерных (масштабных) эффектов, харак-1грпых для высокодисперсных и ультрадисперсных частиц.
8. Объясните принцип седиментационного анализа дисперсных си-итем.
9. Объясните принцип измерения размера дисперсных частиц мето-юм светорассеяния
ГЛАВА 4
ТЕРМОДИНАМИКА ПОВЕРХНОСТНЫХ ЯВЛЕНИЙ И ДИСПЕРСНЫХ СИСТЕМ
Важнейшее общее свойство всех дисперсных систем заключается в том, что удельная поверхность на границе дисперсной фазы с дисперсионной средой очень велика Поэтому многие свойства дисперсных систем сильно зависят от явлении, которые происходят па поверхности дисперсных частиц. Поверхностные явления разделяют на три большие группы: физические, химические и электроповерхностные.
В случае физических поверхностных явлении определяющую роль играет поверхностное натяжение — избыточная (по сравнению с внутренним объемом жидкости или твердо! о тела) свободная энергия поверхности, отнесенная к единице площади (см. подразд. 4.1). Эта избыточная энергия вызывает в дисперсных системах процессы, которые ведут к уменьшению поверхности.
Яркий пример таких процессов — самопроизвольное слияние (коалесценция) капель в эмульсиях и аэрозолях и газовых пузырьков в пенах (см. подразд. 10.3). Другой пример — в условиях невесомости капли и газовые пузыри приобретают сферическую форму, которая соответствует минимуму их поверхности. Это положение было подтверждено экспериментально в одном из первых космических полетов. Большую группу физических поверхностных явлении составляют капиллярные явления. Для них характерно сильное влияние кривизны жидкой или твердой поверхности (см. гл. 5).
Химические поверхностные явления сопровождаются значительными изменениями состава и структуры тонких поверхностных слоев жидкостей и твердых тел. В коллоидной химии наиболее важное поверхностное явление — адсорбция — самопроизвольное концентрирование определенных поверхностно-активных веществ в поверхностном слое (см. гл. 6)
Основными характеристиками электроповерхностныхявлений (см. гл. 9) служат электрический заряд и электрический потенциал поверхности раздела дисперсной фазы и дисперсионной среды.
Наиболее общее описание поверхностных явлений и дисперсных систем дает термодинамика. Для коллоидной химии особенно важны следующие термодинамические проблемы: термодинамические параметры поверхности раздела фаз; условия термодинамического равновесия поверхности раздела фаз; методы рсгу-
'шрования поверхностных свойств; устойчивость дисперсных смени, методы их стабилизации.
4.1. Поверхностное натяжение
Поверхностное натяжение относится к числу наиболее важных коллоидно-химических свойств. Поверхностное натяжение — основная термодинамическая характеристика поверхности раздела между разными фазами, образующими дисперсные системы, между жидкостью и газом, жидкостью и твердым телом, между ивумя жидкостями, между твердым телом и газом
Рассмотрим самый простои случаи — поверхностное натяжение жидкости на границе с газом. Поясним для этого случая фи-тчсский смысл поверхностного натяжения
4.1.1. Определение поверхностного натяжения
Поверхностное натяжение имеет двойной физический смысл — шергетический (термодинамический) и силовой (механический). Оба эти понятия может пояснить простой опыт (рис. 4.1). На прополочной рамке EDCF помещается подвижная перекладина АВ ।диной /, легко скользящая по рамке. Между перекладиной и верхней стороной рамки DC находится тонкая пленка жидкости, например мыльного раствора (мыло обеспечивает стабильность пленки). Приложим к подвижной перекладине АВ направленную вниз силу /; ее создаст груз G. Под действием силы f перекладина АВ переместится на малое расстояние dv и займет положение А'В'. ( ила/произведет при этом работу d W=/dx, если процесс проис-ходит при постоянной температуре (изотермически), то эта рабо-ia затрачивается только на увеличение поверхности пленки Прирост площади составит dQ = 2/dx; коэффициент 2 учитывает, что пленка имеет две стороны. Таким образом, на увеличение единицы площади пленки затрачивается работа (о, Дж/м2):
а = (41а)
Из уравнения (4.1а) следует энергетическое (термодинамическое) определение поверхностное натяжение — это удельная работа увеличения поверхности при ее растяжении при условии постоянства темнеразуры.
При выполнении этого условия работа о, затраченная на обра-ювание единицы новой поверхности, полностью переходит в свободную энергию поверхности, также рассчитанную на единицу поверхности. При условии постоянства объема пленки новая по-
Рис. 4.1. Схема опыта, поясняющего физический смысл поверхностном» натяжения жидкости (EDCF — про водочная рамка; АВ — подвижная перекладина; ADCB и A'DCB' — мыль ная пленка до и после нагружения соответственно, G — груз, создающий силу/; о — сила поверхностного натяжения)
верхность создается благодаря переходу некоторой массы жидкости из ее внутреннего объема на поверхность. Поэтому используют еще одно определение поверхностною натяжения, как термодинамическою параметра поверхности жидкости: поверхностное натяжение равно избы точной удельной свободной энергии (энергии Гельмгольца) единицы
площади поверхности жидкости
! при определенной температуре.
\-------------------q Термин «избыточность» под-
черкивав!, что энергия Гельм-гол ьца молекул, находящихся на поверхности жидкости больше, чем молекул в се внутреннем объеме. Это различие играет важнейшую роль в термодинамике поверхностных явлений (см. подразд. 4.2).
Рассмотренный выше опыт выявляет еще один физический смысл поверхностного натяжения. Определим условия силового механического равновесия перекладины ЛЯ при приложении к пей силы /(см. рис. 4.1). Такое равновесие может обеспечить сила, направленная в противоположную сторону. Обозначим эту силу (в расчете на единицу длины перекладины АВ) о [Н/м]. Равновесию отвечает условие
° = <416)
коэффициент 2 учитывает, что пленка мыльного раствора на рамке имеет две стороны.
Уравнение (4.16) представляет силовое (механическое) определение: поверхностное натяжение — это сила, действующая на единицу длины линии, которая ограничивает поверхность жидкости (линия АВ на рис. 4.1).
( ила поверхностного натяжения направлена тангенциально поверхности раздела фаз и стремится уменьшить эту поверхность. По лому на рис. 4.1 векторы, изображающие поверхностное натя-i.viihc, направлены в сторону пленки (сжимают ее).
Исторически (середина XVIII в.) вначале было введено силовое определение поверхностного натяжения. Сам термин «поверхностное натяжение» (surface tension) указывает именно на силовой смысл этой величины. Энергетическое определение поверхностного натяжения было введено в термодинамику поверхностных явлений в конце XIX в. Дпя жидкостей численные значения поверхностного натяжения при энергетическом и силовом опре-1елепии одинаковы.
В термодинамике удобнее использовать энергетические, а не < иловые величины. Поэтому в дальнейшем, если нет специальных указаний, нами используется энергетическое (термодинамическое) определение поверхностного натяжения о [Дж/м2].
Поверхностное натяжение — термодинамический параметр, характеризующий поверхность раздела между двумя фазами, не-11ВИСИМО от их агрегатного состояния. Например, на границе магнитных доменов происходит скачок магнитной индукции и для |лких объектов используют термин «магнитное поверхностное натяжение». Реальность магнитного натяжения ярко демонстрируют следующие данные. При определенных условиях в ферромагнитных веществах образуются цилиндрические домены, расположенные параллелы ю друг другу. Устойчивая форма таких доменов обеспечивается натяжением искривленной цилиндрической поверхности — по аналогии с устойчивостью пузырьков газа в жидкости под действием капиллярного давления. Эта аналогия объясняет принятое в англоязычной литературе название цилиндрических доменов — «магнитный пузырек».
Поверхности раздела существуют также в поликристаллах между областями («зернами») с одинаковым химическим составом, но 1Х13НОЙ ориентацией кристаллографических граней по отношению ipyr к другу. Такие поверхности называют границами зерен, а соответствующее натяжение — межзеренной энергией.
4.1.2. Факторы, влияющие на поверхностное натяжение жидкостей
В настоящее время разработано несколько методов, позволяющих с достаточной точностью измерять поверхностное натяжение разных жидкостей в широком интервале давлении и температур от О К (жидкий гелий) до нескольких тысяч градусов (см. подразд. 5.6). На основе большого числа экспериментальных данных установлено, что поверхностное натяжение жидкостей определя-
Qsf, мДж/м
Рис. 4.2 Зависимость поверхностно го натяжения (а) щелочных метал лов при температуре их плавления от удельной теплоты поверхностно го слоя (Qv)
ют в основном следующие факторы: химическая природа вещества, температура, природа граничащих фаз, наличие примесей, заряд поверхности, кривизна поверхности жидкости.
Химическая природа вещества. Самое низкое поверхностное натяжение имеют сжиженные инертные газы. Поверхностное натяжение органических веществ и воды составляет 20— 100 мДж/м2. Самые высокие значения поверхностного натяжения жидкостей (~ I03 мДж/м2) характерны для металлов с высокой температурой плавления. В случае алмаза (самого прочного и тугоплавкого вещества) о> I О4 мДж/м2. Отмеченная закономерность подтверждается тем, что для многих веществ (например, жидких металлов) поверхностное натяжение вблизи температуры плавления пропорционально теплоте плавления Q , (рис. 4.2)*.
Поверхностное натяжение (мДж/м2) жидкостей на границе с газом для некоторых веществ при температуре 298 К составляет:
Гелий*................. 0,2
Азот**................. 8,3
Гексан................ 17,9
Этиловый спирт........22.1
Тетрахлор метан........25,0
Сероуглерод........... 31,5
Бензол................ 28,2
Анилин................ 43,2
Вода.................. 71,95
Ртуть................. 473,5
* При темпера гуре 3 К.
* * При температуре 80 К
Температура. Для всех индивидуальных (однокомпонентных) жидкостей поверхностное натяжение на границе с газовой фазой уменьшается при повышении температуры (Г), т.е. выполняется соотношение
do/d Т < 0.
(4.2)
* На рис. 4.2 приведена зависимость поверхностного натяжения щелочных металлов при температуре их плавления от удельной теплоты поверхностного слоя Qs/, которая связана с теплотой плавления металла следующим соотношением: (?„,рб (р — плотность; 5 — толщина поверхностного слоя (пара-
метр кристаллической решетки))
Для многих однокомпонентных жидкостей зависимость о =f(T) Г» hi 1ка к линейной (рис. 4.3).
Для растворов (особенно растворов поверхностно-активных веществ) температурная зависимость поверхностного натяжения носит более сложный характер. Экстраполяция линейной зависимости о -f(T) вплоть до о = 0 определяет (по Д. И. Менделееву) критическую температуру (Те) данного вещества. При этой температуре двухфазная система жидкость—пар перестает суще-с пювать и становится однофазной, т.е. при Т > Tt поверхность раздела фаз исчезает.
При линейном характере зависимости о = /(7 ) производная if = do/dF для данного вещества имеет постоянное значение, ее на зывают температурным коэффициентом поверхностного натяжения жидкости.
Для многих веществ температурные коэффициенты поверх! ю-». гного натяжения составляют примерно от -О, I до -0,2 мДж/(м2- К).
Для описания температурной зависимости поверхностного на-гяжения предложено несколько эмпирических уравнений. Одно из них — уравнение Ван-дер-Ваальса'.
Q^^{\-T/Tcy\ (4.3)
где Со — поверхностное натяжение данной жидкости вблизи тем
пературы плавления.
Рис. 4.3. Зависимость поверхностного натяжения (о) жидкостей от температуры (Г):
/— вода; 2 — глицерин; 3 — нитробензол: 4 — гексан (пересечение пунктирной линии с осью абсцисс определяет критическую температуру (/’.) гексана)
Межфазное натяжение (<У|2) на границе воды с жидкими органическими веществами
Вещество 7, К о12, мДж/м2
Бензол 292 34,4
Анилин 299 4,8
Хлороформ 291 33,8
Крезол 291 3,9
Для органических веществ показатель степени п = 1,23; для жидких металлов 1. Таким образом, уравнение (4.3) показывает, что с ростом температуры поверхностное натяжение убывает несколько быстрее, чем по линейному закону.
Зависимость поверхностного натяжения жидкостей от температуры (см. уравнение (4.3)) играет важную роль в термодинамике поверхностных явлений (см. подразд. 4.2).
Природа граничащих фаз. Термин «поверхностное натяжение» используют прежде всего для характеристики границы раздела жидкость — газ и твердое тело — газ. Для поверхностей раздела конденсированных фаз (между двумя жидкостями и на границе твердое тело—жидкость) часто применяют другой термин — межфазное натяжение. Его физический смысл такой же, как у поверхностного натяжения: межфазное натяжение равно работе образования единицы площади поверхности раздела конденсированных фаз*.
Поверхностное натяжение (о!2) на границе двух жидкостей I и 2 сильно зависит от их химической природы (табл. 4.1).
Данные табл. 4.1 показывают, что значения поверхностного натяжения на границе воды с органическими жидкостями могут различаться примерно в 10 раз. Поверхностное натяжение на границе двух жидкостей зависит от поверхностных натяжений жидкостей 1 и 2 на границе с газом. При определенных условиях (ограниченная растворимость жидкостей 1 и 2 друг в друге, отсутствие химических реакции между ними, низкое давление насыщенного пара) поверхностное натяжение на границе двух жидкостей подчиняется правилу Антонова (1907):
О|2 = О|-сг2. (4.4)
Сильное влияние природы граничной фазы на межфазное натяжение характерно также для твердых тел, оно проявляется очень ярко при их смачивании (см. подразд. 5.2).
* В данной книге используется, как правило, один термин — «поверхностное натяжение» для характеристики поверхности раздела любых фаз.
Таким образом, информация о поверхностном (межфазном) натяжении любого вещества должна обязательно содержать сведении о граничащей с ним фазе.
Наличие примесей. Во многих случаях поверхностное натяжение очень чувствительно к наличию примесей. Например, поверхностное натяжение ртути реагирует на присутствие цезия в концентрации всего 10_,() моль/л. В связи с этим измерения поверхно-ццого натяжения могут использоваться в качестве чувствительного тсст-мезода для определения чистоты вещества. Вместе с тем • ют пример показывает, что при экспериментальном изучении поверхностных явлений требования к химической чистоте должны выполняться очень строго.
Заряд поверхности. Зависимое! ь поверхностного натяжения от плотности электрического заряда поверхности (pv, Кл/м2) называют электрокапиллярным эффектом (см. полразд. 9.7). Этот эффект обусловлен тем, что взаимное отталкивание одноименных зарядов уменьшает работу, необходимую для увеличения площади данной поверхности. Следовательно, при увеличении заряда поверхности поверхностное натяжение будет уменьшаться. Эту зависимость описывает уравнение Липпмана:
-do/dtp = р5, (4.5)
где (р— электрический потенциал поверхности (см. подразд. 9.7).
Максимальное значение поверхностного натяжения достигается при отсутствии поверхностною заряда (р5= 0); это так называемая точка нулевого заряда. В соответствии с уравнением (4.5) снижение поверхностного натяжения (До) может быть весьма значительным. Например, при катодной или анодной поляризации ртути на границе с растворами электролитов эти изменения составляют ~ 10' мДж/м2, т.е. более 10 % по сравнению с натяжением ртути в точке нулевого заряда (см. подразд. 9.7).
Кривизна поверхности жидкости. Поверхностное натяжение очень малых капель и газовых пузырей (на границе с жидкостью), радиус (г) которых находится в области наноразмсров, меньше, чем поверхностное натяжение более крупных объектов. Это еще один пример размерных эффектов — зависимости свойств высокодисперсных частиц от их размера (см. подразд. 3.4). Приближенно зависимость поверхностного натяжения малых капель и газовых пузырей (ог) от их радиуса (г) описывает уравнение Толмена.
где S — размер, соизмеримый с толщиной поверхностного слоя (примерно несколько нанометров); о — поверхностное натяжение крупных капель и пузырей.
В области очень малых радиусов кривизны используют уровне ние Русанова:
<зг = Аг, (4.7)
где К — коэффициент пропорциональности, зависящий от температуры и состава граничащих фаз.
Уравнение (4.7) имеет следующий физический смысл: при предельном уменьшении радиуса кривизны (г э 0) поверхностное натяжение обращается в нуль.
4.1.3. Молекулярная природа поверхностного натяжения жидкостей
Возникновение поверхностного натяжения вызвано различиями строения и свойств очень тонкого (в пределе — мономолеку-лярного) поверхностного слоя и объемной фазы вещества. На молекулярном уровне указанные различия связаны в первую очередь со следующим обстоятельством. У молекул (атомов, ионов) на поверхности жидкости (твердого тела) число ближайших («своих») соседей (Zs) неизбежно меньше, чем во внутреннем объеме, т.е. координационное число для частиц на поверхности Zs< Zv (Zy— координационное число в объеме; см. подразд. 3.3). Современные экспериментальные методы подтверждают, что структура поверхностного слоя жидкостей действительно существенно отличается от структуры объемных фаз тех же веществ.
Из этой модели следует, что поверхностное натяжение тем больше, чем больше рашость координационных чисел (Zv-Zr) и чем больше энергия межмолекулярных связей данного вещества. Чем сильнее эти взаимодействия, тем больше работа, которую нужно совершить для увеличения поверхности; соответственно гем больше поверхностное натяжение. Эту закономерность иллюстрируют данные, приведенные в подразд. 4.1.2.
Силы взаимодействия, определяющие поверхностное натяжение, могут иметь разную природу: дисперсионные, водородные, диполь-дипольные, диполь-индуцированные, электростатические взаимодействия. В первом приближении поверхностное натяжение представляет сумму вкладов каждого вида взаимодействий. Поскольку для любых веществ существуют дисперсионные взаимодействия, целесообразно выделить их вклад (oj и вклад всех остальных взаимодействий (о,) Тогда в соответствии с положением об аддитивности этих вкладов поверхностное натяжение жидкости определяет уравнение
0 = ^+0,. (4.8)
Для многих веществ наиболее важен вклад полярных взаимодействий (ар) (табл. 4.2).
Дисперсионная (g^) и полярная (ср) составляющие поверхностного натяжения (а) разных жидкостей при температуре 20 С, мДж/м2
Жидкость (5 orf
1ексан 18,4 18.4 0
1-Бром нафталин 44,4 44,4 0
1 - М ети л нафтал и н 36,4 36,4 0
Ди подметан 50,8 50,8 0
} (и MCI илсул ьфоксид 44,0 36,0 8,0
}1 и метил си л о кса н 19.0 16,0 3,0
Эгилснгликоль 48,0 29,0 19,0
Глицерин 64,0 34,0 30,0
Вода 72,8 21,8 51 0
Влияние дисперсионных взаимодействии проявляется особенно наглядно в случае неполярных жидкостей. Например, поверхностное натяжение предельных углеводородов растет с увеличением длины цепи, поскольку дисперсионные взаимодействия между молекулами при этом возрастают.
Поверхностное натяжение (о) предельных углеводородов при гемнературе ОТ в зависимости от числа атомов углерода (п) в молекуле изменяется следующим образом:
п............... 5 6 7 8 9 10
о, мДж/м2..... 18,25 20,00 22,10 23.52 24,72 26,46
п................. 12 14 15 18 20
а, мДж/м2....... 27,12 28,30 29,00 30,00 31,00
Представление поверхностного натяжения в виде суммы вкладов разных взаимодействии используют также для расчета поверхностного натяжения (о13) на границе двух жидкостей 1 и 2, например предельною углеводорода (1) и воды (2). Рассмотрим в соответствии с этим подходом вклад в межфазное натяжение со стороны неполярной жидкости 1. Молекулы поверхностного слоя этой жидкости участвую!.
1) во взаимодействии со своей объемной фазой; этому взаимодействию соответствует поверхностное натяжение О| на границе с газом;
2) с молекулами поверхностного слоя жидкости 2 (воды); это взаимодействие создастся дисперсионными силами, оно равно среднегеометрическому значению дисперсионных взаимодействии
углеводород—углеводород и углеводород—вода, т.е. составляв (ou<T2rf)l/2, где Си, а2</ — дисперсионные составляющие поверхностных натяжений углеводорода и воды на границе с газом.
Указанные взаимодействия направлены в противоположные стороны. Поэтому общий вклад этих двух взаимодействий со стороны поверхностного слоя жидкости 1 (of) в межфазное натяжение о12 равен
а? =О|-(Ок/О^)1 • (4.9а)
Аналогичные рассуждения позволяют определить вклад в межфазное натяжение о12 со стороны поверхностного слоя жидкости 2 (воды):
с*2 = о2 - (olrfo2d )'/2 • (4-96)
Межфазное натяжение о)2 равно сумме указанных двух вкладов, отсюда следует уравнение Фоукса (1964):
Oi2 = ai + n2-2(olt/o2rf)/2. (4.10)
Уравнение (4.10) применимо в основном для расчета поверхностных натяжений при контакте воды с органическими веществами, молекулы которых содержат полярные группы.
Количественную связь поверхностного натяжения с химической природой жидкости характеризует также параметр, который называют парахором.
Парахор (Р) рассчитывают по уравнению
Р = Mo'/VAp ~ Vm^/ph (4.11)
где Л/ — молекулярная масса жидкости; Др — разность плогно-стей жидкости и газа; р, — плотность жидкости; Ут = М/р, — молярный объем жидкости.
Для многих веществ удовлетворительно выполняется следующее правило: парахор молекулы складывается из парахоров образующих ее атомов. Эту связь впервые отметил А.Эйнштейн (1901).
4.2. Термодинамика поверхности раздела фаз в однокомпонентных системах
Поверхности раздела фаз являются сложными и своеобразными объектами. Главная их особенность определяется тем, что на очень малых расстояниях вглубь от межфазной поверхности (в пределах нескольких молекулярных размеров) физические и химические свойства вещества существенно отличаются от аналогич-
пых свойств макроскопической (объемной) фазы того же веще-<. ша (см. подразд. 3.4). С физической и химическом точки зрения поверхность раздела фаз не является чисто геометрическим объск-гом (плоскостью «нулевой» толщины). Реально она представляет юн кий поверхностный слой малой, но конечной толщины 6 > О (см. подразд. 3.3).
Рассмотрим наиболее простой случай — плоскую поверхность раздела фаз для однокомпонентной системы, состоящей из двух фаз А и В. Пример такой системы — индивидуальная жидкость (фаза А) на границе с насыщенным паром (фаза В) (рис. 4.4). Гонкий слой EFGH в непосредственной близости от «геометрической» поверхности CD состоит из двух слоев малой толщины. Один (CEFD) из них — это поверхностный слои фазы А (жидко-ст) толщиной бЛ; другой (CHGD) — тонкий поверхностный слой фазы В (насыщенного пара) толщиной бв.
Область EFGH толщиной 5 = бЛ+ 8В, разделяющую фазы А и В. называют поверхностным слоем, или поверхностью разрыва. Поверхностный слой но строению неоднороден, так как состоит из двух разных областей CEFD и CHGD, примыкающих к двум разным фазам А и В. Положение границ ЕЕ и HG поверхностного слоя (। е. расстояния 8Л и 8В от плоскости CD) несколько условно Обычно принимают, ч то за пределами границ EF и HG какое-либо свой-ciBo вещества (чаше всего — плотность) отличается от такого же свойства макроскопической фазы на очень малую величину, а в пределах поверхностного слоя это различие свойств весьма существенно. Определенный таким способом размер 5 = \ЕН называют эффективной толщиной поверхностного слоя Необходимо подчеркнуть, что эффективная топщина слоя зависит от выбора конкретного свойства (плотности, вязкости, электропроводности п т п.).
Молекулярная толщина поверхностного стоя характерна для (смпсратур, отличных от критической температуры ТС(Т « Тс). При приближении к критической температуре (Г—> Гс) наряду с резким уменьшением поверхностного натяжения (о —» 0) резко растет эффективная толщина поверхностного слоя (5 —> °°). Физиче-
Рмс. 4.4. Схема поверхностного слоя EFGH на границе раздела фаз жидкость—пар в однокомпонептной системе
ски это означает превращение двухфазной системы в однофазную.
Поверхностный слой представляет собой открытую термоди намическую систему, так как через границы EF и HG он можо обмениваться с примыкающими объемными фазами А и В веществом и энергией. Поэтому поверхностный слой — неавтономная фаза; он не может существовать самостоятельно, в «отрыве» от граничащих объемных фаз
Наряду с понятием поверхностный слой (поверхноезь разрыва) в термодинамике поверхностных явлений Гиббса использую! понятие разделяющей поверхности, которая находится внутри поверхностного слоя (поверхности разрыва), воспроизводит ее форму и располагается параллельно ей. Точное положение разделяющей поверхности (координата по нормали к поверхности) не является строго определенным.
Представление о поверхностном слое как о фазовом слое малой толщины имеет принципиальное значение. Такая модель позволяет характеризовать поверхность раздела фаз с помощью тех же термодинамических параметров, что используют для описания объемных (трехмерных) фаз.
Одним из методов, описывающих термодинамические параметры повсрхносшого слоя, является метод избыточных величин, предложенный Дж. Гиббсом (1878). Согласно Дж Гиббсу избыточная поверхностная энергия описывается выражением
AU - аАО + рАд/Ая + TAS'. (4.12)
Отнесем эти изменения к единице площади образованной поверхности, получим следующие удельные величины:
Е = А (//ДО,
Т|=Д5/АО, (4.13)
Га = АлДАЯ/УД.
Параметр е (Дж/м ) представляет полную поверхностную энергию. Параметр ц (Дж/(м К)) называют поверхностной энтропией', он характеризует изменение степени порядка при образовании поверхностною слоя. Параметр ГДмоль/м* ) называют автоадсорбцией', он характеризует изменение плотное! и вещества в поверхностном слое. Термин «автоадсорбция» подчеркивает, что данная с и с те м а од! юком по не нтн а
С учетом выражении (4.13) уравнение (4.12) принимает вид
е = о + цГо + 7г|. (4.14)
Уравнение (4.14) — фундаментальное термодинамическое урав нение Гиббса для равновесною состояния плоской поверхности раздела фаз в однокомпонентной системе. Оно представляет тео-
peiическую основу для описания поверхностных и капиллярных явлений, адсорбции и электроповср.хностных явлений (см. гл. 5, 9). Уравнение Гиббса (4.14) относится к числу основных законов коллоидной химии.
Подчеркнем принципиальное положение термодинамики поверхностных явлений. Термодинамические параметры е, гр Гд — но не абсолюшые значения внутренней энергии, энтропии и массы поверхностною слоя, а избыточные величины по сравнению С соответствующими параметрами кон тактирующих объемных фаз.
Рассмотрим несколько следствий уравнения Гиббса (4.14). Одно из них касается понятия свободная поверхностная энергия По определению свободная энергия объемных фаз (энергия Гельм-гольца) равна Г= U- IS (U — внутренняя энергия, Дж/моль; 5 — штроп ня, Дж/(моль • К)).
По аналогии из уравнения (4.14) следует соотношение
С = <з + рГд, (4.15)
г.е. свободная поверхностная энергия складывается из механической работы образования самой поверхности и химической рабо-гы по изменению плотности вещества в поверхностном слое.
Таким образом, образование поверхности раздела фаз — это механохимическии процесс. Он играет ключевую роль при получении коллоидных частиц методами диспергирования (измельчения), в физико-химической механике, а также в мсханохимии — научном направлении, которое изучает реакции, возникающие при превращении механической энергии в химическую
Если рассматривают двухфазную однокомпонентную систему, а положение разделяющей поверхности выбирают таким образом, чтобы Г = 0, то уравнение (4.14) принимает вид
е = о + Тх\. (4.16)
Многочисленные экспериментальные данные показывают, что поверхностное натяжение уменьшается с температурой. Для нсас-социированных жидкостей эта зависимость в широком темпера-ivpiiOM интервале описывается уравнением
о = о0-а(7’- То), (4.17)
«де Со — по верхи ос I ное натяжение жидкости при температуре Т{); о. — эмпирическая постоянная.
Сопоставление уравнений (4.16) и (4 17) показывает, что г - о + а Г, т. е. ос = т|. Учитывая, что do/d Т = -т], получаем уравнение
r do 5
(4.18)
Это соотношение представляет собой аналог гермодинамичс ского уравнения Гиббса—Гельмгольца для объемных фаз.
В заключение рассмотрим влияние кривизны поверхности раздела фаз на ее термодинамическое равновесие. Уравнение Гиббса (4.14) было выведено для случая плоской поверхности. Однако в коллоидной химии очень часто встречаются искривленные повср хности (капли в аэрозолях и эмульсиях, газовые пузырьки в пенах). Поэтому вопрос о влиянии кривизны поверхности на термо динамику поверхностных явлений имеет для коллоидной химии очень важное значение.
Экспериментальные исследования показывают, что приближение плоской поверхности выполняется достаточно хорошо, если радиус кривизны г поверхности дисперсной частицы существен но больше толщины поверхностного слоя б. Это условие справедливо для высокодисперсных частиц размером <7—100 нм. Для уль традисперсных частиц (паиочастиц) влияние кривизны поверхностного слоя может быть существенным. Одно из наиболее важных следствий — поверхностное натяжение зависит от радиуса кривизны (см. подразд. 4.1).
4.3. Расклинивающее давление
В коллоидной химии большое значение имеют тонкие жидкие пленки, толщина которых составляет 101— 103 нм. Именно это1 интервал размеров характерен для дисперсного состояния веще ства
В зависимости от типа дисперсной системы жидкие пленки разделяют на четыре группы.
1. Пенные пленки — пленки жидкости между газовыми ячейками в пенах.
2. Эмульсионные пленки — пленки жидкой дисперсионной сре ды между каплями дисперсной фазы в эмульсиях.
3. Жидкие пленки между твердыми частицами. Такие пленки разделяют дисперсные частицы после их прилипания друг к другу с образованием крупных рыхлых агрегатов в результате коагуляции (см. подразд. 10.2).
4. Смачивающие пленки на поверхности твердых тел при полном смачивании (см. подразд. 5 2).
Пенные и эмульсионные пленки и пленки в коагуляционных агрегатах называют симметричными, так как они разделяют две фазы или дисперсные частицы, находящиеся в одинаковом агрегатном состоянии. Смачивающие пленки несимметричные.
Термодинамические свойства тонких жидких пленок имени существенные отличия по сравнению с поверхностным слоем большого объема той же жидкости (см. подразд. 4.2). Эти особые свои-
i ma количественно характеризует дополнительный термодинамический параметр — расклинивающее давление (П, Па).
Причины возникновения расклинивающего давления в тонких пленках и его физико-химический смысл рассмотрим на примере плоской пенной пленки (рис. 4.5).
В отличие от поверхностного слоя жидкости, имеющего одну поверхность раздела с граничащей фазой (газом), тонкая пленка имеет две поверхности. К каждой из них примыкает свой поверхностный слой (АВ В'А' и CDD'C') толщиной 8. Между ними находится внутренняя («объемная») часть пленки A'B'D'C\ все свойства жидкости которой такие же, как у макроскопической жид-кои фазы. Таким образом, тонкая пленка имеет структуру «сэндвича» — между двумя одинаковыми поверхностными слоями находится внутренняя прослойка макроскопической жидкой фазы.
В зависимости от толщины пленки (Л) и толщины поверхностного слоя (8) различают два типа жидких пленок.
I. Толстые пленки', h > 28. Термодинамические свойства толстых пленок определяются вкладами внутреннего слоя толщиной (Л- 28) и двух поверхностных слоев толщиной 8. При условии аддитивности расклинивающее давление отсутствует (П = 0).
2. Тонкие пленки', h < 28. Тонкие пленки фактически не имеют самостоятельной внутренней области, так как поверхностные слои \ВВ’А' и CDD'C' перекрывают друг друга. Поэтому свойства внутренней области пленки определяются взаимодействием поверхностных слоев в зоне их перекрывания. Пусть Д/у— дополнительная энергия пленки, равная работе, которую нужно затратить для сближения поверхностных слоев и их перекрывания. Очевидно, чем тоньше пленка, тем больше степень перекрывания поверхностных слоев и соответственно тем больше энергия их взаимодей-стия Д/7.
Рис. 4.5. Возникновение расклинивающего давления в плоской пенной пленке:
а — толстая пленка (Л > 2S); б — тонкая пленка с перекрыванием поверхностных слоев (Л < 25)
Таким образом, из-за малой тол шины h термодинамические свойства топких пленок отличаются от аналогичных свойств (сво бодной энергии Гельмгольца) той же жидкости. Зависимость ДЛ = /(Л) — это еще один пример размерного (масштабного) эффекта, характерного .тля дисперсного состояния вещества (см. подразд. 3.4, 3.5). Для увеличения поверхности тонкой пенной плен ки на единицу площади при постоянной температуре нужно затратить работу /у= 2о + Д/у(о — поверхностное натяжение жидко сти; коэффициент 2 учитывает, что пленка имеет две стороны). Величину Ff по физическому смыслу можно рассматривать как энергетическое определение поверхностного натяжения тонкой пленки.
В качестве термодинамического параметра гонких пленок вместо энергии /у обычно используют силовой параметр — расклинивающее давление П (Па), которое определяется уравнением
d/z
(4-19)
(4.20)
В англоязычной научной литературе расклинивающее давление обозначают «disjoining pressure». Положительное расклинивающее давление препятствует утоньшению пленки. В некоторых случаях (например, если в зазоре между пластинами находится не жидкость, а газ) расклинивающее давление отрицательно (П < 0); это означает, что оно способствует утончению пленки.
Физический смысл расклинивающего давления заключается в следующем. Расклинивающее давление — это избыточное давление в тонкой пленке по сравнению с гидростатическим давлением (pG) в большом обьеме жидкости. Отсюда следует определение раскл и и и ваюпieго давлен ия
П(Л) = pt-p^ где pf — давление в тонкой пленке.
Это определение расклинивающего давления можно пояснить на следующем примере. Стеклянную пластинку помещаю! в жидкость, которая хорошо смачивает данный материал и вместе с тем нс растворяет его. В жидкость вводят небольшой газовый пузырек радиусом г<а (а — капиллярная длина). При всплывании пузырек приближается к пластинке, его верхняя часть деформируется и становится плоской. В результате между пузырьком и твердой поверхностью формируется плоская пленка толщиной Л, которая сообщается с «объемной» жидкостью вокруг пузырька, а его радиус становится равен г0. В этом случае разность давлений (Pf~P(d, входящая в уравнение (4.20), равна капиллярному давлению рс = 2в/Го (о — поверхностное натяжение жидкости), определение капиллярного давления и его расчет приведены в
подразд. 5.1. В этом случае расклинивающее давление составляет II = 2о/г0. Это соотношение позволяет найти экспериментально шачение расклинивающего давления. Данный опыт также позволяет определить зависимость расклинивающего давления от голщины жидкой пленки //. т.е. изотерму расклинивающего дав-1С11ня П = f(h).
Рассмотренный опыт иллюстрирует важную роль расклинивающего давления в тонких жидких пленках в коллоидной химии. Например, устойчивая пленка препятствует непосредственному контакту газового пузырька с твердой поверхностью, что играет важную роль в различных технологических процессах. Например, идя обогащения полезных ископаемых часто используют флотацию. Этот процесс основан на том, что дисперсные частицы руды помещают в воду и в образовавшуюся суспензию вводят газовые пузырьки. Они оттесняют волу с поверхности несмачиваемых части (пленка между пузырьком и частицей неустойчива), прилипают к частицам и обеспечивают их всплывание. Напротив, дру-ше частицы, которые хорошо смачиваются водой (обычно это частицы «пустой» породы) не могут вступить с газовыми пузырьками в непосредственный контакт, так как расклинивающее дав-1снис положительно и препятствует утончению пленки. Поэтому шкие частицы под действием силы тяжести оседают. В результате флотации достигается весьма эффективное отделение полезных ископаемых от пустой породы.
Расклинивающее давление открыл Б В. Дерягин (1935). Он ввел и само понятие расклинивающего давления как термодинамического параметра тонких жидких пленок. Возникновение расклинивающего давления связано с поверхностными силами разной природы (электрическими, магнитными, молекулярными), которые действуют в тонком поверхностном слое между двумя граничащими фазами. В коллоидной химии особенно важны электрические (электростатическое отталкивание) и молекулярные силы
Электрические силы определяются поверхностными зарядами и строением двойного электрического слоя возле поверхности раздела фаз. Эти вопросы рассматриваются в гл. 9.
Молекулярные силы делятся на несколько составляющих. Основную роль играют дисперсионные взаимодействия.
Энергия дисперсионных взаимодействий (U h Дж/м ) в случае одинаковых по составу параллельных пластинеразделенных тонкой прослойкой вакуума толщиной /?, определяется уравнением Гамакера (1936):
Ud
4i 12л/г '
(4-21)
। де — константа Гамакера (Дж), которая зависит от числа молекул п в единице объема взаимодействующих фаз, поляризус-
мости а молекул и энергии ионизации Z?v0 (Л — постоянная План ка; v0 — характеристическая частота колебаний зарядов):
Ли = — K/zvoa2«2.
(4.22)
Более сложным считается случай, когда твердые пластины на ходится в жидкости. Тогда для характеристики энергии межмолс кулярного взаимодействия используют сложную константу Гама'-кера (Л12), которая определяется уравнением:
2
^12 - ~ -/Л) ,
(4.23)
где Ль А2 — константы Гамакера для твердой фазы и жидкости соответственно.
Для веществ, в которых определяющий вклад в поверхностное натяжение вносит дисперсионное взаимодействие, простая и сложная константы Гамакера определяются уравнениями
(4.24)
(4.25)
где гп — межмолекулярное расстояние.
4.4. Термодинамика дисперсных систем
Термодинамика дисперсных систем представляет важное направление коллоидной и физической химии, которое изучает термодинамические свойства ансамбля большого количества дисперсных частиц, статистически распределенных в дисперсионной среде.
Термодинамическое описание дисперсных систем зависит от интенсивности взаимодействия между дисперсной фазой и дисперсионной средой. Поэтому оно проводится раздельно для л ио фильных и лиофобных систем.
Термодинамическая (агрегативная) неустойчивость лиофобных дисперсных систем. Для обсуждения термодинамики дисперсных систем рассмотрим наиболее простой случай — свободнодиспер-сную систему со сферическими частицами диаметром d=2r. Удель ная поверхность дисперсной фазы (Qj, м2/кг) определяется уравнением
=6/(Р^) = 3/(Р^Г)> где pj — плотность вещества дисперсной фазы.
Свободную поверхностную энергию в расчете на единицу массы дисперсной фазы такой дисперсной системы находим по соотношению
^.=0^=3—, (4.26)
где о — поверхностное натяжение на границе дисперсных частиц с дисперсионной средой; г — радиус частиц.
По определению свободная поверхностная энергия (Fst Дж/кг) представляет избыток энергии Гельмгольца вещества в дисперсном состоянии по сравнению с энергией Гельмгольца этого же вещества, но в виде компактного макроскопического объекта. Эта избыточная энергия является причиной термодинамической (или агрегативной) неустойчивости дисперсных систем. В соответствии С уравнением (4.26) отклонение дисперсной системы от состояния термодинамического равновесия с минимальной энергией Гельмгольца ТГП1П (при данном объеме дисперсной фазы) будет возрастать по мере уменьшения размера дисперсных частиц. Таким образом, свободная поверхностная энергия представляет количественную меру отклонения дисперсной системы от состояния равновесия.
Термодинамическая неустойчивость дисперсных систем выбывает протекание таких процессов, которые с течением времени (/) ведут к уменьшению поверхностной энергии (см. гл. 10). Следовательно, в свободнодисперснои системе должна происходить определенная эволюция, которая соответствует условию d/\/dr< 0, т.е. постепенному уменьшению избыточной энергии Fs. Это условие в сочетании с уравнением (4.26) показывает, что люлюция дисперсной системы может приводить к ряду следит вий.
1. Увеличение размера частиц (dr/d/> 0) при неизменном поверхностном натяжении. Такой процесс реализуется при слиянии мелких дисперсных частиц в более крупные; этот процесс называют коалесценцией. Коалесценция (см. подразд. 10.3) характерна для систем с жидкой и газовой дисперсной фазой (эмульсии и газовые эмульсии соответственно). В результате коалесценции капли или газовые пузырьки становятся настолько крупными, что их размер превышает верхнюю границу дисперсного состояния вещества (см. подразд. 1.1), т.е. дисперсная фаза прекращает существование и превращается в макроскопическую фазу.
2. Уменьшение поверхностного натяжения (do/d/< 0) при постоянном размере дисперсных частиц. Такой процесс происходит при слипании дисперсных частиц в крупные агрегаты; этот процесс называют коагуляцией. Коагуляция (см. подразд. 10.2) характерна для систем с жидкой дисперсионной средой и твердой дис-
перепой фазой — коллоидных растворов (золей) и аэрозолей (дымов). При определенных условиях коагуляция возможна и в системах с жидкой дисперсной фазой — в эмульсиях и туманах. Эволю ция дисперсной системы в процессе коагуляции приводит к образованию связнодисперсной системы, в которой исходные дисперсные частицы разделены тонкими прослойками жидкой дисперсионной среды. Из-за малой толщины пленок их поверхностное натяжение при определенных условиях может быть меньше, чем толстых слоев той же жидкости. Зависимость поверхностного натяжения от толщины пленки о = /(/?) является размерным эффектом. Его термодинамической причиной служит перекрывание поверхностных слоев и возникающее в результате этого расклинивающее давление (см. подразд. 4.3).
Термодинамическая неустойчивость дисперсных систем — важ нейшая проблема коллоидной химии. Для достаточно продолжительной стабильности любой дисперсной системы необходимо прежде всего обеспечить ее устойчивость, т.с. предотвратить или, по крайней мере, сильно замедлить процессы коагуляции и коалесценции (см. подразд. 10.1). Термодинамика позволяет указать общий метод обеспечения устойчивости дисперсных систем. Он заключается в создании энергетических барьеров, препятствующих агрегации дисперсных часгиц. Механизмы создания таких барьеров различны. Основные факторы стабилизирующего действия связаны с расклинивающим давлением (см. подразд. 4.3) и защитным действием адсорбционных слоев поверхностно-активных веществ (структурно-механический барьер; см. подразд. 2.4).
Уравнение (4.26) показывает, что при одинаковых размерах частиц термодинамическая неустойчивость выражена тем сильнее, чем больше поверхностное натяжение на границе диспср сной фазы и дисперсионной среды. Поэтому проблема стабилизации особенно важна для лиофобных дисперсных систем, которые характеризуются высоким поверхностным натяжением. Продолжительность «жизни» лиофобных систем без применения стабилизирующих методов обычно очень мала — не более нескольких секунд.
Термодинамика лиофильных систем. Важное свойство дисперсных частиц заключается в том, что благодаря своим малым раз мерам они активно участвуют в тепловом (броуновском) движении (см. подразд. 2.4). При термодинамическом описании диспер сных систем следует учитывать это свойство. В свою очередь броуновское движение способствует равномерному распределению дисперсных частиц в дисперсионной среде и тем самым увеличению энтропии дисперсной системы. Поэтому полный анализ условии устойчивости дисперсных систем должен включать и анализ изменения энтропии дисперсной системы при изменении размеров дисперсных частиц.
Рассмотрим в связи с этим процесс самопроизвольного дисперсирования. Начальное состояние системы — макроскопический образец вещества А помещен в жидкость В; конечное состояние — коллоидный раствор дисперсной системы с частицами вещества А о шнакового размера d = 2г.
Термодинамику образования коллоидного раствора можно описать по анало! ии с образованием регулярных (молекулярных) растворов. Изменение энергии Гельмгольца АЛ при образовании дисперсной фазы будет определяться вкладом двух величин: поверхностной энергии ДЛ, = о^и энтропийного вклада T&S(T— температура; А5 — увеличение энтропии сиаемы при диспергировании вещества А).
В начальном состоянии системы все вещество А сосредоточено в одной области. В конечном состоянии оно равномерно распределено в дисперсионной среде. Соответственно энтропия системы будет больше, чем в начальном состоянии. В соответствии с термодинамическим определением имеем
АЛ = cQd - T&S. (4.27)
Для регулярных растворов приращение энтропии AS соответствует комбинаторному вкладу. Приближенно получим
(4.28)
1де р = 15 — 30. кп — постоянная Больцмана.
Термодинамическому равновесию дисперсной системы соответствует условие б(АЛ) = 0. После подстановок получаем, чго это равновесие достигается при определенном соотношении между поверхностным натяжением и размером частиц (г/), а именно, 1ОЛЖНО выполняться неравенство
о <
РАдТ г/2
(4.29)
Уравнение (4.29) показывает, что термодинамическое равновесие дисперсной системы, в принципе, возможно и без стаби-щзирующих барьеров. Необходимое условие — поверхностное натяжение должно быть достаточно мало. Для комнатной температуры этот порог в соответствии с уравнением (4.29) составляет около 0,1 мДж/м2.
Малое поверхностное натяжение на границе дисперсной фазы с дисперсионной средой представляет характерный признак лиофильных дисперсных систем Поэтому условие (4.29) следует считать критерием (условием) лиофильности. По имени авторов его принято называть критерием-лиофильности Ребиндера—Щукина. или критерием самопроизвольного диспергирования.
Контрольные вопросы
1. Приведите силовое и энергетическое (термодинамическое) опреде ление поверхностного натяжения.
2. Назовите основные факторы, которые определяют поверхностное натяжение жидкостей.
3. Объясните правило Антонова для поверхностного натяжения на границе двух жидкостей, укажите условия его выполнения.
4. Приведите уравнение Фоукса, проанализируйте его физико-химический смысл.
5. Укажите основные представления термодинамической теории Гиббса о поверхностном слое на границе раздела двух фаз.
6. Приведите основные термодинамические параметры поверхностного слоя, объясните их физико-химический смысл. Почему эти пара метры являются избыточными?
7. В чем заключается физико-химический смысл температурного коэффициента поверхностного натяжения жидкости? Приведите (примерно) интервал изменения значений этого коэффициента.
8. Объясните причины возникновения расклинивающего давления в тонких жидких пленках.
9. В чем заключается физико-химический смысл константы Гамакера?
10. Охарактеризуйте общую причину термодинамической неустойчивости дисперсных систем.
11. В каком направлении протекает эволюция дисперсных систем?
12. Приведите и проанализируйте условие самопроизвольного диспергирования лиофильных дисперсных систем.
ГЛАВА 5
КАПИЛЛЯРНЫЕ ЯВЛЕНИЯ
Капиллярные явления относят к группе физических поверхно-ш пых явлении. Примеры капиллярных явлений — смачивание твердых тел, слияние капель в аэрозолях и эмульсиях и газовых пузырьков в пенах, подъем смачивающих жидкостей в узких трубках (капиллярах) выше уровня в широком сосуде (пример — вода в стеклянной трубке) и противоположный эффект — опускание несмачивающих жидкостей в капиллярах ниже их уровня в широком сосуде (пример — ртуть в стеклянной трубке).
В капиллярных явлениях основную роль играют два фактора: поверхностное натяжение жидкости и кривизна поверхности жидкости.
5.1. Капиллярное давление
Капиллярным давлением (рс, Па) называют разность давлений (±Др). возникающую вследствие искривления поверхности жидкости. Такую поверхность имеют, например, капли в эмульсиях и туманах, капиллярные мениски. Обозначим давление под искрив-,пен ной поверхностью жидкости — рп давление под плоской поверхностью — /;0. Капиллярное давление определяется уравнением
Рс —Рг Ро-
(5-1)
Знак капиллярною давления («плюс» или «минус») зависит от знака кривизны. Выпуклые поверхности имеют положительную кривизну. Центр кривизны выпуклой поверхности находится внутри соответствующей фазы (в данном случае — внутри жидкости). Тогда согласно уравнению (5.1) капиллярное давление рс> 0, т.е давление под выпуклой поверхностью жидкости больше, чем давление под плоской поверхностью: рг> р0. Пример дисперсной частицы с выпуклой поверхностью — капля жидкости в аэрозоле или эмульсии. Выпуклую поверхность имеет мениск несмачивающей жидкости в капилляре.
Вогнутые поверхности имеют отрицательную кривизну, поэтому капиллярное давление рс< 0 (этому случаю отвечает знак «минус» в уравнении (5.1)). Давление жидкости рг под вогнутой поверхпо-
стью меньше, чем под плоской: рг< р(}. Пример вогнутой поверхности — мениск смачивающей жидкости в капилляре.
Капиллярное давление — это скачок давления (Др) на границе двух фаз, разделенных искривленной поверхностью.
Капиллярное давление зависит от поверхностного натяжения и кривизны поверхности. Эту связь описывает закон Лапласа (1805). Для вывода уравнения капиллярного давления найдем условие, при котором газовый пузырек объемом К внутри жидкости сохраняется неизменным, т.е. не расширяется и не сжимается. Равновесной форме соответствует минимальное значение энергии Гиббса. При увеличении радиуса пузырька на малую величину dr изменение энергии Гиббса dG будет равно
dG = ptdK + odQ. (5.2)
Слагаемое pcd К определяет работу изобарического расширения, слагаемое adQ — затрату работы на увеличение поверхности пузырька; Q = 4лг — поверхность сферического пузырька радиусом г.
При гермодинамическом равновесии фаз должно выполняться условие минимума энергии Гиббса: AG = 0; отсюда получаем
4тсг2/;( + 8лгсу = 0.
В итоге находим связь между капиллярным давлением и радиусом кривизны г для вогнутой сферической поверхности:
2о г
(5.3а)
Отрицательный знак капиллярного давления показывает, что внутри газового пузырька давление рг больше, чем давление в окружающей его жидкости. Именно по этой причине пузырек нс «схлопывается» под давлением окружающей его жидкости.
Аналогично выводится уравнение капиллярною давления для выпуклой поверхности жидкости, например для капли аэрозоля (тумана) в газовой фазе. Для выпуклой сферической поверхности получим
2су _
Л= +—. (э.Зб)
г
Положительное капиллярное давление сжимает каплю. В качестве примера рассчитаем капиллярное давление для капли ртутц радиусом 10 нм. Поверхностное натяжение ртути при комнатной температуре составляет о = 473,5 мДж/м2-. Тогда из уравнения (5.36) находим, что для напоразмерной капли (г = 10 нм) капиллярное давление равно 947 МПа, т.е. оно на несколько порядков превышает атмосферное давление. Таким образом, дчя капель и пузырь
ков дисперсных размеров влияние капиллярного давления весьма шачительно.
Уравнения (5.3а) и (5.36) представляют закон капиллярного (явления Лапласа для сферической поверхности. Для поверхности произвольной формы закон Лапласа имеет вид
д. = ±о - + —
I й г2
(5.4)
где г2 — главные радиусы кривизны.
Для цилиндрической поверхности радиусом второй главный радиус кривизны г2 = °°, поэтому рс = ±о/Г|, т.е. в 2 раза меньше, чем для сферической! поверхности радиусом г,.
Величина 0,5(1/?-] + 1/г2) = Н определяет среднюю кривизну поверхности. Таким образом, уравнение Лапласа (5.4) связывает капиллярное давление со средней кривизной поверхности жидкости’
д.= 2о//.
Закон Лапласа имеет определенные ограничения. Он выполняется достаточно точно, если радиус кривизны поверхности жидкости г » b (Ь — молекулярный размер). Для нанообъектов это условие не выполняется, гак как радиус кривизны соизмерим с молекулярными размерами.
Закон капиллярного давления имеет большое научное значение. Он устанавливает фундаментальное положение о зависимости физического свойства (давления) от геометрии, а именно — от кривизны поверхности жидкости. Теория Лапласа оказала значительное влияние на развитие физикохнмии капиллярных явлений, а также на некоторые другие дисциплины. Например, магматическое описание искривленных поверхностей (основы дифференциальной геометрии) было выполнено К Гауссом именно в связи с капиллярными явлениями.
Закон Лапласа имеет много практических приложений в химической технологии, фильтрации, течении двухфазных потоков и т.д. Уравнение капиллярного давления используют во многих метода,\ измерения поверхностного натяжения жидкостей. Закон Лапласа часто называют первым законом капиллярности.
5.2. Смачивание
Смачивание — физико-химическое явление, происходящее при контакте жидкостей с поверхностью твердых тел. Основную роль в смачивании играют поверхностные свойства жидкости и твердого тела. В зависимости от сочетания этих свойств степень смачивания может различаться очень сильно. Например, вода хорошо смачивает очищенную от жировых загрязнений поверхност ь стекла: капля
волы быстро растекается на достаточно большой площади и образует тонкую плоскую пленку. Напротив, ртуть не смачивает стекло: небольшая капля ртути на стеклянной пластине имеет сферическую форму. Ртуть хорошо смачивает многие металлические поверхности (цинк, свинец, олово, золото).
Важнейшая особенность смачивания заключается в том, что в процессе участвуют три разных фазы. Первая фаза — это твердое тело, вторая фаза — смачивающая жидкость. Кроме гою, ирису! ствует третья фаза, с которой граничила твердая фаза до подвода к ее поверхности смачивающей жидкости. Третьей фазой часто является газ (воздух). В технике и природе достаточно распространен случай, когда третья фаза — это другая жидкость, которая не смешивается со смачивающей жидкостью. Этот случай называю! избирательным смачиванием. Типичным примером избирательного смачивания является контакт с поверхностью твердого тела полярной (вода, водные растворы) и неполярной (предельные углеводороды, ароматические вещества и т.д.) жидкости.
Количественной характеристикой смачивания служит краевой угол 0 (рис. 5.1), который измеряют по наклону касательной АВ к поверхности капли, находящейся на твердой поверхности. Вершина краевою угла (ючка А) находи гея на линии контакта всех трех фаз, участвующих в смачивании. В данном примере это окружность АС в основании капли. По ней соприкасаются друг с другом твердое тело, жидкость и окружающая их третья фаза — газ. Эзу общую границу называют линией смачивания, или линией трехфазного контакта.
Краевой угол О отсчитывают в сторону смоченной поверхно-сги. При избирательном смачивании краевой угол отсчитывают в сторону более полярной жидкости.
Краевой угол 0 определяется законом Юнга (1804). Для трехфаз-пых систем твердое тело — жидкость — газ закон Юнга имеет вид
COS0 = —-----—. (5.5)
Уравнение (5.5) следует из условия термодинамического рав новссия на линии смачивания. В правую часть уравнения (5.5) вхо-
Рис. 5.1. Краевой угол О капли жидкости на твердой поверхности, окруж ность АС — линия смачивания (линия трехфазного контакта)
тят три параметра: удельные поверхностные энергии твердого гела на границе с газом (оп.) и жидкостью (оГА) и поверхностное натяжение жидкости на границе с газом (ожг). Все три параметра являли ся термодинамическими. Поэтому каждой трехфазной системе соответствует (при определенной температуре Т) единственное значение равновесного краевого угла.
При избирательном смачивании закон Юнга имеет вид
- ---1------
(5.6)
• де аТЖ| — удельная поверхностная энершя на границе твердого гела с жидкостью 1; о,ж? — удельная поверхностная энергия на границе твердого тела со смачивающей жидкостью 2; <тЖ1Ж, — поверхностное натяжение на границе жидкость I—жидкость 2.
Закон Юнга, как и закон Лапласа, относится к числу основных законов капиллярных явлений, поэтому его часто называют вторым законом капиллярности. Закон Юнга можно вывести па основе известного положения термодинамики о том, что равновесию системы (при постоянном объеме) соответствует минимальная энергия Гельмгольца. В случае смачивания минимальное значение должна иметь свободная поверхностная энергия, которая определяется вкладами грех поверхностей раздела контактирующих фаз.
Рассчитаем изменения (вариации) поверхностной энергии, которые произойдут при перемещении линии смачивания на расстояние dr при постоянном объеме Инебольшой капли, нанесенной на плоскую поверхность (г — радиус линии смачивания; см. рис. 5.1). 11лощадь смоченной поверхности £2ГЖ увеличится при этом на кольцевую площадку 2nrdr, а изменение поверхностной энергии твердого тела составит 2яг(оп - о|Ж) dr. Перемещение линии смачивания также увеличивает площадь поверхности капли £>Ж1 па границе с газом. Небольшая капля на плоской подложке имеет форму сферического сегмента, так как при условии г< а влиянием силы тяжести на форму капли можно пренебречь (а — капиллярная длина: см. подразд. 2.2). Поэтому приращение площади поверхности капли составит dQ*r= 2;rrd/ (d/ = cos ©dr — удлинение дуги АС) Соответствующее приращение свободной поверхностной энергии на границе жидкость—газ составляет 2rcraAfcosOdr. Таким образом, условие гермодинамического равновесия определяется уравнением
(ап - отж)2лгйг + 2ягожгсо8 ©dr = О
Отсюда следует уравнение равновесною краевого узла (5.5).
На основе закона Юнга построена классификация различных случаев контактного взаимодействия жидкостей с поверхностью звердых тел.
Несмачивание (0 > 90°). В этом случае удельная поверхностная энергия отж на границе твердого тела с жидкостью больше, чем на границе твердого тела с газом (отж > отг). Физический смысл этого неравенства заключается в том, что самопроизвольное растекание жидкости по данной твердой поверхности термодинамически невозможно. Из закона Юнга (5.5) следует, что в случае отж> °т1 имеем cos0 < 0, т.е. краевые углы тупые: 0 > 90°. Поверхности, несмачивасмыс водой, называют гидрофобными. Гидрофобную поверхность имеет, например, парафин. Капля воды на парафине имеет краевой угол 0 = 105° —108°. На тефлоне краевой угол воды еше больше (120°—130°)-
Смачивание (0 < 90°). При смачивании поверхностная энергия отж на границе жидкости с твердой поверхностью меньше поверхностной энергии отг на границе твердое тело—газ: о(ж< о|(. Тогда в соответствии с законом Юнга (5.5) имеем cos0 > 0, т.е. краевые углы острые (0 < 90°). При условии (он -о^) < ожг краевой угол изменяется в интервале 0° < © < 90°. Смачиваемые поверхности называют лиофильными, поверхности, которые смачиваются водой, гидрофильными. Пример гидрофильной поверхности — оксидная пленка на поверхности металла. На окисленном алюминии или цинке краевой угол для воды составляет 60° — 65°.
Полное смачивание или растекание (0 = 0°). Этот случай реализуется при выполнении условия о1Т> о1Ж + оЖ1. Тогда правая часть уравнения (5.5) (отг - отж)/оЖ1. > 1. Это означает, что равновесный краевой угол сформироваться не может. При этом условии капля растекается по твердой поверхности и превращается в плоскую тонкую пленку. Толщина смачивающих пленок обычно составляет 1—10 нм. Пример полного смачивания — ртуть в контакте с нс-окисленпой поверхностью цинка или свинца.
Использование уравнения Юнга (5.5) для расчета равновесных краевых углов 0 требует сведений об удельных поверхностных энергиях отг и отж на границе данного твердого тела с газом и со смачивающей жидкостью. По определенным причинам поверхностные энергии большинства твердых тел не удается измерить достаточно точно. В связи с этим в современной теории смачивания применяют способы, которые позволяют это преодолеть. Рассмотрим два таких способа.
1. Использование молекулярной теории поверхностного натяжения (см. подразд. 4.1). Поверхностную энергию ота можно рассчитать с помошью уравнения Фоукса (4.10). Для контакта жидкости с поверхностью твердого тела его записывают в следующем виде:
^тж — от + ож — 2(от ) . (5.7)
Подставив выражение (5.7) в уравнение Юнга (5 5), получаем
1/2
cos О =
(5-8)
Для жидкостей, у которых поверхностное натяжение создается главным образом дисперсионными силами (органические вещества, сжиженные инертные газы), получаем
(5.9)
Таким образом, краевые углы 0 можно рассчитать теоретически, если известны дисперсионные составляющие о, d и ожд/ для твердых тел и жидкостей соответственно. Эти данные имеются для многих материалов и жидкостей.
В качестве примера приведем дисперсионные составляющие удельной поверхностной энергии некоторых твердых тел (о, /). мДж/м2:
Тефлон............................... 19,5
Полиэтилен........................... 35,0
Парафин.............................. 44,0
Для ряда жидкостей удельная поверхностная энергия (ож) и ее дисперсионный вклад (ож d) составляют, мДж/м2:
Жидкость ож аж d
Подметан......................50,8 48,5
Формамид......................58,2 39,5
Глицерин......................63 4 39,5
Вода..........................72,8 21,8
Ртуть.........................484 200
2. Использование правила Антонова. Правило Антонова (4.4) связывает межфазное натяжение О|2 на границе двух несмешикающихся жидкостей 1 и 2 с их поверхностными натяжениями и о2 на границе с газом: о(2 = О] -о2. Основное условие выполнения правила Антонова — граничащие жидкости I и 2 не должны растворяться друг в друге или химически взаимодействовать. При выполнении этих условий правило Антонова применимо и для контакта жидкости с твердой поверхностью.
Дл таких систем возможны два случая:
1) о|Ж = от-ож (при от > ож); после подстановки этого выражения для удельной поверхностной энергии отж в уравнение Юнга (5.5) при а, > ож получаем cos О = 1, т.е. происходит полное смачивание:
2) сггж= ож-от (при от< ож); в эюм случае подстановка огж в уравнение Юнга дает
Уравнение (5.10) хорошо согласуется с рядом экспериментальных данных: для одной и той же твердой поверхности, контактирующей с разными жидкостями, значения cosG растут линейно в зависимости от величины 1 /<5Ж (рис. 5.2). Тангенс угла наклона этих прямых равен поверхностной энергии твердых тел.
Зависимость смачиваемости от свойств твердой поверхности. Уравнение равновесного краевого угла (5.5) выведено для смачивания идеализированной твердой поверхности. Она считается гладкой, плоской, химически однородной и жесткой (нсдсформиру-смой). В большинстве реальных случаев какое-либо из указанных условий не выполняется.
Рассмотрим влияние на смачиваемость некоторых свойств твердой поверхности.
Влияние шероховатости поверхности. Реальные подложки не бывают идеально гладкими, они имеют достаточно сложный рельеф с микроскопическими и наноразмерными выступами и впадинами разной формы и размера — от 1 нм до 10 мкм. Основное следствие шероховатости заключается в том, что плошадь такой поверхности (Ош) будет больше, чем плошадь идеальной (О0)
Рис. 5.2. Зависимость краевого угла (0) воды (/), глицерина (2). формамида (5). парафинового масла (4), ундекана (5), декана (6). этиленгликоля (7) на тефлоне (сплошная линия) и на парафине (пунктирная линия) от поверхностного натяжения (о/) жидкости
гладкой подложки. Отношение Qlu/Qy = К называют коэффициентом шероховатости. По определению коэффициент К > 0. Для многих твердых поверхностей (полированных, шлифованных) К~ 1,05—1,15. Коэффициент шероховатости измеряют с помощью специальных приборов (микропрофилографов), чувствительный элемент которых выявляет микровыступы и микровпадины высотой или глубиной примерно до 0,1 мкм.
С учетом шероховатости изменение поверхностной энергии твердого тела при перемещении линии смачивания на расстояние dr будет больше, чем для гладкой поверхности (см. выше вывод закона Юнга) и составит 2тиг/С(огг - о1Ж)дг.
На площадь поверхности жидкость — газ шероховатость не влияет, и условие термодинамического равновесия принимает вид
2лгЛ1отг - cOdr = 2я/пж, cos ©...dr,
где 0lu — краевой угол, определяемый по наклону касательной к поверхности жидкости к условной горизонтальной плоскости.
Сопоставление с законом Юнга (5.5) дает уравнение краевого угла на шероховатой твердой поверхности:
сох0ш = Acos0. (5.11)
Уравнение (5.11) называют уравнением Венцеля—Дерягина.
Из уравнения (5.1) следует, что для смачивающих жидкостей (cos 0 > 0) шероховатость приводит к уменьшению краевых углов (cos0U1 > cos©). В случае несмачивания шероховатость вызывает увеличение краевых углов: со5 0ш < cos©. Характерный пример: ворсинки на перьях водоплавающих птиц значительно увеличивают шероховатость их поверхности; это способствует тому, что перья практически не смачиваются водой.
Влияние химической неоднородности поверхности. Химическая неоднородность (гетерогенность) твердой поверхности — важный фактор, влияющий на смачивание. Рассмотрим самый простой случай: твердая поверхность представляет собой мозаику, состоящую их небольших — примерно несколько микрометров — участков («доменов») только двух типов: А и В. Введем обозначение (р относительная доля площади, занимаемой веществом А; тогда (1 - ср) — доля плошади, которая занята веществом В. Удельные поверхностные энергии твердого тела на границе с газом отг и со смачивающей жидкостью огж состоят из вкладов поверхностных энергий каждого материала. Тогда из уравнения Юнга (5.5) получаем уравнение краевого угла (0П) для неоднородной бинарной твердой поверхности:
cos 0„ = tpcos 0Л + (1 - (p)cos 0В, (5.12)
где Од. 0В — краевые углы данной жидкости на однородных твердых поверхностях А и В соответственно.
Уравнение (5.12) называют уравнением Ребиндера— Кассъе (1931, 1946). Оно показывает, что по краевым углам смачивания можно получить информацию о химическом составе поверхности, например о соотношении площадей, занимаемых полярными и неполярными группами на поверхности биополимеров.
Влияние деформации. Фактором, осложняющим смачивание, являются деформационные изменения поверхности твердого тела при контакте со смачивающей жидкостью. Рассмотрим влияние таких деформаций на следующем примере. Капля жидкости нанесена на материал со сравнительно малым модулем упругости (£). К числу таких материалов относятся некоторые полимеры, каучук, а также полимерные и белковые гели. Капля на подложке имеет выпуклую поверхность. Поэтому на подложку в пределах смоченной площади Л/? (рис. 5.3) действует капиллярное давление р1{ - 2<л.^/гх (оЖ1 — поверхностное натяжение жидкости; г, — радиус кривизны поверхности под каплей). Если модуль упругости мал, то под действием капиллярною давления pfI подложка под каплей прогнется и образуется лунка в виде сферическою сегмента с радиусом кривизны г2. В результате на нижней поверхности капли возникает капиллярное давление рЛ = 2сутж/г2.
Обозначим радиус смоченной площади ОА — г, а углы, образованные верхним сегментом капли ВСА и нижним сегментом BDA с горизонтальной плоскостью, — аир соответственно (см. рис. 5.3), тогда радиус кривизны r3 = r/cosa, а радиус кривизны = r/cosp. Из баланса капиллярных давлении = рс2 получаем
ож1 cos a = oTXcos р.
Механическое равновесие на линии смачивания определяют два уравнения:
о.... = огжг cos a + alxcos р, (5.1 За)
1 I 1 I v '
aArsin a = a1)Ksin p. (5.136)
Уравнение (5 13а) следует из баланса сил, действующих в горизонтальной плоскости, а уравнение (5.136) — из баланса сил, направле! 111 ых вертикально.
Рис. 5.3 Краевой угол на деформируемых поверхностях: под каплей формируется лунка в виде сферического сегмента ADB
Краевой угол равен О = а + 0 (см. рис. 5.3). Из уравнении (5.13) следует, что условие равновесия определяется из баланса сил поверхностного натяжения; в векторной форме оно имеет вид
+оЖг+ОжГ = 0. (5 14)
Это соотношение называют уравнением Неймана', оно определяет также краевой угол капли одной жидкости на поверхности другой жидкости (при отсутствии взаимного растворения).
Смачивание нанокаплями. В связи с интенсивным развитием нанохимии и нанотехнологии большой интерес представляет смачивание каплями наноразмеров (нанокаплями). Для небольших капель существенную роль играет еще один термодинамический параметр — линейное натяжение (к, Дж/м). Физический смысл линейного натяжения заключается в следующем. Молекулы жидкости, которые находятся на линии смачивания АС (см. рис. 5.1), непосредственно контактируют с молекулами двух соседних фаз (газ и твердое тело). Поэтому их энергетическое состояние отличается от энергетическою состояния молекул жидкости на поверхности капли, где она граничит только с одной фазой (газом). Избыточную энергию линии трехфазного контакта по сравнению с энергией поверхности жидкости (для одинакового числа молекул) называют линейным натяжением. В отличие от поверхности раздела фаз эту энергию относят не к единице плошади, а к единице длины, т.е. размерность линейного натяжения — джоуль на метр (Дж/м). Поскольку 1 Дж = 1 Н • м линейное натяжение имеет размерность силы — ньютон (Н). Эта сила направлена вдоль линии смачивания, она создает двухмерное давление:
р* - к/г, (5.15)
которое действует по нормали к линии смачивания (см. рис. 5.1).
Соотношение (5.15) называют уравнением Веселовского—Перцова (1936) При выводе закона краевого угла О для малых капель в уравнение равновесия па линии смачивания нужно вводить дополнительное слагаемое к/r. С учетом этой поправки равновесный краевой угол 0* определяет уравнение Шелудко— Гошева — Пла-тиканова (1980):
COS0* = COS0-—. (5.16)
от
Обычно линейное натяжение составляет не более 10~7—10-6 мН. Поэтому влияние поправки к/гстановится существенным только для наноразмерных капель радиусом примерно 1 — 10 нм. Линейное натяжение играет важную роль только в определенных коллоидно-химических процессах, например при гетерогенном образовании зародышей новой фазы, конденсации капель на твердой
поверхности. Для достаточно больших (г> 0,1 мкм) капель вклад слагаемого к/г становится очень малым и уравнение (5.16) принимает обычную форму закона Юнга (5.5).
5.3. Адгезия
Адгезия (дал. adhaesio — притяжение, сцепление, прилипание) — поверхностное явление, которое заключается в возникновении механической прочности при контакте поверхностей двух разных веществ. Причиной адгезии является молекулярное притяжение кон-пак тирующих веществ или их химическое взаимодействие
Термодинамической характеристикой адгезии служи! работа, которую необходимо совершить для разделения двух контактирующих фаз, например для полного отделения капли жидкости от твердой поверхности. Процесс разрыва адгезионного контакта должен происходить при постоянной температуре (изотермически). При этом условии удельную работу, отнесенную к единице площади поверхности, которая образуется при разрыве адгезионного контакта, называют работой адгезии (Дж/м )
Д 1я расчета работы адгезии нужно определить изменение поверхностной энергии при изотермическом отрыве слоя жидкости от твердой подложки. В начальном состоянии капля жидкости контактирует с твердой подложкой. Межфазная поверхность жидкость—твердое тело имеет удельную энергию отж. Для отрыва капли затрачивается работа И' (в расчете на единицу площади). При этом поверхность жидкость—твердое тело исчезает, но вместо нее возникают две новые поверхности: жидкость—газ и твердое тело-газ с удельными поверхностными энергиями ож, и о соответственно Из закона сохранения энергии следует
~ 0>КГ "* ^ТГ'
Таким образом, работу адгезии определяет уравнение
— сутг + сужг — <у.гж.
Это соотношение называют уравнением Дюпре (1869). Заменим числитель в уравнении Юнта (5.5):
отг — — СГЛг.
Тогда получим другое термодинамическое уравнение для краевого угла смачивания:
IV
cosO =------1. (5 17)
аЖ|
Уравнение (5.17) показывает, что краевой угол определяется конкуренцией двух факторов. Первый фактор — молекулярное
притяжение жидкости к твердой поверхности: чем сильнее это притяжение, тем больше работа адгезии. Второй фактор — взаимное притяжение молекул жидкости, количественной мерой которою является поверхностное натяжение жидкости (см. подразд. 4.1).
Из уравнения (5.17) следует, чго при преобладании притяжения жидкости к твердой подложке ( > оЖ1) имеем cos© > 0, т.е.
жидкость смачивает эту твердую поверхность.
Напротив, если сильнее взаимное притяжение молекул жидкости (ожг> то cos О < 0 и О > 90°; это случай нссмачивания.
Из уравнения (5.17) следует, что жидкости с небольшим поверхностным натяжением лучше смачивают твердые тела, чем жидкости с большим натяжением. Эксперименты подтверждают этот вывод. Многие органические жидкости и сжиженные газы полностью смачивают большинство твердых материалов.
Уравнение (5.17) для расчета работы адгезии можно преобразовать с учетом уравнения Фоукса (4.10) для поверхностного натяжения на границе конденсированных фаз. Уравнение (4 10) позволяет выразить натяжение о|Ж следующим образом:
Огж ~ <*т + г/ )
тогда получаем
^=2(ОтЛД,/2. (5.18)
Представление о работе адгезии очень полезно для выявления связи между краевыми углами смачивания и природой сил взаимодействия между жидкостью и поверхностью твердою тела. В расчете на 1 моль вещества твердого тела энергия (Ц.ж) такого взаимодействия равна
= WaNt/>h.
|дс —число Авогадро; пх — число молекул (атомов, ионов) на единице площади твердого тела.
Примем, что толщина поверхностного слоя (5) соответствует мономолекулярпому слою, тогда
«, = (рЛл/Я)2/3.
где р,. Л/, — соответст венно плотность и молекулярная масса твердого вещества.
При этих упрощениях энергия взаимодействия равна
У,ж= WP,W (5.19)
Уравнение (5.19) приводит к следующему выводу. Для жидкостей со сравнительно небольшим (о < 100 мДж/м2) поверхностным натяжением необходимое условие смачивания Wa> может выполняться при сравнительно небольших энергиях взаимо
действия (/тж -1 — 10 кДж/моль. Такие небольшие энергии характерны для молекулярных сил, в том числе и для дисперсионных вза и моде йстви й.
Другая ситуация возникает при контакте твердых тел с жидкостями, поверхностное натяжение которых велико — К)2— К)3 мДж/м2. К ним относятся жидкие металлы, расплавы солей и оксидов и т. п. Для смачивания такими жидкостями необходимы большие энергии взаимодействия - 101—102 кДж/моль. Такие энергии характерны для химических взаимодействий с образованием ионных, ковалентных и металлических связей.
На основе этих представлений проводится классификация твердых тел в зависимости oi их поверхностной энергии оТ1 на границе с газом. Различают две группы твердых поверхностей:
1) низкоэнергетические (ог< 100 мДж/м2);
2) высокоэнергетические (ап > 100 мДж/м2).
При смачивании низкоэнср1етических поверхностей преобладают дисперсионные взаимодействия между жидкостью и твердым телом. При смачивании высокоэнергетических поверхностей большую роль могут играть взаимодействия другой природы, особенно при контакте с жидкостями с высоким поверхностным натяжением.
Для расчета работы адгезии разработано несколько теорий: молекулярная, электрическая и диффузионная.
Согласно молекулярной (или адсорбционной) теории причиной адгезии служат силы Ван-дер-Ваальса. Эта теория используется для описания адгезии полимеров (адгезивов) к различным твердым подложкам (субстратам). Адгезия рассматривается как поверхностный процесс, в котором решающее значение имеет адсорбция молекул вещества (адгезива) па определенных участках i вер-до го субстрата.
Электрическая теория основана на явлении контактной электризации при соприкосновении двух диэлектриков или металла и диэлектрика Система адгезив—субстрат рассматривается как конденсатор, роль обкладок которого играют две контактирующие поверхности. В рамках этой модели при раздвижении обкладок конденсатора возникает разность электрических потенциалов. Поэтому работа адгезии определяется электрической энергией конденсатора:
(5.20)
где р, — плотность поверхностного заряда, Кл/м2; h — расстояние между обкладками, при котором возникает электрический разряд; е — диэлектрическая проницаемость среды.
Диффузионная теория связывает возникновение адгезионной прочности с диффузией молекул адгезива в субстрат. При контак-
re полимерных материалов возможно также диффузионное проникновение в субстрат отдельных сегментов. При достаточно продолжительном контакте происходит «спайка» адгезива и субсгра-га и поверхность раздела между ними исчезает. Прочноегь адгезионного соединения определяется молекулярными взаимодействиями между «переплетенными» макромолекулами адгезива и субстрата.
Адгезия играет большую роль во многих технологических и природных процессах, например в процессах склеивания различных материалов (металлов, пластмасс, дерева), нанесения красок и других покрытии. Адгезия смазок сильно влияет на трение Для существенного снижения коэффициента трения на твердой поверхности должна образоваться мономолекулярная пленка поверхностно-активного вещества. Чем длиннее молекула ПАВ, тем меньше работа адгезии по отношению к этой модифицированной подложке.
Минимальный уровень работы адгезии достшается при максимально упорядоченной структуре смазочного адсорбционного слоя. Высокая степень порядка обеспечивается при бомбардировке одной из трущихся поверхностей атомами гелия. После такой обработки обнаруживается явление сверхнизкого трения', коэффициент трения между двумя твердыми поверхностями уменьшается в десятки раз но сравнению с необработанными материалами. Уменьшение работы адгезии необходимо также для предотвращения прилипания льда к твердым поверхностям (обледенение самолетов, гололед и ряд других нежелательных процессов).
Одно из ключевых требований к тромборезистентным материалам, применяемым в медицине для протезирования кровеносных сосудов и сердечных клапанов, заключается в минимальной адгезии компонентов крови к поверхности выбранного материала.
5.4. Влияние кривизны поверхности на давление пара и растворимость
Кривизна поверхности жидкости существенно влияет на условия равновесия между жидкостью и сс насыщенным паром Рассмотрим каплю радиусом г о поверхностным натяжением о, находящуюся в газовой среде. При условии г< а (а2 — капиллярная постоянная жидкости; см. подразд. 2.2) капля имеет сферическую форму. Поскольку поверхность капли выпуклая, давление в капле повышено на величину капиллярного давления рс = 2о/г(см. подразд. 5.1) Соответственно химический потенциал жидкости в капле больше, чем гой же жидкости с плоской поверхностью. Избыток потенциала равен
АНж = АЛ, = 2аГ„,/г.
где Vm — молярный объем жидкости.
Для равновесия фаз химический потенциал пара (uu) должен возрасти на величину Дцп = Дцж. Химический потенциал идеального газа равен
ц = Но + ЯТ1пр0,
где ц0 — потенциал стандартного состояния; р$ — давление пара над плоской поверхностью: R — универсальная газовая постоянная; Т — температура.
Для искривленной поверхности химический потенциал составляет
Мг= Но + ЯЛпрп
где рг — давление пара над искривленной поверхностью.
Из условия Дрг, = Дцж получаем зависимость давления насыщенного пара рг от кривизны поверхности жидкости (закон Кельвина. 1846):
Рг
~ = ехр
А)
(5.21)
Таким образом, чем меньше радиус капли г, тем больше равновесное давление насыщенного пара рг над ее поверхностью. Поэтому зависимость давления пара ог кривизны особенно важна для высокодиспсрсных и нанодиснерсных систем (аэрозолей).
Для вогнутой поверхности (например, мениск смачивающей жидкости в капилляре) давление насыщенного пара будет меньше, чем над плоской поверхностью. Поэтому уравнение (5.21) принимает вид
Ао 2а Vm
— = схр----—.
Рг rRT
(5.22)
Величина
2оУт = рс rRT ро
представляет отношение капиллярного давления р к давлению пара нал плоской поверхностью р[}. При условии р« получим
2оГт . 2а V„, ехр —— ~ 1 +---—.
rRT rRT
Toiaa из уравнения (5.22) следует
А ~ А) _ 2а И,„
A) rRT
(5.23)
Уравнение (5.23) показывает, что относительное изменение давления насыщенного пара, вызванное кривизной поверхности, обратно пропорционально радиусу кривизны г.
Зависимость давления насыщенного пара от кривизны поверхности жидкости играе т весьма важную роль в стабильности аэрозолей (туманов). В туманах всегда содержатся капли разных размеров. В соответствии с законом Кельвина (5.21) давление пара над мелкими каплями больше, чем над крупными. Разность давлений вызывает перенос молекул пара к крупным каплям с последующей конденсацией перенесенной массы. Этот процесс называют изотермической перегонкой. Он приводит к росту объема больших капель за счет исчезновения малых капель в исходном аэрозоле. В результате высокодисперсные аэрозоли со временем превращаются в грубодисперсные. Этот процесс играет большую роль, например, в выпадении атмосферных осадков (дождя).
Другой физико-химический процесс, определяемый законом Кельвина, — капиллярная конденсация. Поясним суть этого процесса с помощью следующей схемы. Рассмотрим плоскую твердую поверхность в контакте с газом (воздухом), в котором содержатся пары какой-либо жидкости (например, воды). Пусть на этой поверхности имеется углубление — канавка с поперечным сечением в форме треугольника. Температура этой системы такова, что на плоской поверхности нары не конденсируются. Однако в канавке конденсация может произойти, если образующаяся жидкость смачивает данную твердую поверхность, а кривизна обеспечивает необходимое для конденсации давление насыщенного пара. Конденсацию паров смачивающей жидкости па углубленных искривленных участках твердой поверхности называют капиллярной Конденсацией.
Причина капиллярной конденсации заключается в том, что внутри углубления смачивающая жидкость образует вогнутый мениск. В соответствии с уравнением Кельвина давление насыщенных паров над вогнутой поверхностью меньше, чем над плоской. Для капиллярной конденсации необходимо, чтобы радиус лунки г был достаточно мал, его значение определяется уравнением (5.22).
Капиллярная конденсация играет значительную роль в ряде технологических процессов, например при конденсации паров летучих растворителей па пористых адсорбентах.
Кривизна твердой поверхности влияет также на растворимость данною материала в жидкости. Для выступов с радиусом кривизны г предельная растворимость (С,.) определяется уравнением Гиббса— Оствальда'.
Сг=С„схр^. (5.24)
ГК1
где Со — растворимость материала с плоской поверхностью.
Уравнение (5.22) даст такую же зависимость растворимости от радиуса кривизны поверхности, как уравнение Кельвина (5.21) для давления насыщенного пара. Локальное увеличение растворимости имеет большое значение; например, оно может приводить к растворению границ зерен в поликристалличсских материалах с образованием на поверхности двугранных канавок (так называемых канавок термического травления).
5.5. Течение жидкостей в капиллярах и пористых средах
Еще в XVII в. было обнаружено (Р. Бойль, Р. Гук, Г. Галилей), что в узких трубках (капиллярах) жидкость может подниматься вверх — значительно выше уровня жидкости в широком сосуде. Необходимое условие капиллярного подъема — жидкость должна смачивать материал трубки, т.е. краевой угол должен быть острым (0 < 90 ). При этом условии поверхность жидкости в трубке имеет вогнутую форму, т е. образуется вогнутый мениск. Наиболее известный пример капиллярного течения — подъем воды в вертикальных стеклянных трубках.
Высота капиллярного подъема (//) обратно пропорциональна диаметру трубки (J); эту зависимость установил экспериментально Ж.Жюрсн (1718). Чем уже капиллярная трубка, тем больше высота подъема жидкости. Например, вода в стеклянном капилляре диаметром d = 1 мм поднимается примерно на 1,5 см; при d= 1 мкм высота подъема составляет Н = 15 м.
Несмачивающая жидкость, напротив, опускается в капилляре ниже уровня жидкости в широком сосуде. Этот случай называют капиллярной депрессией. Яркий пример депрессии — опускание ртути в стеклянных трубках. Несмачивающая жидкость образует с внутренней поверхностью трубки тупой краевой угол (0 > 90°). Соответственно капиллярный мениск имеет выпуклую форму.
Таким образом, существует однозначное соответствие между знаком кривизны капиллярного мениска и направлением течения жидкости в капиллярной iрубке:
Характеристика
Смачивание
Несмачивание
Краевой угол........................ 0 < 90°
Мениск.............................. Вогнузый
Капиллярное давление................ /<< 0
Капиллярный подъем (депрессия)...... //> 0
О > 90° Выпуклый Р< > 0 Н < 0
Выведем зависимость высоты капиллярного подъема (//) от диаметра капилляра (d). Определим прежде всего причину течения жидкоети вверх, т.е. против действия силы тяжести. В вогну
том мениске давление рг меньше, чем давление р(} под плоской поверхностью в широком сосуде на величину капиллярного давления рс, т.е. перепад давления составляет Др = р0~Рг- +Рс- Положительная разность давлений в мениске и под плоской поверхностью (Др > 0) и является причиной капиллярного подъема смачивающей жидкости.
Высота капиллярного подъема определяется из равенства давлений
АР = Рю
где рн — гидростатическое давление, создаваемое капиллярным столбиком: ри = (рж - pr)gH (рж, рг — плотность жидкости и газа соответственно; g — ускорение свободного падения).
Для расчета капиллярного давления нужно знать форму мениска, которая определяется поверхностным натяжением жидкости и действием силы тяжести. В достаточно тонких капиллярах г« а (а — капиллярная длина) влияние силы тяжести мало. Поэтому можно считать, что мениск имеет форму сферического сегмента. Тогда в соответствии с законом Лапласа (5.3а) капиллярное давление ре = -2g/г. Радиус мениска кривизны г и радиус капиллярной трубки г0 связаны соотношением r = ro/cos0.
Из условия рн=+Рс после соответствующих подстановок получаем уравнение высоты капиллярною подъема
Н = 2acos0/|ro(px-pr)g].
(5.25)
Плотность жидкости гораздо больше плотности газа (рж>> р,), поэтому уравнение (5.25) можно заменить соотношением
2ocos0
Рж&'Ь
(5.26)
Уравнение (5.26) объясняет обратно пропорциональную зависимость высоты капиллярного подъема от диаметра капиллярной трубки d = 2г0 (закон Жюрена). Из уравнения (5.26) также следует, что произведение высоты капиллярного подъема на радиус трубки представляет для данной жидкости постоянную величину:
HrQ = tf2cos0.
Отношение
называют капиллярной постоянной жидкости; сс значение зависит только от физических свойств жидкости (плотности и поверхностного натяжения) и ускорения свободного падения. Капилляр-
пая постоянная а2 имеет размерность площади; например, при комнатной температуре капиллярная постоянная воды а = 14,8 10-6м2. Величину а (м) называют капиллярной длиной. Для воды капилляр ная длина составляет а = 3,85 мм.
Капиллярная длина а определяет границу капиллярных явлений. Эти явления играют существенную роль в области размеров г« а. Напротив, для больших объектов (/» а) доминирует влияние силы тяжести, поэтому капиллярные явления отступаю! на второй план В космосе ускорение свободного падения во много раз меньше, чем на Земле, поэтому в космосе роль капиллярных явлении очень велика. Развитие космонавтики потребовало детального изучения законов капиллярного течения.
Уравнение (5.26) описывает статику капиллярного поведения смачивающих жидкостей. Важное значение имеют также кинетические закономерное!и капиллярного течения. Рассмотрим самый простой случай — течение смачивающей жидкости но горизонтальному капилляру диаметром d - 2г0 (г0 — радиус трубки).
Течение жидкости происходит под действием разности давлений Ар = р0-рм = А на плоской поверхности (р0) и в вогнутом мениске (рм = Ро~Рс)- Силу сопротивления создаст вязкость жидкости (г|, Па с). Для ньютоновских жидкостей вязкость не зависит от скорое!и течения (z\ м/с).
При сравнительно небольших скоростях течение происходит в ламинарном (безвихревом) режиме. Условием ламинарного течения по трубке является неравенство Re < 2 300 (Re — безразмерный критерии (число Рейнольдса): Re = тт/рж/тр рж — плотность жидкости).
Скорость ламинарного течения в трубке радиусом r0 = d/2 определяется законом Пуазейля (1842):
v
8т]/ ’
(5.28)
где / — длина трубки; Ар — перепад давления на концах трубки.
При капиллярном течении имеем Ар = 2ocosO/r0. Следовательно, скорость течения в горизонтальном капилляре описывается уравнением
0/0 cos О
4т|х
(5.29)
где л — расстояние от начала трубки.
Поскольку скорость течения v = dx/dr, зависимость перемещения (а) мениска от времени (/) определяется параболическим уравнением
ог0 cos О
2п
(5.30)
Таким образом, по мере удаления от входа (увеличения расстояния) в капилляр скорость перемещения мениска убывает.
В пористых телах капиллярное течение происходит значительно сложнее, чем в цилиндрической трубке Это связано со сложной геометрией порового пространства. Поры могут иметь различную форму и размер; кроме того, их расположение в пределах пористой среды может бьпь весьма разнообразным. По указанным причинам для количественной характеристики структуры пористых тел используют несколько усредненных параметров; важнейшими из них являются:
• пористость (е) — отношение объема порового пространства к общему объему пористого тела, т е. суммарному объему, который занимают поры и твердые частицы; очевидно, что. е < Г,
• удельная поверхность пор (Пул) — отношение суммарной пло-щади поверхности пор к общему объему пористого тела:
• средняя высота подъема (77, м) смачивающей жидкости в пористом теле определяется но аналогии с уравнением (5.26) из условия равновесия гидростатическою давления и капиллярною давления:
oQv,;cos0
(5.31)
где рж — плотность жидкости; g — ускорение свободного падения;
• скорость течения (Q, м’/с) жидкости в пористой среде определяется уравнением Дарси — аналогом закона Пуазейля (5.28):
ДрЛуд
(5.32)
где Др — разность давлении на входе жидкости в пористую среду длиной L и на выходе из нее, q — вязкое!ь жидкое!и, К — коэффициент проницаемости, который зависит главным образом от формы пор, м2.
Для теоретической оценки коэффициента проницаемости используют различные упрошенные модели поровою пространства
Одна из распространенных моделей — капиллярная: поровое пространство рассматривают как пучок параллельных капиллярных трубок одинаковою радиуса г0. Для такой модели коэффициент проницаемости равен К = сг2/8.
Необходимо отметить, что уравнение Дарси применимо только для ламинарного режима гечения жидкости в пористой среде. Для этого необходимо выполнение условия. Re < I.
В более точных методах расчета учитывают дополнительные факторы: изменение поперечного сечения потока в пористом пространстве, извилистость пор и др.
Практическое значение капиллярного течения жидкостей очень велико. В качестве примера отметим движение воды в почве, течение крови, пропитку разнообразных материалов (например, текстильных), топливные элементы и т д.
5.6. Измерение поверхностного натяжения и краевых углов смачивания
Поверхностное натяжение. Это важнейшая характеристика поверхности раздела фаз. Для решения многих задач коллоидной химии, химической технологии, металлургии, нефтедобычи и ряда других научных дисциплин и областей промышленности необходима подробная информация о поверхностном натяжении различных веществ и о температурной зависимости поверхностного натяжения
Из-за огромного разнообразия физических и химических свойств различных веществ (сжиженные газы, органические жидкости, вода, расплавы солей и оксидов, жидкие металлы и т.д.) и широкого температурного интервала измерение поверхностного натяжения с необходимой достоверностью и точностью представляет сложную методическую проблему, особенно в области очень низких (криогенных) и достаточно высоких (выше 400 К) температур. Сейчас такие методики существуют. Все они основаны на использовании законов капиллярных явлений, рассмотренных в гл. 5.
Большинство методов измерения поверхностного натяжения жидкостей основано на законе капиллярного давления Лапласа (см. подразд. 5.1), связывающем натяжение с кривизной поверхности жидкости. Каждый из этих методов имеет достоинства и недостатки Выбор оптимального метода зависит от конкретных условии — прежде всего от химической природы жидкости и граничащей с ней фазы, а также от температуры.
Рассмотрим наиболее распространенные методы.
Метод капиллярного подъема. Метод основан на использовании уравнения (5.25) высоты капиллярною подъема жидкости. Для повышения точности измерений нужно использовать достаточно тонкие капилляры, т.с. обеспечить выполнение условия г< а (а2 — капиллярная постоянная жидкости). Обычно этот метол применяют для жидкостей, которые полностью смачивают материал капиллярной трубки; тогда cosO= 1. Для обеспечения полного смачивания необходима тщательная очистка внутренней поверхности капилляра. Измерение высоты капиллярного поднятия проводят катетометром. Основной источник погрешностей — измерение радиуса капиллярной трубки, так как обеспечить сю постоянство вдоль всей длины трубки весьма сложно.
Метод капиллярного подъема позволяет измерять поверхностное натяжение индивидуальных однокомпонентных жидкостей с погрешностью до нескольких сотых процента. Для растворов (особенно для растворов поверхностно-активных веществ) этот метод менее пригоден из-за адсорбции растворенных веществ на внутренней поверхности капиллярной трубки (см. гл. 6).
Метод сидящей капли. Равновесная форма капли жидкости на твердой горизонтальной поверхности (рис. 5.4) определяется следующим условием: давление во всем объеме капли должно быть одинаковым. Давление (р) в плоскости основания капли АВ складывается из двух величин: гидростатического давления столба жидкости plf= рж££ (г — высота CD столба жидкости в точке С; рж — плотность жидкости; g — ускорение свободною падения) и положительного капиллярного давления (рД определяемого законом Лапласа (5.4), т.с. для поверхности с двумя радиусами кривизны имеем
Р = Рн + Ре= const.
После подстановки величин рн и pi получаем уравнение равновесной формы сидящей капли
+ Рж«г = С.
(5.33)
где Г|, г2 — главные радиусы кривизны; С — константа, зависящая от объема капли.
Уравнение (5.33) позволяет оценить, как изменяется кривизна поверхности достаточно большой капли. Около оси капли 0< гидростатическое давление рн максимально. Соответственно капиллярное давление в верхней точке Е будет минимальным. Это означает, что радиус кривизны гЕ поверхности капли в точке Е имеет максимальное значение. На линии смачивания АВ гидростатическое давление рИ = 0, поэтому радиус кривизны поверхности капли здесь минимален Таким образом, поверхность сидящей капли имеет переменную кривизну. Профиль капли зависит от краевого угла 0 и капиллярной постоянной жидкости а2 = 2о/(рж#) Для определения поверхностного натяжения достаточно найти размеры (радиус rt и высоту zi) капли в 3 — 4 сечениях. Далее по
Рис. 5.4. Схема для расчета профиля капли жидкости на твердой горизонтальной поверхности
определенным формулам проводят расчет поверхностного натяжения. Метод сидящей капли используют главным образом для измерения поверхностного натяжения жидких металлов и других тугоплавких веществ. Каплю расплава помещают в термостатированной камере (обычно в достаточно глубоком вакууме) на нс-смачиваемую подложку. В современных установках профиль капли и углы измеряют с помощью видеотехники с переводом изображения на компьютер. Минимальная ошибка метода составляем несколько десятых процента.
Метод максимального давления. При выдавливании газового пузырька из капиллярной трубки под действием внешнего давления в жидкость кривизна поверхности пузырька постепенно меняется. С ростом давления радиус кривизны г сначала уменьшается, его минимальное значение rmin = r0 (/у —радиус трубки). В этот момент капиллярное давление рс = 2о/г достигает максимального значения ртах. При дальнейшем раздувании пузырька радиус кривизны возрастает, капиллярное давление уменьшается и уже не может уравновесить внешнее давление на пузырек. Поэтому пузырек газа становится неустойчивым — он быстро увеличивается и отрывается от капилляра. В момент отрыва происходит скачкообразное уменьшение давления Др, которое фиксируется прибором для измерения давления, например жидкостным манометром. Для достаточно тонких капилляров (г0< а: а — капиллярная длина) мениск в момент отрыва пузырька имеет форму, близкую к полусфере. Тогда поверхностное натяжение рассчитывают по уравнению о = &ргц. Этот принцип используют и для измерения межфазного натяжения на границе двух нссмсшивающихся жидкостей I и 2. В этом случае небольшую каплю одной жидкости (например, предельного углеводорода) выдавливают в другую жидкость (воду) и измеряют скачок давления Др при отрыве капли от кончика капиллярной трубки.
Достоинства метода максимального давления — не нужно учитывать влияние краевого угта смачивания О и, главное, можно измерять поверхностное натяжение непрозрачных жидкостей, например жидких металлов и расплавленных солей. Метод пригоден также для измерения поверхностного натяжения растворов поверхностно-активных веществ при условии, что процесс адсорбции происходит сравнительно медленно (гл. 6). Основной недостаток метода — влияние различных флуктуаций (вибрации, неровности среза капилляра и т.п.), в рсзулыагс которых газовый пузырек может оторваться несколько раньше, чем он достигнет формы полусферы; поэтому измеряемый скачок давления может быть меньше максимального. Соответственно и расчетное значение поверхностного натяжения будет несколько заниженным.
Минимальная погрешность данного метода составляет 0,1 — 0,2 мДж/м2.
Метод пластинки Вильгельма. Измерение поверхностного натяжения согласно этому методу проводят следующим образом. Тонкую пластину подводят вертикально к поверхности жидкости, смачивающей материал пластины (обычно используют стекло, алюминий, сталь, платину). После контакта с жидкостью по обеим сторонам пластины образуются вогнутые мениски высотой h. В результате нижняя часть пластины оказывается погруженной в жидкость и ее вес уменьшается в соответствии с законом Архимеда на величину Аб= 2(рж — плотность жидкости; Им — объем одного мениска). Для сохранения механического равновесия на пластину в противоположном направлении (вверх) должна действовать сила/= А <7; эту силу / измеряют достаточно чувствительным динамометром или весами.
Для установления связи между измеряемой силой /и поверхностным натяжением жидкости о нужно рассчитать объем капиллярного мениска Им. Высота капиллярного подъема Н жидкости вдоль вертикальной поверхности определяется (как и для капилляра) капиллярным давлением в вогнутом мениске. Из закона Лапласа следует, что в данном случае Н = а(\ -sin0)l/2 (а — капиллярная длина; 0 < 90° — краевой угол смачивания). Объем мениска равен Им ~ {I — ширина пластины); соответственно 1 змепенпс веса пластины после образования двух менисков составляет
Д(? = H2lp*g = 2o/cos0.
При полном смачивании имеем cos0 =1 и о = У/(2/).
Точность определения поверхностного натяжения методом пластины Вильгельм и может быть доведена до 0,01 мДж/м2. Необходимое условие такой точности — краевой угол смачивания 0 материала пластины данной жидкостью должен быть измерен независимо и достаточно точно.
Метол пластинки Вильгельм и позволяет получить информацию об изменении уравновешивающей силы/в процессе формирования мениска, т.е. найти зависимость/(г) (/— время). Тем самым можно получить необходимые сведения о кинетике смачивания, т.е. о зависимости краевого угла 0 от времени / контакта жидкости с твердой поверхностью. В современных приборах кинетическая зависимость/(/) записывается автоматически.
Метод вращающейся капли. Этот метод используют для измерения очень малых поверхностных натяжений на границе двух нс-смешивающихся жидкостей А и В. Рассмотрим случай, когда капля более легкой жидкости А помещена в трубку, заполненную жидкостью В. При вращении трубки вокру! ее оси центробежные силы перемещают каплю к оси и «расплющивают» ее — превращают в эллипсоид. Равновесная форма такой капли зависит от угловой скорости вращения ю, которая связана с поверхностным
натяжением (о12) на границе двух несмешивающихся жидкостей I и 2 уравнением
с12 = Дрогг3/4, (5.34)
где Др = (рв - рА) — разность плотностей жидкостей А и В; г — радиус трубки.
Метод вращающейся капли позволяет измерять поверхностные натяжения до 10 ’ мН/м, что особенно важно для микроэмульси-онных систем и для процесса самопроизвольного диспергирования (см. подразд. 4.4).
Измерение поверхностной энергии твердых тел. Экспериментальное измерение удельной поверхностной энергии твердых тел связано с большими трудностями. Главное затруднение заключается в том, что образование твердой поверхности представляет, как правило, необратимый процесс из-за неизбежной пластической деформации поверхностного слоя. Для пластичных материалов (металлов) работа, связанная с пластической деформацией, значи-гельно превышает работу, необходимую для разрыва образца. Методы, применяемые для определения поверхностной энергии твердых тел на границе с газом (отг) и жидкостями (отж), по указанной причине менее точны, чем методы измерения поверхностного натяжения жидкостей В основном используются два метода.
1. Метод нулевой ползучести. Измерения проводят вблизи температуры плавления данного материала, когда он обладает высокой пластичностью и легко удлиняется под действием небольших нагрузок. Вертикально подвешенные образцы из металлической фолы и не растягиваются при условии / = оТ|/ (/ — экспериментально определяемая сила, при которой отсутствует ползучесть (отсюда название метода), /— ширина образца).
2. М е т о д хрупкого р а з р у ш е н и я. При достаточно низких температурах (в жидком азоте) пластическая деформация, предшествующая разрушению, очень мала. Поэтому по измерениям разрушающей силы можно рассчитать поверхностную энергию Он данного материала.
Для низкоэнергетических 1вердыхтел поверхностные энергии оп и <э|Ж можно определить, измеряя краевые углы 0 разных жидкостей в контакте сданным материалом (см. подразд. 5.2, уравнение (5.10)). Достоинство этого метода заключается в том, что он не требует механического деформирования и разрушения образцов, гем самым исключаются погрешности, вызванные пластической деформацией и необратимостью процесса образования поверхности.
Краевые углы. Прямой (гониометрический} метод. Наиболее распространен метод сидящей капли. Схема опыта такая же, как при измерении поверхностною натяжения жидкостей этим методом.
Дозированная капля жидкости наносится на горизонтальную твердую пластинку, помещенную в закрытую термостатированную камеру. Для измерения краевого угла используют гониометр — микроскоп, окуляр которого имеет угловую шкалу. Основной источник погрешностей — некоторая неопределенность в положении вершины угла О, что особенно существенно при измерении небольших (менее 10°) краевых углов В современных установках вместо визуального отсчета краевого угла изображение капли передается на экран компьютера.
Косвенные методы. Для уменьшения погрешности краевой угол определяют косвенным путем — по измерению размеров капли: высоты h и радиуса г основания. Для небольшой капли (г « а) влияние силы тяжести на ее форму пренебрежимо мало и каплю можно рассматривать как сферический сегмент, соответственно h = rtgO/2. Для небольших углов (О < 10 ) достаточную точность дает соотношение h = rQ/2, т.е 0 = 2h/r.
Для жидкостей с известным поверхностным натяжением краевые углы можно найти путем расчета по высоте капиллярного подъема или по силе, уравновешивающей пластину Вильгельм и.
Контрольные вопросы
1. Назовите факторы, которые играют основную роль в капиллярных явлениях.
2. Дайте определение понятия «капиллярное давление». Напишите закон капиллярного давления Лапласа для случая сферической поверхности и для общего случая.
3. Рассчитайте капиллярное давление для сферической капли воды радиусом 1 мкм при компа!нои температуре. Поверхностное натяжение воды при температуре 20 С равно 72,88 мН/м.
4 Дайте определение понятии «краевой угол смачивания», «линия смачивания (линия трехфазового контакта)»
5 Какое капиллярное явление называют избирательным смачиванием?
6. Напишите закон Юнга для краевого угла смачивания и рассмотрите основные случаи контактного взаимодействия жидкостей с поверхностью твердых тел. Объясните связь правила Антонова с законом Юнга.
7. Назовите основные отличия процесса смачивания реальных поверхностей от идеализированных Приведите уравнения, учитывающие влияние этих отличий на краевые углы смачивания
8. Объясните физико-химический смысл линейного натяжения. В каком интервале размеров капель влияние этого фактора на краевые углы смачивания становится существенным?
9. В чем состоит явление адгезии? Дайте определение понятия «работа адгезии» и укажите связь этого параметра с краевым углом смачивания.
10 Объясните смысл классификации твердых поверхностей на низко- и высокоэнергетические.
11. Напишите закон Кельвина для связи давления насыщенного пара с кривизной поверхности жидкости. Объясните на основе этого закона процесс изотермической перегонки.
12. Напишите уравнение капиллярного подъема для смачивающих жидкостей. Рассчитайте высоту подъема октана (поверхностное натяжение при температуре 20°C равно 21,76 мН/м; плотность 0,87 г/см3) в стеклянной вертикальной трубке радиусом 0,1 мм.
13. Какую величину на?ывают капиллярной постоянной жидкости? Почему влияние капиллярных явлений в условиях космических полетов гораздо больше, чем на Земле?
14. Назовите и охарактеризуйте основные методы измерения поверхностного натяжения жидкостей.
15. Укажите основные методы измерения поверхностной энергии твердых тел. В чем заключается основная трудность в проведении этих измерений?
ГЛАВА 6
АДСОРБЦИЯ ПОВЕРХНОСТНО-АКТИВНЫХ
ВЕЩЕСТВ
Адсорбция относится к числу наиболее распространенных и важных поверхностных явлений. Адсорбцией называют процесс самопроизвольного концентрирования вещества на границе раздела двух фаз. В результате адсорбция приводит к значительному увеличению концентрации этого вещества на поверхности жидкости или твердого тела.
В коллоидной химии адсорбция играет чрезвычайно важную роль. Причина этого заключается в том, что поверхностные явления на границе дисперсной фазы и дисперсионной среды определяют многие свойства дисперсных систем. Адсорбция определенных веществ является самым эффективным и гибким регулятором поверхностных свойств, в первую очередь — поверхностного натяжения.
Вещества, способные к адсорбции на поверхности раздела фаз, называют поверхностно-активными. Необходимо подчеркнуть, что способность к адсорбции зависит не только от химической природы самого адсорбирующеюся вещества, но и от природы фаз, образующих данную поверхность. Например, ацетон поверхностно активен по отношению к хлороформу на границе с воздухом и нс активен по отношению к гексану. По отношению к ртути большой поверхностной активностью обладают щелочные металлы, особенно цезий.
Таким образом, важнейшее положение теории адсорбции заключается в том, что поверхностная активность — эго не индивидуальное свойство какого-либо вещества, а его свойство в определенной гетерогенной системе.
Во многих природных и технических системах дисперсной фазой или дисперсионной средой является вода или водные растворы и неполярные органические жидкости. По отношению к ним к числу эффективных поверхностно-активных всшсств относят многие органические вещества, молекулы которых имеют дифилъную структуру. Они содержат достаточно длинную неполярную часть (например, углеводородную цепь) и концевую полярную группу. Таковы, например, спирты, карбоновые кислоты и их соли.
Подробно строение, свойства и номенклатура поверхностноактивных веществ рассматриваются в гл. 7.
6.1. Поверхностное натяжение растворов
Основные закономерности адсорбции выявляются наиболее отчетливо для водных растворов ПАВ на границе с газом (воздухом). Главное следствие адсорбции — уменьшение поверхностного натяжения растворителя (воды). Поэтому в коллоидной химии используют сше одно определение: ПАВ — это вещества, которые уменьшают поверхностное натяжение (свободную поверхностную энергию) данной фазы — жидкости или твердого тела. Этот принцип позволяет экспериментально, по результатам измерения поверхностного натяжения растворов разделить вещества на две группы:
1) поверхностно-активные вещества, которые снижают поверхностное натяжение растворителя;
2) поверхностно-инактивные вещества, которые вызывают увеличение поверхностного натяжения растворителя (рис. 6.1).
По отношению к воде па границе с воздухом поверхностно инактивны электролиты (кислоты, шелочи, соли), т.е. вещества более полярные, чем вода.
Рассмотрим самый простой случай: влияние ПАВ со сравнительно коротким неполярным радикалом, т.е. с числом атомов углерода менее К). Влияние более длинных молекул ПАВ имеет свою специфику (см. гл. 8).
Зависимость поверхностного натяжения раствора от его концентрации при постоянной температуре называют изотермой поверхностного натяжения. Поверхностное натяжение уменьшается наиболее резко на начальном участке изотермы — в области малых концентраций ПАВ. Далее ход изотермы становится более пологим (см. рис. 6.1). При достаточно больших концентрациях ПАВ поверхностное натяжение уменьшается примерно в 2 раза по сравнению с поверхностным натяжением растворителя (воды).
Зависимость поверхностною натяжения водных растворов ПАВ от их концентрации (С) достаточно точно описывает эмпирическое уравнение Шиш ков с кого'.
о0-<5 = Я1п(1 + АС), (6.1)
где (у0 — поверхностное натяжение воды (при С- 0); В — постоянный для данного класса ПАВ параметр, мДж/м2; значения нара-
а, мДж/м 80
70
60
50
40----1------1--1------1---
0 0,2 0,4 0,6 0,8 С, моль/л
Рис. 6.1. Изотермы поверхностного натяжения водных раст воров:
/ — поверхностно-активное вещество (масляная кислота); 2 — повсрхностно-инактивное всщесгво (сульфат натрия)
Рис. 6.2. Изотермы поверхностного натяжения водных растворов органических кислот:
/ — муравьиная; 2 — уксусная; J — пропионовая; 4 — масляная; 5 — изо-валериановая
метра В различны для спиртов, карбоновых кислот и других классов ПАВ, но они одинаковы для одного гомологического ряда, А — параметр, характеризующий адсорбционную активность индивидуального ПАВ (определение термина «поверхностная активность» дано ниже), моль1.
В каждом гомологическом ряду ПАВ параметр А увеличивается в среднем в 3 — 3,5 раза при переходе к следующему члену ряда. Физико-химически и смысл параметров А и В в уравнении Шиш-ковского (6.1) разъясняют теория адсорбции ПАВ (см подразд 6.2) и молекулярные модели строения адсорбционных слоев ПАВ (см. подразд. 6.4).
Для сравнения эффективности различных поверхностно-активных веществ используют специальную количественную характеристику — поверхностная активность, которая определяется начальным наклоном изотермы поверхностного натяжения, т.е. равна -do/dC при ДС —> 0.
В гомологических рядах ПАВ поверхностная активность растет по мере перехода к более высоким гомологам (рис. 6.2). При этом выполняется следующая закономерность: при удлинении углеводородной цепи в молекуле поверхностно-активного вещества на одну группу —СН2— поверхностная активность i омолога возрастает в 3 — 3,5 раза, т.е. так же как и параметр А в уравнении Шиш конского (6.1). Это правило было установлено опытным путем в конце XIX в. (правило Дюкло—Траубе).
Таким образом, с ростом углеводородной цепи в арифметической прогрессии поверхностная активность ПАВ увеличивается в геометрической прогрессии.
6.2. Уравнение адсорбции Гиббса
Снижение поверхностною натяжения растворов ПАВ является следствием их адсорбции на поверхности растворителя. Это принципиальное положение следует из термодинамической теории адсорбции Гиббса (1878).
Важнейший гермодинамический параметр теории Гиббса — удельная адсорбция (Г) — самопроизвольное концентрирование вещества на поверхности раздела фаз А и В но сравнению с количеством этою вещества во внутренних объемах фаз А и В. Как и при определении автоадсорбции Го (см. подразд. 4.2). такое сравнение проводят для одинаковых объемов поверхностного слоя на границе фаз А и В и объемов этих фаз.
В коллоидной химии вместо термина «удельная адсорбция» обычно для краткости используют термин «адсорбция». По определению избыточное число молекул (Ад) в поверхностном слое равно
Ал = Д5-(нЛ + лв),
где дх — число молекул ПАВ в поверхностном слое толщиной 6; дЛ, /7в — число молекул ПАВ в объемах фаз А и В соответственно.
Как и в однокомпонентной системе, соответствующие внутренние объемы ИА и Кв равны объемам двух областей, образующих поверхностный слой толщиной 5 (см. рис. 4.4).
Таким образом, удельная адсорбция (Г, моль/м2) определяется уравнением
Г = [//, - (иЛ + HB)l/(Q/yxl), (6.2)
где Na — число Авогадро.
Для гетерогенных систем, в которых одной из граничащих фаз является «аз, при определении адсорбции ПАВ можно пренебречь концентрацией ПАВ в газовой фазе. Тогда для раствора ПАВ на границе с воздухом можно пользоваться приближенным уравнением
Г=(С5-С)б, (6.3)
где Cs — концентрация ПАВ в поверхностном слое толщиной 5; С — концентрация раствора.
Таким образом, здесь снова используется фундаментальное положение теории Гиббса о том, что тонкий поверхностный слой представляет самостоятельную фазу.
Дж.Гиббс показал, что для двухкомпонентной двухфазной системы, в которой один из компонентов адсорбируется на границе раздела фаз А и В, связь между адсорбцией и поверхностным натяжением описывается уравнением
Г = -do/dp. (6.4)
Дчя разбавленных растворов ПАВ уравнение адсорбции Гиббса (6.4) можно привести к форме, позволяющей проводить расчеты изотермы адсорбции ПАВ, т.с. зависимости удельной адсорбции (Г) от концентрации раствора (С). Химический потенциал рас
творенного вещества равен р = р° + RT1п<7 (,u° — стандартный химический потенциал; R — универсальная газовая постоянная; а — активность компонента, в данном случае — активность ПАВ). Для идеальных растворов вместо активности в выражение химического потенциала можно вводить концентрацию С. Тогда dp = = (RT/C)dC и уравнение (6.4) принимает вид
Г = (-do/dC)C/(AT). (6 5)
Таким образом, для расчета адсорбции нужно знать изотерму поверхностного натяжения о = f(C) раствора данного ПАВ (см. рис. 6.1). В нескольких точках этой изотермы находят значение производной -da/dC. Далее по уравнению (6.5) рассчитывают удельную адсорбцию Г при этих концентрациях и строят график Г=/(С). На рис. 6.3 показаны типичные для многих ПАВ изотермы адсорбции Г =/’((?).
Наряду с поверхностной активностью (-do/dC при С -+ 0) для характеристики ПАВ используют еще один термодинамический параметр — работу адсорбции (И^ис), которая равна работе переноса I моля ПАВ из внутреннего объема раствора на его поверхность, граничащую с соседней фазой, при постоянной температуре. Для расчета работы адсорбции снова рассмотрим самый простой случай — разбавленный водный раствор ПАВ на границе с газом Тогда химический потенциал ПАВ в объеме раствора концентрацией С равен и = + RT\nC. Для разбавленных растворов
удельная адсорбция (Г) значительно меньше предельной адсорбции (Гтзх), соответствующей насыщенному монослою ПАВ па поверхности раствора (см. подразд. 6.4). При условии Г « ГП1АХ поверхностный слой толщиной 5 представляет собой разбавленный поверхностный раствор. Тогда химический потенциал ПАВ в поверхностном слое вещества можно определить обычным соотношением = и° + RThiC, (Q — поверхностная концентрация; ,и° — стандартный химический потенциал ПАВ в поверхностном слое раствора). Условие термодинамического равновесия между расгво-
Рис. 6.3. Изогермы адсорбции водных растворов органических кислот:
1 — пропионовая, 2 — масляная; 3 — изовалериановая; 4 — капроновая (Г!11;|Ч — максимальная адсорбция)
ром ПАВ и поверхностным слоем заключается в равенстве их химических потенциалов: ц = Подчеркнем, что стандартные потенциалы ПАВ в объеме раствора (ц°) и поверхностном слое (р5°) отличаются (ц ф ц°). После подстановки выражений для указанных химических потенциалов р и р? получим
СА./С=ехр[(ц°-ц;)/(/?П]. (6-6)
Разность стандартных химических потенциалов данного ПАВ (ц° - ц°) но определению равна работе переноса 1 моля ПАВ из объема на поверхность раствора в стандартных условиях. Именно эта разность определяет работу адсорбции:
ид = ,и° - и;. (6.7)
Из уравнения (6.6) получим
ИД = /?Пп(С,/С).
Для разбавленных растворов ПАВ выполняется соотношение Cs» С, поэтому уравнение (6.3) можно упростить:
Г = СД
где 5 — толщина поверхностного слоя.
Отсюда уравнение работы адсорбции принимает вид
ИД = /?Пп[Г/(5С)]. (6.8)
Уравнение (6.8) используют для расчета работы адсорбции различных: ПАВ по экспериментальным данным. В гомологических рядах ПАВ при переходе к очередному гомологу работа адсорбции возрастает примерно на 3 кДж/моль. Этот результат позволяет объяснить причину увеличения поверхностной активности примерно в 3 — 3,2 раза при удлинении углеводородной цепи в молекуле ПАВ на одну группу —СН2— (см. выше).
6.3. Уравнение Ленгмюра
Важнейшее следствие адсорбции ПАВ заключается в том, что концентрация ПАВ в поверхностном слое раствора (Cv) значительно больше концентрации в объеме раствора (С): С5» С. Поскольку молекулы ПАВ нс закреплены на поверхности, возможен их переход из поверхностного слоя в объем раствора.
Адсорбция ПАВ, таким образом, определяется конкуренцией двух процессов, которые идут в противоположных направлениях:
1) движение молекул ПАВ из объема на поверхность раствора (адсорбционный поток ia)\ этот процесс стимулируется энергетически — уменьшением поверхностного натяжения раствора в соответствии с уравнением адсорбции Гиббса (6.4);
2) движение молекул ПАВ с поверхности в объем раствора, обусловленное более высокой концентрацией ПАВ в поверхностном слое раствора {десорбционный поток id)\ поскольку десорбция ПАВ направлена на выравнивание концентраций, термодинамически этот процесс обусловлен возрастанием энтропии (поверхностный слой + раствор) системы при более равномерном распределении ее компонентов.
Таким образом, адсорбционное равновесие на поверхности раздела фаз раствор ПАВ —газ носит не статический, а динамический характер. Соответственно для молекул ПАВ в адсорбционном слое существует некоторое статистическое «время жизни» на поверхности, после которого вероятность их десорбции становится весьма бочьшой и они покидают поверхностный слой и возвращаются в раствор. Поэтому количество ПАВ в поверхностном слое определяется соотношением потоков адсорбции и десорбции.
Представления о динамической природе адсорбционно-десорбционного равновесия в поверхностном слое составили основу теории Ленгмюра (1920). Следует отметить, что уравнение Ленгмюра впервые было получено для границы раздела твердое тело—газ. Оно устанавливает в явной форме связь между адсорбцией Г и концентрацией С молекул в газовой фазе. За развитие теории адсорбции па разных границах раздела фаз и исследование строения адсорбционных с поев ПАВ (см. подразд. 6.4) И.Ленгмюр в 1932 г. был награжден Нобелевской премией по химии.
Теория Ленгмюра построена на упрощенной модели процессов адсорбции и десорбции ПАВ.
Первое допущение — молекулы ПАВ могут адсорбироваться из раствора только на свободных от ПАВ участках поверхности раствора. Те участки, которые к данному моменту времени уже заняты молекулами ПАВ, для адсорбции новых молекул недоступны. Таким образом, адсорбция ПАВ происходит только на тех участках поверхности, которые заняты молекулами растворителя.
Второе допущение относится к молекулярному строению адсорбционного слоя ПАВ. Принимают, что он состоит только из одного слоя молекул ПАВ (монослой). Поэтому удельная адсорбция имеет предельное значение (ГП1ах), которое достигается при образовании насыщенного монослоя ПАВ. При мопослойном строении доля поверхности ф, занятая молекулами ПАВ, будет равна ф = Г/Гтах. Доля «свободной» поверхности, которая доступна для адсорбции дополнительных молекул ПАВ, равна 1 -ф = 1 -Г/Гтах.
В рамках этой модели можно рассчитать скорость адсорбции (гй, моль/(м2 с)) — количество ПАВ, которое выносит адсорбционный поток на единицу площади поверхности раствора в единицу времени. Скорость адсорбции пропорциональна концентрации ПАВ (С) в растворе и доле (1 - ф) доступной поверхности:
vtJ= ад1-г/гП1;1Х), (6.9)
где K(l — конетанта скорости адсорбции, л/(м2-с).
Скорость десорбции (vd) пропорциональна только доле поверхности ф, занятой адсорбированными молекулами, так как в объеме раствора всегда есть свободные места для размещения дссор-би рова1111 ы х моле кул:
vd=Kd^ (6.10)
где Kd — константа скорости десорбции. моль/(м2с).
Условие динамического равновесия между поверхностным слоем раствора ПАВ и самим раствором заключается в равенстве скоростей потоков адсорбции и десорбции: va = vd. После подстановки уравнений (6.9) и (6.10) получаем уравнение адсорбции Ленгмюра.
где А — коэффициент, равный отношению констант скоростей адсорбции и десорбции:
>4 — KJ
коэффициент А имеет размерность обратной концентрации Рассмотрим два предельных случая
I. В области малых конценграций, когда выполняется условие АС « 1, из уравнения Ленгмюра (6.11) следует, что удельная адсорбция (П прямо пропорциональна объемной концентрации ПАВ (С):
Г = ГП1ахЛС. (6.12)
Значит, начальный участок изотермы адсорбции Г = f(C) (см рис. 6.3) представляет область Генри для поверхностною раствора ПАВ, а произведение Г|1тЛ = КИ можно рассматривать как константу Генри (КИ) для адсорбции ПАВ.
2 В области больших концентраций, когда выполняется условие АС> I, из уравнения Ленгмюра (6.11) получаем Г = Гтах, т.е. при больших концентрациях достигается предельная, или максимальная, адсорбция (Гп1.и). Причина существования такого предела заключается в образовании насыщенного монослоя молекул ПАВ. который занимает всю поверхность раствора (см. подразд. 6.4).
Динамическое уравнение адсорбции Ленгмюра (6.11) в сочетании с термодинамическим уравнением Гиббса (6.5) для разбавленных растворов позволяет теоретически описать зависимость поверхностного натяжения раствора от концентрации ПАВ. Введем в уравнение адсорбции Гиббса для разбавленных растворов
(6.5) адсорбцию Г, определяемую уравнением Ленгмюра, получим соотношение
-do/dC = ГтахЯ7>1С/(1 +ЯС),
отсюда следует
о0-су = Гтах/?7’1п(1 + АС). (6.13)
Это выражение совпадает с уравнением Шишковского (6.1), параметр В при этом равен
£=rmax/?r. (6.14)
Предельная адсорбция ГП1ач определяется площадью Г20, которую занимает одна молекула ПАВ в насыщенном монослос:
Г„^= 1/(^о А,), (6.15)
• где Ал — число Авогадро.
Для гомологического ряда площадь £20, занимаемая молекулой ПАВ, определяется размерами ее полярной группы, поэтому она одинакова для всех гомологов. Этим объясняется постоянство параметра В в уравнении Шишковского (6.1):
B=RJ/(^N4).
6.4. Строение адсорбционных слоев ПАВ
Адсорбционные слои ПАВ являются важнейшим регулятором многих поверхностных явлений и коллоидно-химических процессов. Поэтому свойства и строение адсорбционных слоев играют большую роль во многих дисперсных системах.
Строение адсорбционных слоев зависит от двух факторов.
Первый фактор — химический состав и молекулярная структура самих ПАВ.
Второй важный фактор — химическая природа и агрегатное состояние граничащих фаз. Поверхности твердых тел обычно весьма неоднородны как в геометрическом отношении (разнообразные неровности), так и в отношении поверхностной энергии отдельных участков (яркий пример — поверхности поликристаллов) (см. подразд. 5.2). Поэтому адсорбция ПАВ на твердых телах имеет существенные отличия от адсорбции на поверхности жидкостей (последнюю можно считать однородной) Очно из таких отличий — на твердых поверхностях часто происходит не монослой ная, а по-лислоиная адсорбция, т.е. толщина адсорбционных слоев больше размера молекул ПАВ.
Наиболее простое строение имеют адсорбционные слои низкомолекулярных ПАВ на поверхности водного раствора, граничащего с газом. Главный вопрос относится к расположению молекул
ПАВ в поверхностном слое. Энергетически самое выгодное расположение соответствует нахождению полярной группы молекулы (гидроксильной. карбоксильной и т.д.) в поверхностном слое воды. В этом положении полярные «руины гидратированы. Неполярные «руины (у«леводородные цепи), напротив, находятся в газе.
Такое расположение молекул ПАВ следует из весьма общего принципа организации адсорбционных слоев — правила уравнивания полярности (П. А. Ребиндер, 1936). Сузь этою правила заключается в следующем. Адсорбция ПАВ имеет два следствия:
I) уменьшение поверхностного натяжения раствора (do/dC < 0);
2) уменьшение разности полярностей граничащих фаз А и В (например, воды и воздуха).
Количественной мерой полярности вещества может служить его диэлектрическая проницаемость (е). В соответствии с правилом уравнивания полярностей диэлектрическая проницаемость ПАВ (гПЛВ) должна занимать промежуточное место между диэлектрическими проницаемости ми граничащих фаз А и В: ел> епап> ев.
Другой принципиальный вопрос — как ориентированы молекулы ПАВ по отношению к поверхности жидкости (воды) Ориентация мопскул существенно зависит от концентрации раствора ПАВ. При этом выделяют три случая, которые соответствую! разным участкам изотермы адсорбции Г = /(С).
1. Разреженный монослой. Такой слой формируется в области Генри, когда концентрация ПАВ мала: С« 1/Л (см. подразд. 6.3). Характерная черта разреженного монослоя — среднее расстояние L между молекулами ПАВ гораздо больше их длины /: L » /. При этом условии полярные группы молекул ПАВ взаимодействуют с водой, но нс друг с другом. Предпочтительная ориентация углеводородного радикала — параллельная или под малым у «лом к поверхности жидкости.
В разреженном монослое молекулы ПАВ активно участвуют в двухмерном тепловом движении. Они интенсивно перемещаются но поверхности воды в произвольных направлениях. Такая молекулярно-кинетическая картина позволяет рассматривать разреженный адсорбционный слои как двухмерный аналог идеального газа. Отличие от «аза заключается в меньшем числе степеней свободы (четыре вместо шести), так как перемещения перпендикулярно поверхности запрещены в обоих направлениях — как вверх (в газ), так и вглуб«> (в раствор). Аналогия с двухмерным газом позволяет описать адсорбционный слой ПАВ уравнением Гиббса (6.5) для разбавленных растворов ПАВ. В области Генри поверхностная активность равна
-do/dC= До/С,
где До = (о0 - о) — разность поверхностных натяжений чистого растворителя и раствора ПАВ концентрацией С.
Тогда из уравнения Гиббса получаем Г = &o/(RT). Обозначим = 1/Г — площадь, которую занимает на поверхности 1 моль данного ПАВ. Разность поверхностных натяжений (оп - ст) = тс представляет двухмерное давление (я), которое создают адсорбированные молекулы. После подстановки выражений для и я в уравнение адсорбции получаем уравнение состояния разреженного монослоя:
яйя, = RT, (6.16)
которое представляет полный двухмерный аналог уравнения Менделеева-Клапейрона для идеальных газов pV = RT (р — давление; И — молярный объем).
2. Плотный монослой. При концентрациях С~ \/А область Генри на изотерме адсорбции Г=/(С) заканчивается. На молекулярном уровне это происходит потому, что расстояние L между адсорбированными молекулами стало меньше их осевой длины / (L < /). Поэтому увеличение удельной адсорбции с ростом концентрации требует такой ориентации молекул ПАВ, при которой будет возможна их более плотная упаковка на поверхности. Таким образом, необходима переориентация молекул ПАВ. Некоторое количество молекул должно расположиться перпендикулярно поверхности жидкости. Тогда занимаемая ими площадь станет минимально возможной. На этом участке изотермы Г = ./(С) адсорбционный слой частично состоит из перпендикулярно ориентированных молекул ПАВ, а частично — из молекул, расположенных параллельно поверхности раствора.
По сравнению с разреженным монослоем возникает важное отличие. Углеводородные радикалы перпендикулярно ориентированных, близко расположенных молекул взаимодействуют друг с другом. Такое боковое (латеральное) взаимодействие усиливается с ростом длины радикала. Соответственно двухмерный поверхностный раствор перестает быть идеальным и для его описания вводя! поправки, аналогичные учету собственного объема молекул и взаимодействия между ними в уравнении реального газа Ван-дер-Ваальса. Состояние плотною адсорбционною слоя описывает уравнение Фрумкина’.
(л + a/Q’)(П„-Йо)= ЯГ, (6.17)
где а — параметр, характеризующий латеральное притяжение молекул ПАВ; По = М^пав ~ площадь, занимаемая 1 молем ПАВ при перпендикулярной ориентации всех молекул; ППАВ — плошать, занимаемая в таком слое одной молекулой ПАВ (площадь полярной группы).
Уравнение (6.17) хорошо выполняется для достаточно длинных молекул ПАВ — с числом атомов углерода п > 8.
3. Предельно упакованный монослой. Плотнейшая упаковка молекул в адсорбционном слое ПАВ реализуется при перпендикулярной ориентации всех молекул, причем каждая из них занимает минимальную плошадь £1пав- Такой упаковке соответствует предельная (максимальная) адсорбция:
Г„т = WWA (6.18)
Максимальной адсорбции соответствует плато на изотерме адсорбции Г = /(С).
Такое строение насыщенного монослоя объясняет посюянство параметра В= ГтахАГв уравнении Шишковского (6.1). Действительно, в пределах одного гомологического ряда площадь £2ПАВ является постоянной; она определяется размером полярной группы. Для карбоновых кислот НПлв = 0,216 нм2, спиртов — 0,205 нм2. Поэтому предельная адсорбция и соответственно параметр В имеют постоянные для гомологического ряда ПАВ значения.
Плотно упакованный адсорбционный монослой полиэлектролитов и высокомолекулярных соединений приобретает механические свойства, характерные для твердых тел и структурированных жидкостей, — высокую вязкость и предельное напряжение сдвига. Благодаря этим свойствам адсорбционные слои макромолекул эффективно препятствуют непосредственному контакту дисперсных частиц в коллоидных растворах, пенах, эмульсиях и создают тем самым структурно-механический барьер, обеспечивающий стабилизацию (устойчивость) дисперсных систем.
Еще одно важное свойство адсорбционных слоев заключается в том, что они представляют самоорганизованную структуру, поскольку при достижении предельной адсорбции все молекулы ПАВ имеют одинаковую ориентацию — параллельно друг другу7 и перпендикулярно поверхности раздела фаз. Такая самоорганизация адсорбционных слоев имеет большое значение в биологии. Имеются убедительные экспериментальные данные о возникновении подобных упорядоченных структур органических молекул на поверхности раздела фаз в океанской и морской воле. Такие структуры способны к эволюции и рассматриваются поэтому как предшественники простейших живых организмов. Известный физико-химик Ф.Дж.Доннан отмечал, что ориентация молекул и ионов на поверхности раздела фаз имеет первые признаки организованной структуры жизни.
6.5. Адсорбция ПАВ на поверхности раздела жидких фаз
Адсорбция ПАВ на поверхности раздела жидких фаз представляет собой более сложное физико-химическое явление, чем ад
сорбция на поверхности жидкость —газ Основной осложняющий фактор заключается в том, что во многих случаях ПАВ растворяются в граничащей жидкости. Это влияет на удельную адсорбцию и строение адсорбционного слоя молекул ПАВ.
Широко распространены системы, в которых дисперсной фазой и дисперсионной средой являются полярные и неполярные жидкости, например вода и предельные или ароматические углеводороды. К числу таких дисперсных систем относя тся прямые и обратные эмульсии Другой важный случай — избирательное смачивание поверхности твердого тела, когда с ней одновременно контактируют полярная и неполярная жидкости (см. подразд. 5.2). В данном подразделе рассмотрены закономерности адсорбции ПАВ на плоской поверхности раздела полярной и неполярной жидкостей.
В зависимости от растворимости ПАВ в граничащих жидкостях следует различать три характерных случая.
I. Поверхностно-активные вещества хорошо растворяются в воде и не растворяются в неполярной жидкости. В эту группу входят ПАВ, которые хорошо диссоциируют в воде. Полярная группа в молекуле ПАВ имеет положительный или отрицательный заряд. Такие ПАВ называют ионогенными (см. подразд. 7.1). Другое условие растворимости только в воде — углеводородный радикал должен быть «промежуточной длины», т е. не слишком коротким (в этом случае ПАВ будут в определенной мерс растворимы и в неполярной жидкости) и не слишком длинным (иначе ПАВ не будут растворяться в воде). Оптимальное число атомов углерода (/?с) в алифатической цепи таких ПАВ — от 10 до 18.
2. Поверхностно-активные вещества хорошо растворяются в неполярной жидкости, но не растворяются в воде. Молекулы таких ПАВ имеют достаточно длинную неполярную часть: ис> 20.
3. Поверхностно-активные вещества растворяются в обеих граничащих жидкостях — в полярной и неполярной. В эту группу входят низшие гомологи (лс< 10) жирных кисло г, их солеи, спиртов и некоторые другие вещества.
Рассмотрим характерные черты адсорбции ПАВ этих грех групп на межфазной поверхности полярная жидкость—неполярная жидкость.
Поверхностпо-активпые вещества, растворимые в полярной жидкости. Для этих ПАВ адсорбция на поверхности раздела водною раствора с органической жидкостью имеет значительное сходство с адсорбцией на поверхности раствор ПАВ—газ (см подразд. 6.1 — 6.4). Отметим эти сходные черты.
1. Зависимости поверхностного натяжения оАВ на границе полярной (А) и неполярной (В) жидкости от концентрации ПАВ (С) в водном растворе (изотермы поверхностного натяжения) имеют такую же форму (рис. 6.4), как изотермы поверхностного
С, ммоль/л г
Рис. 6.4. Изогермы поверхностного натяжения на границе водный раствор ПАВ —органическая жидкость:
а — вода —бензол. Т = 35 С: / — масляная кислота; 2 — валериановая кислота, 3 — капроновая кислота; 4— каприловая кислота; б — вода —гептан. Т = 20С: / — этиловый спирт; 2 — бутиловый спирт; 3 — гексиловый спирт; в — вода — парафиновое масло. Т - 50 С: / — лаурат натрия; 2 — миристат натрия; 3 — пальмитат натрия; 4 — стеарат натрия; г — вода —гептан, Г= 50 С: 1 — октил-сульфат натрия; 2 — дснилсульфат натрия; 3 — додецил сульфат натрия, 4 — тетраденилсульфат натрия: 5 — гексадецилсульфа i натрия; 6 — октадецилсульфа г натрия
натяжения о =/(С) водных растворов ПАВ с короткой углеводородной цепью (см. рис. 6.2). Изотермы оАВ = /(С) хорошо описываются уравнением Шишковского (6.1), но с другими значениями параметров А и В, чем для поверхности раздела раствор ПАВ—газ.
2. Тангенс угла наклона начального участка изотерм -doAB/dC при С -> 0 представляет количественную меру поверхностной ак-
тивнос I и данного ПАВ на границе раздела жидкостей А и В. В каждом гомологическом ряду поверхностная активность увеличивается примерно в 3 раза при переходе к очередному, более высокому гомологу. Таким образом, в этих системах также выполняется правило Дюкло—Траубе.
3. Строение адсорбционных слоев ПАВ соответствует правилу уравнивания полярностей Рсбиндера: полярная группа молекулы находится в поверхностном слое воды (т.е. она гидратирована), а неполярная часть находится в неполярной жидкости. В соответ-ствии с теорией Ленгмюра в насыщенном монослое молекулы ПАВ имеют одинаковую ориентацию — перпендикулярно межфазной поверхности. Поэтому для расчета удельной адсорбции Глв =f(C) можно использовать уравнения Гиббса (6.5) и Ленгмюра (6.11).
Отмеченное сходство обусловлено тем, что механизмы адсорбции ПАВ на поверхностях полярная жидкость—газ и полярная жидкость—неполярная жидкость одинаковы. В обоих случаях работа адсорбции определяется главным образом переносом неполярной части молекулы ПАВ из объема водною раствора на его поверхность. Поэтому чем длиннее радикал, тем больше поверхностная активность данного ПАВ (см. рис. 6.4).
Вместе с icm адсорбция водорастворимых ПАВ на поверхности раздела двух жидкостей имеет и определенные отличия ог их адсорбции на поверхности раствор —газ.
Во-первых, поверхностное натяжение, соответствующее предельной адсорбции Гтах, существенно меньше, чем минимальное поверхностное натяжение раствора того же ПАВ на границе с газом (см. рис. 6.4, г).
Во-вторых, значения поверхностной активности —doAB/dCn предельной адсорбции Гтах обычно отличаются от аналогичных параметров для адсорбции ПАВ на поверхности раствор —воздух. Причина этих различий заключается в молекулярных взаимодействиях у тле водородных радикалов ПАВ с неполярной жидкостью. Взаимное притяжение неполярных радикалов и молекул неполярной жидкости увеличивает энергию адсорбции тем сильнее, чем длиннее неполярная часть молекулы ПАВ. Соответственно поверхностная активность -doAB/dC при С -> 0 становится больше, чем при адсорбции па поверхност и раствор —газ. В то же время молекулы неполярной фазы В сгерически препятствуют сближению молекул ПАВ в адсорбционном мопослое. Поэтому площадь приходящаяся на одну молекулу ПАВ при ее верти калькой ориентации, может быть заметно больше, чем на поверхности раствор-таз, для которой такие стеричсские препятствия отсутствуют. Соответственно предельная адсорбция Гтах данного ПАВ на границе двух жидкостей будет существенно меньше, а площадь которая приходится на одну молекулу, напротив, будет больше.
Например, при адсорбции валериановой и каприловой кислот на межфазной поверхности водный раствор —бензол Гтах = = 2,2 • I0-6 моль/м2 и = 0,76 нм2, тогда как на границе раствор — воздух Гпт = 8,0 10 ” моль/м и Q = £20 = 0,21 нм2.
Поверхностно-активные вещества, растворимые в неполярной жидкости. В этом случае решающую роль играет перенос полярной группы молекулы ПАВ из объема неполярной жидкости на межфазную поверхность вплоть до достижения прямого контакта этой группы с водой. Поэтому работа адсорбции в пределах гомологического ряда постоянна и не зависит от длины углеводородного радикала. Соответственно изотермы поверхностного натяжения растворов ПАВ в неполярном растворителе практически совпадают для разных 1 омологов.
Поверхностно-активные вещества, растворимые в граничащих полярной и неполярной жидкостях. В этом случае адсорбционный слой ПАВ граничит с двумя растворами — раствором ПАВ в полярной жидкости А с объемной кон цен грацией СА и раствором ПАВ в неполярной жидкости В с объемной концен грацией Св. Равновесная адсорбция ГАВ должна соответствовать равновесию поверхностного слоя с обоими растворами. Для разбавленных растворов из уравнения Гиббса (6.5) следует
Гдв = (-doAB/dCA)CA/(/?D = (-d(yAB/dCB)CB/(/?D. (6.19)
Отсюда определяется соотношение поверхностных активностей данного ПАВ по отношению к обеим жидкостям:
(-doAB/dCB)c=0/( -doAB/dCA)c^0 = С^/Св = К, (6.20)
где К — коэффициент распределения ПАВ между полярной и неполярной жидкостями; коэффициент К пропорционален отношению предельных растворимостей данного ПАВ в полярной и неполярной жидкостях.
Таким образом, распределение ПАВ между полярной и неполярной жидкостями и соответственно адсорбция ПАВ на поверхности между ними во многом определяются строением молекулы ПАВ, а именно — соотношением размеров ее полярной группы и неполярной части. Эта связь лежит в основе важной количественной характеристики свойств ПАВ — гидрофильно-липофильного баланса (ГЛ Б) (см. подразд. 7.3).
При высоких концензрациях ПАВ и образовании насыщенного адсорбционного слоя различия составов поверхностного слоя и прилегающих к нему объемов полярной и неполярной фаз становятся малыми. Поэтому в таких системах поверхностное натяжение удается снизить до очень низкою уровня — вплоть до 10 3— К)2 мДж/м .
При таких сверхнизких натяжениях свойства дисперсных систем существенно меняются — подобно изменению физических
свойств в области очень низких температур (сверхтекучесть, резкое падение электрическою сопротивления и теплоемкости и др.). Один из важных и актуальных примеров таких систем — микроэмульсии.
6.6. Адсорбция ПАВ из растворов на поверхности твердых тел
Твердые тела используют для адсорбции различных веществ из газов и растворов очень широко — во многих технологических процессах, в области охраны среды, в медицине. Для коллоидной химии наиболее важна адсорбция ПАВ из водных и неполярных растворов. Она играет определяющую роль в управлении смачиванием твердых тел (i идрофильных и гидрофобных), в регулировании свойств коагуляционных структур и механических свойств материалов. Адсорбция ионов имеет ключевое значение в формировании двойного электрического слоя на поверхности дисперсных частиц (см. гл. 9).
Общие закономерности адсорбции (термодинамические и кинетические) выполняются и для твердых адсорбентов. Однако по ряду причин этот случай гораздо сложнее, чем адсорбция ПАВ на поверхности жидкости.
Во-первых, это различие вызвано неоднородностями поверхности твердых тел, которые разделяют на три группы: геометрические, структурные и химические.
Геометрическая неоднородность (шероховатость) обусловлена тем. что поверхность твердых тел нс бывает гладкой, на ней присутствует множество неровностей разнообразной формы (выступы, впадины, ступеньки и т.д.). Размеры неровностей составляют от нескольких нанометров до сотен микрометров. Шероховатость вызывает увеличение реальной поверхности (Q,.) твердою тела но сравнению с площадью Шо) идеально гладкой поверхности. Отношение A'=Qr/£20> 1 называют коэффициентом шероховатости (см. подразд. 5.3).
Для более точного теоретического описания шероховатой поверхности твердых тел сейчас используют весьма совершенные методы (фрактальная геометрия).
Шероховатости влияют нс только на удельную поверхность адсорбента, но и на его локальную поверхностную энергию. Например. возле выступа локальная поверхностная энергия больше, чем на гладкой поверхности, так как в этом случае уменьшается число ближайших соседних атомов. Соочвстственно и локальные значения адсорбции будут сильно различаться. Участки с повышенной адсорбционной способностью называют активными центрами.
Возможен и противоположный эффект. Очень узкие впадины, щели, поры являются стерическим препятствием для проникновения достаточно крупных молекул
Структурная неоднородность характерна для поликристаллов Она обусловлена наличием множества монокристаллов («зерен») разного размера — oi 1 — 10 нм до 10 — 100 мкм. Наружной поверхностью отдельного зерна может быть любая кристаллографическая грань. Вследствие анизотропных свойств кристаллов поверхностная энергия в пределах каждого зерна различна.
Химическая неоднородность поверхности присуща композиционным материалам. Такая поверхность состоит из отдельных участков (доменов) разного химического состава.
Во-вторых, особенность адсорбции ПАВ из растворов заключается в том, что наряду с растворенным веществом может происходить адсорбция растворителя. Она проявляется наиболее заметно в лиофильных системах, когда жидкоегь хорошо смачивает твердую поверхность и поэтому поверхностная энергия (отж) на границе твердое тело —жидкость меньше, чем на границе твердое тело —газ (он). Таким образом, адсорбция на твердой поверхности может иметь конкурентный характер. Эта конкуренция идет между молекулами растворителя и молекулами ПАВ. Она выражена гем сильнее, чем выше поверхностная энергия адсорбента (Сп)-
В-третьих, особенность адсорбции из растворов на твердой поверхности связана с гем, что эти поверхности часто имеют положительный или отрицательный электрический заряд (см. гл 9). Поэтому механизмы адсорбции неэлектролитов и ионов различны. Соответственно принято выделять два случая адсорбции из растворов на твердой поверхности: молекулярную адсорбцию и ионную адсорбцию.
Молекулярная адсорбция. Поскольку молекулярная адсорбция носит физический характер, т. с происходит обратимо в соответствии с теорией Ленгмюра об адсорбционно-десорбционном равновесии, для расчета зависимости удельной адсорбции Г от концентрации ПАВ в растворе в первом приближении применимо уравнение Ленгмюра (6.11). Приближение заключается в том, что в рамках этой теории оценивают среднее значение удельной адсорбции. Из-за неоднородности поверхности адсорбентов локальные значения адсорбции могут сильно отличаться от среднего уровня. Другое ограничение теории Ленгмюра относится к адсорбции в пористых телах с каналами и порами нанометрового размера, поскольку они являются стерическими препятствиями.
В наиболее общей форме адсорбцию из разбавленных растворов ПАВ описывает термодинамическое уравнение Гиббса (6.5). В данном случае оно имеет вид
г = (-dorjk/dC)C7(/?T). (6.21)
где С — концентрация ПАВ в растворе.
Применение уравнения (6 21) для практических расчетов осложняется тем, что измерение удельной поверхностной энергии отж на границе твердое тело —раствор крайне затруднительно. В некоторых случаях такие оценки все же возможны.
Рассмотрим следующий метод. Из уравнения краевою угла Юнга (5.5) следует
d(oxcos0)/d С = d(on - отж).
Для низкоэнергетических материалов удельная поверхностная энергия о|Г на границе с газом не зависит от концентрации ПАВ, поэтому производная равна
dolx/d С = d(oxcos0)/d С.
Ее можно найти экспериментально, измерив изо гермы поверхностного натяжения о =/(С) растворов ПАВ и изотермы краевых углов 0 = /(С) для тех же растворов.
Для про1ноза адсорбции какою-либо вещества на межфазной поверхности твердое тело — жидкость часто используют правило уравнивания полярности Ребиндера (см. подразд. 6.4). В соответствии с ним для адсорбции ПАВ из водных растворов следует применять неполярные адсорбенты (например, уголь). Для адсорбции ПАВ из неполярных растворителей большой эффективностью обладают полярные адсорбеты (силикаты, глина).
Строение адсорбционных слоев ПАВ на твердой поверхности определяется теми же положениями, что и для адсорбции на поверхности жидкости. Адсорбционные слои мономолекулярны и с увеличением концентрации ПАВ в растворе достигается предельное значение удельной адсорбции Гтах. В насыщенном монослое молекулы ПАВ располагаются перпендикулярно поверхности адсорбента. В гомологических рядах спиртов, жирных кислот и других водорастворимых ПАВ адсорбция на границе водный раствор-адсорбент растет но мере удлинения углеводородной цепи в соответствии с правилом Дюкло—Траубе (см. подразд. 6.1).
Однако при адсорбции из неполярной жидкости удлинение углеводородной цепи приводит к противоположному результату, а именно — к уменьшению адсорбции. Причина заключается в повышении растворимости ПАВ с большей молекулярной массой в неполярной жидкости и соответствующем изменении коэффициента распределения ПАВ между граничащими фазами.
Ионная адсорбция. Адсорбция ионов из растворов часто происходит на твердых поверхностях, состоящих из ионов — катионов и анионов. Такое строение имеют, например, поверхности щелочных гало1енидов. Поэтому решающее значение для ионной ад
сорбции имеют силы электростатического притяжения и отталкивания между ионами раствора и ионами на поверхности адсорбента. В связи с этим при ионной адсорбции выполняется ряд закономерностей.
1. При одинаковом заряде ионов их адсорбционная способность растет по мере увеличения ионного радиуса (г,). Эта закономерность объясняется тем, что чем больше радиус иона, тем слабее он гидратируется водой Гидратная оболочка иона уменьшает его притяжение к противоположно заряженному иону на твердой поверхности. По способности к адсорбции катионы одно- и двухвалентных металлов располагаются в следующей последовательности:
Li+ < Na+ < К+ < Rb+ < Csl
Mg2’ < Ca ‘ < Sr24 < Ba2+
Для однозаряженных анионов возрастание адсорбции происходит в таком порядке:
СГ < Вг < NO3- < Г< NCS-
Привсденные последовательности ионов называют лиотропными рядами.
2. Адсорбционная способность ионов растет при увеличении их заряда, гак как при этом возрастает их электростатическое притяжение к соответствующим (по знаку заряда) ионам на твердой поверхности. Примером такого влияния заряда иона может служить следующая последовательность адсорбции катионов металлов:
К+ « Са2+ « АГ4 « Th41
Одна из важных форм ионной адсорбции — ионный обмен. Он происходит между ионами, адсорбированными па поверхности данного адсорбента, и другими ионами, находящимися в растворе. Ионный обмен сильно зависит от строения двойного электрического слоя вблизи поверхности адсорбента. Закономерности ионного обмена изложены в гл. 9 при описании электроповсрхност-ных явлений.
6.7. Химическое модифицирование твердых поверхностей
Одна из основных задач коллоидной химии заключается в регулировании поверхностных свойств различных веществ. Для жидкостей наиболее эффективными регуляторами являются поверхностно-активные вещества. Основным механизмом служит физическая адсорбция ПАВ на поверхностях раздела жидкость— газ. жидкость—другая жидкость и жидкость—твердое тело (см. подразд. 6.2—6.6).
Другой эффективный метод регулирования многих поверхностных явлении основан на использовании электрических свойств твердой поверхности — се заряда и потенциала (см. гл. 9).
Еще один принцип регулирования заключается в химическом модифицировании поверхности различных материалов. Этот метод основан на использовании химических реакций, в результате которых на поверхности твердых тел образуется очень тонкий (в пределе — молекулярный) слом химических соединений. Такие слои закрепляются гораздо прочнее, чем адсорбционные слои ПАВ. Поэтому они обладают большой устойчивостью по отношению к различным внешним воздействиям: течению жидкости, изменению температуры и др. Благодаря этим достоинствам химическое модифицирование стало очень распространенным метолом регулирования свойств коллоидно-химических систем. В качестве примера отмстим применение химически модифицированных материалов в нанотехнологии, медицине и фармакологии, хроматографии, биотехнологии, для получения химических и биологических сенсоров и др. В коллоидной химии химическое модифицирование поверхности применяют для направленного изменения таких важных свойств твердой поверхности, как ее гидрофильность и гидрофобность.
Методы химического модифицирования весьма разнообразны. К числу наиболее перспективных относится метод прививки поверхностных соединений.
Химическим модифицированием поверхности называют химические превращения в поверхностном слое твердого тела при сохранении неизменным его внутреннего состава. В этом заключается принципиальное отличие модифицирования от гетерогенных химических реакций и процессов, которые постепенно распространяются в глубь твердой фазы (растворение, химическая реакция) или приводят к увеличению объема твердой фазы (например, при осаждении).
Модификатором, или модифицирующим веществом, называют вещество, которое химически реагируете поверхностными функционалы Iыми труппами
Привитый слой — это совокупность функциональных групп привит ых веществ, закрепленных ковалентно на поверхности твердой подложки.
Привитый слой включает следующие элементы (рис. 6.5):
• якорная группа, обеспечивающая химическую фиксацию привитого соединения;
• «ножка» — I руппа, отделяющая привитое соединение от поверхности;
• функциональная ipynna, определяющая требуемые поверхностные свойства (I идрофильность, гидрофобность, адсорбционные и каталитические свойства и т.п.).
Подложка
Привитый слой
Рис. 6 5. Схема химического модифицирования твердой поверхности
Важной характеристикой модифицирующего действия служит плотность прививки — число функциональных групп на единице площади. Максимальная плотность прививки определяется либо размером прививаемой молекулы, либо расстоянием между реакционными центрами на поверхности.
Привитый слой геометрически представляет собой двухмерное образование (толщина слоя составляет несколько нанометров). В этом отношении его можно рассматривать как типичный коллоид! ю-химический объект.
Важная особенность привитою слоя но сравнению с адсорбционными слоями ПАВ на поверхности жидкости заключается в большой прочности их закрепления. Поэтому привитые слои называют также иммобилизованными.
Модификаторы должны удовлетворять нескольким требованиям:
I) валентность элемента, образующего якорную группу, должна быль не менее двух;
2) реакция между поверхностными группами подложки и модификатором должна протеказъ селективно;
3) образующиеся поверхностные соединения должны быть стабильными.
В соответствии с этими условиями в качестве модификаторов использую! преимущественно кремнийорганичсские, фосфорорганические, оловоорганические и борорганические соединения, различные тиолы, мсталлокомплсксные (в том числе — металлоорганические) соединения, а также различные полимеры.
При химическом модифицировании в ряде случаев образуются монослои с высокоупорядоченной структурой, т.е. большинство молекул имеют одинаковую ориентацию (рис. 6.6). Поскольку такие структуры образуются самопроизвольно, их называют самосо-бирающимися монослоями. Структура таких привитых слоев аналогична насыщенному адсорбционному мопослою ПАВ на поверх-нос!И жидкости («частокол» Ленгмюра). Высокоуиорялочснные монослои образуются и при адсорбции ПАВ на твердых поверхностях, например при адсорбции длинноцепочечных спиртов и жирных кислот па металлах.
Упорядоченные монослои разного типа (адсорбционные и привитые) являются эффективными регуляторами многих коллоид
но-химичсских процессов (смачивание, адгезия, структурообра-зованис). Самосборка упорядоченных монослойных структур играет ключевую роль в образовании клеточных мембран и в ряде биологических процессов. Такие организованные поверхности используют для создания сенсоров, в нанотехнологии, для моделирования взаимодействии биополимеров и клеток с различными поверх н остя м и (субстрата ми).
Одно из главных направлений применения химическою модифицирования — создание гидрофильных и гидрофобных поверхностей, что особенно важно для регулирования смачивания (см. подразд. 5.2). Для этой цели наиболее эффективен метод ковалентного закрепления органических молекул на твердой поверхности.
Для гидрофобизании используют вещества, функциональные группы которых образуют поверхность с небольшой поверхностной энергией ог1 (см. подразд. 5.2). Для достижения максимальной гидрофобности модифицирующий монослой должен быть плотно упакован, чтобы обеспечить полную «экранизацию» подложки. Эти условия выполняются, например, при гидрофобизации кремнезема с помощью самособираюшихся монослоев длинноцепочечных алкилтрихлорсиланов CMH2,I+|SiCl3 и перфторалкилтрихлор-силанов CwF2,T+|(CH2)2SiC13. При плотной упаковке молекул получаются гидрофобные поверхности, состоящие либо из СН3-,
Функциональная группа
Подложка
Рис. 6.6. Упорядоченный монослой длинноцепочечных молекул, химически закрепленных на твердой поверхности
либо из CF3-rpynn. Краевые углы воды 0 на таких поверхностях весьма велики: для СН3-групп 0 = 110°—115°, для CF3-rpynn О= 118°-125°.
Важное достоинство химического модифицирования заключается также в том, что гидрофобизуюшис слои обеспечивают химическую однородность и молекулярную гладкост ь образующейся поверхности.
Гидрофильные поверхности должны содержать полярные группы, которые сильно взаимодействуют с водой, например ОН-, SO3H-, СООН-группы. Гидрофил изующие слои получают с использованием реакций окисления двойных связей, восстановления эфиров и др. В отличие от гидрофобных модифицирующих слоев гидрофильные слои неустойчивы. Поэтому стечением времени происходит постепенная конформационная перестройка, приводящая к изменению числа гидрофильных групп на наружной поверхности модифицирующего монослоя.
Другую группу модифицирующих веществ представляют полимеры и белки. Модифицирующее действие белков имеет несколько важных особенностей по сравнению с низкомолекулярными ПАВ.
Во-первых, макромолекулы белков содержат большое количество гидрофильных и гидрофобных групп, которые расположены на поверхности глобулы, а не локализованы на концах дифиль-ных молекул ПАВ. Белки относятся к классу амфотерных ПАВ.
Во-вторых, большую роль играет конформационное состояние макромолекул в водном растворе, из которого они адсорбируются на твердую поверхность. Достаточно жесткие макромолекулы обычно нс могут (но стерическим причинам) образовать плотно упакованный монослой.
Для глобулярных белков (а-химотрипсин, бычий сывороточный альбумин) процесс модифицирования различных материалов с разной степенью гидрофильности и гидрофобности поверхности (стекло, полупроводники, полимеры) протекает очень быстро. Необходимое время контакта материала с модифицирующим раствором составляет несколько секунд.
Для макромолекул (например, макромолекул желатины) процесс модифицирования идет в две стадии.
На первой стадии быстро формируется адсорбционный слой на твердой поверхности.
На второй длительной (несколько часов) стадии дополнительная адсорбция нс происходит, но осуществляюich медленные конформационные процессы перестройки адсорбционного слоя, которые приводят к дополнительной гидрофилизации.
Модифицирующее действие белков сильно зависит от концентрации водного раствора. При этом характерна следующая закономерность. Адсорбция белков на гидрофобных материалах (ноли-
этилен, фторопласт) приводит к их гидрофил изаци и. Напротив, адсорбция белка на гидрофильной подложке (стекло, германий) вызывает заметное увеличение краевых углов волы, т.е. приводит к частичной гидрофобизании подложки (рис. 6.7).
Таким образом, белки обладают универсальным модифицирующим действием. Они могут быть как гидрофил и заторам и, так и гидрофобизаторамп. Модифицирующее действие сильно зависит от гибкости макромолекул. Желатина в конформации статистического клубка обладает универсальным модифицирующим действием но отношению к материалам разной химической природы. При определенной концентрации раствора желатины на модифицируемой подложке образуется сплошная пленка желатины, которая полностью экранирует подложку, поэтому степень смачивания определяется только строением и составом макромолекул и строением модифицирующею слоя.
Глобулярные белки обладают гораздо меньшим модифицирующим действием. Основная причина заключается в том. что адсорбционные слои оказываются разреженными, а не сплошными. Например, при адсорбции сывороточного альбумина человека на полипропилене максимальная степень заполнения составляет только 70 %.
Модифицирующее действие белков сильно зависит от pH раствора. Наибольшая степень модифицирования обычно соответствует изоэлектрическому состоянию белка в растворе. При отклонении от изоэлектрической точки макромолекулы белка в растворе приобретают электрический заряд одного знака. Взаимное отталкивание макромолекул приводит к уменьшению плотности упаковки адсорбционного слоя.
Модифицирующее действие белков можно значительно усилить с помощью следующей методики. Вначале формируют адсорбционный слой белка на границе с воздухом или неполярной жидкостью. Такие межфазные слои обычно образуют гелеобразные структуры. Благодаря этому межфазные слои белков обладают
Рис. 6.7. Краевой угол (О) капли воды на подложке, модифицированной адсорбционными слоями белка (бычьего сывороточною альбумина), в зависимости от концентрации белка (С) в водном растворе:
/ — стекло: 2 — германий; 3 — полиэтилен; 4 — фторопласт
достаточно большой механической прочностью и их после формирования можно переносить на различные твердые поверхности. Такие слои могу!' полностью экранировать поверхность материала, на которую они перенесены Поэтому степень смачивания определяется поверхностными свойствами самого белковою слоя. Эти свойства в свою очередь сильно зависят от природы той фазы, с которой граничит водный раствор белка в период формирования адсорбционного слоя.
Яркий пример такого влияния дают межфазные слои желатины, сформированные на границах водных растворов с воздухом и тетрахлорметаном при температуре 40 С (табл. 6.1). При этой температуре макромолекулы желатины в воде имеют конформацию статистического клубка. Слои, образованный на границе с водой, гидрофобен: краевой угол воды О = 108 (как на фанице с парафином) Напротив, межфазный слой желатины, который был сформирован на фанице с тетрахлорметаном, имеет совершенно другие поверхностные свойства. Он обладает значительной гидрофильностью: краевой угол воды 0 = 60 Это качественное различие обусловлено разной ориентацией гидрофильных и гидрофобных сегментов макромолекулы желатины. Контакт раствора с сильно неполярной жидкостью (тетрахлорметаном) способствует конформационной перестройке, при которой в сторону тетрахлорме-тана обращено возможно большее количество неполярных сегментов.
Влияние конформационного состояния макромолекул белка на поверхностные свойства межфазных адсорбционных слоев может быть очень существенным. В этом отношении показателен следующий пример. Конформация макромолекул желатины в водном растворе зависит от температуры. При температуре ниже 16 Смак-
Таблица 6.1
Краевые углы (0) капель воды на поверхности межфазных адсорбционных слоев белка, перенесенных на пластинки германия
белок Граничная фаза 0. |рад
Альбумин сывороточный бычий Воздух Бензол Тстрахлорметан 50 32 32
Желатина Воздух 52
Белок в конформации коллагеноподобной спирали Бен юл Тстрахлорметан 52 52
Белок в кон(|)ор.мации клубка Воздух Бензол Толуол 108 93 60
ромолекулы имеют конформацию жесткой коллагеноподобной спирали, а при более высоких температурах (выше 30 С) — конформацию статистического клубка со средним радиусом инерции 30 — 45 нм. Температурная зависимость краевых углов воды на межфазных адсорбционных слоях желатины характеризуется резким переходом от гидрофильности к i идрофобности именно при температуре 20 С, т.е. при температуре конформационного перехода.
Модифицирующие свойства межфазных слоев белка, так же как и в случае адсорбционных слоев, сильно зависят от pH раствора. Эго влияние особенно сильно проявляется для достаточно гибких макромолекул
Например, для межфазных слоев желатины в конформации статистического клубка обнаруживается резкий максимум краевых углов при изоэлектрическом состоянии раствора. Причина этого заключается в том, что при отклонении от изоэлектрической точки макромолекулы белка приобретают одинаковый заряд, и электростатическое отталкивание препятствует формированию максимально плотного слоя.
Контрольные вопросы
I. Дайте общую характеристику явления адсорбции. Почему адсорбция является наиболее эффективным регулятором поверхностных свойств дисперсных систем?
2. Объясните принцип классификации веществ на поверхностно-активные и иоверхностно-инактивныс
3 Объясните смысл термина «поверхностная axiивноегь». Как измеряют поверхностную активность водорастворимых ПАВ?
4. Напишите уравнение Шишковского и объясните физико-химическим смысл входящих в него величин.
5. Напишите адсорбционное уравнение Гиббса для общею случая и для разбавленных растворов ПАВ.
6. Объясните метод расчета изотермы адсорбции ПАВ на основе измерений поверхностного натяжения растворов ПАВ.
7 Дайте определение работы адсорбции Используйте это понятие для обоснования связи поверхностной активности ПАВ с длиной углеводородного радикала в молекуле ПАВ (правило Дюкло—Траубе).
8. Напишите уравнение адсорбции Ленгмюра Дайте краткую характеристику динамической модели процесса адсорбции в теории Ленгмюра.
9. Объясните строение адсорбционных слоев ПАВ на основе теории Ленгмюра Рассмотрите два случая: разбавленные (разреженные) слои, предельно упакованные монослои.
10. Укажите наиболее существенные особенности адсорбции ПАВ па границе раздела двух несмешивающихся жидкостей.
11 Укажите наиболее важные особенности адсорбции ПАВ на поверхности твердых тел
12. Назовите особенности молекулярной адсорбции и ионной адсорбции.
13. В чем заключается правило уравнивания полярностей Ребиндера?
14. Объясните понятие «лиотропные ряды» при адсорбции ионов.
15. В чем заключается процесс химического модифицирования твердой поверхности?
16. Укажите принципы химического модифицирования методом поверхностных соединений.
17. Приведите примеры эффективных гидрофобизаторов и гидрофи-лизаторов.
18. В чем преимущества высокомолекулярных ПАВ при их использовании для модифицирования твердых поверхностей?
ГЛАВА 7
ПОВЕРХНОСТНО-АКТИВНЫЕ ВЕЩЕСТВА
Поверхностно-активными называют вещества, которые могут адсорбироваться на поверхности раздела различных фаз (жидкость—газ, жидкость—жидкость, жидкость—твердое тело, твердое тело—газ) и снижать ее свободную поверхностную энергию (см. гл. 6). Наряду со способностью адсорбироват ься на межфазной поверхности многие ПАВ обладают еще одним важным свойством — при определенных условиях в растворах ПАВ образуются самоор-ганизованные наноагрегаты (мицеллы), состоящие из десятков и сотен молекул или ионов ПАВ (см. гл. 8). Благодаря этим свойствам (адсорбция на межфазных поверхностях и образование мицелл в растворах) ПАВ используют очень широко во многих технологических процессах и в быту. Общий объем производства ПАВ — примерно 10 млн т в год.
Поверхностно-активные вещества применяют:
• для регулирования поверхностных свойств коллоидных систем, основанных на адсорбции ПАВ (см. гл. 6, 8);
• обеспечения устойчивости (стабильности) коллоидных систем, основанной на структурно-механических свойствах адсорбционных слоев ПАВ (см. гл. 10);
• изменения механических свойств материалов за счет адсорбционного понижения прочности (см. гл. 10);
• в коллоидно-химических технологиях (флотация, нанесение пленок, полиграфия, эмульсионная полимеризация и др.), в биотехнологии и многих других областях.
7.1. Классификация поверхностно-активных веществ
Термин «поверхностно-активные вещества» относится к определенным классам органических веществ, молекулы которых содержат неполярную часть (углеводородную цепь, ароматические группы и т.п.) и полярную группу (обычно концевую). Такие молекулы называют дифтьвыми, потому что они состоят из двух частей — полярной (гидрофильной) и неполярной (гидрофобной). В зарубежной литературе часто используют термин «амфифильные молекулы».
В промышленности, медицине, сельском хозяйстве, в научных цепях применяется несколько тысяч различных поверхностно-активных веществ.
Поверхностно-активные вещества классифицируют по нескольким признакам:
• растворимость (или нерастворимость) в полярной (воде) и неполярной жидкости,
• диссоциация в воде,
• молекулярная масса;
• способность к агрегации в растворе (к образованию ми пел л);
• происхождение (синтетические и природные ПАВ);
• физико-химические механизмы действия ПАВ;
• биоразлагаемость.
Рассмотрим некоторые варианты классификации ПАВ.
Классификация ПАВ по растворимости. По растворимости ПАВ деляг на три группы:
I) растворимые в воде и мало растворимые в неполярной жидкости; например, низшие гомологи спиртов и карбоновых кислот;
2) не растворимые в воде, но хорошо растворимые в неполярной жидкости; например, высшие гомологи (число атомов углерода более 20) спиртов и жирных кислот;
3) растворимые и в воде, и в неполярной жидкости; таким свойством обладают ПАВ с числом атомов углерода в углеводородном радикале 10—18
Соотношение растворимости ПАВ в полярной и неполярной жидкостях определяется соотношением размеров гидрофильной и гидрофобной (липофильной) частей молекулы ПАВ Количественной мерой этою соотношения служит гидрофильно-липофильный баланс (см подразд. 7 3).
Друюй характеристикой соотношения размеров полярной и неполярной частей молекулы ПАВ является критический параметр упаковки (см. подразд. 7.4).
Классификация ПАВ по диссоциации в воде. Промышленные ПАВ классифицируют главным образом по их диссоциации в воде. По этому признаку выделяют пять групп ПАВ.
1 Анионные ПАВ В воде анионные ПАВ диссоциируют и образуют поверхпостно-активныи анион. Наряду с катионными ПАВ анионные ПАВ относятся к ионогенным (диссоциирующим) ПАВ
Производство анионных ПАВ доспиаст около 6 млн т в год — примерно 60% от общего количества ПАВ. Главное достоинство анионных ПАВ — относительная простота их производства и небольшая стоимость. Анионные ПАВ входят в большинство моющих средств.
Типичные полярные группы в составе анионных 11АВ — RCOO , SO/-, SOR, РО4 ; противоионы — Na+, К , NH4+, Са +, Mg2 . Ионы
натрия и калия увеличивают растворимость ПАВ в воде; двухвалентные катионы способствуют растворимости ПАВ в неполярной жидкости (в «масле»).
Главный недостаток анионных ПАВ — они плохо работают в жесткой воде. Чувствительность анионных ПАВ к жесткости воды уменьшается в ряду: карбоксилаты > фосфаты > сульфаты, сульфонаты.
2. Катионные ПАВ. Эти ПАВ диссоциируют в воде, образуя поверхностно-активные катионы. К ним относятся алифатические и ароматические амины и их соли, четырехзамещенные аммониевые основания, пиридиниевые соединения. Основную массу катионных ПАВ составляют азотсодержащие соединения. Достаточно широко используют также фосфониевые, сульфониевые, сульфоксон новые ПАВ
Катионные ПАВ хорошо адсорбируются на отрицательно заряженных поверхностях. Такой знак заряда характерен для металлов, многих минералов, пластиков, клеточных мембран. Поэтому области использования катионных ПАВ очень разнообразны: в качестве антистатиков и антикоррозионных средств, для флотации минералов, в бактерицидных препаратах и т.д. Высокая адсорбируемость катионных ПАВ обеспечивает модифицирование (изменение поверхностных свойств) твердых поверхностей различной химической природы (см подразд. 6 7).
По потреблению катионные ПАВ занимают третье место после анионных и неионогенных ПАВ и составляют около 10 % от общего количества производимых ПАВ.
3. Неионогенные ПАВ. Эти ПАВ не диссоциируют в воде. По потреблению они занимают второе место после анионных ПАВ (около 30 % общего производства). Они не чувствительны к жесткости воды и могут использоваться в сочетании с ПАВ других типов. В качестве полярных групп неионогенные ПАВ содержат полиэфирные или полигилроксильные фрагменты.
Наиболее важный тип неионогенных ПАВ — этоксилированные алифатические спирты; они входят в состав многих моющих средств. Другая важная группа — этоксилированные метиловые эфиры жирных кислот. Этоксилаты метилового спирта сочетают высокую растворимость в воде с достаточно большой поверхностной активностью. Весьма перспективны плюроники — блоксопо-лимеры окиси этилена и окиси пропилена с общей молекулярной массой до 20000. При повышении температуры растворимость нс-ионогенных ПАВ в воде уменьшается
4. Амфотерные (амфолитные) ПАВ. Включают в состав как кислотные группы (например, карбоксильные), так и основные группы (замещенные аминогруппы). Поведение этих ПАВ зависит от pH раствора" при pH < 4 они диссоциируют как катионные ПАВ, при pH > 9 — как анионные; в интервале pH 4 —9 — не диссоци
ируют, т.е. ведут себя как неионогенные ПАВ. Типичные примеры — аминокислоты, белки и многие другие природные ПАВ.
5. Цвиттер-ионные ПАВ. Молекулы цвиггер-ионных ПАВ содержат положительно заряженную (обычно аммониевую) и отрицательно заряженную (обычно карбоксилат-ион) группу. Типичные цвиттер-ионные ПАВ — N-алкилпроизводные аминокислот (глицин, бетаин, аминопропионовая кислота). Другие представители этих ПАВ — имидазолины. Цвиттер-ионные ПАВ занимают промежуточное положение между ионными и неионогенными ПАВ. Основное достоинство цвиттер-ионных ПАВ — они нс вызывают раздражения кожи и глаз, поэтому их широко используют в моющих средствах (шампунях), лекарственных кремах и т.д.
Классификация ПАВ по способности к образованию мицелл. Общее свойство всех ПАВ — способность адсорбироваться на поверхности жидкостей и твердых тел и снижать их поверхностную энергию. Вместе с тем многие ПАВ обладают еще одним важным свойством. При определенных условиях (концентрация, температура) они образуют в растворе наноразмерные агрегаты из десятков и сотен молекул или ионов ПАВ. Эти агрегаты называю! мицеллами (см. гл. 8), а вещества, их образующие, — мицеллярными IIAB. Используют также термин «коллоидные ПАВ», так как мицеллы представляют собой наноразмерную дисперсную частицу. К ним относятся ПАВ с определенной молекулярной структурой — число атомов углерода в углеводородной цепи составляет 10— 20. По механизму физико-химического действия мицеллярные ПАВ относятся к группе, обладающей моющим действием.
Классификация ПАВ по физико-химическому воздействию на поверхность раздела между фазами. По этому признаку выделяют несколько групп ПАВ.
1. Смачиватели и пенообразователи — ПАВ, обладающие умеренным снижением поверхностного натяжения водного раствора на границе жидкость —газ (средние и высшие гомологи спиртов и жирных кислот).
2. Диспергаторы — ПАВ, которые адсорбируются на любых границах раздела фаз (жидкость —газ, жидкость —жидкость, жидкость—твердое тело) и существенно снижают их поверхностное натяжение. В результате значительно облегчаются процессы диспергирования и разрушения (см. гл. 10).
3. Стабилизаторы — адсорбционные слои этих ПАВ обладают механической прочностью и поэтому создают структурно-механический барьер, обеспечивающий устойчивость эмульсии, пен и других дисперсных систем. Такими свойствами обладают высокомолекулярные ПАВ с большим числом полярных групп: белки, производные целлюлозы, поливиниловый спирт.
4. ПАВ моющего действия — вещества, обладающие всеми указанными выше свойствами (см. пп. 1 — 3) и вместе с тем способ
ные при определенных условиях к образованию в растворе мицелл. Способность к мицеллообразован и ю характерна для различных типов ПАВ (ионогенных и неионогенных) с числом атомов углерода в углеводородной цени 10 — 20. Наличие мицелл — важное условие моющего действия ПАВ. так как отмываемые органические загрязнения коллоидно растворяются (солюбилизируются) именно в мицеллах (см. гл. 8).
Классификация ПАВ по происхождению. Согласно указанному признаку ПАВ деля г на синтетические и природные.
Синтетические ПАВ получают главным образом на основе нефтехимических технологий. Ассортимент современных синтетических ПАВ очень широк — несколько тысяч веществ. Одна из основных групп синтетических ПАВ — моющие средства.
Природные ПАВ — это полярные липиды: гликолипиды, фосфолипиды и др. Они выполняют много функции в коллоиднохимических процессах в биологических системах. Например, соли холевых кислот обеспечивают эффективное коплоилное растворение (солюбилизацию) гидрофобных компонентов крови. Природные ПАВ получают из естественных продуктов с помощью ферментативных реакций и некоторыми другими способами.
7.2. Некоторые особенности поверхностно-активных веществ
Большинство промышленных ПАВ (см. подразд. 7.1) химически стабильны. Среди немногих исключений — алкилсульфаты, которые при обычных условиях хранения химически неустойчивы из-за кислотного гидролиза. Проблема устойчивости ПАВ возникла в связи с загрязнением окружающей среды. Широкое использование ПАВ в промышленности и в быту (например, моющие средства) приводит к попаданию их в больших количествах в сточные воды и далее — в водоемы и почвы. Масштабы последствий загрязнения этими ПАВ весьма велики. Во многих странах законодательно определены нормы ПАВ в воде.
Скорость биоразложения (биодеградации) ПАВ в значительной степени зависит от их молекулярной структуры. Биодеградация происходит быстрее, если ПАВ растворимо в воде. Другой важный фактор — молекула ПАВ должна содержать связи, которые легко разрушаются при ферментативном катализе. Поэтому для увеличения скорости биоразложения в молекулу ПАВ вводят слабые, легко разрываемые связи. Обычно их вводят между гидрофобным радикалом и полярной группой. К числу слабых относят эфирные н амидные связи Их разрушение катализируют липазы и пептидазы.
Ешс один фактор, связанный с проблемой биодеградации, — разветвленность неполярной части молекулы ПАВ. Сильная раз-
вствлснность затрудняет разложение из-за стсрических препятствий, создаваемых боковыми группами.
Показателен здесь следующий пример. Разветвленные алкил-бензолсульфонаты ранее использовались как один из основных компонентов бытовых моющих средств. Однако из-за стабильности этих ПАВ они были заменены аналогами с линейными алкильными цепями, последние хорошо разрушаются в анаэробных условиях.
Дня решения экологических проблем синтезирован ряд поверхностно-активных веществ с молекулами, расщепляющимися в природных условиях. Основные типы этих веществ — амфифильные ПАВ, а также ПАВ, разлагаемые под действием ультрафиолетового излучения и озона.
В последнее время проблему биодеградации ПАВ пытаются решить следующим образом: соответствующую реакцию проводят так, чтобы получить полезные вещества. Примером может служить разложение ПАВ с хорошей моющей способностью с получением спирта.
Поверхностно-активные полимеры стали широко применяться только в последние 20 — 25 лет. В основном используют три типа полимерных ПАВ; они применяются в промышленности и в то же время входят в состав растений и животных.
1. Полимерные ПАВ, в которых гидрофильные боковые цепи привиты к гидрофобному скелету. К ним относятся липополисахариды — природные и синтетические (например, гидрофобизо-ванные крахмал и целлюлоза, алкилзамещенные полиуретаны). Широкое применение в промышленности имеет препарат«Эмуль-сан» — полианионный липополисахарид. Он является весьма эффективным эмульгатором и энергично адсорбируется па поверхности раздела вода —нефть. Благодаря этому «Эмульсан» применяют, например, для очистки почвы от нефтяных загрязнений, для стабилизации прямых эмульсий.
Отметим особенности полимерных поверхностно-активных веществ этого типа:
а) эффективность влияния ПАВ на поверхностное натяжение значительно возрастает при добавлении пептидов; например, поверхностное натяжение на границе раздела вода —гексаде кап под действием полимерного ПАВ удается снизить до 30 мДж/м2, однако при добавлении пептидов оно снижается до 14 мДж/м2;
б) действие «Эмульсана» очень специфично, г.с. сильно зависит от химического состава граничащих фаз; например, стабилизация нефтяных эмульсий в воде особенно эффективна при определенном составе сырой нефти (смесь алифатических углеводородов и алкиларильных соединений).
Синтетические полимерные ПАВ этого типа получают гидро-фобизацией крахмала и целлюлозы.
2. Полимерные ПАВ, в которых гидрофобные боковые цепи привиты к гидрофильному скелету. Природные полимерные ПАВ этого типа — гликопротеины; синтетические ПАВ — сополимеры с полиэтиленгликолем: этоксилированные полиакрилаты, этоксилированные лигпосульфопаты и смолы. К этой группе относятся также силиконовые ПАВ (на основе полидиметилсилоксана). Они сильно снижают поверхностное натяжение воды (примерно до 20 мДж/м2), являются хорошими смачивателями (обеспечивают смачивание гидрофобных поверхностей), эффективными антивспенивателями. Основной недостаток — плохая биоразлагаемость и высокая стоимость. Особо следует отмстить использование полимерных ПАВ этого типа в биологии и медицине. При адсорбции таких ПАВ из водного раствора на твердой гидрофобной поверхности образуется монослой, в котором в соответствии с правилом уравнивания полярностей гидрофильные полиэтилен-гликолевые цени обращены в сторону раствора. Такой адсорбционный слой очень эффективно снижает адсорбцию белков и обеспечивает малую адгезию клеток. В результате существенно подавляется адсорбция белков плазмы крови и кровяных телец, что резко снижает возможность образования тромбов.
3. Полимерные ПАВ с гидрофильными и гидрофобными блоками. К природным ПАВ этого типа относятся многие белки: например, белки молока, многие белки слюны. Полярные сегменты макромолекул имеют высокую концентрацию фосфатных групп, а гидрофобность обеспечивают гидрофобные аминокислоты. Синтетические ПАВ — это, например, сополимеры полиэтилен гликоля. Для сополимеров характерно уменьшение раствори юсти при повышении температуры, т.е. они лучше растворяются в холодной воде, а не в теплой. ПАВ с большим содержанием окиси пропилена весьма эффективно регулируют смачивание различных материалов. Эти полимерные ПАВ и белки обладают высокой поверхностной активностью и могут адсорбироваться на поверхностях различной химической природы, т.е. представляют универсальные ПАВ (см. гл. 6).
Поверхностная активность белков и их адсорбция па различных поверхностях раздела фаз играют важную роль во многих природных процессах и в биотехнологии. В связи с этим отметим наиболее существенные свойства белков как поверхностно-активных веществ:
а) на границе водный раствор белка —воздух поверхностное натяжение снижается в пределе примерно до 20 мДж/м2;
б) скорость образования адсорбционного слоя и его строение сильно зависят от конформационного состояния молекул белка;
в) из-за большой молекулярной массы белков процесс их адсорбции на поверхности раствора протекает очень медленно; обыч
но лимитирующей стадией является диффузия макромолекул из объема раствора к поверхности.
Для многих белков удельная адсорбция (Г) связана с коэффициентом диффузии (D) уравнением
Г = 1,1(Рг)|/2, где / — время.
При комнатной температуре для достижения равновесной адсорбции необходимо длительное время (1 ч и более).
7.3. Гидрофильно-липофильный баланс
Способность ПАВ адсорбироваться на различных поверхностях определяется прежде всего спецификой строения их молекул, а именно — их дифил ьностью. Молекула ПАВ состоит из полярной и неполярной частей. Поверхностная активность и другие свойства ПАВ зависят от соотношения свойств этих частей. Показателен следующий пример. В гомологическом ряду низкомолекулярных ПАВ (смачивателей и пенообразователей) поверхностная активность гомологов (спиртов, жирных кислот) на границе раствор—газ растет с увеличением длины углеводородного радикала по правилу Дюкло—Траубе (см. подразд. 6.1). При этом низшие гомологи (СП3ОН, С2Н ОН) поверхностную активность не проявляют. Из-за преобладающею влияния гидроксильной группы они очень хорошо растворяются в воде и поэтому для них пег термодинамического стимула для концентрирования на поверхности (адсорбции). В го же время спирты и карбоновые кислоты с очень длинной углеводородной цепью (число атомов углерода более 10) плохо растворяются в воде из-за преобладающего влияния неполярной части молекулы. Поэтому их нельзя применять в качестве смачивателей или пенообразователей.
Таким образом, оптимальному действию ПАВ соответствует определенная сбалансированность полярной и неполярной частей молекулы ПАВ. Количественной мерой коллоидно-химических свойств поверхностно-активных веществ служит гидрофи 1ыю-липофильный баланс (ГЛБ. (р). определяемый действием полярной (гидрофильной) и неполярной (липофильном) частей молекулы ПАВ.
Существует несколько эмпирических методов определения ГЛ Б. Наиболее распространен метод групповых чисел. Каждой группе, входящей в состав молекулы ПАВ, приписывают определенное групповое число (<?,)• Гидрофильно-липофильный баланс определяется как сумма групповых чисел (подобно расчету парахора молекулы по парахорам составляющих ее атомов; см. подразд 4 1)
<р=7 + Е<7,. (7.1)
Для гидрофильных групп qt> 0; для гидрофобных — 0. Со-
ответственно при ср > 7 преобладает действие полярных групп, и такие ПАВ растворяются преимущественно в воде. Напротив, при <р < 7 преобладает растворимость ПАВ в неполярной жидкости («масле»). В связи с этим значения ГЛ Б используют при подборе ПАВ дпя стабилизации прямых эмульсий (капли «масла» в воде) и обратных эмульсии (капли воды в «масле»). Именно ПАВ, имеющие большие значения ГЛ Б, используют для получения прямых эмульсий. Напротив, для получения обратных эмульсий эффективны ПАВ с низкими значениями ГЛ Б.
Таким образом, по физико-химической сут и ГЛ Б отражает соотношение растворимостей ПАВ в воде (С„) и в «масле» (См) и формулу (7.1) можно представить в виде
Ф = 7 + 0,361п(Св/См). (7.2)
Таким образом, в уравнениях (7.1) и (7.2), определяющих значения ГЛ Б разных ПАВ, «пограничное» значение ГЛ Б равно семи. Этот выбор, конечно, условен.
Уравнение (7 2) имеет термодинамическое обоснование. Равновесное распределение ПАВ между полярной и неполярной фазами определяется равенством их химических потенциалов в этих фазах. Отсюда следует выражение для работы переноса ( И7) молекулы ПАВ из полярной фазы в неполярную:
^Лп(Св/См), (7.3)
где кв — постоянная Больцмана; Т — абсолютная температура.
Отношение Св/См представляет равновесную константу распределения ПАВ между полярной и неполярной фазами. С учетом выражения (7.3) уравнение (7.2) принимает вид
Ф= 7 + 0,36 W/(kB Т). (7.4)
Таким образом, аддитивность значений ГЛ Б является следствием аддитивности работы переноса молекул ПАВ из одной фазы в другую. Становится ясен физико-химический смысл групповых чисел — они пропорциональны парциальным работам переноса отдельных групп:
£<7 =0,36И//(^7’). /
Приведем групповые числа но Дэвису для некоторых гидрофильных и гидрофобных групп:
*
Гидрофильные группы
-SO4Na .................................... 35.7
-COOK...................................... 21.1
—COONa..................................... 19,1
NR4 (аммониевые основания)........................ 9,4
Эфир (сорбитановое кольцо)...................... 6,3
Эфир (свободный).................................. 2,4
—О—............................................... 1,3
-СООН .............................................. 2,1
—ОН (свободная) .................................... 1,9
—ОН (сорбитановое кольцо)...................... 0,5
Гидрофобные групп ы
—CF3..............................................-0,870
-СВ-...............................................-0,870
-СП,..............................................-0.475
-СП,-.............................................-0,475
-СН-..............................................-0,475
Работа переноса определяется по коэффициенту распределения, поэтому ее значения известны для многих веществ. Для гомологических рядов выполняется линейная зависимость работы переноса от числа атомов углерода (дс) в углеводородном радикале;
0,36И//(АгвГ) = a-bnCi (7.5)
где а, b — постоянные; константа а характеризует вклад в работу переноса полярной группы.
Из уравнении (7.4), (7.5) следует
(р = 7 + а - Ьпс. (7.6)
Приведенные выше уравнения показывают, что значение ГЛБ определяется термодинамическими свойствами растворов ПАВ (водного и неполярного). Поэтому ГЛБ характеризует строение молекулы ПАВ, но не отражает непосредственно его адсорбцию на межфазной поверхности вода—«масло». Работа адсорбции зависит не только от строения молекулы ПАВ, но и от структуры поверхностных слоев (см. подразд. 6.4). Поэтому общая количественная связь между ГЛБ и адсорбцией отсутствует.
7.4. Критический параметр упаковки
Важное свойство ПАВ — способность создавать самоорганизо-ванныс наноструктуры.
• насыщенные адсорбционные монослои (см. гл. 6) на поверхности раздела фаз;
• прямые и обратные мицеллы в водных растворах и в растворах неполярных жидкостей;
• жидкокристаллические структуры (см. гл. 8).
В образовании таких структур важную роль играет стерический фактор, а именно — соотношение размеров гидрофобной (липо
фильной) и гидрофильной частей молекулы ПАВ. Это соотношение количественно характеризует критический параметр упаковки (а):
а=Г/(Ш), (7 7)
где V — объем, занимаемый неполярной группой (углеводородным радикалом); / — длина развернутой углеводородной цепи; Q — площадь поперечною сечения молекулы ПАВ.
Во многих случаях площадь поперечного сечения молекулы ПАВ определяется площадью полярной группы (Q ). Отношение V/! определяет площадь сечения неполярной части (£2Л) молекулы.
Таким образом, критический параметр упаковки — это отношение площадей поперечных сечений yiлеводородпой цепи и полярной группы.
Физико-химическое значение критического параметра упаковки заключается в следующем На образование структур из молекул ПАВ (мицелл и др.) сильное влияние оказывают два фактора; взаимное отталкивание головных полярных групп соседних молекул и притяжение между гидрофобными цепями. Взаимодействие головных групп характеризует их эффективная площадь. Например, для ионных ПАВ это взаимодействие сильно зависит оз концентрации электролита: с увеличением концентрации площадь поперечного сечения молекулы уменьшается. Для неионных ПАВ эта площадь си 1ьно зависит от температуры.
Критический параметр упаковки связан с кривизной поверхности агрегата ПАВ: при а < I поверхность выпуклая, при а > 1 поверхность вогнутая.
Контрольные вопросы
I Какие вещества называют поверхностно-активными? Каковы главные области применения ПАВ в коллоидной химии?
2. Назовите основные группы ПАВ при их классификации по степени диссоциации ПАВ в воде
3. Назовите основные типы полимерных ПАВ
4 Объясните, по каким причинам важна проблема биодеградации промышленных ПАВ.
5. Охарактеризуйте классификацию ПАВ по физико-химическому механизму их действия.
6. Охарактеризуйте понятие «гидрофильно-липофильный баланс». Объясните принцип его расчета
7. Напишите уравнение для расчета работы переноса молекулы ПАВ из полярной фазы в неполярную.
8 Какрй фишко-химический смысл имеет критический параметр упаковки?
ГЛАВА 8
МИЦЕЛЛЯРНЫЕ СИСТЕМЫ
Многие поверхностно-активные вещества обладают замечательной способностью к самоорганизации в водных растворах и неполярных растворителях. Самоорганизация заключается в том, что при определенных условиях в растворе самопроизвольно образуется ансамбль наноразмерных агрегатов, которые состоят из десятков и сотен индивидуальных молекул или ионов ПАВ; эти агрегаты называюг мицеллами, а ПАВ, которые способны к образованию таких агрегатов, называют мицеллярными, или мицеллообразующими. Число мицеллярных систем составляет несколько тысяч.
Мицелла представляет собой самоорганизовапную структуру с определенным расположением и упаковкой образующих ее молекул ПАВ. Форма мицелл весьма разнообразна — известны сферические, цилиндрические, пластинчатые мицеллы. Вместе с тем сами мицеллы, располагаясь относительно друг друга в определенном порядке, создают более сложные упорядоченные структуры — кубические, гексагональные и др.
Мицеллярные системы обладают несколькими уникальными свойствами. Одно из них — солюбилизация (коллоидное растворение). Солюбилизация заключается в растворении внутри мицеллы веществ, которые плохо растворяются в жидкой дисперсионной среде. Солюбилизация играет очень важную роль во многих технологических и биологических процессах. Другое важнейшее свойство этих систем — мицеллярный катализ. Таким образом, мицеллярные системы представляют один из важнейших классов дисперсных систем.
8.1. Критическая концентрация мицеллообразования
Образование мицелл в растворах ПАВ доказано разными методами. Большая группа л их методов основана на изучении зависимости различных свойств paciворов ПАВ от их концентрации (С). Для растворов мицеллярных ПАВ такие зависимости обычно имеют два разных участка.
При малых концентрациях ПАВ свойства раствора меняются монотонно — примерно так же, как свойства растворов солей или других электролитов (первый участок). Однако при определенной
для каждого ПАВ концентрации ход зависимости свойство —концентрация резко изменяется.
На рис. 8.1 показана типичная изотерма поверхностного натяжения о =/(С) водного раствора мицеллярных ПАВ. На начальном участке АВ поверхностное натяжение монотонно уменьшается по мере увеличения концентрации в соответствии с уравнением Шишковского (6.1), т.е. так же, как в случае ПАВ с коротким неполярным радикалом. В точке В характер зависимости о =/(С) резко меняется — вместо дальнейшего уменьшения поверхностное натяжение остается постоянным (плато BD). Плато возникает, поскольку в отличие от индивидуальных молекул или ионов ПАВ мицеллы не обладаю! поверхностной активностью (см. подразд. 8.2) и поэтому не могут вызывать снижения поверхностного натяжения растворителя Точка D отвечает предельной растворимости данного ПАВ. Другой пример — резкое уменьшение эквивалентной электрической проводимости (X) при определенной концентрации раствора ПАВ (рис 8.2). Это указывает на появление в растворе крупных агрегатов, подвижность которых значительно меньше, чем индивидуальных ионов или молекул ПАВ.
Зависимости а = /(С) и X = /(С) (см. рис. 8.1, 8.2) косвенно подтверждают образование в растворе ПАВ агрегатов (мицелл) более крупных, чем молекулы (ионы). Прямым доказательством такою агрегирования является зависимость интенсивности светорассеяния от концентрации раствора. При определенной концентрации раствора светорассеяние резко возрастает (рис. 8.3). В соответствии с уравнением Рэлея (3.11) этот скачок свидетельствует о появлении в растворе дисперсных частиц (мицелл), размеры которых больше размера молекул или ионов ПАВ.
Рис. 8.1. Изотерма поверхностного натяжения раствора мицеллярного ПАВ
Рис. 8.2. Зависимость эквивалентной электропроводности (X) водного раствора ионного мицеллярного ПАВ oi концентрации (С) расгворгт
Рис. 8.3. Зависимость интенсивности светорассеяния (/) раствора ПАВ от концентрации (С) раствора
Приведенные примеры скачкообразною изменения свойств раствора при достижении определенной концентрации ПАВ убедительно свидетельствуют об образовании компактных агрегатов из десятков и сотен молекул (ионов) ПАВ — мицелл. Концентрацию, при которой начинается образование мицелл, называют критической концентрацией мицелпообра зования (ККМ, Сккм)-
Мицеллы ПАВ, образующиеся в водной фазе, называют прямыми, а в неполярной жидкости — обратными. Для одного и того же ПАВ значения ККМ при образовании прямых и обратных мицелл различны.
Таким образом, растворы мицеллообразующих ПАВ разделяют на две большие группы.
1. Гомогенные (истинные) растворы при малых концентрациях, т.е. при С< Сккм.
2. Микрогетерогспиые двухфазные системы при С> Сккм- Одна из этих фаз — молекулярный (ионный) раствор ПАВ, концентрация которого равна ККМ, другая фаза — мицеллы. Таким образом, мицеллярная система представляет собой ультрадисперсный лиофильный коллоидный раствор. Причина лиофильности заключается в определенной ориентации молекул ПАВ внутри мицеллы.
Влияние природы ПАВ. Значение ККМ зависит прежде всего от природы ПАВ. Для большинства ПАВ значения ККМ изменяются в достаточно широком интервале Ю^1— 10 ’ моль/л.
В качестве примера приведем значения ККМ для некоторых ПАВ при температуре 25 С, моль/л:
Бромид дсцилтримсгиламмопия................... 6,50 10
Бромид додецилтрнметиламмония................. 1,56 10 2
Бромид гексадецилтриметиламмония.............. 9,20 10-4
Хлорид додециламмония......................... 1,47 10 2
Хлорил додецилтрнметиламмония................. 2.03 10
Хлорил додецил и иридии и я................... 1,47 10
Тстрадецилсульфат натрия...................... 2,10 10
Доде и. и л сульфат натрия.................... 8,30 10
Октилсульфат натрия........................... 1,33 10“
Деканоат натрия.............................. 1,09 101
п-Додецилбепзолсульфонат натрия.............. 1,20 10 3
Димстилдодециламиноксид...................... 2,10 !0-3
СЩ(СН2)н(ОСН7СН)6ОН.......................... 8,70 105
СН3(СН7)7С6Н4(СН7СН2О)(, 2,05 10 4
Псрфтороктана! калия......................... 2.88 10“
Экспериментально установлены определенные закономерности в зависимости ККМ от состава и строения молекул ПАВ.
1. При увеличении длины углеводородной цени в гомологическом ряду ПАВ значения ККМ резко убываю!. Для многих ПАВ выполняется зависимость
lgCKKM=^-^c, (8.1)
где /?с — число атомов углерода в углеводородной цепи; А — константа, приблизительно постоянная для гомологов с разным числом дс, зависящая от природы и числа гидрофильных групп или заместителей в углеводородной цепи; lg2 (табл. 8.1).
2. Значения ККМ неионных ПАВ обычно гораздо меньше (примерно на два порядка), чем ионных ПАВ.
3. Значения ККМ катионных ПАВ несколько больше, чем анионных ПАВ Для неионных оксиэтилированных ПАВ характерно незначительное увеличение ККМ с ростом размера полярной группы.
4. Значение ККМ сильно зависит от валентности противоиона. Для неорганических противоионов увеличение валентности от I до 2 вызывает уменьшение ККМ. Органические противоионы снижают ККМ сильнее, чем неорганические. Этот эффект возрастает с увеличением размера неполярной части органического противоиона.
5. Наличие боковых цепей в молекуле ПАВ, двойных связей или ароматических групп приводит к значительным изменениям ККМ.
Т а б л п па 8.1
Значения констант Л, В в уравнении (8.1) для поверхностно-активных веществ различной природы
ПАВ 1емпература, °C А В
Карбоксила in натрия (мыла) 20 1.85 0,30
Карбоксилаты калия 25 1,92 0,29
Хлориды ал кил аммония 25 1,25 0,27
Бром иды ал килтри метилам мои ия 25 1,77 0,30
Бромиды а'л кил п и р ид и н и я 30 1,70 0,31
i(OC2H/|)f)OR 25 1,82 0,49
Особенно резкое влияние оказываем фторирование алкильной цепи: происходит снижение ККМ примерно на 1—2 порядка.
Влияние электролита. Сильное влияние на ККМ ионных ПАВ оказывают дополнительные вещества, растворяемые в воде вместе с ПАВ. особенно добавки электролитов. Рисунок 8.4 иллюстрирует самый простой и вместе с тем наиболее важный случай — влияние добавок электролита (NaCl) на ККМ мицелл ©образующего анионного ПАВ с таким же противоионом. Введение инертного электролиза приводит к нескольким следствиям:
• добавление электролита вызывает резкое (на один порядок) снижение ККМ;
• влияние электролита на ККМ растет с увеличением длины углеводородного радикала в молекуле мицеллообразующего ПАВ;
• при высоких концентрациях электролита ККМ гораздо сильнее зависит оз длины углеводородной цепи, чем в отсутствие соли;
• значения ККМ неиопных ПАВ от добавок электролитов практически не завися!.
Сильное влияние на ККМ оказывает введение спиртов — они вызывают значительное снижение ККМ (рис. 8.5). Спирты менее полярны, чем вода, они распределяются между раствором и мицеллами. Чем длиннее углеводородный радикал молекулы спирта, 1см выгоднее ее включение в состав мицеллы. Соответственно при этом уменьшав гея агрегатное число (т) — число молекул (ионов) ПАВ, необходимое для образования мицеллы.
Влияние температуры. Еще один фактор, влияющий на ККМ, — это температура. Особенно сильно температура влияет на ККМ многих ионных ПАВ.
При низких температурах растворимость ионных ПАВ в воде очень мала и достижение ККМ невозможно. При достаточном повышении температуры происходит резкое увеличение растворимости, которое вызывает образование мицелл (рис. 8.6).
Температуру, соответствующую началу образования мицелл, называют точкой Крафта (Гк). Фактически переход истинный
Рис. 8 4. Зависимость критической кон цептраци и мицеллообразования (Сккм) растворов ал к ил сульфатов натрия от числа атомов углерода (л) в углеводородной цепи радикала при концентрации водного раствора NaCl Q = 0 (/), 10 (2), 30 (5), 100 (4), 300 (5) ммоль/л
Qkm< моль/ji
^KKAb MOJlb/jl
Рис 8.5. Зависимость критической концентрации мицеллообразован и я (Qkm) додеканоата кадия от концентрации (С) спиртов в водном растворе;
а — этанол (/), пропанол (2), « бутанол (3); б — изопентанол (/). //-гексанол (2), «-гептанол (3); в — н-окганол (/), « нонанол (2), «-деканол (3)
Рис. 8.6. Зависимость растворимости ПАВ в воде от температуры: / — кривая растворимости; 2 — температурная зависимость ККМ
раствор ПАВ -> мицеллярный раствор совершается в узком температурном интервале вблизи точки Крафта. Влияние температуры на ККМ обусловлено температурной зависимостью растворимости ПАВ При низких темпера1урах Т < Тк предельная растворимость С()< Сккм, поэтому мицеллообразование в растворе не происходит. При достижении точки Крафта и более высоких температурах Со> Сккм в растворе образуются мицеллы данного ПАВ (эффект коллоидного растворения).
Точка Крафта является весьма важным фактором, поскопьку для образования мицелл нужны достаточно высокие температуры раствора- Т>ТК Между тем по разным причинам часто требуется обеспечить мицеллообразование при сравнительно невысокой (комнатной) температуре. Поэтому большое практическое значение имеет ряд факторов, влияющих на точку Крафта.
1. Точка Крафта резко повышается при удлинении углеводородной цепи в молекуле ПАВ (при четном числе атомов углерода).
2. Положение точки Крафта сильно зависит от природы полярной группы молекулы ПАВ и химической природы противоиона. Добавление электролитов обычно повышает точку Крафта. Для бромидов точка Крафта обычно выше, чем для хлоридов и иолидов Как правило, для ПАВ с многовалентными противоионами точка Крафта юраздо выше, чем для ПАВ с одновалентными противоионами.
Для получения ПАВ с низкими точками Крафта вводят:
1) дополнительную метильную группу или боковые заместители в алкильную цепь;
2) двойную связь в алкильную цепь;
3) полярный сегмент (обычно — оксиэтиленовую группу) между ионной (полярной) фуппой и алкильной цепью.
8.2. Строение и форма мицелл
Важнейшее свойство мицелл заключается в том, что они имеют высокоорганизованную внутреннюю структуру. Расположение молекул ПАВ в мицеллах соответствует правилу уравнивания полярностей (см. подразд. 6.4).
В прямых мицеллах неполярные части молекул поверхностно-активных веществ (обычно углеводородные цепи) располагаются во внутренней част мицеллы (рис 8.7). Они образуют так называемое ядро прямой мицеллы. Наружную часть мицеллы называют оболочкой. В прямой мицелле оболочка образована полярными группами. При такой структуре полярные группы молекул ПАВ обращены в сторону полярной дисперсионной среды (водный раствор), а неполярные (гидрофобные) группы экранируются полярной оболочкой от прямого контакта с водой В мицеллах ионных ПАВ обо-
Рис. 8.7. Схема прямом мицеллы ПАВ сферическом формы
ломка несет электрический заряд. Она представляет собой слой потенциалопределяюших ионов в двойном электрическом слое (см. подразд. 9.1).
В обратных мицеллах распочожение ПАВ диаметрально противоположно по сравнению с прямыми мицеллами. Ядро обратной мицеллы состоит из полярных групп, а в сторону неполярной дисперсионной среды обращены углеводородные цепи. Такое строение обратных мицелл также соответствует правилу уравнивания полярностей.
Структура ядра мицеллы представляет большой научный интерес Необходимо подчеркнуть, что строго радиальное расположение углеводородных цепей в прямой мицелле, конечно, в определенной степени идеализировано. Фактически ансамбль молекул или ионов, образующих ядро, надо рассматривать как статистический. Поэтому «агрегатное» состояние вещества в ядре следует считать жидким или жидкокристаллическим. Это положение подтверждено с помощью резонансных методов исследования прямых мицелл, а также расчетами методами молекулярной динамики. Соответственно и оболочка прямых мицелл нс является геометрически строгой поверхностью сферы. В молекулярном масштабе она «шероховата», т.е. одни полярные группы расположены над усредненной сферической поверхностью, а другие — ниже нее. Таким образом, мицеллы ПАВ по структуре относятся к псевдофазовым дисперсным частицам (см. гл. 3).
Форма мицелл весьма разнообразна. Вблизи К.КМ и в достаточно широком интервале концентраций выше ККМ (примерно на один порядок) образуются мицеллы, форма которых близка к сферической (рис. 8.8). Диаметр мицеллы (dm) определяется длиной молекулы ПАВ (/„,): dm~ 21т. Радиус ядра мицеллы близок к длине алкильной цепи (примерно 1,5 —3,0 нм). При дальнейшем
повышении концентрации происходит изменение формы мицеллы (см. рис. 8.8). Прямые мицеллы ПАВ в водном растворе вначале приобретают форму гибких вытянутых цилиндров, диаметр которых по-прежнему определяется длиной молекулы (d„,~ При более высоких концентрациях цилиндрические мицеллы располагаются в определенном порядке — возникает гексагональная упаковка. Дальнейшее увеличение концентрации ПАВ приводит к появлению пластинчатых (ламеллярных) мицелл, разделенных тонкими пленками воды. В результате возникает самоорганизованная биконтинуальная дисперсная система. Важно отмстить, что при образовании цилиндрических и пластинчатых мицелл выполняется тог же принцип самосборки, что и для сферических мицелл: ориентация молекул (ионов) ПАВ в мицелле соответствует правилу уравнивания полярностей Ребиндера.
Существование разных форм мицелл в зависимости от концентрации ПАВ называют полиморфизмом мицелл.
Размеры индивидуальных мицелл могут быть разными, т.е. мицеллярная система характеризуется распределением мицелл по размерам в некотором интервале — от минимального до максимального.
Рис. 8.8. Полиморфизм мицелл ПАВ
а — отдельная молекула ПАВ; б — прямая сферическая мицелла; в — прямая цилиндрическая мицелла; г — гексагональная упаковка прямых цилиндрических мицелл; д — пластинчатая (ламеллярная) мицелла; е — гексагональная упаковка обратных цилиндрических мицелл
Важнейшей количественной характеристикой мицелл является агрегатное число (т) — число молекул (ионов) ПАВ в одной мицелле. В прямых мицеллах агрегатное число составляет несколько десятков (как правило, не менее 20); в обратных мицеллах — в несколько раз меньше.
Агрегатное число зависит от строения и размеров молекул ПАВ, а также от размеров мицеллы:
(8.2)
где И,„ — объем углеводородного ядра прямой мицеллы; — объем неполярной части молекулы ПАВ.
Обьем сферической мицеллы равен
К„ = 4я/3/3. (8.3)
где /—длина развернутой углеводородной цепи.
Из уравнений (8.2), (8.3) следует
т = 4л/3/(3 И„). (8.4)
Безразмерное соотношение Vn/(Q.pl) = сх — это критический параметр упаковки (см. подразд 7.4), где Llp — площадь, приходящаяся на одну полярную группу в оболочке прямой мицеллы.
Оболочка мицеллы имеет очень большую кривизну: ее радиус составляет несколько нанометров. Теоретически показано, что условием устойчивой формы мицеллы является постоянное значение обоих главных радиусов кривизны. Из этого положения следует. что возможны три формы устойчивой поверхности мицеллы: сферическая, цилиндрическая и плоская. Они соответствуют основным формам мицелл: сферической, цилиндрической и ламеллярной.
Для сферических мицелл а < %; для цилиндрических мицелл параметр КПУ изменяется в интервале */3 < а < */2; для пластинчатых мицелл а = I.
* Таким образом, строение мицелл ПАВ весьма своеобразно. Мицеллы сочетают определенные свойст ва жидкости и твердого тела. Данные о коэффициентах диффузии молекул ПАВ в ядре мицелл и особенно о способноеги мицелл к солюбилизации (коллоидному растворению) позволяют рассматривать мицеллу ПАВ как нанокаплю углеводородной жидкости. Это свойство обеспечивает низкое поверхностное натяжение на границе мицелл с дисперсионной средой. В то же время мицеллы имеют определенную геометрическую форму, что присуще твердым телам. Модель строения мицелл как образований, сочетающих свойства жидкости и твердого тела, лежит в основе современной теории мицелл (А. И. Русанов).
8.3. Рост мицелл
При концентрации ПАВ выше ККМ в растворе происходят следующие процессы:
I) увеличивается число мицелл при неизменном агрегатном числе, т.е. растет концентрация частиц;
2) растет объем мицелл и соответственно агрегатное число;
3) по мере роста размера и концентрани и мицеллы начинают контактировать друг с другом с образованием различных мицеллярных структур
Процесс укрупнения мицелл происходит в растворах многих ПАВ и протекает по-разному для ионогенных и неионогенных ПАВ
Рост мицелл ионогенных ПАВ. Процесс укрупнения мицелл ионоюнных ПАВ определяется рядом факторов.
1. Удлинение углеводородной цепи способствует росту мицелл Для ПАВ с небольшим числом атомов углерода рост мицелл обычно не наблюдается.
2 Понижение температуры способствует pociy мицелл ионо-генных ПАВ. Например, мицеллы бромида гексадсцил три метил аммония при температуре 30 С растут при достаточном повышении концентрации ПАВ, но при 50 С рост мицелл не происходи!.
3. Влияние природы противоиона, образующегося при диссоциации ионогенного ПАВ, весьма избирательно. Например, мицеллы кати он активного бромида !ексадснилтриметиламмония быстро расту! с увеличением концентрации ПАВ выше ККМ. При замещении в молекулах этого ПАВ ионов брома на ионы хлора рост мицелл не происходит. Аналогичная ситуация характерна и для анионных ПАВ.
Мицеллы додецилсульфата лития не увеличиваются в размерах, но при замене лития на калий или на цезий происходит значительное увеличение мицелл. Неионные ПАВ с органическими противоионами образуют мицеллы, которые обычно резко увеличиваются в размерах с ростом концентрации.
Рис. 8.9. Зависимость числа агрегации (/и) мицелл долей ил сульфат а натрия от концентрации (С) добавляемой соли (NaCl) при температуре Т = 60 ( /), 25 (2) С
4. Действие растворимых добавок может быть противоположным. Соли обычно инициируют рост мицелл (рис. 8.9). Спирты и ароматические вещества, коюрые концентрируются вблизи оболочки прямых мицелл, способствуют их увеличению Например, растворение гсксанола вызывает резкий рост мицелл бромида гек-садепилтриметиламмония. Напротив, вещества, которые концентрируются в ядре мицелл, препятствуют их росту.
Рост мицелл неионогенных ПАВ. Мицеллы производных окиси этилена с увеличением концентрации ПАВ растут тем сильнее, чем короче их полярная группа Например, если число полиэтиленовых групп равно 4—6, то происходит значительный рост мицелл. Если число этих групп >8, мицеллы при концентрациях выше ККМ уже не меняют своих размеров.
Другая важная особенность мицелл нсионогенных ПАВ — повышение температуры способствует росту мицелл, тогда как нагрев мицелл ионогенных ПАВ приводит к обратному результату — подавлению роста мицелл.
Увеличение размера мицелл вызывает изменение их формы По мере роста концентрации ПАВ сферические мицеллы сменяются эллипсоидными, потом появляются цилиндрические и пластинчатые мицеллы (см. подразд. 8.2).
Изменение формы мицелл влияет на многие их свойства. Например, длинные цилиндрические мицеллы («стержни») имеют определенное сходство с линейными макромолекулами: они могут обладать большой гибкостью, которая варьирует в широких пределах. Некоторые цилиндрические мицеллы достаточно жесткие; другие мицеллы очень гибкие. Гибкость можно регулировать составом раствора ПАВ. Например, добавление электролитов вызывает переход от жесткой стержнеобразной к очень гибкой мицелле
При достаточно высокой концентрации ПАВ цилиндрические мицеллы начинают ветвиться. Кроме того, при высоких концентрациях происходит переход системы от коллоидного раствора отдельных мицелл, не взаимодействующих друг с другом, к связнодисперсной мицеллярной структуре. Такая структура возникает, когда мицеллы приходят в непосредственный контакт друг с другом. Появлению таких контактов способствует разветвление цилиндрических мицелл В результате во всем объеме раствора формируется трехмерная сетка из стержневых мицелл и дисперсная система в целом становится биконтинуальной (рис. 8.10). Она состоит из двух взаимно проникающих дисперсных фаз. I) непрерывная сетка из наноразмерных элементов (цилиндрических мицелл); 2) жидкость, заполняющая наноразмерные поры и каналы внутри сетки.
Объемная доля дисперсной фазы (ф), при достижении которой начинается образование сетки, составляет примерно 10~\ Переход
Рис. 8 10. Биконтинуальная структура мицеллярных ПАВ
к биконтинуальной структуре приводит к значительному увеличению эффективном вязкости системы.
В концентрированных растворах ПАВ из ансамбля мицелл образуются различные самоор!авизованные упорядоченные структуры, которые следует рассматривать как мезофазы. Мицеллы представляют собой своеобразные «строительные элементы» таких структур. Строение мезофаз определяется формой мицелл. Сферические мицеллы образуют кубическую упаковку, цилиндрические — гексагональную, пластинчатые — ламеллярную мезо-фа зу. Перечисленные структуры представляют предельные случаи и далеко не исчерпывают многообразия мицеллярных мезофаз-ных структур.
Дня каждого мицеллообразующего ПАВ в зависимости от концентрации ПАВ и темпера гуры могут существовать несколько типов структур.
8.4. Термодинамика образования мицелл
Сложное строение мицелл ПАВ вызывает большие трудности при создании теории их образования. Для расчета термодинамических условии перехода раствора IIAB от молекулярной (ионной) формы к мицеллярной применяют главным образом квазихимически й подход. Согласно другому подходу мицеллу рассматривают как фазовую частицу и соответственно процесс образования мицеллы считают фазовым переходом.
Основное положение квазихимического подхода: мицеллы представляют собой продукт обратимой химической реакции между отдельными молекулами (ионами) ПАВ
/«[ПАВ] |ПАВ]да, (8.5)
где т — агрегатное число; [ПАВ] — конкретное поверхностно-активное вещество; [ПАВ]т — мицелла из т молекул ПАВ.
Другое принципиальное допущение квазихимического подхода: агрегатное число не зависит oi концентрации ПАВ, т.е. с увеличением концентрации растет число мицелл в единице объема раствора (концен грация мицелл v,„), но размеры и форма мицелл при этом не меняются.
В рамках квазихимического подхода агрегирование молекул ПАВ в мицеллу рассматривают как прямое направление «реакции» ми-целюобразования, а дезагрегирование мицелл (распад на исходные молекулы) — как обратное направление данной реакции. Этот подход соответствует экспериментальным результатам. По данным спектроскопии ядерного магнитною резонанса время «жизни» индивидуальной молекулы ПАВ в мицелле составляет около К) с, а период полураспада мицеллы — от 10 3 до 1 с.
Квазихимический подход позволяет охарактеризовать процесс мицеллообразования по аналогии с законом действующих масс константой скорости реакции {Кт)\
= N Ст ’ ^-6)
где ут — концентрация мицелл (число мицелл в единице объема); Na — число Авогадро: С — концентрация ПАВ; т — агрегатное число
Уравнение (8.6) показывает, что при концентрациях выше ККМ число мицелл возрастаем как степенная функция с показателем tn » I, т.е. очень резко. При С< СККм имеем vm = 0, т.е. все повер-хноезно-активное вещество находится в растворе в виде индивидуальных молекул или ионов
Квазихимический подход позволяет рассчитать термодинамические параметры процесса мицеллообразования. Изменение энергии Гиббса (AG, Дж/моль) при переходе молекулярного раствора ПАВ в мицеллярную систему с агрегатным числом т равно
А(7 = -(RT/tn)hiKm. (8 7)
По определению имеем
G= Н- TS,
1дс Н — энтальпия; S— энтропия; Т — температура.
Уравнение (8.7) позволяет по экспериментально определяемой температурной зависимости AG = f(T) рассчитать изменение эн-
тальлии А// и энтропии А5 при мицеллообразовапии. Константа скорости «реакции» мицеллообразовапии соответствует ККМ.
Таким образом, формирование мицелл описывается следующими термодинамическими уравнениями:
А// =-Л Г d—Скк- , (8.8)
ат
АУ = -А1пСккм I (8.9)
\ d/ у
где Д5 — стандартная энтропия процесса минеллообразования.
Энтальпийный эффект А//равен теплоте минеллообразования с обратным знаком. Для прямых мицелл величина А//определяется взаимодействием между углеводородными радикалами и водой.
Табл ица 8.2
Термодинамические функции мицеллообразования ПАВ в водной среде
Сккм моль/л Температура, °C -д//, кДж/моль 7Д5°, кДж/моль
Капронат натрия
13,0 5 4,57 5,49
14,6 20 5,07 5,24
17,8 60 4,52 6,62
21,8 90 5,32 6,24
Каприлат натрия
6,39 20 4,11 8,21
7,23 40 4,61 8,21
8,09 60 4,40 8,92
Додецилсулъфат натрия
0,170 20 4,56 16,60
0,180 30 2,76 18,94
0,183 60 2,30 21,54
0,204 90 2,72 22,92
Олеат натрия
0,0148 5 14,58 11,10
0,0196 25 14,46 10,89
0,0356 60 14,50 13,87
0,0623 90 15,08 14,16
В большинстве систем А// > 0, т.е. процесс мицеллообразования происходит эндотермически. Термодинамическое условие самопроизвольного мицеллообразования заключается в выполнении неравенства AG <0. При положительных значениях АТ/ > 0 оно выполняется, если основное значение имеет изменение энтропии, т.е. Та5°| > АТ/ . Такое соотношение характерно для многих мицеллообразующих ПАВ (табл 8 2).
Увеличение энтропии при мицеллообразовапии А5 > 0 обусловлено гидрофобным эффектом (см. подразд. 8.8). Переход молекул ПАВ из водного раствора в прямые мицеллы включает в тепловое движение те молекулы воды, которые прежде взаимодействовали с углеводородными цепями. Эта модель наглядно объясняет уменьшение ККМ при увеличении числа атомов углерода в неполярной части молекулы ПАВ.
Для обратных мицелл, напротив, главное значение имеет изменение энтальпии при замене связи полярная i руппа — неполярная дисперсионная среда на взаимодействия полярных групп в ядре обратной мицеллы.
8.5. Смешанные мицеллы
Во многих технологических процессах используют не одно поверхностно-активное вещество, а смеси двух или нескольких ПАВ. Такие композиции обладают более высокой эффективностью, чем индивидуальные ПАВ. По этой причине изучение свойств смесей ПАВ представляет одно из наиболее перспективных направлений современной коллоидной химии. Образование мицелл, состоящих из двух разных ПАВ, характеризуется рядом закономерностей.
Критическая концентрация мицеллообразования в растворе смеси ПАВ (СЙЛ) зависит от значений ККМ обоих компонентов (обозначим их и С^м ). Наиболее просты для расчета смеси ПАВ с одинаковыми полярными группами, но разными углеводородными радикалами. В этом случае в мицелле не возникают дополнительные виды межмолекулярпых взаимодействии и поэтому ККМ смеси можно считать аддитивным свойством. Обозначим xIm — мольная доля компонента I в составе мицеллы; значение xIm, конечно, отличается от концентрации Q этого компонента в растворе. Соот ветственно мольную долю второго компонента в мицелле обозначим х2т. Очевидно, что х1/я + х2т = 1. Тогда из условия адди гивност и получаем
ГЧП1Х _ v гЧ!) , v Л’(2) /О 1()\
VKK\1 - л1от'-ККМ + л2шьККМ- vo.iv/
Для расчета ККМ смеси в зависимости от концентраций двух ПАВ (С] и С2) используют уравнение
-----------=----------!— + -—. (б. 11) z^inix-------------------------------гЛ2)
VKKM VKKM сккм
Для расчета состава мицеллы, т.е. долей обоих ПАВ и х2/н в самой мицелле, используют уравнение
г С1СККМ /о
lw С С^2* "
е1'- ККМ С2С ККМ
Уравнение (8.12) показывает, что особенно сильное изменение состава мицелл (по сравнению с раствором ПАВ) происходит в тех случаях, когда критические концентрации компонентов ^ккм и ^ккм сильно отличаются. Например, при отношении ^ккм /^ккм = 0,01 смешан н ые миi1еллы состоят поч т и iюлностью из компонента с меньшей ККМ. Если С^м =^ккм' то доли компонентов 1 и 2 в составе смешанной мицеллы одинаковы: .vlZ7; = х2т (рис. 8.11). Таким образом, даже очень малые примеси ПАВ с сильно выраженными гидрофобными свойствами резко меняют состав мицелл и условия их образования.
Сложнее рассчитать критические концентрации мицеллообра-зования смеси и составы смешанных мицелл в тех случаях, когда смесь состоит из ПАВ разных классов. Например, молекулы неионогенных ПАВ, располагаясь между анионами второго ПАВ, ослабляют их оггалки ван не. Еше более сильное взаимодействие происходит в смесях анионного и катионного ПАВ.
Рис. 8.11. Зависимость критической концентрации миисллообразования смеси (а) и состава смешанных мицелл (б) от содержания второго ПАВ (х2) в растворе при соотношении критических концентраций мицеллооб-разования второго и первого компонентов смеси /С^м = 1 (7), 0,1 (2), 0,01 (5) (л2„, — содержание второго компонента в мицелле)
В таких системах нужно учитывать коэффициенты активности fm и/ъи обоих компонентов в смешанной мицелле. Уравнения (8.11) и (8.12) будут иметь вид
"’inix
ККМ
’(I)
ККМ
2____
’(2)
ККМ
(8.13)
ККМ
=
(8.14)
'-U2mvKKM т v2./lm'-'KKM
Для расчета коэффициентов активности используют уравнения, применяемые в теории регулярных растворов:
ln/2m = (AiJ2₽, (8.16)
где р — параметр взаимодействия между молекулами (ионами) двух ПАВ внутри мицеллы.
Параметр р имеет следующий физико-химический смысл. Положительные значения р > 0 указывают на отталкивание молекул двух ПАВ в смешанной мицелле; если Р < 0, между ними существует притяжение. При р - 0 коэффициенты активности равны f\m -fim- Тогда вступают в силу уравнения (8.10)—(8.12) для невзаимодействующих ПАВ. В применяемых на практике смесях ПАВ обычно р < 0.
Расчеты по приведенным уравнениям приводят к следующим заключениям. Если параметр взаимодействия отрицателен (р < 0). то чем больше он по модулю, тем сильнее уменьшается ККМ смеси ПАВ.
Для смесей неионогенных и ионогенных ПАВ р « -2; для смесей анионных и катионных ПАВ -20 < р <-10. По мере увеличения параметра р (по абсолютной величине) состав смешанной мицеллы приближается к равной доле каждого ПАВ.
Для практических целей важно провести расчет состава смеси, при котором достигается минимальное значение ККМ смеси. Для этого должны выполняться следующие условия: 1) р < 0; 2) 1П (^ккмЛлскм) < I РI • Тогда оптимальная (минимальная) концентрация компонента 2 (C2niin) будет равна
2 nun
(8.17)
При больших концентрациях ПАВ в смешанных мицеллярных системах часто происходят эффекты взаимного усиления действия компонентов смеси (синергизм) или обратный эффект (антагонизм) — ослабление действия индивидуальных ПАВ.
8.6. Солюбилизация
Солюбилизация относится к числу наиболее важных коллоидно-химических явлении. Солюбилизация заключается в способности мицелл аккумулировать внутри себя значительное количество веществ, которые нс растворяются в жидкой дисперсионной среде. Поэтому солюбилизацию называют коллоидным растворением. Прямые мицеллы солюбилизируют неполярные вещества, а обратные мицеллы — полярные вещества. Поглощенное мицеллами вещество называют солюбилизатом.
Наиболее наглядно солюбилизация характеризуется зависимостью концентрации солюбилизата от концентрации поверхностно-активного вещества (рис. 8.12). Участок при малых концентрациях ПАВ (С< С’ккм) соответствует истинной растворимости данною ПАВ в воде при выбранной температуре. При С> Сккм происходит линейное увеличение коллоидной растворимости (С) при возрастании концентрации ПАВ Тангенс угла наклона этой прямой равен солюбилизационной емкости мицелл.
Количественной характеристикой солюбилизации служит со-любилизационная емкость (s), которая равна
'А
s = (8.18)
лгПАП
где и, — число молей солюбилизата в мицелле; дПАВ — число молей ПАВ, образующих данную мицеллу.
Рис. 8 12. Зависимость солюбилизации органическою красителя в мицеллах алканоатов калия от концентрации ПАВ
1 — октаноат: 2— деканоат: 3 — лолеканоаг: 4 — тетрилеканоат (;V — количество солюбилизированного красителя в относительных единицах)
Линейный характер зависимости количества солюбилизированного вещества от концентрации ПАВ (см. рис. 8.12) показывает, что солюбилизационная емкость постоянна. Это положение справедливо при сохранении сферической формы мицелл. Солюбилизирующая способность ПАВ зависит от их гидрофильно-липофильного баланса (см. подразд. 7.3). Для наиболее эффективных солюбилизаторов ГЛ Б составляет примерно 15—18.
Солюбилизационная емкость может быть весьма значительной. Например, предельные углеводороды растворяются в воде очень мало. Так, растворимость н-октана при комнатной температуре составляет всего 0,0015%. Однако в прямых мицеллах олеата натрия поглощается до 2 % октана, т.е. коллоидная растворимость на 3 порядка превышает истинную.
Солюбилизирующая способность мицелл зависит от нескольких факторов, в гом числе и от их формы. Например, солюбилизация алканов в цилиндрических мицеллах выше, чем в сферических Солюбилизация неполярных веществ в прямой мицелле вызывает се удлинение с переходом от эллипсоидной формы к цилиндрической (рис. 8.13). Однако солюбилизация веществ, содержащих полярную Iруппу (например, спиртов), в цилиндрических мицеллах, напротив, ниже. Размещение солюбилизированных веществ внутри мицелл зависит от их химической природы. Предельные углеводороды концентрируются в ядре прямых мицелл, что подтверждает модель ядра как жидкой нанокапли. Вода, солюбилизируемая в обратных мицеллах, также аккумулируется их ядром.
Ароматические соединения распределяются между ядром мицеллы и ее периферической частью вблизи обо точки в зависимости от природы мицеллообразующего ПАВ. Например, в мицеллах катионного ПАВ (бромида цетилтриметиламмония) бензол собирается вблизи оболочки, а в мицеллах анионного ПАВ (доле ни л сульфата натрия) — в ядре мицеллы.
Полярные вещества (спирты с короткой углеводородной цепью) при солюбилизации в прямых мицеллах закрепляются вблизи оболочки. При удлинении углеводородного радикала спирты перемешаются в ядро мицеллы. Солюбилизационная емкость этих систем составляет около 5.
Рис. 8 13 Деформация прямой мицеллы при солюбилизации неполярной жидкое 1 и
Солюбилизация происходит как в мицеллах, состоящих из одного ПАВ, так и в смешанных мицеллах. Солюбилизация некоторых веществ (циклогексана, гексана, додекана и других неполярных соединений) в смешанных мицеллах проходит эффективнее, чем в мицеллах индивидуальных ПАВ, т е. смеси ПАВ обнаруживают в этих случаях синергизм по отношению к солюбилизации.
Солюбилизация полярных веществ (спиртов) в смешанных мицеллах, напротив, происходит хуже, чем в мицеллах одного ПАВ.
При теоретическом описании солюбилизации одна из основных задач заключается в расчете солюбилизационной емкости. Для этого используют следующую модель. В прямой мицелле ядро рассматривают как жидкую неполярную фазу. Условие термодинамического равновесия заключается в равенстве химических потенциалов солюбилизата в мицелле (цда) и в виде самостоятельной жидкой фазы (цж):
= + KHns, (8.19)
где р/п — стандартный химический потенциал солюбилизата; R — универсальная газовая постоянная; Т — абсолютная температура.
Сферическая мицелла находится под действием капиллярного давления рс = 2о/г(см. подразд. 5.1; о — поверхностное натяжение на границе оболочки мицеллы с дисперсионной средой; г — радиус оболочки). Соответственно стандартный химический потенциал вещества в мицелле и °, больше, чем в объемной фазе:
=Иж + 2аГт/(г/?Г), (8.20)
где — молярный объем солюбилизата.
Из уравнений (8.19) и (8.20) следует
5 = схр[-2оКЛ^Г)|. (8.21)
Выражение (8.21) представляет собой аналог уравнения Гиббса—Кюри—Фрейндлиха для расчета растворимости вещества в зависимости от кривизны поверхности (см гл 5).
Уравнение (8.21) позволяет рассчитывать нс только солюби-лизационную емкость мицелл, но и поверхностное натяжение Эти опенки дают значение о - 28 — 30 мДж/м2.
Другая важная задача заключается в расчете энтальпии солюбилизации (ДЯ5). которая определяется работой переноса I моля солюбилизата из макрофазы этого вещества в мицеллу. Этот расчет строи 1ся на основе квазихимическои модели мицеллообразования (см. подразд. 8.5). По аналог ни с температурной зависимое 1ъю ККМ энтальпия солюбилизации рассчитывается по уравнению
(8 22)
d7 RT2' ' ’
В теоретическом и прикладном отношении важным параметром солюбилизации является скорость этого процесса. Ес характеризуют количеством солюбилизата, перенесенного в мицеллу из раствора в единицу времени через единицу площади Скорость солюбилизации растет с увеличением концентрации ПАВ Для мицелл, образованных нсионнымн ПАВ, скорость солюбилизации pacrci с увеличением температуры.
Прикладное значение солюбилизации очень велико. Это явление используют в эмульсионной полимеризации, мицеллярном катализе, нефтедобыче. Солюбилизация жиров в мицеллах желчных холпевых кислот определяет усвоение жиров в организмах.
8.7. Ассоциаты поверхностно-активных веществ с полимерами и белками
Наряду с мицеллообразованием в растворах ПАВ действует еще один механизм образования самоорганизованных структур. Он основан на взаимодействии молекул и ионов ПАВ с водорастворимыми полимерами и белками.
Поясним это влияние полимеров на поверхностную активность ПАВ и мицеллообразовапие на примере поливинилпирролидона при его добавлении к раствору типичного анионного ПАВ — до-денилсульфата натрия (рис. 8.14). При некоторой концентрации С* < Сккм на изотерме поверхностного натяжения о = f(C) возникает излом. При дальнейшем повышении концентрации повер-
Рис. 8 14 Изотермы поверхностного натяжения растворов додецилсульфа га натрия при добавлении поливинилпирролидона в концентрации О (7), 1 (2), 3 (5), 10 (4) г/л
хностное натяжение раствора остается постоянным. После окончания этого участка поверхностное натяжение снова уменьшается до значения, отвечающего мицеллярному раствору данного ПАВ в отсутствие полимера. Критические кон цен грации мицелл ©образования растворов ПАВ с добавками полимера выше, чем раствора ПАВ; это различие растет по мере увеличения концентрации растворенного полимера.
Возникновение первою плато на изотерме поверхностного натяжения при концентрации С* вызвано тем, что ионы ПАВ начинают группироваться (ассоциироваться) возле молекулы полимера. Поэтому концентрацию С* называют критической концентрацией ассоциации (ККА, СККА). Верхняя граница плато соответствует предельной степени ассоциации, которая зависит от длины макромолекулы.
Существую! экспериментальные методы (селективные электроды, спектроскопические и диффузионные методы), позволяющие измерить степень ассоциации (а) — относительную долю ассоциированных молекул ПАВ. Типичная зависимость степени ассоциации от концентрации ПАВ (рис. 8.15) имеет несколько участков. При малых концентрациях влияние растворенного полимера отсутствует При достижении ККА степень ассоциации резко возрастает. При дальнейшем увеличении концентрации степень ассоциации возрастает мало. При концентрациях выше ККМ начинает доминировать другой процесс ассоциирования молекул ПАВ — образование мицелл.
Присутствие полимеров в растворах ПАВ влияет на солюбилизацию — она начинается при концентрациях близких к ККА, т.е. до начала мицеллообразования.
Таким образом, для растворов ПАВ, содержащих полимеры, наряду с ККМ важной характеристикой служит ККА. Отношение концентраций Сккл/Сккм слабо зависит от концентрации полимера. Концентрационный интервал, соответствующий ассоциации молекул ПАВ возле макромолекул, увеличивается пропорционально концентрации растворенного полимера. Способность к ассоциации зависит от свойств ПАВ. Ассоциацию анионных ПАВ вызыва-
Рис. 8 15. Зависимость степени ассоциации (а) молекул ПАВ от концентрации ПАВ
/ — в присутствии полимера (ассоциация ПАВ—полимер); 2 — в отсутствие полимера (мицеллообразование ПАВ)
ют большинство гомополимеров. Kai ионные ПАВ взаимодействуют с полимерами несколько слабее. Цвиттер-ионные и неионогенные ПАВ взаимодействуют с гомополимерами очень редко.
Свойства смешанных растворов, содержащих ПАВ вместе с полимерами, можно рассматривать на основе двух подходов. Один подход ставит во главу угла именно ассоциацию ПАВ с макромолекулами, т.е. образование своеобразных комплексов ПАВ —полимер. Этот подход более продуктивен для полимеров с гидрофобными группами Другой подход рассматривает ассоциацию молекул ПАВ на основе минсллообразования. Этот подход оказывается более предпочтительным для гидрофильных гомополимеров.
Особо следует выделить взаимодействие ПАВ с поверхностноактивными полимерами. Гидрофобные полимеры moiут формировать мицеллы или гидрофобные микродомсны при очень малых концентрациях. Эти мицеллы могуч солюбилизировать гидрофобные молекулы. Такие полимеры в смеси с ПАВ часто образуют смешанные мицеллы. Основную роль в этом процессе играют два фактора: длина углеводородной цепи и распределение электрических зарядов вдоль нее. Совместное действие мицеллообразующих ПАВ и полимеров позволяет эффективно регулировать многие свойства ко гиоидных систем: солюбилизацию, устойчивость, вязкость. Поэтому такие смеси используют очень широко.
Процессы ассоциации происходят также в растворах низкомолекулярных ПАВ (в том числе и липидов) с макромолекулами белков. Во многом эти системы сходны с растворами ПАВ и полимеров. Важная особенность взаимодействия ПАВ с белками заключается в том, что ассоциация ПАВ, особенно ионных, приводит к необратимым конформационным изменениям макромолекулы белка. Таким образом, ПАВ могут вызывать денатурацию белков, что имеет большое практическое значение.
8.8. Гидрофобные взаимодействия
Механизмы адсорбции ПАВ на поверхности растворов и образования мицелл в объеме раствора весьма своеобразны. Оба процесса — адсорбция и мицеллообразование — можно рассматривать как самоорганизацию. Действительно, и адсорбционные слои ПАВ, и мицеллы представляют структуры с высокой степенью организации. Естественно, возникает вопрос о причинах самопроизвольного образования (самосборки) таких упорядоченных наноразмерпых структур
Прежде всего следует рассмотреть гермодинамический аспект этой проблемы. Из термодинамики поверхностных явлений (см. гл. 5) и мицеллообразования (см. гл. 8) следует принципиально важный вывод — адсорбционные слои ПАВ и мицеллы прсдстав-
ля ют собой открытые системы, так как они существую! в условиях постоянного обмена массой и энергией с окружающей их средой — раствором ПАВ. В этом oi ношении данные коллоиднохимические объекты сходны с объектами биофизики, в том числе с живыми организмами, которые функционируют благодаря обмену энергией и массой с окружающей их средой. В таких системах общее изменение энтропии составляет
65 = 65 + 65/,
где 65, — изменение энтропии внутри объекта (мицеллы или адсорбционного слоя); 65/ — изменение энтропии при взаимодействии объекта с окружающей его средой (см. подразд. 2.4).
В соответствии с вторым началом термодинамики любой самопроизвольный процесс приводит к увеличению общей энтропии системы: 65> 0. Эго условие может выполняться и при уменьшении энтропии объекта (65/ < 0), если при этом изменение энтропии среды положительно 65/> 0, причем |65/| > |65|.
Таким образом, формирование упорядоченных адсорбционных слоев и мицелл ПАВ имеет энтропийную природу. Одним из подтверждений этого вывода является зависимость критической концентрации мицеллообразования от температуры
На молекулярном уровне это означает, что переход молекул (ионов) ПАВ на поверхность или в мицеллу обусловлен выталкиванием этих молекул из внутреннего объема раствора, что приводит к увеличению энтропии растворителя (воды).
Для водных растворов причина увеличения энтропии заключается в том, что нарушается упорядоченное расположение молекул воды возле гидрофобной части молекулы ПАВ (углеводородного радикала).
Гидрофобными взаимодействиями называют специфические взаимодействия, вызывающие выталкивание неполярных молекул из воды и имеющие энтропийную природу.
Специфика гидрофобных взаимодействий проявляется, например, в том, что плохая растворимость углеводородов в воде обусловлена не повышением энтальпии системы, а уменьшением энтропии. Поэтому при повышении температуры растворимость углеводородов в воде уменьшается, а не растет как для ионных кристаллов. Это положение иллюстрирует следующий пример. При растворении буганола в воде при температуре 298 К уменьшение энтропии составляет А5 = -96,6 Дж/(моль К), а увеличение энтальпии А// = 4 200 Дж/моль. Общее изменение энергии I иббса будет равно
А (7 = А//- 7Д5= -4 200 + 298-96,6 - 33 000 Дж/моль.
Таким образом, в этой системе уменьшение энтальпии примерно в 7,5 раз меньше энтропийного вклада 7Д5.
Гидрофобные взаимодействия играют важную роль не только в адсорбции ПАВ и образовании мицелл, но и в свойствах растворов водорастворимых полимеров и бепков. Гидрофобные взаимодействия вносят определенный вклад в формирование глобулярной структуры белковых макромолекул. Поскольку именно эти структуры обладают биологической функциональностью, значение гидрофобных взаимодействий в биологии и медицине очень велико.
Для многих практических приложений коллоидной химии важно регулировать гидрофобные взаимодействия. Для этой цели используются три способа.
I. Воздействие на микроструктуру воды. Повышение температуры увеличивает гидрофобные взаимодействия (растет энтропийное слагаемое 7Л5 энергии Гиббса); при понижении температуры они ослабевают. Сильное влияние на ci рук гуру воды оказывают растворяемые в ней электролиты. Так, катионы по способности увеличивать число водородных связей располагаются в ряды:
Н+> ЬГ > Na'
Вс2+> Cd2' > Zi?> Mg2"> Ca2
АГ+> Cr3+
По способности разрушать водородные связи ионы располагаются следующим образом:
Cs+> Rb+> К?
С1О4> I > Вг > NO;> СГ
2. Замена воды другими растворителями (неэлектролитами). Например, добавление спиртов к воде приводит с ростом их концентрации к ослаблению и полному нарушению гидрофобных взаимодействий. С этим связано сильное влияние спиртов па ККМ.
3. Добавление поверхностно-активных веществ. Адсорбция ПАВ на гидрофобных частицах может модифицировать (гилрофилизи-ровать) их поверхность, что ведет к ослаблению гидрофобных взаимодействии Напротив, адсорбция ПАВ на гидрофильных веществах приводит к появлению гидрофобных взаимодействий.
Контрольные вопросы
I Опишите, «по представляют собой мицеллы повсрхносгио-актив-ных веществ. Какие мицеллы называют прямыми, какие — обратными?
2 Дате определение критической концентрации мицедлбобразова-ния Как зависит этот показатель от длины углеводородной цени в гомологическом ряду ПАВ?
3. Назовите основные экспериментальные методы определения критической константы мицеллообразования различных ПАВ. Объясните суть э I их методов.
4. Объясните физико-химический смысл точки Крафта. Какие способы применяют для получения ПАВ с низкими точками Крафта?
5 Охарактеризуйте строение прямых и обратных мицелл ПАВ Что представляет собой ядро мицеллы?
6. Назовите основные формы мицелл ПАВ.
7. Определите число агрегации при образовании мицелл ПАВ.
8. Назовите основные положения квазихимической теории образования мицелл.
9 Приведите термодинамические уравнения квазихимической теории образования мицелл.
10. В чем заключается явление солюбилизации (коллоидного растворения)?
11 Дайте определение солюбилизанионной емкости мицелл ПАВ Приведите термодинамическое уравнение для ее расчета.
12. Объясни тс образование ассоциатов ПАВ с полимерами и белками Охарактеризуйте понятие «критическая концентрация ассоциации».
13. Охарактеризуйте гидрофобные взаимодействия, объясните природу их возникновения.
ГЛАВА 9
ЭЛЕКТРОПОВЕРХНОСТНЫЕ ЯВЛЕНИЯ
Поверхность дисперсных частиц часто имеет электрический заряд. Наличие заряда сильно влияет на свойства поверхностей раздела фаз. Большая группа явлении, происходящих в дисперсных системах, определяется в основном именно электрическими поверхностными зарядами; такие явления называют электропо-вёрхностными. Значение электроповерхностных явлении в коллоидной химии очень велико.
Отметим наиболее важные следствия наличия электрических зарядов на поверхности жидких и твердых дисперсных частиц.
1. Одноименные заряды на поверхности дисперсных частиц вызывают электростатическое (кулоновское) отталкивание и препятствуют сближению и слипанию частиц. Поэтому электрический заряд представляет важнейшее условие агрегативной устойчивости дисперсных систем (см. гл. 10).
2. При наличии поверхностного заряда дисперсные частицы и дисперсионная среда (жидкость) могут направленно перемешаться в электрическом поле. Эти два процесса называют электрофорезом и электроосмосом (см. подразд. 9.5).
3. Электрические заряды вызывают значительное уменьшение поверхностного натяжения жидкостей и поверхностной энергии твердых тел на границе с растворами электролитов. Этот эффект называют электрокапиллярным (см. подразд. 9.6).
Главную роль во всех электроповерхностных явлениях играет распределение ионов вблизи поверхности раздела дисперсной фазы с жидкой дисперсионной средой. В этой области катионы и анионы разделяются на два отдельных слоя и образуют двойной электрический слой (ДЭС) (см. подразд. 9.2). Строение и свойства ДЭС имеют для коллоидной химии такое же важное значение, как строение адсорбционных слоев ПАВ на поверхности раздела различных фаз.
9.1. Поверхностный заряд
Существует несколько механизмов образования электрического заряда на поверхности твердых тел и жидкостей в дисперсных системах.
I
-Si-OH
I
О
I
-Si-OH -------
I
О
I
-Si-OH
I
a
-Si-O’ H4
I
О
-Si-O’ ТГ
I
О
—Si-О" H +
Рис. 9.1. Образование поверхностного электрического заряда по механизму поверхностной диссоциации (а) и механизму адсорбции потенциал-опрсделяющих ионов (б)
I. Катодная или анодная поляризация поверхности электропроводного материала (металла) на границе с раствором электролита. Этот процесс характерен для электрокапиллярного понижения поверхностного натяжения (см. подразд. 9.7).
2. Механохимическая активация. При диспергировании твердых тел и жидкостей на свежеобразованной поверхности могут возникнуть электрические заряды из-за эмиссии электронов, образования свободных радикалов и некоторых других факторов.
3. Столкновение частице заряженными ионами. Этот механизм действует в аэрозолях. С ним связано появление зарядов на каплях воды в облаках.
4. Поверхностная диссоциация. Этот механизм характерен, например, для силикатных частиц в воде (рис. 9.1, а). При взаимодействии поверхности SiO2 с водой образуется кремни» вая кислота, которая далее диссоциирует: H2SiO3 <=> 2Н++ SiOj . Протоны переходят в воду, анионы S О? остаются на поверхности твердых частиц, сообщая им отрицательный заряд; поверхность SiO2 заряжается отри цател ьн о.
5. Ионная адсорбция. Этот процесс происходит, например, в коллоидном растворе (золе) Agl. Его получают обменной реакцией, смешивая водные растворы AgNO3 и KI при избытке одного из реагентов (см. подразд. 9.3). При избытке KI на поверхности кристаллов Agl адсорбируются анионы 1_, так как они входят в состав кристаллов Agl. В результате адсорбции ионов 1 (правило Фаянса достройки кристаллов) поверхность твердых частиц приобретает отрицательный заряд (рис. 9.1, б). Если же золь Agl был получен при избытке AgNO3, то на твердой поверхности будут адсорбироваться катионы Ag+, так как именно они (а нс катионы К+) достраивают кристаллическую решетку Agl. В этом случае поверхностный заряд будет положительным.
9.2. Строение двойного электрического слоя
Рассмотрим строение двойного электрического слоя (ДЭС) на конкретном примере — золе Agl с отрицательно заряженной поверхностью (рис. 9.2).
На поверхности кристалла Agl возле положительно заряженных ионов Ag+адсорбируется слой анионов I. Знак заряда адсорбированных ионов определяет знак заряда поверхности частиц (в данном случае — отрицательный). Эти ионы называю! потен-циалопределяющими. Они формируют первый слой ДЭС, который находится непосредственно у твердой поверхности (слой I на рис. 9.2).
Плотность поверхностного заряда (pv, Кл/м2) определяется количеством потенциалопределяющих ионов на единице площади п, = r,NA (Г, — удельная адсорбция этих ионов, моль/м2; NA — число Аво1адро) и их валентностью (<):
рл. = zent= zFr^ (9.1)
где е — заряд электрона; Е= 96 485 Кл/моль — число Фарадея.
Потснциалопредсляющис ионы I- входят в состав твердой частицы и образуют ее наружный слой I (см. рис. 9.2). За счет кулоновского взаимодействия они притягивают к себе из раствора электролита (KI) ионы другого знака — катионы К+. Эти ионы называ-
Рис. 9.2. Схема строения двойного электрического слоя:
I — ногенцшпопрсдсляюшис ионы: II — ближайший слой противоииов
юг противоионами. Они находятся в жидкой дисперсионной среде и образуют второй слой ионов в ДЭС.
Часть противоионов располагается па расстоянии их диаметра (d) от слоя потен циалопрсделяющих ионов. Эти противоионы образуют плоскость (слой) II наибольшего приближения. Другая часть противоионов находится на более удаленном расстоянии а> d. Распределение противоионов определяют два конкурирующих процесса:
1) электростатическое притяжение к потенциалопрсдсляюшим ионам;
2) диффузия из области высокой концентрации противоионов (плоскости наибольшего приближения) в глубь раствора, где концентрация противоионов равна концентрации электролита в растворе (С+°); поскольку с увеличением расстояния х притяжение катионов к слою I потенциалопределяющих ионов убывает, концентрация противоионов С (а) постепенно уменьшаемся от максимального значения при х= t/до С? (см. рис. 9.2); эта часть ДЭС 11азы вас i ся диффузной.
Наряду с противоионами в диффузной части ДЭС находятся ионы одного знака с нотенциалопределяюшими ионами, их называют коионами (в данном примере — анионы Г). Вследствие электростатическою отталкивания от слоя I потенциалопределяющих ионов I па поверхности концентрация коионов (С_) вблизи поверхности частицы меньше, чем в глубине раствора (С°).
Таким образом, распределение ионов в диффузной части ДЭС соответствует динамическому равновесию адсорбционного и десорбционного процессов на поверхности раздела фаз — аналогично динамическому равновесию при адсорбции ПАВ па поверхности раствора (см. подразд. 6.3).
Двойной электрический слой по составу и строению резко отличается от жидкой фазы (раствора К1). Как и адсорбционный слои ПАВ, ДЭС представляет собой самоорганизованную упорядоченную структуру — потенциал определяющие ионы и противоионы в плоскости наибольшего приближения II располагаются в определенном порядке. Эта способность к самоорганизации имеет очень важное значение дня образования и функционирования биологических структур (клеток, мембран и др.).
9.3. Электрический потенциал и плотность заряда в двойном электрическом слое
При наличии электрических зарядов термодинамическим параметром поверхности раздела фаз служит электрохимический потенциал р:
Й = р,- + <F<p, (9.2)
где ц, — химический потенциал данного иона; z/чр — электрическая компонента электрохимического потенциала, т.е. работа переноса I моля ионов с зарядом гГ(Кл/моль) в точку электрического поля с потенциалом ср (В).
Из-за наличия диффузной части электрический потенциал ДЭС (ф) зависит от расстояния (л) от поверхности раздела фаз (рис. 9 3). Максимальный межфазный скачок потенциала Дф = ф0 определяется равенством электрохимических потенциалов соприкасающихся фаз (твердого тела и раствора). Из уравнения (9.2) следует
Дц
Дф = -~, (9.3)
где Дц, = цм -ц,7: Дф = ф$ -ф/ = ф0; ЦаДй/ — химический потенциал /-го иона в твердой и жидкой фазе соответственно; ф„ ф, — электрический потенциал твердой и жидкой фазы соответственно.
При выполнении условия (9 3) достигается электрохимическое равновесие на поверхности раздела фаз, и переход ионов из одной фазы в другую прекращается. По мере увеличения расстояния х от поверхности электрический потенциал уменьшается от максимального значения ф() на границе раздела фаз. В глубине раствора, где концентрации всех ионов достигают своего значения Со в объеме раствора, потенциал ф = 0.
В нескольких разделах коллоидной химии (устойчивость, электрохимические явления) важную роль играет зависимость потенциала ф от расстояния х. Расчет этой зависимости ф = f(x) основан на нескольких положениях.
Рис. 9.3. Зависимость электрического потенциала (ф) от расстояния (х) от поверхности раздела фаз; пунктиром показана зависимость ф(х) для предельно сжатого слоя противоионов (модель Гельмгольца); d — толщина предельно сжатого ДЭС; Д — толщина ДЭС на уровне гидродинамической границы скольжения; 8 — толщина ДЭС
1. Условие электро!нейтральности ДЭС — поверхностный заряд ри создаваемый потен циалопрсделя юти ми ионами, численно равен и противоположен по знаку суммарному заряду противоионов в ДЭС. Плотность заряда противоионов в ДЭС (рк, Кл/м ) зависит от расстояния лот заряженной поверхности, поэтому
Р» =-Jpi'd*. (9.4)
О
В уравнении (9.4) знак «минус» учитывает противоположные знаки заряда потенциалопрсделяюших ионов и противоионов.
2. Распределение противоионов и коионов в диффузной части ДЭС определяется статистическим законом Больцмана, т.е. отношением электрохимической составляющей электрохимического потенциала <Ар(х) к тепловой энергии ионов RT (R — универсальная газовая постоянная; Т — температура; ф(х) — электрический потенциал на расстоянии х). В этом случае распределение противоионов С_(х) и коионов С+(х) в диффузной части ДЭС описывается экспоненциальной функцией:
С (х) = СР схр[-г/г<р(л)/(Л7’)]. 5)
СДл) = С+° ехр[+гГ<р(.г)/(/?7')].
Здесь рассматривается самый простой случай, когда потенциал-определяюшие ионы и противоионы имеют равную валентность.
3. Плотность обьемного заряда противоионов на расстоянии л в диффузной части ДЭС равна разности между суммарными зарядами противоионов и коионов на этом расстоянии:
рг(х) = zFC (х) - z,FC+(x).
(9.6)
4. Электрический потенциал ф(х) и плотность заряда рДл) связаны уравнением Пуассона:
V ф = -рг/(ее0), (9.7)
где V2 — оператор Лапласа; е — диэлектрическая проницаемость жидкости (дисперсионной среды); е()= 8,854 10~,2Ф/м — диэлек-грическая проницаемость вакуума.
Для плоской поверхности раздела фаз потенциал ф меняется только в одном (перпендикулярном к ней) направлении х. Поэтому уравнение Пуассона принимает вид
d Ф = рг (х)
dx Е£о
(9.8)
Таким образом, задача сводится к решению уравнения (9.8).
Будем далее рассматривать наиболее простой случай — раствор бинарною симметричного электролита. Для него объемные концентрации погенциалонределяющих ионов и противоионов рав
ны: С? = С?. Валентности противоионов и коионов также равны: ZT= <_ = < Этим двум условиям удовлетворяет раствор К1 в рассмотренном выше примере получения дисперсных частиц А^1 в коллоидном растворе.
При указанных упрощениях с учетом соотношений (9.5) и (9.6) уравнение (9.S) принимает вид
(9.9)
Далее следует определить граничные условия. Одно из них относится к границе диффузной части ДЭС с внутренним объемом раствора При х-э <*> (за пределами ДЭС) концентрации всех ионов равны, т е. С? = С? = Сп. Следовательно, при х—»<*> потенциал ср = О Кроме того, при X —> оо производная dtp/dx = 0.
Другое граничное условие относится к плоскости наибольшего приближения х = г/, здесь потенциал ф = ср, (<р, < <р0), производная dcp/cLx = pj/(c£0) (pj — плотность заряда в диффузном слое на расстоянии d}. После первого интегрирования уравнения (9 9) получаем
^ = -[8/<7C0/(K(1)rshM (9.10)
CLV ZK1
где sh — символ гиперболическою синуса; знак «минус» подчерки васI, что при положительном потенциале поверхности <р0> 0 про и з вол ная d(p/dx < 0.
Использование граничных условий для плоскости максимального приближения дает выражение для плотности заряда
Р" =£E,,(S1 * (9.11)
Интегрирование уравнения (9.11) даст искомую зависимость (р(л).
th — = th expГ-к(х - d)l (9 12)
4RT 4RT PL v 71 17
где th — символ гиперболического тангенса; параметр 1 /к = 5 — толщина ионной атмосферы; физико-химический смысл этого параметра обсуждается далее (случай слабозаряженной поверхности). Из приведенных выше соотношений получим
(9.13)
Д 1я разбавленных расiворов электролитов толщина ионной атмосферы 5 » d (d — диаметр ионов). Поэтому (л - d) ~ г/, а потенциал ср, = (р0. Тогда уравнение (9.13) принимает вид
th^₽w=th^exp(_KX). (9 14) 4RT 4RT
Для плотности заряда диффузного слоя при толщине 8 из уравнения (9.11) следует
p6=-(8/?ree0C0),/2sh|^. (9.15)
Уравнения (9.13)—(9.15) представляют теоретическую основу для описания электроповерхностных и других коллоидно-химических явлений, связанных с влиянием электрического поля дисперсных частиц. Важнейшее из этих явлений — устойчивость лиофобных дисперсных систем (см. гл. 10).
Уравнение (9.14) описывает зависимость электрического потенциала ф от расстояния л от поверхности раздела фаз (твердая частица—раствор электролита) (см. рис. 9.3). Наиболее важны потенциалы в следующих точках:
I) при х = 0 имеем ф = ф0, т.е. потенциал в этой точке соответствует скачку потенциала на поверхности раздела фаз;
2) при х = d имеем ф = фн эю потенциал плоскости макси-мал ьного приближения.
Расстояние х = А соотвстст вует положению границы скольжения при движении дисперсной частицы в дисперсионной среде, например, под действием электрического поля (см. подразд. 9.5). Вследствие адгезии к поверхности твердой частицы прилипает слой жидкости, толщина (А) которого превышает молекулярный (ионный) размер (<7); этот слои называют пограничным. Электрический потенциал (Q, который соответствует границе скольжения, на-зывают электрокинетическим потенциалом (^-потенциалом).
Расстояние х = 8, определяемое уравнением (9.13), называют, как отмечалось выше, толщиной ионной атмосферы, эффективной толщиной двойного электрического слоя.
Расстояние х, на котором ф = 0, — это граница, отделяющая диффузную часть ДЭС от раствора электролита, в котором концентрации ионов уже нс зависят от влияния заряженной поверхности.
Из уравнения (9.13) следует, что толщина ионной атмосферы сильно зависит от концентрации ионов в растворе (Со). Рассмотрим два случая.
1. В концентрированном растворе (Со = 0,1 моль/л) при валентности ионов г= I и комнатной температуре (300 К) имеем 8= I нм, т.е. слой противоионов располагается в плоскости максимального приближения. В этом случае монослои потенциалопределяющих ионов и противоионов располагаются параллельно друг другу на минимальном расстоянии 8 ~ d. Такую модель ДЭС называют моделью Гельмгольца (моделью плоскопараллельного конденсатора). В модели Гельмгольца диффузный слой противоионов сжат до
предельно возможною расстояния между ним и слоем погснциа-лопределяюших ионов. Зависимость электрического потенциала Ф(х) характеризует пунктирная прямая на рис. 9.3.
2. При сильном разбавлении (Со = 10 ' моль/л) имеем 8~ 100 нм. Таким образом, толщина диффузного слоя 5 меняется в очень широком интервале. Увеличение концентрации раствора ведет к сжатию ДЭС (вплоть до ДЭС, отвечающего модели Гельмгольца). Разбавление раствора вызывает увеличение толщины диффузного слоя.
Для многих проблем коллоидной химии (агрегативная устойчивость дисперсных систем, электрохимические явления) основное значение имеет электрический потенциал <р(х) на сравнительно большом расстоянии от твердой поверхности х > d. В этой области потенциал может быть достаточно мал, тогда энергия электростатического притяжения противоионов становится меньше их тепловой энергии: гАр(х) « ART В этом случае с достаточной точностью возможно упрощение:
th[?F(pU)/(4/?n] = гГ<р(х)/(4/?Г).
Тогда уравнение (9.15) принимает вид
. ART <Г<р0 z .
ф(х) = th ехр(-кк). (9.16)
Интерес представляют два предельных случая.
I. Малый потенциал <р0 поверхности: zЛр0« ART. Тогда из уравнения (9.16) следует экспоненциальная зависимость:
<р(х) = фоехр(-кх). (9.17)
Из уравнения (9.17) следует, что расстояние 8 = 1/к соответствует уменьшению потенциала ф0 в е (е ~ 2,71828...) раз.
Для плотности заряда из уравнения (9.15) получаем для этого случая упрошенное соотношение
рб = ееофо/6.
(9 18)
Эго уравнение связываш плотность заряда рй и потенциал ф0 для конденсатора, пластины которою находятся на расстоянии 5 друг от друга. Таким образом, при малом потенциале поверхности диффузную часть ДЭС можно заменить плоским слоем («пластиной конденсатора») на расстоянии б, которое называют приведенной, или эффективной, толщиной ДЭС.
2. Большой потенциал ф0 поверхности; гГфо » ART. При этом условии th[£/4p0/(4/?T)| = 1. Соответственно из уравнения (9.16) получаем
Ф(х) =
ART
ехр(-кх).
(9.19)
т.е потенциал поверхности ср0 не влияет на значение <р(х) в диффузной части ДЭС. Причина заключается в том, что противоионы в этом случае си ieho экранируют слои потенниалопредсляюших ионов.
9.4. Влияние электролитов на двойной электрический слой
Строение ДЭС и приведенные в подразд. 9.3 уравнения позволяют прогнозировать влияние электролитов на свойства ДЭС. По действию электролиты разделяют на две группы.
Индифферентные электролиты. К этой группе относят электролиты, которые нс содержат ионы, входящие в состав твердой частицы, или изоморфные ионы. Поэтому они не влияют на скачок потенциала на границе раздела фаз и на плотность поверхностного заряда. Такие электролиты оказывают влияние прежде всего на эффективную толщину ДЭС. Если в состав электролита входят противоионы, то добавление электролита вызывает сжатие ДЭС и уменьшение ci о толщины При вводе дополнительных ионов одного знака с противоионами значительную роль играет их адсорбционная способность, которая растет с увеличением размеров и валентности ионов (лиотропные ряды; см. подразд. 6.7). Круп-
Рис 9 4. Изменение потенциала ДЭС в зависимости от расстояния от заряженной поверхности в исходной системе (/) и при перезарядке дисперсных частиц в результате адсорбции многовалентных ионов (2)
ные многовалентные ионы .могут вызывать сжатие ДЭС и, кроме того, проникать в слой потен циалопрсделяющих ионов и изменять знак заряда ^-потенциала (рис. 9.4). Этот эффект называют перезарядкой. Перезарядка вследствие адсорбции многовалентных ионов ведет к изменению знака электрокинстического потенциала, однако скачок потенциала на границе раздела фаз сохраняет свою величину и знак.
Неиндифферентные электролиты. Такие электролиты содержат ионы, которые могу г достраивать кристаллическую решетку твердой фазы. Возможны два варианта:
I) вводимые дополнительно ионы имеют одинаковый знак с потенциалонределяющими ионами, тогда при небольшой концентрации электролита происходит увеличение потенциала ф0; дальнейший рост концентрации приведет к сжатию диффузной части;
2) вводимые дополнительно ионы имеют знак противоионов, тогда они вначале вызывают сжатие ДЭС, а при достаточно большой концентрации приведут к перезарядке поверхности.
Таким образом, подбирая соответствующие электролиты, можно весьма эффективно регулировать электроповерхностные свойства дисперсных частиц. Этот метод широко используют в процессах, связанных с устойчивостью коллоидных систем (например, при коагуляции золей электролитами) и электрокинетическими явлениями (см. подразд. 9.6).
9.5. Электростатическая составляющая расклинивающего давления
Диффузное распределение противоионов приводит к возникновению электростатического отталкивания при сближении дисперсных частиц. Поэтому ДЭС играет определяющую роль в устойчивости коллоидных растворов и предотвращении их коагуляции (см. подразд. 10.2). Для развит ия количественной теории устойчивости дисперсных систем необходимо установит ь связь между толщиной пленки раствора электролита, разделяющей дисперсные частицы, и свойствами ДЭС в этой прослойке.
Рассмотрим в связи с этим следующий случай (рис. 9.5). Две частицы расположены гак, что их грани, обращенные друг к другу, образуют плоскопараллельный зазор толщиной h < 2d (б — толщина ионной атмосферы; см подразд. 9.2, 9.3). В разбавленных растворах электролитов толщина 6 достигает 0,1 — 1 мкм, т.е. опа соизмерима с размером дисперсных частиц t/или превосходит сю. При условии h < 26 диффузные слои противоионов, сформированные возле каждой частицы, неизбежно перекроют друг друга. Поэтому концентрация противоионов в такой пленке электролита будет отличаться от случая одной частицы (см. рис. 9.2). В резуль-
Рис. 9.5. Распределение потенциала (р в тонкой пленке раствора электролита толщиной Л
тате меняются обе характеристики диффузного слоя: зависимость потенциала ф от расстояния х от поверхности частицы и зависимое гь плотности электрического заряда рДх).
Особый интерес для расчета электростатического отталкивания частиц представляют значения потенциала ф и плотности заряда рг в середине пленки, т.е. при х = Л/2. Для расчета этих величин используют принцип суперпозиции. В соответствии с ним потенциал ф(У2) в середине пленки равен сумме потенциалов Ф|(У>) (ф| ~ потенциал для ДЭС вокруг отдельной частицы, который определяется уравнением (9.14)). Аналогично рассчитывают плотность объемного заряда р>(Л/2). Таким образом, из принципа суперпозиции следуют соотношения:
Ф(У>) = 2ф,(У2); р,ХУ2) = 2Рп(У2).
Наиболее важная особенность строения ДЭС тонкой пленки заключается в том, что потенциал ср в ней не падает до нуля, как в случае ДЭС около отдельной частицы (см. рис. 9.2).
Для расчета потенциала ф|(У2) используем уравнение (9.16), которое относится к случаю больших расстояний от твердой поверхности. В данном случае уравнение (9.16) принимает вид
Ч>(%) = 2<р, ('/) = ~ th |р^ехр(-кЛ/2). (9.20)
Z.F
Для плотности заряда уравнение (9.15) преобразуется к виду
рЛ,У) = 2ри(%) = -2
(<:с)2С()ф(%)
RT
(9.21)
Произведение
определяет в соответствии с законами электростатики плотность энергии электростатического отталкивания (У, Дж/м3) в середине зазора между дисперсными частицами.
По физико-химическому смыслу энергия Ue представляет собой плотность дополнительной энергии тонкой пленки, обусловленной перекрыванием поверхностных слоев, в данном случае — диффузных слоев противоионов. Таким образом, плотность энер-гии Ue, которая зависит от толщины пленки /?, создает в пленке дополнительное давление — расклинивающее давление (П ) В отличие от молекулярной составляющей расклинивающего давления Г1,„< 0 (см. подразд. 4.3) электростатическая составляющая положительна Пе> 0. Физический смысл положительного знака Пе заключается в том, что электростатическая составляющая препятствует сближению дисперсных частиц. В соответствии с термодинамическим определением расклинивающего давления (см. уравнение (4.19)):
где Ue — электростатическая составляющая избыточной поверхностной энергии пленки.
Отсюда следует зависимость энергии U€, обусловленной электростатической составляющей расклинивающего давления, от толщины пленки /?:
64С0/?Г Г zeepo
схр(-кЛ).
(9.22)
Представление об электростатической составляющей расклинивающего давления в гонких пленках относится к числу основных положений теории поверхностных сил и теории устойчивости коллоидных систем (см. подразд. 10.2).
9.6. Электрокинетические явления
Наличие электрического заряда на поверхности дисперсных частиц приводит к нескольким следствиям. Одно из них заключается в том, что при помещении дисперсной системы в электрическое поле заряженные частицы начинают двигаться в направлении вектора электрической напряженности; такое движение называют электрофорезом. Другое следствие — течение жидкой дисперсионной среды через пористые материалы под действием внешнего электрического ноля; такое течение называют электроосмосом.
Электрофорез и электроосмос относят к электрокинетическим явлениям. В число электрокинстических я в пений входя г также обратные эффекты — возникновение электрического заряда па дисперсных частицах при их движении в неподвижной дисперсионной среде (жидкости или газе) и возникновение электрического
заряда жидкости при ее течении через пористую среду или мембрану. Эти два эффекта называют потенциалом оседания (или потенциалом седиментации) и потенциалом течения соответственно.
Рассмотрим основные закономерности перечисленных элект-рокинстических явлений.
Электрофорез. Эго направленное движение заряженных дисперсных частиц коллоидного раствора в постоянном электрическом поле. В зависимости от знака заряда частицы перемешаются к катоду или к аноду.
Электрофорез открыл Ф.Ф. Рейсс в 1808 г. Опыт был поставлен следующим образом. Во влажную глину погружали вертикально две стеклянные трубки с водой. В них вводили электроды. После подачи на электроды напряжения вода в грубке с катодом начинала мутнеть; граница мутности постепенно перемешалась вверх. Причина помутнения — мелкие частицы глины под действием электрического поля отрывались от основной массы и перемешались к положительному электроду.
По перемещению границы области помутнения можно определить скорость движения (г) дисперсных частиц (метод подвижной границы). Для бесцветных золей положение границы определяют с помощью ультрафиолетового освещения или некоторых других методов.
Основная теоретическая задача заключается в расчете скорости электрофореза (г) в зависимости от напряжения (U) на электродах.
Рассмотрим коллоидный раствор (золь) с млчои концентрацией частиц (v, м-2). При этом условии все частицы будут двигаться независимо друг от друга.
Введем два допущения, касающиеся дисперсных частип:
I) размер частиц (d) должен быть мал по сравнению с толщиной ДЭС (6): d < 6; это условие выполняется при малой концентрации противоионов С° (см. подразд. 9.2).
2) материал частицы не обладает электропроводностью; в этом случае заряд, создаваемый потенниалопределяюшими ионами, является только поверхностным.
На каждую дисперсную частицу действуют две силы. Одна сила f,создастся электрическим полем и вызывает движение заряженной частицы к противоположно заряженному электроду, другая сила Д/—это сопротивление жидкости, обусловленное ее вязкостью (т), Па-с).
В соответствии с законом электростатики движущая сила равна Л = Pi л
где Е — напряженность электрического поля (/?= U//.). В/м; L — расстояние между электродами, м; ps — плотность поверхностного заряда, Кл/м2.
Величина р зависит от электрического потенциала <р(х). Для модели ДЭС по Гельмгольцу (плоский конденсатор) имеем
р5 = -Е£оф/Х,
где х — расстояние между пластинами конденсатора.
Принципиален поэтому вопрос: на каком расстоянии хот поверхности частицы расположена граница скольжения. Граница скольжения устанавливается при перемещении твердой частицы в жидкости на некоторое расстояние А > d от ее поверхности (d — положение плоскости максимального приближения противоионов).
В интервале d < х < А жидкость вовлекается в течение при перемещении твердой частицы. На расстоянии х = d скорость этого течения такая же, как у частицы (v); при х> А скорость равна нулю (жидкость неподвижна).
Граница скольжения проходиi внутри диффузной части ДЭС. Потенциал который соответствует границе скольжения, называют жктрокинетическим, или потенциалом (см. рис. 9.4). Поэтому при расчете силы 4, действующей со стороны электрическою поля на дисперсную частицу, принимают, что плотность заряда pv соответствует электрокинетичсскому потенциалу £ и положению плоскости скольжения А; соответственно ps.= -E£(£/A. Тогда сила электрического притяжения (f,) равна
A
Сила вязкого сопротивления (/],) направлена противоположно силе электрического притяжения (f>) и определяется законом Ньютона:
r dr
/к =п —, dx
где dr/dx — градиент скорости течения в направлении х, перпендикулярном направлению течения.
Для тонкого слоя имеем dr/dx~ r/д, поэтому сила вязкого сопротивления жидкости равна
fi =П-7- (9-24)
А
При стационарном режиме электрофореза, т.е. при постоянной скорости движения частиц, силы равны: fe =fv. Тогда из уравнений (9.20) и (9.21) получаем
П
(9-25)
Отношение v/E- г’п называют электрофоретической подвижностью'.
t’0=E€0-- (9 26)
И
Из уравнений (9.22) и (9.23) следует, что скорость электрофореза не зависит от размера дисперсных частиц. Это положение справедливо для электрофореза частиц размером примерно до 100 мкм. Для наночастиц элсктрокинетичсский потенциал зависит от их размера. Поэтому уравнение (9.25) для скорости электрофореза грсбует корректировки, учитывающей этот масштабный эффек г.
Измерение скорости электрофореза представляет один из основных методов экспериментального определения электрокине-тического потенциала, его расчет проводят по уравнению (9 22).
Электрофорез широко используют в научных целях, во многих технологических процессах: нанесение полимерных покрытий на металлы, производство дисплеев и др. Одно из основных применений электрофореза — разделение и анализ белков, аминокислот, антибиотиков, ферментов За разработку этого метода шведский биохимик А.Тизелиус быи награжден Нобелевской премией по химии (1948). Электрофореграммы плазмы крови используют для диагностики различных заболеваний.
Электроосмос. Это направленное течение раствора электролита в капиллярах и пористых телах (в том числе и в фильтрах и мембранах) под действием внешнего электрическою поля. Электроосмос также открыл Ф Ф. Рейсс (1808): U-образную стеклянную трубку заполняли водой, в ее нижнюю часть помешали кварцевый песок В воду опускали положительный и отрицательный электроды. После подачи напряжения вода поднималась к катоду. При напряжении 1 200 В этот подъем достигал 20 см.
Причина электроосмоса заключается в следующем Слой кварцевого песка представляет собой пористую дисперсную систему. На границе кварцевых частиц с водой возникает ДЭС и поверхность частии приобретает электрический заряд. Такой же заряд, но противоположного знака, приобретает диффузная часть ДЭС, находящаяся в жидкости Внешнее электрическое поле создает силу, определяемую уравнением (9.23).
В отличие от электрофореза внешнее поле вызывает течение жидкости. Твердая фаза (частицы песка) остаются неподвижными. Силой сопротивления по-прежнему остается вязкое трение. Существенно, что в течение вовлекается только тонкий слой жидкости, соответствующий плоскости скольжения, т.е. па расстоянии А от поверхности дисперсных частиц. Поэтому сила вязкого трения определяется уравнением (9.24). Поскольку обе действующие силы /1 и/, определяются теми же уравнениями, что и при
электрофорезе, скорость течения жидкости при электроосмосе определяется уравнением (9.25). Для того чтобы вода перемешалась со скоростью v = 1 см/с при потенциале cpj = 100 мВ, необходимо приложить поле напряженностью Е- I 500 В/см.
Однако при электроосмосе из-за сложной структуры пористо-го пространства возникают большие трудности при расчете напряженное! и элсктрическою поля Е= U/L\ так как расстояние L может сильно отличаться от кратчайшего расстояния между электродами. Поэтому уравнение (9.25) преобразуется к форме
(9.27)
где / — сила тока (/ = Uk), А; к — удельная электропроводность жидкости.
Уравнение (9.27) используют при определении электрокине-тических потенциалов по скорости электроосмоса. Основные приложения электроосмоса связаны с осушкой различных пористых сред и материалов.
Потенциал течения (потенциал протекания). Это электрокапил-лярное явление, обратное электроосмосу. Оно заключается в том, что при течении воды через пористую среду (фильтр, мембрану) на ее противоположных сторонах возникает разность электрических потенциалов. Такой же эффект наблюдается при течении по стеклянному капилляру.
Возникновение потенциала течения, как и другие электрока-пиллярные явления, связано со строением ДЭС. а именно — с наличием диффузного слоя противоионов.
Рассмотрим механизм этою явления для наиболее простого случая — течение воды по стеклянному или кварцевому капилляру радиусом г под действием разности давлений Др = р{- р2 на концах трубки. Пусть характер течения будет ламинарным. Условие ламинарпости имеет вид
Re = ^ < 2 300, Ч
где Re — число Рейнольдса; v — средняя скорость течения, р, ц — соответственно плотность и вязкость жидкости; d=2r — диаметр трубки.
При ламинарном течении локальные скорости v(x) постепенно возрастают от v=0 при х:= Д (расстояние от стенки до плоскости скольжения) до максимального значения вдоль оси трубки.
При контакте стеклянной трубки с водой около нее формируется ДЭС (по механизму поверхностной диссоциации; см. подразд. 9.1) с электрокинетическим потенциалом и приведенной толщиной 5; причем б « г. Диффушая часть ДЭС (Д < х < 8),
двигаясь вместе с потоком жидкости, переносит электрический заряд — избыточное число противоионов по сравнению со средней частью потока. Таким образом, в пределах диффузного слоя в направлении потока протекает электрический ток (/5,); его называют током течения. или конвективным током.
Избыточный заряд, создаваемый противоионами, накапливается на выходе из трубки. В результате между концами капилляра возникает разность потенциалов (U). Под действием этого напряжения в трубке возникает противоположно направленный электрический ток (/ ). При динамическом равновесии оба тока равны lsf = /с. Из этого условия рассчитывают потенциал течения U. Для этого используют уравнение для электрокинетического потенциала С при электроосмосе и электрофорезе. Кроме того, используют уравнение Пуазейля, которое описывает объемную скорость течения (г, м3/с) жидкости вязкостью т| по трубке радиусом г и длиной / под действием перепала давления Д/г
v = яг4Др/(8т)/). (9.28)
При этих условиях уравнение, описывающее потенциал течения, имеет вид
и=-^г- (9-29>
где к — удельная электропроводность жидкости.
Потенциал течения необходимо учитывать, например, при течении нефти, бензина и других топлив по трубопроводам. Для неполярных жидкостей, удельная электропроводность которых на мною порядков меньше, чем водных растворов, напряжение достигает нескольких тысяч киловольт. Такое высокое напряжение оказывается причиной взрывов и пожаров на нефтеперерабатывающих заводах и танкерах. Эго требует осторожности при заливке топлива в самолеты.
Данное явление также следует принимать во внимание при разработке медицинской аппаратуры — возможно возникновение биопоте! титиов при течении крови.
Потенциал оседания (седиментационный потенциал). Этот потенциал возникает при оседании частиц суспензии под действием силы тяжести. При наличии ДЭС вместе с част ицей перемещаются противоионы, которые находятся около нес на расстоянии а < Д (Д — положение границы скольжения). В результате при накапливании на дне сосуда осадка создается электрический заряд одного знака с противоионами. При этом между верхним и нижним слоями дисперсионной жидкой среды возникает разность напряжений (Z/), которая в свою очередь создаст встречный электрический ток, направленный снизу вверх (по аналогии с возникновением потенциала течения). Динамическому равновесию соот
ветствует равенство двух токов: седиментационного тока и тока в жидкости. Из это 1'0 баланса следует уравнение, определяющее потенциал оседания для монодиспсрспой суспензии со сферическими частицами радиусом г:
,, (ps -pJgV'A
31*1 к
где р , р/ — плотность вещества дисперсных частиц и жидкости соответственно; g— ускорение свободного падения; v — концентрация частиц, м“3; h — расстояние между электродами, помещенными в суспензию, м; г, — вязкость жидкости; к — удельная электропроводност ь жидкости.
Потенциал оседания является причиной возникновения зарядов на каплях воды при их движении в атмосфере.
Потенциалы оседания возникают не только в суспензиях, но и в эмульсиях. В этом случае они могут создавать большую пожароопасность.
9.7. Электрокапиллярные явления
Наличие электрического заряда на поверхности жидкости или твердого тела ведет к еще одному важному следствию. Поверхностное натяжение зависит в этом случае не только от химической природы граничащих фаз, но и от электрических свойств: плотности поверхностного заряда (ps, Кл/м2) и скачка электрического потенциала (ф, В) на поверхности раздела фаз. Зависимость и = /(ср) называют электрокапиллярной кривой. Поверхностные явления. которые обусловлены этой зависимостью, называют элек-трокапиллярными. Их роль в коллоидной химии весьма велика. Электрокапиллярность позволяет регулировать поверхностное натяжение жидкостей и твердых тел. Благодаря электрокапиллярным явлениям можно эффективно влиять па многие коллоидно-химические процессы: смачивание, адгезию, диспергирование и др.
Рассмотрим причины элсктрокапиллярной зависимости поверхностного натяжения. На качественном уровне зависимость о =/(ф) объясняют следующим образом Термодинамика определяет поверхностное натяжение как работу изотермического процесса образования единицы поверхности (см. подразд. 4.1). Если на данной поверхности находятся одноименные электрические заряды, то они будут отталкиваться друг от друга. Силы электростатического (кулоновского) отталкивания направлены тангенциально поверхности и стремятся увеличить се площадь. Поэтому работа, которая необходима для увеличения заряженной поверхности, будет меньше, чем для электронейтралыюй поверхности.
Элсктрокапиллярная зависимость проявляется достаточно явно для жидких металлов. Для металлов с низкой тем пера турой плавления (ртуть, галлий) в качестве граничной фазы используют водные растворы различных электролитов. Для других металлов электрокапиллярпые явления реализуются на границе с расплавами солеи и оксидов.
В качестве примера на рис. 9.6 представлена электрокапиллярная кривая ртути в водных растворах электролитов при комнатной температуре. В соответствии с приведенной выше причиной электрокапиллярности максимальное значение поверхностного натяжения соответствует электронейтралыюй поверхности ртути, т.е. плотность поверхностного заряда рл. = 0. Поэтому потенциал. при котором поверхностное натяжение электрода максимально, называют потенциалом нулевого заряда (ППЗ, Фпнз)- Значение потенциала нулевого заряда зависит от природы жидкого электрода (металла) и состава раствора. Левую часть элсктрокапиллярной кривой (при потенциалах ф < Фпнз) называют анодной ветвью, правую часть этой кривой (ф > фпнз) — катодной ветвью. Сравнительно небольшие изменения электрического потенциала ф (на десятые доли вольта) могут приводить к большим изменениям поверхностного натяжения. Для ртути эти изменения составляют - К)1 мДж/м2. Еще сильнее меняются поверхностные натяжения жидких металлов на границе с расплавленными солями. Например, поверхностное натяжение олова в контакте с жидкой эвтектикой (КС1 + LiCl) при изменении потенциала ф уменьшается почти в 2 раза.
Приведем теоретический вывод электрокапиллярпои кривой о =/(ф). Используем для этого положения термодинамики поверхностных явлений (см. гл. 4). Изменение поверхностною натяже-ния (do) связано с удельной адсорбцией (Г,) различных компонентов и их химическими потенциалами (р;) уравнением Гиббса
к
<5о = -Х(Г,ф,), (9.31)
/=|
где к — число компонентов в двухфазной системе.
Рис. 9 6. Электрокапиллярная кривая ртути в водном растворе электролита
При выводе уравнения элсктрокапиллярнои кривой компонентами являются заряженные частицы (ионы, электроны). Поэтому условие термодинамического равновесия на межфазной поверхности заключается в равенстве электрохимических потенциалов (Ц,) компонента в граничных фазах А и В: у/А = р(В. Электрохимический потенциал связан с химическим потенциалом (у,) уравнением
И, =И/
где г,— валентность иона; F — число Фарадея; ф — потенциал.
Плотность поверхностных зарядов psA и psB со стороны каждой фазы (т.е. на пластинах конденсатора в модели ДЭС Гельмгольца; см. подразд 9.2) можно выразить через удельные адсорбции компонентов (Г,):
А , А
P.sA — ^/А 1 PsB ~ /Г, £/В-^Св
/-1 /=!
Из условия элсктронейтральности имеем
Р sA = “PsB •
Адсорбционное уравнение Гиббса (4.15) для заряженной поверхности принимает вид
А А
do = rZAdp,A + ГвФ/в- (9.32)
/=1 /=1
Замена химического потенциала (у,) на электрохимический потенциал (у,) вызвана наличием у частиц электрических зарядов.
Преобразуем уравнение (9.32), заменив электрохимические ц, потенциалы на химические у,. Учтем также, что разность потенциалов <рА - = ф. т.е. равна скачку потенциала на межфазной
поверхности. В итоге получим общее уравнение элсктрокапилляр-ной кривой {уравнение Фрумкина, 1923):
А А
= “ХСаФ Л -Ег/вф/в — pAdcp. (9-33)
/=1 z-l
Если изменение потенциала ф не сопровождается ионным обменом между электродом и электролитом, химические потенциалы компонентов не меняются. Такой электрод называют идеально поляризуемым. При условии dy/A = 0 и dy;B =0 уравнение (9.32) упрощается и принимает следующий вид:
-do = рд!ф. (9.34)
Это соотношение называют уравнением Липпмана (1875).
Из уравнения Фрумкина (9.33) следует, что на положение точки нулевого заряда и форму элсктроканиллярпой кривой могут сильно влиять вещества, способные адсорбироваться на поверхности электрода. Аиионактивныс ПАВ влияют на ход анодной ветви элек-трокапиллярнои кривой, а катионные ПАВ — на ее катодную ветвь.
Электрокапиллярный эффект может оказывать сильное влияние на различные поверхностные явления. Характерным примером служит смачивание. В соответствии с законом Юнга (5.5) краевой угол 0 зависит от поверхностного натяжения смачивающей жидкости. При поляризации электрода и отклонении отточки нулевою заряда поверхностное натяжение смачивающей жидкости уменьшается, что ведет к уменьшению краевых углов. В ряде случаев изменения краевых углов очень велики. Например, при контакте капли толуола с поверхностью ртути в I N растворе сульфата натрия краевой угол меняется от 165° вТНЗ до 40 при анодной или катодной поляризации ртути (рис. 9.7). Напомним, что при избирательном смачивании (см. подразд. 5.2) краевой угол 0 принято определять по отношению к полярной жидкости, в данном случае — к водному раствору. При сильной поляризации электрод (ртуть) полностью очищается от органической жидкости. Таким образом, поляризация металлов вереде раствора электролита представляет эффективный метод очистки поверхности от органических загрязнений.
Большие возможности открывает применение электрокапиллярности для регулирования смачивания твердых металлов жидкими металлами. Поскольку гемпература пдавления большинства жидких металлов достаточно высока (сотни градусов и выше) в качестве электролитов в таких случаях используют расплавы солей, например эвтектики (LiCI + КО) или (КС1 + NaCI). Изменяя электрический потенциал можно обеспечить полное смачивание меди, железа, стали различными жидкими металлами: кадмием,
20° 90 160 90’ 20
Рис. 9.7. Зависимость краевого угла (О) капли толуола па поверхности ртути в I N раст воре Na2SO4 от потенциала ртутного электрода <<р), в верхней части рисунка показаны формы капли при разных значениях потенциала ф и краевого угла 0
оловом, свинцом. Этот эффект успешно используют для нанесения тонких металлических покрытии.
Элсктрокапиллярныс явления происходят не только на поверхности жидкостей, но и на поверхности твердых тел. При поляризации твердых тел их поверхностная энергия может изменяться достаточно сильно. Поверхностная энергия твердых тел влияет на их механические свойства, в гом числе и на твердость и прочность. Поляризация поверхности различных материалов (например, графита, цинка, свинца) в водных растворах электролитов изменяет их поверхностное натяжение, что в свою очередь приводит к изменению их т вердости. При этом максимальная твердость материалов отвечает точке нулевого заряда.
Контрольные вопросы
I. Назовите основные механизмы образования поверхностного электрического заряда.
2. Дайте определение понятия «потен цпалопредсляющие ионы». Приведите пример потенциалопрсделяюпт.х ионов для дисперсной частицы.
3. Дайте определение понятия «противоионы в составе двойного электрического слоя». Какую часть двойного электрического слоя называют диффу зной?
4 Какие условия определяю! зависимость электрического потенциала ог расстояния ю границы раздела фаз?
5. Охарактеризуйте понятие «электрокинетичсский потенциал».
6. Дайте характеристику двум предельным случаям: а) малый потенциал поверхности; б) большой потенциал поверхности.
7. В чем заключается основная причина влияния электролитов настроение двойного электрического слоя? Рассмотрите отдельно влияние индифферентных и неиндифферентны.х электролитов.
8. Объясните причину возникновения электростатической составляющей расклинивающего давления в тонких жидких пленках.
9. Назовите известные элсктрокинстические явления.
10. Какие силы определяют скорость электрофореза?
II. Приведите примеры использования на практике потенциала оседания.
12. Дайте характеристику электрокапиллярный явлениям. Какую величину называют потенциалом нулевого заряда?
13. Напишите уравнение Липпмана для элсктрокапиллярной кривой.
ГЛАВА 10
УСТОЙЧИВОСТЬ и эволюция ДИСПЕРСНЫХ СИСТЕМ
Важнейшее общее свойство всех дисперсных систем заключается в том, что они обладают избыточной свободной энергией. Поэтому в дисперсных системах протекают различные самопроизвольные процессы. В соответствии с законами термодинамики эти процессы направлены так, чтобы приблизить систему к равновесному состоянию.
Самопроизвольные процессы, протекающие в дисперсных системах, вызываются различными причинами и протекают с разной скоростью. Некоторые из них происходят очень медленно. Например, седиментационно-диффузионное равновесие (см гюд-разд. 2.4) в коллоидных растворах с дисперсными частицами размером К) нм уста на вл и вас гея при комнатной температуре через зри года.
Таким образом, многие свойства дисперсных систем не постоянны, они изменяются с течением времени но мере эволюции данной системы.
10.1. Причины и формы неустойчивости дисперсных систем
Рассмотрим наиболее важные в коллоидной химии формы неустойчивости дисперсных систем.
Термодинамическая (агрегативная) неустойчивость. Агрегативная неустойчивость проявляется в постепенном увеличении размеров дисперсных частиц или образовании агрегатов из слипшихся частиц
Эволюцию агрегативно неустойчивой дисперсной системы количественно характеризуют зависимостью размера частиц и их распределения но размерам от времени (/), а также зависимостью от времени концентрации частиц (v). Очевидно, что укрупнение частиц при неизменной общей массе дисперсной фазы (nid) ведет к уменьшению концентрации частиц: dv/dr < 0.
Термодинамической причиной агрегирования служиз избыточная поверхностная энергия (Г) дисперсной фазы. По определению (см. подразд. 4.1, 4.2) избыточная поверхностная энергия — эго избыток свободной энергии (энергии Гельмгольца) на поверх
ности частиц дисперсной фазы по сравнению со свободной энергией в объеме вещества.
Для монодисперсной системы частиц одинакового размера (d) избыточная поверхностная энергия определяется поверхностным натяжением на границе дисперсных частиц с дисперсионной средой и общей площадью поверхности раздела фаз (£2), образующих дисперсную систему (см. гл. 2, 4):
Г„ = а£2^, (10.1)
где Qf/ — удельная поверхность дисперсных частиц; md — масса дисперсной фазы.
Удельная поверхность дисперсной фазы (см. уравнение (2.3)) рассчитывается по формуле
где К— коэффициент (для сферических и кубических частиц К=6)-pd — плотность вещества дисперсной фазы.
Тогда уравнение (10.1) принимает вид
К<зт(1 1
F"=~d' <,0-2>
т с. избыточная энергия /^дисперсной системы обратно пропорциональна размеру дисперсных частиц, так же как удельная поверхность (см. рис. 2.1).
Уравнение (10.2) показывает, что возможны два разных процесса уменьшения поверхностной энергии дисперсной системы.
1. Укрупнение дисперсных частиц, приводящее к увеличению их размера. Этог процесс называют коалесценцией (слиянием). Он характерен для дисперсных систем с жидкими и газовыми дисперсными частицами (эмульсии, туманы, пены). Коалесценция происходит при постоянном поверхностном натяжении (о = const).
2. Уменьшение удельной поверхностной энергии (поверхностного натяжения). Соответствующий процесс называют коагуляцией (слипанием). Он заключается в образовании агрегатов из многих дисперсных частиц, разделенных тонкими пленками дисперсионной среды. Коагуляция характерна для систем с твердыми дисперсными частицами (коллоидные растворы, дымы).
В отличие от коалесценции при коагуляции размер дисперсных частиц со временем не меняется (d = const), однако поверхностное натяжение уменьшается (do/d/<0). Снижение поверхностного натяжения связано с расклинивающим давлением в тонких жидких пленках (см. подразд. 4.3).
Седиментационная неустойчивость. В случае дисперсных систем седиментационная неустойчивость вызывается различием плотно
стей вешеств дисперсной фазы (р(/) и дисперсионной среды (р0). Это различие приводит к постепенному оседанию (седиментации) более тяжелых дисперсных частиц (если pf7> р0) или их всплыванию (если рГ|> рД
Таким образом, седиментация приводит к переходу свободно-дисперсных систем в связнодисперсные; при этом концентрация частиц уменьшается: dv/d/ < 0.
Скорость седиментации возрастает при увеличении размеров дисперсных частиц (см. уравнение (2.9)). Поэтому седиментационная неустойчивость гем выше, чем крупнее частицы. Высокодисперсные системы могут быть седиментационно устойчивыми вследствие достижения динамическою седиментационно-диффузионного равновесия (см. подразд 2.4).
Таким образом, размер дисперсных частиц влияет на агрегативную и седиментационную устойчивость противоположным образом. Чем выше степень дисперсности (меньше размер частиц J), тем сильнее проявляется их агрегативная неустойчивость, однако при этом растет их устойчивость по отношению к седиментации.
Седиментационная неустойчивость дисперсных систем играет очень важную роль во многих природных процессах. Например, процесс оседания частиц в воде и в воздухе очень важен при очистке от вредных примесей.
Фазовая неустойчивость. Во многих случаях структура твердых дисперсных частиц в момент их образования оказывается неравновесной (особенно при использовании конденсационных методов их получения; см. подразд 3.2). В таких системах происходит постепенное изменение структуры частиц при сохранении их размеров. Например, при синтезе коллоидных растворов (золей) металлов, оксидов и гидроксидов дисперсные частицы обычно вначале аморфны.
Далее внутри частиц происходит процесс кристаллизации и через определенный период времени возникает присущий данному веществу тип кристаллической решетки, т.е. достигается термодинамически равновесная структура. Длительность кристалнизании зависит от температуры и химической природы дисперсной фазы. Для частиц золя золота при комнатной температуре кристаллизация продолжается всею несколько минут, для золя гидроксида алюминия — несколько суток, для золя кремниевой кислоты — около двух лет. Повышение температуры резко ускоряет этот процесс.
Поверхностная неустойчивость. Во многих дисперсных системах, особенно при наличии поверхностно-активных вешеств, поверхность раздела дисперсной фазы с дисперсионной средой может находиться исходно в неравновесном состоянии. Поэтому па поверхности будут происходить самопроизвольные процессы.
в результате которых термодинамические параметры поверхности в уравнениях Гиббса для однокомпонентной и многокомпонентной систем (см. подразд. 4.1,6.1) достигну! равновесных значений или приблизятся к ним.
Причины поверхнос! ной неустойчивости различны. Достаточно часю она связана с процессом адсорбции ПАВ, особенно полимерных и белковых ПАВ (см. гл. 6, 7). Такие поверхностно-активные вещества из-за большой молекулярной массы диффундируют из объема дисперсионной среды (раствора) на поверхность дисперсных частиц весьма медленно. Кроме того, для образования адсорбционного слоя ПАВ с равновесной структурой (оптимальной ориентацией макромолекул по оiношению к поверхности и максимально плотной упаковкой слоя) также требуется значительное время.
Другой возможный механизм изменения поверхностных свойств связан с растворением вещества дисперсных частиц в жидкой дисперсионной среде. Это обусловливает несколько процессов. Во-первых, изменение химического состава раствора вблизи поверхности дисперсных частиц, что в свою очередь может вызвать изменение строения двойного электрического слоя (см. гл. 9). Во-вюрых, изменение микрорельефа твердой поверхности Гранины зерен на поверхности поликрпсталлических твердых тел растворяются в данной жидкости сильнее, чем кристаллиты (зерна). Поэтому со временем вдоль межзеренных границ возникают микроканавки. Это ведет к увеличению шероховатости твердой поверхности и можег оказывать существенное влияние на краевые углы смачивания (см. подразд. 5.2).
Анализ причин и форм неустончивости дисперсных систем приводит к следующему принципиальному заключению: неравно-весностъ вызывает эволюцию дисперсных систем. Таким образом, характеристики дисперсных систем могут существенно изменяться во времени.
Способность дисперсных систем к эволюции позволяет сопоставлять их с живыми биологическими системами, использовать для описания и анализа данной эволюции идеи и методы биофизики.
С этих позиции дисперсную фазу следует рассматривать как открытую систему, поскольку она существует в условиях взаимодействия с дисперсионной средой Изменение энтропии такой системы составит = 65,+ dS? (d5;, d5z — изменение энтропии фазы и среды соответственно). Согласно второму закону термодинамики в закрытой системе d.S’> 0, т.е. энтропия дисперсной фазы должна возрастать Однако изменение энтропии дисперсионной среды может быть отрицательным (d.S/ > 0). Такой случай реализуется, например, при самопроизвольном диспергировании (см. подразд. 4.4).
При условии d5= 0 достигается стационарное (но нс термодинамически равновесное) состояние дисперсной системы, когда ее свойства уже не зависят от времени.
Теория эволюционных процессов в неравновесных системах развита сейчас очень глубоко. Один из важных выводов заключается в возможной эволюции систем от хаоса к порядку, т.е. к образованию самоорганизованных структур. Процесс хаос —> порядок реализуется во многих дисперсных системах и приводит к об-ргззованию упорядоченных связнодисперсных структур, например периодических коллоидов (см. подразд. 10.4).
10.2. Коагуляция
Коагуляцией называют самопроизвольный процесс слипания твердых частиц в коллоидных растворах (золях). В результате коагуляции образуются крупные рыхлые агрегаты Они состоят из десятков и сотен исходных дисперсных частиц, разделенных тонкими (наноразмерными) пленками жидкой дисперсионной среды.
Из-за большого размера такие агрегаты седиментационно неустойчивы. Если плотность вещества дисперсной фазы (рЭ больше плотности дисперсионной среды (р0), коагуляционные агрегаты оседают. В противоположном случае (prf< р0) агрегаты постепенно всплывают вверх.
Осадки, образованные агрегатами дисперсных частиц, называются коагулятами. Они могут иметь различную структуру: рыхлую, упорядоченную (так называемые периодические или само-организованные структуры), плотную.
Коагуляция относится к числу основных коллоидно-химических процессов. Она играет определяющую роль в эволюции дисперсных систем, во многих технологических процессах, в формировании почв, грунтов и горных пород, очистке воды.
Формы коагуляции весьма разнообразны.
1. Гомогенная коагуляция (обычно используют сокращенное название «коагуляция») — агрегаты состоят из дисперсных частиц одного вещества.
2. Гетерогенная коагуляция (гетерокоагуляция) — коллоидный раствор содержит дисперсные частицы разных веществ. Элементарный акт гетеро коагуляции заключается в прилипании друг к другу дисперсных частиц разного химического состава.
3. Адагуляционная коагуляция (адагуляция) — прилипание дисперсных частиц к различным твердым поверхностям (к стенкам сосуда, волокнах! фильтра и г. п.).
4. Флокуляция — коагуляционные агрегаты образуются под действием полиэлсктролитов, находящихся в дисперсионной среде. Крупные молекулы или ионы полиэлектролитов, адсорбируясь
противоположными концами на двух разных частицах, «скрепляют» их в один общий агрегат.
В зависимости от прочности контактов между дисперсными частицами различают обратимую и необратимую коагуляцию. При обратимой коагуляции контакты обеспечиваются молекулярными (ван-дер-ваальсовыми) взаимодействиями. Поэтому агрегаты легко могут распадаться па исходные дисперсные частицы под действием тепловых колебаний. Процесс распада (дезагрегирования) коагуляционных агрегатов, приводящий к превращению коагулята в золь, называют пептизацией.
При необратимой коагуляции контакты дисперсных частиц обеспечиваются сильными взаимодействиями (обычно химическими связями). Поэтому образующиеся агрегаты достаточно прочны и устойчивы.
Коагуляция сопровождается резким изменением ряда свойств дисперсной системы. Это позволяет достаточно точно определить условия, при которых начинается коагуляция. Один из характерных признаков коагуляции — выпадение осадка дисперсной фазы. Другой признак — резкое увеличение светорассеяния. Оно характерно именно для коллоидных растворов с высокой степенью дисперсности (см. подразд. 3.6).
Коагуляцию золей вызывают различные воздействия. Их разделяют на две группы: физические и химические. К физическим воздействиям относят перемешивание, механические вибрации, воздействие ультразвука, электрического или магнитного поля, изменение температуры, различные виды излучения (ультрафиолетовое, рентгеновское, радиоактивное). Основным химическим воздействием, вызывающим коагуляцию коллоидных растворов, является растворение в дисперсионной среде определенных электролитов. Характерна высокая чувствительность коагуляции к внешним физическим воздействиям. В ряде случаев небольшие изменения указанных факторов могут вызвать интенсивную коагуляцию золей.
Коагуляция золей электролитами. Коагуляция коллоидных растворов представляет большой теоретический и практический интерес. Примеры таких коллоидных систем — гидрозоли металлов (АЦОН)з, Fc(OH)3, Sn(OH)4).
Коагуляцию гидрофобных золей могут вызвать определенные электролиты при их растворении в дисперсионной среде (воде). Для большого числа коллоидных растворов установлены определенные закономерности коагулирующего действия электролитов.
Правило знака заряда. Коагуляция электролитами характерна для таких коллоидных растворов, частицы которых имеют электрический заряд. Между знаком заряда дисперсных частиц и знаком заряда ионов, вызывающих коагуляцию золя, существует следующее соответствие: коагулирующей способностью обладают ионы
с противоположным знаком заряда по отношению к заряду дисперсных частиц (правило Гарди, 1909).
Для иллюстрации правила Гарди рассмотрим золь йодистого серебра Agl, его получают обменной реакцией
AgNO3 + KI -> Agl + KNO3
Для золя Agl с отрицательно заряженными частицами (полученными при избытке KI) коагулирующими ионами являются катионы (NaT, Mg2+, Fe3+ и др.).
Таким образом, правило Гарди может иметь такую формулировку: коагулирующие ионы имеют одинаковый знак заряда с противоионами двойною электрического слоя около поверхности дисперсных частиц.
Правило порога коагуляции. Для коагуляции золей под действием электролитов, удовлетворяющих правилу Гарди, необходимо выполнение дополнительного условия — концентрация электролита должна превысить определенное критическое значение (Ссг). Эту концентрацию называют порогом коагуляции.
При меньших концентрациях (С< Ссг) коагуляция не происходит и коллоидный раствор остается устойчивым (стабильным) в течение длительного времени
В качестве примера приведем значения порога коагуляции для некоторых электролитов, вызывающих коагуляцию отрицательно заряженных частиц золя As_>S;, ммоль/л:
LiCl...............58,0
NaCl...............51.0
КС!............... 49,5
KNOi...............50,0
MgCl2\............ 0.71
ВаСЬ............... 0,69
СаС12.............. 0,65
А1С13.............. 0,09
AI(NO3),.............. 0,09
Поро! коагуляции не являшея строго определенным единственным значением дня данного электролита. Он зависит от концентрации частиц золя, наличия примесей других веществ, температуры и некоторых других факторов, влияющих на коагуляцию
Правило валентности. Порог коагуляции резко уменьшается при увеличении валентности коагулирующих ионов (правило Шульце. 1882). Данные, приведенные выше, показывают, что дня золя As2S3 с отрицательным зарядом дисперсных частиц порог коагуляции двухвалентных катионов (Mg2', Ва2\ Са2+) примерно в 75 — 80 раз ниже, чем для одновалентных; для трехвалентных катионов это соотношение еще больше (примерно в 550 раз).
Правило размеров коагулирующего иона. При одинаковой валентности коагулирующая способность ионов растет с увеличением их размеров. Например, для ионов щелочных металлов nopoi коагуляции уменьшается в ряду Lr< Na+< К . 1акая же последовательность наблюдается и для двухвалентных катионов. 11о коагулирую-
тему действию катионы и анионы одинаковой валентности располагаю! ся в том же порядке, что и в лиотропном ряду при их адсорбции на твердой поверхности (см. подразд. 6.6).
В соответствии с правилом размеров сильным коагулирующим действием обладают органические ионы, например ионы алкалоидов и красителей.
Кинетика коагуляции. Процесс коагуляции приводит к постепенному уменьшению концентрации частиц коллоидного раствора: dv/dr < О (Г — время после начала коагуляции). Скорость коагуляции (г, м3 с ) определяется соотношением
r=-dv/d/. (10.3)
Теория кинетики коагуляции (М Смолуховскии, 1914) использует несколько упрощающих положений.
I. Сближение дисперсных частиц на достаточно близкое для ai регирования расстояние вызывается броуновским движением. Этот механизм сближения частиц соответствует случаю перикине-тическои коагуляции. Возможен и другой механизм сближения — под действием гидродинамических сил. В сю основе — различие скоростей движения дисперсных частиц разного размера. Такая ситуация характерна для седиментации и флокуляции. Этот случаи называюз ортокинетической коагуляцией. Теория Смолуховского рассматривает случаи перикинетической коагуляции, когда сближение частиц определяется коэффициентом диффузии и средним расстоянием между частицами /= v_| .
2. В каждом элементарном акте коагуляции участвуют только две дисперсные частицы. Это положение мотивировано тем, что вероятность одновременного столкновения трех или большего числа частиц юраздо меньше, чем двух частиц. При учете только парных столкновении скорость коагуляции считают пропорциональной квадрату концентрации частиц, т.е. процесс коагуляции можно рассматривать как аналог химической реакции второго порядка.
3 Исходное состояние коллоидного раствора (до начала коагуляции) — это монодисперсная система, т.е. все дисперсные частицы имеют одинаковый размер и поэтому обладают одинаковой броуновской подвижностью. Форму частиц считают сферической.
4. При столкновении дисперсных частиц возможны два варианта дальнейшего поведения:
1) образуется устойчивый агрегат из этих частиц — элементарный акт коагуляции осуществился;
2) после сближения частицы через достаточно малое время удаляются друг от друга — в этом случае коагуляция не произошла.
Эти два варианта соответствуют двум разным режимам коагуляции. Первый вариант, когда все столкновения дисперсных частиц приводят к образованию устойчивых агрегатов, называют бы
строй коагуляцией. При быстрой коагуляции эффективность столкновений (т.е. доля числа столкновений, завершающихся образованием коагуляционных агрегатов) равна 100%. При медленной коагуляции (второй случай) эффективность таких столкновений существенно меньше 100%.
Быстрая коагуляция. Рассмотрим случай быстрой коагуляции (М.Смолуховский, 1918). Аналогия с реакцией второго порядка позволяет описать кинетику коагуляции следующим уравнением:
dv „ -)
~fr=Kv' (Ю.4)
где К — константа скорости коагуляции, м1/с; для перикинсти-ческой коагуляции К - 4nDl (D — коэффициент диффузии; / — расстояние между центрами дисперсных частиц).
Интегрирование уравнения (10.4) позволяет найти в явной форме зависимость концентрации частиц от времени (/) после начала быстрой коагуляции:
Уо
1 + К v^t'
(10.5)
где v() — исходная концентрация частиц золя (до начала коагуляции); произведение Av0 имеет размерность с ’.
Введем величину /0 =l/(Av0), тогда уравнение (10.5) принимает вид
v _ I
v0 1 + / / /0
(10.6)
Отсюда следует, что за время / = исходная концентрация v0 уменьшается в два раза. Поэтому период называют временем половинной коагуляции.
Теория быстрой коагуляции позволяет оценить критическое расстояние /, при ко юром становится возможным слипание двух дисперсных частиц.
Рассмотрим самый простой случай — обе частицы представляют собой сферы одинакового радиуса г. Коэффициент диффузии D определяется уравнением Эйнштейна (2.13):
D= квТ/(6тя]г).
Время половинной коагуляции равно
или
1 _ 1 Зг|г
Kvq 4tcZ)/v0 2v0As77
/ _ Зт|
г Ъ^кБТ^ ‘
Расчеты на основе экспериментальных данных показывают, что //г - 2,0-2,5.
Теория быстрой коагуляции для более сложных систем принимает во внимание некоторые дополнительные факторы. Один из них — форма дисперсных частиц. Для анизометричных частиц (например, в форме «палочек») скорость коагуляции в десятки раз больше, чем для сферических частиц равного объема. Причина заключается в том, что для вытянутых частиц повышается частота столкновений при броуновском движении.
Медленная коагуляция. Рассмотрим теперь более сложный случай — медленную коагуляцию. Медленная коагуляция характеризуется тем, что к образованию коагуляционных ацэегатов приводят нс все соударения дисперсных частиц, а только некоторая доля (е) от общего числа соударении. Безразмерный коэффициент е< 1 называют эффективностью слипания. При быстрой коагуляции е = 1; при полном отсутствии коагуляции е = 0 Причина неэффективности некоторой части соударении заключается в том. что процесс слипания дисперсных частиц связан с преодолением определенного энергетического барьера ((/) (см. далее теорию устойчивости гидрофобных золей)
Теория медленной коагуляции (Н.А.Фукс, 1934) вводит также коэффициент замедления (IV), который показывает, во сколько раз скорость медленной коагуляции (г\) меньше, чем скорость быстрой коагуляции (v(f): W = vs/vq.
Коэффициент замедления определяется уравнением
И^-ЦехрКШяГ)], (10.7)
где к = 1/5 (5 — приведенная толщина диффузного слоя противоионов в двойном электрическом слое); / — расстояние между центрами сферических дисперсных частиц.
Уравнение (10.7) показывает, что режим медленной коагуляции наступает в двух случаях, при большой высоте энергетического барьера U и при увеличении толщины диффузного слоя 5.
Сопоставление этих закономерностей с электроповсрхност-пыми свойствами коллоидных систем (см. гл. 9) показывает, что существует определенная связь между порогом коагуляции и строением двойного электрического слоя. Действительно, для многих золей nopoi коагуляции соответствует снижению элскгроки-нетического потенциала до одного и того же уровня — примерно 30 мВ.
Теория устойчивости коллоидных растворов (золей). Основная проблема теории устойчивости дисперсных систем заключается в определении конкретных причин и механизма объединения отдельных дисперсных частиц в более крупные агрегаты и в выясне
нии факторов, которые препятствуют их агрегированию. Такую теорию детально разработали для гидрофобных золей Б. В. Дерягин и Л.Д.Ландау (1940) и независимо Э.Фервей и Т.Овсрбек (1946). По начальным буквам фамилий авторов для обозначения этой теории используют сокращение ДЛФО (в зарубежной литературе — DLVO).
Центральное место в теории ДЛФО занимает расчет сил. действующих на твердые дисперсные частицы, которые разделены тонкой пленкой раствора электролита. Основное упрощающее положение теории ДЛФО — на частицы действуют только две силы. Одна сила (fd) — взаимное притяжение частиц; эта сила создается дисперсионными взаимодействиями (см. подразд. 4.3). Другая сила (/,) — электрическое отталкивание частиц, которое возникает при одинаковом знаке поверхностного электростатического заряда. В зависимост и от соотношения эт их сил возможны два варианта поведения коллоидного раствора.
1. Если преобладает сила притяжения (> |/,|), ю дисперсные частицы сближаются, между ними возникает контакт, и они объединяются в более крупный агрегат (коллоидный «димер»). Таким образом, в этом случае элементарный акт процесса коагуляции может состояться.
2. Если преобладает электростатическое отталкивание ( fd\ < |/,|), то частицы не Moryi вступить в непосредственное соприкосновение, и коагуляция золя не происходит.
Таким образом, в качестве основного фактора термодинамической устойчивости дисперсной системы в теории ДЛФО принимают электростатическое (кулоновское) отталкивание дисперсных частиц.
Для расчета условия коагуляции нужно ввести дополнительные допущения. Одно упрощение относится к форме част иц и их взаимному расположению. Рассматривается самый простой случай — частицы имеют при ^магическую форму и разделены плоскопараллельным зазором шириной h (см рис. 9.5). Другое допущение относится к перемещению частиц. В принятой модели частицы могут перемещаться только в перпендикулярном направлении относительно зазора и только под влиянием указанных выше сил. Броуновское движение, таким образом, из рассмотрения исключается. Для расчета условии устойчивости удобнее сопоставлять не силы притяжения (fd) и отталкивания (/'), а соответствующие им энер-гии взаимодействия (Ud, Ue).
Энергия дисперсионного взаимодействия (Ud, Дж/м2). отнесенная к единице плошали поверхности дисперсной частицы, определяется уравнениями Гамакера (4.21). (4.23). (4.25):
TI 4*2 и"=-п^- (,0-8)
где — сложная константа Гамакера; h — расстояние между частицами: знак «минус» указывает на взаимное притяжение частиц под действием дисперсионных сил.
Энергия электростатического взаимодействия (t/(,) создается вследствие перекрывания диффузных слоев противоионов в гонкой пленке раствора электролита в зазоре между частицами. Величина Ue определяется уравнением (9.22).
Полная энергия (U) взаимодействия дисперсных частиц представляет алгебраическую сумму энергий притяжения и отталкивания U = Ud + Ue. После подстановки уравнений (10.8) и (9.22) получаем
(Ю.9)
Графически зависимость U=f(h), соответствующая уравнению (10.9), показана на рис. 10.1.
Энергии отталкивания и притяжения имеют разные знаки и по-разному зависят ог толщины разделяющей их жидкой пленки. Энергия отталкивания Ut, меняется по экспоненциальному закону, а энергия притяжения Ud — по степенному.
Поэтому на малых расстояниях (при h —-> 0) будет преобладать притяжение (t/j-э <~). На больших расстояниях также должно преобладать притяжение, так как степенная функция I///2 убывает медленнее, чем экспонента ехр(-кЛ). На средних расстояниях на кривой U=f(h) возможен локальный («дальний») максимум (точка М). Он соответствует энергетическому (потенциальному) барьеру, который препятствует сближению частиц и их коагуляции. Чем выше высота этого барьера, гем выше агрегативная устойчивость коллоидного рас i вора.
Рис 10 1 Изменение энергии (U) топкой пленки электролита в зависимости от ее толщины (/?)
Анализ уравнения (10.9) и рафика U-f(h) на рис. 10.1 позволяет выделить три случая поведения дисперсной системы в зависимости от высоты энергетического барьера (UM) и глубины потенциальной «ямы» (Uy) на больших расстояниях.
1. Высота энергетического барьера и глубина минимума малы по сравнению с энергией тепловых колебаний: т.е. UM<< кп/\ UN^>k[}T. При этих условиях дисперсные частицы могут сблизиться, и в результате произойдет коагуляция дисперсной фазы. Таким образом, этот случай соответствует агрегативной неустойчивости коллоидного раствора.
2. Высота барьера велика (£/,v» Ад?’), а глубина минимума мала (6/Л.« кдТ). В этом случае частицы не могут сблизиться на достаточно малое расстояние, необходимое для коагуляции; поэтому дисперсная система будет устойчива.
3. Большая глубина минимума: UNy> киТ. В этом случае при любой высоте энергетического барьера (UM) частицы находятся на большом расстоянии (примерно 100 нм) друг от друга. Этот случай соответствует образованию периодических коллоидных структур (см. подразд. 10.4).
Уравнение (10.9) позволяет рассчитать nopoi коагуляции. Эта концентрация соответствует исчезновению барьера: U= 0; дополни гельнос условие: dU/dh = 0. С учетом этих условий из уравнения (10.9) получаем
г 4
(«„)2(ЯГ)5 th^
с„ = А, ----—л-----------Т, (10.10)
(42) гМ
где А| — безразмерный коэффициент, равный примерно 105.
Рассмотрим случай сильного и слабого заряда поверхности (см. подразд. 9.3). В случае сильного заряда th[#>(p0/(4/?T)l « I, поэтому концентрация Ссг обратно пропорциональна заряду противоиона (ze)b. Этот вывод дает теоретическое обоснование правилу Шульце—Гарди лая порога коагуляции. При сильном заряде поверхности снижение энергетического барьера вызывает и сжатие диффузного слоя противоионов при введении электролита в достаточно высокой концентрации. Этот случай называют концентрационной коагуляцией.
Для слабозаряженной поверхности имеем
th 5^21 = *е(Ро
4/?Г 4/?Т
Поэтому получаем другую связь между порогом коагуляции и зарядом противоиона (гс):
(ее0)
ЛЭ Z
где к2~ 800.
Этот случай реализуется, если добавление электролита приводит к нейтрализации заряда коллоидной частицы. Поэтому данный процесс называют нейтрализационной коагуляцией.
10.3. Коалесценция
Коалесценцией называю! самопроизвольный процесс слияния капель жидкости в прямых и обратных эмульсиях, в аэрозолях (туманах), а также слияния газовых пузырьков в пенах и газовых эмульсиях.
В результате коалесценции дисперсные частицы (капли и пузырьки) постепенно укрупняются. Поэтому дисперсная система становится седиментанионно неустойчивой. Крупные газовые пузырьки быстро всплывают вверх. То же самое происходит с каплями, ппотность которых pj меньше плотности дисперсионной среды р0. Так, например, ведут себя капли углеводородов при коалесценции в водной дисперсионной среде. Если плотность капель, напротив, больше плотности дисперсионной среды (р^ > р0), то коалесценция стимулирует процесс седиментации капель. Этот случай характерен для обратных эмульсий.
Таким образом, коалесценция приводит к расслоению фаз, образующих дисперсную систему — эмульсию, пену или аэрозоль. В результате происходит гибель дисперсной системы. Она теряет свои основные свойства: малые размеры дисперсных частиц, их равномерное распределение в дисперсионной среде и микрогстс-poi енность дисперсной фазы.
Для обеспечения длительного времени существования эмульсий и пен необходимы эффективные методы их стабилизации, которые предотвращают коагуляцию. Во многих практических случаях требуется решить противоположную задачу — вызвать быструю коагуляцию и обеспсчть разделение дисперсной фазы и дисперсионной жидкой среды. Один из важных примеров такой задачи — вытеснение нефти из се пластов водными растворами поверхностно-активных веществ. Вначале на поверхность поступает эмульсия капель нефти в воде. Для их отделения в виде сплошной нефтяной фаты нужны эффективные методы дестабилизации прямых эмульсии.
Термодинамическая причина коалесценции, как и коагуляции, заключается в агрегативной неустойчивости всех лиофобных дисперсных систем. Мерой неустойчивости является избыточная энер-
гия Гельмгольца (F„) дисперсной системы, т.е. се свободная поверхностная энергия
Л/ - ° 12^12w</,
где о12 — поверхностное натяжение па границе дисперсной фазы с дисперсионной средой; £2]2 — удельная поверхность, md — общая масса дисперсных частиц.
При коагуляции уменьшается избыточная энергия Гельмгольца: d/^/dz < 0.
Хотя термодинамическая причина коагуляции и коалесценции одинаковая, механизмы этих коллоидных процессов разные. При коагуляции твердые частицы слипаются в более крупные агрегаты, но при этом исходная форма и размер отдельной частицы остаются прежними — такими же, как до коагуляции. Соответственно уменьшение поверхностной энергии F„ происходит нс из-за сокращения удельной поверхности, а вследствие понижения поверхностною натяжения в тонких жидких пленках между сблизившимися частицами. Напротив, при коалесценции капли жидкости или газовые пузырьки могут легко сливаться друг с другом с образованием более крупных капель и пузырьков. Поэтому » термодинамическая причина коагуляции заключается в уменьшении удельной поверхности.
Поясним этот вывод о механизме коалесценции расчетом. Рас-смотрим простейший случаи коалесценции — слияние двух сферических капель одинакового объема
Г|
где г( — радиус капли.
Тогда их обшая поверхность будет равна £2, = 8л:/]2. После слияния образуется более крупная капля объемом К2 = 2Г|, радиусом r2 = V2r| и поверхностью П2 = 4х/47гг2. Таким образом, в результате коалесценции двух капель их поверхность уменьшается на (8 -4д/4)лг2, т.е. примерно на 40% от ее начального значения.
Рассмотрим теперь как протекает многостадийный процесс коалесценции.
Первая сталия — сближение двух капель (или пузырьков газа) дру1 с другом. Пузырьки газа в газовых эмульсиях сближаются преимущественно при их всплывании вверх. Очевидно, что скорость подъема различна дьчя крупных и мелких пузырьков В итоге при сближении капель или пузырьков возникает точка касания.
Вторая стадия — возле точки касания образуется узкий «перешеек», имеющий форму вогнутою мениска. Образование такою перешейка связано с увеличением общей поверхности и поэтому требует затраты работы. Такая работа совершается за счет кинети
ческой энергии капель. В момент столкновения двух капель хотя бы одна из них движется и ее скорость определяется коэффициентом диффузии (уравнение (2.13)).
Трщья стадия — перетекание жидкости из одной капли в другую. Причина перетекания — разность капиллярных давлении в соприкоснувшихся каплях. При этом существенную роль играет го обстоятельство, что радиусы капель могул быть не равны. Пусть гЛ< Ль тогда в момент образования перешейка капиллярное давление рЛ> рв и жидкость дисперсной фазы начнет перетекать из капли А в каплю В. В результате объем капли А и ее радиус гЛ уменьшаются: d Ил< 0 и drA< 0 Напротив, в случае капли В благодаря притоку жидкости объем и радиус гв увеличатся: dKB> 0 и d/B > 0. По мере перетекания разность капиллярных давлений Др = Ра~ Ри увеличивается Поэтому перетекание идет с нарастающей скоростью и слияние капель маловязких жидкостей (после их соприкосновения) происходит очень быстро — за десятые доли секунды В среднем такой .малый «период жизни» типичен для многих прямых и обратных эмульсий.
Таким образом, лимитирующей стадией процесса коалесценции обычно является первая стадия — столкновение капель. Чем выше дисперсность исходной эмульсии, тем больше вклад броуновского движения, гем выше частота столкновений. В этих случаях кинетику коалесценции, т.е. зависимость числа капель (v) от времени (/) после начала процесса, можно описать (по аналогии с быстром коагуляцией) как реакцию второго порядка. Скорость коагуляции количественно характеризует производная dv/d/. Константу скорости коалесценции К (по аналогии с уравнением 10.6) можно рассчитать по уравнению
V
(10.12)
где v0 — число капель в единице объема исходной эмульсии.
Поскольку скорость быстрой коалесценции капель определяется частотой их столкновений, большое влияние оказывает вязкость дисперсионной среды; при се увеличении сближение капель замедляется.
Показателен следующий пример. Капли воды, диспергированные в толуоле, сливаются очень быстро — коалесценция завершается за десятые доли секунды При растворении в толуоле 10% полистирола вязкость дисперсионной среды значительно увеличивается, и время жизни такой эмульсии составляет уже несколько минут.
Анализ кинетики коалесценции в эмульсиях чистых жидкостей приводит к выводу: предотвратить столкновение капель в принципе невозможно. Поэтому обеспечить устойчивость эмульсий
можно при резком затруднении второй стадии — образования перешейка между каплями, но которому далее происходит перетекание жидкости из одной капли в другую.
Наиболее эффективным метод стабилизации эмульсий заключается в создании структур!ю-механического барьера на поверхности капель. Такие барьеры образуются разными способами. Основные из них — адсорбция ПАВ с соответствующим гидрофильно-липофильным балансом; образование тонких структурированных слоев высокомолекулярных ПАВ и биополимеров; нанесение на поверхность капель мелких твердых частиц (твердых эмульгаторов) из вещества, которое лучше смачивается в избирательных условиях жидкостью дисперсной фазы.
10.4. Коллоидные структуры
Термодинамическая неустойчивость дисперсных систем с твердыми дисперсными частицами (коллоидные растворы, аэрозоли) приводит к коагуляции — образованию крупных агрегатов из многих отдельных частиц. Из-за больших размеров такие агрегаты быстро оседают в дисперсионной среде. В нижней части дисперсной системы образуется осадок, в котором концентрация частиц намного больше, чем в исходной свободподиспсрсной системе (золе). Таким образом, главный результат коагуляции — превращение свободподиспсрсной системы в связнодисперсную. Многие свойства (особенно механические) таких систем весьма своеобразны и существенно отличаются от свойств обычных (макроскопических) материалов такого же химического состава, как дисперсные частицы. Эти свойства определяются главным образом структурой связнодисперсных систем — взаимным расположением частиц и агрегатов и концентрацией частиц. В зависимости от энергии взаимодействия между частицами связнодиспсрсные системы классифицируют на коагуляционные структуры и структуры с фазовыми контактами между дисперсными частицами.
Коагуляционные структуры. Характерный признак коагуляционных структур — дисперсные частицы разделены тонкими пленками жидкой дисперсионной среды или контактируют друг с другом только в отдельных точках; такие контакты между частицами называют коагуляционными. Механическая прочность коагуляционных контактов мала. При наличии тонких жидких пленок связь между частицами в соответствии с теорией ДЛФО (см. подразд. 10.2) создается слабыми молекулярными силами. Низкая прочность точечных коагуляционных контактов определяется их малой площадью.
Коагуляционные структуры обычно образуются вследствие потери агрегативной устойчивости свободподиспсрсной системы
(золя) и последующей коагуляции. На образование коагуляционных структур и их свойства существенное влияние оказывают несколько факторов: концентрация частиц в коллоидном растворе, форма дисперсных частиц, температура, механические воздействия (например, перемешивание). Рассмотрим влияние этих факторов.
I. Увеличение концентрации частиц вызывает возрастание прочности коагуляционной структуры (Р). Это происходит благодаря росту числа контактов между дисперсными частицами. Приближенно выполняется соотношение
Р-дд (10.13)
где п — число контактов (в расчете на единицу площади поперечного сечения структуры); р — прочность индивидуального контакта.
В гидрофобных системах прочность кон такта между высокодисперсными частицами изменяется в интервале 10“’°— К)"9 Н.
2. Влияние формы частиц сказывается особенно резко, если они сильно вытянуты, т.е. имеют форму стержней, волокон и т.п. В этих случаях коагуляционные контакты образуются преимущественно между торцевыми (узкими) участками частиц. В результате возникает зрехмерная структура («сетка») из дисперсных частиц, в ячейках которой находится жидкая дисперсионная среда; такие коагуляционные структуры называют гелями.
3. Температура влияет на скорость формирования дисперсных структур следующим образом: чем выше температура, тем быстрее происходит коагуляция (см. подразд. 10.2). Вместе с тем с повышением температуры прочность коагуляционных структур уменьшается, поскольку под влиянием тепловых колебаний часть коагуляционных контактов распадается — уменьшается число контактов.
4. Механические воздействия па коагуляционную структуру приводят к разрушению контактов между частицами, поэтому прочность структуры уменьшается. При достаточно интенсивном воздействии структура может разрушиться полностью и система станет свободподиспсрсной: она утратит прочность, но зато приобретет текучесть. Такое изменение свойств пол действием нагрузок характерно для наст, а также для глинистых горных пород.
Замечательное свойство коагуляционных структур заключается в том, что после их разрушения и устранения вызвавшей его нагрузки происходит постепенно восстановление структуры и ее механической прочности. Этот эффект называют тиксотропией. Тиксотропные свойства коагуляционных структур играют ключевую роль во многих технологических процессах. Например, масляные краски должны «разжижаться» при нажиме кистью, а после этого они должны затвердеть. Тиксотропными свойствами облада
ют многие биологические системы (протоплазма, мышечные волокна).
Другое физико-химическое явление, характерное для коагуляционных структур, называют синерезисом, или коллапсом. Оно заключается в самопроизвольном уменьшении размеров геля, которое сопровождайся выделением жидкости, находящейся в ячейках структуры. Термодинамическая причина синерезиса — неравновесное состояние гелей. Для этих систем оно связано не только с большой поверхностью дисперсной фазы, но и с тем, что при образовании геля его структура является рыхлой и число коагуляционных контактов меньше, чем в случае уплотненной структуры. При определенных условиях объем геля резко уменьшается (в некоторых случаях — в согни раз), и благодаря этому возникает более плотная структура.
Возможен сшс один (химическим) механизм синерезиса (коч-лапса) гелей. Характерный пример — полиакриламидные гели. При помещении в воду таких гелей происходит медленный гидролиз с образованием заряженной полиакриловом кислоты, т.е. гель становится полиэлсктролитным. Электрическое отталкивание противоионов ведет к сильному увеличению объема геля (набуханию).
-Лри добавлении к воде ацетона при определенной концентрации (-40%) объем геля резко уменьшается — происходит коллапс (рис. 10.2) В этой системе причиной коллапса геля является плохая растворимость полимера в ацетоне. Заряженные сегменты полиакриловой кислоты не образуются, между макромолекулами полимера преобладаю!’ силы притяжения. Эгог эффект выражен тем сильнее, чем дольше гель находился в воде до добавления анетона.
Структуры с фазовыми контактами. Определяющий признак фазовых контактов заключается в том, что их плошадь значительно превышает молекулярные размеры (г.е. площадь «точечных» коагуляционных контактов) и взаимодействие между частицами определяется силами когезии (см. подразд. 3.4). По этим причинам прочность фазовых контактов гораздо выше, чем коагуляционных.
Теоретические расчет ы оценивают прочность фазовых контактов между дисперсными частицами ионных кристаллов и между металлическими частицами примерно в 10 Н Для расчета прочности структуры с фазовыми контактами можно использовать соотношение (10 13). Эго соотношение охватывает очень широкий интервал значений прочности — от Ю4 до 10s Н/м .
Фазовые контакты делятся на две группы: конденсационные и кристаллизационные. Соот ветственно различают два тина дисперсных структур с фазовыми контактами: конденсационные и кристалл и за ц и о н н ы е.
Рис. 10.2. Зависимость объема (И) полиакриламидного геля от доли (х) ацетона в водно-анетоноиой смеси, в которой набухает гель в течение различного времени:
1 — свежий гель (/ = 0); 2 — t = I нед; 3 — г = 1 мес
Конденсационные структуры образуются при выделении твердых аморфных частиц из переохлажденных расплавов или из пересыщенных растворов. Термин «конденсационный» используется но аналогии с названием соответствующих методов получения дисперсных частиц (см. подразд. 3.3).
Типичными представителями конденсационных дисперсных систем являются неорганические гели — силикагели и алюмосиликаты. Силикагели образуют аморфную фазу при реакции силиката натрия с кислотой.
Образующийся золь кремниевой кислоты после коагуляции образует гель. Благодаря пересыщению дисперсионной среды (раствор Na2SiO3) дисперсные частицы геля срастаются с образованием фазовых контактов. Этот процесс используют для приготовления некоторых катализаторов для переработки нефти.
Кристаллизационные структуры возникаю! в результате сцепления кристаллических дисперсных частиц. Типичный пример таких структур — поликристалл ичсские металлы и минералы.
Поликристаллическис структуры можно получать разными способами. Во многих случаях используют химические реакции между дисперсными частицами и жидкой дисперсионной средой. Примером может служить отверждение полугидрата гипса; при сю контакте с водой идет реакция
CaSO4 X Н2О + % Н2О = CaSO4 2Н2О
с образованием дигидрата, который термодинамически устойчивее, чем полугидрат. Существенно, что при комнатной темпсрату-
ре растворимость дигидрата гипса в несколько раз меньше. Поэтому раствор, насыщенный по отношению к полугидрату, сильно пересыщен по отношению к дигидрату. В результате выделяются кристаллы дигидрата гипса. Вместе с дисперсными частицами полу гидрата они образуют коагуляционную структуру. Далее кристаллы дшидрата гипса растут и постепенно возникает структура с кристаллизационными фазовыми контактами.
Другой способ получения кристаллизационных структур заключается в спекании дисперсных частиц. Обычно этот процесс проводят при высоких температурах и давлениях, чтобы скорость спекания была достаточно высокой. Спекание широко используют в порошковой металлургии С помощью этою процесса получают многие современные материалы, в том числе и композитные.
Структуры с фазовыми контактами обычно гораздо прочнее коагуляционных структур и имеют большое сходство с прочными, но хрупкими материалами. Они мало способны к пластическим деформациям У них совершенно отсутствует тиксотропия — способность к восстановлению структуры после ее разрушения.
Периодические коллоидные структуры. При определенных условиях образуются высокоорганизованные коллоидные структуры, в которых дисперсные частицы расположены в определенном порядке относительно друг друга. Такие самоорганизованные системы называют периодическими коллоидными структурами. Периодическое расположение структурных элементов свойственно кристаллам, поэтому периодические коллоидные структуры представляют собой квазикристаллы, т.е. структуры с дальним порядком. Периодические коллоидные структуры распространены достаточно широко. Их роль особенно важна в биологии. Они, например, существуют в некоторых тинах почв.
Образование периодических структур объясняет теория устойчивости гидрофобных золей — теория ДЛФО (см. подразд. 10.2). Одно из следствий этой теории состоит в том, что взаимодействие дисперсных частиц в коллоидном растворе определяет соотношение между высотой потенциальною барьера, обусловленного электрическим о пал киванием, и глубиной потенциальных ям на графике зависимости энергии взаимодействия от расстояния между частицами (см. рис. 10.1). В данном случае основную роль играет соотношение этих факторов в дальнем минимуме. Если его глубина велика (намного больше тепловой энергии квТ), то при любой высоте потенциального барьера реализуется дальнее взаимодействие двух дисперсных частиц на расстоянии 2/i друг от друга; это расстояние составляет примерно 100 нм. Частицы, оказавшиеся в этих потенциальных ямах, уже не могут сблизиться или удалиться друг от друга Однако к этой «зародышевой» паре moi у г
присоединяться (на таком же удалении) другие дисперсные частицы
Приведем несколько примеров периодических коллоидных структур.
Слои Шиллера. Эти слои представляют собой коагуляционные осадки пластинчатых частиц, например оксидов железа и ванадия. Слои Шиллера толщиной около 100 нм располагаются параллельными горизонтальными слоями. Строение слоев можно регулировать добавлением электролитов. Увеличение их концентрации вс-дс! к уменьшению «периода» структуры, т с среднего расстояния между слоями (в соответствии с теорией ДЛФО). Слои Шиллера возникают также в золях, которые находятся в зоне действия других внешних полей: центробежного, электрического, магнитного.
Тактоиды. Эго анизотропные стержнеобразные агрегаты дисперсных частиц. Такие агрегаты образуются при достаточно высокой концентрации золей, частицы которых сами имеют анизотропную форму Таковы, например, золи Fe(OH)3 и биоколлоид-ные частицы (некоторые вирусы и бактерии). Дисперсные коагуляционные структуры, содержащие тактоиды, состоят из двух областей. Одна из них — это локальные участки («домены») с упорядоченной структурой, т.е. собственно тактоиды; другая — неупорядоченная дисперсная система.
Биконтинуальные дисперсные системы. Периодические коллоидные структуры образуются нс только при коагуляции золей, но и в результате других коллоидно-химических процессов. Примером может служить набухание глины в водных растворах щелочных металлов. Находясь в воде, образцы глины постепенно набухают; кристаллические слои толщиной около 1 нм раздвигаются и между ними образуются пленки раствора толщиной около 30 нм. В результате возникает периодическая биконтинуалызая дисперсная система, в которой твердые дисперсные частицы и жидкие пленки имеют толщину в интервале наноразмеров.
Кольца и слои Лизеганга. Этот тип периодической коллоидной структуры был открыт Р Лизсгангом (1896) на гелях желатины. Раствор желатины с добавлением небольшого количества дихромата калия наливали тонким слоем на стеклянную пластинку, на которой через некоторое время формировался гель. На его поверхность наносили каплю концентрированного раствора соли серебра. Далее вокруг места нанесения капли появлялись концент-рично расположенные кольца дихромата серебра (рис. 10.3).
Другой вариант этого опыта проводят не с тонким слоем геля, а в пробирке с гелем желатины. В этом случае после нанесения на наружную поверхность геля капли раствора соли серебра в глубине геля постепенно образуются параллельные тонкие слои дихромата серебра. Такие структуры называют «ритмическими» осадками.
Рис. 10.3. Периодическая коллоидная структура (кольца Лизеганга)
Основная причина возникновения периодических структур в гелях заключается в том, что в геле не возникаю! конвективные потоки, переносящие продукты реакции но всему объему системы.
Кольца и слои Лизеганга часто встречаются в природе (слоистые минералы, некоторые мускульные ткани).
10.5. Методы стабилизации дисперсных систем
Седиментационная и агрегативная неустойчивость представляют фундаментальное свойство всех дисперсных систем. Поэтому для сохранения дисперсных систем в течение достаточно длительного времени необходимо обеспечить их устойчивость или, но крайней мере, существенно замедлить эволюционные процессы (коагуляцию, коалесценцию и др.), которые приводят к увеличению размеров дисперсных частиц. Наряду с обеспечением устойчивости (стабилизации) дисперсных систем во многих случаях возникает противоположная задача — вызвать максимально быстрое разрушение дисперсных систем и их разделение на две макроскопические фазы дисперсионную среду и дисперсную фазу. Такая проблема возникает во многих технологических процессах, например при разделении водно-нефтяпых эмульсий или при гашении пен.
Таким образом, методы стабилизации и разрушения дисперсных систем представляю!, по сути, основу регулирования различных свойств этих систем.
Рассмотрим ряд факторов, обеспечивающих достаточно длительную стабилизацию дисперсных систем.
Электрическое отталкивание одноименно заряженных дисперсных частиц. Это1 фак гор играет определяющую роль в обеспечении устойчивости свободнодисперсных систем. Устойчивость гидрофобных золей к коагуляции электролитами определяется потенциальным барьером, создаваемым электростатическим отталкиванием: его высота ((/) рассчитывается ио уравнению (10 9), которое следует из теории устойчивости гидрофобных золей ДЛФО (см подразд. 10.2).
В дисперсных системах с газовой дисперсионной средой (аэрозолях) также наиболее существенным фактором устойчивости является электрический заряд дисперсных частиц — твердых или жидких. Причина возникновения зарядов заключается в столкновении движущихся аэрозольных частиц с ионами, присутствующими в газах. Такие столкновения носят случайный характер. Поэтому мерой электрических свойств аэрозольной частицы служит среднеквадратичный заряд (q2):
q2 =4nwkfJ\ (10.14)
где е0= 8,8э КС1 Ф/м; г — радиус аэрозольной частицы: кр — постоянная Больцмана: Т — температура.
В соответствии с уравнением (10.14) аэрозольные частицы радиусом 1 мкм при комнатной температуре несут заряд около 7 10 |(> Кл, т.е. несколько элементарных нарядов.
Другая причина возникновения электрического заряда на частицах и каплях аэрозолей заключается в их электризации при седиментации, т.е. в возникновении потенциала оседания (см. подразд 9.6). Этот эффект может быть весьма значительным. Например, на поверхности капель воды размером около 10 мкм при их оседании возникает сильное электрическое поле напряженностью примерно 10 В/м Поэтому в облаках, которые представляют собой аэрозоль капель воды, возможны мощные электрические разряды (молнии).
Адсорбция поверхностно-активных веществ. Поверхностно-активные вещества применяют преимущественно для стабилизации дисперсных систем с электрически нейтральными дисперсными частицами. К их числу относятся такие распространенные и важные классы дисперсных систем, как пены и эмульсии Стабилизирующее действие ПАВ обусловлено 1ремя механизмами.
Первый механизм заключается в уменьшении удельной поверхностной энергии (поверхностного натяжения) на границе дисперсных частиц с дисперсионной средой (см. гл. 6). В соответствии с уравнением (10.2) чем ниже поверхностное натяжение, тем меньше избыточная поверхностная энергия дисперсной системы, т.е. тем меньше отклонение oi термодинамического равновесия.
Обычно адсорбция поверхностно-активных вешеств снижаем поверхностное натяжение до I — 10 мДж/м2. Поэтому термодинамическая (агрегативная) неустойчивость имеет место и в таких дисперсных системах.
Принципиально важен случай очень сильного снижения поверхностного натяжения — до 10 3— 10 2 мДж/м2. Такой способностью обладают определенные смеси ПАВ (см. гл. 8). Дисперсные системы, в которых поверхностное натяжение очень мало, в соответствии с критерием Ребиндера—Щукина (4.29) термодинамически устойчивы, поэтому агрегация дисперсных частиц в них нс происходит. Важный пример таких систем — микроэмульсии (см. гл. 8).
Второй механизм обеспечения агрегативной устойчивости с помощью ПАВ заключается в том, что насыщенные адсорбционные слои многих ПАВ обладаю! механической прочностью и высокой вязкостью. Поэтому они образуют возле поверхности дисперсных частиц своеобразную защитную оболочку, которая эффективно препятствует процессам коагуляции и коалесценции. Причина защитного действия насыщенных адсорбционных слоев поверхностно-активных веществ заключается в том, что они представляют собой двухмерную коагуляционную структуру с предельным напряжением сдвига и высокой эффективной вязкостью (см. подразд. 4.4).
Таким образом, адсорбционные слои ПАВ могут создавать структурно-механический барьер, обеспечивающий метастабилыюс равновесие дисперсной системы и се длительную агрегативную устойчивость. Этот фактор стабилизации ввел в коллоидную химию П. А. Ребиндер.
Для образования достаточно прочного струю урно-механического барьера в наибольшей мерс подходят ПАВ 3-й и 4-й групп в классификации ПАВ по физико-химическому влиянию на поверхностные свойства дисперсных систем, т.е. стабилизаторы и моющие вещества (см. подразд. 7.1). В качестве примеров таких ПАВ можно назвать производные целлюлозы (карбоксиметилцеллюло-зу), другие высокомолекулярные ПАВ. Очень эффективны в этом 01 ношении белковые ПАВ (желатина и др.).
Структурно-механический барьер может быть создан и другим способом — частицами высокодисперсных порошков, находящихся на поверхности капель в эмульсиях. Необходимое условие этой формы стабилизации состоит в том, чтобы краевой угол избирательного смачивания был больше нуля.
Третий механизм влияния ПАВ на агрегативную устойчивость дисперсных систем связан с упругими свойствами жидких пленок растворов ПАВ Упругость таких пленок возникает но следующей причине: при растяжении пленки площадь ее поверхности увеличивается на величину сЮ. Удельная адсорбция ПАВ при этом умснь-
шается. Соответственно поверхностное натяжение возрастает на величину da, т.е. возникает тангенциально направленная сила, препятствующая растяжению пленки и ее дальнейшему утоньше-нию. Модуль упругости пленки (£п1) определяется уравнением Г иббса
^пл (10.15)
dO
Способность пленок растворов ПАВ сопротивляться их растяжению и угон мнению представляет, таким образом, еще один фактор стабилизации дисперсных систем с жидкой дисперсионной средой (пен и эмульсий).
Вязкое сопротивление дисперсионной среды. Необходимое условие агрегации дисперсных частиц (коагуляции или коалесценции) заключается в установлении непосредственного контакта между твердыми частицами, каплями или газовыми пузырьками. Для возникновения такого контакта необходимо в свою очередь вытеснение дисперсионной среды и з зазора между частицами. В дисперсных системах с жидкой дисперсионной средой (коллоидные растворы, эмульсии, пены) таким процессом является вязкое течение тонких пленок жидкоети. Скорость этого течения обратно пропорциональна вязкости жидкости. Поэтому для повышения устойчивости таких дисперсных систем следует увеличивать вязкость дисперсионной среды. Например, устойчивость пен значительно возрастает при добавлении к воде высоковязкого глицерина. Очень эффективны в этом плане и некоторые высокомолекулярные соединения.
Контрольные вопросы
1. Объясните причины, которые вызывают неустойчивость дисперсных систем и их постепенную эволюцию.
2. Какие два процесса вызывают уменьшение свободной энергии дисперсных систем?
3. Какой фактор вызывает седиментационную неустойчивость дисперсных систем?
4. Охарактеризуйте процесс коагуляции. В чем заключается основное отличие коагуляции от коалесценции?
5. Перечислите основные стадии коагуляции.
6. Охарактеризуйте основные закономерности коагуляции коллоидных растворов под действием электролитов. Назовите основные положения теории быстрой коагуляции М. Смолуховского.
7. Объясните физико-химический смысл времени половинной коагуляции.
8. Приведите основные положения теории устойчивости гидрофобных золей — теории Дерягина—Ландау—Фервея—Овсрбека.
9. Выделите три основных случая поведения дисперсных систем в зависимости от высоты энергетического барьера и глубины потенциальной ямы на больших расстояниях между дисперсными частицами
10 Дайте определение копнен грационной и нейтрализационной коагуляции.
11. Назовите основные стадии процесса коалесценции в эмульсиях.
12 Приведите определения коллоидных структур
13. В чем заключается тиксотропный эффект? Приведите примеры его применения на практике.
14. В чем заключается эффект синерезиса гелей?
15 Дайте общее определение фазовых контактов в дисперсных системах.
16. Приведите примеры периодических коллоидных структур.
17 Назовите основные методы стабилизации дисперсных систем
18 Объясните, в чем заключается стабилизирующее действие структурно-механического барьера (по П.А. Рсбинлеру).
СПИСОК ЛИТЕРАТУРЫ
Абрамзон А. А. Поверхностно-акгивныс вещества/ А. А. Абрамзон. — Л.: Химия, 1984.
Адамсон А Физическая химия поверхностей /А. Адамсон. — М. : Мир, 1979.
Воюцкий С С. Kvpc коллоидной химии / С.С. Воюцкий. — М.: Химия. 1976.
Дамаскин Б. Б. Электрохимия / Б. Б. Дамаскин. О А. Петри й, Г А. Цир-лина. — М.: Химия, 2001.
Дерягин В В. Теория устойчивости коллоидов и тонких пленок / Б. В Дерягин. — М.: Наука. 1986.
Ефремов И.Ф Периодические коллоидные структуры / И.Ф. Ефремов. — Л. : Химия. 1971.
Измайлова В. Н. Поверхностные явления в белковых системах / В. Н. Измайлова, Г. П.Ямпольская. Б.Д Сумм. — М. : Химия, 1988.
Ребиндер П.А. Избранные труды. Поверхностные явления в дисперсных системах. Коллоидная химия / П.А. Ребиндер — М. : Наука. 1978.
Русанов А. И. Фазовые равновесия и поверхностные явления / А. И. Русанов. — Л.: Химия. 1967
Сергеев Г. Б Нанохимия / Г Б.Сергеев. — М. : Изд-во МГУ, 2003.
ФридрихсбергД. А Курс коллоидной химии / Д.А.Фридрихсбсрг. — М : Химия. 1995.
Фролов Ю.Г Курс коллоидной химии / Ю. Г.Фролов. — М. : Химия. 1989.
Шелудко А. Коллоидная химия / А. Шелудко. — М. : Мир, 1984.
Щукин Е.Д. Коллоидная химия / Е.Д. Щукин, А. В. Псрнов, Е. А. Амелина. — М.: Высш, шк., 2004.
Holmberg К. Surfactants and Polymers in Aqueous Solution / К Holmberg. В Jonsson, B. Kronberg, B. Lindman. — West Sussex : John Wiley and Sons, 2004.
ОГЛАВЛЕНИЕ
Предисловие.................................................3
Глава 1. Введение в коллоидную химию........................6
Глава 2. Дисперсные системы................................19
2.1. Количественные характеристики дисперсных систем.......19
2.2. Классификация дисперсных систем.......................21
2.3. Получение, стабилизация и очистка дисперсных систем...25
2.4. Движение частиц в дисперсных системах.................29
2.5. Течение дисперсных систем.............................36
Гл a Bci 3. Дисперсные частицы.............................38
3.1. Классификация дисперсных частиц.......................38
3.2. Получение дисперсных частиц...........................40
3.3. Строение дисперсных частиц и поверхностных слоев жидкостей и твердых тел...............................................50
3.4. Физические и химические свойства дисперсных частиц....56
3.5. Дисперсное состояние вещества.........................64
3.6. Измерение размеров дисперсных частиц..................65
Глав а 4. Термодинамика поверхностных явлений и дисперсных систем.....................................................70
4.1. Поверхностное натяжение...............................71
4.1.1. Определение поверхностного натяжения.............71
4.1.2. Факторы, влияющие на поверхностное натяжение жидкостей...............................................73
4.1.3. Молекулярная природа поверхностного натяжения жидкостей...............................................78
4.2. Термодинамика поверхности раздела фаз в однокомпонентных системах....................................................80
4.3. Расклинивающее давление...............................84
4.4. Термодинамика дисперсных систем.......................88
Глава 5. Капилпарные явления...............................93
5.1. Капиллярное давление..................................93
5.2. Смачивание............................................95
5.3. Адгезия..............................................104
5.4. Влияние кривизны поверхности на давление пара и растворимость............................................107
5.5. Течение жидкостей в капиллярах и пористых средах.......110
5.6. Измерение поверхностного натяжения и краевых углов смачивания.................................................114
Г л а в а 6. Адсорбция поверхностно-активных веществ.......121
6.1. Поверхностное натяжение растворов.....................122
6.2. Уравнение адсорбции Гиббса............................123
6.3. Уравнение Ленгмюра....................................126
6.4. Строение адсорбционных слоев ПАВ......................129
6.5. Адсорбция ПАВ на поверхности раздела жидких фаз.......132
6.6. Адсорбция ПАВ из растворов на поверхности твердых тел.137
6.7. Химическое модифицирование твердых поверхностей.......140
Глава 7. Поверхностно-активные вещества....................149
7.1. Классификация новерхностно-активных веществ...........149
7.2. Некоторые особенности поверхностно-активных веществ...153
7.3. Гидрофильно-липофильный баланс........................156
7.4. Критический параметр упаковки.........................158
Глава 8. Мицеллярные системы...............................160
8.1. Критическая концентрация мицеллообразования...........160
8.2. Строение и форма мицелл...............................166
8.3. Рост мицелл...........................................170
8.4. Термодинамика образования мицелл......................172
8.5. Смешанные мицеллы.....................................175
8.6. Солюбилизация.........................................178
8.7. Ассоциаты поверхностно-ак ивных веществ с полимерами и белками..................................................181
8.8. Гидрофобные взаимодействия............................183
Глава 9. Электроповерхностные явления......................187
9.1. Поверхностный заряд...................................187
9 2. Строение двойного электрического слоя...............189
9.3. Электрический потенциал и плотность заряда в двойном электрическом слое.........................................190
9.4. Влияние электролитов на двойной электрический слой ...196
9.5. Электростатическая составляющая расклинивающего давления .... 197
9.6. Элсктрокинстические явления...........................199
9.7. Электрокапиллярные явления............................205
Гл а в а 10. Устойчивость и эволюция дисперсных систем.....210
10.1. Причины и формы неустойчивости дисперсных систем.....210
10.2. Коагуляция...........................................214
10.3. Коалесценция.........................................223
10.4. Коллоидные структуры ................................226
10.5. Методы стабилизации дисперсных систем................232
Список литературы .........................................237
Учебное издание
Сумм Борис Давидович
Основы коллоидной химии
Учебное пособие
Редактор В. Г. Буханцов Технический редактор Н. И. Горбачева Компьютерная верстка: Л. М. Беляева Корректоры В. М. Малек., Л. В. Гаврилина
Изд. № 102109716. Подписано в печать 23.03.2007. Формат 60 х 90/16.
Гарнитура «Таймс». Печагь офсетная. Бум га офсетная № 1. Усл. печ. л. 15,0.
Тираж 2500 зкз. Заказ № 8000.
Издательский центр «Академия», www.3cademia-moscow.ru
Санитарно-эпидемиологическое заключение № 77.99.02.953.Д.0047963.07.04 от 20.07.2004.
117342. Москва, ул. Бутлерова, 17-Б, к. 360. Тел./факс: (495)330-1092. 334-8337.
ОАО "Тверской полиграфический комбинат", 170024, г. Тверь, пр- Ленина, 5.
Телефон: (4822) 44-52-03.44-50-34, Телефон/фа1 с: (4822) 44-42-15
I loine page - www.tverpk.ru Электронная почта (E-mail) -saies@(.verpk.rii