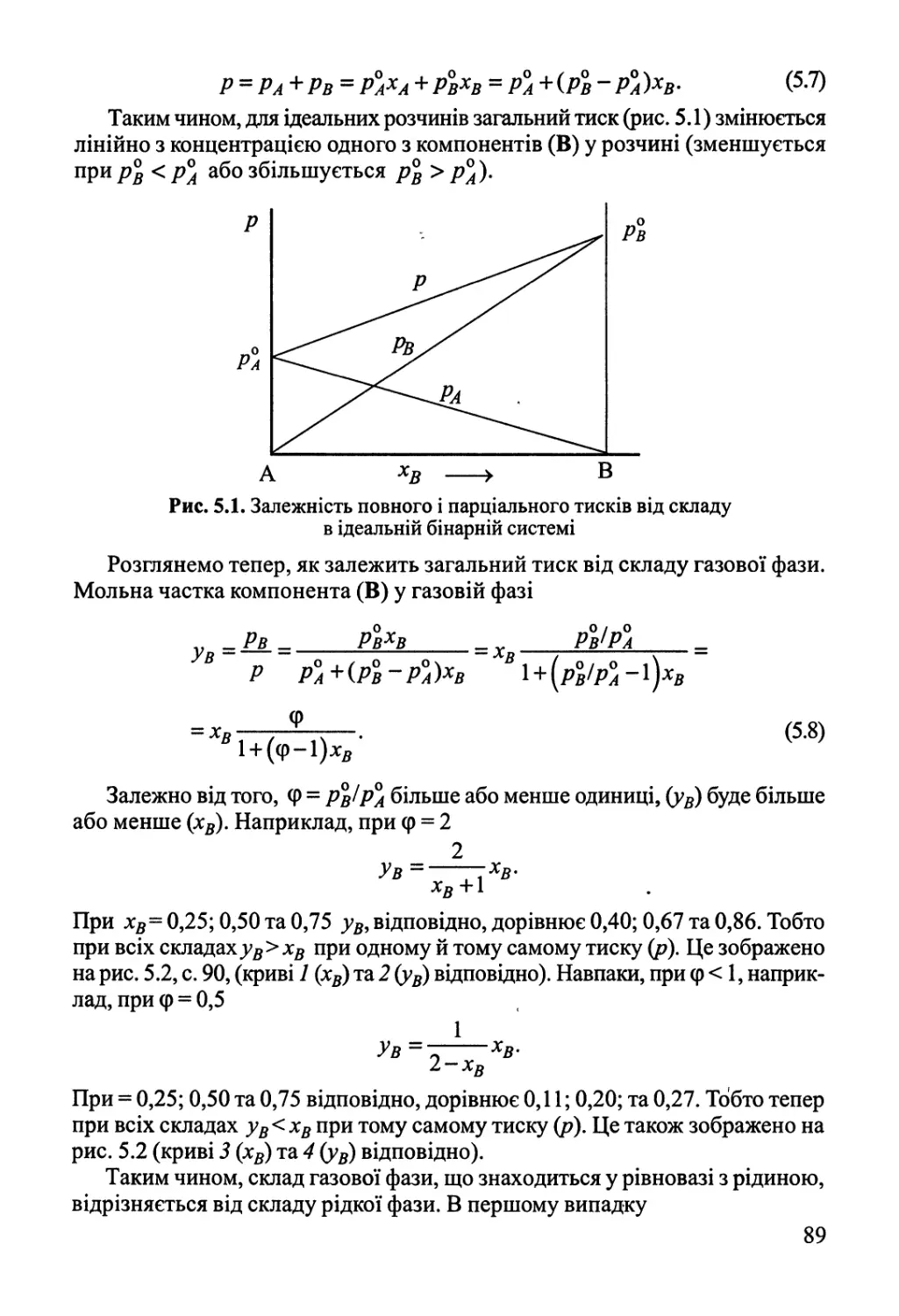
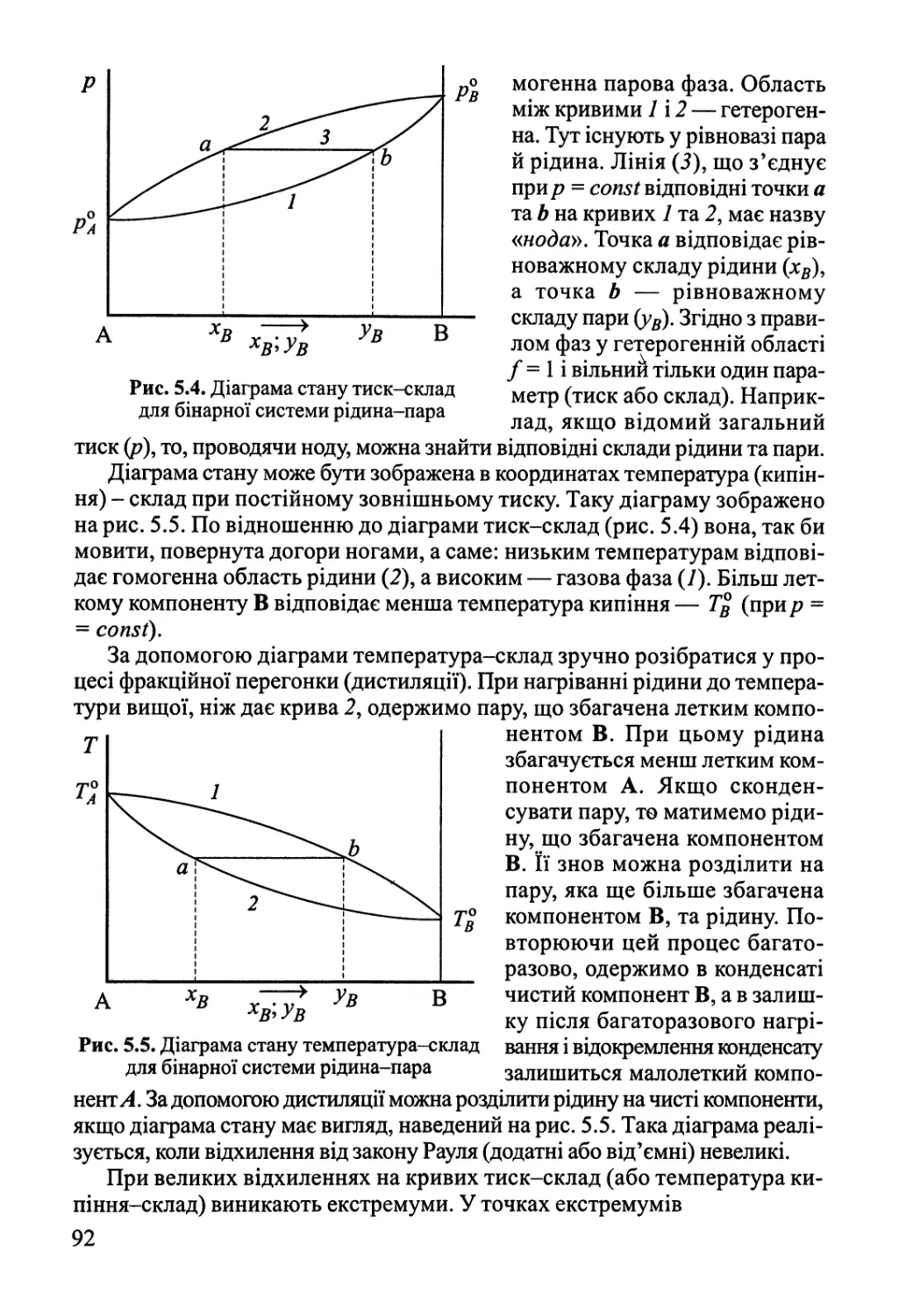
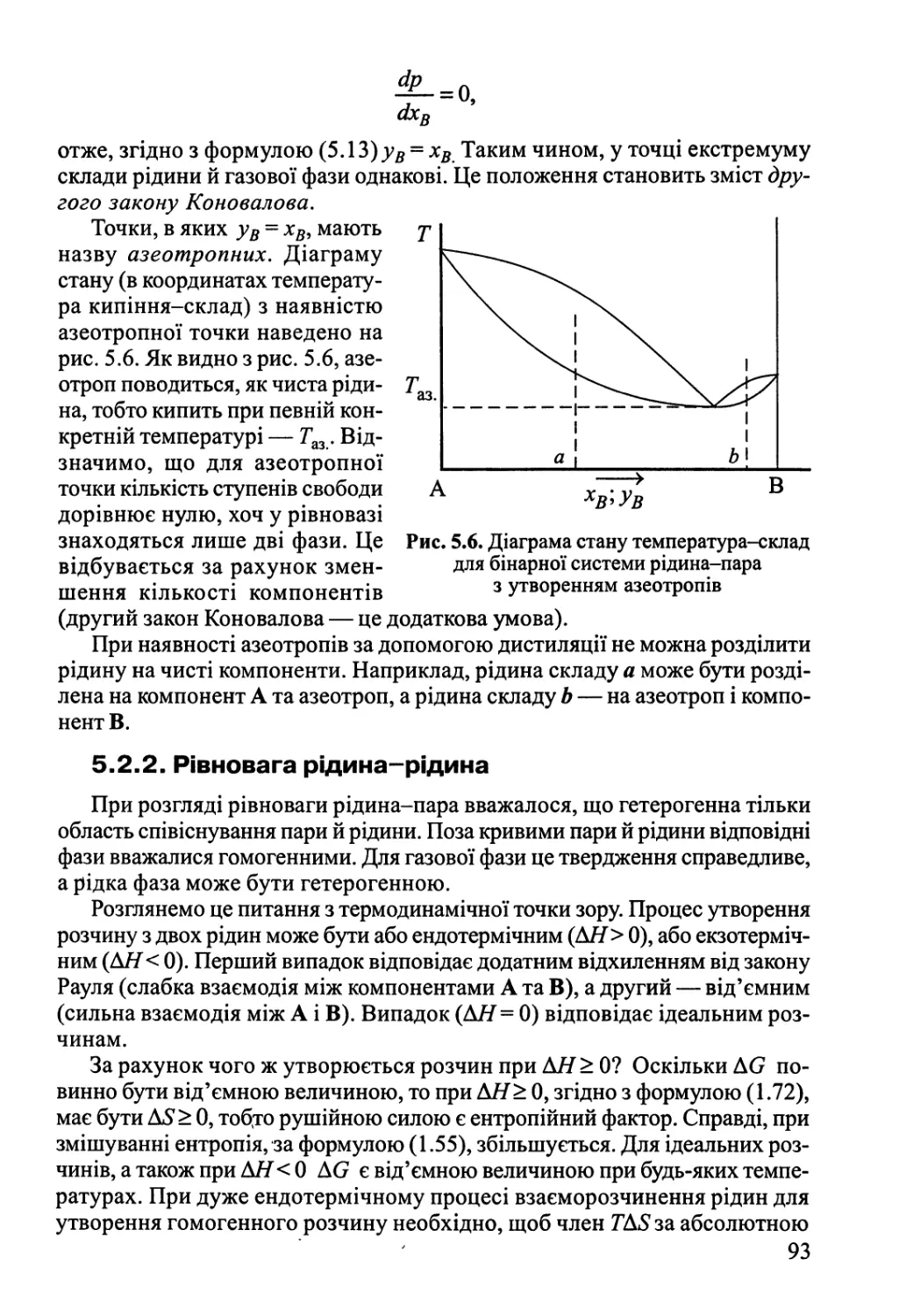
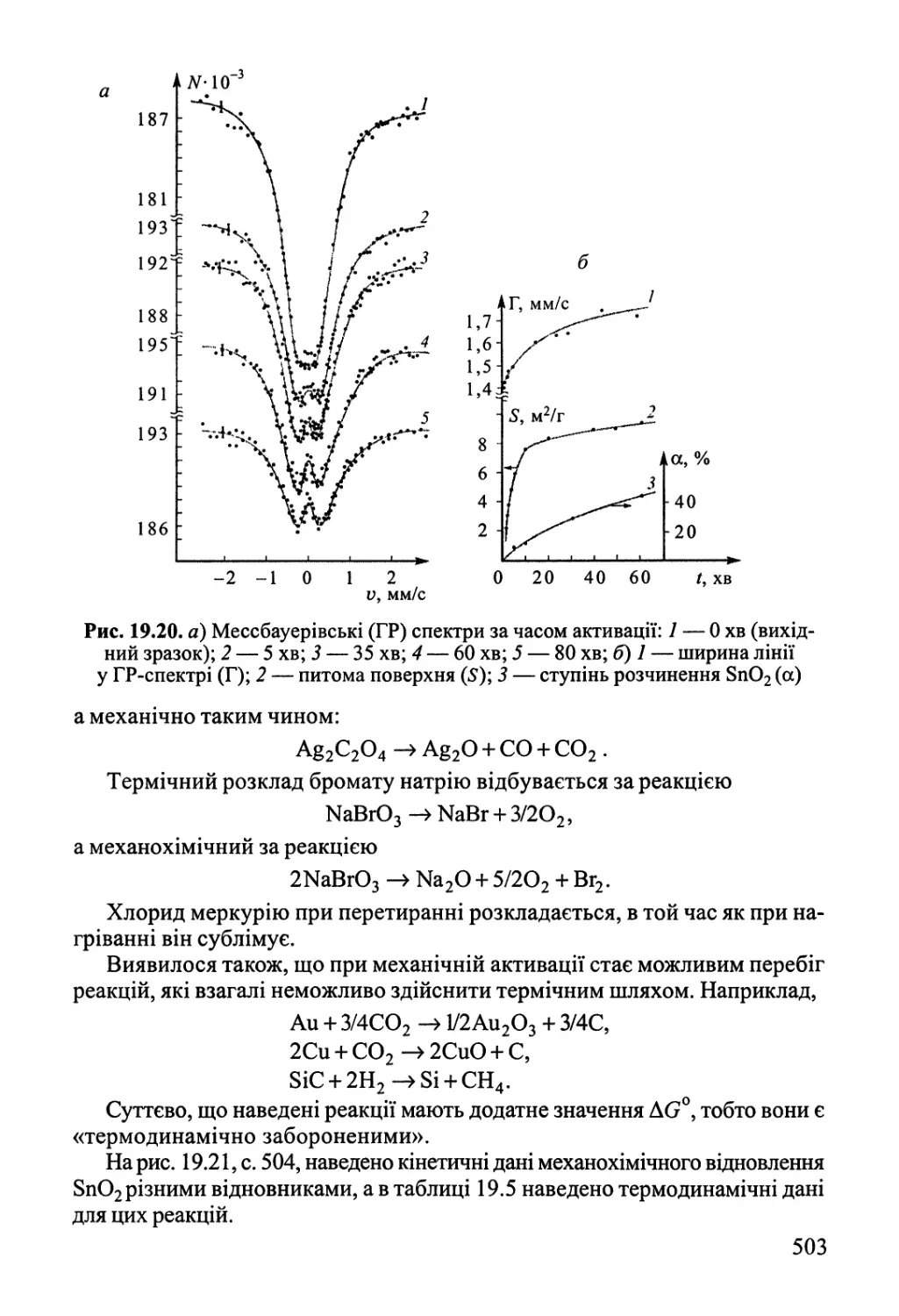
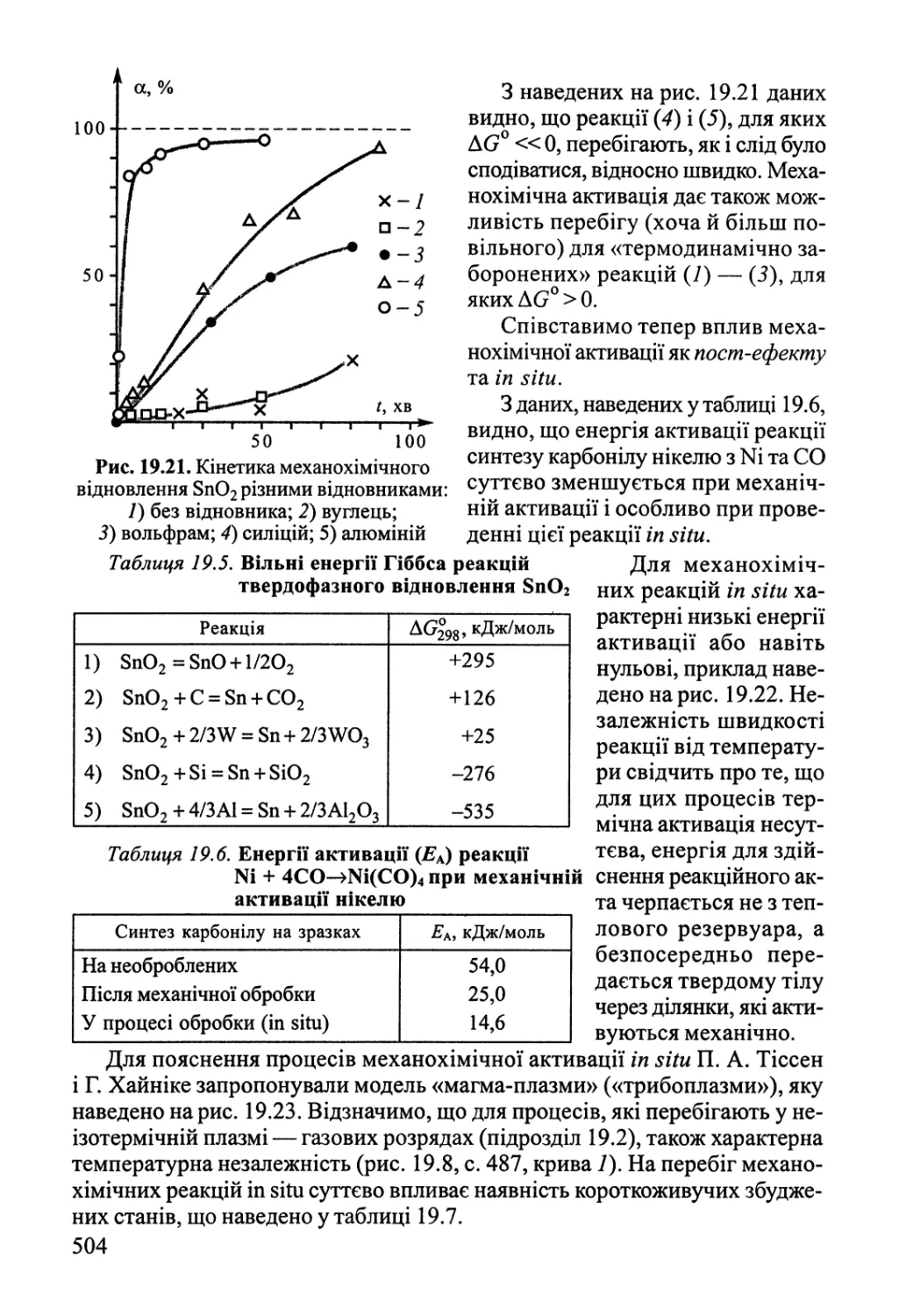
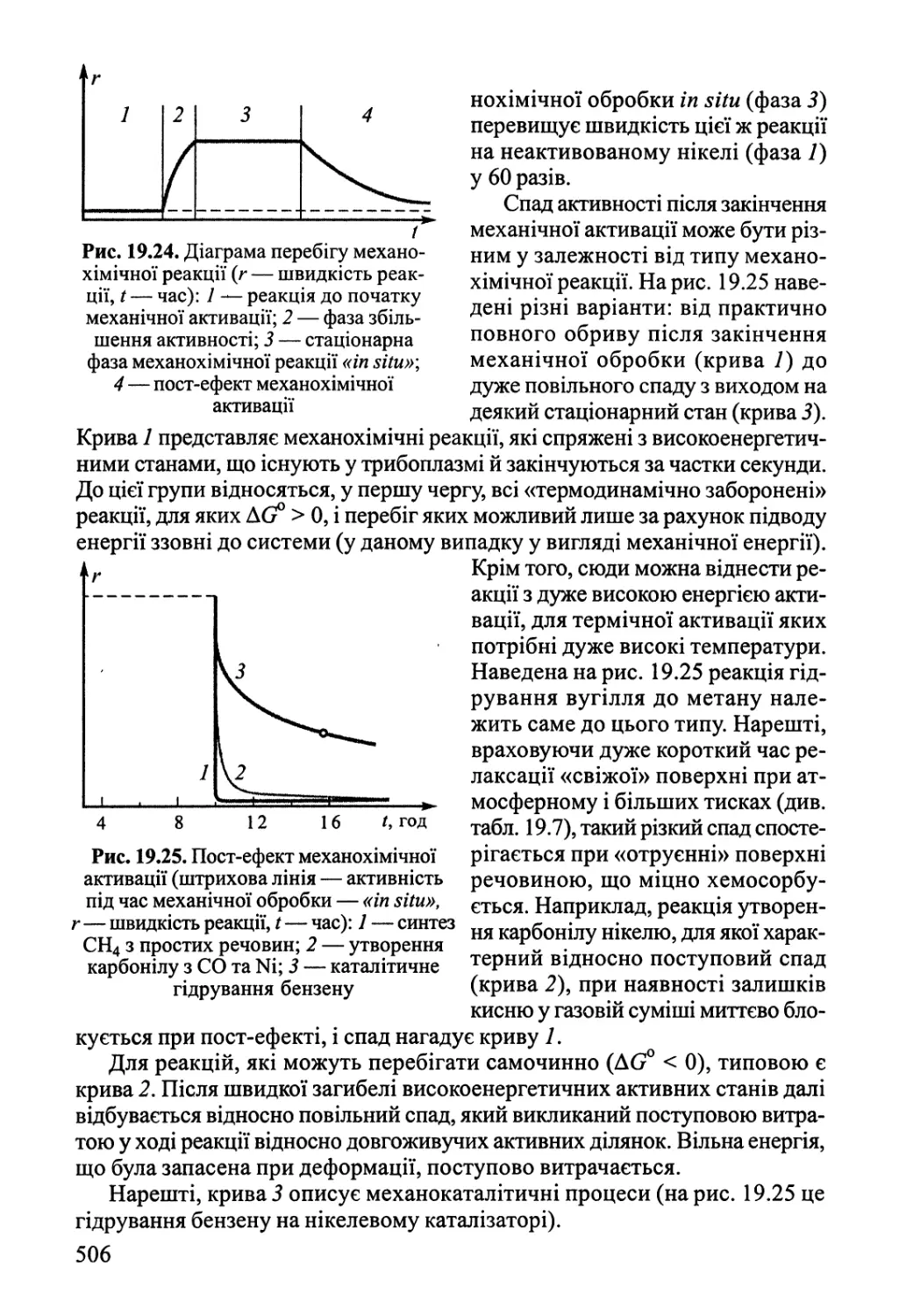
/



















































































































































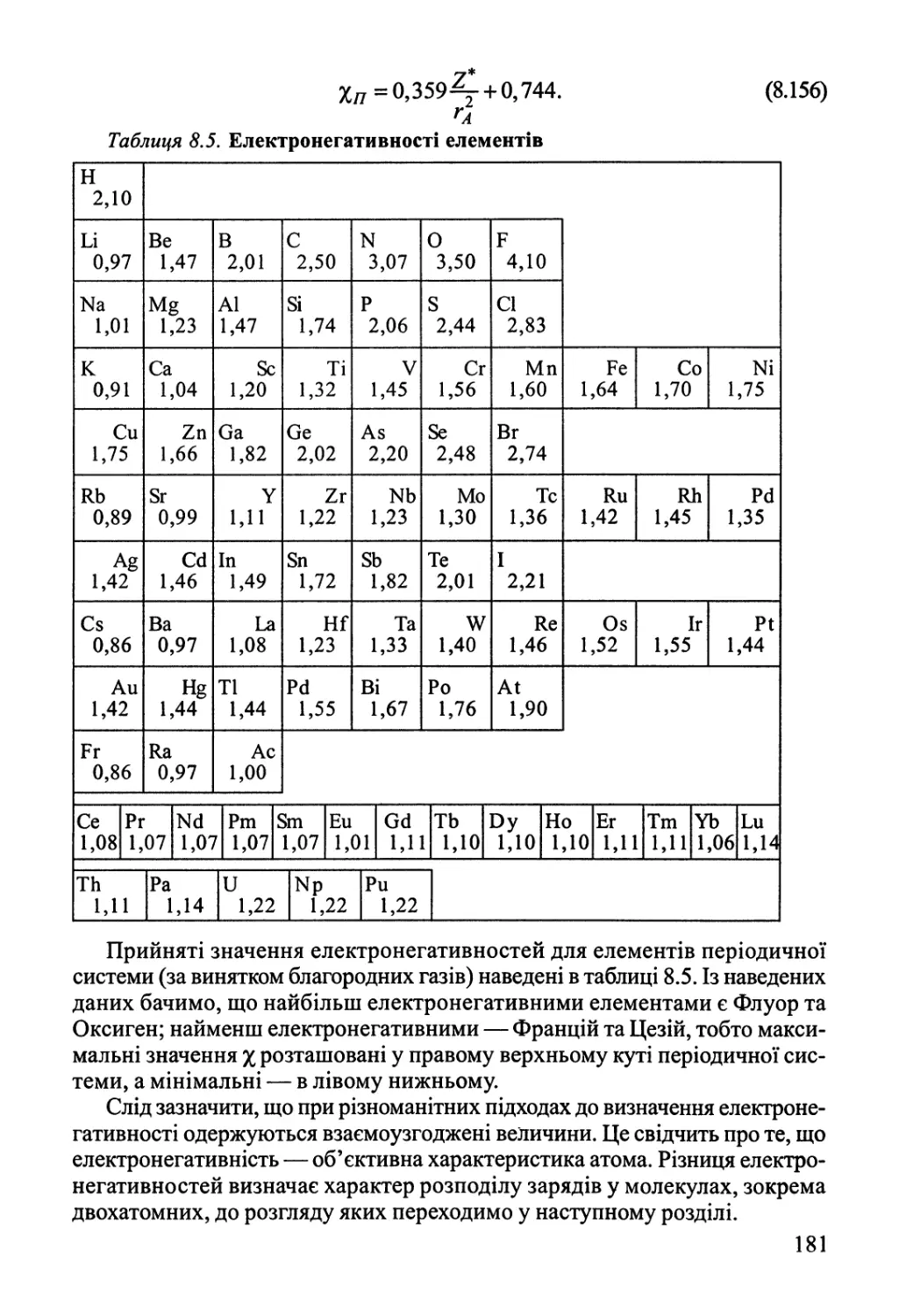









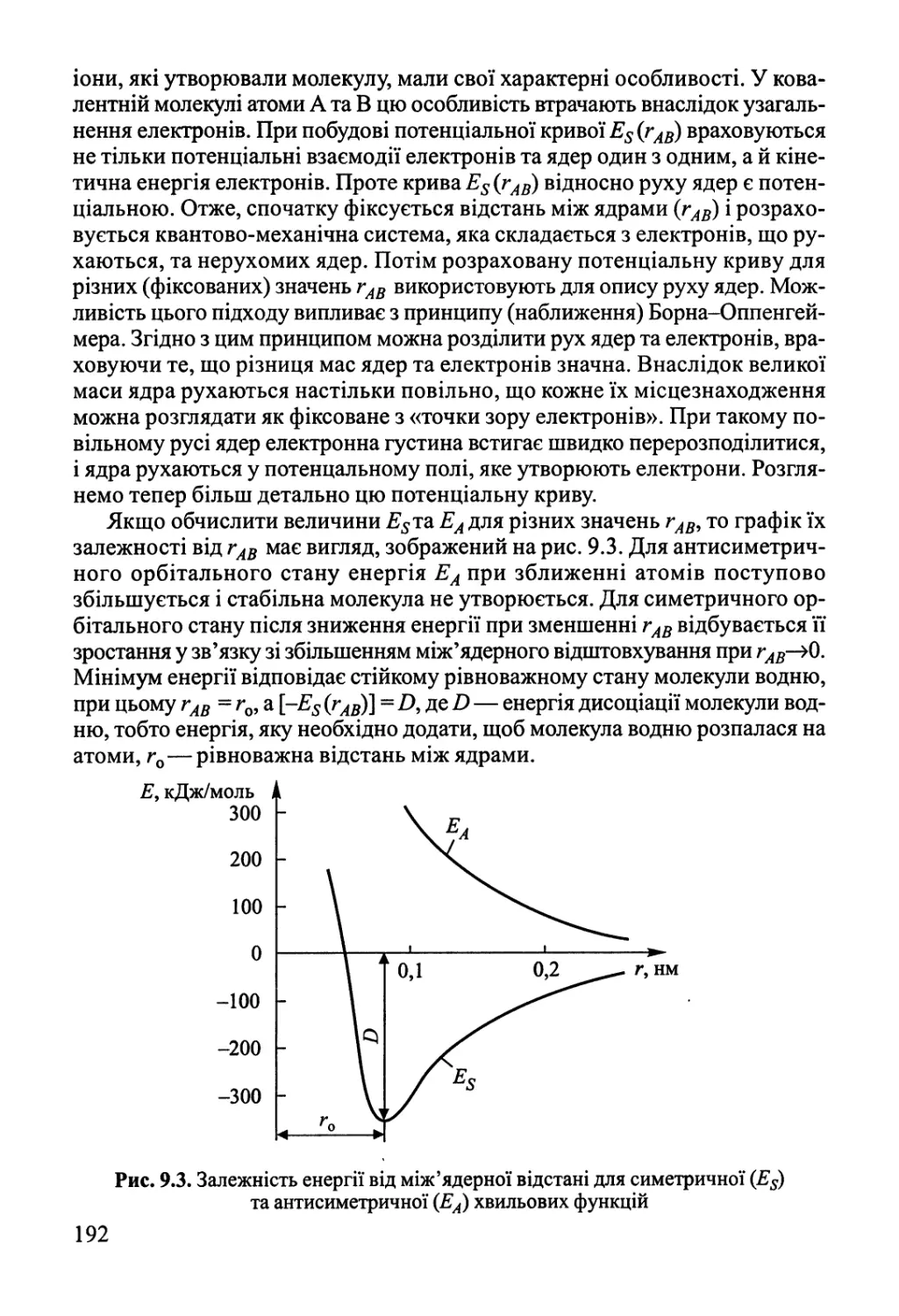
































































































































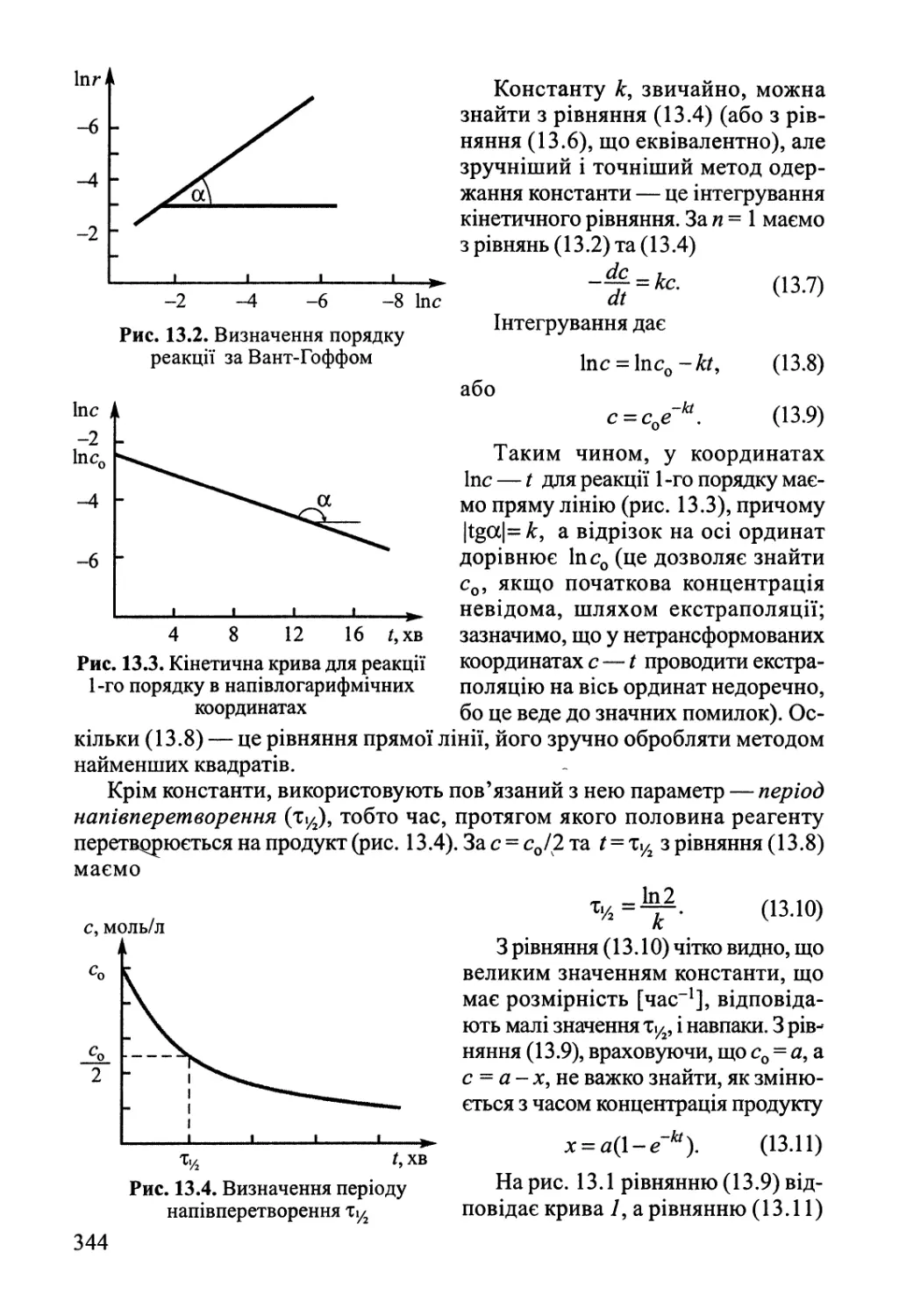


































































































































Author: Яцимирский В.К.
Tags: фізика фізична хімія хімія термодинаміка підручник з хімії видавництво перун
Year: 2007




Text
ЬБК 24 5 я 73
Я 93
Затверджено Міністерством освіти і науки України
(протокол рішення колегії № 14/18 2-1852 від 27.07.2002)
Увага! ©
ЛвіОрські іа видавничі права ВТФ «ІІсрун» захищені Законом України
«Про авторське право і суміжні права»
Яцимирський В. К.
Я 93 Фізична хімія: Підруч. для студ. вищ. навч. закл. К.; Ірпінь:
ВТФ «Псрун», 2007.— 512 с: іл. -
IЗВN 978-966-569-224-9
БІЖ 24.5. я 73
(с) ВТФ «Псруи», 2007
Навчальне видання
Яцимирський Віталій Констянтинович
ФІЗИЧНА ХІМІЯ
Підручник для вищих навчальних закладів
Відповідальна за випуск Т II Боброва
Редактор-коректор Н М. Руденко
Художник обкладинки Є О Іпмшцький
Художік виконання портретів В М Кущ
Комп'ютерна верстка В І Перехрест
Коордипаїорп поліграфічного виконання В Д Ковальчук. Д О Петксаич
Підписано до друку 19 03 07 Формат 60x84/16 Гарнітура Шкільна. Папір офеешпй
Друк офсетнпіі Умов друк арк 29,76 Ум форбовідб 31.15 Обл вид арк 32,5
Зам № 7-43
Видавімічо-торгова фірма «Псрун». 08200 Ірпінь, вул. Київська, 73-а.
Свідоцпю про внесення до державного реєстру- серія ДК № 18 від 20.03 2000 р
Віддруковано у ТОВ «Друкарня «Бізнесполіграф»
02094, м Київ, вул Віскозна, 8
Свідоцтво про внесення до дсржаїиіого рссстру № 1000 17705 іид йод № 334051926593
Видано комі і ом видавництва ВТФ «Псрун».
Видрукувано в Україні Ргіпгесі іп ІЛсгаїпе
ЗМІСТ
Передмова 5
Розділ 1. Основи термодинаміки 7
1.1. Предмет термодинаміки 7
1.2. Перше начало термодинаміки 10
1.3. Термохімія 15
1.4. Ентропія 20
1.5. Вільна енергія 27
1.6. Багатокомпонентні системи. Парціальні мольні величини 31
1.7. Умови рівноваги. Напрям процесів. Друге начало термодинаміки 34
Розділ 2. Однокомпонентні системи 41
2.1. Термодинаміка фазових переходів у однокомпонентних системах 41
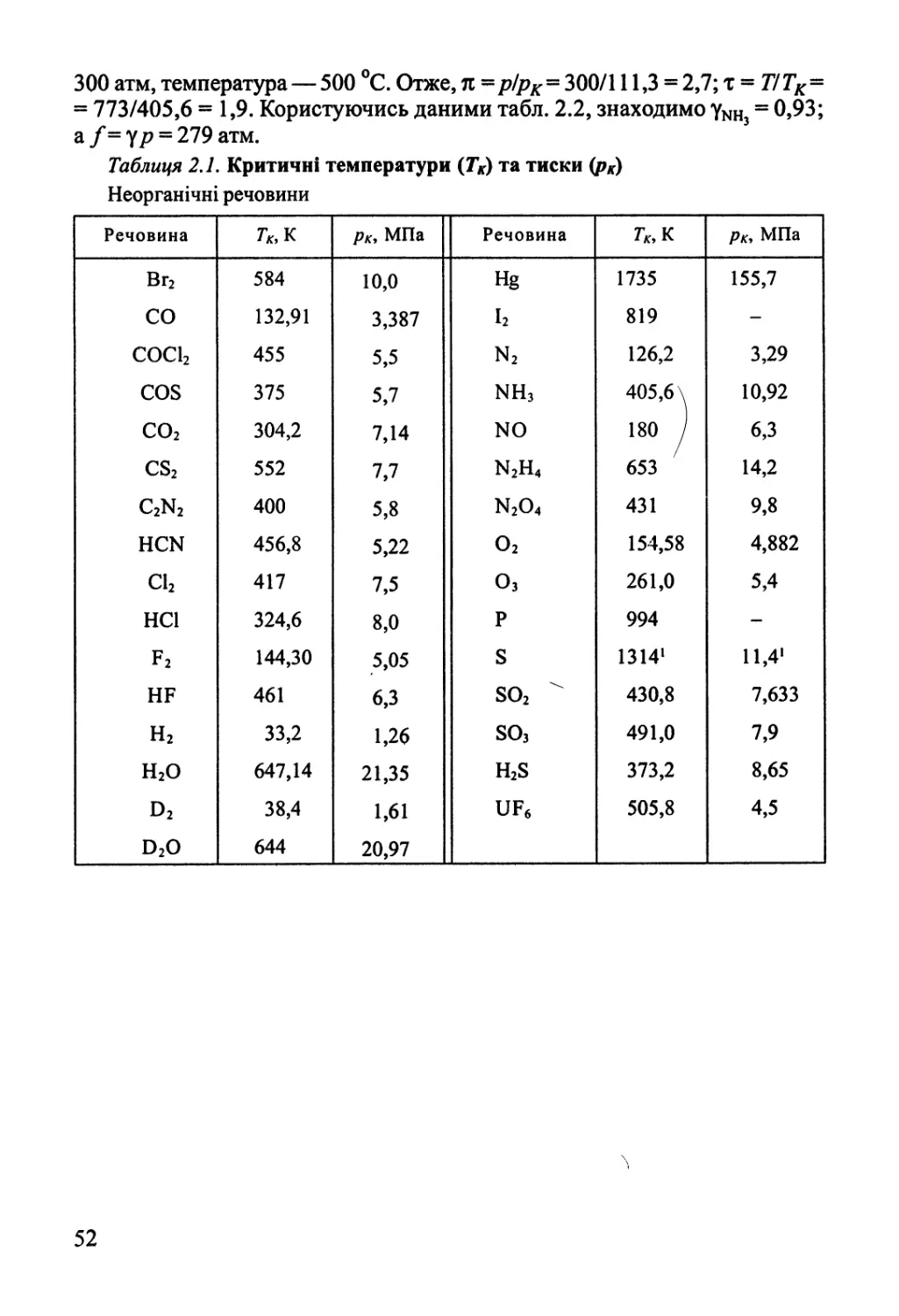
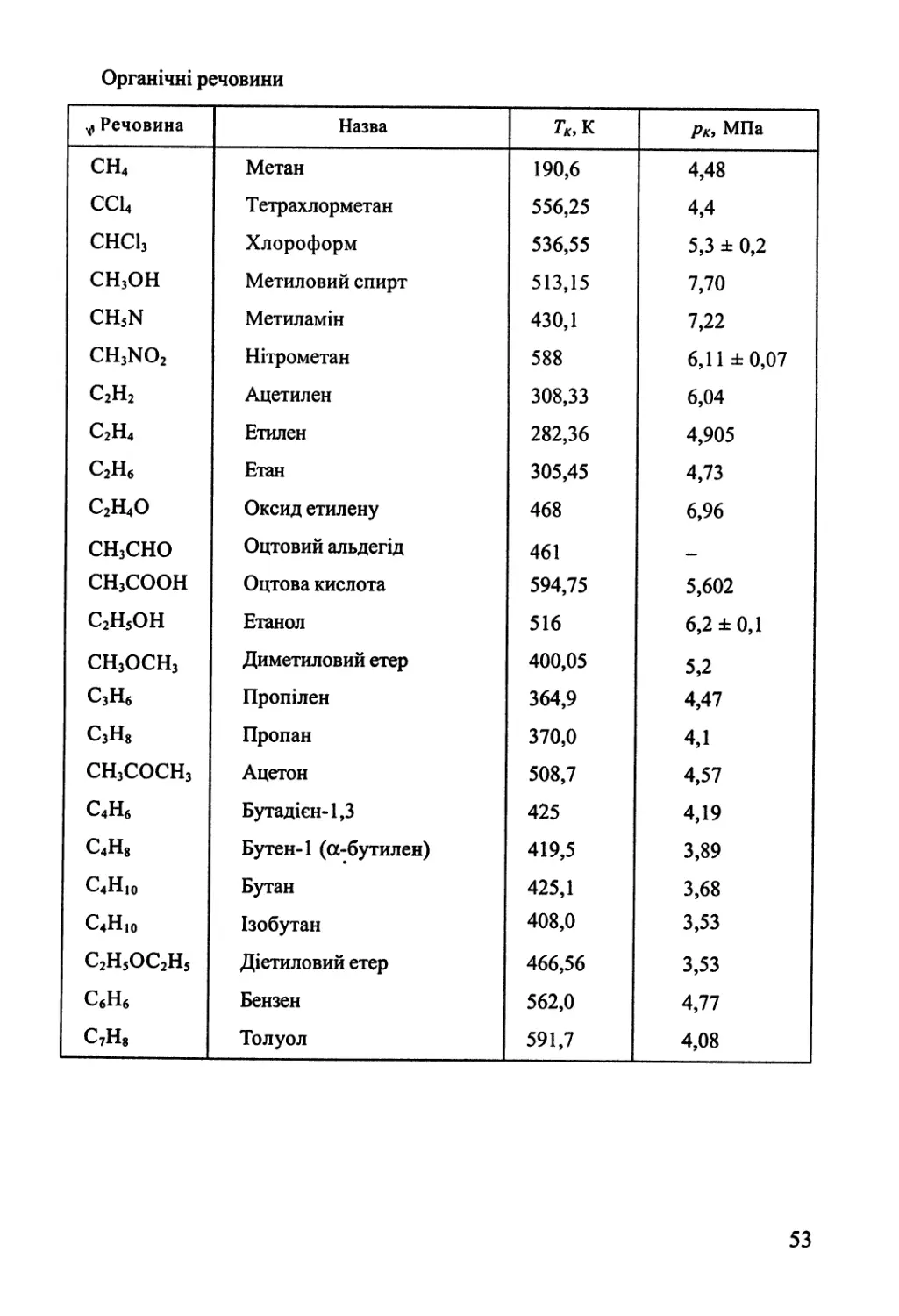
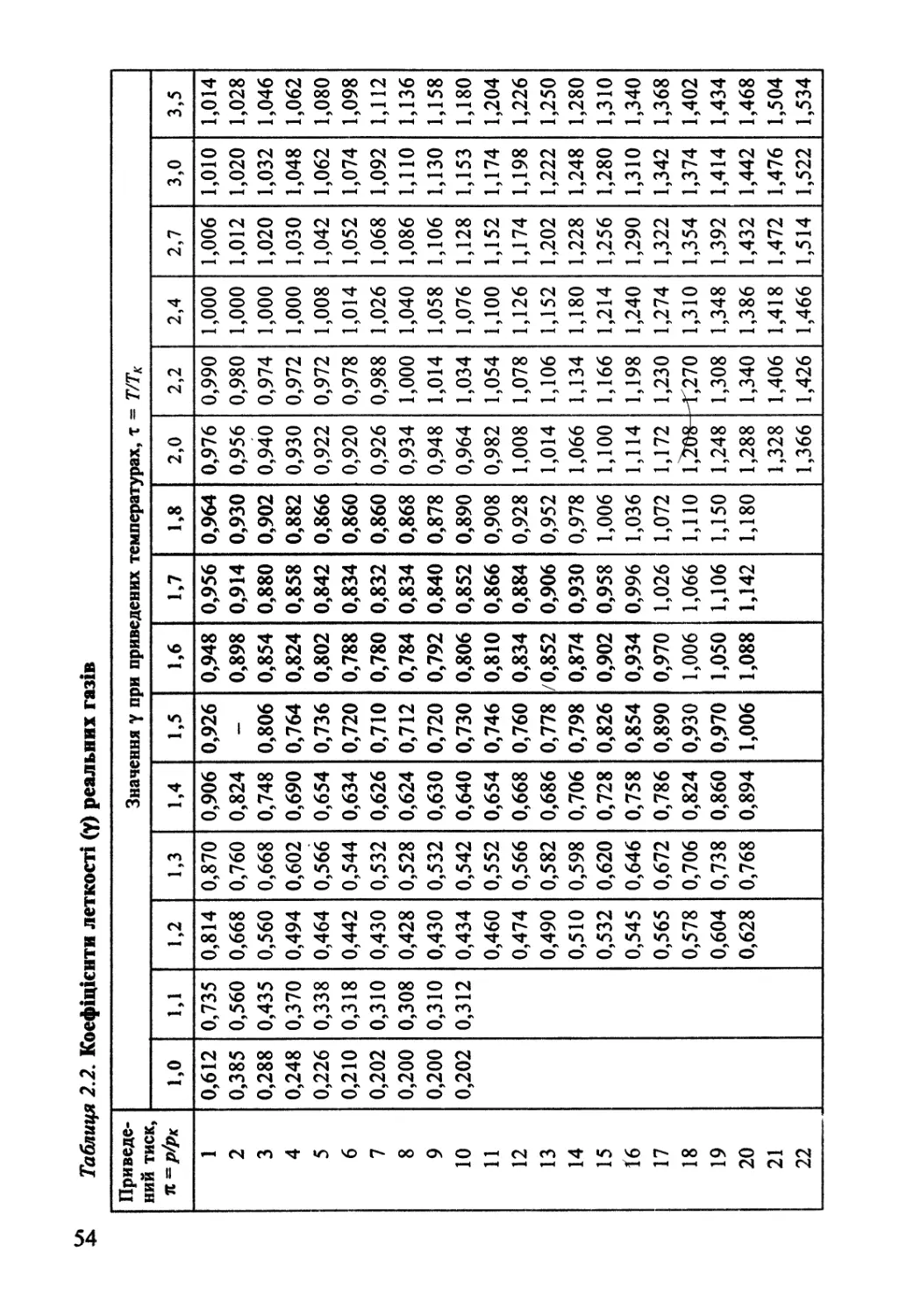
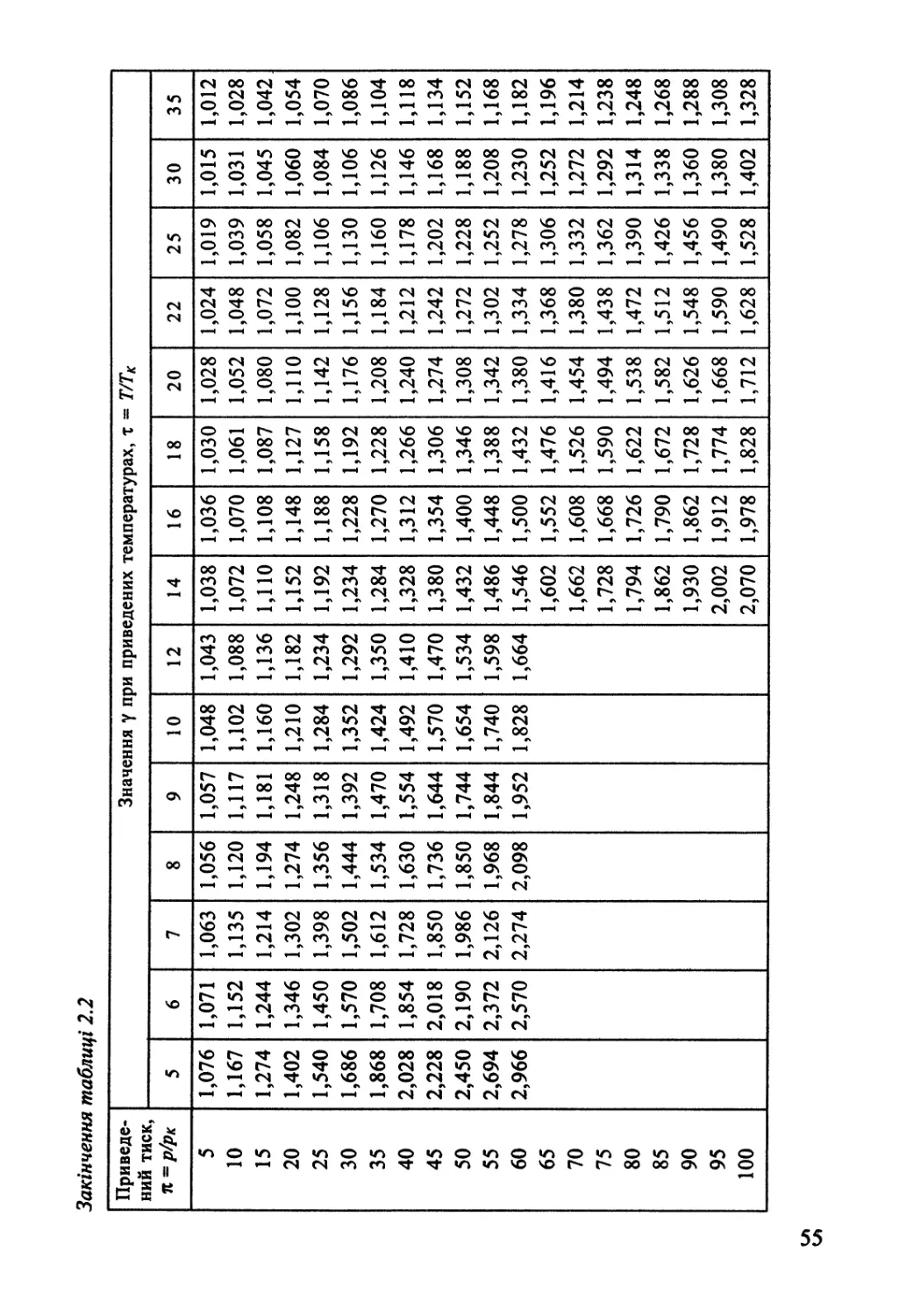
2.2. Реальні гази. Леткість 45
2.3. Термодинаміка конденсованого стану однокомпонентних систем 56
Розділ 3. Розчини неелектролітів 59
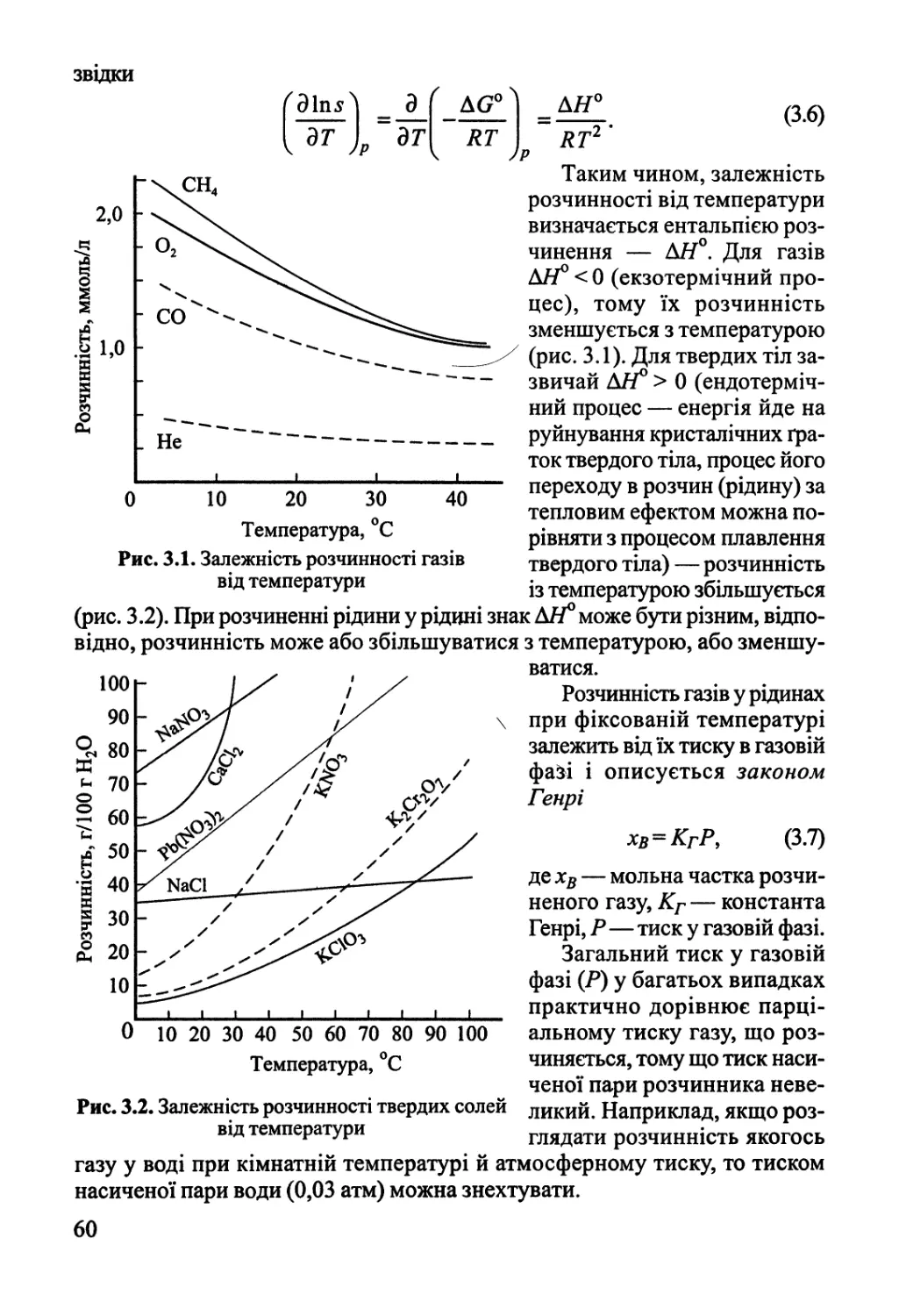
3.1. Закони Генрі та Рауля. Розчинність 59
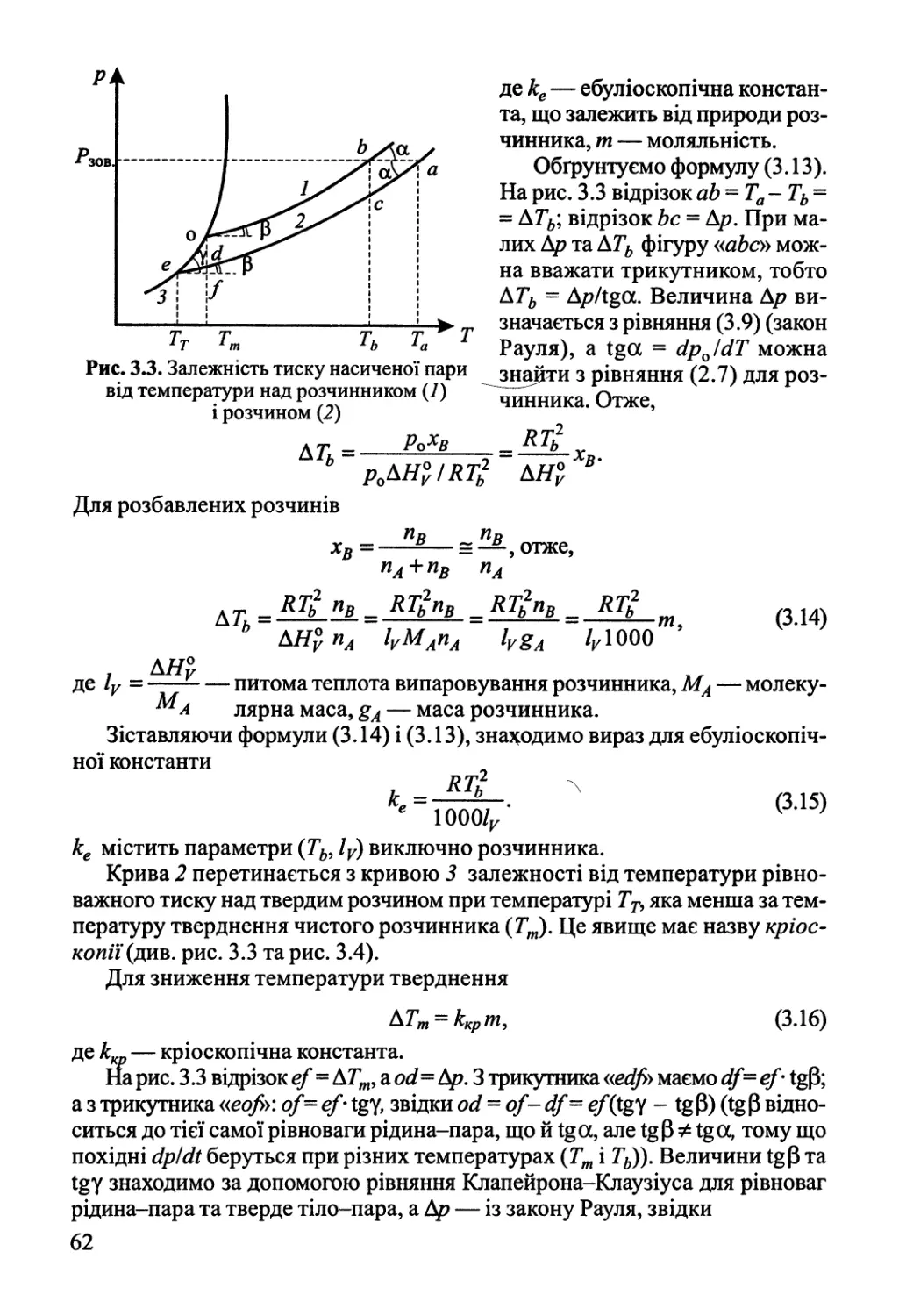
3.2. Ебуліоскопія, кріоскопія, осмос 61
3.3. Активності у розчинах неелектролітів 64
Розділ 4. Розчини електролітів 68
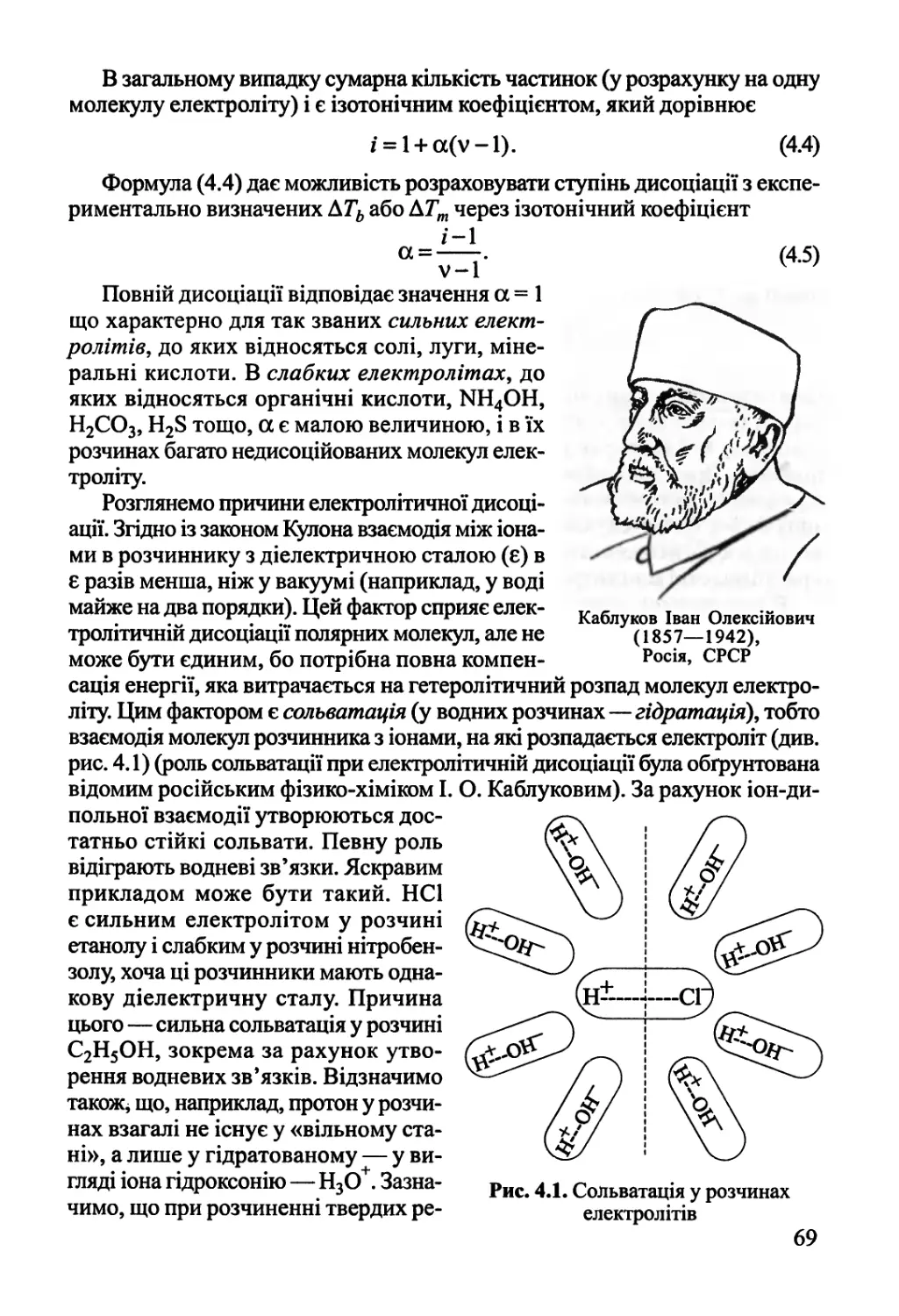
4.1. Електролітична дисоціація 68
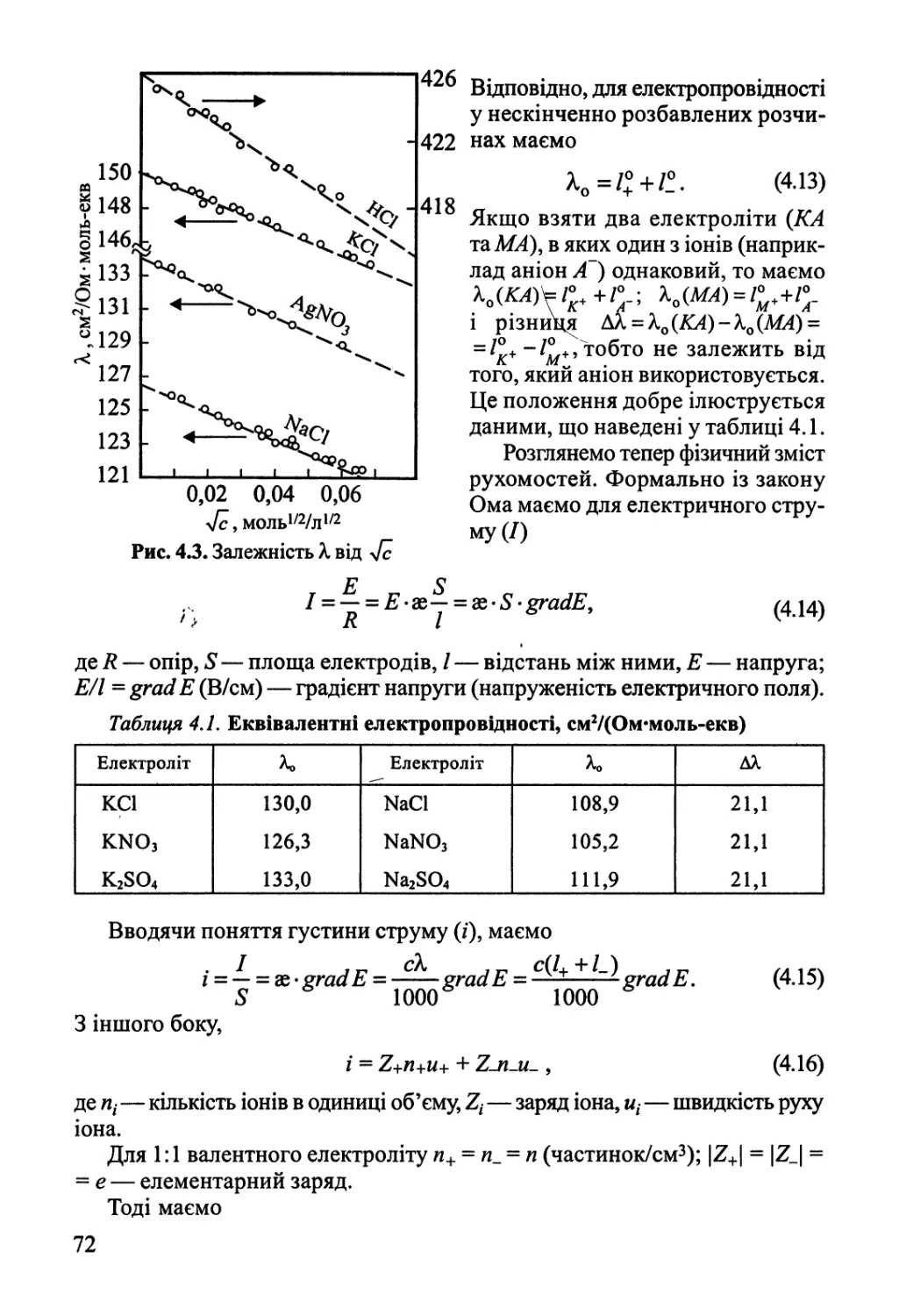
4.2. Електропровідність розчинів електролітів 70
4.3. Основи теорії Дебая-Гюккеля 76
4.4. Електропровідність сильних електролітів 81
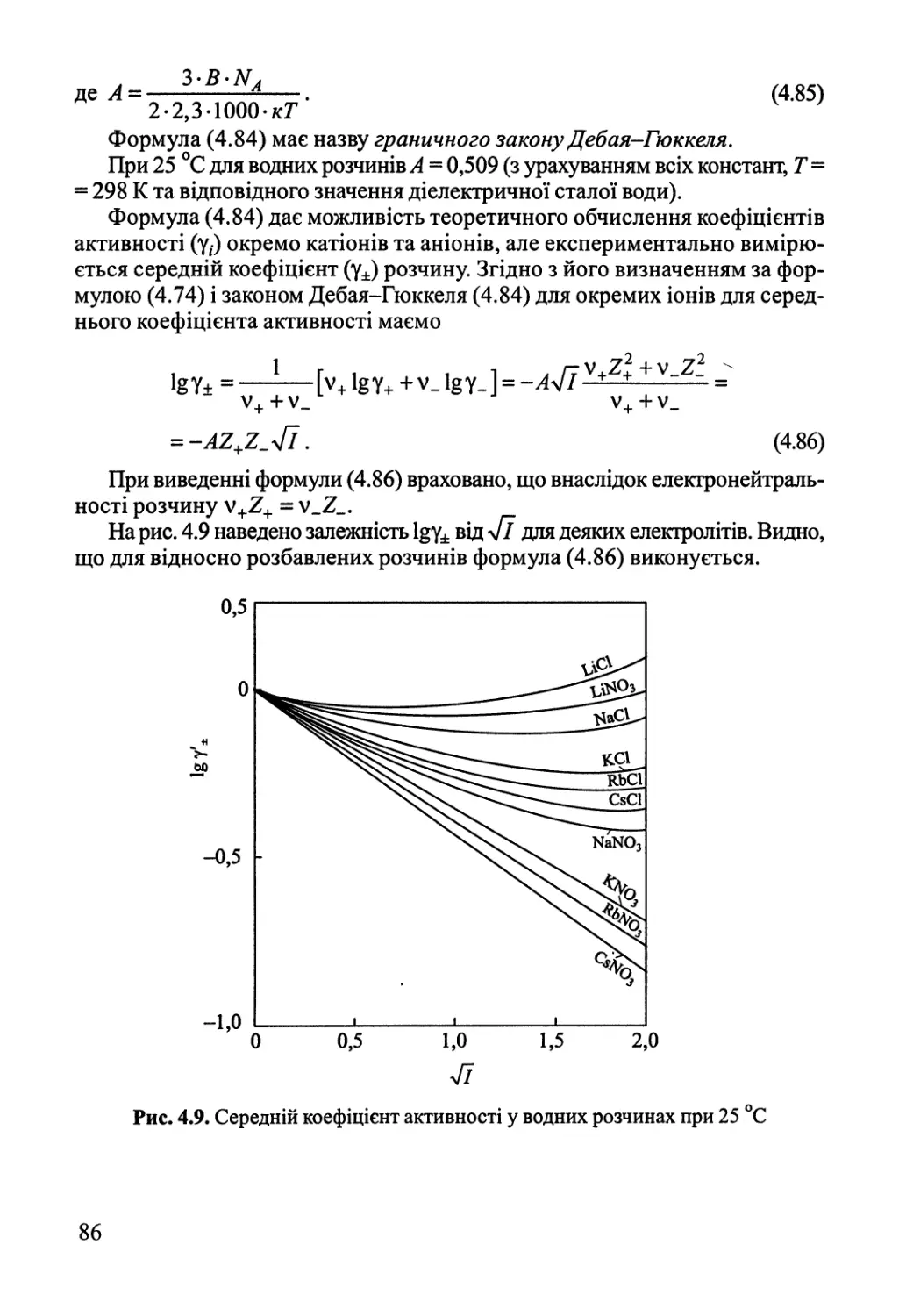
4.5. Активності у розчинах електролітів 84
Розділ 5. Фазові рівноваги у багатокомпонентних системах 87
5.1. Правило фаз Гіббса 87
5.2. Двокомпонентні системи 88
5.2.1. Рівновага рідина-пара 88
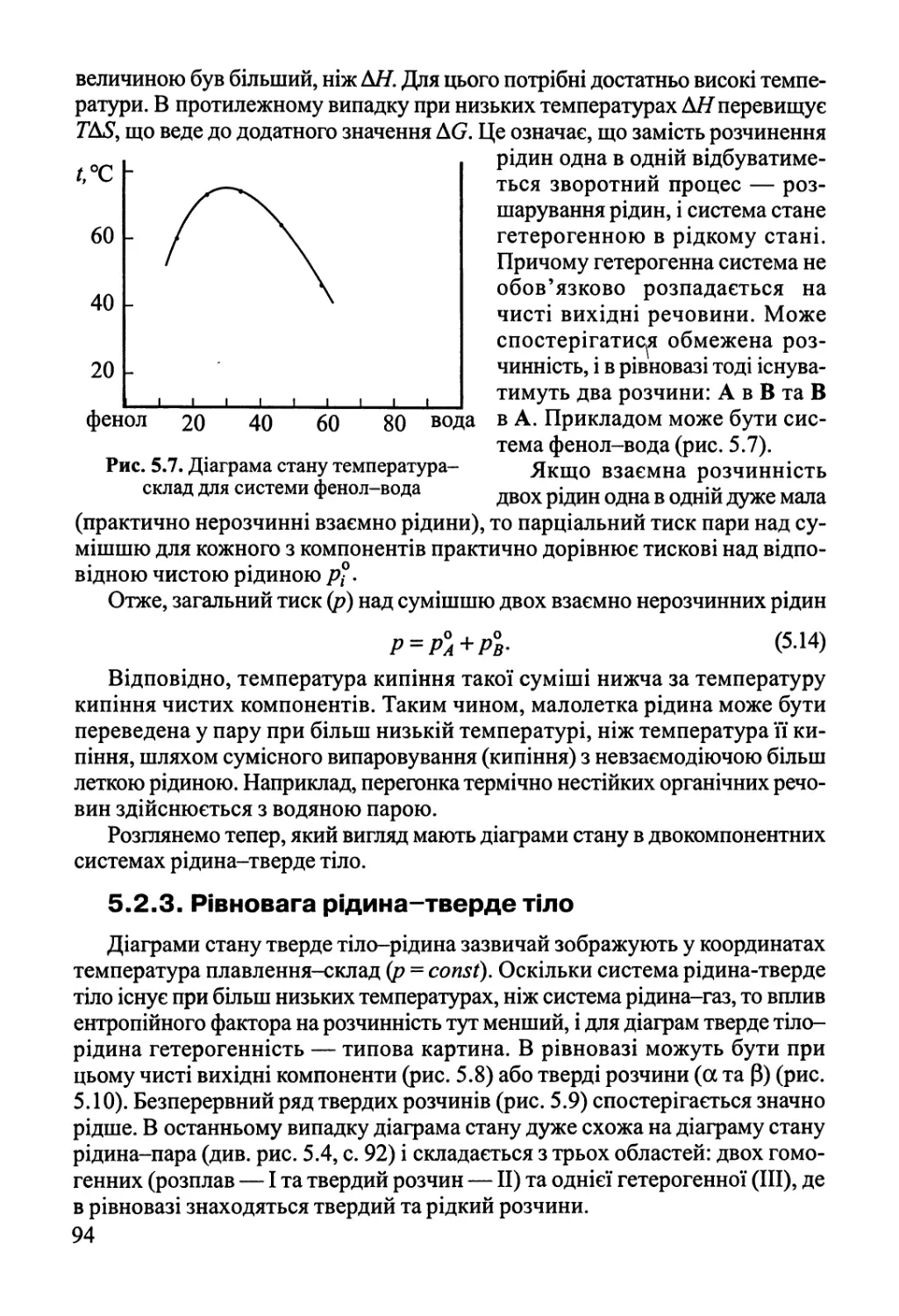
5.2.2. Рівновага рідина-рідина 93
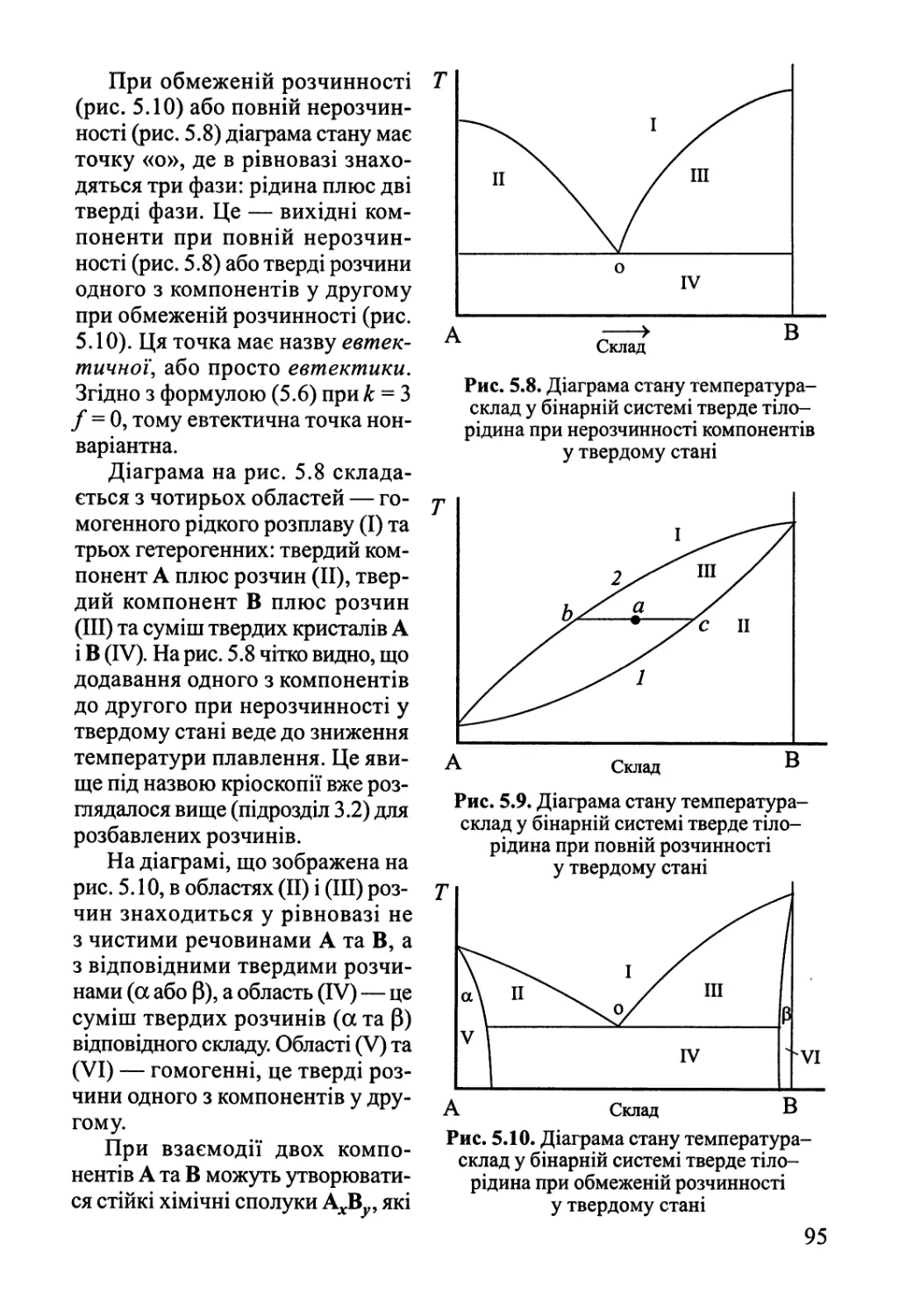
5.2.3. Рівновага рідина-тверде тіло 94
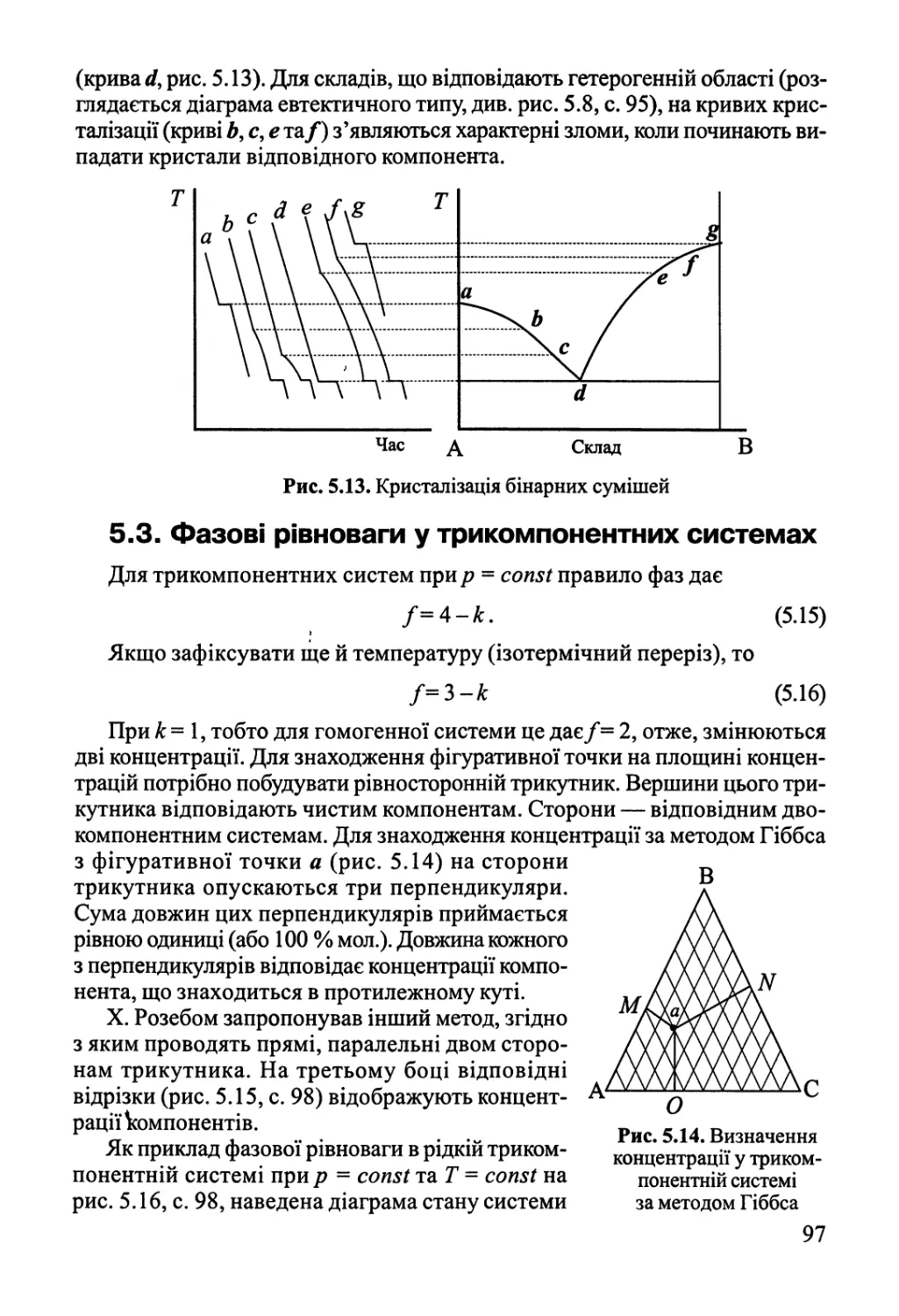
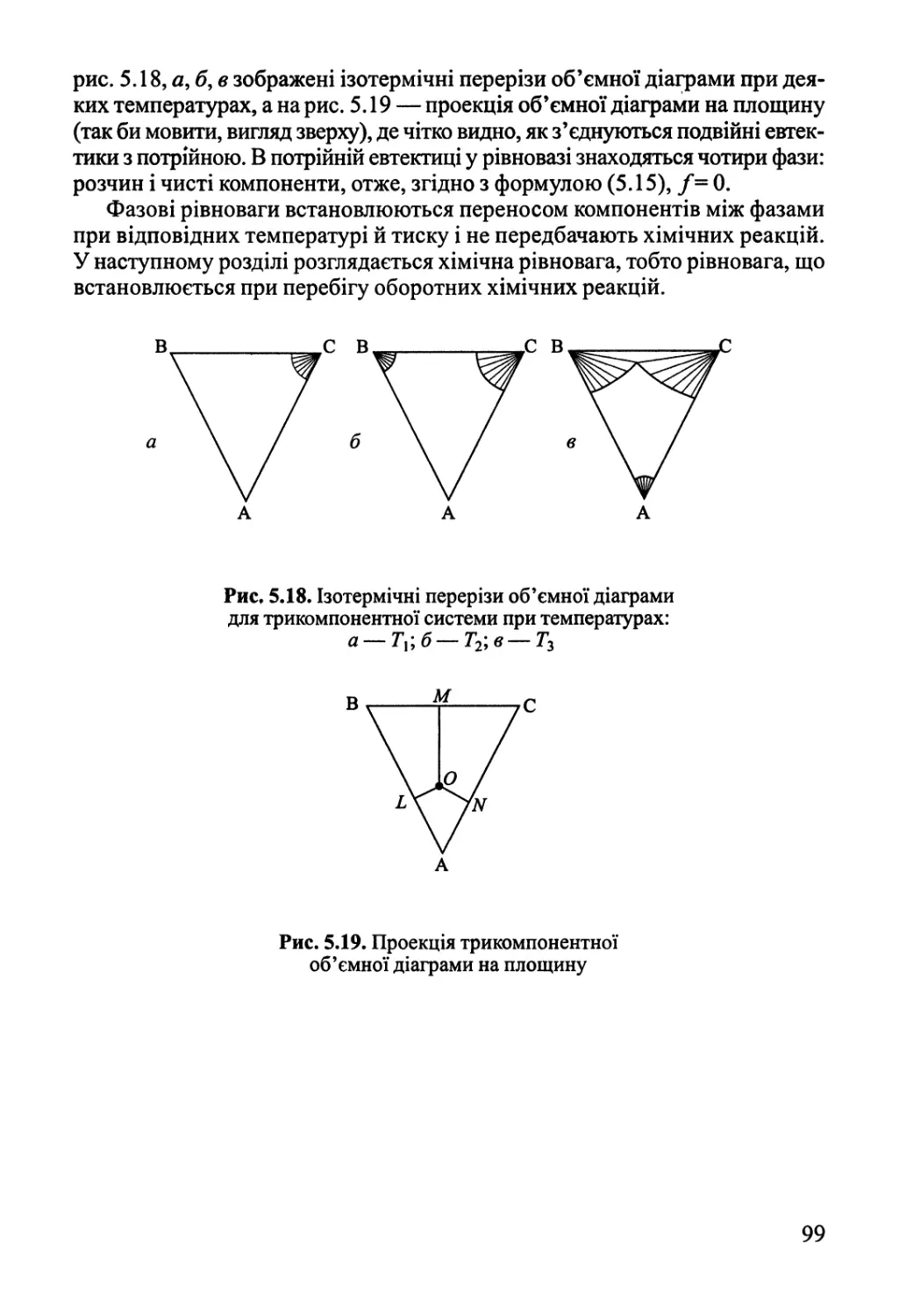
5.3. Фазові рівноваги у трикомпонентних системах 97
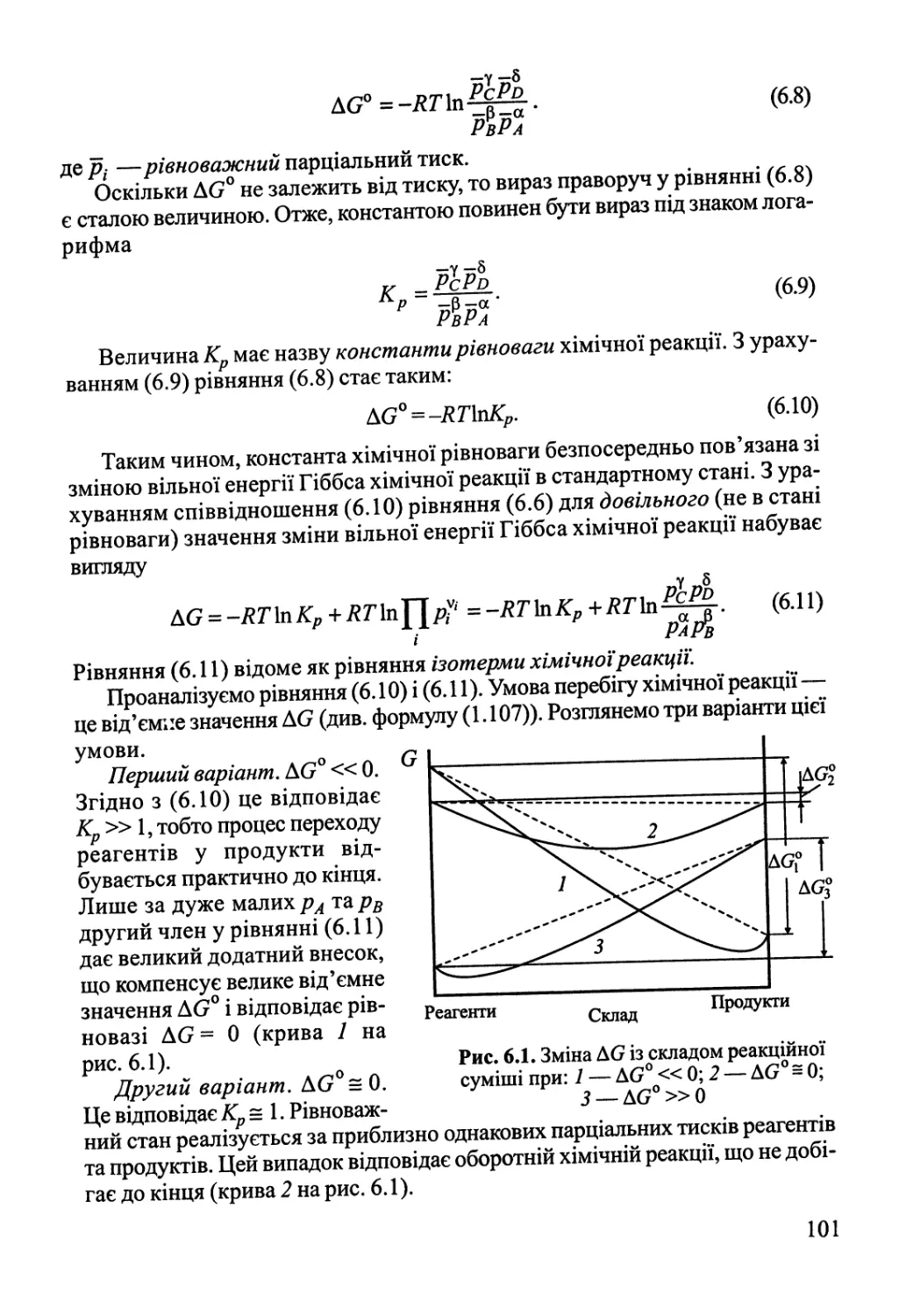
Розділ 6. Хімічна рівновага 100
6.1. Хімічна рівновага у гомогенних системах 100
6.2. Вплив зовнішніх факторів на хімічну рівновагу.
Принцип Ле Шательє 105
6.3. Хімічна рівновага у розчинах. Гетерогенна хімічна рівновага 106
6.4. Обчислення константи рівноваги за термохімічними даними 107
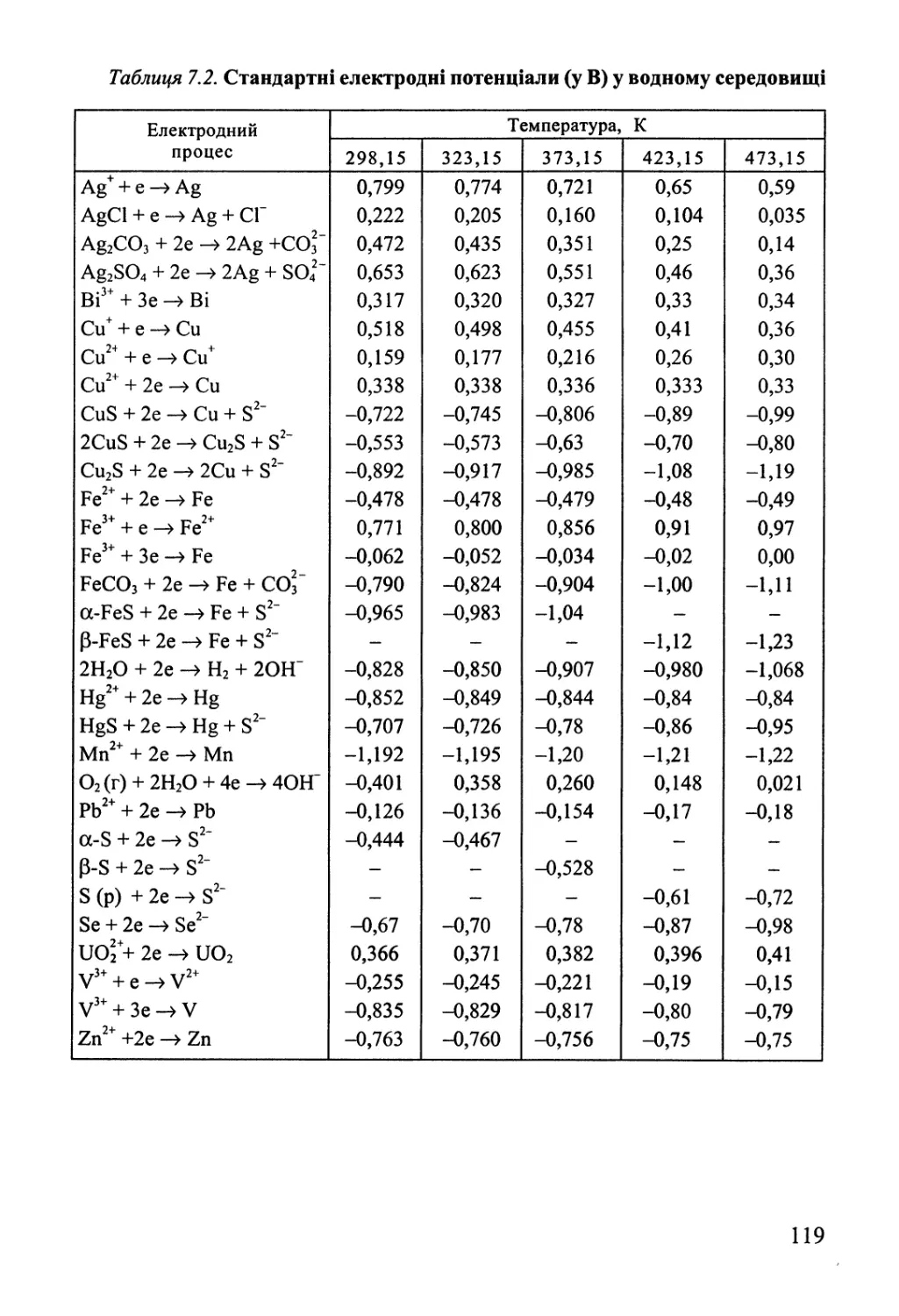
Розділ 7. Електрохімічна рівновага 112
7.1. Термодинаміка гальванічного елемента 112
7.2. Електроди та їх потенціали 117
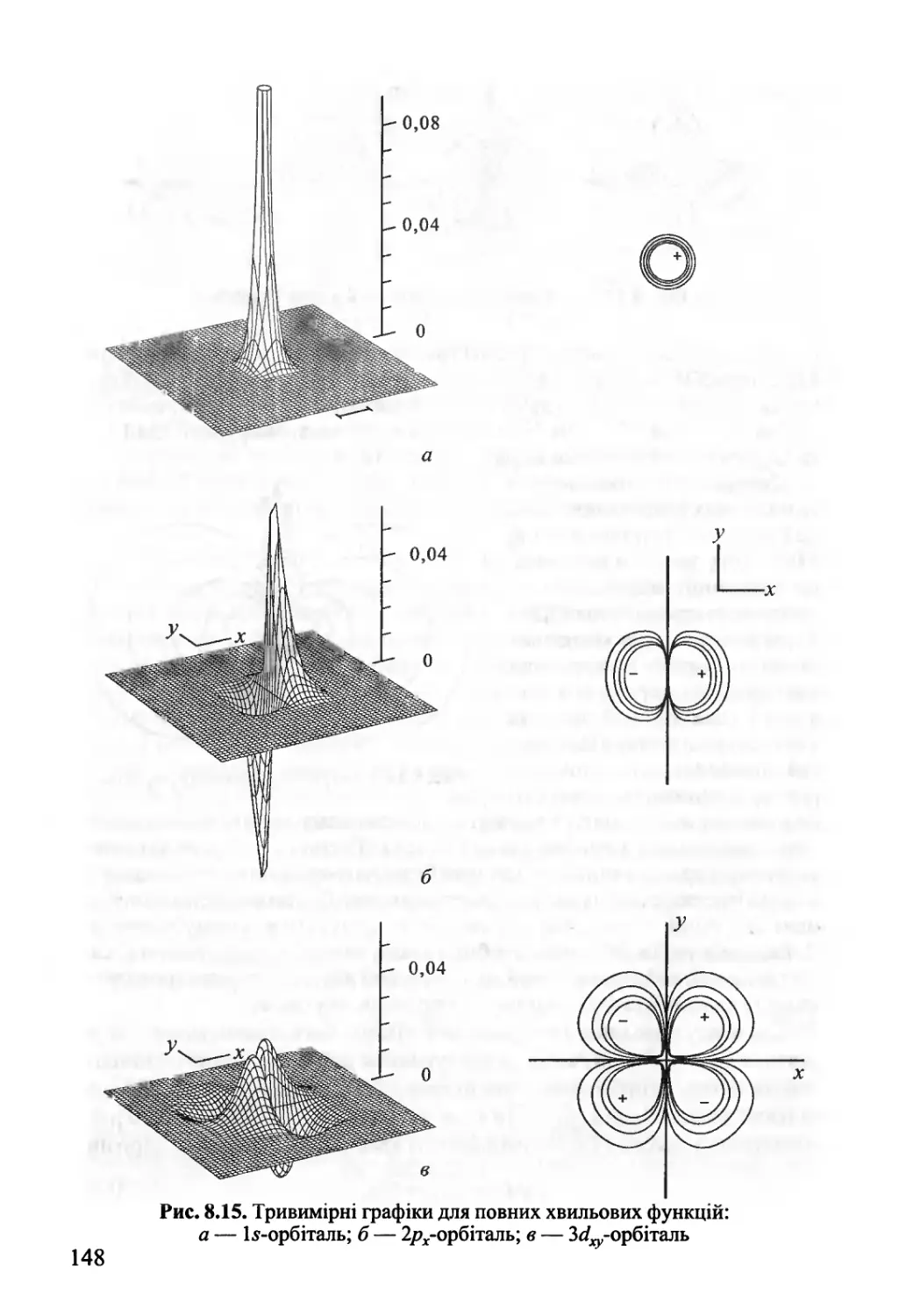
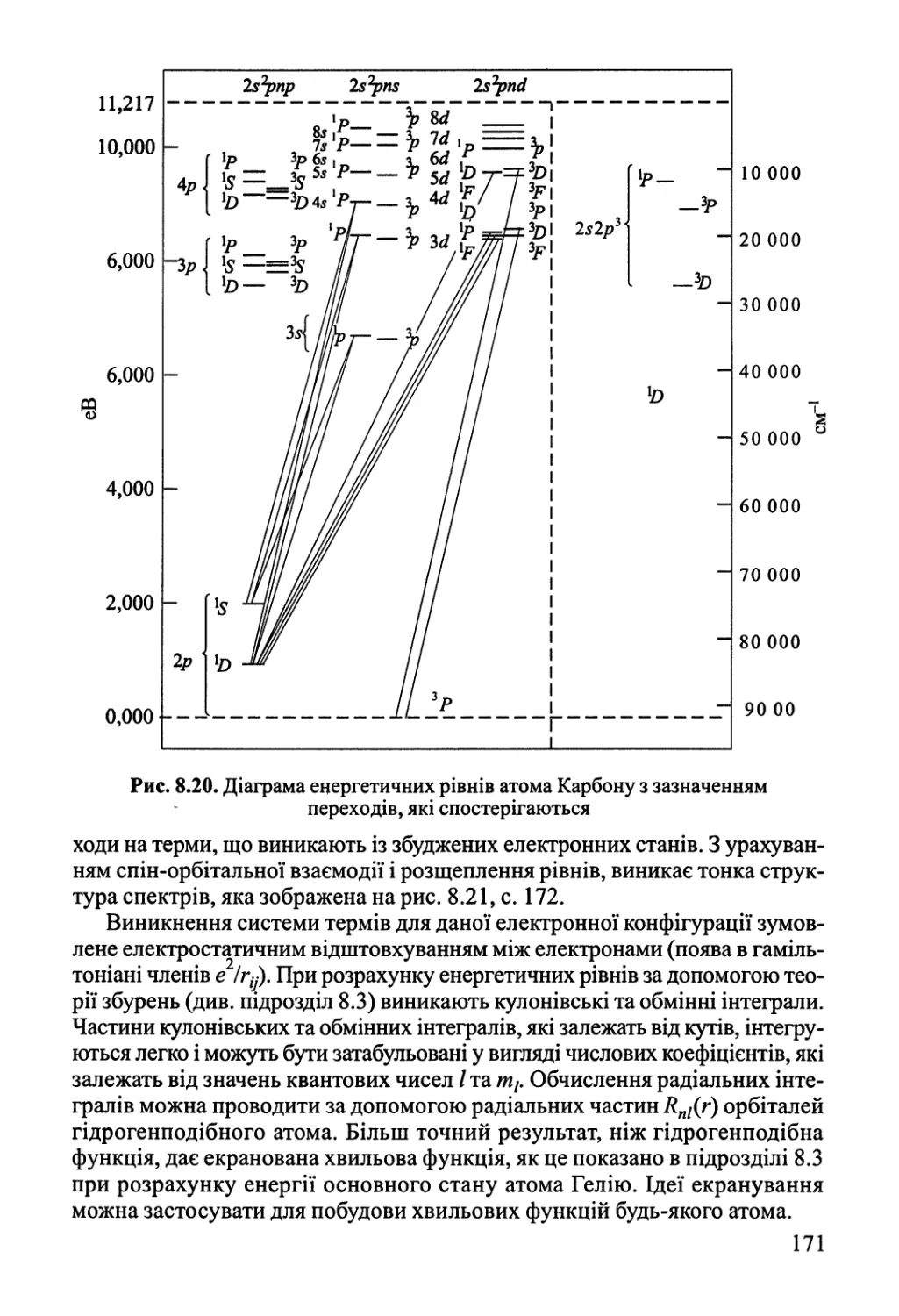
Розділ 8. Квантова теорія, будова атома 124
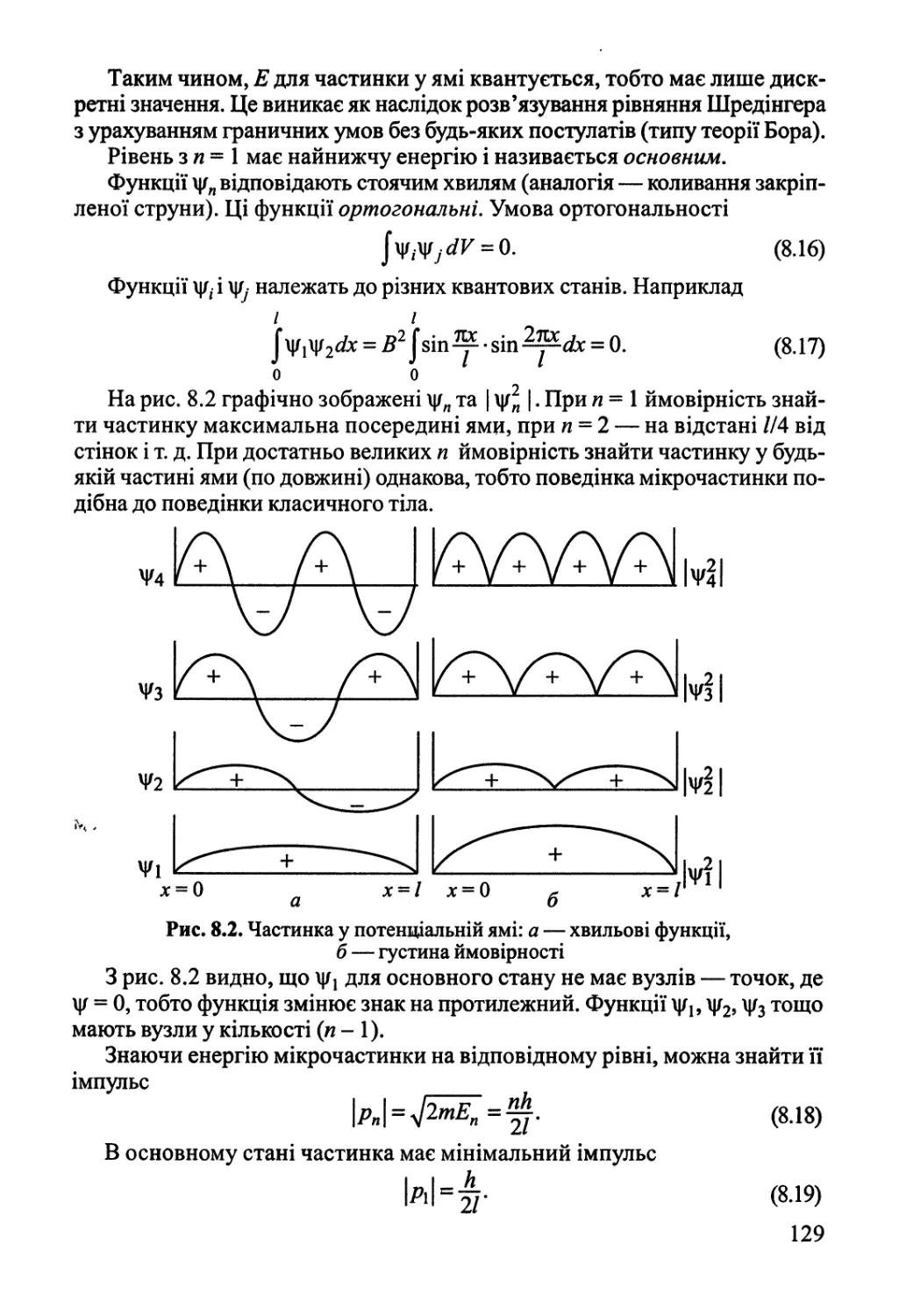
8.1. Основні положення квантової теорії 124
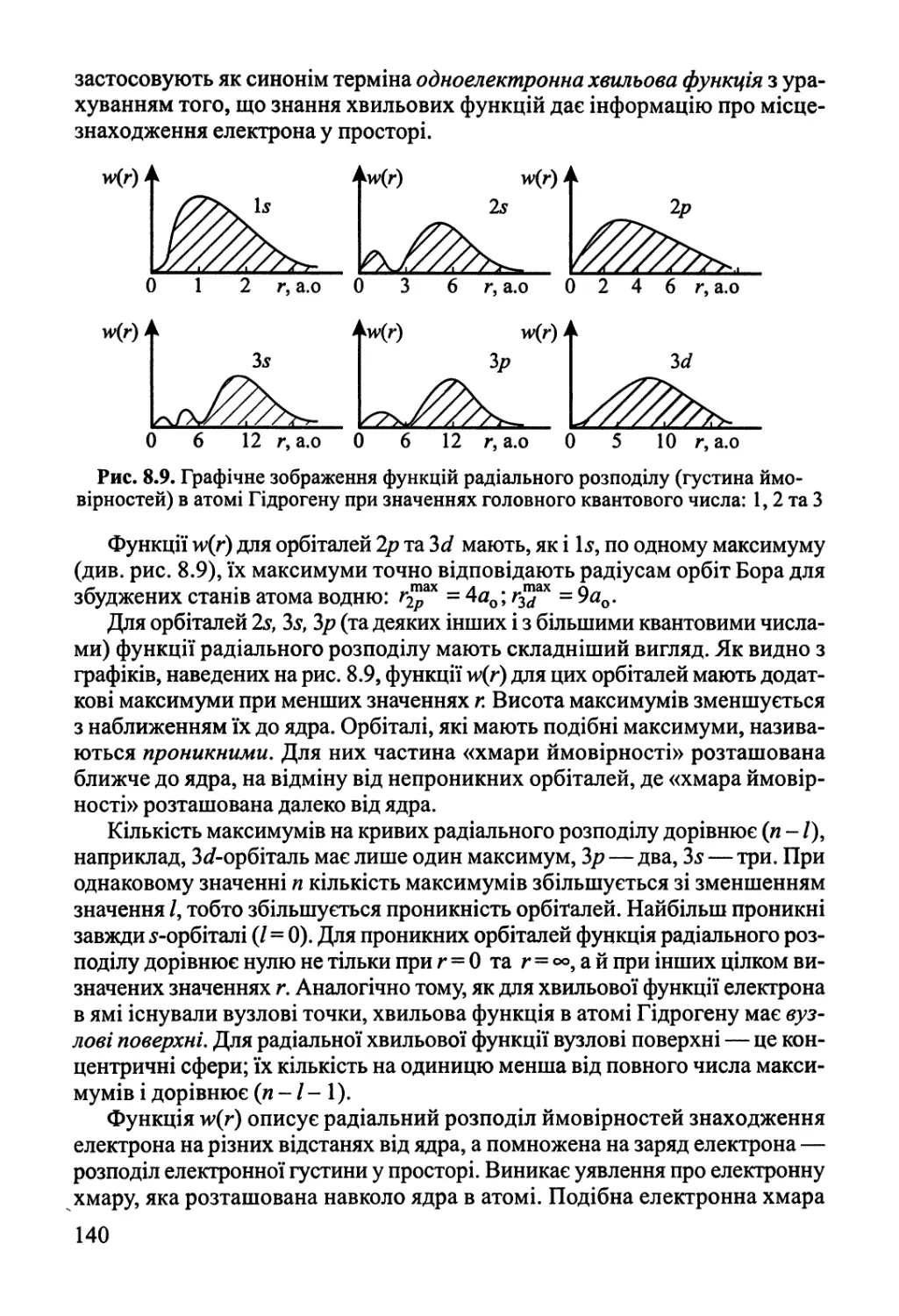
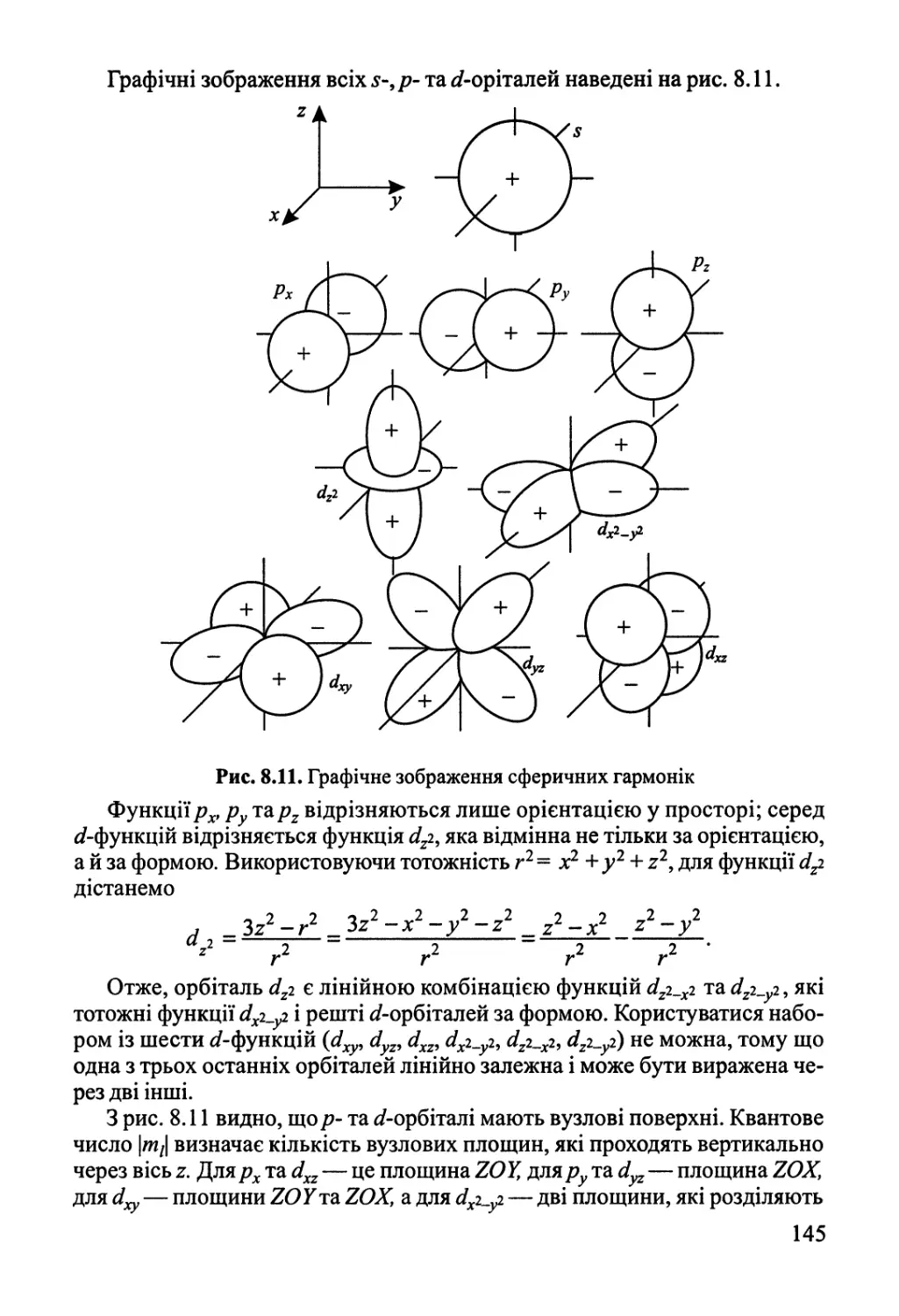
8.2. Одноелектронна система — атом Гідрогену 135
8.3. Двохелектронна система — атом Гелію 149
8.4. Багатоелектронні атоми 163
Розділ 9. Будова молекул 182
9.1. Двохатомні молекули 182
9.2. Багатоатомні молекули з локалізованими зв'язками 219
9.3. Молекули з делокалізованими зв'язками 245
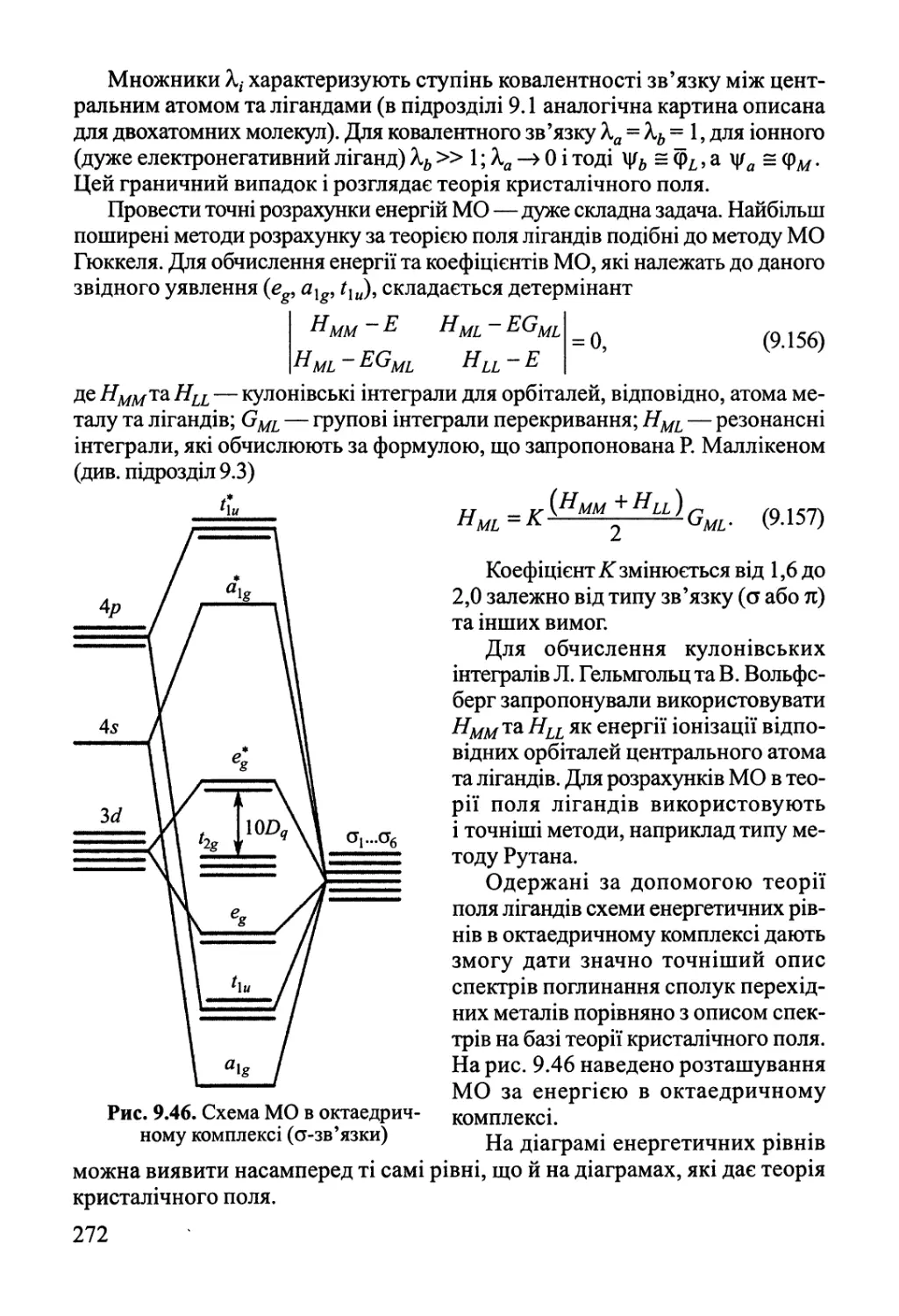
9.4. Електронна будова та властивості координаційних сполук 262
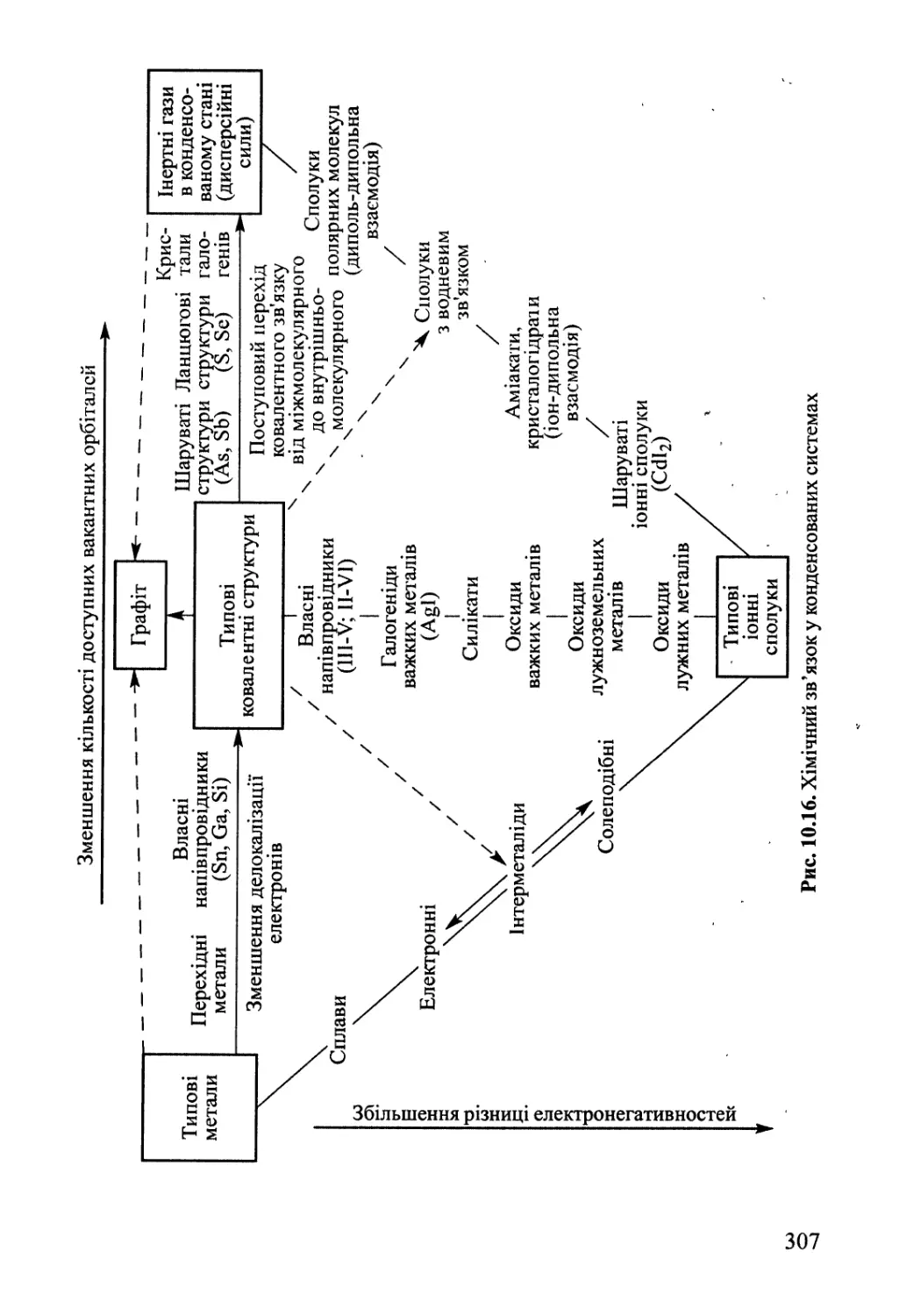
Розділ 10. Хімічний зв'язок у конденсованому стані 276
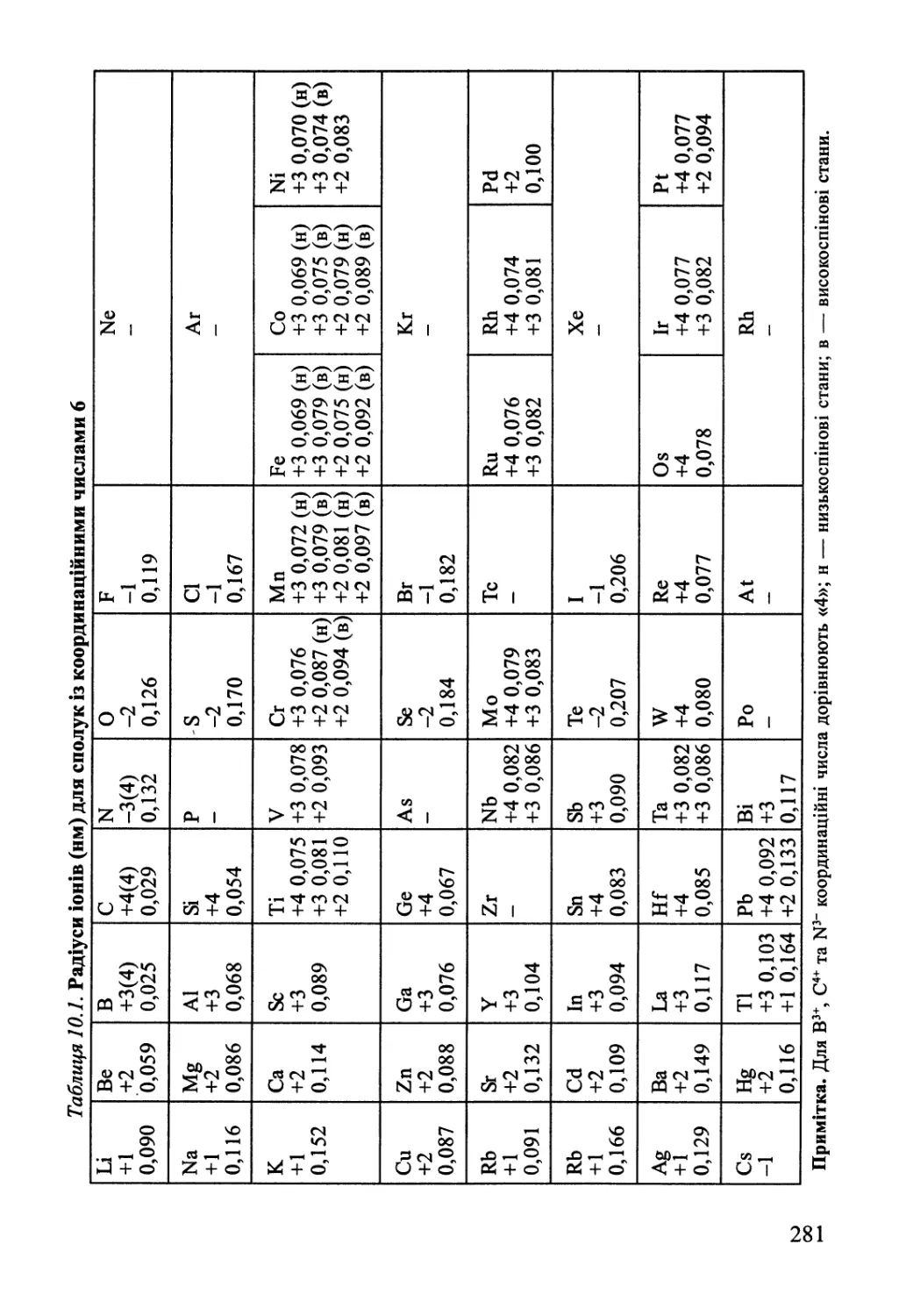
10.1. Іонні сполуки 277
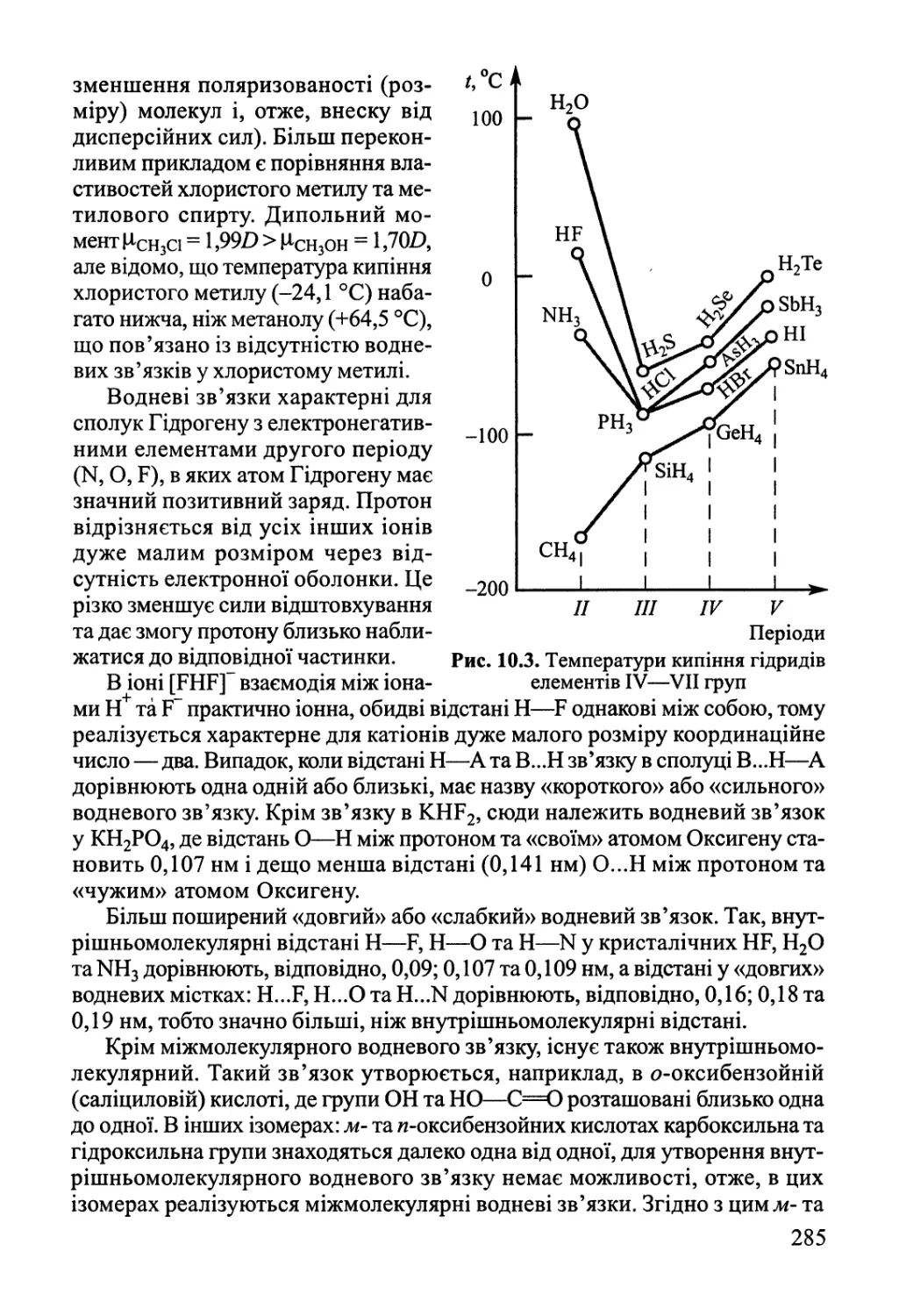
10.2. Міжмолекулярна взаємодія, водневий зв'язок 282
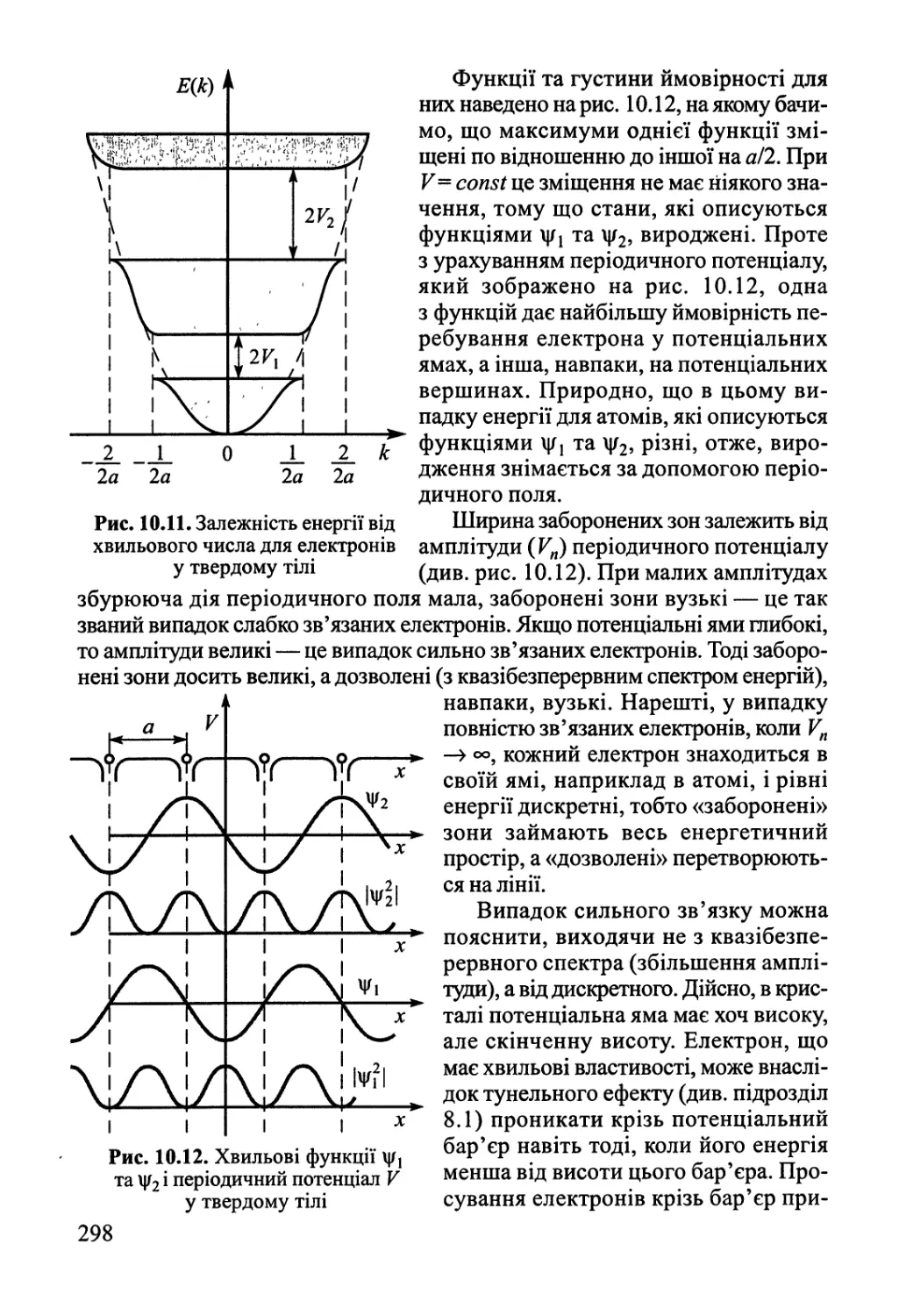
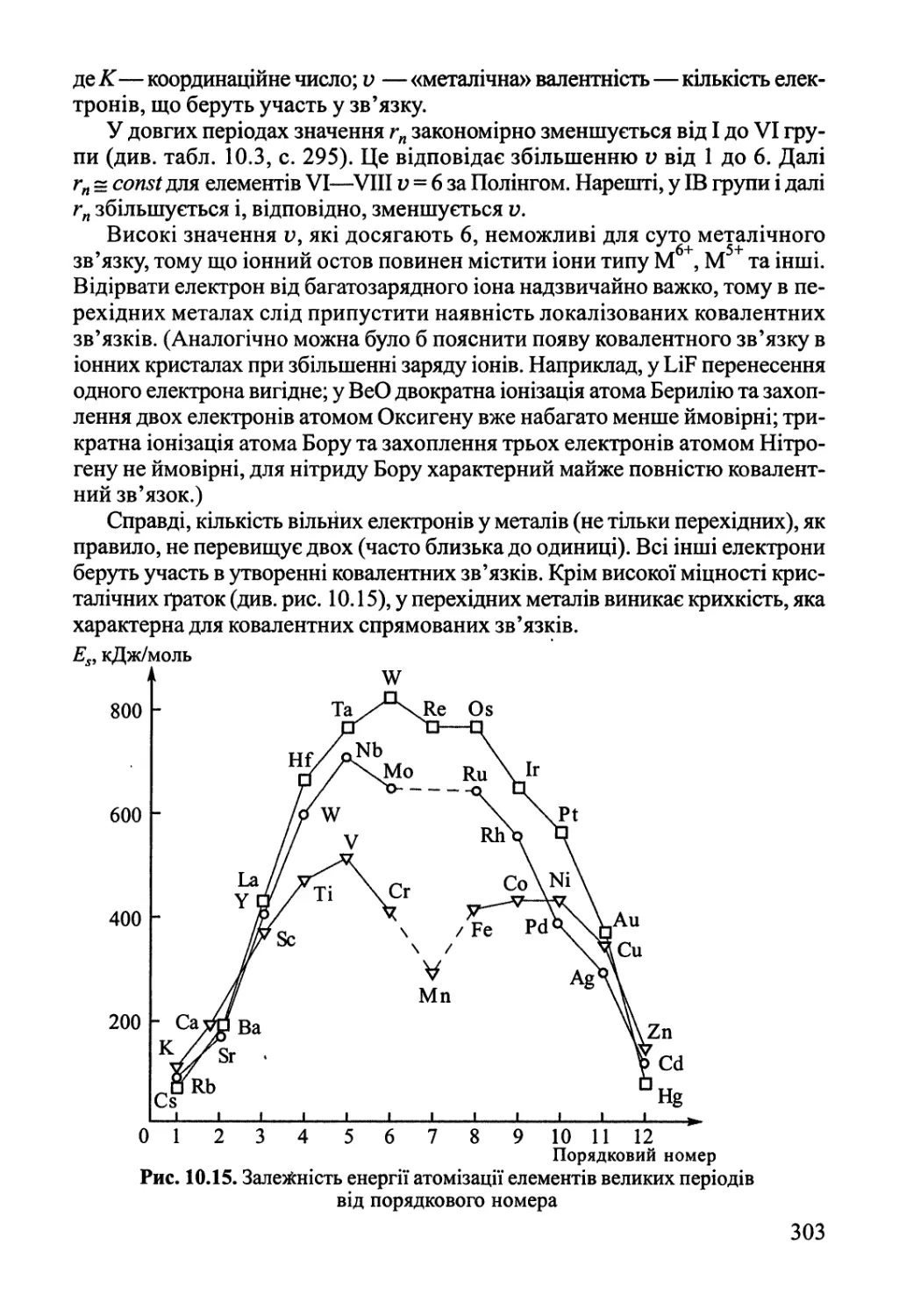
10.3. Ковалентний та металічний зв'язок у твердому тілі 288
Розділ 11. Статистична термодинаміка 308
11.1. Розподілення Больцмана 308
11.2. Зв'язок термодинамічних функцій зі статистичною сумою 312
11.3. Поступальний рух 313
11.4. Електронна статистична сума 316
11.5. Обертальний рух 317
11.6. Коливальний рух. Теорія теплоємності '. 319
11.7. Обчислення константи рівноваги статистичним методом 323
11.8. Статистична термодинаміка реальних газів 325
Розділ 12. Теорія флуктуацій. Статистичні та філософські аспекти
2-го начала термодинаміки 331
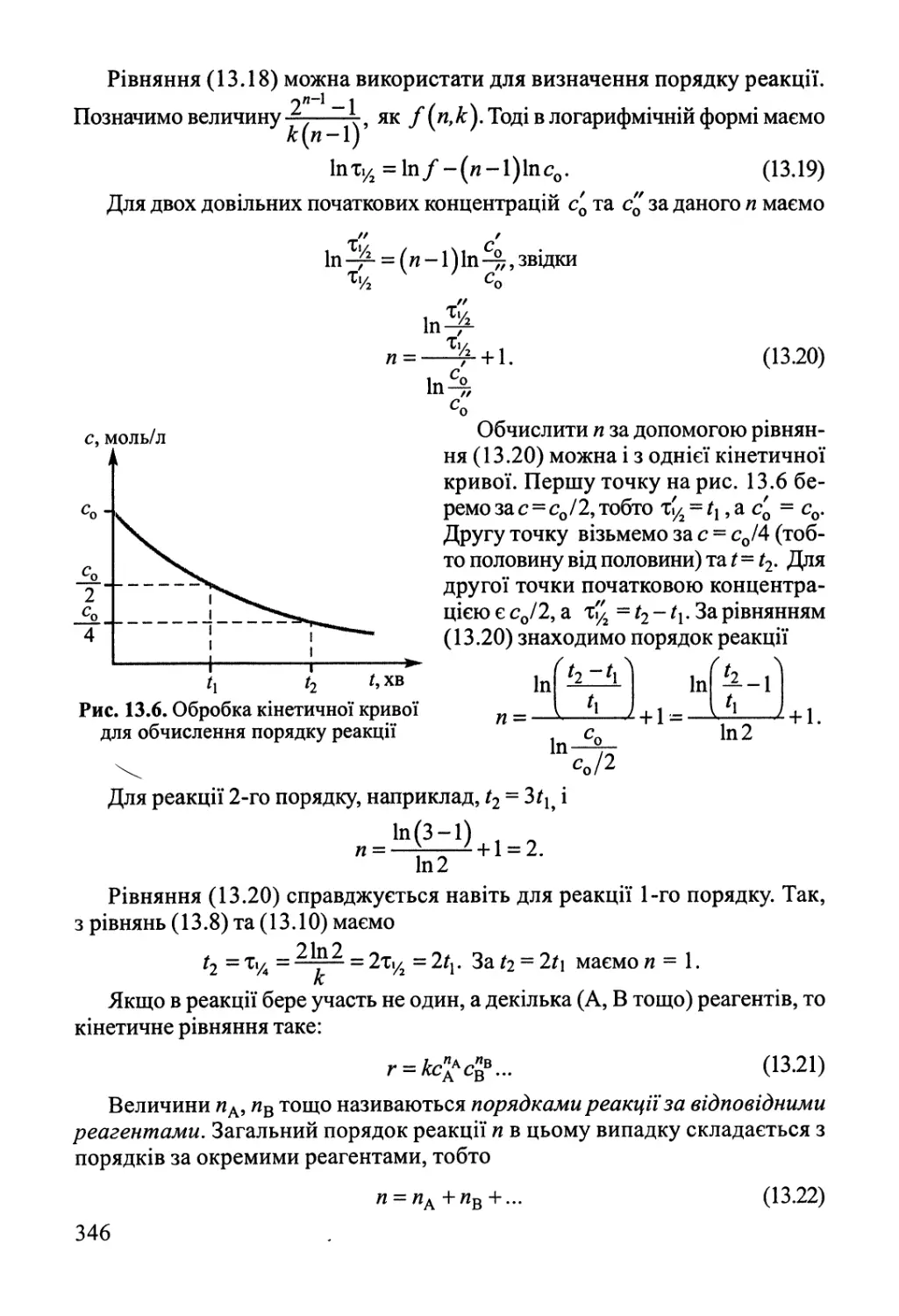
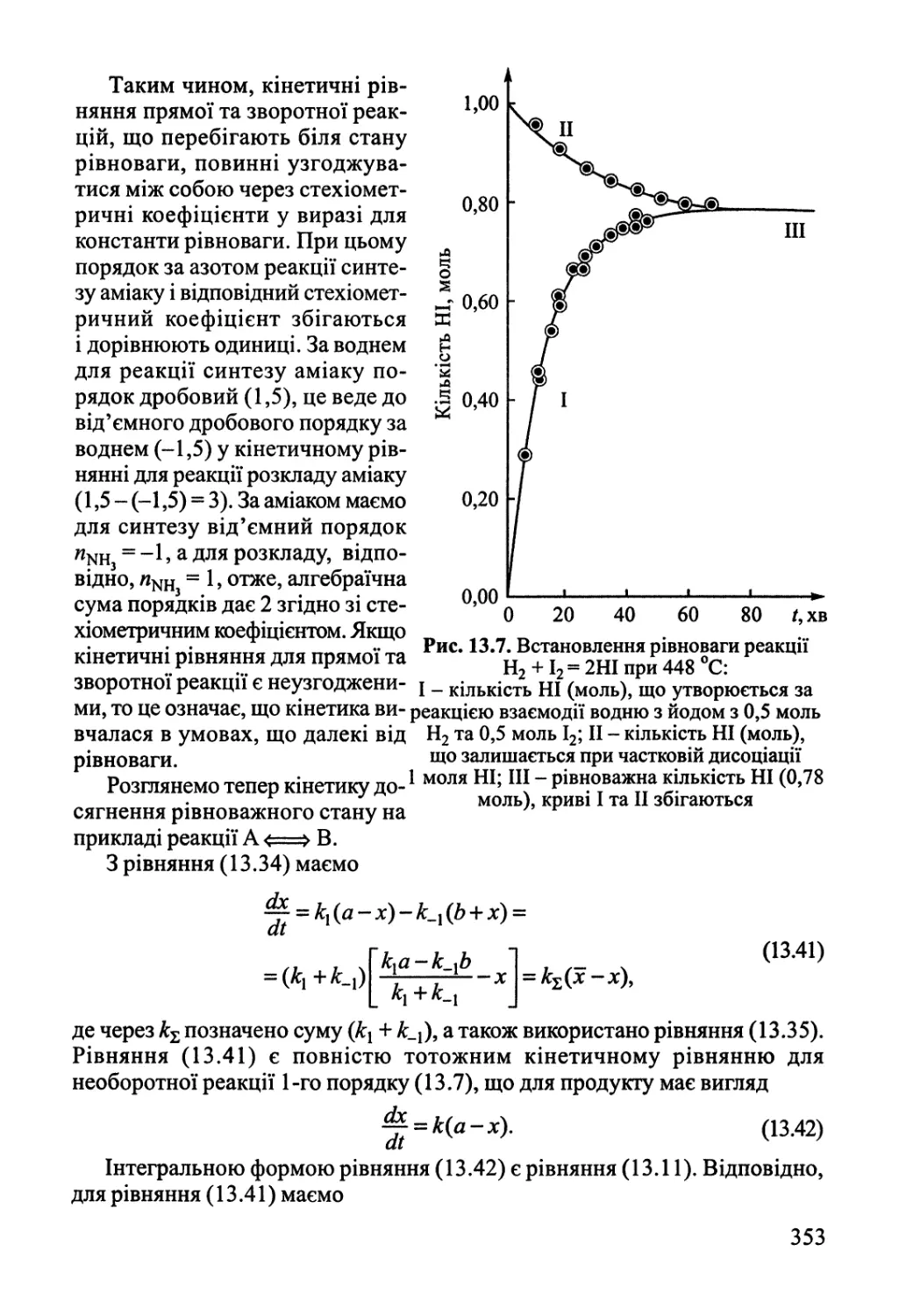
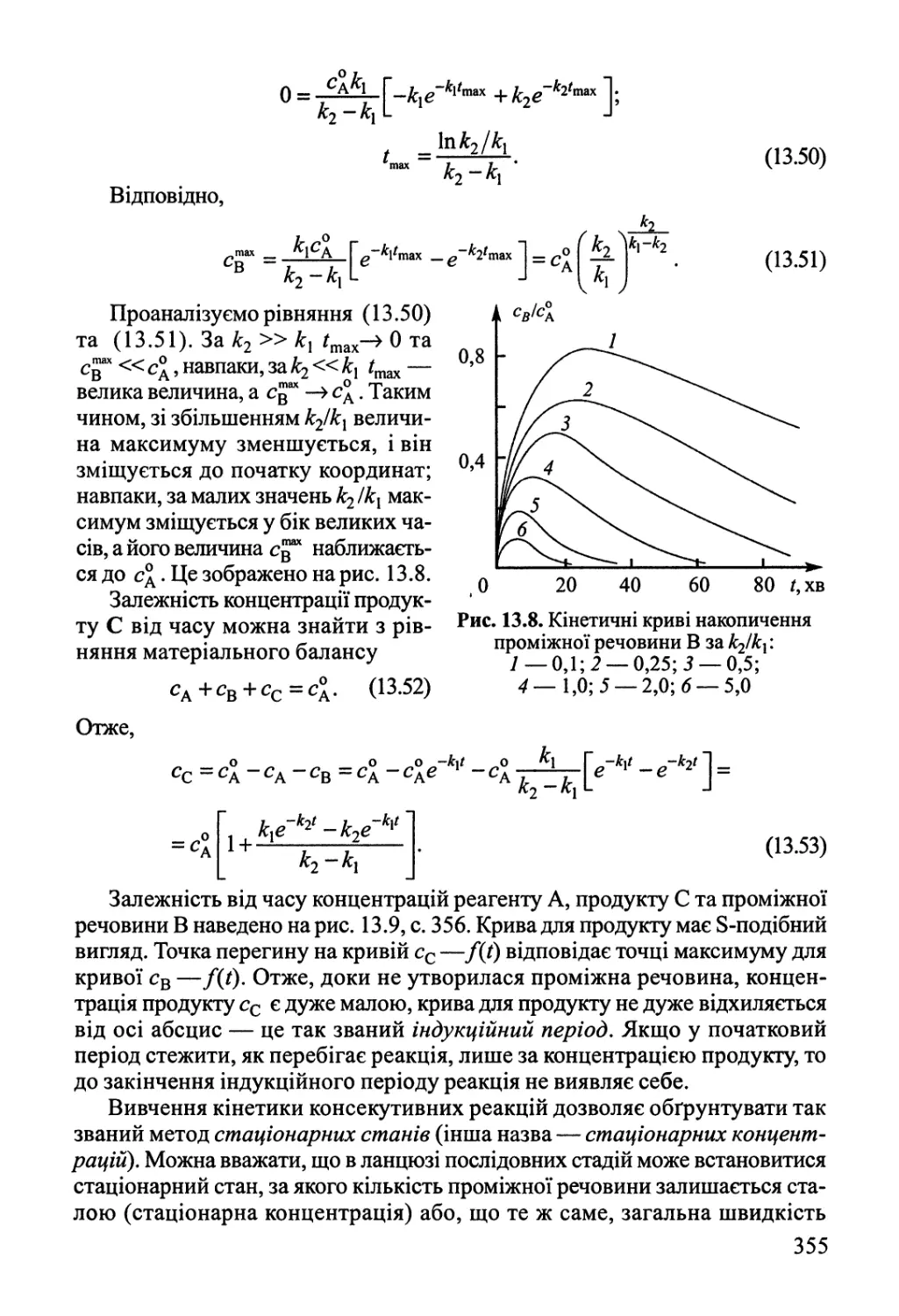
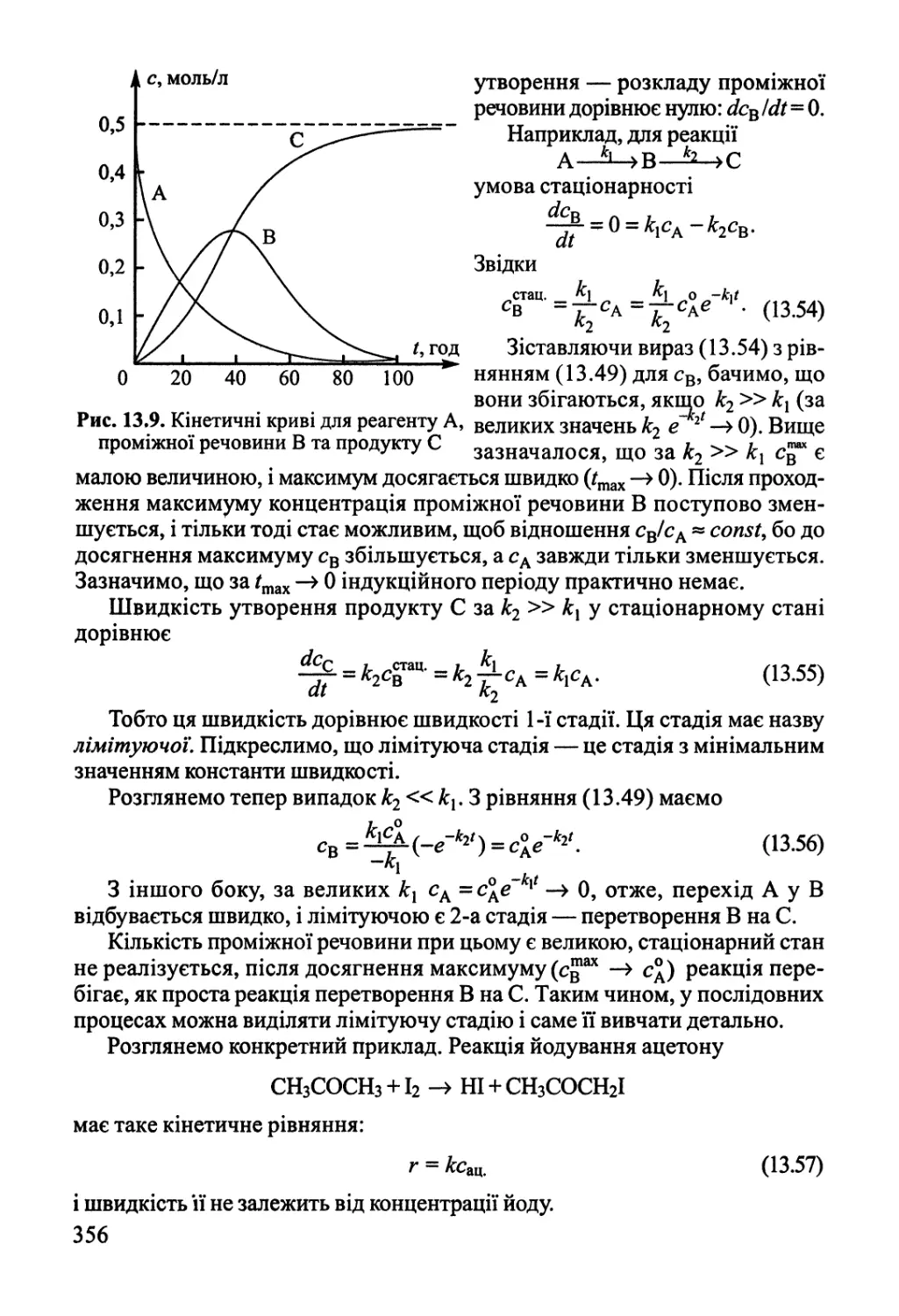
Розділ 13. Основи хімічної кінетики 342
13.1. Основні поняття хімічної кінетики 342
13.2. Кінетика складних реакцій 351
Розділ 14. Кінетика елементарних процесів 362
14.1. Молекулярно-кінетична теорія газів 362
14.2. Явища переносу в газах 367
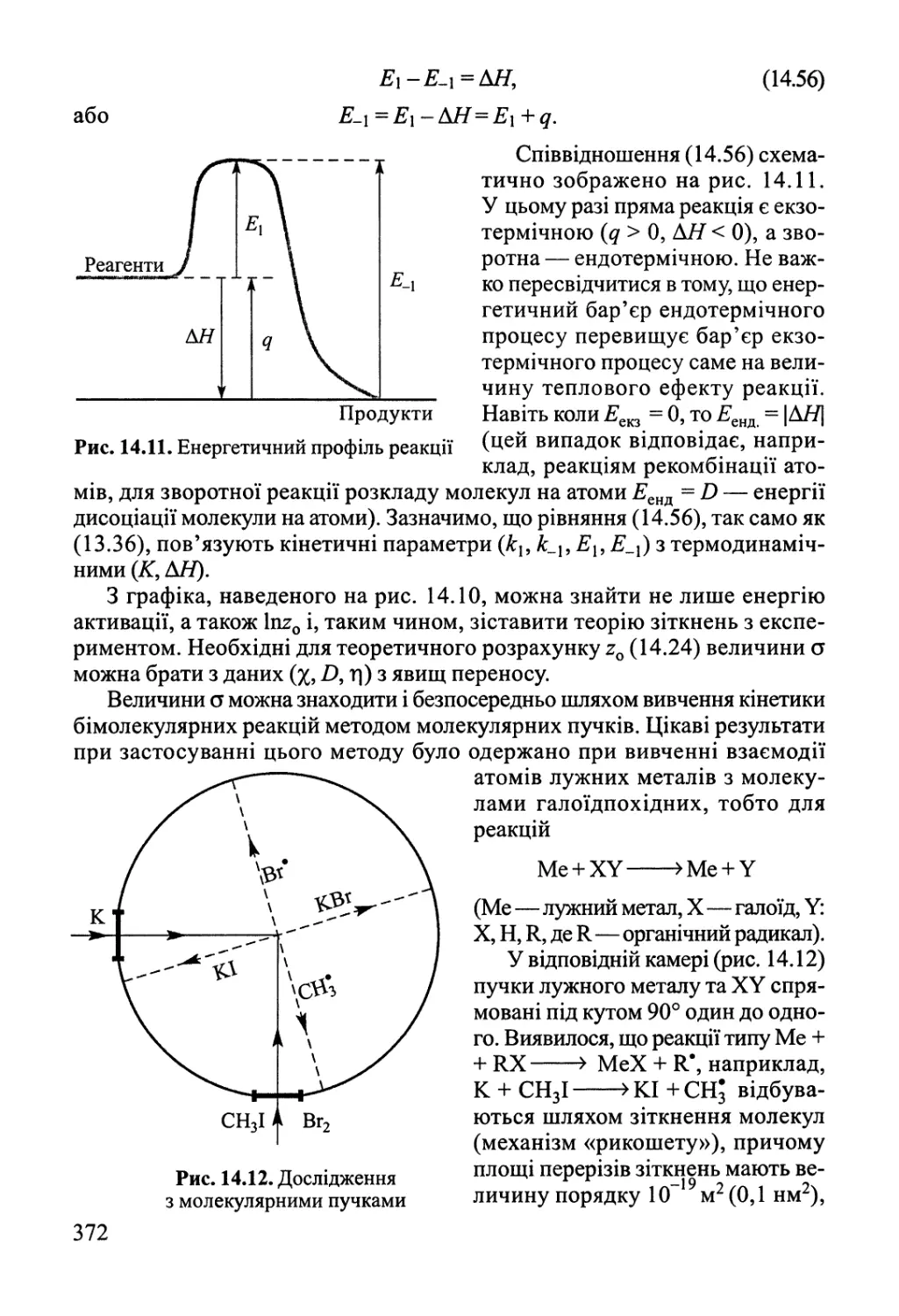
14.3. Теорія зіткнень 370
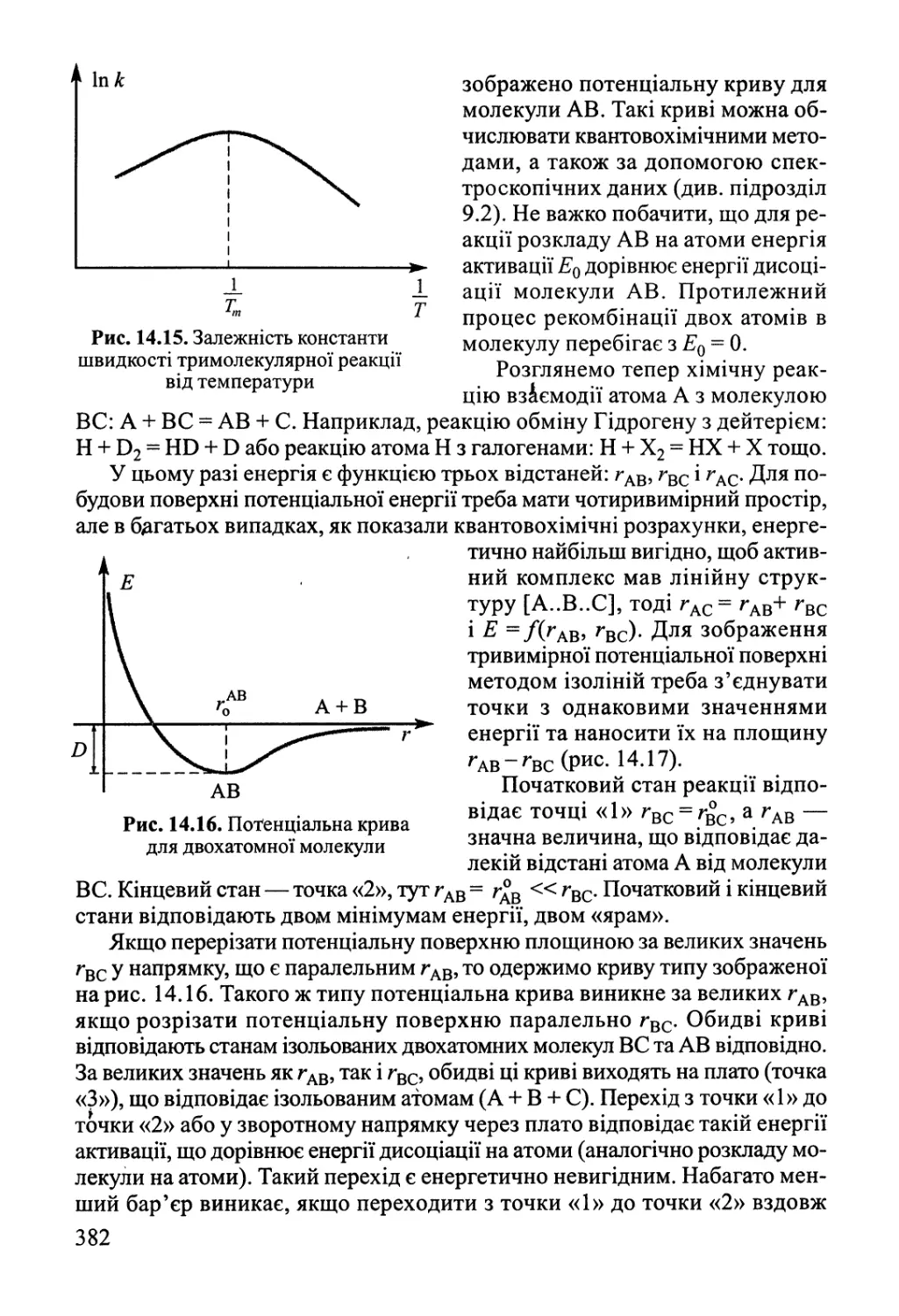
14.4. Теорія перехідного стану (активного комплексу) 376
Розділ 15. Кінетика реакцій у гомогенних системах 388
15.1. Мономолекулярні реакції у газовій фазі 388
15.2. Ланцюгові реакції 391
15.3. Реакції у розчинах неелектролітів 400
15.4. Реакції у розчинах електролітів 405
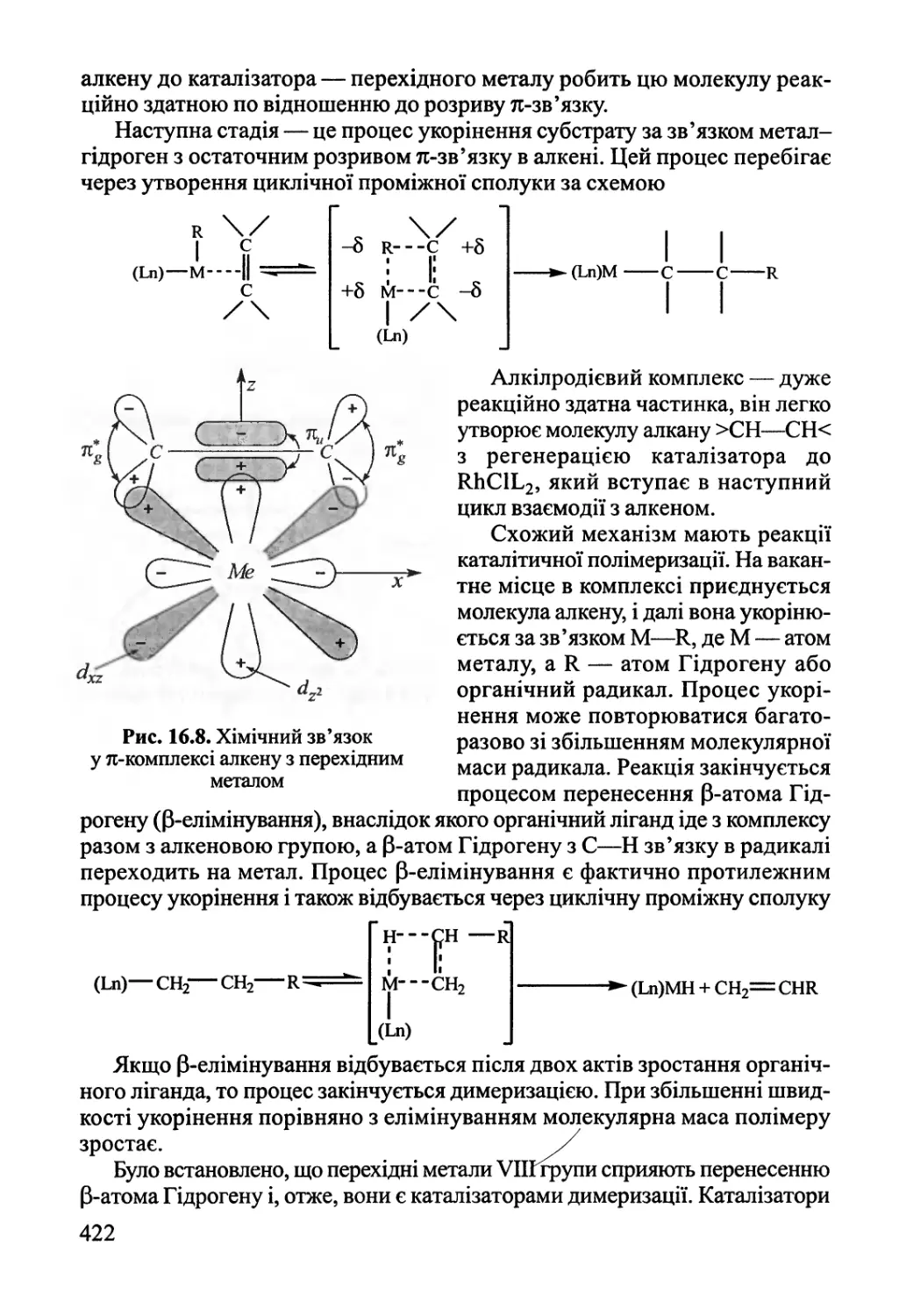
Розділ 16. Гомогенний каталіз 409
16.1. Загальні принципи каталізу. Окисно-відновний каталіз 409
16.2. Кислотно-основний каталіз 414
16.3. Коливальні каталітичні реакції 418
16.4. Каталіз комплексами 420
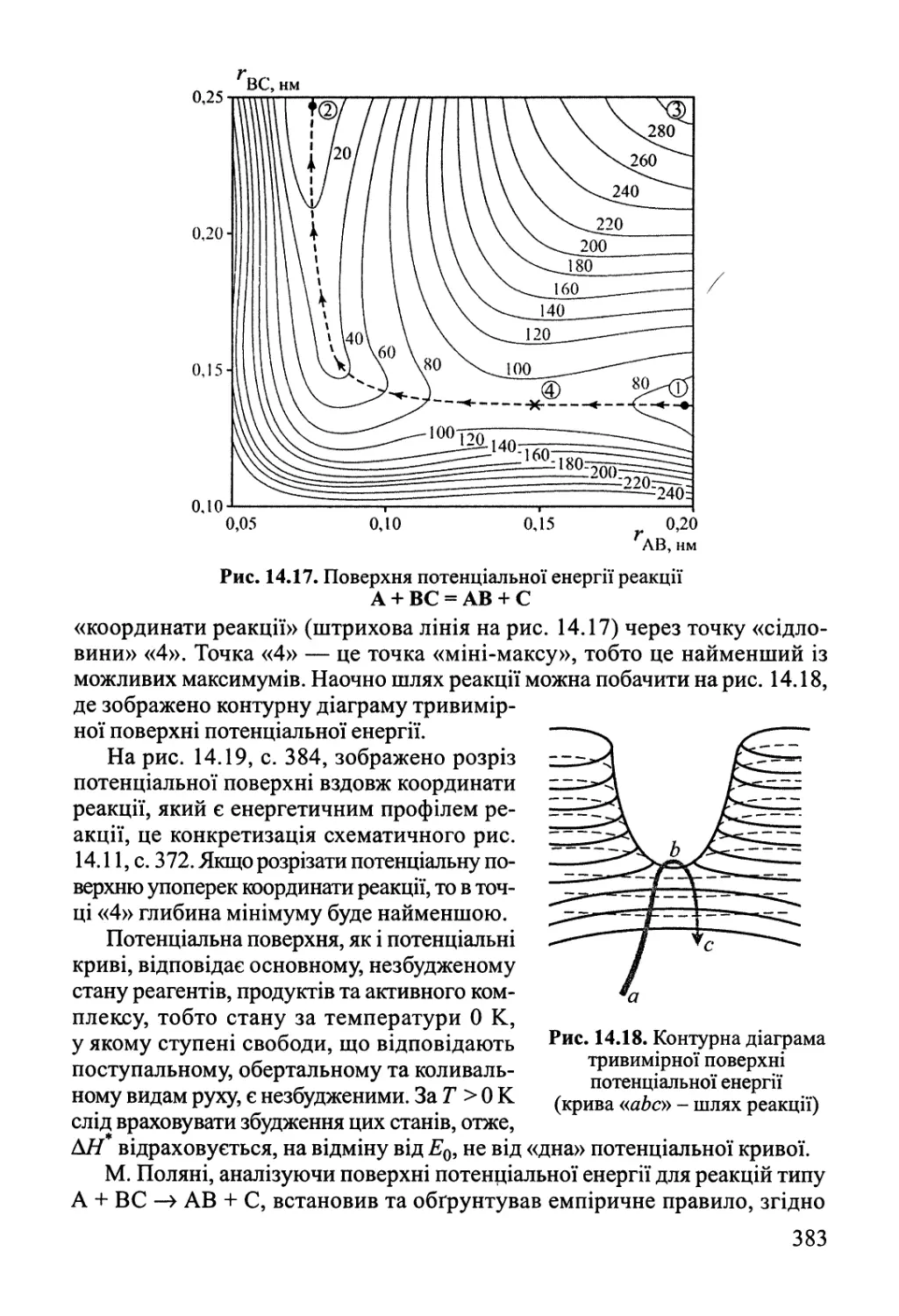
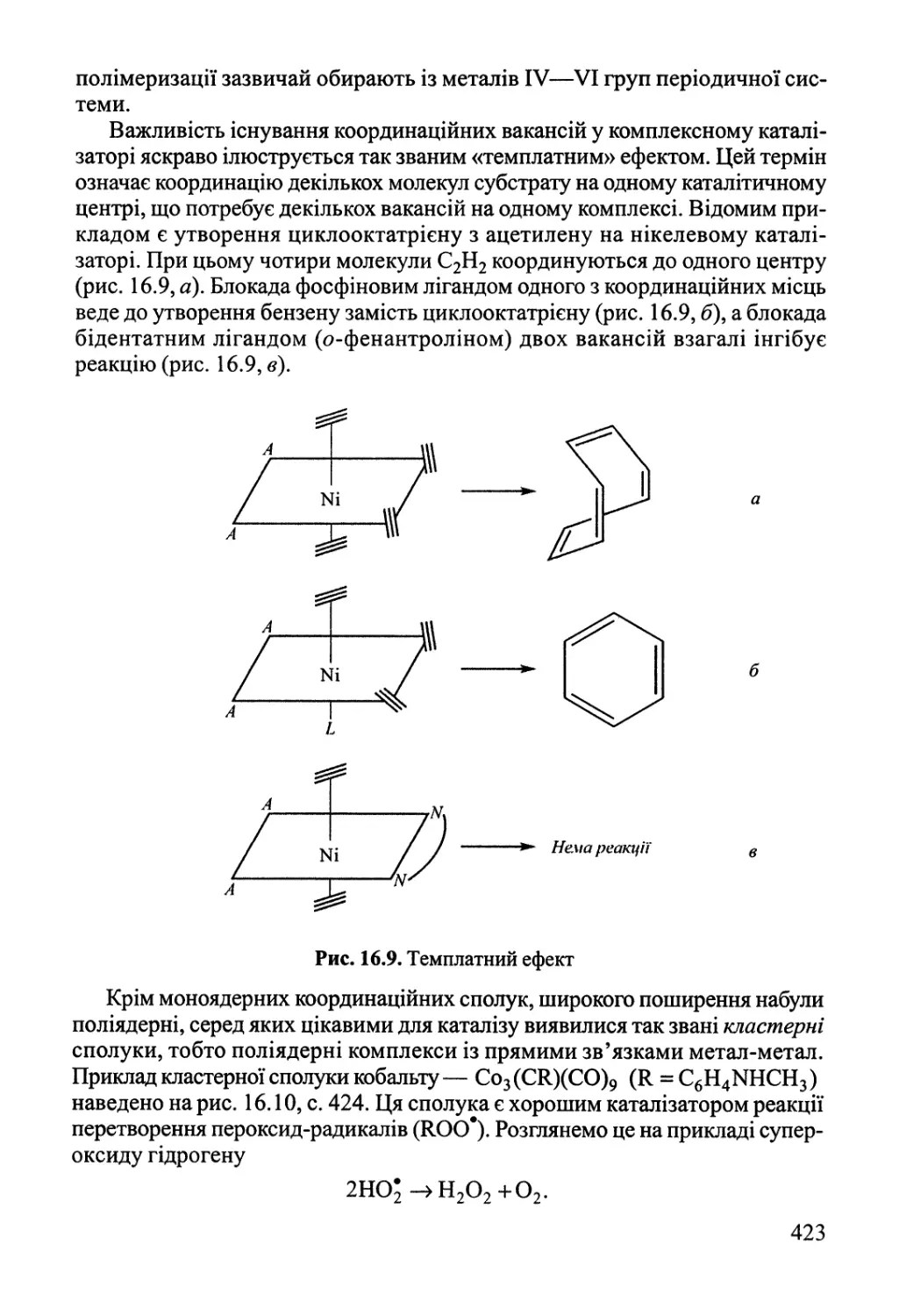
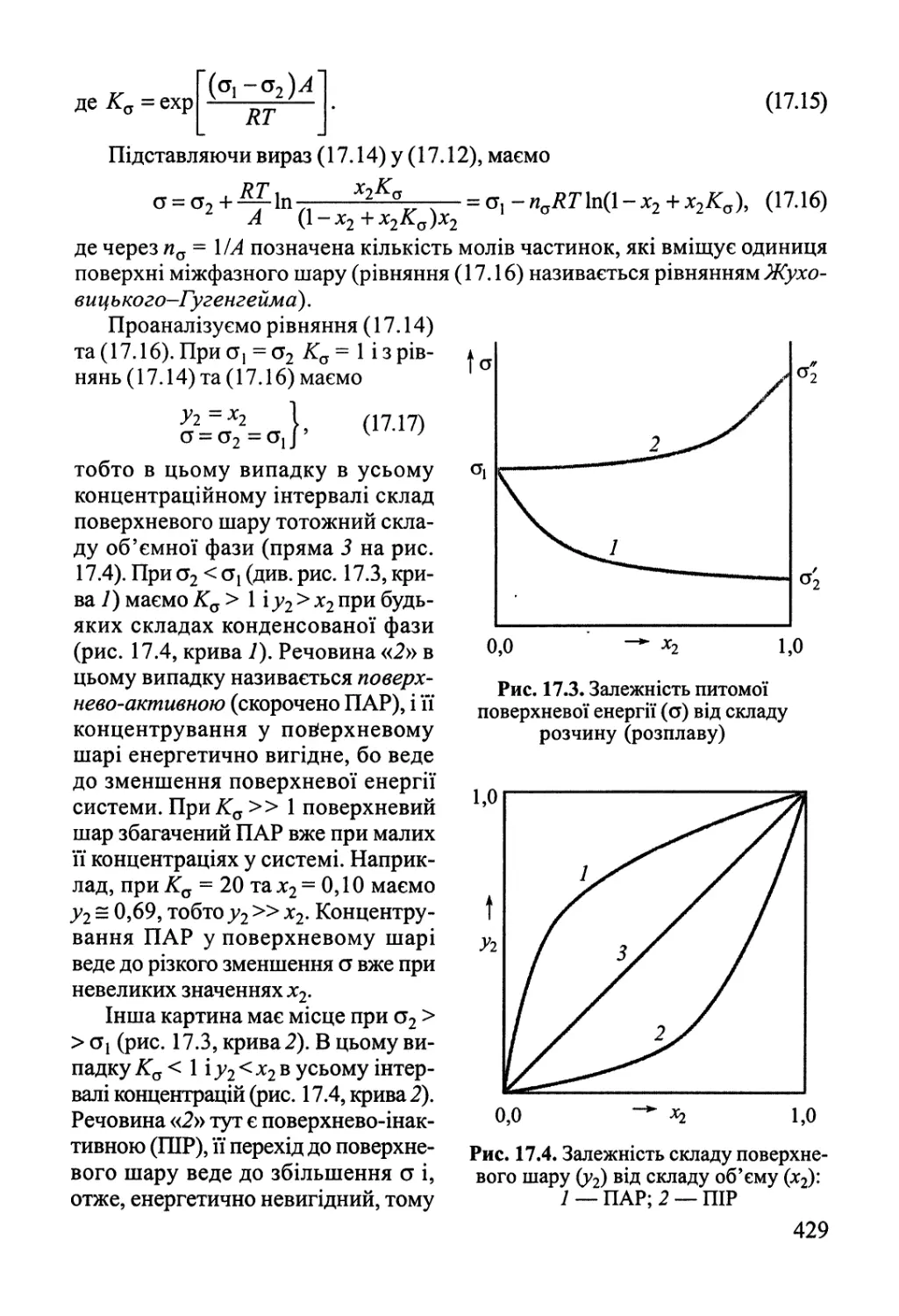
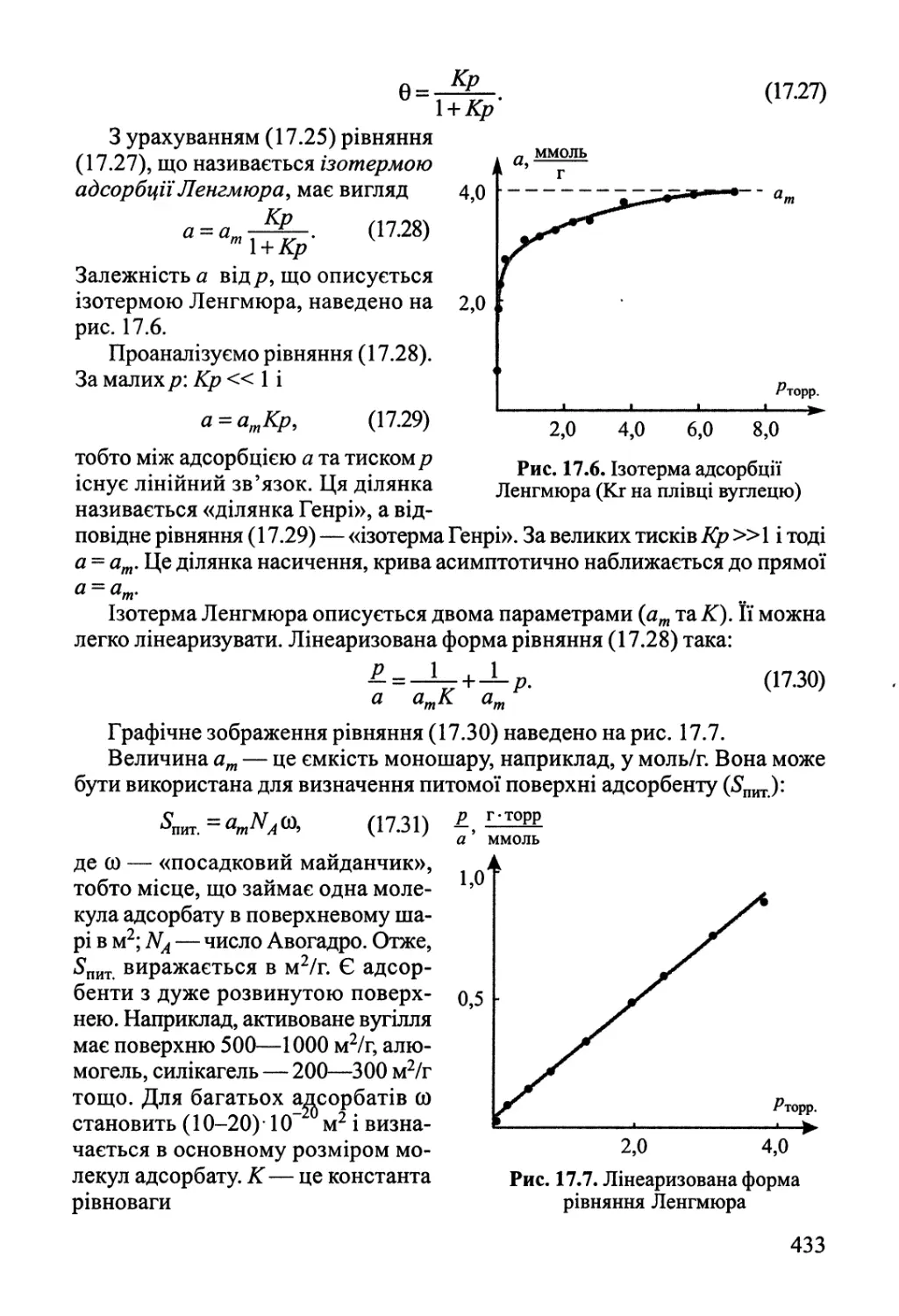
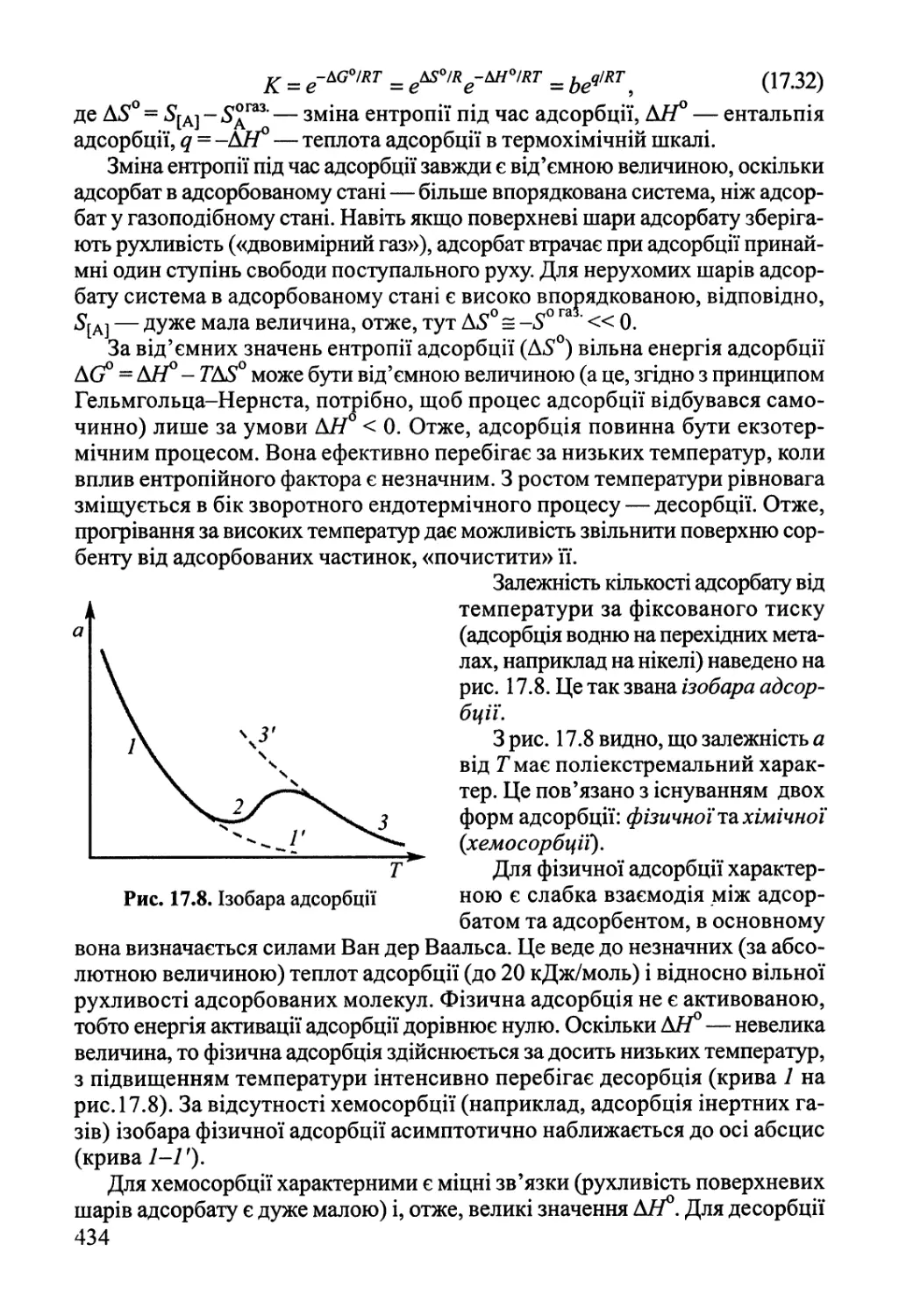
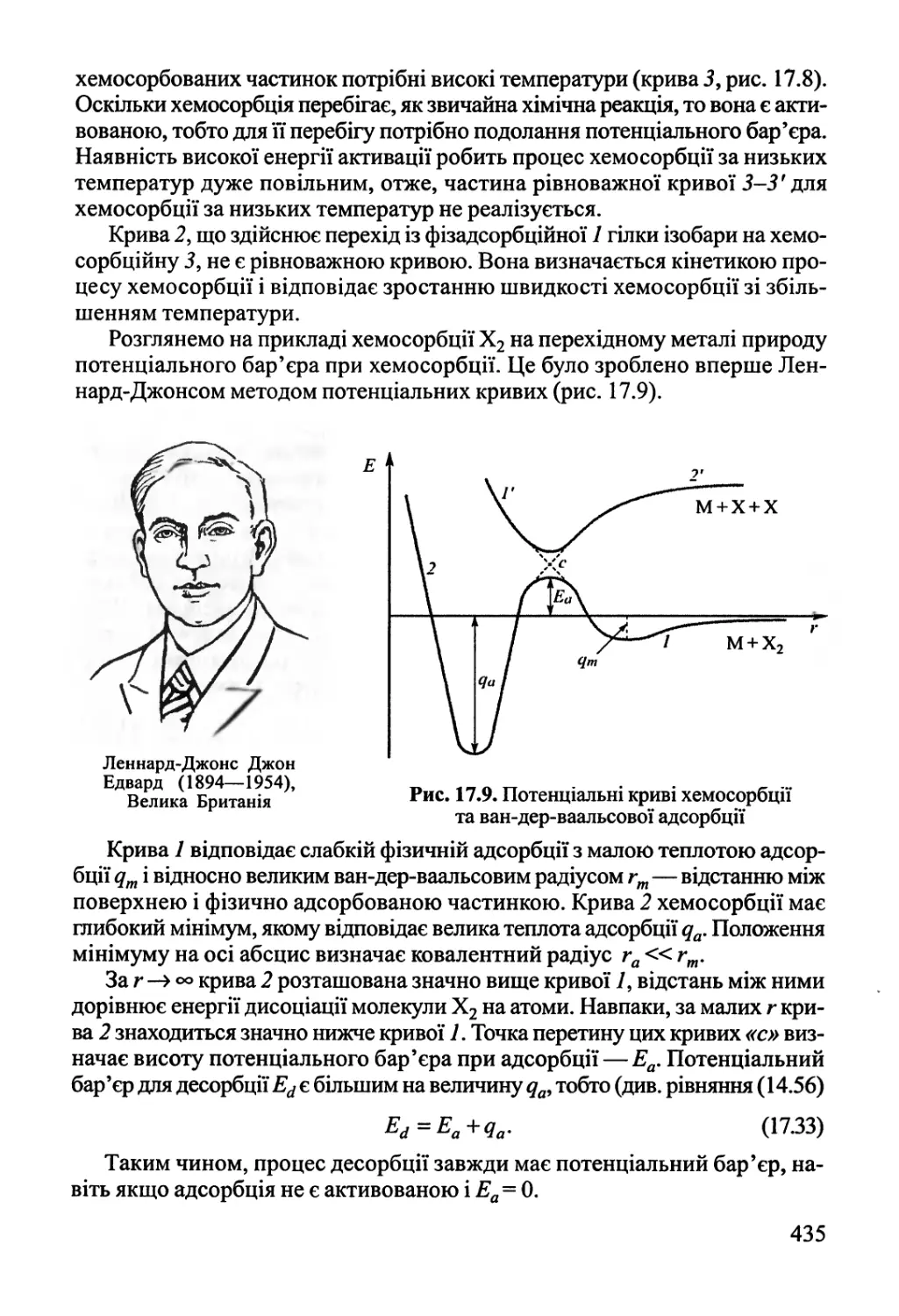
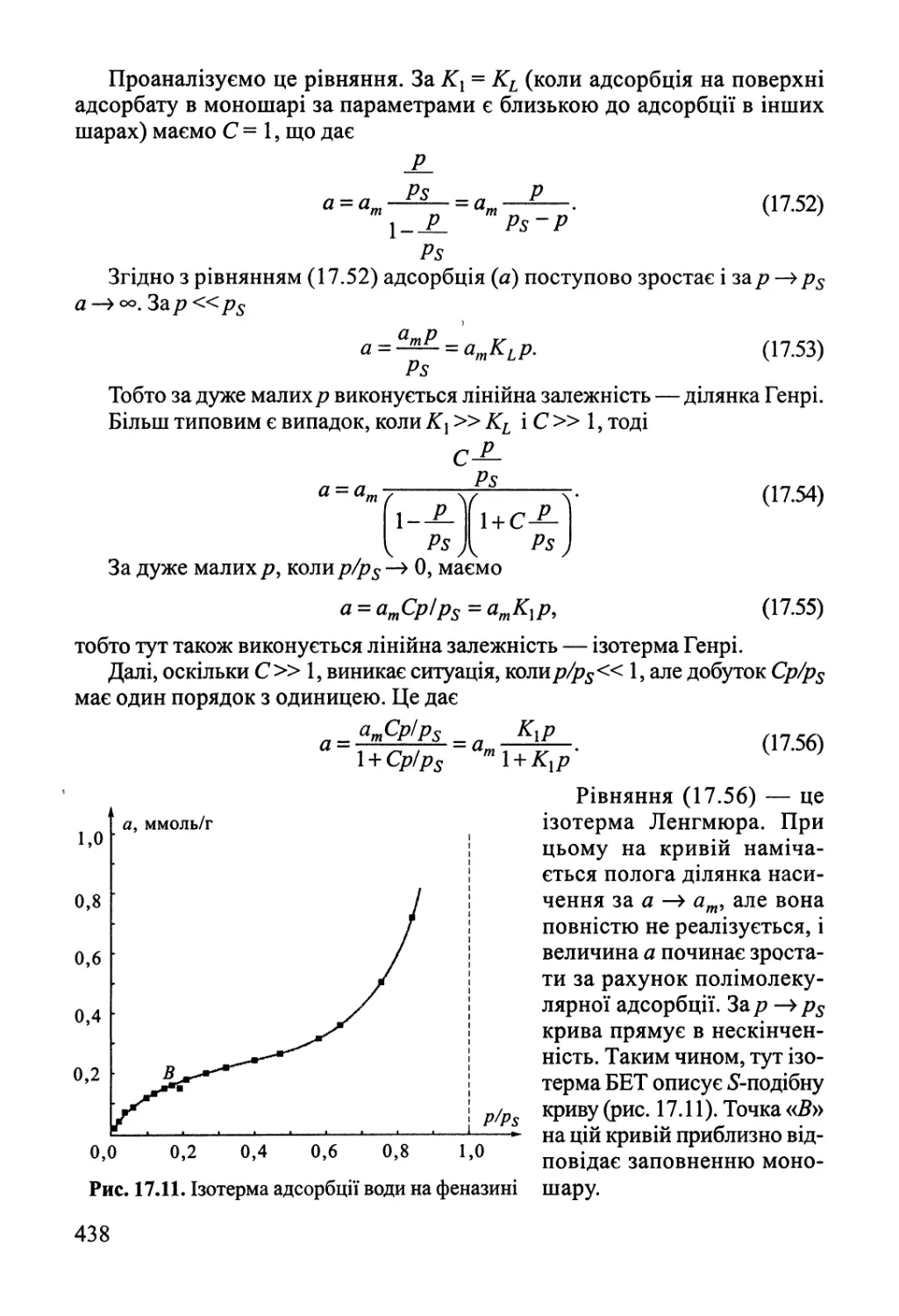
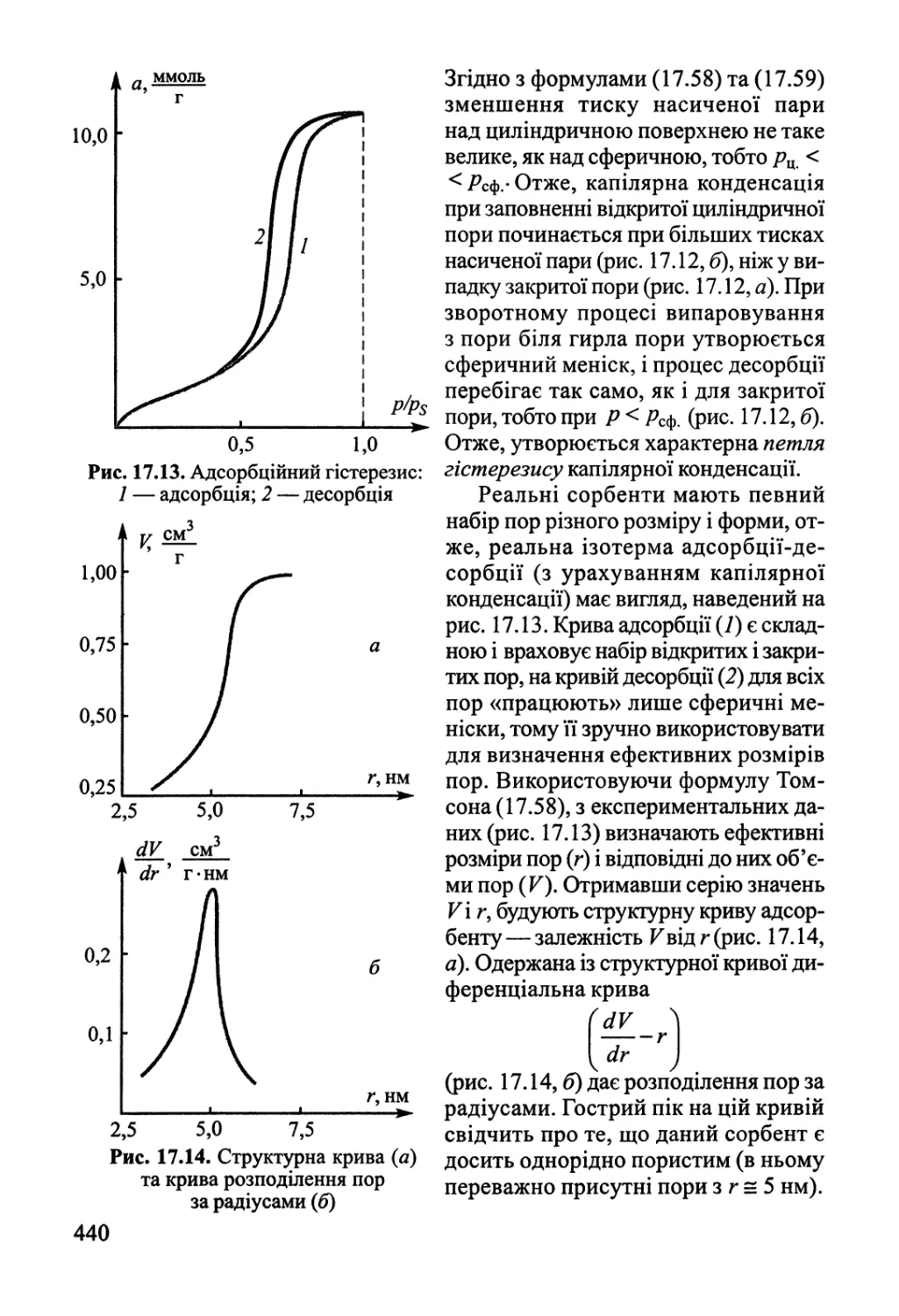
Розділ 17. Фізична хімія міжфазних явищ 425
17.1. Поверхневі явища у конденсованих фазах 425
17.2. Адсорбція газів на поверхні твердого тіла 432
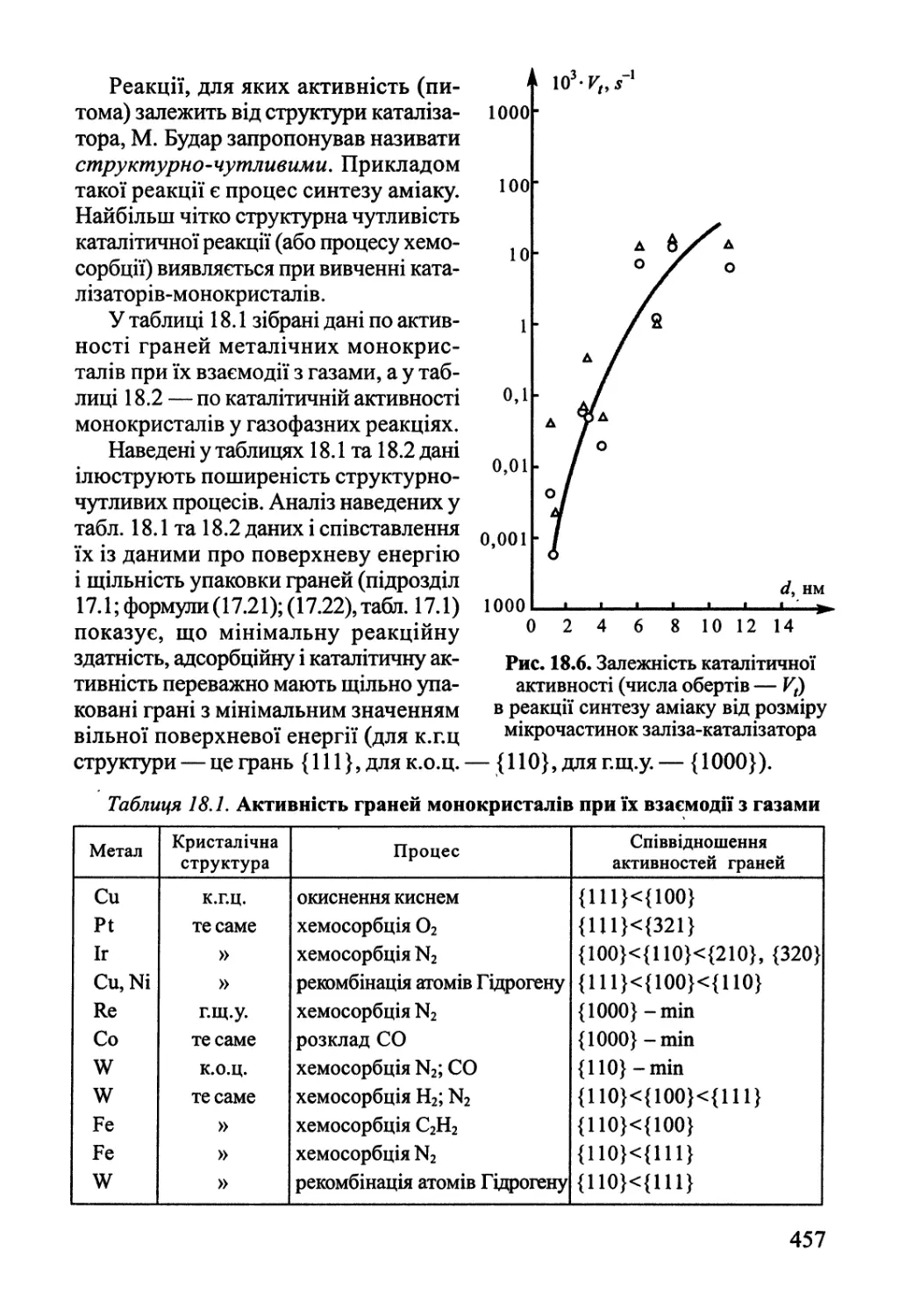
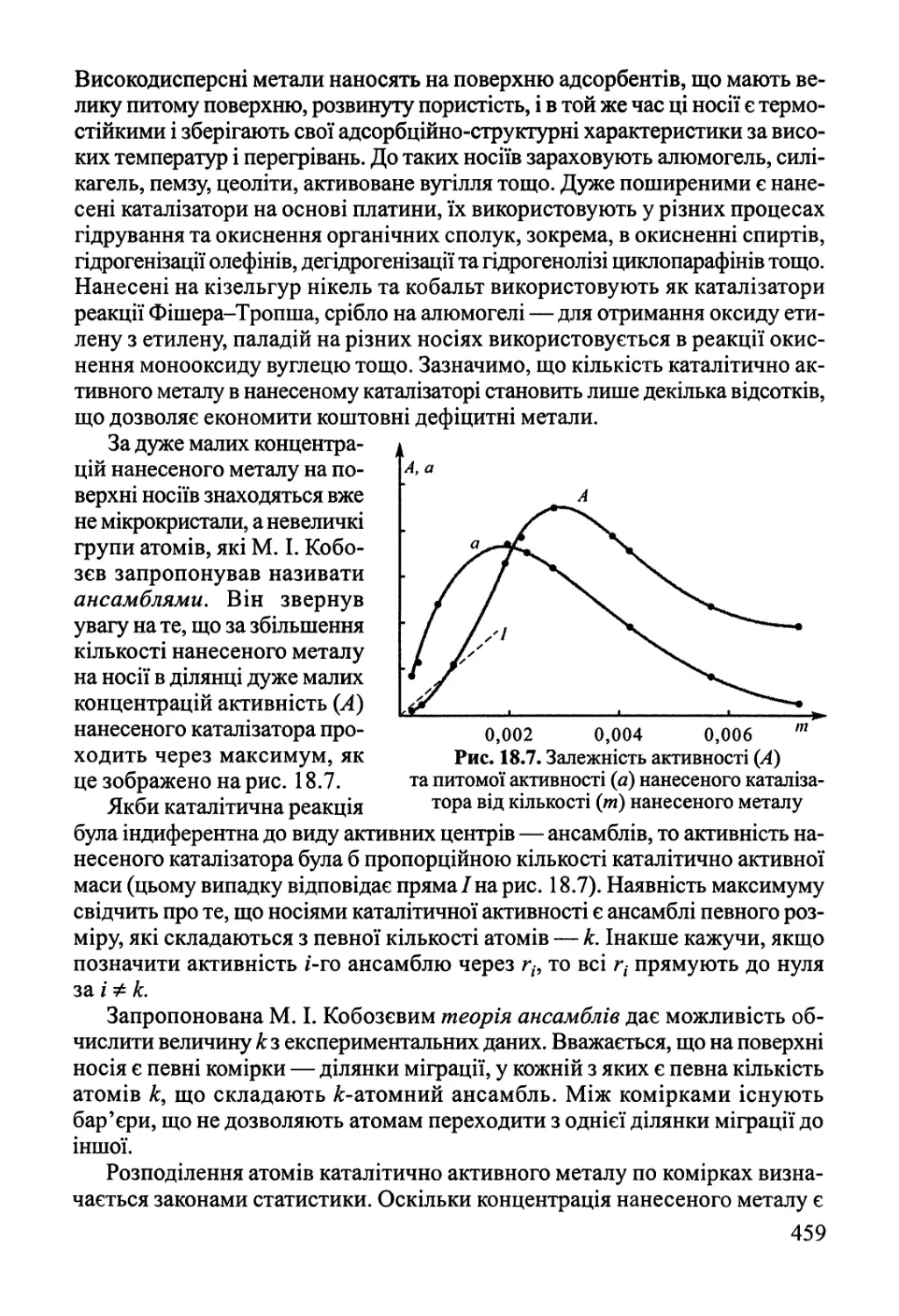
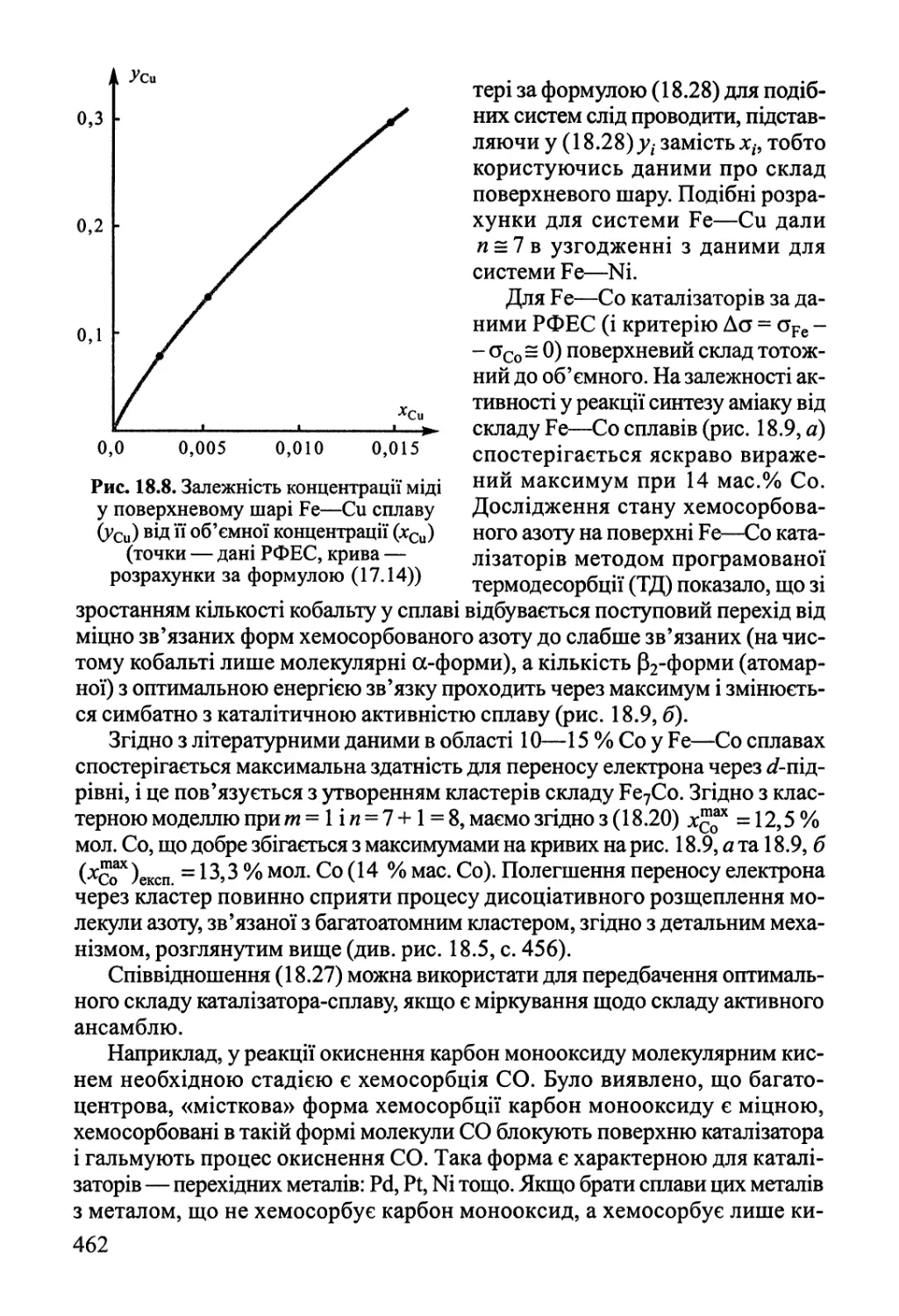
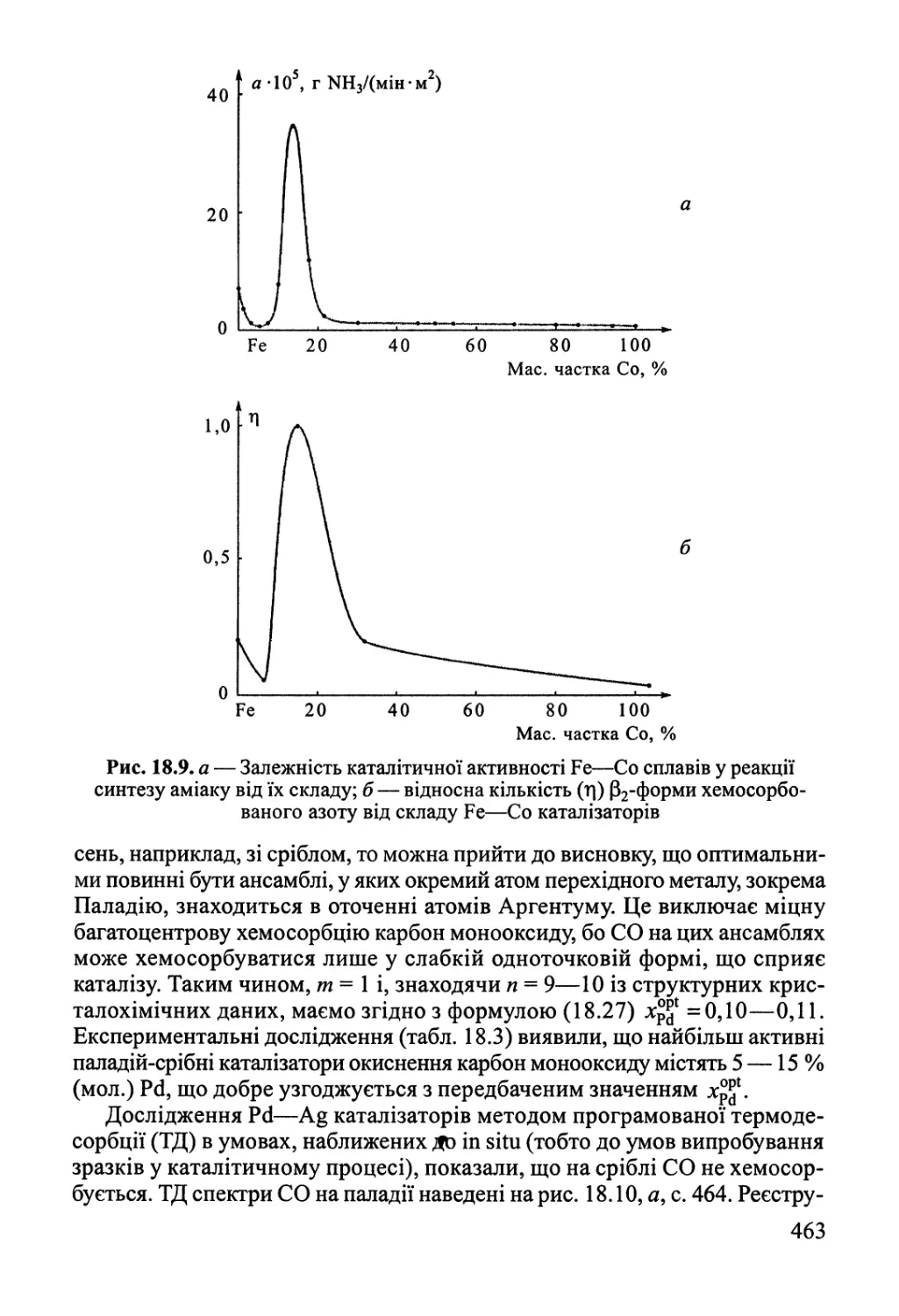
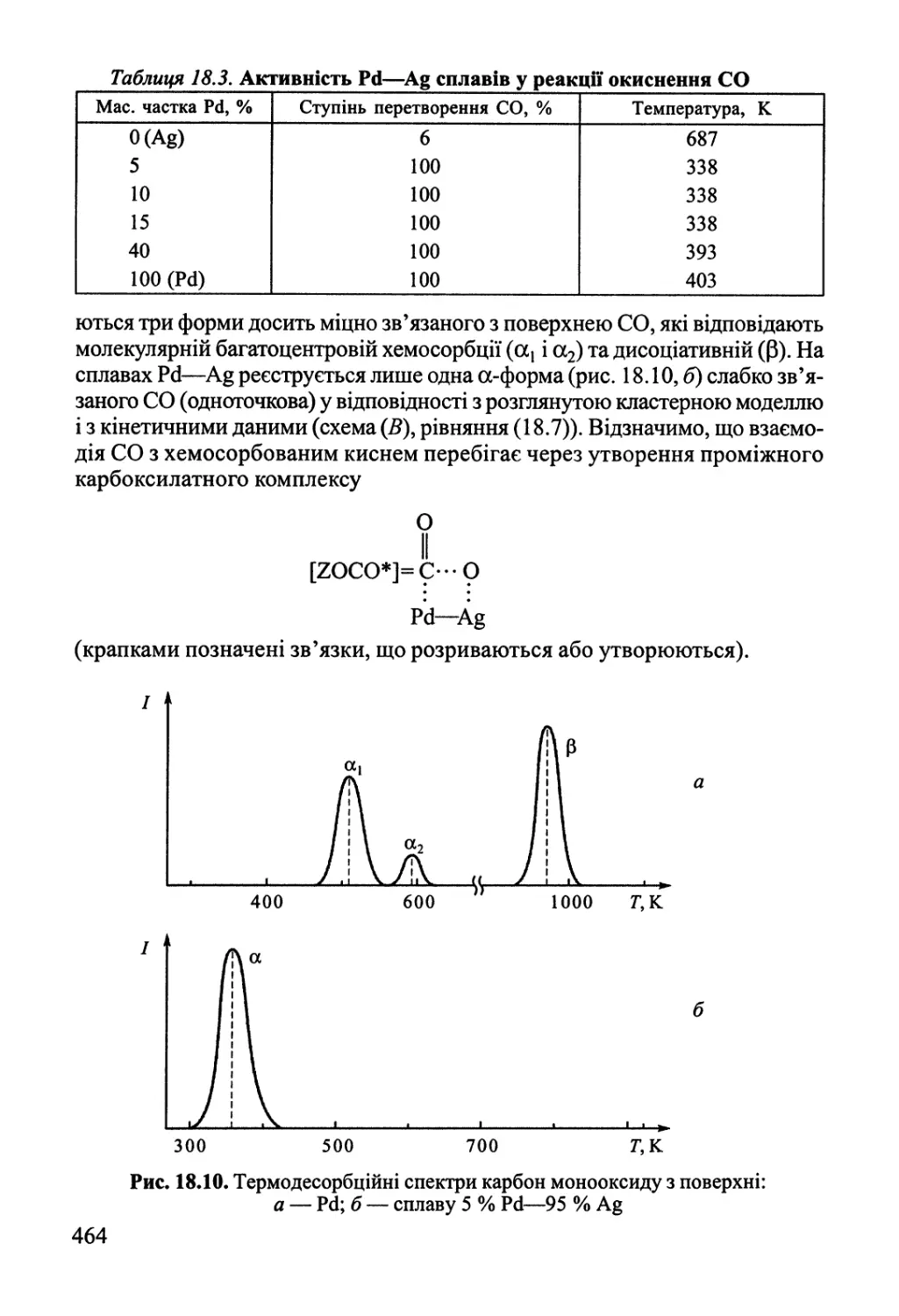
Розділ 18. Гетерогенний каталіз 446
18.1. Кінетика гетерогенно-каталітичних реакцій 446
18.2. Активні центри гетерогенних каталізаторів 455
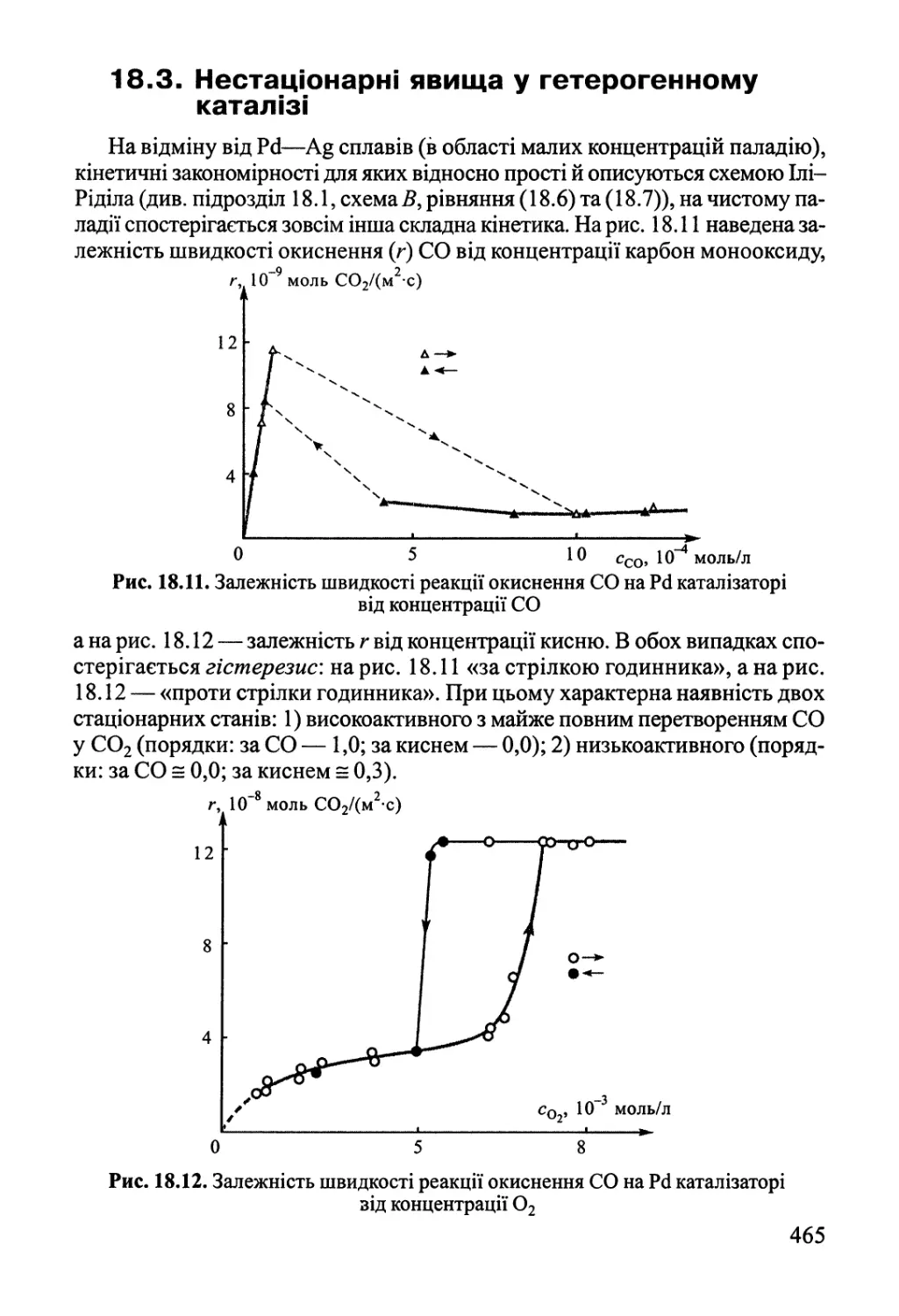
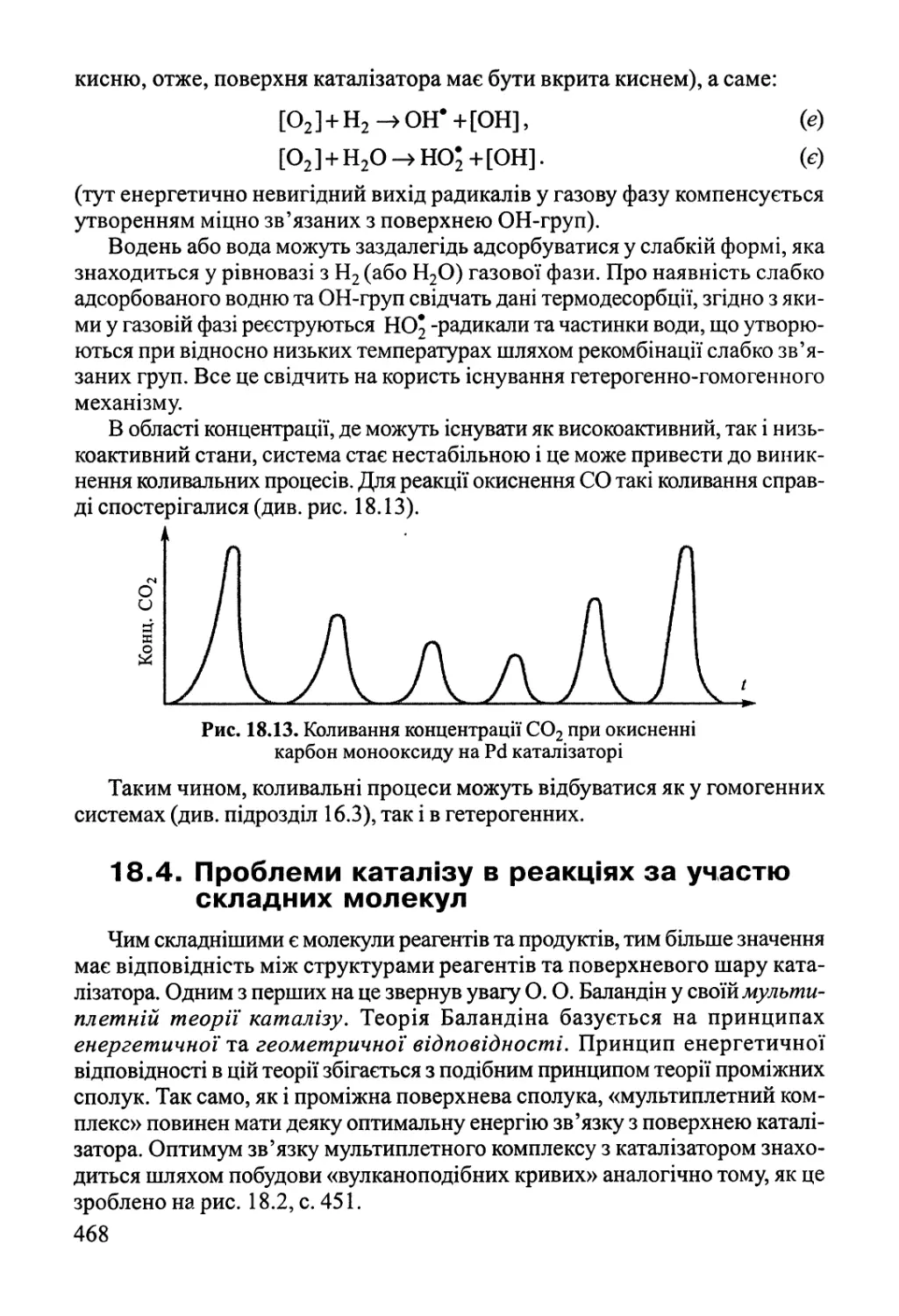
18.3. Нестаціонарні явища у гетерогенному каталізі 465
18.4. Проблеми каталізу в реакціях за участю складних молекул 468
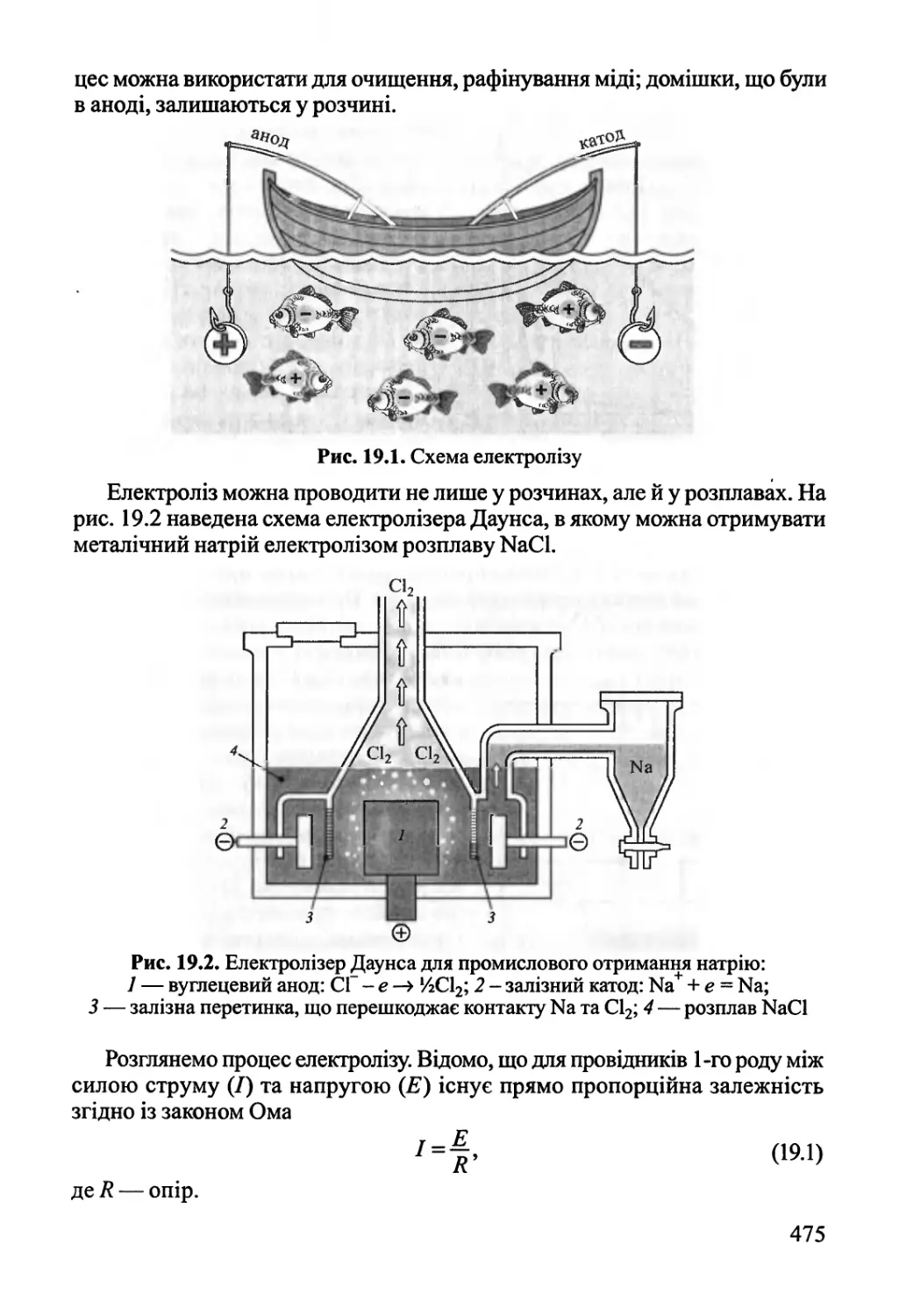
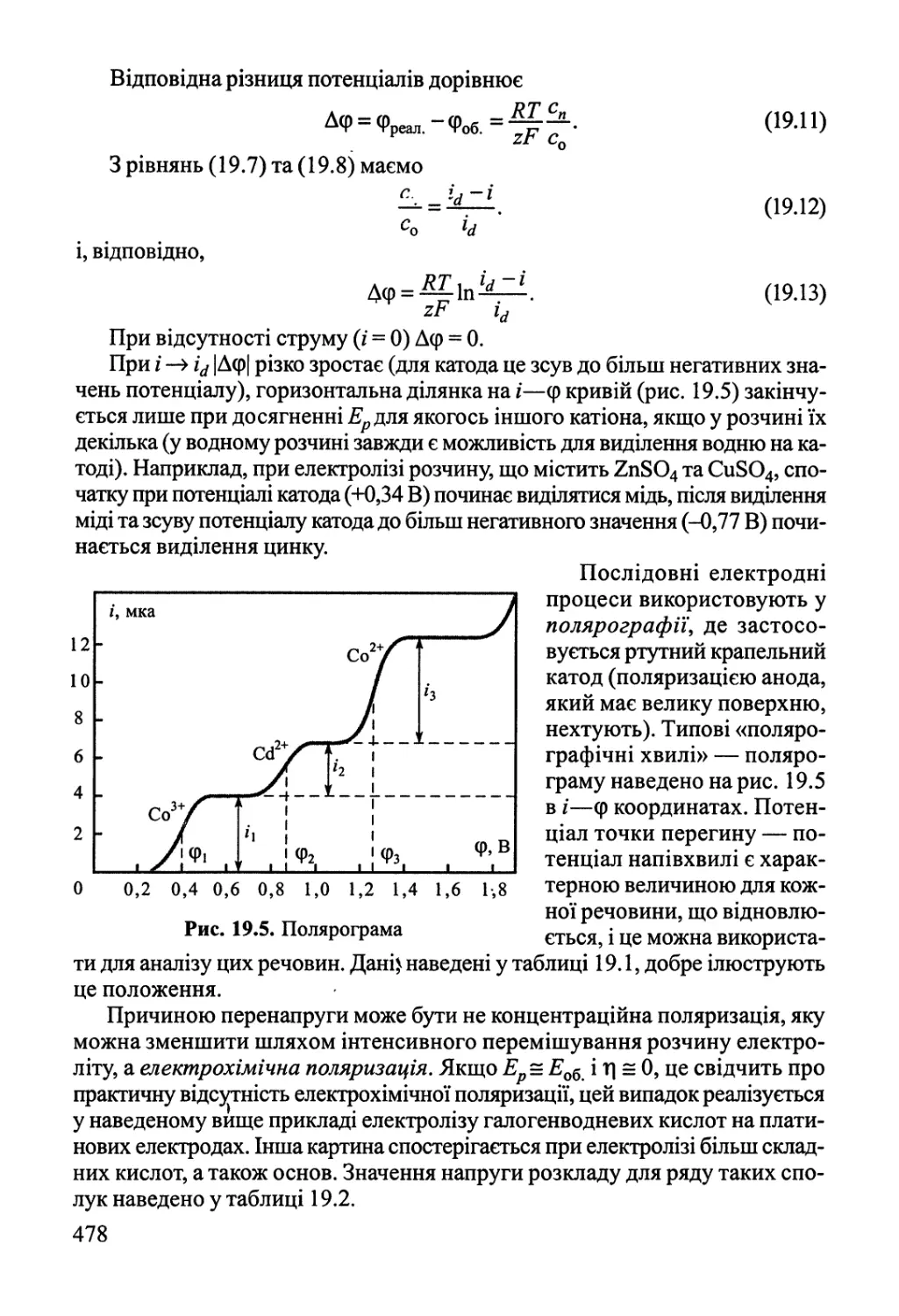
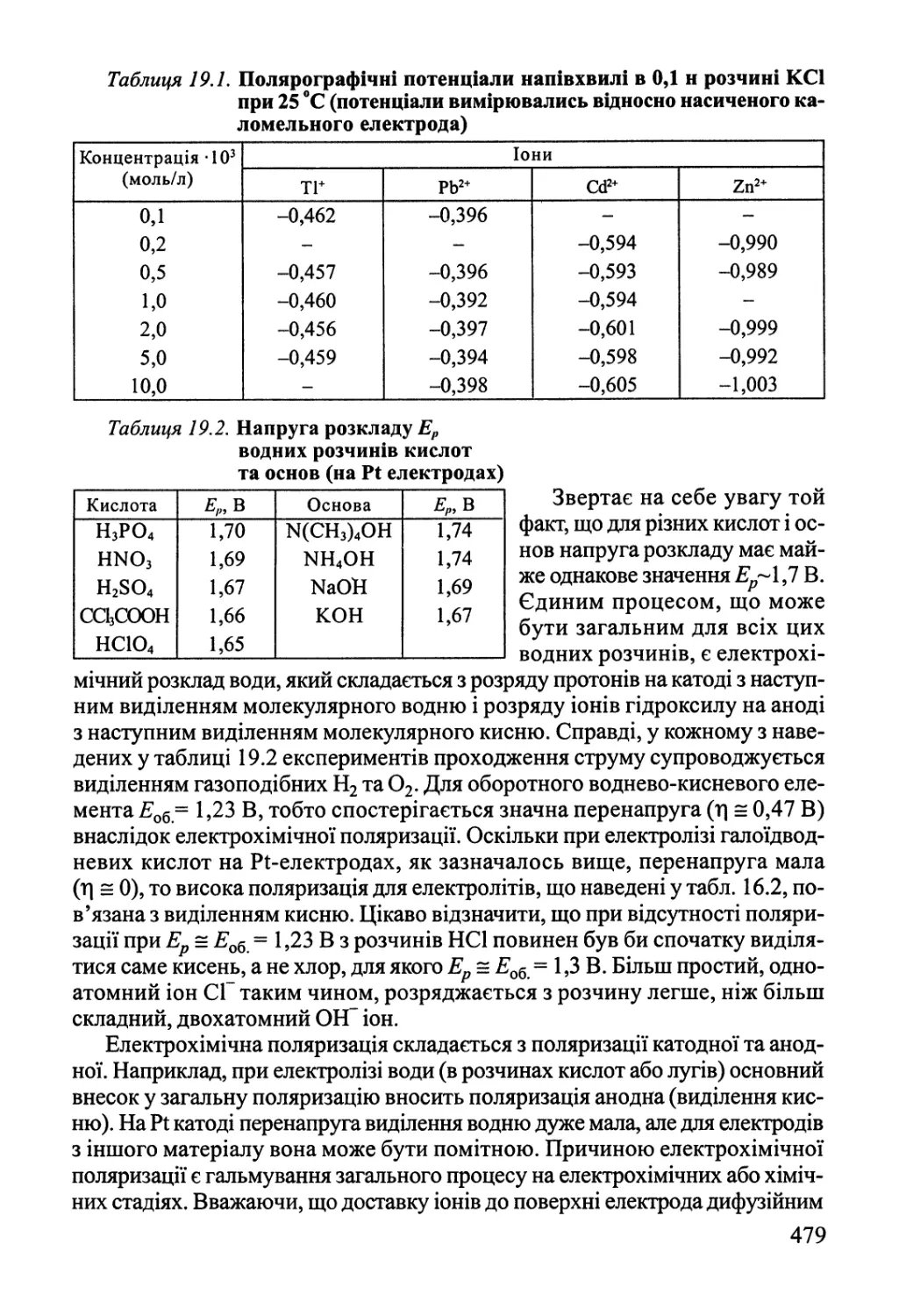
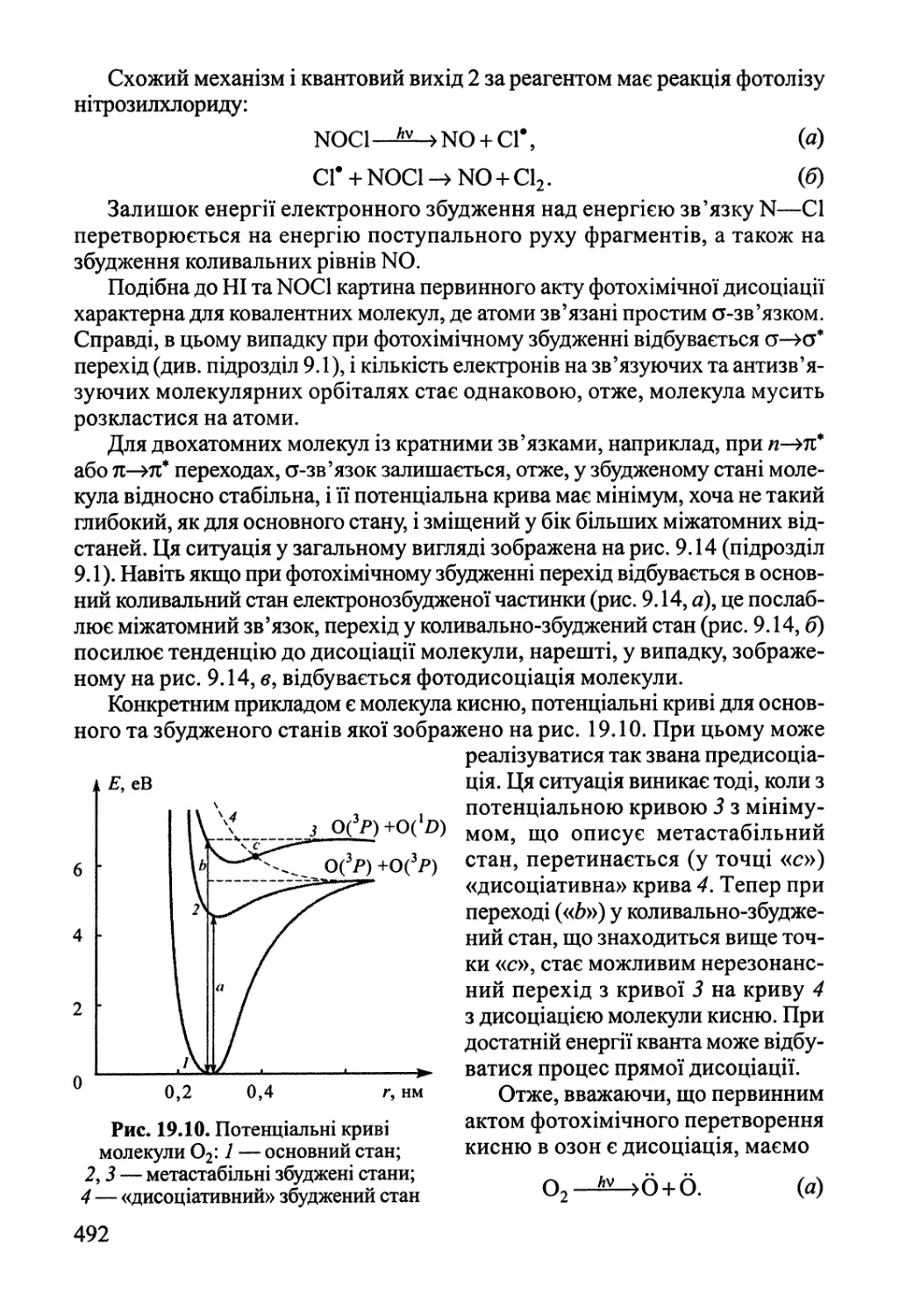
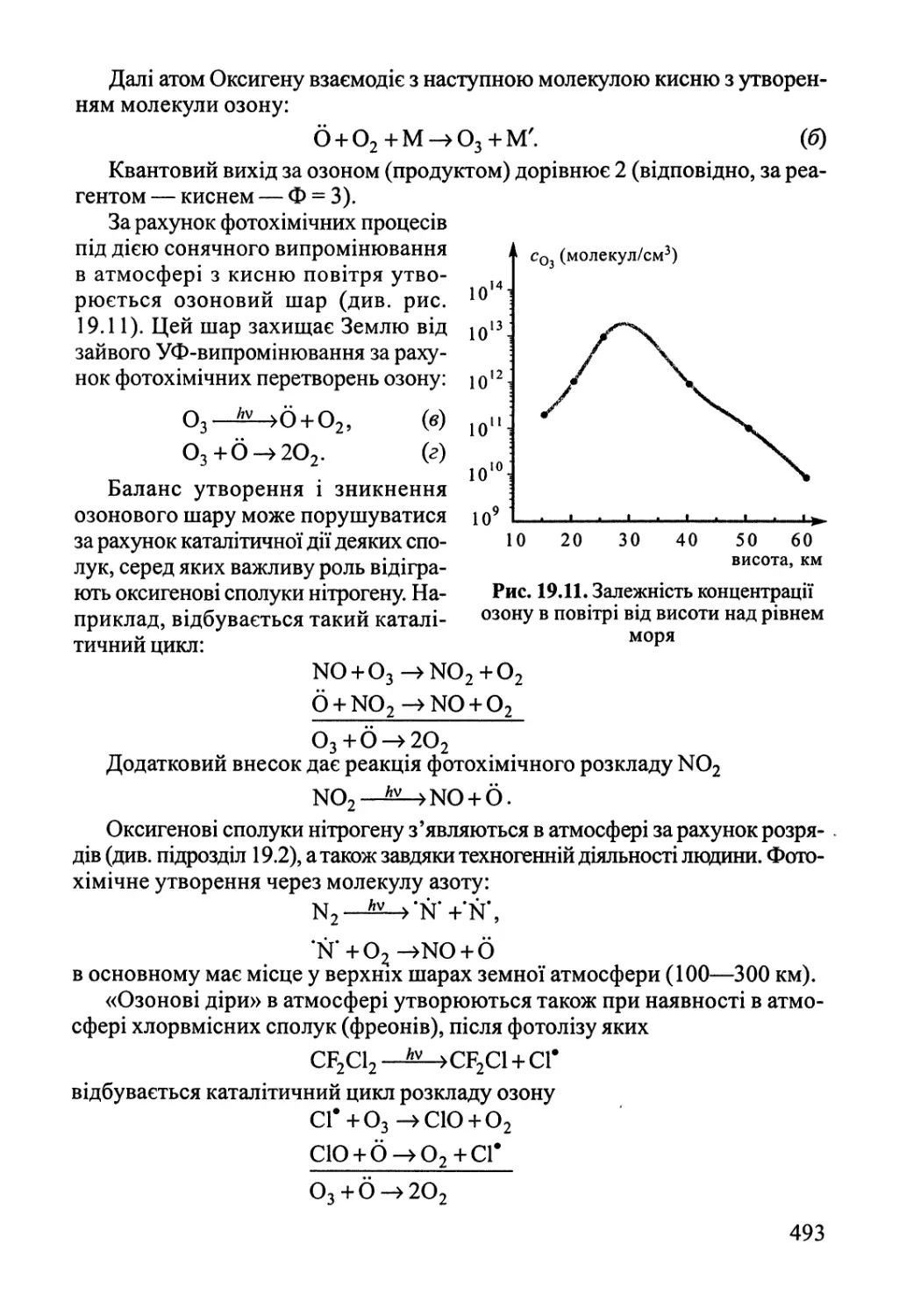
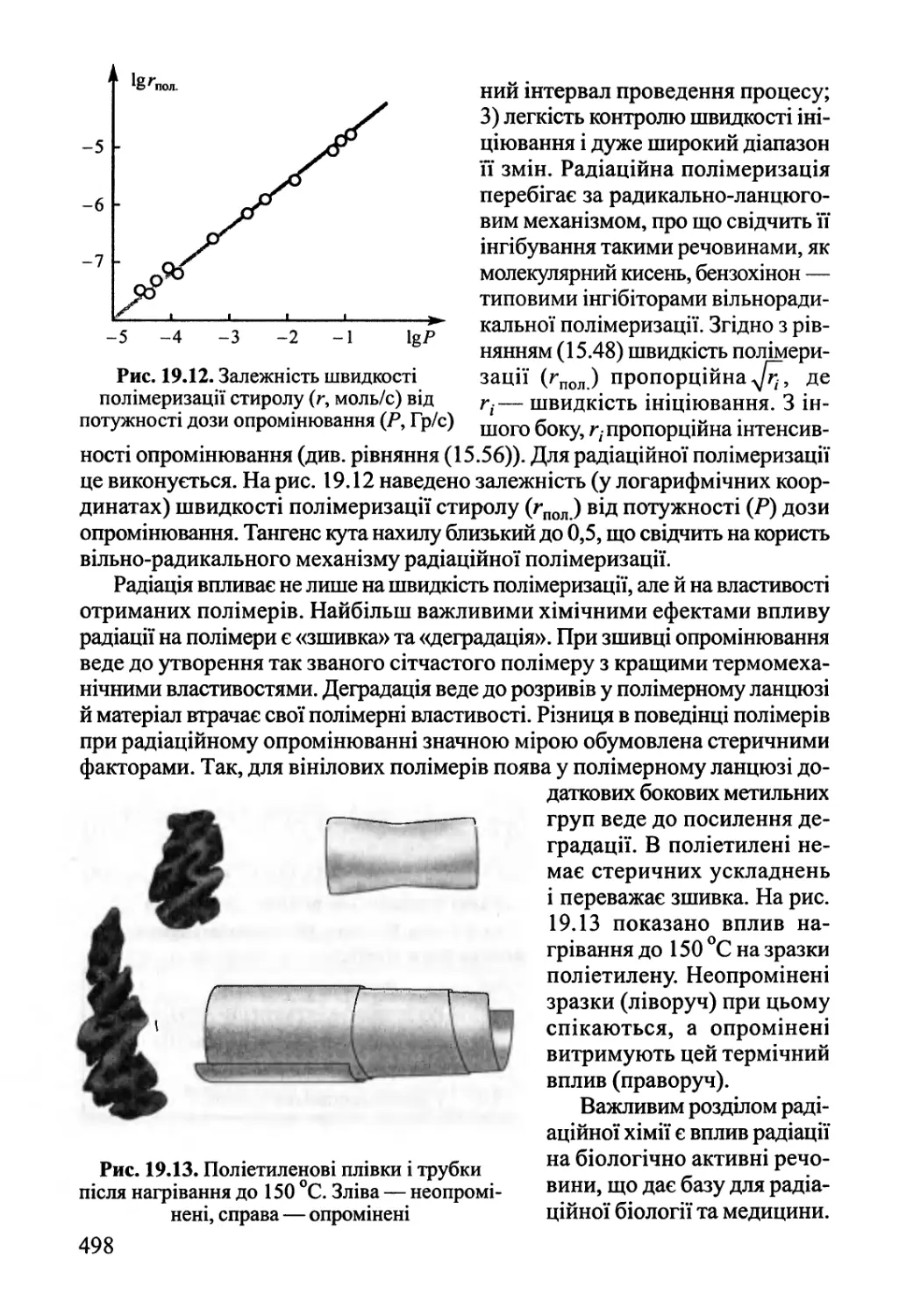
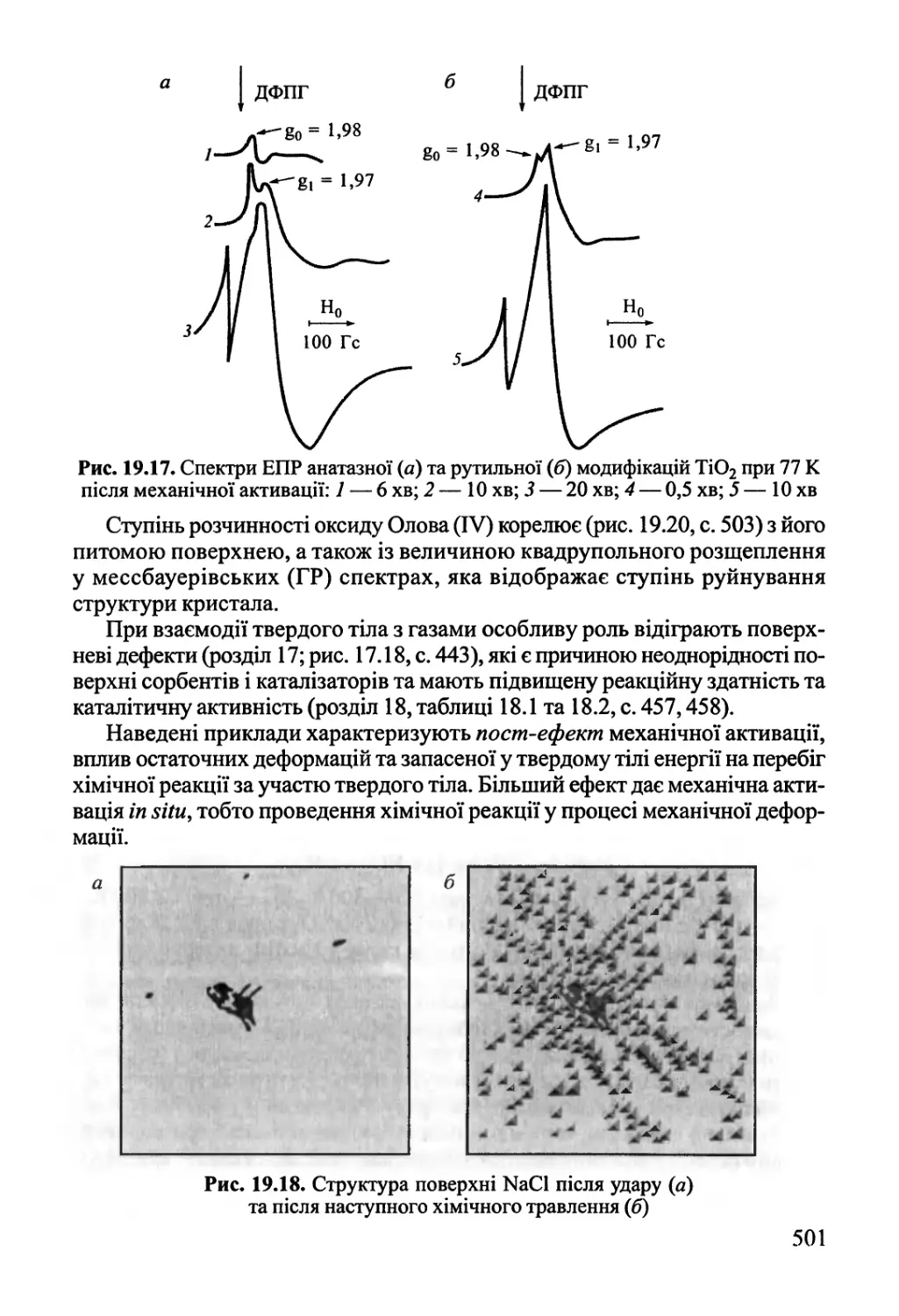
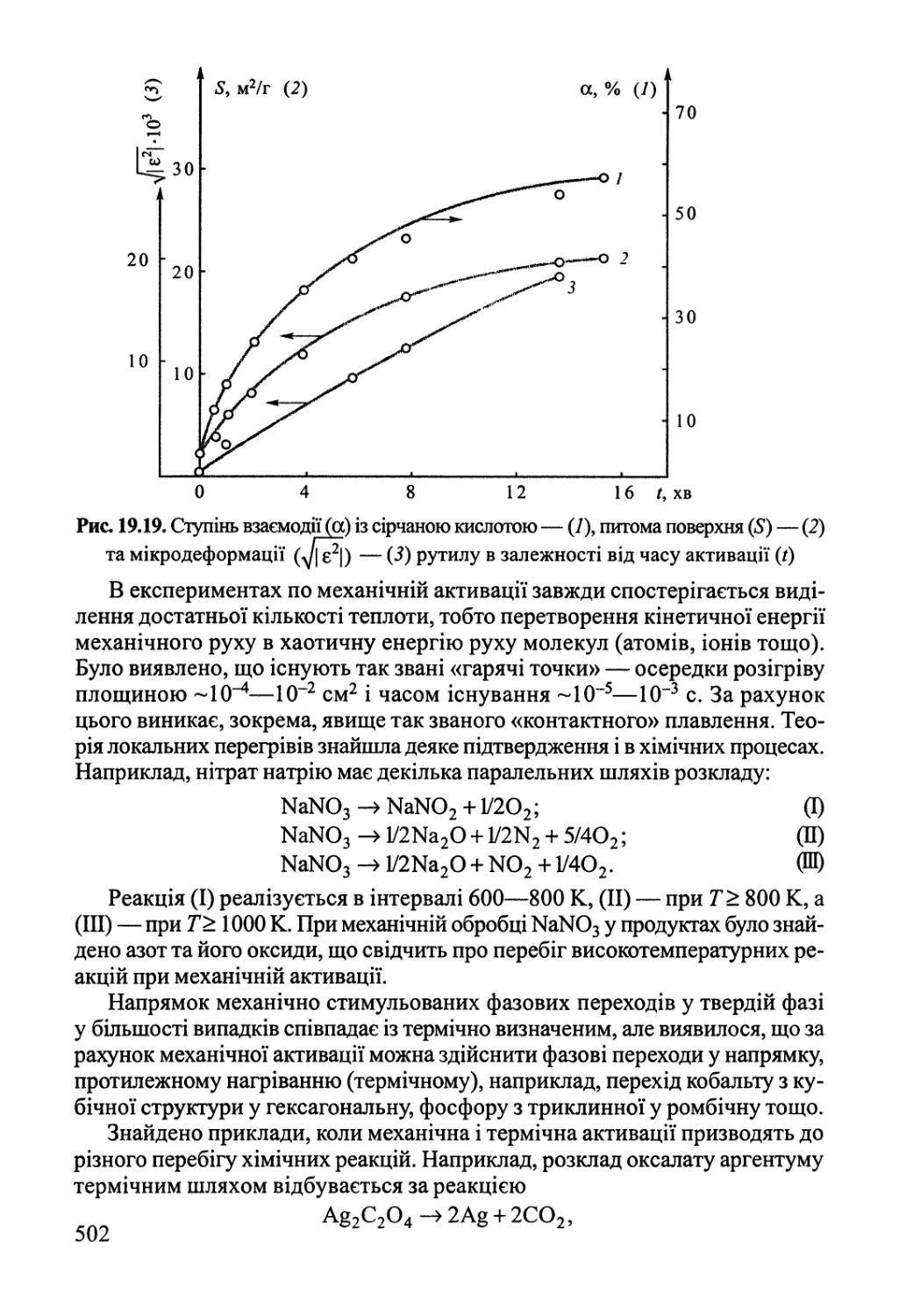
Розділ 19. Хімічні реакції під дією зовнішніх фізичних факторів 474
19.1. Електроліз, електродні процеси 474
19.2. Реакції у розрядах — газова електрохімія 484
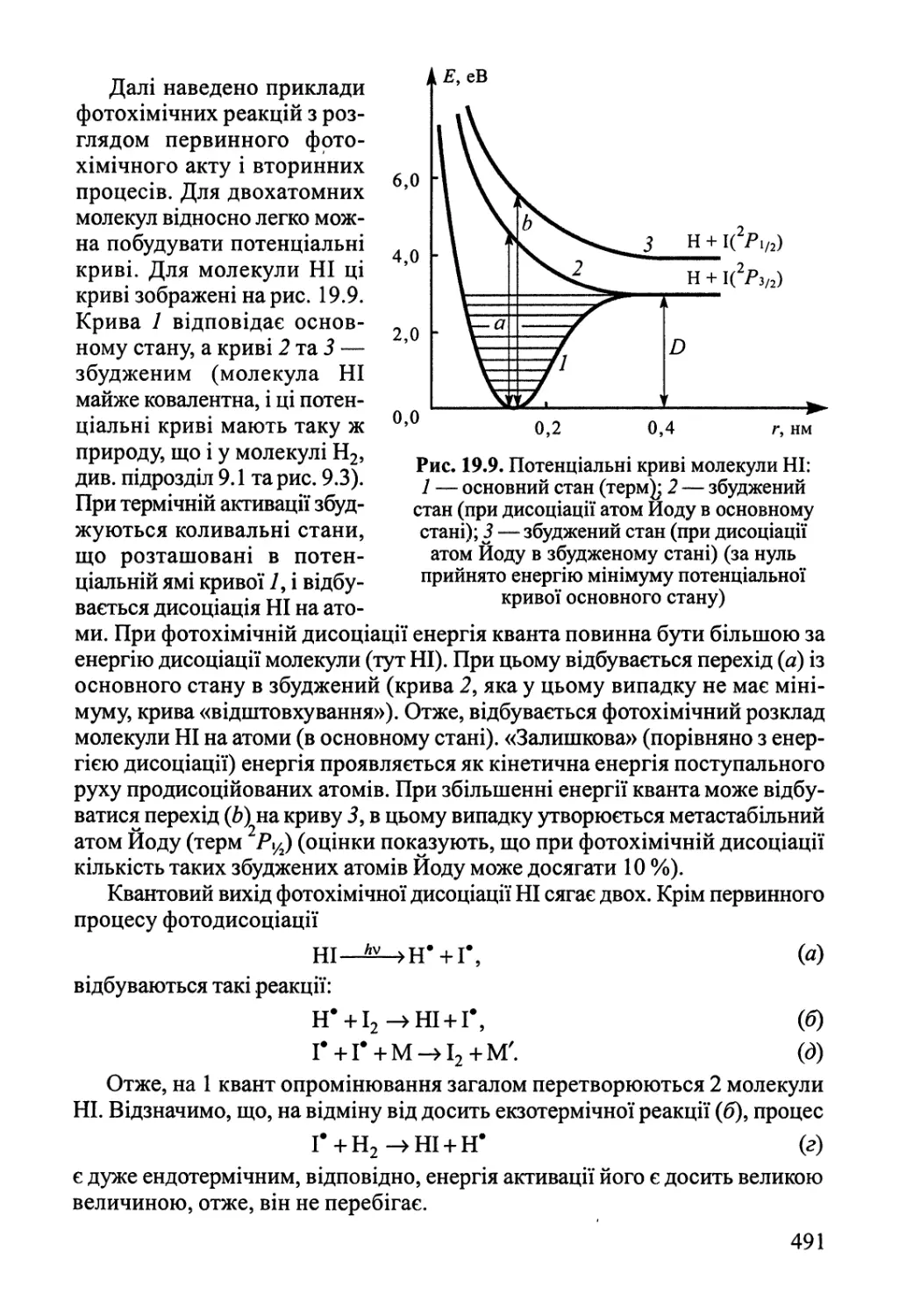
19.3. Фотохімія 489
19.4. Радіаційна хімія 495
19.5. Механохімія 499
Література 508
Додатки 510
ПЕРЕДМОВА
Фізична хімія — це наука, що вивчає проблеми, які лежать на межі між
фізикою й хімією. Таке визначення обґрунтовується двома чинниками. По-перше, це
застосування фізичних концепцій, теорій, математичного апарату фізики до
розгляду загальних положень хімії. Розділи, де вивчаються ці питання, варто було б
назвати теоретичною хімією. До речі, саме таку назву мав перший підручник із
фізичної хімії, виданий В. Нернстом. По-друге, фізична хімія повинна вивчати
вплив на хімічні процеси фізичних факторів: електрики, світла тощо. До цих
розділів фізичної хімії належать електрохімія, фотохімія, механохімія тощо.
Теоретична хімія стоїть на трьох «китах»: хімічній термодинаміці (вчення про
рівновагу), будові речовини (хімічний зв'язок, міжмолекулярна взаємодія,
структура твердих тіл, рідин тощо), хімічній кінетиці (вчення про швидкості та механізми
хімічних реакцій).
Явища на межі поділу фаз мають свою специфіку і виділяються в окремий
розділ — фізичну хімію міжфазних (поверхневих) явищ. Особливо сильний вплив
поверхневі явища мають у дисперсних системах, тому фізична хімія дисперсних
систем відокремлюється від фізичної хімії під назвою колоїдна хімія.
У цьому підручнику матеріал викладено у такому порядку.
Спочатку подано основи хімічної термодинаміки. Потім із термодинамічних
позицій розглянуто однокомпонентні системи, включаючи термодинаміку фазових
переходів.
Вивчення рівноважних багатокомпонентних систем починається з
висвітлення властивостей розчинів неелектролітів, а потім електролітів,
продовжується розглядом фазових рівноваг і завершується розділами «Хімічна
рівновага» та «Електрохімічна рівновага».
У розділах, присвячених питанням будови речовини та хімічного зв'язку,
викладено основи квантової теорії, розглянуто одноелектронну систему — атом
Гідрогену, двохелектронну — атом Гелію та багатоелектронні атоми. Будову
молекули подано на основі методів молекулярних орбіталей та валентних зв'язків,
починаючи з двохатомних молекул, а потім багатоатомних з урахуванням вчення про
симетрію. Окремі підрозділи присвячено хімічному зв'язку в органічних
молекулах, у координаційних сполуках та у твердому тілі.
Статистична термодинаміка завершує розгляд рівноважних систем. На
відміну від феноменологічного підходу класичної термодинаміки, статистична
ґрунтується на законах взаємодії великої кількості частинок, з яких складається система.
У термодинаміці, яка вивчає рівноважні системи, поняття процес
використовується, так би мовити, «за кадром», тобто вивчаються початковий та кінцевий
стани системи і визначається лише напрямок процесу. Кінетика вивчає механізм
процесу в його перебігу, тут з'являється така змінна, як час.
Викладання кінетики починається з розгляду «Основ хімічної кінетики» та
«Кінетики елементарних процесів». Після детального аналізу кінетики в
гомогенних системах, у тому числі гомогенного каталізу, наводяться необхідні відомості
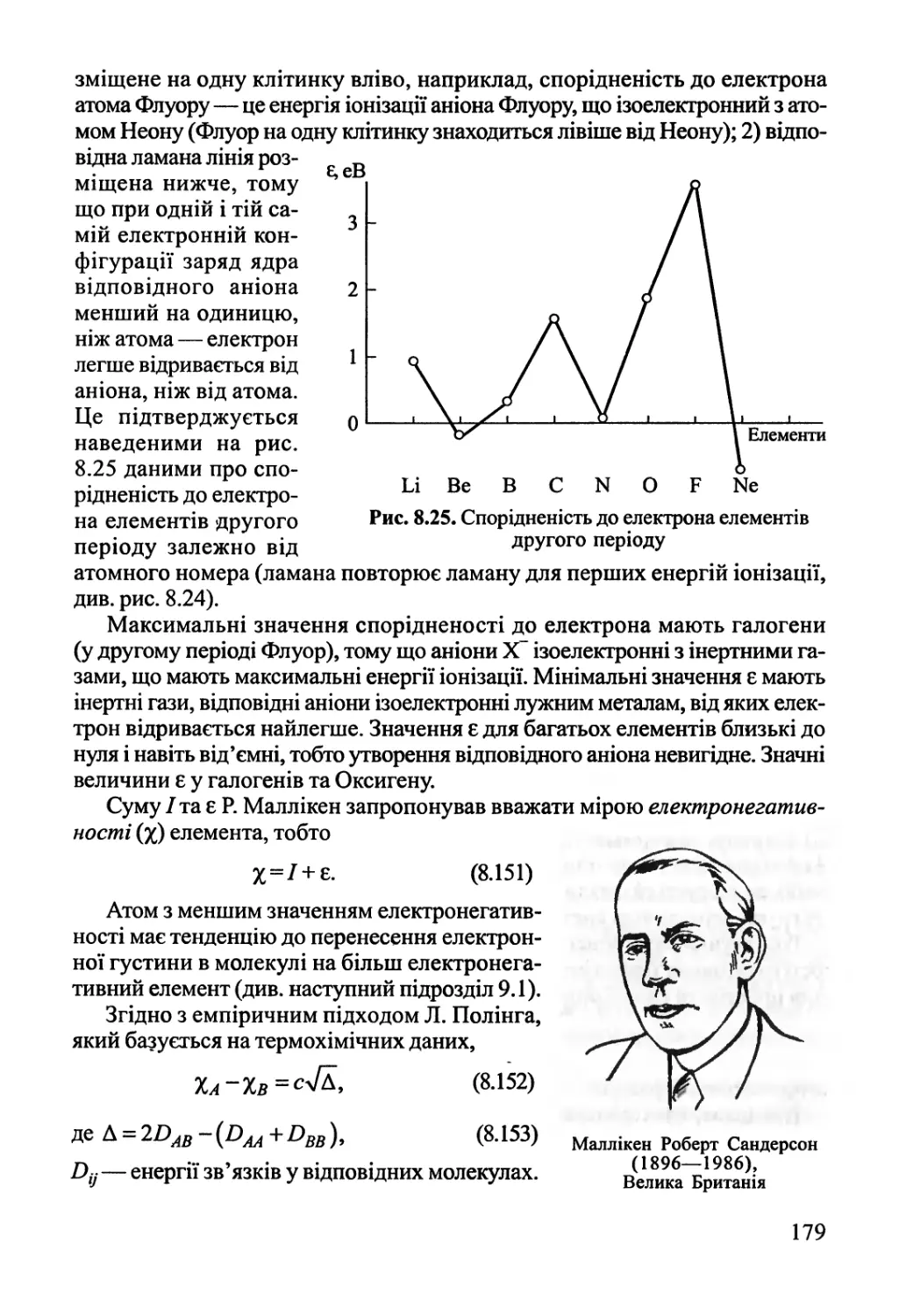
з фізичної хімії міжфазних явищ, а далі іде гетерогенний каталіз.
Підручник завершується викладом перебігу хімічних реакцій під дією
зовнішніх фізичних факторів: електрики — електродні процеси, світла —
фотохімія, радіації—радіаційна хімія, механічних сил —механохімія, а також перебігу
хімічних реакцій у плазмі.
РОЗДІЛ 1.
ОСНОВИ ТЕРМОДИНАМІКИ
Якщо одержання роботи є метою,
А не насичення системи теплотою,
Вільною має енергія бути,
Щоб ентропії здолати всі пути.
1.1. Предмет термодинаміки
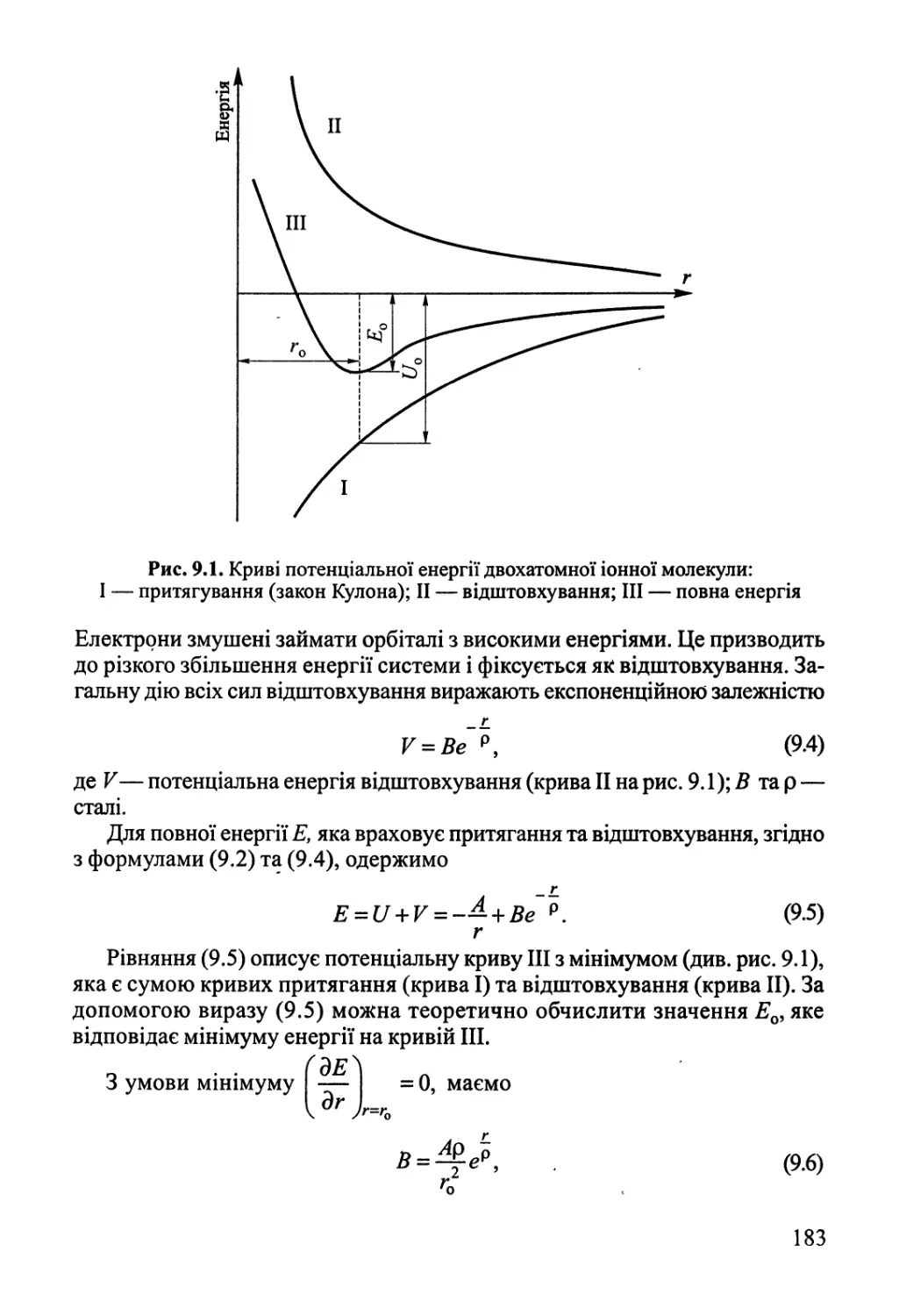
Термодинаміка як наука виникла зі спроб теоретичного обґрунтування
роботи теплових машин, тобто агрегатів, що перетворюють теплоту в
роботу (рис. 1.1). Про це свідчить і назва цієї науки: термо — теплота,
динаміка — рух, сила. Подальший її розвиток показав, що за допомогою
термодинаміки можна передбачити напрям самочинного перебігу процесів як
фізичних, так і хімічних і ретельно вивчати рівноважні стани різних систем.
Рис. 1.1. Схема теплової машини
Термодинаміка спирається на ряд загальних положень, що випливають
із багатого практичного досвіду, експериментальних фактів і мають назйу
начал (постулатів) термодинаміки. Ці положення пов'язані з перетворенням
різних видів енергії, причому особливе місце займає теплота — хаотичний
рух частинок, що складають систему.
Термодинаміка має дуже широке коло своїх застосувань: термохімія,
термодинаміка розчинів, фазові й хімічні рівноваги, термодинаміка міжфаз-
них явищ, термодинаміка гальванічних елементів тощо.
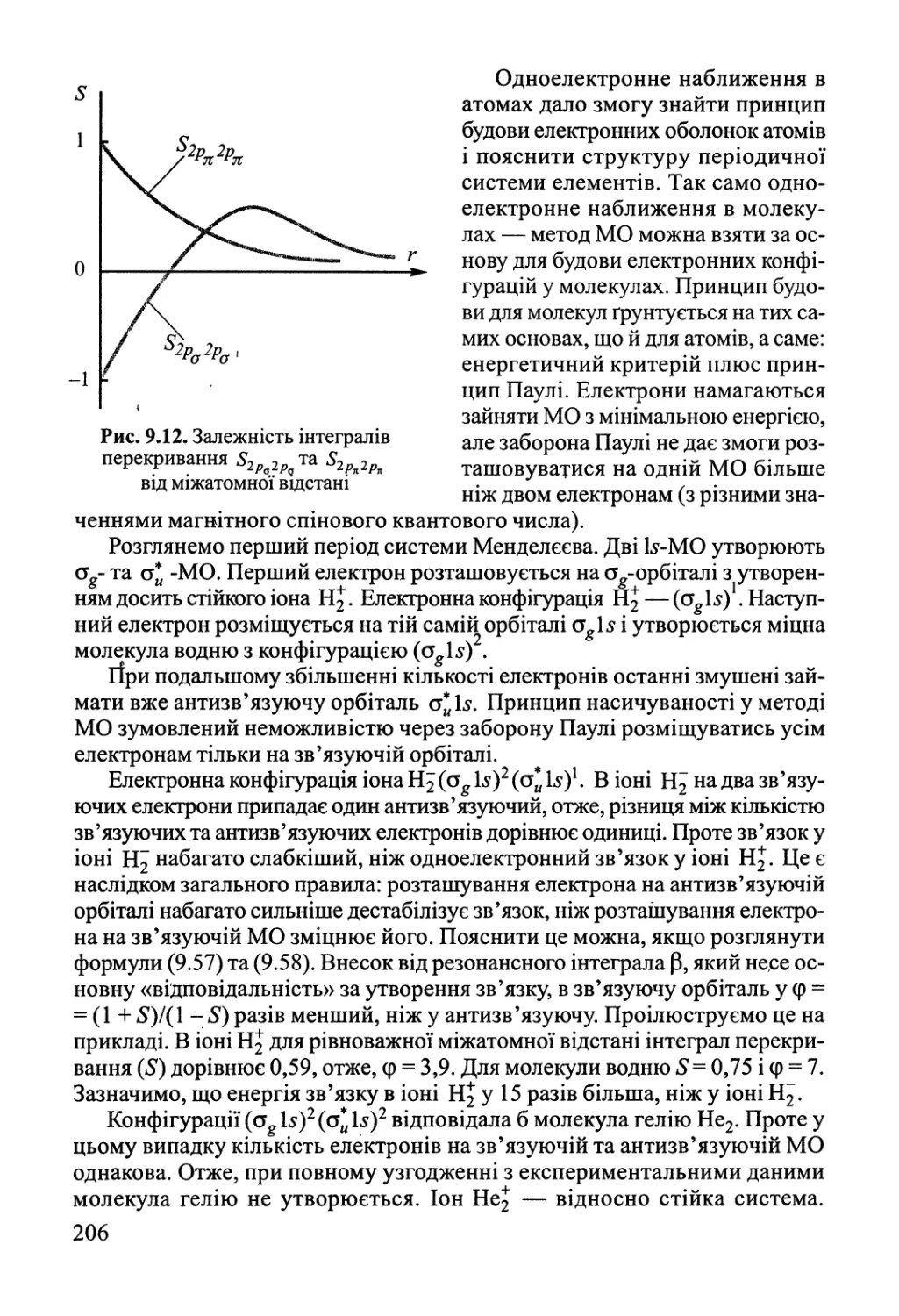
Одним з основних понять термодинаміки є система. Під системою
розуміють тіло або сукупність тіл, відокремлених від зовнішнього середовища,
довкілля поверхнею поділу. Система, що обмінюється із зовнішнім
середовищем речовиною та енергією, має назву відкритої. Закрита система
обмінюється із довкіллям лише енергією. Система, що не обмінюється із
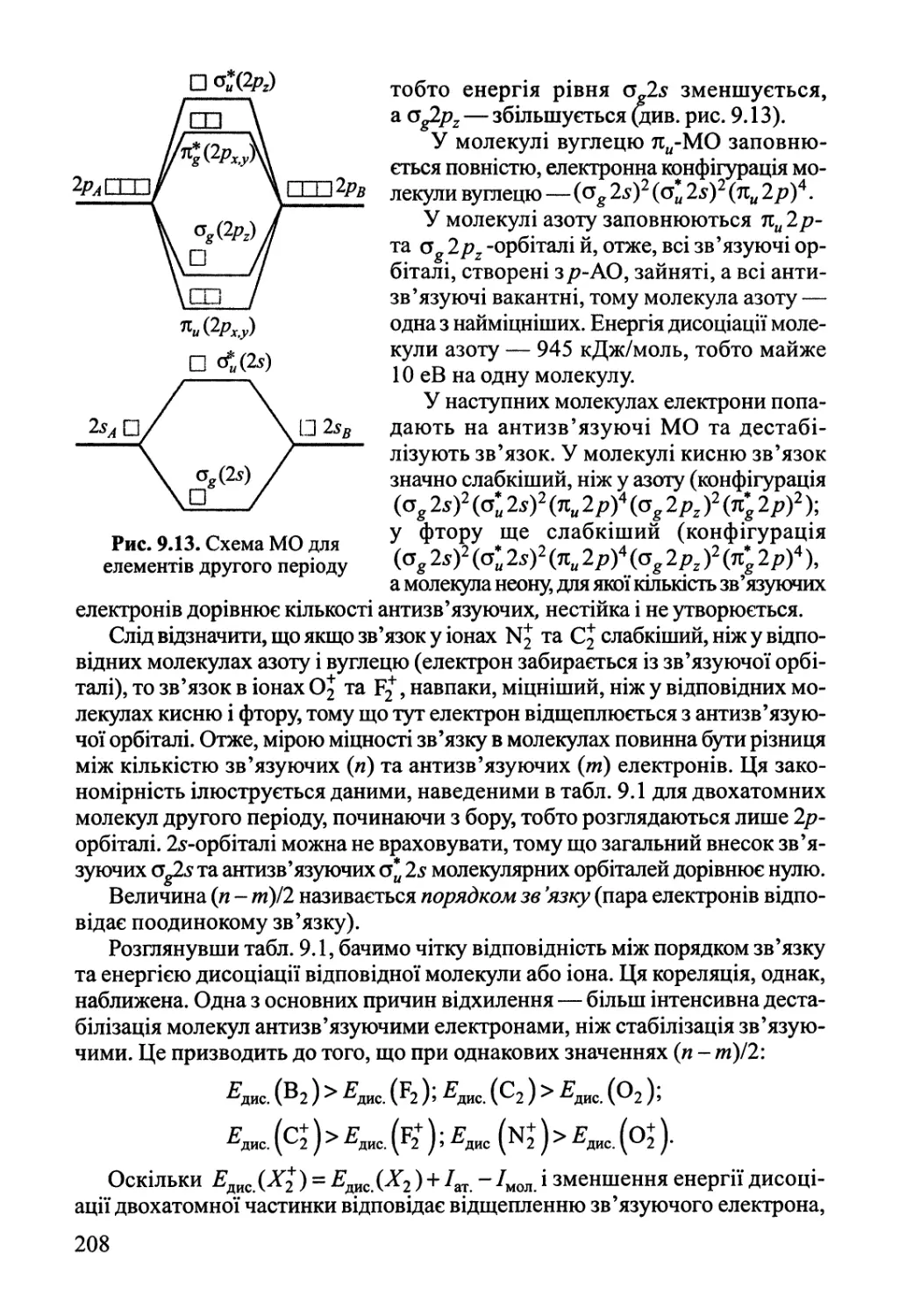
навколишнім середовищем ані речовиною, ані енергією, має назву ізольованої.
1
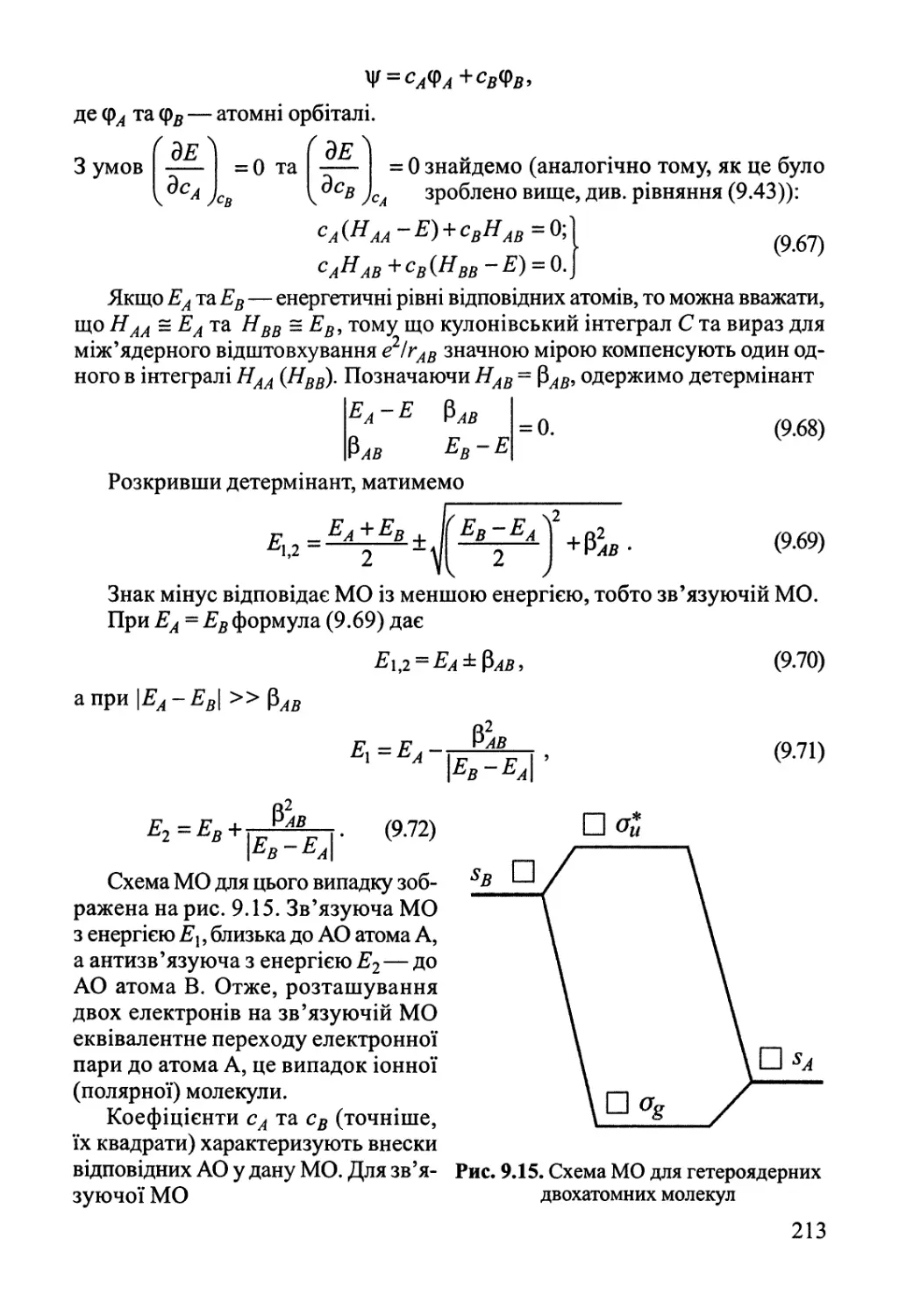
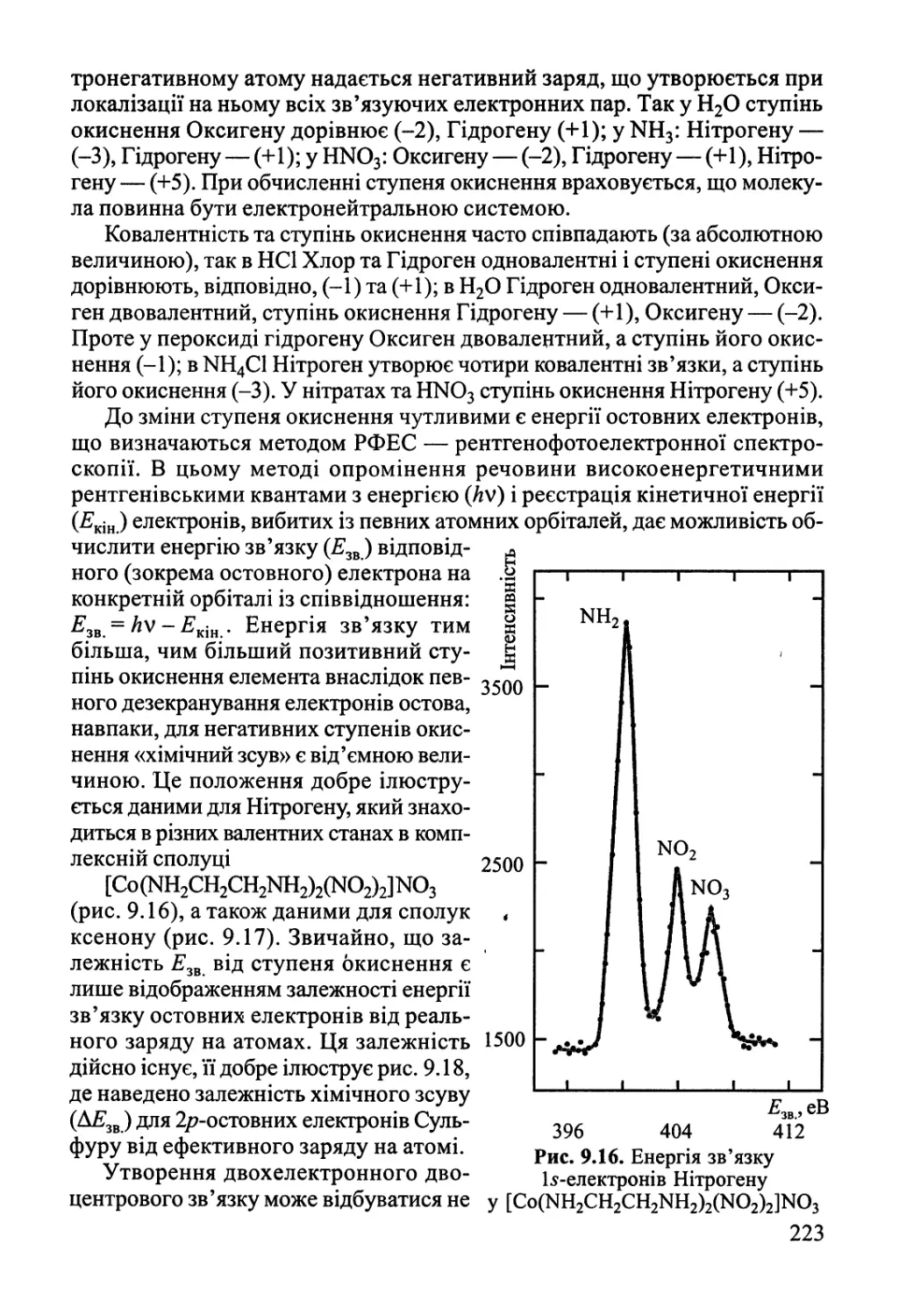
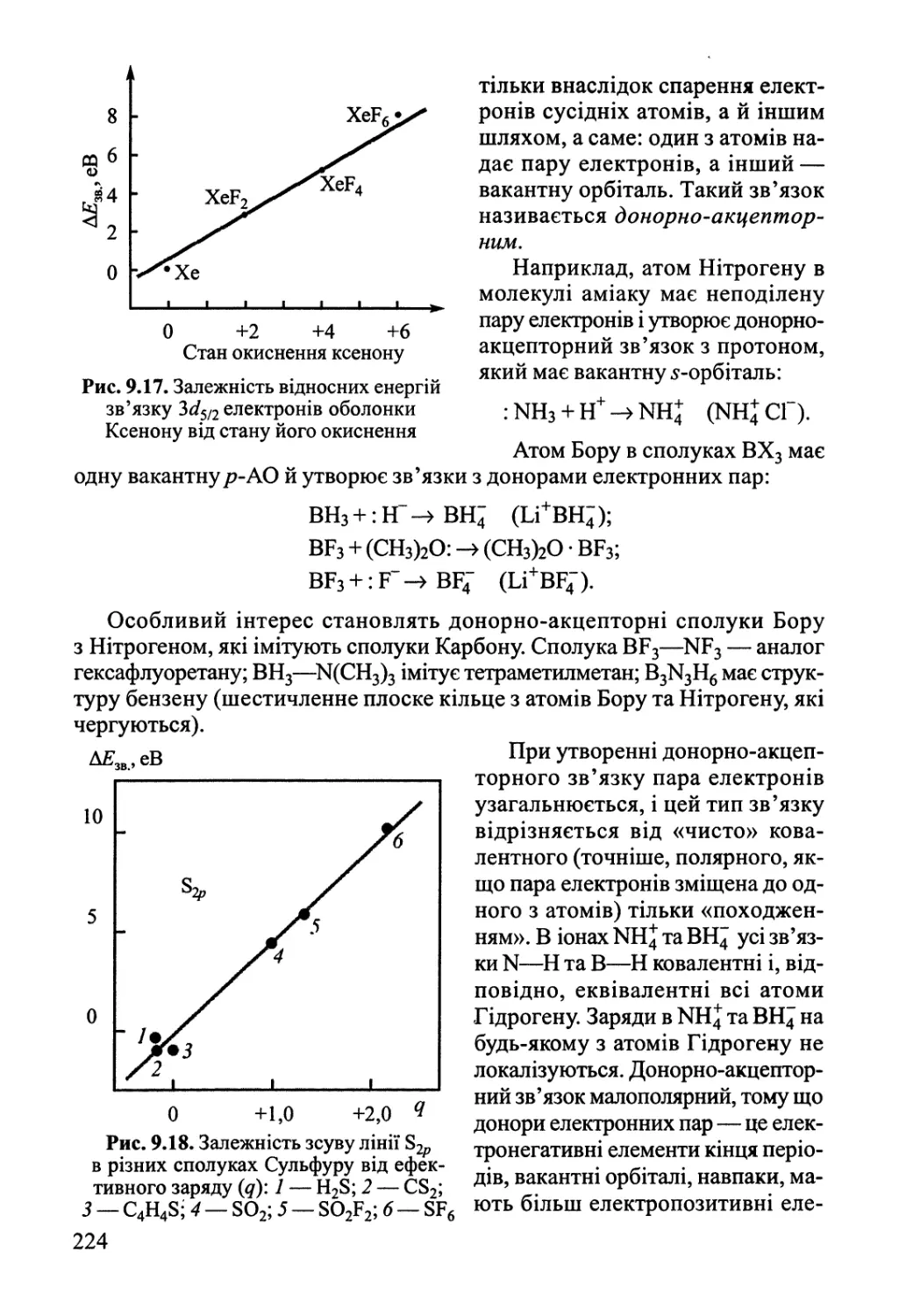
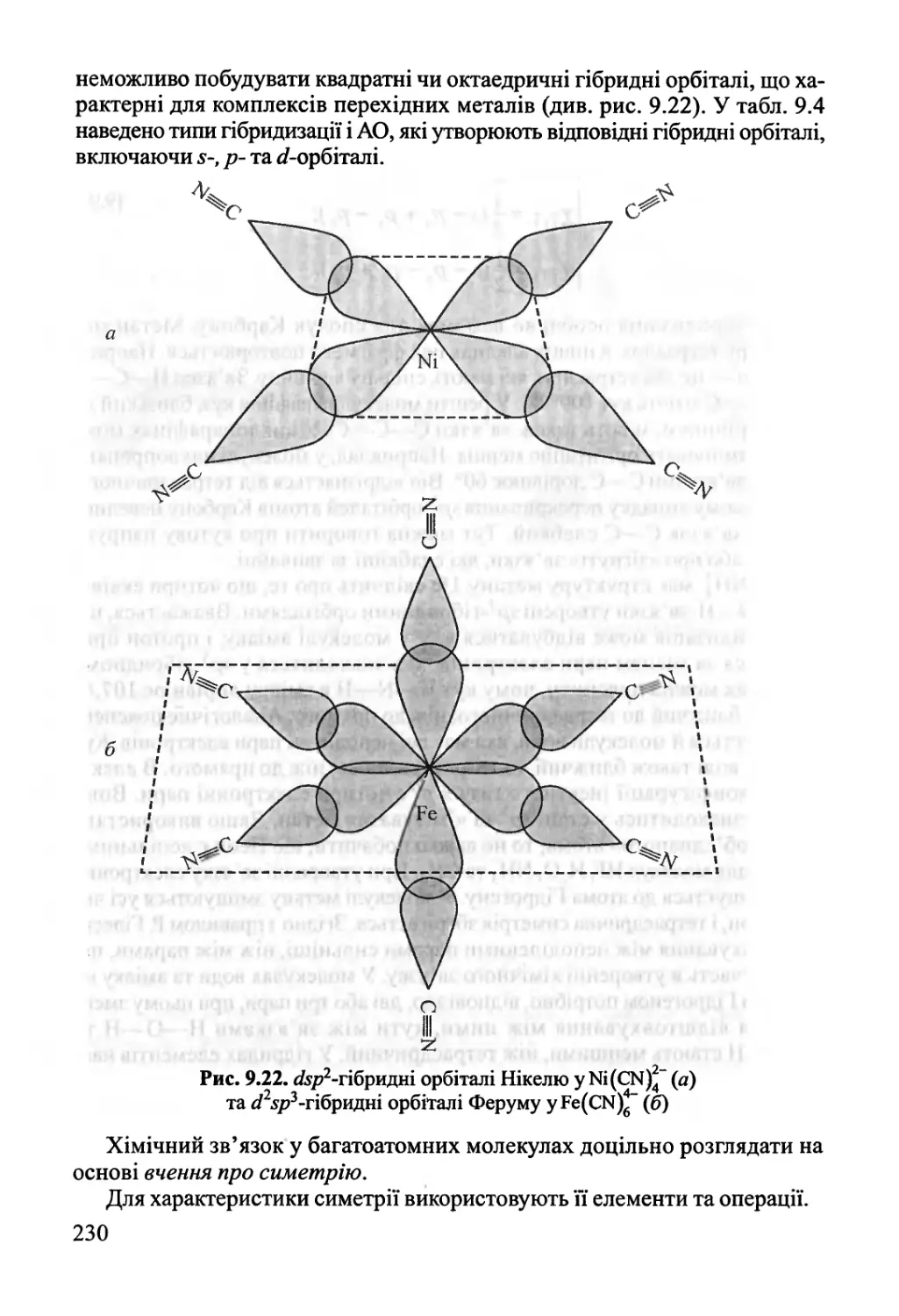
Кожна система описується за допомогою змінних, які називаються
параметрами системи. До параметрів системи належать тиск, температура,
концентрація речовин і ряд інших змінних.
Частина системи, що має однакові фізичні й хімічні властивості та
відокремлена від решти системи поверхнею поділу, має назву фази.
Прикладом двох різних фаз можуть бути рідина та пара над нею; дві рідини, що не
змішуються, наприклад вода та бензен тощо. Система, що складається тільки
з однієї фази, називається гомогенною, а з кількох фаз — гетерогенною.
До кожної системи може входити одна або декілька складових речовин,
що мають назву компонентів (уточнення поняття «компонент» буде
наведено далі у підрозділі 5.1). Відрізняють відповідно одно-, дво-, три- і т. д.
компонентні системи. Наприклад, суміш льоду з водою — це однокомпонентна
система, а розчин сахарози у воді — двокомпонентна.
Далі буде доведено (підрозділ 5.1), що для рівноважної системи діє так
зване правило фаз Гіббса:
/=п+2-к9 (1.1)
де п — кількість компонентів; к—кількість фаз; /— кількість ступенів
свободи, тобто кількість незалежних параметрів, за допомогою яких описується
стан системи.
Для гомогенної однокомпонентної системи п = 1 і к= 1, отже,/= 2. Тому,
якщо для опису системи користуватися такими трьома параметрами, як
тиск, об'єм і температура, то тільки два з них є незалежними. Отже, між
цими параметрами повинен існувати зв'язок, що забезпечується рівнянням
стану.
Одним із найпростіших рівнянь стану є рівняння Менделєєва-Клапей-
рона для ідеального газу, яке є узагальненням емпіричних законів Гей-Люс-
сака, Бойля-Маріотта і Шарля:
рУ8 = пКТ, (1.2)
дер—тиск, У8—об'єм системи, Т—температура, п — кількість молів газу,
К —універсальна газова стала {К = 8,31 Дж/моль-К = 0,082 л-атм/моль-К).
Температура (Т) у термодинаміці визначається шкалою Кельвіна (К), яка
пов'язана зі шкалою Цельсія (°С) простим співвідношенням
Т = 1 + 213. (1.3)
Якщо ввести мольний об'єм газу V = У8/п, то рівняння (1.2) набуває
вигляду
р¥ = КТ. (1.4)
За допомогою параметрів можна описувати функції стану системи.
Особливою рисою функцій стану є те, що при переході від одного стану
до іншого зміна функції стану не залежить від шляху процесу, а визначається
лише кінцевим і початковим станами системи. З математичної точки зору,
8
інтегральна функція, що не залежить від шляху процесу, повинна бути
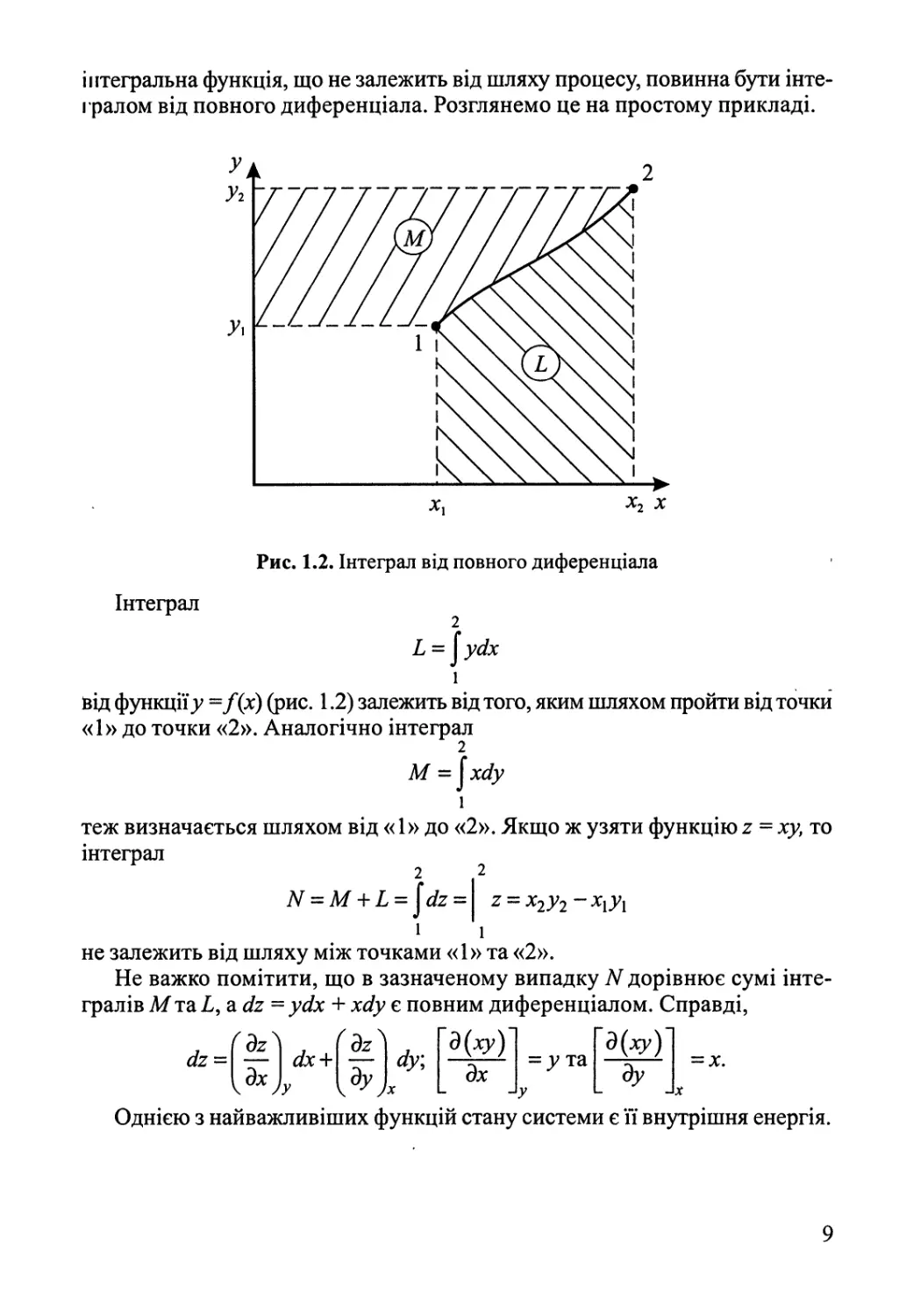
інтегралом від повного диференціала. Розглянемо це на простому прикладі.
Х2 X
Рис. 1.2. Інтеграл від повного диференціала
Інтеграл
Ь= \уйх
і
ьід функції;; =Дх) (рис. 1.2) залежить від того, яким шляхом пройти від точки
«1» до точки «2». Аналогічно інтеграл
2
М = \хйу
теж визначається шляхом від «1» до «2». Якщо ж узяти функцію т, = ху, то
інтеграл
М = М + Ь = $сІ2 =
2 = Х2у2~Х1у1
1
не залежить від шляху між точками «1» та «2».
Не важко помітити, що в зазначеному випадку N дорівнює сумі
інтегралів Мта І, а йі = усіх + хйу є повним диференціалом. Справді,
<І2-\ — йхЛ-
Ьх)у
(
II*
дх
= ута
ду
= х.
Однією з найважливіших функцій стану системи є її внутрішня енергія.
9
1.2. Перше начало термодинаміки
Внутрішня енергія ({/) системи визначається як сумарна енергія всіх
видів руху частинок у системі: поступального, обертального, коливального
тощо. До внутрішньої енергії не належать потенціальна енергія (Еп)
взаємодії системи з навколишнім середовищем (наприклад гравітаційна
енергія) та кінетична енергія (Ек) руху системи як цілого. Суму (Еп + Ек), на
відміну від внутрішньої енергії (II), можна було б назвати «макроенергією»
системи, а суму (Еп + Ек+ II) — повною енергією системи.
Закрита система може змінювати свою внутрішню енергію шляхом
обміну енергією із зовнішнім середовищем. Існує два способи такого обміну:
теплота (#) і робота (и>).
Теплопередача від системи до системи здійснюється за рахунок
хаотичного руху частинок, що складають систему. Якщо система віддає теплоту
(екзотермічний процес), то д вважається від'ємною величиною, а якщо
одержує (ендотермічний процес), то ^ є додатною величиною. Зауважимо,
що, крім термодинамічної шкали, існує також інша, так звана термохімічна,
де знаки я протилежні. Не важко помітити, що термохімічна шкала
розглядає систему з погляду зовнішнього спостерігача, в той час як
термодинамічна — з точки зору системи.
Іншим способом передачі енергії є виконання роботи самою системою,
або виконання роботи над системою. Логічно для роботи використовувати
таку ж систему знаків, що й для теплоти, а саме: коли система виконує
роботу (і при цьому втрачає енергію), то вважати м> від'ємною величиною, а
коли робота виконується над системою, то м> є додатною величиною.
Якщо система не обмінюється енергією із довкіллям (ізольована система),
то її внутрішня енергія залишається сталою величиною: І] = сопзі. Це
положення відоме як закон збереження енергії.
Перше начало термодинаміки узагальнює це положення й розглядає,
крім внутрішньої енергії, теплоту # і роботу м>, а саме
ДС/ = ? + и>, (1.5)
де Д(/ = (Ї72 ~ ^і) — зміна внутрішньої енергії системи.
Таким чином, приріст внутрішньої енергії (ДС/> 0) у системі виникає за
рахунок підведення до системи теплоти (+#) та виконання роботи над
системою (+м>), або навпаки, кількість роботи, що виконується системою (-м>),
та кількість відданої нею теплоти (-#) дорівнює витраті внутрішньої енергії
(АII < 0). За відсутності обміну енергією між системою та довкіллям д = 0
IV = 0 та ДС/= 0, тобто V = соті, як зазначалося вище для ізольованої системи.
Для нескінченно малих величин перше начало термодинаміки має такий
вигляд
</і7 = 8? + 5м;. (1.6)
Внутрішня енергія є функцією стану і, отже, <Ш— повний диференціал.
10
Позначка «8» означає, що 8# та 8м> не є повними диференціалами, тому що
<7 і м> залежать від шляху перебігу процесу.
Для ізотропних систем (рідина, газ, аморфне тверде тіло, полікристал,
складений із дуже маленьких кристалів) найбільш важлива робота
стиснення (розширення) системи
8и> = -/?</К. (1.7)
У процесі стиснення маємо сІУ<0, отже, 8м; > 0 — робота виконується
над системою, при розширенні (IV> 0 і 8м> < 0 — робота виконується самою
системою в повній відповідності до встановленої системи знаків для и>. Якщо
підставити (1.7) в (1.6) (тобто вважати, що, крім роботи розширення
(стиснення), інші види роботи відсутні), то одержимо
Л/ = 8д-/и/Г. (1.8)
За допомогою цього рівняння розглянемо різні процеси.
Ізохорний процес — У= соті, отже, дУ = 0 і, відповідно, з рівняння (1.8):
</С/ = 8^. (1.9)
У процесі ізохорного нагрівання системи її температура збільшується
на величину */Г(при розгляді нескінченно малих величин). Між кількістю
теплоти 8ду, що одержала система у процесі ізохорного нагрівання, та
зміною температури йТіснує зв'язок
^у=Су<ІТ. (1.10)
Величина Су має назву теплоємності (при фіксованому об'ємі). 3(1.10)
та (1.9) одержимо
(Ш=СусІТ. (1.11)
Рівняння (1.10) та (1.11) можна використати для визначення теплоємності:
^ 8?г (дЦЛ
с"=!=[з?ф <1Л2>
Якщо Су = соті, то рівняння (1.10) або (1.11) легко інтегруються від стану
«1» до стану «2», звідки
^ А^ Чу
<^дГ;&. 0-із)
деАС/=С/2-І71,ДГ=772-Г1.
Якщо Су по стала величина, то рівняння (1.13) дає так звану середню
теплоємність у температурному інтервалі від Тх до Т2, на відміну від істинної
теплоємності, що дається рівнянням (1.12). Теплоємність (як істинну, так
і середню) можна віднести до всієї системи або до будь-якої кількості
речовини: до одиниці маси — питома, до одного моля —мольна. З рівняння
(1.13) випливає також наочне визначення теплоємності. При ДГ= 1 С^чисель-
но дорівнює ду, тобто теплоємність — це кількість теплоти, що потрібна для
11
нагрівання системи (одиниці маси, моля) на один градус. Зазначимо, що
калориметричні вимірювання в ізохорних процесах дають можливість
безпосередньо оцінити зміну внутрішньої енергії системи.
Ізобарний процес. Робота ізобарного процесу визначається
інтегруванням рівняння (1.7) за р = сопзі
м = -рАГ. (1.14)
Для ідеального газу, використовуючи рівняння Менделєєва-Клапейрона
(1.4), маємо для роботи
м/ = -ДД7\ (1.15)
Згідно з першим началом термодинаміки
М/ = ф,-рДК, (1.16)
де др — теплота за постійним тиском, звідки
Ф = ДС/ + рДК=С/2-І/і+р(Г2-Кі) = (С/2+/?Р2)-(С/і-рГі). (1.17)
Рівняння (1.17) показує, що величина др (як і #г = Д £/) не залежить від
шляху процесу, а визначається кінцевим «2» та початковим «1» станами
системи. Це означає, що існує функція стану
Н=Ц + рГ, (1.18)
різниця значень якої для двох станів дає величину др, тобто
др = АН = Н2-Н1. (1.19)
Ця функція має назву ентальпія.
У диференціальній формі
Ш = сН/ + сІ(рУ). (1.20)
сШ, як і сШ, є повним диференціалом (диференціал добутку сі(рУ) також
є повним диференціалом, що було у загальній формі розглянуто вище у
підрозділі 1.1).
За рівнянь, аналогічних (1.10) — (1.12), маємо
5др = сіН=Срс1Т, (1.21)
або
с=^=
днЛ
р ат
де Ср—теплоємність (істинна) за сталим тиском. Ср9 як і Су, може бути
питомою, мольною або віднесеною до всієї системи.
Залежність Ср або Су від температури можна врахувати емпірично як
поліном
С(р. V) = а + ЬТ + сТ2 + ... (1.23)
12
Теорію теплоємності газів і твердих тіл буде наведено далі на основі
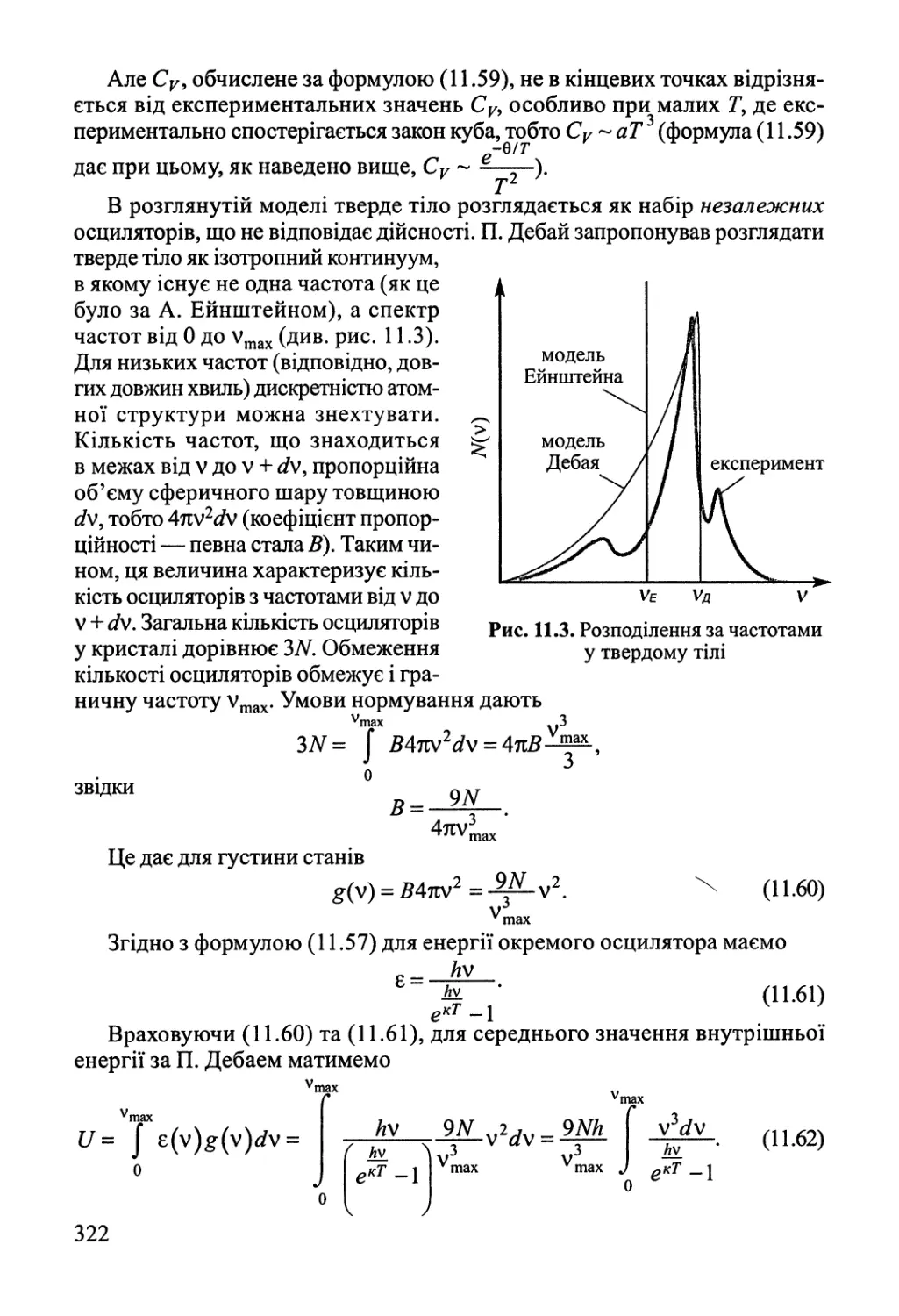
статистичної термодинаміки (підрозділ 11.6).
За допомогою співвідношень (1.18) та (1.19) рівняння (1.17) можна
записати таким чином:
АН = АЦ+рАУ, (1.24)
або з урахуванням Ср (1.22) та Су (1.12)
СрАТ = СуАТ + рАУ (1.25)
(за невеликого АГ Ср та Су можна вважати сталими величинами).
Проаналізуємо ці рівняння. У ході нагрівання від Тх до Г2 за постійного
об'єму величина (/збільшується на СУАТ. У процесі нагрівання від Тх до Г2
за постійного тиску внутрішня енергія змінюється на ту ж саму величину,
але система забирає більше теплоти (др = АН) з навколишнього
середовища, частина якої іде не на збільшення внутрішньої енергії системи, а на
виконання нею роботи (м> = -рАУ), ця частина енергії для системи
втрачається: АІ/ = АН-рАУ
Для ідеального газу, згідно з рівняннями (1.14), (1.15) та (1.25), маємо
СРАТ=СУАТ + КАТ.
Якщо це рівняння скоротити на АГ, то одержимо
СР=СУ + К. (1.26)
Загальний зв'язок між Ср та Су для будь-яких систем та його виведення
розглядатимуться далі (підрозділ 2.3). Відзначимо тільки, що для
конденсованих систем зазвичай А У дуже мала величина, тому при не досить великих
тисках добуток^АКтеж малий, отже, АН = АІ/і, відповідно, різниця між Ср і
і Су невелика.
Ізотермічний процес. При розгляді ізотермічного процесу обмежимося
тільки ідеальним газом як прикладом системи. Якщо Г= соті, робота
розширення ідеального газу дорівнює
Уг У2
м> = -їрс1У = -КТЇ — = -КТІп^ = ЯТІп^. (1.27)
-1 * V У р,
ух ух г у\ У\
За У2 > У\ (розширення) Іп(У2/У1) > 0, отже, м> < 0; за У2 < Ух (стиснення)
1п(К2/ Ух) < 0 і IV > 0, відповідно до прийнятої системи знаків.
В ідеальному газі частинки взаємодіють одна з іншою лише шляхом
зіткнення, вони мають тільки кінетичну енергію. Частинки не взаємодіють
потенціально, тобто не притягуються і не відштовхуються одна від одної за
рахунок якихось сил. Якби ті сили були, то енергія взаємодії залежала б, як
і сили, від відстані між частинками. Якщо сили відсутні, ця відстань не має
значення, тобто внутрішня енергія ідеального газу не залежить від об'єму,
який він займає,
13
\дУ)г .
і є функцією лише температури, яка визначає кінетичну енергію частинок.
Незалежність Д V ідеального газу від об'єму була експериментально
встановлена Ж. Гей-Люссаком . а Д/к. Джоулем. Теоретичне обґрунтування
цього положення буде ііаведено далі на базі статистичної термодинаміки
(розділ 11). Отже, за Г = соті для ідеального газу ДС/= 0 і згідно з першим
началом термодинаміки
0 = -и/=ДЛіД. (1.28)
Таким чином, теплота, що підводиться до
такої системи, повністю витрачається на
роботу її розширення. Навпаки, коли система
стискується, енергія виділяється у зовнішнє
середовище у вигляді теплоти.
Для ізотермічного процесу в ідеальному
газі сталою є не тільки внутрішня енергія
(ДС/= 0), але й ентальпія (АН = 0). Справді,
ДЯ = Я2-Я1=(і/2+ірК2)-(С/1+/7К1) =
(іІ^тТ&^^ш^ <ЗГІДН0 І3 3ак0Н0М Бойля-Маріотта при Т =
= сопзідяя ідеального газурУ = соті).
Адіабатичний процес. В адіабатичному процесі система не
обмінюється із зовнішнім середовищем теплотою (д = 0). Тоді
Д£/=и;.
(1.29)
Таким чином, коли над системою виконується робота (м> > 0), внутрішня
енергія системи збільшується (Д£/> 0), при цьому температура її
підвищується. Навпаки, якщо Д£/< 0, то м> < 0, коли система виконує роботу за
рахунок своєї внутрішньої енергії, то при цьому знижується температура
системи. Прикладом може служити зниження температури газу під час різкого
його розширення (швидкоплинність процесу робить його адіабатичним
тому, що система не встигає обмінюватися теплотою з навколишнім
середовищем).
Для нескінченно малих величин із рівнянь (1.8) та (1.11) одержимо для
адіабатичного процесу (8# = 0)
СусіТ = -рсіУ. (1.30)
Далі, з урахуванням формул (1.4) та (1.26)
Суйт | му_Суйт | (ср-су)іу_^
14
звідки
Т Су V Т к } V
де відношення СрІСу позначено літерою у.
В інтегральній формі
ТУ*-х=соп8Ї. 0-31)
Це і є рівняння адіабати у координатах Т— V. Для переходу в
координати/? — Гтреба виразити Тз рівняння Менделєєва-Клапейрона (1.4), що
дає
рУу=сотґ. (1.32)
Конкретний вираз для роботи адіабатичного процесу можна одержати
інтегруванням
> = -\рйУ
1
з виключенням тиску за допомогою рівняння адіабати (1.32)
г 1Т_ } соті" „, уі\Ух у -соті" уг
Vу
1-у
У-1
= 2^ ^' = 2Р 1=Су(т2-тЛ = м; (і.зз)
у відповідності до (1.29). Нагадаємо, що коли м/ > 0, то Т2 >ТЇ9 а якщо
^<0,тоГ2<Г1.
1.3. Термохімія
Застосування положень 1-го начала термодинаміки до хімічних реакцій
розвинулось в окремий напрямок термодинаміки під назвою термохімія.
Відповідна інформація про теплові ефекти хімічних реакцій отримується при
їх проведенні в калориметрах. Калориметричні експерименти проводяться
за умов постійного тиску (або постійного об'єму) і отримуються конкретні
значення ^р (або ду). Згідно з формулою (1.19) ^р = АН (відповідно,
^у- А1)\ а ентальпія і внутрішня енергія є функціями стану, отже, зміни АН
і ДС/хімічної реакції не повинні залежати від шляху процесу. Таким чином,
приходимо до встановленого Г. Гессом закону: тепловий ефект хімічної
реакції, проведеної при;? = соті (або при V = соті), не залежить від шляху
переходу від реагентів до продуктів.
Наприклад, для реакції конверсії метану до СО і Н2 маємо
СН4 + Н20 = СО + ЗН2 + Д#, (+206,3 кДж).
15
Для конверсії СО результат такий:
СО + Н20 = С02 + Н2 + Д#2 (-41,1 кДж).
Конверсія метану до С02 і Н2 безпосередньо дає
СН4 + 2Н20-СС^ : 4Н2 + Д#з (+165,2 кДж).
Останню реакцію можна провести постадійно: конверсія СН4 до СО і далі
конверсія СО, тобто іншим шляхом.
Не важко пересвідчитись у тому, що, згідно із законом Гесса,
ДЯ3=АЯ1+АЯ2.
Термохімічні рівняння записуються з
урахуванням стехіометричних коефіцієнтів (у
наведеному вище прикладі розрахунки
проведено на 1 моль метану).
Використовуючи закон Гесса, можна
обчислити теплові ефекти хімічних реакцій, які
важко або неможливо провести у
калориметрі. Прикладом такої реакції може бути
утворення кристалогідратів при взаємодії
твердих солей із парами води. Наприклад
сазо4 (з)+н2о (8)=сазо4-н2о (з)+аяь ©
Проведемо реакцію (І) обхідним шляхом.
Воду сконденсуємо
Гесс Герман Іванович
(1802—1850), Росія
Н20(з) = Н20(/) + Д#2.
(П)
Тверду сіль (С(1804) розчинимо у великому надлишку (наприклад
400 моль) води
С(І804 (з) + 400 Н20 (/) = С(Ї804 (щ) + Д#3. (Ш)
Те саме зробимо з кристалогідратом
С<1304 • Н20 (з) + 399 Н20 (/) = С(1804 (щ) + АЯ4. (IV)
Позначки відповідають: § — §аз (газ); /—Іідиісі (рідина); 8 — зоїісі
(тверде тіло); щ — водний розчин солі.
Виходячи з відомих величин Д#2 = -40,6 кДж, Д#з= -45,9 кДж і Д#4 =
= -25,5 кДж, маємо, згідно із законом Гесса,
Д#! = Д#2 +Д#3 -Д#4 =-61,0 кДж.
Цікавим прикладом застосування термохімічних циклів є цикл Борна-
Габера.
М. Борн і Д. Майєр запропонували формулу для розрахунку енергії
кристалічної ґратки іонних кристалів (див. підрозділ 10.1)
16
1--Р
(1.34)
АЯ/(а)= 108,6 кДж/моль
АНщ= 121,3 кДж/моль
Тут 170—енергія, яку треба витратити для розпорошення іонного
кристала на окремі ізольовані іони (у газоподібному стані); г0 — міжатомна
(міжіонна) відстань. Константа ^ дорівнює ,4 = аг2, де а — стала Маделунга,
є — елементарний заряд. Параметр (р) для багатьох кристалічних ґраток
є сталою величиною і дорівнює 0,0345 нм. Для кристалічного КаСІ маємо:
го = (го)№+ + (го)сі- = 0*283 нм. Стала Маделунга (а) для кристала типу КаСІ
(проста кубічна структура) дорівнює 1,7476.3 використанням цих величин
обчислення енергії кристалічної ґратки ШС1 дає величину (£/0)теор. =
= 755 кДж/моль.
М. Борн і Ф. Габер запропонували для
обчислення С/0 такий цикл (вихідними є прості
речовини: металічний натрій і молекула С12):
1) сублімація натрію і дисоціація С12 на
атоми
Иа(8)->№(§)
1/2С12(Е)->С1(8)
АЯ7= АНІ(а)+АНт=229,9 кДж/моль;
2) іонізація атомів Натрію і приєднання
електрона до атома Хлору
Иа(§) -> На+(§) + є ЬНЩа)=495,7 кДж/моль
С1(§) + є -> СГ(§) АНІЩ = -364,9 кДж/моль
АНп= АНш„\+АНттґк\ — 130,8 кДж/моль:
<лііп і*ілща) ілчщь) і-^оіуд, ш , Борн Шкс (1882_1970),
3) об'єднання газоподібних ІОНІВ у твердий Німеччина
кристал ИаСІ
Иа+(е) + СГ(8) = ИаСІ (з) + ЬНШ
(АЯ/Я = -1/0 за визначенням);
4) розклад кристалічного ИаСІ на прості речовини
№С1 (8) = № (з) + ЙС12 (в) + Щу,
АНІУ — це тепловий ефект реакції утворення ШС1 з простих речовин,
взятий зі зворотним знаком, АНІУ = 410,5 кДж/моль.
Для замкнутого циклу 2Д#/ = 0, отже АНІ+АНП + АНШ + АНІУ = 0, звідки
АНШ=-(АЯ7 + АНП + АНІУ), або (С/0) = АЯ7 + АНП + АНІУ = 771,2 кДж/моль
У НеПОГаНОМу УЗГОЖДЄННІ 3 (£/0)теор/
Закон Гесса дає можливість обчислювати теплові ефекти хімічних
реакцій через теплоти (ентальпії) утворення речовин. Під ентальпією
(теплотою) утворення розуміють тепловий ефект (зміну ентальпії) хімічної реакції
утворення 1 моля даної сполуки з простих речовин (АЯутз). Наприклад ДЯ^в
аміаку є тепловий ефект реакції
1/2К2 + 3/2Н2 = Шз + Д#і.
17
АЯутв метану відповідає реакція
СІр. + 2Н2 = СН4 + ДЯ2.
Відзначимо, що прості речовини беруться у станах, що є стійкими при
кімнатних температурах, для Карбону це графіт (але не алмаз), для Окси-
гену — молекула кисню (02), але не озон, тощо. Ентальпії утворення
відносять до стандартного стану. Оскільки ентальпія — ізобарна величина, то
якр = соті вибирають атмосферний тиск;? = 1 атм. Стандартне значення
ентальпії позначають індексом — ДЯ°. За стандартну температуру
обирають Т= 298 К (25 °С), отже, позначка виглядає так: ДЯ£98-
Розглянемо конкретний приклад. (ДЯ^)^103 силікату кальцію є
тепловий ефект реакції
Зі + Са + з/2о2 = СаЗіОз + Д#/.
Цю реакцію можна провести постадійно
Са+1/202 = СаО + АЯ//.
8і + 02 = 8Ю2 + ДЯ/7/.
СаО + 8і02 = Са8і03 + АНІУ.
Згідно із законом Гесса це дає
Д#7 = ДЯ7/ + Д#//7 + АНІУ.
Для реакції утворення силікату з відповідних оксидів маємо
АНІУ = Д#7 - Д#7/ - Д#/7/.
Оскільки ДЯ/? ДЯЯ, АНШ є ентальпіями утворення, відповідно, силікату
кальцію, оксиду кальцію і оксиду силіцію, це дає
ДЯ/г=ДЯутав/ з-ДЯу^з -ДЯ^В2.
Наведений приклад ілюструє 1-ий наслідок із закону Гесса, згідно з яким
тепловий ефект (зміна ентальпії) хімічної реакції дорівнює сумі теплот (ен-
тальпій) утворення продуктів мінус сума теплот (ентальпій) утворення
реагентів (з урахуванням стехіометричних коефіцієнтів), тобто
ДЯ = 5>,(ДВД* -3>у(ДЯ,)5її\ (1.35)
Величини ДЯутз для стандартних умов визначено для багатьох сполук,
в першу чергу неорганічних, і наведено у відповідних довідниках.
Визначення ДЯ^з у деяких випадках зустрічає труднощі, особливо для
органічних сполук. Наприклад для реакції, що відбувається у доменному
процесі:
С02 + С = 2СО + ДЯі, (1)
ДЯ1=2ДЯ^-ДЯ^2.
18
(відзначимо, що для простих речовин ДЯ^. тотожно дорівнюють нулю).
Визначення Д#утв2 не зустрічає складностей, бо реакція
С + 02=С02+Д#£°2(Д#2) (2)
перебігає до кінця. Що ж до реакції
с+і/2о2 = со +дя£° (дя3), (3)
то її практично неможливо реалізувати, бо монооксид карбону відразу окис-
нюється молекулярним киснем далі до діоксиду.
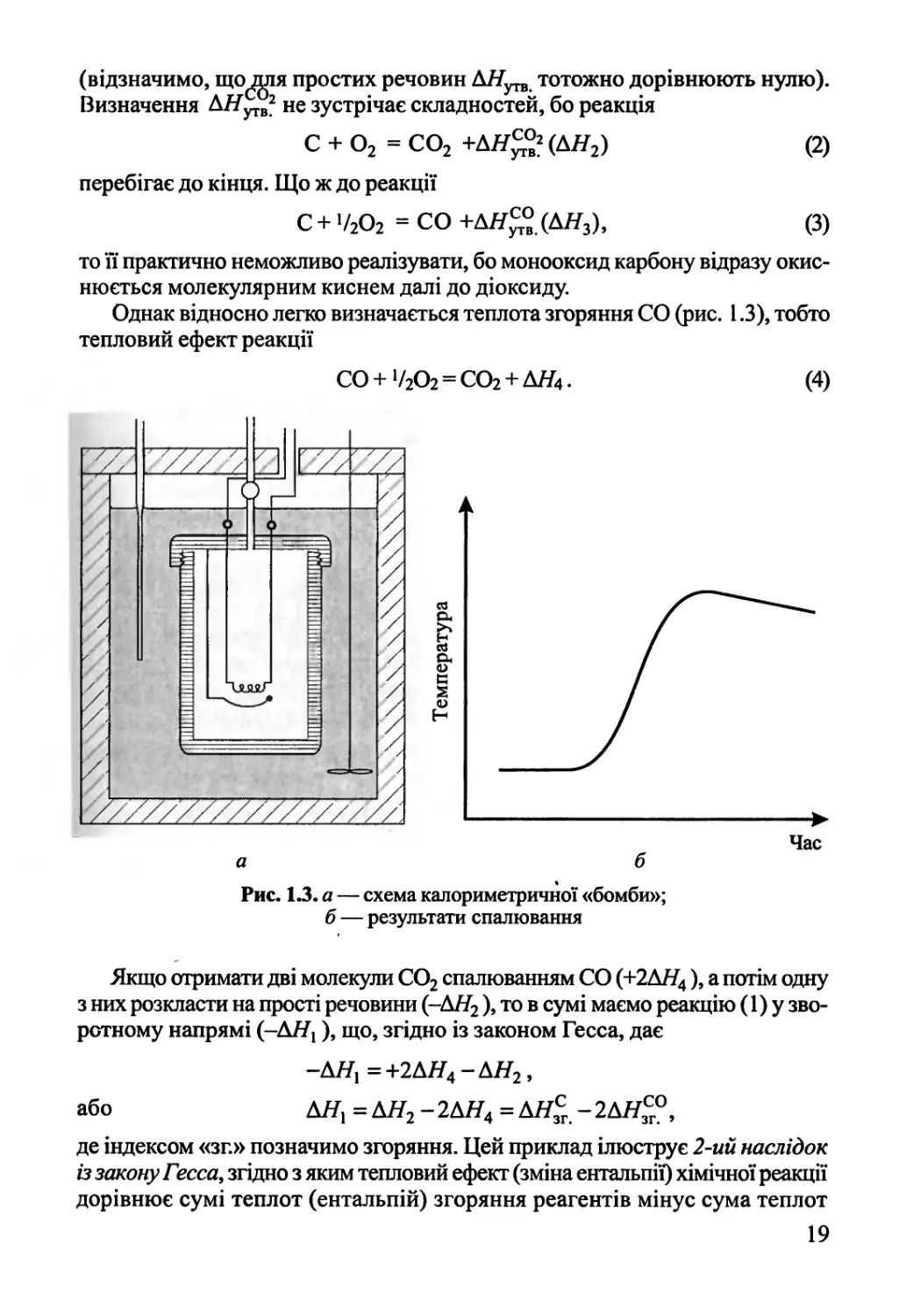
Однак відносно легко визначається теплота згоряння СО (рис. 1.3), тобто
тепловий ефект реакції
СО + 1/202 = С02 + АЯ4. (4)
X
З
0^
в ЧадяГ
і
а б
Рис. 1.3. а — схема калориметричної «бомби»;
б — результати спалювання
Час
Якщо отримати дві молекули С02 спалюванням СО (+2Д#4 )> а потім одну
з них розкласти на прості речовини (-Д//2), то в сумі маємо реакцію (1) у
зворотному напрямі (-Д#!), що, згідно із законом Гесса, дає
або
~ДЯ1=+2ДЯ4-ДЯ2,
АН{ = Д#2 -2Д#4 = Д#3СГ. -2Д#зСг°,
де індексом «зг.» позначимо згоряння. Цей приклад ілюструє 2-ий наслідок
із закону Гесса, згідно з яким тепловий ефект (зміна ентальпії) хімічної реакції
дорівнює сумі теплот (ентальпій) згоряння реагентів мінус сума теплот
19
(ентальпій) згоряння продуктів, тобто
ДЯ = 2>,(ДДіХГ- -^(ЩХГ: (1.36)
1 і
Для органічних сполук використання теплот згоряння є надійним
способом визначення із застосуванням закону Гесса інших теплових ефектів.
Наприклад теплоту утворення метану з простих сполук
Сір. + 2Н2 = СН4 + АЯуст^
легко визначити через теплоти згоряння
АЯ™4 = Д#зСг< + 2Д#зНг2 - Д#3СГН4.
Таким чином, за допомогою табульованих значень ентальпій утворення
або згоряння реагентів і продуктів можна обчислити тепловий ефект
хімічної реакції за стандартних умов — Д#298-
Для визначення Д#р_ції при будь-якій температурі застосовується
формула Кірхгофа
Ті
Д#?2 = АЩ + | АСрсІТ, (1.37)
де АСр — різниця між сумами теплоємностей продуктів та реагентів (з
урахуванням стехіометричних коефіцієнтів). Формула (1.37) легко виводиться
інтегруванням рівняння (1.22) і дозволяє знаходити Д#° при довільній
температурі (Г2), якщо тепловий ефект відомий при заданій температурі (Т{).
Якщо взяти за нижню межу Г= 298 К, а верхню зробити змінною, то для
зміни ентальпії хімічної реакції при довільній температурі маємо
т
Д#£ = Д#£98 + | ЬСрйТ. (1.38)
298
Якщо АСр= сопзі, тобто теплоємність від температури не залежить, то
це дає
ДЯ?=ДЯ?98+ДСі,(Г-298), (139)
тобто ентальпія хімічної реакції в цьому випадку лінійно змінюється з
температурою.
1.4. Ентропія
Серед розглянутих вище величин деякі залежать від кількості речовини,
наприклад, внутрішня енергія, ентальпія, теплоємність, об'єм тощо. Ці
величини мають назву екстенсивних. На відміну від них, такі величини, як тиск,
температура не залежать від кількості речовини. Це — інтенсивні величини.
Якщо розглянути роботу розширення (стиснення), то не важко помітити,
що вона виникає як добуток інтенсивного фактора (тиску) на екстенсивний
(об'єм). Це стосується не тільки роботи розширення (стиснення), а й будь-
20
якої роботи. Наприклад, електрична робота
виникає як добуток різниці потенціалів
(інтенсивний фактор) на кількість електрики
(екстенсивний фактор).
Якщо розглянути іншу енергетичну
характеристику — теплоту, то інтенсивний фактор для
неї відомий — це температура (Г). Відповідний
екстенсивний фактор має назву ентропія і може
бути введений згідно зі співвідношенням
І5 = 5^/Г, (1.40)
тобто
8# = Ш (1.41)
Карно Нікола Леонар Саді
за аналогією до рівності (1.7) для роботи. (1796—1832),
ПОНЯТТЯ еНТрОПІЇ буЛО ВВедеНО у ТерМОДИНа- Франція
міку С. Карно і далі Р. Клаузіусом шляхом аналізу роботи теплових машин,
тобто приладів, які пристосовані для перетворення теплоти у роботу.
Перехід теплоти у роботу можливий у процесі ізотермічного розширення,
наприклад ідельного газу (див. формулу (1.28)), теплота, що підводиться до такої
системи, повністю витрачається на роботу її розширення, але це можливо,
так би мовити, одноразово, бо при цьому робоче тіло (тут: ідеальний газ)
змінює свій об'єм. Щоб знову отримати роботу, потрібно робоче тіло
регулярно повертати у вихідний стан, тобто теплова машина повинна бути
періодично, циклічно діючим агрегатом. У найпростішій ідеальній тепловій
машині здійснюється так званий цикл Карно, який складається з двох ізотерм
і двох адіабат. При температурі нагрівача (Т{) відбувається ізотермічне
перетворення теплоти д^ = -м?аЬ = КТх\п{ Уь/Уа) (див. рис. 1.4, с. 22) у роботу за
рахунок розширення робочого тіла (газу) у системі. Можна довести, що
природа робочого тіла у циклі Карно не має
значення, тобто можна скористатися для
визначення теплоти й роботи в ізотермічному процесі
формулою (1.28). Далі система розширюється
адіабатично, температура при цьому падає до
Т2 (робота утворюється за рахунок зменшення
внутрішньої енергії робочого тіла (див.
формулу (1.29)). Після цього треба повернути
систему у вихідний стан. Для цього стискуємо
робоче тіло при температурі Г2, при цьому
частина теплоти (#2) перейде до зовнішнього
середовища (холодильника) #2 = ~^са = ЯГ21п(^/ Кс),
і далі адіабатичним нагріванням повертаємо
ЙЙь^ішУЖії системУ до ™шФатУРи Г, у вихідний стан.
Німеччина Проаналізуємо результат. На адіабатичних
21
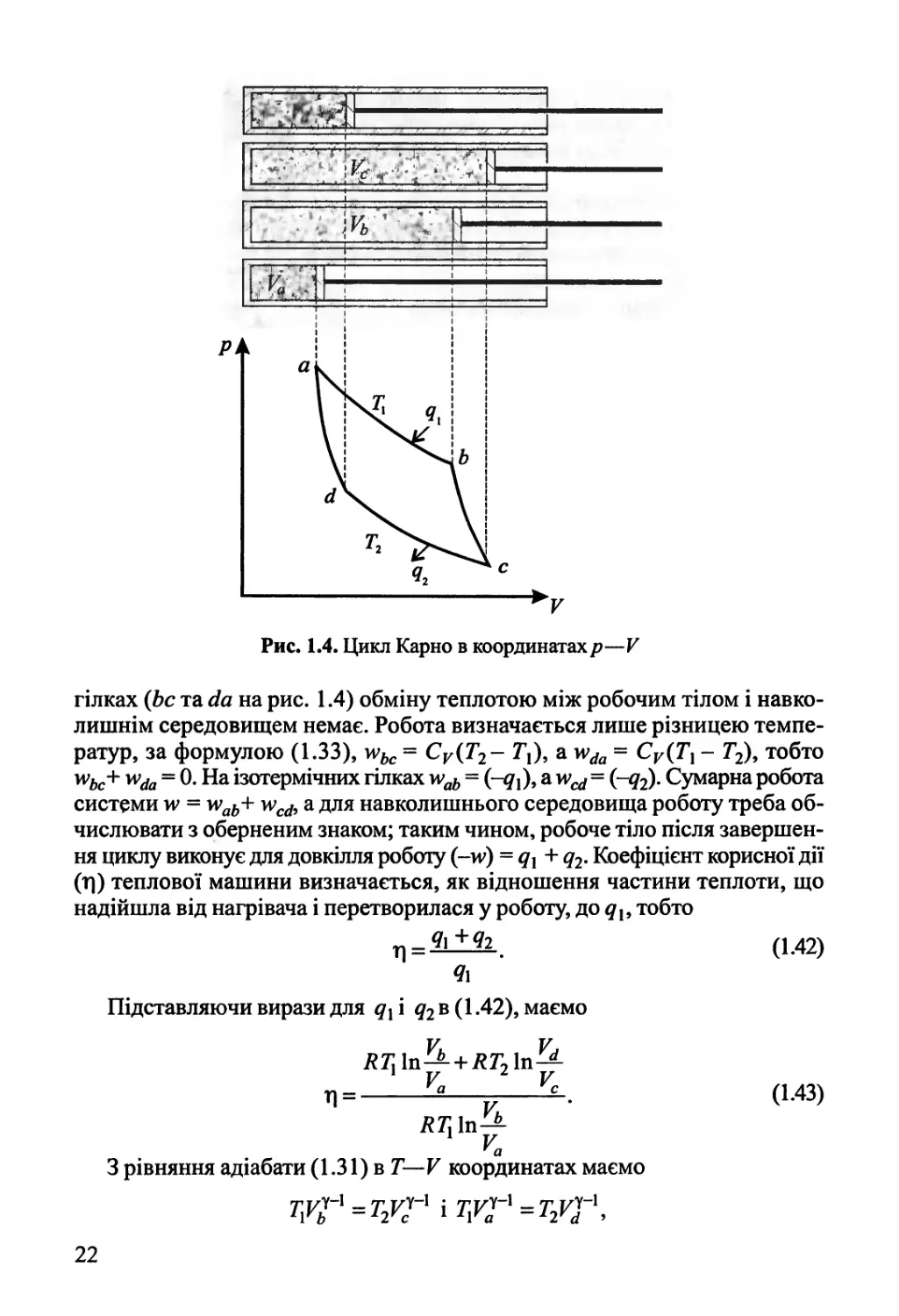
Рис. 1.4. Цикл Карно в координатах/?—V
гілках (Ьс та йа на рис. 1.4) обміну теплотою між робочим тілом і
навколишнім середовищем немає. Робота визначається лише різницею
температур, за формулою (1.33), м?Ьс = СУ(Т2- Г,), а и^ = СУ(Т{ - Г2), тобто
™Ьс+ ™<іа = 0. На ізотермічних гілках м?аЬ = (-д{)9 а и^ = (-^)- Сумарна робота
системи м> = м>а£+ \усф а для навколишнього середовища роботу треба
обчислювати з оберненим знаком; таким чином, робоче тіло після
завершення циклу виконує для довкілля роботу (-и>) = дх + д2. Коефіцієнт корисної дії
(г|) теплової машини визначається, як відношення частини теплоти, що
надійшла від нагрівача і перетворилася у роботу, до дь тобто
(1.42)
Підставляючи вирази для дх\ д2в (1.42), маємо
V V
г__а [_с_
Д7]1іД
1 V.
(1.43)
З рівняння адіабати (1.31) в Т— V координатах маємо
туіх=т2уу\тхуу=т2УІ\
22
2 Чі _ Т2
*1+*1 = 0.
Чг_
т2
Ч\
Уи V
звідки 1Г = 1Г- (Ь44)
Підстановка (1.44) в (1.43) приводить до простого виразу для коефіцієнта
корисної дії циклу Карно
Л = ^^. (1.45)
7]
Формула (1.45) показує, що Г| зростає зі збільшенням різниці (Т{ - Т2).
Теоретично Г| —> 1, якщо Т2 —> 0, тобто холодильник має температуру
абсолютного нуля. Співставляючи формули (1.42) та (1.45), маємо
Ч\
і нарешті, ^2.+ 21 = 0. (1-46)
Т2 7]
Отже, у циклі Карно сума величин (<7//Г,), які називають приведеними
теплотами, дорівнює нулю.
Розбиваючи будь-який цикл на певну кількість малих циклів Карно, маємо
£^ = 0. 0-47)
і *і
Нарешті, можна зробити ці цикли Карно нескінченно малими і перейти
від суми до інтеграла по замкненому контуру, що дає
і**=0. 0-48)
Т
Якщо ф дорівнює нулю, це означає, що підінтегральна функція (5#/Г) є
повним диференціалом. Вводячи позначку для цієї функції 5
(диференціал — сІ8) і назву ентропія, приходимо до формули (1.40).
На відміну від інших екстенсивних факторів, таких як об'єм, кількість
електрики (заряд) тощо, ентропію не можна безпосередньо
експериментально виміряти. Єдиний спосіб її визначення — це обчислення за
допомогою рівності (1.40). Оскільки ентропія є функцією стану, то її зміни
задаються кінцевим (2) та початковим (1) станами системи
Д5 = 52-51. (1.49)
Для ізотермічних процесів інтегрування рівності (1.40) дуже легке і дає
Д£ = ?/7\ (1.50)
Конкретними прикладами ізотермічних процесів є фазові переходи.
Наприклад, при плавленні твердого тіла
Ь8м = Зі-8я = Шт/Тт, (1.51)
а при випаровуванні (кипінні) рідини
23
фі
Д5> = 88 - 5/ = АНу/ Ть. (1.52)
(Індекси відповідають термінам: ^ — «&о1ісі», тверде тіло; /—«Іідиісі»,
рідина; § — «§а8», газ; т — «теШ炙, плавлення; V— «уарогігаїіоп»,
випаровування; Ь — «Ьоі1іп§», кипіння.)
Ізотермічним є також процес розширення ідеального газу при Т = соті,
для якого
ДЛгД
д*=* =—^=кіп^=я\п^. о-53)
Т Т Ух р2
Розглянемо, як змінюється ентропія у процесі змішування ідеальних газів
при Т = соті. Візьмемо ємкість, що розділена перетинкою на дві частини.
В одній частині об'ємом Ух знаходиться пх молів деякого газу «1», а у другій
п2 молів газу «2» займають об'єм У2. Якщо ліквідувати перетинку, то кожен
із газів займе об'єм всієї ємкості, тобто (У\ + У2). Тоді, згідно з формулою
(1.53) для загальної зміни ентропії системи, одержимо
Врахуємо, що
12 1 У{ 2 Ух
У\ = п\ = х а У2 = п2 =х
Ух + У2 щ+п2 и У\ + уі щ+п2 2'
де *,-— мольна частка /-го газу в суміші.
Тоді зміна ентропії при змішуванні двох газів дорівнює
А5 = -К(П\ІПХ\ + П2ІПХ2) .
За умови довільного числа компонентів
А3 = -В£пі1пхі. (1.54)
і
Віднесемо цю величину до 1 моль суміші газів. Для цього потрібно А5
поділити на суму молів (]£ л,-)> і в розрахунку на 1 моль маємо
А8 = -КУ£хі\пхі. (1.55)
і
Для адіабатичного процесу 8# = 0, отже, адіабата є ізоентропою (5 =
= соті).
Цикл Карно, який складається з 2-х ізотерм і 2-х адіабат, у координатах
Т— 5 має дуже простий вигляд прямокутника (рис. 1.5). У процесі
ізотермічного розширення при Тх Уь> Уа,тоді82>8\ і Д5'>0.^1 = ^(^-^і) = ТХА8.
У процесі адіабатичного розширення ентропія залишається сталою (£2),
а температура падає від Тх до Т2. Далі у процесі ізотермічного стиснення
при Г2 ^2 = Т2(8Х - 82) = - Т2А8. Нарешті, у процесі адіабатичного стиснення
ентропія залишається сталою (5!), а температура зростає від Т2 до Тх. Отже,
для коефіцієнта корисної дії маємо
24
^+#2 ТЛ8-Т2А8 Т{-Т2 .„ . . . ,
Ц = — = — — = — у повній відповідності з формулами (1.42)
Ч\ ТА$ т\ та (1.45).
— г
\яя
8д = 5ду— СусІТ,
На рис. 1.5 І^І— це площа «аЬ/е», а |^2| — площа «ссіе/».
Для ізохорного процесу
ї'і
г,
г2
а
й
є
X
%
ь
с
отже,
Д5 = |^-^Г, (1.56)
Ч Т
і коли Су = сопзі,
р істГ іг тЛ А8 = СуІп^. (1.57)
Рис. 1.5. Цикл Карно в координатах Т—5 у т
Аналогічно для ізобарного процесу зміна ентропії визначається
формулою
Ь8 = ]-Є-(ІТ9 (1.58)
а коли Ср = со>Ш, Гі
Д$ = С,1іД. (1.59)
Формули (1.51) — (1.59) наведено з розрахунку на 1 моль речовини.
Зауважимо, що ентропія має таку ж розмірність, що й теплоємність та
універсальна газова стала К, а саме — Дж/мольК.
Формули для обчислення ентропії дають змогу з'ясувати її фізичний
зміст. Теплота — це хаотичний рух частинок. Температура показує
інтенсивність цього руху, а ентропія визначає міру молекулярного хаосу,
безладдя. Розглянемо конкретні приклади.
На плавлення кристала треба витратити енергію (А#т > 0, ендотермічний
процес) і, отже, за формулою (1.51) А5т > 0. Таким чином, 81 > 88. Справді,
рідина, в якій відсутній дальній порядок, — менш упорядкована система, ніж
кристалічне тверде тіло. Аналогічно АНУ» 0, А8У» 0 і 5^ » 5/. Рідина,
в якій є ближній порядок, більш упорядкована система, ніж газ, де частинки
мають можливість вільно рухатися у будь-якому напрямі. Згідно з
формулою (1.53), якщо У2 > Уь Д5> 0, тобто ентропія зростає у процесі переходу
від стисненого газу до розрідженого. Тут також менш упорядкована система
(розріджений газ) має більшу ентропію.
З формул (1.56) і (1.58) випливає, що ентропія під час нагрівання тіл
зростає. Справді, наприклад, у твердому тілі у процесі нагрівання посилюється
коливальний рух, і частинки, що утворюють кристалічні ґратки, зі
збільшенням температури все більше відхиляються від своїх позицій у вузлах
ґраток, тобто зростає невпорядкованість системи.
Перемішування газів призводить до зростання безладдя в системі та,
25
відповідно, до збільшення її ентропії. Це видно з формули (1.55), згідно з якою
Д5> 0 (усі Хі < 1, отже, всі Іпх,- — від'ємні величини).
Таким чином, усі приклади наочно ілюструють фізичний зміст ентропії
як міри молекулярного хаосу.
Якщо розглядати зміну ентропії у бік її збільшення, то тут немає меж.
Зменшуючи ж хаос, можна дійти до стану максимальної впорядкованості.
М. Планк запропонував вважати таким станом ідеальне тверде тіло при
найнижчій можливій температурі — нуль за шкалою Кельвіна і припустити, що
в цьому стані ентропія мінімальна і дорівнює нулю (у будь-яких інших станах
ентропія — додатна величина). Це положення відоме як постулат Планка:
5о=0. (1.60)
Доцільно вважати, що коли Т= 0 К, всі рухи
припиняються і кожна частинка чітко займає
свою позицію у відповідному вузд?
кристалічних ґраток. Зазначимо, що постулат Планка
вимагає від кристала ідеальності. Якщо
у твердому тілі є дефекти, то його ентропія
внаслідок виникнення безладдя зростає.
Наприклад, наявність у кристалі незайнятих
місць веде до появи так званої
конфігураційної ентропії, яку можна обчислити за
формулою, аналогічною (1.55), якщо прийняти за
Планк Макс Карл Ернст (х{) частку вакансій (від загальної кількості
Людвіг (1858—1947), кристалічних вузлів у ґратках), а за (х2) —
частку вузлів, зайнятих атомами
(молекулами, іонами тощо). У цьому випадку безладдя виникає за рахунок того, що
та чи інша вакансія може знаходитися у будь-якому місці кристалічних
ґраток і, відповідно, зростання ентропії пов'язане з «перемішуванням» атомів
і вакансій.
Постулат Планка дає можливість обчислювати абсолютне значення
ентропії, на відміну від енергії, для якої є можливість знаходити тільки її відносні
зміни. Щоб обчислити абсолютні ентропії твердих тіл, можна скористатися
формулою (1.58) (або (1.56) для ізохорних процесів), позначивши нижню
границю інтеграла Т{ = 0, а верхню зробити змінною — Г, тобто
35=І^-<ІТ. (1.61)
0
Константа інтегрування при цьому, згідно з постулатом Планка,
дорівнює нулеві.
Для обчислення абсолютних ентропій рідин, газів треба скористатися
відповідними формулами (1.51)—(1.57) для обчислення зміни ентропії.
Наприклад, абсолютна ентропія рідини за умови довільної температури
26
-І^лч^+|£іл-. <ш)
о тт
Відповідно, для газу, що має температуру (Т) і тиск (р)
Т Т
) т тт ^ т
■я
о г
г (1.63)
т„
(Згідно з формулою (1.53) взялир2 =р, ар, = 1 атм).
Зв'язок ентропії з іншими функціями стану дається комбінуванням
рівняння (1.41) з рівнянням (1.8) першого закону термодинаміки, звідки
аіі=т<і8-рау. (1.64)
Ця формула є одним з основних фундаментальних співвідношень у
термодинаміці.
Зв'язок між ентропією та ентальпією знаходимо за допомогою формул
(1.64) та (1.20)
М = (IV + <і(р V) = Ш-рсІУ + рйУ + Усір = Ш + Усір. (1.65)
Оскільки </І/та Ш— повні диференціали, то для часткових похідних
Таким чином, 17 =/(*Я К), а Н =/(5, р), відповідно, <Ю = 0 коли 5 = соті
та V = соті, а Ш - 0 коли 5 = соті тар = соті. Використання ентропії як
незалежного параметра незручне, тому що ентропія безпосередньо не
вимірюється експериментально.
1.5. Вільна енергія
У математиці перехід від «усіх» до «хсіу» проводиться із застосуванням
повного диференціала від добутку (ху); а саме усіх = сі(ху) -хсіу. Це так зване
перетворення Лагранжа. Вище воно було застосовано у рівностях (1.64) та
(1.65) (перехід від (-рсіУ) до (Усір) здійснено із застосуванням сі(рУ)).
Щоб перейти від 7Ж до &/Г, де змінною величиною була б не ентропія,
а температура, потрібно застосувати функції, які б містили добуток Т8. Такі
функції були запропоновані Г. Гельмгольцом
А = Ц-Т8 (1.67)
27
та Дж. Гіббсом
С = Н-
Т5.
(1.68)
Гельмгольц Герман Людвіг Фердинанд
(1821—1894), Німеччина
Для функції Гельмгольца маємо при
диференціюванні
(ІА=(ІІ/-Т(13-&ІТ
і з урахуванням фундаментального
рівняння (1.64)
М = Ш-рсіГ-Ш-5(ІТ =
= -рсіУ-Ш. (1.69)
Відповідно, для диференціала функції
Гіббса
сІС = -8сіТ+МІр. (1.70)
З формул (1.69) та (1.70) видно, що А =/( У,Т), а О =/(р, Т) і, відповідно,
сіА = 0, якщо V = сопзі та Т = соті, а йС = 0, якщо/? = соті та Т = соті.
Через це деякі автори пропонують називати функцію (А)
ізохорно-ізотермічним потенціалом, а (О) — ізобарно-ізотермічним потенціалом.
3(1.67) і (1.68) видно, що функції (А) та (<7) мають розмірність енергії, тому
їх називають енергія Гельмгольца (А) та енергія Гіббса (С). З точки зору
фізичного змісту, обидві ці функції найкраще відповідають терміну вільна
енергія. Щоб їх не ототожнювати, А — це вільна енергія Гельмгольца, а О —
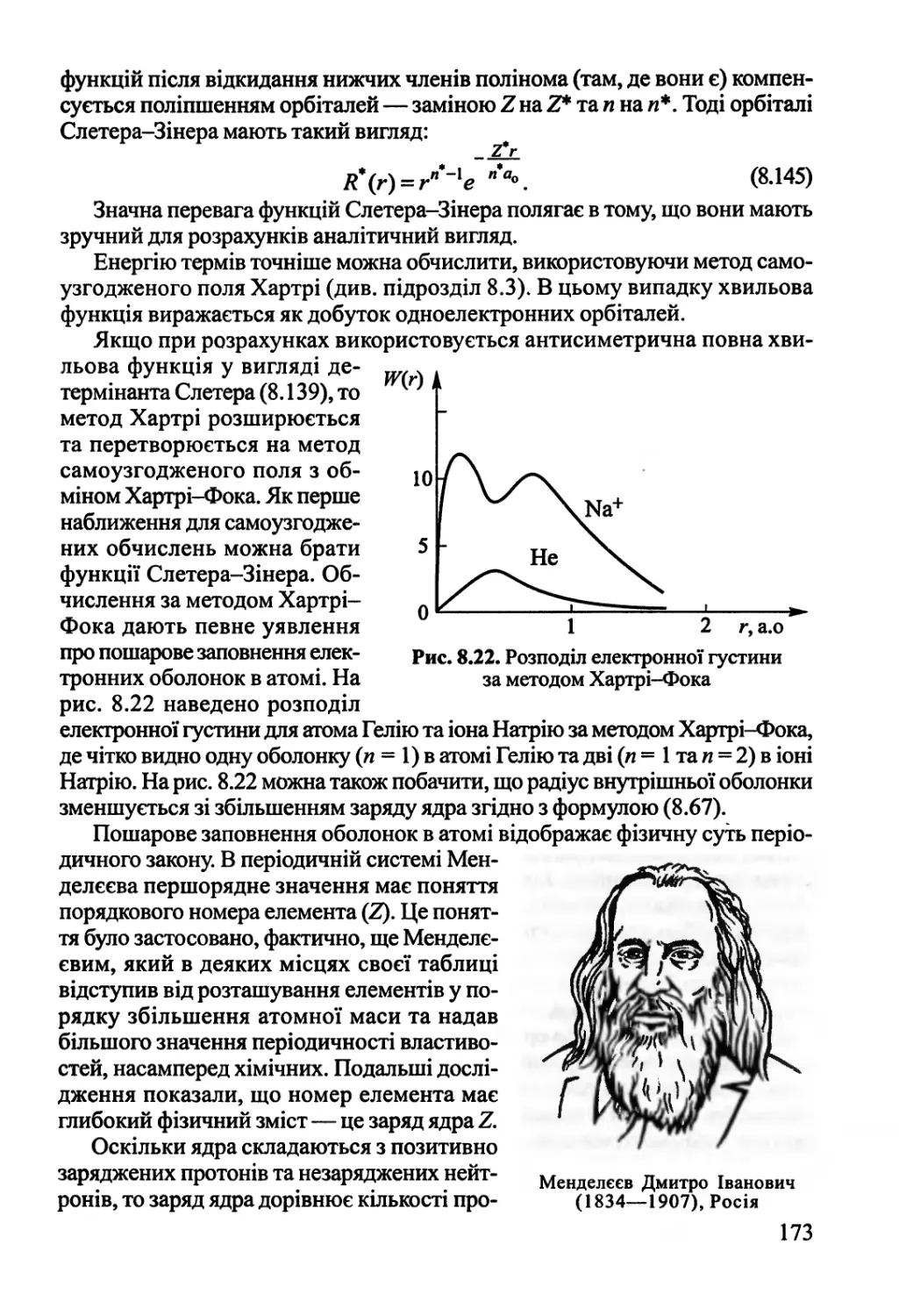
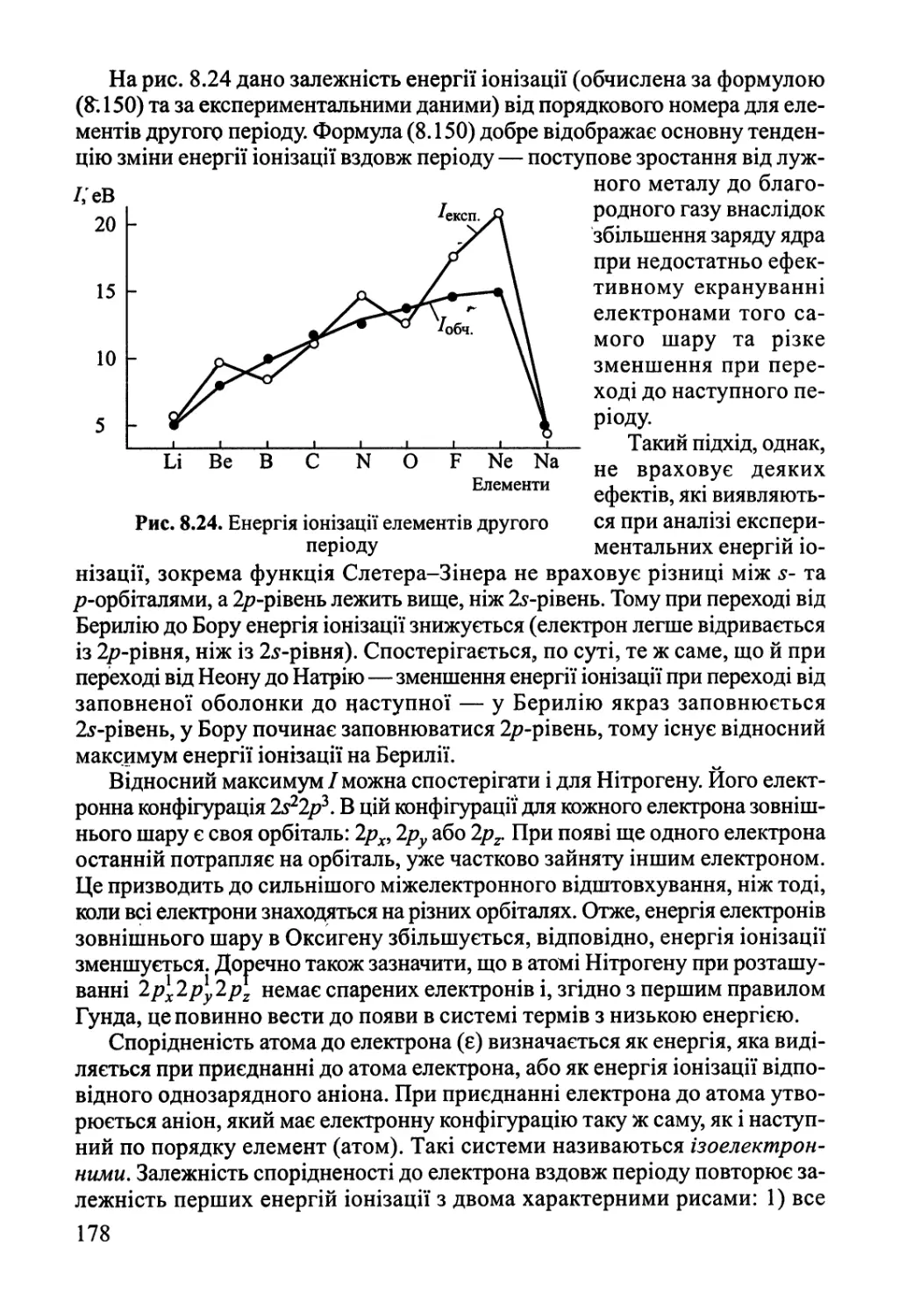
вільна енергія Гіббса.
Розглянемо спочатку функцію Гельмгольца. Рівність (1.67) показує, що
внутрішня енергія (Ц) складається з двох частин: А та Т8. А — вільна
енергія, а Т8 — зв 'язана енергія.
Для будь-яких ізотермічних процесів
А17 = АА + ТА8. (1.71)
Величина (ТА8) показує, яка частина
енергії під час зміни внутрішньої енергії
перетворюється виключно на теплоту
(віддається чи одержується системою залежно від
знака М). Вільна енергія показує, яка
частина АС/ перетворюється на роботу.
Одним з найкращих прикладів може
служити процес у гальванічному елементі (див.
розділ 7). Хімічна енергія (АС/) реакції, що
відбувається в елементі, перетворюється
на електричну роботу частково, а саме у
кількості, що дорівнює (ДЛ). Виконуючи
роботу, гальванічний елемент нагрівається
28
Гіббс Джозайя Уїллард
(1839—1903), США
або охолоджується залежно від кількості одержаної або витраченої теплоти
ц = ТА8.
Для вільної енергії Гіббса, відповідно,
АС = АН-ТА5. (1.72)
Для конденсованих систем АН = АС/, відповідно, АС= АА (зокрема,
процес у гальванічному елементі можна вважати як ізохорним, так і ізобарним).
Для газового стану між АА і АС існує різниця. Зв'язок між АтаС можна
легко знайти на основі формул (1.65), (1.66) та (1.18) (зв'язок між Ята Ц),
тобто
С=А+рУ. (1.73)
Для ідеальних газів/>К = КТ і
С=А+КТ. (1.74)
Вище доводилося, що добуток функцій, наприклад рУ9 обов'язково
є функцією стану й, відповідно, й(рУ) є повним диференціалом. Аналогічно
с!(Т8) також є повним диференціалом, а добуток (ТЗ) є функцією стану.
Оскільки внутрішня енергія та ентальпія є функціями стану, то вільні енергії
Гельмгольца (А) та Гіббса (О) також є функціями стану.
Відповідно йА та АС—повні диференціали, що дає для похідних:
%\-*
Перехресне диференціювання та незалежність других змішаних похідних
від порядку диференціювання дає дві корисні формули, відомі як
співвідношення Максвелла:
ву)т-[вт)
[ФІН- ™
Ізобарні процеси більш поширені, ніж ізохорні. Відповідно, вільна
енергія Гіббса (О) застосовується частіше, ніж енергія Гельмгольца (А).
Розглянемо О докладніше.
Якщо Т = сопзґ, сіТ = 0 і
сІСт= Уйр (1.80)
(1.78)
29
Для ідеального газу, якщо Т = сопзі,
КТ
йС = йр = КТй 1п/?. (1.81)
Р
Інтегрування від/?! до/?2 Дає
^--(^ДО-ДПіА (1.82)
А
Формула (1.82) збігається з формулою (1.27) для роботи ізотермічного
процесу розширення ідеального газу.
Коли/?2 >Р\ (Уг < ^і) ^ = м>> 0, тобто при ізотермічному стисненні газу
робота спрямована на збільшення його вільної енергії (С2 > С{).
Якщо у формулі (1.82) взяти/?! =/?0= 1 атм і вважати цей стан
стандартним, то при довільному значенні тиску (р)
0 = (? + КТІпр9 у (1.83)
де С° — стандартне значення вільної енергії Гіббса.
Якщо/? = соті, згідно з (1.75),
Однак безпосереднє інтегрування цього виразу незручне, оскільки
залежність 5 від температури досить складна. Знайдемо залежність від
температури не безпосередньо вільної енергії Гіббса, а частки СІТ
дТ{т)р [дТ)рТ' "{ Т2) Т2 Т2
згідно з формулами (1.68) та (1.75).
Для зміни вільної енергії—ДО
±ГАвЛ Ш
д7\ Т )р Т2
Формули (1.84) та (1.85) мають назву співвідношень Гіббса-Гельм-
гольца. Інтегрування рівняння (1.85) буде наведено далі для конкретних
випадків (фазові переходи, хімічні реакції тощо) і дає можливість визначати АС
за будь-якої температури.
Зміни вільних енергій Гіббса і Гельмгольца знаходять або за допомогою
формул (1.71), (1.72) через ентальпії, внутрішні енергії та ентропії, або
безпосередньо з експерименту (див. далі у підрозділі 7.1 «Термодинаміка
гальванічного елемента»).
Вільні енергії мають величезне значення у визначенні напряму перебігу
того чи іншого процесу, передбачають можливість його перебігу
(підрозділ 1.7).
ЗО
1.6. Багатокомпонентні системи.
Парціальні мольні величини
У підрозділі 1.4 відзначалося, що всі термодинамічні величини можна
поділити на екстенсивні та інтенсивні, причому екстенсивні залежать від
кількості речовини. Про те, як змінюються екстенсивні величини в
багатокомпонентних системах зі зміною кількості речовини, дають інформацію
парціальні мольні величини.
За визначенням парціальна мольна величина (§)і — це часткова похідна
від відповідної екстенсивної величини (§) за її кількістю при фіксованих
значеннях інтенсивних параметрів і кількості інших речовин, тобто
Ґ3£
>
(1.86)
Якщо залежність £ від пі подана формулою, наприклад £ = ащ + Ьщ +
+с, де а,Ь,с — певні чисельні параметри, то не важко аналітично знайти
парціальну мольну величину: ^ = Іащ+Ь.
Якщо експериментальні дані залежності £ від щ зображено у вигляді
графіка, то 8і знаходять графічним диференціюванням як тангенс кута нахилу
дотичної у відповідній точці.
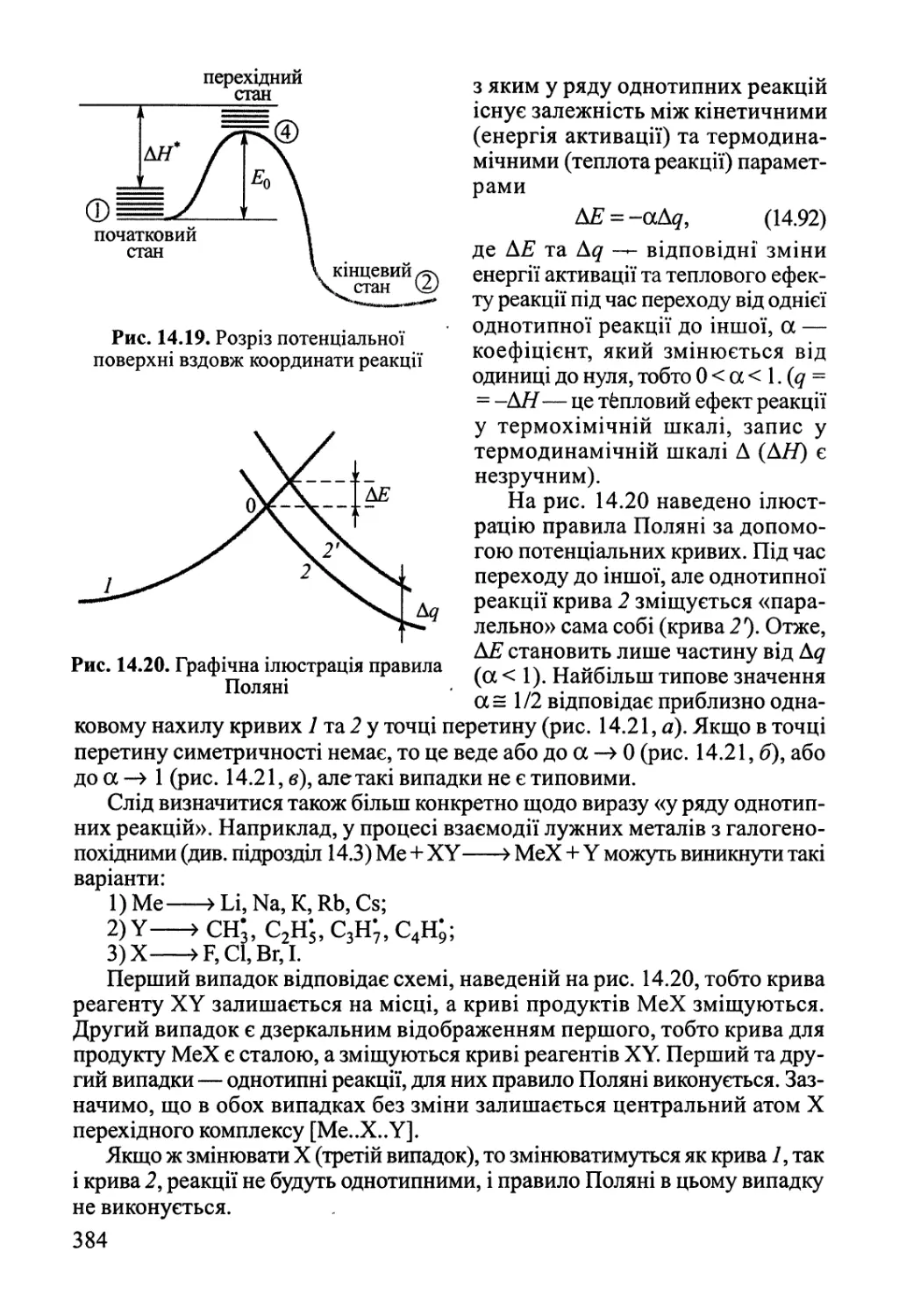
Екстенсивна властивість (#) складається з парціальних мольних величин
таким чином:
£ = £&",- (1-87)
І
Для двокомпонентних систем широко застосовується так званий метод
відрізків. Експериментально визначену величину
£ = £і"і+£2"2- 0-88)
можна віднести до одного моля, що дає
8м = —Т- = ^ + &*2> (1.89)
щ+п2
де хх тах2 — мольні частки відповідних компонентів.
Відзначимо, що мольна величина (£м) та парціальна мольна (§)і мають
однакову розмірність і віднесені до 1 моля.
На графіку залежності %м від мольної частки якогось компонента
(наприклад х2, рис. 1.6, с. 32) проводять дотичну у відповідній точці М. Відрізки,
які ця пряма відтинає на вертикалях х{ = 1 тад:2= 1, і є відповідними
парціальними мольними величинами. Доведемо це за допомогою рис. 1.6 та
формули (1.89)
АВ = ЕГ = МГ-МЕ = МЕ-ВЕі^а = ^м-х2^- =
йх2
31
(враховано, що йххІ<ІХ2 = -Х).
Далі, СІ) = СН + Ш> =АВ +
+ Я#і§сс = гі+1(&-й) = &-
Таким чином, відрізки АВ та
СІ) відповідають парціальним
мольним величинам, відповідно,
£і і £2. Нарис. 1.6відрізкиАКта
СЬ — це &? та %2 > тобто це
значення величини £ для чистих
компонентів, відповідно, «1» та «2».
Взагалі, £і * £і°, а £2 * £2 >але в
окремому випадку, коли #м
змінюється з х2 лінійно, не важко
довести, що ^ = §?/, а £2 = %2 ,
^ ^->*2 С
Рис. 1.6. Метод відрізків для визначення
парціальних мольних величин
тобто ^
8м=*1!Ї+хг8г- (19°)
У такому випадку вживають термін «адитивний», тобто ^адитивно
складається з £° та £2 •
Формула (1.90) для довільної кількості компонентів і на довільну кількість
молів має вигляд
£ = £&Ч. (1.91)
і
Наприклад, об'єм системи може адитивно складатися з об'ємів
компонентів, що входять до складу системи
г = £*5Ч ' О-92)
і
Адитивні властивості має внутрішня енергія ідеального газу (І/) при
сталій температурі. Справді, при Т= соті Ш = 0, тобто внутрішня енергія
ідеального газу не залежить від того, який об'єм він займає. Отже, якщо в
початковому стані маємо л, молів різних газів, розділених перетинками, то
після їх змішування при Т= соті зміна внутрішньої енергії
І І
йі=Ц?, (1-93)
тобто парціальна внутрішня енергія /-го компонента дорівнює внутрішній
енергії відповідного чистого газу.
Для ентальпії при Т= соті також АН = 0 (див. підрозділ 1.2) і не важко
довести, що
Ні=Щ. (1.94)
На відміну від внутрішньої енергії та ентальпії, ентропія при змішуванні
32
газів змінюється на величину А8, що дається формулою (1.54). Отже, для
зміни ентропії маємо
і і і
ЗВІДКИ
Л^-ДІпх,.. (1.95)
У формулах (1.93) — (1.95) величини Ц?, Я?, 5° залежать від
температури.
Співвідношення (1.95) показує, що парціальна мольна величина може
залежати від концентрації. Ці залежності, однак, обмежені співвідношенням
Гіббса-Дюгема
$>/<§/=0. (1.96)
Для обґрунтування рівняння (1.96) достатньо продиференціювати
рівняння (1.87) як добуток
^ = Е&А+ЕЛ^
і далі співставити цей результат із виразом для повного диференціала (сІ£),
який має вигляд
СІ§ =
(
дпл
(
йпх +
;пі*\
ьЛ
дпо
^2+... = Яі^і+Я2^2+- = Е^А- О'97)
/ЛУ*2
В розрахунку на 1 моль для співвідношення Гіббса-Дюгема маємо
2*/<§і=0 (1.98)
і
(вираз (1.96) поділено на 2^щ)-
і
Для парціальної мольної ентропії, з урахуванням (1.95), маємо, наприклад,
( ^ \ Ґ ^
— і гіг \ І
= 0,
оскільки за умовами Х** = *= С0П8*-
і
Для двокомпонентної системи рівняння Гіббса-Дюгема (1.98) набуває
вигляду
*і4й+*2<£2=0- 0-99)
Однією з найважливіших парціальних мольних величин є парціальна
мольна вільна енергія Гіббса (Сі), яка має спеціальну позначку ((і,-) і
назву — хімічний потенціал,
и(. =(<;,)=
Зи,
<Р,Т,пм
(1.100)
33
Хімічний потенціал ідеального газу з урахуванням формул (1.94) та (1.95)
має вигляд _ _ _
ц, = £. = #, -Щ = Я° -Г(5° -Л1шс,) =
= Я/°-Г5,/о+ЛГ1шсі=0і?)/+ЛПііх/. (1Л01)
Для хімічного потенціалу розчину (ідеального) також можна
користуватися цією формулою, у якій за стандартний стан обрано стан
індивідуального («чистого») компонента (лс,- = 1).
Для газу, спираючись на закон Дальтона, можна записати
*/=7> О-102)
де Р — загальний тиск у системі; рі — парціальний тиск і-то газу.
Підставляючи (1.102) у (1.101) і враховуючи, що загальний тиск у
системі сталий, маємо /
ц, =іі°+КТ\пРі. (1.103)
Вираз (1.103) для хімічного потенціалу ідеального газу адекватний виразу
(1.83) для вільної енергії Гіббса ідеального газу в однокомпонентній системі.
Для однокомпонентної системи (ц) і (О) — тотожні величини. Так само, як
і у формулі (1.83), при^= 1 атм \і. = |і?, отже, \і° — стандартний хімічний
потенціал, який залежить від температури. Значення (ц? )' у формулі (1.101)
залежить не лише від температури, а й від загального тиску (Р) в системі
Оі?)' = ц?+ДПпР. (1.104)
З урахуванням хімічного потенціалу вираз для йовного диференціала
вільної енергії Гіббса для багатокомпонентної системи має вигляд
(ІС = -8(ІТ + Уйр + £МЛ/- (1.105)
1.7. Умови рівноваги. Напрям процесів.
Друге начало термодинаміки
У фізиці досить чітко діє принцип мінімуму енергії, наприклад, під дією
гравітаційного поля каміння падає вниз, вода тече згори додолу, під дією
електричного поля заряд переноситься із місць із високим потенціалом до
місць із низьким потенціалом тощо. Застосувавши це положення у
термодинаміці, П. Бертло сформулював принцип, згідно з яким система прагне
позбавитися зайвої енергії, тобто, згідно з П. Бертло, спонтанно перебігають
тільки екзотермічні процеси. Для ізохорних процесів це означає, що
самочинно ідуть процеси, для яких АС/ < 0, а для ізобарних, відповідно, ДЯ < 0.
Справді, за умови низьких температур самочинно перебігають екзотермічні
хімічні реакції, а також такі процеси, як конденсація, тверднення. У принципі
Бертло не враховано, однак, зміни ентропії, і тому це положення виявилося
обмеженим.
34
Г. Гельмгольц і В. Нернст вдосконалили цей принцип таким чином, що
система прагне позбавитися вільної енергії, тобто самочинно перебігають
процеси, для яких
АА<0, (1.106)
при У-сопзі, або
АС<0 (1.107)
\\1рИр=СОП5І.
Знак рівності відповідає рівноважному стану системи. Відповідно до
(1.71) (або (1.72)) від'ємне значення АЛ (або А С) досягається за рахунок
від'ємного значення А С/(або АН) і додатного А5. За низьких температур
Т —> 0 і АА = АІІ (відповідно, А С = А Я). За цих умов принцип Гельмгольца-
Нернста збігається з принципом Бертло (крім загальної вимоги Г-» 0, ці
положення збігаються також в окремому випадку, коли для деякого процесу
А5=0).
Зі зростанням температури все більшого значення набуває ентропійний
фактор. Наприклад, для фазового переходу тверде тіло-рідина
АСт = Сі-С* = АНт-ТА8т = А8т(Тт-Т) (1.108)
(згідно з формулою (1.51) А 5т=5/ - 5у > 0), тому коли Т > Тт, АС < 0 і
процес переходу твердого тіла у рідину перебігає спонтанно за рахунок ентро-
пійного фактора (|ГД 5да| > |Д#т|). Навпаки, якщо Т < Тш, то АС > 0, що
робить перехід твердого тіла в рідину неможливим. Коли Т=Тт АНт = ТтА8т
і АСт = 0, згідно з умовами рівноваги.
Якщо А £/= 0 (або Д#= 0), ентропійний фактор стає вирішальним.
Наприклад для ідеального газу, коли Т = сопзі, Д£/= АН= 0 і
АС =-ТАЗ. (1.109)
Таким чином, Д С < 0 тільки коли А £ > 0, і спонтанно перебігають процеси
\\ збільшенням ентропії. Наприклад, під час ізотермічного розширення
ідеального газу У2 > У\ (відповідно/?2 <Рі) і ДС1 < 0 згідно з формулою (1.82),
а Д5 > 0 згідно з формулою (1.53).
Більш загальний випадок А11= 0, коли система не обмінюється енергією
і навколишнім середовищем. Якщо V = сопзі та є додаткова умова V =
соп8і, то сШ= 0 та (IV'= 0 і, згідно з фундаментальним
співвідношенням (1.64),
</5 = 0. (1.110)
Рівність (1.110) відповідає стану рівноваги. Для спонтанних процесів у таких
системах за Клаузіусом
</5>0, (1.111)
або для кінцевих величин
35
А5>0.
(1.112)
Нерівність (1.111) — це одне з найбільш загальних формулювань другого
начала термодинаміки* згідно з яким у системах із постійними
внутрішньою енергією та об'ємом спонтанно перебігають процеси зі збільшенням
ентропії.
Згідно зі співвідношенням (1.109) зростання ентропії системи — це
втрата нею вільної (але зі збереженням повної) енергії, тобто здатності
здійснювати роботу.
Нерівності (1.106), (1.107) та (1.112) вказують напрям, у якому реальний
процес може проходити самочинно, тобто вони характеризують необоротні
процеси. Різницю між оборотними й необоротними процесами можна
визначити таким чином. Для оборотності необхідно, щоб система повернулася
у вихідний стан з тими ж функціями й параметрами стану. Ця умова
необхідна, але недостатня. Треба також, щоб після повернення системи у
початковий стан не сталося ніяких змін у навколишньому середовищі.
Розглянемо конкретний приклад. Нехай за певної температури (Т)
ідеальний газ розширився від об'єму Ух до У2 (У2 > У\)- При цьому виконується
робота
и> = ДС = -ДГ1п^<0
Ух
і поглинається з навколишнього середовища теплота
а = ТА8 = КТІп^>0.
Для того щоб цей процес був оборотним, необхідне виконання деяких
умов. По-перше, температура навколишнього середовища повинна бути
такою, як і температура системи (точніше, перевищувати її на сіТ, щоб
відбувався процес передачі теплоти). По-друге, треба, щоб тиск газу в системі
дорівнював тиску зовнішнього середовища (точніше, перевищував його на ф,
щоб відбувався процес розширення системи). По-третє, процес зміни
об'єму системи повинен відбуватися дуже повільно, щоб не порушувалась
термічна (Гсистеми = Гдовкілля) та механічна (рСИСТШИ =рД0ВКІЛЛЯ) рівновага між
системою та зовнішнім середовищем. Тому оборотні процеси повинні бути
квазістатичними й рівноважними. Такими ж поступовими кроками можна
повернути систему в початковий стан. При цьому робота, що була
накопичена в зовнішньому середовищі, піде на стиснення системи до початкового
об'єму (У{), ентропія системи зменшується на величину А8 = К\п(У1/У2),
і відповідна кількість теплоти ^=КТІп(У{/У2) < 0 віддається системою
навколишньому середовищу.
Тепер розглянемо необоротний процес — розширення газу у вакуум,
де зовнішній тиск дорівнює нулеві й, отже,/?системи Ф ^довкілля- Оскільки
система не зустрічає опору із зовнішнього середовища, то робота не виконується
і, відповідно, теплота не поглинається системою. Але ентропія як функція
стану, коли Т = соті залежить від об'єму, і якщо кінцевий об'єм взяти таким
36
самим, як і під час оборотного квазістатичного процесу, то А^ = ЯІп(У2/У{).
Щоб повернути систему у вихідний стан, треба її стиснути до об'єму (Уг).
Нехай цей процес перебігає оборотно. При цьому, як було вже наведено
вище, над системою виконується робота м> = -КТІп(У1/У2) =і?71п(К2/К1) > 0,
система віддає теплоту # = КТ\п(У1/У2) < 0, і її ентропія зменшується на
величину А82 = КІп(У1/У2) = -А^. Таким чином, система повертається у
вихідний стан з тими самими об'ємом, температурою, тиском, ентропією, але
у навколишньому середовищі відбуваються зміни — воно «обмінює» деяку
кількість енергії з роботи (-IV) на теплоту (+д) (для зовнішнього
середовища знаки протилежні знакам системи). У цьому й полягає різниця між
оборотними та необоротними процесами.
Оборотні процеси не тільки квазістатичні, але й завжди рівноважні. Вони
перебігають через низку рівноважних станів (або в системі, наприклад під
час фазового переходу, або при взаємодії системи з навколишнім
середовищем — оборотне ізотермічне розширення-стиснення газу). Необоротні
процеси ведуть систему до рівноваги, вони завжди нерівноважні. Майже всі
реальні процеси в природі є необоротними, нерівноважними. Оборотні,
квазістатичні процеси — це ідеалізація, але якщо зосередити увагу на
системі й розглядати саме її стани, які визначаються параметрами та функціями
стану для рівноважної системи, то у випадку з нерівноважними процесами
всі зміни можна віднести до навколишнього середовища. Прикладом може
бути вже розглянутий необоротний процес розширення газу в вакуум.
Розглянемо ще один приклад — хімічний процес у гальванічному
елементі, наприклад реакцію £п + Си804 = 2п804 + Си в елементі Даніеля-
Якобі. Якщо процес перебігає оборотно, то, як було визначено вище,
частина хімічної енергії — ДС перетворюється на електричну роботу, а
частина — ТАЗ переходить у навколишнє середовище у вигляді теплоти. При
цьому, коли АС дорівнюватиме нулеві, у системі встановляться деякі
рівноважні концентрації Купруму та Цинку (в розчині). Тепер за умови таких же
маси речовин, температури й зовнішнього тиску проведемо експеримент
необоротно, а саме — опустимо гранулу цинку у розчин Си804. При цьому
иідбуватиметься така ж хімічна реакція, як і в гальванічному елементі, поки
Л6 < 0. При АС = 0 процес зупиниться, а рівноважні концентрації Цинку
Іі Купруму будуть такими ж, як і в гальванічному елементі. Але в
зовнішньому середовищі пройдуть зміни: не буде запасу ніякої електричної енергії,
и вся повна енергія А#= А(7хімічної реакції перетвориться на хаотичний
тепловий рух довкілля. У першому випадку систему і навколишнє середо-
нище можна оборотно повернути у вихідний стан — перетворити елек-
і ричну енергію в хімічну електролізом, у другому випадку (в необоротному
процесі) це неможливо.
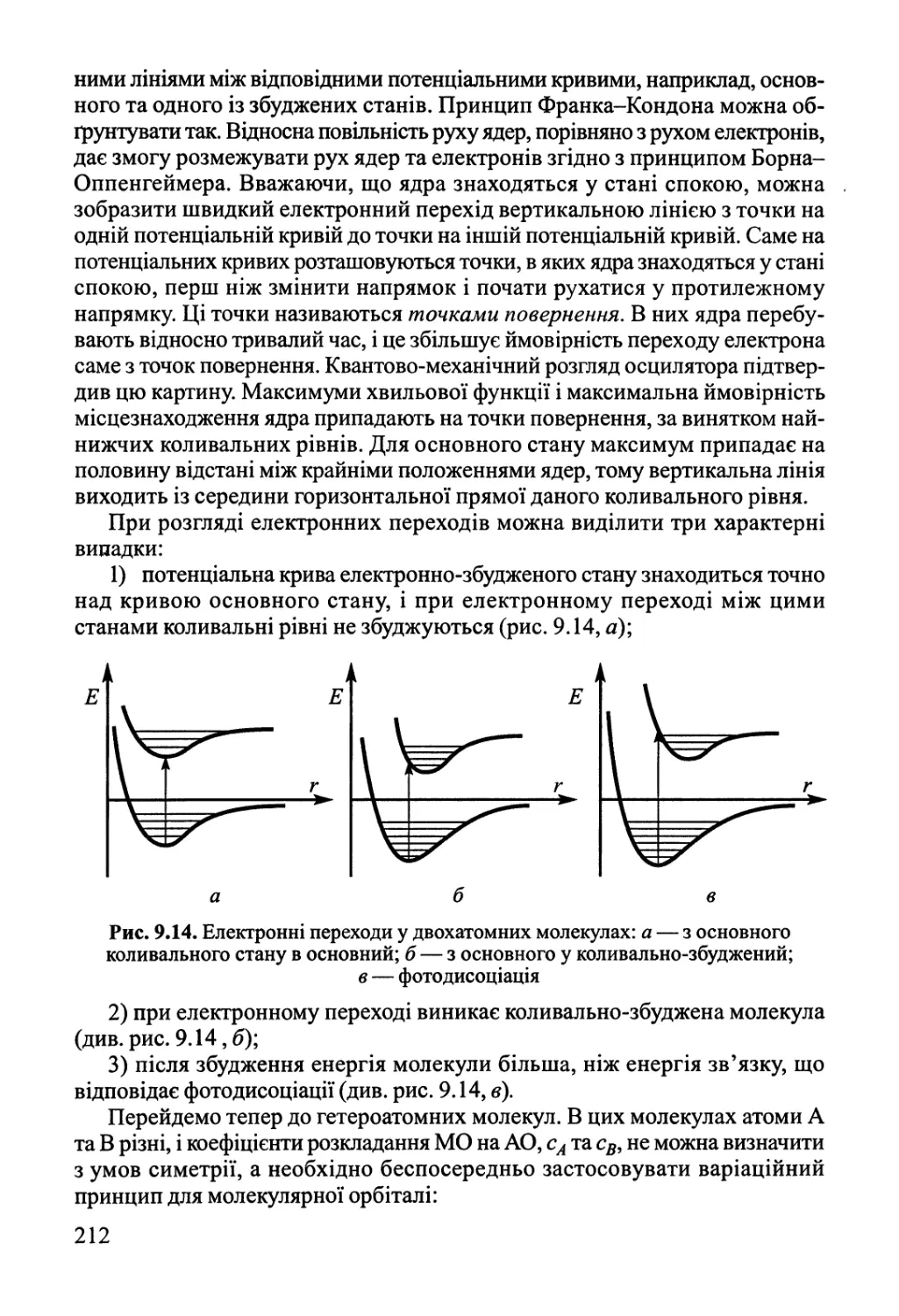
Історично друге начало термодинаміки виникло під час розгляду тепло-
іпїх процесів, зокрема у теплових машинах, тому, крім загального його
формулювання через ентропію (нерівності (1.111) або (1.112)), також існують
окремі формулювання. Розглянемо одне з них, яке запропоновано Р. Клау-
37
зіусом: «Теплота не може спонтанно переходити від холодного тіла до
гарячого». З цього пояснення випливає, що самочинно можуть перебігати
процеси теплопередачі тільки від тіла з більшою температурою до тіла
з меншою.
Розглянемо конкретний приклад. Приведемо у контакт два тіла з
однаковою масою з однакового матеріалу, які мають температури Тх та Т2, і нехай
Тх > Т2. У процесі теплопередачі перше тіло буде охолоджуватися, а друге
нагріватися, доки їх температури не зрівняються і не стануть дорівнювати
деякій температурі Т. Цю температуру не важко обчислити. Перше тіло
віддало теплоту цх = СХ(Т- Тх), а друге прийняло ^2 = С2(Т- Т2). Оскільки
Чг = ~#і>а КРІМ того? вважаємо, що маси тіл та матеріал, з якого вони
виготовлені, є однаковими: Сх = схит'тх =с2ит'т2 =С2 = С, тої це дає Тх - Т=
= Т- Г2, звідки Т = (ТХ + Т2)/2. Загальна зміна ентропії при цьому
А8 = А8х+А82=СЛп— + С21п— = С1п— = СЬ^Гі+Г^ .
1 2 1 Тх 2 Т2 ТХТ2 4ТХТ2
Дріб і-!—2' завжди більший за одиницю, коли Тх Ф Т2. Наприклад, якщо
47]Г2
Тх = 2Г2, то дріб дорівнює 1,125;якщо Тх= 10Г2,то—3,02 тощо. Завдяки цьому
Д5 завжди додатна величина, тобто під час перебігу необоротного процесу
теплопередачі від гарячого тіла до холодного ентропія системи збільшується
в повному узгодженні із загальним принципом (1.112). Слід підкреслити, що
перехід теплоти від тіла з високою температурою до тіла з низькою може бути
проведений не шляхом теплопередачі (необоротно), а_ідляхом використання
цього процесу в тепловій машині, який може бути оборотним. У
найпростішій тепловій машині здійснюється цикл Карно. Оборотний цикл Карно
можна провести у зворотному напрямі (на рис. 1.4, с. 22, проти
годинникової стрілки). При цьому робота витрачається на те, щоб перенести деяку
кількість теплоти від холодного тіла до гарячого (теплова машина працює як
холодильний агрегат). Якщо і прямий, і зворотний цикли Карно оборотні,
рівноважні, то як система, так і навколишнє середовище повертаються у
вихідний стан без будь-яких змін. Для реальних процесів коефіцієнт
кориснім дії перетворення теплоти на роботу завжди менший, ніж (г|) циклу Карно.
Це пов'язане з тим, що у необоротних процесах частина теплоти може
переходити від нагрівача до холодильника шляхом теплопередачі без
виконання роботи, крім того, частина одержаної роботи за рахунок тертя
переходить знову в теплоту.
Таким чином, необоротність реальних процесів так чи інакше пов'язана
зі збільшенням молекулярного хаосу, тобто мовою термодинаміки — зі
зростанням ентропії. Коли б вдалося якось вилучити теплову, молекулярну
форму руху, то всі інші перетворення різних форм енергії були б оборотними.
Наприклад, під час падіння пружного тіла потенціальна енергія
гравітаційного поля переходить у кінетичну енергію руху тіла. Під час удару об тверду
поверхню кінетична енергія переходить у пружну енергію твердого тіла, яка
38
Томсон Вільям (лорд Кельвін)
(1824—1907),
Велика Британія
потім знову переходить у кінетичну и примушує тіло рухатися в
протилежному напрямі — вгору. Коли тіло підіймається, кінетична енергія переходить
у потенціальну енергію гравітації і так далі. Насправді цей процес
поступово затухає, тому що енергія поступово розсіюється і переходить у тепловий
молекулярний рух частинок, що утворюють тіло, а також частинок, що
знаходяться під твердою поверхнею в місці удару тіла об неї.
Другий приклад — перехід електричної
енергії конденсатора в магнітну енергію
індуктивної котушки й навпаки.
Електромагнітні коливання в контурі котушка-конден-
еатор теж поступово затухають. Дріт, по якому
проходить електричний струм, чинить йому
опір і нагрівається, внаслідок чого частина
електричної енергії переходить у
молекулярну, теплову. Усі ці процеси, під час яких інші
види енергії (механічна, електрична та ін.)
переходять у теплову, у молекулярний рух
частинок, що складають систему, одержали назву
дисипаційних. Дисипація означає
розсіювання. Вона не означає кількісної втрати енергії,
оскільки, згідно з першим началом
термодинаміки, енергія зберігається. Енергія змінюється якісно, згідно з В. Томсо-
иом — у замкненій системі енергія прямує до розсіювання, тобто до
переходу в рівномірно розподілену теплову енергію; вона вироджується,
шецінюється. Ентропія і є мірилом цього виродження енергії, тобто її
переходу в стан, з якого вона не може самочинно перейти в інші форми.
Робота виникає за рахунок спрямованого потоку енергії: механічний рух,
електричний струм, гравітаційне поле, сили пружності тощо. Теплота—це
■^е*— хаотичний молекулярний рух, і мірило його
/ґ 2^-^І^ хаотичності — ентропія. В тандемі «робо-
та-теплота» учасники нерівноправні —
робота може перейти у теплоту повністю,
наприклад, механічна робота під час тертя
повністю переходить у теплоту, а теплота
переходить у роботу лише частково. Це
зауважили ще В. Томсон: «Неможливі процеси,
єдиним результатом яких було б здійснення
механічної роботи за рахунок охолодження
теплового резервуара» та В. Оствальд:
«Неможливо побудувати вічний двигун 2-го
роду», тобто машину, що повністю
перетворює теплоту на роботу, наприклад,
шляхом охолодження якогось великого
водоймища. Ці вислови В. Томсона та В. Оствальда
39
Оствальд Вільгельм Фрідріх
(1853—1932), Німеччина
теж вважаються одними з формулювань 2-го начала термодинаміки. Однак
неповне перетворення теплоти в роботу має два аспекти. По-перше, всі
необоротні процеси ведуть до перетворення роботи в теплоту (при необоротній
теплопередачі від гарячого тіла до холодного втрачається потенціальна
можливість перетворення теплоти в роботу). Цей аспект—дисипація енергії
повністю відповідає згаданим щойно формулюванням 2-го начала і,
насамперед, у загальній формі Клаузіуса (збільшення ентропії в ізольованих
системах при перебігу необоротних процесів). Другий аспект пов'язаний з
аналізом циклу Карно. Навіть якщо користуватися лише оборотними
процесами, теплота, згідно зі співвідношеннями (1.42) та (1.45), не може повністю
переходити в роботу, частину її (#2) необхідно віддати холодильнику
(зазначимо, що для нерівноважних, необоротних процесів ця частина
збільшується і, відповідно, коефіцієнт корисної дії теплової машини падає, тобто
Лреал.< Лтеор.= (Ті ~ Т2)ІТ\)-За формулою (1.45) ї] = 1 лише при Т2 = 0 К, тобто
в тому абстрактному випадку, коли температура холодильника дорівнює
абсолютному нулеві (в реальних теплових машинах, наприклад, у двигунах
внутрішнього згоряння, Т2 — це температура зовнішнього середовища, яка
не дорівнює нулю).
Для процесів на молекулярному рівні (хімічні реакції, фазові переходи
тощо) дуже корисним виявився підхід, пов'язаний із розподілом внутрішньої
енергії системи, що складається з різних молекулярних рухів
(поступального, обертального, коливального, електронного тощо) на вільну (А або О)
і зв'язану (Т5) енергію. Нагадаємо, що вільна енергія визначає частку
внутрішньої енергії, що може перетворитися на роботу (в оборотних процесах).
Критерій ((1.106) або (1.107)) Гельмгольца-Нернста дає чітку відповідь на
те, в якому напрямі піде той чи інший процес у відповідних умовах (за
визначеними параметрами: температурою, тиском тощо), і за яких умов
виникає стан рівноваги в системі.
У наступному розділі розглядається, як застосовується цей принцип та
інші положення термодинаміки в однокомпонентних системах.
40
Розділ 2.
ОДНОКОМПОНЕНТНІ СИСТЕМИ
Пишався газ: «Я— ідеальний»,
А стиснули і став реальний.
2.1. Термодинаміка фазових переходів
у однокомпонентних системах
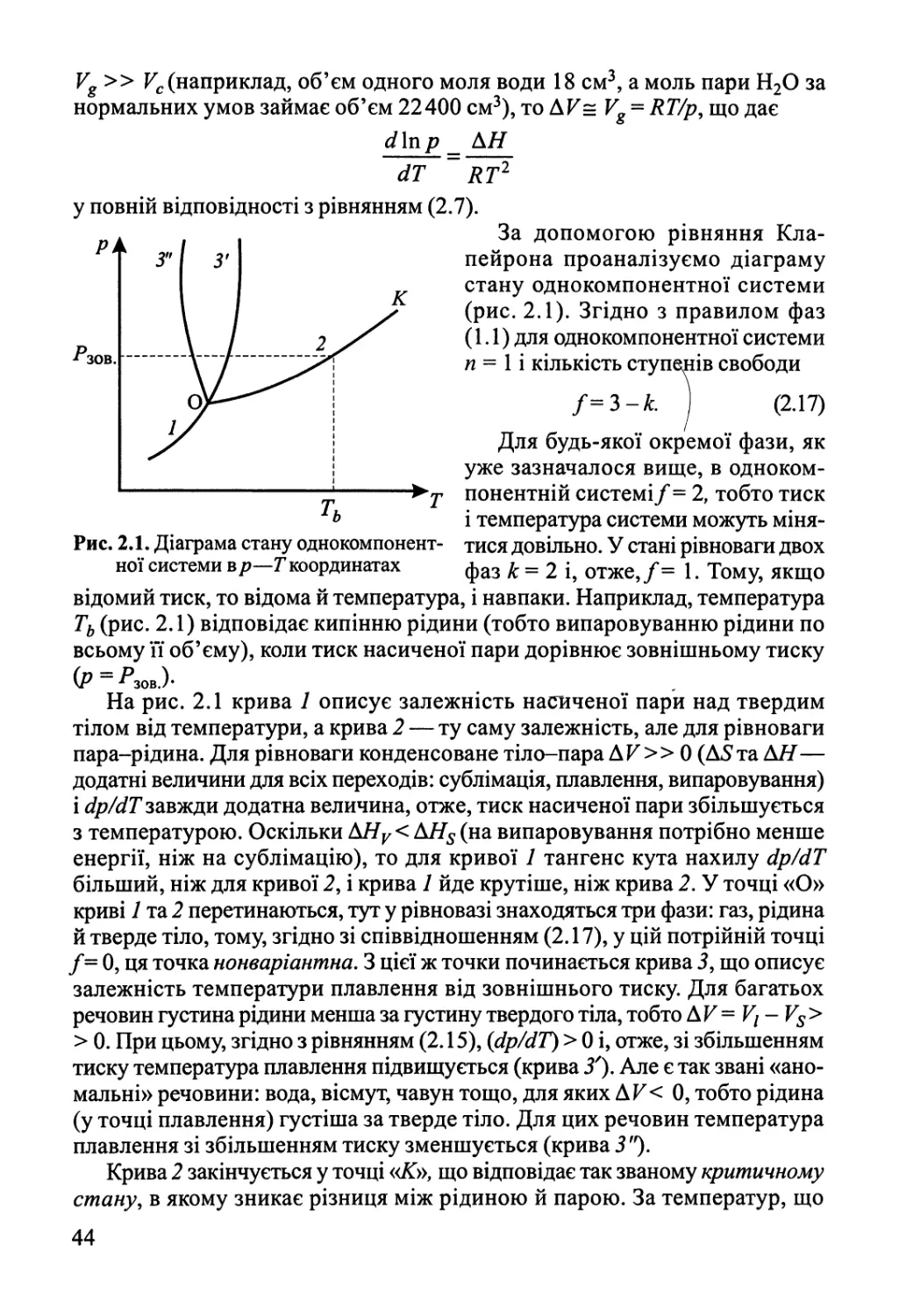
Якщо дві фази (а) та (Р) знаходяться в стані рівноваги, то для них
ДЄ = 0,абоСа = Ср. (2.1)
Для конденсованих фаз О = С°, де 0° — стандартне значення вільної
енергії Гіббса, тому для фазового переходу в конденсованому стані
(плавлення, поліморфне перетворення)
АС=АН°-ТАЗ°=09 (2.2)
звідки випливає формула (1.51), якою користувалися при обчисленні зміни
ентропії під час фазового переходу (плавлення).
Для газу О2 = 0° + КТІпр за формулою (1.83), тобто
ДС = Є*- С°= ДС° + ЯТІпр = 0 (2.3)
(індекс «с» означає «конденсований»).
Кипіння або сублімація настає тоді, коли зовнішній тиск (Р) дорівнює
тиску насиченої пари над конденсованою фазою (р). Якщо розглядати
кипіння за умови атмосферного тиску, то Р =р = 1 атм і ДС = ДО0 = Д#°-
- Д5,0= 0, тобто отримуємо рівність (1.52), яка є аналогічною (1.51). Варто
підкреслити, що теплові ефекти ДЯт, АНУ та ентропії фазових переходів,
що наведені у формулах (1.51) та (1.52), належать до стандартного тиску,
тому їх треба позначити як Д5° та Д#°.
Між стандартними теплотами та температурами фазових переходів
існує залежність: чим більше треба витратити енергії для фазового переходу,
тобто чим міцніші зв'язки між молекулами (атомами), тим вищою має бути
температура переходу. Згідно з правилом Трутона для випаровування
Д#£
Ть
а згідно з правилом Річардсона для плавлення
АНІ
'їй ~
т
= со«^ = 90, (2.4)
і плавлення
соня/= 8,4. (2.5)
Якщо зіставити співвідношення (2.4) і (2.5) з формулами (1.51) і (1.52),
то не важко помітити, що константи в (2.4) та (2.5) — це зміни стандартних
ентропій А8у та Д5° для відповідних фазових переходів. Це означає, що
41
для широкого класу речовин зміна впорядкованості під час відповідного
фазового переходу приблизно однакова. Крім того, видно, що А8У » А8т.
Таким чином, невпорядкованість значно більше зростає під час
випаровування, ніж під час плавлення, тобто, якщо 5/ > 55, то 8§ » 5/. Інакше
кажучи, газ є дуже невпорядкованою системою порівняно з конденсованим
станом.
За умови довільної температури (не за температури кипіння або
сублімації) з (2.3) одержимо
= Ктр.
(2.6)
Якщо продиференціювати (2.6) за температурою і врахувати рівняння
Гіббса-Гельмгольца (1.85), одержимо \
(2.7)
сіїпр _Д#°
йТ ЯТ2
Рівняння (2.7) описує залежність від температури тиску пари, що
знаходиться у рівновазі з конденсованою фазою. Воно має назву рівняння Кла-
пейрона-Клаузіуса.
Інтегруючи це рівняння на невеликому
температурному інтервалі, тобто припустивши,
що АН = соп8І, маємо
Д#°
НІ V = + СОП8І.
кт
(2.8)
Клапейрон Бенуа Поль Еміль
(1799—1864),
Франція
У координатах Іпр — ХІТ маємо пряму лінію,
тангенс кута нахилу якої дорівнює (-АН°/К).
Отже, вивчення залежності тиску насиченої
пари від температури дає можливість обчислити
АН° — теплоту випаровування рідини, або
сублімації твердої фази.
Над конденсованою фазою, крім насиченої
пари, може бути якийсь газ (або суміш газів),
що не взаємодіє з конденсованою фазою і не
конденсується за таких умов, наприклад, гелій, неон. Цей газ створює
додатковий тиск (Р) над конденсованою фазою, яка при цьому стискується. Це
веде до відповідної зміни вільної енергії Гіббса: сІС = УссІР, де Ус — об'єм
(мольний) конденсованої фази. Для того щоб система залишилась у стані
рівноваги, необхідно, щоб у газовій фазі вільна енергія Гіббса змінилася
на таку ж величину сЮ" = У^сір, де У§— об'єм (мольний), ар — тиск
насиченої пари. Вважаючи пару ідеальним газом, одержимо
—с!р = Усс!Р. (2.9)
Р
Інтегрування цього рівняння, коли Т = соті, дає
42
р
'КТ
1
-4р= [УС4Р,
Р
Ро "°
тобто
КТІп-£- = Ус(Р-р0), (2.10)
Ро
де/?0 — тиск насиченої пари за відсутності інертного газу (враховано, що Ус
практично не залежить від тиску).
Оскільки зазвичай Р» р0, маємо
(РУ \
р = Лехр№1. (2.11)
Це рівняння показує, що з підвищенням загального тиску в газовій фазі
тиск насиченої пари збільшується. Інертний газ, стискуючи конденсоване
тіло, сприяє переходу частини речовини в парову фазу, але цей ефект
проявляється лише за умови достатньо високих тисків. Наприклад, щоб
збільшити/^ в є разів, необхідно, щоб РУС = КТ. Для водневої пари при 300 К це
відбувається, лише коли Р = 1300 атм.
Рівняння (2.7) непридатне для опису рівноваги між конденсованими
фазами. Розглянемо процес фазового переходу в однокомпонентних
системах у загальному вигляді. У рівноважній системі енергії Гіббса фаз а та {З,
що співіснують, однакові згідно з формулою (2.1). Будь-яка зміна вільної
енергії в одній із фаз сІСа повинна компенсуватися відповідною зміною
в іншій фазі яЮр. При цьому система знову перейде в стан рівноваги, для
якого
в'а = Оа +сІва =С'р = Ор + сіСр. (2.12)
Якщо порівняти (2.12) та (2.1), одержимо
<Юа=с!Ср. (2.13)
Для кожної з фаз справедливе рівняння (1.70) для сІС, що дає
-8аЛТ + Уа(ір = -8рсІТ + У^р. (2.14)
Якщо позначити 5р - £а= А8, а У$ - Уа= АУ, то з (2.14) одержимо
Ф.= Д£ (2.15)
ЛТ АУ'
Рівняння (2.15) має назву рівняння Клапейрона. Рівняння Клапейрона-
Клаузіуса (2.7) — частковий випадок рівняння (2.15), якщо одна з фаз є
ідеальним газом. Справді, Д5 = АЯ/Г, тобто
&=***-. (2.16)
йТ ТАУ
Для фазового переходу конденсоване тіло-газ АУ= У§-Ус. Оскільки
43
У8 » ^(наприклад, об'єм одного моля води 18 см3, а моль пари Н20 за
нормальних умов займає об'єм 22400 см3), то АУ= У8 = ЯТ/р, що дає
сііпр _ АЯ
ат " кт2
у повній відповідності з рівнянням (2.7).
За допомогою рівняння Кла-
пейрона проаналізуємо діаграму
стану однокомпонентної системи
у (рис. 2.1). Згідно з правилом фаз
< (1.1) для однокомпонентної системи
п = 1 і кількість ступенів свободи
м
/=3-& ] (2.17)
Для будь-якої окремої фази, як
уже зазначалося вище, в одноком-
>т понентній системі/= 2, тобто тиск
1 ь і температура системи можуть міня-
Рис. 2.1. Діаграма стану однокомпонент- тися довільно. У стані рівноваги двох
ноїсистемивр—Ткоординатах фаз £= 2 і, отже,/= 1. Тому, якщо
відомий тиск, то відома й температура, і навпаки. Наприклад, температура
Ть (рис. 2.1) відповідає кипінню рідини (тобто випаровуванню рідини по
всьому її об'єму), коли тиск насиченої пари дорівнює зовнішньому тиску
<Р=Лов.)-
На рис. 2.1 крива 1 описує залежність насиченої пари над твердим
тілом від температури, а крива 2 — ту саму залежність, але для рівноваги
пара-рідина. Для рівноваги конденсоване тіло-пара АУ» 0 (А5та АЯ—
додатні величини для всіх переходів: сублімація, плавлення, випаровування)
і йр/йТзавжди додатна величина, отже, тиск насиченої пари збільшується
з температурою. Оскільки АНУ<АН8 (на випаровування потрібно менше
енергії, ніж на сублімацію), то для кривої 1 тангенс кута нахилу йр/йТ
більший, ніж для кривої 2, і крива І йде крутіше, ніж крива 2. У точці «О»
криві 7 та 2 перетинаються, тут у рівновазі знаходяться три фази: газ, рідина
й тверде тіло, тому, згідно зі співвідношенням (2.17), у цій потрійній точці
/= 0, ця точка нонваріантна. З цієї ж точки починається крива 3, що описує
залежність температури плавлення від зовнішнього тиску. Для багатьох
речовин густина рідини менша за густину твердого тіла, тобто АУ=¥і-У8>
> 0. При цьому, згідно з рівнянням (2.15), (йр/йТ) > 0 і, отже, зі збільшенням
тиску температура плавлення підвищується (крива ЗГ). Але є так звані
«аномальні» речовини: вода, вісмут, чавун тощо, для яких АУ< 0, тобто рідина
(у точці плавлення) густіша за тверде тіло. Для цих речовин температура
плавлення зі збільшенням тиску зменшується (крива 3 ").
Крива 2 закінчується у точці «АГ», що відповідає так званому критичному
стану, в якому зникає різниця між рідиною й парою. За температур, що
44
перевищують критичну (Тк),
неможливо сконденсувати пару
стисненням. Це особливо наочно видно з
діаграму— У (рис. 2.2). Коли Т< Г^на
ізотермі відбувається скачок об'єму
А К = У§ - К/ під час випаровування
рідини (або конденсування пари).
Величина А V зменшується,
наближаючись до критичної температури; коли
Т> Тк, скачка об'єму немає, газ
неможливо сконденсувати його
стисненням (або, інакше кажучи, якщо
Т > Тк, то зникає різниця між рідиною
та її парою).
Ізотерми на діаграмі;?—V, так само,
як ізохори на діаграмі р—Г, — це
проекції на площину об'ємної діаграми
у тривимірному просторі (р—У—Т).
Зразок такої діаграми наведений на
рис. 2.3. Зазначимо, що для
рівноважної системи тільки два з трьох (р,
У, Т) параметрів є незалежними (див.
підрозділ 1.1) внаслідок існування
рівняння стану. Вище згадувалось
лише одне рівняння стану —
рівняння Менделєєва-Клапейрона для
ідеального газу (1.2), (1.4). И. Ван дер Ва-
альс запропонував і фізично
обґрунтував рівняння стану, за допомогою
якого можна описувати поведінку
реальних газів, а також рідин.
у і
у
у \
1 \
1 \^\?'4
т^Гхх
г, -\\\
1 1 1 1 1 1 Л^>1
Рис 2.2. Діаграма стану
однокомпонентної системи вр—V координатах
Г2<Г3<Г4)
(ТЇ<Т2<Т3<
Рис. 2.3. Об'ємна діаграма стану
однокомпонентної системи
(координати р—У—Т)
2.2. Реальні гази. Леткість
Для ідеального газу тиск за формулою (1.4) дорівнює
КТ
Він створюється завдяки ударам об стінку молекул (інших частинок), що
мають лише кінетичну енергію і не взаємодіють одна з одною. Нагадаємо,
що для ідеального газу ґ-\Тт\
Щ =о.
Це є кінетичний внесок у тиск —ркін. У реальному газі між молекулами існує
взаємодія, яка на відносно великих відстанях (г) між молекулами (тобто при
45
г »*/, де */ — розмір молекули) має характер тяжіння. За Й. Ван дер Вааль-
сом, внесок потенціальної взаємодії у тиск
- а
Рпот. ~~ ~ тг2 '
де а—деяка константа. Крім того. Й. Ван дер Ваальс урахував, що
молекули рухаються не у всьому об'ємі, а тільки у вільному просторі: У0 = V- Ь,
де параметр Ь враховує об'єм, що займають молекули. Це веде до поправки
у кінетичну складову тиску
= КТ _ КТ
Отже,
о у-ь
_ КТ а
Р ~ Ркін + Рпот.~ у_, у 2
(2.18)
Це і ерівняння Ван дер Ваальса. Частіше його записують у такому
вигляді:
Р+^гІУ-Ь^КТ.
(2.19)
Ван дер Ваальс Йоханнес
Дідерік (1837—1923),
Нідерланди
Зазначимо, що, якщо об'єм (V) дуже
великий, тобто у розрідженому газі, то поправка
прямує до нуля, аК»Ь, тобто реальний газ
перетворюється на ідеальний, а рівняння (2.19)
вироджується у рівняння (1.4) Менделєєва-
Клапейрона. Аналогічний ефект дає
збільшення температури. Параметри (а) та (Ь) від
температури не залежать. Збільшення правої
частини (КТ) із підвищенням температури веде
до відповідного збільшення лівої частини
рівняння (2.19), що сприяє виконанню умов
У»Ьта р»^т.
Vі
За умови великих (V) асимптотою рівняння Ван дер Ваальса служить вісь
абсцис (так само, як і для рівняння Менделєєва-Клапейрона), а за умови
великих тисків і малих об'ємів асимптотою є не ордината (V = 0), а
вертикальна пряма (У= Ь). Це означає, що об'єм, який займають молекули, далі
не піддається стисненню.
Залежність р від V, що відповідає рівнянню Ван дер Ваальса, зображено
на рис. 2.4 (крива 7, при Т < Тк). Якщо зіставити цю криву \аЬсс!/^\ зр—V
кривою діаграми стану \аЬй[^\у то не важко помітити, що ділянка^/*
відповідає стисненню газу, а Ьа — стисненню рідини. Відрізок Ь/ відповідає
фазовому переходу, що здійснюється за умови сталого тиску. Рівняння Ван дер
Ваальса тут описує поліекстремальну залежність. Можна вважати, що
відрізки Ьс та /є мають фізичний зміст, а саме: відрізок Ьс відповідає
перегрітій рідині, ь/е — переохолодженій одрі.
46
Поліекстремальний характер
залежності/? від V випливає з того, що
рівняння Ван дер Ваальса є кубічним
по відношенню до Кі, отже, повинно
мати три корені: У{, У2 та К3, що
відповідають його розв'язку. Справді,
рівняння (2.19) можна подати у вигляді
Vі-
КТ .
—+ь
\у2 +
(2.20)
Р Р
Якщо розкрити кубічне рівняння
(У- У{)(У- У2)(У- У3) = 0і зіставити
його з (2.20), то одержимо:
РТ а
Уі+У2 + Уз= — + Ь;Г1-У2 + УгУ3 + У2-У3=-;
р р
аЬ
угу2.у3=—.
р
У критичній точці Ух = У2 = К3 = Ук (рис. 2.4, крива 2, якщо Т = Тк).
ач ^>тл2 а тлЗ аЬ
Отже, маємо ЗУ£ =—, а У^ =—, звідки
Рис. 2.4. Рівняння Ван дер Ваальса
в координатах р—V
(2.21)
Рк
Рк
а = 3ркг£, Ь = Ц-.
(2.22)
Ці залежності дають зв'язок між параметрами рівняння Ван дер Ваальса
і критичними параметрами. Відповідно, критичні параметри можна
виразити константами рівняння Ван дер Ваальса:
К
УК=ЗЬ; рк=-
■Рк=:
(2.23)
276'' " К '" 27ЬК
Обидва максимуми, що існували, коли Т<ТК (рис. 2.4, крива 1), у
критичному стані зливаються в один (крива 2, рис. 2.4). Точка К є водночас
точкою перегину (на кривій 1 це точка я"), та точкою екстремумів (точки с та
є на кривій 1). Таким чином,
йУ
= 0 та
ґ ^2 \
ар
(IVі
= 0.
Ж
За цих умов можна знайти критичні параметри, використовуючи похідні від
рівняння (2.18). Результат уже отриманий вище — рівняння (2.23). Не важко
також встановити, що між критичними параметрами існує зв'язок
РкУк=\кТк.
(224)
47
При Т«ТК корені рівняння Ух та У3 — це об'єми, відповідно, рідини й газу.
Якщо вважати, що Ух « У2« У3, то не важко пересвідчитись у тому, що
у{ = ух=ь,&ує = у,=^-.
Рівняння Ван дер Ваальса ста? універсальним для широкого кола
речовин, якщо застосувати т?" звані приведені координати: ^ ,
Т
Р У
Рк Ук
т = -
(2.25)
Використовуючи ці координати, за допомогою співвідношень (2.23) не
важко надати рівнянню (2.19) універсального вигляду
(
11 + -
ф'
(Зф-І) = 8т.
(2.26)
У критичному стані значення всіх параметрів рівняння (2.26) дорівнюють
одиниці. /
Для ідеального газу значення внутрішньої енергії не залежить від об'єму,
тобто С (іц \
(
ЛУ
= 0.
Знайдемо цю похідну в загальному вигляді
йУ
дУ
-іг
дУ)т [дУ
т [дт;г
(2.27)
(при виведенні рівняння (2.27) використані співвідношення (1.76) та (1.78)).
Знаходячи для газу Ван дер Ваальса
ии\ КТ а „ К а
ЬрЛ _ К
У-Ь'
одержимо
ЛУ
+^г + Т = —=■. (228)
У-Ь У2 У-Ь V2 К }
Таким чином, додаток у тиск (рПОІ) якраз визначає залежність внутрішньої
енергії реального газу від об'єму.
За допомогою рівняння Ван дер Ваальса можна визначити
термодинамічні функції. Наприклад, інтегруючи співввідношення
дА)
дУ
т-р-^-
КТ
У-Ь'
дістаємо для вільної енергії Гельмгольца
А = А°(Т)-^-КТЩУ-Ь),
для ентропії
Внутрішня енергія
48
5 =
дА)
дТ)„
= 3°(Т)+КІп(У-Ь).
ІГ = А + Т8 = ІІ0(Т)--.
У
(2.29)
(2.30)
(2.31)
(2.32)
Оскільки для ідеального газу V = £/°(Г), то член — , що описує
залежність внутрішньої енергії від об'єму, показує, що взаємодія між
частинками, їх взаємотяжіння зменшують їх внутрішню енергію.
Ентропія, порівняно з ентропією ідеального газу, змінюється на величину
А5 = 5'-5'ід =8°(Т)+КІп(Г-Ь)-8°(Т) + КІпГ = ЯІп?^<0. (2.33)
За рахунок того, що простір для руху частинок у реальному газі дещо
зменшений (тільки вільний об'єм), його ентропія зменшується. Отже,
реальний газ — більш упорядкована система, ніж ідеальний. Зазначимо, що для
рідини (У-Ь)« Ггазу, тобто в процесі конденсації А5« 0, що
зазначалося вище.
Рівняння Ван дер Ваальса хоч і достатньо фізично обґрунтоване, але у
точних розрахунках термодинамічних властивостей його застосування дає
лише певне наближення. Точні розрахунки можливі при використанні більш
точних, але емпіричних і досить громіздких рівнянь стану. Наприклад,
широко використовуються рівняння стану з так званими віріальними
коефіцієнтами:
рГ = КТ\1 + £ + -Цг + -Цг + ..А; (2.34)
^ V у1 V3 )
рГ = ЯТ(\ +В'р + С'р2 + В'р*+ ...). (2.35)
Перше наближення цих рівнянь — це рівняння Менделєєва-Клапейрона
(у відносно розрідженому газі при малих тисках V—> «>,/? —» 0).
Г. Льюїс та М. Рендалл запропонували метод, що дозволяє для
реального газу зберегти простий вигляд термодинамічних функцій, який вони мали
для ідеального газу. Для цього ці автори ввели поняття леткості (деякі
автори вживають замість терміна «леткість» інший — «фугітивність»). За
Г. Льюїсом і М. Рендаллом, леткість (/*) у реальних газах замінює тиск у газах
ідеальних. Одна з основних термодинамічних величин — вільна енергія
Гіббса — має за рівнянням (1.83) вигляд О - 0° + КТІпр для ідеального газу.
Для реального
С = С° + КТІп/. (2.36)
Це рівняння дає змогу визначати леткість. У диференціальному вигляді
аС = КТсіїп/. (2.37)
З іншого боку, якщо температура стала, то йСт = Уйр (співвідношення
(1.80)), отже,
Лп/ = £;<Ір. (2.38)
Інтегрування рівняння (2.38) дає можливість обчислити леткість
р
1п/=І^ф- (239)
49
Позначка (*) означає, що газ дуже розріджений і, таким чином, зникає
різниця між реальним газом та ідеальним, отже, /*=/**.
Один із способів обчислення леткості — застосування допоміжного
параметра — а.
р ЇД-
(2.40)
Цей параметр описує відхилення об'єму, який займав би за даних
температури та тиску ідеальний газ, від реального об'єму газу. Для ідеального газу
(а) тотожно дорівнює нулю. Підставляючи в (2.39) а замість
Додержимо
V р
р*
( кт
7-
КТ
ар.
К. Р
. Г ^_ґл
') Р ) КТ
р* р*
ф:
Гаф
= 1п^_£І_.
р* КТ
Оскільки 111/*= Іпр*, то
Іп/ = 1пр-—]ас1р
ЛГ-0
(дуже малу величинур* в інтегралі замінено на^нуль).
(2.41)
Інтеграл їасір зазвичай знаходять графічним інтегруванням. Якщо у
досить широкому інтервалі тисків а = соті, то з (2.41)
ар
р КҐ р КТ КТ рід'
ЗВІДКИ
Рід.
(2.42)
де/7ід — тиск, що його мав би газ, якби був ідеальним за умови відповідних
ар
значень V і Г. Розклад експоненти в ряд можливий, тому що — «*
К1
{рУ = КТ).Таким чином, реальний тиск виявляється середньою
геометричною величиною між леткістю та «ідеальним» тиском.
Часто зручно обчислювати не саму леткість, а її відхилення від тиску, так
званий коефіцієнт активності
У = ^- ^ (2.43)
50
Наприклад, із рівняння (2.41)
1пу = ~}аф. (2.44)
Зручною і досить точною формою обчислення (у) виявилося
застосування приведених параметрів: <р, %, т. Пояснимо це, використовуючи
рівняння Ван дер Ваальса для обчислення (у). Рівняння (2.39) в цьому
випадку зручніше інтегрувати, використовуючи як змінну V замість р.
Зрівняння (2.18)
Ар _ КТ | 2а
АУ (У-Ь)г Vі
(2.45)
V
іп-4=
/
V
КТ
КТ +2а
(У-Ь)2 V3
АГ, (2.46)
де У^ — об'єм розрідженого газу при тиску р*, тобто р*У„ = КТ.
Інтегрування (2.46) дає
1п/-1пГ=1п(К„-б)-1п(К-*)+^-|^-^+^:.
У^ — дуже велика величина, що дає
1п/-1пГ = 1п^-1п(Г-б)+^--^ =
= ІпДГ-1пр'-1п(К-6)+^--і^.
Остаточно
1п/ = Ш^+|А_-^, (2.47)
а для коефіцієнта активності, використовуючи приведені координати, маємо
шу [пр(у-ьуу-ь кту ш (тг к >
У_к_ ®М)
3 2х*РкГк _1п 8т 1 9_
Ук КтТкуУк я(3ф-1) Зф-1 4тф
Фгя з"
(див. рівняння (2.22)—(2.24)).
Рівняння (2.48) показує, що коефіцієнт активності є функцією приведе»
них координат (ф, т, ті). Рівняння стану (2.26) дозволяє вилучити один із цих
трьох параметрів. На практиці можна не використовувати рівняння Ван дер
Ваальса, але ідея, що у =/(т, я), залишається і дає можливість знайти у для
будь-якого газу для відповідних тиску і температури. Для визначення Т
і я необхідно, крім;? та Г, знати критичні параметри реального Т№ур%9 Т%
(див. табл. 2.1). Наприклад, синтез аміаку проводять за умовами: тиек —■
51
300 атм, температура—500 °С. Отже, п =р/рк=300П 11,3 = 2,7; т = Т/Тк=
= 773/405,6 = 1,9. Користуючись даними табл. 2.2, знаходимо умн = 0,93;
а /= УР = 279 атм.
Таблиця 2.1. Критичні температури (Тк) та тиски (рк)
Неорганічні речовини
Речовина
Вг2
СО
СОС12
СОЗ
С02
С32
с2>ї2
нси
С12
неї
р2
НР
н2
н2о
£>2
В20
ТК,К
584
132,91
455
375
304,2
552
400
456,8
417
324,6
144,30
461
33,2
647,14
38,4
644
рКі МПа
ю,о
3,387
5,5
5,7
7,14
7,7
5,8
5,22
7,5
8,0
5,05
6,3
1,26
21,35
1,61
20,97 1
Речовина
н§
і2
N.
ИН3
N0
м2н4
и2о4
о2
03
р
3
зо2 "
303
Н28
Ш*
ТкЛ
1735
819
126,2
405,6 \
180 )
653
431
154,58
261,0
994
1314і
430,8
491,0
373,2
505,8
рк, МПа
155,7
-
3,29
10,92
6,3
14,2
9,8
4,882
5,4
-
11,4і
7,633
7,9
8,65
4,5
52
Органічні речовини
^ Речовина
СН4
СС14
СНСІз
СНзОН
СНзИ
СНзИОг
с2н2
СгН4
СгНб
с2н4о
СНзСНО
СНзСООН
С2Н5ОН
СНзОСНз
СзНб
СзН8
СНзСОСНз
СдНб
С4Н8
С4Н10
С4Н10
С2Н5ОС2Н5
с«н6
с7н8
Назва
Метан
Тетрахлорметан
Хлороформ
Метиловий спирт
Метиламін
Нітрометан
Ацетилен
Етилен
Етан
Оксид етилену
Оцтовий альдегід
Оцтова кислота
Етанол
Диметиловий етер
Пропілен
Пропан
Ацетон
Бутадієн-1,3
Бутен-1 (а-бутилен)
Бутан
Ізобутан
Діетиловий етер
Бензен
Толуол
тк,к
190,6
556,25
536,55
513,15
430,1
588
308,33
282,36
305,45
468
461
594,75
516
400,05
364,9
370,0
508,7
425
419,5
425,1
408,0
466,56
562,0
591,7
рк, МПа
4,48
4,4
5,3 ± 0,2
7,70
7,22
6,11 ±0,07
6,04
4,905
4,73
6,96
-
5,602
6,2 ±0,1
5,2
4,47
4,1
4,57
4,19
3,89
3,68
3,53
3,53
4,77
4,08
53
І
В
м
Я
щ
«А
ч
ж
І
з-
й
54
к
їх
II
и
*■
1 й
се
них темпер
**
§
КІП
при
Значенії
Л *
вед
тип
СІ
«О
со
О
СП
г-
ся
^г
СЧ
СЧ
сч
о
сч
00
*
г*
ЧО
*
•л
ч*
*
сп
•*
сч
*
-
1,0
і X
р/р
р*
т-Ч
о
о
Т-Н
о
ЧО
о
о
о
о
о
о
Оч
ОЧ
о
ЧО
г-
оч
о
3
ОЧ
о
ЧО
»л
Оч
О
00
тг
оч
о
ЧО
гч
о>
о
ЧО
о
о\
о
о
г-
00
о
«3-
*—■І
00
о
«о
0,73
гч
0,61
*«ч
ОО
гч
о
о
сч
о
гч
»—н
о
о
о
о
о
00
(^
о
ЧО
«п
оч
о
о
сп
ОЧ
о
ТГ
««4
о\
о
00
ОЧ
00
о
1
тГ
гч
00
о
о
ЧО
г-*
о
00
ЧО
ЧО
о
о
0,56
</">
0,38
гч
ЧО
тг
о
гч
сп
о
о
сч
о
о
о
о
т
г*>
Оч
о
о
"*■
Оч
о
£1
о
Оч
о
о
00
00
о
чі-
«л
00
о
V©
о
00
о
00
"*•
Г-»
о
оо
ЧО
ЧО
о
о
ЧО
ю
о
чп
0,43
00
0,28
со
ГЧ
ЧО
о
00
"3-
о
о
СП
о
о
о
о
сч
г^
оч
о
о
СП
оч
о
сч
00
00
о
00
»л
00
о
"І"
гч
00
о
3
г-
о
о
оч
ЧО
о
о*
о
ЧО
о
«*•
ОЧ
-ЧІ-
о
о
0,37
00
0,24
тГ
о
00
о
гч
ЧО
о
гч
чГ
о
00
о
о
гч
г-
Оч
о
гч
гч
Оч
о
ЧО
ЧО
00
о
гч
гг
00
о
гч
о
00
о
ЧО
т
г^
о
тґ
«л
ЧО
о
ЧО
ЧО
«п
о
тґ
ЧО
тГ
о
00
0,33
ЧО
0,22
<л
оо
ОЧ
о
ті-
г-
о
сч
ю
о
^1*
*—1
о
оо
г^
Оч
о
о
гч
Оч
о
о
ЧО
00
о
ЧІ-
сп
оо
о
00
00
Г-*
о
о
гч
І-»
о
"З"
сп
ЧО
о
тГ
-*-
чп
о
гч
-*-
-ч-
о
оо
0,31
о
0,21
ЧО
гч
Г-Н
гч
ОЧ
о
00
ЧО
о
ЧО
гч
о
00
00
Оч
о
ЧО
гч
Оч
о
о
ЧО
00
о
гч
гл
00
о
о
00
г^
о
о
*-н
г^
о
ЧО
гч
ЧО
о
гч
сп
«о
о
о
сп
^г
о
о
0,31
гч
0,20
|>
ЧО
сп
о
Т-*
г—<
ЧО
00
о
о
"3-
о
о
о
о
г~~і
ТІ-
ГП
оч
о
оо
ЧО
00
о
гГ
ГЛ
00
о
ч*
00
г*-
о
гч
Т-4
г^
о
<ч-
гч
ЧО
о
00
гч
ип
о
00
гч
^
о
оо
0,30
о
0,20
00
оо
ип
о
гп
ЧО
о
00
»п
о
^-
о
*"*
оо
^г
Оч
о
оо
г*»
00
о
о
гГ
00
о
гч
ОЧ
г^
о
о
гч
г»
о
о
гп
ЧО
о
гч
гп
<г>
о
о
т
"^
о
о
0,31
о
0,20
Оч
о
00
т
»о
00
гч
чо
г-
о
«3-
т
о
,—.
ТГ
чо
ОЧ
о
о
Оч
00
о
гч
ип
00
о
чо
о
00
о
о
т
г**
о
о
^
ЧО
о
гч
41-
«г>
о
"*
гп
^ґ
о
гч
0,31
гч
0,20
о
<<г
о
гч
<^-
г^
гч
«п
о
о
^Ґ
^п
о
*""<
гч
00
ОЧ
о
00
о
Оч
о
ЧО
ЧО
00
о
о
00
о
N0
ТГ
с-
о
гґ
«о
ю
о
гч
«п
»п
о
о
ЧО
гг
о
-
ЧО
гч
гч
00
оч
^г
г-
ЧО
гч
00
с^
о
г"н
00
о
о
00
гч
ОЧ
о
<*•
00
00
о
ТГ
т
00
о
о
ЧО
Г"*
о
00
чо
чо
о
чо
чо
«л
о
ч*
г^
ТІ-
о
гч
о
«п
гч
гч
гч
гч
гч
о
гч
ГЧ
**п
чо
о
1—1
'"■'
тГ
»—«
о
гч
ип
Оч
о
ЧО
о
ОЧ
о
гч
«п
00
о
00
г-
г^
о
ЧО
00
чо
о
гч
00
«о
о
о
оч
-<*•
о
т
о
оо
гч
00
ГГ
гч
00
гч
гч
о
00
гг
т
г-Н
-*
чо
чо
о
00
г^
Оч
о
о
т
Оч
о
Т*
г*
оо
о
00
ОЧ
г-
о
чо
о
г-
о
00
оч
ип
о
о
і-Н
«о
о
ТГ
о
»—і
т
о
00
гч
ЧО
«п
гч
^3-
гч
чо
чо
*—1
*"*
о
о
»—1
ЧО
о
о
оо
»г>
Оч
о
гч
о
Оч
о
ЧО
гч
00
о
00
гч
г-
о
о
гч
ЧО
о
гч
т
«п
о
«о
о
<ч*
гп
о
1—1
т
о
Оч
гч
о
чг
гч
00
ОЧ
1—1
""Ч
ч^-
ч-Н
г—1
ЧО
т
о
ЧО
ОЧ
Оч
о
гг
т
ол
о
гґ
»л
00
о
00
ип
г*»
о
ЧО
^*
ЧО
о
»/->
-^г
*о
о
чо
оо
чо
т
ГЧ
-ч-
т
гч
гч
т
"^Г
г^
гч
о
т
гч
*"'
гч
г^
г—і
гч
г-
о
чо
гч
о
'-"'
о
г-
Оч
о
о
Оч
00
о
ЧО
00
г-
о
гч
г^
чо
о
ип
чо
</п
о
г^
гч
о
ТІ-
^1*
г*
гп
'^*
ип
т
о
<—н
т
о
г-
гч
V
4
С5
/ГЧ
о
»—і
т-Н
чо
ю
о
*"•
ЧО
о
о
^
о
гп
Оч
о
ТГ
гч
00
о
ЧО
о
г-
о
оо
г^
ип
о
00
^*
т
гГ
'^Г
^!І*
гч
оч
т
00
"Ч-
т
00
о
т
'-н
00
^1-
гч
о
ю
чо
о
^
о
«/п
о
~
о
г-
Оч
о
о
ЧО
00
о
00
гп
г-
о
"<1-
о
ЧО
о
Оч
00
чо
*<!•
гч
41-
-^г
гч
т
^*
чо
00
т
о
гґ
т
""•
00
оо
гч
о
оо
г—«
гч
^
♦-М
00
00
о
^
чо
о
о
*ч
'41-
Оч
00
о
оо
чо
г^
о
00
гч
ЧО
о
о
гч
ТГ
о
»Г)
ЧО
г-
ТГ
гч
г^
^г
00
1—1
тГ
ЧО
о
"Ч*
1—1
00
гч
т
гч
ТГ
т
*°
гч
гч
ип
^*
1-Н
»п
ЧО
ЧО
^
чо '•
гч
'Ч-
Г-.
ЧО
ЧО і
т і
гч
гч
ІЗ»
«з
5
з»
іі
1 •>
урах
5
О.
емпє
них т
ри приведе
Б
>■"
Значення
о> *
Привед
ний тис
о
со
<м
сч
СЧ
о
сч
00
ЧО
т*
сч
о
о\
оо
г-»
чо
•о
г *
см
о
»л
»—*
о
<^
»—і
о
•*■
см
о
оо
см
о
о
сп
о
чО
ся
о
оо
ся
о
ся
"*
о
оо
;£
о
г-
«л
о
ЧО
»л
о
ся
ЧО
о
^ц
с-
о
ЧО
г^
о
«о
00
см
о
_
ся
о
Оч
гя
о
00
^г
о
см
»л
о
_
ЧО
о
о
г-
о
см
Г-*
о
00
00
о
СМ
о
г*
«—і
о
см
«—<
«л
ся
т—<
см
«л
1-М
г-
ЧО
*-н
о
см
^-
о
іл
ТІ-
о
00
«о
о
сч
г^
о
о
00
о
г-
оо
о
00
о
о
^■ч
»—Н
ЧО
СЯ
о
ЧО
^
оо
,^
Оч
1—ч
^г
сч
ч*
^ґ
сч
тг
г-
<ч
»л
»—1
гГ
«о
о
о
ЧО
о
сч
00
о
о
о
о
1—1
г-
<ч
оо
тГ
сч
«п
г—4
СЧ
00
о
«-Ч
сч
оо
^
ся
тГ
г*
сч
сч
о
ся
ЧО
^*
ся
сч
о
^
о
см
о
г-
о
гг
оо
о
ЧО
о
»—•
00
<ч
сч
п-
00
»л
00
оо
С>|
Оч
«-Н
тг
ся
сч
ТІ-
оо
сч
00
*—Ц
ся
ЧО
«л
ГО
00
оч
ся
о
»л
тГ
о
■ч-
«л
«л
сч
ЧО
оо
о
ЧО
о
о
ся
ЧО
«о
ЧО
г-
сч
о\
оо
сч
сч
"*-
СЯ
сч
сч
Оч
сч
сч
«л
ся
СЧ
Оч
о
"3-
"3*
тГ
см
о
«о
о
г^-
«л
ЧО
оо
ЧО
о
ся
тГ
о
ЧО
сч
о
ЧО
чГ
00
00
о
сч
00
сч
сч
о
г-
сч
^
00
гм
о
«л
СЯ
ті-
сч
^г
о
г^
тг
"<*•
СЯ
«о
гм
ЧО
00
о
г-
оо
ЧО
оо
«л
ся
00
Г-*
ЧО
41-
оо
г-
гч
гч
о
^г
гч
ЧО
ЧО
сч
гч
«—<
ГО
00
гч
гл
о
»—«
тГ
гм
Оч
тГ
^І-
«п
«/■>
о
го
ЧО
00
см
г*
тГ
«о
00
00
см
о
см
о
тГ
ТГ
сл
00
ЧО
см
о
см
см
тг
см
пг
г^
см
чо
о
сп
^
»п
сп
о
00
ГО
о
г-
'«г
о
г^
»п
гг
ч*-
ЧО
ЧО
т
г-
о
«п
00
00
<-н
о
см
оо
см
см
см
«л
чГ
см
«о
00
оо
00
см
см
см
г-
см
00
о
сл
чО
•*•
сл
о
о
-*•
см
сл
гГ
^г
со
»п
гГ
«л
ЧО
<*
^ґ
с^
о
»л
00
ЧО
00
Оч
о
Оч
*—4
см
о
«л
ч^-
см
о
<л
оо
ЧО
00
о
см
см
»л
см
см
о
со
см
тг
СЛ
оо
оо
ся
оо
"3-
тГ
чо
00
тГ
00
Оч
«л
о
тГ
г-
тГ
"*•
00
00
чо
Оч
чо
см
*—і
см
см
г^
сл
см
^
Оч
чо
см
«л
«л
см
оо
о
сп
см
00
г-
см
^г
го
со
о
00
со
см
со
^І-
о
о
»л
чо
^г
»л
тґ
ЧО
ЧО
оо
см
00
см
<л
Оч
00
Оч
о
см
чГ
с^-
см
см
о
г^
•л
см
чо
ЧО
Оч
см
о
ЧО
ЧО
оч
см
»л
см
ЧО
о
со
00
чо
ГО
чо
1—1
^1-
ЧО
г^
гг
см
»л
»л
см
о
чо
«л
чо
ГГ
см
см
г-
см
см
ГО
го
о
00
сп
тГ
•л
-^
ЧО
см
»л
00
о
ЧО
см
ЧО
чо
о
г-
00
со
см
см
Оч
см
см
ЧО
со
оо
со
'|^
4Ї"
Оч
ТҐ
о
ОЧ
»л
оо
ЧО
ЧО
оо
см
1^
«л
г-
00
^
см
^-
ч—(
ГП
о
Оч
со
см
Г"»
тГ
00
т
•л
см
см
ЧО
ЧО
см
г-
^г
Оч
г-*
о
00
оо
чо
см
00
сл
ся
чо
см
'Ч-
гч
*—н
«л
см
оо
<л
см
г^
ЧО
о
ОЧ
г-
см
ЧО
00
»л
оо
оо
00
см
о
ЧО
ся
чо
»л
тг
00
тГ
»л
ЧО
см
ЧО
00
см
г-
см
ЧО
оо
о
СЛ
Оч
о
Оч
00 00
О СМ
сл т
о см
00 О
го та-
О 00
ОЧ СМ
Т «л
О 00
Оч СМ
«л чо
оо см
чо *-і
ЧО Г*»
^ 00
г- см
Г^ 00
СМ 00
—* г-
Оч Оч
см о
о г-
о о
см" см"
«л о
ол о
1—1 1
55
2.3. Термодинаміка конденсованого стану
однокомпонентних систем
Важливими характеристиками ізотропних конденсованих тіл (рідина,
аморфне тверде тіло, полікристал) є стисненість
1 (*Г) (2.49)
і коефіцієнт термічного розширення
а = Цд±) . (2.50)
ав =—
V
Зі збільшенням температури об'єм збільшується, відповідно, —
додатна величина. Навпаки, при збільшенні тиску об'єм зменшується, відпо-
ҐдУЛ
- від'ємна; тому, щоб зробити стисненість додатною
.р
відна похідна
др
)І
величиною, в правій частині формули (2.49) поставлено мінус.
Застосування експериментально визначених параметрів (а) та (ав) дає
можливість використати одне зі співвідношень Максвелла для обчислення
змін ентропії в конденсованому тілі за зміною об'єму
(дУ) ,„ а ...
-— аУ =—аУ. (25\Л
<18 =
Враховано, що
ав
(2.52)
Величини (ав) та (а) за своїм фізичним змістом означають
сприйнятливість тіла до зміни його об'єму внаслідок механічних або термічних
навантажень. Вони залежать від міцності сил зв'язку в конденсованій фазі: міцні
сили зв'язку чинять більший опір дії зовнішніх механічних або термічних сил,
і, відповідно, таке тіло має невеликі значення (ав) та (а). Навпаки, тіло із
слабкими зв'язками між частинками (молекулами, атомами тощо) має великі
значення (ае) та (а). Отже, між ав та а існує певна кореляція. Тому відношення
а/ав є приблизно сталою величиною. Це дає можливість проінтегрувати
рівняння (2.51), замінивши диференціали на відповідні прирости
ав
(2.53)
Оскільки а та ав є додатними величинами, то стиснення конденсованої
фази веде до зменшення ентропії (АУ< 0, Д5 < 0) і, навпаки, при АК> 0
і Д5'> 0. Пропорційність між змінами ентропії та об'єму\арактерна для
конденсованих систем.
56
Для ідеального газу ае = \/р, аа= 1/Гзарівняннями (2.49), (2.50) та (1.4).
Отже, рівняння (2.51) для ідеального газу має такий вигляд:
& = £&=*&. (2.54)
Т V
Інтегрування (2.54) дає вже відомий результат — рівняння (1.53), тобто
залежність Д5 від зміни об'єму не лінійна, хоча зі збільшенням об'єму газу
ентропія його також збільшується.
Експериментальні значення а та ж для конденсованого тіла можна
використати для знаходження зв'язку між Ср та Су. Розглянемо це питання
в загальному вигляді. За визначенням (формула (1.22)) теплоємності при
постійному тиску
дт
(дИЛ (дУЛ
дт
4тт+№\-с.ш\
)у
ІаН^ГЧ^ГПНІШ
ди
+р
формулами (2.50) та (2.27) —— = а.У, а вираз у дужках дорівнює
„,Л ^ >' а
— , що, згідно з формулами (2.49), (2.50) та (2.52), дає Т —. Таким чином,
дТ ]у ж
За
дР
С„-Су =
а2УТ
ае
(2.55)
Це загальне співвідношення між Ср і Су для будь-якої фази.
Для ідеального газу а = 1/Г, ае = \Ір і, отже,
Р У грі гр
згідно з раніше одержаним результатом (формула (1.26)).
Різниця між Ср та Су обумовлена тим, що при нагріванні за постійного
тиску додаткова енергія йде на виконання роботи розширення.
При невеликих змінах температури, згідно з рівнянням (1.25), маємо
С-Су=^-. (2.56)
Для ідеального газурАУ= КАТ і знову маємо рівняння (1.26).
Для конденсованого тіла: а = (\/У)(АУ/АТ); оУ = АУ/АТ; аГ= (Г0 + АТ) =
= аДГ= АУ/У(при Т0« ДГ). Тоді маємо
г _аУаТ (АУ^
ае
1 АУ
АТ Іае V '
57
женні з рівнянням (2.56) Ср-Су--
За теорією пружності для ізотропного тіла відносна зміна об'єму (АУ/У)
пропорційна тиску та стисненню, тобто (АУ/У) =/?ае. Остаточно в узгод-
рАУ
АГ *
Таким чином, якщо відоме Су, то можна знайти Ср.
Для визначення Су простих речовин у твердому стані П. Дюлонг і А. Пті
знайшли просте правило. Вони помітили, що добуток питомої теплоємності
Спит при постійному об'ємі на атомну масу (М) є приблизно сталою
величиною
СПИТМ = С^0Л=ЗД. (2.57)
Рівняння (2.57) свого часу дало можливість оцінювати атомні маси
елементів. Обґрунтування цього рівняння дає квантова теорія теплоємності
(підрозділ 11.6).
58
Розділ 3.
РОЗЧИНИ НЕЕЛЕКТРОЛІТІВ
Чай, кока-кола, різні лимонади...
Цим розчинам, напевно, кожен радий.
3.1. Закони Генрі та Рауля. Розчинність
У розбавлених розчинах рідина завжди знаходиться у надлишку і має
назву розчинника. Концентрація речовини, що розчиняється, визначається
різними способами. В фізичній хімії найчастіше використовуються такі
способи вираження концентрації розчинів: молярність, моляльність і мольна
частка.
Молярність (сі) визначається кількістю молів і-го компонента (пі) на
одиницю об'єму (V) розчину, моль/л,
Сі = Пі/У. (3.1)
Моляльність (ті) визначається кількістю молів і-го компонента на
одиницю маси (%) розчинника, моль/кг,
ті = Пі/£. (3.2)
їіольна частка (х,) визначається кількістю молів /-го компонента по
відношенню до загальної кількості молів розчину
хі ~ Угу > (3.3)
X; — безрозмірна величина.
На відміну від (сі) та (т,), мольна частка може бути застосована не тільки
для розбавлених, а й для розчинів будь-якої концентрації (0 <*,- < 1)
(зазначимо, що при х( > 0,5 вже немає сенсу вживати терміни «розчинник» і
«розчинена речовина»).
Розчинність (з) — це концентрація насиченого розчину, тобто розчину,
що знаходиться у рівновазі з надлишком речовини, що розчиняється (при
визначенні розчинності спосіб вираження концентрації може бути різним).
Розглянемо, як залежить розчинність речовин від температури.
Візьмемо вихідну речовину в стандартному стані (при тиску 1 атм) і переведемо
один моль її в розчин. Зшна вільної енергії (АС) при такому переході
АС = А(?+КТІпхв (3.4)
(для розчиненої речовини у розчині враховано, що Св = \хв = [і°в + КТІпхв>
згідно з формулою (1.101)).
Для насиченого розчину, згідно з умовами рівноваги, АС = 0 і, отже,
маємо
АС0
ІП5 = 1п(хв)рівн = —— , (3.5)
59
ЗВІДКИ
дТ
_3/
дТ
АО°]
КТ
)р
дя°
КТ1'
(3.6)
2,0
Є
.а 1,0
X
І
Он
Не
10 20 ЗО
40
Таким чином, залежність
розчинності від температури
визначається ентальпією
розчинення — Д#°. Для газів
АН° < 0 (екзотермічний
процес), тому їх розчинність
зменшується з температурою
(рис. 3.1). Для твердих тіл
зазвичай АН° > 0
(ендотермічний процес — енергія йде на
руйнування кристалічних
ґраток твердого тіла, процес його
переходу в розчин (рідину) за
тепловим ефектом можна
порівняти з процесом плавлення
твердого тіла) — розчинність
із температурою збільшується
Температура, С
Рис. 3.1. Залежність розчинності газів
від температури
(рис. 3.2). При розчиненні рідини у рідині знак АН° може бути різним,
відповідно, розчинність може або збільшуватися з температурою, або
зменшуватися.
Розчинність газів у рідинах
при фіксованій температурі
залежить від їх тиску в газовій
фа^і і описується законом
Генрі
хв = КгР, (3.7)
де хв — мольна частка
розчиненого газу, Кг — константа
Генрі, Р—тиск у газовій фазі.
Загальний тиск у газовій
фазі (Р) у багатьох випадках
практично дорівнює
парціальному тиску газу, що
розчиняється, тому що тиск
насиченої пари розчинника
невеликий. Наприклад, якщо
розглядати розчинність якогось
0 10 20 30 40 50 60 70 80 90 100
Температура, °С
Рис. 3.2. Залежність розчинності твердих солей
від температури
газу у воді при кімнатній температурі й атмосферному тиску, то тиском
насиченої пари води (0,03 атм) можна знехтувати.
60
Розглянемо тепер ситуацію, коли в рідині у малій кількості розчинено
тверде тіло або малолетка рідина. Тоді можна вважати, що у газовій фазі є
лише пара розчинника. Ф. Рауль установив, що тиск пари над розчином (р)
менший, ніж відповідний (при такій самій температурі) тиск над чистим
розчинником (р°), згідно зі співвідношенням
рА= р°АхА, (3.8)
де хА — мольна частка розчинника у розчині.
Оскільки хА + хв = 1, де хв — мольна частка речовини, що розчиняється,
то закону Рауля можна надати такого вигляду:
&Ра _Р0а-Ра_ІУ-ха)Р°а _г ПОч
Т""" о ~ о "хв- у?*)
Ра Ра Ра
Отже, відносне зниження тиску пари над розчином дорівнює мольній частці
розчиненої речовини.
Наявність розчинених частинок у розчиннику заважає процесу його
випаровування, водночас на процес конденсації це не впливає, отже,
частка пари сконденсується, тиск її зменшиться і, таким чином, рівновага між
розчином і газового фазою встановиться при тиску, меншому, ніж тиск пари
над чистим розчинником.
Зменшення тиску пари розчинника у газовій фазі призводить, згідно
з формулою (1.82), до зміни вільної енергії Гіббса газової фази на величину
Ьвг=КТ\пЦ. (ЗЛО)
Ра
Для розчинника у розчині маємо, згідно з формулою (1.101),
О а = \іа = Іі°А + КТІпхА = С° + КТІпха . (3.11)
Відповідно, зміна вільної енергії Гіббса у розчині
Щ = ^розчин ~^розчинник =С?+КТІПХА-С/° = КТІПХА . (3.12)
При рівновазі АСВ = АС/, що, згідно з формулами (3.10) та (3.12),
приводить до закону Рауля (3.8).
3.2. Ебуліоскопія, кріоскопія, осмос
Зниження тиску над розчином веде до збільшення температури кипіння
розчину порівняно з чистим розчинником. Це явище має назву
ебуліоскопії. На рис. 3.3, с. 62, зображено залежність від температури тиску
насиченої пари над розчинником (крива 7) та розчином (крива 2). Крива 2
перетинає лінію зовнішнього тиску (Р3ов.) (нагадуємо, що рідина кипить за
умови/?нас= Р3ов.) ПРИ температурі (Та), більшій, ніж температура кипіння
чистої рідини (Ть — точка перетину кривої 1 з лінією Р30в.= соті). Величина
Та-Ть = АТЬ залежить від концентрації розчину
АТь=кет, (3.13)
61
РЬ
р І І Л^-А--
де ке — ебуліоскопічна
константа, що залежить від природи
розчинника, т — моляльність.
Обґрунтуємо формулу (3.13).
На рис. 3.3 відрізок аЬ = Та-Ть =
= АТЬ\ відрізок Ьс = Ар. При
малих Ар та АТЬ фігуру «аЬс»
можна вважати трикутником, тобто
АТЬ = АрІХ%а. Величина Ар
визначається з рівняння (3.9) (закон
Рауля), а \%а = <ір0ІсІТ можна
Рис. 3.3. Залежність тиску насиченої пари знайти з рівняння (2.7) для
розвід температури над розчинником (/) винника Отже
і розчином (2)
АТЬ =
Р0хв
р0АН°у/ЯТь*
_Щ
АНГ
Для розбавлених розчинів
«в
_ пв _пв
отже,
АТ„ =
де Іу =
АНУ
пА+пв пА
„ пв ^ ЯТ?пв = ЯТ?пв ^ ЯТ?
АНгпА ІуМАпА Іу8л /к1000
ЯК
ш,
(3.14)
- питома теплота випаровування розчинника, МА —
молекулярна маса, %А — маса розчинника.
Зіставляючи формули (3.14) і (3.13), знаходимо вираз для
ебуліоскопічної константи
*-=■
кп
(3.15)
1000/г
ке містить параметри (Ть, Іу) виключно розчинника.
Крива 2 перетинається з кривою 3 залежності від температури
рівноважного тиску над твердим розчином при температурі Гг, яка менша за
температуру тверднення чистого розчинника (Тт). Це явище має назву
кріоскопії (див. рис. 3.3 та рис. 3.4).
Для зниження температури тверднення
АТт = ккрт, (3.16)
де ккр — кріоскопічна константа.
На рис. 3.3 відрізок є/=АТт а осі'=Ар. З трикутника «еф> маємо сІ/= є/- їф;
а з трикутника «ео/»: о/= є/*- ї§у, звідки осі = о/- сі/= е/(Щу - іф) (Хф
відноситься до тієї самої рівноваги рідина-пара, що й 1§ос, але Хф Ф Х§а, тому що
похідні дрійі беруться при різних температурах (Тт і Ть)). Величини 1§(3 та
і§у знаходимо за допомогою рівняння Клапейрона-Клаузіуса для рівноваг
рідина-пара та тверде тіло-пара, а Ар — із закону Рауля, звідки
62
АГ =
АНІ
Хв =
1000/.
-от,
(3.17)
де
Іт =
АН°„
питома теплота плавлення
розчинника.
МА
Враховано, що Д#£ = АНу + Д#°, тобто
мольна теплота сублімації (АН$) складається з
мольних теплот випаровування (АНу) та плавлення
(Д#£). Відповідно,
ТУТ*
(3.18)
к = КТ>«
кр 1000/,/
тобто кріоскопічна константа, як і ебуліоскопічна,
залежить лише від властивостей розчинника.
Зазначимо, що вимірювання АТЬ або АТт дає
можливість визначати молекулярну масу тієї
речовини, що розчиняється (обчисленням т, за
формулами (3.16) або (3.13) і далі кількості молів
розчиненої речовини при відомій її масі).
Одним із цікавих явищ у розчинах є осмос.
Якщо взяти розчинник і занурити в нього ємкість
із розчином, який відокремлено від розчинника
напівпроникною перетинкою, то молекули
розчинника проникатимуть крізь цю перетинку (молекули розчиненої речовини не
можуть пройти крізь перетинку з тим, щоб зменшити концентрацію
розчину). Сила, яка гонить молекули розчинника крізь цю перетинку,
одержала назву осмотичного тиску (я). Оскільки маса розчину за рахунок
надходження додаткових порцій розчинника збільшується, то рівень рідини в
ємкості з розчином підвищується. Цей процес
відбувається, доки осмотичний тиск не буде
врівноважений гідростатичним тиском «зайвої» маси
розчину. Я. Вант-Гофф показав, що осмотичний
тиск пропорційний концентрації (молярній)
розчину:
Рис. 3.4. Схема приладу
для визначення
температури тверднення:
1 — льодова баня;
4 — термометр Бекмана
(точність до 0,001 °С)
П = сКТ.
(3.19)
Обґрунтуємо формулу Я. Вант-Гоффа з
термодинамічних позицій.
У розчині вільна енергія розчинника С = С° +
+К Т 1п хА + п Ус, де хА — мольна частка
розчинника, Ус—його мольний об'єм. Поява члена пУс
еквівалентна внеску у вільну енергію
зовнішнього тиску, що було розглянуто у підрозділі 2.1
Вант-Гофф Якоб Хендрік
(1852—1911),
Нідерланди
63
(дир. формули (2.9) — (2.11)). Для чистого розчинника С = О0. При рівновазі
ДС для розчинника дорівнює нулеві. Таким чином,
0 = Ав = С° +КТІпхА+пГс-С° = КТІпхА+пГс,
ЗВІДКИ
Ус
кт
ЯТп
-хв) в хв = 2- = ЯТ-2- = сЯТ,
Ус УспА V
тобто одержали формулу (3.19) Я. Вант-Гоффа.
При її виведенні враховано, що хв — мала величина. Це дозволяє
розкласти 1п(1 - хв) в ряд і далі застосувати сшвщдвойїення хв = — . Добуток пА Ус
пА
в розбавлених розчинах практично дорівнює об'єму розчину.
Осмотичний тиск відіграє важливу роль у житті організмів на клітинному
рівні (рис. 3.5).
Рис. 3.5. Осмотичний тиск у клітині (А — мембрана). /— вода проникає
у клітину з розбавленого розчину, клітина розбухає (наприклад, тиск на очі
у прісній воді); II— вода залишає клітину, переходячи у концентрований
розчин, — це явище плазмолізу; НІ— ізотонічний розчин, обмін водою
і осмотичний тиск відсутні, рівновага
3.3. Активності у розчинах неелектролітів
Відхилення від ідеальності у розчинах можна описати, користуючись
прийомами, запропонованими для реальних газів. Як було показано
в розділі 2, термодинамічні закономірності, одержані для ідеальних газів,
можна зберегти для реальних газів, користуючись поняттям леткості (замість
тиску для ідеальних систем). Аналогічний прийом можна застосувати для
розчинів. Для реального розчину можна одержати термодинамічні
співвідношення, які мають такий самий вигляд, що і для ідеального
розчину, якщо замість концентрації користуватися активністю — а. Це поняття
для реального розчину можна ввести через хімічний потенціал, згідно з
рівнянням, аналогічним рівнянню (2.36), а саме:
64
(320)
(321)
Для ідеального розчину, відповідно,
ц;.д- = ц? + ДПпх,..
Відхилення системи від ідеальності
Дц = & -ц)д- = КТ ІіА = ЯПпу,.. (з.22)
Величина (у,.) має назву коефіцієнта активності.
Для ідеальних розчинів Дц = 0, а, = х{ і у,- = 7. Відзначимо, що в рівняннях
(3.21), (3.22) як одиниця концентрації використовується мольна частка (*,-)
/-го компонента (для розбавлених розчинів можна також користуватися
молярною або моляльною концентрацією). Звичайно, визначення активності
залежить від способу вираження концентрації.
Оскільки хімічний потенціал і-го компонента в розчині дорівнює
хімічному потенціалу того самого компонента в газовій фазі, то можна записати
(з урахуванням (1.103))
(3.23)
Аіі = (\і?+КТІпрі')-(іі(;+КТІпр\д') = КТІп
що в зіставленні з (3.22) дає
&
%- = у.=-Щ-
••Уі
Рі
ІД.
(324)
Величину рІд' можна обчислити за законом Рауля для ідеальних систем
(рівняння (3.4))
її
РГ
Ріхі
(3.25)
На рис. 3.6 зображено залежністьрх та/?2 ВІД складу розчину. Штрихові
лінії і та 2 відповідають ідеальним системам (лінійна залежність згідно із
законом Рауля). Додатні
відхилення від закон Рауля дають р\
для уІ та у2 величини, більші за
АС
одиницю (на рис. 3.6 Уі -~77Г
АЕ
і у? = ). Не важко помітити,
2 АО
що у і —» 1 при Хі —> 1, тобто
коефіцієнт активності кожного з
компонентів наближається до
одиниці при наближенні до
чистого і-т компонента.
Ці умови зручні, коли
йдеться про розчинність рідини в
рідині і можна змінювати мольні
1-
І^5^0
Г в
\-
к
Г"
1 д.
\*ііх"\
\>^ *.-"""
4%х ^^^
'''' ^г*-^*****^ >у
й і і і і і ьЗ
♦
Рг
0
Рг
І
м
Склад
N
Рис. 3.6. Визначення коефіцієнтів
активності для бінарної системи
(неелектроліт)
65
частки обох компонентів від нуля до одиниці. Коли ж у рідині розчиняється
газ або тверде тіло, то умову у,- —» 1 при х( —» 1 можна використати лише для
розчинника. Для розчиненої рідини вважається, що у,- -> 1 при х( —> 0, тобто
використовується умова, що нескінченно розбавлений розчин — ідеальна
система.
Дотична кривої тиску для розчиненої речовини (пряма 3 на рис. 3.6)
відповідає закону Генрі (для газів і летких рідин). При таких умовах коефіцієнт
активності другого компонента (у2) на рис. 3.6 відповідає відношенню
АЕ/АР. Тиск р\ відповідає тиску пари над чистим другим компонентом у
гіпотетичному стані нескінченно розбавленого розчину. Не важко
пересвідчитися, що )
* 1 1
кг /
де Кг — константа Генрі, визначена за рівнянням (3.7). Для ідеальної
системи двох летких рідин р\ = р\ і прямі МО та МР збігаються як між собою,
так і з кривою (яка в даному випадку пряма) пари для компонента 2.
Для обчислення коефіцієнта активності твердої речовини в розчині
можна скористатись даними кріоскопії. Введемо величину
І =1-"Г^- (3.26)
КРт
Для ідеальної системи (/) тотожно дорівнює нулеві, відхилення (/) від нуля
характеризують відхилення системи від ідеальності. Знайдемо зв'язок між (/')
та коефіцієнтом активності (ув) розчиненої твердої речовини. Диференціал
(/') дорівнює
ДГ , сІ(Тт-Т) ,л ..сіт йТ П0т
4г=«—гаи—V2—-=а-д—+-—• (3-27)
Активність розчиненої речовини пов'язана з активністю розчинника
співвідношенням Гіббса-Дюгема (1.98), яке для хімічного потенціалу в
двокомпонентній системі має вигляд хА<1\іА + хвй\ів = 0, звідки
сЛ$ав=-^(ІІпаА. (3.28)
хв
Поділивши (3.28) на сіТ, маємо
4\пав _ хА Л\паА ^29)
сіТ хв сіТ
Для розчинника
ЛпаА = АК (330)
сіТ КТІ
(Ентальпія розчинення твердого розчинника в розчині — це практично
ентальпія плавлення.)
З рівнянь (3.29) та (3.30) маємо
66
ЗВІДКИ
(І1п(увт) _ хА Д#° _ пА Д#° _
сіТ хв КТІ пв КТІ
,. йт сІТ
й?1пув= .
тп ккрт
.31) і (3.27), маємо
йИпув =-—<іт-ф',
т
1
к^т'
(3.31)
(3.32)
звідки т
1пул=-]^Ля-у. (333)
0
Інтеграл — сіт береться графічною побудовою в координатах — т .
) т т
Якщо вважати, що відхилення від ідеальності (/') збільшуються пропорційно
концентрації (т) (це приблизно виконується, наприклад, для розчину
цукрози у воді), то можна вважати, що ~ = соті, і тоді
т
1пуд=-2у. (3.34)
Рівняння (3.34) дає можливість відносно просто оцінювати коефіцієнти
активності в розчинах неелектролів.
Розчини електролітів мають свої специфічні властивості.
67
Розділ 4.
РОЗЧИНИ ЕЛЕКТРОЛІТІВ
Електроліту, щоб пройшла дисоціація,
Насамперед потрібна сольватація.
4.1. Електролітична дисоціація
Вивчаючи фізичну хімію розчинів електролітів, Я. Вант-Гофф установив,
що для таких властивостей, як осмотичний тиск (я)^ебуліоскопія (АТЬ),
кріоскопія (АТт), величини тс, АТЬ, АТт виявляються більшими, ніж це дається
формулами (3.1 3), (3.16) та (3.19). Для того щоб ліквідувати цю невідповідність,
Я. Вант-Гофф запропонував ввести так званий ізотонічний коефіцієнт — / > 1.
Таким чином, для електролітів формули (3.13), (3.16) та (3.19) набувають
вигляду:
АТь = ікет;
АТт = іккрт;
п=ісКТ.
(4.1)
(4.2)
(4.3)
Слід відзначити, що перелічені вище фізико-хімічні властивості розчинів
відносяться до так званих колігативних, тобто визначаються загальною
кількістю частинок у розчині. Збільшення кількості частинок у розчинах
електролітів відбувається за рахунок, як це запропонував С. Арреніус,
електролітичної дисоціації.
При гетеролітичному розкладі полярні (іонні) молекули розпадаються на
іони,наприклад: >
СН3СООН <-> СН3СОО~ + Н+; (а)
СООН—СООН о [(СОО)2~] + 2Н+. (б)
Позначимо через V кількість іонів, на які
розпадається молекула. Для оцтової кислоти V = 2,
для оксалатної — V = 3 тощо.
Якщо полярна молекула дисоціює частково,
то це визначається ступенем її дисоціації (а).
В розчині оцтової кислоти маємо: (1 - а) —
молекул СН3СООН, що не продисоціювали;
а — іонів Н ; а — іонів СН^СОО" і загальна
кількість частинок (в розрахунку на одну
молекулу СН3СООН) дорівнює: 1-а+а+ос=1+сс
■ (У = 2).
Арреніус Сванте Для оксалатної кислоти, відповідно, маємо
(18цьІІ27)' 1-ос + а + 2а=1+2сх(у = 3).
68
В загальному випадку сумарна кількість частинок (у розрахунку на одну
молекулу електроліту) і є ізотонічним коефіцієнтом, який дорівнює
/ = 1 + ос(у-1).
(4.4)
Формула (4.4) дає можливість розраховувати ступінь дисоціації з
експериментально визначених АТЬ або АТт через ізотонічний коефіцієнт
/-1
<х = -
V-!
(4.5)
Каблуков Іван Олексійович
(1857—1942),
Росія, СРСР
Повній дисоціації відповідає значення а = 1
що характерно для так званих сильних
електролітів, до яких відносяться солі, луги,
мінеральні кислоти. В слабких електролітах, до
яких відносяться органічні кислоти, КН4ОН,
Н2С03, Н28 тощо, а є малою величиною, і в Їх
розчинах багато недисоційованих молекул
електроліту.
Розглянемо причини електролітичної
дисоціації. Згідно із законом Кулона взаємодія між
іонами в розчиннику з діелектричною сталою (є) в
є разів менша, ніж у вакуумі (наприклад, у воді
майже на два порядки). Цей фактор сприяє
електролітичній дисоціації полярних молекул, але не
може бути єдиним, бо потрібна повна
компенсація енергії, яка витрачається на гетеролітичний розпад молекул
електроліту. Цим фактором є сольватація (у водних розчинах—гідратація), тобто
взаємодія молекул розчинника з іонами, на які розпадається електроліт (див.
рис. 4.1) (роль сольватації при електролітичній дисоціації була обґрунтована
відомим російським фізико-хіміком І. О. Каблуковим). За рахунок іон-ди-
польної взаємодії утворюються
достатньо стійкі сольвати. Певну роль
відіграють водневі зв'язки. Яскравим
прикладом може бути такий. НС1
є сильним електролітом у розчині
етанолу і слабким у розчині
нітробензолу, хоча ці розчинники мають
однакову діелектричну сталу. Причина
цього — сильна сольватація у розчині
С2Н5ОН, зокрема за рахунок
утворення водневих зв'язків. Відзначимо
також, що, наприклад, протон у
розчинах взагалі не існує у «вільному
стані», а лише у гідратованому — у
вигляді іона гідроксонію — Н30 .
Зазначимо, що при розчиненні твердих ре-
Рис. 4.1. Сольватація у розчинах
електролітів
69
човин (як і для неелектролітів) певний внесок у зменшення вільної енергії
Гіббса процесу розчинення дає також ентропійний фактор.
Розглядаючи електролітичну дисоціацію як рівноважний процес, маємо
(на прикладі 1:1 валентного електроліту з початковою концентрацією — с)
МАвМ+ + А".
с(1-ос) сос сос
Для константи рівноваги (яка в даному випадку має назву константа
дисоціації — К^) маємо ч
^=!а^^ = ^£^ = ^ (4,б)
смд с(1-сс) 1-а
(для спрощенні візьмемо досить розбавлені розчини, так, щоб можна було
користуватися саме рівноважними концентраціями, а не активностями).
Рівняння (4.6) відоме як закон розбавлення Оствальда. Проаналізуємо це
рівняння. При с —> 0 маємо а -» 1 (невизначеність типу 0/0 повинна давати
Ка = соті). При с —> ©о, а —» 0 {Ка = соті реалізується через невизначеність
типу 0 х ©о). Отже, існуючи в межах 0 < а < 1, ступінь дисоціації змінюється
від одиниці в нескінченно розбавленому розчині до дуже малих величин
при збільшенні концентрації.
Більш зручний, ніж через колігативні властивості, спосіб обчислення
ступеня дисоціації був запропонований С. Арреніусом — через
вимірювання електропровідності.
4.2. Електропровідність розчинів електролітів
Питому електропровідність (ае) зручно виразити як електропровідність
1 см3 електроліту (відстань між електродами / = 1 см, їх площа 5=1 см2).
Відповідно, ае має розмірність Ом-1 • см-1 (використовують також
розмірність: См/м, де См (сименс) = Ом-1).
Залишаючи / = 1 см і змінюючи Отаким чином, щоб об'єм електроліту
між електродами вміщував 1 еквівалент електроліту, отримуємо
еквівалентну електропровідність — X. х і X пов'язані простим співвідношенням
л = 1000а, (47)
с
де с — концентрація електроліту в моль-екв/л = моль-екв/1000 см3
(розмірність X — см2/Ом • моль-екв).
На рис. 4.2 наведено залежність еквівалентної електропровідності оцтової
кислоти від концентрації розчину. При збільшенні концентрації X
асимптотично наближається до осі абсцис, тобто при с —> °° X —> 0, так само, як
і а —> 0. Отже, причиною зменшення X зі збільшенням концентрації розчину
є зменшення ступеня дисоціації електроліту, і між Я та а існує прямо
пропорційна залежність
70
А —ССЛо,
(4.8)
де коефіцієнтом пропорційності виступає
Х0 — еквівалентна електропровідність
нескінченно розбавленого розчину (при с —> °о
X —» 0). Величину Х0 не зручно визначати
екстраполяцією в координатах X — с, тому що
при с —> 0 X змінюється різко і точність екст- і 200
раполяції є низькою. Відомо, що екстраполя- ^ 150
ція дає хороші результати, якщо залежність <^
лінійна. Підставляючи а з виразу (4.8) у ^ 10°
рівняння (4.6), маємо
сХ2
К= , , ч, (4.9)
(
ЗВІДКИ
1-:
сХ = -КК +
кхі
(4.10)
Рис. 4.2. Залежність
еквівалентної
електропровідності (X) від концентрації
розчину
Отже, в координатах сХ матимемо
А
пряму лінію з тангенсом кута нахилу (§$ = КХ0, а відрізком на осі ординат:
(-а) = (-КХ0), звідки Х0 = —.
а
Для сильних електролітів а = 1 (відзначимо, що такі сполуки, як ИаСІ, вже
у твердому стані існують у вигляді іонів, з яких побудовані їх кристалічні
ґратки), отже, на перший погляд, для них еквівалентна електропровідність не
повинна змінюватися з концентрацією. Тим не менш, X і для цих електролітів
не так швидко, як для слабких, але змінюється зі збільшенням концентрації.
Ф. Кольрауш показав, що для сильних електролітів залежність X від
концентрації дається співвідношенням
•А>[с.
ь=к
(4.11)
Формула (4.11) дає можливість знаходити Х0 сильних електролітів
лінійною екстраполяцією в координатах X — у[с (див. рис. 4.3, с. 72).
Обґрунтування формули (4.11) (і, відповідно, причин зміни X з концентрацією)
буде наведене далі в теорії сильних електролітів (підрозділи 4.3 та 4.4).
Електропровідність у розчинах електролітів здійснюється за рахунок
руху як катіонів, так і аніонів. Ф. Кольрауш логічно запропонував, що
катіони і аніони рухаються незалежно один від одного, отже, згідно з правилом
Кольрауша незалежності руху іонів еквівалентну електропровідність
електроліту можна розділити на окремі іонні електропровідності (/,), які мають
назву рухомостей, тобто
Х = 1+ + 1.
(4.12)
71
426
422
418
0,02 0,04 0,06
у/с, МОЛЬ1/2/л1/2
Рис. 4.3. Залежність X від -7с
І.І.Е-Л
>> К І
Відповідно, для електропровідності
у нескінченно розбавлених
розчинах маємо
х0=/:+/_°. (4.із)
Якщо взяти два електроліти (КА
таМ4), в яких один з іонів
(наприклад аніон А~) однаковий, то маємо
М^)\=/°++/°_; Х0(МА) = 1°М++ГА_
і різнися £к = Х0(КА)-Х0(МА) =
= /£+ -/^+,^гобто не залежить від
того, який аніон використовується.
Це положення добре ілюструється
даними, що наведені у таблиці 4.1.
Розглянемо тепер фізичний зміст
рухомостей. Формально із закону
Ома маємо для електричного
струму со
= х-8-§гасіЕ9
(4.14)
де і? — опір, 5— площа електродів, / — відстань між ними, Е — напруга;
Е/1 = &гад,Е (В/см) — градієнт напруги (напруженість електричного поля).
Таблиця 4.1. Еквівалентні електропровідності, см2/(Ом*моль-екв)
Електроліт
КС1
К>Юз
К2804
к
130,0
126,3
133,0
Електроліт
№С1
МаИОз
Ш28О4
к
108,9
105,2
111,9
АХ
21,1
21,1
21,1
Вводячи поняття густини струму (/'), маємо
і = — = ае • ггасІЕ = ягайЕ = -^ -ггаЛЕ.
5 1000 1000
(4.15)
З іншого боку,
і = 2+п+и+ + 2-П-іі- , (4.16)
де П;—кількість іонів в одиниці об'єму, 2І—заряд іона, щ—швидкість руху
іона.
Для 1:1 валентного електроліту п+ = п_ = п (частинок/см3); |2+| = |2_| =
= є — елементарний заряд.
Тоді маємо
72
і = п-е-(и++и_) =
с-Ил-е, ч с-Р
—(и++и_) =
(и++и_),
(4.17)
1000 т " 1000
де ИА—число Авогадро, Ил • є = Р— число Фарадея (96485 Кул/моль-екв),
с — концентрація розчину в моль/л = моль-екв/л.
Співставляючи рівняння (4.15) і (4.17) і вважаючи £гас! Е=\ В/см, маємо
/, = /?•«,. (4.18)
Отже, рухомості (/,) пропорційні абсолютним швидкостям (щ) руху іонів у
розчині при градієнті потенціалу 1 В/см, а коефіцієнтом пропорційності є
число Фарадея (Р). Згідно з (4.18) розмірність рухомостей така:
,2
см
см
"в"
Кул
моль-екв
см
= см /Ом • моль-екв,
моль-екв і
тобто, як і треба, співпадає з розмірністю еквівалентної електропровідності.
Відносні значення рухомостей (*,.)
/+ _/+ /_ _/_
Анод Катод \
(4.19)
мають назву числа переносу
іонів.
Існує декілька методів
експериментального визначення
чисел переносу. Один з них був
запропонований І. Гітторфом і
полягає у вимірюванні зміни
концентрацій електроліту в
катодному (Ак) та анодному (Да)
просторі при електролізі. В
розрахунку на 1 еквівалент
електроліту М А~ маємо таку
+ +!
В
++
+ + + +
+ + + +
+ + + +
+++++
І++++++++
+++
/ II III
Рис. 4.4. Метод Гітторфа для визначення
чисел переносу
картину (рис. 4.4).
У катодному шарі:
1 екв. катіонів М розрядився на катоді;
/_ аніонів А~ перейшло з католіту в середню частину електролізера;
1+ катіонів М прийшло з середньої частини до католіту.
Зменшення кількості катіонів дорівнює (1 -1+ = 0> аніонів теж (*_)> отже,
загальне зменшення кількості електроліту в катодному шарі (Ак) дорівнює
ЦМА) (на 1 екв).
В анодному шарі:
1 екв аніонів А~ розрядився на аноді;
1_ аніонів А~ прийшло з середньої частини до аноліту;
і+ катіонів М прийшло з аноліту в середню частину.
Зменшення кількості аніонів (!-*_ = 1+), відповідно, катіонів теж (/+),
73
отже, загальне зменшення кількості електроліту в анодному шарі (Аа)
дорівнює (+ (МА) (на 1 екв).
Ак (_
Отже, маємо — =—, звідки:
Аа (+
Аа л Ак
'+ = 1 Г7; 1- = И 17- (4-2°)
Аа + Ак Аа + Ак у '
(Не важко пересвідчитися, що у центральній частині електролізера зміни
концентрації електроліту не відбувається.) Кількість еквівалентів електроліту,
що розкладається в електролізері (Ак + Аа), можна обчислити через
пропущену кількість електрики і число Фарадея.
Числа переносу можна визначати також методо]
рухомої межі (див. рис. 4.5). Використовують два єлеї
троліти з однаковим аніоном (А): КА тьМА. Електроліті
вибирають таким чином, щоб Ік+ було менше, ніж
Ім+ (іон К+ так званий «відстаючий»). Отже, швидкість
руху межі визначається швидкістю руху (рухомістю)
іонаМ . При проходженні ^ кулонів електрики
катіонами АҐ переноситься: ($ІР)і+ еквівалентів електроліту,
(а). З іншого боку, візуально спостерігаючи за
зміщенням межі між КА та МА на / см, маємо для кількості
перенесених катіонами (М ) еквівалентів: (с+57)/1000, (Ь\
де с+— концентрація катіонів (М ) в моль-екв/л; 5 —
площа перерізу в см2. Прирівнюючи (а) і (Ь), маємо
и:
М + А~
К+
Рис. 4.5. Метод
рухомої межі для
визначення чисел
переносу
= с+81Е
'+~1000?'
(4.21)
Визначивши числа переносу ^) і X розчину, можна знайти рухомості (/,-)
катіонів і аніонів. Швидкість руху іонів, а отже, і рухомості, повинні залежати
від в'язкості (г|) розчинника. Це можна обґрунтувати таким чином. Сила, що
рухає і-ий іон, дорівнює
/рух=8гм1Е-Єі9
де є, — заряд іона.
Сила опору (тертя) визначається формулою Стокса
де г,-— радіус іона (з урахуванням сольватної оболонки).
В стаціонарному стані^,^ =/оп., і отже, 6тгг| г^=&га<1 Е- е(.
Вважаючи%гас1Е = 1 В/см і враховуючи, що щ= //і7, маємо
Л/,=-^ = со/иґ. (4.22)
6щ
З таблиці 4.2, де наведено значення добутку Лн2о* 'сн3соо~ ПРИ різних
температурах, видно, що закономірність, яка відзначена співвідношенням
(4.22), справді виконується.
74
Таблиця 4.2. Значення добутку Т|н2о#/сн -соо~ при різних температурах
і, °С
Лн2о'/снзСоо-
0
0,366
18
0,368
25
0,366
59
0,368
75
0,369
Застосовуючи співвідношення (4.22) до електроліту в цілому, маємо
у\-\о = сомі. (4.23)
Співвідношення (4.23) відоме як правило Вальдена.
Дані, що наведені в таблиці 4.3, підтверджують це правило.
Таблиця 4.3. Значення (Т| *Х0) для різних
полярних розчинників
(електроліт — М(СН3)}1~)
В наступній таблиці 4.4
наведено значення рухомостей
(І? ) для деяких поширених іонів
при кімнатній температурі у
водних розчинах.
Звертають на себе увагу
аномально високі значення
рухомостей для Н та ОН" іонів.
Це пояснюється існуванням
специфічного протолітичного
механізму переносу цих іонів.
Іон Н існує у водних розчинах
лише в сольватованому вигляді,
тобто як іон гідроксонію Н30 .
Таблиця 4.4. Рухомості іонів у водних розчинах
Розчинник
СНзОН
СНзСОСНз
СНзСИ
С2Н4СЛ2
СН3Ш2
СбНзШг
С6Н5ОН
Ц'Хо
0,63
0,66
0,64
0,60
0,69
0,67
0,63
Катіон
/°, см2/Ом • моль-екв
Аніон
/°, см2/Ом • моль-екв
Н+
350
ОН"
198
К+
73,5
сг
76,3
№+
50,1
Вг"
78,4
ІЛ+
38,7
Г
76,8
Ва2+
63,6
Ш3"
71,4
м§2+
53,1
80Г
79,8
При цьому можливий перенос протона до сусідніх молекул води
н ©"
н—о—н
+
н н
І — І
о—н н—о
+
" н ©1
н—о—н
ч—'(А)
(В)
75
ЕЗ [ТЗ [ТЗ [ТЗ [+3
+_; [±3[±ЗС±3 |+-
[ЗЕ [ЗЕ Е±] Е±]
[+ЗС+ЗЕЗЕЗ
А
В
С
°
Молекула води, що утворилася (В), має іншу орієнтацію, ніж попередня (А),
і для прийому протона вона повинна переорієнтуватися (переорієнтація
відбувається під дією зовнішнього електричного поля). Таким чином,
реалізується естафетний механізм, який значно прискорює перенос протона.
В лужному середовищі перенос протона відбувається від молекули води до
гідроксил-іона, що забезпечує високу рухомість ОИГ аніону (перехід протона
у деякому напрямі еквівалентний переходу гідроксил-іона в протилежному).
Цікаво відзначити, що цей протолітичний, естафетний механізм нагадує
першу спробу пояснення механізму електролітичної провідності в
розчинах, запропоновану російським ученим Ф. Гроттусом ще на початку XIX
століття. Ф. Гроттус вважав, що при прикладанні до електроліту різниці
потенціалів полярні молекули
електроліту вишиковуються в
ланцюжки таким чином, що їх
позитивні-кійцівки орієнтовані
до катода, а негативні — до
анода (рис. 4.6, А). Після
нейтралізації частини зарядів крайніх
Рис. 4.6. Механізм електропровідності молекул (позитивного на катоді
за Ф. Гроттусом й негативного на аноді (рис.
4.6, В) їх заряджені залишки обмінюються партнерами із сусідніми
молекулами і виникає новий ланцюжок, але орієнтований протилежно (рис. 4.6, С).
Далі під дією поля відбувається переорієнтація диполів, і новий ланцюжок
(рис. 4.6, В) вже є повторенням первісного (рис. 4.6, А). Ф. Гроттус і разом
з ним інші вчені вважали, що дисоціація полярних молекул на іони
обумовлена саме дією зовнішнього електричного поля. Однак це суперечило
наявності електропровідності вже при дуже малій різниці потенціалів. Після
появи теорії електролітичної дисоціації С. Арреніуса та її обґрунтування
І. О. Каблуковим (дисоціація при розчиненні електроліту) від механізму
Ф. Гроттуса відмовилися, але згодом виявилося, що застосований для
пояснення аномально високої рухомості Н і ОН~ іонів протолітичний
естафетний механізм багато в чому співпадає з механізмом Ф. Гроттуса.
Розглянемо тепер теорію сильних електролітів Дебая-Гюі^келя та її
застосування до явищ електропровідності, а також до пояснення причин
відхилення від ідеальності в розчинах сильних електролітів.
4.3. Основи теорії Дебая-Гкжкеля
Для сильних електролітів, таких як КС1, ИаМОз, №ОН тощо, ступінь
дисоціації дорівнює одиниці, тобто вони існують у розчинах лише у вигляді іонів.
Тому для пояснення залежності еквівалентної електропровідності сильних
електролітів від концентрації (правило Кольрауша (4.11)), а також
відхилення розчинів електролітів від ідеальності слід шукати іншу причину, крім
дисоціації. П. Дебай і Е. Гюккель запропонували, що такою причиною є
76
електростатична взаємодія між іонами. Кожний
з іонів за рахунок електростатики намагається
«обгорнутися» іонами протилежного знака і
створити навколо себе так звану іонну
атмосферу (рис. 4.7). Дифузія, броунівський рух
перешкоджають утворенню цієї впорядкованої
системи з іонів, руйнують іонну атмосферу.
Розглянемо це детальніше.
Загальновідоме з електростатики рівняння
Пуассона
гт2 4Тф
(4.24)
Дебай Петер Йозеф Вільгельм
(1884—1966), Нідерланди
дає зв'язок між потенціалом (\|/) і густиною
заряду (р). Тут є — діелектрична стала, V2 — лапласіан.
Для того щоб розв'язати рівняння (4.24), потрібен додатковий зв'язок між
\|/ та р. Саме його і дає модель іонної атмосфери.
Відхилення кількості іонів в одиниці об'єму від середнього значення (щ)
при дії силового поля дається формулою Больцмана (див. розділ 11, фор-
мупа(11.21)) ^ фф
щ = піЄ~г, (4-25) ©
де І/— потенціальна енергія поля. ^рч
Для електростатичного поля
[/ = 2^X1/, (4.26) © ©
де є—елементарний заряд, 2,—цілочисельний Рис*4*7, Іонна атмосФеРа
заряд /-го іона. ,
Підставляючи (4.26) у (4.25), маємо
кТ
(4.27)
Внесок у загальну густину заряду від іона /-го сорту дорівнює
р. =2,«%= ЇіЄщє кт . (4.28)
Підсумовуючи за всіма іонами, маємо для загальної густини заряду в розчині
р=£р(=3>2Лє~и\ (429)
і і
Для розбавлених розчинів \|/ є відносно малою величиною і тому можна
вважати: е2$ « кТ, що дає можливість розкласти експоненту в ряд таким
чином:
і кт 2і_£М. (4.30)
кТ
11
Підставляючи (4.30) у (4.29), маємо
,.^-£а.уї«л-Ьу&.-*іг. (4.з,)
Сума 2)е2,и(- = 0 в силу електрс::сґ:іральності розчину. Величину
Г = 2>,.22 (4.32)
І
називають іонною силою розчину. Підставляючи (4.31) у рівняння Пуассона,
маємо
у^ = ^^Г. (4.33)
є кТ
Позначимо
= Ь\ (4.34)
4яе2Г ,2
гкТ
тоді \
У2у = &2¥- (4-35)
Іонна атмосфера має сферичну симетрію, отже, у лапласіані (V ) у
сферичних координатах залишається тільки радіальна частина
у2=Т2+1Т- (436)
дг г дг
Таким чином, диференційне рівняння Пуассона набуває вигляду
**+2*.^. (4.37)
дг г дг
Рівняння (4.37) розв'язується підстановкою^ = \|/г, що дає
у=Ае*г (4.38)
І, ВІДПОВІДНО,
А +Ьг
У = -* • ^ (4.39)
г
На великому віддаленні від центру іонної атмосфери потенціал прямує до
нуля (\|/ -» 0 при г —> °°), згідно з цією умовою експоненційно зростаючий
розв'язок у рівнянні (4.39) слід відкинути, що дає
у = -е~Ьг. (4.40)
г
В нескінченно розбавленому розчині щ-ь 0, відповідно: Г -» 0 і Ь —> 0. Згідно
з рівнянням (4.40) це дає
\|/ =-. (4.41)
78
З іншого боку, в нескінченно розбавленому розчині іонна атмосфера
відсутня і, отже, потенціал створюється лише центральним іоном
гг
Співставляючи (4.41) та (4.42), маємо
Є
отже, ц = ^е~Ьґ. (4.43)
гг
Для розбавленого (але не нескінченно) розчину Ь є малою величиною (але
не нулем) і тоді експоненту в рівнянні (4.43) можна розкласти у ряд, тобто
е~ г = 1 - Ьг.
Це дає
2,Є /л , ч 2:Є 2:Є ,
У = —(1-^) = -* -Ь. (4.44)
гг гг г
2е
Оскільки, згідно з (4.42), —1- це потенціал центрального іона, то другий
гг член у (4.44) — це внесок у загальний
потенціал (\|/) від потенціалу іонної атмосфери —
і|/а,отже,
2:ЄЬ 2;Є ,А А^
¥*=-т-=-^- (445)
Величина га = \ІЬ є усередненим радіусом іонної атмосфери. Справді, згідно
з (4.34),
—її (4-46)
Га=\
і має розмірність
енергія
4пеТ
І
2
(енергія х довжина) • (довжина)
= довжина.
В наведених вище формулах концентрація (пг) виражалась у частинках на
одиницю об'єму. З практичної точки зору, зручніше користуватися
молярною концентрацією (с,-) і, відповідно, визначати іонну силу через (/), тобто
/=1>д2 (4-47)
(/ пов'язане з Г через число Авогадро ШЛ, п, = ' А і Г = —).
' 1000 1000
Для водних розчинів при Т= 298 К, підставляючи значення
діелектричної сталої води та інших констант, маємо
4,3-10"10 0,43
Г„ г= М=—^-НМ.
V/ 41
79
В наведеній нижче таблиці 4.5 приведено значення (га) для іонів різної
валентності при різній концентрації розчинів.
Таблиця. 4.5. Розміри іонної атмосфери
Валентність
іона
1-1
2-1
0~2)
2-2
3-1
1 (і-з)
Га (НМ)
ііри концентрації (моль/л)
0,100
0,96
0,56
0,48
0,39
0,010
3,05
1,93
1,53
1,36
0,001
9,6
5,6
4,8
3,9
З наведених у таблиці 4.5 даних видно, що збільшення валентності іона
і його концентрації (збільшення іонної сили) веде до стиснення, зменшення
розмірів іонної атмосфери.
Роботу утворення іонної атмосфери можна обчислити на основі
наступного уявного експерименту. Припустимо, що всі іони втратили свій
заряд, а потім поступово набирають його, змінюючи від нуля до 2, є, а
відповідну частку заряду позначимо через <а» (0 <х < 1). Тоді
ХіЄХ
~ . соті соті га
Оскільки га = , то г^ = = —, отже,
є ех х
¥«=-
2іех
Нескінченно мала робота, як функція (х), дорівнює
&У*х = УахїіЄ(ІХ.
Інтегруючи (4.50) з урахуванням (4.49), маємо
722 1 72 2
щ = ІЯіЄУпЛс = —-— [х2<1х = —*-
Зєг„
Підсумовування по всіх іонах дає
IV
= ]У>/Л/=-£и/2/
■2 Є
Зєгл
IV
Зєг„
(4.48)
(4.49)
(4.50)
(4.51)
(4.52)
80
4.4. Електропровідність сильних електролітів
Як показано вище (розділ 4.2), при відсутності іонної атмосфери на іон,
що знаходиться в електричному полі з напруженістю §гас!Е, діє сила
/0=8ли/£.2,е (4.53)
При наявності іонної атмосфери, симетрично розташованої навколо
центрального іона, виникають гальмуючі ефекти: електрофоретичний та
релаксаційний.
Електрофоретичний ефект можна пояснити тим, що іонна атмосфера
екранує центральний іон від зовнішнього поля, оскільки її потенціал
протилежний потенціалу центрального іона, а саме
2{е 2{е 2{е
гГ: гГп ЕГ:
(. Л
(4.54)
Щ
тобто дія атмосфери еквівалентна переходу до деякого ефективного заряду
1-^-
г„
2°*е
/
2,еф- = 2;
( Л
1--5-
(4.55)
На цей ефективний заряд діє в зовнішньому полі сила
/ = 8гас1Е.2**'е = 2гас1Е- 2Лі-і) = /0
Величину
ґІ--) = /о-/о-- (4-56)
г„ гп
п
/о /ел.-ф.
г
'а
можна вважати електрофоретичною силою, яка діє в напрямі,
протилежному зовнішньому полю (/0), і, таким чином, гальмує рух іона.
Релаксаційний ефект викликаний порушенням сферичної симетрії
іонної атмосфери. Це виникає внаслідок того, що іони, які утворюють іонну
атмосферу, є протилежно зарядженими по відношенню до центрального
іона і рухаються у напрямі, протилежному напряму руху центрального іона.
Якщо, наприклад, центральним є аніон, то внаслідок асиметрії іонної
атмосфери позаду аніона виникає позитивно заряджений (-р^/Фи^
«хвіст», який намагається затягнути аніон назад до ат- ^"^^^^
мосфери (рис. 4.8), і, таким чином, гальмує його рух. Ш^г^ч Щ)~^
Вважаючи напрямом руху /-го іона вісь (х), маємо для /^~\~~>/
повертаючої сили релаксаційного ефекту (~Ь)^ С£Ь^
ах Рис. 4.8. Асиметрія
де щ — робота утворення іонної атмосфери. іонної атмосфери,
Згідно з рівнянням (4.51) маємо релаксаційний ефект
81
2 „2
л=
_ 2іе *
Зє йх
ґ
ґ і Л
чг«у
_^е2
6є
Зє йЬс
2,2е2
</Ш*
СОП8ІУІГ = — С0Я.У/ р=- =
^ ' 6є 7г
С0Ю.У/
л/Г-^-
6егп <1х Г
(4.57)
З 2 для Г виділяємо лише іони протилежного до центрального /-го іона
знака. Для одного такогоу-іона маємо
/_2,2е2 І йпі
їі =
6ега П] йх
(4.58)
За Л. Больцманом
§га<іЕ2:ЄХ , .
. 2}2)Єг2гшіЕ
отже, и =——і .
6екТга
Для одно-одновалентного електроліту 2і=:+\;2: = -\
.у _ е3§гасіЕ _
1 /х бєЩ, •/релакс-'
Остаточно маємо
/ = /о + /ел.-ф. + /релакс. = &-айЕе\ 1 —і -
(4.59)
(4.60)
г„ бє&Гг,
= /о
1-І-ІП+ *
6єА:Г
(4.61)
Якщо и0—це швидкість руху іона в нескінченно розбавленому розчині,
а и — в присутності іонної атмосфери, то ^
' .2 Л
Х = Р(и+ +и_) = Ри0
"о /о
Г,+-
бекТ
1
У
1-
г++
бєкТ
1
У
+ ^
1-
г_ +
бекТ
)
1
(4.62)
І- , (4.63)
де и+іи_—швидкості руху катіона і аніона, &г+іг_ — відповідно, радіуси.
82
Для нескінченно розбавленого розчину
І Х = ^(«о+«0)
1 —
1
6є£Г гЛ
-/7
и0г++и0г_
(4.64)
(4.65)
ЗаСтоксом/0 =^гайК-^ = 6ягі^0.При %гайЕ = \ В/см и*г+ =и0г_ =
отже, А, = ^с
1 —
1
6є£Г гп
еР
6щ
(4.66)
Оскільки \Іга -сопзіу/с (для 1:1 валентного електроліту), то рівняння (4.66)
при сталій температурі набуває вигляду
\ = Х0(і-а^)-ЬуІс=Х0-(Х0а + Ь)у[с=\0-А>/с. (4.67)
Для водних розчинів при 18 °С а = 0,2266; 6 = 51,01. Дані ддяА, обчислені
за допомогою цих параметрів (а і Ь), і значення Х0при цій температурі
наведені у таблиці 4.6 (2-й стовпчик). В 3-му стовпчику наведено Аекси .
Порівняння показує, що теорія Дебая-Гюккеля для розбавлених розчинів не лише
обґрунтовує рівняння Кольрауша (4.11), але й дає непогані результати для
константи А цього рівняння.
Таблиця 4.6. Константи рівняння
Дебая-Гюккеля
Електроліт
ІлСІ
N301
КС1
Іл*Ю3
Л0бч
73,4
75,7
80,4
72,6
А
Лексп
74,7
74,6
80,5
72,4
На утворення і, відповідно,
руйнування іонної атмосфери потрібен
деякий час релаксації—т. Оцінка дає при
кімнатній температурі у водному
розчині для с = 0,1 моль/л т = 10" сі для
с = 0,001 моль/л т = 10" с . Отже, т
обернено пропорційний концентрації
розчину. Це випливає з теорії броунів-
ського руху, згідно з якою
т = -
2П
де А — зміщення, О — коефіцієнт дифузії.
Вважаючи Д = га, маємо при сталій температурі
„2
Т = -
СОП8І
Ю І
де / = ^с^?, і при фіксованих 2/ іонна сила визначається концентрацією
і розчину.
Якщо на розчин діяти струмом високої частоти (V), то при V > 1/т іонна
атмосфера зберігає свою симетрію, отже, релаксаційне гальмування
83
відсутнє (електрофоретичне залишається). Це веде до збільшення
електропровідності й відоме як ефект Дебая-Фалькенгагена, причому ^ знаходиться
у межах між X для постійного струму і Х0.
Повністю позбутися релаксаційного і електрофоретичного гальмування
можна, переходячи до дуже розбавленого розчину, або за рахунок ефекту
Віна, який встановив, що під дією дуже високої напруги X—»А,0, тобто
швидкість руху іонів у полі високого градієнта електростатичного
потенціалу настільки зростає, що іонна атмосфера не встигає утворюватись
навколо іона, що рухається.
4.5. Активності у розчинах електролітів
Хімічний потенціал для окремого /-го іона має вигляд
Н^+КТЬіаі, (4.68)
де а і— активність /-го іона у розчині.
Для електроліту в цілому маємо
І
= ч+(ііІ+КТІпам+^_(іі°_+КТІпаА.) =
= (VX+V^-) + ЛГь(а^Vа^) = |і0+ЛГ1пач' (4.69)
де а — активність електроліту; V,- — стехіометричні числа.
Таким чином, маємо
V V
а — а, -а__
(4.70)
Усереднена активність іона має вигляд
і
а±=|у.яГ]у++у-. (4.71)
Для одно-одновалентного електроліту у+ = V, = 1
а± = уІа+-а_, (4.72)
тобто а± є середнім геометричним активностей окремих іонів.
Для розбавлених розчинів електролітів прийнято користуватися моляль-
ною (т) концентрацією (для водних розчинів ш чисельно дорівнює молярній
(с) концентрації). Отже,
щ = у//и/, (4.73)
де у,-— коефіцієнт активності /-го іона.
Підставляючи (4.73) у (4.71), відповідно, маємо
і
у± = (уї+-У--)у*+у- (4-74)
84
Для 1:1 валентного електроліту:
(4.75)
у±=7у7-у-;
(4.76)
т+ = т+=т_=т. (4.77)
Відхилення від ідеальності (Дц.) у розчинах електролітів обумовлено
електростатичною взаємодією між іонами (цГ')> тобто
Дц = цГЛ' -Ц}ие№ =(ц? + кПпа()-(ц° + кПпт,) =
= к7їіА = кГ1пу,.=ц;л- (4-78)
/я,
(у формулі (4.78) хімічний потенціал віднесено до одного іона).
З іншого боку,
'зо. ї
цГ =
ЗЮ;
(4.79)
Внесок у вільну енергію Гіббса від електростатичної взаємодії (Сел)
визначається роботою утворення іонної атмосфери (м>), що надається
формулою (4.52). Згідно з формулою (4.46) (1/га) пропорційно >/г, отже,
формулу (4.52) можна записати у вигляді
3/2
<?«,.=* =-ЯГ
Згідно з формулами (4.52) та (4.46)
В
, = І_ І4пе2
Зє\ гкТ
Підставляючи (4.80) у (4.79), маємо
ц™=-В-Г2
2
аг
^
З урахуванням (4.78) це дає
\ЬпЧР,т,п„
2
Ь 2 кТ
(4.80)
(4.81)
(4.82)
(4.83)
Переходячи від концентрацій (и,) в частинках/см3 до молярних (с,),
і, відповідно, від іонної сили (Г) до іонної сили (/) (4.48), а також від
натуральних логарифмів до десятинних, маємо
і8у,-=-л2?77,
(4.84)
85
Де А = -
з-я-лг.
(4.85)
2-2,3-1000-кГ
Формула (4.84) має назву граничного закону Дебая-Гюккеля.
При 25 °С для водних розчинів А = 0,509 (з урахуванням всіх констант, Т=
= 298 К та відповідного значення діелектричної сталої води).
Формула (4.84) дає можливість теоретичного обчислення коефіцієнтів
активності (у,) окремо катіонів та аніонів, але експериментально
вимірюється середній коефіцієнт (у±) розчину. Згідно з його визначенням за
формулою (4.74) і законом Дебая-Гюккеля (4.84) для окремих іонів для
середнього коефіцієнта активності маємо
І8У±=;
[у+ 1§у+ + V. 1§у_] = -Ау/і -^ =
= -А2+2_Л. (4.86)
При виведенні формули (4.86) враховано, що внаслідок електронейтраль-
ності розчину у+2+ = У_2_.
Нарис. 4.9 наведено залежність 1§у± від у/І для деяких електролітів. Видно,
що для відносно розбавлених розчинів формула (4.86) виконується.
1,5 2,0
Рис. 4.9. Середній коефіцієнт активності у водних розчинах при 25 °С
86
Розділ 5.
ФАЗОВІ РІВНОВАГИ У БАГАТОКОМПОНЕНТНИХ
СИСТЕМАХ
Лиш створили систему, відразу
Компоненти розбіглись по фазах.
5.1. Правило фаз Пббса
У підрозділі 1.1 використовувалося поняття фази і відзначалося, що
система з декількох фаз є системою гетерогенною. Правило фаз Гіббса якраз
і стосується в основному гетерогенних систем. Термін «фазові рівноваги»
практично ідентичний терміну «гетерогенні рівноваги».
Складові речовини, що входять до системи, можуть бути пов'язані
якимись додатковими умовами. Це потрібно враховувати при визначенні
кількості компонентів. Тому кількість компонентів визначається як кількість
складових речовин мінус кількість умов, якими вони пов'язані. Якщо
додаткових умов немає, то кількість компонентів дорівнює кількості складових
речовин.
Пояснимо це на прикладах. Маємо сахарозу і воду. Тут два компоненти,
відповідно, до двох складових речовин при відсутності додаткових умов.
Другий приклад. Маємо азот, водень і аміак. Якщо перемішати ці три
речовини при відносно низькій температурі, то маємо три компоненти. Якщо
перебігають хімічні реакції синтезу та розкладу аміаку і встановилася
хімічна рівновага, то рівноважна концентрація будь-якого з трьох газів може
бути обчислена за допомогою константи рівноваги (див. підрозділ 6.1) та
концентрації двох останніх газів. Отже, маємо два компоненти (при трьох
складових). Далі, якщо взяти водень та азот у співвідношенні 3:1, то
протягом реакції И2 + ЗН2 = 2]ЧН3 це співвідношення зберігається, і в цьому
випадку достатньо знати концентрацію тільки одного з газів, таким чином, тут
число компонентів дорівнює одиниці.
Обґрунтуємо тепер правило фаз (1.1). Якщо система прийшла до стану
рівноваги, то в неї встановлюється термічна, механічна і хімічна рівновага.
Термічна рівновага в гетерогенній системі відповідає рівності
температур у всіх фазах
Г = Т"=Ґ"= ... =Тк = Т9 (5.1)
Т— температура системи.
Аналогічно механічна рівновага — це рівність тисків у кожній фазі
р>=р»=р~=ииш = рк = р9 (52)
Р — тиск у системі.
Рівність вільних енергій Гіббса для різноманітних фаз за
співвідношенням (2.1) була умовою рівноважного стану в однокомпонентних системах.
87
Для багатокомпонентних систем умовою рівноваги стає рівність хімічних
потенціалів кожного з компонентів у всіх фазах, тобто
щ'=|і,' = мГ=... = Иі*
|4=Ц$=М£ = - = Ц2
(53)
Кількість змінних, що описують систему, дорівнює (п - \)к + 2. Це тиск,
температура і (п - 1) концентрацій у кожній фазі (в силу тотожності (£х, = 1)
для концентрацій необхідна (п-І) змінна). Кількість рівнянь, що зв'язують
змінні, за (5.3) дорівнює (к- 1)п. Отже, кількість незалежних параметрів —
ступенів свободи
/=(л-1)* + 2-л(*-1) = /і + 2-*,
що повністю узгоджується з (1.1).
5.2. Двокомпонентні системи
5.2.1. Рівновага рідина-пара
Згідно з правилом фаз для двокомпонентної системи
/=4-£. (5.4)
Якщо зафіксувати тиск або температуру, то рівність (1.1) набуває вигляду
/=л + 1-* (5.5)
і, відповідно, замість (5.4) маємо
/-3-*. (5.6)
У гомогенній області к=1 і /= 2, тобто систему можна описувати двома
змінними: концентрацією і тиском (або температурою). У гетерогенній
області рідина-пара к=2 і, отже, /= 1. При/< 2 можна для зображення
рівноваги рідина-пара скористатися плоскими діаграмами стану. При цьому вісь
абсцис — це концентрація, а вісь ординат — тиск (при Г= соті) або
температура (при р = соті).
Розглянемо спочатку ідеальні розчини, тобто такі, для яких закон Рауля
виконується не тільки для розбавлених розчинів, а й при будь-якій
концентрації. Тоді парціальний тиск компонента А рА = р°А хА, а компонента В рв =
= Р°аХв. Загальний тиск
88
Р = Ра+Рв= Р°аха+Рвхв = Р°а +(Р°в-Р°а)хв- С5-7)
Таким чином, для ідеальних розчинів загальний тиск (рис. 5.1) змінюється
лінійно з концентрацією одного з компонентів (В) у розчині (зменшується
при рв < р°А або збільшується р°в > р°А).
Рис. 5.1. Залежність повного і парціального тисків від складу
в ідеальній бінарній системі
Розглянемо тепер, як залежить загальний тиск від складу газової фази.
Мольна частка компонента (В) у газовій фазі
_Л_
Л= —
Рвхв
— хв
Р РаНр°в-Ра)хв
ф
: Хд-
Рв'Рл
1+
(рУрл-і)
ХВ
1 + (ср-і)х/
(5.8)
Залежно від того, ф = рУр°а більше або менше одиниці, (ув) буде більше
або менше (хв). Наприклад, при ф = 2
2
Ув =——хв.
хв+1
При хв= 0,25; 0,50 та 0,75 ув, відповідно, дорівнює 0,40; 0,67 та 0,86. Тобто
при всіх складах Ув>хв при одному й тому самому тиску (р). Це зображено
на рис. 5.2, с. 90, (криві 1 (хв) та 2 (ув) відповідно). Навпаки, при ф < 1,
наприклад, при ф = 0,5
1
Ув=-ї хв-
і-хв
При = 0,25; 0,50 та 0,75 відповідно, дорівнює 0,11; 0,20; та 0,27. Тобто тепер
при всіх складах ув< хв при тому самому тиску (р). Це також зображено на
рис. 5.2 (криві 3 (хв) та 4 (ув) відповідно).
Таким чином, склад газової фази, що знаходиться у рівновазі з рідиною,
відрізняється від складу рідкої фази. В першому випадку
89
Ф=4>і
Рл
Ув>хв>
л х&Ув
Рис. 5.2. Залежність повного тиску'
в бінарній системі від складу рідини (7,3)
та газової фази (2,4)
тобто газова фаза збагачена
більш летким компонентом В,
який має більший тиск при
фіксованій температурі, ніж
компонент А. У другому ви-
о
падку ф = —~ < 1 і ув < хв, тобто
газова фаза збіднена
компонентом В, який має менший
тиск. Нерівність у^<хв
відповідає нерівності^ >хА. Оскільки
в цьому випадку р°А > р°в, то
газова фаза збагачується знову більш летким компонентом (у даному
випадку це компонент А). Таким чином, приходимо до висновку: пара,
порівняно з рідиною, збагачена більш летким компонентом, тобто компонентом,
додавання якого до системи збільшує її загальний тиск. Це правило відоме
як перший закон Коновалова.
Вище перший закон Коновалова був розглянутий для ідеальних систем.
Розглянемо тепер його більш строго для будь-яких систем. На кривих
залежності тиск пари — склад рідини можуть
спостерігатися відхилення від лінійної залежності
(закону Рауля). При р < рїд маємо від'ємні
відхилення від закону Рауля (рис. 5.3, крива 2)
пряма 1 відповідаєрщ закону Рауля, а при/? >
> рїд —додатні (рис. 5.3, крива 3). Від'ємні
відхилення від закону Рауля виникають тоді,
коли взаємодія між різнорідними частинками А
і В досить велика і перевищує взаємодію між
однаковими частинками. За рахунок сильної
взаємодії частинки міцно утримуються в
рідині, що веде до зниження загального тиску.
При додатних відхиленнях, навпаки, взаємодія
А і В слабка, компоненти, так би мовити,
виштовхують один одного з рідини у газову фазу,
що веде до збільшення загального тиску. Таким
чином, залежність загального тиску пари від складу рідини може бути
зображеною будь-якою з кривих (1—3) на рис. 5.3.
Зростання тиску зі зміною складу відповідає додатному значенню
похідної —-— і умові Рв > Р°а- Отже, щоб обґрунтувати перший закон Конова-
лова, потрібно довести, що при —£— > 0 ув>хв.
сЬсв
90
Коновалов Дмитро Петрович
(1856—1929), Росія, СРСР
Оскільки загальний тиск дорів- р
нює сумі парціальних, то маємо
-£=АА (5.9)
ахв ахв ахв
Величини йрА та йрв залежні р°А
одна від одної. Ця залежність
дається співвідношенням Гібб-
са-Дюгема (1.99), яке для
хімічних потенціалів має вигляд
Рв
В
Рис. 5.3. Відхилення від закону Рауля
у бінарній системі
ха<і\іа + хвсїіів = 0. (5.10)
З використанням залежності
хімічного потенціалу від
парціального тиску компонента щ = |ьі? + КТІпр; (рівняння (1.103)) це дає
хАс11прА+хв(іІпрв =0. (5.11)
Рівняння (5.11) має назву рівняння Дюгема-Маргуліса. З цього рівняння
Фл°-*■&■*>,.
*аРв
(5.12)
Таким чином,
СІХв
= Фв
сЬсв
Фв
СІХв
( \
1_х±Ра_
\-*£УА.
{ ■
*аУв)
_Фв(
сіхву
х *вУаР
хаУвР
(5.13)
Фв
Похідна —— завжди додатна (парціальний тиск якогось компонента зав-
<*хв ф
жди зростає при збільшенні його кількості). Отже, умові —— > 0 відповідає
ахв
умова
і, нарешті,
1 >и> або > > що дає —> —
*лУв
хв
Ув
*в Ув
Ув>хв.
Діаграму стану тиск-склад при > 0 (і, відповідно, рв > р°А) наве-
<1хв
дено на рис. 5.4. Лінії/? =/(ув) (1) та/? =/(хв) (2) мають, відповідно, назви
«крива пари» (7) та «крива рідини» (2). Вище від кривої 2 (великий тиск)
лежить гомогенна область — рідина, а нижче кривої 1 (малий тиск) — го-
91
в'*Ув
Рис. 5.4. Діаграма стану тиск-склад
для бінарної системи рідина-пара
могенна парова фаза. Область
між кривими 1\2 —
гетерогенна. Тут існують у рівновазі пара
й рідина. Лінія (І), що з'єднує
при р = соті відповідні точки а
та Ь на кривих 1 та 2, має назву
«нода». Точка а відповідає
рівноважному складу рідини (хв)9
а точка Ь — рівноважному
складу пари (ув). Згідно з
правилом фаз у гетерогенній області
/ = 1 і вільний тільки один
параметр (тиск або склад).
Наприклад, якщо відомий загальний
тиск (р), то, проводячи ноду, можна знайти відповідні склади рідини та пари.
Діаграма стану може бути зображена в координатах температура
(кипіння) - склад при постійному зовнішньому тиску. Таку діаграму зображено
на рис. 5.5. По відношенню до діаграми тиск-склад (рис. 5.4) вона, так би
мовити, повернута догори ногами, а саме: низьким температурам
відповідає гомогенна область рідини (2), а високим — газова фаза (/). Більш
леткому компоненту В відповідає менша температура кипіння — ТЦ (при/? =
= СОП8Ї).
За допомогою діаграми температура-склад зручно розібратися у
процесі фракційної перегонки (дистиляції). При нагріванні рідини до
температури вищої, ніж дає крива 2, одержимо пару, що збагачена летким
компонентом В. При цьому рідина
збагачується менш летким
компонентом А. Якщо
сконденсувати пару, то матимемо
рідину, що збагачена компонентом
В. її знов можна розділити на
пару, яка ще більше збагачена
компонентом В, та рідину.
Повторюючи цей процес
багаторазово, одержимо в конденсаті
чистий компонент В, а в
залишку після багаторазового
нагрівання і відокремлення конденсату
залишиться малолеткий компо-
:в'*Ув
Рис. 5.5. Діаграма стану температура-склад
для бінарної системи рідина-пара
нентЛ. За допомогою дистиляції можна розділити рідину на чисті компоненти,
якщо діаграма стану має вигляд, наведений на рис. 5.5. Така діаграма
реалізується, коли відхилення від закону Рауля (додатні або від'ємні) невеликі.
При великих відхиленнях на кривих тиск-склад (або температура ки-
піння-склад) виникають екстремуми. У точках екстремумів
92
отже, згідно з формулою (5.13)^ = хв Таким чином, у точці екстремуму
склади рідини й газової фази однакові. Це положення становить зміст
другого закону Коновалова.
Точки, в яких ув = хв, мають у |
назву азеотропних. Діаграму
стану (в координатах
температура кипіння-склад) з наявністю
азеотропної точки наведено на
рис. 5.6. Як видно з рис. 5.6, азе-
отроп поводиться, як чиста
рідина, тобто кипить при певній
конкретній температурі — Газ.
Відзначимо, що для азеотропної
точки кількість ступенів свободи
дорівнює нулю, хоч у рівновазі
знаходяться лише дві фази. Це
відбувається за рахунок
зменшення кількості компонентів
(другий закон Коновалова
хв->Ув
Рис. 5.6. Діаграма стану температура-склад
для бінарної системи рідина-пара
з утворенням азеотропів
це додаткова умова).
При наявності азеотропів за допомогою дистиляції не можна розділити
рідину на чисті компоненти. Наприклад, рідина складу а може бути
розділена на компонент А та азеотроп, а рідина складу Ь — на азеотроп і
компонент В.
5.2.2. Рівновага рідина-рідина
При розгляді рівноваги рідина-пара вважалося, що гетерогенна тільки
область співіснування пари й рідини. Поза кривими пари й рідини відповідні
фази вважалися гомогенними. Для газової фази це твердження справедливе,
а рідка фаза може бути гетерогенною.
Розглянемо це питання з термодинамічної точки зору. Процес утворення
розчину з двох рідин може бути або ендотермічним (Д#> 0), або
екзотермічним (А//< 0). Перший випадок відповідає додатним відхиленням від закону
Рауля (слабка взаємодія між компонентами А та В), а другий — від'ємним
(сильна взаємодія між А і В). Випадок (Д#= 0) відповідає ідеальним
розчинам.
За рахунок чого ж утворюється розчин при Д#>0? Оскільки АС
повинно бути від'ємною величиною, то при Д#> 0, згідно з формулою (1.72),
має бути Д5> 0, тоб(то рушійною силою є ентропійний фактор. Справді, при
змішуванні ентропія, за формулою (1.55), збільшується. Для ідеальних
розчинів, а також при АН<0 АС є від'ємною величиною при будь-яких
температурах. При дуже ендотермічному процесі взаєморозчинення рідин для
утворення гомогенного розчину необхідно, щоб член ГД5за абсолютною
93
величиною був більший, ніж Д#. Для цього потрібні достатньо високі
температури. В протилежному випадку при низьких температурах Д# перевищує
7М, що веде до додатного значення ДС Це означає, що замість розчинення
рідин одна в одній
відбуватимете Н
60 І
40 к
20
фенол 20 40 60 80 вода
ться зворотний процес —
розшарування рідин, і система стане
гетерогенною в рідкому стані.
Причому гетерогенна система не
обов'язково розпадається на
чисті вихідні речовини. Може
спостерігатися обмежена
розчинність, і в рівновазі тоді
існуватимуть два розчини: А в В та В
в А. Прикладом може бути
система фенол-вода (рис. 5.7).
Якщо взаємна розчинність
двох рідин одна в одній дуже мала
Рис. 5.7. Діаграма стану температура-
склад для системи фенол-вода
(практично нерозчинні взаємно рідини), то парціальний тиск пари над
сумішшю для кожного з компонентів практично дорівнює тискові над
відповідною чистою рідиною р°.
Отже, загальний тиск (р) над сумішшю двох взаємно нерозчинних рідин
Р = Р°а+Р°в- (5-14)
Відповідно, температура кипіння такої суміші нижча за температуру
кипіння чистих компонентів. Таким чином, малолетка рідина може бути
переведена у пару при більш низькій температурі, ніж температура її
кипіння, шляхом сумісного випаровування (кипіння) з невзаємодіючою більш
леткою рідиною. Наприклад, перегонка термічно нестійких органічних
речовин здійснюється з водяною парою.
Розглянемо тепер, який вигляд мають діаграми стану в двокомпонентних
системах рідина-тверде тіло.
5.2.3. Рівновага рідина-тверде тіло
Діаграми стану тверде тіло-рідина зазвичай зображують у координатах
температура плавлення-склад (р = соті). Оскільки система рідина-тверде
тіло існує при більш низьких температурах, ніж система рідина-газ, то вплив
ентропійного фактора на розчинність тут менший, і для діаграм тверде тіло-
рідина гетерогенність — типова картина. В рівновазі можуть бути при
цьому чисті вихідні компоненти (рис. 5.8) або тверді розчини (а та Р) (рис.
5.10). Безперервний ряд твердих розчинів (рис. 5.9) спостерігається значно
рідше. В останньому випадку діаграма стану дуже схожа на діаграму стану
рідина-пара (див. рис. 5.4, с. 92) і складається з трьох областей: двох
гомогенних (розплав — І та твердий розчин — II) та однієї гетерогенної (III), де
в рівновазі знаходяться твердий та рідкий розчини.
94
При обмеженій розчинності
(рис. 5.10) або повній
нерозчинності (рис. 5.8) діаграма стану має
точку «о», де в рівновазі
знаходяться три фази: рідина плюс дві
тверді фази. Це — вихідні
компоненти при повній
нерозчинності (рис. 5.8) або тверді розчини
одного з компонентів у другому
при обмеженій розчинності (рис.
5.10). Ця точка має назву
евтектичної, або просто евтектики.
Згідно з формулою (5.6) при к = З
/ = 0, тому евтектична точка нон-
варіантна.
Діаграма на рис. 5.8
складається з чотирьох областей —
гомогенного рідкого розплаву (І) та
трьох гетерогенних: твердий
компонент А плюс розчин (II),
твердий компонент В плюс розчин
(III) та суміш твердих кристалів А
і В (IV). На рис. 5.8 чітко видно, що
додавання одного з компонентів
до другого при нерозчинності у
твердому стані веде до зниження
температури плавлення. Це
явище під назвою кріоскопії вже
розглядалося вище (підрозділ 3.2) для
розбавлених розчинів.
На діаграмі, що зображена на
рис. 5.10, в областях (II) і (III)
розчин знаходиться у рівновазі не
з чистими речовинами А та В, а
з відповідними твердими
розчинами (а або Р), а область (IV) — це
суміш твердих розчинів (а та Р)
відповідного складу. Області (V) та
(VI) — гомогенні, це тверді
розчини одного з компонентів у
другому.
При взаємодії двох
компонентів А та В можуть
утворюватися стійкі хімічні сполуки АдВу, які
Склад
Рис. 5.8. Діаграма стану температура-
склад у бінарній системі тверде тіло-
рідина при нерозчинності компонентів
у твердому стані
Склад
Рис. 5.9. Діаграма стану температура-
склад у бінарній системі тверде тіло-
рідина при повній розчинності
у твердому стані
А Склад
Рис. 5.10. Діаграма стану температура-
склад у бінарній системі тверде тіло-
рідина при обмеженій розчинності
у твердому стані
95
Рис. 5.11. Діаграма стану температура-
склад у бінарній системі тверде
тіло-рідина при утворенні хімічної
сполуки, що плавиться конгруентно
при плавленні поводять себе як
чисті речовини. Діаграма стану в
цьому випадку має вигляд,
зображений на рис. 5.11 (вважається,
що компоненти А, В та сполука
АхВу у твердому стані
нерозчинні). Діаграма на рис. 5.11,
фактично, складається з двох діаграм
(типу зображених на рис. 5.8,
с. 95): А + А^та АХВ^+ В.
Сполука АхВу плавиться при певній
температурі, склад її теж відомий,
отже, система в точці В, яка має
назву дистектики, нонваріантна.
Ситуація тут схожа з ситуацією в азеотропних точках — хоча в рівновазі
тільки дві фази, кількість компонентів (два) зменшується за рахунок
додаткової умови — в дистектиці склад твердої фази такий самий, як склад
рідини. В цьому випадку вважається, що стійка сполука А^В^ плавиться
конгруентно.
Діаграму із нестійкою сполукою, що плавиться інконгруентно, наведено
на рис. 5.12. Гомогенна тільки область І — рідкий розчин. У гетерогенних
областях (II), (III) та (IV) рідкий розплав знаходиться у рівновазі з
компонентами А (II), В (III) та сполукою А^Ву (IV). Область (V) — це гетерогенна
суміш А та АдВу, а (VI) — А^ та В. Відзначимо, що для всіх розглянутих
діаграм розчинність компонентів у рідкому стані необмежена.
На всіх діаграмах у гетерогенних областях діє правило важеля. Якщо
взяти якусь фігуративну точку, наприклад а на рис. 5.9, с. 95, і Провести ноду
до перетину з межами області (тут: кривими 1 — солідусу (твердого) та 2 —
ліквідусу (рідини)) в точках Ь та с, то, згідно з правилом важеля,
маса твердого розчину _ аЬ
маса рідкого розчину ас
Інформацію для побудови
діаграм стану одержують за
допомогою кривих кристалізації. Готують
суміш відповідного складу,
плавлять її і потім спостерігають
охолодження системи протягом
деякого часу. Для чистих компонентів
на кривих температура-час
з'являються полиці при температурах
плавлення чистих речовин (криві
ата^нарис. 5.13). Евтектика теж
твердне при сталій температурі
II
V
І
ІУ^Ч
/ Ш
VI
Склад
В
Рис. 5.12. Діаграма стану температура-
склад у бінарній системі тверде
тіло-рідина при утворенні хімічної
сполуки, що плавиться інконгруентно
96
(крива й, рис. 5.13). Для складів, що відповідають гетерогенній області
(розглядається діаграма евтектичного типу, див. рис. 5.8, с. 95), на кривих
кристалізації (криві Ь, с, є та/) з'являються характерні зломи, коли починають
випадати кристали відповідного компонента.
Час Д Склад
Рис. 5.13. Кристалізація бінарних сумішей
5.3. Фазові рівноваги у трикомпонентних системах
Для трикомпонентних систем при р = соті правило фаз дає
/=4-*. (5.15)
Якщо зафіксувати ще й температуру (ізотермічний переріз), то
/=3-* (5.16)
При к = 1, тобто для гомогенної системи це дає/= 2, отже, змінюються
дві концентрації. Для знаходження фігуративної точки на площині
концентрацій потрібно побудувати рівносторонній трикутник. Вершини цього
трикутника відповідають чистим компонентам. Сторони — відповідним
двокомпонентним системам. Для знаходження концентрації за методом Гіббса
з фігуративної точки а (рис. 5.14) на сторони
трикутника опускаються три перпендикуляри.
Сума довжин цих перпендикулярів приймається
рівною одиниці (або 100 % мол.). Довжина кожного
з перпендикулярів відповідає концентрації
компонента, що знаходиться в протилежному куті.
X. Розебом запропонував інший метод, згідно
з яким проводять прямі, паралельні двом
сторонам трикутника. На третьому боці відповідні
відрізки (рис. 5.15, с. 98) відображують
концентрації компонентів. рис 5 14 Визначення
Як приклад фазової рівноваги в рідкій триком- концентрації у
трикомпонентній системі при р = соті та Т = соті на понентній системі
рис. 5.16, с. 98, наведена діаграма стану системи за методом Гіббса
97
в
Рис. 5.15. Визначення концентрації
в трикомпонентній системі за методом Розебома
вода- ацетон-хлороформ. Крива виділяє гомогенну частину в
«ацетоновому куті» від гетерогенної суміші розчинів: «хлороформ у воді» та «вода у
хлороформі» (при нульовій концентрації ацетону система хлороформ-вода
гетерогенна).
Для рівноваги тверде тіло-рідина візьмемо простий випадок —
компоненти не розчиняються в твердому стані й розчиняються необмежено у
рідкому. Для трикомпонентної системи при Тф соті потрібно над
трикутником концентрацій побудувати об'ємну діаграму. Ця діаграма наведена на
рис. 5.17. Бокові площини діаграми відповідають бінарним діаграмам (типу
зображеної на рис. 5.8) з подвійними евтектиками (точки М, іУта Ь на рис.
5.17). Потрійна евтектика (точка О) знаходиться нижче від усіх подвійних. На
(СН3)2СО
СНСЬ
Н90
Рис. 5.16. Трикомпонентна
система ацетон-хлоро-
форм-вода
Рис. 5.17. Об'ємна діаграма стану
температура-склад у
трикомпонентній системі рідина-тверде тіло
98
рис. 5.18, я, б, в зображені ізотермічні перерізи об'ємної діаграми при
деяких температурах, а на рис. 5.19 — проекція об'ємної діаграми на площину
(так би мовити, вигляд зверху), де чітко видно, як з'єднуються подвійні
евтектики з потрійною. В потрійній евтектиці у рівновазі знаходяться чотири фази:
розчин і чисті компоненти, отже, згідно з формулою (5.15), /= 0.
Фазові рівноваги встановлюються переносом компонентів між фазами
при відповідних температурі й тиску і не передбачають хімічних реакцій.
У наступному розділі розглядається хімічна рівновага, тобто рівновага, що
встановлюється при перебігу оборотних хімічних реакцій.
£ в
Рис, 5.18. Ізотермічні перерізи об'ємної діаграми
для трикомпонентної системи при температурах:
а — Г,;б — Т2;в — Тг
Рис. 5.19. Проекція трикомпонентної
об'ємної діаграми на площину
99
Розділ 6.
ХІМІЧНА РІВНОВАГА
Якщо в системі чи в довкіллі щось міняється,
Система змінам опір чинить, не вагається.
6.1. Хімічна рівновага у гомогенних системах
Умова хімічної рівноваги подана у виразі (1.107), тобто АС = 0. Потрібно
тепер з'ясувати, що являє собою АС для хімічної реакції в умовах сталої
температури.
Згідно з рівнянням (1.105), якщор = сопзі тьТ = сотІ, то маємо
Ю^щЛц. (6.1)
В інтегральній формі, згідно (1.87), маємо
С = 5>,и,- (6.2)
І
Зміна вільної енергії Гіббса для хімічної реакції дорівнює
А^ = ЕуЛ-ЕуЛ- (63)
і З
продукти реагенти
Використовуючи вираз (1.103) для хімічного потенціалу, отримуємо
де = 5>,ц?-2>/;+дг
5>>а-і>у1пл
(6.4)
В стандартному стані всі парціальні тиски дорівнюють 1 атм, отже,
ДЄ0=£у,К-ХуХ-. (6.5)
' З
З урахуванням (6.5) рівняння (6.4) набуває вигляду
АС = ДС° + КТ\п\\р11, (6.6)
у
де V/ беруться зі знакрм (+) для продуктів і зі знаком (-) для реагентів.
Для оборотної хімічної реакції
0сА + рв=уС + 8О,
де а, Р, у та 8 — стехіометричні коефіцієнти,
ш*-44- (б.т)
РаРв
При рівновазі АС = 0, отже,
100
РвРа
(6.8)
де Рі —рівноважний парціальний тиск.
Оскільки АС0 не залежить від тиску, то вираз праворуч у рівнянні (6.8)
є сталою величиною. Отже, константою повинен бути вираз під знаком
логарифма
К>
= РсРр
РвРа
(6.9)
Величина Кр має назву константи рівноваги хімічної реакції. З
урахуванням (6.9) рівняння (6.8) стає таким:
АС0 = -ШпКр. (6.10)
Таким чином, константа хімічної рівноваги безпосередньо пов'язана зі
зміною вільної енергії Гіббса хімічної реакції в стандартному стані. З
урахуванням співвідношення (6.10) рівняння (6.6) для довільного (не в стані
рівноваги) значення зміни вільної енергії Гіббса хімічної реакції набуває
вигляду
ДО:
РсРр
-КПпКр+КПпПр? =-КТ\пКр+КТ]п^£
і РаРв
(6.11)
Рівняння (6.11) відоме як рівняння ізотерми хімічної реакції.
Проаналізуємо рівняння (6.10) і (6.11). Умова перебігу хімічної реакції—
це від'ємне значення АС (див. формулу (1.107)). Розглянемо три варіанти цієї
умови.
Перший варіант. АС° « 0.
Згідно з (6.10) це відповідає
Кр » 1, тобто процес переходу
реагентів у продукти
відбувається практично до кінця.
Лише за дуже малих рА та рв
другий член у рівнянні (6.11)
дає великий додатний внесок,
що компенсує велике від'ємне
значення ДО0 і відповідає
рівновазі ДС= 0 (крива 1 на
рис. 6.1).
Другий варіант. ДО°=0.
Це відповідає Кр = 1. Рівноваж-
Реагенти
Склад
Продукти
Рис. 6.1. Зміна АС із складом реакційної
суміші при: 1 — АС0 « 0; 2 — АС0 = 0;
З — ДС°»0
ний стан реалізується за приблизно однакових парціальних тисків реагентів
та продуктів. Цей випадок відповідає оборотній хімічній реакції, що не
добігає до кінця (крива 2 на рис. 6.1).
101
Третій варіант. АС0 » 0. При цьому Кр « 1. Завдяки великому
додатному внеску стандартного значення вільної енергії Гіббса в АС реакція
практично не перебігає. Другий член у рівнянні (6.11) може бути від'ємним
лише задосить малих парціальних тисків продуктів/?ста/?£). Зауважимо, що
третій варіант—це, фактично, дзеркальне відображення першого. Справді,
умова АС0 » 0 для прямої реакції означає ДО0 « 0 для зворотного процесу,
отже, реакція перебігає до кінця у зворотному напрямі. Реакції, для яких
ДС° — велика додатна величина, називають «термодинамічно
забороненими» (крива 3 на рис. 6.1).
Константу рівноваги можна виразити не тільки через парціальні тиски,
а й через одиниці концентрації, якими можуть бути мольна частка—де,- або
молярна концентрація — с(.
Парціальний тиск/?, = х(Р, де Р — загальний тиск, отже, з рівняння (6,9)
маємо
Кр-{хАРТ{хврГМ •
ДЄ
—у—5
хсх0
хАхв
є константою рівноваги, що виражається через мольні частки.
Розглянемо конкретний приклад — реакцію окиснення 802 до 803.
Позначимо ступінь перетворення сірчаного газу через (а). Тоді маємо в
розрахунку на 1 моль
2302 + 02 <->230з
2(1-а) 1-а 2а
Сума молів дорівнює 2(1 - а) + 2а + (1 - а) = 3 - а.
2(1-а) _ 2а _ 1-а
Мольні частки х^ =~Т—Г; ^°з =Г^; х°2 =Г^'
2 3-а 3 3-а 2 3-а
Згідно з (6.13) для Кх маємо
к (*ю3)2 ^а2(3-а)
г (*ю2)2'*Ь2 (1-а)3 "
Оскільки Ду = 2 - 3 = -1, то, згідно з формулою (6.12), це дає для Кр
к = а2(3-<х)
р (1-а)3Р'
Для різних типів реакцій вирази для Кр через ступінь перетворення (а)
наведено у таблиці 6.1.
102
(6.12)
(6.13)
Таблиця 6.1. Вирази Кр через а
Тип реакції
\АЬ2В
\лигв
2АЬВ
\а +вис
ІАЛ-В^С
за + вигс
а + вигс
А+вигс
Вигляд рівняння
4а2Я
*'_1-а2
_ 27а3?2
*Р (1^а)(1 + 2а)2
К _ а(2"а)
' 4(1-а)2Р
_а(2-а)
Р~(1-а)2Р
_ а(3-2а)2
Р_4(1-а)2Р2
* _ «2(2-а)2
Р 1,3(1 -а)4/»2
г - 4а2
(1-а)2
_ 27а3Р
^ (1-а)2(2 + а)
Тип реакції
іАигв
А*В + С
А*В + 2С
А+ВиС+П
А +3/2#і;С + І>
2А + ВиїС
А +2ВЬС + 0
2А*В + С
Вигляд рівняння
_ 27<Х3Р
'" 4(1-а)2 (2 +а)
у _ 4а3Р2
И (1-а)(1 + 2а)2
г- «2
(1-а)2
_ а2у/5-За
2(1-а)^9(1-а)2Р°-5
*• -а2(3-а)
Л.р — г
(і-а)3Я
а2 (3-а) |
4(1 - а) Р
г- а1
4(1-а)2 |
За рівнянням Менделєєва-Клапейрона (1.2)
що дає
Рі=-±КТ = СіКТ,
ч8 /_ \ї/_ ч8
= (ссКТ){соЯТ) =М(^) т+8-Р-а = (І?Г)АУ (614)
де Кс — константа рівноваги, що виражається через молярні концентрації,
(ссПсо)5
Кс=-
(6.15)
(слТісв?
Для реакцій у гомогенних газових фазах зазвичай користуються Кр (а
також Кх). Величина Кс (поруч із Кх) зручна для опису хімічних рівноваг
у гомогенних розчинах.
Якщо газ неідеальний, то замість тиску зручно користуватися леткістю
(У), що дає для константи —V—я
Ґ Ґ
/=г7
; а; в
(б.іб)
103
Константа К^, зв'язана з Кр таким чином:
_(РсУс)УЩо)* _ „ у
(раУа) (рвУвУ
/>ЛУ
(6.17)
(враховане співвідношення (2.43) між тиском і леткістю), де
Для ідеальних газів усі У,- = 1, звідки Ку= 1 і, отже, Щ = Кр.
Константа рівноваги залежить від температури. З рівнянь (6.10) та (1.85)
маємо
(д\пКр^
дТ
)р
КдТ
ҐАСоЛ
АН°
КТ2
(6.19)
(нагадаємо, що величини АС0 та Д#° належать до стандартного стану, при
якому всі/?,- = 1 атм).
Рівняння (6.19) має назву рівняння ізобари хімічної реакції. Це рівняння
дуже схоже з рівнянням Клапейрона-Клаузіуса (2.7) для залежності тиску
рівноважної пари над конденсованою фазою від температури. Ця схожість
не випадкова, тому що тиск насиченої пари — це фактично константа
рівноваги в системі конденсована речовина-пара.
Коли АЯ°=соті, рівняння (6.19) легко інтегрується від температури Тх
до Т2, що дає
1п5^_ДЯ° АГ
Кп
к
тхт2
(6.20)
де ДТ = Т2 - Тх, а КР2 тьКРі — значення Кр при температурах Т2 та Тх
відповідно.
З рівнянь (6.19) та (6.20) видно, що константа збільшується з
температурою для ендотермічних процесів: Д#°> 0.
За допомогою рівняння (6.20) можна обчислити КР2 при температурі Т2,
якщо відома константа рівноваги КРі при температурі Тх та стандартна зміна
ентальпії в хімічній реакції. Можна розв'язувати і зворотне завдання, тобто
обчислювати Д#°, якщо відомі значення Кр при різних температурах.
Якщо константу виразити через мольні концентрації—Кс, то її
залежність від температури дається рівнянням ізохори хімічної реакції
(дІпКс
Ш°
(6.21)
дТ )у ВТ'
Д&АІҐ—стандартна зміна внутрішньої енергії хімічної реакції (нагадаємо,
що стандартний стан у цьому випадку — всі с, = 1 моль/л).
104
6.2. Вплив зовнішніх факторів на хімічну
рівновагу. Принцип Ле Шательє
А. Ле Шательє сформулював принцип, згідно з яким система, що
перебуває в стані рівноваги, чинить опір дії зовнішніх факторів, тобто при зміні
якогось параметра в рівноважній системі починають відбуватися такі
процеси, що цю зміну якоюсь мірою компенсують. Розглянемо, як впливають
на хімічну рівновагу зміни температури, тиску та концентрації компонентів.
Згідно з рівнянням (6.19) з підвищенням температури константа
рівноваги збільшується для ендотермічного процесу, тобто з підвищенням
температури (наприклад за розрахунок підводу теплоти з довкілля) в системі
починають проходити ендотермічні процеси, що поглинають енергію.
Прикладом такої реакції є процес взаємодії азоту з киснем з
утворенням N0
При високих температурах, наприклад, у низькотемпературній плазмі
(в тому числі у грозових розрядах) або у двигунах внутрішнього згоряння
вихід шкідливого N0 різко
Навпаки, при АН < О
значення Кр збільшується зі
зниженням температури,
і в цьому випадку система
виробляє теплоіу, щоб
компенсувати зниження
температури.
Збільшення тиску
система компенсує за рахунок
перебігу хімічної реакції,
яка характеризується
зменшенням загального числа
молів газу. Це наочно
видно з рівняння (6.12), яке
можна записати так:
КХ=КРР~*\ (6.22)
При Ау = ОКх не залежить від тиску. Якщо значення Ду від'ємне, то Кх
збільшується із підвищенням тиску. Навпаки, якщо Ду > 0, то рівновага зі
збільшенням тиску зміщується у напрямі зворотної реакції. Для перебігу
прямої реакції в цьому випадку вигідне зменшення тиску.
Вплив зміни концентрації (парціального тиску) якогось із компонентів на
рівновагу можна простежити за допомогою рівняння ізотерми хімічної
реакції. При рівновазі ДО = 0. Збільшимо концентрацію одного з реагентів,
наприклад А. При цьому Ра>Ра> що веде Д° збільшення знаменника дру-
збільшується (див. рис. 6.2).
1-Ю"5
но-10 Н
110"15
Температура
вихлопних
газів
Температура
у циліндрі
при згорянні
палива
1000
2000
Температура, К
Рис. 6.2. Залежність константи рівноваги реакції
Уіії? + БОЛІМО від температури
105
гого члена у рівності (6.11) і, відповідно, дає від'ємний внесок у вільну
енергію Гіббса. Отже, ДС набуває від'ємного значення і відбувається прямий
процес, за рахунок чого концентрація компонента А зменшується. Навпаки,
якщо збільшити концентрацію якогось із продуктів, наприклад Б (р0 > рй),
то це дасть додатний внесок у вільну енергію, а якщо АС > 0, то відбувається
зворотна реакція, що зменшує концентрацію компонента Б.
6.3. Хімічна рівновага у розчинах. Гетерогенна
хімічна рівновага
Для хімічних реакцій, що відбуваються у розчинах, доцільно
користуватися константою рівноваги, яка виражається через концентрації, тобто Кс
(молярні концентрації) або^ (мольні частки). Але ці вирази (див.
формули (6.12) та (6.15)) правильні лише для ідеальних систем. Для реальних
розчинів треба замість концентрацій користуватися активностями, а саме:
—8—у —5—у —5—у
V — аРас __ хРхс УрУс _ іг у ,,ЛЛЧ
а" г¥ " ИЧ* т^ " х г (6-23)
аАаВ ХАХВ УаУв
Для ідеальних систем Ка =Кх,Ку = 1.
Гетерогенні хімічні рівйоваги охоплюють компоненти, що знаходяться у
різних фазах. Наприклад:
Ш4С1(тв)=Ш<гЧнС1(г);
СиО(тв)+Н^=Си(тв)+Н20^>.
Для реакції
Е«Л-+ІРД. = Хуа+£5А,
/ і і і
в якій беруть участь газові компоненти А; та С, з леткостями^ та компоненти
розчинів В, та Ц-з активностями аь вираз для константи рівноваги має вигляд
гогік
К, = ' (6-24)
* гоню'
і і
Наприклад, для відновлення воднем оксиду феруму (II), розчиненого у
рідкому залізі,
к _ /н2о%е
^ Ун2аРеО
Для ідеальних систем/ =рь а а( = хі9 отже,
ІК'ІК
К/а - Крх ~ -Л-гьі^т-В, ' (6'25)
Сі
106
Коли тверді або рідкі фази, що беруть участь у реакції, є чистими
індивідуальними хімічними сполуками, то їх хімічний потенціал залежить лише від
температури, а значення х( для них дорівнюють одиниці (при х$ —> 1 У/—> 1,
отже, я,-» 1).Таким чином, у виразі для константи лишаються лише
газоподібні речовини. Наприклад, для розкладу карбонату кальцію:
СаС03(тв)=СаО(т)+СО<г);
кР=Рсо2-
Для реакції добування генераторного газу:
СО(2г)+С(т) = 2СО(г);
-2
Кр-~—.
Рсо2
6.4. Обчислення константи рівноваги
за термохімічними даними
Безпосереднє обчислення константи рівноваги потребує знання
рівноважних концентрацій (парціальних тисків) для всіх реагентів, а також гарантії
того, що система досягла стану рівноваги. В окремих випадках це питання
вирішується.
Як конкретний приклад розглянемо реакцію дисоціації йоду І2 = 21.
Відповідні експериментальні дані щодо хімічної рівноваги цієї реакції
наведено у таблиці 6.2.
Таблиця 6.2. Дані для рівноважного стану реакції дисоціації йоду при 1274 К
Загальний тиск,
/?3аг> атм
0,П22
0,4742
0,7531
0,9308
Початкова концентрація йоду,
с0, ммоль/л
0,7045
3,537
5,866
7,376
Ступінь дисоціації
йоду, а
0,523
0,283
0,229
0,208
Кру
атм
0,169
0,165
0,166
0,169
/Гр=0,167
І2 = 21
1-а 2а
с0(1-а) 2с0а
Рзаґ=Рх+Рі2іРі = сіКТ;р1 = 2с0аКТ;р1 = с0(\ -а)КТ;Рзаг = с0КЦ1 + а),
Лаг і гг А2 4а
СоКТ ' а2 1-а2
107
При розрахунках враховано, що К = 0,082 л • атм/моль • К; КТ= 104,468
л • атм/моль. Розраховані значення констант для кожного вимірювання та їх
середнє значення наведено у крайньому стовпчику таблиці 6.2.
Частіше, однак, виникає потреба у розв'язанні протилежної задачі, а саме
обчислення рівноважних виходів за відомими значеннями Кр. Тому Кр
обчислюють за допомогою рівняння (6.10), тобто з використанням
стандартної зміни вільної енергії Гіббса. Одну з можливостей обчислення АС0 дає
термохімія, згідно з рівнянням (1.72), яке для стандартних величин має вигляд
АО° = АН°-ТА8°. (6.26)
Величина А5° — це сума стандартних абсолютних ентропій продуктів мінус
сума стандартних абсолютних ентропій реагентів (з урахуванням
стехіометричних коефіцієнтів). Методи обчислення абсолютних ентропій наведені
в підрозділі 1.3 — див. формули (1.60)—(1.63). Зазвичай табулюються
абсолютні стандартні ентропії при кімнатній температурі — 298 К, тобто 8%9$.
Перехід до іншої температури здійснюється за допомогою формули (1.58).
При обчисленні зміни ентальпії в хімічному процесі Д#°
використовується закон Гесса та його наслідки. За допомогою табульованих значень
ентальпій утворення або горіння реагентів і продуктів можна обчислити
тепловий ефект хімічної реакції за стандартних умов Д//£98 * Для визначення
АЯпри будь-якій температурі застосовується формула Кірхгофа у вигляді
(1.38).
Таким чином, якщо взяти за нижню межу Г=298 К, а верхню зробити
змінною, то для зміни вільної енергії Гіббса в хімічній реакції при довільній
температурі маємо
г Г АСП
ДО? = ДЯ? - ТАЗ} = Д#£98 + | АСр(іТ - ГА52°98 - Т —£- йТ. (6.27)
298 298 Т
Якщо вважати ентальпію та ентропію незалежними від температури,
тобто АСр = 0, то
ДО£ = Ш°2П - ГД£2°98. (6.28)
Формулу (6.28) можна використати для оцінки Дб£. Це, так би мовити,
нульове наближення при обчисленні АС%.
Далі можна припустити, що АСр = соті, тобто вважати, що від
температури не залежить теплоємність — перше наближення. Це дає
ДС? = АН°т + АСр(Т-298)-ГД52°98 -ТАСр 1п^ =
( Т 298 ^
= ДЯ2098-ГД52°98-ДСрГ|іп—+ —-її. (6.29)
Друге наближення {метод Тьомкіна-Шварцмана) полягає в тому, що Ср
задається у вигляді степеневого ряду
108
--4
Ю
О
О
ю
ЧО
<л
•-4
О
І—*
1ч>
и>
оч
о
чо
и>
о
1 1160 1
о
"о
і—»
<л
Оч
о
"Іо
ю
р
ю
о
и>
»-^
^ о
о "
чо
--4
О
о
о
к>
<1
чо
■и
р
1—>
»—»
*л
и>
о
00
ІЛ
4^
2
О
О
"о>
о
ю
-^
о
"и»
►—»
о
ЧО
о
и>
о
оч
-*4
^
оо
о
о
Іч)
оч
ю
чо
о
»—»
о
-4
Ю
О
-«4
^
и>
| 1120
р
1л
00
ЧО
-<!
Р
1к)
О
Оч
о
и>
о
м
00
<^
Оч
о
о
к>
4*.
2:
о
о
ЧО
ЧО
ю
о
Оч
ЧО
о
1 иоо
о
1л
^1
Оч
<л
О
"ю
ЧО
ю
и>
о
ю
чо
00
чо
<^
4^
о
о
ю
ю
ЧО
^
о
о
ЧО
1—*
и>
о
<^
о
о
1 1080
р
"Ід
ОЧ
и>
ю
о
іо
00
и>
о
о
ю
ЧО
4*>
00
Оч
ю
о
о
к>
1—»
и>
о
о
о
00
и>
<л
о
<л
•—*
<л
0901 1
о
1л
4^
чо
^4
О
1о
--І
и>
00
о
ю
ЧО
2
оч
о
о
о
ЧО
оч
ю
о
о
^
«У»
чо
р
4*
ю
и>
1 1040
о
ІЛ
и>
Оч
О
1о
оч
.&>
Оч
О
ю
00
оч
н"*
<л
00
о
о
о
ЧО
ІЛ
о
о
оч
00
<л
р
и>
ю
-о.
| 1020
р
1л
ю
ю
и>
о
"ю
<л
<л
<л
о
ю
00
>—»
^а
іл
оч
о
о
Оч
ю
^4
О
О
оч
н-»
ю
о
ю
ю
ЧО
0001 1
р
1л
о
00
и>
о
То
4*>
о\
и>
о
ю
^4
«^
1—1
О»
4^
О
О
-и
Оч
и-
о
о
ІУ»
-и
ю
р
1—>
ю
и>
| 980
о
V
ЧО
4^
Ю
О
1о
и>
<і
ю
о
ю
^4
Ю
и>
<л
ю
о
р
ю
ЧО
Оч
о
о
4^
^
и>
р
о
ю
4^
096 1
о
V
^л
ЧО
ЧО
О
ІО
М
00
*""*
о
ьо
оч
-о
и>
<л
О
О
о
н—
и>
и>
о
о
4^
О
--4
О
О
ЧО
^1
| 940
о
V
Оч
<Л
<л
о
То
1—*
чо
о
ю
Оч
ю
ю
4^
ОО
о
о
о
ЧО
-о
и>
р
о
и>
4*.
<-*
о
о
оо
о
^4
| 920
о
V
(л
О
ЧО
О
"ю
1—«
н~'
о
ьо
ІЛ
-4
О
4^
Оч
О
О
о
оо
1—*
00
о
о
ю
оо
<л
о
о
С\
ЧО
С\
006 І
о
V
и>
ОЧ
""^
о
Іо
о
ю
о
ьо
сл
1—»
сл
4^
■и
о
о
о
Оч
о>
00
о
о
ю
ю
ЧО
о
о
^л
00
4ь
1 880
о
V
ю
ьо
р
чо
ю
4*
о
"ю
4^
(Л
чо
4^
ю
о
о
о
<л
ю
Оч
о
о
^4
-^
о
о
4*
^4
к>
1 860
о
V
о
Оч
о
р
оо
и>
<л
о
ю
4^.
о
4^
о
о
о
о
и>
ЧО
ю
о
о
и>
о
о
о
и>
о^
<л
1 840
р
1*>
чо
о
-4
р
^4
4^
00
о
То
и>
4^
о
и>
00
о
о
о
ю
^л
ю
о
о
о
00
00
о
о
к>
ол
00
1 820
р
"и>
-4
^Л
и>
р
Оч
о>
р
Іо
ю
•^1
00
и>
о\
о
о
о
оч
^4
О
о
о
1^1
и>
о
о
о\
ІЛ
1 800
р
!*>
<л
ЧО
-4
Р
*л
^4
4*
О
"ю
ю
и>
и»
4^
о
о
о
о
00
ю
о
о
о
ю
Оч
о
о
о
00
Оч
1 780 І
р
1*>
4^
и>
ЧО
р
4^
00
00
р
Іо
Е
Оч
и>
ю
о
о
о
о
ю
4^
О
о
о
о
^4
О
о
о
ю
ЇЛ
1 760
о
и>
ю
оо
о
р
■и
о
и>
о
ю
о
«^
^1
и>
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
1 740 1
р
І**
)—*
1—»
ЧО
р
и>
*•+
ЧО
О
Іо
о
о
<л
^
7*
£
§
^— 1
о
ш
Ч
о
•^
К
^\
^1
-01
їм
•^
, *.
о
в
о
5^ її 8
^ §
3 Юо
^ 1
+ й °
£м,
[О
+
£
•Г>
II
£
+
£>
^
+
а
ю
N
ч-
р
чо
к^
о
) .
о
(—к
-^
00
о
Оч
оо
о»
о
о
45»
о
и>
ю
оч
оо
о
и>
о
оч
чо
о
І/І
00
<-л
о
4*.
45»
45»
и>
чо
о
о
| ■
о
о
оо
V©
о
Оч
-4
<-*
ю
о
<^
чо
ЧО
оо
ю
оч
ОЧ
о
и>
о
о
оч
2
00
Оч
о
45»
45»
і>і
45»
00
00
о
■ ■
о
о
о
о
о
(^
оч
Оі
<У1
о
и>
ЧО
оо
ІЧ)
ю
оч
45»
о
ю
ЧО
и>
оо
о
и>
00
•^4
О
45»
45»
Іч)
С\
00
Оч
о
о
ЧО
ЧО
о
о
ОЧ
<-к
о»
00
о
и>
чо
Оч
оч
ІЧ)
Оч
ю
о
го
00
-^
1—1
о
Іч)
00
ЧО
о
45»
45»
^4
00
4*
О
о
-8
ЧО
О
£
Оч
О
о
и>
ЧО
4*
ЧО
ю
оч
о
о
Іч)
00
о
4*
О
ЧО
О
О
45»
45»
О
ЧО
00
ю
о
о
ЧО
«*4
ю
00
о
Оч
и>
Оч
и>
о
и>
ЧО
и>
и>
ю
<-*
00
о
ІЧ)
-^
и>
о\
о
о
ЧО
•""'
О
і
8
00
о
о
о
ЧО
оч
и»
оч
о
оч
ю
Оч
ОЧ
о
и>
ЧО
<-*
ю
^
оч
о
к>
оч
Оч
-^
о
ЧО
чо
ЧО
и>
о
4*»
и>
ЧО
о
-^
00
о
о
ЧО
оі
4*»
и>
о
оч
і—*
оч
ЧО
О
и>
00
чо
00
ю
<-*
45»
о
ю
и>
ЧО
-^
О
ЧО
00
ЧО
4*
О
4*
и>
00
<і
ОЧ
о
о
ЧО
45»
45»
ЧО
О
Оч
О
^
•""'
о
и>
00
00
о
ю
<у»
іч>
о
Іч)
сл
ю
^4
о
чо
«^
ЧО
<-*
о
45»
и>
-4
ю
<і
4*
О
о
ЧО
и>
<-*
и>
о
<-л
чо
^і
4*»
О
и>
оо
Оч
и>
ю
і^і
о
о
ю
4*»
иі
00
о
ЧО
С\
чо
^4
О
4*»
и>
оч
и>
<і
Іч)
О
О
ЧО
Гч)
<Уі
00
о
<-д
оо
•^
-4
о
и>
00
4*
45»
Іч)
4*»
00
о
ю
и>
00
оч
о
ЧО
<-*
ЧО
00
о
4ь
и>
^
и>
^
о
о
о
чо
<^
ю
о
<-*
^4
00
о
о
и>
00
Іч)
^л
ю
45»
о\
о
к>
и>
►—»
<-*
о
ЧО
<-*
О
о
о
45»
и>
і
Оч
00
о
о
ЧО
о
оч
<-к
о
<-*
Оч
00
4^
о
и>
00
о
ОЧ
ю
4^
4^
О
Іч)
Іч)
^
О
ЧО
4*
О
О
4ь
и>
и>
4^
ОЧ
С\
о
о
оо
ЧО
^
<^
о
о»
<-ГІ
00
^
о
и>
^і
оо
и»
ю
4^
Іч)
О
Гч)
^
^"^
о
ЧО
и>
о
и>
о
4ь
и>
Іч)
4^
ОЧ
4ь
О
О
00
00
0\
^4
О
и»
4^
ЧО
О
о
и>
-^4
(^
С\
Іч)
4^
О
О
Іч)
О
чо
00
о
ЧО
ю
о
4ь
О
4ь
и>
ь^
4^
С\
ю
о
о
00
--4
<^
-4
О
О»
и)
чо
и>
о
и>
^4
4^
и»
ю
и>
00
о
Іч)
О
ю
С\
о
чо
о
<^
о
4ь
и>
о
и)
ач
о
о
о
00
о\.
ОЧ
и»
о
сл
ІЧ)
ЧО
0\
о
и>
-^
Іч)
4^.
Іч)
и)
ач
о
ЧО
^
|--
о
ЧО
О
о
^4
О
4ь
Іч)
ЧО
и>
<-к
оо
о
о
00
<-л
ач
и>
о
сл
ІЧ)
О
о
о
и>
^4
о
о
ю
и>
£»
о
00
"^
•^
о
00
чо
о
чо
о
£ь
Гч)
00
К)
<-*
Оч
о
о
00
4*
а»
ЧО
о
^
1—1
2
о
и>
Оч
00
о
^
и>
ю
о
00
о
ю
о
оо
00
Ь—к
н—
о
4ь
Гч)
•^
ю
^
4^
о
о
00
и>
СЛ
^г»
о
^
о
о
00
о
и>
Оч
и>
00
Іч)
и>
о
о
-*4
N4
-4
О
00
^
(—І
їч)
О
4ь
Іч)
Оч
^
Ю
О
о
00
Іч)
^
о
о
4*
ЧО
■-*
О
и>
Оч
и)
4^
ю
Іч)
00
о
Оч
^
н-
о
00
Оч
4ь
О
4^
Ю
и»
О
СЛ
О
о
о
оо
І—»
£
о
4*
00
Н-*
иі
о
и>
ач
1—4
ю
ю
Оч
о
а»
-*4
4^
О
00
<-л
і—»
ач
о
4*
Ю
и>
ЧО
4^
00
о
о
00
о
и)
ач
о
4*
-^
чо
О
и>
^
00
ач
ю
ю
4^
О
4*>
ЧО
^4
О
00
4ь
(—»
--4
О
4^
Ю
Ю
•^4
4^
Оч
О
О
-0
ЧО
ю
00
о
4*
Оч
Гч)
и>
о
и)
^Л
Оч
ю
ю
Гч)
Ю
О
£ь
Іч)
О
О
00
и>
•—»
ЧО
О
4*
Іч)
<-*
^
О
О
-^
00
Н—'
00
о
4*
ІЛ
Іч)
оо
о
и>
<-л
и>
^
ю
Іч)
о
о
и>
4*.
Іч)
О
оо
ю
Іч)
|"-
о
4^
Іч)
О
и>
4^
Іч)
О
О
-*1
^4
О
ОО
о
і
и>
Іч)
о
и>
сл
Ь—к
|-—
ю
00
о
Іч)
ач
и)
о
00
і—»
ю
и>
о
4*
ЧО
*""'
4*
О
О
О
-4
О»
ЧО
Оч
О
4*
и>
и>
Оч
О
о^
4ь
00
4і.
^о
Оч
О
к—І
оо
и>
о
оо
о
Гч)
V*
О
4*
^4
ЧО
и>
00
о
о
^1
4^
00
ю
о
4^
ю
4^
О
о
и>
ЧЛ
4^
<І
Іч)
і—>
4^
О
і—*
О
и>
о
-4
ЧО
Іч)
•^
О
4^
Оч
Оч
и>
Оч
О
О
^4
и>
ОЧ
оо
о
4^
к—к
4^
оч
О
и>
4^
Іч)
оо
Іч)
►—к
Іч)
О
О
Іч)
Ю
О
-^1
оо
Іч)
чо
О
4^
и»
и>
и>
4^
О
о
^4
Іч)
1-^
и>
о
4^
О
^
"""'
О
и)
4^
о
о
Іч)
»—к
О
о
о
ЧО
4^
О
^4
->4
и>
о
о
4*
4*
1—1
и>
ІЧ)
о
о
-4
и>
-4
О
и>
чо
^
Оч
О
и>
и>
<і
ю
о
оо
о
о
оо
^л
00
о
-4
Оч
и>
и>
о
4^.
Ю
ОО
и>
о
о
о
-4
О
к—к
ЧО
О
и>
оо
ОЧ
о
о
и)
и>
45-
о
Іч)
О
оч
о
о
^1
--4
оч
о
^4
^
и>
и»
о
4^
і—к
»—к
4^
Ю
ОО
о
о
Оч
оо
ЧО
чо
о
и>
-*4
ач
оч
О
и>
и>
к—к
о
Іч)
О
4^
О
О
оч
чо
ю
о
-^1
4^
и>
^4
О
4^
і—к
О
*~-
Іч)
Оч
О
О
Оч
<І
^4
ЧО
О
и>
ОЧ
^і
Гч)
О
и>
Іч)
^4
ОО
Іч)
О
Іч)
О
о
ач
о
чо
о
^4
^
и>
ЧО
о
45»
О
ОО
--4
Іч)
45*
О
О
ач
оч
с*
^і
о
и>
сл
~-4
-4
О
СО
ю
45-
4^
Ю
О
О
о
о
^
Іч)
4^
О
-4
Ю
4і-
О
45*
О
-4
и>
Іч)
го
о
о
Оч
<-л
и)
45*
о
и>
45»
оо
о^
о
и-
ю
»—к
ІЧ)
ЧО
ОО
О
2
и>
ЧО
О
--4
»—к
45»
и>
О
45»
О
<-К
00
Іч)
О
о
о
£
О
ЧО
о
и>
и)
00
ЧО
о
и)
і—к
^4
^І
ЧО
Оч
О
о
и>
^
Іч)
О
^4
2
ач
о
45»
О
45»
45»
00
о
о
Оч
Ю
00
45»
О
и>
ю
чо
ач
о
и)
►—*
45»
Ю
"Р
45»
О
О
Іч)
ач
іл
о
ач
чо
45»
оо
о
45»
О
ю
ЧО
^
*
н
*:
к—»
О
«і*
*;
•
О
^
*
£
*
о
^
N1
|~-
О
о
о
1
42
Закінчення таблиці 6.3
г,к
2700
2720
2740
2760
2780
2800
2820
2840
М0
1,3138
1,3204
1,3269
1,3334
1,3398
1,3463
1,3526
1,3590
Л/,-10-3
1,0684
1,0782
1,0881
1,0980
1,1079
1,1178
1,1277
1,1375
М_2'105
0,4451 1
0,4459
0,4467
0,4475
0,4483
0,4491
0,4498
0,4506 |
Т\К
1 2860
2880
2900
2920
2940
2960
2980
3000
Л/0
1,3652
1,3714
1,3777
1,3838
1,3899
1,3960
1,4021
1,4082
Л/,-10-3
1,1474
1,1573
1,1672
1,1771
1,1870
1,1969
1,2068
1,2167
М.210М
0,4513
0,4520
0,4527
0,4535
0,4542
0,4549
0,4555
0,4562
Безпосереднє експериментальне вимірювання ДС можливе
електрохімічним методом, якщо реакцію можна провести в гальванічному
елементі. Електрохімічна рівновага розглядається в наступному розділі.
111
Розділ 7.
ЕЛЕКТРОХІМІЧНА РІВНОВАГА
Як енергію хімічну
Перевести в електричну?
Зробить це в один момент
Гальванічний елемент.
7.1. Термодинаміка гальванічного елемента
Окисно-відновну (гесіох) реакцію можна провести у гальванічному
(електрохімічному) елементі. Розглянемо один з таких елементів — елемент
Даніеля-Якобі (рис. 7.1). Цей елемент складається з двох напівелементів:
цинк у розчині солі Цинку (2п804) та мідь у розчині солі Купруму (Си804).
Схематично це записується таким чином:
~2п|2п804||Си804|Си+.
В цьому елементі перебігає така хімічна
реакція:
2п + Си804 = 2п804 + Си.
На лівому електроді цинк віддає електрони
у зовнішнє коло, переходячи в іони Цинку, а на
правому електроді іони Купруму, відповідно,
отримуючи електрони, відновлюються до
металічної міді. Таким чином, напівреакції окисно-
відновного процесу такі:
Якобі Борис Семенович
(1801-1874), Росія
2п°
► 2п2+ + 2е;
Си2+ + 2е-
► Си°
2п(тв.)-»2п (водн.) + 2е
(водн.) + 2е -»Си(тв.)
Рис. 7.1. Елемент Даніеля-Якобі
Ці напівреакції у гальванічному
елементі розділені у просторі й
перебігають окремо.
Напруга на розімкнених кінцях
елемента відповідає його
електрорушійній силі (ЕРС)—Е. Для
працюючого гальванічного елемента Е є
додатною величиною (Е> 0).
Гальванічний елемент можна визначити як
прилад для перетворення хімічної
енергії в електричну. Електрична
енергія, що виробляється
гальванічним елементом (робота
гальванічного елемента), дорівнює зміні віль-
112
ної енергії Гіббса (або вільної енергії Гельмгольца, бо для конденсованих
фаз ДС? = АА) хімічної реакції, що перебігає в елементі. Отже,
АС = -гРЕ,
■ число Фарадея (96485 Кул/моль-екв); і -
(7.1)
де Р— число Фарадея (96485 Кул/моль-екв); 2 — кількість електронів, що
приймаються (віддаються) в розрахунку на 1 іон (атом). Умові самочинного
перебігу гесіох процесу в гальванічному елементі (АС < 0) відповідає, таким
чином, Е > 0.
В гальванічному елементі не вся хімічна енергія перетворюється на
електричну, частина (так звана зв'язана енергія) перетворюється на теплоту.
Таким чином, гальванічний елемент дає яскравий приклад розділення
повної енергії (Д#= АЦ) на вільну (АС = М) та зв'язану (ГА5), остання не може
перетворюватися на електричну роботу. Зазначимо, що все це стосується
оборотного гальванічного елемента і, відповідно, оборотного гесіох
процесу. Хімічну реакцію між цинком та розчином солі Купруму можна
провести необоротно, наприклад, вкинувши шматок цинку у розчин сульфату
Купруму, при цьому вся хімічна енергія перетвориться на теплоту.
Проведення хімічної реакції у гальванічному елементі дає можливість
визначити не лише вільну енергію, а й інші термодинамічні функції.
Наприклад, для зміни ентропії (А5) гесіох реакції, використовуючи співвідношення
(1.75) та (7.1), маємо
^ .-&(*>>,-*(«
02)
Таким чином, необхідно додатково мати інформацію про
температурний коефіцієнт ЕРС. Далі можна розрахувати зміну ентальпії за відомим
співвідношенням (1.72)
Ш = АС + ТА8 = 2Р\тШ;\ -Е
(7.3)
Для ентальпій є можливість співставлення результатів електрохімічного
і термохімічного вимірювань. Відповідні дані для деяких реакцій при
кімнатній температурі наведено у таблиці 7.1.
Таблиця 7.1. Обчислення АЯ через ЕРС і термохімічно
Реакція
Сгі+РЬС12=СсіС12+РЬ
ІА§+1/2Н§2С12=А§С1+Н§
Р<і+2А§С1 = РсіС12+2А8
£, В
0,1880
0,0455
0,4900
Ц-ю4,в/к
-4,80
+ 3,38
-1,86
АЯ, кДж/моль
електрохімічно
-63,75
+ 5,33
-105,2
термохімічно
-61,24
+ 7,94
-101,0
Результати АЯдля наведених реакцій, що отримані різними методами,
непогано збігаються між собою. Вважається, що електрохімічний метод дає
більш точні результати, ніж термохімічний.
113
Величина ДС?, згідно з рівнянням ізотерми хімічної реакції (6.6), залежить
від концентрацій реагентів та продуктів
АС = АС°+КТАІпа; = АС°+КТ\а1[Іа^. (7.4)
З урахуванням (7.1) це дає для ЕРС '
Ав =
2р
АС°
2р
-^и^=^-тпЦаг
2р
(7.5)
Рівняння (7.5) відоме під назвою рівняння Нернста для ЕРС. Величина Е°
має назву стандартної ЕРС, тобто це значення Е при всіх аІ, = 1.
При 298 К рівняння Нернста має спрощений вигляд
Е = Е°-
0,059
і§ГК
С7.-6)
Стандартна ЕРС безпосередньо пов'язана
з константою рівноваги відповідної окисно-
відновної реакції
АО° = -КТІпК = -гРЕ0, звідки
гРЕ0
ІпК = ;
КТ
(7.7)
Нернст Вальтер Фрідріх Герман
(1864—1941), Німеччина
Для кімнатної (298 К) температури,
відповідно
1вК = ~ф§- о (7-8)
Наприклад, для елемента Даніеля-Якобі при 25 С маємо Е= 1,10В
відповідно,
37
^=|іг37'2^=1'9'
10'
Таке велике значення константи рівноваги означає, що реакція взаємодії
металічного цинку з Си804 практично необоротна.
Елемент Даніеля-Якобі складається, як уже зазначалось, із двох напів-
елементів типу: метал—розчин солі того ж металу Такі напівелементи
мають назву електроди 1-го роду. Властивості електродів буде розглянуто
у наступному підрозділі, а зараз перелічимо лише типи електродів. Крім
електродів першого роду, існують також електроди 2-го роду, газові та окисно-
відновні.
Електроди 2-го роду — це метал плюс важкорозчинна сіль того ж
металу плюс легкорозчинна сіль з однаковим аніоном.
х Наприклад, хлор-срібний електрод — А§ | А§С11КС1; каломельний
електрод — Н§ | Н§2С121КС1.
Газовий електрод — електрод з інертного металу (наприклад Рі), в якому
розчиняється газ (наприклад водень) і відповідна форма того ж елемента
в розчині.
Приклади: водневий електрод — Рї (Н2) | Н (НС1); хлорний електрод —
Рі(С12)|СГ(НС1).
114
Фактично, всі електроди є окисно-відновними системами, але
домовились окисно-відновними (гесіох) електродами називати лише ті, де обидві
форми елемента (окиснена та відновлена) знаходяться у розчині, а електрод
з інертного металу лише надає їм можливість обмінюватися електронами.
Приклади окисно-відновних електродів: Рі | Ре , Ре ; Рі | 8п , 8п тощо.
Складемо елемент з водневого та хлор-срібного електродів
РІ(Н2)|НС1|А8С1|А8.
Для цього елемента при кімнатній температурі
£ = £°-0,0591§аНСІ =
= £°-0,0591§(а±)2=£о-0,1181§т-0,1181§у±.
(7.9)
0,276 г
Вимірювання ЕРС при відомому значенні Е° дає можливість обчислити
середній коефіцієнт активності (у±).
Позначимо через /= Е + 0,118 \%т. Ця функція прямує до Е°, коли у±—> 1
(1§У±—»0), тобто у нескінченно розбавлених розчинах. Оскільки за законом
Дебая-Гюккеля (4.86) 1§у± пропорційний V/, то для 1:1 валентного
електроліту Е° можна знайти екстраполяцією в
координатах / — у[т. Дані, що
наведені на рис. 7.2, добре ілюструють це
положення. Таким чином, вимірювання
ЕРС дає можливість
експериментального визначення коефіцієнтів активності
електролітів.
Якщо гальванічний елемент
складено з двох однакових електродів, то
тотожно Е° = 0, але Е може не
дорівнювати нулеві за рахунок різних
концентрацій електроліту в напівелементах.
Складені таким чином гальванічні
елементи називають концентраційними.
Для концентраційних елементів при
Е°= 0 з рівняння Нернста (7.5) маємо
0,268
Рис. 7.2. Визначення Е°
екстраполяцією
р= ЯТ і уДі /продукти __
2р (а.)У*
\іЛ^ /реагенти
реагенти
\ ' /по
(7.10)
/продукти
Процес перебігає самочинно, коли у (7.10) Е > 0, тобто чисельник
більший за знаменник — концентрація «реагентів» більша за концентрацію
«продуктів». Як «реагент» у концентраційному елементі виступає напівеле-
мент із більшою концентрацією розчину, а як «продукт», відповідно, з
меншою. Процес перебігає до повного вирівнювання концентрацій в обох
напівелементах, при цьому Е = 0 (АС = 0 — умова рівноваги).
115
Розглянемо конкретні приклади. Нижче наведено електрохімічне коло,
складене з двох гальванічних елементів, що розглядалися вище при
визначенні коефіцієнтів активності, з різною концентрацією НС1
А§ | А§С11НС11 Рі (Н2) — Рі (Н2) | НС11А8С11 А§. (а)
Якщо то а{>а2, то
£ = Ж1п^ = Ж1п(Ц = 2|І1п(^.. (7.11)
Р а2 Р {а±)\ Р {а±)г
При роботі елемента концентрація НС1 ліворуч (а{) поступово
зменшується за рахунок реакції
НС1 + А§ = А§С1 + 1/2Н2.
Відповідно, праворуч концентрація НС1 (а2) поступово збільшується за
рахунок протилежної реакції
^Нг + АзСІ^Аз + НСІ.
Розчини НС1 різної концентрації не знаходяться у безпосередньому
контакті, тому це є електрохімічне концентраційне коло без переносу іонів.
Для концентраційних елементів з переносом
Рї(Н2)|НС1|НС1|(Н2)Рі (б)
між розчинами НС1 різної концентрації є контакт. Це приводить до того, що,
крім процесів на електродах, слід враховувати перенос іонів з одного розчину
в інший. Вважаємо знову ах > а2. Тоді ліворуч відновлення 1-го моль-екві-
валента Н іонів (розрахунок на 1Р електрики) частково компенсується
приходом /+ моль-екв. Н з правої частини елемента, отже, зменшення кількості
іонів Н+ дорівнює 1 -/+ = /_. Відповідно, і_ іонів СГ, переходить направо.
Отже, зменшення концентрації НС1 ліворуч дорівнює 1_ НС1. Праворуч
приходить с СГ, переходить у розчин 1 моль-екв водню, що, однак,
зменшується на /+Н за рахунок переносу. Отже, збільшення кількості Н
дорівнює 1 - і+ = 1_ і загалом кількість НС1 праворуч збільшується на (_
еквівалентів. Таким чином, для ЕРС концентраційного кола з переносом маємо
£<=2^11пі£±1 (7Л2)
Р (я±)2
При контакті розчинів за рахунок різної швидкості переносу іонів на межі
поділу розчинів різної концентрації виникає дифузійний потенціал
*-^ь^-^"^- (7ЛЗ>
гЛ (а-)і (°+)і (я+)і
Вважаючи т—г- = ; ч = / ч , маємо
(а_)2 (а+)2 (а±)2
116
Ф,=(<--0^1п£^, (7.14)
При і+ = (_ (р0 = 0, отже, дифузійний потенціал можна усунути за
рахунок використання електроліту, для якого рухомості катіона та аніона
однакові. До таких електролітів відносяться КС1 та КМ03. При розміщенні між
розчинами сольового містка з КС1 або КЖ)3 у концентраційному елементі
з переносом усувається дифузійний потенціал. Це позначається таким
чином:
Р*(Н2)НС1||НС1(Н2)РІ. (в)
а\ а2
ЕРС такого елемента дорівнює
£» = £'-Ф0=(2,.-,.+О^Ь^І = Ж,„і^. (7|5)
Цікаво відзначити, що для концентраційного кола (в) ЕРС (£") удвічі
менша, ніж для концентраційного кола (а). Це є наслідком того, що в першому
колі (а) зміна концентрації всього електроліту (НС1) відбувається повністю
за рахунок гесіох процесів, для кола (в) окисно-відновні процеси
забезпечують зміну концентрації лише Н іона, а зміна концентрації іона СГ
відбувається шляхом безпосереднього переносу з розчину в розчин.
Специфічним концентраційним елементом є такий:
Р* (Н2) | НС1 (0,01 М) Ц КОН (0,01 М) | Р* (Н2).
Для цього елемента, згідно з формулою (7.15), маємо
£ = Ж1п^н!)і,але(ан+)2=7-^г,
Р (ян*Ь Н Чж-Ь
де Кю—іонний добуток води. Отже, маємо
£ = Ж1п(У)'>Н-)2
Визначення ЕРС цього елемента при Т = 298 К дало величину Е -
= 0,5874 В, а визначення середнього коефіцієнта активності для 0,01 М
розчину дало у± = 0,93 (відзначимо, що розрахунки за формулою Дебая-Гюк-
келя (4.86) дають у цьому випадку близьке значення у± = 0,89). З
використанням отриманих значень ЕРС та активностей маємо для іонного добутку води
при 25 °С К„ = 1,02 -10". Отже, вимірювання ЕРС дає досить точний метод
визначення^.
7.2. Електроди та їх потенціали
Стандартна ЕРС (Е°) може бути розбитою на стандартні потенціали
(<р°) окремих електродів. При визначенні стандартного потенціалу, як і для
стандартної ЕРС, всі а( = 1. Нулем відліку для стандартних потенціалів
вважають стандартний потенціал водневого електрода, тобто Фн+/н = ^*
2 117
Таким чином, стандартний потенціал деякого електрода—це ЕРС
гальванічного елемента, який складається з даного електрода (справа) і водневого
електрода (зліва) при всіх я, = 1. Наприклад, Ф2п2+/2п — це ЕРС елемента
РІ(Н2) | Н+ || 2п2+ | 2п.
(ан+=1) (я2п2+=1)
Для будь-якого гальванічного елемента стандартну ЕРС можна скласти
зі стандартних потенціалів (ф°) таким чином:
£0=Фправ.-ф°лів, (7Л6)
(Якщо Флів. =Фн+/н2 ==0'ТО Е° =Фправ.> як зазначалося вище.)
Наприклад, для елемента Даніеля-Якобі
2п | 2п2+ Ц Си2+ | Си
при 298 К маємо
Е° =сС-/си "Ф^+/2п =+0,337-(-0,763) = 1Д0В.
У лівого негативно зарядженого електрода перебігає процес окиснення
(2п° —» 2п + 2е), у правого позитивно зарядженого електрода відбувається
процес відновлення (Си + 2е —> Си°).
Оскільки Е° > 0, то АО°=-гРЕ° < 0 і в стандартних умовах (я2п2+ = аСи2+ =
= 1) реакція 2п° + Си = 2п + Си° перебігає самочинно.
Для реакції Си° + 2п = 2п° + Си , в гальванічному елементі
Си | Си2+ Ц 2п2+ | 2п.
(ЯСи2+=1) («2п2+=1)
£о=-1,10 В і, отже, АО°> 0, тобто ця реакція в стандартних умовах не
перебігає самочинно («термодинамічно заборонена»).
У таблиці 7.2 наведено стандартні електродні потенціали для
електродів різних типів. Слід зазначити, що стандартні потенціали наведено для
процесу відновлення, наприклад, для електродів 1-го роду це процеси типу:
2п2 + 2е = 2п°; Си2+ + 2е = Си° тощо.
Розглянемо тепер електродні потенціали для довільних концентрацій
електроліту. Так само, як і для стандартних ЕРС і потенціалів (формула (7.16))
ЕРС у загальному вигляді складається з потенціалів електродів таким чином:
£ = Фправ.-<Рлів, (7Л7>
Складемо елемент з електрода 1-го роду з довільною концентрацією
(активністю) електроліту (#Ме2+) і стандартного водневого, тобто
Рі(Н2) | Н+ || Ме2+ | Ме,
(%+=1) (ЯМе2+)
де Ме — деякий метал.
118
Таблиця 7.2. Стандартні електродні потенціали (у В) у водному середовищі
Електродний
процес
ІА£+ + е->А§
А&С1 + є -> А§ + СГ
А§2С03 + 2е -> 2А§ +С032~
А§2804 + 2е -> 2А& + 8042~
Ві3+ + Зе -> Ві
Си+ + є -> Си
Си2+ + є -» Си+
Си2+ + 2е -> Си
Си8 + 2е -> Си + 82~
2Си8 + 2е -> Си28 + 82~
Си28 + 2е -> 2Си + 82~
Ре2+ + 2е->Ре
і- 3+ . і- 2+
Ре + є —> Ре
Ре3+ + Зе->Ре
РеСОз + 2е -> Ре + СОІ"
ос-Ре8 + 2е -> Ре + 82"
Р-Ре8 + 2е -> Ре + 82"
2Н20 + 2е -> Н2 + 20Н"
Н§2+ + 2е-^Н§
Не8 + 2е-^Н§ + 82"
Мп2+ + 2е -> Мп
02(г) + 2Н20 + 4е-^40Н"
РЬ2+ + 2е -» РЬ
ос-8 + 2е -» 82"
(3-8 + 2е -> 82"
8 (р) + 2е -^ 82"
8е + 2е -> 8е2"
иоГ+2е-^ио2
у3+ + е->У2+
У3+ + Зе-^У
2п2+ +2е -> 2п
298,15
0,799
0,222
0,472
0,653
0,317
0,518
0,159
0,338
-0,722
-0,553
-0,892
-0,478
0,771
-0,062
-0,790
-0,965
-
-0,828
-0,852
-0,707
-1,192
-0,401
-0,126
-0,444
-
-
-0,67
0,366
-0,255
-0,835
-0,763
Температура,
323,15
0,774
0,205
0,435
0,623
0,320
0,498
0,177
0,338
-0,745
-0,573
-0,917
-0,478
0,800
-0,052
-0,824
-0,983
-
-0,850
-0,849
-0,726
-1,195
0,358
-0,136
-0,467
-
-
-0,70
0,371
-0,245
-0,829
-0,760
373,15
0,721
0,160
0,351
0,551
0,327
0,455
0,216
0,336
-0,806
-0,63
-0,985
-0,479
0,856
-0,034
-0,904
-1,04
-
-0,907
-0,844
-0,78
-1,20
0,260
-0,154
-
-0,528
-
-0,78
0,382
-0,221
-0,817
-0,756
К
423,15
0,65
0,104
0,25
0,46
0,33
0,41
0,26
0,333
-0,89
-0,70
-1,08
-0,48
0,91
-0,02
-1,00
-
-1,12
-0,980
-0,84
-0,86
-1,21
0,148
-0,17
-
-
-0,61
-0,87
0,396
-0,19
-0,80
-0,75
473,15
0,59
0,035
0,14
0,36
0,34
0,36
0,30
0,33
-0,99
-0,80
-1Д9
-0,49
0,97
0,00
-1,11
-
-1,23
-1,068
-0,84
-0,95
-1,22
0,021
-0,18
-
-
-0,72
-0,98
0,41
-0,15
-0,79
-0,75
119
При 298,15 К
Електродний
процес
1 А§Вг + є -> А§ + ВГ
А§СN + є -» А§ + СК~
А§1 + є —» А§ + Г
А13+ + Зе->А1
Аи3+ + Зе -> Аи
Ва2+ + 2е -> Ва
Вг2(р) + 2е->2ВГ
Вг2(вод) + 2е->2ВГ
Са2+ + 2е -> Са
Са(ОН)2 + 2е -> Са + 20Н"
С12(г) + 2е->2СГ
С12(вод) + 2е->2СГ
Со2+ + 2е -» Со
Со3+ + є -> Со2+
Сг2+ + 2е->Сг
Сг3+ + е->Сг2+
Сг3+ + Зе-»Сг
СиСІ + є -» Си + СГ
Р2(г) + 2е->2Р~
Ое2+ + 2е -» Ое
2Н+ + 2е-»Н2
Н&С12 + 2е -> 2Н§ + 2СГ
Н§2804 + 2е -» 2Н§ + 804~
І2(тв) + 2е-»2Г
К+ + є -» К
М§2+ + 2е -> М§
Т= 1
= 298,15 К
+0,071
-0,017
-0,152
-1,663
+1,498
-2,905
+1,065
-1,087
-2,866
-3,02
+1,359
+1,395
-0,277
+1,808
-0,913
-0,407
-0,744
+0,137
+2,87
0,000
0,000
+0,2676
+0,615
+0,536
-2,924
'-2,363
Електродний
процес
Мп3+ + е-»Мп2+
МпСОз + 2е -> Мп + СО2"
Мп04 + є -»Мп04~
МпО>8Н++5е->Мп2++4Н20
Иа+ + є —> Ш
Ші2+ + 2е-»№
ШіСОз + 2е -> № + СО2"
РЬСОз + 2е-^ РЬ + СО2-
РЬС12 + 2е -> РЬ + 2СГ
РЬ804 + 2е-» РЬ + 804~
РЬ4++ 2е-> РЬ2+
ра2++ 2е-> ра
Р*2+ + 2е->Рі
Ка2+ + 2е -> Ка
28 + 2е-»82~
Зп2+ + 2е -> 8п
8п4+ + 2е -> 8п2+
8п8 + 2е -> 8п + 82~
Ті2+ + 2е->Ті
Ті3+ + е->Ті2+
Ш3+ + 3е->и
\\Ґ+ + е->1]3+
У2+ + 2е->У
2пС03 + 2е -> 2п + СОз"
2п8 + 2е-> 2п + 82~
Т =
= 298,15 К
+1,509
-1,48
+0,564
+1,507
-2,714
-0,250
-0,45
-0,506
-0,268
-0,356
+1,694
-0,98
+1,188
-2,92
-0,470
-0,13
+0,15
-0,94
-1,63
-0,36
-1,79
-О,60
-1,17
-1,06
-1,44
ЕРС цього елемента дорівнює потенціалу правого електрода. Для ЕРС
з рівняння Нернста (7.5) маємо
Е = Ео_Щ1п(аОщ>оЯу™ (718)
(а Л *
ч^у /реагенти
Для електродного потенціалу, відповідно, для напівреакції Ме2 + ге = Ме°
ф = Фо-^1п-^-. (7.19)
2Ґ амег+
Оскільки аМсо є константою, то для потенціалу електрода 1-го роду маємо
120
ф = Фо+|£іпаМе2+. (7.20)
Ф = Фо+ІЬ^І8«м*+. (7.21)
2р Ме:
При 298 К рівняння (7.20) спрощується
0,059
2
Для водневого електрода відповідною напівреакцією є Н + є = !/2Н2.
Отже,
(Р = (Р°н+/н2 +1Г1п V =1^ V' (7-22)
ПриТ = 298К (Фн+/н2 = 0) маємо
ф = 0,0591§ян+ = -0,059рН. (7.23)
Отже, вимірювання потенціалу водневого електрода дає можливість
електрохімічного кількісного вимірювання рН розчину.
Для водневого електрода в стандартному стані стандартною повинна
бути не лише концентрація розчину (ян+= 1), але й тиск молекулярного
водню (Рщ = 1 атм). В загальному випадку зміну Рщ слід враховувати при
визначенні потенціалу водневого електрода (в рівнянні (7.22)), тобто
<р=^7^- (724)
Для іншого газового електрода — хлорного — маємо такий процес
відновлення на електроді:
!/2СІ2 + е-»СП
Це дає для потенціалу хлорного електрода
рУ2
Ф = Ф°+^1п-^. (7.25)
сг
При РС[ = 1 атм це рівняння спрощується
Ф = Ф°-^1пасг. (726)
Таким чином, для електродів 1-го роду і для водневого потенціал
залежить від концентрації певного катіона, тобто ці електроди є оборотними
відносно катіона.
Для хлорного електрода потенціал визначається концентрацією хлорид-
іона, цей електрод є оборотним відносно аніона.
Аніони є потенціалвизначаючими іонами і для електродів 2-го роду.
Справді, наприклад, для хлор-срібного електрода А§ А§С11КС1 маємо
Ф = (ф°)Ч^1п V =«Р°)'+Ір1п^ = Ф0-^1пас1- (7-27)
Тут через ДР = аА„+ • аСҐ позначено добуток розчинності А§С1.
121
Оскільки всі потенціали є відновлюючими, то «реагентом» виступає
окиснена форма, а «продуктом» — відновлена. Це призводить до того, що
в рівнянні Нернста для потенціалу окиснена форма (аох) завжди має перед
логарифмом знак «плюс» (знаходиться в чисельнику), а відновлена (аге{і)
«мінус» (або пишеться у знаменнику), тобто
о КТг Ш*<»)У/
Цей запис зручно використовувати для окисно-відновних електродів.
Наприклад, для електрода Рі | Ре2+, Ре3+ маємо
ф = ф0+^1іп^!і. (7.29)
Розглянемо далі, як приклад окисно-відновного електрода, хінгідронний
електрод.
Реакція відновлення хінону (х) до гідрохінону (гх)
С6Н402 + Н2 = СбН4(ОН)2
у розчині перебігає таким чином:
С6Н402 + 2Н+ + 2е = С6Н4(ОН)2.
Потенціал хінгідронного електрода дорівнює (при Т= 298 К)
ф = фо + М591§^Чі. (730)
Гідрохінон у розчині дисоціює, але слабо: С6Н4(ОН)2 <=> 2Н+ + С6Н4 О^".
В дослідах використовують хінгідрон
О ОН
о он
який є молекулярною сполукою зі співвідношенням хінону до гідрохінону
як 1:1. Отже, виконується умова ах = агх. Це призводить до спрощення
рівняння (7.30)
ф = фо+М59,Еа2+ =(рО+05059ІЕян+ =ф°-0,059рН. (7.31)
Використовуючи стандартне значення електродного потенціалу
хінгідронного електрода (ф° г =0,699 В), за допомогою вимірювання його
потенціалу можна визначити рН. Цей електрод більш зручний у користуванні,
122
ніж водневий, але його застосування обмежене кислими та нейтральними
розчинами.
В сучасних рН-метрах для вимірювання рН використовують скляний
електрод. Вважається, що в поверхневому шарі скла при дії кислоти або води
відбувається часткове заміщення катіонів лужних металів у силікатному
кістяку скла на іони водню, що робить скляний електрод оборотним по
відношенню до іонів водню.
Деякі електродні потенціали важко або неможливо визначити
експериментально. Наприклад, рівноважний потенціал заліза відносно іонів
тривалентного заліза (Фрез+/Ре) У водному розчині безпосередньо визначити
неможливо, тому що у контакті з металічним залізом Ре відразу
відновлюється до Ре . Добре відомі, проте, електродні потенціали для електрода
1 -го роду Ре/Ре та для окисно-відновного Ре /Ре . Розглянемо це
детальніше. Процес відновлення Ре можна провести стадійно:
Ре3+ + е->Ре2+; (Г)
Ре2+ + 2е->Ре° (П)
або безпосередньо
Ре3++Зе->Ре°. (Ш)
Вільна енергія Гіббса є функцією стану системи, отже, її зміна не
залежить від шляху процесу, тому можна записати для стандартних АС0
З рівняння (7.1) маємо
2ІИфРе3+/Ре = *ІфРе2+/Ре + ^Фре^/Ре2*'
Використовуючи значення ФРе2+/Ре = -0,478 В і ФРез+/Ре2+ = 0,771 В,
отримуємо
2(РрЄ2+/Ге+(РрЄ3+/Ре2+
<Р°Ре-/Ре = 3 = -°'°62 В"
Такий спосіб розрахунку стандартних потенціалів має назву правило
Лютера.
Таким чином, електрохімічний підхід дає корисну інформацію, зокрема
для обчислення термодинамічних функцій, але цей підхід обмежується
окисно-відновними процесами.
Крім експериментальних способів визначення термодинамічних
функцій, їх можна обчислити теоретично за допомогою статистичної
термодинаміки, але для цього потрібна інформація про будову речовини, зокрема
спектроскопічні дані. До розгляду будови атомів, молекул, конденсованих тіл
з точки зору квантової теорії переходимо у наступному розділі.
123
Розділ 8.
КВАНТОВА ТЕОРІЯ, БУДОВА АТОМА
8.1. Основні положення квантової теорії
Частинка з хвилею зустрілися
І на дует перетворилися.
Виклад сучасних уявлень про будову речовини та хімічний зв'язок
логічно починати з відкриття електрона Дж. Томсоном. Це відкриття сприяло
появі іонної теорії хімічного зв'язку Косселя, згідно з якою утворення
хімічного зв'язку відбувається у два етапи:
1) відщеплення електрона від атома і перехід його на інший атом з
утворенням, відповідно, катіона й аніона;
2) сполучення протилежно заряджених іонів у молекули або кристали.
В. Коссель використав ідеї теорії валентності: кількість відщеплених або,
відповідно, приєднаних електронів і є валентністю елемента. Оскільки заряд
іона — це число, кратне заряду електрона, то знання величин зарядів
давало можливість кількісно розраховувати енергію зв'язку.
Наступний етап розвитку електронної теорії валентності — це гіпотеза
Дж. Льюїса про те, що в ковалентних сполуках рискам у структурних
формулах відповідає пара електронів. Наприклад, при утворенні молекули
водню з атомів кожний атом Гідрогену віддає по одному електрону і
виникає одна спільна електронна пара (Н : Н). У гомоатомних молекулах пара
електронів знаходиться на однакових відстанях від кожного атома, у гетеро-
атомних зміщена до більш електронегативного атома з утворенням
полярного зв'язку, наприклад, у НС1. Іонний зв'язок — це крайній випадок
полярного зв'язку, коли пара зв'язуючих електронів належить цілком одному
з атомів, наприклад, Хлору в молекулі Иа СГ, що еквівалентне переходу
електрона від атома Натрію до атома Хлору.
Електронна теорія дала змогу класифікувати хімічний зв'язок за типами:
ковалентний, полярний, іонний. Суттєвим у цій теорії було висунуте
Й. Штарком припущення, що в утворенні хімічного зв'язку беруть участь
не всі електрони атома, а лише зовнішні, так звані валентні.
Подальший прогрес став можливим після вивчення внутрішньої
структури самого атома і, отже, динаміки поведінки електронів в атомах
і молекулах на основі квантовохімічних уявлень. Формування сучасних
уявлень про атом почалося з праць Е. Резерфорда та Н. Бора. Проведені Е.
Резерфордом досліди з розсіювання позитивно заряджених а-частинок
довели, що основна маса атома сконцентрована у позитивно зарядженому ядрі,
розміри якого (~1(Г м) набагато менші, ніж розміри самого атома (-10" м).
Згідно з одержаними даними Е. Резерфорд запропонував планетарну
модель атома — у центрі розташоване масивне позитивно заряджене ядро,
навколо якого обертаються негативно заряджені електрони.
124
105 000 і
100 000 -^
95 000 і
90 000
2
85 000
25 000
20 000
15 000 {
10 000 Н
5 000-Г
А,, нм
-97,253
■102,572 Ч
■121,567 -1
серія
Лаймана
-434,05
-486,13
-656,28
Ну
_]серія
Бальмера
(видима
І область)
на
Найпростішим з атомів є атом
Гідрогену. У нього на орбіті знаходиться
лише один електрон, який притягується
ядром, причому це притягання
урівноважується кінетичною відцентровою
силою. В моделі Резерфорда можливі
орбіти з будь-яким радіусом, тому
частота електромагнітного
випромінювання, яке взаємодіє з атомом, є
довільною, відповідно, спектр
випромінювання (поглинання) є безперервним.
Спектроскопічні дослідження, однак,
показали, що спектри атома Гідрогену
(рис. 8.1) та й інших атомів лінійчасті.
Н. Бор пояснив ці закономірності
у межах планетарної моделі атома,
використовуючи ідеї квантування. Н. Бор
вважав, що коли електрон рухається по
орбітах, то поглинання і
випромінювання енергії не відбувається, тобто ці
орбіти стаціонарні. Випромінювання
(поглинання) енергії відбувається лише при
переході електрона з однієї стаціонарної
орбіти на іншу, тобто в атомі Гідрогену
енергія квантується і її рівні дискретні.
Теорія Бора мала напівкласичний,
напівквантовий характер і
використовувала ідею квантування енергії у постула-
тивному вигляді. Вперше цю ідею
висунув М. Планк. За її допомогою він зміг
пояснити властивості випромінювання
чорного тіла. ^ис* 8-І- Спектр атома Гідрогену
Другим явищем, яке не можна було пояснити, виходячи з чисто
хвильової теорії, був фотоефект — виривання електронів із металу під дією
електромагнітного випромінювання (світла). Було встановлено, що існує деяка
порогова частота (у0), нижче за яку фотоефект не спостерігається.
Класична теорія пов'язувала енергію електромагнітного випромінювання лише
з його інтенсивністю, і тому не могла пояснити, чому низькочастотне
(довгохвильове) випромінювання не дає фотоефекту. Розвиваючи ідеї Планка
щодо квантування енергії, А. Ейнштейн вважав електромагнітне
випромінювання (світло) струмом незвичайних частинок—фотонів, енергія (є) яких
пов'язана з їх частотою (V) співвідношенням
є = йу, (8.1)
-820,4
-1093,8 -і
-1281,8
-1458,4
.1875,1
-2278,8
-2625,1
-4051,2
-7400,0
серія
Пашена
серія
Брекета
серія
Пфунда
125
де А — стала Планка (6,626176-10~34 Дж-с).
Фотон спроможний вибити з твердого тіла
електрон, якщо його енергія не менша за роботу
виходу електрона з металу (ф), тобто є > ф = НV0.
Повна енергія електромагнітного випроміню-
?°.:*ня пропорційна кількості фотонів.
«Частинка» світла має не тільки енергію, а
також масу т та імпульс;?. Оскільки фотон рухається
зі швидкістю світла, то згідно з теорією відносності
Ейнштейна
ку к
с X'
тобто імпульс фотона залежить від його частоти
(V) або довжини хвилі (X). Те, що фотон має
відповідний імпульс, було підтверджено відкритим А. Комптоном ефектом
розсіювання рентгенівського випромінювання на електронах.
Наявність ефектів, пов'язаних із квантовою природою світла, дала змогу
встановити подвійну природу електромагнітного випромінювання: хвилі-
частинки. Луї де Бройль запропонував вважати цю подвійну природу хвилі-
частинки характерною рисою всієї матерії. З цього випливало, що не тільки
хвилі (електромагнітне випромінювання) мають корпускулярні властивості,
а й «звичайні» частинки речовини повинні виявляти хвильові властивості,
тобто для них повинно існувати співвідношення, аналогічне виразу (8.2),
Ейнштейн Альберт
(1879—1955),
Німеччина, СІЛА
є = тс2 та т = -%, а р = тс = — =
с2 с
(8.2)
Х = К
(8.3)
де/? = ти — імпульс частинки; X — довжина хвилі де Бройля.
Стала Планка — дуже мала величина, тому хвильові властивості можна
спостерігати лише для частинок з незначними масами: електронів, мюонів
тощо. Справді, передбачені Л. де Бройлем
хвильові властивості частинок підтвердилися
дослідами К. Девіссона та Л. Джермера, які відкрили
явище дифракції електронів на кристалічних
ґратках твердого тіла. Дифракція — це суто
хвильове явище і спостерігається лише тоді, коли
розміри об'єкта такого самого порядку, що й
довжина хвилі. !
Розділ фізики, який описує властивості
мікросвіту, називається квантовою механікою.
Основним шляхом розвитку квантової механіки
є хвильова механіка,/яку заснував Е. Шредінгер.
Головне рівняння хвильової механіки —
порівняння Шредінгера
#\|/ = £\|/, (8.4)
126
Шредінгер Ервін
(1887—1961),
Австрія
де \|/—хвильова функція; Е—повна енергія системи; Н — оператор*
повної енергії (гамільтоніан).
Рівняння (8.4) описує консервативні, незмінні у часі системи. Це так
зване стаціонарне рівняння Шредінгера. Крім того, існує також часове
рівняння Шредінгера, в якому враховується зміна властивостей системи у часі.
Повна енергія (Е) складається з кінетичної (Г) і потенціальної (II)
відповідно
Н = Т + й, (8.5)
де Т та Ц — відповідно, оператори кінетичної та потенціальної енергій.
Оператор її для вільного руху частинки дорівнює нулю. Для кулонів-
ської взаємодії в одноелектронному атомі
0 = -^-, (8-6)
г
де 2е—заряд ядра; (-є) — заряд електрона; г—відстань між ядром та
електроном.
Для багатоелектронних атомів у гамільтоніан потрібно додавати вираз,
що відповідає за міжелектронне відштовхування.
На відміну від оператора (£/), дія якого зведена до множення хвильової
функції на відповідний вираз, оператор кінетичної енергії (Г) — диферен-
ційний. Для однієї частинки у тривимірному просторі він має вигляд
де т — маса частинки; у2 — оператор Лапласа (лапласіан); х,у,2 —
координати Декарта; Й = А/271.
Головна «дійова особа» у рівнянні (8.4) — це хвильова функція (\|/). її
фізичний зміст ґрунтується на теорії ймовірності. Вважається, що модуль
квадрата хвильової функції пропорційний ймовірності знаходження
мікрочастинки в одиниці об'єму (якщо \|/ — комплексна величина, то замість |\|/ |
використовується добуток V}/ на комплексно сполучену величину \|/*). Згідно
з таким трактуванням хвильової функції ймовірність знайти частинку будь-
де у навколишньому просторі повинна дорівнювати одиниці, тобто
ймовірності вірогідної події. З цього випливає умова нормування хвильової
функції
/|у2|</Г = 1, (8.8)
де (IV— елемент об'єму V.
* Оператор — це символ математичних дій, що перетворюють одну функцію на
іншу. Якщо при дії деякого оператора (а) на функцію (/) маємо: ос/= а/, де а — деяке
число, то функція (/) має назву власноїі функції оператора (а), а число «а» називається
власним значенням оператора (а). Власним значенням оператора (Я) є повна енергія (Е),
а хвильова функція (ц/) є власною функцією (Я).
127
Крім умови нормування (8.8), хвильова функція має ще ряд обмежень —
так звані вимоги регулярності, згідно з якими хвильова функція повинна
бути скінченною, неперервною та однозначною. Конкретний вигляд
хвильової функції, а також гамільтоніана залежить від специфіки задачі, яку
розв'язують.
Один із найпростіших прикладів застосування принципів квантової
механіки — це поступальний рух мікрочастинки в ямі з нескінченно
високими стінками, тобто за межами ями потенціальна енергія V = °°, а в ямі (7=0.
Ця проблема цікава не тільки своєю простотою, але є доброю моделлю для
систем, у яких електрони можуть вільно рухатися: метали, ароматичні
сполуки тощо.
При вільному русі частинки у просторі всі напрями руху рівноцінні,
тому замість тривимірної системи можна розглянути одновимірну. В цьому
випадку від оператора Лапласа залишиться тільки похідна зах, і рівняння
Шредінгера (при 11= 0) матиме такий вигляд:
Розв'язком рівняння (8.9) є функція
\|/=Я$іп(сах). (8.10)
Частинка не може вийти з ями, тому що за її межами 17= °°, і в зв'язку
з цим на хвильову функцію накладаються граничні умови
У(* = 0) = 0| (а) (8П)
¥(* = 0 = 0| (б),
де / — довжина ями (початок координат (х = 0) припадає на її «передню»
стінку). Функція (8.10) задовольняє умові (а) (зіп 0 = 0).
З умови (8.11) (б) маємо: 8Іп(ш/) = 0, що дає
Ш=пп, (8.12)
де п = 1,2,3,... — ціле додатне (квантове) число.
Продиференціюємо двічі рівняння (8.10)
^ = -(02¥,отже,£ = -^< (8.13)
З урахуванням (8.12) маємо для енергії
8/и/
и2А2
і для хвильової функції
,Яй .
\|/„=Я8Іп^х, (8.15)
де Еп— власні значення оператора Я, а \|/„—власні функції.
128
Таким чином, Е для частинки у ямі квантується, тобто має лише
дискретні значення. Це виникає як наслідок розв'язування рівняння Шредінгера
з урахуванням граничних умов без будь-яких постулатів (типу теорії Бора).
Рівень з п = 1 має найнижчу енергію і називається основним.
Функції \|/Л відповідають стоячим хвилям (аналогія — коливання
закріпленої струни). Ці функції ортогональні. Умова ортогональності
/^•11/^^ = 0. (8.16)
Функції і|/, і \|/у належать до різних квантових станів. Наприклад
/ /
\\ц]у2(1х = В1 /зіп^у -8Іп^яЬс = 0. (8.17)
о о
На рис. 8.2 графічно зображені і|/л та | \|/* |. При п = 1 ймовірність
знайти частинку максимальна посередині ямц, при п = 2 — на відстані НА від
стінок і т. д. При достатньо великих п ймовірність знайти частинку у будь-
якій частині ями (по довжині) однакова, тобто поведінка мікрочастинки
подібна до поведінки класичного тіла.
К\АА/^
л; = 0
х=/ х=0
* = /'
а б
Рис. 8.2. Частинка у потенціальній ямі: а — хвильові функції,
б — густина ймовірності
З рис. 8.2 видно, що \\г{ для основного стану не має вузлів — точок, де
\|/ = 0, тобто функція змінює знак на протилежний. Функції щ, \|/2, \|/3 тощо
мають вузли у кількості (п - 1).
Знаючи енергію мікрочастинки на відповідному рівні, можна знайти її
імпульс
\Рп\ = 72^ = ^. (8.18)
В основному стані частинка має мінімальний імпульс
А
2/*
ІАІ4-
(8.19)
129
З рис. 8.2 видно, що по довжині ями укладається ціле число (п) півхвиль
(А/2), тобто
п-ХІ2 = 1. (8.20)
Використовуючи формули (8.18) та (8.20), не важко довести, що X = к/р,
тобто розв'язання рівняння Шредінгера автоматично веде до співвідношення
де Бройля (8.3). Це свідчить про те, що хвильове рівняння Шредінгера має
фундаментальний характер.
Розмістимо центр системи з мікрочастинкою на початку координат, тоді
середнє значення координати х = 0, а відхилення х від середнього
д*4-НИ (8-21)
тобто дорівнює довжині ями.
Припустимо, що відносно цих координат система з мікрочастинкою не
рухається, тоді середня швидкість V =0ір = 0 і зміни імпульсу частинки в
ямі можна вважати відхиленнями (Арх) від середнього значення. Для
основного стану, згідно з (8.19),
Добуток Ах на Арх дає
Арх-Ах = к. (8.23)
Співвідношення (8.23) показує, що в мікросвіті неможливо одночасно
точно визначити координату та імпульс мікрочастинки. Воно називається
співвідношенням невизначеностей Гейзенберга.
Це співвідношення є наслідком хвильових
властивостей мікрочастинок. Для звичайного
тіла у класичній механіці імпульс є функцією
координат і може бути визначений точно разом
з координатою тіла. У квантовій механіці, згідно
зі співвідношенням де Бройля, імпульс
залежить від значення Я і не є функцією координат.
Для монохроматичної хвилі значення X
визначене точно і Д^ = 0, але ця хвиля делокалізова-
на в усьому просторі і Ах = °°. Якщо зробити
спробу зафіксувати координати, пропускаючи
електрони крізь щілину, то можна зменшити
значення Ах, але при цьому пучок частинок
Гейзенберг Вернер Карл внаслідок дифракції вже не буде монохрома-
(1901—1976), тичним і значення Архзбільшується.
Німеччина Співвідношення Гейзенберга має
фундаментальний, об'єктивний характер і пов'язане зі специфікою руху
мікрочастинок, воно справедливе для будь-якої системи (не лише для «частинки в ямі»).
130
Для частинки в ямі потенціальна енергія II - 0, тому повна енергія (Е)
дорівнює кінетичній
Е = ти /2 -р 12т.
(8.24)
Після диференціювання виразу (8.24) маємо
дЕ = 2рйр/2т = уф = йр(йх/(1І), або йЕйі - йхйр. (8.25)
Замінивши диференціали на скінченні прирощення, дістанемо
&ЕА( = Ах-Арх=к. (8.26)
Співвідношення (8.26)) свідчить про те, що для стаціонарних систем, які
існують тривалий час, А і —> ©о і Д £—> 0, тобто енергетичні рівні точно
визначені. Для короткоживучих станів вимірювання енергії не може бути довшим,
ніж тривалість існування відповідного стану (т), отже,
АЕ = к/і.
(8.27)
Формула (8.27) визначає так звану природну ширину спектральних ліній.
Співвідношення невизначеностей дає змогу пояснити так званий
тунельний ефект — проходження мікрочастинки крізь потенціальний бар'єр (рис.
8.3), висота якого перевищує кінетичну енергію частинки. Класична
частинка не може подолати енергетичний бар'єр, якщо її кінетична енергія
менша від висоти бар'єра. Мікрочас- потенціальний бар'єр
тинка може проходити крізь бар'єр
навіть тоді, коли має енергію меншу, ніж
його висота. Треба тільки, щоб ширина
бар'єра не була занадто великою. Чим
вужчий бар'єр, тим більша, згідно зі
співвідношенням Гейзенберга,
невизначеність в імпульсі частинки, і саме це дає змогу мікрочастинці проникати
крізь високий потенціальний бар'єр. Тунельний ефект — поширене явище.
З ним пов'язана холодна емісія електронів із металу, проходження окисно-
відновних реакцій, ос-розпад радіоактивних нуклідів та інші процеси.
Модель «частинка в ямі» описує поведінку мікрочастинки з
урахуванням її хвильових властивостей у випадку поступального руху в заданому
напрямку на обмеженій ділянці (одновимірний випадок). Одновимірною
також є задача для обертального руху частинки вздовж кола радіусом к
В сферичних координатах (г, 0, ф) лапласіан має вигляд
Рис. 8.3. Тунельний ефект
у2=-2;
г дг г2
.д— + сйгО— + — ^~
дв2 § М \т2вд<р2
= У2
+ -І-У2
(8.28)
131
Для одновимірного ротатора г = соті та 6 = я/2 =соп$1 і рівняння Шре-
дінгера набуває вигляду (змінюється тільки значення азимутального кута ф)
2^ г1 </ф2
(8.29)
Розв'язок рівняння (8.29) для хвильової функції (Ф) шукаємо у
комплексному вигляді
Ф(ф) = Ле/ш/ф, (8.30)
ДЄ І = л/-ї.
За формулою Ейлера е/ф = созф + /зіпф і вираз (8.30) еквівалентний
тригонометричній формі розв'язку рівняння типу (8.10). Вимоги однозначності
і неперервності хвильової функції накладають на Ф(ф) додаткові умови, а
саме: зміна кута ф на 2я не повинна приводити до зміни Ф(ф), тобто
е2пщ = 1. Ці вимоги виконуються лише тоді, коли ти, — ціле (додатне або
від'ємне) число, тобто
т/=0;±1;±2;±3. (8.31)
Отже, стаціонарними є лише ті стани, для
яких у довжину кола вкладається ціле число
хвиль (рис. 8.4, а). Інакше виникають
послаблення і затухання хвиль під час інтерференції
з хвилями, які набігають при послідовному
обертанні уздовж кола (рис. 8.4, б).
Підстановка Ф(ф) у рівняння Шредінгера
(8.29) з урахуванням дискретності т1 показує,
що для обертального руху енергія також кван-
тується
Е = П2т}І2тг2. (8.32)
Формула (8.32) повністю відповідає
формулі (8.14) для енергії частинки в одно-
вимірній ямі, якщо врахувати, що
матеріальна точка попадає в початкове положення, пройшовши в ямі Ь = 2/, а вздовж
кола—Ь=2пк
Квантування енергії веде до квантування моменту кількості руху (М).
В загальному вигляді (М) = [гхр]9 де р — вектор імпульсу; г — радіус-
вектор. Коли частинка рухається по колу, кут між векторами г і р дорівнює
я/2. Оскільки зіп(я/2) = 1, то |А/| = |г||р|. Вектор М орієнтований
перпендикулярно до площини обертання, тобто вздовж осі 2 (М2). Е=р 12т і, відповідно,
(8.33)
Рис. 8.4. Хвилі де Бройля
для обертального руху:
а — стаціонарна орбіталь;
б — нестаціонарна орбіталь
\М =М2 = \р\ • \г\ = ^2тЕг = /Ю;Й.
Розглянутий вище випадок відповідає плоскому ротатору.
Просторовий ротатор—це рух частинки по поверхні кулі зі сталим
радіусом. Для опису її поведінки треба, крім азимутального (ф), ввести полярний
132
кут (6). Рівняння Шредінгера (8.4) при г = соті і введенні безрозмірної
величини (Р)
$ = 2тЕг2ІП2 (8.34)
набуває вигляду
<ф\|/ + р\|/ = 0 (8.35)
(лапласіан залежить тільки від значення кутів 9 і ф, але не від значення г).
Розв'язок виразу (8.35) знаходять у вигляді добутку \|/ = 0(9)Ф(ф) двох
функцій, кожна з яких залежить лише від однієї змінної: 8 чи ф. Якщо
використати подібну хвильову функцію, то у рівнянні (8.35) змінні розділяються.
Рівняння для Ф(ф) є аналогічним рівнянню (8.30) для одновимірного
випадку. Потім визначають вигляд хвильової функції (7(6) і виявляється, що
величина (Р) квантується
Р = /(/+1), (8.36)
де / — квантове число (/ = 0,1,2,3,...).
Не важко побачити^ що чисельник виразу (8.34) є квадратом моменту
кількості руху, отже, М квантується згідно з виразом (8.36), тобто
\М\ = ТРИ = ^//(/ + 1)Й. (8.37)
Розв'язування Ф(ф) у випадку просторового ротатора описує
поведінку проекції матеріальної точки на площину ХОУі умова (8.33) відповідає
квантуванню проекції моменту кількості руху на вісь 2. Оскільки М2 не
більше, ніжМ, то виникає умова
щ</.
(8.38)
Якщо /, наприклад, дорівнює 2
(рис. 8.5, а), то т1 набуває значень
(-2), (-1), 0,1,2, тобто (2/+1) різних
значень. Величина | М\ в цьому
випадку дорівнює у]2 -3_=2,45 в
одиницях Й.Значення \М\ таМ2
задають цілком визначене
положення вектора М у просторі
відносно ПОЛЯрНОЇ ОСІ 2.
Отже, квантується не тільки
абсолютне значення вектора М
(квантове число /), а і його
можлива орієнтація у просторі (квантове
число ті), тобто спостерігається просторове квантування.
На відміну від проекції М2, проекції Мх та Му не визначені, і набір
можливих положень задається конусами відповідного розхилу (див. рис. 8.5, б для
випадку, коли 1 = 2). Неможливість одночасного точного визначення век-
133
Рис. 8.5. Квантування моменту кількості
руху: а — вектор М та його проекції при
/ = 2; б — прецесія вектора М
тора М та всіх його проекцій (Мх, Му9 М2) є ще одним виявом
співвідношення невизначеностей.
З формул (8.36) і (8.34) випливає, що
В.ІЄ1!^.І£І|^, (839)
2тг2 2/
де /— момент інерції, тобто енергія просторового ротатора визначається
тільки квантовим числом / і не залежить від значення т{. Всі стани, зображені
на рис. 8.5, з різними значеннями т1 мають однакову енергію. Наявність
кількох (тут 2/ + 1) станів з однаковою енергією називається виродженням.
У даному .випадку виродження пов'язане з рівноцінністю всіх напрямків
вектора М у просторі. Виродження можна зняти, якщо систему піддати
зовнішньому впливу, наприклад подіяти на систему постійним магнітним
полем.
Електрон, рухаючись по сфері, створює магнітний момент Д. Між
механічним і магнітним моментами існує пропорційна залежність, коефіцієнт
якої називається гіромагнітним відношенням
у = е/2тс, (8.40)
де є — заряд електрона; т — маса електрона; с — швидкість світла.
Оскільки механічний момент квантується й одиницею його
вимірювання є Й, то магнітний момент теж квантується
[ів=уП = еП/2тс, (8.41)
де \ів— найменша універсальна одиниця магнітного моменту (магнетон
Бора).
Для проекції магнітного моменту на вісь і, згідно з виразами (8.41) та
(8.33), отримаємо
Цг=т/|іЛ. (8.42)
Візьмемо магнітне поле напруженістю Я, що орієнтоване вздовж осі 2.
Тоді енергія взаємодії орбітального магнітного моменту з полем дорівнює
Е = Я\ь = трів!Н:. (8.43)
Отже, стани з різними значеннями т1 мають різну енергію, і тому
магнітне поле «знімає» виродження. Відповідно до цього /Я/ називається
магнітним орбітальним квантовим числом, а / — просто орбітальним
квантовим числом. Слід зазначити, що експериментально визначаються саме
магнітні властивості атома, в теорії ж насамперед важливі механічні
властивості. Тісний зв'язок між магнітними і механічними властивостями атома
є своєрідним «містком», який поєднує теорію з експериментом.
134
8.2. Одноелектронна система — атом Гідрогену
В системі електрон лише один,
Яким же чином рухається він?
Орбіт-шляхів немає тут, на жаль,
Але залишилась принаймні орбіталь.
Одне з найважливіших завдань квантової теорії, що має пряме
відношення до теорії хімічного зв'язку, — це вивчення властивостей атомів. Атом
Гідрогену — найпростіший з усіх атомів і складається тільки з одного
електрона та ядра.
Стаціонарне рівняння Шредінгера для атома Гідрогену має такий вигляд:
Я\|/ = [г + [/]\|/ =
П2 ^2 2е2
2т г
\|/ = £у.
(8.44)
Потенціальну енергію визначають за формулою (8.6). Рівняння (8.44)
використовується не лише для атома Н, а й для інших гідрогенподібних
систем: Не , Ьі тощо. В атомі, який є сферично симетричним, зручно
користуватися полярною системою координат: 0 та ф — полярний та
азимутальний кути, г — відстань від початку координат. Хвильову функцію \|/, яка
залежить від параметрів г, 0 та ф, можна подати у вигляді добутку трьох
функцій: К{г); С(0) та Ф(ф), кожна з яких залежить тільки від однієї змінної (г,
0 або ф)
\|/(г,0,ф) = Л(г).С?(0).ф(ф). (8.45)
Лапласіан, згідно (8.28), ділиться на дві частини
V2=4V2>ф+V;
Перша частина цього виразу відповідає відцентровим силам при г - соті.
Рівняння Шредінгера для кутової частини хвильової функції С(0) • Ф(ф) атома
Гідрогену точно відповідає рівнянню (8.35) для просторового ротатора.
Друга частина цього виразу відповідає оператору кінетичної енергії для
радіального руху
Після ділення на
Т =-А_у2
2т ґ'
Ь1
(8.46)
2т
розділення змінних та врахування розв язку для кутової частини, рівняння
Шредінгера для радіальної частини має такий вигляд:
• ( чл\ /(/ + 1)"
І2
(і2К + 2М^к
йґ
йг
2т
£ +
&
= 0.
(8.47)
135
Крім повної (Е) та потенціальної (-2е Іг) енергій, в рівнянні (8.47)
враховується член ,,, ..
? '
який зумовлений відцентровими силами. При великих г зникають члени, що
містять г у знаменнику, і це рівняння спрощується
Л = _2МЛ = Х2Л (8.48)
В атомі повна енергія Е — від'ємна величина, тому параметр X —
додатне число. Рівняння (8.48) має простий розв'язок
/^ (г) = Ще'Хг + М2е+Хг. (8.49)
Експоненційно зростаючий розв'язок фізичного змісту не має, тому що
при г -» оо Щг) —> «% тобто хвильова функція не скінченна. Повний
розв'язок рівняння (8.47) є добутком Я0(г) на поліном/(Хг)
К(г) = Щ(\г)е-х\ (8.50)
де N— нормуючий множник.
Вимоги скінченності хвильової функції К(г) накладають певні умови на
поліном/(Хг) і приводять до співвідношення
/ + лг+1 = ^, (8.51)
де пг= 0,1,2,3,..., а
а = т2е2ІП2. (8.52)
Ввівши квантове число п = 1 + пг+ 1, дістанемо
п = а/Х. (8.53)
п має значення 1,2,3... (при/ = 0талг = 0 л = 1).
Із рівнянь (8.48) та (8.53) можна одержати вираз для повної енергії гідро-
генподібного атома
Е = -Я2Й212т = а2П212тп2 = -2 V т/іп2 П2. (8.54)
Величина
«о88-8!- (8-55)
те
має розмірність довжини і називається «борівським радіусом», тому що
точно відповідає радіусу найменшої за енергією орбіти в атомі Гідрогену
згідно з теорією Бора. Якщо ввести величину а0 у формулу (8.54), то ця
формула набуває такого вигляду:
г2 є2
Для параметра а, згідно з виразами (8.52) і (8.55), маємо
а=І/а0. (8.57)
136
Рівняння (8.56) має зовсім простий вигляд, якщо енергію вимірювати в
так званих атомних одиницях енергії (а.о.е.) (1 а.о.е. = е2/а0 = 27,2131 еВ =
= 4,359814- 10~18Дж)
Е = -г2І2п. (8.58)
Борівський радіус також належить до атомних одиниць — це атомна
одиниця довжини (а0= 5,2917 -10" м = 52,9167 пм), відповідно, атомна одиниця
заряду — заряд електрона (\е\ = 1,6021892 • 10~ Кл).
Квантове число (п) називається головним квантовим числом. Воно, як це
видно із формули (8.58), визначає енергетичні рівні у гідрогенподібних
атомах. Найменше можливе значення (п) дорівнює одиниці. Цьому значенню
п відповідає основний стан атома Гідрогену. Енергія цього стану в атомних
одиницях дорівнює
Е0=-2?/2. (8.59)
Е0 — це енергія, яку треба витратити, щоб видалити електрон із системи
(атома) у нескінченність. Цю енергію можна визначити експериментально.
Вона дорівнює чисельно енергії іонізації системи. Експериментальні та
теоретичні значення енергії іонізації гідрогенподібних систем добре
узгоджуються між собою. Енергія іонізації іона Не (54,4 еВ) в чотири рази більша
(2= 2), ніж атома Гідрогену Н (13,6 еВ), для якого 2= 1 і т. д.
Енергія гідрогенподібних систем залежить тільки від значення п; значення
/ та т1 на неї не впливають. Стани з однаковими значеннями я, але різними
значеннями / та ть мають однакову енергію, тобто вони є виродженими.
Оскільки п = / + пг + 1 і мінімальне значення пг- 0, то максимально
можливе значення /дорівнює (п -1). При кожному значенні /, згідно зі
співвідношенням (8.38), ті приймає значення від (-/) до (+/), всього
(2/ + 1).
Ступінь виродження (§) енергетичних рівнів у атомі Гідрогену дорівнює
Я = Е(2/ + і) = 1 + 3 + 5 + ... + (2«-і) = «2. (8.бо)
Отже, рівень при п = 1 невироджений, при п = 2 — чотирикратно
вироджений і т. д. Таким чином, дискретність енергетичних рівнів атома
Гідрогену і поява цілих квантових чисел виникає не у вигляді постулатів, як це було
у теорії Бора, а природно, як наслідок розв'язку рівняння Шредінгера з
урахуванням вимог скінченності та однозначності хвильових функцій. Згідно
з умовою частот Бора
АЕ = ЕІ-Е^=^^V, (8.61)
тобто дискретність значень Е{ (£,) в атомі Гідрогену приводить до того
(рис. 8.6, с. 138), що частоти V випромінених (поглинутих) атомом фотонів
виявляються точно визначеними, відповідно, спектр атомарного Гідрогену—
лінійчастий (рис. 8.1, с. 125) і повністю узгоджений з експериментом.
137
14к
12
10
м
О)
4\-
0
1 00
континуум
1 п = 3 1
їй = 2
/2=1
і п н
Мі
її .а 2 .а 5 і
серія
Пашена
сер
Бреке
сер
Пфун
II в; «$ 1
1 ^
з
°
20000
140000
о
60000
80000
100000
Радіальна частина К(г)
повної хвильової функції,
згідно з рівнянням (8.50),
визначається як добуток
експоненти на поліном.
Згідно з формулою (8.53)
параметр X залежить від
головного квантового числа
п. Поліном/ріг), крім того,
залежить від орбітального
квантового числа /. Отже,
радіальна частина повної
хвильової функції залежить
від квантових чисел п та /
і має такий вигляд:
аг
п
Кп1=Ве «/„/, (8'62)
де стала В визначається в
кожному конкретному
випадку з умов нормування
оо
Л^|г2Л- = 1. (8.63)
Рис. 8.6. Енергетичні рівні та походження
спектра атома Гідрогену
Множник г йг — це
радіальна частина повного
елемента об'єму у сферичних координатах, який дорівнює добутку йг, гсів
та г§іп8<Лр, тобто
^=Л/г8ІпЄ<Шф. (8.64)
Наочно це можна побачити на
рис. 8.7.
Квантове число / прийнято
позначати відповідною літерою
латинського алфавіту. Хвильова
функція при / = 0 називається
^-функцією, при / = 1 —р-функцією, при
/ = 2 — ^-функцією, / - 3
—/функцією, / = 4—^-функцією тощо.
Походження цих позначень має
історичний характер і випливає із
спектроскопії.
Значення К(г) наведені у таблиці
Рис. 8.7. Елемент об'єму у сферичних 8-1 Для головних квантових чисел:
координатах га = 1,2таЗ.
138,
Г/ у
Іе /Оу,
$/\
і /'
чгс/8
ур<йр
/ ^
Позначення
орбіталі
*Лг)
2^2е~аг
аз/2 !_^: и-с-/2
Таблиця 8.1. Радіальні хвильові функції
гідрогенподібного атома (К„і)
Графічно радіальні
хвильові функції К(г) для п = 1,
2 та 3 зображені на рис. 8.8.
Згідно зі статистичною
інтерпретацією хвильової
функції ймовірність
знаходження електрона на
відстані г від ядра в
елементі об'єму (IV
пропорційна |/^|. Якщо напрям,
у якому шукається
електрон, довільний, то кутову
частину у виразі (8.64) для
елемента об'єму можна
проінтегрувати, що дає 4я.
Отже, ймовірність
знаходження електрона на відстані від г до (г + дг\ тобто у сферичному зазорі
завтовшки (сіг) і площею 4пг2, становить
І*
2з
2р
Зз
за
72
* а3'2 (агVа''2
—а3/2
зТз
1--(<хг) +— (аг)2
3К ' 21х '
,-а/З
8 3/2
21у[б
4
81>/30
(аг)
а3/2(аг)2,
(аг)2
,-аг/З
-аг/3
Нг) = \К2п1\4пг2.
(8.65)
+0,05 М
Функція м>(г) називається функцією радіального розподілу. Для
основного стану гідрогенподібного атома «, ч
Цг) = со^г2^22г/й°. (8.66)
Функція має максимум за умови
<Ьн(г)І<1г = 0, звідки
гтах = а0/2=\/а. (8.67)
Для атома Гідрогену 2=1, тоді
^тох = «о. (8.68)
Ймовірність знайти електрон
максимальна на відстані (а0) від ядра, що
відповідає першій борівській орбіті. Повний -0,05
вигляд функції м>(г) для 1$-стану атома 5 10 г/ао
Гідрогену наведено на рис. 8.9, с. 140, з рИс. 8.8. Графічне зображення
якого видно, що м?(г) не дорівнює нулю радіальних хвильових функцій
і на відстанях, які не дорівнюють борів- атома Гідрогену
ському радіусу, електрон «розмазаний» у просторі. Виходячи з цього, від
уявлення про локалізовані орбіти борівського типу слід відмовитися.
Оскільки місце найбільш ймовірного перебування електрона все ж таки
відповідає орбітам Бора, користуються терміном орбіталь. Це поняття найчастіше
139
застосовують як синонім терміна одноелектронна хвильова функція з
урахуванням того, що знання хвильових функцій дає інформацію про
місцезнаходження електрона у просторі.
м<г)А
м<г)А
Ьм(г)
Мг)
25
к^^у.
2р
12 г,а.о
2 4 6 г, а.о
3<1
УпШ>^ У^М^ \уїШ^
12 г, а.о
5 10 г,а.о
Рис. 8.9. Графічне зображення функцій радіального розподілу (густина
ймовірностей) в атомі Гідрогену при значеннях головного квантового числа: 1,2 та З
Функції \у(г) для орбіталей 2р та Зеї мають, як і Із, по одному максимуму
(див. рис. 8.9), їх максимуми точно відповідають радіусам орбіт Бора для
збуджених станів атома водню: г2™ах = 4а0; г™* = 9а0.
Для орбіталей 2з, 3$, Зр (та деяких інших і з більшими квантовими
числами) функції радіального розподілу мають складніший вигляд. Як видно з
графіків, наведених на рис. 8.9, функції м>(г) для цих орбіталей мають
додаткові максимуми при менших значеннях к Висота максимумів зменшується
з наближенням їх до ядра. Орбіталі, які мають подібні максимуми,
називаються проникними. Для них частина «хмари ймовірності» розташована
ближче до ядра, на відміну від непроникних орбіталей, де «хмара
ймовірності» розташована далеко від ядра.
Кількість максимумів на кривих радіального розподілу дорівнює (п - /),
наприклад, 3</-орбіталь має лише один максимум, Зр — два, 3^ — три. При
однаковому значенні п кількість максимумів збільшується зі зменшенням
значення /, тобто збільшується проникність орбіталей. Найбільш проникні
завжди 5-орбіталі (/=0). Для проникних орбіталей функція радіального
розподілу дорівнює нулю не тільки при г = 0 та г=°°, ай при інших цілком
визначених значеннях г. Аналогічно тому, як для хвильової функції електрона
в ямі існували вузлові точки, хвильова функція в атомі Гідрогену має
вузлові поверхні. Для радіальної хвильової функції вузлові поверхні — це
концентричні сфери; їх кількість на одиницю менша від повного числа
максимумів і дорівнює (п - /- 1).
Функція м>(г) описує радіальний розподіл ймовірностей знаходження
електрона на різних відстанях від ядра, а помножена на заряд електрона —
розподіл електронної густини у просторі. Виникає уявлення про електронну
хмару, яка розташована навколо ядра в атомі. Подібна електронна хмара
140
виникає внаслідок усереднення різноманітних рухів електрона у різних
напрямах у просторі. «Перетворення» частинки (електрона) на хмару є
наслідком її хвильових властивостей.
На розміри «радіуса» орбіталі істотно впливає заряд ядра (згідно з
формулою (8.67)). При збільшенні заряду ядра електронна хмара стискується,
Наприклад, якщо в атомі Гідрогену гтах=а0= 52,9 пм, то для Ія-орбіталі атома
Урану гтах = 0,6 пм (1 пм = 10~ м).
Ймовірний характер хвильової функції не дає змоги точно визначати
деякі величини, а визначає їх лише як середні. Оскільки «точна відстань»
електрона від ядра невідома (немає напівкласичних борівських орбіт), то
потенціальну енергію електрона у гідрогенподібному атомі можна
визначити лише як середню величину
Я*
0 = -2е2\-\, (8.69)
(Г\ ^
де - — середнє значення величини, оберненої до відстані від електрона
Іг ) до ядра.
Згідно з теорією ймовірностей, середні величини визначаються за
формулою
\х/(х)сіх
'•\ш- <8'70)
деДх) — густина ймовірності.
У квантовій механіці рольДх) виконує функція |і|/| . Оскільки хвильова
функція \|/ нормована, то знаменник виразу (8.70), згідно з умовою (8.8),
перетворюється на одиницю.
Для середнього значення потенціальної енергії гідрогенподібного атома
в основному стані дістанемо
і і
О = -2е2 \\чі\-<іУ = -2е2а = -— = -22 (а.о.е.). (8.71)
а° Г*І 21 Г -
Оператор О є оператором множення, і вирази ] V Щ \сіУ чи ] \|/£Л|/*/К
для нього ідентичні. Отже, формула (8.70) для визначення V справедлива.
У загальному випадку з урахуванням комплексності і|/ та складного
характеру оператора формула (8.70) потребує узагальнення. У квантовій механіці
для визначення середніх величин користуються формулою
Гі|/*х\|/</К г л
*= Г * л, =К^^ (8.72)
(враховано, що хвильова функція нормована).
Середнє значення кінетичної енергії гідрогенподібного атома в
основному стані дорівнює
Т=\ч*иТуисІУ = \а2е\ =+£-^ = +І- (а.о.е.). (8.73)
2а0 2
141
Якщо порівняти формули (8.59), (8.71) та (8.73), то не важко побачити, що
|І7| = |2Г| = |2£|та (8.74)
Е = Т + 0 = +£г-22=-Ц- = -Т. (8.75)
2 2
Рівняння (8.74) і (8.75) ілюструють так звану теорему віріала.
Відповідно до системи, де силами притягання є кулонівські сили, ця теорема
стверджує, що повна енергія стаціонарної системи дорівнює половині
потенціальної. Вона також протилежна за знаком і дорівнює за величиною
кінетичній енергії цієї системи. Зазначимо, що теорема віріала виконується для
середніх значень кінетичної, потенціальної і повної енергій. Подібне
співвідношення між енергіями (Е, II, Т) виконується не тільки для атома
Гідрогену, а й для всіх стаціонарних систем (атом, молекула тощо).
Для ^-станів кутова складова хвильової функції С(0)Ф(ф) — стала
величина. Це означає, що залежність хвильової функції від кутів 9 та ф відсутня,
у ^-стані електронна хмара має сферичну симетрію. Для .у-стану немає на-
півкласичної аналогії типу орбіт Бора, де електрон обертається навколо ядра,
перебуваючи в одній площині. При / = 0 М = 0, тобто відсутній момент
кількості руху, отже, електрон не здійснює орбітального руху, а тільки
радіальний (до ядра і назад), причому в довільному напрямі. Усереднення
радіального руху електрона і дає розмиту сферично-симетричну хмару
ймовірності.
При розгляді радіальної складової для ^-орбіталей (див. табл. 8.1) видно,
що на відміну від/?-, сі- та інших орбіталей, ^-орбіталі єдині, для яких хвильова
функція не дорівнює нулю при г=0, тобто на ядрі. Виникає питання, чому ж
електрон «не падає» на ядро, а .у-стани стійкі, незважаючи на відсутність
моменту кількості руху. Відповідь на це питання дає співвідношення неви-
значеностей, яке для радіального руху має такий вигляд:
Аг-Арг=П. (8.76)
При спробі локалізувати електрон в області біля ядра, Аг —> 0 і рг
необмежено зростає, що приводить до збільшення ДГ = Ар2/2т. АГ повинно
бути одного порядку з Т, тому необмежене зростання Арп неможливе,
а отже, Аг не дорівнює нулю. Оцінити значення Аг можна без особливих
труднощів. Якщо
№№
згідно з виразами (8.74) і (8.75), то з урахуванням рівняння (8.55) маємо
Арг=ртЕІ5=^~. (8.77)
а° -п
Зіставивши вирази (8.76) та (8.77) дістанемо Аг = а0 = 5 • 10 м, що на
кілька порядків більше, ніж розміри ядра (= 10~15 м).
2ап
142
Кутова складова хвильової функції, яка залежить від полярного кута 0,
має вигляд
%,\ (е) = *8ІпМ Исозб), (8.78)
де В — нормуючий множник.
При І/и/І = / поліном Р(соз0) перетворюється на одиницю і (8.78)
спрощується
С?(Є) = 58Іп|т/І9 = ^8Іп/Є (8.79)
Хвильові функції для/?-, сі- та /-станів дорівнюють (з урахуванням
відповідних значень В)
4-^^022=^зіп2в;С33=&
8ІП3 Є.
г А
б
4 ™ — 4^
На відміну від 5-стану, де 10 (0)| = соті для будь-яких кутів 0, ймовірність
\С (0)| для/?-, сі-, /- та інших станів залежить від значення кута 0. Знаючи
значення С?(0), не важко обчислити \0 (0)| для різних значень кута 0.
Результат для С(0) зручно зобразити графічно так. Від вертикальної осі г
відраховується кут 0 і від початку координат проводиться радіус-вектор, що
дорівнює (або пропорційний) значенню С(0). Потім кінці таких радіусів-векторів
з'єднують плавною лінією. Побудовані таким чином графіки для я-, /?-,
сі- та /функцій наведені на рис. 8.10. Із графіків видно, що зі збільшенням
значення / орбіталі стають все більш сплющеними.
Графічні зображення на рис. 8.10 не
враховують залежності від кута ф; це проекції повних
кутових функцій С(0)Ф(ф) на площину Х02
(при ф = 0 е/т/ф= 1). Для того щоб була повна
картина, треба одержані зображення обертати
навколо осі і. Заміна т7 на (гтї) відповідає
обертанню електрона навколо осі т. у
протилежному напрямі (заміна т1 на (-т^ — це
перехід від М2 до (-М2)).
«Розмитість» орбіталей у кутах 0 пов^язана
з невизначеністю положення вектора М
відносно осей х та^ (див. вище). Можна вважати,
що вона зумовлена прецесією плоскої орбіталі
(тобто вектора М) навколо осі 2. Чим більше
квантове число /, тим менший кут між
вектором М та віссю 2, менша прецесія і більш
плоска орбіталь, що поступово перетворюється на
орбіту-траєкторію планетарної моделі Бора-Резерфорда. Навпаки, чим
менше значення /, тим більший кут конуса прецесії. Якщо спробувати
використати ці уявлення для ^-орбіталей, то «прецесія» була б такою, що
«розхил» конуса досягав би я/2, а це еквівалентне знищенню вектора і залежність
від кута 0 зникає.
143
€*а
Рис. 8.10. Кутовий розподіл
при |т/| = / для функцій:
а — з\ б —р; в — сі; г —/
Розглянемо тепер детальніше повні хвильові кутові функції для/7-орбіта-
лей: для/= 1 і/И/ = ±1
\|/^=58ІП0Є+/ф
ХІ/Г1 =В8Іпве""Іф
Ці функції, як зазначався вище, відповідають двом подібним
тороїдальним «тілам», але з різним напрямом обертання електрона. Однак для задач
хімічного зв'язку (див. розділ 9) частіше використовують не ці функції, а їх
лінійні комбінації, що відповідають дійсним частинам (созф та зіпф)
розв'язку рівняння Шредінгера
рх =2?8ІП0СО8ф]
О • А • • (8«81)
ру =#81пЄ81Пф І
Функції/^ тар відповідають не визначеним магнітним квантовим
числам, а лише їх лінійним комбінаціям. На рис. 8.11 наведені графічні
зображення орбіталей рх та ру, з яких видно, що ці функції витягнуті вздовж
відповідних декартових осей. В полярних координатах х = г зіпб созф, ьу =
= г 8ІП0 8Іпф, отже, рх пропорційна хіг, ьру —уїк
При т1 = 0 Ф(ф) = 1, 8Іп' ''0 = 1, а для / = 1 Р = созв і, згідно з виразом
(8.78),
\|/?=5со80 = р2. (8.82)
Оскільки 2 = г СО80, то функціяр2 пропорційна ііг. Вона орієнтована
вздовж осі і і подібна до функцій/^ і ру Загальна кількість функцій, що
відповідають даному значенню /, дорівнює 2/ + 1, тобто для ^-орбіталей — одна,
/7-орбіталей—три, ^/-орбіталей—п'ять і т. д. Ці функції—добуток 0(6)Ф(ф) —
називають сферичними гармоніками (або повною кутовою складовою). Для
значень / = 0,1 та 2 вони наведені у таблиці 8.2.
Таблиця 8.2. Сферичні гармоніки
Оропаль
Х|/оо= ^
УіО = Рж
Уи=<
Рх
Ру
4/20= <*22
¥21 =
\|/22=,
*хг
<*ху
^х'-у2
Координати
полярні
1
СО80
8ІП0
С08ф
ЗІПф
ЗСО820-1
8ІП0СО80
С08ф
8ІПф
. 2п[8Іп2(Р
81П 0<
[С08 2ф
декартові
1
г/г *
х/г
у/г
Зг2-г2
хг/г1
уг/г2
ху/г2
{х^уУг2
(8.80)
144
Графічні зображення всіх $-, /?- та </-оріталей наведені на рис. 8.11.
Рис. 8.11. Графічне зображення сферичних гармонік
Функції/^, ру та/?2 відрізняються лише орієнтацією у просторі; серед
^/-функцій відрізняється функція ф, яка відмінна не тільки за орієнтацією,
а й за формою. Використовуючи тотожність г2 = х2 + у2 + г2, для функції й^
дістанемо
И -322-г2_3г2-х2-у2-г2_2*-х> г2-у2
* г2 г2 г2 г2 *
Отже, орбіталь й2і є лінійною комбінацією функцій сі22_х2 та сі22_у2, які
тотожні функції йх2_уі. і решті я?-орбіталей за формою. Користуватися
набором із шести ^/-функцій (сіХу, йу„ сІХ2, сіх2_у2, сі22_х2, с122_у2) не можна, тому що
одна з трьох останніх орбіталей лінійно залежна і може бути виражена
через дві інші.
З рис. 8.11 видно, що/?- та ^/-орбіталі мають вузлові поверхні. Квантове
число \гпі\ визначає кількість вузлових площин, які проходять вертикально
через вісь 2. Для рх та йХ2 — це площина 20¥, дляру та йуг—площина 20Х,
для йф — площини 20 У та 20Х, а для сІх2_уі — дві площини, які розділяють
145
І і II (відповідно III і IV) квадранти. Різниця /- |т7|
визначає кількість вузлових поверхонь — конусів із
вершиною на початку координат та віссю, яка
збігається з віссю 2. Дпярх9 ру9 сІху та сІхг_уг кількість
таких конусів дорівнює нулю. Функції/>г, йХ2 та <1у2
мають по одному такому «конусу» з розхилом я/2,
тобто конуси перетворються на площину Х02. Для
функції й22 /-|/и/| = 2, тобто вузлових конусів два.
При переході крізь них функція змінює знак (рис.
8.12).
Загальна кількість вузлових поверхонь для куто-
Рис. 8.12. Вузлові В01- частини хвильової функції дорівнює квантовому
поверхні а-2-орбіталі . _ л Тг- - і г- ~
2 числу /, тобто 0 для л-орбіталеи, 1 — для/?-орбіталеи,
2 — для </-орбіталей і т. д.
У сферичних гармоніках (див. рис. 8.11) не врахована залежність
хвильової функції від відстані до ядра. Для того щоб одержати повну картину,
необхідно об'єднати кутову та радіальну частини хвильової функції
Уп,ті=Кп,С1тіФтг (8-83)
Вище згадувалося, що кількість вузлових поверхонь (сфер) для радіальної
частини хвильової функції дорівнює (п -1 - 1). Для сферичних гармонік
кількість вузлових поверхонь дорівнює /. Отже, повна кількість вузлових
поверхонь дорівнює (п - 1), тобто визначається лише головним квантовим числом.
Слід зазначити, що для частинки в ямі повна кількість вузлових поверхонь теж
дорівнювала (п -1), причому хвильові функції як для частинки в ямі, так і для
атома Гідрогену в основному стані вузлових поверхонь не мають.
Повна хвильова функція залежить від трьох квантових чисел. Головне
квантове число (п) задає власні значення енергії, повне число вузлових
поверхонь, а також середню відстань електрона від ядра. Орбітальне квантове
число (/) визначає власні значення моменту кількості руху та задає форму
орбіталі. Магнітне орбітальне квантове число т/ визначає власні значення
проекції (на вісь г) моменту кількості руху та задає просторову орієнтацію
орбіталі.
Функції \|/л/т не тільки нормовані, але й ортогональні (ортонормовані).
Вимогу ортогональності (8.16) можна в даному випадку записати так:
\^пІт^пТт^^ = 0 (8.84)
за умови, що порушена хоча б одна з рівностей: п = п'; І = /'; т = т{.
Ортогональність двох хвильових функцій свідчить про те, що ці орбіталі
або не перекриваються, наприклад,рхтару (рис. 8.13, я), або перекривання
відбувається так, що додатні внески цілком компенсують від'ємні,
наприклад при перекриванні 5і- ір2- (див. рис. 8.13, б) або І5- і 2^-орбіталей (див.
рис. 8.13, в).
Повну хвильову функцію найбільш наочно можна зобразити за допомо-
146
Рис. 8.13. Ортогональність орбіталей атома Гідрогену:
а —рх та/зу; б — 5 та/>г; в — 1$ та 2$
гою так званих контурних діаграм. На рис. 8.14 наведені контури 2/^-орбіталі
в площині ІОХ. Контурні лінії, для яких \|/—> 0 з одного боку, поступово
сягають до ± оо вздовж осі х, а з другого — концентруються поблизу вузлової
поверхні (площини 20У). Ще більш наочно повні хвильові функції для 1$,2рх
та Зя^-орбіталей наведено на рис. 8.15, с. 148.
Детальне вивчення спектрів
та магнітних властивостей
атома Гідрогену (та інших атомів)
дало змогу зробити висновок,
що наведений вище опис за
допомогою хвильової функції, яка
залежить від трьох квантових
чисел, неповний.
Найпереконливіше це підтверджують
досліди О, Штерна та В. Герлаха
з вивчення магнітних
властивостей атомних пучків. Атом Гід- Рис 8Л4. Контурна діаграма 2^-орбіталі
рогену в основному стані (1$)
не повинен взаємодіяти з магнітним полем, тому що його орбітальний
магнітний момент дорівнює нулю. Проте О. Штерн та В. Герлах
встановили, що при пропусканні пучка атомів Гідрогену крізь потужне
неоднорідне магнітне поле цей пучок розділяється на два. Оскільки орбітальний
момент відсутній, то магнітні властивості слід шукати в самому електроні.
С. Гаудсміт та Дж. Юленбек зробили припущення, що електрон має свої
власні магнітний і механічний моменти. Цю властивість електрона вони
назвали спіном (від англ. «8ріп» — обертання, крутіння).
Для опису поведінки електрона, пов'язаної з його спіном, можна
скористатися формалізмом, яким користувалися для орбітального моменту
кількості руху та орбітального магнетизму. Згідно з формулою (8.37),
орбітальний момент Ід/ І = М{1 + \)Ь (все, що належить до орбітального руху,
позначимо індексом /). Спіновий момент електрона, відповідно, дорівнює
|М,| = 7фТЇ)Й, (8.85)
де 5і — спінове квантове число.
147
ио,о4
■
., .».
а
ш
148
Рис. 8.15. Тривимірні графіки для повних хвильових функцій:
а — Ь-орбіталь; б — 2/?х-орбіталь; в — Зй^-орбіталь
Проекція спіна (у) на визначений напрямок (вісь і) задається аналогічно
виразу (8.33)
М*2=гп5П, (8.86)
де т8— магнітне спінове квантове число.
Аналогічно з орбітальним рухом, для якого |іи/| < /, для спінового руху
\т8\ <з і т8 набуває всіх значень від (-.у) до (+5), тобто всього (2$ + 1) значень.
Для окремого електрона в атомі Гідрогену досліди О. Штерна та В. Гер-
лаха дали 2$ + 1 = 2, звідки 5 = 1Л, а можливі значення т8 дорівнюють (Н-/4) та
(-Уг). Отже, на відміну від цілих орбітальних квантових чисел, спінове
квантове число має напівціле значення. Крок просторового квантування, однак,
тут такий самий, як і в орбітальному русі (Дт/= 1) і дорівнює одиниці: Ат, =
= (+■/*)-(44) = 1.
П. Дірак обґрунтував гіпотезу спіна шляхом розв'язування
релятивістського хвильового рівняння. У звичайному (нерелятивістському) рівнянні
Шредінгера релятивістські ефекти не враховуються, а спінове квантове число
вводиться ззовні як наслідок експериментально обґрунтованої гіпотези.
Врахування спіна в доповнення до відомої хвильової функції \|/й/т дає
змогу повністю пояснити всі властивості атомних систем, зокрема атомні
спектри. З урахуванням спіна з'являється додаткове спінове виродження,
зокрема основний стан (1$) атома Гідрогену виявляється виродженим. Це
виродження знімається при накладанні зовнішнього магнітного поля, при
цьому лінії у спектрі атома Гідрогену стають дублетами.
8.3. Двохелектронна система — атом Гелію
Два електрони вже штовхаються, нервують,
Вони за місце в орбіталі конкурують.
Одержані у попередньому розділі результати про атом Гідрогену є
основою при вивченні багатоелектронних атомів. Розв'язати рівняння
Шредінгера для багатоелектронних систем можливо лише наближеними методами
(точний розв'язок неможливий, тому що не можна розділити змінні). Один
з таких методів — теорія збурень (див. додаток 2). Ця теорія вперше
виникла у небесній механіці при дослідженні динамічної задачі руху багатьох тіл.
Щоб розв'язати подібну задачу, наприклад для системи Сонце — Земля —
Венера, спочатку розглядають окремо рухи Сонце — Земля та Сонце —
Венера і розв'язують відповідні задачі точно, а потім на одержані розв'язки
накладають більш слабку збурюючу взаємодію Земля — Венера і
розв'язують уже задачу для трьох тіл.
Аналогічний підхід можна застосувати і у квантовій механіці; Основні
труднощі при вивченні багатоелектронних атомів виникають уже при
наявності двох електронів, тобто в атомі Гелію. Атом Гелію — це найпростіша
149
багатоелектронна система, тому для нього наближені розрахунки можна
довести до високого ступеня точності. Крім того, на атомі Гелію можна
проілюструвати ряд закономірностей, які стосуються всіх багатоелектронних
систем.
Гамільтоніан для атома Гелію має такий вигляд:
Я = Я1°+Я2°+Аи, (8.87)
де Ні = —-—V,. — одночастинковий (одноелектронний) гамільто-
г* ніан; г—відстань від ядра до і-го електрона (/ має
значення 1 або 2); 2 — заряд ядра; А, 2 = —
' . Г1,2
потенціальна енергія взаємодії електронів; г12 —
відстань між електронами.
У нульовому наближенні нехтують взаємодією між електронами, тобто
відкидають у гамільтоніані член Нх 2. Якщо взяти повну хвильову функцію
(Ч^) у вигляді добутку одночастинкових у? та ^, то у рівнянні Шредінгера
змінні не важко розділити, і для енергії в нульовому наближенні маємо
Е0=Е? + Е%. (8.88)
валентні і д
істеми (іон 1
ням (8.59), маємо
Електрони еквівалентні і для кожного з них задача подібна до задачі для
гідрогенподібної системи (іон Не ). Для основного стану Не , згідно з
рівняная =£?=-£:, (8-89)
що дає
У2
£° = -2Г. (8.90)
Урахування збурюючої дії к12 дає поправку є (перше наближення)
є = /^Х2Ч/0^. (8.91)
Оскільки Ч*0 = \|/° • \|/2, то вираз (8.91) набуває такого вигляду:
є
є =
Ч>Г
|2
-Є\
¥2
42
2
-йУ. (8.92)
І пі2 І оі2
Величини \|/° та \щ\ —Це густини ймовірностей знаходження
першого та другого електронів, а добутки е\|/° таеш/^ —відповідні
електронні густини. Отже, поправка (є) для енергії має наочний фізичний зміст —
це усереднене за всім об'ємом кулонівське відштовхування двох
електронних хмар. Інтеграл г, а і а
ЛУ (8.93)
К =
150
42
називається кулонівським. У атомній системі одиниць значення є у формулі
(8.92) і Ку (8.93) збігаються.
Для основного стану (1$) атома Гелію гідрогенподібна функція має
такий вигляд (табл. 8.1):
у°и=Ме-Іг/а% (8.94)
де N— нормуючий множник.
Розрахунок є із використанням функцій (8.94) дає (в атомних одиницях)
є = * = |2. (8.95)
Енергія основного стану атома Гелію
Е = Е° + є = -22 +|г. (8.96)
о
Для Гелію 2 = 2 та Е = -2,75 а.о.е. Експериментальне значення Е
одержують як суму першої та другої енергій іонізації (для гелію Есксп = -2,90
а.о.е.).
Для того щоб поліпшити узгодження між теорією та експериментом,
можна використати друге та наступні наближення теорії збурень, але це
призводить до дуже громіздких обчислень. Інший шлях — це уточнення
поправок першого наближення удосконаленням хвильових функцій. Це
можна зробити на основі варіаційного принципу. Якщо до хвильової
функції ввести кілька параметрів (а, Ь, с тощо) та обчислювати за допомогою
цієї функції \|/(а, Ь,с.) енергію системи, варіюючи ці параметри, то
найближче до дійсного значення енергії (£д) знаходиться Ет{п — мінімальне з
усіх одержаних значень енергії. Гранично Ет{п = ЕД9 у решті випадків Етіп > Ед.
Для знаходження параметрів а, Ь, с використовують відомі умови
визначення екстремуму
(їі...=о;(іі,..=° ^
іт.д.
Продемонструємо застосування варіаційного принципу на прикладі
атома Гідрогену, для якого відомий точний розв'язок. Для основного стану
атома Гідрогену застосуємо таку пробну хвильову функцію:
^(с) = Ме-сг, (8.98)
де с — невідомий параметр.
Якщо в формулах (8.71) та (8.73) а замінити на с, то для повної енергії
маємо
Е(с) = Ц(с)+Т(с) = -2е2с + ±с2а0е2. (8.99)
З умови сіЕ/сіс = 0 не важко знайти
Стіп=2/а0, (8.100)
151
ап 2 аг
що повністю узгоджується з формулою (8.57). Отже, Уь = Ме~2г/а°, що
збігається з точним розв'язком для \|/І5 (8.94).
Використовуючи значення стіп для повної енергії, згідно з формулами
(8.99) та (8.100), маємо
-о; 2 «о 2
що збігається з виразом (8.59) для енергії основного стану гідрогенподібного
атома.
Оскільки а0 — стала величина, то зміна с, по суті, зводиться до
варіювання 2 в показнику хвильової функції.
Позначаючи змінне значення 2через 2, маємо згідно з (8.101) для
основного стану гідрогенподібного атома
£°(2) =-22+^ а.о.е. (8.102)
Для повної енергії основного стану атома Гелію маємо, згідно з
формулами (8.88) та (8.96),
ЕНе(2) = 2Е°(2) + е(2) = -222 + 22+Ї2. (8.103)
Варіюючи параметр 2, з умови мінімуму с!Е(2)/с12 = 0 знаходимо:
2тіп=^-^ = ^еф. та (8.104)
Яне (2+) = ~22 +|2 -Ц. (8.105)
При 2= 2 з (8.105) маємо ЕІ5(Н.е) = -2,85 а.о.е., що дуже близько до
експериментального значення (-2,90 а.о.е.).
Варіаційний принцип дає результат, якому легко надати фізичну
інтерпретацію. Формула (8.104) показує, що внаслідок відштовхування кожний
з електронів притягується до ядра слабше, інший електрон екранує його від
ядра; це видно з позірного зменшення заряду ядра Гелію від реального (2 = 2)
до ефективного (2еф =2 — 5/16= 1,6875).
Яким чином здійснюється екранування, якщо обидва електрони Гелію
еквівалентні і знаходяться на подібних орбіталях? Це виникає внаслідок
розмитості електронної хмари. Частина її для одного електрона завжди
екранує інший від ядра і навпаки. Електрони рухаються незалежно один від
одного, тому в деякі моменти часу «перший» електрон може опинитися між
ядром та «другим» електроном та екранувати його від ядра, а потім вони
міняються ролями. При цьому враховується, що кожний електрон може
бути ближче до ядра, ніж на відстані радіуса орбіталі (максимум
електронної густини). Ймовірність знайти його там відрізняється від нуля. Величина
а = 2-2еф.. (8.106)
152
називається константою екранування (для Гелію о = 5/16 = 0,3125). Якщо
один електрон завжди знаходиться на великій відстані від ядра, а другий —
на близькій, то константа екранування другого електрона першим близька
до одиниці. Це спостерігається, наприклад, в атомі Літію. Два електрони на
Ія-орбіталі розташовані набагато ближче до ядра, ніж третій електрон на 2з-
орбіталі. Перші два електрони досить ефективно екранують третій від ядра,
так що він рухається у полі приблизно однозарядного іона Літію. Отже, дія
одного електрона на інший полягає не тільки в кулонівському
відштовхуванні між ними, а й у тому, що кожний з електронів впливає на взаємодію
іншого з ядром та його кінетичну енергію. Слід зазначити, що найбільш
чутлива до зміни значення 2 (тобто до зміни вигляду хвильової функції)
саме кінетична енергія, тому що вона залежить від 2 квадратично
(потенціальна — лінійно).
Удосконалення хвильової функції за допомогою варіаційного принципу
приводить не тільки до поліпшення результату обчислення, а й дає
результати, які відповідають теоремі віріалу. В нульовому наближенні теорема
віріалу для атома Гелію виконується так, як вона виконується для будь-якого
гідрогенподібного атома. В першому наближенні теорії збурень величина
є є поправкою до потенціальної енергії, тобто теорема віріалу
порушується. При застосуванні варіаційного принципу
Г=+2е2ф.=(2-1|)2=22-|2 + ^ = -£, (8.107)
згідно з виразом (8.105), тобто тепер теорема віріалу виконується.
Цей результат можн^проілюструвати графічно. Графічне зображення
Еь а.о.
ЕНе(2) та її складових Г(2)та
[/(^наведено на рис. 8.16. Функція Еш (2) проходить
через мінімум, причому Е(2тії) найближче
лежить до справжнього значення енергії,
згідно з вимогами варіаційного принципу.
Незважаючи на деяку складність
обчислень (теорія збурень плюс варіаційний
принцип) простота кінцевого результату
Е = -{І- а)2 (8.108)
приводить до наочних ідей екранування,
тобто міжелектронне відштовхування
виявляється в ослабленні взаємодії кожного
електрона з ядром шляхом зменшення
ефективного заряду ядра.
Варіаційний принцип можна
застосувати у більш загальному вигляді. У хвильову функцію ніяких параметрів не
вводять, а задають її у вигляді добутку одноелектронних хвильових функцій.
Мінімізація енергії ведеться лише за однієї додаткової умови — вимогами
Рис. 8.16. Залежність повної (Е),
кінетичної (Г) та потенціальної
(£/) енергій від параметра 2
в атомі Гелію
153
нормування. Після виконання всіх операцій одержимо таке рівняння
■^Ь¥і+т=т-
(8.109)
Рівняння (8.109) називається рівнянням Хартрі. Воно записане для і-го
електрона і його розв'язання дає значення енергії цього електрона. Роль
потенціалу в цьому рівнянні виконує функція Уі
2
К=-^- +
и
ГУ
-щ.
(8.110)
Перша частина потенціалу Уі — це кулонівська взаємодія
' 2е2
даного (/-то) електрона з ядром. Друга частина
(
V/
ї
-аг.
ГЧ
це потенціальна енергія взаємодії /-го електрона зу-м, усереднена по всіх
координатаху-го електрона. Тобто /-й електрон рухається в ефективному
полі, яке створюєтьсяу-м електроном (а також ядром — перша частина У{).
Виходячи з цієї фізичної ідеї ефективного середнього потенціалу, Д. Хартрі
вивів це рівняння. Пізніше В. А. Фок довів, що це рівняння випливає із
узагальненого варіаційного принципу.
Для того щоб розв'язати рівняння Хартрі, наприклад, для першого
електрона, треба знати Уь але для обчислення Ух необхідно знати функцію \|/2.
Цю функцію можна було б знайти з подібного рівняння для і = 2, але для
цього необхідно знати потенціал У2 і т. д. Щоб розв'язати рівняння, треба
застосувати метод послідовних наближень — ітерацій. Задаючи певне
значення \|/І5 обчислюють значення УІ9 потім £2та \|/2 За допомогою і|/2
визначають Ух та уточнюють \у{ та Ех тощо. Кінцева функція називається само-
узгодженим полем і-го електрона, відповідно, метод Хартрі називається
методом самоузгодженого поля, а процедура ітерацій —
самоузгодженням. Метод самоузгодженого поля дає результати, які не залежать від вибору
початкової функції. Якщо замість простих гідрогенподібних функцій взяти
кращі, екрановані, то самоузгодження потребує менше циклів. Розрахунки
із самоузгодженням дають, однак, лише чисельні результати, які, на відміну
від аналітичних, не можна використати в подальших розрахунках,
наприклад, у розрахунках молекулярних систем.
Якщо брати повну хвильову функцію у вигляді добутку одноелектрон-
них, то метод Хартрі дає найкращий результат. Але цей результат (Ех = -2,86
а.о.е.) відрізняється від експериментального 0ЕеКСГІ. = -2,90 а.о.е.) більш ніж
на 1 еВ (0,04 а.о.е.).
154
Для того щоб поліпшити результат обчислення, слід відмовитися від
одноелектронного наближення. З трактування фізичного змісту хвильової
функції випливає, що ймовірність знаходження першого електрона,
пропорційна ш/Л, другого — \|/2 , першого та другого одночасно — Жі 4*2 •
Отже, ймовірність для повної хвильової функції дорівнює добутку
ймовірностей її складових. З теорії ймовірності відомо, що ймовірність складної
події дорівнює добутку ймовірностей складових подій у тому випадку, коли
останні незалежні. Тому, коли повна хвильова функція (Т) задається у вигляді
добутку одноелектронних функцій, то припускається, що рух кожного з
електронів відбувається незалежно від іншого. Це припущення слушне і дає
цілком задовільні результати.
Проте, якби рух електронів був скорельований так, щоб вони якомога
рідше підходили близько один до одного, то це зменшило б кулонівське
відштовхування між ними і знизило б енергію.
Кореляцію можна врахувати, якщо ввести в хвильову функцію члени, які
містять відстань між електронами (г{ 2) у явному вигляді. Наприклад,
Е. Хіллераас обрав функцію у такому вигляді:
хР = \|/1\|/2(1 + аг1>2) = ^(гі+Г2)(і + аг1)2). (8.111)
Параметр 2 має той самий зміст, що й раніше, і враховує екранування,
а параметр а враховує кореляцію (при а = 0Т дорівнює добутку \\?х та \|/2
і кореляція відсутня). Обчислення Е. Хіллерааса дало такі результати:
2еф. = 1,849, а = 0,364, £=-2,891 а.о.е.,
що відрізняється від експериментального значення лише на 0,3 %.
Корельована функція, порівняно з некорельованою, відображає
зменшення ймовірності перебування електронів на близькій відстані один від
одного і, навпаки, збільшує ймовірність перебування одного електрона
подалі від іншого. Як наслідок, знижується енергія системи, тому що
зменшується міжелектронне відштовхування. Азимутальна кореляція
синхронізує рух взаємодіючих електронів таким чином, щоб вони знаходилися по
різні боки від ядра. Радіальна кореляція враховується додаванням члена
Ь{гх - г2) і враховує тенденцію електронів не знаходитися одночасно на
одній і тій самій відстані від ядра. Врахування кореляції при кількох
параметрах дає змогу обчислити енергію основного стану атома Гелію з точністю,
що узгоджується з експериментом і навіть ще кращою. Якщо для Гелію та
катіонів Ьі , Ве , В та інших врахування кореляції дає змогу уточнити
результати, то для аніона Н~ (також гелієподібна система) воно є вирішальним.
Обчислення без введення кореляції давали від'ємні значення енергії іонізації,
тому аніон Н~ вважався нестійким. Врахування кореляції дало змогу Е. Хілле-
раасу отримати додатні значення енергії іонізації та довести, що аніон Н~—
стабільна система. В аніоні Н~ заряд ядра дорівнює (+ 1) і взаємодія
електронів з ядром набагато слабкіша порівняно з Гелієм та гелієподібними
катіонами. Тому в аніоні Н~ зменшення міжелектронного відштовхування
155
в результаті врахування кореляції руху електронів значне порівняно з іншими
внесками в енергію. Отже, чим слабкіше зв'язані електрони, тим більще
значення має кореляція. Зазначимо, що саме кореляція і синхронізація руху
електронів відіграють основну роль у так званих дисперсійних силах, на яких
ґрунтується міжмолекулярна взаємодія (див. розділ 10).
Розглянемо тепер збуджений стан атома Гелію, коли один з електронів
знаходиться у стані з п = 2 (2$ чи 2р\ а інший в основному (Ія) стані. В
нульовому наближенні енергія атома Гелію обчислюється за формулою (8.88),
наприклад:
£5е(Ь,2*) = £Йе(І5)+£Йв(2*) 1
Е°Не(и,2р) = Е0Пе(із)+Е0Ие(2р)\
Для гідрогенподібного атома Е(І8) = Е(2р)9 тому в нульовому
наближенні і?не (2,ї) = £не (2/?), тобто рівні з однаковими значеннями п
залишаються виродженими.
Розрахунок у першому наближенні теорії збурень, який враховує міже-
лектронне відштовхування, дає для кулонівських інтегралів такі чисельні
значення:
(8.112)
Кіз -
К2Р =
к
УІ
І2
Ш
№*
дУ = 0,420 (а)
(IV = 0,486 (б)
1,2
(8.113)
Із цих результатів можна зробити такі висновки. По-перше, значення К^
і К2р набагато менші, ніж КІ5 (Ки = 1,25) (див. вираз (8.95) при 2=2), тобто
кулонівське відштовхування між електронами істотно зменшується тоді, коли
вони знаходяться на різних орбіталях (Іу та 2$ або 2р\ а не на одній і тій самій
(в основному стані обидва електрони на Ія-орбіталі). По-друге, КІ8 < К2р,
тому рівень ЕІ8 знаходиться глибше, ніж Е2р (кулонівські інтеграли дають
додатний внесок в енергію). Таким чином, збурення знімає виродження
по /. В усіх багатоелектронних атомах енергія стає функцією не тільки
головного квантового числа (л), а й орбітального (/), причому при однакових
значеннях п нижчу енергію мають рівні з мінімальним значенням / (у
розглянутому вище випадку Е2з (/ = 0) < Е2р (/ = 1) при п = 2). Цей результат може
бути розтлумачений за допомогою уявлень про екранування. Глибше
розташовані електрони (тут 1$) екранують зовнішні (тут Із та 2р) від ядра.
Електрони, розташовані на більш проникних зовнішніх орбіталях (2$ більш
проникна, ніж 2р), екрануються менше, ближче підходять до ядра і сильніше
притягуються, що призводить до зниження енергії. Проникність орбіталей
залежить від значення квантового числа /: чим менше значення /, тим більша
проникність (див. підрозділ 8.2). Це призводить до того, що із ростом / енергія
156
також зростає (при сталому головному квантовому числі п). В атомі
Гідрогену, де є лише один електрон, ефект екранування відсутній (немає міжелек-
тронного відштовхування) і рівні з різними значеннями / мають однакову
енергію. Першому збудженому стану (п = 2) в атомі Гідрогену відповідає
один рівень, в атомі Гелію внаслідок зняття виродження по / орбіталям 2$
і 2р відповідають різні рівні.
В результаті спектроскопічних досліджень було виявлено, що першому
збудженому стану атома Гелію відповідають не два, а чотири рівні.
Аналогічна картина спостерігається і для більш високих збуджених станів атома
Гелію. Щоб пояснити це явище, треба детальніше розглянути хвильові
функції.
Для основного стану (без урахування кореляції) хвильова функція — це
добуток (1$)і# (іу)2 (тут 1 та 2 — «номери» електронів; одноелектронні орбі-
талі позначені квантовими числами п та /). Перестановка електронів 1 та 2
місцями нічого не змінює, тому що обидва електрони еквівалентні та
знаходяться на тій самій орбіталі — 1$.
Для конфігурації 1^ • 2$, крім функції Ч^ = (Ь)^ (2$)2, можна використати
також функцію Тц = (Ь)2- (2з){9 де перший електрон знаходиться у
збудженому стані, а другий — в основному. У природі ніяких фізичних позначок,
за допомогою яких можна було б відрізнити один електрон від іншого, не
існує, всі електрони тотожні. Тому функція ХРП повинна бути розв'язком
рівняння Шредінгера, як і функція Ч^. Якщо деякі функції є розв'язками дифе-
ренційного рівняння, то розв'язком є також їхня лінійна комбінація
Ч^СіТі + СЛ (8.114)
Коефіцієнти С1 та Сп відображають внески функцій Ч^та Ч^ у повну
хвильову функцію Т.
Використання теорії збурень (див. додаток 2) дає такі рівняння для
коефіцієнтів Сх та Сп:
С1(К-е) + СиА = 0)
С1А + Си(К-г) = 0\
де
А = І^Ал^п^ = І^іА^і^ (б) \
є — поправка першого порядку до енергії нульового наближення.
Рівняння (8.115) мають нетривіальні (відмінні від нуля) розв'язки для
коефіцієнтів Сі та Сп лише тоді, коли наступний детермінант дорівнює нулю
\К-е А
\А К-г\
(8.115)
(8.116)
= 0. (8.117)
157
Його розв'язок дає
гАі8 = К±А (8.118)
(значення індексів А та 5 буде з'ясовано далі).
Розглянемо інтеграли Кта А, які входять у формулу (8.118). Пам'ятаючи,
що Нх 2 = — і є = 1 в атоній системі одиниць та підставляючи вирази для
г1,2
4х! (або Ч^) у формулу (8.116, а), одержимо
К =
[НІНІ
г1,2
АУх<іУ2 ■■
гНІН
г1,2
аухау2
(8.119)
(тут і далі атомні орбіталі позначені лише своїми квантовими числами).
Внаслідок еквівалентності 1- та 2-го електронів обидва інтеграли в
рівнянні (8.119) тотожні. Не важко побачити, що К— це відомий кулонівський
інтеграл (див. вирази (8.93) та (8.113)). Кулонівський інтеграл в атомах завжди
додатна величина, тому що всі величини |(Ь) |, |(2у) |, з яких він складається,—
додатні.
Підставляючи Ч^та Ч*и у формулу (8.116, б), для А одержимо
А =
Ш2*)2ШЧ
1-сікш.
(8.120)
г1,2
Цей інтеграл називається обмінним. В нього входять не квадрати хвильових
функцій, а їх добутки, які можуть бути як додатними, так і від'ємними.
Обмінні інтеграли в атомах завжди додатні. Це можна пояснити, не
обчислюючи А у кожному конкретному випадку. Відповідний вираз (8.120)
обмінного інтеграла без значення г{2 в знаменнику дорівнює нулю внаслідок
ортогональності 1$- та 2^-орбіталей. Ділянки з додатним перекриванням
компенсують відповідні ділянки з від'ємним перекриванням. Не важко
помітити, що внески в обмінний інтеграл тоді додатні, коли обидва електрони
знаходяться у одній і тій самій ділянці (додатній чи від'ємній). Якщо
електрони потрапляють у різні ділянки, то внески в обмінний інтеграл від'ємні.
Тепер врахуємо, що коли електрони знаходяться в різних ділянках, то
відстань гх1 — велика, а коли в одній — мала. Тому від'ємні внески в
інтеграл менші, ніж додатні, і обмінний інтеграл в цьому випадку — додатна
величина. Повна енергія дорівнює
Е = Е°+еА8=Е0+К±А.
Оскільки А > 0, то нижче розташований рівень з енергією
а вище
ЕА=Е°+К-А,
Е8=Е°+К + А.
(8.121)
(8.122)
(8.123)
Підставляючи формулу (8.118) у вираз (8.115), можна знайти Ст та Сп
158
і, отже, хвильові функції, що відповідають енергіям^ таЕ8. З урахуванням
ортогональності Ч^та Тц і нормування маємо:
^=^№+^11); (8.124)
^=^№-^н)- (8.125)
Індекси хвильових функцій відповідають індексам енергій. Хвильова
функція Ч^ — симетрична, а функція Х¥А — антисиметрична щодо
перестановки першого та другого електронів місцями. При подібній процедурі
функція хРІ=(1^)і(25,)2 перетворюється на функцію 4^!= (25)1(І5)2, тому
симетрична функція ¥$ залишається без змін, а антисиметрична функція Х¥А —
змінює знак на протилежний. У нульовому наближенні симетрична й
антисиметрична хвильові функції відповідають виродженим станам.
Внаслідок ортогональності 1$- та І8 -орбіталей ортогональні також
функції Ч^та Ч'ц. З урахуванням цього отримаємо
І ->І І о І ЇМ +
\І/2 __ \т/2 __ І 1
Г*$ Г" Г Л Г~ о
ш2
МІ
У нульовому наближенні ймовірністі знаходження системи в
симетричному або антисиметричному стані однакові. Цей особливий тип
виродження, що раніше не зустрічався, називається обмінним. Подібне
виродження є наслідком тотожності, нерозрізненості мікрочастинок (електронів)
і можливе тільки в багатрелектронних системах.
Під дією збурення (кх 2) обмінне виродження знімається і симетрична
та антисиметрична хвильові функції починають описувати стани з різною
енергією. Зняття обмінного виродження зовсім не означає, що при цьому
з'явилась можливість відрізняти електрони. Як показує наведений вище
розрахунок енергії, антисиметричній функції відповідає нижчий рівень ЕА,
а Ч^ — вищий рівень Е8.
Дійти висновку про те, що розв'язки рівняння Шредінгера повинні бути
симетричними або антисиметричними, можна і не розв'язуючи рівняння.
Уявімо процес перестановки електронів місцями як дію деякого оператора
перестановки Р12 на хвильову функцію. Якщо електрони двічі поміняти
місцями, то все повинно повернутися до початкового стану, тобто, якщо
двічі подіяти оператором Р12 на хвильову функцію 4х, то одержимо
тотожність
(р12)2Ч/ = Ч/. (8.126)
Із рівняння (8.126) випливає, що Рї2 = ±1, тобто при перестановці
електронів хвильова функція або змінює знак (антисиметрична функція Ч^) або
не змінює (симетрична функція Ч/і5).
Розглянемо властивості функцій У¥А та Ч^. Якщо обидва електрони
розташувати на тій самій орбіталі, наприклад, Ь,тоЧ/І=Ч/п = (Ь^І^ісимет-
159
рична функція при цьому є простим добутком (1^)!(1у)2 одноелектронних
хвильових функцій (з точністю до нормувального множника), а антисимет-
рична — нулем. Отже, тотожні стани неможливо описати за допомогою
антисиметричної функції. Ця властивість антисиметричної функції наочно
виявляється, якщо її написати у вигляді визначника. Наприклад, для вихідних
Ія- та 2^-орбіталей
що повністю узгоджується з виразом (8.125). Якщо вихідні орбіталі тотожні
(Іята 1$),то
(і,)2(і,)2
= 0
(8.128)
(однією з основних властивостей визначника є рівність його нулю при
наявності тотожних рядків або стовпчиків).
На даній стадії вивчення теорії хімічного зв'язку корисно з'ясувати
поняття «орбіталь». Раніше воно було визначене як синонім терміна «хвильова
функція». Для систем, які містять більше одного електрона, поняття орбіталь
застосовується як синонім поняття одноелектронна хвильова функція.
Наприклад, можна говорити про 1$- та І8- орбіталі, але для функцій У¥А та Ч'д
можливі лише терміни: симетрична та антисиметрична хвильові функції.
Як було показано у підрозділі 8.2, врахування спіна електрона дає
додаткове виродження станів у атомах. Виродження станів у атомі Гідрогену
двократне відповідно до двох значень спінового магнітного квантового
числа т8 одного електрона. Позначимо спіновий стан із т5 = (+!/г) через а,
а з т8 = (-Л) через р. ос та Р можна назвати спіновими орбіталями. Із цих спін-
орбіталей треба скласти повні спінові функції для двох електронів. Якщо
розташувати два електрони (1 та 2) на двох орбіталях (а та Р), то всього
матимемо чотири комбінації Щ(Х2; «іР2;ос2Рі; РіР2. Повні спінові функції, як
і орбітальні, повинні бути або симетричними, або антисиметричними
відносно перестановки електронів. Функції а^2та Р^2 — симетричні
внаслідок еквівалентності електронів 1 і 2 (аналогічно координатній функції,
яка описує основний стан гелію *Р5 = (к)1(к)2). Позначимо ці функції так:
5 1 2 . (8.129)
^Ч=РіР2)
Індекс справа зверху показує значення проекції сумарного спіна, тобто
спінового магнітного числа М8 = Хт5 всього гелію. Спінові функції а^2 та
а2Р! — ні симетричні, ні антисиметричні. Вони перетворюються одна на
одну при перестановці електронів. З них необхідно створити лінійні
комбінації:
симетричну
160
та антисиметричну
«-^(«А-оД). <8131)
де $£ не змінює знак при перестановці електронів, а 5^ змінює знак на
протилежний.
В атомі Гелію з високим ступенем точності можна прийняти, що
енергія не залежить від орієнтації спіна, тому всі чотири спінові функції
відповідають однаковій енергії і є виродженими (зняття спінового виродження
і так звана тонка структура спектрів розглядаються далі у підрозділі 8.4). Але
експеримент показує, що в атомі Гелію чотирикратно вироджених рівнів
немає, а існують лише трикратно вироджені — триплети та невироджені —
синглети. Якщо придивитися до цих чотирьох спінових функцій, то не важко
побачити, що їх можна розділити на дві групи. 1) Я^1; $?; Зу1 — для цих
функцій повне спінове квантове число атома дорівнює 1, а його проекція М8
має відповідно три значення: +1,0, — 1 (через одиницю). Ця група утворює
триплет. 2) 8°А — антисиметрична спінова функція, для якої 5 = 0 та М8 = 0.
Це синглет. 5$ та 8А функції зображені на рис. 8.17.
?!?
І5'1, / \\ 5=о'
М4-0 /_іу\ Д£ = о
р <4 Ж А
' а ' б
Рис. 8.17. Симетрична 5$ (а) та антисиметрична 5^ (б) спінові функції
Згідно з експериментальними даними для всіх збуджених станів атома
Гелію триплети знаходяться нижче, ніж синглети, тобто триплети
відповідають станам, які описуються антисиметричною орбітальною хвильовою
функцією, а синглети — станам із симетричною орбітальною функцією.
Отже, спостерігається така закономірність: симетричній орбітальній функції
(Чу відповідає антисиметрична спінова (5^) і, навпаки, антисиметричній
орбітальній 0¥А) відповідає симетрична спінова (5$). При такому сполученні
161
орбітальних та спінових функцій повна
хвильова функція атома, яка дорівнює добутку
орбітальної функції на спінову, завжди є тільки ан-
тисиметричною. Це твердження називається
принципом Паулі і було встановлене В. Паулі
шляхом аналізу атомних спектрів.
Для збуджених станів атома Гелію, які
описуються симетричними та антисиметричними
орбітальними хвильовими функціями,
можливі як триплетні, так і синглетні спінові стани.
В основному стані, який описується лише
симетричною орбітальною функцією, спінова
функція може бути тільки антисиметричною
згідно з принципом Паулі. Тому основний стан
Гелію невироджений, синглетний.
Антисиметричні хвильові функції (Ф) слід записувати у вигляді
детермінанта, наприклад, для основного стану атома Гелію
|(І5)іаі 0*)2а2І
Паулі Вольфганг
(1900—1958), Швейцарія
1
Ф =-7=г
° >/2
/1"! \"/2
|04 Р, (і5)2р2
Для збудженого стану (1$2$):
1
= -^(1*)1(І5)2[а,р2-р1<х2]. (8.132)
Ф,=
72
Ф,=
Л
(Ь\щ (1*)2а2
(2*),аі (25)2а2
(1*), Р, (1*)2Р2
(25), р, (25)2Р2
= <£'¥
А<>
= ^,
(8.133)
(8.134)
Щоб отримати функції Ф3 = 5^ ^а та Ф4 = ^а ^$> треба брати лінійні
комбінації
Фз = ^ (Ф5 + фб ) та Ф4 = -±= (Ф5 - Фб) (8.135)
таких детермінантів, складених зі спінових та координатних орбіталей:
1(15), а, (і^)2ос2|
|(25), Р, (25)2р2|
|(25), а, (25)2а2|
Детальніше принцип Паулі та висновки з нього розглядаються далі у
підрозділі 8.4.
Ф>-Т2
Ф,=Ч=
72
(8.136)
(8.137)
162
8.4. Багатоелектронні атоми
Гей, електрони! Спін — наполовину!
Не можна лізти всім в одну клітину!
Тут зайвого не втиснеш електрона,
Бо пильно Паулі працює заборона!
Для системи з N електронів та А^орбіталей повну хвильову функцію Ф
можна було б скласти, розміщуючи перший електрон на першій орбіталі
(ф!), другий на другій (ф2) і т. д., а потім перемножити ці одноелектронні
хвильові функції
Ф = ф,(1) • ф2(2) -фз(3)... ф*(Л0. (8.13?)
Як можна було вже переконатися, розглядаючи будову атома Гелію, така
хвильова функція має принаймні два істотні недоліки. По-перше, вона не
враховує нерозрізненості мікрочастинок, неможливості приписати
окремому електрону відповідну орбіталь. По-друге, повна хвильова функція
повинна задовольняти принципу Паулі, тобто бути антисиметричною щодо
перестановки електронів. Обидві ці умови можна врахувати, якщо повну
хвильову функцію записати у вигляді детермінанта
\<Рі(і)^1(2):.,(і>1(М)
Ф2(1)-Ф2(2)---Ф2(^)|
Ф =
1
(8.139)
К(і)-Ф"(2).-.-Ф*(Л0|
Цей детермінант називається визначником (детермінантом) Слетера.
Кількість різних добутків, які виявляються при розкладанні визначника,
дорівнює N1, отже, множник і/-7м враховує нормування. В атомі Гелію
N = 2 і цей множник дорівнює 1/-\/2 .
Кожному номеру орбіталі відповідає свій набір квантових чисел.
Наприклад, орбіталі атома Берилію в основному стані можна пронумерувати
так: 1 — 1 з0а; 2 — І50Р; 3 — 2*0а; 4 — 2$0Р.
Квантове число / позначається відповідною літерою, а т, індексом
справа внизу біля цієї літери, а та Р відповідають значенням спінового
квантового числа т5 (+Уі) та (-'/г). Отже, кожна з одноелектронних орбіталей
задається чотирма квантовими числами: п, І, ті та т5 і відрізняється від іншої
одноелектронної функції принаймні одним із чотирьох квантових чисел. Для
атома Берилію повна хвильова функція має такий вигляд:
ІЦ,ос(1) 1^0а(2) І50а(3) Ц,а(4)|
_|_ ЦД1) 1*0Р(2) Ц,Р(3) ЦД4)|
УІ4І 250а(1) 2.у0а(2) 2я0а(3)
|2*0р(1) 2*0Р(2) 250Р(3)
Ф =
2*0а(4)|
2*0Р(4)
(8.140)
163
ф-^
Л5І
Повна хвильова функція однозначно записується у вигляді відповідного
детермінанта, коли кількість електронів дорівнює кількості орбіталей. Ця
ситуація здійснюється для заповнених оболонок: .у \р ; сі1 ;/ 4.
Подивимось, що вийде, коли кількість електронів перевищує кількість
орбіталей. Наприклад, спробуємо розташувати в атомі Гелію три
електрони, використовуючи тільки дві орбіталі: \з0а та Ь0Р. У цьому випадку два із
трьох електронів попадуть на ту саму орбіталь, наприклад, на ІяД і повна
хвильова функція матиме такий вигляд:
|ца(і) Цд(2) ца(3)|
ЦДІ) ЦД2) ЦДЗ). (8.141)
|і*0р(1) ЦДО) 1*0Р(3)|
У детермінанті (8.141) другий рядок дорівнює третьому, і тому цей
визначник тотожно дорівнює нулю. Отже, кількість електронів в атомі не
повинна перевищувати кількості орбіталей, і тому в атомі не може бути двох
і більше електронів з чотирма однаковими квантовими числами. Останнє
твердження є іншим (менш загальним) формулюванням принципу Паулі. Це
формулювання і було насамперед запропоновано В. Паулі і називалося
забороною Паулі. Воно дуже зручне при вивченні розподілу електронів
в атомі, побудові періодичної системи елементів і повинно враховуватися
також при вивченні системи атомних термів.
При вивченні атома Гелію було встановлено, що електронна
конфігурація Ь2^ (збуджений стан атома Гелію) складається із двох енергетичних
рівнів: синглету та триплету. Аналогічна картина спостерігається в усіх
багатоелектронних атомах — кожній електронній конфігурації відповідає
певний набір енергетичних рівнів — термів.
Розібратися у складній картині атомних термів можна за допомогою
векторної моделі атома, використовуючи принципи просторового
квантування. В одноелектронних атомах момент кількості руху дорівнює
моменту кількості руху окремого електрона і визначається квантовим числом /,
а його проекція — квантовим числом /Я/. Вектор спіна та його проекції
визначаються квантовими числами 8 та т5. Для характеристики орбітального
і спінового моментів багатоелектронного атома та їх проекцій
використовують аналогічні квантові числа: /, та М^ — для орбітального квантування;
5таМ$—відповідно, для спінового. Момент кількості руху для орбітального
руху в атомі задає квантове число І, яке приймає значення: 0,1,2,3,...
Проекцію Ь на вісь 2 характеризує квантове число Мь, яке набуває значень від
(-£) до (+1) через одиницю. Спінове квантове число £ має або цілі (0,1,2,
З,...), або напівцілі (1/2,3/2,5/2...) значення через одиницю. Відповідно М8
змінюється від (-5) до (+5) через одиницю. Кількість значень Мь дорівнює
(2Ь +1), а М8 —(28+ 1). Остання величина називається мультиплетністю
і позначається літерою М, тобто: М = 25'+1. Наприклад, при«5'=0 М= 1
(синглет), а при 5 = 1 М= 3 (триплет). Аналогічно тому, як для окремого
164
електрона замість / використовувалися відповідні літери, для визначення І
використовуються великі літери:
Величина! 0 12 3 4 5
Позначення 5 Р В Р ОН
Для атома Гідрогену терм ототожнювався з енергетичним рівнем і
визначався лише головним квантовим числом. При вивченні найпростішого
багатоелектронного атома Гелію встановлено, що енергетичні рівні
залежать від величини / (Ь) (для конфігурацій 1$2,у, Із2р, Ь-1 другого електрона,
тому що перший (1 ^-електрон) має / = 0) і різні для станів із різною мульти-
плетністю М(сумарним спіном). Згідно з даними для Гелію (див. підрозділ
8.3) триплет має меншу енергію, ніж синглет (для конфігурації Хзів). Тому
в багатоелектронних атомах термом називається енергетичний рівень (або
точніше, група близько розташованих рівнів) із заданими значеннями Ь та 5.
Враховуючи позначення для Ь та 5, терми називають так: Р — триплет Р,
8 — синглет 5 тощо.
Розглянемо, які терми можуть утворювати два/7-електрони із різними
головними квантовими числами, наприклад конфігурація 2р Зр (такі
електрони називаються нееквівалентними).
Згідно з векторною моделлю атома моменти кількості руху для окремих
електронів (А//) складаються та утворюють момент кількості руху атома:
М = £М/. Це співвідношення використовується як для орбітального, так
і для спінового руху. Отже, для орбітального руху М визначається
квантовим числом І, а Мі9 відповідно, квантовими числами /,. Для спінового руху
М визначається квантовим числом 5 (мультиплетністю), а
М{—квантовими числами зі%
Для/7-електронів Іх = /2 = 1 і \М{\ = \М2\ = л/2. При 1 = 0 \М\ = 0; приЬ = 1
|М|=7Д^+Ї)=>/Г:2 = 1,41; при/, = 2 \М\ = ,/2(2 + 1) =л/б = 2,45.
Згідно з векторною схемою, М утворюється з Мх та М2:
1 = 0 М 1=1 м 1=2
Значенням Ь = 0,1,2 відповідають терми: 5, Р, В.
Аналогічно, якщо додати спіни двох електронів (5і! = 1Л; $2= У*)> то для
спіна атома отримаємо значення: 5 = 0 та 1, відповідно, мультиплетність
дорівнює 1 або 3. Отже, кожний з наведених вище термів може бути синг-
летом або триплетом. Повний набір термів для конфігурації 2р Зр такий: 5;
35; 1Р; 3Р;'/); 3/Х
Виведення системи термів можливо робити через мікростани. Кожен
мікростан задається квантовими числами т/ та т5. Для т3, як і раніше,
застосовуємо позначки: а — (+14); Р — (-У*)- Наприклад, мікростан для
окремого електрона з т7 = (-1) та т8 - (+!/г) позначається як (-1а). Для окремого
електрона нар-орбіталі можливі такі мікростани: 1а; 0а; (-1а); 1|3; 0р; (-1|3),
тобто всього 6 мікростанів (для кожного електрона можливе (2/+ 1)-кратне
165
виродження за / (для/7-електронів — трикратне) та двократне виродження
за спіном). Комбінуючи шість мікростанів одного/7-електрона з кожним із
шести мікростанів іншого/7-електрона, дістанемо 6-6 = 36 мікростанів (в
конфігурації 2/7 3/7 електрони нееквівалентні, отже, заборони на комбінування
мікростанів немає).
Алгебраїчна сума т/ визначає Мь а сума т8—М8. Наприклад, для мікро-
стану (-1 а) 1 а Мь = -1 +1 = 0, а М8 = +1А + Уг = 1. У табл. 8.3 зображені всі 36
мікростанів для двох нееквівалентних/?-електронів, згрупованих за Мь та М8.
Таблиця 8.3. Мікростани для двох/^електронів
2
1
0
-1
-2
1
ІОСІОС
ІаОа
Оосіос
ОаОа
(1а)(-1а)
(-1а)(1а)
0а(-1а)
(-Іа)Оа
(-1а)(-1а)
0
іаїр
іріа
ІаОР оріа
Оаїр ІрОа
Оавр 0Р0а
(-іа)ір 1р(-1а)
(-іР)Іа 1а(-1р)
(-1а)0Р 0р(-1а)
0а(-іР) (-іР)Оа
(-1а)(-ір)
(-іР)(-1а)
~*
1Р1Р
ірор
орір
орор
(-ІР)ІР
ІРПР)
(-тор
0Р(-1р)
(-1Р)(-1Р)
Користуючись таблицею 8.3, не важко вивести систему термів для
конфігурації 2/? 3/7. Для цього потрібно зробити ряд послідовних операцій.
Спочатку шукаємо максимально можливе значення Мь. В даному випадку
д^тах = 2 Оскільки М™8* не може бути більше, ніж Ь, то мікростани з Мь=
= 2 відповідають терму О. Для даного значення Ь є стан М™3* = 1, отже, 5 =
= 1 таМ= 3 і маємо терм В. Відбираємо для нього 15 мікростанів (Мь від
(+2) до (-2), тобто в першому-п'ятому рядках; М8 від (+1) до (-1), тобто
в першому-третьому стовйцях). У клітинці з Мь = 2 та М8=0 ще залишається
один мікростан, який відповідає терму О. Відбираємо для цього терма ще
п'ять мікростанів у другому стовпці.
Після цього залишається М™** =1 при М™3* =1. Відбираємо дев'ять
мікростанів для терма Р. Із клітинки Мь = 1 та М8 = 0 відібрано три
мікростани (по одному для термів Д £>та Р). У цій клітинці залишається ще
один мікростан, який входить до терма Р Відберемо для цього терма три
мікростани у середньому стовпчику. Після цього залишається всього
чотири мікростани в рядку з Мь = 0. Три із них утворюють 5-терм. Останній
мікростан, розташований у клітинці з Мь = 0 та М8 = 0. належить терму «У.
Отже, отримаємо наведену вище систему термів: Д Д Р, Р, 8, 8.
Для Д-термів виродження по Ь п'ятикратне: для Р-термів — трикратне,
терми 5 не вироджені. Відповідно, виродження за спіном у триплетних
166
термів трикратне, синглети не вироджені. Виродження отриманих термів
таке: ^ — 1; 35 — 3; 1Р — 3; 3Р — 9; О — 5; 3£> — 15. Сумарна кількість
вироджених станів для конфігурації 2р Зр така: 1 + 3 + 3 + 9 + 5 + 15 = 36, що
збігається з наведеним вище числом.
Розглянемо тепер конфігурацію 1$ 2$ . Для цієї конфігурації мікростанів
чотири: ОаОос, ОаОР, 0Р0а та 0|30р. Тут Хт/ = 0 і терми—лише & Один з них —
терм 5, для якого М™ах = 1,5= 1 та М= 3. До нього належать мікростани:
ОаОос, ОРОР та один з двох: ОаОР або 0Р0ос. Другий з цих двох останніх
мікростанів дає терм 5. Мікростани ОаОос та ОРОР відповідають розглянутим у
підрозділі 8.3 функціям (8.133) та (8.134), а мікростани ОаОР та
ОрОос—функціям (8.135).
Для нееквівалентних електронів схема виведення системи термів через
мікростани складніша, ніж векторні уявлення, але для еквівалентних
електронів вона є єдиною можливою. Еквівалентні електрони мають одне і те
саме значення головного (п) та орбітального (/) квантових чисел, тому згідно
із принципом (забороною) Паулі деякі мікростани для них заборонені.
Наприклад, для двох еквівалентних ^-електронів заборонений мікростан
0а0а[І5,оа(1); 1,у0а(2)], тому що тут у обох Ь-електронів однакові спіни (т8 =
= + Уг) і, отже, однакові чотири квантових числа. Для нееквівалентних
електронів мікростан ОаОос можливий — заборона Паулі не порушується
внаслідок різних значень головного квантового числа п (1$0а; 2у0а). Із 36
комбінацій двох нееквівалентних/7-електронів для еквівалентних залишаються
тільки 15. Зазначимо, що кількість можливих комбінацій скорочується не
тільки через заборону Паулі, а й тому, що деякі з комбінацій є тотожними
внаслідок еквівалентності електронів, наприклад, Іаірта іріа. Кількість
різних комбінацій без повторів та перестановок дорівнює кількості
сполучень з п елементів по т:
™ пі
т\{п-т)\
де т — кількість електронів; п — кількість спін-орбіталей — число
можливих комбінацій для кожного електрона. Для двох /7-електронів т = 2, п = 6 та
Сб~2!4Г15'
У таблиці 8.3 ці п'ятнадцять мікростанів виділені курсивом. Вони
відповідають Д Рта 5-термам п'яти-, дев'яти-та однократно виродженим. Ці
терми входять до числа термів, які зустрічалися для нееквівалентних/7-елек-
тронів.
Для двох еквівалентних сі -електронів маємо таку систему термів: О; Р;
1А3Р;Ї5'. '
Розглянемо будову р -оболонки. У цьому випадку принципу Паулі
відповідає тільки один мікростан, а саме: 1а, Оа, (-1а), 1р, ОР, (-1Р). Для нього
Мь = 1ип1 = 0 та М8 = 2>% = 0 тобто одержимо невироджений терм 5. Це
справедливо для будь-яких замкнутих оболонок: я ,р ,й , / . Задати стан
167
Гунд Фрідріх
(1896—1997), Німеччина
для замкнутої оболонки можна лише одним способом (при т = п С™ = 1).
Невиродженість замкнутих оболонок спрощує атомні спектри, оскільки
внутрішні електрони в атомах групуються, утворюючи замкнуті оболонки
і, отже, важливим для хімії стає лише вивчення зовнішніх електронів.
У системі термів для даної конфігурації терм
^найменшою енергією називається основним.
Його можна знайти, використовуючи правіша
Гунда.
Правило 1. У системі термів найменшу
енергію має терм із максимальною мульти-
плетністю.
Правило 2. Якщо мультиплетність термів
однакова, то найменшу енергію має терм з
максимальним значенням Ь.
Результати, що одержані для атома Гелію,
підтверджують, що для конфігурації 1$ 2з
Гелію триплет має меншу енергію, ніж синглет.
Згідно з першим правилом Гунда, для
конфігурації^ основний терм— Р Для
конфігурації яҐ за допомогою першого правила знаходимо терми і7 та Р, згідно
з другим правилом, знаходимо основний терм Е Аналогічна ситуація і для
двох нееквівалентних /^-електронів. За допомогою першого правила
знаходимо О-, Р-та 5-терми, а за допомогою другого правила— £>-терм.
Правила Гунда дають можливість знайти основний терм, не виводячи
всю систему термів. Для цього потрібно розташувати електрони на орбіта-
лях так, щоб була максимальна можлива кількість неспарених електронів
(максимальне значення М8 і, отже, 15 та мультиплетність) і значення М^
і, отже, і.
Як приклад, розглянемо два еквівалентні /^-електрони. Позначимо
умовно орбіталі клітинками із значенням т7. Розміщуємо на цих орбіталях
два електрони так, щоб вони по одному займали клітинки з максимальним
значенням /Я/. Два електрони з однаковими спінами неможливо
розміщувати в одній клітинці внаслідок заборони Паулі. Електрони позначаємо
стрілками: вгору (Т), якщо т5=+1Л, та вниз (і), якщо т8--хА\
&!/=+ 1+0=1 =М£ШХ;І = 1;термР.
Іт =+1/2 + 1/2=1=МГх;5=1;М = 2-1+1=3.
+1 0 -1 лі/
Отже, основний терм для двох еквівалентних/^-електронів — Р.
Для двох нееквівалентних/^-електронів, наприклад, конфігурації 2р Зр
маємо:
т т
Зр
Т
Іт, = + 1 + 1 = 2=М£МХ; І = 2; терм Б.
+1 0 -1 т,
168
2Р І Т 1 1 1 2т5= + >/2 + »/2 = 1=М^іах;5=1;М = 2-1+1=3.
+1 0 -1 т1
Основний терм — Д що узгоджується з викладеним вище.
Як було показано у підрозділі 8.3, розщеплення електронних
конфігурацій на мультиплетні терми виникає, коли у гамільтоніан введено вираз
і*} У
(гу— відстань між і'-м тау-м електронами), тобто внаслідок міжелектронного
електростатичного відштовхування. Спін електрона виявляв свій вплив тоді,
коли це вимагає принцип Паулі — обмінне виродження існує для повної
хвильової функції. Енергетичною взаємодією, пов'язаною із наявністю спіна
в електрона, до цього часу нехтували. Урахування цієї взаємодії, яка
називається спін-орбітальною, пояснює тонку структуру атомних спектрів.
У багатоелектронних атомах сумарний спіновий момент
£ = ]££„,
і
2і сумарний орбітальний момент
і
Величина тонкого розщеплення пропорційна скалярному добутку Ь
та 5. Введемо повний момент кількості руху всіх електронів згідно зі
співвідношенням
^ = ^+5.
(8.142)
Векторна рівність (8.142)
зображена на рис. 8.18. Тоді
для скалярного добутку І8
одержимо
І5 = У2-ї?-52. (8.143)
Повний момент кількості
руху залежить від квантового
числаУ
1/1 = ^/7(7+1)», (8.144)
де 3 має цілі або напівцілі
значення (через одиницю).
Квантове число М^
визначає проекцію 1 на
обраний напрямок у просторі
і має значення від (-/) до
(+7), тобто всього (27+1)
значень.
Електростатична
взаємодія
5
Спін-орбітальна
взаємодія
Рис. 8.18. Ь-8 зв'язок
169
Внаслідок спін-орбітальної взаємодії кожний терм розщеплюється на
ряд енергетичних рівнів з різними значеннями У. Оскільки для даного терма
Ь та 5 визначені, то Утах=Ь + 8, а Утіп=1-5, інші значення /розташовані між
Лііп та Ліах- При 5 = 0*Утах = Утіп = І, тобто синглетні терми не
розщеплюються. При Ь = 0 Утах = Утіп = 5, тобто 5 терми також не розщеплюються.
Для електронної конфігураціїр розщеплюється тільки Р-терм. Для
ньогоУтах=1 + 1=2, Уті=1-
1 = 0 і можливі три значення У: 0,1 та 2, тобто
Р-терм розщеплюється на три енергетичні рівні: Р0, Рх та Р2або
скорочено Р0}12- Кількість можливих значень У дорівнює мультиплетності терма.
Порядок розташування рівнів з різними У в термі визначається третім
правилом Гунда: якщо оболонка заповнена менш ніж наполовину, то рівні
енергії розташовуються в порядку збільшення У; якщо ж оболонка
заповнена більш ніж наполовину, тоді в протилежному порядку.
Для Р-термар -конфігурації порядок такий: Р0, Рх, Р2; для
аналогічного Р-терма, але/? -конфігурації — порядок протилежний.
Рівень, який відповідає даному значенню У, вироджений (2У+ 1 )-кратно
згідно з можливими значеннями М7. Виродження за У знімається при
накладанні магнітного поля. Загальна кількість рівнів при цьому дорівнює
повному ступеню виродження терма
•Лтіах
£(2У + 1) = (2І + 1)(25 + 1).
Наприклад, для Р-терма: (2Ь + 1) = 3; (28 + 1)=М = 3, і терм вироджений
дев'ятикратно. Рівень Р0 — не вироджений; рівень Рх— вироджений
трикратно; рівень Р2 — п'яти-
'5 '5° .і#.-о}*
кратно, що дає у сумі 9. Схему
енергетичних
Нульове Міжелектронне Спін-орбі- Дія зовнішнього
набли- відштовхування тальна магнітного поля
ження взаємодія
Рис. 8.19. Розщеплення енергетичних рівнів
для /^-конфігурації
170
розщеплення
рівнів для/? -конфігурації зоб
ражено на рис. 8.19.
Система термів є дуже
важливою при розшифровці
спектрів. Як приклад, на рис. 8.20
наведено діаграму
енергетичних рівнів атома Карбону,
який як основну електронну
конфігурацію має 2з2р\
Оскільки 2з — відповідає
повністю заповненому рівню, то
реалізується розібрана вище
конфігурація/? , яка
в.основному стані розшеплюється на
терми: Р, В та 5. Саме з цих
термів, як це видно з рис. 8.20,
відбуваються електронні пере-
11,217
10,000
6,000
6,000
4,000
2,000
0,000
Ар\ 15
І^рпр Ів^пв Із^рпсі
=_%ьр——т> иЬ
——Ьая'р-
-і юооо
20 000
30 000
4 40 000
50 000
4 60 000
Н 70 000
"І 80 000
90 00
Рис. 8.20. Діаграма енергетичних рівнів атома Карбону з зазначенням
переходів, які спостерігаються
ходи на терми, що виникають із збуджених електронних станів. З
урахуванням спін-орбітальної взаємодії і розщеплення рівнів, виникає тонка
структура спектрів, яка зображена на рис. 8.21, с. 172.
Виникнення системи термів для даної електронної конфігурації
зумовлене електростатичним відштовхуванням між електронами (поява в гаміль-
тоніані членів є Іг^. При розрахунку енергетичних рівнів за допомогою
теорії збурень (див. підрозділ 8.3) виникають кулонівські та обмінні інтеграли.
Частини кулонівських та обмінних інтегралів, які залежать від кутів,
інтегруються легко і можуть бути затабульовані у вигляді числових коефіцієнтів, які
залежать від значень квантових чисел /та т/. Обчислення радіальних
інтегралів можна проводити за допомогою радіальних частин Кп1(г) орбіталей
гідрогенподібного атома. Більш точний результат, ніж гідрогенподібна
функція, дає екранована хвильова функція, як це показано в підрозділі 8.3
при розрахунку енергії основного стану атома Гелію. Ідеї екранування
можна застосувати для побудови хвильових функцій будь-якого атома.
171
Відносні рівні енергії
3Р0
64 093,18 см"1
64 092,01 см"1
64 088,56 см-]
43,50 слг1
16,40 см"1
0,00
Рис. 8.21. Переходи з підрівнів основного стану гР атома Карбону
на підрівні першого збудженого стану 3£>
Дж. Слетер та К. Зінер запропонували прості правила для розрахунку
константи екранування та обґрунтували їх за допомогою варіаційного
методу.
Константа екранування (а) включає такі складові частини:
1) для й- та/-орбіталей по 1,00 від кожного електрона з головним
квантовим числом, меншим від даного;
2) для 5- тар-орбіталей по 0,85 від кожного електрона з головним
квантовим числом, на одиницю менше від даного, і по 1,00 від усіх більш глибоко
розташованих електронів;
3) по 0,35 від кожного електрона з тим самим значенням п (для 1$ по
0,30 — останній результат—а = 0,3125 одержаний у підрозділі 8.3
варіаційним методом).
Електрони з орбіталей більш зовнішніх, ніж дана орбіталь, внесок у а не
дають.
Дж. Слетер та К. Зінер запропонували також для одержання більш
точних результатів застосовувати замість головного квантового числа п
ефективне числоп*. Прип = 1,2та3 п* = п; прип = 4 п* = 3,7; прип = 5 я* = 4
і при я = 6 п* = 4,2. Відмінність між значеннями п та п* виникає внаслідок
поляризації остова зовнішнім електроном.
Згідно з підрозділом 8.2 при п > 1 та / < (п - 1) поліном /й/(г) містить
більше, ніж один член, і обчислення з такими багатовузловими функціями
стають важкими. Дж. Слетер та К. Зінер показали, що в поліномі істотне
значення має лише член з максимальним степенем відносно г. Спрощення
172
функцій після відкидання нижчих членів полінома (там, де вони є)
компенсується поліпшенням орбіталей — заміною 2 на 2* та п на п*. Тоді орбіталі
Слетера-Зінера мають такий вигляд:
2 г
К*(г) = гп*-Іе
Щг)к
(8.145)
Значна перевага функцій Слетера-Зінера полягає в тому, що вони мають
зручний для розрахунків аналітичний вигляд.
Енергію термів точніше можна обчислити, використовуючи метод само-
узгодженого поля Хартрі (див. підрозділ 8.3). В цьому випадку хвильова
функція виражається як добуток одноелектронних орбіталей.
Якщо при розрахунках використовується антисиметрична повна
хвильова функція у вигляді
детермінанта Слетера (8.139), то
метод Хартрі розширюється
та перетворюється на метод
самоузгодженого поля з
обміном Хартрі-Фока. Як перше
наближення для самоузгодже-
них обчислень можна брати
функції Слетера-Зінера.
Обчислення за методом Хартрі-
Фока дають певне уявлення
про пошарове заповнення
електронних оболонок в атомі. На
рис. 8.22 наведено розподіл
електронної густини для атома Гелію та іона Натрію за методом Хартрі-Фока,
де чітко видно одну оболонку {п = 1) в атомі Гелію та дві (п = 1 та п - 2) в іоні
Натрію. На рис. 8.22 можна також побачити, що радіус внутрішньої оболонки
зменшується зі збільшенням заряду ядра згідно з формулою (8.67).
Пошарове заповнення оболонок в атомі відображає фізичну суть
періодичного закону. В періодичній системі
Менделєєва першорядне значення має поняття
порядкового номера елемента (2). Це
поняття було застосовано, фактично, ще
Менделєєвим, який в деяких місцях своєї таблиці
відступив від розташування елементів у
порядку збільшення атомної маси та надав
більшого значення періодичності
властивостей, насамперед хімічних. Подальші
дослідження показали, що номер елемента має
глибокий фізичний зміст—це заряд ядра 2.
Оскільки ядра складаються з позитивно
заряджених протонів та незаряджених
нейтронів, то заряд ядра дорівнює кількості про-
1 2 г, а.о
Рис. 8.22. Розподіл електронної густини
за методом Хартрі-Фока
Менделєєв Дмитро Іванович
(1834—1907), Росія
173
тонів. Для багатьох елементів кількість нейтронів у ядрі приблизно дорівнює
кількості протонів. Маси протона та нейтрона близькі, тому атомна маса (А)
елементів (у вуглецевих одиницях) приблизно дорівнює подвійному заряду
ядра: А = 22. Завдяки цьому розташування елементів у порядку
збільшення атомної маси (як вважав Д. І. Менделєєв) дає (за деякими випадками)
практично такий самий результат, що й розташування у порядку
збільшення заряду ядра. Оскільки атом — це електронейтральна частинка, то повна
кількість електронів в атомі дорівнює 2, і періодичний закон насамперед
проявляється у періодичності зміни структури електронних оболонок
атомів.
При заповненні електронних оболонок в атомі треба враховувати два
принципи. По-перше, кожний електрон намагається зайняти найнижчий
вільний рівень енергії, по-друге, діє заборона Паулі — електрони на
орбіталях повинні відрізнятися хоча б одним квантовим числом. Ці два
принципи (заборона Паулі та вимога мінімуму енергії) є основою
«принципу побудови».
Якщо розташовувати електрони на гідрогенподібних орбіталях, то
заповнення проходило б по шарах, що відповідають значенням головного
квантового числа—п. Максимальне виродження (див. підрозділ 8.2) по п
дорівнює п2 без урахування спіна, а з його урахуванням — 2л2. Отже, в першому
шарі є можливість розташувати два електрони, в другому—вісім, у
третьому —вісімнадцять і т. д. Певною мірою це відповідає дійсності. Спочатку
заповнюється шар з п = 1, на Ія-орбіталі розташовуються послідовно два
електрони,— це Гідроген (І51) та Гелій (1я2). Згідно з принципом Паулі,
подальше заповнення цього шару не відбувається, і електрони розташовуються в
другому шарі, який налічує вісім елементів (Ьі, Ве, В, С, И, О, Р, Ие). У
третьому шарі ця закономірність порушується. Він вміщує тільки вісім
електронів (№, М§, А1,8і, Р, 8, СІ, Аг) замість очікуваних вісімнадцяти. У
наступного за Аргоном елемента—Калію — вже починає заповнюватися
четвертий шар. Порушення «ідеальної» картини (на основі гідрогенподібних орбі-
талей) викликано взаємодією електронів, яка знімає виродження по /. Рівні
з різними орбітальними квантовими числами (при однакових п) мають у
багатоелектронних атомах різну енергію, причому більш висока енергія
відповідає більшим значенням /. Наприклад, для атома Гелію доведено, що
збуджений 2у-рівень має меншу енергію, ніж 2р. Залежність енергії від
квантового числа / приводить до того, що навіть у другому періоді, де «ідеальна»
картина підтверджується, спочатку заповнюється електронами 2^-орбіталь
(Ьі, Ве), а тільки потім 2/?-орбіталь (В, С, >І, О, Р, №).
Отже, в багатоелектронних атомах треба розглядати енергію як функцію
двох квантових чисел: п та /. Заповнення електронних оболонок в атомах
майже без винятків відповідає правилу «п + /, л», згідно з яким оболонки в
атомах заповнюються електронами в порядку збільшення суми головного
та орбітального квантових чисел, а при заданій сумі (п + /) — в порядку
збільшення п. Наприклад, для суми (п +1) = 7 порядок буде такий: 4/1 5сІ, 6р,
174
І8. Найбільш чітко правило «п + /, п» сформулював та розвинув у своїх
працях В. М. Клєчковський.
У випадку близькості величин енергій електронних оболонок можливі
деякі порушення закономірності їх заповнення. Наприклад, можливі зміни
в електронній структурі типу (п -1)д, V —> (п -1 )сІч 8 при ($ + 1) =5 або
10, тобто для заповнених повністю або наполовину оболонок, які мають
підвищену енергетичну стабільність і, відповідно, нижчу енергію (атоми
Хрому, Купруму тощо). Дуже близько розташовані за енергією 4/- та 5<1~
оболонки і особливо 5/- та ^/-оболонки. Це призводить до порушення
правила «п + /, п». На початку рядів лантаноїдів спочатку з'являється 5а?-елек-
трон в атомі Лантану і лише потім заповнюється 4/-оболонка у наступних
членів цієї групи, аналогічна картина спостерігається для актиноїдів.
Електронні конфігурації атомів позначаються відповідними орбіталями
та кількістю електронів на них. Наприклад, електронна конфігурація Гелію —
Ь2, Нітрогену— І522з22р3 тощо. Електронні конфігурації для внутрішніх
електронів зазвичай замінюють на позначення відповідного атома
інертного газу, наприклад Нітроген — (Не) 2з22рг; Ферум — (Аг) 4у23</6; Цезій —
(Хе) б.?1 іт.д.
Інформацію про найбільш стійку електронну конфігурацію атома дає
дослідження системи термів. Наприклад, для конфігурації (п - 1)сі5П51
основний терм — 5; для конфігурації (п - \)<іА№2 основний терм — £>. Основний
терм 5 характерний для Хрому, а В — для Вольфраму, отже, Хром має
електронну конфігурацію зовнішньої оболонки Зс154519 а Вольфрам —
5(146з2. Нікелю з електронною конфігурацією ЗсіНз2 відповідає основний
терм Р, Паладію з 4я?10— терм 5, а Платині з 5с196з — £>.
Важливою характеристикою атома є енергія його іонізації— енергія,
яку необхідно затратити, щоб відірвати електрон від атома (іона) і перевести
його у нескінченність. При цьому вважається, що атом (іон) знаходиться в
основному стані (замість терміна «енергія іонізації» деякі автори вживають
менш коректний термін «потенціал іонізації»; потенціал іонізації
пропорційний енергії іонізації і чисельно дорівнює їй, якщо енергію вимірювати в
електрон-вольтах, а потенціал у вольтах). Приклади: енергія іонізації атома
Літію — це енергія процесу Ьі—> Ьі + є, яка дорівнює 5,39 еВ; енергія
іонізації іона Ьі — це енергія процесу Ьі —> Ьі + е> яка дорівнює 75,62 еВ.
Використовують такі терміни: перша енергія іонізації (І{), друга (І2) і т. д.,
наприклад для Літію І{ = 5,39 еВ, І2=75,62 еВ і т. д.
Для Літію І2» І\т Можна припустити, що збільшення енергії іонізації
обумовлене тим, що другий електрон відривається не від нейтрального атома,
а від позитивно зарядженого іона, і це призводить до збільшення кулонівсь-
кого притягання між частиною атома (іона), яка залишається, і тим електроном,
що відривається. У тому, що цей фактор відіграє важливу роль, можна
переконатися, якщо розглянути кількісні дані для атома Гелію. Енергія
іонізації гідрогенподібного іона Не згідно з формулою (8.59) 2/2=2 а.о.е. =
= 54,4 еВ = І2. Енергія основного стану атома Гелію (див. підрозділ 8.3)
175
дорівнює -2,90 а. о. є. = -78,88 еВ, звідки /, = £0(Не+) - £0(Не) = (-54,4) +
+ 78,88 = 24,58 еВ. Отже, /2>Л>Ш1& різниця між значеннямиІ2 таІх для Гелію
не така велика, як для Літію.
Для Берилію Іх = 9,32 еВ, І2 = 18,21,13 = 153,85 еВ. Збільшення від Іх до І2
того самого порядку, що й для Гелію, зумовлене зростанням заряду ядра,
проте значення /3 значно більше, ніж Іх та І2. Причина цього різкого
зростання при переході від і, до І2 у Літію та від І2 до І3 у Берилію пов'язана з
переходом від іонізації 2$ -електронів до іонізації глибше розташованих Ь-елек-
тронів.
У таблиці 8.4 наведені значення енергії іонізації від першої до восьмої
групи перших дев'яти елементів періодичної системи. При переході від шару
з п = 2 до шару з п = 1 завжди спостерігається різке збільшення відповідної
(/#+і> де N— номер групи) енергії іонізації. Отже, різке (на порядок і більше)
зростання енергії іонізації свідчить про те, що має місце іонізація глцбше
розташованих внутрішніх електронів.
Таблиця 8.4. Енергії іонізації елементів 1-го та 2-го періодів
Атомний
номер ;
1
2
3
4
5
6
7
8
9
Елемент
Н
Не
Ьі
Ве
В
С
N
0
Р
/і
13,60
24,58
5,39
9,32
8,30
11,26
14,54
13,61
17,42
1г
54,40
75,62
18,21
25,15
24,38
29,61
35,15
34,98
Енергія іонізації, еВ
/з
и
п
Д
122,42
153,85
37,92
47,86
217,66
259,30
64,48
47,43 77,45
340,13
391,99
97,86
54,93 77,39 113,87
62,65
87,23
114,21
489,84
551,93
138,08
157,12
її
666,83
739,11
185,14
/8
871,12
953,60
Енергія іонізації дає змогу розділяти електрони на внутрішні та зовнішні.
Енергії іонізації внутрішніх електронів дуже великі, тому вони не беруть
участі у процесах утворення хімічного зв'язку. В хімічній перебудові беруть
участь лише зовнішні валентні електрони. Внутрішні електрони
називаються електронами остова. Остов атома майже не змінюється при
утворенні атомом хімічного зв'язку. У багатьох випадках остов має структуру
заповнених електронних оболонок (благородних газів), тобто пз2пр6(для п > 2)
чи Із2 (для п = 1).
Найпростіше можна оцінити енергію іонізації для атомів лужних металів,
які мають на зовнішній орбіталі лише один електрон. Енергія іонізації
лужного металу дорівнює енергії електрона на зовнішній орбіталі з
протилежним знаком. Після видалення електрона залишається однозарядний іон.
Тому енергію іонізації можна обчислювати за формулою (8.58),
використовуючи ефективні 2* та п * і враховуючи, що /=-Е, тобто
176
І = -Е--
(2*)2
*Ч2"
2(іГ>
(8.146)
Невелике збільшення Ґ при переході від Літію до Натрію не компенсує
різкого зниження /внаслідок збільшення и*; при русі далі вниз по ІА групі
2* залишається сталим, а п*
збільшується. Отже, енергія
іонізації лужних металів
зменшується у групі зверху вниз.
Це можна побачити на рис.
8.23, де наведено
експериментальні значення енергії
іонізації атомів лужних
металів. Зниження енергії
іонізації елементів характерне і для
інших груп періодичної
системи при русі зверху вниз.
Розглянемо тепер, як змі-
ДеВА
6,0 к
5,0
4,0
Ьі Иа К КЬ
С§
Лужні метали
Рис. 8.23. Енергія іонізації лужних металів
нюється енергія іонізації вздовж періоду. Формула (8.146) придатна тільки
для розрахунку енергії іонізації елементів, у яких на зовнішній оболонці є
лише один електрон. Якщо електронів кілька, то обчислення енергії іонізації
здійснюється за формулою
/ = Е^-Е^ (8.147)
7=1 /=1
де Л^та к—відповідно кількість електронів в атомі та іоні (для першої енергії
іонізації к:=И-\)\ЕігїдіЕ] — енергія електрона в атомі (і) або іоні (/).
Оцінимо £,(£,) за формулою типу (8.146). Для другого періоду п* = п =
= 2 = сопві. Константа екранування кожного електрона в шарі дорівнює 0,35,
отже,
= 2Г.
(8.148)
^ атома "™5-^-
При переході від Літію до наступних елементів зміщення на одну
клітинку вправо збільшує заряд ядра на одиницю, а екранування додатковим
електроном становить усього 0,35, тому упродовж періоду значення 2*
збільшується (враховано, що 2и = 1,3):
<тома =2и+(#-і). 0,65 = 0,65.(^ + 1). (8.149)
Використовуючи співвідношення (8.148 — 8.149) з урахуванням п = 2 та
к = И- 1, одержимо просту формулу для обчислення /як функції тільки
кількості електронів
/ = 3,385 + 2,446ЛГ-0,1ЦГ
(у формулі (8.150) значення/наведено в електрон-вольтах).
(8.150)
177
На рис. 8.24 дано залежність енергії іонізації (обчислена за формулою
(8:150) та за експериментальними даними) від порядкового номера для
елементів другого періоду. Формула (8.150) добре відображає основну
тенденцію зміни енергії іонізації вздовж періоду — поступове зростання від
лужного металу до
благородного газу внаслідок
збільшення заряду ядра
при недостатньо
ефективному екрануванні
електронами того
самого шару та різке
зменшення при
переході до наступного
періоду.
Такий підхід, однак,
не враховує деяких
ефектів, які
виявляються при аналізі
експериментальних енергій іо-
Р Ие №
Елементи
Рис. 8.24. Енергія іонізації елементів другого
періоду
нізації, зокрема функція Слетера-Зінера не враховує різниці між .?- та
р-орбіталями, а 2/?-рівень лежить вище, ніж 2$-рівень. Тому при переході від
Берилію до Бору енергія іонізації знижується (електрон легше відривається
із 2/7-рівня, ніж із 2^-рівня). Спостерігається, по суті, те ж саме, що й при
переході від Неону до Натрію—зменшення енергії іонізації при переході від
заповненої оболонки до наступної — у Берилію якраз заповнюється
2^-рівень, у Бору починає заповнюватися 2/?-рівень, тому існує відносний
максимум енергії іонізації на Берилії.
Відносний максимум /можна спостерігати і для Нітрогену. Його
електронна конфігурація 2з22р3. В цій конфігурації для кожного електрона
зовнішнього шару є своя орбіталь: 2рХ9 2ру або 2р2. При появі ще одного електрона
останній потрапляє на орбіталь, уже частково зайняту іншим електроном.
Це призводить до сильнішого міжелектронного відштовхування, ніж тоді,
коли всі електрони знаходяться на різних орбіталях. Отже, енергія електронів
зовнішнього шару в Оксигену збільшується, відповідно, енергія іонізації
зменшується. Доречно також зазначити, що в атомі Нітрогену при
розташуванні 2р1х2р1у2р2 немає спарених електронів і, згідно з першим правилом
Гунда, це повинно вести до появи в системі термів з низькою енергією.
Спорідненість атома до електрона (є) визначається як енергія, яка
виділяється при приєднанні до атома електрона, або як енергія іонізації
відповідного однозарядного аніона. При приєднанні електрона до атома
утворюється аніон, який має електронну конфігурацію таку ж саму, як і
наступний по порядку елемент (атом). Такі системи називаються ізоелектрон-
ними. Залежність спорідненості до електрона вздовж періоду повторює
залежність перших енергій іонізації з двома характерними рисами: 1) все
178
зміщене на одну клітинку вліво, наприклад, спорідненість до електрона
атома Флуору—це енергія іонізації аніона Флуору, що ізоелектронний з
атомом Неону (Флуор на одну клітинку знаходиться лівіше від Неону); 2)
відповідна ламана лінія роз- „
міщена нижче, тому
що при одній і тій
самій електронній
конфігурації заряд ядра
відповідного аніона
менший на одиницю,
ніж атома — електрон
легше відривається від
аніона, ніж від атома.
Це підтверджується
наведеними на рис.
8.25 даними про спо-
рідненістьдоелектро- Іл Ве В С N О Р Ке
на елементів другого Рис» 8.25. Спорідненість до електрона елементів
періоду залежно від другого періоду
атомного номера (ламана повторює ламану для перших енергій іонізації,
див. рис. 8.24).
Максимальні значення спорідненості до електрона мають галогени
(у другому періоді Флуор), тому що аніони Х~ ізоелектронні з інертними
газами, що мають максимальні енергії іонізації. Мінімальні значення є мають
інертні гази, відповідні аніони ізоелектронні лужним металам, від яких
електрон відривається найлегше. Значення є для багатьох елементів близькі до
нуля і навіть від'ємні, тобто утворення відповідного аніона невигідне. Значні
величини є у галогенів та Оксигену.
Суму /та є Р. Маллікен запропонував вважати мірою електронегатив-
ності (%) елемента, тобто
% = / + є.
(8.151)
Атом з меншим значенням електронегатив-
ності має тенденцію до перенесення
електронної густини в молекулі на більш
електронегативний елемент (див. наступний підрозділ 9.1).
Згідно з емпіричним підходом Л. Полінга,
який базується на термохімічних даних,
%а-Хв=су/К9 (8.152)
де А = 2ВАВ - {рм + Вт), (8.153)
енергії зв'язків у відповідних молекулах.
щ
Маллікен Роберт Сандерсон
(1896—1986),
Велика Британія
179
Співвідношення (8.152) дає змогу
знаходити різницю електронегативностей. Л. По-
лінг узяв за початок відліку
електронегативність найбільш електронегативного
елемента — Флуору, що дорівнює 4,0.
Електронегативність інших елементів має значення
менші, ніж 4,0. Наприклад, для Гідрогену —
2,1; Хлору — 3,0; Брому — 2,8; Йоду — 2,5.
Близькість електронегативностей Хлору та
Брому призводить до того, що молекула ВгСІ
малополярна, те ж саме можна сказати про
молекули ІВг і НІ.
Між електронегативністю за Полінгом
(%я) та за Маллікеном (%м) існує лінійний
зв'язок
Хп = а%м + Ь. (8.154)
Л. Полінг вдало обрав початок відліку для своєї системи — константа (Ь)
у рівнянні (8.154) близька до нуля. Коефіцієнт а дорівнює 0,178. Дехто з
авторів пропонує електронегативність за Маллікеном рахувати не як суму, а
як півсуму енергії іонізації та спорідненості до електрона, тоді значення
константи а вдвічі більше.
Кожна шкала електронегативностей має свої переваги та недоліки. Шкала
Маллікена—абсолютна, шкала Полінга—відносна. Цей недолік шкали По-
лінга, однак, несуттєвий, оскільки завжди мають справу саме з різницею
електронегативностей елементів. Шкала Маллікена теоретично
обґрунтована, шкала Полінга емпірична. Теоретична обґрунтованість дає змогу
розвивати поняття електронегативності Маллікена, наприклад, здійснити
перехід від електронегативності як властивості цілого атома до
електронегативності окремих орбіталей даного атома. Емпіричний підхід Л. Полінга,
що потребує інформації про енергії зв'язків, має і свої переваги. Шкала
Маллікена практично обмежена одновалентними елементами, це
обмеження не стосується шкали Полінга. Він побудував свою шкалу, яка містить
усі групи періодичної системи.
Розрахунки електронегативностей, згідно з Полінгом, вимагають
наявності ґрунтовних термохімічних даних. А. Л. Оллред та Е. Рохов
запропонували простіший спосіб оцінки електронегативностей
Ха=^іг> (8Л55>
гл
де гА — атомний радіус.
При цьому електронегативність визначається силами електростатичної
взаємодії між електронами та екранованим ядром на відстані гА. Якщо у
рівнянні (8.155) ввести відповідні числові коефіцієнти, то
електронегативність за Оллредом та Роховим можна зіставити із шкалою Полінга.
180
Полінг Лайнус Карл
(1901—1994), США
ХП = 0,359^ + 0,744. (8.156)
гл
Таблиця 8.5. Електронегативності елементів
|н
2,10
ІІл
0,97
N3
1,01
к
0,91
1 Си1
1,75
0,89
1,42
С8
0,86
Аи
1,42
Рг
0,86
Ве
1,47
1,23
Са
1,04
2п
1,66
Зг
0,99
Ссі
1,46
Ва
0,97
1,44
Яа
0,97
В
2,01
А1
1,47
8с
1,20
Са
1,82
У
1,11
Іп
1,49
Ьа
1,08
ТІ
1,44
Ас
1,00
Се
1,08
Рг
1,07
N(1
1,0/
ІРт к
І 1,07|
С
2,50
Зі
1,74
Ті
1,32
Се
2,02
2г
1,22
Зп
1,72
т
1,23
ра
1,55
N
3,07
Р
2,06
V
1,45
А8
2,20
1,23
ЗЬ
1,82
Та
1,33
Ві
1,67
0
3,50
8
2,44
Сг
1,56
8е
2,48
Мо
1,30
Те
2,01
1,40
Ро
1,76
¥
4,10
СІ
2,83
Мп
1,60
Вг
2,74
Тс
1,36
І
2,21
Ке
| 1,46
\Аі
1,90
Ре
1,64
Со
1,70
N1
1,75
Ки
1,42 |
Кіі
1,45
Реї
1,35
08 !
1,52
Іг
1,55
1,44
5т
1,07
Еи
1,01
Ссі
1,11
ІТЬ 1
11>Щ
ТЬ
1,11
Ра
1,14
1 1,22
1 1,22
ІРи
1 1,22
оу
1,10
Но
1,10
Ег
1,11
Тт
1,11
УЬ
1,06
Ьи
1,И
Прийняті значення електронегативностей для елементів періодичної
системи (за винятком благородних газів) наведені в таблиці 8.5. Із наведених
даних бачимо, що найбільш електронегативними елементами є Флуор та
Оксиген; найменш електронегативними — Францій та Цезій, тобто
максимальні значення % розташовані у правому верхньому куті періодичної
системи, а мінімальні — в лівому нижньому.
Слід зазначити, що при різноманітних підходах до визначення
електронегативності одержуються взаємоузгоджені величини. Це свідчить про те, що
електронегативність — об'єктивна характеристика атома. Різниця
електронегативностей визначає характер розподілу зарядів у молекулах, зокрема
двохатомних, до розгляду яких переходимо у наступному розділі.
181
Розділ 9.
БУДОВА МОЛЕКУЛ
Неповторну молекул будову
Зв 'язок хімічний створює чудово!
9.1. Двохатомні молекули
Найпростіші серед молекул — двохатомні. Вони, як і атоми, є в цілому
електронейтральними системами. Проте всередині цих систем можливий
нерівномірний розподіл зарядів: на одному з атомів може концентруватися
позитивний заряд, а на іншому — негативний. Протилежні за знаком та
однакові за величиною заряди Ц;, які розташовані на відстані (/) один від одного,
створюють дипольний момент молекули
\іі = Яіі (9.1)
Молекули з великим значенням дипольного моменту належать до
іонних. Таку молекулу можна уявити собі як складену з двох іонів: катіона (К )
та аніона (А~). Кулонівську енергію (£/) взаємодії цих іонів легко обчислити
за формулою
ц = 9а~9к+ = ^А-^к+в _ А (9.2)
г г г'
де— #к+ =+2к+е, <7А- =-2А-е — відповідно, заряди катіона та аніона; є—
абсолютна величина заряду електрона; г — відстань між центрами іонів;
А — стала, яка об'єднує величини, незалежні від г.
Залежність потенціальної енергії від відстані г зображена на рис. 9.1,
крива І. Кулонівська взаємодія між різнойменно зарядженими іонами стягує
іони до деякої відстані (г0). Якщо вважати іони частинками, що не
стискаються (кулями), які мають, відповідно, радіуси гк+ та гА-, то не важко
побачити, що г0 = гк+ + гА-. Енергія £/0, яка виділяється при утворенні молекули з
іонів, дорівнює
С/0=-А (9.3)
Необхідна для обчислення І/0 рівноважна відстань г0 між центрами іонів
повинна визначатися безпосередньо з експерименту. Розрахунок £/0 дає
відносно непогані результати, проте недоліки цієї простої моделі очевидні.
В основному, це стосується припущення про абсолютно тверді іони.
Насправді між іонами на коротких відстанях (при г < г0) діють сили
відштовхування. Ці сили насамперед зумовлені відштовхуванням між негативно
зарядженими електронними хмарами іонів, а при малих відстанях також
між'ядерним відштовхуванням. Крім того, діють так звані сили Паулі. Річ у тім, що при
зближенні двох іонів із заповненими електронними оболонками принцип
Паулі забороняє електронам розташовуватися на вже зайнятих орбіталях.
182
Рис. 9.1. Криві потенціальної енергії двохатомної іонної молекули:
І — притягування (закон Кулона); II — відштовхування; III — повна енергія
Електрони змушені займати орбіталі з високими енергіями. Це призводить
до різкого збільшення енергії системи і фіксується як відштовхування.
Загальну дію всіх сил відштовхування виражають експоненційною залежністю
Г = Ве р, (9.4)
де V— потенціальна енергія відштовхування (крива II на рис. 9.1); В та р —
сталі.
Для повної енергії Е, яка враховує притягання та відштовхування, згідно
з формулами (9.2) та (9.4), одержимо
Е = 1/ + У = -4 + Ве
г
(9.5)
Рівняння (9.5) описує потенціальну криву III з мінімумом (див. рис. 9.1),
яка є сумою кривих притягання (крива І) та відштовхування (крива II). За
допомогою виразу (9.5) можна теоретично обчислити значення Е09 яке
відповідає мінімуму енергії на кривій III.
умови мінімуму — = 0,
маємо
в=4е~р,
(9.6)
183
ЗВІДКИ
Е--А+Ве-£=-А\1-Р.
(9.7)
Отже, врахування сил відштовхування зменшу є значення І)0 до Е0
'-і
разів.
Досліди свідчать про те, що для багатьох іонних молекул р є приблизно
сталою величиною, яка дорівнює 0,0345 нм. Оскільки г0 = 0,2—0,3 нм, то
Е0 = (0,85—0,90)£/о (див. рис. 9.1, с. 183). Величина £0 дорівнює кільком
електрон-вольтам.
Утворення іонної молекули з іонів є енергетично вигідним процесом,
але спочатку треба, щоб ці іони утворилися з атомів. Розглянемо процес
утворення іонної молекули з атомів за стадіями з урахуванням
енергетичних внесків на прикладі хлориду Натрію:
Ка-е->№++/; (І)
С1 + е-+СГ-є; (її)
Ка++СГ->На+СГ + £0; (III)
№ + С1 = Ма+СГ+Д#. (IV)
Тут АЯ— тепловий ефект (ентальпія) реакції між атомами Натрію та
Хлору; є — спорідненість до електрона атома Хлору; /— енергія іонізації
атома Натрію. З рівнянь (І)—(IV) одержимо
Д#=/-є + £0. (9.8)
Перебіг реакції (IV) утворення іонної молекули з атомів енергетично
вигідний, коли
ДЯ<0, (9.9)
тобто ця реакція повинна бути екзотермічною.
Вимозі (9.9) сприяють великі (за абсолютною величиною) значення Е0,
які завжди від'ємні, а також невелика різниця (/- є). Для цього треба
підбирати атоми так, щоб один з них мав малу енергію іонізації, а другий
відносно велику спорідненість до електрона. Оскільки лужні метали (Ме) мають
низькі енергії іонізації, а галогени (X) — велику спорідненість до електрона,
то вони легко утворюють іонні молекули типу Ме Х~. Зазначимо, що навіть
за найбільш сприятливих умов різниця (/- є) все ж таки додатна величина
і вирішальний внесок в АНдає велика від'ємна величина Е0. Молекули типу
Ме~ Х+ не утворюються, тому що для цього потрібна велика витрата енергії
на відривання електрона від атома галогену (7гал велика), яка майже не
компенсується при утворенні аніона Ме" (спорідненість до електрона лужного
металу дуже мала). Не важко показати, що іонні молекули утворюються між
184
атомами з великою різницею електронегативностей. Візьмемо за основу
критерій (9.9). Якщо з атомів А та В утворюється молекула А В", то
АН' = ІА-гв + Е'0, (9.10)
а якщо молекула А~В , то
АН" = Ів-еА+Е"0. (9.11)
Якщо вважати, що міжатомні відстані (г0) у молекулах А В~таА В
однакові, то, згідно з формулою (9.7), Е'0 = Е^, тому
АН'-АН" = (ІА-ев)-(Ів-£А) = (ІА+єА)-(Ів+£в) = хА-Хв (9.12)
відповідно до формули (8.151) визначення електронегативності (за Малліке-
ном).
Якщо %А>%В, то А#' > ДЯ" і, отже, АН" — більш від'ємна величина,
згідно з критерієм (9.9) більш вигідне утворення А~В молекули (тобто мінус
на більш електронегативному елементі). При %А < %в, навпаки, вигідніше
утворення молекули А В~. Отже, різниця електронегативностей дає
відповідь, чи можливе утворення іонної молекули. При %А = %в Д#' = А//'
й іонна молекула не утворюється.
У молекулах з малою різницею електронегативностей, а також у тих
випадках, де утворюються багатозарядні іони, наприклад, М§ О ~, А1 N ~,
іонний зв'язок існує тільки частково. Для таких молекул характерні, по-
перше, низькі експериментальні значення дипольних моментів порівняно
з розрахованими для іонних молекул; по-друге, розрахунки енергії іонного
зв'язку (Е0) дають нижчі значення, ніж експериментальні. Такі відхилення
значною мірою можна пояснити поляризацією іонів. На відстані г іон із
зарядом д, створює напруженість електричного поля
/г = % (9.13)
г
Кожний з іонів у дипольній молекулі може поляризувати інший. При
цьому зміщуються заряди та наводиться новий диполь
ІіРоі = аЕ, (9.14)
де а — поляризовність іона.
Поляризовність визначає, як зміщуються електронні оболонки атома
(іона) під дією електричного поля і, очевидно, вона повинна бути більшою
для великих атомів. Якщо вважати, що негативний заряд, який дають 2
електронів, розподілений рівномірно у середині сфери радіуса Д, то,
користуючись законами електростатистики, не важко довести, що
сс = Д3. (9.15)
Формула (9.15) приблизна, але вона показує, що фактором, який
визначає поляризовність, є розмір частинки.
185
Більш пухкі, великі частинки легше поляризуються. Розміри аніонів, як
правило, більші, ніж нейтральних атомів, і значно більші, ніж катіонів. Тому
поляризуються в основному аніони.
Величина напруженості (Р), яку створює іон, залежить від його заряду
та розміру, згідно з формулою (9.13). Високу напруженість створюють іони
невеликого розміру і з великим зарядом. Багатозарядні катіони утворюються
легше, ніж багатозарядні аніони, і мають досить малі розміри, тому вони
виявляють значну поляризуючу дію.
Дипольні моменти, які наводять катіон в аніоні та аніон в катіоні,
напрямлені проти дипольного моменту всієї молекули та призводять до його
зменшення. Згідно з формулами (9.1) та (9.14) сумарний дипольний момент (для
однозарядних іонів)
,К+ „А"
Враховуючи, що Рі = д( 112 (9.13) та ос, = г? (9.15), а також те, що / =
маємо
:Г*г+
[І = ЄІ
1-
г3 +г3
(у+у)
(9.16)
+ >А->
(9.17)
При г + = г _ \і = 0,15еІ. Для малого катіона та великого аніона, коли
поляризація істотна, формула (9.17) дає при гк+ « гА- |ьі—> 0, тобто зникає
основна характеристика іонної молекули — дипольний момент.
Внаслідок поляризації частка негативного заряду зміщується до
позитивного іона і молекула стає менш полярною. Зменшення полярності
молекули можна розглядати як зміщення пари електронів («свого» та
«захопленого») від аніона до катіона. В тому випадку, коли атоми однакові, наприклад,
у молекулах водню, хлору, кисню тощо, електронегативність їх тотожно
дорівнює одна одній і електронна пара повинна знаходитися «посередині».
Утворення ковалентного зв'язку за допомогою
пари електронів було запропоновано ще Дж.
Льюїсом. Проте, якщо іонний (пара електронів
локалізована на одному з атомів) і полярний
зв'язки (іонний зв'язок та поляризація) можна
пояснити, виходячи з суто електростатичних
уявлень, то таке пояснення для гіпотези Льюїса
непридатне. Обґрунтувати цю гіпотезу можна
лише з урахуванням динаміки руху електронів
на основі квантово-механічної теорії.
Вперше це було зроблено В. Гайтлером та
Ф. Лондоном для молекули водню —
найпростішої з двохатомних молекул. Розглянемо
спочатку два ізольовані атоми Гідрогену А і В,
відповідно, з електронами 1 та 2. Для кожного
з цих атомів гамільтоніан має вигляд
Гайтлер Вальтер Генріх
(1904—1981), Німеччина,
Велика Британія, Швейцарія
186
Л„=-2-^-£- =
1А\
2т
ГА\
(9.18)
2т " гВ2
де г^ та гВ2 — відповідно, відстані між ядром та
електроном.
При зближенні атомів Гідрогену в гамільто-
ніані з'являються члени
/
.1)
гв\
(
є
гА2
+—
І2
З. \
Я
АВ
Лондон Фріц
(1900—1954),
Німеччина, СІІІА
які відповідають притяганню між електронами
та «чужими» ядрами і відштовхуванню — між-
електронному та між'ядерному. Ці члени
можна об'єднати та позначити їх суму як НтХ (іпіе-
гасііоп — взаєм.одія).Повний гамільтоніан молекули водню складається
з#іттасуми(#^+#52).
Хвильова функція молекули водню може бути зображена як добуток
одноелектронних хвильових функцій атомів
Т,= 1^(1) Лзв{2).
(9.19)
Проте через нерозрізненість електронів повну хвильову функцію не
можна записати як добуток одноелектронних функцій (для атома Гелію —
(І5)і • (2«у)2, для молекули водню — 1^(1) * 1$я(2)), а слід використовувати
симетричну та антисиметричну лінійні комбінації (див. формули (8.124) та
(8.125));
(9.20)
(9.21)
де Ч*! зазначено вище; Ч^ = 1^(2) • 1$в(\).
У підрозділі 8.3 для атома Гелію показано, що симетрична (*¥§) та анти-
симетрична 0¥А) функції описують стани з різною енергією, яка включає
кулонівські та обмінні інтеграли. Аналогічно виразам (8.123) та (8.122)
одержимо:
Е8 = <2 + Р; (9.22)
ЕА = а-Р. (9.23)
За рівень відрахунку прийнято 2Е°Н, тобто суму енергій двох
ізольованих атомів Гідрогену.
Згідно з різними складовими, які входять до Нш, кулонівський (0 та
обмінний (Р) інтеграли молекули водню складаються з кількох частин.
187
Додатний внесок у £? та Р дають кулонівський та обмінний двохелект-
ронні інтеграли К та А, які відповідають за міжелектронне відштовхування
(для Кт&А збережені їх колишні позначення, див. (8.3)):
1 ^У М ^ Щ4У2; (9.24)
г\,г
К =
,.Г^(')-^Р)-ь.0)-^ИДІД,, (925)
Формули (9.24) та (9.25) ідентичні формулам (8.119) та (8.120).
Притягання ядром одного атома електронної хмари, розташованої біля
іншого атома, описується від'ємним за величиною кулонівським одноелек-
тронним інтегралом, який входить до складу інтеграла ()
ги |2
С = -
н ,.
>А\
(IV = ■
м
сіУ. (9.26)
Зниження енергії забезпечує також від'ємний
одноелектроннийрезонансний інтеграл, який входить до складу інтеграла Р
§=(]їА±ііау =(]±А±ііау^ (927)
^ гА ^ гв
Зазначимо, що одноелектронні інтеграли Ста Р описано також далі при
розгляді молекулярного іона Н^.
На малих відстанях між ядрами істотний додатний внесок дає між'ядерне
відштовхування
є2 1
+ (або —— в атомних одиницях), де гАВ— між'ядерна відстань.
ГАВ ГАВ
Величини К, С, А, р також залежать від значення гАВ і при гАВ-*>о прямують
до нуля.
Додавання всіх внесків показує, що інтеграли ()і Ре від'ємними
величинами. Проте додатний кулонівський інтеграл К, який характеризує
міжелектронне відштовхування, значно більший, ніж обмінний інтеграл А
взаємодії електронів (див. підрозділ 8.3). Це призводить до того, що сумарний
обмінний інтеграл Р є значно більшою від'ємною величиною, ніж інтеграл ().
У формулі (9.22) інтеграл 0, дає лише близько 15 % від загального внеску в
утворення зв'язку при об'єднанні атомів Гідрогену в молекулу.
Оскільки обмінний інтеграл Р—від'ємна величина (|Р| »|£?|), то для
стану, який описується антисиметричною орбітальною функцією 4іА,
характерне збільшення енергії і ЕА — додатна величина, тобто в цьому випадку
при зближенні атомів Гідрогену молекула Н2 не утворюється. Чому ж
функція Ч^ забезпечує утворення зв'язку, а функція Ч*А ні? Для цього треба
розглянути властивості функцій і згадати, що функція \Ч* | виражає ймовірність
знаходження електрона. Для симетричної хвильової функції (з точністю до
нормувального множника)
188
рІІ^Р/І + І^яІ + г^Тя, (9.28)
а для антисиметричної
1^11 = 1^/1 + 1^1-2^^. (9.29)
Добуток І^/Ч'//1 = |1^ Ш • Ьр (2) • 1?д (1) • \8А (2)| у функцію |Ч^ | входить
зі знаком «+», а у функцію І^^Гзі знаком «-». Цей добуток великий тільки
у просторі між ядрами, де функції 1^ та \зв значно перекриваються (поза
цим простором або \зА, або Ід^чи обидві ці атомні функції малі). Отже,
густина ймовірності і, відповідно, електронна густина для стану, який
описується симетричною функцією (Ч^), підвищена, а для стану, який описується
антисиметричною функцією С¥А), занижена у просторі між ядрами. На
рис. 9.2 зображені криві однакової густини електронного заряду для станів,
які описуються функціями 4*$ (а) і *¥А (б). Для симетричного стану
підвищена електронна густина між ядрами забезпечує зв'язок між атомами
Гідрогену, для антисиметричного — електронна густина у між'ядерному
просторі дуже низька, тому молекула водню в цьому випадку не утворюється.
Рис. 9.2. Контурні діаграми електронної густини в молекулі водню
для симетричної (а) та антисиметричної (б) хвильових функцій
(більшому значенню електронної густини відповідає більше число)
Електронна густина у просторі між ядрами для антисиметричної функції
зменшується настільки, що є місця, де вона взагалі дорівнює нулю.
Нульове значення функції \^а\ припадає на площину, яка перпендикулярна до
лінії зв'язку і знаходиться на відстані гАВІ2 від кожного з ядер, тому ця
площина вузлова. Отже, підтверджується згадане вище положення про те, що
хвильові функції з вузлами (тут функція Ч*А) належать до станів із вищою
енергією, ніж безвузлові (тут функція Чу.
Функції ЧРу та Ч?А — це орбітальні хвильові функції. Повна хвильова
функція повинна мати спінову частину. Згідно з принципом Паулі (див. підрозділ
8.3), симетричній орбітальній частині функції (Ч^) відповідає антисимет-
рична спінова (8А), а антисиметричній орбітальній функції 0¥А) — симет-
189
рична спінова (5у), причому стан 8А — синглетний
^=-^(аір2-а2рі),
а 5*5— триплетний; згідно з формулами (8.129)-(8.131):
55 =
5^=а1ос2;
5? =
-)=(аір2 + р1а2);
[55-'=рір2,
де а та Р — спінові стани окремих електронів, які відповідають т5 = +х/г та
т8 = -У2.
Отже, у стабільному стані молекули спіни електронів, які з'єднують
атоми в молекулу, спарені й сумарний спін (для стану 8А) дорівнює нулю. Це
добре узгоджується з відомим експериментальним фактом відсутності
парамагнетизму у більшості насичених двохатомних молекул.
Розрахунки Гайтлера-Лондона обґрунтували гіпотезу Льюїса про те, що
ковалентний зв'язок утворює пара електронів, та показали, що двохатомні
молекули діамагнітні внаслідок спарювання електронів. Заснований В. Гайт-
лером та Ф. Лондоном і розвинутий потім Л. Полінгом та Дж. Слетером
метод, у якому кожній валентній рисці відповідають два спарені електрони,
дістав назву метод валентних зв 'яків (ВЗ), або локалізованих пар (ЛП). На
основі цього методу вдається пояснити насичуваність хімічного зв'язку.
Експериментально хіміками було встановлено насичуваність хімічного
зв'язку, але цей факт було важко теоретично обґрунтувати. Не випадково ще
Дж. Дальтон, критикуючи позиції А. Авогадро, дивувався з приводу того,
що якщо до одного атома Гідрогену може приєднатися другий, то що
заважає зробити це третьому, четвертому і т. д.
Згідно з теорією валентних зв'язків міцний зв'язок утворюють електрони
у синглетному спіновому стані, а у триплетному спіновому стані
відбувається їх відштовхування. Третій атом С, який наближається до молекули
АВ, не може мати спіна, який одночасно спарювався б з обома
зв'язуючими електронами. Тому можлива тільки реакція обміну: АВ + С = ВС + А,
а триатомна молекула АВС не утворюється. Слід зазц^чити, що власне спін
енергетично не бере участі в утворенні зв'язку (спін-орбітальна взаємодія
в легких атомах дуже мала і нею можна знехтувати, див. підрозділ 8.4).
Коли один атом Гідрогену підходить до іншого, то відбувається їх
притягування і утворюється стійка молекула водню. При цьому орбітальна
хвильова функція симетрична і, отже, згідно з принципом Паулі, спінова
функція повинна бути антисиметричною. Саме орбітальна взаємодія орієнтує
спіни так, щоб утворювалася спінова функція 8Лі тобто спіни спарювалися.
Спіни лише «виявляють», як зорієнтовані просторові орбіталі. Припустимо,
що при зіткненні атоми Гідрогену не встигають переорієнтуватися. Оскільки
стан Е8за спіном невироджений, а стан ЕА — вироджений тричі, то в серед-
190
ньому з кожних чотирьох зіткнень атомів Гідрогену тільки в одному
утворюється стабільна молекула водню, а в трьох останніх атоми Гідрогену не
утворюють молекули. Нехай тепер два електрони (перший та другий)
знаходяться в молекулі АВ, а третій — в атомі С, що розміщений на деякій
відстані від молекули АВ. При наближенні атома С до молекули АВ третій
електрон починає взаємодіяти з першими двома. Розглядаючи кожний з двох
зв'язуючих електронів окремо по відношенню до третього, дійдемо того
самого висновку, що й при аналізі поведінки двох електронів, які
знаходилися у різних ізольованих атомах Гідрогену, а саме: співвідношення внесків
симетричного та антисиметричного орбітальних станів дорівнює 1:3. Тоді
для енергії взаємодії третього електрона з першими двома можна записати
1/17 \ , З,
Ч^1^8)Л(еЛ
(9.30)
Якщо у формулу (9.30) підставити вирази для Е8 та ЕА через кулонівську
(0 та обмінну (Р) енергії, то
^і,з ~4^с + 4^с+ 4^с 4^с~^с 2
р
аналогічно: Еіг = <2ВС —~1
(9.31)
Оскільки обмінний інтеграл значно перевищує кулонівський за
абсолютною величиною і має від'ємний знак, то величини Ехг та Е17) додатні. Тоді
повна енергія системи (молекула АВ + атом С)
Р + Р
Е = Еи2 +£1>3 +£2,з = Оав +0,вс +(?ас + рав —АС 2 В =
хп 1 ^ (932)
Сума за Сума за неспареними
зв'язками електронами
Оскільки Ех з та £2,з — додатні величини, то енергія системи молекула
АВ плюс атом С вища, ніж енергія ізольованої молекули АВ, яка становить
^1,2= Оав + ?ав- Отже, на атом С при його наближенні до молекули АВ діють
сили відштовхування. Зазначимо, що, згідно з формулою (9.32), кулонівські
сили не насичуються. Кулонівські інтеграли £) знижують повну енергію,
підвищення енергії відбувається тільки внаслідок обмінних інтегралів. Якби
природа ковалентного зв'язку ґрунтувалась тільки на кулонівській взаємодії,
то насичення не відбувалось би.
Для випадку з іонними молекулами потенціальна крива виникла як
сумарний результат дії сил кулонівського притягання та сил відштовхування
(міжелектронне, між'ядерне відштовхування, «сили Паулі») і при цьому
191
іони, які утворювали молекулу, мали свої характерні особливості. У
ковалентній молекулі атоми А та В цю особливість втрачають внаслідок
узагальнення електронів. При побудові потенціальної кривої Е8(гАВ) враховуються
не тільки потенціальні взаємодії електронів та ядер один з одним, а й
кінетична енергія електронів. Проте крива, Е8(гАВ) відносно руху ядер є
потенціальною. Отже, спочатку фіксується відстань між ядрами (гАВ) і
розраховується квантово-механічна система, яка складається з електронів, що
рухаються, та нерухомих ядер. Потім розраховану потенціальну криву для
різних (фіксованих) значень гАВ використовують для опису руху ядер.
Можливість цього підходу випливає з принципу (наближення) Борна-Оппенгей-
мера. Згідно з цим принципом можна розділити рух ядер та електронів,
враховуючи те, що різниця мас ядер та електронів значна. Внаслідок великої
маси ядра рухаються настільки повільно, що кожне їх місцезнаходження
можна розглядати як фіксоване з «точки зору електронів». При такому
повільному русі ядер електронна густина встигає швидко перерозподілитися,
і ядра рухаються у потенцальному полі, яке утворюють електрони.
Розглянемо тепер більш детально цю потенціальну криву.
Якщо обчислити величини Е8та ЕА для різних значень гАВ, то графік їх
залежності від гАВ має вигляд, зображений на рис. 9.3. Для антисиметрич-
ного орбітального стану енергія ЕА при зближенні атомів поступово
збільшується і стабільна молекула не утворюється. Для симетричного
орбітального стану після зниження енергії при зменшенні гАВ відбувається її
зростання у зв'язку зі збільшенням між'ядерного відштовхування при гАВ-*0.
Мінімум енергії відповідає стійкому рівноважному стану молекули водню,
при цьому гАВ = г0, а [-Е8 (гАВ)] = Д де В — енергія дисоціації молекули
водню, тобто енергія, яку необхідно додати, щоб молекула водню розпалася на
атоми, г0—рівноважна відстань між ядрами.
Е, кДж/моль
300
200
100
0
-100
-200
-300
Рис. 9.3. Залежність енергії від між'ядерної відстані для симетричної (Е8)
та антисиметричної (ЕА) хвильових функцій
192
Точний теоретичний розрахунок потенціальної кривої навіть для такої
простої молекули, як водень, важкий (уточнення розрахунку після
вдосконалення методу ВЗ та використання інших методів буде наведено далі). Для
складніших молекул теоретичні розрахунки проводити ще важче, тому
потенціальні криві двохатомних молекул часто описують, використовуючи
емпіричне рівняння Морзе
Е(г) = -2Ве'а{г-^ + Ое-2а(ґ-г°\ (9.33)
де а — емпіричний параметр (постійний для даної молекули); Б та г0—
характеристики точки мінімуму на кривій.
Функція Морзе при г —> ©о перетворюється на нуль, а при г = г0 дорівнює
(-£>), тобто її вигляд відповідає потенціальній кривій для Е8.
Енергія дисоціації може бути знайдена термохімічним шляхом при
вивченні реакції Л 2 <-> 2Л. Рівноважні міжатомні відстані можуть бути визначені
за допомогою рентгено- чи електронографії. Для обертального руху
можна скористатися формулою (8.39) енергії просторового ротатора у вигляді
*.4!ІШ1. (934)
де і — обертальне квантове число, /— момент інерції, який для лінійної
молекули має вигляд
і • » • ' • ап тАтВ
де І — між ядерна відстань у молекулі АВ; и = ——— приведена маса.
тА +тв
Вивчення обертальних спектрів дає інформацію про моменти інерції
Молекул і, отже, про міжатомні відстані г09 оскільки маси ядер відомі. При
невеликих відхиленнях г від г0 потенціальну криву (9.33) можна апроксиму-
вати параболою і це відповідає лінійному гармонійному осцилятору.
Справді, якщо відраховувати енергію від точки мінімуму Е(г0)=-Д то маємо
^(г) = £(г) + £ = я[і~2е-"(г^ (9.36)
При невеликих (г-г0), розкладаючи експоненту в ряд, маємо
С/(г) = ^[і-1 + а(г-г0)]2=^2(г-г0)2=А-, (9.37)
де х = г - г0—відхилення від рівноважного стану (дна потенціальної ями).
/ = 2£>а2=4ті2у2т, (9.38)
де/— квазіпружна стала; V — частота осцилятора; т — його маса.
Розв'язок рівняння Шредінгера для гармонійного осцилятора показує,
що енергія квантується
де пу = 0,1,2,3... — коливальне квантове число.
193
З урахуванням асиметричності потенціальної кривої коливання стають
ангармонійними і для Еп маємо
Еп, = «V + " \^-х\ п^+Ц йу, (9.40)
де х — константа ангармонійності.
Дані про частоту (у) коливань зв'язку та константу ангармонійності (х)
одержуємо із коливальних спектрів. За допомогою цих величин можна
обчислити енергію дисоціації молекули
£> = /гу/4х, (9.41)
а також квазіпружну (силову) константу зв'язку (/) згідно з формулою (9.38).
Квазіпружна константа, як і Д характеризує міцність хімічного зв'язку.
Знання /та В дають змогу за допомогою формули (9.38) знайти емпіричний
параметр а формули Морзе. Зазначимо, що переходи між коливальними або
обертальними рівнями відповідають випромінюванню (поглинанню)
квантів електромагнітного випромінювання в інфрачервоній області
(скорочено ІЧ-спектри). Електронні спектри буде розглянуто далі. Суттєво
зазначити, що оскільки Уел » укол » уоб, повна система смуг у спектрі може
охоплювати області спектра від далекої ультрафіолетової до далекої
інфрачервоної (аж до мікрохвильової). Схематично це зображено на рис. 9.4.
Вертикальна лінія на схемі показує перехід між електронно збудженим станом
В, коли коливальне квантове число иу= 1, а обертальне / = 8, і основним
електронним станом А з яу= 0 та і = 7. Потенціальні криві молекул корисно
знати не тільки для порівняння теорії з експериментом, а й для можливості
використання напівемпіричних методів для розв'язування складніших задач,
наприклад, побудови потенціальних поверхонь, що дуже важливо,
наприклад, у теорії перехідного стану (див. підрозділ 14.4).
Гомоядерні молекули можуть бути тільки ковалентними, для гетероядер-
них все залежить від різниці електронегативностей, зі збільшенням якої
електронна пара поступово зміщується до електронегативного атома, і має
місце поступовий перехід від ковалентного зв'язку до іонного. Вище
показано, що поступовий перехід від іонних молекул до полярних був наслідком
поляризації іонів. Тепер після з'ясування природи ковалентного зв'язку
полярні молекули можна розглядати як проміжний випадок між суто
ковалентними та іонними молекулами.
Хвильову функцію полярної молекули 0¥) можна умовно розглядати як
«суперпозицію» (накладання) іонної (Ч^-) та ковалентної (Ч*с) хвильових
функцій
Ч = с>¥і + ссЧс. (9.42)
Коефіцієнти сг-та сс можна знайти за допомогою варіаційного принципу,
тобто з умов мінімуму енергії системи
194
•/
♦
10
0 =
_і_
_ь
=
10
5
0
♦
_і_
^^^=
в
' ■
=
л_
_і_:
.
•
♦
'-з
-2
- 1
-о
_і_
_{_
А
_і_
•
-і-
•- ь
— 4
_ о
3
2
1
л
І/
Рис. 9.4. Схематичне зображення електронних, коливальних та обертальних
термів (лу — коливальне квантове число; і — обертальне квантове число)
дЕ
де,
= 0;
дЕ_
= 0.
З цих умов матимемо два рівняння:
с,.(#,.-£)+ссЯ,.с=0;1
сІ.Я/с+сс(Ясе-£) = 0)]' (943)
де Нсс = \*¥сНЧ>ссІГ; Ни = /^ЯЧ^/Г; Я,с = Нсі = /^ЯЧ^Г =
Рівняння (9.43) мають нетривіальні розв'язки, якщо наступний
детермінант дорівнює нулю
195
Нн-Е Ніс
ніс Нсс-Е\
= 0. (9.44)
Розв'язуючи детермінант, для Е дістанемо
г^ніі-Нсс + \{ніі-нсс) Н2 (9.45)
2 "V 4 *"
Величини #„ та Нсс— це з достатнім ступенем точності енергії іонного
(Е;) та ковалентного (Ес) гіпотетичних станів. Вони відомі для довільних
відстаней між атомами, якщо відоміпотенціальні криві для іонного і
ковалентного станів. Інтеграл Ніс = \х¥іНх¥ссІ¥ — від'ємна величина, його
поява пов'язана зі змішуванням іонного та ковалентного станів. Тут х¥с—
симетрична зв'язуюча функція Гайтлера-Лондона С¥5); Т, — функція, в якій
обидва електрони знаходяться біля одного (більш електронегативного)
атома. Наприклад, якщо атом В більш електронегативний, то Ч*,- = (рв (1) • (рв (2),
де фя— відповідна атомна орбіталь.
Якщо інтеграл Ніс дорівнює нулю, то згідно з виразом (9.45) (це видно
також і з рівняння (9.44)), Ех - Ни = і^та Е2 = Нсс = Ес і кожний із станів
(іонний та ковалентний) існують окремо. Якщо Ніс Ф 0, то кожний
енергетичний рівень розщеплюється на два, які розташовані вище та нижче. У цьому
випадку вживають термін «резонанс» станів (іонного та ковалентного), хоча
аналогія між квантово-хімічним «резонансом» та класичним резонансом
осциляторів стосується в обох випадках тільки розщеплення енергетичних
рівнів.
Розглянемо кілька випадків. Перший випадок. Молекула малополярна,
наприклад НІ. Електронегативності атомів А та В майже однакові, %А = %в,
тому перенесення електрона від А до В потребує витрати енергії (ІА - ев). При
Ха - Хв можна вважати, що гА = гв отже, ІА-ев = ІА -гА. Спорідненість
до електрона (є^) для даного атома (А) завжди набагато менша, ніж енергія
іонізації (ІА), тому іонний стан молекули АВ (А В~) повинен мати набагато
вищу енергію, ніж ковалентний, тобто при будь-яких КАВ справедлива
нерівність Е}» Ес (Ни » Нсс).
Для систем, які дуже відрізняються за своєю енергією, енергія взаємодії
незначна: \НІС\«\НІЇ-НСС\ = |£,— Ес\. З урахуванням цієї нерівності формулу
(9.45) для енергії основного стану можна записати так:
Е -Е і**—. (9.46)
1 с Еі-Ее
Оскільки \НІС\« {Еі-Есі, то Е{ = Ес9 тобто «справжня» крива
розташується лише трохи нижче, ніж крива для ковалентного стану, і вони практично
співпадуть. Для збудженого стану матимемо
Е^Е'+~Вг- (9-47)
196
З урахуванням нерівності \НІС\ « |£/-£с| з формули (9.47) одержимо
Е2 = Ег. Отже, майже ковалентна молекула у збудженому стані
перетворюється на іонну, і перехід електрона з рівня ЕІ9 на рівень Е2 фактично
еквівалентний зміщенню його з атома А на атом В.
Підставляючи рівняння (9.46) та (9.47) у (9.43), одержимо значення
коефіцієнтів с,та ссі переконаємося, що сі« сс. Величина
ЛІ
-у-100% (9.48)
к2
називається ступенем іонності зв 'язку. В даному випадку Р = 0, тобто
молекула АВ в основному стані практично ковалентна.
Другий випадок. Молекула полярна, наприклад, НС1, НР. Криві іонного
і ковалентного станів при рівноважних відстанях підходять близько одна до
одної або перетинаються. Тоді Ес = Еі і, вважаючи Ни = Нсс = Нт, з
формули (9.45) визначимо
Еіа = Нт±Ніс (9.49)
В цьому випадку інтеграл Ніс за абсолютною величиною значний, і
оскільки за знаком Ніс < 0, то значення Ех набагато менше, ніж Нт9 тобто
зменшення енергії внаслідок «резонансу» між іонним і ковалентним атомами
велике.
Не важко довести, що при Ес = Еі9 с{ = сста Р = 50 %, тобто цей зв'язок
проміжний між іонним і ковалентним.
Ступінь іонності змінюється симбатно зі значеннями різниці електро-
негативностей атомів. Емпіричне співвідношення між величинами Р та
ІЗСл-ХяІтаке:
Р=Щ%а-Хв\ + \5\Ха-ХвЇ. (9.50)
Симбатно зі значеннями Р та \%А - %в\ змінюється також дипольний
момент ц. У неполярних молекул ц = 0, у полярних він збільшується. Найбільші
значення Р, |іі, \%А - %в\ мають іонні молекули.
Третій випадок. Для іонних молекул, наприклад, ЬіР, №С1 тощо, Е(« Ес
при рівноважних відстанях (рис. 9.5, с. 198). Математично цей випадок
аналогічний першому, слід тільки поміняти індекси «с» та «і» місцями. Тому для
іонних молекул Сі» ссі величинаРмає значення, близьке до ста процентів.
Перенесення електрона в рівноважному стані (г = г0) виникає під дією
випромінювання. При цьому (див. рис. 9.5) для іонної молекули, наприклад
№ СГ, збудженою формою є ковалентна (КаСІ). Отже, електронне збудження
супроводжується перенесенням заряду між атомами. Ця картина є
дзеркальним відображенням першого випадку (для малополярних молекул).
Процеси електронного перенесення в молекулах краще описувати на
базі моделі молекулярних орбіталей (МО) — «місць» можливого (та
реального) перебування електронів у молекулі (з відповідним значенням енергії).
197
Ме X"
Рис. 9.5. Потенціальні криві для іонної
Ме+Х~(І) та ковалентної МеХ (II) молекул
Атомна орбіталь (АО) була
визначена як одноелектронна
хвильова функція в атомі (див.
підрозділ 8.3). Аналогічно МО
визначається як
одноелектронна хвильова функція у
молекулі. Метод молекулярних орбі-
талей створений працями
Ф. Гунда, Р. Маллікената інших
авторів. Основою для
побудови атомних орбіталей було роз-
в'язування рівняння
Шредінгера для відповідної одноелек-
тронної системи — гідроген-
подібного атома. Аналогічно,
основою для побудови
молекулярних орбіталей є
розв'язування рівняння Шредінгера для найпростішої одноелектронної
молекулярної системи — молекулярного іона Н^.
Вважаючи, що, згідно з принципом Борна-Оппенгеймера, ядра
(протони) можна розглядати як закріплені на місцях, запишемо гамільтоніан для
найпростішої молекулярної системи — іона Н^:
2т
„2 „2
є . є
(9.51)
ГА ГВ ГАВ
Оскільки електрон однаковою мірою належить як атому А, так і атому В,
то для розв'язку рівняння Шредінгера слід взяти лінійну комбінацію АО
функцій І^та \зв
у = сіІ5А + С2Ізв, (9-52)
де \|/ — молекулярна орбіталь; с( — коефіцієнти розкладання МО на АО.
Навіть не розв'язуючи рівняння типу (9.43), легко показати, що повинні
існувати дві молекулярні орбіталі:
симетрична
щ=М(\зА + Ьв) (9.53)
та антисиметрична
ув=ЛГ(Ц*-1.у*),
(9.54)
де N— нормуючий множник.
Справді, з умов симетрії маємо (атомні функції ІзА та, \$в нормовані,
тому від квадратів хвильових функцій залишаються тільки квадрати
коефіцієнтів)
к2Н4 (9.55)
198
Тоді сх = с2 або сх = -с2> що і веде до виразів (9.53) та (9.54). Якщо за
допомогою хвильової функції \уь знайти повну енергію, то
Еь =^ьНщсіГ = Е0н+^ + ^, (9.56)
' ГАВ 1 + ^
а для енергії зв'язування (утворення іона Н^ з атома Гідрогену та іона Н )
Щ=Е-Е°н=£- + %±£. (9.57)
гАВ і + ^
Для функції і|/й маємо
^т-^т^- <9-58>
ГАВ і &
Інтеграл
3 = \{\зл)(\8в)<1У (9.59)
називається інтегралом перекривання.
Одноелектронний кулонівський (С) і резонансний (Р) інтеграли визначені
за допомогою формул (9.26) та (9.27). Розглянемо їх докладніше.
Інтегралу С можна надати наочне тлумачення. Він відповідає
притяганню між точковим позитивним ядром (+е) одного з атомів, наприклад В, та
електронною хмарою, яка описується Ід-орбіталлю сусіднього атома А.
Очевидно, що при досить великих відстанях між ядрами протбн із
нейтральним атомом не взаємодіє (слабкою поляризаційною взаємодією можна
знехтувати), тобто притягання протона до електронної хмари сусіднього
атома компенсується відштовхуванням між ядрами. Справді, залежність С
від відстані між ядрами має такий вигляд:
С = -М\-е-2ґ**(1 + гАВ)1 (9.60)
ГАВ1 А
У формулі (9.60) і далі всі величини наведені в атомних одиницях, тобто
енергія у (е2/а0), а гАВ у частках а0.
Зі збільшенням гАВ експоненційний член швидко наближається до нуля і
ГАВ
При цьому між'ядерне відштовхування дорівнює саме цій величині, але
з протилежним знаком
ГАВ
Отже, кулонівська енергія притягання та енергія між'ядерного
відштовхування значною мірою компенсують одна одну, і зв'язок виникає в основному
завдяки «резонансній» енергії (3, згідно з формулою (9.56) (нагадаємо, що
Р — від'ємна величина). Резонансний інтеграл (див. рівняння (9.27))
визначається притяганням «заряду перекривання» [{-є )\зАЛ8в\ позитивними
ядрами остова. Очевидно, що його величина повинна суттєво залежати від
перекривання атомних орбіталей 1^ та \зв.
199
При розгляді будови атомів
зазначалося (див. розділ 8), що ортого-
нальність хвильових функцій — це
наслідок нульового (сумарного)
перекривання. Для утворення
хімічного зв'язку, навпаки,
необхідне перекривання. У просторі між
ядрами електронна густина при
перекриванні атомних орбіталей
збільшується, і саме цей
негативний заряд притягує позитивні ядра,
утворюючи стабільну систему.
Перекривання АО залежить від
відстані між атомами. Дляя-орбіта-
лей воно значне при малих
відстанях між атомами (рис. 9.6), при
великих відстанях воно невелике для
будь-яких атомних орбіталей.
Відповідно, змінюється й інтеграл
перекривання. При гАВ—>°о $ —» 0,
тому що хвильові функції кожного
з атомів експоненційно зменшуються при віддаленні від свого ядра (див.
підрозділ 8.2). При гАВ—> 0 5—> 1. Верхньою межею для інтеграла
перекривання є одиниця, тому що при накладанні атома А на атом В функції \зА та
\зв співпадають, добуток (ІзА • 1зв) стає рівним |ь£| або |1у||, що при
нормуванні АО дорівнює одиниці. Кількісні розрахунки інтеграла перекривання
між І5-орбіталями атомів Гідрогену це підтверджують. Значення 8\зА,\*в
залежить від гАВ згідно з формулою
( ^2 ^
Рис. 9.6. Перекривання І5-АО в іоні н£:
а — малі між'ядерні відстані;
б — великі між'ядерні відстані
3і*а№і
= е
-ГАВ
1 + ^* +
ГАВ
З
(9.61)
Як бачимо, при гАВ -> 0 5 —»1. При гАВ —> °° член е~Г/4В зменшується
швидше, ніж збільшується багаточлен у дужках, отже, 5і—»0.
Обчислення резонансного інтеграла показує, що інтеграл Р залежить від
відстані гАВ таким чином:
Р = -^*(1 + ^). (9.62)
Якщо порівняти формули (9.61) і (9.62), то можна дійти висновку, що між
інтегралами Р та 5 існує тісна залежність. Ця залежність природна, оскільки
взаємодія нерухомих позитивно заряджених ядер із «зарядом перекривання»
в основному визначається саме величиною цього заряду і тому повинна
характеризуватися інтегралом перекривання.
Зазначимо, що наявність перекривання для утворення зв'язку між
атомами має суттєве значення, проте інтеграл перекривання та енергія зв'язку
200
не завжди змінюються симбатно зі зміною між'ядерної відстані. В цьому
можна переконатися на прикладі молекулярного іона Гідрогену. При гАВ-^ 0
інтеграл перекривання збільшується, але при дуже малих гАВ внаслідок
між'ядерного відштовхування зв'язок стає слабкішим, а повна енергія
системи підвищується.
Для стану, який описується
функцією \|/а, внески від ядерного
відштовхування та кулонівського
інтеграла (С) також компенсують один
одного, але резонансний інтеграл у
вираз (9.58) для АЕа входить зі знаком
мінус. Оскільки Р — від'ємна
величина, то енергія системи у цьому
випадку збільшується. Отже, зв'язок у
стані, який описується функцією і|/а
не утворюється. Тому ця
молекулярна орбіталь називається антизв 'язу-
ючою МО. Відповідно, молекулярна
орбіталь і|/6 називається зв 'язуючою
МО (від англ. апШюпсііпз — анти-
зв'язуючий (індекс «а») і ЬопсІіп§ —
зв'язуючий (індекс «6»).
При розгляді функцій \|/дта \|/а(див. формули (9.53) та (9.54) і рис. 9.7)
бачимо, що орбіталь \|/а має вузлову площину якраз посередині відстані між
ядрами. При проходженні через цю площину функція \|/а змінює знак на
протилежний, отже, |\|/^| на половині відстані між ядрами дорівнює нулю.
При г = гАВ/2 \8А = \8В\ згідно з виразом (9.54), \|/а = 0,тому електрон, який
знаходиться на молекулярній орбіталі \|/а «уникає» простору між ядрами.
Порівняємо густину ймовірності для зв'язуючої та антизв'язуючої
молекулярних орбіталей (функції \|/6 та \|/атут розглядаються без
нормувального множника):
Рис. 9.7. Зр'язуюча (щ)
та антизв'язуюча (\|/а) МО іонаН^
Уь
фА + Ьв\2фА\2+\ізв\2 +2(1^-15,);
(9.63)
|Уа2| =К -1**| =М +КГ -Ч^А -1*В)- (9.64)
Добуток (І5АЛзв) значно відрізняється від нуля там, де орбіталі 1^ та Ізв
перекриваються, тобто в просторі між ядрами. Із формул (9.63) і (9.64)
можна зробити висновок про те, що в просторі між ядрами ймовірність знайти
електрон на МО порівняно із сумою ймовірностей для окремих атомів |1^|
та \\$в\ для зв'язуючої орбіталі більша, а для антизв'язуючої, навпаки,
менша.
Кількісне обчислення енергії зв'язку у молекулярному іоні Н£ за
формулою (9.57) з використанням значень для інтегралів Р, Ста 5, наведених у
формулах (9.60)—(9.62), дало занижені значення для АЕЬ. Для атома Гелію
201
істотне поліпшення енергетичних розрахунків давала варіація ефективного
заряду 2 (зміна вигляду хвильової функції, див. підрозділ 8.3)^. Варіаційні
розрахунки для іона Н£ складніші, ніж для гелію, тому що крім 2 необхідно
враховувати зміну міжатомної відстані, щоб обчислити всю потенціальну криву
і знайти Д£тіпта гАВ - гАВ. Проведені розрахунки дали рівноважну
міжатомну відстань, яка майже точно збігається з експериментальною (гексп )0 =
= 0,106 нм, та енергією зв'язку в іоні Н£ — 2,25 еВ порівняно з 1,77 еВ при
2 = 1 = соп8І. Значення 2 для рівноважної конфігурації іона Н^ дорівнює
1,24, тобто значно відрізняється від одиниці. Значення 2 міняється зі зміною
міжатомної відстані від 2 = 2 при гАВ = 0 до 2 = 1 при гАВ —>°о.
Крайні значення 2 можна було б передбачити без розрахунків. Справді,
при гАВ= 0 система Н^ «перетворюється» у гідрогенподібний іон Не , для
якого 2=2 (якщо можна було б насправді об'єднувати ці системи, то
молекула водню при гАВ —> 0 перетворилася б на атом Гелію, відповідно, іон Н^ при
гАВ —> 0 переходить в іон Не ). При гАВ —> ©о другий протон знаходиться на
значній віддалі від першого і не впливає на атом Гідрогену, тому 2 = 1.
У атомі Гелію 2еф = 27/16 було меншим, ніж 2Не= 2. Це було наслідком
міжелектронного відштовхування і трактувалося як екранування одного
електрона іншим, тобто електрон рухався в слабкішому полі, ніж те, яке
створювало неекрановане ядро атома. У системі Н£ картина протилежна —
2еф = 1,24 більше, ніж 2Н = 1. Причиною цього є додаткове (порівняно з
одним протоном) притягання електрона іншим ядром, тобто друге ядро
посилює кулонівське поле, яке було створене першим.
Зміна вигляду хвильової функції іона Н£ пов'язана не тільки з
варіюванням ефективного заряду, а й з поляризацією атома Гідрогену протоном,
який до нього наближається. Поляризація викликає додаткове зміщення
електронної густини у простір між ядрами, її врахування приводить до
зміцнення зв'язку, й енергія дисоціації Н£ —» Н + Н дорівнює 2,71 еВ, що
дуже близько до експериментального значення.
Усі ці уточнення проводилися з огляду на те, що молекулярна орбіталь
будується з атомних — це так зване наближення МО-ЛКАО (лінійна
комбінація атомних орбіталей). Для іона Н^ точне розв'язування рівняння Шре-
дінгера для цієї одноелектронної задачі можна одержати шляхом введення
еліптичних координат, що дає можливість розділити змінні (так, як це було
зроблено для атома Гідрогену, див. підрозділ 8.2) і мати точні значення
енергії і хвильових сункцій. Можливість одержання МО, єдиної для всієї системи
(без розкладання її на АО), доводить фізичну реальність самої ідеї існування
молекулярних орбіталей.
Розгляд системи Н£ свідчить про те, що навіть один електрон, який
знаходиться на зв'язуючій молекулярній орбіталі, може досить міцно з'єднувати
ядра одне з одним. Тому зв'язок у молекулярних утвореннях не обов'язково
повинен бути двохелектронним, як це передбачалося методом валентних
зв'язків. Можливість існування одноелектронних зв'язків також показує, що
утворення хімічного зв'язку не є наслідком якогось «обміну» електронами
202
між атомами, а зумовлене притяганням між ядрами та електронами
(електроном), які знаходяться у між'ядерному просторі. Зазначимо, що в
кулонівську (0 та обмінну (Р) енергію, згідно з Гайтлером-Лондоном,
внесок, що забезпечує хімічний зв'язок, дають саме одноелектронні інтеграли С
та (3, які відповідають і за одноелектронний зв'язок у молекулярному іоні Н£.
Перейдемо тепер до класифікації і позначення молекулярних орбіталей,
причому будемо користуватися наближенням ЛКАО. Це наближення дуже
зручне для якісних висновків, до того ж, як це показано на прикладі іона Н^,
на його базі й кількісні розрахунки можна довести до високого ступеня
точності. Атомні орбіталі характеризуються (див. розділ 8) значеннями трьох
квантових чисел: я, /та т1 (без урахування спіна). Гідрогенподібні АО при
відсутності зовнішнього магнітного поля (2/ + 1)-кратно вироджені. Якщо
накласти магнітне поле вздовж обраної осі, наприклад осі 2, то виродження
знімається, проте не повністю. Орбіталі з т/ = (+1) та (-1), (+2) та (-2) і т. д.,
як і раніше, залишаються двократно виродженими. Наприклад, АО/?+1 та/?_1?
або, відповідно,рхтару\ сІ+1 тасІ_х\ сі+2т2ісі_2 або, відповідно, (іХ2тасіу2; сі^тз,
іїх2_у2. ^-орбіталі, для яких т1 = 0 тотожно, а також орбіталі р0 (р2); й0 (<£Д які
витягнуті вздовж заданого напрямку (осі г), у цьому випадку невироджені.
В двохатомній молекулі вже існує обраний напрямок — лінія зв'язку між
атомами. Позначимо її як вісь 2. В атомі квантове число т1 визначало
проекцію моменту кількості руху на обрану вісь і. Аналогічно у двохатомній
молекулі існує квантове число А,, яке визначає проекцію моменту кількості
руху на вісь — лінію, яка з'єднує ядра. Стани з однаковими значеннями |А,|
двократно вироджені (за винятком, коли X = 0) аналогічно розглянутому
вище випадку з атомними орбіталями. Замість числових значень \Х\
використовують грецькі літери: 0 — а; 1 — я; 2 — 8 і т. д.
Квантове число А, молекулярної орбіталі відповідає квантовому числу т/
для атомної орбіталі. Відомо, що |т7| < / (див. формулу (8.38)), тому із я-атом-
них орбіталей можна утворювати тільки а-молекулярні орбіталі (а-МО); зр-
атомних орбіталей а- та тг-молекулярні орбіталі (я-МО); з б?-атомних
орбіталей, відповідно, а-, п- та 8-молекулярні орбіталі (5-МО). Не важко
встановити відповідність між грецькою назвою максимально можливого значення |А,|
для даної молекулярної орбіталі та латинською назвою відповідної атомної
орбіталі.
У молекулярному іоні Н^ є атомні орбіталі тільки типу Із, тому
молекулярні орбіталі в основному стані Н£ — це молекулярні орбіталі а-типу. На
рис. 9.8, с. 204, зображені зв'язуюча та антизв'язуюча молекулярні а-орбіталі
іона Н£ . Обидві молекулярні орбіталі мають циліндричну симетрію
відносно осі 2, проте їх поведінка щодо вузлової площини (перпендикулярної до лінії
зв'язку на однаковій відстані між ядрами) різна. Зв'язуюча а-МО не змінює
знака при відображенні у вузловій площині, а антизв'язуюча змінює.
Зв'язуючі МО далі будемо позначати відповідною грецькою буквою,
наприклад а-МО, а антизв'язуючі а*-МО. В позначеннях МО вказується
також їх поведінка відносно центра інверсії (точки перетину вузлової площини
203
Рис. 9.8. МО а-типу, які
утворюються з 5-АО: а — зв'язуюча;
б — антизв'язуюча
©•© ©-0
2л, 2л.
з лінією зв'язку). Орбіталь, що
зберігає свій знак при інверсії,
позначається індексом «£», а та, що змінює
знак, — індексом «и» (від нім. §е-
гас-е — парний та ші^егасіе —
непарний). Для основного стану іона Н2
зв'язуюча орбіталь зберігає знак при
інверсії—це о^-орбіталь;
антизв'язуюча молекулярна орбіталь змінює
знак — це а* -орбіталь.
Молекулярні орбіталі, які
утворені р-АО, можуть бути як а-, так і тг-
типу. />2-орбіталі утворюють с8- та
а* - МО (рис. 9.9, а), ьрх- та/уатомні
орбіталі утворюють пи- та я*-МО
(див. рис. 9.9, б), я-молекулярні
орбіталі двократно вироджені, інакше
кажучи, кожна я-МО (як зв'язуюча, так
і антизв'язуюча) складається з двох
молекулярних орбіталей (пх та пу), які
мають однакову енергію. На відміну
від а-МО, зв'язуюча я-МО має м-ха-
рактер, а
антизв'язуюча—^-характер, що добре видно на рис. 9.9.
0^00^0 ^
А . .В
©•С+>е
0
б А*
0
2л,
0 /
• В<
0 ч
2л,
А • • В
/ е%
\СО
А • • В
204
па
пи
Рис. 9.9. МО, які утворюються з/?-АО: а — а-типу; б — я-типу
Рис. 9.10. МО, які
утворюються з */-АО: а — а-типу;
б — я-типу; в — 5-типу
(зображені тільки зв'язуючі МО)
й-АО можуть утворювати зв'язки а-, я- та
8-типу (для зв'язуючих МО це зображено на
рис. 9.10).
Квантове число / у позначення
молекулярних орбіталей не входить. Проте оскільки
МО утворюється з АО (за схемою ЛКАО), то
вказують головне та орбітальне квантові
числа тих АО, з яких утворюється МО. Так, з
атомних к-орбіталей утворені а§\з та а* Ь
молекулярні орбіталі в іоні Н^.
Кількість утворених МО дорівнює
кількості АО, з яких складаються молекулярні
орбіталі. Наприклад, для іона Н£ дві МО:.
а§1з та а* ^утворюються з двох АО: І^та
Ізв (рис. 9.11, а). Для/7-АО з шістьох атомних
орбіталей: р2л \ РХа \ РУа \ Р2в І РХв \ РУв
утворюється шість М0:о8 ;а* ;пих;тси ;я* ;я*
(рис. 9.11, б). Із рис. 9.11,6 бачимо, що
розщеплення енергії більше для а-МО, ніж для
я-МО. Це пов'язано з більшим
перекриванням /7-АО тоді, коли вони утворюють зв'язок
а-типу порівняно з перекриванням лхгипу.
На рис. 9.12 зображено залежність інтегралів
перекривання 82р 2р та $2рп2рп ВІД
міжатомної відстані. 82р 2Р > $2рп2р за
винятком малих гАВ (< 0,1 нм). ( 82р 2р ПРИ гав """* 0 прямує до одиниці, як і інте
грал 8\зллзв (див. формулу (9.61)), але $2ра2ра ПРИ гля~* 0 прямує до
(-1). Це виникає внаслідок того, що при суміщенні точок А і В (див. рис. 9.9)
2рх- (2ру) функції накладаються одна на одну з однаковими знаками (як
функції 1$ в іоні Н2), а 2рг -функції — з різними знаками.
Ш л|\
Рл
РхРуРгІ
РхРуРі
ш
Рв
а б
Рис. 9.11. Схеми МО для 5-АО (а) і дляр-АО (б)
205
^Рх2ря
2Ра2Ра>
Рис. 9.12. Залежність інтегралів
перекривання 82ра2ря™ $гРі12рп
від міжатомної відстані
Одноелектронне наближення в
атомах дало змогу знайти принцип
будови електронних оболонок атомів
і пояснити структуру періодичної
системи елементів. Так само
одноелектронне наближення в
молекулах — метод МО можна взяти за
основу для будови електронних
конфігурацій у молекулах. Принцип
будови для молекул ґрунтується на тих
самих основах, що й для атомів, а саме:
енергетичний критерій плюс
принцип Паулі. Електрони намагаються
зайняти МО з мінімальною енергією,
але заборона Паулі не дає змоги
розташовуватися на одній МО більше
ніж двом електронам (з різними
значеннями магнітного спінового квантового числа).
Розглянемо перший період системи Менделєєва. Дві іу-МО утворюють
о^- та а* -МО. Перший електрон розташовується на сг^-орбіталі з
утворенням досить стійкого іона Н£. Електронна конфігурація Н^ — (о^ія)
.Наступний електрон розміщується на тій самій орбіталі а§\з і утворюється міцна
молекула водню з конфігурацією (а^Ь) .
ГІри подальшому збільшенні кількості електронів останні змушені
займати вже антизв'язуючу орбіталь а*І5. Принцип насичуваності у методі
МО зумовлений неможливістю через заборону Паулі розміщуватись усім
електронам тільки на зв'язуючій орбіталі.
Електронна конфігурація іона Щ (ст^ іу)2 (а* І?)1 . В іоні Щ на два
зв'язуючих електрони припадає один антизв'язуючий, отже, різниця між кількістю
зв'язуючих та антизв'язуючих електронів дорівнює одиниці. Проте зв'язок у
іоні Щ набагато слабкіший, ніж одноелектронний зв'язок у іоні Н^. Цеє
наслідком загального правила: розташування електрона на антизв'язуючій
орбіталі набагато сильніше дестабілізує зв'язок, ніж розташування
електрона на зв'язуючій МО зміцнює його. Пояснити це можна, якщо розглянути
формули (9.57) та (9.58). Внесок від резонансного інтеграла Р, який несе
основну «відповідальність» за утворення зв'язку, в зв'язуючу орбіталь у ф =
= (1 + 5)/(1 -5) разів менший, ніж у антизв'язуючу. Проілюструємо це на
прикладі. В іоні Н£ для рівноважної міжатомної відстані інтеграл
перекривання (5) дорівнює 0,59, отже, ф = 3,9. Для молекули водню 5= 0,75 і ф = 7.
Зазначимо, що енергія зв'язку в іоні Н£ у 15 разів більша, ніж у іоні Щ.
Конфігурації (сг^іу)2 (а* Із)2 відповідала б молекула гелію Не2. Проте у
цьому випадку кількість електронів на зв'язуючій та антизв'язуючій МО
однакова. Отже, при повному узгодженні з експериментальними даними
молекула гелію не утворюється. Іон Не^ — відносно стійка система.
206
Порівняно із молекулою Не2 у нього не вистачає одного антизв'язуючого
електрона, і це добре стабілізує систему та сприяє зміцненню зв'язку.
Молекула Ьі2 — перша в ряду гомоядерних молекул другого періоду.
Перекривання між Ь-орбіталями у літію дуже мале, тому розщеплення 1,у-
АО на зв'язуючі та антизв'язуючі практично не виникає. У зв'язку з цим для
Літію та інших елементів другого періоду внутрішні Ія-електрони не
розглядатимуться, тому що вони не беруть участі в утворенні хімічного зв'язку.
Електронна конфігурація молекули літію (<з§ 2з) . Обидва електрони
знаходяться на зв'язуючій орбіталі, тому Ьі2 — стабільна молекула. Проте
міцність зв'язку у молекули літію набагато менша, ніж молекули водню. Це
пов'язано зі збільшенням головного квантового числа. 2$-електрон Літію
розташований далі від ядра і притягується ним слабкіше (енергія іонізації
атома Літію значно менша, ніж Гідрогену). Аналогічно спарені 2£-електрони
Літію слабкіше притягуються позитивними іонами остова молекули літію,
ніж Ія-електрони Гідрогену протонами. Збільшення розміру атома (як
наслідок збільшення головного квантового числа) призводить до збільшення
міжатомної відстані і менш ефективного перекривання. Як зазначалося
вище, слабкий зв'язок відповідає невеликому перекриванню.
Молекула берилію має електронну конфігурацію (а^ 2з)2 (а* Із)2, тому
ця молекула дуже нестабільна.
Електронна конфігурація молекули бору (а§ Із)2 (а*и$)2 (пи 2р)2.
Оскільки рівень тім-орбіталі двічі вироджений, то обидва електрони на цій МО не-
спарені, отже, молекула бору в повному узгодженні з експериментом, є
парамагнітною. Якщо дотримуватися схеми МО, зображеної на рис. 9.11,
то електронна конфігурація молекули, бору мала б мати такий вигляд:
(а^ 2$) (аи 2з) (с8 2р2) , але рівень пи 2р може бути нижчим, ніж (о§ 2р2).
Причиною зміщення орбіталі а 2р2 вгору є її взаємодія (відштовхування)
з а 2^-орбіталлю. ки2р- та а 2?-МО мають різну симетрію і тому не
взаємодіють (згідно з так званим правилом неперетину).
Взаємодія 0^2?- та а 2р2-М0, які мають однакову симетрію,
описується детермінантом
\Е(ое2*)-Е
= 0.
(9.65)
На
Н52 Е(а$2Рг)-Е\
Зазвичай Е(о 2з)«Е(о 2р2). Ця ситуація математично аналогічна
описаній вище взаємодії між іонним і ковалентним станами у полярній
молекулі. Аналогічно формулам (9.46) і (9.47) дістанемо
Ех=Е{вя28)-
Нс.
Е(оя2рг)-Е(о.2з)
Е2=Е(ае2рг) +
Н'
Е(ая2р2)-Е(ая25)
(9.66)
207
Р^и\2Р2) тобто енергія рівня оЛз зменшується,
а ор.р2 — збільшується ^ив. рис. 9.13).
У молекулі вуглецю пи-МО
заповнюється повністю, електронна конфігурація мо-
,4
лекули вуглецю—(ов 2з)2 (а* 2з)2 (пи 2р)
У молекулі азоту заповнюються пи 2р-
та о 2р2 -орбіталі й, отже, всі зв'язуючі
орбіталі, створені з^-АО, зайняті, а всі анти-
зв'язуючі вакантні, тому молекула азоту —
пи 0-Рх,у) °Дна 3 найміцніших. Енергія дисоціації моле-
* (2 \ кули азоту — 945 кДж/моль, тобто майже
и 10 еВ на одну молекулу.
У наступних молекулах електрони попа-
2зА{2/ \О 2зв дають на антизв'язуючі МО та дестабі-
/ лізують зв'язок. У молекулі кисню зв'язок
значно слабкіший, ніж у азоту (конфігурація
£ / (ря2з)2{рІ2в)2(пи2р)\ог2р2)2{%*&2р)2);
Рис 9 13 Схема МО шія У ФТ0РУ ЩЄ слабкіший (конфігурація
гис. у.і з. ^хема іуіи для / _ ч?/ *•■* \2/_ о \4/_ о \2/_* о \4\
елементів другого періоду (082зУ(0и2зУ(пи2рУ(082р2У(п82рУ\
а молекула неону, для якої кількість зв'язуючих
електронів дорівнює кількості антизв'язуючих, нестійка і не утворюється.
Слід відзначити, що якщо зв'язок у іонах N2 та С£ слабкіший, ніж у
відповідних молекулах азоту і вуглецю (електрон забирається із зв'язуючої
орбіталі), то зв'язок в іонах 0£ та Р2+, навпаки, міцніший, ніж у відповідних
молекулах кисню і фтору, тому що тут електрон відщеплюється з антизв'язую-
чої орбіталі. Отже, мірою міцності зв'язку в молекулах повинна бути різниця
між кількістю зв'язуючих (п) та антизв'язуючих (т) електронів. Ця
закономірність ілюструється даними, наведеними в табл. 9.1 для двохатомних
молекул другого періоду, починаючи з бору, тобто розглядаються лише 2/?-
орбіталі. 25-орбіталі можна не враховувати, тому що загальний внесок
зв'язуючих о^2$ та антизв'язуючих а* 2з молекулярних орбіталей дорівнює нулю.
Величина (п - т)І2 називається порядком зв 'язку (пара електронів
відповідає поодинокому зв'язку).
Розглянувши табл. 9.1, бачимо чітку відповідність між порядком зв'язку
та енергією дисоціації відповідної молекули або іона. Ця кореляція, однак,
наближена. Одна з основних причин відхилення — більш інтенсивна
дестабілізація молекул антизв'язуючими електронами, ніж стабілізація
зв'язуючими. Це призводить до того, що при однакових значеннях (п - т)І2:
(с2)>£д„с.(с>2);
Ядис. (СІ) > Етс, (Р2+ ); Етс (Щ ) > £дис. (о;).
Оскільки Етс(Х2) = Етс{Х2) + /ат." Люл.1 зменшення енергії
дисоціації двохатомної частинки відповідає відщепленню зв'язуючого електрона,
208
а збільшення — антизв'язуючого, то ^а величиною А/ = /ат -/мол. можна
зробити висновок про те, який електрон знаходиться на вищій орбіталі.
Наприклад, для водню та азоту Д/< 0, отже, з молекули відщеплюється зв'язуючий
електрон; навпаки, для кисню та фтору А/> 0 і відщеплюється
антизв'язуючии електрон.
Таблиця 9А. Електронні конфігурації та енергії зв'язку двохатомних
молекул елементів другого періоду
Молекула
або
і ІОН
в2
<3
с2
с~2
І мї
N2
°2
о2
о-2
р?
р2
р2"
№£
Ие2
Кількість
зв'язуючих
електронів
п
2
3
4
5
5
6
6
6
6
6
6
6
6
6
Кількість
антизв'язуючих
електронів
т
0
0
0
0
0
0
1
2
3
3
4
5
5
6
п-т
2
1,0
1,5
2,0
2,5
2,5
3,0
2,5
2,0
1,5
1,5
1,0
0,5
0,5
0
Енергія
дисоціації,
кД ж/моль
263
531
602
784
842
940
644
497
394
318
163
117
67
Молекула не
утворюється
Міжатомна
відстань,
нм
0,159
-
0,124
-
0,112
0,110
0,112
0,121
0,133
0,133
0,142
-
-
Молекула не
утворюється
Підтвердженням істинності схеми молекулярних орбіталей було
пояснення на її основі парамагнетизму бору і особливо поширеного у природі
кисню. Із позицій методу валентних зв'язків це було незрозуміле, тому що,
згідно з цим методом, утворення зв'язку супроводжується спарюванням
електронів.
Поступове заповнення зв'язуючих МО електронами приводить до
збільшення енергії дисоціації (С2 —> С2 —> С^; див. табл. 9.1), навпаки,
заповнення антизв'язуючих МО електронами дестабілізує зв'язок, наприклад,
С>2 ->02->С>2та р2+-ч>р2->р-.
У гомоядерних молекул рівноважна міжатомна відстань (див. табл. 9.1)
зменшується у ряду від бору до азоту (збільшення кількості зв'язуючих
електронів) і збільшується в ряду від азоту до фтору (збільшення кількості
антизв'язуючих електронів).
В атомі кожній електронній конфігурації відповідає своя система термів.
Експериментальне вивчення термів дає можливість встановити електронну
конфігурацію системи. Для визначення молекулярних термів можна скорис-
209
татися символікою, аналогічною тій, яка була використана при позначенні
атомних термів.
Як для атомів, так і для легких молекул спін-орбітальна взаємодія значно
слабкіша, ніж міжелектронна. Отже, загальний спін (5) у молекулі, як і в
атомі, знаходять шляхом додавання спінів окремих електронів і позначають
мультиплетністю терма—М Для молекул більше, ніж для атомів, характерні
стани зі спареними спінами — синглетні терми. Повністю заповнені
оболонки відповідають невиродженим станам. Для позначення молекулярних
термів використовують великі грецькі літери X, П, А, Ф..., які відповідають
значенням 0,1,2,3,... квантового числа Л. Це число визначає проекцію
орбітального моменту всієї молекули на лінію зв'язку. Для одноелектронних
систем Л = |А,| окремої орбіталі. Отже, а-орбіталь утворює Е-терм, я-орбіталь
П-терм, 5-орбіталь А-терм і т. д.
Для багатоелектронних систем Л = \ІХ\. Для оррбіталей X = 0, тому а-
орбіталі утворюють виключно Е-терми. Комбінації а-орбіталей з я-, 8- та
іншими орбіталями утворюють, відповідно, терми П, А тощо. Електрони на
двох нееквівалентних я-орбіталях можуть утворювати Л = 0 та Л = 2 і,
відповідно, терми £ та А. Для двох електронів спінові квантові числа 5
дорівнюють 0 або 1, тому можливі синглетні та триплетні терми. Отже, система
термів для двох електронів на нееквівалентних я-орбіталях така: Е, 2, А, А.
Парність термів (#- або м-характер) визначається кількістю непарних
електронів, тобто електронів, які знаходяться на непарних аи, ям-орбіталях.
Якщо кількість цих електронів парна, то терми мають ^-характер, якщо
непарна, то м-характер. Усі терми даної електронної конфігурації або парні
(#), або непарні (и).
Терми з Л Ф 0, як і одноелектронні стани з X Ф 0, двократно вироджені
відповідно до двох значень 1Х(.
Виродження Е-термів залежить від їх «походження». Х-терми, утворені
електронами на а-орбіталях, невироджені, тому що будь-яка комбінація
нулів (чисел X) дає тільки нуль. Навпаки, Е-термд, утворені електронами на
я-орбіталях, двократно вироджені. Це виродження, однак, відразу знімається
обертанням молекули навколо осі, яка з'єднує ядра, і утворює терми 2 та
5Г з різною енергією. Знаки плюс та мінус характеризують симетрію терма
(симетричний чи антисиметричний) по відношенню до площини, яка
проходить через згадану вище вісь. Отже, для двох електронів на
нееквівалентних я-орбіталях терми такі: 21 , 2Г, £ , 5Г, А, А.
Якщо я-орбіталі еквівалентні, то для виведення системи термів, так як
і для атомів, необхідно розглянути всі можливі мікростани. Для двох
електронів на еквівалентних я-орбіталях можливі такі три терми: Е~, А, 2. Ці
терми є також серед тердоіц для двох електронів на нееквівалентних орбіта-
лях. Тут, як і для атомних термів, спостерігається скорочення кількості термів
при переході від нееквівалентних орбіталей до еквівалентних внаслідок
заборони Паулі і тотожності електронів.
Основні стани всіх двохатомних гомоядерних молекул із заповненими
210
оболонками описуються термом Е^. Такий терм мають молекули водню,
літію, азоту, фтору. Молекули бору і кисню мають по два неспарені
електрони на еквівалентних я-орбіталях і система термів для них наведена вище.
Згідно з першим правилом Гунда основний терм має найбільшу мульти-
плетність — це терм 3 Е^ • Кількість електронів парна (два) і, незважаючи на
те, що у бору електрони на я„-орбіталях, а в кисню — на я*, терми в обох
випадках мають ^-характер.
Цікавою молекулярною системою є синглетний кисень. Для
перетворення звичайного (триплетного) кисню в синглетний шляхом
опромінювання світлом необхідно витратити 92 кДж/моль для переходу у Д^-стан
і 155 кДж/моль для переходу у ! Е£ - стан. Згідно з другим правилом Гунда
з двох збуджених станів молекули кисню нижче повинен бути терм А^
(більше значення Ь). Синглетний кисень можна добути і хімічним шляхом,
діючи хлором чи хлорноватистою кислотою на пероксид водню. Синглетний
кисень фотохімічно активний (характеризується фосфоресценцією) і легко
окиснює багато різних сполук з ненасиченими зв'язками, наприклад
бутадієн. В іоні 0£ є один неспарений електрон на я-орбіталі, терм, відповідно
П^; в іоні N2 неспарений електрон знаходиться на ст^-орбіталі, терм — Е^;
в іоні Не2 неспарений електрон на а* -орбіталі й основний терм *Е„.
Спектральні дані повністю підтверджують висновки, що одержані при розгляді
електронних конфігурацій. Особливо слід підкреслити, що основний стан для
кисню Е^, згідно зі спектральними даними, легко трактується на основі
методу МО (як і парамагнетизм молекули кисню), на відміну від методу ВЗ.
Довгий час точилася дискусія, яі^ий терм є основним для молекули
вуглецю: 3П„ чи] Е^. Терм1Е^ відповідає електронній конфігурації
(а^2^)2(а*25,)2(я1/2/7)4. Терм 3Пи відповідає електронній конфігурації
(а^2^)2(а* 25)2(пи2р)3((У§2р2У. Оскільки два електрони на я-орбіталі
спарені, то конфігурація апг еквівалентна конфігурації ая. Для електрона на
а-орбіталі X = 0, тому Л = Мя-орбіталі= Ь отже, це терм П. Два неспарені
електрони утворюють синглет та триплет, тобто терми Пта П. Згідно з першим
правилом Гунда основний терм — 3П. Кількість неспарених електронів на
ям-орбіталі дорівнює трьом, тому терм має и-характер. Отже, маємо терм
3Пи. Хоча збудження пи2р -» аЛр2 потребує затрати енергії, це
компенсується виграшем енергії внаслідок розпарування електронів, тобто
енергія термів 3ПМ та Е^ майже однакова. Сучасні дослідження спектральних
даних довели, що основним у молекулі С2 є терм Е^.
Переходи електронів у молекулах, як і в атомах, відбуваються не з будь-
якого рівня на будь-який інший, а підлягають правилам відбору: при
переході електрона квантове число не повинно змінюватися більш, ніж на
одиницю, мультиплетність, парність, знак (плюс чи мінус) і характер повинні
зберігатися. Оскільки в молекулі (на відміну від атома) слід враховувати
взаємне розташування та рух ядер, то необхідні додаткові умови, які
формуються у вигляді принципу Франка-Кондона. Згідно з цим принципом
електронні переходи з максимальною інтенсивністю зображуються вертикаль-
211
ними лініями між відповідними потенціальними кривими, наприклад,
основного та одного із збуджених станів. Принцип Франка-Кондона можна
обґрунтувати так. Відносна повільність руху ядер, порівняно з рухом електронів,
дає змогу розмежувати рух ядер та електронів згідно з принципом Борна-
Оппенгеймера. Вважаючи, що ядра знаходяться у стані спокою, можна
зобразити швидкий електронний перехід вертикальною лінією з точки на
одній потенціальній кривій до точки на іншій потенціальній кривій. Саме на
потенціальних кривих розташовуються точки, в яких ядра знаходяться у стані
спокою, перш ніж змінити напрямок і почати рухатися у протилежному
напрямку. Ці точки називаються точками повернення. В них ядра
перебувають відносно тривалий час, і це збільшує ймовірність переходу електрона
саме з точок повернення. Квантово-механічний розгляд осцилятора
підтвердив цю картину. Максимуми хвильової функції і максимальна ймовірність
місцезнаходження ядра припадають на точки повернення, за винятком
найнижчих коливальних рівнів. Для основного стану максимум припадає на
половину відстані між крайніми положеннями ядер, тому вертикальна лінія
виходить із середини горизонтальної прямої даного коливального рівня.
При розгляді електронних переходів можна виділити три характерні
випадки:
1) потенціальна крива електронно-збудженого стану знаходиться точно
над кривою основного стану, і при електронному переході між цими
станами коливальні рівні не збуджуються (рис. 9.14, а);
\&
а б в
Рис. 9.14. Електронні переходи у двохатомних молекулах: а — з основного
коливального стану в основний; б — з основного у коливально-збуджений;
в — фотодисоціація
2) при електронному переході виникає коливально-збуджена молекула
(див.рис. 9.14,6);
3) після збудження енергія молекули більша, ніж енергія зв'язку, що
відповідає фотодисоціації (див. рис. 9.14, в).
Перейдемо тепер до гетероатомних молекул. В цих молекулах атоми А
та В різні, і коефіцієнти розкладання МО на АО, сА та св, не можна визначити
з умов симетрії, а необхідно беспосередньо застосовувати варіаційний
принцип для молекулярної орбіталі:
212
Ж^фл+сдфл,
де (рА та срв — атомні орбіталі.
З умов
ґа/О
= 0 та
'**!-<
(9.67)
,_„.„. . - 0 знайдемо (аналогічно тому, як це було
А )св V в )сА зроблено вище, див. рівняння (9.43)):
сА(НАА-Е) + свНАВ=0;}
Санав+Св(нвв-е) = 0-
Якщо ЕА та Ев—енергетичні рівні відповідних атомів, то можна вважати,
що НАА = ЕА та Нвв - Ев, тому що кулонівський інтеграл Ста вираз для
між'ядерного відштовхування е*/гАВ значною мірою компенсують один
одного в інтегралі НАА (Нвв). Позначаючи НАВ = $АВ, одержимо детермінант
а-Е Рав
\$ав Ев ~ Е\
Розкривши детермінант, матимемо
= 0.
£,,2=^4^±і
Ео —Ел
+Р:
АВ
(9.68)
(9.69)
Знак мінус відповідає МО із меншою енергією, тобто зв'язуючій МО.
При ЕА = Ев формула (9.69) дає
Е\чг -ЕА±$АВ,
апри\ЕА-Ев\»$АВ
Еі =ЕВ+-
й.
ЕВ~ЕА
ЕХ=ЕА~]
(9.72)
Раз
ев-еа\
(9.70)
(9.71)
По*и
Схема МО для цього випадку
зображена на рис. 9.15. Зв'язуюча МО
з енергією Ех, близька до АО атома А,
а антизв'язуюча з енергією Е2—до
АО атома В. Отже, розташування
двох електронів на зв'язуючій МО
еквівалентне переходу електронної
пари до атома А, це випадок іонної
(полярної) молекули.
Коефіцієнти сА та св (точніше,
їх квадрати) характеризують внески
відповідних АО у дану МО. Для
зв'язуючої МО
Рис. 9.15. Схема МО для гетероядерних
двохатомних молекул
213
с\ЛЕА-Ех)2= $АВ = У>АВ
сі &ав &ав{Ев-Еа)2 (ЕВ-ЕА)2- <9-73>
Оскільки виконується умова \ЕА -Ев\ » $АВ, то сА » св, тобто орбіталь
атома А дає основний внесок у зв'язуючу МО.
Знаючи коефіцієнти сА та св, можна оцінити заряди д на атомах
( \
<ІА=КА-
£&4
(9.74)
)
де ИА — кількість електронів на атомі до участі його в утворенні зв'язку;
/ — номер МО; & — кількість електронів на даній МО (&= 0, 1 чи 2); сІА —
коефіцієнт ЛКАО (/-та МО атома А).
Для молекули ЬіН та їй подібних (Ил = ИВ=\) зайнята лише одна
молекулярна орбіталь, £ = 2, тому дА = 1 - 2с2А, а дв = 1 - 2с2в. При сА —> 1 та св —> 0
(сл + св = 13 Умов нормування) дА —> (-1), а #5—> (+1), що характерне для
іонної чи дуже полярної молекули.
Розглянемо тепер антизв'язуючу орбіталь для полярної молекули, тоді
|2
\2
М = Рлв Лев-Еа) (975)
\сА\2 (Ев-Е2)2 Й,
За умовою \ЕА-ЕВ\ » $АВ'\ |с^|»|с^|. Отже, якщо обидва електрони
розташувати на антизв'язуючій орбіталі, то молекула змінює свою
полярність, наприклад, Ьі Н —»Іл Н , де 8 близька до одиниці.
Оскільки, як правило, збуджується один електрон, то цікаво розглянути
молекулу, в якої один електрон знаходиться на зв'язуючій, а інший — на
антизв'язуючій МО. Оцінимо для цього випадку заряди на атомах А та В
за формулою (9.74):
і і і2 І * і2
ча-і-\сл\ -\са\ ;
і І І2 І * І2
Яв=і-\св\ -\св\ •
Внаслідок нормування АО \сА| + \с*А\ =1 та \св\ +\с*в\ = 1,тому^ =
=дв = 0.
Схема МО, зображена на рис. 9.15, підтверджує цей висновок: кожний
електрон знаходиться на МО, яка розташована біля свого атома. Отже,
збуджений стан іонної молекули відповідає неполярній частинці, це співпадає
з результатами методу ВЗ. Згідно з методом МО, можна передбачити зміну
полярності при дальшому збудженні молекули. Щоб одержати цей
результат у методі ВЗ, необхідно його передбачити заздалегідь і ввести додаткову
іонну структуру.
З одержанних вище співвідношень бачимо, що полярність молекули
визначається в основному різницею енергій (Ев - ЕА) вихідних атомів. Навіть
214
якщо НАВ є малою величиною (для ковалентних молекул це відповідає
відсутності зв'язку) та Ех = ЕА, а Ех = Ев, то може утворюватися відносно міцна
іонна молекула, тому що електрон переходить з АО атома В на МО з
низькою енергією Е{=ЕА. Це дає виграш у енергії - (Ев - ЕА) (для ковалентних
молекул цей ефект неможливий, тому що ЕА = Ев). Оскільки Ев = -Ів, а
ЕА = -ІА, то | Д£| = | А/| = \іА - Ів |, де ІА та Ів — енергії іонізації відповідних
атомів. Величина А/ близька до Д% (різниці електронегативностей), тому що
основний внесок у % робить саме енергія іонізації (див. підрозділ 8.4). Отже,
метод МО приводить до відомого висновку про те, що полярність зв'язку
зумовлена різницею електронегативностей атомів, які його утворюють.
У термах та електронних конфігураціях гетероядерних молекул відсутні
індекси «£» та «м», тому що у гетероатомних молекулах немає центра
інверсії. Інші позначення повністю зберігаються. Якщо не звертати уваги на
індекси «£» та «ш>, то серед гетероядерних молекул можна знайти такі, які
мають таку ж електронну конфігурацію, що й відповідна гомоядерна
молекула. Такі молекули називаються ізоелектронними.
Ізоелектронні молекули та молекулярні іони, утворені з атомів елементів
другого періоду, та електронні конфігурації, що їм відповідають, такі:
1) (п2р)4 — С2, ВИ, ВеО, ЬІР, СК+, ВО+;
2) (п2р)4 (о2р)1 — Щ,СК СО+, ВО, ВР+, ВеР;
3) (п2р)4 (а2Рг)2 — N2, СО, ВР, С^Г, Ш+;
4)(п2р)4(о2рг)2(п2р)1 -КО,0£,СР;
5) (п2р)4 (а2р2)2 (п2р )2 - 02, ОТ, РО+;
6)(я2р)4(а2;?г)2(7і*2/?)3-ОР, Р2+, 02;
1)(п2р)4(а2рг)2(п2р)4-¥ьО¥-,
Ізоелектронні молекули близькі за міцністю зв'язку, тому що різниця.між
кількістю зв'язуючих та антизв'язуючих електронів у них однакова.
Наприклад, енергії дисоціації молекул першої серії дорівнюють, кДж/моль: С2 —
598; ВИ — 389; ВеО — 443; ЬіР — 573. Хоча ці молекули значно
відрізняються за полярністю (С2— ковалентна, ЬіР — майже чисто іонна), проте енергії
їх дисоціації у межах даної серії дуже близькі. Найміцнішими є молекули
третьої серії, які мають найбільшу кількість зв'язуючих електронів. Енергія
дисоціації молекул цієї серії становить, кДж/моль: И2 — 940; СО — 1072;
N0 — 1050; СКҐ — 1000. Дана серія характеризується також близькістю
інших властивостей. Так, міжатомні відстані молекул у цій серії такі, нм: г#2—
0,110; гсо— 0,113; гВР — 0,126; гш+ —0,106; гга- —0,115.
Дещо несподіваним явищем є подібність дипольних моментів—для М2,
СО та ВР вони близькі до нуля (для И2 дипольний момент тотожно дорівнює
нулю). Майже повна відсутність полярності у молекул СО та ВР, на перший
погляд, незрозуміла, тому що різниця електронегативностей атомів Окси-
215
гену та Карбону, і особливо Флуору та Бору, значна. Відповідно, внески
(коефіцієнти сІА) АО Флуору та Оксигену у зв'язуючій МО помітно
перевищують внески для Бору та Карбону. Однак перенесення заряду в молекулах СО
та ВР не відбувається, тому що зв'язуючі МО, які розташовані близько до
більш електронегативних атомів (Оксигену чи Флуору), заповнюють в
основному електрони із цих самих атомів. Справді, якщо шість зв'язуючих
електронів конфігурації (п2р) (^2Р) в молекулі азоту беруться по три від кожного
атома, то в молекулі СО — чотири від атома Оксигену і лише два від атома
Карбону, а у ВР — п'ять від Флуору і лише один від Бору. Зв'язок, у якому
один атом надає вільні орбіталі (Карбон — одну, Бор — дві), а другий,
відповідно, електронні пари (Оксиген — одну, Флуор—дві), називається донорно-
акцепторним (див. далі підрозділ 9.2). У молекулах СО та ВР «компенсація»
зумовлена тим, що донорами є більш електронегативні елементи. Якщо
згадати, що у періоді (див. підрозділ 8.4) збільшується як енергія іонізації, так
і кількість електронів на зовнішній оболонці атома, то розглянуті вище
молекули не слід вважати чимось екзотичним. Компенсація дуже наочно
виявляється при розгляді формули (9.74) для визначення заряду атомів. У атомів
Оксигену та Флуору великі значення як Ил, так і сІЛ, а в атомів В та С, навпаки,
значення Ив і сІВ — малі.
Тепер настала черга зробити порівняння методів МО та ВЗ. Обидва ці
методи є певним наближенням. Для якісного опису властивостей молекул
метод МО має деякі переваги перед методом ВЗ. В першу чергу тому, що
він є хорошою основою для опису електронних спектрів. За допомогою
методу МО можна вивчати як молекули з парним числом електронів
зв'язку, так і з непарним, які незручні для опису методом ВЗ; за допомогою
методу МО вивчаються як гомо-, так і гетероядерні молекули; метод ВЗ
в основному придатний для вивчення властивостей ковалентних молекул,
для опису полярних молекул необхідна додаткова концепція резонансу.
Метод МО має, однак, і свої недоліки. Розрахунки за методом Гайтлера-
Лондона дають для енергії зв'язку в молекулі водню 3,14 еВ, а простий
метод МО ЛКАО тільки 2,68 еВ (експериментальне значення АЕщ = 4,747 еВ),
тобто метод МО дає гірший результат для АЕН, ніж метод ВЗ.
Метод МО в його простому варіанті може іноді призвести і до якісно
неправильних результатів. При збільшенні кількості електронів у молекулах
насичуваність зв'язку відображається методом МО правильно, але при
взаємодії між атомом та молекулою картина порушується. Розглянемо
систему (Н2 + Н) як конфігурацію Н3, тобто коли атом Гідрогену підходить
до молекули водню з утворенням рівностороннього трикутника. У методі
МО з трьох атомних орбіталей легко сконструювати три МО з енергіями
£і=Ян+2р 1 (9?6)
Е2 =Е3=ЕН -$)
де р — резонансний інтеграл між атомами Гідрогену (для конфігурації
рівнобедреного трикутника усі рн_н між собою однакові).
216
Повна енергія комплексу Н3 з трьома електронами така (кожна МО
вміщує не більше, ніж два електрони):
Ещ = 2Е{ +Е2= ЗЕп + ЗР. (9.77)
Енергія системи молекула Н2 плюс атом Н у цьому наближенні така:
£н2+н =2(ЕН +р) + £н =3£н +2Р- (9-78)
Оскільки Р — від'ємна величина, то Ещ < £н2+н> тобто, згідно з методом
МО, відбувається зменшення енергії при утворенні комплексу Н3 з
молекули Н2 та атома Н, що суперечить експерименту та методу ВЗ (див. вище).
Для того щоб при використанні методів МО та ВЗ результати були
правильними, необхідно їх уточнювати.
У методі МО молекулярна орбіталь с§Ь = і|/6 може мати два електрони
з різними спінами, тому МО всієї молекули водню матиме такий вигляд:
¥мо=Х|/6(1)¥б(2). • (9.79)
Обчислення енергії зв'язку із застосуванням функції (9.79) дає 2,70 еВ, що
значно відрізняється від експерименту. Для поліпшення результатів можна
скористатися підходами, які були використані при розрахунках енергії
молекулярного іона Гідрогену (див. вище). Так, варіювання ефективного заряду
в методі МО дає набагато кращий результат Д£Нг = 3,49 еВ при 2еф =1,197.
Метод МО у розглянутому варіанті — це одноелектронне наближення. Для
атомних систем у рамках одноелектронного наближення найкращий
результат дає метод самоузгодженого поля. Енергія зв'язку в молекулі Н2,
розрахована за допомогою самоузгоджених МО, становить 3,62 еВ.
Врахування екранування та поляризації можна провести і в рамках
методу ВЗ, що приводить до поліпшення результатів, а саме: екранування дає
для енергії зв'язку в молекулі водню 3,78 еВ при 2еф = 1,166, а екранування
плюс поляризація дає 4,04 еВ порівняно із 3,14 еВ при використанні
найпростішого варіанта методу Гайтлера-Лондона.
Величини 2еф, розраховані за методами МО та ВЗ, близькі між собою
і значно менші, ніж для іона Н^. Зменшення 2еф у молекулі водню
порівняно з іоном Н2 враховує міжелектронне відштовхування зв'язуючих
електронів (межею для 2 в іоні Н£ є 2Не+ = 2, а межею для 2 у молекулі водню
є 2еф атома Гелію, що дорівнює 1,6875). Хоча поліпшення хвильової функції
дає кращі результати для обох методів, проте за методом МО енергія зв'язку
на 0,3—0,4 еВ менша, ніж за методом ВЗ. Зіставимо тепер хвильові функції
методів МО та ВЗ. Якщо підставити у вираз (9.79) значення \|/6 з формули
(9.53), то (без урахування нормуючого множника):
^мо =|>л (0+Ц, 0)]|>л (2)+Ь, (2)] =
= 1^(1)1^(2) + 1^(1)1^(2)+1^(1)1^(2)+1^(1)1^(2)= (9.80)
217
Перші два члени цієї функції (Ч^) повністю збігаються з відповідною
функцією Гайтлера-Лондона (9.20), а два інші (¥,) — це іонні стани: Н^Нд
та Н^Щ. Отже, функція Гайтлера-Лондона відрізняється від повної
хвильової функції методу МО відсутністю іонних станів. Ці стани можна врахувати
у методі ВЗ (як це було зроблено вище для полярних молекул), а саме
У = скУк +с,ЧІ = М(Ч'к +ХТ,), (9.81)
де X = Сі Іск\ N— нормувальний множник.
Внесок іонних станів такий:
ф = _^ = _А^. (9.82)
с]+с\ \ + Х2
Енергія зв'язку після розрахунків основного стану молекули водню,
згідно з функцією (9.81) та варіацією параметра X, становить 4,12 еВ. Це
кращий результат, ніж дає функція Гайтлера-Лондона без урахування
іонних станів (але з урахуванням поляризації та екранування). Параметр X
невеликий, максимальне його значення, яке майже точно відповідає
рівноважній відстані у молекулі водню, дорівнює 0,25 (при К —> °° X —> 0). Внесок
іонних станів ф при такому значенні Я, згідно з формулою (9.82), малий і
дорівнює приблизно 0,06.
Функція МО (9.80), для якої X = 1, дає неправильні результати, причому
при К —» 0 вона передбачає рівномірне розщеплення молекули водню як на
атоми Н, так і на іони Н та Н~. Оскільки енергія іонізації атома Гідрогену
набагато більша, ніж його спорідненість до електрона, то енергія системи
Н + Н~— набагато вища, ніж для системи Н + Н. Функція МО дає гірший
результат, ніж функція Гайтлера-Лондона, тому що рух електронів на МО не
скорельований | Ч^оІ = | Ч/^ (і) | • | Уь (2) |, тобто електрони рухаються
незалежно один від одного. У функції Гайтлера-Лондона х¥к = \зА (і) • Ізв (2) +
+ Ізв (і)• І8А (2) рух електронів не незалежний — коли перший електрон
знаходиться біля ядра А, то другий — біля ядра В, і навпаки. Електрони
уникають один одного і це призводить до зниження енергії. Коли ж обидва
електрони знаходятся біля одного ядра, міжелектронне відштовхування зростає,
і енергія збільшується. Зазначимо, що саме міжелектронне відштовхування
призводить до того, що іон Нне дуже стабільний, і це є причиною високої
енергії іонних станів. Властивість електронів розташовуватися біля різних
ядер називається право-лівою кореляцією. Врахування кореляції дає для
енергії зв'язку кращі результати за методом валентних зв'язків порівняно
з методом МО, де кореляція не враховується. Право-ліва кореляція — це
тенденція, а не закономірність. У методі ВЗ вимога до електронів (вони
повинні знаходитися біля різних ядер) надто сувора, тому запровадження
іонної складової, яка певною мірою дає змогу обом електронам перебувати
біля одного ядра, дає дещо кращі наслідки. Проте, як показують розрахунки,
іонна складова дуже мала, тому функція МО, яка взагалі не враховує
тенденцію електронів знаходитися біля різних ядер, дає гірші результати, ніж
функція ВЗ, яка перетворює тенденцію на закономірність.
218
Для того щоб поліпшити результат у методі ВЗ, необхідно вийти за межі
суто ковалентного зв'язку та врахувати іонну складову. Для дальшого
поліпшення методу МО необхідно враховувати взаємодію різних МО, так звану
конфігураційну взаємодію.
Основний стан молекули водню — (о^ія) »терм 2^ (див. вище).
Цьому терму відповідає також двічі збуджений стан — (а* Із)2 (кількість
електронів на и-орбіталі парна). Як показано вище, орбіталі з однаковою
симетрією взаємодіють, і функція, яка враховує цю взаємодію, має такий вигляд:
^ = М1,2+Х(¥0)1(2=[1^(1) + 1^(1)][1^(2)+ЬВ(2)] +
+ х[ад)-ьв(і)][і*Л2)-і*я(2)}
Розкривши дужки та звівши подібні члени, матимемо
^=(і-%)^+(і+х№ = ^
.1-Х
(9.84)
При X - (1 + зс)/(1 - %) функція (9.84), яка враховує конфігураційну
взаємодію еквівалентна функції (9.81) методу ВЗ, що враховує резонанс
ковалентного та інного станів, тобто якщо уточнити метод ВЗ додаванням
іонних станів, а метод МО шляхом конфігураційної взаємодії, то результати
обох методів збігаються.
9.2. Багатоатомні молекули з локалізованими
зв'язками
Для характеристики зв'язку в двохатомних молекулах використовують
такі параметри, як енергія, довжина, полярність зв'язку тощо. У
багатоатомних молекулах ситуація складніша. Наприклад, у молекулі С02 хімічним
зв'язком сполучені три атоми. Молекула С02 має лінійну конфігурацію,
причому атом С знаходиться посередині. Атоми Оксигену зв'язані з атомом
Карбону і практично не зв'язані один з одним, тому у молекулі С02 є два
зв'язки Карбон—Оксиген і немає зв'язку Оксиген—Оксиген. Отже, є
можливість ділити багатоатомну молекулу на окремі фрагменти, у яких зв'язок
здійснюється між сусідніми атомами. Фрагментація багатоатомної
молекули є припущенням, проте для багатьох молекул воно наближається до
дійсності. Сполуки, у яких зв'язки є багатоцентровими, і де не завжди можна
провести фрагментацію, будуть розглянуті у наступному розділі. У цьому
розділі розглядаються багатоатомні молекули, до яких можна застосувати
наближення двоцентрових локалізованих зв'язків між атомами. Як і для
двохатомних молекул, у цьому випадку окремі зв'язки можна описати
деякими кількісними параметрами. Проаналізуємо характеристики зв'язку між
даною парою атомів (у фрагменті) при переході від однієї багатоатомної
молекули до іншої (табл. 9.2). Із табл. 9.2 видно, що в обраних фрагментах
різних молекул практично зберігається сталість відстаней між атомами
(довжина зв'язків).
219
Таблиця 9.2. Довжина деяких зв'язків у різних сполуках
Фрагмент
X—У
*Г—Н
С—0
>Х-V,
НМ
0,1017
0,1016
0,1014
0,1010
0,1030
0,1162
0,1164
0,1174
0,1160
0,1166
0,1210
Сполука, яка
містить зв'язок
г>ГН3
^Н4
Ш2С1
Ш2ОН-цис
КН2ОН-транс
С02
СОЗ
СОР2
СОРС1
СОСІ2
Н2СО
Фрагмент
X—У
С—СІ
'Х—V,
НМ
0,1766
0,1750
0,1767
0,1770
0,1780
0,1770
0,1759
0,1746
0,1750
Сполука, яка
містить зв'язок
ССЦ
СР3С1
СНСІз
СН2С12
СН3С1
СНРСІВ
СН2РС1
СОС12
СОРС1
Характерна величина для даного зв'язку — це його енергія.
Гомологічний ряд насичених вуглеводнів — це добрий матеріал для перевірки сталості
енергій зв'язків С—С та С—Н. Ці енергії можна розрахувати за теплотою
утворення сполук (табл. 9.3). Як видно з табл. 9.3, енергії зв'язків С—С та
С—Н у різних алканів практично сталі.
Розглянемо ще один приклад, який ілюструє можливість фрагментації у
молекулах. Візьмемо три такі лінійні молекули: С02, СОЗ та С82. У молекулі
С02 гс=0 = 0,1162 нм, у молекулі СОЗ — 0,1164 нм. У молекулі С82 гс=8 =
= 0,1553 нм, а у молекулі СОЗ — 0,1559 нм. По реакції С02 + С82 = 2С08
молекули розщеплюються і потім із них утворюються по два зв'язки: С=0
та С== 8. Якщо енергія цих зв'язків стала величина, то Д#° наведеної реакції
не повинна значно відрізнятися від нуля. Термохімічні дані це
підтверджують — АН° = 3,3 кДж/моль.
При відсутності кратних зв'язків між кожною парою атомів у
багатоатомних молекулах існує двохелектронний зв'язок, який, згідно з методом
ВЗ, описується функцією Гайтлера-Лондона (функцією зв'язку).
Відповідно до методу ВЗ валентність атома — це кількість зв'язків, які він може
утворювати, і визначається кількістю неспарених електронів. Елементи першої
та сьомої груп мають один неспарений електрон (конфігурації, відповідно,
5і тая2/?5), отже, вони одновалентні, елементи шостої групи (конфігурація
$2рА має два неспарені електрони) — двовалентні; п'ятої групи
(конфігурація 82рг має три неспарені електрони) — тривалентні.
Якщо розглянути електронні конфігурації елементів (неперехідних) IV, III
та II груп, відповідно: з2р2,82рх та я2, то можна допустити, що елементи IV
групи повинні бути двовалентними, III групи — одновалентними, а II
групи — нульвалентними (аналогічно Гелію, який має конфігурацію Із2).
Валентність 2 зустрічається у елементів IV групи, а валентність 1 — у елементів
220
Таблиця 9.3. Енергії зв'язків С—С та С—Н
Зв'язок
С—Н
Середнє
значення єс_н
С—С
Середнє
значення Єс-н
Енергія, кДж/моль
430 ± 12
457 ± 12
442 ± 12
425 ±4
443 ±9
410±4
436 ± Ш
369 ±8
353 ±6
342 ±8
357 ±8
360 ±6
347 ±8
343 ±8
353 ±8
Реакція і
СН^СЬҐ+Н*
сн;->сн2,+н#
сн;-^сн2+н2
сн4->сн;+н#
с2н4->с2н;+н-
с2н6-+с2н;+н#
с2н6->сн;+сн;
с3н8-*с2н;+сн;
С4Н10 —> С2Н5 +С2Н5
н-с4н10->с2н;+сн;
«-с5н12-^«-с4н;0+сн;
и-сбн|4->с5н?і+сн;
«-с6н,4 -> «-с3н; +«-с3н;
III групи. Проте для цих груп, відповідно, більш характерні валентності 4 та
3. Цей факт можна пояснити переходом атомів у збуджений стан: зр3 —
у елементів IV групи та зр2—для III групи. Чотири та, відповідно, три неспа-
рені електрони дають шукані валентності 4 та 3. Витрата енергії на перехід
електрона до збудженого стану компенсується утворенням двох додаткових
зв'язків. Наприклад, енергія 2$ —» 2/?-переходу в атомі Карбону дорівнює 418
кДж/моль, а енергія двох С—Н-зв'язків (див. табл. 9.3) дорівнює 872 кДж/
моль. З цієї самої причини елементи II групи не нульвалентні, а двовалентні
(збудження з2 —з1р1 дає два неспарені електрони). Оскільки збудження в усіх
випадках дає два додаткових неспарених електрони, то для багатьох
елементів характерні валентні стани, що відрізняються саме на два, наприклад,
ТІ— 1таЗ;8п,РЬ — 2 та 4; Аз, Р, 8Ь — 3 та 5; 8, 8е, Те — 2,4,6; СІ, Вг, І —
1,3,5,7.
При розпаровуванні електронів необхідна наявність відносно близько
розташованої (за енергією) орбіталі. Для того щоб збільшити валентність
Оксигену чи Флуору, необхідно перевести електрон із 2р-АО на Зз-АО, що
енергетично невигідно, і не компенсується енергією хімічних зв'язків, що
утворюються, отже, ці елементи ніколи не виявляють валентності, більшої
ніж 2 або 1. Проте для їх аналогів 8 та СІ (та інших халькогенів та галогенів)
збудження пов'язане з переходом пр —> псі, що потребує значно меншої
енергії. Тому Сульфур та Хлор можуть реалізувати максимальну (а також
проміжні) валентність, яка дорівнює кількості електронів у зовнішньому
шарі (6 та 7). Відносно близько розташовані вакантні ^/-орбіталі вміщують до
п'яти неспарених електронів, що разом з одною з- та трьома/7-орбіталями
221
дає змогу розмістити шість електронів Сульфуру та сім Хлору. Аналогічна
ситуація характерна і для елементів V групи: Фосфор, а також Арсен, Стибій,
Бісмут мають валентності п'ять поряд із валентністю три, наприклад, відомі
хлориди РС13 та РС15, але сполука N015 невідома. Наявність у елементів V-
VII груп з п > 3 вакантних ^-орбіталей з однаковим головним квантовим
числом сприятлива для збудження валентності не тільки тому, що зі
збільшенням п зменшується енергетична відстань пр —» псі, ай унаслідок
близькості радіальних розподілів пр- та и<і-орбіталей, що дає змогу їм ефективно
перекриватися і залучає тим самим л<і-орбіталі до зв'язку. Практична
відсутність перекривання Зя-АО з 2з- та 2р-А0 і велика енергетична відстань
2р —> Зя не дають можливості алемштам другого періоду (Нітрогену, Окси-
гену, Флуору) мати більшу валентність.
Однократно іонізовані Нітроген (N-1-) та Оксиген (0+) мають, відповідно,
конфігурації 2зр3 та 2з2р3 і можуть виявляти валентності 4 та 3, наприклад,
у іонах ІЧНд та Н30+. Особливо слід зупинитися на «п'ятивалентності»
Нітрогену. Всі молекули, в яких Нітроген «п'ятивалентний»: N205, НК03
тощо мають великі дипольні моменти. П'ятиковалентних зв'язків Нітроген
не утворює (вище вже зазначалося, що сполука ЖЛ5 не існує).
У тому випадку, коли збудження електрона потребує великих витрат
енергії, компенсувати це можна лише тоді, коли в утворенні зв'язку беруть
участь дуже електронегативні атоми, що здатні захопити та міцно
утримувати слабко зв'язаний електрон. З дуже електронегативними елементами
(Оксиген, Флуор) утворюють сполуки навіть благородні гази. Для них
енергія збудження пр —> (п + 1)^ максимальна (як і енергія іонізації) серед
елементів даного періоду, проте вона зменшується, як і для всіх елементів, зі
збільшенням п. Тому Гелій, Неон та Аргон залишаються благородними, а
Криптон і особливо Ксенон утворюють відносно стійкі сполуки: КгР2, ХеР2,
ХеР4, ХеОР4, ХеР6. Слід зазначити, що у Ксенону з'являється можливість
збудження валентності за схемою 5р —> 4/, що, певно, і відрізняє Ксенон від
інших благородних газів за можливістю утворювати хімічні зв'язки.
Валентність, що зумовлена кількістю локалізованих пар електронів,
називається ковалентністю. Для іонного зв'язку можна ввести поняття
ступеня окиснення. Кількість зовнішніх електронів, яку атом може віддати,
дорівнює заряду катіона, який утворюється при іонізації, і називається
позитивним ступенем окиснення. Кількість електронів, яку він може прийняти,
називається негативним ступенем окиснення. Позитивні ступені
окиснення характерні для металів, наприклад, (+1)—для Натрію, (+2)—для Кальцію,
(+3)—для Алюмінію і т. д. Негативні ступені окиснення зустрічаються у
неметалів, наприклад, (-1) — для Флуору, Хлору, (-2) — для Оксигену, (-3) —
для Нітрогену і т. д. Позитивний ступінь окиснення елемента часто
співпадає з номером (И) групи періодичної системи Менделєєва, де він
розташований (та співпадає з «валентністю за Оксигеном»). Негативний ступінь
окиснення часто дорівнює (8 - И) (так звана «валентність за Гідрогеном»).
Це поняття можна поширити на полярні молекули. При цьому більш елек-
222
тронегативному атому надається негативний заряд, що утворюється при
локалізації на ньому всіх зв'язуючих електронних пар. Так у Н20 ступінь
окиснення Оксигену дорівнює (-2), Гідрогену (+1); у МН3: Нітрогену —
(-3), Гідрогену — (+1); у НК03: Оксигену — (-2), Гідрогену — (+1),
Нітрогену— (+5). При обчисленні ступеня окиснення враховується, що
молекула повинна бути електронейтральною системою.
Ковалентність та ступінь окиснення часто співпадають (за абсолютною
величиною), так в НС1 Хлор та Гідроген одновалентні і ступені окиснення
дорівнюють, відповідно, (-1) та (+1); в Н20 Гідроген одновалентний, Окси-
ген двовалентний, ступінь окиснення Гідрогену — (+1), Оксигену — (-2).
Проте у пероксиді гідрогену Оксиген двовалентний, а ступінь його
окиснення (-1); в >Ш4С1 Нітроген утворює чотири ковалентні зв'язки, а ступінь
його окиснення (-3). У нітратах та НИ03 ступінь окиснення Нітрогену (+5).
До зміни ступеня окиснення чутливими є енергії остовних електронів,
що визначаються методом РФЕС — рентгенофотоелектронної
спектроскопії. В цьому методі опромінення речовини високоенергетичними
рентгенівськими квантами з енергією (Ну) і реєстрація кінетичної енергії
(Дсін.) електронів, вибитих із певних атомних орбіталей, дає можливість
обчислити енергію зв'язку (Езв) відповід- й
ного (зокрема остовного) електрона на
конкретній орбіталі із співвідношення:
Езв=Ну-Екін. Енергія зв'язку тим
більша, чим більший позитивний
ступінь окиснення елемента внаслідок
певного дезекранування електронів остова,
навпаки, для негативних ступенів
окиснення «хімічний зсув» є від'ємною
величиною. Це положення добре
ілюструється даними для Нітрогену, який
знаходиться в різних валентних станах в
комплексній сполуці
[Со(НН2СН2СН2НН2)2(К02)2]Н03
(рис. 9.16), а також даними для сполук ,
ксенону (рис. 9.17). Звичайно, що
залежність Езв від ступеня бкиснення є
лише відображенням залежності енергії
зв'язку остовних електронів від
реального заряду на атомах. Ця залежність 1500
дійсно існує, її добре ілюструє рис. 9.18,
де наведено залежність хімічного зсуву „ ~
(Д£зв.) ДО* 2/7-остовних електронів Суль- -96 404 ^
фуру від ефективного заряду на атомі. Рис 9 16 Енергія зв,язку
Утворення двохелектронного дво- Ь-електронів Нітрогену
центрового зв'язку може відбуватися не у [Со(НН2СН2СН2]ЧН2)2(Ш2)2]Шз
223
я
о
я
8
я
3500 Н
2500 Н
_,— ,.
мн2.
1 1-
1'" "" Т
N02
м
і і
і і
Ч
Ч
^ 1
і
н
Г ХеР^Х
к^Хе
1 і і і
ХеР^
/ХХеР4
і і і і—
—►
00 6
г §4
0
0 +2 +4 +6
Стан окиснення ксенону
Рис. 9.17. Залежність відносних енергій
зв'язку Зй?5/2 електронів оболонки
Ксенону від стану його окиснення
тільки внаслідок спарення
електронів сусідніх атомів, а й іншим
шляхом, а саме: один з атомів
надає пару електронів, а інший —
вакантну орбіталь. Такий зв'язок
називається донорно-акцептор-
ним.
Наприклад, атом Нітрогену в
молекулі аміаку має неподілену
пару електронів і утворює донорно-
акцепторний зв'язок з протоном,
який має вакантну 5-орбіталь:
:ш3+н+->>ш; (>щ;сг).
Атом Бору в сполуках ВХ3 має
одну вакантнур-АО й утворює зв'язки з донорами електронних пар:
ВН3 +: Н" -> ВЩ (Іл+ВЩ);
ВРз + (СН3)20: -> (СН3)20 • ВР3;
ВР3 +: Р"-> ВР4" (П+ВР4").
Особливий інтерес становлять донорно-акцепторні сполуки Бору
з Нітрогеном, які імітують сполуки Карбону. Сполука ВР3—№3 — аналог
гексафлуоретану; ВН3—К(СН3)3 імітує тетраметилметан; В3^Н6 має
структуру бензену (шестичленне плоске кільце з атомів Бору та Нітрогену, які
чергуються).
При утворенні донорно-акцеп-
торного зв'язку пара електронів
узагальнюється, і цей тип зв'язку
відрізняється від «чисто»
ковалентного (точніше, полярного,
якщо пара електронів зміщена до
одного з атомів) тільки
«походженням». В іонах №і4 та ВЩ усі
зв'язки N—Н та В—Н ковалентні і,
відповідно, еквівалентні всі атоми
Гідрогену. Заряди в КГН4 та ВЩ на
будь-якому з атомів Гідрогену не
локалізуються. Донорно-акцептор-
ний зв'язок малополярний, тому що
0 +10+20^
.. донори електронних пар — це елек-
Рис. 9.18. Залежність зсуву лінії 82р тронегативні елементи кінця періо-
в різних сполуках Сульфуру від ефек- . . -. .
.тгптІЛгЛоо««т,м. / и с. 7 гс . дів, вакантні орбіталі, навпаки, ма-
тивного заряду (у): 1 — Н2а; 2 — СЬ2; ' . г .
З — с4Н43; 4 — 802; 5 — 802Р2; 6 — 8Р6 ють більш електропозитивні еле-
224
АЕ
10
5
0
зв.>еВ
§2р
/і
1 і
г4
\_ .
5
/б
-і \
менти, це приводить до своєрідної компенсації (див. вище підрозділ 9.1,
властивості молекул СО та ВР), а саме: донор, який дає електронну пару (в
результаті цього на ньому повинен виникнути позитивний заряд), відтягує її до
себе внаслідок різниці в електронегативностях. '
Донорно-акцепторний зв'язок збільшує валентні можливості атомів
і відіграє суттєву роль у координаційних сполуках (див. далі підрозділ 9.4).
Теорія хімічного зв'язку в багатоатомних молекулах повинна
пояснювати не тільки валентні властивості атомів, а й геометричну конфігурацію
молекул.
Двохатомні молекули можуть бути лише лінійними. Для трьохатомних
існує вибір — молекула може бути або лінійною, або зігнутою.
Розглянемо трьохатомні молекули типу Н2Х, де X — 0,8,8е, Те. Ці
молекули мають зігнуту структуру з кутом між зв'язками, близьким до 90°. Це
можна пояснити таким чином.
В елементах X з атомною
конфігурацією з2р4 є по два неспарених
електрони, наприклад, рх тару (з та/?2 —
електрони спарені й не беруть участі
в утворенні хімічного зв'язку, це не-
зв'язуючі електрони). Хмари
ймовірності для рх- та/уорбіталей витягнуті
вздовж відповідних осей, це і
зумовлює зігнуту конфігурацію молекул
Н2Х. Перекривання/^-хмари з хмарою
1$-орбіталі атома Гідрогену
максимальне, якщо атом Гідрогену
розмістити на відповідній відстані на осі х9
і дорівнює нулю, якщо розмістити
його на осі;; (в останньому випадку всі
додатні внески повністю скомпенсо-
вані від'ємними, рис. 9.19). Відповідно
до перекривання змінюється і
резонансний інтеграл, який визначає міцність зв'язку.
Врахування взаємодії між атомами Гідрогену вносить поправку. Ця
«обмінна» взаємодія має характер відштовхування. Це приводить до збільшення
кута Н—X—Н. У молекулі води відстань між атомами Гідрогену
мінімальна (зі збільшенням головного квантового числа збільшується також
протяжність р-орбіталей) і кут між зв'язками максимальний— 104,5°. В інших
молекулах Н2Х кут Н—X—Н наближається до прямого; Н—8—Н — 92°;
Н—8е—Н — 91°; Н—Те—Н — 90,25°. Додатковий внесок у
відштовхування дають кулонівські сили. Молекула води полярна (різниця електронегатив-
ностей Оксигену та Гідрогену велика), і відштовхування між позитивно
зарядженими атомами Гідрогену досить значне, навпаки, інші молекули
майже неполярні, і кулонівська взаємодія у них незначна. Слід зазначити, що
225
Рис. 9.19. Перекривання/7х-орбіталі
з $-орбіталлю, яка розташована
на осі у
у молекулах Ме20, де Ме—лужний метал, кулонівське відштовхування між
катіонами таке велике, що ці молекули мають лінійну конфігурацію при
мінімумі кулонівської взаємодії.
Елементи V групи мають електронну £2/?3-конфігурацію. Три неспарені
електрони повинні утворювати три взаємно перпендикулярні зв'язки.
Справді, молекули УН3 мають структуру піраміди з атомом У у вершині та
кутами між зв'язками, близькими до 90°: Н—N—Н — 107,8°; Н—Р—Н —
93,5°; Н—А§—Н — 91,8°; Н—8Ь—Н — 91,3°. При переході від Нітрогену до
Стибію міжатомні відстані Н—У збільшуються, ступінь іонізації водню
зменшується і, відповідно, відхилення від валентного кута 90° зменшуються.
У розглянутих молекулах Н2Х та УН3 усі зв'язки між Гідрогеном та
центральним атомом для даної молекули еквівалентні (за довжиною, енергією,
полярністю тощо). Ця еквівалентність є наслідком рівноцінностір-орбіталей
(Рх'РуРг) центрального атома.
У елементів І—IV періодів внаслідок збудження валентностей, крім/?-
орбіталей, в утворенні зв'язку беруть участь я-орбіталі. Хоч/?- та £-АО мають
різну форму, всі зв'язки у молекулах типу СН4, ВН3 та інших еквівалентні. Це
має місце тому, що зв'язок утворюють не самі я- та^-орбіталі, а так звані
гібридні орбіталі. Процес гібридизації атомних орбіталей призводить до
того, що орбіталі, які беруть участь у формуванні зв'язку, є
еквівалентними.
Розглянемо найпростіший випадок, коли відбувається гібридизація однієї
.у-та однієїр-АО. лр-конфігурація реалізується з конфігурації'з2 при збудженні
електрона з ^-орбіталі на/7-орбіталь. Згідно з правилами Гунда більш
стабільний терм має максимальний спін, тобто спінова частина його хвильової
функції симетрична (55), отже, згідно з принципом Паулі, орбітальна частина
повинна бути антисиметричною
\з(2) р(2)[
Додавши та віднявши стовпчики визначника, матимемо
(9.85)
72
х(і)
Х(2)
Х(1)
Х(2)|
де
х2 = ^-р).
(9.86)
(9.87)
Функції X] та %2 — ортонормовані (з урахуванням цього вони виводяться
з .у- т&р-АО). Обидві ці функції відповідають однаковій енергії.
Е\,2 ="
Е.+Е„
(9.88)
226
Справді, Ех = \гхНгхЛУ = 1/(5 + />)Я(* + р)с/К = да2 ^ *= —
+ £„
тому що Я5/7 = їзНрсІУ = 0 -
Аналогічно
внаслідок ортогональності ^- та/7-орбіталей
одного атома.
Ег=\%2нг2йУ =
н.
"*" . ДР __ 5
+ £„
2 2
Гібридні %г та %2-орбіталі зображені на рис. 9.20. Вони еквівалентні
і відрізняються лише орієнтацією
«лопатей», ^-гібридизація добре пояснює
будову молекул МХ2, де X — галоген
або Гідроген, М — лужноземельний
метал. Усі ці молекули лінійні, зв'язки
М—X у них еквівалентні. Орбіталі %г
та %2" мають найбільше поширення
вздовж лінії зв'язку і забезпечують
ефективне перекривання з орбіталями Рис'9Ж ^-гібРиДні °Рбі™і
атомів галогенів або Гідрогену (див. рис. 9.20).
5/7-гібридизацію слід враховувати і у двохатомних молекулах. Наприклад,
при розгляді двохатомних молекул, що утворюються металами ІА та ПА,
врахування гібридизації сприяє зміцненню зв'язків у молекулах Ме2 (Ме —
лужний метал) порівняно з ^-^-зв'язком, а також дає стабільність молекулам
М2 (М — лужноземельний метал). Без урахування ^-гібридизації
молекули М2 взагалі не повинні були б існувати (аналогічно молекулі гелію). З
позицій методу МО ^-гібридизацію у молекулах Ме2 та М2 можна тлумачити
як розширення базису АО шляхом включення/7-орбіталей.
Для пояснення геометрії 4-х- та 5-атомних молекул також
використовуються уявлення про гібридизацію АО. Як і у випадку з ^-гібридними
орбіталями зр2- та£/?3-гібридні орбіталі одержують у вигляді лінійних
комбінацій АО.
При ^-гібридизації три атомні з-, рх
гібридні орбіталі:
1
та /у орбіталі утворюють три
■І'
'*Тгр>
(9.89)
1
**-іг8-кр'-кр>
Оскільки/^- та/у-АО лежать в одній площині, то й гібридні орбіталі
розташовуються у цій площині (ХОУ). Орбіталь %х орієнтована вздовж осіх. Не
важко довести, що кут між %г та %2-, а також між %2- та %3 дорівнює 120°.
227
Отже, при ^-гібридизації три гібридні орбіталі утворюють плоску
фігуру з кутами 120° (рис. 9.21, а). Таку будову мають молекули ВХ3, А1Х3, ЬаХ3,
де X — галоген. Нагадаємо, що електронна конфігурація атомів III групи
у збудженому стані є зр2.
а б
6
Рис. 9.21. а) зр2-, б) 5/?3- та в) 5/?3-гібридні орбіталі (альтернативний варіант)
Внаслідок .ур3-гібридизації утворюються чотири еквівалентні орбіталі
з тетраедричною структурою і кутом між зв'язками 109° 28!:
Хі"2* 2 Р"
1 ^73
5С2=х* +
*а;
(9.90)
2"' 2Ух у/й"9
У цьому випадку орбіталь %і зорієнтована вздовж осі 2, орбіталь %2
знаходиться в площині Х02 (див. рис. 9.21, б).
Альтернативним є варіант, колид/Лгібридні орбіталі зорієнтовані вздовж
діагоналей квадрантів (див. рис. 9.21, в). У цьому варіанті у позначення
гібридних орбіталей можна внести відповідні індекси, що позначають
квадранти:
228
\%т=^(* + Рх + Ру+РіУ>
хтіт=^-а+^-л); (9,91)
[хїїі=-^-Л-/^ + /?г)-
5,/?3-гібридизація особливо важлива для сполук Карбону. Метан має
структуру тетраедра, в інших алканах цей фрагмент повторюється.
Наприклад, етан — це два тетраедри, які мають спільну вершину. Зв'язки Н—С—Н
та Н—С—С мають кут 109°28\ У решти молекул парафінів кут, близький до
тетраедричного, мають також зв'язки С—С—С. У циклопарафінах
можливість змінювати орієнтацію менша. Наприклад, у молекулі циклопропану
кут між зв'язками С—С дорівнює 60°. Він відрізняється від тетраедричного,
тому в цьому випадку перекривання 5/?3-орбіталей атомів Карбону невелике
і, отже, зв'язок С—С слабкий. Тут можна говорити про кутову напругу
зв'язків або про «зігнуті» зв'язки, які слабкіші за звичайні.
Іон ІЧН^ має структуру метану. Це свідчить про те, що чотири
еквівалентні N—Н-зв'язки утворені .ур5-гібридними орбіталями. Вважається, що
£/?3-гібридизація може відбуватися вже у молекулі аміаку, і протон
приєднується за місцем пари електронів, яка знаходиться у ^/?3-гібридному
стані. Так можна пояснити, чому кут Н—N—Н в аміаку дорівнює 107,8°
і значно ближчий до тетраедричного, ніж до прямого. Аналогічне
пояснення стосується й молекули води, яка має дві неподілені пари електронів. Кут
104,5° у воді також ближчий до тетраедричного, ніж до прямого. В
електронній конфігурації інертного газу з2р6 є чотири електронні пари. Вони
можуть знаходитись у стані зрг та «імітувати» метан. Якщо використати
метод «об'єднаного» атома, то не важко побачити, що Неон є «спільним»
атомом для молекул НР, Н20, ИН3 та СН4. При утворенні зв'язку електронна
пара зміщується до атома Гідрогену. У молекулі метану зміщуються усі
чотири пари, і тетраедрична симетрія зберігається. Згідно з правилом Р. Гілеспі
відштовхування між неподіленими парами сильніші, ніж між парами, що
беруть участь в утворенні хімічного зв'язку. У молекулах води та аміаку на
зв'язок з Гідрогеном потрібно, відповідно, дві або три пари, при цьому
зменшується відштовхування між ними, кути між зв'язками Н—О—Н та
Н—N—Н стають меншими, ніж тетраедричний. У гідридах елементів
наступних періодів періодичної системи електронні пари знаходяться далеко
одна від одної і відштовхування ще слабкіше. Крім того, зі збільшенням
головного квантового числа 5- та/ьорбіталі «розмазуються» у просторі,
слабкіше перекриваються і менше піддаються гібридизації.
Підключення до гібридизації й?-орбіталей дає можливість для реалізації
більш різноманітних структур. Наприклад, обмежуючись лише 5- та/?-АО,
229
неможливо побудувати квадратні чи октаедричні гібридні орбіталі, що
характерні для комплексів перехідних металів (див. рис. 9.22). У табл. 9.4
наведено типи гібридизації і АО, які утворюють відповідні гібридні орбіталі,
включаючи 5-, р- та ^/-орбіталі.
а
і
о
_-_ ____..
І
І
б і
і
;
і \%&
І—„.і ^
піні я .і^і^&вШ'^^Л
с
-г-і
- І
1
І
І
-----
« *.—і
З ■: .:■
О
III
Рис. 9.22. с/$7?2-гібридні орбіталі Нікелю у №(СН)4~ (а)
та Л/?3-гібридні орбіталі Феруму уРе(С>ї)*~ (б)
Хімічний зв'язок у багатоатомних молекулах доцільно розглядати на
основі вчення про симетрію.
Для характеристики симетрії використовують її елементи та операції.
230
Таблиця 9.4. Типи гібридизації (а-зв'язок)
Молекула
МХ2
мх2
МХ3
МХ3
МХ4
МХ4
мх5
мх5
мх6
Конфігурація
молекули
Лінійна
Зігнута
Трикутник
Трикутна піраміда
Тетраедр
Квадрат
Тригональна біпіраміда
Тетрагональна піраміда
Октаедр
АО, що утворюють
гібридні орбіталі
ЗРг\Л^Р
РхРу> Л^-у^Ху
ЗРхРу\<*2гРхРу\
^х2_у2^ху'^22СІх2_у2СІху
РхРуРг'^х2.у2^хуР2
ЗРхРуРг'^хуЛхгЛуг
^^рхру',ах2_у2сі22РхРу
Лг2 *РхРуРг; й22^хуАх2_у2 Рг
(І22(Іх2-у28РхРу'^г2^х2_угРгРхРу
*?йУ-у**РхРуРг
Елементи симетрії— це уявні точки, лінії або площини у тих об'єктах,
що розглядаються. До елементів симетрії належать: площини, центр, осі
тощо. Осі симетрії мають різний порядок, тобто кількість суміщень самого
об'єкта з собою при повороті його навколо осі на 360°. Зустрічаються
і складніші елементи симетрії — дзеркально-поворотні осі симетрії
(суміщення самого об'єкта з собою при повороті на визначений кут навколо цієї
осі та відображення на площині, що перпендикулярна до осі повороту).
Операція симетрії— це дія над об'єктом, яка приводить його до
суміщення з самим собою. До операцій симетрії належать: обертання (С„),
відображення у площині (а) та невласне обертання (5Л), тобто обертання
з одночасним відображенням у площині, що перпендикулярна до осі
обертання. Операцію 8п можна уявити як добуток двох операцій
$п = Сп'Он. (9.92)
Між елементами та операціями симетрії існує зв'язок; операції
обертання (Сп) припускають наявність осей симетрії, операції відображення (вн)
пов'язані з наявністю площин симетрії, а операція 8п можлива при
дзеркально-поворотних осях визначеного порядку.
Операції обертання С™. Індекс «я» вказує на кількість суміщень
самого об'єкта з собою при обертанні на 360°. Кут 360% називають
елементарним кутом обертання. Для С2 він дорівнює 180°, для С3 —120°, С4 — 90°
і т. д. Індекс т вказує кількість оборотів на елементарний кут: С\ означає
поворот на 120°; СІ —на 240°; С3 —на 360°. Отже, операція С^ тотожна
з операцією спокою, яка позначається символом Е.
Операції відображення а доповнюються індексами А, V та сі, тобто о»/,,
а„, оа. Операція ан — це відображення у горизонтальній площині, тобто у
площині, яка перпендикулярна до осі симетрії найвищого порядку. Площини
відображення для операції а1) проходять через вісь симетрії вертикально.
231
Операція оа—це відображення у вертикальних площинах симетрії, які
проходять через бісектриси координатної площини ХОУ.
Операції невласного обертання 8п є «добутком» двох операцій
симетрії: Сп та сн. Іноді вони мають спеціальні позначення. Так, операцію 82
позначають буквою / та називають інверсією
і = С2ан. (9.93)
Справді, при наявності осі другого порядку та перпендикулярної до неї
площини у тілі можна виявити точку, де всі лінії, які через неї проходять,
з'єднують еквівалентні точки фігури. Така точка називається центром
інверсії, або центром симетрії. Центр інверсії знаходиться посередині
відрізків, які з'єднують еквівалентні точки фігури. У молекулі інверсія
кожного атома через центр симетрії призводить до суміщення його з таким
самим атомом.
Наприклад, для ромбічної піраміди (симетрія С2у) відомо чотири операції
симетрії: Е — спокій, С2 — обертання на 180°, о'„ та а^ — відображення
у площинах. При всіх операціях на місці залишається висота піраміди
(загальна вимога — щоб при будь-якій операції залишалася незмінною хоча б
одна точка).
Кількість операцій симетрії для даної молекули називається числом
симетрії. Наприклад, молекула води (точкова група С2і)) має чотири операції
симетрій Е, С2, СУу та а^, отже, число симетрії для неї дорівнює чотирьом.
Молекула аміаку — тригональна піраміда (точкова група С3і;) має такі
операції симетрії: Е, С3, С|, сг^,, а^, а" і, отже, число симетрії для неї дорівнює
шести.
Точкові групи симетрії діляться на чотири типи:
1) ізотропні — сферичні (Я5)9 ікосаедричні (/та/Л), октаедричні (Ота Он)
і тетраедричні (Г, Та та Тн);
2) діедричні містять п взаємно перпендикулярних осей симетрії £>„, ВпН
таД^(« = 2,3,4,...,оо);
3) осьові мають тільки одну вісь симетрії порядку п: Сп, СпЬ Сп1) (« = 1,2,
З,...,-);
4) невласні осьові, або «альтернуючі» 8п (п = 1,2,3,..., °°), при п = 1 групи
позначаються як С5, при п > 2 як С, (52 = 0-
Кожну багатоатомну молекулу можна віднести до певної точкової групи.
У табл. 9.5 наведені приклади найпоширеніших точкових груп і молекул, що
до них належать.
Послідовне виконання кількох операцій симетрії має назву їх «добутку».
Наприклад у групі С2х)\ С2- С2 = Е; о'„ • а'„ = Е. Можна одержати ще кілька
добутків, наприклад С2 о'и=оІ; Оу-оі^ С2. Отже, добуток будь-яких двох
операцій симетрії еквівалентний третій операції, яка характерна для
точкової групи. У розглянутому випадку площини симетрії взаємно
перпендикулярні, вісь симетрії знаходиться на перетині двох площин симетрії. Якщо
добуток двох операцій симетрії не залежить від порядку їх виконання, то
232
Таблиця 9.5. Точкові групи
Точкова
група
с,
с2
Сгн
Сіо
Сзи
Сао
с
Операції
симетрії
Е
Е,С2
Е, С2, аА
Е, С2,2ау
Е, 2С3, Зау
£, 2С4, С2,
4ау
Е, Соо, ооау
Приклад
ЗіВгРСІІ; 1
н2о2
транс-С2Н2С1
Н20
Ш3
[СиСВДзГ
неї 1
Точкова
1 група
г>«
£з*
І>4*
£~*
Ям
Г"
| о*
Операції
симетрії
£, 302,2а,,, аА
£, 2С3, ЗС2, Зау, аА
Е, 2С4,4С2,4ау, аА
£, Соо, °°С2, ^а^, аА
£,ЗС2,2а„,84
2Г, 6<$4> ЗС2, 8Сз, 6а^,
48 операцій
Приклад
ї<204(плоска)
ВСІз (плоска)
[Р*С14]2-(плоска)
N2, С02
СН=С=СН2
СН4
зр6 •
З З
2 2
кажуть, що операції симетрії комутують, наприклад, С2ои = ауС2. Проте
якщо осі симетрії розташовані одна до одної під іншими, ніж 180° кутами,
наприклад, під кутом 120° у тригональній біпіраміді чи тригональній призмі,
то багато операцій симетрії не комутують, отже, результат залежить від
порядку проведення операцій.
У рівнобедреному трикутнику 2 1 З
(рис. 9.23), де площини симетрії
розташовані під кутами 120° один до
одного, результат залежить від
порядку проведення операцій симетрії.
Так, результат повороту на 120° (С3)
та наступного відображення у
площині, що проходить через лівий
нижній кут (а^), еквівалентний від- і кі X 2 З
ображенню у площині, яка
проходить через вершину 2 (а^). Якщо
спочатку провести відображення у
площині, що проходить через нижню
Е
з
Сз
2
сі
1 2
Рис. 9.23. Операції симетрії в точковій
групі С3и
ліву вершину (аи), а потім повернути фігуру на 120° (С3), то результат буде
еквівалентним відображенню у площині, яка проходить через правий нижній
кут (О- Отже, о'„С3 = о£ С^ = сґ0; о„С3 Ф С3о'„
Щоб кожного разу не робити послідовно певні операції на тій чи іншій
фігурі, можна заздалегідь скласти таблицю множення операцій симетрії та
з її допомогою швидко знаходити відповідний результат. Таблицю множення
для точкової групи симетрії С2у наведено у табл. 9.6.
Набір операцій симетрії для будь-якої точкової групи утворює множину,
яка називається групою. Відповідний математичний апарат — теоретико-
груповий аналіз можна застосувати для розв'язування багатьох задач теорії
хімічного зв'язку.
Для того щоб множина була групою, необхідно, щоб виконувалися такі
вимоги:
Таблиця 9.6. Таблиця множення 1) добуток будь-яких двох опе-
для^гочкової групи С2„ рацій, що входять у групу, повинен
також бути операцією, яка належить
до даної групи. У цьому, зокрема,
можна пересвідчитися на прикладі
операцій для точкової групи С2и
(операціїЕу С2, ст^та а^). Так,
добуток С2 с/у дає операцію а^, яка
також належить до групи С2и;
2) група повинна мати одиничну операцію — операцію спокою Е.
Добуток одиничної операції на іншу завжди комутативний, наприклад, ЕС2 =
= С2Е — С2\
3) повинен виконуватися закон асоціативності відносно добутку трьох
операцій симетрії. Згідно з цим законом добуток результату множення
перших двох операцій на третю повинен дорівнювати добутку першої
операції на результат множення другої та третьої операцій. Наприклад, добуток
С2 • Су • с>у можна подати двояко: (С2 • с£)с£ та С, (а'^-с^). У першому
випадку дістанемо (див. табл. 9.6): (С2 • о'^ві = <^у <з0 = Е; а у другому
випадку — С2 (сГу-СУу) =С2 - С2 = Е. З розглянутого прикладу бачимо, що
результат множення однаковий як у першій, так і в другій послідовності. Закон
асоціативності для операцій симетрії виконується без винятків;
4) для кожної операції повинна бути зворотна операція. Добуток прямої
та зворотної операцій залишає молекулу у вихідному положенні. У групі С2у
для операції С2 зворотна операція також С2, для о'и — &'„ і т. д. (див. табл. 9.6).
У групі С3и для операції С3 зворотна операція — С\. У будь-якій точковій
групі є операції симетрії, які зворотні кожній з операцій, що існують у групі.
Дуже важливо встановити, як ведуть себе координати даної молекули,
а також її атомні та молекулярні орбіталі при виконанні тих чи інших операцій
симетрії. Ця проблема вирішується за допомогою таблиць характерів. Як
приклад розглянемо таблицю характерів для точкової групи С2х) (табл. 9.7).
Таблиця 9.7. Таблиця характерів груп С2и та С3„
Сги
Ах
Аг
В\
Вг
Сзо
Аг
Е
Е
1
1
1
І
Е
1<
1
2 .
С?
1
1
-1
-1
2С3 ,
, 1
І
-1
о$«>
1
-1
1
-1
о*.)
1
-1
-1
1
Зау
1
-1
0
Координати або АО
2, 22, 5
ху
Х,Х2
У,У2
Координати або АО
2, 229 £
(х>уУ, (хг9уг)і {хг~у1,ху)
У стовпцях таблиці характерів наведені відповідні операції симетрії,
а рядки містять характери незвідних уявлень. Уявлення А х — повносимет-
234
Е
С2
<
<
Е
Е
Сг
<
<
Сг
Сг
Е
<
<
<
<
<
Е
Сг
<
<
<
Сг
Е
ричне, описує поведінку .у-орбіталей, координати і (орбіталі/?2) та орбіталі
с12г. Ці орбіталі при будь-яких перетвореннях симетрії точкової групи С2і;
незмінні (усі характери дорівнюють (+1)). Уявлення А2 описує поведінку
орбіталі <4>, (див. рис. 8.11, с. 145) при перетвореннях симетрії групи С2у5 і має
характери: 1,1,-1,-1. Справді, при операції спокою (Е) та обертанні навколо
осі 2 на 180° (С2) орбіталь сі АО залишається без змін. При відображенні
у площинах Х02 та У02 орбіталь ^змінює свій знак на протилежний.
Символами В позначають зображення, що змінюють свій знак при повороті
навколо головної осі (в даному випадку осі С2).
Простежимо за поведінкою/^-,ру- та/?2-орбіталей при дії на них операцій
симетрії точкової групи С2у. В спокої всі три орбіталі залишаються на місці
і характер уявлення дорівнює трьом. При повороті навколо осі 2 на 180°
(операція С2) знак орбіталі/?2 не змінюється, а орбіталірх та/?^ змінюють знак
на протилежний. Сума всіх цифр дорівнює: +1-1-1 =-1, отже, характер
уявлення при дії оператора дорівнює (-1). При відображенні в площині Х02
знак змінює тільки орбіталь^, а при відображенні в площині У02 лише/7^.,
отже, характери уявлень для операцій а^ та а^ дорівнюють (+1). Отже, для
/?-орбіталей одержуємо уявлення П з характерами: 3, -1, +1, +1. У таблиці
характерів (див. табл. 9.7) такого уявлення немає, проте його можна зобразити
як суму простіших уявлень. Складне уявлення називається звідним, а прості
уявлення, які наведені у таблиці характерів,—незвідними. Для зведення звідного
уявлення (П) до суми незвідних (Г,) використовують формулу
П = 5>/Г'> (9.94)
і
де а{ — цілочисельді коефіцієнти, які знаходять за формулою
«і-^Х&ХпХгі/- (9.95)
* і
Тут §—число симетрії; & — число операцій симетрії у класі і;
%Г/—характер незвідного уявлення; %П(. — характер звідного уявлення.
У наведеному вище прикладі число симетрії дорівнює 4, у кожному класі
міститься лише одна операція симетрії й обчислення коефіцієнтів а( дає
(9.96)
Отже,
а, =І[М-3 + 1-Н)-1 + М-1+1-М]=1
а2=і[і1-3 + 1-(-1)1 + 1-(-1)1 + 1-(-1)і] = 0
а3=і[і1-3 + 1-(-1)-(-1) + ЬЬ(-1)+1М] = 1
а4=І[і.1-3+1.(-1).(-1) + И-1 + 1-Ь(-1)] = 1
П=Лі+Я,+£2. (9.97)
235
У справедливості цього результату не важко переконатися, якщо додати
характери трьох незвідних уявлень.
Із таблиці характерів 9.7 бачимо, щоАьВ{ тз,В2 відповідають уявленням,
за якими перетворюються координати х,утаг при всіх операціях симетрії
групи С2і;. У таблиці характерів для точкової групи симетрії С3и операції
симетрії згруповані за класами. Шість операцій поділені на три класи: спокій,
дві операції обертання навколо осі третього порядку, три операції
відображення у трьох вертикальних площинах. Серед незвідних уявлень з'явилося
двократно вироджене уявлення Е (не плутати з операцією спокою Е). Справді,
при обертанні на 120° координата х не просто змінює свій знак, а
«змішується» з координатою^. Незалежний розгляд поведінки орбіталей/^ та/?^ або
сІХ2та, сІу2 неможливий.
Для високосиметричної точкової групи Та наводимо таблицю характерів
(табл. 9.8). Молекули та іони, що мають симетрію Тф такі: СН4, СС14, Н§ С14~,
2п С14~ тощо. Зазначимо, що тут з'являється новий тип незвідних уявлень —
Г(тричі вироджене). У цій таблиці, як і в попередніх, сума квадратів
розмірностей уявлень дорівнює числу симетрії, тобто £ = 1 +1 +2+3 +3=24.
Таблиця 9.8. Таблиця характерів точкової групи Тй
! т<
\ М
Аг
Е
Ті
1 Гг
Е
1
1
2
3
3
8С3
1
1
-1
0
0
гсг
і
і
2
-1
-1
65*4
1
-1
0
1
-1
ге,
і
-і
0
1
-1
Координати або АО
5
—
22,Х2-у2
(х,у9г);(х2,у29ху)
Хімічний зв'язок у різних молекулах типу АВЙ можна розглянути з
позицій молекулярних орбіталей. Використання вчення про симетрію дає змогу
уніфікувати і до певного ступеня спростити будову МО. При цьому
використовується така загальна схема розгляду молекул типу АВП:
1) вибір придатного за енергією базису атомних орбіталей атома А;
2) класифікація за допомогою таблиць характерів відібраних базисних АО
за симетрією;
3) складання придатних за енергією та симетрією лінійних комбінацій АО
атомів В;
4) виведення МО як лінійних комбінацій атомних орбіталей (ЛКАО);
5) приблизне розташування МО за величиною збільшення енергії;
6) «заселення» МО валентними електронами.
Як перший приклад розглянемо молекулу ВеН2.
1. Електронна структура атома Берилію І822з22р0. 1^-АО розташована
дуже глибоко (внутрішня АО) і суттєвої участі в утворенні хімічного зв'язку
не бере. 2^-АО належить до «придатних» за енергією орбіталей, тобто її
енергія близька до енергії Ь-АО Гідрогену. Вакантні 2р-АО атома Берилію
також можуть брати участь в утворенні хімічного зв'язку з атомами Гідро-
236
гену. Атоми Гідрогену мають єдину Ь-орбіталь, яка бере участь в утворенні
хімічного зв'язку. Отже, як базисні АО для побудови МО ВеН2 вибрані 2з- та
2/7-АО атома Берилію та 1^-АО атома Гідрогену.
2. Симетрія лінійної молекули ВеН2—Д^ (зазначимо, що до тієї самої
групи симетрії належать усі двохатомні молекули типу А2; відповідні
позначення для МО вже були використані у підрозділі 9.1). Значення характерів
групи Д^ наведені в табл. 9.9.
Таблиця 9.9. Таблиця характерів групи А*,/,
й-к
\К
^
\К
Е'<
г*
пи
А«
К
2К(2п, 2),
Е
+ 1
+ 1
+ 1
+ 1
+2
+2
+2
+2
2Д((р, 2)
+1...
+1...
+1...
+ 1...
+2...
+2...
+2...
+2...
©ОСТу
+ 1
-1
+ 1
-1
0
0
0
0
і
+ 1
+ 1
-1
-1
+2
-2
+2
-2
25(ф, і)
+ 1...
+ 1...
-1...
-1...
+2 созф...
-2 С08ф...
+2 соз2ф...
-2соз2ф...
ооС2
+ 1
-1
-1
+1
0
0
0
0
Координати або АО
Х2+У + 22(5)
яг
7.
уг, хг; Кх, Яу
х,у
Х2-У2,Ху\
Віднесення АО Берилію за симетрією наведено на рис. 9.24, с. 238. Ор-
біталі 2$ та 2р2 належать до незвідних уявлень X* та X*, відповідно (вісь 2
проходить через атом Берилію та два атоми Гідрогену), орбіталі 2рх та 2ру
належать до двічі виродженого незвідного уявлення Пм (див. табл. 9.9).
3. Лінійні комбінації з Ія-АО атомів Гідрогену складаються так, щоб знаки
Ь-АО атомів Гідрогену співпадали зі знаками тих АО Берилію, з якими вони
перекриваються. Тоді для ст^-МО слід використовувати (поряд з 2^-АО
Берилію) лінійну комбінацію (і/+ Із'") — скорочено (Н1 + й2), а для ам-МО (поряд
з 2р2-АО Берилію)—лінійну комбінацію (Н{ - к2). Орбіталі типу пи (2рх та 2р-
АО Берилію) з Ія-орбіталями атомів Гідрогену дають нульове перекривання
(область додатного перекривання компенсується областю від'ємного
перекривання), отже, пи орбіталі є незв'язуючими.
4. Лінійні комбінації АО для перелічених МО такі:
(іа§ =2^ + Х1(А1+А2)
\іои=2р2+Х2(Н1-к2)
\2с§ = 28-\г{ку+К1)
І2ои=2р2-Х4(к1-к2).
Наведені рівняння для МО не нормовані. Коефіцієнти X для зв'язуючих
МО більші за одиницю (електронна густина локалізована на атомах
Гідрогену), а для антизв'язуіочих, навпаки, вони менші за одиницю.
237
Рис. 9.24. Орбіталі атома Берилію
та Гідрогену в молекулі ВеН2
ВеН2
Рис. 9.25. Енергії МО ВеН2
5Єв& Шз®
ІОо
\аи
(Засю
2аи
Рис. 9.26. Молекулярні орбіталі ВеН2
238
5. Розташування МО за енергією
приблизно відповідає схемі,
наведеній на рис. 9.25.
6. Чотири валентні електрони (два
від атома Берилію та по одному від
кожного атома Гідрогену) займають
дві нижні за енергією МО.
Електронна структура молекули ВеН2 —
(1а/(1аи)2.
Електрони, які знаходяться на
МО, рухаються у полі ядерного
остова всієї молекули, тобто молекулярні
орбіталі належать усій молекулі,
вони — багатоцентрові, в даному
випадку МО о§- та аи — трицентрові.
З наведеного на рис. 9.26
зображення МО можна побачити, що на
зв'язуючій Іа^-МО немає вузлових
площин, а на 1ам-МО вузлова
площина припадає на центральний атом.
На відміну від цього, для антизв'язу-
ючих МО вузлові площини (де
функція змінює знак) перетинають саме
лінію зв'язку.
На перший погляд, метод МО,
у якому кожна МО належить усій
молекулі, не відповідає концепції про
утворення локалізованих хімічних
зв'язків між сусідніми атомами. Проте
Дж. Е. Леннард-Джонс та Дж. Попл
показали, що уявлення про
локалізовані двоцентрові зв'язки не є
характерними тільки для методу ВЗ, а
можуть бути виявлені і при
використанні методу МО. Розглянемо це
питання на прикладі молекули ВеН2.
Повна хвильова функція
молекули ВеН2 зображується
детермінантом, який містить спін-орбіталі для
чотирьох електронів. Величина
детермінанта не змінюється, якщо
додавати та віднімати його рядки (або
стовпці). Якщо зробити ці
процедури, то можна мати такі орбіталі (на-
водиться тільки орбітальна частина без спінових функцій):
(9.99)
Гх,=ЛГ(1а^+1аи)
[%2=Лґ(1ая-1а„).
Ці молекулярні орбіталі називаються еквівалентними, або
локалізованими. Якщо взяти для \ав та 1 си конкретні вирази із формули (9.98) та
підставити їх у (9.99), то одержимо (вважаючи Х]=Х2 = X):
\хг^[^2Рж) + Щ] (9Л0())
\х2 = м[(2,-2р2) + ХН2]
Функції Хі та %2 — еквівалентні. При відображенні у перпендикулярній
площині вони не зберігаються незмінними (з точністю до знака), як \ая та
\аи, а перетворюються при цьому одна на одну.
Еквівалентні МО добре передають локалізацію електронів на лінії зв'язку.
При співставленні методів МО та ВЗ, як це робилося для двохатомних
молекул (див. підрозділ 9.1), з функціями Гайтлера-Лондона необхідно співстав-
ляти локалізовані еквівалентні, а не делокалізовані багатоцентрові МО.
Якщо співставити %х та %2-°рбіталі з гібридними .ур-орбіталями (9.87), то
не важко пересвідчитися, що локалізовані еквівалентні МО відповідають
лінійним комбінаціям гібридних ^р-орбіталей Берилію та І5-АО атомів
Гідрогену. Гібридні 5/7-орбіталі також еквівалентні (вони відрізняються (див.
рис. 9.20, с. 227) тільки орієнтацією лопатей і перетворюються одна на одну
при відображенні в площині, яка перпендикулярна до лінії зв'язку та
проходить через центральний атом Берилію).
Як інший приклад застосування методу МО розглянемо молекулу води.
1. Електронна будова атома Оксигену 1$ Із 2р . Ь-АО розташована дуже
глибоко і в утворенні МО суттєвої участі не бере. Придатні за енергією 2з-
та 2р-АО; вакантна Зя-АО розташована дуже високо за енергією і не бере
участі в утворенні МО. Придатними за енергією орбіталями Гідрогену
можуть бути тільки Ь-орбіталі. Отже, як базис для будови МО води відібрані 2з-
та 2/7-АО атома Оксигену та І5-АО атома Гідрогену.
2. На рис. 9.27, с. 240, показано, як розміщена молекула води на площині
Х02. У цьому випадку орбіталь;?^ відповідає незвідному уявленню аьр2—
незвідному уявленню Ьи ару — незвідному уявленню Ь2.
3. Лінійні комбінації І5--АО атома Гідрогену складаються так, щоб вони
мали такі самі знаки, що й орбіталі, з якими вони перекриваються. Очевидно,
що для агорбіталі підходить лінійна комбінація (Нх + Н2) (обидві орбіталі
перекриваються з додатною частиною/?х-орбіталі), а для Ь х -орбіталі —
лінійна комбінація (Нх - А2). 62-°рбіталь (ру), як це бачимо на рис. 9.27, не
перекривається з 1 ^-орбіталями атомів Гідрогену і, отже, є незв'язуючою.
2^-АО атома Оксигену належить до незвідного уявлення аи тобто до того
самого, що й/^-орбіталь, і тому вона може комбінуватися з (Н{ + Н2) ЛКАО
Гідрогену. При утворенні МО енергетичний рівень 2.У-АО Оксигену трохи
239
(9.101)
Рис. 9.27. Орбіталі атома Оксигену
та Гідрогену в молекулі Н20
знижується і орбіталь стає слабко
зв'язуючою.
4. Запишемо МО у вигляді ЛКАО
у порядку збільшення енергії:
ІІЩ = І8 + Х1 (/*! + /*2 )
\\Ь1=2р2+Х2(к1-к2)
І2а! =2/?х+Х3(А| +Аг)
)\Ь2=2ру
\2Ь{ = 2р2-Х4(к] -к2)
[За{=2рх-Х5(к{+к2)
Коефіцієнти Хі9 як правило, не
дорівнюють одиниці, рівняння МО не
нормовані.
Зв'язуючі(\аь2а1 і Іб^таантизв'я-
зуючі (З^! і 2ЬХ) МО зображені на рис.
9.28. Усі МО—трицентрові. Для зв'язуючих МО на лінії зв'язку (ОН) МО не
мають вузлів, навпаки, у антизв'язуючих МО зміна знака функції має місце
на лініях зв'язку.
Зазначимо, що як і для молекули ВеН2, для води можна також з даних МО
сконструювати двоцентрові двох-
електронні зв'язки О—Н.
5. Точне розташування МО за
рівнями енергії досить складна справа,
вона вирішується після відповідних
розрахунків. У першому наближенні
можна вважати, що рівні зв'язуючої
та антизв'язуючої орбіталей
визначаються насамперед величиною
інтегралів перекривання. Кут Н—О—Н
дорівнює ~ 105°, тобто перекривання Ь-
АО атомів Гідрогену значно більше
3 І^і (/?2)-орбіталлю, ніж з 2ах (рх)-ор-
біталлю (кут*—О—Н дорівнює -53°,
кут 2—О—Н дорівнює -37°). Саме зав-
дякищьому орбіталь \Ь{ знаходиться
нижче, ніж орбіталь 2ах (рис. 9.29).
Правильність виведення МО
можна перевірити за допомогою такого
правила: кількість МО повинна
дорівнювати кількості базисних АО. Для
Рис. 9.28. Молекулярні орбіталі Н20 конструювання МО молекули води
240
було використано шість АО: чотири
АО атома Оксигену (2з, 2рх, 2ру9 2р2)
та дві АО (Із) атомів Гідрогену. У
результаті одержали шість МО
молекули води: Іаь 1й1,2а1, ІЬ2,2Ь1,За1.
6. Загальна кількість валентних
електронів дорівнює восьми (шість
від атома Оксигену та два від атомів
Гідрогену). Електрони «заселяють»
нижні за енергією молекулярні
орбіталі. Тоді електронна конфігурація
молекули води матиме такий вигляд:
(Іа{) (16,) (2а{) (\Ь2) .Найвищою
зайнятою орбіталлю є незв'язуюча
МО ІЬ2.
Отже, на прикладі молекул Н20
та ВеН2 можна пересвідчитися, що
хімічний зв'язок у трьохатомних
молекулах можна описати або три-
центровими МО, які охоплюють усі Рис* 9,29# ЕнеРпя мо н2°
ядра молекули, або еквівалентними двоцентровими МО, які локалізовані на
зв'язках (аналогічно тому, як описує зв'язок метод ВЗ — двохелектронний,
двоцентровий). Еквівалентні МО (а також метод ВЗ) добре узгоджуються з
ідеєю фрагментації молекули і зручні для опису таких властивостей
молекул, як довжина зв'язків, їх енергія, полярність тощо. Багатоцентрові МО, що
ґрунтуються на одноелектронному підході до всієї молекули, необхідні для
опису властивостей молекул, які пов'язані з перенесенням електронів та
належать до всієї молекули в цілому. Це енергія іонізації, спорідненість до
електрона молекули, електронні переходи з основного стану у збуджений і т. д.
Розглянемо тепер молекули із кратними зв 'язками.
Найпростіші вуглеводневі молекули з кратними зв'язками — це
молекули ацетилену та етилену. Енергія зв'язку Карбон—Карбон в
ацетилені близька до енергії зв'язку Нітроген—Нітроген у молекулі азоту і
відповідає потрійному зв'язку; в етилені зв'язок Карбон—Карбон подвійний,
міцність його близька до міцності зв'язку в молекулі кисню.
Молекула ацетилену — лінійна, етилену — плоска. Аналогічно
двохатомним молекулам потрійний зв'язок в ацетилені слід розглядати як
комбінацію одного а- та двох я-зв'язків, а подвійний в етилені — як одного а- та
одного тс-зв'язку. Будова та симетрія молекул С2Н2 та С2Н4 дають змогу
утворювати такі зв'язки (рис. 9.30, с. 242).
а- та 7і-орбіталі нееквівалентні, але за допомогою концепції еквівалентних
орбіталей можна побудувати сит2- та ая-еквівалентні орбіталі. Проекції цих
орбіталей на площину, перпендикулярну до лінії зв'язку, орієнтовані під
кутом 180° для ая-еквівалентних орбіталей та під кутом 120° для сш2-орбіта-
241
а б
Рис. 9.30. Схема утворення а- та я-зв'язків: а — в етилені; б — в ацетилені
лей, тобто вимальовується картина, аналогічна зр- та ^-гібридним орбіта-
лям. ая2- та ая-еквівалентні орбіталі в молекулах ацетилену та етилену зігнуті
(рис. 9.31). Як уже зазначалося, етан за будовою — це два тетраедри, які
мають загальну вершину. Уявлення про еквівалентні орбіталі в ацетилені та
етилені добре відповідає «класичній» картині, згідно з якою для опису
геометрії ацетилену та етилену, як і для етану, використовувалися ті самі два
тетраедри, які з'єднуються основами (ацетилен) або ребрами (етилен).
Властивості етилену та ацетилену можна вивчати за допомогою обох моделей.
а
Рис. 9.31. Еквівалентні орбіталі: а — в етилені; б — в ацетилені
а-зв'язок дає змогу обертатися навколо осі, яка з'єднує атоми, тому
кутова орієнтація груп СН3 в етані довільна, я-електрони закріплюють
зв'язок, роблять його «жорстким». В етилені, на відміну від етану, можливі
циста, транс-ізомери. Два еквівалентні зігнуті зв'язки також не дають
можливості групам СН2 в етилені вільно обертатися. Подвійний зв'язок С=С в
етилені слабкіший, ніж сума двох зв'язків С—С в етані. Це можна пояснити
слабкістю я-зв'язку порівняно з а-зв'язком (яя-перекривання для даних
міжатомних відстаней менше, ніж аа-перекривання). Модель еквівалентних
зв'язків пояснює це напругою і зменшенням перекривання зв'язків, які
спрямовані під кутом один до одного.
Кратні зв'язки в ацетилені міцніші, ніж в етилені, тому що додатковий
зв'язок скорочує міжатомну відстань, яя-перекривання збільшується і, відповідно,
міцніє я-зв'язок. аа-перекривання, навпаки, зменшується (див. підрозділ 9.1,
242
рис. 9.12) і а-зв'язок дещо слабне. У моделі еквівалентних зв'язків зменшення
перекривання внаслідок орієнтації орбіталей під кутом компенсується
збільшенням перекривання внаслідок скорочення міжатомної відстані.
Проте деякі факти дають змогу зробити вибір між моделями. По-перше,
це геометрична конфігурація молекул. Розгляд ацетилену в цьому випадку
нічого не дає, тому що обидві моделі передбачають лінійну структуру. Для
молекули етилену обидві моделі завбачають плоску структуру, але з різними
валентними кутами. Модель еквівалентних зв'язків повинна утворювати
^НСН = 109°28' (тетраедричний), ^НСС = 125° 16'. В іншій моделі а-зв'язки
утворюються зр2 ($-,рх- тару — АО)-гібридними орбіталями, отже, ^НСН =
= ,/НСС = 120° (я-зв'язок здійснюється/?2-орбіталями, які перпендикулярні
до площини молекули). Експериментальні значення кутів НСН = 117° та
НСС = 121,5° знаходяться в проміжку між значеннями, які передбачають
обидві моделі, але значно ближче до 120°, тобто до моделі окремих а- та тг-
зв'язків у молекулі етилену.
Іншим фактом, який свідчить про недоцільність моделі еквівалентних
зв'язків, є нееквівалентність зв'язків С—Н у молекулах етану, етилену та
ацетилену. У моделі локалізованих еквівалентних орбіталей зв'язки С—Н в усіх
сполуках — це а-зв'язки з я-АО атомів Гідрогену та ^-гібридних орбіталей
атомів Карбону. Проте міцність зв'язків С—Н така (кДж/моль): в етані—405,
в етилені — 435, в ацетилені — 481, тобто збільшується від етану до
ацетилену. Це збільшення не можна пояснити в моделі еквівалентних зв'язків, але
досить легко пояснюється в моделі а-я-зв'язків, що пов'язано зі зміною типу
гібридизації атома Карбону. Міцність зв'язків С—Н збільшується при
переході від 5/73- до зр2- та до ^-гібридизації. Це пов'язано з тим, що ^р-гібридна
орбіталь має максимальне значення інтеграла перекривання з І5-АО атома
Гідрогену. Отже, при дослідженні кратних зв'язків слід надати перевагу
моделі з а- та я-орбіталями над моделлю еквівалентних зв'язків.
Розглянемо тепер молекулу С02. Ця молекула має лінійну геометричну
структуру. Центральний атом Карбону утворює по одному а-зв'язку з
атомами Оксигену за участю .ур-гібридних орбіталей (рис. 9.32), подвійний
зв'язок утворюється за
допомогою я-орбіталей, які
розташовані на взаємно
перпендикулярних площинах. Якщо
замінити атоми Оксигену на
групи СН2, то цю картину
можна «проявити» — в
молекулі алену СН2=С=СН2
атоми Гідрогену
розташовані попарно у взаємно пер-
, /ч 1* ~ лл^ Рис. 9.32. Схема утворення о- та я-зв'язків
(рис. 9.33, с. 244). у молекулі С02
243
І
/
/н \|
н
У0 \
\ґ^]\/
н
/
Розглянемо електронну будову
молекули С02 детальніше. Як базис
для побудови МО відбираємо 2з- та
2/?-АО Карбону (я, рх, ру, р2) та 2/7-
АО Оксигену (рх, ру, р2 від кожного
атома). Десять вихідних АО-повинні
перетворитися на десять МО.
Розмістимо вісь і так, щоб вона
проходила через усі атоми молекули С02
Рис. 9.33. Схема будови молекули алену (рис. 9.34). Віднесення АО Карбону
за симетрією таке:
2$ 2р2 2рх 2ру
V у /
Згідно з прийнятою системою координат (див. рис. 9.34) лінійні
комбінації атомів Оксигену (О' та О") такі:
о8=(р2о,-рго.)
а*=[-(р,0,+А0.)]
ХРхп+Рхг)
х<у
ХО"
двократно вироджена орбіталь;
я,
двократно вироджена орбіталь.
'и[(Ру0-+Ру0.)
''{(Руа-Руо-)
Лінійні комбінації АО для МО молекули С02 такі:
Іаіщ=зе+\і(рЖ(Г-рЖ(у)
1стиі|/2 =р2с -Х2(р20, +р20.)
Уз=Рхс+*-з(Рх0-+Рх0')
У4=Рус+^і(Ру<у+Ру0-)
Ч5=Рх0.-РХ0.
Уб=РУ0.-Ру0.
\Уі=Рхс-К(Рхо.+Рхо-)
\щ = Рус-^рУолруо.)
2а8щ=5с-Х5(рго,-рго.)
2°нУ 10 ^Рхс+КіРге+Рцг)
ІЯ,
ІЯ
2я,
зв'язуючі МО
незв'язуюча МО
антизв'язуючі МО
244
Рис. 9.34. /?-А0 Карбону та Оксигену у молекулі С02 (у площині Х02)
Наведені вище хвильові функції АО не нормовані. Для зв'язуючих МО
(1а^, \аю Іпи) коефіцієнти X більші за одиницю (зміщення електронної
густини до атомів Оксигену від атома
Карбону), для антизв'язуючих (2ям,
2с8, 2ои) ці коефіцієнти менші за
одиницю.
Розташування МО молекули С02
за енергією наведено на рис. 9.35
(схематично). Дванадцять валентних
електронів (чотири від атома
Карбону та по чотири від атомів
Оксигену) заповнюють молекулярні ор-
біталі так: (\ав)2 (\аи)2 (\пи)4 (Іп8)4.
Переваги методу МО, його більш
гнучкий підхід, порівняно з
методом ВЗ, особливо чітко
виявляються при розгляді сполук із делокалізо-
ваними зв'язками, які розглянуто
в наступному підрозділі.
Рис. 9.35. Молекулярні орбіталі С02
9.3. Молекули з делокалізованими зв'язками
У ненасичених вуглеводнях, які мають кілька подвійних зв'язків, ці
зв'язки можуть бути відділені один від одного більш ніж одним простим зв'язком,
наприклад у 1,4-пентадієні (СН2= СН—СН2—СН= СН2). У таких алкенах
тс-зв'язки локалізовані, їх властивості майже не відрізняються від
властивостей окремого подвійного зв'язку, наприклад в етилені. Алкени, в яких прості
та подвійні зв'язки чергуються, називаються спряженими. Найпростіший їх
представник—це 1,3-бутадієн (СН2=СН—СН=СН2). Зазначимо, що
існують також сполуки, в яких подвійні зв'язки розташовані один за одним, це
кумулени, найпростіший із них — ален (див. підрозділ 9.2).
Якщо спряжений вуглецевий ланцюг замкнути і утворити один чи
декілька циклів, то одержимо сполуки, які називаються ароматичними.
Найпростіший серед них бензен (С6Н6). Ароматичні сполуки значно відрізня-
245
ються від сполук із локалізованими я-зв'язками, наприклад, для етилену та
його аналогів характерні реакції приєднання внаслідок відносної нестійкості
я-зв'язку, навпаки, для ароматичних сполук реакції приєднання не типові.
Розглянемо на прикладах бутадієну та бензену геометричні та
енергетичні властивості спряжених та ароматичних сполук порівняно з «етилено-
подібними».
Спробуємо використати концепцію адитивності енергій зв'язків (див.
підрозділ 9.2) для бутадієну та бензену. Із термохімічних даних для сполук із
локалізованими я-зв'язками можна визначити АН^, АНс_с та ДНс=с —
теплоти згоряння, які припадають на відповідний зв'язок. У моделі окремих
зв'язків теплота згоряння бутадієну така:
АН^ = бДНЕн + 2АН£С + АН£С. (9.102)
Експериментальна теплота згоряння бутадієну на 14,7 кДж/моль менша,
ніж обчислена за формулою (9.102). Відповідні розрахунки для бензену
дають ще більшу різницю між експериментальною та обчисленою за
моделлю окремих зв'язків теплотою згоряння, а саме на 155 кДж/моль.
Аналогічні результати одержуються і при використанні термохімічних даних за
теплотами гідрування.
Добуті результати свідчать про те, що енергії реальних бутадієну і
особливо бензену нижчі, ніж для сполук із локалізованими я-зв'язками.
Міжатомні відстані у молекулі бутадієну між атомами Карбону такі, нм:
г{ 2 — 0,135; г34 — 0,135; г23—0,148. У молекулі бензену відстань між усіма
атомами Карбону однакова і дорівнює 0,140 нм. Отже, модель із
чергуванням простих та подвійних зв'язків для бензену непридатна.
За міжатомними відстанями можна оцінити порядки зв'язків між
атомами Карбону в бутадієні та бензені. Для етану (гс_с= 0,154 нм) порядок
зв'язку приймемо за одиницю, для етилену—два (гс=с=0,134 нм) та для
ацетилену — три (гс=с = 0,120 нм). По-
V
»Етан
будуємо за цими даними графік
залежності відстані зв'язків С—С від їх
0,15&^1сШ порядку (рис. 9.36), а потім за відо-
[\ Бутадієн - (2,3-зв'язок) мими відстанями у бензені та бу-
х*х Графіт тадієні оцінимо відповідні порядки
0,14
0,13
0,12
^х^Бензен цих зв'язків. Така оцінка дає у бута-
Бутадієн**^ Етилен дієні: 1 >95—№* зв'язків «І"2» та «3~
(І^-зв'язокрЧ^ 4» і 1,35 —для зв'язку «2-3», у
бензені 1,65 —для всіх зв'язків. Отже,
у бутадієні подвійний зв'язок дещо
Ацетилен ^о послаблений, а «простий» зв'язок
{ 2 У С—С значно зміцнений порівняно
Порядок зв'язку з неспряженими сполуками. У бен-
Рис. 9.36. Міжатомна відстань С—С зені зв'язок Карбон—Карбон міц-
залежно від порядку зв'язку ніший за «полуторний».
246
Спряжені та ароматичні сполуки мають плоску геометричну структуру,
а-зв'язки у них утворюються, як і в етилені, атомами Карбону в
^-гібридному стані. Всі кути зв'язків С—С—С у молекулах бензену та бутадієну
дорівнюють 120°. Відповідно, молекула бензену — це плоский рівносто-
ронній шестикутник, а бутадієн може мати транс- та цис- форми.
я-зв'язки у цих сполуках утворюють/^-орбіталі атомів Карбону, які
розташовані перпендикулярно до площини ХОУ молекули. р2-А0 всіх атомів
Карбону можуть ефективно перекриватися із сусідами, отже, виникає
спряження.
Якщо подвійні зв'язки розташовані далеко один від одного, то
перекривання можливе тільки між сусідніми зв'язками, а між далеко розташованими
р2-А0 воно практично відсутнє. Крім того, поява проміжних груп —СН2—,
у яких Карбон знаходиться у стані ^-гібридизації, призводить до не-
лінійності молекули, що також порушує спряження.
Особливо чітко спряження відбувається в ароматичних сполуках. Для
бензену при спробі записати його формулу за допомогою валентних рисок
ще класична органічна хімія наштовхнулася на суттєві труднощі. Наприклад,
формулі (А) можна протиставити формулу (В) з іншим розташуванням
«простих» та «подвійних» зв'язків:
^Ч
б
*^ч
6
А В
Тому ще Ф. А. Кекуле, який запропонував формулу бензену, вважав, що
подвійний зв'язок у бензені осцилює шляхом динамічного співіснування
обох форм: А та В. Усереднення форм А та В пояснює еквівалентність усіх
зв'язків С—С у бензені, але вище вже було доведено, що С—С зв'язок у
молекулі бензену міцніший від «полуторного». Це видно після розгляду як
геометричних, так і енергетичних параметрів бензену (шість «полуторних»
зв'язків за енергією були б еквівалентні сумі трьох подвійних та трьох
простих зв'язків Карбон-Карбон). Крім того, Д. Дьюар довів, що для бензену,
крім двох структур Кекуле, можна навести ще декілька структурних формул:
С
247
Труднощі класичної органічної хімії стали труднощами методу ВЗ, який
замінив валентну риску на два спарені електрони. Для опису делокалізова-
них систем краще підходить метод МО, тому що молекулярні орбіталі бага-
тоцентрові й описують поведінку електрона у полі ядерного остова всієї
молекули.
Вивчення полієнів почнемо з етилену в я-електронному наближенні
методу МО. В цьому наближенні взаємодія двох однакових р2-А0 атомів
Карбону повністю аналогічна взаємодії двох І^-АО атомів Гідрогену (див.
підрозділ 9.1). Позначимо недіагональні елементи (інтеграли) НАВ = НВА
через Р та збережемо термін резонансний інтеграл (нагадаємо, що р —
від'ємна величина). Діагональні елементи НАА=НВВ позначимо через а. До
а входять власна енергія атомів (тут 2/?-станів атомів Карбону) та кулонівсь-
кий інтеграл С, тому часто всю величину а називають кулонівським
інтегралом, я-перекривання зазвичай менше, ніж а, і у першому наближенні
можна знехтувати інтегралом перекривання.
Для енергії основного стану (зв'язуюча орбіталь) маємо
£і=а + Р, (9.103)
а для збудженого (антизв'язуюча орбіталь)
£,2 = а-р. (9.104)
Відповідні МО такі:
У\=-П^(Ра+Рв)-
У2=-^(Ра-Рв)-
-зв язуюча;
- антизв язуюча.
(9.105)
(9.106)
Розрахунки у даному наближенні, яке називається методом Гюккеля, для
складніших, ніж етилен, молекул, мають ще одне припущення—резонансні
інтеграли враховуються лише між сусідніми атомами, там, де є перекривання.
Для молекули бутадієну метод Гюккеля дає такий детермінант:
Р 0 0
а-Е Р 0
р а-Е Р
0 Р а-Е\
\а-Е
Р
0
0
Введемо позначення
= 0.
(9.107)
х = (а-Е)/$.
Тоді визначник (9.107) має такий вигляд:
х 1 0 0
1 х 1 0|
0 1x1
0 0 1 х|
= 0.
(9.108)
(9.109)
248
х\,2 -
(9.110)
(9.111)
Розкривши детермінант та розв'язавши відповідні алгебраїчні рівняння,
маємо
1±У5. г _1±л/5
Відповідно, маємо чотири рівні енергії
Г^=а + 1,618Р
І£2=а + 0,618Р
|£3=ос-0,618|3
|£4=а-1,618Р
Рівні Ех та Е2 — зв'язуючі (Еі < ос), аЕ3 таЕ4 — антизв'язуючі {Е( > а).
Використовуючи величини Еі9 можна знайти вирази для відповідних МО
|>, =0,3111 рА + 0,60\5рв +0,6015/?с + 0,3717/7^
І \|/2 =0,6015/^+0,3717/^-0,3717/^-0,6015/?/,
\|/3 =0,6015/7^-0,3717/75-0,3717^+0,6015/?^ (9.112)
[\|/4= 0,3111 рА - 0,6015 рв +0,6015 рс -0,3111 рр
Молекулярні орбіталі \у{—\|/4 нормовані та ортогональні одна до одної
(рис. 9.37). \|/гМО вузлів не має, це сильно зв'язуюча орбіталь; і|/2-МО має
один вузол, вона зміцнює два крайні зв'язки, але дестабілізує центральний,
тому ця МО зв'язує слабкіше, ніж \|/горбі-
таль. і|/3-МО має два вузли та діє
протилежно, ніж і|/2-орбіталь; вона слабко антизв'я-
зуюча. \|/4-МО має три вузли, вона
дестабілізує усі зв'язки С—С і, отже, це сильно
антизв'язуюча орбіталь.
Подивимось, яка у бутадієні делокаліза-
ція електронів. Визначимо енергію делока-
лізації як різницю між повною енергією
системи (у я-наближенні), яка розрахована
за методом МО, та сумарною енергією всіх
локалізованих я-зв'язків. У бутадієні чотири
я-електрони розташовані на двох
зв'язуючих (\|/! та і|/2) МО і повна енергія системи
дорівнює
£ = 2(а + 1,618|3) + 2(а + 0,618Р) =
= 4а + 4,472р. (9.113)
У наближенні локалізованих зв'язків
енергія бутадієну дорівнює подвійній
енергії етилену, тобто, згідно з формулою
(9.103), 4а + 4р. Отже, енергія делокалізації
Рис. 9.37. я-молекулярні
орбіталі бутадієну
249
становить 0,472(3, інакше кажучи, делокалізація я-електронів у молекулі
бутадієну призводить до зниження енергії (р-від'ємна величина).
Неможливість використати наближення локалізованих зв'язків для опису
бутадієну наочно виявляється при спробі сконструювати еквівалентні орбі-
талі. У молекулі бутадієну \\ї{ МО симетрична, а \|/2 МО — антисиметрична
(по відношенню до площини, яка проходить через середину зв'язку між
другим та третім атомами у і/ис-бутадієні або до центру інверсії в транс-бута-
дієні). Спробуємо скомбінувати з і^ та \|/2 дві еквівалентні локалізовані
орбіталі:
1
(9.114)
(9.115)
Підстановка виразів для \|/, та \|/2 із формули (9.112) у (9.114) дає:
[Хл=0>6*ЩРл+Рв) + 0Л625(рс-ро)
\Хв=0>$*ЩРс + Ро)-0ЛбЩРл-Рв)
При повній локалізації одного з/?-зв'язків між атомами А та В та
другого — між атомами С та В коефіцієнти при (рА + рв) у %А та при (рс + рв)
у Хв дорівнювали б одиниці, а при (рс-Ро) У Ха та ПРИ ІРа ~Рв) У Хв
Дорівнювали б нулю. Це не відповідає дійсності, тому що я-зв'язки не повністю
локалізовані на відповідних С—С зв'язках, хоча деяка локалізація існує.
Для полієну з и-Карбоновими атомами відповідний детермінант має
такий вигляд:
1 0 0 ... 0|
1 х
0 1
1
х
0
1
= 0.
(9.116)
|0 0 0 0 ... х\
Його розв'язок можна записати у загальному вигляді
пі
X: =-2соз
або, відповідно,
л + 1
£;=а + 2|Зсо8|
т
и + 1
Для коефіцієнтів МО справедлива така формула:
л + 1
•81П
ггп
/2 + 1
(9.117)
(9.118)
(9.119)
де і — номер МО; г — номер АО.
250
Формули (9.103)—(9.106) та (9.111)—(9.112) для етилену та бутадієну є
окремими випадками формул (9.118) та (9.119), відповідно, при п = 2 та п = 4.
Для найпростішої ароматичної сполуки — бензену — детермінант має
вигаяд
X
1
0
0
0
1
1
X
1
0
0
0
0
1
X
1
0
0
0
0
1
X
1
0
0
0
0
1
X
1
1
0
0
0
1
X
■0.
(9.120)
Розкриваючи детермінант та розв'язуючи відповідні рівняння,
запишемо
хі,6 = ±2;л:2,з;4,5 = ±1, (9.121)
звідки
£1 = а + 2Р; Е2,з = а + $; Е4,5 = ос-р; £,6 = ос-2р. (9.122)
Рівні Ех та Е6 не вироджені, а £2,зта ^4,5 — двократно вироджені.
Для відповідних ортонормованих МО дістанемо
¥2 = ^(2Л +/72 -А -*2/?4 -Л + л)
Уз =^Ср2+л-л-а>)
4/5 = 2^3 (2Рі ~Рі ~Рз + 2РА ~Р$ ~Рб^
Уб=4=:(Р\-Р2+Рз-Р4+Р5-Рб)
В основному стані шість я-електронів займають першу, другу та третю
МО. Сумарна енергія бензену 2Е{ + 4£2,з= 6а + 8Р, що на 2Р відрізняється
від енергії 6а + 6Р, яка розрахована у наближенні локалізованих 7Т-зв'язків.
Енергія делокалізації (2Р) у бензені більша, ніж у бутадієні (0,472Р), що
повністю узгоджується з експериментальними даними.
Для бензену та інших циклічних одноядерних полієнів можна одержати
загальний розв'язок для детермінанта л-го порядку
(9.123)
Я^а + грсо/—
п
(9.124)
251
При /і = 6 формула (9.124) дає наведені вище значення Е( для бензену. При
п = 4 (циклобутадієн) Ех = а + 2|3; £2,з = а; £4 = ос - 2Р- Загальна кількість
я-електронів чотири: два розташовані на зв'язуючій орбіталі з енергією
(а + 2(3), а два—на незв'язуючій двократно виродженій орбіталі з енергією
а. Якщо п = 8 (циклооктатетраєн), то Ех = а + 2Р; £2,з= а + Ь414Р; Е4^5 = а;
^6,7 = а~ 1,414(3; £8 = а-2р. З восьми я-електронів шість займають три
зв'язуючі орбіталі (першу, другу та третю), а останні два розміщуться, як і в цик-
лобутадієні, на незв'язуючих. Лише у бензені всі електрони розташовані
тільки на зв'язуючих орбіталях, що забезпечує високу стабільність молекули
бензену. Е. Гюккель, аналізуючи формулу (9.124), дійшов висновку, що
стабільними повинні бути циклічні полієни з п = Ат + 2, де т — ціле число.
Відповідно до правила Гюккеля (правила ароматичності) циклобутадієн
нестійка молекула; справді, ця сполука у вільному стані не синтезована.
Циклооктатетраєн існує, але має неплоску структуру і не має ароматичного
характеру — це типова сполука з локалізованими я-зв'язками.
Крім бензену, по шість я-електронів мають також аніон С5Н^ — (цикло-
пентадієніл-іон) та катіон СуН? (цикло-гептатрієніл-(тропілій)-іон). Ці іони
стійкі, їх комбінація дає молекулу азулену:
©І ©
Ця молекула має досить великий дипольний момент.
Правило Гюккеля одержане в я-електронному наближенні. Стійкість
бензену пов'язана також з його геометричною конфігурацією: а-остов най-
стабільніший, коли кути зв'язків С—С—С дорівнюють 120°
(^-гібридизація), в інших випадках зв'язки згинаються, виникає напруга і енергія
молекули зростає. Для циклобутадієну все назване вище несприятливе.
Важливими характеристиками молекул є порядки зв 'язку та індекси
вільної валентності.
Порядок я-зв'язку (рГ8) між атомами г та ^ розраховують за формулою
п
Рг5=^8іСігсі5, (9.125)
/=і
де і — номер МО; сіг (сіз) — коефіцієнти розкладання МО за базовими АО;
£(= 0,1 або 2 — кількість електронів на /-й МО.
Для етилену А,2 = 2"гг *"7г = *•
Для бутадієну, згідно з формулою (9.125), порядкирГ5 дорівнюють
(р12 = 2-0,3717 -0,6015 +2 -0,6015 -0,3717 =0,894 = л>4
[Рг* = 2-0,6015 -0,6015 +2 -0,3717 -(-0,3717) =0,447
252
Повний порядок (Р) зв'язку (а + п) знаходять додаванням одиниці до
я-порядку (порядок а-зв'язку вважається рівним одиниці).
Для бутадієну зв'язки 1,2 та 3,4 майже подвійні (Рх 2 = Р3,4 = 1>894), а
зв'язок 2,3 набагато міцніший від простого (Р2,з= 1 >447). Ці величини добре
узгоджуються з порядками цих зв'язків (відповідно, 1,95 та 1,35), які були
одержані за допомогою калібрувального графіка для довжин зв'язків (рис. 9.36,
с. 246). Для бензену згідно з формулою (9.123), для усіх я-зв'язків запишемо:
/7і,2=Р2,з=/7з,4=/74,5=/?5,б==/7і,б=0'667. Повний порядок зв'язку Р= 1,667. Це
добре узгоджується з цифрою 1,65, яку можна знайти для бензену з
калібрувального графіка (рис. 9.36).
Сума порядків усіх а- та я-зв'язків
(£рГ5) даного атома визначає його
валентну насиченість. Різниця між
максимально можливою насиченістю атома
та 2<рГ5 називається індексом вільної
валентності (Гг) і характеризує
валентну ненасиченість атома Карбону.
Максимально можлива валентна
насиченість реалізується для центрального
атома Карбону в молекулі триметилен-
метану — С(СН2)3. У цій сполуці
взаємодіють чотири /?2-орбіталі Карбону
(рис. 9.38), які утворюють одну
зв'язуючу МО з Ех =а + Рл/з, одну антизв'я-
зуючу з Е4 = а-Рл/з та дві незв'язу-
ючі з £2,з = а- Розрахунки дають для
порядків я-зв'язків центрального атома
з крайніми/?! 2=/?, з =/?і}4= 1/>/3. Крім
я-зв'язків, центральний атом з'єднаний з крайніми трьома а-зв'язками, отже,
для повного порядку кожного зв'язку одержимо
Рис. 9.38. Взаємодія р2-А0
в триметиленметані
1 +
7з
а для сумарного зв язку центрального атома з трьома крайніми
З 1 +
_1_
= 3 + л/з=4,732.
Отже, для індексу вільної валентності маємо
^=4,732-2>„.
(9.126)
Для будь-якого з атомів Карбону в бензені (порядок а-зв'язку С—Н
також дорівнює одиниці)
/7=4,732-2 • 1,667-1 • 1 =0,398.
253
Для кінцевих атомів бутадієну
І7=4,732-3-1-0,894 = 0,838,
а для середніх
/7=4,732-3-1-0,894-0,447 = 0,391.
Найбільш валентноненасичені у бутадієні кінцеві атоми Карбону, тому
бромування бутадієну відбувається з утворенням 1,4-дибромбутену-2, а не
1,2-дибромбутену-З, як це можна було б чекати, якби бутадієн був
сполукою з локалізованими я-зв'язками.
Порядки зв'язків та індекси вільної валентності прийнято зображувати на
молекулярних діаграмах. Для бутадієну така діаграма має вигляд
0,835 0,391
Т Т 0,447 0,894
сн2— сн — сн — сн2
а для бензену
Порядки я-зв'язків зображують цифрою над зв'язком, а індекс вільної
валентності — стрілкою над відповідним атомом із значенням Рг За
формулою, аналогічною (9.74), можна розрахувати я-електронну густину (єг) та
заряд (#>.) на атомах (г)
п
£г = Х&4 * Яг = #-єг, (9.127)
і=і
де N— кількість я-електронів на атомі г (для Карбону вона дорівнює
одиниці). Для бутадієну
?і=1-Єі=1-(2-0,37172 + 2-0,60152) = 0.
Аналогічний результат маємо для інших атомів Карбону в молекулі
бутадієну, а також для всіх атомів Карбону в бензені та етилені. Як і слід було
чекати, ніякого перенесення електронної густини у полієнах з одного атома
Карбону на інший не відбувається. Інша картина спостерігається для
сполук з гетероатомами.
При розрахунках полієнів за методом Гюккеля необхідно було знати
лише дві величини: а та р. Точний розрахунок їх — складна справа, тому а
та Р використовують як параметри.
Величину Р можна оцінити з енергій делокалізації (тобто з термохімічних
даних) або з електронних спектрів; Рс^ — від'ємна величина (1—2 еВ за
модулем).
254
Якщо гетероатомів немає, то всі відносні енергії у методі Гюккеля
виражають тільки через (3, а а беруть за початок відліку. Взагалі вважається, що
кулонівський інтеграл ас дорівнює енергії іонізації (із протилежним знаком)
атома Карбону в 2р-валентному стані; осс — значна від'ємна величина
порядку 8 еВ. Кулонівські інтеграли ах гетероатомів для зручності виражають
через ас та деяку поправку 8Х, яка залежить від Рс_с
ах=осс + 5хрс-с. (9.128)
Аналогічно резонансні інтеграли між Карбоном та гетероатомом
виражаються так:
Рс-х=*с-хРс-о (9.129)
При перетворенні відповідних детермінантів після введення х = (ас - Е)І
Рс_с замість діагональних елементів для гетероатомів з'являться величини
(х - 8Х), а недіагональних — кс_х замість одиниці. Величина 5Х залежить від
різниці в електронегативностях (%) гетероатома X та Карбону
8х=*0Сх-Хс). (9.130)
У першому наближенні а = 1. При використанні електронегативностей
за Полінгом: для Оксигену 8^ = 1,0; для Нітрогену 8^ = 0,5. Якщо атоми
Нітрогену та Оксигену віддають у я-систему не один електрон (як,
наприклад, у піридині), а два (як у піролі), то 8^ та 8^ збільшуються приблизно на
одиницю: 8ф =2,0; 8^ = 1,5.
Для галогенів, згідно з їх електронегативностями, 8Х також додатні
величини, але для Бору 8Х — величина від'ємна.
Значення кс_х мало відрізняється від одиниці. Для Нітрогену та
Оксигену, які зв'язані з Карбоном простим зв'язком, &с_н = кс_0- 0,8. Для цих
самих гетероатомів, коли зв'язок подвійний, £с=к= 1,0; кс=0 = 1,4.
Поява гетероатома викликає зміну кулонівських інтегралів атомів
Карбону (сусідніх з гетероатомом) внаслідок так званого індуктивного ефекту
аС' = ссс+8сгРс_с. (9.131)
Величина 8С' залежить від гетероатома і становить (0,1—0,3)8Х. Значення
8Х, кс_х та 8С' — наближені, тому метод Гюккеля для сполук із
гетероатомами дає гірші результати, ніж для вуглеводнів.
Це примушує шукати досконаліші методи та відмовитися від деяких
спрощених припущень, зроблених у методі Гюккеля.
У більш точних розрахунках насамперед треба враховувати залежність
параметрів від міжатомної відстані. Це особливо важливо для полієнів, у
яких чергуються короткі та довгі міжатомні відстані. У підрозділі 9.1
показано, що у молекулі водню при взаємодії Ія-АО резонансний інтеграл
змінюється симбатно з інтегралом перекривання. Така сама залежність
спостерігається і при взаємодії 2/?л-АО атомів Карбону — Рс^ змінюється
симбатно з 5с-с- Це дає змогу обчислити Рс_с за формулою
255
Знаючи (30 та 50 при визначеній відстані г0, можна обчислити коефіцієнт Ь.
Р. С. Маллікен для обчислення Р(6) запропонував таку формулу:
Р = НАВ = К^Л.8ав = КЕаа±Еш5ав_ (9ЛЗЗ)
Коефіцієнт АГ у формулі (9.133) близький до двох. Тут доречно зіставити
значення |3 та 5, що обчислені для молекули водню за формулами (9.61) та
(9.62) (див. підрозділ 9.1). Якщо врахувати, що для водню НАА = Нвв=-(1/2)
(в атомних одиницях), то АГ дорівнює точно двом при нехтуванні членом
ЯАВ/3 у інтегралі 5. З урахуванням цього члена 5 збільшується і К слід
брати дещо меншим двох.
Формула Маллікена дає можливість обчислювати резонансні інтеграли
не тільки для я-систем, а й для а-зв'язків. Відповідний метод має назву РМГ
(розширеного методу Гюккеля), або методу Р. Гофмана.
Якщо на атомі виникає великий позитивний заряд, то електрон важче
відірвати від нього, це повинно вести до збільшення орбітальної енергії і,
отже, до росту (за абсолютною величиною) кулонівського інтеграла а. Цей
факт враховує формула Уейленда-Манна
аг = осо-^сор, (9.134)
де ^^ — заряд атома Карбону (вважається, що атом Карбону віддає в
систему один/^-електрон); со — сталий коефіцієнт.
При ^^ > 0 аг< ос0, оскільки р — від'ємна величина, а при ^^ < О, навпаки,
аг > ос0, що узгоджується зі сказаним вище.
Для обчислення ^^ і аг необхідно знати коефіцієнти сіг МО ЛКАО. Однак
обчислити сіг неможливо, якщо аг невідоме, отже, розрахунки ведуть
методом ітерацій, який ще називається «со-технікою».
У методі Гюккеля інтегралами перекривання нехтують, якщо їх
враховувати, то відповідні недіагональні елементи детермінанта матимуть вигляд
(Р - Е5) замість р. Позначаючи
х= а~Е (9.135)
Р-£5'
для полієнів матимемо детермінант (9.120). Після обчислення детермінанта
енергію визначають за формулою
Еі-т^-а~Хіт^-а-Хі\^' (9Л36)
Отже, замість Р використовують інший емпіричний параметр у, і
врахування величини 5 суттєвих змін не дає.
До недоліків методу Гюккеля слід віднести також різні значення
параметра Р при розрахунках його з різних фізико-хімічних характеристик.
Наприклад, емпіричне значення Р, яке добре описує електронні спектри
256
полієнів, істотно відрізняється від |3, одержаного за допомогою
термохімічних даних.
Нарешті, існують ефекти, які не описуються методом Гюккеля. Це,
наприклад, синглетні та триплетні стани в електронних спектрах, які мають різну
енергію. При переході електрона у молекулі етилену з \ух МО на вакантну \|/2
МО утворюється конфігурація у^І) • 4/2(2) і виникає ситуація, аналогічна
збудженому стану атома Гелію: (1$,)1 • (2$)2 (див. підрозділ 8.2). Врахування
обмінного виродження та міжелектронного відштовхування дає змогу
пояснити синглет-триплетне розщеплення термів (як в атомі Гелію, так і в
молекулі етилену), однак при цьому необхідно вийти за рамки чисто одноелек-
тронного підходу методу Гюккеля (можна вважати, що для ефективного
одноелектронного гамільтоніана методу Гюккеля міжелектронне
відштовхування враховано лише як деяке «усереднення»).
У методі, який запропонували Р. Паризер, Р. Г. Пар та Дж. Попл (ППП-
метод), під час розрахунків міжелектронне відштовхування враховується.
Гамільтоніан записується так:
Я = 2#+І7-' (9137)
і і/ У
де Н? — гамільтоніан остова (індекс с означає соге (англ.) — остов), який
містить кінетичну енергію /-го електрона та енергію його потенціальної
взаємодії з а-остовом (притягання «своїм» та «чужими» ядрами).
Молекулярні орбіталі, як і в методі Гюккеля, записують у вигляді лінійної
комбінації атомних орбіталей (ЛКАО). У методі ППП використовується
наближення нульового диференціального перекривання (НДП), тобто
нехтують не тільки інтегралами перекривання, а й усіма інтегралами, які містять
добутки типу (рАсрвсІУ9 де (рА та срв — атомні орбіталі.
Щоб уяснити ідею методу ППП, розглянемо простий приклад —
врахуємо міжелектронне відштовхування в основному стані молекули етилену.
На найнижчій МО \|/ = сА<рА + св(рв розташуємо два електрони, тоді повна
хвильова функція молекули
гР = \|/(1)-і|/(2). (9.138)
Згідно з формулою (8.92) поправка до енергії внаслідок міжелектронного
відштовхування така:
ГкО)Гк(2)12
є =
(IV =
І2
Г [са<і>а 0)+СдФд (І)]2 -[саУа (2)+сдфд (2)]2 (9В9)
'1,2
З урахуванням НДП дістанемо: є = с^у^ + 2^0^^ + с\увв, (9.140)
257
Г ,Л
Уаа =
Увв =
Уав =
фиО-фи2)
г\л
С ,Л
фИ0-фИ2)
йУ
йУ
(9.141)
г1,2
ґ~2
фПО-фИ2)^_
Г,л2
г1,2
^ = їаг
фнО-фи2)
£/Г
г1,2
де уАА та у55 — одноцентрові, а у^д — двоцентровии кулонівські інтеграли
міжелектронного відштовхування.
Повну енергію в цьому наближенні запишемо так:
Е = °А^АА +2сАсвНАВ + свНвв +є ,д 1^2)
ДЄ
НсАА=\чАНсчАсІУ
Нсвв=\щНсщйУ
НсАВ=\<рАНс<?в4У
(9.143)
Наа ' &ВВ > ^л# — одноелектронні інтеграли остова {Нсм та Нвв —
кулонівські, НАВ —резонансний).
Варіюючи формули (9.142) за сА та св, одержимо такий детермінант для
обчислення енергії:
Гаа-Е
АВ
1 АВ
Рвв - Е\
= 0,
(9.144)
де РАВ = НАВ; РАА = НАА + ІсІЧм + 2с2АуАВ (для Рвв — аналогічно).
Оскільки коефіцієнти ЛКАО входять у вирази для елементів детермінанта,
то розв'язати проблему можна лише ітераційним методом. У спрощеному
рівнянні (9.144) враховане тільки відштовхування між електронами на одній
МО, врахування міжелектронного відштовхування (кулонівського та
обмінного) в усіх МО даної молекули приводить до таких виразів для
діагональних (РАА) та недіагональних (РАг) елементів детермінанта в методі ППП:
^ЬІЧлг+СиІлл
гФА
РАА -#І4+]£
і І.
^Аг = НАг ~2*<СігСіаУгА>
і
де / — номер МО; А та г — індекси атомів.
(9.145)
(9.146)
258
Рівняння ППП для розрахунку коефіцієнтів сіг таке:
Хс<> (рлг ~ еіК ) = 0. (9.147) '
Г
де Еі— один із коренів вікового детермінанта
1^-8^1 = 0, (9.148)
ЬАг — символ Кроннекера (ЬАг= 1 при А = г та 8^г= 0 при А Ф г).
Розрахунки за методом ППП дають набагато кращі результати, ніж
метод Гюккеля при обчисленні фізико-хімічних характеристик молекули
(дипольні моменти, електронні спектри тощо). Це досягається більш коректною
постановкою задачі та введенням додаткових, порівняно з ос та Р методу
Гюккеля, параметрів уАА та уАп які враховують міжелектронне
відштовхування.
Метод ППП є напівемпіричним варіантом методу Рутана. В свою чергу,
метод Рутана — це ЛКАО наближення до методу самоузгодженого поля
Хартрі-Фока.
У плоских системах з електронами на я-орбіталях має місце делокаліза-
ція, якщо електрони не дуже віддалені один від одного. Як приклади я-елек-
тронної делокалізації в неорганічних сполуках розглянемо ізоелектронні
іони N0^ та СОз". Це плоскі системи. Атоми Оксигену в них розташовані
в кутах рівностороннього трикутника, в центрі якого знаходяться атоми
Карбону або Нітрогену. Це свідчить про те, що центральні атоми знаходяться в
стані 5/72-гібридизації. На а-зв'язки припадає шість електронів: три від
центрального атома та по одному від кожного атома Оксигену.
При взаємодії чотирьох р-АО (орієнтованих перпендикулярно до
площини трикутника) утворюється чотири МО. Схема 71-МО іонів N0 ^ та СО2"
подібна до схеми МО триметиленметану з тією різницею, що рівні вихідних
АО не співпадають. Утворюються, як і у триметиленметані, одна зв'язуюча,
одна антизв'язуюча та дві незв'язуючі МО. У системах з іонами N0^ та
СОз" є всього дванадцять валентних електронів, по два від кожного атома
Оксигену; п'ять (від Нітрогену) плюс один в іоні N0^; чотири (від Карбону)
плюс два в іоні СО3". Шість із цих дванадцяти електронів беруть участь в
утворенні а-зв'язків. Останні шість розміщуються на зв'язуючій та двох незв'я-
зуючих МО. Тому антизв'язуючі орбіталі залишаються вільними і системи
повинні бути стабільними. Стійкість іонів N0^ та СОз" добре відома і
пов'язана саме з їх стабілізацією при тс-електронній делокалізації.
Приєднання протонів порушує тс-електронну делокалізацію, тому
молекули НИОз і особливо Н2С03 — нестійкі.
Делокалізація можлива також у системі а-зв'язків. Прикладом можуть
бути борани. Відомо, що ВН3 димеризується з утворенням В2Н6. Спроба
описати зв'язки в В2Н6за допомогою методу ВЗ приводить до концепції
«резонансу» структур типу
259
н
у
н н н н н
а б
або до ще складніших та надуманих побудов.
У ВН3 є шість валентних електронів, у В2Н6— відповідно, дванадцять.
Мінімальна кількість зв'язків у В2Н6 — сім, отже, для опису за методом ВЗ
«не вистачає» двох електронів. Структури типу В2Н6, в яких кількість
валентних електронів менша, ніж 2п (п — число формальних ковалентних зв'язків),
називають сполуками з «дефіцитом електронів». Такі сполуки добре
описуються методом МО, для якого припущення про двоцентрові двохелект-
ронні зв'язки не є необхідним.
Геометрична структура В2Н6 така. Дві групи ВН2 розташовані в одній
площині (рис. 9.39), причому кут НВН дорівнює 120°. Останні два атоми
Гідрогену розташовані над та під цією площиною і утворюють так звані
Рис. 9.39. Структура диборану та орбіталі атомів Бору (а{—с4) і(ки к2),
які беруть участь в утворенні місткових зв'язків
«місткові» зв'язки. Ця схема нагадує схему еквівалентних орбіталей етилену
(див. рис. 9.31, с. 242). Площину, в якій розташовані групи ВН2, вважаємо
площиною ХОУ. Зв'язки між атомами Бору та Гідрогену в цій площині —
це звичайні двоцентрові двохелектронні а-зв'язки, утворені 1^-АО
Гідрогену тая/?2-гібридними орбіталями Бору. Із трьох гібридних орбіталей Бору
в а-зв'язках з атомами Гідрогену в площині ХОУ беруть участь дві. Третя
разом з/?2-орбіталлю використовується для утворення місткових зв'язків
з двома атомами Гідрогену, які залишилися. Комбінація %х та/?2-орбіталей
утворює дві еквівалентні а-орбіталі атомів Бору:
Сті=7б5+;кл+^г:
260
■^
Гг'
°*'Тб1+Тзр'~Т2р- (9Л49)
Оскільки р2 пропорційне сов 6, арх — 8Іп 0 (соз ф вздовж осі х дорівнює
іА/з_/2 о .
одиниці), то І£ 0 = - / гг - л/"2 і 0 = 39 14 . Кут між а{ та с2 дорівнює
180° -20=1О1°32,що добре узгоджується з експериментальним значенням
кута НВН у місткових зв'язках (96° 36').
Орбіталі а! та а2, а також відповідні орбіталі а3 та а4 другого атома Бору
разом з АО місткових атомів Гідрогену кх та Н2 зображені на рис. 9.39.
З а- та й-орбіталей можна побудувати молекулярні орбіталі:
симетричну (відносно площини ХОУ) (рис. 9.40, а)
4/5 = сх (Ні + Н2) + с2(о\ + а2 + а3 + а4), (9.150)
та антисиметричну (рис. 9.40, б)
\|/А =с3(Аі - А2) + С4(<*і - СТ2 + сг3 - а4). (9.151)
де \|/5та \уА — зв'язуючі МО.
Відповідні антизв'язуючі МО можна одержати при заміні плюса перед
с2 та с4 на мінус. Оскільки кількість вихідних орбіталей становить шість, то
й МО повинно бути шість, отже, крім двох зв'язуючих та двох антизв'язую-
чих, повинно бути ще дві незв'язуючі МО
Фл, = ^(аі +а2 -°з ~а4); <Рп2 =М(ох -о2 -а3 +а4).
Загальна кількість валентних електронів — дванадцять: по три від двох
атомів Бору та по одному від шести атомів Гідрогену. З них вісім електронів
зайняті в чотирьох двоцентрових двохелектронних зв'язках В—Н. Останні
чотири розміщуються на двох зв'язуючих \|/5- та х^-МО і стабілізують
сполуку. Незв'язуючі та антизв'язуючі
МО вакантні. Отже, термін «дефіцит
електронів» не існує в підході МО, він
має сенс лише з позицій класичної
теорії валентності або з точки зору
методу ВЗ.
У молекулах із локалізованими
двоцентровими двохелектронними
зв'язками можна побудувати
локалізовані еквівалентні орбіталі.
Для місткових зв'язків у В2Н6
можна також побудувати еквівалентні
зв'язки, але вони трицентрові (див.
рис. 9.40, в). Можна вважати, що
місткові зв'язки в В2Н6 утворюються
двома еквівалентними трицентровими рис# 9.40. Зв'язуючі (а б)
двохелектронними орбіталями. та еквівалентні (в) МО в диборані
261
Слід відзначити, що галогеніди бору, на відміну від ВН3, не димери-
зуються. Це пов'язано зі стабілізуючою дією я-взаємодії такого типу, як в
іонах Ж>з та СОз~. Справді, атом Бору має вакантну акцепторну ;?-орбі-
таль, а галогеніди мають донорнір-орбіталі (з трьома парами електронів).
7і-взаємодія, як і в іонах N0^ та С0\~, стабілізує систему і робить димери-
зацію зайвою. Відзначимо, що А1С13 димеризується в А12С16. Розмір атома
Алюмінію набагато більший, ніж Бору, перекривання р-орбіталей за тг-типом
зменшується, 7і-взаємодія слабне і її стабілізуючої дії вже недостатньо, щоб
перешкодити димеризації.
9.4. Електронна будова та властивості
координаційних сполук
У попередніх розділах розглядалися переважно хімічні зв'язки, які
утворювалися 5- та/?-атомними орбіталями. При взаємодії ^/-орбіталей можуть
утворюватися не лише а- та я-зв'язки, а й 5-зв'язки (див. підрозділ 9.1). У 8-
зв'язках перекривання здійснюється в чотирьох областях, які розташовані на
перпендикулярах до лінії зв'язку (див. рис. 9.10, с. 205). Якщо вважати, що
лінія зв'язку співпадає з віссю2, то с122-АО утворює сх-зв'язки; <^г- тайу2-
АО — утворюють 7і-зв'язки, а ^2 2 - та сі^-АО — утворюють 5-зв'язки.
На відміну від 8- тар-орбіталей, на окремих д?-орбіталях атомів та іонів
електрони можуть розміщуватися по одному чи по декілька, при цьому
утворюються частинки з відкритими (незаповненими) електронними
оболонками з високими значеннями сумарного спінового числа. Для .у- та^-
орбіталей таке заповнення скоріше виняток, а ті частинки, що утворюються,
як правило, нестійкі, вони мають вільнорадикальний характер.
Іони з частково заповненою ^-оболонкою, як правило, парамагнітні
й часто утворюють забарвлені сполуки. Забарвлення сполук перехідних
елементів пов'язане з їх спектральними властивостями, а саме: з присутністю
розташованих відносно недалеко один від одного рівнів енергії, переходи між
якими призводять до поглинання світла у видимій частині спектра. Для
пояснення походження спектрів поглинання комплексних сполук можна
застосувати теорію кристалічного поля.
Основні її положення такі:
а) між складовими частинами комплексу існує тільки електростатична
взаємодія: іонна, наприклад, в [СоС14] ~, або іон-дипольна, наприклад,
в[КІ(МН3)6]2+чи[Со(Н20)6]2+;
б) ліганди розглядаються як точкові заряди, що створюють
електростатичне поле;
в) центральний іон розглядається детально з урахуванням його
електронної будови.
Як приклад розглянемо Ті , що має один електрон на ^/-орбіталі у полі
октаедричної симетрії, що створене шістьма аніонами Р". Нумерація
лігандів дана на рис. 9.41 (7 та 3 — на осі х; 2 та 4 — на осі у\ 5 та 6 — на осі г).
262
4 5
з
з+
В окремому іоні Ті всі п 'ять я?-орбі-
талей вироджені й утворюють терм Б
(див. підрозділ 8.4). В октаедричному
комплексі ^/-орбіталі мають різну
енергію, сі22- та сіх2-у2 - орбіталі, які
розташовані на координатних осях, зазнають
сильнішого електростатичного
відштовхування від лігандів, ніж сІху-, й^- та йХ2-
орбіталі, що зорієнтовані між
координатними осями. Перша група(сі22, сІхг-уг)
називається е§- орбіталями, а друга (сІху,
й2у, сІХ2) — /2£~°рбіталями. Таким чином,
вихідний 5-кратно вироджений (без ура- Рис. 9.41. Розташування та нумерація
хування спіна) терм розщеплюється на лігандів в октаедричному комплексі
тричі вироджений нижчий за енергією Г2£-терм та розташований вище двічі
вироджений £^-терм (відштовхування від лігандів збільшує енергію), як це
зображено на рис. 9.42.
♦ б
У
*
/
/
Газоподібний
іон (Ті )
В
1
/
І
І
г
Сферичне
поле
■А
N
___5_х
ч
'* ^
Октаедричне
поле (0/,)
»Ь
± А
Аху
—" Я,
П28
====== Ек
<*Х2> Луг
Тетрагональне
ПОЛе (Д^)
.3+
Рис. 9.42. Вплив електричного поля на розщеплення ^/-орбіталі іона Ті
Енергетична відстань між 2? - та ^-термами позначається через 10£>#>
іноді А. Єдиний ^/-електрон іона Ті знаходится або на основному рівні Т2§,
або на збудженому Е8. Електронному переходу Т2§ -> Е§ відповідає лише
одна смуга у спектрі поглинання Тір|~. Нагадаємо, що для окремого
електрона поняття терм і орбіталь співпадають, тобто можна говорити як про Е§-
та Г2£-терми, так і про е§- та ^-орбіталі. При зниженні симетрії відбувається
подальше розщеплення термів. Якщо перейти до тетрагональної біпіраміди
(іони Флуору в положенні 5 та 6 зміщуються в протилежні боки вздовж осі
2 або замінюються на інші іони, що створюють інше електростатичне поле),
263
то координатна вісь і відрізнятиметься від осей х тау. У зв'язку з цим двічі
виродженими залишаються лише орбіталі й^ та й^ і в тетрагональному полі
(див. рис. 9.42) рівень Т2§ розщеплюється на два Е8 (й^ сі^) та В2§ (4^); рівень
Е8 октаедричного поля також розщеплюється на два: В1§ (сІхг-уг) та Аї§
(с12г). У спектрі з'являються три смуги, що відповідають переходам: Е§ —>
В2р Е§-^А1§ та Е8 —> В]8 (Е8 — основний рівень). Відповідні орбіталі такі:
<Іхг-уг -> Ь[§; й22 -> а\8; сіху -> Ь2я; (сІху, >!г) -» ^. Отже, кількість смуг у
спектрах поглинання може дати інформацію про симетрію найближчого
оточення іона.
При розташуванні лігандів у вершинах куба чи тетраедра кількість
компонентів £)-терма залишається сталою, однак рівні Т2 та Е обертаються:
нижчим за енергією стає £-рівень (е-орбіталь), а вищим — Т2 (/-орбіталь).
Відзначимо, що в групі симетрії тетраедра (7^) відсутній центр інверсії, тому
індекс § опущений. У тетраедрі ^-орбіталі (сіху, сі^ та сі^) знаходяться ближче
до лігандів, ніж е-орбіталі (йгі ть<іх2-уї). Величина розщеплення в тетраедрі
становить 4/9 від величини розщеплення в октаедрі з тими самими
лігандами. Пояснюється це меншою кількістю лігандів (4 замість 6).
При кількох ^/-електронах необхідно враховувати не тільки взаємодію
^/-електронів з електростатичним полем лігандів, а й взаємні відштовхування
я?-електронів між собою. В багатоелектронних системах як наслідок між-
електронного відштовхування з'являється система термів (див. підрозділ 8.4).
Ці терми під дією електростатичного поля лігандів розщеплюються, проте
слабкі поля не порушують до кінця взаємодії, що існувала в окремому
газоподібному іоні.
Задача про розщеплення даного терма в полі відповідної симетрії
розв'язується за допомогою теорії груп (див. підрозділ 9.2). Кожному терму в полі
заданої симетрії відповідає певне уявлення. Для термів сферично
симетричних іонів уявлення складається за допомогою такої формули:
зіп(2І + і)ф/2
Л 8ІПф/2 '
де х—характер уявлення; Ь — орбітальне квантове число атома (іона); ф —
елементарний кут обертання при операції Сп або 8п (для операції
відображення в площині величина ф дорівнює 180°).
Як приклад розглянемо розщеплення £>-терма у полі тетраедра Та.
Таблицю характерів точкової групи Тй наведено у табл. 9.8. При 1 = 2 формула
(9.152) матиме такий вигляд:
Х = ^£. (9.153)
81Пф/2 Ч '
Для операції спокою (Е) ф = 0 і для відповідного характеру дістанемо
невизначеність
%[Щ = 0/0.
264
Розкриємо невизначеність за допомогою правила Лопіталя
хЮ.І-їМ.І^ч^-а (9.154)
(зіпф/2) ^совф/2
Для операції С3 ф = 120°, отже, %(С3) = -1; для операцій С2 та С5а ф = 180°
Х(С2) = Х(оа) = 1, а для операції 5п ф = 90° %(5;) = -1.
Отже, звідне уявлення для £)-терма має такий вигляд:
Е 8С3 ЗС2 65„ 6а^
П 5 -1 1-1 1
За допомогою формул (9.94) та (9.95) це звідне уявлення можна звести
до суми двох незвідних: П (П) = Т2 + Е, як це наведено вище на основі
елементарних міркувань. Проте для двох та більшої кількості електронів без
застосування теорії груп розв'язати задачу про розщеплення термів у полях
різної симетрії неможливо. При розв'язуванні цієї задачі маємо два крайні
випадки:
1) наближення слабкого поля: головну роль відіграє міжелектронна
взаємодія, електростатична дія лігандів на ^-електрони мала, у спектрі
поглинання комплексного іона можна виявити «вихідні» терми окремого
газоподібного іона;
2) наближення сильного поля: електростатична дія лігандів набагато
перевищує міжелектронне відштовхування, з'являється нова система орбі-
талей, наприклад, і28-та е^-орбіталі замість п'яти <і-орбіталей.
Спочатку розглянемо наближення слабкого поля на і -конфігурації
(У3+,ТІ2+,сЛмЬ3+,Та3+тощо).
д, - конфігурація дає такий набір термів: Р9 Р, О, Д 5 (див.
підрозділ 8.4). Крім основного терма Р9 ту ж саму мультиплетність (М= 3) має терм
Р (найбільшу інтенсивність мають спектральні переходи між термами з
однаковою мультиплетністю).
Розглянемо розщеплення термів і7та Р в октаедричному полі, наприклад
у комплексному іоні [УС16] ~. Застосування рівняння (9.152) для
визначення уявлень ЩР) та ЩР) в полі октаедричної симетрії О дає результати,
наведені у табл. 9.10.
Таблиця 9.10. Таблиця характерів точкової групи О
Е
Аі(1)
А2(1)
Е{2)
43)
Тг(?)
1 П(І0(7)
П(Р)(3)
8С3
1
1
-1
0
0
1
0
зс2
1
1
2
-1
-1
-1
-1
6С2
1
-1
0
-1
1
-1
-1
6С4
1
-1
0
1
-1
-1
1
Координати
або АО
5
^х'-у1
Х,У>2
Ху,Х2,у
265
У точковій групі О центра симетрії немає, тому індекси £ та и незвідних
уявлень відсутні, ці індекси є в точковій групі Он. Кінцеві результати для груп
О та Он аналогічні. Оскільки всі <£-орбіталі мають парний характер, то
компоненти, які утворюються при розщепленні термів, повинні мати індекс £.
Застосування формул (9.94) та (9.95) дає можливість звести звідне
уявлення і^-терма (див. табл. 9.10) до суми незвідних уявлень
ЩГ)=А2§+Т1§+Т28.
Уявлення Р-терма (див. табл. 9.10)
незвідне і відповідає уявленню Т1§-
терма. Розщеплення термів для
сі -конфігурації зображено на рис.
9.43.
з-
Є2Л)1
У спектрі [УС16] повинні епос
терігатися три смуги, які відповіда
ють таким переходам:
3Г,^28(18^);
Т,„(^-
2г
а б в г д
Рис. 9.43. Рівні енергії й -іона:
а — газоподібний іон (триплетні рівні);
б — слабке поле; в — проміжні випадки;
г — сильне поле з урахуванням міжелек
тронного відштовхування; д — сильне : з
поле без урахування міжелектронного 11 000 та 18 000 см. Перехід Т1§—>
відштовхування А2§ замаскований іншими
спектральними смугами (смуги перенесення заряду, внутрішньолігандні смуги).
Через енергію цих двох переходів можна обчислити спектральні
параметри
(Щ[);'Ті-ї'А2в(1№д);3ТІІ
3Г1г (Р) (бЬ? + 15В'). Параметр 5'
називається параметром Рака і
визначає різницю між І^-та Р-терма-
ми з урахуванням зміщення цих
рівнів під дією кристалічного поля.
В експерименті для [УС16] ~ вдалося
спостерігати лише два переходи:
%->% та % (Р)^% (Р) з
відповідними хвильовими числами
КШя =
1100010
= 13800 см-1; В' =
18000-0,6-13800
15
= 650 см"
З З
Т\8 (Р)- та Т1§ (Р)-терми мають однакову симетрію і тому вони
відштовхуються один від одного. Енергія відштовхування між термами Ек=2800 см"1.
З урахуванням енергії відштовхування енергія переходу Тх (Р) -^ Т1§ (Р)
дорівнює (6£># +\5В/+Ек), апереходу 3Г^-> Г2^—(8£)^ +Ек/2). Це дає 10Д? =
: 530 см"1. Енергію смуги Т1§ (Р)-* А2§ можна тепер
12 000 см"1 та Я"
обчислити так:
18£>?+£* = 1,8 -12000+ 1400 = 23 000 см"1.
Значення параметра В для ізольованого іона V дорівнює 850 см"1, тобто
більше, ніж значення В'. Величина Р = В'/В має назву «нефелауксетичного
відношення». Воно завжди менше одиниці, для [УС16]
266
3-
Р = 0,62. Зменшення
параметра Р при комплексоутворенні свідчить про зменшення міжелект-
ронного відштовхування в комплексному іоні порівняно з вільним іоном.
Чим менша величина Р (порівняно з одиницею), тим більше відрізняється
хімічний зв'язок від іонного.
«Сила» поля лігандів визначається параметром ІОЛ7. Умовною межею
між слабкими та сильними полями вважається величина 10А? = 20000 см_1.
Для [УСІ6] " 10£># < 20 000 см_1, тобто підхід з позицій слабкого поля
виправданий.
Процедура, яка наведена вище для й -конфігурації, може бути
застосована до будь-якої ^^-конфігурації.
В наближенні сильного поля вважається, що дія кристалічного поля
істотно перевищує міжелектронну. Електрони розміщуються на орбіталях, що
виникають внаслідок розщеплення й?-рівнів у кристалічному полі.
Наприклад, в октаедричному полі утворюються два типи орбіталей: трикратно
вироджені /2£ (розташовані за енергією нижче) та двократно вироджені е§-
орбіталі.
Розглянемо в наближенні сильного поля сі -конфігурацію в іоні
[У(СИ)6] ~. Для нього спостерігаються такі спектральні переходи: ТІ8 -> Т2§
(22200 см"1) та 3Т1§ (Е) -> 3Г^ (Р) (28 600 см"1). Перехід 3Т1§ -> 3А2§
знаходиться в ультрафіолетовій частині спектра.
/2£-орбіталі розташовані нижче барицентра («центра ваги») на величину
40#, а орбіталі е§ — на в^^ вище барицентра (різниця в енергіях (2§ та V
орбіталей — 10£>#). Згідно з правилом барицентра сума добутків енергій на
кратність виродження терма повинна дорівнювати енергії нерозщепленого
терма. Якщо енергію терма і7 взяти за нуль відліку, то (~40ф • 3 + 6£><7 * 2 = 0.
Для й -іона можливі три електронні конфігурації: (і28Г — найнижча за
енергією, (Іі8)1 (е8)1 та (е§) — найвища за енергією. Не важко визначити, що
різниця в енергіях [(і{§){ (е8){ - (і2§)2] та [(е8)2- ^2§У(е8У] конфігурацій
дорівнює ІОЛ?.
Для визначення симетрії одержаних електронних конфігурацій
користуються так званими «прямими добутками» незвідних уявлень, які визначають
як добутки характерів відповідних уявлень, наприклад, для добутка (Г2 х Е)
в точковій групі О за допомогою табл. 9.10 знаходимо
Е 8С3 ЗС2 6С2 6С4
П 6 0 -2 0 0
Це уявлення звідне і виражається як сума двох незвідних уявлень Тх та Г2,
тобто ЕхТ2 = Т{+ Т2. Отже, електронній конфігурації (12Л (ее) відповіда-
3 3 £ £
ють терми Т1§ та Т2г Один з термів (Т1§) відповідає розташуванню
^-орбіталей в одній площині (наприклад, сІх2-у2 та а^-орбіталі знаходяться в
площині ХОУ), другий терм (Т2§) відповідає розташуванню ^/-орбіталей у різних
площинах (наприклад, сі х2_у2- та 4^-орбіталі зорієнтовані в різних площинах).
Природно, що в першому випадку міжелектронне відштовхування більше,
ніж в другому, і Т1§- та Г2я-терми мають різну енергію. Для інших конфігу-
267
рацій прямі добутки дорівнюють:
Т2§ х Т2§ = X + 1Е8+ х Т2§ + 3ТЇ8,
Прямі добутки можуть бути симетричними або антисиметричними.
Згідно з принципом Паулі антисиметричній орбітальній функції відповідає
симетрична спінова (триплет) і, навпаки, синглет відповідає
антисиметричній спіновій функції. Отже, відповідно до заданих електронних
конфігурацій одержимо чотири триплетні терми:
(І28) — Т\2,
(*2*) (е8) — Т2§; Т\е;
(е8)2 — 3А28.
Зазначимо, що кількість компонентів розщеплення та їх симетрія
однакові при обох наближеннях — слабкого і сильного полів.
Енергія електронів у найбільш енергетично вигідному стані (і2§) така:
-8£><7 = 2(-4£>#), якщо не враховувати міжелектронного відштовхування,
у збудженому стані (і2§) (е§) енергія така: -4Вд + 6£)# = +2£>#; нарешті, для
конфігурації (еЛ енергія дорівнює (+ 6 Всі)! = + 12Л?. Різниця в енергії між
термами 7^ та Т1& що виникли в електронній конфігурації (/2«) (е§) в
наближенні слабкого поля, дорівнювала 15 ^(різниця енергії термів Рта Р—
15 Я). Параметр В' менший, ніж параметр В (нефелоауксетичний ефект), в
наближенні сильного поля різниця між Т{8 та Т2в зменшується. Це добре
можна побачити на рис. 9.43, де наведена кореляційна діаграма між двома
крайніми випадками — сильне та слабке поле. Перша смуга в спектрі
[У(С>06] ~ (22 000 см"1), яка відповідає переходу Т1§-> Т2§9 повинна
дорівнювати (див. рис. 9.43) приблизно 10£><7 (різниця між (і2е) ~та (^Уі^)1-?™'
нями). В дійсності параметр 10£># дещо більший (23 800 см"1). Різниця в цих
величинах свідчить про відхилення від «чистого» випадку сильного поля.
Інші ^"-конфігурації в наближенні сильного поля розглядаються
аналогічно.
Теорія кристалічного поля широко використовується при поясненні
спектрів сполук перехідних елементів. Для відповідних електронних
конфігурацій відомі системи термів і, знаючи симетрію комплексів, визначають
енергетичні відстані між термами через емпіричні параметри£># та/?' (з
поправками на взаємодію між термами однакової симетрії).
Величина параметра £>#, який визначає «силу» лігандів, залежить
насамперед від їх природи. В порядку збільшення І><7 ліганди можна розташувати
в ряд, відомий під назвою електрохімічної серії: Г < Вг~ < СГ < Р~ < С204~ <
< Н20 < >Ш3 < Ж)2 <<: С>Г. Одержана зі спектрів поглинання сполук А-
перехідних металів емпірична величина ЮЛ7 широко використовується для
пояснення деяких інших фізико-хімічних властивостей цих комплексних
268
сполук, зокрема магнітних
властивостей.
Розглянемо як приклад сполуки
з електронною конфігурацією сі .
Існують два крайні випадки
розташування електронів на і2£- та е§-
орбіталях: (і2§) > (е2§) та (*28) (Рис-
9.44). В першому випадку в
сполуці є чотири неспарені електрони,
в другому — всі спарені. Сполуки
першого типу парамагнітні,
другого— діамагнітні. Наприклад, іон
Ре має електронну конфігурацію
сі . Акваіон Феруму [Ре(Н20)6]
ІНІН
г
НІ
<2*
Рис. 9.44. Розподіл електронів на 12„-
та е^-орбіталях для сі -конфігурації
в слабкому і сильному полях
та деякі інші його сполуки —
типові парамагнетики, проте іон
[Ре(СИ)6] — діамагнітна сполука.
Різний розподіл електронів за
/2£- та ^-рівнями залежить від різної сили поля лігандів, яка визначається
параметром 1(Ю#. В першому випадку (і2я) (е§) два електрони займають
високо розташований рівень е^ отже, енергія системи збільшується на 20£>#.
В другому випадку (і2§) енергія (2Ер) витрачається на спарення електронів
на рівні і2е. Отже, якщо 1 (Ю# < Ер>то реалізується конфігурація {і2^ (е§) —
високоспінові комплекси, а якщо 10^ > Ер, електрони спарюються на
нижчому рівні, та реалізується система з максимально можливим числом
спарених електронів (тут (і2§) ) — це випадок низькоспінових комплексів.
Відмінність між високо- та низькоспіновими октаедричними
комплексами може проявлятися при електронних сі ,сІ ,сі ,сі -конфігураціях. В решті
випадків розподіл електронів по орбіталях типу 12§ та е§ однаковий незалежно
від сили поля. Всі -9сі -,сі -конфігураціях усі електрони розміщуються на
нижньому рівні і2§ незалежно від сили поля; всі -та сі -конфігураціях нижній
рівень заповнений (і2§) незалежно від сили поля.
Енергія спарювання (Ер) дорівнює приблизно 20000 см~1, отже, величина
10£><7 дая низькоспінового комплексу повинна бути більшою, ніж 20000 см"1.
Розглянемо тепер деякі термохімічні характеристики комплексних
сполук ^/-елементів. Для цих комплексів в ряду однаково заряджених іонів зі
зміною кількості ^/-електронів такі термохімічні властивості, як теплоти
сольватації простих іонів, енергії кристалічних ґраток однотипних сполук,
теплоти утворення комплексних іонів та деякі інші, змінюються за так
званою «двогорбою» кривою. Як ілюстрація на рис. 9.45, с. 270, наведені
теплоти гідратації іонів 3<і-елементів, розташовані в порядку збільшення
атомного номера. Складається враження, що значення термохімічних величин для
іонів з сі -,сі -тасі -конфігурацією укладаються на одну пряму, а
значення для решти конфігурацій завжди більші (за абсолютною величиною), ніж
269
Са8с Ті V СгМпРеСо№Си2п
Елементи
Рис. 9.45. Теплота гідратації двозарядних
іонів 3*/-елементів
знайдені в результаті
інтерполяції.
Надлишкова зміна
термодинамічної величини, порівняно
з інтерпольованим значенням,
називається «енергією
стабілізації» кристалічного поля. Вона
знаходиться як різниця між
експериментальними та
інтерпольованими значеннями
відповідних енергій.
Походження енергії
стабілізації можна з'ясувати з позицій
теорії кристалічного поля.
Справді, при заповненні електронами
/2£-рівня повинен бути
додатковий виграш енергії А^^ на
кожний електрон. При
заповненні електронами е^-рівня,
який розташований над
барицентром, повинен спостерігатися програш енергії 6£># на кожний електрон
(див. рис. 9.42, с. 263). Енергію стабілізації полем лігандів можна знайти для
будь-якої електронної конфігурації за формулою Е8 = 4(£>#)и/ - 6(£>#)лг, де
щ та пе—відповідно, кількість електронів на і28- та е^-орбіталях. Термохімічні
величини (Е5) розраховуються за спектральними даними.
Основний недолік теорії кристалічного поля — це нехтування
можливістю утворення різного типу ковалентних зв'язків між центральним атомом
та лігандами. Цей недолік усувається при застосуванні методу
молекулярних орбіталей до комплексів перехідних металів. Метод МО в цьому
випадку одержав назву теорії поля лігандів.
Розглянемо основи цієї теорії на прикладі комплексу [МЬ6]Й , наприклад
[Со (КН3)6] , із симетрією октаедра Ок та хімічним зв'язком тільки а-типу.
Перший етап конструювання МО — вибір базисних АО. В базис атомних
орбіталей від центрального атома Зс/-металу можна взяти п'ять 3^-, одну Аз-
та три 4р-АО. Від лігандів включаємо шість атомних орбіталей, які
утворюють озв'язки: сь а3 (вздовж осіх); а2, а4 (вздовж осі>>) та а5, а6 (вздовж осі і).
Для молекули аміаку це можуть бути/?-орбіталі Нітрогену (чи зр -гібридні
орбіталі) з неподіленою електронною парою.
Другий етап МО — класифікація АО базису за їх симетрією. 3</-АО в ок-
таедричному полі поділяються на два типи: 12§ та е§- З них тільки е^-орбіталі
{й2і іьйхг-уг) можуть утворювати а-зв'язки, орієнтовані вздовж
координатних осей, ^-орбіталі, орієнтовані вздовж бісектрис координатних площин,
в утворенні а-зв'язків участі не беруть. 4я-АО належать до незвідного
уявлення аІ8 і можуть брати участь в утворенні а-зв'язків, як і 4р-орбіталі, які
270
належать до незвідного уявлення ї1и. Орбіталі лігандів утворюють групові
орбіталі, тобто лінійні комбінації а-орбіталей. Кожна а-орбіталь у групі
повинна перекриватися з відповідною АО центрального атома (з
урахуванням знака її лопаті). Відповідні лінійні комбінації а-АО лігандів (групові
орбіталі) наведені в табл. 9.11.
Таблиця 9.11. Симетрія та групові орбіталі в комплексі типу МЬ» (симетрія
Он) із а-зв'язками
АО ^-металу
3(1
45
4р
Симетрія АО металу
*и
а,е
Чи'
Рх
Ру
Рг
Групові орбіталі лігандів
—
^(0|-ст2 + а3-а4)
-^=(2<т5 + 2<т6-а, -а2-аг-а4)
-^(о,+о2+аз+ст4 + 05+Об)
-^(°2-<*4)
Виведення ЛКАО лігандів здійснюється так, щоб знак коефіцієнта
відповідав знаку хвильової функції центрального атома.
При виконанні цих вимог для повністю симетричної 4.У-АО одержуємо
всюди додатні знаки та однакове перекривання з усіма а-АО. Для/?-АО, які
орієнтовані вздовж координатних осей та мають протилежні знаки плюс та
мінус на своїх лопатях, одержимо в груповій орбіталі тільки по дві а-орбіталі
з протилежними знаками. Орбіталь йхі-уг розташована в площині ХОУ та
перекривається з чотирма (а1? а2, а3 та а4) орбіталями. Вздовж осі х знаки
(1х2-у2 орбіталі додатні, а вздовж осі у—від'ємні. Нарешті, орбіталь й2і
(повністю сІ222-гі) може бути зображена, як д,І2г_хі_уг (г2 = х2 + у2 + г2) і тому
має знак плюс вздовж осі і і знак мінус — вздовж осей х тау. Після виведення
групових орбіталей слід переходити до складання МО як лінійних комбінацій
АО металу та групових орбіталей лігандів. При цьому в кожному випадку
повинна утворюватися пара МО: зв'язуюча \рь та антизв'язуюча \|/а. Вони
мають такий вигляд:
де фм— атомна орбіталь металу; ф^ — групові орбіталі лігандів; Иа тьИь —
нормувальні множники.
271
Множники Х( характеризують ступінь ковалентності зв'язку між
центральним атомом та лігандами (в підрозділі 9.1 аналогічна картина описана
для двохатомних молекул). Для ковалентного зв'язку Ха = Хь = 1, для іонного
(дуже електронегативний ліганд) Хь » 1; Ха -> 0 і тоді \|/6 = ф^, а \|/а = фм.
Цей граничний випадок і розглядає теорія кристалічного поля.
Провести точні розрахунки енергій МО—дуже складна задача. Найбільш
поширені методи розрахунку за теорією поля лігандів подібні до методу МО
Гюккеля. Для обчислення енергії та коефіцієнтів МО, які належать до даного
звідного уявлення (е§, а1§, 11и), складається детермінант
= 0,
(9.156)
Нмм % НМІ - ЕОМІ
нмь~ЕСмь Нц-~Е І
де Нммтг. НІЬ — кулонівські інтеграли для орбіталей, відповідно, атома
металу та лігандів; ОМІ — групові інтеграли перекривання; НМІ — резонансні
інтеграли, які обчислюють за формулою, що запропонована Р. Маллікеном
(див. підрозділ 9.3)
Я
мь
= к(нмм +ни){
тмь-
(9.157)
Рис. 9.46. Схема МО в октаедрич-
ному комплексі (а-зв'язки)
Коефіцієнт АГ змінюється від 1,6 до
2,0 залежно від типу зв'язку (а або я)
та інших вимог.
Для обчислення кулонівських
інтегралів Л. Гельмгольц та В. Вольфс-
берг запропонували використовувати
Нммтз, Нц як енергії іонізації
відповідних орбіталей центрального атома
та лігандів. Для розрахунків МО в
теорії поля лігандів використовують
і точніші методи, наприклад типу
методу Рутана.
Одержані за допомогою теорії
поля лігандів схеми енергетичних
рівнів в октаедричному комплексі дають
змогу дати значно точніший опис
спектрів поглинання сполук
перехідних металів порівняно з описом
спектрів на базі теорії кристалічного поля.
На рис. 9.46 наведено розташування
МО за енергією в октаедричному
комплексі.
На діаграмі енергетичних рівнів
можна виявити насамперед ті самі рівні, що и на діаграмах, які дає теорія
кристалічного поля.
272
Розглянемо такий комплекс — [Сг(КН3)6] . сі -конфігурація
центрального іона Сг та шість пар електронів від шести молекул аміаку утворюють
п'ятнадцять валентних електронів. Заповнимо МО в порядку збільшення їх
енергії: (аІ8) (11и) (е8) (128) . ^-°рбіталь, яка по а-типу не перекривається
з орбіталями лігандів і є незв'язуючою, заповнена електронами частково.
Вакантна орбіталь є* — антизв'язуюча. Вона локалізована переважно на
атомі металу.
Параметр \^^^ теорії кристалічного поля можна інтерпретувати як
різницю енергій між вищою заповненою незв'язуючою МО (і2$) та нижчою
антизв'язуючоюе*-орбіталлю. Для приблизної оцінки параметра 10£><7
можна скористатися формулою
ЮЛ7 = #^%Ч (9.158)
деК—коефіцієнт, який приблизно дорівнює одиниці (Ни, Нмм, Сж
визначені вище).
Формула (9.158), однак, не враховує електростатичної взаємодії між
центральним іоном та лігандами (при електростатичній взаємодії Смь = 0).
Проте навіть при іонному характері зв'язку параметр КЮд не дорівнює нулю.
Спектральні переходи між і2§-та ^-орбіталями називаються
^/-^-переходами та описуються аналогічно переходам, розглянутим вище на основі
теорії кристалічного поля. Інтенсивність </-г/-переходів відносно невелика.
У короткохвильовому діапазоні спектра повинні спостерігатися смуги,
які відповідають переходу електронів від зв'язуючих МО, які локалізовані
в основному на атомах лігандів, на вакантні антизв'язуючі орбіталі, що
локалізовані в основному на атомах металів. Як приклад можна навести перехід
електрона з орбіталей а-типу (е§9 іХи та аІ8) на вакантні або частково
заповнені орбіталі, що локалізовані на атомі
металу (12§ або є*).
Крім а-зв'язків, ліганди можуть
утворювати з центральним атомом
зв'язки я-типу. На рис. 9.47 наведено
схему утворення я-зв'язків у площині
ХОУ. З орбіталлю й^ я-зв'язки
утворюють такі лінійні комбінації ті-орбіталей
лігандів:
^(щу+п2х-п3у-п4ху
Цифра в індексі тс-орбіталі означає
номер ліганда, літера — орієнтацію
орбіталі вздовж визначеної
координатної осі. Таких лінійних комбінацій
можна побудувати три (в площинах рис. 9.47. Орієнтація орбіталей, які
Х02 та У 02, крім ХОУ, і, відповідно, утворюють я-зв'язки в площині ХОУ
273
використати йХ2 та йу2-К0 центрального атома). Сукупність усіх цих
орбіталей відповідає незвідному уявленню Т2£.
Не важко показати, що групова орбіталь
^(п1у-п2х+п3у-п4х)
ортогональна по відношенню до групової орбіталі
^(щу+п2х-п3у-п4х).
Ця орбіталь незв'язуюча, тому що її сумарне перекривання з орбіталлю йху
дорівнює нулю. Сукупність таких групових орбіталей відповідає незвідному
уявленню Т{§.
Кожний із лігандів для утворення я-зв'язків може дати по дві взаємно
перпендикулярні орбіталі. Отже, загальна кількість орбіталей лігандів, які
можуть утворювати я-зв'язки, дорівнює дванадцяти. Вони об'єднуються в
чотири типи групових орбіталей: Т2§, Т1и, Т{§, Т2и (кожний із типів належить до
трикратно вироджених уявлень). Перший тип (група Т2§) утворює зв'язуючі
та антизв'язуючі я-орбіталі {12? та і* між груповими орбіталями лігандів та
а?-орбіталями металу (сІху, сіХ2, сіу2). Другий тип (група Т2и) утворює слабко
зв'язуючі та антизв'язуючі 1Хи МО з групових я-орбіталей та комбінаціїр-
орбіталей (рх,ру,р2) металу. Третій і четвертий типи групових орбіталей (11§
та і28) належать до незв'язуючих лінійних комбінацій орбіталей лігандів.
Якщо я-орбіталі лігандів за енергією розміщуються нижче ^/-орбіталей
центрального атома (рис. 9.48), то ^-орбіталі, які локалізовані переважно
на лігандах, стають зв'язуючими орбіталями я-типу. ^-обіталі металу
стають антизв'язуючими 1*^. При цьому параметр 10£># зменшується
порівняно з системою, де ліганди утворюють а-зв'язки з центральним атомом за
абсолютною величиною (відстань і2і —» є* менша, ніж 12§—> є*; в системі
з а-зв'язками орбіталь і2§—незв'язуюча).
Якщо я-орбіталі лігандів мають рівень енергії вищий, ніж й?-орбіталі
центрального атома, то рівень ^-орбіталі металу знижується і відстань 12§ —> є*
збільшується. Зв'язуюча ^-орбіталь локалізується переважно на атомі
металу, і частина електронної густини передається з металу на ліганд. Такий
тип хімічних зв'язків називають я-дативним. Він реалізується у комплексних
сполуках металу з монооксидом карбону (карбоніли) та ціанід-іоном
(ціаніди). Аномально велике значення параметра 10£># для комплексів з цим
іоном (див. вище) пояснюється не його електростатичними
характеристиками, а можливістю утворення дативних я-зв'язків між іоном металу та
ціанід-іоном.
При переході електронів з/?-орбіталей лігандів (11и, і1§, 12и, 12§) (див. рис.
9.48) на 12&- або є* -орбіталі має місце часткове перенесення електронної
густини з лігандів на центральні атоми (внаслідок різної локалізації
орбіталей). Ці смуги називаються «смугами з пересенням заряду». Вони
характеризуються великою інтенсивністю і знаходяться, як правило, в короткохвильо-
274
3</
(\и(\8*2и \ Щ—Щ:
вій ділянці спектра. Інтенсивність
спектральних смуг визначається
так званими правилами відбору.
Найінтенсивніші переходи
спостерігаються при зміні квантового
числа / на одиницю (Л/ = ±1),
наприклад, 5 —>р,р —> (і, сі-їр і т. д.
Для смуг «перенесення заряду»
перехід електрона здійснюється з/?-
орбіталі ліганда на ^/-орбіталь
центрального атома. Інтенсивні
смуги можна спостерігати також при
внутрішньоатомних переходах
типу 4/*—» 5сі; такі смуги
називаються рідберговими. Значно менш
інтенсивні смуги при А/ = 0. Такі
смуги називають «заборонені за
Лапортом». Заборона за Лапор- \ %
том може бути частково знята при
утворенні ковалентних зв'язків „„„ 0 .в ^.««ш-л^о^ттлт,
, т , „ч гис. 9.4». Схема МО в октаедричному
(«змішування» й- тар-орбіталеи), комплексі (я-зв'язки)
тому в спектрах сполук перехідних
металів можна спостерігати ^/-ч/'-переходи. Саме ці переходи розглядає
теорія кристалічного поля. Усі ці смуги реалізуються при переходах між
термами з однаковою мультиплетністю (Д5=0). Нарешті, дуже слабкі смуги
можуть спостерігатися при ще сильнішій забороні — забороні за спіном
(Д5 Ф 0). Такі смуги спостерігаються, наприклад, у спектрі сполук
двовалентного марганцю, де основний терм - 5 (спін 5/2), а інші терми — квартети
(спін 3/2) або дублети (спін 1/2).
Теорія поля лігандів природно пояснює спектральні та інші фізико-
хімічні властивості сполук перехідних металів. Теорія кристалічного поля—
це граничний випадок теорії поля лігандів, який не враховує структури
лігандів та делокалізації електронів центрального атома.
Свою специфіку має хімічний зв'язок у конденсованому стані, що
розглянуто у наступному розділі.
275
Розділ 10.
ХІМІЧНИЙ ЗВ'ЯЗОК У КОНДЕНСОВАНОМУ СТАНІ
Чотири типи в фазі кристалічній
Зв 'язків існує, ось ці побратими:
Іонний, ковалентний, металічний,
Молекулярний, є містки між: ними.
Конденсований стан — це рідкі і тверді фази. Рідка фаза характеризується
лише близьким порядком, тверда фаза більш упорядкована і має дальній
порядок. Оскільки хімічні сили діють тільки на коротких відстанях, то зв'язки
в рідкому і твердому станах близькі за своїми властивостями. Краще
описувати зв'язки для упорядкованого твердого стану.
За типом хімічних зв'язків та за деякими фізико-хімічними
характеристиками конденсовані системи можна поділити на чотири класи: іонні,
молекулярні, ковалентні, металічні.
В іонних сполуках зв'язок здійснюється внаслідок електростатичної
взаємодії протилежно заряджених іонів. Він принципово не відрізняється
від зв'язку між іонами в двохатомних або багатоатомних молекулах (див.
розділ 9).
Електростатичні кулонівські сили характеризуються ненасиченістю і
досить значні, внаслідок чого іонні ґратки досить міцні. Це зумовлює такі
властивості іонних кристалів, як механічна міцність, твердість. У полярних
розчинниках, наприклад, у воді, іонні кристали легко розчиняються з
утворенням сольватованих іонів.
Іонні кристали—діамагнетики (іони перехідних металів можуть
зумовлювати парамагнетизм (див. підрозділ 9.4)). Структурні одиниці, з яких
складається конденсована іонна система, — це іони. Типовими представниками
цього класу сполук є ИаС1, ПР, СаР2 тощо. Іонні кристали—діелектрики, але
при розплавленні вони перетворюються на провідники другого роду.
У молекулярних кристалах та рідинах зв'язок між молекулами
здійснюється силами Ван дер Ваальса. Це слабкі сили, тому молекулярні кристали
плавляться при невисоких температурах. Найміцнішими молекулярними
кристалами є парафіни з довгим карбоновим ланцюгом, лід; найслабкі-
шими — гелій. Рідкий гелій перетворюється на твердий лише при тиску
13,8 МПа та температурі 3,15 К (потрійна точка). При звичайному тиску
гелій кипить при 4,2 К.
Молекулярні сполуки діамагнітні і не проводять електричного струму,
наприклад, це рідкі та тверді інертні гази, вода, аміак, парафіни та інші
органічні сполуки.
Молекулярні кристали та рідини — це єдині конденсовані системи, для
яких зберігається поняття «молекула». Вони складаються з окремих
молекул, усередині яких атоми зв'язані міцним хімічним зв'язком, як правило,
ковалентним.
276
У ковалентних кристалах зв'язки між атомами локалізовані. Такі двоцент-
рові двохелектронні зв'язки вже були розглянуті у розділі 9. Ці зв'язки дуже
міцні, тому ковалентні кристали також тверді та механічно міцні. Один з
типових представників ковалентних сполук — алмаз — є найтвердішою
речовиною. Спрямованість валентних зв'язків зумовлює також крихкість
ковалентних кристалів. При нагріванні та плавленні спрямованість ковалентних
зв'язків внаслідок невпорядкованості рідкої структури слабне і система
«металізується» (див. підрозділ 10.3). Вважають, що структурною одиницею
в ковалентних кристалах є атом, але це не саме істотне, тому що основна
особливість ковалентних кристалів (як і багатоатомних молекул із
локалізованими зв'язками, див. підрозділ 9.2) — це можливість виділення окремих
двоцентрових двохелектронних зв'язків.
У металічних системах зв'язок здійснюється делокалізованими в усьому
об'ємі електронами. Це найбільш яскраво виражений випадок багатоцент-
рового зв'язку—кількість центрів дорівнює кількості атомів у кристалі, тобто
приблизно числу Авогадро. Тому металічний кристал — це єдина квантово-
механічна система (відзначимо, що будь-яка система повинна розглядатися
як єдина квантово-механічна, проте можливість фрагментації на іони,
молекули, атоми, зв'язки диктується не лише міркуванням зручності, а й
обґрунтовується хімічною будовою систем).
З присутністю делокалізованих електронів у металах пов'язані такі їхні
властивості, як висока електро- та теплопровідність, ковкість, пластичність.
Звичайні метали — це слабкі діа- або парамагнетики, перехідні — сильні
пара- або феромагнетики. Багато елементів періодичної системи Д. І.
Менделєєва у конденсованому стані є металами.
Між класами конденсованих систем немає різких переходів, оскільки
завжди можна знайти сполуки з проміжними типами зв'язку.
Вивчення хімічного зв'язку в конденсованих системах, як і в молекулах,
доцільно починати з розгляду найпростішого типу зв'язку — іонного.
10.1. Іонні сполуки
Для багатьох конденсованих систем характерна тенденція до найбільш
компактного пакування (за винятком ковалентних кристалів зі
спрямованими зв'язками). В іонних сполуках, які складаються з окремих катіонів та
аніонів, крім цієї тенденції, слід враховувати також намагання кожного іона
оточувати себе іонами протилежного знака. Поєднання цих двох умов
призводить до того, що для іонних кристалів найхарактернішими для рівнозаряд-
них іонів є такі структури:
а) СзСІ—кожний катіон (аніон) оточений вісьмома аніонами (катіонами),
що розташовані у вершинах куба (див. рис. 10.1, а, с. 278); підґратки катіонів
та аніонів — прості кубічні, а повні ґратки — кубічні об'ємноцентровані;
б) №С1 — кожний катіон (аніон) оточений шістьма аніонами (катіонами),
що розташовані у вершинах октаедра (див. рис. 10.1, б); підґратки катіонів
та аніонів — кубічні гранецентровані, а повні ґратки — прості кубічні.
277
а б
Рис. 10.1. Кристалічні ґратки СзСІ (а) та ИаСІ (б)
Кулонівські сили, які діють між іонами у ґратках, ненасичені й можуть
діяти на значних відстанях, тому в іонних сполуках треба враховувати
взаємодію не тільки із сусідніми, а й з далі розташованими іонами. Так, у
ґратках ИаС1 кожний іон взаємодіє з шістьма іонами, що мають протилежний
знак і розташовані на відстані г\ з дванадцятьма іонами того самого знака—
на відстані з вісьмома іонами — на відстані гу/з і т. д. Відповідна куло-
нівська енергія (із розрахунку на один іон) дорівнює
М >/2 л/3 ) г г
(10.1)
де а — константа Маделунга (залежить лише від геометрії ґраток і може бути
обчислена для будь-якої кристалічної структури). Для ґраток типу ИаСІ
а = 1,7476, дая СзСІ а = 1,7627.
Сумування енергії відштовхування відбувається так само, але в цьому
випадку всі члени додатні і ряд швидко збігається
Г = Ье р
6 + 12е
+ 8е
= $Ье р=Ве
(10.2)
Другий член (в дужках) дорівнює 0,19 (при р = 34,5 пм), тобто малий
порівняно з першим, інші взагалі дуже малі, тому Р = К, де К—
координаційне число (для ИаС1 К=6).
Для енергії ґраток одержуємо вираз, який тотожний формулі (9.5) для
двохатомних іонних молекул (див. підрозділ 9.1), але з іншими значеннями
константи та В. При г = г0 для розрахунку Е0 можна використовувати
формулу (9.7) (з тим самим значенням р = 34,5 пм), яка в цьому випадку
називається формулою Борна-Майєра (див. формулу (1.34), де 1/0 = -Е0).
Експериментальні значення енергії ґраток можна визначити за
допомогою термодинамічного циклу Борна-Габера (див. підрозділ 1.3).
278
Величина г0 — міжатомна відстань відома з рентгеноструктурних даних.
Л. Полінг та В. Гольдшмідт показали, що г0 можна поділити на дві частини —
іонні радіуси катіона та аніона (наприклад, для ИаСІ г0 = гКа+ + гсг). Для того
щоб побудувати систему іонних радіусів, необхідно визначити спочатку
радіус одного з іонів. В. Гольдшмідт прийняв гк+ = Гр-, виходячи з рівності по-
ляризованості іонів К та Р~ і зв'язку між поляризованістю та розміром
частинок (див. формулу (9.15)); для КР г0 = 0,266 нм, звідки гк+ = Гр- = 0,133 нм.
Л. Полінг запропонував свою систему іонних радіусів. Він взяв за основу
слетерівські орбіталі (8.145), згідно з якими функція радіального розподілу
22* г
м;(г) = 4пг2К2(г) = 4пг2п*е п (Ю.З)
з максимумом при (г в атомних одиницях)
г^ = {п)2І2\ (10.4)
Дж. Слетер запропонував вважати радіусом іона (г,) відстань від центра
іона, при якій
Чп) = 0,1и<гтах), (10.5)
тобто шукати г( у такому вигляді:
г = аг = а\п / (Ю.6)
Згідно з формулами (Ю.З)-(Ю.б) параметр а можна обчислити зі
співвідношення
ае1~а = 2фл. (10.7)
Отже, параметр а залежить лише від п*. Для ізоелектронних іонів значення
а та п* однакові, відрізняються лише значення 2*. В №Р іони Иа та Р~—
ізоелектронні (конфігурація І522з22р6). 2*Ка=2Ка - а = 11 - (2 • 0,85 + 7 • 0,35) =
= 6,85 (див. підрозділ 8.4). Для Флуору константа екранування а та ж сама, але
заряд ядра на дві одиниці менший (2Р = 9), отже, 2р = 4,85. Тому, згідно
з Полінгом, гКа+/гр-=4,85/6,85 = 0,71. Оскільки гМа++ Гр- = г0Мар = 0,231 нм, то
знаходимо гКа+ = 0,096 нм, а гР- = 0,135 нм, що добре узгоджується з гР- за
В. Гольдшмідтом.
Формула (10.6) пояснює основні закономірності в іонних радіусах.
У групі періодичної системи іонні радіуси завжди збільшуються зверху вниз
(табл. 10.1), тому що збільшується значення п . Для ізоелектронних систем,
як це показано вище, значення 2* для катіона завжди більше, ніж значення
Ґ для аніона, тому катіони мають менший радіус, ніж аніони. Зі
збільшенням заряду катіона значення 2* також збільшується, а значення гі9 відповідно,
зменшується. При зміщенні у періодичній системі на одну клітинку вправо
значення 2* збільшується, а при зміщенні на одну клітинку вниз зростають
п* та а. Ці фактори компенсують один одного при зміщенні по діагоналі
зверху вниз та одночасно зліва направо. Значення гКа+ = 0,116; гСа2+ = 0,114;
279
гуз+ = 0,104 (див. табл. 10.1) добре ілюструють приблизну сталість г{ при русі
по діагоналі — відоме в періодичній системі емпіричне правило діагоналі.
Завдяки широкому застосуванню високопрецезійних рентгенівських
дифракційних методів з'явилася можливість побудувати «карти
електронної густини» і з'ясувати, як змінюється електронна густина вздовж ліній, які
з'єднують атомні ядра катіонів та аніонів у кристалі, і визначити відстані від
атомних ядер до мінімумів електронної густини на цих лініях. Слід зазначити,
що величина іонного радіуса залежить від координаційного числа іона: чим
більша кількість іонів з протилежним зарядом оточує іон, тим сильніше
вони відштовхуються один від одного, внаслідок чого відстань між катіоном
та аніоном збільшується. Наведені у табл. 10.1 дані належать (за винятком В +,
С та N ~) до найбільш розповсюдженого шестикоординаційного оточення.
Для будь-якого елемента радіус іона зменшується при збільшенні його
заряду, наприклад, Ті — 0,100 нм, Ті — 0,081 нм, Ті — 0,075 нм. Якщо іон
може знаходитися в різних спінових станах (наприклад, Ре + (НС), Ре + (ВС),
Со (НС), Со (ВС) і т. д.), то радіус іона в низькоспіновому (НС) стані
менший, ніж у високоспіновому (ВС). Знаючи іонні радіуси, можна
обчислювати енергії іонних ґраток тих сполук, для яких міжіонні відстані невідомі.
Крім іон-іонної взаємодії досить великі електростатичні сили виникають
при взаємодії іонів з дипольними молекулами. Енергія іон-дипольної
взаємодії
£/„=-11/7 = -ц4 = -ц% (10.8)
г г
де \х — дипольний момент; г—відстань між іоном та центром диполя; ^ =
2е — заряд іона; Р = #/г2 — напруженість поля, яке створює іон.
Для взаємодії іона з наведеним диполем маємо
^о=~ = ~^-, (Ю.9)
де а — поляризованість.
Із формул (10.8) та (10.9) видно, що енергія взаємодії насамперед
залежить від заряду іонів (2), особливо при взаємодії іона з наведеним диполем.
Для сталого диполя суттєвим є також значення ц, тобто І)гш збільшується зі
збільшенням полярності молекули, яка взаємодіє з іоном. Іон-дипольна
взаємодія забезпечує досить міцний зв'язок у кристалогідратах, аміакатах,
комплексах типу 1^ і т. д.
Системи з іон-дипольною взаємодією — це проміжний випадок між
системами з досить міцним іонним зв'язком (іон-іонна взаємодія) та
молекулярними сполуками (диполь-дипольна взаємодія), до розгляду яких ми
і переходимо.
280
Таблиця 10.1. Радіуси іонів (нм) для сполук із координаційними числами 6
ІЬі
1+1
і 0,090
1+1
0,116
К
+1
0,152
Си
+2
0,087
КЬ
+1
0,091
КЬ
+1
0,166
А£
+1
0,129
С8
-1
Ве
+2
0,059
+2
0,086
Са
+2
0,114
2п
+2
0,088
Зг
+2
0,132
Ссі
+2
0,109
Ва
+2
0,149
+2
0,116
В
+3(4)
0,025
А1
+3
0,068
8с
+3
0,089
Са
+3
0,076
У
+3
0,104
Іп
+3
0,094
Ьа
+3
0,117
ТІ
+3 0,103
+1 0,164
с
+4(4)
0,029
Зі
+4
0,054
Ті
+4 0,075
+3 0,081
+2 0,110
Се
+4
0,067
їх
Зп
+4
0,083
т
+4
0,085
РЬ
+4 0,092
+2 0,133
N
-3(4)
0,132
Р
V
+3 0,078
+2 0,093
А8
N1)
+4 0,082
+3 0,086
ЗЬ
+3
0,090
Та
+3 0,082
+3 0,086
Ві
+3
0,117
0
-2
0,126
8
-2
0,170
Сг
+3 0,076
+2 0,087 (н)
+2 0,094 (в)
8е
-2
0,184
Мо
+4 0,079
+3 0,083
Те
-2
0,207
+4
0,080
Ро
Р
-1
0,119
СІ
-1
0,167
Мп
+3 0,072 (н)
+3 0,079 (в)
+2 0,081 (н)
+2 0,097 (в)
Вг
-1
0,182
Тс
І
-1
0,206
Ке
+4
0,077
А1
" _Ие "" 1
Аг 1
Ре
+3 0,069 (н)
+3 0,079 (в)
+2 0,075 (н)
+2 0,092 (в)
Со
+3 0,069 (н)
+3 0,075 (в)
+2 0,079 (н)
+2 0,089 (в)
N1
+3 0,070 (н)
+3 0,074 (в)
+2 0,083
Кг
Ки
+4 0,076
+3 0,082
КЬ
+4 0,074
+3 0,081
Реї
0,100
Хе
08
+4
0,078
Іг
+4 0,077
+3 0,082
+4 0,077
+2 0,094
КЬ
Примітка. Для В3+, С4+ та № координаційні числа дорівнюють «4»; н — низькоспінові стани; в — високоспінові стани.
10.2. Міжмолекулярна взаємодія, водневий
зв'язок
Дипольна молекула створює навколо себе електростатичне поле та
орієнтує решту диполів системи, що приводить до зниження енергії.
Розрахована П. Кізомом середня енергія орієнтаційної
диполь-дипольноївзаємодії між полярними молекулами складає
Сог=4-£г, (10-10)
-> г кТ
де |і — дипольний момент молекули; г — відстань між центрами молекул;
к — константа Больцмана; Т— температура за Кельвіном.
Множник (кТ) в знаменнику відображує вплив розупорядкування на
орієнтацію диполів внаслідок теплового руху, який зростає зі збільшенням
температури. Крім орієнтаційного, слід враховувати індукційний ефект
(С//т/), тобто взаємодію диполя з наведеним диполем, яка, згідно з П. Дебаєм,
дорівнює
иш=-^- (Ю.П)
Г
Орієнтаційні та індукційні сили виникають між полярними молекулами
і не можуть пояснити міжмолекулярну взаємодію між неполярними.
Врахування слабкої квадруполь-квадрупольної взаємодії не вирішує проблеми,
тим більше, що молекули типу СН4 та атоми інертних газів не мають взагалі
квадрупольного моменту (відзначимо, що квадрупольний момент (без
дипольного) мають молекули типу С02, ВеН2; квадруполями можна також
вважати двохатомні гомоядерні молекули — Н2, 1М2, 02 тощо).
Природа міжмолекулярних сил у неполярних системах була з'ясована
Ф. Лондоном за допомогою квантової механіки. У підрозділі 8.3 показано,
що врахування кореляції під час руху атомних електронів призводить до
зниження енергії. Якщо рух електронів у різних атомах скорельований, то це
також сприяє зниженню енергії. Атоми з рухливими електронами можна
вважати своєрідними диполями, які осцилюють із деякою частотою У0. При
синхронному русі електронів миттєві диполі орієнтуються завжди так, що
це призводить до зниження енергії
_ зК«2 ПП19,
ийьр - т ~- (10.12)
Замінивши й\>0 на/, де /— енергія іонізації молекули (атома), одержимо
^=-|т£- (10ЛЗ)
Формулу (10.13) можна одержати більш послідовно (не застосовуючи
модель осцилюючих диполів) на основі теорії збурень.
Дж. Слетер та Дж. Кірквуд для взаємодії багатоелектронних атомів вивели
таку формулу:
282
де N— кількість електронів на зовнішній оболонці; т — маса електрона;
є — його заряд.
Формули (10.12) та (10.14) співпадають при N = 1, якщо замість У0
підставити його вираз: у0 = е/2лу/ат. З наведених формул можна зробити
висновок про те, що основна характеристика, яка визначає величину сил
Лондона,— це поляризованість (ос) атомів (молекул). У зв'язку з тим, що поляри-
зованість тісно пов'язана з коефіцієнтом заломлення світла та характеризує
здатність речовини до розсіювання (дисперсії) світла, сили Лондона часто
називають дисперсійними (Т/^).
Поляризованість залежить від розміру частинки, тому міцність
молекулярних ґраток повинна зростати зі збільшенням розмірів атомів та молекул,
які взаємодіють. Ця закономірність добре ілюструється збільшенням
температур кипіння (аналогічні залежності спостерігаються для теплот і
температур плавлення, сублімації, випаровування тощо, тобто для величин, які
залежать від міцності молекулярних зв'язків) у групі інертних газів та в гомо-
логічному ряду па]
Речовина
Т °С
х КИП.5 ^
Не
-267
Ие
-246
рафінів.
Аг
-186
Кг
-153
Хе
-108
Кп
-63
СН4
-161
с2н6
-88
С3Н8
-42
СДію
-0,5
С5Н12
+36
СбНи
+69
Атом Гелію настільки малий і дисперсійні сили при взаємодії атомів
Гелію такі слабкі, що гелій не може існували в кристалічному стані навіть при
звичайному тиску і 0 К. Причина цього — існування нульової кінетичної
енергії, яка для гелію більша, ніж енергія зв'язку. Наявність кінетичної енергії
ядер у зв'язаних атомів (при 0 К) є наслідком співвідношення невизначе-
ностей Гейзенберга.
Енергія зв'язку для гелію Езв= 0,105 кДж/моль. Міжатомна відстань
у рідкому гелію становить 0,25 нм. Невизначеність у положенні атома не
повинна перевищувати міжатомну відстань, отже, Ах = 0,25 нм, тоді
Д£™
.(АрГ
ДЄИ2
2пг
маса атома Гелію.
2т (Ах)"
= 0,200 кДж/моль,
(10.15)
Тому АЕКІН > Езв і кристалічний стан не може реалізуватися навіть при
0 К. Лише при великому зовнішньому тискові гелій може перейти у
кристалічний стан.
Всі міжмолекулярні взаємодії (їх часто об'єднують загальною назвою —
взаємодія Ван дер Ваальса) можна виразити у такому вигляді:
(10.16)
"--*
Орієнтаційна, індукційна та дисперсійна взаємодії дають різний внесок
у повну енергію зв'язку. Для атомів та неполярних молекул 1/ог та Цш дорів-
283
нюють нулю і залишається тільки дисперсійна взаємодія. Внесок орієнта-
ційних та індукційних сил збільшується з ростом дипольного моменту
молекул. У молекулі СО ц = 0,12 В (1£>-дебай = 3,33564 10"30 Кл • м) 1]ог дає
внесок 0,005 %, а 1/ш—0,085 %; в НВг ц = 0,78 Д Цог—3,3 %, 1]ш—2£ %;
в НС1 ц = 1,03Д 1]ог— 14,4 %, С/.^—4,2 %. З наведених даних можна
зробити висновок, що навіть для дуже полярних молекул дисперсійна
взаємодія дає суттєвий внесок.
Дисперсійна взаємодія адитивна й
універсальна. Вона дає свій внесок у будь-які
зв'язки, зокрема в розглянуту в попередньому
параграфі енергію іонних ґраток. Сполуки, в
яких зв'язок проміжний між іонним та
зв'язком Ван дер Ваальса, частково розглядалися
вище (гідрати, аміакати тощо — іон-дипольна
взаємодія). Інший випадок проміжного типу
зв'язку — це шаруваті структури типу С(И2.
Кожний шар в С<Н2, складається з трьох іонних
сіток, які чергуються у такій послідовності:
Йод, Кадмій, Йод (рис. 10.2). Шарувата
структура, що утворюється при цьому,
енергетично вигідна, незважаючи на аніон-аніонний
контакт, внаслідок того, що іон Ссі має
невеликий розмір та достатню поляризуючу дію,
а іон Г має великий розмір і легко
поляризується. Тому всередині шарів, крім іон-іонної,
виникає сильна іон-дипольна взаємодія між
іонами Ссі та наведеними в атомах Йоду
диполями (цих сил немає у високосиметричних
Рис. 10.2. Структура С(ії2 іонних ґратках типу ИаСІ чи СзСІ). Аніон-ані-
онне відштовхування між шарами
компенсується значними дисперсійними силами внаслідок високої поляризова-
ності великих за розміром іонів Г.
Проміжним між іонним та молекулярним є також водневий зв'язок.
Щоб проілюструвати роль водневого зв'язку в міжмолекулярній взаємодії,
розглянемо температури кипіння гідридів елементів IV—VII груп. З графіка
(рис. 10.3) бачимо, що НР, Н20 та ИН3 випадають із загальної закономірності.
Високі температури кипіння НР, Н20 та ИН3, порівняно з іншими гідридами,
свідчать про те, що в цих сполуках існують додаткові зв'язки, які зміцнюють
структуру. Чому ж високу міцність структур в НР, Н20, >Ш3 не можна
пояснити значним внеском орієнтаційних сил і знадобилося уявлення про
водневі зв'язки? Справді, НР, Н20 та МН3 мають великі дипольні моменти
(відповідно 2,0; 1,84 та 1,44), тому сили орієнтації повинні бути значними. Проте
в ряду НІ -» НС1 дипольний момент значно збільшується, а міжмолекулярна
взаємодія, судячи із температур кипіння, все ж таки зменшується (внаслідок
284
зменшення поляризованості
(розміру) молекул і, отже, внеску від
дисперсійних сил). Більш
переконливим прикладом є порівняння
властивостей хлористого метилу та
метилового спирту. Дипольний
момент Исн3сі = 1,99/) > Цснзон = 1,700,
але відомо, що температура кипіння
хлористого метилу (-24,1 °С)
набагато нижча, ніж метанолу (+64,5 °С),
що пов'язано із відсутністю
водневих зв'язків у хлористому метилі.
Водневі зв'язки характерні для
сполук Гідрогену з
електронегативними елементами другого періоду
(И, О, Р), в яких атом Гідрогену має
значний позитивний заряд. Протон
відрізняється від усіх інших іонів
дуже малим розміром через
відсутність електронної оболонки. Це
різко зменшує сили відштовхування // /// IV V
та дає змогу протону близько набли- Періоди
жатися до відповідної частинки. рИс. 10.3. Температури кипіння гідридів
В іоні [РНР] взаємодія між іона- елементів IV—VII груп
ми Н та Р~ практично іонна, обидві відстані Н—Р однакові між собою, тому
реалізується характерне для катіонів дуже малого розміру координаційне
число—два. Випадок, коли відстані Н—А та В...Н зв'язку в сполуці В...Н—А
дорівнюють одна одній або близькі, має назву «короткого» або «сильного»
водневого зв'язку. Крім зв'язку в КНР2, сюди належить водневий зв'язок
у КН2Р04, де відстань О—Н між протоном та «своїм» атомом Оксигену
становить 0,107 нм і дещо менша відстані (0,141 нм) О...Н між протоном та
«чужим» атомом Оксигену.
Більш поширений «довгий» або «слабкий» водневий зв'язок. Так, внут-
рішньомолекулярні відстані Н—Р, Н—О та Н—N у кристалічних НР, Н20
та ІЧНз дорівнюють, відповідно, 0,09; 0,107 та 0,109 нм, а відстані у «довгих»
водневих містках: Н...Р, Н...0 та Н...И дорівнюють, відповідно, 0,16; 0,18 та
0,19 нм, тобто значно більші, ніж внутрішньомолекулярні відстані.
Крім міжмолекулярного водневого зв'язку, існує також внутрішньомо-
лекулярний. Такий зв'язок утворюється, наприклад, в о-оксибензойній
(саліциловій) кислоті, де групи ОН та НО—С=0 розташовані близько одна
до одної. В інших ізомерах: м- та л-оксибензойних кислотах карбоксильна та
гідроксильна групи знаходяться далеко одна від одної, для утворення внут-
рішньомолекулярного водневого зв'язку немає можливості, отже, в цих
ізомерах реалізуються міжмолекулярні водневі зв'язки. Згідно з цим м- та
285
л-ізомери утворюють міцніші ґратки, ніж о-ізомер — саліцилова кислота
(температури плавлення ізомерів такі, °С: мета — 201, пара — 210, орто —
159). Саліцилова кислота гірше розчиняється у воді (один протон зв'язаний),
але краще в органічних розчинниках, ніж п- тал*-оксибензойні кислоти.
Оскільки водневий зв'язок утворюють дуже електронегативні елементи,
і він виникає у полярних мо-скулах, то відповідний внесок до енергії повинна
давати електростатична взаємодія, чому також сприяють малі розміри
атомів Нітрогену, Оксигену та Флуору. Чим більше «оголений» протон, тим
міцнішим повинен бути водневий зв'язок. Для груп ОН (водневий зв'язок
О—Н...О) повинна існувати кореляція між полярністю зв'язку О—Н та
міцністю водневого зв'язку. Наприклад, енергія водневого зв'язку при
утворенні димеру мурашиної кислоти більша (29,4 кДж/моль), ніж у воді
(18,8 кДж/моль). Водневий зв'язок набагато міцніший, ніж звичайний
міжмолекулярний (< 4 кДж/моль), але значно поступається іонному (сотні
кілоджоулів на 1 моль). Слід зазначити, що в КНР2 міцність водневого зв'язку
досягає 126 кДж/моль.
Електростатична модель, однак, має ряд недоліків. Вона неспроможна
пояснити відсутність кореляції між дипольним моментом та енергією
водневого зв'язку в деяких основах, не може пояснити наявність кутів між
диполями у кристалах з водневим зв'язком. Так, ланцюжки ...Н—Р...Н—Р... у
кристалічному НР не лінійні, а зигзагоподібні, причому кут РНР дорівнює
120°. Цей результат можна пояснити, якщо припустити, що водневий
зв'язок має донорно-акцепторний характер.
У підрозділі 9.2 показано, що вільний протон є добрим акцептором та
утворює донорно-акцепторний зв'язок із донорами електронних пар,
наприклад, з Оксигеном в Н30 , з Нітрогеном в >Щ £. У сполуках з
електронегативними елементами (для яких і характерний водневий зв'язок) атом Гідрогену
частково іонізований, електронна пара знаходиться ближче до
електронегативного елемента, і Гідроген проявляє акцепторні властивості. Атоми
Нітрогену, Оксигену та Флуору є донорами електронних пар. Розташування
електронних пар, які не беруть участі в утворенні зв'язку, згідно з правилом
Гілеспі (див. підрозділ 9.2), близьке до тетраедричного (кут 109° 28і). До цього
значення наближається у кристалічному стані фтороводень, величина кута
між молекулами якого дорівнює 120° (відзначимо, що водневий зв'язок
(особливо слабкий) все ж таки близький до зв'язку Ван дер Ваальса і поняття
«молекула» в кристалах з водневими зв'язками ще зберігає свою суть).
Донорно-акцепторна схема водневого зв'язку добре узгоджується з
будовою кристалів льоду — атоми Оксигену знаходяться у тетраедричному
оточенні атомів Гідрогену: двох «своїх» — зв'язки ОН води, та двох
«чужих» — водневі зв'язки «сусідніх» атомів Гідрогену з тетраедрично
розташованими неподіленими парами Оксигену (рис. 10.4). Молекула води має
дві неподілені пари електронів та два атоми Гідрогену. Це оптимальний
випадок порівняно з аміаком (три атоми Гідрогену — одна неподілена пара
електронів) та фтороводнем (один атом Гідрогену — три неподілені пари
286
електронів). Тому температура кипіння ^ ^Оо (*&~0~~~~'$&
води вища, ніж аміаку та фтороводню, о^^^^х^У^-^^СУо^
незважаючи, наприклад, на більшу
електронегативність Флуору
порівняно з Оксигеном.
Водневий зв'язок має відповідну
подібність з містковими зв'язками, які
атоми Гідрогену утворюють в дибо-
рані (див. підрозділ 9.3). Розглянемо іон
[РНР]~ із позицій методу МО.
Використаємо 2/? та 2р2 орбіталі атомів
Флуору (позначених А та В) та Ь-АО
Гідрогену. Всі атоми розташовані
вздовж осі 2 (лінія зв'язку), причому
атом Гідрогену посередині між
атомами Флуору. Симетрія іона [РНР]~ —
^ооЛ. Використовуючи три АО,
одержимо три трицентрові МО:
-V
Vу IV
,оОсгі-
с^р
Рис. 10.4. Кристалічна структура
льоду
а8=М
(2р2А-2р2в) + Х\з І —зв'язуюча;
\Х(2р2А-2р2в) - іу І — антизв'язуюча;
аи = Щ2р2А+ 2р2в) — незв'язуюча.
Сумарне перекривання незв'язуючої ам-молекулярної орбіталі з 1$-АО
Гідрогену дорівнює нулю. Схема цих трицентрових МО (с^ о*§ та аи) подібна
до схеми МО для одного з «містків» диборану (МО побудована за
допомогою еквівалентних орбіталей). В обох випадках антизв'язуючі МО вільні,
тому енергії місткових зв'язків Н близькі між собою. Поряд із подібністю МО
в диборані та іоні [РНР]~ існує і відмінність між ними. По-перше, МО цих
частинок мають різне походження: в диборанах Гідроген — донор, а Бор —
акцептор, в іоні [РНР]~, навпаки, Гідроген — акцептор, а флуорид-іон —
донор. По-друге, відмінність полягає в тому, що в місткових зв'язках
диборану незв'язуюча МО вільна (два електрони приходяться на три МО), а в іоні
[РНР]~—зайнята (чотири електрони на три МО), що призводить до зайвого
негативного заряду на атомах Флуору, оскільки, як було показано вище,
незв'язуюча а„-МО складається виключно з АО атомів Флуору.
Хоча при опису водневого зв'язку необхідно враховувати ковалентну
донорно-акцепторну складову, в основному, водневий зв'язок слід
розглядати як проміжний між іонним та Ван дер Ваальсовим (ближче до
міжмолекулярного).
287
10.3. Ковалентний та металічний зв'язок
у твердому тілі
Прикладом взаємодії, яка визначається лише дисперсійними силами,
можуть бути міжатомні сили в інертних газах. Щоб позбавитися іонного
внеску в хімічний зв'язок, резтіпсмо сполуки, які складаються з однакових
атомів. Сполуки почнем^ розглядати по періодах періодичної системи
справа наліво, починаючи від інертних газів. Далі ідуть галогени. Вони
витрачають свою єдину валентність на утворення двохатомних молекул. У
конденсованому стані ці двохатомні молекули галогенів зв'язані між собою тими
самими малими дисперсійними силами, що й інертні гази. Внутрішньо-
молекулярні відстані між атомами в молекулах, де діють потужні ковалентні
сили, набагато менші, ніж відстані між атомами в сусідніх молекулах, які
визначаються слабкими міжмолекулярними силами. В елементів VI групи
є дві вільні валентності, в елементів V періоду—три; в елементів IV періоду —
чотири. Згідно з цим для ковалентних структур діє правило: К=%-И,д&К—
координаційне число, тобто кількість близько розташованих сусідніх атомів;
N— номер групи. Для інертних газів N = 8 і К = 0; для галогенів N = 7
і К = 1 (є тільки один близько розташований атом).
Для елементів VI групи
К = 2 і для них характерні
ланцюгові структури (ланцюги
можуть замикатися в кільця).
Зигзагоподібні ланцюги
характерні для кристалічних
структур сірки, селену та
телуру (рис. 10.5). Зв'язки
утворюються р-орбіталями, тому
кути між ними бувають від
90° («чисті» /^-зв'язки) до
109° 28і (у/?3-гібридні зв'язки)
(див. підрозділ 9.2). Ланцюги
зв'язані один з одним
відносно слабкими силами Ван дер
Ваальса і відстані між
атомами у різних ланцюгах
набагато довші, ніж усередині ланцюгів (табл. 10.2).
Для елементів V групи АГ= 3, тому для них характерні шаруваті структури
(рис. 10.6, а). Як і для ланцюгових структур, кути між зв'язками близькі до
прямих, тому шари виникають «гофрованими». Шари між собою зв'язані
відносно слабко і відстані між атомами різних шарів перевищують відстані
між атомами всередині кожного шару (див. табл. 10.2). Крім шаруватих
структур, при АГ = 3 можуть утворюватися неміцні молекулярні структури
у вигляді правильного тетраедра (Х4) (білий фосфор, жовтий арсен). Зв'язки
Рис. 10.5. Ланцюгова структура ґраток
халькогенідів
288
в тетраедрах (див. мал. 10.6, б) сильно напружені (кути 60° замість 90°), тому
ці структури дуже нестійкі.
а б
Рис. 10.6. Структури ґраток елементів V групи:
а — шарувата; б — молекулярна
Таблиця 10.2. Міжатомні відстані в кристалічних ґратках елементів V—VII
груп
Елемент
СІ
Вг
І
8
8е
Координаційне
число
1
1
1
2
2
Внутрішньо-
молекулярна
відстань, нм
0,199
0,227
0,270
0,210
0,232
Міжмолекулярна
відстань, нм
0,279
0,330
0,354
0,330
0,346
Елемент
Те
Р
А8
8Ь
Ві
Координаційне
число
2
3
3
3
3
Внутрішньо-
молекулярна
відстань, нм
0,286
0,220
0,251
0,290
0,310
Міжмолекулярна
відстань, нм
0,346
0,387
0,315
0,336
0,347
Нарешті, для елементів IV групи (АГ=4) характерні об'ємні структури.
Оскільки 5- тар-рівні окремих атомів розташовані близько один до одного,
то має місце л,/?3-гібридизація (як у парафінах), і структури елементів IV
групи тетраедричні. Це так званий тип алмазу (рис. 10.7, а, с. 290).
Подібність між зв'язками С-С у парафінах і в алмазі не тільки в симетрії,
а й у міжатомних відстанях та енергії. Відстань між зв'язками С—С в алмазі
дорівнює 0,154 нм, тобто точно відповідає одинарному зв'язку С—С у
парафінах. Теплота атомізації (сублімації) алмазу 1^ = 714 кДж/моль.
Враховуючи, що координаційне число дорівнює 4, а кожний зв'язок С—С належить
двом атомам Карбону, то Ес_с = (і^/4)2 =Ь5/2 = 357 кДж/моль, що добре
узгоджується з даними для Ес^с парафінів (див. підрозділ 9.2).
При з'єднанні різних атомів у ковалентних структурах кожний із них
зберігає властиве йому координаційне число. Так, у карборунді 8іС атоми
289
Рис. 10.7. Структурний тип алмазу (а) та сфалериту (цинкової обманки) (б)
Силіцію та Карбону зберігають тетраедричне оточення, в 8і82 Силіцій
(К=4) оточений тетраедрами з атомів Сульфуру, а Сульфур (К= 2) утворює
містки між атомами Силіцію і т. д.
Чим більша різниця в електронегативностях між атомами в сполуці, тим
більша «іонна складова». Оксиген значно більш електронегативний, ніж
Сульфур, тому в оксиді Силіцію (IV) 8і02 позитивний заряд Силіцію значно
більший, ніж у 8і82. Відповідно змінюється структура. В 8і82 тетраедри [8і84]
«з'єднувалися» ребрами, в 8і02 подібна будова привела б до невигідного
сильного відштовхування близько розташованих та однойменно заряджених
атомів Силіцію, тому в 8і02 тетраедри [8і04] з'єднуються вершинами, що
відповідає більшій віддалі атомів Силіцію один від одного.
Поступовий перехід від ковалентного зв'язку до іонного можна
простежити на сполуках типу А В ~ . Ці сполуки ізоелектронні відповідним
ковалентним кристалам атомів IV групи (С, 8і, Ое тощо). Для них характерна
тетраедрична структура — кожний атом оточений тетраедром з атомів
іншого елемента (тип цинкової обманки 2п8, рис. 10.7, б). До подібних
сполук належать ВИ, АЇР, ОаА8,2п8 тощо.
Для сполук А^В8"^характерна наявність донорно-акцепторних зв'язків
(див. підрозділ 9.2). Так, алюміній в ^-конфігурації має одну вакантну р-
орбіталь, а фосфор — зайву пару електронів; при утворенні донорно-акцеп-
торного зв'язку обидва елементи знаходяться в стані з/?3-гібридизації та
утворюють відповідну тетраедричну структуру. В ряду 8і—»А1Р—»М§8--»]МаС1
відбувається поступовий перехід від ковалентних структур до іонних. Для
останніх вже характерне координаційне число не 4, а 6 (чи 8). Зазначимо, що
зменшення координаційного числа від 6 до 4 можна було б пояснити і на
ОСНОВІ СуТО ІОННОГО ПІДХОДУ Зменшенням ВІДНОШеННЯ ?*кат /ган, ОСКІЛЬКИ
при переході від одно- до багатозарядних іонів радіус катіона повинен
зменшуватися, а аніона — відповідно збільшуватися. Проте такий підхід не зав-
290
жди приводить до правильних результатів. Наприклад, гСи+ =0,091 нм, >*Вг- =
= 0,196 нм. Відношення >си+/гВг- =0,51. Межею переходу від
координаційного числа 6 до 4 вважається відношення гкат/ган = 0,41. В даному випадку
гСи+/ гВт- > 0,41, але СиВг кристалізується у вигляді тетраедричної
структури (АГ =4).
Невиправданість іонного підходу до кристалів типу А§1,2п8 виявляється
також у занижених розрахункових значеннях енергії іонних ґраток. Слід
зазначити, що іонний характер зв'язку в кристалах типу А§1,2п8 послаблений
саме внаслідок донорно-акцепторної взаємодії, тому що донори — це більш
електронегативні елементи (наприклад, Йод в А§1 віддає в донорно-акцеп-
торний зв'язок три пари електронів). Кристали А§1,2п8 та інші згідно зі
своїм ковалентним характером, не розчиняються у воді (на відміну від
типових іонних сполук типу КІ).
Донорно-акцепторна взаємодія змінює координаційне число не тільки
в структурах типу А В8"^, айв інших сполуках. Так, у хлориді паладію в
результаті донорно-акцепторної взаємодії (дві вакантні орбіталі Паладію та
по парі електронів від кожного з атомів Хлору) координаційне число Хлору
збільшується від одиниці до двох, а Паладію від двох до чотирьох (структура
РсіС12 стає подібна до структури 8і82).
Збільшення частки ковалентного зв'язку в іонних сполуках може
сприяти утворенню як ковалентних кристалів типу АЇР, 2п8, так і молекулярних
(при малих координаційних числах), де ковалентні сили зосереджені тільки
всередині молекули. Характерним прикладом можна вважати перехід від
А1Р3 до ВР3. В першій сполуці в силу меншої електронегативності
алюмінію зв'язки АЇР значною мірою іонні і температура плавлення А1Р3 досить
висока (1040 °С), хоч і менша, ніж у М§Р2 (1400 °С) (якби А1Р3 був суто
іонною сполукою, то його температура плавлення була б більшою, ніж М§Р2,
тому що заряд А1 більший, ніж М§ ). В сполуці ВР3 з Флуором з'єднується
Бор, який більш електронегативний, ніж Алюміній. Тому зв'язки В—Р більш
ковалентні, ніж А1—Р, і вони «замикаються» в молекулі ВР3, температура
плавлення якої (-127 °С), тобто в конденсованому стані ВР3 має типові
міжмолекулярні зв'язки Ван дер Ваальса.
Розглянемо тепер відхилення від правила К = 8 -И. Ці відхилення
спостерігаються для елементів другого періоду: Оксигену, Нітрогену та
Карбону. Так, Оксиген та Нітроген кристалізуються в молекулярних ґратках
(К= 1) замість утворення ланцюгових чи шаруватих структур, які характерні
для елементів VI та V груп. С02 та С82, на відміну від 8і02 та 8і82,
кристалізуються у молекулярних структурах.
Причину такої поведінки елементів другого періоду не важко з'ясувати,
якщо згадати, що вони можуть утворювати міцні 7С-зв'язки (див.
підрозділ 9.3). Атоми наступних періодів мають великий розмір,
тому/^-/^-перекривання у них мале і утворення кратних я-зв'язків невигідне. Для них
а-зв'язки міцніші, ніж я-зв'язки, і при цьому реалізується максимально
можливе координаційне число згідно з правилом К=8-М.
291
Утворенням міцних я-зв'язків зумовлена здатність Карбону
кристалізуватися не тільки в структурі алмазу (К= 4), а й у структурі графіту (К= 3)
(рис. 10.8). У графіті атоми Карбону утворюють плоскі сітки з правильних
шестикутників з відстанями між
атомами 0,142 нм. Атоми Карбону
знаходяться в стані ^-гібридизації
та утворюють три а-зв'язки з
кутами 120°. Четвертий електрон
кожного атома Карбону знаходиться на
/?-орбіталі, яка розташована
перпендикулярно до площини а-зв'яз-
ків. Ці/?-АО утворюють я-зв'язки.
Аналогія з ароматичними
сполуками (див. підрозділ 9.3) повна.
Використовуючи експериментальне
значення міжатомної відстані С—С
в площині /*£_£ = 0,142 нм, за допо-
Рис. 10.8. Кристалічні ґратки графіту ^огою калібрувального графіка
(див. рис. 9.36, с. 246) знаходимо, що
порядок зв'язку С—С в графіті дорівнює 1,5.
Щільні шари в графіті знаходяться один від одного на відстані 0,335 нм та
пов'язані між собою силами Ван дер Ваальса. Тому графіт легко
відшаровується і використовується як мастило. Слід відзначити, що нітрид бору
повністю імітує вуглець. Він має дві модифікації: боразон — дуже твердий
алмазоподібний матеріал та білий графіт — м'який шаруватий матеріал.
Завдяки делокалізації тс-електронів графіту по всьому кристалу він є
добрим провідником електрики. Це наближає графіт до металів, у яких
електрони також делокалізовані у всьому кристалі, проте (на відміну від графіту)
не лише в одній площині, а й по всьому об'єму тіла
В елементів І—III груп періодичної системи кількість валентних
електронів (1—3) менша від кількості атомних орбіталей (4) (перехідні ^/-метали
розглядатимуться далі). Для завершення октету (з2р6) атоми третьої групи,
згідно з правилом К = 8 -Иповинні були б мати координаційне число п'ять,
другої групи—шість, першої—сім. Ці координаційні числа в елементів І—III
не реалізуються, проте для їх ґраток характерні великі координаційні числа:
8 — кубічна об'ємноцентрована структура (к.о.ц.) (К, Иа, КЬ тощо), 12 —
кубічна гранецентрована (к.г.ц.) (А 1, Са тощо), 12 — гексагональна щільна
упаковка (М§, Тлу тощо). Якщо на вісім-дванадцять зв'язків припадає 1—З
електрони, то неможливе утворення двоцентрових двохелектронних зв'язків,
які характерні для ковалентних структур. При дефіциті електронів
порівняно з наявними орбіталями вони можуть вільно пересуватися по кристалу.
На це вказують насамперед високі електро- та теплопровідність металів.
Розглянемо застосування моделі вільних електронів до металів.
Поведінка частинки (зокрема електрона) в ямі з високими потенціальними стін-
292
•^^^М
ками описана в підрозділі 8.1. Вважаючи, що «довжина ями» / дорівнює
лінійному розміру кристала/,, для кінетичної енергії електрона в
металічному кристалі одержимо
Е =
п2Н2
(10.17)
Формула (10.17) справедлива для одновимірного випадку, наприклад, для
руху електрона вздовж осі х, тобто п = пх. За її допомогою не важко
порахувати, що пх — це кількість квантових станів (§г1), які мають енергію від нуля
до заданої величини Е, тобто
8\
(10.18)
Перш ніж перейти до розгляду руху електрона у тривимірному просторі,
розглянемо менш складний рух у двовимірному просторі. Оскільки рух у
кожному напрямку незалежний, то для енергії частинки, яка рухається в
площині ХО¥,
£ = -
-К2 + "*) = ;
, . Л у. Гг • (10Л9)
Кількість атомів з енергією від 0 до Е порахуємо так. Кожний стан атома
зобразимо точкою на площині для двох ступенів свободи, відкладаючи на
осях значення пх та пу. У цьому випадку умові 0 < 8, < Е задовольняє кожна
точка, яка лежить посередині сфери з радіусом г = Лг?х + п2у (див. рис. 10.9).
Кількість таких точок дорівнює площі квадранта цього кола (радіуса г), отже,
кількість станів (#2) з енергією від 0 до £ дорівнює (аналогічно обчисленню
за формулою (10.18) для одновимірного випадку)
&=^=-А[тЬітЕ)=^2тЕ)- (шо)
Для тривимірного простору г =
-і
2 2 2
Пг+Пу+ПІ9
а кількість точок, що
відповідають станам з енергією 0 < є,-
< Е і знаходяться посередині сфери
радіуса г, дорівнює об'єму
відповідного октанта, тобто
іГз
й=г=оІ4ЯГ І =
2І)(^Г-
чЗ/2
|^(2^Г =
Рис. 10.9. Власні стани енергії для
двох ступенів свободи
293
_ АпУ (2тЕ)3/2
—3 ^з—> (10.21)
де V = Ь — об'єм кристала (індекс 3, який означає розмірність простору,
далі випустимо).
Кількість атомів, що мають енергію від Е до Е +АЕ, де АЕ—дуже мала
величина порівняно з Е, знайдемо диференціюванням формули (10.21)
^ = 22гЕ(2/й)з/2£І/2
^ , ...... - . (Ю-22)
Перенісши Уь ліву частину, одержимо формулу для визначення густини
станів
1^_2«(2шУ2)
V <ІЕ нг
Е{1\
(10.23)
(10.24)
У формулах (10.21) та (10.23) не враховано, що кожний стан двічі
вироджений за спіном. З урахуванням цього кількість станів (спін-орбіталей)
збільшується вдвічі, отже,
Шу(2тЕ)г/2
8 3 н3 '
/(*)-££-^**. <Ю*>
Залежність/^ від Е наведена на рис. 10.10. Частина станів зайнята
електронами, а частина вільна. Межа між зайнятими та вільними станами
відповідає максимально можливій кінетичній енергії системи і називається
енергією Фермі — Ер. Величину Ер можна оцінити за формулою (10.24), якщо
вважати, що кількість електронів (И) дорівнює кількості зайнятих
спін-орбіталей (%)
(10.26)
е'-^-
Кінетична енергія електронів пропорційна їх концентрації в одиниці
об'єму в степені 2/3. На рис. 10.10 заштрихована частинаДЕ) при Е<ЕР
відповідає кривій електронної гус-
ЛЕ)к
0
Рис. 10.10. Залежність густини станів
від енергії у моделі вільних електронів
294
тини. При Е> Ер відповідні стани
вільні та/(£) відповідає кривій
густини вільних станів.
Використовуючи вираз для
кінетичної енергії електронів, можна
побудувати модель для розрахунку
енергії зчеплення у металічних
системах. Атоми, які віддали електрони,
перетворюються на іони. Ці іони
досить компактні, про що можна пересвідчитись, якщо порівняти іонні
радіуси (див. табл. 10.1, с. 281) з так званими «металічними» (див. табл. 10.3).
Таблиця. 10.3. Таблиця атомних (металічних) радіусів, нм
н
0,037
п
0,155
0,189
К
0,236
Си
0,128
ЯЬ
0,242
А8
0,144
Сз
0,268
Аи
0,144
Ве
0,113
0,160
Са
0,197
2п
0,139
8г
0,215
са
0,156
Ва
0,221
0,160
в
0,089
А1
0,143
8с
0,164
Са
0,139
Уг
0,181
Іп
0,166
Ьа
0,187
ТІ
0,171
С
0,077
8і
0,117
Ті
0,146
Се
0,122
2г
0,160
8п
0,158
Ж
0,159
РЬ
0,175
N
0,071
Р
0,110
V
0,134
А8
0,121
N1)
0,145
8Ь
0,161
Та
0,146
Ві
0,182
0
0,073
8
0,104
Сг
0,127
8е
0,117
Мо
0,139
Те
0,137
0,140
Р
0,071
СІ
0,099
Мп
0,130
Вг
0,114
Тс
І
0,133
Ке
0,137
Не
0,122
Ие
0,160
Аг
0,192
Ре
0,126
Кг
0,108
Ки
0,134
Хе
0,218
08
0,135
Со
0,125
Мі
0,134
Іг
0,135
N1
0,124
ра
0,137
Рі
0,138
Металічний радіус (гт) — це половина найкоротшої відстані між
атомами в кристалічних ґратках металів. Для неметалів наведені відповідні
ковалентні радіуси (для інертних газів ван-дер-ваальсівські). Оскільки г(« гт,
то, по-перше, електрони мають досить вільного простору для руху, а по-
друге, іон-іонна взаємодія в металах незначна і відштовхування
забезпечується в основному кінетичною енергією вільних електронів. Згідно з
формулою (10.26), кінетична енергія пропорційна К~2/3, тобто обернено
пропорційна г2, де г—відстань між атомами. Притягання — це кулонівська
взаємодія між катіонами та негативно зарядженим електронним газом
(враховуючи те, що концентрація вільних електронів у металі дуже висока, деякі
автори використовують термін «електронна рідина»). Для повної енергії
£ = -^ + 4> (10-27)
г г2
де параметр В визначається за допомогою формули (10.26), а параметр А за
допомогою формули, що аналогічна формулі (10.1).
Для одновалентних металів кількість вільних електронів (ІУ) дорівнює
кількості атомів, отже, параметр В визначається однозначно. На рівноважній
295
відстані
= 0 та Е = Е0, яке можна визначити кількісно:
(10.28)
Для лужних металів умова г(« гт добре виконується. Використовуючи
значення гт для лужних металів (див. табл. 10.3), за допомогою формули
(10.28) одержимо відповідні значення {-Е^ч'\ які наведені в табл. 10.4.
Експериментально енергію металічних ґраток, тобто енергію, яку необхідно
витратити для перетворення металу на вільні катіони та окремі електрони,
можна обчислити за формулою
(-£00бч-) = /м+4,
де Ім — енергія іонізації атома металу; Ь,
ного металу.
Відповідні значення (-££б4*) також наведені в табл. 10.4,
Таблиця 10.4. Енергії металічих ґраток лужних металів
(10.29)
теплота сублімації кристаліч-
Метал
Ьі
К
кДж/моль
781
639
514
(-£оКСП), 1
кДж/моль |
681
606
506
Метал
КЬ
Сз
(-£о°бЧ-Х
кДж/моль
468
451
(-£оексп'Х
кДж/моль
485
456
Значення (-Е°бц) та (-Е™™) непогано узгоджуються між собою,
незважаючи на наближений характер моделі.
Точніше розрахувати енергію зв'язку в металічних кристалах можна за
методом комірок Вігнера-Зейтца. В окремому атомі хвильова функція \|/
при г —> ©о прямує до нуля, одночасно до нуля наближається і похідна <Л|//і/г.
В металі хвильова функція на межі комірки плавно переходить на хвильову
функцію сусідньої комірки. Отже, на стінках комірки (при г = г0, де г0 —
радіус комірки) значення \|/ та й\ф/йг сусідніх комірок співпадають.
Розрахунок можна провести методом самоузгодженого поля Хартрі-Фока. При
точніших розрахунках треба враховувати поправку на кулонівське
відштовхування електронів, на обмінну взаємодію електронів з однаковими спінами
та на електронну кореляцію.
У теорії вільних електронів енергетичний спектр практично
безперервний — квазібезперервний. Справді, якщо взяти розмір кристала 1 см та
обчислити різницю енергії (А£) двох рівнів, для яких квантове число змінюється
на одиницю, то АЕ= Н2/тЬ2= 10 еВ, тобто надзвичайно мала величина.
Тому обмеження, пов'язані з тим, що кристал має скінченні розміри,
практично не позначаються на русі вільних електронів.
Проте в твердому тілі потенціал не стала величина, а змінюється
періодично від атома до атома. Періодичний потенціал (V), який створюється
у ґратках, можна зобразити у вигляді набору періодичних функцій (ряду
296
Фур'є), що залежать від періодів ґраток (а) та амплітуди (Уп)
^=2Х
81П
2ппх
(10.30)
де п — ціле додатне число.
Періодичний потенціал можна розглядати як збурення (А). Якщо
збурення немає, то розв'язком хвильового рівняння Шредінгера (8.9) є
періодична функція (див. підрозділ 8.1)
\|/ = А 8Іп (сод:) + В соз (сох) = А\ух + 2?\|/2, (10.31)
десо = 2я/А, = 27і±
Цей вираз співпадає з формулою (8.10), при 5 = 0. Тоді для енергії
справедлива формула (8.13)
Е =
_ йУ _ н2к2
(10.32)
2т 2т
яка дає квадратичну залежність енергії від хвильового числа (к) (це
зображено на рис. 10.11, с. 298, штриховою лінією). При відсутності збурення
хвильові функції іі/^зіп сох) та і|/2(соз сох) вироджені й відповідають станам з
однаковою енергією.
Згідно з теорією збурень (див. додаток 2) енергетичні рівні для двократно
виродженої системи розщеплюються на величину 2/*! 2, де
42=1^1^1/2^ =
І
1 АВ , . /о ч.
зіп(2сох) 2,ги8іп \ах= (103з)
: \А&іп((дх)УВсо$(ш)(іх =
АВх^тг Г • (~2пхЛ . 2ппх ,
о
У формулі (10.33) відповідний інтеграл не дорівнює нулю лише при умові
2/Х = п/а або пХ = 2а. З урахуванням від'ємних значень со одержимо таку
загальну умову нерівності Нх 2 нулю:
пХ = ±2а.
При п = 1 X = ±2а такх 2 = 2У1 прип = 2 Х= ±а та Нх>2 :
(10.34)
:2К2іт. д. В цих
точках функція Е - Дсо) =/(£) зазнає розриву, причому величина
енергетичної «щілини» дорівнює IVп (див. рис. 10.11). При великих значеннях X довгі
хвилі де Бройля вільно розповсюджуються у ґратках. Перший розрив у
енергетичному спектрі відбувається при X = ±2а. Хвильові функції \уї та і|/2 при
цьому мають такий вигляд:
Щ =^8ІП(С0Х) = ^8ІП^ = ^8ІП —
\|/2=5С08 —
а
2а
(10.35)
297
Функції та густини ймовірності для
них наведено на рис. 10.12, на якому
бачимо, що максимуми однієї функції
зміщені по відношенню до іншої на а/2. При
К= соті це зміщення не має ніякого
значення, тому що стани, які описуються
функціями у{ та \|/2, вироджені. Проте
з урахуванням періодичного потенціалу,
який зображено на рис. 10.12, одна
з функцій дає найбільшу ймовірність
перебування електрона у потенціальних
ямах, а інша, навпаки, на потенціальних
вершинах. Природно, що в цьому
випадку енергії для атомів, які описуються
функціями \\гх та \|/2, різні, отже,
виродження знімається за допомогою
періодичного поля.
Рис. 10.11. Залежність енергії від Ширина заборонених зон залежить від
хвильового числа для електронів амплітуди ( Уп) періодичного потенціалу
у твердому тілі (ДИВ рис 10 12). При малих амплітудах
збурююча дія періодичного поля мала, заборонені зони вузькі — це так
званий випадок слабко зв'язаних електронів. Якщо потенціальні ями глибокі,
то амплітуди великі — це випадок сильно зв'язаних електронів. Тоді
заборонені зони досить великі, а дозволені (з квазібезперервним спектром енергій),
навпаки, вузькі. Нарешті, у випадку
повністю зв'язаних електронів, коли Уп
—» °°, кожний електрон знаходиться в
своїй ямі, наприклад в атомі, і рівні
енергії дискретні, тобто «заборонені»
зони займають весь енергетичний
простір, а «дозволені»
перетворюються на лінії.
Випадок сильного зв'язку можна
пояснити, виходячи не з квазібезпе-
рервного спектра (збільшення
амплітуди), а від дискретного. Дійсно, в
кристалі потенціальна яма має хоч високу,
але скінченну висоту. Електрон, що
має хвильові властивості, може
внаслідок тунельного ефекту (див. підрозділ
8.1) проникати крізь потенціальний
бар'єр навіть тоді, коли його енергія
менша від висоти цього бар'єра.
Просування електронів крізь бар'єр при-
\;/Л;/К|/\|І
/Ті /і\ /!\ /і\™
І¥і
І
І
Рис. 10.12. Хвильові функції і|/,
та \|/2 і періодичний потенціал V
у твердому тілі
298
зводить до розмивання енергетичних рівнів та появи дозволених зон у випадку
сильного зв'язку.
Нарешті, утворення зонної моделі можна пояснити і на базі методу МО.
При створенні МО діє правило: кількість створених МО дорівнює кількості
вихідних АО (див. розділ 9). Два атоми при з'єднанні утворюють дві МО
(мається на увазі, що кожний атом утворює тільки одну АО) — зв'язуючу
та антизв'язуючу. Чотири АО утворюють чотири МО, шість АО — шість МО
(див. рис. 10.13) і т. д. Нарешті, 6-Ю атомів утворять 6-Ю МО, тобто квазі-
безперервний спектр. Кожний атомний рівень вихідного атома утворює свою
енергетичну зону з квазібезпербрвним спектром енергії. Ці зони можуть
перекриватися, якщо вихідні атомні рівні розташовані енергетично недалеко
один від одного, в протилежному випадку з'являються заборонені зони. *
Розглянемо структуру зон (так
звані зони Бріллюена), що
визначається умовою відбиття
(рентгенівських хвиль або електронів) Вуль-
фа-Брегга. Для к.г.ц. та к.о.ц.
структур зонами Бріллюена є кубоокта-
едр та ромбододекаедр відповідно.
Зони Бріллюена будуються в А:-прос-
торі хвильових векторів (так
званому «оберненому» просторі), тому
вони не співпадають з комірками
Вігнера-Зейтца, які будуються у
звичайному просторі. Зокрема, для
к.г.ц. структури комірка Вігнера-
Зейтца — ромбододекаедр, а для
к.о.ц. — кубооктаедр, тобто
структури (к.г.ц.) та (к.о.ц.) ніби
обмінюються багатогранниками при
переході від комірок Вігнера-Зейтца до
зон Бріллюена.
Є8В-
Є88&
Рис.
10.13. Утворення зонної структури
при об'єднанні атомів
Якби гранична зона була сферою, то залежність густини станів у зоні від
енергії була б такою самою, як і для квазібезперервного спектра вільних
електронів, тобто збільшувалася як Е (див. формулу (10.25) та рис. 10.10)
тільки при Е = Е межі зониДЕ) обривалася б. Проте зона
Бріллюена—багатогранник і залежністьДЕ) від Е дещо інша. При малих значеннях Е (малих
к) форма багатогранника не має значення, залежністьДЕ) від і? така, як і для
вільних електронів. Після того як сфера наблизиться до меж зон Бріллюена
(цей момент відповідає максимумуДЕ) у даній зоні), починають
заповнюватися «куточки» багатогранника і густина станів поступово (не стрибком)
зменшується до нуля (рис. 10.14, с. 300).
Оскільки внутрішні орбіталі атомів практично не перекриваються, то їх
рівні майже не розщеплюються і в утворенні зонної структури (як і в утво-
299
АЕ)\
О Е0 ЕА Ек Ев Ес Ен Е
Рис. 10.14. Залежність густини станів від енергії згідно із зонною теорією
ренні хімічного зв'язку) беруть участь лише валентні електрони. Відповідна
зона називається валентною. Інформацію про зонну структуру кристалів
можна одержати з рентгенівських спектрів. Переходи з внутрішніх рівнів,
енергії яких фіксовані, у валентні зони (та назад) відповідають за енергією
м'якому рентгенівському випромінюванню й описують енергетичний
спектр зони (див. рис. 10.14).
Залежно від зонної структури тверде тіло є провідником або ізолятором.
Якщо зона заповнена частково, то перенесення електронів здійснюється
легко і тверде тіло має металічну провідність (на рис. 10.14 поверхні Фермі
відповідає енергія ЕА). Для металів валентна зона є зоною провідності. Якщо
валентна зона повністю заповнена (Е = Ев) і порожня зона, яка відповідає
збудженим станам атома, відділена від неї широким інтервалом (ДЯ = (Ес -
- Ев) » 0 на рис. 10.14), то таке тверде тіло є ізолятором.
Між провідниками та ізоляторами існуюють тверді тіла, які називаються
напівпровідниками. Якщо поверхня Фермі знаходиться на дні зони (Е0 та Ен
на рис. 10.14), то це означає, що кількість носіїв заряду—електронів — мала
і речовина є електронним напівпровідником «-типу. Для майже заповненої
зони (Ек на рис. 10.14) щільність вакантних станів/(£) мала, якщо робити
відлік від верхньої межі зони. Тому цей випадок є дзеркальним
відображенням попереднього — малої кількості електронів на дні зони. Вакансії у
майже повністю заповненої зони називають «дірками». Дірки ведуть себе
як позитивні квазічастинки із зарядом, що дорівнює (за величиною) заряду
електрона. Оскільки концентрація їх невелика, то речовина, яка містить дірки,
є напівпровідником, але не електронним, а «дірковим» (р-типу). Нарешті,
якщо ширина забороненої зони між заповненою валентною і вакантною
зонами провідності мала, то електрони можуть збуджуватися, наприклад, під
дією теплової енергії, і переходити з одної зони до іншої. Провідність
з'являється за участю електронів у зоні провідності та дірок у валентній зоні. Така
речовина називається власним напівпровідником.
Розглянемо різні тверді тіла з точки зору їх зонної структури. В елементів
І групи періодичної системи 5-зона заповнена наполовину, тому що число
300
станів у зоні з урахуванням спіна дорівнює 2И, де N— число атомів, а
кожний атом має по одному електрону. Отже, ці елементи — типові метали.
Електронні конфігурації інертних газів {$2рв) утворюють повністю
заповнені валентні зони, тому кристали інертних газів — ізолятори. В іонних
кристалах (типу ИаС1 чи СзСІ) електрони локалізовані на іонах, які мають
конфігурацію інертних газів. Цілком заповнені валентні зони катіонів та аніонів
розділені великими енергетичними «щілинами» (до 10 еВ), тому іонні
кристали — ізолятори. Проте, якщо іонний кристал розплавити, то іони стають
рухомими і з'являється так звана іонна провідність.
Атоми елементів II групи періодичної системи мають заповнену
.^-оболонку, але вони — метали. Це пов'язано з тим, що я-зона перекривається
з близько розташованою/7-зоною (нагадаємо, що саме близькість $•- та/?-
рівнів в атомах (див. підрозділ 9.2) дає цим елементам можливість стати
двовалентними внаслідок збудження валентності).
У ковалентних кристалах електрони локалізовані на зв'язках. Початкове
розщеплення на зв'язуючі та антизв'язуючі рівні при утворенні двоцентро-
вих двохелектронних зв'язків у них настільки велике, що зв'язуючі та
антизв'язуючі рівні розщеплюються на окремі зони, між якими залишається
велика енергетична щілина. Зона зв'язуючих станів (,у/?3-зона) заповнена
повністю, а антизв'язуючих вільна, отже, ковалентні кристали типу алмазу —
ізолятори.
Введення в кристали елементів IV групи домішок елементів V групи,
наприклад Стибію в Силіцій, перетворює їх на ^-напівпровідники, тому що
чотири електрони з елемента V групи витрачаються на утворення
ковалентних зв'язків, а зайвий (п'ятий) надходить для «колективного використання».
Навпаки, домішки елементів III групи, наприклад Оа в Ое, перетворюють
кристали елементів IV групи на/7-напівпровідники, тому що для утворення
чотирьох ковалентних зв'язків елементам III групи не вистачає одного
електрона і вони використовують його з валентної зони, внаслідок чого в цій зоні
утворюються «дірки».
Елементи IV групи можуть і самі стати напівпровідниками. Це має місце
під час руху вниз у IV групі. В алмазу ширина забороненої зони ДЕ = 7,0 еВ,
8і — 1,21; Ое — 0,75; 8п (сірого) — 0,08 еВ. Отже, АЕ при переході від
вуглецю до олова різко зменшується. Тому алмаз — типовий ізолятор, германій
і силіцій — власні напівпровідники, олово має дві модифікації: сіре —
ковалентний напівпровідник, але з достатньо високою провідністю, та біле, що
має металічні властивості. Свинець уже є типовим металом.
Збільшення металічних властивостей елементів спостерігається при русі
вниз по групі і в елементів V, VI груп. Отже, перехід від ковалентних
структур до металічних може здійснюватися внаслідок звуження заборонених зон
і появи вакантних рівнів близько до заповнених.
Ще один фактор, який сприяє «металізації» напівпровідникових
систем, — це нагрівання. Кількість носіїв струму у власних напівпровідників
пропорційна фактору Больцмана (~е~АЕ/кТ), отже, електропровідність
301
напівпровідників різко збільшується із підвищенням температури. В рідкому
стані вона досягає значної величини, яка характерна для провідності
«металічного» типу.
Для перехідних елементів великих періодів характерні незаповнені сі-
орбіталі в атомів і, відповідно, незаповнені */-зони в кристалічному стані,
отже, всі перехідні елемент?- — метали. Наявність ^-орбіталей зумовлює ряд
специфічних властивостей ^-елементів у кристалічному стані.
Особливості, пов'язані з роллю я?-орбіталей, мають місце вже в елементів
ІВ групи, для якої </-зона щойно заповнилася. З даних, наведених у табл. 10.1
та 10.3, бачимо, що радіуси однозарядних іонів ІВ-елементів не набагато
менші, ніж металічні (на відміну від лужних металів, для яких гі« гт). Отже,
в металах групи ІВ ^/-оболонки перекриваються та взаємодіють, на відміну
від лужних металів, у яких іони відділені один від одного досить товстим
шаром із вільних електронів. Тому метали типу лужних називаються
«відкритими» (для електронного газу), а метали типу ІВ, де «вільного місця»
мало, — «закритими». Взаємодія електронів на </-орбіталях зміцнює
міжатомний зв'язок у металах ІВ-групи, наприклад, теплота атомізації міді —
340 кДж/моль майже в чотири рази більша, ніж калію — 88 кДж/моль.
Метали ІА та ІВ груп відрізняються і за магнітними властивостями.
Лужні метали слабко парамагнітні. їх магнітні властивості зумовлені в
основному магнітними властивостями вільних електронів. У зоні електрони
в основному спарені. Електрони поблизу рівня Фермі можуть переміщатися
на вакантні рівні, але не дуже високо, тому що це призведе до збільшення їх
кінетичної енергії. Розпарування частини електронних спінів поблизу рівня
Фермі і зумовлює слабкий парамагнетизм електронного газу, який частково
компенсується діамагнетизмом іонів остова та діамагнетизмом при русі
вільних електронів. У металів ІВ групи діамагнітний внесок іонів остова, які
мають великі розміри, значно збільшується, отже, мідь та її аналоги —
діамагнітні системи.
Розглянемо тепер перехідні елементи, в яких ^-оболонка заповнена
частково. Нарис. 10.15 наведено теплоти атомізації (Ь5) елементів великих
періодів. Включення </-орбіталей у міжатомний зв'язок різко зміцнює кристалічні
ґратки перехідних металів. Для оцінки кількості електронів, які беруть участь
у міжатомному зв'язку, Л. Полінг використав дані міжатомних відстаней
аналогічно тому, як це було зроблено (див. підрозділ 9.3) для оцінки порядку
зв'язку С—С. Міжатомні відстані зменшуються зі збільшенням порядку
зв'язку. Для металів цю залежність можна апроксимувати такою формулою:
гп =Г!-с1ое«, (10.36)
де п — порядок зв'язку; гх — міжатомна відстань при п = 1.
Полінг запропонував у формулі (10.36) значення с = 0,3 (для зв'язку С—С
стала с дорівнювала 0,353) та визначив порядок зв'язку між атомами у кріста-
лічних ґратках таким чином:
п = ьІК9 (10.37)
302
де К— координаційне число; V — «металічна» валентність — кількість
електронів, що беруть участь у зв'язку.
У довгих періодах значення гп закономірно зменшується від І до VI
групи (див. табл. 10.3, с. 295). Це відповідає збільшенню V від 1 до 6. Далі
гп = соті для елементів VI—VIIIV = 6 за Полінгом. Нарешті, у ІВ групи і далі
гп збільшується і, відповідно, зменшується V.
Високі значення V, які досягають 6, неможливі для суто металічного
зв'язку, тому що іонний остов повинен містити іони типу М , М та інші.
Відірвати електрон від багатозарядного іона надзвичайно важко, тому в
перехідних металах слід припустити наявність локалізованих ковалентних
зв'язків. (Аналогічно можна було б пояснити появу ковалентного зв'язку в
іонних кристалах при збільшенні заряду іонів. Наприклад, у ЬіР перенесення
одного електрона вигідне; у ВеО двократна іонізація атома Берилію та
захоплення двох електронів атомом Оксигену вже набагато менше ймовірні;
трикратна іонізація атома Бору та захоплення трьох електронів атомом
Нітрогену не ймовірні, для нітриду Бору характерний майже повністю
ковалентний зв'язок.)
Справді, кількість вільних електронів у металів (не тільки перехідних), як
правило, не перевищує двох (часто близька до одиниці). Всі інші електрони
беруть участь в утворенні ковалентних зв'язків. Крім високої міцності
кристалічних ґраток (див. рис. 10.15), у перехідних металів виникає крихкість, яка
характерна для ковалентних спрямованих зв'язків.
ЕХ9 кДж/моль
Порядковий номер
Рис. 10.15. Залежність енергії атомізації елементів великих періодів
від порядкового номера
303
Схема валентностей Л. Полінга дає явно завищені (5 та 4) значення V для
металів ІВ- та ІІВ-груп, судячи із теплот атомізації ІВ- та ІІВ-елементів. Крім
того, вона не пояснює збільшення міцності ґраток елементів V—VIII груп
при переході від першого великого періоду до другого та третього,
наприклад у ряду Сг —> Мо —> \У значення Ья збільшується: 400 < 664 < 840 кДж/моль;
в ряду Ре —> Ки —> 08, відпооідпи, 416 < 651 < 786 кДж/моль. В. Юм-Розері
та деякі інші автори запропонували уточнити систему Полінга. Вони
вважали, що «металічні» валентності близькі до валентностей відповідних атомів
у неорганічних сполуках. Для елементів І—V груп валентність співпадає
з номером групи і з валентністю за Полінгом. В елементів VI—VIII груп між
першим та іншими періодами існує відмінність. Так, для Хрому більш
характерна валентність 3, а для Молібдену та Вольфраму — 6; для Феруму — 2
та 3, а для Осмію — 4,6 та 8; Нікелю — 2, а Платини—4. Для мангану
характерна валентність 2 (іони, Мп дуже стійкі) і, відповідно, манган має теплоту
атомізації меншу, ніж хром та залізо (для цих елементів характерна
валентність 3). Метали групи ІІВ—двовалентні (на відміну від V = 4 за Полінгом),
метали групи ІВ виявляють валентність від одиниці до трьох, їхні ґратки
міцніші, ніж лужних металів, але значення і; = 5 за Полінгом явно завищене.
Оскільки, починаючи з VI групи періодичної системи, електрони в
утворенні зв'язків участі не беруть, вони повинні бути локалізовані на атомних
орбіталях. Згідно з правилами Гунда (див. підрозділ 8.4), електрони в атомах
намагаються бути неспареними. Тому перехідні метали, починаючи з VI
групи, повинні бути парамагнетиками. Експеримент це підтверджує.
Магнітний момент (у магнетонах Бора) хрому дорівнює 0,22, мангану — 1,22,
заліза—2,22. Збільшення магнітного моменту при переході від хрому до
мангану та від мангану до заліза відповідає збільшенню кількості електронів у цих
атомах. Повна кількість електронів на атом така: Хрому — 6, Мангану — 7,
Феруму—8, отже, середня кількість зв'язуючих електронів на атом дорівнює:
6 - 0,22 = 7 -1,22 = 8 - 2,22 = 5,78, що близько до граничного значення V = 6.
Максимальне значення магнітного моменту 2,44|іі5 досягається у сплаві
(22 % Со — 78 % Ре), тобто при 8,22 електронах на атом (8,22 - 2,44 = 5,78
зв'язуючих електронів, як у чистих (Сг, Мп, Ре) металів). Величина 2,44 — це
максимально можлива кількість електронів, яку можна розташувати на
незв'язуючих АО без спарення, тобто це кількість незв'язуючих й?-орбіталей
у кристалічних перехідних металах (для ізольованих атомів дробові числа
орбіталей неможливі, але у конденсованих тілах дробові числа — це
усереднення станів різних атомів). Після максимуму відбувається зменшення
магнітного моменту. В атома Кобальту в валентній оболонці 9 електронів,
з них 5,78 є зв'язуючими, а 2,44 можна розташувати на АО без спарення,
решта 9 - (5,78 + 2,44) = 9 - 8,22 = 0,78 електронів залишається також на
незв'язуючих АО, але вже з протилежним знаком спінового магнітного
моменту. Отже, сумарний магнітний момент металічного кобальту повинен
бути 2,44 - 0,78 = 1,66, що відповідає експериментальному значенню. При
переході до нікелю магнітний момент зменшується на одиницю (кількість
304
електронів збільшується на одиницю) і становить 0,66|ід. У міді всі
електрони в ^/-оболонці спарені і вона діамагнітна. Не важко розрахувати, що
магнітний момент повинен зникати при 10,66 електронах на один атом.
Експеримент дає близьке значення — магнітний момент зникає у сплавах N1 — Си
при вмісті міді 0,62 молярних часток. Магнітний момент паладію дорівнює,
як і для нікелю, 0,66\хв. При насиченні паладію воднем парамагнітна
сприйнятливість зникає при вмісті водню 0,66 молярних часток.
Якщо частинки речовини (атоми, молекули, іони) мають власні магнітні
моменти (діамагнетики не мають), то речовина може бути: а) парамагнети-
ком при розупорядкованих магнітних моментах (часто у парамагнетиках
кількість носіїв магнітних моментів невелика і вони знаходяться в
діамагнітній матриці, наприклад, іони ^/-металів у комплексних сполуках, іони
Сг в А1203 (рубін); іони перехідних металів в алюмосилікатній матриці
(металізовані цеоліти) тощо); б) феромагнетиком, коли магнітні моменти
упорядковані в просторі і зорієнтовані в однаковому напрямку в межах
визначених зон — доменів; в) антиферомагнетиком, коли магнітні
моменти впорядковані антипаралельно.
Для деяких перехідних металів характерний прояв феромагнетизму, але
при нагріванні до температури, яка називається точкою Кюрі в (в = 769 °С —
для заліза, 360 °С — для нікелю, 1075 °С—для кобальту) впорядкована
система магнітних моментів руйнується і речовина стає звичайним парамаг-
нетиком. Нагрівання (до точки Нееля) руйнує впорядкування і в антиферо-
магнетиків.
При розгляді періодичної системи бачимо, що метали займають більшу її
частину. У зв'язку з цим цікаво розглянути, як вони взаємодіють один з одним.
Для металів характерні щільно упаковані структури. Якщо атоми двох
металів мають приблизно однакові розміри і не дуже відрізняються за
хімічною природою, то вони утворюють тверді розчини, наприклад сплав
А§—Аи (міжатомні відстані А§—А§ — 0,2883 нм, а Аи—Аи — 0,2877 нм).
При невеликих відхиленнях у розмірах атомів тверді розчини можуть
утворювати так звані надструктури (інша назва — сполуки Курнакова).
Наприклад, у сплавах Си—Аи утворюються надструктури СиАи та Си3Аи,
в яких впорядковане розташування атомів знімає напруги в ґратках, які
виникають внаслідок невідповідності розмірів атомів вихідних компонентів.
Атом Алюмінію, однак, має однакові розміри з атомами Ауруму та
Аргентуму, а в системах Аи—А1 та А§—А1 немає безперервного ряду
твердих розчинів, а утворюються інтерметалічні сполуки. В. Юм-Розері
довів, що, крім геометричного фактора, важливу роль відіграє електронний.
Інтерметалічні сполуки з однаковою кількістю електронів на один атом (/V)
мають подібні кристалічні структури (табл. 10.5). Наприклад, для Р-фази
лг г> гг 2 + 1 3 л лі 3 + 3_3.~ с 5±4_3. ,
N дорівнює: Си£п ^5^ = 7о А§3А1 —т- = Х' Си53п —т~-^, дляу-фази
г^ с 5 + 16 21 о лі 9 + 12,21. А с 31+-32 _ 21. .
Си58п8 ^^ = ^;Си9А14 -^--^, А§3іЗп8 39 ~ЇЗ'ДЛЯ є"Фази:
305
А пА 1 + 6_7.„ с 3 + 4_7.А А1 5+9_7
А§Са3 -^—4'Сиз8п ~4~~~4'Аи5А1з "^_~4-
Таблиця 10.5. Електронні інтерметалічні сполуки
Фаза
Р
У
Є
Кількість електронів
на один атом
3
2
21
13
7
4
Інтерметаліди
Си2п, СиВе, А§СМ, Аи2п, А&А1, Си3А1, Си58п
Си52п8, А§5Н§8, Аи52п, Си9А14, А§9А14, Си9Са4,
Си3,8п8, А§3]5п8
Си2п3, А§Сс13, АиН§3, Аи2п3, СиВе3, Си28п,
Си3Се, А§5А13, Аи5А13
Правила Юм-Розері були пояснені Г. Джонсом при розгляді зонної
структури відповідних фаз.
Кількість різних інтерметалідів дуже велика. Відзначимо так звані
солеподібні. Ці інтерметаліди утворюються між елементами з помітною
відмінністю в електронегативностях. У цих сполуках елементи мають
«нормальні» значення валентності, наприклад, М§28і, М§28п, Са2Рсі, Іл3Ві,
Са38Ь2 та інші. Такі інтерметаліди можна вважати проміжними сполуками
між інтерметалідами металів, які близькі за своїми властивостями, та
іонними сполуками. До того ж, у цих сполуках є і ковалентні зв'язки. Всі три типи
зв'язку можна спостерігати в інтерметаліді КаОа. Іон Натрію відносно
легко віддає свій електрон, що дає можливість атому Галію добудуватися до
конфігурації^/?3 та створити ковалентну тетраедричну структуру. Ця
структура, однак, побудована не з нейтральних атомів, як у силіцію чи германію,
а з іонів Оа~. Іони Иа , які віддали електрон, утримуються в ґратках завдяки
кулонівському притяганню, тобто силами іонного типу.
Як уже відмічалося на початку цього розділу, в природі рідко
реалізується який-небудь зв'язок у чистому вигляді. Це ілюструється схемою,
наведеною на рис. 10.16, де зображені поступові переходи між різними типами
зв'язку в конденсованих системах. При цьому слід зазначити два
найважливіші фактори:
1) при зменшенні кількості доступних вакантних орбіталей
спостерігається перехід від металічного зв'язку до ковалентного і потім до
міжмолекулярного (ковалентний залишається як внутрішньомолекулярний);
2) зі збільшенням різниці електронегативностей усі три типи зв'язків
поступово переходять в іонний.
Дані по теорії хімічного зв'язку, в тому числі спектроскопії, дуже важливі
взагалі і, зокрема, при статистичному підході до розгляду рівноважних
систем, до якого переходимо у наступному розділі.
306
Зменшення кількості доступних вакантних орбіталсй
Типові
метали
Власні
Перехідні напівпровідники
метали (8п, Оа, 8і)
Графіт
Зменшення делокалізації
електронів
Типові
ковалентні структури
Крис-
Шаруваті Ланцюгові тали
структури структури гало-
(Аз, 5Ь) (5,8е) генів
Поступовий перехід
ковалентного зв'язку
Інертні гази
в
конденсованому стані
(дисперсійні
сили)
Сплави
00
о\
є
І л
я
»
я
8
°
я
Із
°
Кс
Електронні
Власні
напівпровідники
(II 1-У; 1І-У1)
І
Галогеніди
важких металів
(А§1)
.1
Силікати
к ч від міжмолекулярного
ч до внутрішньо- Сполуки
\ молекулярного полярних молекул
4 (диполь-дипольна
4 \ взаємодія)
/
\
А
Інтерметаліди
Солеподібні
Оксиди
важких металів
і
Оксиди
лужноземельних
металів
І
Оксиди
лужних металів
X Сполуки
з водневим
зв'язком
/
Аміакати,
кристалогідраї и
(іон-дипольна
взаємодія)
/
Шаруваті
іонні сполуки
(С(ІІ2)
Типові
іонні
сполуки
Рис. 10.16. Хімічний зв'язок у конденсованих системах
Розділ 11.
СТАТИСТИЧНА ТЕРМОДИНАМІКА
Частинок мало —
Явища ймовірні.
Багато стало —
Воює закономірні.
11.1. Розподілення Больцмана
Проблема, яка розглядається у статистичній термодинаміці, — це
розподілення частинок за енергіями. Для будь-якої системи (для простоти поки
що розглядаємо однокомпонентні системи) існує
набір енергетичних рівнів: є0; є^ є2; є3;... є,;...
тощо (рис. 11.1). Рівень з найнижчою енергією
(є0) називається основним. Для
однокомпонентних систем можна прийняти енергію основного
стану за нуль відліку, тобто вважати є0= 0.
Кількість частинок на певному рівні (єу)
позначимо через Иі . Систему вважаємо закритою,
тобто виконується умова
2^=АГ = со/иЛ (И.1)
і
де N— повне число частинок (стала величина).
Больцман Людвіг Вважаємо також, що загальна внутрішня
(1844—1906), Австрія енергія системи (Ц) є сталою величиною
(ізольована система)
£єД.=[/ = а^. (П.2)
і
При абсолютному нулі заселений лише основний рівень; зі збільшенням
температури поступово починають заповнюватися збуджені стани з
більшою енергією. Фактором, що, з термодинамічної точки зору, сприяє
заселенню збуджених рівнів, є ентропія (нагадаємо, що при абсолютному нулі
за шкалою Кельвіна ентропія, згідно з постулатом Планка (1.60), дорівнює
нулю). Заселення збуджених рівнів веде до збільшення ентропії, тому що
кожна частинка має вибір при розташуванні на енергетичних рівнях, інакше
кажучи, внаслідок того, що частинки не розрізняються за своїми
властивостями, перестановки їх між рівнями ведуть до збільшення ентропії. Врахувати
це можливо за допомогою формули (1.55) для ентропії змішування (тут
відбувається «перемішування» частинок між рівнями енергії)
А5 = -^л:/1пх/. (11.3)
В даному випадку '
Хі^Иі/И. (11.4)
308
^ = 5-50 = 5, (11.5)
оскільки, згідно з постулатом Планка, 50= 0.
Вводячи сталу Больцмана
-23
де Я—універсальна газова стала; ИА — стала Авогадро (к = 1,3 8 • 10 Дж/К),
маємо К = кИА. Переходячи у формулі (11.3) від одного моля до довільної
кількості частинок (И) і враховуючи формули (11.4) та (11.5), одержимо для
ентропії:
Я^СЛПпЛГ-^.Іп^.). (Ц.6)
4 Формулу (11.6) можна також от-
е римати, спираючись на постулат
* Больцмана, який дає зв'язок між ен-
тропією та термодинамічною ймо-
є вірністю (Щ
Єо
8 = кІпт (11.7)
Рис. 11.1. Набір енергетичних Термодинамічна ймовірність —
рівнів частинки це число мікростанів, якими
реалізується макростан системи. Мікростани визначаються своєю енергією — є,-.
Перестановки частинок на одному енергетичному рівні не вносять змін у
макростан системи внаслідок нерозрізненості мікрочастинок. Коли всі
частинки у системі перебувають в основному стані при 0 К, цей стан
реалізується єдиним можливим способом і тому для нього IV- 1. Це дає 50=0,
що узгоджується з гіпотезою Планка для абсолютних ентропій.
За Л. Больцманом термодинамічна ймовірність
і
Зазначимо, що згідно з формулою (11.8), коли всі частинки перебувають
в основному стані, то Л^ = ІУ0= N і IV = 1.
Підставляючи (11.8.) у (11.7) і користуючись формулою Стірлінга для
обчислення факторіала при великих N
N1
•№■
одержимо
5 = /с(1пЛГ!-^1п^,!) = к(ЛГ1п^-Л^-^Л^1пЛГ1. + £^І.) =
= к(ЛПпЛГ-£лГ,.1пЛГ,.)
І
у повному узгодженні з формулою (11.6).
309
Для диференціала ентропії, згідно з формулою (11.6), маємо
Ж^-кЦіпЩ + Мі^Щ^-к^ігіМі + ^Щ. (11.9)
Для системи з II = соті умова рівноваги дається співвідношенням
(1.110) с18= 0. Враховуючи вираз (11.9) та скорочуючи на (-«•), отримуємо
Х(1пДГ- +1)^=0. (11.10)
і
Рівності (11.1) та (11.2) дають додаткові умови (якщо N = соті, <ІИ = 0,
а коли V = соті, (Ш= 0)
2>Г=0 (11.11)
та
£є^=0. (11.12)
і
Використовуючи метод невизначених множників Лагранжа, домножи-
мо рівності (11.11) та (11.12) на певні параметри ос та Р і додамо до рівністі
(11.10)
20пЯІ +1)^. + %аЩ + ^РєЖ = Х(1п ^ +1 + а + Ре/)й^/ =0. (11.13)
/ / / /
Оскільки <Ш( Ф 0, то нулеві дорівнює вираз у дужках, тобто
1пМ+1+а + Рє/ = 0. (11.14)
Це дає для кількості частинок в /-му стані
Мі=е-1-ае*е'=Ве*Еі. (11.15)
Знайдемо тепер фізичний зміст констант Р та В (або а). Якщо продифе-
ренціювати рівняння (11.2) як добуток, то
Ш = ^Щ +Х^єі- <11Л6)
і і
Перша сума відповідає зміні кількості частинок на різних енергетичних
рівнях під час нагрівання (охолодження) системи, але самі рівні залишаються
незмінними: тут змінна температура, а об'єм фіксований, тобто
^Щ^ІсіЩу. (11.17)
і
Друга сума у (11.16) відповідає зміні саме енергетичних рівнів при
стисненні (розширенні) системи, частинки ж залишаються на тих самих рівнях,
тобто тут Т = соті, а об'єм змінний. Згідно з класичною термодинамікою
(див. формулу (1.64))
(<Я/)к=Г</5,
звідки
^ = ^£М^- (НЛ8)
і
310
Згідно з рівностями (11.9) та (11.11) маємо
<Ю = -кХ(1пЛІ + ІЩ =-к^(-а-$єіЩ =крХМ^,- (П-19)
І І
Зіставляючи (11.18) та (11.19), одержимо
Р = ^, (П-20)
звідки
Мі=Ве-Є!'кТ. (П-21)
Формула (11.21) має назву розподілення (за енергіями) Больцмана.
Константу В можна виразити через кількість частинок в основному стані.
Якщо є,- = є0 = 0, то е~е°/кТ = 1 і Щ = И0 = В, отже,
#/ = лг0«геА7'. (и-22)
Другий спосіб вираження В — це використання умов нормування (11.1),
згідно з якими
звідки Я = т^;
Величина
ЛГ = 5>,=2?5>-^ = Я£,
М.=Ке-^г_ (1123)
е = £е-єлг
(П-24)
має назву суми станів або статистичної суми. Формули (11.23) та (11.24)
правильні, коли стани невироджені. Для вироджених станів
і, відповідно,
X, =%№-**". (Н-26)
Величина & характеризує ступінь виродження і-го стану й має назву
статистична вага. Вироджені стани відрізняються не енергією, а якоюсь
іншою характеристикою. Наприклад, в атомі орбіталі можуть мати різну
форму, але однакову енергію. Через статистичну суму можна виразити
основні термодинамічні функції.
311
11.2. Зв'язок термодинамічних функцій
зі статистичною сумою
Для ентропії, згідно з формулами (11.6) та (11.23), маємо
5 = к
ЛПпЛГ-^^Іп^.
= к
лпп#-]£#/(Ьлг-ье--^)
= к
N 1п N - 5) ЛГ- 1п N + £ ТУ,- 1п б + -і——
Знайдемо похідну від статистичної суми за температурою
$1-*-
гіІкТ\ _££
1
І«*
-Є//кГ
кГ
(11.27)
(11.28)
Тепер знаходимо вираз для внутрішньої енергії через статистичну суму
" = £е,Д. =^гЛе-^т =^еіЄ-^т =
N
Ш) кТ^шАд-Ш\.
[дТ)у
[** >
Для вільної енергії Гельмгольца
А = ІІ-Т5 = і;-кМТ\^-тЦ; = -кИТ\п(2.
, то маємо можливість перевірити результат
(11.29)
(11.30)
Оскільки «5 = - —
(11.27) 1 дТ
у повному узгодженні з формулою (11.27).
У формулах (11.21)—(11.30) N— це число частинок у системі. Якщо
розрахунки вести на 1 моль, то N = Л^ і кИА = К.
Тоді формули (11.27)—(11.30) мають вигляд:
5-Л1пЄ + ^ = ЛЬС + і?гГ^
и = кт
І аг У
А=-кт\й^.
(11.31)
(11.32)
(11.33)
Інші термодинамічні функції та параметри системи розглядатимуться
далі для конкретних статистичних сум та видів руху. Почнемо з
поступального руху.
312
11.3. Поступальний рух
Для одновимірного поступального руху енергія квантується і задається
формулою (8.14). Індекс сумування, згідно з вищенаведеними формулами,
позначений через «/», відповідно, формула (8.14) набуває вигляду
*-$■ (,и4>
де і приймає значення 0,1,2,3,....
Згідно з формулою (11.34)
А2/2
а,ост.=2>8т"сГ- (ц.з5)
Для поступального руху енергетичні рівні розташовані дуже близько
один до одного і утворюють квазібезперервний спектр. Оцінка для різниці
енергій сусідніх станів АЄ;= Н /&тІ дає, коли / = 10" м та т = 10" кг,
Ає = 5,5-10~28 Дж. Водночас кТ= 4,14-10"21 Дж (при 300 К). Отже, маємо
Ає « кТ. Для квазібезперервного спектра суму (11.35) можна замінити
інтегралом, що дає
2пост.=ф Ш2кТЛ- (Н.36)
о
оо у—
Табличний інтеграл Ге""* сіх = ^-а~У2, отже, з (11.36) маємо
Рух у кожному з напрямів х,утаг незалежний, отже, для кожного з
напрямів матимемо той самий результат, а для тривимірного простору
_(2ттгкТ)ш 3 _(2шпкТ)ш
Цюст. 7з 1 " 7з х)' (11.3»)
де у — об'єм, що припадає у просторі на одну частинку.
Результат (11.38) був би правильним, якби кожна частинка рухалася
в межах своєї частки простору, однак для газу доступний весь об'єм ємності,
у якій він знаходиться, отже, частинки обмінюються «комірками» одна з
одною. Розглянемо це спочатку на прикладі двох частинок. Позначимо частину
статистичної суми, що не залежить від об'єму, літерою #, тобто
0юсіі = ?у. (Н39)
Для двох частинок об'єм системи У= 2и і для всієї системи
Єсист. = 01& = №)№) = (д2о)\
З іншого боку, через нерозрізненість частинок невідомо, яка з них
займає «свій» об'єм, а яка «чужий», тобто результат потрібно поділити на
кількість перестановок з двох частинок — 2!
313
У загальному випадку для системи Л^частинок
ЛГ! ґ„
Є І
враховуючи, що загальний об'єм системи У=М;, а також формулу Стірлінга.
У розрахунку на одну частинку
_ (2пткТ)
Єпост. = V* = ~ ТТ2-*0- О Ь4°)
п
Таким чином, якщо врахувати можливість для кожної частинки рухатися
по всьому об'єму, то одержаний вище результат для бпост> (11.38) треба
збільшити в є разів. Зазначимо, що в деяких випадках слід користуватися все ж
таки формулою (11.38), наприклад, при застосуванні статистичного підходу
для опису елементарного акта хімічної реакції (теорія перехідного стану
в хімічній кінетиці, див. підрозділ 14.4), де частинка розглядається саме в
своїй «комірці» простору.
Знайдемо тепер внутрішню енергію, зумовлену поступальним рухом.
Згідно з формулами (11.40) та (11.32) маємо
и = кт2-§г[1п(аТУ2)]у=КТ22Т = 2КТ' (1141)
де а — температурно незалежна частина статистичної суми.
Відповідно, молярна теплоємність при постійному об'ємі
с-(!)Л* (1,'42)
Для вільної енергії Гельмгольца маємо
А = -ЯТ\п
(2пткТ)
н3
(11.43)
Звідки можемо знайти тиск ідеального газу, зумовлений поступальним
рухом.
р = 4^\ =яТ-2-ЩЬГ)т=—9 (И.44)
де Ь — частина статистичної суми, що не залежить від об'єму (в неї входить
також \ІИ, оскільки У= і;АГ).
Таким чином, для ідеального газу внутрішня енергія залежить лише від
температури, згідно із законом, встановленим емпірично Джоулем
(підрозділ 1.2). Тиск газу підпорядковується рівнянню Менделєєва-Клапейрона.
Якщо виразити КТ з рівняння (11.41) та підставити його у (11.44),
одержимо
211
Р = Іу> (П.45)
314
тобто тиск — це густина кінетичної енергії (в одиниці об'єму простору).
У формулі (11.40) об'єм V можна замінити на тиск
" = ^г = -"і£-=—• О1-46)
Отже,
(2пткТ) „т
0с^=К ,, еЦ-. (11.47)
п Р
(У стандартному стані/? = 1 атм.)
Для ентропії поступального руху, згідно з (11.31) і (11.47),
(2птк)
8 = К\п
Н3
+ ^КІпТ + Я + ЯІпк + К\пТ-
2
-Д1пр + |^ = |діпГ-Д1пр + 5г (11.48)
Зіставляючи з класичним виразом, згідно з формулами (1.65) і (1.21),
маємо
Срс1Т = Ш = Ш + Усір,
звідки:
Т Т у Т р и'
8=СрІпТ-Мпр + 8у. (11.49)
Рівняння (11.49) повністю відповідає рівнянню (11.48), оскільки при
цьому
Сг=|д, згідно (11.42), Ср=Су+К = ^К.
У класичному випадку константу інтегрування 8у знаходять за допомогою
термохімічних даних та рівняння (1.60) (постулату Планка). Статистична
термодинаміка дає можливість теоретично обчислити 8у і, отже, абсолютні
ентропії. Крім фундаментальних констант К, й, к, тс в 8у входить маса
частинки. Виділивши залежність ентропії від маси та підставивши числові
значення констант, рівняння (11.48) можна записати так:
8 = СрІпТ-К\пр + ^КІпМ-9,675, (Ц.50)
де 5—ентропія, Дж/(моль • К); р—тиск, атм; М = гпИл — молярна маса
частинки.
Співвідношення (11.50) має назву формули Закура-Тешроде і дозволяє
обчислювати абсолютні ентропії одноатомних газів (для яких немає внесків
обертального і коливального рухів) з високою точністю. У таблиці 11.1
наведені стандартні абсолютні ентропії 8^8 окремих одноатомних газів.
Для інертних газів і ртуті різниця між теоретично обчисленими та
експериментально визначеними значеннями абсолютних ентропій знахо-
315
Таблиця 11.1. Стандартні мольні ентропії одноатомних газів
Елемент
Не
Ие
Аг
К
Н8
3^98,Дж/(Моль-К)
експериментальна
127,0
146,3
153,8
155,5
160,1
176,4
обчислена
125,9
146,0
154,6
147,7
154,2
174,6
А5-£>експ. -§обч
1,1
0,3
-0,8
7,8
5,9
1,8
Д1П£о
0
0
0
5,76
5,76
0
диться в межах похибки експерименту. Для лужних металів теорія дає
занижені результати. Це означає, що в останньому випадку ще якийсь рух робить
внесок до статистичної суми і, відповідно, до ентропії. Для атомів інертних
газів та Меркурію характерні заповнені електронні оболонки, а для лужних
металів — незаповнені. Отже, на статистичну суму впливає електронна будова
атомів. Розглянемо, який внесок до статистичної суми дає рух електронів.
11.4. Електронна статистична сума
Рівні енергії (терми) в атомах знаходяться відносно далеко один від
одного, і не важко довести, що в цьому випадку Ає » кТ. За таких умов уже
другий після основного рівень дає дуже малий внесок у статистичну суму (він
пропорційний е~(Лє/* \ а за Ає » кТ— це дуже мала величина).
Вважаючи, що в основному стані є0= 0, одержимо £ел = 1 і відповідний внесок —
Л1п0ел в ентропію дорівнює нулеві. Але така ситуація існує тоді, коли
основний рівень невироджений (як буде показано далі, ця ситуація реалізується
в атомах інертних газів і Меркурію). У загальному випадку основний стан
може бути виродженим, і тоді
2ел. = £0. (11.51)
де £0— ступінь виродження основного стану.
Величину £0 можна знайти з електронної конфігурації атомів (див.
підрозділ 8.4). За рахунок взаємодії електронів в атомі виникають енергетичні
рівні (терми), що відповідають певним значенням квантових чисел Ь та 5.
У позначках термів замість Ь фігурують літери, а замість 5— мультиплет-
ність. Наприклад, 3Р (триплет Р) — це терм з Ь = Іта 5 = 1; 5 (синглет 5) —
терм з £ = Ота5=0.
Користуючись правилами Гунда, можна знайти основний терм
безпосередньо з електронної конфігурації атома.
В процесі виведення системи термів для деякої конфігурації атомів
доцільно розглядати тільки зовнішні, не повністю заповнені оболонки.
За рахунок спін-орбітальної взаємодії спіновий та орбітальний моменти
дають загальний момент атома: 7 = 1 + 5.
316
Кожному значенню У відповідає (2У +1) значень проекцій загального
моменту на обраний напрям і, відповідно, ступінь виродження
£о = 2У + 1. (11.52)
Таким чином, за допомогою двох перших правил Гунда знаходимо
основний терм; а третє правило Гунда дає можливість знайти найнижчий
рівень у цьому термі і ступінь його виродження.
Заповнені повністю оболонки мають 5=0 та £ = 0, відповідний терм —
5. Оскільки 5 та Ь дорівнюють нулеві, то /= 0.
Отже, для таких станів, зокрема інертних газів, основний стан невиро-
дженийі£ел =£0=1.
Знайдемо £0 для атомів лужних металів. Електронна конфігурація атома
лужного металу 5і. Терм 5. Оскільки Ь = 0, то У = 5 = 1/2 і £0 = 2• 1/2 + 1 = 2.
Внесок до ентропії за рахунок електронної складової для атомів лужних
металів дорівнює: 5ел = КІіі£0=КІп2 = 5,76 Дж/ моль-К. Ця величина якраз
і пояснює розбіжність між теорією та експериментом (див. табл. 11.1) при
обчисленні абсолютної ентропії.
Для молекул за рахунок утворення хімічного зв'язку між атомами
характерна взаємокомпенсація спінових та орбітальних моментів атомів, що
складають молекулу, тобто для молекул найбільш характерний невироджений
терм — X (§0 = 1). Такий терм мають молекули Н2, N2, С12, СО, С02 та
багато інших, але зустрічаються молекули, де є неспарені електрони (ці молекули
парамагнітні). Наприклад, N0 має один неспарений п — електрон і,
відповідно, терм П зі значеннями 3 = 1/2 та 3/2; основний терм П1/2, ступінь його
виродження £0=2. Кисень 02 має два неспарені я-електрони, але з
протилежними значеннями /и/(я+та я~), отже, сумарний спін дорівнює одиниці, а
сумарний орбітальний момент — нулю і, відповідно, основний терм X
(тричі вироджений).
У молекулах уже треба враховувати обертальний та коливальний рухи.
11.5. Обертальний рух
Для двохатомної молекули енергія обертального руху
(див. формулу (8.39)), де і—обертальне квантове число; /— момент інерції,
/ = V гп( г} = и/2; ц = —— приведена маса; /—відстань між атомами
і тх+тг
(ядрами). Кожен з рівнів (2/ + 1)-кратно вироджений, отже,
І(І+\)П2
йл=Е(и+і)в"2,кТ ■
і
Для обертального руху, як і для поступального, виконується умова
Де « кТ, отже, спектр енергій квазібезперервний, і від суми можна перейти
317
до інтеграла
°Г і(і+1)П2
Єоб.=](2/ + 1)е" ™ Ли
о
Вводячи змінну;; = /(/ + 1) *-'":мо йу = (2/ + 1)Л і, отже,
' П2
*2
— ґ ІІкТХ ,2ІкТ Ш2ІкТ
о
П
2
п2 н2
0
Для одержання остаточного результату треба додати число симетрії а,
яке дорівнює одиниці для гетероядерних молекул (НС1, СО, НВг, N0 та ін.)
і двом — для гомоядерних (N2, С12,02, Н2 та ін.).
Отже, остаточний вираз для статистичної суми обертального руху
двохатомної молекули такий:
Єоб.=^-" 01.53)
Якщо вважати лінію, що зв'язує ядра, віссю 2, то моменти інерції навколо
осей л: та.у дорівнюють один одному: Іх=Іу = І,аІ2 = 0.
У багатоатомній нелінійній молекулі є так звані головні осі А, В, С,
навколо яких існують три різні моменти інерції: ІА; Ів; Іс.
Отже, для нелінійної молекули
0*.=^Щ^. (П.54)
Для несиметричних молекул а = 1, для симетричних число симетрії
істотно відрізняється від одиниці, наприклад, для молекул метану і бензену
а = 12; етилену а = 4; аміаку а =^3 тощо.
Для лінійної молекули внесок до внутрішньої енергії за рахунок
обертальних ступенів свободи дорівнює КТ, а для нелінійної — т: КТ. У цьому не
важко пересвідчитися за допомогою формул (11.32), (11.53) та (11.54). Ці
розрахунки велися на моль частинок, а на одну частинку енергія, відповідно,
дорівнює кТтз, х кТ для лінійної та нелінійної молекул. Оскільки лінійна
молекула має два обертальні ступені свободи, а нелінійна — три, то можна
зробити висновок, що на один ступінь свободи припадає — енергії. За
допомогою формули (11.41) не важко пересвідчитися в тому, що на один
ступінь свободи поступального руху припадає також ^-- внутрішньої
енергії. Цей висновок має назву закон рівномірного розподілу енергії за
ступенями свободи. Відповідно, при виконанні цього положення на кожен
ступінь свободи припадає внесок до теплоємності — £
318
11.6. Коливальний рух. Теорія теплоємності
частота гармонійного осциля-
Для коливального руху 8,= іку, де V
тора, / = 0,1,2,3,...
&ш,.=І> КТ=1 + Є «Т+е
2Ау
кТ
+ ... = ■
1
(11.55)
1-е кТ
Проаналізуємо три варіанти.
1) Ну » кТ. Низькі температури, високі частоти. Тодіе~(ШкТї «і
і <2К0Л = 1. Ця ситуація схожа з £ел Для невироджених станів — високо
розташовані рівні при низьких температурах не збуджуються, всі частинки
перебувають в основному стані.
2) Иу << кТ. Високі температури, низькі частоти, є (ІіУ/кТ) = і -
— = ^— » 1, оскільки за умовою кТ» Ну.
Ну .
кГ
;&,
1-1 + -
кТ
Це випадок, коли спектр коливальних енергій практично безперервний.
Справді, якщо в (11.55) замінимо суму на інтеграл, то дістанемо
&
*■■-/•""*-(-£]
Ну
Ну
„кТ
Ну
недійсні
3) Якщо Ну = кТ, то обидва наближення £>кол = 1 та ()кол .
й потрібно користуватися формулою (11.55).
Для коливального руху закон рівномірного розподілу енергії за
ступенями свободи діє при відносно високих температурах, тобто за умови
Ну « кТ. Для внутрішньої енергії коливального руху в цьому випадку
маємо, згідно з (11.32),
Ц = КГ
31п
Ну
дТ
= КТ2- = КТ
Т
(11.56)
або кГна одну частинку. На відміну від обертального й поступального рухів,
коливальний, крім кінетичної енергії, містить внесок від енергії
потенціальної. Отже, кТ припадає на два ступені свободи відповідно до внесків
кінетичної та потенціальної енергії, а енергія на один ступінь свободи
дорівнює ^.
Таким чином, закон рівномірного розподілу енергії за ступенями
свободи діє лише тоді, коли енергетичний спектр квазібезперервний. Для
дискретного спектра цей закон не виконується.
Внутрішня енергія коливального руху для дискретного спектра, згідно
з формулами (11.32) та (11.55), така:
319
дТ
1п
1-е кТ
Далі зручно ввести параметр 0 =—, що має розмірність температури
и називається характеристичною температурою.
и = кт2^
_кве-»/т _
-в/г
1п-
1
1-е
де
-в/г
= КГ
1-е
„е/г
1
V
0-«-е/г)(-о
(ї-е-ш)2
К9/Г)н)[-І
(1157)
■1
Зазначимо, що умова йу «кТ відповідає умові 9 « Г, за якої
^і+!і£/=
Д0
1+1-1
т
= кт
у повному узгодженні з наведеним вище виразом (11.56).
Для теплоємності
/0л2
в/г
С =
— = д-
зг^ "(^в/г-і)2.
(11.58)
Не важко помітити, що теплоємність є функцією тр- Формула (11.58) дає
можливість затабулювати функції Су = /(^) — це так звані функції
Ейнштейна для гармонійного осцилятора.
Загальну теплоємність молекули знаходять так. За наявності п атомів
молекула має загальну кількість ступенів свободи — Зп. З них 3 поступальні,
для лінійних молекул: 2 обертальні і (Зп - 5) коливальних; для нелінійних
молекул: 3 обертальні і (Зп - 6) коливальних.
Наприклад, молекула Н28 нелінійна; 9 ступенів свободи складаються
з трьох поступальних, трьох обертальних і трьох коливальних (коливання
вздовж 8—Н зв'язків, рис. 112 (а) СО! = 2611 смг1 та (б) со2=2684 см"1 і
деформаційне коливання (в) (03 = 1290 см-1). Кожний поступальний та обертальний
ступені дають внески до теплоємності ~ (у розрахунку на 1 моль — ^).
Для молекули Н28 це буде
|д + !д = ЗД; 0 = ^ = ^0) = 1,4324(0,
2 2 к к
звідки маємо б! = 3740; 92= 3845; Є3 = 1848. За стандартних умов (Т= 298 К)
0і л л „,. 0о л л лл 0з
^- = 12,55; %- = 12,90;
= 6,17.
320
,
Рис. 11.2. Молекула Н28. Коливання: а, б — валентні (вздовж зв'язку);
в— деформаційне; г — обертання
З формули (11.58) або, користуючись таблицями функцій Ейнштейна, маємо
С,кол =0,010; С2К0Л =0,007; С3кол' =0,690 Дж/моль-К.
Оскільки Ср = Су + К, остаточно маємо Ср = ЗК + Д + С*0*' + СТ1" +
+ С3К0Л' = 33,95 Дж/моль • К, що добре узгоджується з С*ксп- =34,06 Дж/
моль-К.
Формула (11.58) дає також можливість обчислити теплоємність твердого
тіла. Якщо вважати кристал тривимірним осцилятором, то
\2
Су =ЗД
еш\ -
(е«,т-\)2'
(11.59)
Для кристалів Су прямує до нуля, коли Г—> 0, а при великих Т прямує
до ЗК згідно з правилом Дюлонга-Пті. Формула (11.59) задовольняє ці
вимоги. Справді, коли Т—> 0, то
е/г(|)2
в/г
є- »\ісу=зк* е/гУ2 = зке~ш(І) =зде2^
-е/г
0
(е0/г)2
Невизначеність т: легко розкривається за допомогою правила Лопіталя,
що дає Су -> 0. 0/г е
При високих температурах 0/Г —»0 і є = 1 +—1
Отже,
1+вув*
Т
ср=зк±
ч Т
гі-=зл(і+|і=зя.
321
Але Су, обчислене за формулою (11.59), не в кінцевих точках
відрізняється від експериментальних значень Су, особливо при малих Г, де
експериментально спостерігається закон куба, тобто Су~аТ (формула (11.59)
—9/7"
дає при цьому, як наведено вище, Су ~ е 0 ).
Т2
В розглянутій моделі тверде тіло розглядається як набір незалежних
осциляторів, що не відповідає дійсності. П. Дебай запропонував розглядати
тверде тіло як ізотропний континуум,
в якому існує не одна частота (як це
було за А. Ейнштейном), а спектр
частот від 0 до Утах (див. рис. 11.3).
Для низьких частот (відповідно,
довгих довжин хвиль) дискретністю
атомної структури можна знехтувати.
Кількість частот, що знаходиться
в межах від V до V + гЛ>, пропорційна
об'єму сферичного шару товщиною
<Л>, тобто 4тгу2<Л/ (коефіцієнт
пропорційності — певна стала В). Таким
чином, ця величина характеризує
кількість осциляторів з частотами від V до
V + сіу. Загальна кількість осциляторів
у кристалі дорівнює ЗЖ Обмеження
кількості осциляторів обмежує і
граничну частоту Утах. Умови нормування дають
Рис. 11.3. Розподілення за частотами
у твердому тілі
звідки
•тах -.
ЗЛҐ= І В4тм2сіу = 4пВ-
о
»__ 9ЛГ
з
тах
В-
4яу„
Це дає для густини станів
£(у) = #4т™2=-^у2.
(11.60)
Згідно з формулою (11.57) для енергії окремого осцилятора маємо
г_ Ну
"в£_Г (1Ш)
Враховуючи (11.60) та (11.61), для середнього значення внутрішньої
енергії за П. Дебаем матимемо
утах
йу
9М^ = Щ_
V
утах
у3Лу
екТ -1
(11.62)
322
Зробимо підстановку х = —9 тоді й\ = ~-<1х і
V3 А4 -1 є*-І
утах" 0
За високими температурами Ау <<кГі х« 1,щодаєе*-1 = 1+д:-1=хі
V КТ ) 3
(Кчах) 0 (^тах)
і, відповідно, Су= ЗК згідно з правилом Дюлонга-Пті.
За низькими температурами Ну» кТ , отже,хш >> 1, і можна верхню
межу вважати °°. Це дає для інтеграла
°° 1 А
С X СІХ = 71
^-1~15
(^тах)3 15
1, ВІДПОВІДНО,
Позначаючи —^^^ = 0^ як температуру Дебая, маємо
_ЗМк(кТ)34п4 _пп4к( ТЛ'
У 5(ЙУ3тах)
V
(11.65)
Таким чином, теорія Дебая дає в узгодженні з експериментом закон куба
температур для теплоємності при Г—» 0 К. Зазначимо, що характеристична
температура за Ейнштейном вЕ становить приблизно т^я*
За допомогою статистичної термодинаміки можна розглядати не лише
властивості окремих речовин, а й хімічні рівноваги.
11.7. Обчислення константи рівноваги
статистичним методом
Розглянемо спочатку простий приклад рівноваги А<-»В (наприклад,
реакція ізомеризації). У даному випадку константа рівноваги є безрозмірною
величиною, отже, можна користуватися будь-яким способом для виразу
концентрації (парціального тиску). Виразимо константу рівноваги через
кількість частинок _
К = 2£. (11.66)
Оскільки компонентів два, то енергії основного стану різні (є^ Ф ев) і не
можуть обидві дорівнювати нулю. Отже, (11.22) запишемо як
е<— Е0 л. еі~е0
И^Ще кТ =У-е кТ . (П-67)
323
Цю формулу можна використати для порівняння кількості частинок А і В
в основному стані, а саме:
ЕДо~Ч А£р
\="4>е КТ =ЛЧе *Т- <1Ш)
Підставляючи (11.67) та (11.68) у (11.66), одержимо для константи
рівноваги _
їїА м^в й/ Є/ * (1169)
У загальному вигляді для обчислення Кр скористаємося формулою (6.10)
Кр=е~ *т . (П.70)
За формулами (1.74) та (11.33)
АС°=М°+АуКТ = А4-КТАІпд + АуКТ = АН^КТ\пІІд^+АуКТ,
і
де Ду — різниця стехіометричних коефіцієнтів. Враховано, що коли Г—» 0,
ДС00=ДЛо0=Д#о0=ДС/о-
Отже,
_м
Кр^ІІ&еПе-". (11.71)
і
Наприклад, для реакції а А + рВ=уС + 80 Ду=у + 8-а-Р,
Як конкретний приклад розглянемо реакцію дисоціації йоду І2 = 21 і
зіставимо теоретичні розрахунки з експериментальними даними з хімічної
рівноваги для цієї реакції, що наведені у табл. 6.2 (підрозділ 6.4).
Для молекули І2 маємо з обертальних спектрів Ь = —У-— = 0,0373 см~ ;
сокол = 213,17 см ; єДИс. і2 =148,3 кДж/(моль • К) = АН$. Терм І2 £, отже,
(£о)ь=1.
Відносна молекулярна маса Йоду 126,92.
Шукаємо основний терм атома Йоду. Конфігурація/?5
«5
гЧгЧт
иі/ +1 0 ^Г Х/и/ = 2х1+2хО-1 = 1=М]™х.Терм/>
Іт5 = 1/2; М - 2 • (1/2) +1=2. Отже, терм 2Р; 7^ = 3/2; Утіп = 1/2.
Згідно з третім правилом Гунда основний терм Рш. Отже, §0 = 2 • (3/2) +
+ 1=4.
&л.-0 і~-16.
324
Єкол =77-*—= 1-е"9/г;Є = -^ш = 1,4385-213,7 = 306,65;
' й2кол. к
^ = 0,24; \-е~ш =0,213.
ехр
КТ
, 148300 і . .. ]П_7
= ЄХр|-8Ж42741 = 8'25-10 ;
бобер. -
ай2
ду = 2-1 = 1;е
іІ>бер.І2 8я2кГ кГ
Є
2НЬс.а = 2
і^ПОСТ.
_(8пост.і) _ (2птікТ)3 є2 (кТ)2 Н3 _(тіт1кТ)е(кТ)
бпост.і2 Н6 (іпіШікТ)212 е(кТ)
Шс
кТ
\3/2
п П <гА* - 2НЬс(ПтікТ) е{кТ)\ _ ?ьНг(]Щ^У
^обер.^пост.^ - ^ ^3 є" І А2 І
Абс = 6,624 • 10~34 • 0,0373 • 102 • 3 • 108 = 7,408 • 10"25 Дж.
-іЗ/2
пт{кТ
3/2
3,14'126,9'1,66»10"274,38-10"23'1274
зз
= 4,31-10" м-3.
(6,624-Ю"34)
1Дж/м3 = 1Н/м2=1Па;
(^)атм=16-0,213-2-7,408-10""25-8,25-10''7-4,ЗЬ1033 *—г = 0,177 атм.
ра™ 1,013-105
Це значення розрахованої константи знаходиться у доброму узгодженні
з експериментальними даними, що наведені у таблиці 6.2 (підрозділ 6.4).
Все викладене вище у цьому розділі відносилося до ідеальних газів, для
реальних газів картина дещо складніша.
11.8. Статистична термодинаміка реальних
газів
Усі ступені свободи можна умовно поділити на внутрішні та зовнішні. До
перших належать обертальний, коливальний рух та електронні переходи. Для
цих ступенів свободи статистична сума всієї системи 2 є добутком
статистичних сум <2 окремих частинок
2 = 0^., (11.72)
де N— кількість частинок у системі.
Формула (11.72) ґрунтується на незалежності руху для внутрішніх
ступенів свободи окремих частинок. Використання повної статистичної суми
системи 2 в цьому випадку не дає жодної переваги над використанням
325
молекулярної суми (?. Наприклад, для вільної енергії Гельмгольца маємо
А =-кТІп2 = -кТІп(£Ґ) =-МсГЙі0 (11.73)
у повному узгодженні з формулою (11.30).
Для поступального руху раніше використовувалась модель ідеального
газу, тобто було знехтувано взаємодією між частинками. Якщо врахувати
таку взаємодію в розподіленні Больцмана за енергіями, необхідно брати до
уваги не тільки кінетичну, а й потенціальну енергію. Для статистичної суми
системи тоді маємо
*=2«
кТ
і=1
або для квазібезперервного спектра
-ї,і'
,+Уп,
кТ
йрхйр1..Лри(іУх(іУ1..ЛУи.
(11.74)
Оскільки частинки не відрізняються одна від одної, у формулі (11.74)
врахована кількість можливих комбінацій (перестановок з N частинок), яка
дорівнює N1. 2
За відсутності взаємодії £/пот = 0 та інтегрування по імпульсах (єкін = тр-)
дає відомий результат для статистичної суми поступального руху
(формула (11.38), частина, що не залежить від об'єму)
Ґ =
ґ \3/
ІшпкТ і
І *2 )
N
а інтегрування за просторовими координатами для кожної частинки дає
об'єм системи У. Відповідний внесок до статистичної суми системи є V .
Таким чином, із формули (11.74)
2 =
[ ІпткТ і
І *2 )
-і#
ґ Л3/
[ ІпгпкТ і
І *2 )
N1
аг
І ІпгпкТ
{ ь2 )
3/2
Vе
(11.75)
де і; — об'єм, що припадає на одну частинку, і; = УІИ9 а статистична сума на
одну частинку повністювідповідає формулі (11.40) (нагадаємо, що при
виведенні формули (11.40) була врахована можливість для частинок
обмінюватися своїми «комірками»).
Позначимо далі результат, що дає формула (11.75), через 2ІД. Для
реального газу, на відміну від ідеального, слід враховувати взаємодію між
частинками і, відповідно, потенціальну енергію. Це веде до появи у статистичній
сумі так званого конфігураційного інтеграла
є кТ сіУ^У^.ЛУ^. (11.76)
326
Коли взаємодія між частинками відсутня (£/пот.= 0) — ідеальний газ,
/ід.= ^, (11.77)
як це було доведено вище.
Розрахунок сукупної взаємодії ТУ частинок дуже складна задача, але
ймовірність уже потрійних взаємодій дуже мала, не кажучи вже про
одночасну взаємодію чотирьох або більшої кількості частинок. Тому цілком
слушно можна обмежитися лише парними взаємодіями.
Розглянемо спочатку взаємодію окремих двох частинок. Нехай між ними
діють ван-дер-ваальсові сили, основною складовою частиною яких є
дисперсійні (лондонівські) сили. У цьому випадку потенціальна енергія
залежить від відстані г між частинками за законом
А.
.„6 *
С/ = -
V
Рис. 11.4. Потенціальна енергія
взаємодії двох частинок
(11.78)
У моделі жорстких сфер дві
частинки А і В не можуть підійти одна до
одної на відстань <1, меншу, ніж сума
їх радіусів, тобто сі=гА + гв Для
однакових частинок гА = гв = г0ісІ = 2г0,
тобто ця відстань дорівнює діаметру
частинки. Зміна потенціальної
енергії з відстанню між частинками для
цього випадку зображена на рис.
11.4.
Обчислюючи конфігураційний
інтеграл, зручно використовувати
сферичні координати із центром на
одній із частинок. При цьому
інтегрування за кутовими координатами
дає 4я, і від елемента об'єму йУ
залишається г^йг. Відповідний внесок до конфігураційного інтеграла
К Ц(г)
у = 4ті|е кТ Л/г, (11.79)
о
де і?—лінійний розмір (радіус) комірки (V = УІИ), що припадає на одну
частинку. Ц{г)
Якщо 0<г<сІ Щг) —>©о [9 відповідно, е кТ —> 0; якщой<г<К,то можна
вважати £/(г) «кТ, і тоді
кТ
Таким чином,
у = 4тс
■■№.
А?Г:
. 4пК3 4тиі3
-%№У*:
327
= у-Р~ = у-& (11.80)
Через 8 позначено
5 = Р + ^;, (11.81)
де V — простір, що припадає на окрему частинку; Р — зона недосяжності
однієї частинки другою.
Для обчислення інтеграла а можна скористатися формулою (11.78), яка
показує залежність потенціальної енергії від відстані між частинками
СІ <*
Якщо деяка Л'-на частинка взаємодіє з рештою (А/"— 1) частинок, то згідно
з (11.80) відповідний внесок до конфігураційного інтеграла
/^ = (Л^-1)((;-5) + і; = ^і;-(^-1)8=Г-(Л^-1)8 (11.83)
(інтегрування по координатах ЛЧ частинки, що сама з собою не взаємодіє,
дає V, для всіх інших (/V- 1) частинок одержуємо у = V - 8). Для взаємодії
(Лґ- 1)-ї частинки з рештою (/V- 2) частинок відповідно Ім_х = У- (М- 2)8
тощо.
Загальний конфігураційний інтеграл визначається як добуток
/ = /^/^_і/^-2..^і = .4^-(^-1)5][К-(^-2)8][Г-(^-3)8]...К (11.84)
Об'єднуючи Імта Іь маємо
[Г-(ЛГ-і)8]К = Г2[і-(ЛГ-і)|].
Член (Лґ -1)~ суттєво менший від одиниці, справді, V = М; і ІУ- 1=7/,
отже,
(ЛГ-1)АЛ5«и
Далі знехтуємо членами, які містять (Л^ -1)-^- степенях, більших від одиниці.
Об'єднуючи Ім_х та І2, маємо
[У-(М-2)8}[У-Ь]^[і-(М-2 + іЩ = ^[і-(М-іЩ.
Далі, об'єднуючи 7#_2та/3, 4г-зта А»— > Амста^л:+і> маємо ідентичний
результат
V І-(іУ-і)—І Всього таких множників є -т*, отже,
7-{ка[і-(^-1)і]]?=^[і-(АГ-1)^^
враховуючи, що N » 1, остаточно маємо з урахуванням (11.77)
= ^[і-Л^|-]2 =/ід.[і-Л^|:]2. (11.85)
328
Відповідно
2 = 2,„
"$1
Згідно з формулами (1.76) та (11.73) тиск у системі дорівнює
Для ідеальної системи
кТ
дУ
(11.86)
(11.87)
Р = кТ(Щ^} =КТ±\Ь(ВУ»)] =Мкт(Ц¥.) =^£. (11-88)
и { ду )т дуі \ >\т у ду )т у
В розрахунку на 1 моль маємо формулу Менделєєва-Клапейрона у
повному узгодженні з формулами (1.4) та (11.44).
Для реальних газів
р = кТ
дІп2
дУ
1-—кТ-—
У 2 дУ
дУ
1п| 1-М
2 V
ЛГ6
Розкладаючи логарифм у ряд при малих ^у-, маємо
р—Г+-ТдУ
І 2 У\т
№Т ШТ б _
У 2 у2
_ИкТ
№}
У розрахунку на 1 моль частинок (ІУ = ИА)
Р =
КТ
1 + —4—
2У
КТ
У
1+ш
(11.89)
(11.90)
Рівняння (11.90) можна порівняти з рівнянням з віріальними
коефіцієнтами (2.34), в якому залишився тільки другий—В(Т) віріальний коефіцієнт.
Враховуючи (11.81), для віріального коефіцієнта маємо
ВІТ)
Величина —г— дорівнює
= ^8 = ^Р|^а
кТ
Ял$_МА4мР _2Нл%аг
3-2
= Ь.
(11.91)
(11.92)
те/3
Оскільки об'єм однієї частинки дорівнює —-г-, то стала Ь — це
збільшений учетверо об'єм, який займають саме частинки. Згідно з (11.91) і (11.82)
329
N.
Аа_НА( АпАЛ АіОі\А \ _ а
кТ кТ
гаг)
Зеї3 КТ КТ'
Вводячи (11.92)та (11.93) в (11.91), одержимо
В{Т) = Ь--£-
1, ВІДПОВІДНО,
КТ
V
1 + -
Ь--
кт
;-1
(11.93)
(11.94)
(11.95)
Рівняння (11.95) практично збігається з рівнянням Ван дер Ваальса.
Справді, з (11.95) маємо
Р = -
ЯТ
V
а КТ
КТУ V
КТ
И1--
V
КТ
У-Ь V
2'
(11.96)
1 А
(при малих х (1+х) = т^—, а величина — «1).
Формули (11.92) та (11.93) давали б можливість теоретично обчислити
константи а та Ь рівняння Ван дер Ваальса, але невідоме точне значення сі.
Комбінація рівнянь (11.92) та (11.93) дозволяє виключити цей параметр.
Справді, добуток
а-Ь--
З Зеї3 9 а
(11.97)
не містить сі.
Якщо відома величина Ь, то з рівняння (11.97) можна теоретично
обчислити а і порівняти з аексп з рівняння Ван дер Ваальса (таблиця 11.2).
Таблиця 11.2. Експериментальні та обчислені значення
сталої а рівняння Ван дер Ваальса
Газ
Не
N6
Аг
N2
С12
СО
#обч
4,8
26
163
147
680
334
#експ
3,5
21
135
135
632
361
Статистична термодинаміка є одним із джерел осмислення 2-го начала
термодинаміки.
330
Розділ 12.
ТЕОРІЯ ФЛУКТУАЦІЙ. СТАТИСТИЧНІ
ТА ФІЛОСОФСЬКІ АСПЕКТИ 2-го НАЧАЛА
ТЕРМОДИНАМІКИ
Начало друге, про нього новела,
Тут флуктуації і демони Максвелла.
Розширюючи поняття системи до Всесвіту, Р. Клаузіус сформулював 1-е
та 2-е начала термодинаміки так: «енергія Всесвіту є константою, ентропія
Всесвіту прямує до максимуму». Перше начало показувало кількісне
збереження енергії, друге — її якісну деградацію. Визначався загальний
напрямок процесів у Всесвіті — поступове зростання ентропії, тобто
молекулярного хаосу. Це означало, що всі види вільної («хорошої») енергії:
електрична, магнітна, хімічна тощо поступово переходять у зв'язану («погану»)
теплову енергію атомно-молекулярного руху. Оскільки вільну енергію
частково можна відтворювати з теплової, маючи різницю температур («нагрі-
вач» — «холодильник»), то повинні зникнути і системи, де є така різниця
(наприклад, Сонячна система з «гарячим» Сонцем та «холодними»
планетами і «космічним пилом»). Поступово, згідно з концепцією Р. Клаузіуса,
Всесвіт повинен перетворитися на великий рівноважний термостат, де всі
температури, а також тиски однакові, зникають також інші різниці
потенціалів: хімічні, електричні тощо. Цей стан Всесвіту з розсіяною, дисипованою
енергією називають «тепловою смертю». Система—Всесвіт—із цього
стану сама вийти не може, отже, у Всесвіту є кінець.
Якщо Всесвіт має кінець, то повинен бути і початок Всесвіту, тобто
такий його стан, у якому він знаходився з низькою ентропією. Це підштовхує
до ідеї створення Всесвіту, до ідеї Творця, хоча не ясно, чому Творець,
створивши Всесвіт, дозволяє йому рухатися у напрямку деградації згідно з 2-м
началом термодинаміки. Виникає також питання: чи можна пояснити суть
«теплової смерті» і 2-го начала в цілому з матеріалістичних позицій.
Порівняно з класичною термодинамікою, кроком уперед є підходи, що
розробляє статистична термодинаміка. Згідно зі статистичним
тлумаченням 2-го начала термодинаміки зростання ентропії ізольованої системи не
є жорстко обумовленим процесом, а лише найбільш ймовірним. Система
поступово переходить до станів, які мають більшу ймовірність
реалізуватися. При цьому не заперечується можливість переходу з більш ймовірного
до менш ймовірного стану, це реалізується шляхом виникнення у системі
флуктуацій. Лише наявність флуктуацій дозволяє пояснити такі фізичні
явища, як броунівський рух, блакитний колір неба, зародження фази,
шумовий «фон» теле-, радіоапаратури тощо.
Розглянемо в основних рисах теорію флуктуацій.
Для статистичного середнього деякої фізичної величини (х) можна
написати
331
*=5>,=5>,4, 02.1)
і
лг
де х(—значення х в /-му стані; р{—ймовірність цього стану; А/, — кількість
частинок в /-му стані (або кількість вимірювань, що веде до цього стану);
N—загальна кількість частинок (вимірювань). Відхилення х від середнього
значення в обидві сторони (збільшення або зменшеннях, відбувається
однаково часто, тому середнє відхилення (Ах) дорівнює нулю
Ах = х-х =х-х =0 (12.2)
і використовується квадратичне відхилення — квадратична флуктуація
(Ах)2 = (х-х)2 =х2-(3с)2. (12.3)
Відносна флуктуація визначається як
6 7^7 (124)
X
Припустимо, що система складається з 2-х незалежних підсистем; тоді
з урахуванням (12.2) маємо
[А(х{ + х2)] =(Ах1)2+2Ах1 'Ах2+(Ах2)2=(Ах{)2 +(Ах2)2. (12.5)
Отже, квадратична флуктуація 2-х незалежних величин дорівнює сумі
квадратичних флуктуацій цих величин.
Узагальнюючи (12.5) на N незалежних підсистем, маємо для
квадратичної флуктуації системи
N N
(Ах)2 =[Д(]>,)]2 = £(Л**)2. (12-6)
А:=1
Оскільки всі підсистеми рівноправні між собою, то можна вважати, що
2 ~ГЛ^ \2 = ~ї
(Дх,)^ (Ах2У = (ЬхкУ = ... = (Ах„У,
отже, (Дх, )2 пропорційне N.
Середнє значення х системи складається із середніх значень хк, тобто
Число доданків у (12.7) дорівнює N і при умові, що вони не дуже
відрізняються один від одного, величинах також пропорційна N. Отже, для
відносної флуктуації маємо
Розглянемо тепер деякі більш конкретні приклади. Почнемо з флуктуацій
об'єму при сталій температурі. При малій ізотермічній зміні об'єму (АУ) для
вільної енергії Гіббса маємо
332
АС = р0АУ + АА = роАУ +
ґдАЛ .„ (д2А^
удУ)т [дУ
АУ2+...=
/
^° ^ {дУ)т 2 2кУ (12.9)
(враховано, що флуктуаційний процес є квазістатичним, отже,/?0 =р (тиск
. . . , \(дУ) .
у підсистемі дорівнює тиску довкілля); ае = — — —ізотермічна стис-
. У[ ар )т
неність). V г у і
Ймовірність (сім) того, що система має об'єм між (У) та (У+сі У), дається
формулою Больцмана
(Іп = Ве кТс1У = Ве 2*кТсіУ. (12.10)
Знаходячи за допомогою формули (12.10) квадратичну флуктуацію
об'єму, маємо
(АУ)2=кТяУ. (12.11)
Для ідеального газу ж = —, отже,
г2 т,2
^^кїУ^кїУІ^ (1212)
Відносна флуктуація об'єму дорівнює
_^|(АV^ _ у _ !
Ьу=^-' =-^и- = -7і= (12.13)
14)
у повному узгодженні з формулою (12.8).
Квадратична флуктуація густини (р) дорівнює
'ш1=т2[А^ = ^УЇ = ^-кТ^У = ^гкТ^ (12.
де гп — маса.
З флуктуаціями густини пов'язане явище розсіювання світла при
проходженні його крізь шар речовини. При проходженні світла через речовину,
наприклад газ із строго однорідноюгустиною, ця речовина не розсіює
світло і залишається прозорою і невидимою. Розсіювання відбувається лише за
рахунок відхилення від однорідності завдяки флуктуаціям густини. З
урахуванням формули (12.14) маємо для інтенсивності світла (7), що розсіяне
в об'ємі (У)
І = сопії^УкТх, (12.15)
де І0—інтенсивність променя, що падає; X — довжина хвилі.
Розсіювання світла різко зростає при зменшенні довжини хвилі,
наприклад, при переході від червоної до фіолетової частини видимого спектра інтен-
333
сивність розсіювання зростає у 5 разів. При проходженні сонячного світла
через атмосферу більш інтенсивне розсіювання у блакитній частині
спектра надає атмосфері блакитного кольору. Коли сонце знаходиться біля
горизонту і товщина шару, крізь який проходять сонячні промені, суттєво
збільшується, то за рахунок інтенсивного розсіювання блакитних променів
лишається, в основному, т:~рвона частина спектра, що надає диску сонця
червоного кольору.
Яскравим прикладом існування флуктуацій є броунівський рух —
хаотичний рух малих частинок у рідині або газі (рис. 12.1). Броунівський рух
перебігає протягом як завгодно великого проміжку часу, не затухаючи.
Частинки, що беруть участь у броунівському русі, описують невпорядковані
траєкторії, причому характер руху не залежить від зовнішніх умов і хімічної
природи частинок.
Природа броунівського руху
полягає у передачі імпульсу
малим частинкам при
зіткненні з нщш молекул, що
знаходяться у постійному
тепловому русі. Для
великих частинок імпульси від
багатьох молекул з різних
боків врівноважуються і
така частинка не рухається.
Для частинок з розміром
10" см і менше загальна
сила від переданих молеку-
Рис. 12.1. Діаграма броунівського руху лами імпульсів уже відріз-
за Перреном няється від нуля,
безперервно змінюючись за величиною і напрямком, і вона примушує частинку
хаотично рухатися. Фіксуючи положення частинки через певний інтервал часу
(декілька секунд), можна пересвідчитися, що частинка змінює своє
положення, що є сумарним результатом хаотичних рухів, напрям і величина яких
змінюється приблизно 10 разів за секунду. Таким чином, броунівський рух
обумовлений флуктуаціями тиску і є своєрідним індикатором
молекулярного теплового руху.
Кількісна теорія броунівського руху була розроблена А. Ейнштейном
і М. Смолуховським. Середнє квадратичне зміщення (А) частинки за час (т)
пов'язане із коефіцієнтом дифузії (О) таким співвідношенням:
0 = £. (Ш6)
З іншого боку, з використанням формули Дж. Стокса для зовнішньої
сили (/)
/ = 6щги, (12.17)
334
де и — швидкість руху частинки; г—її розмір; ті — в язкість середовища;
отримуємо для В співвідношення
£> = -^-. (12.18)
втщг
Співставлення (12.16) і (12.18) дає для середнього квадратичного
зміщення
А= /-^--т. (12.19)
(Зтгг|г
Таким чином, інтенсивність броунівського руху збільшується зі
збільшенням температури і зі зменшенням розміру частинки та в'язкості
середовища. Зазначимо, що свого часу за допомогою формули (12.19) була досить
точно визначена константа Больцмана (к).
Для флуктуації енергії відповідним чином одержимо
АІ/2=1/2-(У)2=кТ2С„. (12.20)
Для квазібезперервного спектра на кожен ступінь свободи припадає
енергія £+-; отже, для середньої енергії маємо
" = /*$: (12.21)
а, відповідно, для теплоємності
Су=/|. (12.22)
З урахуванням формул (12.4), (12.20—12.22) маємо для відносної
флуктуації енергії
о _ УАЕГ _ V " 2 _ соті Л9 9Тк
Ьи"~її~~ гкг "ТГ (1123)
-* 2
Оскільки/пропорційне ІУ, то знову маємо підтвердження загальної
формули (12.8).
Зі збільшенням числа ступенів свободи (кількості частинок) величина
відносної флуктуації (об'єму, енергії тощо) стає дуже малою. Наприклад,
для одного моля при N = 6,02* 10
5У= , 1 =10"10%.
Л
23
6-Ю
Таким чином, при великих N 5^. практично дорівнює нулю і статистичні
закони мають фактично достовірний характер. Навпаки, у системах з малим
числом ступенів свободи відносні флуктуації можуть бути великими і деякі
поняття, зокрема, ентропії, температури, розмиваються.
Є, однак, випадки, коли флуктуації енергії виявляються великими навіть
у макроскопічній системі. Мова йде про системи, що мають вельми велику
(гранично-нескінченну) теплоємність, тобто системи, в яких температура
335
не підвищується при підведенні теплоти з довкілля. Як приклад можна
навести систему, що складається з 2-х рівноважних фаз, наприклад, рідина-пара.
Підведення теплоти до такої системи іде не на її нагрів, а на випаровування
рідини і теплоємність системи рідина-пара можна вважати нескінченно
великою. Згідно з формулою (12.20) у такій системі повинні існувати великі
флуктуації енергії та інших величин. Так, наявність великих флуктуацій у
рідині, щЬ кипить, легко спостерігати, весь процес кипіння має хаотичний
характер.
Іншим цікавим випадком є системи при досить низьких температурах.
Для низьких температур класичні" формули для енергії (12.17) та
теплоємності не працюють, спектр енергій не квазібезперервний, а дискретний,
і згідно з квантовою теорією теплоємності маємо за П. Дебаєм при низьких
температурах (див. формули (11.64) та (11.65)):
17 = соті'N7*; (12.24)
С»=соті'т*. ' (12.25)
Це дає для відносної флуктуації енергії, згідно з (12.20), (12.24) та (12.25),
Ьи= ™™< (12.26)
уШ-уіТ3
Таким чином, флуктуації енергії зменшуются із кількістю частинок, як
і для класичного випадку, як ІД/ЇУ. Однак зі зниженням температури,
особливо при Т—» 0, флуктуації зростають і стають великими навіть для систем
з великою кількістю частинок.
Л. Больцман використав можливість існування флуктуацій великого
розміру і побудував так звану «флуктуаційну модель Всесвіту», згідно
з якою в окремих ділянках Всесвіту — флуктуаціях — можливі процеси, які
перебігають як зі збільшенням (ділянки «аЬ» та «ссі» на рис. 12.2), так і зі
зменшенням (ділянки «6с» та «сіє» на рис. 12.2) ентропії. Оскільки Всесвіт
безмежний, то ці «флуктуації» можуть
бути відносно великого розміру —
Галактики, Метагалактики тощо. В тій
частині Всесвіту, де знаходиться Сонячна
система, еволюція у неживій природі
перебігає у бік збільшення ентропії, а
десь в іншому місці, в іншій флуктуації
можливий зворотний процес.
На перший погляд, статистичне
тлумачення 2-го начала термодинаміки, а
також «флуктуаційна модель Всесвіту»
Рис. 12.2. Зміна ентропії Всесвіту За Л. Больцманом дозволяють позбути-
з часом: 1) за Р. Клаузіусом 5-»лтах ,.
(крива /V ся метаФ13ИЧН01 концепції «теплової
2) за Л. Больцманом — флуктуації смерті». А!ие за Л. Больцманом вихо-
при 5 = £тах (крива 2) дить, що рух можливий лише у флукту-
336
аціях, а інша основна частина Всесвіту вже знаходиться у стані рівноваги,
тобто спокою. «Теплова смерть», таким чином, переноситься з майбутнього
у сучасність, спокій абсолютизується, рух, навпаки, набирає відносного
характеру в протиріччі з матеріалістичними і діалектичними поглядами на
явища Всесвіту.
Відповідь на ці питання лежить у розгляді можливості застосування 2-го
начала термодинаміки до Всесвіту у цілому. Неможливо вважати Всесвіт
ізольованою системою, як того потребує формулювання 2-го начала згідно
з Р. Клаузіусом. Застосування 2-го начала і поняття «ізольована система»
відбувається при абстрагуванні від одного з основних понять, що існують
у природі, — взаємодії. Справді, саме невзаємодіючі частинки — ідеальний
розріджений газ — мають максимальну ентропію, конденсація за рахунок
взаємодії речовини у рідину і поява ближнього порядку вже різко зменшує
ентропію, далі перехід у твердий стан і поява дальнього порядку веде до
подальшого зменшення ентропії. Введення поняття «ізольована система»
ігнорує саме взаємодію. Е. Борель, підкреслюючи всепроникаючу роль
взаємодії, навів такий приклад. Збурення гравітаційного поля, яке виникає при
переміщенні 1 г речовини на відстань 1 см на Сіріусі через деякий час
порушує детермінізм у русі молекул у певному місці на Землі. Наявність
«ізольованої» системи у відносно впорядкованому стані примушує
констатувати, що перед цим мала місце взаємодія системи з довкіллям. Реальна
картина Всесвіту скоріш за все якраз протилежна «флуктуаційній моделі»
Л. Больцмана, а саме не «віковий спокій» і окремі невеличкі флуктуаційні
зміни, а навпаки, віковий рух, а в локальних, відносно слабко зв'язаних з іншим
світом ділянках — квазіізольованих—є прагнення до рівноваги,
встановлення тимчасового спокою. Як висловлювався Ф. Енгельс: «Окремий рух прямує
до рівноваги, сукупний рух знищує її». Таким чином, екстраполяція 2-го
начала термодинаміки на Всесвіт непридатна, призводить до казусу.
Для звичайних макросистем 2-ге
начало термодинаміки виконується
добре, але Дж. Максвелл показав, що
і тут можливі питання. Дж. Максвелл
придумав «демона», тобто якусь
істоту, яка має можливість розрізняти
молекули за швидкостями (див. рис.
12.3). Якщо уявити собі посудину,
заповнену газом при певній
температурі й розділену перетинкою із зас-
лонкою, яку демон може відчиняти,
то, відчиняючи заслонку певної миті
й пропускаючи швидкі молекули,
скажімо, зліва направо, а повільні
навпаки, справа наліво, поступово
демон між частинами посудини ство- Рис. 12.3. Демон Максвелла працює
337
*
Рис. 12
1 1
.4. «Одномолекульна»
Сциларда
"*
машина
рює різницю температур, за рахунок якої можна отримати роботу. Таким
чином, «температурний демон» створює вічний двигун 2-го роду, всупереч
2-му началу термодинаміки. Крім «температурного», можливий «демон
тиску», який просто пропускає молекули з однієї частини посуду в іншу і не
пропускає їх у зворотному напрямку, створюючи, таким чином, різницю
тисків, за рахунок чого знову можна отримати роботу.
Суть дій демона полягає в тому, що він має певну інформацію:
«температурний» — про швидкість молекул, демон «тиску» — про розташування
частинки, отже, виходить, що за рахунок інформації можна зменшувати
ентропію.
Щоб розібратися з діями демона
Максвелла, Л. Сцилард спростив
задачу, змоделювавши «одномолекуль-
ну» теплову машину (рис. 12.4). В
циліндрі об'ємом 2Уе два поршні, які
можуть рухатись без тертя до
середини циліндра. В циліндрі є всього одна
молекула розрідженого ідеального
газу при сталій температурі (Г). Якщо один з поршнів просувати до
середини циліндра, то при цьому стисненні, згідно з формулою (1.27), в якій треба
замінити К на -^- = к, над системою виконується робота
А (^\=-кТІп^ = кТІп2 (12.27)
з точки зору довкілля, ця робота витрачається
(ии)і=-кГ1п2. (12.28)
При розширенні газу до об'єму всього циліндра (IV) молекула рухає
поршень, виконуючи роботу, для довкілля
М2=-М2=+кТІп2. (12.29)
Сумарний результат (оборотний процес!)
К )і + К )г = К), + К )2 = °- (12-3°)
Простежимо, що відбувається з ентропією.
При стисненні ентропія системи змінюється на величину (див. формулу
(1.53) з заміною К на к)
А81=кІп-^- = -кЬі2.
При розширенні, відповідно,
А5,2=+/с1п2.
В циклічному оборотному процесі
д^+д^о.
(12.31)
(12.32)
(12.33)
338
Тепер на 1-му етапі дамо змогу «попрацювати» демону, який має
інформацію, в якій частині системи («зліва» чи «справа») знаходиться молекула
і, отже, поршень можна просунути до середини посудини без опору, тобто
не витрачаючи при цьому роботу. Оскільки кінцевий об'єм є V, то ентропія
зменшується при цьому на величину (-к1п2), згідно з формулою (12.31). Далі
система за рахунок зіткнення молекули з поршнем повертає його у вихідний
стан, виконуючи роботу розширення (за рахунок теплоти довкілля).
Цей процес можна повторювати неодноразово. Отже, всупереч другому
началу термодинаміки, відбувається за допомогою демона реалізація
вічного двигуна роду 2-го, що перетворює теплоту на роботу при сталій
температурі, черпаючи енергію (теплоту) з довкілля. При цьому зменшення
ентропії відбувається не за рахунок віддачі теплоти у довкілля, а за рахунок
одержання інформації, в якій саме частині циліндра знаходиться молекула.
Отже, Л. Сцилард на чисельному прикладі показав, що за рахунок
отримання певної інформації можливе зменшення ентропії.
В теорії інформації справедлива формула Шеннона, яка пов'язує
кількість отриманої інформації (/) з ймовірністю (/?,-) конкретних подій
/-2АІ082А- <1234)
І
Для достовірних подій Рі = 1 і, отже, інформація дорівнює нулю. Для
рівноймовірних подій Рі=— і формула (12.34) спрощується
/ = -£ііо§2(і) = 1о§2«. (12.35)
При п = 2 (наприклад, підкидання монети дає^ = 1Л для випадання герба
або цифри) 1=1 — ця одиниця називається «Ьіпагу йі&іі» або скорочено
«біт». Біт — це відповідь на питання: «так — ні», «герб — цифра» тощо.
При п = 2т /= І08 і2т = т. (12.36)
Наприклад, щоб напевно знайти певну карту в колоді з 32 карт, потрібно
5 біт інформації, тобто 5 питань типу «так — ні», щоб упізнати обрану карту.
З точки зору теорії інформації, єдиний спосіб підвищити порядок у системі
і, відповідно, знизити її ентропію, це отримання інформації. Можна вважати,
що ентропія є мірою нестачі інформації про систему. Для найбільш
упорядкованої системи — кристала при Г= 0 К координати всіх структурних
одиниць (атомів, іонів, молекул) чітко визначені, стан системи достовірний,
додаткова інформація не потрібна і ентропія мінімальна (згідно з
постулатом Планка 50 = 0 (див. формулу 1.60)). Якщо в системі відбуваються
необоротні процеси (скажімо, при нагріванні), це веде до збільшення хаосу і не
вистачає достовірної інформації про систему (про положення і швидкість
частинок, з яких вона складається).
Повертаючись до «одномолекульної» моделі Сциларда, спробуємо
«вигнати» демона. Сцилард намагався це зробити за рахунок врахування
339
енергії, що потрібна для передачі інформації. Так, при кожному циклі своєї
діяльності в машині Сциларда демон передає рівно один біт інформації.
Для передачі інформації зі швидкістю С біт/секунду потрібен сигнал з
потужністю (Р), який би дозволяв передавати інформацію на фоні теплового
шуму (Рш)
( р Л ґ
С = м;1о§2
1+р
= И>І0£
(12.37)
1 Ш.
де Рш = кТм? потужність шуму, м> — ширина полоси частот сигналу (Гц).
При Р « Рш (тобто, розглядаючи найменшу можливу потужність
сигналу) з (12.37) маємо (з урахуванням 1п(1 + х) = х при малих х)
и>1п
С =
кТм?
Р Р
-,отже,
1п2 ХпІкТм; кГ1п2
Р = (кТІп2)С. (12.38)
З формули (12.38) видно, що якщо для передачі С біт/с потрібна
потужність Р, то для передачі 1 біт інформації потрібна як мінімум робота
™ = кТ\п2. (12.39)
Таким чином, демон втрачає на передачу 1 біт інформації саме стільки
енергії, скільки отримає система при одержанні цієї інформації. Сцилард
вважав, що таким чином відбувається «вигнання» демона, але слід зазначити,
що при обґрунтуванні цього положення припускалося, що способи
передачі інформації базуються на електромагнітних сигналах, можливість
існування у демона деякого «миттєвого» почуття для передачі інформації при
цьому ігнорується.
Таким чином, 2-му началу термодинаміки можна надати тлумачення як
закону деградації, втрати пам'яті про систему зі збільшенням її ентропії.
Існують, однак, системи, які еволюціонують у протилежному
напрямку — впорядкування, це живі організми. Процес життя прямо пов'язаний
із насиченням інформацією. Генетична інформація, що існує вже у
найпростіших мікроорганізмах, доповнюється інформацією, яка отримується
в процесі онтогенезу (умовні рефлекси) і, нарешті, у найбільш розумної
істоти — людини — виходить за межі окремого організму і доповнюється
зовнішніми носіями інформації (книги, журнали тощо), процес
накопичення і обміну якої особливо прискорився останнім часом
(комп'ютеризація). Прогрес живого відбувається, таким чином, у напрямку зменшення
ентропії за рахунок накопичення інформації. Якщо у неживій природі
межа зменшення ентропії завершується, згідно з постулатом Планка,
нульовою ентропією, то для живих систем вводиться поняття негативної
ентропії — «негентропія» за Л. Бріллюеном. Накопичення негентропії і є
прогресом живих систем.
340
Таким чином, еволюція організмів і еволюція неживої матерії
перебігають у протилежних напрямках (рис. 12.5). Життя намагається піднятись на
той схил, з якого скочується речовина. Живий організм — це приклад
складної хімічної системи в нестійкому (з термодинамічної точки зору) стані
рівноваги, оскільки він має дуже тонку організацію і надто малоймовірну
структуру з надзвичайно низькою ентропією (рівновага організму
забезпечується за рахунок зворотних зв'язків кібернетичного плану). Однак у живому
організмі перебігають різні фізико-хімічні процеси, що веде до зростання
позитивної ентропії і врешті-решт до
рівноваги, до смерті. Щоб вижити,
кожен організм повинен постійно
позбавлятись цієї додаткової ентропії,
віддаючи її в довкілля, або, що те ж
саме, черпати негентропію з
навколишнього середовища. Для низькооргані-
зованих живих систем для цього
вистачає вільної енергії харчування (для
рослин основне джерело вільної енергії —
сонячне випромінювання), чим більш
високоорганізована жива істота, тим
більшого значення набуває зовнішня
інформація. 2-е начало термодинаміки
можна розглядати як «смертний
вирок» для неживої природи, життя
використовує ту обставину, що цей вирок
оголошено без вказівки про терміни
його виконання. Відзначимо, що
довкілля платить дуже велику ціну за про- Рис. 12.5. Еволюція неживої (а)
грес людини шляхом збільшення своєї та живої (б) матерії
ентропії. Так, людина у великих масштабах переводить цінну хімічну
енергію у теплову спалюванням нафти, газу, вугілля та інших горючих речовин;
людина добуває корисні копалини, що сконцентровані у певних місцях, і
виробляє з отриманих речовин різні речі, агрегати тощо, а далі все це
розпорошується по всій земній кулі, теж збільшуючи ентропію і т. д.
Завершуючи тему ентропія і життя, цікаво звернутись до зв'язку ентропії
і часу. Необоротність явищ у природі характеризується у часі причинно-
наслідковими зв'язками, коли минуле визначає сучасне. В той же час
необоротність, з точки зору 2-го начала термодинаміки, визначається зростанням
ентропії. Можна вважати, що причинно-наслідкове зростання часу
визначається саме необоротним зростанням ентропії, що характерно для
неживої природи. «Прив'язуючи» позитивний напрямок часу до ентропії, можна
вважати, що при зменшенні ентропії час змінюється у зворотному напрямку.
В якомусь сенсі діяльність людини це ілюструє, використовуючи поняття
мети, коли людина щось планує на перспективу, то її сучасне значною мірою
341
визначається майбутнім. Студенти прийшли в аудиторію не тому, що їх
привіз тролейбус (автобус, метро, пішки), а тому, що вони спланували
відвідування лекції і мали про це інформацію.
На закінчення розділу ще один простий приклад про зв'язок інформації
і роботи — яку треба виконати роботу, щоб вийти з приміщення, якщо
відсутня інформація про розташування дверей?
Завершуючи розділи, які присвячені термодинаміці та її застосуванням,
визначимо, що термодинаміка розглядає процес лише через низку станів,
описуючи стан рівноваги і визначаючи для нерівноважних процесів лише
напрямок їх перебігу. Механізм різних процесів, їх реальне проходження
з часом визначається кінетикою хімічних реакцій, виклад якої наводиться
в наступних розділах.
Розділ 13.
ОСНОВИ ХІМІЧНОЇ КІНЕТИКИ
Термодинамікою визначено напрям,
Дозволено процесу перебігти,
Як це реально все здійснити з часом,
Кінетика покаже, що зробити.
13.1. Основні поняття хімічної кінетики
Під час перебігу будь-якої хімічної реакції відбувається зміна
концентрацій речовин, що беруть у ній участь, причому концентрація реагентів
зменшується, а концентрація продуктів збільшується. Це графічно
зображується у вигляді кінетичної кривої. На рис. 13.1 наведено кінетичні криві
для реагенту (7) та продукту (2).
с, моль/л
'і Н
4 8 12 16 ',хв
Рис. 13.1. Кінетичні криві для реагенту (7)
та продукту (2)
Прийнято позначати
початкову концентрацію
реагенту с0 = а, поточну
реагенту : с = а - х, продукту — х.
Якщо взяти на кінетичній
кривій 7 дві довільні точки з
координатами сь Іх та с2, і2, то
зміна концентрації Ас = с2-сх
відбувається протягом часу
Аі = і2 - і\. Відношення цих
двох величин з урахуванням
того, що Ас є від'ємною
величиною, можна вважати серед-
342
ньою швидкістю г реакції на відрізку А*, тобто
г=-%- (Ш)
Поступово зменшуючи с та І до нескінченно малих величин, приходимо
до визначення істинної швидкості хімічної реакції
г = ~ % (13.2)
Оскільки с = а - х, то існує відповідне визначення швидкості через
продукт хімічної реакції
' = §• (13.3)
Зазначимо, що в рівняннях (13.1)—(13.3) швидкість реакції є додатною
величиною.
Рівняння (13.2) та (13.3) стверджуються лише для гомогенних систем
за сталого об'єму. В дійсності, у хімічну реакцію вступають окремі
частинки (атоми, молекули тощо), тому швидкість хімічної реакції слід визначати
похідною ШІсіІ, де N— кількість частинок. Для гомогенних систем логічно
віднести швидкість ЛШдХ до одиниці об'єму, і тоді сІИ!сії є пропорційним йс/йі
(якщо с вимірюється в моль/л, то у коефіцієнт пропорційності входить, крім
У= соті, також число Авогадро). Для процесів на межі поділу фаз швидкість
відносять до величини поверхні поділу, тобто замість об'ємних
використовуються поверхневі концентрації. Не важко пересвідчитися в тому (див. рис.
13.1), що швидкість реакції змінюється з часом, кінетична крива для реагенту
асимптотично наближається до осі абсцис. Таким чином, коли реакція
завершується, її швидкість прямує до нуля. Логічно пов'язати зменшення
швидкості реакції зі зменшенням концентрації реагенту. Фізично і
математично це зручно зобразити як степеневу залежність, тобто
г = ксп, (13.4)
де п — порядок реакції; к — константа швидкості', г чисельно дорівнює
к за с = 1 моль/л. Константа швидкості має розмірність: [час-1 • конц.1_и] і є
швидкістю реакції у стандартних умовах.
Для реакцій у газовій фазі замість концентрацій можна користуватися
парціальними тисками реагентівр{ = с{КТ, і рівняння (13.4) тоді буде таким:
г = крп (13.5)
(константа в цьому випадку має розмірність [час-1 -тиск1 ~п]).
Найбільш загальний метод визначення порядку реакції запропоновано
Я. Вант-Гоффом. Логарифмуючи рівняння (13.4), маємо
Іпг = \пк + піпс. (13.6)
Таким чином, п = Ща на графіку, що побудований у координатах Іпг—Іпс
(рис. 13.2). Величини г знаходять із кінетичної кривої (рис. 13.1) шляхом
графічного або чисельного диференціювання.
343
Константу к, звичайно, можна
знайти з рівняння (13.4) (або з
рівняння (13.6), що еквівалентно), але
зручніший і точніший метод
одержання константи — це інтегрування
кінетичного рівняння. За п = 1 маємо
з рівнянь (13.2) та (13.4)
Рис. 13.2. Визначення порядку
реакції за Вант-Гоффом
Лі
іання дає
1пс = 1пс0 -к(,
с = с0е .
(13.7)
(13.8)
(13.9)
або
Таким чином, у координатах
Іпс — і для реакції 1 -го порядку
маємо пряму лінію (рис. 13.3), причому
|1§сс|=£, а відрізок на осі ординат
дорівнює 1пс0 (це дозволяє знайти
с0, якщо початкова концентрація
невідома, шляхом екстраполяції;
зазначимо, що у нетрансформованих
координатах с — І проводити
екстраполяцію на вісь ординат недоречно,
бо це веде до значних помилок). Ос-
4 8 12 16
Рис. 13.3. Кінетична крива для реакції
1-го порядку в напівлогарифмічних
координатах
кільки (13.8) — це рівняння прямої лінії, його зручно обробляти методом
найменших квадратів.
Крім константи, використовують пов'язаний з нею параметр — період
напівперетворення (Ті/2), тобто час, протягом якого половина реагенту
перетворюється на продукт (рис. 13.4). За с = с0/2 та І = Ту2 з рівняння (13.8)
маємо
т,/2=^. (13.10)
З рівняння (13.10) чітко видно, що
великим значенням константи, що
має розмірність [час-1],
відповідають малі значення Ті/2, і навпаки. З
рівняння (13.9), враховуючи, що с0 = а, а
с = а - х, не важко знайти, як
змінюється з часом концентрація продукту
х = а(1-е~к1). (13.11)
Нарис. 13.1 рівнянню (13.9)
відповідає крива 1, а рівнянню (13.11)
т./2 и хв
Рис. 13.4. Визначення періоду
напівперетворення Ті/2
344
крива 2, які є дзеркальними відображеннями одна одної (зазначимо, що
точка їх перетину відповідає с = с0/2 та / = Ті/2).
Для реакції 2-го порядку кінетичне рівняння має вигляд
Його інтегрування дає
І = і- + &,
або
с =
\ + спкґ
(13.12)
(13.13)
(13.14)
Зазначимо, що за / = 0 рівняння (13.14) та (13.9) дають с = с0. Період
напівперетворення для реакції 2-го порядку дорівнює
1
4 =
кс0'
1/с, моль/л
4
(13.15)
Таким чином, для реакції 2-го
порядку період напівперетворення
залежить не лише від константи
швидкості, що в цьому випадку має
розмірність [час-1 • концентрація-1], але
й від початкової концентрації с0. Для
реакції 2-го порядку, згідно з
рівнянням (13.13), кінетична крива
випрямляється в координатах 1/с — і,
в цих координатах і§ос = £, а відрізок
на осі ординат дорівнює 1/с0 (рис.
13.5). Зазначимо, що випрямлення
кінетичної кривої у відповідних
координатах можна використати для
визначення порядку реакції, а саме, випрямлення в координатах Іпс
дить, що п = 1, а в координатах 1/с — і, що п = 2.
За довільного п кінетичне рівняння
*, хв
Рис. 13.5. Кінетична крива для реакції
2-го порядку в координатах 1/с — і
£довоюй
можна проштегрувати, що дає
і-
-сп
Л-п
/1-1
- = **.
(13.16)
(13.17)
Не важко пересвідчитися в тому, що за п
у рівняння (13.13).
Зас = с0/2та і-Ту2 з рівняння (13.17) маємо
2 рівняння (13.17) переходить
Ті / = -
2лЧ-1
ЬГ(п-і)
(за п = 2 рівняння (13.18) переходить у рівняння (13.15)).
(13.18)
345
Рівняння (13.18) можна використати для визначення порядку реакції.
Позначимо величину
2""1 -1
, ч, як /(п,к). Тоді в логарифмічній формі маємо
к(п-\)
1пТі/2 = 1п/-(л-і)1пс0. (13.19)
Для двох довільних початкових концентрацій с0 та с^ за даного п маємо
ІП-Т^ = («-^ІП-т?, ЗВІДКИ
1п-
п = -
1'/2
1п%
сл
+1.
(13.20)
Обчислити п за допомогою
рівняння (13.20) можна і з однієї кінетичної
кривої. Першу точку на рис. 13.6
беремо за с = с0/2, тобто т'/2 = ({, а с0 = с0.
Другу точку візьмемо за с - с0ІА
(тобто половину від половини) та і - і2- Для
другої точки початковою
концентрацією є с0/2, а Ху2 = 12 -1\. За рівнянням
(13.20) знаходимо порядок реакції
1п
Рис. 13.6. Обробка кінетичної кривої
для обчислення порядку реакції
л = -
1п
+ 1 = -
-1
1п
1п2
+ 1.
^о/2
Для реакції 2-го порядку, наприклад, і2 = 3^ і
ЦЗ-1) 1 о
и= ; „ ; + 1 = 2.
1п2
Рівняння (13.20) справджується навіть для реакції 1-го порядку. Так,
з рівнянь (13.8) та (13.10) маємо
Іг =Ті/4 =Щ^ = 2тУ2 =2*\- За/2 = 2^і маємо я = 1.
/с
Якщо в реакції бере участь не один, а декілька (А, В тощо) реагентів, то
кінетичне рівняння таке:
г = кспА*с£... (13.21)
Величини пА, пв тощо називаються порядками реакції за відповідними
реагентами. Загальний порядок реакції п в цьому випадку складається з
порядків за окремими реагентами, тобто
(13.22)
п = пА +лк+...
346
Наприклад, для реакції Н2 +І2 = 2НІ кінетичне рівняння таке:
г = ксИ2%. (13.23)
Ця реакція загального 2-го порядку і 1-го за кожним із реагентів (Н2 та І2).
Для зворотної реакції кінетичне рівняння має вигляд
г = кс2ш. (13.24)
Ця реакція 2-го порядку за НІ, і загальний порядок її теж другий.
Для реакції 2Т>Ю + 02 = 2И02 кінетичне рівняння має такий вигляд:
г = ке10с02. (13.25)
Ця реакція 3-го порядку: 1-го за киснем та 2-го за N0.
Щоб знайти порядок за кожним із реагентів, застосовують такий
експериментальний прийом: початкові концентрації всіх реагентів, крім одного,
беруться з великим надлишком, отже, практично змінюється лише
концентрація того з реагентів, який присутній у невеликій кількості. Іноді ця
умова виконується майже автоматично, наприклад, якщо один із реагентів
є розчинником. Так, реакція інверсії цукрози відбувається за рівнянням
С12Н22О11 + НгО = С6Н1206 + С6Н1206.
цукроза глюкоза фруктоза
Кінетичне рівняння цієї реакції: г = ксцукр (швидкість інверсії не залежить
від концентрації води, яка є у великому надлишку). Обґрунтуємо це
положення на прикладі реакції 2-го порядку з різними початковими
концентраціями реагентів (за кожним із реагентів порядок 1-й (див., наприклад,
рівняння (13.23)). Позначимо початкові концентрації реагентів через а та Ь,
а поточні, відповідно, через (а - х) та (Ь - х), де х — концентрація продукту.
Диференціальне рівняння тоді є таким:
сіх,
Ж
Розділення змінних та інтегрування за початкових умов х = 0 за і = О дає
і 1 (Ь-х)а 1
Якщо Ь » а, то 6-а- Ь і Ь-х =Ь, що дає
І1п(^)Г*'' <Ш8>
Іп(а-х) = Іпа-Ш. (13.29)
Рівняння (13.29) є інтегрального формою кінетичного рівняння для
реакції 1-го порядку (див. рівняння (13.8) з урахуванням того, що с0 = а, а
с = а -х), але з тією різницею, що замість константи швидкості реакції 1-го
порядку [час-1] тут фігурує добуток константи швидкості реакції 2-го
порядку [час-1 • концентрація-1] на початкову концентрацію реагенту, який
знаходиться у надлишку.
347
• = к(а-х)(Ь-х). (13.26)
Цікаво простежити за тим, що відбувається з рівнянням (13.27) за а = Ь.
Безпосередня підстановка а = Ьу рівняння (13.27) дає невизначеність типу
0/0. Розкриваючи її за допомогою правила Лопіталя за Ь —> я, маємо
а Іп(ь-Х)а
1 * (Ь-х)а ,. (1а (а-х)Ь
Ііт --^— 1п 7 тт = 1іт—т-^ —-
ь->аЬ-а уа-х)Ь ь^а а п \
Ь(а - х)[(Ь - х)(а - х)Ь - а (р - х)Ь~^
а(Ь-х)Ь2(а-х)2(-\)
= а-(а~х) = -т^—- = ки (13.30)
а\а-х) а\а-х)
Цьому рівнянню не важко надати вигляд рівняння (13.13), враховуючи,
що а = с0, а -х = с тах = с0 - с. Таким чином, рівняння 2-го порядку з різними
реагентами, якщо виконується умова сА = св (для виконання цієї умови
необхідно, щоб Сд = Сз та реакція відбувалась зі стехіометричними
коефіцієнтами 1:1), виявляється еквівалентним рівнянню 2-го порядку для
одного реагенту.
Для наведених вище реакцій порядки за відповідними реагентами
збігаються зі стехіометричними коефіцієнтами, тому рівняння (13.21) деякі
автори називають законом діючих мас у кінетиці за аналогією з відповідним
законом у термодинаміці, де в степенях фігурують саме стехіометричні
коефіцієнти.
Розглянемо це питання детальніше. Усі реакції можна поділити на прості
(або елементарні) та складні. Прості реакції відбуваються в одну стадію, за
один елементарний акт, складні реакції комбінуються з елементарних стадій
і поділяються на паралельні, послідовні та оборотні (див. підрозділ 13.2).
Будь-яка складна реакція або належить до одного з названих типів, або може
бути представлена як комбінація різних типів.
Прості реакції, в свою чергу, поділяються на моно-, бі- та
тримолекулярні. Молекулярність простої реакції — це кількість частинок (атомів,
молекул, радикалів тощо), що беруть участь в елементарному акті реакції.
Наприклад, реакція взаємодії водню з йодом в газовій фазі відбувається
шляхом зіткнення двох молекул Н2 та І2, після чого виникають дві молекули
НІ. Це є бімолекулярна реакція. Найбільш поширені моно- та бімолекулярні
реакції. Тримолекулярні реакції, до яких належить, наприклад, реакція
взаємодії монооксиду нітрогену з молекулярним киснем, зустрічаються
дуже рідко. Молекулярність вище трьох не спостерігається через практичну
неможливість одночасного зіткнення чотирьох та більшої кількості частинок
(ймовірність такої події практично дорівнює нулю).
Для простих реакцій молекулярність збігається із загальним порядком
реакції. Тому бімолекулярні реакції (наприклад, взаємодія Н2 і І2 та розклад
348
НІ) описуються кінетичними рівняннями (відповідно (13.23) та (13.24.)) 2-го
порядку (зазначимо, що тут мається на увазі загальний, сумарний порядок,
а не порядки за кожним із реагентів). Тримолекулярні реакції (наприклад,
взаємодія монооксиду нітрогену з молекулярним киснем) відносяться до
реакцій 3-го порядку (див. кінетичне рівняння (13.25)).
До мономолекулярних відносяться реакції ізомеризації, розкладу та деякі
інші. Вони описуються кінетичними рівняннями реакцій 1-го порядку.
Таким чином, типові значення порядків хімічних реакцій — це 1-й та 2-й
внаслідок поширеності моно- та бімолекулярних процесів.
Реакції із великими стехіометричними коефіцієнтами, наприклад, реакція
синтезу аміаку N2 + ЗН2 = 2>Ш3, не можуть проходити в одну стадію, отже,
вони повинні бути складними реакціями.
Серед реакцій із малими стехіометричними коефіцієнтами також можуть
бути складні. Наприклад, реакція Н2 + С12 описується таким кінетичним
рівнянням:
г = ксН2с%*2. (13.31)
Наявність дробових порядків свідчить про те, що реакція є складною.
Справді, реакція взаємодії водню з хлором є ланцюговим процесом (див.
підрозділ 15.3).
Таким чином, для складних реакцій порядок (як за відповідним
реагентом, так і загальний) носить формальний характер і може бути як цілим
числом, так і дробом, навіть від'ємним числом та нулем.
Одним з важливих понять хімічної кінетики є механізм хімічної реакції.
Знайти механізм складної реакції означає розкласти цю реакцію на
елементарні (з відповідними константами). Це так званий стехіометричний
механізм. Наведемо приклад.
Розглянемо реакцію окиснення двовалентного Феруму киснем до
тривалентного у кислому середовищі
4Н+ + 4Ре2+ + 02 = 4Ре3+ + 2Н20.
Якщо б ця реакція була простою, то в елементарному акті потрібно було б
зіткнення дев'яти частинок, із яких вісім мають однаковий позитивний заряд,
тобто відштовхуються одна від одної. Для цієї реакції можна запропонувати
такий стехіометричний механізм:
Ре2++02—^->Ре3++02,
02+Н+ —^->Н02,
Ре2+ + Н02 —^->Ре3+ + Н02,
но2+н+—^->н2о2,
Ре2+ + Н202 —^->Ре3+ + ОН# + ОН",
ОН~+Н+ —^->Н20
349
Ре2+ +ОН#—^->Ре3+ +0Н",
ОН~+Н+ —^->Н20.
Кожна стадія в цьому механізмі є бімолекулярною, причому на непарних
стадіях відбувається перехід електрона з Ре на нейтральну частинку, а на
парних стадіях катіон Н реагує з відповідним аніоном.
Поняття механізм може бути застосовано і для простої реакції
(відповідно до окремої стадії складної реакції). У цьому разі йдеться про механізм
елементарного акту. Цей механізм, на відміну від стехіометричного,
називають детальним, глибоким, інтимним.
Цікавий приклад різних варіантів детального механізму дає реакція валь-
денівського обернення — заміщення в оптично активних галоїдпохідних
ХСК^Кз+У >Х + УСК,Я2К3,
де К1? К2, К3 — радикали; X та У — атоми галоїдів.
Сполуки ХСК^Кзта ^СК1К2К3 мають форму тетраедрів, усі чотори
замісники є різними, центральний атом вуглецю є оптично активним. При
взаємодії атома У з ХСК^К^ можливі три варіанти:
1) атом У при зіткненні вибиває атом X з його місця, в цьому разі
симетрія частинки зберігається і, відповідно, реагент і продукт мають однакову
оптичну активність;
2) атом У підходить до тетраедра ХСК1К2К3 з боку, протилежного щодо
X, при цьому зв'язок С—X поступово рветься, а С—У поступово
утворюється, у цьому разі тетраедр «обертається», відбувається зміна оптичної
активності центрального атома вуглецю;
3) якщо зв'язок С—X є дуже полярним, то може відбуватися дисоціація
на іони: ХСК^К^ >Х"+[СК1К2К3], катіон [С^К^] має плоску
структуру. Отже, маємо ситуацію, коли ймовірність приєднання У з обох
боків є однаковою і продуктом є рацемічна суміш.
Досліди довели, що найчастіше реалізується другий механізм, отже,
у процесі заміщення відбувається обертання і, відповідно, зміна оптичної
активності (за високих значень діелектричної сталої розчинника реалізується
третій механізм). У даному разі додаткова інформація, що дозволяє обрати
той чи інший механізм, — це вивчення оптичної активності продуктів
реакції.
Для складної реакції спочатку вивчають її стехіометричний механізм,
а потім переходять до детального аналізу механізму елементарних стадій.
Основою для вивчення механізму складної реакції є дослідження її
кінетики. Вище вже зазначалося, що будь-яку складну реакцію, наприклад,
ланцюговий процес, каталітичну реакцію тощо, можна скласти, як із
цеглинок, з набору паралельних та послідовних стадій, враховуючи при цьому, що
кожна стадія може бути оборотною.
Отже, як найбільш прості серед складних реакцій слід розглянути
оборотні, паралельні та послідовні процеси.
350
13.2. Кінетика складних реакцій
Складні реакції, як зазначалося вище, поділяються на паралельні,
послідовні {консекутивні) та оборотні. Усі інші можуть бути представлені
комбінаціями цих трьох типів складних реакцій.
Прикладом паралельноїреакції є розклад мурашиної кислоти, що може
перебігати шляхом дегідратації або дегідрування:
НСООН—^С02+Н2;
НСООН—^->СО + Н20.
Обидві ці реакції є мономолекулярними і, відповідно, є реакціями 1-го
порядку
ІП =£іСНСОон
г2 =^2сНСООН-
Загальна швидкість дорівнює
г ~ г\ + г2 - ^1сНСООН + ^2СНСООН = (^1 + ^2 )сНСООН = ^сНСООН«
Отже, дослідження кінетики дає величину А: — суму констант кх та к2
елементарних стадій. Визначити їх окремо можна, аналізуючи склад
продуктів. Так,
сЬсх
г\ __ кхсИСООИ _ кх _ & _ (Іхх
г2 ^2сНСООН ^2 "х2 "х2
Л
Неважко пересвідчитися в тому, що за йЬс1/йЬс2 = сопзі хх = соп8Іх2.
Отже,
*- = сопз( = ^. (13.33)
х2 *2
Простішим прикладом оборотних реакцій є реакція ізомеризації. Для
реакції
маємо
-Л = г = Г] _ Г{ = кхСА _ ^_іСв =к1(а-х)~ к_х (Ь + х), (13.34)
де а та Ь — початкові концентрації реагенту та продукту.
Відповідно, початкові умови: х = 0 за / = 0.
Розглянемо спочатку за допомогою рівняння (13.34) стан рівноваги.
За рівноваги гх= г_х (г=0) і, отже, кх (а - х) = к_х (х + Ь)9 де (а - х ) та (х + Ь)
рівноважні концентрації реагенту та продукту відповідно. Звідки
ш-Ь (ш5)
351
Дріб __ є не що інше, як константа рівноваги оборотної реакції, отже,
маємо
^ = К. (13.36)
Для оборотних реакцій термодинаміка накладає також обмеження на
вигляд кінетичних рівнянь для прямого та зворотного процесів. Наприклад,
для константи рівноваги реакції Н2 +12 = 2НІ маємо
К =
• _ сні
(13.37)
сн2ч2
Кінетичні рівняння тут відомі (див. рівняння (13.23) та (13.24))
г1 =^1СН2СІ2' Г-\ =^-1СНІ*
Умова г, = г_ї у стані рівноваги дає
к\Сн2с\2 = £-існі
і _2
^і _ сні _^
£-1 сн2сі2
у повній відповідності до рівнянь (13.36) та (13.37).
На рис. 13.7 показано, як досягається рівноважний стан за концентрацією
НІ, в рівнянні (13.37) концентрації всіх реагентів рівноважні.
У цьому разі порядки прямої та зворотної реакцій збігаються зі
стехіометричними коефіцієнтами.
Для складних оборотних реакцій порядки часто не збігаються із
відповідними стехіометричними коефіцієнтами. Наприклад, для
гетерогенно-каталітичної реакції синтезу аміаку (див. розділ 18) маємо
^н5
^"БГ1-- (13.38, а)
^н3
Для зворотної реакції розкладу аміаку кінетичне рівняння є таким:
р
г=*-^. (13.38,6)
%
Загальне рівняння має вигляд
г=кР« -Ь-
За рівноваги г = 0, звідки
к рШіРш, _ рпщ _К
ї р'.5р1>5 5 рЗ 5 (13.40)
к ГН7 ГН, ГЩ ГКг ГЩ
352
Таким чином, кінетичні
рівняння прямої та зворотної
реакцій, що перебігають біля стану
рівноваги, повинні
узгоджуватися між собою через
стехіометричні коефіцієнти у виразі для
константи рівноваги. При цьому
порядок за азотом реакції
синтезу аміаку і відповідний
стехіометричний коефіцієнт збігаються
і дорівнюють одиниці. За воднем
для реакції синтезу аміаку
порядок дробовий (1,5), це веде до
від'ємного дробового порядку за
воднем (-1,5) у кінетичному
рівнянні для реакції розкладу аміаку
(1,5 - (-1,5) = 3). За аміаком маємо
для синтезу від'ємний порядок
Лш =-1, а для розкладу,
відповідно, яш = 1, отже, алгебраїчна
сума порядків дає 2 згідно зі
стехіометричним коефіцієнтом. Якщо
кінетичні рівняння для прямої та
/,хв
Рис. 13.7. Встановлення рівноваги реакції
Н2 + І2=2НІпри448°С:
зворотної реакції є неузгоджени- ! _ кількість НІ (моль)) що утворюється за
ми, то це означає, що кінетика ви- реакцією взаємодії водню з йодом з 0,5 моль
вчалася в умовах, що далекі від Н2 та 0,5 моль І2; II - кількість НІ (моль),
рівноваги Щ° залишається при частковій дисоціації
Розпіянемо тепер кінетику до-1 моля Щ ш - Рівноважна кількість НІ (0,78
сягнення рівноважного стану на
прикладі реакції А <=> В.
З рівняння (13.34) маємо
моль), криві І та II збігаються
& = к[(а-х)-к_1(Ь + х) =
= (*і+*-і)
кха~
к.хЬ
кх+к_
-х
— к^{х —х),
(13.41)
де через £х позначено суму (кх + к_х), а також використано рівняння (13.35).
Рівняння (13.41) є повністю тотожним кінетичному рівнянню для
необоротної реакції 1-го порядку (13.7), що для продукту має вигляд
& = к(а-х).
(13.42)
Інтегральною формою рівняння (13.42) є рівняння (13.11). Відповідно,
для рівняння (13.41) маємо
353
х = *(1-<Г**). (13.43)
За кх » к_х кг ^ кх , а за Ь = 0 х дорівнює кхаІ(кх + к_х) = а. У цьому
випадку реакція є практично необоротною, і рівняння (13.43) та (13.11)
збігаються.
За кх = к_х х = а/2 (за й = 0), а Л2 = 2^.
Розглянемо тепер послідовні (консекутивні) реакції. Візьмемо дві
послідовні необоротні реакції 1-го порядку
А—^->В—^С.
Для першої стадії маємо
сії -*,Са'
звідки сА=с°Ае~к]і. (13.44)
Це рівняння є ідентичним рівнянню (13.9) і описує перехід реагенту А
у проміжну речовину В.
Для проміжної речовини В швидкість складається із швидкості її
утворення (гх) та швидкості її витрачання (г2), тобто
сіс
—2. = гх-г2= кхсА - к2св. (13.45)
Розв'язок рівняння (13.45) шукаємо у вигляді
св=уе-**9 (13.46)
де у — деяка змінна величина.
Тоді маємо
^ = ^+Уе-кН-к2) = ^-к2сь. (13.47)
Порівнюючи рівняння (13.47) та (13.45) та враховуючи (13.44), маємо
звідки % = к1сІеік>-к')<
і в інтегральной формі
у = кА\е^^ = ^4і-[е^'-і]. (13.48)
о 2 *
Підставляючи (13.48) у (13.46), остаточно маємо
За І —> 0 та за / —> °° маємо св —> 0. Отже, концентрація проміжної
речовини повинна проходити через максимум. Знайдемо положення
максимуму з умови сісв/сіІ = 0, що дає
354
Л о — лі І- -І
с =
Відповідно,
_ 1п А:2 / Агі
Ку /Сі
сшах =_*з£А_Гв-*Апах -
/С9 /Сі І-
Проаналізуємо рівняння (13.50)
та (13.51). За к2 » кх ІтйХ-> 0 та
с тах « ^о ^ навпакИ5 за £2 « ^! /тах —
велика величина, а с™ —» Сд. Таким
чином, зі збільшенням Аг2/А:і
величина максимуму зменшується, і він
зміщується до початку координат;
навпаки, за малих значень к2 Ікх
максимум зміщується у бік великих
часів, а його величина с^
наближається до Сд. Це зображено на рис. 13.8.
Залежність концентрації
продукту С від часу можна знайти з
рівняння матеріального балансу
Са+св+сс=с°а. (13.52)
Отже,
(13.50)
(13.51)
Рис. 13.8. Кінетичні криві накопичення
проміжної речовини В за к21кх\
1— 0,1; 2 — 0,25; 3 — 0,5;
4—1,0; 5 — 2,0; 6 — 5,0
СС - СА СА СВ ~ СА САЄ СА £ _£ Iе е ]-
-Я
1 +
кр-Ь-к*-*
Ку /Сі
(13.53)
Залежність від часу концентрацій реагенту А, продукту С та проміжної
речовини В наведено на рис. 13.9, с. 356. Крива для продукту має 8-подібний
вигляд. Точка перегину на кривій сс —/(і) відповідає точці максимуму для
кривої св —/(і). Отже, доки не утворилася проміжна речовина,
концентрація продукту сс є дуже малою, крива для продукту не дуже відхиляється
від осі абсцис — це так званий індукційний період. Якщо у початковий
період стежити, як перебігає реакція, лише за концентрацією продукту, то
до закінчення індукційного періоду реакція не виявляє себе.
Вивчення кінетики консекутивних реакцій дозволяє обґрунтувати так
званий метод стаціонарних станів (інша назва — стаціонарних
концентрацій). Можна вважати, що в ланцюзі послідовних стадій може встановитися
стаціонарний стан, за якого кількість проміжної речовини залишається
сталою (стаціонарна концентрація) або, що те ж саме, загальна швидкість
355
і
0,5
0,4
0,3
0,2
0,1
к с, моль;
\а
£_ і_
Оі
С^-
/\В
і ^^Т~—==ьд
^^, *,год
Й?СВ
Л ^^""^СА ~^2СВ-
утворення — розкладу проміжної
речовини дорівнює нулю: йсв Ійі=0.
Наприклад, для реакції
А—^->В—£*->С
умова стаціонарності
Звідки
стац. #і Л| 0 _£./
Зіставляючи вираз (13.54) з рів-
0 20 40 60 80 ШО *~ нянням (13.49) для св, бачимо, що
вони збігаються, якщо к2 » кх (за
Рис. 13,9. Кінетичні криві для реагенту А, великих значень к2 є 2' -> 0). Вище
проміжної речовини В та продукту С зазначалося, що за к2 » кх с™ є
малою величиною, і максимум досягається швидко (/тах —»0). Після
проходження максимуму концентрація проміжної речовини В поступово
зменшується, і тільки тоді стає можливим, щоб відношення св/сА « соті, бо до
досягнення максимуму св збільшується, а сА завжди тільки зменшується.
Зазначимо, що за /тах -» 0 індукційного періоду практично немає.
Швидкість утворення продукту С за к2 » к{ у стаціонарному стані
дорівнює
^ = к2сГ=к2^сА=кісА. (13.55)
Тобто ця швидкість дорівнює швидкості 1-ї стадії. Ця стадія має назву
лімітуючої. Підкреслимо, що лімітуюча стадія — це стадія з мінімальним
значенням константи швидкості.
Розглянемо тепер випадок к2 «кх.З рівняння (13.49) маємо
^в=^(^"М) = с^-^. (13.56)
З іншого боку, за великих кх сА =сАе {1 —> 0, отже, перехід А у В
відбувається швидко, і лімітуючою є 2-а стадія — перетворення В на С.
Кількість проміжної речовини при цьому є великою, стаціонарний стан
не реалізується, після досягнення максимуму (св ** —> сА) реакція
перебігає, як проста реакція перетворення В на С. Таким чином, у послідовних
процесах можна виділяти лімітуючу стадію і саме її вивчати детально.
Розглянемо конкретний приклад. Реакція йодування ацетону
СН3СОСНз + І2 -» НІ + СН3СОСН2І
має таке кінетичне рівняння:
г = кст. (13.57)
і швидкість її не залежить від концентрації йоду.
356
Зазначимо, що якщо б це була проста бімолекулярна реакція, то
кінетичне рівняння було б таким:
г = ксац сІ2. (13.58)
Отже, реакція йодування ацетону є складною. Вона перебігає у дві Стадії.
Перша стадія — ізомеризація ацетону в енольну форму
СНз—С—СНз —**-> СНз—С=СН2
II І
О ОН
Друга стадія — приєднання молекули йоду до подвійного зв'язку і
відщеплення НІ, тобто
І
СНз—С=СН2 +12 —Ь-* СНз—С—СН2І > СНз—С—СН2І + НІ
І І II
ОН ОН О
За к{ « к2 перша стадія є лімітуючою, і швидкість загальної реакції
дорівнює швидкості першої стадії
г = г1=^,сац, (13.59)
що збігається з емпіричним рівнянням (13.57).
Поняття лімітуючої стадії та застосування методу стаціонарних станів
дозволяють краще розібратися в кінетиці складних реакцій. Оскільки похідні за
концентраціями для стаціонарних станів дорівнюють нулю, то метод
стаціонарних станів надає можливість перейти від диференціальних рівнянь до
алгебраїчних. Умовою застосування цього методу є висока реакційна
здатність проміжних речовин і, відповідно, мала їх стаціонарна концентрація (як
вже доведено вище, для реакції А—^—>В—&—>С це відповідало к2 » кх).
Розглянемо тепер застосування методу стаціонарних станів до дво-
стадійної реакції, у якій перша стадія є оборотною
С *-1
Метод стаціонарних станів дає для проміжної речовини В
—&- = кхсА -к2св -*_,св = 0. (13.60)
Звідки для стаціонарної концентрації проміжної сполуки маємо
сстац.= кхСА (1361)
К2 "Ь £_і
Швидкість отримання продукту С в стаціонарному стані дорівнює
357
За к2 » к_х маємо г=А^Сд, тобто лімітуючою є перша стадія, і кінетичне
рівняння є повністю тотожним рівнянню (13.55) для необоротних реакцій.
За к2 « к_{ маємо
г = М2£а=*є/Сд. (13.63)
Згідно з рівнягіням (13.36) відношення констант швидкості прямої та
зворотної реакції дорівнює константі рівноваги, отже,
ке/=к2К{. (13.64)
Таким чином, у цьому випадку лімітуючою є друга стадія, а на першій
стадії встановлюється рівновага між реагентом А та проміжною
сполукою В. Так, згідно з рівнянням (13.61) стаціонарна концентрація В при
^-і >:> ^2 визначається константою рівноваги першої стадії, тобто
с-«=сГ=-^-сА=^сА. (13.65)
Таким чином, с^1вн' є рівноважною концентрацією, точніше, квазірівно-
важною, бо рівновага встановлюється лише на першій стадії, і сА не є
рівноважною концентрацією, а змінюється з часом, поступово
перетворюючись на продукт (через проміжну сполуку).
Суворо кажучи, «стаціонарна» концентрація Сзтац* € насправді квазі-
стаціонарною, і частина авторів використовує саме термін «метод квазі-
стаціонарних станів». Цей термін є більш точним, але в роботах та
підручниках з кінетики хімічних реакцій частіше вживається термін «метод
стаціонарних станів». Нагадаємо, що цей метод широко використовується при
встановленні стехіометричного механізму різних реакцій.
Специфічним прикладом складних реакцій є спряжені реакції, тобто дві
реакції, що перебігають в одній фазі, одна (І) з яких (первинна)
А + В —> продукти (І)
перебігає самочинно, а друга (II) (вторинна)
А + С —» продукти (П)
не є самочинною, та її перебіг можливий лише за наявності первинної
реакції.
Загальна для обох спряжених реакцій речовина А називається актором.
Речовина В, яка, реагуючи з актором у первинній реакції (І), індукує перебіг
вторинної реакції (II), називається індуктором, а речовина С, що приймає
ефект первинної реакції (І) і бере участь лише у вторинній реакції (II),
називається акцептором. Явище перебігу несамочинної реакції (II) під впливом
спряженої самочинної реакції (І) отримало назву хімічної індукції.
Прикладом спряжених реакцій є спільне окиснення бромноватою
кислотою Н2803 та Н3А803. НВЮ3 безпосередньо не окиснює арсенатну
кислоту, але окиснює сульфітну і останній процес індукує окиснення Н3А803.
358
Спряження реакцій є можливим лише в тому випадку, коли є проміжні
речовини, які можуть бути зв'язуючими ланками обох реакцій. Наприклад,
для вищенаведених спряжених реакцій маємо
[НВгОз + Н2303 -> Н2304 + НВЮ2
|нВг02+Н2503-»Н2304 + НВгО .
[нВгО + Н2303 -» Н2504 + НВг
Проміжні речовини: НВг02 та НВгО є активними окисниками, і вони
окиснюють Н3Аз03. В даному випадку НВЮ3 є актором, Н2803—
індуктором, а Н3Аз03 — акцептором.
Спряженими можуть бути не лише окисно-відновні, а й інші, наприклад,
кислотно-основні реакції. Так, утворення аміду з карбонової кислоти
і аміну (МН2К') у водному розчині практично не перебігає. Додавання до
розчину карбодиіміду (X'—К=С=К—X"), який може гідратуватися за
реакцією
(Xі—Н=С=И—Xм) + Н20 -> X—Ш—СО-N11—X",
веде до спряженого утворення аміду відповідно до рівняння
К'СООН+МЬЯ" +Х—№=0=ї*-,Хи^К'СО№Ж" +Х—Ш—СО—№1—X".
Активною проміжною речовиною тут є заміщена о-ацилізомочевина,
яка утворюється за реакцією
к'соон+х'—и=с=к—хи-йс—ш—с(ос(Ж')=:к—хи,
а далі або гідратується з утворенням заміщеної сечовини
X'—Ш—С(ОСОК')=К—X" + Н20 -> X'—Ш—СО—ИН—X" + К'СООН,
або реагує з аміном з утворенням аміду
X—ИН—С(ОСОК') =И—X" ^-NН2К,,-^X,—NН—СО—NН—X" 4-К'СОКНК".
Тут карбодиімід є індуктором, амін — акцептором, а карбонова
кислота — актором.
Загальну схему спряжених реакцій можна написати у такому вигляді:
А + В->Р (1)1
? + тВ-*(т + 1)В (2)1, (13.66)
Р + иС->Е + Б (3)]
де А — актор; В — індуктор; С — акцептор; Р — проміжна речовина; В —
продукт перетворення індуктора; Е — продукт перетворення акцептора;
т — кількість молекул індуктора, що можуть додатково реагувати з Р; п —
кількість молекул акцептора, що реагує з одною активною проміжною
частинкою Р.
У цій схемі спряжені реакції утворюють двомаршрутний процес.
Перший (І) маршрут — індукуючий — реакція актора з індуктором (без
акцептора), другий (II) маршрут — індукована реакція перетворення акцептора.
359
Рівняння маршрутів такі:
А + (іп + 1)В->(іи + 1)П (1)1
А + В + лС->Е + Б (П)|*
Розглянемо конкретний приклад. Реакція окиснення бензену (акцептор)
пероксидом Гідрогену (актор) індукується іонами Ре (індуктор). Реакційна
схема така:
Ре2+ + Н202 + Н+ -> Ре3+ + ОН* + Н20 (а)
Ре2+ + ОН* + Н+ -> Ре3+ + Н20 (б)
он-+с6н6 -> с6н;+н2о (в) (1168)
[с6Н5#+ОН*-->С6Н5ОН (г)
Брутто реакція така:
2Ре2+ + 2Н202 +С6Н6 + 2Н+ ->2Ре3+ +ЗН20 +С6Н5ОН
(крім фенолу, може утворюватися також дифеніл шляхом рекомбінації
фенільних радикалів, але для спрощення цей процес не розглядаємо).
Активною проміжною частинкою тут є радикал ОН*, який у першому маршруті
(без акцептора) реагує з Ре , а в другому — з акцептором — бензеном. Для
цих реакцій, згідно зі схемою (13.67), маємо
2Ре2+ + Н202 + 2Н+ -» 2Ре3+ + 2Н20 (І) 1
, (13.69)
Ре2+ +Н202 +Н+ +0,5С6Н6 -> Ре3+ +1,5Н2О + 0,5С6Н5ОН (П)]
Тут т = 1 і п = 0,5 (згідно зі схемою (13.68) один радикал ОН*
перетворює: або 1 іон Ре + на Ре +, або півмолекули бензену — на фенол (на
перетворення 1 молекули бензену згідно зі стадіями (в) і (г) рівняння (13.68)
потрібно 2 радикали ОН*)).
Кількісною мірою ефективності хімічної індукції є фактор індукції (Ф),
який визначається як відношення швидкості витрати акцептора до швидкості
витрати індуктора. Згідно зі схемою (13.67)
ф = КС) = пгг
г(В) (т + і^+га' (117°)
де гх та г2 — швидкості рівнянь (І) та (II) схеми (13.67).
гх не залежить від концентрації акцептора (Сс), а г2 збільшується з ростом
Сс. З рівняння (13.70) видно, що коли г2 дуже велика величина (при великих
значеннях Сс), то г2» (т + 1)гь і фактор індукції прямує до граничного
значення Фит=>л. Отже, рівняння (13.70) можна записати таким чином:
ф Фіип'*2 (13в?1)
(т + 1)г,+г2 ч '
Хімічна індукція є одним із засобів реалізації процесів, які перебігають
з ростом вільної енергії Гіббса (ДС? > 0) («термодинамічно заборонені»
360
процеси). Для того щоб у системі могла перебігати реакція, для якої АС > 0,
треба над системою виконувати роботу, тобто мати джерело вільної енергії.
Таким джерелом у даному випадку є вільна енергія (АС{) первинної реакції
(І), яка індукує вторинну, для якої АСП > 0. Сумарне значення вільної енергії
у системі спряжених реакцій повинно бути від'ємним, тобто
ДОе = ДЄі + ДОн<0. (13.72)
Спряжені реакції відіграють велику роль у біохімії. Біосинтез білків та
нуклеїнових кислот у клітині перебігає зі збільшенням вільної енергії, і це
компенсується спряженням синтезу з гідролізом пірофосфатних зв'язків
молекул АТФ — аденозинтрифосфорної кислоти. В свою чергу, утворення
АТФ перебігає як реакція, що спряжена з процесами окиснення.
Після встановлення стехіометричного механізму, визначення лімітуючої
стадії складного процесу, отримання інформації про константи
елементарних стадій необхідно вже вивчати саме елементарні реакції. Логічно
починати з газів, де чітко визначені реагуючі частинки: атоми, молекули тощо.
Базою для вивчення елементарних реакцій у газоподібному стані є, в першу
чергу, молекулярно-кінетична теорія газів.
361
Розділ 14.
КІНЕТИКА ЕЛЕМЕНТАРНИХ ПРОЦЕСІВ
Щоби бар 'єр енергетичний подолати,
Частинка змушена себе активувати.
14.1. Молекулярно-кінетична теорія газів
Молекулярно-кінетична теорія газів розглядає лише поступальний рух
частинок. Згідно з формулою Больцмана кількість частинок Иь що мають
енергію є/9 дорівнює
Л^Л^-6'^. (14.1)
Для поступального руху спектр енергій є квазібезперервним, отже,
умову нормування ЕЛ^- = к9 де N— загальна кількість частинок, можна
записати таким чином:
|<ЯУ = М
(14.2)
Тут <ІИ— кількість частинок, що мають швидкості від их до их+ сІих
(в одновимірному випадку), тобто
(Ш = Ве-т*/2кТ(Іиг
(14.3)
З умови (14.2) можна знайти нормувальний множник В, який дорівнює
В = и\-^\ . (14.4)
[2пкТ
Таким чином, відносна частка частинок, що мають швидкість від их до
1/2
і^ + сІих, є такою:
М =( т 1 е'тх)
N 2пкТ
^2кТсіих. (14.5)
Вираз (14.5) є розподіленням Максвелла-Больцмана для одновимірного
поступального руху. При обчисленні В і надалі використовуються табличні
інтеграли типу
/л
-"Ос.
п
оо
1
0
0
£ґ
1
1
2а
2
ІґтО
1/2
3
1
2а2
4
ьт
5
1
2а3
За парного п / = 2І, за непарного п 1 = 0.
З розподілення за швидкостями можна знайти середню швидкість. Для
362
безперервних величин (х) теорія ймовірності дає для середнього (х)
значення вираз (8.70), який при умові нормуванняДх) на одиницю має вигляд
х = І ІЇМ^х*
деДх) — густина ймовірності.
В даному випадку
Таким чином, для середнього значення швидкості дх маємо
їїх = ] VX^{VX)<ІVX = ї (т/2шТ){/\хе-т^2кТ^х = 0.
—оо —оо
Це означає, що система, як ціле, не рухається.
Знайдемо тепер середнє значення их в обраному напрямку х
\2пкТ) уіпт
(14.6)
(14.7)
(14.8)
(14.9)
У тривимірному просторі частинка рухається незалежно вздовж будь-
якої декартової осі (рх9 іл, та V2), отже, ймовірність того, що вона має
швидкість від их до их + йох, від иу до иу + йоу та від V; до ь2 + йо2, дорівнює
добутку відповідних густин ймовірностей для одновимірних рухів, тобто
4-
т
3/2
п(иІ+и^+о^\/2кТ
(14.10)
уІпкТ
Добуток Аихйиу(1\)г =сіт— це
елемент об'єму в просторі швидкостей, а
Vx+Vу+V2 =V , ДЄ V радІуС-ВЄКТОр
у тому самому просторі (рис. 14.1).
Таким чином,
Формула (14.11) показує, що для
тривимірного простору раціонально
користуватися сферичною системою
координат. У цій системі елемент
об'єму (див. формулу (8.64) та рис. 8.7,
с. 138) у просторі швидкостей набуває вигляду
^хсіиусІи2.
Щр
^-
Рис. 14.1. Простір швидкостей
Л = и ^і>8Іп6йШф.
(14.12)
363
Рух частинки у просторі є довільним, тому залежність від кутів не грає
ролі, і за кутами можна проінтегрувати, що дає 4я.
Отже, ймовірність того, що швидкість частинки має абсолютну
величину ВІД V ДО V + <Й>, ДОрІВНЮЄ
Я")^ = ґ;гг^1 е-т"2/2кТ4™2<іи. (14.13)
уіпкТ ^
Зазначимо, що 4пи2<1и — це об'єм (у просторі швидкостей) кільцевого
зазору, що дорівнює добутку площі сфери 4пи2 на товщину зазору сій.
Знайдемо тепер середню швидкість частинок у тривимірному просторі
0=^и/(и)іІи =
V4п\
(—І
3/2
■2/2«Г2
и^о =
топ )
(14.14)
Порівняння (14.14) з (14.9) показує, що V = 45^.
Оцінимо середню швидкість молекули азоту за температури 298 К.
Згідно з(14.14), враховуючи, що т = МІЙ, де М— молярна маса, маємо
V =
8-1,38.1(Г23.298
3,14-
2840
-з
ч23
= 470 м/с.
6,02-10'
Крім середньої швидкості, використовують також середню квадратичну
швидкість (укв). Знайдемо спочатку середнє значення квадрата швидкості
оо
\3/2
у2=]УДі,)</у= 4яГ
т
2шТ
72кГ.,=ЗкГ.
и4в"шГ/ї*і'Л> =^-; (14.15)
тп
,.,РГ=(
т )
(14.16)
Відношення середньої квадратичної швидкості до середньої не дуже
відрізняється від одиниці, а саме:
V
ЗкТ/т
\У2
Зя
У2
т1 ■'■
ікТ/пт
У виразі дляДі;) (14.13) є множники, один з яких (и2) збільшується зі
зростанням і;, а другий {е"тхз1і2кТ) зменшується. Це веде до того, що функція
Ар) має максимум. Знайдемо швидкість (ит)9 що відповідає цьому
максимуму. Цю величину можна назвати найбільш імовірною швидкістю. от
знаходимо з умови
—— = 0, звідки
(XV
364
,„=ґм|
1/2
т
(14.17)
Таким чином, Vт є меншою від
ита йкв.На рис. 14.2 зображено
розподілення Максвелла-Больцма-
на і нанесено відповідні значення
швидкостей.
Характер кривоїДи) суттєво
залежить від температури—за низьких
температур максимум є гострим
і наближеним до осі ординат, зі
збільшенням температури розподі- Рис. 14.2. Розподілення Максвелла-
лення/(у) розмивається, як це зоб- Больцмана для швидкостей
ражено на рис. 14.3. У тривимірному просторі
На відміну від розподілення Ді>), яке є асиметричним, розподілення/^)
є симетричним, причому віссю симетрії є вісь ординат, відповідно,
максимум/^) припадає на середнє значення их = 0. Відповідна крива (рис. 14.4,
с. 366) має назву кривої Гаусса^
Середнє значення квадрата у2 не дорівнює нулю
оІ=\иІГ(рх)^х =
—1
2пкТ )
Ї/2КТ2
кТ
т
(14.18)
Відповідно, середнє значення
кінетичної енергії для одновимірного
руху дорівнює
2
-*!.. (14.19)
Оскільки напрямки в просторі є
незалежними, то для є2 та є
отримаємо той самий результат, тобто
- кТ
сх ьу с2 .
(14.20)
Формула (14.20) ілюструє закон
рівномірного розподілу енергії за
ступенями свободи.
Для тривимірного руху
Рис. 14.3. Залежність/^)
від температури
Р = С X С X С —^
Є = Є
+ гу + єг
= ±кТ.
Цей результат збігається з формулою (14.15), оскільки
є=^-
пвкТ __ 3 кТ
2т 2
(14.21)
(14.22)
Розглянемо тепер зіткнення між частинками. Дві частинки, що руха-
365
-V*
О
ються назустріч одна одній, зіткну-.
ться, якщо вони потраплять у
середину циліндра з площею перерізу
(рис. 14.5)
а = яД2=ті(гА+гв)2, (14.23)
де гА та гв — радіуси частинок.
При цьому частинки одна
відносно іншої рухаються з відносною
швидкістю увідн>. Тоді об'єм циліндра
Рис. 14.4. Розподілення Максвелла-
Больцмана для одновимірного руху д Р
<™відн. = *о- (14.24)
Ця величина має назву фактор зіткнення і відповідає ймовірності
зіткнення частинок в одиниці об'єму в одиницю часу. Не важко
пересвідчитися в тому, що г0 має
розмірність [об'єм-час-1]. Відносна
швидкість обчислюється за
формулою, схожою з (14.14)
У формулі (14.25) замість маси
частинки (т) фігурує так звана
приведена маса (ц,)
Рис. 14.5. Зіткнення двох частинок
И =
_ "*а"*в
(14.26)
тА+тв
де тА та /ив — маси частинок.
За тА »тв ц = тв, тобто можна вважати, що частинка з великою масою
тА практично не рухається відносно частинки з дуже малою масою.
ЗатА=:тв = т, тобто для однакових частинок, \і = тії і
^відн.=^. (14.27)
Для однакових частинок гА = гв = г, отже, площа перерізу дорівнює
а = п(2г)2 =т/2,
(14.28)
де й—діаметр частинки.
Частота зіткнень V між частинками дорівнює добутку 20 на кількість
частинок в одиниці об'єму, тобто
(14.29)
Відповідно, довжина вільного пробігу А,, тобто шлях, що проходить
частинка між зіткненнями, дорівнює
366
V
*>вщн.те
(14.30)
де т = І/у — час між зіткненнями.
Для однакових частинок, згідно з формулою (14.27), маємо
Я = -^=. (14.31)
Величина VIИ= ф — це об'єм комірки, що припадає в середньому на
одну частинку. Оскільки ИІУ= с, то формулу (14.31) можна записати так:
а>/2
(14.32)
За стандартних умов (298 К, 1 атм) об'єм 1 моля ідеального газу дорівнює
приблизно 25 л = 0,025 м3 і, отже,
Ф =
0,025
6,02-10
23
= 4.10"26м3.
Вважаючи */ = 5 • 10~ м, маємо а = 8 • 10~ м2 і X = 3 • 10~ м. Середня
відстань між частинками: / = (ф) = 3 • 10" м. Таким чином, X »/, і кожна
частинка пролітає через 10 комірок, перш ніж зіткнеться з іншою.
14.2. Явища переносу в газах
Інформацію про молекулярно-кінетичні параметри (а, X) можна
отримати, вивчаючи явища переносу в газах.
Знайдемо спочатку кількість частинок (со), що перетинають в обраному
напрямку плоску поверхню, площа якої дорівнює одиниці. Усі частинки, що
мають компоненту швидкості дх в напрямку, перпендикулярному поверхні,
досягнуть її (рис. 14.6). Враховуючи, що концентрація частинок дорівнює с,
одержимо
(О = с0х. (14.33)
Поверхня може бути уявною, а
може бути реальною; в останньому
разі со — це кількість зіткнень
частинок зі стінкою (за одиницю часу на
одиницю поверхні).
З рівняння рУ = ИкТ маємо с =
= ИІУ = рІкТ. ї)х визначимо з
формули (14.9). Отже,
Рис. 14.6. Зіткнення частинки
з поверхнею
(0 =
р ( кТ ]
1/2
ч1/2'
(14.34)
кТ{2пт) (2пткТ)и
Формула (14.34) відома як формула Кнудсена-Герца.
Явище ефузії дає можливість вимірювати масу (#) частинок, що вилі-
367
тають крізь отвір у вакуум із посудини,
тиск усередині якої дорівнює р (рис.
14.7).
Віднесена до одиниці поверхні, ця
маса (#) є пропорційною потоку
частинок (со), тобто
£ = сот, (14.35)
де т — маса окремої частинки.
З формул (14.34) та (14.35) маємо
р = Ц2птКтГ=4^Т.(Н36)
пг у М )
Формула (14.36) дає можливість
за даними з ефузії визначити тиск. Це
так званий метод Кнудсена, він
застосовується для точного вимірювання малих тисків.
Розглянемо тепер процес переносу у загальному вигляді. Позначимо
через П — потік, тобто швидкість переносу деякої властивості (І). Потік
через поверхню в обраному напрямку дорівнює
П = соІ = оу:, (14.37)
де со визначено формулою (14.33).
Сумарний потік П2 через поверхню у двох протилежних напрямках, якщо
немає ніяких градієнтів і газ є однорідним, повинен дорівнювати нулю
ПХ=П-П = (^-^)І = 0. (14.38)
Якщо є градієнт деякої властивості в обраному напрямку (наприклад,
уздовж осі х), сумарний потік через обрану поверхню вже відрізняється від
нуля.
Оберемо деяку поверхню (АВСБ на рис. 14.8), що розташована
перпендикулярно напрямку змінення властивості Ь (§га<іІ). На відстані X в обидва
боки від АВСО візьмемо ще дві поверхні. Тоді
П2 = П - П = Ці - А,§гасі£) ~0)(і + Х%т<М) =
= -2А,со§гасі£ = -2Хсдх&дАЬ.
Розглянемо тепер конкретні властивості. Нехай Ь — це енергія частинки
(вважається, що існуюча неоднорідність суттєво не порушує розподілення
Максвелла-Больцмана)
Ц = 3/2КТ=СиТ. (14.40)
Тоді 8гаіІ= Си§гас1Г і
. П = -2ХсихСи&гайТ. (14.41)
Рис. 14.7. Ефузія газів. 1 — посудина;
2 — отвір, розмір (0) якого малий
порівняно з відстанню (X) між
молекулами (якщо отвір великий,
то газ просто витікає, що вже не є
ефузією)
368
З іншого боку, феноменологічно
для потоку теплоти маємо
(14.42)
2 4
П = ^ = -%2гасі7\
Отже, коефіцієнт
теплопровідності (%) для ідеальних газів з
урахуванням формули (14.31) дорівнює
х = 2ХсохС1)=^£±. (14.43)
і—
А
і
1
її
X
В
т—
X
А
с
и
1
НІ
У
Коефіцієнт теплопровідності має
розмірність [Дж/К-м-с].
Тепер розглянемо внутрішнє тертя
(в'язкість). На рис. 14.9 зображено дві паралельні
поверхні. Одна з них АВ є нерухомою, друга
СВ рухається вздовж осі у. Шари, що межують
із площиною АВ, є нерухомими (і; = 0), а
шари, що знаходяться біля площини СБ,
рухаються з максимальною швидкістю вздовж
осі у. Таким чином, виникає (вздовж осі х)
градієнт швидкості V)» згасІЦу = д\)уІдх.
Віднесена до одиниці площі сила зсуву, тобто
тангенціальна сила, що рухає одну площу
відносно іншої, дорівнює
Рис. 14.8. Процес переносу в газах
С
О
хк
А
В
Рис. 14.9. Внутрішнє тертя
в газах
/=-Л8га(1^ С14-44)
де Т] — внутрішнє тертя, в'язкість. Віднесена до одиниці площі сила в системі
СІ вимірюється у паскалях (Па), розмірність ^гасії^ =1/с, отже, розмірність
ті [Па-с].
Властивість Ь, що розглядається тут, це імпульс (тоу), внутрішнє тертя —
це перенос імпульсу і &т&Ь = т£га.йиу.
З рівняння (14.39) маємо
П = -2Хсихт §гасі иу. (14.45)
Швидкість переносу імпульсу (потік імпульсу) — це і є сила (тут
тангенціальна). Отже, порівнюючи (14.44) та (14.45), маємо
ц = 2Хсйхт = ^^. (14.46)
Нарешті, розглянемо дифузію, тобто потік речовини. Згідно з першим
законом Фіка
= -£)§гас1с,
(14.47)
тут Б — коефіцієнт дифузії, ШІЛі—це потік частинок, що перетинають оди-
369
ничну площину за одиницю часу. Розмірність АИІАі [м"2-с-1], £гасі с має
розмірність [м~3-м-1], отже, коефіцієнт дифузії має розмірність [м2-с_1].
З позиції молекулярної теорії відповідна властивість Ь, що
розглядається, — це концентрація частинок с. У формулі (14.37) с вже фігурує. Отже,
в даному разі дотік П — це со, тобто потік речовини <ПЯІйІ\ формула (14.39)
використовується у вигляді
П = -2Ат;х§пі(Іс. (14.48)
Таким чином, для коефіцієнта дифузії маємо
0 = 2Хи=^^. (14.49)
ос
Наведені вище формули (14.43), (14.46) та (14.49) для коефіцієнтів
теплопровідності, внутрішнього тертя та дифузії містять такий параметр, як
а, отже, вони дають можливість експериментального визначення площі
перерізу. Зазначимо, що згідно з моделями кінетичної теорії газів
коефіцієнти %, і] та £> пов'язані один з одним. Наприклад
ті 2дхХтс мт % ґлл СЛЧ
о=-%Х = ст = ІТ = у=^ (1450)
де § — маса газу; р — його густина.
14.3. Теорія зіткнень
На кінетичній теорії газів безпосередньо базується теорія зіткнень.
Ця теорія хімічної кінетики стосується бімолекулярних реакцій. Можна
вважати, що швидкість бімолекулярної реакції залежить від кількості зіткнень
між частинками за одиницю часу в одиниці об'єму. Це число 2 можна
знайти, домножуючи частоту зіткнень V між частинками на їх концентрацію с,
тобто
2 = ус = 20с2 (14.51)
Якщо частинки різні (А та В), тоді
7 = 20сАсв, (14.52)
де сА та св — концентрації частинок А та В, відповідно, тут розмірність с
[об'єм-1]. Для переходу до молярних концентрацій треба врахувати число
Авогадро.
Якщо зіставити отримані рівняння (14.51) та (14.52) з відповідними
рівняннями формальної кінетики для реакцій 2-го порядку (13.12) та (13.21) (за
пА = пв= 1), то можна зробити висновок, що константа бімолекулярної
реакції — це якраз і є фактор зіткнення — 20 (14.24).
Оцінимо час перебігу бімолекулярної реакції, вважаючи, що кожне
зіткнення веде до хімічного перетворення, тобто до переходу молекул
реагентів у продукти. Для реакції 2-го порядку період напівперетворення
370
виражається формулою (13.15). У цьому разі, згідно з формулами (14.29) та
(14.30),
1 = 1 -1-^
ІСС„ 2пСп V V*
Т1/2 - ■
Підставляючи одержані вище оцінки для V та X, маємо т1/2 = 3 • 10~ /470 =
= 6,4-10" с. Таким чином, якщо б усі зіткнення приводили до утворення
продуктів, то бімолекулярні реакції закінчувалися б за надзвичайно
короткий час. У дійсності, основна частина зіткнень — це так звані пружні
зіткнення, після яких частинки розлітаються без внутрішніх змін.
С. Арреніус припустив, що у хімічну реакцію вступають лише так звані
активні частинки, які мають енергію, достатню для подолання
потенціального бар'єра. Цей бар'єр має назву енергії активації. Він
виникає внаслідок необхідності розриву зв'язків у молекулах реагентів.
Наприклад, у реакції Н2 + І2 необхідно розірвати відносно міцні зв'язки
Н—Н та І—І. Частку активних частинок N ЇЙ, що мають енергію, вищу за
означену, можна знайти, користуючись формулою Больцмана
N1
N
є належить одному елементарному акту, а£, відповідно, 1 молю частинок.
Вважаючи, що кількість активних зіткнень пропорційна експоненті
(14.53), маємо для константи швидкості бімолекулярної реакції
-Е/ЯТ
2е-е/кТ=е-Е/ЯТя
(14.53)
к = 20е
Отже, з графіка в координатах
Арреніуса (\пк— 1/Г) можна знайти
енергію активації (рис. 14.10, і§ (3 =
= ЕІЕ). Можливості теоретичного
обчислення енергії активації теорія
Арреніуса не дає.
Якщо рівняння Арреніуса (14.54)
прологарифмувати та знайти
похідну за температурою, то воно
набуває вигляду
<£"&■ 0"*>
Застосувавши диференціальну
форму рівняння Арреніуса (14.5) до
оборотних реакцій, згідно зі
співвідношенням (13.36), маємо
(14.54)
-15 г
Рис. 14.10. Визначення енергії
активації для реакції 2НІ = Н2 +12
Звідки
а\хікх (Нпк^ = Ех-Е_х = сіїпК , ЛЯ
371
або
і
Реагенти і
і
Щ!\ Т
Е> \
і
>
І \
я \
Е-і
Продукти
Рис. 14.11. Енергетичний профіль реакції
£і-£_і=Д#, (14.56)
Е-{=Еі-АН=Еу+д.
Співвідношення (14.56)
схематично зображено на рис. 14.11.
У цьому разі пряма реакція є
екзотермічною (# > 0, А#< 0), а
зворотна — ендотермічною. Не
важко пересвідчитися в тому, що
енергетичний бар'єр ендотермічного
процесу перевищує бар'єр
екзотермічного процесу саме на
величину теплового ефекту реакції.
Навіть коли Еекз = 0, то £енд = |АЯ|
(цей випадок відповідає,
наприклад, реакціям рекомбінації
атомів, для зворотної реакції розкладу молекул на атоми £енд = В — енергії
дисоціації молекули на атоми). Зазначимо, що рівняння (14.56), так само як
(13.36), пов'язують кінетичні параметри (кь к_ь Е{, Е_х) з
термодинамічними (К, АН).
З графіка, наведеного на рис. 14.10, можна знайти не лише енергію
активації, а також ІП20 і, таким чином, зіставити теорію зіткнень з
експериментом. Необхідні для теоретичного розрахунку г0 (14.24) величини а
можна брати з даних (%, Дт])з явищ переносу.
Величини с можна знаходити і безпосередньо шляхом вивчення кінетики
бімолекулярних реакцій методом молекулярних пучків. Цікаві результати
при застосуванні цього методу було одержано при вивченні взаємодії
атомів лужних металів з
молекулами галоїдпохідних, тобто для
реакцій
Ме + ХУ >Ме + У
(Ме—лужний метал, X—галоїд, У:
X, Н, К, де К—органічний радикал).
У відповідній камері (рис. 14.12)
пучки лужного металу та ХУ
спрямовані під кутом 90° один до
одного. Виявилося, що реакції типу Ме +
+ КХ > МеХ + К", наприклад,
К + СН3І > КІ + СН;
відбуваються шляхом зіткнення молекул
(механізм «рикошету»), причому
площі перерізів зіткнень мають
величину порядку 10" м2 (0,1 нм2),
СНЛА Вг2
Рис. 14.12. Дослідження
з молекулярними пучками
372
—^-—■*--
• Н> 1—•
тобто це саме та величина, що задається формулою (14.23). Це звичайна
ситуація, коли перерозподіл зв'язків та електронної густини між
частинками реагентів та продуктів відбувається саме при зіткненні.
Зовсім інша картина спостерігається, коли У — не органічний радикал,
а галоїд, наприклад, для реакції К + Вг2—»КВг + Вг#. У цьому разі реалізується
так званий механізм «зриву». Молекули КВг при вивченні розподілення за
кутами знаходяться саме там, де атоми Калію, що не прореагували.
Відповідно зберігають «свій» напрямок, що заданий реагентом ХУ, частинки У
Тобто реакція відбувається таким чином, ніби атом Калію, пролітаючи повз
молекулу ХУ, «знімає» з неї один з атомів Брому, інший атом Брому
продовжує свій шлях, а зіткнення не відбувається. Встановлено, що в механізмі
«зриву» площі перерізів зіткнення на порядок більші, ніж під час
«рикошету», тобто досягають 10" м2 (1 нм2) і більше.
Дуже великі значення о свідчать про те, що між частинками існують
далекодіючі сили притягування. Дійсно, модель для визначення а (рис. 14.5,
с. 366) побудовано для невзаємодіючих частинок, тобто за умови, що між
частинками немає далекодіючих сил притягування або відштовхування
(взаємодія в цьому разі зводиться
тільки до зіткнень, і частинки мають
лише кінетичну енергію). Якщо ж
між реагуючими частинками є
взаємодія, то у разі відштовхування
а « п(гА + гв)2, а у разі
притягування а » п(гА+гв)2. Останній
випадок зображено на рис. 14.13.
Траєкторії за рахунок взаємодії є скрив- Рис. 14.13. Площа перерізу (а)
леними, притягування спрямовує Для частинок, що притягуються одна
шляхи частинок один до одного (за до °ДНШ
відсутності притягування ті частинки, що знаходяться за межами циліндра
я(гА + гв)2, не зустрінуться). За рахунок чого ж виникає притягування, якщо
атоми К та молекули Вг2 є нейтральними частинками?
Це можна пояснити за допомогою так званого «гарпунного» механізму.
Атом Калію, знаходячись на деякій відстані від молекули брому, закидає на
неї електрон. Нестійкий молекулярний іон ВрГ, що утворюється при
цьому, швидко розпадається на атом Брому та бромід-іон, катіон К+ підтягує до
себе Вг~ з утворенням молекули КВг, а інший атом Брому продовжує свій
шлях у просторі відповідно до встановленого явища механізму «зриву».
Розглянемо це питання докладніше. Спочатку простежимо взаємодію
атома лужного металу з атомом галоїду. На рис. 9.5, с. 198, зображено
потенціальні криві іонної Ме Х~ (І) та ковалентної МеХ (II) молекул. За г —> °°,
тобто для ізольованих атомів, крива І завжди розташована вище кривої II.
Справді, величина АЕ = І-г(І— енергія іонізації лужного металу, а є —
спорідненість до електрона галогену) більша нуля, оскільки [/] більше [є] для
будь-якої пари атомів. Отже, утворення двох ізольованих іонів із двох
373
ізольованих атомів є процесом енергетично невигідним. Навпаки, при
зближенні іонів Ме та X" між ними виникають великі кулонівські сили,
а крива II для ковалентних структур має дуже неглибокий мінімум. Отже, за
малих відстаней крива І розташована нижче кривої II, і утворення іонної
молекули є енергетично вигідним. На деякій відстані гс » г0 (де г0 —
рівноважна відстань у молекулі Ме X") криві І та II перетинаються. Саме ця
точка визначає момент включення «гарпунного» механізму, за г = гс
електрон перекидається з атома лужного металу на атом галогену. Оцінимо
величину гс. Оскільки гс » г0, то можна вважати, що права частина
кривої І — це суто кулонівська взаємодія, тобто
1/ = _е2(гк.^А-) (14.57)
г
де г — відстань між іонами; 2К+, 2А- — заряди іонів; є — заряд електрона.
Крива II на великій відстані практично не відхиляється від осі абсцис,
отже, за г = гс кулонівська взаємодія повністю компенсує АЕ
[*У] = [Д£] = [/-є]. (14.58)
Звідки маємо
'•• V-.]- (і4-59>
Для одного і того ж самого лужного металу /— стала величина, отже,
координати точки перетину визначаються спорідненістю до електрона,
збільшення є веде до збільшення гс. Дуже велика спорідненість до електрона
у молекули N02 О"1' називають за це «надгалогеном») і, справді, для реакції
К + N02 = К^02 встановлено, що а має дуже велике значення 10" м2
(10 нм2).
Молекули галогенів мають великі значення спорідненості до електрона,
отже, для них гс є відносно великою величиною. При цьому «закинутий» на
Х2 електрон потрапляє на антизв'язуючу молекулярну орбіталь і робить
молекулярний іон Х2 дуже нестійким. Розрахунки показують, що загс-г0 =
= 4 • 10~ м і й = 5 • 10 м/с, час між переходом електрона з атома лужного
металу на молекулу галогену і зіткненням іонів Ме і X"—дорівнює -10" с,
що на порядок більше, ніж час (10" с) розпаду іона Х^ на X" та X. Для
сполук КХ, наприклад СН3І, спорідненість до електрона — від'ємна величина,
це, згідно з рівнянням (14.59), дуже зменшує гс, аж до значень г0. Отже,
перенесення електронної густини відбувається разом із зіткненням
частинок (звичайний механізм «рикошету»).
Теорія зіткнень дає непогані результати при обчисленні 20 для відносно
нескладних безструктурних частинок (атоми, невеличкі молекули та
радикали). Для складних молекул між 2®ксп' та ^еор- виникають значні
розбіжності.
Розглянемо реакцію димеризації бутадієну. Кінетика цієї реакції
відповідає 2-му порядку
374
Г — лГбуг,
(14.60)
що є характерним для бімолекулярних реакцій.
Згідно з (14.24) та (14.25) г0 залежить від температури, як \ІТ, тому для
зіставлення теорії з експериментом візьмемо не 20, а
2°"4т'
(14.61)
На рис. 14.14 наведено експериментальні дані в координатах 1§-т=—уг
(константа швидкості 2-го порядку, виражена в см3/моль • с).
Екстраполяція прямої на вісь ординат дає: І£ 20' = 8,4, звідки маємо (20')експ=
= 2,5-10 см3/моль-с-К1/2. Згідно з формулами (14.24)—(14.28)
м
_ ^відн. _
псі'
теор.
V
8*Г
піп 12
2л/г 2уіТ
Цей результат домножуємо на
число Авогадро, щоб перейти до
РОЗМІРНОСТІ (20')експ.- ВиКОрИСТОВу-
ючи значення М = 54, сі = 5 • 10 см,
одержимо/2(20*)теор = 6,5-1012 см3/
С'МОЛЬ-К . Отже, МаЄМО (Оексп/
Ютеор. = 4-10-5«1.
Ця велика розбіжність виникає
внаслідок того, що для зіткнення
складних молекул необхідна їх
відповідна орієнтація у просторі. У
нашому випадку дві молекули
бутадієну СН2=СН—СН=СН2 у процесі
димеризації повинні орієнтуватися
«голова до хвоста». Умовно це
можна зобразити таким чином:
[• • • •]. Ймовірність такої орієнтації є дуже малою величиною,
але її можна врахувати шляхом використання так званого стеричного
фактора —р. У рівнянні Арреніуса (14.54) величину, що стоїть перед
експонентою, позначимо к0 і назвемо її передекспонентою, тоді це рівняння набуває
вигляду
Рис. 14.14. Залежність константи
швидкості димеризації бутадієну
від температури
к - к0е
ГЕ/КТ
Таким чином,
де р — стеричний фактор.
%о Р%о
(14.62)
(14.63)
__ теор.
Оскільки к0 =20 , а і0 =2^ , тор дорівнює відношенню цих величин,
що для реакції димеризації бутадієну становить 4-10~ , тобтор « 1. Для усіх
реакцій величина/? знаходиться емпірично, як у наведеному вище прикладі
для реакції димеризації бутадієну.
375
Теорія зіткнень не дає можливості для теоретичного обчислення сте-
ричного фактора. Отже, для реакцій між складними молекулами теорія
зіткнень не може нічого передбачити, з експерименту емпірично
знаходяться як енергія активації, так і передекспонента к0. Теорію зіткнень можна
застосувати до тримолекулярних реакцій, але вона не придатна для
мономолекулярних процесів. Отже, ця теорія поступається місцем більш
універсальній теорії перехідного стану {активного комплексу), розробленої
вперше Г. Ейрінгом, М. Евансом та М. Поляні спочатку під назвою «теорія
абсолютних швидкостей реакцій».
14.4. Теорія перехідного стану (активного
комплексу)
Теорія перехідного стану розглядає систему
на вершині (рис. 14.11) потенціального бар'єра
(енергетичного профілю реакції). Сама система
має назву активного комплексу (АК ) (тут і
надалі все, що відноситься до активного комплексу,
позначаємо зірочкою). Швидкість реакції
визначається концентрацією с*АК та часом т* існування
активних комплексів, тобто
г = ^-. (14.64)
Вважається, що в реакції між активними
комплексами та реагентами встановлюється
статистична рівновага, тобто виконується
розподілення Максвелла-Больцмана між активними
частинками та реагентами. Ця ситуація схожа з розглянутою у розділі 13 для
квазірівноважної проміжної сполуки. Різниця
полягає в тому, що активний комплекс є
лабільною структурою, а проміжна сполука —
метастабільним утворенням, тобто є різниця в їх
реакційній стійкості та часі існування (далі буде
показано, що активний комплекс існує дуже короткий
час). З кінетичного погляду, ситуація є
аналогічною, і тому для визначення концентрації
активного комплексу можна скористатися рівнянням
(13.65).
Для довільної реакції
осА + |ЗВ <=> АК* -» продукти
маємо
(14.65)
Ейрінг Генрі
(1901—1981), США
иАК
-А САСВ.
Поляні Майкл
(1891 — 1976),
Велика Британія
376
З (14.65) та (14.64) для швидкості реакції маємо
Ф& (14.66)
Г = ДІС?СЙ.
т
З позиції формальної кінетики
г = ксІсі. (14.67)
<-АНВ
Отже, константа швидкості дорівнює
т
Константу рівноваги А^* можна знайти статистичним методом
і
(14.68)
' ~ейГ • (1469)
Величина £0 має назву нульової енергії активації, тобто це висота
потенціального бар'єра за температури 0 К.
Статистична сума £?* вміщує рух уздовж координати реакції. Якщо цей
рух вважати одновимірним поступальним рухом, то відповідна
статистична сума є такою:
£=- ^-^-5, (14.70)
де 8 — довжина шляху вздовж координати реакції в перехідному стані,
тобто ширина енергетичного бар'єра; т* — маса активного комплексу.
Тоді маємо:
С-Є*Й; О4-71)
О*О* * ,„,. * (2пт*кТ)
к. = У_ихе-Ео/ят = к*\ —!—Ь. (14 72)
У константі іС та у відповідній статистичній сумі £?* враховано, що
активний комплекс має на один ступінь свободи менше, ніж звичайна частинка
(цей ступінь свободи припадає на рух уздовж координати реакції). Швидкість
одновимірного руху в обраному напрямку (див. формулу (14.9)) дорівнює
*»-[&) • (14.73)
Отже, ^ і/2
З урахуванням (14.72) та (14.74) маємо з (14.68) для константи швидкості
А_ЛГ(2яіи*КГ)1/28_кГ^
нЬ(2пт*Т Ь " (14.75)
І кТ
377
Формула (14.75) — це основне рівняння теорії перехідного стану. Його
можна отримати також іншим шляхом. Перехід через бар'єр можна
вважати одним коливанням одновимірного осцилятора з частотою (у*). Тоді т* =
= і/у*, а
£ =ЇТ-^ = І7 (3аНу*<<кТ^' (14.76)
Отже, к = К* Щ^ \ = %І-К*у повному узгодженні з (14.75).
Ну і V п
Величина кТІН має розмірність с~{, тобто розмірність частоти. За Т= 500 К
(к = 1,38-10~23 Дж/К, Н = 6,62-10"34 Дж-с) маємо кТІН = 1013 с"1. Цю
величину можна вважати частотою переходу активних комплексів через
потенціальний бар'єр. Відповідно, зворотна величина 10_1 с є часом існування
активного комплексу (зазначимо, що це надзвичайно мала величина).
Проаналізуємо з позиції теорії перехідного стану бімолекулярні процеси
і зіставимо результати з теорією зіткнень. Для реакції
А + В <=> АВ -» продукти
маємо для передекспоненти
к=Ш&- <м77>
Розглянемо спочатку ситуацію, коли А та В — атоми. Тоді:
п -^пост._(2ягеА*Г)3/2
(2птвкТ)
нг ;
Є* _ ґі* пост.уг-ч* обер..
АВ ~ ^АВ ^АВ >
Єв=Єв°ст- 7Г
п
ЄГ пост.
і
_[2п(пгА+тв)кТ]
3/2
АВ " А3
п*обеР. __ 8п2ІкТ
^АВ " 7Т~'
Для поступальних сум враховано, що т* =тА + тв; а в стандартному
стані У= 1 (концентрація в частинках на одиницю об'єму).
Активний комплекс є гетероядерною лінійною двохатомною частинкою,
отже, число симетрії а = 1, а момент інерції /=\іі ,
де / = гА + гв, ц = —^—- приведена маса.
тА+тв
Підставляючи всі ці вирази в (14.77), маємо для передекспоненти
константи швидкості бімолекулярної реакції
378
кп =
кТ[2п(тА+тв)кТ]
$п2ц-12кТ
(2топАкТ)ш (2тапвкТ)ш
ґ8пкТл
(^А+'в)2
(14.78)
Теорія зіткнень у цьому разі дає (14.24)
К=2о=п(га+гв)2
8кТ
ЛІ/2
87РсгУ'
Ц )
('а+'в)2
Таким чином, для безструктурних частинок результат виявляється
ідентичним. З позиції теорії перехідного стану, під час зіткнення та утворення
активного комплексу відбувається перехід шести поступальних ступенів
свободи (по три від кожного атома) реагентів у три поступальні та два
обертальні ступені свободи активного комплексу. Позначаючи статистичні суми
на один ступінь свободи для відповідних видів руху, як/,п /т1 та /УІЬг (від
Ігапзіагіоп — поступальний рух, трансляція; гоШіоп — обертання та уіЬга-
іїоп — коливання), маємо для всіх частинок (2пост= /її > Для лінійних
частинок £обер. = /І,, 0Кол. = /ЗГ5. а ДЛЯ НеЛІНІЙНИХ бобер. = /го,, 0кол. = /^> ДЄ
п — кількість атомів у частинці. Для активних комплексів кількість
коливальних ступенів свободи є меншою, ніж для звичайних частинок на одиницю
(один ступінь свободи в активному комплексі витрачається на рух уздовж
координати реакції), отже, для лінійних активних комплексів маємо
2кол. = /%?, аДЛЯ НеЛІНІЙНИХ Є*ол. =/уҐ
'уіЬг
Тоді під час зіткнення двох атомів маємо
_ кЧгоіМг _ кЧгоі
ЬЇІЇІ
МІ
(14.79)
Розглянемо тепер, який вигляд має к0 для реакції між складними
молекулами бутадієну. У цьому разі реагенти та активний комплекс мають близьку
до лінійної структуру і передекспонента визначається таким чином:
/ту3 г2 ^3(йа+лв)-6
кЩ
АВ
кЧгоі*уіЬг
\іЬг .
= 20Р-
Отже,
ґ
^^о^
(14.80)
Таким чином, теорія зіткнень (з позиції теорії перехідного стану)
враховує, що під час зіткнення відбувається перехід поступального руху
в обертальний, але не враховує, що для складних частинок відбувається
379
також перехід обертальних ступенів свободи реагентів у коливальні ступені
свободи активного комплексу.
Оцінимо величину/? за допомогою формули (14.80).
ЗаГ=400Ктау=1012с-1:
ЛіЬг - Ну ю12 '
ІГОІ ~
ШкТ
ак2
1/2
= ^(2рсГ)1/2.
Вважаючи / = 5 • 10~ м, |И = 10~ кг, а = 1 маємо/.0, = 100. Отже,р = 10 ,
що непогано збігається з експериментальними значеннями р для реакції
димеризації бутадієну.
Основному рівнянню (14.75) теорії перехідного стану, крім
статистичного вигляду
к=кі:к*=^І^е-Ео/хт (1481)
можна надати так званий термодинамічний вигляд
к = аГК'= Щ-е-^'І™ = кГ^/я^-иґ/ят (14 82)
п п п ч '
де величини АС*, А5* та Д#* — це вільна енергія Гіббса, ентропія та
ентальпія активації, відповідно.
Рівняння (14.81) та (14.82) містять залежність константи швидкості від
температури. Крім того, існує ще емпіричне рівняння Арреніуса (14.62).
Отже, маємо три величини Е0, А#* та £експ>. Проаналізуємо, як вони
пов'язані між собою. Для цього використаємо диференціальну форму рівняння
Арреніуса (14.55), що в такому випадку має вигляд
йШ. = ^ш,т (14.83)
сіТ кт2
У формулах (14.81) та (14.82) передекспонента дещо залежить від
температури. Запишемо це емпірично таким чином:
к = АТте-Е>/ІІТ, (14.84)
де Ех — це Е0 або Д#*.
Логарифмуючи (14.84), маємо
\пк = \пА + т\пТ-^. (14.85)
Відповідна похідна за температурою дорівнює
а\пк = т , Ех ^Ех+тКТ
ЛТ Т ЯТ2 КТ2
Зіставляючи (14.83) та (14.86), маємо
(14.86)
380
£експ. —Ех + тКТ.
(14.87)
За£=Д# т = 1,отже,
= Д# +КТ.
(14.88)
Для простих бімолекулярних реакцій при обчисленні статистичних сум
видно, що ш = 1/2, отже, у цьому разі
Ерлсги -Ег\+—ЯТ.
(14.89)
Звичайно, точність вимірювання £експ. приблизно становить саме КТ,
тобто з достатнім ступенем точності можна вважати ЛЯ = £експ<, а також,
КОЛИ Ш НЄ Дуже ВІДРІЗНЯЄТЬСЯ ВІД НуЛЯ, Е0 = ^експ.-
Однак існують випадки, коли член тК Т суттєво впливає на загальну
енергію активації. Ця ситуація виникає, якщо Е0 є невеликою величиною, а ш
суттєво відрізняється від нуля. Цей випадок реалізується у тримолекулярних
реакціях, наприклад, у реакції окиснення N0, якій відповідає кінетичне
рівняння (13.25). Активний комплекс для цієї реакції можна уявити таким:
О О
і
N
О
І
І
N
О
Пунктиром позначено зв'язки, що рвуться або утворюються.
Теорія перехідного стану для цієї реакції дає
к= кТОак с-Еьікт
АЄЙ0&2
За Ну » кТ статистична сума коливального руху ^ко^ = ■
(14.90)
1
'\-е-Ч* мало
відрізняється від одиниці й практично не залежить від температури. Отже,
оцінимо т, враховуючи залежність від температури тільки поступальних
(Т ) та обертальних (Т — нелінійні частинки, Т— лінійні) сум. Це дає
т=1+3/2 + 3/2-(3/2+1)-2-(3/2+1) = -7/2.
Отже, згідно з рівнянням (14.87), маємо
^експ. -Е0--КТ.
(14.91)
За деякої температури Тт Е0 = 7/2 КТт; Еексп = 0, за більш високих
температур ^експ. < 0. Це наочно видно з рис. 14.15, с. 382: у координатах
Арреніуса за Т = Тт крива змінює нахил.
На відміну від теорії зіткнень, де енергія активації знаходиться емпірично,
теорія перехідного стану дає можливість обчислити £0. На рис. 14.16, с. З 82,
381
♦ ІпА:
1
1
Т
Рис. 14.15. Залежність константи
швидкості тримолекулярної реакції
від температури
зображено потенціальну криву для
молекули АВ. Такі криві можна
обчислювати квантовохімічними
методами, а також за допомогою
спектроскопічних даних (див. підрозділ
9.2). Не важко побачити, що для
реакції розкладу АВ на атоми енергія
активації Е0 дорівнює енергії
дисоціації молекули АВ. Протилежний
процес рекомбінації двох атомів в
молекулу перебігає з Е0 = 0.
Розглянемо тепер хімічну
реакцію взаємодії атома А з молекулою
ВС: А + ВС = АВ + С. Наприклад, реакцію обміну Гідрогену з дейтерієм:
Н + Т>2 = НБ + В або реакцію атома Н з галогенами: Н + Х2 = НХ + X тощо.
У цьому разі енергія є функцією трьох відстаней: гАВ, гвс і гАС. Для
побудови поверхні потенціальної енергії треба мати чотиривимірний простір,
але в бдгатьох випадках, як показали квантовохімічні розрахунки,
енергетично найбільш вигідно, щоб
активний комплекс мав лінійну
структуру [А..В..С], тоді гАС= гАВ+ гвс
і Е =/(гАВ, гвс). Для зображення
тривимірної потенціальної поверхні
методом ізоліній треба з'єднувати
точки з однаковими значеннями
енергії та наносити їх на площину
гАВ-гвс (рис. 14.17).
Початковий стан реакції
відповідає точці «1» гвс = гвс, а гАВ —
значна величина, що відповідає
далекій відстані атома А від молекули
ВС. Кінцевий стан — точка «2», тут гАВ = гАВ « гвс. Початковий і кінцевий
стани відповідають двом мінімумам енергії, двом «ярам».
Якщо перерізати потенціальну поверхню площиною за великих значень
гвс у напрямку, що є паралельним гАВ, то одержимо криву типу зображеної
на рис. 14.16. Такого ж типу потенціальна крива виникне за великих гАВ,
якщо розрізати потенціальну поверхню паралельно гвс. Обидві криві
відповідають станам ізольованих двохатомних молекул ВС та АВ відповідно.
За великих значень як гАВ, так і гвс, обидві ці криві виходять на плато (точка
«З»), що відповідає ізольованим атомам (А + В + С). Перехід з точки «1» до
точки «2» або у зворотному напрямку через плато відповідає такій енергії
активації, що дорівнює енергії дисоціації на атоми (аналогічно розкладу
молекули на атоми). Такий перехід є енергетично невигідним. Набагато
менший бар'єр виникає, якщо переходити з точки «1» до точки «2» вздовж
382
Рис. 14.16. Потенціальна крива
для двохатомної молекули
0,25
ВС, нм
0,20
0,15
0,10
0,05
0,10
0,15
г 0,20
АВ, нм
Рис. 14.17. Поверхня потенціальної енергії реакції
А + ВС = АВ + С
«координати реакції» (штрихова лінія на рис. 14.17) через точку
«сідловини» «4». Точка «4» — це точка «міні-максу», тобто це найменший із
можливих максимумів. Наочно шлях реакції можна побачити на рис. 14.18,
де зображено контурну діаграму
тривимірної поверхні потенціальної енергії.
На рис. 14.19, с. 384, зображено розріз
потенціальної поверхні вздовж координати
реакції, який є енергетичним профілем
реакції, це конкретизація схематичного рис.
14.11, с. 372. Якщо розрізати потенціальну
поверхню упоперек координати реакції, то в
точці «4» глибина мінімуму буде найменшою.
Потенціальна поверхня, як і потенціальні
криві, відповідає основному, незбудженому
стану реагентів, продуктів та активного
комплексу, тобто стану за температури 0 К,
у якому ступені свободи, що відповідають
поступальному, обертальному та
коливальному видам руху, є незбудженими. За Т > 0 К
слід враховувати збудження цих станів, отже,
АН відраховується, на відміну від Е0, не від «дна» потенціальної кривої.
М. Поляні, аналізуючи поверхні потенціальної енергії для реакцій типу
А + ВС —» АВ + С, встановив та обґрунтував емпіричне правило, згідно
Рис. 14.18. Контурна діаграма
тривимірної поверхні
потенціальної енергії
(крива «аЬс» - шлях реакції)
383
перехідний
стан
початковий
стан
Рис. 14.19. Розріз потенціальної
поверхні вздовж координати реакції
Рис. 14.20. Графічна ілюстрація правила
Поляні
з яким у ряду однотипних реакцій
існує залежність між кінетичними
(енергія активації) та
термодинамічними (теплота реакції)
параметрами
Д£* = -аД#, (14.92)
де АЕ та А^ — відповідні зміни
енергії активації та теплового
ефекту реакції під час переходу від однієї
однотипної реакції до іншої, а —
коефіцієнт, який змінюється від
одиниці до нуля, тобто 0 < а < 1. (# =
= -АН— це тепловий ефект реакції
у термохімічній шкалі, запис у
термодинамічній шкалі Д (АН) є
незручним).
На рис. 14.20 наведено
ілюстрацію правила Поляні за
допомогою потенціальних кривих. Під час
переходу до іншої, але однотипної
реакції крива 2 зміщується
«паралельно» сама собі (крива 2 *). Отже,
АЕ становить лише частину від А^
(а < 1). Найбільш типове значення
ос= 1/2 відповідає приблизно
однаковому нахилу кривих 1 та 2 у точці перетину (рис. 14.21, а). Якщо в точці
перетину симетричності немає, то це веде або до а —» 0 (рис. 14.21, б), або
до а —> 1 (рис. 14.21, в), але такі випадки не є типовими.
Слід визначитися також більш конкретно щодо виразу «у ряду
однотипних реакцій». Наприклад, у процесі взаємодії лужних металів з
галогенопохідними (див. підрозділ 14.3) Ме+ХУ > МеХ+У можуть виникнути такі
варіанти:
1)Ме >Ьі,Ма,К,КЬ,С8;
2) У > СН3, С2Н5, С3Н7, С4Н9;
3)Х >Р,С1,Вг,І.
Перший випадок відповідає схемі, наведеній на рис. 14.20, тобто крива
реагенту ХУ залишається на місці, а криві продуктів МеХ зміщуються.
Другий випадок є дзеркальним відображенням першого, тобто крива для
продукту МеХ є сталою, а зміщуються криві реагентів ХУ. Перший та
другий випадки — однотипні реакції, для них правило Поляні виконується.
Зазначимо, що в обох випадках без зміни залишається центральний атом X
перехідного комплексу [Ме..Х..У].
Якщо ж змінювати X (третій випадок), то змінюватимуться як крива 7, так
і крива 2, реакції не будуть однотипними, і правило Поляні в цьому випадку
не виконується.
384
Рис. 14.21. Перетин потенціальних кривих
При виведенні основного рівняння (14.75) теорії перехідного стану
вважалося, що кожен активний комплекс перетворюється на продукти. Це
припущення не є обов'язковим, і для врахування інших можливостей в рівняння
(14.75) вводиться так званий трансмісійний коефіцієнт (ае)
к = в*£к*. (14.93)
п
Для багатьох реакцій ае = 1, що відповідає обов'язковому перетворенню
активного комплексу на продукти, але є реакції, для яких ае « 1.
Розглянемо декілька прикладів.
Виявилося, що за низьких тисків реакція рекомбінації атомів Гідрогену
Н + Н > Н2 перебігає лише на поверхні реактора, тобто шляхом адсорбції
атомів Гідрогену на поверхні з наступною рекомбінацією та десорбцією
молекули Н2. Чому ж ця реакція обирає гетерогенний шлях, якщо енергія
активації під час об'єднання атомів у молекулу дорівнює нулю? Виявилося,
що для цієї реакції трансмісійний коефіцієнт менший за 10" . Цей ефект
виникає внаслідок того, що активний комплекс Н2 не має можливостей
позбутися зайвої енергії. Під час рекомбінації, наприклад радикалів
СНз + СНз > С2Н6, енергія, що виділяється, може перейти на збудження
коливальних ступенів свободи. Процес радіаційної віддачі енергії шляхом
висвічування фотона для молекули Н2, яка не є навіть диполем, проходить
повільно порівняно з часом існування (10~ с) активного комплексу, і його
можна не брати до уваги. Отже, реакція рекомбінації відбувається на
поверхні, і тверде тіло забирає надлишок енергії. При збільшенні тиску зро-
385
стає можливість потрійних зіткнень, і рекомбінація атомів Гідрогену
перебігає, як тримолекулярна реакція. Третьою частинкою, що бере на себе
надлишок енергії, може бути як атом Гідрогену, так і атом будь-якого
інертного газу, що уведений у систему.
Другий приклад, коли х «1, це реакція розкладу геміоксиду нітрогену:
И20 > ]Ч2+ О, для якої ж = 10" . Це приклад так званих неадіабатичних
реакцій (термін «адіабатичний» в цьому разі має інше значення, ніж у
термодинаміці, і базується на принципі Борна-Оппенгеймера, що дозволяє
розділити рух ядер та електронів, див. розділ 9).
Пояснити, чому ае « 1 для неадіабатичних реакцій, можна за
допомогою потенціальних поверхонь. Розглянемо утворення координати реакції як
перетин двох потенціальних кривих (цей прийом уже застосовували вище
(рис. 14.20), коли роз'яснювали правило Поляні). У точці перетину
потенціальних кривих (точка 0 на рис. 14.22) стани, що описуються різними
кривими (7 та 2), мають однакову енергію, тобто є виродженими. Якщо хвильові
функції Х|Х! та \|/2, що описують стани 1 та 2, взаємодіють, то виродження
знімається, і виникає так звана енергія резонансу
Єр=/жіНіп1\|/2Л, (14.94)
де Ніт — гамільтоніан, що відповідає взаємодії. Наявність гр веде до
розщеплення енергетичних рівнів (точки а та а') довкола точки О, отже,
відбувається плавний перехід з кривої 1 на криву 2 через точку а. Потенціальна
поверхня для основного стану (її зображено на рис. 14.17) відокремлюється
від поверхні для збуджених станів (на рис. 14.22 це крива 2'а7').
Таким чином, якщо гр » 0, то внаслідок відокремлення потенціальної
поверхні для основного стану перехід із початкового стану 1 в кінцевий стан
2 через перехідний стан а відбувається поступово, в цьому випадку ге = 1,
тобто усі активні комплекси перетворюються на продукти.
Для неадіабатичних процесів стани 7 та 2 не взаємодіють, отже, гр = 0,
і в точці О розщеплення не відбувається. Це ускладнює перехід з кривої 7
на криву 2. Система, замість того, щоб «спуститися» у мінімум по кривій 2,
продовжує рух по кривій 77', а потім, маючи дуже високу енергію, знову
скочується по кривій 7 7 у
зворотному напрямку, активний комплекс
перетворюється знов у реагенти,
що веде до х « 1.
До неадіабатичних належать
реакції, за яких відбувається зміна
спінового стану (мультиплетності).
Так, молекули И20 та И2 мають
терми X (синглети), а в атомі Окси-
гену основний терм— Р.
Рис. 14.22. Взаємодія потенціальних Таким чином>коли « « 1, це оз-
кривих начає, що перетворення активного
386
комплексу на продукти не є вірогідною подією (ймовірність вірогідної події
дорівнює одиниці).
З цього погляду, дещо парадоксальними є випадки, для яких ае > 1. За ае = 1
всі активні комплекси перетворюються на продукти, ае > 1 означає, що
продукти утворюються не тільки через активний комплекс, а якимось іншим
шляхом.
Цю можливість дає «тунельний ефект», тобто здатність частинки, що має
енергію меншу, ніж висота бар'єра (див. підрозділ 8.1), «перетинати» бар'єр.
Цей ефект виникає лише для мікрочастинок, що мають хвильові властивості.
До таких частинок належить електрон. В окисно-відновних реакціях, де
головним є перехід електрона, подолання бар'єра за достатньо низьких
температур можливе за рахунок тунельного ефекту, і тоді трансмісійний
коефіцієнт може бути більшим за одиницю.
Як відзначалося вище, теорія перехідного стану пояснює елементарні
реакції. З позиції цієї теорії було розглянуто бімолекулярні та
тримолекулярні реакції. Мономолекулярні реакції мають свою специфіку.
Виявилося, що в деяких випадках ці реакції не є елементарними процесами і їх слід
вважати складними реакціями, тому ці реакції розглядаються у наступному
розділі разом з іншими складними реакціями у газовій фазі.
387
Розділ 15.
КІНЕТИКА РЕАКЦІЙ У ГОМОГЕННИХ СИСТЕМАХ
— Холмс, вибухне ця суміш кисню з воднем?—
У Ватсона питання постає.
— Елементарно, Ватсон! На сьогодні
В науці відповідь на це питання є:
В координатах тиск-температура
«Спалахнення півострів» постає,
Якщо опинимось за межами фігури,
То щоб не стався вибух, шанси є.
15.1. Мономолекулярні реакції у газовій фазі
До мономолекулярних належать реакції ізомеризації, розкладу тощо.
Зазвичай це реакції 1-го порядку. Якщо мономолекулярний процес є
елементарним, то застосування теорії перехідного стану дає
наАе - (15Л)
Якщо вважати, що активний комплекс у цьому разі не дуже відрізняється
за геометрією від молекули реагенту, то це дає приблизно однакові
значення моменту інерції (для нелінійної молекули — моментів інерції навколо
відповідних осей), отже, обертальні суми активного комплексу і реагенту
є близькими, тобто (бдк)об - (6а)о6.- Оскільки маси активного комлексу
і реагенту є однаковими, то це веде до тотожності поступальних сум
(£?ак)пост = (2а)пост • Коливальні суми зв'язків, що не беруть участі у
перетворенні, теж є однаковими, але, як це зазначалося у розділі 14, активний
комплекс має на один ступінь коливального руху менше. Отже, після
відповідного скорочення передекспонента в рівнянні (15.1) набуває такого
вигляду:
де V — частота коливань зв'язку, що рветься.
За Ну « кТ, тобто за відносно високих температур і низьких частот
є = 1 -Ну/кТі, отже,
За Ну » кТ, тобто за великих частот і відносно низьких температур є У * —»
->0/у/6г=1,отже,
*о=-у' (15'4)
Зазначимо, що за Ну = кТ статистична сума /^теж не дуже відрізняється
від одиниці (/уіЬг = е/(е- 1) = 1,58).
388
Отже, типове значення передекспоненти дорівнює 10 с"1 (за Ну = кТ
V = 10 с"1). Це значення передекспоненціального множника насправді є
типовим для багатьох мономолекулярних реакцій.
Але трапляються випадки, де к0 »10 с"1 або к0 « 10 с"1. Наприклад,
для реакції ізомеризації вінілалілового етеру у відповідний альдегід к0 =
= 10 с-1, а при ізомеризації циклопропану в пропен к0 = 10 с-1.
Розглянемо ці процеси. Для реакції
СН2
/V
СгІ2—СгІ2
необхідно, щоб розірвався цикл. Отже, система в перехідному стані є більш
невпорядкованою, ніж у вихідному. Це веде до збільшення ентропії, і
оскільки 5АК*» 5д, то і ентропія активації
->СНг=СН—СН,
А5 „
АК*
є великою додатною величиною.
Відповідно, з рівняння (14.82)
- 5Д* 5А
к0=е'
А?м кТ
(15.5)
(15.6)
За Д5 » 0 к0 » кТІН. Зазначимо, що припущення подібності активного
комплексу і вихідної речовини дає: 5АК* = 5д, Д5 = 0 і к0 = кТІН, як це було
наведено вище.
У реакції
Н
/
сн2=сн—о-<:н2—сн=сн2 —> сн2=сн-<:н2—сн2—с
\\
о
необхідно перенести атом Оксигену з середини ланцюга на його край. Це
можна зробити, згортаючи реагент у шестичленний цикл, тоді активний
комплекс має таку будову:
Г сн *
0^' -СН,
1 1 *•
1 1
1 1
1 1
С/Н2 \^ х^ СН2
[ сн'
Отже, тут активний комплекс — більш упорядкована система, ніж вихідна
речовина, і Д5* << 0.
Мовою статистичних сум, в обох випадках відбувається перехід між
обертальними та коливальними ступенями свободи. Оскільки, як було показано,
389
Лої >>ЛіЬг> то перехід від коливальних (реагент) до обертальних (АК )
ступенів свободи (ізомеризація циклопропану) веде до збільшення пере-
декспоненти; навпаки, поява коливальних (у циклі АК ) ступенів свободи
замість обертальних веде до зменшення к0 (порівняно з кТІк).
До мономолекулярних процесів можна застосовувати теорію
перехідного стану, якщо вважати ці реакції елементарними, але виявилося, що за
дуже низьких тисків змінюється кінетика мономолекулярних реакцій, і вони
перебігають як реакції 2-го порядку.
Це явище пояснив Ф. Ліндеман, який для мономолекулярних реакцій
запропонував таку кінетичну схему:
а + а-^а* + а5 (і)
А —£—> продукти, (її)
А* + А к(і >А + А. (Ш)
Першою стадією, згідно з цією схемою, є активація молекул А. Активація
відбувається шляхом зіткнення молекул з перерозподілом енергії між ними.
Далі активна частинка (А ) може подолати потенціальний бар'єр і
перетворитися на продукт (стадія II) або може, зіткнувшись з іншою частинкою,
віддати їй свою енергію і дезактивуватися (стадія III). Застосувавши метод
стаціонарних станів до активних частинок А , маємо
сісА*
—г-~ = 0 = касА - КСА* ~~ ^асксА* >
звідки 2
а кг+касА V >
Швидкість отримання продукту
За високих тисків концентрація молекул А є значною і можна вважати
касА»кп що дає
К(1СА Кй
де к—емпірична константа швидкості.
Кінетичне рівняння (15.9) відповідає 1-му порядку. Це звичайний
порядок для мономолекулярних реакцій за не досить низького тиску. Не важко
пересвідчитися в тому, що відношення каІка = К , тобто це константа
рівноваги процесу активації
А + А < , } А + А (об'єднання стадій І та III).
У цьому разі існує рівновага між активними частинками і реагентами,
а це і є однією з важливих умов теорії перехідного стану. Таким чином, згідно
з рівнянням (15.9) маємо
390
* = кгК*
(15.10)
і воно за структурою є таким самим, як і основне рівняння теорії перехідного
стану (14.75).
Інша картина спостерігається за малих тисків. За сА —> 0 касК « кг і, отже,
г=кг*А=каСі (15.ц)
Таким чином, реакція перебігає за 2-им порядком, і швидкість загальної
реакції дорівнює швидкості лімітуючої 1-ої стадії—активації, яка є
бімолекулярним процесом. У цьому випадку немає рівноваги між активними та
неактивними частинками, розподілення Максвелла-Больцмана не
виконується, і теорію перехідного стану не можна застосувати.
Активація молекул А може відбуватися не лише за рахунок зіткнення
виключно між ними. До системи можна додати, наприклад, інертний газ. Це
веде до збільшення швидкості активації молекул А, яка дорівнює
Г = каСА+КСАС1> (15Л2)
де с{ — концентрація інертного газу. Цей факт спостерігається
експериментально і підтверджує схему Ліндемана.
15.2. Ланцюгові реакції
Далі як приклад складних реакцій розглянемо ланцюгові процеси у
газовій фазі. Розгляд почнемо з нерозгалужених ланцюгових реакцій.
Прикладом таких реакцій є реакція взаємодії молекулярних хлору та
водню. Кінетичне рівняння цієї реакції наведено в розділі 13(13.31), вона
має 1-й порядок за воднем і дробовий (0,5) за хлором.
Механізм будь-якого ланцюгового процесу містить як мінімум три стадії:
1) зародження ланцюга, ініціювання;
2) зростання ланцюга;
3) обрив ланцюга.
Зародження ланцюга — це утворення активних частинок —радикалів,
зокрема атомів Хлору або Гідрогену. Енергія зв'язку є(Н—Н) = 430 кДж/
моль » є(С1—СІ) = 238 кДж/моль, отже, зв'язок у молекулі водню є
міцнішим за зв'язок у молекулі хлору, і зародження ланцюга відбувається
шляхом розриву слабкішого зв'язку СІ—СІ.
Існують різні можливості утворення атомів Хлору з молекули С12.
Фотохімічне ініціювання умовно можна зобразити таким чином:
сь-^гсґ.
Молекула хлору має таку електронну конфігурацію: (с2)2(пХгу)4(п*у)4.
Під дією кванта світла молекула хлору переходить у збуджений стан:
(а2) (пху) (к*у) (с/2) , де кількість електронів на зв'язуючих орбіталях така
сама, як на антизв'язуючих, отже, молекула С12 розпадається на атоми.
391
Хімічне ініціювання. Одна з можливих реакцій хімічного ініціювання
розглядалася у підрозділі 11.3, це взаємодія атомів лужних металів із
молекулами галогенів: Ме* + Х2 = МеХ + X*. Наприклад: Иа* + С12 = ИаСІ + СІ*.
Друга можливість хімічного ініціювання — це розпад нестійких сполук,
наприклад N001 -> N0 + СГ.
Термічне ініціювання. Зі зростанням температури рівновага
ендотермічної реакції С12 = 2СГ зміщується у бік атомізації молекули С12.
Нарешті, можливе гетерогенне ініціювання. Позначимо символом [ ]
активний центр на твердій поверхні, що може захоплювати атом Хлору.
Взаємодія цього центра з молекулою С12 відбувається таким чином: [ ] + С12—>
->[С1]адс. + Сі'.
Зростання ланцюга складається з двох елементарних реакцій:
СГ + Н2 ки > НС1 + Н#;
н*+сі2 кгь > нсі+сГ.
Для нерозгалужених ланцюгових процесів під час взаємодії радикала
(атома) з молекулою радикал відтворюється, але кількість радикалів на стадії
подовження ланцюга не збільшується.
Обрив ланцюга. Розрізняють лінійний та квадратичний види обриву. За
лінійного обриву його швидкість залежить від концентрації радикалів
лінійно, тобто у цьому разі реакція обриву має 1-й порядок за активними
частинками (атомами, радикалами). За малого тиску довжина вільного
пробігу (Х9 див. підрозділ 14.1) є відносно великою, отже, у радикала є можливість
досягти стінки без зіткнення з іншою частинкою і адсорбуватися на поверхні
за схемою
Н#+[]-^[Н]адс.;С1# + []^[С1]адс,
За високих тисків обрив є квадратичним, тобто відбувається шляхом
бімолекулярної рекомбінації атомів:
Н#+Н#->Н2;
Н#+СГ->НС1;
сГ+сГ->сі2.
Зазначимо, що, фактично, за високих тисків відбуваються
тримолекулярні зіткнення, наприклад
СГ+СГ + М > С12+ М*,
де третя частинка М забирає надлишок енергії.
Розглянемо кінетику нерозгалужених ланцюгових процесів,
використовуючи метод стаціонарних станів. За будь-якого способу ініціювання стадія
ініціювання є реакцією 1-го порядку за хлором, отже, її можна записати так:
СІ2—^->2С1#.
Нижче буде доведено, що в стаціонарному стані виконується умова
392
стац. ^ ^ стац. Тт
сс1. » сн. . Це веде до того, що з трьох наведених вище можливих
реакцій рекомбінації, фактично, реалізується лише одна, а саме:
ОГ + СҐ—^->С12.
Метод стаціонарних станів, застосований до атомів Гідрогену, дає
йс .
-^- = 0 = к2асН2сс]. -к2ЬсСІ2сц., (15.13)
а застосований до атомів Хлору
йс . 2
-^- = 0 = *,сСі2 +А:26сСІ2сн. -*2всН2сс1. -*3ссГ 05.14)
З урахуванням рівняння (15.13) з формули (15.14) маємо
сста
СІ"
*ЇСС12 ~"з(с™- ) '
(15.17)
отже, > (15.15)
^ц=^з),/24,52-
У разі термічного ініціювання відношення кхІкг дорівнює К — константі
рівноваги
с™={КГс%Гс^: (15.16)
Загальна швидкість утворення продукту становить
^НСІ 7 7 лі
—^- = *2<ісН2сс1- + *26са2сн- = 2к2асН2са- =
= ^^2а СЮ СН2 ССІ2 = *еф.сН2 СС\2 •
У виведенні рівняння (15.17) враховано рівняння (15.13) та (15.16).
Рівняння (1^.17) збігається з експериментальним (13.31), причому
*експ = *еф. = 2к2а (К)У1 = 2к2а ІКІк^. (15.18)
Проаналізуємо стадію зростання ланцюга. Згідно з умовами
стаціонарності (15.13)
Г1а = *2а%<£аЦ- = Г2Ь = к^С^с'Р'. (15.19)
Вважаючи вихідну суміш стехіометричною (с£І2 = Сщ), маємо впродовж
усієї реакції Сщ = сСІ2, отже, з (15.19) одержимо
_а^_%_(^о)2^"и— (1520)
стац. Ь п ч -Е2аІКТ *
<?н. ^ (А:0)2ае 2«
Реакції (2а) та (26) — це прості бімолекулярні процеси між атомами
і двохатомними молекулами, близькими за розміром. Отже, можна вважати,
що площі перерізу (а) в обох реакціях є близькими, а це дає (к0)2а = (к0)2Ь
згідно з формулами (14.24) та (14.63) (стеричний фактор у цьому разі є близь-
393
ким до одиниці). Отже, різниця в константах реакцій (2а) та (2Ь) визначається
енергіями активації. Енергії активації цих елементарних реакцій суттєво
відрізняються: Е^сп = 28 кДж/моль, в той час як Е^сп = 8 кДж/моль. За Т=
= 500 К це дає
стац.
СГ _ е{Е1а-Е1ь)ІКТ _ е(28-8)103/8,31-500 = ^л
стац.
Н*
^ стац. ^ ^ стац.
Отже, виконується умова сС[. » си. ц.
При цьому к2а« к2Ь, отже, реакція взаємодії атома Хлору з молекулою
водню є лімітуючим процесом. Зазначимо, що терміни «повільна» та
«швидка» вжиті стосовно до елементарних реакцій і визначають саме значення
констант швидкостей. У стаціонарному стані швидкість «повільної» стадії
(2а) дорівнює швидкості «швидкої» стадії (2Ь) (див. рівняння (15.19)).
Велике значення константи швидкості к2Ь «компенсується» малим значенням
концентрації с£ац і навпаки, в реакції (2а) малому значенню константи к2а
відповідає значна концентрація атомів Хлору.
Необхідно тепер відповісти на питання, чому Е2а » Е2Ь. Це можна
зробити, спираючись на правило Поляні (14.92). Запишемо його у такому
вигляді:
Еі-Е] = -а&і -?,) -> Еі +Щі = Е] +щ = соті. (15.21)
Аналізуючи результати за реакціями взаємодії атомів з молекулами,
М. М. Семенов запропонував такі значення параметрів для екзотермічних
процесів: а = 0,25; соті = 48 кДж/моль, тобто
ЕеК3' =48-0,25?. (15.22)
Для ендотермічних процесів, враховуючи рівняння (14.56), що пов'язує
енергії активації прямого та зворотного процесів, з (15.22) одержуємо
£енд-=48 + 0,75|?|. (15.23)
Для реакції взаємодії атома Хлору з молекулою водню
Чіа = єн~сі - єн-н = 426 - 430 = -4кДж/моль.
а для реакції взаємодії атома Гідрогену з молекулою хлору
Чіь = єн-сі ""єсі-сі = 426 -238 = 188 кДж/моль.
Отже, реакція взаємодії атома Гідрогену з С12 виявляється дуже
екзотермічною, а реакція взаємодії атома Хлору з Н2—слабко ендотермічною.
Відповідні оцінки для енергій активації є такими:
Е£ = 48 + 0,75 - 4 = 51 кДж/моль;
Е°£ = 48 - 0,25 • 188 = 1 кДж/моль.
Хоча ці оцінки не досить чітко збігаються з експериментальними зна-
394
ченнями Е2а та Е2Ь вони дозволяють зрозуміти, чому Е2а » Е2Ь. Це, врешті-
решт, випливає з нерівності єн_н » єс1_с1.
У виведенні кінетичного рівняння ланцюгового процесу було
використано умову стаціонарності щодо атомів Хлору та Гідрогену. Згідно з
розглянутим в розділі 14 обґрунтуванням методу стаціонарних станів активні
проміжні частинки повинні бути в невеликих концентраціях. Оскільки сС{* »
» сн., то достатньо оцінити стаціонарну концентрацію атомів Хлору.
Найпростіше це зробити у разі термічного ініціювання, тоді згідно з (15.16) сс1.
визначається концентрацією молекулярного хлору та константою рівноваги.
Покладемо сСІ2 = 0,1 моль/л та Т= 500 К. Тоді
-АО° ( -АЯ° Д5° ^
/ \У1 05 "
СС\2
Д#° = єСі_с1 = 238 кДж/моль; оцінка для ДО0 = 25°1# - 5£І2 дає приблизно
80 кДж/моль • К, отже,
ссг=(0,1)°'5ехр
238-103
8,31-2-500
ехр
80
2-8,31
= 5-10 12 моль/л.
Це дуже мала величина, отже, умова стаціонарності виконується.
Виникає питання, чому реакція Н2 + С12 —> 2НС1 перебігає, як
ланцюговий процес, а схожа з нею реакція Н2 +12 —> 2НІ, як проста бімолекулярна
реакція? Зіставимо ці реакції. Ефективна константа ланцюгового процесу
задається рівнянням (15.18). Відповідно, ефективна енергія активації така:
£еФ.=£2а+у, (15-24)
де О — енергія дисоціації молекули галогену.
Для реакції Н2 + С12 маємо
£Єф. = 28 + 238/2 = 147 кДж/моль.
Якби реакція Н2 +12 перебігала за ланцюговим механізмом, то
лімітуючою (2а) стадією була б реакція взаємодії атома Йоду з Н2, тобто
І#+Н2->НІ + Н\
Тепловий ефект цієї реакції дорівнює
Чіа = Єн-і - Вн-н= 297 - 430 = -133 кДж/моль.
Для цієї дуже ендотермічної реакції оцінка енергії активації за рівнянням
Семенова (15.23) дає: Е%% = 48 + 0,75-133 = 148 кДж/моль, що непогано
узгоджується з експериментальною величиною Еексп'= 138 кДж/моль.
В = єм = 150 кДж/моль. Отже, £еф = 138 + 150/2 = 213 кДж/моль.
Експериментальна енергія активації простої бімолекулярної реакції
Н2 +12 дорівнює 167 кДж/моль, тобто є набагато меншою, ніж для
ланцюгового процесу. Тому реакція Н2 + І2не перебігає за ланцюговим механізмом.
395
Зазначимо, що вирішальний внесок у £еф робить енергія активації
лімітуючої стадії (2а), яка за таких обставин є дуже ендотермічним процесом.
Для реакції Н2 + С12 енергія активації (Ебім) У припущенні, що ця реакція
перебігає, як проста бімолекулярна, невідома, але можна вважати, що
оскільки єси:і» єм, то Ебім для реакції взаємодії водню з хлором повинна
бути більшою, ніж для реакції Н2 +І2, тобто Е6т > 167 кДж/моль. Отже, для
реакції Н2 + С12 £бім > £еф . Це надає перевагу ланцюговому механізму.
Характерною величиною для ланцюгових реакцій є середня довжина
ланцюга. її можна визначити як кількість молекул продукту (НС1), що
утворюється від появи однієї активної частинки до її загибелі під час обриву.
Інакше кажучи, це кількість елементарних реакцій на стадії зростання
ланцюга після ініціювання. Якщо позначити через а ймовірність зростання, а
через Р = 1 - а—ймовірність обриву ланцюга, то ймовірність того, що
ланцюг має рівно п ланок, становить
^(«) = а"р.
Середня довжина ланцюга V є такою:
(15.25)
V = %пр(п) = £пап$ = (ф%паГ1 =
л=0 л=0 л=0
оо оо
= сф£<Ша(сс") = а#Ша^а" =
л=0
л=0
= ар^аР-1 = -^2- = ^ = сс/р. (15.26)
[1-а] (і_а)2 р2
Вважаючи а та Р пропорційними відповідним швидкостям, матимемо
у = а _ г2а _ к2°ССГСН2 _ к2аСС\2 _ к2а
к3с2а. кзС<
1/2СС12-
(15.27)
'з к,сХ. *зса. (к&У
У виведенні рівняння (15.27) враховано рівняння (15.18) та умови сн =сс1.
Вважаючи, що к%аЦк°кІ =1, матимемо
1а 2 2
у = ехр
-ь.ЛЛ
КТ
(15.28)
Оцінимо V для реакції Н2+С12.3 наведених вище даних маємо Е2а = 28 кДж/
мояь;Е1=Оа =238кДж/моль;£,3 = 0?ЩОзаГ=500Кдає У = 3-109.Отже,
для реакції Н2+С12 ланцюг має величезну довжину.
Оцінимо тепер довжину ланцюга для реакції Н2 +12. У цьому випадку
£2а = ШкДж/моль,^/)^ ^ОкДж/моль^з^О.Цедає у= 10"6« І.Таке
396
мале значення V яскраво підтверджує, що реакція Н2 +12 не є ланцюговим
процесом.
Швидкість ланцюгової реакції можна визначити як добуток швидкості
ініціювання г( на довжину ланцюга, тобто
г = пч. (15.29)
Для реакції Н2+ С12 це дає
г = гр = кіСс, ^^ = к^Ік^Жс» , (1530)
;і2 ; 2 '^а^і'^/ ~СІ2~Н2'
' СС1#
Ьрі
що збігається з наведеними вище рівняннями (15.17) та (15.18). Зазначимо,
що згідно з рівняннями (15.13)—(15.15) швидкість ініціювання ^дорівнює
швидкості обриву гоб, отже, з (15.29) та (15.27) маємо
г = гу = ^К5^ = гюст., (15.31)
гг
об.
* зрост. з
де гзрост — швидкість зростання ланцюга. Отже, загальна швидкість реакції
дорівнює швидкості лімітуючої стадії.
Реакція Н2 + С12 — дуже екзотермічний процес із тепловим ефектом: ^ =
= 2єн_с1 - єн_н - єСІ_с1= 184 кДж/моль. Якщо не забезпечено відведення
тепла, то це веде до зростання температури в реакторі. У свою чергу, це
прискорює термічну дисоціацію молекули С12 і збільшує концентрацію атомів
Хлору, що веде до прискорення ланцюгового процесу і збільшення теплоти,
що виділяється; посудина розігрівається далі, отже, процес набуває
лавиноподібного характеру, це так званий тепловий вибух.
Однак вибух може мати й ланцюговий характер. Це явище
спостерігається для розгалужених ланцюгових процесів, для яких є характерним
збільшення кількості активних частинок не лише в процесі ініціювання, а й
протягом перебігу ланцюгового процесу на стадії зростання ланцюга.
Розглянемо такі реакції на прикладі взаємодії водню з киснем. Серед молекул
Н2 та 02 зв'язок є міцнішим у молекулі кисню, і ланцюгова реакція
починається з утворення атомів Гідрогену (реакцію ініціювання не розглядаємо
детально, позначаючи її швидкість через г,). Далі проходять такі стадії:
Н# + 02 —^-> ОН* + *0*, (1)
*0*+Н2 —Ь-> ОНЧ Н#, (2)
ОН* + Н2 —**-> Н20 + Н#. (3)
Таким чином, один атом Н\ вступаючи у реакцію, дає три атоми Н*
(враховуючи, що радикали ОН* перетворюються на атоми Н ), тобто має
місце розмноження активних частинок. Обрив ланцюга відбувається або на
стінці (за низьких тисків)
Н-+[]_*»_>[ Н], (4)
397
або (за високих тисків) шляхом потрійних зіткнень з утворенням відносно
стабільного радикала Ж>2
н +о2+м
■* м + но;.
(5)
Якщо енергії вистачає і третьої частинки немає, то згідно з елементарною
реакцією (1) при взаємодії атома Гідрогену з молекулярним киснем
утворюються частинки ОН* та *0*. Для реакції Н2 + С12= 2НС1 виконувалася
умова сс1. » сн., тобто атом (радикал), що утворюється легше за інші,
присутній у більшій кількості, ніж інші. Так само в реакції 2Н2 + 02 = 2Н20 з усіх
активних радикалів максимальну
концентрацію мають атоми Гідрогену, тобто сн# »
>> сон#' с#о* *
Для розгалужених ланцюгових реакцій
М. М. Семенов запропонував метод напів-
стаціонарних концентрацій, згідно з яким
концентрації всіх активних частинок, крім
присутніх у найбільшій кількості, є
стаціонарними. Для реакції окиснення водню метод на-
півстаціонарних концентрацій дає
Ач
'о#
йі
= 0;
йс.
*.ш%
(іси
але-
йі
■*0.
Семенов Микола Миколайович
(1896—1986), СРСР
А\
З цих умов маємо
о*
Лсй
йі
= к{сИ.с02 - к2с.0.сН2 = 0.
он*
А
Ік\сн.с02 +к2с.0.сН2 -к3сш.сН2 =0.
сісИ.
—- = Гі -к1сн.с02 + к2с.0.сН2 ~к3сон.сН2 -к4сн. -к5с02сн.см.
З урахуванням (15.32) та (15.33) маємо
йсл
н# _
Л
= г( + 2к{сн.с02 -к4сн. -к5с0існ.см = г( + <рсн.,
де ф = 2кхс0і -к4 -к5с0ісм.
Інтегрування (15.36) за початкових умов сн. = 0 за / = 0 дає
-н#
= п
ф
За дуже малих тисків с0 —> 0, отже, ф = -к4
Тоді
і за І —> ©о
= П
[і-ехр(-*40]
->с'
,стац. _
= Гіікц.
(15.32)
(15.33)
(15.34)
(15.35)
(15.36)
(15.37)
(15.38)
(15.39)
398
Таким чином, за цих умов реакція окиснення водню проходить
стаціонарно. Залежність сн. від часу зображено для цього випадку на рис. 15.1
(крива І). Зі збільшенням с0 ф зменшується за абсолютною величиною, але
якщо ф < 0, то реакція дійде до стаціонарного стану (крива II на рис. 15.1), але
за більших значень с^т.ац'
стац. _ і
|ф| к4
= Ае*'
(15.40)
(15.41)
|СН'
III
А^—
и
і
Рис. 15.1. Зміна концентрації атомів Н
з часом у реакції окиснення водню
Якщожф>0^то
с . = г, - -
Н ' ф
і концентрація сн. експоненціально,
лавиноподібно зростає, тобто
відбувається ланцюговий вибух (крива III
нарис. 15.1).
Перехід від стаціонарного (за
малих тисків) до вибухового (за
збільшення тиску) перебігу реакції окиснення водню спостерігається
експериментально. У координатахр — Тможна розрізнити (рис. 15.2) дві ділянки
перебігу реакції: стаціонарну (біле поле) та вибухову (штриховка).
За малих тисків реакція перебігає
стаціонарно. Зі збільшенням тиску
відбувається перехід до вибухового
режиму, але за дуже високих тисків
реакція знову переходить у
стаціонарний режим. Це пов'язано зі
збільшенням потрійних зіткнень і,
відповідно, зі зростанням обриву ланцюга
за рахунок утворення неактивних
радикалів НО^. Так, за високих
тисків у рівнянні (15.36) можна знехту- та 1в1 п. .
* ^ ч , ' * Рис. 15.2. Півострів спалахування
вати величиною &4, що дає
Ф = с02[2*,-*5см]- (15.42)
За дуже високих тисків внаслідок зростання си вираз у дужках стає
від'ємним, а, як було вже зазначено вище, значення ф < 0 відповідає
стаціонарному стану. За дуже низьких температур вибуховий режим не
реалізується, отже, у координатах/? — Гвиникає так званий «півострів
спалахування» (рис. 15.2). Зазначимо, що положення нижньої межі «півострова»
(крива І) залежить від розміру посудини та від стану стінок. Це підтверджує,
що в цьому разі реакція обриву є гетерогенним процесом. Далі переходимо
до реакцій у розчинах.
399
15.3. Реакції у розчинах неелектролітів
Реакції в неполярних розчинах схожі на реакції у газовій фазі, тому що
вакуум можна вважати «неполярним розчинником». При цьому активація
частинок відбувається за рахунок обміну енергією з молекулами
розчинника, отже, можна вважати, що для цих реакцій ніяких відхилень від
розподілення Максвелла-Больцмана за енергіями не відбувається.
У таблиці 15.1 наведено дані для двох мономолекулярних реакцій у рідких
неполярних розчинниках, а також у газовій фазі. Видно, що зміна
розчинника суттєво не впливає на значення константи швидкості, причому для
газової фази константа теж не дуже відрізняється від значень для
неполярних розчинів.
Таблиця 15.1. Константи швидкості мономолекулярних реакцій
у неполярних розчинниках та в газовій фазі
Реакція
а-пінен
діпентен
розклад
н2о5
Середовище
газ
петролейний ефір
газ
СС14
СНСІз
к-103, є4
1,43
2,13
0,16
1,16
1,28
Атмосферному тиску в газах відповідають деци-сантимолярні розчини.
Справді, маємо/? = сКТі за температури Т= 300 К,р = 25 с, якщо с
вимірюється в моль/л, а/7 в атм. Таким чином, концентрації 0,01—0,1 моль/л
відповідає тиск 0,25—2,5 атм.
Для реакцій у розчинах може виникати так званий «комірковий» ефект.
Наприклад, розклад органічного пероксиду відбувається у газовій фазі
таким чином:
СНз—€—О—О—С—СНз -> 2СН3СОО#-> 2СН; + 2С02 -» С2Н6 + 2С02
О О
Якщо додати до системи пари йоду, то молекула І2 реагує з радикалом
сн;+і2->сн3і+і#.
У розчиннику радикали СЩ, що утворилися з однієї молекули
пероксиду, опиняються в «комірці» з молекул розчинника і відразу рекомбінують.
Додавання йоду в цьому разі не впливає на хід реакції.
У неполярних розчинах відбуваються і реакції вільнорадикальної
полімеризації. Ці реакції проходять як нерозгалужені ланцюгові процеси.
Зародження радикалів, ініціювання відбувається шляхом розкладу нестійких
органічних сполук, наприклад пероксидів. Приклад такої реакції наведено
вище.
Далі радикал реагує з мономером СН2=СНК. Приєднання молекули
мономеру до радикала знову дає радикал. Наприклад:
400
СН; +СН2=СНК-^СН3—СНК—СНз іт.д.
Це можна записати таким чином:
я;+м—^—>к;_
/+і
Реакційна здатність радикалів практично не змінюється зі зростанням
вуглецевого «хвоста», отже, можна вважати, що
к\ = к2 = к3 =.... */=.... кр, (15.43)
де кр— константа зростання полімерного ланцюга.
Обрив відбувається або шляхом рекомбінації двох радикалів
К| +К-2 —^ К| — К^2'
або шляхом диспропорціювання
К,—СН2—СН2 + К2 ->Кі—СН=СН2+К2Н
(при цьому атом Гідрогену переходить з одного радикала на інший, а в
першому виникає подвійний зв'язок). Зазначимо, що, з кінетичного погляду,
обидва ці процеси (рекомбінація та диспропорціювання) є
бімолекулярними реакціями. Застосовуючи метод стаціонарних станів, для стаціонарної
концентрації радикалів маємо
сіс
0 = ~? = Г/~*°Ск-' (15,44)
де Гі— швидкість ініціювання: к0—константа реакції обриву. Нагадаємо, що
на стадії подовження ланцюга концентрація радикалів не змінюється.
З рівняння (15.44) знаходимо стаціонарну концентрацію радикалів
(15.45)
Швидкість полімеризації дорівнює
г,°>5
:крсК* 'см-^7?см7оТ' (15.46)
к"
де см— концентрація мономеру.
Довжина реакційного ланцюга дорівнює (див. рівняння (15.27))
і стац. , , ,
_. ^ крсК. см ^ крси ^ крсм = Крси
К(с™ц')2 к0с™«' к0(П/к0)У2 (Пк0)У2'
Відповідно, швидкість полімеризації дорівнює добутку гі на V (див.
(15.29)), отже,
401
_ крси _ кр У2
гпоп. - П у/2 - -~У2СМП (15.48)
у повному узгодженні з рівнянням (15.46).
Для реакцій полімеризації цікаво зіставити довжину реакційного
ланцюга V з довжиною полімерного ланцюга Ь, яку можна визначити через
молекулярну масу полімеру. Якщо обрив здійснюється шляхом рекомбінації,
то Ь = 2у , а якщо диспропорціюванням, то Ь = V. Враховуючи обидві
можливості, позначимо через х частку радикалів, що обриваються шляхом
рекомбінації. Тоді кор = хк0, відповідно, код = (1 -х)к0, де кор та код —
константи рекомбінації та диспропорціювання. Тоді маємо
І = (1 - х)у + 2ху = у(1 + х). (15.49)
Якщо відомі V та Ь, то за рівнянням (15.49) можна визначити х.
Величину х можна визначити і незалежним шляхом за кількістю
подвійних зв'язків у полімері (подвійні зв'язки утворюються лише при дис-
пропорціюванні).
Користуючись рівняннями стаціонарної кінетики, можна з рівняння
(15.48) знайти відношення кр/к10 (процес ініціювання можна вивчити
окремо і мати інформацію про г{ у відповідних умовах). Для того щоб знайти
окремо константи (кр та к0) елементарних стадій, потрібна додаткова
інформація. її можна одержати, вивчаючи кінетику полімеризації в
нестаціонарних умовах. Залежність концентрації радикалів від часу зображено
на рис. 15.3. Ділянка II відповідає стаціонарному перебігу ланцюгової
реакції. Ділянка І (зростання концентрації радикалів до стаціонарної) має назву
«пре-ефекту», а ділянка III (зменшення концентрації радикалів після
«відключення» ініціювання) є так званим «пост-ефектом». Кінетика пост-ефекту
є простою — це бімолекулярний процес
*- = -к0сІ. (15.50)
або в інтегральній формі
III
ск'=-
_стац.
V (15.51)
(початку пост-ефекту відповідає ср. =
т =4?ц).
Рис. 15.3. Залежність концентрації Якщо діяти короткочасними ім-
вільних радикалів від часу пульсами, наприклад, вмикаючи на
дуже короткий час потужний опромінювач, що стимулює процес фотоіні-
ціювання, то кінетична крива для ск. є такою, як це зображено на рис. 15.4.
Величина А*—це частки секунди, отже, кількістю полімеру, що утворився
за цей час, можна знехтувати і вважати, що весь полімер утворюється в
процесі пост-ефекту. Позначаючи кількість синтезованого полімеру через
7, маємо
402
У = \гаоя.<Н = крс™си \ * =^см1п(і + к0с™*). (15.52)
У/ • лстац. ч
двох різних дослідах (за різних значень сК. )
, , * 1 + к0(с™ц)'(
Д7 = Г-Г = -^см1п ° к .
к„ М \ + к0(с™Уі
(15.53)
За відносно великих і можна знехтувати одиницею порівняно з добутком
,СТГ"
7 стац.^
к0с /, що дає
■«**„ ^Ь^їі-кр, і-<«е
ДУ = -^см1п
')'
— =—см1п-
М^")' *о (сСТаЦ)'
(15.54)
(<?"•)'
(ссхац.Г
можна замінити на
^'
V У
1/2
, отже,
клг ікР і Г'
2*Л
Г/
(15.55)
У фотохімічному ініціюванні
Гі = соп8ііі, (15.56)
де /— інтенсивність опромінювання.
Отже, з (15.56) та (15.55) маємо
ДГ=4^см1іД. (15.57)
З показниками, отриманими з пост- _
ефекту, знаходячи ДУза різних Іх та/2,
можна знайти відношення кр/к0. Це
разом з відношенням крІк^2 дає
можливість оцінити елементарні константи зростання кр та обриву к0 процесу
полімеризації.
Знання елементарних констант дозволяє простежити вплив будови
вихідних речовин на їх реакційну здатність та виявити детальний механізм
реакції полімеризації. Розглянемо, як впливає спряження вуглеводневих
груп (&! та К2) з неспареним електроном та з подвійним зв'язком на енергію
активації елементарної реакції зростання ланцюга
Рис. 15.4. Полімеризація
в імпульсному режимі
сн; +сн2=сн-
->СН2—СН2—СН
Гсн2...сн2...сн#]
И МІ І
&1 &2 ^1 ^2 ^1 ^2
Зіставимо реакції приєднання метильного радикала (К^ = Н) до етилену
(К2=Н) та стиролу (К2 = РЬ — фенільна група). За рахунок енергії спряження
403
з фенільною групою знижується енергія я-підсистеми і, відповідно, всієї
системи як у початковому стані (у стиролі порівняно з етиленом), так і в
кінцевому Енергія спряження РЬ з подвійним зв'язком є набагато меншою,
ніж з неспареним електроном. Отже, тепловий ефект (у термохімічній
шкалі) реакції приєднання радикала до мономера під час переходу від
етилену до стиролу збільшується. У перехідному стані енергія спряження
фенільної групи з квазіалільним радикалом має проміжне значення між
енергіями спряження в початковому та кінцевому станах, отже, енергія
активації реакції приєднання СН3 до мономера при переході від етилену до
стиролу зменшується. Таким чином, Д£ та Д# змінюються антибатно,
причому [АЕ] становить частину від [Д#], що є ще однією ілюстрацією
правила Поляні (див. співвідношення (14.92)).
В процесі взаємодії бензильного радикала (К^ = РЬ) з етиленом (К2 = Н)
енергія спряження (гіперкон'югація) фенільної групи в кінцевому стані, де
РЬ-група відокремлену від неспареного електрона двома СН2-групами,
набагато менша, ніж у початковому, і це веде до зменшення теплового
ефекту при заміні в метильному радикалі Гідрогену на феніл. У перехідному
стані енергія спряження теж зменшується порівняно з бензильним
радикалом, і це веде до збільшення енергії активації при заміні фенілу в
метильному радикалі. Отже, тут також спостерігаємо антибатну зміну енергії
активації та теплового ефекту реакції і, таким чином, маємо ще одну
ілюстрацію правила Поляні.
За відсутності гетероатомів ефект спряження дозволяє достатньо чітко
пояснити вплив замісників (К^ та К2) у радикалі й мономері, відповідно, на
процес радикальної полімеризації, зокрема для кополімеризації, коли К^ та
К2 належать різним частинкам. У гомополімеризації К^ К2= К,
позначаючи полімерний ланцюг через «оооооо», згідно з наведеною вище схемою
взаємодії радикала з мономером, маємо
оооооо СНК* + СН2=СНК -> схх>оооСНК—СН2—СНК#.
У наведеній схемі мономер приєднується до полімерного радикала так
званим способом «голова до хвоста». При приєднанні за схемою «голова до
голови» маємо
оооооо СНК# + СНК=СН2 -> ооооооСНК—СНК— СН2.
Якщо вважати, що К — це фенільна група, то при цьому способі в
кінцевому продукті втрачається структура бензильного радикала, отже,
порівняно зі схемою «голова до хвоста» тепловий ефект реакції зменшується
на величину енергетичного ефекту спряження в бензильному радикалі,
відповідно, зростає енергія активації. Таким чином, енергетично вигідним
є механізм «голова до хвоста», і це забезпечує регулярну будову полімеру,
зокрема полістиролу.
404
15.4. Реакції у розчинах електролітів
Реакції радикальної полімеризації в неполярних розчинах відбуваються
між незарядженими частинками. Для них вплив розчинника (див. початок
цього розділу) є незначнимТНавпаки, для реакцій у полярних розчинах між
зарядженими частинками (іони, дипольні молекули) велике значення має
електростатична взаємодія, що суттєво переважає всі інші неелектро-
статичні сили (зазначимо, що вода має велику діелектричну сталу і є
класичним полярним розчинником). Це ілюструється даними, наведеними в
таблиці 15.2, згідно з якими передекспоненціальні множники для реакцій між
іонами одного знака є аномально низькими, а для реакцій між іонами
протилежного знака — дуже великими.
Таблиця 15.2. Передекспоненти (к0) та ентропії активації (Д51*) для реакцій
між іонами
Реагенти
1)[Сг(Н20)6]3+ + СМ5-->
2) [Со(МН3)5Вг]2++ ОН" ->
3) СН2ВгСООСН3+ 320^->
4) СН2С1СОСГ + ОН~ ->
5) СЩЗгСОСГ + 320^"->
6)$202і + 8205"->
л0,
л/с-моль
1019
1017
1014
6-Ю10
109
2-Ю4
Д5*скс,
Дж/моль-К
130
92
24
-50
-76
-172
ДЯ'ел,
Дж/моль-К
126
84
0
-42
-84
-168
Спираючись на теорію перехідного стану і вважаючи, що вільна енергія
активації АС* отримує суттєвий внесок від електростатистичної взаємодії
(АС*Л), маємо
ДС*=ДС£Л+ДЄ0*. (15.58)
Обчислимо Д(?*л , користуючись простою моделлю «наближених куль».
Іони А та В, що реагують, мають заряди 2ке та 2ве і, згідно із законом Кулона,
взаємодіють із силою
2д2в£_
(15.59)
гг
де г— відстань між іонами; є — діелектрична стала розчинника.
ДС*Л дорівнює роботі утворення активного комплексу при зближенні
іонів з нескінченності до деякої відстані сІАВ
АС
"АВ 2 «АВ
(15.60)
Позначаючи всі складові в константі швидкості, що не залежать від
електростатичної взаємодії, через кИ, маємо
1п* = 1п£н-%^-.
(15.61)
405
0,00 Н
-0,25 [
-0,50 Ь
0,010
0,015
*АВ
З рівняння (15.61) видно, що в
координатах Іпк— І/є маємо пряму
лінію. Справді, як це видно з рис.
15.5, при взаємодії іонів ВгСН2СОО~
та 82Оз~ експериментальні точки
добре відповідають лінійній
залежності між І£&та І/є. Зі збільшенням
діелектричної сталої (зменшення І/є)
константа швидкості зростає, бо при
цьому зменшується сила
відштовхування між однаково зарядженими
іонами. З тангенса кута нахилу
можна оцінити */АВ, який тут дорівнює
0,6 нм.
Формула (15.60) дає можливість
оцінити ентропію активації (А5?), яка
дорівнює (в розрахунку на 1 моль)
2,1 '?§• <і5й)
І2Ае'Мл
Рис. 15.5. Залежність 1§ к від І/є
для реакції між іонами бромацетату
та тіосульфату
З експериментальних значень є та сіє/сіТ для води та фундаментальних
констант (є — заряд електрона, ИА — число Авогадро) та оцінки сіАВ маємо
А5*ел.=-А2А2В, (15.63)
де А — стала величина (мінус з'являється у формулі (15.63) тому, що йгІйТ—
від'ємна величина).
У таблиці 15.2 наведено розраховані за формулою (15.63) та
експериментальні значення ентропії активації для реакцій між іонами. Непогане
узгодження між ДЯ*^ та Д£*л свідчить про те, що електростатичні сили в
цьому випадку роблять вирішальний внесок в ентропію активації.
Множник ем ІК є пропорційним ймовірності взаємодії між частинками.
Природно, що для іонів протилежного знака ймовірність взаємодії збільшується
(Д5* » 0), а для іонів однакового знака зменшується (Д5* << 0) порівняно
з електронейтральними частинками.
Зі зміною концентрації іонів сі у розчині змінюється його іонна сила (див.
розділ 4)
/ = 2>,2,2. (15.64)
і
Іонна сила, за теорією Дебая-Гюккеля, визначає коефіцієнт активності
у і іона в розчині
ІЗУ^-^Т/. (15.65)
Швидкість реакції між іонами А та В визначається їх концентраціями
г = ксАсв. (15.66)
406
Але константа рівноваги К між активними комплексами та реагентами
визначається через активності {а( = у^), тобто
а * *
савУав
Звідки
£* _ аАВ
*асвУаУв
-АВ
= К'
УаУв
*
Уав
САСВ-
Оскільки, згідно з (14.64), швидкість г є пропорційною сАВ, то
* = *:.
УаУв
*
Уав
(15.67)
(15.68)
(15.69)
У рівнянні (15.69), яке носить назву рівняння Бренстеда-Б'єррума, &ід —
це значення константи швидкості за всіх у,- =1, тобто за с{ - а{ (в ідеальному
розчині). Використовуючи рівняння (15.65), маємо з (15.69)
18^ = ^77Г(2А+2В)2-2І-2Л = 2^2А2В77. (15.70)
На рис. 15.5 в координатах \%— >/7 нанесено дані для реакцій:
1)[Со(НН3)5Вг]2+ + Н£2+-->
л2+
2)820|~+Г-> 3)СНзСООС2Н5 + ОБГ->
5)Ре2++[Со(С204)з]3"
к
-»
4) [Со(Ш3)5ВгГ + ОНГ-
3 наведених на рис. 15.6 даних
видно, що експериментальні точки
добре відповідають теоретичним
прямим. Для реакції між
однойменно зарядженими іонами швидкість
збільшується зі зростанням іонної
сили. Фізично це означає, що іонна
атмосфера, яка утворюється
навколо іона з іонів протилежного знака,
за великих / більш ефективно
екранує відповідний іон, і це зменшує
відштовхування між однойменно
зарядженими іонами. Для
нейтральних частинок швидкість не залежить
від іонної сили, а при взаємодії іонів
протилежного знака зменшується зі
зростанням /.
Цікаву інформацію про механізм реакцій у розчинах можна отримати,
вивчаючи швидкість реакції за різних тисків. З відомого термодинамічного
співвідношення
ЛІД.
0,6
0,4
0,2
0,0
-0,2
-0,4
і
\о б^ч^
1
уі
^^2
"^оЗ
1
7
0
1
—^
0,1
0,2
0,3 V/
Рис. 15.6. Зв'язок між константою
швидкості реакцій між іонами
та іонною силою розчину
сір
V
)т
(15.71)
407
можна отримати рівняння, що описує залежність константи рівноваги від
зовнішнього тиску
Застосовуючи це рівняння до активного комплексу, маємо
АК* (15.73)
сііпк
{*)
т
КТ '
Величина (АУ) називається об'ємом активації.
Інтегрування рівняння (15.73) дає
1п* = 1п*0-^^, (15.74)
де к° — константа швидкості за/> —> 0, практично зар = 1 атм.
Як приклад залежності константи швидкості від тиску для реакцій у
полярних розчинах можна навести реакцію М. О. Меншуткіна
СзНз^СзНзІ^СзНзНСгНз)^,
для якої АК * — значна від'ємна величина (-17 см3/моль). Оскільки в реакції
виникають заряджені частинки, то від'ємні значення ЬУ можна пояснити
впливом електростатичного поля створених іонів на полярний розчинник
(ацетон). У сольватних оболонках, що виникають навколо іонів, розчинник
знаходиться у більш стисненому стані, ніж у середовищі розчинника.
Зазначимо, що контракція (стиснення) молекул розчинника під впливом
електростатичних сил впорядковує систему. Так, для реакцій Меншуткіна
характерними є великі від'ємні значення ентропії активації (А5 ).
Відносно великі від'ємні значення об'єму активації притаманні також
реакціям гідролізу складних естерів
КСООК'+Н20 > КСООН + К'ОН
та амідів
КСОШг + Н20 > КСООН+ІЧНз,
що, на перший погляд, не зрозуміло, бо це є реакції між нейтральними
частинками. Проте виявляється, що такі реакції безпосередньо у воді
перебігають дуже повільно, а для збільшення швидкості необхідно їх проводити
у лужному або кислому середовищі. Таким чином, у лімітуючих стадіях цих
реакцій беруть участь іони ОН~ або Н , які і створюють електростатичне
поле, що діє на полярний розчинник—воду. Прискорення реакцій гідролізу
іонами Н або ОН" є прикладом гомогенного каталізу, до розгляду якого
переходимо у наступному розділі.
408
Розділ 16.
ГОМОГЕННИЙ КАТАЛІЗ
Каталізатор не має спокою,
З тим, що гальмує, готовий до бою,
Фігаро там, Фігаро тут,
І реагент переходить в продукт.
16.1. Загальні принципи каталізу.
Окисно-відновний каталіз
У ланцюгових процесах важливою є стадія ініціювання, яка в
основному здійснюється за рахунок уведення в систему ініціаторів. Наприклад,
ініціаторами реакцій вільнорадикальної полімеризації є нестійкі органічні
сполуки: діазосполуки, органічні пероксиди тощо. На відміну від ініціаторів,
каталізатори прискорюють реакцію таким чином, що після (або в процесі)
її проходження вони регенеруються. Проілюструємо різницю між
каталізатором та ініціатором на прикладі реакції розкладу ацетальдегіду
СН3СОН->СН4 + СО.
Ця реакція перебігає за ланцюговим механізмом:
сн3сон->сн3со#+н#-+сн; +со+н#, (а)
н#+сн3сон -> сн; +со+н2, (б)
сн;+снзсон -> сни* сн; +со, (в)
СН;+Н2-->СН4+Н#, (г)
сн;+н#->сн4. (д)
Основною елементарною реакцією є стадія (в), в ході якої утворюються
обидва продукти та регенерується радикал СН;.
Ініціатор — нестійка органічна сполука, наприклад біацетил, генерує
радикали СН; за реакцією з малою енергією активації
СН3—С—С—СН3 -» 2СН: +2СО
II II
О О
При цьому біацетил зникає як сполука і не регенерується.
По-іншому діє каталізатор, у ролі якого в цій реакції виступає пара йоду.
Слабкий зв'язок між атомами Йоду дає молекулі І2 можливість легко
дисоціювати на атоми
І2 —> І* -ь Г. (а)
Далі перебігають такі реакції:
409
ґ+снзсон -> ш + сн; + со,
сн;+і2->сн3і+і#,
сн3і + ні->сн4 + і2,
(б)
00
(г)
(д)
І +Ґ-П2.
Отже, молекула І2 після перебігу реакції регенерується.
Таким чином, каталізатори — це речовини, що прискорюють хімічну
реакцію, беручи участь в елементарних стадіях хімічного процесу і
регенеруючись у ході реакції. Саме явище прискорення хімічних реакцій таким
шляхом має назву каталізу. У наведеному вище прикладі каталізатор та
реагенти знаходилися в одній фазі. Цей приклад належить до гомогенного
каталізу. Крім того, існує каталіз гетерогенний, де процеси на межі поділу
фаз є суттєвими; переважно це каталіз твердими речовинами хімічних
процесів у газовій та рідкій фазах. Гетерогенний каталіз розглядатиметься далі
(розділ 18) після викладу основ фізичної хімії поверхневих явищ (розділ 17).
У механізмі каталізу характерною є так звана двоступінчаста схема, яку
умовно можна зобразити таким чином:
А + К—^->АК
АК + В *2 )АВ(продукти) + К
(1)
(2)
Розглянемо застосування двостадійної схеми на конкретному
прикладі окиснення 802 молекулярним киснем, що каталізується оксидами
нітрогену.
Діоксид сульфуру погано
окиснюється молекулярним
киснем, але відносно легко діокси-
дом нітрогену за реакцією:
802 + Ш2->803 + Ш. (а)
Далі N0 легко реокиснюється
до Ж)2 молекулярним киснем
2Ш + 02->2Ш2. (б)
Реакції (а) та (б) утворюють
каталітичний цикл, який
схематично зображено на рис. 16.1.
Кожна з цих стадій має
набагато меншу енергію активації,
ніж некаталітична реакція,
2802 + 02->2803. (в)
Рис. 16.1. Каталітичний цикл у реакції . Енергетичний профіль цієї
окиснення 802 кінетичної схеми наведено на
410
Рис. 16.2. Енергетичний профіль
для двостадійної каталітичної реакції:
Ех та Е2 — енергії активації стадій,
Е — енергія активації некаталітичної
реакції
рис. 16.2. Двостадійний шлях через
стадії (1) та (2) є більш ефективним,
ніж некаталітичний одностадійний,
що має дуже високу енергію
активації (Е).
Таким чином, дія каталізатора
в основному зводиться до того, що
він надає реакції можливість
перебігати іншим шляхом з набагато
меншими потенціальними
бар'єрами. Тут доречно зупинитись на
так званому «негативному
каталізі». В. Оствальд початково
визначив каталізатор як речовину, що
змінює швидкість реакції. Це визначення має на увазі, крім прискорення,
також і гальмування реакції. Речовини, які галі^мують реакцію, існують.
Наприклад, хінони і деякі інші сполуки реагують з активними радикалами,
переводячи їх у неактивні, і, таким чином, гальмують ланцюговий процес.
Ці речовини називаються інгібіторами. Проте в ході реакції інгібітори не
відновлюються, як й ініціатори. Отже, інгібітори є антиподами ініціаторів, їх
можна було б назвати «негативними ініціаторами», але не каталізаторами.
Принципово важко уявити собі дію негативного каталізатора. «Позитивний»
каталізатор прискорює реакцію, надаючи інший шлях з малою енергією
активації. Відповідно, негативний мусив би створювати повільний шлях із
високою енергією активації, але при цьому реакція просто обминала б цей
шлях, перебігаючи некаталітично.
Прикладом двостадійної каталітичної реакції у розчині є реакція
розкладу пероксиду гідрогену, що каталізується бромом. На першій стадії Вг2
виступає як окисник, а Н202 — як відновник
Н202 + Вг2 -> 2НВг + 02.
(а)
Далі бромід окиснюється вже іншою молекулою пероксиду гідрогену,
тобто на стадії (б) бром регенерується
Н202 + 2НВг -> Вг2 + 2Н20.
Некаталітична реакція
2Н202->2Н20 + 02.
(б)
(в)
має високу енергію активації і перебігає повільно.
Згідно з двостадійною схемою каталізатор повинен знаходитися
принаймні у двох станах. Для окисно-відновних реакцій, до яких належать
розглянуті вище реакції, це є різні ступені окиснення каталізатора. Наприклад,
нітроген (в оксидах) по черзі міняє ступінь окиснення з (+2) до (+4) і
зворотно, а бром з (0) до (-1) і зворотно.
411
Таким чином, для участі у гесіох реакціях каталізатор повинен мати
принаймні два відносно стабільні ступені окиснення. Наприклад, у процесі
розкладу пероксиду гідрогену в розчині, Ферум, що має два ступені
окиснення (+2) та (+3), є добрим каталізатором цієї реакції на відміну від Нікелю
та Кобальту, для яких характерним є лише один стабільний ступінь
окиснення (+2).
Механізм каталітичної дії іонів Феруму на пероксид гідрогену можна
зобразити таким чином:
Ре2+ + Н202 -> Ре3+ + ОН* + ОН~,
Ре3+ + Н202 -> Ре2+ + Н20 %,
що\ + он~-> н2о + но;,
Ре2++ОН#->Ре3+ + ОН",
Ре3++Н02->Ре2++НС4,
НО£ + ОН~->Н20 + 02.
Ферум у ході реакції по черзі змінює свій ступінь окиснення.
Цікавим є випадок, коли каталізатор одночасно є продуктом реакції. Це
явище має назву автокаталізу. До автокаталітичних належить окисно-
відновна реакція окиснення оксалату перманганат-іоном, що каталізується
іонами Мп , які одночасно є продуктом реакції
5С2024~ +2М11О4 + 16Н+ Мп2+ > 2Мп2+ + 10СО2+8Н2О.
МпС>4 та С204~ іони мають однакові заряди й відштовхуються один від
одного, але при взаємодії МПО4 з Мп утворюється іон Мп , який і окис-
нює оксалат. Щоб реакція почалася, треба в реакційну посудину внести
невелику кількість каталізатора — Мп або чекати, доки цей продукт
утвориться за рахунок некаталітичної реакції.
Розглянемо кінетичну схему автокаталізу, де каталізатор і реагент входять
до кінетичного рівняння у 1-му порядку
^ = к(а-х)(<1 + хІ (16.1)
аі
де а — початкова концентрація реагенту, й— каталізатора, (й + х) —
поточна концентрація продукту — каталізатора.
За відсутності деякої кількості каталізатора у початковому стані треба
враховувати некаталітичний процес
§ = *>-*)• (16.2)
Для двох паралельних процесів загальна швидкість дорівнює
г = & = к(а-х)((і + х) + кп(а-х) =
Ж (16.3)
= к(а - х)(сі + к„/к + х) = к(а - х)ф + х).
412
Вираз (16.3) кінетично є ідентичним формулі (16.1). Інтегрування
рівняння (16.3) дає (див. схожі рівняння (13.26) та (13.27))
1 -1п^±^ = &
Ь + а {а- х)Ь
(початкові умови х = 0 за і = 0).
Розв'язуючи це рівняння щодо х9 маємо
х =
аЬ[е^а)к'-\]
(16.4)
(16.5)
Рис. 16.3. Залежність концентрації х
та швидкості реакції г від часу
для автокаталітичної реакції
а+Ье{Ь+а)к1
Оскільки практично завжди
виконується умова а »Ь (умова кпІк «
« 1 виконується завжди, оскільки
некаталітична реакція є набагато
повільнішою, ніж каталітична, умову
й« а легко створити штучно), то
(16.5) трохи спрощується
аЬ(еа -1) ґлс с.
* = , аЬ ' О6'6)
Залежність концентрації про-
дукту-каталізатора* від часу
зображено на рис. 16.3.
Кінетична крива має 5-подібний
вигляд. Спочатку є індукційний період, потім різке
зростання х, нарешті, коли значна частина
реагенту прореагувала, крива асимптотично
наближається до лінії а = соті (з рівняння (16.5) маємо
за і -» ©о х —> а). На тому самому графіку
наведено залежність г {йх/йі) від часу. Точці перегину (р)
на кривій х =Ді) ((і хШ = 0) відповідає точка
максимуму на кривій г =Д0 (йг/йі = 0). Так, за
рівнянням (16.3) можна пересвідчитися в тому, що
дх/йі проходить через максимум. За х —> 0 дх/йі =
= каЬ, але Ь є малою величиною, далі виникає
така ситуація, коли а-х=а, але Ь+х = х (х
суттєво перевищує 6), отже, тут дхШ = ках, і оскільки
х >> Ь, то йх/йі зростає. За х —> а йх/йі —> 0.
З умови йг/йі - 0 не важко знайти хр = (а - Ь)І2 =
= а 12. До точки перегину (р) можна вважати, що
еакі»\, але оскільки а » 6, то за відносно неве- рис# і$.4. Ядерний вибух
ликих/ у знаменнику (16.6) можна знехтувати як приклад розгалуженого
другим членом. Це дає ланцюгового процесу
з позитивним зворотним
х = Ьеакі. (16.7) зв'язком
413
Тобто кількість каталізатора (продукту) на цьому етапі зростає експонен-
ційно. Рівняння (16.7) корисно зіставити з рівнянням (15.41) для розгалужених
ланцюгових процесів, де також кількість активних частинок (радикалів)
зростала експоненційно, лавиноподібно. В обох випадках реалізується так
званий «позитивний зворотний зв'язок», коли наслідок посилює причину, що
його викликала. Це веде ^, лавиноподібного наростання процесу. Одним
з найбільш яскравих прикладів такого лавиноподібного процесу є ядерний
вибух (рис. 16.4, с. 413). Зазначимо, що в межах фізичної хімії приклад
«негативного зворотного зв'язку», коли система намагається ліквідувати наслідки
зовнішньої дії, надає принцип Ле-Шательє (див. підрозділ 6.2).
16.2. Кислотно-основний каталіз
Реакції гомогенного каталізу — це переважно
реакції у розчинах. Серед них поширеними є
реакції, що каталізуються кислотами або
основами. Прикладами таких реакцій є інверсія
цукрози, мутаротація глюкози, омилення естерів
тощо.
Розглянемо механізми кислотно-основного
каталізу, користуючись теорією кислот (основ)
Й. Бренстеда. Згідно з цією теорією кислота — це
донор протонів, а основа—акцептор протонів. Для
кожної кислоти є відповідна основа. Тобто існує
рівновага
Бренстед Йоханнес Ніколаус
(1879—1947), Данія кислота <==> основа + протон.
Наприклад: Н30+<==> Н20 + Н+, НСк==>Н++ СГ, НН+4<==^Н3 + Н+,
Н20<==ЮН~ + НҐ тощо. Ці напівреакції не існують окремо, а об'єднуються
попарно з переносом протона від кислоти на відповідну основу. Наприклад:
НС1 + Н20ф==»Н30+ + СГ
к-та осн. к-та осн.
Н30++ ОН~*=>Н20 + Н20
к-та осн. к-та осн.
Ш| + Н20«=*Ш3 + Н30+
к-та осн. осн. к-та
Механізм кислотно-основного каталізу розглянемо на прикладі дії
кислоти НА. На першій стадії відбувається перехід протона з каталізатора на
реагент (субстрат 8)
5 + НА( *' >8Н++А~
*-' +
Друга стадія — це взаємодія протонізованої форми субстрату (8Н ) з
розчинником (В)
414
8Н+ + В —Ь-> ВН+ + продукти.
Нарешті, завершальна стадія (регенерація) — це обмін протонами між
каталізатором та розчинником
ВН++А"—^->В + НА
(якщо водні розчини, то В — це Н20, а ВН — Н30 ).
Конкретним прикладом може бути реакція гідратації карбодиіміду
к—н=с=:к—к+н2о—> к—ин—со—ин—к,
що перебігає повільно. Кислота-каталізатор (НА) дає протон, як^й
приєднується до диіміду /
К—М=С=И—К + НА >К—Ш— С+=И—К + А~, (а)
і робить цю молекулу зарядженою і реакційно здатною. До неї тепер легко
приєднується молекула води, використовуючи неподілені пари електронів
Оксигену
К—Ш— С+=К—К+Н20 >[К—Ш—С+=Н—К ], (б)
6-Й
Н
Далі після перерозподілу зв'язків у проміжній сполуці від неї
відщеплюється протон з регенерацією каталізатора
[К—Ш— С—Ш—К] + А" >К—Ш—СО—Ш—К + НА. (в)
ОН+
Використовуючи наведений вище стехіометричний механізм та метод
стаціонарних станів, маємо
-*н- = 0 = А:,с8сНА -к_{сш+сА -к2сш+св, звідки
&
сстац. = ^1С5СНА
зн+ к_хсА_+к2св'
Швидкість утворення продукту дорівнює
~ _ 1г ^.стац. _ ^1^2С3СНАСВ
г-к2с5н+с»-к_іСА_+к2св- (16.8)
Розглянемо різні варіанти. Якщо к_хсА-» к2св, то в знаменнику (16.8)
можна знехтувати величиною к2св, що дає
_ кхк2с^сЯксв _ кхк2сьсяксвсш+ _ _
Г~ , - к С С -^2^1ЛЗсЗсвн+-^еф.^вн^ У1™'
К-1СА~ ,С-іСА-СВН+
Для водних розчинів
г = кЄфС$сп3о+. (16.10)
415
Це так званий випадок специфічного кислотно-основного каталізу, коли
каталізатором виступає протон (приєднаний до молекули розчинника,
наприклад, іон гідроксонію Н30 у водних розчинах). Не важко
пересвідчитися в тому, що активна проміжна частинка 8Н (протонізована форма
субстрату) при цьому знаходиться у рівновазі (квазірівновага на 1-й стадії)
із субстратом та каталізатором. Лімітуюча стадія — взаємодія 8Н з
розчинником.
Другий варіант: к_хск- « к2св, що дає, згідно з (16.8),
г = М2£з£на£в=/:іСнаС8 (1611)
К2С&
Це так званий випадок загального кислотно-основного каталізу.
Каталізатором виступає кислота НА (або відповідна основа). Лімітуюча стадія —
це взаємодія субстрату (8) з каталізатором (НА).
Для загального каталізу Й. Бренстедом було встановлено емпіричним
шляхом залежність між константою швидкості каталітичної реакції (тут кх)
та силою кислоти (основи), що визначається відповідною константою
дисоціації (К^),
^=^!н1 (16.12)
СНА
Константа дисоціації характеризує здатність кислоти (основи)
передавати (брати) протон розчиннику, тобто відповідає рівновазі
НА + В«=»ВН+ + А~
або для водних розчинів
НА + Н20«==»Н30++А"
Концентрацію розчинника (наприклад Н20) в розчині можна для не дуже
концентрованих розчинів вважати сталою величиною, отже, вона входить
у константу КЛ.
Співвідношення Бренстеда має вигляд:
к = §(К,)а, (16.13)
де 0 < ос < 1, £ — деяка стала в ряду однотипних каталізаторів (наприклад,
у ряду однотипних кислот).
Таким чином, здатність каталізатора—кислоти (НА) — віддавати протон
розчиннику (термодинамічний параметр — К^) корелює зі здатністю
кислоти віддавати протон субстрату (кінетичний параметр, який у даному разі
є константою швидкості (к{) лімітуючої стадії—взаємодії субстрату з
каталізатором НА).
Співвідношенню (16.13) можна надати вигляд
Іпкі = соті - аЩКсі)» (16.14)
або
Д1п£, = осДВД/)/ (16.15)
416
ідалі
Д(ДС*) = аД(ДС°).
(16.16)
Таким чином, зміна вільної енергії активації в ряду однотипних реакцій
у процесі утворення активного комплексу (8Н ) складає частину (0 < а < 1)
зміни вільної енергії при переносі протона від НА до розчинника.
Співвідношення Бренстеда (16.16) для вільних енергій є ідентичним
співвідношенню Поляні (14.92) для повних енергій і виступає своєрідним «містком»
між кінетичними та термодинамічними параметрами.
Конкретний приклад, що ілюструє співвідношення Бренстеда, наведено
на рис. 16.5 для реакції йодування ацетону
НА
СНз—СО—СН3 + І2-
що каталізується відповідними
органічними кислотами (НА).
Стехіометричний механізм цієї
реакції було розглянуто в розділі 13
(кінетичне рівняння (13.57) дає 1-й
порядок за ацетоном і 0-й за йодом),
де було доведено, що лімітуюча
стадія реакції йодування ацетону —
це кето-енольне перетворення. Саме
ця стадія прискорюється кислотою
шляхом приєднання протона до СО-
групи з наступним його
відщепленням з СН3-групи.
Приєднання протона до
молекули субстрату та наступне його
відщеплення можуть відбуватися
синхронно за так званим «пуш-пульним»
механізмом.
->СН3—СО—СН2І + НІ,
2 ґ
1 Ь
Рис. 16.5. Співвідношення Бренстеда
для каталізу кислотами:
1 — (СН3)3ССООН; 2 — СН3СН2СООН;
З — СН3СООН; 4 — СН3СНС1СООН;
5 — СН2ОНСООН; б — СН2С1СООН;
7 — СН2ВгСНВгСООН; 8 — СНС12СООН
Проілюструємо дію цього механізму на реакції мутаротації глюкози. Для
цієї реакції було встановлено, що в неводних розчинах суміш кислоти
(крезолу) та основи (піридину) діє набагато ефективніше, ніж кожен з
каталізаторів (кислота або основа) окремо. Це пояснюється «пуш-пульним»
механізмом, що схематично зображено на схемі:
сн2он НА
І /
сн—о
он—сн сн—он—►в
\ /
сн—сн
он он
417
СН2ОН
І
сн—он 0
/ " ♦
ОН СН СН + ВН +А~
\ / ^—
сн—сн
І І
он он
Ще більш ефективним каталізатором реакції ізомеризації глюкози є
оксипіридин. Цей каталізатор є біфункціональним і має кислотну та основну
групи в метаположенні, що дає йому змогу ефективно діяти за «пуш-пуль-
ним» механізмом.
16.3. Коливальні каталітичні реакції
Розгляд механізмів гомогенного каталізу (як окисно-відновного, так і
кислотно-основного) показує, що каталізатор може знаходитися у різних
(принаймні двох) квазірівноважних або квазістаціонарних станах. Виявилося,
однак, що у нерівноважній хімічній системі стаціонарний стан може
втратити стабільність, в результаті можуть виникнути коливання між
стаціонарними станами.
Підбираючи каталізатор для окиснення бромату цитратом, В. П. Бєлоу-
сов вибрав церій, який відносно легко переходить із ступеня окиснення (III)
до (IV) і навпаки, причому відновлена форма каталізатора (Се ) відносно
легко реагує з окисником загальної реакції—броматом, а окиснена (Се ) —
з відновником цитратом. Церій насправді виявився хорошим каталізатором
цієї реакції, але при цьому спостерігалися коливання концентрації окисне-
ної і відновленої форм церію. Робота В. П. Бєлбусова була продовжена
А. М. Жаботинським, і було виявлено цілий клас коливальних каталітичних
хімічних реакцій окиснення броматом органічних сполук, які отримали
назву—реакції Бєлоусова-Жаботинського (БЖ). Однією з найбільш
відомих реакцій цього класу, яка ілюструє дію хімічних осциляторів, є реакція
окиснення малонової кислоти броматом
(Се3+ Се4+)
ЗСН2(СООН)2 +2НВЮ3 = 2СНВг(СООН)2 +ЗС02 +4Н20, (а)
що каталізується іонами церію.
Бромат-іон не реагує безпосередньо з органічною кислотою, але
відновлюється іоном Се
2ВЮ^+12Н++10Се3+ =Вг2+6Н2О + 10Се4+. (б)
Далі відбувається бромування малонової кислоти
н+
Вг2 + СН2 (СООН)2 = СНВг(СООН)2 + Н+ + Вг" (в)
418
Цей процес каталізуєтся іонами Н і перебігає через енольну форму
кислоти
СН2(СООН)2 ( н+ )ОН—С(ОН)=СНСООН.
(аналогічним до розглянутої вище реакції (в) є процес йодування ацетону,
див. підрозділи 13.2 та 16.2).
Малонова та броммалонова кислоти відновлюють Се за реакціями
6Се4+ + СН2 (СООН)2 + Н20 = 6Се3+ + НСООН + 2С02 + 6Н+. (г)
(відзначимо, що якщо вихідною формою каталізатора — церію є Се , а не
Се , то слід починати саме з реакції (г)),
4Се4+ +СНВг(СООН)2 + 2Н20 =4Се3+ +НСООН+2С02 +5Н+ + Вг~. (д)
Мурашина кислота у кінцевих продуктах відсутня, вона окиснюється до
С02 бромом
НСООН + Вг2 = С02 + 2Н+ + 2Вг". (є)
Реакції (д) та (є) є постачальниками у систему бромід-іонів. які є
хорошими відновниками, і вони починають конкурувати з іонами Се у процесі
відновлення бромату
5Вг_+ВЮ^ + 6Н+ =ЗВг2 +ЗН20. (є)
Реакція відновлення бромату (є), так само, як і реакція (б), не є
елементарною, а перебігає через утворення проміжних сполук, серед яких є НВгО
та НВЮ2. Наприклад, для реакції (є) маємо
ВЮ^ + ВГ + 2Н+ = НВЮ2 + НВгО]
НВЮ2 + Вг" + Н+ = 2НОВг 1
НОВг + Вг" + Н+ = Вг2 + Н20
Після певного індукційного періоду накопичення броммалонової
кислоти і далі Вг~ (введення в систему СНВг(СООН)2 знімає індукційний період),
починається різке зростання концентрації бромід-іонів (рис. 16.6, с. 420). Це
веде до того, що реакція (є) стає домінуючою в утворенні брому, а реакція
(б) пригнічується, домінуючою формою каталізатора при цьому стає Се
(див. рис. 16.6).
Зникнення форми Се веде до гальмування реакцій (г) і (д), і далі (є),
отже, зникає джерело постачання іонів Вг~, які продовжують інтенсивно
витрачатися за реакцією (є). Коли концентрація бромід-іону стає меншою від
деякої критичної величини [сВг-]крит., реакція (є) в утворенні Вг2 вже не
відіграє головної ролі, яка переходить знову до реакції (б). У реакції (б)
регенерується Се , концентрація якого зростає (див. рис. 16.6). Після появи
Се за реакціями (г), (д) і (є) починає зростати концентрація бромід-іону
і після переходу через [сВг- ]крит. знизу реакція (є) знову починає відігравати
основну роль в утворенні Вг2.
419
Се(ІУ)/Се(Ш)
ЧЛЛЛЛЛЛЛЛ
вг
Рис. 16.6. Зміна 1§(ВГ) та 1§[Се(ІУ)/Се(Ш)] з часом
у реакції Бєлоусова-Жаботинського
Таким чином, відбуваються коливання між двома формами
каталізатора Се /Се , причому в антифазі з відновленою формою каталізатора (Се )
Се(ІУ)
знаходиться концентрація Вг~
(відновника).
Математичне моделювання
реакції БЖ дає так званий граничний цикл
коливань (атрактор) (рис. 16.7). Час як
змінна при цьому виключений,
максимальні та мінімальні значення
концентрацій (тут Се та Вг~) визначають
амплітуду коливань.
Крім реакцій БЖ, відомі інші
коливальні реакції, наприклад, розклад
Н202 у присутності йодату (реакція
Брея-Лібавські); періодичний
«йодний годинник» (реакція Бріггса-Рау-
шера).
Коливальні процеси
спостерігаються і при перебігу гетерогенно-каталітичних реакцій (підрозділ 18.3).
Відзначимо, що коливальні хімічні реакції є хорошими моделями коливальних
процесів у біохімічних системах.
ВГ
Рис. 16.7. Граничний цикл (атрактор)
у площині координат концентрацій Вг~
та Се(ІУ) у реакції БЖ
16.4. Каталіз комплексами
Важливим підрозділом гомогенного каталізу є каталіз розчиненими
комплексами перехідних металів. Перехідні метали мають частково
заповнені й?-орбіталі, що дає можливість координації центральним іоном металу
відповідних молекул субстратів. Оскільки на й?-орбіталях кожна розташувати
різну кількість електронів, то для того самого металу можливі різні стабільні
ступені окиснення та різні координаційні числа, що різко збільшує його
потенційні можливості бути застосованим як каталізатор.
420
Розглянемо дію металокомплексних каталізаторів на прикладі реакцій
гідрування олефінів
>С=С< + Н2 -> >СН—СН<.
Конкретний приклад — це гідрування алкенів у неводних розчинах із
застосуванням як каталізаторів комплексів родію — КЬС1Ь3, де Ь —
ароматичний третичний фосфін. Комплекс КЬС1Ь3 має квадратну структуру, а
для приєднання молекули водню з дисоціацією на атоми треба мати
вакантні місця не в транс-, а в ї^с-положенні. Отже, першою стадією
каталітичного процесу є рівноважна дисоціація комплексу з відщепленням одного
з лігандів Ь
ШіС1Ь3 = КЬС1Ь2 + Ь.
Концентрація комплексу ЯЬС1Ь2 є дуже низькою, але він має високу
реакційну здатність і приєднує водень за схемою
Н
Ь _ Ь | Н
сіх ь сі рь
□
Отриманий комплекс є недобудованою октаедричною структурою, і на
вакантне місце (Д) приєднується молекула алкену з утворенням так званого
я-комплексу
Розглянемо характер хімічного зв'язку у цьому я-комплексі відповідно
до моделі Дьюара-Чатта-Дункансона (рис. 16.8, с. 422).
ям-зв'язуюча орбіталь алкену перекриваться з вакантною <іа-орбіталлю
центрального атома (і2і) з утворенням донорно-акцепторного зв'язку. При
цьому відбувається перенесення електронної густини з алкену на метал, що
веде до послаблення я-зв'язку в алкені, оскільки зменшується електронна
густина на зв'язуючій ям-МО. Вакантна я* -антизв'язуюча орбіталь алкену
перекривається із заповненою й^-орбіталлю металу (с!Х2) з утворенням да-
тивного (зворотного донорно-акцепторного) зв'язку. При цьому
відбувається перенесення електронної густини з металу на алкен, причому, оскільки
електрони потрапляють на антизв'язуючу молекулярну орбіталь алкену, то
це також веде до послаблення я-зв'язку. Таким чином, координація молекули
421
алкену до каталізатора — перехідного металу робить цю молекулу
реакційно здатною по відношенню до розриву я-зв'язку.
Наступна стадія — це процес укорінення субстрату за зв'язком метал-
гідроген з остаточним розривом я-зв'язку в алкені. Цей процес перебігає
через утворення циклічної проміжної сполуки за схемою
-6 и---с +5
- (Ьп)М ■
Алкілродієвий комплекс — дуже
реакційно здатна частинка, він легко
утворює молекулу алкану >СН—СН<
з регенерацією каталізатора до
КЬС1Ь2, який вступає в наступний
цикл взаємодії з алкеном.
Схожий механізм мають реакції
каталітичної полімеризації. На
вакантне місце в комплексі приєднується
молекула алкену, і далі вона
укорінюється за зв'язком М—К, де М — атом
металу, а К — атом Гідрогену або
органічний радикал. Процес
укорінення може повторюватися
багаторазово зі збільшенням молекулярної
маси радикала. Реакція закінчується
процесом перенесення |3-атома
Гідрогену (Р-елімінування), внаслідок якого органічний ліганд іде з комплексу
разом з алкеновою групою, а Р-атом Гідрогену з С—Н зв'язку в радикалі
переходить на метал. Процес Р-елімінування є фактично протилежним
процесу укорінення і також відбувається через циклічну проміжну сполуку
Рис. 16.8. Хімічний зв'язок
у я-комплексі алкену з перехідним
металом
(Ьп)—СН2—СН2"
Пн
м---сн2
—к
(Ьп)
(Ьп)МН + СН2=СНК
Якщо Р-елімінування відбувається після двох актів зростання
органічного ліганда, то процес закінчується димеризацією. При збільшенні
швидкості укорінення порівняно з елімінуванням молекулярна маса полімеру
зростає. /
Було встановлено, що перехідні метали УІІГгрупи сприяють перенесенню
Р-атома Гідрогену і, отже, вони є каталізаторами димеризації. Каталізатори
422
полімеризації зазвичай обирають із металів IV—VI груп періодичної
системи.
Важливість існування координаційних вакансій у комплексному
каталізаторі яскраво ілюструється так званим «темплатним» ефектом. Цей термін
означає координацію декількох молекул субстрату на одному каталітичному
центрі, що потребує декількох вакансій на одному комплексі. Відомим
прикладом є утворення циклооктатрієну з ацетилену на нікелевому
каталізаторі. При цьому чотири молекули С2Н2 координуються до одного центру
(рис. 16.9, а). Блокада фосфіновим лігандом одного з координаційних місць
веде до утворення бензену замість циклооктатрієну (рис. 16.9, б), а блокада
бідентатним лігандом (о-фенантроліном) двох вакансій взагалі інгібує
реакцію (рис. 16.9, в).
N1
~Ш
Нема реакції
Рис. 16.9. Темплатний ефект
Крім моноядерних координаційних сполук, широкого поширення набули
поліядерні, серед яких цікавими для каталізу виявилися так звані кластерні
сполуки, тобто поліядерні комплекси із прямими зв'язками метал-метал.
Приклад кластерної сполуки кобальту — Со3 (СК)(СО)9 (К = С6Н4КНСН3)
наведено на рис. 16.10, с. 424. Ця сполука є хорошим каталізатором реакції
перетворення пероксид-радикалів (КОО*). Розглянемо це на прикладі супер-
оксиду гідрогену
2Н02->Н202+02.
423
Окисно-відновний каталізатор (наприклад моноядерний комплекс МІлі
перехідного металу) діє в цій реакції за схемою
НО^+МЬп-
а)
б)
в)
а)
► Н++02+Ке</,
де Косі— відновлена форма металокомплексу і далі
НО;+Ке</-*НС>2+МЬп
Н02+Н+->Н202.
Кластерца сполука [К] може координувати реагент
НО; + [Я1-»[£<-: О-Н],
І
О
що підвищує електрофільність Н02 і полегшує його взаємодію з наступною
частинкою супероксиду
[К<-:0-Н] + Н02-^[К] + 02 + Н+ + Н02
І * „ ' б)
О н2о2
Відзначимо, що на стадії (б) друга частинка Н02 також координується
до кластера з утворенням лабільного проміжного комплексу, при цьому
через кластер можливий перенос електронної густини з однієї координованої
частинки Н02 до іншої.
Кластерні сполуки набули поширення як каталізатори досить широкого
спектра реакцій, серед яких окиснення спиртів, гідрування, олігомеризація
та полімеризація олефінів, гідрування СО, карбонілювання метанолу та
нітросполук, ацетоксилювання етилену та пропілену тощо.
Металокомплексні та особливо кластерні каталізатори мають певну
структуру, будову (порівняно, скажімо, з іонами металів в
окисно-відновному каталізі). В цій структурі є
місця, де координуються та
перетворюються молекули
реагентів, ці ділянки отримали
назву активних центрів. Поняття
активний центр виникає і чітко
формулюється саме в метало-
комплексному кластерному
каталізі. Це поняття є дуже
важливим у гетерогенному каталізі,
але для опанування гетерогенно-
каталітичними процесами, які
перебігають на межі поділу фаз,
необхідні певні знання з області
фізичної хімії поверхневих
явищ, що викладені у наступному
розділі 17.
Рис. 16.10. Кластерна сполука Кобальту
424
Розділ 17.
ФІЗИЧНА ХІМІЯ МІЖФАЗНИХ ЯВИЩ
Обличчя в кожній фазі є своє,
Тому межа між фазами встає.
17.1. Поверхневі явища у конденсованих фазах
Згідно з прийнятою термінологією міжфазні явища на межі
конденсована фаза-газ (вакуум, пара), тобто на межах поділу рідина-газ та тверде
тіло-газ, називають поверхневими явищами. Термін міжфазні явища
зберігся для меж між конденсованими фазами: рідина-тверде тіло; рідина-
рідина; тверде тіло-тверде тіло. В даному підрозділі розглядатимуться саме
поверхневі явища.
Візьмемо ізотропну однокомпонентну систему (рідина, аморфне тверде
тіло) і розглянемо межу конденсована фаза-пара (вакуум). |Сожна система
намагається позбутися зайвої енергії, з урахуванням поверхневої енергії
цього можна досягнути за рахунок скорочення межі поділу. При
фіксованому об'ємі мінімальну поверхню з усіх геометричних фігур має сфера,
отже, для ізотропних тіл рівноважною формою є куля. Якщо для кулі
радіуса (г) збільшити радіус до (г + сіг), тобто на нескінченно малу величину, то
при цьому її поверхня збільшується на величину
а об'єм на величину
4п(г + сіг)2 - 4пг2 = 8пгсІг,
^(г + ^)3_^=4яЛ/г
(нехтуючи членами, що містять (сіг)2 і (сіг)3).
Загальна зміна поверхневої енергії &пгсіг • а (а), де а — питома вільна
поверхнева енергія, повинна компенсуватися роботою стиснення
(розширення) системи АР4пг2сіг — (б), де АР — різниця зовнішнього і
внутрішнього тисків. Прирівнюючи (а) і (б), маємо
АР = ^. (17.1)
Формула (17.1) відома як рівняння Юнга-Лапласа. Для несферичних тіл
формула (17.1) має вигляд
АР = а[ -І- + -Ї- І, (17.2)
де г{ та г2 — радіуси кривизни.
Длясфери^ = г2 = г і формула (17.2) перетворюється на (17.1). Для
циліндра радіусом г г1 = г,г2 = °° і (17.2) набуває вигляду (
ДР = Я (17.3)
425
Формули (17.2) та (17.1) справедливі для рухомих меж поділу ізотропних
фаз і обґрунтовують явища капілярності, на базі яких можливе
експериментальне визначення поверхневої енергії.
Розглянемо деякі конкретні приклади.
Метод сталагмометри базується на вимірюванні ваги краплин, що
витікають із капіляра радіусом г. Сила, що відриває краплю, дорівнює:/! = т%,
де т — маса краплини; £ — прискорення вільного падіння. Сила, що
утримує краплю, дорівнює: /2 = сг27гг, де 2кг — довжина периметра, а тут
трактується як сила на одиницю довжини, тобто як «поверхневий натяг». При
відриві краплини/! =/2 і звідти маємо
[сила] . . . [енергія]
Відзначимо, що розмірність^—^ дор.внює розмірності [шющина]»
наприклад, —=^~- Історично поняття а з'явилося саме як «поверхневий
натяг» (див. рис. 17.1), поняття «поверхнева енергія» виникло згодом. Для
рухомих меж поділу в ізотропних системах ці поняття адекватні. Поняття
«поверхнева енергія» виявилося більш універсальним, і для твердих тіл
(монокристалів) слід користуватися саме їм (про що мова піде далі).
Рис. 17.1. Поверхневий натяг тримає водоміра на поверхні води
Розглянемо далі капілярне підняття рідини, що зображене на рис. 17.2, а.
В цьому випадку центр кривизни поверхні, що поділяє газову та рідку фази,
лежить у газовій фазі. Згідно з рівнянням (17.1) маємо
2в = 2ощЄ = АР = (р )ф е н (1? 5)
г К *
426
У
г
» >\
К А
V У\
гт
-^
[ і'Ар-о]
Рис. 17.2. Капілярне підняття (а) та опущення (б)
(АР дорівнює гідростатичному тискові), де И — висота капілярного
підняття; р/— густина рідини; р^— густина газу (зазвичай р;» р8)9 г со§ 6 = Я, де
Я — радіус капіляра, г—радіус кривизни, 6 — кут змочування (при
повному змочуванні 0 = 0, со80= 1і саме такі рідини використовують для
визначення а). Користуючись формулою (17.5), можна також вимірювати радіус
капіляра, якщо застосовувати рідину з відомою поверхневою енергією.
Для рідин, які не змочують стінки капіляра, соз9 і, відповідно, Н мають
від'ємні значення, меніск рідини стає не ввігнутим, а випуклим, і центр
кривизни знаходиться у рідині (див. рис. 17.2, б). Наприклад, ртуть не
змочує скло, і це можна використати для визначення розмірів пор у пористих
тілах шляхом втиснення ртуті у пори-капіляри і вимірювання відповідного
тиску (АР). Ртуть має а=465 * 10~ Дж/м2 і при 450 атм починає входити у пори
розміром -15 нм.
У розділі 2.1 було показано, що додатковий тиск на поверхню рідини веде
до збільшення тиску насиченої пари над рідиною (див. формулу (2.11)). Цей
додатковий тиск у випадку випуклого меніска може виникати за рахунок
кривизни поверхні («тиск Лапласа») згідно з формулою (17.1).
Підставляючи (17.1) у (2.11), одержимо рівняння Томсона (Кельвіна) для тиску (Рп)
насиченої пари над випуклою поверхнею
2оУт
Рп=РоЄгЯТ > 07.6)
де Ут—молярний об'єм рідини; Р0—тиск насиченої пари над плоскою
поверхнею (нагадаємо, що для плоскої поверхні гх = г2=°° і, згідно з формулою
(17.2), АР = 0).
Реально залежність Рп від кривизни поверхні починає виявлятися лише
при дуже малих г. Наприклад, для води при г = 10 мРп /Р0 =1,001; при г =
= 10" м Рп /Р0 = 1,011; при г = 10~ м РпІР0 =1,114. Підвищення тиску
насиченої пари над маленькими краплинами, порівняно з великими, призводить
до «перегонки» речовини з маленьких крапель на великі, і це є однією з
головних причин росту крапель у туманах та хмарах з наступним їх випадан-
427
ням під дією сили тяжіння. Зазначимо, що над ввігнутим меніском рідини
тиск насиченої пари менший, ніж над плоскою поверхнею, у формулі (17.6)
в цьому випадку в експоненті слід змінити знак на «мінус», тобто
Рп=Р0е *т. (17.7)
Розглянемо тепер рівновагу між поверхневим шаром та об'ємом для
багатокомпонентної гомогенної конденсованої системи (розчин, розплав).
Вважаючи систему ідеальною і позначаючи мольну частку компонента
в об'ємі через хі9 а в поверхневому шарі через уь напишемо вирази для
відповідних хімічних потенціалів, а саме:
для об'єму
^=(ІЇУ+КТІпхі; (17.8)
для поверхневого шару
ііГ = Юа + КТЬУі. (17.9)
Стандартний стан (\х°)у та (ц°)ст віднесено до відповідної чистої
речовини. Хімічний потенціал у поверхневому шарі відрізняється від хімічного
потенціалу в об'ємі на величину роботи утворення поверхні, тобто
іх?=ііГ+<ц, (17Л°)
де а — вільна поверхнева енергія розчину (розплаву); А(—площа, яку
займає розташований у вигляді моношару 1 моль частинок (молекул, атомів
тощо) (Аі= со^, де (О,-— площа, що припадає на 1 частинку — так званий
«посадковий майданчик»).
Співвідношення (17.10) справедливе і для хімічних потенціалів у
стандартному стані
(ц°)а = (И,°)К+М- (17.11)
Об'єднуючи вирази (17.8)—(17.11), маємо для поверхневої енергії
гомогенної конденсованої фази
о = о/+^1п^-. (17.12)
А *
Розглянемо далі двокомпонентну систему з частинками, що не дуже
відрізняються за розміром, тобто (д{ = со2 = со і, відповідно, А1=А2 = А. Тоді
маємо
0і+ЕГ1аУі=^тпУі. (17.13)
А X* А Ху
Розв'язуючи це рівняння відносноу2та х2 (з урахуванням х1+л:2= 1 та
Уі+У2= 1), маємо
1 — Л>> + -^2^4*
428
де Ка=ехр\
(а,-ст2)л"
КТ
Підставляючи вираз (17.14) у (17.12), маємо
о = а і КТ\п ХіК°
2 А (1-х2+х2К<:!)х2
(17.15)
= а1-паКТЇп(1-х2+х2Ка), (17.16)
де через па = І/А позначена кількість молів частинок, які вміщує одиниця
поверхні міжфазного шару (рівняння (17.16) називається рівнянням Жухо-
вицького-Гугенгейма).
Проаналізуємо рівняння (17.14)
та (17.16). При СГ| = а2 К0= 1 і з
рівнянь (17.14) та (17.16) маємо
Л=*2 X
о = о2=в\у
(17.17)
тобто в цьому випадку в усьому
концентраційному інтервалі склад
поверхневого шару тотожний
складу об'ємної фази (пряма 3 на рис.
17.4). При а2 < о і (див. рис. 17.3,
крива 7) маємо Ка > 1 іу2 > х2 при будь-
яких складах конденсованої фази
(рис. 17.4, крива 7). Речовина «2» в
цьому випадку називається поверх-
нево-активною (скорочено ПАР), і її
концентрування у поверхневому
шарі енергетично вигідне, бо веде
до зменшення поверхневої енергії
системи. При Кс » 1 поверхневий
шар збагачений ПАР вже при малих
її концентраціях у системі.
Наприклад, при Кс = 20 та х2 = 0,10 маємо
у2 = 0,69, тобто 72>:>*2-
Концентрування ПАР у поверхневому шарі
веде до різкого зменшення а вже при
невеликих значеннях х2.
Інша картина має місце при а2 >
> а{ (рис. 17.3, крива 2). В цьому
випадку Ка < 1 і у2 < х2 в усьому
інтервалі концентрацій (рис. 17.4, крива І).
Речовина «2» тут є поверхнево-інак-
тивною (ПІР), її перехід до
поверхневого шару веде до збільшення а і,
отже, енергетично невигідний, тому
Рис. 17.3. Залежність питомої
поверхневої енергії (а) від складу
розчину (розплаву)
Рис. 17.4. Залежність складу
поверхневого шару (у2) від складу об'єму (х2):
7 — ПАР; 2 — ПІР
429
ця речовина концентрується в об'ємі. Суттєві зміни (збільшення)
поверхневої енергії в цьому випадку відбуваються лише при великих концентраціях
компонента «2» (при х2 —> 1), тобто спочатку треба, щоб ПІР був
«насичений» об'єм. При Кс « 1 рівняння (17.16) набуває вигляду
а = а{ -п0КТЩ\-х2) = ох+паЯТх2 (17.18)
(при малих х2 1п(1 -х2) = (-х2)9 тобто а зростає зі збільшенням х2 практично
лінійно і дуже повільно.
Таким чином, при Аа = а{ - а2 > 0 речовина «2» є ПАР по відношенню
до речовини «7», і навпаки, при Аа < 0 речовина «2» є ПІР. Критерій Аа > 0
{критерій Жуховицького) для визначення поверхневої активності речовин
працює добре, згідно з експериментальними даними для великої кількості
бінарних систем він виправдовується у 96 %.
Для поверхнево-активних речовин у розбавлених розчинах відомі
рівняння Ленгмюра для адсорбції
г=г-їш <Ш9>
та Шишковського для поверхневого натягу
а = а0- Г^ЯТЩ + Ьс). (17.20)
В цих рівняннях а0 — поверхнева енергія розчинника, Г — поточна, а
Г^ — максимальна (при с —> ©о) кількість ПАР у поверхневому шарі. Ці
рівняння можна вважати наближеннями відповідних рівнянь (17.14) та
(17.16). Справді, при малих концентраціях х2 « 1 і вираз (1 - х2 + Ксх2) =
= 1 + Ках2 (нагадаємо, що для ПАР константа Ка суттєво більша від одиниці).
Величина Г^ практично тотожна з па, а відношення Г/Г^ = 9 — це частка
місць у поверхневому шарі, зайнятих ПАР, при щ = (02 = со 0 співпадає зу2.
Нарешті, у розбавлених розчинах х2 пропорційна молярній концентрації—с.
На відміну від ізотропних тіл (рідина), тверді монокристали мають різні
значення поверхневої енергії для різних граней, тобто а для них є
анізотропною величиною. Наприклад, для кубічної гранецентрованої (к.г.ц.)
структури мінімальну поверхневу енергію має грань {111} октаедра, для інших
граней значення а вищі. Для граней з малими індексами Міллера порядок
розташування а для речовин, що кристалізуються в к.г.ц. структурі, такий:
аГігІц<а^оц<<іоц- (17-21)
(мінімальну енергію має грань {111} октаедра).
Для кубічної об'ємноцентрованої структури (к.о.ц.) маємо такий порядок:
<іГ<<оЦ<<іЦ- (17-22)
(мінімальну енергію має грань {110} ромбододекаедра).
Для ізотропних тіл принцип мінімуму вільної енергії при V = соті і а =
= соті реалізувався мінімумом поверхні поділу 5 = тіп (рівноважна фор-
430
ма — сфера). Для монокристала форма визначається принципом мінімуму
вільної енергії Гіббса-Кюрі
^аД. = тіп при V = сопзі,
де а і — поверхнева енергія /-Ї грані, £, — її площа.
З умов Гіббса-Кюрі випливає теорема Вульфа, згідно з якою
—[- = -± = ... = -+ = соті,
пх п2 щ
(17.23)
(17.24)
де Ні— висота перпендикуляра, що опущений з центра кристалізації на /-ту
грань монокристала. Співвідношення (17.24) показує, що грані з малими
значеннями а, розташовані у безпосередній близькості до центрів
кристалізації і отримують максимальний розвиток.
Розглянемо побудову рівноважної форми методом Вульфа на
гіпотетичному двовимірному кристалі, для якого а10 = 250 • 10" Дж/м2 (аналог а100 —
грані куба); а с1 {= 225 • 10~ Дж/м2 (аналог стИІ — грані октаедра). Якщо
розглядати лише «площини» (10), то повна поверхнева енергія кристала
площиною 1 м2 (площина у двовимірному випадку є аналогом об'єму у
тривимірному) дорівнює: 4 • 1 • 250 • 10~ =1 Дж (рис. 17.5, а). Якщо є лише «площини»
(11), то повна енергія дорівнює 4 • 1 • 225 • 10" = 0,9 Дж (рис. 17.5, б). Тепер
використаємо побудову Вульфа. З центра проводимо вектори з довжиною, що
пропорційна аі9 і на кінці кожного вектора проводимо «площину» (тут
лінію) перпендикулярно вектору. Перетин цих «площин» (ліній у
двовимірному випадку) дає шукану геометричну фігуру. Для двовимірного випадку це
зображено на рис. 17.5, в, а принцип побудови за Вульфом — нарис. 17.5, г.
У випадку «в» маємо для повної енергії (4-0,32-250+4-0,59-225)-10" =0,851 Дж,
що менше, ніж у випадках «а» та «б» (за \ ... 0,32
рахунок певного зменшення периметра
кристала).
Таким чином, відповідно до
співвідношень (17.21) та (17.22) рівноважною
формою, згідно з принципом Пббса-Кюрі-
Вульфа, для к.о.ц. структури є
ромбододекаедр, а для к.г.ц. структури—октаедр (або куб-
октаедр).
Природним доповненням принципу
Гіббса-Кюрі-Вульфа є принцип Браве,
згідно з яким рівноважними і максимально
розвинутими повинні бути грані з
максимальною ретикулярною щільністю.
Доцільно співставити порядок розташу- ^
вання граней по мірі росту о, з послідовніс- Рис п#5# Конформації гіпо.
тю щільності упаковки цих граней. Для к.г.ц. тетичного двовимірного
та к.о.ц. структур це зроблено у табл. 17.1 кристала
431
1
О (У
І "УА Ч "і
1< ^ж^ м
' X У X і
. X т \>г
(за одиницю прийнято щільність упаковки атомів для найщільніше
упакованих граней: {111} в к.г.ц. структурі і {110} в к.о.ц. структурі).
Таблиця 17.1. Щільність упаковки атомів на поверхні (р5)
Структура
кубічна гране-
центрована
(к.г.ц.)
Площина
{пі}
{100}
{110}
{210}
{211}
{221}'
Рз' Ртах
1,000
0,866
0,612
0,387
0,354
0,289 1
Структура
кубічна об'ємно-
центрована
(к.о.ц.)
Площина
{110}
{100}
{111}
{211}
{210}
{221}
Р*/Ртах
1,000
0,707
0,409
0,578
*0,316
0,236
Співставлення наведених у таблиці 17.1 даних з нерівностями (17.21) та
(17.22) показує, що щільність упаковки і поверхневі енергії граней
змінюються антибатно, тобто найщільніше упаковані грані мають мінімальні
значення Сі і, навпаки, пухкі грані мають великі значення а,. Таким чином,
принципи Пббса-Кюрі-Вульфа і Браве є узгодженими.
17.2. Адсорбція газів на поверхні твердого тіла
У даному розділі обмежимось розглядом поверхневих явищ на межі
поділу тверде тіло-газ. У цьому разі тверде тіло має назву адсорбент, а газ
(пара), що концентрується на його поверхні, — адсорбат.
Розглянемо спочатку рівновагу адсорбції, яку можна зобразити таким
чином:
адсорбат у газовій фазі + вільний центр на поверхні =
адсорбат у поверхневому шарі,
аоо умовно
А + [] = [А].
Позначимо ступінь заповнення поверхні
адсорбатом через 6.
9 = -^, (17.25)
ат
де а — кількість адсорбату в поверхневому
шарі, ат — кількість адсорбату, що потрібна для
покриття всього адсорбенту моношаром
адсорбату.
Для константи рівноваги адсорбції АГ, згідно
з І. Ленгмюром, маємо
Є
К = -
(17.26)
Ленгмюр Ірвінг
(1881—1957), США
(1-6)/?'
дер — тиск адсорбату в газовій фазі.
Розв'язуючи рівняння (17.26) щодо 0, отримуємо
432
6=-^-
З урахуванням (17.25) рівняння
(17.27), що називається ізотермою
адсорбції Ленгмюра, має вигляд
—•&;■ <іта)
Залежність а від/?, що описується
ізотермою Ленгмюра, наведено на
рис. 17.6.
Проаналізуємо рівняння (17.28).
За малих р: Кр«\і
І + Кр
(17.27)
4,0
2,0 1
а,'
а = атКр,
(17.29)
-і ►
2,0 4,0 6,0 8,0
Рис. 17.6. Ізотерма адсорбції
Ленгмюра (Кг на плівці вуглецю)
тобто між адсорбцією а та тиском/?
існує лінійний зв'язок. Ця ділянка
називається «ділянка Генрі», а
відповідне рівняння (17.29) — «ізотерма Генрі». За великих тисків Кр »1 і тоді
а = ат. Це ділянка насичення, крива асимптотично наближається до прямої
а = ат.
Ізотерма Ленгмюра описується двома параметрами {ат та К). її можна
легко лінеаризувати. Лінеаризована форма рівняння (17.28) така:
/?_ 1 , 1
а
атК а„
(17.30)
Графічне зображення рівняння (17.30) наведено нарис. 17.7.
Величина ат — це ємкість моношару, наприклад, у моль/г. Вона може
бути використана для визначення питомої поверхні адсорбенту (5ПИТ3:
Зїит^^О), (17.31)
де 0) — «посадковий майданчик»,
тобто місце, що займає одна
молекула адсорбату в поверхневому
шарі в м2; Ил — число Авогадро. Отже,
8ПИТ виражається в м2/г. Є
адсорбенти з дуже розвинутою
поверхнею. Наприклад, активоване вугілля
має поверхню 500—1000 м2/г,
алюмогель, силікагель — 200—300 м2/г
тощо. Для багатьох адсорбатів ш
становить(10-20) 10" м2 і
визначається в основному розміром
молекул адсорбату. К — це константа рИс. 17.7. Лінеаризована форма
рівноваги рівняння Ленгмюра
433
К = е-АС°/КТ = еМО/Ке-Ш°/КТ = Ьед/КТ ^ (17.32)
де А50 = 5^ - іїд1^3' — зміна ентропії під час адсорбції, Д#° — ентальпія
адсорбції, ^ = -АН° — теплота адсорбції в термохімічній шкалі.
Зміна ентропії під час адсорбції завжди є від'ємною величиною, оскільки
адсорбат в адсорбованому стані — більше впорядкована система, ніж адсор-
бат у газоподібному стані. Навіть якщо поверхневі шари адсорбату
зберігають рухливість («двовимірний газ»), адсорбат втрачає при адсорбції
принаймні один ступінь свободи поступального руху. Для нерухомих шарів
адсорбату система в адсорбованому стані є високо впорядкованою, відповідно,
£[А] — дуже мала величина, отже, тут А£ =-5 '«0.
За від'ємних значень ентропії адсорбції (А5°) вільна енергія адсорбції
АС° = АЯ° - ТА8° може бути від'ємною величиною (а це, згідно з принципом
Гельмгольца-Нернста, потрібно, щоб процес адсорбції відбувався
самочинно) лише за умови Д#° < 0. Отже, адсорбція повинна бути
екзотермічним процесом. Вона ефективно перебігає за низьких температур, коли
вплив ентропійного фактора є незначним. З ростом температури рівновага
зміщується в бік зворотного ендотермічного процесу — десорбції. Отже,
прогрівання за високих температур дає можливість звільнити поверхню
сорбенту від адсорбованих частинок, «почистити» її.
Залежність кількості адсорбату від
і температури за фіксованого тиску
(адсорбція водню на перехідних
металах, наприклад на нікелі) наведено на
рис. 17.8. Це так звана ізобара
адсорбції.
З рис. 17.8 видно, що залежність а
від Гмає поліекстремальний
характер. Це пов'язано з існуванням двох
форм адсорбції: фізичної та хімічної
, =_ї (хемосорбції).
Т Для фізичної адсорбції характер-
Рис. 17.8. Ізобара адсорбції ною є слабка взаємодія між адсор-
батом та адсорбентом, в основному
вона визначається силами Ван дер Ваальса. Це веде до незначних (за
абсолютною величиною) теплот адсорбції (до 20 кДж/моль) і відносно вільної
рухливості адсорбованих молекул. Фізична адсорбція не є активованою,
тобто енергія активації адсорбції дорівнює нулю. Оскільки А#° — невелика
величина, то фізична адсорбція здійснюється за досить низьких температур,
з підвищенням температури інтенсивно перебігає десорбція (крива 1 на
рис. 17.8). За відсутності хемосорбції (наприклад, адсорбція інертних
газів) ізобара фізичної адсорбції асимптотично наближається до осі абсцис
(крива 1-1').
Для хемосорбції характерними є міцні зв'язки (рухливість поверхневих
шарів адсорбату є дуже малою) і, отже, великі значення ДЯ°. Для десорбції
434
хемосорбованих частинок потрібні високі температури (крива З, рис. 17.8).
Оскільки хемосорбція перебігає, як звичайна хімічна реакція, то вона є
активованою, тобто для її перебігу потрібно подолання потенціального бар'єра.
Наявність високої енергії активації робить процес хемосорбції за низьких
температур дуже повільним, отже, частина рівноважної кривої 3-3' для
хемосорбції за низьких температур не реалізується.
Крива 2, що здійснює перехід із фізадсорбційної 7 гілки ізобари на хемо-
сорбційну 3, не є рівноважною кривою. Вона визначається кінетикою
процесу хемосорбції і відповідає зростанню швидкості хемосорбції зі
збільшенням температури.
Розглянемо на прикладі хемосорбції Х2 на перехідному металі природу
потенціального бар'єра при хемосорбції. Це було зроблено вперше Лен-
нард-Джонсом методом потенціальних кривих (рис. 17.9).
Леннард-Джонс Джон
Едвард (1894—1954), _ лп л .__ . . . - ...
Велика Британія ^ис# ^.9. Потенціальні криві хемосорбції
та ван-дер-ваальсової адсорбції
Крива 7 відповідає слабкій фізичній адсорбції з малою теплотою
адсорбції цт і відносно великим ван-дер-ваальсовим радіусом гт — відстанню між
поверхнею і фізично адсорбованою частинкою. Крива 2 хемосорбції має
глибокий мінімум, якому відповідає велика теплота адсорбції #а. Положення
мінімуму на осі абсцис визначає ковалентний радіус га « гт.
За г —> ©о крива 2 розташована значно вище кривої 7, відстань між ними
дорівнює енергії дисоціації молекули Х2 на атоми. Навпаки, за малих г
крива 2 знаходиться значно нижче кривої 7. Точка перетину цих кривих «с»
визначає висоту потенціального бар'єра при адсорбції — Еа. Потенціальний
бар'єр для десорбції £,/ є більшим на величину #а, тобто (див. рівняння (14.56)
Е,=Еа+да. (17.33)
Таким чином, процес десорбції завжди має потенціальний бар'єр,
навіть якщо адсорбція не є активованою і £а= 0.
435
Коли хемосорбція відбувається з дисоціацією на атоми, то рівняння
адсорбції Ленгмюра має інший вигляд. Так, для дисоціативної хемосорбції
Х2 + 2[ ]<==>2[Х] маємо
К= в\ . (17.34)
0-Є)2р
Отже,
в = _д_= (*Р)І/2 (Ш5)
ат \ + {Кр)уг
За низьких тисків маємо з (17.35)
а = ат(Кр)У2. (17.36)
Для фізичної адсорбції характерним є утворення полімолекулярних
шарів. Схематично полімолекулярну адсорбцію зображено нарис. 17.10.
Теорію полімолекулярної адсорбції було розроблено трьома авторами:
С. Брунауером, П. X. Емметом та Е. Теллером (скорочено — БЕТ).
Позначимо через 0І5 02,0з>... 6,-, —
частки поверхні, що припадають на
моношар, подвійний, потрійний шари
Г\ Л тощо. 90 — частка поверхні, що зали-
-^—^— шається вільною від адсорбату. Вва-
„ -_ <л ~ .. жаємо, що між /-тим та (і + 1) шаром
Рис. 17.10. Схема полімолекулярної '. . у у .
адсорбції існує рівновага, що підкоряється
рівнянню Ленгмюра, а саме:
*і=-|-, (1737)
К2=\у (17.38)
РЄ}
З рівнянь (17.37)—(17.39) маємо
0! = КіРЄ0; 02 = К2рЄх; 03 = К3рЄ2... (17.40)
Можна вважати, що адсорбція адсорбату на будь-якому шарі з адсорбату
практично однакова, тобто, починаючи з другого шару,
£2=А:з =••• = */• (17.41)
З великим ступенем точності можна вважати, що при адсорбції
адсорбату на адсорбаті константа адсорбції дорівнює константі конденсації Кь того
самого адсорбату, тобто
Кі=Кі=у^ (17.42)
де р5—тиск насиченої пари адсорбату.
436
Позначаючи
з рівняння (17.40) маємо
= *,
Рз
(17.43)
(17.44)
Загальна кількість адсорбату на поверхні адсорбенту дорівнює
а = атЄ1+2аиЄ2+ЗатЄ3+... = ат[еі+2Є1х + ЗЄ1х2 ] =
= атЄ1[\ + 2х + Зх2 +..] = атв1-ї-[і + х + х2 +х3 +....] =
= <**% —
1
_ Д«в)
(іх{\-х) (1-хУ
де ат — як і раніше, кількість адсорбату, що є необхідною для покриття
адсорбенту моношаром.
Щоб виключити в) з рівняння (17.45), скористаємось тотожністю
Є0 + Є,+Є2+Єз+... + Є,+... = 1. (17.46)
З урахуванням (17.40) та (17.44) маємо
1 = —1— + в,+в,х + в,х2+.
К\Р
'Є,
1 +(1 + х+х2 +....)
К\Р
= 6,
^+^
1-х-К^р
1 К{р(\-хУ
Кхр 1 - х
З урахуванням (17.47) формула (17.45) стає такою:
кхР
ат Кхр(\-х)
а = , „ „ \ = а„
(17.47)
(17.48)
(1-х)2(1-* + ВД т(1-х)(1-х+КіРУ
Позначимо відношення константи рівноваги в першому шарі до будь-якої
іншої константи через С, тобто
звідки
кх=скь=-£-.
Рз
(17.49)
(17.50)
Використовуючи (17.50) та (17.43), остаточно маємо з формули (17.48)
а = а„
Рз
Рз
1--Р- + С-Р-
Рз Рз
= аа
Рз
Рз
1 + (С-1)-2-
Рз
Рівняння (17.51) має назву ізотерма адсорбції БЕТ.
• (17.51)
437
Проаналізуємо це рівняння. За Кх = Кь (коли адсорбція на поверхні
адсорбату в моношарі за параметрами є близькою до адсорбції в інших
шарах) маємо С = 1, що дає
Р
а = а„
£^ = .
1--
Рз-Р
(17.52)
Рз
Згідно з рівнянням (17.52) адсорбція (а) поступово зростає ізар-
а—>°о. Зар«р8
'Рз
а =
атР
_ ~тг _
Рз
*п£іР-
Тобто за дуже малих р виконується лінійна залежність -
Більш типовим є випадок, коли КХ»КЬ і С » 1, тоді
(17.53)
-ділянка Генрі.
а = а„
Рз
Рз
За дуже малих/?, копи р/р8^> 0, маємо
1 + С-
Рз
(17.54)
(17.55)
(17.56)
тобто тут також виконується лінійна залежність — ізотерма Генрі.
Далі, оскільки С» 1, виникає ситуація, кояи р/р8« 1, але добуток Ср/р8
має один порядок з одиницею. Це дає
атСрІр8 Кхр
\ + СрІр8 т\ + Кхр'
Рівняння (17.56) — це
а, ммоль/г ізотерма Ленгмюра. При
цьому на кривій
намічається полога ділянка
насичення за а —> ат, але вона
повністю не реалізується, і
величина а починає
зростати за рахунок полімолеку-
лярної адсорбції. За/? —>р8
крива прямує в
нескінченність. Таким чином, тут
ізотерма БЕТ описує 5-подібну
криву (рис. 17.11). Точка «В»
на цій кривій приблизно
відповідає заповненню моно-
Рис. 17.11. Ізотерма адсорбції води на феназині шару.
438
Для визначення параметрів рівняння БЕТ його слід лінеаризувати. Ліне-
аризована форма ізотерми БЕТ має вигляд
1
С-1
Р_
атС Р8
Р
(17.57)
а(Р8~Р) атС
Р . . .
Таким чином, у координатах / \ ~ маємо пряму лінію,
обробкою якої знаходимо параметри ат та С.
Згідно з рівнянням БЕТ при/? —>р5 а —> °°, тобто полімолекулярна ад-
сорбія переходить у конденсацію пари у рідину (див. також рис. 17.11). Ця
конденсація має свою специфіку на пористих сорбентах, які мають велику
кількість пор, і називається капілярною конденсацією. У порах сорбенту
утворюються плівки рідини, меніски яких мають увігнутий характер. Згідно з
рівнянням Томсона (17.7) це веде до зниження тиску насиченої пари над
увігнутою поверхнею (оскільки радіуси кривизни для ввігнутої поверхні
лежать у газовій фазі), тобто для сферичного меніска
2сУ
(17.58)
'еф.
кт
Рсф. = Р8е
а для циліндричного (згідно з рівнянням (17.3))
Рц.=Р8е
оУ
г*ЯТ
(17.59)
де/?5 — тиск насиченої пари над плоскою поверхнею.
Розглянемо процес конденсації пари у порах на модельних системах. Для
закритої циліндричної пори (рис. 17.12, а) у закритого кінця пори
утворюється сферичний меніск і при
2аУ
г.,1, КТ
Рсф. =Р8е < Р8
починається конденсація. При
цьому тиску пора поступово
заповнюється й ізотерма
конденсації пари має вигляд
вертикальної кривої (рис. 17.12, а).
Випаровування (десорбція) з
наповненої пори відбувається за тою ж
кривою у зворотному напрямі
(рис. 17.12, а).
Інша картина спостерігається
для відкритої {з обох кінців)
циліндричної пори (рис. 17.12, б).
В цьому випадку сферичний
меніск не утворюється, і капілярна
конденсація починається на
циліндричній поверхні стінок пори.
%
■А
а
а
■■■■
о
і
с
І
/
'
{
г
-*РФ*
1
~~*ч
р/р
[
3
Рис. 17.12. Схема капілярної конденсації
у порах різної форми: а — закрита
циліндрична; б — відкрита циліндрична
439
0,5 1,0
Рис. 17.13. Адсорбційний гістерезис:
1 — адсорбція; 2 — десорбція
4 V см3
1
0,2
0,1
1 Л-'
V
см2
Г-Н]
/І
/1
/
1
VI
б
V
г, нм
1 ►
2,5 5,0 7,5
Рис. 17.14. Структурна крива (а)
та крива розподілення пор
за радіусами (б)
Згідно з формулами (17.58) та (17.59)
зменшення тиску насиченої пари
над циліндричною поверхнею не таке
велике, як над сферичною, тобто /?ц. <
</?сф.-Отже, капілярна конденсація
при заповненні відкритої циліндричної
пори починається при більших тисках
насиченої пари (рис. 17.12, б), ніж у
випадку закритої пори (рис. 17.12, а). При
зворотному процесі випаровування
з пори біля гирла пори утворюється
сферичний меніск, і процес десорбції
перебігає так само, як і для закритої
пори, тобто при Р< РСф. (рис. 17.12,6).
Отже, утворюється характерна петля
гістерезису капілярної конденсації.
Реальні сорбенти мають певний
набір пор різного розміру і форми,
отже, реальна ізотерма адсорбції-де-
сорбції (з урахуванням капілярної
конденсації) має вигляд, наведений на
рис. 17.13. Крива адсорбції (і) є
складною і враховує набір відкритих і
закритих пор, на кривій десорбції (2) для всіх
пор «працюють» лише сферичні
меніски, тому її зручно використовувати
для визначення ефективних розмірів
пор. Використовуючи формулу Том-
сона (17.58), з експериментальних
даних (рис. 17.13) визначають ефективні
розміри пор (г) і відповідні до них
об'єми пор {V). Отримавши серію значень
У\ г, будують структурну криву
адсорбенту— залежність Квідг(рис. 17.14,
а). Одержана із структурної кривої
диференціальна крива
йг
(рис. 17.14, б) дає розподілення пор за
радіусами. Гострий пік на цій кривій
свідчить про те, що даний сорбент є
досить однорідно пористим (в ньому
переважно присутні пори з г = 5 нм).
- —г
440
При розгляді як мономолекулярної, так і полімолекулярної адсорбції
вважалося, що всі ділянки поверхні адсорбенту є однаковими за своєю
адсорбційною здатністю, тобто поверхня адсорбенту є однорідною. Це
припущення виправдовує себе не завжди. Критерієм однорідності поверхні є
незалежність теплоти адсорбції від ступеня заповнення поверхні адсорбатом
у моношарі. Для однорідної поверхні рівняння (17.32) дає
*Щ£ = ШІя (1760)
лт кт2
За фіксованого значення 9, згідно з рівнянням (17.26), маємо
д\пр
дТ
= -%. (17.61)
КТ2
Величина #е має назву ізостеричної теплоти адсорбції {у термохімічній
шкалі). Якщо вона не змінюється залежно від ступеня заповнення моно-
шару, то це свідчить про те, що поверхня є однорідною.
Для неоднорідних поверхонь залежність #е від 0 може бути різною.
Розглянемо досить поширений випадок, коли теплота адсорбції лінійно
зменшується зі збільшенням 0, це так звана рівномірно-неоднорідна поверхня,
для якої
?е=<7о-С0. (17.62)
У рівнянні (17.62) #0 — теплота адсорбції на місцях із максимальною
адсорбційною здатністю за 0 —»0, тобто на майже чистій поверхні. Стала С
визначає інтервал неоднорідності й дорівнює
С = д0-яи (17.63)
де ^ — теплота адсорбції за 0 —> 1 за майже повністю заповненого моно-
шару на місцях із мінімальною адсорбційною здатністю.
Підставляючи вираз (17.62) у рівняння (17.32), отримаємо для константи
рівноваги на неоднорідній поверхні
Кв = Ье«*ге-С*'*Т = К0е~'е. (17.64)
При цьому вважається, що ентропія адсорбції не змінюється при
переході від однієї ділянки неоднорідної поверхні до іншої. Параметр/ = СІКТ
характеризує ступінь неоднорідності поверхні і може бути значною
величиною. Наприклад, для хемосорбції азоту на залізі маємо
#о = 170 кДж/моль, #і = 84 кДж/моль, отже, за 300 °С /=18.
Вважаючи, що для кожної окремої ї'-ї ділянки д,- = сопзі і, отже,
виконується рівняння Ленгмюра (17.27), отримаємо для загального ступеня
заповнення поверхні
Є = т=2т^~- (17-65)
У формулі (17.65) можна замінити И на /, якщо вважати спектр теплот
441
адсорбції квазібезперервним. Якщо також К^К?» отримуємо, згідно з (17.64),
1 _гв *о/
коРе } іП_ і Г 4у - і ,„1+коР
е=
^0 = і. Г_^_ = ^іп^±оіі. (17.66)
. 1 + К0ре~/в І ) 1+У / і + КіР
0 К\Р
Тут зроблено підстановку у = К0ре~^в і враховано, що Я^ = К0е~^.
Рівняння (17.66) називається квазілогарифмічна ізотерма Тьомкіна.
Воно описує залежність ступеня заповнення поверхні адсорбатом від тиску
за сталої температури на рівномірно неоднорідній поверхні. Це рівняння дає
за/? -> 0 0 —> 0, а за/? —> °° К0р » 1 та Кхр » 1, отже,
За середніх ступенів заповнення, оскільки константа #0 завжди є
набагато більшою за константу Кх, виконуються такі умови: К0р » 1, але Кхр «
«1, що дає
Є = і:1п(^0/?). (17.67)
Таким чином, у ділянці середніх заповнень квазілогарифмічна ізотерма
для рівномірно неоднорідної поверхні перетворюється на логарифмічну
і на цій ділянці суттєво відрізняється від ізотерми Ленгмюра, яка є
характерною для однорідної поверхні.
Зазначимо, що за відносно низьких тисків квазілогарифмічна ізотерма,
як і ізотерма Ленгмюра, перетворюється на ізотерму Генрі. Справді,
нехтуючи К\р порівняно з одиницею, розкладаємо 1п(1 + К0р) в ряд, що дає
® = у~Р- (17-68)
Рівнянням (17.66) та похідними від нього (17.67) та (17.68) добре описується
хемосорбція азоту на неоднорідній поверхні заліза та інші системи, для яких
доведено, що поверхня є неоднорідною.
Фізичні причини неоднорідності поверхні можуть бути різними. Якщо
обмежитися випадками, коли адсорбент є чистою речовиною, то
неоднорідність має структурне походження. Мається на увазі, що реальні кристали
завжди мають недосконалості, дефекти структури. Це можуть бути точкові
дефекти: вакансії, міжвузлові атоми (рис. 17.15); лінійні дефекти —
дислокації (рис. 17.16), двовимірні — дефекти упаковки, нерівноважні нещільно
упаковані грані (див. підрозділ 17.1), які роблять суттєвий внесок у
неоднорідність поверхневого шару. В полікристалах слід також враховувати розупо-
рядковані міжкристалічні межі (рис. 17.17), а в монокристалах — тераси та
сходинки (рис. 17.18). На рис. 17.18 представлені різні типи поверхневих
дефектів: тераси, сходинки, виступи, вакансії, поверхневі атоми тощо.
Дефекти у твердому тілі утворюються у процесі росту кристалів, при
механічній деформації (див. підрозділ 19.5), при опромінюванні матеріалу
частинками з високою енергією (див. підрозділ 19.4) тощо.
442
п
Г-і
р
1_
г
1 1
гт
2) *
•
4 •-—• •—-»-~
Рис. 17.15. Викривлення ґраток біля вакансії (1) та міжвузлового атома (2)
Перейдемо тепер до розгляду кінетики адсорбції. Для однорідної
поверхні швидкість адсорбції повинна визначатися кількістю зіткнень з
поверхнею (за одиницю часу на одиницю поверхні), що дається формулою
Кнудсена-Герца (14.34), тобто вона є пропорційною тиску адсорбату в
газовій фазі. Це справедливо в припущенні, що після кожного зіткнення
молекули адсорбату з поверхнею адсорбенту вона залишається в
поверхневому шарі. Якщо розглядати адсорбцію в моношарі, то молекули повинні
адсорбуватися лише на тій частці поверхні адсорбенту, що не зайнята в
моношарі адсорбатом: 60= 1 - 0. Крім того, слід враховувати, що процес
адсорбції (хемосорбції) може мати відповідну енергію активації, про що
йшлося вище. Отже, для швидкості адсорбції справедливим є співвідношення:
„ооооооооо
о°Л°0оооооооо
о°л°о0 оооо о о о
о°л°0 ооооо о о о
°^о ооооо о о о
о°л°0оохооо о о
о°л00о ооооо
о°л00о ооооо
о°л00оооооо
о°л0ооооооо
о°л00оооооо
° лО ОООО О О О
° оооохоооо
°оооо оооо
° оооо оооо
° оооо оооо
°л°0ооооооо
°оооооооо
°000£0000
°ооохоооо
°о0оо оооо
Рис. 17.16. Крайові дислокації
на малокутових межах зерен
Рис. 17.17. Полікристал, складений
із когерентно зсунутих ділянок
Рис. 17.18. Типи поверхневих дефектів
443
га=каР{\-в), (17.69)
Іса=(К)оЄ-ЕМТ, (17-70)
де ка — константа швидкості адсорбції: Еа — енергія активації адсорбції.
Відповідно, для десорбції маємо
*=**Є = (*,)0е~£'/ЯГЄ. (17-71)
За рівноваги га = г(і, що дає
АвР(1-Є) = *„Є. (17.72)
Розв'язування рівняння (17.72) відносно 6 з урахуванням, що каІкА = К
приводить до рівняння ізотерми Ленгмюра (17.27). Відповідно, між
енергіями активації адсорбції та десорбції існує співвідношення (17.33).
Для кінетики хемосорбції двохатомних молекул (И2,02, Н2 тощо)
справедливим є рівняння
гв=*в^Р = *в(1-в)2Рта (17.73)
О=^02. (17.74)
Комбінація рівнянь (17.73) та (17.74) у рівноважному стані дає відповідні
рівняння (17.34) та (17.35) для адсорбції з дисоціацією.
Для адсорбції на неоднорідній поверхні різні значення теплот
хемосорбції повинні приводити до різних значень енергії активації на відповідних
ділянках через співвідношення лінійності (14.92), яке для цього випадку має
вигляд
АЕа=-аАда. (17.75)
Рівняння (17.75) можна записати таким чином:
(Ев)а + ос^е = (Е0)а + сси, = сомі. (17.76)
Враховуючи рівняння (17.62), маємо для енергії активації на неоднорідній
поверхні за довільного 0
(Ев)а=(Е0)а+аСв. (17.77)
Отже, на рівномірно неоднорідній поверхні енергія активації адсорбції
зростає із заповненням поверхні адсорбатом. Це веде до зменшення
константи швидкості адсорбції і, відповідно, до гальмування процесу адсорбції
на неоднорідній поверхні при переході до місць із меншою адсорбційною
здатністю у міру заповнення поверхні адсорбатом. Таким чином, рівняння
для константи швидкості адсорбції на неоднорідній поверхні має вигляд
(*в).-(*о)««-аСаГИ'-(*о)^-4^. <17-78>
де (к0)а — константа швидкості адсорбції на місцях із мінімальною енергією
активації за 0 —> 0.
444
Для енергії активації десорбції з рівнянь (17.33), (17.63) та (17.76) маємо
(ЯеХ/^о^-рСе, (17.79)
де Р = 1 - а.
Таким чином, енергія активації десорбції є максимальною на місцях із
максимальною адсорбційною здатністю й поступово зменшується із
заповненням неоднорідної поверхні адсорбатом. Відповідно, для константи
швидкості десорбції маємо
(*е)^=(*о)^. (17-8°)
Таким чином, на неоднорідній поверхні константа швидкості адсорбції
експоненціально спадає, а константа швидкості десорбції експоненціально
зростає зі збільшенням ступеня заповнення поверхні адсорбатом і
поступового блокування місць із максимальною адсорбційною здатністю.
Відношення константи швидкості адсорбції до константи швидкості
десорбції для неоднорідної поверхні дає, як і у випадку однорідної поверхні,
константу рівноваги адсорбції, у чому не важко пересвідчитися, якщо
поділити рівняння (17.78) на рівняння (17.80) і отримати рівняння (17.64)
(з урахуванням, що а + Р = 1).
Вирази для швидкості адсорбції на однорідній поверхні (17.69) та (17.73) такі:
га=каРДв), (17.81)
причому ка не залежить від 0.
Для неоднорідної поверхні константа швидкості адсорбції різко,
експоненціально (див. рівняння (17.78)) залежить від ступеня заповнення
поверхні адсорбатом. Цією залежністю від 6 можна обмежитись і для
швидкості адсорбції маємо
га=І.К)аР = {к,)аРе-^. 07.82)
Відповідний вираз для швидкості десорбції є таким:
^=(*о)^Р/Є- (17-83)
Рівнянням (17.82) та (17.83) можна надати інший вигляд на ділянці середніх
заповнень поверхні адсорбенту адсорбатом, для яких на рівномірно
неоднорідній поверхні виконується логарифмічна ізотерма адсорбції.
Підставляючи в (17.82) та (17.83) значення 0 з рівняння (17.67), маємо:
га=(ке/)аРр~а, (17.84)
^=(М^Р' (17.85)
Де(ке/)а = (ко)а(КоУа> (кє/)сі = (коЬ(к<уР> Р — рівноважний тиск
адсорбату, що відповідає ступеню заповнення 0, а Р — реальний тиск адсорбату
в газовій фазі.
Адсорбція та десорбція — важливі стадії гетерогенно-каталітичних
процесів, до розгляду яких переходимо у наступному розділі.
445
Розділ 18.
ГЕТЕРОГЕННИЙ
КАТАЛІЗ
Багато синтезів хімічних:
Спирт, оксид сірки, аміак...
Проходять лиш каталітично,
Бо ж без каталізу ніяк.
18.1. Кінетика гетерогенно-каталітичних реакцій
Гетерогенно-каталітичні реакції перебігають на межі поділу фаз.
Зазвичай гетерогенні каталізатори є твердими тілами, а реакційне середовище
є рідиною (розчином) або газом.
Вивчаючи кінетику гетерогенно-каталітичних реакцій, розрізняють
кінетичну та дифузійну ділянки перебігу реакції. До кінетичних відносяться
адсорбційно-десорбційні процеси та реакції між адсорбованими
частинками в поверхневому шарі. Для кінетичної ділянки характерними є відносно
високі енергії активації, тому
кінетичні стадії є лімітуючими за
відносно низьких температур.
Дифузійні стадії мають невеликі енергії
активації, особливо для газової фази,
тому вони стають лімітуючими за
відносно високих температур. У
координатах Арреніуса (1п&- ХІТ) при
переході від кінетичної ділянки до
дифузійної спостерігається
характерний злам, як це зображено на
рис. 18.1.
Якщо каталізатор дуже
пористий, особливо з малими радіусами пор, то
лімітуючою може стати не зовнішня, а внутрішня
(в порах) дифузія. Внутрішньодифузійного
гальмування можна позбутися шляхом подрібнення
гранул каталізатора. Далі розглядатимуться лише
суто кінетичні закономірності гетерогенного
каталізу (без дифузійних ускладнень).
І. Ленгмюр та С. Гіншельвуд запропонували
таку кінетичну схему: адсорбція всіх реагентів,
далі реакція між адсорбованими частинками у
поверхневому шарі і, нарешті, десорбція
продуктів. За такою схемою, наприклад, може пере-
Пншельвуд^ірил Норман бігати реакція ізотопного обміну водню з дей-
Велика Британія терІЄМ
2,2 2,3 2,4
Рис. 18.1. Кінетична (/) та дифузійна (2)
ділянки перебігу гетерогенно-
каталітичної реакції
446
Н2+2[] = 2[Н]
В2 + 2[ ] = 2[0]
[Н] + [Б] = НБ + 2[ ]]
де [Н] та [Т)]—адсорбовані атоми водню та дейтерію, відповідно; [ ] — вільна
ділянка поверхні адсорбенту.
Е. Ріділ та Д. Ілі запропонували більш гнучку кінетичну схему, згідно
з якою адсорбовані частинки можуть реагувати не тільки між собою, але і з
молекулами газової фази. Наприклад, для реакції ізотопного обміну водню
з дейтерієм можливі такі елементарні процеси:
[Н] + Б2=ЬГО + [В]\
[0] + Н2=НО + [Н]]-
Таким чином, у схемі Ілі-Ріділа адсорбційні та десорбційні стадії існують
не окремо, як у схемі Ленгмюра-Гіншельвуда, а об'єднуються з реакціями
в поверхневому шарі.
Розглянуті вище кінетичні схеми відносяться до моделі однорідної
поверхні каталізатора. Розглянемо деякі конкретні приклади кінетики та
стехіометричного механізму гетерогенно-каталітичних реакцій на однорідній
поверхні каталізатора.
При окисненні СО на паладій-срібних каталізаторах порядки як за СО, так
і за 02 змінні від 1 при малих концентраціях реагенту (СО або 02) до 0 при
великих концентраціях реагенту. Таку залежність можна описати таким
емпіричним кінетичним рівнянням:
г =
^ЛусР2ССО
кхсо7 + куссо
яке при фіксованих концентраціях СО або 02 має вигляд
У = —
/Сф + Ь '
де сі— концентрація реагенту, що змінюється.
При с(—» 0 маємо 1-й порядок за сі9 при с,—5
зі схемою Ленгмюра-Гіншельвуда маємо
22 + 02«->220
2 + СО<->2СО
(18.1)
(18.2)
»маємо нульовий. Згідно
(А)
20 + 2ХЮ —^-> 22 + С02 ]
де 2— активна ділянка поверхні каталізатора. В лімітуючій 3-й стадії маємо
Г = Г3=£302О02СО> (183)
де 620 і 02СО — ступені заповнення поверхні проміжними речовинами.
Враховуючи, що 92СО + 920+0^ = 1, і використовуючи рівняння (17.27) та
(17.35) для залежності 6;від тисків реагентів у газовій фазі, маємо
447
г = _Ц/5^со_ (184)
(і + ^о2+^со)
—1 —2
Рівняння (18.4) при великих значеннях К2ссо та Кхс0 дає від'ємні порядки
за карбон монооксидом (-1) та киснем (-0,5), що не збігається з
експериментом (рівняння (18.1)). Отже, механізм (А) Ленгмюра-Гіншельвуда не описує
експериментальні кінетичні дані окиснення СО на Реї—А§ сплавах.
За схемою Ілі-Ріділа, враховуючи можливість хемосорбції кисню як в
атомарній (20), так і в молекулярній (202) формах, маємо
2 + 02<->20;>
к_і
202+СО—^->20 + СЮ2
(В)
20 + СО—^->2 + С02
У цій схемі вважається, що СО реагує безпосередньо з газової фази. Це
припущення відповідає слабкій (порівняно з киснем) адсорбції СО на цих
каталізаторах. Далі буде показано, що дійсно карбон монооксид на Р<і-А§
каталізаторах хемосорбується слабо.
Для отримання кінетичного рівняння із кінетичної схеми (В) можна
скористатися методом стаціонарних станів, але більш ефективним є
застосування методу графів. Запишемо наведену вище кінетичну схему (В) у
вигляді графа (проміжні сполуки є вершинами графа)
со2 Гг) і
З Г 20) ^ « (20Л 2
^~^С02 2Ссо ^"^
Рівняння швидкості реакції знаходиться з рівняння Мезона
—Еа"- (18'5)
де Д — базові детермінанти; к( — константи швидкості стадій, де
утворюються продукти.
У даному випадку: £)1 = к2ссо • к3ссо + к3ссо • к_х; В2 - к3ссо • кхс0;
^з =^ісо2 '^2ссо-
Умовно це можна зобразити так:
З» 4 «2 39 #2 З* *2 Зі М2
448
Тобто для Д шлях проходить через всі вершини графа та закінчується
та ї'-й.
Підставляючи Д у (18.5) згідно із наведеною схемою, маємо
_ ^2ссоА + ^зссо^з ^\^2со2ссо (\%б\
Логічно припустити, що взаємодія СО із адсорбованим киснем
відбувається швидше, ніж його десорбція, тобто к2ссо » к_ь тоді маємо
г = 2^2^СС° . (18.7)
к\со2\ 1 + д~ +^2ссо
Рівняння (18.7) з точністю до позначень констант співпадає з емпіричним
рівнянням (18.1) і, отже, механізм (В) Ілі-Ріділа правильно описує
експериментальні кінетичні дані для окиснення СО на РсІ-А§ каталізаторах.
Реакція гідрування алкенів на перехідних металах (№, Реї, Рі тощо)
відбувається через утворення відносно міцних поверхневих комплексів алкену
з поверхнею каталізатора, водень за цих умов адсорбується слабко, і тому
можна вважати, що він вступає у реакцію з газової фази. Отже, швидкість
реакції гідрування є пропорційною тиску водню в газовій фазі та ступеню
заповнення однорідної поверхні каталізатора хемосорбованим алкеном
', = «н2вс2н4- (18.8)
Вважаючи, що ступінь заповнення поверхні хемосорбованим алкеном
рівноважний, і для однорідної поверхні виконується рівняння Ленгмюра
(17.27), отримуємо
г = к%^°кТ ' (18'9)
За відносно низьких температур та високих тисків етилену маємо
КаРс2п4 >:>1>і рівняння (18.9) перетворюється на таке:
г = кРщ. (18Л°)
Воно відповідає взаємодії молекул водню з поверхнею каталізатора, яка
є майже повністю заповненою міцно хемосорбованим етиленом. За малих
тисків етилену та відносно високих температур КаРс н «1 і рівняння (18.9)
має вигляд
г = Щі2КаРС2щ = МнД2н4- (18.11)
Рівняння (18.11) відповідає реакції на майже чистій поверхні адсорбенту
між воднем та слабко хемосорбованим етиленом.
Величина Ка залежить не лише від температури, а й від природи поверхні
каталізатора. Метали, які дуже міцно зв'язують алкен, не є добрими
каталізаторами, тому що повільно перебігає процес гідрування хемосорбова-
449
ного етилену. Цей випадок відповідає кінетичному рівнянню (18.10). Метали,
які слабко взаємодіють з алкеном або взагалі не хемосорбують його, теж не
є добрими каталізаторами, тому що в цьому разі загальний процес
гальмується стадією активації алкену, поверхня каталізатора при цьому є майже
вільною. Оптимальним є каталізатор із деякою проміжною міцністю зв'язку
хемосорбованого етилену з поверхнею так, щоб ані стадія утворення
проміжної поверхневої сполуки, ані стадія її
гідрування не були лімітуючими. При цьому
оптимальний ступінь заповнення поверхні
проміжною хемосорбованою сполукою
дорівнює половині (0ОПТ = Уі). Для реакцій
гідрування алкенів оптимальними
каталізаторами серед перехідних металів є нікель та його
аналоги — паладій та платина.
Викладена вище концепція про
оптимальну міцність проміжних хемосорбованих
частинок має назву теорії проміжних
поверхневих сполук, яка була вперше
запропонована у працях П. Сабатье та В. М. Іпатьєва.
Проілюструємо дію цієї теорії на реакції з відносно
простою кінетикою — дегідруванні
мурашиної кислоти
Сабатье Поль
(1854—1941),
Франція
НСООН->С02+Н2.
Перша стадія цієї реакції—це взаємодія пари кислоти з металом-каталі-
затором з утворенням поверхневого форміату
НСООН + [ ] —*-> 1/2Н2 + [НСОО].
Далі відбувається розклад форміату з
утворенням продуктів та регенерацією каталізатора
[НСОО]-
->С02+1/2Н2 + [].
Позначаючи через 0 ступінь заповнення
поверхні каталізатора поверхневим форміатом,
маємо з умов стаціонарності
сів.
Лі
= 0 = к{Р(1-в)-к2в,
звідки
Є,
кхР
(18.12)
Іпатьев
Володимир Миколайович
(1867—1952),
Росія, США
кхР + к2
Для швидкості каталітичної реакції маємо
12
г = *2Єс
кхР + к2
(18.13)
450
Проаналізуємо рівняння (18.13)
за різних співвідношень констант кх
та к2. За к{Р « к2 маємо г = кхР
і 0 —> 0, тобто лімітуючою стадією є
утворення поверхневого форміату, і
реакція перебігає на майже чистій
поверхні каталізатора. За кхР » к2
маємо г- к2 і 0 —> 1, тобто
лімітуючою стадією є розклад форміату,
і нульовий порядок відповідає
майже повному заповненню поверхні
форміатом. Очевидно, обидва крайні
випадки не є оптимальними.
Нарис. 18.2 наведено залежність
швидкості каталітичної реакції
дегідрування мурашиної кислоти від
гг,к
400
500
600
му
/•А§
Аи
• Іг
Юі
£и
Си4
і
\Иі
Со\Ре
кДж/моль
1 »
250
350
450 0
Рис. 18.2. Вулканоподібна крива
для реакції дегідрування НСООН
теплоти утворення проміжного форміату, так звана «вулканоподібна»
крива. На лівій гілці цієї кривої розташовані метали, що недостатньо міцно
зв'язують форміати, на правій—ті, що утворюють занадто міцні поверхневі
сполуки. Максимуму відповідає кхР = к2 і 0ОПТ.= Уг> тобто лімітуюча стадія
відсутня, і поверхня оптимального каталізатора заповнена поверхневим
форміатом саме наполовину. Термін «оптимальний» тут використовується
відносно активності каталізатора.
На прикладі реакції розкладу мурашиної кислоти можна проілюструвати
ще одну властивість каталізатора — його вплив на селективність процесу.
Крім вищезазначеного процесу дегідрування, мурашина кислота може
розкладатися за схемою
НСООН = СО + Н20.
Якщо для прискорення дегідрування потрібні каталізатори — перехідні
метали, то для прискорення реакції дегідратації потрібні каталізатори
кислотно-основного типу, наприклад А1203. Обидва шляхи розкладу мурашиної
кислоти є термодинамічно можливими, отже, можна регулювати напрямок
реакції, тобто керувати селективністю процесу, використовуючи відповідні
каталізатори.
Як і мурашину кислоту, спирти можна дегідрувати з утворенням
альдегідів, так і дегідратувати з утворенням алкенів.
Напрямок (селективність) процесу визначається застосуванням певних
гетерогенних каталізаторів. Так, при дії на етиловий спирт
кислотно-основного каталізатора (наприклад А1203) відбувається дегідратація з утворенням
етилену, а при дії дегідруючого металічного (наприклад мідного)
каталізатора утворюється альдегід. С. В. Лєбєдєву вдалося поєднати обидва ці
процеси на каталізаторах, що мають одночасно як дегідратуючу, так і дегідру-
ючу дію — це оксиди урану (торію) або багатокомпонентна суміш, напри-
451
с.
(1874-
Етилен
(СН2=-СН2)
Рис. 18.3. Отримання різних продуктів з одного реагенту
під дією різних каталізаторів
клад А1203 (дегідратуюча дія) та Мп02 (дегідру-
юча дія). За рахунок цього С. В. Лєбєдєв
реалізував з високою селективністю синтез із
етилового спирту бутадієну (дивінілу), який є
важливою сировиною для виробництва
синтетичного каучуку. Селективність дії каталізаторів
перетворень С2Н5ОН схематично зображено
нарис. 18.3.
Важливе значення проблема селективності
має при окисненні органічних сполук. Замість
глибокого окиснення до С02 та Н20 є
можливість отримати цінні продукти неповного
окиснення за допомогою каталізаторів м'якого
селективного окиснення. Наприклад, шляхом
м'якого окиснення етилену на срібному
каталізаторі можна отримати оксид етилену.
В. Лєбєдєв У деяких випадках у кінетичних рівняннях
-1934), СРСР гетерогенно-каталітичних реакцій спостеріга-
452
ються від'ємні порядки за продуктами, тобто ці реакції гальмуються
продуктами. Наприклад, реакція дегідрування циклогексану на нікелевому
каталізаторі гальмується бензеном, і відповідне кінетичне рівняння має
вигаяд
Рс6н6
Розглянемо механізм цієї реакції. Його можна уявити як двостадійну
схему:
С6Н™+[]—^[С6Н6] + ЗН2;
[СбНбІ^^СвН^+Е],
де [ ] — вільне місце на поверхні каталізатора.
Вважається, що водень хемосорбується слабко й швидко переходить у
газову фазу при взаємодії циклогексану з поверхнею каталізатора. Хемосор-
бований на поверхні бензен знаходиться у рівновазі з бензеном у газовій фазі
(температури кипіння бензену та циклогексану є значно нижчими за
температури проведення реакції). Рівноважна стадія не може бути лімітуючою,
отже, процес взаємодії циклогексану з поверхнею каталізатора є
лімітуючою стадією, для якої маємо
г=^в°=>Шг- (Ш5)
Частку поверхні каталізатора 60, що не зайнята хемосорбованим
бензеном, визначено за допомогою рівняння Ленгмюра (17.27). Якщо вважати, що
бензен хемосорбується міцно, то К2Рс н >:>1 і рівняння (18.15) збігається з
емпіричним (18.14) за к = кх/К2. Таким чином, міцна хемосорбція продукту
(тут бензену) веде до гальмування гетерогенно-каталітичної реакції.
У реакції синтезу аміаку також спостерігається від'ємний порядок за
продуктом (див. рівняння (13.39)), але аміак не хемосорбується у вигляді
молекул на поверхні заліза та інших перехідних металів. Отже, причина
гальмування реакції синтезу МН3 аміаком полягає не у міцній хемосорбції
продукту.
Розглянемо механізм каталітичного синтезу аміаку на перехідних
металах. Азот і водень на поверхні цих каталізаторів хемосорбуються як у
молекулярній, так і в атомарній формі. За відносно низьких температур
переважає молекулярна форма, бо її утворення проходить з малою енергією
активації, дисоціативна форма для свого утворення потребує значної енергії
активації, особливо для азоту, молекула якого є найміцнішою серед гомо-
ядерних двохатомних молекул. Отже, за відносно високих температур азот
та водень існують на поверхні каталізатора у вигляді атомів, які можуть
реагувати один з одним з утворенням проміжних поверхневих сполук —
хемосорбованих частинок імідів та амідів. На ці самі частинки
перетворюється молекула аміаку, взаємодіючи з поверхнею, тобто молекула аміаку
453
з поверхнею реагує (маються на увазі відносно великі температури) так
само, як молекули водню й азоту—дисоціативно з відщепленням атомарно
хемосорбованого водню. Отже, механізм каталітичного синтезу аміаку
можна записати через такі стадії:
Н2 + 2[]->2[М],
Н2 + 2[]?±2[Н],
[Н] + [Н]?±[МН] + [],
[Ш] + [Н]<±[Ш2] + [],
[НН2] + [Н]?±2[] + МНз.
Усі стадії, крім першої, є оборотними і рівноважними, отже, їх можна
об'єднати в одну квазірівноважну стадію адсорбційно-хімічної рівноваги
і, таким чином, зобразити механізм гетерогенно-каталітичного синтезу
аміаку двостадійною схемою:
1)Н2 + 2[]->2[М],
2)2[К] + ЗН2<=>2[] + 2Ш3.
При адсорбції азоту на залізі (див. підрозділ 17.2) диференційна теплота
адсорбції зменшується із заповненням поверхні лінійно, отже, поверхня за-
лізгіого каталізатора є рівномірно неоднорідною, і для кінетики
хемосорбції азоту можна застосувати рівняння (17.84)
га=КРщРщ- (18.16)
Тут Рм — тиск азоту в газовій фазі, що не знаходиться в рівновазі з хемо-
сорбованим азотом, а/?^2 — рівноважний тиск азоту. Величину/?^ можна
визначити через адсорбційно-хімічну рівновагу на другій стадії каталітичної
реакції, що дає
( »2 Vа ( оз Vх
^ ~ каРщ
Р* Ч
= кР„
Р2
(18.17)
Для зворотного процесу — розкладання аміаку лімітуючою є стадія
десорбції хемосорбованого азоту (з рекомбінацією адсорбованих атомів
Нітрогену в молекулу). Використовуючи для кінетики десорбції на
неоднорідній поверхні рівняння (17.85) і враховуючи адсорбційно-хімічну рівновагу
на стадії взаємодії хемосорбованого азоту з воднем та аміаком газової фази,
маємо
(&..
лР
г = к
'ш3
(18.18)
де Р = 1 - а.
За типового значення а = Р = 0,5 рівняння (18.17) та (18.18) переходять,
відповідно, у наведені вище у підрозділі 13.2 рівняння (13.38, а) та (13.38, б).
454
Таким чином, гальмування аміаком реакції синтезу аміаку та гальмування
воднем реакції розкладу аміаку пов'язано з встановленням адсорбційно-
хімічної рівноваги між хемосорбованим азотом та газоподібними воднем
та аміаком.
Для реакції синтезу аміаку теорія проміжних поверхневих сполук також
дає можливості для пошуку оптимального каталізатора. Для металів, що
дуже міцно хемосорбують азот, енергія активації хемосорбції азоту, згідно
зі співвідношенням (17.75), повинна знижуватися, але, оскільки міцно хемо-
сорбований азот погано гідрується воднем, то поверхня таких каталізаторів
майже повністю заблокована азотом, що гальмує загальний процес. До
таких каталізаторів належать, наприклад, перехідні метали IV—VII груп
періодичної системи, такі як Ті, 2г, V, Сг тощо. Метали, що дуже слабко
хемосорбують азот або не можуть його хемосорбувати взагалі, також не є
добрими каталізаторами внаслідок дуже високої енергії активації
хемосорбції. Це такі метали, як А§, Си, №, Рі, Реї тощо. Оптимальними
виявилися метали з проміжною здатністю до хемосорбування азоту — це Ре,
Ки та Оз. Зазначимо, що на оптимальних каталізаторах ступінь заповнення
поверхні проміжною сполукою — хемосорбованим азотом приблизно
дорівнює половині.
18.2. Активні центри гетерогенних каталізаторів
У металів, що є оптимальними для синтезу аміаку, я?-орбіталі частково
заповнені, і це має важливе значення
при взаємодії молекулярного азоту в
лімітуючій стадії з поверхнею
каталізатора, точніше, з її окремою
ділянкою, яку X. С. Тейлор запропонував
називати активним центром.
Розглянемо спочатку взаємодію
молекули азоту з окремим атомом
^/-металу, наприклад Феруму. При
цьому можливі дві орієнтації
молекули азоту до атома перехідного металу:
лінійна (рис. 18.4, а) та у вигляді
я-комплексу (рис. 18.4, б). Частково
заповнені */-орбіталі надають
можливість утворення як донорно-акцеп-
торного зв'язку з переносом
електронної густини з молекули азоту на
вакантні й?-орбіталі металу, так і датив-
ного зв'язку з переносом електронів з рис 18 4 Взашодія мод
металу на вакантні антизв язуючі мо- 3 атомом перехідного металу:
лекулярні я-орбіталі молекули азоту. а—лінійний комплекс; б—я-комплекс
455
Характер дативного зв'язку в обох випадках (рис. 18.4, а та б) схожий,
а в утворенні донорно-акцепторного зв'язку в лінійному комплексі бере
участь неподілена пара електронів, а в я-комплексі електрони зв'язуючих
молекулярних орбіталей И2.
Квантовохімічні розрахунки, що базуються на цих моделях, показують,
що лінійний комплекс є енергетично вигіднішим, а в я-комплексі
спостерігається більша дестабілізація зв'язку Нітроген—Нітроген у молекулі И2.
Оптимальною є взаємодія молекули азоту не з окремим атомом металу,
а цілою групою — кластером (рис. 18.5, а). При цьому спочатку
утворюється більш стійкий торцевий комплекс, а потім по мірі занурення молекули
азоту у западину кластера підключаються бокові атоми, які діють на
молекулу азоту по я-типу і ефективно дестабілізують №№ зв'язок (рис. 18.5, б).
Отже, оптимальним повинен бути активний центр у поверхневому шарі
каталізатора, який складається з групи атомів (кластер) і має вигляд
невеличкої западини молекулярного розміру. Така атомна група досить міцно
тримає молекулу азоту і в той самий час ефективно дестабілізує зв'язок
Нітроген—Нітроген. Це може бути вакансія в поверхневому шарі, вихід
дислокації, а найголовніше — наявність нещільно упакованих граней. Справді,
збільшення дефектності полікристалічних металів — каталізаторів (Ре, Со, АУ, Ке)
веде до зростання їх каталітичної активності в реакції синтезу аміаку. На
монокристалах Ре, АУ, що мають кубічну об'ємноцентровану структуру,
щільно упакована грань [110] не синтезує аміак і взагалі не хемосорбує азот,
на відміну від нещільно упакованих граней [111], [100] та граней з високими
індексами, на яких саме реалізуються зазначені вище активні
центри—западини молекулярного розміру. Кількість таких западин-кластерів
зменшується при зменшенні розмірів мікрокристалів в інтервалі від 10 до 2 нм. Так,
зменшення розміру мікрочастинок заліза (рис. 18.6) призводить до падіння
їх каталітичної активності у реакції синтезу аміаку.
456
Реакції, для яких активність
(питома) залежить від структури
каталізатора, М. Будар запропонував називати
структурно-чутливими. Прикладом
такої реакції є процес синтезу аміаку.
Найбільш чітко структурна чутливість
каталітичної реакції (або процесу
хемосорбції) виявляється при вивченні ката-
лізаторів-монокристалів.
У таблиці 18.1 зібрані дані по
активності граней металічних
монокристалів при їх взаємодії з газами, а у
таблиці 18.2 — по каталітичній активності
монокристалів у газофазних реакціях.
Наведені у таблицях 18.1 та 18.2 дані
ілюструють поширеність структурно-
чутливих процесів. Аналіз наведених у
табл. 18.1 та 18.2 даних і співставлення
їх із даними про поверхневу енергію
і щільність упаковки граней (підрозділ
17.1;формупи(17.21);(17.22), табл. 17.1)
показує, що мінімальну реакційну
здатність, адсорбційну і каталітичну
активність переважно мають щільно
упаковані грані з мінімальним значенням
вільної поверхневої енергії (для к.г.ц
структури — це грань {111}, для к.о.ц.
Таблиця 18.1. Активність граней монокристалів при їх взаємодії з газами
Метал
Ґси
?і
Іг
Си,№
Ке
Со
\У
^
Ре
Ре
XV
Кристалічна
структура
к.г.ц.
те саме
»
»
г.щ.у.
те саме
К.О.Ц.
те саме
»
»
»
Процес
окиснення киснем
хемосорбція 02
хемосорбція N2
рекомбінація атомів Гідрогену
хемосорбція N2
розклад СО
хемосорбція И2; СО
хемосорбція Н2; И2
хемосорбція С2Н2
хемосорбція N2
рекомбінація атомів Гідрогену
Співвідношення
активностей граней
{111}<{100}
{Ш}<{321}
{100}<{110}<{210}, {320}
{111}<{100}<{110}
{1000}-тіп
{1000} -тіп
{110}-тіп
{110}<{100}<{111}
{110}<{100}
{110}<{111}
{110}<{П1}
і
1000
100
10
1
0,1
0,01
0,001
1000
* Ю3
А
о /
• /
1
•УІ9Ї
А /
! о
1
-1
А Ь/
о /
/й
і і
А
О
І 1
СІ, НМ
—■ >
0 2 4 6 8 10 12 14
Рис. 18.6. Залежність каталітичної
активності (числа обертів — У{)
в реакції синтезу аміаку від розміру
мікрочастинок заліза-каталізатора
-{110}, для г.щ.у.—{1000}).
457
Таблиця 18.2. Каталітична активність граней монокристалів
Метал
Си
те саме
»
№
те саме
?і
Рі
№
Ре
Кристалічна
структура
к.г.ц.
те саме
»
»
»
»
»
к.о.ц.
те саме
Реакція
окиснення Н2
дегідрування спиртів
розклад Н2Н4
гідрування Н2Н4
гідрогеноліз С2Н6, С4Н10
Н2+02 обмін
окиснення ИНз
гідрування С2Н4
синтез МНз
Співвідношення
активностей граней
{111}<{100}<{110}, {311}, {211}
{111}-тіп
{111}<{100}
{100}<{321}
{111}<{100}
{111}<{100}<{211}<{110}
{111} - тіп; ребра - тах
{110}<{100}<{111}<{2П}
{110}<{100}<{111}
к.г.ц. —кубічна гранецентрована структура;
к.о.ц.—кубічна об'ємноцентрована структура;
г.щ.у. —гексагональна щільна упаковка.
Якщо каталізатор працює в умовах високих температур з локальними
перегрівами (наприклад, реакція синтезу аміаку є дуже екзотермічним
процесом), то це веде до знищення дефектів, крім того, внаслідок рекристалізації
зменшується поверхня каталізатора. Усе це веде до зменшення як питомої
(на одиницю межі поділу тверде тіло-газ), так і загальної активності
каталізатора. Щоб запобігти цьому, до каталізатора додають так звані
структуроутворюючі промотори. Наприклад, для залізного каталізатора
синтезу аміаку таким промотором є оксид алюмінію. Він розташовується
тонкою острівковою плівкою в поверхневому шарі каталізатора і захищає
його від рекристалізації, зберігаючи дефекти у поверхневому шарі.
Промотори, що впливають на питому активність, називають
модифікуючими. Каталізатор Ре—А1203 можна модифікувати домішками оксидів
лужних металів, зокрема К20, додавання якого суттєво збільшує питому
активність каталізатора.
Домішку, що зменшує питому активність, називають отрутою.
Отруєння може бути оборотним чи необоротним. Сполуки, що містять сірку,
зокрема, сірководень, меркаптани тощо, сполуки арсену, отруюють багато
металічних каталізаторів необоротно. Прикладом оборотного отруєння
може бути вплив пари води або молекулярного кисню на активність
залізного каталізатора синтезу аміаку. Якщо після отруєння на каталізатор
подавати азото-водневу суміш, що ретельно очищена від домішок води та кисню,
то питома активність залізного каталізатора поступово зростає до величини,
що передувала отруєнню. Слід зазначити, що навіть незначна кількість
отрути суттєво впливає на активність каталізатора, що підтверджує висунуту
X. Тейлором концепцію активних центрів, тобто наявність на поверхні
каталізатора окремих ділянок з підвищеною каталітичною активністю.
Одним із засобів захищення каталізаторів від рекристалізації, зберігання
їх високорозвинутої поверхні є створення нанесених каталізаторів.
458
Високодисперсні метали наносять на поверхню адсорбентів, що мають
велику питому поверхню, розвинуту пористість, і в той же час ці носії є
термостійкими і зберігають свої адсорбційно-структурні характеристики за
високих температур і перегрівань. До таких носіїв зараховують алюмогель,
силікагель, пемзу, цеоліти, активоване вугілля тощо. Дуже поширеними є
нанесені каталізатори на основі платини, їх використовують у різних процесах
гідрування та окиснення органічних сполук, зокрема, в окисненні спиртів,
гідрогенізації олефінів, дегідрогенізації та гідрогенолізі циклопарафінів тощо.
Нанесені на кізельгур нікель та кобальт використовують як каталізатори
реакції Фішера-Тропша, срібло на алюмогелі — для отримання оксиду
етилену з етилену, паладій на різних носіях використовується в реакції
окиснення монооксиду вуглецю тощо. Зазначимо, що кількість каталітично
активного металу в нанесеному каталізаторі становить лише декілька відсотків,
що дозволяє економити коштовні дефіцитні метали.
За дуже малих
концентрацій нанесеного металу на по- М, а
верхні носіїв знаходяться вже І А
не мікрокристали, а невеличкі
групи атомів, які М. І. Кобо-
зєв запропонував називати
ансамблями. Він звернув
увагу на те, що за збільшення
кількості нанесеного металу
на носії в ділянці дуже малих
концентрацій активність (А)
нанесеного каталізатора
проходить через максимум, як
це зображено на рис. 18.7.
Якби каталітична реакція
0,002 0,004 0,006
Рис. 18.7. Залежність активності (А)
та питомої активності (а) нанесеного
каталізатора від кількості (т) нанесеного металу
була індиферентна до виду активних центрів — ансамблів, то активність
нанесеного каталізатора була б пропорційною кількості каталітично активної
маси (цьому випадку відповідає пряма / на рис. 18.7). Наявність максимуму
свідчить про те, що носіями каталітичної активності є ансамблі певного
розміру, які складаються з певної кількості атомів — к. Інакше кажучи, якщо
позначити активність і'-го ансамблю через гі9 то всі г,- прямують до нуля
за і Ф к.
Запропонована М. І. Кобозєвим теорія ансамблів дає можливість
обчислити величину із експериментальних даних. Вважається, що на поверхні
носія є певні комірки — ділянки міграції, у кожній з яких є певна кількість
атомів к, що складають і-атомний ансамбль. Між комірками існують
бар'єри, що не дозволяють атомам переходити з однієї ділянки міграції до
іншої.
Розподілення атомів каталітично активного металу по комірках
визначається законами статистики. Оскільки концентрація нанесеного металу є
459
дуже малою, то можна застосувати закон рідкісних подій — розподілення
Пуассона. Згідно з цим розподіленням імовірність (рк) утворення £-атом-
ного ансамблю дорівнює
Р*=|^Л 08-19)
де V — середня кількість атомів у комірці.
Величина V пов'язана з масою т нанесеного металу простим
співвідношенням
т = уЛ^ = сотІУ, (18.20)
де N— кількість комірок на поверхні носія; # — маса атома металу.
Активність А нанесеного каталізатора, що містить ансамблі з А: атомів,
дорівнює
А = гкРк. (18.21)
Положення максимуму на кривій залежності А від т визначається
умовою йАІйт = 0. Оскільки т є пропорційною V, а А —рь то ця умова
переходить у сІркШ = 0. Відповідно до цієї умови з рівняння (18.19) маємо
у;Г=£. (18-22)
Проте з графіка залежності А від т (рис. 18.7) маємо не У™**, а лише
пропорційну їй величину тя™ах. Додатково можна використати положення
максимуму залежності від маси нанесеного металу питомої активності а,
що визначається відношеннями до т. Отже, маємо
а = А = !кК = сотіу!^_е-ч (18 23)
т уЛ/£ к\
і з умов йаійт = 0 і, відповідно, сіа/сіу = 0 знаходимо
у^^-І. (18-24)
З рівностей (18.22), (18.24) та (18.20) легко знайти
тах тах
к = ^ = ^ . (18.25)
утах_утах ттах ^^тах
Дослідження активності нанесених каталізаторів у реакціях різного типу
показало, що реакції гідрування-дегідрування перебігають на двохатомних
ансамблях, у реакціях окиснення типове значення к = 1 (зазначимо, що при
цьому /я™3* = 0, тобто на кривій а =/(т) максимум збігається з віссю
ординат), для реакції синтезу аміаку теорія ансамблів дає к = 3. Таким чином,
теорія Кобозєва дозволяє визначити кількість атомів, що складають активний
центр.
Д. Дауден поширив ідеї М. І. Кобозєва на каталітичні властивості
гомогенних металічних сплавів. За моделлю Даудена, сплав є статистичною
сукупністю кластерів (ансамблів) різного складу. Якщо у бінарному сплаві
460
(АВ) існують кластери з п атомів, що містять т атомів певного компонента А,
то ймовірність (Р™) утворення таких кластерів дається біноміальним
розподіленням
Рпт=С:х^І-хАГт, (18.26)
де хА — мольна частка компонента А у сплаві.
Якщо у якійсь реакції переважно працюють кластери певного складу, що
містять т атомів компонента А, то каталітична активність є пропорційною
Рлт, і на кривій залежності активності від складу сплаву з'являється
максимум, положення якого на осі абсцис визначається співвідношенням
*Г=*. (18.27)
За т = я, тобто, якщо максимальну активність мають кластери, що
складаються з атомів лише одного сорту, активність сплаву (а) пропорційна
Р„=х"А=-^, (1828)
ао
де а0 — активність чистого компонента А.
Побудувавши графік у координатах 1§— ^2>ха> можна знайти п.
Подібна ситуація реалізується, наприклад, при проведенні реакції
синтезу аміаку на Ре—№ сплавах, де нікель є інертним у каталізі компонентом.
Оцінка величини п за експериментальними даними з каталітичної активності
Ре—№ сплавів в області гомогенності дала для середнього значення п = 6
атомів, що непогано збігається з даними, отриманими за теорією Кобозєва
для нанесених каталізаторів при дуже малих концентраціях заліза, й
підтверджує висловлену вище думку про те, що активним центром у реакції
синтезу аміаку є певна група атомів у поверхневому шарі каталізатора.
У системі Ре—N1 склади об'єму і поверхневого шару ідентичні, що
підтверджено методом РФЕС (інтенсивність піків в РФЕС спектрах пропорційна
кількості атомів елемента в поверхневому шарі). В загальному випадку вони
відрізняються внаслідок збагачення поверхневого шару одним із
компонентів — ПАР (див. підрозділ 17.1), який має меншу поверхневу енергію (а).
Зміни у складі поверхневого шару повинні впливати на каталітичну
активність сплавів. При цьому можна виділити два типи систем: «А»—поверхнево-
активний компонент одночасно є каталітично активним; «В»—ПАР —
інертна або малоактивна у каталізі речовина. Системи типу «А» сприятливі для
каталізу, в системах типу «В» активно діюча ПАР фактично виконує функції
отрути. Прикладом систем типу «В» є сплави Ре—Си Ас = аРе - аСи » 0,
отже, мідь є ПАР по відношенню до заліза. Це наочно видно з рис. 18.8, с. 462,
де наведено залежність поверхневого складу (уСи) Ре—Си системи від її
об'ємного складу (хСи) — вже при досить малих значеннях хСи у
поверхневому шарі є достатня кількість міді. Відзначимо, що дані теоретичного
розрахунку д:Си за формулою (17.14) добре збігаються з експериментальними
даними, отриманими методом РФЕС. Обчислення кількості атомів у клас-
461
А Ус*
0,005
0,010
0,015
Рис. 18.8. Залежність концентрації міді
у поверхневому шарі Ре—Си сплаву
(уСи) від її об'ємної концентрації (л:Си)
(точки — дані РФЕС, крива —
розрахунки за формулою (17.14))
тері за формулою (18.28) для
подібних систем слід проводити,
підставляючи у (18.28)^ замість лс,-, тобто
користуючись даними про склад
поверхневого шару. Подібні
розрахунки для системи Ре—Си дали
п = 7 в узгодженні з даними для
системи Ре—№.
Для Ре—Со каталізаторів за
даними РФЕС (і критерію Аа = аРе -
- аСо=0) поверхневий склад
тотожний до об'ємного. На залежності
активності у реакції синтезу аміаку від
складу Ре—Со сплавів (рис. 18.9, а)
спостерігається яскраво
виражений максимум при 14 мас.% Со.
Дослідження стану хемосорбова-
ного азоту на поверхні Ре—Со
каталізаторів методом програмованої
термодесорбції (ТД) показало, що зі
зростанням кількості кобальту у сплаві відбувається поступовий перехід від
міцно зв'язаних форм хемосорбованого азоту до слабше зв'язаних (на
чистому кобальті лише молекулярні а-форми), а кількість (32~Ф°РМИ
(атомарної) з оптимальною енергією зв'язку проходить через максимум і
змінюється симбатно з каталітичною активністю сплаву (рис. 18.9, б).
Згідно з літературними даними в області 10—15 % Со у Ре—Со сплавах
спостерігається максимальна здатність для переносу електрона через */-під-
рівні, і це пов'язується з утворенням кластерів складу Ре7Со. Згідно з клас-
терною моделлю при т=1іи = 7+1 = 8, маємо згідно з (18.20) х™™ = 12,5 %
мол. Со, що добре збігається з максимумами на кривих на рис. 18.9, а та 18.9, б
(л:СоХ)експ = 13,3 % мол- Со (14 % мас. Со). Полегшення переносу електрона
через кластер повинно сприяти процесу дисоціативного розщеплення
молекули азоту, зв'язаної з багатоатомним кластером, згідно з детальним
механізмом, розглянутим вище (див. рис. 18.5, с. 456).
Співвідношення (18.27) можна використати для передбачення
оптимального складу каталізатора-сплаву, якщо є міркування щодо складу активного
ансамблю.
Наприклад, у реакції окиснення карбон монооксиду молекулярним
киснем необхідною стадією є хемосорбція СО. Було виявлено, що багато-
центрова, «місткова» форма хемосорбції карбон монооксиду є міцною,
хемосорбовані в такій формі молекули СО блокують поверхню каталізатора
і гальмують процес окиснення СО. Така форма є характерною для
каталізаторів — перехідних металів: Реї, Рі, № тощо. Якщо брати сплави цих металів
з металом, що не хемосорбує карбон монооксид, а хемосорбує лише ки-
462
40
20
І а-105, гШ3/(мін-м2)
Ре 20 40 60 80 100
Мас. частка Со, %
80 100
Мас. частка Со, %
Рис. 18.9. а — Залежність каталітичної активності Ре—Со сплавів у реакції
синтезу аміаку від їх складу; б — відносна кількість (ті) р2-форми хемосорбо-
ваного азоту від складу Ре—Со каталізаторів
сень, наприклад, зі сріблом, то можна прийти до висновку, що
оптимальними повинні бути ансамблі, у яких окремий атом перехідного металу, зокрема
Паладію, знаходиться в оточенні атомів Аргентуму. Це виключає міцну
багатоцентрову хемосорбцію карбон монооксиду, бо СО на цих ансамблях
може хемосорбуватися лише у слабкій одноточковій формі, що сприяє
каталізу. Таким чином, т = 1 і, знаходячи п = 9—10 із структурних
кристал охімічних даних, маємо згідно з формулою (18.27) х^ =0,10—0,11.
Експериментальні дослідження (табл. 18.3) виявили, що найбільш активні
паладій-срібні каталізатори окиснення карбон монооксиду містять 5 —15 %
(мол.) Реї, що добре узгоджується з передбаченим значенням х^.
Дослідження ЇМ—А§ каталізаторів методом програмованої термоде-
сорбції (ТД) в умовах, наближених до іп зіїи (тобто до умов випробування
зразків у каталітичному процесі), показали, що на сріблі СО не хемосор-
бується. ТД спектри СО на паладії наведені на рис. 18.10, я, с. 464. Реєстру-
463
Таблиця 18.3. Активність Реї—А§ сплавів у реакції окиснення СО
Мас. частка Реї, %
0(А§)
5
10
15
40
100 (Рсі)
Ступінь перетворення СО, %
6
100
100
100
100
100
Температура, К
687
338
338
338
393
403
ються три форми досить міцно зв'язаного з поверхнею СО, які відповідають
молекулярній багатоцентровій хемосорбції (а{ і а2) та дисоціативній ((3). На
сплавах Реї—А§ реєструється лише одна а-форма (рис. 18.10, б) слабко
зв'язаного СО (одноточкова) у відповідності з розглянутою кластерною моделлю
і з кінетичними даними (схема (В), рівняння (18.7)). Відзначимо, що
взаємодія СО з хемосорбованим киснем перебігає через утворення проміжного
карбоксилатного комплексу
О
II
[20СО*]=С"0
(крапками позначені зв'язки, що розриваються або утворюються).
/ І
400
600
1000
г,к
300
500
700
т,к
Рис. 18.10. Термодесорбційні спектри карбон монооксиду з поверхні:
а — Р<1; б — сплаву 5 % Рсі—95 % А§
464
18.3. Нестаціонарні явища у гетерогенному
каталізі
На відміну від Реї—А§ сплавів (в області малих концентрацій паладію),
кінетичні закономірності для яких відносно прості й описуються схемою Ьіі-
Ріділа (див. підрозділ 18.1, схема 2?, рівняння (18.6) та (18.7)), на чистому
паладії спостерігається зовсім інша складна кінетика. На рис. 18.11 наведена
залежність швидкості окиснення (г) СО від концентрації карбон монооксиду,
-9 2
г, 10 моль С02/(м с)
0 5 10 ссо, Ю моль/л
Рис. 18.11. Залежність швидкості реакції окиснення СО на Рсі каталізаторі
від концентрації СО
а на рис. 18.12 — залежність г від концентрації кисню. В обох випадках
спостерігається гістерезис, на рис. 18.11 «за стрілкою годинника», а на рис.
18.12 — «проти стрілки годинника». При цьому характерна наявність двох
стаціонарних станів: 1) високоактивного з майже повним перетворенням СО
у С02 (порядки: за СО — 1,0; за киснем — 0,0); 2) низькоактивного
(порядки: за СО = 0,0; за киснем = 0,3).
г,
і
12
8
4
10~8
•
моль
С02/(м2
•с)
і ■ ■
с0 , 10" моль
1 ^_
Рис. 18.12. Залежність швидкості реакції окиснення СО на Рсі каталізаторі
зід концентрації 02
465
Наявність гістерезисів, множинності стаціонарних станів, коливань
швидкості, критичних переходів відносять до нестаціонарних явищ.
Однозначного пояснення цих явищ немає, але для всіх випадків характерна
наявність зворотного зв'язку, тобто впливу результатів каталітичного процесу
на закономірності його перебігу. Зворотний зв'язок може бути
обумовлений низкою причин, серед яких:
— наявність стадії взаємодії проміжних речовин;
— зміна швидкості елементарних стадій при зміні концентрацій реагентів
у поверхневому шарі;
— перегрів поверхневого шару каталізатора;
— ланцюгові процеси на поверхні каталізатора;
— зміна структури поверхні під дією реакційної суміші, зокрема фазові
перетворення у поверхневому шарі каталізатора;
— гетерогенно-гомогенний механізм — гомогенне подовження
гетерогенно-каталітичної реакції.
Розглянемо деякі пояснення нестаціонарних явищ у реакції окиснення СО.
Множинність стаціонарних станів може виникати при окисненні карбон
монооксиду за двомаршрутним механізмом:
Стехіометричне число стадії
| Маршрут І І Маршрут II І
1) 02 + 22< Д )220 11
2) 2 + С0( к% >2СО 2 0
3) 20 + 2СО—^-»22 + С02 2 0
| 4)20 + СО—**->2 + С02 0 2
Цей 2-маршрутний механізм є «гібридизацією» механізмів Ленгмюра-
Гіншельвуда (маршрут І, стадії 1,2,3) та Ілі-Ріділа (маршрут II, стадії 1,4).
Взаємодія двох маршрутів дає нелінійність, і це може обумовити
нестаціонарні явища, зокрема множинність станів і гістерезиси.
Наявність низькоактивного стану обумовлена блокуванням поверхні Реї
каталізатора формами міцної багатоцентрової хемосорбції СО при
збільшенні співвідношення СО/02 до певної величини. В цьому стані реакція має
малу швидкість і перебігає за гетерогенним механізмом переважно по краях
«острівців» хемосорбованого СО. Цьому відповідає нульовий порядок за
карбон монооксидом і позитивний за киснем.
Зі збільшенням концентрації кисню (цей же ефект дає підвищення
температури) СО поступово знімається з поверхні, реагуючи з киснем, який
покриває поверхню каталізатора. Кисень в адсорбованому стані достатньо
активний, і реакція перебігає з підвищеною швидкістю за 1-им порядком за
СО і нульовим за киснем.
У високоактивному стані окиснення може перебігати не лише за
«ударним» механізмом Ілі-Ріділа, але й за гетерогенно-гомогенним механізмом,
466
який полягає у тому, що поверхня каталізатора генерує радикали, які
переходять в об'єм газової фази, де ініціюють ланцюговий процес. На користь
гетерогенно-гомогенного механізму свідчать дані методу роздільного кало-
риметрування, згідно з яким спостерігається підвищення температури
газової суміші над каталізатором у проточному реакторі «назустріч» газовій
суміші, а також досліди з використанням спеціальної насадки, яка сприяє
обриву ланцюгів.
Реалізація гетерогенно-гомогенного механізму обумовлена двома
факторами:
1) можливістю перебігу ланцюгового процесу у газовій фазі;
2) можливістю генерації радикалів на поверхні каталізатора.
При окисненні СО обмеження лише двома реагентами: СО та 02 робить
ланцюговий процес неможливим. Справді, утворення радикала
СО + 02-^С02+6
має своє логічне продовження лише у реакції
СО + 6->С02,
тобто маємо «ланцюг» лише з 2-х ланок.
Окиснення СО за ланцюговим механізмом, однак, можливе у
присутності радикалів ОН*, Н* та Н02 за рахунок таких реакцій:
ОН#+СО->С02 + Н#, (а)
Н#+02->ОН#+6, (б)
Н#+02+М->Н02+М. (в)
При досить високих (атмосферних) тисках процес перебігає вище
верхньої межі «півострова спалахування», отже, реакція (б), що веде до
розгалуження ланцюга, не перебігає.
Таким чином, реакція може перебігати лише як нерозгалужена
ланцюгова через радикал Н02
НО!.+СО->С02+ОН\ (г)
Радикал Н02 досить інертний і реакція (г) перебігає лише при високих
температурах. При відносно низьких температурах, окрім тримолекулярної
реакції (в), слід врахувати таку бімолекулярну стадію:
Н#+02->Н02, (д)
а далі Н02 реагує з СО за реакцією типу (г).
Подібні механізми ланцюгових процесів через збуджені молекули відомі,
наприклад, для реакції водню з фтором. Реакції (а), (д) та (г) складають нероз-
галужений ланцюг.
Зародження радикалів можливе при взаємодії водню (або води) з хемо-
сорбованим киснем (реакція у високоактивному стані перебігає у надлишку
467
кисню, отже, поверхня каталізатора має бути вкрита киснем), а саме:
[02] + Н2->ОН#+[ОН], (є)
[02] + Н20 -> Н02 +[ОН]. (є)
(тут енергетично невигідний вихід радикалів у газову фазу компенсується
утворенням міцно зв'язаних з поверхнею ОН-груп).
Водень або вода можуть заздалегідь адсорбуватися у слабкій формі, яка
знаходиться у рівновазі з Н2 (або Н20) газової фази. Про наявність слабко
адсорбованого водню та ОН-груп свідчать дані термодесорбції, згідно з
якими у газовій фазі реєструються Н02 -радикали та частинки води, що
утворюються при відносно низьких температурах шляхом рекомбінації слабко
зв'язаних груп. Все це свідчить на користь існування гетерогенно-гомогенного
механізму.
В області концентрації, де можуть існувати як високоактивний, так і низь-
коактивний стани, система стає нестабільною і це може привести до
виникнення коливальних процесів. Для реакції окиснення СО такі коливання
справді спостерігалися (див. рис. 18.13).
уіЛллЛА.
Рис. 18.13. Коливання концентрації С02 при окисненні
карбон монооксиду на Реї каталізаторі
Таким чином, коливальні процеси можуть відбуватися як у гомогенних
системах (див. підрозділ 16.3), так і в гетерогенних.
18.4. Проблеми каталізу в реакціях за участю
складних молекул
Чим складнішими є молекули реагентів та продуктів, тим більше значення
має відповідність між структурами реагентів та поверхневого шару
каталізатора. Одним з перших на це звернув увагу О. О. Баландін у своїй мульти-
плетній теорії каталізу. Теорія Баландіна базується на принципах
енергетичної та геометричної відповідності. Принцип енергетичної
відповідності в цій теорії збігається з подібним принципом теорії проміжних
сполук. Так само, як і проміжна поверхнева сполука, «мультиплетний
комплекс» повинен мати деяку оптимальну енергію зв'язку з поверхнею
каталізатора. Оптимум зв'язку мультиплетного комплексу з каталізатором
знаходиться шляхом побудови «вулканоподібних кривих» аналогічно тому, як це
зроблено на рис. 18.2, с. 451.
468
Баландін Олексій
Олександрович
(1898—1967), СРСР
Згідно з принципом геометричної відповідності
легкість перебігу каталітичної реакції залежить від
просторового співвідношення між будовою
активного центру на поверхні каталізатора і геометричними
параметрами тієї частини молекули, що
перетворюється у ході реакції (ця частина молекули в мульти-
плетній теорії має назву «індексної групи»).
Геометрична відповідність повинна зумовлювати
мінімальну напругу зв'язків, що залишаються в молекулі
у процесі адсорбції або виникають під час взаємодії
молекули з поверхнею.
Наприклад, для реакції дегідрування циклогексану
в мультиплетній теорії припускається утворення сек-
стетного комплексу за участю шести атомів поверхні
каталізатора, як це наведено на рис. 18.14. Згідно з цією схемою активними
каталізаторами повинні бути метали, що мають відповідну симетрію граней,
а саме метали, що кристалізуються в кубічній гранецентрованій структурі
(грань {111}) та в гексагональній
щільній упаковці (грань {1000}).
Справді, метали, які кристалізуються
в кубічній об'ємноцентрованій
структурі, наприклад, залізо або
вольфрам, погано каталізують реакцію
дегідрування циклогексану, бо не
мають граней відповідної симетрії.
Крім відповідності у симетрії, Рис. 18.14. Утворення секстетного
принцип геометричної відповідності комплексу в процесі дегідрування
вимагає певних міжатомних відста- циклогексану
ней у кристалічних ґратках каталізатора (тут ці відстані повинні знаходитися
в межах 0,25—0,28 нм), які б відповідали певним міжатомним відстаням у
молекулах реагентів та продуктів.
Слід зазначити, що принцип геометричної відповідності має велике
значення у ферментативному каталізі і є одним з найважливіших факторів
високої активності та селективності біологічних каталізаторів — ферментів.
За образним висловлюванням Е. Фішера «фермент підходить до субстрату,
як ключ до замка» (умовно це зображено на рис. 18.15, с. 470), тобто
важливим є процес «впізнавання» ферментом субстрату (реагенту). Розвиваючи
ідею впізнавання, Д. Кошланд запропонував модель індукованої
відповідності ферменту та субстрату, згідно з якою субстрат спричиняє зміну
конформації ферменту. Це призводить до зближення активних груп ферменту
та субстрату і створює умови для їх ефективної взаємодії. Можливо також,
що фермент має декілька різних конформацій, а субстрат обирає серед них
найбільш активні.
469
, Активний центр
^~—-^-___-уС ферменту
\ І ~*
/ I І ОН—СН2 |
) | "|сн-сн2^к|
\ | ОН—СН2 Г
І Субстрат
Рис. 18.15. «Впізнавання» ферментом субстрату
Для реакцій між складними молекулами, особливо для реакцій їх
синтезу, характерні великі від'ємні значення ентропій активації лімітуючої стадії.
В цьому випадку основним «завданням» каталізатора є подолання не
стільки енергетичного, скільки інформаційного бар'єра.
Приклад такої дії каталізатора можна знайти у кластерному каталізі (про
каталіз кластерними сполуками див. підрозділ 16.4). У таблиці 18.4 наведені
дані для реакції відновлення метиленового блакитного (МБ) спиртами, що
каталізується карбонілами заліза.
Таблиця 18.4. Кінетичні параметри реакції відновлення метиленового
блакитного спиртами »
Каталізатор
Ре(СО)5
Рез(СО)12
Реагент (спирт)
СН3ОН
£-10~3, л/мольс
20
500
Е, кДж/моль
90
92
С2Н5ОН
к- 10і"3, л/мольс
2,5
46
Е, кДж/моль
65
69
Як видно з даних, наведених у табл. 18.4, енергії активації у присутності
пента- та додекакарбонілу заліза близькі за величиною, відповідно, більша
каталітична активність Ре3(СО)12 пов'язана з ентропійним фактором.
Наявність для цієї реакції ізотопного кінетичного ефекту (при заміні протію на
дейтерій) свідчить про те, що в лімітуючій стадії відбувається перенос атома
Гідрогену від спирту до МБ. Активний комплекс лімітуючої стадії є
циклічним і передбачає одночасну координацію молекул МБ та спирту на
активному центрі каталізатора. Багатоядерна кластерна сполука Ре3(СО)12 робить
це кращим чином, ніж моноядерний комплекс Ре(СО)5 і, отже, дозволяє
краще подолати інформаційний (ентропійний) бар'єр.
При синтезі складних молекул саме подолання інформаційного бар'єра
починає відігравати вирішальну роль. Наприклад, ймовірність синтезу
молекули білка шляхом хаотичного з'єднання певних амінокислот настільки мала,
що для утворення лише однієї молекули білка в цьому випадку потрібно
470
10 років (для порівняння — число атомів у Сонячній системі дорівнює
10 ). Якщо врахувати, що Земля існує лише 10 років, то можна прийти до
висновку, що синтез складних молекул некаталітичним шляхом просто
неможливий. З цією задачею, однак, успішно справляються біокаталізатори —
ферменти (див. рис. 18.16).
Рис. 18.16. Фермент орієнтує молекули і здійснює реакцію синтезу
Створені у живій природі протягом її еволюції біокаталізатори є набагато
активнішими та селективнішими, ніж штучні звичайні гомогенні та
гетерогенні каталізатори. Наприклад, гомогенний каталізатор Ре прискорює
розклад Н202 у 10 разів, колоїдна платина—у 10 разів, а фермент каталаза —
у 1013 разів. Залізний промотований каталізатор синтезує аміак з азоту та
водню при 450-500 °С та 300 атм, а фермент нітрогеназа — при кімнатній
температурі й атмосферному тиску.
Біокаталізатори, таким чином, є зразками для створення активних
каталітичних систем, і це дало поштовх до розвитку нового напрямку в каталізі —
біоміметиці, тобто до створення синтетичних каталізаторів, здатних
моделювати ферменти і виконувати їхні функції. Прикладом таких каталізаторів
можна вважати стереоспецифічні каталізатори Циглера-Натта, побудовані
з комплексних сполук перехідних металів та металорганічних сполук. У
каталітичних реакціях стереоспецифічної полімеризації на цих каталізаторах
утворюються так звані регулярні полімери, де молекули мономерів
розташовані впорядковано (наприклад, «голова до хвоста»), на відміну від
звичайних полімерів, де розташування мономерів хаотичне.
Каталізатори Циглера-Натта складаються на основі комплексних сполук
титану та галогенідів алкілалюмінію. Вихідний комплекс титану—бісцикло-
пентадієніл-етилтитанхлорид — має тетраедричну структуру без вакантних
координаційних місць, що потрібні для активації молекул і каталізу. При
приєднанні алкілгалогенідів алюмінію утворюється октаедрична структура
з вакантним координаційним місцем □, як це зображено на схемі:
471
ь
Тут К—алкіл або зростаючий ланцюг, М — атом перехідного металу
(зокрема титану), Ь — відповідні ліганди.
Прикладом біоміметичного підходу (точніше, біоміметичного напряму)
можна також вважати каталіз закріпленими металокомплексами. Закріплення
металокомплексів на поверхні твердого тіла призводить до появи нових
властивостей та переваг. Носій може сприяти певній просторовій орієнтації
субстрату біля активного центру каталізатора, що веде до підвищення
активності та селективності. Закріплення у ряді випадків дозволяє стабілізувати
нестійку структуру комплексу. Поверхня носія також певним чином
захищає активні центри металокомплексного каталізатора від дії отрути. Природу
поверхні носія можна попередньо змінювати шляхом її хімічного
модифікування (наприклад, модифікування неорганічного носія металорганічни-
ми сполуками). Для закріплення комплексів перехідних металів можуть бути
використані різноманітні функціональні групи (Ф) на поверхні носія, що
можна схематично зобразити таким чином:
де П — поверхня носія; Я — «якір» (група, яка відповідає за фіксацію на
поверхні частинки, що прищеплюється до неї); Н — «ніжка» (група, що
забезпечує зв'язок між функціональною групою та якорем). Внаслідок високої
реакційної здатності деяких металокомплексів вони можуть бути також
використані як прекурсори (попередники) різних поверхневих сполук.
Наприклад, відновлення поверхневих металорганічних комплексів
елементів VI групи веде до утворення низьковалентних станів перехідного
металу, що мають координаційну ненасиченість і є активними центрами
ряду реакцій, зокрема гідрування.
Таким чином, закріплені металокомплекси об'єднують переваги суто
гетерогенних каталізаторів із позитивними якостями гомогенних
металокомплексів. Однією з переваг прищеплених комплексів є можливість спільної
координації декількох реагентів та їх вигідної взаємної орієнтації, що дозволяє
долати як енергетичний, так й інформаційний бар'єри.
Особливий інтерес становлять закріплені багатоядерні, зокрема клас-
терні комплексні сполуки. За їх допомогою можна вивчати вплив кількості
атомів в активному центрі на каталітичну активність та селективність,
виявляти оптимальну кількість атомів. Таким чином, з'являється можливість
створювати складні високоактивні каталітичні системи, здатні конкурувати
з біокаталізаторами.
Закінчуючи розділи про каталітичні процеси, поглянемо на хімічну
реакцію «з точки зору каталізатора». Для каталізатора хімічний процес, яким він
управляє, є, в першу чергу, джерелом вільної енергії, за рахунок якої він
472
може існувати у нерівноважному метастабільному стані. Для каталізу
можна побудувати логічний ланцюг, аналогічний тому, що побудував
К. Маркс в економіці:
Економіка
1) Простий товарний обмін:
товар <-» товар
2) Товарно-грошовий обмін:
товар 4-> гроші <-» товар
3) Капіталізм:
гроші <-» товар <-» гроші*
Хімія
1) Некаталітична реакція:
реагенти <-> продукти
2) Каталітична реакція:
реагенти <-> каталізатор <-> продукти
3) «Існування каталізатора»:
каталізатор <-> хімічна реакція <->
каталізатор*
У товарно-грошовому обміні (2-й етап) гроші — це каталізатор обміну
товарів. На 3-му етапі гроші* — це зростання кількості грошей, і це є перехід
від товарно-грошового обміну до капіталізму. При цьому мета та засіб
міняються місцями, тобто розвиток відбувається шляхом опосередкування.
Аналогічно, перехід від некаталітичної реакції до каталітичної не змінює
мети — отримання цільового продукту. Якщо ж мета — це існування
каталізатора у метастабільному стані, то засіб підтримання його — це
використання вільної енергії хімічної реакції. При цьому каталізатор* — це
каталізатор у більш високоорганізованому стані, а також у більшій кількості, тобто
можна говорити про «розмноження» каталізаторів. Один з можливих
варіантів цього розмноження — це явище автокаталізу (див. підрозділ 16.2), коли
продукт є каталізатором.
Таким чином, життя, яке теж є існуванням високоорганізованих систем
у нерівноважному метастабільному стані, виникло не лише за допомогою
каталізу, але й через каталіз. Кожна жива істота, включаючи вінець
природи — людину, своїм існуванням зобов'язана каталізу, пам'яті ферментів-біо-
каталізаторів, які здійснюють усі найскладніші процеси в організмі,
черпаючи вільну енегію і необхідні матеріали з довкілля, і конструюють усі
необхідні для життя елементи організму. Звичайні хімічні каталізатори були
попередниками складних біокаталізаторів, і зараз біоміметика якраз і дає
можливість повчитися у живої природи творчо синтезувати моделі ферментів,
які будуть здатні конкурувати зі створеними еволюцією живої природи біо-
каталізаторами. Тут також варто нагадати, що прогрес живої природи
відбувається (як це визначалося у розділі 12) шляхом накопичення інформації
високоорганізованими системами, до яких, у першу чергу, відносяться фер-
менти-біокаталізатори (а далі: клітина, живий організм і, нарешті,
суспільство).
Детальне ознайомлення з кінетикою та механізмом ферментативного
каталізу виходить за межі підручника фізичної хімії і належить до біохімії.
Наступний розділ підручника присвячено дії різних фізичних факторів
на перебіг хімічних реакцій.
473
Розділ 19.
ХІМІЧНІ РЕАКЦІЇ ПІД ДІЄЮ ЗОВНІШНІХ
ФІЗИЧНИХ ФАКТОРІВ
Згідно з термодинамікою самочинно перебігають лише ті процеси,
зокрема хімічні реакції, для яких, згідно з принципом Гельмгольца-Нернста,
АС < 0. Реакції, для яких АС > 0, є «термодинамічно забороненими». Для
того щоб здійснився їх перебіг, необхідне джерело вільної енергії. Таким
джерелом може бути спряжена хімічна реакція, і цей випадок розглядався
у розділі 13.
Джерелом вільної енергії може бути також фізичний фактор. До цих
факторів відносяться: електрика, світло, радіаційне випромінювання, механічна
енергія тощо. Відповідно, маємо такі підрозділи фізичної хімії, як
електрохімія, фотохімія, радіаційна хімія, механохімія тощо.
Відзначимо, що до електрохімії традиційно відносять також розглянуті
вище розчини електролітів (розділ 4) та електрохімічну рівновагу (розділ 7).
Якщо ж розглядати лише дію фізичних факторів на хімічну реакцію (як для
фото-, механо-, радіаційної хімії), то слід обмежитися процесами
електролізу, що і зроблено у наступному підрозділі.
19.1. Електроліз, електродні процеси
Напругу дати ледве встигли —
До електродів іони побігли.
У гальванічному елементі (див. розділ 7) хімічна енергія
перетворювалася на електричну за рахунок самочинно перебігаючої хімічної реакції.
Наприклад, в елементі Рі (Н2) | НС11 (С12) Рі перебігає самочинно реакція
Н2 + С12 = 2НС1, для якої АС < 0 (відповідно, Е > 0). Для протилежної реакції
розкладу НС1 на елементи АС > 0, і вона є «термодинамічно забороненою».
Електрохімія, однак, дає можливість для перебігу таких заборонених
реакцій (наприклад 2НС1 = Н2 + С12) шляхом електролізу. При цьому
відбувається зворотний процес, а саме, електрична енергія перетворюється на
хімічну. Цей процес відбувається в електролізері, який є ємкістю з
відповідним розчином і зануреними у нього електродами, до яких прикладена
зовнішня різниця потенціалів (рис. 19.1). Наприклад, маємо розчин НС1 і
платинові електроди. Під дією напруги до позитивно зарядженого електрода—
катода — пересуваються позитивно заряджені іони — катіони Гідрогену, які
потім розряджаються на поверхні катода: Н + є —> /4Н2; до негативно
зарядженого електрода — анода, відповідно, прямують аніони Хлору, які
там розряджаються: СГ-е-ї УгС\2. Якщо анод зроблено не з такого
стійкого матеріалу, як платина, то він може розчинятись. Наприклад, при
електролізі розчину Си304 з мідними електродами анод розчиняється: Си - 2е
—»Си2+, відповідно, на катоді мідь осаджується: Си + + 2е —> Си . Цей про-
474
цес можна використати для очищення, рафінування міді; домішки, що були
в аноді, залишаються у розчині.
Рис. 19.1. Схема електролізу
Електроліз можна проводити не лише у розчинах, але й у розплавах. На
рис. 19.2 наведена схема електролізера Даунса, в якому можна отримувати
металічний натрій електролізом розплаву №С1.
Рис. 19.2. Електролізер Даунса для промислового отримання натрію:
1 — вуглецевий анод: СГ - є —»1ЛС12; 2 - залізний катод: № + є = №;
З — залізна перетинка, що перешкоджає контакту № та С12; 4 — розплав І^аСІ
Розглянемо процес електролізу. Відомо, що для провідників 1-го роду між
силою струму (/) та напругою (Е) існує прямо пропорційна залежність
згідно із законом Ома
де К — опір.
475
Напруга
Ця залежність наведена на рис.
19.3 (пряма 7). Реальна крива 2 (рис.
19.3) в координатах/—Еддя
процесу електролізу має зовсім інший
вигляд. При поступовому зростанні
напруги сила струму лишається
дуже малою величиною, крива 2 на
початковій ділянці майже не
відхиляється від осі абсцис. Лише після
досягнення певної величини
напруги {Ер), яка називається напругою
розкладу, струм починає зростати
і процес електролізу стає явним
(наприклад, при електролізі НС1
починається виділення Н2 на катоді й С12
на аноді). Причиною зменшення
сили струму при електролізі є паді-
Рис. 19.3. Залежність /—Е для
провідників першого (7) та другого (2) роду
ння напруги в електролізері внаслідок виникнення ЕРС, що діє в напрямі,
протилежному до прикладеної напруги. Ця ЕРС має назву ЕРС поляризації
(Еп). Наявність такої ЕРС можна проілюструвати таким простим
експериментом. Схема досліду показана на рис. 19.4. При замиканні контура І
(перемикач у положенні ГЕ) із зовнішнього джерела (В) струм проходить через
електролізер (А). Якщо через деякий час перемкнути перемикач у
положення /<Д то контур /буде розірвано, а в
контурі // гальванометр (С) покаже
наявність струму у напрямі, зворотному
відносно першого струму в контурі /.
Ясно, що після початку електролізу
електролітична ванна перетворилася на
гальванічний елемент, який деякий час дає
свій струм, що реєструється
гальванометром. Враховуючи ЕРС поляризації,
що виникає при електролізі, для сили
струму під час електролізу можна напи-
!7/ ІІЖІГ І /=^- (ш)
о
Рис. 19.4. Виникнення ЕРС
поляризації
При малих значеннях напруги (Е)
ЕРС поляризації майже повністю її
компенсує і згідно з (19.2) /= 0. Еп, однак
зростає лише до певної величини (Ер),
яка далі залишається сталою і це
дозволяє струму зростати при подальшому
зростанні Е.
476
Величини Ер при електролізі 1 н розчинів галоїдводневих кислот такі:
Др(нсі)= 1,31 В;£,р(нвГ)=0,94В;і^(ні)=0,52В.
Ці значення дуже близькі до значень ЕРС оборотних гальванічних
елементів, що складаються з водневого та відповідного галоїдного електродів.
Отже, межею зростання Еп є значення £об для певних гальванічних елементів.
Відзначимо, що в цих гальванічних елементах перебігають процеси, зворотні
тим, що відбуваються при електролізі (при електролізі — розклад НХ на Н2
та Х2, в гальванічному елементі — синтез НХ з Н2 та Х2). В свою чергу, Еоб
є мінімально можливим значенням для напруги розкладу 2? . Якщо Ер
більше, ніж Еоб, то ця величина називається перенапругою (ті)
ї\=Ер-Еоб, (19.3)
Причиною перенапруги може бути сповільнення деякої стадії процесу
електролізу. Якщо лімітуючою стадією є дифузія, доставка іонів, що
розряджаються, до поверхні електрода, то спостерігається явище
концентраційної поляризації. Розглянемо це детальніше на прикладі катодної поляризації.
Згідно із законом Фіка швидкість дифузії (г^) дорівнює
г^В-З'^гайс, (19.4)
де О — коефіцієнт дифузії, 5—площа перерізу (тут площа катода).
£ш^с = ^рЧ (19.5)
де с0 — концентрація електроліту в об'ємі розчину; сп — концентрація
електроліту біля поверхні електрода; / — товщина дифузійного шару (для
багатьох електролітів / за порядком величини дорівнює 0,05 см).
У стаціонарному стані швидкість дифузії дорівнює швидкості розряду (гр)
гР=ір = ^ 09-6)
де / — густина струму. Співставивши (19.4) та (19.6), з урахуванням (19.5)
маємо
/ = ^(с°"Сл)* (19/7>
Максимально можливий струм іа (граничне значення і) виникає при
сп -» 0, отже,
<</=іуЧ. (19.8)
Оборотний потенціал катода дається рівнянням Нернста
Фоб.=Ф°+|^1пс0. (19.9)
Реальний потенціал визначається концентрацією іонів біля поверхні
електрода (сп)
Фреал.=Ф°+^1ПСп. (19.10)
477
Відповідна різниця потенціалів дорівнює
Аф = Фреал.-фоб.=|^|-- 09.11)
З рівнянь (19.7) та (19.8) маємо
і- = ^—і. (19.12)
і, відповідно,
дф = Жіп^і. (19.13)
іР іа
При відсутності струму (/ = 0) Аф = 0.
При / —> ій | Аф| різко зростає (для катода це зсув до більш негативних
значень потенціалу), горизонтальна ділянка на /—ф кривій (рис. 19.5)
закінчується лише при досягненні Ер для якогось іншого катіона, якщо у розчині їх
декілька (у водному розчині завжди є можливість для виділення водню на
катоді). Наприклад, при електролізі розчину, що містить 2п804 та Си804,
спочатку при потенціалі катода (+0,34 В) починає виділятися мідь, після виділення
міді та зсуву потенціалу катода до більш негативного значення (-0,77 В)
починається виділення цинку.
Послідовні електродні
процеси використовують у
полярографії, де
застосовується ртутний крапельний
катод (поляризацією анода,
який має велику поверхню,
нехтують). Типові
«полярографічні хвилі» —
полярограму наведено на рис. 19.5
в і—ф координатах.
Потенціал точки перегину —
потенціал напівхвилі є харак-
0 0,2 0,4 0,6 0,8 1,0 1,2 1,4 1,6 1,8 тернОЮ ВЄЛИЧИНОЮ ДЛЯ
КОЖНОЇ речовини, що відновлю-
Рис. 19.5. Полярограма ється, і це можна
використати для аналізу цих речовин. Дані} наведені у таблиці 19.1, добре ілюструють
це положення.
Причиною перенапруги може бути не концентраційна поляризація, яку
можна зменшити шляхом інтенсивного перемішування розчину
електроліту, а електрохімічна поляризація. Якщо Ер=Еоб і Г| = 0, це свідчить про
практичну відсутність електрохімічної поляризації, цей випадок реалізується
у наведеному вище прикладі електролізу галогенводневих кислот на
платинових електродах. Інша картина спостерігається при електролізі більш
складних кислот, а також основ. Значення напруги розкладу для ряду таких
сполук наведено у таблиці 19.2.
478
12
10
8
6
/|
2
і, мка
\-£« і и
СогГ
С42у~>
*-УІ :
/1
і
ь
'
'2 1
1
, І*»Ч ,!^, .
У\
Ф,в
1 1
Таблиця 19.1. Полярографічні потенціали напівхвилі в ОД н розчині КС1
при 25 °С (потенціали вимірювались відносно насиченого
каломельного електрода)
Концентрація • 103
(моль/л)
0,1
0,2
0,5
1,0
2,0
5,0
1 ю,о
ТГ
-0,462
-
-0,457
-0,460
-0,456
-0,459
-
Іони
РЬ2+
-0,396
-
-0,396
-0,392
-0,397
-0,394
-0,398
Ссі2+
-
-0,594
-0,593
-0,594
-0,601
-0,598
-0,605
2п2+
-
-0,990
-0,989
-
-0,999
-0,992
-1,003
Кислота
Н3Р04
Н>Ю3
! н2зо4
ССІзСООН
НС104
Е,„ В
1,70
1,69
1,67
1,66
1,65
Основа
М(СН3)4ОН
>Ш4ОН
ИаОН
КОН
ЕР,В
1,74
1,74
1,69
1,67
Таблиця 19.2. Напруга розкладу Ер
водних розчинів кислот
та основ (на Р* електродах)
Звертає на себе увагу той
факт, що для різних кислот і
основ напруга розкладу має
майже однакове значення Ер~\,1 В.
Єдиним процесом, що може
бути загальним для всіх цих
водних розчинів, є
електрохімічний розклад води, який складається з розряду протонів на катоді з
наступним виділенням молекулярного водню і розряду іонів гідроксилу на аноді
з наступним виділенням молекулярного кисню. Справді, у кожному з
наведених у таблиці 19.2 експериментів проходження струму супроводжується
виділенням газоподібних Н2 та 02. Для оборотного воднево-кисневого
елемента Еоб = 1,23 В, тобто спостерігається значна перенапруга (ті = 0,47 В)
внаслідок електрохімічної поляризації. Оскільки при електролізі галоїдвод-
невих кислот на Рї-електродах, як зазначалось вище, перенапруга мала
(ті = 0), то висока поляризація для електролітів, що наведені у табл. 16.2,
пов'язана з виділенням кисню. Цікаво відзначити, що при відсутності
поляризації при Ер = Еоб = 1,23 В з розчинів НС1 повинен був би спочатку
виділятися саме кисень, а не хлор, для якого Ер = Еоб = 1,3 В. Більш простий,
одноатомний іон СГ таким чином, розряджається з розчину легше, ніж більш
складний, двохатомний ОН" іон.
Електрохімічна поляризація складається з поляризації катодної та
анодної. Наприклад, при електролізі води (в розчинах кислот або лугів) основний
внесок у загальну поляризацію вносить поляризація анодна (виділення
кисню). На Рі катоді перенапруга виділення водню дуже мала, але для електродів
з іншого матеріалу вона може бути помітною. Причиною електрохімічної
поляризації є гальмування загального процесу на електрохімічних або
хімічних стадіях. Вважаючи, що доставку іонів до поверхні електрода дифузійним
479
шляхом забезпечено, розглянемо це на найбільш детально вивченому процесі
катодної поляризації при виділенні водню.
Дж. Тафель показав, що перенапруга виділення водню не є константою,
а залежить від струму (і) при не дуже малих / таким чином
х\ = а + Ь\&. (19.14)
У рівнянні Тафеля параметр Ь є приблизно константою для багатьох
електродів і при температурі 20 °С дорівнює у середньому 0,116 В. Параметр
а визначається природою металу електрода, що підтверджується даними,
наведеними у таблиці 19.3 і на рис. 19.6.
Таблиця 19.3. Значення параметрів а і Ь
рівняння Тафеля при
катодному виділенні водню
на різних металах при 20 °С
Одне з найменших значень
а і, відповідно, ц, має
платина. Ер = Еоб саме тому, що
при електролізі галогенвод-
невих кислот на платинових
електродах Г| є малою
величиною.
Обґрунтовуючи своє
рівняння, Дж. Тафель вважав, що
лімітуючою стадією процесу
електрохімічного виділення
водню є молізація —
рекомбінація сорбованих на поверхні
електрода атомів Гідрогену,
що з'явилися при
розряді Н іонів, у молекулу
водню з наступним
виділенням Н2 у газову фазу.
Безпосереднє
вимірювання швидкості
рекомбінації (грек) атомів
Гідрогену на поверхні
металу показало, що вели-
Метал
Свинець
Ртуть
Цинк
Олово
Мідь
Залізо
Нікель
Кобальт
Паладій
Вольфрам
Платина
а
1,51
1,40
1,24
1,24
0,79
0,77
0,64
0,62
0,64
0,55
0,10
Ь
0,118
0,116
0,116
0,116
0,117
0,130
0,100
0,140
0,125
0,110
0,130
а
в
се
X
а>
о«
с
1,2
1,0
0,8
0,6
0,4
0,2
0
'т
?ь.
У,
с
у
У Аи
?Іу
А§
0
-7 -6 -5 -4 -3 -2 -1
1§ і, а/см2
Рис. 19.6. Перенапруга водню на металах
чина грек збільшується у
ряду:РЬ = Н§<Ре <Р(1<
< Рі, що чітко корелює
зі зменшенням
перенапруги виділення водню
(див.табл. 19.3, значення
параметра а).
480
Рекомбінація є бімолекулярним процесом по відношенню до
поверхневої концентрації (А^н) атомів Гідрогену, отже, можна записати
Гра,=ШІ (19.15)
У стаціонарному стані швидкість рекомбінації дорівнює швидкості
розряду (Гр0зр.), яка виражається через густину струму (і)
'розр.=4:- 09.16)
•розр. 2р-
3(19.15) та(19.16) маємо
стац.
(*н)с
УгРк]
(19.17)
Для рівноважного оборотного електрода всі стадії рівноважні, отже,
адсорбований водень знаходиться у рівновазі з молекулярним воднем газової
фази, а також з іонами Гідрогену у розчині. Якщо рекомбінація є повільною
стадією, відповідно, розряд — швидкою, то рівновага між адсорбованим
воднем і іонами Гідрогену зберігається, а рівновага між адсорбованим
воднем і молекулярним газової фази порушується, отже, (Л^н)стац Ф (Л^н)рівн.-
За рахунок різниці концентрації між (ІУН )стац і (ІУН )рівн виникає різниця
потенціалів на електроді, яка і обумовлює перенапругу
Л рш[п]а ' (19л8)
1- п -фівн.
Оскільки [#н ] . є константою, то (19.18) можна переписати таким
чином:
Л = сотҐ + ^Іп[Ми]стац . (19.19)
Підставляючи в (19.19) вираз [Л/ц ] з (19.17), маємо
ц = соп5Ґ+^Іпі. (19.20)
Переходячи до десятинних логарифмів, маємо
Л = в + 2^г18»=в + *І8і'- (19.21)
Таким чином, рекомбінаційна теорія приводить до рівняння Тафеля
(19.14), але обчислення коефіцієнта Ь при 20 °С дає величину 0,029 В, що
вчетверо менша від експериментальної.
Недоліки рекомбінаційної теорії змусили повернутися до розряду іонів
Гідрогену як лімітуючої стадії. Хоча думка про те, що ця стадія може бути
повільною, висловлювалася давно, це здавалося малоймовірним, бо що, на
перший погляд, може заважати електрону приєднатися до протона? Т. Ердей-
Труз і М. Фольмер довели, однак, що розряд протонів потребує значної
енергії активації і не в останню чергу це пов'язане з тим, що протон міцно
зв'язаний у іон гідроксонію Н30 .
481
Для швидкості електрохімічного процесу можна написати звичайне
кінетичне рівняння
грозР- Н30+ •
(19.22)
Враховуючи зв'язок між грозр і густиною струму (19.15), а також
температурну залежність константи швидкості, маємо
грозр. гГ К Є Сн30+
І ■■
(19.23)
Для розчину сталого складу с + = сопзі, отже, (19.23) можна записати
таким чином:
і = С-е-ЕІКТ, (19.24)
де С—деяка константа.
Роль напруги зводиться до зниження енергії активації (Е0) процесу
розряду
Е = Е0-аг\Е9 (19.25)
де Е0— енергія активації для неполяризованого електрода; а — коефіцієнт
співвідношення лінійності, типу того, що застосовувався в однотипних
реакціях (див. розділи 14,16,17,18).
2 Співвідношення (19.25), як і
інші співвідношення подібного
типу (Бренстеда-Поляні), можна
обґрунтувати методом
потенціальних кривих. Нарис. 19.7
крива 2 відповідає потенціальній
кривій адсорбованого атома
Гідрогену, а крива 1 — іону гідро-
ксонію. Поляризація катода
призводить до зсуву кривої 1
(крива Ґ) на величину цЕ, що веде до
зниження енергії активації на
величину аг\Е. Для симетричного
бар'єра а = 0,5. Крива 2 при цьому залишається на місці, тому що атом
водню незаряджений і поляризація не впливає на енергію зв'язку Ме—Н.
Підставляючи (19.25) у (19.24), маємо
/ = С-е-ЕІКТ =С-е-Е°Ікте^РІКТ. (19.26)
Рис. 19.7. Потенціальні криві іона гідроксо-
нію (7) і сорбованого атома Гідрогену (2)
Розв'язання цього рівняння відносно х\ дає
ТУТ1
1 аР
(19.27)
Переходячи до десятинних логарифмів, отримуємо рівняння Тафеля
Л = а + ар?231&і = а + Ь]&і. (19.28)
482
Оскільки типовим значенням для коефіцієнта а є 0,5, то для параметра Ь
маємо при І = 20 °С значення 0,116 у доброму узгодженні з
експериментальним.
Вплив природи металу на перенапругу визначається енергією активації
(Е0) неполяризованого електрода, яка входить до константи а рівняння Та-
феля. Справді, перехід до металів, які міцно хемосорбують водень (Реї, Рі),
веде до зміцнення зв'язку Ме—Н, отже, крива 2 зміщується вниз (крива
2'), що веде до зменшення енергії активації (Е0) і, відповідно, до зменшення
параметра а. Це пояснює малу перенапругу на металах, які добре
хемосорбують водень.
Навпаки, на металах, які погано хемосорбують водень або зовсім його
не хемосорбують, маємо високі значення перенапруги водню. Це можна
використати у технологічних цілях. Наприклад, завдяки високій перенапрузі
водню на ртуті при електролізі водного розчину ИаСІ на ртутному катоді
виділяється не водень, а металічний натрій, який розчиняється у ртуті,
утворюючи амальгаму.
Дію електричного струму можна використати не лише для електролізу
(«ліз» — розкладання, розпад сполук), але й для електрохімічного синтезу
в гесіох реакціях. Наведемо декілька прикладів.
Одним з найкращих відновників є катод, причому, чим більший (за
абсолютною величиною) катодний потенціал (ф^), тим більша відновлююча
здатність електрода. Наприклад, при відновленні нітробензену маємо такі
продукти: ф^= (-0,6 В) — в основному азоксибензен; ф^= (-1,0 В) — з'являється
гідроазобензол; ф^=(-1,5 В)—до ЗО % аніліну. При цьому слід врахувати, що
електрод, крім того, що надає електрони, може впливати на загальний
перебіг процесу, діючи як каталізатор. Наприклад, на мідному та свинцевому
катодах не перебігає процес електролітичного відновлення подвійного зв'язку,
який з легкістю здійснюється на платиновому та нікелевому електродах.
Відомо, що Рі та №, на відміну від Си та РЬ, є хорошими каталізаторами гідрування.
Анодне окиснення реалізується в основному на Рі електродах, які є
електрохімічно інертними, на відміну від багатьох інших металів, які при
збільшенні потенціалу анода починають розчинятися. При збільшенні
анодного потенціалу (ф^) збільшується вихід більш окисненої форми продукту.
Наприклад, при окисненні етанолу при (рА < 0,9 В утворюється тільки аце-
тальдегід, при (рА = 1,0 В — 85 % ацетальд егіду, 15 % оцтової кислоти, при
збільшенні (рА до 1,5—1,7 В вихід ацетальдегіду зменшується до 40 %, а
оцтової кислоти, відповідно, зростає до 60 %.
На аноді можна здійснювати процеси димеризації аніонів, а саме:
сульфату в персульфат: 28О42 = $2®1 + 2е;
сульфіту в дитіонат: 280з~ = §2^6" + 2е;
тіосульфату в тетратіонат: 282Оз~ = З^б" + ^е'
Цікавою є електрохімічна реакція Кольбе (на прикладі ацетату)
2СН3СОО" = С2Н6 + 2С02 + 2е,
483
в процесі якої окиснення КСОО" аніона до С02 дає можливість рекомбіну-
вати групам СН3 з утворенням етану. За допомогою реакції Кольбе можна
отримати з солей аліфатичних кислот алкани аж до С34Н70. Вважається, що
механізм реакції Кольбе складається з розрядження на аноді не відносно
складного ацетат-іона, а більш простого — гідроксил-іона
ОН~->ОН#+е
з наступними вже суто хімічними процесами:
СН3СОО" +ОН* -> ОН" + СН3СОО#,
2СН3СОО# -> 2С02 +С2Н6.
Схожим за механізмом з реакцією Кольбе є електросинтез Брауна-Воке-
ра — синтез складних 2-основних кислот із більш простих. Сумарний
анодний процес реакції Брауна-Вокера такий:
2(СН2)„-СОО-С2Н5-> (СН2)„-СОО-С2Н5
І _ І + 2С02 + 2е
СОО (СН2)„—СОО—С2Н5
Продуктом є естер 2-основної кислоти з подвійною порівняно з
вихідною кислотою кількістю (СН2)-груп. Реакція Брауна-Вокера дозволила
синтезувати (через естери) 2-основні кислоти, що містять до 32 (СН2)-груп.
19.2. Реакції у розрядах — газова електрохімія
Температуру і напругу дали газу,
Перетворивсь на плазму він одразу.
Електричний струм може впливати на хімічні процеси не лише у
системах, які є провідниками 2-го роду (розчини електролітів, іонні розплави), але
й у діелектричній газовій фазі. У газі під дією зовнішніх іонізаторів (світла,
радіоактивного опромінювання тощо, див. далі підрозділи 19.3; 19.4)
виникають іони, і тоді між електродами з'являється електричний струм — це
явище має назву несамостійного розряду, який зникає, коли припиняється дія
зовнішнього іонізатора.
При високих значеннях напруги градієнт потенціалу «пробиває»
заповнений газом простір, і виникає самостійний розряд, у якому заряджені
частинки утворюються шляхом лавиноподібної іонізації, і зовнішній
іонізатор уже не потрібен. Самостійні розряди бувають різних видів. При
потужному джерелі струму, відносно невеликому опорі у зовнішньому ланцюзі
й достатньо високому тиску (атмосферний і вище) виникає дуговий розряд
(при не досить потужному джерелі струму замість дугового утворюється
іскровий розряд). При низьких тисках (декілька мм рт. ст.) і достатньому
опорі зовнішнього ланцюга виникає тліючий розряд. У різко неоднорідних
484
полях на електродах малого радіуса кривизни (наприклад, тонкий дріт
усередині коаксіального циліндра) у відносно тонкому шарі біля дроту, де
градієнт потенціалу достатньо високий, виникає коронний розряд (світло —
«корона» лише біля дроту) або взагалі без висвічування — темний, тихий
розряд. У цьому випадку розряд є самостійним лише біля дроту, а в іншій
частині об'єму він є несамостійним та існує за рахунок дифузії іонів із зони
самостійного розряду.
Перелічені вище види розрядів реалізуються під дією постійної
електричної напруги. Крім цього, газові розряди можуть виникати і під дією змінного
електричного поля — це високочастотний (ВЧ) розряд (та його різновид —
бар 'єрний розряд).
У розрядах газ знаходиться у частково іонізованому стані. Для
несамостійного розряду суттєвий внесок в іонізацію газу дає фотоіонізація
(іонізація під дією випромінювання). У самостійному розряді іонізація
відбувається, в першу чергу, за рахунок ударного механізму: електрони та іони у
полі високої напруги набувають великої кінетичної енергії і спроможні при
зіткненнях іонізувати нейтральні частинки; катіони також здатні вибивати
електрони з катода. Великі кінетичні енергії частинок відповідають
підвищеній температурі, отже, можлива термічна іонізація, а також
термоелектронна емісія електронів з поверхні катоду. Нарешті, у полях високої напруги
створюються умови для виходу електронів із нейтральних частинок за
рахунок тунельного ефекту — процес автоіонізації.
Стан речовини у частково або повністю іонізованому вигляді називається
плазмою. Цей стан можна досягти не лише у розряді під дією електричного
поля, але й шляхом нагрівання речовини до високої температури. Так,
нагрівання твердого тіла веде до його топлення, а далі до повного розриву
хімічних зв'язків і переходу до газового стану. При подальшому нагріванні газу
кінетична енергія частинок збільшується до величин, що співрозмірні з
енергією іонізації, отже, газ починає іонізуватися й переходить у стан плазми.
Розрізняють «низькотемпературну» (Т = 10 К) та «високотемпературну»
(Г= 10 —10 К) плазму. У високотемпературній плазмі газ повністю
іонізований, у цьому стані знаходиться значна частина речовини Всесвіту, що
зосереджена у зірках. Низькотемпературна плазма частково іонізована, вона
складається з іонів (переважно катіонів), електронів, атомів та молекул,
значна частина яких перебуває у збудженому стані. Саме у стані
низькотемпературної плазми знаходиться речовина у розрядах. Середні енергії
частинок різного типу можуть відрізнятися одна від одної. Відповідно,
розрізняють електронну температуру — Те, іонну температуру — Т( та
температуру нейтральних частинок — Та. Якщо Те= Т{ = Тф то плазма називається
ізотермічною. Таку плазму можна отримати шляхом нагріву газу до високої
температури, вона характерна для стану речовини у дуговому розряді. Для
деяких видів розрядів (тліючий, коронний тощо) кінетична енергія електронів
набагато перевищує кінетичну енергію іонів та нейтральних частинок (Те>>
» Ть Та), отже, ця плазма називається неізотермічною.
485
Розглянемо термодинамічні та кінетичні критерії перебігу хімічних
реакцій у розряді — низькотемпературній плазмі. Згідно з принципом Ле-Ша-
тельє (див. підрозділ 6.2) зі збільшенням температури хімічна рівновага
зміщується у бік ендотермічних процесів. Отже, у першу чергу, у
низькотемпературній плазмі слід проводити сильно ендотермічні процеси. До таких
реакцій належить синтез оксиду азоту з простих речовин. Цей процес є прямим
шляхом зв'язування атмосферного азоту (у природі він реалізується у
грозових розрядах), застосування його у промислових масштабах обмежується
лише високими енерговитратами. Практичне застосування знайшов сильно
ендотермічний процес електрокрекінгу метану з утворенням ацетилену
в дуговому розряді: 2СН4 = С2Н2 + 2Н2. Реакція відбувається при
температурі -1600 °С, для запобігання термічній дисоціації ацетилену і водню
реакційну суміш після проходження розрядного простору різко охолоджують до
150 С. Ендотермічним є процес синтезу озону з молекулярного кисню,
який реалізується у бар'єрному ВЧ-розряді.
З кінетичної точки зору, основним є механізм активації частинок у
розряді для участі їх у хімічній реакції. Первинним є процес іонізації. Самі по
собі іони не завжди є реакційно здатними частинками. Наприклад, якщо при
іонізації молекули азоту електрон знімається із зв'язуючої орбіталі, й
міжатомний зв'язок в іоні N2 набагато слабкіший, ніж у молекулі N2, то при
іонізації 02 електрон знімається з антизв'язуючої орбіталі й міжатомний
зв'язок у С>2 міцніший, ніж у 02 (див. підрозділ 9.1). Основну роль в активації
частинок відіграють вторинні процеси, в яких іони шляхом непружних
зіткнень віддають енергію, яку вони набрали, «розігрівшись» в електричному
полі. При цьому утворюються атоми, радикали, частинки у збудженому
стані тощо. Активні частинки можуть утворюватися і при безпосередньому
зіткненні з електронами. Наприклад, К2 + є = N2 + є.
Якщо при цьому один з атомів має велику спорідненість до електрона,
то утворюються іон і атом, наприклад:
е+Х2-*Х" + Х" або
е + НХ->Х"+Н#,
де X — атом галогену.
Суттєву роль при цьому можуть відігравати так звані удари другого
роду, при яких електронозбуджена частинка при зіткненні передає свою
енергію, яка перетворюється на інший вид енергії (наприклад, сприяє
дисоціації молекули на атоми). Це явище має назву сенсибілізації (тоді
використовують термін «енергетичний каталіз»). Так, при розряді у суміші аргону
та кисню відбувається процес
Аг*+02->Аг + б + 6.
Присутність пари ртуті сенсибілізує дисоціацію Н2, знижуючи порогову
енергію іонізуючих електронів з 11,4 до 7,7 еВ.
Утворення озону з молекулярного кисню перебігає через стадії:
486
02+е-»0+0+е,
02+0 + М-
>03+М
(в)
(б)
(частинка Мзабирає надлишок енергії).
Сенсибілізуючу дію дає присутність молекулярного азоту:
Щ+е->Н*2+е, (в)
К2+02 ->М2+6 + 6. (г)
Стадії (а) та (в) є важливими і при синтезі N0 з И2 і 02, який далі перебігає
за схемою:
^2+6->Ш + ^, (д)
^+02->Ш + 6. (є)
Суттєво, що для цієї реакції введення у реакційний простір інертного
твердого наповнювача різко зменшує вихід N0 за рахунок «гасіння»
активних частинок на поверхні твердого тіла.
Розглянемо тепер реакцію синтезу аміаку. Це екзотермічна реакція, отже,
проведення її в ізотермічній плазмі (дуговому розряді) при високій
температурі неможливе, бо термічно нестійкий аміак відразу розкладається на азот
та водень. Синтез аміаку, однак, можна здійснювати у неізотермічній плазмі,
тобто в коронному (тихому) або тліючому розрядах, де температура
частинок (Ті9 Та) відносно низька, а велика лише електронна температура, і це веде
до збудження молекул шляхом електронного удару. Суттєвим є те (як і при
гетерогенно-каталітичному синтезі N113, див. підрозділи 18.1; 18.2), що
основна задача при синтезі аміаку —
це активація N2. Виявилося, що аміак
синтезується при пропусканні через 0 2
розрядний простір азото-водневої
суміші або лише азоту, який потім
змішується з неактивованим воднем.
Активація у розряді лише водню не
веде до утворення аміаку. Виявилося
також, що додавання у реакційний 0,1 |-
простір інертного наповнювача веде
до збільшення виходу аміаку при
синтезі N113 у коронному (тихому)
розряді при атмосферному тискові.
Цікавим виявився результат
синтезу N113 при атмосферному тиску
при спільній дії коронного (тихого)
розряду і гетерогенного (на основі про-
мотованого заліза, див. підрозділ 18.2)
каталізатора. Ці дані наведено на рис.
19.8. При відносно низьких (до 200 °С)
200
400
600
Рис. 19.8. Спільна дія розряду
та каталізатора на процес синтезу
аміаку: 1 — (О) - розряд; 2 — (□) —
каталізатор; 3 — (V) — загальна дія
розряду та каталізатора
487
температурах вихід >ЇН3 обумовлений синтезом у розряді (крива 7).
Відзначимо, що вихід аміаку в розряді в цьому температурному інтервалі не
залежить від температури завдяки високій електронній температурі, яка
забезпечує цей процес. При високих (> 400 °С) температурах практично весь
аміак синтезується на гетерогенному каталізаторі (крива 2). У певному
температурному інтервалі (200—400 °С), де виходи №13 у розряді та на
каталізаторі одного порядку, спостерігається антагонізм (крива 3). Ці явища
можна пояснити за допомогою наступного механізму, в якому вирішальна роль
відводиться активним молекулам азоту:
Ь12+е~>ЬІ*2+е,
Ц+Н2-»2№і\
Ш*+Н2->Шз,
Ш;+[]->[Ш3],
[ЛН3]-»НН3+[],
(а)
(б)
(«)
(г)
(д)
де [] — адсорбційна ділянка на поверхні твердого тіла. Стадія (б) може
включати також активацію водню, тобто
Н2+е->Н2+е, (б') ]
ї^ + Н^гШ*. (б")\
Збільшення виходу аміаку при введенні інертного твердого тіла
обґрунтовує наявність стадій (г) та (д) для «гасіння» збудженої молекули аміаку.
При високих (> 400 °С) температурах каталізатор достатньо активний в
реакції синтезу ИН3 з азоту та водню. У певному температурному інтервалі
(200-400 °С) каталізатор ще недостатньо активний для синтезу ИН3 з N2 та Н2,
але вже може розкладати збуджені молекули аміаку, тобто замість стадії (г)
маємо:
КН3+2->2МН + Н2, (є)
22КН->22 + М2+Н2, (є)
де 2 — активний центр поверхні каталізатора.
Наведений механізм підтвердився при вивченні реакції розкладу аміаку:
ш3+е-->нн3+е, (ж)
Ш3->НН*+Н2, (з)
Ш*+Ш1* ->К2+И2. (")
Введення твердого інертного тіла при цьому повинно гальмувати
процес за рахунок дезактивації збуджених молекул №І3 на поверхні сорбенту
(стадія (г)). Це спостерігалося в експерименті. З іншого боку, у певному
температурному інтервалі (400—425 °С) спостерігався синергізм дії розряду
і каталізатора за рахунок розкладу ]ЧН3 на активних центрах каталізатора
(стадії (є) та (є)).
488
Нарешті, якщо при реакції синтезу аміаку зменшувати тиск, то
збільшується довжина вільного пробігу, і активна молекула N2, не встигнувши
прореагувати з воднем, гаситься на стінці. Тоді введене інертне тіло гальмує
утворення продукту (>Шз) у розряді, а при попаданні на поверхню
каталізатора N2 швидше розкладається на атоми, ніж неактивна молекула И2, отже,
при спільній дії розряду і каталізатора антагонізм повинен поступатися
місцем синергізму. Цей сценарій реалізується при синтезі аміаку у тліючому
розряді при низьких тисках азотоводневої суміші.
19.3. Фотохімія
Частинка десь фотона прихопила,
Активувалась і в реакцію вступила.
Фотохімія вивчає вплив на хімічні реакції електромагнітного
випромінювання в області видимого та УФ-спектра. Саме у цій області енергія
фотонів сягає величин енергій хімічних зв'язків та електронних переходів у
молекулах та атомах. Кванти з меншою енергією (інфрачервоні та
радіохвилі) не мають специфічного впливу на хімічні реакції і діють як теплове
опромінювання (збудження поступальних, обертальних та коливальних
ступенів вільності). Дію на речовину більш високо енергетичних
рентгенівських та у-квантів вивчає радіаційна хімія (див. наступний підрозділ).
Відповідно до закону Ламберта-Бера при проходженні через шар
речовини товщиною (/) і концентрацією (с) інтенсивність електромагнітного
випромінювання зменшується з початкової (І0) до певного значення на виході (І)
І = І0е-ш. (19.29)
Кількість енергії, що поглинається,
А/= /„-/ =/„(1-е-**), (19-30)
а відповідна кількість поглинених (за одиницю часу) фотонів дорівнює
/о0^£—)
Згідно із законом фотохімічної еквівалентності Штарка-Ейнштейна
кожному поглиненому кванту відповідає один елементарний хімічний акт (одна
змінена частинка), отже, для суто фотохімічних, первинних процесів
величина я, повинна визначати швидкість фотохімічної реакції (г). З
урахуванням можливості вторинних процесів маємо
г=Фм/0(і-^)
Ну
де Фм — квантовий вихід фотохімічної реакції за речовиною М, тобто
відношення кількості частинок, що утворилися (за продуктом) або прореагували
(за реагентом), до кількості поглинених квантів.
489
Для тих реакцій, у яких вторинні процеси несуттєві, Фм =1. Прикладом
може слугувати фотоліз ацетону, для якого Фсо = 1 (відповідно, Фацетону =
= 1). У цьому випадку монооксид карбону утворюється у первинній
фотохімічній реакції
сн3сосн3 —^->со+ген;
і, оскільки він є молекулою стабільною, то у подальших перетвореннях участі
не бере.
Випадок Ф < 1 відповідає дезактивації збуджених частинок. Наприклад,
для реакції фотохімічного розкладу аміаку маємо Ф = 0,2-*-0,5, що
пояснюється таким механізмом:
НН3-^->НН2+Н#, (а)
Ш2+Н#->Ш + Н2, (б)
Ш + Ш->Щ+К2, (в)
Ш2+ЬҐ->Ш3. (г)
Тут із реакціями (б) та (в), які ведуть до утворення продуктів (ТМ2та Н2),
конкурує реакція (г), яка шляхом рекомбінації відтворює вихідну речовину
(Ш3).
Значення Ф < 1 часто спостерігається у розчинах за рахунок дезактивації
фотохімічно збуджених частинок молекулами розчинника або внаслідок
«коміркового ефекту» (див. підрозділ 15.3). Ф < 1 також типове для реакцій
у газовій фазі при малих тисках за рахунок дезактивації збуджених частинок
на поверхні стінок реактора (довжина вільного пробігу (X) тут є достатньо
великою величиною) або процесів флуоресценції (випромінювання квантів
при переході із збудженого стану в основний) (велике значення X відповідає
збільшенню часу між зіткненнями і це зменшує ймовірність хімічного
перетворення фотохімічно збудженої молекули).
Поширеним є випадок, коли Ф > 1 і дорівнює невеликому числу, тобто
є одна чи декілька вторинних стадій, що ведуть до утворення продукту
(приклади розглянуті далі).
Нарешті, є випадки, коли Ф » 1. Це реалізується, якщо вторинним є
ланцюговий процес із великою довжиною ланцюга, прикладом може бути
розглянута у підрозділі 15.2 ланцюгова реакція взаємодії водню з хлором або
ланцюгові реакції радикальної полімеризації (підрозділ 15.3).
Для отримання інформації про квантовий вихід із рівняння (19.32)
необхідні дані про абсолютну інтенсивність (І0) падаючого монохроматичного
світла. /0 вимірювати важко, тому користуються методом хімічної
актинометрії. Система, яку при цьому використовують (так званий хімічний
актинометр), повинна мати надійно встановлене значення квантового виходу
і можливість легко вимірювати концентрації продуктів фотолізу.
Визначивши І0 через хімічний актинометр, знаходять далі Фм для фотохімічної
реакції, що вивчається.
490
Далі наведено приклади
фотохімічних реакцій з
розглядом первинного
фотохімічного акту і вторинних
процесів. Для двохатомних
молекул відносно легко
можна побудувати потенціальні
криві. Для молекули НІ ці
криві зображені на рис. 19.9.
Крива / відповідає
основному стану, а криві 2 та 5 —
збудженим (молекула НІ
майже ковалентна, і ці
потенціальні криві мають таку ж
природу, що і у молекулі Н2,
див. підрозділ 9.1 та рис. 9.3).
При термічній активації
збуджуються коливальні стани,
що розташовані в
потенціальній ямі кривої 7, і
відбувається дисоціація НІ на
атоми. При фотохімічній дисоціації енергія кванта повинна бути більшою за
енергію дисоціації молекули (тут НІ). При цьому відбувається перехід (а) із
основного стану в збуджений (крива 2, яка у цьому випадку не має
мінімуму, крива «відштовхування»). Отже, відбувається фотохімічний розклад
молекули НІ на атоми (в основному стані). «Залишкова» (порівняно з
енергією дисоціації) енергія проявляється як кінетична енергія поступального
руху продисоційованих атомів. При збільшенні енергії кванта може
відбуватися перехід (Ь) на криву 3, в цьому випадку утворюється метастабільний
атом Йоду (терм Р«/2) (оцінки показують, що при фотохімічній дисоціації
кількість таких збуджених атомів Йоду може досягати 10 %).
Квантовий вихід фотохімічної дисоціації НІ сягає двох. Крім первинного
процесу фотодисоціації
НІ-^->Н#+Ґ, (а)
відбуваються такі реакції:
Н#+І2->НІ + Ґ, (б)
Ґ+Ґ+М->І2+М'. (д)
Отже, на 1 квант опромінювання загалом перетворюються 2 молекули
НІ. Відзначимо, що, на відміну від досить екзотермічної реакції (б), процес
Ґ+Н2->НІ + Н# (г)
є дуже ендотермічним, відповідно, енергія активації його є досить великою
величиною, отже, він не перебігає.
6,0
4,0
2,0
0,0
кЕ, еВ
и \-а\
Р4^
І4^
1 "^^ "
/
н + і(2рі/2)
Н + і(2Л/2)
І
о
& 4 *.
0,2
0,4
г, нм
Рис. 19.9. Потенціальні криві молекули НІ:
1 — основний стан (терм); 2 — збуджений
стан (при дисоціації атом Йоду в основному
стані); 3 — збуджений стан (при дисоціації
атом Йоду в збудженому стані) (за нуль
прийнято енергію мінімуму потенціальної
кривої основного стану)
491
Схожий механізм і квантовий вихід 2 за реагентом має реакція фотолізу
нітрозилхлориду:
N001—&-»ш + СҐ, (а)
СҐ + N00 ->Ш + С12. (б)
Залишок енергії електронного збудження над енергією зв'язку N—СІ
перетворюється на енергію поступального руху фрагментів, а також на
збудження коливальних рівнів N0.
Подібна до НІ та М)С1 картина первинного акту фотохімічної дисоціації
характерна для ковалентних молекул, де атоми зв'язані простим а-зв'язком.
Справді, в цьому випадку при фотохімічному збудженні відбувається а—>а*
перехід (див. підрозділ 9.1), і кількість електронів на зв'язуючих та антизв'я-
зуючих молекулярних орбіталях стає однаковою, отже, молекула мусить
розкластися на атоми.
Для двохатомних молекул із кратними зв'язками, наприклад, при п—>тс*
або п-їп* переходах, а-зв'язок залишається, отже, у збудженому стані
молекула відносно стабільна, і її потенціальна крива має мінімум, хоча не такий
глибокий, як для основного стану, і зміщений у бік більших міжатомних
відстаней. Ця ситуація у загальному вигляді зображена на рис. 9.14 (підрозділ
9.1). Навіть якщо при фотохімічному збудженні перехід відбувається в
основний коливальний стан електронозбудженої частинки (рис. 9.14, а\ це
послаблює міжатомний зв'язок, перехід у коливально-збуджений стан (рис. 9.14, б)
посилює тенденцію до дисоціації молекули, нарешті, у випадку,
зображеному нарис. 9.14,в, відбувається фотодисоціація молекули.
Конкретним прикладом є молекула кисню, потенціальні криві для
основного та збудженого станів якої зображено на рис. 19.10. При цьому може
реалізуватися так звана предисоціа-
£, еВ
О^іЧ+СН1/))
0(3/>) +0(3/>)
г, нм
Рис. 19.10. Потенціальні криві
молекули 02:1 — основний стан;
2,3 — метастабільні збуджені стани;
4 — «дисоціативний» збуджений стан
ція. Ця ситуація виникає тоді, коли з
потенціальною кривою 3 з
мінімумом, що описує метастабільний
стан, перетинається (у точці «с»)
«дисоціативна» крива 4. Тепер при
переході («6») у
коливально-збуджений стан, що знаходиться вище
точки «с», стає можливим нерезонанс-
ний перехід з кривої 3 на криву 4
з дисоціацією молекули кисню. При
достатній енергії кванта може
відбуватися процес прямої дисоціації.
Отже, вважаючи, що первинним
актом фотохімічного перетворення
кисню в озон є дисоціація, маємо
/IV
о,
->0 + 0.
(а)
492
Далі атом Оксигену взаємодіє з наступною молекулою кисню з
утворенням молекули озону:
6 + 02+М->03+М'. (б)
Квантовий вихід за озоном (продуктом) дорівнює 2 (відповідно, за
реагентом — киснем — Ф = 3).
За рахунок фотохімічних процесів
під дією сонячного випромінювання
в атмосфері з кисню повітря
утворюється озоновий шар (див. рис.
19.11). Цей шар захищає Землю від
зайвого УФ-випромінювання за
рахунок фотохімічних перетворень озону:
10"
10і
ю'Ч
03
03+6-
->0 + 02,
->20..
(в)
(г)
ю1Ч
ю'
10у
с0з (молекул/см3)
Баланс утворення і зникнення
озонового шару може порушуватися
за рахунок каталітичної дії деяких
сполук, серед яких важливу роль
відіграють оксигенові сполуки нітрогену.
Наприклад, відбувається такий
каталітичний цикл:
Ж) + 03->Ж)2+02
6 + К02->Ж) + 02
10 20
30 40
50 60
висота, км
Рис. 19.11. Залежність концентрації
озону в повітрі від висоти над рівнем
моря
03+0-
20,
Додатковий внесок дає реакція фотохімічного розкладу >Ю2
Ш2—^N0 + 6.
Оксигенові сполуки нітрогену з'являються в атмосфері за рахунок
розрядів (див. підрозділ 19.2), а також завдяки техногенній діяльності людини.
Фотохімічне утворення через молекулу азоту:
N2—^'й"+':й\
"й'+Од-жо+б
в основному має місце у верхніх шарах земної атмосфери (100—300 км).
«Озонові діри» в атмосфері утворюються також при наявності в
атмосфері хлорвмісних сполук (фреонів), після фотолізу яких
СР2С12--^->СР2С1 + СҐ
відбувається каталітичний цикл розкладу озону
СГ+03->СЮ + 02
СЮ + б->02+СГ
03+0-
20,
493
Розглянемо з точки зору фотохімії атмосферу Марса, яка більш ніж на
80 % складається з діоксиду карбону (до 20 % — це інертні аргон та И2). Під
дією світла молекула С02 повинна розкладатися
С02-^СО + 6. (а)
Процес рекомбінації атомів Отсгигену
С-гО-^-Ю2 (б)
на три порядки швидший, ніж процес, зворотний процесу (а):
СО + 0-^->С02 (в)
З цієї точки зору, в атмосфері Марса повинні бути 02та СО. Вони
присутні, але у кількості лише 0,1 %. Нескладні розрахунки показують, що таких
концентрацій монооксиду карбону та кисню в атмосфері Марса можна
досягнути всього за 2 роки. Цей несподіваний результат можна пояснити
каталітичним прискоренням процесу (в) гідрогенвмісними активними
частинками (Н*,ОН*,Н02), які утворюються при фотолізі води
Н20—^->ОН#+Н# (г)
(кількість Н20 в атмосфері Марса -0,1 %).
СО + ОН#->С02+Н# (д)
Н#+02-^->Н02 (є)
Н02+6->ОНЧ02 (Є)
Сумарний процес
СО + 6->С02. (ж)
Відзначимо, що, як це зазначалося у підрозділі 18.3, саме наявність
частинок Н#,ОН*,Н02 створює можливість перебігу реакції окиснення СО за
ланцюговим механізмом.
Фотохімічні реакції відіграють найважливішу роль у процесах запису
інформації за допомогою світла. Фотоліз галогенідів Аргентуму є основою
класичних галогенсрібних фотоматеріалів. Поступово розвиваються без-
срібні процеси реєстрації інформації, наприклад діазотипний процес, в
основі якого лежить реакція фотолізу ароматичних діазосполук
> И-<0>--Й= ИЬУ,Н2°» >И -<§>—ОН + И2 + Н+
На неекспонованих ділянках, де зберігається діазосполука, проводять
реакцію азосполучення з фенолом із метою отримання кольорового
(позитивного) зображення.
Крім фотолізу (розкладу під дією опромінювання), можливі також інші
фотохімічні перетворення. Наприклад, у випадку стимульованого світлом
я—ж* переходу стає можливим вільне обертання навколо міжатомного
зв'язку, і це полегшує процес ізомеризації, наприклад цис-транс-ізомери-
зації. Так, під дією світла легко відбувається ізомеризація азобензену:
<о> <о>-^<о>
<о>
494
Відзначимо, що найважливішим етапом зорового процесу є фотохромне
перетворення — фотохімічна цис-транс ізомеризація каротиноїдного
ланцюга.
За рахунок поглинання променевої енергії можливим стає перебіг
«термодинамічно заборонених» реакцій, для яких АС > 0, і саме фотохімічні
процеси дають можливість хімічного акумулювання променевої енергії.
Яскравим прикладом є фотосинтез вуглеводнів з води та діоксиду карбону
пС02 + иН20 -> (СН20)„ + п02.
Безпосередньо ця реакція не є фотохімічною, вона бере енергію для
свого перебігу від високоенергетичних молекул аденозинтрифосфату (АТФ)
та відновленої форми нікотинамідаденіндинуклеотидфосфату (НАДФ-Н2).
В свою чергу, НАДФ-Н2 утворюється при фотохімічному відновленні НАДФ,
а АТФ — при фотохімічному фосфорилюванні АДФ (аденозиндифосфату).
19.4. Радіаційна хімія
Рентген і гамма-кванти швидко діють,
З речовиною як взаємодіють,
І електрони з атомів виймають,
Як з килима пилинки вибивають.
Дію на хімічні реакції випромінювань великої енергії вивчає
радіаційна хімія. Енергія радіаційного випромінювання багатократно перевищує
енергію квантових рівнів валентних електронів, отже, на відміну від
фотохімічних процесів, первинний акт дії радіаційного випромінювання не має
вибіркового характеру і, в першу чергу, призводить до іонізації реагентів
(крім того, утворюються також вільні радикали (атоми) та збуджені частинки).
До іонізуючого випромінювання відносять не лише (як у фотохімії)
електромагнітне випромінювання: фотони з енергією більшою, ніж 50 еВ
(рентгенівські та у-промені); але й корпускулярне випромінювання — потік
частинок високої енергії: швидкі електрони (Р-частинки), протони, ос-частинки
(ядра атомів Гелію) тощо. Доза опромінювання — це відношення
поглинутої енергії іонізуючого випромінювання до одиниці маси речовини, що
опромінюється. Дозу вимірюють у греях (Гр). 1 Гр = 1 Дж/кг.
Характеристикою активності джерел випромінювання є кількість розпадів радіоактивного
ізотопу за 1 с. Одиницею активності є бекерель (Б). 1 Б = 1 с"1 (один розпад
за секунду).
У первинному акті заряджені частинки (ос-частинки, протони тощо)
вибивають із валентних зон електрони, які набувають великої кінетичної енергії,
р-частинки (електрони) при взаємодії з речовиною додатково утворюють
велику кількість вторинних електронів із достатнім запасом кінетичної енергії.
у- та рентгенівські кванти діють за механізмом фотоефекту — вибивання
електронів з атомів, а також за ефектом Комптона — пружне розсіювання
495
квантів на електронах, при якому енергія електрона збільшується, а кванта —
зменшується. Дію радіації на речовину характеризують через радіаційно-
хімічний вихід (О) — кількість змінених (утворених або зіпсованих) частинок
(молекул, радикалів, атомів тощо) на 100 еВ поглиненої енергії.
Серед а-, р- та у-опромінювання найбільшу іонізуючу здатність мають
ос-частинки, а найбільшу проникність при взаємодії з речовиною мають у-
промені. При у-опромінюванні первинні продукти (активні частинки)
виникають по всьому об'єму опромінюваної речовини, при ос-опромінюван-
ні — лише вздовж траєкторії руху (треки, шпори) ос-частинки. При
опромінюванні конденсованої фази а- та р-частинки взаємодіють лише з
поверхневим шаром речовини. Характерною величиною є ЛВЕ (лінійна витрата
енергії) — витрата енергії на одиницю шляху при проходженні іонізуючої
частинки через речовину. ЛВЕ вимірюється в еВ на 10" м (на 1 А).
Розглянемо деякі конкретні приклади радіолізу, починаючи з газової
фази. Первинна активація кисню в основному перебігає через іонізацію 02
02 м;->02+е (а)
(позначка м>—> означає радіаційне опромінювання).
Молекула 02 має достатнє значення спорідненості до електрона, отже,
може його захоплювати
02+е->02. (б)
Нейтралізація молекулярних іонів відбувається з утворенням певної
кількості атомарного Оксигену
02+02 ->02+б + б. (в)
Не виключається також пряма дисоціація кисню під дією опромінювання
02 м/->6 + 6. (г)
Атомарний Оксиген з молекулярним киснем утворює озон, аналогічно
вторинним актам фотолізу (див. підрозділ 19.3)
02+6-^->03. (д)
Озонування повітря під дією радіації спостерігалося експериментально.
На відміну від кисню, молекула К2 стійка до дії радіації.
Радіоліз водневої пари призводить до утворення Н2,6 , Н* та ОН*
частинок. Це відбувається через безпосередній розрив зв'язків у молекулі води
Н20 м;->Н#+ОН# (а)
і в основному через процеси іонізації, первинним із яких є такий:
Н20 м;->Н20++е. (б)
При зіткненні іоца Н20 з молекулою води відбувається утворення
стабільного іона — гідроксонію та ОН-радикала
Н20 + Н20+ -» Н30+ + ОН*. (в)
Електрон з молекулою води утворює нестабільний іон Н20~, який
розпадається на стабільний іон ОН" і атом Гідрогену
496
Н20~->ОН~+Н*. (г)
Рекомбінація атомів Гідрогену дає Н2, а рекомбінація Н* та ОН*
радикалів або Н30 та ОН" іонів відтворює реагент — воду.
При радіолізі рідкої води початкові стадії такі ж самі, як і для водяної пари.
Електрон, що приєднався до молекули води, прийнято називати гідратова-
ним (сольватованим) і позначати через егідр. Тоді замість реакції (г) у
кислому середовищі маємо
Єгідр. + Н30+->Н-+Н20. (д)
У рідкої води стабільним стає пероксид водню (в газовій фазі він термічно
нестійкий), отже, крім рекомбінації атомів Гідрогену
Н#+Н*->Н2, (є)
спостерігається рекомбінація ОН радикалів з утворенням Н202
ОН*+ОН*->Н202. (є)
Наявність у воді розчиненого молекулярного кисню збільшує вихід Н202
за реакціями
Н#+02->Н02 (ж)
та далі
но;+но2 -> н2о2 + о2. (з)
Завдяки присутності 02 у розчині система набирає окиснювального
характеру (у газовій фазі Н* є відновником, а ОН* — окисником).
Це можна використати у хімічному дозиметрі Фріке, який являє собою
розчин ферісульфату в кислому середовищі (з додаванням Н2804). При
цьому перебігають такі реакції окиснення Ре до Ре :
Ре2+ + ОН* +н+ )Ре3+ + Н20, (и)
Ре2+ + НО*2 +н* )Ре3+ + Н202, (/)
Ре2++Н202 +Н+ )Ре3+ +Н20+ОН*. (й)
У таблиці 19.4 зібрані дані, що ілюструють дію дозиметра Фріке.
Таблиця 19.4. Радіаційно-хімічний вихід Ре3+при дії опромінювання
на дозиметр Фріке (в 0,8 н Н2804)
Вид випромінювання
а-частинки (Ро-120)
(3-частинки (тритій)
Р-частинки (Р-32)
Рентгенівські кванти (50 кеВ)
0 (Ре3+)
5,2
13,0
15,5
13,0
Вид випромінювання
Рентгенівські кванти (100 кеВ)
Рентгенівські кванти (200 кеВ)
у-випромінювання (Сз-137)
у-випромінювання (Со-60)
О (Ре3+)
14,2
14,6
15,5
15,5
Широкого застосування набула радіаційна хімія полімерів.
Радіаційний метод ініціювання полімеризації має певні переваги, до яких
відносяться: 1) можливість отримання полімерів високого ступеня чистоти
без домішок залишків ініціаторів або каталізаторів; 2) широкий температур-
497
ний інтервал проведення процесу;
3) легкість контролю швидкості
ініціювання і дуже широкий діапазон
її змін. Радіаційна полімеризація
перебігає за
радикально-ланцюговим механізмом, про що свідчить її
інгібування такими речовинами, як
молекулярний кисень, бензохінон —
типовими інгібіторами вільноради-
кальної полімеризації. Згідно з
рівнянням (15.48) швидкість полімери-
Рис. 19.12.Залежність швидкості зації (гпол ) пропорційнаф), де
полімеризації стиролу (г, моль/с) від г._ швидкість ініціювання. З ін-
потужності дози опромінювання (/>, Гр/с) шого боку? Г/ПрОПОрцІЙНа
інтенсивності опромінювання (див. рівняння (15.56)). Для радіаційної полімеризації
це виконується. На рис. 19.12 наведено залежність (у логарифмічних
координатах) швидкості полімеризації стиролу (гпол) від потужності (Р) дози
опромінювання. Тангенс кута нахилу близький до 0,5, що свідчить на користь
вільно-радикального механізму радіаційної полімеризації.
Радіація впливає не лише на швидкість полімеризації, але й на властивості
отриманих полімерів. Найбільш важливими хімічними ефектами впливу
радіації на полімери є «зшивка» та «деградація». При зшивці опромінювання
веде до утворення так званого сітчастого полімеру з кращими
термомеханічними властивостями. Деградація веде до розривів у полімерному ланцюзі
й матеріал втрачає свої полімерні властивості. Різниця в поведінці полімерів
при радіаційному опромінюванні значною мірою обумовлена стеричними
факторами. Так, для вінілових полімерів поява у полімерному ланцюзі
додаткових бокових метальних
груп веде до посилення
деградації. В поліетилені
немає стеричних ускладнень
і переважає зшивка. На рис.
19.13 показано вплив
нагрівання до 150 °С на зразки
поліетилену. Неопромінені
зразки (ліворуч) при цьому
спікаються, а опромінені
витримують цей термічний
вплив (праворуч).
Важливим розділом
радіаційної хімії є вплив радіації
Рис. 19.13. Поліетиленові плівки і трубки на біологічно активні речо-
після нагрівання до 150 °С. Зліва — неопромі- вини, ЩО дає базу для радіа-
нені, справа — опромінені цінної біології та медицини.
498
При цьому слід враховувати не лише безпосередньо дію опромінювання на
ці речовини, але й вплив на них продуктів радіолізу води.
19.5. Механохімія
Механохімія — наука стародавня.
Відомо, що тертям досягнеш нагрівання.
Щоб без вогню від холоду не вмерти,
Потрібно тільки терти, терти, терти...
Механохімія вивчає вплив механічних навантажень на фізико-хімічний
стан речовин і перебіг хімічних реакцій. Термін «механохімія» був введений
В. Оствальдом, а дію механохімічних факторів людина використовувала ще
на світанку своєї цивілізації,
добуваючи вогонь шляхом тертя (рис. 19.14).
Механохімія в основному
стосується процесів у твердих тілах або за
участю твердих тіл (для цього
основного розділу механохімії П. А. Тіссен
та Г. Хайніке навіть запропонували
спеціальну назву — «трибохімія»;
звичайно, що до механохімічних
можна віднести і процеси у рідкій
фазі, наприклад, фізико-хімічні явища
при кавітації у рідині під дією
ультразвуку). Механічна обробка твердих тіл
може бути різноманітною: тертя,
різка, ковка, пластична деформація,
удар, диспергування тощо. Механіч- рИс. 19.14. Механохімічне
на дія супроводжується змінами у добування вогню
структурі твердого тіла і цілою низкою фізичних (про хімічні далі) процесів,
до яких відносяться:
— утворення та міграція структурних та електронних дефектів;
— збудження коливань ґраток;
— пластична деформація, утворення тріщин;
— локальні розігріви у твердій фазі;
— збільшення поверхні, утворення «свіжої» поверхні;
— аморфізація;
— електризація, п'єзоефект;
— емісія електронів та фотонів тощо.
На рис. 19.15, с. 500, схематично показано стадії ударної деформації:
зліва — проникнення інструмента, що ударяє, у незбуджену поверхню
твердого тіла, справа—процеси релаксації при видаленні цього інструмента. Вид-
499
но, що дуже дефектний стан,
який виникає безпосередньо при
механічній обробці, частково
зберігається і після її закінчення.
За оцінками Г. Таммана до
5—15 % витраченої механічної
енергії перетворюється на
потенціальну енергію твердого тіла
і підвищує його
термодинамічний потенціал. При
диспергуванні енергія в основному
запасається у вигляді поверхневої
енергії межі поділу фаз. Крім того,
залишкова деформація
зберігається у вигляді дефектів
кристалічної структури, до яких
відносяться: точкові дефекти (вакансії,
міжвузлові атоми), лінійні
дефекти — дислокації (крайові,
гвинтові), двовимірні дефекти
(дефекти пакування, межі зерен у
полікристалах, нерівноважні,
нещільно упаковані грані тощо).
Приклади деяких структурних
дефектів було наведено у розділі 17,
рис. 17.15—17.18, с. 443. Крім
суто структурних дефектів,
можливе також утворення електрон-
I II
Рис. 19.15. Стадії ударної деформації:
І -— проникнення інструмента у кристалічні
ґратки твердого тіла; II — фази руйнування,
що відповідають видаленню інструмента
з твердого тіла з утворенням остаточної
деформації
них дефектів: нерівномірне розподілення зарядів у діелектриках (рис. 19.16);
виникнення неспарених електронів згідно зі спектрами ЕПР — електронного
парамагнітного резонансу (рис. 19.17). Наявність структурних дефектів та
запасеної енергії у твердому тілі підвищує його реакційну здатність.
На рис. 19.18 наведена дислокаційна структура поверхні №С1 після
удару (а) та після наступного хімічного травлення (б). Місця скупчення
дислокацій мають підвищену реакційну здатність і першими видаляються при
хімічному травленні. Відзначимо, що
травлення є одним із методів виявлення
дислокацій.
Нарис. 19.19, с. 502, показано, що
реакційна здатність рутилу по відношенню до
лл\/^ . - його взаємодії із сірчаною кислотою
Рис. 19.16. Заряджені області при збільшується симбатно зі збільшенням
згинанні кристала ІлР, декоровані ->«"і»ш/і-і»^л ^"™««^п« ->■ ^швшмішііі
діелектриком—свинцевим дисперсності (питомої поверхні) та ступе-
суриком ня деформації ґраток (мікродеформації).
500
ДФПГ
8і = 1.97
Рис. 19.17. Спектри ЕПР анатазної (а) та рутильної (б) модифікацій Ті02 при 77 К
після механічної активації: 1 — 6 хв; 2 — 10 хв; 3 — 20 хв; 4 — 0,5 хв; 5 — 10 хв
Ступінь розчинності оксиду Олова (IV) корелює (рис. 19.20, с. 503) з його
питомою поверхнею, а також із величиною квадрупольного розщеплення
у мессбауерівських (ГР) спектрах, яка відображає ступінь руйнування
структури кристала.
При взаємодії твердого тіла з газами особливу роль відіграють
поверхневі дефекти (розділ 17; рис. 17.18, с. 443), які є причиною неоднорідності
поверхні сорбентів і каталізаторів та мають підвищену реакційну здатність та
каталітичну активність (розділ 18, таблиці 18.1 та 18.2, с. 457,458).
Наведені приклади характеризують пост-ефект механічної активації,
вплив остаточних деформацій та запасеної у твердому тілі енергії на перебіг
хімічної реакції за участю твердого тіла. Більший ефект дає механічна
активація і/і зіш, тобто проведення хімічної реакції у процесі механічної
деформації.
А А А**
Щґ* -*>Чр *
*.■■
АІАІ
%фф->
Рис. 19.18. Структура поверхні №С1 після удару (а)
та після наступного хімічного травлення (б)
501
70
50
30
10
О 4 8 12 16 і, хв
Рис 19.19. Ступінь взаємодії (а) із сірчаною кислотою — (7), питома поверхня (5) — (2)
та мікродеформації (у| є2|) — (•?) рутилу в залежності від часу активації (/)
В експериментах по механічній активації завжди спостерігається
виділення достатньої кількості теплоти, тобто перетворення кінетичної енергії
механічного руху в хаотичну енергію руху молекул (атомів, іонів тощо).
Було виявлено, що існують так звані «гарячі точки» — осередки розігріву
площиною ~10^—10"2 см2 і часом існування ~10~5—10~3 с. За рахунок
цього виникає, зокрема, явище так званого «контактного» плавлення.
Теорія локальних перегрівів знайшла деяке підтвердження і в хімічних процесах.
Наприклад, нітрат натрію має декілька паралельних шляхів розкладу:
ШЖ)3 -» ШЖ)2 +1/202; (І)
ШШз -> У2Ш20 + У2ІЇ2 + 5/402; (її)
КаЖ)3 -> 1/2Ка20 + К02 +1/402. (Ш)
Реакція (І) реалізується в інтервалі 600—800 К, (II) — при Т> 800 К, а
(III) — при Т> 1000 К. При механічній обробці №N03 У продуктах було
знайдено азот та його оксиди, що свідчить про перебіг високотемпературних
реакцій при механічній активації.
Напрямок механічно стимульованих фазових переходів у твердій фазі
у більшості випадків співпадає із термічно визначеним, але виявилося, що за
рахунок механічної активації можна здійснити фазові переходи у напрямку,
протилежному нагріванню (термічному), наприклад, перехід кобальту з
кубічної структури у гексагональну, фосфору з триклинної у ромбічну тощо.
Знайдено приклади, коли механічна і термічна активації призводять до
різного перебігу хімічних реакцій. Наприклад, розклад оксалату аргентуму
термічним шляхом відбувається за реакцією
А§2С204-»2А§ + 2С02,
АЛМО"
187
181
193
192
188
195
191
193
186
ІГ, мм/с
1,7"
1,6-
1,5-
1,4:
-
8 -
6 -
4 -
2 -
ґ'
5, м2/г
ґ~
\ ^
С—і І>—і І__
І
•
2
*<х,%
_4 1
-40
-20
1 1 1 1»!
-2 -1 0
1 2
V, ММ/С
0 20 40 60
/, хв
Рис. 19.20. а) Мессбауерівські (ГР) спектри за часом активації: 1 — 0 хв
(вихідний зразок); 2 — 5 хв; 3 — 35 хв; 4 — 60 хв; 5 — 80 хв; б) 7 — ширина лінії
у ГР-спектрі (Г); 2 — питома поверхня (8); 3 — ступінь розчинення 8п02 (а)
а механічно таким чином:
А§2С204 -> А£20 + СО + С02 .
Термічний розклад бромату натрію відбувається за реакцією
НаВЮ3->КаВг + 3/202,
а механохімічний за реакцією
2ИаВг03 -> Ма20 + 5/202 + Вг2.
Хлорид меркурію при перетиранні розкладається, в той час як при
нагріванні він сублімує.
Виявилося також, що при механічній активації стає можливим перебіг
реакцій, які взагалі неможливо здійснити термічним шляхом. Наприклад,
Аи + 3/4С02 -> 1/2Аи203 + 3/4С,
2Си + С02->2СиО + С,
8іС + 2Н2->Зі + СН4.
Суттєво, що наведені реакції мають додатне значення АС0, тобто вони є
«термодинамічно забороненими».
На рис. 19.21, с. 504, наведено кінетичні дані механохімічного відновлення
8п02 різними відновниками, а в таблиці 19.5 наведено термодинамічні дані
для цих реакцій.
503
100
50Н
50 100
Рис. 19.21. Кінетика механохімічного
відновлення 8п02 різними відновниками:
7) без відновника; 2) вуглець;
3) вольфрам; 4) силіцій; 5) алюміній
Таблиця 19.5. Вільні енергії Гіббса реакцій
твердофазного відновлення 8п02
З наведених на рис. 19.21 даних
видно, що реакції {4) і (5), для яких
АС° « 0, перебігають, як і слід було
сподіватися, відносно швидко. Меха-
нохімічна активація дає також
можливість перебігу (хоча й більш
повільного) для «термодинамічно
заборонених» реакцій (7) — (3), для
яких АС?0 >0.
Співставимо тепер вплив меха-
нохімічної активації як пост-ефекту
та іп 5ІШ.
З даних, наведених у таблиці 19.6,
видно, що енергія активації реакції
синтезу карбонілу нікелю з № та СО
суттєво зменшується при
механічній активації і особливо при
проведенні цієї реакції іп 8Ііи.
Для механохіміч-
них реакцій іп 8ІІи
характерні низькі енергії
активації або навіть
нульові, приклад
наведено на рис. 19.22.
Незалежність швидкості
реакції від
температури свідчить про те, що
для цих процесів
термічна активація несут-
Таблиця 19.6. Енергії активації (ЕА) реакції тєва, енергія для здій-
ІЧі + 4СО->№(СО)4 при механічній снення реакційного ак-
активації нікелю та черпається не з
теплового резервуара, а
безпосередньо
передається твердому тілу
через ділянки, які
активуються механічно.
Для пояснення процесів механохімічної активації іп зііи П. А. Тіссен
і Г. Хайніке запропонували модель «магма-плазми» («трибоплазми»), яку
наведено на рис. 19.23. Відзначимо, що для процесів, які перебігають у не-
ізотермічній плазмі — газових розрядах (підрозділ 19.2), також характерна
температурна незалежність (рис. 19.8, с. 487, крива 7). На перебіг механо-
хімічних реакцій іп 8ІШ суттєво впливає наявність короткоживучих
збуджених станів, що наведено у таблиці 19.7.
504
Реакція
1) 8іЮ2=8пО + 1/202
2) 8п02 + С = 8п + С02
3) 8п02 + 2/3\У = 8п + 2/3\\ГО3
4) 8п02+8і = 3п + 8і02
5) 8п02 + 4/3 А1 = 8п + 2/3 А1203
^^298» КДЖ/М0ЛЬ
+295
+126
+25
-276
-535
Синтез карбонілу на зразках
На необроблених
Після механічної обробки
У процесі обробки (іп зіїи)
ЕА, кДж/моль
54,0
25,0
14,6
Таким чином, в залежності від
характеру дисипації енергії на основі
«ієрархії» енергетичних станів, яка
визначається перебігом релаксаційних
процесів, механохімічні реакції можна
поділити на такі групи. До першої групи
відносяться реакції, що викликані ко-
роткоживучими високоенергетичними
збудженими станами, які не
знаходяться у тепловій рівновазі із довкіллям і
перебігають в ударній «трибоплазмі» (див.
50
40
зоь
20 [
10
0
У,г
Т,К
293 313 333 353 423
Рис. 19.22. Температурна залежність
виходу (У) механохімічної реакції
Зп + 2С6Н5СН2С1 -> (С6Н5СН2)28пС12
рис. 19.23). До другої групи можна віднести реакції, які запускаються або
пришвидшуються за рахунок наявності у твердому тілі запасеної енергії та
дефектів, що виникли при механічній активації, тобто механохімічний пост-
ефект. Можливий також «проміжний» варіант між цими групами за
рахунок метастабільних
збуджених «постплазматичних»
станів (див. табл. 19.7), що
існують відносно довго
порівняно зі станом трибоплазми,
релаксацією коливань,
«гарячими точками» тощо.
Загальний хід
механохімічної реакції можна описати
діаграмою, що представлена
нарис. 19.24, с. 506. Реакція до
початку механічної обробки
визначається термічною
активацією і залежить від
температури, для багатьох твердофаз-
Таблиця 19.7. Час релаксації (т) при механохімічній обробці
Рис. 19.23. Модель «магма-плазми»:
1 — недеформована структура; 2 — дефектна,
розупорядкована структура; 3 — стан
«трибоплази»; 4 — екзоемісія електронів
Процес збудження
Удар
Стан трибоплазми
«Гарячі точки»
Коливання ґраток
Екзоелектронна емісія
т, с
ю-6
~ю-7
ю~мо-3
іо10-ю-9
іо-м о51
Процес збудження
Механолюмінесценція
Збуджені метастабільні стани
Утворення «свіжої» поверхні
л
Дефекти структури
X, С
іо-м о3
ю-м о-2
10* при 105 Па
1-100 при 10^ Па
ю-м о6
них реакцій при кімнатній температурі це дуже мала величина (фаза / на
рис. 19.24). Механічна активація зазвичай різко прискорює твердофазну
реакцію і після деякого періоду її зростання (фаза 2) виходить на
стаціонарний рівень (фаза і), причому швидкість реакції збільшується у багато разів,
наприклад, швидкість синтезу карбонілу нікелю з СО та N1 у процесі меха-
505
[г
1 \2
і'
} 1 3
ч
4
і ►
/
Рис. 19.24. Діаграма перебігу
механохімічної реакції (г — швидкість
реакції, і — час): 1 — реакція до початку
механічної активації; 2 — фаза
збільшення активності; З — стаціонарна
фаза механохімічної реакції «іп 8ІШ»\
4 — пост-ефект механохімічної
активації
нохімічної обробки іп зііи (фаза 3)
перевищує швидкість цієї ж реакції
на неактивованому нікелі (фаза 7)
у 60 разів.
Спад активності після закінчення
механічної активації може бути
різним у залежності від типу
механохімічної реакції. Нарис. 19.25
наведені різні варіанти: від практично
повного обриву після закінчення
механічної обробки (крива 1) до
дуже повільного спаду з виходом на
деякий стаціонарний стан (крива 3).
Крива 1 представляє механохімічні реакції, які спряжені з
високоенергетичними станами, що існують у трибоплазмі й закінчуються за частки секунди.
До цієї групи відносяться, у першу чергу, всі «термодинамічно заборонені»
реакції, для яких АС0 > 0, і перебіг яких можливий лише за рахунок підводу
енергії ззовні до системи (у даному випадку у вигляді механічної енергії).
Крім того, сюди можна віднести
реакції з дуже високою енергією
активації, для термічної активації яких
потрібні дуже високі температури.
Наведена на рис. 19.25 реакція
гідрування вугілля до метану
належить саме до цього типу. Нарешті,
враховуючи дуже короткий час
релаксації «свіжої» поверхні при
атмосферному і більших тисках (див.
табл. 19.7), такий різкий спад
спостерігається при «отруєнні» поверхні
речовиною, що міцно хемосорбу-
ється. Наприклад, реакція
утворення карбонілу нікелю, для якої
характерний відносно поступовий спад
(крива 2), при наявності залишків
кисню у газовій суміші миттєво
блокується при пост-ефекті, і спад нагадує криву 1.
Для реакцій, які можуть перебігати самочинно (АС° < 0), типовою є
крива 2. Після швидкої загибелі високоенергетичних активних станів далі
відбувається відносно повільний спад, який викликаний поступовою
витратою у ході реакції відносно довгоживучих активних ділянок. Вільна енергія,
що була запасена при деформації, поступово витрачається.
Нарешті, крива 3 описує механокаталітичні процеси (на рис. 19.25 це
гідрування бензену на нікелевому каталізаторі).
506
І, год
Рис. 19.25. Пост-ефект механохімічної
активації (штрихова лінія — активність
під час механічної обробки — «іп зііи»,
г—швидкість реакції, /—час): І — синтез
СН4 з простих речовин; 2 — утворення
карбонілу з СО та №; 3 — каталітичне
гідрування бензену
Дія пост-ефекту при перебігу реакцій з відносно низькою енергією
активації полягає у створенні при механічній активації відповідних дефектних
структур, що слугують активними центрами для конкретної структурно-
чутливої реакції (див. підрозділ 18.2). При не досить високих температурах
проведення гетерогенно-каталітичної реакції процеси дифузії у твердому
тілі відбуваються повільно, отже, дефекти — активні ділянки, що утворилися
при механічній активації, на стадії пост-ефекту можуть зберігатися досить
тривалий час.
Звичайно, якщо каталітичний процес має дуже високу енергію активації,
то при дуже низьких температурах він можливий лише при постійній
механічній активації каталізатора (цей випадок описується кривими 1 або 2).
Прикладом може бути механохімічний синтез аміаку при кімнатній
температурі, який здійснюється шляхом постійного бомбардування поверхні
залізного промотованого каталізатора (див. підрозділ 18.2) частинками
твердого карборунду (8іС), що додаються в азотоводневу суміш.
Ефективність дії механохімічних процесів, можливість їх застосування
на практиці можна охарактеризувати так званим енергетичним виходом
(Ягп) — кількістю молів утворених або прореагувавших частинок на 1 МДж
витраченої механічної роботи. Деякі приклади наведено у таблиці 19.8.
Таблиця 19.8. Енергетичний вихід (%„) механохімічних процесів
Процес
Утворення вільних радикалів
Розклад неорганічних твердих матеріалів (А£2С204,
НаШ3,СаС03тощо)
Генерація поверхневих атомів (8Ю2,ВМтощо)
Хемосорбція (СО на М§0; Н2 на 8і02 та графіті тощо)
Реакції у твердій фазі (приклад: ВаС03 + ^03 = Ва\\Ю4 + С02)
Поліморфні переходи в оксидах
£т, моль/МДж
ю-3
10"2—10"'
10"'
5-Ю"'
2—3
102
З даних, наведених у табл. 19.8, видно, що енергетичні виходи різних
механохімічних процесів знаходяться у дуже широкому інтервалі й
визначаються механізмом ініціювання певного механохімічного процесу.
Незважаючи на те, що механічна активація — дуже енергоємний процес,
є певні інші технологічні переваги механохімічних процесів. Розглянемо це
на прикладі реакції синтезу карбонілу нікелю. Збільшення реакційної
здатності при механічній обробці збільшує швидкість реакції на порядки. Це веде
до скорочення технологічного часу, наприклад, механохімічна реакція
синтезу карбонілу нікелю дозволяє отримати ту ж саму кількість продукту за
час, у 45 разів менший, ніж у класичному суто хімічному варіанті.
Збільшення швидкості при механохімічній дії дає можливість проводити реакцію
507
при знижених температурах і тисках (класичні хімічні синтези карбонілів
металів реалізуються при температурах 400—500 К і тисках порядку 107 Па),
що дозволяє зменшити енерговитрати. При механохімічному синтезі
руйнуються плівки, поверхневі шари, що блокують поверхню і перешкоджають
перебігу хімічної реакції. Наприклад, класичний хімічний варіант синтезу
№(СО)4 потребує використання ретельно очищеного монооксиду карбону,
оскільки вже залишкові кількості кисню зупиняють реакцію, міцно хемосор-
буючись на поверхні нікелю. При механохімічному синтезі карбонілу
нікелю постійно утворюється свіжа поверхня твердого тіла, і присутність у
газовій фазі до 3 % кисню лише незначно зменшує швидкість механохімічної
реакції синтезу №(СО)4.
Перспективним також може вважатися механохімічна активація
гетерогенних каталізаторів, оскільки, як було зазначено вище, при цьому
утворюються довгоживучі дефектні стани, що є активними центрами каталізу.
Корисним для збільшення питомої поверхні сорбентів і каталізаторів є їх механічне
диспергування (з паралельною механічною активацією їх поверхневого
шару).
Нарешті, вивчення механізму механохімічних реакцій дає можливість їх
пригальмувати там, де вони приносять шкоду, наприклад, це спрацювання
машин та приладів за рахунок тертя, механокорозія, механохімічне
спалахування тощо.
Література
Основна
І.Данизльс Ф., Олберти Р. А. Физическая химия.— М: Мир, 1978.— 645 с.
2. Жуховицкий А. А., ШварцманЛ. А. Физическая химия.— М.: Металлургия, 2001.—
688 с.
3. Киреев В. А. Курс физической химии.— М.: Химия, 1975.— 775 с.
4. Курс физической химии: В 2 т. / Под ред. Я. И. Герасимова.— М.: Химия.— Т. І.—
1970.— 592 с; Т. II.— 1973.— 623 с.
5. Мелвин Хьюз Е. А. Физическая химия: В 2 т,— М: ИЛ, 1962.— 1148 с.
6. Стромберг А. Г., Семченко Д. П. Физическая химия.— М.: Вьісш. шк., 1999.—
527 с.
7. Физическая химия: В 2 т. / Под ред. К. С. Краснова.— М.: Вьісш. шк., 2001.—Т. 1.—
512 а; Т. 2.— 319 с.
8. Товбин М. В. Физическая химия.— К.: Вища шк.— 1975.— 488 с.
9. Зткинс П. Физическая химия: В 2 т— М.: Мир, 1980.— Т. 1.— 580 с; Т. 2.—
584 с.
За окремими розділами
1. Аввакумов Е. Г. Механические методьі активации химических процессов.— Н-ск:
Наука, СО, 1986.—305 с.
2. Адамсон А. Физическая химия поверхностей.— М.: Мир, 1979.— 568 с.
508
3. Гончарук В. В., Камалов Г Л., Ковтун Г А., Рудаков Е. С, Яцимирский В. К. Ка-
тализ. Механизмьі гомогенного и гетерогенного катализа. Кластерньїе подходьі.—
К.: Наук, думка, 2002.— 541 с.
4. Гомонай В., Гомонай О. Фізична хімія. Хімічна термодинаміка.— Ужгород:
Мистецька лінія, 2000.— 290 с.
5. Гомонай В., Гомонай О. Фізична хімія. Хімічна кінетика. Каталітичні реакції. Фізико-
хімія поверхневих явищ. Фото- та радіаційнохімічні процеси. Електрохімія.—
Ужгород: Мистецька лінія, 2003.— 478 с.
в.Денисов Е. Т. Кинетика гомогенних химических реакций.— М.: Вьісш. шк.,
1978.—367 с.
1. Драго Р. Физические методи в химии: В 2 т.— М.: Мир, 1981.— Т. 1.— 422 с;
Т. 2.—456 с.
8. Еремин Н. А. Злементьі газовой злектрохимии.— Изд. МГУ, 1968.— 212 с.
9. Карапетьянц М. X. Химическая термодинамика.— М.: Химия, 1975.— 576 с.
10. Киттель Ч. Введение в физику твердого тела.— М.: Наука, 1978.— 791 с.
ХХ.Козман У. Введение в квантовую химию.— М.: ИЛ, 1960.— 560 с.
12. Коулсон Ч. Валентность.— М.: Мир, 1965.— 426 с.
13. МаррелДж., Кеттл С, ТеддерДж. Химическая связь.— М.: Мир, 1980.— 382 с.
14. Робертс М., Макки Ч. Химия поверхности раздела металл-газ.— М.: Мир,
1981.— 539 с.
15. РотинянА. Л., ТихоновК. И., Шошина И. А. Теоретическая злектрохимия.— Л.:
Химия, 1981.—424 с.
16. СмирноваН. А. Методи статистической термодинамики в физической химии.—
М.: Внсш. шк., 1982.—456 с.
17. ТомасДж., Томас У Гетерогенний катализ.— М.: Мир, 1989.— 452 с.
18. Хайнике Г Трибохимия.— М.: Мир, 1987.— 582 с.
19. Хзйли 3., Джонсон 3. А. Радиационная химия.— М.: Атомиздат, 1974.— 415 с.
20. Змманузль Н. М., КнорреД. Г Курс химической кинетики.— М.: Внсш. шк.,
1984.—463 с.
21. Яцимирський В. К. Фізична хімія рівноважних систем.— К.: МОУ, КУ, 1992.—
110 с.
22. Яцимирський К Б., Яцимирський В. К Хімічний зв'язок.— К.: Вища шк., 1993.—
309 с.
23. Яцимирський В. К Фізична хімія процесів.— К.: КУ, 1999.— 143 с.
509
Додаток 1. Фізичні сталі та одиниці енергії
Число Авогадро ИА = 6,0220943 • 1023 моль"1.
Універсальна газова стала К = 8,31441 Дж/моль • К.
Стала Больцмана к = ША = 1,380662 • 10~23 Дж/К.
Стала Планка Н = 6,626176-10" Дж • с.
Й = 1Г- = 1,0545887 • 10" Дж • с (атомна одиниця дії).
Атомна одиниця довжини (радіус Бора) а0 = 5,2917706 • 10"11 м.
2
Атомна одиниця енергії 1 а.о.е= — = 4,359814-10~18 Дж = 27,2131 еВ.
ао
-27
Атомна одиниця маси 1 а.о.м. = 1,6605655-10 кг.
Заряд електрона \е\ = 1,6021892 -10" Кл (атомна одиниця заряду).
Маса електрона те = 9,\ 09534 • 10" кг.
Маса протона тр = 1,6726485 • 10"27 кг = 1836,15152 те.
Швидкість світла с = 2,99792458 • 108 м/с.
Магнетон Бора \ів = -^- = 9,274078 • 10~24 Дж/Тл.
1еВ= 1,6021-10~19Дж. Є
1 еВ/атом = 96,48 кДж/моль.
Ісм"1 = 1,9861 -10"23Дж.
1 нм= 10" м; 1 пм= 10" м; 1 А = 0,1 нм= 10" м.
1 атм= 1,0133 -105 Па.
1 мм рт. ст. (торр) = 1,3332 • 10 Па.
1л-а™ =1,0133- 10 Дж.
Число Фарадея Р=ИА • є = 9,648456 • 104 Кул/моль.
Додаток 2. Теорія збурень
Повний гамільтоніан Н дорівнює
Н = Н°+Хк, (1)
де Н° — незбурений гамільтоніан; Хк — оператор збурення.
Розкладемо енергію та хвильову функцію за степенями малого параметра X:
Еп=Е°п+\Е'п+Х2Е: + ... (2)
уп=Ч,°п+Хч,'п+),2у"п+... (3)
При відсутності збурення
Н°Уп=Е°пу°п, (4)
де Е„ — власні значення; і|/° — власні значення незбуреного оператора Я0.
Точний розв'язок має такий вигляд
НЧп=Епч/п. (5)
510
Підставимо вирази (1), (2) та (3) в рівняння (5)
(я+ял)(<+х<+хуя +...)=
=(е°п+\е'п+\2еп+...) (<+х¥;+х2<+4 о (6)
У формулі (6) згрупуємо члени однакової величини за X. При Х° маємо
рівняння (4). При Xі маємо
яХ+^ = £Х+£>0„> (7)
а при А
ях|/;7+м/„=£>;+£>;+£>;. ©
Виразимо \|/'7 у вигляді лінійної комбінації ортогональних незбурених
функцій
< = 2Х<- (9)
Діючи оператором #° на\|/^, згідно з виразом (9), одержимо
£Х = 2>тчС. (Ю)
Вирази (9) та (10) підставимо у рівняння (7), потім помножимо зліва
рівняння (7) на і|/° (\|/° вважаємо дійсною) та проінтегруємо.
З урахуванням ортонормування незбурених хвильових функцій, тобто
умови/\|/° \|/°*/К = 5т„, де 5тя — символ Кроннекера (8^ = 0 при т Ф п та
5т„ = 1 при т-гі) дістанемо
апЕ°п+\<Ь<<ІУ = апЕ0п+Е'п,
звідки
К=кп, (П)
Кп=\<Ьч/УГ. (12)
Отже, поправка до енергії у першому порядку теорії збурень дорівнює
середньому значенню оператора збурення, що обчислене за допомогою
незбурених хвильових функцій.
Якщо рівняння (7) помножити зліва на \ц°т та проінтегрувати, то
ат^т + "тп = ^пат->
ЗВІДКИ
ат=~Е^¥ (13)
при тФп,де п т
ктп=\у°тЩуУ. (14)
Формула (13) дає змогу обчислити всі коефіцієнти ат при розкладанні
і|/^ (крім аю який знаходиться з умов нормування), отже, знайти поправку
до хвильової функції у першому порядку теорії збурень.
Рівняння (7) можна записати у такому вигляді:
(Н°-Е°„уп=[іг-Е'пуп. (15)
511
Помноживши на \|/° та проінтегрувавши, маємо у правій частині кпп - Е'п = 0
(згідно з (11)), отже, інтеграл у лівій частині також дорівнює нулю
/<(я-£„°у„^=о, (іб)
тобто і|/„ ортогональна до \|/°.
Добуті вище співвідношення справедливі, якщо незбурена хвильова
функція \|/° єдина, тобто л-й стан невироджений. При виродженні
(/-кратному) власному значенню енергії Е° відповідає набір /-лінійно незалежних
хвильових функцій \|/°£5 кожна з яких є розв'язком незбуреного рівняння
Шредінгера.
Оскільки функції \|/°£ лінійно незалежні, то візьмемо їх лінійну
комбінацію 1
V* =!>*<*• (17)
Підставивши вираз (17) у рівняння (15) і позначивши далі Еп через є, мати-
мемо 1
(н0-Е°п)ч'п=(и-е)%скУп
'пк-
к=\
(18)
Помноживши рівняння (18) зліва на у°пХ та проінтегрувавши, одержимо
зліва нуль (згідно зі співвідношенням (16)), а справа
с1(А11-є) + с2А12+... + с/Л1/=0.
Далі беремо знову рівняння (18), домножуємо його зліва вже на\|/°2 та
інтегруємо. Тоді
схк2Х + с2(к22-е) + ... + с1к21 = 0.
Потім робимо ту саму процедуру, домножуючи послідовно на \|/°3 > 4^4 >—
та \у°п1 інтегруючи. Одержимо систему /-лінійних однорідних рівнянь з
/-невідомими (с1;с2;с3,...9с1). Ця система має нетривіальні розв'язки, якщо
відповідний детермінант дорівнює нулю
Ап-є
А21
А9? -є
к
2/
= 0.
(19)
'22
Ч\ А/2 ... А//-ЄІ
При розкриванні детермінанта дістанемо / різних значень є, внаслідок
чого виродження знімається.
Для атома Гелію в детермінанті другого порядку кхх=к22=К, а кХ2 =
= к2\ = А. Тоді
І*-є А
А АГ-єІ
= 0,
тобто маємо формулу (8.117).
512