/







































































































































































Author: Макарова В.Г. Песков Д.Д.
Tags: функциональные расстройства расстройства обмена веществ антропология медицина метаболизм патологии учебное пособие
ISBN: 5-8423-015-X
Year: 2001




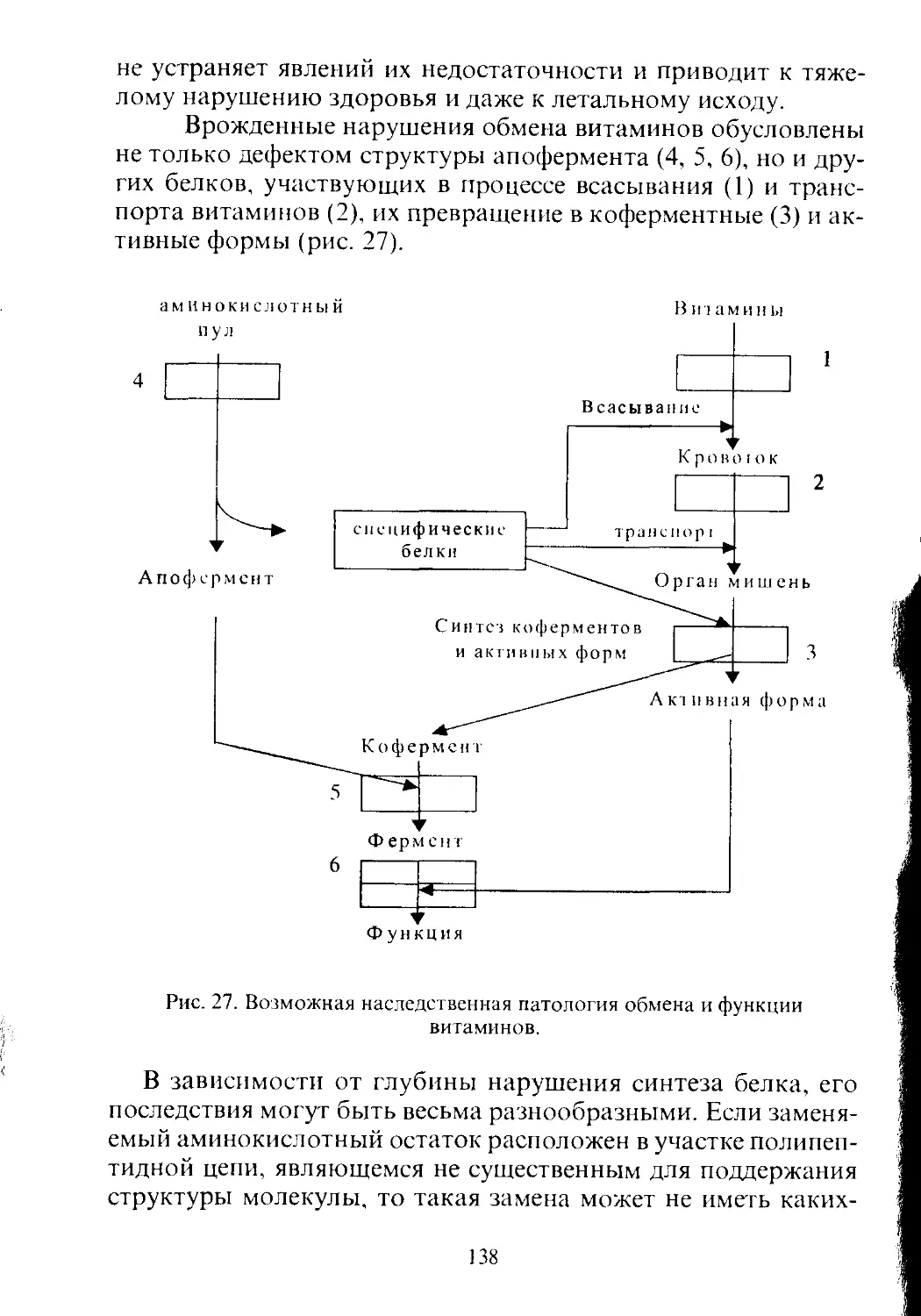
Text
УДК 616-008.9
ББК 28.707.2
П-205
Молекулярная патология метаболизма: Учеб, пособие / Под ред.
В.Г.Макаровой, Д.Д.Пескова. - Рязань: Изд-во Рязан. гос. мед. ун-та,
2001. - 141 с., ил.
В учебном пособии изложены современные представления о молекуляр-
ных механизмах нарушений процессов пищеварения и основных видов обме-
на - белков, аминокислот, липидов, углеводов и витаминов, их возможные
клинические признаки.
Предлагаемое издание - первое учебное пособие по молекулярной пато-
логии метаболизма, предназначенное для студентов, аспирантов, преподава-
телей медицинских вузов и практических врачей.
Рис. 30. Табл. 4. Библиогр. 27 назв.
Печатается по решению Научно-методического Совета Рязанско-
го государственного медицинского университета имени академика
И.П.Павлова.
Рецензенты: Н. В. Дмитриева, зав. кафедрой детских болезней с кур-
сом детской хирургии РязГМУ, д.м.н., профессор
Д.Г.Узбекова, д.м.н., профессор кафедры фармаколо-
гии с курсом фармакотерапии ФПДО РязГМУ
RzGMU.Narod.Ru
ISBN 5-8423-015-Х
© Рязанский государственный
медицинский университет
им. акад. И.П.Павлова, 2001.
ПЕРЕЧЕНЬ СОКРАЩЕНИЙ
АЛК-синтаза - 3-аминолевулинатсинтаза
АС - атеросклероз
АХАТ - ацил-КоА-холестерол-ацилтрансфераза
ВФ - внутренний фактор
ВДЗР - витамин D-зависимый рахит
ВДРР - витамин D-резистентный рахит
ГАМК - у-аминомасляная кислота
ГГФРТ - гипоксантингуанинфосфорибозил-трансферазгх
ГЛП - гиперлипопротеинемии
ГМГ-КоА-редуктаза - (З-гидрокси-Р-метилглутарил-КоА-
редуктаза
ДЛП - дислипопротеинемии
ДОФА - дигидроксифенилаланин
И МФ - инозинмонофосфат
КП Г - копропорфириноген
ЛП - липопротеины
ЛПОНП - липопротеины очень низкой плотности
ЛПНП - липопротеины низкой плотности
ЛППП - липопротеины промежуточной плотности
ЛПВП - липопротеины высокой плотности
ЛП(а) - липопротеины а
ЛПЛ - липопротеинлипаза
ЛПХ - липопротеины X
(3-ЛП - липопротеины низкой плотности
пре-(3-ЛП - липопротеины очень низкой плотности
ЛХАТ - лецитинхолестеролацилтрансфераза
ос-ЛП - липопротеины высокой плотности
ММК - метилмалоновая кислота
НАД+ - никотинамидадениндинуклеотид
НАДФ+ - никотинамидадениндинуклеотидфосфат
ПАЛФ - пиридоксальфосфат
ПАМФ - пиридоксаминфосфат
ПБГ - порфобилиноген
ПЗСС - пиридоксинзависимый судорожный синдром
ПЛ - печеночная липаза
ПНЭ - подострая некротизирующая энцефаломиелопатия
ПОЛ - перекисное окисление липидов
СГХС - семейная гиперхолестеринемия
3
СД - сахарный диабет
СЖК - свободные жирные кислоты
СМЖ - спинно-мозговая жидкость
ТГ - триглицериды
ТГФК - тетрагидрофолиевая кислота
ТДФ - тиаминдифосфат
TK-I - транскобаламин I
ТК-П - транскобаламин II
ТТФ - тиаминтрифосфат
ФЛ - фосфолипиды
ФРПФ - фосфорибозилпирофосфат
ХМ - хиломикроны
ХС - холестерин
ЦНС - центральная нервная система
ЭХ - эфиры холестерина
GalCer - галактозилцерамид
GlcCer - глюкозилцерамид
NeuAc - N-ацетилнейраминовая кислота
4
ПРЕДИСЛОВИЕ
Метаболизм представляет собой совокупность специфических
последовательностей ферментативных реакций (метаболических
путей), обеспечивающих функционирование живой системы.
Основной результат метаболизма заключается в синтезе
сложных молекул, которые специфически взаимодействуют
друг с другом, образуя необходимые для организма структуры.
В биохимии принято различать анаболические и катаболи-
ческие пути, которые составляют соответственно анаболичес-
кую и катаболическую фазы обмена веществ. Наличие слож-
ной системы взаимосвязанных между собой метаболических
путей в клетке осуществляет быстрый ферментативный синтез
и распад различных соединений, однако, их концентрация ос-
тается, как правило, в любой момент почти без изменений.
Однако, даже незначительные изменения в системе биохи-
мических процессов могут приводить как к нарушению обмена
процессов, так и функциональному состоянию клеток органов
и тканей и организма в целом.
Выявлены различные причины, приводящие к возникнове-
нию метаболического блока обмена веществ, из которых наи-
более частыми являются: отсутствие, недостаток или ненор-
мальная структура энзима, регуляторные нарушения его био-
синтеза или усиление расщепления и выделения.
По своему характеру метаболические блоки могут быть
врожденными (наследственными) или приобретенными. В ос-
нове наследственных болезней лежит генетически обусловлен-
ное отсутствие определенных белков или их модификация. Ка-
ковы бы ни были причины наследственных болезней обмен ве-
ществ, их клиническое проявление непосредственно вытекают
из тех дефектов метаболизма, которые являются следствием
нарушения структуры соответствующих белков.
Следует отметить, что эти формы патологии встречаются
довольно редко, но их значение может быть весьма велико и
последствия многих из них - тяжелые, но в ряде случаев их мож-
но избежать, если диагноз поставлен на ранней стадии и назна-
чено соответствующее лечение.
Наследственность носит в большинстве случаев рецессивно-
аутосомный характер и у гетерозиготных обычно отсутствуют
фенотипические отклонения.
5
Большие успехи, достигнутые биохимией за последние годы,
приблизили изучение жизненных процессов к молекулярному
уровню, и накапливается все больше фактического материала
о возможных связях между отдельными функциями и молеку-
лярным строением определенных соединений в живом организ-
ме. Решением этих вопросов занимается молекулярная биоло-
гия и биохимическая генетика.
Идеи молекулярной биологии нашли свое отражение в па-
тологии. Были установлены патологические состояния, связан-
ные с ненормальной молекулярной структурой важных в физи-
ологическом отношении соединений.
Отсутствие фермента приводит к трем различным послед-
ствиям - уменьшению образования продукта реакции, накоп-
лению субстрата или усилению образования других метаболи-
тов, - и может проявиться в комбинации друг с другом.
С точки зрения энзимопатологии, возникновение данного
патологического процесса можно представить как следствие
нарушения действия одной или нескольких энзимных систем.
Блокируется течение соответствующих метаболических процес-
сов и как следствие возникает заболевание с характерными для
него симптомами.
6
Глава 1
ПАТОЛОГИЯ ПИЩЕВАРЕНИЯ И ВСАСЫВАНИЯ
1.1. НАРУШЕНИЕ ПРОЦЕССОВ ПИЩЕВАРЕНИЯ
В основе возникновения нарушения пищеварения лежит ге-
нетически обусловленная или приобретенная недостаточная
выработка пищеварительных ферментов. Может наблюдаться
либо отсутствие синтеза одного или нескольких ферментов,
либо снижение их активности.
Среди врожденных энзимопатий наиболее часто встречают-
ся недостаточность дисахаридаз (лактазы, сахаразы, изомаль-
тазы), пептидаз, энтерокиназы.
Приобретенные энзимопатии наблюдаются при хроничес-
ких энтеритах, болезни Крона, резекции тонкой кишки, панк-
реатите, гепатите, циррозе печени, при заболеваниях органов
эндокринной системы (диабет, гипертиреоз), а также при при-
еме некоторых лекарственных препаратов (антибиотики, ци-
тостатики) и лучевой болезни. Из приобретенных ферменто-
патий наиболее часто встречается алиментарная ферментопа-
тия, при которой возникновение нарушений выработки и ак-
тивности ферментов связано с характером питания. Так дефи-
цит белков, витаминов, микроэлементов в рационе, несбалан-
сированное питание (аминокислотный дисбаланс, нарушение
соотношения между жирными кислотами, водорастворимы-
ми и жирорастворимыми витаминами, минеральными соля-
ми) могут приводить к стойким расстройствам процесса пи-
щеварения.
Угнетение активности и биосинтеза ферментов может быть
обусловлено токсическим воздействием некоторых естествен-
ных компонентов пищи или загрязняющими их чужеродными
примесями. Так, в таких пищевых продуктах, как бобовые, зла-
ковые, рис, яйца и др. обнаружены термостабильные специ-
фические белковые ингибиторы, образующие стойкие комп-
лексы с протеиназами желудочно-кишечного тракта и вызы-
вающие угнетение их активности. Вследствие этого наруша-
ются процессы переваривания и усвоения белка пищи. Био-
синтез некоторых ферментов нарушается при недостаточнос-
ти коферментов, в состав которых входят водорастворимые
7
витамины. Это связано с наличием в пищевых продуктах ан-
тивитаминов, которые разрушают или замещают витамины в
структуре молекулы фермента. Например, антагонистом ни-
котиновой кислоты является ниацитин, выделенный из куку-
рузы, пиридоксина - линатин, содержащийся в семенах льна.
Пресноводные рыбы содержат фермент тиаминазу, который
расщепляет тиамин.
Загрязнение пищевых продуктов солями тяжелых металлов
(ртуть, мышьяк, свинец и др.), пестицидами, микотоксинами
(афлотоксин, трихотеценовые микотоксины), которые реагиру-
ют с SH-группами белковых молекул, подавляет биосинтез бел-
ка, вызывает угнетение ферментативной активности.
В зависимости от вида пищеварения выделяют недостаточ-
ность полостного пищеварения, пристеночного (мембранного)
пищеварения и внутриклеточного пищеварения. Кроме того,
существуют смешанные формы недостаточности пищеварения.
Все эти разновидности форм сопровождаются кишечными
расстройствами. Вместе с тем каждая из них имеет свои, преж-
де всего патогенетические особенности.
При нарушении преимущественно полостного пищеварения
возникает некомпенсированное снижение секреторной функции
желудка, кишечника, поджелудочной железы, желчеотделения.
Существенную роль в ее возникновении играет нарушение дви-
гательной функции желудочно-кишечного тракта: застой содер-
жимого вследствие спазма, стеноза или сдавления кишки. По-
явлению диспепсии способствуют перенесенные кишечные ин-
фекции, изменение кишечной микрофлоры и активизация па-
тогенной флоры, вызывающей процессы брожения и гниения в
толстой кишке. В результате этого образуются такие токсич-
ные продукты, как индол, аммиак и другие метаболиты, кото-
рые раздражают слизистую оболочку кишки, усиливают ее дви-
гательную активность и, всасываясь, вызывают интоксикацию
организма. Повышение ферментативной активности микроор-
ганизмов в толстой кишке сопровождается повышенным обра-
зованием указанных токсических продуктов и в дистальных
отделах кишечника. К диспепсии приводят алиментарные на-
рушения: переедание, несбалансированное питание с употреб-
лением чрезмерного количества углеводов, белков, жиров; упот-
ребление пищи, лишенной витаминов. Особенно опасна чрез-
мерная пищевая нагрузка в сочетании с психическим и физи-
8
ческим перенапряжением, перегреванием, переохлаждением, т.е.
с факторами, приводящими к торможению секреторной функ-
ции пищеварительных желез.
Недостаточность пристеночного пищеварения - характер-
ный признак хронических заболеваний тонкой кишки, возни-
кают дистрофические и склеротические изменения слизистой
оболочки, уменьшение числа и изменение структуры ворсинок
и микроворсинок на единицу поверхности. Возникновению не-
достаточности пристеночного пищеварения способствуют из-
менение ферментного слоя кишечной поверхности и расстрой-
ства кишечной перистальтики, при которых нарушается пере-
нос пищевых веществ из полости кишки на поверхность энте-
роцитов.
Наследственные нарушения ферментативной функции
кишечника (интестинальные энзимона гии)
Недостаточность ^-галактозидазы (лактазы)
Это наиболее распространенная форма наследственной эн-
зимопатии, обусловленная дефицитом в слизистой оболочке
тонкой кишки фермента (3-галактозидазы (лактазы), который
гидролизует дисахарид лактозу на глюкозу и галактозу.
Недостаточность лактазы у детей была опубликована в 1957
году, у взрослых - в 1963 г.
Первичную лактазную недостаточность разделяют па 2 тина:
врожденную, наблюдающуюся у грудных детей, и лактазную
недостаточность взрослых (поздний тип), проявляющуюся пос-
ле периода вскармливания, чаще всего после 4-5 лет. Основным
клиническим проявлением недостаточности лактазы являются
поносы после приема в пищу молока, обычно через 1-2 часа
после употребления. При исключении из рациона молока (боль-
ные могут почувствовать себя здоровыми) клинические прояв-
ления прекращаются.
Одним из основных методов диагностики лактазного дефи-
цита является проба с нагрузкой лактозой - низкий подъем уров-
ня глюкозы в крови свидетельствует о наличии дефицита лак-
тазы (рис. 1).
9
Рис. 1. Гликемические кривые после нагрузки лактозой:
1 - у здорового человека; 2 - у больного с лактазной недостаточностью.
Кроме врожденной лактазной недостаточности, где этот де-
фицит возникает первично и является основным нарушением,
существуют и вторичные, приобретенные формы дефицита лак-
тазы при различных патологических процессах: инфекционные
заболевания (дизентерия), пострезекционные (удаление боль-
шой части тонкого кишечника), облучение и т.д.
Недостаточность ^-фруктофуранозидазы (сахаразы)
Заболевание, связанное с дефицитом сахаразы, встречается
гораздо реже, чем дефицит лактазы. Так в Северной Америке
частота данной патологии составляет 0,2%, а у населения Грен-
ландии до 5%.
Сахараза гидролизует сахарозу на глюкозу и фруктозу.
У грудных детей, питающихся материнским молоком, нет
клинических проявлений. Они возникают тогда, когда детей
переводят на искусственное или смешанное вскармливание и
дают им продукты, содержащие сахарозу. Неабсорбирующая-
ся в кишечнике сахароза способствует увеличению объема ки-
шечного содержимого и ускорению передвижения пищи по ки-
шечнику, вызывая понос, упорное вздутие живота, рвоту.
Одним из методов диагностики сахаразного дефицита явля-
ется проба с нагрузкой сахарозы. (У больных не наступало
подъема уровня глюкозы в крови, в то время как прием смеси
глюкозы и фруктозы вызывал нормальное повышение уровня
глюкозы в крови).
Плоские кривые гликемии после нагрузки сахарозой помо-
гают установлению диагноза.
10
Недостаточность трегалазы
Трегалаза гидролизует трегалозу на две молекулы глюкозы.
Дисахарид содержится в грибах (особенно молодых - до 1,4%)
и водорослях.
При расстройстве кишечника после приема грибов можно
думать о дефиците фермента трегалазы. В случае непереноси-
мости многих дисахаридов чаще всего выявляется вторичный
дефицит дисахаридаз, обусловленный приобретенными изме-
нениями слизистой оболочки тонкого кишечника.
Глютеновая болезнь (глютеночувствительная энтеропатия)
Глютен является белковым компонентом клейковины зла-
ковых (пшеницы, ржи, ячменя, овса). При эстрагировании глю-
тена алкоголем выделяются две фракции: глю геин- относитель-
но безвреден и глиадин, оказывающий токсическое действие на
слизистую оболочку тонкой кишки. Для гидролиза пептида
глиадина нужны соответствующие ферменты из группы пепти-
даз.
Основным патогенетическим механизмом глютеновой болез-
ни является врожденный дефицит специфической пептидазы,
необходимой для гидролиза глиадина и, следовательно, для его
детоксификации. Такими ферментами могут быть N-глютами-
нил-дипептидаза, дипептидилдипептидаза, глиадинамидаза.
Имеется большой экспериментальный и клинический мате-
риал, свидетельствующий о роли иммунных механизмов в па-
тогенезе глютеновой болезни. Сущность этой гипо тезы сводился
к тому, что при глютеновой болезни имеет место патологичес-
кая иммунная реакция на глютен. Гипотеза предполагает на-
личие в ткани тонкой кишки повреждающих иммунных факто-
ров. К циркулирующим иммунным факторам, обнаруживаемым
при данной патологии, относятся антитела к глютену.
Исследования в последние годы предполагают, что иммун-
ные расстройства при глютеновой патологии являются скорее,
проявлением болезни, чем изначальным патогенетическим ме-
ханизмом. Подобное предположение дает основание для объе-
динения энзимопатической и иммунологической гипотезы. Со-
гласно такой гипотезы, нерасщепленный вследствие энзимных
нарушений глютен взаимодействует с иммуноцитами слизис-
11
той оболочки кишки, что приводит к сенсибилизации их и дру-
гим иммунным реакциям, с образованием различных патоло-
гических продуктов иммуногенеза.
Заболевание может начаться в любом возрасте. Существует,
однако, два пика, когда эта болезнь начинается чаще всего: в
раннем детском возрасте и в возрасте 30-40 лет. В случаях, ког-
да болезнь появляется в зрелом возрасте, следует полагать, что
различные внешние факторы способствуют реализации генети-
ческих дефектов.
Клинической триадой глютеновой болезни считают: диарею,
стеаторею, потерю массы тела. Течение заболевания характе-
ризуется чередованием обострений с периодами удовлетвори-
тельного самочувствия, продолжительность “светлых” проме-
жутков колеблется от нескольких месяцев до нескольких дней.
Основными диагностическими критериями глютеновой бо-
лезни являются:
1. Малабсорбция (недостаточность всасывания) - синдром,
который сочетает симптомы, включающие диарею, потерю
массы тела, белковую недостаточность и признаки гиповита-
миноза.
2. Субтотальная или тотальная атрофия слизистой оболоч-
ки тонкой кишки.
Основным методом лечения глютеновой болезни является
назначение безглютеновой диеты. Клинический эффект безглю-
теновой диеты начинает проявляться через 6-7 дней после на-
чала применения. Исключаются все пищевые продукты, в со-
став которых при приготовлении входят злаки, содержащие
глютен: хлеб, сосиски, колбаса, каши из coo i ветствующих круп,
печенье, макароны и др. Пиво также содержит глютен. Не со-
держат глютен и могут использоваться при приготовлении без-
глютеновой диеты рисовый, кукурузный хлеб, овощи, фрукты,
молочные продукты, рыба, маргарин, растительное масло, са-
хар, мед и др.
Энтерокиназа является ключевым ферментом протеолити-
ческих процессов в кишечнике. Она активирует трипсиноген,
переводя его в активный протеолитический фермент - трипсин
(рис. 2).
12
Рис. 2. Активация протеолитических проферментов кишечника
(Строев Е.А., 1986).
Вследствие дефицита энтерокиназы [1] нарушается перева-
ривание белков в кишечнике, что приводит к тяжелой энтераль-
ной недостаточности, которая характеризуется выраженными
нарушениями белкового обмена, гипопротеинемией, отеками
и поносами.
Болезнь Крона
Неспецифическое воспалительное поражение различных от-
делов желудочно-кишечного тракта, характеризующееся ссг-
ментарностью, рецидивирующим течением с образованием вос-
палительных инфильтратов и глубоких продольных язв.
Этиология и патогенез болезни Крона остаются невыяснен-
ными. Предполагались различные теории происхождения за-
болевания, в том числе и генетическая.
При болезни Крона выявляются значительные нарушения
микробиоценоза кишечника. Наиболее характерно уменьшение
количества бифидобактерий и одновременное увеличение эн-
теробактерий с признаками патогенности. Воспалительные
процессы и изъязвления в пищеварительном тракте приводят к
интоксикации, профузным поносам, кишечному кровотечению,
13
анемии, потере массы тела, диспротеинемии, нарушениям элек-
тролитного обмена.
Ведущий клинический симптом - боль в животе, которая
носит схваткообразный характер. Существенного улучшения
при болезни Крона можно добиться с помощью диеты.
Кишечная липодистрофия (болезнь Уиппла)
Редкое системное заболевание, при котором в патологичес-
кий процесс вовлекается тонкая кишка. В основе заболевания
лежит нарушение метаболизма липидов.
Этиология и патогенез неясны. Возможной причиной болез-
ни Уиппла считают бактериальную инфекцию, входные воро-
та которой неизвестны. Полагают, что в развитии болезни важ-
ную роль играет фактор восприимчивости.
Кардинальными клиническими проявлениями заболевания
являются диарея, боли в мезогастрпи, нарастающая слабость,
снижение массы тела, лимфоаденонатия. Из лабораторных дан-
ных характерны стеаторея, а при выраженном расстройстве
всасывания - снижение концентрации сывороточного альбуми-
на, холестерина, калия, кальция, магния, железа.
Положительные результаты лечения дают антибиотики ши-
рокого спектра действия.
Характеризуется повышенной потерей белка с калом. В фи-
зиологических условиях ежедневно через пищеварительный тракт
организм теряет около 1/3 от общего количества вырабатывае-
мого в нем белка (пищеварительные ферменты, гормоны, белки
слущенного эпителия и даже неизменные белки, плазма крови).
Причина эксудативной энтеропатии неясна, определенную
роль в возникновении заболевания играет семейная предрас-
положенность. Патогенетической основой заболевания является
усиленный белковый катаболизм и одновременно с этим повы-
шенная потеря белка плазмы (альбумина, у-глобулина) через
эктазированные лимфатические сосуды кишечника.
Основные клинические признаки заболевания: диарея и мас-
сивные отеки (ног, лица, рук, поясницы), асцит, резкая слабость,
14
быстрая утомляемость, потеря массы тела. Главными биохи-
мическими показателями являются стойкая гипопротеинемия,
гипоальбуминемия, гипогаммаглобулинемия.
Лечение должно быть направлено на устранение диареи,
коррекцию метаболических расстройств, назначение щадящей
и богатой белком диеты.
1.2. НАРУШЕНИЕ ПРОЦЕССОВ ВСАСЫВАНИЯ
В основе развития недостаточности всасывания лежат мор-
фологические изменения слизистой оболочки тонкого кишеч-
ника, изменение ферментных систем, а также расстройство спе-
цифических транспортных механизмов, кишечный дисбактери-
оз, нарушение двигательной функции кишечника.
Выделяют врожденные-первичные (наследственно обуслов-
ленные) и приобретенные (вторичные) нарушения всасывания.
При первичных нарушениях происходят наследственные изме-
нения в строении слизистой оболочки тонкого кишечника. При этом
нарушается всасывание моносахаридов и аминокислот, липидов.
Вторичные нарушения недостаточности всасывания связа-
ны с приобретенными изменениями структуры слизистой обо-
лочки тонкого кишечника, возникающие при тех или иных ее
заболеваниях, а также болезнях других органов брюшной по-
лости с вовлечением в патологический процесс тонкой кишки.
Среди заболеваний тонкой кишки, характеризующихся на-
рушением процесса кишечной абсорбции, выделяют хроничес-
кий энтерит, глютеновую энтеропатию, болезнь Крона, болезнь
Уиппла, эксудативную энтеропатию, а также обширную резек-
цию тонкой кишки.
Недостаточность всасывания может усугубляться при сопут-
ствующих заболеваниях поджелудочной железы, при амилои-
дозах, склеродермии, лимфоме, сердечной недостаточности,
тиреотоксикозе и др.
Всасывание нарушается также при отравлениях, кровопоте-
рях, авитаминозе, лучевом поражении. При острых состояниях
нарушение всасывания связано, прежде всего, с патологией пе-
реваривания пищевых веществ и ускоренным передвижением
содержимого по кишечнику.
При хронических состояниях расстройство процесса всасы-
вания в кишечнике обусловлено дистрофическими, атрофичес-
15
кими и склеротическими изменениями эпителия слизистой обо-
лочки тонкого кишечника. При этом нарушаются крово- и лим-
фообращение, уменьшается число микроворсинок, тем самым
уменьшается общая всасывающая поверхность. Вследствие это-
го в организм в недостаточном количестве поступают продук-
ты гидролиза белков, жиров, углеводов, а также минеральных
солей и витаминов. Развивается картина, напоминающая али-
ментарную дистрофию.
Следовательно, заболевания тонкой кишки, при которых
нарушаются процессы всасывания, являются часто и причиной
недостаточного питания. Это в свою очередь сказывается на
белково-энергетической недостаточности кишечного эпителия,
период обновления которого составляет 2-3 дня. Создается по-
рочный круг. Патологический процесс в тонком кишечнике,
возникающий при белковой недостаточности, напоминает та-
ковой при его заболевании. Он характеризуется истончением
слизистой оболочки, потерей дисахаридаз щеточной каймы,
нарушением всасывания моно- и дисахаридов, уменьшением
переваривания белков, увеличением времени транспорта содер-
жимого по кишечнику, заселением бактериями верхних отде-
лов тонкого кишечника.
Кишечная микрофлора может изменяться под влиянием раз-
личных патологических процессов или экзогенных факторов,
что проявляется нарушением нормальных соотношений между
различными видами микроорганизмов и их распределением но
разным отделам кишечника. Появление измененной дисбиоти-
ческой микрофлоры и характеризует состояние, называемое
дисбактериозом. Дисбактериоз наиболее часто характеризует-
ся уменьшением общего числа микробов, иногда до полного
исчезновения отдельных видов нормальной микрофлоры с од-
новременным преобладанием видов, которые в норме присут-
ствуют в минимальных количествах.
При дисбактериозе нарушается антагонис тическая функция
нормальной микрофлоры кишечника по отношению к патоген-
ным и гнилостным микробам, отсюда нарушается витамино-
образующая и ферментативная функции. Нарушая нормальную
ферментативную активность пищеварительного тракта, изме-
ненная микрофлора приводит к образованию токсических про-
дуктов, которые всасываются в кишечнике.
Причинами дисбактериоза могут быть также заболевания
16
желудка, протекающие с ахлоргидрией, заболевания кишечни-
ка, поджелудочной железы, печени, почек, опухоли, резекция
желудка и тонкой кишки, нарушение перистальтики.
Возникновению дисбактериоза способствует необоснован-
ное и бессистемное применение антибиотиков и других анти-
бактериальных препаратов, которые уничтожают нормальных
симбионтов и приводят к размножению устойчивой к ним па-
тогенной микрофлоры.
врожденные нарушения всасывательной функции кишечника
(Малабсорбция) (первичная энтеральная недостаточность)
Нарушениевсасывания моиосахаршкав
Врожденная глюкозо-галактозная непереносимость впервые
была описана в 1962 году. У больных с первичной малабсорб-
цией глюкозы и галактозы слизистая оболочка тонкого кишеч-
ника активно не поглощает эти моносахариды или данный про-
цесс резко снижен. Причиной такой патологии является дефект
транспортного переносчика в связи с генетически обусловлен-
ными нарушениями его синтеза. Установлено, что эта патоло-
гия обязана своим происхождением мутации гена, локализован-
ного в 22-ой хромосоме.
Под действием бактериальной флоры кишечника неабсор-
бирующиеся глюкоза и галактоза подвергаются метаболичес-
ким превращениям с образованием неорганических кислот и
других углеродсодержащих фрагментов обмена углеводов.
Повышается осмотическое давление в полости кишечника, что
способствует секреции жидкости. Клиническая картина харак-
теризуется поносами, болями в животе, вздутием.
Основным диагностическим признаком являются низкие гли-
кемические кривые в крови после нагрузки глюкозой и галак-
тозой, в то время как после нагрузки фруктозой они становятся
нормальными.
Исключение из пищи продуктов, содержащих глюкозу, га-
лактозу, крахмал, лактозу приводит к нормализации патоло-
гических процессов.
Первичная малабсорбция фруктозы была описана в 1978 г.
Полагают, что нарушение всасывания фруктозы обусловлено
генетическим дефектом транспортных систем кишечника.
При нарушении абсорбции фруктозы наблюдаются вздутие
живота, боли, диарея. При соблюдении диеты с исключением
продуктов, содержащих фруктозу, все эти явления исчезали.
Первичные нарушения всасывания аминокислот и липидов
описаны в соответствующих разделах патологии белкового и
липидного обмена.
Глава 2
ПАТОЛОГИЯ ОБМЕНА ПРОСТЫХ БЕЛКОВ И
АМИНОКИСЛОТ
Белковый обмен в норме включает большое количество раз-
личных реакций и отсюда весьма разнообразные формы его
нарушений.
Общее представление о нарушениях белкового обмена дает
определение азотистого баланса.
У здорового взрослого человека количество азотистых ве-
ществ, поступающих с пищей, равняется их количеству, выво-
димому из организма (с мочой и фекалиями). Это - состояние
азотистого равновесия и в определенной степени свидетельству-
ет о динамическом единстве анаболических и катаболических
процессов.
В ряде случаев возможно возникновение положительного
азотистого баланса, когда азота из организма выводится мень-
ше, чем поступает и подобное явление свидетельствует об уси-
лении анаболических процессов. В частности, может наблю-
даться в молодом растущем организме, после тяжелых истоща-
ющих заболеваний, при беременности и при увеличении синте-
за ряда гормонов (инсулина, соматотропина, андрогенов и эст-
рогенов).
В случае, если азота выводится больше, чем его поступает,
речь идет об отрицательном азотистом балансе и может на-
блюдаться при голодании, инфекционных заболеваниях, про-
теинурии, диарее, ожогах кожи и действии ряда гормонов - глю-
кокортикоидов, йодтиронинов (последние активируют катеп-
сины и этим самым увеличивают их протеолитическое действие).
Это свидетельствует о катаболических процессах.
Нарушения обмена белков могут быть на различных этапах
превращений и среди них можно выделить следующие:
- при поступлении и переваривании белков в желудочно-ки-
шечном тракте и всасывании продуктов его гидролиза,
- синтезе и распаде белков в клетках и тканях организма,
- на различных этапах его регуляции,
, - белковом составе плазмы крови,
J-J,- на конечном этапе белкового обмена,
- метаболизме отдельных аминокислот.
2*
19
Одной из форм нарушений белкового обмена выступает раз-
витие алиментарной недостаточности белка, которая может
развиваться в силу разных причин - нарушения процессов пи-
щеварения (см. соответствующий раздел), белковом голода-
нии, несбалансированном питании, когда нарушены количе-
ственные соотношения в содержании отдельных незаменимых
аминокислот. Недостаточное поступление в организм незаме-
нимых аминокислот вызывает не только общее нарушение
синтеза белка, но и избирательное нарушение образования
отдельных белков.
В результате недостаточного поступления белка и его усвое-
ния возможны следующие последствия для организма - наблю-
дается гипопротеинемия и, как следствие, возникновение оте-
ков, развитие анемии и снижение иммунитета, сопровождаемое
склонностью к инфекционным заболеваниям. Наблюдается так-
же диарея и нарушение транспорта гормонов.
Вследствие активации катаболизма белков происходит ат-
рофия мышечной ткани, лимфоидных узлов, желудочно-кишеч-
ного тракта с последующим усугублением процессов гидроли-
за и всасывания не только белков, но и углеводов, витаминов,
минеральных веществ и др. При обследовании - отрицатель-
ный азотистый баланс.
Возможно всасывание нерасщепленного белка с развитием
аллергической реакции различной степени выраженности.
При поступлении нерасщепленного белка в толстом кишеч-
нике усиливаются процессы бактериального распада (гниения) с
образованием аминов (кадаверина, путресцина) и циклических
токсических соединений (индол, скатол, крезол), которые при
всасывании оказывают токсическое действие на организм.
2.1. ПРОТЕОЛИЗ И ЕГО НАРУШЕНИЯ
Протеолитические ферменты катализируют расщепление
пептидных связей в белках и пептидах. По современной клас-
сификации они относятся к классу гидролаз, подклассу пептид-
гидролаз, который разделяется на два подподкласса - пептида-
зы (экзопептидазы) и протеиназы (эндопептидазы, пептидил-
пептид-гидролазы). Пептидазы либо отщепляют от белков и
пептидов N- и С-концевые аминокислоты, либо гидролизуют
дипептиды. Протеиназы расщепляют внутренние пептидные
20
связи в белках, пептидах и составляют наибольшую группу пеп-
тид-гидролаз.
Протеолитические ферменты не только участвуют в неспе-
цифическом распаде белковых молекул, но имеют и регулятор-
ное значение, являясь одним из механизмов биологического
контроля функций органов и тканей организма. Регуляторный
механизм действия протеиназ включает два типа реакции. Пер-
вый связан с полным расщеплением белков до аминокислот,
которые впоследствии вовлекаются в общий метаболический
обмен. Второй обусловлен реакциями ограниченного протео-
лиза и заключается в расщеплении одной или нескольких пеп-
тидных связей в молекулах белков, что приводит к появлению
активных форм белков или пептидов. В отличие от общего про-
теолиза при этом не происходит общего расщепления белко-
вых молекул. Ограниченный протеолиз играет решающую роль
в образовании активных форм ферментов, синтезе и инактива-
ции биоактивных пептидов (ангиотензинов, кининов, пейропеп-
гидов), имеющих значение в реакциях сосудистого тонуса, ар-
териального давления, деятельности мозга, воспалительных и
аллергических реакциях.
Защитные функции организма -свертывание крови, фибри-
нолиз, иммунный ответ осуществляются в каскадных реакциях
при угнетении протеиназ с ограниченной специфичностью.
Протеолитические ферменты играют важную роль в опло-
дотворении, морфогенезе, межклеточных взаимодействиях,
онкогенной трансформации, патогенности вирусов.
Протеолитические ферменты, обладающие высокой биоло-
гической активностью, представляют потенциальную опасность
для большинства белковых структур тканей. Однако в организ-
ме существуют механизмы, контролирующие их свойства. К
одним из важнейших физиологических регуляторов протеоли-
за следует отнести специфические белки - ингибиторы крови и
тканей, которые связывают протеолитические ферменты, ли-
шая их полностью или частично каталитической активности.
В норме существует динамическое равновесие между проте-
олитическими ферментами и их ингибиторами. При ряде пато-
логических процессов происходит избыточная активация про-
теолиза, что является важным патогенетическим звеном в раз-
витии деструктивных, воспалительных, аллергических реакций,
нарушении процессов гемостаза, а также одним из факторов.
21
способствующим метастазированию клеток злокачественных
опухолей.
Изучение протеолиза и механизмов его регуляции способ-
ствует решению задач практической медицины - для диагнос-
тики ряда заболеваний, а также возможности использования
протеиназ, их активаторов и ингибиторов в качестве эффектив-
ных лечебных средств.
Все вышесказанное дает основание для рассмотрения ряда
вопросов взаимосвязи протеолиза с развитием конкретных
форм патологических процессов.
Кининовая система и протеолитические ферменты
Кинины являются вазоактивными, низкомолекулярными
пептидами, образующимися под действием различных протео-
литических ферментов с ограниченным специфическим действи-
ем, участвующими в регуляции сосудистого тонуса, процессах
микроциркуляции, воспалительных и аллергических реакциях.
Концентрация отдельных компонентов кининовой системы
в плазме крови 0,03-0,2 г/л и в физиологических условиях их
образование происходит под действием протеолитического
фермента калликреина, который в плазме крови находится в
неактивной форме в виде прекалликреина. Его активация -
сложный протеолитический процесс, осуществляющийся в кас-
кадном механизме и требующий участия нескольких белков.
Функция калликреин-кининовой системы нарушается при
ряде патологических состояний. Исследование содержания в
плазме крови контактного прекалликреина в динамике экспе-
риментального острого панкреатита показало, что в течение
первых 3-х дней заболевания наблюдается нарастание актив-
ности калликреина и повышение образования кининов.
При остром инфаркте миокарда обнаруживается резкая ак-
тивация кининовой системы, сопровождающаяся повышением
уровня кининов, увеличением активности калликреинов и сни-
жением концентрации кининогена. Особенно сильно эти изме-
нения выражены при крупноочаговых инфарктах миокарда.
Активирование кининовых систем наблюдается также при
ожоговых травмах. Излишнее содержание компонентов кини-
новой системы и ингибиторов протеолиза, способствующих
активации прекалликреинов и образованию кининов, выяви-
22
лись при нефротическом синдроме в крови, интерстициальной
жидкости, абдоминальных и плевральных экссудатах.
Большую ценность представляет изучение протеолитических
ферментов и их ингибиторов, участвующих в функционирова-
нии кининовой системы при заболеваниях печени у взрослых и
детей. Установлено, что вирусный гепатит сопровождается сни-
жением в крови уровня прекалликреина и ингибиторов каллик-
реина, степень которого зависит от тяжести болезни. При за-
тяжных и хронических формах вирусного гепатита отсутствие
нормализации этих показателей указывает на продолжающий-
ся процесс в печени. Содержание исследуемых компонентов
кининовой системы не изменяется при гепатохоленгатах и ге-
мостатических анемиях, что можно использовать для функци-
ональной диагностики вирусного гепатита и этих заболеваний.
Описаны случаи наследственной недостаточности ряда ком-
понентов калликреин-кининовой системы, сопровождающей-
ся нарушением процессов кининогенеза. Известны генетически
обусловленный дефицит протеолитического фермента кинина-
зы I с более чем четырехкратным увеличением содержания ки-
нинов в плазме крови и случаи наследственной недостаточнос-
ти высокомолекулярного кининогена.
В настоящее время проводятся работы по разработке низко-
молекулярных синтетических ингибиторов калликреина, кото-
рые могли бы использоваться в медицинских целях при пато-
логических состояниях, характеризующихся активацией кини-
новой системы.
Протеолиз и активация комплемента
Реакции ограниченного протеолиза участвуют в активации
системы комплемента, который представляет собой многоком-
понентную систему белков, которые в определенных случаях
преобразуются в активный протеолитический комплекс с мно-
гообразными физиологическими свойствами - освобождение
организма от микробов и чужеродных клеток, инактивация
вирусов, активация фагоцитоза, образование биологически
активных веществ (биогенных аминов, анафилатоксинов и др.).
Как и другие протеолитические системы крови, система ком-
племента активируется в каскадном механизме, причем ее от-
дельные компоненты приобретают способность к активации
23
после присоединения к определенной поверхности. Эта систе-
ма включает 19 плазменных белков, которые образуют 9 ком-
понентов, реагирующих друг с другом в определенной после-
довательности с формированием высокоспецифических проте-
аз, входящих в состав многокомпонентного белкового комп-
лекса.
Компоненты системы комплемента являются высокомоле-
кулярными белками, входящими в состав а-, 0- и у-глобулинов
и составляют около 5-10% от всех белков плазмы крови.
Общая концентрация комплемента в крови обычно опреде-
ляется по его гемолитической активности в отношении эритро-
цитов барана, сенсибилизированных кроличьим гемолизатом. В
последнее время широко применяются методы с использовани-
ем антисыворотки к гомогенным компонентам комплемента.
В настоящее время известен ряд заболеваний (например, си-
стемная красная волчанка), которые являются результатом ге-
нетических нарушений в системе комплемента и в основном
дефицит компонентов комплемента передается по аутосомно-
рецессивному типу.
Недостаточность компонентов комплемента и их ингибито-
ров может быть также вторичной (приобретенной). Так, в час-
тности, дефицит комплемента обнаруживается при аллергичес-
ких состояниях, инфаркте миокарда, гломерулонефрите, кол-
лагенозах, болезнях печени (цирроз, гепатит), ожогах (особен-
но перед летальным исходом).
Определение количественного содержания комплемента в
динамике патологического процесса позволяет оценить имму-
нологическое состояние организма, может иметь прогностичес-
кое значение и служить объективным критерием эффективной
терапии.
Протеиназы и ренин-ангиотензиновая система
Является наиболее важной гуморальной протеолитической
системой, участвующей в регуляции артериального давления.
Ее конечный продукт - октапептид ангиотензин II представля-
ет собой самое мощное из известных сосудосуживающих
средств. По силе вазоконстрикторного эффекта он значитель-
но превосходит норадреналин.
К ферментам, участвующим в образовании ангиотензина,
24
относятся ренин и пептидилдипептидаза. Последний является
гликопротеином и способствует не только образованию анги-
отензина II, но и инактивирует брадикинин, действие которо-
го направлено на расширение кровеносных сосудов. Следова-
тельно, данный фермент участвует в регуляции в организме
концентрации пептидов, от которых зависит уровень сосудис-
того тонуса и артериального давления.
Определение основных компонентов ренин-ангиотензиновой
системы показало возможности использования биохимических
показателей в диагностике ряда патологических процессов.
Так, у больных инфарктом миокарда изменяется активность
пептидилдипептидазы, причем интенсивность сдвигов опреде-
ляется наличием гипертонического компонента. При развитии
болезни на фоне коронарного атеросклероза без гипертоничес-
кого синдрома активность фермента находится на нижнем уров-
не нормы. При осложнении инфаркта миокарда гипертоничес-
кой болезнью - повышается. Осложнения инфаркта миокарда,
сопровождающиеся легочной патологией, вызывают значитель-
ное снижение активности фермента.
Активность пептидилдипептидазы увеличивается в плазме
крови и лимфатических узлах больных саркомой п лепрой.
При циррозе печени активность ренина повышается в зави-
симости от тяжести заболевания: в стадии компенсации она
увеличивается в 3 раза, при декомпенсированном циррозе с на-
личием асцита - в 4 раза, при циррозе печени с желтушностью
кожи в 9 раз.
Наряду с диагностическим значением пептидилдипептида-
зы в последнее время изучается возможность использования
препаратов ее ингибиторов в качестве терапевтических средств
при гипертонической болезни.
ПАТОЛОГИЯ АМИНОКИСЛОТНОГО ОБМЕНА
Одной из форм нарушения обмена веществ является патоло-
гия аминокислотного обмена. Нарушения бывают приобретен-
ные и генетические, и могут быть связаны с поступлением, рас-
пределением, синтезом и распадом аминокислот. Причины на-
рушений поступления могут быть обусловлены избытком или
недостатком субстрата (полное или белковое голодание, несба-
лансированный состав пептидов и т.д.) и белка, участвующего
25
в его превращении, т.е. фермента (см. раздел “Патология про-
цессов пищеварения и всасывания”). Недостаточное поступле-
ние или полное отсутствие незаменимых аминокислот приво-
дит к серьезным последствиям для организма. Так, в регионах,
где употребляют в пищу преимущественно злаки (которые бед-
ны триптофаном и лизином) распространены такие заболева-
ния как кахексия и квашиоркор (детская дистрофия). Кваши-
оркор развивается при переводе ребенка с грудного молока на
обедненную белком крахмальную диету. Недостаток незамени-
мых аминокислот вызывает не только общие нарушения био-
синтеза белка в организме в целом, но и избирательно наруша-
ет образование отдельных белков. Дефицит гистидина приво-
дит к снижению концентрации гемоглобина, недостаток вали-
на вызывает задержку роста, похудание и т.д. Заменимые ами-
нокислоты существенно влияют на потребность в незаменимых.
Некоторые заменимые аминокислоты становятся незаменимы-
ми, если они не поступают с пищей, потому что организм не
справляется с быстрым их синтезом. Можно предположить, что
заменимые аминокислоты могут оказаться более важными, так
как зависимость их поступления с пищей является более благо-
приятной для выживания, чем способность организма синтези-
ровать их. В организме человека девять заменимых аминокис-
лот образуются из различных метаболитов, три - из незамени-
мых аминокислот. При их биосинтезе центральное место при-
надлежит трем ферментам: глутаматдегидрогеназе, глутамин-
синтетазе и трансаминазам, которые осуществляют включение
аммиака в а-аминогруппы аминокислот.
Выделяют “врожденные ошибки обмена” или протеинопа-
тии - это результат либо полного отсутствия белка, либо сни-
жения его биологической активности. В большинстве случаев
речь идет о дефектах ферментов. Когда фермент, в норме рабо-
тающий на определенном этапе обмена, функционально не ак-
тивен, можно ожидать либо накопления субстрата, вовлечен-
ного в аномальную ферментативную реакцию, либо отсутствия
или снижения образования продукта в ходе определенной ре-
акции. На самом деле в организме реализуются обе возможно-
сти. Помимо этого могут образовываться необычные метабо-
литы, редко встречающиеся у нормальных индивидуумов, т.к.
организм стремится избавиться от субстратов, накапливающих-
ся вследствие дефекта.
26
В основе других нарушений обмена лежат наследственные
дефекты структурных и транспортных белков, а также субъе-
диниц функционирующих белков.
Научные основы учения о врожденных заболеваниях были
заложены в 1899 году английским врачом Garrod (Гаррод). Он
ввел понятие “inborn errors of metabolism” (врожденный дефект
обмена веществ).
Нарушения обмена аминокислот могут быть связаны не толь-
ко с дефектами генетических структур, но и с системами, регу-
лирующими эти процессы на клеточном уровне и уровне орга-
нов и систем в целом. Речь идет о различных метаболитах и
нервно-гуморальной регуляции, оказывающих существенное
влияние на обмен аминокислот.
Нарушения распределения аминокислот могут быть обуслов-
лены дефектом транспорта - внутриклеточного, межклеточно-
го и мембранного.
2.2. НАСЛЕДСТВЕННЫЕ НАРУШЕНИЯ ТРАНСПОРТА
АМИНОКИСЛОТ
Перенос ряда молекул через плазматическую мембрану клет-
ки осуществляется транспортными системами, специфичность
которых определяется мембранными рецепторами и белками-
переносчиками. Эти компоненты мембраны распознают отдель-
ные молекулы или структурно близкие к ним вещества и ката-
лизируют их трансмембранное перемещение с помощью меха-
низмов, недостаточно изученных на настоящий момент.
У человека обнаружено более 20 наследственных нарушений
мембранного транспорта, 10 из которых являются нарушения-
ми транспорта аминокислот.
Пять из них (цистинурия, повышенная экскреция двухоснов-
ных аминокислот, болезнь Хартнупа, иминоглицииурия и по-
вышенная экскреция дикарбоновых аминокислот) сопровож-
даются нарушением транспорта близких по структуре амино-
кислот, что свидетельствует о групповой специфичное! и мемб-
ранных рецепторов или переносчиков. При остальных пяти
дефектах нарушается транспорт только одной аминокислоты,
что говорит о существовании специфических транспортных
систем для одного субстрата. Каждое из этих 10 состояний про-
является нарушением процессов транспорта в почках, кишеч-
27
нике или в обоих органах одновременно, но при этом практи-
чески не выявлено нарушения процессов транспорта в других
тканях.
Существует точка зрения, что причина дефекта транспорта
может быть связана и с нарушениями энергетического метабо-
лизма, обеспечивающего процессы обратной резорбции ами-
нокислот путем активного транспорта.
Выделяют дефекты транспорта:
1) двухосновных аминокислот (т.е. тех аминокислот, кото-
рые имеют две аминогруппы в молекуле: это цистин, орнитин,
аргинин, лизин),
2) нейтральных аминокислот (в молекуле которых одна -
NH, и одна СООН группа),
3) дикарбоновых аминокислот (аспарагиновая и глутамино-
вая кислота),
4) иминокислот (пролин и гидроксипролин) возможно в со-
четании с глицином.
Наиболее типичное наследственное нарушение транспорта
аминокислот - цистинурия, известное с начала 19 столетия -
это выведение наименее растворимой из природных аминокис-
лот цистина с мочой (иногда до 2 г в сутки). Цистинурия встре-
чается в клинической практике чаще других наследственных
дефектов транспорта аминокислот (частота 1:600). Хотя в моче
одновременно появляются и другие аминокислоты, в частно-
сти, аргинин, лизин, орнитин, считалось, что здесь имеется на-
следственный дефект транспорта, главным образом, цистина в
извитых канальцах почек. В дальнейшем оказалось, что при
цистинурии наблюдается также нарушение активного транспор-
та цистина и основных (диаминокарбоновых) аминокислот че-
рез мембраны энтероцитов, в результате чего эти аминокисло-
ты недостаточно всасываются в тонком кишечнике.
Цистинурия вызывает образование цистиновых камней в
мочевыводящих путях и сопровождается болями, гематурией и
иногда обструкцией.
Заболевание может протекать бессимптомно. Это объясня-
ется тем, что при данной патологии не нарушено всасывание
олигопептидов, при распаде которых в дальнейшем образуют-
ся диаминомонокарбоновые аминокислоты.
С целью диагностики в качестве скрининг-теста используют
пробу с цианид-нитропруссидом (вишнево-красная окраска),
28
которая бывает положительной, когда концентрация цистина
в моче достаточно высока. Диагностическое значение имеет
прямое определение цистина и диаминокарбоновых аминокис-
лот в моче. Цистиновые камни рентгеноконтрастны.
Заболевание проявляется обычно на втором, третьем деся-
тилетии жизни. Кристаллурия и камнеобразование более веро-
ятны во время сна, когда из-за отсутствия поступления в орга-
низм жидкости сокращается объем мочи. Поэтому лечение на-
правлено на предотвращение образования камней путем введе-
ния в организм днем и ночью большого количества жидкости.
Сделаны попытки повысить растворимость цистина подщела-
чиванием мочи. Однако при pH ниже 7,5 его растворимость
существенно не изменяется, а поддерживать pH мочи выше 7,5
на протяжении длительного времени трудно. И это способствует
преципитации солей Са, что еще больше снижает обоснован-
ность такого подхода. В настоящее время эффективным мето-
дом лечения является образование смешанных дисульфидных
соединений с помощью введения Д-пеницилламина ([3-(3-
диметилцистеина). В результате с мочой экскретируется про-
дукт дисульфидного обмена, который лучше растворяется, чем
цистин. Возможность применения пеницилламина ограничена
в связи с его токсическими эффек тами. Опубликовано сообще-
ние японских исследователей о том, что глутаминовая кисло та
(до 8 г ежедневно) значительно снижает экскрецию с мочой
цистина.
К наследственным нарушениям транспорта нейтральных
аминокислот относится болезнь “Hartnup”,
Заболевание впервые было описано в 1956 году и названо по
фамилии семьи, у членов которой оно было впервые обнаруже-
но.
Причины заболевания точно не установлены и высказыва-
ется несколько точек зрения.
Большинство клинических симптомов связывают с наруше-
нием обмена триптофана. Считают, что при данном заболева-
нии нарушается транспорт триптофана и других нейтральных
аминокислот в почках и пищеварительном тракте, триптофан
подвергается гнилостному разложению в кишечнике и с мочой
выделяются производные индикана. Последние, вследствие ток-
сического действия на почки, нарушают процессы реабсорбции,
что сопровождается гипераминоацидурией. Кроме того, про-
29
исходит блок метаболизма этой аминокислоты по пути обме-
на, приводящего к образованию никотиновой кислоты из-за
врожденного дефекта фермента триптофанпирролазы. В резуль-
тате активируется боковой путь распада триптофана и проис-
ходит выделение индоловых веществ (рис. 3).
Триптофан-
пирролаза
Триптофан __________
и
И ндолил пируват
►
Индолиллактат Индолилацетат
Формилкинуренин
Никотиновая кислота
Рис. 3. Нарушение катаболизма триптофана при болезни “Hartnup”.
Из-за недостатка никотиновой кислоты возникают пеллаг-
роидные изменения кожи. Индоловые производные или/и не-
достаток никотиновой кислоты вызывают нарушения со сто-
роны нервной системы и аминоацидурию почечного типа.
Клиника:
а) поражение кожи (красная чешуйчатая сыпь);
б) нарушения функций ЖКТ (коликоподобные боли в живо-
те, диарея);
в) поражение нервной системы (проявляется обратимой моз-
жечковой атаксией, спутанностью сознания).
Диагностика: гипераминоацидурия (без повышения содер-
жания аминокислот в крови), повышение выделения с мочой
индольных соединений (индикан до 400 мг/сут., норма до 100
мг/сут.), отсутствие триптофана в крови.
Лечение: введение никотиновой кислоты или ее амида, дие-
та, фруктово-сахарные дни.
Нарушения транспорта отдельных аминокислот
Это довольно тяжелая патология. Речь идет о непереноси-
мости белка, обусловленной дефектом транспорта лизина, ар-
гинина и орнитина.
30
Заболевание было впервые описано в 1965 году в Финлян-
дии. Болезнь известнг! под различными названиями: "врожден-
ная лизинурия”, “лизинурическая непереносимость белка”, "се-
мейная непереносимость белка с нарушением транспорта ами-
нокислот”.
Дефект транспорта лизина при этом заболевании локализо-
ван на базолатеральной мембране кишечного эпителиоцита.
Транспорт основных аминокислот при врожденной лизинурии
нарушен и в почечных канальцах, вследствие чего повышается
мочевая экскреция аминокислот, особенно лизина. У некото-
рых больных отмечается отставание в психическом развитии.
Диагностика: повышенное выделение диаминокарбоновых
кислот с мочой, в первую очередь - лизина, гипераммониемия
после белковой нагрузки.
Нарушение всасывания триптофана
В 1964 году был впервые описан синдром, характеризующий-
ся изолированным дефектом всасывания триптофана в тонком
кишечнике, реабсорбция его в почечных канальцах оставалась
в пределах нормы. Это довольно редкая патология.
В неонатальном периоде дети начинают лихорадить, отста-
вать в развитии, для них характерна повышенная возбудимость.
Со стулом экскретируются в значительных количествах трип-
тофан и продукты его бактериальной деградации - индол и
триптамин. Имеет место пндиканурия. Индикан окисляется в
индиготин, который окрашивает пеленки больных детей в го-
лубой цвет, поэтому заболевание имеет еще название синдрома
“голубых пеленок”. Болезнь диагностируется на основании это-
го характерного окрашивания.
Лечение: метод лечения не разработан.
Эта патология впервые была описана французскими авто-
рами. При данном заболевании нарушен транспорт глицина,
пролина и гидроксипролина. Происходит повышенная экскре-
ция этих аминокислот с мочой, уровень их в крови в норме или
несколько снижен, реабсорбция иминокислот и глицина в по-
чечных канальцах уменьшена. Транспорт указанных аминокис-
31
лот нарушен не только в почках, но и в кишечнике. Иминогли-
цинурия обычно протекает бессимптомно, т.к. транспортный
дефект в кишечнике, вероятно, компенсируется абсорбцией
иминокислот из дипептидов. Диагноз ставится окончательно
при выявлении гипериминоацидурии спустя 6 месяцев после
рождения ребенка.
Нарушение всасываниямет нонина
Это заболевание впервые было обнаружено в 1958 г. Нару-
шается транспорт метионина в почках и тонком кишечнике. У
больных детей наблюдаются отеки, одышка, судорожные при-
падки, отставание в умственном развитии. Заболевание встре-
чается довольно редко.
Диагностика: в моче обнаруживается метионин и а-гидрокси-
масляная кислота - побочный продукт бактериального разру-
шения невсосавшегося в кишечнике метионина. Это вещество
придает моче запах “сушеного хмеля” и обуславливает, веро-
ятно, белый цвет волос таких больных.
Лечение: диета с ограничением метионина.
Гистидинурия
В 1976 году испанскими авторами был описан дефект транс-
порта гистидина в тонком кишечнике и почках у детей 9 и 11
лет с признаками слабоумия. Эта патология должна рассмат-
риваться как доказательство существования специфического
переносчика, участвующего в процессе транспорта гистидина.
Причина заболевания - нарушение структуры переносчика этой
аминокислоты, который обеспечивает транспорт гистидина
через мембраны кишечника.
2.3. НАРУШЕНИЯ ОБЩИХ ПУТЕЙ ПРЕВРАЩЕНИЙ
АМИНОКИСЛОТ
Известно, что аминокислоты могут вступать в общие пути
превращений - трансаминирование, дезаминирование, декар-
боксилирование.
32
Нарушение трансаминирования
Для реакции характерен перенос 1ЧН,-группы с аминокис-
лоты на кетокислоту без промежуточного образования NHj в
присутствии трансаминаз с коферментом ПАЛФ. ПАМФ.
Нарушение трансаминирования может иметь место:
- при недостаточности пиридоксина (беременность, подав-
ление кишечной микрофлоры);
- при торможении синтеза пиридоксальфосфата (например,
при лечении фтивазидом);
- при ограничении синтеза белка (голодание, заболевание
печени).
В изменении скорости трансаминирования существенная
роль принадлежит нарушению соотношений между субстрата-
ми реакции, а также гормонам, особенно глюкокортикоидам и
тироксину, оказывающим стимулирующее влияние на этот про-
цесс.
Нарушение окислительного дезаминирования
Окислительное дезаминирование - процесс, идущий с обра-
зованием кетокислот и свободного NHr
Нарушение дезаминирования возникает при недостач ке ком-
понентов, прямо или косвенно участвующих в реакции (недо-
статок пиридоксина, рибофлавина, никотиновой кислоты, ги-
поксия, белковая недостаточность при голодании).
Угнетение окислительного дезаминирования ведет к повы-
шению концентрации аминокислот в крови (гипераминоаци-
демия) и выделению их с мочой (гипераминоацидурия). При
этом создаются неблагоприятные условия для синтеза белков.
Нарушение декарбоксилирования
Декарбоксилирование - это процесс, протекающий с обра-
зованием СО, и аминов, ряд из которых являются биогенными
(гистамин, серотонин, ГАМК, таурин).
Накопление биогенных аминов в тканях и крови и проявле-
ние их токсического действия возникает при:
Г - усилении активности декарбоксилаз;
| - торможении активности аминооксидаз;
В. Эи к. 49 33
- нарушении связывания их с белком.
Содержание биогенных аминов в тканях и крови увеличива-
ется при гипоксии и деструкции тканей.
При патологических процессах, сопровождающихся угнете-
нием окислительного дезаминирования, превращение амино-
кислот в большей степени происходит путем декарбоксилиро-
вания с накоплением биогенных аминов.
2.4. НАРУШЕНИЯ КАТАБОЛИЗМА ОТДЕЛЬНЫХ
АМИНОКИСЛОТ
2.4.1. Нарушения распада циклических аминокислот
Наруш£ние.ката6олизма^фенилаланина
В норме из фенилаланина синтезируется тирозин.
Фенилаланингидроксилазный комплекс является оксигена-
зой со смешанной функцией. В результате реакции один атом
молекулярного кислорода включается в пара-положение фени-
лаланина, а другой восстанавливается, образуя воду. Гидрокси-
лирование осуществляется с участием биоптерина. Окисленный
биоптерин восстанавливается за счет НАДФН, (рис. 4) (I - фе-
нилаланингидроксилаза); (II - дигидробиоптеринредуктаза).
' НАДФ’
НДЦФН2
фенилаланин
тетрагидробиоптерин дигидробиоптерин
О2 Н2О ОН
тирозин
Рис. 4. Образование тирозина из фенилаланина.
Выделяют следующие причины нарушения метаболизма фе-
нилаланина:
34
1) недостаточность фенилаланингидроксилазы (гиперфени-
лаланинемия I типа, классическая фенилкетонурия, болезнь
Феллинга),
2) недостаточность дигидробиоптеринредуктазы (гиперфе-
нилаланинемия II и III типа),
3) нарушение биосинтеза дигидробиоптерина (гиперфенила-
ланинемия IV и V типа).
Наиболее часто встречающимся заболеванием среди гипер-
фениаланинемий является гиперфениаланинемия 1 типа или
классическая фенилкетонурия.
Фенилкетонурия (фени.шщювишлри.шаяо.ппофрелия.
ба 1езн ь Фелли и га)
Болезнь была открыта в 1934 году норвежским врачом и
биохимиком Феллингом (частота 1:10000). Присутствие фенил-
пировиноградной кислоты в моче больных детей он связал с
обменом фенилаланина и развивающимся у таких пациентов
слабоумием. Через 20 лет Jerves доказал, ч то при болезни Фел-
линга выпадает фенилаланингидроксилазная реакция. В резуль-
тате из фенилаланина не образуется тирозин.
При гиперфенилаланинемии 1 типа отсутствуе т фермент фе-
нилаланин-4-гидроксилаза, который в значительных количе-
ствах обнаружен только в печени и почках. Это приводит к
повышению содержания фенилаланина с 0,0015-0,004 мг до 200
мг на 1 л крови. Высокая концентрация этой аминокислоты в
течение недели - 1 месяца вызывает индукцию фермента ами-
нотрансферазы, которая катализирует превращение фенилала-
нина в фенилпируват. Путем восстановления он превращается
в фениллактат, а затем путем декарбоксилирования в фенила-
цетат (рис. 5).
Большая часть фенилацетата в печени конъюгируется с глу-
тамином и экскретируется с мочой в виде фенилацетилглута-
мина.
Поражение мозга, приводящее к умственной неполноценно-
сти, раньше объяснялось накоплением и токсическим влияни-
ем этих метаболитов, отсюда и название - фенилпировиноград-
ная олигофрения. Однако, в настоящее время считают, что ток-
сическое повреждение мозга вызывают, главным образом, вы-
сокие концентрации фенилаланина. По-видимому, происходит
35
конкурентное ингибирование активного транспорта многих
аминокислот в нервные клетки высокими концентрациями фе-
нилаланина. Нарушение аминокислотного состава ведет к ог-
раничению биосинтеза белка. Аналогично избыток фенилала-
нина влияет на транспорт аминокислот в слизистой оболочке
тонкого кишечника или/и на обратную резорбцию их в почеч-
ных канальцах. Нарушение транспорта 5-гидрокситриптофа-
на приводит к уменьшению количества нейромедиатора серо-
тонина. Ингибирование тирозиназы фенилаланином или “ток-
сическими” метаболитами снижает образование меланина, чем
объясняется неестественно светлый цвет волос и кожи у таких
больных.
Ранние симптомы болезни - повышенное усиление рефлек-
сов, возбудимость, запах плесени от пота и мочи, экземоподоб-
ная сыпь. Нервные клетки коры головного мозга разрушаются
и замещаются разрастанием микроглиальных клеток, развива-
ется олигофрения.
Дефицит тирозина усиливается еще торможением фенила-
ланином транспорта этой аминокислоты через биологические
мембраны. Это сокращает в свою очередь источник нейроак-
тивных производных тирозина, которые синтезируются в нор-
ме, в частности, катехоламинов (адреналин, норадреналин).
Экскреция фенилпирувата с мочой достигает 1-2 г/сутки и
фенилацетилглутамина до 2-3 г/сутки.
Диагностика: снижение содержания тирозина и повышение
фенилаланина в крови, а также повышение уровня фенилпиру-
вата в моче.
Свежая моча + несколько капель 5% хлорида железа (III) -
оливково-зеленая окраска.
36
Фенилаланин
глутамат
а-кетоглутарат
сн-соон
фснилацетат
н соон
CONH2
фенил ацетилглугамин
Рис. 5. Боковой путь катаболизма фенилаланина.
37
Лечение: диета с ограничением фенилаланина. Поскольку все
природные белки содержат 3-6% фенилаланина, производится
не содержащий фенилаланина гидролизат. В зависимости от
возраста ребенка необходимо принимать еще около 2-6 г био-
логически полноценного белка, чтобы компенсировать мини-
мальную потребность в фенилаланине - 15-30 мг на 1 кг массы
тела. Считается, не стоит прерывать лечение до 10 летнего воз-
раста.
Введение тетрагидробиоптерина при определенном типе ги-
перфенилаланинемии снижает уровень фенилаланина в крови,
но не влияет на общее состояние ребенка, т.к. он не проникает
через гематоэнцефалический барьер, поэтому эффективность
заместительного введения птеридинового кофактора находит-
ся в стадии изучения.
Алкашонурия
Это наследственное заболевание описано в литературе еще
в 16 веке. (Частота 1:200000).
Оно проявляется в потемнении (иногда почернении) мочи
при длительном стоянии на воздухе. Откуда же появляется тем-
ное красящее вещество - алкаптон? Тирозин, подвергаясь даль-
нейшим превращениям в организме, образует гомогентизино-
вую кислоту (рис. 6). В норме ее окисляет фермент оксидаза,
содержащийся, главным образом, в печени. Разрывая кольцо в
молекуле гомогентизиновой кислоты, он превращает ее в мале-
илацетоуксусную кислоту, а затем в фумарилацетоуксусную
кислоту, расщепляющуюся с образованием фумарата и ацетоа-
цетата. В 1962 году были получены доказательства, что причи-
ной заболевания является врожденное отсутствие фермента го-
могентизинатоксидазы в печени и почках. При алкаптонурии
этот фермент не синтезируется, и гомогентизиновая кислота
выделяется с мочой в больших количествах. При стоянии мочи
кислота, особенно в присутствии щелочи, присоединяет кисло-
род и образует алкаптон (что означает “жадно поглощающий
щелочь”). Признаки недостаточности фермента могут наблю-
даться вскоре после рождения. Так как ферментативный дефект
находится ниже того уровня, на котором тирозин утилизирует-
38
ся для синтеза белка и превращения, например, в нейромедиа-
торы, то недостаточность оксидазы не оказывает быстропро-
являющегося влияния на обмен. В детском возрасте потемне-
ние мочи - единственное проявление дефекта (клинически он не
обнаруживается). Позднее, в связи с накоплением в тканях про-
дукта полимеризации гомогентизиновой кислоты - алкаптона
- развивается охроноз.
Охроноз - пигментация соединительной ткани. Пигмент тем-
ного цвета откладывается в хрящах носа, ушных раковинах,
эндокарде, крупных кровеносных сосудах, почках, легких. При
значительных отложениях пигмента в суставах наблюдается
нарушение их подвижности.
Предполагают, что механизм охроноза обусловлен окисле-
нием гомогентизината полифенолоксидазой, в результате чего
образуется бензохиноацетат, который далее полимеризуется и
связывается с макромолекулами соединительной ткани (рис. 6).
Гомогентизиновая кислота и ее окисленные полимеры свя-
зываются коллагеном. Алкаптонурии часто сопутствует почеч-
но-каменная болезнь.
Диагностика: моча больных темнеет на воздухе или, особен-
но, при добавлении щелочи. Желчь, порфирины, миоглобин
также делают мочу темной, поэтому необходимы дополнитель-
ные исследования.
I. Реакция Бенедикта (с водным раствором CaSO4. Na,СО, и
цитратом натрия) коричневая окраска с оранжевым осадком.
П. Реакция с хлоридом железа - фиолетово-черная окраска.
111. Реакция с нитратом серебра - черная окраска.
Лечение: у мужчин заболевание встречается в два раза чаще,
чем у женщин. Радикальных способов лечения пока не разра-
ботано. Рекомендуется диета с ограничением фенилаланина и
тирозина, но на короткие периоды времени, т.к. фенилаланин
и тирозин незаменимая и полузаменимая аминокислоты. Име-
ются данные, что большие дозы витамина С предупреждают
отложение темного пигмента.
К этой же группе заболеваний (нарушения катаболизма ти-
розина) относят альбинизм. (Частота - около 5 случаев на 100000
человек).
г
II и
39
СН2-СН-СООН
^н2
он
Тирозин
тирозинтрансаминаза
ОН
п-гидроксифенилпируватгидроксилаза
Фумарилацетоуксусная кислота
гидролаза
Фумарат ацетоацетат
Рис. 6. Основной путь катаболизма тирозина и его нарушения.
40
Альбиносы - люди с белыми волосами, кожей и розово-крас-
ными глазами (из-за просвечивающихся капилляров), что яв-
ляется следствием отсутствия красящего вещества - меланина.
Причиной заболевания является недостаток фермента тирози-
назы, который окисляет тирозин и ускоряет дальнейшие пре-
вращения ДОФА (рис. 7).
Меланоциты Тирозин Надпочечники
Дигидроксифснилаланип (ДОФА)
^1 Тирозиназа
ДОФА-хинон
ДОФА-хром
Меланин
ДОФА
ДОС^амин
Норадреналин
л
Адреналин
Рис. 7. Схема метаболических нарушении при альбинтме.
Серьезных нарушений это заболевание нс вызывает, лишь
приходится избегать действия прямого солнечною света.
К нарушениям катаболизма тирозина относятся: тпрозине-
мия I типа (тирозиноз), тирозинемия II типа (синдром Рихщра-
XaH.xapia) и юрозинемия новорожденных Пранзизорпая! про-
зинемия).
При тирозинозе дефектным ферментом является фумарнла-
цетоацстат-гидролаза (рис. 6).
Содержание тирозина в плазме повышается до 6-12 мг/100
мл, увеличено содержание и некоторых других аминокислот,
особенно метионина (до 10-50 мг/л). В крови накапливаются и
выделяются с мочой предшественники фумарилацетоуксусной
кислоты, а также образующиеся из них сукцинилацетон и сук-
цинилацетоацетат. Эти соединения накапливаются в печени и
почках, вызывая повреждение этих органов. Сукцинилацетон
ингибирует порфобилиногенсинтетазу (фермент синтеза гема),
что способствует накоплению и экскреции 8-аминолевулино-
вой кислоты. Известны острая и хроническая формы тирози-
ноза. Острое проявление заболевания характеризуется гепато-
мегалией, “капустным” запахом, задержкой в развитии, асци-
41
том и характерно для младенческого возраста. Если не прово-
дится лечение, то летальный исход наступает в возрасте 6-8 ме-
сяцев. При хроническом течении заболевания наблюдается узел-
ковый цирроз печени, синдром Фанкони. Летальный исход на-
ступает в возрасте примерно 10 лет.
Диагностика: реакция Бенедикта - положительна, тирозину-
рия, аминоацидурия, гиперметионинемия, повышение содержа-
ния 5-аминолевулиновой кислоты в моче.
Лечение: диета с пониженным содержанием фенилаланина и
тирозина, в ряде случаев (если необходимо) и метионина.
При тирозинемии II типа предполагаемой причиной мета-
болических нарушений является недостаточность тирозинтран-
саминазы печени (рис. 6). Заболевание характеризуется повы-
шением содержания тирозина в плазме до 4-5 мг/100 мл, харак-
терными поражениями глаз и кожи, умеренной умственной от-
сталостью. Отмечается тирозинурия, ио клубочковая фильтра-
ция и реабсорбция тирозина остаются в пределах нормы. С
мочой экскретируются n-гидроксифенилпируват, п-гидрокси-
фсниллактат, n-гидроксифенилацетат и тирамин.
Диагностика: тирозинемия, тирозинурия (тирозин является
единственной аминокислотой, концентрация которой в моче
повышена).
Лечение: диета.
Причиной транзиторной тирозинемии считается недостаточ-
ность п-гидроксифенилпируватгидроксилазы - фермента, ката-
лизирующего образование гомогентизиновой кислоты (рис. 6).
Обычно заболевание проявляется у недоношенных детей отста-
ванием в развитии. Существует мнение, что активность этого
фермента у таких детей подавляется тирозином, содержащим-
ся в их пище в избытке.
Диагностика: тирозинемия (концентрация тирозина в 2-3 раза
выше нормы), повышенное содержание п-гидроксифенилпиру-
вата в крови. Реакции Бенедикта и с хлоридом железа - поло-
жительны. В моче повышено содержание тирозина, п-гидрокси-
фенилацетата, N-ацетилтирозина и тирамина.
Лечение: диета со сниженным содержанием белка.
42
Нарушения катаболизма гистидина
Гистидинемия
Превращения гистидина в организме достаточно изучены.
Основной путь ферментативного распада гистидина начинает-
ся с образования уроканиновой кислоты, которая подвергает-
ся дальнейшим превращениям (эта кислота была впервые вы-
делена из мочи собак) (рис. 8).
CH-NH,
СН
СООН
СООН
СООН
Гистидин
уроканат
И м и дазолi1 ро и I io новая
кислота
> N-формилглутаминовая кислота ------> глутамат
Рис. 8. Катаболизм гистидина и его нарушения.
При гистидинемии имеет место врожденный дефицит гисти-
дазы в печени.
У больных возрастает уровень гистидина в плазме и частич-
но в моче. Заболевание диагностируют к концу первого года
жизни или позднее: дефект речи, связанный с нарушением слу-
ховой памяти, снижение интеллекта. Ребенок чаще других под-
вержен инфекционным заболеваниям, отличается малым рос-
том, появляются судороги и изменения на ЭКГ.
Диагностика: норма гистидина в крови 4-10 мг/л, у больных
от 20 до 270 мг/л.
Лечение: рациональных способов лечения не разработано.
43
Мастоцитоз
Наследственное заболевание, причина которого повышение
активности гистидиндекарбоксилазы. Образующийся гистамин
накапливается в печени, селезенке и других органах. Заболева-
ние сопровождается усиленной пролиферацией тучных клеток,
нарушениями деятельности сердца, функции ЖКТ и пораже-
нием кожи.
Диагностика: повышение экскреции гистамина с мочой.
2.4.2. Нарушения распада ациклических аминокислот
Циепшоз
При цистинозе малорастворимая аминокислота - цистин как
бы “выпадает из обмена” и откладывается во всех органах и
тканях. Это очень тяжелое наследственное заболевание, кото-
рое не нужно путать с цистинурией.
Болезнь характеризуется накоплением свободного цистина
в лизосомах разных тканей организма. Кристаллы цистина по-
являются в роговице, конъюнктиве, костном мозге, лимфати-
ческих узлах, лейкоцитах и внутренних органах. Существуют
три формы болезни:
инфантильная (нефропатическая), характеризующаяся раз-
витием почечной недостаточност! i в течение первых 10 лет жиз-
ни;
ювенильная (промежуточная), при которой поражение по-
чек проявляется в течение второго 10-летия жизни;
взрослая (доброкачественная), сопровождающаяся отложе-
нием цистина в роговице, но не в почках.
Причиной болезни является нарушение оттока цистина из
лизосом, но не нарушение его распада. Этот “отток” представ-
ляет собой активный АТФ-зависимый процесс. Внутриклеточ-
ный цистин локализуется в лизосомах и не обменивается с дру-
гими внутри- и внеклеточными пулами этой аминокислоты.
Концентрация цистина в плазме и моче существенно не изме-
няется.
При инфатильной форме содержание цистина в тканях мо-
44
жет превышать норму более чем в 100 раз, а при взрослой фор-
ме более чем в 30 раз. При взрослой форме болезни развивается
только глазная патология, которая характеризуется проявле-
нием фотофобии из-за отложения цистина в роговице, чувством
жжения или зуда в глазах.
Заболевание проявляется в потере аппетита, усиленной жаж-
де, рвоте, светобоязни, повышенной температуре, задержке раз-
вития. Возникает картина тяжелого рахита. Характерный при-
знак - гипераминоацидурия - повышенное выделение аминокис-
лот с мочой, особенно лейцина, пролина и валина.
Диагностика: присутствие гексагональных или прямоуголь-
ных кристаллов цистина в роговице, слизистой оболочке пря-
мой кишки, костном мозге. Диагноз подтверждают с помощью
количественного определения цистина в лейкоцитах перифери-
ческой крови. Моча - гипераминоацидурия.
Лечение: симптоматическое. В течение последнего времени
с целью снижения внутриклеточного уровня цистина применя-
ется препарат цистеамин, связывающий сульфгидрильные груп-
пы.
Витамины Е и Вр также способствуют восстановлению ди-
сульфидных групп в молекуле цистина и лучшему использова-
нию образовавшегося цистеина.
Гомопистинурия (см. “Нарушение обмена витаминов”).
Нарушение катаболизма разветвленных аминокислот
Лейниноз или болезнь “кленового сиропа”
Возникает при недостаточном декарбоксилировании окси-
кислот, которые образуются в результате трансаминирования
трех аминокислот с разветвленной цепью атомов углерода в их
молекуле: валина, лейцина, изолейцина.
Причиной болезни является отсутствие или снижение актив-
ности декарбоксилазы, катализирующей превращение трех раз-
ветвленных а-кетокислот в ацилКоА-тиоэфиры.
Диагностика: в крови повышается уровень разветвленных
аминокислот и их кетопроизводных, увеличивается выделение
их с мочой. Поэтому заболевание иногда называют кетонури-
ей разветвленных кетокислот. Отмечено также присутствие в
45
моче разветвленных а-гидроксикислот, которые образуются
при восстановлении а-кетокислот. Моча и выдыхаемый воздух
имеют запах “кленового сиропа” или жженого сахара.
У больных отмечается мышечная слабость с задержкой ум-
ственного развития, слепота, сильная рвота.
Лечение: диета с низким содержанием белка с добавлением в
пищу незаменимых аминокислот.
Скачкообразная кетонурия разветвленных кетокислот
Это одна из форм болезни “кленового сиропа”, которая свя-
зана с меньшими структурными изменениями декарбоксилазы
а-кетокислот. Активность этого фермента в лейкоцитах и фиб-
робластах несколько снижена, но выше, чем в классических слу-
чаях болезни “кленового сиропа”. Больные обладают неболь-
шой способностью к катаболизму валина, лейцина и изолейци-
на, и симптомы болезни наблюдаются лишь эпизодически.
2.5. НАРУШЕНИЯ ЦИКЛА МОЧЕВИНООБРАЗОВАНИЯ
У человека в сутки подвергается распаду около 70 г амино-
кислот, при этом в результате реакции дезаминирования и рас-
пада биогенных аминов освобождается большое количество
аммиака, который является высокотоксичным соединением.
Одним из путей обезвреживания этого вещества является об-
разование амидов глутамина и аспарагина. Так как в голов-
ном мозге это основной способ детоксикации, следовательно,
любые нарушения этого процесса несовместимы с жизнью.
Главный путь удаления аммиака из клеток - синтез мочевины
в печени. Существуют наследственные нарушения, вызывае-
мые частичным блокированием одной из реакций цикла мо-
чевины. Полное отсутствие какого-либо фермента урогенеза
вызывает кому и приводит к смерти сразу после рождения.
Частичная недостаточность характеризуется задержкой ум-
ственного развития, рвотой, нарушением координации дви-
жения, сонливостью, отвращением к белковой пище. При ар-
гинино-сукцинатной ацидурии нарушается еще рост волос. Все
заболевания вызывают гипераммониемию, которая сдвигает
равновесие реакции, катализируемой глутаматдегидрогеназой
46
в сторону образования глутамата. Это в свою очередь снижа-
ет уровень 2-оксоглутарата, который является метаболитом
цикла Кребса. В конечном итоге это приводит к уменьшению
продукции АТФ.
Выявлены нарушения, обусловленные недостатком каждо-
го из 5 ферментов синтеза мочевины.
Гипераммониемия I типа
Заболевание, вероятно, наследственное, связано с недоста-
точностью фермента карбомоилфосфатсинтетазы. Снижение
образования карбомоилфосфата приводит не только к наруше-
нию синтеза мочевины, по и к нарушению образования пири-
мидиновых нуклеотидов в организме.
Диагностика: высокий уровень аммония в крови, в некото-
рых случаях имеет место гиперглутаминемия, гипералашшемия.
гиперлизинемия. Диагноз подтверждают путем определения ак-
тивности фермента в печени и лейкоцитах.
Лечение: строгое ограничение белка, гемодиализ, назначе-
ние бензойной и фенилуксусной кислот (300-350 mi7кг) для сти-
муляции образования продуктов ацетилирования накопивших-
ся аминокислот. Эти продукты ацетилирования и фенилацетил-
глутамин, вероятно, служат акцепторами аминогрупп, в норме
удаляющихся из организма в виде мочевины. Таким образом,
применение этих веществ облегчает выведение из организма
избытка аминогрупп аминокислот.
Гипераммониемия II типа
Это наиболее распространенное заболевание среди вышеука-
занной группы болезней, обусловленное недостатком фермен-
та орнитинкарбамоилтрансферазы.
Диагностика: повышение содержания глутамина в крови,
моче и спинномозговой жидкости, т.к. из-за высокого содер-
жания аммиака в тканях происходит усиленное образование
этого вещества под действием глутаминсинтетазы. Для заболе-
вания характерно наличие кристаллов оротовой кислоты в
моче. При недостаточности орнитинкарбамоилтрансферазы на-
капливается карбамоилфосфат, который вовлекается в путь
синтеза пиримидинов. Избыток оротовой кислоты образует в
47
моче игольчатой формы кристаллы (это основное отличие ги-
пераммониемии II типа от гипераммониемии I типа).
Лечение: ограничение белка, гемодиализ, введение цитрата для
стимуляции образования 2-оксоглутарата, редко-бензоат натрия.
Это очень редкое наследственное заболевание, характеризу-
ющееся снижением активности фермента аргининосукцинат-
синтетазы. Описаны случаи полного отсутствия активности
этого фермента.
Диагностика: повышение содержания цитруллина в крови и
моче.
Лечение: ограничение белка, назначение аргинина для по-
вышения экскреции цитруллина.
Заболевание связано с отсутствием фермента аргининосук-
цинат-лиазы в мозгу, печени, почках, эритроцитах и фиброб-
ластах кожи.
Диагностика: обнаружение в моче аргинино-сукцината (2-9
г/сут), иногда используют определение аргининосукцинат-ли-
азы в эритроцитах. Для ранней диагностики анализируют кровь,
взятую из пупочного канатика. Так как фермент содержится в
клетках амниотической жидкости, то иногда проводят пункцию
плодного пузыря (амниоцентез). В некоторых случаях имеет
место гипераммониемия и гипераминоацидурия (глутамин, ала-
нин, лизин).
Лечение: назначение аргинина и бензоата натрия с целью
увеличения экскреции азотсодержащих метаболитов, ограни-
чение белка.
Г ипераргининемия
Это нарушение синтеза мочевины, обусловленное снижени-
ем содержания в эритроцитах аргиназы.
Диагностика: повышение содержания в моче аргинина, а так-
же лизина и редко цистина.
Лечение: низкобелковая диета. ’
48
Глава 3
ПАТОЛОГИЯ ОБМЕНА СЛОЖНЫХ БЕЛКОВ
Сложные белки представляют собой большую группу хими-
ческих соединений с различной структурой ее небелковой час-
ти, характер которой определяет вид сложного белка.
Сборка определенного вида протеида представляет собой
сложный биохимический процесс, который условно может быть
разделен на два этапа - формирование белковой и небелковой
частей с последующей трансформацией в структуру сложного
белка.
Нарушение каждого этапа биосинтетического механизма
определенным образом сказывается и на структурной органи-
зации в целом и ведет к формированию дефектного протеида,
что в свою очередь вызывает изменения на различных путях
метаболизма и, как следствие, изменение обменных процессов,
возникновение той или иной форм патологии, получивших в
целом название “молекулярных болезней”.
В данном разделе рассмотрим возможные патологические
процессы, связанные с нарушением синтеза геминовой группи-
ровки и белковой, входящих в хромпрогеид гемоглобин. Рас-
смотрим основные этапы синтеза протопорфирина.
3.1. НАРУШЕНИЯ ОБМЕНА ГЕМПРОТЕИДОВ
Биосинтез гема
Начало исследований принято относить к 1945 году, когда
американский ученый Шемин поставил эксперимент на соб-
ственном организме, приняв порцию глицина, содержащего
изотоп N15. Затем было выявлено его включение в гемоглобин,
а позднее показано, что глицин конденсируется сукцинил-КоА,
давая 5-аминолевулиновую кислоту (5-АЛК). Данная реакция
катализируется АЛК-синтазой с коферментом пиридоксальфос-
фатом.
Схематически биосинтез гема можно представить следую-
щим образом.
4. 11Щ.«
49
глицин + сукцинил-S-KoA
3-аминолевулинатсинтаза
(8-АЛК-синтаза)
СО, + HS-KoA
2х 8-аминолевулиновая кислота (8-АЛК)
порфобилиногенсинтаза
(ПБГ-синтаза)
2Н,О
4х порфобилиноген (ПБГ)
* уропорфириноген I
УПГ-синтаза
УПГ-косинтаза
копропорфириноген I
уропорфириноген III (УПГ-Ш)
УПГ-декарбоксилаза
4СО3
копропорфириноген Ill (КПГIII)
КПГ-декарбоксилаза
КПГ-оксидаза
протофорфириноген IX (ППГ IX)
ПБГ
дезаминаза
ППГ-оксидаза
протопорфирин IX
УПГ-1
косинтаза . j
гемсинтетаза (феррохелатаза)
Fe-
УПГ-Ш
гем
Как следует из представленной схемы, конденсация двух
молекул АЛК с участием порфобилиногенсинтазы приводит к
образованию одного пиррольного кольца - порфобилиногена
(ПБГ). Из четырех молекул ПБГ в присутствии УПГ-синтазы
50
образуется уропорфириноген (УПГ) и это соединение может
присутствовать в виде четырех изомеров.
Согласно имеющимся на сегодня представлениям из порфо-
билиногена под действием ПБГ-дезаминазы (УПГ-синтазы)
образуется первая изомерная форма (УПГ-1).
Конденсация ПБГ в УПГ-Ш состоит из двух этапов. На пер-
вом происходит полимеризация ПБГ под действием фермента
ПБГ-дезаминазы в линейный тетрапиррол. На втором в при-
сутствии фермента косинтазы происходит не только замыка-
ние билана в макроцикл, но и поворот одного из пиррольных
колец (пиррола Д) с образованием природного III типа изоме-
ра - УПГ-Ш. При отсутствии косинтазы билан замыкается в
другой изомер-УПГ-I, который в последующем биосинтезе не
участвует. Только при наличии двух ферментов дезаминазы и
косинтазы ПБГ замыкается в изомер III типа. Следует отме-
тить. что отдельно взятая косинтаза не полимеризует ПБГ и не
в состоянии изомеризовать УПГ-I в УПГ-Ш.
Из УПГ III образуется гем посредством двух типов химичес-
ких превращений: изменения в боковых цепях или окисления
(дегидрирования) самого кольца.
Согласно принятой номенклатуре для обозначения произ-
водных, различающихся по составу боковых цепей добавляют
приставки уро-, копро- и прото-. Природу кольца обозначают
второй частью слова: более восстановленные называются пор-
фириногенами, более окисленные - порфиринами.
В ходе реакций всегда образуется некоторое количество
УПГ I, который, как и его изомер типа III, подвергается декар-
боксилированию и восстановлению, а образовавшийся копро-
порфирин I выводится с мочой.
Следует отметить, что наиболее растворимый уропорфири-
ноген не попадает в кал, но обнаруживается в моче. Менее ра-
створимый КПГ присутствует в моче и кале.
Нарушение биосинтеза геминовой группировки ведет к раз-
витию патологического процесса вследствие энзимопатии на
определенных уровнях.
4*
51
3.1.1. Порфирии и порфиринурии
Порфирии
Под порфириями понимают группу заболеваний, ведущие
симптомы которых связаны с накоплением в организме проме-
жуточных продуктов синтеза порфиринового кольца. По свое-
му характеру могут быть врожденными или приобретенными.
Классификацией, принятой в 1963 году на международной
конференции в Кейптауне, заболевания этой группы подразде-
ляются на эритропоэтические и печеночные порфирии в соот-
ветствии с двумя основными центрами синтеза ггорфиринов
эритробластами костного мозга и печенью.
Эритропоэтические порфирии
К данной группе заболеваний относятся уро-, прото- и коп-
ропорфирии.
Уропорфирия (болезнь Гюнтера, врожденная порфирия)
В патогенезе болезни, по всей видимости, имеет значение
повышение активности АЛК-синтазы и несколько понижается
активность косинтазы УПГ III.
В результате из избыточного количества порфобилиногена
образуется УПГ 1, который не может использоваться для даль-
нейших превращений.
Следует отметить, что количество образующегося УПГ III
достаточно для синтеза гема, поэтому у больных гипохромная
анемия отсутствует.
Заболевание характеризуется поражением почек, гемолити-
ческой анемией и внутриклеточным гемолизом. Связано с тем,
что избыточное отложение уропорфирина в эритроцитах при-
водит к укорочению продолжительности их жизни и повышен-
ному гемолизу. При этом из эритроцитов освобождается боль-
шое количество уропорфириногена, который окисляется в уро-
порфирин и откладывается в коже, обуславливая фотосенсиби-
лизацию.
При биохимическом исследовании в моче значительное ко-
личество УПГ I и в меньшей степени КПГ I. Наблюдается на-
копление УПГ I в эритроцитах, которые в УФ лучах светятся
52
красным светом. В костном мозге больных обнаруживаются
интенсивно флюорисцирующие в УФ лучах нормобласты (пор-
фиробласты).
Лечение: так как гемолиз происходит в основном в селезен-
ке, то выраженный гемолиз является показанием для спленэк-
томии.
В патогенезе - отсутствие феррохелатазы и возможное уве-
личение активности 8-АЛК-синтазы.
Из клинических признаков отмечается повышенная чувстви-
тельность к солнечному облучению (отек, зуд, покраснение
кожи), при длительном пребывании на солнце - геморрагичес-
кие высыпания.
При биохимическим исследовании возрастает содержание
протопорфирина IX в эритроцитах (в 20-100 раз при норме до 50
мкг/100 мл) при нормальном содержании уро- и коиропорфири-
нов. У некоторых больных обнаруживается значительное увели-
чение количества протопорфирина и копропорфирнна в кале.
Эритропоэтическая копропорфирия
Встречается исключительно редко и патогенез точно неиз-
вестен.
Клинические проявления те же, что и при эритроиоэгичес-
кой протопорфирии.
Биохимические показатели - содержание протопорфирина
в эритроцитах повышается в 30-80 раз по сравнению с нормой.
Количество порфиринов в моче возрастает незначительно в
основном за счет копропорфирнна.
Печеночные порфирии
К данной группе порфирий относятся следующие формы:
Острая перемежающая порфирия (пироллопорфирия. порфи-
рия шведская)
Возникает вследствие увеличения активности 8-АЛК-синта-
зы и метаболического блока ПБГ-дезаминазы.
Проявляется в раннем детском возрасте. Характеризуется
приступами острых болей в животе с диспепсией, сопровожда-
53
ющихся нерезким подъемом температуры, лейкоцитозом, что
может симулировать клинику “острого живота”. Наряду с эти-
ми признаками формируются неврологические нарушения в
виде полиневритов. Поражение нервной системы может прояв-
ляться эпилептическими припадками, с расстройствами психи-
ки (бредовые идеи, галлюцинации, нарушение сознания).
Клинические проявления связаны, согласно имеющимся дан-
ным, с накоплением в нервных клетках токсического вещества
- 8-АЛК. Показано, что аминолевулиновая кислота накапли-
вается в гипоталамусе и тормозит активность Na+, К+-зависи-
мой АТФазы. что ведет к нарушению транспорта ионов через
мембраны и нарушает функцию нервного волокна. Развивает-
ся его демиелинизация, аксональная невропатия, что и обус-
лавливает клинические проявления болезни.
При обследовании выявляется значительное выделение с
мочой порфобилиногена и его предшественников, а также
8-АЛК. Моча таких больных приобретает красную окраску.
Лечение состоит в назначении диеты, содержащей избыток
углеводов и аденозиновых препаратов (фосфаден), тормозящих
синтез порфиринов.
Наследственная печеночная копропорфирия - относится к чис-
лу довольно редких форм порфирий и, вероятнее всего, связана
с нарушением активности КПГ-оксидазы в печени, при этом
обнаруживается повышение активности АЛК-синтазы.
Клинические проявления во многом характерны как для ос-
трой перемежающей порфирии.
В период обострения процесса в моче обнаруживается по-
вышение количества АЛК и ПБГ, которые, однако, бывают
ниже, чем при острой перемежающей порфирии. В моче и кале
значительно возрастает количество копропорфирина.
В период ремиссии содержание ПБГ и АЛК может быть нор-
мальным, при повышенном содержании копропорфирина в
моче и кале.
Копропротопорфирия (вариегатная пестрая порфирия)
В основе патогенеза нарушение активности протопорфири-
ногеноксидазы (ППГ-оксидазы) и наблюдается нарастание ак-
тивности АЛК-синтазы.
54
Заболевание характеризуется неврологической симптомати-
кой как при острой перемежающей порфирии. Наблюдается од-
новременно и кожная симптоматика: отек, везикулы, гиперт-
рихоз, склеродермия. Отмечено, что кожные проявления чаще
встречаются у мужчин, невралогические нарушения-у женщин.
При биохимическом обследовании в период обострения в моче
понижено содержание порфобилиногена, копропорфирина I и
III, уропорфирина. В межприступный период в отличие от ост-
рой порфирии повышено содержание копропорфирина в кале.
В качестве лечения рекомендуются те же мероприятия, как и
при острой перемежающейся порфирии.
Урокопропорфирия (поздняя кожная порфирия)
Относится к числу наиболее часто встречающихся форм пор-
фирий.
Наблюдается у лиц, имевших длительные контакты с гепа-
тотропными ядами, перенесших гепатит, злоупотребляющих
алкоголем. Во всех этих случаях развивается нарушение функ-
ционального состояния печени. Это позволило предположить,
что урокопропорфирия - приобретенная форма порфирии. В
то же время высказывается мнение и о наследственной пред-
расположенности к болезни, что возможно связано с наруше-
нием активности УПГ-декарбоксилазы. Возможно также и на-
рушение восстановления уропорфирина в уропорфириноген,
необходимый для синтеза гема.
У ряда больных в течение всей жизни может наблюдаться
высокое содержание порфиринов, при нормальном состоянии
печени происходит их выделение с желчью и содержание пор-
фиринов в коже и моче остается в норме. Однако при наруше-
нии функции печени порфирины не выводятся в достаточном
количестве, остаются в организме и откладываются в печени и
коже.
Заболевание характеризуется выраженной кожной симпто-
матикой, которая чаще у лиц старше 40 лет. Основные призна-
ки - повышение чувствительности к легкой механической трав-
ме и солнечному облучению, увеличение или уменьшение пиг-
ментации, диффузное истончение или утолщение кожи (харак-
терно для псевдосклеродермии).
55
Порфиринурии
Представляют собою вторичные нарушения обмена порфи-
ринов и являются следствием основного заболевания.
В качестве примера рассмотрим некоторые из них:
При гипохромной анемии вследствие дефицита железа в эрит-
робластах у больных нарушается синтез гемоглобина. Порфи-
рины не используются для синтеза гема, а отсюда в эритроци-
тах возрастает содержание протопорфирина и в меньшей сте-
пени копропорфирина. Считается, что повышение количества
протопорфиринов в эритроцитах является ранним признаком
дефицита железа в организме.
Прианемиях, вызванных длительными инфекционными про-
цессами (абсцесс, туберкулез, остеомиелит и др.), содержание
железа в эритроцитах снижено, хотя общее количество железа
в организме не изменяется, имеет место, так называемый, пере-
распределительный дефицит. В эритроцитах отмечается нарас-
тание концентрации копропорфиринов и в меньшей степени
протопорфиринов.
При гемолитических анемиях изменения в порфириновом
обмере обусловлены активацией эритропоэза. В эритроцитах
увеличено содержание прото- и копропорфиринов, в моче -
АЛК и ПБГ.
Повышение уровня копропорфиринов может наблюдаться
также и в других случаях - инфаркте миокарда (одновременно
в моче также увеличивается концентрация АЛК), лихорадоч-
ных состояниях, ревматизме, авитаминозах (тиамина, рибоф-
лавина, пиридоксина, ниацина и др.).
Во всех этих случаях содержание в моче копропорфирина
всегда выше, чем уропорфиринов, что является основным от-
личием порфиринурии от порфирий, при которых в моче пре-
обладают уропорфирины I, III и их предшественники АЛК и
ПБГ.
3.1.2. Метаболизм билирубина и его нарушения
Билирубин образуется главным образом из гема, который
56
высвобождается из гемоглобина, когда эритроциты по мере
старения выводятся ретикуло-эндотелиальной системой из кро-
вотока. Железо гема реутилизируется, а тетрапиррольное коль-
цо разлагается до билирубина. Другими источниками билиру-
бина являются миоглобин и цитохромы.
Свободный билирубин нерастворим в воде и в крови транс-
портируется в связанной с альбумином форме. В печени он зах-
ватывается гепатоцитами при участии специфических белков-
переносчиков. Билирубин доставляется в гладкий эндоплазма-
тический ретикулум, где подвергается конъюгации главным
образом с глюкуроновой кислотой, что сопровождается обра-
зованием диглюкуронида. Этот процесс катализируется фермен-
том УДФ-глюкуронозилтрансферазой. Коньюгированный (свя-
занный) билирубин растворим в воде и секретируется в желч-
ные канальцы, попадая в конечном итоге через протоки желче-
выводящей системы в тонкую кишку.
Секреция билирубина в желчные канальцы ограничивает
скорость его обмена в целом. В кишечнике билирубин под
действием бактерий превращается в бесцветный уробилино-
ген и стеркобилиноген. Некоторое количество уробилиноге-
на всасывается в кишечнике и попадает в портальную кровь.
Печень поглощает его не полностью, и небольшое количе-
ство уробилиногена попадает в системную циркуляцию и
выводится с мочой. Большая часть образующегося в кишеч-
нике уробилиногена (стеркобилиногена) далее экскретиру-
ется с фекалиями и под влиянием кислорода окисляется до
стеркобилина.
Нарушения в обмене билирубина и его содержании наибо-
лее часто связаны с заболеванием печени и проявляется в виде
желтух - пожелтения тканей из-за отложения билирубина. Кли-
нически желтуха может не определяться до тех пор, пока кон-
центрация билирубина в плазме не превысит верхний предел
нормы более чем в 2,5 раза.
Гипербилирубинемия может быть результатом повышенно-
го образования билирубина, нарушения его метаболизма, сни-
жения экскреции или сочетания этих факторов.
Желтуха по своим этиопатологическим причинам может
быть нескольких видов:
57
Предпеченочные (надпеченочные) Постпеченочные (подпеченочные)
Г емолиз Неэффективный эритропоэз Желчные конкременты Стеноз желчных протоков Холангит Опухоли поджелудочной железы или желчных протоков
Печеночные (гепатоцеллюлярные)
Предмикросомальные: Постмикросомальные:
лекарства (например, римфапицин). которые мешают поглощению билирубина Микросомальные: гепатит лекарства (метилтестостерон, римфапицин) синдром Дабина-Джонсона
недоношенность гепатит(вирусный или ятрогенный) синдром Жильбера синдром Криглера-Найяра Внутрипеченочная обструкция: гепатит цирроз инфильтрация(например, лимфома, амилоид) атрезия желчных протоков опухоли
Следует отметить, что при гепатите метаболизм билируби-
на может нарушаться на нескольких этапах. Желтуха обычно
возникает из-за накопления конъюгированного билирубина.
Гипербилирубинемия необязательно наблюдается у пациен-
тов с заболеваниями печени и не является их исключительным
признаком. Так ее может не быть у больных с хорошо компен-
сированным циррозом, но она частый признак опухоли подже-
лудочной железы на поздних стадиях.
Рассмотрим возможные причины повышения концентрации
свободного и связанного билирубина в крови.
Повышение содержания свободного билирубина
Если избыток билирубина не конъюгируется, его концент-
рация в плазме у взрослых редко превышает 100 мкмоль/л (при
норме 20 мкмоль/л). При отсутствии заболеваний печени не-
конъюгированиая гипербилирубинемия чаще всего может быть
результатом либо гемолиза, либо синдрома Жильбера (наслед-
ственный дефицит метаболизма билирубина).
58
При гемолизе гипербилирубинемия возникает вследствие
повышенной продукции билирубина, которая превышает спо-
собность печени выводить и связывать этот пигмент. Тем не
менее, большое количество билирубина экскретируется в желчь,
возрастает количество уробилиногена, поступающего в пече-
ночный кровоток, и больше уробилиногена выводится с мочой.
Активность конъюгирующих ферментов печени обычно
бывает низкой при рождении, но потом быстро возрастает.
Отражением этого является преходящая “физиологическая”
желтуха новорожденных.
При чрезмерном гемолизе, как при резус-несовместимости
или недостаточной активности ферментов, как у недоношен-
ных или при синдроме Криглера-Найяра, повышение концент-
рации свободного билирубина в плазме может быть весьма зна-
чительным. Если содержание билирубина превышает 340
мкмоль/л, то поступление билирубина в головной мозг может
вызвать его сильное поражение (билирубиновую энцефалопа-
тию).
Повышение содержания связанного билирубина
Это состояние возникает при поступлении билирубина в
кровоток либо из гепатоцитов, либо из желчевыводящей сис-
темы, когда нарушен нормальный путь его экскреции. Раство-
римый в воде конъюгированный билирубин, поступающий в
системное кровообращение, экскретируется с мочой, придавая
ей насыщенный оранжево-коричневый цвет. При полном зак-
рытии желчных протоков билирубин по поступает в кишечник,
уробилин не образуется и цвет фекалий оказывается бледным.
На основании определения содержания конъюгированного
билирубина проводится дифференциальная диагностика жел-
тух.
Гипербилирубинемия может быть следствием избытка как
связанного, так и свободного билирубина.
Измерение их концентраций по отдельности может быть
полезным при постановке диагноза желтуха новорожденных,
когда есть сомнения относительно вклада нарушенного конъ-
югирования и других аномалий в развитие этого патологичес-
кого состояния.
Третья фракция билирубина, состоящая из конъюгирован-
59
ного билирубина, ковалентно связанного с альбумином, обна-
руживается в плазме пациентов с длительным повышением со-
держания конъюгированного билирубина в крови. Время по-
лусуществования этого пигмента такое же, как у альбумина. Его
содержание в плазме в период выздоровления после заболева-
ния печени или после устранения обструкции желчевыводящих
путей объясняет наличие желтухи при отсутствии билирубину-
рии.
Наследственные нарушения метаболизма билирубина
Известны следующие заболевания, при которых желтуха
вызывается наследственными нарушениями метаболизма били-
рубина.
Синдром Жильбера - желтуха обычно бывает слабой, возни-
кает периодически, часто после инфекционных заболеваний или
периодов плохого питания и в основе-снижение конъюгации.
Так, в частности, при голодании снижается содержание УДФ-
глюкуроновой кислоты в печени и, возможно, вследствие кон-
куренции свободных жирных кислот с билирубином за связы-
вание с альбумином и поглощение клетками печени.
Печень в гистологическом отношении нормальна и ослож-
нений это заболевание не имеет.
Диагностика - при введении никотиновой кислоты (50 мг
внутривенно в течение 30 сек с последующим взятием крови для
анализа каждые полчаса в течение 2 часов и еще ежечасно в
течение трех часов) отмечается повышение концентрации би-
лирубина в плазме на 20 мкмоль/л.
I тип - аутосомно-рецессивный и отмечается отсутствие конъ-
югирующего фермента. Клинически-тяжелая гипербилируби-
немия, обусловленная свободным билирубином. Довольно ран-
ний летальный исход из-за ядерной желтухи. Частичная реак-
ция на фототерапию, но не на фенобарбитал.
II тип - аутосомно-доминантный, частичная недостаточ-
ность конъюгирующего фермента. Клинически - тяжелая ги-
пербилирубинемия, обусловленная свободным билирубином,
хорошо поддается фототерапии и лечению фенобарбиталом.
Синдром Дабина-Джонсона - аутосомно-рецессивный, сни-
жена экскреция билирубина печенью. Клинически - легкая ги-
60
пербилирубинемия, обусловленная конъюгированным билиру-
бином. Отложение пигмента меланина в печени. Нормальная
продолжительность жизни.
Синдром Ротора - аутосомно-рецессивный, дефект точно не
известен. Клинически - признаки сходны с наблюдаемыми при
синдроме Дабина-Джонсона, но пигментация печени отсутству-
ет.
3.1.3. Гемоглобинопатии (гемоглобииозы)
Представляют собой группу заболеваний, в основе которых
лежит образование аномальных форм гемоглобинов вследствие
нарушения образования его белковой части. Нарушение син-
теза а-, (3-, у-, S-цепей глобина может быть следствием мутации
соответствующих структурных генов, либо снижением синтеза
нормальных цепей глобина.
Талассемия мишеневидная (болезнь Кули)
В норме в крови взрослого человека определяется три типа
гемоглобинов:
Нв А - а2 (3,
Нв А, - d2 S2
Нв F - а2 у,
В основе заболеваний - изменение синтеза а- или [3-цепей гло-
бина и вследствие этого различают a-и [3-формы талассемии.
а-талассемии
Полностью выключается синтез a-цепей и, следовательно,
не может образовываться Нв А, Нв А, и Нв F. Однако у взрос-
лых происходит избыточный синтез [3-цепей и возникает тетра-
мер Р4 - Нв Н.
У новорожденных при а-талассемии избыточно образуются
у-цепи фетального Нв вместо a-цепей, что ведет к появлению в
эритроцитах тетрамера у4 (гемоглобин Барта).
Q-талассемии
Прекращается синтез [3-цепей, что ведет к прекращению об-
разования Нв А, в состав которого входит [3-цепь. В результате
компенсаторно синтезируется в эритроцитах Нв F и Нв А,.
61
Талассемия встречается в трех клинических формах - боль-
шая, малая и минимальная.
Аномалия синтеза Нв при талассемии приводит к дегенера-
тивным изменениям эритроцитов в периферической крови, по-
явлению мишеневидных эритроцитов с центрально расположен-
ным гемоглобином, уменьшению жизни эритроцитов (до 30
дней).
Серповидно-клеточная анемия
Развивается в результате образования аномального HbS.
Структура данного гемоглобина формируется в результате на-
рушения аминокислотной последовательности в P-цепи. В час-
тности, вместо глютаминовой кислоты в шестом положении в
P-цепи физиологического гемоглобина происходит ее замена
на валин. Высказывается предположение, что замена отрица-
тельно заряженного глютамата на нейтральную структуру ва-
лина и является одной из причин изменения физико-химичес-
ких свойств гемоглобина и, как следствие, биологической ха-
рактеристики. В частности, значительно хуже растворим по
сравнению с гемоглобином А, происходит его кристаллизация
и выпадение в эритроцитах в осадок, что, в свою очередь, вы-
зывает изменение формы эритроцита - из дисковидной в сер-
повидную. Подобная форма эритроцитов способна к агрега-
ции (склеиванию), становится менее устойчивой, в большей сте-
пени подвержена распаду в клетках РЭС и приводит к разви-
тию гемолитического процесса.
Клиническое течение заболевания характеризуется чередо-
ванием состояния относительной компенсации и кризов, про-
воцируемых различными факторами (инфекция, лихорадка,
резкое изменение относительной влажности окружающей сре-
ды и др.). При увеличении числа кризов возможен кумулятив-
ный эффект, сопровождающийся различными осложнениями.
В стабильном состоянии больной серповидно-клеточной
анемией, в основном, не требует лечения. Необходимо, однако,
постоянное наблюдение, профилактика инфекции, усиленное
питание и постоянное введение фолиевой кислоты.
Показано, что ацетилирование гемоглобина с помощью ас-
пирина приводит к снижению тяжести анемии.
62
3.2. НАРУШЕНИЯ ОБМЕНА НУКЛЕОПРОТЕИДОВ
В зависимости от структуры небелковой части нуклеопроте-
иды представлены дезокси- и рибонуклеопротеидами. Нуклеи-
новые кислоты - полинуклеотидные цепи, состоящие из пури-
новых и пиримидиновых нуклеотидов. В клетках постоянно
осуществляются процессы их синтеза и распада.
3.2.1. Нарушение метаболизма пуриновых нуклеотидов
Пурины, синтезируемые в организме, поступающие с про-
дуктами питания и высвобождаемые при распаде эндогенных
нуклеиновых кислот, подвергаются дальнейшим превращени-
ям. Они могут быть использованы либо для синтеза нуклеино-
вых кислот, либо окисляться до мочевой кислоты (уратов).
Факторы, ведущие к развитию гиперурикемии, могут быть
сведены к следующим:
1. повышение синтеза пуринов, потребление их с пищей, и.
как следствие, образование мочевой кислоты,
2. стимуляция обновления нуклеиновых кислот,
3. снижение скорости экскреции уратов.
Различают первичную и вторичную гиперурикемию.
Первичная гиперурикемии
Причиной первичной гиперурикемии может быть стимуля-
ция синтеза пуринов и угнетение секреции уратов в почках.
Наиболее распространенными формами выступают: синдром
Леша-Нихена, недостаточность глюкозо-6-фосфатазы, подагра.
Синдром Леша-Нихена (ювенильная гиперурикемии)
Врожденное, наследственное заболевание. У маленьких маль-
чиков развивается тяжелая гиперурикемия.
В этиопатогенезе заболевания недостаточность фермента
гипоксантингуанин-фосфорибозилтрансферазы (ГГФРТ), ката-
лизирующего повторное использование пуриновых оснований
для синтеза соответствующих нуклеотидов.
63
гуании + фосфорибозилпиросфосфат
(ФРПФ)
ГГФРТ
-----1ГМФ + пирофосфат
(РР)
ГГФРТ
гипоксантин + ФРПФ
ИМФ + PPi
В физиологических условиях гуанин распадается до моче-
вой кислоты через стадии гипоксантина и ксантина.
В результате ферментного блока происходит накопление гу-
анина, гипоксантина, ксантина и, как следствие, мочевой кис-
лоты, а также ФРПФ, являющегося ключевым соединением, за-
пускающим образование пуринов.
Метаболизм при болезни Леша-Нихена можно представить
следующим образом.
рибозо-5-фосфат
ФРПФ + глутамин
ГМФ ◄----- ИМФ
мочевая кислота
При данной патологии на ранних стадиях наблюдается: час-
тая рвота, гематурия, жажда, полиурия. В крови - прогресси-
рующая анемия, устойчивая к терапии витамином Вр и желе-
зом. В эритроцитах - высокое содержание ФРПФ. В крови и
моче - повышенное количество мочевой кислоты. На поздних
64
стадиях - умственная отсталость, склонность к самоистязанию,
агрессивность поведения, спастическая параплегия, атетоз.
Для лечения - аллопуринол, ингибитор ксантиноксидазы.
Обязателен контроль за состоянием почек.
Недостаточность глюкозо-6-фосфатазы
Отмечается нарушением превращения глюкозо-6-фосфата в
свободную глюкозу и, в связи с этим, содержание глюкозо-6-
фосфата нарастает, в то же время данный субстрат занимает
ключевую роль, запуская различные метаболические пути:
1. Реакции пентозо-фосфатного цикла и вследствие актива-
ции апотомического пути распада углеводов увеличива-
ется содержание рибозо-5-фосфата, который является ис-
ходным субстратом для синтеза ФРПФ и, соответствен-
но, синтеза пуринов и, следовательно, образования уратов.
2. Включается в гликолитические процессы с образованием
молочной кислоты, которая может уменьшать экскрецию
уратов с мочой и отмечается гиперурикемия.
Подагра
Из этиологических факторов, вызывающих развитие подаг-
ры, следует, прежде всего, назвать избыточное поступление
пуринов в организм, а также избыточное поступление молиб-
дена, который входит в состав ксантиноксидазы, катализиру-
ющей образование мочевой кислоты.
К подагре имеется и конституциональная предрасположен-
ность в виде гиперурикемии. Пол и возраст также играет опре-
деленную роль в возникновении подагры (болеют в основном
мужчины, женщины - обычно после климакса). Возможно, что
уровень половых гормонов оказывает влияние на экскрецию
уратов с мочой. Заболевание чаще в пожилом возрасте в связи
с развитием возрастной гиперурикемии.
Патогенез подагры включает:
1) Накопление мочекислых соединений в организме может
быть связано с недостаточным выделением их почками, а так-
же вследствие их усиленного образования.
2) Отложение солей мочевой кислоты, видимо, зависит от
повышенного связывания этих солей тканями (уратогистехия).
В суставах, суставных сумках, хрящах кровоснабжение слабое
и всегда имеется тенденция к закислению среды. В кислой среде
5, Зак. 49
65
натриевая соль мочевой кислоты легче выпадает в осадок. Ме-
стный воспалительный процесс, обусловленный преципитаци-
ей уратов, вызывает увеличение числа лейкоцитов в очаге вос-
паления. Образование этими клетками молочной кислоты ло-
кально понижает величину pH. Эти процессы приводят к умень-
шению растворимости уратов - установлению порочного цик-
ла, следствием чего является дальнейшая преципитация уратов.
3) Острое подагрическое воспаление суставов, являющееся
следствием отложения в них уратов. Суставы резко болезнен-
ны, отечны, гиперемированы. В мелких суставах отложение
солей мочевой кислоты образует подагрические узлы, пораже-
ние крупных суставов приводит к их деформации.
4) Приступы подагры часто сопровождаются аллергически-
ми явлениями (крапивница, эозинофилия).
Причина - либо увеличение выведения (встречается чаще)
или снижение образования мочевой кислоты.
К гипоурикемии ведет недостаточность ксантиноксидазы,
вызванная генетическим дефектом, но наблюдается и при тя-
желом поражении печени.
При этом с мочой увеличивается выведение гипоксантина и
ксантина (ксантинурия), что может вести к образованию ксан-
тиновых камней.
Вторичная гиперурикемия
Высокий уровень уратов в плазме крови может наблюдаться
при выраженном обмене нуклеиновых кислот, быстро растущих
злокачественных новообразованиях и их лечении. Терапия опу-
холей методами радиотерапии или с помощью цитотоксических
лекарственных средств приводит к высвобождению нуклеиновых
кислот и их последующему распаду. В результате блокирования
почечных канальцев кристаллами мочевой кислоты может раз-
виться острая почечная недостаточность. Во время указанных ле-
чебных процедур рекомендуется назначение аллопуринола.
Голодание и повреждение тканей может сопровождаться
повышением количества образующихся уратов.
В этом случае, по-видимому, развивается ацидоз, причем
кислоты, очевидно, подавляют экскрецию уратов с мочой, усу-
66
губляя проявление гиперурикемии. Установлено, что при пол-
ном голодании концентрация уратов может достигать 0,9
ммоль/л и более.
Гиперурикемия наблюдается также при недостаточности
почечных клубочков, псориазе, лечении диуретиками и т.д.
3.2.2. Нарушение обмена пиримидиновых нуклеотидов
Катаболизм пиримидинов протекает в основном в печени с
образованием хорошо растворимых конечных продуктов: при
распаде урацила, цитозина - NH,, СО,, (3-аланин, а при распаде
тимина - СО,, NH, и (3-аминоизомасляная кислота. В силу хо-
рошей растворимости данных соединений клинические симп-
томы при их избыточном образовании слабо выражены.
Экскреция (3-аминоизомасляной кислоты увеличивается при
лейкемии и после рентгеновского облучения, что указывает на
ускорение гибели клеток и деструкцию их ДНК.
карбамоилфосфат + аспартат ------------► карбамоиласпартат
дигидрооротаза дигидрооротовая
► кислота
Н2О
днгидрооротатдегидрогеиаза
НАД+ НАДН:
оротидин-5-фосфат-
декарбоксилаза
оротовая
кислота
оротатфоефо-
рибозиллраисфераза
ФРПФ PPi
оротидин-5-
фосфат
уридин-5-фосфат
(уридиновая кислота)
УМФ + АТФ ---------► ЦДФ + АДФ
УДФ + АТФ --------► ЦТФ + АДФ
УТФ
ЦТФ
глутамин глутамат
тимидилатсинтаза
УМФ J ► ТМФ
№-М|Ометилен- Н2-фолат
Н4-фолат
5*
67
Нарушение катаболизма пиримидинов
Наследственная оропювая ацидурия
Описано два возможных типа этой патологии.
I тип
Является более распространенной формой и возникает при
дефиците оротатфосфорибозилтрансферазы и оротидин-5-фос-
фатдекарбоксилазы. В детском возрасте характерна задержка
физического и психического развития и мегалобластическая
анемия. В моче обнаруживаются кристаллы оротовой кислоты
оранжевого цвета (оранжевая кристаллоурия).
Заболевание поддается лечению глюкокортикоидами и пи-
римидиннуклеотидами. При отсутствии лечения больные бо-
лее подвержены инфекциям.
II тип
Развивается при недостатке только одного фермента - оро-
тидин-5-фосфатдекарбоксилазы.
При обследовании у таких пациентов в моче обнаруживает-
ся оротидин в отличие от I типа, при котором в большей степе-
ни экскретируется оротовая кислота.
Возникает при поражении митохондрий печени. В результа-
те нарушается утилизация образующегося карбомоилфосфата
и он включается в цикл синтеза оротовой кислоты.
68
Глава 4
ПАТОЛОГИЯ ЛИПИДНОГО ОБМЕНА
Метаболизм липидов может нарушаться, в основном, на двух
этапах:
1) транспорт жиров по крови;
2) распад сфинголипидов, триацилглицеринов, жирных кис-
лот, эфиров холестерина в тканях.
4.1. ТРАНСПОРТ ЛИПИДОВ И ЕГО НАРУШЕНИЯ
Основными липидами плазмы крови человека являются сво-
бодные жирные кислоты (СЖК), триглицериды (ТГ), фосфо-
липиды (ФЛ), холестерин (ХС) и его эфиры (ЭХ). СЖК связа-
ны с альбумином, остальные липиды - с а- и (3-глобулинами.
образуя липопротеины (ЛП). ЛП состоят из гидрофильной на-
ружной оболочки, формируемой белками (апопротеинами),
фосфолипидами и свободным ХС, и гидрофобного ядра, кото-
рое образуют ТГ и эфиры ХС (рис. 9).
Периферический апобелок (например, Апо-С)
Рис. 9. Схема строения липопротеина (по Р.Марри и др.. 1993).
69
Выделяют следующие классы ЛП (табл. 1): хиломикроны
(ХМ), липопротеины очень низкой плотности (ЛПОНП или
пре-Р-ЛП). липопротеины промежуточной плотности (ЛППП
или Р-ЛПОНП), липопротеины низкой плотности (ЛПНП или
Р-ЛП), липопротеины высокой плотности - 2 и 3 (ЛПВП2,
ЛПВПЗ или а-ЛП), липопротеины а (ЛП(а)).
Таблица 1
Характеристика основных классов липопротеинов
Класс липопротеинов Основные липиды Апопро- теины Плотность, г/мл Диаметр, мкм Электрофоре- тическая подвижность
Хиломикро- ны и их остатки Пищевые триглицериды A-I, А-П, В-48, С-1, С-И, С-Ш, Е <1,006 80-500 Остаются на старте
ЛПОНП Эндогенные триглицериды В-48, С-1, С-П, С-Ш, Е <1,006 30-80 Пре-р-липо- протеины
ЛППП Эфиры хо- лестерина, триглицериды В-100. С-Ш, Е <1,019 25-35 Медленные пре-Р-липо- протеины
ЛПНП Эфиры холестерина В-100 1,019-1,063 18-28 р-ЛИПО- протеииы
ЛПВП То же A-I. А-П 1.063-1,210 5-12 а-лиио- протеины
4ЛЛ. Характеристика и метаболизм липопротеидов
Триацилглицерины синтезируются преимущественно в пече-
ни, жировой ткани и тонком кишечнике. Основная транспорт-
ная форма экзогенных (пищевых) ТГ - хиломикроны, а эндо-
генных - ЛПОНП. Распад ТГ в составе ХМ и ЛПОНП внутри
капилляров жировой ткани, скелетных и сердечной мышц ка-
тализирует внепеченочная липаза - липопротеинлипаза (ЛП Л),
которая активируется апо С-П, гепарином и инсулином. У жен-
щин ЛПЛ в жировой ткани более активна, поэтому уровень
ЛПОНП у них ниже, а ЛПВП выше, чем у мужчин. Активность
ЛПЛ в жировой ткани мужчин может повыситься при регуляр-
ном умеренном употреблении алкоголя, а в мышцах - при фи-
зической нагрузке. В эндотелии печени находится печеночная
липаза (ПЛ), которая действует на ТГ и фосфолипиды в соста-
ве ЛППП и ЛПВП2.
70
Фосфолипиды, в основном фосфатидилхолин (лецитин) и
сфингомиелин, поставляются в плазму печенью и тонким ки-
шечником, обладают амфифильностью, входят в состав обо-
лочки ЛП.
Холестерин может существовать как в свободном виде (1/4),
так и в форме эфира с жирными кислотами (3/4). В плазме кро-
ви (как и в атероматозной бляшке) преобладают эфиры ХС.
Почти весь ХС в норме синтезируется в печени и дистальной
части тонкого кишечника из ацетил-КоА. Ключевым фермен-
том является р-гидрокси-Р-метилглутарил-КоА-редуктаза
(ГМГ-КоА-редуктаза), активность которой регулируется, во-
первых, конечным продуктом (ХС) по механизму отрицатель-
ной обратной связи и, во-вторых, фосфорилированием (тормо-
зится) и дефосфорилированием (стимулируется).
В печени из ХС образуются желчные кислоты; ключевой
фермент их синтеза - 7-а-гидроксилаза. Этерификация ХС в ос-
новном происходит в плазме крови под действием лецитинхо-
лестеро л ацил тра нсфераз ы (ЛХАТ):
ЛХАТ
Лецитин + Холестерин —Лизолецитин + Эфир холестерина
и в небольшом количестве - в тканях (например, в тонком ки-
шечнике и печени) с участием ацил-КоА-холестерол-ацилтран-
сферазы (АХАТ):
АХАТ
Холестерин + Ацил-КоА —Эфир холестерина + HSKoA
ЛХАТ синтезируется в печени, активируется апо A-I и ката-
лизирует этерификацию ХС в составе ЛПВПЗ.
В плазме большая часть ХС обнаруживается в ЛПНП, суще-
ственно меньшая - в ЛПОНП и ЛПВП.
Характеристика апопротеинов
Некоторые апобелки постоянно входят в состав ЛП, другие
могут переноситься между ними. Апопротеины выполняют не
только структурную, но и регуляторную функцию: активиру-
ют ферменты, действующие на ЛП, и связываются с рецептора-
ми на поверхности клеток, определяя таким образом места зах-
вата и скорость распада ЛП.
71
Апопротеины А (апо A-I, апо А-П, ano-IV) синтезируются в
печени и кишечнике в виде пре-про-белка и окончательно со-
зревают в токе крови. В большом количестве входят в ЛПВП.
Апо A-I активирует ЛХАТ.
Апопротеины В (апо В-100 и апо В-48). Апо В-100 образуется
в печени и входит в состав ЛПОНП, ЛППП, ЛПНП, ЛП(а). На
плазматических мембранах паренхиматозных и соединитель-
нотканных клеток есть специальные апо В,Е-рецепторы, кото-
рые узнают этот белок и захватывают содержащие его Л П. Апо
В-48 входит в состав хиломикрон и не распознается апо В,Е-
рецепторами клеточных мембран.
Апопротеины С (апо С-1, апо C-II, апо С-Ш) являются ос-
новными компонентами ЛПОНП и минорными компонента-
ми ЛПВП. Апо С-П активирует ЛПЛ, расщепляющую ТГ в хи-
ломикронах и ЛПОНП.
Апопротеин Е- компонент ЛПОНП, ЛППП, ЛПНП, ЛПВП.
В наибольших количествах образуется в печени и поступает в
плазму, в основном, в составе новосинтезированных ЛПВП и
ЛПОНП. Важнейшая функция апо Е - регуляция переноса ХС
между тканями и плазмой, так как:
1) на мембранах клеток печени и периферических тканей есть
два вида рецепторов, распознающих этот белок: апо В,Е-рецеп-
тор и апо Е-рецептор. Апо Е-рецепторы связывают ЛП, содер-
жащие апо Е, а апо В,Е-рецепторы - ЛП, содержащие апо Е,
апо В-100 или оба апопротеина. Число апо В,Е-рецепторов (но
не апо Е-рецепторов) снижается при старении, употреблении
ХС с пищей, а возрастает при недолгом голодании, недостатке
желчных кислот, под влиянием ингибиторов ГМГ-КоА-редук-
тазы, трийодтиронина и эстрогенов;
2) апо Е может связывать ХС и его эфиры, что способствует
удалению ХС из мембран клеток, в том числе из клеток арте-
рий;
3) может активировать ЛХАТ, что также ускоряет обратный
транспорт ХС.
Характеристика липопротеинов
Хиломикроны - самые крупные ЛП, образуются в кишечнике
и содержат преимущественно ТГ и небольшие количества фос-
фолипидов, ХС, эфиров ХС и белка: апо В-48, апо А, Си Е. У
72
здоровых людей после 12-14-часового голодания плазма крови
не содержит ХМ.
ЛПОНП (npe-fi-ЛП) содержат несколько меньше ТГ, чем
ХМ, но больше ХС, фосфолипидов и белка: апо С, Е и В-100.
Образуются главным образом в печени, особенно активны в тех
условиях, когда повышается синтез эндогенных ТГ:
- при потреблении богатой углеводами пищи,
- при высоком содержании СЖК в крови,
- при потреблении этанола,
- при высокой концентрации инсулина и низкой - глюка-
гона.
ЛПОНП бывают большие и маленькие. Маленькие ЛПОНП
распадаются до ЛППП, которые затем превращаются в ЛПНП.
Из больших ЛПОНП образуются такие ЛППП, которые уда-
ляются из плазмы без превращения в ЛПНП. При избыточном
поступлении углеводов с пищей растет количество и размер
ЛПОНП, поэтому уровень ЛПНП снижается.
ЛПНП ($-ЛП) - главные из ЛП плазмы, доставляющих ХС
клеткам периферических тканей. В них мало ТГ; основной бе-
лок - апо В-100. В течение дня разрушается 30-40‘%> ЛПНП, из
них половина - рецептор-опосредованным путем через ано В,Е-
рецептор (другое название его - ЛПНП-рецептор). Известны
две основные функции Л ПНП-рецептора: 1) обеспечить клетки
ХС, который используется в синтезе мембран, желчных кислот,
стероидных гормонов, витамина D3 (поэтому клетки печени,
половых желез, надпочечников имеют большое количество ре-
цепторов ЛПНП); 2) поддерживать уровень ХС и Л ПНП в кро-
ви в пределах нормы.
ЛПВП (а-ЛП) имеют сложное происхождение: липидный
компонент формируют свободный ХС и ФЛ, высвобождающи-
еся при липолизе ХМ и ЛПОНП, а также свободный ХС, по-
ступающий из периферических клеток. Основной апопротеин,
апо A-I, синтезируется в печени и в тонком кишечнике; другим
апобелком, апо С, на долю которого приходится до 5%, ЛПВП
постоянно обмениваются с ХМ и ЛПОНП. ЛПВП подразделя-
ются на ЛПВП2 и ЛПВПЗ. Уровень болеелегких ЛПВПЗ в плаз-
ме выше, они содержат больше белка, но меньше ТГ и ЭХ и
превращаются в ЛПВП2 после захвата поверхностных компо-
нентов распадающихся ХМ и ЛПОНП. Концентрация ЛПВП2
в плазме крови, с одной стороны, и концентрация ХМ и
73
ЛПОНП, с другой стороны, изменяются реципрокно, то есть
повышение ЛПВП в крови сопровождается снижением ЛПОНП
и наоборот. В норме ЛПВП играют ведущую роль в удалении
тканевого ХС, так как способны забирать его с плазматичес-
ких мембран периферических клеток и с поверхности липопро-
теинов, богатых триглицеридами. Забрав ХС, ЛПВП транспор-
тируют его в печень для окисления в желчные кислоты.
ЛП(а) крупнее ЛПНП. но имеют большую плотность. Ли-
пидный состав - как у ЛПНП, а белка больше - апо В-100 и
апо(а). Апо(а) является гликопротеином и образуется только в
печени. Именно с ним связывают повышенную атерогенность
Л П (л). так как соединение апо(а) с апо В нарушает узнавание и
захват этих ЛП апо В,Е-рецепторами. В результате ЛП(а) дли-
тельно циркулируют в крови, модифицируются и поступают в
клетки путем нерегулируемого эндоцитоза. Таким образом, чем
выше уровень ЛП(а), тем больше риск развития АС. При низ-
ком содержании ЛП(а) никакой патологии не выявлено.
ЛП разных классов могут обмениваться друг с другом от-
дельными компонентами.
Метаболизм липопротеинов
Метаболизм липопротеинов - это сложный динамический
процесс, в ходе которого постоянно происходят перемещения
липидов и апопротеинов между разными классами ЛП и целый
ряд ферментативных реакций (рис. 10).
ХМ доставляют липиды пищи из кишечника через лимфу в
кровь. Под действием внепеченочной ЛПЛ, активируемой апо
С-П, ХМ в плазме превращаются в ремнанты. Ремнанты ХМ
узнаются рецепторами печени по апо Е, захватываются и рас-
падаются в гепатоцитах. (Хиломикроны не захватываются этим
рецептором из-за наличия в них апо С).
В печени образуются ЛПОНП, которые переносят эндоген-
ные ТГ в кровь, где они расщепляются той же ЛПЛ до ремнан-
тных ЛПОНП, т.е. ЛППП. ЛППП либо узнаются по апо Е или
апо В-100 и захватываются рецепторами, либо под действием
ПЛ превращаются в ЛПНП, содержащие апо В-100, но уже не
имеющие апо Е.
74
Рис. 10. Упрощенная схема метаболизма липопротеинов (по Томпсону. 1991).
Катаболизм ЛПНП (рис. II) связан с ЛПНП-рецепторами,
которые узнают ЛП по апо В-100 и захватывают его. В клетке,
внутри эндосом, комплекс “ЛПНП + рецептор" диссоциирует.
Рецептор возвращается в плазматическую мембрану, а ЛПНП
разрушается в лизосомах. Освободившийся ХС вызывает три
регуляторных эффекта: 1) ингибирует ГМГ-КоА-редуктазу и,
следовательно, свой собственный синтез, 2) подавляет синтез
новых молекул ЛПНП-рецепторов, 3) активирует АХАТ, быс-
тро этерифицируется и используется на нужды клетки.
75
Рис. 11. Схема захвата и деградации ЛПНП в фибробласчах с участием
ЛПНП-рецепторов (по Томпсону, 1991).
Лекарственные средства, конкурентно ингибирующие ГМГ-КоА-
редуктазу, блокируют эндогенный синтез ХС и посредством этого сти-
мулируют синтез рецепторов ЛПНП. В итоге уровень ХС ЛПНП в
плазме снижается.
Новосинтезиро ванные частицы ЛПВП представлены в плаз-
ме Л ПВПЗ, но в конечном итоге под действием ЛХАТ, активи-
руемой апо A-I, они превращаются в ЛПВП2. Обратное обра-
зование ЛПВПЗ возможно в результате гидролиза ТГ, содер-
жащихся в ЛПВП2, печеночной липазой. В механизме антиате-
рогенного действия а-ЛП можно выделить два момента:
1) рецепторы периферических клеток узнают ЛПВП по апо
A-I и захватывают их. В клетке а-ЛП обогащаются ХС и апо Е,
выходят в кровь и направляются в печень, где взаимодейству-
ют с апо Е- или апо В, Е-рецепторами, поглощаются гепатоци-
тами и там распадаются;
2) ЛХАТ, активность которой связана с а-ЛП, активно эте-
рифицирует захваченный ими в периферических клетках ХС.
Эфиры ХС, теряя полярность, погружаются внутрь ЛПВП, ос-
вобождая место в поверхностном слое для новой молекулы ХС.
В результате в тканях не накапливается избыток ХС.
76
4.1.2. Нарушение транспорта липопротеидов
Нарушение транспорта липидов проявляется в форме дисли-
попротеинемий (ДЛП). ДЛП - это изменение содержания ЛП в
плазме (повышение, снижение, отсутствие) или появление нео-
бычных ЛП. Если ДЛП имеют генетическую природу, то это
первичные заболевания семейного характера. Выраженность их
подвержена влиянию факторов окружающей среды. Если ДЛП
сопутствуют другим заболеваниям, то они называются вторич-
ными.
Первичные гипср.шпоиротеинемии
Первая классификация гиперлшюпротеинемий (ГЛП) была
предложена Фредриксоном и одобрена ВОЗ в 1970 г. (табл. 2).
Таблица 2
Основные типы гиперлипопротеинемий
Тип лило- протеинемии В плазме повышен уровень в основном
липопротеинов ЛИПИДОВ
1 Хиломикроны Триглицериды
2а ЛПНП Холестерин
26 ЛПНП иЛПОНП Холестерин и триглицериды
3 Остатки (ремнанты ХМ и ЛППП) - ' Триглицериды и холестерин
4 ЛПОНП Триглицериды
5 ЛПОНП и хиломикроны Триглицериды и холестерин
I тип - семейная гиперхиломикронемия - характеризуется
повышением ТГ и ХМ уже в детстве. В основе патологии лежит
снижение активности ЛПЛ в результате нарушения ее синтеза
или дефицита ее активатора апо С-11. Это приводит к резкому
повышению ХМ в плазме с последующим развитием панкреа-
тита, т.к. ткань поджелудочной железы повреждается СЖК,
которые высвобождаются из ТГ ХМ под действием панкреати-
ческой липазы. Снижение поступления пищевых ТГ в печень
уменьшает секрецию ЛПОНП, но концентрация их в крови ос-
тается нормальной, так как расщепление их ЛПЛ тоже нару-
шено. В результате уровни ЛППП, ЛПНП и ЛПВП снижают-
ся. Клинически 1 тип ГЛП проявляется повторяющимися при-
77
ступами боли в животе вследствие острого панкреатита, ксан-
томами, гепатоспленомегалией. Риск атеросклероза (АС) не
увеличивается. Среди лечебных мероприятий - снижение по-
требления ТГ до 50 г/д.
IV тип ГЛП: повышение ТГ и ЛПОНП вследствие усиления
их образования и замедления распада. ЛПОНП большого раз-
мера, в них много ТГ; образование апо В-100 повышается. Уро-
вень ЛПВП снижен. Увеличение синтеза ЛПОНП сопровожда-
ется снижением их превращения в ЛПНП. Однако ЛПНП мел-
кие и плотные, хуже связываются с ЛПНП-рецепторами, но
очень хорошо -- с протеогликанами аорты, то есть обладают
атерогенными свойствами. Механизм повышенного образова-
ния ТГ ЛПОНП неясен, но у больных определяется высокий
уровень инсулина и инсулинорезистентность. Целесообразно
рекомендовать диету для снижения массы тела, ограничение
сахара и алкоголя, физическую нагрузку.
V тип ГЛП сочетает черты IV и I типов, так как в крови по-
вышается уровень и ЛПОНП, и ХМ вследствие дефицита апо
С-П. Редко возникает в детстве. Проявляется приступами бо-
лей в животе (острый панкреатит), ксантомами, снижением то-
лерантности к глюкозе; могут быть гиперурикемия, перифери-
ческая нейропатия. Часто после применения диеты V тип мо-
жет перейти в IV.
II тип - семейная гиперхолестеринемия (СГХС) - наиболее
серьезная патология в обмене ЛП, так как при этом риск разви-
тия ИБС повышается в 10-20 раз по сравнению с нормой. В ос-
нове данного состояния - нарушение функции рецепторов
ЛПНП. В 1985 г. американцам Brown и Goldstein была присуж-
дена Нобелевская премия за открытие рецептора ЛПНП и ус-
тановление причин семейной гиперхолестеринемии (СГХС, На
тип ГЛП).
II тип ГЛП наследуется по аутосомно-доминантному типу и
поражает 0,2% населения Земли. Гомозиготная форма СГХС
встречается довольно редко, чаще - гетерозиготная. Известны
четыре класса мутаций, разделенных на две группы:
- рецептор-негативные мутации (не синтезируются рецепто-
ры),
- рецептор-дефицитные мутации (есть рецепторы, но они не
могут правильно функционировать, так как нарушается транс-
порт апо В,Е-рецепторов из эндоплазматического ретикулума
78
в комплекс Гольджи, расположение их на мембране или связы-
вание ими ЛПНП) (рис. 12).
Рис. 12. Четыре классг! мутаций по локусу ЛПНП-рсцептора
(по Томпсону, 1991).
Уже с рождения в плазме больных накапливаются ЛПНП.
Концентрация ХС у гетерозигот в два раза больше нормы, у
гомозигот - в четыре. Содержание ТГ при Па в пределах нор-
мы, при Пб повышается. Уровень ЛПВП может быть в норме,
но у гомозигот снижается. У 60% рецептор-негативных гомо-
зигот ИБС развивается в возрасте до 10 лет и потом прогресси-
рует, у рецептор-дефицитных гомозигот симптомы коронарной
недостаточности возникают всегда после 10 лет. К другим яр-
ким проявлениям раннего атеросклероза относят формирова-
ние уже в детстве и даже при рождении кожных и сухожильных
ксантом, а позже - липоидной дуги роговицы. Больные могут
внезапно умереть в возрасте до 30 лет от острой коронарной
недостаточности. При гетерозиготной форме СГХС все симп-
томы проявляются позже, чем у гомозигот.
Наиболее эффективное лечение - ЛПНП-аферез, то есть по-
стоянное удаление ЛПНП из крови.
79
При недостаточности ЛПНП-рецепторов не только повы-
шается уровень ЛПНП в крови, но и продлевается время их
циркуляции, что способствует образованию измененных, мо-
дифицированных форм ЛПНП, обладающих высокой атероген-
ностью. Рецепторы ЛПНП связывают также ЛППП и тот под-
класс ЛПВП, в котором есть апо Е. Более медленное удаление
ЛППП при II типе ГЛП (только через нормально функциони-
рующие апо Е-рецепторы) приводит к более значительному
превращению их в ЛПНП (рис. 13).
Рис. 13. Механизм развития семейной гиперхолестеринемии: нарушен
захват и катаболизм как ЛППП, так и ЛПНП; одновременно активируется
превращение ЛППП в ЛПНП (по Томпсону, 1991).
К ГХС может вести и структурный дефект апо В-100, зат-
рудняющий узнавание и связывание такого ЛПНП нормально
функционирующими апо В,Е- рецепторами. В крови повыша-
ется уровень ЛПНП, а ЛППП выводятся нормально, так как
связываются по апо Е с рецепторами ЛПНП. Клинические про-
явления близки тем, что наблюдаются при гетерозиготной фор-
ме СГХС.
III тип - семейная дис-[3-липопротеинемия - характеризуется
80
накоплением ремнантов ХМ и ЛПОНП (ЛППП) из-за наруше-
ния их метаболизма по рецепторному пути (рис. 14).
J Рис. 14. Механизм развития гиперлипоиротепнемии III типа: нарушен
ji захват ремнантов хиломикрон и ЛППП печенью, снижена скорость
; превращения ЛППП в ЛПНП. повышен синтез ЛПОНП
(по Томпсону, 1991).
Причина - апо Е либо отсутствует, либо представлен не нор-
мальным фенотипом ЕЗ/ЕЗ, а фенотипом Е2/Е2, реже Е2/ЕЗ или
Е2/Е4; могут быть редкие изоформы апо Е2 и апо ЕЗ. Струк-
турные аномалии апо Е или его отсутствие нарушают рецеп-
торное связывание ремнантных форм ХМ и ЛПОНП с апо Е- и
апо В,Е-рецепторами, что ведет к раннему атеросклерозу. Од-
нако для развития болезни нужен не только дефект апо Е, но и
метаболические нарушения, при которых повышается синтез
ЛПОНП (ожирение, сахарный диабет, гипотиреоз и т.д.). У
больных замедлено превращение ЛППП в ЛПНП, но атероск-
лероз развивается, так как ремнантные частицы атерогенны и
захватываются макрофагами. Клиническая картина: липоидная
ду। а роговицы, ксантелазмы, локтевые ксантомы, ладонные
стрпн, возможны снижение толерантности к глюкозе, острый
6. З.о. 19
81
панкреатит. При электрофорезе выявляется расширение “0-по-
лосы’', характерной для ремнантных частиц.
К первичным нарушениям транспорта липидов, помимо пяти
типов ГЛП, относят ещё целый ряд состояний, не имеющих чет-
кой классификации.
Неклассифицированные дислипопротеинемии
Наследственный дефицит печеночной липазы, или гипер-а-
триглицеридемия, характеризуется накоплением ТГ во фракции
a-ЛП. В результате торможения катализируемых печеночной
липазой реакций в крови, во-первых, фракция ЛПВП представ-
лена только крупными ЛПВП2, обогащенными триацилглице-
ринами, а ЛПВПЗ отсутствуют; во-вторых, накапливаются
ЛППП. Проявления: липоидная дуга роговицы, ксантомы, ла-
донные стрии, ИБС.
Семейная гипер-а-липопротеинемия выявляется, как прави-
ло, случайно - по повышению в крови ЛПВП2 и ЛПВПЗ вслед-
ствие усиленного синтеза апо А-1. Клинических проявлений нет,
более того, как правило, снижается вероятность развития ИБС
и увеличивается продолжительность жизни.
Болезнь накопления эфиров ХС обусловлена дефицитом гид-
ролазы эфиров ХС в лизосомах фибробластов, где в норме про-
исходит распад ЛПНП. К постоянным признакам относят по-
вышение ХС ЛПНП и снижение ХС ЛПВП в плазме, увеличе-
ние печени и селезенки, преждевременное развитие АС.
Гиперапо-В-липопротеинемия возникает как результат акти-
вации синтеза апо В. Проявляется повышением ЛПОНП и
ЛПНП при нормальной концентрации ХС ЛПНП, повышени-
ем ТГ, предрасположенностью к АС.
Наследственный дефицит ЛХАТ характеризуется накоплени-
ем ХС и лецитина, снижением ЭХС и лизолецитина во всех фрак-
циях ЛП. Уровень ЛПВП снижен, а ЛПОНП - повышен. Про-
исходит массивное отложение липидов в почках. Находящий-
ся в крови неэтерифицированный ХС вызывает гибель эндоте-
лиоцитов в клубочках и образование патологических гломеру-
лярных мембран. Накопление ХС в эритроцитах сокращает
время их жизни. Болезнь проявляется диффузным помутнени-
ем роговицы, гемолитической анемией, протеинурией, гиперт-
риглицеридемией. С целью предупреждения почечной недоста-
82
точности больным рекомендуют диету с низким содержанием
жиров.
Г иполипопротеипемии
А-^-липопротеинемия (акантоцитоз) это отсутствие р-ЛП в
плазме крови. В основе патологии - невозможность синтеза апо
В, вследствие чего нарушается образование ХМ и ЛПОНП. В ре-
зультате в крови определяется низкий уровень общих липидов и
отсутствие ЛПНП. Жиры накапливаются в кишечнике и печени в
виде капель. Нарушается сгруктура мембран, в том числе клеток
нервной системы. На поверхности эритроцитов появляются вы-
пячивания, делающие их похожими на незрелые плоды каштана;
такие клетки называются акантоцитами. Время их жизни сокра-
щено, поэтому развивается анемия. Организм нс усваивает пище-
вые жиры, возникает стеаторея, дефицит ненасыщенных жирных
кислот, жирорастворимых витаминов (в том числе витамина Е
сильного антиоксиданта, стабилизирующего клеточные мембра-
ны), прогрессирующая атаксия и другие неврологические симп-
томы из-за поражения миелиновых оболочек, офтальмопатия.
Больным рекомендуется ограничение животных жиров, введение
больших доз полиненасыщенных жирных кислот и жирораство-
римых витаминов, в первую очередь витамина Е.
Семейная гипо-Р-липопротеинемия, в отличие от акантоци-
тоза, никакой патологией не сопровождается. Организм спосо-
бен образовывать ХМ и ЛПОНП, но меньше, чем у здоровых,
поэтому содержание ЛПНП составляет от 10 до 50% нормы.
Ан-а-липопротеинемия (Танжерская болезнь) встречается
редко. Впервые её обнаружили у членов семьи, жившей на ост-
рове Tanger. В организме таких больных не синтезируется ано
А-1 и практически отсутствуют ЛПВП, а в тканях, особенно в
РЭС, накапливается много ХС. Самый яркий симптом, позво-
ляющий поставить диагноз - изменение миндалин: они увели-
чены, оранжевого или желто-серого цвета из-за накопления в
них ЭХ. Другие проявления - гепатоспленомегалия, лимфаде-
нопатия, лимфоцитоз, неврологические расстройства.
Наряду с изменением концентрации основных классов ЛП в
организме может происходить модификация этих липид-бел-
ковых комплексов, то есть изменение строения и свойств, что
может коренным образом нарушить их метаболизм.
в*
83
Модифицированные липопротеины
Модифицированные липопротеины образуются как в плаз-
ме, так и в стенке артерий из нормально синтезированных и
секретированных в кровь ЛП. Причиной их модификации мо-
жет быть выброс клетками свободных радикалов и продуктов
перекисного окисления липидов (ПОЛ), повышенная концент-
рация в крови и межклеточной жидкости некоторых метаболи-
тов (например, глюкозы), а также разных ферментов.
В организме могут образовываться:
1) гликозилированные ЛП. Гликозилирование - это нефер-
ментативное ковалентное присоединение моносахаридов (пре-
имущественно глюкозы) к белку по е-аминогруппе лизинового
остатка; особенно активно протекает при сахарном диабете. Ему
подвергаются все классы ЛП, но в большей степени - ЛПНП и
ЛПВП. Гликозилирование ЛПНП ведет к нарушению их взаи-
модействия с апо В,Е-рецепторами клетки, замедлению их вы-
ведения и катаболизма, а значит, к ГХС. Гликозилирование
ЛПВП сопровождается ускорением их катаболизма и гипо-а-
липопротеинемией. В ответ на появление в крови гликозили-
рованных ЛП может развиться аутоиммунный ответ с образо-
ванием комплекса “гликозилированный ЛП-антитело”, кото-
рый способен повреждать артериальную стенку.
Гликозилирование ЛП может протекать с одновременным
их окислением, что получило название глиоксидация, а также
десиалированием (отщеплением молекул сиаловой кислоты от
углеводной части апобелков) и изменением липидного соста-
ва;
2) перекисно-модифицированные ЛП: ЛПНП, ЛПОНП и
ЛП(а). Перекисно-модифицированные ЛПНП могут повреж-
дать эндотелиальный покров артерий. Стабилизировать час-
тицы ЛПНП и защищать их от ПОЛ способны ЛПВП, так как
активная в них ЛХАТ переносит легко окисляемую ненасыщен-
ную жирную кислоту с лецитина на ХС с образованием ЭХС,
перемещающегося с поверхности в ядро частицы. Если соотно-
шение ЛПНП/ЛПВП в крови повышено, то образование моди-
фицированных ЛПНП ускоряется, если понижено - замедляет-
ся;
3) аутоиммунные комплексы ЛП-АТ, обладающие атероген-
ностью. Причиной образования антител к ЛП может служить
84
их модификация (пероксидация, гликозилирование, десиалиро-
вание и др.) и, как следствие этого, приобретение ими аутоан-
тигенных свойств;
4) продукты ограниченного протеолиза ЛП, десиалирован-
ные ЛПНП, комплексы ЛПНП с гликозаминогликанами, про-
теогликанами, коллагеном, эластином, агрегированные ЛП.
Модификация апо В-содержащих ЛП коренным образом ме-
няет их метаболизм. Такие ЛП узнаются и усиленно захватыва-
ются не обычными апо В,Е-рецепторами, а специфическими, так
называемыми ацетил-ЛПНП-рецеиторами, или скевенджер-ре-
цепторадш (от scavenger уборщик мусора). Выделяют два типа
скэвенджер-рецепторов, они являются тримерами и характери-
зуются высокой степенью гликозилирования. Эти рецепторы есть
в разных клетках РЭС, в том числе в макрофагах и эндотелиаль-
ных клетках сосудов. Особенность их в том, что они, в отличие
от апо В,Е-рецепторов, не снижают своей активности при на-
коплении ХС в макрофагах, и содержание ХС в клетке растет
пропорционально уровню его в соответствующих ЛП крови.
Накапливая ЭХС, макрофаги трансформируются в пенистые
клетки, составляющие основу атеросклеротической бляшки.
Таким образом, за развитие АС, вероятно, более ответствен-
ны не столько нативные, сколько модифицированные ЛПНП.
Вторичные гиперлипоиропгеииемии
сопровождают целый ряд заболеваний и клинических состоя-
ний.
Сахарный диабет (СД) вызывает модификацию ЛП (глико-
зилирование), повышение уровня ЛПОНП, в меньшей степени
- ХМ, а также ЛП(а). Увеличивается доля ТГ в ядре ЛП всех
классов, изменяется спектр ЛПНП за счет нарастания менее
плотных частиц.
Как правило, атерогенные сдвиги в обмене ЛП наблюдают-
ся при II типе СД, то есть в условиях повышенного уровня ин-
сулина и резистентности к нему. Инсулин играет сложную роль
в метаболизме липидов:
1) активирует ГМГ-КоА-редуктазу и синтез ХС;
2) стимулирует а-глицеролфосфатацилтрансферазу и обра-
зование ТГ из глюкозы или жирных кислот (в том числе в пече-
ни);
85
3) активирует ЛПЛ и вывод ТГ из плазмы крови;
4) ингибирует тканевую липазу и распад ТГ в жировой ткани.
Можно представить следующую цепь событий, приводящих
к дислипидемии при СД (рис. 15): распад ТГ в жировой ткани и
выход СЖК в кровь; поступление в печень больших количеств
СЖК и глюкозы (проницаемость мембраны гепатоцитов для
глюкозы не зависит от инсулина); активный синтез из них ТГ;
усиление секреции ЛПОНП печенью и замедление их разруше-
ния в крови из-за снижения активности ЛПЛ, которая теряет
чувствительность к инсулину. В итоге - значительное повыше-
ние уровня пре-Р-ЛП в плазме. В клетках инсулин ингибирует
лизосомальный протеолиз и таким образом нарушает распад
ЛП и своевременное выведение их из плазмы.
Рис. 15. Изменения в метаболизме ЛПОНП при ИНСД. В плазме крови
возрастает число обогащенных триглицеридами ЛПОНП
(по Томпсону, 1991).
Присутствуя в крови в большой концентрации длительное
время, инсулин и сам выступает как фактор роста, и активиру-
ет другие ростовые факторы, что приводит к пролиферации
гладкомышечных клеток, синтезу соединительной ткани.
86
Активирующееся при СД гликозилирование белков может
быть ферментативным и неферментативным. Неферментатив-
но гликозилируются:
- ЛП (см. выше);
- Нв А: в результате повышается его сродство к кислороду и
развивается гипоксия клеток, в том числе в артериальной стенке;
- белки крови, что не только нарушает соответствующие им
функции (например, гемостатическую), но и повышает вязкость
крови из-за удлинения периода гюлувыведения гликозилиро-
ванных протеинов;
- структурные белки (коллаген, эластин и др.): базальные
мембраны утолщаются, функция их меняется.
При ферментативном гликозилировании растет скорость
использования глюкозы и образующихся из неё других моно-
сахаридов (глюкуроновая кислота, глюкозамин и т.д.) в синте-
зе гликолипидов, гликопротеинов и протеогликанов -- компо-
нентов межклеточного матрикса, базальных мембран. Актива-
ция сорбитолового пути превращения глюкозы (рис. 16) ведет
к осмотическому набуханию клеток (в том числе - артериаль-
ных стенок) и нарушению их функций. Все эти процессы обус-
ловливают более раннее и ускоренное развитие атеросклероза
у больных СД.
нс=о
неон
носн
неон
неон
н2с — он
н2с — он
неон
альдоз о-
редуктаза
НАДФ'Н2 НАДФ+
носн
I
неон
неон
I
н2с — он
глюкоза сорбитол
Рис. 16. Образование сорбитола из глюкозы.
87
Болезнь фон Гирке (гликогеноз I типа) характеризуется ги-
погликемией, которая снижает секрецию инсулина. Как и при
СД, в крови таких больных повышается уровень ЛПОНП, в
меньшей степени - ХМ, так как нарушено их разрушение мало-
активной ЛПЛ и усилена секреция ЛПОНП печенью.
Обусловленное инсулинорезистентностью повышение секре-
ции ЛПОНП (IV тип ГЛП) наблюдается при липодистрофии,
акромегалии и стрессе (эмоциональном, при остром инфаркте
миокарда, обширных ожогах, остром сепсисе). Недостаточ-
ность гормона роста и синдром Кушинга сопровождаются уси-
лением как образования ЛПОНП, так и превращения их в
ЛПНП с развитием II типа ГЛП.
Синдром Xвключает нарушение толерантности к глюкозе или
ИНСД, ожирение, дислипопротеинемию, гипертензию и пред-
расположенность к развитию АС. Центральное нарушение -
резистентность к инсулину и развивающаяся при этом гиперин-
сулинемия. В липопротеидном спектре крови наблюдаются ате-
рогенные сдвиги: повышается концентрация ЛПОНП и снижа-
ется - ЛПВП; увеличивается число мелких плотных частиц во
фракции ЛПНП.
Гипотиреоз, замедляя разрушение ЛПОНП и ЛППП, может
сопровождаться разнообразными нарушениями в обмене ЛП,
которые чаще соответствуют Па, Пб, иногда III и IV типам ГЛП.
Па тип ГЛП развивается при острой интермиттирующей
порфирии (механизм неясен), при нервной анорексии - из-за сни-
женной экскреции ХС и желчных кислот с желчью.
При нефротическом синдроме чаще встречается ГЛП Па, Пб,
но могут быть IV и V типы; наблюдаются гипоальбуминемия и
гиперхолестеринемия.
Холестаз ведет к поступлению ХС и ФЛ желчи в кровь и
появлению патологического ЛП - ЛП X (лецитин + ХС + аль-
бумин + апо С + относительная недостаточность ЛХАТ, воз-
можно, из-за избытка субстратов).
Злоупотребление алкоголем повышает ТГ и ЛПОНП в плаз-
ме (IV, редко V типы ГЛП). Впервые нарушается реципрокная
связь между уровнем ЛПОНП и ЛПВП, так как концентрация
а-ЛП в крови тоже растет. Предполагают даже целесообраз-
ность “умеренного” употребления алкоголя с целью профилак-
тики атеросклероза. Однако до сих пор не доказано, защища-
ют ли от АС повышенные таким образом ЛПВП. При хрони-
88
ческом потреблении больших доз алкоголя может наблюдать-
ся нормальный и даже сниженный уровень ЛПВП, а в случае
абстиненции развивается гипо-а-липопротеинемия. Часто по-
требление алкоголя ведет к поражениям печени и снижению
обмена липидов и ЛП.
Острый гепатит может протекать с ГЛП IV типа, что выз-
вано снижением секреции ЛХАТ печенью. Сочетание Па типа
ГЛП сгепатомой обусловлено отсутствием ингибирования син-
теза ХС в печени пищевым ХС по механизму обратной связи.
I тип ГЛП сопутствует системной красной/ волчанке, при ко-
торой иммуноглобулины связывают гепарин и снижают тем
самым активность Л ПЛ. Моноклональные гаммапатии (миело-
ма, макроглобулинемия, лимфома) протекают с ГЛП 111 или
IV типа, так как IgG или IgM образуют комплексы с ремнанта-
ми ХМ и/или ЛПОНП и таким образом тормозят их разруше-
ние.
4.2. НАРУШЕНИЯ ОБМЕНА ЛИПИДОВ В ТКАНЯХ
4.2.1. Нарушение процесса окисления жирных кисло г
Недостаточность карнитинпальмитоилтрансферазы печени
приводит к гипогликемии и понижению содержания кетоновых
тел в плазме крови. Снижение активности этого фермента в
мышцах нарушает процесс окисления жирных кислот - основ-
ных энергетических субстратов покоящихся и длительно рабо-
тающих (более 40 мин) миоцитов, в результате чего периоди-
чески возникает мышечная слабость, миалгия, спазмы и миог-
лобинурия. В отличие от гликогенозов, кратковременная мак-
симальная нагрузка не приводит к контрактурам, а нагрузка в
условиях ишемии вызывает повышение уровня лактата в плаз-
ме. Длительное голодание сопровождается увеличением актив-
ности креатинкиназы в сыворотке и миоглобинурией. Больным
рекомендуют регулярный прием богатой углеводами и бедной
жирами пищи, ограничение физической нагрузки.
Похожая клиническая картина наблюдается и при дефици-
те карнитина - одного из субстратов карнитинпальмитоилт-
рансферазы. Встречаются мышечная (миопатическая) и общая
(системная) формы недостатка карнитина. Как известно, это
витаминоподобное вещество синтезируется в печени из лизина
89
с использованием метионина в качестве источника метильных
групп. При миопатической форме уровень карнитина в крови
и печени нормальный, а в мышцах значительно снижен. Веро-
ятно, в основе патологии лежит нарушение его транспорта.
Болезнь проявляется мышечной слабостью без миоглобинурии.
При системной форме патологии уровень карнитина снижа-
ется не только в мышцах, но и в сыворотке крови и печени, что
говорит о нарушении синтеза его в гепатоцитах. Признаками
общего недостатка карнитина, наряду с мышечной слабостью
(миастенией), являются приступы гипогликемии, повышение
содержания СЖК в плазме крови, накопление липидов, умень-
шение образования кетоновых тел. Гипогликемия возникает из-
за нарушения энергетического обмена двух типов:
1) клетка не может нормально окислять жирные кислоты,
поэтому усиленно использует глюкозу в качестве энергетичес-
кого субстрата;
2) скорость глюконеогенеза снижается из-за нехватки АТФ.
Состояние больных улучшается при заместительной терапии
карнитином (по 2 г в день) и включении в диету содержащих
его продуктов.
Ямайская рвотная болезнь возникает у людей после упот-
ребления в пищу незрелых плодов аки (Blighia sapida), которые
содержат токсин гипоглицин. Гипоглицин ингибирует первый
фермент Р-окисления жирных кислот - ацил-КоА-дегидрогена-
зу, в результате чего повышается уровень СЖК в крови и моче,
развивается ацидоз, жировая инфильтрация печени и тяжелая
гипогликемия, обусловленная торможением глюконеогенеза.
При дикарбоновой ацидурии в митохондриях отсутствуют
ацил-КоА-дегидрогеназы среднецепочечных жирных кислот.
При этом нарушается Р-окисление, но активируется ш-окисле-
ние длинноцепочечных жирных кислот. Оно протекает в ЭПС
с участием гидроксилаз и цитохрома Р450 которые превращают
СН^-группу сначала в СН,ОН и потом - в СООН. Образовав-
шиеся дикарбоновые кислоты укорачиваются Р-окислением до
среднецепочечных С6-С10-дикарбоновых кислот, выводимых из
организма. Из-за нарушения окисления СЖК развивается ги-
погликемия.
Нарушение утилизации пропионил-КоА (рис. 17), образую-
щегося при окислении жирных кислот с нечетным числом угле-
родных атомов, наблюдается ^^недостаточностипропионил-
90
КоА-карбоксилазы, обусловленной изменением её апофермен-
та.
пропионил-КоА- метилмалонил-КоА
карбоксилаза(биотин) мутаза
Пропионил-КоА ----—-----------► Метилмалонил-КоА-------------►
f (дезоксиаденозил-
' ' ” кобаламин)
АТФ СО, АДФ + Ф
----► Сукцинил-КоА -----------► Цикл Кребса
Рис. 17. Схема метаболизма пропиопил-КоА.
Пропионил-КоА нс превращается в метилмалонил-КоА,
накапливается в крови, гидролизуется до пропионовой кисло-
ты, которая в большом количестве выделяется с мочой. Про-
нионацидемия сопровождается тяжелым кетоацидозом. Так как
пропионил-КоА образуется и в ходе метаболизма некоторых
аминокислот (метионин, валин, изолейцин, треонин), больным
рекомендуют ограничить белковое питание, а необходимое
количество калорий обеспечить глюкозой и жирными кисло-
тами с четным числом атомов углерода. Назначение биотина
неэффективно, что отличает данную патологию от недостаточ-
ности карбоксилаз при дефиците витамина Н (см. соответству-
ющий раздел).
Недостаточность метилмалонил-КоА-мутазы - следующе-
го фермента, участвующего в подключении пропиопил-КоА к
циклу Кребса, - может быть вызвана как изменением апофер-
мента, гак и (чаще) нарушением образования кофермента дс-
зоксиаденозилкобаламина (см. соответствующий раздел). В
первом случае положительной реакции на введение витамина
В|2 не наблюдается. Лабораторное обследование выявляет по-
вышение уровня метилмалоновой кислоты в крови и моче, аци-
доз.
Болезнь Рефсума - редкое неврологическое заболевание, обус-
ловленное врожденным нарушением а-окисления жирных кис-
лот. В тканях накапливается фитановая кислота, единственным
источником которой служит хлорофилл, содержащийся в зеле-
ных частях растений. Непосредственным субстратом для обра-
зования фитановой кислоты является составная часть хлоро-
филла спирт фитол.
91
Особенность катаболизма фитановой кислоты связана с ме-
тильной группой, которая находится у третьего атома углеро-
да и блокирует Р-окисление. В норме эта СН,-группа удаляется
с помощью ос-оксидазы фитановой кислоты, которая отсутству-
ет при болезни Рефсума. Патология проявляется обычно к 20
годам мозжечковой атаксией, периферической нейропатией,
ночной слепотой, глухотой, ихтиозными изменениями в облас-
ти ладоней и стоп, повышением содержания белка в спинно-
мозговой жидкости. Диагноз подтверждает повышение уровня
фитановой кислоты в крови до 30% от общего количества жир-
ных кислот (в норме - менее 0,3%, т.е. определяются лишь её
следы). Больным рекомендуют как можно раньше исключить
фитановую кислоту из пищи.
Синдром Цсльвегсра (Zellweger), илицереброгепаторенальный
синдром, является редким наследственным заболеванием, при
котором во всех тканях отсутствуют пероксисомы. В этих орга-
неллах происходит окисление длинноцепочечных жирных кис-
лот, поэтому у больных в мозгу накапливаются С26-С38-поли-
еновые кислоты.
4.2.2. Нарушение процесса распада сфинголипидов (липидозы)
Ряд заболеваний характеризуется накоплением избыточ-
ных количеств сфинголипидов (сфингомиелина, гликосфин-
голипидов) в клетках, чаще всего в нервных. Обязательным
компонентом сфинголипидов является сложный аминоспирт
сфингозин:
НО-^Н2
HC-NH2
НО - CH - CH = CH - (CH2)i2 - СН3
сфингозин
Соединение сфингозина с жирной кислотой получило назва-
ние N-ацилсфингозин, или церамид:
92
HO(fH2
НС - NH - CO R
I
HOCH - CH = CH - (CH2)t2 - CH3
церамид
R остатки, главным образом, С24-жирных кислот, напри-
мер, лигноцериновой, цереброновой, нервоновой. Сфингомие-
лины состоят из церамида, фосфорной кислоты и холина:
О
+ II
(CH3)3N—СН2—СН2—О—Р—О—СН2
ОН НС—NH—СО—R
НОСН—CH- СН—(СН2)12—сн3
сфингомиелин
Сфингомиелин является компонентом плазматических мем-
бран, эндоплазматической сети и митохондрий, а также входит
в состав миелиновых оболочек.
Замещение гидроксила у С,-церамида гексозой (галактозой
или глюкозой) приводит к образованию простейших гликос-
финголипидов (илицереброзидов) -галактозилцерамида (GalCer)
и глюкозилцерамида (GlcCer). GalCer и его сульфатированное
производное (сульфогалактозилцерамид) являются главными
липидами миелина (рис. 18):
93
О-СН,
о
II
НС - NH - С - СН(ОН) - (СН2)21 - СН3
HOCH - CH = СН - (СН2)12 - СНз
Рис. 18. Структура галактозилцерамида (R=H) и
сульфогалактозилцерамида (R=SO42).
GlcCer служит основным гликосфинголипидом мембран кле-
ток тканей, отличных от нервной, и предшественником боль-
шинства более сложных гликосфинголипидов - ганглиозидов.
Ганглиозидами называют гликосфинголипиды, дополнитель-
но содержащие одну или несколько молекул сиаловой кисло-
ты, в основном нейраминовой. Ганглиозиды в больших коли-
чествах находятся в нервной ткани и выполняют рецепторную
функцию. Простейшим ганглиозидом является GM3, который
содержит церамид, одну молекулу глюкозы, одну молекулу га-
лактозы и одну молекулу нейраминовой кислоты (NeuAc). В
данном сокращении G означает ганглиозид (ganglioside), М-
моносиаловое соединение (содержащее одну молекулу сиало-
вой кислоты), а индекс 3 - условный номер, присвоенный на
основе положения при хроматографическом разделении. Струк-
тура более сложного ганглиозида GM|, образующегося из GMV
показана на рис. 19. Другие ганглиозиды содержат от одной до
пяти молекул сиаловой кислоты: их называют ди-, трисиало-
ганглиозиды и т.д.
Сег - Glc - Gal - GalNAc - Gal
Neu-Ac
Рис. 19. ОМ1-Ганглиозид. GalNAc-N - ацетил галактозамин.
94
Ганглиозиды синтезируются из церамида путем последова-
тельного присоединения активированных сахаров, поставляе-
мых, например, УДФ-глюкозой и УДФ-галактозой, и одной из
сиаловых кислот, как правило, N-ацетилнейраминовой кисло-
ты.
Таким образом, может образоваться большое число гангли-
озидов с возрастающими молекулярными массами. Большин-
ство ферментов, катализирующих перенос остатков сахаров от
нуклеотидсахаров (гликозил ! рансферазы), находятся в аппарате
Гольджи.
При нарушении обмена сфинголипидов развиваются лини-
дозы.
Сфииголипидозами называют группу наследственных забо-
леваний, появляющихся чаще всего в детском возрасте и от-
носящихся к большой группе лизоеомных болезней. Врожден-
ную лизосомную болезнь определяют как состояние, при ко-
тором: 1) определяется недостаточность какого-либо лизосом-
ного фермента и 2) внутри связанных с лизосомами вакуолей
появляются необычные отложения (субстрат данного фермен-
та). Некоторые лизосомные гидролазы обладают специфич-
ностью к той или иной функциональной группе и се конфигу-
рации (а или Р), то есть эти ферменты способны расщеплять
определенные связи в разных полимерах: гликосфинголипи-
дах, гликозамингликанах, гликопротеинах. В связи с этим при
наличии блока на пути нормального их расщепления возмож-
но накопление не одного, а нескольких нерасщепленных суб-
стратов.
Поражаются те органы, которые являются обычными мес-
тами разрушения той или иной макромолекулы. Например, ге-
нерализованное повреждение тканей наблюдается при наруше-
нии распада вездесущих мукополисахаридов; белое вещество
головного мозга поражается при нарушении распада миелина;
гепатоспленомегалия развивается при нарушении распада эрит-
роцитарных гликолипидов. Накапливающийся материал может
вызвать как увеличение внутренних органов (висцеромегалию
или макроцефалию), так и вторичную атрофию, особенно моз-
га и мышц. Все эти болезни прогрессируют, многие заканчива-
ются смертью в детском или юношеском возрасте. Для оконча-
тельного диагноза важны результаты определения соответству-
ющих ферментов в сыворотке, лейкоцитах или культивируемых
95
фибробластах кожи. Стало возможным также выявлять гете-
розиготных носителей дефектных генов, ответственных за раз-
витие этих заболеваний, и определять сфинголиподистрофию
у плода.
Лизосомные болезни накопления характеризуются клиничес-
кой и биохимической гетерогенностью, которая определяется
разными мутациями структурных генов лизосомных фермен-
тов. Мутация гена может либо полностью лишить фермент ак-
тивности, либо только снизить ее, изменив те или иные свой-
ства фермента. Фенотипические колебания часто связаны с воз-
растом, поэтому выделяют инфантильную, ювенильную и взрос-
лую формы лизосомных болезней. Они различаются по степе-
ни тяжести, по сочетанию висцеральных, скелетных, невроло-
гических, глазных и других проявлений одной и той же болез-
ни.
Основные данные о некоторых болезнях накопления липи-
дов (сфинголипидозах) приведены в табл. 3.
С)и ^ганглиозидоз бгенерализованиьш ганглиозидоз ) - аутосом-
но- рецессивно наследуемая недостаточность (3-галактозидазы,
отщепляющей концевую галактозу от Ом|-ганглиозида. В ре-
зультате ферментного блока накапливается нормальный G и
его асиалопроизводное. У больных инфантильной формой с
рождения выявляется задержка психомоторного развития, ге-
патоспленомегалия и системные поражения, как при мукопо-
лисахаридозах. В костном мозге присутствуют пенистые клет-
ки. С мочой выделяются гликозамингликаны, но меньше, чем
при мукополисахаридозах. Смерть наступает в возрасте 1-2 лет.
При ювенильной (продолжительность жизни более 5 лет) и
взрослой формах болезни внутренние органы не изменяются, а
вовлечение в патологический процесс костной ткани минималь-
но. Представители разных этнических групп заболевают с рав-
ной частотой. Она довольно низкая, всего известно менее 50
лиц с каждым фенотипом болезни.
G Х1-ганглиозидоз (болезнь Тея-Сакса) - сравнительно час-
то встречающаяся врожденная аномалия метаболизма: дока-
зано уже несколько тысяч случаев заболевания. Младенчес-
кая форма (встречается преимущественно среди евреев ашке-
нази) в первые 5-6 месяцев жизни никак не проявляется, но
потом у ребенка повышается реакция на внешние раздражи-
тели, возникают миоклонические судороги, постепенно утра-
96
чиваются приобретенные навыки. К 1,5 годам развивается
центральная слепота, на втором году ребенок гибнет. Юно-
шеская форма встречается в разных этнических группах и ха-
рактеризуется более медленным прогрессированием. Больные
умирают в возрасте 5-15 лет из-за повторных аспирационных
пневмоний. Все формы болезни наследуются как аутосомный
рецессивный признак.
С^гш/г.7г7б>:шс>ш И пита (болезнь Сандхоффа) клинически
проявляется так же, как болезнь Тея-Сакса, только тяжелее.
GM2-ганглиозидоз обусловлен невозможностью отщепления
от Ом2-ганглиозида концевого N-ацетил-Р-галактозамина. Эту
реакцию катализирует гексозаминидаза, представленная дву-
мя изоферментами - А и В. При I типе (болезнь Тея-Сакса) на-
блюдается недостаточность гексозаминидазы А, при II типе
(болезнь Сандхоффа) отмечается недостаточность обоих изо-
ферментов. Это объясняется тем, что гексозаминидаза В состо-
ит из Р-субъединиц, структурный ген которых расположен на
5-й хромосоме, тогда как гексозаминидаза А включает и а-, и
Р-субъединицы, причем структурный ген а-субьединицы лока-
лизуется на 15-й хромосоме. Таким образом, для болезни Тея-
Сакса типичен дефект а-субъединицы, а для болезни Сандхоф-
фа - Р-субъединицы. Нерасщеплённый ганглиозид накаплива-
ется в мозге и, в меньшей степени, во внутренних органах. В
нормальной ткани мозга Ом2-ганглиозид присутствует в сле-
довых количествах, а при болезни Тея-Сакса на его долю при-
ходится от 6 до 12 % сухой массы; происходит центральная де-
миелинизация.
При болезни Сандхоффа в мозге тоже очень много GM2- ган-
глиозида и его асиалопроизводного, а в печени, почках и селе-
зенке накапливается глобозид - липидный компонент эритро-
цитарной и почечной мембран.
Болезнь Краббе (или галактозилцерамидный липидоз. или ша-
ровидно-клеточная лейкодистрофия) возникает из-за недоста-
точности галактозилцерамид-Р-галактозидазы и характеризу-
ется симптомами демиелинизации. Поражение белого вещества
проявляется в младенчестве; в результате периферической де-
миелинизации повышается содержание белка в спинномозго-
вой жидкости, что указывает на лейкодистрофию. Смерть на-
ступает через 1-2 года при резком усугублении неврологичес-
кой симптоматики. При аутопсии выявляют резкое нарушение
7, Зак. 49
97
Таблица 3
Заболевание Фермент, недостаточность которого обусловливает заболевание Накапливающийся липид Клинические симптомы
Г енерализованный ганглиозидоз (G.mi- ганглиозидоз) G.m । -Р-Галактозидаза Сег—Glc—Gal-(NeuAc)—GalNAc---Gal Ганглиозид G\n Умственная отсталость, увеличение пе- чени. деформация скелета
Болезнь Тея-Сакса (Смгганглиозидоз) Гексозаминидаза А Сег—Glc—GaI-(NeuAcH-GaINAc Ганглиозид Gm? Умственная отсталость, слепота, мы- шечная слабость
Вариант болезни Тея- Сакса. или болезнь Сандхоффа Гексозаминидазы А и В Сег—Glc—Gal—Gal-4-GaINAc Глобозид * ганглиозид Gm2 (тетраозилцерамид) Тс же. что и в случае болезни Тея- Сакса, но развиванием быстрее
Болезнь Краббе р-Галактозидаза Сег—Gal Галактозилцсрамид Умственная oicrxioctb, почти полное отсутствие миелина
Болезнь Гоше Р~ Глюкозидаза Сег—Glc Глюкозилцсрамид Увеличение печени и селезенки, эрозия трубчатых костей, умственная отста- лость у детей
Болезнь Нимана-Пика Сфингомиелиназа Сег—Р—холин Сфингомиелин Увеличение печени и селезенки, умст- венная Усталость.
Болезнь Фабри а-Галактозидаза Сег—Glc—Gal—Gal Церамидтригексозид (тригексоозилцерамид) Кожная сыпь, почечная недостаточ- ность (сильно выражена только у муж- чин); рецессивный генетический при- знак. свя зан с Х-хромосомой
Церамидлактозидли- пидоз Цсрамидлаклозидаза ф- галактозидаза) Сег—Glc—Gal Церамидлактозид Прогрессирующее поражение мозга, увеличение печени и селезенки
Met ахрома i ическая лейкодистрофия, суль- фагидный липидоз Арилсульфа газа (церебро- зидсульфатаза) Сег—Gal—OSO3 3-Сульфогалактозилисрамид Умственная oicianoctb и психические нарушения у взрослых: демиелинизация
Болезнь Фарбера Церамидаза Ацил—-Сфингозин Церамид Хрипота, дерматит, деформация сксле- ta. умственная отсталость; фатальна в раннем возрасте
NeuAc - N-аиетилнейраминовая кислота; Сег - церамид; Glc глюкоза; Gal - галактоза; GalNAc - N-аиетилгалактозамин
образования миелина и снижение абсолютного количества его
структурного компонента галактозил-цереброзида. Как объяс-
нить нехватку Gal-Cer, несмотря на отсутствие фермента, необ-
ходимого для его разрушения? Деструкцию миелина связыва-
ют с цитотоксическим действием психозина (галактозилсфин-
гозина), количество которого при болезни Краббе в 10 100 раз
превышает норму. Это вещество повреждает олигодендрогли-
альные клетки, производящие миелиновую оболочку.
Прижизненный диагноз основан на определении активнос-
ти галактозилцерамидазы в лейкоцитах периферической крови
или в культивируемых фибробласгах. Характерным специфи-
ческим признаком служат шаровидные клетки в нервной тка-
ни. Болезнь Краббе встречается относительно редко: выявлено
всего около 150 больных. Наследуется как аутосомный рецес-
сивный признак; несколько чаще болеют жители Скандинавс-
ких стран.
Болезнь Гоше (глюкозилцералшдиьш липидоз! обусловлена не-
достаточностью глюкозилцерамидазы (0-глюкозпдазы) и на-
коплением глюкозилцерамида в ретикулоэндотелиальных клер-
ках печени, селезенки и костного мозга. Эти “нагруженные"
клетки называются клетками Гоше. Главным источником Glc-
Сег являются гранулоциты, их время жизни значительно коро-
че, чем у эритроцитов. Клетки Гоше могут образовываться и
при хроническом миелоидном лейкозе, когда из-за ускоренно-
го круг ооборота лейкоцитов Glc-Cer не успевает вовремя рас-
падаться. Выделяют инфантильную форму (активность глюко-
зилцерамидазы не более 5% от нормы; ранняя смерть) и взрос-
лую (активность фермента - 18-40%), которая относится к наи-
более частой разновидности лизосомной болезни накопления.
Взрослая форма среди евреев ашкенази вст речается примерно
в 30 раз чаще, чем среди представителей других этнических
групп: 1:2500 новорожденных. Взрослая форма представляет
собой одну из тех лизосомных болезней накопления, которые с
наибольшей вероятностью могут встретиться в терапевтичес-
кой практике. Клинически она проявляется либо случайно об-
наруживаемой спленомегалией, либо тромбоцит опенией вслед-
ствие гиперсплении; гепатомегалией; возможны боли в костях
(нередко мучительные) и патологические переломы. Невроло-
гические симптомы отсутствуют. В селезенке, печени и костном
мозге обнаруживаются клетки Гоше. Характерно повышение в
7*
99
сыворотке уровня кислой фосфатазы. Клиническое течение ва-
рьирует: вовлечение в процесс легких или сердца может приве-
сти к ранней смерти, но у многих больных продолжительность
жизни не уменьшается. Диагноз можно поставить пренатально
путем исследования культивируемых амниотических клеток,
полученных во 11 триместре внутриутробного развития. Пред-
приняты попытки заместительного лечения взрослой формы
болезни путем введения очищенного фермента, заключенного
в липосомы.
Болезнь Нимана-Пика (типы А и В) развивается при недоста-
точности сфингомиелиназы, расщепляющей сфингомиелин на
церамид и фосфорилхолин. Активность фермента при типе А
составляет менее 7% от нормы, при типе В - 15-18%. Биохими-
ческая основа типов С, D и Е болезни, при которых активность
сфингомиелиназы сохраняется, неясна. Тип А (или младенчес-
кая форма) проявляется в возрасте до 3 месяцев трудностями
вскармливания, гипотрофией, неврологическими расстройства-
ми, глухотой, слепотой, гепатоспленомегалией. Смерть насту-
пает в 2-3 года. Для диагностики важны вакуолизированные
лимфоциты в мазках периферической крови и пенистые клетки
Нимана-Пика в пунктате костного мозга и ткани печени. Тип
В (висцеральная форма)-хроническоезаболевание, протекаю-
щее без поражения нервной системы. Обычно в возрасте 2 6
лет развивается спленомегалия, позже поражаются печень и
легкие, но продолжительность жизни нормальная. Тип С (сме-
шанная форма) проявляется в возрасте 4-20 лет гепатосплено-
мегалией, к которой присоединяются постепенно прогрессиру-
ющие неврологические симптомы.
Болезнь Фабри наследуется как признак, сцепленный с Х-хро-
мосомой, и особенно выражен у лиц мужского пола; развивает-
ся обычно в зрелом возрасте. Причина - недостаточность
а-галактозидазы с соответствующим накоплением тригексози-
да (GalGalGlcCer), в основном, в эндотелии сосудов. Среди кли-
нических проявлений превалируют признаки поражения почек
(протеинурия, уремия, артериальная гипертензия почечного
происхождения) и сердечно-сосудистой системы (стенокардия,
инфаркт миокарда, поражение клапанов), но не нервной тка-
ни. Для болезни характерны так называемые кризы Фабри, во
время которых пациент ощущает обжигающую, иногда нестер-
пимую боль в ладонях и стопах. Классическим признаком бо-
100
лезни являются ангиокератомы, появляющиеся уже в детском
возрасте и расположенные симметрично на бедрах, боках, спи-
не, ягодицах и наружных половых органах. Смерть чаще на-
ступает от почечной недостаточности после 30-40 лет.
Болезнь Фарбера (липогрануломатоз Фарбера) встречается
редко, обусловлена отсутствием лизосомного фермента цера-
мидазы, которая гидролизует церамид на сфингозин и жирную
кислоту. Церамид накапливается под кожей в виде узлов, в поч-
ках и печени. У детей поражаются суставы, нарушается дыха-
ние, глотание, наблюдаются психические расстройства. Боль-
ные погибают обычно в возрасте до 2 лет от легочных ослож-
нений. Диагноз подтверждают определением активности фер-
мента в культивируемых кожных фибробластах и лейкоцитах
периферической крови. При гистологическом исследовании в
подкожных узлах определяется жирорастворимый матсриал, из
которого с помощью хроматографии можно выделить церамид.
Причиной метахромной лейкодистрофии, встречающейся с
частотой 1:40000, служит недостаточность арилсульфатазы А
(церсброзидсульфатазы) или белкового активатора разрушения
сфинголипидов. Сульфогалактозилцерамид накапливается в
миелиновых оболочках, вызывая их дезинтеграцию, а также в
почках, печени и желчном пузыре, вероятно, без нарушения их
функции. Этот сульфатид действует как хроматрон, то есть из-
меняет свойства некоторых красителей: после связывания с ним
будет отражаться свет с большей длиной волны. Метахромати-
ческие включения накапливаются в почечных канальцах, а в
моче значительно повышена концентрация сульфатидов. Вы-
деляют детскую (возраст 2-5 лет) и взрослую (10-40 лет) формы
болезни. Клинически она проявляется неврологическими нару-
шениями, снижением интеллекта, повышением концентрации
белка в спинномозговой жидкости. У взрослых более выражен
психотический компонент. Диагностика основана на опреде-
лении ферментативной активности в лейкоцитах или фиброб-
ластах.
Множественная недостаточность сульфатаз приводит к на-
коплению сульфогалактозилцерамида, сульфостероидов и про-
теогликанов из-за одновременного недостатка пяти или более
клеточных сульфатаз: арилсульфатаз А, В, С и др. арилсульфа-
таз мукополисахаридов и нелизосомной стероидсульфатазы. Бо-
лезнь проявляется клинически в возрасте 1-2 лет признаками
101
как метахромной лейкодистрофии, так и мукополисахаридозов
(дизостоз, гепатоспленомегалия) и ихтиоза. Последний связан,
вероятно, с недостаточностью сульфатазы стероидов, которая
может быть изолированной, наследуемой как признак, сцеплен-
ный с Х-хромосомой. Обычно больные живут не более 10 лет.
Нарушение распада триглицеридов и эфиров холестерина
К болезням накопления липидов относится и недостаточность
кислой (лизосомной) липазы, катализирующей расщепление
эфиров холестерина и триглицеридов. Дефицит этого фермен-
та лежит в основе двух патологий с разным фенотипом: болез-
ни накопления эфиров холестерина, которая протекает более
легко и может встречаться у взрослых (см. ранее), и болезни
Волмана. Болезнь Волмана - тяжелая аномалия, проявляюща-
яся в самом раннем детстве пернициозной анемией, рвотой, ги-
потрофией, стеатореей. К постоянным признакам относятся
гепатоспленомегалия и повышенный уровень холестерина в
плазме. Рентгенологически выявляется кальцификация надпо-
чечников. Смерть может наступить в возрасте до 6 месяцев.
Нередко встречаются больные с отчетливыми клинически-
ми признаками липидоза, муколипидоза или мукополисахари-
доза, у которых не удается выявить ни одного из известных в
настоящее время биохимических нарушений. В связи с этим
число лизосомных болезней накопления будет, вероятно, уве-
личиваться.
В настоящее время эффективные средства лечения лизосом-
ных болезней накопления отсутствуют, поэтому оно остается в
основном симптоматическим. Продолжается работа по клони-
рованию ДНК, кодирующих ряд лизосомных ферментов, с це-
лью осуществления генной заместительной терапии.
102
Глава 5
ПАТОЛОГИЯ УГЛЕВОДНОГО ОБМЕНА
Основными представителями углеводов в организме чело-
века являются моносахариды (глюкоза, галактоза, фруктоза) и
полисахариды (гликоген). Кроме того, углеводы входят в со-
став смешанных макромолекул, образуя гликопротеиды, про-
теогликаны и гликолипиды. В углеводном обмене различают
катаболические процессы, такие как гликолиз, пентозофосфат-
ный цикл, “путь уроновых кислот”, гликогенолиз, деградацию
гликопротеидов и протеогликанов. Глюконеогенез, синтез гли-
когена, гликопротеидов и протеогликанов относятся к анабо-
лическим процессам. Функциональная активность любого ме-
таболического пути зависит от его обеспеченности субстраза-
ми, ферментами и кофакторами.
Любые дефекты на этапах поступления, переваривания и
всасывания углеводов (подробно рассматриваются в разделе
“Патология пищеварения и всасывания”) создают дефицит суб-
стратов, что снижает скорость протекания обменных реакций.
К различного рода нарушениям может приводить не только
недостаток, но и избыток субстратов.
Недостаточная активность ферментов, участвующих в ме-
таболизме углеводов, приводит к различным нарушениям, одни
из которых протекают бессимптомно (идиопатическая пенто-
зурия, фруктозурия). Другие болезни, при условии ранней ди-
агностики и правильно подобранной диете, позволяют детям
расти и развиваться нормально (галактоземия). Вместе с тем
существуют тяжелые аномалии, многие из которых заканчива-
ются смертельным исходом и часто в раннем детском возрасте.
5.1. НАРУШЕНИЯ МЕТАБОЛИЗМА ГЛЮКОЗЫ
Основными процессами, потребляющими глюкозу в клетке
(а также фруктозу и галактозу), являются гликолиз и пентозо-
фосфатный цикл.
5.1 Л. Нарушения гликолиза
Гликолиз - это путь распада глюкозы до пирувата или лак-
тата. При нормальном снабжении организма кислородом гли-
103
колиз является начальным необходимым этапом превращения
углеводов в пируват на пути их использования в качестве энер-
гетических субстратов.
Глюкоза
Г люкозо-6-фосфат
глюкозофосфатизомераза
Фруктозо-6-фосфат
фосфофруктокиназа
Фруктозо-1,6-бисфосфат
Дигидроксиацетонфосфат 4----->. Глицсральдегид-З-фосфат
1,3-Дифосфоглицерат
юглицерат
3- Фосфоглицерат
фосфо глицератфосфомутаза
2-Фосфоглицерат
Фосфоенолпируват
Щ-АДФ
С пируваткиназа
,>АТФ
/ват « Д ► Лактат
лактатдегидрогеназа
Рис. 20. Схема гликолиза.
| ] - недостаточная активность ферментов.
------ особенность гликолиза в эритроцитах.
104
Кроме того, в аэробных условиях гликолиз служит допол-
нительным путем образования АТФ и НАДН,. В анаэробных
условиях гликолиз становится единственным источником АТФ
для клетки, способным поддерживать ее жизнедеятельность.
При недостаточной активности ферментов гликолиза: глю-
козофосфатизомеразы, фосфофруктокиназы, фосфоглицерат-
фосфомутазы или пируваткиназы (рис. 20) нарушается про-
цесс превращения глюкозы по гликолитическому пути и, сле-
довательно, образование АТФ в ходе гликолитического фос-
форилирования. Из-за отсутствия митохондрий для эритро-
цитов гликолиз является единственным источником АТФ, ко-
торый используется главным образом Na’K’-АТФазой для
создания натрий-калиевого градиента на мембране. Кроме
того, в ходе гликолиза в эритроцитах образуется 2,3-дифос-
фоглицерат, который связывается с гемоглобином, понижая
его сродство к кислороду, что способствует диссоциации ок-
сигемоглобина и переходу кислорода в ткани. При блокиро-
вании гликолиза нарушается транспорт кислорода, происхо-
дит выравнивание трансмембранного градиента ионов Na+ и
К* и гибель эритроцитов. Развивается врожденная гемолити-
Отдельно выделяют недостаточность фосфофруктокиназы,
фосфоглицератфосфомутазы или М-субъединицы лактатдегид-
рогеназы в мышцах (эти нарушения описаны в разделе “Глико-
генозы”).
5.1.2. Нарушение пентозофосфатного цикла
Пентозофосфатный цикл является ответвлением от глико-
лиза на уровне глюкозо-6-фосфата и выполняет важнейшие
биологические функции, связанные с образованием НАДФН,
и рибозо-5-фосфата.
Недостаточность одного из ферментов пентозофосфатного
цикла - глюкозо-6-фосфат-дегидрогеназы сопровождается раз-
витием гемолитической анемии.
В норме глюкозо-6-фосфат-дегидрогеназа катализирует пре-
вращение глюкозо-6-фосфата в 6-фосфоглюконолактон. В этой
реакции образуется НАДФН2.
105
глюко зо-6-фосфат-
глюкозо-6-фосфат
дегидрогеназа
> 6-фосфоглюконолактон
НАДФН
.ДФН + Н
В эритроцитах НАДФН, используется для восстановления
окисленного глутатиона.
глутатионредуктаза
G-S-S-G + НАДФН + Н+ --------------------► 2 G - SH + НАДФ
окисленный
глутатион
восстановленный
глутатион
Восстановленный глутатион разрушает Н2О,. Для этой ре-
акции необходим селен, который является кофактором глута-
тионпероксидазы .
uiyi атионпероксидаза
2 G - SH + Н2О2 ------------------► G-S-S-G + 2 Н2О
При недостаточности глюкозо-6-фосфат-дегидрогеназы воз-
никает ферментативный блок и нарушается образование
НАДФН,. В результате в эритроцитах накапливается Н,О,,
которая сокращает время их жизни из-за повышения скорости
окисления гемоглобина в метгемоглобин. На земле живет око-
ло 100 млн. людей с генетически обусловленным пониженным
уровнем глюкозо-6-фосфат-дегидрогеназы. После приема пре-
паратов, действующих как оксиданты (аспирин, сульфанила-
миды и др.), а также после употребления в пищу бобов (Vicia
faba - конские бобы) у таких людей происходит гемолиз эрит-
роцитов.
5.1.3. Нарушение “пути уроновых кислот”
Кроме гликолиза и пентозофосфатного цикла глюкозо-6-
фосфат превращается по “пути уроновых кислот”, при кото-
ром происходит превращение глюкозы в глюкуроновую кис-
лоту и пентозы. Важность этого пути во многом определяется
глюкуроновой кислотой, которая необходима для обезврежи-
106
вания различных токсических веществ и является структурным
компонентом протеогликанов.
Первые реакции превращения глюкозы в уроновые кислоты
совпадают с начальными стадиями синтеза гликогена (до обра-
зования УДФ-глюкозы). УДФ-глюкоза окисляется по С6 с обра-
зованием УДФ-глюкуроновой кислоты. Затем глюкуроновая
кислота восстанавливается, окисляется и декарбоксилируется, в
результате образуется L-ксилулоза. Под действием L-ксилулозо-
редуктазы L-ксилулоза восстанавливается до ксилитола.
При отсутствии L-ксилулозоредуктазы нарушается восста-
новление L-ксилулозы в ксилитол и наблюдается идиопати-
ческая (эссенциальная) нентозурия. При этом заболевании в
моче появляется большое количество L-ксилулозы. Клини-
ческие симптомы при пентозурии отсутствуют. Недостаточ-
ность фермента может быть определена в эритроцитах, но
обычно в этом не бывает необходимости. Некоторые лекар-
ственные препараты, например, амидопирин и аспирин, уве-
личивают экскрецию L-ксилулозы у больных пентозурисй,
т.к. увеличивают скорость превращения глюкозы ио пути
уроновых кислот. Специального лечения при данном нару-
шении не требуется.
5.1.4. Нарушения глюконеогенеза
Глюконеогенез имеет важное значение для организма чело-
века как способ образования глюкозы из неуглеводных источ-
ников. Для нормального течения глюконеогенеза необходимы
соответствующие субстраты, ферменты, кофакторы и нуклео-
зидтрифосфаты (АТФ, ГТФ).
Выделяют следующие нарушения:
• недостаточность пируваткарбоксилазы,
• недостаточность синтетазы холокарбоксилазы,
• недостаточность фосфоенолпируваткарбоксикиназы,
• недостаточность фруктозе-1,6-бисфосфатазы,
недостаточность глюкозо-6-фосфатазы.
Недостаточность пируваткарбоксилазы
Недостаточность пируваткарбоксилазы - генетически обус-
ловленное нарушение, связанное с ключевым ферментом глю-
107
конеогенеза. В норме пируваткарбоксилаза при участии АТФ,
биотина и СО, превращает пируват в оксалоацетат.
пируват + НСО3
АТФ
пируваткарбоксилаза
АДФ + Н3РО4
оксалоацетат
При недостаточности пируваткарбоксилазы блокируется
синтез глюкозы из неуглеводных источников (лактата, пирува-
та, аминокислот). В крови значительно повышается содержа-
ние пирувата, лактата и аланина. Развиваются тяжелые психи-
ческие нарушения.
Пируваткарбоксилаза относится к биотинзависимым фер-
ментам. Функция биотина заключается в присоединении СО,
к ферменту, далее СО, переносится на пируват. Связывание
биотина с апоферментом катализируется специальным фер-
ментом, который обеспечивает образование амидной связи
через е-аминогруппу остатка лизина и называется синтетаза
холокарбоксилазы. Обнаружена недостаточность этого фер-
мента, которая отражается не только на метаболизме пирува-
та и лактата (нарушается глюконеогенез), но и на других ре-
акциях карбоксилирования (ацетил КоА, Р-метилкротоната,
Р-гидроксиизовалерата), протекающих с участием биотина. В
результате происходит накопление этих субстратов и выделе-
ние их с мочой. У детей развивается дерматит, замедляется
рост, наблюдаются алопеция, расстройство мышечной дея-
тельности, в некоторых случаях ослабление функций иммун-
ной системы.
Фосфоенолпируваткарбоксикиназа, катализирующая декар-
боксилирование оксалоацетата с образованием фосфоенолпи-
рувата, является ключевым ферментом глюконеогенеза.
108
оксалоацетат
фосфоенолпируваткарбоксикиназа
--------------------------► фосфоенолпируват + СО2
ГТФ ГДФ
При недостаточности этого фермента нарушается новооб-
разование глюкозы из таких субстратов как лактат, пируват и
протеиногенные аминокислоты, но сохраняется возможность
ее синтеза за счет глицерина. Наблюдаются гипогликемия, ге-
патомегалия, гипотония, задержка роста и развития.
Недостаточность фруктозой 1, б-басфосфатазы
Фруктозо-1,6-бисфосфатаза обеспечивает превращение фрук-
тозе-1,6-бисфосфата во фруктозо-6-фосфат.
фруктозо-1,6-6nc<|xxx|imna
фруктозо-1,6-бисфосфат + Н 2О----1-------► фруктозо-6-фосфат + Н ,РО4
При блоке на уровне фруктозо-1.6-бисфосфатазы глюконе-
огенез тормозится на одной из последних стадий, поэтому на-
рушается синтез глюкозы из всех нсуглеводных источников, а
также превращенйе фруктозы в глюкозу. Вся поступающая в
организм фруктоза превращается по гликолитическому нуги,
вызывая накопление лактата. Кроме того, фруктозо-1,6-бисфос-
фат ингибирует фосфорилазу гликогена, нарушая, таким обра-
зом, гликогенолиз. В итоге уровень глюкозы в крови полнос-
тью зависит от поступления экзогенной глюкозы. Основными
проявлениями данного нарушения являются лактацидоз, гипог-
ликемия и кетоз. В результате развиваются гепатомегалия, ги-
первентиляция, судороги и кома. Лечение заключается в исклю-
чении продуктов, содержащих сахарозу и фруктозу, при обес-
печении достаточного поступления глюкозы.
Недостаточность глюкозо-6-фосфатазы
Недостаточность глюкозо-6-фосфатазы была первым обна-
руженным случаем врожденной недостаточности ферментов пе-
чени. При этом нарушается не только глюконеогенез, но и гли-
когенолиз в печени как основной механизм поддержания нор-
мального уровня глюкозы в крови, а также синтез гликогена.
109
(Подробная характеристика этого заболевания дана в разделе
“Гликогенозы”).
5.2. НАРУШЕНИЯ МЕТАБОЛИЗМА ГАЛАКТОЗЫ
В норме галактоза, входящая в состав лактозы (основного
углевода молока), превращается в печени в глюкозу при вы-
полнении определенной последовательности реакций.
Сначала галактоза фосфорилируется в галактозо-1-фосфат
под действием галактокиназы.
галактокиназа
галактоза галактозо-1-фосфат
Галактозо-1 -фосфат реагирует с УДФ-глюкозой с образова-
нием УДФ-галактозы и глюкозо-1-фосфата. Фермент, катали-
зирующий данную реакцию, называется галактозо-1-фосфат-
уридил-трансфераза:
галактозо-1-фосфат + УДФ-глюкоза ►УДФ-галактоза + глюкозо-1-фосфат
Взаимопревращения УДФ-галактозы и УДФ-глюкозы про-
исходят под действием эпимеразы:
эпимераза
УДФ-галактоза -4— УДФ-глюкоза
Далее УДФ-глюкоза используется при синтезе гликогена,
при последующем фосфоролизе которого образуется глюко-
зо-1-фосфат. Глюкозо-1-фосфат под действием фосфоглюко-
мутазы превращается в глюкозо-6-фосфат, который гидроли-
зуется глюкозо-6-фосфатазой до свободной глюкозы. При на-
рушении основного пути метаболизма галактозы развивается
наследственное заболевание, которое называется галактозе-
мия.
Галактоземия может быть вызвана недостаточностью га-
лактокиназы. При блокировании основного метаболическо-
го пути на галактозу начинают действовать другие фермен-
ты (рис. 21), активность которых в организме здорового че-
ловека невелика. Галакто дегидрогеназа катализирует окис-
110
ление в галактоновую кислоту, альдозоредуктаза приводит
к восстановлению в галактитол (дульцитол). В крови и тка-
нях происходит накопление галактозы и ее необычных мета-
болитов.
галактозодегидрогеназа
галактоза----------------=^------► галактоновая кислота
НАДФН2
НАД
надн2
НАДФ
альдозоредуктаза
галактитол
Рис. 21. Пути превращения галактозы при галактоземии.
Галактитол является осмотически активным веществом, из-
быток которого представляет наибольшую опасность для хру-
сталика глаза. Мембраны хрусталика непроницаемы для галак-
титола, его накопление и чрезмерная гидратация способству-
ют развитию катаракты, которая является характерным при-
знаком галактоземии. Недостаточность галактокиназы в дру-
гих органах не вызывает повреждения их функций.
Значительно чаще встречается классическая галактоземия,
обусловленная недостаточностью галактозо-1-фосфат-уридил-
грансферазы.
При этом в тканях (хрусталике, печени, почках, мозге) и в
крови накапливаются галактозо-1 -фосфат - соединение токсич-
ное для организма и галактоза. Повышенный уровень галакто-
зы в крови уменьшает печеночную продукцию глюкозы, что
приводит к гипогликемии. В почках и кишечнике накопление
галактозы и галактозо-1-фосфата вызывает торможение транс-
порта аминокислот.
Симптомы классической галактоземии появляются в первые
дни или недели после рождения. Ребенок отказывается от мо-
лочных продуктов, появляется рвота, диарея, развивается ка-
таракта, замедляется развитие.
Диагноз устанавливают, определяя активность ферментов в
эритроцитах, а также по накоплению галактозо-1-фосфата в
эритроцитах и по наличию галактозы в моче.
111
Лечение заключается в исключении из питания продуктов,
содержащих галактозу. При переводе на безгалактозную дие-
ту все симптомы исчезают, кроме уже развившейся катарак-
ты. Несмотря на исключение галактозы, которая необходима
в организме для образования лактозы, гликолипидов (цереб-
розидов), протеогликанов и гликопротеидов, дети могут нор-
мально расти и развиваться, потому что при нормальной ак-
тивности эпимеразы возможно образование УДФ-галактозы
из глюкозы (рис. 22). Если на ранней стадии не исключить лак-
тозу, то развивается гепатомегалия, цирроз печени, пораже-
ние почек и головного мозга, наблюдается отставание в ум-
ственном развитии.
глюкоза—► глюкозо 6-фосфат-----► глюкозо-1-фосфат
I
УДФ-глюкоза
УДФ-гадактоза
лактоза гликопротеиды
гликолипиды протеогликаны
Рис. 22. Схема превращения глюкозы в галактозу.
По данным некоторых авторов возможна галактоземия из-
за блока на уровне эпимеразы.
5.3. НАРУШЕНИЯ МЕТАБОЛИЗМА ФРУКТОЗЫ
У здорового человека фруктоза может превращаться по двум
путям:
1) Под действием гексокиназы (в печени, мышцах, почках,
жировой ткани) образуется фруктозо-6-фосфат. Сродство это-
го фермента к фруктозе гораздо ниже, чем к глюкозе, поэтому
реакция образования фруктозо-6-фосфата ингибируется глю-
козой.
2) Главный путь превращения фруктозы происходит в пе-
чени под действием фруктокиназы (рис. 23). В результате
фруктоза превращается во фруктозо-1-фосфат. Эта реакция
112
глюкозой не блокируется. Фруктокиназа обладает высоким
сродством к своему субстрату и обнаружена также в почках
и кишечнике. Фруктозе-1-фосфат под действием альдолазы
В (фруктозо-1-фосфат-альдолаза) расщепляется на дигидрок-
сиацетонфосфат и глицеральдегид, который фосфорилиру-
ется триокиназой с образованием глицеральдегид-3-фосфа-
та. Два триозофосфата (дигидроксиацетонфосфат и глице-
ральдегид-3-фосфат) превращаются либо по гликолитичес-
кому пути, либо конденсируются под действием фруктозо-
1,6-бисфосфат-альдолазы во фруктозо-1,6-бисфосфат с пос-
ледующим превращением в глюкозу через фруктозо-6-фос-
фат и глюкозо-6-фосфат. Расщепление фруктозы по пути гли-
колиза происходит быстрее, чем глюкозы, т.к. минует ста-
дию, катализируемую фосфофруктокиназой. Однако для пе-
чени более характерным является путь превращения фрук-
тозы в глюкозу.
Известны следующие нарушения метаболизма фруктозы:
идиопатическая фруктозурия и наследственная нетолерантность
к фруктозе.
Идиопатическая фруктозурия или эссенциальная фруктозурия,
или фруктоземия
Идиопатическая фруктозурия - врожденное нарушение об-
мена фруктозы из-за отсутствия фермента фруктокиназы в пе-
чени. В результате превращение фруктозы возможно только
через фруктозо-6-фосфат, но реакция его образования тормо-
зится глюкозой. В результате фруктоза накапливается в кро-
ви. Почечный порог для фруктозы очень низкий, поэтому
фруктоза обнаруживается в моче уже при ее концентрации в
крови 0,73 ммоль/л. Клинические симптомы при фруктозурии
отсутствуют, потому что свободная фруктоза не токсична для
организма.
8. Зак. 49
ИЗ
Фруктоза
АТФ
АДФ
АТФ
АДФ.
гексокиназа
фруктозе-1 -фосфат
фруктозо-6-фосфат
2
альдолаза В
глицеральдегид дигидроксиацетонфосфат
АТФ :
АДФ
гл и 1щрал ьдегид-3 -фосфат
гликолиз
пируват(лактат)
Рис. 23. Нарушения метаболизма фруктозы.
I - блок при идиопатической фруктозурии;
2 - блок при наследственной нетолерантности к фруктозе.
Наследственная нетолерантность к фруктозе или врожденная
непереносимость фруктозы
Причина заболевания заключается в недостаточности альдо-
лазы В (фруктозе-1-фосфат-альдолазы) в печени (рис. 23). Из-за
этого блокируется расщепление фруктозе-1-фосфата до дигид-
роксиацетонфосфата и глицеральдегида. В результате накапли-
вается токсичный для организма фруктозе-1-фосфат, который
ингибирует фруктозе- 1,6-бисфосфат-альдолазу и фосфорилазу
гликогена по аллостерическому механизму. Из-за этого наруша-
ются важнейшие процессы превращения углеводов: гликолиз,
114
глюконеогенез и распад гликогена (гликогенолиз). Заболевание
обычно обнаруживается при переходе от грудного кормления
на пищу, содержащую сахарозу и фруктозу. После ее приема на-
блюдается гипогликемия, рвота, судороги, а в дальнейшем раз-
вивается патология печени и повреждение почечных канальцев
из-за накопления фруктозо-1-фосфата в этих органах. При уст-
ранении фруктозы из пищи дети развиваются нормально.
5.4. НАРУШЕНИЯ МЕТАБОЛИЗМА ГЛИКОГЕНА
Гликоген - это полисахарид, мономерами которого являют-
ся остатки глюкозы, соединенные а-1,4- и а-1,6-гликозидными
связями. В организме человека гликоген выполняет энсргеги-
ческую и резервную функции: избыток глюкозы запасается в
виде гликогена, который при необходимости расщепляется и
поставляет глюкозу.
Выделяют две группы наследственных заболеваний, связан-
ных с нарушениями обмена гликогена:
1. Агликогенозы.
2. Гликогенозы.
5.4.1. Агликогенозы
Агликогенозы - заболевания, обусловленные нарушением
синтеза гликогена из-за недостаточности гликогенсинтазы. При
этом запасы гликогена в тканях отсутствуют или резко сниже-
ны. Самый характерный признак агликогенозов - резкая гипог-
ликемия натощак, особенно после ночного перерыва в кормле-
нии. Возникают рвота, судороги, потеря сознания, отставание
умственного развития. Больные часто погибают в раннем дет-
стве. Частые кормления ослабляют симптомы заболевания.
5.4.2. Гликогенозы
Гликогенозы или болезни накопления гликогена - группа
наследственных заболеваний, при которых в тканях происхо-
дит накопление гликогена нормальной или измененной струк-
туры из-за дефекта ферментов, участвующих в синтезе или рас-
паде гликогена. Первая болезнь накопления гликогена была
описана Эдгаром фон Гирке в 1929 году. Природу этого фер-
8*
115
ментативного нарушения, связанного с глюкозо-6-фосфатазой,
установили Карл и Герти Кори в 1952 году. С тех пор охаракте-
ризован целый ряд болезней накопления гликогена.
В последнее время гликогенозы предложено называть по
недостаточности специфического фермента. Для разделения
гликогенозов на типы также используют обозначения римски-
ми цифрами и названия по классификации Кори, которая име-
ет историческое значение.
Гликогенозы можно разделить на три группы:
1. Печеночно-гипогликемические нарушения.
2. Мышечно-энергетические нарушения.
3. В третью группу объединены гликогенозы, которые име-
ют индивидуальные особенности и поэтому не могут быть
включены ни в первую, ни во вторую группы.
Печеночно-гшюгликемичсские нарушения
К печеночно-гипогликемическим нарушениям относятся:
• недостаточность глюкозо-6-фосфатазы (тип 1а),
недостаточность микросомальной глюкозо-6-фосфат-
транслоказы (тип 1b),
• недостаточность амило-1,6-глюкозидазы (тип III),
недостаточность фосфорилазы в печени (тип VI),
недостаточность киназы фосфорилазы В в печени.
фон Гирке
Недостаточность глюкозо-6-фосфатазы представляет собой
тяжелое наследственное заболевание (частота 1:100000 -
1:400000), которое обычно проявляется в первые 12 месяцев
жизни гипогликемией (сопровождается судорогами) и гепато-
мегалией, а в дальнейшем отставанием в росте и развитии.
В норме гликоген расщепляется под действием двух фермен-
тов: фосфорилазы А и амило-1,6-глюкозидазы (деветвящий фер-
мент). В результате образуется глюкозо-1-фосфат и небольшое
количество свободной глюкозы. Глюкозо-1-фосфат изомеризует-
ся в глюкозо-6-фосфат, который поступает в гликолиз и пентозо-
фосфатный цикл. В печени глюкозо-6-фосфат переносится специ-
фической транслоказой в эндоплазматическую сеть, где гидроли-
зуется глюкозо-6-фосфатазой до свободной глюкозы, которая
116
высвобождается в кровь. Это основной путь поддержания нор-
мального уровня глюкозы в крови (рис. 24). Глюкозо-6-фосфата-
за является также ключевым ферментом глюконеогенеза.
гликоген
гликогенсинтаза,
ветвящий фермент
гликогепфосфорилаза А
амило-1,6-глюкозидаза
глюкозо-1 -фосфаз
глюкоза (немного)
>кровь
глюкозо-6-фосфат
пешозофофосфатпып путь—► рпбозо-5-фосфат
lb
гликолиз
транслоказа
пируват (лактат)
глюкозо-6-фосфа! 4------- глюконеогенез
1а глюкозо-6-
фосфатаза
глюкоза
I
кровь
Рис. 24. Ферментативные блоки
( ) при печеночно-гипогликемических нарушениях.
------- активация гликогенсинтазы глюкозо-6-фосфатом.
При недостаточности глюкозо-6-фосфатазы уровень глюко-
зо-6-фосфата и его метаболитов повышается, гликолиз ускоря-
ется, что приводит к избыточному образованию лактата. Глю-
козо-6-фосфат является аллостерическим активатором глико-
генсинтазы. В результате в печени накапливается гликоген нор-
мальной структуры.
В крови больных резко снижено содержание глюкозы, по-
вышен уровень лактата, холестерина, триглицеридов и моче-
вой кислоты. Обычно развивается кетоз. Активация окисления
жирных кислот как энергетических субстратов в конечном итоге
приводит к кетонемии и кетонурии. Гиперурикемия обуслов-
ит
лена повышением продукции мочевой кислоты и снижением ее
почечной экскреции.
Уровень глюкозы в крови после введения адреналина или
глюкагона, а также после приема галактозы (нарушение теста
толерантности к галактозе) повышается незначительно (за счет
деветвящего фермента), слабее, чем в норме.
Для диагностики заболевания определяют активность глю-
козо-6-фосфатазы и содержание гликогена в биоптатах печени.
Лечение: частый прием пищи днем и ночное кормление через
зонд. Диетотерапия: 60-70% углеводов, сниженное содержание
белка. Требуется ограничение сахарозы и лактозы, т.к. содержа-
щиеся в них фруктоза и галактоза не могут нормально метабо-
лизироваться для поддержания уровня глюкозы в крови. В каче-
стве медленно всасывающегося полимера глюкозы используют
сырой кукурузный крахмал. При необходимости назначают ал-
лопуринол - для снижения уровня мочевой кислоты в плазме.
Недостаточность микросомальной глюкозо-6-фосфат-транс-
локазы, тип 1b
Недостаточность микросомальной глюкозо-6-фосфат-транс-
локазы - редкое заболевание, раньше его называли псевдотип I
(рис. 24). Протекает тяжелее, чем тип 1а.
При этом заболевании нарушается перенос глюкозо-6-фос-
фата в эндоплазматическую сеть. В отличие от типа 1а, при 1b
активность глюкозо-6-фосфатазы в норме. При диагностике тип
lb отличают от 1а по определению активности глюкозо-6-фос-
фатазы в биоптатах печени.
Проявления 1b типа аналогичны типу 1а, но имеются допол-
нительные клинические признаки, такие как нейтропения и по-
вторные рецидивирующие инфекции. Лабораторные данные,
реакция на тесты и лечение одинаковы при типе 1а и lb. Они
отличаются от других печеночно-гипогликемических аномалий
невозможностью поддерживать уровень глюкозы в крови за счет
глюконеогенеза или введения галактозы и фруктозы.
Недостаточность амило-1,6-глюкозидазы (или деветвящего
фермента), тип III или болезнь Кори-Форбса
Недостаточность амило-1,6-глюкозидазы встречается часто.
118
У здоровых людей гликогенфосфорилаза А расщепляет в
гликогене ос-1,4-гликозидные связи с образованием глюкозо-1-
фосфата, амило-1,6-глюкозидаза гидролизует ос-1,6-гликозид-
ные связи до свободной глюкозы (рис. 24).
При недостаточности амило-1,6-глюкозидазы происходит
неполное расщепление гликогена до лимитдекстрина (разветв-
ленного полисахарида), который и накапливается при данной
патологии. В связи с этим заболевание иногда называют ли-
митдекстриноз.
В крови больных уровень глюкозы натощак снижается (у 80%
больных), уровень лактата и мочевой кислоты в пределах нор-
мы, повышены содержание холестерина и триглицеридов, ак-
тивность трансаминаз. Выражен кетоз. Реакция на введение
глюкагона и адреналина нарушена. Тест толерантности к га-
лактозе, фруктозе, глицерину в норме.
Клинические признаки: гипогликемия, гепатомегалия, более
выражена спленомш алия; у некоторых больных малый рост и
задержка развития, легкая миопатия. Клиническое течение за-
болевания менее тяжелое, чем при типе 1а.
Для диагностики заболевания используют определение гли-
когена с измененной структурой в печени и мышцах; определе-
ние фермента в биоптатах печени, мышц или фибробластах.
Лечение: частые кормления в детстве. Глюконеогенез не на-
рушен, поэтому больной может получать галактозу, фруктозу
и белок. Диета: 50% углеводов и 15-20% белка.
Недостаточность фосфорилазы в печени, тип VI или болезнь
Недостаточность фосфорилазы в печени - редкое заболе-
вание.
Фосфорилаза является ферментом, лимитирующим скороеть
гликогенолиза (рис. 24). В печени и мышцах этот фермент ко-
дируется разными генами. При данном нарушении в печени
накапливается гликоген с нормальной структурой. Проявления
в большинстве случаев сходны с гликогенезом III типа. Наблю-
дается гепатомегалия, йепостоянная гипогликемия.
Для диагностики заболевания определяют активность фос-
форилазы (отсутствует) и киназы фосфорилазы В (в норме) в
биоптатах печени.
Лечение: диета как при III типе.
119
Недостаточность киназы фосфорилазы В в печени
Недостаточность киназы фосфорилазы В в печени - само-
стоятельное, очень легкое, но возможно широко распростра-
ненное (т.к. часто не диагносцируется) заболевание. Ранее обо-
значали как тип Via, VIII, IX.
В норме киназа фосфорилазы В фосфорилирует гликоген-
фосфорилазу В, которая при этом переходит в активную фос-
форилазу А. При данной патологии нарушается каскадный
механизм активации гликогенфосфорилазы. В результате в пе-
чени повышен уровень гликогена с нормальной структурой.
Клинически недостаточность киназы фосфорилазы В про-
является гепатомегалией и непостоянной гипогликемией.
Для диагностики заболевания определяют активность фер-
мента в лейкоцитах, фибробластах или биоптатах печени. В
мышцах активность киназы фосфорилазы В в норме.
Лечение: диета как при III типе.
К мышечно-энергетическим нарушениям относятся:
• недостаточность фосфорилазы в мышцах (тип V);
• недостаточность фосфофруктокиназы (тип VII);
• недостаточность фосфоглицератфосфомутазы;
• недостаточность М-субъединицы лактатдегидрогеназы.
Недостаточность фосфорилазы в мышцах, тип V или болезнь
МакгАрдля
Недостаточность фосфорилазы в мышцах встречается редко.
В мышцах накапливается гликоген с нормальной структурой.
Заболевание впервые проявляется в возрасте 20-30 лет: при тя-
желой физической нагрузке отмечаются боли в мышцах, судоро-
ги, миоглобинурия и повышение активности КФК в крови.
Диагностические признаки: неувеличивающийся уровень
лактата и пирувата в крови в ответ на физическую нагрузку,
накопление гликогена нормальной структуры и отсутствие фос-
форилазы в биоптатах мышечной ткани.
Лечение при V типе и трех последующих аномалиях: исклю-
чение физических нагрузок, при необходимости - прием глю-
козы или фруктозы перед нагрузкой.
120
Недостаточность фосфофруктокиназы в мышцах, тип VII
Недостаточность фосфофруктокиназы в мышцах - редкое
заболевание. Фосфофруктокиназа - гликолитический фермент,
катализирующий реакцию превращения фруктозо-6-фосфата во
фруктозо-1,6-бисфосфат:
фосфофруктокиназа
фруктозо-6-фосфат "s’ V ► фруктозо- 1,6-бисфосфат
АТФ АДФ
При отсутствии фермента блокируются все последующие
реакции гликолиза, который закапчивается образованием пи-
рувата или лактата. Поэтому клинические проявления, лабо-
раторные данные при этом заболевании аналогичны типу V.
Болезнь проявляется также незначительной гемолитической!
анемией из-за недостаточности фермента в эритроцитах.
Недостаточность фосфоглицератфосфомутазы в мышцах
Фосфоглицератфосфомутаза катализирует реакцию глико-
лиза, в которой происходит изомеризация 3-фосфоглицерата в
2-фосфоглицерат:
фосфоглицератфосфомугаза
3-фосфоглицерат ----------_-----------------> 2-фосфоглицерат
Возникающий ферментативный блок препятствует дальней-
шим реакциям гликолиза и образованию пирувата и лактата.
Клинические проявления и результаты тестов аналогичны
V типу, но содержание гликогена в мышцах нормальное.
Для диагностики заболевания необходимо определение фер-
мента в биоптатах мышц.
Недостаточность М-субъединипы лактатдегидрогеназы
Недостаточность М-субъединицы лактатдегидрогеназы от-
личается от других мышечных нарушений тем, что при физи-
ческой нагрузке в крови повышается уровень пирувата, а не
121
лактата. Все реакции гликогенолиза и гликолиза при этом на-
рушении протекают нормально до образования пирувата. Но
дальнейшего превращения пирувата в лактат не происходит из-
за дефекта лактатдегидрогеназы.
Клинические проявления аналогичны V типу. Содержание
гликогена в мышцах нормальное.
Для диагностики заболевания определяют изоферменты лак-
татдегидрогеназы в сыворотке, эритроцитах, лейкоцитах и био-
птатах мышц.
В третью группу гликогенозов объединены две патологии,
обусловленные:
недостаточностью а-глюкозидазы (тип П),
• недостаточностью ветвящего фермента.
Недостаточность а-глюкозидазы. тип II или болезнь Помпе
Недостаточность а-глюкозидазы представляет собой лизо-
сомную болезнь накопления, потому что накопление гликоге-
на происходит в лизосомах.
Причина заболевания заключается в недостаточности лизо-
сомального фермента - кислой а-глюкозидазы, которая в норме
в лизосомах расщепляет как а-1,4-, так и а-1,6-гликозидные свя-
зи в молекуле гликогена с образованием свободной глюкозы.
Гипогликемия при данном заболевании отсутствует, т.к. ак-
тивность других ферментов обмена гликогена в норме.
Различают три формы данного нарушения:
- инфантильная форма (0-6 месяцев): гипотензия, мышечная
слабость, кардиомегалия, сердечная недостаточность, увеличе-
ние размеров языка; ранняя смерть от сердечной недостаточ-
ности. Уровень КФК и альдолазы в сыворотке повышен;
- ювенильная форма: прогрессирующая слабость скелетных
мышц, сердечные проявления отсутствуют;
- взрослая форма: прогрессирующая слабость скелетных
мышц, явления легочной недостаточности.
Для установления диагноза определяют фермент в биопта-
тах мышц или фибробластах. Эффективного лечения не разра-
ботано.
122
Недостаточность ветвящего фермента, тип IV или болезнь
Недостаточность ветвящего фермента - редкое заболевание.
Для синтеза нормальноразветвленного гликогена требуют-
ся два фермента: гликогенсинтаза, которая катализирует обра-
зование а-1,4-гликозидных связей, и ветвящий фермент, кото-
рый обеспечивает образование а-1.6-гликозидных связей.
При отсутствии ветвящего фермента происходит синтез гли-
когена нарушенной структуры, который накапливается в пече-
ни и вызывает ее повреждение. Иногда заболевание называют
“амилопектиноз”, т.к. синтезируемый гликоген по структуре
похож на амилопектин.
Способность поддерживать нормальный уровень глюкозы
в крови при данной патологии не нарушается, поэтому гипог-
ликемия отсутствует.
Наблюдается замедленное развитие новорожденных, цирроз
и недостаточность печени, резкая гипотензия и слабость у не-
которых больных, ранняя смерть из-за печеночной или сердеч-
ной недостаточности. Эффективного лечения нс разработано.
5.5. НАРУШЕНИЯ МЕМБРАННОГО ТРАНСПОРТА
ГЕКСОЗ
Трансмембранный перенос моносахаридов осуществляет-
ся в норме с помощью двух механизмов: активного транспор-
та, характерного для глюкозы и галактозы, и простой диффу-
зии для фруктозы. Существуют врожденные нарушения мем-
бранного транспорта моносахаридов, такие как почечная глю-
козурия (гликозурия) и нарушение всасывания глюкозы и га-
лактозы.
Почечная глюкозурия (гликозурия)
У здорового человека в клубочковом фильтрате глюкоза
присутствует в той же концентрации, что и в плазме. В прокси-
мальных почечных канальцах она реабсорбируется с помощью
специфического белкового переносчика с участием Na+. Почки
способны реабсорбировать глюкозу при большей ее концент-
рации в плазме и клубочковом фильтрате, чем в норме, поэто-
123
му она не появляется в моче до тех пор, пока не будет превы-
шен порог реабсорбции.
При почечной глюкозурии (аутосомно-рецессивное заболе-
вание) предполагается мутация транспортного белка для D-глю-
козы в проксимальных почечных канальцах. Отличительные
признаки почечной глюкозурии:
- глюкозурия наблюдается при нормальном уровне глюко-
зы в крови;
- глюкозурия постоянна, с небольшими колебаниями в зави-
симости от питания;
- при проведении перорального теста на толерантность к
глюкозе получается нормальная сахарная кривая;
- в качестве восстанавливающего вещества в моче иденти-
фицируется глюкоза;
- процессы распада и синтеза углеводов в норме.
Различают два типа глюкозурии: тип А, ко горый характери-
зуется снижением максимальной способности канальцев к реаб-
сорбции и тип Б, обусловленный снижением порога глюкозурии.
Нарушение всасывания глюкозы и галактозы
Причина данного нарушения заключается в мутации обще-
го транспортного белка для глюкозы и галактозы в слизистой
оболочке тощей кишки и проксимальных почечных канальцах.
Заболевание проявляется водной диареей после приема глю-
козы, галактозы, лактозы или сахарозы. Фруктоза и безугле-
водная диета хорошо переносятся.
Потери глюкозы с мочой меньше, чем при изолированной
почечной глюкозурии. Вероятно потому, что глюкоза реабсор-
бируется с помощью специфического для нее белка и общего
(для глюкозы и галактозы) транспортного белка.
Аналогичные нарушения возникают при повреждении транс-
портных систем различными факторами, дефектеNa+, К"-АТФа-
зы, действии ингибиторов и уменьшении продукции АТФ.
5.6. НАРУШЕНИЯ МЕТАБОЛИЗМА УГЛЕВОД-
БЕЛКОВЫХ КОМПЛЕКСОВ
Углевод-белковые комплексы подразделяют на две группы:
Е гликопротеиды 2. протеогликаны.
124
5.6.1. Нарушения метаболизма гликопротеидов
Гликопротеиды - это сложные белки, в которых белковая
часть ковалентно связана с олигосахаридными цепями нерегу-
лярного строения. Содержание белка в гликопротеидах обыч-
но составляет 80-90%. Основными компонентами олигосахарид-
ных цепей являются гексозы (манноза, галактоза), ацетилгек-
созамины (N-ацетилглюкозамин, N-ацетилгалактозамин), пен-
тозы (L-арабиноза, ксилоза), метилпентоза(L-фукоза) и сиало-
вые кислоты (N-ацилпроизводные нейраминовой кислоты). В
гликопротеидах углеводный компонент присоединяется к бел-
ковой части молекулы с помощью О-гликозидной связи за счет
гидроксильной группы серина (треонина) или N-гликозидной
связи через амидную группу аспарагина. На основании этого
гликопротеиды подразделяют на О-гликопротеиды и N-глико-
протеиды.
Многообразие гликопротеидов проявляется в функциях,
ко горые они выполняют в организме. Различают структурную,
транспортную, иммунологическую, антифризную, гормональ-
ную, ферментативную, защитную (“смазочную") функции, гли-
копротеиды также обеспечивают специфичность межклеточных
контактов.
Метаболизм гл и ко протеидов складывается из процессов их
синтеза и распада. Образование О-гликопротепдов осуществ-
ляется путем ступенчатого присоединения моносахаридных
остатков к белку, которое начинается во время синтеза поли-
пептидной цепи и продолжается после трансляции в аппарате
Гольджи. Донорами моносахаридов являются их активирован-
ные формы (УДФ-Г<-ацетилгалактозамип, УДФ-галактоза,
ГДФ-манноза и др.), реакции гликозилирования катализиру-
ются соответствующими гликопротеид-гликозил-трансфсраза-
ми. Нарушений синтеза О-гликопротеидов не обнаружено.
Синтез N-гликопротеидов характеризуется следующими
особенностями: сборка олигосахаридной цепи происходит на
долихолпирофосфате, содержание которого влияет на ско-
рость синтеза гликопротеидов. Далее олигосахарид целиком
переносится на синтезируемый на рибосомах белок. Затем с
участием ферментов эндоплазматического ретикулума и ап-
парата Гольджи происходит модификация олигосахаридных
цепей, которая заключается в отщеплении одних углеводных
125
остатков и присоединении других. Существует нарушение син-
теза N-гликопротеидов, известное под названием 1-клеточная
болезнь.
Расщепление гликопротеидов осуществляется с помощью
лизосомальных гидролаз, проявляющих специфичность к уг-
леводным остаткам. При недостаточности ферментов лизосом
развиваются следующие нарушения: фукозидоз, маннозидоз и
аспартилглюкозаминурия.
Нарушение синтеза гликопротеидов
I-клеточная болезнь (клетки с включениями - inclusion cell)
Лизосомальные ферменты по структуре относятся к N-гли-
копротеидам, проходят общие стадии синтеза и посттрансля-
ционного процессинга, но отличаются дополнительным этапом,
который заключается в следующем:
N-ацетилглюкозаминфосфотрансфераза эндоплазматическо-
го ретикулума в норме катализирует присоединение N-ацетил-
глюкозамин-1 -фосфата к ферментам, предназначенным для ли-
зосом. Присоединение М-ацетилглюкозамин-1-фосфата проис-
ходит к остаткам маннозы в составе гликопротеида.
Затем N-ацетилглюкозамин удаляется под действием фосфо-
диэстеразы, а остатки маннозы в составе фермента остаются
фосфорилированными в 6-положении.
В комплексе Гольджи локализованы рецепторы для манно-
зо-6-фосфата, которые связывают остаток маннозо-6-фосфата
в структуре ферментов и направляют их в лизосомы.
Причина I-клеточной болезни заключается в недостаточной
активности N-ацетилглюкозаминфосфотрансферазы. В резуль-
тате нарушается посттрансляционный процессинг лизосомаль-
ных ферментов, а именно процессинг олигосахаридных цепей. У
таких больных синтез ферментов, предназначенных для лизосом,
не нарушен и в клетках для них имеется нормальный рецептор.
Но из-за отсутствия в своей структуре маннозо-6-фосфата эти
ферменты не распознаются и не достигают лизосом. При 1-кле-
точной болезни в фибробластах отсутствуют лизосомальные
ферменты и поэтому накапливаются различные недеградирован-
ные молекулы (гликопротеиды, гликолипиды). I-клеточную бо-
лезнь относят к лизосомным болезням накопления.
126
Заболевание начинается в раннем детстве и проявляется от-
ставанием психического развития, резко выраженной диспла-
зией костей, помутнением роговицы. В сыворотке крови отме-
чается резко повышенный уровень лизосомальных ферментов.
Нарушение распада гликопротеидов
Фукозидоз, маннозидоз и аспартилглюкозаминурия относят-
ся к лизосомным болезням накопления, при которых из-за не-
достаточности лизосомальных гидролаз происходит накопле-
ние соответствующих нерасщепляемых субстратов.
а-Фукозидаза - лизосомальный фермент, который в норме
отщепляет концевые остатки L-фукозы от гликопептидов. При
недостаточной активности этого фермента развивается фуко-
зидоз, при котором накапливаются гликопептиды, гликолипи-
ды и олигосахариды. Наблюдаются отставание в умственном
развитии, нарушения электролитного состава пота и кожные
апгиокератомы. Смерть наступает в детском возрасте. Для ди-
агностики заболевания определяют активность а-фукозидазы.
Маннозидоз
При маннозидозе накапливаются олигосахариды из-за не-
достаточности а-маннозидазы, которая в норме отщепляет ос-
татки маннозы от олигосахаридов. Заболевание характеризу-
ется неврологическими нарушениями, необычными круговыми
катарактами, летальным исходом в детстве. Для диагностики
заболевания проводят определение активности фермента.
Аспартилглюкозаминурия обусловлена недостаточностью
аспартил-глюкозаминамидазы - фермента, который расщепля-
ет амидную связь между олигосахаридом и полипептидом в
структуре гликопротеида. При этом заболевании накапливают-
ся аспартилглюкозамин и гликопептиды. Наблюдается отста-
лость психического развития.
127
Диагностическое значение имеет определение активности
фермента, а также повышенных количеств аспартилглюкоза-
мина в моче.
5.6.2. Нарушение метаболизма протеогликанов
Протеогликаны относятся к смешанным макромолекулам и
состоят из глюкозамингликанов, связанных ковалентно с бел-
ками. Содержание белка в протеогликанах варьирует от 2 до
30%. Глюкозамингликаны (или мукополисахариды) - это гете-
рополисахариды, состоящие из повторяющихся дисахаридных
единиц, в которых всегда присутствуют аминосахара (D-глю-
козамин или D-галактозамин) и уроновая кислота (L-глюкуро-
новая кислота или L-идуроновая кислота). Представителями
мукополисахаридов являются: гиалуроновая кислота, хондро-
итин-сульфаты, кератан-сульфаты, гепарин и гепарансульфа-
ты, дерматансульфаты. Все они, кроме гиалуроновой кислоты,
содержат сульфатированные по разным положениям аминоса-
хара, и ковалентно присоединены к белкам. Кератансульфат
вместо уроновых кислот содержит D-галактозу. Редко в соста-
ве протеогликанов встречаются L-фукоза, D-манноза, D-кси-
лулоза.
Присоединение гетерополисахаридных цепей к белку про-
исходит с помощью связей трех типов: I) О-гликозидная связь
между ксилозой и серином, характерная только для протеогли-
канов; 2) О-гликозидная связь между N-ацетилгалактозамином
и серином (треонином); 3) N-гликозидная связь между N-аце-
тилглюкозамином и аспарагином.
Мукополисахариды осуществляют такие важные функции
как опорную, защитно-механическую, связующую или струк-
турную, гидроосмотическую и ионрегулирующую, кофактор-
ную.
В здоровом организме происходит обновление протеогли-
канов за счет их распада и синтеза. Образование протеогли-
канов протекает по одному из двух путей, описанных для гли-
копротеидов, в зависимости от типа связи между углеводным
и белковым компонентами макромолекулы. После образова-
ния гетерополисной цепи происходят различные модифика-
ции: сульфатирование, ацетилирование, деацетилирование,
превращение L-глюкуроновой кислоты в ее 5-эпимер - L-иду-
128
роновую кислоту. Нарушений синтеза протеогликанов не опи-
сано.
Деградация протеогликанов начинается с нередуцирующе-
го конца молекулы под действием специфических ферментов -
экзогликозидаз, эндогликозидаз и сульфатаз, последовательно
расщепляющих одну связь. Поэтому каждый следующий фер-
мент проявляет свою активность только после успешного дей-
ствия другого фермента. Существует целый ряд заболеваний, в
основе развития которых лежат различные нарушения в рас-
щеплении протеогликанов.
Мукополисахаридозы
Мукополисахаридозы - группа наследственных заболеваний,
обусловленных недостаточностью лизосомных ферментов, раз-
рушающих протеогликаны соединительной ткани. В результа-
те в тканях накапливаются дерматансульфат, гепарансульфат
или кератансульфат в зависимости от дефектного фермента. При
этих заболеваниях мукополисахариды выделяются в повышен-
ных количествах с мочой.
Мукополисахаридозы подразделяют на типы в зависимости
от биохимического дефекта: например, I тип - недостаточность
фермента а-идуронидазы, II тип - идуронатсульфатазы и т.д.
Мукополисахаридозы имеют общие признаки, выраженные
в разной степени в зависимости от типа мукополисахаридоза:
грубые черты лица, помутнение роговицы, патология кост-
ной ткани (дисплазия костей, тугоподвижность суставов, дефор-
мация скелета), сердечно-сосудистая патология, гепатосплено-
мегалия. Встречаются мукополисахаридозы как с тяжелым от-
ставанием в психическом развитии, так и с сохраненным ин-
теллектом. Описаны разные по тяжести формы мукополисаха-
ридозов от легкой, при которой больные доживают до зрелого
возраста, до тяжелой - смертельной в детском возрасте.
Диагностика этих заболеваний заключается в определении
специфических ферментов в фибробластах, лейкоцитах и тка-
нях, а также в определении типа мукополисахаридов, экскре-
тируемых с мочой. Лечение мукополисахаридозов симптома-
тическое.
9. Зак. 49
129
Глава 6
НАРУШЕНИЕ ОБМЕНА ВИТАМИНОВ
Витамины - это низкомолекулярные органические соедине-
ния, необходимые для нормальной жизнедеятельности, синтез
которых у организма данного вида отсутствует или ограничен.
Неспособность организма к синтезу витаминов можно рас-
сматривать как летальную мутацию, патологическое следствие
которой реализуется лишь в особых неблагоприятных услови-
ях внешней среды при недостаточном поступлении витаминов
с пищей, или нарушение их синтеза кишечными бактериями.
Так как запасов витаминов в организме, как правило, нет (ис-
ключением является цианокобаламин и ретинол), то, следова-
тельно, необходимое количество витаминов должно поступать
в организм систематически. Если соответствующее количество
витаминов организм не получает в течение длительного време-
ни, то возникает, и в последующем нарастает витаминная недо-
статочность с определенными клиническими проявлениями.
Витаминная недостаточность может остановиться на любом
начальном уровне. Однако, если преобладание расхода вита-
минов над их поступлением будет продолжаться, закономерно
будут прогрессировать и проявления витаминной недостаточ-
ности по направлению к гипо- и даже авитаминозу.
Существуют приобретенные и врожденные нарушения об-
мена витаминов.
6.1. НАРУШЕНИЯ ОБМЕНА ВОДОРАСТВОРИМЫХ
ВИТАМИНОВ
6.1.1. Приобретенные нарушения обмена витаминов
Приобретенные нарушения обмена витаминов могут быть
как экзогенного, так и эндогенного происхождения.
Экзогенная витаминная недостаточность. К экзогенным ги-
повитаминозам относятся нерациональное питание (однообраз-
ная, бедная витаминами пища), изменение состава нормальной
кишечной флоры (дисбактериоз), обычно вызываемое длитель-
ным применением химиотерапевтических средств (антибиоти-
ков, сульфаниламидов и т.д.). Среди причин экзогенной недо-
статочности витаминов следует выделить большую физическую
130
и нервно-психическую нагрузку, а также пребывание человека
в трудных климатических условиях (в условиях Крайнего Севе-
ра, высокогорных местностях, при кислородном голодании).
Обычное содержание витаминов в пище может оказаться
недостаточным и в ряде других случаев (беременные, кормя-
щие матери, нарушение правил кулинарной обработки продук-
тов). Возрастают потери водорастворимых витаминов с потом
во время пребывания в жарком климате, при работе в горячих
цехах. Установлено, что потребность в тиамине, пиридоксине
и пантотеновой кислоте при температуре 32°С в два раза выше,
чем при 18°С. В целях предупреждения витаминной недоста-
точности во всех этих случаях пища должна содержать витами-
нов больше, чем в обычных условиях.
Общая направленность в развитии витаминной недостаточ-
ности представлена в табл. 4.
Отдельные формы витаминной недостаточности изложены
в учебнике Е.А. Строева "Биологическая химия" (1986 г.).
Таблица 4
Общая направленность в развитии витаминной
недостаточности
Содержание витаминов в суточном рационе Характер влияния на организм Клинические проявления
Количество витаминов в суточном рационе доста- точное. количество по- ступающих витаминов соответствует нагрузке организма Положительное влияние на обмен веществ, обес- печивающее полный объем всех физиологи- ческих процессов Здоров
Содержание витаминов в суточном рационе отно- сительно недостаточное, дефицит витаминов в силу превалирования их расхода над поступлени- ем Начальная витаминная недостаточность: нару- шение обменных процес- сов сочетается с функци- ональной недостаточно- стью отдельных органов и систем, снижение реак- тивности организма Гиповитаминозы: 1. доклиническая стадия - снижение выносливос- ти организма к физичес- ким и нервным нагруз- кам, понижение сопро- тивляемости к инфекци- онным заболеваниям, трофические нарушения, ухудшение аппетита, снижение массы тела 2. клиническая стадия - наличие специфических признаков моно- и поли- гиповитаминозов
9*
131
Содержание витаминов в суточном рационе Характер влияния на организм Клинические проявления
Содержание витаминов в суточном рационе абсо- лютно недостаточное или имеется резкое пре- вышение расхода над по- ступлением; нарушение усвоения витаминов Выраженная недостаточ- ность того или иного ви- тамина (или витаминов) развивается как след- ствие дальнейшего уг- лубления начальной ви- таминной недостаточ- ности Авитаминозы (чаще по- лиавитаминозы) с харак- терной клинической кар- тиной
Эндогенная витаминная недостаточность является чаще все-
го следствием хронических заболеваний. Среди первопричин-
ных факторов эндогенной витаминной недостаточности явля-
ется нарушение всасывания витаминов при заболеваниях же-
лудочно-кишечного тракта (гипоацидный гастрит, энтерит и
ДР-).
Возникновение витаминной недостаточности возможно при
заболеваниях печени (хронические гепатиты, цирроз печени).
С одной стороны при этом нарушается превращение провита-
минов, например, каротина в ретинол, образование фосфори-
лированных форм - коферментов, с другой - недостаточное по-
ступление желчных кислот в кишечник нарушает усвоение жи-
рорастворимых витаминов. Увеличивается потребность в ви-
таминах и при заболеваниях, характеризующихся повышением
обмена веществ (первичный диффузно-токсический зоб, забо-
левания с повышенной температурой). Снижение количества
поступающей пищи, ввиду плохого аппетита у больного, нали-
чие рвоты, боязни принимать пищу из-за боли (язвенная бо-
лезнь и др.), также ведет к уменьшению поступления витами-
нов в организм. Снижает витаминную ценность питания боль-
ных более тщательная обработка продуктов во время приго-
товления диетических блюд. Таким образом, дефицит витами-
нов в организме может быть вызван различными факторами
(см. схему).
Нарушение обмена витамина В12
В процессе переваривания в желудке из пищи высвобожда-
ется витамин Вр, образующий стабильный комплекс с R-свя-
132
Схема возникновения и развития витаминной недостаточности у больных хроническими заболеваниями желудочно-
кишечного тракта.
зывающим гликопротеином. Этот белок относится к группе
сходных по структуре гликопротеинов с неустановленной фун-
кцией, обнаруживаемых в слюне, молоке, желудочном соке,
желчи, плазме. В двенадцатиперстной кишке комплекс витамин
B|2-R -связывающий протеин разрушается, а высвобождающий-
ся витамин образует комплекс с внутренним фактором (фактор
Кастла). Этот гликопротеид с молекулярной массой 50000 Да
синтезируется париетальными клетками желудка.
Комплекс витаминов с внутренним фактором устойчив к
воздействию протеолитических ферментов. Он транспортиру-
ется в дистальные отделы подвздошной кишки, в которых спе-
цифические рецепторы слизистой оболочки захватывают его с
последующей резорбцией. Таким образом, внутренний фактор
играет роль протеина-носителя, обеспечивающего направлен-
ный транспорт витамина В|2 к клеткам; далее витамин Вр транс-
портируется через слизистую оболочку в капиллярную сеть, в
которой первоначально связывается с другим транспортным
белком - транскобаламином II (ТК-П), период полувыведения
которого очень короткий (1 час). Комплекс витамина Вр с ТК-П
быстро поглощается печенью, костным мозгом и другими орга-
нами. В норме около 2 мг витамина В скапливается в печени и
столько же в других органах и тканях. С точки зрения мини-
мальной суточной потребности (2,5 мкг) в витамине Вр, для
развития его дефицита у ранее здорового человека необходимо
3-6 лет при условии полного прекращения поступления вита-
мина В|2 с пищей.
В крови витамин В12 циркулирует преимущественно в комп-
лексе с транскобаламином I (ТК-1), период полувыведения ко-
торого составляет несколько недель.
Поступление витамина Вр с пищей более чем достаточно для
удовлетворения потребностей организма, за исключением орга-
низма истинных вегетарианцев. Таким образом, дефицит вита-
мина Вр развивается вследствие нарушения процессов всасы-
вания в кишечнике (при отсутствии фактора Кастла).
В организме человека витамин В|2 находится в двух метаболи-
чески активных формах: метилкобаламин и дезоксиаденозилко-
баламин. Метилкобаламин является коферментом гомоцистеин-
метилтрансферазы, участвующей в переносе метила с №-метил-
ТГФК на гомоцистеин с образованием метионина (в этой реак-
ции метилкобаламин действует как синергист с ТГФК, см. рис. 25).
134
(ТГФК)
М5-МеТГФК
у/ Клеточная
^^^хмембрана
▼ СН3-В12
Ы5-Ме'П ФК-^=----ТГФК----------► Конъюги-
Гомоцистеин
Метионин
рованные
фолаты
Рис. 25. Взаимосвязь внутриклеточного метаболизма витамина Вр
(метилкобаламин) и фолатов.
При дефиците витамина В,, в результате невозможности нор-
мального течения этой реакции расстраивается система мета-
болизма фолатов, а, следовательно, синтеза ДНК и проявля-
ются признаки мегалобластического кроветворения (перници-
озная анемия), которая характеризуется снижением количества
эритроцитов и гемоглобина в крови, появляются крупные клет-
ки мегалобласты в периферической крови и коса ном мозге. От-
мечаются поражения задних и боковых столбов спинного моз-
га (фуникулярный миелоз), повышение выделения с мочой ме-
тилмалоновой кислоты, которая не усваивается. Нарушение
процесса превращения гомоцистеина в метионин может быть
причиной развития неврологических осложнений при дефици-
те витамина Вр. Образующийся в результате этой реакции ме-
тионин необходим для синтеза холина и холинсодержащих фос-
фолипидов, а также для метилирования белка миелина. Невро-
логические нарушения, по-видимому, обусловливаются анома-
лией именно этих процессов вследствие уменьшения продукции
метионина при недостаточности витамина Вр.
Дезоксиаденозилкобаламин является коферментом метилма-
лонил-КоА-мутазы, катализирующей превращение метилмало-
нил-КоА в сукцинил-КоА. Отсутствие кофермента сопровож-
дается накоплением в тканях метилмалонил-КоА и его пред-
шественника пропионил-КоА. В результате синтезируются не-
физиологические жирные кислоты, содержащие добавочное ко-
135
личество атомов углерода, которые включаются в структуру
нейролипидов. Такие биохимические нарушения могут также
способствовать развитию неврологических осложнений.
Нарушение обмена фолиевой кислоты
Биохимические функции фолиевой кислоты определяются ее
коферментами (рис. 26). Все коферменты взаимопревращаются
друг в друга, образуя одноуглеродный донорский пул. Исклю-
чение составляет реакция образования дТМФ из дУМФ, ката-
лизируемая тимидилатсинтетазой, в результате которой выде-
ляется дигидрофолат. Под влиянием дигидрофолатредуктазы
он вновь превращается в тетрагидрофолат, который и перехо-
дит в донорский пул (рис. 26). Коферменты фолиевой кислоты
участвуют также в биосинтезе 2-го и 8-го углеродных атомов
пуринового кольца. Поэтому фолиевая кислота играет исклю-
чительную роль в биосинтезе нуклеиновых кислот и процессах
деления клетки.
Недостаточность фолиевой кислоты приводит к развитию
мегалобластической анемии (нарушение кроветворения выра-
жается теми же признаками, что и при недостатке витамина Вр).
Рис. 26. Схема метаболизма фолатов.
136
Дефицит фолиевой кислоты может быть вызван различны-
ми причинами.
Недостаточное поступление ее в организм, особенно у боль-
ных алкоголизмом, так как основным источником калорий у
них служат алкогольные напитки, в которых содержится очень
малое количество витамина, не позволяющее удовлетворить
ежедневную потребность в нем. Больные наркоманией также
склонны к развитию дефицита фолиевой кислоты из-за недо-
статочности питания. Дефицит может быть вызван повышен-
ной потребностью организма в фолиевой кислоте. У больных
с хроническими гемолитическими анемиями и другими забо-
леваниями, сопровождающимися активацией костно-мозгово-
го эритропоэза, часто развивается недостаточность фолатов,
если не увеличивается их поступление с пищей. Дефицит фо-
лиевой кислоты может возникать у беременных, поскольку раз-
вивающийся плод требует ее дополнительного количества.
Дефицит фолатов может наступить в детском и юношеском
возрасте при быстром росте организма. Нарушается всасыва-
ние фолиевой кислоты при хронических заболеваниях тонко-
го кишечника. Частой причиной развития мегалобластичес-
кой анемии вслед за дефицитом витамина Вр и фолиевой кис-
лоты служит прием лекарственных препаратов, нарушающих
синтез ДНК. Наиболее типичным препаратом этой группы
является метотрексат - ингибитор дигидрофолатредуктазы,
использующийся при некоторых злокачественных опухолях
(рис. 26).
6.1.2. Врожденные нарушения обмена витаминов
Клинические симптомы, сходные с проявлениями тех или
иных авитаминозов и которые развиваются, несмотря на нор-
мальную обеспеченность организма соответствующими вита-
минами, относятся, как правило, к врожденным нарушениям
обмена витаминов. В некоторых случаях болезнь удается пол-
ностью или частично коррегировать путем постоянного введе-
ния соответствующего витамина в дозах, превышающих физи-
ологическую потребность в 100-1000 раз. Подобные случаи по-
лучили название витаминзависимых состояний. В других слу-
чаях, которые получили название витаминрезистентных, вве-
дение даже крайне высоких доз соответствующих витаминов
137
не устраняет явлений их недостаточности и приводит к тяже-
лому нарушению здоровья и даже к летальному исходу.
Врожденные нарушения обмена витаминов обусловлены
не только дефектом структуры апофермента (4, 5, 6), но и дру-
гих белков, участвующих в процессе всасывания (1) и транс-
порта витаминов (2), их превращение в коферментные (3) и ак-
тивные формы (рис. 27).
ам ин оки с.чотн ы й
В in ам и н ы
Функция
Рис. 27. Возможная наследственная патология обмена и функции
витаминов.
В зависимости от глубины нарушения синтеза белка, его
последствия могут быть весьма разнообразными. Если заменя-
емый аминокислотный остаток расположен в участке полипеп-
тидной цепи, являющемся не существенным для поддержания
структуры молекулы, то такая замена может не иметь каких-
138
либо биохимических последствий. Во многих случаях замена
может вести к изменению физико-химических свойств и стабиль-
ности белковой молекулы. В том случае, если замена затраги-
вает участок пептида, входящего в состав активного центра
фермента, то следствием может являться частичная или полная
утрата им каталитической активности. К нарушениям подоб-
ного типа относятся мутации, затрагивающие аминокислотные
остатки, ответственные за связывание молекулы субстрата или
кофермента. Если в результате такого нарушения снижается
сродство апофермента к коферменту, то при обычной концент-
рации последнего в тканях апофермент не будет насыщен, что
может выражаться в отсутствии или снижении каталитической
активности соответствующего фермента.
В ряде случаев это нарушение может быть частично корре-
гировано путем увеличения концентрации кофермента при вве-
дении повышенных доз витамина, являющегося их предшествен-
ником. Подобные состояния лежат в основе витаминзависимых
врожденных нарушений обмена веществ.
Когда в результате генной мутации биосинтез апофермепта
нарушается полностью или образуется белок аномальной струк-
туры, лишенный каталитической активности, то увеличение
концентрации кофермента при введении повышенных доз вп-
тамина-предшественника не может дать какого-либо эффекта.
Такие дефекты являются причиной витаминрезистен гных врож-
денных нарушений.
То же самое относи тся к генетически обусловленным дефек-
там структуры не только апоферментных, но и любых других
белков, необходимых для реализации специфических функций
того или иного витамина в организме.
Врожденные нарушения обмена и функции витамина В,
Участие тиамина в регуляции метаболизма тканей опреде-
ляется тиаминдифосфатом (ТДФ), который входит в состав
пируватдегидрогеназного, 2-оксоглутаратдегидрогеназного
комплексов и транскетолазы. Тиаминдифосфат принимает уча-
стие также в окислительном декарбоксилировании оксокислот
с разветвленным углеродным скелетом (см. нарушение обмена
разветвленных аминокислот).
Помимо ТДФ в тканях синтезируется и тиаминтрифосфат
139
(ТТФ), который выполняет специфическую роль в функциони-
ровании нервной ткани. В частности, ТТФ может принадлежать
важная роль в механизме проведения нервного импульса, свя-
занная с изменением проницаемости мембраны нервного волок-
на и транспорта через нее ионов.
Подострая некротизирующая энцефаломиелопатия
В 1951 году Leigh описал это заболевание и назвал его подо-
строй некротизирующей энцефаломиелопатией (ПНЭ). Болезнь
проявляется в первые два года жизни, прогноз неблагоприятный.
Наиболее ранними симптомами являются: отсутствие аппе-
тита, рвота, затрудненность глотания, задержка или регрессия
психомоторного развития. Неврологические симптомы ПНЭ
наиболее часто указывают на поражение стволовой части го-
ловного мозга.
Биохимические исследования показали, что концентрация
свободного тиамина, ТМФ и ТДФ в ткани мозга при ПНЭ не
отличалась от нормы. В то же время ТТФ отсутствовал, и это
нарушение носило локальный характер, т.к. в печени и почках
ТТФ находился в норме. В дальнейшем был выявлен ингиби-
тор (гликопротеид с молекулярной массой 30000), который по-
давлял активность тимидиндифосфаткиназы. Этот фермент осу-
ществляет синтез ТТФ из АТФ и ТДФ. Ингибитор, обнаружен-
ный в крови, моче и СМЖ больных ПНЭ, подавлял активность
тимидиндифосфаткиназы мозговой ткани и не оказывал влия-
ния на аналогичный фермент в печени и почках.
Литературные данные свидетельствуют, что развитие ПНЭ
обусловлено врожденным нарушением обмена веществ, приво-
дящим к появлению или активации белкового фактора, инги-
бирующего тиаминдифосфаткиназу мозговой ткани, и что это
заболевание наследуется по аутосомно-рецессивному типу.
Врожденный дефект пируватдегидрогеназного комплекса
Болезнь характеризуется увеличением содержания пирувата,
лактата и аланина в крови и повышением выделения их с мочой,
а также снижением окислительного декарбоксилирования пиро-
виноградной кислоты. Торможение окислительного декарбок-
силирования пирувата обусловлено резким снижением активно-
го
сти ТДФ-зависимой пируватдегидрогеназы. Происходит накоп-
ление пирувата в крови и повышенная экскреция его с мочой.
Часть пирувата подвергается восстановлению, что ведет к уве-
личению концентрации лактата. Поскольку пируват является
одним из продуктов обмена аланина, то торможение его окисле-
ния приводит к тому, что аланин накапливается в крови и увели-
чивается выделение его с мочой. Назначение повышенных доз
тиамина нормализует биохимические показатели.
Нарушение окислительного декарбоксилирования пирови-
ноградной кислоты и ее повышенное накопление в мозговой
ткани лежит в основе неврологических нарушений, в частно-
сти, перемежающей атаксии.
Комбинация клинических признаков и точная природа био-
химического дефекта позволяет дифференцировать перемежа-
ющую атаксию, обусловленную дефектом пируватдегидрогеназ-
ного комплекса от ПНЭ. Изучение кинетических характерис-
тик пируватдегидрогсназного комплекса указывает и на суще-
ствование различных изоферментых форм этого комплекса,
одна из которых может отсутствовать при перемежающей атак-
сии.
Болезнь характеризуется резким снижением гемоглобина,
уменьшением гематокрита, лейкоцитов. Выявляется эритроид-
ная гиперплазия с большим числом мегалобластов.
Предполагают, что при тиаминзависимой анемии может
быть нарушена структура тиаминзависимого фермента, участву-
ющего в биосинтезе ДНК и кроветворении таким образом, что
его потребность в тиамине повышается. В этом случае актив-
ность соответствующего фермента оказывается сниженной даже
при нормальной обеспеченности организма тиамином. Забо-
левание поддается лечению высокими дозами тиамина. Тиамин-
зависимая форма болезни “моча с запахом кленового сиропа”
см. раздел “Нарушение аминокислотного обмена”.
Врожденные нарушения обмена и функции витамина В6
Пиридоксинзависимая анемия
Длительный дефицит витамина В6 ведет к развитию гипох-
ромной микроцитарной, сидеробластической анемии, обуслов-
141
ленной нарушением биосинтеза гема из-за снижения активнос-
ти синтетазы 5-аминолевулиновой кислоты, коферментом ко-
торой является пиридоксальфосфат.
Введение повышенных доз пиридоксина обеспечивает гема-
тологическую ремиссию с быстрой нормализацией гематокри-
та и концентрации гемоглобина.
Развитие судорожных состояний при недостатке витамина
В6 обусловлено нарушением синтеза гамма-аминомасляной кис-
лоты (ГАМК) вследствие снижения активности глутаматдекар-
боксилазы - пиридоксальзависимого фермента, катализирую-
щего декарбоксилирование глутаминовой кислоты. Особенно
чувствительна к этому нарушению нервная система детей, у
которых процессы возбуждения превалируют над тормозными
реакциями. Причиной этого заболевания является наследствен-
ный дефект глутаматдекарбоксилазы, затрудняющий ее взаи-
модействие с пиридоксальфосфатом.
Прямое изучение активности глутаматдекарбоксилазы при
ПЗСС долгое время сдерживалось недоступностью мозговой
ткани для исследования. Положение изменилось после того, как
было установлено, что глутаматдекарбоксилаза, кроме голов-
ного мозга, присутствует также и в почечной ткани, более дос-
тупной для изучения с помощью биопсии.
Проведенные исследования выявили снижение активности
глутаматдекарбоксилазы в почках больного ПЗСС и ее норма-
лизацию при приеме повышенных доз витамина В6.
(синдромКногша-Комровсра)
Это заболевание встречается значительно чаще, чем фенил-
кетонурия и обнаруживается у 0,5-1% населения. Болезнь ха-
рактеризуется психоневрологическими расстройствами с про-
явлениями аллергии. Причиной заболевания является сниже-
ние активности кинурениназы - пиридоксальзависимого фер-
мента, вследствие этого нарушается превращение триптофана
в никотинамид и накапливаются в тканях его метаболиты (ксан-
туреновая кислота, кинуренин, кинуреновая кислота и оксике-
нуренин) (рис. 28).
142
Триптофан
Форм ил кинуренин
Антраниловая
кислота
З-Оксиаи 1 раниловая
кислота
Никотинамид 1 6 Хинолиновая кислота
N-метил никотинамид
I
N-мстилпирпдон
Рис. 28. Обмен триптофана и участие в нем пиридоксалевых ферментов
1 - трансаминаза кинуренина; 2 - кинурениназа.
Проявления заболевания обусловлены избыточным обра-
зованием метаболитов триптофана и недостаточным синте-
зом никотиновой кислоты. Нарушение интеллекта у больных
с наследственной ксантуренурией может быть результатом
внутриутробно развившейся эндогенной недостаточности ни-
котиновой кислоты, являющейся источником коферментов
(НАД, НАДФ), обеспечивающих процессы энергетического
обмена и крайне необходимых для развивающейся нервной
ткани.
Снижение активности кинурениназы при пиридоксинзави-
симой ксантуренурии, по-видимому, обусловлено изменением
структуры апофермента, затрудняющим взаимодействие с ко-
ферментом. Биохимические показатели нормализуются при
приеме повышенных доз витамина Вб.
143
Г омоццспшурия
В настоящее время известно несколько форм гомоцистину-
рии, различающихся природой первичного ферментного дефи-
цита (рис. 29):
1. Недостаточность М5-метил-тетрагидрофолат-гомоцисте-
ин-метилтрансферазы (см. врожденные нарушения обмена ви-
тамина В]2).
2. Недостаточность метилен-тетрагидрофолат-редуктазы
(см. врожденные нарушения обмена фолиевой кислоты).
Гомоцистинурия, обусловленная дефектом пиридоксальза-
висимого фермента цистатионинсинтазы, катализирующей ре-
акцию конденсации гомоцистеина и серина с образованием
цистатионина.
Рис. 29. Обмен серосодержащих аминокислот.
1 - реакции трансметилирования с участием S-аденозилметионина; 2 - гид-
ролиз S-аденозилгомоцистеина; 3 - цистатионинсинтаза; 4 - цистатионаза;
5 - бетаин-гомоцистеин-метилтрансфераза; 6 - N-метил-ТГФК-гомоцисте-
ин-метилтрансфераза; 7 - серин-ТГФК-оксиметил-трансфераза. Заштрихо-
ванным прямогольником показано место ферментативного блока при гомо-
цистинурии. В сплошные рамки заключены накапливающиеся из-за блока
вещества, в пунктирные - метаболиты, образование которых нарушено.
144
Заболевание характеризуется довольно полиморфными кли-
ническими проявлениями: нарушениями строения скелета, из-
менениями нервной системы, расстройствами зрения, измене-
ниями сердечно-сосудистой системы, обычными причинами,
приводящими к гибели больного, являются тромбоэмболии.
Механизм повышенной склонности к тромбообразованию
при гомоцистинурии не ясен. Имеются данные об усилении скле-
ивания тромбоцитов при этом заболевании, а также о способ-
ности гомоцистина ускорять их агрегацию in vitro. Показано,
что гомоцистин активирует фактор Хагемана, ускоряя сверты-
вание крови и образование плазмокининов. Назначение повы-
шенных доз витамина В6 (250 мг в день) нормализует биохими-
ческие показатели крови и мочи.
Причина развития гомоцистинурии - недостаточность или
отсутствие цистатионинсинтазы, при этом нарушается превра-
щение гомоцистеина в цистатионин, что ведет к накоплению
гомоцистеина в тканях, в крови и повышенной экскреции с мо-
чой, главным образом в виде его окисленной дисульфидной
формы - гомоцистина.
Поскольку одним из следствий дефекта циста гиопинсинта-
зы является нарушение синтеза цистатионина и цистеина, то
существенную роль в патогенезе гомоцистинурии и ее клини-
ческих проявлений может сыграть недостаток этих серосодер-
жащих аминокислот. Высокая концентрация цистатионина в
мозговой ткани здоровых людей (0,225-0,565 мг/г) и его отсут-
ствие у больных гомоцистинурией могут служить основанием
для предположения о патогенетической роли недостатка этой
аминокислоты в развитии умственных и психических наруше-
ний при этом заболевании. Недостаток цистеина, входящего в
состав коллагена и ряда других белков соединительной ткани,
может служить одной из причин скелетных нарушений, изме-
нения структуры сосудистой стенки и нарушений фибрилл о ге-
неза, лежащих в основе таких дефектов как подвывих хруста-
лика. В то же время в патогенезе гомоцистинурии существен-
ная роль может принадлежать избыточному накоплению в орга-
низме гомоцистеина и аномальных продуктов обмена (избы-
точно сульфатированных протеогликанов), что может лежать
в основе нарушений фибриллогенеза и других изменений со-
единительной ткани при гомоцистинурии.
Однако, как показывает практика, чувствительными к лече-
10. Зак. 49
145
нию пиридоксином являются не все больные гомоцистинури-
ей. Примерно у половины больных введение витамина В6 не дает
эффекта, несмотря на увеличение его дозы до 1-1,2 г в сутки.
Эти данные дают основание считать, что при гомоцистинурии,
резистентной к пиридоксину отсутствует апофермент цистати-
онинсинтазы или структура его изменена таким образом, что
полностью утрачивает свою активность.
Цистатионинурия - дефект пиридоксальзависимого фермен-
та цистатионазы, катализирующей расщепление цистатионина
на цистеин и оксобутират. Заболевание обусловлено наслед-
ственным повреждением структуры апофермента, снижающим
сродство к ПАЛФ.
Цистатионинурия часто сочетается с другими врожденны-
ми нарушениями обмена веществ (фенилкетонурией, подострой
некротизирующей энцефаломиелопатией и др.). У большинства
больных цистатионинурией отмечалось отставание умственно-
го развития. Основными биохимическими проявлениями забо-
левания является увеличение содержания цистатионина в кро-
ви и повышенная экскреция с мочой. Назначение повышенных
доз пиридоксина (от 25-400 мг в сутки) приводит к полной нор-
мализации уровня цистатионина в крови и экскреции с мочой.
Наследственные нарушения обмена витамина В12
А немия, обусловленная дефекпюдсобразованияля1утреш1его
При этом заболевании нарушение образования ВФ носит
избирательный характер, все остальные функции эпителиаль-
ных клеток желудка остаются нормальными. Генетический де-
фект при этом может выражаться в полном отсутствии ВФ или
в образовании белка с аномальной структурой, лишенного био-
логической активности. Концентрация витамина В)3 в плазме
крови резко снижена. Внутримышечное введение физиологи-
ческих доз витамина или его прием per os вместе с препаратами
ВФ, обеспечивают полную гематологическую и клиническую
ремиссию.
Брлезнь Имерслунда-Гресбека
Характеризуется избирательным нарушением всасывания
витамина В|2 в кишечнике, несмотря на нормальное образова-
146
ние ВФ в желудке. Врожденный дефект при этом заболевании
локализован на этапе между присоединением комплекса ВФ-Вр
к мембране энтероцита и освобождением в кровоток витамина
В|2, связанного с транскобаламином II.
( Для нормализации кроветворения необходимо регулярное
* внутримышечное введение витамина В|2 (50 мкг 1 раз в 10 дней).
Врожденный дефект транскобаламина II
Отсутствие специфического транспортного белка. При этой
патологии концентрация витамина Вр и фолиевой кислоты в
сыворотке крови больных, в отличие от классических форм
мегалобластических анемий, находится в пределах нормы, что
{ указывает на нормальные процессы всасывания этих витами-
НОВ.
Эффективность высоких доз витамина Вр (превышающих
физиологическую в 1 000 раз) при этом заболевании может бы ть
| обусловлена тем, что при значительном повышении его кон-
| центрации в крови в транспорте витамина Вр могут принимать
) участие другие белки, обладающие более низким сродством к
нему, чем транскобаламин II.
| Если врожденное отсутствие транскобаламина 11 является
| серьезным дефектом, требующим обязательной коррекции, то
' врожденное отсутствие транскобаламина I протекает практи-
' чески бессимптомно, без каких-либо гематологических и невро-
1 логических нарушений.
-I Врожденные метшъмсъчонитицидеАищ
Известно несколько форм метилмалонатацидемий, различа-
ющихся молекулярным механизмом дефекта. Клинические сим-
птомы при всех формах во многом одинаковые и проявляются
в первые недели или месяцы жизни ребенка (рвота, вялость, за-
держка роста и психомоторного развития, кетоацидоз).
Метилмалонатацидемия, обусловленная нарушением образо-
вания дезоксиаденозилкобаламина (ДАВр). ДАВ|2 является ко-
ч ферментом метилмалонил-КоА-мутазы, катализирующей пре-
вращение a-метилмалонил-КоА в сукцинил-КоА. При дефекте
кофермента блокируется данная реакция, в результате чего на-
капливается ММК в тканях и крови и повышается ее выделе-
ние с мочой. При введении высоких доз витамина В|2 (в 1000
раз превышающих физиологическую потребность) удается ча-
10*
147
стично нормализовать биохимические показатели, что говорит
о витаминзависимой форме метилмалонатацидемии.
М етилмалонатацидемия, обусловленная дефектом апофер-
мента метилмалонил-КоА-мутазы. При этой форме метилма-
лонатацидемии биохимический дефект также состоит в нару-
шении изомеризации метилмалонил-КоА в сукцинил-КоА, од-
нако, причиной этого дефекта является нарушение структуры
апофермента метилмалонил-КоА-мутазы, и он не устраняется
введением высоких доз витамина Вр (витаминрезистентная ме-
тилмалонатацидемия).
Метилмалонатацидемия, сочетающаяся с гомоцистинурией.
При этой форме патологии нарушено образование как ДАВ|2,
так и метилкобаламина, необходимого для нормального функ-
ционирования М5-метилтетрагидрофолат-гомоцистеин-метил-
грансферазы. Вследствие этого оказывается заблокированной
нс только изомеризация метилмалонил-КоА, но и биосинтез
метионина из гомоцистеина, что ведет наряду с повышенным
выделением ММКк развитию гомоцистинурии.
Основным биохимическим показателем является появление
метилмалоновой кислоты (ММК) в крови (в норме отсутству-
ет) и резкое увеличение выделения ее с мочой (0,2-5,0 г в сутки,
тогда как в норме не превышает 5,0 мг в сутки). Хотя ММК
является малотоксичной, однако, накапливающийся в тканях
метилмалонил-КоА тормозит синтез жирных кислот и включа-
ется в них вместо малонил-КоА, что приводит к появлению
разветвленных жирных кислот необычной структуры. Накап-
ливающийся в тканях проиионил-КоА, являющийся предше-
ственником метилмалонил-КоА, также может включаться в
биосинтез жирных кислот, что приводит к образованию кис-
лот с нечетным числом атомов углерода.
Вролсденные нарушения обмена фолиевой кислоты
Фолатзависимая мегалобластическая анемия обусловлена
врожденным нарушением всасывания фолатов в тонком кишеч-
нике. Дефект всасывания фолиевой кислоты носит избиратель-
ный характер и не связан с какими-либо общими нарушениями
структуры и функции тонкого кишечника. Предполагается, что
причиной патологического процесса является утрата специфи-
ческого механизма активного транспорта фолиевой кислоты в
тонком кишечнике. Мегалобластическая анемия, возникающая
148
при врожденном нарушении всасывания фолатов в тонком ки-
шечнике, как правило, сочетается с задержкой психического
развития и рядом других нарушений со стороны центральной
нервной системы (атаксия, судороги).
Пероральное введение фолиевой кислоты в дозе 10-40 мг в
день, превышающей физиологическую потребность в 200-1000
раз, обеспечивает быструю и полную гематологическую ремис-
сию. Коррекция дефекта при приеме повышенных доз фолие-
вой кислоты может быть обусловлена ее всасыванием за счет
пассивной диффузии.
Однако те изменения ЦНС, которые возникают в первые
месяцы жизни, при несвоевременном начале лечения носят, как
правило, необратимый характер.
Генетические нарушения коферментного состава фолиевой
кислоты
КР-формилТГФК-зависимая мегалобластическая анемия -
дефект дигидрофолатредуктазы, нарушающий превращение
фолиевой кислоты в коферментные формы. Мегалобластичес-
кая анемия развивается, несмотря на нормальную концентра-
цию общих фолатов в сыворотке крови. В го же время нагрузка
гистидином выявляла аномальное увеличение экскреции фор-
миминоглутаминовой кислоты (ФИГК). Введение N'-фор-
милТГФК вдозеОД мгприводило к полной нормализации кро-
ви, костного мозга и экскреции ФИГК.
Недостаточность формиминотрансферавы
Причиной заболевания является снижение активности
формиминотрансферазы, участвующей в образовании форми-
мино-ТГФК из ТГФК и формилглутаминовой кислоты. Нали-
чие этого дефекта хорошо объясняет повышенную экскрецию
ФИГК при нагрузке гистидином у таких больных, несмотря на
высокую концентрацию фолатов в сыворотке крови.
6.2. ВРОЖДЕННЫЕ НАРУШЕНИЯ ОБМЕНА И
ФУНКЦИИ ЖИРОРАСТВОРИМЫХ ВИТАМИНОВ
Витамин D. Помимо обычного витамин D-дефицитного ра-
хита, обусловленного недостаточным поступлением этого ви-
тамина с пищей, сходные патологические изменения могут раз-
149
виваться и при нормальной обеспеченности организма витами-
ном D вследствие тех или иных нарушений его обмена. К по-
добным заболеваниям относится гипофосфатемический вита-
мин D-резистентный рахит (ВДРР), псевдодефицитный вита-
мин D-зависимый рахит (ВДЗР) и синдром Де Тони-Дебре-Фан-
кони.
Гипофосфатемический витамин D-резистентный рахит. У
детей рахитические изменения скелета при ВДРР обнаружива-
ются на 1-2 году жизни в виде деформации черепа, искривле-
ния и укорочения конечностей, задержки роста. В крови резкое
снижение фосфора и незначительное снижение кальция. Эти
изменения возникают, несмотря на прием профилактических доз
витамина D или его активных метаболитов, и нс поддаются
воздействию этого витамина в дозах, используемых для лече-
ния классического рахита. Причиной этого заболевания явля-
ется снижение реабсорбции неорганического фосфора в почеч-
ных канальцах, вследствие чего увеличивается выведение его с
мочой. Дефект почечных канальцев при ВДРР носит ограни-
ченный характер и затрагивает только реабсорбцию неоргани-
ческого фосфора. Все остальные функции почек, в частности,
их способность реабсорбировать глюкозу, аминокислоты, би-
карбонат и др. остаются нормальными.
Нарушение активного транспорта неорганического фосфо-
ра при ВДРР не ограничивается его реабсорбцией в почечных
канальцах, но затрагивает также процессы всасывания фосфо-
ра в тонком кишечнике. По мнению Condon и других авторов,
первичное нарушение всасывания фосфора в тонком кишечни-
ке при ВДРР может играть ведущую роль в патогенезе рахити-
ческих изменений у большей части таких больных.
Врожденньшjamaмин.Р-зднисимыйрахит (ВДЗР) характе-
ризуется теми же клиническими признаками, что и при класси-
ческом алиментарном рахите. Из основных биохимических про-
явлений наблюдается выраженная гипокальциемия, что может
являться причиной судорог и эпилептических припадков.
Первичным метаболическим дефектом при ВДЗР является
нарушение синтеза 1-гидроксилазы в почках (рис. 30), что в свою
очередь приводит к нарушению образования активного мета-
болита витамина D (1,25(OH)2D). Вследствие этого нарушает-
ся всасывание кальция в тонком кишечнике, возникает гипо-
кальциемия, ведущая к клиническим проявлениям.
150
Назначение высоких доз витамина D приводит к полному
выздоровлению с исчезновением всех биохимических и клини-
ческих симптомов заболевания, нормализации роста и развития.
Рис. 30. Обмен и механизм действия витамина D.
ПЩЖ - паращитовидная железа; ПТГ - паратиреоидный гормон;
1 - блок 1-гидроксилаза.
151
Эффективность высоких доз витамина D при витамин D-за-
висимом рахите может быть обусловлена тем, что генетичес-
кий блок образования 1,25 дигидроксихолекальциферола но-
сит не абсолютный характер и при введении повышенных доз
витамина D крайне низкая активность 1-гидроксилазы оказы-
вается достаточной для образования минимально необходимых
количеств 1,25(OH),D.
Другое объяснение может состоять в том, что образующий-
ся в повышенных количествах 25-гидроксихолекальциферол (в
100 раз превышающий необходимую концентрацию 1,25 дигид-
роксихолекальциферола) может заменять последний в системе
транспорта кальция в кишечнике и его мобилизации из кост-
ной ткани.
Синдром Де Тони-Дебре-Фанкони (глюкозоаминфосфатди-
абет). В основе этого синдрома лежат нарушения функций про-
ксимальных почечных канальцев, выражающихся в дефекте
реабсорбции фосфора, глюкозы, аминокислот, бикарбонатов
и калия. Причиной таких дефектов может являться генетичес-
ки обусловленное нарушение систем транспорта соответству-
ющих веществ в почках или повреждение этих систем при та-
ких врожденных заболеваниях, как цистиноз, тирозиноз, псев-
досклероз Вильсона и др. Классической триадой при этом за-
болевании является: гиперфосфатурия, гиперглюкозурия и ги-
пераминоацидурия. При этой форме болезни развивается вто-
ричный рахит, обусловленный, по-видимому, чрезмерными
потерями фосфора с мочой и ацидозом.
152
Литература
1. Балаболкин М.И. Эндокринология. - М.: Медицина, 1989.-416с.
2. Березов Т.Т., Коровкин Б.Ф. Биологическая химия. - М.: Меди-
цина, 1982. - 528 с.
3. Внутренние болезни: В 10 книгах / Под ред. Браунвальда и др. -
М.: Медицина, 1996.
4. Гальперин Ю.М., Лазарев П.И. Пищеварение и гомеостаз. - М.:
Наука, 1986. - 232 с.
5. Геллер Л.И. Основы клинической эндокринологии системы
пищеварения. - Владивосток, 1988. - 128 с.
6. Гребнев А.Л., Мягкова Л.П. Болезни кишечника. - М.: Меди-
цина, 1994. - 170 с.
7. Иванов Е.П. Диагностика нарушений гемостаза. - Минск: Бе-
ларусь, 1983. - 223 с.
8. Климов А.Н., Никульчева Н.Г. Обмен липидов и липопротеи-
дов и его нарушения. - СПб., 1999. 512 с.
9. Клиническая эндокринология / Под ред. Н.Т.Старковой. - М.:
Медицина, 1991.
10. Лебедев Н.Н. Биоритмы пищеварительной системы. - М.: Ме-
дицина, 1987. - 217 с.
11. Левина Л.И. Сердце при эндокринных заболеваниях. - Л.: Ме-
дицина, 1989. - 264 с.
12. Марри Р., Греннер Д., Мейес П., Родуэлл В. Биохимия человека:
В 2-х томах: Пер. с англ. - М.: Мир, 1993. Т.2. 415 с.
13. Мусил Я. Основы биохимии патологических процессов. - М.:
Медицина, 1985.-432 с.
14. Николаев А.Я. Биологическая химия: Учеб, для мед. спец, ву-
зов. - М.: Высш, шк., 1989. - 495 с.
15. Патология пищеварения и проблемы питания: Межвуз. тем. сб./
Отв. ред. А. С. Смагилов. - Актюбинск, 1977. 158 с.
16. Патологическая физиология / Под ред. Н.Н. Зайко. - М.: Элис-
та, 1994.-575 с.
17. Сергеев П.В., Галенко-Ярошевский П.А., Шимановский Н.Л.
Очерки биохимической фармакологии. - М.: РЦ “Фармединфо”, 1996.
18. Спиричев Б.В., Барашнсв Ю.И. Врожденные нарушения обме-
на витаминов. - М., 1977.
19. Старкова Н.Т. Фармакотерапия в эндокринологии. - М.: Ме-
дицина, 1989. - 288 с.
20. Строев Е.А. Биологическая химия. - М., 1986. - 479 с.
21. Тапперман Дж, Тапперман X. Физиология обмена веществ и
эндокринной системы. - М.: Мир, 1989. - 656 с.
22. Томпсон Г.Р. Руководство по гиперлипидемии. - Югославия,
1991.-255 с.
153
23. Файтельберг P.O. Всасывание в желудочно-кишечном тракте. -
М.: Медицина, 1976. - 237 с.
24. Фролькис А.В., Горанская С.В. Интестинальные энзимопатии.
- Кишинев, 1982. - 182 с.
25. Фролькис А.В. Заболевания желудочно-кишечного тракта и
наследственность. - М.: Медицина, 1995. - 242 с.
26. Хорст А. Молекулярные основы патогенеза болезней. - М.:
Медицина, 1982. - 285 с.
27. Яковлев Т.Н. Лечебно-профилактическая витаминология. - М.:
Медицина, 1981. - 186 с.
154
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Агликогенозы - 115
Адреналин - 36
Азотистый баланс - 19
Акантоцитоз - 83
Акромегалия - 88
Аланин - 48,142
Алкаптон - 38
Алкаптонурия - 38
Альбинизм - 39
Алиментарная ферментопатия - 7
Альдолаза В - 113
Амилопектиноз - 123 J
Аминокислотный дисбаланс - 7
Анорексия нервная - 88
Ан-а-липопротеинемия - 83 ' ’
Апопротеины (апобелки) - 70,71 j
АпоА-1 -72,73 -
Апо С-П - 70, 72
Апо В-72, 83
Апо Е - 72, 76
Апо В,Е-рецептор - 72, 80
Апо Е-рецептор - 72
Арилсульфатазы - 101
Арилсульфатазы А и В - 101
Аргиназа - 48
Аргинин - 28
Аргининосукцинат-лиаза - 48
Аргининосукцинат-синтетаза - 48
Аргининосукцинатная ацидурия - 48
Аспартилглюкозаминурия - 127
Афлотоксин - 8
N-ацетилглюкозаминфосфотрансфераза - 126
Ацетил-ЛПНП-рецептор - 85 ‘ ?
N-ацетилнейраминовая кислота - 95 п
N-ацетилтирозин - 42
Ацил-КоА-дегидрогеназа - 90
Ацил-КоА-холестерол-ацилтрансфераза - 71, 75
155
Бензохиноацетат - 39, 40 ?: i
Биотин - 91
Болезнь Андерсена - 123 •. ,
Болезнь Гордона-14
Болезнь Волмана - 101
Болезнь Гоше - 99
Болезнь Гюнтера - 52
Болезнь Имерслунда-Гресбека - 146
Болезнь “кленового сиропа” - 45
Болезнь Кори-Форбса - 118
Болезнь Краббе - 97
Болезнь Крона - 13
Болезнь Кули - 61
Болезнь Лея - 140
Болезнь Мак-Ардля - 120
Болезнь накопления эфиров ХС - 82
Болезнь Нимана-Пика - 100
Болезнь Помпе - 122
Болезнь Рефсума - 91
Болезнь Сандхоффа - 97
Болезнь Таруи - 121
Болезнь Тея-Сакса 96
Болезнь Уиппла - 14
Болезнь Фабри - 100
Болезнь Фарбера - 101
Болезнь фон Гирке - 88, 116
Болезнь Феллинга - 35
Болезнь Хартнуп (“Hartnup”) - 27, 29
Болезнь Эра - 119
Валин-26 ...
-
Вариегатная острая порфирия - 54
Витамин Вр - 132
Врожденная гемолитическая анемия - 104
Врожденный дефект транскобаламина II - 147
Врожденные метилмалонатацидемии - 147
Врожденная непереносимость фруктозы - 114
Вторичная оротовая ацидурия - 68
156
Галактоза - 110
Галактоземия - НО
а-галактозидаза - 100
Р-галактозидаза - 96
; Г алактозо-1 -фосфат-уридилтрансфераза - 110
Галактозилсфингозин - 99
• Галактозилцерамидный липидоз - 97
Галактозилцерамид - 93, 99
Галактозилцерамидаза - 99
Галактозилцерамид-Р-галактозидаза - 97
Галактокиназа - 110
Ганглиозиды - 94
ОМ|-ганглиозид - 94, 96
ОМ2-ганглиозид - 97
Омз-ганглиозид - 94 ?
ОМ|-ганглиозидоз - 96
ОМ2-ганглиозидоз - 96 £ г
Гексозаминидазы А и В - 97 П.
Гемолитическая анемия - 56
Генерализованный ганглиозидоз - 96
Гепатит острый - 89
| Гепатома - 89
| 7-а-гидроксилаза - 71
5 .; Р-гидрокси-Р-метилглутарил-КоА-редуктаза - 71
ч п-гидроксифенилацетат - 42
п-гидроксифениллактат - 42 .л i
Д п-гидроксифенилпируват - 40,42 ' N
п 5-гидрокситриптофан - 36
pi Гипераланинемия - 47
Гипераминоацидемия - 33
•> Гипераминоацидурия - 29, 30, 33, 45
Гипераммониемия - 31,47
Гиперапо-В-липопротеинемия - 82
Гипераргининемия - 48
) Гиперглутаминемия - 47
Гиперлипопротеинемии - 77
I тип - 77 А
II тип - 78 ь
: III тип - 80 <<>,.
• IV тип - 78
157
V тип - 78
Гипер-а-триглицеридемия - 82
Гиперурикемия - 63
Гиперфенилаланинемия - 35
Гипоглицин 90
Г иполипопротеинемии:
- первичные - 77
- вторичные - 85
Гипотиреоз - 89
Гипохромная анемия - 56 = .
Гистидаза - 43
Гистидин - 26, 32, 43
Г истидиндекарбоксилаза - 44
Гистидинемия - 43
Гистидинурия - 32
Глиадин - 11
Глиадинамидаза - 11
Гликопротеиды 125
Гликоген - 115
Гликогенозы 115
- тип la - 116
- тип lb - 118
- тип 11-122
- тип 111 - 118
- тип IV - 123
- тип V - 120
- тип VI 119
- тип VII - 121
Гликогенсинтаза - 115
Гликогеноз I типа - 88
Гликосфинголипиды - 93
Глутаматдегидрогеназа - 26, 46
Глутаматдекарбоксилаза - 142
Глутаминсинтетаза - 26, 47
Глюкоза - 103
Р-глюкозидаза - 99
Глюкозилцерамид - 93, 99
Глюкозил церамидаза - 99
Глюкозилцерамидный липидоз - 99
Глюкозо-6-фосфат-дегидрогеназа - 105
158
Глюкозофосфатизомераза - 105
N-глютаминил-дипептидаза - 11
Глютен - 11
Глютеновая болезнь - 11
Гомогентизинатоксидаза - 38, 40
Гомоцистеинметилтрансфераза - 134
Гомоцистин - 145
Гомоцистинурия - 144
Дезоксиаденозилкобаламин - 135, 147
Дигидробиоптерин - 34
Дигидробиоптеринредуктаза - 34
Дигидроксифенилаланин - 41
Дигидрофолатредуктаза - 136
Дикарбоновая ацидурия - 90
P-0-диметилцистеин - 29
Дипептидилдипептидаза - 11
Дисбактериоз - 16
Дислипопротеинемии - 77
Диспепсия - 8
ДОФА-хинон - 41
ДОФА-хром - 41
Д-пеницилламин - 29
Желтуха - 57
Желчные кислоты - 71
Злоупотребление алкоголем (или алкоголь) - 88
Идиопатическая пентозурия - 103, 107
Идиопатическая фруктозурия - 113
Идуронатсульфатаза - 129
а-идуронидаза - 129
Изолейцин - 45
Иминоглицинурия - 27, 31
Индиготин - 30
159
Индикан -30 /
Индиканурия - 31 t
Индолилацетат - 30
Индолиллактат - 30
Индолилпируват - 30
Карнитин - 89 -И
Карнитинпальмитоилтрансфераза - 89 нс
Катехоламины - 36
Кахексия - 26
Квашиоркор - 26 ш
а-кетоглутарат - 37 -у
Кинуренин - 142
Кинурениназа - 142
Кислая липаза - 102
Кислота
- аспарагиновая - 28
- ацетоуксусная (ацетоацетат) - 38, 40
- 3-аминолевулиновая - 41,49
- бензойная - 47
- а-гидроксимасляная - 32
- гомогентизиновая - 38, 40
- глутаминовая - 28
- имидазолпропионовая - 43
- малеилацетоуксусная - 38, 40
- никотиновая - 30
- уроканиновая- 43
- фенилуксусная - 47
- N-формилглутаминовая - 43
- фумарилацетоуксусная - 38, 40
- фумаровая (фумарат) - 38, 40
Кишечная липодистрофия - 14
Клетки Гоше - 99
I-клеточная болезнь - 126
Копропротопорфирия - 54
Ксантуренурия - 142
Лейцин - 45
Лейциноз- 45
Лецитинхолестеролацилтрансфераза - 71, 76, 82
160
Лизин - 28, 31
Лизинурия - 30
Лизосомные болезни - 95
Лимитдекстриноз - 119
Линатин - 8
Липогрануломатоз Фарбера 101
Липодистрофия - 88
А-Р-липопротеинемия - 83
Липопротеинлипаза - 70, 74
Липопротеины - 69, 70, 77
Липопротеины а - 74
Липопротеины высокой плотности - 72, 73, 76, 82
Липопротеины модифицированные - 84
Липопротеины низкой плотности - 72, 73, 75, 82
Липопротеины очень низкой плотности - 72, 73, 74, 77, 88
Липопротеины промежуточной плотности - 72, 80
Липопротеины X - 88
ЛПНП-рецептор - 73, 75, 78, 80
р-ЛП-70, 73, 83
пре-р-ЛП - 70, 73, 86
L-ксилулозоредуктаза - 107
Малабсорбция - 12, 17
а-маннозидаза - 127
Маннозидоз - 127
Мастоцитоз - 44
Мегалобластическая анемия - 141
Метахромная лейкодистрофия - 101
Метилмалонил-КоА-мутаза - 91, 148
Меланин - 41
Метилкобаламин - 134, 148
Метилмалоновая кислота - 147
Метилмалонатацидемия - 147
Метилен-тетрагидрофолат-редуктаза - 144
№-метил-тетрагидрофолат-гомоцистеин-
метилтрансфераза - 144
Метионин - 32, 42, 134
Метотрексат - 137
Множественная сульфатазная недостаточность - 101
Моноклониальные гаммапатии - 89
II. Зак. 49
161
Мочевина -46
Мукополисахаридозы - 129 ».
Мукополисахариды - 128
Наследственный дефицит ЛХАТ - 82
Наследственная печеночная копропорфирия - 54
Наследственная оротовая ацидурия - 68
Наследственная нетолерантность к фруктозе - 114
Нарушение всасывания - 15
Нарушение всасывания моносахаридов - 17
Нарушение обмена витаминов - 130
Нарушение обмена витамина Bj - 139
Нарушение обмена витамина В6 - 141
Нарушение обмена витамина В|2 - 132, 146
Нарушение обмена витамина D - - 149
Нарушение обмена фолиевой кислоты - 136, 148
Нарушения:
- всасывания глюкозы и галактозы - 124
- гликолиза - 103
- глюконеогенеза - 107
- пентозофосфатного цикла - 105
- “пути уроновых кислот” - 106
Недостаточность:
- амило-1,6-глюкозидазы - 118
- ветвящего фермента - 123
- а-глюкозидазы - 122
- глюкозо-6-фосфатазы - 109
- глюкозо-6-фосфат-транслоказы - 118
- деветвящего фермента - 118
- киназы фосфорилазы В - 120
- М-субъединицы лактатдегидрогеназы - 121
- пируваткарбоксилазы - 107
- синтетазы холокарбоксилазы - 108
- фосфоглицератфосфомутазы - 121
- фосфоенолпируваткарбоксикиназы - 108
- фосфорилазы - 119, 120
- фосфофруктокиназы - 121
- фруктозо-1,6-бисфосфатазы - 109
Недостаточность гормона роста - 88
Недостаточность глюкозо-6-фосфатазы - 65, 116
162
Недостаточность трегалазы - 11
Недостаточность энтерокиназы - 12
Недостаточность [3-галактозидазы - 9
Недостаточность [3-фру ктофуранозидазы - 10
Неклассифицированные дислипопротеинемии - 82
Нелизосомная сульфатаза стероидов - 101
Ниацитин - 8
Норадреналин - 24, 36
а-Окисление жирных кислот - 91
[3-окисление жирных кислот - 90, 92
to-окисление жирных кислот 90
Орнитин - 28
Орнитинкарбамоилтрансфераза - 47
Острая интермиттирующая порфирия - 88
Острая перемежающая порфирия - 53
Охроноз 39
Первичная малабсорбция фруктозы - 18
Пернициозная анемия - 135
Печеночная липаза - 70, 82
Пируватдегидрогеназный комплекс - 140
Пиридоксальфосфат - 49
Пиридоксинзависимая анемия - 141
Пиридоксинзависимый судорожный синдром - 142 л
Пируваткиназа - 105 :
Плазмокинины - 145
Подагра - 65
Подострая некротизирующая энцефаломиелопатия - 140
Поздняя кожная порфирия - 55
Полифенолоксидаза - 39, 40
Почечная гликозурия - 123
Почечная глюкозурия - 123
Порфобилиногенсинтаза - 50
Порфобилиногенсинтетаза - 41
Пристеночное пищеварение - 9
Пролин - 28, 31
Пропионил-КоА - 90, 91
Пропионил-КоА-карбоксилаза - 75
Протеинопатии - 26
ч* 163
Протеогликаны - 128
Протопорфирия - 53
Ремнанты ХМ - 74, 81
Сахарный диабет - 85
Свободные жирные кислоты - 69, 86
Семейная гиперхолестеринемия - 78
Семейная гипо-Р-липопротеинемия - 83
Семейная гипер-а-липопротеинемия - 82
Серповидно-клеточная анемия - 62
Сидеробластическая анемия - 141
Сиаловые кислоты - 94, 125
Синдром “голубых пеленок” - 31
Синдром Дабина-Джонсона - 60
Синдром Де Тони-Дебре-Фанкони - 152
Синдром Жильбера 60
Синдром Криглера-Найяра 60
Синдром Кноппа-Комровера - 142
Синдром Кушинга - 88
Синдром Леша-Нихена - 63
Синдром нефротический - 88
Синдром Рейс 68
Синдром Ротора - 61
Синдром Рихнера-Ханхарта - 41
Синдром X - 88
Синдром Цельвегера - 92
Синтетаза 8-аминолевулиновой кислоты - 142
Системная красная волчанка - 24, 89
Скачкообразная кетонурия разветвленных кетокислот - 46
Скэвенджер-рецептор - 85
Стресс - 88
Сукцинилацетон - 41
Сукцинилацетоацетат - 41
Сульфогалактозилцерамид - 93, 101
Сфинголипиды - 92
Сфингозин - 92
Сфингомиелин - 93
164
Талассемия - 61
Танжерская болезнь - 83
Тетрагидробио птерин - 34
Тиаминаза - 8
Тиаминдифосфат - 139
Тиаминзависимая мегалобластическая анемия - 141
Тимидилатсинтетаза - 136
Тимидинфосфаткиназа - 140
Тирамин - 42
Тирозин - 34
Тирозиназа - 41
Тирозинемия - 41
Тирозинемия транзиторная 41
Тирозиноз - 41
Тирозинтрансаминаза 40
Триглицериды - 70, 82
Триптофан - 26, 29, 31
Тринтофанпирролаза 30
Транскобаламин 1-134
Транскобаламин II - 134
Трипсин - 12
Трихотеценовые микотоксины - 8
УДФ-галактоза - 95
УДФ-глюкоза - 95
УДФ-глюкуронозилтрансфераза - 57
Углеводы - 103
Уроканаза - 43
Урокопропорфирия - 55
Уропорфирия - 52
Фактор Кастла - 134
Фактор Хагемана - 145
Фенилаланин - 34
Фенилаланин-4-гидроксилаза - 35
Фенилацетат - 35, 37
Фенилацетилглутамин - 35, 37, 47
Фенилкетонурия - 35
Фениллактат - 35, 37
Фенилпируват - 35, 37
165
Фенилпировиноградная олигофрения - 35
Фитановая кислота - 91
ФИТОЛ -91
Фолатзависимая мегалобластическая анемия - 148 •_,
Формиминотрансфераза-149
Формилкинуренин - 30
Фосфолипиды - 71
Фосфоглицератфосфомутаза - 105, 121
Фосфофруктокиназа - 105
Фруктоза - 112
Фруктоземия - 113
Фруктозо-1-фосфат-альдолаза - 113
Фруктокиназа - 112
а-фукозидаза - 127
Фукозидоз - 127
Фуникулярный миелоз - 135
Хиломикроны - 70, 72
Холестерин - 71
Холестаз 88
Церамид - 92, 101 (
Церамидаза - 101
Цереброгепаторенальный синдром - 92 /
Цереброзидсульфатаза - 101
Цистеин - 45, 145
Цистеамин - 45
Цистин - 28, 44
Цистиноз - 44
Цистинурия - 27, 28
Цитруллин - 48
Цистатионаза - 146
Цистатионин - 144
Цистатионинсинтаза - 144
Цистатионинурия - 146
Шаровидноклеточная лейкодистрофия - 97
Экзогенная витаминная недостаточность - 130
Эксудативная гипопротеинемическая энтеропатия - 14
166
Эндогенная витаминная недостаточность - 132
Энтеропептидаза - 12
Эпимераза - 110
Эритропоэтическая копропорфирия - 53
Эссенциальная пентозурия - 107
Эссенциальная фруктозурия - 113
Эфиры холестерина - 69, 82, 101
Ювенильная гиперурикемия - 63
Ямайская рвотная болезнь - 90
RzGMU.Narod.Ru
167
СОДЕРЖАНИЕ
Перечень сокращений................................3
Предисловие........................................5
Глава 1. Паталогия пищеварения и всасывания
(Д.Д. Песков)......................................7
1.1. Нарушения процессов пищеварения.............7
1.2. Нарушения процессов всасывания............ 15
Глава 2. Паталогия обмена простых белков и аминокислот
(Т.В. Кременецкая, С.А. Покровский).............. 19
2.1. Протеолиз и его нарушения .................20
2.2. Наследственные нарушения транспорта
аминокислот.................................... 27
2.3. Нарушения общих путей превращений аминокислот 32
2.4. Нарушения катаболизма отдельных аминокислот .... 34
2.4.1. Нарушения распада циклических
аминокислот.................................. 34
2.4.2. Нарушения распада ациклических
аминокислот ....................................
2.5. Нарушения цикла мочевинообразования........46
Глава 3. Патология обмена сложных белков
(С.А. Покровский)................................ 49
3.1. Нарушения обмена гемпротеидов..............49
3.1.1. Порфирии и порфиринурии.............. 52
3.1.2. Метаболизм билирубина и его нарушения.. 56
3.1.3. Гемоглобинопатии (гемоглобинозы)......61
3.2. Нарушения обмена нуклеопротеидов.......... 63
3.2.1. Нарушение метаболизма пуриновых
нуклеотидов.................................. 63
3.2.2. Нарушение обмена пиримидиновых
нуклеотидов.................................. 67
Глава 4. Патология липидного обмена
(И.В. Матвеева) ................................. 69
4.1. Транспорт липидов и его нарушения......... 69
4.1.1. Характеристика и метаболизм
липопротеидов.................................70
168
4.1.2. Нарушение транспорта липопротеидов... 77
4.2. Нарушение обмена липидов в тканях..........89
4.2.1. Нарушение процесса окисления
жирных кислот............................... 89
4.2.2. Нарушение процесса распада сфинголипидов
(липидозы)...................................92
Глава 5. Патология углеводного обмена
(Е.А. Рязанова)..................................103
5.1. Нарушения метаболизма глюкозы.............103
5.1.1. Нарушения гликолиза..................103
5.1.2. Нарушение пентозофосфатного цикла ...105
5.1.3. Нарушение “пути уроновых кислот”.....106
5.1.4. Нарушения глюконеогенеза.............107
5.2. Нарушения метаболизма галактозы..........110
5.3. Нарушения метаболизма фруктозы...........112
5.4. Нарушения метаболизма гликогена..........115
5.4.1. Агликогенозы.........................115
5.4.2. Гликогенозы .........................115
5.5. Нарушения мембранного траспорта гексоз...123
5.6. Нарушения метаболизма углевод-белковых
комплексов.................................124
5.6.1. Нарушения метаболизма гликопротеидов.125
5.6.2. Нарушение метаболизма протеогликанов.128
Глава 6. Нарушение обмена витаминов
(Д.Д. Песков)....................................130
6.1. Нарушения обмена водорастворимых витаминов .... 130
6.1.1. Приобретенные нарушения
обмена витаминов............................130
6.1.2. Врожденные нарушения обмена витаминов.137
6.2. Врожденные нарушения обмена жирорастворимых
витаминов ....................................149
Литература.......................................153
Предметный указатель.............................155
169