/



























































































































































































































































































































































































































































































































































































































Text
ВЫСШЕЕ ПРОФЕССИОНАЛЬНОЕ ОБРАЗОВАНИЕ
ПРАКТИКУМ
ПО МИКРОБИОЛОГИИ
Под редакцией профессора А. И. Нетрусова
Допущено
Министерством образования и науки Российской Федерации в качестве учебного пособия для студентов высших учебных заведений, обучающихся по направлению 510600 «Биология», специальности 012400 «Микробиология» и биологическим специальностям
Москва
ACADEMIA
2005
УДК 579(075.8)
ББК 24.4я73
П69
Авторы:
А.И.Нетрусов, М.А.Егорова, Л.М.Захарчук, Н.Н.Колотилова, И.Б.Котова, Е.В.Семенова,
Н.Ю. Татаринова, Н. В.Уголькова, Е. А. Цавкелова, А. Ф. Бобкова, А. Г. Богданов,
И. В.Данилова, Т.Ю.Динариева, В. В. Зинченко, А.Д. Исмаилов, А. В.Кураков,
В. И. Максимов, Е.С. Милько, Е.П. Никитина, Е.П. Рыжкова, А. М. Семенов,
Д. В. Хомякова, Т.А.Чердынцева, Т. Г. Юдина >
Рецензенты:
проф. Н.С.Егоров (Международный биотехнологический центр МГУ им. М. В.Ломоносова); д-р биол. наук, проф. Ю.Д.Цыганков (ФГУП ГосНИИ генетики и селекции промышленных микроорганизмов)
Практикум по микробиологии: Учеб, пособие для студ. высш. учеб, заве-П69 дений / А.И.Нетрусов, М.А.Егорова, Л.М.Захарчук и др.; Под ред.
А. И. Нетрусова. — М.: Издательский центр «Академия», 2005. — 608 с.
ISBN 5-7695-1809-Х
В практикуме изложены методы оптической и электронной микроскопии, приемы
фиксации и окраски препаратов клеток, методы приготовления и стерилизации сред, 1 современные способы их культивирования, методы выделения и подсчета микроорга- J низмов, правила работы с чистыми культурами микроорганизмов и основные принципы их идентификации. Ряд разделов книги содержит материал по метаболизму и генетике ; микроорганизмов.
Для студентов высших учебных заведений, обучающихся по биологическим специальностям.
УДК 579 (075.8)
ББК 28.4я73
Оригинал-макет данного издания является собственностью
Издательского центра «Академия», и его воспроизведение любым способом без согласия правообладателя запрещается
© Коллектив авторов, 2005
© Образовательно-издательский центр «Академия», 2005
ISBN 5-7695-1809-Х © Оформление. Издательский центр «Академия», 2005
СОДЕРЖАНИЕ
Предисловие (А. И. Нетрусов).......................................... 6
Раздел I
ВВЕДЕНИЕ (НАЧАЛА МИКРОБИОЛОГИИ)
Глава 1. Общая характеристика микроорганизмов {А. И. Нетрусов)...........9
Глава 2. Устройство микробиологической лаборатории и правила работы в ней (Я. В. Семенова).....................................................24
Раздел II
ПРИНЦИПЫ ВЫДЕЛЕНИЯ И ИЗУЧЕНИЯ МИКРООРГАНИЗМОВ
Глава 3. Составление сред и культивирование микроорганизмов (JEB. Семенова).31
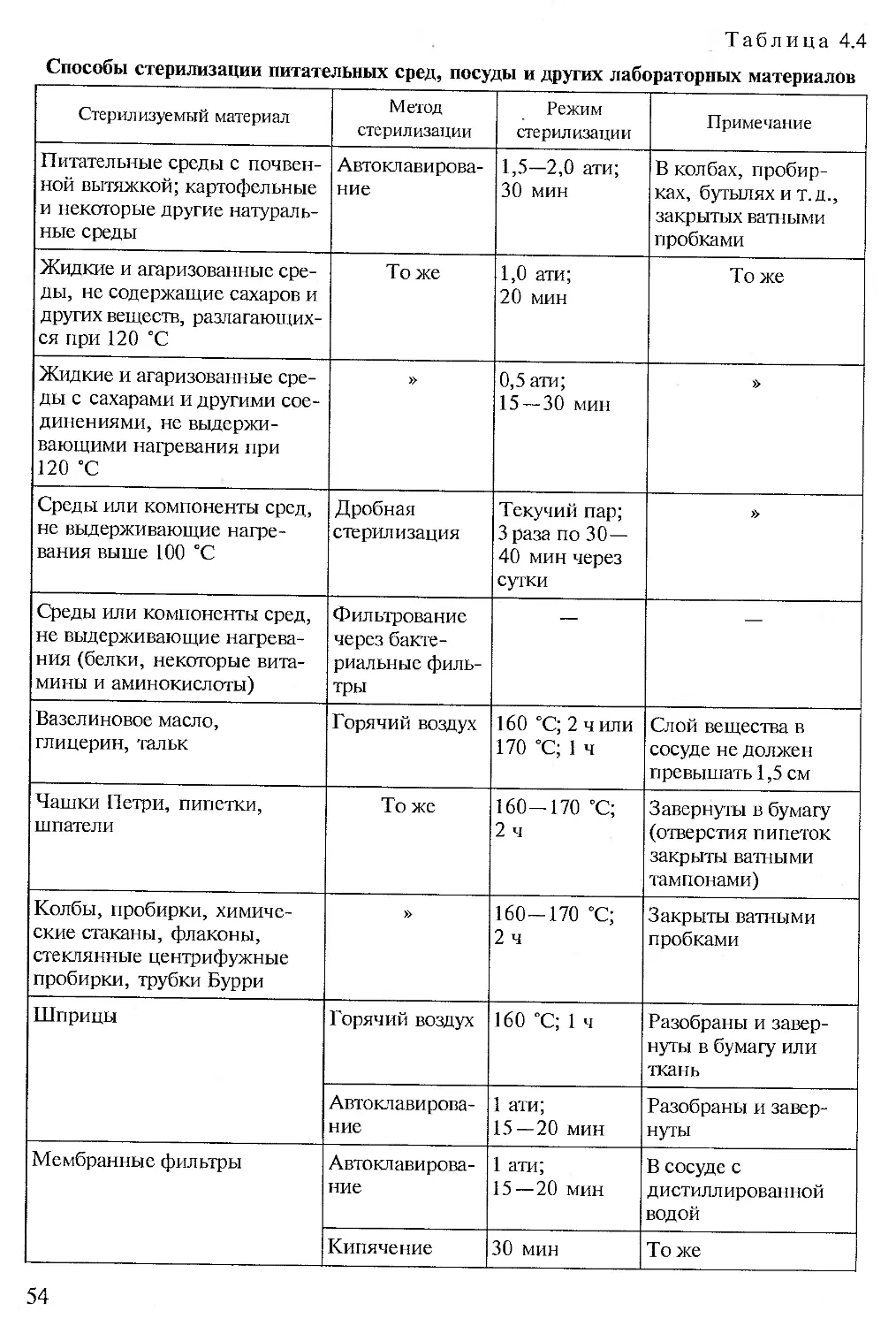
Глава 4. Методы стерилизации {Е. В. Семенова)...........................45
Глава 5. Изучение морфологии и цитологии микроорганизмов {А.М. Семенов, Е. А. Цавкелова, Т. Г. Юдина, А. Г. Богданов).......................56
Глава 6. Выделение чистых культур микроорганизмов (НЮ. Татаринова, Л. М. Захарчук)..............................................93
Глава 7. Методы количественного учета микроорганизмов (Я. Н. Колотилова, А. М. Семенов)..............................................101
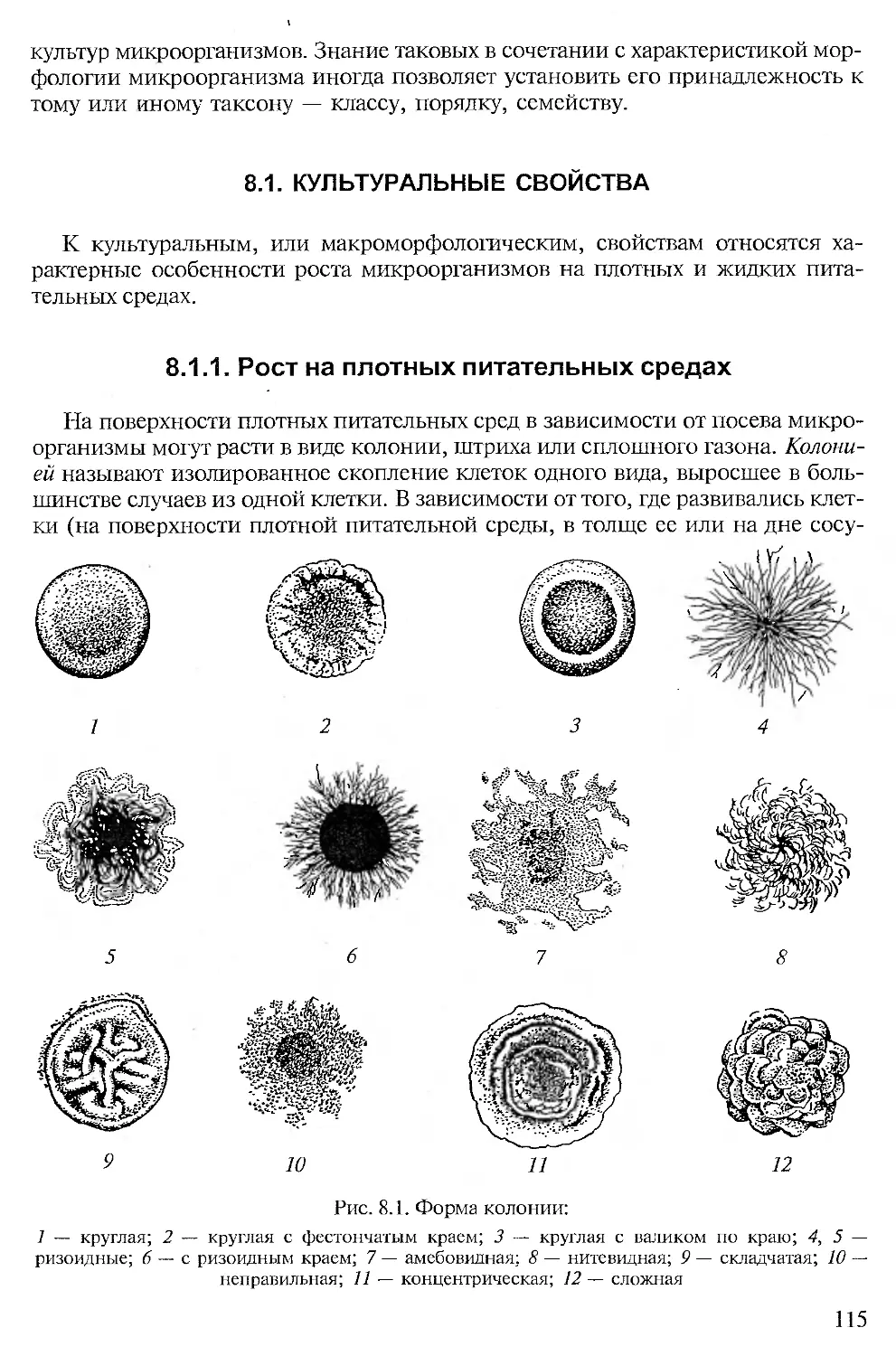
Глава 8. Культуральные и физиолого-биохимические свойства микроорганизмов (Я. В. Уголькова)...........................................114
Глава 9. Систематика и идентификация микроорганизмов (Н.Ю. Татаринова, Л. М. Захарчук).............................................132
Глава 10. Идентификация микроорганизмов из природных сообществ без выделения в чистые культуры (А. М. Семенов, А. И. Нетрусов)..143
Глава 11. Хранение микроорганизмов {Н.Ю. Татаринова, Т.А. Чердынцева) .159
Глава 12. Определение химического состава клеток микроорганизмов (А. И. Нетрусов).....................................................167
Глава 13. Подготовка клеток для энзиматических исследований {А. И. Нетрусов, Т. Ю.Динариева)......................................................180
Раздел III ВЫДЕЛЕНИЕ МИКРООРГАНИЗМОВ ОТДЕЛЬНЫХ ФИЗИОЛОГИЧЕСКИХ ГРУПП
Глава 14. Аэробные органотрофные микроорганизмы (Л.М.Захарчук, Е.В. Семенова, Н. Ю. Татаринова, С. Ф. Герасина)...................................196
Глава 15. Аэробные литотрофные микроорганизмы {А. И. Нетрусов, М.А. Егорова, Т. Я. Лафицкая, А. В. Куликов)......................................203
Глава 16. Получение накопительных и чистых культур анаэробных
микроорганизмов {А.И.Нетрусов, И. Б. Котова, Е. В. Семенова).214
Глава 17. Выделение фототрофных микроорганизмов (Я Н. Колотилова) ....222
Глава 18. Выделение и учет грибов методами посева на питательные среды
и приманок (Л. В. Кураков)...................................231
Глава 19. Изучение биологии простейших {Н.В.Уголъкова, Е.А. Цавкелова).254
Раздел IV
ИЗУЧЕНИЕ ФИЗИОЛОГИИ И МЕТАБОЛИЗМА МИКРООРГАНИЗМОВ
Глава 20. Применение методов планирования многофакторного эксперимента при оптимизации состава питательной среды {В. Н. Максимов,
Е. П. Никитина)...............................................264
Глава 21. Периодическое и непрерывное культивирование микроорганизмов
{Л. М. Захарчук, А.Д. Исламов)................................284
Глава 22. Метаболизм дрожжей (£. В. Семенова) ........................299
Глава 23. Биосинтез и функции корриноидов в физиологии пропионовокислых бактерий {Т.П. Рыжкова, И. В.Данилова)................................307
Глава 24. Получение энергии литотрофными бактериями (А. И. Нетрусов)..315
Глава 25. Сульфидогены. Сульфатное и серное дыхание (А. И. Нетрусов,
Т. Г. Юдина)..................................................327
Глава 26. Метаболическая вариабельность микроорганизмов {А.И.Нетрусов,
М. А. Егорова)................................................331
Глава 27. Ассимиляция углекислоты цианобактериями {А.И.Нетрусов,
И. Н. Колотилова).............................................333
Глава 28. Транспорт субстратов в клетки микроорганизмов {А. И. Нетрусов,
М. А. Егорова)................................................335
Глава 29. Образование микроорганизмами ферментов и методы их учета
(Я. Н. Колотилова, И. Ю. Татаринова, Л. М. Захарчук).........337
Глава 30. Образование и направленный синтез антибиотиков-актиномицинов
{Л. М. Захарчук, Н. Ю. Татаринова)...........................354
Глава 31. Методы изучения образования витаминов микроорганизмами {Л. М. Захарчук, Н. Ю. Татаринова)....................................360
Глава 32. Полифункциональные белки бактерий {Т.Г.Юдина)...............371
Глава 33. Анаэробная биодеградация азокрасителей и их производных (Я. Б. Котова)........................................................376
Глава 34. Изучение явления диссоциации у микроорганизмов {Е. С. Милько,
И. Б. Котова) ................................................387
Глава 35. Генетика микроорганизмов {И.Б. Котова, В. В. Зинченко, А.И.Нетрусов,
Е. А. Цаквслова)..............................................392
Глава 36. Исследования бактериофагов {А. Ф. Бобкова)..................423
Раздел V
НЕКОТОРЫЕ АСПЕКТЫ ПРИКЛАДНОЙ МИКРОБИОЛОГИИ (ЭКОЛОГИЯ, БИОТЕХНОЛОГИЯ, САНИТАРНАЯ И МЕДИЦИНСКАЯ МИКРОБИОЛОГИЯ)
Глава 37. Экология и трофические цепи {Н. И. Колотилова)..............430
Глава 38. Цикл азота и микроорганизмы в нем участвующие (А. М. Семенов, Е. А. Цавкелова).....................................................434
Глава 39. Лабораторные опыты с растениями (Е. А. Цавкелова, Т.А. Чердынцева).445
Глава 40. Бактерии, минерализующие соединения фосфора (77. М. Захарчук)......450
Глава 41. Микроорганизмы, участвующие в разложении силикатов (А. И. Нетрусов, Т. Н. Лафицкая)..............................................452
Глава 42. Бактериальные удобрения (Л. М. Захарчук)...................453
Глава 43. Микроорганизмы — вредители производства (Л. М. Захарчук)...459
Глава 44. Молочнокислые бактерии (77. Г. Стоянова)...................467
Глава 45. Выделение углеводородокисляющих микроорганизмов (Д. В. Хомякова)...478
Глава 46. Санитарная микробиология (77. В. Уголькова)................481
Глава 47. Основы медицинской микробиологии (77. М. Захарчук).........505
ПРИЛОЖЕНИЯ
Приложение 1. Примерная программа практических занятий по микробиологии для студентов биологических факультетов университетов (77. М. Захарчук, Н.Н. Колотилова, А. И. Нетрусов) ..................................520
Приложение 2. Устройство приборов и принципы работы на них (А. И. Нетрусов, М. А. Егорова) ....................................................537
Приложение 3. Работа с изотопами (А. И. Нетрусов, С. Ф. Герасина) ...567
Приложение 4. Среды для культивирования различных микроорганизмов (77. В. Уголькова).................................................569
Приложение 5. Рецепты красителей и индикаторов (77. В. Уголькова)....584
Приложение 6. Рецепты растворов (Н.В. Уголькова).....................587
Приложение 7. Неанглийские выражения и сокращения, встречающиеся в научной литературе (Л.М. Захарчук) ............................ 591
Список литературы....................................................594
Предметный указатель.................................................600
Посвящается 250-летию
Московского государственного университета им. М. В. Ломоносова и 80-летию кафедры микробиологии МГУ
ПРЕДИСЛОВИЕ
Практикум охватывает широкий круг вопросов по выделению и изучению сапротрофных микроорганизмов из окружающей среды, включая экстремальные места их обитания, и продолжает серию издания учебных пособий к практическим занятиям, выпущенных кафедрой микробиологии МГУ им. М. В. Ломоносова с 1976 по 1995 г. Настоящее издание вобрало в себя весь тот материал, который был накоплен преподавателями и научными сотрудниками данной кафедры по дидактике и методологии изложения курсов общей микробиологии для студентов, обучающихся по специальности «Микробиология», а также для студентов других биологических специальностей университетов и вузов.
В начале практикума изложены общие принципы работы с микроорганизмами, их физиолого-биохимические свойства, метаболизм, прикладные аспекты общей микробиологии. Завершает книгу справочный материал. Отдельные задачи для практических занятий по микробиологии включены в соответствующие разделы с необходимыми методиками, однако разработаны и написаны они пока не для всех групп микроорганизмов.
Первый раздел книги содержит обзор общих свойств микроорганизмов, описание их распространения и участия в превращении веществ на Земле, а также роли в процессах, применяемых в биотехнологии. В нем рассмотрены принципы работы в микробиологической лаборатории, подробные инструкции по приготовлению и приемам стерилизации разнообразных питательных сред (большое количество прописей самых распространенных в современной микробиологии сред представлено в приложениях).
Во втором разделе описаны приемы постановки накопительных культур для выделения различных физиологических групп микроорганизмов и получения из них чистых культур. Даны методы окраски и микроскопирования микроорганизмов, включая сканирующую и просвечивающую электронную микроскопию с цифровой обработкой изображений. Изложены приемы и способы изучения физиолого-биохимических свойств чистых культур микроорганизмов и их идентификации, в том числе с применением современных методов молекулярной микробиологии по сравнению последовательностей 16S рРНК, а также по обнаружению и идентификации отдельных филогенетических групп микроорганизмов без выделения их в чистые культуры: использование денатурирующего гель-электрофореза в градиентах температуры или мочевины, или ДНК-микрочипов при изучении структуры популяций микроорганизмов в природных эконишах. Приведены современные методы поддержания и хранения культур микроорганизмов с мониторингом их нужных свойств, методы количественного учета микроорганизмов с применением прижизненных и по
стмортальных окрасок флуоресцентными красителями. Рассмотрены все наиболее часто используемые способы разрушения клеток и подготовки их для энзиматического анализа, методы исследования состава клеток микроорганизмов (клеточных стенок, полисахаридов, белка, нуклеиновых кислот, в том числе определения Г + Ц, мол. %, содержания поли-р-оксимасляной кислоты).
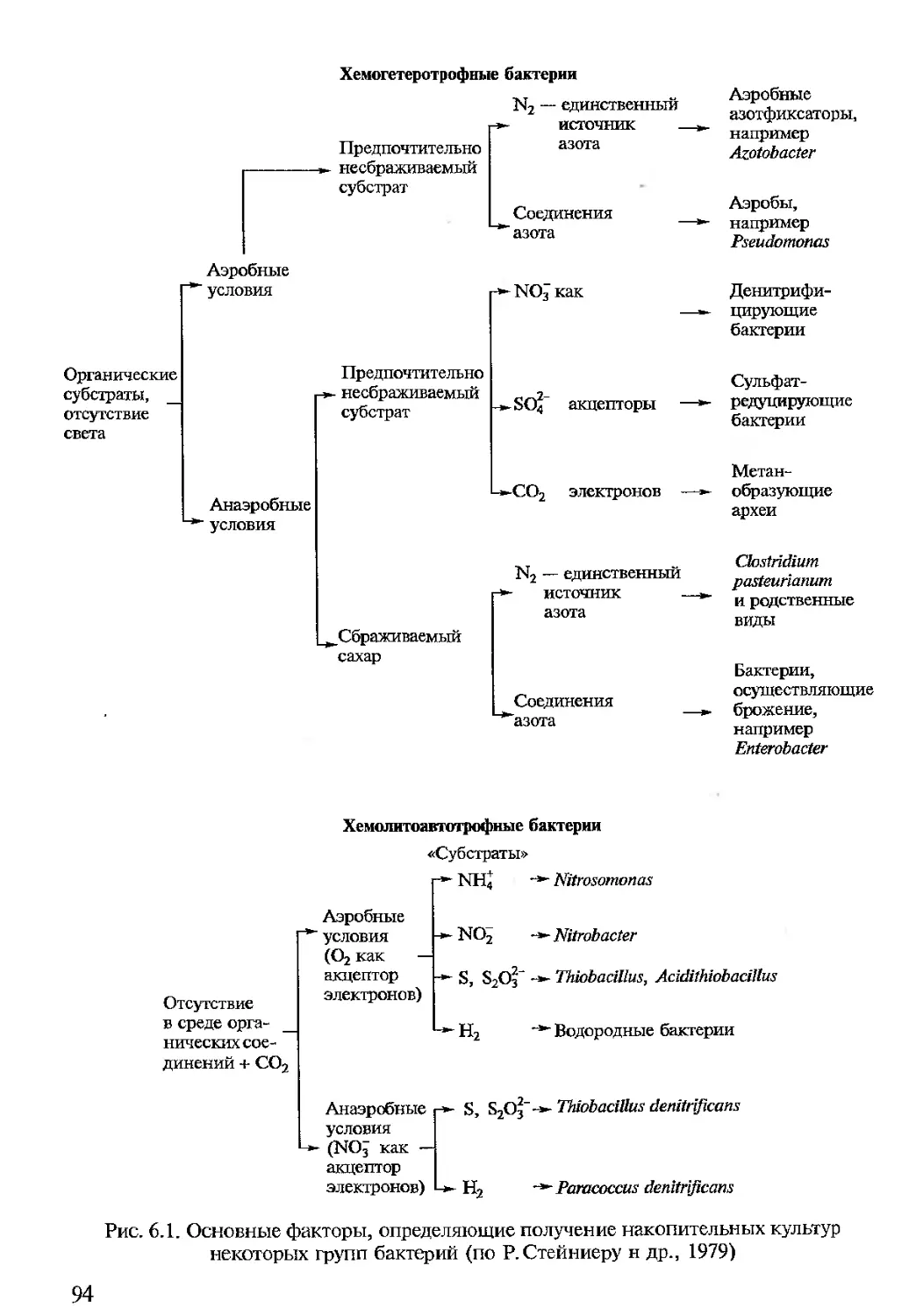
В третьем разделе приведены методы работы с анаэробными и аэробными гетеротрофами, хемолитотрофами и метилотрофами, микроорганизмами, мобилизующими фосфор и кремний, первичными бродильщиками и микроорганизмами, способными к анаэробному дыханию, включая строжайших анаэробов — метаногенов и ацетогенов.
Четвертый раздел состоит в основном из практических задач, предназначенных для выполнения студентами 3 —5-го курсов во время прохождения ими малого и большого практикумов по микробиологии. Такие задачи включают применение методов математического планирования эксперимента по разработке и оптимизации сред для выращивания микроорганизмов с накоплением максимальной биомассы, разработки и испытания параметров непрерывного культивирования светящихся фотобактерий в автоматическом ферментере с расчетом параметров процесса, изучения метаболитической вариабельности отдельных микроорганизмов на примере Paracoccus denitrificans при росте культур аэробно/анаэробно в автотрофных или гетеротрофных условиях. Несколько практических задач посвящены изучению процессов образования микроорганизмами ферментов (протеаз, амилаз), антибиотиков и витаминов (с их количественной оценкой в динамике роста культур), исследованию процессов, проходящих в свободных и иммобилизованных клетках микроорганизмов, определению скорости фотосинтеза по фиксации клетками цианобактерий меченой углекислоты, изучению генетических процессов у микроорганизмов, таких, как получение и идентификация ауксотрофных мутантов, выделение и анализ плазмид, индуцированный мутагенез (с применением теста Эймса), развитие бактериофагов в клетках бактерий.
Пятый методологический раздел (гл. 37—45) знакомит с приемами выделения и изучения микроорганизмов, принимающих участие в глобальном цикле азота, с взаимоотношениями растений и микроорганизмов, с методами изучения простейших и их взаимоотношений с бактериями, с выделением и учетом грибов методами посева на питательные среды и методом приманок, основами медицинской микробиологии, а также с использованием микроорганизмов в пищевой и сельскохозяйственной биотехнологии.
Завершают книгу приложения с подробным изложением примерной программы малого практикума для студентов, изучающих начальный курс микробиологии: программа включает 11 практических занятий, рассчитанных на один семестр. В приложениях описаны также самые необходимые и распространенные приборы, применяемые микробиологами, и правила их использования, приведен большой справочный материал с рецептами самых распространенных сред, красителей и растворов для микробиологических исследований. Книга снабжена также словарем неанглийских выражений и сокращений, встречающихся при чтении научной литературы, и подробным индексом-указателем.
Для уменьшения физического объема книги некоторая часть материала набрана петитом, однако это не означает, что эти разделы учебника имеют подчиненное значение или необязательны для изучения. Авторы постарались дать
материал в максимально сжатом изложении, поэтому все разделы книги одинаково важны для прочтения и запоминания студентами.
* * *
В разработке оригинальных задач, подготовке и написании отдельных глав книги принимали участие бывшие и настоящие сотрудники кафедры микробиологии МГУ: И.В.Авсюк, М. А.Аль-Нури, Л. Г. Азова, Н. Н. Гречушкина, Т. Ю.Динариева, М. А. Егорова, Л. М. Захарчук, Е. И. Козлова, И. Б. Котова, Е.Н.Красильникова, И. Б. Наумова, И.Т. Нетте, М. В. Нефедова, Е. В. Семенова, А.М.Семенов, Н.Ю.Татаринова, Е.А.Цавкелова, Т.Г.Юдина, Т.А.Чер-дынцева. Авторы выражают признательность доценту Н. Н. Колотиловой за помощь в подготовке рукописи к изданию.
Предыдущие выпуски практикумов по микробиологии, подготовленнные профессорами, преподавателями и сотрудниками кафедры микробиологии МГУ, выходили в свет под редакцией профессора Н. С. Егорова и при непосредственном участии академика РАН Е.Н. Кондратьевой, чьи ценные замечания и патронаж способствовали осуществлению данного издания.
Это всеобъемлющее практическое пособие, энциклопедическое по объему, должно стать настольной книгой студента, преподавателя и научного сотрудника, изучающих мир микробов в его разнообразии, а также исследующих свойства микроорганизмов и работающих в области общей или прикладной микробиологии и биотехнологии.
РАЗДЕЛ I
ВВЕДЕНИЕ (НАЧАЛА МИКРОБИОЛОГИИ)
Глава 1
ОБЩАЯ ХАРАКТЕРИСТИКА МИКРООРГАНИЗМОВ
Микроорганизмы — это мельчайшие живые существа, размеры которых в большинстве случаев не превышают 1 — 2 мкм, что делает их невидимыми для человеческого глаза без увеличения. Мир микробов, населяющих нашу планету, велик и разнообразен. Они различаются между собой морфологически, а также физиологическими и биохимическими свойствами. По принципу клеточной организации все микроорганизмы могут быть разделены на два типа — прокариоты и эукариоты. У прокариот ядерный аппарат, называемый часто нуклеоидом, представлен кольцевой молекулой ДНК, соответствующей одной хромосоме. У эукариот ядро содержит набор хромосом и отделено от цитоплазмы мембраной. Различия в организации ядерного аппарата коррелируют с рядом других особенностей эукариот и прокариот (табл. 1.1). Первоначально к микроорганизмам относили и вирусы, однако в настоящее время их чаще рассматривают как особые формы жизни, в связи с тем, что они Не имеют клеточного строения и в отличие от про- и эукариот содержат лишь один тип нуклеиновых кислот (ДНК или РНК).
Таблица 1.1
Некоторые отличительные признаки эукариот и прокариот
Характеристика Прокариоты Эукариоты
Цитологические признаки
Наименьший размер клетки — 0,05 мкм + -
Наличие оформленного ядра - +
Наличие автономных органелл (митохондрии, хлоропласты) - +
Локализация рибосом: распределены в цитоплазме прикреплены на эндоплазматическом ретикулуме + +
Жгутики (если присутствуют): диаметр 0,01 —0,02 мкм, формула среза 8 + 1 диаметр около 0,2 мкм, формула среза 9 + 2 + +
Окончание табл. 1.1
Характеристика Прокариоты Эукариоты
Молекулярно-биологические особенности
Число хромосом 1-2 >1
Кольцевая хромосома + -
Линейные хромосомы - +
Константы седиментации рибосом: 70S 80S + +
Константы седиментации рибосомной РНК: 5S, 16S, 23S 5S, 5,85S, 18S, 28S + 1 +
Признаки, основанные на химических анализах
Присутствие пептидогликана +(-) -
Особенности размножения
Клеточное деление происходит в результате митоза - +
Возможность мейоза - +
Перенос генов и рекомбинация включают гаметогенез и образование зигот - +
Питание
Диффузия или транспорт через мембрану + +
Эндоцитоз - +
Метаболические особенности
Дыхательный и фотосинтезирующий аппарат ассоциирован с плазматической мембраной или ее выростами + -
Возможность хемолитотрофного метаболизма + -
Способность к фиксации молекулярного азота + -
Способность к метаногенезу + -
Способность к аноксигенному фотосинтезу + -
1.1. ПРОКАРИОТЫ
Среди прокариот различают бактерии и археи. Основанием для выделения этих групп, рассматриваемых некоторыми исследователями как отдельные царства или даже империи, послужили результаты сравнения олигонуклео-тидных последовательностей 16S рибосомных РНК, а также выявления различий в составах клеточных стенок, липидов и ряда других особенностей.
Большинство известных прокариот составляют различные группы бактерий. Известные археи включают метаногенов, отдельных сульфатредукторов, экстремальных галофилов, термоплазм, лишенных клеточной стенки, а также экстремально термофильных бактерий, окисляющих и восстанавливающих молекулярную серу. За последние годы обнаружено значительное число новых представителей архей.
Размеры и форма клеток. Большинство прокариот — одноклеточные формы. Величина клеток многих прокариот находится в пределах 0,2—10,0 мкм, однако среди них есть «карлики» (около 0,05 мкм — трепонемы, микоплазмы) и «гиганты» (длиной до 100 мкм — Achromatium, Macromonas). Самой крупной из то сих пор выделенных прокариот является клетка Epulopiscium fishelsoni — до 600 мкм в длину. Формы клеток бактерий не отличаются большим разнообразием. Это чаще всего палочки разной длины, сферические клетки (кокки), а также извитые формы — вибрионы, спириллы и спирохеты. Обнаружены виды с треугольными, квадратными, звездообразными и плоскими (тарелкообразными) клетками, некоторые имеют отростки-простеки (рис. 1.1 —1.7).
3
Рис. 1.1. Сочетания сферических клеток:
— диплококки; 2 — стрептококки; 3 — тетракокки и сардины; 4— стафилококки и микрококки
Рис. 1.2. Палочковидные бактерии:
.' — Pseudomonas aeruginosa', 2 — Bacillus mycoides', 3 — Bacillus megateriunr, 4 — Cytophaga sp.
2
Рис. 1.3. Azotobacter vinelandii.
1 — суточная культура; 2 — 16-суточная; 3 — 10-суточная
Рис. 1.5. Миксобактерии:
1 — вегетативные клетки; 2 — формы, переходные к микроцистам; 3 — микроцисты; 4 — делящиеся клетки
Рис. 1.4. Клубеньковые бактерии:
.’ — палочки и кокки в молодой культуре; 2 — бактероиды
1 2
Рис. 1.6. Извитые клетки:
1 — вибрионы; 2 — спириллы; 3 — спирохеты
3
Рис. 1.7. Бактерии, образующие выросты:
7— Caulobacter, 2— Hyphomicrobiunr, 3—Апса-lomicrobium; 4 — Callionella
Тип группирования клеток иногда помогает определить систематическую принадлежность бактерий. Они могут быть одиночными, образовывать пары, короткие и длинные цепочки правильной (стрептококки) и неправильной (стафилококки) формы, формировать пакеты из 4, 8 и более клеток (сардины), розетки и сети. Значительное число бактерий из актиномицетной группы образуют мицелий. Известны также многоклеточные прокариоты, образующие трихомы, прямые и ветвящиеся (рис. 1.8—1.13).
Строение клеток. Большинство прокариот имеет ригидную клеточную стенку, под которой расположена цитоплазматическая мембрана. Состав и строение клеточной стенки — важный систематический признак, по которому прокариоты подразделяют на следующие группы: грамположительные,
Рис. 1.8. Нитчатые бактерии:
1 — Beggiatocr, 2 — Thiothrix', 3 — Saprospira', 4 — Simonsiellcr, 5 — Caryophanorr, 6 — цианобактерии рода Microcoleus', 7 — Leptothrix', 8 — Sphaerotilus", 9 — Crenothrix
Рис. 1.9. Мицелий актиномицета (7) и гриба (2) при одинаковом увеличении
Рис. 1.10. Нокардии:
1 — 2-суточная культура; 2 — 4—5-суточная; 3 — 7—8-суточная
Рис. 1.11. Микобактерии:
1 — суточная культура; 2 — 2-суточная; 3 — 3 — 4-суточная; 4 — 10-суточная
1
Рис. 1.12. Arthrobacter (1) и Corynebacterium (2)
Рис. 1.13. Микоплазмы. Схема электронной микрофотографии
Рис. 1.14. Типы жгутикования у бактерий:
I — монотрихиальное; 2 — лофотрихиальное; 3 — латеральное; 4 — амфитрихиальное; 5 — перитрихиальное; 6 — «смешанное» полярно-перитрихиальное
грамотрицательные и не имеющие клеточной стенки. Своеобразным строением и составом клеточной стенки характеризуются археи. Грам-положительные бактерии отличаются от грамотрицательных большим (до 40 раз) содержанием муреина (пептидогликана) в клеточной стенке и отсутствием внешней мембраны. Археи пептидогликана не синтезируют, но некоторые образуют псевдо-
муреин.
У многих бактерий на поверхности находят ворсинки (фимбрии, пили), а подвижные формы часто имеют жгутики. На поверхности клеточных стенок многих прокариот можно обнаружить слизистые капсулы различной толщины. Они чаще всего полисахаридной, но бывают и гликопротеидной или полипеп-тидной природы. Обнаружены также S-слои (от англ, surface — поверхность), выстилающие наружную поверхность клеточной оболочки равномерно упакованными белковыми структурами правильной формы.
Прокариоты характеризуются сравнительно простой внутриклеточной организацией и не содержат автономных органелл, хотя многие бактерии имеют включения. Среди них в первую очередь следует отметить различного рода внутриклеточные мембранные пузырьки, образованные в результате инвагинации цитоплазматической мембраны. Развитая цепь внутрицитоплазматических мембран характерна для фототрофных прокариот (хроматофоры, тилакоиды), нитрифицирующих и метанокисляющих бактерий. Некоторые клетки образуют газовые вакуоли (аэросомы), окруженные белковой мембраной, выполняющие у водных организмов роль регуляторов плавучей плотности. Многие бактерии откладывают внутриклеточно запасные вещества (полисахариды, поли-р-оксибутират, полифосфаты, серу). Отдельные виды спорообразующих бактерий иногда имеют параспоральные тельца белковой природы.
Движение клеток. Среди прокариот есть подвижные и неподвижные виды. Движение клеток чаще всего осуществляется за счет вращения жгутиков (рис. 1.14). Еще одним способом движения является скольжение клеток, механизм которого изучен недостаточно. Описано
Рис. 1.15. Схемы роста и деления клеток Caulobacter
«прыгающее» движение, природа которого не выяснена. Подвижные бактерии способны осуществлять реакции таксиса: аэро- и фототаксис, хемо- и магнитотаксис.
Размножение и развитие прокариот. Большинство бактерий размножаются в результате бинарного деления, реже — почкованием, а некоторые (например, актиномипеты) — с помощью экзоспор или обрывков мицелия (рис. 1.15 — 1.18). Известен также способ множественного деле-
Рис. 1.16. Гонидии (7) и гормогонии (2) нитчатых бактерий
Рис. 1.17. Формы воздушных спороносцев у актомицетов
Рис. 1.18. Спорангии актиномицетов:
1— Actinoplanes', 2— Atnorphosporangiiun', 3— Spirillospora
Рис. 1.19. Типы образования эндоспор у бактерий:
.' — бациллярный; 2 — клостридиальный; 3 — плек-тридиальный
Рис. 1.20. Акинеты (А) и гетероцисты (Г) нитчатой цианобактерии Cylindrospermum
нця (образование баеоцитов у ряда цианобактерий). Многоклеточные прокариоты могут размножаться отделением от трихома одной или нескольких клеток. У ряда бактерий обнаружена конъюгация, однако, как правило, она не обеспечивает полной передачи генетического материала из одной клетки в другую.
Некоторые прокариоты характеризуются сложным циклом развития, в процессе которого может меняться морфология клеток и образуются покоящиеся формы: цисты, эндоспоры, акинеты (рис. 1.19—1.20). Известны бактерии, образующие плодовые тела, часто причудливых конфигураций и расцветок.
Бактерии отличаются способностью к быстрому размножению. «Чемпионами» з этом отношении являются фотобактерии: время их генерации около 8 мин. Для Escherichia coli (время удвоения — 20 мин) подсчитано, что потомство
одной клетки в случае неограниченного роста бактерий уже через 48 ч могло бы дать биомассу, в 150 раз превышающую массу Земли.
1.2. ЭУКАРИОТЫ
К эукариотам в отличие от прокариот относятся как микро-, так и макроорганизмы. Эукариотные микроорганизмы представлены грибами, водорослями и простейшими.
1.2.1. Грибы
Грибы — обширная группа гетеротрофных микроорганизмов, широко распространенных в природе. Большинство из них сапрофиты, однако есть и паразитические виды. Грибы подразделяют на два отдела: Eumycota и Oomycota. Характерной особенностью большинства грибов является образование мицелия. Мицелиальные грибы, предмет изучения микробиологов, объединяют представителей в основном трех классов: Zygomycetes, Ascomycetes и Deutero-mycetes (все — из отдела Eumycota).
Рис. 1.21. Спорангии и спорангиеносцы некоторых зигомицетов:
а — Absidia-, б — Rhizopus-, в — Мисог, г — Actinomucor; 1 — плодоносящий мицелий; 2 — спорангиеносцы; 3 — спорангий со спорами
Зигомицеты имеют не разделенный на отдельные клетки многоядерный мицелий (ценоцитный). Они характеризуются особым типом полового размножения, которое включает стадию образования зигоспоры из двух родительских гиф (рис. 1.21). К числу зигомицетов относятся мукоровые грибы Rhizopus, Phycomyces, Absidia и др.
Рис. 1.22. Способы вегетативного размножения дрожжей:
1 — почкование; 2 — деление; 3 — почкующееся деление
Рис. 1.23. Баллистоспоры на отдельных клетках и гифах мицелия:
1 — Sporobolomyces', 2 — Sporidiobolus
Рис. 1.24. Мицелиальные формы дрожжей:
I — псевдомицелий; 2 — истинный мицелий; а — артроспоры; б — эндоспоры
Гаплоидные клетки
Аск с гаплоидными аскоспорами
Рис. 1.25. Жизненные циклы некоторых дрожжей:
1 — Saccharomyces cerevisiac, 2 — Schizosaccharomyces (Octosporomyces) octosporus
Диплоидная фаза
до
Вегетативные и Q делящиеся клетки Н
Аскоспоры, освободившиеся из сумки
Рис. 1.26. Копуляция между аскоспорами одной сумки (2) и разных сумок (2)
Рис. 1.27. Хламидоспоры дрожжей:
7 — на гифах мицелия; 2 — без мицелия. В хламидоспорах видны капли жира
Рис. 1.28. Конидиеносцы и конидии несовершенных грибов:
1 — Trichodenncr, 2 — Cladosporiunr, 3 — Altenaria', 4 — Fusarium', 5 — Stachybotris\ 6 — Stemphylium', 7 — Verticillium; 8 — Oospora', 9 — Cephalosporium; 10— Botrytis; 11 — Phoma (пикнида с конидиями); 12 — Mycogone; a — конидии
Аскомицеты (сумчатые грибы) образуют гифы, которые в отличие от зиго-мицетов разделены поперечными перегородками. Особенностью этих грибов является характерный орган спороношения — аск, в котором формируются аскоспоры. Значительная часть аскомицетов способна размножаться также не-
половым путем — с помощью конидий (рис. 1.22—1.24). К аскомицетам относятся многие дрожжи, которые существуют в виде отдельных неподвижных клеток и размножаются почкованием или (реже) делением. Аскоми-цетные дрожжи могут размножаться и половым путем, образуя аски с 2 — 8 спорами (рис. 1.25—1.26). На определенных стадиях развития некоторые дрожжи образуют мицелий или псевдомицелий, на концах которого можно обнаружить споры (см. рис. 1.23—1.24). Отдельные представители дрожжей (Lipomyces, Crypto-coccus) в качестве покоящейся стадии образуют хламидоспоры (рис. 1.27). К числу широко используемых в различных микробиологических исследованиях дрожжей относятся представители рода Saccharomyces, например S. cerevisiae.
Рис. 1.29. Конидиеносцы у грибов рода Aspergillus (а) и Penicillium (б)\
1 — вегетативный мицелий; 2 — конидиофор; 3 — стеригмы; 4 — конидии
Дейтеромицеты (несовершенные грибы) — разнообразная группа микроорганизмов с отсутствующей (или не обнаруженной) половой стадией (рис. 1.28). Многие несовершенные грибы могут осуществлять парасексуальный процесс,
при котором слияние и последующее деление ядер происходят непосредственно в мицелии. К дейтеромицетам относят представителей родов Penicillium, Aspergillus (рис. 1.29), некоторые дрожжи {Candida, Kloechera, Rhodotorula). Многие дейтеромицеты патогенны для животных и человека.
1.2.2. Водоросли
Водоросли включают фототрофные организмы, среди которых есть макро-и микроформы. Обладая двумя фотосистемами, водоросли осуществляют ок-сигенный фотосинтез. Состав пигментов обусловливает их цвет, который может быть зеленым, бурым, красным, золотистым. Все водоросли образуют каротиноиды и хлорофилл а, а некоторые, в дополнение к ним, хлорофилл b и/или с, а также фикобилины. В клетках водорослей могут накапливаться различные запасные вещества — крахмал, парамилон, ламинарии.
Классификация водорослей основана на их морфологии, подвижности, наборе пигментов, природе запасных веществ, составе клеточной стенки. Они подразделены на ряд групп, из которых объектами микробиологов в основном служат представители следующих отделов.
Зеленые водоросли — обитатели пресных и морских вод, почв и стволов деревьев, одноклеточные и многоклеточные, которые размножаются как бесполым, так и половым путем. Клеточные стенки многих из них содержат целлюлозу.
Диатомовые водоросли — одноклеточные микроорганизмы, многие из которых заключены в кремниевые панцири различной величины и формы и которые составляют основную массу фитопланктона (более 10 тыс. видов). Панцирь, состоящий из двух створок, напоминает чашку Петри; он раскрывается
в процессе бесполого размножения, расходится после деления на две части, каждая из которых достраивает себе недостающие половинки. Диатомеи размножаются и половым путем. Многие представители этой группы обладают билатеральной симметрией и осуществляют скользящее движение за счет выделения слизи из специальных отверстий в створках.
Динофлагелляты — одноклеточные микроорганизмы с двумя боковыми жгутиками. Наряду с диатомеями они составляют существенную часть фитопланктона. Некоторые динофлагелляты образуют токсины, летальные для человека, поэтому в период их массового развития («красное цветение» водоемов) запрещается отлов морских организмов, в мясе которых концентрируется токсин.
Эвгленовые — небольшая группа пресноводных водорослей, лишенная клеточной стенки и имеющая сходство с простейшими. Помимо роста на свету они способны расти в темноте, получая энергию при окислении органических веществ. Некоторые виды эвгленовых передвигаются амебовидным движением, в то время как другие — с помощью жгутиков. Интересной особенностью некоторых видов является наличие светочувствительного органа стигмы, которая ориентирует клетки на источник света.
1.2.3. Простейшие
Простейшие составляют большую группу одноклеточных гетеротрофных микроорганизмов, широко распространенных в природе. Они лишены клеточной стенки (хотя некоторые имеют панцирь) и поглощают питательные вещества абсорбцией через клеточную мембрану или путем эндоцитоза. Размножение этих микроорганизмов осуществляется половым и бесполым путями, иногда оба способа воспроизводства составляют стадии единого жизненного цикла организма. Простейших, представляющих интерес для микробиологов, находят в 4 классах: амеб, жгутиконосцев, реснитчатых и споровиков. По некоторым классификационным схемам к простейшим относят и слизевиков (Myxomycota), которых раньше относили к грибам.
Амебы обитают преимущественно в воде. Они подвижны благодаря псевдоподиям, которые также способствуют захвату пищи с последующим фагоцитозом. Жизненный цикл амеб относительно прост. Одной из его характерных стадий является фаза инцистирования — образование покоящихся цист из активно питающихся клеток, трофозоитов. Как правило, амебы — свободноживущие организмы, однако некоторые виды являются паразитами и вызывают различные заболевания человека, например кариес зубов и амебную дизентерию.
Жгутиконосцы — организмы, включающие паразитирующие и свободноживущие формы. Некоторые их представители вызывают тяжелые бюлезни человека и животных.
Реснитчатые — в основном свободноживущие виды, играющие активную роль в разложении органических загрязнений при очистке воды. Некоторые реснитчатые образуют симбиозы и растут в рубце жвачных животных.
Споровики — класс простейших, образующих споры на определенных стадиях развития. Эти паразитирующие формы простейших имеют сложный жизненный цикл, который включает половую и бесполую стадии размножения. Среди споровиков много возбудителей заболеваний человека и животных.
1.3. ВЛИЯНИЕ ФИЗИЧЕСКИХ И ХИМИЧЕСКИХ ФАКТОРОВ НА РОСТ МИКРООРГАНИЗМОВ
Среди физических факторов, определяющих рост микроорганизмов, следует прежде всего выделить температуру. Оптимальная температура для мезофильных форм составляет 25 — 40 °C. Среди обитателей глубин океанов, почв и болот тундры находят психрофилов, которые растут при более низкой температуре (оптимумы роста при 5 —15 °C). С поверхности снегов Антарктики выделены водоросли, которые растут при -36 °C. Известны экстремальные термофилы, способные расти при 100—ИЗ °C. Споры отдельных микроорганизмов могут выдерживать кратковременное нагревание до 160—180 °C и длительное охлаждение до -196 °C и ниже.
Некоторые виды микроорганизмов хорошо переносят гидростатическое давление до 1000 атм. Выделены облигатные барофилы, которые не способны расти при давлении ниже 500 атм. Но есть виды, в основном образующие газовые вакуоли, не выдерживающие даже незначительного превышения давления над атмосферным. Отдельные представители микроорганизмов чрезвычайно устойчивы к ионизирующей радиации и способны расти даже в воде охлаждающих контуров атомных реакторов (Deinococcus radiodurans, некоторые дрожжи).
Важным фактором, от которого зависит рост микроорганизмов, является осмотическое давление. В то время как большинство организмов не размножаются при концентрации соли (NaCl) в среде более 0,5 М, экстремальные галофилы нуждаются в содержании в среде от 2,5М NaCl и выше до насыщенного раствора (5,2 М).
Микроорганизмы чувствительны к кислотности окружающей среды. Экстремальные ацидофилы могут расти при pH 0,1—0,5, алкалифилы — при pH до 13,0, однако большинство микроорганизмов растут в средах с pH, близким 7,0.
По отношению к молекулярному кислороду микроорганизмы делятся на облигатных аэробов и анаэробов (факультативных, аэротолерантных и строгих, или облигатных). Подавляющая часть известных микроорганизмов относится к аэробам, способным расти только в присутствии молекулярного кислорода, но некоторых угнетает его обычная концентрация в воздухе, и они могут расти лишь при незначительном его содержании в газовой фазе (до 1,0— 5.0 %). Последних называют микроаэрофилами. Факультативные анаэробы могут расти как в присутствии, так и в отсутствие молекулярного кислорода, переключая свой метаболизм, например, с дыхания на брожение (некоторые дрожжи). Рост аэротолерантных анаэробов не угнетается из-за небольшого содержания молекулярного кислорода, однако эти микроорганизмы кислород не используют (например, молочнокислые бактерии). Строгие же анаэробы не выдерживают даже следов молекулярного кислорода в среде, где они растут: он является для них ядом (метаногены, ацетогены, большинство сульфатре-дукторов, некоторые грибы, а также отдельные виды простейших).
1.4. ОБМЕН ВЕЩЕСТВ
Энергетические и конструктивные процессы, осуществляемые микроорганизмами, отличаются большим многообразием (табл. 1.2).
Таблица 1.2
Типы энергетического и конструктивного обмена микроорганизмов
Источник энергии Доноры электронов Источники углерода
органические вещества углекислота
Свет Органические Фотоорганогетеро-трофия Фотоорга ноа втотроф ия
Свет Неорганические Фотолитогетеротро-фия Фотолитоавтотрофия
Органические вещества Органические Хемоорганогетеро-грофия Хемоорганоавтотрофия
Неорганические вещества Неорганические Хемолитогетеротро-фия Хе мол итоа втотроф и я
В зависимости от соединений углерода, которые используются в конструктивном обмене, микроорганизмы подразделены на автотрофов, осуществляющих синтез углеродсодержащих клеточных компонентов целиком из СО2, и гетеротрофов, которые нуждаются для этих целей в готовых органических веществах. Среди гетеротрофов основная часть микроорганизмов представлена сапротрофами, которые используют органические соединения, образуемые в процессе жизнедеятельности или распада других организмов. Гетеротрофные микроорганизмы включают виды, способные расти при наличии простых органических веществ в среде, и облигатных паразитов (паратрофов), которые полностью зависят от метаболизма хозяина, используя синтезируемые им различные сложные органические вещества.
Широкие возможности микроорганизмы проявляют при утилизации соединений азота. Большинство про- и эукариот использует восстановленные соединения азота (чаще всего соли аммония), некоторые нуждаются в готовых аминокислотах, а другие усваивают и окисленные его формы (прежде всего нитраты). Значительное число прокариот, свободноживущих и симбиотических, обладают способностью фиксировать молекулярный азот. Это свойство отмечено только для прокариот и обнаружено у аэробов и анаэробов. Недавно у микроорганизмов, растущих аэробно на средах с СО (Streptomyces thermoautotrophicus), обнаружена нитрогеназа, не чувствительная к кислороду, тогда как активность нитрогеназ других микроорганизмов полностью подавляется в аэробных условиях и фермент часто необратимо инактивируется.
Фосфор, входящий в состав нуклеиновых кислот и других соединений клетки, микроорганизмы получают преимущественно из фосфатов. Источником серы, которая необходима для биосинтеза аминокислот и некоторых кофакторов, чаще всего является сульфат. Лишь некоторые виды микроорганизмов, не способные к ассимиляционной сульфатредукции, нуждаются в восстановленных соединениях серы.
Энергетические процессы, осуществляемые микроорганизмами, включают брожения, фотосинтез, аэробное и анаэробные дыхания. Все они приводят в конечном итоге к запасанию энергии главным образом в форме АТФ, которая расходуется на различные энергопотребляющие процессы.
Синтез АТФ у микроорганизмов может происходить различными путями. При окислительном и фотосинтетическом фосфорилировании, которые связаны с мембранами и поэтому объединены под общим названием мембранное фосфорилирование, преобразование энергии происходит вначале в форме трансмембранного электрохимического потенциала ионов водорода или Na+, в то время как субстратное фосфорилирование приводит к запасанию энергии непосредственно в макроэргических связях АТФ и других химических соединений.
1.5. МИКРООРГАНИЗМЫ В ПРИРОДЕ И БИОТЕХНОЛОГИИ
Сообщество микроорганизмов в естественных местах обитания является важнейшим фактором, определяющим целостность экологических систем в природе. В особых условиях микроорганизмы могут представлять единственную форму жизни. В процессе эволюции выработались различные типы взаимоотношения между микроорганизмами. Тесная связь между ними {симбиоз} строится на различных характерах зависимости партнеров: взаимовыгода {мутуализм), неблагоприятное влияние на одного из партнеров {паразитизм), реже — индифферентные отношения друг с другом {нейтрализм).
Изучение микробного мира расширяет рамки наших представлений о границах живой природы и свидетельствует об активном участии микроорганизмов в кругообороте веществ на Земле. Используя и образуя метан, поглощая СО и СО2, трансформируя всевозможные органические соединения в разных, в том числе экстремальных условиях, микроорганизмы активно участвуют в цикле превращения углерода. Фиксируя молекулярный азот, окисляя аммиак и нитриты, осуществляя денитрификацию, они обеспечивают азотный круговорот в природе, а способность окислять восстановленные серосодержащие соединения и восстанавливать окисленные определяет их роль в круговороте серы.
Микроорганизмы играют значительную роль в превращениях Са, Se, Р, Мп и других веществ и металлов с переходной валентностью.
Влияние микроорганизмов не всегда позитивно: некоторые из них вызывают тяжелые заболевания у человека, животных и растений. Нередки случаи, когда микроорганизмы приводят к порче сельскохозяйственной продукции, разрушению подземных частей зданий, трубопроводов и металлических конструкций шахт. Изучение свойств таких микроорганизмов позволяет разработать эффективные способы защиты от вызываемых ими повреждений. С другой стороны, положительное значение микроорганизмов в хозяйственной деятельности человека невозможно переоценить. С помощью грибов и бактерий готовят хлеб, вино, пиво, квас, молочные продукты, закваски. При участии микробов получают ацетон и бутанол, уксус, лимонную кислоту, некоторые витамины, ряд ферментов, антибиотики и каротиноиды. Микробы участвуют в трансформации стероидных гормонов и других соединений. Их используют для получения белка и ряда аминокислот. Реализуется идея использования микробных ферментов в диагностических целях. Применение микробных комплексов для превращения сельскохозяйственных отходов в биогаз (смесь метана и углекислоты) или этанол открывает возможность создания принципиально новых систем восполнения энергетических ресурсов. Микроорганизмы используют для целей гидрометаллургии, извлекая металлы из бедных руд
или труднодоступных месторождений. Б последние годы микроорганизмы, особенно прокариоты, широко применяют в качестве объектов генной инженерии для клонирования генов и создания векторов, экспрессии гетерологичных белков.
Потенциал микроорганизмов в практическом отношении неисчерпаем. Углубление знаний о жизнедеятельности этих микроскопических, но макро-значимых существ открывает новые направления их эффективного использования в биотехнологии.
Глава 2
УСТРОЙСТВО МИКРОБИОЛОГИЧЕСКОЙ ЛАБОРАТОРИИ И ПРАВИЛА РАБОТЫ В НЕЙ
Микробиологи имеют дело с популяциями (культурами) микроорганизмов, состоящими из миллионов особей. Культуру, содержащую микроорганизмы одного вида, называют чистой. Если в культуре содержится более одного вида микроорганизмов, она носит название смешанной. В микробиологической практике используют главным образом чистые культуры микроорганизмов. Ввиду того что в воздухе и на поверхности предметов (на столах, инструментах, одежде), а также на руках, волосах и т.д. всегда имеется большое количество разнообразных микроорганизмов, следует постоянно заботиться о сохранении чистоты изучаемых культур. Требование чистоты культур в значительной степени определяет специфику устройства микробиологической лаборатории и правила работы микробиолога.
Микробиологическая лаборатория включает ряд помещений, где проводят работу с микроорганизмами или подготовку к ней. Под лабораторные комнаты отводят наиболее светлые, просторные помещения, естественная освещенность которых должно составлять не менее ПО лк. Поверхность столов и пол всех лабораторных помещений покрывают легко моющимся материалом — пластиком или линолеумом, а стены на высоту 170 см от пола окрашивают в светлые тона. Основное рабочее помещение оборудовано столами лабораторного типа, шкафами и полками для хранения аппаратуры, посуды и реактивов. Столы имеют подводку электроэнергии и снабжены газовыми горелками.
Кроме основного рабочего помещения лаборатория имеет стерилизационную, где размещены автоклавы и сушильные шкафы, термостатированную комнату для выращивания микроорганизмов, помещение для хранения культур микроорганизмов, холодильную комнату, моечную и т.д. Пересевы микроорганизмов осуществляют в боксах разных конструкций — от изолированных помещений до настольных камер (ламинаров), чистота атмосферы рабочего пространства в которых обеспечивается циркуляцией стерильного воздушного потока внутри камеры.
Работа в ламинарном боксе. Конструкция ламинарного бокса позволяет стерильно работать с микроорганизмами в нестерильном помещении. Ламинарные боксы бывают двух степеней защиты — класса I и класса II. Ламинары класса I
Рис. 2.1. Ламинарный бокс (пояснение в тексте)
оборудованы притяжной вентиляцией нестерильного воздуха из помещения и выходом этого воздуха в то же помещение после фильтрации (защита от микробных аэрозолей) и в строгом смысле слова не пригодны для стерильной работы. Ламинары с защитой класса II (рис. 2.1) образуют внутри бокса поток стерильного воздуха, забор которого происходит из помещения, и который стерилизуется, проходя через бактериальные фильтры. Таким образом, внутренние поверхности бокса остаются стерильными. Конструкция позволяет также проводить стерильные посевы микроорганизмов в струе стерильного воздуха, прошедшего через бактериальные фильтры и распределенного внутри ла-минара в виде ламинароного потока (без завихрений). Работа в ламинаре, однако, предполагает использование асептической техники (работа у пламени горелки).
Перед началом работы ламинар должен быть вымыт с помощью растворов нейтральных детергентов и все доступные внутренние его поверхности должны быть про-стерилизованы химическими дезинфектантами (70%-й этанол). В течение рутинной работы ламинар необходимо мыть раз в месяц, снимая съемные детали поверхности и прочищая пространство под ними. После промывки панели прибора вновь стерилизуют этанолом.
Бактериальные фильтры ламинара необходимо подвергать процессу химической дезинфекции один раз в течение 1 — 2 мес в зависимости от интенсивности использования. Химическую дезинфекцию внутренней поверхности ламинара и в особенности его фильтров проводят парами формальдегида. С этой целью 50 мл 37%-го раствора формальдегида (формалин) наливают в фарфоровую чашку, ставят на подставку и нагревают чашку для медленного (30 мин) испарения формальдегида. Воздушный насос ламинара должен быть при этом включен для равномерного распределения паров дезинфектанта по внутреннему объему камеры, воздушная заслонка выхода воздуха закрыта, так же как и передняя крышка прибора. При стерилизации ламинара люди должны покинуть помещение. После полного испарения формалина нагреватель и воздушный насос выключают, ламинар оставляют на сутки для дезинфекции и затем проветривают его от остатков паров формальдегида. Для этого открывают переднюю крышку прибора, включают воздушный насос и открывают заслонку выхода воздуха. Прибор оставляют в рабочем состоянии на 15 — 20 мин, и далее он готов к работе. При продувке прибора от паров формальдегида помещение необходимо хорошо проветрить, а люди на это время должны его покинуть.
2.1. ПОДГОТОВКА МИКРОБИОЛОГИЧЕСКОЙ ЛАБОРАТОРИИ К РАБОТЕ
Микробиологическую лабораторию необходимо содержать в чистоте. В ней не должно находиться никаких лишних предметов. Следует регулярно проводить гигиеническую уборку лабораторных помещений. Обеспечить полную стерильность лаборатории очень трудно и это не всегда необходимо, но значительно снизить количество микроорганизмов в воздухе и на различных поверхностях в лабораторных помещениях возможно. Для этого применяют различные способы дезинфекции. Слово «дезинфекция» означает обеззараживание, т.е. уничтожение возбудителей инфекционных болезней на объектах внешней среды. Однако при дезинфекционной обработке погибают не только патогенные, но и сапротрофные бактерии. Иногда процесс дезинфекции оказывает стерилизующее действие.
Обработка помещений микробиологической лаборатории
Пол, стены и мебель в микробиологической лаборатории обрабатывают пылесосом и протирают раствором различных дезинфицирующих веществ. Обработка пылесосом обеспечивает освобождение предметов от пыли и удаление с них значительного количества микроорганизмов. Установлено, что при 4-кратном проведении щеткой пылесоса по поверхности предмета с него удаляется примерно 47 % микроорганизмов, а при 12-кратном — до 97 %. В качестве дезинфицирующих растворов чаще всего пользуются 2 —3%-м раствором соды (бикарбоната натрия), 3 —5%-м водным раствором фенола (карболовой кислоты) или лизола (препарата фенола с добавлением зеленого мыла), 0,5 —3%-м водным раствором хлорамина и некоторыми другими дезинфектантами.
Воздух в лаборатории очищают проветриванием — это наиболее простой способ. Продолжительная вентиляция помещения через форточку (не менее 30 — 60 мин) резко снижает количество микроорганизмов в воздухе, особенно при значительной разнице в температуре между наружным воздухом и воздухом помещения. Более эффективный и наиболее часто применяемый способ дезинфекции воздуха — ультрафиолетовое облучение лучами с длиной волны от 260 нм. Эти лучи обладают высокой антимикробной активностью и могут вызывать гибель не только вегетативных клеток, но и спор микроорганизмов.
Воздействие ультрафиолетовых лучей должно быть непосредственным и длительным. Это связано прежде всего с тем, что ультрафиолетовые лучи обладают слабой проникающей способностью. Например, они не проходят через обычное стекло, легко поглощаются частицами пыли. Кроме того, листы белой бумаги, пластины алюминия и хрома, а также предметы, изготовленные из них, могут заметно отражать ультрафиолетовые лучи. Поэтому в зависимости от степени загрязнения воздуха для его стерилизации требуется облучение от 30 мин до нескольких часов.
В качестве источника ультрафиолетового излучения используются бактерицидные лампы. Излучателем в них служит электрическая дуга, возникающая в парах ртути низкого давления. Более 80 % испускаемого ими спектра приходится на волну длиной 254 нм. Обычно бактерицидные лампы представляют собой трубки различного диаметра и длины, изготовленные из специального стекла, пропускающего излучение с длиной волны 254 нм. Каждая трубка вмонтирована в корпус-держатель и может быть снабжена отражателем. Необходимо иметь в виду, что ультрафиолетовые лучи могут вызывать тяжелые поражения глаз, поэтому при работе с бактерицидными лампами нужно строго следить за тем, чтобы ни прямые, ни отраженные ультрафиолетовые лучи не попадали
в глаза. В небольших помещениях при включенной бактерицидной лампе находиться нельзя. Следует также учитывать, что при длительной непрерывной работе бактерицидной лампы интенсивность излучения снижается. В этих случаях облучение целесообразно вести с перерывами.
Рабочее место, где непосредственно работают с культурами микроорганизмов, требует особенно тщательной обработки. Рабочий стол следует дезинфицировать не только до начала работы, но и после ее окончания. Для протирания поверхности стола можно использовать растворы лизола и хлорамина, а также 70%-е (по объему) растворы изопропилового или этилового спиртов. Спирты весьма эффективны в отношении вегетативных форм микроорганизмов. Названные спирты можно также применять для дезинфекции рук. В тех случаях, когда поверхность стола имеет водоотталкивающее покрытие, особенно удобен лизол. Поверхность рабочего стола можно дезинфицировать и ультрафиолетовыми лучами. При этом следует учитывать, что бактерицидное действие лучей тем выше, чем ближе облучаемая поверхность к источнику излучения.
В лаборатории не разрешается курить, хранить и употреблять еду, напитки, жевательную резинку. Работать следует в халатах.
2.2. ПРАВИЛА РАБОТЫ С КУЛЬТУРАМИ МИКРООРГАНИЗМОВ
В лаборатории микроорганизмы выращивают на плотных и жидких питательных средах, которые разливают в пробирки, колбы, матрасы и чашки Петри (рис. 2.2). Посуду и питательные среды предварительно стерилизуют. Способы приготовления питательных сред и стерилизации подробно описаны ниже (см. гл. 3).
4 5 6
Рис. 2.2. Посуда для культивирования микроорганизмов:
1 — качалочная колба; 2 — качалочная колба с отбойниками; 3 — коническая колба; 4 — чашка Петри; 5 — пробирка; б — матрас
р
Рис. 2.3. Бактериологическая петля (7) и бактериологическая игла (2)
I Рис. 2.4. Значения температуры (°C)
41 Ч! в разных участках пламени газовой
1 2 горелки
Внесение микроорганизмов в стерильную среду называется посевом, или инокуляцией. Посев микроорганизмов требует соблюдения определенных правил, которые необходимо выполнять, чтобы предохранить исследуемую культуру от загрязнения посторонними микроорганизмами. Перед посевом следует тщательно надписать на пробирке (колбе или чашке Петри) название микроорганизма и дату посева.
Клетки микроорганизмов для посева или приготовления препаратов берут бактериологической петлей или иглой (рис. 2.3), если микроорганизмы выращены на плотной среде. В том случае, когда микроорганизмы выращены в жидкой питательной среде, лучше пользоваться не петлей, а стерильной пипеткой. Бактериологические петли и иглы делают, используя тонкую проволоку из вольфрама или нихрома, которую закрепляют в металлическом или стеклянном держателе. Диаметр бактериологической петли — 4 — 5 мм.
Бактериологическую петлю (иглу) перед взятием клеток микроорганизмов стерилизуют. Для этого проволоку накаливают докрасна в пламени горелки и одновременно обжигают примыкающую к петле часть держателя, которую будут вводить внутрь сосуда, содержащего микроорганизмы. Петлю рекомендуется держать в пламени горелки почти вертикально, чтобы проволока была равномерно раскалена на всем протяжении. При прокаливании необходимо помнить, что наивысшая температура развивается в верхней и периферической частях пламени (рис. 2.4), поэтому не следует опускать петлю непосредственно к горелке. Сразу же после стерилизации петлю (иглу) вводят в сосуд с микроорганизмами. Чтобы не повредить клетки микроорганизмов, петлю (иглу) вначале охлаждают, прикасаясь ею к внутренней поверхности сосуда или к питательной среде, свободной от клеток микроорганизмов, и только после этого захватывают небольшое количество микробной массы.
Отбор проб
Отбор клеток микроорганизмов, выращенных на плотной среде в пробирке, осуществляют следующим образом. Пробирку с культурой берут в левую руку так, чтобы поверхность питательной среды с налетом микроорганизмов была обращена кверху и
хорошо видна. Пробирку держат в горизонтальном или несколько наклонном положении. В правую руку берут петлю так, как держат карандаш, и прокаливают в пламени горелки. Затем, не выпуская петли, мизинцем и безымянным пальцем правой руки прижимают ватную пробку к ладони, вынимают ее из пробирки и держат так во время последующих манипуляций. Края открытой пробирки с культурой микроорганизмов обжигают в пламени горелки и после этого вводят в пробирку стерильную петлю. Взяв небольшое количество микробной массы с поверхности субстрата, вынимают петлю из пробирки, следя за тем, чтобы переносимый материал не касался стенок или краев пробирки. Горлышко пробирки снова обжигают в пламени горелки, затем обжигают ватную пробку и закрывают ею пробирку. Если конец ватной пробки загорится, то не следует бросать пробку. Ее нужно быстро ввести в пробирку, где вата сама потухнет.
Ни в коем случае нельзя дуть на загоревшуюся пробку, так как это только усилит горение'. Если в момент пересева ватная пробка упадет на пол или на стол, то не следует снова вставлять ее в пробирку. Нужно взять новую стерильную пробку и начать всю операцию заново. Закрытую ватной пробкой пробирку с культурой ставят в штатив, а извлеченный материал используюз для приготовления препарата или для пересева культуры в свежую среду.
Если культуру пересевают на скошенную агаризованную среду, то петлю вводят в пробирку до конца и, слегка касаясь ею поверхности агара, проводят снизу вверх либо зигзагообразную, либо прямую черту — штрих. При этом стараются не повредить поверхности плотной среды. В случае пересева в жидкую среду (в колбы или пробирки) петлю с микробной массой погружают непосредственно в среду. Оставшиеся на петле после пересева или приготовления препарата клетки микроорганизмов тщательно сжигают в пламени горелки. Прокаливание петли в этом случае начинают с участка проволоки, примыкающего к кольцу, для того чтобы микробная масса, оставшаяся на петле, подсохла. Затем петлю переводят в вертикальное положение и прокаливают докрасна. Такой порядок стерилизации петли необходим потому, что при быстром нагревании влажной микробной массы происходит ее разбрызгивание и образуется аэрозоль, загрязняющий воздух. Только после прокаливания петлю можно положить на место.
Из жидкой среды клетки берут пипеткой. Пипетку за верхний конец вынимают из бумаги или пенала, в которых ее стерилизовали, и вводят в пробирку или колбу с культурой, соблюдая все правила предосторожности, описанные выше. Отбирать жидкую культуру пипеткой можно с помощью резиновой груши. Использованную пипетку следует немедленно перенести в дезинфицирующий раствор, например 3 —5%-й водный раствор фенола или 2%-й раствор хлорамина, не касаясь ею окружающих предметов.
Когда необходимо провести рассев микроорганизмов из жидкой питательной среды на поверхность плотной среды в чашке Петри, поступают следующим образом. Расплавленную на кипящей водяной бане стерильную питательную среду, содержащую агар или желатину, разливают в стерильные чашки Петри. Для этого сосуд со средой берут в правую руку, вынимают из него пробку, зажимая ее мизинцем и безымянным пальцем левой руки, обжигают горло сосуда в пламени горелки и, приоткрыв большим и средним пальцами левой руки крышку чашки Петри, быстро наливают в чашку расплавленную среду в таком количестве (20 — 30 мл), чтобы дно чашки было полностью покрыто. Крышку тотчас закрывают и чашку оставляют на горизонтальной поверхности до тех пор, пока не застынет среда. Для посева приоткрывают крышку чашки Петри и на поверхность плотной среды наносят каплю или «петлю» жидкой культуры, которую осторожно распределяют стеклянным стерильным шпателем (шпатель Дри-гальского) либо петлей.
Все описанные манипуляции следует проводить около пламени горелки, но не в пламени, по возможности быстро, чтобы не загрязнить культуру посторонними микроорганизмами. Не рекомендуется делать резкие движения и ходить около работающего с чистой культурой, так как движение воздуха увеличивает вероятность случайного ее загрязнения.
После пересевов пробирки или другие сосуды, в которых выращивают микроорганизмы, помещают в термостаты, где с помощью терморегуляторов (см. гл. 3) поддерживается постоянная температура.
Посуду с культурами микроорганизмов, подлежащими выбрасыванию, следует автоклавировать, чтобы убить клетки, и только после этого мыть. Культуры на плотных питательных средах можно заливать на сутки дезинфицирующим раствором, после чего их выбрасывают и посуду моют. Неаккуратное обращение с культурами микроорганизмов приводит к возникновению бактериального аэрозоля.
2.3. ВЕДЕНИЕ ЛАБОРАТОРНЫХ ЗАПИСЕЙ
Журнал лабораторных работ является документом, позволяющим контролировать правильность полученных результатов. В нем должны быть записаны сведения, имеющие отношение к выполнению данной работы. Запись необходимо вести четко, аккуратно и в определенном порядке, например:
1. Название опыта и его цель, дата постановки и окончания.
2. Объект исследования.
3. Условия проведения опыта.
4. Основной принцип используемого метода анализа.
5. Полученные результаты.
Цифровой материал приводят в таблицах. Если необходимо, делают графики, диаграммы, рисунки. Каждая лабораторная работа должна заканчиваться собственными наблюдениями и выводами, записанными в журнале. Журнал является собственностью лаборатории, в которой проводилась работа, и всегда хранится в лаборатории.
РАЗДЕЛ II
ПРИНЦИПЫ ВЫДЕЛЕНИЯ И ИЗУЧЕНИЯ МИКРООРГАНИЗМОВ
Глава 3
СОСТАВЛЕНИЕ СРЕД И КУЛЬТИВИРОВАНИЕ МИКРООРГАНИЗМОВ
Культивирование микроорганизмов является одним из основных методов микробиологии. От умения культивировать микроорганизмы в лабораторных условиях в значительной степени зависят успехи их изучения и практического применения. Культивирование основано на знании метаболизма микроорганизмов и понимании значения физико-химических условий среды, необходимых для их жизнедеятельности.
3.1. ПИТАТЕЛЬНЫЕ СРЕДЫ ДЛЯ КУЛЬТИВИРОВАНИЯ МИКРООРГАНИЗМОВ
Потребности микроорганизмов в питательных веществах чрезвычайно разнообразны и определяются особенностями их метаболизма. Во-первых, питательная среда должна включать доступный для клетки источник энергии. Для одних организмов (фототрофов) таким источником служит свет, для других — органический (хемоорганотрофы) или неорганический (хемолитотрофы) субстрат. Во-вторых, среда должна содержать все необходимые компоненты для реализации конструктивных процессов в клетке, причем синтетические способности микроорганизмов могут варьировать от использования СО2 в качестве единственного источника углерода (автотрофы) до потребности в более восстановленных соединениях углерода — кислотах, спиртах, углеводах и др. (гетеротрофы). Последние чаще всего получают и углерод, и энергию, метаболизируя одно и то же органическое соединение, т.е., строго говоря, являются хемоорганогетеротрофами.
По способности использовать те или иные источники азота микроорганизмы также существенно отличаются друг от друга. Одни из них довольствуются молекулярным азотом воздуха и не требуют добавления в среду каких-либо азотсодержащих соединений. Другие требуют присутствия в среде неорганических солей азота. Третьим нужна одна или несколько аминокислот. Отдельные аминокислоты в L- или ДД-форме добавляют к стерильной среде в концентрации от 0,1 до 0,05 г на 100 мл непосредственно перед засевом ее микроорганизмами. Для этого рекомендуется использовать растворы аминокислот, в
которых концентрация превышает содержание аминокислоты в среде в 100 раз. Глицин, аланин, пролин, лизин и орнитин растворяют в дистиллированной воде, фенилаланин и триптофан — в дистиллированной воде, подщелоченной NaOH, остальные аминокислоты — в дистиллированной воде, подкисленной НС1. Аминокислоты — цистин и цистеин, а также амиды — глутамин и аспарагин неустойчивы к нагреванию, поэтому их стерилизуют фильтрованием. Остальные аминокислоты можно стерилизовать при 0,5 ати* в течение 15 мин.
Потребности микроорганизмов в некоторых аминокислотах часто удовлетворяют, добавляя к среде гидролизат белка. Состав гидролизатов неодинаков и зависит от исходного субстрата, а также способа получения. Чаще других используют гидролизат казеина, который готовят в лаборатории, как правило, кислотным гидролизом, еще чаще используют готовый препарат гидролизата казеина или готовят его в лаборатории (см. приложение 4). Его вносят в среду от 1,0 до 0,1 г на 100 мл в зависимости от потребности микроорганизмов.
Наиболее требовательные организмы культивируют на средах, содержащих белки или продукты их неполного расщепления — пептоны, представляющие собой смесь поли- и олигопептидов, аминокислот, органических азотных оснований, солей и микроэлементов. Пептоны получают в результате воздействия протеолитических ферментов на белки животного или растительного происхождения. В питательные среды пептон добавляют в количестве от 1 — 2 до 20 г на 1 л.
Необходимо иметь в виду, что аминокислоты и пептон могут использоваться микроорганизмами не только как источник азота, но и как источник углерода и энергии.
Многие микроорганизмы требуют наличия в среде так называемых факторов роста, к которым относятся пурины, пиримидины и аминокислоты. Чтобы подчеркнуть потребность микроорганизмов в факторах роста, принято использовать термины «прототрофы» и «ауксотрофы». Прототрофы не нуждаются в факторах роста, для ауксотрофов абсолютно необходимо наличие в среде одного или нескольких факторов роста. Этим термином особенно широко пользуются в литературе по генетике. Если потребности микроорганизмов в факторах роста ограничены одним или несколькими витаминами, то рекомендуется вносить их в культуральные среды, используя следующие концентрации: тиамин (витамин В,), пантотенат Са, рибофлавин (витамин В2), никотиновая кислота (ниацин), пиридоксин, пиридоксамин, холин, кобаламин (витамин В12) — по 1 мкг на 1 мл среды; фолиевая кислота и пара-аминобензойная кислота — 0,05 мкг на 1 мл среды; биотин — 0,005 мкг на 1 мл среды.
Витамины добавляют к стерильной среде непосредственно перед ее засевом. Для этого рекомендуется использовать растворы, в которых концентрация витамина превышает его содержание в среде в 100 раз. Растворы готовят в стерильной посуде и используют стерильную дистиллированную воду. Исключение составляют рибофлавин и фолиевая кислота. Рибофлавин растворяют в 0,02 н. уксусной кислоте, а фолиевую кислоту — в 0,01 н. NaOH, доводя затем концентрацию NaON в растворе до 0,001 н. Полученные растворы стерилизуют прогреванием в кипящей водяной бане 3 мин. Раствор тиамина рекомендуется стерилизовать фильтрованием, так как при нагревании тиамин разрушается. При
* «ати» — атмосферное давление избыточное.
температуре 4 °C растворы витаминов сохраняются не менее месяца. Растворы фолиевой кислоты, пиридоксина и рибофлавина хранят в темноте, так как они чувствительны к свету.
Примерами смесей, содержащих различные факторы роста, могут служить дрожжевой экстракт, дрожжевой автолизат, а также кукурузный экстракт. Дрожжевой экстракт вносят в среду для культивирования от 0,05 до 0,5 г на 100 мл, дрожжевой автолизат — в таком количестве, чтобы концентрация аминного азота составляла 5 — 30 мг на 100 мл среды. Дрожжевой экстракт имеется в продаже. Дрожжевой автолизат легко приготовить в лабораторных условиях (см. приложение 4).
Кукурузный экстракт — готовый продукт заводов крахмалопаточной промышленности. Он содержит аминокислоты, витамины, большое количество органических кислот (молочной, уксусной и муравьиной) и минеральные соли. Кукурузный экстракт вносят в среды в количестве от 0,2 до 5%; стерилизуют при 0,5 ати.
Для построения вещества клетки микроорганизмам необходимы фосфор, сера, калий и ряд других элементов. Они должны содержаться в питательной среде в доступной для микроорганизмов форме. Потребности разных групп микроорганизмов в сере, фосфоре и других зольных элементах удовлетворяются обычно за счет минеральных солей. Поэтому «минеральный фон» сред для культивирования многих микроорганизмов может быть близким по составу. Так, потребности значительного числа микроорганизмов в сере удовлетворяются сульфатами, хотя в клетке сера находится в основном в восстановленной форме, в виде сульфгидрильных групп. Значительно реже встречаются микроорганизмы, требующие наличия в среде восстановленной серы. В этом случае в среду вносят сульфиды, чаще всего Na2S, или органические соединения, содержащие сульфгидрильные группы, например цистеин.
Соли фосфорной кислоты удовлетворяют потребности микроорганизмов в фосфоре.
Микроорганизмы все необходимые металлы — К, Na, Са, Mg, Fe, Мп, Со, Си — и другие элементы получают в форме катионов или анионов неорганических солей. Например, источником магния служит MgSO4, источником натрия и хлора — NaCl, кальция — СаСО3 или СаС12. Железо вносят в среды в виде хлорида, сульфата или цитрата.
Чтобы избежать выпадения осадка в результате образования нерастворимых комплексов фосфатов с некоторыми катионами, особенно с железом или кальцием, к средам рекомендуется добавлять от 0,001 до 1 г/л этилендиаминтетраацетата (ЭДТА) или гексаметафосфата натрия в концентрации 4 г/л. Комплексы, образуемые этими соединениями с катионами, служат резервом, из которого в результате диссоциации поступают свободные катионы.
Калий, магний, кальций и железо требуются в относительно больших количествах, поэтому их соли, как правило, включаются в состав питательных сред. Потребности микроорганизмов в марганце, молибдене, цинке, меде, кобальте и др. очень малы. Эти элементы часто называют микроэлементами, их вносят в среды строго определенного состава, приготовленные на дистиллированной воде в количестве от 1 мг до 1 мкг на I л. Об оптимальных концентрациях микроэлементов для разных микроорганизмов известно мало, поэтому предложены различные по составу смеси микроэлементов. Растворы микро-
2 Нетрусов
33
элементов рекомендуется стерилизовать отдельно и вносить в среду непосредственно перед посевом. Среды, приготовленные на водопроводной воде, а также среды, включающие натуральные компоненты, содержат необходимые микроэлементы и не требуют их специального добавления.
По составу среды для культивирования микроорганизмов делят на натуральные (естественные) и синтетические. К натуральным относятся среды, состоящие из продуктов животного или растительного происхождения: овощные или фруктовые соки, молоко, животные ткани, разведенная кровь, вода морей, озер, минеральных источников, а также отвары или экстракты, полученные из природных субстратов — мяса, рыбы, дрожжей, растений, круп, навоза и почвы. К натуральным относятся также и так называемые полусинтетиче-ские среды, состоящие из природных продуктов в комбинации с рядом определенных химических соединений. Натуральные среды богаты органическими веществами, однако имеют сложный и непостоянный состав, поэтому они малопригодны для исследования обмена веществ микроорганизмов. Синтетические среды представляют собой комплекс определенных химически чистых соединений, взятых в точно указанных концентрациях. Синтетические среды наиболее удобны для изучения метаболизма микроорганизмов. Состав синтетических сред может варьировать настолько широко, насколько широко изменяются пищевые потребности культивируемых на них микроорганизмов. В зависимости от задачи исследования синтетические среды готовят на водопроводной или дистиллированной воде. Синтетические среды могут иметь довольно большой набор компонентов (включая микроэлементы), но могут быть и относительно простыми по составу. Рецепты некоторых синтетических сред и способы приготовления натуральных приведены в приложении 4.
По назначению среды подразделяют на элективные и дифференциально-диагностические (индикаторные). Элективные среды обеспечивают преимущественное развитие одного вида или группы микроорганизмов и менее пригодны или даже совсем не пригодны для развития других. Элективные среды применяют главным образом на первом этапе выделения микроорганизмов из естественных субстратов, т.е. для получения накопительных культур (например, среда Эшби является элективной для рода Azotobacter).
Дифференциально-диагностические среды дают возможность быстро отличить одни виды микроорганизмов от других или выявить некоторые их особенности. Примером индикаторной среды для выявления бактерий из группы кишечной палочки в естественных субстратах может служить агаризованная среда Эндо. Состав этой среды приводится в приложении 4. Бактерии рода Escherichia на этой среде образуют малиновые колонии с металлическим блеском. При определении видовой принадлежности бактерий часто используют pH-индикаторные среды, в состав которых входит один из индикаторов — нейтральный красный (0,0005 %), феноловый красный (0,005 %) или бромтимоловый синий (0,0005 %). Если развитие микроорганизма сопровождается образованием кислоты или щелочи, цвет индикатора меняется. Индикаторные среды особенно часто применяются в санитарной и медицинской микробиологии.
По физическому состоянию среды могут быть жидкими, полужидкими, твердыми (плотными) и сыпучими. Жидкие среды широко применяют для выявления физиолого-биохимических особенностей микроорганизмов, накопления биомассы или продуктов обмена, а также поддержания и хранения многих микроорганизмов, плохо развивающихся на плотных средах. Полужидкие среды получают
добавлением к жидким средам 0,5 % агара и используют для специальных целей, например культивирования анаэробных микроорганизмов. Сыпучие (сухие) питательные среды приобретают все большее практическое значение. Стандартность, простота хранения, транспортировки и изготовления делают их весьма удобными для работы. В микробиологическом производстве используются разваренное пшено, отруби. В клинической микробиологии нашли применение дифференциально-диагностические среды — среда Эндо, сухая желчь, среда Леффлера и т.д. Они представляют собой светло-желтые гигроскопичные порошки без комков с влажностью до 10 %. В сухом месте в хорошо закупоренной посуде их можно хранить длительное время. Они хорошо растворяются в воде при комнатной температуре в концентрации 1,5 —6,0 %. Плотные среды используют в микробиологической практике со времен Р. Коха. Они необходимы для выделения чистых культур микроорганизмов, в диагностических целях, для количественного учета микроорганизмов, хранения культур и в ряде других случаев. Уплотняющими агентами являются агар, желатин, силикагель.
Агар используют для уплотнения особенно часто. Он представляет собой сложный полисахарид, в состав которого входят агароза и агаропектин. Кроме того, агар включает небольшое количество легкоассимилируемых веществ и различные соли. Агар получают из некоторых морских водорослей и выпускают в виде стебельков, пластин или порошка. Агар удобен тем, что большинство микроорганизмов не используют его в качестве субстрата для роста. В воде он образует гель, который плавится примерно при 100°С и затвердевает при температуре около 45 °C, поэтому на агаризованных средах можно культивировать значительную часть известных микроорганизмов. Чаще всего агар добавляют к средам в количестве 2 %, реже — 1,5 (более влажная среда) или 3 % (более сухая среда). Необходимо учитывать, что агар сохраняет свои свойства только при pH, близком к нейтральному. Агар всегда содержит органические и неорганические примеси, поэтому для приготовления сред строго определенного состава агар предварительно очищают, «выщелачивают». Экстракцию примесей проводят в течение 2 — 3 нед водопроводной водой при 30 — 37 °C со сменой воды через 1 — 2 сут. Процедуру прекращают, когда вода перестанет мутнеть. «Выщелоченный» агар помещают в двойной марлевый мешок и 2 — 3 сут промывают в проточной водопроводной воде. Затем агар высушивают и используют в количестве 1,5 —2,0% (табл. 3.1).
Желатин — белок, получаемый из костей, хрящей и кожи животных. Образуемый им гель плавится при температуре 24—26 °C, которая ниже обычной температуры инкубации многих микроорганизмов (30 — 37 °C). К тому же многие микроорганизмы образуют протеолитические ферменты, разлагающие желатин. Поэтому желатин главным образом используют для определения протеолитической активности микроорганизмов с целью их идентификации (табл. 3.1).
Кремнекислый гель (силикагель) применяют как твердую основу для синтетических сред строго определенного состава, поскольку он является веществом неорганической природы. Для получения силикагеля к соляной кислоте (плотность 1,1) добавляют при перемешивании равный объем жидкого стекла (раствор Na2SiOj или K2S1O3 той же плотности). Смесь разливают по чашкам Петри по 25 — 30 мл в каждую и оставляют в покое для образования геля. Затем чашки помещают в стеклянный сосуд и промывают гель проточной водой для удаления хлоридов. Наличие хлоридов проверяют качественной реакцией с AgNOj. Завершают процедуру промыванием силикагеля горячей дистиллированной водой (табл. 3.1).
В качестве уплотняющего средства в микробиологическую практику входит каррагинан — подобный агару полисахарид, экстрагируемый из красных водорослей. Каррагинан не используется большинством видов микроорганизмов. Для получения устойчивого при 45 и 60 °C геля используют соответственно 2,0 и 2,4 % каррагинана.
Таблица 3.1
Основные характеристики веществ, употребляемых для уплотнения питательных сред
Характеристика Агар Желатин Кремнекислый гель
Исходный материал для получения Морские водоросли Кожа, кости, хрящи Кремнекислый калий или натрий и соляная кислота
Основные компоненты Полисахариды Белки Кремневая кислота
Температура плавления, °C « 100 22-25 —
Температура застывания, °C ~45 22-25 —
Действие протеаз Не действуют Действуют Не действуют
Конденсационная вода Выделяется Не выделяется Не выделяется
Употребляемая концентрация, % 1,0-3,0 10-20 —
Для некоторых специальных исследований, например для получения хорошо видимых в толще среды изолированных колоний анаэробных микроорганизмов, применяют осветленные среды. Прозрачные среды получают несколькими способами. Наиболее простой способ осветления — фильтрование среды через ватный фильтр. Если этого бывает недостаточно, среды осветляют с помощью белков куриного яйца. Для осветления 500 мл среды достаточно одного яйца. Белок отделяют от желтка и встряхивают с равным объемом воды до образования сплошной пены. Взбитый белок выливают в предварительно расплавленную и остуженную до 45 — 50 °C среду. Перед внесением белка проверяют pH и, если необходимо, доводят его до 7,0 —7,3. Среду с белком тщательно перемешивают и прогревают при 100 °C в автоклаве в течение часа. Белок свертывается и адсорбирует все взвешенные в среде частицы. Когда свернувшийся белок поднимается на поверхность или опускается вниз, среду быстро отфильтровывают в горячем виде через вату. При этом удобно пользоваться специально подогреваемыми подставками для воронок, благодаря которым предотвращается застывание среды во время фильтрования.
Синтетические агаризованные среды, в которые вносить белок нежелательно, осветляют следующим образом. Среду наливают в химический стакан, автоклавируют и оставляют после стерилизации в закрытом автоклаве на 10 —12 ч, обычно на ночь. При таком медленном остывании все взвешенные частицы оседают на дно. Застывшую ага-ризованную среду извлекают из стакана, верхнюю (прозрачную часть) срезают, помещают в колбу и вновь стерилизуют.
3.2. УСЛОВИЯ КУЛЬТИВИРОВАНИЯ МИКРООРГАНИЗМОВ
Для роста микроорганизмов существенное значение имеют не только состав питательной среды, но и такие факторы, как кислотность среды, аэрация, температура, свет, влажность. Развитие микроорганизмов возможно лишь в определенных пределах каждого фактора, причем для различных групп микроорганизмов эти пределы часто неодинаковы.
Активная кислотность среды. Активная кислотность (pH) среды имеет решающее значение для роста многих микроорганизмов. Большинство бактерий лучше всего растет при pH, близком к 7,0, микроскопические грибы, напротив, предпочитают слабокислые среды, поэтому в приготовленных средах всегда следует определить значение pH. Измеряют pH электрометрическим методом на потенциометре. В лабораторной практике удобно использовать различные жидкие или бумажные индикаторы (см. приложение 5).
Широко применяется, например, жидкий двуцветный индикатор, бромтимоловый синий (бромтимолблау). Его цвет изменяется от желтого к синему при сдвиге pH от 6,0 до 7,6. При pH 7,3 индикатор имеет сине-зеленую окраску.
В случае необходимости pH среды доводят до нужного значения растворами кислот (НС1, H2SO4), щелочей (NaOH, КОН) или солей, имеющих щелочную реакцию (Na2CO3, NaHCO3). Для корректировки pH целесообразно иметь стерильные растворы разной концентрации.
Значение pH сред может измениться в процессе стерилизации, поэтому после стерилизации его следует проверить и, если требуется, довести до нужного с помощью стерильных растворов кислоты или щелочи. Активная кислотность питательной среды, благоприятная для начала роста микроорганизмов, часто меняется в процессе их культивирования. Эти изменения могут быть результатом образования продуктов метаболизма или неравномерного потребления отдельных компонентов среды.
Например, при потреблении углеводов в среде накапливаются органические кислоты, снижающие pH среды. В средах с KNO3, как уже отмечалось, pH возрастает за счет более интенсивного потребления нитрат-иона и накопления ионов калия.
Чтобы не допустить чрезмерного изменения pH в культурах микроорганизмов и удержать его на необходимом уровне, используют различные приемы. Иногда в среды добавляют буферные растворы (см. приложение 6). В микробиологической практике чаще других применяют фосфатные буферы. Однако если рост микроорганизмов сопровождается образованием большого количества кислот, то тех количеств буферного раствора, которые можно добавлять к средам (не более 5 г фосфатов на 1 л среды), оказывается недостаточно, так как противодействие любого буфера изменению pH ограничено. В связи с этим для микроорганизмов, активно изменяющих кислотность среды, применение буферов не эффективно. При культивировании таких микроорганизмов в среды вводят избыточное количество мела, который нейтрализует образующиеся кислоты. Для нейтрализации кислот по ходу развития культуры можно применять 10%-й стерильный раствор NaHCO3.
Поддержание определенного значения pH во время роста особенно важно для тех микроорганизмов, которые в процессе жизнедеятельности образуют кислоты, но не обладают устойчивостью к ним. К их числу относятся молочнокислые бактерии, а также многие псевдомонады. Большие затруднения встречаются, когда нужно поддержать pH в слабощелочных средах, так как для диапазона pH от 7,2 до 8,5 подходящих буферов не существует. С этой целью иногда приходится периодически или непрерывно доводить pH до нужной величины, добавляя стерильно в среду растворы кислоты или щелочи при постоянном контроле значения pH. В современных ферментерах это достигается с помощью специальных автоматических устройств.
Аэрация. Кислород входит в состав воды и многих соединений, поэтому поступает в клетки в больших количествах. Значительная часть микроорганизмов нуждается в постоянном притоке молекулярного кислорода. Такие микроорганизмы принято объединять в группу облигатных аэробов. Энергетическим процессом у них является аэробное дыхание, а молекулярный кислород играет роль терминального окислителя.
Среди облигатных аэробов выделяют группу микроаэрофильных микроорганизмов, которые нуждаются в кислороде, но лучше растут, если его парциальное давление меньше, чем в воздухе. Развитие других микроорганизмов, напротив, возможно только в отсутствие кислорода. Получение энергии у них не связано с использованием молекулярного кислорода, а для многих кислород даже токсичен: он угнетает рост или вызывает гибель клеток. Такие микроорганизмы называются облигатными анаэробами. Факультативные анаэробы способны расти как в присутствии, так и в отсутствие молекулярного кислорода. Например, некоторые дрожжи и энтеробактерии в зависимости от наличия кислорода осуществляют аэробное либо дыхание, либо брожение. Неодинаковые потребности микроорганизмов в свободном кислороде учитывают при выборе способа их выращивания.
Культивирование аэробных микроорганизмов. Аэробные микроорганизмы можно выращивать на поверхности жидких и плотных сред — поверхностное культивирование или внутри жидкой питательной среды — глубинное культивирование. При поверхностном культивировании кислород поступает в клетки микроорганизмов непосредственно из воздуха, поэтому важно увеличить площадь соприкосновения среды с воздухом. Для этого среды наливают тонким слоем в посуду с широким дном — чашки Петри, колбы Виноградского, матрасы (см. рис. 2.2).
Растущие в жидких средах аэробные микроорганизмы часто образуют на поверхности пленку. Факультативные анаэробы развиваются не только на поверхности, но и в толще жидкой среды, вызывая более или менее равномерное ее помутнение. Поверхностное культивирование применяется как в лабораторных условиях, так и в промышленности.
Все способы глубинного культивирования аэробных микроорганизмов сводятся к увеличению поверхности соприкосновения питательной среды с кислородом воздуха. Следует иметь в виду, что при глубинном культивировании в жидких средах микроорганизмы используют растворенный кислород. Вместе с тем растворимость кислорода в воде невелика, поэтому, чтобы обеспечить рост аэробных микроорганизмов в толще среды, ее необходимо постоянно аэрировать.
При аэрации среды учитывают два показателя: интенсивность и степень аэрации. Интенсивность аэрации характеризуется скоростью растворения кислорода в единице объема культуральной жидкости за единицу времени. Степень аэрации — это объем воздуха, продуваемый через определенный объем среды за единицу времени.
Наиболее простой и широко распространенный в лабораторной практике способ глубинного культивирования — выращивание на качалках, обеспечивающих встряхивание или вращение колб и пробирок с 100 — 200 об/мин и более. Чем больше частота вращения, тем больше соприкосновение среды с воздухом и выше насыщение ее кислородом. Увеличить аэрацию среды при работе
на одной и той же качалке можно за счет уменьшения объема среды или применения колб с отбойниками — вдавливаниями стекла внутрь в виде 4 — 8 отростков длиной 2 — 3 см (см. рис. 2.2). При вращении колб с отбойниками поверхность соприкосновения среды с воздухом заметно увеличивается благодаря разбрызгиванию жидкости. Чем больше отбойников, тем сильнее разбрызгивание жидкости, тем выше аэрация.
Интенсивность аэрации при выращивании микроорганизмов на качалке косвенно характеризуют сульфитным числом. В основе метода лежит определение скорости окисления сульфита в сульфат в присутствии меди как катализатора.
Сульфит, оставшийся неокисленным, связывают йодом. Количество йода, не вступившего в реакцию с сульфитом, оттитровывают тиосульфатом. Таким образом выясняют, сколько сульфита в определенном объеме его раствора за определенное время окисляется кислородом воздуха. Метод дает лишь сравнительные данные о скорости поглощения кислорода раствором сульфита в условиях аэрации, аналогичных опытным.
Определение сульфитного числа. Готовят следующие реактивы: 0,1 н. раствор тиосульфата (Na2S2O3) с установленным титром; 1%-й раствор крахмала; 0,1 н. свежеприготовленный раствор йода; 0,8 н. свежеприготовленный раствор сульфита Na2SO3 с CuSO4 Количество сульфата меди в растворе сульфита натрия соответствует 0,001 М. Сернокислую медь растворяют отдельно в небольшом объеме воды, а затем по расчету вносят в раствор сульфита.
Следует выяснить, какое количество йода полностью связывает количество взятого для работы сульфита. Для этого к 5 мл свежеприготовленного раствора сульфита в конической колбе на 250 мл приливают из бюретки раствор йода до появления светло-коричневой окраски. Так определяют объем раствора йода а. Затем добавляют 0,5 мл 1%-го раствора крахмала и титруют 0,1 н. раствором тиосульфата до исчезновения синей окраски. На титрование должно пойти 1 — 3 мл тиосульфата. Далее проверяют правильность определения объема йода и титруют его тиосульфатом, так же, как в предыдущем случае. Если объем йода подобран правильно, то на его титрование идет около 30 — 40 мл тиосульфата.
Для определения сульфитного числа используют конические колбы объемом 750 мл без отбойников и с отбойниками. Сульфит в колбы наливают в количестве 300 мл, 100 мл (колбы без отбойников) и 100 мл (колба с отбойниками). Затем из каждой колбы отбирают по 5 мл сульфита в конические колбы на 250 мл, в которые заранее наливают по а мл раствора йода. Колбы закрывают часовыми стеклами и оставляют в темноте на 5—10 мин, затем споласкивают часовые стекла дистиллированной водой в ту же колбу, которую они закрывали, и титруют ее содержимое 0,1 н. тиосульфатом. Количество миллилитров тиосульфата, пошедшего на титрование, П,„ записывают в таблицу.
Колбы с разными объемами сульфита помещают на определенное время на качалку. Время включения и выключения качалки замечают с точностью до 1 мин. К моменту остановки качалки через 15 — 20 мин в три колбы на 100 мл наливают подобранный для работы раствор йода а. Остановив качалку, отбирают из встряхиваемых колб по 5 мл раствора сульфита и сразу же приливают пробы к раствору йода. Снова включают качалку еще на 20 — 25 мин. Отобранные пробы выдерживают в темноте и снова титруют 0,1 н. раствором тиосульфата, как указано выше. Количество миллилитров тиосульфата 77Д| записывают в таблицу. В ту же таблицу вносят результаты Пд.2, полученные титрованием пробы сульфита, отобранной через последующие 20 — 25 мин. Рекомендуемая продолжительность встряхивания колб с сульфитом подобрана специально для условий настоящего опыта. В других случаях она может быть иной.
Рис. 3.1. Схема ферментера для глубинного культивирования аэробных микроорганизмов:
1 — вход воздуха; 2 — выход воздуха; 3 — барботер; 4 — отбойники; 5 — мешалка
Сульфит при аэрации окисляется в сульфат, поэтому на его связывание требуется все меньше йода и соответственно все больше тиосульфата идет на титрование свободного йода. Данные титрования, близкие к данным титрования йода без сульфита, для расчетов не используют, так как они свидетельствуют о полном окислении последнего к моменту отбора проб, и результаты определения будут ошибочными.
Сульфитное число рассчитывают следующим образом:
скорость поступления кислорода в раствор сульфита (мг О2/л/мин) = [(Лх-П„)/Т] • (0,8«), где П„ — количество 0,1 н. раствора тиосульфата (мл), пошедшее на титрование 5 мл раствора сульфата до аэрации; Пх — количество 0,1 н. раствора тиосульфата (мл), пошедшее на титрование 5 мл раствора сульфита после аэрации; Т — время аэрации в минутах; 0,8 — количество кислорода (мг), идущее на окисление 1 мл 0,1 н. раствора тиосульфата; а — коэффициент пересчета на 1 л сульфита (в данном случае он равен 200, так как на титрование берут 5 мл сульфита). Для каждой колбы рассчитывают сульфитные числа после первого и второго периодов аэрации и берут среднее из двух чисел или более достоверное число.
Для аэрации культуры микроорганизмов помимо перемешивания можно применять продувание стерильного воздуха через толщу среды. Этот способ часто используют в лабораторных исследованиях, но особенно широкое применение он нашел в промышленной микробиологии при получении биомассы и различных продуктов жизнедеятельности микроорганизмов — антибиотиков, ферментов, кислот. Скорость потока воздуха через среду необходимо контролировать. Для этого используют различные приборы — газовые часы, ратометры и др. В ферментерах количество пропускаемого воздуха поддерживают на заданном уровне автоматически. Воздух стерилизуют путем прохождения через активированный древесный, уголь, стеклянную вату, пропитанную антисептиком, или специальные ткани из полимеров. В лабораторных опытах, когда объем и скорость поступления воздуха невелики, используют заранее простерилизованные ватные фильтры. Для возможно более сильного распыления воздух пропускают через мелкопористые пластинки — барботеры; в лабораторных опытах с этой целью применяют стеклянные фильтры. Для аэрации культур микроорганизмов, как правило, используют обычный воздух.
Продувание сред кислородом в лабораторных условиях не рекомендуется, так как чрезмерное насыщение среды кислородом (до 40 мг/л) может привести к угнетению роста микроорганизмов. В ферментерах принудительную аэрацию обычно совмещают с механическим перемешиванием среды мешалками, скорость вращения которых может достигать сотен и даже тысяч оборотов в минуту. Схема ферментера для глубинного культивирования аэробных микроорганизмов приведена на рис. 3.1.
Культивирование анаэробных микроорганизмов более сложно, чем культивирование аэробов, так как соприкосновение клеток анаэробов с кислородом воздуха должно быть сведено к минимуму или даже полностью исключено. Для этого используют разные приемы, нередко комбинируя их друг с другом.
Выращивание в высоком слое среды — наиболее простой способ ограничения воздуха к клеткам микроорганизмов. Жидкую среду наливают в сосуды для культивирования высоким слоем. Поскольку нельзя стерилизовать среды, если они занимают более половины высоты сосуда, часть среды стерилизуют отдельно и стерильно доливают ею сосуд для культивирования сразу после посева. Непосредственно перед посевом среду кипятят или прогревают на кипящей водяной бане 30—40 мин, затем быстро охлаждают, чтобы в ней не успел раствориться кислород воздуха, и вносят на дно посевной материал.
Иногда используют метод культивирования в вязких средах. Диффузия кислорода в жидкость уменьшается с увеличением ее вязкости, поэтому в вязких средах, таких, как картофельная или среды с кукурузной либо другой мукой, хорошо развиваются некоторые облигатные анаэробы, например возбудители маслянокислого или ацетонобутилового брожения. Вязкость сред легко увеличить добавлением к ним 0,2—0,3 % агара.
Для получения изолированных колоний при выделении чистых культур или определении их численности анаэробные микроорганизмы выращивают в толще питательной среды. Для этого посевной материал вносят в расплавленную и остуженную до 48 — 50 °C агаризованную, желательно осветленную среду, тщательно перемешивают и оставляют в пробирках или переливают стерильной пипеткой в заранее простерилизованные трубки Бурри или чашки Петри. Поверхность среды в пробирках заливают парафином. Трубки Бурри — это стеклянные трубки длиной 20—25 см, диаметром 1,0—1,5 см. Трубки стерилизуют, закрыв оба конца ватными пробками. Перед посевом ватную пробку у одного конца заменяют стерильной резиновой, через другой конец трубки вносят среду с посевным материалом и закрывают также резиновой пробкой (рис. 3.2). При использовании чашек Петри для выращивания анаэробов засеянную агаризованную среду наливают в крышку чашки и, после того как среда застынет, плотно прижимают к ее поверхности дно чашки. Зазор между стенками дна и крышки, где среда соприкасается с воздухом, заливают стерильным парафином (рис. 3.3).
Анаэробные микроорганизмы можно выращивать в анаэростатах — вакуумных металлических камерах, снабженных манометром. Анаэростатом может служить обычный вакуумный стеклянный эксикатор. Из анаэростата откачивают воздух, а затем, как правило, заполняют его газовой смесью, состоящей из
Рис. 3.2. Трубка Бурри:
7 — трубка, подготовленная к стерилизации; 2 — колонии анаэробных микроорганизмов в толще агаризованной среды
Рис. 3.3. Культивирование анаэробов в чашке Петри:
7 — агаризованная среда; 2 — парафин
азота (90 — 80%) и углекислоты (10—20%), до давления порядка 67- 103 Па (0,7 ати). Избыточное давление исключает возможность диффузии кислорода воздуха.
Некоторые строгие анаэробы, например апетогенные бактерии, метаногены, некоторые грибы, расщепляющие целлюлозу, микроорганизмы рубца жвачных животных, погибают даже при кратковременном контакте с кислородом воздуха. При культивировании таких микроорганизмов следует применять методы, позволяющие исключить молекулярный кислород из сред культивирования, создавать и поддерживать в них низкий окислительно-восстановительный потенциал {Eh). Степень поглощения кислорода и восстановленно-сти среды определяется Eh, который может быть измерен электрометрически на потенциометре или с помощью индикаторных красителей, например реза-зурина, феносафранина, нейтрального красного и т.д., изменяющих окраску при изменении Eh. Особенно удобен резазурин, который добавляют к средам в концентрации 1 мг/л и стерилизуют вместе с минеральными компонентами среды. В окисленной форме он окрашен в голубой цвет, при восстановлении цвет меняется на слабо-розовый, полностью восстановленная форма бесцветна. Резазурин изменяет окраску при значениях Eh, близких к 420 мВ, феносафранин — в области 252 мВ.
Принципы культивирования строгих анаэробов, разработанные профессором Р. Хан-гейтом (США), состоят в следующем: 1) все процедуры — приготовление и разлив сред, посевы, культивирование микроорганизмов и т.д., проводят с максимальной защищенностью от контакта с кислородом (анаэробный бокс, кипячение, работа в токе стерильного газа, использование герметически закрытых сосудов и т.д.); 2) до посева микроорганизмов в среды добавляют восстановители (цистеин, сульфид натрия, тиогликолат в щелочной среде) для химического поглощения следов кислорода, диффундирующего в сосуды для культивирования даже через резиновые пробки; 3) все пересевы и добавки в среды проводят с помощью шприцев, продутых газом, не содержащим кислород; 4) все трубки и шланги, используемые для пропускания газа, должны быть изготовлены из материала, не допускающего (медь) или допускающего в слабой степени диффузию сквозь него кислорода воздуха (бутилированная резина).
В качестве поглотителя кислорода из газов в лабораторной практике используют щелочные растворы пирогаллола или дитионита натрия, металлическое железо, медь, нагретую до 400 °C, или некоторые другие реактивы. При этом необходимо учитывать поглощающую способность реактивов и объем замкнутого пространства, в котором выращивают микроорганизм. Например, на каждые 100 мл емкости используют 1 г пирогаллола и 10 мл 2,5 н. раствора гидроксида натрия. Поскольку многие анаэробы нуждаются в углекислоте, пирогаллол часто растворяют не в щелочи, а в насыщенном растворе бикарбоната натрия. Полноту поглощения кислорода контролируют раствором, содержащим окислительно-восстановительный индикатор. Для приготовления раствора смешивают равные объемы 0,024%-го NaOH, 0,015%-го водного метиленового синего и 6%-й глюкозы; в качестве антисептика к раствору добавляют тимол. Перед использованием в пробирку наливают 5 мл смеси и нагревают на кипящей водяной бане до обесцвечивания, быстро охлаждают и помещают в анаэростат или анаэробный бокс. В анаэробных условиях раствор остается бесцветным.
Для удаления следов кислорода из промышленных газов, которые поставляют в баллонах, содержащих до 1,5% воздуха, используют следующую процедуру: газ пропускают через стеклянную трубку, заполненную медными стружками и нагретую до 400 °C с помошью электронагревателя (для этой цели подойдет спираль от бытовой электроплитки с керамическими изоляторами). Обработанный таким образом газ прак
тически не содержит кислорода, так как медь при высокой температуре эффективно связывает кислород с образованием СиО. Для регенерации меди используют газообразный водород: СиО + Н2 = Си + Н2О. Такой газ необходим при заполнении переходного шлюза в анаэробном боксе.
В качестве восстановителя чаще всего используют сульфид и тиогликолат натрия. Обычно готовят 1%-е растворы этих восстановителей в 5%-м растворе бикарбоната натрия, стерилизуют автоклавированием и добавляют к средам сульфид натрия из расчета 250 — 500 мг/л, а тиогликолат — до 250 мг/л среды. Восстановители используют в концентрациях, не влияющих на рост микроорганизмов.
Работа со строгими анаэробами представляет большие трудности и требует специального оборудования (см. гл. 16).
Температура. Интервалы температур, в которых возможен рост различных микроорганизмов, заметно варьируют. У мезофилов, к которым относится большинство известных нам форм, температурный оптимум лежит в интервале от 25 до 37 °C. У термофилов он значительно выше: от 45 до 80—90 °C. Психрофилы хорошо развиваются в интервале температур 5—10 °C. Отклонение температуры от оптимальной неблагоприятно влияет на развитие микроорганизмов, поэтому микроорганизмы выращивают в термостатах или специальных термостатированных комнатах, где с помощью терморегуляторов поддерживается соответствующая оптимальная температура. Мезофильные бактерии, естественным местом обитания которых являются вода и почва, выращивают в интервале от 20 до 30 °C, тогда как бактерии кожных покровов, слизистой или кишечника человека и животных культивируют при более высокой температуре: 35 —37 °C. Для выращивания психрофилов используют холодильные камеры.
Свет. Для роста подавляющего большинства микроорганизмов освещение не требуется. Напротив, прямые солнечные лучи отрицательно влияют на их развитие, поэтому такие микроорганизмы выращивают в темноте. Свет необходим для роста фототрофных микроорганизмов. Однако естественное освещение используют редко, так как оно непостоянно и плохо контролируемо. Как правило, фототрофов выращивают в люминостатах, т.е. камерах, освещенных лампами накаливания или флуоресцентными лампами дневного света. Необходимая температура в люминостатах создается благодаря вентиляции или холодильному устройству.
Выбор источника освещения определяется спектром его излучения и длинами волн, при которых культивируемые микроорганизмы осуществляют фотосинтез. Для выращивания пурпурных и зеленых бактерий лучше использовать лампы накаливания; для культивирования цианобактерий и микроводорослей нужно применять флуоресцентные лампы дневного света. Помимо спектрального состава света обращают внимание на освещенность, которую измеряют с помощью люксметра.
Вода. Рост микроорганизмов невозможен без присутствия в окружающей среде воды, причем вода должна находиться в доступной для клетки форме, т.е. жидкой фазе. Однако в природных субстратах и питательных средах часть воды ассоциирована с молекулами растворенных веществ и не может быть использована микроорганизмами. Доступность воды в субстрате для роста микроорганизмов выражают величиной активности воды (оД: а„ = Р/Ро, где Р — давление пара над раствором, мм рт. ст.; Ро — давление пара над чистой водой
при данной температуре, мм рт. ст. Для дистиллированной воды aw = 1,00. При растворении различных веществ в воде эта величина уменьшается и соответственно падает доступность для клетки воды. Микроорганизмы могут расти на средах, где aw = 0,99 — 0,61. Потребности в доступной воде у бактерий, как правило, выше, чем у дрожжей и мицелиальных грибов. Так, большинство бактерий, за исключением галофилов, хорошо растет на средах с величиной aw - 0,99 — 0,95; минимальная величина ат обеспечивающая рост дрожжей, находится в пределах от 0,91 до 0,61.
Активность воды в среде можно определить по формуле aw = /1/100, где А — относительная влажность (%) атмосферы, которую измеряют при равновесии в закрытом сосуде, содержащем среду. Различную активность воды в питательной среде или субстрате создают добавлением к ним таких соединений, как NaCl, КО, глюкоза, глицерин, полиэтиленгликоль.
3.3. РЕЖИМ КУЛЬТИВИРОВАНИЯ МИКРООРГАНИЗМОВ
Существует два принципиально разных режима выращивания микроорганизмов в жидкой среде. В одном случае после инокуляции среды в нее не добавляют и из нее не удаляют какие-либо компоненты, кроме газовой фазы. Такой режим культивирования, называемый периодическим, может поддерживать размножение клеток только в течение ограниченного времени, на протяжении которого меняются состав исходной среды и окружающие условия. Непрерывное (проточное) культивирование в отличие от периодического характеризуется постоянной подачей питательной среды со скоростью, равной скорости удаления культуры. При этом объем культуры в ферментере во времени не меняется. Одно из основных условий непрерывного культивирования — хорошее перемешивание культуры в ферментере. Режим непрерывного культивирования может быть реализован по принципу турбидостата или хемостата. Турбидостат — наиболее простой режим проточного культивирования, концентрация клеток в нем выбирается исследователем, а поступление питательных компонентов автоматически регулируется в соответствии с плотностью популяции. Меняя скорость подачи питательной среды («скорость разбавления»), экспериментатор может получать разные значения скорости роста популяции — от близкой к нулю до максимальной, воспроизводя таким образом разные состояния культуры от стационарной фазы до стадии экспоненциального роста.
Для непрерывного культивирования микроорганизмов использован слегка модифицированный ферментер, применяемый при периодическом культивировании (см. рис. 3.3). Для проточного культивирования требуется система двойного насоса (для добавления свежей среды и удаления культуральной жидкости), используемая вместе с регулятором уровня среды. При этом культуральная жидкость удаляется через отверстие для отбора проб. Такие насосы необходимы при проточном культивировании в больших емкостях.
В малых ферментерах удаление жидкости может происходить через боковую отводную трубку, расположенную на уровне, позволяюшем поддерживать определенный объем культуры. Малые ферментеры с боковым отводом жидкости можно изготовить из стандартных стеклянных или стальных сосудов.
Глава 4
МЕТОДЫ СТЕРИЛИЗАЦИИ
Стерилизация является одним из важнейших и необходимых приемов в микробиологической практике. Слово «стерилизация» в переводе с латинского означает «обеспложивание». В практической работе под стерилизацией понимают методы, применяемые для уничтожения всех форм жизни как на поверхности, так и внутри стерилизуемых объектов. Микробиологи стерилизуют питательные среды, посуду, различные инструменты и другие необходимые предметы с целью не допустить развитие посторонних микроорганизмов в исследуемых культурах. Термин «стерильность» имеет абсолютное значение. Можно говорить только либо о стерильности, либо о нестерильности, но не может быть состояния «частичной или неполной стерильности», «близкого к стерильному», «почти стерильного».
Различают термическую и холодную стерилизацию. В микробиологии находят применение следующие способы термической стерилизации: прокаливание в пламени и обжигание, сухожаровая стерилизация (горячим воздухом), стерилизация насыщенным паром под давлением (автоклавирование), дробная стерилизация (тиндализация), кипячение. Из методов холодной стерилизации микробиологи используют стерилизацию фильтрованием, газообразными средствами, ультрафиолетовыми лучами и другими видами излучений.
Возможность и целесообразность применения того или иного способа определяются в первую очередь физико-химическими свойствами материала, подлежащего стерилизации, а иногда и целью исследования.
4.1. СТЕРИЛИЗАЦИЯ ПИТАТЕЛЬНЫХ СРЕД
Стерилизация насыщенным паром под давлением — автоклавирование. Данный способ стерилизации питательных сред является наиболее надежным и чаще всего применяемым. Он основан на нагревании материала насыщенным
водяным паром при давлении выше атмосферного. Известно, что температура пара возрастает при повышении его давления (табл. 4.1).
Совместное действие высокой температуры и пара обеспечивает особую эффективность этого процесса. При этом погибают и вегетативные клетки, и споры микроорганизмов. Установлено, что споры большинства микроорганизмов не выдерживают и 5-минут-ную экспозицию в насыщенном паре при 121 °C. Лишь споры некоторых почвенных бактерий погибают при 1 ати только через 30 мин.
Таблица 4.1
Температура насыщенного пара при разных давлениях
Давление Температура, °C
атм ати кПа
1,0 0,0 101,32 100
1,5 0,5 151,98 111
2,0 1,0 202,65 121
2,5 1,5 251,20 128
з,о 2,0 299,75 134
10
Рис. 4.1. Схема автоклава:
/ — стерилизационная камера; 2 — кран для выхода воздуха; 3 — манометр; 4 — предохранительный клапан; 5 — водопаровая камера; 6 — воронка для заполнения автоклава водой; 7 — водомерная трубка; 8 — отверстия для поступления пара в стерилизационную камеру; 9- защитный кожух; 10 — крышка автоклава; 11 — подставка для размещения стерилизуемых предметов
Стерилизацию паром под давлением осуществляют в специальных герметически закрывающихся толстостенных аппаратах — автоклавах. Автоклавы разнообразны по форме, размерам, рабочему давлению, конструкции и другим показателям. Они могут быть с ручным управлением, полуавтоматические и автоматические, но, поскольку все автоклавы предназначены для выполнения одной и той же задачи — стерилизации, основной принцип их устройства один и тот же.
На рис. 4.1 показана схема вертикального автоклава с ручным управлением. Автоклав представляет собой металлический двустенный резервуар, способный выдержать высокое давление. Его внутренняя часть является стерилизационной камерой (/). В нее помещают стерилизуемый материал. Стерилизационная камера снабжена краном (2) для выхода воздуха, манометром (5) для определения давления пара и предохранительным клапаном (4) для выхода пара при повышении давления сверх необходимого и для предотвращения разрыва автоклава. Пространство между стенками, называемое водопаровой камерой (5), заполняется через воронку (6) водой (лучше дистиллированной, чтобы не образовывалась накипь) до определенного уровня, который отмечен на специальной водомерной трубке автоклава (7). Выше этого уровня воду наливать не следует, так как при бурном кипении вода может попасть в трубку, ведущую к манометру и исказить его показания. В верхней части внутренней стенки водопаровой камеры имеются отверстия (8), через которые пар поступает в стерилизационную камеру. Паровой котел сверху покрыт защитным кожухом (9). Он предохраняет котел от механических повреждений, а работающего около автоклава — от ожогов. Для создания герметичности автоклав плотно закрывают массивной крышкой (10) с резиновой прокладкой. Стерилизуемые предметы помещают на специальную подставку (11).
Процесс автоклавирования. Отдельные операции процесса стерилизации в автоклавах разных типов могут быть несколько различными. Соответственно несколько различается и техника работы с ними, однако общий принцип проведения стерилизации в разных автоклавах одинаковый.
Перед работой осматривают автоклав и контрольно-измерительную аппаратуру. При наличии любой неисправности (смещение стрелки манометра с нуля, трещина на водомерной трубке и др.) работать с прибором нельзя. После осмотра автоклава в водопаровую камеру наливают воду до верхней отметки на водомерной трубке. В стерилизационную камеру на специальную подставку из дерева помещают стерилизуемый материал. Предметы следует размещать не слишком плотно, так как пар должен свободно проходить между ними, иначе они не нагреются до нужной температуры и могут остаться нестерильными. Загрузив стерилизационную камеру, устанавливают и плотно завинчивают крышку (дверь) автоклава. Затем открывают кран, соединяющий стерилизационную камеру с наружным воздухом, и включают нагрев.
После начала парообразования удаляют воздух из стерилизационной камеры. Это необходимое условие стерилизации, так как при одном и том же давлении температура чистого пара выше температуры смеси пара и воздуха. Если в автоклаве останется воздух, материал может не простерилизоваться. Наиболее простой и очень распространенный способ освобождения автоклава от воздуха — вытеснение воздуха паром. Пар и конденсат отводят либо в сосуд с водой, либо в специальное устройство, соединенное с канализацией. В первом случае на кран (2) надевают резиновый шланг, который опускают в воду. Началом продувания считается появление устойчивой непрерывной струи чистого пара. Пока в автоклаве еще имеется воздух, смесь воздуха и пара, проходя через воду, издает сильный треск. Чистый пар выходит с равномерным шипящим звуком. Его пропускают в течение 10 мин. В целом вся операция с момента появления пара с воздухом должна занимать не более 15 — 20 мин, иначе в автоклаве останется мало воды и он может испортиться. Чтобы уменьшить расход пара (воды), кран открывают не полностью. Степень открывания крана устанавливают на практике при эксплуатации автоклава. В наиболее совершенных автоклавах воздух из стерилизационной камеры удаляют с помощью вакуумного насоса.
Когда воздух вытеснен, закрывают пароотводной кран, и давление пара доводят до показания, соответствующего режиму стерилизации. Режим автоклавирования часто выражают в единицах избыточного давления, указывая при этом длительность его поддержания, например стерилизация при 1 ати в течение 20 мин. На манометре автоклава обозначается именно то избыточное давление, которое создается в автоклаве сверх нормального. Нередко режим автоклавирования характеризуется температурой и временем. Как только стрелка манометра дойдет до указателя определенного дополнительного давления и, следовательно, температура пара достигнет соответствующего значения, этот уровень давления пара поддерживают в течение необходимого времени путем ручного или автоматического регулирования подачи пара. В автоматических автоклавах подачу пара регулируют электроконтактным манометром.
По окончании времени стерилизации выключают нагрев автоклава. Давление в автоклаве постепенно падает и сравнивается с атмосферным. Лишь после этого открывают кран, выводящий пар. Преждевременное открывание крана недопустимо, так как перегретые среды при резком снижении давления сразу же бурно закипают, смачивают и даже иногда выталкивают ватные пробки, что нарушает стерильность материала. Когда пар выйдет, открывают крышку (дверь) автоклава, соблюдая при этом осторожность во избежание ожога паром лица и рук. Удаление пара из стерилизационной камеры автоклавов, оснащенных вакуумным насосом, осуществляют с помощью насоса. Одновременно происходит подсушивание стерильного материала.
Поскольку автоклав работает при высоких давлениях и температурах, неправильное обращение с ним может быть причиной несчастных случаев. Установка автоклава и работа с ним производятся при точном и строгом выполнении правил, указанных в прилагаемой к автоклаву инструкции. К работе допускаются только подготовленные лица, имеющие специальное разрешение.
При необходимости проконтролировать температуру в автоклаве пользуются разными веществами, плавящимися при определенной температуре. Эти вещества предварительно смешивают с нейтральными красителями и помещают в автоклав до начала стерилизации. В качестве индикаторов температуры используют фенантрен (температура плавления 98—100°C), бензаурин (115°C), серу (119°C), бензойную кислоту (121 —122 °C), мочевину (132 °C), глюкозу (146 °C), тиомочевину (180 °C), аскорбиновую кислоту (187—192 °C). На 100 г этих веществ берут 0,01 г красителя (фуксин, метиленовый синий), тщательно смешивают, рассыпают в стеклянные трубочки с одинаковым диаметром и толщиной стенок, запаивают и в вертикальном положении раскладывают между стерилизуемым материалом в автоклаве. По достижении в сосуде соответствующей температуры эти вещества расплавляются и окрашиваются в цвет добавленного в них красителя.
Рис. 4.2. Ватные пробки: а — приготовлена правильно; б, в — приготовлены неправильно
Подготовка сред к стерилизации. При автоклавировании 3 — 5 % жидкости теряется в результате испарения, поэтому рекомендуется в приготавливаемые среды добавлять сверх объема примерно 5 % дистиллированной воды. Тогда после стерилизации среда (раствор) будет иметь требуемую концентрацию.
Среды обычно стерилизуют в пробирках, колбах, бутылях. Емкости заполняют средой не более чем на половину их высоты, чтобы предотвратить смачивание пробок. Сосуды со средами закрывают ватными пробками. Они предохраняют среду от заражения микроорганизмами, находящимися в окружающем воздухе.
Пробки должны быть достаточно плотными, чтобы выполнять эту функцию, но с достаточно равномерным распределением волокон ваты, так как через них происходит газообмен культур с окружающей средой. Слишком плотные пробки затрудняют снабжение культур воздухом.
Для приготовления пробки плоский кусок ваты, взятый вдоль волокна, скатывают валиком. Чтобы придать пробке прочность, ее прокатывают между ладонью и чистым стеклом, лежащим на столе. Длина пробки для обычной пробирки примерно 4 см. Пробка должна входить в пробирку на 1,5—2,0 см (рис. 4.2). Для сохранения формы пробку вынимают из горлышка, слегка вращая. Удобно обернуть пробку чистой марлевой салфеткой.
Перед стерилизацией пробки можно прикрыть бумажными колпачками. Нельзя обертывать пробки сосудов, которые будут стерилизоваться в автоклаве, целлофаном, фольгой или другими материалами, не пропускающими пар, так как пар должен проникать через пробку в сосуд, иначе среды не нагреются до нужной температуры и не простерилизуются. При использовании стеклянных, резиновых, корковых и других пробок их завертывают в двойной слой оберточной бумаги и стерилизуют привязанными к склянке, закрытой ватной пробкой. Пробки в сосуде меняют стерильно около пламени горелки.
Выбор режима автоклавирования. В микробиологической практике стерилизацию в автоклавах осуществляют при температуре 111 — 138 °C, т.е. от 0,5 до 2,5 ати. Температура ниже 111 °C не может считаться надежной; температура выше 138 °C, как правило, не является необходимой, к тому же чем выше давление пара, тем сложнее условия эксплуатации автоклава. При использовании автоклавов без вакуумных насосов наиболее надежными считают следующие режимы стерилизации: 15 — 45 мин при 121 °C (1 ати) и 10 — 30 мин при 128 °C (1,5 ати). Микробиологи чаще всего стерилизуют среды при 0,5 и 1 ати. Температура и длительность автоклавирования питательных сред определяются прежде всего их составом, термоустойчивостью или термолабильностью компонентов. Такие легко разрушающиеся субстраты, как молоко или желатиновые среды, а также субстраты, содержащие сахара, витамины (пивное сусло, соки, дрожжевой автолизат и др.), обычно стерилизуют при 0,5 ати в течение 15 — 30 мин. Мясопептонные среды можно стерилизовать при 1 ати 20 мин. Среды, содержащие агар, стерилизуются труднее, потому что стерилизация начинается фактически после того, как агар расплавится. Но и расплавленный агар требует для
стерилизации в два раза больше времени, чем тот же объем воды. С трудом поддаются стерилизации в автоклаве различные порошки (тальк, мел) и вязкие жидкости (глицерин, вазелиновое масло), поскольку они плохо передают тепло и очень медленно прогреваются. Их лучше стерилизовать в сушильных шкафах при 160 °C в течение 2 ч или 1 ч при 170 °C. Слой масла или порошка в сосуде не должен превышать 1,5 см.
Имеются субстраты, в которых могут быть споры, отличающиеся особой термостабильностью. К ним относится почва, причем она, кроме того, нагревается с замедленной скоростью. Ее обычно стерилизуют при 1 ати либо один раз 2 ч, либо два дня подряд по 1 ч, а иногда 2 ч при 2 ати.
Выбирая режим стерилизации, необходимо учитывать pH среды. При кислой реакции многие вещества, входящие в ее состав, могут подвергнуться гидролизу. Чем ниже значение pH, чем выше температура и продолжительнее время стерилизации, тем интенсивнее происходит гидролиз. В результате после стерилизации перестают застывать среды с желатиной и даже с агаром. Если реакция среды щелочная, то при стерилизации выпадают в осадок соли железа; они карамелизуются и сахара становятся непригодными для использования бактериями. В некоторых случаях в процессе стерилизации изменяется pH среды. Так, если pH среды с углеводами выше 7,0, то может произойти ее подкисление до pH 6,0. Чтобы избежать таких явлений, рекомендуется углеводы, фосфаты, соли железа автоклавировать отдельно в виде более или менее концентрированных растворов в дистиллированной воде при том значении pH, которое обеспечивает целостность вещества. После стерилизации растворы объединяют в нужном соотношении. Таким приемом раздельной стерилизации в микробиологии пользуются довольно часто, поскольку многие компоненты сред нельзя стерилизовать одним и тем же способом.
Режим автоклавирования в значительной степени зависит от объема стерилизуемого субстрата. Чем больше объем, тем больше времени при одной и той же температуре (давлении) требуется для обеспечения надежности стерилизации. Имеет значение толщина стенок и форма емкостей (табл. 4.2). Это нужно учитывать в практической работе.
Например, не следует стерилизовать термочувствительный субстрат одновременно в пробирках и больших бутылях. Если среда стерилизуется по режимам, рекомендуемым для малых объемов, содержимое бутыли может не про-стерилизоваться. Если же стерилизация проводится с расчетом на большой объем, среда в пробирке прогревается намного дольше и может испортиться.
После автоклавирования среды для проверки стерильности выдерживают 2—3 сут в термостате при 30 °C. Если в средах обнаруживается рост микроорганизмов, их готовят заново.
Дробная стерилизация (тиндализация) и пастеризация. Тиндализация, или дробная стерилизация, была предложена в 1877 г. Дж. Тиндалем. Она применяется для сред, портящихся при действии температур выше 100 °C. Тиндали-зацию осуществляют текучим паром в автоклаве с незавинченной крышкой или в кипятильнике Коха. Среды прогревают несколько раз по 10—15 мин. Между прогреванием среды ставят в термостат при температуре 30 °C на 8 —12 ч для прорастания жизнеспособных спор. Среды, не выдерживающие нагревания при 100 °C, прогревают более осторожно: при 60 —80 °C через каждые 8 — 12 ч 4 —5 сут подряд.
Таблица 4.2
Зависимость продолжительности стерилизации жидкости от объема сосудов
Емкость Объем Время стерили-зации при 121 — 123 "С, мин
ММ МЛ
Пробирки 18 х 150 12-14
32 х 200 13-17
38 х 200 14-20
Колбы: 50 12-14
ТОНКО- 125 12-14
стенные 200 12-15
500 17-22
900 19-24
1000 20-25
1800 25-30
2000 30-35
толсто- 500 24-28
стенные 1000 25-30
2000 40-45
Матрасы 1000 30-35
Бутыли 9000 50-55
100 13 — 17
Однократный прогрев материала при температуре ниже 100 °C известен под названием пастеризация. Этот метод, предложенный Пастером, предназначен для уничтожения только бес-споровых форм микроорганизмов. Следовательно, в подавляющем большинстве случаев он не обеспечивает стерильности. Пастеризацию проводят при 60—80 °C в течение 10—30 мин. Пастеризацию используют в пищевой промышленности для обработки молока, фруктовых соков, вина, пива и др.
Стерилизация фильтрованием. Фильтрованием стерилизуют синтетические среды строго определенного состава, которые содержат легкоразрушающи-еся или летучие компоненты — витамины, аминокислоты (цистеин и цистин), белки, ароматические углеводороды, антибиотики и др. Фильтрование жидкостей осуществляют через мелкопористые материалы, легко адсорбирующие клетки микроорганизмов: асбест,
целлюлозу, фарфор, каолин и т.д.
Стерилизующими фильтрами теоретически считают такие, размер пор которых не превышает 0,7 мкм. В практике же пригодность фильтров для стерили-
зации устанавливают путем пробной фильтрации через них суспензии какого-нибудь мелкого микроорганизма, например Serratia marcescens. Для проверки на стерильность фильтрат в большом количестве высевают на питательную среду. Если в течение 5 сут тест-организм не вырастет, фильтры могут быть использованы для стерилизации. Широкое распространение получили мембранные фильтры. Это диски разного размера, диаметра, напоминающие бумажные. Их готовят на основе нитроцеллюлозы. Мембранные фильтры в зависимости от величины пор применяют для фильтрования и стерилизации. Для стерилизации используют фильтры с номера 1 до номера 4 фирмы «Владипор» (Россия), фильтры с номера 5 до номера 10 фирмы «СИНПОР» (Чехия) и марок VF, VM, VC, SLGS, SLHA, DA фирмы «МИЛЛИПОР» (США).
Плотные диски, изготовленные из смеси асбеста с целлюлозой, называются фильтрами Зейтца. В зависимости от диаметра пор они обозначаются разными индексами: ЕК — поры диаметром 1,5 —1,8 мкм; EKS — 1,2— 1,5; EKS-1 — 1,0—1,2; ЕКР — 0,8—1,0 мкм. В России выпускают асбестовые фильтры марок Ф2 и СФ. Стерилизующими являются СФ-3 и СФ-4.
Асбестовую пластинку помещают в специальный держатель, который обычно
изготавливают из нержавеющей стали, и крепко зажимают винтами между верхней (цилиндрической) и нижней (воронкообразной) частями держателя. Трубка нижней части держателя через резиновую пробку проходит в колбу Бунзена. Нередко весь этот прибор в собранном виде называют фильтром Зейтца.
а б
Рис. 4.3. Приборы для стерилизации фильтрованием: а — со стеклянным держателем; б — с металлическим держателем
Для стерилизации используют стеклянные пористые фильтры, а также изготовленные из каолина с примесью кварцевого песка — «свечи» Шамберлана и из инфузорной земли — «свечи» Беркефельда. Пористость первых обозначается буквой L с цифрами от 1 до 13 соответственно уменьшению диаметра пор фильтра от 9 до 1,2 мкм. Мелкопористые «свечи» обозначаются маркой В, крупнопористые — F. Фильтры Беркефельда обозначают буквами V, N, W, что соответствует следующим размерам пор (мкм): 8—12, 5 — 7, 3 — 4.
«Свечи» и специальные держатели с закрепленными в них асбестовыми фильтрами герметически соединяют с колбой Бунзена для фильтрации в вакууме (рис. 4.3). Перед употреблением фильтры, их держатели и приемник фильтрата должны быть простерили-зованы.
Мембранные фильтры стерилизуют автоклавированием при 1 ати 15 мин или длительным кипячением. Держатель вместе с резиновой пробкой заворачивают в бумагу и автоклавируют при 1 ати 20 — 30 мин. Фильтры Зейтца автоклавируют в собранном виде.
«Свечи» стерилизуют вместе с резиновыми пробками в автоклаве. Колбу Бунзена закрывают ватной пробкой, в отводную трубку вставляют тампон и стерилизуют горячим воздухом — сухим жаром.
4.2. СТЕРИЛИЗАЦИЯ СТЕКЛЯННОЙ ПОСУДЫ
Основной способ стерилизации стеклянной посуды — обработка сухим горячим воздухом при температуре не выше 180 °C в течение 1 — 3 ч (табл. 4.3). При этом погибают и вегетативные клетки, и споры микроорганизмов. Стерилизацию осуществляют в специальных суховоздушных (сухожаровых) стерилизаторах и сушильных шкафах, приспособленных для стерилизации. Они различаются по форме и способам обогрева, но имеют сходное устройство. Их делают из
термостойких материалов — обычно из металла и асбеста. Внутри стерилизатора имеются полки для размещения посуды, а наверху — отверстие, в котором с помощью пробки укрепляют термометр. У стенки стерилизатора (шкафа) или вблизи греющей поверхности температура всегда значительно выше, чем внутри, поэтому ртутный шарик термометра должен находиться внутри шкафа на расстоянии 6 — 8 см от верхней стенки. В верхней части сушильных
Таблица 4.3
Условия стерилизации стеклянной посуды сухим жаром
Температура, °C Время, мин
140 180
150 150
160 120
170 60
шкафов имеется также отверстие для вентиляции, которое при стерилизации закрывают. Стерилизаторы и шкафы с электрическим обогревом снабжены терморегулятором, обеспечивающим автоматическое поддержание необходимой температуры.
Подготовка посуды к стерилизации. Посуда перед стерилизацией должна быть тщательно вымыта и завернута в бумагу для сохранения стерильности после прогревания. Посуду развертывают непосредственно перед употреблением. В верхние концы пипеток вставляют ватные тампоны. Торчащие из пипеток волокна ваты сжигают в пламени горелки. Пипетки заворачивают в длинные полоски бумаги шириной 4 — 5 см. Обмотку начинают с оттянутого конца и постепенным движением бумаги по спирали заканчивают у конца с ватным тампоном. Завернутые пипетки для предохранения бумаги от загрязнения и разрывов перед стерилизацией упаковывают по несколько штук вместе или помещают в специальные металлические или картонные пеналы. Чашки Петри обычно заворачивают в пакеты по 2—4 штуки, шпатели — по отдельности, но затем; как и пипетки, их объединяют в общий сверток. Колбы, пробирки и трубки Бурри закрывают ватными пробками. На пробки можно надеть бумажные колпачки, предохраняющие горлышко от пыли.
Стерилизация. Посуду, подготовленную для стерилизации, загружают в стерилизатор (или сушильный шкаф) не слишком плотно, чтобы обеспечить циркуляцию воздуха и равномерный надежный прогрев стерилизуемого материала. Стерилизатор (сушильный шкаф) во время работы должен быть плотно закрыт. При отсутствии терморегулятора необходимо строго следить за температурой, так как при ее понижении не осуществится стерилизация, а при нагреве выше 180 °C бумага и пробки начинают обугливаться. По окончании стерилизации шкаф не открывают до тех пор, пока температура в нем не упадет до 80 °C, поскольку при резком охлаждении иногда нарушается стерильность материала, а сильно нагретое стекло может растрескаться. Лучше всего выгружать посуду, когда температура в стерилизаторе сравняется с комнатной.
Посуду можно стерилизовать и в автоклаве. Режим стерилизации в этом случае существенно зависит от объема сосудов и толщины стекла (см. табл. 4.2). Для автоклавирования посуду готовят, как и для сухожаровой стерилизации. Следует иметь в виду, что в автоклаве посуда увлажняется.
4.3. СТЕРИЛИЗАЦИЯ ИНСТРУМЕНТОВ И ПРИБОРОВ
Мелкие металлические инструменты: петли, иглы, пинцеты, ножницы, шпатели — стерилизуют прокаливанием в пламени (т. е. нагреванием докрасна) непосредственно перед использованием. На пламени кратковременно обжигают предметные и покровные стекла, стеклянные шпатели и палочки, фарфоровые ступки и пестики, горлышки колб, пробирок, бутылок, а также ватные пробки при пересевах культур и разливах сред. В пламени погибают и вегетативные клетки, и споры микроорганизмов.
Наряду со стерильными одноразовыми шприцами в лабораторной практике широко распространены традиционные шприцы многократного использования,-Их лучше всего стерилизовать сухим жаром при 160 °C в собранном либо в разобранном виде. В первом случае длительность стерилизации 75 мин, во втором —
60 мин. Собранные шприцы вместе с иглой стерилизуют в пробирке, закрытой ватной пробкой, разобранные заворачивают в бумагу или ткань. Можно стерилизовать шприцы и в автоклаве при 1 ати в течение 15 — 20 мин. Автоклавируют их только в разобранном виде, иначе они повреждаются. Прокаливать шприцы нельзя, так как от этого они портятся.
Термостойкие приборы для культивирования микроорганизмов, а также детали к этим приборам, резиновые пробки и шланги стерилизуют в автоклаве. При этом емкости завертывают в бумагу. Режим автоклавирования выбирают в соответствии с термостойкостью материала, из которого сделан прибор.
Некоторые предметы (металлические инструменты, мелкие стеклянные детали, мембранные фильтры) иногда стерилизуют длительным (в течение 20 — 30 мин) кипячением в дистиллированной воде. Металлические и стеклянные предметы лучше всего кипятить в специальных закрытых сосудах — стерилизаторах. Можно использовать для этой цели и металлическую посуду. Мембранные фильтры обычно кипятят в колбе или химическом стакане, закрытых ватными пробками. Однако этим способом стерилизации в микробиологической практике пользуются редко в связи с тем, что длительное кипячение может повредить обрабатываемый материал, а сокращение времени кипячения может не обеспечить стерильность, так как споры некоторых микроорганизмов способны сохранять жизнеспособность даже после длительного кипячения. Надежность стерилизации при кипячении может быть увеличена внесением в воду какого-либо бактерицидного средства: 2%-го формальдегида, 1 %-й бриллиантовой зелени или 0,1 %-й сулемы. Но в этом случае возможно загрязнение биоцидами стерилизуемых предметов.
Стерилизация газообразными веществами. Аппаратуру, имеющую зеркальное, оптическое и радиоэлектронное оборудование, а также изделия из термолабильных пластмасс, например центрифужные пробирки, стерилизуют газовым методом. Для газовой стерилизации применяются только те соединения, которые обладают спороцидными свойствами, — оксид этилена, метилбро-мид, оксид пропилена, формальдегид, глутаральдегид, бета-пропиолактон, озон и др. Особенно эффективна смесь оксида этилена и бромистого метила в массовом соотношении 1 :1,44 (смесь «ОБ»).
Газовую стерилизацию проводят в специальных герметически закрывающихся аппаратах. Стерилизуемые объекты, помещаемые в камеру, упаковывают как при стерилизации в автоклаве или сушильном шкафу. Для упаковки используют материалы, не портящиеся в газовой среде и пропускающие газ и влагу, — оберточную бумагу, хлорвиниловые и нейлоновые пленки и др. Перед введением в камеру биоцида из нее как можно полнее удаляют воздух, чтобы обеспечить тесный контакт активно действующего вещества со стерилизуемым объектом. При стерилизации строго контролируют концентрацию газа, давление, влажность, температуру и длительность экспозиции. В большинстве случаев процесс проводят в сочетании с некоторым повышением температуры (до 45 — 70 °C). Режимы стерилизации разными газами неодинаковы. Они определяются прежде всего свойствами биоцида. Имеет значение и конструкция стерилизационного аппарата. Оптимальный режим стерилизации смесью «ОБ» при 50 °C в аппарате вместимостью 100 л и более следующий: концентрация смеси 3,36 г/л (давление 1,2 ати), относительная влажность 80—100%, продолжительность 24 ч. По окончании стерилизации удаляют газы из камеры с
Способы стерилизации питательных сред, посуды и других лабораторных материалов
Стерилизуемый материал Метод стерилизации Режим стерилизации Примечание
Питательные среды с почвенной вытяжкой; картофельные и некоторые другие натуральные среды Автоклавирование 1,5—2,0 ати; 30 мин В колбах, пробирках, бутылях и т.д., закрытых ватными пробками
Жидкие и агаризованные среды, не содержащие сахаров и других веществ, разлагающихся при 120 °C Тоже 1,0 ати; 20 мин Тоже
Жидкие и агаризованные среды с сахарами и другими соединениями, не выдерживающими нагревания при 120 °C » 0,5 ати; 15 — 30 мин »
Среды или компоненты сред, не выдерживающие нагревания выше 100 °C Дробная стерилизация Текучий пар; 3 раза по 30 — 40 мин через сутки »
Среды или компоненты сред, не выдерживающие нагревания (белки, некоторые витамины и аминокислоты) Фильтрование через бактериальные фильтры — —
Вазелиновое масло, глицерин, тальк Горячий воздух 160 °C; 2чили 170 °C; 1 ч Слой вещества в сосуде не должен превышать 1,5 см
Чашки Петри, пипетки, шпатели Тоже 160-170 °C; 2 ч Завернуты в бумагу (отверстия пипеток закрыты ватными тампонами)
Колбы, пробирки, химические стаканы, флаконы, стеклянные центрифужные пробирки, трубки Бурри » 160-170 °C; 2 ч Закрыты ватными пробками
Шприцы Горячий воздух 160 °C; 1ч Разобраны и завернуты в бумагу или ткань
Автоклавирование 1 ати; 15—20 мин Разобраны и завернуты
Мембранные фильтры Автоклавирование 1 ати; 15—20 мин В сосуде с дистиллированной водой
Кипячение 30 мин То же
Стерилизуемый материал Метод стерилизации Режим стерилизации Примечание
Фильтры Зейтца Автоклавирование 1—1,5 ати; 20—30 мин Цилиндр держателя закрыт ватной пробкой, в отводной трубке ватный тампон
Горячий воздух 160 °C; 1 ч Верхняя часть завернута в бумагу
Свечи Шамберлана и Беркефельда Автоклавирование 0,5—1 ати; 20—30 мин Можно вместе с держателем
Горячий воздух 160-170 °C; 2 ч Закрыты ватными пробками, завернуты в бумагу
Стеклянные фильтры с держателем или отдельно Автоклавирование 1 ати; 30 мин Закрыты ватными пробками, завернуты в пергамент и алюминиевую фольгу
Стеклянные фильтры без резиновых пробок Горячий воздух 1 ати; 30 мин Завернуты в бумагу
Центрифужные пробирки, изготовленные из термолабильных пластмасс Газовая стерилизация Зависит от применяемого биоцида —
Ультрафиолетовые лучи Время экспозиции устанавливают экспериментально Пробирки после облучения хранят в стерильной посуде
помощью вакуумного насоса и на некоторое время камеру оставляют под вакуумом для десорбции газов из стерилизованных предметов. После этого камеру заполняют стерильным воздухом. Предметами, простерилизованными газами, рекомендуется пользоваться не ранее чем через 24 ч после стерилизации. Это необходимо для полного удаления из них газа. Целесообразно на данный период поместить их в вытяжной шкаф или оставить в хорошо проветриваемом помещении. Некоторые изделия из пластмасс требуют дополнительной аэрации до 9 сут. При проведении газовой стерилизации строго соблюдают правила работы с ядовитыми газообразными веществами.
Стерилизация облучением. Для стерилизации помещений, оборудования, некоторых медицинских принадлежностей, пищевых продуктов используют разные виды излучений: инфракрасное, ультрафиолетовое, рентгеновские лучи, а-, р-, у-лучи радиоактивных элементов. Единицей дозы облучения является радиан (1 рад = 0,01 Гр), который эквивалентен поглощенной энергии примерно в 100 эрг/г. Стерилизующими являются дозы облучения 2—3 Мрад (104 Гр, или рад). Чаще других в микробиологической практике используют ультрафио-
летовое облучение. Следует отметить, что все большее распространение получают посуда и инструменты одноразового использования.
Основные способы стерилизации питательных сред, посуды и других лабораторных материалов обобщены в табл. 4.4. Следует отметить, что все большее распространение получают посуда и инструменты одноразового использования.
Глава 5
ИЗУЧЕНИЕ МОРФОЛОГИИ и цитологии МИКРООРГАНИЗМОВ
5.1. МЕТОДЫ МИКРОСКОПИИ
Изучение морфологии и строения клеток микроорганизмов, величина которых измеряется в большинстве случаев микрометрами, возможно только с помощью микроскопов, обеспечивающих увеличение исследуемых объектов в сотни (световая микроскопия) и десятки тысяч (электронная микроскопия) раз. Изображение в световом микроскопе формируется вследствие того, что объект и различные элементы его структуры избирательно поглощают свет с различной длиной волны (абсорбционный контраст) или вследствие изменения фазы световой волны при прохождении света через объект (фазовый контраст).
Световая микроскопия включает обычную просвечивающую микроскопию (светло- и темнопольную), фазово-контрастную и люминесцентную.
В последнее время разработаны и другие микроскопии и микроскопы — инверсионная и конфокальная лазерная сканирующая микроскопия.
5.1.1. Светлопольная микроскопия
Существуют различные модели учебных и исследовательских световых микроскопов. Внешний вид и принципиальное устройство двух из них (микроскоп биологический рабочий, МБР-1 и Биолам 2) приведены на рис. 5.1 и 5.2. Подобные микроскопы позволяют определить форму клеток микроорганизмов, их размер, подвижность, степень морфологической гетерогенности, а также характерную для микроорганизмов способность к дифференцирующему окрашиванию. Успех наблюдения объекта и надежность получаемых результатов зависят от хорошего знания оптической системы микроскопа, которая включает осветительный аппарат, объектив и окуляр.
Особенности оптической системы светового микроскопа МБР-1. Осветительный аппарат состоит из зеркала и конденсора и предназначен для наилучшего освещения препарата. Регулируемое зеркало укреплено у основания штатива и имеет две стороны: вогнутую и плоскую. Вогнутое зеркало собирает и концентрирует в плоскости препарата пучок лучей, идущих от источника света, поэтому им пользуются только в тех случаях, когда работают без конденсора, т.е. с очень малыми увеличениями. При работе с конденсором, который рассчитан на использование параллельных лучей, следует пользо-
Рис. 5.1. Микроскоп МБР-1:
1 — подковообразное основание микроскопа; 2 — предметный столик; 3 — винты для перемещения предметного столика; 4 — клеммы, прижимающие препарат; 5 — конденсор; 6 — кронштейн конденсора; 7 — винт, укрепляющий конденсор в гильзе; 8 — рукоятка перемещения конденсора; 9 — рукоятка ирисовой диафрагмы конденсора; 10 — зеркало; 11 — тубу-содержатель; 12 — рукоятка микрометрического винта; 13 — рукоятка микрометрического винта; 14 — револьвер; 75 — объективы; 16 — наклонный тубус; 77 — винт для крепления тубуса; 18 — окуляр
Рис. 5.2. Микроскоп с бинокулярной насадкой, встроенным в основание осветителем с галогенной лампой 6 В, 6 Вт или 6 В, 10 Вт и совмещенным с сетевой вилкой источником питания (Биолам-2, ЛОМО, Россия):
7 — окуляры; 2 — бинокулярная насадка; 3 — револьверное устройство; 4 — объектив; 5 — предметный столик; 6 — конденсор; 7 — корпус коллекторной линзы; 8 — патрон с лампой; 9 — шарнир; 10 — рукоятка перемещения кронштейна конденсора; 77 — рукоятка тонкой фокусировки; 72 — рукоятка грубой фокусировки; 13 — тубусодержатель; 14 — выключатель; 75 — источник электропитания; 16 — гнездо для подключения штекера источника электропитания;
17 — штекер
ваться только плоской стороной зеркала. Конденсор, укрепленный непосредственно над зеркалом, состоит из нескольких линз и предназначен для собирания параллельных лучей света, идущих от источника света и отраженных плоским зеркалом в одной точке — фокусе, который должен находиться в плоскости препарата. В конденсор вмонтирована ирисовая (апертурная) диафрагма, позволяющая задерживать излишние лучи света и регулировать апертуру конденсора. Под конденсором находится откидная оправа для светофильтра.
В современных микроскопах источник света вмонтирован непосредственно под конденсором. В таких микроскопах зеркала отсутствуют, а ирисовая диафрагма имеется не только в конденсоре, но и над источником света.
Объектив представляет собой наиболее важную часть микроскопа. Он дает действительное увеличенное и обратное изображение изучаемого объекта. Объектив состоит из системы линз, заключенных в металлическую оправу. Самая главная — наружная (фронтальная) линза, от фокусного расстояния которой зависит увеличение объектива. Чем больше кривизна фронтальной линзы, тем короче фокусное расстояние и тем больше увеличение объектива. Увеличение объектива всегда обозначено на его оправе. От увеличения объектива зависят еще две его характеристики — рабочее расстояние, т.е. расстояние от фронтальной линзы до плоскости препарата при сфокусированном объекте, и площадь поля зрения. Чем больше увеличение объектива, тем меньше его рабочее расстояние и поле зрения.
Микроскопы имеют «сухие» объективы для наблюдения живых микроорганизмов, а также для поиска нужного поля зрения, дающие увеличение в 8 (10) и 40 раз, и иммерсионные, увеличивающие объект в 90 (100) раз и позволяющие провести более детальное изучение формы и строения микроорганизмов.
Окуляр содержит две линзы — глазную (верхнюю) и собирательную и служит для рассмотрения изображения предмета, даваемого объективом, т. е. выполняет роль лупы. Окуляры могут давать увеличение в 5, 7, 10, 12, 15 и 20 раз, что указано на их оправе, например 15х. Общее увеличение окуляра повышается с уменьшением фокусного расстояния составляющих его линз, поэтому более сильные окуляры будут короткими, а более слабые — длинными.
Увеличение, которое дает микроскоп, определяется произведением увеличения объектива на увеличение окуляра. Так, например, использование окуляра 15 х и объектива 90х позволяет увеличить объект в 1350 раз. Однако общее увеличение еще не характеризует всех возможностей микроскопа. Увеличенное изображение может оказаться как четким, так и нечетким.
Отчетливость получаемого изображения определяется разрешающей способностью микроскопа, которая зависит от длины волны используемого света и числовой апертуры оптической системы микроскопа. Разрешающая способность связана обратной зависимостью с пределом разрешения — минимальным расстоянием между двумя точками, при котором еще можно различить каждую из них. Предел разрешения определяется по формуле
/1| + A'i
где d — минимальное расстояние между двумя точками; At — числовая апертура объектива; /12 ~ числовая апертура конденсора; >. — длина волны используемого света. Числовая апертура определяется произведением синуса половины отвесного угла на показатель преломления среды, граничащей с линзой: А = и-sin и. Иными словами, числовая апертура — это оптический охват линзы; она является мерой количества света, попадающего в линзу. Использование объективов с большей апертурой и коротковолнового света позволяет увидеть структурную организацию клетки и даже крупные вирусы.
Числовая апертура любой линзы, граничащей с воздухом, не может быть больше 1, так как показатель преломления воздуха равен 1, а угол и не может быть больше 90° (т. е. sin и < 1). В микроскопе МБР-1 объектив 40х имеет апертуру 0,65, а конденсор — примерно 1. Таким образом, при использовании объектива 40 х и зеленого света с длиной волны 550 нм (0,55 мкм) предел разрешения составляет 0,33 мкм. Повысить разрешающую способность можно двумя путями: либо освещать объект более короткими лучами света, например ультрафиолетом, что требует применения дорогостоящей кварцевой оптики и соответствующих источников света, дорогостоящих ртутных ламп, либо увеличивать показатель преломления среды, граничащей с линзой, с тем
чтобы приблизить его к показателю преломления стекла, на котором находится объект (л стекла = 1,5). Для этого между фронтальной линзой объектива и исследуемым объектом помещают каплю жидкости с показателем преломления, большим показателя преломления воздуха, например каплю воды (п = 1,3), глицерина (и = 1,4) или иммерсионного (например кедрового) масла (и = 1,5). Для каждой указанной жидкости выпускают специальные объективы, которые называются иммерсионными. Числовая апертура этих объективов возрастает благодаря увеличению как значения и, так sin и.
Числовая апертура объектива указана на его оправе. Объективы микроскопа МБР =18* и 40х имеют апертуру соответственно 0,20 и 0,65. У масляного иммерсионного объектива с увеличением 90х апертура 1,25. На оправе этого объектива нанесено также обозначение «МИ» — масляная иммерсия — и черное кольцо. На оправе объектива водной иммерсии имеется обозначение «ВИ» (водная иммерсия) и белое кольцо. Этот объектив увеличивает в 70 раз (70х) и имеет апертуру 1,23. Апертура конденсора должна соответствовать числовой апертуре объектива. Когда она меньше апертуры объектива, оптические возможности линзы последнего не будут использованы полностью из-за слабости попадающего в нее светового потока. Если апертура конденсора больше апертуры объектива (что, в частности, бывает при работе с сухими системами), то необходимо несколько прикрыть ирисовую диафрагму конденсора. Это приведет к устранению рассеянного света и даст нужную контрастность изображения. Неиммергирован-ный конденсор микроскопа МБР-1 имеет апертуру не более 0,95. Помещая иммерсионное масло между верхней линзой конденсора и нижней поверхностью предметного стекла, повышают апертуру конденсора до 1,2.
В работе со световым микроскопом следует помнить, что четкое изображение получается при хорошем освещении, однако диафрагмирование конденсора дает эффект контрастирования живых объектов. Препарат должен быть частью однородной оптической системы, что достигается заливкой объекта водой, маслом или другой подходящей жидкостью. В случае необходимости наблюдения объекта через дно чашки Петри лучше использовать вогнутое зеркало, устраняя эффекты искажения, вызванные неровностью поверхности чашки.
Порядок работы со светлопольным микроскопом при внешнем источнике освещения. Хорошие результаты при работе с микроскопом могут быть получены только при условии правильного освещения объекта. Лучший способ освещения основан на системе Кёлера; ее последовательные этапы описаны ниже.
Установка света по Кёлеру*.
1. Устанавливают микроскоп и осветитель на крестовину, что обеспечивает необходимое расстояние между источником света и зеркалом микроскопа. На предметный столик помещают препарат. Устанавливают объектив 8 х. Поднимают конденсор вверх до упора. Открывают полностью диафрагму' конденсора. Отодвигают матовое стекло. Ставят плоское зеркало. Закрывают диафрагму осветителя, оставив только небольшое отверстие.
2. Включают осветитель. Пользуясь реостатом, регулируют яркость света таким образом, чтобы нить лампы давала слабый накал (иначе можно повредить глаза!). Корпусу осветителя придают такое положение, при котором свет падал бы в центр зеркала.
* Приводимая методика установки света нужна только при работе с микроскопами старых : эразцов, не имеющих вмонтированного в микроскоп источника света, но оснащенных зеркалами для улавливания и направления света в конденсор. При работе с современными микроскопами эта методика не требуется.
3. Закрывают зеркало микроскопа кружком белой бумаги и фокусируют на него изображение витка нити лампы осветителя. Это достигается передвижением патрона лампы осветителя.
4. Глядя в окуляр и слегка вращая зеркало, ловят в поле зрения изображение краев диафрагмы осветителя, которое будет иметь вид светлого пятна с нечеткими краями. Величина пятна зависит от степени раскрытия диафрагмы и положения объектива. Чем больше отверстие диафрагмы и чем выше объектив, тем больше пятно. Если оно занимает значительную часть поля зрения, его уменьшают, несколько опустив объектив или сузив отверстие диафрагмы осветителя, глядя при этом в окуляр. В тех случаях, когда пятно сдвинуто к краю поля зрения, его переводят в центр осторожным поворотом зеркала. По ряду причин (отсутствие идеального точечного источника света, аберрация) под микроскопом чаще всего видно не одно пятно, а несколько четких пятен. Установку света осуществляют по центральному пятну. В случае образования по краям диафрагмы осветителя цветной бахромы центрируют конденсор, смещая его регулировочными ручками в сторону красного цвета бахромы и корректируя при этом центровку зеркалом до момента симметричного распределения цвета бахромы.
5. Используя объектив 8*, фокусируют объект в области светлого пятна.
6. Слегка опуская конденсор, фокусируют в плоскости препарата изображение краев диафрагмы осветителя, т. е. получают изображение светлого пятна с четко очерченными краями.
7. С помощью зеркала переводят полученное яркое пятно в центр поля зрения.
Если все сделано правильно, то это светлое пятно, видимое одновременно с препаратом, будет равномерно освещено. В противном случае нужно добиться его равномерного освещения, слегка поворачивая корпус осветителя.
8. Открывают диафрагму осветителя так, чтобы светлое пятно заняло все поле зрения.
9. Устанавливают объектив 40* для препарата «раздавленная капля» или 90 х (100х) для фиксированного окрашенного препарата, на который предварительно наносят иммерсионное масло, и фокусируют объект.
Яркость освещения следует регулировать только изменением накала лампы осветителя или применением светофильтров. Положение зеркала, конденсора и диафрагмы осветителя больше не изменять! Никогда не следует поднимать и опускать конденсор или необоснованно использовать его ирисовую диафрагму для регулировки яркости поля зрения, так как это снижает разрешающую способность микроскопа, ухудшает изображение, может даже исказить его. Диафрагмой конденсора пользуются только для изменения контрастности изображения. Необходимость использования диафрагмы конденсора отпадает, если заранее привести в соответствие апертуру конденсора с апертурой используемого объектива. Как указывалось, апертура неиммергированного конденсора близка к 1, а числовая апертура объектива 40* составляет 0,65. Практически можно воспользоваться следующим приемом: установив свет по Кёлеру и сфокусировав препарат с объективом 40*, вынуть окуляр и, глядя в тубус, прикрывать диафрагму конденсора до тех пор, пока края диафрагмы не станут видны у границы равномерно освещенной задней линзы объектива. В этот момент числовая апертура конденсора будет примерно равна числовой апертуре
объектива. При работе с объективом 90 х диафрагму конденсора оставляют открытой, поскольку числовая апертура этого объектива 1,25.
Правила работы с иммерсионным объективом. Сухой окрашенный препарат (приготовление см. ниже) помещают на столик микроскопа и, пользуясь объективом 8х, устанавливают свет по Кёлеру. Затем в центр препарата на мазок наносят каплю иммерсионного масла, заменяя тем самым «сухую» систему иммерсионной. С помощью макрометрического винта опускают тубус микроскопа до погружения объектива в масло. Эту операцию нужно проводить очень осторожно, следя сбоку за тем, чтобы фронтальная линза не коснулась предметного стекла и не получила повреждения. После погружения объектива в масло осторожно, также пользуясь макровпнтом, поднимают тубус и, наблюдая в окуляр, находят плоскость препарата. Точная фокусировка достигается с помощью микрометрического винта.
По окончании микроскопирования поднимают тубус, снимают препарат и осторожно протирают фронтальную линзу объектива сначала сухой хлопчатобумажной салфеткой, а затем той же салфеткой, но слегка смоченной бензином или бензолом. Оставлять масло на поверхности линзы ни в коем случае нельзя, так как оно способствует фиксированию пыли и может со временем привести к повреждению оптики микроскопа. Эффективен способ удаления масла как жидкого, так и застывшего, свежеотломленным пенопластом. В отдельных случаях помогает протирка тканью, смоченной дистиллированной водой. Края линз с выступающей оправой очищают с помощью палочки, обернутой тканью.
5.1.2. Микроскопия в темном поле
Микроскопия в темном поле основана на освещении объекта косыми лучами света. Эти лучи не попадают в объектив, поэтому поле зрения выглядит темным. Если препарат содержит клетки микроорганизмов, то косые лучи, проходя через такой препарат, в значительной степени отражаются от поверх-
ности клеток и настолько уклоняются от своего первоначального направления, что попадают в объектив. Тогда наблюдатель видит на черном фоне интенсивно светящиеся объекты, даже если их диаметр в 10 раз меньше, чем предел разрешения объектива. Такое освещение препарата достигается применением специального конденсора. Темнопольный конденсор имеет затемненную среднюю часть, поэтому центральные лучи света, идущие от зеркала, задерживаются, а в плоскость препарата попадают только боковые лучи, отраженные от зеркальных поверхностей, расположенных внутри конденсора (рис. 5.3).
При микроскопировании в темном поле можно увидеть объекты, величина которых измеряется сотыми долями микрометра, т.е.
Рис. 5.3. Конденсор темного поля ОИ-13
Рис. 5.4. Схема хода лучей в конденсоре темного поля:
1 — линза конденсора; 2 — черная пластинка, задерживающая центральные лучи; 3 —объектив
лежит за пределами видимости обычного микроскопа. Однако наблюдение объектов в темном поле позволяет различить только их контуры, но не дает возможности рассмотреть внутреннее строение (рис. 5.4).
Требования при работе с темнопольным конденсором. Успешная работа с темнопольным конденсором возможна только при строгом соблюдении ряда условий.
1. Апертура темнопольного конденсора должна быть на 0,2 — 0,4 единицы больше апертуры объектива (см. схему хода лу
чей в темнопольном конденсоре, рис. 5.4). В противном случае часть боковых лучей попадет в объектив, что вызовет частичное освещение поля зрения и снижение контраста. Поэтому наблюдение в темном поле ведут обычно с иммергированным конденсором, имеющим апертуру около 1,2, и сухими системами с числовой апертурой 0,65 — 0,85. Когда используют объектив с большей апертурой, то для получения четкого изображения объектив обязательно диафрагмируют, т. е. снижают его апертуру. С этой целью в объектив, извлеченный из револьвера, вводят специальную вставную диафрагму, которая входит в комплект выпускаемых темнопольных конденсоров. Еще удобнее пользоваться объективами, снабженными ирисовой диафрагмой, расположенной между линзами. Вращением специального кольца, имеющегося на оправе, изменяют диаметр отверстия диафрагмы и тем самым снижают числовую апертуру объектива.
2. Накал лампы осветителя должен быть максимальным, так как темнопольный конденсор пропускает лишь незначительную часть поступающего светового потока, поэтому особое значение приобретает правильная установка света, максимальное его использование и особенно тщательная центровка.
3. Толщина предметных стекол не должна превышать 1,2 мм, так как все конденсоры темного поля имеют очень маленькое рабочее расстояние. В противном случае фокус конденсора окажется толще предметного стекла, а не в плоскости препарата, и наблюдатель ничего не увидит.
4. Следует обращать внимание на толщину и чистоту препарата. Чем толще препарат и чем больше в нем посторонних частиц, преломляющих свет (пыль, пузырьки воздуха и т.д.), тем менее контрастным получается изображение, так как каждая частица, отражая лучи, освещает поле зрения. В связи с этим для приготовления препарата используют тщательно очищенные предметные и покровные стекла.
Порядок работы с темнопольным конденсором.
L. Препарат «раздавленная капля», приготовленный на тонком и тщательно очищенном предметном стекле, помешают на столик микроскопа и фокусируют с объективом 8х. После этого положение тубуса не меняют до фокусировки препарата с объективом 40 х.
2. Вынимают светлопольный конденсор и окуляр, вывинчивают один из объективов. В некоторых микроскопах может иметься одно свободное от объектива отверстие, закрытое специальной заглушкой, так что можно воспользоваться этим отверстием, вывинтив заглушку.
3. Закрыв диафрагму осветителя, фокусируют изображение нити лампы на зеркале, прикрытом кружком белой бумаги, как при установке света по Кёлеру. Предварительно необходимо с помощью реостата уменьшить яркость света.
4. Открывают диафрагму осветителя. Прикрывают верхний конец тубуса микроскопа матовым стеклом (вместо окуляра) и, слегка поворачивая зеркало, добиваются равномерного освещения поля. После этого зеркало перемещать нельзя!
5. Вставляют окуляр и устанавливают объектив 8*. Осторожно, не задевая зеркала, устанавливают темнопольный конденсор таким образом, чтобы белый винт был обращен в сторону штатива микроскопа, а два регулировочных винта — в сторону осветителя. Надевают на регулировочные винты ключи.
6. Препарат сдвигают в сторону, на верхнюю линзу конденсора наносят каплю иммерсионного масла и, несколько опустив конденсор, снова устанавливают препарат, закрепив его клеммами.
7. Поднимают конденсор вверх до соприкосновения масляной капли с предметным стеклом. Капля должна равномерно заполнить пространство между линзой конденсора и предметным стеклом и не содержать пузырьков воздуха.
8. Отключают реостат осветителя, т.е. получают максимальное осветление.
9. Глядя в окуляр, центрируют конденсор. Для этого с помощью регулировочных винтов приводят точно в центр поля зрения изображение светлого кольца с темным пятном в середине или только светлого пятна.
10. Слегка поднимая или отпуская конденсор, устанавливают его в таком положении, чтобы в поле зрения исчезло темное пятно и осталось только замкнутое светлое пятно.
11. Ставят объектив 40 х и фокусируют препарат.
5,1.3. Микроскопия с фазово-контрастным устройством
Микроскопия с фазово-контрастным устройством основана на том, что с его помощью различия в фазе световых лучей, возникающие при прохождении их через прозрачные объекты, превращаются в амплитудные, в результате чего объекты становятся контрастными. Глаз человека выявляет различия в длине волны света (цвет) и ее амплитуде (интенсивность, яркость), но не в состоянии заметить смещение фазы. Неокрашенные клетки микроорганизмов хорошо видны в проходящем свете обычного светлопольного микроскопа только в том случае, когда значительная часть энергии света, прошедшего через них, поглощается. При этом выходящая из объекта (клетки) световая волна имеет меньшую амплитуду, т.е. яркость, и этот объект воспринимается глазом наблюдателя как более темный, контрастный по сравнению с окружающей средой. Однако многие микроорганизмы, размеры которых лежат в пределах разрешающей способности микроскопа, мало отличаются по прозрачности (плотности) от окружающей среды. Амплитуда световой волны, проходя идей через клетки таких микроорганизмов, почти не меняется, поэтому объекты плохо различимы или даже невидимы в обычный светлопольный микроскоп. Поле зрения кажется наблюдателю почти однородным.
Объект можно сделать более контрастным, либо почти до предела закрывая диафрагму конденсора, либо окрашивая клетки, либо меняя фазово-контрастное устройство. Первое нежелательно, так как снижает апертуру конденсора и тем самым заметно уменьшает разрешающую способность микроскопа. Окрашивание препарата дает хорошие результаты, но в большинстве случаев оно осуществляется после фиксации микроорганизмов, что не всегда желательно. Основная ценность метода фазового контраста состоит в том, что он дает возможность наблюдать живые объекты, не прибегая к их фиксации и окрашиванию. Применение фазово-контрастного устройства не позволяет увеличить разрешающую способность микроскопа, но дает возможность увидеть прозрачные
Рис. 5.5. Схема хода лучей при использовании фазово-контрастного устройства:
1 — кольцевая диафрагма; 2 — конденсор; 3 — объект; 4 — объектив; 5 — фазовая пластинка
объекты более четко и даже выявить некоторые структуры и включения в клетках крупных бактерий.
Оптическая система, используемая для получения фазового контраста, состоит из фазовой пластинки и кольцевой диафрагмы. Фазовая пластинка расположена в задней фокальной плоскости объектива и представляет собой прозрачный диск, на котором напылено кольцо из металлов. Кольцевая диафрагма расположена под конденсором и представляет собой прозрачную щель в виде кольца на непроницаемой для света пластинке. Световая волна, проходя через клетки микроорганизмов, отстает по фазе примерно на */4 X относительно прямых волн, прошедших только через окружающую среду. В объективе микроскопа эти две волны интерферируют. Результирующая волна имеет ту же длину и амплитуду, что и прямая, но несколько отличается от нее по фазе. Этого отличия оказывается недостаточно, чтобы частицу можно было заметить в обычный микроскоп. Для превращения разности фаз в разность амплитуд служит фазовая пластина, которая дополнительно сдвигает дифрагированный луч на '/4 X (рис. 5.5).
Фазовый эффект создается в результате интерференции прямых лучей, не отклонившихся при прохождении через препарат, и боковых дифрагированных лучей, которые в результате прохождения через объект и через фазовую пластинку либо совпадают по фазе с прямыми, либо сдвинуты относительно них на */2 X, т.е. находятся в противофазе. В первом случае обе волны складываются и изображение объекта становится более светлым, чем окружающий фон. Это светлый негативный контраст. По принципу негативного контраста устроен так называемый аноптралъный микроскоп. Во втором случае дифракционная волна вычитается из прямой и объект выглядит более темным — темный, позитивный, контраст (рис. 5.6). Кроме того, кольцевая диафрагма уменьшает интенсивность центрального светового пучка, что также усиливает контрастность.
Широкое применение нашли фазово-контрастные устройства, выполненные по схеме позитивного контраста. Наиболее распространена модель КФ-4 (рис. 5.7). Фазово-контрастное устройство представляет собой приставку к микроскопу, состоящую из вспомогательного окуляра, специальных фазовых объективов и конденсора с набором кольцевых диафрагм, каждая из которых соответствует фазовой пластинке определенного объектива. Кольцевые диафрагмы установлены в револьверном диске под конденсором и поворотом диска могут легко меняться, причем в окне крышки диска появляется цифра, соответствующая увеличению применяемого объектива. Кроме того, в револьверном диске имеется свободное отверстие — «нулевая диафрагма» — для наблюдений обыч-
Рис. 5.6. Схемы, поясняющие принцип фазового контраста:
а — сдвиг фаз между дифрагированной (£>) и прямой (У) волнами; б — темный контраст; в — светлый контраст
Рис. 5.7. Фазово-контрастное устройство КФ-4:
1 — конденсор; 2 — револьверный диск с набором кольцевых диафрагм; 3 — центровочные винты; 4 — вспомогательный микроскоп; 5 — набор фазовых объективов
ным способом. Конденсор снабжен двумя винтами для центровки кольцевой диафрагмы относительно фазового кольца объектива. Все фазовые объективы имеют на оправе обозначение «ф». Ими можно пользоваться и при обычной микроскопии, но из-за наличия фазового кольца они дают изображение пониженного качества.
Прядок работы с фазово-контрастным устройством. Методика исследования живых микроорганизмов с фазово-контрастным устройством сводится к следующему.
1. Удаляют из микроскопа обычный конденсор и на его месте устанавливают фазовоконтрастный. Диск револьвера конденсора поворачивают таким образом, чтобы в окошке стояла цифра «О». Ирисовая диафрагма конденсора должна быть полностью открыта.
2. Заменяют объектив 40х на фазовый объектив 40 х ф.
3. На предметный столик помещают препарат «раздавленная капля». Устанавливают свет по Кёлеру, пользуясь объективом 8х.
4. Устанавливают объектив 40 х ф.
5. Окуляр заменяют на «вспомогательный» и, перемещая тубус последнего, добиваются четкой фокусировки фазовой пластинки объектива. Она имеет вид темного кольца.
з Нетрусов
65
6. Поворотом диска револьвера выбирают объектив 40 хф — с кольцевой диафрагмой. Теперь под микроскопом видно не только фазовое кольцо, но и светлое кольцо — щель диафрагмы.
7. Пользуясь винтами для центровки конденсора, перемещают кольцевую диафрагму так, чтобы она совместилась с фазовым кольцом (см. рис. 5.6). Если ширина темного кольца (фазовой пластинки) больше, чем ширина светлого кольца (щель диафрагмы), то необходимо, чтобы светлое кольцо вписалось в контур темного кольца концентрично.
8. Вспомогательный окуляр заменяют обычным и фокусируют препарат.
9. При микроскопировании с другими объективами устанавливают соответствующие кольцевые диафрагмы и каждый раз проверяют центрировку, пользуясь вспомогательным окуляром.
5.1.4. Люминесцентная микроскопия
Люминесцентная микроскопия основана на способности многих веществ биологического происхождения и красителей светиться под воздействием падающего на них света. Молекулы веществ, способных к люминесценции, поглощают энергию падающего света и переходят в возбужденное состояние, которое характеризуется более высоким энергетическим уровнем. В таком состоянии они находятся непродолжительное время и вновь возвращаются к исходному энергетическому уровню. Этот переход сопровождается отдачей избытка энергии в виде света — люминесценцией. Как правило, для возбуждения люминесценции объект освещают ультрафиолетовыми лучами с длиной волны 300—400 нм или сине-фиолетовыми лучами с длиной волны 400—460 нм.
Ряд веществ биологического происхождения — хлорофилл, витамин В2, алкалоиды, каротиноиды, порфирины, кофакторы, некоторые антибиотоки и другие соединения обладают собственной люминесценцией. В зависимости от содержания таких веществ в клетке группе микроорганизмов, например зеленым водорослям, некоторым дрожжам и бактериям (метаногенам), свойственна первичная люминесценция. Однако клетки большинства микроорганизмов люминесцируют очень слабо, поэтому их обрабатывают специальными красителями — флуорохромами, обладающими люминесценцией. Люминесценцию объекта после обработки флуорохромами называют наведенной, или вторичной.
Люминесцентная микроскопия увеличивает контрастность изображения, дает возможность различить отдельные клеточные структуры и даже отметить их изменения при различных функциональных состояниях клетки. Этот вид микроскопии широко применяют для выявления и учета живых и мертвых клеток про- и эукариот в природных субстратах, почвах, илах, ризосфере растений, а также для цитологических исследований клеток.
Люминесценцию в сине-фиолетовых лучах видимого света можно наблюдать с помощью обычного микроскопа, установив на пути лучей синий стеклянный или жидкий светофильтр, проникающие сине-фиолетовые лучи видимого спектра. Синие лучи, мешающие выявлению люминесценции, убирают желтым светофильтром, который помещают на окуляр микроскопа. В результате наблюдатель видит на темном фоне люминесцирующие объекты. Однако для микробиологических исследований наиболее удобен люминесцентный микроскоп, в котором люминесценция возбуждается сине-фиолетовыми
лучами и ближним ультрафиолетом. Оптическая схема люминесцентных микроскопов позволяет наблюдать объекты при освещении их как в проходящем, так и в падающем свете. В современных люминесцентных микроскопах возбуждающий люминесценцию свет направляется на препарат сверху, через объектив (МИКМЕД 2, ЛЮМАМ РПО 11 —ОМО, Санкт-Петербург, Россия; «Axios-сор» — Zeiss, Jena, Germany). При освещении объектов сверху (для возбуждения люминесценции) одновременно допускается освещение объектов снизу с помощью конденсора темного поля или фазово-контрастного устройства. Люминесцентный микроскоп позволяет также исследовать объекты в видимой области спектра в проходящем и отраженном свете в темном поле. Устройство люминесцентного микроскопа и порядок работы даны в описании каждой конкретной модели и здесь не приводятся.
Большинство микробиологических объектов рассматривают с объективами 40 х или 100 х. При переходе от объектива меньшего увеличения к объективу большего увеличения соблюдают определенную последовательность. Просмотрев объект с меньшим увеличением, ставят объектив большего увеличения. Затем фокусируют препарат и вновь настраивают освещение, проверяя центричность и резкость изображения полевой диафрагмы и источника света. С иммерсионными объективами используют соответственно дистиллированную воду и нелюминесцирующее иммерсионное масло (вазелиновое или сандаловое).
5.1.5. Конфокальная лазерная сканирующая микроскопия
Конфокальная (софокусная) лазерная сканирующая микроскопия (КЛСМ) позволяет наблюдать исследуемые объекты (ткани, клетки как растительные и животные, так и бактериальные) в их естественном состоянии, не подвергая обезвоживанию, и с разрешением не меньшим, чем это достигается при флуоресцентной микроскопии. Микроскоп незаменим при исследовании биопленок, выявлены! пространственного расположения бактерий и грибов на поверхности растений и почвенных частицах, а также для многих других целей, где требуется на микроскопическом уровне проанализировать ненарушенную структуру объекта.
В основе КЛСМ используется схема флуоресцентного светового микроскопа (ФСМ). В отличие от традиционного светового микроскопа, в котором весь спектр светового пучка, отходящего от исследуемого образца, попадает в глаза исследователя или на пленку, лазерный луч в сканирующем лазерном микроскопе, управляемый посредством колеблющегося зеркала, расщепляющего луч, непрерывно и постепенно, линия за линией, сканирует заданную поверхность. При этом каждый участок образца, возбужденный посредством луча лазера, флуоресцирует и испускает свет, который в цифровом виде регистрируется фотоумножителем (обычно в пределах 768 — 512 линий). В качестве лазера используются аргонокриптоновые ионные лазеры, обеспечивающие свет с длинами волн в интервале 488 — 512 нм. Используются лазеры и на основе других инертных газов: гелий-неоновые (543 нм), аргоновый (351 —363 нм). Варьированием вида газа или смеси газов и энергетической интенсивности лазера можно получить необходимую длину возбуждающего света.
Источник света, луч лазера
Распределяющее луч лазера зеркало
Конфокальный микроканал
Линза объектива микроскопа
Сфокусированные на препарате лучи
Детектор (фотоумножитель)
Рис. 5.8. Принципиальная схема конфокального лазерного сканирующего микроскопа
Лучи, сфокусированные выше препарата
В любом случае в фотоумножитель попадают только те лучи, которые были точно сфокусированы на образце. Все другие лучи отсекаются «конфокальным микроканалом». Наличие этого канала наряду с лазерным источником света существенно отличают КЛСМ от традиционного ФСМ. В результате «одного прохода» луча формируется только один плоский вид образца. В результате наложения серии таких «плоских» изображений формируется трехмерное изображение объекта, при этом оно формируется только из полностью сфокусированных на объекте лучей, а все другие лучи отсекаются.
Скорость сканирования каждого участка варьирует от 0,25 до 4 с. Увеличение может быть усилено при изменении фокусного расстояния до объекта, при этом исследуемый участок будет или уменьшаться или увеличиваться соответственно. Создаваемое при сканировании изображение (имидж) аккумулируется в точечной форме. Результаты исследования регистрируются в цифровом виде в компьютере, что позволяет проводить математическую обработку получаемых результатов и реконструировать трехмерную форму объекта. Основными узлами КЛСМ являются: микроскоп, один или несколько лазеров, сканирующая головка и детектор (фотоумножитель). Схема КЛСМ приведена на рис. 5.8*.
В некоторых КЛСМ флуоресценция «расщепляется» на несколько каналов и регистрируется через индивидуальный конфокальный микроканал и детектор, что позволяет наряду с использованием соответствующих светопоглоща
* КЛСМ производят в ряде стран Западной Европы (Великобритании, Германии) и США. КЛСМ является очень дорогим прибором и в зависимости от предоставляемых возможностей может стоить от 80 тыс. до 450 тыс. долларов. Кроме того, программное обеспечение, которое продается отдельно, также может стоить от 5 тыс. до 20 тыс. долларов.
ющих фильтров получать цветное изображение объекта. Для работы с КЛСМ необходимо иметь мощный компьютер с пишущим CD-ROM и двумя мониторами, где на один выводится изображение исследуемого объекта, а с другого осуществляется управление процессом микроскопирования и регистрации изображений.
5.1.6. Учет активных (живых) и неактивных (мертвых) клеток бактерий с помощью флуоресцентных красителей
Традиционно для тотального учета клеток микроорганизмов как мертвых, так и живых используют флуоресцентные красители — акридин оранжевый, DAPI (4,6-diamino-2-phenylindole), FITC (fluorescein isothiocyanate) и др. Для выявления и учета только живых, активных клеток бактерий используют FDA (fluorescein diacetate) и SFDA (5-sulfofluorescein diacetate). Следовательно, для учета тех и других клеток одновременно в одном образце необходимо готовить два препарата.
В настоящее время разработаны комплексы красителей, позволяющие учитывать в одном препарате мертвые и живые клетки одновременно. Были разработаны наборы флуоресцентных красителей, известных под названием Live/Dead .бес-комплект (Haugland, 1996). Комплект содержит набор красителей (например, Syto 9), окрашивающий живые клетки, и иодид пропидия (PI), окрашивающий мертвые клетки. Клетки с неповрежденными мембранами проницаемы для обоих красителей. При промывании клеток краситель Syto9 остается в клетках с ненарушенными мембранами и вымывается из клеток с поврежденными мембранами, тогда как PI остается в клетках в том числе и с поврежденными мембранами.
Оба красителя вносятся в исследуемый образец одновременно, и образец инкубируется в течение 10—15 мин. После этого образец фильтруют через бактериальный (с размером пор 0,2 мкм) поликарбонатный (черный) фильтр и промывают 2 мл изопропанола. Такая промывка усиливает контрастность препарата, живые клетки светятся зеленым светом, мертвые — красным.
Клетки учитывают под люминесцентным микроскопом со светофильтрами: 450-490 FT 510 LP 520.
5.2. ИЗУЧЕНИЕ МИКРООРГАНИЗМОВ С ПОМОЩЬЮ СВЕТОВОЙ МИКРОСКОПИИ
Морфологические особенности клеток микроорганизмов можно изучать с помощью различных методов микроскопии, а также применяя некоторые способы дифференциальной окраски. Выбор методов микроскопического анализа и способов окраски определяется конкретной целью исследования. Однако существует ряд приемов, которые имеют принципиальное значение и лежат в основе большинства специальных методов исследования морфологии и цитологии бактерий, — способы приготовления препаратов, фиксации и окраски клеток.
Препараты готовят, как правило, на предметных стеклах, толщина которых не должна превышать 1,2—1,4 мм. Более толстые стекла не позволяют получить резкое изображение краев диафрагмы осветителя в плоскости препарата, так как оно оказывается в толще стекла, а это нарушает фокусировку конденсора и резко снижает четкость изображения. Толстые предметные стекла недопустимы при работе с иммерсионным объективом, когда необходимо полностью использовать числовую апертуру системы.
Существенным моментом является подготовка поверхности предметных стекол, что особенно важно при изготовлении фиксированных препаратов. Поверхность стекла должна быть тщательно очищена и обезжирена, чтобы капля жидкости равномерно расплывалась по стеклу, а не собиралась в выпуклые, медленно высыхающие капельки. Наиболее надежный способ обезжиривания — обработка стекол хромовой смесью с последующим споласкиванием водой и спиртом. В повседневной работе, однако, вполне достаточно бывает тщательно натереть сухое стекло мылом, после чего вытереть его чистой хлопчатобумажной салфеткой. Хорошее обезжиривание достигается протиранием вымытых и высушенных стекол ватой, смоченной эфиром (после этого промывание водой не требуется), или обжиганием поверхности стекол в пламени горелки (жир при этом сгорает). Запрещается кипячение стекол в растворах щелочей, в том числе моющих средствах, а также длительное выдерживание стекол в таких растворах, так как щелочи разъедают стекло, делая его поверхность матовой. Хранить чистые обезжиренные стекла можно в сухом состоянии или в этаноле.
Покровные стекла, применяемые для приготовления препаратов микроорганизмов, также должны быть тщательно вымыты и высушены. Толщина покровных стекол не должна превышать 0,15 — 0,17 мм. Более толстые стекла резко ухудшают качество получаемого изображения.
5.2.1. Препараты живых клеток микроорганизмов
Для изучения живых клеток микроорганизмов применяют препараты «раздавленная капля», «висячая капля», «отпечаток», «агаровая пленка» («микрокультура»). Препараты живых клеток рассматривают с «сухими системами» микроскопа. Препараты, работа с которыми закончена, прежде чем вымыть, выдерживают в дезинфицирующем растворе.
Препарат «раздавленная капля». На предметное стекло наносят каплю стерилизованной фильтрацией водопроводной воды, помещают в нее небольшое количество клеток изучаемых микроорганизмов, размешивают и накрывают покровным стеклом. Микроорганизмы, выращенные и на плотной, и в жидкой питательной среде, переносят в каплю воды бактериологической петлей; выращенные в жидкой среде можно переносить также стерильной пипеткой. В последнем случае каплю воды на предметное стекло можно не наносить. Капля исследуемого материала должна быть настолько мала, чтобы из-под покровного стекла не выступал избыток жидкости. Если он образовался, его необходимо удалить фильтровальной бумагой. При продолжительном изучении микро-скопируемого объекта края покровного стекла рекомендуется залить лаком для ногтей, что предотвратит быстрое высыхание препарата.
Препарат «висячая капля». Каплю суспензии микроорганизмов петлей или обычным пером наносят на покровное стекло, которое поворачивают каплей вниз и помещают на специальное предметное стекло с углублением (лункой) в центре. Капля должна свободно висеть, не касаясь краев и дна лунки. Края лунки предварительно смазывают вазелином. Капля оказывается герметизированной во влажной камере, что делает возможным многодневное наблюдение за объектом. Для длительных наблюдений используют стерильные стекла, а суспензию микроорганизмов готовят в жидкой питательной среде. При работе с бактериями этот метод используется редко.
Препарат «отпечаток». Из агаризованной среды, на которой микроорганизмы растут сплошным газоном или в виде отдельных колоний, вырезают скальпелем небольшой кубик и переносят его на предметное стекло так, чтобы поверхность с микроорганизмами была обращена вверх. Затем к газону или к колонии прикладывают чистое покровное стекло, слегка надавливают на него петлей или пинцетом и тотчас же снимают, стараясь не сдвинуть в сторону. Полученный препарат помещают отпечатком вниз в каплю воды или метиленового синего (1:40) на предметное стекло. Отпечаток можно получить и на предметном стекле, если касаться поверхности колонии или газона предметным стеклом. Препарат «отпечаток» используют в основном при исследовании спороношения стрептомицетов.
Препарат «микрокультура» (или «агаровая пленка»). На тонкое, простери-лизованное и нагретое предметное стекло наносят стерильной нагретой пипеткой 0,2—0,3 мл горячей агаризованной питательной среды и распределяют по всей поверхности стекла. После застывания среды удаляют петлей лишний агар, оставляя два тонких участка пленки величиной с покровное стекло каждый. В центр квадратов бактериальной петлей или пипеткой наносят каплю жидкой культуры или суспензии клеток микроорганизма. Стекло помещают во влажную камеру (чашка Петри со слоем мокрой фильтровальной бумаги), которую ставят в термостат. Перед микроскопированием на пленку с выросшей микрокультурой наносят каплю красителя или каплю воды в случае подсыхания пленки и затем осторожно накрывают покровным стеклом.
Выращивание микроорганизмов непосредственно на предметном стекле позволяет вести микроскопическое наблюдение за процессами их роста и развития, изучать цикл развития, способ размножения (деление — почкование), влияние на эти процессы каких-либо агентов. На препаратах не нарушается естественное расположение клеток в растущей микроколонии. Выращивание микроколонии можно проводить в аэробных или анаэробных (под покровным стеклом, загерметизированным лаком) условиях.
Агаровую пленку можно нанести на покровное стекло и приготовить препарат «висячая капля». На таком препарате можно наблюдать движение бактерий по типу скольжения.
5.2.2. Препараты фиксированных окрашенных клеток микроорганизмов
Получение фиксированных окрашенных препаратов включает приготовление мазка, высушивание, фиксацию и окраску.
Приготовление мазка. На обезжиренное спиртом предметное стекло помещают маленькую каплю водопроводной воды и переносят в нее петлей небольшое количество исследуемого материала, как для препарата «раздавленная капля». Полученную суспензию равномерно размазывают петлей на площади 1 — 2 см максимально тонким слоем. Мазок должен быть настолько тонок, чтобы быстро высыхал после приготовления.
Высушивание мазка лучше всего производить при комнатной температуре на воздухе. Хорошо приготовленный тонкий мазок высыхает очень быстро. Если высушивание происходит медленно, препарат можно слегка нагреть в струе теплого воздуха высоко над пламенем горелки, держа стекло мазком вверх. Эту операцию следует проводить осторожно, не перегревая мазок, иначе клетки микроорганизмов деформируются.
Фиксация препарата преследует несколько целей: убить микроорганизмы, т.е. сделать безопасным дальнейшее обращение с ними; обеспечить лучшее прилипание клеток к стеклу; сделать мазок более восприимчивым к окраске, так как мертвые клетки окрашиваются лучше, чем живые. Самым распространенным способом фиксации является термическая обработка. Для этого препарат обычно трижды проводят через наиболее горячую часть пламени горелки, держа предметное стекло мазком вверх. Не следует перегревать мазок, так как при этом происходят грубые изменения клеточных структур, а иногда и внешнего вида клеток, например их сморщивание. Для исследования тонкого строения клетки прибегают к фиксации различными химическими веществами (см. приложение 5). Фиксирующую жидкость наливают на мазок, либо препарат на определенное время погружают в стакан с фиксирующим раствором.
Окраска. Клетки микроорганизмов окрашивают главным образом анилиновыми красителями. Различают кислые и основные красители. К кислым относятся красители, у которых красящими свойствами обладает анион, у основных красителей хромофором является катион. Примерами кислых красителей служат эозин, эритрозин, нигрозин, кислый фуксин; все они интенсивно связываются с цитоплазматическими компонентами клетки. Основные красители — метиленовый синий, основной фуксин, генциановый фиолетовый, кристалл-виолет, сафранин — интенсивнее связываются с ядерными компонентами клетки. Высокая концентрация ДНК и рибосомальной РНК в клетке бактерии делает ее более чувствительной к основным красителям. В связи с этим в микробиологической практике применяются почти исключительно основные красители.
Различают простое и дифференциальное окрашивание микроорганизмов. В первом случае прокрашивается вся клетка, так что становятся хорошо видны ее форма и размеры. Дифференциальное окрашивание выявляет только определенные структуры клетки и запасные вещества.
Для простого окрашивания клеток микроорганизмов чаще всего пользуются фуксином, генциановым фиолетовым, метиленовым синим. Фиксированный препарат помещают на параллельные стеклянные палочки, лежащие над лотком, наносят на него раствор красителя и выдерживают в нем в течение 1 — 3 мин. Следят за тем, чтобы во время окрашивания краситель на мазке не подсыхал, и в случае необходимости добавляют новую порцию красителя.
По окончании окраски препарат промывают водой до тех пор, пока стекающая вода не станет бесцветной. Затем препарат высушивают на воздухе или осторожно промокают фильтровальной бумагой, помещают на окрашенный
мазок каплю иммерсионного масла и просматривают с объективом 100*. Для получения более чистых препаратов краситель наносят на мазок, покрытый фильтровальной бумагой. Метод окрашивания в модификации Синева позволяет использовать вместо раствора красителя заранее пропитанную им фильтровальную бумагу. В правильно окрашенном и хорошо промытом препарате поле зрения светлое и чистое, окрашены только клетки.
Фиксировать и окрашивать можно также и препараты-«отпечатки». Фиксированные, окрашенные препараты могут храниться длительное время.
Форму клеток и их расположение (цепочки, розетки, пакеты, тетрады и т.д.) выявляют, как правило, на препаратах «раздавленная капля» при светлопольной или фазово-контрастной микроскопии. Для определения формы клеток мелких палочковидных бактерий, таких как Serratia marcescens, готовят препарат фиксированных клеток и применяют простое окрашивание. Клетки мелких бактерий, имеющих выросты — простеки (роды Caulobacter, Labrys, Prosthecomicrobium, Stella и некоторые другие,), целесообразно исследовать методом фазового контраста или в темном поле. Естественное расположение клеток в колонии микроорганизмов, а также спор и спороносцев у актиномицетов и мицелиальных грибов изучают на препарате «отпечаток».
Необходимо помнить, что возраст культуры, состав среды и условия культивирования существенно влияют на морфологию и цитологию микроорганизмов.
5.2.3. Определение размеров клеток микроорганизмов
Клетки микроорганизмов измеряют под микроскопом с помощью окулярной линейки — микрометра или окулярного винтового микрометра. Для измерения лучше использовать живые, а не фиксированные клетки, так как фиксация и окраска приводят к некоторому изменению истинных размеров клеток. Размеры клетки удобно определять с помощью фазово-контрастного устройства. Если клетки подвижны, препарат слегка подогревают или к капле исследуемой суспензии добавляют каплю 0,1%-го водного раствора агара. Размеры клеток выражают в микрометрах.
Окулярный микрометр (окуляр-микрометр) представляет собой круглую стеклянную пластинку, в центре которой выгравирована линейка длиной 5 мм. Линейка разделена на 50 частей. Окулярный микрометр вставляют в окуляр. Для этого вывинчивают глазную линзу окуляра, помещают на его диафрагму окулярный микрометр делениями вниз и завинчивают линзу. Однако только с помощью окуляр-микрометра нельзя непосредственно измерить величину клетки, так как последние рассматриваются через объектив и окуляр, а деления линейки — только через верхнюю линзу окуляра. Поэтому, прежде чем приступить к измерению величины клеток, необходимо определить цену деления окулярного микрометра для данного увеличения микроскопа. Это делают с помощью объективного микрометра.
Объективный микрометр (объект-микрометр) — это металлическая пластинка с отверстием в центре (рис. 5.9). В отверстие вставлено стекло, на которое нанесена линейка .глиной 1 мм. Она разделена на 100 частей, т. е. деление объективного микрометра соответствует 0,01 мм, или 10 мкм. Для определения цены делений окулярного микрометра объективный микрометр помещают на столик микроскопа и фокусируют при малом увеличении. Изображение линейки перемещают в центр поля зрения и только после этого меняют объектив на тот, при котором будут определяться размеры клеток. Пере-
Рис. 5.9. Объективный микрометр: а — общий вид; б — вид под микроскопом
мещая столик микроскопа и поворачивая окуляр, устанавливают микрометры так, чтобы их шкалы были параллельны и одна перекрывала другую. Цену деления окулярного микрометра определяют по принципу нониуса, т. е. совмещают одно из делений шкалы окулярного и объективного микрометров и находят следующее их совмещение. Устанавливают, скольким делениям объективного микрометра соответствует одно деление окулярного микрометра. Например, 2 деления объект-микрометра (20 мкм) соответствуют 5 делениям окуляр-микрометра. Следовательно, одно деление окуляр-микро-метра равно 4 мкм (20: 5). Если теперь на столик микроскопа поместить препарат с клетками микроорганизмов и рассматривать его при том же увеличении, то можно измерить величину клетки. Для этого определяют, какому числу делений окулярной линейки соответствует величина измеряемого объекта, и умножают это число на цену деления окулярного микрометра.
Удобно определять размеры клеток с помощью винтового окулярного микрометра МОВ-1-15. Винтовой окулярный микрометр закрепляют на тубусе микроскопа, предварительно вынув окуляр. В окуляре винтового микрометра имеется неподвижная шкала с ценой деления 1 мм для определения размеров крупных объектов и подвижная стеклянная пластинка с перекрестием. Пластинка связана с микрометрическим винтом-барабаном и перемешается вместе с перекрестием при его вращении. Для измерения длины клетки вращением микрометрического винта-барабана окулярного микрометра подводят перекрестие к концу клетки и отмечают деление на барабане. Затем, вращая барабан, перемещают перекрестие до другого конца клетки и вновь отмечают деление на барабане. Определяют, скольким делениям микрометрического винта-барабана соответствует длина клетки, и умножают полученное значение на цену деления барабана при данном увеличении микроскопа.
Цену деления барабана для каждого объектива определяют с помощью объективного микрометра. С этой целью подводят перекрестие к началу одного деления объективного микрометра и отмечают деление на барабане. Затем, вращая барабан, перемещают перекрестие до конца деления объективного микрометра и вновь отмечают деление на барабане. Определяют, скольким делениям микрометрического винта-барабана соответствует одно деление объективного микрометра. Например, одно деление объективного микрометра, т.е. 10 мкм, соответствует ^делениям микрометрического винта-барабана; следовательно, одно деление его при данном увеличении микроскопа равно 10: X [мкм].
Для получения достоверных результатов необходимо измерить не менее 20 — 30 клеток. При определении размеров клеток округлых форм измеряют их диаметр, у клеток других форм — длину и ширину; указывают средние размеры клеток и пределы колебаний, т. е. минимальные и максимальные размеры.
5.2.4. Выявление клеточных структур и запасных веществ
Капсулы. Клетки многих микроорганизмов, особенно при росте на средах, богатых углеводами, могут быть окружены рыхлым внешним слоем — капсу
лой или слизью. Эти структуры часто имеют консистенцию геля и плохо видны при микроскопировании живых клеток. Химический состав капсул у разных бактерий неодинаков, поэтому их нельзя выявить каким-либо одним методом окраски. Кроме того, капсулы при окраске легко деформируются, а вещество капсулы слабо связывает краситель, который легко отмывается в процессе обработки препарата. Чаще всего для выявления капсул применяют способ «негативной» окраски (негативного контрастирования) с помощью жидкой туши. Для этого небольшое количество клеток с плотной среды помещают в каплю разбавленного фуксина, смешивают с каплей туши, закрывают покровным стеклом и просматривают с объективом 40 х. На общем темном фоне препарата хорошо видны бесцветные капсулы, окружающие клетки микроорганизмов, окрашенные в розовый цвет. Кроме того, существуют специальные методы окраски капсул, один из них приведен ниже.
Выявление капсул по методу Гипса. На конец предметного стекла микробиологической петлей наносят каплю черной туши, вносят в нее клетки, хорошо перемешивают и ребром покровного стекла делают мазок по всей поверхности предметного стекла. Мазок высушивают на воздухе и фиксируют 5—10 мин смесью Никифорова или 3 мин абсолютным метанолом. Далее мазок окрашивают карболовым фуксином Циля, разбавленным водой в соотношении 1:3. Время окрашивания — 2 — 3 мин. Препарат промывают водой, высушивают на воздухе и микроскопируют с иммерсионной системой. На темно-сером фоне препарата контрастно выделяются розово-малиновые клетки бактерий, окруженные бесцветными капсулами.
Клеточная стенка. Тонкую структуру клеточной стенки хорошо видно лишь при электронной микроскопии. Для наблюдения клеточной стенки при световой микроскопии применяют метод темного поля либо специальную окраску, с помощью которой удается легко выявить границы между отдельными клетками, расположенными в виде длинных нитей или плотных агрегатов. На обезжиренном стекле делают мазок клеток исследуемых бактерий, высушивают его на воздухе и фиксируют в течение 5 мин 5%-м раствором фосфомолибденовой кислоты. Затем препарат промываю !’ водой и окрашивают не более 15 с 0,02%-м раствором кристаллвиолета. Снова промывают водой, высушивают и микроскопируют с иммерсионной системой. Клеточная стенка окрашивается в черный, а цитоплазма в бледно-сиреневый цвет.
Можно использовать метод окраски клеточной стенки по Гутштейну. Мазок выдерживают 15 мин в жидкости Карнуа, протравляют в течение 25 мин 10%-м водным раствором таннина, промывают водой и окрашивают водным фуксином или 0,02%-м раствором кристаллического фиолетового в течение 30 — 60 с. Затем препарат промывают, высушивают и микроскопируют с иммерсией.
Окраска по Граму. С молекулярной организацией и химическим составом клеточной стенки связывают способность бактерий окрашиваться по Граму. Эта окраска является важным диагностическим признаком, она коррелирует со многими другими свойствами бактерий. По способности окрашиваться красителями триметилфенолового ряда все бактерии делятся на две группы: грам-положительные и грамотрицательные. Грамположительные бактерии удерживают комплекс генцианового фиолетового с йодом при обработке препарата спиртом и поэтому окрашиваются в фиолетовый цвет. Грамотрицательные бактерии не обладают такой способностью и обесцвечиваются спиртом. При пос
ледующей обработке фуксином или сафранином они приобретают розовую окраску.
По Граму рекомендуется окрашивать клетки молодых, чаще всего суточных, культур, так как способность удерживать краситель зависит от физиологического состояния бактерий. Например, некоторые бактерии после прекращения активного роста теряют способность окрашиваться по Граму.
Окраска по Граму заключается в следующем. На одном обезжиренном стекле делают мазки разных микроорганизмов: в центре — мазок клеток исследуемой культуры, слева и справа — контрольных культур. Клетки одной контрольной культуры должны быть грамположительными (например, Micrococcus luteus или Bacillus cercus), другой — грамотрицательными (например, Escherichia coli). Мазки следует готовить тонкими, чтобы клетки равномерно распределялись по поверхности стекла и не образовывали скоплений. Препарат высушивают на воздухе, фиксируют над пламенем горелки и окрашивают в течение 1 — 2 мин карболовым генциановым или кристаллическим фиолетовым. Затем краситель сливают и, не промывая мазок водой, обрабатывают его 1 — 2 мин раствором Люголя до почернения. Сливают раствор Люголя, препарат обесцвечивают в течение 0,5— 1,0 мин 96%-м этиловым спиртом, быстро промывают водой и дополнительно окрашивают 1 — 2 мин водным фуксином. Краситель сливают, препарат промывают водой, высушивают и микроскопируют с иммерсионной системой. При правильном окрашивании грамположительные бактерии имеют сине-фиолетовый, грамотрицательные розово-красный цвет.
Выявление кислотоустойчивости клеток. Кислотоустойчивость — свойство, характерное для некоторых микобактерий и нокардий: при обработке кислотой их окраска сохраняется. Кислотоустойчивость обусловлена особенностями химического состава клеточной стенки этих бактерий — высоким содержанием в ней сложных липидов и, в частности, наличием миколовых кислот.
Наибольшее распространение получил способ выявления кислотоустойчивости по Циль—Нильсену. На обезжиренном предметном стекле готовят два мазка: исследуемых клеток и клеток кислотоустойчивых микобактерий. Препарат высушивают на воздухе и фиксируют над пламенем горелки. На мазки помешают фильтровальную бумагу, заливают препарат карболовым фуксином Циля и затем 2—3 раза подогревают до появления паров, держа стекло высоко над пламенем горелки. За появлением паров наблюдают, глядя на мазок сбоку, и при их появлении тотчас отставляют препарат в сторону. Дают препарату остыть, снимают фильтровальную бумагу, сливают краситель и мазок промывают водой. Затем препарат обесцвечивают 5%-м раствором H2SO4. Для этого предметное стекло погружают 2—3 раза в стакан с кислотой, не задерживая его в ней. Вновь тщательно промывают препарат водой и докрашивают 3 — 5 мин метиленовым синим по Леффлеру. Краску сливают, препарат промывают водой, высушивают и исследуют с иммерсионной системой. При строгом соблюдении режима окраски кислотоустойчивые клетки приобретают красный цвет, тогда как некислотоустойчивые — синий. Кислотоустойчивость можно определить у клеток любого возраста.
Жгутики. Способность к движению у большинства микроорганизмов обусловлена наличием жгутиков. Расположение и количество их у различных бактерий варьирует и имеет диагностическое значение. Особенно важен этот признак для идентификации палочковидных грамотрицательных бактерий. По ха
рактеру движения бактерий в препарате можно предположительно судить о типе жгутикования. Если жгутики расположены на одном или на двух полюсах клетки, то движение обычно очень быстрое — «ввинчивающееся», без покачивания из стороны в сторону; при латеральном или перитрихиальном расположении жгутиков клетки двигаются плавно, совершая колебательные отклонения от оси движения.
Отдельный бактериальный жгутик настолько тонок, что неразличим в световом микроскопе, если не окрашен особым способом, который увеличивает его кажущуюся толщину. Так, диаметр отдельных жгутиков колеблется в пределах 0,01—0,02 мкм (у Bdellovibrio 0,04—0,06 мкм), а максимальное разрешение светового микроскопа составляет 0,2 мкм. Лишь у немногих бактерий, например у представителей рода Spirillum, пучки жгутиков достаточно плотны, и их удается обнаружить при наблюдении в светлом поле с помощью фазовоконтрастного устройства или в темном поле. Существует несколько способов окраски жгутиков. Все они основаны на использовании различных «протрав» (состав см. в приложении 4), осаждающихся на поверхности жгутиков, благодаря чему диаметр жгутиков увеличивается и они становятся видимыми под световым микроскопом. Окраска жгутиков требует тщательной подготовки клеток и аккуратности в работе, так как они легко обламываются даже при легком взбалтывании суспензии.
Окраска жгутиков по методу Леффлера в модификации Пешкова. Бактерии, предназначенные для окраски жгутиков, ежедневно в течение 2—3 дней пересевают в свежую жидкую или на плотную среду, содержащую не более 1,5% агара. Для окраски используют клетки 12— 16-часовой культуры. Клетки осторожно берут петлей и переносят в пробирку со стерильной водой, подогретой до температуры, при которой их выращивали. Прежде чем делать мазок, каплю полученной суспензии просматривают под микроскопом и убеждаются в том, что клетки подвижны и плотность суспензии невелика: 5 —10 клеток в поле зрения.
Клетки легко теряют жгутики в момент приготовления мазка, поэтому необходимо обращать внимание на чистоту стекла и способ нанесения на него суспензии. Предметные стекла должны быть тщательно обезжирены. Непосредственно перед приготовлением мазка стекло 3 — 4 раза проводят через наиболее горячую часть пламени горелки. Дают стеклу остыть и пастеровской пипеткой, пером перьевой ручки или петлей наносят 3—4 маленькие капли культуры на стекло. Капли должны хорошо расплываться по стеклу и быстро высыхать. Высушенный мазок заливают протравой. Необходимо следить, чтобы протрава не подсыхала. Через 15 мин протраву смывают дистиллированной водой и препарат окрашивают в течение 5 мин разбавленным фуксином Циля, погружая его мазком вниз в раствор красителя. Затем препарат промывают водой, высушивают на воздухе и исследуют с иммерсионной системой. Обращают внимание на расположение жгутиков, их количество и длину.
По другому методу препарат заливают протравой через воронку с фильтром и трижды з течение 3 — 5 мин нагревают стекло над пламенем горелки до появления паров. Затем протраву сливают и препарат заливают через воронку с фильтром раствором Циля на 5—10 мин.
Нуклеоид. Обнаружить нуклеоид в бактериальной клетке при помогли светового микроскопа трудно. Основные красители, избирательно окрашивающие хроматин ядер эукариотических клеток, равномерно и интенсивно окрашива-?эт всю прокариотную клетку. Для избирательного окрашивания нуклеоида фиксированные клетки предварительно обрабатывают рибонуклеазой или раз
бавленной соляной кислотой, чтобы разрушить рибосомальную РНК. Последующее окрашивание основным красителем позволяет выявить нуклеоид в виде плотных тел, имеющих неправильные очертания и расположенных в центре или на обоих полюсах клетки. Довольно четко нуклеоид обнаруживается у следующих бактерий: Proteus vulgaris, Azotobacter chroococcum, Bacillus megaterium, B. cereus var. mycoides, B. subtilis.
Для выявления нуклеоида на предметном стекле делают мазок суточной культуры бактерий, высушивают его на воздухе и фиксируют в течение 2—3 мин в парах 2%-го раствора осмиевой кислоты. С этой целью на дно чашки Петри наносят 2—3 капли фиксатора, а предметное стекло помещают мазком вниз на обрезки стекла над фиксатором. По окончании фиксации препарат опускают на 2—3 мин в стаканчик с раствором 1 н. НС1 для гидролиза рибосомальной РНК. Стакан держат на водяной бане при 60 °C. После гидролиза препарат немедленно промывают водой. Затем мазок помешают на 15 мин в 1%-й раствор формалина, вновь промывают водой и окрашивают в течение 1 — 2 мин 0,1 — 1,0%-м водным раствором основного фуксина. Препарат промывают, высушивают и микроскопируют с иммерсионной системой. Цитоплазма окрашивается в розовый цвет, нуклеоид — в ярко-малиновый.
Многие микроорганизмы содержат различные внутрицитоплазматические включения: это могут быть продукты метаболизма, «складирующиеся» внутри клетки, структуры, необходимые для энергетического (хлоросомы зеленых бактерий, в которых находятся бактериохлорофиллы, фикобилисомы цианобактерий, содержащие пигменты фикобилипротеины) и конструктивного (карбоксисомы автотрофов, состоящие из ключевого фермента цикла Кальвина — рибулозодифосфаткарбоксилазы) обмена веществ. Некоторые включения имеют приспособительное значение — это, например, газовые вакуоли, необходимые для поддержания плавучей плотности водных микроорганизмов и магнитосомы железобактерий, содержащие магнетит (Fe3O4), который позволяет бактериям перемещаться в направлении линий магнитного поля.
Разнообразными соединениями представлены запасные полимерные гранулярные цитоплазматические вещества микроорганизмов, которые они накапливают внутри клетки. Чаще всего это полисахариды (гликоген, крахмал, гранулёза), липиды, полифосфаты. Некоторые бактерии накапливают в клетках серу. Резервом азота являются цианофициновые гранулы цианобактерий.
Гранулы углеводной природы (полисахариды) выявляют при обработке клеток раствором Люголя. Для этого к капле суспензии клеток на предметном стекле добавляют каплю раствора Люголя. Препарат накрывают покровным стеклом и микроскопируют. Гранулы крахмалоподобных веществ — гранулёзы — окрашиваются в синий, а гранулы гликогеноподобных полисахаридов — в красновато-коричневый цвет. Гликоген при окраске Люголем легко выявляется у дрожжей, гранулёза характерна для бактерий рода Clostridium. Реакция на гликоген хорошо проходит только в кислой среде, поэтому перед выявлением в клетках гликогена среду, в которой выращивали микроорганизмы, подкисляют.
Липидные гранулы. У дрожжей и мицелиальных грибов запасные липиды представлены нейтральными жирами, которые легко обнаруживаются в живых клетках без специальных методов окраски в виде сильно преломляющих свет капель. Бактерии в качестве резервных липидов образуют поли-р-оксимасля-
ную кислоту. Гранулы поли-р-оксибутирата хорошо заметны при микроскопировании живых бактериальных клеток с фазово-контрастным устройством, однако чаще для их выявления клетки окрашивают лиофильными красителями — Суданом III или Суданом черным. Готовят тонкий мазок клеток, высушивают его на воздухе и фиксируют в пламени горелки. Заливают поверхность мазка раствором Судана черного и оставляют краситель на 5 —15 мин. Избыток красителя сливают, просушивают препарат фильтровальной бумагой, просветляют в ксилоле, погружая в него несколько раз предметное стекло. Время просветления препарата не должно превышать 1 мин. После этого клетки дополнительно окрашивают в течение 10 с 0,1%-м водным раствором сафранина. Более длительная обработка сафранином нежелательна, так как маскируется основная окраска. Гранулы поли-р-оксибутирата окрашиваются в темный цвет, остальная часть клетки — в розовый.
Полифосфаты (волютин и метахроматин). В клетках прокариот волютин локализован в цитоплазме, в клетках эукариот — в вакуолях. Окраска волютиновых гранул основана на свойстве метахромазии — способности вызывать изменение цвета некоторых красителей (метиленовый синий, толуидиновый синий). Готовят тонкий мазок клеток, высушивают его на воздухе и фиксируют в пламени горелки. На фиксированный мазок наливают метиленовый синий по Леффлеру и окрашивают клетки в течение 10 мин. Краситель сливают, препарат промывают водой, высушивают и микроскопируют с иммерсионной системой. Клетки окрашиваются в голубой цвет, а зерна волютина — в фиолетово-красный.
Волютин легко выявить окраской по способу Омелянского, основанному на его плохой растворимости в растворах кислот. В этом случае на фиксированный в пламени горелки мазок наливают карболовый фуксин Циля и окрашивают клетки в течение 0,5 —1,0 мин. Краску сливают, промывают препарат водой и обесцвечивают 1%-м раствором H2SO4 в течение 20 — 30 с. Затем кислоту сливают, препарат промывают водой и дополнительно окрашивают 20 — 30 с метиленовым синим (1:40). Снова промывают препарат водой, высушивают и микроскопируют с иммерсионной системой. При правильном окрашивании гранулы волютина имеют красный цвет и хорошо видны на фоне синей цитоплазмы. Для выявления волютина у дрожжей применяют следующий способ. Фиксированный в пламени горелки мазок окрашивают метиленовым синим по Леффлеру в течение 3 мин. Краситель сливают, препарат промывают водой и, не высушивая, наносят на мазок небольшую каплю 1%-го раствора серной кислоты; мазок накрывают покровным стеклом и микроскопируют. Волютин имеет вид капель сине-фиолетового цвета на слабо-голубом фоне цитоплазмы.
Включения серы встречаются у бактерий, чей метаболизм зависит от этого соединения. Так, сера является источником энергии для аэробных тионовых бактерий, окисляющих сероводород, и донором электронов для анаэробных фотосинтезирующих бактерий. Благодаря двойному лучепреломлению включения серы хорошо заметны в клетках без специального окрашивания. Они растворяются при обработке клеток абсолютным спиртом, сероуглеродом или ледяной уксусной кислотой.
Параспоральные включения. Бактерии Bacillus thuringiensis, включающие более 80 подвидов, образуют параспоральные включения примерно в тот же пе-
риод, что и споры. Обычно эти включения состоят из нескольких полифунк-циональных белков — высокоспецифичных токсинов некоторых беспозвоночных животных, также проявляющих менее специфическую антимикробную активность. В состав некоторых включений могут входить и другие белки, например цитолитические. В клетке образуется от одного до нескольких включений разных размеров, часто имеющих форму бипирамид, ромбоэдров, пластинок-параллелепипедов, ромбоидов, а также неправильных глыбок. Поэтому белковые включения часто называют параспоральными кристаллами. Их окрашивают с помощью красителей, хорошо связывающихся с белками, например амидошварцем или анилиновым черным для шерсти.
Для выявления параспоральных включений готовят тонкий мазок спорули-рующей культуры, высушивают его на воздухе, фиксируют над пламенем горелки и в течение примерно 1 — 2 мин окрашивают одним из вышеуказанных красителей, следя за тем, чтобы краситель не подсыхал. Затем осторожно смывают краску водой и мазок в течение 15 — 30 с докрашивают фуксином Циля или сафранином. Препарат промывают водой, высушивают и микроскопируют с иммерсионной системой. При правильной окраске кристаллы белка окрашиваются в черный, а клетки — в розовый цвет. Белковые включения образуют и другие бактерии, например Bacillus popilliae, Xenorhabdus nematophilus и др. Их окрашивают также по вышеописанной методике. Параспоральные тельца можно наблюдать и в живых клетках, используя фазово-контрастное устройство. В этом случае параспоральные тельца выглядят как гранулы, способные сильно преломлять свет.
5.2.5. Эндоспоры
Эндоспоры образуют бактерии родов Bacillus, Clostridium и некоторых других. Для обнаружения способности клеток к спорообразованию лучше использовать старые культуры. Споры можно обнаружить при наблюдении живых клеток, а также путем дифференциального окрашивания цитоплазмы и споры. В случае выявления у микроорганизма способности к образованию спор необходимо обратить внимание на тип спорообразования (бациллярный, клостридиальный, плектридиальный), расположение споры в клетке (центральное, экс-центральное или полярное), форму свободных спор (круглая, овальная или продолговатая) и определить их размеры. С этой целью просматривают клетки 2—3-суточной культуры, так как большинство спорообразующих бактерий проходят за этот период времени все стадии развития — от вегетативной клетки до свободной споры.
Наблюдение спорообразования в микрокультуре. На стерильном предметном стекле, как описано выше (см. 5.2.1), получают тонкую пленку агаризованной питательной среды, благоприятной для спорообразования (например, картофельного агара). В центр пленки наносят петлей каплю жидкой 24-часовой культуры Bacillus megaterium или другой крупной спорообразующей бактерии. Полученный препарат инкубируют при 37 °C во влажной камере в течение 20 — 24 ч. Перед микроскопированием препарат подкрашивают метиленовым синим по Леффлеру и осторожно накладывают покровное стекло. Микроскопируют с иммерсией или методом фазового контраста. Просматривают край колонии,
обращая внимание на расположение клеток и на клетки в разных стадиях спорообразования.
Наблюдение эндоспор в живых клетках. Споры по сравнению с цитоплазмой характеризуются более высоким показателем преломления света, поэтому при микроскопировании в светлом поле они видны как более темные включения округлой или овальной формы. При использовании фазово-контрастного устройства споры имеют вид светлых включений на фоне почти черных клеток.
Метод выявления спор негативным окрашиванием. На предметном стекле готовят тонкий мазок клеток спорулирующих бактерий, подсушивают на воздухе и фиксируют в пламени. Затем на 3 — 5 мин наносят метиленовый синий или на 1 — 3 мин фуксин, после чего препарат осторожно промывают водой и подсушивают на воздухе. Просматривают с иммерсией.
Вегетативные клетки бактерий прокрашиваются, а споры, имеющие многослойную, труднопроницаемую оболочку — нет. Они видны как сильно преломляющие свет сферические или овальные образования, находящиеся в зависимости от стадии спорообразования внутри или вне клеток бактерий. Иногда на поверхности спор откладывается тонкий белковый слой. Благодаря его прокрашиванию споры становятся видимыми, но окрашены они значительно слабее, чем клетки. Например, при окраске фуксином Циля клетки красные, а споры светло-розовые.
Метод удобен при количественной оценке процесса спорообразования путем подсчета количества спор и вегетативных клеток в разных условиях роста бактерий.
Дифференциальная окраска споры и цитоплазмы по методу Пешкова. Споры и цитоплазму окрашивают при нагревании. Промывание препарата водой ведет к обесцвечиванию цитоплазмы, тогда как спора прочно удерживает краситель. На обезжиренном предметном стекле готовят мазок, высушивают его на воздухе, фиксируют в пламени горелки и заливают раствором метиленового синего по Леффлеру. Краситель доводят до кипения, держа предметное стекло над пламенем горелки. По мере испарения красителя добавляют новые его порции. Продолжительность окраски, считая с момента закипания красителя, составляет 10 — 20 с. Затем предметное стекло охлаждают, препарат тщательно промывают водой, после чего клетки в течение 30 с докрашивают 0,5%-м водным раствором нейтрального красного или сафранина. Краситель сливают, препарат промывают водой и просматривают с иммерсионной системой. При правильном окрашивании клетки имеют красный цвет, а споры — синий.
Вместо метиленового синего можно использовать малахитовый зеленый. В этом случае препарат, фиксированный в пламени горелки, заливают на 7—10 мин 7,5%-м раствором малахитового зеленого. Окрашивание проводят с подогревом, помещая препарат над пламенем горелки. По окончании окраски предметное стекло охлаждают, промывают препарат водой и докрашивают клетки 0,25%-м водным раствором сафранина в течение 1 —2 мин. Споры окрашиваются в зеленый цвет, клетки — в розовый.
5.3. ПРИГОТОВЛЕНИЕ ПРЕПАРАТОВ ДЛЯ ЛЮМИНЕСЦЕНТНОЙ
МИКРОСКОПИИ
Как уже отмечалось (см. 5.1.4), некоторые микроорганизмы обладают собственной (первичной) люминесценцией, что связано с наличием в их клетках люминесцирующих веществ — хлорофилла, каротиноидов, рибофлавина, ал-
калоидов, порфиринов, некоторых антибиотиков. Такие микроорганизмы можно изучать с помощью люминесцентного микроскопа на препаратах «раздавленная капля», однако в большинстве случаев применяется предварительная обработка клеток специальными красителями — флуорохромами, что приводит к вторичной люминесценции. Существуют природные и синтетические флуо-рохромы. Широко используются акридин оранжевый, этидиумбромид, приму-лин, родамин, берберинсульфат, флуорескамин, аурофосфин и некоторые другие, избирательно концентрирующиеся на отдельных структурах клетки. Флуорескамин выявляет аминогруппы, этидиумбромид — ДНК, фосфин 3R — липиды. Калькофлуор белый образует соединения с хитином, целлюлозой и используется для учета грибов в почве. Диацетилфлуоресцеин выявляет живые клетки.
Особый интерес вызывает флуоресцеинизотиоцианат (ФИТЦ), образующий связь с белками антител без нарушения их способности соединяться с гомологичными антигенами. Меченные ФИТЦ антитела представляют собой высокочувствительные индикаторы на соответствующие антигены. Антитела получают общепринятыми методами из крови иммунизированных кроликов и затем флуорохромируют. Кроме того, широко используются готовые, меченные флуорохромом определенные антисыворотки, а также готовый комплекс ФИТЦ-у-глобулин. Иммунофлуоресцентный анализ широко применяется в медицинской и санитарной микробиологии, генетике, систематике, экологии микроорганизмов.
Препараты живых клеток. На предметное стекло в каплю раствора акридина оранжевого в воде или 0,5%-го NaCl в разведении 1:104 вносят исследуемую культуру микроорганизмов и накрывают покровным стеклом. Через 2—10 мин микроскопируют при увеличении объектива 40 к или ЮОх с вазелиновым маслом.
Препараты фиксированных клеток. На предметном стекле готовят мазок, фиксируют и окрашивают нейтральным или слабокислым раствором акридина оранжевого в разведении 1 :104 в течение 2—4 мин. Затем препарат отмывают от избытка флуорохрома слабопроточной водой из промывалки в течение 5 — 10 мин, накрывают покровным стеклом и просматривают с объективом 90х.
Иммунофлуоресцентное окрашивание: прямой метод. На предметное стекло помещают суспензию клеток или образец, содержащий искомый микроорганизм, подсушивают и фиксируют нагреванием или другим способом. Препарат окрашивают меченной ФИТЦ антисывороткой, поместив его во влажную камеру (чашка Петри с мокрой фильтровальной бумагой). Время взаимодействия подбирают опытным путем; обычно достаточно 20 мин. Затем препарат о'гмы-вают в стоячей или слабопроточной воде 2 — 3 мин, накрывают покровным стеклом и исследуют с объективом 90 х. Так как оболочка клетки непроницаема для антител, они адсорбируются на поверхности клетки и при этом возникает характерный «эффект ореола» как один из критериев специфичности. Если в сыворотке есть несвязанный флуорохром, он, проникая в клетку, вызывает ее свечение вследствие неспецифической реакции.
Иммунофлуоресцентное окрашивание (непрямой метод) включает два этапа. Сначала препарат фиксируют нагреванием, наносят каплю нелюминесцирую-щей гомологичной иммунной сыворотки определенного разведения и оставляют на 20 — 30 мин во влажной камере, затем осторожно промывают в течение
5 мин. На втором этапе на препарат наносят каплю меченого антикроличьего у-глобулина, разведенного до так называемого окрашивающего титра. Время инкубации на втором этапе и окрашивающий титр определяют экспериментально. После этого препарат осторожно промывают и микроскопируют при увеличении 90 х.
Непрямой метод обладает принципиальным преимуществом, так как даже при наличии лишь одной люминесцирующей сыворотки можно отыскать большое число разных антигенов, используя стандартные кроличьи сыворотки. При непрямом методе, однако, крайне важен контроль, включающий инкубирование препарата на втором этапе непосредственно с меченой сывороткой; обработку препарата гетерологичной меченой сывороткой; обработку препарата на первом этапе нормальной или гетерологичной сывороткой, не содержащей антител к исследуемому антигену.
Обнаружение мелких колоний микроорганизмов. Мембранный фильтр марки «Синпор» с диаметром пор 1 мкм стерилизуют кипячением в дистиллированной воде в течение 20 мин. Через фильтр пропускают исследуемый субстрат (воду, почвенную вытяжку) или суспензию микроорганизмов. Фильтр накладывают тыльной стороной на агаровую пластинку с элективной питательной средой и помещают на 5 — 6 ч в термостат. Затем фильтр с колониями снимают со среды и кладут тыльной стороной на дно чашки Петри, содержащей несколько капель флуорохрома, или диск фильтровальной бумаги, смоченный раствором флуорохрома, например акридина оранжевого, в разведении 1 : 5000. Диффундирующий через фильтр флуорохром в течение 5—10 мин окрашивает колонии микроорганизмов. При необходимости фильтр накладывают на фильтровальную бумагу, смоченную физиологическим раствором, для удаления избытка флуорохрома. Фильтры просматривают в синем свете. Микроорганизмы остаются живыми и могут быть использованы для дальнейшего выращивания и изучения.
5.4. ЭЛЕКТРОННАЯ МИКРОСКОПИЯ И СКАНИРУЮЩАЯ ЗОНДОВАЯ МИКРОСКОПИЯ В МИКРОБИОЛОГИИ
5.4.1. Просвечивающая электронная микроскопия
Электронная микроскопия является неотъемлемой частью микробиологических исследований уже три четверти века. Приборостроительные компании выпускают много типов электронных микроскопов, отличающихся принципом действия, разрешающей способностью и диапазоном рабочих увеличений. В практике микробиологических исследований первыми были применены просвечивающие, или трансмиссионные, электронные микроскопы (ТЭМ), которые дают возможность визуально изучать только очень тонкие срезы или частицы с увеличением до 100 тысяч крат. Можно использовать и более высокие увеличения, но новые структуры биологических микрообъектов при этом уже не обнаруживаются. Вместо светового излучения в ТЭМ используют пучки ускоренных электронов, обладающие волновыми свойствами, причем эквивалентная длина волны таких пучков (длина волны Де-Бройля) на несколько порядков меньше све-
тобой, что обусловливает намного большую разрешающую способность электронных микроскопов этого типа. Практически (для биологических объектов) ее величина для ТЭМ составляет 0,5 нм, причем этот предел определяется не характеристиками самого ТЭМ, а толщиной ультратонких срезов, при которой изображение имеет достаточный контраст. В оптимальных условиях (на сверхтонких монокристаллических пленках) достигается разрешение 0,1 нм.
Основной частью ТЭМ является вакуумная колонна (электронно-оптическая система), в которой последовательно, сверху вниз, расположены и точно съюстированы относительно общей оси симметрии (оптической оси): .
• электронная пушка с катодом и анодом, обеспечивающая эмиссию и ускорение электронов;
• две конденсорные магнитные линзы, управляющие диаметром электронного пучка;
• объектный столик, обеспечивающий медленное и плавное перемещение образца;
• три или четыре магнитные линзы, формирующие увеличенное изображение;
• вспомогательные устройства (дефлекторы и стигматоры) для точной юстировки системы;
• люминесцирующий экран для наблюдения увеличенного изображения. Средняя часть экрана может подниматься для точной фокусировки изображения с помощью дополнительного 10-кратного бинокулярного микроскопа и для пропускания пучка, несущего изображение, в фотокамеру;
• фотокамера (пленочная или цифровая).
В вакуумной колонне с помощью вакуум-насосов создается глубокий вакуум для предотвращения рассеяния электронов и обеспечения условий работы накаленного катода. Источник электронов (катод) — это вольфрамовая нить, накаливаемая электрическим током до высокой температуры, при которой возникает электронная эмиссия. Между катодом и анодом (металлическим диском с отверстием в центре, расположенным под катодом) подается высокое ускоряющее напряжение (50—100 кВ), благодаря чему пучок ускоренных электронов направляется вниз, в сторону образца. При этом электронный луч, сфокусированный конденсорными линзами, направляется на образец.
Биологические объекты, исследуемые в условиях вакуума и электронного облучения, должны быть зафиксированы и обезвожены, а для повышения контрастности изображения еще и обработаны соединениями тяжелых металлов, т. е. химически контрастированы. Прошедшие через тонкий объект электроны попадают в магнитное поле объективной магнитной линзы, формирующей первичное увеличенное изображение объекта. При этом электроны, рассеянные микроучастками образца с более высокой электронной плотностью (насыщенными тяжелыми металлами с целью контрастирования), в большей степени отклоняются магнитным полем и задерживаются объективной диафрагмой. Таким образом, они не попадают на люминесцентный экран, и участки изображения, соответствующие таким микроучасткам объекта, выглядят темными. Контраст изображения может быть обусловлен также и поглощением электронов электронно-плотными микроучастками объекта, но поглощение электронов сопровождается повышением температуры объекта, что приводит
к тепловому дрейфу и даже к разрывам срезов и пленок-подложек, поэтому при подготовке срезов следует избегать их чрезмерной толщины, а также тщательно отмывать неспецифический осадок контрастирующих веществ. Первичное увеличенное изображение, сформированное объективной линзой из электронов, летящих близко к оптической оси (слабо рассеянных объектом и не задержанных объективной диафрагмой), имеет увеличение около 1000 крат, но его разрешение настолько высоко, что позволяет увеличить его дополнительно до 100 тысяч крат с помощью промежуточных линз (обычно их 2) и проекционной линзы. При этом становятся видимыми все новые детали микроструктуры. Проекционная линза, кроме того, обеспечивает большую глубину фокуса, необходимую для сохранения резкости изображения на поднятом экране и на фотопленке.
Любой современный научный прибор снабжается блоком фиксирования полученной с его помощью информации. В электронных микроскопах это — фотографирование объектов на фотопленку и их оцифровка компьютерными системами. Специальные программы обработки изображений позволяют не только сохранять их в виде графических файлов, но и гибко корректировать их контраст. Это очень важно для ТЭМ, где оптимальное контрастирование биологических объектов остается одной из главных проблем. Оцифровка позволяет также увеличить число отснятых кадров за сеанс микроскопии с 2 — 3 десятков негативов, получаемых с помощью фотографирования, примерно до сотни цифровых изображений. Необходимо, однако, отметить, что для получения цифровых изображений высокого разрешения требуются дорогостоящие специализированные цифровые фотокамеры, а объем полученных файлов измеряется десятками мегабайт.
Подготовка образцов для исследования в ТЭМ — это почти всегда достаточно сложный процесс (исключение — простые методы контрастирования микроорганизмов, которые будут описаны далее), состоящий из ряда обязательных стадий, а также множества модификаций, разработанных в целях лучшего выявления особенностей строения отдельных объектов. Обязательны фиксация, обезвоживание, заливка специальными смолами, получение улътратонких срезов, контраст ирование.
Цель фиксации — сохранить вид объекта в состоянии, по возможности близком к прижизненному. Наиболее полно фиксация осуществляется с помощью обработки объекта растворами глутаральдегида и четырехокиси осмия OsO4. Эти вещества образуют поперечные связи между различными молекулами в клетке, благодаря чему возникает прочная сеть, которая предотвращает или замедляет потерю включенных в нее веществ при обезвоживании и дальнейшей обработке. Четырехокись осмия не только фиксирует клетки, но и контрастирует отдельные их компоненты, прежде всего фосфолипидные мембраны и жировые включения, которые во время фиксации поглощают наибольшее количество осмия и поэтому приобретают способность сильно рассеивать ускоренные электроны. Для преимущественного четкого выявления мембран используют перманганат калия КМпО4 связывающийся с фосфолипидами. Перманганат калия плохо фиксирует белки и не удерживает большинство других компонентов клетки, кроме мембран.
Для обезвоживания применяют водные растворы этанола возрастающих концентраций. В процессе фиксации и особенно обезвоживания могут терять не-
которые компоненты клеток. Поэтому часто полезно готовить один и тот же объект к исследованию в ТЭМ, фиксируя его различными способами. Например, для изучения мембранотропного действия на микроорганизмы некоторых антибиотиков и других соединений целесообразно зафиксировать клетки как универсальным способом, описанным далее, — с помощью глутарового альдегида и четырехокиси осмия, так и с помощью 4%-го раствора перманганата калия, и сравнить вид срезов в ТЭМ.
Для получения ультратонких (не более 0,1 мкм) срезов клеток, через которые может пройти электронный луч ТЭМ, фиксированные обезвоженные клетки пропитывают полимерными (часто эпоксидными) компаундами — «смолами». Наиболее употребительны такие компаунды, как Эпон, Аралдит и их смеси.
Улыпратонкие срезы готовят с помощью ультрамикротомов, используя стеклянные или алмазные ножи. Срезы помещают на медные (в особых случаях никелевые или золотые), мелкоячеистые сетки диаметром 3 мм, предварительно покрытые тончайшими пленками-подложками из специальных полимеров — Формвара (поливинилформаль) или Бутвара (поливинилбутираль). (Эпон, Аралдит, Формвар, Бутвар — это не химические названия, а торговые марки фирм, производящих реактивы для электронной микроскопии.) Срезы большой площади и серийные срезы помещают на 3-миллиметровые бленды (диски с одним круглым или овальным отверстием).
В ТЭМ с повышенным (200—300 кВ) и сверхвысоким (1000—3000 кВ) ускоряющим напряжением можно исследовать сравнительно толстые (1 — 5 мкм) срезы. В сочетании с гониометрическими объектными столиками, позволяющими наклонять объектные сетки, эти микроскопы дают возможность получать стереоизображения структур при высоком разрешении.
Изучение срезов клеток в различных видах ТЭМ дает возможность выявить множество деталей внутреннего строения микроорганизмов и особенности их взаимодействия. Для этого применяют предварительную обработку срезов на сетках, позволяющую достигнуть наибольшего контрастирования — усиления контраста изображения различных участков объекта при связывании с ионами тяжелых металлов. Основной механизм достижения контрастности изображения при ТЭМ — рассеяние электронов и их поглощение объективной диафрагмой. Контрастирование биологических объектов проходи! уже в ходе их фиксации и проводки благодаря использованию четырехокиси осмия и уранилацетата, а усиливается при контрастировании срезов. Для увеличения контрастности срезов их обрабатывают следующими соединениями тяжелых металлов: 1) OsO4, взаимодействующим с полярными и С=С-группами липидов, SH-группами белков, что приводит не только к фиксации, но и к возрастанию контрастности компонентов клеток, содержащих белки и фосфолипиды; 2) уранилацетат, реагирующий с белками и фосфатными группами нуклеиновых кислот и, соответственно, увеличивающий их контрастность; 3) свинец при высоких значениях pH и в присутствии хелатообразующих агентов — реактив Рейнольдса (пропись его приготовления дана ниже); реагирующий с РНК и с ОН-группами углеводов, а также с восстановленным осмием; 4) вольфрам, иногда молибден, в виде раствора Na или К соли фосфовольфрамовой кислоты, молибдата аммония, обычно применяются при негативном контрастировании клеток микроорганизмов. Кратковременная обработка срезов уранилацетатом перед их обработкой цитратом свинца приводит к более интенсивному включе
нию свинца. Поэтому наиболее эффективно последовательное совместное применение соединений урана и свинца в качестве контрастирующих веществ, что приводит к значительному возрастанию контрастности срезов клеток, при этом в увеличении контраста принимают участие осмий, уран, свинец.
Метод негативного контрастирования применяется для контрастного выявления деталей поверхности бактерий, вирусов. Это достигается благодаря погружению таких объектов в тонкую, быстро высыхающую пленку электронно-плотного вещества, которое не «окрашивает» объект, а создает вокруг него темный фон. Чаще всего для этих целей используют разбавленные растворы фосфовольфрамовокислого натрия или молибдата аммония. В такую пленку можно заключать, без предварительного обезвоживания, только мелкие клетки или частицы: испарение жидкости из них в условиях вакуума при внесении в ТЭМ незначительно и происходит очень быстро. Метод дает возможность изучать морфологию клеток, их жгутики, пили, реснички, часто определяется толщина клеточной стенки. Это также удобный способ наблюдения за изменением поверхности клеток, их деградацией под действием ферментов, антибиотиков, поверхностно-активных веществ. Метод не позволяет выявить детали внутреннего строения микроорганизмов — их можно изучать только на срезах.
Для негативного контрастирования каплю негустой суспензии клеток наносят на сетки, покрытые формваровой пленкой-подложкой; примерно через 30—60 с лишнюю жидкость убирают фильтровальной бумагой. Далее наносят на сетки приблизительно на это же время 2%-й раствор фосфовольфрамата натрия или калия. Можно использовать и 1%-й раствор уранилацетата, но он способен деформировать клетки при выпадении в осадок. Лишнюю жидкость также убирают с помощью полоски фильтровальной бумаги. Иногда перед негативным контрастированием фиксируют клетки примерно от получаса до нескольких часов в парах 25%-го глутаральдегида.
Если атомы тяжелых металлов присоединяются к структурным компонентам клеток, окрашивая их, говорят о позитивном окрашивании.
Подготовка срезов клеток для ТЭМ. Приведем один из способов. Суспензии микроорганизмов, срезы которых будут исследовать в ТЭМ, помещают в центрифужные пробирки с пробками. Желательно, чтобы количество материала было не меньше 50 мг. Заливают раствором глутарового альдегида в фосфатном буфере из расчета, чтобы концентрация их в пробирке составила 2,5 % глутарового альдегида в 0,1 М фосфатном буфере, pH 7,0—7,2, и оставляют в темноте при комнатной температуре на 1 — 3 ч. Затем суспензии центрифугируют при условиях, обеспечивающих хорошее осаждение всех частиц, и ресуспен-дируют в вышеуказанном буфере для промывки. Промывку проводят трижды с интервалом примерно 15 мин. После каждой смены любого раствора суспензию осаждают центрифугированием. Осадок ресуспендируют в 1%-м водном OsO4 и оставляют обычно на ночь. Затем отмывают четырехокись осмия и проводят обезвоживание материала в этаноле возрастающих концентраций: 30 %, 50 % по 30 мин, затем 70 % с 1%-м уранил ацетатом. Обычно в таком растворе осадок оставляют на ночь в холодильнике. Далее суспензию проводят через 96%-й этиловый спирт, абсолютные спирт и ацетон (в каждом случае по 30 мин) с центрифугированием. По окончании проводки осадки оставляют на дне центрифужных стаканов и начинают их пропитку смолами.
Пропитку смолами часто проводят, используя следующую смесь: на 10 мл смеси: смолы Эпон 812 — 4,5 мл; DDSA — 3,5 мл; MNA — 2 мл; DMP (катализатор) — 0,2 мл. Последний вносится только после пропитки образцов смесью смол без катализатора, когда их помещают в заливочные капсулы. Вначале приготовленную смесь без катализатора смешивают с ацетоном в соотношениях — смола:ацетон: 1:3, 1:1, 3 :1, поочередно заливая этими смесями суспензии и каждый раз оставляя в них не менее чем на 1 — 3 ч, а обычно — на ночь или на сутки. Затем образцы выдерживают 1 — 3 сут в смоле без катализатора при комнатной температуре. Эту смолу сливают, переносят осадки в заливочные капсулы и заливают образцы смолой с катализатором. В постепенно затвердевающей смоле образцы выдерживают при 37 °C (обычно в течение суток) и 60 °C (2 сут). Микроорганизмы, заключенные в смолы, готовы для резки на ультрамикротоме после соответствующей заточки полимерных блоков. Срезы наносят на специальные сеточки, такие же, какие используют и для негативного контрастирования (в этом случае покрытие сеток формваровой пленкой не обязательно, хотя и желательно), и проводят контрастирование срезов 1%-м уранил ацетатом в течение примерно 10 мин (следить, чтобы не выпадал осадок), а затем в реактиве Рейнольдса.
Приготовление реактива Рейнольдса и окрашивание им тонких срезов микроорганизмов для исследования в ТЭМ. 1,33 г нитрата свинца, 1,76 г цитрата натрия Na3(C6H5O2) Н2О и 30 мл дистиллированной (а лучше бидистиллированной) воды взбалтывают в мерной колбе на 50 мл и оставляют на 30 мин, периодически встряхивая. Затем добавляют 8 мл 1 М NaOH и доводят объем раствора водой до 50 мл, перемешивая его переворачиванием колбы. Краситель устойчив при хранении в холодильнике в закрытой колбе в течение полугода. Сетки со срезами промывают перед окрашиванием и после него в 0,02 М растворе NaOH. Время окрашивания реактивом Рейнольдса 5 — 30 мин, при этом нужно следить, чтобы не выпал осадок. После окраски и окончательной промывки 0,02 М NaOH сетки промывают, опуская их примерно 30 раз в дистиллированную воду. Высушенные сетки готовы для исследования срезов микроорганизмов в ТЭМ.
Следует подчеркнуть, что при всех способах подготовки объектов для всех видов микроскопических исследований необходимо: использовать только химически чистые реактивы и качественную дистиллированную воду; нужные растворы хранить в холодильнике; помнить о важности используемых буферов; многие этапы работы проводить в вытяжном шкафу.
5.4.2. Сканирующая электронная микроскопия
В последние три десятилетия достигнуто много новых успехов в развитии сканирующей электронной микроскопии (СЭМ), которые существенно расширили возможности использования этого метода в микробиологии. В основу СЭМ положен принцип формирования изображения на экране электронно-лучевой трубки с помощью развертки, синхронной с разверткой электронного луча, последовательно облучающего сканируемую поверхность изучаемого объекта. При этом одновременно регистрируется ряд вторичных излучений, что обеспечивает высокую информативность и разрешающую способность метода СЭМ, с помощью которого изучаются морфология и структура поверхности различных объектов. Очень важно изучение рельефа поверхности отдельных клеток микроорганиз
мов или составляющих эти клетки структур, а также колоний, конгломератов, консорциумов, включающих большое число микроорганизмов. Одной из замечательных особенностей СЭМ является и большая глубина резко изображаемого пространства (глубина фокуса), которую можно к тому же усилить путем наклона объектного столика (до 45°) и с помощью динамического фокусирования. Это имеет особое значение при изучении морфологии микроорганизмов, выявления их поверхностных структур, формы и архитектоники расположения клеток в пространстве, например в колониях, конгломератах.
При использовании традиционных сканирующих электронных микроскопов объект должен быть фиксирован, обезвожен и высушен с помощью методов, щадящих его поверхность, так как он подвергается воздействию глубокого вакуума (ИГ5 мм рт. ст.), интенсивной бомбардировке ускоренными электронами подобно тому, как и в просвечивающем электронном микроскопе (ТЭМ). Ускоряющее напряжение для СЭМ меньше, чем для ТЭМ: обычно 10 кВ, не более 20 кВ. Фиксация и последующая обработка объектов для изучения их с помощью СЭМ включает меньше стадий, чем для ТЭМ. В еще большей степени облегчается фиксация микроорганизмов, имеющих ригидную клеточную стенку.
При СЭМ в качестве регистрируемого вторичного излучения выбирают прежде всего вторичные электроны, т. е. электроны внешних оболочек атомов вещества изучаемого объекта, напыленного металлом. Такие электроны легко фокусируются и могут покинуть изучаемый объект только из тонкого (10 нм) поверхностного слоя вблизи места бомбардировки, что и обеспечивает высокую разрешающую способность СЭМ, которая,-однако, меньше, чем для ТЭМ. Предельное разрешение в СЭМ обычно достигает порядка 10 нм при использовании вольфрамового термокатода (что соответствует полезным увеличениям в 50 —60 тыс. раз) и порядка 5 нм со специальными электронными пушками повышенной яркости. В большинстве случаев для изучения морфологии микроорганизмов бывает достаточно увеличений в 5 —25 тыс. раз.
Следует помнить, что разрешающая способность при СЭМ в значительно большей степени зависит от выбранного режима работы микроскопа и способа подготовки образца, чем при ТЭМ, так как в формировании изображений участвует не только электронно-оптическая система, но и система регистрации вторичных излучений. Поэтому разрешающая способность для каждого препарата, изучаемого с помощью СЭМ, определяется, помимо параметров оптической системы, величиной отношения сигнал/шум. Для увеличения этого соотношения и соответственно разрешающей способности используют несколько компромиссных приемов, в частности стараются приблизить свойства образца к оптимальным для исследования его поверхности в СЭМ (идеальна твердая, сухая, тепло- и электропроводная поверхность) за счет фиксации. Чаще всего используют химическую фиксацию, подобную применяемой для подготовки образца к ТЭМ. При этом фиксация глутаровым альдегидом обязательна, а применение четырехокиси осмия желательно, но не обязательно при фиксации микроорганизмов, имеющих ригидную клеточную стенку.
Обезвоживание — обязательный этап, если не используются низковакуумные СЭМ, о которых будет сказано далее. Обезвоживание существенно ослабляет действие сил поверхностного натяжения на границе раздела жидкость— воздух и тем самым предохраняет поверхность-мишень от деформации этими силами при высушивании. Высушивание необходимо для последующей подго-
товки объекта к исследованию в СЭМ в условиях высокого вакуума. Оно должно обеспечить удаление всех жидких компонентов из образца. Этот этап может привести к нарушению сохранности поверхности (нарушению ресничек, микроворсинок, общему сжатию клеток, формированию ложных морфологических структур). Поэтому при СЭМ часто высушивают образцы щадящим их поверхность методом высушивания в критической точке (ВКТ). Метод ВКТ основан на переходе жидкости, пропитывающей образец, в газообразное состояние сразу по всему объему, а не испарением с поверхности, как при высушивании на воздухе. Этого обычно достигают при повышенном давлении. Используют специальную толстостенную стальную камеру, куда помешают образец в абсолютном (безводном) ацетоне. Малейшее повышение температуры свыше критической приводит к переходу жидкости в газ, и таким образом образец оказывается высушенным сразу в полном объеме, а не при испарении жидкости через поверхность. Затем медленно, не допуская конденсации газа, снижают давление до атмосферного.
Высушивание объектов возможно и после быстрого глубокого их замораживания в условиях, когда не успевают образоваться крупные кристаллы льда, разрушающие поверхность (например, в жидком азоте в присутствии криопротекторов — этанола, ацетона).
После высушивания образцы прикрепляют к специальным немагнитным объектным столикам (из алюминиевых сплавов или латуни) с помощью лака для ногтей или двусторонней клейкой пленки «скотч», создающих ровный бесструктурный фон. Наиболее удобно прикреплять к объектному столику образцы, фиксированные на покровных стеклах.
Последний этап подготовки к исследованию поверхности образков, который обычно проводят на следующий день после приклеивания, — нанесение на них сверхтонкой (не более 20 нм, чтобы не исказить картину микрорельефа на больших увеличениях) пленки сплавов золото/палладий или платина/палладий. Для нанесения этих или подобных электропроводных покрытий используют метод вакуумного испарения, а в последние годы — более дешевый и быстрый метод ионного распыления, обеспечивающий образование дисперсных электропроводных покрытий за 10 мин при распылении металла в вакууме в среде ионизированного аргона.
Кроме повсеместно применяемой химической фиксации объекта и последующих описанных операций его подготовки к СЭМ разработан и более короткий путь: физическая фиксация объекта путем замораживания и нанесение на замороженную поверхность металлического покрытия. Однако этот путь требует поддержания низкой температуры в течение всего периода исследования. Поэтому чаще образец подметают репликой — точным слепком с его замороженной поверхности.
В последние годы часто используют путь изменения конструкции или режимов работы СЭМ таким образом, чтобы можно было наблюдать объекты без предварительной фиксации. Созданы низковольтные низковакуумные СЭМ (LVSEM), позволяющие исследовать влагосодержащие и неэлектропроводные объекты. Такие приборы еще называют микроскопами с переменным давлением (VPSEM), так как в их электронной пушке и линзовой части колонны давление в мм рт. ст. может изменяться от вакуума, необходимого для традиционной работы СЭМ (на уровне КГ3 мм рт. ст.) до практически атмосферного, что позволяет исследовать в течение нескольких десятков секунд даже поверхности жидкостей
и живых объектов, полупогруженных в жидкую среду. Причем в таких условиях отношение сигнал/шум значительно возрастает за счет «размножения» вторичных электронов и усиления сигнала, несущего информацию о микрорельефе поверхности благодаря ионизации молекул остаточных газов. В результате повышается четкость и детализация изображения поверхности изучаемого объекта.
Возможности СЭМ могут быть расширены с помощью спектрометрических приставок, позволяющих оценивать различия в химическом составе микроучастков поверхности объектов. Рентгеновская и Оже-электронная спектрометрии на базе СЭМ являются экспресс-методами неразрушающего локального химического микроанализа. Использование в СЭМ проходящих электронов для исследования тонкопленочных образцов и срезов известно как трансмиссионно-ска-нирующая электронная микроскопия. Она позволяет получать четкие (с разрешающей способностью до 3 нм) изображения таких срезов, какие невозможно изучить в ТЭМ из-за резкого ухудшения разрешающей способности за счет хроматической аберрации и теплового разрушения срезов. Созданы также сканирующие приставки и для ТЭМ.
С помощью СЭМ можно получать стереомикрофотографии, что позволяет рассчитывать истинные расстояния между точками изображенной поверхности и восстанавливать взаимное расположение структур, в том числе и микробных клеток, в пространстве.
Приготовление препаратов клеток микроорганизмов для их исследования в СЭМ удобнее всего осуществлять, помещая такие клетки, обычно предварительно промытые в 0,1 М фосфатном буферном растворе, pH 7,0—7,2, на обезжиренное покровное стекло. В таком случае не потребуется много раз, после каждой смены раствора, проводить центрифугирование образца, как это делается при обработке препаратов в центрифужных пробирках. Однако последний вариант также используют, особенно когда объекты после фиксации глутаровым альдегидом плохо прикрепляются к покровным стеклам. Влажные объекты размазывают неравномерным слоем на покровных стеклах, которые помещают в стеклянную чашку' Петри, и осторожно заливают их тонким, но полностью покрывающим слоем 2,5—3%-го раствора глутарового альдегида на вышеуказанном фосфатном буфере комнатной температуры. Если исследуемые микроорганизмы имеют достаточно плотную поверхность, их можно высушить на покровном стекле, а затем залить глутаровым альдегидом в чашках Петри. Залитые образцы оставляют для фиксации в темноте, обычно на 1 — 2 ч. Затем очень осторожно сливают раствор глутарового альдегида и дважды промывают образцы, закрепившиеся на покровных стеклах, фосфатным буфером, каждый раз через 10—15 мин сливая его. После этого можно проводить осмиевую фиксацию, особенно желательную при изучении микоплазм, протопластов, сферо-пластов, а также некоторых грамотрицательных бактерий и не имеющих клеточных стенок археи. Для этого образцы заливают 0,5—1%-м водным раствором OsO4 и оставляют при комнатной температуре в темноте на 1 — 2 ч. Затем промывают образцы три раза водой. Далее проводят обезвоживание образцов в водных, растворах этанола возрастающих концентраций (20, 30, 50, 70 %) по 15—30 мин. В 70%-м этаноле образцы можно хранить в холодильнике в течение 1 — 3 нед. В 96%-м этаноле и затем в абсолютном спирте образны выдерживают обычно по 1 ч, после чего помещают их в безводный (абсолютный) ацетон (такой ацетон хранится с добавлением 1 —2 % безводного CuSO4) и хранят в холодильнике до
высушивания в критической точке. Это высушивание и последующие этапы подготовки образцов для СЭМ, описанные ранее, проводят с помощью специальных приборов в лаборатории электронной микроскопии.
В целях изучения архитектоники колоний микроорганизмов их выращивают на поверхности стерильных мембранных фильтров (например, «Владипор» № 2 — 5), помещенных на соответствующую агаризованную питательную среду в чашках Петри. Выросшие колонии подвергают фиксации и обезвоживанию, последовательно помещая фильтры в чашки Петри, крышки которых покрыты внутри фильтровальной бумагой, пропитанной следующими веществами: 25%-й глутаровый альдегид (на 30 мин), безводный ацетон (2 раза по 20 мин). После ВКТ, приклеивания к объектным столикам и напыления металлом колонии исследуют в СЭМ.
Экзоспоры актиномицетов и грибов готовят к исследованию в СЭМ следующим образом: к спороносящему воздушному мицелию осторожно прикасаются поверхностью объектного столика, покрытого лаком. Полученный отпечаток после подсушивания на воздухе напыляют металлом в ионно-распылительной установке.
5.4.3. Сканирующая зондовая микроскопия
В 1982 г. был разработан принципиально новый метод субмикроскопического, с разрешением до долей нанометров, исследования поверхности объектов, расположенных на диэлектрических подложках из слюды, графита и других материалов, благодаря возникновению между изучаемой поверхностью и специальным зондом особых (туннельных) токов. В качестве зондов используют платиновые или вольфрамовые мглы. Различные способы сканирующей туннельной, атомно-силовой и других видов неразрушающей зондовой микроскопии основаны на иных физических закономерностях, чем электронная микроскопия. Методы сканирующей зондовой микроскопии используют для получения предельных разрешений поверхности, вплоть до топографии на атомарном уровне. По достигнутому разрешению (доли нонометров) эти методы сопоставимы лишь с рентгеноструктурным анализом, но в отличие от него не требуют кристаллизации объектов. Образцы не разрушаются электронами высоких энергий, как при традиционной электронной микроскопии. Новые методы исследования в нанометровом диапазоне нашли широкое применение в материаловедении, для исследований металлов, полупроводников, диэлектриков, полимеров, пленочных структур, молекулярных кластеров, порошков, для контроля устройств хранения информации, положив начало бурному развитию нанотехнологий, а также нанобиологии. С помощью этих методов получены уникальные данные о строении нативных биологических объектов — вирусов, бактерий, археи и отдельных структур разных живых клеток, например, жгутиков, пилей, ресничек, а также ферментов, нуклеиновых кислот, антибиотиков, токсинов, о кинетике некоторых биологических процессов.
Для исследований биологических объектов, в том числе и микроорганизмов, с помощью сканирующей зондовой микроскопии разработаны различные модели микроскопов. Например, фирмой НТ-МДТ (Москва) — одним из мировых лидеров в этой области, созданы универсальные «Солвер Р47Н», или «Солвер
Р47БИО» для исследования живых объектов в биологии и медицине. Это компактные (не более 50 см в высот}') относительно простые и недорогие (по сравнению с электронными микроскопами) приборы, соединенные с компьютером, обрабатывающим получаемую с помощью зонда информацию и воспроизводящим ее в виде файла аналогично цветным географическим картам, на которых высот}' изучаемой поверхности обозначает условный цвет. Вывод изображения на печать производится в виде трехмерного изометрического рисунка с указанием величин всех параметров в реальном масштабе времени. Эти и другие подобные приборы обеспечивают широкие возможности неразрушающих исследований живых клеток и некоторых проходящих в них процессов с разрешением от микрометрового до атомарного в различных окружающих условиях.
Разработан подобный прибор и для научно-образовательного процесса в области нанотехнологий и нанобиологии — Nano Educator. С его помощью возможно изучение поверхности клеток различных микроорганизмов, адгезии этих клеток, порядка укладки и величины, ориентации в пространстве белковых и других глобул на поверхности клеток, эндоспор, в составе включений различных бактерий, процессов взаимодействия биологически активных соединений с микробными клетками. Такие исследования можно проводить в капле жидкости с биологическими объектами, расположенной на диэлектрической подложке.
Глава 6
ВЫДЕЛЕНИЕ ЧИСТЫХ КУЛЬТУР МИКРООРГАНИЗМОВ
Физиологию, биохимические свойства и циклы развития микроорганизмов исследуют, как правило, при работе с чистыми культурами. Чистой, или аксе-нической, называют культуру, содержащую микроорганизмы одного вида. Умение выделить микроорганизмы одного вида из смешанной популяции, существующей в природе, и поддерживать чистоту культуры — необходимые условия работы с микроорганизмами. Выделение чистой культуры обычно включает три этапа: 1) получение накопительной культуры, 2) выделение чистой культуры, 3) определение чистоты выделенной культуры.
6.1. ПОЛУЧЕНИЕ НАКОПИТЕЛЬНОЙ КУЛЬТУРЫ
Накопительной называют такую культуру, в которой преобладают представители одной физиологической группы или даже одного вида мшероорганиз-мов. Метод накопительных культур был введен в практику микробиологических исследований С. Н. Виноградским и М.Бейеринком. Сущность его заключается в создании элективных, т.е. избирательных, условий, которые обеспечивают преимущественное развитие желаемых (необходимых исследователю) микроорганизмов или группы микроорганизмов из смешанной популяции.
При создании элективных условий необходимо знать физиологию или четко представлять те особенности, которыми должны обладать выделяемые микроорганизмы. Элективные условия создают чаще всего подбором соответству-
Хемогетеротрофные бактерии
Аэробные условия
Предпочтительно несбраживаемый субстрат
N2 — единственный источник азота
Соединения азота
NO3 как
Аэробные азотфиксаторы, например Azotobacter
Аэробы, например Pseudomonas
Денитрифицирующие бактерии
Органические субстраты, _ отсутствие света
Предпочтительно несбраживаемый субстрат
акцепторы
Сульфат-редущфующие бактерии
Анаэробные условия
-*-СО2 электронов
Метан-образующие археи
^Сбраживаемый сахар
N2 — единственный источник азота
Clostridium pasteurianum и родственные виды
Соединения ^азота
Бактерии, осуществляющие брожение, например Enterobacter
Хемолитоавтотрофные бактерии
«Субстраты»
NH4 -*• Nitrosomonas
Отсутствие в среде органических сое-
Аэробные
"* условия NO2 -*• Nitrobacter
(О2 как —
акцептор S, 82Оз' Thiobacillus, Acidithiobacillus электронов)
Водородные бактерии
динений + СО2
Анаэробные г*- S, SjO*"-*- Thiobacillus denitrificans условия
(NO3 как —
акцептор
электронов) L*- Н2 Paracoccus denitrificans
Рис. 6.1. Основные факторы, определяющие получение накопительных культур некоторых групп бактерий (по Р.Стейниеру н др., 1979)
Фототрофные микроорганизмы
Аэробные условия
Сульфид отсутствует
N2 единственный источник азота
Связанный азот
Циано-бактерии
Водоросли, цианобактерии
СО2 как единствен- _ ный источник углерода
Свет как источник— энергии
Высокая концентрация SH-
Анаэробные Сульфид _ условия присутствует Низкая
!-*- концентрация SH-
Зеленые серобактерии
Пурпур-ные серобактерии
г Органические ____ Анаэробные
соединения условия
Пурпурные несерные бактерии
Рис. 6.1. Продолжение
ющих сред, поскольку различные микроорганизмы для своего развития предъявляют неодинаковые требования к источникам питания. Например, микроорганизмы, способные фиксировать молекулярный азот, могут расти в среде, из состава которой исключены связанные формы азота. Если внести в такую среду почву, то из громадного разнообразия имеющихся в ней микроорганизмов в первую очередь будут развиваться азотфиксаторы. Накопительные культуры автотрофных микроорганизмов получают на средах, где единственным источником углерода служит углекислота. Отсутствие в среде других соединений углерода задерживает развитие гетеротрофов. Такие специфические питательные среды, удовлетворяющие потребности преимущественно одной группы микроорганизмов, носят название элективных. В зарубежной литературе большее распространение получили термины «накопительные», или «селективные», среды.
Иногда при выделении микроорганизмов из природных популяций в среду включают антибиотики, которые отличаются специфичностью действия и позволяют избирательно подавлять рост определенной группы микроорганизмов. Так, элективные условия для развития грамотрицательных бактерий можно создавать внесением в среду пенициллина в концентрации от 0,2 до 100 мг/л, поскольку многие виды грамположптельных бактерий при этом или совсем не развиваются, или развиваются медленно. Чтобы создать благоприятные условия для развития бактерий и, напротив, подавить рост мицелиальных грибов, к средам рекомендуют добавлять нистатин в концентрации от 0,1 до 20 мг/л или гризеофульвин в концентрации от 1 до 20 мг/л.
При создании элективных условий следует учитывать неодинаковое отношение различных микроорганизмов к аэрации, температуре, кислотности среды, освещению и т.д. Поэтому при получении накопительной культуры аэроб
ных микроорганизмов обеспечивают большую поверхность контакта питательной среды с воздухом, а для обогащения среды анаэробными микроорганизмами тем или иным способом создают анаэробные условия. Культивирование при высокой температуре (50 °C и выше) исключает развитие мезофильных микроорганизмов и обеспечивает рост термофилов. Селективным фактором может служить также неодинаковая скорость роста различных микроорганизмов при данной температуре. Например, в ряде случаев из-за различий в оптимальных температурах роста на минеральной среде при освещении и температуре 35 °C удается почти полностью подавить рост зеленых водорослей и получить культуру, обогащенную цианобактериями.
При получении накопительных культур следует учитывать и такую особенность микроорганизмов, как способность к образованию эндоспор. Для накопления спорообразующих бактерий среды инокулируют, как правило, субстратом, который предварительно пастеризуют, т.е. кратковременно прогревают при высокой температуре (10 мин при 75 °C или 2—5 мин при 80°C). Таким образом можно полностью или почти полностью исключить развитие бактерий, не образующих споры.
Следует иметь в виду, что элективные условия далеко не всегда оптимальны для роста выделяемых микроорганизмов, однако они переносятся ими лучше, чем сопутствующими формами.
О развитии накопительной культуры судят по появлению характерных признаков роста выделяемых микроорганизмов — помутнению среды, иногда сопровождаемому пигментацией, появлению пленки, осадка, выделению газов. Помимо визуального наблюдения накопительную культуру микроскопируют и выявляют присутствие желаемых форм. Иногда необходимо определить продукты метаболизма, образование которых свойственно выделяемым микроорганизмам. Например, о развитии нитрифицирующих бактерий свидетельствует появление в среде нитрит- и нитрат-ионов и уменьшение или даже полное исчезновение иона аммония.
Основные условия элективности, позволяющие получить накопительные культуры микроорганизмов с различным типом обмена веществ, см. на рис. 6.1.
6.2. ВЫДЕЛЕНИЕ ЧИСТОЙ КУЛЬТУРЫ
После того как получена накопительная культура, приступают к выделению чистой культуры. Она может быть получена из отдельной колонии или одной клетки.
6.2.1. Выделение чистой культуры из отдельной колонии
Основным методом выделения чистых культур микроорганизмов является метод, предложенный Р. Кохом. Принцип его заключается в получении чистой культуры из отдельной колонии. Однако этот метод неприменим для выделения микроорганизмов, которые не растут или плохо растут на плотных средах. К числу таких микроорганизмов относятся некоторые бактерии, многие водоросли и простейшие.
Рис. 6.2. Рассев культуры микрооршнизмов на поверхность плотной среды шпателем: а — шпатель Дригальского; б — рассев; в — пост микроорганизмов после рассева
При выделении чистой культуры аэробных микроорганизмов накопительную культуру высевают на поверхность плотной среды. Порядок работы следующий. Расплавленную на кипящей водяной бане стерильную и охлажденную до 50 °C питательную среду, содержащую агар или желатину, разливают в стерильные чашки Петри. После того как среда застынет, на ее поверхность из пипетки наносят каплю накопительной культуры или ее разведения в стерильной воде и стерильным стеклянным шпателем Дригальского распределяют каплю по поверхности плотной среды в чашке Петри. Далее этим же шпателем протирают поверхность среды последовательно во второй, третьей и четвертой чашках. Обычно в первых двух чашках после инкубации наблюдается сплошной рост микроорганизмов, тогда как в последующих — рост изолированных колоний (рис. 6.2). Рассевать накопительную культуру можно бактериологической петлей методом истощающего штриха. В этом случае накопительную культуру или ее разведение отбирают петлей и на поверхности плотной среды проводят штрихи в порядке, указанном на рис. 6.3. Перед каждым новым штрихом
петлю стерилизуют в пламени горелки.
После посева чашки Петри помещают в термостат крышками вниз, чтобы конденсационная вода, образовавшаяся на крышке чашки при застывании
агара, не помешала получить изолированные колонии. Чашки выдерживают в термостате в течение 1 —7 сут в зависимости от скорости роста микроорганизмов. Выросшие изолированные колонии отсевают петлей на поверхность скошенной плотной среды в пробирке или в жидкую среду.
Метод глубинного посева. Изолированные колонии микроаэрофильных микроорганизмов и факультативных анаэробов чаше получают методом глубинного посева. Для этого плотную питательную среду предварительно разливают в пробирки по 15 — 20 мл и стерилизуют. Непосредственно перед посевом пробирки помещают в кипящую водяную баню, чтобы среда расплавилась. Высев проводят из разведений накопительной культуры в стерильной водопроводной воде. Разведения готовят с таким расчетом, чтобы при высеве 0,5 — 1,0 мл разведения получить изолированные колонии. Степень разведения определяется
плотностью накопительной культуры. Высевы делают, как правило, из трехчетырех последних разведений. Для этого в пробирку с расплавленной и остуженной до 48 —50 °C средой вносят 0,5—1,0 мл одного из разведений накопительной культуры. Посевной ма-
Рис. 6.3. Схема последовательности (7—5) рассева культуры микроорганизмов на поверхность плотной среды петлей
териал тщательно перемешивают, вращая пробирку' между ладонями. Затем около пламени горелки вынимают из
пробирки пробку, обжигая края пробирки в пламени горелки, и быстро выливают содержимое пробирки в стерильную чашю/ Петри. После того как агари-зованная среда застынет, чашки Петри помещают в термостат. Колонии, выросшие в толще среды, вырезают стерильным скальпелем, извлекают стерильными капиллярными трубками или просто петлей и переносят в жидкую среду, благоприятную для развития выделяемых микроорганизмов.
Особые трудности возникают при выделении чистых культур облигатных анаэробов. Если контакт с молекулярным кислородом не вызывает сразу же гибели клеток, то посев проводят на поверхность среды в чашки Петри, но после посева чашки тотчас помещают в анаэростат. Однако чаще пользуются методом разведения. Сущность его заключается в том, что разведения накопительной культуры проводят в расплавленной и охлажденной до 45 — 50 °C ага-ризованной питательной среде. Делают 6—10 последовательных разведений. Затем среду в пробирках быстро охлаждают и заливают поверхность слоем стерильной смеси парафина и вазелинового масла (в соотношенш-i 3:1), что препятствует проникновению воздуха в толщу агаризованной среды.
Иногда агаризованпую питательную среду после посева и тщательного перемешивания перенося'1 в стерильные трубки Бурри. Можно использовать капиллярные пипетки Пастера, в которые набирают соответствующее разведение накопительной культуры в расплавленной агаризованной питательной среде. Конец капилляра запаивают. При удачно выбранном разведении накопительной культуры в одной из пробирок (пипеток Пастера, трубок Бурри) вырастают изолированные колонии. Чтобы извлечь образовавшиеся колонии, поступают следующим образом. Удаляют стерильной иглой слой парафина, а столбик агаризованной среды осторожно выдувают из пробирки в стерильную чашку Петри, пропуская газ, не содержащий кислород, через капилляр или иглу, помещенные между стенкой пробирки и агаризованной средой. Агаризован-ную среду из трубки Бурри выдувают, пропуская газ через ватную пробку.
В некоторых случаях плотную среду из пробирки извлекают иначе. Пробирку' слегка нагревают, вес время быстро вращая ее над пламенем горелки. При этом агар, непосредственно прилегающий к стенке, плавился и содержимое пробирки в виде агарового столбика легко выскальзывает в стерильную чашку Петри. Столбик агара разрезают стерильным ланцетом и извлекают колонии, захватывая их стерильными капиллярными трубками или петлей. Можно также вырезать их стерильным ланцетом. Извлеченные колонии переносят в жидкую среду, благоприятную для развития выделяемых микроорганизмов. Если изолированные колонии получены в капилляре, то после тщательной дезинфекции поверхности его разламывают стерильным пинцетом и участки капилляра, содержащие изолированные колонии, переносят в стерильную среду.
Для получения изолированных колоний методом глубинного посева и методом разведений рекомендуется использовать осветленные питательные среды (см. гл. 3).
Метод вращающихся пробирок. Когда хотят получить изолированные колонии облигатных анаэробных бактерий, характеризующихся особенно высокой чувствительностью к кислороду (экстремальные анаэробы), используют метод вращающихся пробирок Р.Хайгейта. Сущность его заключается в следующем. Расплавленную агаризован-ную среду? засевают бактериями при постоянном токе через пробирку стерильного инертного газа, освобожденного от примеси кислорода. Затем пробирку закрывают резиновой пробкой и помещают горизонтально в зажим, вращающий пробирку. Агаризо-ванная среда при этом равномерно распределяется по стенкам пробирки и застывает тонким слоем. Тонкий слой агаризованной среды в пробирке, заполненной газовой
Рис. 6.4. Изолированные колонии облигатных анаэробов в пробирках, заполненных газом (по Р. Хайгейту):
/ — агаризованная питательная среда; 2 — конденсационная вода; 3 — колонии бактерий; 4 — смесь газов Н2 и СО2
------1
---3
К
Рис. 6.5. Посев культуры строго анаэробных микроорганизмов с использованием модификации метода вращающихся пробирок Р. Хайгейта:
1 — бактериальная петля; 2 — трубка для подачи стерильного инертного газа; 3 — питательная среда, застывшая на стенках пробирки: 4 — механизм, вращающий пробирку
смесью, позволяет получить изолированные колонии, хорошо видимые невооруженным глазом (см. рис. 6.4). Применяют также модификацию метода Р.Хангейта. При этом расплавленную агаризованную среду при постоянном токе стерильного инертного газа разливают по пробиркам. Затем пробирку закрывают резиновой пробкой и помещают горизонтально в зажим, вращающий пробирку. После того как среда на стенках пробирки застынет, производят засев среды в этой пробирке в токе стерильного газа с помощью бактериальной петли, проводя штрих во вращающейся пробирке в направлении от ее дна к горлышку (рис. 6.5).
Иногда для получения чистой культуры бывает достаточно одного посева в плотную среду, однако чаще посев в плотную питательную среду повторяют 2-3 раза. В качестве посевного материала при этом используют культуру, полученную из отдельной колонии.
6.2.2. Выделение чистой культуры из одной клетки
Чистую культуру из одной клетки можно выделить капельным методом с помощью микроманипулятора, а также с помощью микроселектора.
Капельный метод Линднера. Метод используют при работе с крупными микроорганизмами: дрожжами, мицелиальными грибами, цианобактериями, водорослями. Порядок работы следующий. Накопительную культуру разводят в стерильной среде с таким расчетом, чтобы в небольшой капле были одиночные клетки микроорганизмов. Затем на поверхность нескольких стерильных покровных стекол стерильным стальным пером наносят по капле приготовленного разведения. Готовят препараты «висячая капля». Нанесенные на покровные стекла капли просматривают под микроскопом и отмечают тс, в которых обнаружена только одна клетка. После этого препарат помещают в термостат во влажную камеру, которой обычно служит чашка Петри с увлажненной фильтровальной бумагой на дне. Через 12—24 ч отмеченные капли вновь микроскопируют. Капли, в которых наблюдается образование микроколоний, осторожно снимают с покровного стекла кусочками стерильной фильтровальной бумаги и переносят в пробирки со стерильной средой.
Выделение отдельных клеток с помощью микроманнвулятора. Микрома! шпупятор — прибор, позволяющий с помощью специальной микро пипетки или микропетли извлекать из суспензии одну' клетку. Эту операцию контролируют под микроскопом. Микроманипулятор имеет два операционных штатива, между которыми расположен обычный микроскоп. На предметном столике микроскопа установлена влажная камера, в
которую помещают препарат «висячая капля». В держателях операционных штативов закреплены микропипетки (микропстли), перемещение которых в поле зрения микроскопа осуществляется с микронной точностью благодаря системе винтов и рычагов. Микропипетки вводят во влажную камеру таким образом, чтобы их концы оказались в висячей капле. Исследователь, глядя в микроскоп, извлекает отдельные клетки микропипетками и переносит их в пробирки со стерильной жидкой средой.
Выделение отдельных клеток с помощью микроселектора Перфильева. Наиболее существенной частью микросслсктора Перфильева является стеклянный микрокапилляр, имеющий строго прямоугольное сечение. Благодаря этому капал капилляра хорошо просматривается даже с иммерсионным объективом. Стерильный капилляр заполняют исследуемой суспензией клеток в агаризованной питательной среде и при большом увеличении микроскопа находят участок с одной клеткой. Специальным приспособлением этот участок капилляра стерильно выбивают в приемник, из которого затем переносят в стерильную среду. Микроселектор Перфильева можно использовать для выделения как крупных, так и мелких микроорганизмов.
6.3. ОПРЕДЕЛЕНИЕ ЧИСТОТЫ ВЫДЕЛЕННОЙ КУЛЬТУРЫ
Чистота выделенной культуры микроорганизмов должна быть тщательно проверена. Это осуществляется обычно несколькими способами: визуальным, микроскопическим контролем и высевом на ряд питательных сред. При визуальном контроле просматривается рост микроорганизмов по штриху на поверхности скошенной агаризованной среды. Если рост по штриху неоднороден, культура загрязнена. Такой контроль возможен только для культур, способных расти на поверхности плотных сред.
Чистоту культур микроорганизмов обязательно нужно контролировать под микроскопом. Для этого следует приготовить препарат фиксированных окрашенных клеток и посмотреть его с иммерсионной системой или сделать препарат живых клеток и посмотреть его, используя фазово-контрастное устройство. Чистая культура многих микроорганизмов, как правило, морфологически однородна; допустимо лишь незначительное варьирование размеров клеток. Однако необходимо помнить, что клетки некоторых бактерий, например микобактерий, нокардий и др., очень полиморфны, поэтому определение чистоты таких культур при микроскопировании вызывает некоторые затруднения. Чистоту культур микроорганизмов обязательно проверяют высевом на питательные среды. Прежде всего выделенную культуру высевают на питательную среду, благоприятную для ее роста. Однородность выросших колоний — свидетельство чистоты культуры. Обязателен посев на мясопептонпый агар — среду, которая обеспечивает рост многих хемогетеротрофов. Критерием чистоты культуры является однородность выросших колоний или отсутствие роста, если данные микроорганизмы на мясопептонном агаре не развиваются.
Следует иметь в виду, что заключение о чистоте некоторых культур микроорганизмов нельзя сделать только по результатам высева на МПА. Особенно это касается автотрофных микроорганизмов, а также представителей гетеротрофов, склонных развиваться с одним или несколькими спутниками. Чистоту таких культур микроорганизмов проверяют высевом еще на ряд сред — сусло, мясопептонпый бульон, картофельный агар и др. Набор сред и их состав определяются особенностями метаболизма выделенных микроорганизмов, а также их возможных спутников.
Глава 7
МЕТОДЫ КОЛИЧЕСТВЕННОГО УЧЕТА МИКРООРГАНИЗМОВ
О росте микроорганизмов в естественных субстратах или в питательных средах судят по изменению количества их клеток или биомассы в единице объема. Методы определения этих показателей могут быть прямыми (подсчет клеток под микроскопом, взвешивание) или косвенными. Косвенные методы основаны на измерении параметров, величина которых зависит от количества или биомассы микроорганизмов (число колоний, выросших после посева суспензии клеток на питательную среду, рассеяние или поглощение суспензией света, содержание в ней белка и т.д.). Выбор метода зависит от целей исследования, свойств питательной среды или субстрата, а также особенностей роста и морфологии микроорганизмов. Так, многие методы, используемые для определения числа одноклеточных микроорганизмов, неприемлемы при подсчете многоклеточных (нитчатых, мицелиальных и др. форм).
При оценке численности микроорганизмов, особенно в естественных субстратах (прежде всего в почве), необходимо помнить, что их клетки часто находятся в прикрепленном (адгезированном) состоянии или в виде микроколоний. Поэтому перед началом подсчета их нужно отделить от частиц субстрата и друг от друга (десорбировать). Выбор метода десорбции (механическое перемешивание суспензии клеток, растирание, обработка ультразвуком, применение поверхностно-активных веществ и т.д.) определяется особенностями исследуемого субстрата.
7.1. ОПРЕДЕЛЕНИЕ КОЛИЧЕСТВА КЛЕТОК МИКРООРГАНИЗМОВ ПОД МИКРОСКОПОМ
Для подсчета клеток микроорганизмов под микроскопом можно использовать счетные камеры, препараты фиксированных и окрашенных клеток, приготовленные на предметных стеклах или мембранных фильтрах. Перечисленные методы позволяют определить общее количество клеток в единице объема (как живых, так и мертвых). Основное ограничение большинства указанных методов — необходимость довольно высоких концентраций клеток в единице исследуемого субстрата.
7.1.1. Подсчет клеток в счетных камерах
Это метод рекомендуется использовать для подсчета крупных объектов — дрожжей, одноклеточных водорослей, конидий грибов, некоторых относительно крупных бактерий. В качестве примера можно привести камеру Горяева—Тома (рис. 7.1). Камера внешне представляет собой толстое предметное стекло, разделенное бороздками. Центральная часть стекла содержит выемку глубиной ),1 мм, на дно которой нанесена сетка. Глубина камеры (0,1 мм) и площадь больших и малых квадратов сетки (соответственно !/25 и !/400 мм2) указаны на предметном стекле.
Рис. 7.1. Счетная камера Горяева—Тома:
а — вид сверху; б — сид сбоку; в — при малом увеличении микроскопа
Заполнение камеры и подсчет клеток. При работе с камерой необходимо соблюдать определенные правила ее заполнения. Углубление с сеткой покрывают специальным шлифованным покровным стеклом и, слегка прижимая, двигают покровное стекло в противоположные стороны до появления картины интерференции (колец Ньютона), свидетельствующей о том. что стекло притерто к сторонам камеры. Только при таком условии объем камеры соответствует расчетному. Предварительно камеру заполняют исследуемой суспензией микроорганизмов, которую вносят капилляром или пипеткой. Заполненную камеру помещают на столик микроскопа. Подсчет клеток рекомендуется начинать через 2—3 мин после заполнения камеры, чтобы клетки осели, и при микроскопировании находились в одной плоскости. Подвижные клетки перед заполнением камеры убивают нагреванием или 0,5%-м раствором формалина.
Число клеток подсчитывают с объективом 8* (10х), реже 40х. С иммерсионным объективом работать нельзя, так как его рабочее расстояние меньше толщины стеки камеры. Обычно просчитывают клетки микроорганизмов в 10 больших или 20 малых квадратах сетки, следуя по диагонали. Учитывают все клетки, лежащие в квадрате сетки, а также пересекающие верхнюю и правую стороны квадрата. При подсчете число клеток в большом квадрате не должно превышать 20, а в малом — 10, в противном случае исходную суспензию разводят водопроводной водой. Для получения достоверного результа
та общее число подсчитанных клеток микроорганизмов должно быть не менее 600.
Подсчет клеток повторяют 3—5 раз, каждый раз заново монтируя камеру и заполняя ее суспензией микроорганизмов. Это обеспечивает большую точность, чем подсчет 600 клеток при однократном монтаже камеры. Количество клеток в 1 мл исследуемой суспензии вычисляют по формуле
«103
М -------п,
hS
где М — количество клеток в I мл суспензии; а — среднее количество клеток в квадрате сетки; h — высота камеры, мм; S— площадь квадрата сетки, мм2; I03 — коэффициент перевода [см3] в [мм3]; п — коэффициент разведения исследуемой суспензии.
7Л.2. Капиллярный метод прямого счета микрооорганизмов
Для подсчета микроорганизмов Б. В. Перфильевым были разработаны специальные многоходовые счетные капилляры* с плоскими параллельными стеклами (см. также гл. 37). Они имеют прямоугольное сечение и по принципу подсчета аналогичны счетной камере, их глубина и ширина определяются конструкцией капилляра и представляют известные величины, а длина обычно соответствует диаметру поля зрения микроскопа или может быть измерена. В от
* Перфильев Б.В., Габе Д.Р. Капиллярные методы изучения микроорганизмов. — М.; Л.: Изд-во АН СССР, 1961. - С. 307—332.
личие от камеры Горяева в капиллярах Перфильева можно использовать объективы с большим увеличением, в том числе иммерсионные, что позволяет подсчитывать мелкие микроорганизмы. Поэтому такие капилляры в принципе могут применяться для подсчета микробных клеток и контроля роста бактерий в промышленной, пищевой, а также медицинской микробиологии. Многоходовые капилляры представляют собой конструкцию из нескольких (обычно 5) параллельных капиллярных ячеек, смонтированных на специальном предметном стекле; клетки микроорганизмов подсчитывают в каждой капиллярной ячейке, а для дальнейших расчетов используют усредненное число.
При погружении капилляра в субстрат он заполняется им в силу своих капиллярных свойств. Затем капилляр помещают на предметное стекло, заливают конец расплавленным парафином (что позволяет предохранить содержимое капилляра от высыхания) и подсчитывают клетки, используя объективы 40 х, 90х или фазово-контрастное устройство.
Для получения достоверного результата подсчитывают клетки в 50—100 полях зрения, подсчет ведут во всех капиллярах. Количество клеток в 1 мл исследуемого субстрата определяют по формуле
где М — количество клеток в 1 мл исследуемого субстрата: а — среднее число клеток в капилляре длиной в диаметр поля зрения; h — глубина капилляра, мм; / — ширина капилляра, мм; d — диаметр поля зрения (длина капилляра) при данном увеличении микроскопа, мм; 103 — коэффициент перевода [см3] в [мм3]; п — коэффициент разведения исследуемого субстрата.
7.1.3. Подсчет клеток на фиксированных окрашенных мазках (метод Виноградского—Брида)
Этот метод применяется в различных модификациях для определения численности микроорганизмов в разнообразных естественных субстратах — почве, загрязненной воде, оптически непрозрачных средах, содержащих нерастворимые в воде компоненты, например крахмал, соевую муку и т.д. Преимущество метода заключается также в том, что фиксированные окрашенные препараты могут долго храниться, поэтому подсчет можно производить в удобное для исследователя время.
Приготовление препарата. Хорошо обезжиренное предметное стекло помещают на миллиметровую бумагу, на которой размечен прямоугольник площадью 4 или 6 см2. Затем на стекло наносят точно отмеренный объем исследуемой суспензии (10; 20 или 30 мкл) и в некоторых случаях добавляют каплю 0,03—0,1%-го водного раствора агара. Нанесенную суспензию равномерно распределяют истлей по площади, отмеченной па миллиметровой бумаге. Препарат подсушивают на воздухе, фиксируют 10—20 мин абсолютным спиртом (96°) и окрашивают эритрозином, метиленовым синим, фуксином или другим красителем. Затем препарат промывают, последовательно погружая стекло в 4 — 5 сосудов с водой (промывать под проточной водой не следует) и высушивают на воздухе. В таком виде препараты хорошо сохраняются.
Правила подсчета. Препарат микроскопируют с иммерсионным объективом. При этом подсчитывают количество клеток в квадратах окулярной сетки, которую помеща
ют в окуляр между собирательной и глазной линзами. При отсутствии сетки подсчитывают число клеток в поле зрения микроскопа. Правило подсчета в квадратах окулярной сетки то же, что и в квадратах сетки счетной камеры. Чтобы результат был достоверным, клетки микроорганизмов рекомендуется подсчитывать в 50— 100 полях зрения. Общее количество подсчитанных клеток нс должно быть менее 600. Количество клеток микроорганизмов в 1 мл исследуемого субстрата вычисляют по формуле
где М — количество клеток в I мл исследуемого субстрата; а — среднее количество клеток в квадрате окулярной сетки (поле зрения); л — площадь квадрата окулярной сетки (поля зрения), мм2; V— объем нанесенной на стекло суспензии, мл; 5— площадь мазка, мм2; п — коэффициент разведения исследуемого субстрата.
Плошадь квадрата сетки, или поля зрения, определяют с помощью объект-микрометра, который помещают на столик микроскопа вместо препарата, и при том же увеличении, при котором проводили подсчет, определяют сторону квадрата окулярной сетки или диаметр поля зрения. Площадь поля зрения вычисляют по формуле 5 = яг2.
7.1.4. Подсчет клеток на мембранных фильтрах
Данный метод рекомендуется использовать для определения численности микроорганизмов в субстратах с низкой плотностью клеток. Его применяют при определении количества микроорганизмов в различных водоемах, при санитарно-бактериологических и некоторых других исследованиях. Фильтрование пробы известного объема (от нескольких миллилитров до литров) позволяет сконцентрировать на поверхности фильтра содержащиеся в пробе клетки микроорганизмов. Затем их окрашивают и подсчитывают.
Приготовление фильтра. Для фильтрования выбирают фильтр, размер пор которого позволяет задерживать микроорганизмы, находящиеся в субстрате. Характеристика мембранных фильтров приведена в гл. 4. Перед использованием фильтры кипятят в дистиллированной воде для удаления воздуха и остатков растворителей; воду следует 2—3 раза сменить (кипячение не должно быть слишком бурным, иначе фильтры будут скручиваться). Затем фильтр помешают на специальный держатель матовой стороной вверх и пропускают через него точно измеренный объем пробы (подробнее о технике фильтрования через мембранные фильтры см. гл. 4).
Чем больше плотность клеток в исследуемом материале, гем меньше должен быть исследуемый объем, и наоборот. Клетки микроорганизмов, осевшие на фильтре, окрашивают карболовым эритрозином. Для этого фильтр помещают нижней стороной в чашку Петри на фильтровальную бумагу, увлажненную красителем, чашку закрывают крышкой и оставляют на 3 — 5 ч или на сутки. Для равномерного окрашивания клеток мембранный фильтр должен плотно прилегать к бумажному фильтру с эритрозином. Затем мембранный фильтр отмывают от красителя, перекладывая его в чашки Петри с фильтровальной бумагой, обильно смоченной дистиллированной водой, до тех пор, пока он не перестанет ее окрашивать. После отмывания фильтр высушивают на воздухе и готовят препарат для микроскопии. На предметное стекло наносят каплю иммерсионного масла и помещают на него окрашенный мембранный филыр так, чтобы клетки микроорганизмов были сверху. На поверхность мембранного фильтра наносят еще каплю иммерсионного масла и покрывают фильтр покровным стеклом.
Правила подсчета. Количество клеток микроорганизмов подсчитывают с иммерсионным объективом 90х в квадратах окулярной сетки или в поле зрения микроскопа. Правила подсчета аналогичны тем, которые изложены для .метода Виноградского— Брида. Количество клеток в I мл исследуемого субстрата вычисляют по формуле
sV
где М — количество клеток в I мл исследуемого субстрата; а — среднее количество клеток в квадрате окулярной сетки (паче зрения); F— площадь мембранного фильтра, мм2; V— объем профильтрованной жидкости, мл; 10б — коэффициент перевода [мм2] в [мкм2]; 5 — площадь квадрата окулярной сетки (поля зрения).
При выявлении и количественном учете микроорганизмов широко применяют люминесцентную микроскопию (см. гл. 5). Препараты микроорганизмов готовят непосредственно из исследуемой суспензии или ее разведения либо концентрируют клетки на специально обработанных нефлуоресцируюших фильтрах. Препараты и фильтры с микроорганизмами обрабатывают акридином оранжевым или другими флуоресцирующими красителями. Принципы подсчета при светлопольной и люминесцентной микроскопии одинаковы, однако окрашенные флуорохромами клетки более четко видны на темном фоне препарата, их легче отличить от небиологических объектов (частиц ила, почвы и т.д.) и произвести подсчет более точно.
Люминесцентная микроскопия дает также возможность выявить и оценить в исследуемой пробе численность отдельных групп микроорганизмов. Для этого применяют специальные флуорохромы (например, калькофлуор белый — для выявления грибов), метод иммунофлуоресценции и др.
7.2. ОПРЕДЕЛЕНИЕ ЧИСЛА КЛЕТОК МИКРООРГАНИЗМОВ ВЫСЕВОМ НА ПИТАТЕЛЬНЫЕ СРЕДЫ
В отличие от подсчета микроорганизмов под микроскопом этот метод дает возможность определить только число жизнеспособных клеток в популяции. Поскольку сред, пригодных для роста всех микроорганизмов, не существует, метод высева дает возможность определить лишь число микроорганизмов, способных расти на среде данного состава, причем не позволяет учесть те микроорганизмы, которые не растут (например, так называемые жизнеспособные, но не культивируемые формы) или растут очень медленно. Это важно помнить при анализе таких естественных субстратов, как почва, вода и т.п.
7.2.1. Определение количества клеток высевом на плотные питательные среды (метод Коха)
Метод широко применяют для определения численности жизнеспособных клеток в различных естественных субстратах и в лабораторных культурах. В его основе лежит принцип Коха, согласно которому каждая колония является потомством одной клетки. Это позволяет на основании числа колоний, вырос
ших после посева на плотную питательную среду определенного объема исследуемой суспензии, судить об исходном содержании в ней клеток микроорганизмов. Результаты количественного определения микроорганизмов, проведенного по методу Коха, часто выражают нс в числе клеток, а в условных, так называемых колониеобразуюших единицах (КОЕ).
Определение числа микроорганизмов этим методом включает три этапа: приготовление разведений; посев на плотную среду в чашки Петри и подсчет выросших колоний.
1. Приготовление разведеиий. Численность популяции микроорганизмов обычно велика, поэтому для получения изолированных колоний необходимо приготовить ряд последовательных разведений. Разведения готовят в стерильной водопроводной воде или в 0,85%-м растворе NaCl (физиологическом растворе). В ходе опыта целесообразно использовать один и тот же коэффициент разведения, например 10, что уменьшает вероятность ошибки.
Для приготовления разведений стерильную водопроводную воду разливают по 9 мл в стерильные сухие пробирки. Затем I мл исследуемой суспензии стерильной пипеткой переносят в пробирку с 9 мл стерильной воды — это первое разведение (10"1). Полученное разведение тщательно перемешивают новой стерильной пипеткой, несколько раз вбирая в пипетку и выпуская из нее полученную суспензию клеток. Затем той же пипеткой отбирают I мл суспензии и переносят во вторую пробирку, получая второе разведение (10“ ). Таким же образом готовят последующие разведения. Степень разведения зависит от плотности исследуемой популяции микроорганизмов; соответственно она тем больше, чем больше плотность популяции.
Для приготовления каждого разведения следует обязательно использовать новую пипетку. Пренебрежение этим правилом приводит к получению ошибочного результата вследствие высокой способности клеток микроорганизмов к сорбции на поверхности стекла.
2. Посев. Высевать суспензию можно поверхностным или глубинным способом. Перед посевом поверхностным способом (рис. 7.2) разливают расправленную агаризованную питательную среду в ряд стерильных чашек Петри по 15 — 20 мл в каждую. Чашки оставляют на горизонтальной поверхности, пока среда нс застынет. Поверхность ага-ризованных сред перед посевом рекомендуется подсушить для удаления кондеисаци он нои воды, например поместив чашки в термостат на 2—3 сут крышками вниз.
В чашку Петри с подсушенной средой вносят точно измеренный объем (0,05 или 0,1 мл) соответствующего разведения и распределяют его стеклянным шпателем по поверхности среды. Высевы на плотную среду проводят, как правило, из трех последних разведений, причем из каждого делают 2—4 параллельных высева. Посевы можно делать одной пипеткой, но при этом начинать следует обязательно с большего разведения. Для каждого разведения используют новый стерильный шпатель. После посева чашки Петри помещают в термостат крышками вниз.
При глубинном посеве точно измеренный объем (как правило, 0,1; 0,5 или 1,0 мл) исходной суспензии или разведения вносят в расплавленную и остуженную до 45—50 °C среду, тщательно перемешивают, затем немедленно выливают в чашку Петри и дают среде застыть. В случае глубинного посева пользуются средой, разлитой в пробирки. При больших масштабах работы среду по пробиркам не разливают, а поступают следующим образом. По 1 мл из соответствующего разведения переносят стерильной пипеткой в 2 —4 стерильные чашки Петри. Затем заливают в чашки по 15 — 20 мл среды, расплавленной и остуженной до 45 — 50 СС, и смешивают питательную среду с посевным материалом легким вращательным движением чашки по поверхности стола, после чего чашки оставляют на горизонтальной поверхности до застывания среды. Когда среда застынет, чашки Петри в перевернутом виде помещают в термостат.
Рис. 7.2. Схема приготовления разведений и посева суспензии микроорганизмов шпателем
Для определения количества клеток анаэробных микроорганизмов чашки Петри после посева помещают в анаэростат. Иногда для определения численности анаэробов плотную среду после засева оставляют в пробирках. Поверхность застывшей среды заливают парафином. Для лучшего учета колоний микроорганизмов среды в этом случае рекомендуется осветлять. Определение численности строгих анаэробов требует применения техники Р.Хангейта (см. гл. 3).
3. Подсчет выросших колоний. Колонии микроорганизмов в зависимости от скорости роста подсчитывают через I —15 сут инкубации. Подсчет, как правило, проводят, не открывая чашек Петри. Для удобства каждую просчитанную колонию отмечают точкой на наружной стороне дна чашки. При большом количестве колоний дно чашки Петри делят на секторы, просчитывают колонии в каждом секторе и суммируют результаты. Иногда для подсчета колоний используют специальные полуавтоматические счетчики.
Лучшим разведением следует считать то, из которого при высеве в чашке Петри вырастает от 30—50 до 100— 150 колоний. Если число выросших колоний меньше 10, то эти результаты для расчета количества клеток в исходном материале не используют. Результаты параллельных высевов из одного и того же разведения суммируют и определяют среднее число колоний, выросших при высеве из разведения на одной чашке.
Количество клеток в 1 мл исследуемого субстрата вычисляют по формуле
Л/=^°7
V
где М— количество клеток в I мл; а— среднее число колоний, выросших после посева из данного разведения; И — объем суспензии, взятый для посева, мл; 10й — коэффициент разведения.
7.2.2. Определение количества клеток высевом в жидкие среды (метод предельных разведений)
Метод используют для подсчета микроорганизмов, которые плохо или совсем не растут на плотных питательных средах. В пробирки с жидкой средой вносят строго измеренный объем из различных разведений питательного субстрата. После инкубации, исходя из числа пробирок, в которых наблюдался или отсутствовал рост, рассчитывают наиболее вероятное число клеток, содержащихся в 1 мл исследуемого субстрата. Таким образом, определение количества микроорганизмов методом предельных разведений включает приготовление разведений, посев в жидкую среду, регистрацию наличия или отсутствия роста после инкубации и расчет наиболее вероятного числа клеток в единице объема исходного субстрата.
1. Приготовление разведений. Разведения исходной суспензии готовят каки для чашечного метода.
2. Посев и регистрация результатов. Стерильную среду предварительно разливают в пробирки (колбы) и стерилизуют. В пробирки (колбы) следует наливать одинаковый объем среды. Посев проводят из каждого разведения или из 4 — 5 последних, причем каждое разведение высевают в 3—5 повторностях (параллельных пробирках). Количество посевного материала везде одинаково и, как правило, составляет 1 мл. Засеянные пробирки помещают в термостат. Время инкубации колеблется от 2 до 15 сут и зависит от скорости роста микроорганизмов, численность которых определяют. После инкубации регистрируют рост микроорганизмов, используя различные показатели: помутнение среды, образование пленки, осадка, газа или накопление в среде определенных продуктов метаболизма.
Таблица 7.1
Наиболее вероятное количество клеток микроорганизмов в единице объема исходной суспензии (по Мак-Креди)
Чмеловая характеристика Наиболее вероятное число микроорганизмов при засеве параллельных пробирок в числе Числовая характеристика Наиболее вероятное число микроорганизмов при засеве параллельных пробирок в числе Числовая характеристика Наиболее вероятное число микроорганизмов при засеве параллельных пробирок в числе
2 3 4 5 2 3 4 5 2 3 4 5
ООО 0,0 0,0 0,0 0,0 222 110,0 3,5 2,0 1,4 433 - - 30,0 -
001 002 0,5 0,3 0,2 0,2 223 - 4,0 - - 434 35,0
- 0,5 0,4 230 - 3,0 U7 1,2 440 - - 25,0 3,5
003 - - 0,7 - 231 - 3,5 2,0 1,4 441 - - 40,0 4,0
010 0,5 0,3 0,2 0,2 232 - 4,0 - - 442 - - 70,0 -
ОН 0,9 0,9 0,5 0,4 240 - - 2,0 1,4 443 - - 140,0 -
012 013 - - 0,7 0,6 241 - - 3,0 444 160,0 -
- - 0,9 - 300 - 2,5 1,1 0,8 450 - - - 4,0
020 0,9 0,6 0,5 0,5 301 - 4,0 1,6 1,1 451 - - - 5,0
021 - - 0,7 0,6 302 - 6,5 2,0 1,4 500 - - - 2,5
Окончание табл. 7.1
Числовая характеристика Наиболее вероятное число микроорганизмов при засеве параллельных пробирок в числе Числовая характеристика Наиболее вероятное число микроорганизмов при засеве параллельных пробирок в числе Числовая ха ракте-ристика Наиболее вероятное число микроорганизмов при засеве параллельных пробирок в числе
2 3 4 5 2 3 4 5 2 3 4 5
022 0,9 303 - - 2,5 - 501 - - - 0,3
030 - - 0,7 0,6 310 - 4,5 1,6 1,1 502 - - - 4,0
031 - - 0,9 - 311 - 7,5 2,0 1,4 503 - - - 6,0
040 - - 0,9 - 312 - 11,5 3,0 1,7 504 - - - 7,5
041 - - 1,2 - 313 - 16,5 3,5 2,0 510 - - - 3,5
100 0,6 0,4 0,3 0,2 320 - 9,5 2,0 1,4 511 - - - 4,5
101 1,2 0,7 0,5 0,4 321 - 15,0 3,0 1,7 512 - - - 6,0
102 - и 0,8 0,6 322 20,0 3,5 2,0 513 - - - 8,5
103 - - 1,0 0,8 323 - 30,0 - - 520 - - - 5,0
но 1,3 0,7 0,5 0,4 330 - 25,0 3,0 1,7 521 - - - 7,0
111 2,0 1,1 0,8 0,8 331 45,0 3,5 2,0 522 - - - 9,5
112 - - 1,1 0,8 332 - 110,0 4,0 - 523 - - - 12,0
113 - - 1,3 - 333 - 140,0 5,0 - 524 - - - 15,0
120 2,0 1,1 0,8 0,6 340 - - 3,5 2,0 525 - - - 17,5
121 3,0 1,5 1,1 0,8 341 - - 4,5 2,5 530 - - - 8,0
122 - - 1,3 1,0 350 - - - 2,5 531 - - - 11,0
123 - - 1,6 - 400 - - 2,5 1,3 532 - - - 14,0
130 - 1,6 1,1 1,8 401 - - 3,5 1,7 533 - - 17,5
131 - - 1,4 1,0 402 - 5,0 2,0 534 - - - 20,0
132 - - 1,6 - 403 - - 7,0 2,5 535 - - - 25,0
140 - - 1,4 1,1 410 - - 3,5 1,7 540 - - - 13,0
141 - - 1,7 - 411 - - 5,5 2,0 541 - - - 17,0
200 2,5 0,9 0,6 0,5 412 - - 8,2 2,5 542 - - 25,0
201 5,0 I,4 0,9 0,7 413 - - 8,0 - 543 - - - 30,0
202 - 2,0 1,2 0,9 414 - - 14,0 - 544 - - - 35,0
203 - 1,6 1,2 420 - - 6,0 2,0 545 - - - 45,0
210 6,0 1,5 0,9 0,7 421 - - 6,5 2,5 550 - - - 25,0
211 13,0 2,0 1,3 0,9 422 - - 17,0 - 551 - - - 35,0
212 2,0 3,0 1,6 1,2 423 - - 13,0 3,0 552 - - 60,0
213 - - 2,0 424 - - 20,0 - 553 - - - 90,0
220 25,0 2,0 1,3 0,9 430 - - 11,5 1,5 554 - - 100,0
221 70,0 3,0 1,6 1,2 431 - - 16,5 3,0 555 - - 180,0
432 - - 20,0 4,0
Наиболее вероятное число микроорганизмов в единице объема рассчитывают по таблице Мак-Кредм (табл. 7.1), разработанной на основании методов вариационной статистики. Для этого первоначально составляют числовую характеристику, которая включает три цифры. Первая цифра слева показывает число пробирок в том последнем разведении, при высеве из которого во всех засеянных пробирках был отмечен рост. Две следующие цифры обозначают число пробирок, в которых отмечен рост микроорганизмов при высеве из двух последующих разведений. Затем по таблице находят наиболее вероятное число микроорганизмов, соответствующее данному значению числовой характеристики. Количество микроорганизмов в 1 мл (I г) исходного субстрата соответствует этому числу, умноженному на то разведение, которое было взято для получения первой цифры числовой характеристики. Приведем примеры.
Пример 1
Разведения исходной суспензии 0 ю-' ю-2 КУ3 кг1
Число засеянных пробирок 4 4 4 4 4
Число пробирок, в которых обнаружен рост 4 4 3 I 0
Числовая характеристика 431 — — — —
Наиболее вероятное число клеток микроорганизмов 16,5 — — —
Количество клеток микроорганизмов в 1 мл исходной суспензии 165 — — — —
Пример 2
Разведения исходной суспензии 10-’- I0-3 10 4 ПН 10-6
Число засеянных пробирок 3 3 3 3 3
Число пробирок, в которых обнаружен рост 3 3 2 0 0
Числовая характеристика 320 — — —
Наиболее вероятное число клеток микроорганизмов 9,5 — — — —
Количество клеток микроорганизмов в 1 мл исходной суспензии 9,5 Ю3 - — - —
7.2.3. Общие замечания
Точность любого метода определения числа микроорганизмов ограничена ошибкой метода, которая возникает вследствие случайного распределения клеток в суспензии и является результатом ограниченного числа подсчитываемых клеток, а также связана с техническими ошибками, т.е. неточностью в приготовлении разведений, неправильным монтажом камеры, повторным учетом одной и той же клетки (колонии) и т.д. Ошибки метода неизбежны, тогда как технические ошибки зависят главным образом от качества работы исследователя. Следует помнить, что статистическая обработка результатов возможна только при минимальной технической ошибке. Чашечный метод и метод предельных разведений требуют особой чистоты и аккуратности при выполне-
но
нии всех операций. Необходимо тщательно оберегать пипетки, пробирки и среды от заражения микроорганизмами из воздуха, так как каждая случайно попавшая клетка может заметно завысить число микроорганизмов в исследуемом субстрате.
7.3. ОПРЕДЕЛЕНИЕ БИОМАССЫ ВЗВЕШИВАНИЕМ
Этот метод широко применяют для оценки роста микроорганизмов в жидких питательных средах. Можно использовать его и для определения массы клеток, выращенных на плотной питательной среде, однако в этом случае микроорганизмы необходимо предварительно тщательно смыть с поверхности среды физиологическим раствором или водой и перевести в суспензию. Метод не может быть использован при культивировании микроорганизмов на средах, в состав которых входят соединения, не растворимые в воде.
Определение биомассы состоит из трех последовательных операций: доведение массы центрифужных пробирок или фильтров до постоянного значения; отделение клеток микроорганизмов от культуральной жидкости; определение их массы. Чаше всего определяют массу сухих клеток, хотя иногда можно ограничиться определением сырой биомассы. В последнем случае первый этап отпадает; достаточно только взвесить пустую центрифужную пробирку (фильтр), но не доводить ее массу до постоянного значения. Биомассу обычно выражают в граммах или миллиграммах на литр культуральной жидкости.
1. Доведение массы центрифужных пробирок или фильтров до постоянного значения. С этой целью фильтры, предварительно положенные в открытую чашку Петри или центрифужные пробирки (не пластиковые!), помещают в сушильный шкаф и высушивают в течение 1 —2 ч при температуре 80 — 85 °C или 90—100 °C соответственно. Затем чашку Петри с фильтрами или центрифужные пробирки вынимают из сушильного шкафа и быстро переносят в эксикатор с безводным хлористым кальцием (СаС12) или концентрированной серной кислотой. Эксикатор ставят около аналитических весов, на которых будет проводиться взвешивание. Через час фильтры (центрифужные пробирки) взвешивают с точностью до 0,0001 г. Высушивание и взвешивание повторяют, соблюдая указанную последовательность операций, пока масса не достигнет постоянного значения, т. е. колебания в ее определениях не будут превышать десятых долей мг.
2. Отделение микроорганизмов от среды возможно центрифугированием или фильтрованием. В центрифужную пробирку наливают точно измеренный объем тщательно перемешанной жидкой культуры, который в зависимости от ее плотности колеблется от 5 до 20 мл. Время центрифугирования и число оборотов зависят от размеров клеток. Чем они меньше, тем больше требуется оборотов и тем продолжительнее должно быть время центрифугирования. Чаще всего центрифугируют 15 — 20 мин при 3 — 5 тыс. g. После центрифугирования надосадочную жидкость осторожно сливают, осадок промывают слегка подкисленной дистиллированной водой (1 мл концентрированной НС1 на 1 л воды) и снова центрифугируют при том же числе оборотов. Супернатант сливают тотчас после остановки центрифуги. В противном случае часть осадка может быть потеряна.
Мицелий актиномицетов и грибов отделяют фильтрованием. Бумажный фильтр помещают в стеклянную воронку и фильтруют через него точно измеренный объем культуры — от 5 до 10 мл. Осадок на фильтре многократно промывают подкисленной дистиллированной водой.
Для отделения бактерий используют мембранные фильтры. Размеры пор мембранного фильтра должны быть меньше величины клеток, биомассу которых определяют
(характеристику фильтров см. гл. 4.). Мембранный фильтр помещают на пористую пластинку специального держателя, вставленного в колбу. Чтобы ускорить фильтрование, установку подключают к водоструйному насосу. Осадок несколько раз промывают подкисленной водой. Подробно порядок работы с мембранными фильтрами см. гл. 4.
3. Определение биомассы. Чтобы определить массу сухих клеток, центрифужную пробирку или фильтр с осадком клеток микроорганизмов помещают в сушильный шкаф, высушивают и взвешивают. Режим высушивания и взвешивания тот же, что использовали и при определении массы пробирок или фильтров. Сухую биомассу определяют по формуле
д/ (Л - В) 1000
И
где М — сухая биомасса, г/л; Л — масса центрифужной пробирки (фильтра) с осадком, г; В — масса центрифужной пробирки (фильтра) без осадка, г; V — объем культуральной жидкости, взятый для центрифугирования (фильтрования), мл. Точность метода определяется полнотой отмывания клеток от компонентов среды и тщательностью взвешивания.
7.4. ОПРЕДЕЛЕНИЕ КОЛИЧЕСТВА КЛЕТОК И БИОМАССЫ НЕФЕЛОМЕТРИЧЕСКИМ МЕТОДОМ
Оптический (нефелометрический, турбидиметрический) метод определения биомассы нашел широкое применение в лабораторных микробиологических исследованиях, поскольку позволяет быстро и довольно точно определить концентрацию клеток в суспензии или культуральной жидкости.
В основе метода лежит измерение ослабления светового пучка при его прохождении через суспензию клеток. В определенных пределах оно обусловлено преимущественно рассеянием света клетками и пропорционально их концентрации. Величина этого показателя зависит от многих факторов (формы и размеров клеток, оптических свойств культуральной среды, длины волны света и т.д.), поэтому нефелометрический метод пригоден лишь для тех микроорганизмов, рост которых вызывает равномерное помутнение среды и не сопровождается заметным изменением формы и размеров клеток, образованием мицелия, пленок или других скоплений.
Питательная среда для культивирования микроорганизмов, в которой предполагается определять число клеток по светорассеянию, должна быть оптически прозрачной.
Изменение интенсивности света при прохождении через суспензию клеток измеряют с помощью нефелометра, фотоэлектроколориметра (ФЭК) или спектрофотометра, выбирая длину волны (обычно в интервале 540—650 нм), при которой поглощение света данной суспензией клеток минимальное. При высоких концентрациях клеток в культуральной среде происходит вторичное рассеяние света, что приводит к занижению результатов. Правила работы на фотоэлектроколориметре подробно изложены в инструкции, прилагаемой к прибору.
В некоторых случаях плотность клеточной суспензии выражают в показателях нефелометра. Олнако чаще строят калибровочные кривые между величиной светорассеяния и числом клеток или сухой биомассой в единице объема. Для построения калибровочной кривой поступают следующим образом. Изме-
ряют величину оптической плотности суспензий с различным содержанием клеток и в каждой из них определяют одним из применяемых методов кол ичество клеток или биомассу. Полученную зависимость выражают графически, откладывая на оси ординат показания ФЭК, а на оси абсцисс — количество клеток, содержащихся в 1 мл суспензии, или биомассу в г/л. Для каждого микроорганизма следует строить свою калибровочную кривую.
7.4.1. Стандарты мутности и их применение
В ряде случаев количество клеток в суспензии бывает достаточно определить визуально путем сравнения со стандартом мутности. Стандарты мутности представляют собой взвесь частиц стекла пирекс в дистиллированной воде. За единицу стандарта мутности условно принята мутность суспензии (в физрастворе) бактерий — возбудителей тифа с концентрацией клеток 100 млн/мл. Стандарт мутности включает 4 эталона на 11, 10, 9 и 5 единиц, что соответствует содержанию 1,1 109; 1 • 109; 0,9 • 109 и 0,5 109 клеток в 1 мл взвеси. Для определения количества клеток пробирку с исследуемой суспензией ставят рядом с эталоном 10 и рассматривают их в отраженном и проходящем свете на фоне белого листа бумаги, в центре которого нанесено несколько черных линий. Эталоны 9 и 11 вспомогательные, позволяющие более четко сравнить мутность исследуемой суспензии бактерий с эталоном. Стандартизация мутности суспензии бактерий (особенно часто в случае тест-организмов) имеет существенное значение при приготовлении посевного материала в серийных опытах, например при определении антибиотической активности препаратов методом диффузии в агар.
7.5. ПРОТОЧНАЯ ЦИТОМЕТРИЯ
Первоначально прибор и метод проточной цитометрии (Flow cytometry, или FCM) были разработаны для подсчета форменных элементов крови. После соответствующих модификаций этот прибор стали применять для подсчета клеток микроорганизмов (одноклеточных) сначала в водных образцах, а впоследствии и в почвенных. Принцип метода основан на регистрации сигнала (светового, электрического) при прохождении частицы по капилляру. Если разные клетки рассеивают проходящий луч света по-разному, то можно дифференцированно осуществлять учет разных клеток. Однако в любом случае будет регистрироваться как проходящая клетка (живая или неживая), так и любая другая частица. Чтобы избежать этого, перед учетом клетки в образцах стали окрашивать специфическими флуоресцентными красителями. В этом случае учитываются только флуоресцирующие клетки. Если суспензию окрасить разными флуоресцентными красителями, различно окрашивающими, живые и мертвые клетки, то появляется возможность вести дифференцированный учет живых и мертвых клеток в одном образце.
Современные приборы измеряют несколько характеристик проходящей частицы: переднее и боковое угловое рассеяние и флуоресценцию при разных длинах волн. В качестве источника света при освещении (возбуждении) объек-
та и регистрации испускаемого света используются «холодные» лазеры с длинами волн возбуждающего света, соответствующих голубой части спектра, и красители (чаще всего нуклеиновых кислот), характеристики которых как раз соответствуют этим длинам волн. Переднее угловое рассеяние регистрируют с помощью диодного детектора, боковое угловое рассеяние и флуоресценция регистрируются фотоумножителями. Получаемые результаты анализируют с помощью соответствующих компьютерных программ (WinMDI; Joseph Totter, Salk Institute for Biological Studies, La Jolla, США; http://facs.Scripps.edu/ software, html), которые разрабатываются изготовителями цитометров (Becton Dickinson, США), или другими компаниями (http://jcsmr.anu.edu.au). Результаты подсчета клеток обычно представляют в логарифмическом масштабе после соответствующей статистической обработки.
Перед использованием проточного питометра производятся его настройка и калибровка путем пропускания суспензии, содержащей известное количество клеток, с разными скоростями протока жидкости — обычно от 10 до 90 мкл/мин в течение нескольких минут. Для учета рекомендуется использовать суспензии клеток, содержащие до 106 кл/мл. Проводят учет от 5000 до 10 000 клеток. В качестве внутреннего стандарта в исследуемые образны рекомендуется добавлять (в объеме до 10 мкл при объеме исследуемого образца 1 мл) суспензию, например желто-зеленых частичек латекса размером 0,7—0,9 мкм (Polyscience, Eppelheim, ФРГ), так чтобы конечное их количество составляло до К)6 частичек/мл. Для предотвращения ошибок, возникающих из-за неспецифической или природной флуоресценции объектов (например, флуоресценция хлорофилла и т.д.), используют соответствующие фильтры, которые поглощают такую флуоресценцию.
Проточные цитометры производят в разных странах, в частности в США. Проточный цитометр FACSCalibr (Becton Dickinson Immunocytometry Systems, San Jose, США) снабжен воздушно-охлаждаемым аргоновым лазером мощностью 15 мВт при 488 нм и сортирующей клетки трубкой-ловушкой. Широко используется проточный цитометр EPICS 541 (Coulter Со., Hialeah, США), оборудованный биосенсором проходящих клеток и самонастраивающимся лазером (Coherent Innova 90) мощностью 0,5 Вт, создающий луч света с длиной волн в зависимости от используемых флуоресцирующих красителей в интервале от 488 (для красителей YOYO-1, YO-PRO-1 или PicoGrecn) до 353 — 357 нм (для красителей Hoechst 33342 или DAPI). Скорость учета в таком цитометре достигает 1000 клеток в секунду, однако рекомендуется использовать скорость счета не более 300 клеток в секунду.
Последовательность операций на разных приборах может заметно отличаться, поэтому при выполнении конкретных анализов следует руководствоваться инструкциями и рекомендациями, прилагаемыми к конкретному прибору. Следует отметить, что такая техника довольно дорогая, в связи с чем рекомендуется для приобретения и использования только при необходимости массовых анализов.
Глава 8
КУЛЬТУРАЛЬНЫЕ И ФИЗИОЛОГО-БИОХИМИЧЕСКИЕ СВОЙСТВА МИКРООРГАНИЗМОВ
Для решения многих задач бывает достаточно определять только некоторые, легко выявляемые свойства и физиолого-биохимические особенности
культур микроорганизмов. Знание таковых в сочетании с характеристикой морфологии микроорганизма иногда позволяет установить его принадлежность к тому или иному таксону — классу, порядку, семейству.
8.1. КУЛЬТУРАЛЬНЫЕ СВОЙСТВА
К культуральным, или макроморфологическим, свойствам относятся характерные особенности роста микроорганизмов на плотных и жидких питательных средах.
8.1.1. Рост на плотных питательных средах
На поверхности плотных питательных сред в зависимости от посева микроорганизмы могут расти в виде колонии, штриха или сплошного газона. Колонией называют изолированное скопление клеток одного вида, выросшее в большинстве случаев из одной клетки. В зависимости от того, где развивались клетки (на поверхности плотной питательной среды, в толще ее или на дне сосу-
Рис. 8.1. Форма колонии:
1 — круглая; 2 — круглая с фестончатым краем; 3 — круглая с валиком по краю; 4, 5 — ризоидные; 6 — с ризоидным краем; 7 — амебовидная; 8 — нитевидная; 9 — складчатая; 10 — неправильная; 11 — концентрическая; 12 — сложная
1
2
3
4
Рис. 8.2. Профиль колонии:
1 — изогнутый; 2 — кратерообразный; 3 — бугристый; 4 — врастающий в субстрат; 5 — плоский; 6— выпуклый; 7— каплевидный; 8 — конусовидный
Рис. 8.3. Край колонии:
1 — гладкий; 2 — волнистый; 3 — зубчатый; 4 — лопастной; 5 — неправильный; 6 — реснитчатый; 7 — нитчатый; 8 — ворсинчатый; 9 — ветвистый
да), различают поверхностные, глубинные и донные колонии. Образование поверхностных колоний — наиболее существенная особенность роста многих микроорганизмов на плотном субстрате. Такне колонии отличаются большим разнообразием. При их описании учитывают следующие признаки:
• форму — округлая, амебовидная, неправильная, ризоидная ит.д. (рис. 8.1);
• размер {диаметр) — измеряют в миллиметрах; если размеры колонии не превышают 1 мм. то их называют точечными;
• поверхность — гладкая, шероховатая, бороздчатая, складчатая, морщинистая, с концентрическими кругами или радиально исчерченная;
• профиль — плоский, выпуклый, кратерообразный, конусовидный и т.д. (рис. 8.2);
• блеск и прозрачность - колония блестящая, матовая, тусклая, мучнистая, прозрачная;
• цвет — бесцветная (грязно-белые колонии относят к бесцветным) или пигментированная — белая, желтая, золотистая, оранжевая, сиреневая, красная, черная и т.д.; особо отмечают выделение в субстрат пигмента; при описа-
Рис. 8.4. Структура колонии:
/ — однородная; 2 — мелкозернистая; 3 — крупнозернистая; 4 — струйчатая; 5 — волокнистая
нии колоний актиномицетов отмечают пигментацию воздушного и субстратного мицелия, а также выделение пигментов в среду;
• край — ровный, волнистый, зубчатый, бахромчатый ит.д. (рис. 8.3);
• структуру — однородная, мелко- или крупнозернистая, струйчатая ит.д. (рис. 8.4); край и структуру колонии определяют с помощью лупы или при малом увеличении микроскопа. Для этого чашку Петри помешают на столик микроскопа крышкой вниз:
Рис. 8.5. Рост бактерии по штриху:
7 сплошной с ровным краем; 2 — сплошной с волнистым краем; 3 — четковидный; 4 — диффузный; 5 — перистый; 6 — ризовдный
• консистенцию определяют, прикасаясь к поверхности колонии петлей. Колония может легко сниматься с агара, быть плотной, мягкой или врастающей в агар, слизистой
(прилипает к петле), тягучей, иметь вид пленки (снимается целиком), быть хрупкой (легко ломается при прикосновении петлей).
Глубинные колонии, напротив, довольно однообразны. Чаще всего они по
виду похожи на более или менее сплющенные чечевички, в проекции имеющие форму овалов с заостренными концами. Лишь у немногих бактерий глубинные колонии напоминают пучки ваты с нитевидными выростами в питательную среду. Образование глубинных колоний часто сопровождается разрывом плотной среды, если микроорганизмы выделяют углекислоту или другие
газы.
Донные колонии разных микроорганизмов обычно имеют вид тонких про-
зрачных пленок, стелющихся по дну.
Размеры и многие другие особенности колонии могут изменяться с возрастом и зависят от состава среды. Поэтому при их описании указывают возраст
культуры, состав среды и температуру культивирования.
При описании роста микроорганизмов по штриху отмечают следующие особенности: скудный, умеренный или обильный, сплошной с ровным или волнистым краем, четковидный, напоминающий цепочки изолированных колоний, диффузный, перистый, древовидный или ризоидный (рис. 8.5). Характеризуют оптические свойства налета, его цвет, поверхность и консистенцию.
Для описания колоний и роста по штриху многие микроорганизмы часто выращивают на мясопептонном агаре. Применяют также мясопептонную желатину. Для лучшего рассмотрения глубинных колоний среды с агаром или желатиной рекомендуется осветлять (см. гл. 5)
8.1.2. Рост в жидких питательных средах
Рост микроорганизмов в жидких питательных средах более однообразен и сопровождается помутнением среды, образованием пленки или осадка. Характеризуя рост микроорганизмов в жидкой среде, отмечают степень помутнения — слабая, умеренная или сильная, особенности пленки — тонкая, плотная или рыхлая, гладкая или складчатая, а при образовании осадка указывают — скудный он или обильный, плотный, рыхлый, слизистый или хлопьевидный.
Нередко рост микроорганизмов сопровождается появлением запаха, пигментацией среды, выделением газа. Последнее обнаруживают по образованию пены, пузырьков, а также с помощью «поплавков» — маленьких, запаянных с одного конца трубочек. Поплавок помещают в пробирку запаянным концом вверх перед стерилизацией среды и следят, чтобы он полностью был заполнен средой. В случае выделения газа он скапливается в поплавке в виде пузырька.
Для описания характера роста микроорганизмов в жидких средах их выращивают па МПБ или на другой среде, обеспечивающей хороший рост.
8.2. ФИЗИОЛОГО-БИОХИМИЧЕСКИЕ СВОЙСТВА
Характеристика физиолого-биохимических особенностей микроорганизмов включает в себя описание их способности расти на разных питательных средах и вызывать определенные превращения веществ, входящих в состав этих сред. Учитывают использование различных соединений углерода, азота и серы, отношение к молекулярному кислороду, способность образовывать антибиотические вещества и проявлять ферментативную активность в отношении определенных субстратов. В ряде случаев определяют чувствительность микроорганизмов к тем или иным антибиотикам.
8.2.1. Использование соединений углерода
Микроорганизмы характеризуются неодинаковой способностью использовать различные соединения углерода для конструктивного и энергетического метаболизма. Чтобы выяснить возможность роста микроорганизмов за счет тех или иных углеродсодержащих веществ, их обычно высевают на синтетические среды, содержащие в качестве единственного источника углерода различные моно-, ди- и полисахариды, многоатомные спирты, органические кислоты, углеводороды. Из углеводов и многоатомных спиртов испытывают, как правило, следующие соединения: арабинозу, ксилозу, рамнозу, глюкозу, фруктозу, маннозу, галактозу, сорбозу, сахарозу, лактозу, мальтозу, трегалозу, целлобиозу, рафинозу, декстрин, крахмал, инулин, целлюлозу, глицерол, эритритол, маннитол, дульцитол, сорбитол, инозитол, салицин.
Определение способности микроорганизмов использовать углеводы и спирты. Готовят основной фон среды, который часто имеет следующий состав (г/л): пептон — 5,0; К2НРО4 — 1,0; вода дистиллированная. В зависимости от физиологических особенностей микроорганизмов можно применять иной состав фона. Так, например, для кори-нсподобных бактерий рекомендуется такая фоновая среда (г/л): пептон — 3,0; NaCI — 2,5, вода дистиллированная, а для актиномицетов среда состава (г/л): (blH4)2SO4 — 2,6; КН2РО4 — 2,4; К2НРО4- ЗН2О — 5,6; MgSO4- 7Н2О — 1,0; раствор микроэлементов 0,1 мл; вода дистиллированная. Раствор микроэлементов (г): CuSO4-5H2O — 0,64; FeSO4- 7Н2О — 0,11; МпС12-4Н2О — 0,79; ZnSO4- 7Н2О — 0,15; вода дистиллированная — 100. Раствор готовят отдельно и хранят при температуре 3 — 5 °C.
Рост микроорганизмов на средах с углеводами или многоатомными спиртами часто сопровождается накоплением органических кислот, нейтральных продуктов, газов. Образование кислот регистрируют по изменению pH среды.
Рис. 8.6. Накопление газа в поплавке: а — рост культуры сопровождается образованием газа; б — газ не образуется
Для этого к основному фону добавляют индикатор из расчета 2 мл 1,6%-го спиртового раствора на 1 л среды. В качестве индикатора применяют бромтимолблау, который изменяет окраску от желтой к синей в интервале pH 6,0—-7,6, или бромкрезолпурпур, изменяющий цвет от пурпурного к желтому в интервале pH 6,8 — 5,2. «Основной фон» разливают в пробирки по 8 — 10 мл, опускают на дно каждой пробирки поплавок и стерилизуют при 1,0 ати.
Углеводы и многоатомные спирты готовят в виде 10 —40%-х водных растворов и стерилизуют отдельно от основного фона среды автоклавированием при 0,5 ати или фильтрованием. Раздельная стерилизация рекомендуется в связи с тем, что сахара в присутствии фосфатов и других компонентов среды частично разрушаются и образуют соединения, токсичные для микроорганизмов. Стерильные растворы добавляют к основному фону в таком количестве, чтобы концентрация сахара (спирта) в среде составляла 1 г на 100 мл среды. Среды засевают суспензией клеток микроорганизмов и выдерживают в течение 1 — 5 сут при соответствующей температуре. Медленно развивающиеся микроорганизмы инкубируют в течение 7—10 сут. Рост или его отсутствие на среде с данным источником углерода определяют по помутнению среды, образованию пленки или осадка. Изменение цвета индикатора указывает на образование кислых или щелочных (вследствие разложения пептона) продуктов метаболизма. Об образовании газа свидетельствует его накопление в поплавке (рис. 8.6). Результаты наблюдений сравнивают с показателями роста в контрольной (фоновой) среде, не содержащей испытуемого источника углерода.
Способность микроорганизмов использовать углеводы и многоатомные спирты можно определять и на плотных средах. В этом случае стерильный раствор углевода или спирта добавляют в стерильную агаризованную расплавленную среду, тщательно перемешивают ее и разливают в стерильные чашки Петри. После того как агар застынет, дно чашки Петри с наружной стороны делят на секторы чернилами или карандашом (фломастером) по стеклу. Затем каждую из исследуемых культур микроорганизмов высевают петлей, проводя радиаль-
ный штрих (рис. 8.7). В качестве посевного материала используют густые суспензии клеток, которые готовят смывом культур с поверхности скошенного агара. Продолжительность культивирования 2—10 сут. О результатах судят по интенсивности роста в сравнении с ростом на контрольной среде, не содержащей испытываемых соединений углерода. Этот метод удобен тем, что позволяет в
Рис. 8.7. Схема посева на плотную среду различных микроорганизмов (7, 2, 3, .., 10) для определения способности использовать углеводы
Рис. 8.8. Определение способности микроорганизмов использовать углеводороды: а — схема посева (1—6 штрихи различных микроорганизмов; в центре — лунка с углеводородом); б — рост различных культур
одной чашке Петри одновременно проверить способность нескольких микроорганизмов использовать тот или иной субстрат.
Многие микроорганизмы могут использовать в качестве единственного источника углерода органические кислоты. Для определения способности расти на средах с органическими кислотами рекомендуется плотная среда состава (г/л): (NHd)2HPO4 — 0.5: MgSO4- 7Н2О — 0,2; NaCl — 0,1; агар — 15,0; органическая кислота в виде соли Na или К — 2,0; pH 6,8. До стерилизации к среде добавляют 20 мл 0,04%-го водного раствора индикатора метилрот, который в интервале pH 6,8 — 8,4 изменяет окраску от желтой к красной. Среду разливают в пробирки и стерилизуют при 1 ати. Посев проводят уколом. Продолжительность культивирования от 2 до 14 сут в зависимости от скорости роста микроорганизмов. О потреблении органических кислот свидетельствует рост по уколу и изменение кислотности среды в щелочную сторону, что отчетливо заметно по цвету индикатора.
Некоторые микроорганизмы способны использовать и такие химически устойчивые соединения, как. углеводороды. Выявить способность микроорганизма окислять жидкие нелетучие углеводороды можно на плотной среде состава (г/л): KNO3 - 4.0; КН2РО4 - 0,6; Na2HPO4- 12Н2О - 1,4; MgSO4-7H2O - 0,8; выщелоченный агар — 2,0; pH 7,2. Среду стерилизуют в колбах при 1 ати и разливают в чашки Петри толстым слоем. После того как среда застынет, в центре агаровой пластинки вырезают лунку. Для этой цели можно воспользоваться пробочным сверлом (диаметр 8—10 мм), которое предварительно стерилизуют в пламени горелки. Микроорганизмы высевают радиальными штрихами от лунки к периферии чашки (рис. 8.8). В лунку вносят 2—3 капли исследуемого углеводорода (стерилизуют фильтрованием). На чашке с одним углеводородом можно проверить рост нескольких микроорганизмов. Чашки помешают в термостат строго горизонтально, не переворачивая. Через 7 —10 сут отмечают наличие или отсутствие роста по штриху в сравнении с контролем — ростом на среде без углеводорода.
8.2.2. Использование соединений азота
Для определения способности микроорганизмов использовать те или иные соединения азота их выращивают на соответствующих питательных средах.
Использование органических азотсодержащих веществ
Многие микроорганизмы способны усваивать азот органических соединений, например пептонов, аминокислот и белков. В процессе ферментативного гидролиза белка освобождаются аминокислоты, которые используются клеткой непосредственно в процессах биосинтеза, а также подвергаются расщеплению в результате дыхания или брожения до более простых соединений. Поэтому разложение белка микроорганизмами (в процессе аммонификации) всегда сопровождается образованием побочных продуктов: аммиака, который выделяется при дезаминировании аминокислот, сероводорода, освобождающегося при разрушении серосодержащих аминокислот (цистина, цистеина, метионина), а также индола, который образуется при распаде триптофана. Обнаружение этих продуктов в культурах свидетельствует об использовании аминокислот микроорганизмами. Процесс аммонификации всегда сопровождается повышением щёлочности среды.
Обнаружение аммиака. Способность микроорганизмов образовывать аммиак можно выявить при их росте на мясопептонном бульоне. Для этого МПБ разливают в пробирки по 8—10 мл в каждую и стерилизуют при 1 ати. После посева под ватной пробкой укрепляют узкую стерильную полоску лакмусовой бумаги (стерилизуют, поместив в чашку Петри, в автоклаве при 0,5 ати) так, чтобы она не соприкасалась с питательной средой (рис. 8.9). Пробку рекомендуется заворачивать в полиэтилен, чтобы затруднить улетучивание аммиака. При образовании аммиака красный лакмус синеет.
Если в качестве среды используют 4%-ю пептонную воду, то образование аммиака выявляют с помощью реактива Несслера (приготовление реактива см. в приложении 5). На фарфоровую пластинку помещают несколько капель культуральной жидкости и добавляют к ним каплю реактива Несслера. В присутствии следов аммиака жидкость окрашивается в желтый цвет, при большом количестве аммиака образуется ко-
ричневый осадок.
Обнаружение сероводорода. Выявление способности бактерий продуцировать H2S основано на реакции образования сульфидов металлов. Наиболее
распространенный способ — проба с уксуснокислым свинцом. Микроорганизмы выращивают в МПБ, содержащем 0,01 % цистина и цистеина, которые добавляют к стерильному бульону в виде раствора в подкисленной дистиллированной воде. Растворы цистина и цистеина стерилизуют фильтрованием. После посева исследуемых бактерий над средой помещают стерильную (стерилизуют, поместив в чашку Петри или пробирку, в автоклаве при 0,5 ати) полоску Фильтровальной бумаги, пропитанную раствором уксуснокислого свинца (приготовление см. в приложении 5), укрепив ее под ватной /робкой. Пробку пробирки заворачивают полиэтиленом, чтобы за-
Рис. 8.9. Обнаружение сероводорода, образуемого
микроорганизмами:
с — почернение свинцовой бумаги; б — образование FeS в толще среды
труднить улетучивание H2S. Продолжительность культивирования 7—10 сут. Выделение сероводорода обнаруживают по почернению бумаги вследствие образования сульфида свинца.
Более четкие результаты наблюдаются при использовании среды следующего состава (г/л): пептон — 10,0; NaCl — 5,0; цитрат NH4Fe — 0,30; агар — 15,0; дрожжевой автолизат — 2,0 мл; вода водопроводная, pH среды 7,0. Среду разливают в пробирки и стерилизуют при 0,5 ати, после стерилизации скашивают и делают посев штрихом. О выделении H2S судят по почернению среды вследствие образования сульфида железа (рис. 8.9).
Обнаружение индола. Индол обнаруживают по качественной реакции с реактивом Эрлиха (см. приложение 5). Для выявления индола можно использовать различные среды: МПБ с 0,01 % триптофана; 1%-ю казеиновую воду (г/100 мл: казеин — 1,0; Na2HPO4 — 0,2; NaCl — 0,3) или 2—3%-ю пептонную воду (г/100 мл: пептон — 2,5; Na2HPO4 — 0,2; NaCl — 0,3); pH сред 7,2—7,4. Среды разливают в пробирки по 8 —10 мл, стерилизуют при 1,0 атм (триптофан добавляют к стерильному бульону в виде раствора, который готовят следующим образом: триптофан вносят в воду и прибавляют по каплям 5— 10%-й раствор NaOH до полного его растворения; раствор триптофана стерилизуют 15 мин при 0,5 ати) и засевают суспензией бактерий. Через 5 — 7 сут проводят качественную реакцию на присутствие индола в культуре и в контроле — стерильной среде. Для этого на поверхность среды, нс перемешивая, помещают 1—2 мл реактива Эрлиха; появление красной окраски свидетельствует об образовании индола.
Субстратами аммонификации могут быть и более простые органические соединения азота, например мочевина. Ферментативный гидролиз мочевины до аммиака и углекислоты способны осуществлять микроорганизмы, образующие уреазу. Образовавшийся аммиак используется этими бактериями в качестве источника азота. Процесс аммонификации мочевины сопровождается сильным подщелочеиием среды.
Для выявления способности микроорганизмов использовать мочевину часто применяют среду следующего состава (г/л): (NH2)2CO — 5,0; К2НРО4 — 0,5; яблочнокислый, лимоннокислый или виннокислый Na — 5,0. Среду без мочевины наливают в конические колбы тонким слоем и стерилизуют при 0,5 ати. Отдельно готовят 20%-й раствор мочевины в подкисленной дистиллированной воде и стерилизуют его фильтрованием или автоклавированием при 0,5 ати. Мочевину добавляют к стерильной среде из такого расчета, чтобы ее концентрация в среде составляла 2 %. После засева среды микроорганизмами под ватной пробкой укрепляют стерильную полоску красной лакмусовой бумаги. Продолжительность культивирования 3 — 5 сут. Образование аммиака обнаруживают по изменению цвета лакмусовой бумаги, а также качественной реакцией с реактивом Несслера.
Использование азота минеральных солей
Способность микроорганизмов использовать соли аммония или нитраты в качестве источника азота проверяют на синтетических средах. Готовят два варианта среды. Состав основной среды (г/л): глюкоза — 20,0; К2НРО4 — 1,0; КН2РО4 — 1,0; MgSO4- 7Н2О — 0,5; NaCl — 0,5; агар — 15,0; раствор микроэлементов (см. 8.2.1) — 1 мл; pH 7,1 —7,2. В первом варианте к основной среде добавляют NH4Q — 1,0 и СаСО3 — 5,0, так как хлористый аммоний — физиологически кислая соль, во втором варианте KNO3 — 1,0. Среды разливают в
пробирки, стерилизуют при 0,5 ати и после стерилизации скашивают. Посев проводят штрихом, продолжительность культивирования 2—10 сут. Рост или его отсутствие отмечают визуально.
Способность усваивать аммонийные соли или нитраты можно проверить и на жидких средах того же состава. В этом случае по окончании опыта целесообразно установить полноту использования данной соли. С этой целью в культуральной жидкости определяют присутствие ионов аммония или нитратов качественными реакциями. Для контроля проводят реакции с соответствующей стерильной средой.
8.2.3. Использование молекулярного азота
О способности аэробных микроорганизмов использовать молекулярный азот можно судить по их росту на безазотистой среде Эшби, которая имеет следующий состав (г/л): маннит — 20,0; К2НРО4 — 0,2; MgSO4 * 7Н2О - 0,2; NaCi — 0,2; K2SO4 — 0,1; СаСОз — 5,0; агар 20,0; pH 7,1—7,3. Среду разливают в пробирки, стерилизуют при 0,5 ати и готовят скошенный агар. Посев проводят штрихом. Продолжительность культивирования 7—10 сут. Необходимо отметить, что на среде Эшби могут расти не только микроорганизмы, фиксирующие молекулярный азот, но и олигонитрофилы, т.е. микроорганизмы, способные усваивать ничтожные количества связанного азота, содержащегося в реактивах, воде и воздухе. Обильный рост на среде Эшби может свидетельствовать о принадлежности бактерий к азотфиксаторам.
Молекулярный азот могут фиксировать и анаэробные бактерии. Для выявления этой способности у представителей рода Clostridium используют среду следующего состава Iг/л): дрожжевой экстракт— 0,015; глюкоза — 20,0; К2НРО4 — 1.0: MgSO4-7H2O — 0,5; X'aCl — 0,5; MnSO4 и FeSO4 — следы (по 1 мл 1%-го раствора каждой соли); СаСО3 — 20,0; pH 7,0. Среду разливают в высокие пробирки и стерилизуют при 0,5 ати. После стерилизации пробирки доливают почти до пробки стерильной средой и прогревают на кипящей водяной бане в течение 20 — 30 мин для удаления растворенного в среде кислорода. Затем среду в пробирках быстро охлаждают под струей холодной воды и. проводят посев суспензией микроорганизмов. Посевной материал вносят пипеткой на дно пробирки. Продолжительность культивирования — 2—7 сут. По окончании опыта отмечают рост азотфиксаторов — помутнение среды, образование газа, появление запаха масляной кислоты (признак, характерный для азотфиксирующих клостридий) пли его отсутствие.
8.2.4. Отношение к молекулярному кислороду и рост в анаэробных условиях
По отношению к молекулярному кислороду среди микроорганизмов выделяют группы облигатных аэробое и микроаэрофилов, факультативных, аэротоле-рантных и строгих анаэробов. Чтобы судить о принадлежности микроорганизмов к той или иной группе, микробную суспензию высевают в пробирки с расплавленной и остуженной до 40—45 °C агаризованной питательной средой. Посев можно проводить и уколом. Строгие аэробы растут на поверхности среды и в верхнем слое, микроаэрофилы — на некотором расстоянии от поверхности.
б
Рис. 8.10. Рост микроорганизмов при посеве уколом (а) и при посеве в расплавленную плотную среду (б):
1 — аэробы; 2 микроаэрофилы; 3 — факультативные анаэробы; 4 — анаэробы
Факультативные анаэробы обычно развиваются по всей толще среды. Строгие анаэробы растут только в глубине среды, у самого дна пробирки (рис. 8.10).
Факультативные анаэробы хорошо растут и в аэробных, и в анаэробных условиях. В анаэробных условиях многие из них способны использовать в качестве конечного акцептора электронов не кислород, а нитраты. В ходе этого процесса, называемого нитратным дыханием, нитраты восстанавливаются до нитритов, и далее до N2O и газообразного азота. В случае образования этих газообразных продуктов говорят о денитрификации. Другие факультативные анаэробы могут в анаэробных условиях переключаться с дыхания на брожение, и тогда в средах с углеводами накапливаются значительные количества органических кислот и других продуктов метаболизма.
Определение способности к аэробному дыханию
Тест на дыхание (поглощение молекулярного кислорода) проводят с использованием полярографа и платинохлорсеребряного электрода «закрытого» типа, позволяющего регистрировать дыхание в небольших по объему образцах (см. гл. 3). Для измерения скорости дыхания клетки аэробных микроорганизмов, выращенные на соответствующих средах, собирают центрифугированием (режим зависит от размеров клеток), промывают физиологическим или буферным раствором с оптимальным для данного объекта pH и суспендируют в небольшом объеме буфера из расчета примерно 3 мл на 1. г осадка сырых клеток. Клетки в течение эксперимента хранят на льду. В ячейку полярографа вносят клетки (обычно 0.1 объема ячейки) и добавляют проаэрированный буфер той же температуры. Регистрацию поглощения кислорода следует проводить в термостатированных ячейках при температуре, близкой к температуре выращивания испытуемого микроорганизма. Регистрируют поглощение О2 пробой без субстрата (эндогенное дыхание), затем шприцем добавляют соответ-
ствующий субстрат (обычно ИГ2— Iff ' М). Регистрацию ведут в течение нескольких минут, причем в первый период времени после добавления субстрата дыхание должно быть линейным, т. е. его скорость не должна изменяться со временем. Если такой линейности не наблюдается, то можно попытаться снизить концентрацию клеток или увеличить скорость движения ленты самописца.
В качестве теста на функционирование терминальных оксидаз можно добавить в ячейку с клетками ингибиторы, цианид или азид {Внимание1. Сильнейшие яды1.). Их добавляют до конечной концентрации 10"4—10“3 М, в таких условиях клетки теряют способность поглощать кислород.
Расчет скорости дыхания проводят по ленте самописца, учитывая скорость поглощения клетками кислорода в первые 1 — 3 мин после добавления субстрата; рассчитывают также степень подавления дыхания (%) при той или иной действующей концентрации ингибитора. Данные рассчитывают на I мг общего клеточного белка (см. гл. 12).
Для калибровки шкалы полярографа измеряют содержание кислорода в насыщенной воздухом дистиллированной воде (100% кислорода) и в той же пробе после добавления 10“2 М дитионита натрия (0 % кислорода).
Способность к денитрификации
Это свойство определяют на среде следующего состава (г/л): глицерин — 10,0; дрожжевой экстракт — 5,0; (NH4)2SO4 — 2,0; KNO3 — 10,0; К2НРО4 — 2,0; MgSO4-7H2O - 0,025; NaCl - 0,5; FeSO4-7H2O - 0,001; агар - 1,0. Среду разливают в пробирки по 5 и 10 мл и стерилизуют при 0,5 ати. Исследуемый микроорганизм вначале засевают в пробирки с 5 мл среды (1-й пассаж). Посев делают уколом. Через 24 ч петлю культуры 1-го пассажа переносят в пробирки с 10 мл среды, которую предварительно расплавляют и охлаждают до 40— 45 °C. После внесения микроорганизмов среду перемешивают, остужают и заливают 2 — 3 мл стерильного 1%-го водного агара для создания анаэробных условий. Рост бактерий, способных к денитрификации, сопровождается помутнением среды и выделением газа.
Проверить способность бактерий к восстановлению нитратов можно и на МПБ, к которому добавляют 0,2%-й раствор KNO3. Среду разливают в пробирки по 10 мл, опускают на дно каждой поплавок и стерилизуют при 1,0 ати. Посев проводят суспензией клеток. Продолжительность культивирования — 2 — 5 сут. По окончании опыта отмечают накопление в поплавке газа (N2) и определяют качественными реакциями присутствие в культуральной жидкости нитратов и нитритов.
Реакция на нитраты с дифениламином. На фарфоровую пластинку наносят каплю концентрированной серной кислоты, помещают в нее несколько кристаллов дифениламина и после его растворения добавляют каплю исследуемой жидкости. При наличии ионов NOJ жидкость окрашивается в темно-синий цвет. Следует иметь в виду, что азотистая кислота также дает с дифениламином синее окрашивание, поэтому пробу на нитраты можно проводить только при отсутствии в среде нитритов.
Крахмал-йодная реакция на нитриты основана на том, что нитриты в кислой среде окисляют йодистый цинк с выделением йода, присутствие которого обнаруживают с
крахмалом. Для проведения реакции к капле культуральной жидкости добавляют каплю раствора, содержащего ZnCl2, KI и крахмал (приготовление реактива см. в приложении 5), и каплю раствора HCI. При наличии в среде нитритов появляется синее окрашивание.
Реакция на нитриты с реактивом Грисса основана на образовании в кислой среде в присутствии нитритов и ароматических аминов (сульфофеноловой кислоты и а-на-фтиламипа) азосоединения, окрашенного в красно-розовый цвет. Для проведения реакции к капле реактива Грисса (приготовление реактива см. в приложении 5) добавляют каплю культуральной жидкости. Появление красного окрашивания свидетельствует о присутствии нитритов. Параллельно проводят реакции со стерильной средой.
Определение способности к брожению
Способность к брожению обычно определяют при использовании углеводно-пептонной среды с относительно высокой концентрацией углеводов и небольшим количеством пептона. Состав среды (г/л): углевод — 10,0; пептон — 2,0; NaCl — 5,0; К2НРО4 — 0,3; агар — 3,0. На 100 мл среды добавляют 0,3 мл 1%-го водного раствора бромтимолового синего, pH среды 7,1—7,2. Среду' без углевода стерилизуют при 1,0 ати. Углеводы (глюкозу, сахарозу, лактозу) добавляют к стерильной расплавленной среде в виде 20 — 40%-х растворов в дистиллированной воде, которые стерилизуют при 0,5 ати. Стерильную среду с углеводами разливают в стерильные пробирки слоем в 5 —6 см и после того, как среда застынет, ее засевают исследуемым микроорганизмом. Посев делают уколом. Для каждого углевода используют две пробирки. После посева поверхность среды в одной пробирке заливают стерильным расплавленным парафином или смесью вазелинового масла и парафина (1:1) или водным агаром (1,5 г агара на 100 мл) слоем 1. — 2 см.
Продолжительность культивирования от 2 до 7 сут. По окончании опыта регистрируют изменение pH среды по перемене цвета индикатора. Аэробные микроорганизмы, ^осуществляющие дыхание и не способные к брожению, растут на поверхности среды только в пробирке без парафина и образуют небольшое количество кислот лишь в верхнем слое среды. В случае нейтрализации этих кислот продуктами разложения пептона в поверхностном слое среды наблюдается щелочная реакция. Микроорганизмы, способные сбраживать углеводы, растут и образуют кислоты в обеих пробирках, но кислотность в анаэробных условиях значительно выше, чем в первом варианте. Это хорошо заметно по изменению цвета индикатора. Если брожение сопровождается образованием газов, то происходит разрыв агаризованной среды.
8.2.5. Определение внеклеточных ферментов
Как указывалось выше, некоторые микроорганизмы способны использовать в качестве питательных субстратов самые различные высокомолекулярные соединения: полисахариды, белки, нуклеиновые кислоты, липиды и др. Однако макромолекулы не могут проникать через мембрану клетки. Они подвергаются расщеплению, которое осуществляется под воздействием экзоферментов, относящихся к класс}' гидролаз. Большинство из них катализирует гидро-
лиз полимера до растворимых продуктов, обычно димеров или мономеров, которые поступают в клетку с помощью специфических транспортных механизмов. Образование экзоферментов широко распространено среди различных групп микроорганизмов. При поиске продуцентов ферментов гидролаз в лабораторной практике применяют специальные методы. Сущность их состоит в следующем. Микроорганизмы выращивают на агаризованной среде, содержащей макромолекулярный субстрат. Если клетки выделяют в среду экзоферменты, гидролизуюшйе данное соединение, то выросшие колонии будут окружены зоной, в которой обнаруживаются продукты гидролиза.
Амилолитическая активность
Крахмал подвергается гидролитическому расщеплению под действием амилаз, активными продуцентами которых являются различные виды бацилл, псевдомонад, стрептомицетов и мицелиальных грибов. Для выявления амилолитической активности часто используют среду следующего состава (г/л): пептон — 10,0; КН2РО4 — 5,0; растворимый крахмал — 2,0; агар — 15,0; pH среды 6,8 — 7,0. Среду стерилизуют при 1,0 ати и разливают в стерильные чашки Петри. Когда среда застынет, исследуемые микроорганизмы высевают штрихом по диаметру чашки или уколом. Продолжительность культивирования 2—10 сут. Гидролиз крахмала обнаруживают после обработки агаровой пластинки раствором Люголя (см. приложение 5). Для этого на поверхность среды наливают 3 — 5 мл раствора Люголя. Среда, содержащая крахмал, окрашивается в синий цвет, а зона гидролиза остается бесцветной или приобретает красно-бурую окраску, если крахмал гидролизовался до декстринов. Зону гидролиза крахмала измеряют в миллиметрах от края штриха (колонии) до границы светлой зоны. Чем больше диаметр светлой зоны, тем выше амилолитическая активность (рис. 8.11).
Рис. 8.11. Определение амилолитической активности:
а — схема посева (1—4 — штрихи разных культур); б — среда с культурами после обработки раствором Люголя
Протеолитическая активность
Протеолитические ферменты (протеазы) катализируют расщепление белков на поли- и олигопептиды. Протеазы выделяются различными видами бацилл, актиномицетов, мицелиальных грибов и другими микроорганизмами. Активность внеклеточных протеаз определяют, используя в качестве субстрата желатину, казеин или другие белки.
Протеолиз желатины. Микроорганизмы высевают на мясопептонную желатину (МПЖ). Ее готовят следующим образом. К 100 мл МПБ добавляют 10—15 г желатины, оставляют на 20—30 мин, чтобы она набухла, затем смесь нагревают на водяной бане до полного растворения желатины и разливают полученную МПЖ в пробирки по 8—10 мл. Стерилизуют 15 мин при 0,5 ати. Посев проводят уколом. Продолжительность культивирования 7 10 сут при комнатной температуре. Разжижение желатины отмечают визуально. Если желатина разжижается, указывают интенсивность и форму разжижения — послойное, воронкообразное, мешковидное, пузыревидное и т.д. (рис. 8.12).
Протеолиз казеина. Для выявления этой способности используют молочный агар — среду, состоящую из равных частей стерильного обезжиренного молока и стерильного 3%-го водного агара. Как правило, перед приготовлением среды молоко обезжиривают центрифугированием в течение 15 мин при 650—1500 g. Жиры, которые образуют на поверхности молока достаточно плотную пленку, удаляют, а молоко стерилизуют при 0,5 ати. После стерилизации его подогревают и добавляют при постоянном помешивании к стерильному расплавленному и остуженному до 50 °C водному агару. Полученную среду разливают в стерильные чашки Петри. Микроорганизмы высевают штрихом по диаметру чашки или уколом. Продолжительность культивирования 2—10 сут. Гидролиз казеина обнаруживают по зоне просветления среды вокруг колонии или выросших по штриху микроорганизмов. Особенно четко она видна после обработки среды 5%-м раствором трихлоруксусной кислоты. Зон)7 гидролиза казеина измеряют в миллиметрах от края штриха или колонии до границы светлой зоны. Чем больше диаметр светлой зоны, тем выше казеинолитическая активность бактерий.
Рис. 8.12. Схема разжижения желатины микроорганизмами при посеве уколом:
а — Кратеровидпое; б — реповидное; в — воронковидное; г — мешковидное; д — послойное; е — пузыревидное (а—в и д — разжижение вызвано аэробами; г — факультативными анаэробами; е — анаэробами)
Липолитическая активность
Липиды подвергаются гидролитическому разложению под действием липаз. Продуценты липаз обнаружены среди дрожжей, мицелиальных грибов, бактерий рода Clostridium и других микроорганизмов. Для выявления липолитической активности исследуемые микроорганизмы высевают на среду, содержащую соответствующий липид. Определенная трудность при постановке таких опытов связана с тем, что жиры не смешиваются с водой. Поэтому чаще всего вместо жиров в среду вводят твины — эфиры жирных кислот и сорбита. Твин-40 содержит пальмитиновую, твин-60 — стеариновую, а твин-80 — олеиновую кислоты. Твины хорошо растворимы в воде и имеют нейтральную реакцию. Состав среды (г/л): твин — 10,0; пептон — 10,0; NaCl — 5,0; СаС12- Н2О — 0,1; агар — 20,0; pH среды 7,4.
Готовят среду без твина и стерилизуют ее при 1,0 ати. Водный раствор твина соответствующей концентрации стерилизуют отдельно при 0,5 ати и добавляют к стерильной основной среде. Среду разливают в стерильные чашки Петри. Когда агар застынет, на поверхность агаровой пластинки высевают штрихом или уколом исследуемый микроорганизм. Засеянные чашки Петри выдерживают при соответствующей температуре необходимое для роста микроорганизма время. На наличие липазы указывает образование вокруг штриха или колонии непрозрачной зоны кальциевых солей жирных кислот, освобожденных из твина.
8.2.6. Образование антибиотиков
В процессе жизнедеятельности многие микроорганизмы образуют специфические продукты, которые обладают высокой физиологической активностью по отношению к другим микроорганизмам или вирусам, задерживая их рост или убивая их. Они называются антибиотиками. В отличие от общебиологических ядов антибиотики проявляют свое действие лишь по отношению к отдельным, вполне определенным видам или группам микроорганизмов. Одни антибиотики — пенициллин, фумагиллин, бацитрацин и др. — подавляют рост ограниченного числа видов микроорганизмов. Другие — тетрациклины, хлорамфеникол, эритромицин, карбомицин — имеют широкий спектр действия, т. е. подавляют рост многих грамположительных и грамотрицательных бактерий, а также риккетсий и некоторых вирусов. Как правило, грамположитель-ные бактерии более чувствительны к действию антибиотиков по сравнению с грамотрицательными, что связано со строением их клеточной стенки. Наиболее часто активные продуценты антибиотических веществ встречаются среди стрептомицетов, спорообразующих бактерий и мицелиальных грибов.
Определение антибиотической активности микроорганизмов
Существуют различные способы выявления антибиотических свойств микроорганизмов. Многие из них основаны на способности антибиотиков диффундировать в ага-ризованные среды и образовывать зоны, в которых не растут тест-организмы. Величи-
б
Рис. 8.13. Определение антибиотической активности методом перпендикулярных штрихов:
а — схема посева (1—6 — штрихи тест-культур; 7 — штрих продуцента антибиотического вещества); б — рост продуцента и тест-культур
на зоны отсутствия роста указывает на степень активности данного антибиотика в отношении тест-культуры и зависит от его концентрации, а также от плотности культуры тест-организма, состава и толщины слоя агаризованной среды, температуры ин кубации, длительности диффузии и других факторов. В качестве тест-организмов используют представителей различных микроорганизмов, в первую очередь берут Escherichia coll (грамотрицательные бактерии), Staphylococcus aureus (грамположитель-ные бактерии). Bacillus subtilis или Bacillus cereus (спорообразующие бактерии), дрожжи рода Candida или Saccharomyces. При необходимости этот стандартный набор тест-организмов дополняют другими микроорганизмами. Антибиотическую активность чаще всего выявляют методами перпендикулярных штрихов и агаровых блочков.
Метод перпендикулярных штрихов. На питательный агар в чашке Петри высевают штрихом предполагаемый продуцент антибиотического вещества. Посев штрихом делают по диаметру чашки Петри. Время инкубации продуцента зависит от скорости егс роста. После того как продуцент вырастет и образует антибиотическое вешество, диффундирующее в толщу агара, перпендикулярно к его штриху подсевают штрихами тест-организмы, начиная от периферии чашки. Для посева используют густые суспензии тест-организмов в стерильной водопроводной воде. Чашки выдерживают в термостате при 28—30 °C в течение 2—8 сут в зависимости от скорости роста тест-органпз-мов. Нечувствительные к антибиотическому веществу тест-организмы растут вблизи штриха продуцента. Если антибиотик оказывает' действие на тест-организм, то рост последнего будет наблюдаться вдали от штриха продуцента (рис. 8.13). Чем больше этс расстояние, тем более чувствителен тест-организм к антибиотическому веществу, образуемому изучаемым продуцентом.
Этот метод имеет, однако, существенный недостаток. Продуцент антибиотического вещества и тест-организм выращивают на одной среде, хотя известно, что не всегда одна и та же среда одинаково благоприятна как для продуцента и образования им антибиотика, так и для роста тест-организма.
Метод агаровых блочков предусматривает использование разных питательных сред для выращивания продуцента антибиотика и тест-организмов. Для выявления антибиотических свойств актиномицетов предполагаемый продуцент выращивается на среде следующего состава (г/л): глюкоза — 30,0; KNO3 — 5,5; MgSO4 — 0,5; NaCl — 1,0: К7НРО4 — 0,4; Z11SO4 — 0,002; FeSO4 — 0,002; агар — 25,0; вода дистиллированная; pH 7,1 —7,2. Среду стерилизуют при 0,5 ати и разливают в стерильные чашки Петри.
Рис. 8.14. Определение антибиотической активности методом агаровых блочков:
а — схема постановки опыта (1—4 — блочки с разными продуцентами); б — зоны подавления роста тест-культуры под действием антибиотика, диффундирующего в агар из блочков
После застывания агара продуцент антибиотического вещества высевают сплошным газоном. Для этого споры актиномицета переносят петлей на агаровую пластинку, распределяют по всей поверхности шпателем и инкубируют при 28 —30 °C в течение 8—10 сут. Затем стерильным пробочным сверлом (диаметр 6 — 8 мм) вырезают агаровые блочки с газоном актиномипета и переносят их на поверхность агаризованной среды, например МПА, только что засеянной тест-организмом. Агаровые блочки раскладывают по шаблону на равном расстоянии один от другого и на расстоянии 1,5 — 2,0 см от края чашки мицелием вверх и плотно прижимают к агаровой пластинке. Для лучшей диффузии антибиотических веществ в толщу питательной среды, засеянной тест-организмом, блочки можно закладывать непосредственно в лунки, предварительно вырезанные тем же сверлом. На одной чашке Петри с тест-организмом можно разместить 4—5 агаровых блочков с различными продуцентами антибиотиков (рис. 8.14). Чашки выдерживают 1 ч при комнатной температуре для диффузии антибиотических веществ в толщу агара, затем помещают в термостат при температуре, благоприятной для развития тест-организма на сутки и более в зависимости от скорости его роста. Если тест-организм чувствителен к антибиотическому веществу продуцента, то после инкубации вокруг агаровых блочков образуются зоны отсутствия его роста. Чем больше выделяется антибиотика и чем он активнее, тем больше будет диаметр зоны отсутствия роста тест-организма. Тест-организм, не чувствительный к антибиотическому веществу данного продуцента, растет по всей поверхности среды и даже вблизи блочка продуцента.
Определение чувствительности микроорганизмов к антибиотическим веществам
Чувствительность микроорганизмов к антибиотикам удобно определять с помощью готовых бумажных дисков, пропитанных определенными антибиотиками. Концентрация антибиотиков в дисках подобрана с таким расчетом, чтобы диаметры зон задержки роста стандартных тест-организмов были 28 — 32 мм.
Исследуемые микроорганизмы выращивают на соответствующей плотной питательной среде. Готовят однородную суспензию клеток в стерильной водопроводной воде. В 1 мл суспензии должно содержаться около 2 млрд клеток
(определяют по стандарту мутности). Вносят 1 мл суспензии в пробирку с 20 мл стерильной расплавленной и остуженной до 50 °C агаризованной среды, например МПА. Если микроорганизмы выращивали в жидкой питательной среде, то в агар вносят соответствующий объем культуры. Содержимое пробирки быстро и тщательно перемешивают и выливают в стерильную чашку Петри. Когда среда застынет, на ее поверхности помещают бумажные диски на равном расстоянии друг от друга и на 1,5—2,0 см от края чашки. Чашки выдерживают 2 ч при комнатной температуре для лучшей диффузии антибиотиков в толщу агаризованной среды, а затем 24 ч при 28 —30 °C. Если исследуемые микроорганизмы чувствительны к данным антибиотикам, то вокруг дисков образуются зоны отсутствия роста. Диаметр зоны измеряют миллиметровой линейкой. Зона более 30 мм свидетельствует о высокой чувствительности микроорганизма к антибиотику, а менее 12 мм — о слабой чувствительности.
Когда в распоряжении экспериментатора имеются растворы антибиотических веществ или культуральные жидкости, содержащие антибиотик, используют метод с применением лунок в толще агара. В этом случае в застывшей агаризованной среде, засеянной испытуемым микроорганизмом, стерильным пробочным сверлом (диаметр 6—8 мм) делают лунки на расстоянии 1,5—2,0 см от края чашки. В лунки вносят растворы антибиотиков или культуральную жидкость. Этот метод позволяет также выявить способность к образованию антибиотических веществ микроорганизмами, выращенными в жидкой среде.
Глава 9
СИСТЕМАТИКА И ИДЕНТИФИКАЦИЯ МИКРООРГАНИЗМОВ
9.1. ОСНОВНЫЕ ПОНЯТИЯ И ПРАВИЛА НАИМЕНОВАНИЯ
Описано несколько тысяч видов микроорганизмов, однако считают, что это составляет менее 1 % от реально существующих. Изучение разнообразия микроорганизмов составляет предмет систематики. Основной ее задачей является создание естественной системы, отражающей филогенетические взаимоотношения микроорганизмов. До последнего времени систематика микроорганизмов базировалась преимущественно на фенотипических признаках: морфологических, физиологических, биохимических и др., поэтому существующие системы классификации носят в значительной степени искусственный характер. Однако они позволяют сравнительно легко идентифицировать некоторые вновь выделенные штаммы микроорганизмов.
Систематика включает такие разделы, как классификация, номенклатура и идентификация. Классификация определяет порядок помещения индивидуумов, обладающих заданной степенью однородности, в определенные группы (таксоны). Номенклатура представляет собой свод правил наименования таксонов. Идентификация означает определение принадлежности изучаемого организма к тому или иному таксону.
Термин «таксономия» часто используют как синоним систематики, однако иногда под ним понимают раздел систематики, включающий теорию классификации, учение о системе таксономических категорий, границах и соподчинении таксонов. Основной таксономической категорией в микробиологии, как и в других биологических науках,
является вид — совокупность особей, характеризующихся рядом общих морфологических, физиолого-биохимических, молекулярно-генетических признаков.
Под термином «штамм» понимают чистую культуру микроорганизма, выделенную из определенного места обитания (воды, почвы, организма животного и т.д.). Разные штаммы одного вида микроорганизмов могут различаться по некоторым признакам, например чувствительности к антибиотикам, способности синтезировать некоторые продукты метаболизма и т.д., но эти различия меньше, чем видовые. Понятие «штамм» в микробиологии и генетике несколько различаются: в микробиологии оно является более широким. Виды микроорганизмов объединяют в таксономические категории более высокого порядка: роды, семейства, порядки, классы, отделы, царства. Эти категории называют обязательными. Предусмотрены также необязательные категории: подкласс, подпорядок, подсемейство, триба, подтриба, подрод, подвид. Однако в систематике необязательные категории используются довольно редко.
Номенклатура микроорганизмов подчиняется международным правилам. Так, имеется Международный кодекс номенклатуры бактерий. Для дрожжевых грибов основным руководством является «The Yeasts. A Taxonomic Study», для мицелиальных грибов и водорослей — Международный кодекс ботанической номенклатуры.
Для наименования объектов в микробиологии, как в зоологии и ботанике, используют бинарную или биноминальную (от лат. bis — дважды) систему номенклатуры, в соответствии с которой каждый вид имеет название, состоящее из двух латинских слов. Первое слово означает род, а второе определяет конкретный вид этого рода и называется видовым эпитетом. Родовое название всегда пишется с заглавной буквы, а видовое — со строчной даже в том случае, если видовой эпитет присвоен в честь ученого, например Clostridium pasteurianum. В тексте, особенно с латинской графикой, все словосочетание выделяют курсивом. При повторном упоминании названия микроорганизма родовое название можно сократить до одной или нескольких начальных букв, например С. pasteurianum. Если в тексте встречаются названия двух микроорганизмов, которые начинаются с одной и той же буквы (например, Clostridium pasteurianum и Citrobacterfreundii), то сокращения должны быть разными (С. pasteurianum и Ct. freundid). Если микроорганизм идентифицирован только до рода, вместо видового эпитета пишут слово sp. {species — вид), например Pseudomonas sp. В этом случае при повторном упоминании названия микроорганизма в тексте родовое название следует всегда писать полностью.
Для наименования подвида используют словосочетание, состоящее из названия рода, а также видового и подвидового эпитетов. Для разграничения этих эпитетов между ними пишут буквенное сочетание, представляющее собой сокращенное слово subspecies — «subsp.» или (реже) «ss.». Например, Lactobacillus delbrueckii subsp. bulgaricus.
Для каждого штамма указывают также аббревиатуру названия коллекции культур микроорганизмов, в которой он хранится, и номер, под которым он там значится. Например, Clostridium butyricum АТСС 19398 означает, что штамм хранится в американской коллекции типовых культур (American Type Culture Collection, АТСС) под номером 19398. Список коллекций микроорганизмов, пользующихся мировой известностью, приводится в «Руководстве Берджи по систематике бактерий» (Bergey’s Manual of Systematic Bacteriology, 1984—1989), в каталогах культур микроорганизмов и других справочных изданиях.
Описание любого нового вида микроорганизмов базируется на типовом штамме, который хранится в одной из коллекций микроорганизмов и на основании
совокупности свойств которого данный вид характеризуется в оригинальной статье или определителе. Типовой штамм является номенклатурным типом вида, поскольку за ним закреплено видовое название. Если какие-либо штаммы, первоначально включенные в тот же вид, в дальнейшем будут признаны заслуживающими выделения в особые виды, они должны получить новые названия, а старое видовое название сохраняется за типовым и родственным ему штаммами. При этом номер переименованного штамма сохраняется прежним. Аутентичными называют штаммы, полностью совпадающие по своим свойствам.
Для рода номенклатурным типом является специально обозначенный типовой вид, обладающий набором наиболее характерных для представителей данного таксона признаков. Например, в роде Bacillus типовым видом является В. subtilis.
В некоторых определителях и каталогах указывают старые названия переименованных микроорганизмов, а также приводят фамилии авторов, которые первыми выделили данный микроорганизм, и год публикации, в которой впервые описан этот организм. Например, один из видов дрожжей указывается в каталоге Всероссийской коллекции микроорганизмов (ВКМ) как Candida magnoliae (Lodder et Kreger van Rij, 1952) Meyer et Yarrow 1978, BKM Y-1685. Это означает, что он впервые был описан Lodder и Kreger van Rij в публикации 1952 г., тогда этот вид был назван Torulopsis magnoliae. В 1978 г. Torulopsis magnoliae был переименован такими исследователями, как Meyer и Yarrow, в Candida magnoliae и хранится в настоящее время в ВКМ под номером ВКМ Y-1685. Буква Y перед номером штамма означает «the Yeasts» — дрожжи.
Кроме понятия «штамм» в микробиологии применяют термины «вариант», «тип», «форма». Они обычно используются для обозначения штаммов микроорганизмов, отличающихся по некоторым признакам от типового штамма. Штамм, отличающийся от типового по морфологическим особенностям, называют морфовар (морфотип), физиолого-биохимическим особенностям — биовар (биотип, физиологический тип), способности синтезировать определенные химические соединения — хемовар (хсмоформа, хсмотип), условиям культивирования —- культивар, тип ответа на внедрение бактериофага — фаговар (фаготип, лизотип), антигенным характеристикам — серовар (серотип) и т.п.
В работах по генетике микроорганизмов часто используют термин «клон». под которым подразумевают популяцию генетически родственных клеток, полученную неполовым путем из одной родительской клетки. В молекулярной биологии клоном называют множественные копии идентичных последовательностей ДНК, полученные при их встраивании в клонирующие векторы (например, плазмиды). Под термином «генетически модифицированные», или «рекомбинантные», штаммы понимают штаммы микроорганизмов, полущенные в результате генноинженерных манипуляций. Часто новые штаммы микроорганизмов получают с помощью мутагенов.
Каждый новый штамм микроорганизмов, выделенный из природных или техногенных источников, должен быть охарактеризован для получения полного набора данных о свойствах микроорганизма в чистой культуре. Эти данные могут быть использованы, например, для составления паспорта ценных в промышленном отношении штаммов, а также для их идентификации.
Цель идентификации — установить таксономическое положение исследуемого штамма на основании сравнения его свойств с изученными и принятыми
(официально зарегистрированными) видами. Поэтому результатом идентификации обычно является отождествление исследуемого микроорганизма с каким-нибудь видом или отнесение к определенному роду. Если исследуемый штамм или группа штаммов отличаются по своим свойствам от представителей известных таксонов, то они могут быть выделены в новый таксон. Для этого дают описание нового таксона, включающее, например, в случае бактерий следующее: перечень штаммов, входящих в таксон; характеристику каждого штамма; перечень свойств, рассматриваемых в качестве существенных в таксоне; перечень свойств, которые квалифицируют таксон для представительства в ближайшем более высоком таксоне; перечень диагностических характеристик, дифференцирующих предлагаемый таксон от близкородственных таксонов; отдельное описание типового (для вида) штамма; фотографию микроорганизма.
Чтобы вновь предлагаемый таксон мог быть официально принят, его описание должно быть опубликовано в соответствии с определенными правилами. Например, действительное или узаконенное опубликование таксона бактерий предусматривает помещение статьи с его описанием в Международном журнале по систематике и эволюционной микробиологии «International Journal of Systematic and Evolutionary Microbiology» (IJSEM). Если публикация появляется в другом авторитетном научном журнале (эффективное опубликование), то оттиск статьи из этого журнала направляется в IJSEM. С 1980 г. в IJSEM регулярно публикуются так называемые списки узаконенных наименований бактерий. В них перечисляются все наименования бактерий, которые были опубликованы в IJSEM (действительное или узаконенное опубликование) или эффективно опубликованы прежде в каких-либо других авторитетных журналах. После внесения наименования бактерий в список узаконенных IJSEM наименований это наименование признается действительным, независимо от того, было ли оно ранее опубликовано в IJSEM или другом журнале. Дата появления публикации наименования данного таксона в IJSEM или в списке узаконенных наименований IJSEM является для таксона приоритетной.
Культура типового штамма нового вида .микроорганизмов передается на хранение в одну из коллекций микроорганизмов мирового значения. В случае утери типового штамма возможна его замена на так называемый нести повой штамм. При этом должно быть подтверждено, что свойства нового штамма хорошо совпадают с описанием утерянного. Чтобы показать, что таксон предлагается впервые, после названия нового семейства добавляется сокращенная комбинация «fam.nov.», нового рода — «gen. nov.», а нового вида — «sp. nov.». Например, в 2000 г. О.И.Кеппен с соавт. было предложено новое семейство бактерий — Oscillochloridaceae, fam. nov. Выражение «species insertae sedis» означает, что речь идет о виде, временно не имеющем определенного таксономического статуса, так как не ясно, в какой таксон более высокого порядка — род или семейство — следует поместить данный вид из-за недостатка необходимых для этого экспериментальных данных.
9.2. ОПИСАНИЕ И ИДЕНТИФИКАЦИЯ
Как уже отмечалось, принципы классификации и идентификации разных групп прокариот и эукариотных микроорганизмов имеют существенные раз
личия. Идентификация грибов до классов, порядков и семейств основана на характерных чертах строения и способах образования в первую очередь половых структур. Кроме того, используется характеристика бесполых спороноше-ний, строение и степень развития мицелия (зачаточный, хорошо развитый, септированный или несептированный), культуральные (колония) и физиологические признаки. Дифференциация родов внутри семейств и идентификация видов проводятся с применением морфологических признаков, полученных с использованием электронной микроскопии, а также физиологических и культуральных особенностей. Единого определителя для идентификации всех грибов не существует, поэтому вначале определяют класс или порядок идентифицируемого гриба и далее пользуются соответствующим определителем для этого класса или порядка.
Идентификация дрожжевых грибов, которые относятся к числу широко используемых объектов разных микробиологических исследований, основана на культуральных (макроморфологических), цитологических, физиолого-биохимических особенностях, характеристике жизненных циклов и полового процесса, специфических признаках, связанных с экологией, и проводится с использованием специальных определителей для дрожжей.
В основе систематики микроскопических форм водорослей лежит строение их клеток и состав пигментов. Определение систематического положения простейших проводится с использованием морфологических особенностей и жизненных циклов. Таким образом, идентификация эукариот базируется главным образом на особенностях их морфологии и циклов развития.
Идентификация прокариот, которые морфологически менее разнообразны, чем эукариоты, основана на использовании широкого спектра фенотипических, а во многих случаях и генотипических признаков. Она в большей степени, чем идентификация эукариот, основывается на функциональных признаках, поскольку большинство бактерий можно идентифицировать не по их внешнему виду, а только выяснив, какие процессы они способны осуществлять.
При описании и идентификации бактерий изучают их культуральные свойства, морфологию, организацию клетки, физиолого-биохимические особенности, химический состав клеток, содержание гуанина и цитозина (ГЦ) в ДНК, последовательность нуклеотидов в гене, кодирующем синтез 16S рРНК и другие фено- и генотипические признаки. При этом необходимо соблюдать следующие правила: работать с чистыми культурами, применять стандартные методы исследования, а также использовать для инокуляции клетки, находящиеся в активном физиологическом состоянии.
Культуральные свойства, т.е. характерные особенности роста бактерий на плотных и жидких питательных средах (см. гл. 8), обычно используют для их характеристики, однако для идентификации применяют довольно редко.
Морфологическая характеристика и организация клеток бактерий (см. гл. 5) включает такие признаки, как форма и размеры клеток, их подвижность, наличие жгутиков и тип жгутикования, способность к спорообразованию. Полезным может оказаться также выявление в клетках характерных мембранных систем (хлоросом, карбоксисом, фико-билисом. газовых вакуолей и т.д.), присущих отдельным группам бактерий, а также включений (параспоральных телец, гранул волютина, поли-р-гидроксибутирата, полисахаридов и т.д.). Первостепенное значение для систематики бактерий придается окраске клеток по Граму и строению их клеточных стенок.
Изучение физиолого-биохимических свойств включает, прежде всего, установление способа питания исследуемой бактерии (фото/хемо-, авто/гетеротрофия) и типа энергетического метаболизма (способность к брожению, аэробному или анаэробному дыханию или фотосинтезу). Важно определить такие признаки, как отношение бактерии к молекулярному кислороду, температуре, pH среды, солености, освещенности и другим факторам среды. В данную группу признаков входит также перечень субстратов, утилизируемых в качестве источников углерода, азота и серы, потребность в витаминах и других факторах роста, образование характерных продуктов метаболизма, наличие некоторых ферментов. Для этого используют специальные тесты (см. гл. 8).
Многие тесты, применяемые дня обнаружения перечисленных признаков (их иногда называют рутинными тестами), важны для диагностики и широко используются в медицинской микробиологии. Их постановка требует значительных затрат времени, большого количества сложных сред и реактивов, соблюдения стандартных условий проведения, аккуратности выполнения- Для ускорения и облегчения процесса идентификации некоторых микроорганизмов, имеющих главным образом медицинское значение, разработаны различные тест-системы, например системы Oxi/Ferm Tube, Mycotube и Enterotubc II фирмы Hoffmann-La Roche (Швейцария) и др. Так, система Enterotube 11, предназначенная дтя идентификации энтеробактерий, представляет собой пластиковую камеру с 12 ячейками, содержащими окрашенные диагностические среды. Засев всех сред производится поступательно-вращательными движениями через камеру иглы с посевным материалом. Инкубацию проводят в течение 24 ч при 37 °C. О положительном или отрицательном результате теста супят по изменению цвета среды, разрыву агара (тест на газообразование) или после введения специальных реактивов (тест на образование индола, реакция Фогес—Проскауэра). Каждый признак обозначают определенной цифрой, поэтому полученные данные можно ввести в компьютер с соответствующей программой и получить ответ о таксономическом положении исследуемого штамма.
При идентификации симбиотических и паразитических (патогенных) бактерий важно установить специфичность симбионта к хозяину, а также устойчивость к антимикробным веществам и фагам (фаготипирование).
Определение состава клеток бактерий также имеет значение для их систематики (хемосистематика). Хемотаксономические методы могут быть важными, в частности, для тех групп бактерий, у которых морфологические и физиологические характеристики широко варьируются и недостаточны для проведения их удовлетворительной идентификации. В состав клеточных стенок разных прокариот входит несколько классов уникальных гетерополимеров: муреин (или псевдомуреин), липополисахариды, миколовые и тейхоевые кислоты. Состав клеточной стенки определяет и серологические свойства бактерий. Это лежит в основе иммунохимических методов их идентификации.
В качестве хемотаксономического маркера иногда используют также липидный и жирнокислотный состав клеток бактерий. Интенсивное изучение жирных кислот стало возможным с развитием метода газохроматографического анализа. Различия в составе липидов используют для идентификации бактерий на уровне рода и даже вида. Этот метод, однако, имеет определенные ограничения, поскольку содержание жирных кислот в клетках может зависеть от условий культивирования и возраста культуры.
В систематике некоторых бактерий учитывается состав хинонов и других переносчиков электронов, а также пигментов.
Важная информация о взаимном родстве бактерий может быть получена при изучении клеточных белков — продуктов трансляции генов. На основании
изучения мембранных, рибосомных, суммарных клеточных белков, а также отдельных ферментов сформировалось новое направление — белковая таксономия. Спектры рибосомных белков относятся к числу наиболее стабильных и используются для идентификации бактерий на уровне семейства или порядка. Спектры мембранных белков могут отражать родовые, видовые и даже внутривидовые различия. Однако характеристики химических соединений клетки не могут использоваться для идентификации бактерий изолированно от других данных, описывающих фенотип, поскольку нет критерия опенки значимости фенотипических признаков.
Иногда для идентификации бактерий или других микроорганизмов, например дрожжей, используют метод нумерической (или адансоновской) таксономии. В ее основе лежат идеи французского ботаника М.Алансона (M.Adanson), предложившего различные фенотипические признаки, поддающиеся учету, считать равноценными, что позволяет количественно выразить таксономические дистанции между организмами в виде отношения числа положительных признаков к общему числу изученных. Сходство между двумя исследуемыми организмами определяется путем количественной оценки возможно большего числа (обычно не менее ста) фенотипических признаков, которые подбираются так, чтобы их варианты были альтернативными и могли обозначаться знаками «минус» и «плюс». Степень сходства устанавливается на основании количества совпадающих признаков и выражается в виде коэффициента соответствия 5:
a + d
a+b+c+d’
где а + d — сумма признаков, по которым штаммы А и В совпадают (о — оба штамма с положительными признаками; d — оба с отрицательными); Ь — сумма признаков, по которым штамм А положителен, В отрицателен; с — сумма признаков, по которым штамм А отрицателен, штамм В положителен. Значение коэффициента соответствия может меняться от 0 до 1. Коэффициент 1 означает полную идентичность, 0 — полное несходство. Оценки комбинаций признаков производят с помощью компьютера. Полученные результаты представляют в виде матрицы сходства и/или в виде дендрограммы. Нумерическая таксономия может применяться при оценке сходства между таксонами микроорганизмов только невысокого ранга (роды, виды). Она не позволяет делать непосредственные выводы относительно генетического родства микроорганизмов, однако в известной степени отражает их филогенетические свойства. Так, установлено, что фенотипические признаки бактерий, поддающиеся изучению в настоящее время, отражают от 5 до 20 % свойств их генотипа.
Изучение генотипа микроорганизмов стало возможным в результате успешного развития молекулярной биологии и привело к возникновению геносисте-матики. Исследование генотипа, основанное на анализе нуклеиновых кислот, в принципе дает возможность построить со временем естественную (филогенетическую) систему микроорганизмов. Филогенетические взаимоотношения бактерий оценивают определением молярного содержания ГЦ в ДНК, методами ДНК-ДНКиДНК—рРНК-гибридизации, с помощью ДНК-зондов, а также изучением последовательности нуклеотидов в 5S, 16S и 23$рРНК (см. гл. 16).
Определение молярного содержания ГЦ от общего количества оснований ДНК у прокариот, как уже указывалось, колеблется от 25 до 75 %. Каждый вид бактерий имеет ДНК с характерным средним содержанием ГЦ. Однако поскольку генетический код
вырожден, а генетическое кодирование основано не только на содержании нуклеотидных оснований в единицах кодирования (триплетах), но и на взаимном расположении, то одинаковое среднее содержание ГЦ в ДНК двух видов бактерий может сопровождаться их значительным генотипическим разделением. Если два организма очень близки по нуклеотидному составу, то это может являться свидетельством их эволюционного родства только при условии, что они обладают большим числом общих фенотипических признаков или генетическим сходством, подтвержденным другими методами. В то же время расхождение (более 10—15 %) в нуклеотидном составе ДНК двух штаммов бактерий с общими фенотипическими свойствами показывает, что они относятся по крайней мере к разным видам.
Метод ДНК—ДНК-гибридизации является более важным для оценки генетического родства бактерий. При тщательном проведении экспериментов можно получить ценную информацию о степени их генетической гомологии. Внутри одного вида бактерий степень генетической гомологии штаммов достигает 70 — 100 %. Однако если в результате эволюционной дивергенции последовательности нуклеотидных оснований геномов двух бактерий различаются в большей степени, то специфическая реассоциация ДНК—ДНК становится такой слабой, что не поддается измерению. В таком случае гибридизация ДНК—рРНК позволяет значительно увеличить круг организмов, у которых можно определить степень генетической гомологии благодаря тому, что на относительно небольшом участке бактериального генома, кодирующем рибосомные РНК, исходная последовательность оснований сохраняется значительно полнее, чем на других участках хромосомы. В итоге методом ДНК—рРНК-гибридизации часто обнаруживают довольно высокую гомологию геномов бактерий, у которых реассоциация ДНК— ДНК не выявляет заметной гомологии.
Для идентификации бактерий иногда используют также метод ДНК-зондов (генных зондов) — разновидность метода молекулярной гибридизации ДНК—ДНК. Реакция гибридизации ведется в этом случае не между двумя препаратами тотальной ДНК, а между фрагментом нуклеотидной последовательности ДНК (зондом), включающим ген (генетический маркер), ответственный за какую-то определенную функцию (например, устойчивость к какому-нибудь антибиотику), и ДНК изучаемой бактерии. Самым распространенным способом создания генных зондов является выделение специфических фрагментов путем молекулярного клонирования. Для этого вначале создают банк генов изучаемой бактерии расщеплением ее ДНК эндонуклеазами рестрикции, а затем отбирают нужный клон из суммы фрагментов ДНК методом электрофореза с последующей проверкой генетических свойств этих фрагментов методом трансформации. Далее выбранный фрагмент ДНК лигируют в состав подходящей плазмиды (вектора), а эту комбинированную плазмиду вводят в удобный для работы штамм бактерий (например, Escherichia coli). Из биомассы бактерии, несущей ДНК-зонд, выделяют плазмидную ДНК и метят ее, например, радиоизотопной меткой. Затем осуществляют гибридизацию ДНК-зонда с ДНК бактерии. Образовавшиеся гибридные участки проявляют методом авторадиографии. По относительной частоте гибридизации генетического маркера с хромосомой той или иной бактерии делают заключение о генетическом родстве этих бактерий с исследуемым штаммом.
Для идентификации бактерий и создания филогенетической системы их классификации наиболее широкое распространение и значение получил метод анализа нуклеотидных последовательностей в рибосомальных РНК. Молекулы 5S, 16S и 23S рРНК содержат участки с самой высокой степенью генетической стабильности. Считают, что они находятся вне механизма действия естественного отбора и эволюционируют только в результате спонтанных мутаций, происходящих с постоянной скоростью. Накопление мутаций зависит только от времени, поэтому информация о нуклеотидной последовательности этих молекул считается наиболее объективной для определения филогенетического родства организмов на уровне от подвида до царства. В случае анализа 5S рРНК обычно определяют полную последовательность нуклеотидов, которая в этой молекуле у прокариот составляет 120 нуклеотидов. При исследовании 16S и 23S рРНК,
содержащих 1500 и 2500 нуклеотидов соответственно, часто проводят анализ олигонуклеотидов, полученных из этих молекул с помощью специфических эндонуклеаз рестрикции. Наиболее широкое распространение получило изучение последовательности нуклеотидов в 16S рРНК. Изучение структуры 16S рРНК представителей разных микроорганизмов привело к выявлению среди прокариот группы архей. Значения коэффициента сходства 5ав, отделяющие 16S рРНК бактерий и архей, лежат в пределах 0,1, в то время как значение 5Л[>, равное 1,0, соответствует полной гомологии нуклеотидных последовательностей, а 0,02 — уровню случайного совпадения.
Все чаще для идентификации бактерий предлагают дендрограммы, показывающие взаимоотношения между бактериальными родами, видами или штаммами на основании изучения последовательности нуклеотидов (или олигонуклеотидов) в рРНК, а также ДНК—ДНК и ДНК —рРНК гибридизации. Однако идентификация бактерий до родов на основании только генетических методов без предварительного изучения их фенотипических характеристик часто вообще невозможна. Поэтому лучшим подходом в работе по систематике бактерий считается изучение как генотипических, так и фенотипических свойств. В случае несоответствия между филогенетическими и фенотипическими данными приоритет временно отдают последним.
Особой проблемой является идентификация таких бактерий и архей, особенно морских видов, которые не способны расти на известных лабораторных питательных средах и для которых поэтому нельзя было получить чистую культуру. До недавнего времени эта проблема казалась неразрешимой. Однако около 15 лет назад были разработаны методы, позволившие экстрагировать, клонировать, секвенировать и сравнивать рибосомные РНК прямо из окружающей среды. Это позволило точно сосчитать и идентифицировать микроорганизмы, населяющие данный биотоп без их выделения в чистую культуру. Идентифицированный таким образом «некультивируемый» в лаборатории микроорганизм может быть даже описан, но с присоединением слова «candidatus» (кандидат). Слово «candidatus» будет сопровождать новый вид до тех пор, пока учеными не будут найдены условия культивирования этого организма в лаборатории и не будет получена его чистая культура, что позволит изучить все его свойства и опубликовать в качестве узаконненого.
Идентификацию бактерий проводят обычно с помощью «Определителя бактерий Берджи» (Bergey’s Manual of Determinative Bacteriology). Первое издание этого пособия было осуществлено в 1923 г. под руководством известного американского бактериолога Д. Берджи (D. Н. Bergey, 1860—1937). С тех пор оно регулярно переиздается с участием ведущих ученых-микробиологов мира. В последнем, 9-м издании определителя (1994) все бактерии разделены на 35 групп по легко определяемым фенотипическим признакам. Эти признаки вынесены в названии групп. Таксономическое положение бактерий внутри групп определяется с помощью таблиц и ключей, составленных на основе небольшого числа фенотипических признаков. Дифференцировочные таблицы для дифференциации видов бактерий некоторых родов, например рода Bacillus, не приводятся, а читатель отсылается к «Руководству Берджи по систематике бактерий».
В 4-томном «Руководстве Берджи по систематике бактерий» (Bergey’s Manual of Systematic Bacteriology, 1984 — 1989) содержится более полная информация о таксономическом положении бактерий. Для каждой группы бактерий дается описание входящих в него родов и видов, в том числе с неясным таксономическим статусом. Помимо подробного фенотипического описания, включающего морфологию, организацию и химический состав клеток, антигенные
свойства, вид колоний, особенности жизненного цикла и экологии, в характеристике родов приводятся также сведения о содержании ГЦ в ДНК, результатах гибридизации ДНК—ДНК и ДНК—рРНК. Ключи и таблицы позволяют идентифицировать бактерии не только до рода, но и до вида.
В настоящее время выходит в свет второе издание 4-томного «Руководства Берджи по систематике бактерий» (Bergey’s Manual of Systematic Bacteriology). В 2002 г. вышел первый том. Кроме этого имеется ряд статей и книг, в которых предлагаются оригинальные ключи для идентификации отдельных групп бактерий, например бацилл, псевдомонад, актиномицетов, энтеробактерий. Для иллюстрации приведем схему идентификации некоторых видов бацилл.
Ключ для определения бактерий рода Bacillus
1. Каталаза — есть ... — нет .... 2 17
2. Реакция Фогес-Проскауэра — положительная .. 3
— отрицательная 10
3. Рост в анаэробном агаре — есть ... .. 4
.— нет .... .. 9
4. Рост при 50 °C — есть ... .: 5 '
— нет .... .. 6
Трос?в МПБ с 7 % КаС.Г * — есть В. licheniformis
— нет. В. coagulans
6. Кислота й газ на среде с глюкозой — есть В. polymyxa
— нет ... .. 7
7/Восстановление NO3 до NO? — есть ... .. 8
— нет В. alvei
8. Параспоральные тельца в спорангии — "есть В. thurlngiensis
— нет В. cereus
9. Гидролиз крахмала — есть В. subtiHs
— нет В. pumilus
10. Рост при 65 °C — есть В stearothermophilus
— нет ... 11
11. Гидролиз крахмала — есть .. 12
— нет ... 15
12. Кислот:! й газ на среда с глюкозой — есть В. macerans
— нет ... 13
13. Ширина клеток 10 мкм й оолее — "да ~~Brtnegafenum
— нет ... 14
14. pH среды для образования ацетоина ....... -
менее 6,0 — да В. circulans
— нет В. firmus
15. Рост в анаэробном агаре — есть В. laterosponis
— нет ... 16
16. Кислота на среде с глюкозой — есть В. brevis
— нет В. sphaericus
17. Рост при 65 °C — есть В. stearothermophlTus
— нет ... 18
18. Гидролиз казеина — есть В. larvae
— нет....................... 19
19. Параспоральные тельца — есть В. popilliae
— нет В. lentimorbus
Полные дифференциальные характеристики этих видов бактерий рода Bacillus по нескольким десяткам признаков приводятся в Bergey’s Manual of Systematic Bacteriology, 1984—1989.
В настоящее время накопилось много новых данных, в том числе полученных в результате анализа нуклеотидных последовательностей рибосомальных РНК, об изученных ранее и вновь выделенных видах бактерий. На основании этих сведений видовой состав некоторых групп бактерий, например рода Bacillus, будет пересмотрен: часть видов останется в составе рода Bacillus, а некоторые образуют новые роды или будут отнесены к другим, уже существующим родам бактерий. Следует отметить также, что для описания новых штаммов бактерий изучают, как правило, больше признаков, чем необходимо для их идентификации, так как ключи и таблицы включают не все признаки идентифицируемых бактерий, а только те, которые отличаются у разных видов (табл. 9.1).
Таблица 9.1
Минимальный перечень данных, необходимых для описания новых штаммов бактерий
(по H.Truper, K.Schleifer, 1992)
Свойства Основные признаки Дополнительные признаки
Морфология клеток Форма; размер; подвижность; внутри- и внеклеточные структуры; взаимное расположение клеток; клеточная дифференцировка; тип клеточного деления; ультраструктура клетки Цвет; характер жгутикования; споры; капсулы; чехлы; выросты; жизненный цикл; гетероцисты; гормогонии; ультраструктура жгутиков, оболочки, клеточной стенки
Характер роста Особенности роста на плотных и в жидких питательных средах; морфология колоний Цвет колоний, суспензии
Окраска По Граму На кислотоустойчивость; окраска спор, жгутиков
Состав клетки Состав ДНК; запасные вещества Гомология нуклеиновых кислот; клеточные пигменты; состав клеточной стенки; типичные ферменты
Физиология Отношение к температуре; к pH среды; тип метаболизма (фототроф, хе-мотроф, литотроф, органотроф); отношение к молекулярному кислороду; акцепторы электронов; источники углерода; источники азота; источники серы Потребность в солях или осмотических факторах; потребность в факторах роста; типичные продукты метаболизма (кислоты, пигменты, антибиотики, токсины); устойчивость к антибиотикам
Экология Условия обитания Патогенность; круг хозяев; образование антигенов; серология; восприимчивость к фагам; симбиоз
Глава 10
ИДЕНТИФИКАЦИЯ МИКРООРГАНИЗМОВ ИЗ ПРИРОДНЫХ СООБЩЕСТВ БЕЗ ВЫДЕЛЕНИЯ В ЧИСТЫЕ КУЛЬТУРЫ
В последнее- десятилетие XX в. было разработано довольно много методов для характеристики микробного разнообразия в природных местообитаниях и в первую очередь почвы как наиболее сложной системы методов, основанных на экстрагировании и исследовании ДНК тотального микробного сообщества (Muyzer and Smalla, 1998). Наиболее широко распространенные молекулярнобиологические, а также и микроскопические методы учета и определения активности микроорганизмов без выделения в чистые культуры приведены в сводной таблице (рис. 10.1).
Рис. 10.1. Микроскопические и молекулярно-биологические методы учета и определения активности микроорганизмов без выделения в чистые культуры:
АО — акридин оранжевый [acridine orange]; DAFI — 5-(4,6-дихлортриазин-2-ил) аминофлуоресцеин [5-(4,6-dichlorotriazine-2-yl) aminofluorescein]; DAPI — 4,6-диамидино-2-фенилиндол [4,6-diamidino-2-phenylindole]; FDA — флуоресцеин диацетат [fluorescein diacetate]; FITC — флуоресцеин изотиоцианат [fluorescein isothiocyanate]; SFDA — 5-(и 6-) сульфофлуоресцеин диацетат [5-(and 6-) sulfofluorescein diacetate]; GFP — зеленый флуоресцирующий белок [green fluorescein protein]; FISH — fluorecsence in situ hybridization; LUX — белок люцифераза [luciferase protein]
К таким методам относятся: анализ реассоциации ДНК; гибридизация ДНК микробного сообщества; определение профилей процентного состава ГЦ оснований ДНК; сравнительный анализ секвенсов фрагментов рестрикции ДНК; анализ полиморфизма фрагментов рестрикции 16S рДНК; рестрикционный анализ амплифицированных рибосомальных ДНК 16S рДНК генов; клонирование ДНК-фрагментов, полученных обратной транскрипцией 16S рРНК и
клонирование амплифицированных с помощью ПЦР рДНК фрагментов, денатурирующий градиентный гель электрофорез (ДГГЭ в английской транскрипции — DGGE) 16S рДНК ампликонов (Muyzer et al., 1998) и некоторые другие. В результате появилась реальная возможность осуществлять выявление и идентификацию известных бактерий в природных образцах без выделения этих бактерий в чистые культуры, а также выявлять присутствие в исследуемом образце неидентифицированых (отсутствующих в банке данных, коллекциях), новых организмов.
При выполнении таких исследований необходимо проведение ряда последовательных операций. Первая операция — это разрушение клеток, выделение нуклеиновых кислот (НК) и при необходимости их дополнительная очистка с
помощью соответствующих наборов реактивов. Следующая важнейшая опера-
Микробное сообщество Экстракция НК
| ДНК |
ПЦР
V
FISH
Построение дендрограмм
Рис. 10.2. Схема анализа биоразнообразия микробного сообщества
Синтез проб
ция — амплификация исследуемых участков в выделенной смеси НК для увеличения и выравнивания концентрации этих участков (с применением соответствующих праймеров — затравок) в результате полимеразной цепной реакции (ПЦР). После этого проводится проверка наличия и чистоты продуктов ПЦР электрофоретически в агарозном геле. Полученные ПЦР-продукты — концентрированная смесь амплифицированных участков НК (ампликоны), принадлежащих разным бактериям, разделяют, например, методом ДГГЭ. Если на этой стадии ввести соответствующие контроли, то можно провести и предварительную идентификацию исследуемых микроорганизмов. Однако для достоверной идентификации, как и для выявления новых организмов, нужно будет провести изъятие (вырезание) отдельных полос (ампликонов) из электрофореграммы геля и определение их нуклеотидных последовательностей (секвенирование). Выявленные последовательности сравнивают — идентифицируют с данными, имеющимися в международном банке генов, после чего составляют списки выявленных микроорганизмов и строят филогенетические схемы микробного сообщества (рис. 10.2).
В качестве дополнительной, но обязательной информации при описании новых мик
роорганизмов используют данные о процентном составе ГЦ-оснований ДНК каждой чистой культуры и степень ДНК—ДНК гомологии вновь описываемых штаммов и известных или близкородственных из коллекций культур. Однако обе операции правомерны только лишь при работе с чистыми культурами.
Каждая из перечисленных выше операций достаточно сложна и ответственна и требует определенных навыков, точного следования прописям соответствующих методик, а самое главное — использования стандартных реактивов и специального оборудования. В зависимости от поставленной задачи некоторые операции могут быть исключены, а другие, наоборот, расширены или проведены многократно. В результате таких исследований можно идентифицировать отдельную чистую культуру, а также получить перечень микроорганизмов, выращенных, например, в виде отдельных колоний на агаризованной среде или, более того, определить разнообразие микроорганизмов, обитающих в какой-либо эконише (экотопе) без предварительного культивирования. Существует довольно много и других методов, позволяющих охарактеризовать микробное разнообразие, и в каждом конкретном случае выбор метода должен определяться поставленной задачей.
Как и в традиционных микробиологических методах, в молекулярно-биологических методах имеются свои ограничения и условности. Как невозможно выделить на одной среде и в одних условиях в чистые культуры сильно различающиеся по метаболизму бактерии, так и, например, для выявления эубак-терий и архей молекулярно-биологическими методами нужны разные праймеры. Для изоляции НК рекомендуется использовать минимальное количество образца, например 0,5 г почвы, и, следовательно, выявленное разнообразие будет отражать экотоп размером в 0,5 г.
При идентификации микроорганизмов из природных образцов без выделения в чистые культуры предполагается, что после проведения ПЦР получают примерно равное количество ампликонов всех присутствовавших в образце бактерий. Однако это может быть далеко не так. В результате проведения ПЦР с универсальным праймером получают больше ампликонов тех бактерий, численность которых была больше в исходном образце и НК которых из образца было выделено больше. При разделении продуктов ПЦР-методом ДГГЭ из-за очень низкой концентрации некоторых ампликонов их полосы могут быть трудно различимы на фоне других полос. С другой стороны, высокие концентрации ампликонов близкородственных организмов на недостаточно качественной ДГГЭ-фореграмме могут выглядеть в виде одной объемной полосы. Помимо этих проблем, легче или труднее, но проверяемых и решаемых, существуют и более трудные задачи, такие, как спонтанный синтез химерных ампликонов при проведении ПЦР, которые легко могут быть приняты за наличие в образце новых, неидентифицированных или некультивируемых организмов. В частности, и из-за этого факта Международным комитетом по систематике и таксономии бактерий было принято решение не принимать к рассмотрению описание новых микроорганизмов без выделения чистых культур этих организмов.
В результате проведения всех стандартных операций будут получены данные, отражающие только разнообразие микроорганизмов в исследованном образце по типу «есть—нет», но не количественное присутствие каждого вида. Формально вид, представленный в образце одной клеткой и миллионом клеток, имеет равные шансы быть представленным в списке. Для преодоления
этого существенного неудобства в настоящее время разрабатываются методы количественной ПЦР. В одном из них используется внутренний контроль в виде известных концентраций НК. Такие стандартные контрольные образцы подвергаются амплификации наряду с экспериментальными. После прохождения соответствующего количества циклов контрольные и экспериментальные образцы сравниваются, а количество НК и число циклов дают возможность рассчитать исходное количество того или иного микроорганизма. Однако этот метод имеет ряд существенных недостатков из-за высокой чувствительности конечных этапов ПЦР к разного рода химическим и физическим факторам.
В другом, более точном методе, где используется TaqMan 5'-нуклеаза, имеет место определение накопления ПЦР-продукта в течение реакции амплификации по изменению свечения флуоресцирующей пробы. При этом происходит накопление флуоресцирующего продукта, что приводит к усилению флуоресценции, которая пропорциональна накоплению ПЦР-продукта. В этом методе фактически контролируется количество циклов, а не количество ПЦР-продукта после фиксированного количества циклов. Этот метод и его некоторые модификации получили название Real-Time PCR.
10.1. РАЗРУШЕНИЕ КЛЕТОК, ВЫДЕЛЕНИЕ И ОЧИСТКА ДНК
Первый этап при идентификации микроорганизмов молекулярно-биологическими методами — разрушение клеток и выделение НК. Известно множество методов, позволяющих разрушать бактериальные клетки как чистых культур, так и микробных сообществ в образцах естественных субстратов. В целом следует отметить, что для разрушения клеток чистых культур чаще используют комбинацию детергентов и литических ферментов, а для разрушения клеток в естественных образцах (почве, воде и др.) — механическое разрушение (см. гл. 14). В том случае, если собираются проводить идентификацию бактерий, представленных колониями разных размеров на агаризованной среде, для уменьшения последующих погрешностей рекомендуется не просто смывать все колонии, а отобрать равное количество биомассы каждой колонии. Известно, что из 0,1 г сырой бактериальной биомассы может быть выделено 40—200 мкг ДНК в зависимости от вида бактерии и условий выращивания. При этом с увеличением объема биомассы, используемой для выделения ДНК, эффективность выделения снижается.
При выделении НК из природных образцов, объем образца, используемого для выделения НК, зависит от предполагаемого обилия в нем микроорганизмов, а также типа образца — вода, почва или др. Так как в природных образцах, особенно в почвенных, часто присутствует достаточно много гуминовых веществ, минимизация образца определяется и этим фактором. Обычно в случае почвенных образцов используют 0,5 — 2,0 г почвы (в пересчете на сухое состояние образца).
Последовательность операций при разрушении клеток и экстрагировании НК может быть следующей. В пластиковую пробирку на 15 мл вносят 2 г почвы, заливают 3 мл фосфатного буфера (120 мМ Na2HPO4/NaH2PO4, pH 8,0), добавляют 3 г стеклянных стерильных бус размером 0,1 мм. Закрывают пробирку соответствующей пробкой и помещают ее на 30 с (или два раза по 15 с) во встряхиватель (Bead Beater, Brown, Germany). В отсутствие специального встряхивателя суспензию образца в буфере гото
вят в стерильной фарфоровой ступке. Образец замораживают жидким азотом и пестиком растирают в течение примерно 5 мин до получения гомогенной массы. Метод применим также и для разрушения грибных гиф. Сравнительную информацию об этих и других методах можно найти в гл. 13.
Выделение тотальной фракции нуклеиновых кислот обычно осуществляют после добавления додецилсульфата натрия (от 30 мкл 10%-го раствора), тщательного перемешивания и последующего внесения (от 0,5 мл) холодного водного (30%-го, pH 7,5 — 8,0) раствора фенола (Duineveld et al., 2001). Смесь перемешивают в руках (без использования вортекса) и выдерживают на льду, после чего центрифугируют в течение 5 мин на настольной центрифуге при 15 тыс. об/мин при комнатной температуре. При этом содержимое пробирки расслаивается на верхнюю водную фракцию и нижнюю — фенольную. Пипеткой отбирают верхнюю часть супернатанта, стараясь не захватить нижнюю фенольную часть. Отобранную верхнюю часть смешивают с равным объемом смеси хлороформа и изоамилового спирта (24: 1), перемешивают в руках и отбирают водную фазу. Добавляют к отобранной водной фазе 0,1 объема 5 М NaCl и два объема холодного 96%-го этанола. Смесь выдерживают на льду в течение 20 мин или 1 ч при -20 °C. После этого смесь опять центрифугируют 10 мин на центрифуге при 15 тыс. об/мин, но с охлаждением до -6 °C. Удаляют супернатант и промывают осадок 1,5 мл 70%-го холодного этанола. Осадок выделенной ДНК должен быть бесцветным (белым). Если изолированная ДНК остается окрашенной (различной интенсивности коричневый оттенок), то промывание спиртом повторяют. В этой же операции происходит и удаление соли. Отмытую высокополимерную ДНК подсушивают на воздухе. После этого сухую ДНК растворяют в деионизированной воде и хранят в замороженном виде при -20 °C.
Часть полученного препарата ДНК может быть использована для определения процентного состава ГЦ-оснований ДНК и степени ДНК—ДНК гомологии, если ДНК была выделена из чистой культуры.
Если тотальный препарат ДНК был выделен из образцов естественных субстратов, особенно почвы, компостов и др., фактически обязательной дополнительной стадией является очистка ДНК. Для этой цели лучше использовать наборы реактивов и приспособления (колонки), выпускаемые соответствующими компаниями. Одна из них — Wizard DNA cleanup system (Promega, USA). Инструкция по применению набора реактивов для дополнительной очистки ДНК прилагается к каждому набору.
10.2. АМПЛИФИКАЦИЯ ФРАГМЕНТОВ ВЫДЕЛЕННОЙ И ОЧИЩЕННОЙ ДНК С ПОМОЩЬЮ ПЦР
Для амплификации (увеличения числа копий) отдельных фрагментов или секвенсов (последовательностей) ДНК применяют полимеразную цепную реакцию (ПЦР). Она происходит, когда два олигонуклеотидных праймера гибри-дизуются на двуцепочечную матрицу ДНК с двух разных концов (5' и 3'), что позволяет ДНК-полимеразе синтезировать ДНК-последовательность, находящуюся между этими двумя праймерами (рис. 10.3).
Для проведения ПЦР необходимо иметь: двухцепочечную ДНК-матрицу, содержащую участок, который должен подвергнуться амплификации; праймеры (в избытке!), представляющие собой одноцепочечные фрагменты НК, которые могут «отжигать» (разъединять) нити ДНК, связываясь с того или другого конца с одной из цепей
З'-конец матрицы
Праймер 2
ДНК-матрица
5'-конец
Праймер 1
Синтез участка между двумя праймерами в ПЦР с помощью ДНК-полимеразы
Цикл 1
ПЦР-продукт (ампликоны)
Рис. 10.3. Схема работы полимеразной цепной реакции (ПЦР).
Каждый цикл синтеза участка между двумя праймерами в ПЦР с помощью ДНК-полимеразы включает несколько последовательных ступеней, описанных в тексте
ДНК-матрицы, в комплементарном праймеру участке; смесь (избыток!) четырех дезоксирибонуклеотидфосфатов (ДНФ), из которых будет синтезироваться новая ДНК; ДНК-полимераза — фермент, который осуществляет синтез нового фрагмента ДНК, апликона. В ПЦР обычно используют Taq-ДНК-полимеразу, высокотермостабильный фермент, полученный из термофильной бактерии Termus aquaticus. Эта полимераза запатентована производящей ее фирмой. Кроме этих в реакционную смесь вносят также некоторые необходимые компоненты для стабильности протекания реакций.
ПЦР-продукты, характеризующие большинство почвенных бактерий, можно получить с использованием бактериальных праймеров U968 и L1401 и схемы соединения-отсоединения праймеров в течение 30 тепловых циклов. 17-мерные праймеры U968 и L1401 представляют собой нуклеотидные последовательности, специфичные высококонсервативной 16S рДНК области прокариот и соответствующие положению у Е. coli между 968 и 984; 1385 и 1401 нуклеотидами хромосомной ДНК соответственно.
Структура праймеров: U968 + GC: sequence 5' — (GC clamp)-AAC GCG AAG AAC CTT AC-3'. L140: sequence 5' - CGG TGT GTA CAA GAC CC-3'. Компонент GC clamp (зажим, довесок), представляет собой 40-мерный фрагмент ДНК (5'-CGC CCG GGG CGC GCC CCG GGC GGG GCG GGG GCA CGG GGG G-3'), предназначенный для разделения продуктов ПЦР методом ДГГЭ.
Типичная рабочая смесь (master mix), используемая для амплификации почвенных и ризосферных бактерий, содержит: 23,34 мкл деионизованной воды; 5 мкл специального буфера (stoffel buffer), содержащего 100 мМ трис-НС], 100 мМ КО, pH 8,3; 7,5 мкл (25мМ) MgCl2; 10 мкл смеси четырех нуклеотидтрифосфатов: АТФ, ГТФ, ТТФ и ЦТФ; 1 мкл (10 мМ) U968 GC праймера; 1 мкл (10 мМ) L140J праймера; 0,5 мкл 100%-го формамида; 0,16 мкл белка Т4 гена 32 (белок, способствующий более стабильному протеканию реакции); 0,5 мкл AmpliTaq DNA Polymerase, Stoffelfragment (10 ед/мкл); 1 мкл амплифицируемой ДНК. Общий объем смеси составляет в данном случае 50 мкл (van Diepeningen et al., 2003). Приготовленную смесь до внесения образца амплифицируемой ДНК подвергают УФ-облучению в течение 3 мин для стерилизации раствора.
Синхронность в работе ДНК-полимеразы и праймеров достигается за счет циклического изменения температуры смеси. Циклическое изменение температуры обеспечивает прибор амплификатор, работающий в соответствии с введенной в него про-
Рис. 10.4. Пример электрофореграммы продуктов ПЦР
граммой. После каждого цикла в реакцию включается и продукт (ампликон) предыдущей реакции в качестве ДНК-матрицы. В результате имеет место геометрическая прогрессия увеличения количества ампликонов и после 30 — 40 циклов их концентрация достигает миллионов копий (ампликонов). Точность воспроизведения ампликона обеспечивается в существенной степени качеством праймеров.
Надежным аплификатором, рекомендуемым для работы, является прибор РТС-200 (DNA Engine (Gradient cycler) BiOzym. The Netherlands) и соответствующая программа. Последовательность циклов нагревания—охлаждения и их продолжительность может быть следующая. Начальную денатурирующую стадию проводят при температуре 94 °C в течение 3 мин; далее в каждом цикле денатурация осуществляется при 94 °C в течение 1 мин. Начальный отжиг праймеров происходит при температуре 60 °C в течение I мин и затем температура снижается на 1 °C в каждом втором цикле до 55 °C (в первых 11 циклах). В следующих 19 циклах отжиг проводят при 55 °C и также в течение 1 мин. Время «растяжения» (extension) праймеров равно 2 мин при 72 °C. Заключительная стадия «растяжения» осуществляется при 72 °C в течение 10 мин и затем смесь охлаждается до +4 °C.
Качество продуктов ПЦР (ампликонов), размер которых при описанных выше условиях будет равен 474 парам нуклеотидов (п. н.), проверяют электрофоретически в стандартном (1%-м) агарозном геле с последующим окрашиванием этидиум бромидом (рис. 10.4). Продукты ПЦР можно хранить некоторое время при -20°С.
10.3. ДЕНАТУРИРУЮЩИЙ ГРАДИЕНТНЫЙ ГЕЛЬ-ЭЛЕКТРОФОРЕЗ (ДГГЭ) И ЕГО ВОЗМОЖНОСТИ
Этим методом проводят разделение фрагментов ДНК одинаковой длины, но различных по составу комплементарных пар нуклеотидов (п. н.). Теоретически метод позволяет разделить ампликоны величиной до 500 и. н., различающиеся всего лишь одной парой нуклеоидов (Myers et al., 1998). Следовательно, этот метод особенно полезен как при сравнении, например, микробных сообществ каких-либо образцов, так и при сравнении исходных вариантов и мутантов чистых культур. В последнем случае ПЦР должна быть проведена с использованием специфического для данной мутации праймера (функциональным праймером). Вместе с тем анализ ДГГЭ методом ПЦР амплифицирован-ных экстрактов ДНК с универсальными праймерами, экстрактов, выделенных из природных образцов с высокой численностью и разнообразием микроорганизмов (богатые почвы, компосты и др.), позволяет выявить не все разнообразие, а численно доминирующие виды.
Разделение в ДГГЭ основывается на снижении электрофоретической подвижности частично расплавленной двухцепочечной молекулы ДНК (амплико
на) в полиакриламидном геле, содержащем линейно увеличивающийся градиент денатурантов ДНК, которыми являются смесь мочевины и формамида. Плавление фрагментов ДНК обеспечивается в дискретных местах — доменах плавления, полосах пар оснований с идентичной температурой плавления. Когда домен, подверженный плавлению, достигает «своего» места в денатурирующем градиенте геля, спиральная структура ДНК переходит в частично расплавленное (расщепленное) состояние, и движение молекулы практически останавливается.
Описанный подход позволяет разделить до 50 % всех вариантов исследуемых последовательностей в фрагментах ДНК длиной до 500 п.н. Процент разделения (чувствительность) может быть увеличен практически до 100 %, если прикрепить к разделяемым путем ДГЭ ДНК-фрагментам «фиксаторы» — нуклеотидные последовательности с высоким содержанием ГЦ-оснований, которые затем будут действовать как высокотемпературные домены плавления. Прикрепление таких последовательностей осуществляется или в процессе клонирования, или при проведении ПЦР, где к синтезируемым ампликонам прикрепляются добавочные нуклеотидные последовательности, размером от 40 до 50 п.н., которые в свою очередь были прикреплены к 5'-концу праймера при его синтезе.
Таким образом, в методе ДГГЭ задействованы следующие факторы: 1) одинаковый размер (длина) фрагментов ДНК (одинаковое количество пар нуклеотидов), например 474 пары, с соответствующими «фиксаторами» на концах, полученными в процессе ПЦР; 2) состав нуклеотидов ампликона; 3) повышенная, но стабильная температура, которая инициирует плавление фрагментов куска ДНК, но из-за разного состава нуклеотидов плавление этих фрагментов ДНК происходит сильнее или слабее; 4) электрическое поле, обеспечивающее движение заряженных фрагментов ДНК в геле с определенной скоростью; 5) наличие в геле химических денатурантов, причем эти денатуранты должны образовывать в геле градиент — от очень низкой концентрации в верхней части геля к максимально высокой в нижней части. Наиболее широко используемый вариант ДГЭ — это «параллельный» денатурирующий градиент, параллельный вследствие того, что денатурирующий градиент увеличивается в том же направлении, что и направление электрического поля.
Ниже приводится протокол приготовления пластин геля с денатурирующим градиентом, последовательность внесения продуктов ПЦР в гель, условия проведения электрофореза, фиксирования, окрашивания и других операций (Van Diepeningen et al., 2003). Согласно этому протоколу, можно будет приготовить денатурирующий градиент в пределах от 45 до 60 %, где 100%-й денатурирующий эффект создают 7 М мочевина и 40%-й формамид, а 8%-й акриламид без мочевины и формамида обеспечивает нулевой денатурирующий эффект.
Материалы. Количество посуды и реактивов определяется количеством исследуемых образцов. Перчатки резиновые для работы в лаборатории; пробирки типа Эппен-дорф объемом от 0,2 до 1,0 мл; автоматические пипетки с наконечниками и рабочими объемами от 1 мкл до 5 мл; 4 стеклянные пластинки, две размером 22,3 * 20 см и две — 20 х 20 см; пластиковые термоустойчивые полоски, определяющие толщину геля (Spacers), толщиной 1 мм; целлофановая пленка размером не менее больших стеклянных пластин (Amersham Pharmacia Biotech AG, Uppsala, Sweden); «гребенки»; пластиковые прозрачные (упругие) пластинки (Gelbond®), (Amersham Pharmacia Biotech, AG, Uppsala, Sweden, 80-1129-37); шприцы разного объема с иглами.
Оборудование. Магнитные мешалки, разных размеров и мощности; качалка наклонная, вращающаяся с изменяемой скоростью вращения; pH-метр; холодильник; морозильная камера на -20 °C; ДГГЭ-машина [The Dcode system, BIORad Laboratories, США] для проведения электрофореза; блок питания для ДГГЭ-машины; специальный двухкамерный стакан для приготовления и создания градиента; шланги разного диаметра, в частности 1,7 мм (внутренний диаметр); перистальтический насос с точно программируемой скоростью подачи жидкости (Heidolph PD 5201, The Netherlands).
Реактивы и их подготовка для приготовления геля с денатурирующим градиентом. Ионообменная смола AG 501-Х8 (Bio-Rad, США) и деионизованный формамид (Sigma, США). Последний готовят добавлением 12,5 г ионообменной смолы к 250 мл (100%-го) формамида. Перемешивают смесь при комнатной температуре в течение часа. Пропускают раствор через фильтровальную бумагу, избегая попадания смолы. Хранят в темноте при температуре 4 —6°С. Раствор КОН в метаноле (5%-й) для очистки стекол, используемых для приготовления геля. Мочевина-формамид (МФ), 45%-й раствор в 6%-м растворе акриламида-бисакриламида. Готовят смешиванием 47,3 г мочевины (Bio-Rad, США); 37,5 мл 40%-го акриламида-бисакриламида (37,5:1, Bio-Rad, США); 45 мл деионизованного формамида; 2,5 мл 50 х ТАЕ (/дрпс-ЭДТА) буфера (Bio-Rad, США); 250 стерильной деионизованной воды (ДИВ). Раствор можно хранить в темной стеклянной посуде до 6 мес при 4 °C. Мочевина-формамид, 60%-й раствор в 6%-м растворе акриламида-бисакриламида. Готовят добавлением 63,0 г мочевины, 37,5 мл 40%-го акриламида-бисакриламида (37,5 :1), 60 мл деионизованного формамида, 2,5 мл 50 х ТАЕ буфера, 250 стерильной ДИВ. Раствор можно хранить в темной стеклянной посуде до 6 мес при 4 °C. Мочевина-формамид, 0%-й раствор в 8%-м растворе акриламида-бисакриламида. Готовят добавлением 40 мл 40%-го акриламида-бисакриламида (37,5:1); 2,0 мл 50 х ТАЕ буфера; 200 стерильной ДИВ. Раствор можно хранить в темной стеклянной посуде до 6 мес при 4 °C. Персульфат аммония (АПС, Bio-Rad, США) готовят в виде 10%-го раствора (вес/объем) в стерильной ДИВ. Разливают по 400 мкл в 1,0 — 0,5 мл пробирки (Эппендорф) и хранят при -20 °C, каждый раз используя новую пробирку.
Реактивы и их подготовка для окрашивания нуклеиновых кислот в геле азотнокислым серебром после фореза. Набор (kit) включает реагенты: азотнокислое серебро (BioRad, США), окислитель и проявитель. Приведенного ниже количества достаточно для обработки двух пластин геля. Хранить набор нужно в холодильнике. Раствор для фиксирования геля готовят смешиванием (объем/объем) 10%-го этанола и 5%-й уксусной кислоты. Для фиксирования одного геля требуется 400 мл. Для приготовления раствора используют ДИВ. Окислитель готовят растворением 15 мл (10-кратно концентрирован-' ного) реагента из упомянутого выше набора (Bio-Rad, США) в 150 мл деионизованной воды. Каждый раз необходимо готовить свежий раствор*. Серебряный краситель (на основе азотнокислого серебра) готовят растворением 15 мл (Ю-кратно концентрированного) реагента из упомянутого выше набора (Bio-Rad, США) в 150 мл ДИВ. Каждый раз готовится свежий раствор*. Проявитель готовят растворением 16 г проявителя, взятого из набора (Bio-Rad, США), в 500 мл ДИВ. Хранят при 4 °C. Останавливающий реакцию проявления раствор состоит из 5%-го раствора уксусной кислоты в ДИВ. Консервирующий раствор для геля состоит из смеси 25 % этанола (объем/объем) и 10% (объем/объем) глицерола. Раствор может быть использован многократно. Загрузочный буфер (ЗБ) состоит из 1,5 мл глицерола и 12,5 мг бромфенол голубого (БФГ) в 5 мл ДИВ.
Подготовка материалов и оборудования. 1. Тщательно вымыть большие стеклянные пластинки с моющим средством и мягкой (не царапающей стекло*.) губкой. Промыть водопроводной и ДИВ. Высушить стекла. Промыть чистым (96%) этанолом, протереть чистой хлопковой салфеткой. Стеклянные пластинки меньшего размера начинают мыть сначала смесью КОН/метанол, а далее так же, как и большие. 2. Вырезать пластинки (Gelbond®) по размеру больших стекол, не удаляя покрывающую ее бумагу. 3. Около 0,5 мл ДИВ налить на одну из поверхностей большого стекла и накрыть стекло выре
занной пластинкой гидрофобной стороной к стеклу. Выявление гидрофобной стороны пластинки осуществляется нанесением на нее капли воды: на гидрофобной стороне вода не растекается. Тщательно разгладить пластинку на поверхности стекла (не допускать образования воздушных пузырей) и после этого удалить бумагу. Высушить пластинку с помощью этанола и чистой хлопковой салфетки. Обдуть поверхность подсушенной пластинки струей чистого воздуха. Обдуть поверхность второго (меньшего) стекла. 4. Отмыть 96%-м этанолом пластиковые термоустойчивые полоски, определяющие толщину геля (Spacers). Нанести пальцем (в перчатке) вазелин на край стекла покрытого пластинкой Gelbond®. Ширина вазелиновой полоски ~1 см. Положить на вазелиновый слой спейсеры, покрыть их сверху вазелином и положить меньшее стекло стороной, промытой КОН в этаноле, внутрь. 5. Поместить «сэндвич» из стекол в специальный держатель, компонент ДГГЭ-прибора. Закрутить равномерно зажимы держателя. Все края должны быть идеально ровными и пластинка нигде не должна выступать. Замазать вазелином отверстие между стеклами и нижней частью «сэндвича». Промазать аккуратно вазелином боковые стороны «сэндвича» (там, где вставлены спейсеры). 6. Промазать вазелином нижнюю пластиковую полоску, лежащую в специальном штативе, на которую устанавливают «сэндвич» из стекол, и установить стекла, зажатые в держателе, в этот штатив. Несколько ослабить зажимы держателя и с помощью специальной пластинки (она прилагается к прибору), вставляя ее между стеклами, проверить пространство. При этом нельзя касаться вазелина, который может выступать сбоку, а особенно на дне «сэндвича» из стекол. Зажать зажимы. Если «сэндвич» из стекол окажется не герметичным, то, при заливании в него геля, последний будет просто вытекать. 7. Штатив с «сэндвичем» из стекол должен быть установлен на строго горизонтальной поверхности. При необходимости приготовить два таких «сэндвича» из стекол.
Приготовление пластинок геля с денатурирующим градиентом. 8. Разморозить одну пробирку с АПС. 9. Вставить в шланг для подачи геля из двухкамерного стакана новую иглу. Открыть отверстие, соединяющее камеры стакана, открутив зажим. Промыть обе камеры ДИВ, откачивая ДИВ с помощью перистальтического насоса при скорости ПО мл/мин. Высушить систему, продувая сжатым (чистым1.) воздухом. Закрыть отверстие, соединяющее камеры стакана. 10. Приготовить растворы, содержащие 45%-й раствор мочевина-формамид (низкая концентрация, НК), 60%-й раствор мочевина-формамид (высокая концентрация, ВК) и «закрепитель» геля согласно прописи в табл. 10.1 в 50 мл пластиковых пробирках (подписанных1.). 11. Добавить АПС и ТЕМЕД к низкой и к высокой концентрациям смесей мочевина-формамид (МФ) согласно табл. 10.1 и немедленно приступить к выполнению операций (5 — 8). 12. Перелить раствор МФ высокой концентрации в правую часть двухкамерного стакана, низкой концентрации — в левую. Включить магнитную мешалку. Иглу, соединенную со шлангом, отходящим от двухкамерного стакана и проходящим через перистальтический насос, опустить между стеклами «сэндвича». 13. Открыть отверстие, соединяющее камеры ста-
Таблица 10.1
Порядок смешивания растворов для получения денатурирующего градиента в полиакриламидном геле
Тип раствора МФ Концентрация раствора МФ, % Концентрация раствора акрилам ид-бисакри-ламид, % Объем раствора МФ, мл Объем 10%-го раствора АПС, мкл Объем ТЕМЕД, мкл
НК 45 6 13 78 6
ВК 60 6 13 78 6
Закрепитель 0 8 8 40 11
Схема приготовления образцов для загрузки в гель с денатурирующим градиентом
Объект Объем, мкл
ЗБ 6 х (БФГ/глицерин) ДИВ ПЦР-продукт Вносимый объем, мкл
Образец 5 0 20 25
ДГГЭ-маркер 2 4 6 12
Контроль 2 . 4 6 12
кана, открутив зажим, и немедленно включить перистальтический насос со скоростью 4,0 мл/мин. Внимательно контролировать процесс заполнения пространства между стеклами смесью растворов. Проверить, нет ли утечки растворов} 14. Перекачивание геля занимает 10 мин. Как только растворы будут полностью перекачены между стеклами, сразу же тщательно промыть двухкамерный стакан и трубку ДИВ, перекачивая воду тем же насосом, но со скоростью 10 мл/мин, и высушить сжатым воздухом. После этого иглу заменить новой. 15. Тщательно вымытую, в том числе 96%-м этанолом, и потом высушенную «гребенку», создающую 16 карманов (в которые будут вноситься продукты ПЦР), вставить в пространство между стеклами, которое к этому времени должно быть почти полностью заполненно гелем. 16. Внести 10%-й АПС и ТЕМЕД в закрепитель. Быстро набрать этот раствор в шприц объемом 10 мл, насадив при этом новую иглу, и нанести его на поверхность еще не застывшего геля между стеклами под «гребенку», так чтобы гель едва выступал над низким стеклом. Не допускать образования воздушных пузырей как на предыдущем, так и на данном этапе. Тщательно промыть шприц дистиллированной водой. 17. Оставить гель на 1 ч для полимеризации.
Электрофорез ДНК в денатурирующем градиенте геля. 18. Залить свежеприготовленный 0,5 х ТАЕ буфер в «ванну» ДГГЭ-машины до отметки «Fill» (7 л). 19. Установить верхнюю часть ДГГЭ-машины, содержащую блок подключения к электропитанию и др., на «ванну» с буфером и включить прибор (DCode™, США) по крайней мере за 60 мин до начала электрофореза, с тем чтобы буфер успел нагреться до 60 °C. Мешалку под «ванной» и перемешивающий насос пока не включать} 20. После часа полимеризации геля осторожно удалить «гребенку». Вынуть из штатива держатель с затвердевшим гелем между стеклами. Отмыть горячей водой остатки вазелина с поверхностей. Промыть «карманы» в геле 0,5 х ТАЕ буфером с помощью шприца. 21. Отключить ДГГЭ-маши-ну, подождать 1 мин, снять верхнюю часть прибора. Поместить «сэндвич» из стекол с гелем в держатель, находящийся в «ванне» ДГГЭ-машины с горячим буфером. Оба держателя в «ванне» прибора должны быть заполнены «сэндвичами» с гелем или без (холостой вариант). Установить верхнюю часть прибора, содержащую блок подключения к электропитанию, на «ванну» и подсоединить к блоку управления электропитанием. 22. Включить ДГГЭ-машину, включить верхнюю мешалку и мешалку, находящуюся под «ванной» машины (300 об/мин). Оставить в таком состоянии на период подготовки образцов. 23. Подготовить исследуемые образцы ДНК согласно табл. 10.2. Выключить прибор. Выждать 1 мин. Снять верхнюю часть. Внести исследуемые образцы в «карманы» гелевых пластинок. Если образцов много и их вносят во все «карманы» обоих «сэндвичей», то последовательность заполнения карманов второго геля осуществляют в зеркальном порядке относительно первого геля. Не использовать «карманы» 1, 2 и 15, 16. Для внесения образцов ДНК в «карманы» используют не традиционные, а специальные наконечники с удлиненными носиками. Возвратить верхнюю часть прибора на место. 24. Произвести настройку (при необходимости) программы блока питания (16 ч, напряжение 100 В) и самого ДГГЭ-прибора и включить его. Система надежно работает в течение заданного времени и автоматически отключится по его истечении.
Последовательность операций при окрашивании гелей
Номер операции Раствор Время обработки, мин*
1 Фиксирование 30
2 То же 30
3 Окислитель (/ = 23 — 25 °C) 10
4 ДИВ 10
Повторить операцию 4 не менее 9 раз, до тех пор пока исчезнет желтоватый цвет геля!
5 Серебрение (/ = 23 — 25 °C) 30
6 ДИВ 2
7 То же 2
Раствор проявителя должен быть подогрет до 45° С
8 Проявитель 1
9 То же 5
10 » 5**
11 Стоп-раствор 10
12 То же 10
13 Консервирующий раствор Не менее 1 ч
* Все операции выполняются при слабом покачивании на качалке (Heidolph Unimax), 50 об/мин. Операции 8—10 и операции 11 и 12 — при 90 об/мин. При проявлении гелей лотки должны быть накрыты.
** В некоторых случаях эта операция не нужна, если окрашиваются ПЦР-продукты, полученные из чистых культур. В некоторых случаях операции 9 и 10 в сумме занимают 15 мин, если ПЦР-продукты были получены после экстрагирования ДНК из почвы и т.д.
Окрашивание электрофореграммы для выявления полос ДНК. 1. Выключить прибор. Снять верхнюю часть прибора, изъять оба «сэндвича» с гелем из «ванны». Ополоснуть ДИВ. Промыть два стеклянных лотка (20 х 20 см) не менее 20 мин 0,1 н. HNO3, после ополоснуть ДИВ. 2. Удалить стеклянные пластинки и изъять гель вместе с пластиковой подложкой Gelbond®. Провести обрезку геля. Для этого вырезать гель нужной ширины (примерно 17 х 17 см), отрезав дорожки 1, 2 и 15, 16, и обрезать верх и низ (оставив примерно 15,5 см). Если было два геля, пометить фронтальный. Ополоснуть гели ДИВ. После обрезки гелевые пластинки должны легко вмещаться и двигаться в стеклянных лотках. 3. Приготовить окислитель, серебряный краситель и проявитель. Подогреть проявитель до 45 °C. 4. Провести окрашивание геля согласно табл. 10.3. 5. Операции 1—7 следует проводить, используя один лоток для обоих гелей. При этом один гель следует поместить пластиковой подложкой Gelbond® ко дну лотка, а другой — подложкой вверх. 6. Для переворачивания и изымания гелей из растворов пользоваться только пинцетами соответствующей формы. Раствор AgNO3 после использования следует собирать, помня, что раствор содержит тяжелый металл. 7. После операции 13 верхнюю часть геля (при этом Gelbond® подложка находится снизу) покрыть целлофановой пленкой, обрезать ее по размеру геля и поместить гель в сушильный шкаф при 60 °C на сутки. 8. Высушенный гель протереть 96%-м этанолом, его можно хранить неограниченно долго.
Интерпретация результатов ДГГЭ. После электрофореза продуктов ПЦР получают фореграмму ампликонов в виде горизонтальных полос, по ширине соответствующих ширине зубцов использованной «гребенки» и имеющих «толщину», зависящую от концентрации каждого ампликона (см. пример на рис. 10.1). С учетом некоторых особенностей и ограничений, перечисленных во введении, результаты ДГГЕ-анализа могут быть интерпретированы следующим образом.
Формально каждая полоса (ампликон) принадлежит соответствующему виду. Следовательно, подсчет количества полос в каждой дорожке является одной из характеристик микроорганизмов анализируемого образца. Имеет значение и расстояние каждой полосы от начала фореграммы. Это расстояние зависит от нуклеотидного состава ампликона. Расстояние лучше рассчитывать в относительных единицах, т.е. относительно соответствующих контролей, маркеров, которые обычно располагают по краям фореграммы, а иногда еще и в середине. В качестве маркеров используют синтетическую смесь амплифицированных 16S рДНК бактериальных клонов с известными различиями в денатурирующих характеристиках. В реальных исследованиях хорошо зарекомендовали себя маркеры, являющиеся фрагментами 16S рДНК клонов бактерий: Eubacterium halii/^l, Fusobacteriumprausnitzii-like A10, Butyrivibriofibrisolvens-hke Al 1, Eubacteriumplautii-like A27, Clostridium celerecrescens-like A54, Ruminococcus obeum-like A57, Eubacterium formicigenerans-\ike A71 [39] и Bifidobacterium XII. Существенное значение имеет и оптическая плотность полос. Последний показатель отражает концентрацию идентичных ампликонов и фактически количество ДНК (количество клеток) соответствующей бактерии в исходном образце. Плотность полос на ДГГЭ-фореграмме анализируют после сканирования каждой фореграммы и перенесения информации в компьютер. Для анализа плотности полос используют специальную программу Phoretix ID (NonLinear Dynamics Ltd., Newcastle upon Tyne, UK).
В настоящее время для анализа сложных фореграмм, получаемых в результате анализа природных образцов, используют комплекс специальных компьютерных программ, совмещенных со статистическим анализом результатов. В качестве примера такого комплексного подхода можно рекомендовать кластерный анализ, дискриминантный анализ (SAS Institute, Inc., Cary, USA), индекс Шенона и др.
10.4. ВЫЯВЛЕНИЕ И ОПРЕДЕЛЕНИЕ МИКРООРГАНИЗМОВ FISH-МЕТОДОМ
Для выявления и определения микроорганизмов в природных образцах и после выращивания их на средах разработан метод гибридизации флуоресцирующих олигомерных нуклеотидных последовательностей с нуклеиновыми кислотами целых клеток — fluorescent in situ hybridization (FISH).
Традиционно для выявления присутствия какого-либо микроорганизма в той или иной природной среде обитания или в накопительной культуре нужно было выделить этот микроорганизм в чистую культуру и провести его идентификацию. Позднее были разработаны иммунофлуоресцентные методы (ИФМ), позволяющие выявить интересующий исследователя организм без выделения в чистую культуру. В основе любых модификаций ИФМ используется взаимодействие антиген — антитело, которое соединено с флуоресцирующим красителем. Специфичность реакции антиген — антитело высока, поэтому и вероятность выявления искомого микроорганизма в природных образцах или накопительных культурах тоже высока. Недостатком являются относительно долгая процедура приготовления сывороток с флуоресцирующими красителями, существование перекрестных реакций, а самое главное — «удаленность» реак
ции антиген—антитело от генетического кода исследуемого организма. И хотя ИФМ используются широко, в настоящее время получают преимущество методы, основанные на идентификации специфических для конкретного организма нуклеотидных последовательностей ДНК (рРНК). К таким методам относятся уже упомянутый ДГГЭ, клонирование и секвенс специфических последовательностей ДНК. Однако и эти методы тоже имеют свои недостатки, и главный из них — использование ПЦР, результаты которой пока еще не могут быть точно «конвертированы» в численность конкретного микроорганизма. Недостатков ИФМ и некоторых молекулярно-биологических методов лишен метод, основанный на взаимодействии окрашенных флуоресцентно олигонук-леотидных проб с соответствующими участками 16S рРНК целых клеток (FISH). Более того, с помощью этого метода можно учитывать даже не культивируемые на средах микроорганизмы. Из названия метода ясно, что этот метод предполагает использование флуоресцентной микроскопии, поэтому возможно его применение для количественного учета клеток микроорганизмов и метода проточной флуороцитометрии (ПФЦМ). Естественно, как и любой другой метод, FISH имеет свои особенности. Например, его чувствительность зависит от типа используемого для исследований образца, примененной молекулярной пробы, вида красителя, эффективности окрашивания (связывания) пробы с красителем, условий проведения гибридизации и др.
Для использования FISH-метода необходимо иметь олигонуклеотидные пробы, которые могут быть синтезированы, если исследователь знает последовательности нуклеотидов в 16S рДНК. Полезная информация для создания соответствующих проб может быть получена в базе данных «Ribosomal Database Project»*. Ниже приводится достаточно подробная методика выявления и учета бактерий в образцах, в частности для выявления Ruminococcus obeum-подобных бактерий с помощью флуоресцентной микроскопии. Как отмечалось, условия гибридизации для разных микроорганизмов должны подбираться индивидуально.
Исследуемые образцы должны быть подготовлены, как и в случае традиционного учета микроорганизмов с использованием флуоресцентного микроскопа, т.е. должна быть проведена десорбция клеток, если, например, образец содержит какую-либо твердую фракцию, подобрано оптимальное разведение и т.д. На поверхность предметных стекол, предварительно покрытых тонким слоем желатины, наносится определенный объем исследуемого образца. Стекло с образцом высушивают 20 мин при 45 °C. Подсушенные препараты подвергают дегидратации, погружая препарат на 2 — 3 мин в ряд с увеличивающимися концентрациями (градиент) растворов этанола от 50 до 75 % и далее до 96%. После этого на препарат наносят 10 мкл «гибридизационного» буфера (0,9 М NaCl; 20 мМ трис-НС1; pH 7,5; 0,1 %-й додецилсульфата натрия, ДСН). Используемый объем гибридизационного буфера должен содержать (для данной конкретной методики и с учетом используемого объема), например, 3 нг СуЗ-меченной Urobe63 пробы на каждый микролитр или 5 нг флуоресцеинизотиоционат (ФИТЦ)-меченной Егес482 пробы на каждый микролитр. Затем препарат помещают в термостат {предотвращая его высыхание'.) и инкубируют при 50 °C в течение 3 ч в темноте. После процесса гибридизации препараты промывают с покачиванием, помещая на 10 — 20 мин в 50 мл гибридизационного буфера, но без SDS.
Для того чтобы помимо выявления и учета искомого организма провести учет и тотального количества клеток в исследуемом природном образце, в промывочный бу
* Maidak B.L. et al. The RDP-II // Nucl. Acids Res. 2001. - V. 29. - P. 173- 174.
фер можно добавить флуоресцентный краситель DAPI в концентрации 100 нг/мл. После промывачного буфера препараты следует промыть в воде и немедленно высушить на воздухе. Учет клеток с помошью флуоресцентного микроскопа проводят визуально или с помощью цифровой камеры и компьютера осуществляют имидж-анализ полученных снимков. Расчет количества клеток производят, как и при традиционном учете клеток, под микроскопом. Приготовленные препараты некоторое время можно хранить при -20 °C.
10.5. ПРИМЕНЕНИЕ МИКРОЧИПОВ В МИКРОБИОЛОГИЧЕСКИХ ИССЛЕДОВАНИЯХ
Высокоэффективное и быстрое выявление микроорганизмов и их идентификация в природных образцах — одна из основных задач практической микробиологии. Другое важнейшее направление — выявление мутаций в геномах организмов и тестирование экспрессии генов. При решении этих задач все чаще применяются методы молекулярной биологии. Наибольшее значение такие методы приобретают в случаях, когда требуется в кратчайшие сроки проанализировать образец на присутствие болезнетворного агента, например для ограничения распространения высококонтагиозных заболеваний, либо если диагностика классическими культуральными методами трудоемка и занимает продолжительное время.
Иммунно-флуоресцентный анализ (ИФА) и FISH-метод (см. 10.4) позволяют специфично выявлять и учитывать клетки, содержащиеся в исследуемом образце. Однако ограничение обоих подходов заключается в возможности выявления микроорганизмов только того вида, к которым были приготовлены иммунные или ДНК-пробы (зонды). Для выявления клеток, принадлежащих к разным таксономическим единицам в одном и том же образце, молекулярные зоны (помимо специфичности к разным микроорганизмам) должны нести различные метки, в противном случае их нельзя будет различить. Таким образом, первым лимитирующим фактором широкого использования FISH является число доступных красителей, вторым — проникновение молекулярной пробы в клетку и процесс связывания пробы с комплементарным участком нуклеиновой кислоты внутри клетки, третьим, не менее важным, — макроскопические размеры используемых стекол, а следовательно, большие затраты труда и дорогостоящих реагентов и др.
Одного из первых двух недостатков лишен метод точечной гибридизации (dot-blot), когда на специальной подложке (например, фильтре) закрепляют молекулярную пробу (олигонуклеотидную последовательность, часто радиоактивно меченную), принадлежащую искомому исследователем микроорганизму. После этого на фильтр наносят изолированный из образца раствор тотальной нуклеиновой кислоты (НК). После гибридизации и удаления несвязавшейся НК определяют, была ли в исследованной пробе НК, принадлежащая искомому организму, или нет. Однако этот метод, так же как и FISH, имеет недостаток, связанный с его макроскопичностью и, следовательно, высокой трудоемкостью.
Многих недостатков методов dot-blot и FISH для анализа образцов на наличие микроорганизмов и вирусов удалось избежать при использовании технологии биологических микрочипов (слово «чип» с английского языка переводится как «кристалл» или «микросхема»). Метод биологических микрочипов основан, как и множество других методов, на способности комплементарных одноцепочечных молекул нуклеиновых кислот к гибридизации. Гибридизация — это процесс, в результате которого две комплементарные здноцепочечные нити нуклеиновой кислоты образуют стабильную двуцепочечную спираль. Реакция гибридизации может происходить между двумя комплементарными ни
тями (молекулами) в растворе или между молекулами в растворе и комплементарными молекулами, иммобилизованными на твердой подложке.
Применение гибридизационного анализа на олиго нуклеотидных микрочипах не только упрощает процедуру исследования, но и предоставляет принципиально новые возможности для одновременной идентификации и изучения целого ряда генетических локусов микроорганизмов и вирусов. Технология биологических микрочипов позволяет автоматизировать процесс идентификации биологических объектов.
Биологический микрочип — это некое упорядоченное множество микроскопических ячеек, каждая из которых содержит индивидуальный зонд и является, таким образом, самостоятельной реакционной единицей. Известно несколько принципиально разных подходов к изготовлению микрочипов (DNA Microarrays, 1999). В Институте молекулярной биологии РАН традиционно разрабатываются так называемые трехмерные биочипы, каждая ячейка которых представляет собой микроскопическую каплю полиакриламидного геля (около 200 мкм толщиной), в которой равномерно иммобилизованы ДНК-пробы. Трехмерная пористая структура геля позволяет иммобилизовать пробы в гораздо большем количестве по сравнению с плоской поверхностью, а значит, повысить чувствительность анализа. В качестве ностителя для ячеек используются обычные предметные стекла, т.е. твердые, непористые подложки.
ДНК-пробы — искусственно синтезированные олигонуклеотиды, состоящие обычно из 15 — 20 оснований. Каждая из таких проб полностью соответствует (комплементарна) участку гена одного определенного микроорганизма или вируса, что и позволяет проводить их идентификацию. Чем больше таких проб на биочипе, тем больший спектр микроорганизмов или функций можно проанализировать на их наличие или отсутствие в образце.
Для осуществления анализа на микрочипе НК, содержащаяся в исследуемом образце, должна быть соответствующим образом подготовлена. В случае анализа геномной ДНК применяют подход ПЦР (полимеразная цепная реакция) для того, чтобы увеличить содержание в образце интересующего исследователя фрагмента ДНК. Если же для анализа выбирается РНК, обычно применяют обратную транскрипцию с последующей амплификацией (ОТ-ПЦР). При этом праймеры, используемые в ПЦР, несут метку (чаще флуоресцентную), что позволяет полностью пометить все образующиеся ДНК-мишени. Альтернативным подходом является окрашивание ДНК после ПЦР специальными красителями. В настоящее время имеется возможность готовить чипы с разноцветными флуоресцирующими красителями.
После подготовки исследуемого материала необходимо провести гибридизацию ДНК-мишеней с пробами, иммобилизованными на микрочипе. Процесс гибридизации проводят в гибридизационной камере. Время инкубации, температуру и влажность в камере подбирают экспериментально, осуществляя процесс при комнатной или при повышенной температуре, например при 62 °C, с формамидом или без и т.д. Причем реакция идет лишь с теми пробами, которые соответствуют присутствующей в образце НК. По истечении этого времени препарат промывают и далее наблюдают свечение.
Поскольку НК, содержащиеся в образце, «переведены» в форму сравнительно коротких ДНК-мишеней, несущих флуоресцентную метку, ячейки, где прошла реакция, начинают светиться. Исследователю остается лишь учесть, какие ячейки засветились, и сделать вывод о наличии в образце тех или иных микроорганизмов или вирусов. При анализе (визуальном под флуоресцентным микроскопом или после микросканирования и использования соответствующей программы — имидж-анализа) будут выявляться те светящиеся микропятна, где произошло связывание мишени с пробой. Сигналы рассматривают как значимые, если их свечение превышает некий пороговый уровень, который устанавливается в предварительных экспериментах.
Для изготовления биологических микрочипов должна быть проведена большая подготовительная работа. Первоначально формулируется задача исследования, очерчивается спектр объектов, идентификация которых представляет интерес. После этого проводится выбор генетических мишеней — участков внутри геномов микроорганизмов,
последовательности которых характерны лишь для представителей того или иного вида или рода. Можно избрать наиболее консервативные последовательности в генах, ответственных за синтез, например 16S— 18S или 23S —28S рДНК и, следовательно, в дальнейшем изготовленный чип будет распознавать определенную филогенетическую группу микроорганизмов, поэтому его можно назвать «филочипом». Можно избрать последовательности, которые характеризуют искомый организм по его некоторым, интересующим исследователя функциям, например по гену, ответственному за синтез веро-токсина у бактерии Е. coli 0157: Н7. Тогда изготовленный чип следует называть «функциональным».
Избранные нуклеотидные последовательности в дальнейшем используются для конструирования праймеров. Основное требование, предъявляемое к праймерам, — их специфичность только к представителям одного таксона (вида или рода). Для проверки такой специфичности проводят так называемый BLAST-анализ, т.е. сравнение избранных последовательностей с последовательностями других микроорганизмов и вирусов. Если обнаруживается гомология выбранного праймера с участками геномов других организмов, такой праймер бракуется и процесс выбора повторяется.
После этого осуществляют синтез праймеров, введение в них флуоресцентной метки, а также синтез проб для иммобилизации.
Олигонуклеотидные пробы обычно имеют такие характеристики, как название (условное, дается разработчиком), специфичность, например универсальная (т.е. для всех бактерий), или группоспецифичная (например, для сс-подгруппы протеобакте-рий), или функционально-специфичная и т.д., секвенс пробы в направлении от 5'- к 3'- концу, 16S рДНК связывающий сайт (обычно местоположение указывается по местоположению в гене 16S рДНК Е. coli), процентный состав ГЦ-пробы, а также номер несоответствия с мишенью, в том случае если такое несоответствие имеется.
Дальнейший этап подготовки микрочипа — иммобилизация синтезированных ДНК-проб, по завершении которой чип формально можно считать готовым. Однако с этого момента исследователь приступает к его тестированию, после чего могут быть изменены последовательности проб, схема подготовки материала, условия проведения гибридизации, т.е. формируется окончательный вариант метода анализа.
С помощью технологии микрочипов, при наличии готовых чипов и соответствующего оборудования, квалифицированный специалист может провести анализ достаточно большого количества образцов всего лишь в течение нескольких часов.
Глава 11
ХРАНЕНИЕ МИКРООРГАНИЗМОВ
Необходимым условием успешной работы с микроорганизмами является правильное поддержание их в целях сохранения не только жизнеспособности клеток, но и таксономических, а также любых других, важных для исследователя свойств. Микроорганизмы разных систематических групп и даже различные штаммы и варианты одного вида отличаются чувствительностью к способу их хранения. Поэтому общего метода, одинаково пригодного для хранения многочисленных и разнообразных групп микроорганизмов, пока не существует. В крупных коллекциях разные группы микроорганизмов сохраняются индивидуальными методами. Кроме того, чтобы исключить возможность потери микроорганизма, каждый штамм сохраняется не одним, а несколькими способами.
Известно, что при хранении микроорганизмов в результате их популяционной изменчивости, обусловленной гетерогенностью популяции, изменяются
их физиолого-биохимические особенности и, в частности, снижается антибиотическая или ферментативная активность. В процессе хранения, так же как и при культивировании бактерий в различных условиях, может происходить диссоциация, т.е. расщепление однородной популяции бактерий на варианты, различающиеся генетическими, физиолого-биохимическими и морфологическими свойствами. Поэтому в некоторых случаях целесообразно подбирать условия хранения, оптимальные для определенного варианта, например для варианта, обладающего наибольшей биосинтетической активностью.
К числу наиболее распространенных способов хранения микроорганизмов относятся периодические пересевы на свежие питательные среды, сохранение культур на питательной среде под вазелиновым маслом, хранение клеток в лиофилизированном состоянии. Значительно реже микроорганизмы сохраняют при низких или сверхнизких температурах, в дистиллированной воде или 1%-м растворе хлористого натрия, а также на адсорбентах в высушенном состоянии. Выбор метода хранения во многом зависит от целей, для которых используются микроорганизмы, а также от имеющегося в распоряжении исследователя оборудования.
11.1. ПЕРИОДИЧЕСКИЕ ПЕРЕСЕВЫ НА ПИТАТЕЛЬНЫЕ СРЕДЫ
Этот способ был одним из первых приемов длительного сохранения микроорганизмов в лабораторных условиях и до настоящего времени широко используется в практике микробиологических работ. Аэробные микроорганизмы пересевают чаще всего на поверхность скошенной агаризованной среды, микро-аэрофилы — в полужидкую среду, содержащую 0,2 —0,3 % агара, анаэробы — в толщу плотной среды или в жидкую среду, соблюдая принципы техники Р. Хан-гейта. Культуры пересевают на свежие среды в 2 пробирки (колбы). В дальнейшем микроорганизмы из одной пробирки используют для работы, а культуру во второй пробирке оставляют для хранения и следующего пересева.
Частота пересева на свежую среду различных микроорганизмов неодинакова и в большей степени определяется их свойствами. Многие микроорганизмы можно пересевать один раз в 1 —2 мес, хотя другие, например молочнокислые бактерии, нуждаются в более частых пересевах. Допустимые сроки пересева некоторых микроорганизмов приведены в табл. 11.1. Хранение культур в холодильнике при 4 — 6 °C позволяет увеличить время между пересевами.
Таблица 11.1
Условия хранения и допустимые сроки периодических пересевов некоторых гетеротрофных микроорганизмов
Микроорганизм Питательная среда Температура хранения, °C Число пере- : севов в год j
Acetobacter aceti A. xylinum 6 °Б сусло + 6 % этанола 18-20 12
Azorobacter chroococcum A. vinelandii Агаризованная среда Эшби 18-20 б
Окончание табл. 11.1
Микроорганизм Питательная среда Температура хранения, °C Число пересевов в год
Bacillus cereus В. polymyxa В. subtilis Картофельный агар 18-20 4
Caulobacter sp. Агаризованная среда с дрожжевым автолизатом 4-6 2-3
Citrobacterfreundii МПА 18-20 6
Escherichia coli МПА 18-20 12
МПБ + 0,2 % агара 18-20 6-8
' Lactobacillus casei Снятое молоко 18-20 18-20
Lactobacillus delbrueckii Leuconostoc mesenteroides 8—10 °Б сусло + + дробина + мел 18-20 18-20
Micrococcus luteus M. varians МПА 18-20 6
Mycobacterium flavescens M. fortuitum \M. phlei МПА+3°Б сусло (1:1) 18-20 3-4
M. sm egm at is 4-6 2-3
Nocardia sp. 18-20 3-4
Pseudomonas aeruginosa P. fluorescens P. putida МПА 18-20 6-8
Propionibacterium freudenreichii Среды с кукурузным экстрактом или лактатом 4-6 4-5
Proteus vulgaris P. mirabilis МПА 18-20 9
Staphylococcus aureus МПА 18-20 6
Streptomyces anulatus var. griseus S. lavendulae S. violaceus Агаризованная среда Чапека Овсяный агар 18—20 4
Saccharonryces cerevisiae Candida guilliermondii C. utilis Rhodotorula glutinis’ R. rubra Schizosaccharomyces pombe Saccharomycodes ludwigii 4 — 5 ° Б сусло + агар 18—20 4-5 6 6 6 6 4-5 4-5
Поддержание культур микроорганизмов регулярными пересевами имеет ряд существенных недостатков. Основной из них — возможная утрата некоторых морфологических и физиологических признаков. Кроме того, частые пересевы нередко снижают биохимическую активность культур, повышают опасность инфицирования ее посторонними микроорганизмами. При частых пересевах, особенно на жидкие среды, велика вероятность возникновения спонтанных мутантов и их селекция.
11.2. ХРАНЕНИЕ ПОД МИНЕРАЛЬНЫМ МАСЛОМ
Хранение под минеральным маслом широко используется для бактерий и микроскопических грибов. Этот метод обеспечивает довольно длительное сохранение жизнеспособности и стабильности таксономических и других признаков у микроорганизмов различных систематических групп. Масло предотвращает высыхание среды, замедляет процессы метаболизма и позволяет увеличить время между пересевами.
Микроорганизмы выращивают на благоприятной агаризованной питательной среде; аэробные микроорганизмы — на поверхности коротко скошенной (под углом 45°) среды, микроаэрофилы и факультативные анаэробы — в полужидкой среде, анаэробы — в толще среды (посев уколом или в расплавленную среду с перемешиванием). Хорошо развившиеся культуры заливают маслом. Как правило, аспорогенные бактерии заливают через 2 — 7 сут после посева в зависимости от скорости роста микроорганизма, бациллы и актиномицеты — в стадии сформировавшихся покоящихся форм. Дрожжи рекомендуется заливать маслом через 4— 10, мицелиальные грибы — через 7 — 12 сут. Наиболее пригодно для заливки культур микроорганизмов высокоочищенное медицинское вазелиновое масло с плотностью 0,8 — 0,9. Предварительно масло стерилизуют 1 ч в автоклаве при 1 ати, а затем для удаления влаги прогревают в течение 1 ч в сушильном шкафу при температуре не выше 150 °C или оставляют на двое-трое суток при комнатной температуре. Культуры заливают маслом так, чтобы его слой не превышал 1 см над средой или верхним краем скошенной среды, и сохраняют при комнатной температуре либо в холодильнике при 4 — 6 °C.
Для пересева клетки из-под масла отбирают петлей и, удалив излишек масла проведением петли по стенке пробирки, переносят на свежую питательную среду. Рекомендуется использовать среду того же состава, на которой культуру хранили. Многие микроорганизмы в первом пассаже после хранения под маслом развиваются медленнее, однако при последующих пересевах скорость их роста восстанавливается.
Метод хранения микроорганизмов под вазелиновым маслом прост, удобен в обращении, может быть использован в любой лаборатории. К недостаткам его можно отнести возможность инфицирования помещения микроорганизмами за счет разбрызгивания масла при обжигании петли, а также необходимость специальной очистки посуды от масла.
11.3. ХРАНЕНИЕ В ЛИОФИЛИЗИРОВАННОМ СОСТОЯНИИ
Хранение лиофильно высушенных клеток — широко распространенный метод длительного сохранения микроорганизмов. Лиофилизацией называют процесс высушивания под вакуумом замороженных клеток. Лиофильно-высушенные клетки сохраняют в ампулах, запаянных под вакуумом или в струе стерильного газа (чаще всего азота). Применение этого метода позволяет в течение 10 — 20 лет и более сохранить без заметных изменений жизнеспособность, морфологические, культуральные, физиологические свойства, а также биохимическую активность клеток.
Микроорганизмы, подлежащие лиофилизации, выращивают в оптимальных условиях до начала стационарной фазы роста или окончания формирования покоящихся форм. Затем клетки, или соответственно, покоящиеся формы суспендируют в специальных жидкостях, получивших название защитных сред. В состав защитных сред входят различные вещества, которые предохраняют клетки от повреждений в период замораживания и высушивания.
Ниже приведены рецепты некоторых защитных сред, используемых при лиофилизации клеток различных микроорганизмов:
• желатин — 1 г, сахароза — 10 г, вода дистиллированная — 100 мл;
• молоко обезжиренное — 100 мл, глюкоза — 7 г;
Рис. 11.1. Последовательность {1—6) вскрытия двухкамерной ампулы с лиофильно высушенными клетками микроорганизмов (а) и различных однокамерных ампул (б)
• молоко обезжиренное — 100 мл, NH4C1 — 0,5 г, аскорбиновая кислота — 0,5 г, тиомочевина — 0,5 г;
• лошадиная сыворотка — 75 мл, мясной бульон — 25 мл, глюкоза — 7,5 г.
Для успешной лиофилизации плотность микроорганизмов в защитной среде должна быть как можно более высокой: 109— Ю10 клеток в 1 мл. Полученную суспензию разливают в ампулы из нейтрального стекла по 0,5 —1,0 мл, замораживают при температуре от -20 до -70 °C, затем высушивают и запаивают под вакуумом. Остаточная влажность лиофилизированных клеток колеблется от I до 6 % и определяется составом защитной среды и режимом высушивания. В различных лабораториях режимы замораживания и высушивания заметно варьируют и во многом зависят от имеющегося оборудования. Ампулы с лио-фильно высушенными клетками рекомендуется сохранять в темноте при температуре 4 —6 °C. Хранение при более высокой температуре, особенно превышающей 25 —30 °C, заметно снижает выживаемость клеток.
Для реактивации к лиофилизированным клеткам добавляют по каплям стерильную дистиллированную или водопроводную воду в количестве 0,5 — 1,0 мл. После регидратации (от 10 мин до 2 ч) клетки высевают на более богатые питательные среды (рис. 11.1).
Лиофилизацию широко применяют для длительного хранения различных микроорганизмов. Тем не менее этот метод нельзя считать универсальным. Следует отметить, что к лиофилизации более устойчивы грамположитель-ные, чем грамотрицательные бактерии. Очень плохо переносят ее фототроф-ные и хемолитотрофные бактерии, микоплазмы, многие облигатные анаэробы. Выживаемость спор после лиофилизации заметно выше, чем вегетативных клеток. Дрожжи с мелкими клетками и аскоспорами родов Pichia и Lipomyces выдерживают лиофилизацию лучше, чем слабоспорулирующие или неспорулирующие крупные клетки дрожжей родов Saccharomyces, Kluyveromyces, Rhodotorula.
11.4. ХРАНЕНИЕ ПРИ НИЗКИХ И СВЕРХНИЗКИХ ТЕМПЕРАТУРАХ
Хранение микроорганизмов в замороженном состоянии при низких и сверхнизких температурах по сравнению с другими методами характеризуется наибольшей универсальностью. Однако этот метод требует специального оборудования и большой осторожности в работе с жидким азотом, поэтому используется лишь для сохранения микроорганизмов, не выдерживающих лиофилизацию.
Клетки замораживают в широком диапазоне температур (от -10 до -196 °C) и при различных скоростях замораживания. Для защиты от повреждающего действия низких температур клетки предварительно суспендируют в растворах криопротекторов. Чаще других применяют 10 — 20%-й раствор глицерина, 7 — 10%-й раствор диметилсульфоксида или 20%-й раствор сахарозы. Суспензию клеток с высокой плотностью (109—1010 клеток в 1 мл) разливают в ампулы или флаконы с завинчивающейся крышкой. Ампулы запаивают и помещают в холодильник с температурой -70 °C (скорость охлаждения 1 °С/с), а затем переносят в жидкий азот, где и сохраняют при -196 °C. Оттаивание замороженных клеток должно быть как можно более быстрым. Поэтому ампулы погружа
ют на 2 мин в водяную баню с температурой 35 — 45 °C. Клетки из ампул высевают на богатые питательные среды.
При температуре хранения от -20 до -40 °C хорошо выживают немногие микроорганизмы; значительно эффективнее хранение при -70 °C в твердой углекислоте и особенно в условиях сверхнизких температур: при -196 °C (жидкий азот) или при -210 °C (газовая фаза жидкого азота).
11.5. ХРАНЕНИЕ В ГЛИЦЕРОЛЕ
Одним из самых удобных методов хранения микроорганизмов является их содержание при низких температурах (-20 °C) в растворах глицерола, который служит криопротектором. Данный способ широко распространен, однако не все микроорганизмы выдерживают такую обработку. Поэтому считается, что для клеток, подвергаемых замораживанию в глицероле, необходимы предварительные эксперименты по определению степени их выживаемости. Наибольшее распространение этот способ консервации микроорганизмов получил при хранении суспензий различных спор.
Для проведения экспериментов по хранению готовят 50%-й раствор глицерола в дистиллированной воде и стерилизуют его при I ати в течение 30 мин. Клетки из выросших культур (обычно из середины экспоненциальной фазы роста), предназначенные для хранения, смешивают в пропорции 1:1с 50%-м глицеролом, перемешивают и ставят в морозильник. Хранить суспензии удобно в стерильных пробирках Эппендорф, а для их приготовления использовать автоматические пипетки со стерильными наконечниками и готовить смеси в ламинарном шкафу. Из сохраняемых клеток периодически (2 — 6 раз в год) отбирают пробы для проверки на выживаемость. В зависимости от результатов проверки рассчитывают схемы консервации той или иной культуры и периодичности их пересева.
Для культур микроорганизмов с твердых сред перед смешиванием с глицеролом готовят суспензии в жидкой среде или в оптимальном для хранения буферном растворе (проверяется экспериментально). Можно также суспендировать клетки с твердых сред непосредственно в 25%-м стерильном глицероле.
11.6. ХРАНЕНИЕ В ДИСТИЛЛИРОВАННОЙ ВОДЕ ИЛИ 1%-м РАСТВОРЕ ХЛОРИДА НАТРИЯ
Этот метод не требует специального оборудования и доступен любому экспериментатору.
Микроорганизмы предварительно выращивают в оптимальных условиях, при необходимости центрифугируют, после чего клетки суспендируют в дистиллированной воде или 1%-м растворе хлорида натрия. Успешному сохранению клеток способствует высокая плотность суспензии: не менее 108 — 109 клеток в 1 мл.
Суспензию разливают в стерильные пробирки или флаконы и сохраняют в холодильнике или при комнатной температуре. Рекомендуется оставлять на
Допустимые сроки хранения (мес) микроорганизмов в холодильнике (4 — 6 °C) в дистиллированной воде и 1%-м растворе NaCl
Микроорганизм Дистиллированная вода 1%-й раствор NaCl
Arthrobacter globiformis 6 6
Escherichia coli 12 12
Gluconobacter oxydans 12 6
Bacillus subtilis 12 12
Micrococcus varians 12 12
Pseudomonas aeruginosa 6 6
Micrococcus luteus 6 6
Serratia marcescens 12 12
хранение клетки начала стационарной фазы роста культуры или сформировавшиеся покоящиеся формы — споры, цисты.
Допустимые сроки хранения некоторых микроорганизмов этими методами в холодильнике при 4—6 °C приведены в табл. 11.2.
11.7. ХРАНЕНИЕ В ВЫСУШЕННОМ СОСТОЯНИИ
НА АДСОРБЕНТАХ
Этот метод применяют главным образом для актиномицетов, микроскопических грибов и анаэробных бактерий, образующих споры. В качестве адсорбентов используют почву, кварцевый песок, силикагель, вату, фильтровальную бумагу. Разработанной стандартной техники этот способ не имеет. В самом общем виде он сводится к тому, что стерильный адсорбент, помещенный в ампулы, смешивают с густой суспензией клеток и высушивают под вакуумом или при комнатной температуре. Имеются данные, что у актиномицетов после хранения в почве или кварцевом песке восстанавливаются некоторые таксономические признаки (окраска воздушного и субстратного мицелия), которые были утрачены в процессе длительного культивирования в лаборатории.
11.8. ОЦЕНКА ЖИЗНЕСПОСОБНОСТИ МИКРООРГАНИЗМОВ ПОСЛЕ
ДЛИТЕЛЬНОГО ХРАНЕНИЯ
Жизнеспособность микроорганизмов после различных сроков хранения определяют путем высева их на богатые питательные среды с последующим подсчетом выросших колоний. Процент выживаемости микроорганизмов находят по отношению числа сохранившихся клеток к первоначальному числу жизнеспособных клеток (до начала хранения), принятому за 100 %.
Глава 12
ОПРЕДЕЛЕНИЕ ХИМИЧЕСКОГО СОСТАВА КЛЕТОК МИКРООРГАНИЗМОВ
12.1. ОПРЕДЕЛЕНИЕ БЕЛКА
Содержание белка можно измерять непосредственно в нерассеивающих (истинных) растворах или после проведения гидролиза клеток и отдельных клеточных структур. Для каждого эксперимента выбирают методы, которые удовлетворяют по скорости, точности и удобству измерений. При определении строят калибровочные кривые, анализируя известное количество белка в тех же условиях, что и опытные пробы. Если перед проведением определения содержания белка требуется гидролиз материала, необходимо в тех же условиях провести и гидролиз стандарта, используемого для построения калибровочного графика. Во всех случаях необходимо учитывать только те значения графика, которые лежат в области 0,1 —0,7 оптической единицы в кювете с длиной оптического пути 10 мм, при этом значения поглощения опытных образцов должны находиться в пределах полученных экспериментальных точек калибровочного графика. В необходимых случаях проводят кратные разведения исходного материала. Калибровочные кривые строят с использованием растворов альбуминов — бычьего сывороточного, яичного или из сыворотки человека.
12.1.1. Измерение поглощения при длине волны 280 нм
Преимущество этого наиболее простого и быстрого метода измерения количества белка в содержащих его нерассеивающих растворах — быстрота и неизменность пробы до и после анализа. Метод применим для измерения концентраций белка и в растворах смесей белков. К недостаткам следует отнести то, что метод не является строго количественным, так как основан на поглощении тирозиновых, фенилаланиновых и триптофановых остатков. Вследствие этого белки отличаются коэффициентами поглощения. Если белок не содержит вышеуказанных аминокислот, то он не будет поглощать при 280 нм.
Для большинства рутинных измерений можно с большой вероятностью допустить, что поглощение при длине волны (X) 280 нм раствором белка с концентрацией 1 мг/мл даст значение оптической плотности Л28о около 1 единицы в кювете с рабочим расстоянием 10 мм. Более скрупулезные исследования показывают, однако, что поглощение в растворах с одинаковой концентрацией белков может значительно различаться (табл. 12.1).
Область чувствительности метода лежит в пределах от 0,2 до 2,0 мг белка/мл, а в микрокюветах возможно измерение -0,05 мг белка (0,1 мл пробы). Для измерения необходим спектрофотометр, позволяющий регистрировать ближнюю ультрафиолетовую область спектра, кварцевые кюветы и пипетки для переноса образцов.
При измерении выставляют значение спектрофотометра при X = 280 нм на 0 поглощения в кювете с экспериментальным буферным раствором, затем в той
Различия в поглощении растворами некоторых белков
Белок 4г80> мг/мл Белок Z^gg, МГ/МЛ
Бычий сывороточный альбумин (БСА) 0,70 у-Глобулин 1,38
Рибонуклеаза А 0,77 Трипсин 1,60
Яичный альбумин 0,79 Хемотрипсин 2,02
Энтеротоксин 1,33 а-Амилаза 2,42
же или аналогичной кювете измеряют поглощение белком в таком же буфере. При измерении с помощью двухлучевого спектрофотометра устанавливают значение прибора на 0 при длине волны X = 280 нм и измеряют образец против контроля с соответствующим буферным раствором.
Стеклянные и пластиковые кюветы сами по себе значительно поглощают УФ-лучи, поэтому при измерении вышеописанным методом их не используют. Если поглощение слишком велико, можно использовать кюветы с более короткими рабочими расстояниями, когда разбавление пробы нежелательно.
Оптический метод широко используется для быстрого неразрушающего определения концентрации белков, например при колоночной хроматографии белков. Однако не любое поглощение лучей с длиной волны 280 нм можно принимать за белковое, так как значительный вклад в абсорбцию могут вносить присутствующие в пробе нуклеиновые кислоты (максимум их поглощения при X = 260 нм). Если измеряемый образец сильно загрязнен нуклеиновыми кислотами, то более точно концентрация белка вычисляется по формуле, учитывающей вклад абсорбции при 260 нм:
концентрация белка (мг/мл) = 1,5 Л28о - 0,75 ^260-
Результаты эксперимента показывают, что если величина Л28о _ Ф«) приблизительно равна 2,0, то содержание нуклеиновых кислот в исследуемой пробе незначительно. При высоком содержании нуклеиновых кислот гораздо более точным является определение белка по Брэдфорд.
12.1.2. Определение белка по Брэдфорд
Это быстрый, и удобный метод измерения концентрации растворенных белков, основанный на количественном связывании белков с красителем. Он неприменим для измерения количества белка в рассеивающих образцах. Хотя число соединений, мешающих определению белка этим методом, сравнительно невелико, краситель в большей или меньшей степени реагирует с различными очищенными белками. Поэтому концентрацию различных по составу белков определяют лишь количественно до некоторой степени. К преимуществам метода можно отнести быстроту определения (10 мин) и высокую чувствительность, к недостаткам — варьирование результатов при определении концентраций различных белков и необратимое денатурирование порции белка, пошедшей на определение.
Определение проводят с помощью спектрофотометра или ФЭК (длина волны максимума поглощения 595 нм). Для работы потребуются кюветы (можно пластиковые), пипетки, пробирки, штатив для пробирок. В качестве стандарта применяют бычий сывороточный альбумин (1 мг/мл) или яичный альбумин, который дает более точные результаты.
Стандартный исходный раствор содержит 350 мг краски (Serva Blue G или Brilliant Blue R, Sigma); 100 мл 95%-го этанола и 200 мл 85%-й ортофосфорной кислоты. Данный раствор стабилен при комнатной температуре. Рабочий раствор состоит из 30 мл стандартного раствора; 15 мл 95%-го этанола, 30 мл 85%-й ортофосфорной кислоты и 425 мл дистиллированной воды. Рабочий раствор пропускается через фильтр № 1 (Whatman). Он хранится при комнатной температуре в затемненной бутыли в течение нескольких недель с периодическим фильтрованием.
Процедура определения. В пробу белка (100 мкл максимум) добавляют буфер до 100 мкл (если требуется) и 1 мл рабочего раствора краски, перемешивают и после 2 мин инкубации определяют оптическую плотность раствора при 595 нм (Л595). Оптическая плотность может измениться через 1 ч с момента добавления краски.
Стандартную кривую строят по растворам яичного альбумина (или бычьего сывороточного) с содержанием белка от 2 до 20 мкг/100 мкл пробы (20— 200 мкг/мл). Значения оптической плотности в этом случае должны быть в пределах 0,1 — 0,7 ед. при 595 нм.
12.1.3. Определение белка по методу Лоури
Данный метод был предложен Лоури с соавт. в 1951 г. и с тех пор широко используется в лабораторной практике. К преимуществам метода относятся универсальность и точность, к недостаткам — отрицательное влияние на точность измерения многих соединений (однако этот недостаток может быть преодолен осаждением белка из раствора перед определением), медленное развитие цветной реакции, нестабильность некоторых реактивов, а также необратимая денатурация порции белка, пошедшей на определение. Чувствительность метода от 5 до 100 мкг белка/мл.
Определение проводят с помощью спектрофотометра или ФЭК (длина волны максимума поглощения 750 нм). Для работы потребуются кюветы (можно пластиковые), пипетки, пробирки, штатив для пробирок. В качестве стандарта можно применять бычий сывороточный альбумин (1 мг/мл), однако яичный альбумин дает более точные результаты. Ниже приведена современная модификация метода.
Для определения готовят рабочие растворы реактивов А, В, С и D. Раствор А содержит 0,5 г CuSO4- 5Н2О и 1 г Ка3С6Н5О7- 2Н2О (цитрат натрия) на 100 мл дистиллированной воды. Раствор В: 20 г Na2CO3 и 4 г NaOH в 1 л дистиллированной воды. Растворы А и В устойчивы при комнатной температуре. Раствор С: 50 мл раствора В и 1 мл раствора А (готовят перед определением). Раствор D\ разведенный в 2 раза дистиллированной водой перед определением реактив Фолина (приготовление реактива Фолина см. в приложении 5).
Процедура определения. Смешивают 0,5 мл пробы белка и 2,5 мл раствора С. После перемешивания и инкубации при комнатной температуре 5—10 мин добавляют 0,25 мл раствора D, перемешивают, выдерживают при комнатной температуре 20 — 30 мин и измеряют оптическую плотность раствора при 750 нм.
Проба белка может быть отделена от мешающих определению веществ осаждением трихлоруксусной кислотой (ТХУ). Этим же способом можно пользоваться при определении белка в сильно разбавленных растворах (менее 1 мкг/мл). Для осаждения к пробе
белка в 1 мл добавляют 0,1 мл 0,15%-го раствора дезоксихолата натрия (ДХН), перемешивают и выдерживают при комнатной температуре 10 мин. К смеси добавляют 0,1 мл 72%-й ТХУ, перемешивают и выпавший осадок отделяют центрифугированием при 1 — 3 тыс. g в течение 5 — 30 мин. При использовании угловых роторов, при низких температурах или больших объемах время осаждения увеличивают. После осаждения надосадочную жидкость сливают и отбрасывают, осадок перерастворяют в реактиве С.
Метод Лоури применим также для определения белка в гидролизатах клеток микроорганизмов. Для гидролиза берут 2 М КОН и ведут гидролиз при 37 °C в течение 2 ч, в течение ночи при комнатной температуре или 10 мин на кипящей водяной бане.
Время развития цветной реакции обычно не превышает 20 — 30 мин после добавления раствора D, затем оптическая плотность уменьшается примерно на 1 % в час. Большинство мешающих определению веществ снижает развитие окраски, однако некоторые детергенты могут, напротив, ее усилить.
12.2. АНАЛИЗ НУКЛЕИНОВЫХ КИСЛОТ
Анализ нуклеиновых кислот получил широкое распространение при их идентификации в систематике прокариот, поскольку вся или основная генетическая информация заключена в одной молекуле ДНК. Это позволяет изучать ее, не опасаясь загрязнения другими ДНК — митохондриальной, хлоропластной, как может быть в случае эукариотных организмов.
Геномы различных бактерий сравнивают по их размерам, общему содержанию оснований ДНК [молярная доля гуанина и цитозина (ГЦ) в ДНК, %] по последовательностям нуклеотидов рибосомных РНК и степени гибридизации нуклеиновых кислот. Для разных прокариот установлены границы молярного содержания ГЦ в ДНК от 23 до 75%. Значение ГЦ постоянно для данного микроорганизма. Если по этой характеристике штаммы отличаются значительно (более чем на 10%), то это свидетельствует, что они относятся к разным родам. Однако близкие значения ГЦ в ДНК не обязательно говорят о филогенетическом родстве, поскольку организмы могут существенно различаться по последовательностям нуклеотидов в ДНК. В связи с этим более важным является сравнение гомологии участков ДНК или РНК изучаемых видов. Достаточно просты и доступны методы гибридизации сравниваемых ДНК—рРНК, которые проводят после их выделения и очистки.
12.2.1. Выделение и очистка ДНК
Для выделения нуклеиновых кислот обычно применяют мягкие методы разрушения клеток, включающие использование детергентов и лизоцима, хотя для разрушения некоторых микроорганизмов с прочными клеточными стенками могут понадобиться пресс, стеклянные бусы или растирание клеток с абразивами (см. 13.1). В некоторых случаях для «ослабления» клеточных стенок в растущие культуры (например, грамположительных бактерий) добавляют глицин, лизин или треонин. Иногда для тех же целей клетки грамположительных бактерий в растущих культурах обрабатывают антибиотиками, такими, как пенициллин С или метициллин, которые подавляют биосинтез пептидоглика
на клеточной стенки. Чувствительность к лизоциму микобактерий, содержащих высокий процент липидов и полисахаридов в клеточной стенке, может быть усилена их обработкой изопропанолом. При лизисе микроорганизмов, имеющих особенно прочные клеточные стенки, применяют поли(этиленгли-коль) с последующим удалением из лизата, так как его присутствие мешает проведению дальнейшей очистки ДНК.
Очистку ДНК из разрушенных клеток ведут последовательным отделением от белков обычно с использованием смесей хлороформ—изоамиловый спирт или фенол—хлороформ и многократным переосаждением ДНК этанолом. От следов РНК препараты ДНК освобождают с помощью РНКазы, а от следов белка — с помощью протеаз. ДНК сушат под вакуумом и хранят при 4 °C.
12.2.2. Определение нуклеотидного состава ДНК
Для определения молярного содержания ГЦ (%) в ДНК чаще всего применяют два метода: один основан на измерении плавучей плотности, другой — на анализе кривых температуры плавления (Д„) выделенных ДНК. В качестве стандарта в обоих случаях применяют ДНК известного состава, выделенную тем же методом. Оба метода быстрые и дешевые. Более точным является метод фракционирования ДНК на основания и количественное определение каждого нуклеотида в отдельности с использованием хроматографии на бумаге. Однако этот метод более длителен, трудоемок, и для получения воспроизводимых результатов необходима тщательная очистка препарата ДНК от РНК.
Различия в спектрах поглощения очищенных ДНК могут быть использованы также при определении их состава в сравнении с препаратом ДНК известного процентного содержания ГЦ. Этот метод дает быстрое и точное определение. Он считается удобной альтернативой, если оборудование для определения плавучей плотности или термальной денатурации ДНК недоступно.
Метод тепловой денатурации основан на разрушении водородных связей между комплементарными цепями нативной ДНК и их расхождении друг от друга при повышении температуры раствора ДНК. Однонитевые ДНК поглощают при Л. = 260 нм примерно на 40 % больше, чем двунитевые. При нагревании раствора ДНК процент однонитевых ДНК увеличивается и наблюдается увеличение поглощения при 260 нм (гиперхромизм). Термостабильность водородных связей между гуанином и цитозином в ДНК выше по сравнению со связями между аденином и тимином. Поэтому, чем выше содержание Г + Ц-пар в молекуле ДНК, тем больше энергии требуется для расхождения нитей. После расхождения цепей ДНК поглощение при 260 нм раствора с повышением температуры далее не увеличивается. Значение температуры плавления (Тт) соответствует температуре, при которой в растворе содержится 50 % разошедшихся нитей ДНК (температура 50%-го гиперхромизма). Эти значения линейно соответствуют молярному содержанию ГЦ в ДНК между 30 и 70 %.
Широко используют следующую модификацию этого метода. Раствор ДНК доводят до плотности приблизительно 25 мкг/мл (/12бо = 0,50) в 10-миллиметровых кварцевых кюветах с герметичными крышками и термопарой (фиксирующей температуру), конец которой должен быть непосредственно под поверхностью раствора. Кюветы помещают в регистрирующий спектрофотометр с
термостатирующим устройством. Температуру устанавливают равной 25 °C и регистрируют поглощение в опытной кювете при 260 нм против контрольной (с растворителем). Температуру медленно повышают (0,5 °С/мин) и проводят постоянную регистрацию изменения поглощения. Значение температуры, дающее 50%-е увеличение поглощения, будет соответствовать значению Тп, для данной ДНК.
Поскольку значения молярного содержания ГЦ (%) и Т1П связаны линейно, расчет для неизвестной ДНК производится сравнительно просто. На расчеты влияют концентрации буферных растворов, в которых проводят измерения, так как значение Тт логарифмически зависит от концентрации иона натрия в растворе. При измерении значений тепловой денатурации наиболее широко применяется цитратно-солевой буфер (1 • SSC, 0,015 М трехзамещенного цитрата натрия в 0,15 М NaCl, pH 7,0). Для этого буферного раствора установлено следующее соотношение:
молярное содержание ГЦ, % = 244 Тт - 169,00.
В случае ДНК с высоким молярным содержанием ГЦ используют более разбавленные буферные растворы, например 0,33- или 0,1-кратный раствор SSC. Для таких растворов уравнения выглядят следующим образом:
0,33SSC: молярное содержание ГЦ, % = 2,47 Тт - 135,14;
0,33SSC: молярное содержание ГЦ, % = 2,08 Тт - 106,40.
В целях воспроизводимости результатов, проверки метода и аппаратуры необходимо регулярно определять данным методом содержание ГЦ в ДНК из Е. coli АТСС 11775, поскольку этот штамм является международным стандартом и Тт его ДНК в 1 • SSC равна 90,5 °C, а значение плавучей плотности — 1,710.
Метод определения состава ДНК по плавучей плотности основан на том, что, если раствор ДНК подвергнуть высокоскоростному центрифугированию в градиенте плотности CsCl, то через определенное время она займет в градиенте место, соответствующее своей плавучей плотности. Значение плотности ДНК в середине пика, формируемого в центрифужной пробирке, называется плавучей плотностью ДНК. Она линейно зависит от молярного содержания ГЦ, %. Метод был впервые описан в 1962 г. и с тех пор не претерпел значительных модификаций.
Измерение плавучей плотности ДНК производят следующим образом. Сухой CsCl растворяют в буфере (50 мМ inpuc-HCl, pH 7,3) до плотности 1,7 г/см3 и вносят 2 — 3 мкг исследуемой и реперной (с известным составом) ДНК. Растворы переносят в чистые и сухие центрифужные пробирки, помещают в аналитическую центрифугу и центрифугируют при 150 тыс. g в течение 20 ч при 25 °C. За это время градиент обычно формируется полностью.
Плавучая плотность неизвестной ДНК рассчитывается с использованием расстояния между серединой ее пика и серединой пика реперной ДНК на градиенте плотности CsCl. Маркерная ДНК должна быть выбрана заранее, чтобы ее пик не перекрывал пик определяемой ДНК. Нуклеотидный состав ДНК и ее плотность (р) связаны зависимостью:
р = 1,660 + 0,098(Г + Ц).
12.2.3. Определение нуклеотидного состава ДНК методами хроматографии
Для определения нуклеотидного состава используют препараты ДНК, получение путем депротеинизации. Если выделение нативного препарата ДНК по каким-либо причинам невозможно, то для изучения нуклеотидного состава используют навеску в 200 — 500 мг биомассы. Исследуемый материал без предварительного удаления кислоторастворимой фракции подвергают щелочному гидролизу 0,75 н. NaOH при 37 °C в течение 18 ч. Использование едкого натра вместо едкого калия для гидролиза обусловлено тем, что образующийся при последующей нейтрализации перхлорат натрия хорошо растворяется и полностью удаляется при отделении рибонуклеотидов от ДНК. После гидролиза суспензию охлаждают до 0°С, щелочь нейтрализуют 57%-й НС1О4 на холоде, затем доводят концентрацию кислоты до 1 — 2 %. При этом в растворе остаются кислоторастворимые соединения (свободные нуклеотиды, мононуклеотиды РНК, фосфосахара и др.), а в осадок выпадают белки и ДНК. Осадок, содержащий ДНК и белки, тщательно отмывают от кислоторастворимых веществ холодной 0,2 н. НС1О4 при центрифугировании, а затем обрабатывают спиртом, смесью спирт—эфир и эфиром и помещают для полного высушивания в вакуум-эксикатор. Высушенный остаток, содержащий ДНК, переносят в толстостенную центрифужную пробирку, заливают 8 —10 мл 10%-го раствора NaCl, тщательно размешивают, добавляют несколько капель насыщенного раствора NaHCO3 до pH 7,5 — 8,0 и нагревают на кипящей водяной бане в течение часа. При нагревании постоянно следят за тем, чтобы реакция суспензии оставалась нейтральной или слабощелочной, поскольку при подкислении отщепляются пуриновые основания. При сильном вспенивании суспензии к ней добавляют несколько капель изоамилового спирта. В результате нагревания с NaCl ДНК переходит в раствор в виде натриевой соли и происходит ее частичная депротеинизация (освобождение от белков).
Раствор, содержащий ДНК, отделяют от денатурированных белков центрифугированием при 5 —10 тыс. ув течение 20 мин. Осадок дважды промывают небольшими порциями 10%-го раствора NaCl и отбрасывают. Промывные воды объединяют с основным солевым раствором ДНК. Раствор нейтрализуют 0,5 н. НС1 до pH 6,0 — 6,5, прибавляют 3 объема 96%-го спирта и оставляют на ночь на холоду для выпадения осадка ДНК. Выпавший осадок отделяют центрифугированием, дважды промывают 70%-м спиртом для удаления избытка соли, а затем дважды — 96%-м спиртом и один раз — эфиром для высушивания. Высушенный осадок ДНК хранится долго. Для лучшего высушивания и удаления эфира осадок ДНК распределяют стеклянной палочкой по стенкам центрифужного стакана, затем помещают в вакуум-эксикатор.
Гидролиз ДНК. Для гидролиза ДНК до оснований высушенную ДНК переносят в стеклянную ампулу и добавляют 72%-ю НС1О4 (на 1 мг ДНК берут около 30 мкл хлорной кислоты). Большой избыток хлорной кислоты приводит к значительному разложению тимина, поэтому для гидролиза ДНК используют минимальное количество кислоты, добавляя ее с таким расчетом, чтобы только полностью смочить осадок ДНК. Затем ампулу запаивают {глаза обязательно должны быть защищены очками!) и проводят гидролиз на кипящей водяной бане или в термостате при 105 °C в течение часа. Под действием кон
центрированной НС1О4 ДНК полностью гидролизуется с отщеплением азотистых оснований.
По окончании гидролиза ампулу охлаждают, заворачивают в полотенце, осторожно вскрывают (аккуратно надпиливая и надламывая) и добавляют к гидролизату 0,1 —0,5 мл воды. Содержимое ампулы тщательно перемешивают и нейтрализуют по универсальному индикатору, прибавляя по каплям 40%-й раствор КОН. Слабокислый гидролизат отделяют от осадка путем фильтрования через стеклянный фильтр или центрифугированием, при необходимости упаривают до небольшого объема и используют для хроматографического разделения азотистых оснований на бумаге или в тонком слое целлюлозы. Гидролизат ДНК можно наносить на хроматографическую бумагу и без предварительной нейтрализации кислоты КОН. В этом случае к гидролизату ДНК добавляют несколько капель воды и перемешивают. В чашку Петри наливают 14%-й раствор аммиака. На нее кладут хроматограмму так, чтобы стартовая линия находилась над аммиаком. Затем наносят гидролизат ДНК.
Хроматография оснований на бумаге. Для разделения оснований используют отечественную «медленную» или «среднюю» хроматографическую бумагу, а также сходные импортные сорта. Бумагу предварительно промывают 2 н. раствором СН3СООН. На прямоугольную полоску хроматографической бумаги на расстоянии 4 см от ее верхнего края наносят с помощью капилляра гидролизат ДНК в виде штрихов длиной 1 —2 см. В один штрих наносят обычно объем гидролизата, соответствующий 300—-400 мкг ДНК. Расстояние между образцами должно быть не менее 2 см. Одну полосу шириной 5 см, контрольную, оставляют на бумаге свободной.
На дно хроматографической камеры наливают растворитель слоем 1,5 — 2 см, а затем помещают в нее лист бумаги таким образом, чтобы ее верхний край был неподвижно закреплен, а нижний, на котором нанесен гидролизат, погружен в растворитель на 1 —1,5 см. Для разделения оснований на бумаге или в тонком слое целлюлозы используют следующие системы растворителей:
1) кислый растворитель Кирби: метанол —концентрированная НС1—вода (75:20: 15);
2) изо пропанол —концентрированная НС1 —вода (170:45:35);
3) щелочной растворитель Уайта: и-бутанол—вода —концентрированный аммиак (60: 10:0,1);
4) //-бутанол —пода (43:7), в газовой фазе кон центр ированнный аммиак (наливают аммиак в чашку Петри и помещают на дно камеры).
Растворитель пропускают через бумагу или слой сорбента на пластинке обычно один раз: при этом происходит удовлетворительное разделение оснований. В кислых системах (1 и 2) разделение оснований при комнатной температуре происходит за 18 —20 ч. На хроматограмме в направлении от старта к фронту основания располагаются следующим образом: Г, А, Ц, Т. В системе 2 отделяется также 5-метилцитозин (МЦ), который располагается между Ц и Т. В щелочной системе 3 при комнатной температуре разделение оснований происходит обычно за 36 — 48 ч, при этом основания располагаются в направлении от старта к фронту в следующем порядке: Г, Ц, МЦ, А и Т. Этот растворитель (3) очень удобен для определения небольших количеств оснований, так как на хроматограммах всегда обнаруживаются компактные пятна всех оснований, за исключением гуанина. В кислых растворителях пятна оснований оказываются гораздо более размыты. Для отделения 6-метилаланина (6-МА) от других оснований применяют систему 4.
Существенным недостатком щелочного растворителя является то, что в нем не удается количественно отделить и определить гуанин. Это объясняется тем, что гуанин хорошо растворим только в кислых растворах, поэтому на хроматограммах после про
пускания растворителя Уайта гуанин образует «хвосты» и не вымывается полностью со старта. В связи с этим для определения количества всех азотистых оснований в ДНК проводят параллельно разделение одного и того же гидролизата в двух системах растворителей (чаще всего 2 и 3). При разделении оснований в системе 3 содержание гуанина не учитывают, а определяют только содержание других оснований: Ц, А, Т и иногда МЦ. При разделении оснований в кислом растворителе 2 определяют только количества Г и А, а затем рассчитывают отношение Г/А. Используя это отношение и полученные ранее данные по содержанию Ц, А и Т, окончательно рассчитывают нуклеотидный состав данного препарата ДНК.
Пятна оснований обнаруживают по поглощению УФ-света, очерчивают простым карандашом, нарезают на полоски 2 — 3 мм и элюируют 5 мл 0,1 н НС1 при 37 °C в течение ночи. Против каждого пятна основания из контрольной полоски вырезают участок бумаги такого же размера и элюируют его в тех же условиях. Гуанин растворяется в 0,1 н. НС1 несколько хуже, чем другие основания, поэтому для его элюции обычно используют 0,5 н. НО. Тем же раствором элюируют и контроль для гуанина.
После элюции содержимое пробирок осторожно перемешивают, дают осесть ворсинкам бумаги и спектрофотометрируют против элюатов из соответствующих контрольных участков хроматограмм.
Хроматография оснований в тонком слое целлюлозы. Для приготовления хроматографических пластин используют стеклянные пластинки, размеры которых определяются параметрами хроматографической камеры. Их тщательно моют стиральным порошком (до равномерного стекания воды с поверхности), промывают дистиллированной водой и высушивают при комнатной температуре. Мелкодисперсную целлюлозу фирмы «Фильтрак» в количестве 10 г суспендируют в дистиллированной воде с таким расчетом, чтобы объем полученной суспензии составил 100 мл, и перемешивают с помощью магнитной мешалки в течение 20 — 30 мин. Полученную суспензию целлюлозы равномерно наносят на горизонтально расположенные стеклянные пластинки из расчета 20 мл на 120 см2, которые затем высушивают при комнатной температуре.
Нейтрализованный гидролизат ДНК наносят на слой целлюлозы с помощью капилляров в виде штрихов длиной 1 см на линии, отстоящей от нижнего края пластинки на 3 см. Чтобы не допустить разрушения слоя целлюлозы, гидролизат наносят в виде серии расположенных друг за другом точек. В каждый штрих наносят объем гидролизата, соответствующий от 200 до 600 мкг ДНК. На каждой пластинке оставляют один свободный (контрольный) участок.
Разделение оснований проводят в тех же системах растворителей, что и в случае хроматографии на бумаге (чаще всего 2 и 3).
Пластинку помешают в хроматографическую камеру, на дно которой наливают растворитель слоем в 1 —2 см, камеру герметически закрывают пришлифованной крышкой и проводят хроматографическое разделение до тех пор, пока растворитель не дойдет до верхнего края пластинки. Высушенную пластинку просматривают на ультрахе-москопе при 260 нм в отраженном свете и очерчивают карандашом УФ-поглощение пятна оснований. Для каждого основания на свободном участке очерчивают равные по площади контрольные участки. Затем все очерченные зоны соскабливают ланцетом или бритвой в пробирки, наливают в каждую пробирку по 3 — 5 мл элюирующего раствора и проводят элюцию оснований в течение 2 — 5 ч при 37 °C. В качестве элюирующего раствора используют (для всех оснований, кроме гуанина) 0,1 н. НС1, а для гуанина и соответствующего контроля — 0,5 н. НС1. По окончании элюции суспензию тщательно перемешивают и центрифугируют: супернатанты спектрофотометрируют против контрольных элюатов.
Спектрофотометрия и проведение расчетов. Элюаты оснований, полученные в результате того или иного способа хроматографии, спектрофотометрируют при следующих длинах волн (нм): гуанин — 250 и 290; аденин — 260 и 290; цитозин — 276 и 290; 5-метилцитозин — 280 и 300; тимин — 260 и 290.
Для расчета количества оснований (мкмоль в 1 мл элюата) используют коэффициенты:
гуанин 0,1428 (у425о — /129о)1 аденин — 0,0792 (/Ьбо — /129о)1 цитозин — 0,1880 (Л27б — ^290); 5-метилцитозин — 0,1786 (>4280 - ^зоо); тимин — 0,1486 (А2№ - Л2до); 6-метиладе-нин — 0,1005 (Л267 - Дгдо)-
Количество всех оснований суммируют, сумму принимают за 100% и определяют процентное содержание каждого основания в ДНК. Нуклеотидный состав ДНК выражают в молярных процентах Г + Ц.
12.3. ГИБРИДИЗАЦИЯ НУКЛЕИНОВЫХ КИСЛОТ
Методы гибридизации ДНК—ДНК и ДНК—рРНК получили в последние годы широкое распространение при идентификации новых штаммов бактерий. Они позволяют установить степень генетического родства изучаемого штамма с определенным родом, видом или выявить штаммовые различия. Принцип метода заключается в денатурации выделенной двухцепочечной ДНК нагреванием и фиксации разошедшихся цепей на фильтрах. При понижении температуры возможна ренатурация цепей, причем соединиться с цепью может только комплементарная ей последовательность оснований, а температура связывания во многом определяет результаты опыта. Количество ренатурированной двуспиральной ДНК служит прямой мерой сходства геномов сравниваемых организмов, причем последовательности, образующие двуцепочечные комплексы, не обязательно комплементарны по всем нуклеотидам.
При гибридизации фрагменты одноцепочечной ДНК известного микроорганизма (ДНК-репер) закрепляют на фильтре и добавляют радиоактивно меченные фрагменты одноцепочечной ДНК изучаемого штамма. Контролем служит связывание гомологичной реперу ДНК (100 %-я гибридизация). Меченая ДНК (зонд) должна составлять в опыте не более 1: 300 — 1:500 части от прикрепленной к фильтру немеченой ДНК-репера. Связывание зонда в пределах 100 — 70% считают штаммовыми различиями, 70 — 50% — различиями видовыми, менее 50 % — различиями на уровне рода. Данный метод удобен и объективен, однако может применяться лишь для определения сходства бактерий на уровне рода и более низком таксономическом уровне.
Для проведения гибридизации клетки изучаемого штамма разрушают тем или иным способом (см. 13.1), выделяют ДНК, фрагментируют ее ультразвуком на кусочки длиной 300 — 350 пар оснований (определяется электрофорезом в 1%-й агарозе), денатурируют нагреванием и закрепляют на нитроцеллюлозных фильтрах. ДНК сравниваемого штамма готовят таким же способом, вводят реактивную метку ник-трансляцией с помощью меченого нуклеозид-5'-трифосфата при одновременном действии ДНКазы-1 и ДНК-полимеразы-1 Е. coli и используют для гибридизации. Ее чаще всего проводят по методу Ден-хардта в 20%-м формамиде при 62 °C в течение 18 — 24 ч («мягкая ренатурация»), отмывают от несвязавшейся меченой ДНК и радиоактивность фильтров просчитывают в сцинтилляционном счетчике. Гибридизацию гомологичных ДНК принимают за 100 % и рассчитывают проценты связанной исследуемой ДНК с ДНК реперного штамма. Процент связанной ДНК отражает степень гомологии молекул и служит показателем родства штаммов. Аналогичная реассоциация может быть осуществлена между молекулами ДНК и рРНК, поскольку двой
ные спирали образуются также между одноцепочечными ДНК и комплементарными цепями РНК. Подробнее эти методы описаны в справочном руководстве (Ф. Герхард и др., 1984).
12.4. КАТАЛОГИЗАЦИЯ НУКЛЕОТИДНЫХ ПОСЛЕДОВАТЕЛЬНОСТЕЙ 5S И 16S рРНК
. Методы гибридизации ДНК-ДНК и ДНК-рРНК требуют прямого экспериментального сравнения изучаемых микроорганизмов. В отличие от этого методы анализа генома, построенные на определении последовательностей нуклеотидов в высококонсервативных полимерных молекулах клетки, таких, как 5S, 16S (18S) или 23S (28S) рРНК, дают возможность оценивать данные, полученные в разное время и в разных лабораториях. С помощью сравнения последовательностей нуклеотидов можно проводить филогенетический анализ любых организмов, как про-, так и эукариотных, и строить эволюционные деревья, что важно для определения степени их родства и путей эволюции жизни.
Метод изучения последовательностей нуклеотидов 5S рРНК по сравнению с анализом 16S (или аналогичной 18S рРНК у эукариот) дает меньше информации (это понятно, если учитывать длину цепи молекул — 120 и 1500— 1600 нуклеотидов соответственно), однако он более прост, быстр и дешев. Последовательности нуклеотидов сравнивают между собой для получения коэффициента сходства (5АВ), характеризующего подобие нуклеотидов у организмов А и В. Коэффициент (5'лв) рассчитывают путем деления суммы нуклеотидных остатков, находящихся в общих для двух организмов нуклеотидах, на сумму нуклеотидных остатков, содержащихся во всех сравниваемых нуклеотидах, определенных у организмов А и В. Коэффициенты ,8ЛВ используют для построения дендрограмм, которые рассматривают как филогенетические деревья. Подробнее эти методы описаны в справочном руководстве (Ф.Герхард и др., 1984).
12.5. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ПОЛИ-р-ОКСИМАСЛЯНОЙ КИСЛОТЫ
Многие бактерии образуют поли-р-оксимасляную кислоту (ПБОМК) и другие полимеры на основе оксижирных кислот и их производных в качестве внутриклеточного запасного продукта. Иногда способность к такому синтезу служит диагностическим признаком.
Проведение определения. Для экстракции ПБОМК из клеток биомассу после осаждения высушивают смесью диэтилового эфира и метанола в соотношении объемов 1:1. Сухую биомассу освобождают от остатков растворителей в вытяжном шкафу. Осадок растирают в ступке до порошкообразного состояния и взвешивают. Если для анализа берут сухую биомассу, то этап высушивания смесью растворителей пропускают.
ПБМОК экстрагируют из навески биомассы смесью хлороформа и метанола (в объемном соотношении 2:1) при 60 °C. Экстрагирование проводят в колбе или пробирке с притертой и прижатой пробкой (во избежание ее выталкивания при испарении раство
рителей и подъема давления внутри колбы или пробирки), помещенных в водяную термостатированную баню. Для экстракции берут 5 мл смеси растворителей на 0,5 г сухих клеток. Экстракцию ведут в течение 20 мин при периодическом покачивании содержимого, затем экстракт сливают с осадка и экстракцию повторяют. После экстракции осадок биомассы отбрасывают.
Объединенные экстракты фильтруют через стеклянный фильтр и упаривают досуха под вакуумом или в вытяжном шкафу в фарфоровой чашке на кипящей водяной бане. Осадок после упаривания растворяют в 1 н. серной кислоте (5 мл на каждый грамм высушенной исходной биомассы) и ПБОМК гидролизуют в течение 2 ч на кипящей водяной бане или в термостате при 100 — 105 °C. Гидролизат фильтруют через стеклянный фильтр под вакуумом, доводят его объем до 5 мл 1 н. серной кислотой и измеряют экстинкцию при 235 нм против серной кислоты на спектрофотометре. Калибровочную кривую строят по 3-гидроксимасляной кислоте с кислотной обработкой ее параллельно опытной пробе.
12.6. ВЫДЕЛЕНИЕ И АНАЛИЗ ПОЛИСАХАРИДОВ
Полисахариды, образуемые микроорганизмами, входят в состав ряда компонентов клеток, в частности в клеточную стенку, служат запасными веществами, локализуются внутри и вне клетки. Внеклеточные полисахариды могут быть капсульными или свободными. Часто полисахариды определяют антигенную специфичность штамма, выполняют защитную функцию, играя роль барьера проницаемости для молекул, ионов или препятствуют потере клеткой-влаги.
Проведение определения. Из культуральной жидкости полисахариды выделяют осаждением 3 — 5 объемами этанола или изопропанола; осадок, образующийся за 24 ч при комнатной температуре, отделяют центрифугированием (3 тыс. g, 15 мин) и сушат на воздухе.
Для определения моносахаридного состава проводят гидролиз 1—2%-го препарата полисахарида в 1 н. НС1 при 100 °C в течение 3 ч в герметическом сосуде и затем смесь доводят сухим бикарбонатом натрия до нейтральной реакции по индикатору. Раствор гидролизе ванного полисахарида хранят в холодильнике.
Количество редуцирующих сахаров в гидролизате полисахарида определяют по восстановлению 2,3,5-трифенилтетразолия хлористого (ТТХ), используя в качестве стандарта раствор D-глюкозы. Качественный моносахаридный состав полисахарида определяют методом хроматографии на бумаге с соответствующими свидетелями.
Для определения образования внутриклеточных полисахаридов в качестве запасных веществ проводят кислотный гидролиз клеточной массы и в гидролизате определяют количество редуцирующих сахаров.
12.7. ИЗУЧЕНИЕ СОСТАВА КЛЕТОЧНЫХ СТЕНОК
МИКРООРГАНИЗМОВ
Главный полимер клеточной стенки большинства бактерий — пептидогликан (ПГ). В современной классификации грамположительных бактерий для характеристики рода и вида учитывают его строение. Известно около 100 типов ПГ. Все грамотрицательные бактерии содержат, за небольшим исключением, один тип ПГ. У этих бактерий его строение не является таксономическим признаком.
Выделение и очистка фракций клеточных стенок. Фракции клеточных стенок для анализа ПГ получают после механического разрушения клеток бактерий (ультразвуковой дезинтегратор с бусами или без; декомпрессионный шок, см. выше) и центрифугирования при 15 тыс. g в течение 20 мин (условия осаждения могут несколько различаться в зависимости от вида). Полученный осадок делится на два слоя: нижний — неразрушенные клетки, и верхний, студенистой консистенции, — клеточные стенки. Верхний слой осадка отделяют и несколько раз промывают водой, пока расслаивание осадка не прекратится (нижний слой каждый раз отбрасывают). После последнего промывания (всего 5 — 6 промываний) верхний слой отмытых клеточных стенок лиофилизируют или высушивают при обработке спиртом (3 раза), эфиром (2 раза), затем на воздухе или в вакуум-эксикаторе.
Высушенную массу стенок грамположительных бактерий экстрагируют трихлоруксусной кислотой (ТХУ) для удаления тейхоевых кислот. С этой целью к 100 — 200 мг сухих клеточных стенок добавляют 10 — 20 мл 5%-й ТХУ и суспензию нагревают при 100 °C в течение 20 мин. Смесь охлаждают и осадок стенок отмывают водой при центрифугировании до кислотности, близкой к нейтральной. Осадок суспендируют в 50 мМ /прис-НСГбуфере (pH 7,8), препарат обрабатывают трипсином (2,5 мг сухого фермента в 2,5 мл буфера на каждые 10 мг стенок) в течение 20 мин при 37 °C при постоянном перемешивании. Осадки стенок промывают 3 раза буфером и обрабатывают 2%-м дезоксихолатом натрия в том же буфере для удаления белков. Обработку ведут 5 мин при 100 °C. Затем детергент отмывают сначала 3 раза 1н. NaCl, а после 3 — 5 раз дистиллированной водой. Полученный препарат ПГ высушивают при обработке спиртом (3 раза), эфиром (2. раза) и досушивают в вакуум-эксикаторе.
Анализ состава пептидогликана. Выделенный и очищенный препарат ПГ гидролизуют в 4 н. НС1. Для этого 2 — 3 мг ПГ помещают в ампулу, добавляют 0,5 мл кислоты и запаянную ампулу инкубируют в течение 16 ч при 100 °C. В редких случаях бывает необходим гидролиз препарата 6 н. НС1 в течение 18 ч при 100 °C или 3 н. трифторуксусной кислотой в течение 4 ч при 100 °C. После проведения гидролиза и охлаждения ампулы ее вскрывают и гидролизат переносят на чашечку из фторопласта. Из гидролизата НС1 удаляют в вакуум-эксикаторе, многократно добавляя в чашечку по 0,5 мл дистиллированной воды и высушивая в эксикаторе. В дальнейшем гидролизат количественно переносят в мерную пробирку, объем доводят бидистиллированной водой до 1 мл и при необходимости центрифугируют. Анализ гидролизата проводят на аминокислотном анализаторе, после чего вычисляют мольное отношение аминокислот, мурамовой кислоты и глюкозамина. В том случае, когда в гидролизате обнаружена диаминопимелино-вая кислота, определяют ее стереоизомер.
Для определения стереоизомера диаминопимелиновой кислоты (ДАП) гидролизат ПГ (из расчета 1 мг исходного ПГ; при отсутствии белка в клеточной стенке можно использовать ее гидролизат) разделяют хроматографией на бумаге в системе растворителей: метанол —вода — НС1—пиридин (объемное соотношение 40 : 13:2: 5). Хроматограмму высушивают, опрыскивают 0,5%-м нингидрином и нагревают в течение 5 мин при 100— 110°С. ДАП проявляется в виде желтых пятен на белом фоне. Полученные пятна сравнивают по Rrco стандартными образцами мезо- и /.-формами ДАП, которые должны быть нанесены на ту же хроматограмму в качестве свидетелей.
В некоторых случаях для таксономических целей необходимо определить только природу диаминокислоты. ДАП определяют вышеописанным способом, лизин — хроматографически в системе бутанол —уксусная кислота —вода (4:1:1) и сравнивают со стандартом.
Идентификацию редких диаминокислот проводят несколькими способами: хроматографически в разных системах растворителей или на аминокислотном анализаторе с соответствующими свидетелями.
В гидролизате определяют аминосахарный и аминокислотный состав ПГ. В большинстве случаев для целей таксономии достаточно определить мольное отношение аминосахаров и аминокислот. В ПГ может быть от 3 до 6 различных аминокислот,
однако в их составе никогда не встречаются разветвленные и ароматические аминокислоты.
Предположим, что в пробе гидролизата ПГ получено следующее количество аминосахаров и аминокислот (мкмоль): мурамовая кислота (М) — 4,03; глюкозамин (ГН) — 4,52; диаминопимелиновая кислота (ДАН) — 5,03; аланин (А) — 9,81; глутаминовая кислота (ГК) - 5,40 (М-ГН-ДАП-А-ГК-0,80:0,90:1,00:1,95:1,02). Учитывая, что мурамовая кислота и глюкозамин при кислотном гидролизе частично подвергаются разрушению, истинное мольное отношение в природном полимере данного типа составляет, по-видимому, 1:1:1 : 2:1.
Выяснив, что ДАП имеет мезо-форму, приходим к выводу, что ПГ имеет А1у-тип, т.е. все остатки мурамовой кислоты замещены тетрапептидом, состоящим из двух остатков аланина, ДАП, глутаминовой кислоты, и перекрест между соседними пептидными единицами осуществляется напрямую (без аминокислот в мостике) между ДАП одной цепи и последним аланином другой.
Глава 13
ПОДГОТОВКА КЛЕТОК ДЛЯ ЭНЗИМАТИЧЕСКИХ ИССЛЕДОВАНИЙ
13.1. ОБРАБОТКА КЛЕТОК И МЕТОДЫ ДЕЗИНТЕГРАЦИИ
Цель энзимологических исследований заключается в установлении путей получения клеткой энергии и синтеза основных метаболитов для построения новой клетки. Лучше всего проводить эксперименты на целых клетках, которые метаболически и структурно интактны, и во многих случаях такие оптимальные условия могут быть достигнуты. Однако в большинстве случаев это сделать трудно, и исследователям приходится работать с менее идеальным материалом. В каждом отдельном случае необходимо выбрать, какой тип клеточного препарата более подходит для проведения нужного эксперимента: растущие или покоящиеся целые клетки микроорганизмов, разрушенные препараты клеток или клетки с нарушенной клеточной стенкой или мембраной (методы лизиса и дезинтеграции описаны ниже).
Интактные клетки
Растущие клетки. В активно растущих бактериях идут все основные жизненные процессы, характерные для высокоорганизованных и отрегулированных систем, включая репликацию, и поэтому они являются наилучшим материалом для энзиматического анализа. Где возможно, рекомендуется использование таких интактных растущих клеток.
Покоящиеся клетки. Рост может быть остановлен удалением клеток (центрифугированием или фильтрованием) из среды культивирования с последующим суспендированием их в нейтральной осмотически приемлемой среде, например в буферном физиологическом растворе или в минеральной среде без источников углерода и азота. Обработанные таким образом клетки называют покоящимися. Они содержат весь набор ферментов, необходимых для роста, однако ферменты находятся в относительно
неактивном или покоящемся состоянии. В таких клетках обычно наблюдают за одной реакцией или группой реакций, представляющих интерес. Можно проследить, например, поток электронов от субстратов, например D-лактата или сукцината, через дыхательную систему в условиях, когда затраты на рост минимальны. Покоящиеся клетки можно использовать при изучении синтеза белка, транспортных систем для переноса субстратов или ионов. Для этих экспериментов клетки обычно суспендируют в осмотически пригодной среде, содержащей 50—100 мкг/мл хлорамфеникола, препятствующего синтезу белков. В некоторых случаях, например в опытах по исследованию транспорта веществ в клетку, нужно быть уверенным, что покоящиеся клетки снабжены источником энергии.
Голодающие покоящиеся клетки. Для некоторых экспериментов важно, чтобы клетки были освобождены от эндогенных запасов энергии. Примером использования голодающих покоящихся клеток является эксперимент по измерению пула внутриклеточного АТФ, образующегося за счет мембранного потенциала. Эндогенный уровень образования АТФ в неголодающих клетках затрудняет измерение АТФ, синтезируемого за счет хемиосмотического градиента. Для Е. colt разработана процедура истощения энергетических запасов в циклической реакции фосфорилирования/дефосфорилиро-вания а-метилглюкозида, который в этих клетках фосфорилируется в результате действия фосфоенолпируват-фототрансферазной системы. Клетки осаждают центрифугированием при 4 °C и отмывают 0,12 М пгрпс-НС1-буфером (pH 8,0). Затем клетки суспендируют до плотности 5 мг/мл в том же буфере, содержащем 20 мМ сс-метилглюко-зида и 40 мМ NaN3, и инкубируют при 37 °C от 45 до 120 мин в зависимости от вида бактерий. Затем клетки отмывают от ингибитора и используют в дальнейших экспериментах.
В большинстве случаев для уменьшения эндогенного метаболизма аэробных или факультативно-аэробных бактерий проводят интенсивную аэрацию плотной бактериальной суспензии в течение 2 —4 ч в минеральной среде без источника углерода или буфере. Уменьшение клеточных резервов можно контролировать по снижению эндогенного дыхания полярографически, сравнивая его с уровнем дыхания в присутствии энергетического субстрата, например сукцината.
13.1.1. Лизис клеток
Лизис клеток с применением ферментов. Разрушение (лизис) клеточных стенок с применением детергентов и ферментов проводят обычно с небольшими порциями биомассы, причем эти методы дают более стандартную обработку для каждой клетки в суспензии, так как концентрации детергентов и ферментов практически одинаковы в любой порции пробы. Как правило, время лизиса не превышает 15 — 30 мин. Рассмотрим этот метод на примере Escherichia coli. Для лизирования клетки суспендируют в ТЕ-буфере (50 мМ трис-НС\, pH 8,0 и 10 мМ ЭДТА) из расчета 3 мл буфера на 1 г сырой биомассы и нагревают до 37 °C. Добавляют 1 мл раствора лизоцима (свежеприготовленный раствор в ТЕ-буфере, 10 мг/мл) на каждые 5 мл суспензии или эквивалентное количество сухого лизоцима и инкубируют 10 — 20 мин при 37 °C с легким покачиванием. Быстрее клетки лизируются при повышении действующей концентрации лизоцима до 10 мг/мл. В таких условиях удовлетворительного лизиса можно достичь в течение 5 мин даже при 4 °C.
Для получения сферопластов дрожжей суспензии их клеток могут быть подвергнуты лизису с помощью препарата ферментов, выделенных из содержимого желудка виноградной улитки. Такой «улиточный фермент» в течение короткого времени при 37 —40 °C лизирует, например, клеточную стенку дрож
жей рода Saccharoniyces. Препарат фермента готовят в лаборатории из свежесобранных улиток и хранят в замороженном состоянии, используя по потребности.
Лизис клеток с помощью детергентов. Этот наиболее мягкий и быстрый способ разрушения клеток по сравнению с другими методами (занимает от 20 до 90 мин) применим лишь для микроорганизмов, лишенных клеточной стенки. Перед лизисом клетки промывают несколько раз забуференным солевым раствором (например, 10 мМ mpuc-HCl, pH 7,5, с 150 мМ NaCl). После последнего центрифугирования биомассу суспендируют до плотности 107— 108кл/мл или 3 —4 мг сухих клеток на 1 мл в том же буфере, с добавлением 0,1 —0,3 % тритона X-100. Содержимое перемешивают и инкубируют на льду от 10 до 90 мин. Для стабилизации белков в экстракт добавляют 0,2 объема 50%-го глицерола. Вместо тритона Х-100 могут быть использованы и другие неионные детергенты.
Лизис клеток в присутствии органических растворителей. Визирование клеток органическими растворителями обычно применяют для микроорганизмов, осажденных на фильтрах в целях проведения реакций с антителами или гибридизаций с пробами нуклеиновых кислот. Для проведения такой обработки чаще всего используют невысокие концентрации толуола (0,1 —1,0 %) в соответствующем буфере.
Лизис с помощью замораживания—оттаивания. Этот метод (многократное замораживание, например в ванне с жидким азотом, и оттаивание, например в теплой воде) быстрый, удобный и относительно дешевый. Он позволяет обрабатывать одновременно большое количество биомассы и дает легко воспроизводимые результаты, однако имеет свои ограничения. Клетки, разрушаемые таким образом, должны обладать не очень прочной стенкой или не содержать ее, а интересуемые внутриклеточные компоненты должны быть относительно стабильны к подобного рода обработке, не подвержены денатурации и защищены от протеолиза.
13.1.2. Методы дезинтеграции
В большинстве экспериментов для выделения и определения внутриклеточных компонентов применяют различные методы разрушения микроорганизмов. Это могут быть ручные и механические гомогенизаторы, растирание клеток с абразивами, растирание замороженных клеток, перемешивание биомассы со стеклянными бусами. Наиболее полное разрушение клеток достигается в случае применения декомпрессионных методов (Х-пресс, Френч-пресс) или ультразвука. Реже применяют осмотический шок и метод замораживания—оттаивания. Описаны также комбинированные методы разрушения микроорганизмов, сочетающие обработку клеточных стенок ферментами с последующим осмотическим шоком полученных сферопластов (протопластов) или растиранием их в гомогенизаторе.
Перед разрушением клетки необходимо отделить от среды выращивания. В зависимости от вида микроорганизма применяют различные способы отделения, такие, как использование сепараторов для крупных микроорганизмов, осаждение их фильтрованием через ткань (грибной мицелий) и применение
различного рода центрифуг, позволяющих осаждать клетки с охлаждением и без него. В зависимости от величины объектов, целей эксперимента и термолабильности выделяемых структур применяют низко- или высокоскоростное центрифугирование в периодическом или непрерывном режимах. Для крупных и тяжелых клеток бывает достаточным короткое осаждение при 2 — 3 тыс. g, тогда как мелкие и легкие клетки требуют длительного центрифугирования при 15 — 20 тыс. g. Необходимо учитывать возможный нагрев биологического материала при высокоскоростном центрифугировании объектов без соответствующего охлаждения.
Отделенную от культуральной жидкости биомассу 2 — 3 раза промывают для удаления следов продуктов метаболизма, суспендируя каждый раз в свежей среде (обычно без источника углерода) или в буферном растворе. Экстракт, полученный после разрушения клеток, отделяют от неразрушенных клеток и крупных обломков центрифугированием с охлаждением в течение от 10 мин при 15 тыс. ддо 1 ч при 40 тыс. g (в зависимости от целей эксперимента). Супернатант, называемый грубым, или исходным экстрактом, можно затем повторно центрифугировать при 150 — 200 тыс. g в течение 1,5 — 2 ч для получения прозрачного экстракта клеток и осадка клеточных мембран. Если при этом исходный экстракт содержит плавающие на поверхности частицы, их можно удалить фильтрованием через ткань или стеклянную вату.
Для полного разрушения клеток применяют обработку ультразвуком, разрушение на механических мельницах со стеклянными бусами, разрушение на гидравлическом прессе, механическое растирание замороженных в жидком азоте клеток с кварцевым песком в керамической ступке. Ниже приводится описание нескольких способов, которые будут применяться на практикуме.
Гомогенизация. Этот способ можно использовать для микроорганизмов с тонкой клеточной стенкой, не имеющих клеточной стенки или для животных и некоторых растительных клеток. Порции биомассы, суспендированные в буферном растворе (3 — 5 объемов), переносят в ручной гомогенизатор и пропускают через него биомассу 10 — 20 раз. При использовании механических гомогенизаторов с быстро вращающимися ножами (блендеров) время обработки обычно не превышает 1—2 мин интервалами по 20 с с промежуточным охлаждением биомассы на льду.
Зазор между стеклянной стенкой и тефлоновым пестиком ручного гомогенизатора должен быть в пределах 0,35 — 70 мм. Механический гомогенизатор можно охлаждать в воде со льдом для поддержания низкой температуры при разрушении клеток (в этом случае резко снижается активность внутриклеточных протеиназ) или проводить эксперимент в холодной комнате. Степень разрушения контролируют фазово-контрастной микроскопией или по белку, высвобожденному из клеток.
Растирание клеток. Одним из простых, но достаточно надежных способов разрушения клеток микроорганизмов является растирание их суспензий с абразивами. Для этой цели применяют промытый кварцевый песок, толченое кварцевое стекло, пудру окиси алюминия или другие твердые вещества. При разрушении к клеточной пасте постепенно добавляют двойное количество (по массе) абразива и растирают с силой до появления щелкающих звуков (обычно 10—15 мин после добавления последней порции абразива). В процессе растирания клеточная паста подвергается нагреву, поэтому дно ступки следует охлаждать (на лотке со льдом).
Особо следует отметить способ растирания клеток микроорганизмов, замороженных в жидком азоте, где в качестве разрушающего абразива выступают кристаллики льда, образовавшиеся при быстром замораживании биомассы (вкапывание суспензии
в буфере в толстостенную ступку с налитым жидким азотом). Для растирания применяют обычно фарфоровые ступки и пестики. Время обработки зависит от толщины клеточных стенок разрушаемого микроорганизма. Этими способами можно растирать до 30 г сырых клеток за один прием. После растираний к полученной массе добавляют равный объем буферного раствора и центрифугируют для удаления абразивов, неразрушенных клеток и их крупных обломков.
Разрушение со стеклянными бусами. Данный метод можно применять даже для разрушения клеточных стенок дрожжей, у которых она очень прочная, поскольку он основан на механическом растирании клеток, попадающих между двумя мелкими стеклянными шариками, вращающимися с большой скоростью в механических мельницах. Малые порции биомассы можно разрушать и в пробирке, используя пробирочный смеситель («вортекс»). Для больших порций клеток необходимо применять специальные механические смесители («мельницы»).
Отмытые клетки ресуспендируют в равном объеме буфера и помещают в прочную центрифужную пробирку с навинчивающейся пробкой для предотвращения вытекания жидкости при встряхивании. В пробирку добавляют 1 — 3 г охлажденных стеклянных шариков на каждый грамм клеточной массы и встряхивают на смесителе с максимальной скоростью 3 — 5 раз в течение 1 мин. В интервалах суспензию охлаждают на льду. Для микроколичеств применяют пробирки Эппендорф.
Стеклянные шарики перед опытом отмывают концентрированной соляной кислотой (или хромовой смесью), водопроводной, а затем дистиллированной водой до нейтральной реакции и высушивают в сушильном шкафу. К недостаткам метода относятся сильный разогрев содержимого при встряхивании.
13.1.3. Ультразвуковой дезинтегратор и обработка клеток ультразвуком
Разрушение клеток ультразвуковым методом происходит в результате микроколебаний суспензии бактерий или других микроорганизмов, помещенных в стеклянный стакан с излучателем ультразвуковых волн. Ультразвук создает высокую степень вибрации суспензии, вследствие чего происходит механический разрыв клеток. Для достижения максимального эффекта необходимо применять высокую мощность излучателя и тщательно настраивать его в резонанс с пробой. Следует контролировать уровень пенообразования и перемешивая при обработке, поскольку интенсивное пенообразование вызывает денатурацию белков в образующихся тонких пленках на разделе фаз жидкость/газ. Внутренние структуры клеток при этом также подвержены разрушению.
Внешний вид отечественного ультразвукового дезинтегратора УЗДН-1 приведен на рис. 13.1. При подготовке прибора к работе излучатель ультразвуковых колебаний присоединяют к соответствующему гнезду прибора. Кончик пестика излучателя должен быть опущен в обрабатываемую суспензию на глубину 1 — 2 см. Перед началом обработки излучатель должен быть настроен в резонанс при выбранной мощности на порции подвергаемой обработке пробы или другой сходной по вязкости суспензии в сосуде, используемой для озвучивания. Обычно обработку проводят в пластиковых (полиэтиленовых) центрифужных стаканах, но можно использовать стеклянные стаканы или стаканы из нержавеющей стали. Необходимым условием является строгий контроль за нагревом излучателя и температурой материала. Излучатель охлаждают холодной проточной водопроводной водой, а пробу — в воде со льдом.
Имеется три типа излучателей: на 15, 22 и 35 кГц. Для разрушения бактерий чаще всего используют излучатель на 22 кГц, к которому необходимо подвести для охлажде-
ния воду от водопровода (вход — снизу, слив — сверху). Прибор включают в сеть. После 5-минутного прогрева включают анодное напряжение и ручкой «Анодный ток» устанавливают на шкале амперметра ток желаемой величины (обычно 0,7 А). Далее ручкой «Резонатор» вводят прибор в резонанс, что можно проконтролировать по резкому возрастанию уровня звука и минимальному значению тока. При выбранном значении облучают суспензию клеток (1г сырой биомассы и 5 мл среды разрушения) последовательно 6—10 раз с интервалами 30 с при промежуточном охлаждении излучателя и суспензии в воде со льдом. Разрушенные клетки затем осаждают на скоростной центрифуге (примерно 20 тыс. g в течение 10—15 мин при 4 °C) и надосадочную жидкость используют для определения ферментативных активностей. В целях безопасности при облучении суспен-
Рис. 13.1. Схема ультразвукового дезинтегратора УЗДН-1 (справа — панель управления):
1 — тумблер и лампочка общего включения прибора; 2 — тумблер и лампочка включения анодного напряжения; 3 — ручка и шкала мощности прибора; 4 — ручка выбора частоты излучения; 5 — ручка подстройки разонансной частоты; 6 — выходной разъем; 7 — излучатель; 8 — патрубки входа и выхода охлаждающей воды; 9 — шнур подключения излучателя к выходному разъему прибора; 10 — штатив крепления излучателя
зии ультразвуком прибор должен быть заключен в кожух1. Описанным методом можно разрушать многие грамположительные бактерии, но клетки актиномицетов, дрожжей и других грибов таким методом разрушаются с трудом. Иногда для усиления эффекта ультразвуковой обработки к суспензии клеток добавляют кварцевый песок, стеклян-
ные бусы малого диаметра или пудру окиси алюминия.
13.1.4. Разрушение клеток с помощью пресса
Френч-пресс. Ячейка Френч-пресса предназначена для разрушения микроорганизмов, основанного на перепаде давления в пробе от высокого (550 — 1400 ати) до атмосферного. Быстрое изменение давления взрывает клетки изнутри и таким образом разрушает их. Метод наиболее применим к суспензиям объемами от 10 до 30 мл; разрушение меньших объемов сложно технически, а больших — занимает много времени. Время, требуемое для разрушения одной порции клеток небольшого объема, обычно не превышает 10 — 15 мин. В перерывах необходимо тщательно промывать ячейку от предыдущей пробы и постоянно контролировать состояние полиэтиленового шарика, регулирующего на ячейке степень перепада давления.
Перед разрушением клеточную пасту смешивают с буферным раствором в соотношении от 1; 1 до 1:4 г/мл. Суспензию помещают в ячейку и ставят в гидравлический пресс. Медленным открыванием клапана устанавливают требуемую скорость вытекания разрушенного гомогената (обычно 1 капля/с). Для некоторых микроорганизмов необходима двойная или даже тройная обработка, чтобы достичь необходимой степени разрушения. Ячейку перед заполнением суспензией охлаждают в ледяной воде. Степень разрушения контролируют микроскопированием или по увеличению вязкости жидкости (в результате выхода ДНК).
го разрушения
Рис. 13.2. Пресс для декомпрессионного разрушения клеток: 1 — поршень цилиндра; 2 — цилиндр с отверстием для поршня; 3 — нижняя часть пресса с приемным стаканом для разрушенных клеток; 4 — отверстие для выхода клеток
Х-пресс. Разрушение клеток с применением Х-пресса основано на том же принципе, что и на Френч-прессе, за исключением состояния разрушаемой суспензии. Перед разрушением приготовленную суспензию клеток в буфере замораживают в ячейке (рис. 13.2) при -70 °C и затем продавливают через тонкую фильеру с помощью гидравлической системы. Микроорганизмы при этом подвергаются перепаду давления от 500 до 1000 ати и дополнительно абразивному действию кристалликов льда внутри клеток. Под действием давления (обычно это усилие до 20 т на шкале гидравлического пресса) происходит постепенно разогрев замороженного образца. Когда температура суспензии поднимается до -10 — 15 °C, начинается продавливание пробы через фильеру. На этом этапе для максимальнонеобходимо поддерживать давление по манометру на уровне
10— 15 т. О конце процесса судят по степени вхождения поршня в ячейку. Пос-
ле продавливания всей порции клеток (поршень входит в верхнюю часть стакана до риски) давление сбрасывают, отделяют верхнюю часть стакана от нижней и размораживают разрушенные клетки. Гомогенат центрифугируют при 20 тыс. g в течение 10—15 мин при 4 °C для осаждения неразрушенных клеток и их крупных обломков. Этим методом можно разрушить за один прием до 10 г сырых клеток (количество зависит от размеров ячейки для разрушения). Метод неудобен для многократного разрушения клеток, поскольку приходится замораживать и оттаивать ячейку. Обычно время разрушения одной пробы не превышает 15 — 20 мин.
По степени устойчивости к механическому разрушению на прессе клетки микроорганизмов можно выстроить в следующий ряд: дрожжи > мицелиальные грибы > грамположительные кокки > грамположительные палочки > грамотрицательные палочки > галобактерии > микоплазмы. Данный метод разрушения по степени его действия на клетку наиболее эффективен и позволяет разрушить даже клетки дрожжей.
13.2. ИЗУЧЕНИЕ ФЕРМЕНТОВ МИКРООРГАНИЗМОВ
По характеру связи с протоплазменными структурами различают эндо- и экзоферменты, или внутри- и внеклеточные. Для полной характеристики ферментной системы микроорганизма определяют ферменты в биомассе и культуральной жидкости. Однако следует иметь в виду, что в среде могут находиться не только экзоферменты, но также эндоферменты, высвободившиеся в результате лизиса отмерших клеток.
Для выделения высокоактивных препаратов ферментов необходимо знать условия культивирования микроорганизма, оптимальные для биосинтеза и активности желае
мого фермента. В случае объектов с неизученной физиологией лучше брать активно растущие культуры, клетки которых богаче ферментами, а их состав разнообразней.
Для определения экзоферментов получают культуральную жидкость, осаждая клетки центрифугированием при 5 — 10 тыс. g в течение 10 — 30 мин. Режим центрифугирования варьируют в зависимости от микроорганизма. В целях выделения эндоферментов биомассу разрушают одним из описанных ранее способов (см. 13.1), затем получают грубые, или бесклеточные, экстракты осаждением неразрушенных клеток (18 тыс. g, 30 мин, 4 °C). В случае необходимости бесклеточные экстракты подвергают дальнейшему фракционированию на растворимую и мембранную фракции ультрацентрифугированием при 140 тыс. g в течение 2 —4 ч. В зависимости от локализации изучаемого фермента в качестве анализируемого материала используют культуральную жидкость, бесклеточный экстракт, растворимую или мембранную фракцию.
Активность ферментов выражают в единицах. За единицу активности любого фермента {Е) принимают такое его количество, которое катализирует превращение 1 мкмоля субстрата (или образование 1 мкмоля продукта) в 1 мин при определенных условиях реакции — pH и температуре. Удельная активность выражается числом единиц активности фермента, приходящихся на 1 мг белка.
13.2.1. Липаза
Липаза (НФ 3.1.1.3) принадлежит к группе эстераз, катализирующих гидролиз сложных эфиров глицерола с образованием глицерина и жирной органической кислоты.
Способность разлагать жиры широко распространена среди микроорганизмов — бактерий, мицелиальных грибов {Aspergillus, Rhizopus) и дрожжей (Candida). У микроорганизмов липаза встречается в виде эндо- и экзофермента.
Полагают, что оптимум pH липазы микроорганизмов находится в области 8,0. Однако при выяснении наличия у объектов липолитической активности определение ведут как при кислом (4,5 — 5,6), так и при щелочном (7,6 — 8,5) значениях pH.
Определение. Липазную активность измеряют спектрофотометрически с «-нитро-фенилпальмитатом («-НФП) в качестве субстрата по увеличению поглощения при 405 нм в результате освобождения «-нитрофенола (коэффициент молярной экстинкции 18,3 мМ^-см-1). Субстрат («-НФП) растворяют в смеси, состоящей из ацетонитрила, изопропанола и 50 мМ трис-HCl буфера (pH 8,0) в объемном соотношении 1:4:95, до конечной концентрации 100 мкМ. Реакционная смесь (1 мл) содержит 985 мкл субстратной смеси, 10 мкл 0,5 М СаС12 и 5 мкл липазы в концентрации 0,2 мкг/мкл. За единицу активности принимают количество фермента, необходимое для образования 1 мкмоля «-нитрофенола в минуту при указанных условиях реакции.
Липазную активность можно также определить, титруя щелочью свободные жирные кислоты, образующиеся из оливкового масла в результате действия фермента. Реакционную смесь, содержащую 0,5 мл оливкового масла, 5 мл ацетатного буфера 'pH 5,6), 10 мМ СаС12 и 50—100 мкл раствора фермента, инкубируют при 30°C в течение 30 мин на магнитной мешалке. Реакцию останавливают добавлением 20 мл этанола. Количество образовавшихся жирных кислот определяют титрованием 0,1 н. КОН с индикатором фенолфталеином. Контролем служит проба с прокипяченным раствором фермента. Результаты титрования контроля вычитают из данных опытных проб. Активность липазы выражают в миллилитрах 0,1 н. КОН. За единицу липазной активности принимают количество 0,1 н. щелочи, пошедшей на титрование 1 мкмоля жирных кислот, образующихся за 1 мин.
13.2.2. Инвертаза
Инвертаза (Р-фруктофуранозидаза, НФ 3.2.1.26) катализирует гидролиз дисахарида сахарозы на два мономера — глюкозу и фруктозу. Инвертаза действует и на трисахарид раффинозу, при этом образуется молекула фруктозы и молекула дисахарида мелибиозы. Фермент гидролизует р-фруктофуранозидные остатки в p-фруктофуранозидах с нередуцирующего конца. Причем мальтаза (сс-Д-глюкозидаза), отщепляющая терминальные остатки глюкозы, тоже может катализировать эту реакцию. При определении фермента следует учитывать, что сахароза достаточно легко гидролизуется и неферментативным путем при кислой реакции среды.
Инвертаза широко распространена среди различных групп микроорганизмов, причем многие культуры имеют более одной формы фермента: внеклеточный и внутриклеточный.
Инвертаза обладает достаточно высокой активностью в широком диапазоне pH (3,5 —5,5) с оптимумом активности при pH 4,5; температурный оптимум фермента находится около 55 °C.
Выделение. Дрожжи, пленку плесени или бактериальную массу обезвоживают сушкой при 40 °C или водоотнимающими реактивами. Сухой препарат растирают в ступке с трехкратным количеством кварцевого песка или стеклянных бус, прибавляют десятикратное количество воды, несколько капель толуола и оставляют при 35 °C на 2 ч. После этого смесь фильтруют через бумажный фильтр и прозрачный фильтрат употребляют в качестве препарата инвертазы. Альтернативно получают бесклеточный экстракт любым из приведенных в гл. 15 методов и используют его в качестве источника фермента.
Определение. В пробирку наливают 3 мл раствора фермента в 100 мМ ацетатном буфере (pH 4,5) и инкубируют в течение 5 мин на водяной бане при желаемой температуре (23 — 40 °C). Затем прибавляют 3 мл предварительно подогретого до той же температуры 5%-го раствора сахарозы и оставляют на 5 мин. По окончании экспозиции гидролиз сахарозы останавливают прибавлением 6 мл щелочного реагента на основе динитросалициловой кислоты (ДИСК). Контрольную пробу ставят с прокипяченным раствором фермента. Количество редуцирующих сахаров определяют колориметрическим методом с ДНСК-реагентом. С этой целью пробирки инкубируют в течение 10 мин на водяной бане при 95 °C для развития характерной красно-коричневой окраски, охлаждают реакционную смесь и измеряют поглощение при 540 нм. Количество гидролизованной сахарозы определяют по калибровочной кривой, построенной на основании поглощения эквимолярного раствора глюкозы и фруктозы. За единицу инвертазной активности принимают такое количество фермента, которое гидролизует 1 мкмоль сахарозы в 1 мин при pH 4,5 и температуре реакции.
ДНСК-реагент готовят, смешивая в 1 л 10 г 3,5-динитросалициловой кислоты, 16 г NaOH и 300 г K-Na-тартрата тетрагидрата.
13.2.3. Амилаза
Амилаза — фермент, катализирующий гидролиз крахмала и гликогена. Растительный крахмал состоит из двух полимеров — амилозы (15 —25 %) и амилопектина. Амилоза представляет собой линейную цепь из остатков Д-глюкозы, связанных 1,4-а-связями. Амилопектин — это тоже поли-1,4-а- Д-глюкоза, но его молекула сильно разветвлена благодаря наличию 1,6-ос-связей. Амилоза малорастворима в воде и окрашивается при взаимодействии с йодом в синий
цвет. Амилопектин, напротив, растворяется в горячей воде с набуханием, образуя крахмальный клейстер, а йодом окрашивается в коричневый цвет.
а-Амилаза (1,4-а-Д-глюканглюканогидролаза, НФ 3.2.1.1) расщепляет крахмал по эндотипу, гидролизуя 1,4-а-связи в амилозе и амилопектине во всех частях молекулы. Это приводит к быстрому снижению вязкости раствора крахмала и его способности окрашиваться йодом в синий цвет. Гидролиз крахмала сопровождается образованием низкомолекулярных олигосахаридов, «-декстринов, ограниченных 1,6-а-связями, а также мальтозы и глюкозы.
Р-Амилаза (1,4-а-Д-глюканмальтогидролаза, НФ 3.2.1.2) расщепляет крахмал по экзотипу: гидролизует 1,4-а-связи через одну с нередуцирующих концов цепей, образуя мальтозу с редуцирующей группой без промежуточных продуктов, а также высокомолекулярные декстрины, ограниченные p-связями. При воздействии р-амилазы крахмал долгое время сохраняет способность окрашиваться йодом.
р-Амилаза разрушается при нагревании до 70 °C в течение 15 мин, а термостабильная а-амилаза почти не снижает своей активности при этой температуре, зато теряет активность при кратковременном снижении pH среды до 3,3. Этим пользуются для раздельного определения амилаз при их совместном присутствии.
а-Амилаза широко распространена у плесневых грибов (Aspergillus), бактерии же образуют амилазы обоих типов (Bacillus).
Активность а-амилазы определяют методом йодной пробы, а также вискозометрически по скорости разжижения крахмального клейстера. р-Амилазу, как и суммарное действие обоих ферментов, определяют по количеству образовавшейся мальтозы.
Определение. Активность а-амилазы измеряют по методу Смита и Роя в модификации Маннинг и Кемпбелла. Метод основан на фотометрическом определении скорости изменения окраски крахмала с йодом под действием фермента. Реакционная смесь (1 мл) содержит 50 мМ Na-ацетатный буфер (pH 5,5), 0,1 М СаС12 и 1%-й раствор растворимого крахмала (навеску крахмала растворяют в небольшом объеме холодной воды, после чего вливают при постоянном помешивании в доведенную до кипения воду). Реакцию гидролиза крахмала начинают добавлением 50 мкл раствора фермента. Смесь инкубируют в течение 10 мин при 25 °C, затем реакцию останавливают добавлением 1 мл 1 М НС1, прибавляют 0,2 мл 0,5%-го раствора йода в 5%-м водном растворе KI и измеряют оптическую плотность на спектрофотометре при 620 нм или на ФЭК (фильтр № 7) против воды. Контрольную пробу ставят с прокипяченным раствором фермента. Действие фермента учитывают по разнице в окраске между контрольной и опытной пробами. За единицу амилолитической активности условно принимают такое количество фермента, которое вызывает снижение оптической плотности при 620 нм на 0,01 ед. в 1 мин при pH 5,5 и температуре 25 °C.
Другой метод измерения а-амилазной активности основан на количественном определении образованной в результате гидролиза крахмала глюкозы. Реакционная смесь содержит 1%-й растворимый картофельный крахмал в 50 мМ Na-ацетатном буфере (pH 5,5) и раствор фермента. Реакцию останавливают после 10 мин инкубации при 55 °C добавлением равного объема ДНСК-реагента (см. 13.2.2). Контрольную пробу ставят с прокипяченным раствором фермента. Количество редуцирующих сахаров определяют колориметрическим методом с ДНСК-реагентом. Для этого пробирки инкубируют в течение 5 мин в кипящей водяной бане для развития характерной красно-коричневой окраски, охлаждают реакционную смесь и измеряют поглощение при 540 нм. Количество гидролизованного крахмала определяют по калибровочной кривой, построенной на основании поглощения раствора глюкозы. За единицу активности фермента принимают количество амилазы, которое образует 1 мкмоль редуцирующих сахаров (глюкозы) в минуту при указанных условиях реакции.
13.2.4. Мальтаза
Мальтаза (а-глюкозидаза, НФ 3.2.1.20) гидролизует 1,4-связи концевого нередуциующего остатка а-D-глюкозы с освобождением «.-глюкозы. Фермент содержится в плесневых грибах, дрожжах и бактериях. Плесневые грибы содержат истинную мальтазу, а дрожжи — менее специфичную сс-глюкозидазу, действующую и на другие о-глюкозиды.
Получение. Мальтазу получают из дрожжей одним из следующих способов.
1. Густую суспензию свежих дрожжей в 0,1 М цитратном буфере (pH 6,5), содержащем ингибиторы протеаз (0,1 М ЭДТА; 1 мМ ДТТ; 0,17 мг/мл фенилметилсульфонил-фторида; 0,7 мкМ пепстатина), растирают с кварцевым песком или со стеклянным порошком в фарфоровой ступке в течение 5 мин.
2. Прессованные дрожжи растирают с сухим (NH4)2HPO4 и экстрагируют фермент 10-кратным количеством воды.
3. Прессованные дрожжи измельчают, расстилают тонким слоем на листы фильтровальной бумаги и медленно высушивают в токе воздуха при 40 °C. Из сухих дрожжей мальтазу извлекают настаиванием с водой. Полученные гомогенизаты центрифугируют (11 тыс. g, 10 мин, 4 °C), супернатанты используют для определения «.-глюкозидазной активности.
Определение. Мальтазную активность измеряют спектрофотометрически с «-нитро-фенил-а-Д-глюкопиранозидом в качестве субстрата по увеличению поглощения при 405 нм в результате освобождения «-нитрофенола (коэффициент молярной экстинкции 18,3 мМ-1 • см-1). В спектрофотометрическую кювету вносят 1 мл раствора и-нитро-фенил-глюкопиранозида в 50 мМ К-фосфатном буфере с pH 6,8 (1 мг/мл) и измеряют оптическую плотность при 405 нм. Затем добавляют 50—100 мкл бесклеточного экстракта и продолжают измерение. Контрольную пробу ставят с прокипяченным раствором фермента. Удельную активность выражают в наномолях «-нитрофенола, образующегося в минуту на 1 мг белка. За единицу мальтазной активности принимают такое количество фермента, которое освобождает 1 мкмоль «-нитрофенола в 1 мин при pH 6,8 и 25 °C. В бесклеточных экстрактах «-глюкозидазу можно определять спектрофотометрически в системе, сопряженной с глюкозо-6-фосфатдегидрогеназой и гексокиназой. В присутствии АТФ и гексокиназы происходит фосфорилирование образовавшейся в результате мальтазной активности глюкозы в глюкозо-6-фосфат. Далее глюкозо-6-фос-фат окисляется глюкозо-6-фосфатдегидрогеназой с образованием эквивалентных количеств НАДФН, который определяют спектрофотометрически. В спектрофотометрическую кювету вносят реакционную смесь, содержащую 50 мМ имидазольный буфер (pH 7,0), 1 мМ MgCl2, 3 мМ АТФ, 0,4 мМ НАДФ+, по 0,5 единиц глюкозо-6-фос-фатдегидрогеназы и гексокиназы на 1 мл и необходимое количество экстракта. Пробу перемешивают и измеряют оптическую плотность при 340 нм до ее стабилизации. После этого добавляют сахарозу до конечной концентрации 50 мМ. Содержимое кюветы быстро перемешивают и продолжают измерение оптической плотности при 340 нм до прекращения ее изменения. Количество глюкозы в пробе рассчитывают по количеству образовавшегося НАДФН, используя коэффициент молярной экстинкции 6220 мМ“'-см_|. Растворы ферментов, НАДФ* и АТФ должны находиться в ледяной бане. За единицу мальтазной активности принимают такое количество фермента, которое образует 1 мкмоль глюкозы в 1 мин при pH 7,0 и температуре 25 °C.
13.2.5. Протеазы
Протеазы — ферменты, гидролизующие белки и полипептиды по месту пептидной связи. В зависимости от степени сложности гидролизуемого суб
страта различают протеиназы, полипептидазы и дипептидазы. Протеиназы действуют на нативные белки с образованием пептидов. Полипептидазы и дипептидазы действуют соответственно на полипептиды и дипептиды с образованием аминокислот. Протеолитические ферменты широко распространены у микроорганизмов. Микробные протеазы делят на три класса: сериновые, металло-протеазы и кислые, имеющие оптимум pH при щелочной, нейтральной и кислой реакции среды соответственно.
В силу отсутствия белков и полипептидов строго определенного состава раздельное определение протеаз затруднено, поэтому проводят суммарное определение протеаз.
Определение. Для определения суммарной активности протеаз используют колориметрический метод, основанный на измерении поглощения пигмента, высвободившегося в раствор в результате гидролиза окрашенного нерастворимого аналога белка ферментами. В качестве пигментированных аналогов белков используют азоказеин или азоколлаген, имеющие сульфаниламидные группы, ковалентно присоединенные к пептидным связям. Для приготовления раствора субстрата 500 мг окрашенного белка растирают в фарфоровой ступке, постепенно добавляя 100 мл 20 мМ mpuc-HCl буфера (pH 7,8) до получения равномерной взвеси. Реакционная смесь содержит 1 мл раствора субстрата; 0,5 мл 0,2 М трис-HCl буфера (pH 7,8); 0,5 мл препарата фермента. Пробы инкубируют при 37 °C в течение 10 —15 мин, после чего реакцию останавливают добавлением 2 мл 10%-й трихлоруксусной кислоты (ТХУ). В контрольную пробу 0,5 мл препарата фермента вносят после добавления ТХУ. Не подвергнутый протеолизу субстрат отфильтровывают через бумажные фильтры (Whatman № 1). В фильтратах спектрофотометрически определяют при 366 нм количество высвободившегося под действием протеаз красно-оранжевого пигмента (е = 900 Мг1 • см '). За единицу протеолитической активности принимают количество фермента, необходимое для освобождения 1 мкмоля пигмента в минуту при указанных условиях реакции.
Суммарную активность протеаз можно определить, используя более доступный субстрат — 1%-й раствор казеина в 50 мМ К-фосфатном буфере (pH 7,5). В целях нахождения линейного участка зависимости протеолитической активности от времени ставят серию проб: пять опытных и одну контрольную. В каждую пробу вносят по 1,0 мл раствора казеина и 0,5 мл культуральной жидкости, затем реакционные смеси инкубируют при 37 °C. В пробах последовательно, с интервалами в 2 мин останавливают реакцию добавлением 1,5 мл холодной 10%-й ТХУ для преципитации не подвергнутого ферментативному гидролизу белка. В контрольную пробу вносят 0,5 мл культуральной жидкости после добавления ТХУ. Пробы выдерживают при комнатной температуре в течение 20 — 30 мин для формирования преципитатов, после чего суспензии фильтруют через бумажные фильтры (Whatman № 1). В фильтратах определяют белок методом Лоури. За единицу протеолитической активности принимают такое количество фермента, которое гидролизует казеин с образованием окраски (с реактивом Фолина — Чокальтеу), эквивалентной освобождению 1 мкмоль (181 мкг) тирозина в 1 мин при pH 7,5 и температуре 37 °C.
13.2.6. Уреаза
Уреаза (НФ 3.5.1.5) катализирует гидролиз мочевины до аммиака и углекислоты:
(NH2)2CO + ЗН2О -> 2NH4OH + СО2
Фермент специфичен к мочевине. Уреаза обнаружена у многих бактерий, плесневых грибов и некоторых дрожжей. Оптимум pH уреазы близок к 7,0.
Определение. Уреазную активность измеряют по скорости образования клетками или грубыми экстрактами аммиака из мочевины. Для этого отмытые клетки или грубый экстракт помешают в доведенный до нужной температуры 50 мМ К-фосфатный буфер (pH 7,0). Реакцию инициируют добавлением мочевины до конечной концентрации 10 мМ, отбирают нулевую пробу, затем инкубируют реакционную смесь при желаемой температуре, отбирая пробы через определенные промежутки времени. Клетки в пробах осаждают центрифугированием (10 тыс. g, 5 — 10 мин). Контрольную пробу ставят с прокипяченной аликвотой клеточной суспензии или раствора фермента. Ам-моний-ион в пробах определяют по методу Фосетта и Скотта. Определение основано на реакции между аммиаком, фенолом и гипохлоритом, протекающей в щелочном растворе с образованием интенсивной синей окраски индофенола. К 0,2 мл анализируемого материала добавляют последовательно 0,8 мл раствора А и 0,8 мл раствора Б. Реакционную смесь тщательно перемешивают и инкубируют при 37 °C в течение 30 мин. Образование окрашенного комплекса регистрируют при 578 нм в термостатированной спектрофотометрической кювете при 37 °C против контроля на реактивы. Количество образовавшегося аммония определяют по калибровочной кривой, которую строят по стандартному раствору сернокислого аммония, содержащему 1 — 15 мкг аммоний-иона в 1 мл. За единицу уреазной активности принимают такое количество фермента, которое образует 2 мкмоля NH4 в 1 мин при pH 7,0 и температуре реакции.
Раствор А для хромогенной реакции содержит (г/л): фенол — 10; нитропруссид Na — 0,05. Раствор Б содержит 37 г/л Na2HPO4; 10 г/л NaOH и 10 мл/л 1%-ro NaOCl. Растворы А и Б хранят в сосудах из темного стекла не более двух месяцев.
13.2.7. Целлюлаза
Целлюлаза — сложный ферментный комплекс, участвующий в разложении целлюлозы у микроорганизмов. Целлюлоза является основным структурным компонентом растительных клеток и одним из наиболее распространенных органических соединений. Целлюлоза — линейный полимер из глюкозных остатков, соединенных 1,4-р-связями, число которых достигает нескольких тысяч. Индивидуальные цепочки полимера могут соединяться друг с другом водородными связями, образуя кристаллическую структуру, при этом участки, имеющие кристаллическое строение, чередуются с аморфной целлюлозой.
Целлюлозолитической активностью обладают многие аэробные микроорганизмы: миксобактерии (Polyangium, Sporangium), представители группы Cytophaga, а также грибы {Fusarium, Trichoderma, Penicillium). В анаэробных условиях целлюлозу разлагают в основном клостридии. На грибах было показано, что ферментная система для разложения целлюлозы включает 1,4-р-эндог-люконазы, неупорядоченно разрывающие 1,4-р-связи внутри макромолекулы, 1,4-р-экзоглкжоназы, отщепляющие с нередуцирующих концов ди сахарид целлобиозу, и 1,4-р-глюкозидазы, гидролизующие целлобиозу с образованием глюкозы. Грибные целлюлазы имеют оптимум pH в кислой области (4,5 —5,5), а оптимум температуры около 40 °C.
Определение. О целлюлазной активности судят по скорости образования редуцирующих сахаров под действием фермента. В качестве субстрата для целлюлазы можно использовать фильтровальную бумагу (Whatman № 1), карбоксиметилцеллюлозу (аморфная целлюлоза), хлопок (кристаллическая структура). К смеси из 1 мл 0,1 М Na-аце-татного буфера (pH 5,2) и 1 мл раствора фермента (культуральная жидкость) добавляют 20 мг измельченного субстрата. Реакцию проводят при 37 —40 °C в течение 1 ч.
затем пробы помещают в кипящую водяную баню на 5 мин для прекращения гидролиза. Пробы доводят до комнатной температуры и определяют в них количество образовавшейся глюкозы колориметрическим методом с ДНСК-реагентом (см. 13.2.2). Контроль ставят с прокипяченным раствором фермента.
13.2.8. Алкогол ьоксидаза
Алкогольоксидаза (АО, НФ 1.1.3.13) — растворимый фермент, отвечающий за первую реакцию окисления метанола у метилотрофных дрожжей. Он локализован в пероксисомах, выросших на среде с метанолом клеток, и его содержание в клетке может достигать 20 % от всех растворимых белков. Активность фермента изучают в гомогенатах, полученных после растирания клеток, замороженных в жидком азоте, или после обработки их на прессе. Определение основано на количественном измерении изменения концентрации перекиси водорода, образующейся в реакции окисления метанола АО:
СН3ОН + О2 -> неон + Н2О2
Реакционная смесь содержит в 1 мл: КРО4-буфер с о-дианизидином (pH 7,0, 50 мМ) — 970 мкл; раствор пероксидазы хрена (10 ME) — 10 мкл; метанол (10 мкмоль) — 10 мкл, раствор фермента (0,5 — 1 мг белка) — 10 мкл. Буферный раствор с о-дианизидином готовят следующим образом. К 9 мг о-дианизидина приливают 10 мл ацетона, растворяют реактив и приливают 90 мл КРО4-буфера (pH 7,0, 50 мМ). Раствор стабилен при 4 °C в течение недели. Реакцию начинают добавлением фермента (АО) и изменение поглощения раствора регистрируют при 436 нм в течение 1—2 мин. За единицу активности АО принимают количество фермента, которое окисляет 1 мкмоль метанола в минуту при pH 7,0 и 20 °C.
Расчет активности фермента проводят с учетом коэффициента молярной экстинкций о-дианизидина — 8,3 мМ-1 см-1.
13.2.9. Глюкозооксидаза
Глюкозооксидаза (ГО, НФ 1.1.3.4) — растворимый внеклеточный фермент, образуемый многими микромицетами. Определение основано на количественной оценке изменения концентрации перекиси водорода, образующейся в реакции окисления глюкозы ГО:
глюкоза + О2 -> глюконолакгон + Н2О2
Реакционная смесь содержит в 1 мл (мкл): КРО4-буфер с о-дианизидином (pH 7,0, 50 мМ) — 970 (приготовление см. выше); раствор пероксидазы хрена (10 ME) — 10; глюкоза (10 мкмолей) — 10; раствор фермента (0,5—1 мг белка) — 10. Реакцию начинают добавлением фермента (ГО) и регистрируют изменение поглощения раствора при 436 нм в течение 1 — 2 мин. За единицу активности ГО принимают количество фермента, которое окисляет 1 мкмоль глюкозы в 1 мин при pH 7,0 и 20 °C.
13.2.10. Глутаматоксидаза
Глутаматоксидаза (ГлутО, НФ 1.4.3.11) — растворимый внеклеточный фермент, образуемый некоторыми стрептомицетами. Определение основано на
количественном учете изменения концентрации перекиси водорода, образующейся в реакции окисления глутаминовой кислоты ГлутО:
L-глутаминовая кислота + О2 + Н2О -> сс-кетоглутаровая кислота + + NH3 + Н2О2
Реакционная смесь содержит в 1 мл (мкл): КРО4-буфер с о-дианизидином (pH 7,0. 50 мМ) — 970 (приготовление см. выше); раствор пероксидазы хрена (10 ME) — 10: L-глутамат (10 мкмолей) — 10; раствор фермента (0,5—1 мг белка) — 10. Реакцию начинают добавлением фермента (ГлутО) и регистрируют изменение поглощения раствора при 436 нм в течение 1—2 мин. За единицу активности ГлутО принимают количество фермента, которое окисляет 1 мкмоль глутамата в 1 мин при pH 7,0 и температуре 20 °C.
13.2.11. Оксидаза
Оксидаза — конечный переносчик электронов от восстановительных эквивалентов по дыхательной цепи, передающий их непосредственно на кислород с последующим его восстановлением до воды. Терминальная оксидаза присуща всем аэробным микроорганизмам и даже некоторым анаэробным, в частности сульфатредукторам. Бактерии и археи зачастую имеют разветвленные дыхательные цепи с двумя или более оксидазами. На основании природы донора электронов — цитохром с или хинол — различают соответственно цитохром- или хинол оксидазы. Оптимумы pH и температуры ферментов существенно варьируют в зависимости от объекта.
Определение. Оксидазную активность определяют полярографически. Реакционная смесь для измерения цитохромоксидазной активности содержит соответствующий буфер; исследуемый материал (грубый экстракт или мембранную фракцию); 2 мМ аскорбат и 0,2 мМ Д', Л'',Л,'',Л''-'1е'|раме'1ИЛ-«-(]|енилендиамин (ТМФД). В случае измерения хинолоксидазной активности вместо аскорбата/ТМФД в качестве субстрата используют восстановленные 5 мМ ДТТ хинолы или их аналоги (дурохинол).. Оксидазную активность выражают в наномолях потребленного кислорода в 1 мин на 1 мг белка.
Цитохромоксидазную активность можно измерить также спектрофотометрически по скорости образования окисленной формы ТМФД (синий Вурстера). В спектрофотометрическую кювету вносят соответствующий буфер, 0,2 мМ ТМФД и измеряют увеличение оптической плотности при 560 нм в результате окисления субстрата кислородом воздуха, затем добавляют раствор фермента и продолжают измерение. Оксидазнук активность определяют по разнице между двумя скоростями окисления (опытной и контрольной), учитывая, что коэффициент молярной экстинкции ТМФД при 560 нм составляет 12,1 мМ’1 • см’1. За единицу оксидазной активности принимают такое количество фермента, которое окисляет 1 мкмоль ТМФД в 1 мин при соответствующих условиях реакции.
13.2.12. Пероксидаза
Пероксидаза (НФ 1.11.1.7) катализирует окисление полифенолов в присутствии органических перекисей или перекиси водорода. Образуя с перекиськ комплексное соединение, она активирует кислород перекиси и с его участием окисляет субстрат:
Н2О2 + ДН2 -> 2Н2О + Д
С кислородом воздуха пероксидаза не реагирует. В составе каталитического центра фермент содержит железо, обладает сравнительно высокой термостабильностью и ограниченной специфичностью: способен действовать на пирогаллол, гидрохинон, пирокатехин, ортокрезол и некоторые другие фенолы и ароматические амины.
Оптимум pH пероксидазы находится в нейтральной или слабощелочной области и несколько колеблется в зависимости от субстрата. Оптимальная температура для активности около 20 °C.
Определение. Пероксидазную активность измеряют колориметрическим методом по скорости развития окраски в индикаторной реакции разложения Н2О2 ферментом. Освобожденный в пероксидазной реакции кислород окисляет индикатор (например, о-дианизидин), переводя его в окрашенное соединение Д: Н2О2 + ДН2-> 2Н2О + Д. В спектрофотометрическую кювету вносят 840 мкл 0,5 М К-фосфатного буфера (pH 7,0); 10 мкл 3%-й Н2О2; 100. мкл хромогена (5 мг о-дианизидингидрохлорида/мл) и измеряют поглощение при 436 нм. Затем добавляют 50 мкл раствора фермента и фиксируют увеличение оптической плотности за счет развития желто-оранжевой окраски. Контролем служит проба, содержащая прокипяченный раствор фермента. Пероксидазную активность определяют по разнице между двумя скоростями окисления (опытной и контрольной), учитывая, что коэффициент молярной экстинкции окисленной формы о-дианизидина при 436 нм составляет 8,3 мМ-1 • см-1. За единицу пероксидазной активности принимают такое количество фермента, которое окисляет 1 мкмоль о-дианизидина в 1 мин при pH 7,0 и температуре 25 °C.
13.2.13. Каталаза
Каталаза (НФ 1.11.1.6) катализирует разложение перекиси водорода на воду и кислород:
2Н2О2 -> 2Н2О + О2
Каталаза широко распространена в природе; она содержится как у аэробных, так и у анаэробных микроорганизмов. У большинства микроорганизмов каталаза обладает широкими оптимумами pH (4,5 —9,0) и температуры (15 — 40 °C). Однако каталазную активность принято измерять при нейтральном значении pH и комнатной температуре.
Определение. Каталазную активность определяют спектрофотометрически по скорости разложения перекиси водорода ферментом. В спектрофотометрическую кювету вносят 50 мМ К-фосфатный буфер (pH 7,0), 50 мМ Н2О2 и измеряют поглощение при 240 нм. Затем добавляют раствор фермента и фиксируют уменьшение оптической плотности в результате разложения Н2О2 (е240 = 43,6 М-1 • см-1). За единицу каталазной активности принимают такое количество фермента, которое разлагает 1 мкмоль Н2О2 за 1 мин при pH 7,0 и температуре 25 °C.
РАЗДЕЛ III
ВЫДЕЛЕНИЕ МИКРООРГАНИЗМОВ ОТДЕЛЬНЫХ ФИЗИОЛОГИЧЕСКИХ ГРУПП
Глава 14
АЭРОБНЫЕ ОРГАНОТРОФНЫЕ МИКРООРГАНИЗМЫ
Микроорганизмы редко развиваются в виде чистых культур в природных популяциях, однако свойства отдельных видов изучают на чистых культурах. Поэтому необходимо выделять чистые культуры из природных сообществ, где различные микробы сосуществуют в одном и том же микроокружении. Постановка накопительных культур позволяет преимущественно увеличить численность желаемой физиологической группы микроорганизмов перед выделением чистых культур (подробнее см. гл. 6).
Аэробные хемоорганогетеротрофы в природе ответственны за основную массу минерализуемого отмершего органического вещества (например, листового опада). Они перерабатывают также белки, жиры, полисахариды, нуклеиновые кислоты и другие мономеры и полимеры мортмассы. В данной главе рассмотрены некоторые примеры и приемы, позволяющие выделить аэробных органо-трофов из природных сообществ. Другие примеры выделения приведены в тексте задач малого практикума по микробиологиии для студентов-биологов (см. приложение 1).
14.1. АММОНИФИЦИРУЮЩИЕ БАКТЕРИИ
Аммонификация — это процесс аэробного или анаэробного превращения органического азота в аммиачную форму. Его осуществляют микроорганизмы различных физиологических и таксономических групп, включая бактерии, ак-тиномицеты и грибы. Процесс аммонификации происходит с помощью внеклеточных протеаз различной природы, которые могут гидролизовать белки в широком диапазоне pH — от кислых значений до щелочных. Аминокислоты, получающиеся в результате такого гидролиза, поступают в клетку в результате диффузии или с помощью специфических транспортных процессов, где дезаминируются с выделением аммиака обычно в газообразной форме. Клетки используют органические кислоты в качестве источника углерода и энергии, часть образованного аммиака может усваиваться также в анаболических пре -цессах, однако большая его часть выделяется в свободном виде. Аммонификацию в средние века использовали в процессе получения селитры, компонента дымного пороха, после окисления аммиака в аэробном процессе нитрифика-
торами первой и второй фазы. При аммонификации белков выделяются также сероводород, меркаптаны, индол и скатол — все они имеют резко неприятный запах гниющих яиц. Аммиак, выделяющийся при аммонификации, поступает в глобальный цикл азота, где может быть окислен нитрификаторами, ассимилирован в белки или усвоен растениями в виде нитратов (с последующей ассимиляционной нитратредукцией). В последнее время аммонификацией называют также процесс, который приводит к восстановлению нитратов до аммиака с получением при этом метаболитической энергии.
Для изучения процесса аммонификации подходят простые среды (МПБ с 3 % пептона), которые засевают пробами любой почвы. Для обнаружения выделяющегося аммиака между стенками колбы и ватной пробкой закрепляют красную лакмусовую бумажку (проба на аммиак) и фильтровальную полоску бумаги, смоченную раствором уксуснокислого свинца (проба на сероводород и меркаптаны). Опытные колбы обычно инкубируют при 28 — 30 °C и анализируют на 3 —5-е сутки. Подробнее о постановке опыта см. в приложении 1.
14.2. УРОБАКТЕРИИ
Уробактерии являются распространенными представителями микробиоты, использующими органический азот и, в частности, разлагающими мочевину. Мочевина — продукт разложения органических соединений азота в организме животных и человека, посредством образования этого соединения животные выводят избыток аммиака через мочевую систему. Мочевина разлагается ферментом уреазой, образуемой многими представителями различных физиологических групп микроорганизмов — как про- так и эукариотических. Фермент может быть вне- и внутриклеточным относится к классу гидролаз:
CO(NH2)2 + Н2О -> 2NH3 + СО2
Уробактерии используют некоторые углеводы или органические кислоты в качестве источника углерода и энергии. Принципы постановки накопительных культур для выделения уробактерий — те же, что и для аммонификаторов । с лакмусовым тестом на образование аммиака). Подробнее о постановке опыта см. в приложении 1.
14.3. ВЫДЕЛЕНИЕ БАКТЕРИЙ — ПРОДУЦЕНТОВ ПЕКТОЛИТИЧЕСКИХ ФЕРМЕНТОВ
Пектиновые (межклеточные) вещества (от греч. pectys — студень) представляют собой полисахариды, состоящие из остатков £>-галактуроновой кислоты, соединенных а-1,4-гликозидными связями. Карбоксильные группы полигалак-туроновой кислоты могут быть полностью или частично этерифицированы метанолом. Пектиновые вещества содержатся в любом растительном материале, но больше всего их в землянике, яблоках, грушах, крыжовнике, персиках, свекле, луке, моркови.
Пектиновые вещества подразделяются на несколько групп. Протопектин представляет собой нерастворимое в воде соединение сложного химического
строения. Пектиновая кислота — полигалактуроновая кислота, в малой степени этерифицированная остатками метанола; пектин — почти полностью эте-рифицированная пектиновая кислота; пектовая кислота — полигактуроновая кислота; пектинаты — соли пектиновой кислоты; пектаты — соли пектовой кислоты. В присутствии сахара, кислоты и под действием поливалентных ионов металлов пектины образуют гель.
Пектиновые вещества являются субстратами для ряда пектолитических (пектинолитических) ферментов — пектинлиазы (КФ. 4.2.2.10), экзополиталакту-ронатлиазы (КФ. 4.2.2.9), полигалактуроназы (полигалактуронидгликаногид-ролаза, КФ. 3.2.1.15), пектинэстеразы (пекгингидролаза, КФ. 3.1.1.11). Пек-тинлиаза деполимеризует пектин путем элиминирования остатков 6-метил-Д-4,5-Л-галактуроната. Экзополигалактуронатлиаза элиминирует Д-4,5-7)-галак-туронозо-£>-галактуронатные остатки с редуцирующего конца деэтерифици-рованного пектина. Полигалактуроназа осуществляет гидролитическое расщепление <х-1,4-гликозидных связей в цепи пектиновых веществ. Пектинэстераза атакует этерифицированные карбоксильные группы.
Пектолитические ферменты образуют комплексы, отдельные компоненты которых расщепляют молекулу пектина в различных местах. Комплекс ферментов, синтезируемый микроорганизмами, различен и зависит от вида продуцента, состава среды и условий культивирования.
Значительное число микроорганизмов образуют ферменты, разлагающие пектины. Они широко распространены у грибов и бактерий.
Бактерии рода Clostridium, например С/, pectinofermentans, Cl. butyricum, продуцируют комплекс пектолитических ферментов и получают энергию, осуществляя маслянокислое брожение пектиновых веществ. Химизм брожения пектиновых веществ состоит из двух последовательных стадий. В первой стадии они гидролизуются до сахаров, во второй идет сбраживание отдельных продуктов гидролиза — галактозы и арабинозы до масляной кислоты, СО2 и Н2 Бактерии рода Clostridium играют важную роль в процессе мацерации волокон при мочке льна. Несколько пектинолитических ферментов обнаружены у Bacillus polymyxa. Фито-патогенные виды бактерий рода Erwinia вызывают распад тканей ряда растений также благодаря высокой активности комплекса пектолитических ферментов.
Грибные пектолитические ферменты наиболее стабильны и имеют оптимум pH от 3,5 до 5,0. Некоторые фитопатогенные грибы, например Fusarium oxysporum, обладают высокоактивным пектолитическим комплексом ферментов, обусловливающим их патогенность. В промышленных условиях пектолитические ферменты получают в основном из Aspergillus awamori, A. niger и других видов грибов при поверхностном и глубинном способах культивирования.
Для выделения культуры бактерий, образующих пектолитические ферменты, из гниющей моркови, используют пектиновый агар состава: полипектат натрия — 18,0 г/л; кристаллвиолет (0,075%-й водный раствор) — 2,0 мл г/л; NaNO3 — 2,0 г/л; СаС12- 2Н2О (10%-й водный раствор) — 14 мл; цитрат натрия — 5,0 г/л; триптон — 1,0 г/л; новобио-цин (1%-й водный раствор натриевой соли) — 4 мл; агар — 4,0 г/л; вода водопроводная — до 1л. Значение pH среды до стерилизации доводят до 7,0. Стерилизацию проводят 30 мин при 0,5 ати. Новобиоцин (pH 7,0) стерилизуют фильтрованием и добавляют в среду после ее автоклавирования.
Полипектат натрия включают в состав среды в качестве основного источника углерода. При выращивании микроорганизмов на твердой среде пектолитиче-
ские ферменты диффундируют в агаровый гель вокруг колонии. Колонии бактерий, образующих пектолитические ферменты, можно обнаружить по глубоким впадинам или ямкам, которые образуются за счет ферментативного расщепления пектинового геля и по характерной каплевидной морфологии колоний. Цитрат натрия добавляют в среду, чтобы уменьшить развитие бактерий рода Pseudomonas в пользу преимущественного роста бактерий рода Erwinia. Антибиотик новобио-цин подавляет рост многих грамположительных и некоторых грамотрицатель-ных бактерий. Кристаллвиолет также подавляет развитие грамположительных почвенных бактерий, в основном принадлежащих к роду Bacillus.
Для опытов используют подверженную начальному этапу процесса гниения морковь. Питательную среду, предназначенную для работы по выделению чистой культуры, расплавляют и разливают в стерильные чашки Петри из расчета 6 чашек (около 120 мл) на один объект и подсушивают при 30 —35 °C в течение 24 —48 ч.
Выделение продуцента пектолитических ферментов. Перед выделением культуры микроорганизма из моркови объект осторожно моют водой с мылом, осторожно протирают ватой, смоченной 70%-м этиловым спиртом, и помещают в стерильный стеклянный сосуд. Стерильной иглой или скальпелем делают в кожице моркови в месте гнилостного поражения отверстие диаметром 3 — 4 мм. Из этого отверстия стерильно, с помощью бактериологической петли, берут небольшой комочек гниюшей ткани и переносят в пробирку с 6 — 7 мл стерильной водопроводной воды. Содержимое пробирки энергично перемешивают встряхиванием. Выделение культуры возбудителя процесса гниения проводят с помощью бактериологической петли методом истощающего штриха. Используют 3 чашки Петри с соответствующей стерильной средой. Кроме этого, для получения чистой культуры используют стеклянные шпатели Дригальского. С этой целью петлю («зеркальце») суспензии гнилостных микроорганизмов наносят на поверхность стерильной среды в чашке Петри, а затем распределяют микроорганизмы стерильным шпателем и, не обжигая шпатель в пламени горелки, распределяют материал по 2-й и 3-й чашкам Петри со стерильной средой. Засеянные чашки инкубируют в термостате при 22 °C в течение 3 дней. Выросшие колонии обнаруживают по глубоким впадинам или ямкам. Бактерии рода Erwinia образуют характерные каплевидные полупрозрачные колонии. Проводят изучение морфологии клеток бактерий, образующих пектолитические ферменты, используя окрашенный фуксином препарат (объектив 100х).
Определение активности пектинэстеразы. Для определения активности пектинэсте-разы получают биомассу бактерий. Для этого с помощью бактериологической петли производят посев бактерий из полученных характерных колоний бактерий рода Erwinia на поверхность пектинового агара в 3 чашках Петри частыми штрихами. Засеянные чашки инкубируют в термостате при 22 °C в течение 2 сут. Биомассу с чашек смывают стерильным физиологическим раствором, pH 7,0. Разрушение клеток проводят на ультразвуковом дезинтеграторе УЗДН при 22 кГц в течение 6 циклов по 30 с с перерывами по 1 мин при охлаждении. Полученный гомогенат центрифугируют 20 мин при 40 тыс. g. Супернатант используют для определения активности пектинэстеразы.
Пектинэстераза представляет собой гидролазу эфиров карбоновых кислот. Ионы Са2+ и Mg2' активируют фермент. Пектинэстераза омыляет сложные эфирные связи в пектиновой кислоте и пектине и отщепляет метиловый спирт. Отщепление СН3-групп происходит последовательно, начиная со свободной СООН-группы. В результате действия фермента в реакционной среде накапливаются частично или полностью деме-токсилированная полигалактуроновая кислота и метиловый спирт. В 3 мл реакционной смеси содержится: калий-фосфатный буфер (0,1 М, pH 6,5) — 2,0 мл; полипектат натрия — 0,05 г; СаС12 — 4 мМ; MgCl2-6H2O — 5 мМ; экстракт бактерий — 0,1 мл; дистиллированная вода — до 3 мл. Время опыта — 20, 40, 60 мин при 30 °C. В конт
рольную пробу вместо экстракта добавляют воду. Реакцию останавливают 0,3 мл 50%-й ТХУ. Активность фермента определяют по количеству образовавшегося метанола. Метанол определяют на газовом хроматографе (см. гл. 23).
14.4. АКТИНОМИЦЕТЫ (КЛАСС ACTINOBACTERIA)
Актиномицеты — это чрезвычайно разнообразная группа грамположитель-ных бактерий, включающая в настоящее время более 60 родов. Среди представителей актиномицетов есть организмы, обладающие самой разной морфологией — это кокки, плейоморфные палочки, палочковидные организмы, которые на определенном этапе своего развития образуют нитевидные формы, и, наконец, истинные мицелиальные актиномицеты, образующие как субстратный, или вегетативный, так и воздушный мицелий. Размножаются эти бактерии бинарным делением, фрагментацией нитевидного мицелия или путем образования спор.
Актиномицеты характеризуются высоким содержанием гуанина и цитозина в молекуле ДНК: 58 — 77 мол. %. Все они являются хемоорганотрофными микроорганизмами, и большая их часть принадлежит к строгим аэробам. Однако среди представителей этой группы бактерий встречаются также анаэробные и факультативно-анаэробные формы. Экологическая ниша большинства актиномицетов — это хорошо аэрируемая зона почвы, где они ведут сапротрофный образ жизни. Количество актиномицетов в почве очень велико и может достигать миллиона колониеобразующих единиц (КОЕ) на 1 г сухой почвы. Некоторые представители являются важными компонентами нормальной микрофлоры кишечника, кожи и слизистых оболочек человека и животных. Так, виды рода Actinomyces считаются естественными обитателями полости рта человека. Однако помимо сапротрофных форм и комменсалов, среди актиномицетов встречается достаточно большое количество патогенных форм.
Среды для выделения актиномицетов. Используют различные среды, содержащие органические вещества растительного и животного происхождения, например МПА, сусло-агар, картофельный агар, а также синтетические среды. Оптимальная температура для роста мезофильных почвенных актиномицетов составляет 23 — 30 °C, а диапат зон pH — 4,5 —9,0 при оптимуме pH 6,8 —7,5. Ниже приведены составы 5 сред, на которых выявляются различные почвенные актиномицеты, в особенности те, что принадлежат к роду Streptomyces.
Среда Г. крахмал - 20 г; КНРО4 - 0,5 г; MgSO4- 7Н2О - 0,5 г; KNO3 - 1,0 г; NaCl -0,5 г; FeSO4 — 0,01 г; агар — 30 г; вода — 1000 мл; pH 7,2 —7,4.
Среда 2: пептон — 1 г; глюкоза — 5 г; MgSO4- 7Н2О — 0,4 г; К2НРО4 — 0,4 г; агар — 30 г; процеженный отвар картофеля — 100 мл (20 г картофеля варят в 100 мл воды, фильтруют и доводят водой объем до 100 мл); вода — до 1000 мл; pH 7,2 —7,4.
Среда 3: гороховая мука — 10 г; глюкоза — 10 г; NaCl — 5 г; СаСО3 — 1 г; агар — 30 г; вода — до 1000 мл; pH 7,2 —7,4.
Среда 4: мясной экстракт — 50 мл; пептон — 4 г; NaCl — 2,5 г; дрожжевой экстракт — 5 мл; глюкоза — 10 г; агар — 30 г; вода — до 1000 мл; pH 7,2 —7,4.
Среда 5: крахмал — 10 г; дрожжевой экстракт — 5 г; кукурузный экстракт — 10 г; NaCl — 2 г; агар —30 г; вода — до 1000 мл; pH 7,2—7,4.
Наиболее распространенной средой для учета стрептомицетов в почве является крахмало-аммиачная среда: (NH4)2SO4 — 2 г; К2НРО4, MgSO4-7H2O и NaCl по 1 г; СаСО3 — 3 г; крахмал — 10 г; агар — 20 г; вода — 1000 мл.
При засеве питательных сред разведение делают в зависимости от предполагаемого количества актиномицетов в исследуемом субстрате. Часто готовят взвесь из расчета 1 г почвы на 10 мл воды, а посев производят обычно из 2—4 разведений, т.е. 1:100, 1:1000, 1:10000. При этом на поверхность среды в чашки Петри наносят 0,05 мл взвеси и растирают шпателем по поверхности. Засеянные чашки культивируют при температуре 25 — 28 °C в течение 7 — 10 сут. Колонии стрептомицетов и некоторых других актиномицетов хорошо распознаются невооруженным глазом. Большей частью они окрашены в разные цвета — бурый, черный, желтый, красный и т.д. Колонии многих стрептомицетов покрыты пушистым, бархатистым или мучнистым налетом и практически: не снимаются с поверхности агара бактериологической петлей. Для подтверждения принадлежности выделенных бактерий классу Actinobacteria необходимо исследовать их морфологию, физиолого-биохимические и хемотаксономиче-ские особенности, а если это необходимо, провести молекулярно-генетические тесты.
14.5. УКСУСНОКИСЛЫЕ БАКТЕРИИ
Накопительную культуру уксуснокислых бактерий получить достаточно просто. Во-первых, они способны выдерживать высокие (до 10 %) концентрации спирта, который используют в качестве окисляемого субстрата. Во-вторых, они менее других бактерий чувствительны к уксусной кислоте, которую образуют из спирта при дыхании. Для получения накопительной культуры уксуснокислых бактерий среду, содержащую 40 мл этилового спирта и 10 г дрожжевого экстракта в 1 л воды (pH доводят до 6,0), инокулируют плодами, цветками, листьями плодовых растений.
Пиво и вина при нарушении технологии их хранения также служат прекрасным субстратом для активного развития и получения накопительной культуры уксуснокислых бактерий. Так, легко получить накопительную культуру уксуснокислых бактерий в колбе, содержащей 30 —50 мл пива, слегка подкисленного уксусной кислотой (можно использовать 5 — 10 мл 9 %-го столового уксуса). Через двое суток инкубации на поверхности пива появится сероватобелая пленка уксуснокислых бактерий. Их видовой состав будет зависеть от температуры инкубации: при 25 — 30 °C уксуснокислые бактерии будут представлены видом Acetobacter pasteurianus, при 20 — 22 °C преимущественное развитие получат A. aceti и A. xylinum. Выделение уксуснокислых бактерий в чистую культуру проводят на мясопептонном агаре с добавлением 2 % глюкозы или на сусло-агаре, в который после стерилизации вносят 3 % этилового спирта.
14.6. МЕТИЛОТРОФНЫЕ БАКТЕРИИ
Бактерии, выделяемые а аэробных условиях на простых минеральных средах с метанолом в качестве единственного источника углерода и энергии для роста, относят к семейству Methylobacterium. Это розовые факультативные метилотрофные грамотрицательные подвижные палочки, способные к росту
на средах с метанолом или метиламином, так же как и на средах с полиуг-леродными субстратами (сукцинатом, лактатом, фруктозой), хотя обычно на богатых средах (МПА) они не развиваются. Культуры таких микроорганизмов могут быть выделены практически из любых местообитаний: воздуха, почвы, воды — так как в любом из таких мест может находиться метанол — продукт природной биодеградации пектинов и лигнинов. Метилобактерии прекрасно переживают перенос с частичками пыли по воздуху и их выделяют из большого количества часто необычных мест: из осадков озер, поверхности веток и листьев деревьев, зерен риса, воздуха больниц, даже из кремов для лица и с поверхности компьютерных чипов. В силу своей способности метаболизировать различные продукты разложения отмерших растений метанол, метилированные амины и различные другие метилированные субстраты играют важную экологическую роль в глобальном цикле углерода. Способность переживать высушивание и расти на средах с минимальными концентрациями углерода и азота делают их идеальными кандидатами на выживание в стрессовых условиях. Обнаружены также внутри- и внеклеточные ассоциации метилобак-терий и некоторых растений, особенно с мхами (печеночники), плаунами и лишайниками, хотя прямые симбиозы и обмен органическим веществом между организмами пока не доказаны.
Метилобактерии сравнительно легко выделяются из проб окружающей среды (см. также приложение 4). Обычная для роста метилобактерии среда с метанолом (MMS) содержит (г/л): К2НРО4 - 1,2; КН2РО4 - 0,62; СаС12-6Н2О - 0,05; MgSO4-7H2O -0,20; NaCl — 0,10. Микроэлементы (мкг/л, конечная концентрация в среде): FeCl2- 6Н2О — 1000,0; (NH4)2SO4 - 0,5; CuSO4- 5Н2О - 5,0; MnSO4- 5Н2О - 10,0; Na2MoO4- 2Н2О -10,0; Н3ВО3 — 10,0; ZnSO4- 7Н2О — 70,0; СоС12- 6Н2О — 5,0. Стерилизация осуществляется при 1 ати. В стерильную среду добавляют стерильный (фильтрация) метанол до 0,1—0,2%, pH среды 7,0. В агаризованную среду добавляют 0,5 —2,0% очищенного агара (выщелоченного). Метилобактерии не требуют витаминов.
Культуры развиваются при 5 —37 °C, но лучше при 30 °C, обычно необходимо 2 — 3 сут для появления видимых окрашенных колоний, а к 7-м сут колонии'могут достигать 1 —3 мм в диаметре. Колонии появляются быстрее и их пигментация заметнее на глицеролпептонном агаре (г/л): глицерол — 10,0; пептон — 10,0; агар — 15,0. Стерилизация осуществляется при 1 ати, pH 7,0. В случае появления на чашках больших количеств грибов при выделении метилобактерий из некоторых местообитаний рекомендуют добавлять 20 мкг/мл циклогексимида.
Выделенные метилобактерии обычно представлены палочками (1—8 мкм), одиночными или формирующими розетки, часто они разветвлены или плейо-морфны, особенно в старых культурах. Все штаммы подвижны благодаря наличию полярных, субполярных или латеральных жгутиков. Часто клетки содержат большие, окрашиваемые Суданом включения (поли-р-оксибутират), а также гранулы волютина. Подавляющее большинство штаммов граммотрицательны, некоторые — грамвариабельны. Пигменты, образуемые метилобактериями, не диффундируют в агар, не флуоресцируют и, возможно, относятся к каротиноидам. В стационарных жидких культурах большинство метилобактерий образуют розовое кольцо на поверхности среды. Все штаммы — аэробы, каталазо- и оксидазоположительны. Они являются хемоорганотрофами и факультативными метилотрофами, способными окислять целый ряд Сгсоединений. Метилобактерии растут на средах с формальдегидом (в микромолярных концентраци
ях), формиате и метаноле, некоторые используют метилированные амины. Субстраты С, ассимилируют через ицл-сериновый цикл и проявляют активность всех ферментов цикла лимонной кислоты при росте на сложных органических субстратах. Рост оптимален при нейтральных значениях pH среды, однако некоторые штаммы способны расти при pH 4 и до pH 10. Все штаммы образуют уреазу (см. 13.3), некоторые способны к нитратредукции, а тест Фо-геса—Проскауэра для всех метилобактерий отрицателен.
Большинство штаммов чувствительны к антибиотикам: канамицину, аль-бамицину Т, стрептомицину, фрамицетину и особенно к тетрациклину. Многие штаммы, однако, устойчивы к цефалоспорину, налидиксовой кислоте, пенициллину, бацитрацину, карбенциллину, колистинсульфату, полимиксину В и нитрофурантоину.
Наряду с С,-субстратами большинство метилобактерий используют для роста глицерол, малонат, сукцинат, фумарат, 2-оксоглутарат, DZ-лактат, DZ-малат, ацетат, пируват, пропиленгликоль, этанол. Некоторые штаммы могут расти, кроме названных, на средах с арабинозой, глюкозой, ксилозой, фукозой, галактозой, фруктозой, аспартатом, глутаматом, цитратом, цитракона-том, моно-, ди- и триметиламинами, этаноламином, бутиламином, диметил-глицином и бетаином. Ни один из изученных штаммов не использует дисахариды или сахароспирты. Аммоний, нитрат и мочевина могут служить источниками азота. Состав жирных кислот обычно представлен С,8. ,-мононенасыщен-ными кислотами с прямой цепью, а мол. % ГЦ в ДНК варьирует от 68 до 72.
В некоторых штаммах метилобактерий обнаружен бактериохлорофилл, что говорит о их возможной связи с предшественниками современных фототроф-ных бактерий. По современной филогенетической классификации метилобак-терии размещены в ветке филогенетического древа, образуемой комплексом Agrobacterium — Rhizobium.
Хотя метилобактерий рассматривались в качестве кандидатов на продуценты белка, их степень конверсии (преобразование углерода субстрата в углерод биомассы) не выдержала конкуренции с другими метилотрофами. Поэтому метилобактерий в настоящее время рассматривают в качестве потенциальных продуцентов кофермента Ql0, аминокислот (Z-лизина, Z-тирозина, Z-фени-лаланина, Z-глутамата), витаминов (В|2) и как источник поли-р-оксибутира-та. Их каротиноидные пигменты применяют в качестве природного красителя для пищевой индустрии (производство маргарина). Высокая устойчивость метилобактерий к облучению гамма-лучами делает эти организмы возможными кандидатами для тест-культур при контроле качества производства пищевых продуктов и упаковочной продукции.
Глава 15
АЭРОБНЫЕ ЛИТОТРОФНЫЕ МИКРООРГАНИЗМЫ
Целый рад микроорганизмов способен окислять различные неорганические соединения с получением метаболической энергии для роста клеток. Прежде всего это две группы нитрификаторов, железоокисляющие бактерии, водород
ные бактерии, метаногены, сулъфидогены, денитрификаторы и ацетогены, использующие водород в качестве источника энергии, карбоксидобактерии и некоторые другие микроорганизмы, окисляющие ионы некоторых металлов (сурьмы, марганца). В данной главе будут рассмотрены методологические приемы, позволяющие выделять интересующие хемолитотрофы из природных образцов и изучать их. Кроме того, в настоящем практикуме имеются задачи по изучению метаболизма чистых культур некоторых литотрофов (см. гл. 24).
15.1. ВЫДЕЛЕНИЕ И ИЗУЧЕНИЕ ВОДОРОДНЫХ БАКТЕРИЙ
Аэробные хемолитоавтотрофные водородокисляющие бактерии — физиологически сходная, но филогенетически разнородная группа организмов, включающая таксономически различных представителей. Общим свойством членов этой группы является способность использовать газообразный водород как донор электронов и кислород как акцептор, фиксируя СО2, т. е. расти хемолито-автотрофно. Такие водородные бактерии в строгом смысле отличаются от других микроорганизмов, способных метаболизировать Н2 (некоторые диазотро-фы, энтеробактерии и др.), однако не фиксирующих при этом СО2. К тому же они отличаются от микроорганизмов, использующих водород в анаэробных условиях при сульфатном, серном или карбонатном дыхании (например, Desulfovibrio, Thermoproteus, Pyrodictium, Clostridium aceticum, Acetobacterium woodii и Methanosarcina barker!).
Водородные бактерии, в строгом смысле, являются в основном факультативными автотрофами, способными расти гетеротрофно на средах с различными органическими субстратами в качестве доноров энергии и углерода. Возможен также миксотрофный рост. Большинство водородных бактерий растут на средах с органическими веществами быстрее, чем на средах с водородом. Наряду с этим выделены и описаны три вида облигатных хемолитотрофных водородных бактерий: два термофила (Hydrogenobacter thermophilus и Caldero-bacterium hydrogenophilum) и один галофил (штамм МН ПО).
В окисление водорода вовлечены две различные гидрогеназы. Одна из них растворимая цитоплазматическая, НАД+-зависимая (водород: НАД+-оксидо-редуктаза), другая — мембраносвязанная, не-НАД+-зависимая. Некоторые водородные бактерии содержат одну из этих гидрогеназ, другие — обе.
Метаболизм углерода у водородных бактерий протекает с участием восстановительного рибулозобисфосфатного цикла (цикла Кальвина), и комбинация этих двух активностей (использование водорода и фиксация СО2) уникальна в биологическом мире.
15.1.1. Методы выделения водородных бактерий
Принципы, лежащие в основе выделения водородных бактерий, просты по определению: только они могут расти аэробно и автотрофно, используя водород в качестве единственного источника энергии. Преимущественно применяют два способа выделения водородных бактерий — прямой чашечный метод и метод получения жидких накопительных культур.
Прямое выделение водородных бактерий на чашках с селективной твердой средой приводит к появлению колоний различных микроорганизмов как быстрорастущих, так и медленнорастущих культур.
Около 1 г почвы или ила суспендируют в 10 мл стерильного раствора NaCl (0,9%-го). Смешивают 1 мл этой суспензии (или ее серийные разведения в том же растворе) в чашке Петри с 20 мл минеральной среды с агаром высокой степени чистоты, расплавленной и остуженной до 45 °C (состав см. в приложении 4). После застывания агара чашки помещают в эксикатор, в котором воздух заменяют на газовую смесь, содержащую 5 % О2, 85 % Н2 и 10 % СО2. Альтернативно этому небольшие аликвоты суспензий могут быть распределены шпателем по поверхности плотной среды.
Для посева из жидких проб, где концентрация водородных бактерий невелика (пруды, озера), можно применять предварительную концентрацию образцов (2 — 500 мл в зависимости от концентрации клеток) на мембранных фильтрах (0,22—0,45 мкм). Мембраны затем раскладывают на поверхности чашек со средой, причем чашки должны быть предварительно подсушены в ламинаре.
В таких условиях выращивания многие олигокарбофильные бактерии будут также развиваться на чашках. Для выявления колоний водородных бактерий можно применить технику обнаружения их на фильтрах с подращенными колониями с использованием трифенилтетразолия хлористого (ТТХ). Фильтры переносят на несколько слоев фильтровальной бумаги в чашке Петри, смоченных свежеприготовленным раствором 0,1%-го ТТХ и инкубируют в течение 10 мин на воздухе в темноте. Некоторые колонии приобретают при этом красный цвет вследствие осаждения формазана. Такие колонии должны быть отброшены.
Последующая инкубация мембран в атмосфере 90 % Н2 и 10 % О2 приводит к окрашиванию гидрогеназположительных колоний. Такая обработка не вызывает гибель клеток, и колонии водородных бактерий могут быть пересеяны на свежую среду для выделения чистой культуры.
Выделение водородных бактерий с использованием жидких накопительных культур. В противоположность прямому чашечному методу выделения, постановка жидких накопительных культур приводит к возникновению конкуренции среди водородных бактерий и селекции наиболее быстрорастущих форм. Эта техника удобна для выделения быстрорастущих и образующих большую биомассу водородных бактерий, что важно для применения в биотехнологии. Накопительные культуры следует пересевать каждые 1 — 5 дней.
Для одновременного выделения быстро- и медленнорастущих клеток из природных образцов делают серию 10-кратных разведений и засевают из них среды с 9 мл жидкости в пробирке (+1 мл посевного материала из соответствующего разведения). Из каждого разведения необходимо засевать не менее 5 пробирок. Пробирки помещают в эксикатор, в котором создают соответствующую газовую среду и инкубируют в термостате стационарно или с покачиванием. Через 1 —2 нед отмечают рост в пробирках и пересевают их для выделения чистых культур. Одновременно этим же способом можно определить наиболее вероятное число водородных бактерий в пробе (по таблицам МакКреди: см. гл. 13).
15.1.2. Выделение чистых культур
Выделения чистых культур добиваются многократным пересевом из отдельной колонии как на минеральную, так и на органическую среду. Поскольку большинство водородных бактерий являются факультативными автотрофами, они должны формировать идентичные колонии на чашках с гетеротрофными и автотрофными средами.
Выделение различных физиологических подгрупп водородных бактерий. Используя различные среды для накопления, можно преимущественно выделять водородные бактерии, различающиеся по физиологическим свойствам, таким, как способность к фиксации атмосферного азота, использование нитрата как альтернативного кислороду акцептора электронов, рост на СО, пропане и других углеводородах, образование спор, рост при высокой температуре. Другие возможности водородных бактерий включают устойчивость к высоким концентрациям фосфата, магния или NaCl, способность расти при пониженной или повышенной (до 40 %) концентрации кислорода, устойчивость к антибиотикам и токсичным субстратам.
Хотя Alcaligenes eutrophus и Pseudomonas pseudoflava могут расти гетеротрофно с нитратом в качестве акцептора электронов, их рост в этих условиях очень незначительный. Автотрофно в анаэробных условиях с нитратом, нитритом или N2O в качестве акцепторов электронов и водородом в качестве донора из выделенных до настоящего времени водородных бактерий может развиваться только Paracoccus denitrificans. Поэтому все накопительные культуры, поставленные в таких условиях, приводят к выделению лишь штаммов Paracoccus.
15.1.3. Составление газовых смесей
Внимание*. Работа с водородом должна проводиться в хорошо проветриваемом помещении при соблюдении норм безопасности. Следует учитывать, что при 20 °C и нормальном давлении нижняя и верхняя точки воспламенения Н2 в воздухе — 4 % и 74,5 %; соответствующие пределы в кислороде — 4 % и 94 % Н2. Это значит, что смесь 95 % Н2 и 5 % О2 безопасна. Смесь 65 % Н2 + 25 % О2 + 10 %СО2 — чрезвычайно взрывоопасна, но с ней можно работать, если соблюдать все меры предосторожности.
Водород — бесцветный нетоксичный газ без запаха. Его растворимость в воде при 30 °C составляет 16,3 мл газа/л (коэффициент Бунзена). Вследствие низкой плотности (м. в. 2,016 г/моль) газ обычно собирается поверх массы воздуха, кислорода, СО2 и других газов или газовых смесей. Обычно для рутинной работы с водородными бактериями используют смесь 0,05 атм кислорода, 0,1 атм СО2 и остальное (до 1 атм) — Н2.
15.1.4. Хранение и поддержание водородных бактерий
Некоторые штаммы водородных бактерий сохраняют автотрофные свойства, несмотря на многочисленные пересевы на среды с органическими веществами (например, Paracoccus denitrificans, выделенный М. Бейеринком в 1910 г.). Другие виды, однако, легко теряют способность к водородной автотрофии при выращивании гетеротрофно, что может объясняться нестабильностью плазмид (Hydrogenophaga flava, Nocardia ораса). Поэтому водородные бактерии рекомендуется поддерживать в автотрофных условиях, где они также более стабильны по сравнению с гетеротрофными и остаются жизнеспособными до 6 и более месяцев при 4 °C. Выживаемость клеток на скошенном агаре может быть повышена при заливке его парафиновым маслом.
Водородные бактерии хорошо переносят лиофилизацию в 5 %-м глутамате, снятом молоке с 5 %-м глутаматом, мезо-инозитолом (5 %) или медом (10%). Для процедуры лиофилизации берут автотрофно выросшие культуры клеток.
15.1.5. Определение гидрогеназной активности
Для определения гидрогеназной активности в автотрофно выросших на чашках колониях используют методы, основанные на образовании нерастворимого в воде формазана красного цвета, появляющегося при восстановлении ТТХ гидрогеназой за счет водорода.
Метод А выявляет наличие растворимых и мембраносвязанных гидрогеназ, тогда как метод Б — только мембраносвязанных гидрогеназ вследствие ингибирования активности всех цитоплазматических гидрогеназ в присутствии мо-нойод-ацетата.
Метод А. Помещают стерильный мембранный фильтр с диаметром пор 0,22 — 0,45 мкм на чашку с минеральным агаром. Переносят штаммы, проверяемые на активность гидрогеназы, штрихами петлей, под номерами, на фильтр. Если колонии не имеют тенденции расплываться, то на одном фильтре можно разместить до 10 точек разных колоний. Инкубируют фильтр на чашке в атмосфере: 5 % О2 + 10 % СО2 + 85 % Н2. После развития колоний помещают фильтр на смоченную 0,1%-м ТТХ фильтровальную бумагу (см. метод выше).
Иногда метод дает положительные результаты при инкубации в анаэробной атмосфере без Н2, видимо, вследствие эндогенного дыхания за счет пула запасных органических веществ.
Метод Б. Клетки выращивают аэробно на агаризованной минеральной среде с глюконатом (0,1% Na-глюконата), с тем чтобы на чашке было от 80 до 100 колоний (48 ч). Диск фильтровальной бумаги опускают на поверхность чашки и слегка прижимают к ней, чтобы колонии дали отпечатки на диске. Диск помечают карандашом и совмещают с меткой на чашке. Обычно через 3 мин диск снимают и помещают в другую чашку Петри, стороной с отпечатками колоний вверх, на другой фильтр, пропитанный 0,4 мл 0,2 М ацетатом и 0,8 мл 4%-м ТТХ, растворенными в 0,067 М К-фосфатном буфере (pH 7,0). После 30 мин инкубации чашку помещают в прозрачный эксикатор, который заполняют N2 для обнаружения эндогенного восстановления краски. После 20 — 30 мин инкубирования при комнатной температуре атмосферу в эксикаторе замещают на Н2. Колонии, содержащие мембраносвязанные гидрогеназы, окрашиваются при этом в пурпурно-красный цвет. Реакция обычно проходит за 30 мин.
Метод В. Активность гидрогеназы можно определить в бесклеточных экстрактах клеток, выросших под водородом. Для этого клетки разрушают одним из способов, описанных в гл. 13, и получают их бесклеточные экстракты. Гидрогеназу определяют при 570 нм в кювете с длиной оптического пути 1 см. Добавление глюкозы, глюкозо-оксидазы и каталазы препятствует спонтанному окислению восстановленной метиленовой сини. Реакционная смесь содержит: К-фосфатный буфер (50 мМ, pH 7,0) с 167 мкМ метиленовой сини — 2,5 мл; глюкозу (4 мМ) — 50 мкл; глюкозооксидазу (60 мг/мл) — 10 мкл; каталазу (20 мг/мл) — 10 мкл; экстракт — 0,1 —0,2 мл (1 — 2 мг) белка и воду дистиллированную — до 3,0 мл. До добавления экстракта кюветы закрывают резиновыми пробками и продувают через две иглы чистым водородом. Коэффициент молярной экстинции для метиленовой сини — 13,1 см2/мкмоль. При определении можно использовать целые клетки разбавленных суспензий бактерий.
Метод Г. Этим методом определяют активности гидрогеназы (НАД+-зависимой) в бесклеточных экстрактах. Экстинцию измеряют при 365 нм с НАД+ как акцептором электронов. Реакционная смесь (3 мл) содержит 50 мМ К-фосфатный буфер (pH 8,0), 0,8 мМ НАД+ и соответствующее количество экстракта клеток. До добавления экстракта кювету продувают водородом (см. метод В). Коэффициент молярной экстинции НАДН — 6,22 см/ммоль.
15.2. КАРБОКСИДОБАКТЕРИИ
К карбоксидобактериям (термин предложен Г. А. Заварзиным в 1972 г.) относят группу аэробных водородных бактерий, способных окислять СО. Все до настоящего времени выделенные карбоксидобактерии способны также лито-трофно окислять водород, тогда как не все водородные бактерии могут использовать СО как источник углерода и энергии для роста. Карбоксидобактерии — факультативные автотрофы и обычно мезофилы. Однако есть сведения об умеренно термофильном стрептомицете Streptomyces thermoautotrophicus, который, помимо способности расти автотрофно и аэробно за счет окисления СО, обладает нечувствительной к кислороду нитрогеназой. Кроме СО карбоксидобактерии способны использовать для роста и некоторые многоуглеродные соединения: ацетат, малат, сукцинат, этанол, пропанол и др. Некоторые штаммы растут в присутствии одноуглеродных соединений — метанола и формиата.
В зависимости от вида карбоксидобактерии требуют различного содержания СО в газовой фазе — от 20 до 95 об. %. Окисление СО проходит по уравнению
СО + -> СО2, AG°' = -283,0 кДж/моль
Для фиксации 1 моля СО2 клетки должны окислить 7 молей СО по уравнению
7СО + 2*/2О2 + Н2О -> (Н2О) + 6СО2
Реакцию окисления СО проводит особая СО-дегидрогеназа, часто она бывает НАД+-зависимой:
СО + Н2О + А -> СО2 + Н2А,
где А — окисленная форма акцептора электрона. В тех случаях, когда природный акцептор электрона СО-дегидрогеназы неизвестен, реакцию проводят в присутствии метиленовой сини. Молекулярная масса большинства выделенных и очищенных СО-дегидрогеназ карбоксидобактерий — около 230 кДа. Бактерии, окисляющие СО, формируют дыхательную цепь, участвующую в переносе электронов от СО к кислороду, заканчивающуюся особой цитохромокси-дазой, нечувствительной к СО.
Известен рад микроорганизмов, среди которых хорошо изучены фототроф-ные пурпурные бактерии, растущие в темноте в анаэробных условиях со 100 % СО в качестве газовой фазы с образованием молекулярного водорода по уравнению
СО + Н2О -> со2 + н2
Некоторые метаногены (Methanosarcina barken) способны использовать СО, преобразуя его в метан:
СО + 2Н2О -> СН4 + СО2
Выделение карбоксидобактерий. Карбоксидобактерии выделяют из верхних слоев почвы вдоль оживленных городских магистралей, из дерновых матов, которыми углежоги обкладывают поленницы дров при традиционном способе производства древес-
него (и активированного) угля, из почв областей с высокой вулканической активностью или с частыми лесными пожарами. Среды — обычно минеральные с добавлением или без добавления витаминов (в виде раствора смеси витаминов или дрожжевого автолизата). Газовая фаза герметических сосудов для культивирования должна содержать смесь СО и воздуха в различных концентрациях (см. с. 206). Обычно культивирование проводят в качалочных колбах с резиновыми пробками, которые постоянно перемешивают на качалке для лучшего газообмена. Примеры сред для культивирования карбоксидобактерий приведены в приложении 4.
15.3. БАКТЕРИИ, ОКИСЛЯЮЩИЕ МАРГАНЕЦ
Марганецокисляющие микроорганизмы — филогенетически разнообразная группа, объединенная способностью окислять двухвалентный растворимый ион марганца (Мп2+) до нерастворимых соединений общей формулы МпОА., где х — некоторое число между 1 и 2, в которых марганец находится в четырехвалентном состоянии. Такое окисление в природе приводит к накоплению легко обнаруживаемых нерастворимых осадков оксидов марганца (IV) черного или коричневого цвета. К окислению марганца способны многие микроорганизмы — бактерии, грибы, водоросли. Среди бактерий такая способность обнаружена у многих филогенетически различающихся групп микроорганизмов: цианобактерий, гетеротрофных палочек и кокков, образующих чехлы (подобно Leptothrix} и почкующихся (подобно Hyphomicrobium) бактерий, некоторых автотрофных штаммов, близких псевдомонадам, и среди представителей все еще загадочного рода Metallogenium. Анаэробные лактобациллы не включены в их число, так как окисление Мп служит у них защитой от вредного действия кислорода, оксиды Мп не откладываются вне-клеточно, а накапливаются до миллимолярного уровня в цитоплазме в связанном с белками виде.
Окисление Мп2+ в Мп4+ термодинамически выгодно в аэробных условиях (Дб°' = -67,2 кДж/моль), однако высокая энергия активации процесса делает Мп2+ довольно стабильным элементом в водных средах. Энергетический барьер может быть преодолен повышением pH или добавлением Мп-связывающих компонентов, включая сами оксиды Мп, которые являются активными хелаторами для Мп2+. Поэтому катализ окисления Мп2+ оксидами Мп (аутоокисление) затрудняет точное разделение микробного и химического окисления, особенно в природных экосистемах, где органические хелаторы и частицы оксидов Мп присутствуют в большом количестве.
15.3.1. Распространение Mn-окисляющих микроорганизмов
Окисляющие Мп2+ микроорганизмы вездесущи, их можно выделить почти из любой пробы почвы или воды. Особенно большие их количества связаны с глубоководными осадками оксидов Мп, выходами гидротерм, окислительно-восстановительными переходами осадков фьордов, пленками осадков в пустынях, осадками Мп в водопроводных трубах, богатых Мп поверхностных биопленках неглубоких озер, осадками пресноводных озер и железомарганцевыми осадками. Особенно Mn-окислители распространены в зонах переходов от окис
ления к восстановлению. В анаэробных зонах Мп3+ или Мп41 могут быстро восстанавливаться за счет анаэробного Мп-дыхания от органических или неорганических доноров электронов. Растворимый Мп2+ может затем диффундировать в окисленную зону, где подвергается окислению Мп-окислителей. Такие процессы заноса Мп2+ в окисленные зоны постоянно встречаются в гидротермальных источниках, а также в местах выхода горячих подземных источников. В стратифицированных водных местообитаниях микробное окисление марганца может быть процессом значительной биогеохимической важности. Свежеобразованные оксиды Мп возвращаются в виде твердого преципитата в осадки или в анаэробную водную зону, где они могут послужить акцепторами электронов в анаэробном окислении Fe2+, H2S и органического вещества. Мп хорошо подходит для этой роли, так как не образует нерастворимых сульфвдов, и поэтому остается в растворе в анаэробных зонах. В таких местах наблюдается развитие бактерий, заключенных в чехлы (подобно Leptothrix), пропитанные оксидами Fe или Мп.
Выделение Мп-окислителей. В местах с высоким содержанием оксидов Мп обычно наблюдают массовое развитие клеток, напоминающих виды Hyphomicrobium, что делает вероятным участие этих бактерий в формировании таких осадков. В других случаях, хотя Mn-окисляющие бактерии можно легко выделить, они не составляют большинства в местообитании и не могут быть ответственны за образование наблюдаемых осаждений оксидов. Таким образом, можно заключить, что многие организмы, выделяемые как Mn-окисляющие в культуре, никогда не принимают участия в формировании осадков оксидов марганца в природе. Лабораторные накопительные культуры с различными пробами морских осадков, в которые добавлен МпС12, обычно приводят к появлению зон темного осадка оксидов Мп после 3—12 мес инкубации. Вокруг зон таких осадков обычно концентрируются морфологически одинаковые и таксономически схожие бактерии. Mn-окислители могут быть легко выделены в чистые культуры и хорошо растут на твердых и жидких средах, если предприняты необходимые меры к снятию токсичного действия Мп2+. Для выделения используют обычные источники углерода (ацетат, сукцинат, глицерол и т.д.). Во многих культурах окисление Мп2+ начинается после того, как культуры войдут в стационарную фазу роста. Некоторые источники углерода могут снижать pH среды, другие подавляют окисление Мп. В большинстве сред используют низкие концентрации углерода, а в других в качестве источника углерода применяют аминокислоты. Поскольку окисление Мп строго зависит от pH среды и с наибольшей термодинамической эффективностью происходит при pH 7 —8, в среды обычно добавляют 10—100 мМ HEPES-буфера и контролируют pH в этом интервале. Как правило, Mn-окислители — прото-трофы, но некоторые исследователи добавляют в среды смесь витаминов или дрожжевой экстракт. Форма добавки источника Мп2+ может быть разная (ацетатные, карбонатные, хлоридные или сульфатные соли), однако добавлять эти соли в виде растворов необходимо после автоклавирования среды в виде профильтрованного раствора во избежание выпадения осадка оксидов Мп при автоклавировании и связанного с этим процесса дальнейшего химического самоокисления Мп . Концентрации Мп2+ в природе редко превышают 1 —5 мкМ, а выше 10 мкМ становятся токсичными для бактерий. В некоторых средах Мп2* добавляют в существенно больших концентрациях (в мил-лимолярном диапазоне), что, однако, не ведет к токсичному эффекту. Для вновь выделенных микроорганизмов обычно рекомендуют использовать 10 — 100-микромолярные концентрации Мп2'.
Один из способов снятия токсичности Мп включает использование непрерывных культур в хемостатах. Начинают эксперимент с культивирования на гетеротрофной среде с источниками углерода — ацетатом или сукцинатом — и низкими концентра-
днями Mn2+ (5 — 20 мкМ). Концентрацию Мп2+ измеряют в культуральной среде, выходящей из хемостата. Когда она станет ниже 1 мкМ, концентрацию Мп2+ в питающем резервуаре медленно поднимают до 1 мМ или выше. Это позволяет изучать взаимоотношения между потреблением источника углерода и Мп2+ при их различных концентрациях. Непрерывные культуры позволяют также изучить высокообогащенные накопительные культуры Мп2+-окислителей, выделенных из природных местообитаний, и получить культуры, использующие окисление Мп2+ с целью запасания метаболической энергии.
15.3.2. Среды для культивирования Мп-окислителей
Среда Крумбайна. Это богатая среда, на которой растут многие гетеротрофные окислители Мп24. В качестве раствора может быть использована морская, пресная или дистиллированная вода. Состав (г/л): FeSO4- 7Н2О — 0,001 г (можно его исключить); MnSO4-4H2O (или МпС12) — 0,2 г; пептон — 2,0 г; дрожже-зой экстракт — 0,5 г; HEPES-буфер — 10 мМ; pH 7,0; стерилизация — при 1 ати.
Минимальная среда с сукцинатом. Это основная среда для изучения связывания и метаболизма Мп. Готовят ее на дистиллированной или искусственной морской воде. Состав'. NH4C1 (или KNO3) — 9,0 мМ; К2НРО4 — 0,4 мМ; NaHCO3 — 2,0 мМ; FeSO4 — 5,0 мкМ; HEPES-буфер — 10 мМ; раствор витаминов — 10 мл; Na-сукцинат — 10 мМ; МпС12 — 10 мкМ. Готовят 1 л раствора витаминов на дистиллированной воде двойной перегонки при перемешивании, стерилизуют фильтрованием и хранят при 5 °C в темноте. Состав (мг): биотин — 2,0; ниацин — 5,0; тиамин-НС1 — 5,0; и-аминобензойная кислота — 5,0; пантотенат Са — 5,0; пиридоксин-НС1 — 5,0; В12 — 0,1; рибофлавин — 5,0; фолиевая кислота — 2,0.
Среда PC. Эту низкоконцентрированную среду используют для выделения Мп2+-окисляющих видов Hyphomicrobium. Ее готовят на дистиллированной воде или на воде источника, который изучают. Состав (г/л): MnSO4- 4Н2О — 0,02; дрожжевой экстракт — 0,05.
Среда PYGV. Это еще одна среда с низкими концентрациями питательных веществ для выделения медленнорастущих почкующихся и чехольчатых бактерий (Pedomicrobium и Leptothrix). Обычно видимые колонии развиваются на этой среде в течение нескольких дней, причем быстрорастущие гетеротрофы могут «перерасти» Мп-окислителей. Среду готовят на дистиллированной или морской воде. Состав (г/л): пептон — 0,25; дрожжевой экстракт — 0,25; глюкоза — 0,25; раствор витаминов — 10 мл (см. выше); раствор минеральных солей — 20 мл.
Раствор минеральных солей: 10 мг нитрилоацетата растворяют при нейтрализации КОН в 900 мл дистиллированной воды. Затем добавляют следующие соли: СаС12-2Н2О - 3,34 г; FeSO4-7H2O - 99 мг; MgSO4-7H2O - 29,7 г; NaMoO4- 2Н2О — 12,67 г и 50 мл раствора металлов «44». Объем доводят до 1 л дистиллированной водой, и раствор, который должен быть прозрачным, хранят при 5 °C.
Раствор солей «44». В 1 л дважды перегнанной дистиллированной воды, к которой прибавляют несколько капель концентрированной H2SO4, добавляют последовательно (мг): ЭДТА — 250; ZnSO4- 7Н2О — 1,095; MnSO4- 4Н2О — 154; СоС13-6Н2О - 20,3; FeSO4-7H2O - 500; CuSO4-5H2O - 39,2; NaB4O7- ЮН2О -17,7.
15.3.3. Идентификация Mn-окислителей
Выявление Mn-окислителей обычно проводят по обнаружению черных или коричневых осадков твердых оксидов Мп, которые откладываются вне или внутри клеток. Однако это свойство не является определяющим: многие бактерии образуют темные пигменты; известны также несколько кристаллических форм оксидов Мп, которые имеют разный цвет и консистенцию. К тому же вместе с Мп-оксидами часто откладываются и другие металлы, особенно железо, привнося свой цвет и консистенцию к осадкам Мп. Для преодоления этих трудностей применяют два специфических теста на Мп. Первый тест основан на использовании бензидина-HCl, который дает голубую окраску в присутствии оксидов Мп. Реактив готовят, растворяя 5 г бензидина-HCl в 35 мл ледяной уксусной кислоты. Раствор доводят до 500 мл дистиллированной водой и хранят при 5 °C. Колонии можно напрямую проверить на наличие вокруг них оксидов Мп, заливая чашку 10 мл реактива. Колонии с осадком окрашиваются в темно-синий цвет. При работе с этим реактивом, однако, следует учитывать следующие особенности: 1) реакции мешают оксиды железа, которые дают слабое голубое окрашивание; 2) реагент токсичен и канцерогенен; 3) реагент проявляет бактерицидные свойства, поэтому колонии перед его применением должны быть реплицированы на новые чашки. Поскольку реактив токсичен, рекомендуется не использовать его, если есть альтернатива.
Во втором тесте используют реактив лейкобербелин голубой (ЛБГ), окислительно-восстановительный индикатор, который, реагируя с Мп3+ или Мп4+, окисляется и из бесцветного становится голубым. Оксиды Мп восстанавливаются в этой реакции, так что ее можно использовать для мягкого удаления оксидов из окружения клеток. Реакция с ЛБГ меньше подвержена влиянию оксидов железа по сравнению с бензидиновой и, поскольку она менее бактерицидна, может быть применена непосредственно на клетки в колониях без существенного подавления их физиологических функций.
Приготовление реагента. Растворяют 4 г лейкобербелина голубого-I (N,N'-диметил-амино-л,и'-трифенилметан-о-сульфоновая кислота) в 80 мл кипящей дистиллированной воды. Перед кипячением в воду добавляют несколько капель концентрированной фосфорной кислоты. Затем добавляют 0,3 мл концентрированного NH4OH и доводят раствор до 100 мл. Этот слабо кислый исходный раствор можно хранить до года при 5 °C в темноте. Рабочий раствор, используемый для тестов, сохраняет стабильность в течение лишь нескольких дней. Готовят его, разводя исходный раствор в 100 раз в 1 мМ уксусной кислоте.
Окисление Мп можно контролировать количественно по исчезновению Мп2’ из раствора колориметрически с формальдоксимом или атомно-абсорбционной спектроскопией. Количество оксидов Мп можно определить, отфильтровав их от культуральной жидкости на мембранных фильтрах, растворив (восстановив) их до Мп2+ и измерив количество получившегося Мп2+. Растворение можно провести несколькими методами: 1) обработкой сильной кислотой (6 М НО) в течение нескольких дней; 2) обработкой смесью 5 частей концентрированной HNO3 с тремя частями концентрированной НО в тефлоновой посуде в течение 3 ч при 105 °C; 3) выщелачиванием в течение нескольких часов концентрированной азотной кислотой, содержащей 1 % (NH2OH)C1 (гидро
ксиламин-HCl); 4) выщелачиванием концентрированной азотной кислотой, содержащей 3 % (вес/объем) Н2О2. Выбранный метод зависит от природы анализируемого осадка и проведения последующих анализов. Как правило, хороший эффект достигается при действии 1 % (вес/объем) NH2OH без НС1, поскольку это быстрый и легкий метод обработки осадков, не дающий побочных эффектов при атомной адсорбции.
Mn-окисляющие бактерии могут быть идентифицированы и микроскопически, однако при таком подходе следует учесть некоторые потенциальные трудности. Почкующиеся или образующие выросты Mn-окислители легко обнаружить внутри и около оксидов Мп, в других случаях оксиды Мп должны быть удалены, прежде чем клетки будут идентифицированы. Обработка ЛБГ приводит к удалению осадков Мп. Она рекомендуется при работе со световым микроскопом с пробами, взятыми из природных экосистем, в которых идентифицируют Mn-окислителей. Осадки оксидов могут быть разрезаны и просмотрены в поле просвечивающего электронного микроскопа на предмет установления степенгт и формы контакта клеток с оксидами. При этом во многих случаях исследователь может столкнуться с тем, что оксиды Мп не имеют ничего общего с клетками, поэтому результаты микроскопических исследований следует интерпретировать с особой осторожностью.
15.3.4. Применение Мп-окислителей
Высокий уровень Мп в водопроводной гиги колодезной воде может быть опасен для человека и животных. Повышение концентрации Мп может произойти в летние месяцы, когда уровень кислорода в воде снижается вследствие усиления дыхания водных организмов, что создает благоприятные условия для анаэробного восстановления оксидов Мп. Как только Мп восстанавливается до Мп2+, он становится чрезвычайно устойчив в присутствии кислорода, и одна из потенциальных возможностей применения Mn-окислителей —использование для снижения концентрации Мп2+ в питьевой воде. Оксиды Мп также являются великолепными акцепторами электронов в анаэробном Мп-дыхании. Поэтому возможно применение Mn-окислителей для стимулирования минерализации органических остатков в осадках.
15.3.5. Значение окисления Мп2* для Мп-окислителей
Имеется лишь одно сообщение о том, что морской штамм Pseudomonas sp. S-36 мог расти в хемостате непрерывно с лимитом по Мп, окисляя Мп2+ и фиксируя СО2. В клетках обнаружен ген, кодирующий синтез большой субъединицы RuBisCO, и карбоксисомы, в которых, как полагают, содержится большая часть этого фермента. Данный организм поэтому является отличным кандидатом на изучение хемолитотрофного роста для Мп-окислителей.
Есть сведения, что постоянный приток добавляемого Мп2+ существенно удлиняет стационарную фазу роста культур Leptothrix, что может играть значительную роль для клеток в природных местообитаниях. Возможно также, что окисление Мп2+ проводят многие группы микроорганизмов в неспецифических реакциях просто для того, чтобы снять высокий, потенциально токсич
ный уровень этого катиона в микроокружении. Ясно, однако, что работа по выделению настоящих, облигатных Мп-окислителей, использующих эту реакцию для получения метаболической энергии, еще впереди, но совершенно очевидно, что данный раздел книги поможет исследователям, начинающим изучение интересной и важной в геохимическом плане группы микроорганизмов.
Глава 16
ПОЛУЧЕНИЕ НАКОПИТЕЛЬНЫХ И ЧИСТЫХ КУЛЬТУР АНАЭРОБНЫХ МИКРООРГАНИЗМОВ
16.1. НАКОПИТЕЛЬНЫЕ КУЛЬТУРЫ ПЕРВИЧНЫХ БРОДИЛЫЦИКОВ
Получение накопительных культур микроорганизмов, обладающих сложными пищевыми потребностями, рассмотрим на примере организмов, осуществляющих брожение (дрожжи, молочнокислые, пропионовые, маслянокислые бактерии).
Первое условие для выделения таких организмов — химическая природа органического субстрата. Чтобы субстрат мог сбраживаться, он не должен быть ни слишком окислен, ни слишком восстановлен. Лучшим субстратом для брожения служат сахара, но сбраживаться могут также органические кислоты, аминокислоты и другие соединения такой же степени окисленности. Второе необходимое требование — анаэробные условия выращивания организмов, не только потому, что многие из бродильщиков являются строгими анаэробами, но и для предотвращения конкуренции со стороны аэробных форм.
При выделении микроорганизмов-бродильщиков используют неодинаковую чувствительность клеток смешанной популяции к продуктам обмена веществ, накапливающимся в среде, в первую очередь к кислотам и спиртам. В ряде случаев в качестве условий элективности снижают pH среды и/или добавляют этанол.
Молочнокислые бактерии можно накопить, используя солодовое сусло без мела, т.е. среду, первоначально не обладающую элективностью. После внесения природного материала, содержащего молочнокислые бактерии, в среде наряду с молочнокислыми бактериями хорошо развиваются представители родов Enterobacter и Escherichia. Однако по мере накопления в среде молочной кислоты и этилового спирта, образуемого гетероферментативными видами, условия для развития энтеробактерий постепенно ухудшаются, тогда как молочнокислые бактерии, которым свойственна высокая кислото- и спиртоустойчи-вость, продолжают расти. Таким образом, в результате развития молочнокислых бактерий среда приобретает необходимую степень элективности, что и обеспечивает получение накопительной культуры этих бактерий.
Молочнокислые бактерии можно выделять и на среде другого состава (%): растительный (морковный, капустный) отвар — 10; дрожжевой автолизат — 1,0; пептон — 1,0; глюкоза — 2,0; этиловый спирт — 8—16. Спирт следует вносить в инокулированную субстратом жидкую среду через 18 —24 ч культивирования. Из жидкой среды производят высев на агаризованную среду того
же состава, но без спирта, в которую добавляют 4 % измельченного мела. Молочнокислые бактерии при росте на данной среде образуют вокруг колоний зоны просветления, обусловленного превращением нерастворимого углекислого кальция в растворимый лактат кальция.
Среда обитания молочнокислых бактерий — филлосфера (листья, стебли, цветы и плоды растений), почва — ризосфера и верхние горизонты, где содержится много растительных остатков, желудочно-кишечный тракт животных и человека. Все перечисленные места обитания могут быть использованы для выделения молочнокислых бактерий. Но, конечно, лучшим источником выделения является молоко и молочные продукты. Хранят молочнокислые бактерии на средах того или иного состава с добавлением 8 — 12 % этанола (см. гл. 11).
По практической значимости с молочнокислыми бактериями конкурируют только дрожжи (см. гл. 22). Как и молочнокислые бактерии, они широко распространены на поверхности листьев, цветков и плодов. Дрожжи — неизменные обитатели молока и молочных продуктов. Они появляются в молоке уже через несколько часов после дойки и составляют более 10 % от общего числа микроорганизмов. Дрожжи в молоке представлены видами Pichia, Rhodotorula, небольшим количеством сахаромицетов.
Дрожжи встречаются в лесной подстилке и почве, но их численность в почве обычно ниже, чем бактерий, актиномицетов и мицелиальных грибов. Дрожжи легко обнаруживаются в почвах с высоким содержанием свежих растительных остатков, умеренно водно-температурным режимом и низким значением pH. Чаще других в почвах встречаются представители родов Lypomyces, Torulopsis, Cryptococcus и Candida.
Накопительную культуру дрожжей получают путем высева исследуемого материала на сусло-агар, подкисленный молочной кислотой до pH 4,0 — 4,5 1что исключает рост большинства бактерий), и культивирования при температуре 6 °C в течение двух недель.
Для накопления пропионовокислых бактерий, которые растут довольно медленно, не выносят кислых условий и не могут конкурировать при росте на богатых средах с такими организмами, как молочнокислые или энтеробактерии, в качестве фактора элективности используют молочную кислоту. В отличие от других бродилыциков, пропионовокислые способны сбраживать этот субстрат. Для получения накопительной культуры этих бактерий используют нейтральную среду, содержащую 20 г лактата натрия и 10 г дрожжевого экстракта на 1 л воды, засевают природным материалом, содержащим пропионовые бактерии. Для инокуляции среды лучше всего использовать твердые сорта сыров. Инкубируют засеянные среды при 30 °C в анаэробных условиях в течение двух недель. Для получения колоний пропионовокислых бактерий накопительную культуру высевают на молочно-сывороточный агар в чашки Петри. Для того чтобы отличить колонии пропионовых бактерий от колоний молочнокислых, которые также могут появиться на этой среде, выделенные штаммы проверяют на образование кислот в молочно-кальциевом бульоне, выращивая их в течение 30 сут.
Кроме сыров и молочных продуктов пропионовокислые бактерии можно выделить также с поверхности кожных покровов человека и из желудка жвачных. При выделении последних необходимо помнить, что оптимальная температура для их роста 37 °C.
Возбудителями маслянокислого и его разновидности ацетонобутилового брожения являются представители рода Clostridium. Они широко распространены в природе. Их можно выделить из почвы, навоза, силоса, с поверхности зерен злаков и даже из молока и сыра. Для получения накопительных культур видов этого рода можно воспользоваться некоторыми их особенностями. Поскольку споры клостридий терморезистентны, инокулят предварительно пастеризуют. Создавая анаэробные условия, исключают рост всех аэробных бактерий. Клостридиям доступны многие природные субстраты. Они способны гидролизовать полисахариды, белки, нуклеиновые кислоты и сбраживать мономеры, образующиеся в результате гидролиза. На этом основании в качестве источника углерода и энергии для сахаролитических клостридий используют углеводы или полисахариды (чаше всего крахмал), а для пептолитических — аминокислоты или белки. Некоторые клостридии используют молекулярный азот как единственный источник этого элемента, что может стать дополнительным фактором элективности при постановке накопительных культур.
В приложении 4 приводится ряд сред для получения накопительных культур клостридий: картофельная среда с мелом для сахаролитических клостридий, среда Виноградского для клостридий, фиксирующих молекулярный азот и т.д. Накопительную культуру возбудителя ацетонобутилового брожения Clostridium acetobutylicum можно получить на средах с кукурузным (ржаным, картофельным) затором. Пробирки со стерильным 7 — 8%-м кукурузным затором заражают зернами хлебных злаков или комочками почвы и пастеризуют на кипящей водяной бане в течение 45 с. Выращивают при 35 — 37 °C в запаянных ампулах или пробирках под слоем вазелинового масла. Для обогащения среды клостридиями делают несколько последовательных пересевов культуры на кукурузный затор, а затем высевают ее на чашки с картофельным агаром и выращивают в анаэробных условиях — в анаэростатах.
Муравьинокислое брожение или, как его еще называют, брожение смешанных кислот, осуществляют энтеробактерии. Эпидемиологическое значение этих бактерий известно, а их представитель Escherichia coli является общепризнанным в санитарной микробиологии тест-организмом, поэтому выделение этой группы бактерий описано в соответствующем разделе практикума (см. гл. 46).
16.2. МЕТАНОГЕНЫ
Метаногены— строгие анаэробы, обитающие в донных осадках пресноводных и морских зон, богатых органикой, или в рубце жвачных. Это самая большая группа архей из филума Euryarchaeota. Обычно они являются конечным звеном анаэробной пищевой цепи разложения биологических полимеров и выделяют биогаз в качестве конечного продукта (60 % СН4 + 40 % СО2). Биогаз образуется на дне озер и болот, поднимаясь вверх, попадает в аэробную зону и используется метанокисляющими микроорганизмами. Метан образуется также в рубце жвачных (корова выделяет до 200 л метана в день) и в кишечнике термитов.
По морфологии метаногены варьируют от простых палочек, кокков и сар-цин до спиральных форм и неправильных кокков. Рост — от автотрофного и гетеротрофного до метилотрофного, температурные интервалы роста — от
мезофильных до экстремально термофильных. Метаногены используют довольно узкий набор субстратов. К первой группе субстратов относят Н2/СО2, СО. При этом 95 % фиксированного СО2 используется для получения энергии и выделяется в виде СН4 и только 5 % идет на биосинтез. Ко второй группе относят ацетат и некоторые спирты (ацетокластический метаногенез). Третью группу составляют одноуглеродные субстраты (формиат, метанол, метиламин, 2- и 3-метиламины). В морских водоемах сульфатредукторы опережают метаногенов в борьбе за общие субстраты (ацетат и молекулярный водород), однако многие морские метаногены способны использовать метиламины, которые не являются субстратами для сульфатредукторов.
Классы Methanobacteria, Methanococci, Methanopyri включают метаногенные археи, образующие метан на Н2/СО2, формиате, ацетате, метаноле, метиламинах и Н2/метаноле. Клетки содержат коферменты М, F420, F430, мета-ноптерин. При освещении светом с длиной волны 420 нм они флуоресцируют сине-зеленым светом (свойство кофакторов F420 и F430).
Для получения накопительных культур метаногенов следует использовать минеральные среды, содержащие азот в форме NH4n серу в форме S2-. Необходимые микроэлементы добавляют в виде стерильного концентрированного раствора, а потребность в факторах роста удовлетворяется добавлением дрожжевого экстракта. При приготовлении сред и растворов добиваются условий строгого анаэробиоза путем кипячения среды, замены газовой фазы на азот или аргон, добавления редуцирующих агентов и герметизации сосудов для культивирования. Поддержанию низкого окислительно-восстановительного потенциала способствуют дрожжевой экстракт, добавляемый в среду в качестве источника витаминов и других факторов роста, а также сульфид натрия и цистеин — источники серы. В концентрациях до 100 мг 52 /л сульфид не оказывает отрицательного влияния на рост метаногенных архей. Для избавления от бактерий в среду добавляют антибиотики широкого спектра действия (циклосерин, пенициллин, эритромицин, стрептомицин и др.) в количестве -0,2 мг/мл. В качестве субстрата в зависимости от потребностей выделяемой группы метаногенных архей вносят ацетат, формиат, метанол и метиламин в концентрации -10 мМ. Для накопления и выделения водородиспользующих метаногенов газовую фазу в сосудах для культивирования заменяют на смесь Н2 + СО2 (80 % + 20 %).
Источником биологического материала может служить образец любого анаэробного донного осадка или рубцовой жидкости, отобранный без нарушения анаэробных условий, но предпочтительнее использовать анаэробные осадки, активно образующие биогаз (например, из метантенков).
Среды и растворы. Для преимущественного развития, выделения, очистки и роста культур метаногенов используют среду следующего состава (г/л): КН2РО4 — 0,38; Na2HPO4 - 0,53; NH4C1 - 0,30; NaCl - 0,30; СаС12- 2Н2О - 0,11; MgCl2- 6Н2О - 0,10; резазурин — 0,001 (индикатор восстановительных условий); раствор микроэлементов — 1,0 мл/л; раствор витаминов — 0,5 мл/л.
Конечная концентрация микроэлементов в среде должна составить (мг/л): FeCl2 4Н2О — 9,944; Н3ВО, - 0,062; ZnCl2 - 0,068; CuCl2 - 0,013; MnCl2- 4Н2О - 0,061; СоС12- 6Н2О -9,066; NiCl2 - 0,013; НС1 - 0,002; NaOH - 0,400; Na2SeO3 - 0,017; Na2WO4 - 0,029; Na2MoO4 — 0,021.
Конечная концентрация витаминов в среде составляет (мг/л): биотин — 0,01; фолиевая кислота — 0,01; никотинамид — ОДО; л-аминобензойная кислота — 0,05; тиамин В,) — 0,10; пантотеновая кислота — 0,05; пиридоксамин — 0,25; цианкобаламин (В12) — 9,05; рибофлавин (В2) — 0,05.
Минеральную среду кипятят для избавления от кислорода, затем охлаждают до комнатной температуры и разливают под током азота (свободного от кислорода) для
предотвращения диффузии кислорода в среду. Среды разливают во флаконы, закрывают резиновыми пробками и зажимают алюминиевыми колпачками. Затем газовую фазу во флаконах дополнительно заменяют на требуемую: Ar, N2, Н2, N2/CO2 (80/20) или Н2/СО2 (80/20). Конечное давление во флаконах составляет —1,6 ат. Среды стерилизуют автоклавированием (1 ати), а раствор витаминов — фильтрованием (бактериальный фильтр диаметром 0,2 мкм). После стерилизации в среды добавляют раствор Na2S • 9Н2О до конечной концентрации 278 мг/л, витамины, а также требуемое количество стерильного субстрата (см. выше).
Все пересевы микроорганизмов осуществляют с помощью стерильных шприцев, не нарушая условий анаэробиоза.
Накопительные культуры метаногенов. Для получения накопительных культур в анаэробную стерильную среду вносят до 20 % (объем/объем) гомогенизированной пробы анаэробного осадка. После стерилизации среды устанавливают pH 6,8 — 7,0.
Культивирование накопительных культур проводят при 30 или 37 °C. Для накопления метаногенов в среду вносят 0,04 % дрожжевого экстракта. Для накопления Л/е//гл//ол«е/«-подобных архей дрожжевой экстракт в среду не вносят. Культивирование водородиспользующих метаногенов проводят на орбитальной качалке или шейкере для лучшего газообмена.
Развитие накопительной культуры контролируют люминесцентной (отмечают флуоресценцию при 420 нм) и световой микроскопией (отмечают наличие клеток разной морфологии, преимущественно метанококков, обладающих наиболее быстрым ростом). С помощью газожидкостной хроматографии определяют убыль субстрата и появление метана. Описывают изменения культуральных признаков («вспухание» анаэробного осадка, появление пузырьков газа и др.).
Для получения чистых культур метаногенов используют рассев накопительной культуры на поверхность или в толщу агаризованной минеральной среды того же состава (с 1,5 — 2% агара). Стерильную анаэробную агаризованную среду в плотно закупоренных сосудах расплавляют, остужают до 45 °C и стерильным шприцем вносят аликвоту культуральной жидкости из накопительной культуры с соблюдением условий анаэробиоза. После тщательного перемешивания содержимого сосуда его либо оставляют в горизонтальном положении для застывания, либо раскатывают рукой по ровной поверхности до затвердевания агара по стенкам сосуда в виде тонкого слоя. В обоих случаях добиваются получения максимальной поверхности агаризованной среды. Допустим рассев накопительной культуры на поверхность твердой среды в чашках Петри петлей или шпателем при работе в анаэробном боксе и последующей инкубации посевов в анаэростате. Культивирование ведут при 30 или 37 °C до появления отдельных колоний. Для выделения Methanosaeta-подобных метаногенов, которые практически не растут на твердых средах, применяют метод предельных разведений в жидкой среде с ацетатом в качестве субстрата.
Чистоту выделенных культур оценивают по однородности полученных колоний и микроскопической картине, а также путем рассева на твердые среды в анаэробных условиях (в анаэробном боксе). Материал берут из отдельной колонии с помощью микробиологической иглы или петли и осуществляют поверхностный посев на среду в чашках Петри. При глубинном посеве в толщу расплавленной агаризованной среды стерильным шприпем вносят суспензию клеток из отдельной колонии, приготовленную в стерильном анаэробном физрастворе. Свидетельством чистоты культуры является однородность выросших колоний и совпадение их признаков с описанными ранее.
16.3. ГОМОАЦЕТОГЕНЫ (АЦЕТОГЕНЫ)
Микроорганизмы, осуществляющие гомоацетатное брожение, продуктом которого является только ацетат, широко распространены в анаэробной зоне. Это строгие анаэробы, способные перерабатывать большой набор субстратов. Они связаны синтрофными связями с ацетокластическими метаногенами, которые проводят реакцию: ацетат —> метан + СО2, так как в пресных водоемах нет других возможностей удалить ацетат в анаэробных условиях. Процесс хотя и называется брожением, однако часть реакций происходит за счет анаэробного дыхания. Можно сказать, что гомоацетогены стоят на перепутье между бродиль-щиками и организмами, дышащими анаэробно, поскольку две молекулы ацетата образуются из глюкозы за счет брожения, а третья получается за счет анаэробного карбонатного дыхания. Ацетогены могут расти автотрофно на СО2 + Н2, образуя ацетат, при этом водород служит донором, а углекислота — акцептором электронов. В этом случае АТФ генерируется с помощью хемиоосмотиче-ского механизма, сопряженного с восстановлением углекислоты до ацетил-КоА (путь Вуда—Льюнгдала). Возможен рост в атмосфере СО с образованием ацетата (4СО + 2Н2О —> СН3СООН + 2СО2). Большинство ацетогенов способны к росту на одноуглеродных субстратах. Другие соединения, используемые некоторыми ацетогенами, — галактоза, рибоза, глюкуроновая кислота, глюконат, маннит, глицерин, метанол, многие спирты, органические кислоты и аминокислоты, целлобиоза и целлюлоза. Acetobacterium wooclii и некоторые клостридии в присутствии СО2 (СО) используют О-метильные группы большого количества метоксилированных ароматических кислот, включая ванильную, сиринговую, 3,4,5-триметоксибензойную, ферулиновую и др., для синтеза ацетата.
Гомоацетогенные микроорганизмы распространены во многих таксономических группах. Это представители родов Clostridium, например С. thermoaceticum, С. formicoaceticum, С. thermoautotrophicum (рост при 70 °C), Sporomusa (S. ovata), Acetobacterium (A. woodii), Acetoanaerobium, Acetogenium, Acetitomaculum, Acetohalobium, Acetonema, Moorella, Eubacterium, Peptostreptococcus, Ruminococcus, Syntrophococcus, Natroniella. Все они относятся к домену Bacteria. Среди архей и эукарий ацетогенов пока не обнаружено.
Для получения накопительных культур гомоацетогенов следует использовать минеральные среды с витаминами и микроэлементами на карбонатном буфере. Для выделения ряда культур следует добавлять дрожжевой экстракт. Поскольку в окислении Н2 и формиата гомоацетогены конкурируют с метаногенами и сульфатредукгорами, необходимо создать условия, «отсекающие» обе эти группы. Гомоацетогены получают преимущества перед метаногенами в слабокислых условиях (pH 6,1) и при пониженных температурах (ниже 20 °C). Для подавления метаногенеза добавляют соответствующие ингибиторы (BESA). В средах для гомоацетогенов не используют сульфаты, чтобы не давать преимуществ для развития сульфатредукторов. Поскольку гомоацетогены могут потреблять широкий спектр органических субстратов, в качестве селективного фактора можно применять их способность к деметилированию метоксилированных ароматических соединений. При этом следует учитывать, что использование таких субстратов может потребовать длительного периода адаптации.
Источником биологического материала может служить образец любого анаэробного донного осадка или рубцовой жидкости, отобранный без нарушения анаэробных условий.
Среды и растворы. Для преимущественного развития, выделения, очистки и роста культур гомоацетогенов используют среду следующего состава (г/л): КН2РО4 — 0,2; NH4C1 — 0,5; NaCl — 1,0 (до 20,0 для образцов из соленых местообитаний); СаС12 — 0,15; MgCl2-6H2O — 0,4 (до 3,0 для образцов из минерализованных вод); КС1 — 0,5; резазурин — 0,001 (индикатор восстановительных условий); раствор микроэлементов — 1,0 мл/л; раствор витаминов — 0,5 мл/л.
Конечная концентрация микроэлементов в среде должна составить (мг/л): FeCl2 • 4Н2О — 0,944; Н3ВО3 - 0,062; ZnCl2 - 0,068; CuCl2 - 0,013; MnCl2- 4Н2О - 0,061; СоС12- 6Н2О -0,066; NiCl2 - 0,013; НС1 - 0,002; NaOH - 0,400; Na2SeO3 - 0,017; Na2WO4 - 0,029; Na2MoO4 - 0,021.
Конечная концентрация витаминов в среде составляет (мг/л): биотин — 0,01; фолиевая кислота — 0,01; никотинамид — 0,10; и-аминобензойная кислота — 0,05; тиамин (В]) — 0,10; пантотеновая кислота — 0,05; пиридоксамин — 0,25; цианкобаламин (В,2) — 0,05; рибофлавин (В2) — 0,05.
Метоксилированные ароматические субстраты (ванилат, ферулат, сирингат и др.) вносят в концентрациях 5—10 мМ. В случаях использования в качестве субстратов спиртов и органических кислот (метанола, этанола, и-бутанола, и-пропанола, метоксиэтанола, малата и т.д.) их добавляют в количестве 10 — 20 мМ.
Минеральную среду кипятят для избавления от кислорода, затем охлаждают до комнатной температуры и разливают под током азота (свободного от кислорода) для предотвращения диффузии кислорода в среду. Среды разливают во флаконы, закрывают резиновыми пробками и зажимают алюминиевыми колпачками. Затем газовую фазу во флаконах дополнительно заменяют на требуемую: Ar, N2, Н2, N2/CO2 (90/10) или Н2/СО2 (80/20). Конечное давление во флаконах составляет ~ 1,6 ат. Среды стерилизуют автоклавированием (1 ати), а раствор витаминов — фильтрованием (бактериальный фильтр диаметром 0,2 мкм). После стерилизации в среды вносят витамины, 30 мл/л 1 М NaHCO3 и добавляют раствор Na2S • 9Н2О до конечной концентрации 278 мг/л. В качестве ингибиторов метаногенеза в среду вносят 2 мМ BESA или 0,5 % ацетилена в газовую фазу. Для образцов из желудочно-кишечного тракта добавляют в среду 1 % рубцовой жидкости.
Все пересевы микроорганизмов осуществляют с помощью стерильных шприцев, не нарушая условий анаэробиоза.
Накопительные культуры гомоацетогенов. Для получения накопительных культур в анаэробную стерильную среду вносят до 20 % (объем/объем) гомогенизированной пробы анаэробного осадка. После стерилизации среды устанавливают pH 7,1 — 7,3.
Культивирование проводят при 20—30 °C. Инкубировать накопительные культуры рекомендуется в полузаполненных сывороточных бутылях при их горизонтальном положении. Культивирование водородиспользующих гомоацетогенов проводят на орбитальной качалке или в шейкере для лучшего газообмена.
Развитие накопительной культуры контролируют после 1 —2 пассажей путем микроскопирования и наиболее часто отмечают наличие палочковидных нефлуоресцирующих клеток, обладающих активной подвижностью. С помощью газожидкостной хроматографии определяют убыль молекулярного водорода и спиртов (без существенного образования метана), а также накопление ацетата. Трансформацию метоксилирован-ного ароматического субстрата регистрируют с помощью ВЭЖХ.
Для получения чистых культур гомоацетогенов используют рассев накопительной культуры на поверхность или в толщу агаризованной минеральной среды того же состава (с 1,5 —2 % агара). Стерильную анаэробную агаризован-ную среду в плотно укупоренных сосудах расплавляют, остужают до 45 °C и стерильным шприцем вносят аликвоту культуральной жидкости из накопительной культуры с соблюдением условий анаэробиоза. После тщательного перемешивания содержимого сосуда его либо оставляют в горизонтальном
положении для застывания, либо раскатывают сосуд рукой по ровной поверхности до затвердевания агара по стенкам в виде тонкого слоя. В обоих случаях добиваются получения максимальной поверхности агаризованной среды. Допустим рассев накопительной культуры на поверхность твердой среды в чашках Петри петлей или шпателем при работе в анаэробном боксе и последующей инкубации посевов в анаэростате. Мел в агаризованной среде может служить индикатором образования ацетата (зоны просветления вокруг колоний). Культивирование ведут при 20 — 30 °C до появления отдельных колоний. При использовании в качестве субстрата молекулярного водорода очистку культуры проводят либо в тонком слое агаризованной среды, либо методом предельных разведений в жидкой среде при перемешивании.
Чистоту выделенных культур оценивают по однородности полученных колоний и микроскопической картины, а также путем рассева на твердые среды в анаэробных условиях (в анаэробном боксе). Материал берут из отдельной колонии с помощью микробиологической иглы или петли и осуществляют поверхностный посев на среду в чашках Петри. При глубинном посеве в толщу расплавленной агаризованной среды стерильным шприцем вносят суспензию клеток из отдельной колонии, приготовленную в стерильном анаэробном физрастворе. Через 2 пассажа проверяют чистоту выделенной культуры на богатых средах, содержащих 0,1 % дрожжевого или солодового экстракта и пептона. Свидетельством чистоты культуры является однородность выросших колоний и совпадение их признаков с описанными в литературе.
16.4. ЖЕЛЕЗОВОССТАНАВЛИВАЮЩИЕ МИКРООРГАНИЗМЫ
Микроорганизмы, восстанавливающие ионы окисленного железа (ферри-ионы, Fe3+), широко распространены в анаэробных зонах на Земле. Они играют значительную роль в изменении окружающей среды в анаэробных осадках, содержащих окисленное железо. Полагают, что эти микроорганизмы озветственны за большой процент окисленного органического вещества в таких экологических нишах. Ре3+-восстановители играют важную роль в биогеохимических циклах и проводят биоминерализацию ароматических органических соединений. Организмы, способные к восстановлению Fe3+, относят к разным филогенетическим группам, однако одна группа облигатных анаэробных бактерий сем. Geo-bacteriaceae образует тесный таксон. Все члены этой группы восстанавливают Fe3+, а некоторые способны также восстанавливать серу, нитраты и гуминовые кислоты. Эти акцепторы электронов интересны тем, что их можно рассматривать в качестве акцепторов электронов в ранний, бескислородный период развития жизни на Земле. Изучение восстановления железа и других практически нерастворимых акцепторов электронов представляет значительный интерес с точки зрения физиологии.
Физиологию сем. Geobacteriaceae изучали в основном на двух представителях: G. metalUreducens и G. sulfurreducens. G. metallireducens был первым облигатно-анаэробным организмом, для которого было показано восстановление природных форм Fe3+. Организм полностью окисляет ряд органических субстратов до СО2, используя при этом замкнутый цикл трикарбоновых кислот, и восстанавливает Fe3+, возможно, через менахиноны и цитохромы, которые были
обнаружены в клетках. Недавно описана система окисления фумарата с восстановлением Fe3+ через тригемовый цитохром с3 с оксидазой (Ре3+-редуктазой), локализованной на внешней мембране грамотрицательных G. sulfurreducens. Представителей рода Geobacter выделяют анаэробно на пресноводной среде с ацетатом (см. ниже), куда в качестве акцептора электронов добавляют 50 мМ Fe3+. Культивирование ведут на жидких средах в пенициллиновых пузырьках, закрытых резиновыми пробками, снабженными алюминиевыми колпачками. При культивировании соблюдают принципы анаэробного выращивания культур по Хангейту (см. 16.2). За ростом культур следят по появлению Fe2' феррозиновым методом, определением общего количества клеток по Виноградскому и определением числа живых клеток с акридином оранжевым (см. гл. 5).
Среда для Geobacter metallireducens (г/л): Ре(Ш)-цитрат — 13,70; NaHCO3 — 2,50; NH4C1 — 1,50; NaH2PO4 — 0,60; КО — 0,10; Na-ацетат — 2,50; раствор витаминов — 10,00 мл; раствор микроэлементов — 10,00 мл; Na2WO4-2 Н2О — 0,25 мг; вода (диет.) — 980,00 мл. Довести pH до 6,7 —7,0. Среду готовят анаэробно под струей газовой смеси: 80 % N2 + 20 % СО2. Вначале цитрат железа растворяют в воде при нагревании и доводят pH до 6,0, затем добавляют остальные компоненты среды. Используют 3— 10%-й посевной материал.
Раствор микроэлементов (г/л): нитрилотриацетат — 1,5; MgSO4-7H2O — 3,0; MnSO4-2H2O - 0,5; NaCl - 1,0; FeSO4-7H2O - 0,1; CoSO4-7H2O - 0,18: CaCl2-2H2O - 0,1; ZnSO4-7H2O - 0,18; CuSO4-5H2O - 0,01; KA1(SO4)2-12H2O -0,02; H3BO3- 0,01; Na2MoO4 • 2H2O - 0,01; NiCl2 • 6H2O - 0,025; Na2SeO3 • 5H2O - 0,3 мг: вода (диет.) — 1000 мл.
Раствор витаминов (мг/л): биотин — 2; фолиевая кислота — 2; пиридоксин-НС1 — 10; тиамин-НС1 2Н2О — 5; рибофлавин — 5; никотиновая кислота — 5; D-Са-панте-тонат — 5; витамин В|2 — 0,1; н-аминобензойная кислота — 5; липоевая кислота — 5: вода (диет.) — 1000 мл.
Среда для Geobacter sulfurreducens (г/л): NH4C1 — 1,50; Na2HPO4 — 0,60; КО — 0,10; Na-ацетат — 0,82; раствор микроэлементов — 10,00 мл; раствор витаминов — 10,00 мл: селенит-вольфрамовый раствор — 1 мл; резазурин — 0,5 мг; NaHCO3 — 2,50; вода (диет.) — 980 мл.
Растворы витаминов и микроэлементов — те же, что и для Geobacter metallireducens. Селенит-волъфрамовый раствор-. 0,5 г NaOH, 3 мг Na2SeO3- 5Н2О; 4 мг Na2WO4- 2Н2О в 1 л воды (диет.) Растворяют все компоненты среды (за исключением NaHCO3), кипятят, остужают до комнатной температуры под током газа (80 % N2 + 20 % СО2) и добавляют бикарбонат. Доводят pH среды, барботируя газ, до 6,8. Разливают по анаэробным флаконам подтоком газа. После автоклавирования (1 атм) добавляют фумарат из анаэробного концентрированного раствора (16 % вес/об.), простерилизованного фильтрованием, в количестве 0,5 мл на 10 мл среды, проверяют pH (должен быть между 6,9 и 7,1).
Глава 17
ВЫДЕЛЕНИЕ ФОТОТРОФНЫХ МИКРООРГАНИЗМОВ
Способность к фотосинтезу свойственна нескольким группам микроорганизмов. Известно два основных типа фотосинтеза, основанных на фотохимических реакциях пигментов хлорофилльной природы: оксигенный (т. е. с выделением О2), при котором донором электронов служит вода, и аноксигенный
(без выделения 02), при котором используется не вода, а другие доноры электронов, например восстановленные соединения серы, органические вещества, Н2
Оксигенный тип фотосинтеза характерен для цианобактерий и эукариотических водорослей. Эти организмы в целом занимают сходные экологические ниши (что и отражено в общебиологическом понятии «водоросли»). Как правило, они являются аэробными фотоавтотрофами, часто довольно близкими друг другу по физиологии, пищевым потребностям, отношению к физико-химическим факторам среды. Выделение и культивирование цианобактерий и эукариотических водорослей в общем основано на одних принципах, поэтому эти вопросы целесообразно рассмотреть в одном разделе.
Аноксигенные фототрофы существенно отличаются от оксигенных по своим экологическим нишам, физиологии, пищевым потребностям и отношению к условиям среды, поэтому они рассматриваются в отдельном разделе. Типичными аноксигенными фототрофами являются пурпурные серные и несерные, зеленые серные, зеленые нитчатые бактерии и гелиобактерии. При всем разнообразии метаболизма и организации фотосинтезирующего аппарата это, в общем случае, анаэробы, которые могут быть облигатными или факультативными, лито- или органо-, авто- или гетеротрофами.
17.1. ВЫДЕЛЕНИЕ ОКСИГЕННЫХ ФОТОТРОФОВ
Основными элективными условиями для выделения цианобактерий и водорослей являются: освещение, наличие в среде необходимых для роста минеральных компонентов, а также в большинстве случаев отсутствие органических веществ, стимулирующих рост органогетеротрофов.
Отбор проб. Цианобактерии и эукариотические водоросли развиваются в самых разнообразных местообитаниях — в водоемах, почве, на поверхности камней и деревьев, часто образуя хорошо заметные невооруженным глазом окрашенные пятна. Обычно сбор образцов проводят в период их активного размножения (для большинства это время с весны по осень) и в местах с максимальной численностью. Пробы воды и ила для выделения цианобактерий и водорослей отбирают в стерильные флаконы, обычно заполняя их не более чем на 2/3 объема. Образцы почвы или соскобы с твердых поверхностей, сделанные простерилизованным ножом или лопаточкой, помещают в стерильные флаконы, банки или пакеты. До посева пробы хранят при рассеянном освещении и температуре не выше 20 °C. Рекомендуется проводить посев непосредственно в день отбора пробы; если же предстоит длительная транспортировка и хранение, то допустимо часть времени держать пробы в холодильнике, хотя для многих видов (особенно термофильных цианобактерий) это нежелательно.
Приготовление питательных сред. Имеется большое число рецептов простых и достаточно универсальных сред для выделения и культивирования цианобактерий и водорослей. Наиболее универсальными являются: среда BG-11 и среда Громова № 6, которые поддерживают активный или умеренный рост большинства выделенных штаммов цианобактерий, а также среда Кратца — Майерса для водорослей и цианобактерий (см. приложение 4). Рецепты боль
шинства других известных сред можно найти в специальных руководствах, приведенных в списке литературы*. Для почвенных водорослей рекомендуют среду Бристоль в модификации Голлербаха, иногда используют среды Бейеринка, Кнопа, Бенеке, Чу, среду с почвенной вытяжкой и др. Для морских цианобактерий предложены среды, приготовленные на искусственной морской воде. При выделении организмов из соленых или содовых озер часто используют среды, имитирующие химический состав воды этих водоемов. Другим примером могут служить разработанные Е. Е. Успенским среды для водорослей, близкие по составу к природной чистой и загрязненной воде.
Помимо относительно универсальных сред для фототрофов разработано немало сред для культивирования отдельных таксонов. Например, рост Chlorella хорошо поддерживают среда Крейга и Трилиза, среда Тамия; для выращивания Spirulina рекомендована среда Громова № 16 с высоким значением pH. Для отдельных групп водорослей разработаны среды, учитывающие их специфические потребности в определенных элементах (например, потребность в кремнии у диатомовых водорослей). Наконец, некоторые водоросли нуждаются в витаминах, факторах роста или органических веществах как основном источнике углерода (среди водорослей имеется немало гетеротрофов), что также учитывают при оптимизации сред для их выделения и культивирования.
Следует еще раз напомнить, что многие фототрофы очень чувствительны к качеству (составу) воды. Еще в начале XX в. Е.Е. Успенский обнаружил, что в природных водоемах в зависимости от концентрации ряда элементов внешний вид (габитус) водорослей (например, представителей родов Drapalnaldia, Stigeoclonium, Spirogyrd) может сильно меняться. Поэтому некоторые из них он даже предлагал использовать как индикаторные виды на содержание в водоемах железа, азота и фосфора**. Избыток фосфора в среде иногда может вызывать у цианобактерий потерю чехлов, появление инволюционных форм. Рост ряда цианобактерий подавляют ничтожные концентрации ионов меди, что приходится учитывать при использовании металлических дистилляторов.
Как правило, среды для цианобактерий и водорослей готовят на дистиллированной (или бидистиллированной) воде, однако при первых посевах часто рекомендуют разводить среду простерилизованной природной водой (взятой из того водоема, откуда отобрана проба), постепенно уменьшая ее долю в среде.
Для приготовления твердых сред для цианобактерий и водорослей чаше всего используют хорошо очищенный 2%-й агар-агар (или 1,5%-й агар марок Bacto, Difko), иногда силикагель. Культивирование на твердых средах проводят в чашках Петри или пробирках со скошенным агаром. Кроме того, циано-
* Рецепты этих и других упоминаемых в главе сред для цианобактерий и водорослей можно найти в руководствах: Культивирование коллекционных штаммов водорослей / Под ред проф. Б.В.Громова. — Л.: Изд-во ЛГУ, 1983; Успенская В.И. Экология и физиология питания пресноводных водорослей. — М.: Изд-во МГУ, 1966; Голлербах М.М., Штина Э.А. Почвенные водоросли. — Л.: Изд-во АН СССР, 1968; Методы физиолого-биохимического исследования водорослей в гидробиологической практике. — Киев: Наукова думка, 1975. Зенова Г.М., Штина Э.А. Почвенные водоросли. — М.: Изд-во МГУ, 1990; Андреева М. В. Почвенные и аэрофиль-ные зеленые водоросли. — СПб.: Наука, 1998; Bergey’s Manual of Systematic Bacteriology L.R.Boone, R.W.Castenholz, eds. Vol.l. — New York; Heidelberg; Berlin: Springer-Verlag, 2001. — P. 485-487; и др.
** Успенский E. Железо как фактор распределения водорослей. — М.: Изд-во МГУ, 1925. См. также: Успенская В. И. Экология и физиология питания пресноводных водорослей. — М.: Изд-вс МГУ, 1966.
бактерии рекомендуют выращивать в пробирках с полужидкой агаризованной средой (0,5 —0,8 % агара), в которой их клетки меньше подвержены высыханию и дольше сохраняются.
Получение накопительной культуры. В наиболее распространенном случае небольшое количество отобранного образца (пробы воды, ила, почвы, зеленого налета с поверхности и т.д.) вносят в колбу, на объема заполненную жидкой средой, и помещают ее на свет, в условия естественного освещения 'как правило, на северное окно) или в люминостат. Прямого солнечного света желательно избегать. Засеянные колбы выдерживают при умеренной освещенности и температуре 20 — 25 °C от нескольких дней до 2 — 3 мес, отмечая появление визуально заметных признаков роста. Далее в зависимости от целей исследования полученную накопительную культуру описывают, определяют, рассевают на питательные среды для получения чистых культур и т.д. Для определенных групп организмов создают соответствующие элективные условия: например, протококковые водоросли получают преимущество перед другими фототрофами при высокой интенсивности света; цианобактерии (в ряде случаев) — при повышенной температуре (35 °C); азотфиксируюшие цианобактерии — при отсутствии в среде соединений азота и т.д.
Однако не все цианобактерии и водоросли (особенно почвенные) удается сразу вырастить и выделить в жидкой среде, поэтому иногда их выделяют на различных твердых субстратах. Некоторые из таких методов разработаны очень давно, но могут оказаться полезными и в настоящее время. Наиболее простой из них, метод почвенных культур, заключается в том, что образец почвы помещают в чашку Петри, равномерно увлажняют дистиллированной или водопроводной водой и выдерживают на свету при постоянной влажности в течение 2 — 4 мес. За это время почвенные водоросли и цианобактерии разрастаются внутри и на поверхности почвы, а также на внутренней поверхности чашки Петри, откуда их можно взять для приготовления препаратов и посева в питательную среду.
Удобным вариантом описанного метода является почвенная культура со стеклами збрастания. В этом случае исследуемую почву ровным слоем вносят в чашки Петри, увлажняют, раскладывают на ней несколько стерильных покровных стекол, слегка прижимая их к почве пинцетом или стеклянной палочкой, и помещают на свет. Мелкие свободные пространства под стеклами представляют собой своеобразные влажные камеры, в которых создаются условия для развития водорослей. Через разные промежутки времени (от 2 — 3 нед до 2 — 3 мес) покровные стекла последовательно снимают и готовят препараты для микроскопирования или отсевают выросшие организмы в питательные среды. Последовательный просмотр стекол обрастания дает возможность выявить сукцессию в развитии микроорганизмов (обычно первыми вырастают зеленые водоросли, затем цианобактерии, позднее диатомовые водоросли) и выбрать подходящий момент для посева.
Предложенный М. М. Голлербахом метод выделения водорослей в песчаных камерах позволяет изолировать водоросли с разным отношением к влажности. В коническую кол-5у помещают предварительно стерилизованный кварцевый песок и засевают почвенной суспензией, которую распределяют по всей поверхности песка. После этого колбу наклоняют, чтобы одна часть песка была лишь влажной, а другая часть находилась под слоем жидкости. В наклонном положении колбу помещают на свет. После развития водорослей их пересевают в чашки Петри на среду, содержащую почвенную вытяжку с 1 % згар-агара. Развитие водорослей становится заметным по прошествии 2 — 3 нед.
Метод почвенных культур с фильтровальной бумагой заключается в следующем. В чашку Петри помещают исследуемую почву слоем около 1 см, хорошо увлажняют дистиллированной водой, выравнивают поверхность и покрывают кружком фильтровальной бумаги. Чашку Петри выдерживают при рассеянном свете и 20 — 25 °C, увлажняя фильтровальную бумагу по мере ее высыхания. В пределах от 2 до 60 сут через поры фильтровальной бумаги прорастают различные цианобактерии (хуже проникают более крупные водоросли). Недостаток метода связан с тем, что на фильтровальной бумаге могут обильно развиваться целлюлозолитические бактерии и грибы, изменяя ее свойства и делая непригодной для роста водорослей.
Образец исследуемой почвы можно также рассеять непосредственно на поверхность твердой среды (в чашке Петри) в виде почвенного мелкозема. На свету вокруг комочков почвы хорошо разрастаются цианобактерии, диатомовые и зеленые водоросли, которые затем удается подцепить петлей и выделить в культуру. Аналогичный посев «россыпью» на твердую среду дает хорошие результаты при выделении цианобактерий и водорослей с поверхности камня и других твердых субстратов.
Выделение чистой культуры. В отношении водорослей и цианобактерий используют устоявшиеся, хотя и не совсем точные, понятия альгологически чистая культура (т.е. культура только одного вида цианобактерий или водорослей) и аксеничная, бактериально чистая культура (т.е. культура водоросли, не содержащая бактерий, и культура цианобактерии, не содержащая других бактерий).
Для получения альгологически чистой культуры используют обычные микробиологические методы: разведение и пересев накопительной культуры или посев клеток, которые отделяют с помощью микроманипулятора. В первом случае небольшое количество накопительной культуры микроорганизмов переносят в чашку Петри с твердой средой; петлей или шпателем распределяют ее по поверхности среды и далее инкубируют на свету до появления видимых признаков роста. Из выросших изолированных колоний или скоплений трихомов переносят часть материала на новую среду, многократно (10 — 20 раз) повторяя пересевы. При пересевах используют как твердые, так и жидкие среды. Кроме того, иногда рекомендуют использовать двуслойную среду (на ага-ризованную скошенную среду в пробирке наливают 2 — 5 мл жидкой среды и вносят в нее культуру), развитие как бы на поверхности раздела двух фаз интенсифицирует рост водоросли и облегчает выделение ее в культуру.
Для выделения подвижных организмов, например Oscillatoria, можно воспользоваться их способностью к фототаксису. Небольшой комочек нитей цианобактерии вносят в чашку Петри с агаризованной средой. Чашку помещают на свет, прикрыв одну ее часть (куда произведен посев) черной бумагой. Нити цианобактерии начинают двигаться по направлению к свету; через несколько часов инкубации их можно подцепить петлей или тонкой иглой на некотором расстоянии от места посева и поместить в свежую среду. Процесс проводят под контролем микроскопа (при малом увеличении).
Выделение альгологически чистых культур довольно крупных микроорганизмов, видимых под лупой (10 — 20*), можно осуществить с помощью пипеток или капилляров с последующим высевом в питательную среду. Для получения культур ряда водорослей {Stigeoclonium, Drapalnaldia, Oedogonium, Ulothrix) рекомендуют рассевать их зооспоры.
Получение бактериально чистых культур цианобактерий и водорослей сопряжено обычно с немалыми трудностями. Это обусловлено их значительно
более крупными размерами клеток по сравнению с клетками бактерий-спутников, образованием чехлов и слизи, в которых эти бактерии часто обитают и питаются, а также собственно биологической природой устойчивости альго- и цианобактериальных ассоциаций. Тем не менее в коллекциях микроорганизмов имеется большое количество аксеничных штаммов цианобактерий и водорослей, а в литературе описан ряд методов их выделения.
Самым распространенным является классический метод выделения чистых культур из изолированной колонии, позволяющий получить в чистой культуре, прежде всего, одноклеточные формы. Для успешного выделения рекомендуется отбирать микроколонии фототрофов, как можно более удаленные от колоний сопутствующих микроорганизмов. Они хорошо различимы при малом увеличении микроскопа. Вначале просматриваю!’ (не открывая!) перевернутые чашки с выросшими микроколониями, отмечая их на донышке чашки, а затем из открытых чашек выбранные микроколонии отсевают в пробирки с 2— 5 мл жидкой питательной среды. Процесс проводят под контролем микроскопа. Пробирки помещают на свет и по мере роста культур контролируют их чистоту как микроскопически, так и высевом на богатую среду, например МПБ с 0,1 % глюкозы.
Один из способов выделения чистой культуры Chlorella sp. заключается в чередовании пересевов водоросли на минеральную и богатую (например, с глюкозой) твердую среду, обеспечивающую интенсивный рост и быстрое появление колоний.
Методы очистки подвижных нитчатых цианобактерий, как и в случае получения альгологически чистых культур, основаны на том, что нити, расползаясь по агаризованной среде, с одной стороны, механически очищаются от сопутствующих бактерий, а с другой стороны, как бы «обгоняют» их, отползая на значительное расстояние. В чашку Петри с агаризованной средой, иногда наполовину затемненную черной бумагой, помещают небольшой комочек нитей цианобактерий, инкубируют на свету, а через несколько часов находят под микроскопом наиболее далеко отползшие и наименее загрязненные бактериями нити и отсевают их в пробирки с жидкой средой. Однако, как правило, нитчатые микроорганизмы особенно трудно поддаются очистке.
Помимо методов, связанных с механическим отделением клеток или колоний, предложено большое количество методов очистки культур цианобактерий и водорослей, основанных на обработке их химическими соединениями: этанолом, риванолом, фенолом, различными детергентами, йодом (для диатомовых водорослей), антисептиками, а также различными антибиотиками и их сочетаниями. Например, для очистки Chlorella была предложена обработка левомицетином (1 мг/мл) и стрептомицином (1000 ед./мл) в течение 4 ч с последующим отмыванием от антибиотика и посевом на свежую среду. При очистке Dunaliella положительные результаты отмечены при обработке в течение суток пенициллином (15 000 ед./мл) и стрептомицином (12 500 ед./мл), для очистки разных водорослей предложено использовать также актиномицин, ко-лимицин, хлортетрациклин, окситетрациклин, эритромицин*. Возможность применения антибиотиков обусловлена тем, что их альгицидные концентра
* См., например: Методы физиолого-биохимического исследования водорослей в гидробиологической практике. — Киев: Наукова думка, 1975.
ции, как правило, выше бактерицидных (например, левомицетин подавляет рост бактерий в концентрации 50 мг/мл, а хлореллы — в концентрации 200 мг/мл). Во многих случаях, однако, разница между этими концентрациями очень мала и обработка антибиотиком оказывается неэффективной.
Из физических методов наиболее эффективным способом очистки водорослей и особенно цианобактерий считается обработка ультрафиолетом, поскольку цианобактерии более устойчивы к облучению, чем сопутствующие микроорганизмы. Однако эти микроорганизмы часто оказываются защищены слизью и чехлами цианобактерий, поэтому очистить культуру удается не всегда. Кроме того, при УФ-облучении существует вероятность получить мутантные формы выделяемых организмов.
Выделенные чистые культуры цианобактерий и водорослей периодически пересевают в жидкую, полужидкую или на твердую агаризованную среду и выращивают при постоянном или периодическом освещении. В некоторых коллекциях их поддерживают в условиях пониженной температуры (15 °C) и освещенности. На твердой среде водоросли обычно можно длительно хранить в холодильнике, цианобактерии хранить в холодильнике не рекомендуется.
17.2. ВЫДЕЛЕНИЕ АНОКСИГЕННЫХ ФОТОТРОФНЫХ БАКТЕРИЙ
Основными элективными условиями, которые надо создать при выделении культур большинства аноксигенных фототрофных бактерий, являются: наличие света, присутствие в среде подходящего донора и акцептора электронов, а также отсутствие О2. Изменяя эти и другие параметры (спектральный состав света и его интенсивность, окислительно-восстановительный потенциал и концентрацию сульфида, наличие и состав органических соединений, температуру и значение pH среды), можно добиться преимущественного роста представителей определенных групп.
Отбор проб
Большинство аноксигенных фототрофных бактерий обитают в анаэробной зоне различных водоемов: в илах и придонной воде озер, в морских прибрежных водах, серных и горячих источниках, сточных водах, некоторые виды встречаются в почве. Нередко пурпурные серобактерии образуют на поверхности ила и разлагающихся водорослей заметные невооруженным глазом розоватые пятна или окрашивают слои воды в пурпурный и розовый цвет. Пробы воды и ила отбирают в стерильные флаконы (заполняя их не доверху) и закрывают пробками. Анализ проб и посев микроорганизмов рекомендуется проводить как можно скорее, в случае длительного хранения пробы можно держать в холодильнике.
Приготовление сред и получение накопительных культур
Исторически первые накопительные культуры аноксигенных фототрофов были получены в 1888 г. С. Н. Виноградским при постановке так называемой колонки Вино
градского (см. гл. 37). В результате противоположно направленных процессов диффузии О2 и H2S в колонке образуются градиенты их концентраций, а также градиент окислительно-восстановительного потенциала. При этом создаются зоны, условия в которых соответствуют экологическим нишам разных групп бактерий. Фототрофные бактерии развиваются на разной высоте, образуя окрашенные слои воды или окрашенные пятна на границе между илом и стеклом цилиндра на стороне, обращенной к свету. Как модель, демонстрирующая развитие микробиологических процессов в природной системе, колонка Виноградского сохраняет значение до наших дней, однако для выделения фототрофных бактерий чаще используют синтетические среды, позволяющие выращивать микроорганизмы в контролируемых условиях.
Синтетические среды для фототрофных бактерий были впервые разработаны М. Бейе-ринком и немного позднее К. Б. ван Нилем. В настоящее время известно большое число рецептур сред, предложенных разными исследователями*. Примеры типичных по составу сред приведены в приложении 4.
Пурпурные и зеленые серные бактерии обладают сходными пищевыми потребностями. Одной из наиболее универсальных для них является минеральная среда, предложенная Г.Библом и Н. Пфеннигом**. Основные компоненты (г/л): КН2РО4 — 1,0; NH4C1 — 0,5; MgSO4 — 0,4; СаС12 — 0,05; микроэлементы, а также Na2S (360 мг/л для пурпурных и 600 мг/л для зеленых бактерий) и NaHCO3 (1,5 г/л), которые готовят отдельно в виде 10%-х растворов и вносят в среду после стерилизации. В зависимости от того, откуда выделяют бактерии, в среду вносят 1—20 г/л NaCl и до 3 г/л MgSO4. Морские штаммы бактерий также часто нуждаются в витамине В12; его вносят в количестве 0,002 мг/л.
Зеленые серобактерии по сравнению с пурпурными часто предпочитают несколько более низкие значения pH среды и более устойчивы к сульфиду, что можно использовать для получения их преимущественного роста. Кроме того, зеленые серобактерии более эффективно используют свет и могут расти при более низкой освещенности. Одним из элективных условий для роста пурпурных бактерий может быть освещение длинноволновым светом (800 нм или более), что достигается с помощью соответствующих светофильтров. Преимущественного развития отдельных представителей пурпурных серобактерий можно также достичь, варьируя интенсивность и режим освещения или температуру. Так, освешенность 2000 — 3000 лк и температура 30 °C более способствует доминированию быстрорастущих видов (например, Allochomatium vinosum, прежнее название Chromatium vinosum), а более низкие освещенность (50 — 300 лк) и температура (20 °C), а также чередование периодов света и темноты более благоприятны для медленнорастущих {Chromatium okenii или Isochromatium warmingi, прежнее название Chromatium warmingii).
Примером среды для несерных фототрофных бактерий может служить среда Орме-рода, содержащая (г/л): MgSO4- 7Н2О — 0,20; СаС12- 2Н2О — 0,08; FeSO4- 7Н2О — 0,01; К2НРО4 — 0,90; КН2РО4 — 0,60; (NH4)2SO4 — 1,25; ЭДТА— 0,02; микроэлементы — 1 мл; малат Na — 2,0; дрожжевой экстракт — 0,10. Состав микроэлементов (г/л): Н3ВО3 — 2,80; MnSO4-4H20 - 2,10; Na2MoO4- 2Н2О - 0,75; ZnSO4- 7Н2О - 0,24; Cu(NO3)2- ЗН2О -0,04.
Пурпурные несерные бактерии предпочитают фотоорганогетеротрофные условия роста и часто чувствительны к сульфиду. В среды для их выделения вносят органический субстрат (обычно ацетат, малат, пируват или другие Na-соли органических кислот) в качестве донора электронов и основного или дополнительного источника углерода. Обязательным компонентом среды для большинства несерных бактерий является раствор витаминов или дрожжевой экстракт. Иногда в среду вносят тиосульфат Na (0,2 г/л). Сульфид в большинстве случаев не вносят и даже часто снижают концентра
* Подробные сводки имеются, например, в руководствах «The Prokaryotes» и «Bergey’s Manual of Systematic Bacteriology».
** Biebl H., Pfennig N. 1978. Цит. no: The Prokaryotes. 1992. — V. IV. — P. 3204.
цию сульфата в среде, чтобы избежать развития в накопительной культуре сульфатре-дукторов, однако известны пурпурные несерные бактерии, особенно морские штаммы, которые устойчивы к сульфиду и способны хорошо его использовать как донор электронов. Элективные условия для развития представителей отдельных родов и видов можно создать внесением в среду различных субстратов (например, бензоата или формиата для Rhodopseudomonas palustris) и/или вариацией pH среды (например, при pH 5,2 и наличии в качестве субстрата сукцината Na создаются условия для преимущественного выделения Rhodopseudomonas acidophila и Rhodomicrobium vannieliij.
При простоте состава многие среды довольно сложны в приготовлении, в частности, поскольку содержащиеся в них восстановленные вещества, устойчивые в анаэробных условиях, легко взаимодействуют и окисляются в присутствии О2. Поэтому среды часто готовят, сливая несколько отдельно простерилизованных растворов непосредственно перед посевом, и затем доводят pH до нужного значения с помощью стерильных растворов кислоты или щелочи (соды).
Для получения накопительной культуры фототрофных бактерий обычно используют жидкие среды. Небольшое количество отобранной пробы ила или воды вносят в пробирку или флакон, доверху заливают питательной средой и герметично закрывают пробкой. После посева пробирки и флаконы помещают в люминостат и выдерживают в течение нескольких суток, пока среда не приобретет характерный для фототрофных бактерий красноватый или зеленоватый оттенок. Полученную накопительную культуру используют в качестве инокулята для дальнейшего культивирования и выделения чистых культур бактерий.
Другой способ посева при получении как накопительных, так и чистых культур фототрофных бактерий — это непосредственный посев в пробирки с агаризованной средой для получения изолированных колоний. Небольшое количество посевного материала вносят в пробирку, заливают расплавленной теплой питательной средой и быстро перемешивают. Для получения отдельных колоний рекомендуется сделать 8 — 10 серийных разведений посевного материала (с шагом приблизительно 1:10). Разведения можно готовить в воде или жидкой среде, рассевая затем по несколько капель ь пробирки с расплавленной агаризованной средой, либо последовательно использоват:-в качестве инокулята небольшое количество засеянной агаризованной среды (такой посев надо проводить быстро, чтобы среда не успела застыть). Существует несколько модификаций этого метода. После посева пробирки герметично закрывают резиновыми пробками и помещают в люминостат. Через несколько дней в них можно увидеть отдельные окрашенные колонии.
Для получения культур пурпурных несерных бактерий, многие из которых устойчивы к небольшим концентрациям кислорода, рекомендуют также посев петлей ид шпателем на поверхность агаризованной среды в чашке Петри. Засеянные чашки пс-мещают в люминостат и инкубируют в атмосфере воздуха либо помещают в анаэросте? из прозрачного стекла и инкубируют при освещении в атмосфере, обогащенной 5 СО2 и 3 % Н2. На поверхность агаризованной среды можно также разложить фильтры на которых сконцентрированы пробы исследуемой воды. Отдельные колонии, выросшие на среде или на фильтрах, в дальнейшем используют для получения чистой культуры пурпурных несерных бактерий.
Получение чистых культур. Наиболее эффективным методом для получени чистых культур фототрофных бактерий является их выделение из отдельны изолированных колоний. Из пробирки извлекают столбик агаризованной среды с выросшими в ней окрашенными колониями (обычно для этого у нее отпиливают дно и выталкивают агар, вместо пробирок удобно также использовать трубки Бурри), помещают его в чашку Петри и с помощью капилляр, достают выбранную колонию. Переносят колонию в стерильную пробирке раздавливают пипеткой, растирают на внутренней стенке пробирки и суспен
дируют бактерии в небольшом количестве среды. Суспензию используют в качестве инокулята для засева серии пробирок с агаризованной средой, как описано выше. Все операции проводятся в боксе или ламинарном шкафу со строгим соблюдением условий стерильности. Выделение культуры из отдельной колонии обычно повторяют несколько раз, обязательно проверяя культуры на чистоту как микроскопированием, так и высевом в питательные среды, в частности, в среду для сульфатредуцирующих бактерий.
Культуры фототрофных бактерий выращивают в люминостате, а хранить их можно при естественном освещении или в холодильнике.
ГЛАВА 18
ВЫДЕЛЕНИЕ И УЧЕТ ГРИБОВ МЕТОДАМИ ПОСЕВА НА ПИТАТЕЛЬНЫЕ СРЕДЫ И ПРИМАНОК
18.1. ОТБОР ОБРАЗЦОВ ПОЧВ И РАСТЕНИЙ
Важную роль при проведении микологического анализа почв и растений играет отбор проб. Процедура отбора, хранения и транспортировки образцов должна гарантировать уверенность, что выделяемые на завершающем этапе работы грибы действительно являются обитателями данных почв.
Для получения достоверной характеристики комплекса почвенных микроскопических грибов в биогеоценозах необходимо проведение нескольких анализов за вегетационный сезон (не менее трех) при достаточном в каждом случае числе повторностей для статистической обработки результатов. Почву анализируют, как правило, на основе 5—10 отдельно взятых образцов, отобранных методом случайных проб. В зависимости от цели работы образцы берут из верхнего минерального горизонта, пахотного горизонта (в случае агроценоза) или по горизонтам почвенного профиля. Для отбора образцов необходимо иметь стерильные пакеты из водопрочной бумаги или полиэтиленовые пакеты, этикетки, почвенный нож, лопату, спирт, вату, спички, полевой дневник и карандаш. Для взятия образца делается прикопка лопатой и затем почвенным ножом, протертым ваткой, смоченной спиртом, и обожженным в пламени, отбирается навеска, представляющая исследуемый горизонт почвы или подстилки. В полевых условиях возможна и более грубая стерилизация ножа — путем предварительного многократного втыкания его в исследуемый горизонт. При анализе грибов по генетическим горизонтам образцы берут начиная с нижнего к верхнему горизонту почвенного профиля, что уменьшает возможность внесения микроорганизмов в нижние слои из верхних. Каждую пробу (10—100 г в зависимости от целей работы) помещают в стерильный пакет, этикетируют и транспортируют в лабораторию в термостатируемом ящике. Образцы анализируют в день отбора. Если образцы анализируются по истечении нескольких суток (до 3—4), то их в этот период хранят в холодильнике при 5 °C. Долго хранить при положительной температуре влажные образцы нельзя, так как в них меняются биомасса и соотношение различных групп
микромицетов. Такие образцы хранят в замороженном состоянии в холодильнике. Аналогичным образом хранят образцы растительных остатков и надземных частей растений.
Если нет возможности произвести анализы свежих образцов и хранить в холодильнике, их необходимо быстро высушить до воздушно-сухого состояния при температуре не выше 30 °C, не подвергая воздействию солнечных лучей.
Для проведения анализа корней и ризосферной почвы растения отбирают вместе с монолитом окружающей их почвы. Для сравнения берут образцы контрольной почвы (из междурядий или с аналогичного по почвенным свойствам участка без растительности). Подготовку почвы и анализы необходимо проводить в день отбора проб. Удаляют большие комки, оставляя только ризосферную почву — слой почвы вокруг корней до 3 — 5 мм.
18.2. ОПРЕДЕЛЕНИЕ ЧИСЛЕННОСТИ, ВИДОВОГО СОСТАВА
И СТРУКТУРЫ БИОТЫ МИКРОМИЦЕТОВ В ПОЧВАХ
И ДРУГИХ ПРИРОДНЫХ ОБЪЕКТАХ МЕТОДАМИ ПОСЕВА НА ПЛОТНЫЕ ПИТАТЕЛЬНЫЕ СРЕДЫ
Точность анализа в значительной степени зависит от использованных методических приемов. Для определения численности, видового состава и структуры комплексов микроскопических грибов производят посев из разведений почвенной суспензии.
Предпосевная обработка образцов. Учет мицелиальных микроорганизмов на питательных средах имеет более условный характер по сравнению с учетом бактерий и дрожжевых грибов, так как здесь более неопределенными являются величина и характер пропагулы, давшей колонию. Это могут быть различные генеративные образования, фрагменты гиф, разнообразные мицелиальные структуры. Предварительная обработка дает возможность разбить гифы на куски, размер которых хотя и продолжает различаться в несколько раз, но уже не в десятки и сотни, как это было до диспергирования почвы.
При подготовке почвы для посева разведений необходимо разрушить почвенные агрегаты, десорбировать споры и мицелий с поверхности почвенных частиц, дезагрегировать скопления спор и разбить гифы на достаточно однородные по размерам жизнеспособные фрагменты. Эти цели достигаются механическими и химическими воздействиями. При учете грибов целесообразно использовать пропеллерную мешалку, растирание почвенного образца, увлажненного до пастообразного состояния, или интенсивное встряхивание почвенной суспензии. При неэнергичном перемешивании суспензии в колбах на качалке почвенные агрегаты совершают плавное поступательно-вращательное движение и не подвергаются существенному диспергированию. Грибы больше, чем бактерии и актиномицеты, страдают от жестких способов обработки почв. Так, при учете почвенных грибов ультразвуковая обработка оказывает явно отрицательное действие. Известно, что чем крупнее клетка, тем больше она повреждается ультразвуком. Крупные клетки грибов повреждаются ультразвуком сильнее, чем мелкие клетки бактерий. При использовании ультразвуковой обработки, применении алкилсульфата и щелочи определяемое посевом количество грибов часто не увеличивается, а иногда даже уменьшается.
Эффективность предпосевных обработок зависит от свойств почвы и наиболее действенны они при работе с сухими образцами в сравнении с влажными или замороженными.
Проведение обработки. Почвенную суспензию (1 — 10 г почвы на 100 мл стерильной водопроводной воды) обрабатывают путем 15 — 20-минутного встряхивания в колбах на качалке (180 об/мин) или на пропеллерной мешалке-миксере (микроразмельчитель тканей РТ-2) с предварительным растиранием почвы, увлажненной до пастообразного состояния пестиком с резиновым наконечником в фарфоровой ступке. Время обработки на микроизмельчителе тканей — 3 — 5 мин при 5000 об/мин. Затем готовят нужное для посева разведение. Стержень и пропеллер мешалки тщательно очищают от почвенных частиц механически. Далее стержень протирают спиртом, дают ему испариться и производят обработку новой пробы почвы. Стаканчики, в которых проводится диспергирование почвы, стерилизуют в автоклаве или спиртом.
Поверхностно-активные вещества (алкилсульфат — 0,01 — 0,05%, пирофосфат натрия — 0,1 %, твины — 0,0001 %) в указанных концентрациях добавляют в водные суспензии при предпосевной обработке почв с водопрочными агрегатами, образцов выветриваемых горных пород и минералов, десорбции мицелия и спор с поверхности растений.
18.2.1. Выбор сред и условий культивирования для выделения различных групп грибов
Грибы разнообразны по систематическому положению, пищевым потребностям и ростовым характеристикам, поэтому для их выделения из природных объектов не может быть одной универсальной среды. Сапротрофные микроми-цеты по способности усваивать те или иные субстраты объединяют в различные пищевые группы: сахаролитические грибы; грибы, разлагающие целлюлозу, лигнин, кератин, хитин, пектин, и т.д. Не отрицая наличия существенных физиологических различий среди сапротрофных микромицетов, необходимо подчеркнуть, что деление их на специфические трофические группы в некоторой степени условно. Так, в отношении «сахарных» грибов нужно заметить, что вообще нет микроорганизмов, которые не имели бы гидролаз. Поэтому наряду с использованием сахаров и других простых органических соединений «сахарные» грибы могут потреблять и полимеры — крахмал, ксилан, пектин.
Для выделения из почв и других местообитаний определенных эколого-трофических и систематических групп грибов важнейшим этапом является выбор соответствующей питательной среды и условий культивирования посевов. Использование различных методических приемов и питательных сред позволяет выделить большее или меньшее число видов микроскопических грибов. Наиболее часто из почвы на различные питательные среды выделяются грибы (царство Eumycota) отделов Ascomycota, Zygomycota, формальной систематической группы несовершенных грибов Deuteromycota, представляющих чаще всего анаморфы аскомипетов. Значительно реже и, как правило, только при использовании специальных сред и методов приманок изолируются представители отделов Chytridiomycota, Basidiomycota и грибоподобные протоктисты царства Chromista отдела Oomycota.
Подавление роста бактерий при выделении грибов из почвы и других природных источников достигают установлением кислой реакции питательной среды (pH 4,5 — 5,5). Для роста многих грибов оптимален pH около 5,5. Среды подкисляют различными
кислотами. В среднем на 1 л агаризованной среды добавляют 15 —20 мл 0,1 н. раствора серной или соляной кислоты. Молочную кислоту обычно применяют в концентрированном виде в количестве 4 мл на 1 л среды, лимонную — в виде 50%-го раствора в количестве 10 мл на 1 л среды. Подкисление агаровых сред до pH около 4,0 не оказывает отрицательного влияния на их способность к переходу в твердое состояние при остывании. Концентрированные кислоты стерилизовать не надо, 0,1 и. растворы серной и соляной кислот следует предварительно простерилизовать. Наиболее часто при проведении посевов из почвы используют молочную или лимонную кислоту.
При выделении грибов в качестве ингибиторов бактерий широко применяются различные антибиотики — стрептомицин сульфат (40 — 50 мг/л); тетрациклин, окситетрациклин, хлортетрациклин (20 — 50 мг/л); пенициллин, неомицин, полимиксин, бацитрацин (50 мг/л среды), хлорамфеникол (100 мг/л) и красители — роданистый натрий (0,25 — 0,4 моль/л), бенгальский розовый (70 мг/л). Добавление кислот и других ингибиторов бактерий производится асептически в стерильные, охлажденные до 45 °C питательные агаровые среды перед их разливом в чашки Петри.
Подавляющее большинство грибов — мезофилы, оптимальная температура для их выделения 22 — 26 °C. Для изоляции группировки грибов, способных к росту при пониженных температурах, инкубацию посевов проводят при 4— 8-°С, для термофильных— 42 — 55 °C, чаще всего 45 °C, и термотолерантных грибов — 35 — 42 °C. Чашки Петри с посевом помещают в эксикаторы или пластиковые пакеты для предотвращения высыхания среды.
Многие микроорганимы, в том числе грибы, меняют pH среды в зоне роста на оптимальный для своего развития. Поэтому для выделения алкалотолерант-ных и ацидофильных грибов в среды вводят соединения, обеспечивающие буферность среды. Используют двузамешенный фосфат натрия и однозамещенный фосфат калия, которые в различных пропорциях позволяют создать и поддерживать pH среды в широком диапазоне (от кислой до щелочной области). При изоляции алкалотолерантных грибов в среды рекомендуется добавлять соли Na2CO3, NaHCO3, К2СО3, Na3PO4 в концентрациях от 0,5 до 2 %, которые поддерживают pH в интервале 8,5 — 11.
Инкубация посевов при дневном свете резко снижает спорообразование у некоторых видов грибов (Chaetomium и Ophistoma), а в большинстве случаев свет не вреден или оказывает благоприятное воздействие (за исключение прямого солнечного света). Обычный дневной свет и близкий к ультрафиолетовому (в черной области — 310 — 360 нм) усиливают образование плодовых тел у аскомицетов, пикнид, целомицетов и конидий у гифомицетов (особенно тем-ноокрашенных).
. Для выделения быстрорастущих и эффективно усваивающих легкодоступные углеводы грибов используют сусло-агар 3 — 4° по Баллингу (как правило, сусло разбавляют водой в соотношении 1:3), картофельно-глюкозный агар и синтетические среды с простыми углеводами — среды Чапека, Чапека с дрожжевым экстрактом, Мартина, Ролэна, пептоно-глюкозный агар Ваксмана, почвенный агар с добавкой углеводов.
Для обнаружения медленнорастущих и слабоконкурентных видов в среды рекомендуется добавлять вещества, ограничивающие рост быстрорастущих форм микромицетов, такие, как бенгальский розовый (30 мг/л), пропионат натрия (0,1 %), дихлоран (2,6-дихлор-4-нитроанилин) для подавления мукоровых, фундазол (беномил) — пенициллов, и ряд других веществ — циклоспорин, детергенты.
Олиготрофные грибы и те, которые не выдерживают конкуренции за моносахара при высоких их концентрациях в среде и способны к росту при их следовом количестве, выделяют на «голодные» среды — водный агар, агар с почвенной вытяжкой, агар с разведенным в 8—10 раз суслом. На этих средах большинство микромицетов развивается медленно, образуют мелкие (2 — 3 мм в диаметре) колонии, при оптимальном разведении удаленные друг от друга, что удобно для выделения грибов в чистые культуры.
Для приготовления почвенного экстракта 1 кг почвы автоклавируется в 1 л воды, полученная суспензия фильтруется и доводится до 1 л. Если фильтрат мутный, добавляют немного CaSO4H фильтруют снова. Для приготовления среды в почвенный экстракт добавляют агар (1,5 %), а также используют варианты почвенного агара с добавкой других питательных элементов, КН2РО4 (0,02 %) или глюкозы (0,1 %).
Для выделения грибов, разлагающих целлюлозу, гемицеллюлозу, пектин, хитин и другие соединения, их вводят в состав питательных сред в качестве источника углерода, минеральная основа которых аналогична среде Гетчинсона (возможна замена NaNO3 на NH4NO3). Если микромицет продуцирует фермент, расщепляющий данный субстрат, то проявление этой ферментативной активности можно обнаружить в виде зоны просветления в среде. В случае синтеза ферментов, связанных с клеткой, необходимо удалить колонию с пластинки агара, чтобы обнаружить пятно на месте гидролиза субстрата. Нерастворимые субстраты (хитин, целлюлоза и др.) вносят в среду в виде тонко размолотого порошка. После инкубации посева вокруг колоний грибов, продуцирующих фермент, появляются зоны просветления. Для выявления зон просветления растворимых субстратов их осаждают или окрашивают. Необходимо учитывать, что краситель часто убивает микроорганизмы, поэтому, если требуется выделить культуру, предварительно делают реплику колоний. Существует альтернативный способ, заключающийся в присоединении красителя к субстрату. Для этого обычно используют ремазол бриллиантовый синий или азур. Эти приемы широко используют при первичном скрининге продуцентов различных ферментов (табл. 18.1).
Целлюлозоразрушающие грибы изолируют на питательные среды Гетчинсона, Частухина, которые содержат в качестве источника углерода целлюлозу или карбоксиметилцеллюлозу (в концентрациях 0,8 —1,0 %). Стерильную фильтровальную бумагу раскладывают на поверхность питательной среды или вносят в нее в измельченном виде (растертой в ступке или шаровой мельнице в порошок). Фильтровальную бумагу следует предварительно прокипятить в дистиллированной воде и проверить отсутствие в ней примеси крахмала по отрицательной йодной реакции. Для приготовления среды необходимо брать очищенный агар. К минеральной основе среды можно также добавлять микрокристаллическую целлюлозу или аморфную целлюлозу (обработанную предварительно фосфорной кислотой и промытую дистиллированной водой). Проводят посев разведений или почвенных комочков (по 50 комочков на чашку). Эти среды часто используют для выделения сумчатых грибов (рода Chaetomium и др.).
Для выделения грибов, разлагающих лигнин, в среды вносят в 1 %-й концентрации препарат лигнина или субстраты, содержащие его в большом количестве. Разрушающие лигнин микромицеты можно выделить, используя силика-
Среды и реагенты для выявления внеклеточных и связанных с клетками ферментов микроорганизмов
Фермент Субстрат Реагент Критерий для отбора штаммов
Амилаза Крахмал/гликоген Раствор йода Прозрачные (а-амилаза) или красные (р-амилаза) зоны на окрашенном фоне
а-Амилаза Крахмальный азур — Бледно-синий ореол
Целлюлоза Целлюлозный азур Фильтровальная бумага Аморфная или микро-кристалическая целлюлоза Бледно-синий ореол Разложение фильтровальной бумаги Зоны просветления
Хитиназа Хитин — Прозрачные зоны в нерастворимом хитине
Пектатлиаза Яблочный пектин или полигалактуроновая кислота Гексадецилме-тиламмоний бромид Прозрачные зоны в осажденном субстрате
Ксиланаза Ксилан Этанол Прозрачная зона в осажденном субстрате через 16ч
Пуллуланаза Пуллулан Этанол или ацетон Прозрачная зона в субстрате через 16 ч
Протеазы Снятое молоко Желатин Измельченная кожа с азуром Насыщенный раствор сульфата аммония Зоны просветления Зоны просветления в осажденном субстрате Бледно-синие зоны гидролиза
Липаза, эстераза Твин-60, 80 Оливковое масло Триолеин Твин вводят в подходящую среду; опалесцирующий ореол Зоны просветления
гельные пластинки, пропитанные минеральными солями, на поверхность которых нанесен порошкообразный лигнин в количестве 0,25 г на чашку. Чашки инокулируют 1 мл почвенной суспензии и инкубируют при 28 °C во влажной камере. Колонии микромицетов появляются на поверхности лигнина через 3 — 4 нед. В качестве единственного источника энергии в среды добавляют также различные производные лигнина (ванилин, сирингальдегид, л-гидроксибенз-альдегид и др.). Эти вещества в концентрации не выше 0,01 % вводят в минеральную основу среды Чапека 10-кратноразбавленную. Повышать концентрацию производных лигнина не рекомендуется, поскольку они, как и все фенолы, токсичны для микроорганизмов.
Для выделения гумусоразрушающих грибов используют гумат натрия (в концентрации до 0,01 %) в качестве источника углерода в минеральной основе
среды Гетчинсона с NH4NO3; применяют также силикагелевые пластинки, пропитанные минеральным раствором Виноградского (2 — 3 мл на чашку) и гуматом натрия (8 — 10 мг на чашку' в 0,02 н. растворе гидроокиси натрия).
При учете и выделении дрожжевых грибов методом посева используется разведение 1:5 — 1:50 для минеральных горизонтов и 1:100—1:1000 для подстилки, поверхности растений. Наиболее широко применяется сусло-агар (6 — 7° по Баллингу с молочной кислотой — 4 мл/л), глюкозопептонный агар, глюкозоминеральная среда. Для ингибирования роста бактерий применяют также антибиотики — стрептомицин сульфат (20 мг/л) с левомицитином (20 мг/л) и хлортетрациклином солянокислым (100 мг/л). Развитие мицелиальных грибов подавляют циклогексимидом (50 — 500 мг/л) и пропионатом натрия (0,15 — 0,25 %). Эти соединения подавляют рост и отдельных групп дрожжей (циклогексимид уже в концентрации 50 мг/л — представителей рода Cryptococcus, пропионат натрия — рода Rhodotoruld). Поэтому для этих целей используются и другие ингибиторы: дифенил (0,005 — 0,01 %), красители бенгальский розовый (0,003 %), кристаллический фиолетовый (0,001 %), бромкрезол пурпурный (0,002 %). Олигонитрофильные дрожжи, относящиеся главным образом к липомицетам, учитывают на среде Эшби и других безазотистых средах. Для липомицетов рекомендована также среда Юргенсена и Даниелсона. Посевы разведений инкубируют при 18 —20 °C, или при 0°С для психрофилов и 5 — 7 °C для мезофилов, для выделения патогенных видов при 37 °C. Учет и выделение проводят на 3 — 4-е сут инкубирования при 37 °C, на 7 — 10-е сут — при комнатной температуре, 14-е сут — при 5 —7 °C и после 20 сут — при 0°С. Рекомендуется применять низкотемпературное инкубирование, которое снимает необходимость добавки ингибиторов роста микроскопических мицелиальных грибов при температурах выше 24 — 26 °C. При низкой плотности популяций липомицетов используют посев на среду Эшби со стептомицином рассыпкой почвенного мелкозема и метод почвенных комочков, приготовленных из густой водно-почвенной пасты. Из 100 мг почвы готовят около 100 комочков, раскладывают по 50 на чашку и инкубируют 3 — 4 нед при 18 — 22 °C. Край слизистых обрастаний просматривают под микроскопом и выделяют из него культуры липомицетов.
Для выделения фитопатогенных грибов из почвы используют среды с анти-грибными антибиотиками (табл. 18.2). Наиболее часто применяют полиеновые антибиотики (эндомицин, нистатин, пимарицин) и циклогексимид, обладающие широким фунгицидным действием, но вместе с тем не подавляющие рост и развитие отдельных видов фитопатогенных микромицетов. Широко используются для этих целей и такие химические соединения, как красители (бенгальский розовый, кристалл- и генцианвиолет, малахитовый зеленый), спирты, хлораты, фенолы, пентахлорбензол и его производные — коммерческие фунгициды (каптан, пентахлорнитробензол, беномил, ботран и др.). При изоляции фитопатогенных грибов родов Bipolaris, Curvularia, Drechslera, Exseophilum для подавления быстрорастущих сапротрофных грибов можно использовать дезоксихолат натрия (1 % от состава среды), желчные кислоты (1 — 5 %), сорбит (1 %). Для выделения фитопатогенных грибов из почв и растений предложено множество сред. Широко используют различные растительные среды: картофельный, картофельно-морковный, мучной (овсяный, кукурузный и др.) агар, среды на основе овощного сока V-8).
Химические агенты для создания селективных сред
Грибы Антибиотики Фунгициды Другие соединения
Pytium, Phytophthora Пимарицин, 20— 100 ppm; эндомицин, 5 ppm Беномил, 5—20 ppm; PCNB, 25-100 ppm Галовая кислота, 425 ppm
Aspergillus — Ботран, 10 ppm, для A. flavus Танин 20%-й для A. niger
Fusarium Стрептомицин, 300 ppm PCNB, 100—750 ppm, в 0,5 %-м пептонном агаре К- мета-бисулъфмт, 300 ppm
Verticillium dahliae Стрептомицин, 200 ppm, и др. PCNB, 75 ppm, вкар-тофельно-декстрозном агаре Полигалактуроновая кислота (0,2%-я) в почвенном агаре или этанол 0,5 %-й в водном агаре
Basidiomyce-tes — Беномил, 1 ppm о-Фенилфенол, 6 ppm
Дерматофиты Циклогексимид, 500 ppm; хлорамфеникол, 30—100 ppm — Волосы или перья
В качестве примера можно привести среду для выделения и учета в почве Fusarium oxysporum — возбудителя фузариозного увядания и корневых гнилей растений, в состав которой входят (г/л): NaNO3 — 2,0; К2НРО4 — 1,0; КС1 — 0,5; MgSO4 — 0,5; FeSO4 — 0,01; галактоза — 20,0; агар — 20,0 и в качестве селектирующих агентов — медицинская желчь — 15 мл/л, пентахлорбензол — 1 г/л, стрептомицин сульфат — 0,3 г/л. Производят посев 1 мл почвенной суспензии из разведения 10 3 глубинным способом. Засеянные чашки инкубируют 7 — 10 сут на свету при 26 — 28 °C. По истечении этого времени на среде образуются характерные колонии Fusarium oxysporum с пленчато-пушистым мицелием телесно-розового цвета, хорошо дифференцирующиеся от других видов грибов.
Для изоляции из почвы и зараженных нематод энтомопатогенного гриба Paecylomyces lilacinus применяют среду следующего состава: картофельно-дек-строзный агар (фирма «Дифко») — 39 г; NaCl — 10 г; фунгициды пентахлорбензол и беномил — по 50 мг; вода деионизированная — до 1 л. Среду автоклавируют 15 мин при 121 °C; охлаждают до 45 — 50 °C; добавляют 100 мг сульфата стрептомицина; 50 мг хлортетрациклина монохлоргидрата; 1 мл тергитола и разливают в чашки Петри.
Для выделения хищных микромицетов из почвы предложен водньш агар (4 — 5 %) без добавки питательных веществ. Добавка нематод или их личинок повышает эффективность обнаружения нематофаговых грибов. Обнаруженные грибы пересевают на арбузно-пептонный агар. Одновременно таким способом можно выявить в почве микопаразитические виды зигомицетов. Амебофаговые грибы выделяют, используя газон Azotobacter chroococcum на чашке Петри, который инокулируют исследуемой почвой. На нем развиваются амебы, инфи
цированные или служащие приманкой для амебофаговых грибов. Энтомопато-генные грибы обычно выделяют на сусло-агар, картофельно-декстрозный агар и среду Сабуро. В случае аскомицетов рода Cordyceps образцы насекомых помещают в чашку Петри и отстреленные споры переносят на агаровые среды. Другие грибы довольно успешно выделяют из тканей инфицированных беспозвоночных.
Копрофильные грибы часто выделяют, инкубируя образцы субстратов на влажной фильтровальной бумаге в чашке Петри и периодически просматривая под стереоскопическим микроскопом. С помощью препаровальной иглы или оттянутой стеклянной пипетки спорулирующие дейтеромицеты, аскомицеты и зигомицеты переносят на питательные среды. Споры Pilobolus и отстреливающих аскомицетов часто можно обнаружить на крышке чашки Петри и пересеять на питательные среды иглой.
Для выделения термофильных грибов в качестве стандартной среды предложен YpSS-arap; применяют также сусло-агар, овсяный агар, среду Сабуро (которая широко используется для выявлении оппортунистических и патогенных микромицетов) и ряд других сред с повышенной (1,5 — 3 раза) концентрацией антибактериальных антибиотиков (стрептомицина) (50 — 100 мг/л), аурео-мицина (до .800 мг/л).
Для изоляции грибов, способных к росту в анаэробных условиях, — фузариев, мукоро-вых, триходерм, используют модификации методики Хангейта. Посев проводят на глюкозоминеральную агаризованную (3%-й агар) среду следующего состава (г/л): КН2РО4 — 1,0; КС1 — 0,5; MgSO4 — 0,5; FeSO4 — 0,01; (NH4)2SO4 — 2,0; пептон — 5,0; глюкоза — 5,0; дрожжевой экстракт — 0,5; 1 мл/л раствора микроэлементов (мг/л): ЭДТА — 500; FeCl2- 7Н2О - 200; ZnCl2- 7Н2О - 10; MgCl2- 4Н2О - 3; Н3ВО4 - 30; СоС12 - 6Н2О - 2; CuCI2- 2Н2О — 1; Na2MoO4 — 3; NiCl2- 6Н2О — 2 и 1 мл/л раствора витаминов (мг/л): 4-аминобензойной кислоты — 25; Д-биотина — 100; никотиновой кислоты — 25; пантотената кальция — 25; пиридоксина гидрохлорида — 25; фолиевой кислоты — 10; рибофлавина — 25; В12 — 0,5; липоевой кислоты — 25. Применяются также ряд других сред (Чапека, сусло-агар).
Модификации для выделения грибов', посев на агаризованную среду, разлитую тонким слоем в небольшие матрасы (объемом 100 мл); внесение большого количества антибактериальных антибиотиков, использование низких разведений (1:1—1:10) или прямого посева мелкозема. Раствор минеральных компонентов кипятят для удаления растворенного кислорода, потом остужают в кристаллизаторе с холодной водой. При этом через раствор продувают молекулярный азот, пропущенный предварительно через колонку с нагретой медью для удаления из него следов кислорода. После охлаждения в раствор добавляют органические компоненты, витамины и микроэлементы. Среду анаэробно (в токе N2) разливают в стеклянные матрасы, в которых находились навески агара. В расплавленную, стерильную, негорячую среду добавляют стрептомицин и хлорамфеникол в концентрации 200 мг/л среды. Посев мелкозема или почвенного разведения инкубируют в матрасах в атмосфере азота или аргона в течение 5 — 12 сут при 24—26 °C.
При выявлении группы почвенных грибоподобных протоктистов царства Chromista класса Oomycetes семейства Pythiaceae порядка Peronosporales (среди них много фитопатогенных видов) применяют пептоно-декстрозную среду Мартина. Селективность среды для этих грибов создается введением антибиотика (10 мкг/мл) пимарицина или эндомицина. Для выделения питиевых грибов из почв и растительных субстратов используют картофельно-декстрозный
агар с добавлением 300 мкг/мл натриевой соли ампициллина и 17 мкг/мл фунгицида этаконазола с широким спектром действия.
Для выделения грибов класса Chytridiomycetes в среды дополнительно вводятся микроэлементы и витамины.
Серьезные методические трудности сохраняются при попытках выделить из почв, подстилок и разрушающейся древесины грибы, которые играют важную роль в разложении органических веществ и формируют эктомикоризу у древесных растений. Белую гниль (грибами используются лигнин и целлюлоза) и бурую гниль (используется в основном целлюлоза) вызывают базидиомицеты. Аскомицеты и дейтеромицеты, разрушающие преимущественно целлюлозу, вызывают мягкую гниль и сине-зелено-черные окрашивания древесины. Помимо непосредственного выделения из плодовых тел эти грибы могут быть изолированы непосредственно из древесины на сусло-агаре с беномилом (1 ppm), и споры аскомицетов и гифомицетов с помощью оттянутой стеклянной пипетки перенесены на агаровые среды.
Для изоляции эктомикоризных грибов необходимо существенное усложнение сред, обусловленное их потребностями в специфических веществах. Кроме того, из-за низкой скорости роста на традиционно используемых средах они не могут успешно конкурировать с более быстро растущими видами сапро-трофных грибов. Показано, что беномил (в концентрации 5 ppm) может быть использован для ограничения этих грибов при выделении базидиомицетов с эктомикоризных корней.
Среды, предназначенные для грибов, образующих эктомикоризу, обычно содержат какую-либо растительную вытяжку, чаше всего солод, в качестве источника углерода служат глюкоза, крахмал, пектин, источниками азота — соли аммония, гидролизат казеина, различные аминокислоты. В их состав входят также неорганические соли и во многих случаях — микроэлементы и витамины. Одними из наиболее обычных сред для выделения эктомикоризных грибов из корней является среда Хагема и Мелина—Норканса (D. Parkinson, 1982). Микоризные грибы довольно чувствительны к реакции питательной среды. Большинство из них активно образует воздушный мицелий при pH 4,2 —5,0, хотя многие способны развиваться в широком интервале pH (от 2,8 до 7,1). Температурный интервал, в котором грибы сохраняют способность к росту, также достаточно широк. Минимальная допустимая температура около 5 °C, максимальная — около 30 °C, оптимальная в большинстве случаев составляет 18 —22 °C.
Для выделения эктомикоризных грибов корни не должны быть подсушены и их температура не должна повышаться при доставке в лабораторию. Корни промывают водопроводной водой для удаления почвы, разрезают на куски 2 — 10 мм и помещают в банку с раствором детергента и встряхивают, промывают водой и обрабатывают дезинфицирующим раствором (NaOCl, 1%-м раствором в течение 10 мин, 30 %-й перекисью водорода (Н2О2) — 5 — 20 с или 0,01 %-м раствором сулемы (HgCl2) — 2 — 3 мин). Затем корни промывают дистиллированной водой и переносят на агаризованные среды.
Для грибов, образующих эндомикоризу (везикулярно-арбускулярную микоризу), к настоящему моменту не разработаны способы культивирования на питательных средах в чистой культуре. Существует несколько методов учета и изоляции спор эндомикоризных грибов из почвы, основанных на их крупных размерах — влажное просеивание, центрифугирование в растворах сахарозы, при-
липание-флотация, дифференциальное осаждение в растворах желатина или глицерина. Выделенные споры обрабатывают 5—20 мин 2 — 5 %-м раствором хлорамина для уничтожения сопутствующих микроорганизмов. Споры эндо-микоризных грибов способны прорастать на 0,75%-м агаре с добавлением экстракта почвы или водном агаре с дрожжевым экстрактом. Для установления принадлежности спор к эндомикоризным грибам используют тест-растения, которые выращивают в стерильном песке или в пробирках на агаризованной среде и инфицируют их отдельными спорами.
18.2.2. Характеристика комплексов микроскопических грибов
Для характеристики структуры комплекса сапротрофных микромицетов наиболее часто используют метод посева водных разведений почв на среды с простыми углеводами — среды Чапека и Мартина, сусло-агар и картофельно-декстрозный агар, почвенный агар с сахарами, подкисленные молочной кислотой (4 мл/л) или с добавлением антибиотиков для подавления роста бактерий. Применение этих сред позволяет выявлять, как правило, максимальное разнообразие сапротрофных микроскопических грибов, и одновременно они удобны для дифференциального учета колоний грибов в посевах, так как на этих средах дано описание культур в определителях. Наиболее детально на этих средах выявляется комплекс сахаролитических быстрорастущих грибов, хотя в него входят и грибы, обладающие разнообразными гидролитическими ферментами (целлюлазами, амилазами, пектиназами, липазами и др.). Для создания более оптимальных условий для выявления медленнорастущих грибов в среды вводятся соединения (бенгальский розовый, беномил), сдерживающие развитие мукоровых грибов, триходермы и других микромицетов, образующих широкорастущие, обильно спороносящие колонии. Выбор сред с другими источниками углерода (целлюлозой, крахмалом, пектином и т.д.) для выделения грибов позволяет полнее выявить микроскопические грибы, активно синтезирующие необходимые для роста на этих соединениях ферменты (т. е. группы целлюлозолитических, амилолитических, пектинолитических грибов и др.). Применение сред с гуматами, лигнином и их производными или «голодного» агара дает возможность установить группу олигокарботрофных грибов и разрушающих гумусовые вещества.
Посев на агаровые питательные среды рекомендуется проводить из разведений 1:10, 1:100; -1:1000; 1:10000 в зависимости от содержания грибов в почве. Последнее разведение используется для лесных подстилок. Для большинства почв оптимальным является разведение в интервале 1:100 — 1:1000. На поверхность твердой питательной среды наносят одну каплю (50 мкл) почвенной суспензии и с помощью стерильного шпателя распределяют ее по всей поверхности агаровой пластинки. При глубинном посеве в стерильные чашки Петри вносят по 1 мл водно-почвенной суспензии различных разведений, затем наливают охлажденную среду и, совершая осторожно круговые движения чашкой, распределяют суспензию по объему среды.
Каждый из 5—10 образцов изучаемого биогеоценоза высевают на 3 — 5 чашек Петри и инкубируют при 25 —26 °C. Чашки Петри периодически просматривают, обычно начиная с 4 —5-х сут, и отдельные колонии грибов в случае угрозы их зарастания мицелием быстрорастущих микромицетов отсевают в пробирки или чашки Петри. Оптимальным считается разведение, когда на чашках Петри выросло по 10 — 30 ко
лоний микроскопических грибов. Окончательный учет производится в зависимости от среды выделения — на сусло-агаре и синтетических средах с простыми углеводами на 6 — 12-е сут, на средах с целлюлозой, КМЦ через 2—3 нед, на голодном и почвенном агаре через 2 — 4 нед. Численность грибных зачатков в почве, выделяемых методом посева на твердую среду, рассчитывают по формуле
а = бвг/б,
где а — число грибных зачатков на 1 г почвы; б — среднее количество колоний на чашках; в — разведение, из которого сделан посев; г — количество капель в I мл суспензии; д — масса почвы, взятой для анализа. Полученные данным методом значения численности грибных зачатков (пропагул) позволяют составить представление о степени заселенности исследуемой почвы микроскопическими грибами.
Для определения состава и представленности различных видов в изучаемой почве проводят выделение культур микромицетов из одной или группы одинаковых колоний. Одновременно подсчитывают количество колоний в посеве из данной почвы, которые представлены изолированными для идентификации штаммами. Выделение почвенных грибов для видовой идентификации осуществляют на среды Чапека—Докса, сусло-агар или другие среды в соответствии с систематическим положением организма. Каждый штамм нумеруют, записывают в лабораторный журнал принадлежность его к определенному образцу и варианту опыта, что необходимо для дальнейшей обработки данных и характеристики структуры комплекса грибов.
При характеристике микромицетного населения почв часто используется понятие комплекса, которое подразумевает совокупность популяций разных видов микроскопических грибов, обитающих в почве или другом компоненте экосистемы (Т. Г.Мирчинк, 1988). При описании структуры комплекса грибов изучаются состав и представленность видов, т. е. в отличие от изучения структуры сообществ не выявляются функциональные (трофические и регуляторные) связи между видами. При этом исходят из того, что участие грибов в круговоротах веществ в природе обычно тесно взаимосвязано с другими организмами. Вместе с тем, при наличии признаков взаимодействия видов (или его возможности) вполне правомочно характеризовать биоту сапротрофных микроскопических грибов в данном местообитании как сообщества.
Оценку представленности микроскопических грибов в почвах и других компонентах наземных экосистем проводят на основе показателей обилия (доля колоний данного вида от общего числа колоний в посеве исследуемого объекта), временной частоты встречаемости (число моментов времени, когда вид обнаружен, от общего числа сроков анализа) и пространственной частоты встречаемости видов (доля индивидуальных образцов, в которых вид выявлен от общего числа проанализированных образцов). Типичными для изучаемого местообитания считают виды с временной частотой встречаемости не менее 30% (Т. Г. Мирчинк, 1988). При сравнительном анализе комплексов грибов в различных компонентах биоценозов необходимо использовать данные, полученные при сходных методических подходах, в частности при посевах на одни и те же среды — с легкодоступными углеводами (среды Чапека, Мартина, Ролэна, сусло-агар), с целлюлозой (среда Гетчинсона и «голодный агар») и равном числе образцов из каждого местообитания.
Для характеристики и сравнения комплексов микроскопических грибов в разных местообитаниях применяют экологические индексы — индексы разно-
эбразия и выравненное™ Шеннона, доминирования Симпсона, коэффициент сходства Серенсена; рассчитывают соответствие различным моделям рантового распределения относительного обилия видов, среднюю радиальную скорость роста типичных видов и некоторые другие показатели (Т. Г. Мирчинк, 1988; Д.Г.Звягинцев, 1991; Э.Мэгарран, 1992; А.В.Кураков, 2001).
18.3. МЕТОДЫ ПРЕДПОСЕВНОЙ ОБРАБОТКИ ОБРАЗЦОВ
ДЛЯ БОЛЕЕ ПОЛНОГО ВЫЯВЛЕНИЯ РАЗНООБРАЗИЯ МИКРОСКОПИЧЕСКИХ ГРИБОВ
Существуют специальные приемы предпосевной обработки и предынкуба-пии почв и водно-почвенных суспензий, позволяющие более детально охарактеризовать видовой состав и выделить отдельные группы грибов в почвах.
Методы прямого посева мелкозема на питательные среды. Для более полного представления о микобиоте почвы рекомендуется производить посев почвенных частиц, рассыпая их по поверхности агара, что дает возможность судить о количестве микроколоний и качественном составе грибов. Предпосевная обработка почв в этом случае сведена к минимуму.
Для выделения грибов методом прямого посева из средней пробы образца стерильным ланцетом берутся небольшие навески почвы (от 20 до 200 мг в зависимости от плотности грибных популяций в почве) и равномерно наносятся на поверхность питательных сред.
Существует прием, когда стерильным пинцетом переносят точно взвешенные навески мелкозема почвы (5 — 50 мг) в стеклянную трубку или маленькую воронку, затянутые снизу сеткой из мельничного газа с диаметром пор 0,25 мм. Навеску просеивают через сетку, равномерно распределяя почвенные частицы по поверхности агаризованной питательной среды. Из каждого отдельно взятого образца делают высев на 3 — 5 чашек Петри. При посеве мелкозема на богатые среды — сусло-агар, среды Чапека и Мартина — навеска, как правило, не превышает 5 — 20 мг, при посеве на бедные среды — водный и почвенный агар — берут 50 мг почвы. Анализ состава грибов и выделение их в чистые культуры можно провести на 5 — 10-е сут на богатых средах и через 2—3 нед на бедных. Для этого необходимо установить оптимальную для исследуемой почвы массу образца, чтобы на чашке Петри вырастало не более 15 — 25 колоний.
Мелкозем можно также внести на дно стерильной чашки Петри, смешать с каплей стерильной воды, добавить охлажденную питательную среду и затем легкими круговыми движениями чашки частицы почвы равномерно распределить по всей ее площади. В сравнении с методом разведений мы имеем дело с практически ненарушенными частицами почвы и получаем дополнительные возможности для выявления видового разнообразия грибов.
Известен ряд близких к описанному методу способов посева почвенного мелкозема на питательные среды для выделения микромицетов: метод прямых отпечатков почвенной пробы и электростатический метод, — суть которых сводится к различным приемам нанесения почвенных частиц на поверхность питательных сред.
Сукцессионный и обогатительный подходы. Сукцессионный подход заключается в инкубировании почвы в течение 1—4 нед и более при различных
температуре, влажности, с обогащением почвы и без него различными соединениями (глюкозой, целлюлозой и др.) и периодическим проведением посевов. Инкубация образцов после их увлажнения с добавкой или без добавки субстрата ведет к развитию и смене разных групп грибов (сукцессии) в ходе трансформации субстратов в почве, что позволяет более полно описать качественный состав грибов в изучаемой почве.
Принцип накопительного подхода заключается в обеспечении благоприятных условий для роста грибов с определенными свойствами, что приводит к увеличению их численности в почве. Следует, однако, учитывать, что при секреции внеклеточных ферментов в процессе разложения субстрата, применяемого в качестве селектирующего фактора для конкретной группы грибов, продукты их ферментативного гидролиза могут быть использованы и другими микроорганизмами. Поэтому важно выявить ранние стадии повышения плотности популяций грибов, интересующих исследователя. Так, при внесении фильтровальной бумаги и инкубации увлажненной почвы при температурах более 45 °C в период 7 — 14 сут можно добиться преимущественного развития целлюлозолитических и термофильных микромицетов.
Индукция прорастания пропагул. Для повышения разнообразия выделяемых видов целлюлозолитических микромицетов в качестве индуктора можно использовать добавку целлобиозы (0,3 — 30 мМ), D-сорбозы (0,05 — 2 мМ), арабинозы (0,05 — 0,5 мМ) или лактозы (0,1—2,0 мМ) в водно-почвенные суспензии за несколько часов до посева на агаровые среды с микрокристаллической или аморфной целлюлозой. Прием основывается на том, что невысокие концентрации этих соединений могут индуцировать синтез целлюлаз у микроорганизмов.
Методы водной отмывки почвенных частиц. Промывание или отмывка почвенных частиц, корней, различных растительных остатков представляют собой приемы снижения в посевах колоний обильноспороносящих видов грибов и повышение числа видов, находящихся в мицелиальном состоянии. Сущность метода составляет проведение серии промываний почвы или многократных смывов с корней стерильной водой в целях удаления максимально возможного количества спор. По завершении процедуры почвенные частицы помещают на стерильную фильтровальную бумагу для устранения избытка воды и затем их раскладывают на агаровые среды (по 2—4 частицы на чашку Петри). Оптимальное число промываний и их продолжительность для достижения эффективного удаления спор должно определяться в предварительных экспериментах.
Промывка почв может проводиться простыми приемами (на качалке с последовательной заменой воды) и с использованием специальной аппаратуры. Они основаны на инструментальном разделении почвенных частиц на фракции разных размеров с дальнейшим суспендированием их в стерильной воде. Существуют различные модификации специальных аппаратов для такого отмывания почвенных частиц. Принцип их конструкции — наличие внутри полости аппарата нескольких закрепленных сит с отверстиями разного диаметра, наверху — с более крупными, а ниже — с более мелкими. Проба почвы помещается на поверхность верхнего сита, а затем через аппарат пропускают порции воды. Образец почвы (20 г) помещают в аппарат на верхнее сито (размер отверстий 1,5 мм). Плотно закрывают прибор пробкой. Через отверстие в проб
ке заливают аппарат наполовину водой. Скорость потока воды составляет 1,5 л/мин. Промывную воду собирают в ведро. Первоначально можно использовать 4 л водопроводной воды, а затем 2 л стерильной воды. После отмывания аппарат открывают и почву, которая аккумулировалась на нижнем сите (с порами 0,5 мм), переносят в стерильные чашки Петри с небольшим количеством воды. Органические частицы, которые всплывают на поверхность, собирают первыми при помощи тонкого пинцета, помещают на стерильную фильтровальную бумагу, а затем переносят по 4 частицы на чашку на кукурузный агар или почвенный агар с добавлением антибиотиков. В пробу сливают супернатант и минеральные частицы обрабатывают таким же образом. Результаты могут быть подсчитаны для каждого вида гриба как процент от общего числа частиц или общего числа изолятов. Первый метод дает сравнительную оценку грибной плотности для отдельных почв — «коэффициент колонизации». Сравнение результатов для различных почвенных образцов может проводиться только для почв, сходных по структуре. Эффективность водных промывок связана с физическими свойствами почв (легче промыть песчаную почву, чем тяжелосуглинистую).
Тепловая обработка почв. Образцы увлажненной почвы или водно-почвенной суспензии выдерживают в термостате при температуре 60 — 80 °C в течение нескольких часов или на водяной бане в течение 10 — 30 мин с последующим быстрым охлаждением в тающем льде. Эффективность обработки можно усилить, погрузив образец почвы на 6 —8 мин в 60%-й этанол, а затем приготовить суспензию и прогреть ее, как сказано выше. Можно образцы почвы прогреть паром при 60 — 65 °C в течение 30 мин и затем высевать на агаризованные питательные среды. Тепловую обработку в течение нескольких часов (до 12 ч) можно провести и уже засеянных методом разведений или (что эффективнее) мелкоземом агаризованных сред в чашках Петри. Эти приемы используют для изолирования термотолерантных грибов и индукции прорастания спор (например, аскоспор у сумчатых грибов).
Фумигация почвы. Метод рекомендуют для выделения сумчатых грибов. Для фумигации чаще всего используют формалин, метилбромид, хлороформ, пары которых легко диффундируют в исследуемые образцы и могут быстро удаляться из них проветриванием. Просеянную почву (2—4 мм) помещают в стеклянную трубку с несколькими боковыми отверстиями, вводят через них нужную дозу фумиганта и закрывают резиновыми пробками. Через несколько часов экспозиции резиновые пробки заменяют на ватные. Обработку почвы парами хлороформа можно провести в эксикаторе аналогично процедуре фумигаци-онного метода определения микробной биомассы. Фумигация почвы этими веществами в определенной дозе не убивает аскоспоры многих видов родов Thielavia, Chaetomium, Sordaria, телеоморф аспергиллов и пенициллов. Дозировка и время экспозиции зависят от типа почвы и вида гриба и подбираются экспериментально. Так, виды из песчаной почвы выделяют после ее обработки метилбромидом в течение 4 ч в дозе 0,6 мл на 1000 мг почвы.
Метод флотации. Метод применяется для учета и выделения из почвы грибов родов Bipolaris, Curvularia, Drechslera, Exserohilum и др., которые имеют крупные споры. 1 —10 г почвы суспендируют в 90 мл стерилизованной воды и добавляют 5 —10 мл стерильного вазелинового или другого минерального масла. Колбы помещают на качалку (150 — 180 об/мин) на 1 — 6 ч. После взбалты
вания суспензии дают отстояться и с помощью стерильной пипетки берут несколько капелек из поверхностной масляной пленки для учета в камере Горяева и посева на питательные среды для выделения грибов в чистые культуры. Эмульсия, собирающаяся на поверхности воды, содержит большую часть спор, имеющихся в почве.
Центрифугирование водно-почвенной суспензии. Метод центрифугирования дает возможность изолировать пиреномицеты, имеющие крупные споры (Chaetomium murorum, С. megalocarpon, Sordaria fimicola и др.), многочисленные стерильные изоляты, хламидоспоры фузариев.
Почву предварительно просеивают через сито 2 мм, затем образец массой 10 г взбалтывают энергично в 100 мл стерильной воды, отстаивают 30 с и сливают надосадочную суспензию, осадок удаляют. Процедуру повторяют 4 раза, что позволяет удалить песок и другие крупные частицы. После этого взвесь центрифугируют 5 мин при 500 об/мин. Осадок переносят в 20 мл стерильной воды, взбалтывают, отбирают 10 мл и пропускают через колонку с растворами сахарозы. Затем центрифугируют 30 мин при 350 об/мин для освобождения от минеральных частиц. Один объем суспензии разбавляют тремя объемами стерильной воды и центрифугируют 5 мин при 1000 об/мин. Полученный осадок переносят в пробирку с 3 мл стерильной воды и засевают по 0,25 мл в чашку Петри на агаризованную среду.
УФ-облучение. Метод применяют для выделения темноокрашенных грибов. Засеянные почвенной суспензией или мелкоземом чашки Петри облучают ультрафиолетовыми лучами в течение 15 — 240 с. Облучение чашек Петри в течение 15 с уменьшает количество грибов на 20 %, 240 с — на 50 %. Уменьшение количества выросших колоний происходит за счет гибели грибов с палевыми и тонкостенными спорами (виды родов Penicillium, Aspergillus, Mucof).
Предложена методика, позволяющая подавлять развитие быстро- и широ-корастущих грибов порядка Mucorales и семейства Mucedinaceae, стимулировать развитие видов родов Aspergillus, Fusarium, не изменяя состава выявляемых темноокрашенных гифомицетов. Для этого почвенную суспензию в разведении 1:10 разливают в пробирки по 5 —8 мл и облучают при постоянной интенсивности дозой у-лучей 100 крад. Разведение для посева из облученных образцов на 1 —2 порядка ниже, чем в случае необлученных проб.
18.4. ВЫДЕЛЕНИЕ ГРИБОВ, НАХОДЯЩИХСЯ В АКТИВНОМ
СОСТОЯНИИ И РАЗВИВАЮЩИХСЯ НА РАЗЛИЧНЫХ СУБСТРАТАХ В ПОЧВЕ
18.4.1. Выделение грибов, находящихся в мицелиальной форме
Методы прямого выделения гиф грибов из почвы. Небольшие образцы почв (почвенных агрегатов) насыщают стерильной водой и аккуратно разбивают тонкой струей воды. Удаляют тонкодисперсную часть взвеси суспензии и крупные минеральные частицы. Повторяют эту операцию в целях получения фракции
тяжелых почвенных частиц, которые распределяют в тонком слое стерильной зоды и просматривают в стереоскопическом микроскопе для обнаружения гиф.
Существует прием по отделению гиф от почвенных частиц путем фильтрации через фильтр с порами 50 мкм.
Отдельные гифы переносят в капле стерильной воды на кончике препаровальной иглы под контролем микроскопа в стерильные чашки Петри и затем наливают расплавленную, но охлажденную питательную среду. Можно переносить фрагменты гиф непосредственно на поверхность застывшей питательной агаровой среды и отмечать маркером на обратной стороне чашки место инокуляции. Проводят ежедневные наблюдения под микроскопом, чтобы быть уверенными в том, что рост грибов начался из посеянной гифы.
Метод почвенных отмывок. Этот метод описан в предыдущем разделе. В связи с тем, что процедура водных отмывок направлена на удаление спор с почвенных частиц, он может также использоваться для выделения грибов из мицелия.
Иммерсионные методы. Для выделения грибов, присутствующих в почве в виде растущего мицелия, в почву помещают специальные пробирки или стекла с водным агаром или другой средой (трубки Честерса, стекла-ловушки Ля-Туш, экранированные стекла Торнтона, пластинки с ячейками), которые, согласно конструктивному решению, имеют воздушный промежуток между средой и почвой. Наличие воздушного зазора дает преимущество в заселении среды активно растущим в почве гифам грибов.
Один из наиболее простых методов заключается в использовании пластиковых автоклавируемых пробирок, в стенках которых делаются отверстия (диаметр 0,25 см), пробирки обматывают пластиковой лентой, заполняются питательной средой (воздушный зазор сверху 4 см), закрывают пробкой и автоклавируют. Перед закладкой в почву большой стерильной иглой прокалывают отверстия в ленте напротив уже имеющихся в пробирках и помещают в почвенном образце или желаемой позиции почвенного профиля обычно на 5 — 7 дней. Пробирки затем осторожно достают из почвы и в лаборатории вырезается сердцевина агара из каждой пробирки, делится на небольшие кусочки, которые помещаются на выбранную среду для выделения грибов.
18.4.2. Методы приманок
Для выявления микроскопических грибов, развивающихся на различных субстратах в почве, применяют методы приманок, в качестве которых в почву помещают различные соединения, растительные или животные субстраты. Эти методы широко распространены в фитопатологии и почвенной микологии. В последние десятилетия методы приманок применяют для выделения грибов-деструкторов промышленных материалов. Они используются как в полевых, так и в лабораторных исследованиях.
Выделение целлюлозоразлагающих грибов в полевых условиях. Стерильное хлопчатобумажное полотно, фильтровальную бумагу, целлофан, прикрепленные к стеклянным пластинам (аппликационный метод), погружают в почву на необходимую глубину, предварительно сделав прикопку или разрез. После определенного периода инкубации (от нескольких дней до месяцев) пластинки осторожно извлекают из почвы, просматривают целлюлозный субстрат под
бинокулярной лупой и появившиеся грибы (мицелий, репродуктивные структуры) отсевают с помощью препаровальной иглы на агаризованные питательные среды. При закладке пластинок в достаточной повторности можно исследовать динамику заселения различными грибами целлюлозы и их состав при различных погодных условиях.
Существует способ изучения состава грибов на целлюлозе с последующей «проявкой». Стерильные куски фильтровальной, крафтовой бумаги или целлофана размером 5 х 5 см закрепляли в диапозитивных рамках для проектора и помещали вертикально в соответствующий горизонт исследуемого почвенного профиля. Повторность для каждого варианта не менее трех раз. После экспозиции, время которой определяется предварительно экспериментальным путем (например, для фильтровальной бумаги в верхнем горизонте почвы 7—10 дней), куски целлюлозы извлекают из почвы, очищают и помещают на твердую питательную среду с бактериальным ингибитором, а затем на неделю в термостат. Целесообразно использовать минеральную среду Гетчинсона без добавок органического вещества. Экспозиция на питательной среде и выдерживание в термостате как бы «проявляют» субстрат, так как стимулируют развитие спо-роношений у грибов и дают возможность их родовой идентификации. Пластинку микроскопируют целиком, оценивая заселенность отдельными грибами от общей площади пластинки и частоту их встречаемости на нескольких (5 — 10) пластинках в каждом варианте. Затем при визуальном микроскопическом контроле грибы выделяют стерильной иглой в чистую культуру для видовой идентификации. При использовании этого метода наряду с обычными целлюлозоразлагающими удается выделить значительно большее разнообразие грибов, особенно медленнорастущих темноокрашенных форм, обычно плохо выделяющихся на твердые питательные среды.
Выделение целлюлозоразрушающих грибов из почв в лабораторных условиях. Почву увлажняют до пастообразного состояния (60 % от полной влагоем-кости) в чашках Петри. С помощью слегка увлажненного шпателя ее поверхность выравнивают и помещают полоску стерильной фильтровальной бумаги, хлопчатобумажного полотна или природные материалы с высоким содержанием целлюлозы (измельченную солому злаков, древесные опилки). На поверхность почвенной пластинки можно нанести тонкий слой порошковидной микрокристаллической целлюлозы или карбоксиметилцеллюлозы. Необходимо плотно прижать наносимый субстрат к почве и следить, чтобы он был увлажнен. Почвенные пластинки помещают во влажную камеру и инкубируют в термостате при 25 — 27 °C в течение нескольких недель. Если используются пластиковые чашки Петри диаметром 3 см, то их можно поставить в стеклянные чашки Петри диаметром 10 см, в которые налита вода (1—2 мл). Появление грибного мицелия на целлюлазных материалах, как правило, происходит после недельной инкубации почвы, образование спороносных структур — на 2 —4-й нед. Выделение грибов осуществляют непосредственно с почвенной пластинки с субстратом на питательные среды Гетчинсона, сусло-агар и Чапека (кусочек мицелия или споры берут препаровальной иглой под контролем бинокулярного микроскопа).
Выделение амилолитического микробного сообщества. Метод предназначен для выяснения и оценки степени антропогенного воздействия на комплекс почвенных микроорганизмов (В. С. Гузев, 1991; Д. Г. Звягинцев, 1991). Одно
временно он может быть использован для выделения микромицетов, обладающих амилазами, и грибов, развивающихся на продуктах гидролиза крахмала в почве. Готовят почвенную пластинку в чашках Петри (3 — 4 см диаметром) и на ее поверхность наносят полоску тонкого слоя крахмала (в несколько зерен толщиной). Удобно наносить крахмал напылением или легким прижатием почвенной пластинки к нанесенному на лист бумаги порошку крахмала. Постоянную влажность почвы (60 % от полевой влагоемкости) поддерживают, помещая чашки во влажную камеру (чашки Петри диаметром 10 см). В течение следующих 2 — 3 нед инкубирования почвы при 25 °C наблюдают за развитием .микроорганизмов на поверхности крахмала и под контролем бинокулярной лупы препаровальной иглой выделяют их на питательные среды.
Выделение хитинразлагающих грибов. Для выделения грибов, разлагающих хитин, используют препараты хитина или специально готовят приманку из покровных тканей насекомых, крабов или каракатицы. Предварительно ткани декальцинируют в 10%-й НС1. Полученный хитинопротеиновый комплекс гидролизуют в течение 6 ч 10%-й NaOH при 105 °C в автоклаве. Хитозан удаляют 5%-й НС1. Полученный хитин в виде пленки толщиной 0,25 мм разрезают на квадраты 0,5 х 0,5 см, приклеивают на покровное стекло, помещают в почву и выделяют через определенные периоды инкубации грибы, развившиеся на хитине.
Выделение кератинофильных грибов и дерматофитов. Кератины — специфические серосодержащие белки, характеризующиеся высокой устойчивостью к различным биологическим и химическим воздействиям. В почву кератины могут попадать с покровных тканей животных (эпидермиса кожи, роговых образований, перьев, шерсти). Ряд грибов может разлагать кератины, причем одни виды используют кератин мертвых тканей, другие способны развиваться и на живых тканях, вызывая заболевания — дерматомикозы. Поэтому выявление качественного состава и плотности популяций этих грибов может быть важным в специфических экологических местообитаниях — почвах животноводческих угодий, на территориях переработки кератинсодержащих соединений.
Образцы почв (20 —100 г) в нескольких повторностях отбирают с исследуемого участка, помещают в чашки Петри и увлажняют стерильной водой. На поверхность почвы наносят стерилизованные волосы лошади, овец, человека (лучше детские) (в виде кусочков длиной 4 — 7 мм) или другие приманки — шерсть животных, кусочки роговых тканей и т. д. Чашки термостатируют в темноте при 20 — 25 °C в течение 1—6 нед. Контроль осуществляют микроскопически при малых увеличениях (20 х Ю). При появлении мицелия приманки извлекают из почвы и переносят на среду Сабуро, содержащую циклогексимид (0,5 г/л) и хлорамфеникол (0,05 г/л). Затем мицелий, выросший вокруг приманок, пересевают на свежие косяки со средой Сабуро. Как характеристика плотности в почвах отдельных видов кератинофильных грибов учитывается процент приманок, заросших определенными грибами.
При работе с кератинофильными грибами следует соблюдать особую осторожность, так как наряду с сапротрофными микромицетами возможно выделение патогенных дерматофитов.
Выделение фитопатогенных и микопаразитических грибов. Для выделения фитопатогенных грибов, которые определенный период своего жизненного цикла функционируют в почве как сапротрофы, в качестве приманок исполь
зуют различные органы растений. При выявлении видов рода Fusarium хорошими приманками служат стерильные кусочки клубней картофеля, для Verticillium dahliae — отрезки стеблей, черенки и листья шелковицы, ивы, томатов, для Thielaviopsis basicola — стерильные ломтики моркови, для фитофторовых грибов (Phytophthora, Pythiuni) — недозрелые яблоки и ягоды земляники, семена конопли, зрелый лимон. Для подавления роста типичных сапротрофных грибов на приманках в почву могут вносить фунгицид. Так, беномил добавляют для обнаружения грибов рода Phytophthora. Исследуемую почву (не менее 10 г) раскладывают в стерильные чашки Петри или стеклянные стаканчики, заливают стерильной водой (в ряде случаев необходимо увлажнять до 60 % от полной влагоемкости), помещают приманки и инкубируют в термостате, поддерживая установленную влажность почвы. Через 2 — 3 сут начинают просматривать приманки под бинокулярным микроскопом и появившиеся грибы отсевают на агаризованные среды. Используют, как правило, среды, в состав которых входят растительные отвары — овсяный, картофельный, кукурузный и дрожжевой экстракты и т.д. (Билай, 1982).
Так, при выделении грибов рода Verticillium свежесобранный растительный материал разрезают на фрагменты, очищают от коры, стерилизуют в 96%-м спирте и помещают в стаканчики с почвой на глубину 7 — 8 см. После 3 сут экспонирования при 24—26 °C палочки вынимают и микроскопируют, регистрируя наличие конидиального налета Verticillium dahliae на участке, расположенном на границе почвы с воздухом. По количеству отрезков с конидиаль-ным спороношением можно характеризовать относительную инфекционность образцов почвы. Однако следует учитывать, что такой тип конидиального спо-роношения свойствен не только V. dahliae, но и другим видам вертициллов, обитающим в почве. Поэтому в данном случае, как и с другими фитопатогенами, необходимо их выделение в чистую культуру на питательные среды, которые существенно различаются по составу для разных фитопатогенных видов грибов.
Микопаразитические грибы обычно легко обнаружить в природной обстановке на плодовых телах по их деформации и изменению цвета. С помощью препаровальной иглы или оттянутой стеклянной пипетки небольшое количество конидий или аскоспор выделяется в чистую культуру непосредственно на среды с антибактериальными антибиотиками. Для выделения почвообитающих грибов, обладающих микопаразитическими свойствами (Coniothyrium minitns, видов Trichoderma, Gliocladium, Talaromyces), в качестве приманки используют инкубируемые в почве склероции Sclerotinia.
18.4.3. Метод «ловчих растений» для выделения фитопатогенных грибов
Метод «ловчих растений», т. е. вегетирующих растений-хозяев, их проростков или укорененных пасынков, с успехом применяется для выявления фитофторовых грибов. Почву в чашках Петри заливают водой до полного насыщения. Здоровые сеянцы восприимчивого растения на стадии семядольных листьев, которые предварительно были пророщены в чашках Петри на увлажненной фильтровальной бумаге, раскладывают в чашки Петри с почвой. Для
стимулирования инфицирования сеянцев их перед высадкой в почву накалывают препаровальной иглой. При таком способе на растениях через 5 — 7 дней инкубирования при 20 —22 °C в зараженной почве обычно хорошо заметны первые признаки развития возбудителя.
На зародышевых корешках сеянцев обнаруживаются участки с побуревшей и размягченной тканью. При микроскопическом анализе хорошо просматриваются образующиеся на поверхности пораженных корней спорангии. Органы полового размножения (ооспоры) гриба образуются спустя 10—15 сут.
18.5. ВЫДЕЛЕНИЕ ГРИБОВ ИЗ РИЗОСФЕРЫ, РИЗОПЛАНЫ, НАДЗЕМНЫХ ЧАСТЕЙ РАСТЕНИЙ И РАСТИТЕЛЬНЫХ ОСТАТКОВ
Подготовка образцов. Выделение грибов из ризосферы и ризопланы и надземных частей растений имеет ряд специфических особенностей, что вызывает необходимость дать отдельное описание этих методов.
Подготовку почвы и корней необходимо проводить в день отбора проб. Корни растений отбирают вместе с монолитом почвы. Для сравнения берут образцы контрольной почвы (из междурядий или с аналогичного по почвенным свойствам участка без растительности). В качестве ризосферной почвы берут слой почвы вокруг корней до 3 — 5 мм.
Ризосферную почву можно удалить с корней посредством водных смывов. Навеску корней с почвой (5—10 г) помещают с помощью простерилизованного над пламенем пинцета в заранее подписанные колбы со 100 мл стерильной воды и встряхивают на качалке в течение 15 мин. Корни предварительно разрезают на фрагменты 3 — 5 см. Отмытые корни вынимают микробиологическим крючком из колбы, помещают между листами фильтровальной бумаги, подсушивают для удаления остатков влаги и взвешивают. По разности массы корней с почвой и отмытых корней определяют массу ризосферной почвы (при полевой влажности почвы на день отбора проб). Для представления данных в расчете на воздушно-сухой вес проводят определение полевой влажности почвы и затем соответствующие корректировки.
Более точные расчеты можно провести, если после посева водно-почвенную суспензию отфильтровать через предварительно взвешенный фильтр, который сушат с почвой до постоянной массы и таким образом устанавливают массу ризосферной почвы. Ризосферную почву возможно собрать с корней, не прибегая к водным смывам, с соблюдением условий стерильности, осторожно удаляя ее с поверхности корней скальпелем и кисточкой. Затем почву взвешивают, и навески (не менее 0,5 — 1 г) вносят в колбы со 100 мл стерильной воды. Несколько навесок используют для определения влажности почвы.
Десорбцию микромицетов с почвенных частиц проводят на качалке (180 об/мин) в течение 15 мин, микроразмельчителе тканей РТ-2 в течение 5 мин или ультразвуковой установке до 2 мин. Для посева ризосферной почвы готовят серию разведений: 102, 103, 104, 105.
Для выделения микроскопических грибов, населяющих ризоплану (поверхность корней) и тех, что внедряются во внутренние ткани корня, разработаны следующие методические приемы.
Методы водных смывов. Для изучения состава микромицетов, обитающих на поверхности корней, навески (не менее 0,5—1 г) кусочков корней (3 — 5 см) после удаления ризосферной почвы помещают в колбы со 100 мл стерильной воды. Колбы встряхивают на качалке (180 об/мин) в течение 5—10 мин. Затем корни с помощью стерильного пинцета или крючка переносят в следующую серию колб со стерильной водой и проводят дальнейшую десорбцию грибов на
качалке. Проводят до 5 — 10 смывов с корней. Посев на питательные среды осуществляют из ряда последовательных смывов. Более жестким способом десорбции микроскопических грибов с корней является 1 — 5-кратная их обработка в стерильной воде на микроразмельчителе тканей (5000 об/мин, 5 мин).
Водные смывы (без и с проведением центрифугирования) можно подвергнуть прямому микроскопическому анализу на содержание спор и мицелия и их непосредственному выделению приемом, аналогичным для почв.
Посев растертых корней. Тщательно промытые стерильной водой от почвы корни режут на кусочки и просушивают между двумя листами стерильной фильтровальной бумаги. Берут навески корней по 1 г и растирают их в стерильной фарфоровой ступке с кварцевым песком. Растертую массу переносят в колбу со 100 мл стерильной воды и встряхивают в течение 5 мин. Далее готовят серию разведений и проводят посев на питательные среды.
Посев фрагментов корней. На поверхность питательных сред помещают отрезки корня, с которого удалена ризосферная почва. Если раскладывают фрагменты размером 1 — 3 см, то часто отдельные быстрорастущие виды могут покрыть поверхность всего кусочка корня и среды в чашке Петри. Это не позволяет обнаружить другие виды микромицетов, населяющих данный фрагмент корня. Для устранения этого необходимо проводить раскладывание мелких кусочков корней (0,5—1 мм; толстые корни предварительно расщепляют). Для избежания нарушений пространственного распределения грибов вдоль корня рекомендуется проводить удаление ризосферной почвы не водными смывами, а скальпелем, препаровальной иглой или кисточкой. Небольшие кусочки корня стерильной препаровальной иглой или пинцетом с заостренными концами переносят на поверхность среды в чашке Петри. 5 — 6 кусочков распределяют равноудаленно друг от друга. В случае раздельного размещения на чашках кусочков с различных зон корня (корневого кончика, зоны растяжения, зоны корневых волосков, базальной части корня) можно изучать также локальные различия в составе микромицетов ризопланы. Раскладывают порядка 50—100 кусочков корня на различные питательные среды. Чашки просматривают в течение 3 — 10 сут и выделяют микромицеты в чистые культуры с помощью препаровальной иглы, при необходимости под контролем светового микроскопа.
Непосредственное выделение гиф грибов с корней. Корни, с которых предварительно удалена ризосферная почва, тщательно просматривают под бинокулярной лупой при максимальном увеличении и обнаруженные гифы переносят с помощью тонко заостренных препаровальных игл или пинцета на поверхность агаровой среды.
Получение спор сухоспоровых аскомицетов и пикнидиальных гифомицетов. Выделение в чистые культуры сумчатых и пикнидиальных грибов, плодовые тела и пикниды которых образуются внутри или на поверхности корней, других растительных и животных тканях, экскрементах животных, может быть серьезно затруднено из-за высокой плотности популяций быстрорастущих и обильноспороносящих грибов и других организмов. Проведение стерилизации образцов часто убивает и интересующие исследователя виды грибов.
Существует метод получения спор сухоспоровых аскомицетов с активным споро-отделением, плодовые тела которых снабжены отверстием, с последующим перенесением этих спор на питательные среды. Метод не пригоден для выделе
ния грибов, споры которых погружены в слизь или чьи плодовые тела лишены отверстия. Им можно получать споры некоторых пикнидиальных грибов.
В стерильные чашки Коха или Петри диаметром 10 — 18 см помещают маленькие стерильные чашки Петри (3 — 5 см), предварительно сняв с них крышки. Свежие образцы с плодоношениями грибов (корни, стебли, листья) осторожно очищают от частичек почвы и растительных остатков, кладут на края маленьких чашек Петри так, чтобы они не касались их дна, или помещают на стерильную сетку (металлическую или пластиковую) с крупными отверстиями. Образцы можно поместить и на стерильные стеклянные палочки в большой чашке Петри. Плодовые тела или пикниды обязательно должны располагаться отверстиями вниз. Разложенные над чашками образцы накрывают кружками (2 — 4 шт.) или полосками стерильной увлажненной фильтровальной бумаги, или в одну из чашек наливают стерильную воду для повышения влажности воздуха. Через 3 —48 ч маленькие чашки вынимают и заменяют новыми в случае продолжающегося отделения спор. Под малым увеличением микроскопа находят места с одиночными спорами и с помощью препаровальной иглы переносят их на питательные среды.
Метод накопительной культуры во влажных камерах. Метод используют для выявления грибов, развивающихся на поверхности корней и инфицирующих их внутренние ткани. В качестве влажных камер применяют чашки Петри, на дно которых помещают стерильную фильтровальную бумагу. Влага в чашках держится дольше, если под бумагу подкладывают стерильную марлю или вату. Фильтровальную бумагу увлажняют стерильной водой, не создавая при этом избытка воды на поверхности. Отрезки корней (3 — 5 см) раскладывают на поверхность бумаги, и чашку Петри ставят в термостат для инкубации при температуре 18 — 24 °C. Периодически, начиная со 2-х сут, корни просматривают под бинокулярным микроскопом и с помощью препаровальной иглы выделяют грибы в чистую культуру.
При исследовании состава микромицетов ризопланы параллельно выявляют инфицированность грибами внутренних тканей корня.
Выделение микромицетов из внутренних тканей растений. Для установления патогенных видов микромицетов, внедряющихся во внутренние ткани корня, стебля, листьев и семян, и эндофитных грибов проводят стерилизацию поверхности отмытых от почвы образцов растения различными антисептиками. Проводят погружение исследуемого объекта в 0,1%-й раствор сулемы с последующей многократной промывкой 50%-м этиловым спиртом и стерильной водой; обработку в течение 1 мин 0,1 —1 %-м раствором азотнокислого серебра; 1 — 2%-м раствором медного купороса в течение 2 — 5 мин и затем тщательной отмывкой образцов стерильной водой; 1%-м раствором в течение 1 мин или парами формалина, 0,1 — 1%-й бромной водой; 10 — 15%-м раствором перекиси водорода; 2%-м раствором гипохлорита натрия. Экспериментально подбирают оптимальный для конкретного объекта реагент, его концентрацию и время экспозиции (наиболее часто оно составляет от 10 с до 2 мин). После стерилизации корни промывают стерильной водой, меняя ее 3—4 раза для удаления стерилизующего агента. Отмывка от сильнодействующих соединений контролируется по качественным реакциям (сулемы — по качественной реакции на хлор с азотнокислым серебром). Затем корни или другие части растений разрезают стерильным скальпелем на мелкие кусочки и помещают во влажную
камеру. Инкубацию ведут до появления на их поверхности мицелия и спорообразования. Выделение в чистую культуру проводят препаровальной иглой под контролем светового микроскопа. Плотность заселения мицелия (численность грибов в тканях корня) определяют путем посева на сусло-агар определенной навески растертых корней.
Отрезки корней после стерилизации поверхности можно вместо влажной камеры раскладывать на разные питательные среды. При этом лучше использовать маленькие фрагменты корней (0,5—1 мм), чтобы снизить вероятность их инфицирования грибами, сохранившими жизнеспособность, но колонизирующими только поверхность корня. Вместо раскладывания корней на агаризо-ванных питательных средах можно поместить их в стерильный увлажненный песок на 2 —4 нед и затем обнаруженные грибы (при необходимости под контролем микроскопа) перенести на питательные среды.
При посеве ризосферной почвы и выделении сапротрофных грибов с корней используют различные среды: сусло-агар, среды Чапека, Мартина, глюкозопептонные и среды, приготовленные на растительных отварах, — овсяную, картофельную и др. Инкубируют чашки при температуре 24 — 26 °C в течение 1 — 3 нед. Для изоляции фитопатогенных видов применяют различные селективные среды, состав которых зависит от вида изучаемого патогена.
Все методы, описанные для выделения грибов с корней могут быть использованы для изучения микобиоты надземных частей растений и растительных остатков на различных стадиях разложения.
Глава 19
ИЗУЧЕНИЕ БИОЛОГИИ ПРОСТЕЙШИХ
Простейшие (Protozoa, протисты) — богатая по видовому составу (до 200 тыс видов) группа одноклеточных животных, распространенных повсеместно. Он;: встречаются в различных географических условиях, заселяя океаны, моря, пресные и солоноватые водоемы, почву. Очень многие простейшие от свободного образа жизни перешли к обитанию в других организмах и стали симбионтами или паразитами. Простейшие лишены клеточной стенки (хотя некоторые имеют панцирь) и поглощают питательные вещества, абсорбцией через клеточную мембрану или путем эндоцитоза. Для простейших наиболее характерна гетеротрофный и миксотрофный типы питания; облигатных автотрофов, по-видимому, нет. Обычной пищей свободноживущих протистов служат водоросли, бактерии, дрожжи, разлагающийся опад и остатки микроорганизмов, : также другие простейшие и мелкие членистоногие. Размножение простейших осуществляется половым и бесполым путями, иногда оба способа воспроизводства составляют стадии единого жизненного цикла организма.
Простейшие играют значительную роль в продукции и деструкции органических веществ, в цепях питания животных. Велика их роль и в разложение органических загрязнений при биологической очистке сточных вод и самоочищении водоемов. Возможно использование биомассы простейших в качестве нового источника кормового белка. Некоторые представители Protozoa проду
цируют ценные биологически активные вещества. В то же время многие паразитические простейшие являются возбудителями тяжелых заболеваний человека, животных и растений.
19.1. МЕТОДЫ ИЗУЧЕНИЯ ПОЧВЕННЫХ ПРОСТЕЙШИХ
Почвенные простейшие колонизируют разнообразные типы почв, но наибольшее богатство видов и высокая их численность характерны для почвы лесов. В 1 г почвы содержится от десятков до сотен тысяч простейших, которые представлены жгутиконосцами (Mastigophora), саркодовыми (Sarcodina) и инфузориями (Infusoria, Ciliata).
Наиболее распространены жгутиконосцы родов Euglena (фитомастигины), Monas, Bodo, Cercomonas, Oicomonas (зоомастигины). Среди саркодовых в почве встречаются корненожки: голые амебы представлены родами Amoeba, Hya/odiscus, Naegleria, Vahlkampfia, раковинные — родами Centropyxis, Plagiopyxis, Nebela, Difflugia, Arcella, Corythion, Euglypha, Tinema. Почвенные инфузории — это в основном виды рода Colpoda, а также некоторые виды Hypotrichida и Prostomatida.
Для простейших характерны разнообразное морфологическое строение и значительное отличие по физиологическим функциям. В связи с этим необходимы различные приемы и способы их исследования. На сегодняшний момент предложено множество методов прямого подсчета почвенных протестов, предназначенных для выявления находящихся в почве простейших без их предварительного размножения. Однако прямым подсчетом удается обнаружить лишь немногих почвенных простейших и не всегда, поэтому широко используются методы количественного учета, требующие предварительного размножения простейших. Таким образом, все способы обнаружения простейших в почве могут быть сведены к двум основным: 1) прямое обнаружение в почве или почвенной суспензии; 2) косвенное обнаружение в различных питательных средах, в которые были внесены пробы почвы. Несмотря на то что большинство методик было разработано довольно давно, они не утратили своей актуальности и в настоящее время.
19.1.1. Определение количества простейших, обнаруженных непосредственно в почве
Подсчет простейших в почвенной суспензии проводят методами количественного учета, принятыми в микробиологии. Наиболее прост, хотя и несовершенен, следующий метод. Почву, смешанную с небольшим количеством воды, помещают на предметное стекло с лункой и сразу же микроскопируют. Для замедления движения быстродвигающихся форм и их подсчета добавляют 1 — 2 %-й раствор желатины или 0,001 % агара. Недостатком этого метода является то, что с его помощью учитывают только свободноплавающих простейших и не учитывают всю массу организмов, оставшуюся среди почвенных частиц. Необходимо иметь в виду, что некоторые простейшие при смачивании быстро (через несколько минут) выходят из цист и, следовательно, для обнаружения активных форм пробу почвы нужно просматривать незамедлительно. Этим способом удается обнаружить главным образом крупные формы активных простейших при условии их высокой численности в почве. Сравнительно легко различимы раковинные корненожки.
Для учета раковинных амеб используют микроскопирование водной суспензии почвы. Для этого к навеске почвы (0,1 г) добавляют 20 — 25 мл воды, оставляют на несколько часов до размокания почвенных частиц и взбалтывают в течение 10 мин. Дно стерильной чашки Петри расчерчивают по трафарету на квадраты 1x1 см. В приготовленную чашку выливают полученную суспензию, распределяя мелкозем по дну чашки тонким слоем. Крупные комочки почвы разрушают препаровальной иглой. Взвесь микроскопируют с объективами 8х или 40 х. Число раковинок подсчитывают по диагонали в 14 квадратах, расположенных крестообразно. Общее количество амеб (М) в навеске определяют по формуле
М= (N/14)S,
где N — число раковинок в 14 квадратах; S — плошадь дна чашки.
После фиксации микроорганизмов на предметном стекле можно использовать окраску карболовым эритрозином (рецепт красителя см. в приложении 5). В результате живые амебы окрашиваются в интенсивно-малиновый цвет, а их пустые раковинки — в розовый.
19.1.2. Обнаружение и подсчет почвенных простейших с использованием питательных сред
Принцип косвенных методов заключается в предварительном культивировании почвенных простейших в питательной среде, для чего в стерильную среду вносят навеску почвы, взятую из естественных условий. Среда должна быть также благоприятной и для развития бактерий, которыми питаются простейшие. Считается, что в жидких средах лучше развиваются инфузории и жгутиконосцы, а на плотных средах — амебы.
Метод проращивания почвенного мелкозема во влажных камерах на агари-зованных покровных стеклах. Для этого метода необходимо приготовить водную почвенную вытяжку (1 кг почвы/л воды), которую затем кипятят 1 ч. отстаивают в течение суток и фильтруют. К полученному фильтрату добавляют 1 % агара, очищают белком куриного яйца (см. гл. 3) и наносят тонким слоем на обезжиренные покровные стекла. Стекла помещают на влажную фильтровальную бумагу в чашки Петри. Почву размельчают и просеивают через сито 0,25 мм; почвенный мелкозем через трубку, один конец которой закрыт сеткой, равномерно высевают на поверхность агара на стекле. Чашки инкубируют при 20 —24 °C в течение 7 — 10 сут. Этот метод позволяет наблюдать за развитием и активным движением простейших in situ, а также выделять их в чистую культуру.
Для окрашивания стекол молибденовым гематоксилином (протрава — молибденово-кислый аммоний MoO4(NH4)2, гематоксилин — 0,5%-й водный раствор) микроорганизмы необходимо предварительно зафиксировать, для чего стекла помещают в парь: 1%-й осмиевой кислоты на 1 — 2 мин или в жидкость Шаудина (приготовление реактива см. в приложении 6). Эксперимент проводят в 10-кратной повторности. Учитывая площадь стекла, поля зрения микроскопа и вес почвенного мелкозема, вычисляют количество протистов в 1 г почвы. Особенно хорошо этим методом учитываются амебы.
Метод предельных разведений. Навеску почвы разводят жидкой питательной средой. Далее проводится математическая обработка полученных результатов пс таблице Мак-Креди (см. гл. 7). Этот метод используют для количественного учета
активнодвигающихся простейших. Увлажняют 10 г почвы до пастообразного состояния стерильной питательной средой из сенного отвара с почвенной вытяжкой 1: 1 (рецепты см. в приложении 4) и растирают 5 мин в фарфоровой ступке резиновым напалечником. Затем готовят ряд последовательных 10-кратных разведений (не менее пяти). Для каждого разведения берут по три пробирки с 9 мл среды. Исходную суспензию готовят, помещая навеску почвы в колбу с 90 мл жидкой питательной среды (остальное количество среды пошло на смачивание почвы). Суспензию взбалтывают в течение 10—15 мин, затем оставляют на 30 с для осаждения крупных почвенных частиц и готовят из нее второе разведение. Культивирование микроорганизмов осуществляют при 22 —24 °C, проводя микроскопический контроль в течение месяца, учитывая ин- и экс-цистирование различных видов простейших, происходящее в разные сроки.
Для количественного учета простейших с помощью таблицы Мак-Креди подсчитывают число пробирок, в которых они размножились. На основании полученных результатов составляют числовую характеристику, состоящую из трех цифр. На первом месте отмечают число пробирок того разведения, где во всех повторностях развилась культура простейших (обычно это 3 пробирки). Следующие две цифры обозначают пробирки с развившейся культурой из двух последовательных разведений. По таблице Мак-Креди находят вероятное число, соответствующее числовой характеристике. Для пересчета количества простейших на 1 г почвы полученное число умножают на разведение, в котором во всех пробирках был отмечен рост простейших. При анализе свежей почвы необходимо учитывать влажность субстрата, для определения которой можно использовать формулу
т = (а - Ь)100/(а - с),
где а — масса влажной земли и тары; b — масса высушенной земли и тары; с — масса тары; т — влажность почвы, %.
Разведение по Кетлеру. Желательно приготовить не менее 15 разведений (1: 10; 1:100; 1: 1000; 1: 2500 и т.д.). Для приготовления разведения 1:10 берут 10 г почвы и разводят их в 100 мл стерильной водопроводной воды. Затем из каждого разведения берут 1 мл суспензии, которую необходимо предварительно взболтать, и помещают в жидкую питательную среду сенного настоя с почвенной вытяжкой (приготовление см. в приложении 4). Культивирование проводят при 22 — 24 °C.
В течение месяца через определенные промежутки времени проводят микроскопические исследования (при увеличениях 8х и 40*) и устанавливают наибольшее разведение, при котором обнаружено развитие простейших. Количество простейших должно соответствовать числу, находящемуся между двумя ближайшими наибольшими разведениями, из которых последующее не дало роста простейших. Например, 1 мл суспензии из разведения 1 : 2500 дал развитие простейших, а 1: 5000 не дал его, следовательно, число простейших лежит между 2500 и 5000 на 1 г почвы.
Метод Сингха. В чашки Петри помещают по 8 стеклянных колец (внутренний диаметр 2 см, глубина 1 см, толщина 1—2 мм) и стерилизуют. Затем в чашки Петри разливают 20 мл стерильного расплавленного «голодного» агара (1 —1,5%-й агар, содержащий 5 г/л NaCl и 1 г/л СаСО3). Кольца стерильным пинцетом размещают по периферии чашки на одинаковом расстоянии так, чтобы среда попала в каждое из них. После застывания агара в каждое кольцо вносят каплю густой суспензии одной или нескольких бактериальных культур, потребляемых простейшими, например Е. coli или Azotobacter chroococcum. Чашки инкубируют в течение суток при температуре 37 или 30 °C соответственно.
Для приготовления почвенной суспензии 10 г почвы смачивают стерильной водой, растирают в стерильной ступке напалечником и переносят в 50 мл стерильного физиологического раствора или в стерильную водопроводную, прудовую или дождевую воду. Суспензию встряхивают в течение 5 мин, оставляют на 30 с для осаждения крупных частиц почвы и готовят ряд разведений (не менее 15).
Из исходного разведения ('/5) делают большие разведения — от */10 до '/81920, путем разведения 5 мл суспензии, взятой из предыдущего более слабого разведения, в 5 мл стерильного физиологического раствора или водопроводной воды. На чашке Петри 8 колец заражают 0,05 мл из определенного разведения. Затем чашки инкубируют 15 — 30 сут при 22 —24 °C. Через .4 — 6 сут культуру микроорганизмов просматривают под микроскопом с увеличением 8* или 40* для обнаружения и идентификации простейших. Численность простейших определяют путем подсчета колец, имеющих простейших и лишенных их, из каждого разведения (табл. 19.1; 19.2).
Таблица 19.1
Количество колец с простейшими и без них при разных разведениях
Разведение Амебы Жгути коносцы Инфузории
+ - + - + -
1/10 8 0 8 0 6 2
1/15 8 0 8 0 8 0
1/20 8 0 8 0 3 5
1/40 8 0 8 0 3 5
1/80 8 0 8 0 4 4
1/160 8 0 8 0 0 8
1/320 8 0 8 0 2 6
1/640 7 1 8 0 0 8
1/1280 5 3 6 2 0 8
1/2560 8 0 8 0 0 8
1/5120 2 6 4 0 0 8
1/10240 1 7 2 6 0 8
1/20480 0 8 1 7 0 8
1/40960 0 8 0 8 0 8
1/81920 1 7 1 7 0 0
Всего 80 40 86 34 26 94
В итоге имеем
Культуры с простейшими (+)...
Культуры без простейших (-)..
Число на 1 г почвы (см. табл. 19.2)
Амебы Жгутиконосцы Инфузории
....80..........86............ 26
....40..........34............ 94
.41400.........70500........... 377
Таблица 19.2
Определение численности простейших методом разведений
(на 1 г почвы при разведении от 5 до 81920 раз в 8 повторностях на 15 чашках)
Отрицательные культуры Организмы (на 1 г) Отрицательные культуры Организмы (на 1 г) Отрицательные культуры Организмы (на 1 г) Отрица-тельные-культуры Организмы (на 1 г) Отрицательные культуры Организмы (на 1 г)
4 1690000 п 132000 50 17300 73 2330 96 317
5 1430000 28 121000 51 15800 74 2140 97 290
6 1230000 29 110000 52 14500 75 1960 98 265
7 1060000 30 101000 53 13300 76 1800 99 243
8 931000 31 92000 54 12200 77 1650 100 223
9 824000 32 84200 55 11100 78 1510 101 203
10 729000 33 77100 56 10200 79 1390 102 185
П 650000 34 70500 57 9380 80 1270 103 169
12 581000 35 64500 58 8570 81 1170 104 154
i 13 520000 36 59000 59 7860 82 1070 105 140
; 14 467000 ' 37 54000 60 7210 83 979 106 126
15 421000 38 49400 61 6600 84 898 107 113
16 380000 39 45200 62 6040 85 823 108 101
17 344000 40 41400 63 5540 86 755 109 90,2
18 311000 41 37900 64 5080 87 693 НО 79,4
19 282000 42 34700 65 4670 88 635 111 69,5
20 256000 43 31800 66 4280 89 582 112 60,2
21 232000 44 29200 67 3920 90 534 113 51,3
22 211000 45 26700 68 3600 91 490 114 42,9
23 192000 46 24500 69 3300 92 450 115 34,8
24 175000 47 22400 70 3020 93 412 116 27,4
25 159000 48 20500 71 2770 94 377
26 145000 49 18800 72 2540 95 346
19.1.3. Определение числа активных форм и цист
Метод Северцовой. Для количественного учета амеб чашки Петри с агаризованной средой заражают бактериальной культурой в форме креста; таких крестов наносят 10 —15 на каждую чашку. Развивающиеся культуры не должны соприкасаться между собой. В центр каждого креста вносят около 0,12 мл почвенной суспензии (10 г почвы в 100 мл стерильной воды). Приготовление почвенной суспензии и последовательных разведений осуществляют как было
указано выше. Чашки помещают в термостат при температуре 22 —24 °C и наблюдают за появлением роста протист на крестиках. Исходя из положения, что одна амеба дает начало одной колонии, подсчитывая число крестиков с амебами, можно определить их число в исходной пробе. Например, на крестики внесено по 0,12 мл суспензии почвы с разбавлением в 100 раз. Из 20 крестиков 10 оказались с простейшими, а 10 без них. Отсюда можно предположить, что лишь в половине порций по 0,00012 г находилось по одному простейшему, давшему начало культуре. Следовательно, в 1 г находилось от 4166 до 8332 простейших:
1 г/0,00012 г = 8332; 1 г/(0,00012 • 2) г = 4166.
На основании подсчетов, произведенных на нескольких чашках, берут среднее из всех разведений почвенной суспензии, опуская минимальные и максимальные числа.
Для определения числа активных форм и цист, находящихся в суспензии, последнюю нагревают до 60 —70 °C, т.е. до температуры, при которой гибнут активные простейшие. К недостаткам этого метода можно отнести вероятное переползание простейших из одного креста в другой, а также возможное повреждение цист уже при 58 °C.
Метод Кетлера. Одновременно с приготовлением серий разведений берут 10 г той же почвы и обрабатывают ее аликвотой 2 %-го раствора НС1 так, чтобы создать некоторый избыток кислоты после нейтрализации карбонатов. Почву оставляют на ночь, в течение этого времени все активные формы погибают, а цисты сохраняют жизнеспособность. Затем почву разбавляют таким же способом, как и не обработанную соляной кислотой. Дальнейшее развитие на питательной среде обнаруживает лишь тех простейших, которые развились из цист. Разность между первым и вторым подсчетом дает число активных простейших. Просмотр питательной среды при этом следует производить очень часто — вначале до двух раз в сутки, так как численность активных форм и цист быстро меняется.
19.1.4. Физиологическая активность и распространение простейших в почве
Изучение распространения простейших в почве. Для проведения эксперимента почву доводят до воздушно-сухого состояния, отсеяв крупные частицы, размельчают, просеивают через сито 0,25 мм и помещают в чашки Петри так, чтобы она равномерно располагалась по дну. Если изучают активность простейших в зависимости от механического состава почвы, в чашки Петри помещают отдельные фракции увлажненной до полной влагоемкости почвы. На почве, находящейся в чашке, делают отметки через 1 см от центра, после чего чашки стерилизуют при 2 ати в течение 30 мин. Затем в центр чашки с почвой в небольшое углубление помещают 1 — 2 петли культур простейших или комочек почвы, зараженной простейшими. Зараженные чашки инкубируют при 22— 24 °C в камере или под стеклянным колпаком для избежания испарения. Ежедневно с каждого участка почвы, начиная от центра к периферии, берут пробу почвы и переносят в стерильную питательную среду (в чашки с плотной средой или в колбы с жидкой средой).
Ежедневно, начиная со следующего дня, среду используют для обнаружения простейших.
Если, например, через два дня после заражения почвы простейшими, последние будут обнаружены в колбах, в которые были внесены пробы почвы с расстояния 1 и 2 см от места заражения, а проба с расстояния 3 см в колбе окажется стерильной, то, следовательно, в течение 2 сут простейшие распространились на 2 см или несколько больше 2 см. Таким же способом выясняется распространение почвенных бактерий. Распространение простейших в стерильной почве показывает их активное движение и размножение, что зависит в свою очередь от механического состава почвы и ее влажности.
Изучение питания простейших. Для выявления пищевых предпочтений разных видов простейших необходимо иметь чистые культуры протист (амеб, инфузорий или жгутиконосцев), а также бактерий, принадлежащих к олигони-трофильным микроорганизмам, нитрификаторам, аммонификаторам, дени-трификаторам, аэробным целлюлозолитикам, спорообразующим бациллам. Широкий выбор бактериальных культур объясняется их избирательным выеданием различными представителями простейших.
Перед началом эксперимента определяют плотность популяции простейших (микроскопически в камере Горяева, см. гл. 7) и бактерий (методом предельных разведений или измеряя оптическую плотность культуры, см. гл. 7). Затем в каждую пробирку с развивающимися бактериями вносят по 2 мл культуры простейших. Культивирование проводят при 26 °C. Интенсивность развития простейших оценивают в динамике в течение недели и выражают в процентах, принимая за 100% рост протистов на олигонитрофильных микроорганизмах. Количественный учет простейших проводят микроскопически.
Для наблюдения за питанием простейших дрожжами готовят густую суспензию дрожжей (Saccharomyces cereviseae) в стерильной водопроводной воде, которую затем по трафарету наносят штрихами длиной 2 см в двойной повторности на поверхность «голодного» агара (15,0 г агара на литр водопроводной воды) в стерильные чашки Петри. В конец каждого штриха инокулируют цисты и вегетативные клетки амеб. Наблюдения проводят на 3 —4-е сут, отмечая степень выедания штриха дрожжевых клеток амебами, а также инцистирование амеб. Необходимо учитывать, что иногда амебы инцисти-руются до того, как выедают штрих полностью.
Возможно также использование другого способа наблюдения за питанием простейших. Он заключается в том, что в большие пробирки наливают по 1 мл стерильного физиологического раствора, в который вносят смешанные культуры различных дрожжей и амеб. Стерильно отбирают пипеткой каплю смеси и помещают в камеру Горяева для отдельного подсчета клеток амеб и дрожжей под микроскопом. Пробирки инкубируют при 25 °C. Подсчеты делают через каждые 12 ч, учитывая изменение числа дрожжей, вегетативных клеток и цист амеб.
Для микроскопического наблюдения за питанием простейших несколько инфузорий {Paramecium caudatum) отсаживают на часовое стекло в стерильную питательную среду (см. приложение 4). Через 1 ч, в течение которого инфузории освобождаются от имевшихся пищевых вакуолей, в среду помешают суспензию чистой культуры бактерий. Через 30 мин под бинокулярным микроскопом (увеличение 8х) простейших вылавливают капилляром из часового стекла и помещают на предметное стекло. Для замедления движения инфузорий в прижизненный препарат добавляют 1—2%-й раствор агара и подсчитывают вакуоли при увеличении 40 х. Для фиксации парамеций используют пары ледяной уксусной или осмиевой кислоты. При этом предметное стекло с каплей, в которой находятся инфузории, перевернув вниз, держат над парами кислоты в тече
ние 1 мин. Вместо бактериальной культуры можно также использовать суспензию туши, каплю которой вносят в питательную среду простейших. Вакуоли, содержащие черную тушь, хорошо видны при малом увеличении микроскопа.
Обнаружение пищеварительных ферментов простейших на примере кислой фосфатазы. Для выявления кислой фосфатазы (маркерный фермент лизосом) перед фиксацией в течение 10 мин инфузорий кормят олигонитрофильными бактериями на среде из сенного отвара с почвенной вытяжкой (см. приложение 4). Затем простейших фиксируют 30 мин раствором Карновского на ледяной бане, четырехкратно (по 10 мин на холоду) отмывают 100 мМ Na-ацетат-ным буфером (pH 5,5), инкубируют в среде Гомори (pH 5,5) при 37 °C на водяной бане в течение 25 мин. Среда Гомори {готовится в день использования^'. 0,06 г нитрата свинца растворяют в 45 мл Na-ацетатного буфера (100 мМ, pH 5,5). Затем при помешивании добавляют по каплям 10 мл 3%-го раствора Na в глицерофосфате (субстрат) и доводят водой до 90 мл. Доводят pH до 5,5 с помощью 10%-й уксусной кислоты. Готовый раствор хранят при 37 °C (в термостате). Перед инкубацией проверяют pH среды и, если нужно, доводят до 5,5; затем доводят объем до 100 мл водой и фильтруют через бумажный фильтр. Контролем служит среда Гомори, содержащая 10 мМ NaF. Клетки трижды отмывают от среды в 100 мМ Na-ацетатном буфере при комнатной температуре (по 10 мин), затем осаждают непрореагировавший нитрат свинца в виде PbS с помощью 0,5 %-го сульфида аммония. Обработку проводят не более 1 мин, после чего клетки дважды промывают водой. Опытный и контрольный варианты микроскопируют. Следует иметь в виду, что после каждого этапа обработки суспензию клеток центрифугируют, а надосадочную жидкость осторожно отсасывают. Во время инкубации при 37 °C и pH 5,5 (оптимумы температуры и pH кислой фосфатазы) от субстрата (Na в глицерофосфате) с помощью кислой фосфатазы отщепляется РО^“, который немедленно осаждается нитратом свинца в виде фосфата свинца, тем самым предотвращается диффузия фосфата из мест ферментной активности. Выпавший белый фосфат свинца виден только при электронной микроскопии, поэтому для световой микроскопии фосфат свинца нужно перевести с помощью сульфида аммония в хорошо видимый черно-коричневый сульфид свинца. В контрольной среде NaF блокирует расщепление субстрата, так как ингибирует все активные в кислой среде ферменты.
19.1.5. Видовое определение простейших
Видовому определению простейших предшествуют: выделение их в чистую культуру, морфологическое описание вегетативных форм, стадий покоя и цист, развивающихся на агаризованных и жидких средах, приготовление прижизненных и постоянных окрашенных препаратов.
Выделение чистых культур простейших. Для этого их выращивают на культуре олигонитрофильных бактерий, затем втягивают в стерильную пипетку или капилляр, после чего проводят через ряд (не менее 5) часовых стекол с питательной средой. После очистки культуры простейших помещают в чистую культуру бактерий.
Можно применять и другой, более удобный способ получения чистых культур простейших. Расплавленную агаризованную среду разливают в стерильные чашки Петри, инокулируют одним из видов бактерий, располагая их в виде креста. В центр помещают простейших или частицы почвы с простейшими. Последние мигрируют к концам крестообразной колонии бактерий. После 2 — 3 пересевов имеют полное освобождение простейших от всех сопровождавших их ранее бактерий, за исключением посеянной микробной культуры.
Видовое определение простейших проводят при их микроскопировании и выделении клоновых культур, которые поддерживают на жидкой и плотной питательных средах. Для идентификации обнаруженных форм готовят препарат «раздавленная капля». Движение инфузорий и жгутиконосцев замедляют 1 —2%-м раствором желатины или агара. Для прижизненного окрашивания применяют водные растворы метиленового синего, метиленового зеленого и нейтрального красного. Работу ресничек, жгутиков и пищеварительных вакуолей наблюдают в слабых разведениях туши.
Для фиксирования и выявления цитологических структур объекты обрабатывают парами осмиевой кислоты 1—2 мин (1%-й водный раствор OsO4) или фиксатором Шаудина (приготовление реактива см. в приложении 6). Ядро выявляется при окрашивании 0,1%-м раствором метиленового зеленого в 1%-й уксусной кислоте (фиксатор Роскина). Реснички и жгутики окрашивают йодной тинктурой (несколько капель настойки йода в 10 мл воды). При прибавлении 2 —4%-го раствора соды рельефно выступают ресничный аппарат, различные эктоплазматические образования, строение рта и глотки.
Все обнаруженные формы инфузорий, жгутиконосцев и амеб, их цисты зарисовывают. Рисунки выполняют с живых объектов при увеличении микроскопа — 900* и 1500*.
Для распознавания трофически активных стадий вегетативных клеток очень важно появление жгутиконосных форм в жидкой среде. Безошибочно отличить амебовидные стадии жгутиковых от амеб можно при исследовании форм в «висячей капле» во влажной камере, где формируются плавающие стадии жгутиконосцев. Однако одних лишь морфологических признаков недостаточно для точной видовой идентификации, поэтому большое внимание уделяется и тонкому цитологичесому строению протозойных клеток. Для этого готовят постоянные окрашенные препараты по методу Добелла, выявляют особенности кариокинеза на препаратах, обработанных по Фельгену и др. (точное описание этих методов можно найти в специальных руководствах).
РАЗДЕЛ IV
ИЗУЧЕНИЕ ФИЗИОЛОГИИ И МЕТАБОЛИЗМА МИКРООРГАНИЗМОВ
Глава 20
ПРИМЕНЕНИЕ МЕТОДОВ ПЛАНИРОВАНИЯ МНОГОФАКТОРНОГО ЭКСПЕРИМЕНТА ПРИ ОПТИМИЗАЦИИ
СОСТАВА ПИТАТЕЛЬНОЙ СРЕДЫ
Методы математического планирования эксперимента нашли широкое применение для подбора питательных сред в целях повышения выхода биомассы или накопления определенных продуктов обмена веществ микроорганизмов. Они позволяют не только одновременно изучить действие нескольких факторов на интересующий нас процесс, но и количественно оценить степень этого влияния.
Цель работы, поиск оптимальных концентраций компонентов среды, обеспечивающих максимальное накопление биомассы исследуемого вида. Практические занятия сопровождаются курсом лекций, который знакомит студентов с различными типами планов многофакторных экспериментов и методами математической обработки их результатов.
ЗАДАЧА. ОПТИМИЗАЦИЯ СОСТАВА ПИТАТЕЛЬНОЙ СРЕДЫ
ДЛЯ НАКОПЛЕНИЯ БИОМАССЫ САПРОТРОФНЫМ ШТАММОМ PSEUDOMONAS AERUGINOSA
При постановке задачи оптимизации предполагается, что для изучаемого процесса выбран (на основании теоретических представлений или чисто практических соображений) показатель интенсивности этого процесса у, о котором заранее известно, что
1) он зависит от некоторого числа п переменных (факторов): хь х2, х3, ..., хп, т.е. существует функция (называемая функцией отклика):
у = /(х1,х2,х3,...,х„);
2) эта функция имеет единственный максимум.
Тогда решение задачи оптимизации сводится к нахождению этого максимума по всем п переменным. В теории эксперимента у называют обычно параметром оптимизации, а переменные х-, — факторами.
Вид функции у = f(x}, хъ х3, ..., х„), как правило, неизвестен, и при экспериментальном изучении подобной зависимости для ее описания используют уравнения регрессии. В простейшем случае, когда параметр у связан с каждым из факторов х, линейно, такое уравнение имеет вид:
У — bo + bixi + b2x2 +... + b„x„ — bo + '^biXj, (20.1)
i
где коэффициенты b\, b2, ..., bn характеризуют влияние каждого фактора на величину у. Нетрудно понять, что при увеличении х, на единицу значение у увеличивается или уменьшается на | Ь, | в зависимости от того, какой знак («+» или «-») стоит перед членом Ь-,х,. Однако большинство биологических зависимостей имеет нелинейный характер и уравнение (20.1) может удовлетворительно («адекватно») описывать функцию отклика только в достаточно узкой области изменений х,. Ясно также, что функция отклика (20.1) не имеет максимума. Поэтому при оптимизации биологических процессов более эффективным оказывается использование уравнения регрессии 2-й степени (квадратичное уравнение):
У = А) + X b>xi + X bux'xJ + X bux? ('2°'2-)
i i< j H
Члены вида b,jX-,Xj оценивают так называемое взаимодействие факторов, т. е. ситуацию, когда влияние одного из факторов xt на у зависит от уровня второго, взаимодействующего с ним фактора Xj. Квадратичные члены Ьпх] отражают нелинейность зависимости у от фактора х„ которая выражается в том, что влияние Xj на у зависит от уровня самого этого фактора, вследствие чего на кривой, изображающей зависимость у от xh обнаруживается минимум (если Ьц > 0) или максимум (если by < 0).
Для определения величин коэффициентов в линейном уравнении (20.1) достаточно поставить опыты по плану, который предусматривает исследование каждого из факторов всего на двух уровнях: верхнем и нижнем. К числу таких планов относятся планы полного факторного эксперимента (ПФЭ) 2й, где п — число изучаемых факторов. Реализация таких планов позволяет получить также оценки взаимодействий факторов, т. е. величины коэффициентов by в уравнении (20.2).
Чтобы оценить квадратичные эффекты факторов (коэффициенты Ьц в уравнении 20.2), следует применять планы экспериментов, в которых каждый фактор варьируют по меньшей мере на трех уровнях. Такие планы называются планами 2-го порядка. Они требуют постановки большего числа опытов по сравнению с планами 1-го порядка, но с их помощью задачу оптимизации микробиологического процесса можно решить в два этапа. На первом этапе ставят серию опытов, выбирая интервалы варьирования факторов так, чтобы охватить всю область их действия — от уровней, лимитирующих изучаемый процесс, до уровней, ингибирующих его. В этом случае можно ожидать, что оптимум по каждому фактору находится внутри исследуемой области варьирования факторов.
По результатам этой серии опытов вычисляют коэффициенты квадратичного уравнения (20.2) и находят координаты точки максимума функции, описываемой этим уравнением. Даже в том случае, если уравнение (20.2) аппроксимирует изучаемую функцию отклика не слишком точно, можно рассчитывать на то, что найденный максимум квадратичной функции находится в око-лооптимальной области. Поэтому на втором этапе оптимизации следует поставить новую серию опытов по плану 2-го порядка с центром эксперимента в точке максимума, рассчитанной по результатам первой серии опытов. На ос
нове квадратичной модели, полученной на 2-м этапе, уточняют положение оптимума для изученной совокупности факторов.
При современном уровне компьютеризации научных исследований кажется вполне естественным использование персональных компьютеров для построения планов эксперимента и статистического анализа полученных результатов. Соответствующие программы имеются в разнообразных прикладных статистических пакетах, таких как STADIA, STATISTICA, SYSTAT, SPSS и др. Однако в ходе выполнения данной задачи предусматривается выполнение всех вычислений «вручную», т. е. с помощью обычного калькулятора. Многолетний опыт преподавания убеждает в том, что в этом случае достигается более глубокое понимание смысла применяемых вычислительных процедур и выводов, получаемых на их основе. Следует также отметить, что в данной задаче все необходимые расчеты можно проделать, используя табличный процессор EXCEL — обязательный компонент программного обеспечения в пакете Microsoft Office, который имеется теперь практически в каждом персональном компьютере.
1. Опыты по композиционному плану 2-го порядка
Опыты проводят, используя среды, содержащие следующие компоненты: глюкозу; NaNO3; NaH2PO4-2Н2О; КО; MgSO4-7H2O. Варьируют 4 фактора: концентрации глюкозы [С] — xh азотнокислого натрия [N] — х2, фосфата [Р] — х3 и хлористого калия [К] — х4. В табл. 20.1 приведены единицы варьирования (Л.) этих факторов и их концентрации, соответствующие нижнему (-1), среднему (0) и верхнему (+1) уровням.
План эксперимента приведен в табл. 20.2. Первые 16 опытов — это план полного факторного эксперимента (ПФЭ) 24. В нем представлены все возможные комбинации 4 изучаемых факторов на 2 уровнях. Следующие 8 опытов — так называемые звездные точки — дополняют ПФЭ 24 до композиционного плана бокса 2-го порядка (план Б4). Последний, 25-й опыт в центре эксперимента используется для независимой проверки адекватности квадратичной модели (см. ниже).
Таблица 20.1
Единицы варьирования (X) и концентрации (г/100 мл) компонентов сред на нижнем, среднем и верхнем уровнях
Компонент среды Фактор Уровень Единица варьирования (X)
НИЖНИЙ «—» средний «0» верхний «+»
Глюкоза 0,2 2,0 3,8 1,8
NaNO3 A 0,05 0,5 0,95 0,45
NaH2PO4-2H2O A 0,005 0,03 0,055 0,025
КС1 0,01 0,06 0,11 0,05
MgSO4-7H2O Постоянный уровень — 0,02
Матрица композиционного плана 2-го порядка
Номер опыта (среды) Уровни факторов Номер опыта (среды) Уровни факторов
х, Х2 Г ХА х4
1 -1 -1 -1 -1 14 +1 -1 +1 +1
2 +1 -1 -1 -1 15 -1 +1 +1 +1
3 -1 + 1 -1 -1 16 +1 +1 +1 +1
4 +1 + 1 -1 -1 17 -1 0 0 0
5 -1 -1 +1 -1 18 +1 0 0 0
6 +1 -1 +1 -1 19 0 -1 0 0
7 -1 +г +1 -1 20 0 +1 0 0
8 +1 +1 +1 -1 21 0 0 -1 0
9 -1 -1 -1 + 1 22 0 0 +1 0
10 + 1 -1 -1 + 1 23 0 0 0 -1
И -1 +1 -1 + 1 24 0 0 0 +1
12 +1 +1 -1 + 1 25 0 0 0 0
13 -1 -1 +1 + 1
Значения кодированных переменных х, в плане эксперимента и концентрации компонентов среды связаны простыми соотношениями:
ГС1 - 2,0 л = =
1,8 2
[N]-0,5 [Р]-0,03 [К]-0,06
--------, Лз —---------, Ха —---------
0,45 0,025 0,05
где [С] — концентрация глюкозы; [N] — концентрация нитрата натрия; [Р] — концентрация фосфата натрия; [К] — концентрация хлористого калия (все концентрации даны в граммах на 100 мл).
Приготовление сред. Готовят по 25 мл минерального фона каждой среды. Для составления сред удобно приготовить растворы каждой соли в более высоких концентрациях, чем они содержатся в средах, и сливать растворы в определенных объемах. Например, если в 25 мл среды количество NaNo3 на нижнем уровне соответствует 0, 05 г, на среднем в 10, а на верхнем в 19 раз больше, то, имея раствор, в 1 мл которого содержится 0,05 г NaNo3, берут для приготовления сред соответственно 1, 10 и 19 мл этого раствора. Делают подобные расчеты для других компонентов сред.
Рассчитывают количество воды, необходимое для доведения общего объема каждой среды до 25 мл, а также объем раствора каждой соли, требуемый для приготовления всех сред, и соответствующую ему навеску вещества. Результаты записывают в табл. 20.3, которой пользуются при составлении сред. Каждую среду разливают по 9 мл в 2 широкие пробирки и стерилизуют при 1 ати.
Готовят 3 раствора глюкозы, в 1 мл каждого из которых количество глюкозы соответствует содержанию ее на нижнем, среднем и верхнем уровнях в 10 мл среды. Рассчитывают объем каждого раствора, необходимый для приготовления соответствующих сред. Растворы готовят на дистиллированной воде. Их
Схема приготовления сред для эксперимента по композиционному плану 2-го порядка (план Бокса, Р)
Вариант приготовления среды Матрица планирования Объем раствора и воды, мл
Х2 -*3 Х4 NaNO3 NaH2PO4- 2Н2О КС1 MgSO4-7H2O Вода водопроводная
Номер среды 1 2 3 4 25 -1 -1 1 1 0 -1 -1 -1 -1 0 -1 -1 -1 -1 0
Общий объем раствора каждого компонента, мл
Объем раствора для опытов, мл
Навеска вещества, г
стерилизуют при 0,5 ати и добавляют по 1 мл к 9 мл стерильного минерального фона в пробирках перед посевом.
Посев и условия культивирования. Две пробирки с каждой средой засевают суспензией клеток односуточной культуры Р. aeruginosa в стерилизованной водопроводной воде. Суспензию получают смывом клеток с поверхности ско-
Таблица 20.4
Рост Р. aeruginosa в показаниях нефелометра (*100) (фильтр 6, рабочее расстояние кюветы 0,5 см)
шейного МПА. Количество посевного материала — 0,3 мл. Пробирки с засеянными средами помещают на качалку с 200 — 300 об/мин в термостат (30 °C). Продолжительность культивирования — 2 — 3 сут.
Регистрация результатов. Рост бактерий оценивают нефелометрически и представляют в виде таблицы (табл. 20.4). Перед нефелометрирова-нием в каждую пробирку добавляют 1 — 2 капли концентрированной соляной кислоты для растворения осадка солей и содержимое пробирок тщательно перемешивают. Контролем при измерении служит стерильная среда или вода. Культуры с высокой плотностью, если это необходимо, разбавляют водой. Показания нефелометра с учетом разведения для удобства умножают на 100 и записывают в табл. 20.4.
Номер опыта У, А У„
1
2
3
25
Обработка результатов ПФЭ 24
Расчет коэффициентов регрессии. Коэффициенты регрессии (£>,) рассчитывают по алгоритму Йейтса. Схема расчета приведена в табл. 20.5. Результаты всех опытов (значения уь у2, уи) располагают в столбец обязательно в том порядке, в каком представлены первые 16 строк в матрице планирования (см. табл. 20.2). Для расчета коэффициентов регрессии значения уи следует округлить до целых значений.
Как видно из табл. 20.5, вычисляют сначала парные суммы у( + у2; у3 + у4 и т.д. для всех у„ , а затем разности у2 ~ Уь У 4 - Уз для всех У и, записывая результаты в столбец рядом со столбцом результатов у„. Чтобы не пропустить ни одного из значений уи, каждую пару после сложения и после вычитания отмечают точками. Таким образом, по окончании всех вычислений около каждой цифры у„ должны стоять две точки. Получают первый столбец (см. графу 7: 1-й этап расчета), в котором первые 8 чисел представляют собой парные суммы, а
Таблица 20.5
Схема расчета коэффициентов регрессии для ПФЭ 24 по алгоритму Йейтса
Номер опыта (среды) Матрица планирования Результаты опытов Расчет коэффициентов регрессии Обозначение эффектов
х, Х2 ХА Уп 1-й этап 2-й этап 3-й этап К b.
1 2 3 4 5 6 7 8 9 10 и 12
1 — - - - и й =yf +у2 W, = г, + v2 bo «1»
2 + - - - у2 Ь =Уз+У4 w2 = v3 + v4 bi a
3 - 4- - - Уз Гз =У5+Уб w3 = v5 + v6 Ь2 b
4 + 4- - - У4 1’4=у7+у8 % = Ъ + v8 Ь12 ab
5 - - 4- - Уз й> =У9+У1О »v5 = r9 + v10 Ь3 c
6 4- - 4- - Уб Уб=Уц+У12 w6 = vH + vI2 Ь,3 ac
7 - 4- 4- - У1 Й = У13 + У14 W2 = VI3 + VI4 Ь23 be
8 4- 4- 4- - У8 Ч = У15 + У16 = VI5 + V,6 Ь123 a be
9 - - - 4- У9 1’9 = у2 + У] w9 = v2 - V, Ь< d
10 4- - - 4- Уш йо = У4+Уз W!0 = V4 - V3 bl4 ad
11 - 4- - 4- Уп Й1 =У6+Уз wn = V6-V5 Ь1А bd
12 4- 4- - 4- У12 Й2=У8+У7 W,2= V8-V? Ь\2Л abd
13 - - 4- 4- Ув йз=Ую+У9 wi3 = 4o-v9 Ь34 cd
14 4- - 4- 4- У14 Й4 =У12 +У11 M'l4 = VI2-V„ b<34 acd
15 - 4- + 4- У15 Й5 =У14 +У13 wi5 = vM - vI3 Ь234 bed
16 4- 4- + 4- У16 йб =У16 +У15 ^6 = VI6 - V13 ЬЮ4 abed
вторые 8 чисел — парные разности результатов опытов. С числами этого столбца делают те же операции парного сложения и вычитания и получают следующий столбец (см. графу 8: 2-й этап) и т.д. Число этапов расчета должно быть равно числу факторов в плане эксперимента. Следовательно, для ПФЭ 24 следует получить 4 столбца (см. графы 7 —10 в табл. 20.5).
Чтобы проверить правильность расчетов, необходимо убедиться в справедливости равенств:
Е^« = йб •16; Е^« '16 = ЕКи • и и и
Результаты последнего шага (контрасты Ки, графа 10 в табл. 20.5) делят на число опытов в плане ПФЭ 24 N= 16 и получают коэффициенты регрессии (bh графа И). Определить, к каким эффектам они относятся, можно с помощью столбца «Обозначение эффектов» (графа 12 в табл. 20.5), в котором приведены условные обозначения строк в матрице планирования: 1-ю строку, соответствующую опыту, в котором все факторы исследовали на нижнем уровне, обозначают «1». В этой строке в столбце Ь, находится оценка Ьо в уравнении регрессии. Остальные обозначения получены в соответствии с тем, какой фактор находится в данной строке на верхнем уровне. Здесь использованы обозначения, часто встречающиеся в литературе по планированию эксперимента. Верхний уровень xj обозначают буквой о; х2 - b; х3- с, х4 - d. Поэтому во 2-й строке мы видим обозначение а, и в этой строке в столбце Ь, находится оценка Ь{. Точно так же, например, в 4-й строке, где в матрице планирования на верхнем уровне находятся факторы х, и х2, стоит обозначение ab, и это означает, что в этой строке в столбце коэффициентов регрессии Ь, находится оценка коэффициента Ь]2 (следует читать «Ь один—два», а не «Ь двенадцать»!).
Оценка значимости коэффициентов регрессии. Как было сказано выше, коэффициенты регрессии Ь, являются мерой влияния фактора на процесс. Оценка их значимости позволяет установить, какие из исследуемых факторов существенны (в статистическом смысле) для данного процесса. Если величина Ь, незначимо отличается от нуля, можно утверждать, что в нашем эксперименте изменение выхода процесса уи при изменении уровня соответствующего фактора Xj соизмеримо с ошибкой измерения уи.
Значимость коэффициентов регрессии проверяют двумя способами: графическим методом и с помощью обычного статистического анализа. При использовании графического метода абсолютные величины коэффициентов регрессии (| Ь,\) или соответствующих им контрастов (|Ки\~) ранжируют, т.е. располагают в порядке возрастания их абсолютной величины. Поскольку коэффициент Ьо не относится к какому-либо эффекту, его для ранжировки не используют. Для построения графика на оси абсцисс откладывают абсолютные величины bj или Ки, масштаб которых выбирают произвольно, а на оси ординат — их порядковые номера на расстояниях от начала координат, которые приведены в табл. 20.6.
На графике все незначимые коэффициенты должны оказаться на одной прямой, проходящей через начало координат. Коэффициенты, соответствующие значимым эффектам, заметно отклоняются от этой прямой, т. е. выделяются из совокупности остальных, меньших эффектов (т.е. из совокупности
Таблица 20.6
Данные для построения х/г нормального графика (ПФЭ 24)
Номер в ранжировке Расстояние от начала оси ординат, мм 1*1 или |A"J Номер в ранжировке Расстояние от начала оси ординат, мм 1*1 или [A7J
1 2 9 31
2 5 10 36,5
3 8 11 41,5
4 12 12 47,5
5 15,5 13 55,5
6 19 14 66
7 23 ' 15 86
8 27
нормально распределенных оценок ошибки воспроизводимости), и являются статистически значимыми.
Традиционный статистический анализ проводят в такой последовательности. Сначала рассчитывают построчные дисперсии:
shy} =
Е(л«-т«)2 к________
т -1
где т — число повторностей опыта в каждой строке, к = 1, 2, ..., т. Построчные дисперсии рассчитывают для всех 25 опытов в композиционном плане. Если т = 2, эта формула упрощается, так как уи1 -уи = - (y[l2 - уи), и достаточно рассчитать только уи1 - уи, а затем {yu} = 2(yul - у,,)2. При этом не следует округлять значения у и. Результаты представляют в виде табл. 20.7.
Находят дисперсию единичного значения как среднее арифметическое построч-
_ s2 {у} ных дисперсий:52 {у} = —---; дисперсию среднего значения 52{у}=-—.
Таблица 20.7
Последовательность расчета построчных дисперсий
Номер опыта УИ| - У„ (к, у,У 5<?{к} = 2(л1 - у,,)2’
1 2 3 25
Примечание. * При т = 2.
Значения критерия Стьюдента (/-критерия)
Число степеней свободы Уровни значимости Число степеней свободы Уровни значимости
5%-й 1%-й 5%-й 1 %-й
1 12,706 63,656 17 2,110 2,898
2 4,303 9,925 18 2,101 2,878
3 3,182 5,841 19 2,093 2,861
4 2,776 4,604 20 2,086 2,845
5 2,571 4,032 21 2,080 2,831
6 2,447 3,707 22 2,074 2,819
7 2,365 3,499 23 2,069 2,807
8 2,306 3,355 24 2,064 2,797
9 2,262 3,250 25 2,060 2,787
10 2,228 3,169 26 2,056 2,779
11 2,201 3,106 27 2,052 2,771
12 2,179 3,055 28 2,048 2,763
13 2,160 3,012 29 2,045 2,756
14 2,145 2,977 30 2,042 2,750
15 2,131 2,947 40 2,021 2,704
16 2,120 2,921 60 2,000 2,660
Дисперсию воспроизводимости коэффициентов регрессии находят по фор-
муле 52 {Ь,} = - М
10
, так как в ПФЭ 24 каждый коэффициент рассчитывали на
основе 16 значений уи.
Коэффициенты регрессии считают отличными от нуля, т. е. значимыми при 5%-м уровне значимости, если выполняется следующее неравенство:
где5{Д} = ^2 {bi} — средняя квадратичная ошибка определения коэффициентов регрессии в эксперименте; — критерий Стьюдента при 5%-м уровне значимости и числе степеней свободы f„ равном N(n - 1). Находят /-критерий по табл. 20.8.
Используя значимые коэффициенты, составляют уравнение регрессии. Полученное в факторном эксперименте уравнение регрессии связывает концентрации компонентов в среде с ростом бактерий в изученной области поверхности отклика.
Значимость коэффициентов регрессии при линейном члене указывает на то, что концентрация соответствующего компонента находится в лимитиру
ющей области, если коэффициент регрессии имеет знак «+», или в ингибирующей области, если коэффициент регрессии имеет знак «-».
Обработка результатов опытов по плану 2-го порядка
Расчет коэффициентов регрессии. Для расчета коэффициентов регрессии квадратичного уравнения (20.2) используют опыты 1 — 24.
Значение Ьо определяют по формуле
пЕл -зЕЕх£й
А. -_«______L_«____
где Е>’« — сумма всеху„ и ЕЕХ/Ь« = Ех1«й/ +ЕхлД« + Ехз«К + ЕХ4«Т«-и i и и и и и
Суммы х?иУи рассчитывают следующим образом:
Exi2 Уи = Kq + у17 + Ив;
ЕХ2 Уи = Ко + у19 + у2(Ъ
ЕХзЛ = К0 + Й1 + У22',
ХХ4Уи =^0 + У23 + У24-
ЗдесьKq = У] + у2 + Уз +... + Дб, т.е. это первое число в столбце, полученном на 4-м этапе расчетов коэффициентов Ь; для ПФЭ 24 по схеме Йейтса (см. графу 10 в табл. 20.5).
Таблица 20.9
Схема расчетов для определения в плане 2-го порядка
Номер опыта (среды) Матрица планирования Результаты опытов Расчеты
X, *2 *3 Х4 Уи *1 Х2 хз Х4
1 -1 -1 -1 -1 У1 -й -й ~Й -й
2 +1 -1 -1 -1 У2 -У2 —й -й -й
3 -1 + 1 -1 -1 Уз -Уз +Уз -Уз -Уз
4 +1 + 1 -1 -1 У4 -У4 +У4 -й -Й4
5 -1 -1 + 1 -1 Уз ~Уз -Уз +й -Уз
24 0 0 0 + 1 Й4 0 0 0 +У24
и
Коэффициенты регрессии при линейных членах находят по формуле ЪХшУи
гдеХЛ/«Л — алгебраическая сумма значений уи со знаками, определяемыми по графам матрицы планирования, соответствующим данному фактору. Таким образом, уи приобретает знак «+» или «-», если х, в данной строке равен +1 или -1, и становится равным 0, если х,- = 0. Результаты записывают в табл. 20.9.
Значения коэффициентов регрессии при квадратичных эффектах рассчитывают по формуле
ЗХк +5££х?у„
Ьи = 0,5£х? у„ - -----.
Значения коэффициентов регрессии при парных взаимодействиях by берут из графы Ь,в табл. 20.5 (расчеты для ПФЭ 24).
Оценка значимости коэффициентов регрессии. Для проверки значимости коэффициентов регрессии определяют их дисперсии воспроизводимости по формулам
S2 {Z>0} = 0,2295S2 {у};
S2 {/>,} = 0,56S2 {у};
S2 {6Й} = 0,396S2 {у};
S2 {^} = 0.0625S2 {у}.
Находят условия значимости коэффициентов регрессии:
М t0M\!$2{b0};
|4| toMXls2^};
N t0My]s2 [Ьиу,
IM {bv}.
Определив значимые коэффициенты, записывают уравнение регрессии, учитывая значимые линейные коэффициенты факторов и их взаимодействий и все квадратичные члены.
Проверка адекватности модели. Проверка необходима, чтобы убедиться в том, что полученное уравнение действительно с достаточной точностью описывает изучаемый процесс. Величина погрешности, с которой уравнение регрессии предсказывает результаты опытов, определяется дисперсией адекватности. Ее рассчитывают по формуле
8ad ~
Цуи-уи)2
и_______
N-N}
Последовательность расчета дисперсии адекватности
Номер опыта Матрица планирования Результаты ОПЫТОВ Уи Рассчитанные значения Уи Уи-Уи 1- - \2 (Л
А *2 хз Х4
1 2 3 25
£(л -у и? и
где уи - уи j — сумма квадратов отклонений экспериментальных значений уи от предсказанных значений уи , вычисленных по уравнению модели для всех тех опытов, которые были предусмотрены планом; (N- Nj) — число степеней свободы, которое равно числу опытов в плане N за вычетом числа коэффициентов bj в уравнении регрессии.
Расчеты делают в такой последовательности. Находят значения уи по уравнению регрессии. Чтобы вычислить уи для данной строки (опыта), умножают каждый коэффициент в уравнении на величину кодированной переменной (-1,0 или +1) для данного фактора, или произведения факторов в этой строке, и суммируют полученные значения. Алгебраическая сумма соответствует уи для данной строки (опыта). Затем определяют отклонения экспериментальных значенийуи от рассчитанных уи , т.е. уи-уи-> возводят каждую разность в квадрат, суммируют квадраты отклонений. Результаты расчетов записывают в табл. 20.10. Записывают условие неадекватности:
где jF0 05(^,— критерий Фишера при 5%-м уровне значимости. Значения F находят по табл. 20.11, учитывая число степеней свободы для дисперсии воспроизводимости среднего значения^ = N(n - 1) и число степеней свободы f = = N - Nx для дисперсии неадекватности.
Если полученная дисперсия неадекватности превышает дисперсию воспроизводимости среднего результата более чем в Араз, значит, уравнение модели непригодно для описания изучаемого процесса. В ином случае принимают, что найденное уравнение модели адекватно описывает процесс («уравнение адекватно»), Убедившись в адекватности модели, приступают к вычислению оптимальных значений для каждого фактора.
Расчет оптимальных уровней факторов. При расчете координат точки оптимума по уравнению 2-й степени (20.2) надо иметь в виду, что, как было ска-
Значения критерия Фишера для 5%-го уровня значимости
Число степеней свободы для большей дисперсии (числителя) ft Число степеней свободы для меньшей дисперсии (знаменателя) f2
1 2 3 4 5 6 7 8 12 24
3 10,13 9,55 9,28 9,12 9,01 8,94 8,89 8,85 8,74 8,64
4 7,71 6,94 6,59 6,39 6,26 6,16 6,09 6,04 5,91 5,77
5 6,61 5,79 5,41 5,19 5,05 4,95 4,88 4,82 4,68 4,53
6 5,99 5,14 4,76 4,53 4,39 4,28 4,21 4,15 4,00 3,84
7 5,59 4,74 4,35 4,12 3,97 3,87 3,79 3,73 3,57 3,41
8 5,32 4,46 4,07 3,84 3,69 3,58 3,50 3,44 3,28 3,12
9 5,12 4,26 3,86 3,63 3,48 3,37 3,29 3,23 3,07 2,90
10 4,96 4,10 3,71 3,48 3,33 3,22 3,14 3,07 2,91 2,74
11 4,84 3,98 3,59 3,36 3,20 3,09 3,01 2,95 2,79 2,61
12 4,75 3,89 3,49 3,26 з,и 3,00 2,91 2,85 2,69 2,51
13 4,67 3,81 3,41 3,18 3,03 2,92 2,83 2,77 2,60 2,42
14 4,60 3,74 3,34 з,п 2,96 2,85 2,76 2,70 2,53 2,35
15 4,54 3,68 3,29 3,06 2,90 2,79 2,71 2,64 2,48 2,29
16 4,49 3,63 3,24 3,01 2,85 2,74 2,66 2,59 2,42 2,24
17 4,45 3,59 3,20 2,96 2,81 2,70 2,61 2,55 2,38 2,19
18 4,41 3,55 3,16 2,93 2,77 2,66 2,58 2,51 2,34 2,15
19 4,38 3,52 3,13 2,90 2,74 2,63 2,54 2,48 2,31 2,11
20 4,35 3,49 3,10 2,87 2,71 2,60 2,51 2,45 2,28 2,08
22 4,30 3,44 3,05 2,82 2,66 2,55 2,46 2,40 2,23 2,03
24 4,26 3,40 3,01 2,78 2,62 2,51 2,42 2,36 2,18 1,98
26 4,23 3,37 2,98 2,74 2,59 2,47 2,39 2,32 2,15 1,95
28 4,20 3,34 2,95 2,71 2,56 2,45 2,36 2,29 2,12 1,91
30 4,17 3,32 2,92 2,69 2,53 2,42. 2,33 2,27 2,09 1,89
32 4,15 3,29 2,90 2,67 2,51 2,40 2,31 2,24 2,07 1,86
34 4,13 3,28 2,88 2,65 2,49 2,38 2,29 2,23 2,05 1,84
36 4,11 3,26 2,87 2,63 2,48 2,36 2,28 2,21 2,03 1,82
38 4,10 3,24 2,85 2,62 2,46 2,35 2,26 2,19 2,02 1,81
40 4,08 3,23 2,84 2,61 2,45 2,34 2,25 2,18 2,00 1,79
50 4,03 3,18 2,79 2,56 2,40 2,29 2,20 2,13 1,95 1,74
60 4,00 3,15 2,76 2,53 2,37 2,25 2,17 2,10 1,92 1,70
80 3,96 3,11 2,72 2,49 2,33 2,21 2,13 2,06 1,88 1,65
100 3,94 3,09 2,70 2,46 2,31 2,19 2,10 2,03 1,85 1,63
зано выше, квадратичная функция у = b0 + bjXj + ЬцХ? может иметь либо максимум, либо минимум в зависимости от того, какой знак имеет коэффициент Ьц при х]. Знак «+» показывает, что величина у в центре эксперимента (при х, = 0) меньше, чем следует ожидать по линейной модели у = Ьо + Л,х,. Таким образом, точка экстремума этой функции представляет собой минимум. Естественно, что при поиске максимума функции расчет величины х,, соответствующей минимуму функции, не имеет практического смысла.
Вообще говоря, появление в уравнении регрессии квадратичных членов с Ьц > 0 свидетельствует о неудачном выборе интервала варьирования для соответствующих факторов, и эксперимент следовало бы повторить, изменив эти интервалы с учетом результатов, полученных в исходном эксперименте. Однако, поскольку в данном случае речь идет об учебной задаче практикума и время работы ограничено, мы считаем возможным использовать некоторый искусственный прием. Предлагается фиксировать факторы, для которых получены положительные квадратичные коэффициенты Ьц, на уровнях, которые были у этих факторов в наилучшем варианте исходной серии опытов. Поскольку при использованном нами подходе наилучший результат чаще всего достигается в центре эксперимента, достаточно подставить в уравнение регрессии значение х,- = 0 для всех х;, у которых Ьц > 0.
По уравнению регрессии для оставшихся факторов Xj находят частные производные и, приравнивая их к 0, составляют систему линейных уравнений ДЛЯ Xj.
—~ — b\ + 2Z?j]X] т Ь^2х2 + bl3x3 + bi4x4 — 0; дхх
~— — Ь2 + 2/?; 2Л'| + Ь22Х2 + Ф3Х3 + />24X4 = 0; дх2
~~ = b3 + 2bl3x{ + b23x2 + b33x3 + b34x4 = 0; Sx3
——- = b4 + 2b[4X\ + b24x2 + b34x3 + b44x4 — 0. Sx4
Решение этой системы дает величины XjOpt, т. е. координаты точки максимума в кодированных переменных. Решение системы линейных уравнений для 3 или 4 переменных представляет собой не слишком сложную задачу, но требует довольно много времени, если пользоваться для необходимых вычислений обычным калькулятором. Поэтому в данном случае предпочтительнее обращаться к помощи персонального компьютера. При этом не требуется обращение к каким-либо специализированным статистическим пакетам, поскольку достаточно использовать упомянутый выше табличный процессор EXCEL.
Переход к натуральным переменным осуществляется по формуле
Xjopt ~ Xjopt'hj + XjG,
где xjV — значение натуральной переменной в центре эксперимента; Ху — величина единицы варьирования в натуральных переменных (см. табл. 20.1).
2. Опыты по плану ПФЭ З3
В целях проверки полученных результатов и уточнения оптимальной области в этой серии опытов варьируют те факторы, для которых в предыдущих экспериментах были определены концентрации, обеспечивающие максимальный прирост оптической плотности культуры. Иными словами, выбирают 3 фактора, для которых были получены значимые отрицательные коэффициенты при квадратичных членах Ьи.
Значения этих концентраций принимают за средний (0) уровень. Единицу варьирования каждого фактора выбирают такой же, как в опытах по композиционному плану. Если при анализе результатов 1-й серии опытов было найдено, что какой-либо фактор имеет положительный квадратичный эффект, для него в качестве среднего уровня выбирают уровень в лучшем варианте опытов 1-й серии. Данные рекомендации следует рассматривать как предварительные. Окончательное решение принимается после обсуждения результатов 1-й серии опытов совместно с преподавателем.
План ПФЭ З3 приведен в табл. 20.12. В качестве контроля используют 2 — 3 среды композиционного плана, в которых был отмечен наилучший рост бактерий. Приготовление сред, подготовка посевного материала, проведение посева, условия и продолжительность культивирования бактерий такие же, как в опытах по композиционному плану Д4.
Таблица 20.12
Матрица ПФЭ З3
Номер опыта (среды) Матрица планирования Номер опыта (среды) Матрица планирования
х\ Х2 хз Х, Х2 хз
1 -1 -1 -1 15 +1 0 0
2 0 -1 -1 16 -1 +1 0
3 +1 -1 -1 17 0 +1 0
4 -1 0 -1 18 +1 +1 0
5 0 0 -1 19 -1 -1 +1
6 +1 0 -1 20 0 -1 +1
7 -1 +1 -1 21 +1 -1 +1
8 0 +1 -1 22 -1 0 +1
9 +1 +1 -1 23 0 0 +1
10 -1 -1 0 24 +1 0 +1
11 0 -1 0 25 -1 +1 +1
12 +1 -1 0 26 0 +1 +1
13 -1 0 0 27 +1 +1 +1
14 0 0 0
Обработка результатов ПФЭ З3
Расчет коэффициентов Ви. Результаты опытов выписывают в столбцы (графы) табл. 20.13 в том порядке, в котором расположены соответствующие строки в матрице ПФЭ З3 (см. табл. 20.12). Все 27 значенийуи разбивают на 9 последовательных групп по 3 значения в каждой. После этого проводят следующие расчеты (табл. 20.13):
1. Для каждой группы подсчитывают сумму трех значений уи и записывают ее в следующий столбец (графа 6 в табл. 20.13). Таким образом, заполняют первые 9 мест в этом столбце.
2. Рассчитывают разности между третьим и первым значениями в каждой группе. Полученные числа записывают в тот же столбец, заполняя следующие 9 мест в этом столбце.
3. Для каждой группы рассчитывают величины у, - 2у2 + у3 и записывают их в графе 6 на оставшиеся 9 мест.
Полученный столбец снова разбивают на 9 групп по 3 значения в каждой и с ними проводят те же операции в том же порядке, заполняя результатами расчетов следующий столбец (графа 7). С числами этого столбца поступают точно так же, и в результате получают 27 чисел (графа 8 в табл. 20.13), которые представляют собой суммы, соответствующие эффектам факторов (линейным и квадратичным) и эффектам взаимодействий. Эти суммы, называемые контрастами (как и в опытах по плану ПФЭ 24), обозначаются Ки. Чтобы получить коэффициенты регрессии Ви, нужно разделить соответствующие контрасты Ки на делители du, приведенные в столбце 9.
Для определения того, к какому именно эффекту относится тот или иной контраст, введем новые обозначения уровней факторов: нижний уровень обозначим «0», средний — «1», а верхний — «2». Тогда план эксперимента может быть переписан так, как это показано в графе 5 табл. 20.14. Полученные обозначения уровней представляют собой степени независимых переменных в произведениях вида: х“х2*з- Однако следует иметь в виду, что коэффициенты Ви, рассчитанные по алгоритму Йейтса (см. табл. 20.13), относятся к так называемым ортогональным полиномам Д. При этом
Ро = хр = 1; Д = х,.; Д = Зх,2 - 2.
Например, в 1-й строке в табл. 20.14 произведение х“х^Хз дает x^x®. Это указывает, что в этой строке в графе 2 оценка В} относится к полиному Ро. Соответствующее обозначение поставим в 1-й строке графы 8 в табл. 20.14. Во второй строке получим: х{х°х° = хь поэтому в графе 8 во 2-й строке поставим обозначение Д. Аналогично в 3-й строке xlx2x3=xl, т.е. Ви в этой строке относится к квадратичному эффекту 1-го фактора. В соответствии с принятыми обозначениями в графе 8 в 3-й строке поставлено обозначение ДР
Уравнение регрессии с ортогональными полиномами выглядит так:
у = Д + ДД + ДД1 + ДД + ДД Д + ДД1Д + ••• + Д?Д1Д2Дз-
Если подставить в это уравнение х,- вместо Д и 3xf - 2 вместо Д, раскрыть скобки и сделать приведение подобных, мы получим уравнение регрессии, связывающее величину у с кодированными переменными х,. Проверка правильно-
Порядок расчетов коэффициентов рецессии по результатам ПФЭ З3
Номер опыта Матрица планирования Результаты опытов Расчет коэффициентов регрессии
и Х2 И Уи 1-й шаг 2-й шаг Ku Bu
1 2 3 4 5 6 7 8 9 10
1 - - - Й И = И + Уз + У2 Wj = V, 4- У2 + V3 27
2 0 - - У2 v2 = >4 + >5 + >б W2 = v4 + v5 + V6 18
3 4- - Уз И = Уз + Уя + У9 W3 = V7 + vg + v9 54
4 - 0 - Уь v4 = Ио + >11 + >12 W4 = V10 + Vu + H2 18
5 0 0 - Уз И = Из + >14 + >15 w5 = v13 + v14+ v15 12
6 4- 0 - Уб И = >16 + >17 + >18 W6= V16 + H7+ Й8 36
7 - 4- - Уз У7 = >|9 + >20 + >21 w7= v19 + v20+v21 54
8 0 4- - .Й И = У22 + У23 + >24 W8 = V22 + V23 + V24 36
9 4- + - У9 v9 = >25 + У26 + >27 W9 = V25 + V26+V27 108
10 - - 0 Йо Ио = Уз + И W!0=V3-V1 18
И 0 - 0 Й1 И1 = >б+>4 WH = H- V4 12
12 4- - 0 Й2 И2 = У9 + Уз w12 = V9 - V7 36
13 - 0 0 Из Из = >12 + Ио И'13=Й2-Й0 12
14 0 0 0 Й4 И4 = >15 +ЙЗ VVI4 = V15 “ V13 8
15 4- 0 0 Й15 Из = >18 + Йб И'15 = Й8-Й6 24
16 - + 0 Йб Иб = >21 + >19 W!6 = V2I - Й9 36
17 0 + 0 Й17 +17 = У14 + У22 VV17 — ^24 “ V22 24
18 + + 0 >18 Ив = У21 + >25 WI8 = V27 - V25 72
19 - - 4~ >19 Иэ - >1 + 2>г + Уз W19=V|-2V2 + V3 54
20 0 - 4~ Узо +20 = >4 + 2Й> + Уб W20=V4-2V5+V6 36
21 + - 4~ У11 +21 = Уз +-2>8 + Уд W21 = V7 - 2VS + V9 108
22 - 0 + У22 v22 - Ио + 2 >11 + >12 W22 ~ V10 " 2VI1 + V12 36
23 0 0 4- >23 +23 = >13 + 2Й4 + >15 >% = VI3'2VI4+ V!5 24
24 4- 0 4- >24 +24 = Иб + 2>17 + >18 VV24= Иб-2Й7+И8 72
25 - 4- 4- >25 +25 = >19 + 2 >20 + >21 W25=V19-2V20+V21 108
26 0 4- 4- >26 +26 = У12 + 2Из + р24 VV26 - V22 ~ 2v23 + V24 72
27 4- 4- 4- >27 }23 = Из + 2 >’26 + >27 W27=V25-2V26 + V27 216
сти расчетов сводится к вычислению суммы всех 27 значений Ви. Эта сумма, рассчитанная с точностью по крайней мере до 4 —5 знаков после запятой, должна быть равна величине у21, т. е. последнему значению в графе с исходными данными (графа 5 в табл. 20.13).
Оценка значимости коэффициентов Ви. Определяют величину дисперсии воспроизводимости S2 {у}, как это делали в первой серии опытов, т. е. рассчитывают построчные дисперсии для всех 27 опытов, находят дисперсию еди-2 _ £2{у}
ничного результата: S2 {у} = ———— и дисперсию среднего: S2 {у} =-----.
N т
Затем рассчитывают величины: |5„| = для всех Ки, кроме К], который, как и в предыдущей серии опытов, в плане ПФЭ 24, представляет собой сумму исходных результатов опытов. Значения |S„| записывают в графу 4 табл. 20.14. Отличными от 0 при заданном уровне значимости следует считать те коэффициенты Ви, для которых выполняется неравенство
Проверку значимости величин Ви можно провести и с помощью '/з-нор-мального графика. Для этого нужно ранжировать значения Blt в порядке возра-
Таблица 20.14
Расчеты для статистического анализа результатов ПФЭ З3
Номер опыта £ и Степени полиномов Р. Обозначения эффектов Ь. для квадратичной модели
а Р Y
1 2 3 4 5 6 7 8 9
1 В\ — 0 0 0 Ре b0= Bt - 2(Д + В1 + Т?19)
2 в2 4,243 1 0 0 в, й,=Б2
3 В. 7,348 2 0 0 = зд
4 В4 4,243 0 1 0 Л va
5 В5 3,464 1 1 0 дд Z>12 = В5
6 В6 6,000 2 1 0 ЛЛ
7 В-, 7,348 0 2 0 Вц ^22 = ^В7
8 В* 6,000 1 2 0
9 В9 10,392 2 2 0 ВцВгг
10 В10 4,243 0 0 I Р. ~ В10
11 вц 3,464 1 0 1 РА ^1з = Btl
12 В» 6,000 2 0 1
Номер опыта в„ 1Д1 Степени полиномов Р Обозначения эффектов Д для квадратичной модели
а Р У
1 2 3 4 5 6 7 8 9
13 Дз 3,464 0 1 1 ДД Дз = Дз
14 Д4 2,828 1 1 1 ддд
15 Д5 4,899 2 1 1 д.дд
16 Д6 6,000 0 2 1 р р 22 3
17 Д7 4,899 1 2 1 Р Р Р Г 22* 3
18 Д8 8,485 2 2 1 Д.ДЛ
19 д. 7,348 0 0 2 Дз Дз= ^д?
20 До 6,000 1 0 2 ДДз
21 д. 10,392 2 0 2 Р р 11 33
22 д2 6,000 0 1 2 р р * 33
23 Дз 4,899 1 1 2 Р Р Р 11 2* 33
24 Д< 8,485 2 1 2 Р Р Р 11 2* 33
25 Дз 10,392 0 2 2 Р Р 22* 33
26 До 8,485 1 2 2 Р Р Р * 1 22^ 33
27 Д7 14,697 2 2 2 Р Р Р * 11 22* 33
стания их абсолютной величины и затем нанести на график, на котором величины |Д| откладываются по оси абсцисс в произвольном линейном масштабе, а соответствующие номера в ранжировке наносятся на ось ординат на расстояниях от начала координат, которые приведены в табл. 20.15.
Величины коэффициентов Ви записывают в 10-й столбец табл. 20.13, приравнивая к 0 коэффициенты при незначимых эффектах. Составляют уравнение регрессии, включающее только члены с Ви, значимо отличными от 0. Используя коэффициенты при значимых эффектах, записывают уравнение регрессии в виде:
у = Д + В2Р\ + В3Рп + В4Р2 + BSPXP2 + ... и т.д.
Как было указано выше, Р,- = х,-; Рц = Зх? - 2. Заменяют в уравнении Р, и Р„ соответствующими значениями. Получают
у = В} + В2х{ + В3 (Зху - 2) + В4х2 + Р5Х|Х2 +... и т.д.
Возможна ситуация, когда наряду с эффектами 1-го и 2-го порядка оказываются значимыми эффекты более высоких порядков, так что в уравнение регрессии нужно добавить, например, члены вида: Р6РцР2 (член 3-й степени) или даже В№Р{{Р22Р3 (член 5-й степени). Это означает, что квадратичная модель заведомо неадекватна, как описание экспериментальных данных. В этом случае
Таблица 20.15
Данные для построения полунормального графика (ПФЭ З3)
Номер в ранжировке Расстояние от начала оси ординат, мм Ви Номер в ранжировке Расстояние от начала оси ординат, мм Ви
1 1 14 28
2 3 15 30,5
3 5 16 33
4 7 17 35,5
5 9 18 39
6 10,5 19 42
7 13 20 45,5
8 15 21 49,5
9 17 22 53,5
10 19 23 58,5
11 21 24 65,5
12 23,5 25 74,5
13 26 26 93
поиск экстремума на поверхности отклика представляет собой достаточно сложную задачу даже при использовании компьютера. Однако, как было сказано выше, имеет смысл найти максимум для уравнения регрессии 2-й степени, поскольку он может находиться ближе к «истинному» оптимуму, чем, к примеру, условия опытов с верхними или нижними уровнями факторов.
Уточнение оптимального состава среды по результатам ПФЭ З3. Для получения квадратичного уравнения регрессии следует выписать из графы 9 табл. 20.14 приведенные в ней значения коэффициентов bh bih by и составить на их основе систему уравнений (см. первый этап работы). Решают полученную систему и находят кодированные значения переменных для точки максимума на поверхности отклика. Определяют реальные концентрации каждого фактора, представляющие состав среды, который можно считать оптимальным на данном этапе исследования.
Полученные результаты полезно проиллюстрировать с помощью графиков. Например, для любых 2 факторов при фиксированных концентрациях других компонентов можно построить трехмерный график, отложив на горизонтальных осях реальные значения концентраций соответствующих компонентов среды, а на вертикальной оси — величины оптической плотности культуры.
Результаты оформляют в виде отчета с подробным описанием всех экспериментальных и вычислительных процедур, обсуждением результатов и выводами.
Материалы, оборудование, посуда, реактивы
Пробирки широкие — 60; пипетки на 1, 2, 5, 10 мл — по 2; цилиндры мерные на 25 мл — 2; реактивы для приготовления сред (см. текст).
План выполнения задачи
Для выполнения задачи необходимо 45 — 50 ч.
Занятие 1. Лекция. Расчет сред для опытов по композиционному плану. Приготовление и стерилизация МПА в пробирках.
Занятие 2. Посев бактерий на скошенный МПА. Приготовление и стерилизация сред. Подготовка к стерилизации сред и пипеток, водопроводной воды в колбах.
Занятие 3. Посев культуры в опытные среды. Установка пробирок на качалку. Лекция.
Занятие 4. Лекция. Оценка роста культуры нефелометрически. Расчет коэффициентов регрессии по результатам ПФЭ 24 и оценка их значимости.
Занятие 5. Лекция. Расчет коэффициентов регрессии в уравнении 2-го порядка и оценка их значимости. Проверка адекватности модели и расчет оптимальной области.
Занятие 6. Обсуждение результатов опытов по композиционному плану. Расчет сред для постановки ПФЭ З3.
Занятие 7. Пересев культуры на скошенный МПА. Приготовление и стерилизация сред для ПФЭ З3.
Занятие 8. Посев культуры в опытные среды. Установка пробирок на качалку. Лекция.
Занятие 9. Лекция. Оценка роста культуры нефелометрически.
Занятие 10. Расчет коэффициентов регрессии, определение значимости эффектов и уточнение оптимальной области по результатам ПФЭ З3.
Занятие 11. Оформление и обсуждение результатов задачи.
Глава 21
ПЕРИОДИЧЕСКОЕ И НЕПРЕРЫВНОЕ КУЛЬТИВИРОВАНИЕ МИКРООРГАНИЗМОВ
При выращивании микроорганизмов в периодической культуре условия, в которых происходит их рост, меняются: увеличивается плотность популяций, уменьшается концентрация субстратов и ростовых факторов. Для многих исследований желательно, чтобы культура длительное время находилась в одной фазе роста при постоянной концентрации субстратов и неизменных прочих условиях. К этим условиям можно в какой-то мере приблизиться, многократно перенося микроорганизмы (через короткие промежутки времени) в новую питательную среду. Той же цели можно достичь, вводя новую питательную среду в сосуд для культивирования, удаляя одновременно соответствующее количество микробной суспензии. Такой метод положен в основу непрерывного культивирования микроорганизмов в хемостатах и турбидостатах. Периодическую культуру можно рассматривать как замкнутую систему, подобную многоклеточному организму, которая в своем развитии проходит четыре фазы — начальную, экспоненциальную, стационарную и фазу отмирания (юность, расцвет, старение и смерть). Условия существования культуры во всех этих фазах различны, а автоматическое регулирование невозможно. Непрерывная культура представляет собой открытую систему, стремящуюся к установлению динамического равновесия. Фактор времени в ней в известной мере исключается. Для организмов создаются неизменные условия среды, а установка легко поддается автоматическому регулированию.
Хемостат представляет собой прибор для непрерывного культивирования микроорганизмов. Он состоит из сосуда-культиватора (ферментера), в который постоянно поступает свежая среда. Благодаря перемешиванию питательных веществ, поступающих с новыми порциями раствора, а также подаче воздуха при необходимости, в ферментере создаются оптимальные концентрации питательных субстратов и кислорода, необходимых для роста микроорганизмов. По мере поступления в сосуд свежей среды из сосуда удаляется такой же объем микробной суспензии.
Турбидостат основан на поддержании постоянной плотности суспензии микроорганизмов в ферментере. Фотоэлемент, измеряющий мутность микробной суспензии, через систему специальных реле регулирует поступление в ферментер свежего питательного раствора. В сосуде для культивирования все питательные вещества содержатся в избытке и скорость роста микроорганизмов приближается к максимальной.
21.1. ПАРАМЕТРЫ МИКРОБНОГО РОСТА
Многие одноклеточные микроорганизмы размножаются делением пополам и их число в оптимальных условиях растет в геометрической прогрессии: 2°—21 — 22—... — 2". Если на единицу объема растущей культуры приходится 7V0 клеток, то после п делений число клеток станет N = No 2". Логарифмируя, получим lgN = lgNo + nlg2, откуда число клеточных делений:
IgA'-lgA'o lg2
(21.1)
Число клеточных делений в единицу времени называют коэффициентом скорости деления (К) и определяют по формуле
п IgTV-lgTVp 3,22(lgjV-lgiV0) t-h t-t0
(21.2)
Время, требующееся для одного цикла делений, или время генерации равно:
= ~ = 1 п ~ V'
(21.3)
Скорость прироста клеток пропорциональна начальной их концентрации.
Коэффициент пропорциональности (ц.) называют удельной скоростью роста. Он различен для разных культур микроорганизмов и даже для одного микроорганизма может меняться в зависимости от условий выращивания клеток.
При сбалансированном росте прирост числа клеток или биомассы в единицу времени рассчитывается по формулам
dN dX
= цА и — = щГ, dt dt
(21-4)
где N — число клеток в 1 мл; X — масса клеток в 1 мл; t — время; ц — удельная скорость роста. Интегрируя, получаем
ln7V-ln7V0 = p(/-/0), (21.5)
а после перехода к десятичным логарифмам:
2,303
t-t0
(21.6)
Например, если культура содержит 104 клеток в 1 мл в момент времени 10 и 108 клеток в 1 мл через 4 ч, то
(8-адзоз и 4
Взаимосвязь между ц (удельной скоростью роста) и g (временем генерации) можно вывести из вышеприведенных уравнений, так как, если рассматриваемый интервал времени будет д, то Л'= 2N0 (количество клеток в популяции увеличилось вдвое за одну генерацию). Подставляя эти соотношения в уравнение (21.5), получаем
In 2 0,693
В =---=-------
g g
(21.7)
Для выбранного примера g = —---_ _л---j. е. время удвоения числа клеток в
р 2,303 культуре около 20 мин.
Рассмотренные уравнения предопределяют линейную зависимость между логарифмом числа клеток (или любым другим измеряемым параметром куль
туры) и временем с наклоном прямой, соответствующим , отсекающей на ординате отрезок, равный lg7V0. Потенциируя уравнение (21.6), можно написать его в экспоненциальной форме:
N= No-1(Н' - 'o’/2-303.
(21-8)
В этом уравнении выражена экспоненциальная связь между числом клеток (или концентрацией биомассы) и временем. О популяции микроорганизмов, развитие которой подчиняется этим уравнениям, говорят, что она находится в экспоненциальной фазе роста. Такой рост обычно долго не поддерживается, дальше начинаются несбалансированный рост, затем стационарная фаза и фаза отмирания.
В некоторых случаях кинетика микробного роста является линейной. В этих условиях скорость прироста компонентов популяции постоянна:
t/7V , х
—— = c(const), dt v ’
или, интегрируя,
N=c-t.
(21-9)
(21.10)
Линейной функцией времени здесь является число клеток, а не логарифм этого числа. Это бывает, если поступление какого-либо компонента среды к клеткам ограничено.
21.2. ЭФФЕКТИВНОСТЬ РОСТА МИКРООРГАНИЗМОВ, ЭКОНОМИЧЕСКИЙ КОЭФФИЦИЕНТ
Если рост микроорганизмов в культуре ограничен количеством внесенного субстрата, то между начальной концентрацией внесенного в среду лимитирующего субстрата и полученным общим урожаем существует постоянная линейная зависимость (при условии ограничения роста одним параметром). Поэтому масса клеток, образованная на единицу использованного субстрата, представляет собой величину, которую называют экономическим коэффициентом (или выходом биомассы) — Y. Величина Y может быть вычислена из определения количества использованного субстрата и общего урожая по уравнению
Yx/S = ^\ (21.11)
где X — масса сухого вещества клеток (г/мл культуры), вступившей в стационарную фазу роста; Хо — масса сухого вещества клеток в 1 мл среды после инокуляции; 5 — количество использованного лимитирующего субстрата.
Различают также понятие молярного экономического коэффициента, которое определяется как количество биомассы (г), образованное на 1 моль использованного субстрата. Близко к этому понятию стоит коэффициент выхода биомассы в расчете на количество использованной АТФ (Ух/атф)- Этот параметр определяется по формуле
где п — число молей АТФ, образующихся при расщеплении 1 моля энергетического субстрата; М— молекулярная масса (г); MY, — экономический коэффициент в молях, когда YE выражается в граммах образованной сухой биомассы на 1 г использованного энергетического субстрата.
21.3. ВЛИЯНИЕ КОНЦЕНТРАЦИИ ПИТАТЕЛЬНЫХ ВЕЩЕСТВ НА СКОРОСТЬ РОСТА КУЛЬТУР
Кривые, отражающие зависимость скорости роста от концентрации питательных веществ, представляют собой гиперболы и описываются уравнением
5
Р — Ртах ла с- ’ (21.12)
+ О
где ц — удельная скорость роста при концентрации лимитирующего субстрата 5; Ртах — скорость роста при насыщающей концентрации субстрата; Ks — вели
чина, аналогичная К„, в уравнении Михаэлиса—Ментен, т.е. та концентрация субстрата, при которой ц достигает половины максимального значения. Величина К$ наряду с величинами У и цтах — важнейшие параметры, характеризующие рост микрорганизмов в хемостатах и в периодической культуре.
21.4. ПРОГНОЗИРОВАНИЕ УСЛОВИЙ НЕПРЕРЫВНОГО
КУЛЬТИВИРОВАНИЯ
Объем культуры в хемостате поддерживается на постоянном уровне путем непрерывного отлива части культуры и заполнения освободившегося объема культиватора свежей средой. Задача количественной теории хемостата состоит в том, чтобы предсказать значение скорости роста, концентраций биомассы и субстрата в различных условиях. Пусть ц — удельная скорость роста; V — объем культуры; F — скорость подачи свежей среды. Значение F/V = D известно как скорость разбавления. Оно характеризует скорость потока среды на единицу объема. Увеличение концентрации биомассы можно рассчитать по уравнению баланса биомассы:
общее увеличение биомассы = прирост - отток.
Для полной культуры при бесконечно малом промежутке времени dt этот баланс будет выглядеть следующим образом:
VdX = VpXdt - FXdt. (21.13)
Поделив это уравнение на Vdt, получим:
dX/dt = (у - D)X. (21.14)
В стационарном состоянии, когда dX/dt = 0, мы имеем у. = D, т. е. скорость роста равна скорости разбавления.
21.5. КОНЦЕНТРАЦИЯ БИОМАССЫ И ЛИМИТИРУЮЩЕГО РОСТ СУБСТРАТА
Баланс для лимитирующего рост субстрата дается уравнением:
общее увеличение субстрата = поступление - отток - субстрат, использованный на рост.
Для бесконечно малого промежутка времени dt этот же баланс для полной культуры выражается уравнением
VdX = FSr dt-FS- VyXdt/Y, (21.15)
где У— экономический коэффициент. Следовательно,
dX/dt = D(S,. - S) - yX/Y. (21.16)
В стационарном состоянии dX/dt = dS/dt = 0, тогда значения стационарных концентраций 5 и X даются уравнениями
Рис. 21.1. Области роста культур при выращивании в хемостатах и турбидостатах.
Объяснение в тексте
(21.17)
(ц-Т>)Х = 0
и
Я(5г-5)-цХ/У = 0, (21.18)
где знак «~» означает значение в стационарном состоянии. Чтобы получить X и 5, подставим выражение для удельной скорости роста:
И =цтах5/(^+5). (21.19)
Подставив в уравнение (21.19) ц = D, получим уравнение для стационарного состояния:
S = KsD/^-D), (21.20)
а из уравнения (21.18) при подстановке ц = D получим:
X = Y(Sr-S) = Y{Sr-KsD/^maK-D)}. (21.21)
В этом уравнении выражена основная зависимость между концентрацией клеток в стационарном состоянии (У) и концентрацией лимитирующего субстрата в ферментере (5). Хемостаты обычно работают в областях кривых по Хи 5 (рис. 21.1), не доходящих до прерывистой линии, турбидостаты — в областях, близких к критическим уровням разбавления, т.е. когда D ~ цтах.
21.6. УРАВНЕНИЕ МАТЕРИАЛЬНОГО БАЛАНСА И РАСЧЕТ
СКОРОСТИ ПРОТОКА ПРИ НЕПРЕРЫВНОМ КУЛЬТИВИРОВАНИИ
Для перехода от периодического процесса культивирования клеток к непрерывному необходимо построить график зависимости продуктивности периодического процесса по выходу биомассы (или оптической плотности абсолютного прироста биомассы за единицу времени, т. е. dX/dt от плотности популяции X(рис. 21.2).
Касательная к кривой роста характеризует скорость роста клеток в экспоненциальной фазе. Тангенс угла наклона этой касательной к оси абсцисс представляет собой величину максимума удельной скорости роста в условиях перио-
Рис. 21.2. Пример графика расчета продуктивности периодического процесса по выходу биомассы: а — Ь — лаг-фаза: d—c — экспоненциальная фаза
дического культивирования. Прямые от начала координат, проведенные через точки кривой роста, называют рабочими линиями. Точка пересечения рабочей линии с кривой роста соответствует состоянию динамического равновесия при концентрации клеток, равной величине отрезка, отсекаемого перпендикуляром, опущенным из этой точки на ось абсцисс. Тангенс угла наклона рабочей линии к оси абсцисс равен скорости разбавления в одном из состояний динамического равновесия. При этом числовые значения при определении тангенса угла наклона рабочей линии к оси абсцисс берутся из числовых значений dX/dt (величина отрезка перпендикуляра, опущенного на ось абсцисс из точки пересечения соответствующей рабочей линии с кривой роста) и X (величина отрезка, отсекаемого от оси абсцисс этим перпендикуляром), в тех же единицах. Например:
1 млрд 1,8 млрд
tgQ - -0,2ч-1; tgQ2 =-^ИЧ_ = 0,4ч").
5,35 млрд 4,5 млрд
мл мл
В приведенном графике (рис. 21.2) в расчетах можно вместо dX/dt и X брать D (значение оптической плотности) и dD/dt.
21.7. ГРАФОАНАЛИТИЧЕСКИЙ МЕТОД РАСЧЕТА УСЛОВИЙ НЕПРЕРЫВНОГО КУЛЬТИВИРОВАНИЯ МИКРООРГАНИЗМОВ
Для предсказания изменения концентрации продуктов метаболизма при непрерывном культивировании микроорганизмов существует графоаналитический метод, принцип которого состоит в сопоставлении графиков зависимости накопления биомассы и продукта метаболизма от времени культивирования с кривой продуктивности по выходу биомассы.
По данным периодического культивирования строят график зависимости образования биомассы и накопления продукта от времени культивирования и на этот же график в правой половине наносят кривую зависимости продуктивности культуры по выходу биомассы от ее концентрации, т. е. dX/dt от Хв тех же единицах.
Рис. 21.3. Пример графоаналитического расчета параметров непрерывного культивирования клеток в ферментере. Объяснение в тексте
Д, ед./мл X, ед. опт. пл. X, ед. опт. пл.
Из графика в левой части рис. 21.3 определяют ту концентрацию биомассы, при которой наблюдается максимальная производительность процесса. Для этого точку максимального наклона кривой графика накопления продукта P(t) проецируют на кривую X(t) накопления биомассы и далее на кривую удельной продуктивности биомассы dX/dt(X). При пересечении перпендикуляра к оси ординат, опущенного из точки Б на кривую dX/dt(X), находят искомую точку А, которая соответствует состоянию динамического равновесия при непрерывном культивировании, в котором концентрация микроорганизмов X равна в данном случае 3 ед. оптической плотности, концентрация продукта равна 5 ед./мл и скорость разбавления:
_ dX 1 1,5 ед.опт.пл./ч „ с ,
V = ц =-------=------------------= U,5 ч .
dt X 3 ед.опт.пл.
Таким образом, находим, что если при оптической плотности культуры, равной 3,0, в ферментере включить проток со скоростью 0,5 ч-1, то производительность процесса будет максимальна и концентрация искомого продукта в культиваторе сохранится на постоянном уровне — 5 ед./мл.
21.8. НЕКОТОРЫЕ ПРИНЦИПЫ ПОВЕДЕНИЯ КУЛЬТУР ПРИ ПЕРЕХОДЕ ОТ ПЕРИОДИЧЕСКОГО ПРОЦЕССА К НЕПРЕРЫВНОМУ
При переводе культуры с ограничением роста одним субстратом на непрерывный процесс в момент времени t, возможны три исхода (рис. 21.4). Предпо-
Рис. 21.4. Три возможных варианта (7, 2, 3) поведения культуры в ферментере при переходе от периодического процесса выращивания к непрерывному:
Хо — начальное число клеток; Sr — концентрация субстрата в резервуаре для стерильной среды; Z, — время переключения режима выращивания клеток
ложим, что вначале добавления среды не происходит и культура растет в течение времени 0-/7, как и в периодической культуре. Если скорость подачи среды окажется большей, чем максимальная скорость роста культуры (цтах), то скорость вымывания клеток окажется выше скорости их прироста и концентрация клеток будет падать со временем, а концентрация неиспользованного субстрата будет расти {кривая 1 на рис. 21.4, а, б) и приближаться к концентрации субстрата в резервуаре, из которого поступает среда (У,). Вторая возможность заключается в том, что скорость вымывания будет точно соответствовать скорости роста клеток, и при этом организмы будут расти с их максимальной удельной скоростью роста цтах. При таком исходе в ферментере устанавливается стационарное состояние {кривая 2 на рис. 21.4, а, б), при котором концентрации биомассы и субстрата остаются постоянными, но это состояние не будет устойчивым, поскольку любое временное изменение условий приведет к изменению концентрации биомассы и субстрата. Третье состояние {кривая 3 на рис. 21.4, а, б) может наступить, если скорость протока станет меньше максимальной скорости роста. Тогда концентрация биомассы начнет непрерывно увеличиваться, при этом концентрация субстрата снижается {кривая 3 на рис. 21.4, а, б), скорость прироста клеток постепенно уменьшается, пока не станет равной скорости вымывания. Дальнейшие изменения концентраций лимитирующего субстрата и биомассы невозможны без изменения скорости протока. Такое стационарное состояние культуры является саморегулирующимся. Уменьшение концентрации субстрата приводит к установлению концентрации клеток на более низком уровне. Увеличение концентрации субстрата вызовет повышение концентрации биомассы в ферментере.
ЗАДАЧА. ПОЛУЧЕНИЕ НЕПРЕРЫВНОЙ КУЛЬТУРЫ СВЕТЯЩИХСЯ БАКТЕРИЙ
Цель работы, изучение особенностей периодического и непрерывного культивирования микроорганизмов. Определение активности некоторых ферментов в клетках и содержания АТФ.
Ход выполнения задачи
Микроорганизм и его культивирование. Изучение периодического и непрерывного культивирования бактерий проводится с использованием морской светящейся бактерии Vibrio flscheri из коллекции кафедры микробиологии МГУ. Это грамотрицательная факультативно-анаэробная хемоорганотрофная бактерия, клетки которой представляют собой прямые или слегка изогнутые подвижные палочки размером 0,5 х 1,5 мкм. Микроорганизм растет в интервале температур от 10 до 28 °C, при 30 °C роста нет.
Культуры хранят на среде, содержащей (г/л): пептона — 10; агара — 20; морской воды — 400 мл; мясной воды — 600 мл; pH 7,4. Выращивание культур в колбах или ферментере ведут на жидкой среде следующего состава (г/л): дрожжевой экстракт — 0,5; NaCl — 30,0; Na2HPO4 — 5,3; КН2РО4 — 2,1;
(NH4)2HPO4 — 0,5; MgSO4 — 0,1; FeSO4- 7H2O — 0,001; пептон — 10,0; глицерин — 3,0; pH 7,4 до стерилизации. Среду стерилизуют при 0,5 ати 30 мин.
Для получения посевного материала V. fischeri выращивают в течение 8 —12 ч на качалке (200 об/мин) при 26 °C в колбах на 750 мл с объемом среды 100 мл. Среду в колбах засевают смывом стерильной питательной средой клеток бактерии, выращенной на твердой питательной среде, из расчета одна пробирка со скошенным агаром на две колбы. Качество посевного материала считают хорошим, если в темноте визуально определяется интенсивное свечение культуры. В целях изучения кривых роста и интенсивности свечения бактерии выращивают в аналогичных условиях в течение 24 ч.
Периодическое и непрерывное культивирование V. fischeri в ферментере проводят на жидкой среде, состав которой приведен выше.
Устройство и принцип работы ферментера. Для обучения студентов принципам проточного культивирования подходит любой ферментер, в котором при соблюдении стерильности проводимого процесса имеется возможность автоматически регулировать температуру, скорость перемешивания, количество подаваемого стерильного воздуха, значение pH и скорость подачи свежей среды.
Ферментер «Ультроферм 1601» (LKB, Швеция) представляет собой компактный настольный прибор (рис. 21.5), предназначенный для массового культивирования микроорганизмов в периодическом и непрерывном режиме. Прибор позволяет выращивать большое число различных видов микроорганизмов в строго контролируемых условиях. Ферментер оборудован устройствами для поддержания и контролирования температуры, pH, скорости перемешивания и аэрации. Клетки культивируют в толстостенном стеклянном сосуде, выдерживающем автоклавирование. Сосуд фиксируется на приборе с помощью двух направляющих рельсов и зажимов с винтами, позволяющими быстро отделять культиватор от стенда. Для уменьшения возможности инфицирования ферментера посторонней микрофлорой мешалка связана с приводом мотора при помощи двух постоянных магнитов. Лопасти мешалки обеспечивают эффективное перемешивание содержимого ферментера в радиальном направлении и могут быть легко приспособлены к любому уровню среды в сосуде.
Постоянное перемешивание среды в ферментере достигается благодаря применению прецизионного мотора с калиброванным электрическим тахометром, позволяющим устанавливать скорость вращения мешалки от 10 до 2000 об/мин. Контролирующий прибор поддерживает скорость вращения на заданном уровне, независимо от скачков напряжения в сети или вязкости культуры, по типу обратной связи.
Стенд снабжен газометром, позволяющим учитывать и регулировать количество подаваемого в прибор воздуха или другого газа (СО2, аргона). Регулирование температуры жидкости в ферментере осуществляется с помощью миниатюрного нагревателя и термодатчика, которые находятся в нижней части ферментера внутри сосуда. Охлаждение ферментера осуществляется водопроводной водой.
Кислотность среды регулируется автоматически специальным прибором для контроля pH. Комбинированный электрод (измерительный и сравнения) перед стерилизацией монтируется на верхней крышке сосуда. Устройство электродов данного прибора позволяет стерилизовать их в автоклаве при давлении 1 ати, однако для других типов электродов может быть с успехом применена стерилизация спиртом. Перед каждой стерилизацией необходимо проверять уровень жидкости внутри электрода и при необходимости доливать свежий раствор электролита. Регулирование pH осуществляется подачей щелочи или кислоты через два перистальтических насоса. Кислота и щелочь стерилизуются в сосудах с заранее подобранными силиконовыми шлангами, концы которых снабжены медицинскими иглами, завернутыми в бумагу или фольгу. Аналогично готовятся и стерилизуются питательная среда и другие растворы, подающиеся в ферментер после его стерилизации.
Рис. 21.5. Принципиальная схема ферментера «Ультроферм 1601» (LKB, Швеция), работающего в режиме непрерывного культивирования микроорганизмов:
1 — подача магистрального воздуха через ротаметр; 2 — бактериальный фильтр для стерилизации поступающего воздуха; 3 — вход воздуха в ферментер через мелкие отверстия; 4 — магнитная мешалка; 5 — обратный холодильник для предотвращения испарения питательной среды; 6 — бактериальный фильтр для улавливания клеток микроорганизмов в отходящих газах; 7 — сосуд с дезраствором (5%-й раствор NaHCO3) для обеззараживания отходящих газов; 8 — шприц со стерильным пеногасителем; 9 — шариковый пробоотборник; 10 — система нагрева питательной среды в ферментере; 11 — отвод суспензии микроорганизмов через перистальтический насос; 12 — сосуд для сбора суспензии микроорганизмов; 13 — сосуд со стерильной питательной средой; 14 — подача стерильной питательной среды через перистальтический насос; 15 — перистальтический насос; 16 — блок управления: поддержание значения pH среды (необходим датчик pH) температуры, количества растворенного в среде О2 (необходим датчик О2), аэрации
Крышка стеклянного сосуда фиксируется с помощью шести винтов-«барашков». К отверстиям входа и выхода воздуха присоединяют шланги с воздушными фильтрами, наполненными стерильной стеклянной ватой (воздух через фильтры должен проходить свободно). Устройство прибора позволяет пользоваться для аэрации обычным магистральным сжатым воздухом, который стерилизуется, проходя через фильтры со стеклянной ватой. Воздух, выходящий из ферментера, также автоматически стерилизуется через фильтр.
Ферментер стерилизуют в вертикальном положении в автоклаве при давлении 1 ати от 30 до 45 мин в зависимости от объема среды в нем. По окончании стерилизации сосуд укрепляют на основном приборе, зажимают прижимными винтами и присоединяют к пульту управления с помощью специальных проводов датчики pH, кислорода, термодатчик, мешалку. При необходимости подводят охлаждающую воду. Питательную среду и другие растворы, включая посевной материал, подсоединяют к ферментеру с помощью медицинских игл, используя специальные отверстия, закрытые вакуумной резиной и находящи
еся на верхней крышке ферментера, строго соблюдая правила асептики (протирают отверстие спиртом до и после засева ферментера).
При достижении заданной температуры производят посев культуры. Закончив перемешивание засеянной культуры, из ферментера через выходной шланг отбирают пробу, где регистрируют начальную оптическую плотность клеток, значение pH и начальный уровень продукта (свечение). В дальнейшем пробы отбирают каждые 15 — 30 мин, измеряют названные параметры и строят график.
После проведения периодического культивирования клеток в ферментере, а также построения графика роста и свечения культуры строят вспомогательный график для определения необходимых параметров непрерывного культивирования. Задача вспомогательного графика — помочь установить параметры непрерывного процесса для получения максимального количества продукта (в нашем случае — свечения) при максимальной скорости роста и протока среды. Пример построения вспомогательного графика приведен в разделе «Графоаналитический метод расчета условий непрерывного культивирования микроорганизмов» (см. рис. 21.3).
Графически вычислив параметры непрерывного культивирования, необходимые для проведения процесса с максимальной эффективностью, производят стерилизацию и очередной засев ферментера. По достижении необходимой плотности клеток (рассчитанной графически для максимального свечения) переводят процесс на непрерывный режим выращивания при соответствующей скорости разбавления, рассчитанной графоаналитическим методом. В дальнейшем через каждые 15 — 30 мин отбирают пробы, измеряют оптическую плотность культуры и уровень свечения клеток и убеждаются в том, что на протяжении 10 — 12 ч оптическая плотность клеток и уровень их свечения практически не изменяются и соответствуют максимальной производительности культуры при проточном культивировании. Небольшие изменения в концентрации клеток и свечении в ферментере объясняются в теоретическом разделе задачи.
Методы анализов
В процессе выращивания клеток в колбах и ферментере студенты отбирают пробы, проводят измерения в них оптической плотности и биолюминесцент-ной активности (свечения) для построения кривой роста и кривой интенсивности свечения культуры от времени. Кривые строят, откладывая по оси ординат оптическую плотность (в единицах оптической плотности) по ФЭК-56М (фильтр № 6, длина волны 540 нм) или интенсивность свечения (в условных единицах), которую определяют с применением специального прибора, а по оси абсцисс — время культивирования в часах (на 24 ч роста).
1. Определение биолюминесцентной активности бактерий. Клетки И. fischeri имеют активную люминесцентную систему, испускающую кванты света в присутствии альдегида, восстановленного флавина (ФМН) и кислорода. Для измерения интенсивности свечения клеток используют специальный прибор — люминометр Биотокс-6 (Россия).
Суспензии V. fischeri, взятые из колбы или ферментера, разводят в 10, 100 или 1000 раз 0,1 М К-фосфатным буфером или 3%-м раствором NaCl (pH 7,0) в зависимости от интенсивности свечения и концентрации клеток. Разведенную суспензию клеток вносят в кювету в количестве 1,0 мл, помещают кювету в гнездо кюветного отделе
ния светонепроницаемой камеры и производят измерение интенсивности свечения. Результаты представляют в виде таблицы и графика.
Результаты экспериментов по периодическому и проточному культивированию светящихся бактерий в управляемом процессе в ферментере представляют в виде соответствующих таблиц и графиков. Из графика по периодическому культивированию рассчитывают параметры непрерывного культивирования и на основании полученных данных проводят непрерывное выращивание.
2. Измерение активности бактериальных ферментов. При определении активности бактериальных ферментов необходимо учитывать, что она должна быть измерена в оптимальных условиях, неодинаковых для разных видов бактерий. Поэтому для определения активности ферментов каждой конкретной культуры необходимо подбирать условия, которые включают оптимум pH, количество субстрата, внесенного в пробу, оптимум температуры для проведения реакции, наличие в биологической пробе возможных ингибиторов реакции.
Если активность фермента в экстракте клеток не проявляется, то это может быть следствием нескольких причин: 1) необходимо быть уверенным, что искомая активность вообще в клетках присутствует; 2) фермент может присутствовать в неактивной форме, и его активность проявится только лишь при добавлении активаторов; 3) если возможно, полезно провести позитивный контроль искомой реакции, добавив в реакционную смесь источник фермента с заведомо имеющейся активностью — такой контроль может указать на наличие в экстракте клеток ингибитора, мешающего определению искомой активности.
При определении активности ферментов в биологических источниках рекомендуется выражать скорость реакций в международных единицах. Для ферментов такой единицей является микромоль использованного субстрата (или образованного продукта) за минуту при 30 °C. Специфическая активность препарата выражается в количестве международных единиц на 1 мг белка пробы. Ниже приведено описание метода определения оксидазной активности клеток, используемых в задаче.
3. Определение активностей НАДН- и НАДФН-оксидаз бактерий. Работа проводится с двумя препаратами разрушенных с помощью ультразвука или пресса клеток. Для работы необходим регистрирующий спектрофотометр, способный измерять изменение оптической плотности при 340 или 365 нм, однако реакцию можно проводить, используя и обычный ФЭК при соответствующей длине волны.
Клетки отделяют от среды центрифугированием при 10 тыс. g, промывают 50 мМ К-фосфатного буфера, pH 7,5, и разрушают в том же буфере с помощью ультразвука при 22 кГц в течение 4 мин по 1 мин с перерывами для охлаждения. Гомогенат центрифугируют при 40 тыс. g 25 мин и супернатант (экстракт) используют для определения ферментов. Готовят две пробы (если для измерения активности используют ФЭК, то контроль готовят в двойном объеме), содержащие в 3 мл: К-фосфатного буфера (0,05 М, pH 7,5) — 2,5 мл; НАДН (или НАДФН) — 0,5 мкмоль; экстракта — 0,1 мл; воды дистиллированной до 3 мл. В контрольную пробу вместо экстракта добавляют воду. Реакцию начинают внесением экстракта и проводят в течение 3 — 5 мин. Необходимо выбирать такое количество экстракта (по белку), чтобы реакция за время измерения была линейной. Рассчитывают активность НАД(Ф)Н-оксидазы в мкмолях субстрата, окисленного в 1 мин, на I мг белка экстракта, принимая, что коэффициент экстинкции НАД(Ф)Н при 340 нм равен 6,22 см/мкмоль.
4. Определение содержания АТФ в клетках бактерий. Аденозинтрифосфат является важнейшим компонентом энергодающих и энерготранспортных систем клетки, универсальным донором макроэргических фосфатных групп для многих транспортных, биосинтетических и других процессов. В подавляющем большинстве в клетках микроорганизмов АТФ представляет собой основной макроэргический компонент, его содержание обычно превышает содержание других трифосфатов вместе взятых. Активности некоторых ключевых аллостерических биоэнергетических и биосинтетических ферментов регулируются в зависимости от энергетического заряда клетки. Энергетический
заряд определяется суммой молярного содержания АТФ и половины молярного содержания АДФ клетки (так как 2 моля АДФ могут быть превращены в 1 моль АТФ с помощью аденилаткиназы), отнесенной к молярному содержанию всех аденилатов:
энергетический заряд = (АТФ + 1/2АДФ)/(АТФ + АДФ + АМФ).
Из этого видно, насколько важно знать количество АТФ клетки для изучения некоторых аллостерических ферментов. Наиболее специфичны и удобны энзимологические методы определения количества АТФ в клетках микроорганизмов. В большинстве из них используются сопряженные системы из двух и более ферментативных реакций.
Содержащийся в пробе АТФ в присутствии гексокиназы (ГК) фосфорилирует глюкозу; образующийся глюкозо-6-фосфат вступает в глюкозо-6-фосфатдегидрогеназную реакцию (Г-6-ФДГ):
глюкоза + АТФ -> глюкозо-6-фосфат + АДФ;
глюкозо-6-фосфат + НАЦФ+ -> 6-фосфоглюконовая кислота + НАДФН + Н+-
Как видно из уравнений, количество прореагировавшего АТФ эквимолярно количеству НАДФН, образующемуся в результате глюкозо-6-фосфатдегидрогеназной реакции; его количество определяют спектрофотометрически при длине волны 340 или 365 нм.
Реактивы. 6%-й раствор НС1О4 (свежеприготовленный); 5 М раствор К2СО3; 60 мМ триэтаноламиновый буфер (pH 7,5); 0,1 М раствор MgCl2; 7,5 мМ раствор НАДФ+ (свежеприготовленный); 0,5 М раствор глюкозы; раствор гексокиназы (НФ 2.7.1.1) — 10 мг препарата кристаллической гексокиназы с активностью 10 ед/мг растворяют в 1 мл дистиллированной воды непосредственно перед определением; раствор глюко-зо-6-фосфатдегидрогеназы (НФ 1.1.1.49) — 40 ед. фермента в 3,3 М растворе сульфата аммония растворяют в 1 мл дистиллированной воды. Растворы ферментов готовят непосредственно перед употреблением и в течение эксперимента хранят их на льду.
Ход определения. Навеску клеток, разрушенных на прессе при замораживании (см. гл. 13), соответствующую 500 мг сырой биомассы, помещают в центрифужную пробирку, куда предварительно наливают 1 мл 6%-й НС1О4. Пробу перемешивают на льду 5 мин, добавляют холодную хлорную кислоту из расчета примерно '/3 объема клеток и оставляют на 10 мин на льду для лучшей экстракции. Осадок белка затем удаляют центрифугированием в течение 10 мин при 6 тыс. g (центрифуга ЦУМ-1 —8 тыс. об/мин). Безбелковый экстракт переносят в другую центрифужную пробирку и удаляют избыток хлорной кислоты, добавляя 5 М раствора К2СО3 из расчета 0,05 мл К2СО3 на 1 мл кислого экстракта. Для более полного осаждения хлорного калия пробу оставляют на 10 мин во льду, а затем отделяют осадок центрифугированием при 5 тыс. g в течение 5 мин.
Нейтрализованный экстракт клеток нагревают до комнатной температуры и немедленно используют для энзиматического анализа, поскольку при стоянии содержащийся в пробе АТФ гидролизуется.
Для проведения ферментативного анализа в кювету спектрофотометра (1 см) наливают 2,5 мл триэтаноламинового буфера, 0,05 мл раствора НАДФ+ и 0,35 мл раствора MgCl2. Добавляют 0,1 мл клеточного экстракта, перемешивают и через 3 мин после установления равновесия измеряют исходную величину оптической плотности (Аt). К пробе добавляют 0,05 мл раствора глюкозо-6-фосфатдегидрогеназы и через 5 мин измеряют оптическую плотность (А2). Нарастание оптической плотности пробы после добавления глюкозо-6-фосфатдегидрогеназы связано с окислением содержащегося в клеточном экстракте глюкозо-6-фосфата. В кювету вносят 0,4 мл раствора глюкозы и через 30 с измеряют оптическую плотность (А3); небольшое уменьшение ее объясняется разбавлением пробы при добавлении раствора глюкозы. Добавляют 0,05 мл раствора гексокиназы и после окончания реакции (через 12 —15 мин) записывают конечное показание спектрофотометра (Л4). Увеличение оптической плотности пробы после до-
давления гексокиназы связано с вовлечением в ферментативные реакции содержащегося в экстракте АТФ.
Для учета изменений оптической плотности, вызванных добавлением ферментативных препаратов, в кювету дополнительно вносят по 0,05 мл препаратов глю-козо-6-фосфатдегидрогеназы или гексокиназы и при расчете учитывают соответствующие поправки. Количество АТФ в пробе (микромоль на 1 г сырых клеток) вычисляют по формуле Х= Д/1 • И • А/6,22, где АЛ — изменение оптической плотности пробы, вызванное использованием АТФ в сопряженной системе ферментативных реакций (АЛ = ДЛ4 - АЛ3); V — конечный объем пробы в кювете (3,5 мл); К — коэффициент разведения пробы по отношению к 1 г клеток, в данном случае равный 41,6; 6,22 — коэффициент микромолярной экстинкции восстановленной формы пиридиновых нуклеотидов при длине волны 340 нм и длине оптического пути 1 см. По разнице экстинкции АЛ = ДЛ2 - можно аналогичным образом рассчитать содержание в пробе глюкозо-6-фосфата. Данный метод специфичен и дает хорошо воспроизводимые результаты.
Материалы, оборудование, посуда, реактивы
Ферментер «Ультроферм 1601»; люминометр; пипетки на 1 мл — 25; на 2 мл — 15; пробирки с агаризованной средой для поддержания культуры — 10; ФЭК-56М; К-фос-фатный буфер (pH 7,0, 0,1 М), реактивы для приготовления питательной среды, определения НАД(Ф)Н-оксидаз и количества АТФ (см. в тексте), дезинтегратор УЗДН-1, пресс для разрушения бактерий, центрифуги ЦУМ-1 и Beckman, спектрофотометр.
План выполнения задачи
Для выполнения задачи необходимо 60 ч.
Занятие 1. Подготовка посуды и сред к стерилизации.
Занятие 2. Посев культуры микроорганизма в колбы для получения посевного материала и периодического выращивания, отбор проб и определение биомассы по оптической плотности и свечения на специальной установке.
Занятие 3. Отбор проб культуры из колб в процессе периодического выращивания бактерий, определение их оптической плотности и интенсивности свечения. Семинар.
Занятие 4. Знакомство с устройством и принципом работы ферментера, подготовка его к стерилизации и стерилизация в автоклаве. Приготовление и стерилизация питательной среды для периодического процесса в ферментере.
Занятие 5. Посев культуры в ферментер для периодического выращивания бактерий, отбор проб и определение в них интенсивности свечения и оптической плотности. Знакомство с методом графоаналитического расчета условий непрерывного культивирования.
Занятие 6. Отбор проб из ферментера, определение свечения и оптической плотности. Слив ферментера, подготовка к стерилизации и стерилизация ферментера. Расчет условий непрерывного культивирования клеток.
Занятие 7. Посев культуры в ферментер для непрерывного культивирования бактерий, отбор проб для определения оптической плотности и свечения. Перевод культуры на непрерывный процесс выращивания, отбор проб, проведение анализов, построение графиков кривой роста культуры.
Занятие 8. Слив ферментера, разборка и мойка. Отделение биомассы, разрушение на прессе или ультразвуком, определение ферментативных активностей и количества АТФ. Построение таблиц и графиков. Оформление результатов задачи с использованием компьютера. Зачет.
Примечание. Поскольку выполнение задачи требует каждодневной работы, целесообразно проводить ее во время учебной практики студентов.
МЕТАБОЛИЗМ ДРОЖЖЕЙ
Дрожжи имеют большое значение в хозяйственной деятельности и жизни человека. Их традиционно используют в хлебопечении, виноделии, пивоварении и т. д. Биомасса дрожжей является источником белка, витаминов, липидов и других ценных веществ.
Термин «дрожжи» не несет таксономической нагрузки, а объединяет одноклеточные эукариотные микроорганизмы, которые в зависимости от наличия и типа полового процесса относятся к трем разным классам грибов. Дрожжи, используемые в производстве, относятся к классу Ascomycetes, роду Saccha-romyces.
В зависимости от степени аэрации среды дрожжи получают энергию либо в результате спиртового брожения, либо в результате аэробного дыхания. Таким образом, изменяя условия культивирования, возможно направлять метаболизм дрожжей на получение метаболитов (этанол, СО2) или биомассы. Условия выращивания дрожжей сказываются также на скорости их роста, времени генерации, скорости потребления субстрата, а также на их морфологии и цитологии.
ЗАДАЧА. ИЗУЧЕНИЕ МЕТАБОЛИЗМА ДРОЖЖЕЙ В АЭРОБНЫХ И АНАЭРОБНЫХ УСЛОВИЯХ
Цель работы', сравнить параметры роста дрожжей, их морфологические и физиолого-биохимические особенности при культивировании в разных условиях аэрации.
Ход выполнения задачи
Микроорганизмы и условия культивирования. В работе используют разные расы дрожжей Saccharomyces cerevisiae: пекарские, винные, спиртовые, пивные. Посевной материал выращивают в пробирках на скошенном сусло-агаре (6 °Б, 2 % агара) в течение 2 сут при 30 °C. Затем биомассу смывают стерильной водой и переносят по 3—4 мл в каждый сосуд для культивирования.
Дрожжи для опыта выращивают на среде следующего состава (г/л): (NH4)2SO4 — 5,0; КН2РО4 — 1,0; КС1 — 0,15; MgSO4- 7 Н2О — 0,2; СаС12 — 0,05; дрожжевой автолизат — 50 мл; pH 6,0. Вода водопроводная. Каждый студент готовит 750 мл среды, разливает ее по 150 мл в 2 качал очные колбы (объем колбы 750 мл) и в 3 флакона (объем флакона 250 мл). Среды стерилизуют при 1 ати. Отдельно готовят необходимое количество 50%-го раствора глюкозы и стерилизуют его при 0,5 ати. Перед посевом во все опытные сосуды вносят стерильный раствор глюкозы из расчета: 2 % (по массе) — для аэробного культивирования и 6 % (по массе) — для анаэробного культивирования. Посевной материал в количестве 3 — 4 мл клеток (см. выше) добавляют в каждый сосуд для культивирования.
Выращивание дрожжей в аэробных условиях (7-й вариант) проводят при интенсивной аэрации. Для этого колбы помещают на качалку (скорость вращения 180—200 об/мин).
Анаэробные условия (2-й вариант) обеспечиваются высоким слоем среды во флаконах и стационарными условиями культивирования. Для определения скорости брожения в одном флаконе ватную пробку заменяют стерильной резиновой пробкой с затвором, заполненным до половины H2SO4. На верхний конец затвора необходимо надеть резиновый клапан. Затвор обеспечивает поглощение воды, испаряющейся из среды, и свободное выделение СО2 образовавшейся при брожении.
Продолжительность эксперимента 96 ч. Температура культивирования — 25 — 30 °C. Периодически (раз в сутки) из культуральных сосудов (из колбы и флакона без затвора) стерильно отбирают пробы для анализов, одновременно флакон с затвором взвешивают.
Концентрацию дрожжей в культуральной среде определяют нефелометрически, используя кювету с длиной оптического пути 0,5 см и зеленый светофильтр № 6 (Л. = 540 нм). Для перехода от показаний ФЭК к количеству клеток строят калибровочную кривую зависимости между величиной светорассеяния и числом клеток в единице объема. Для ее построения готовят 5 — 6 суспензий дрожжей разной плотности. В этом случае можно воспользоваться оставшимся инокулятом либо сделать дополнительный смыв клеток со скошенного сусло-агара. Оптическую плотность каждой суспензии измеряют на ФЭКе. Наиболее точные результаты получаются при работе с суспензиями, которые дают показания на средней шкале прибора — от 0,1 до 0,7 D. Затем в каждой суспензии подсчитывают число клеток дрожжей, пользуясь камерой Горяева. Результаты подсчета записывают в табл. 22.1.
Количество клеток дрожжей в 1 мл соответствующей суспензии вычисляют по формуле
М=а- (Ш/h-s,
где М — число клеток в 1 мл суспензии; а — среднее число клеток в квадрате сетки; h — глубина камеры (0,1 мм); х — площадь квадрата сетки (0,04 мм2); 1000 мм2 = 1 мл. Тогда количество клеток дрожжей в 1 мл суспензии = а 25 • 104. Полученные результаты (млн/мл) записывают в табл. 22.2.
На основании данных табл. 22.2 строят калибровочную кривую, откладывая на оси абсцисс количество дрожжевых клеток, а на оси ординат — оптическую плотность соответствующей суспензии.
Полученным графиком пользуются при определении числа клеток дрожжей в пробах, взятых из 1-го и 2-го вариантов опыта. Перед отбором проб содержимое сосудов необходимо тщательно взбалтывать для перевода клеток
Таблица 22.1
Количество клеток дрожжей в квадратах счетной камеры
Номер суспензии Квадраты счетной камеры
1 2 3 4 10
1 2 3 И т.д.
Количество клеток дрожжей в суспензиях и соответствующие показания ФЭК
Показатель Номер суспензии дрожжей
1 2 3 4 5 6 7 8
Показания ФЭК Количество клеток, млн/мл ( М)
Таблица 22.3
Количество клеток дрожжей и удельная скорость роста в разных условиях культивирования для двух вариантов опыта
Время культивирования, ч Показания ФЭК Количество клеток Удельная скорость роста
1 2 1 2 1 2
0 7 24 48 и т.д.
во взвешенное состояние. Следует подчеркнуть, что культуральную среду с высокой концентрацией клеток необходимо разводить, пользуясь стерильной средой из запасного 3-го флакона. Разведение учитывают при расчетах. Результаты вносят в табл. 22.3.
Зная прирост биомассы в единицу времени, можно определить удельную скорость роста:
ц = 2,3(/gn2 - - tx,
где ц — удельная скорость роста; ах и <з2 — количество клеток в начале и конце промежутка времени соответственно (/2 - /|); 2,3 — коэффициент перевода натуральных логарифмов в десятичные.
Определение редуцирующих сахаров. Для определения концентрации редуцирующих углеводов, в частности глюкозы, используют 2,3,5-трифенилтетра-золийхлорид (ТТХ), который восстанавливаясь глюкозой, превращается в окрашенный трифенилформазан. Последний, растворяясь в органических растворителях, окрашивает раствор в вишнево-красный цвет.
_ К 2 мл раствора (культуральной среды), содержащего глюкозу, добавляют 1 мл бесцветного водного 0,3%-го раствора ТТХ и 1 мл 2 н. NaOH. Смесь нагревают на кипящей водяной бане 3 мин, затем вынимают из бани, немедленно подкисляют 1 мл 2,1 н. раствора СН3СООН для прекращения реакции. После охлаждения раствора объем жидкости доводят до 20 мл этанолом или изопропанолом. При этом развивается устойчивая окраска, интенсивность которой измеряют на ФЭК с зеленым светофильтром (X = 485) в кювете с длиной оптического пути 3 мм.
Для перехода от показаний ФЭК к выражению количества глюкозы в единицах массы строят калибровочную кривую, используя растворы, содержащие от 20 до 200 мкг глюкозы на 1 мл.
Определение глюкозы в культуральной среде проводят, как описано выше, предварительно освободив ее от клеток центрифугированием или фильтрованием, а также сделав соответствующие разведения при содержании глюкозы выше 200 мкг/мл. Супернатант (фильтрат) до определения глюкозы можно сохранять в холодильнике в течение нескольких суток, прибавив к нему несколько капель толуола. Результаты определений вносят в табл. 22.4.
На основании полученных данных об урожае дрожжей и количестве потребленной глюкозы рассчитывают экономический коэффициент в аэробных и анаэробных условиях: у = x/s, где х — количество клеток к моменту окончания опыта; s — количество использованной к концу опыта глюкозы.
Определение скорости брожения. О скорости брожения g судят, во-первых, по количеству СО2, выделившегося в единицу времени из определенного объема культуры, во-вторых, по количеству этанола, образовавшегося за определенный интервал времени.
Убыль СО-i определяют, взвешивая периодически флакон с затвором. Для этой цели можно пользоваться техническими весами (взвешивают с точностью до 0,01 г). Рассчитывают, сколько углекислоты (г) выделилось из 1 л культуральной жидкости в 1 ч за каждый промежуток времени между взвешиваниями флакона. Результаты записывают в табл. 22.5.
Определение количества этанола химическим методом. Количество этанола, образовавшегося при брожении, определяют оксидометрическим методом, основанным на окислении этилового спирта до уксусной кислоты и воды смесью бихромата калия с серной кислотой:
ЗСН3СН2ОН + 2К2Сг2О7 + 8H2SO4 = ЗСН3СООН + 2Cr(SO4)2 + K2SO4 + 11Н2О
Таблица 22.4
Содержание глюкозы в культуральной среде в процессе роста дрожжей
Условия роста Параметр Время, ч
0 7 24 48 72 96
Аэробные Разведение Показание ФЭК Глюкоза, г/л
Анаэробные Разведение Показание ФЭК Глюкоза, г/л
Таблица 22.5
Выделение СО2 в процессе брожения, осуществляемого дрожжами
Время, ч Масса сосуда, г Количество выделившегося СО2
от начала опыта, г/л за интервал времени, г/л • ч, (g)
0 7 24 48 и т.д.
По окончании окисления спирта избыток бихромата оттитровывают раствором соли Мора:
K2Cr2O7 + 6FeSO4(NH4)2SO4 + 7Н2О = Cr2(SO4)3 + Fe2(SO4)2+ 6(NH4)2SO4 + + 7Н2О + K2SO4
По количеству бихромата, израсходованного на окисление, вычисляют концентрацию спирта. Наиболее точные результаты метод дает при содержании спирта в жидкости в пределах 1 — 2 %. Поэтому при более высоких концентрациях спирта разбавляют культуральную жидкость, а при меньших — растворы соли Мора и бихромата.
Ход анализа. Культуральную жидкость в объеме 2 мл, освобожденную от клеток фильтрованием или центрифугированием, разводят дистиллированной водой в пять раз и переносят в круглодонную колбу на 50 — 75 мл.
В трехшариковый приемник наливают 25 мл раствора бихромата калия и 10 мл конц. H2SO4. Колбу плотно закрывают резиновой пробкой с отводной трубкой, суженный конец которой должен доходить почти до дна приемника. Затем в течение 10 — 15 мин отгоняют 2/з содержимого колбы. Раствор бихромата за это время меняет окраску от оранжевой до грязно-бурой. После отгонки во избежание перебрасывания жидкости из приемника убирают отводную трубку и только потом отставляют горелку.
Содержимое приемника переносят в мерную колбу на 100 мл. Дистиллированной водой споласкивают приемник и отводную трубку, промывные воды выливают в ту же мерную колбу. После этого раствор доводят до метки дистиллированной водой, перемешивают, отбирают из него 20 мл в коническую колбу на 250 мл, добавляют 10 мл дистиллированной воды и титруют солью Мора. Параллельно ведут контрольное титрование, чтобы определить количество соли Мора, идущее на титрование 5 мл раствора бихромата. Для этого в коническую колбу помещают 5 мл раствора бихромата, 2 мл конц. H2SO4, 20 — 30 мл дистиллированной воды и титруют солью Мора. Соль Мора — нестойкое соединение, поэтому контрольное титрование обязательно делают при каждом определении. Конец титрования устанавливают капельной пробой с феррицианидом {K3Fe(CN)6}. Для этого на чистую белую фарфоровую или керамическую пластинку стеклянной палочкой наносят каплю титруемой жидкости и рядом из капельницы — каплю раствора феррицианида. Интенсивное синее окрашивание, получаемое при слиянии капель, указывает на конец реакции. Опытный и контрольный растворы титруют по два раза. Первое титрование служит для получения ориентировочных результатов, поэтому реакцию титруемой жидкости с раствором феррицианида проводят после добавления каждого миллилитра раствора соли Мора, которое было определено первым титрованием, а затем делают качественную реакцию каждый раз после добавления 0,1 мл раствора соли Мора.
Количество этанола X (в г/10 мл культуральной среды) рассчитывают по формуле
Х= [(о - б) 25 5о]0,01,
где а, б — количество соли Мора, пошедшее на титрование соответственно контрольного и опытного вариантов, мл; 25 — количество К2Сг2О7, мл. Полученные результаты представляют в виде табл. 22.6.
Определение количества этанола ферментативным методом с помощью набора реактивов «Фотоэтанол». Определение этанола основано на реакции, катализируемой ал-когольоксидазой:
этанол + О2 -> ацетальдегид + Н2О2
Образующаяся в ходе данной реакции перекись водорода окисляет субстрат пероксидазы с образованием окрашенного продукта. С помощью данного метода можно определить от 1 до 80 % этанола в среде.
Образование этанола при росте дрожжей в анаэробных условиях
Время,ч Разведение Количество соли Мора, мл Содержание этанола
ОПЫТ контроль г/10 мл г/л г/л • ч
0 7 24 48 и т.д.
Ферменты для определения этанола выпускаются в виде специального набора «Фотоэтанол» («Импакт» Москва, Россия), который включает три реагента.
Реагент 1 — фосфатный буфер (pH 7,0), субстрат пероксидазы; перед использованием содержимое флакона растворяют в 100 мл дистиллированной воды; раствор стабилен при хранении в темном флаконе при температуре 2 — 6 °C в течение 48 ч.
Реагент 2 — калибровочный раствор этанола.
Реагент 3 — лиофильно-высушенный порошок, содержащий фермент алко-гольоксидазу и его стабилизаторы; стабилен при 2 — 6 °C в течение 3 мес.
Рабочий реактив готовят добавлением к реагенту 1 содержимого флакона 3 и 1 мг пероксидазы. Рабочий реактив стабилен при 2 — 6 °C в темноте не дольше 48 ч.
Для анализа культуральную жидкость разбавляют водой в 20 раз, тщательно перемешивают, отбирают из них аликвоты объемом 20 мкл и вносят в пробирки, содержащие 2 мл рабочего реагента. Параллельно в одну пробирку вносят 20 мкл калибровочного 1%-го раствора этанола, предварительно разбавленного, как и исследуемые пробы.
Оптическую плотность реакционной смеси измеряют через 30 мин инкубации при комнатной температуре. Измерение проводят на ФЭК при длине волны 470 — 540 нм относительно рабочего реактива, в который вносят 0,04 мл дистиллированной воды, в кювете с длиной оптического пути 10 мм.
Концентрацию этанола рассчитывают по формуле
С= (£„//•„) Сст-1 %,
где Ео — оптическая плотность в кювете с образцом, измеренная относительно холостой пробы; Е„ — оптическая плотность в кювете со стандартом; Сст — концентрация этанола в стандартном растворе.
Определение этилового спирта методом газожидкостной хроматографии. Определение этилового спирта можно проводить на хроматографе с колонкой, содержащей неподвижную фазу полиэтиленгликольадипината, нанесенного на хромосорб W, и пламенно-ионизационным детектором.
Ход анализа. В одноразовую пластиковую пробирку вносят 100 — 200 мкл образца. К пробе приливают 1 мл дистиллированной воды и гомогенизируют смесь путем энергичного встряхивания. Пробирку с гомогенной смесью центрифугируют 10 мин при 7000 об/мин. Полученный таким образом препарат должен быть прозрачным. Хроматографирование и обработку результатов проводят так же, как при определении ЛЖК (см. гл. 33).
Для расчета концентрации используется метод внутреннего стандарта, который состоит в добавлении к определенной массе исследуемого материала этанола с известной массой и известной площадью пика. Для более точного и быстрого расчета концентрации по площадям пиков следует использовать компьютерную программу «ЭКОХРОМ» (разработка ИОХ, Россия).
Недостатком данной методики является определение суммарной фракции этилового и метилового спирта в образцах. Однако в связи с тем, что культуральная жидкость Saccharomyces cerevisiae метанола не содержит, описанный выше метод в этой работе может быть использован для определения этанола.
Определение интенсивности дыхания клеток. Дыхание дрожжей (поглощение клетками молекулярного кислорода) определяют полярографическим методом.
Около 100 мг сырой дрожжевой биомассы ресуспендируют в 5 мл фосфатного буфера (0,05 М; pH 7,0), встряхивая пробирку. Суспензию в количестве 2,5 мл вводят шприцем в ячейку полярографа. При заполнении ячейку наклоняют таким образом, чтобы воздух полностью вытеснялся из камеры через верхний штуцер. Кинетику поглощения О2 записывают в течение 1 — 2 мин. Прописывается кривая, соответствующая эндогенному дыханию. Затем в ячейку добавляют 0,1 мл раствора глюкозы до конечной концентрации 10-3 М и измеряют скорость поглощения кислорода. Для получения окончательного результата из найденной величины скорости дыхания вычитают величину скорости эндогенного дыхания. Результаты вносят в табл. 22.7
Изучение морфологии и цитологии дрожжей. Каждую пробу первого и второго вариантов опыта микроскопируют, определяя размеры дрожжевых клеток и наличие в них гликогена, а также подсчитывают процент живых и почкующихся клеток.
Размеры дрожжевых клеток определяют в препарате «раздавленная капля» с объективом 40х, используя винтовой окулярный микрометр. Измеряют не менее 30 клеток и средние величины записывают в табл. 22.8.
Гликоген может накапливаться в цитоплазме дрожжей в виде гранул. При обработке клеток раствором Люголя он приобретает красно-коричневый цвет. Для наблюдения за содержанием гликогена в клетках готовят препарат «раздавленная капля» в растворе Люголя и просматривают его с объективом 40 х.
Таблица 22.7
Интенсивность дыхания дрожжевых клеток в зависимости от условий их выращивания
Условия выращивания Скорость потребления О2, нмоль/мг мин
эндогенное дыхание дыхание в присутствии глюкозы
Аэробные Анаэробные
Таблица 22.8
Размеры клеток дрожжей (длина и ширина, мкм) при росте в аэробных и анаэробных условиях
Время, ч Аэробные Анаэробные
длина ширина длина ширина
0 7 24 48 и т.д.
В каждом препарате следует просчитать не менее 100 клеток (в нескольких полях зрения). Результаты записывают в табл. 22.9.
Количество дрожжевых клеток, имеющих почки, подсчитывают в препарате «раздавленная капля» с объективом 40х. Результаты записывают в табл. 22.10.
Количество живых и мертвых клеток дрожжей подсчитывают в препаратах, обработанных раствором метиленового синего. Для этого в пробирке смешивают равные объемы метиленового синего (10 мг/100 мл) и суспензии дрожжей и встряхивают в течение 10—15 мин. Мертвые клетки легко адсорбируют краску и окрашиваются в синий цвет. Живые клетки, восстанавливая метиленовый синий, остаются бесцветными. Подсчет ведут в препарате «раздавленная капля» с объективом 40*. Результат записывают в табл. 22.11.
На основании полученных данных составляют отчет, включающий краткий обзор литературы, касающийся метаболизма дрожжей в аэробных и анаэробных условиях и их цитологических особенностей, сводную таблицу результатов
Таблица 22.9
Количество клеток дрожжей, содержащих гликоген, при росте в разных условиях
Время, ч Аэробные условия Анаэробные условия
1 2 3 1 2 3
0 7 24 и т.д.
Примечание. 1 — всего клеток, 2 — клеток с гликогеном, 3 — % клеток с гликогеном.
Таблица 22.10
Количество почкующихся клеток дрожжей при росте в разных условиях
Время, ч Аэробные условия Анаэробные условия
1 2 3 1 2 3
0 7 24 и т.д.
Примечание. 1 — всего клеток, 2 — клеток с почками, 3 — % почкующихся клеток.
Таблица 22.11
Количество живых клеток дрожжей при росте в разных условиях
Время, ч Аэробные условия Анаэробные условия
1 2 3 1 2 3
0 7 24 ит.д.
Примечание. 1 — живые клетки, 2 — мертвые клетки, 3 — % живых клеток.
опытов, соответствующие графики, а также обсуждение полученных результатов в сравнительном аспекте.
Материалы, посуда, оборудование, реактивы
Скошенный сусло-агар — по 2 пробирки на каждого студента; глюкоза; трифенил-тетразолий хлористый — 0,3%-й водный раствор; 2 н. раствор NaOH; 2,1 н. раствор уксусной кислоты; этанол; раствор Люголя; толуол; раствор метиленового синего (10 мг/100 мл); растворы: К2Сг2О7 — 42,6 г/л; FeSO4(NH4)2 (соли Мора) — 92 г и H2SO4 (пл. 1,84) — 20 г на 1 л воды; K3Fe(CN)6 — 0,2 г /л; фосфатный буфер 0,05 М, pH 7,0; набор реактивов для приготовления среды, набор реактивов «Фотоэтанол»; колбы: на 1 л — 1, на 150 мл — 2, колбы качалочные — 2, колбы мерные на 100 мл — 2, на 50 мл — 1; пробирки обычные 12—14 шт.; флаконы на 250 мл — 3; затвор — 1; резиновые пробки; пипетки: на 1 — 2 мл — 2, на 10 мл — 10, на 5 мл — 5; пипетки Мора на 5 мл — 1, на 1 —2 мл — 2; пипетки Пастера с резиновым шлангом и грушей; микропипетки — 2; бюретки на 50 мл — 1; фарфоровая пластинка; воронка; стеклянная палочка; шприц с изогнутой иглой; микроскоп; предметные и покровные стекла; винтовой окулярный микрометр; камера Горяева; качалка; центрифуга; весы аналитические; весы технические; ФЭК; полярограф; прибор для отгонки спирта; водяная баня.
План выполнения задачи
На выполнение задачи требуется 55 — 60 ч.
Занятие 1. Пересев дрожжей для получения посевного материала. Приготовление среды и необходимых растворов. Подготовка посуды к сухой стерилизации.
Занятие 2. Постановка опыта. Построение калибровочной кривой для определения числа клеток.
Занятие 3 — 6. Отбор и анализ проб.
Занятие 7. Отбор и анализ проб. Измерение интенсивности дыхания клеток из аэробного и анаэробного вариантов опыта.
Занятие 8. Определение глюкозы в пробах. Определение этанола в пробах с помощью набора реактивов «Фотоэтанол».
Занятие 9. Написание отчета о работе.
Занятие 10. Коллоквиум. Обсуждение результатов, сравнение параметров, полученных с разными расами дрожжей — пекарскими, спиртовыми, винными, пивными.
Глава 23
БИОСИНТЕЗ И ФУНКЦИИ КОРРИНОИДОВ В ФИЗИОЛОГИИ ПРОПИОНОВОКИСЛЫХ БАКТЕРИЙ
Пропионовокислые бактерии (ПКБ), известные с начала прошлого века и выделяемые из ферментируемых растительных остатков, молока и твердых сыров, в настоящее время рассматриваются как компактная филогенетическая линия в домене «грамположительные бактерии с высоким содержанием ГЦ-пар в хромосоме». Они интересны как организмы, неоднозначно относящиеся к молекулярному кислороду и образующие в норме большие количества кор-риноидов — соединений группы витамина В12. Это аэротолерантные анаэробы (микроаэрофилы), основным способом получения энергии которых является пропионовокислое брожение (ПБ), при этом ПКБ имеют редуцированную
дыхательную цепь переноса электронов на О2 и систему ферментов антиокисли-тельной защиты. Синтез цитохрома d, действующего в качестве терминальной оксидазы у бактерий, индуцируется молекулярным кислородом (при культивировании бактерий на богатых средах без добавления соли кобальта), но для этого концентрация кислорода в среде должна быть вдвое ниже насыщающей. В биологии прокариот корриноиды проявляют множественные функции. В метаболизме Р. freudenreichii витамин В12 участвует в четырех биохимических реакциях: в качестве коферментов метилмалонил-КоА-мутазы — ключевого фермента ПБ, метионинсинтазы и рибонуклеотидредуктазы (РНРазы), а также как субстрат (метилированный кобаламин) в трансметилировании ДНК по цитозину. ПКБ практически значимы как пробиотики, как источники консервантов, применяемых в пищевой промышленности и сельском хозяйстве, а также как продуценты биологически активных веществ, прежде всего витамина В12
ЗАДАЧА. ИЗУЧЕНИЕ РОЛИ КОРРИНОИДОВ В ФИЗИОЛОГИИ PROPIONIBACTERIUM FREUDENREICHII
Цепь работы, выявление корреляции между содержанием корриноидов в клетках Р. freudenreichii, которое зависит от концентрации ионов кобальта в среде, и ростом бактерий, интенсивностью ПБ, образованием и содержанием ДНК в клетках.
Для выполнения задач исследования необходимо:
• культивировать бактерию на глюкозоминеральной и обогащенной триптоном (пептоном) средах с добавлением и без добавления соли кобальта; сравнить динамику роста вариантов в зависимости от обеспеченности среды ионами кобальта; ответить на вопрос, в каких условиях культивирования ионы кобальта существенны для развития бактерии;
• в случае вариантов, для которых содержание соли кобальта в среде не влияет на рост культуры, измерить содержание суммарных корриноидов (витамина В12) в клетках; выявить зависимость содержания витамина В,2 от концентрации соли кобальта в среде; сделать заключение о необходимости кобальта для биосинтеза витамина В12 в клетках;
• оценить интенсивность ПБ как основного энергетического процесса в полноценных (СоГ) и дефицитных (Сог") по содержанию корриноидов в клетках бактерии; ответить на вопрос, требуется ли высокое содержание витамина В12 в клетках для эффективного ПБ;
• измерить интенсивность образования и содержание ДНК в полноценных (Сог+) и дефицитных (Сок) по содержанию витамина В|2 в клетках; сделать заключение о наличии корреляции между количеством корриноидов в клетках и эффективностью образования ДНК.
Ход выполнения работы
Микроорганизм и его культивирование. Р. freudenreichii subsp. shermanii, ВКМ-103 выращивают на средах следующего состава. Среда 1 — глюкозоминеральная (минимальная, или основная): глюкоза (г-л-1) — 20,0; макроэлементы («соле-
вой фон» среды - СФС, г-д-1): (NH4)2SO4- 3,0; КН2РО4 - 1,5; MgSO4- 7Н20 -0,25; микроэлементы (мг л"1): NaCl — 5,0; MnSO4 • 5Н2О — 5,0 или МпС12 • 4Н2О — 4,0; ZnSO4-7H2O - 0,01; FeCl3-6H20 - 0,005; СоС12-6Н2О - 1,0-1,5; витамины (мкг • л"1): пантотенат кальция — 1000; тиамин — 200; биотин — 1,0; вода дистиллированная; pH среды 6,8 —7,0. Среда 2 — среда 1 без добавления кобальта. Среда 3 — среда 1, дополненная 0,05%-м триптоном или 0,1%-м пептоном. Среда 4 — среда 2, дополненная 0,05%-м триптоном или 0,1%-м пептоном.
Раствор микроэлементов готовят отдельно (из расчета 1 мл на 1 л среды) и добавляют в среду до стерилизации. Глюкозу, витамины и хлорид кобальта стерилизуют по отдельности, готовя растворы из расчета: глюкоза — 100 мл на 1 л среды, витамины и хлорид кобальта — 1 мл на 1 л среды. Режим стерилизации: основная среда, глюкоза и хлорид кобальта при 0,5 атм (30 мин), витамины — 0,5 атм (15 мин). Культуру выращивают на средах 1 и 2 в колбах Эрлен-мейера объемом 100 мл с 50 мл среды и на средах 3 и 4 в колбах Эрленмейера объемом 750 мл с 500 мл среды. Культивирование проводят при 30 °C периодическим способом в стационарных условиях (при свободном доступе воздуха: концентрация кислорода -0,024 мМ, или -0,7 мг-л-1). В качестве посевного материала используют 48-часовую культуру, выращенную на той же среде. Объемная доля посевного материала — 10 %. На средах 2 и 4 культуру предварительно пассируют 2—3 раза. Ежедневно подводят pH культур до значения 6,8 —7,0 (зеленый цвет индикатора бромтимолового синего в капле раствора на поверхности белой кафельной плитки) с помощью стерильных 10%-х растворов NaHCO3 и H2SO4 Время культивирования — 72 ч. Пипетки, используемые при работе с вариантами 1 и 3, необходимо готовить и содержать отдельно во избежание попадания ионов кобальта в остальные растворы. Рост бактерий оценивают нефелометрически, измеряя поглощение при 540 нм дважды в сутки. Отдельно строят калибровочную кривую для перевода значений оптической плотности культур в граммы абсолютно сухой биомассы (АСБ). АСБ определяют гравиметрически, используя клетки, оставшиеся после завершения опыта.
Измерение содержания суммарных корриноидов (витамина В12) проводят в клетках 72-часовых культур (варианты 3 и 4, которые соответствуют номерам сред).
Определение микробиологическим методом общего содержания кобаламинов в виде их цианоформ проводится с помощью тест-культуры Escherichia coli 113-3 по Каноп-кайте (1978) в модификации. Штамм Е. coli 113-3 является ауксотрофом по кобаламину. Кобаламин необходим бактерии для синтеза метионина. Метод основан на том, что в определенном диапазоне количество кобаламина в среде пропорционально интенсивности роста бактерии, которую измеряют нефелометрически. Чувствительность метода: 2 —4 нг витамина В|2 в образце. Культуру Е. coli 113-3 поддерживают, выращивая в течение 24 ч при 37 °C в пробирках со скошенной агаризованной средой состава (г • л_|): триптон — 6,0; К2НРО4 — 0,2; FeSO4-7H20 — 0,005; /.-аспарагин — 0,2; глицерин — 2,0; цианкобаламин — 0,0001; агар — 15,0; вода дистиллированная, pH 7,0. Культуру хранят при 6 °C. Частота пересева — 1 раз в 3 мес. Для определения витамина готовят основную среду (ОС) состава (г-л_|): глюкоза — 2,0; К2НРО4 — 7,0; КН2РО4 — 3,0; натрий лимоннокислый — 0,5; (NH4)2SO4 — 1,0; MgSO4- 7Н2О — 0,1; NaCl — 0,5; NaNO2 — 0,01; вода дистиллированная, pH 6,8 —7,0.
Для получения калибровочной кривой используют стандартный водный раствор витамина В,2 (цианкобаламина, CN-Cbl) с концентрацией 5 • I О5 мкг • мл-1. За сутки до проведения опыта готовят посевной материал. Для этого Е. coll (штамм 113-3) пересевают с агаризованной среды в пробирку, содержащую 5 мл ОС и 5 мл стандартного раствора цианкобаламина, и выращивают 20 — 24 ч при 37 °C. В испытуемом образце кобаламины предварительно переводят в раствор и стабилизируют в виде нитритной формы. Для этого к 1 мл хорошо перемешанной культуры в пробирке, градуированной на 10 мл, добавляют 4 мл дистиллированной воды и 2 мл 0,1%-го раствора NaNO2. Затем доводят pH до 4 — 5 с помощью 0,1 н. раствора НС1 и нагревают ее в кипящей водяной бане в течение 15 мин. Пробирки охлаждают, растворы нейтрализуют сухим NaHCO3 (pH 7,0), объем жидкости доводят дистиллированной водой до 10 мл. Образцы хранят в холодильнике.
Исследуемые образцы могут содержать метионин, который также поддерживает рост Е. coli 113-3, поэтому параллельно рекомендуется проводить контрольный анализ образцов, в которых кобаламины предварительно разрушены. Для этого в подготовленные образцы добавляют NaOH до концентрации 0,1 н., нагревают их 20 мин на водяной бане, охлаждают и нейтрализуют 0,1 н. НО. Для получения калибровочной кривой в широкие пробирки со стерильной ОС (5 мл) добавляют стандартный стерильный раствор CN-Cbl (5-10-5 мкг-мл-1) в количестве: 0,5; 1,0; 1,5; 2,0 мл, что соответствует 0,025; 0,05; 0,075 и 0,1 мкг CN-Cbl. Подготовленные опытные образцы добавляют в количестве от 0,5 до 4,0 мл (например, по 0,5; 2,0 и 4,0 мл). Объем среды в пробирках доводят стерильной дистиллированной водой до 10 мл. Культуру (0,1 мл), используемую в качестве посевного материала, суспендируют в 10 мл 0,9%-го стерильного раствора NaCl. Посевной материал вносят в количестве 1 % (по объему). Выращивание проводят в течение 20 — 24 ч при 37 °C. Рост определяют, измеряя оптическую плотность при 540 нм.
Для проведения спектрофотометрического определения кобаламины освобождают из клеток в водный раствор путем гидролиза, переводят в гидроксильные производные и измеряют поглощение при длине волны максимального поглощения соответствующих производных кобаламина. Полученное значение пропорционально концентрации кобаламина. Чувствительность метода — 10— 100 мкг кобаламина в пробе. Клетки из 200 мл 72-часовых культур вариантов 3 и 4 собирают центрифугированием (6 тыс. g, 15 мин), количественно помещают в колбы на 250 мл путем суспендирования клеток в подкисленной до pH 4,6 —5,0 дистиллированной воде (~30-кратный объем по отношению к объему сырой биомассы). Корриноиды экстрагируют путем нагревания суспензии в кипящей водяной бане в течение 45 мин. После охлаждения осадок отделяют центрифугированием (6 тыс. g, 15 мин). Супернатант должен иметь pH 4,6—5,0. В суммарном препарате могут присутствовать разные производные кобаламина, различающиеся по коэффициентам экстинкции и максимумам поглощения. Поэтому корриноиды переводят в форму гидроксильных производных путем помещения раствора под лампу накаливания (60 Вт) на расстоянии 25 см на 30 мин. Измеряют конечный объем раствора. Определяют поглощение при длине волны, соответствующей максимуму поглощения гидроксикобаламина (ОН-СЫ) — 530 нм. Суммарное содержание коррино-идов в 1 л культуры рассчитывают по формуле
„ _ 4s30 • ю3
K-V2 ’
где С — содержание корриноидов, ммоль • л_| исследуемого раствора; /53о — оптическая плотность при Z = 530 нм, толщина кюветы 1 см; К — коэффициент миллимолярной экстинкции (для ОН-СЫ К= 56); Ь) — объем раствора (гидролизата), мл; И2 — объем культуры, из которой собрана биомасса, мл.
Для выражения содержания корриноидов в весовых единицах необходимо использовать величины их молекулярной массы. Для ОН-СЫ она равна 1341. Однако большая часть корриноидов Р. freudenreichii представлена аналогом кобаламина — кобинами-дом, не содержащим сс-лиганд (нуклеотид с основанием 5,6-диметилбензимидазол, 5,6-ДМБ). Молекулярная масса производных этого аналога -1000— 1100. Поэтому обычно за несколько часов до измерения в культуру вводят 5,6-ДМБ (10 мг-л-1).
Интенсивность и характер пропионовокислого брожения (ПБ) оценивают по количеству и соотношению выделяемых бактерией в культуральную жидкость пропионовой и уксусной кислот. Качественное и количественное определение летучих кислот проводят методом газожидкостной хроматографии (Методы общей бактериологии, 1984). Калибровку колонок ГЖХ проводят растворами пропионовой и уксусной кислот. Анализ кислот проводят в культуральной жидкости 72-часовых культур вариантов 3 и 4. Культуральную жидкость отделяют от клеток центрифугированием (6000 g, 15 мин). К 100 — 200 мкл образца приливают 1 мл 0,02 н. НС1. Раствор вводят через фильтрующую насадку. Колонка размером 1000 х 4 мм заполнена полиэтиленгликольадипатом на хромосорбе W. Скорость потока водорода в пламенно-ионизационном детекторе — 40 мл-мин-1; температура — 240 °C в испарителе и 130 °C в колонке. Газ-носитель (азот) подается в колонку под давлением 70 кПа. Результаты хроматографии регистрируют и обрабатывают с помощью компьютерной программы.
Интенсивность образования ДНК оценивают по скорости включения меченого аденина во фракцию ДНК в процессе инкубирования суспензий клеток (60—90 мин). В опыте сравнивают варианты Р. freudenreichii (Сог+ и Сог“), выращенные на средах 3 и 4 в течение 65—72 ч. Для измерения интенсивности образования ДНК готовят следующие растворы, раствор [8~14С] аденина (ПО «Изотоп», С.-Петербург; удельная радиоактивность 10—12 ГБк- г~*) 37 МБк в 500 мкл дистиллированной воды; раствор аденина 2 мг в 1,0 мл дистиллированной воды (порошок аденина растворяют при нагревании, по каплям подкисляя раствор с помощью 0,1 н. НС1); 200 мл раствора СФС + 0,5 % глюкозы, pH 7,2 (хранить в холодильнике); 20 мл раствора 1,2 н. КОН; 40 мл раствора 0,6 н. НО; 70 мл раствора 2 н. НС1О4; 200 мл раствора 0,3 н. НСЮ4; 250 мл 70%-го водного раствора этилового спирта; 200 мл сцинтилляционной жидкости ЖС-106. В 5 чашек Петри помещают бумажные фильтры, разделенные пополам (карандашом), обозначенные цифрами 3 и 4; на крышках пишут время: 0, 20, 40, 60, 80 мин.
Клетки из 40 мл 65 — 72-часовых культур вариантов 3 и 4 собирают центрифугированием (6 тыс. g, 15 мин), ресуспендируют на холоду (+6 °C) в 12 мл СФС с 0,5%-й глюкозой (pH 7,2), измеряют оптическую плотность Ц540) при разведении в 10 раз. В 10 мл суспензии вводят [8“14С]аденин (74 • 103 Бк • мл-1) и немеченный аденин (5 мкг мл-1), инкубируют 5—10 мин при 37 °C и отбирают пробы по 0,5 мл с интервалом в 20 мин в трех повторностях (в точках 0, 20, 40, 60, 80 мин). Пробы помещают в пробирки с 0,5 мл 1,2 н. раствора КОН и оставляют на ночь при 37 °C для щелочного гидролиза РНК. Щелочь в образцах с радиоактивной ДНК нейтрализуют добавлением 1 мл 0,6 н. НС1 (при комнатной температуре). Растворы ДНК охлаждают до 0 °C и приливают к ним по 2 мл холодного раствора 2 н. НС1О4. Пробирки оставляют при 0 — 6 °C не менее чем на 4 ч. Осадки, содержащие ДНК, собирают на мембранных фильтрах,
которые с помощью пинцета (матовой стороной вверх) помещают в воронку Бюхнера, соединенную с колбой Бунзена и водоструйным насосом. Осадки промывают растворами холодной 0,3 н. НС1О4 (3 объема по отношению к исходному раствору, 12 мл на каждую пробу) и 70%-го этилового спирта (2 объема, 8 мл).
Затем фильтры высушивают на воздухе, помещают во флаконы (осадком вверх) и заливают сцинтилляционной жидкостью ЖС-106 (по 5 мл). Радиоактивность измеряют на автоматическом счетчике.
Содержание ДНК определяют в клетках 72-часовых культур вариантов 3 и 4 одним из следующих методов.
Колориметрическое определение ДНК с дифениламиновым реактивом в экстракте нуклеиновых кислот из клеток хлорной кислотой (Методы общей бактериологии, 1984). Чувствительность метода 5 — 50 мкг ДНК • мл-1. Для приготовления хлорного экстракта клетки из 10 мл культуры собирают центрифугированием, ресуспендируют в 5 мл 10%-й НС1О4, нагревают в течение 15 мин в кипящей водяной бане, центрифугируют и в супернатанте определяют содержание ДНК. Для определения используют следующие реактивы:
Реактив. А: 1,5 г дифениламина растворяют в 100 мл ледяной уксусной кислоты и добавляют 1,5 мл концентрированной H2SO4.
Реактив В: 0,5 мл уксусного альдегида растворяют в 25 мл дистиллированной Н2О.
Реактив С\ непосредственно перед определением смешивают 20 мл реактива А и 0,1 мл реактива В. К 1 мл экстракта, содержащего ДНК, добавляют 2 мл реактива С и выдерживают 14 ч при 37 °C. Измеряют поглощение при 600 нм. Калибровочную кривую строят, используя стандартный раствор ДНК в 10%-й НС1О4. Диапазон концентраций ДНК 20 — 100 мкг-мл-1.
Определение ДНК в бесклеточном экстракте с коммерческим реактивом «Хёкст» (Hoechst 333258) проводят на специализированном минифлуориметре или спектро-флуориметре. Для приготовления бесклеточного экстракта клетки суспендируют в СФС, pH 7,5 (нестерильно) в соотношении вес (сырой биомассы): объем СФС =1:4. Затем производят ультразвуковую обработку суспензий на УЗДН-2Т в режиме: 22 кГц, 40 мкА, 10 раз по 30 с при двухминутном охлаждении (0 °C) между озвучиваниями.’ Фрагменты неразрушенных клеток удаляют центрифугированием (30 тыс. g, 20 мин,' 6 °C, центрифуга фирмы «Beckman», США, модель J2-21, центрифужные стаканы на 50 мл с крышками). Рабочий раствор реактива «Хёкст» должен содержать 1 мг сухого вещества в 1 мл буферного раствора (БР): 0,01 М трис-НС1 (pH 7,4) + 1 мМ ЭДТА + 0,1 М NaCl. В мерную колбу, содержащую 100 мл БР, следует внести 10 мкл рабочего раствора реактива «Хёкст».
При измерении концентрации ДНК с помощью спектрофлуориметра проводят его калибровку с помощью стандартного раствора ДНК в буферном растворе реактива «Хёкст». Предварительно следует найти значения длин волн максимального поглощения и максимального испускания света (флуоресценции) стандартным раствором. После этого можно проводить измерение интенсивности флуоресценции в опытных растворах ДНК и рассчитывать ее концентрацию (мкг мл-1) по стандартной кривой. Перед измерением прибор устанавливают в 0-положение. Для этого в камеру ставят пустую кювету, поворачивают ручку «scale» вправо, до упора; ручкой «zero» выставляют «000» на табло. Затем в кювету вносят 2 мл буферного раствора с реактивом «Хёкст», добавляют 2 мкл стандартного раствора ДНК (100 мкг-мл~') и ручкой «scale» выводят на табло это значение. Для измерения концентрации ДНК в опытных образцах в кювету также вносят 2 мл буферного раствора с реактивом «Хёкст» и добавляют 2 мкл исследуемого препарата.
Показание (концентрация ДНК, мкг-л-1) высвечивается на табло. Чувствительность метода — 10 мкг - мл-1.
После проведенных измерений сравнивают концентрацию ДНК в клетках вариантов 3 и 4. Содержание ДНК рассчитывают в 1 г АСБ (с учетом разбавления суспензии). Для того чтобы точнее сравнить содержание ДНК в препаратах вариантов 3 и 4 следует измерить в них концентрацию белка и рассчитать отношение ДНК/белок. Концентрацию белка можно измерить любым из известных способов (см. гл. 14).
Материалы, посуда, оборудование, реактивы
Оборудование (в расчете на группу студентов): центрифуга с охлаждением (6 тыс. g); центрифуга с охлаждением (30 тыс. g); воздушный и водяной термостаты с температурой 30 и 37 °C; водоструйный насос; газожидкостный хроматогроф (ГЖХ, например CHROM 4, Чешская Республика), оборудованный пламенноионизационным детектором и колонкой, размером 1000 * 4 мм), заполненной полиэтиленгликоль-адипатом на хромосорбе W; pH-метр; ультразвуковой дезинтегратор УЗДН-2Т (Россия); спектрофотометр с диапазоном длин волн 260 — 600 нм; спектрофлуориметр (например, фирмы «Jobin Ivon», Франция) и/или минифлуориметр (ТКО 100-dedicated mini fluorimeter, Hoefer Scientific Instruments, США); автоматический счетчик радиоактивности (например, фирмы «Pharmacia-LKB», Швеция).
Посуда и инструменты (в расчете на 1 студента): для приготовления стерильных сред 1—4: колба или другая посуда объемом 2 л. Посуда с ватными пробками: цилиндр или градуированная колба на 50 мл, 4 колбы на 250 мл, одна обычная пробирка, две широкие пробирки; колбы Эрленмейера (две на 100 мл и две на 750 мл или на 1 л), одна колба на 100 мл (на группу). Стерильные пипетки: на 10, 5 (по 2 шт.), 2 и 1 мл (по 5 шт.). Для определения суммарных корриноидов (витамина В|2) в культурах и клетках: колба (на 250 мл) для приготовления «основной» среды (ОС); 2 стерильные пробирки (одна из них со стерильной дистиллированной водой, 5 мл), 20 широких пробирок с ватными пробками, градуированных на 10 мл; пипетки на 2, 5 и 10 мл (по 4 шт.); пробирки, градуированные на 10 мл (2 шт.); центрифужные стаканы (на 250 мл, 4 шт.), 2 колбы (на 250 мл, желательно с притертыми крышками). Для регистрации биосинтеза (образования) ДНК в клетках: химически чистые пробирки (32 шт.); автоматические пипетки: на 1 мл с переменным объемом и с фиксированными объемами на 50 и 20 мкл; носики на 1 мл, 50 и 20 мкл; штативы для пробирок и автоматических пипеток; пипетка на 10 мл (со специальным устройством для отбора агрессивной жидкости); мембранные фильтры диаметром 25 мм, с порами диаметром 0,4 или 0,6 мкм; колба Бунзена с воронкой Бюхнера; 5 чашек Петри с бумажными фильтрами на дне, разделенными (карандашом) на две части; флаконы для счета радиоактивности (30 шт.); 4 колбы на 100 — 250 мл (желательно с притертыми крышками); 2 цилиндра на 50 или 100 мл; пинцет; секундомер; резиновые перчатки. Для получения гомогенатов клеток (при измерении ДНК): 2 химических стакана объемом 50—100 мл, секундомер, центрифужные стаканы с крышками (на 50 мл, 4 шт.), колба на 1 л.
Реактивы, растворы и штамм теап-культуры (для определения витамина В12):
. глюкоза (чда); соли (хч или чда); (NH4)2SO4; КН2РО4; MgSO4-7H2O; триптон, например Tripton Bacto («Ferae», Berlin); пептон (Serva, Германия) или гидролизат казеина (Россия); NaCl; MnSO4- 5Н2О; ZnSO 7Н2О; FeCl3- 6Н2О; биотин; тиамин; пан-тотенат-Са; 10%-е растворы NaHCO3 и H2SO4; индикатор pH бромтимолблау;
• Е. coli, штамм 113-3; цианокобаламин (CN-Cbl, Россия); NaNO2; К2НРО4; натрий лимоннокислый; раствор 0,1 М НС1, 0,9%-й NaCl (10 мл — на группу);
♦ газы в баллонах: азот и водород; водные растворы уксусной и пропионовой кислот;
. нестерильный СФС + 0,5%-я глюкоза (50 мл, pH 7,0); аденин («Химреактив», Москва), [8-14С]аденин (ПО «Изотоп», С.-Петербург; удельная радиоактивность 10—12 ГБк г *);
растворы: 1,2 н. КОН; 0,6 н. НС1; 2 н. НСЮ4; 0,3 н. НСЮ4; 70%-й водный раствор этилового спирта; сцинтилляционная жидкость ЖС-106 (ПО «Изотоп», С.-Петербург);
• СФС (без глюкозы), pH -7,5; мелкие кусочки льда; реактив «Хёкст» (Hoechst 333258, Hoefer Scientific Instruments, San Francisco, USA, 100 мг); буферный раствор 0,01 M /ирпс-HCl, содержащий I мМ ЭДТАи 0,1 М NaCl.
План проведения работы
На проведение работы необходимо 60 ч.
Занятие 1. Приготовление и стерилизация сред и посуды для культивирования Р. freudenreichii.
Занятие 2. Завершение приготовления сред для культивирования Р. freudenreichii. Посев культуры. Приготовление и стерилизация основной среды и среды для предварительного культивирования Е. coli 113-3. Приготовление стандартного раствора витамина В12 (цианокобаламина).
Занятие 3. Измерение оптической плотности и подведение pH растущих культур. Приготовление буферного раствора для измерения ДНК в гомогенате клеток с реактивом «Хёкст» и льда для разрушения клеток на ультразвуковом дезинтеграторе (хранить в холодильнике). Подготовка растворов для измерения интенсивности образования ДНК по включению меченого аденина в щелочестабильную кислотонерастворимую фракцию клеток.
Занятие 4. Измерение оптической плотности и подведение pH растущих культур. Приготовление растворов меченого и немеченого аденина. Калибровка колонок ГЖХ пропионовой и уксусной кислотами.
Занятие 5. Измерение оптической плотности и подведение pH во всех вариантах культур. Проведение предварительного посева тест-культуры (Е. coli 113-3) для определения витамина В12. Подготовка и стерилизация образцов для определения витамина В|2 в культурах вариантов 3 и 4 микробиологическим методом. Проведение с этими же вариантами опыта по измерению интенсивности образования ДНК до стадии щелочного гидролиза РНК. Отбор по 200 мл культур этих же вариантов для определения витамина В12 спектрофотометрическим методом и для измерения содержания ДНК в гомогенатах клеток, которые хранятся в холодильнике до следующего занятия.
Занятие 6. Нейтрализация щелочи в образцах с радиоактивной ДНК. Образцы хранятся в холодильнике до следующего занятия. Посев Е. coli 113-3 в образцы для проведения микробиологического определения витамина В12. Сбор биомассы вариантов 3 и 4 центрифугированием для спектрофотометрического измерения содержания суммарных корриноидов и для измерения содержания ДНК в гомогенатах клеток. Отбор культуральной жидкости для определения летучих жирных кислот.
Занятие 7. Измерение роста тест-культуры с препаратами витамина Bt2. Расчет содержания корриноидов по стандартной кривой. Продолжение обработки образцов радиоактивной ДНК. Определение радиоактивности образцов.
Занятие 8. Определение содержания корриноидов спектрофотометрическим методом. Расчет суммарного содержания корриноидов в 1 л культуры.
Занятие 9. Определение содержания ДНК в гомогенатах клеток вариантов 3 и 4 с реактивом «Хёкст» (Hoechst 333258). Определение количества пропионовой и уксусной кислот в культуральной жидкости.
Занятие 10. Оформление результатов. Построение кривых роста культур вариантов 1, 2, 3, 4. Выявление различий. Сравнение данных по общему содержанию корриноидов, полученных микробиологическим и спектрофотометрическим методами. Расчет радиоактивности ДНК. Построение кривых включения меченого аденина в ДНК для вариантов, различающихся по уровню образования корриноидов. Расчет количества летучих кислот, образованных вариантами культур Р. freudenreichii, различающихся по содержанию корриноидов в клетках. Обсуждение полученных результатов.
Глава 24
ПОЛУЧЕНИЕ ЭНЕРГИИ ЛИТОТРОФНЫМИ БАКТЕРИЯМИ
ЗАДАЧА 1. ОКИСЛЕНИЕ СОЕДИНЕНИЙ СЕРЫ СЕРНЫМИ (ТИОНОВЫМИ) БАКТЕРИЯМИ
Микроорганизмы, окисляющие соединения серы, можно разделить на три группы: фотосинтезирующие (окрашенные бактерии), бесцветные серобактерии и тионовые бактерии. Фотосинтезирующие бактерии окисляют сероводород и серу в анаэробных условиях на свету. Серобактерии окисляют сероводород кислородом воздуха и могут откладывать капли серы внутри клетки. Предполагают, что сера, которую откладывают в клетках бесцветные серобактерии, может быть использована как акцептор электронов при окислении органических веществ в анаэробных условиях, например при нахождении бактерий в илах. Считают также, что окисление сероводорода иногда может иметь защитное значение для бесцветных серобактерий, не обладающих каталазной активностью. В этом случае образующаяся в клетках перекись водорода выступает окислителем сероводорода и восстанавливается до воды.
К серобактериям относят микроорганизмы различного происхождения: нитчатые скользящие серобактерии родов Beggiatoa, Thiothrix, Thioploca, Thiospirillum и др., серобактерии с гигантскими одиночными клетками (Achromatium, Macromonas и Thiovulvum) и относительно мелкие формы (Thiospira и Thiobacterium).
Лишь немногие штаммы серобактерий удается выращивать в виде чистых культур на минеральной среде с сероводородом. Чаще они требуют присутствия небольших количеств органических веществ. Таким образом, среди серобактерий имеются хемо-литоавтотрофные и хемолитогетеротрофные виды.
В отличие от серобактерий, группа тионовых бактерий изучена лучше. Эти формы при окислении сероводорода не откладывают серу внутри клеток, большинство из них — хемолитоавтотрофы. Они представляют собой единую в морфологическом и биохимическом отношении группу организмов. Все тионовые бактерии, за исключением небольшого числа денитрифицирующих видов, — аэробы и способны использовать энергию окисления восстановленных соединений серы в серную кислоту для ассимиляции углерода, образования веществ клетки и для других функций. Некоторые тионовые бактерии способны кроме окисления серы использовать для своей жизнедеятельности окисление других соединений, например органических веществ или закисного железа.
Морфологически тионовые бактерии представляют собой мелкие палочки с округлыми концами и одним полярным жгутиком, этим они напоминают типичные псевдомонады. Различают группу автотрофных тиобацилл, развивающихся в кислой среде (например, Acidithiobacillus thiooxidans), окисляющих ионы железа (A. ferrooxidans), развивающихся в нейтральной среде {Thiobacillus thioparus) и денитрифицирующих (Т. denitrificans). К этой группе относятся основные виды тиобацилл. Существуют также гетеротрофные тиобациллы, развивающиеся в нейтральной или щелочной среде (Т. novel his).
Конечным продуктом окисления серы у всех тионовых бактерий является сульфат. Ниже приведена схема окисления тиосульфата у различных тиобацилл, подтвержденная экспериментальными данными. По этой схеме тиосульфат либо превращается в тетратионат под действием тиосульфатокисляющего фермента (J), либо расщепляется на серу и сульфит при участии тиосульфатрасгцепляющего фермента — роданезы (2).
S—SO3 2
O3S—s—s—so / /3
+ SOj"
I 6<-АМФ
SO3“ + АФС
I / 7«—Фн
SO?” —>ДЦФ
По-видимому, при низкой концентрации сульфата идет прямое расщепление, а при высоком его содержании — образование тетратионата. Под действием неизученного фермента (5) тетратионат гидролизуется с восстановлением в тиосульфат, серу и сульфит. Окисление серы ведет к образованию сульфита, но не тиосульфата. Реакцию осуществляет сероокисляющий фермент (4), который, возможно, является оксигеназой. Окисление сульфита может осуществляться сульфитоксидазой (сульфитцито-хром с-оксидоредуктазой (5) или проходит с учас
тием АМФ, аденилилсульфатредуктазы (6), АДФ-сульфурилазы (7) и идет с субстратным фосфорилированием. Акцептором электрона сульфатредуктазы может быть цитохром с, окисление которого, по-видимому, связано с окислительным фосфорилированием, однако неизвестно, какое количество АТФ синтезируется в результате потери атомом серы 8ё при окислении сульфида до сульфата. Ферментная система, окисляющая сульфид до элементарной серы, неизвестна (8). Однако легко можно наблюдать быстрое внутриклеточное образование элементарной серы хемолитотрофами, использующими сероводород.
Пель работы, ознакомление студентов с морфологией и физиологией сероокисляющих и тионовых бактерий, а также процессом образования клетками сульфатов при развитии на средах с восстановленными соединениями серы.
Ход выполнения задачи
Микроорганизмы и их культивирование. Работу проводят с представителями бесцветных серобактерий Beggiatoa sp., Macromonas sp. и Thiospira sp., а также тионовыми бактериями Thiobacillus thioparus, A. thiooxidans, A. ferrooxidans. Штаммы тионовых бактерий, используемые в задаче, получают энергию в результате окисления минеральных восстановленных соединений серы или свободной серы, при этом сера в качестве промежуточного продукта окисления откладывается вне клеток. Акцептором электронов служит молекулярный кислород.
Для Т. thioparus готовят 500 мл среды следующего состава (г/л): NH4C1 — 0,1; Na2HPO4- 2Н2О — 0,2; MgCl2- 6Н2О — 0,1; вода водопроводная — 800 мл. Среду стерилизуют при 0,5 ати 30 мин. После стерилизации в среду стерильно вносят 100 мл 5%-го раствора Na2S2O3- 5Н2О и 100 мл 1%-го раствора NaHCO3 в дистиллированной воде, доводят pH до 8 — 8,5 с помощью стерильных 10%-х растворов NaHCO3 или НС1.
Агаризованная среда для Т. thioparus имеет следующий состав (г/л): Na2S2O3- 5Н2О - 5,0; К2НРО4 - 0,1; NH4C1 - 0,1; NaHCO3 - 0,2; агар - 20,0; вода водопроводная — 1 л. До стерилизации в среду добавляют избыток СаСО3. После стерилизации при 1 ати 30 мин доводят pH до 8 — 8,5 и разливают среду в стерильные чашки Петри.
Состав среды для A. thiooxidans (г/л): (NH4)2SO4 — 0,2; КН2РО4 — 3,0; MgSO4 • 7Н2О — 0,5; СаС12-6Н2О — 0,25; FeSO4-7H2O — следы; S° (порошок) — 10,0; вода дистиллированная —1л. Готовят 500 мл среды, разливают по 100 мл в кача-лочные колбы на 750 мл, стерилизуют 30 мин при 0,5 ати, после стерилизации доводят pH до 4,0 с помощью стерильного 10%-го раствора Н3РО4.
A. ferrooxidans культивируют в той же среде, значение pH которой доводят перед посевом до 3,5 стерильным 10%-м раствором.
В процессе выращивания отбирают пробы культур A. thiooxidans, Т. thioparus л A. ferrooxidans через 2, 3 и 5 сут культивирования.
Микроскопирование. Готовят препарат «раздавленная капля» и просматривают его в микроскопе с фазово-контрастным устройством. Визуально фиксируют наличие капель серы в колониях Т. thioparus при росте на чашках Петри с агаризованной средой и зарисовывают их в отраженном свете. Из культур бесцветных серобактерий, отобранных из пробирок, готовят препарат «раздавленная капля». В микроскопе с фазово-контрастным устройством наблюдают за скользящим движением Beggiatoa sp., ее морфологией, отложением капель серы в клетках, морфологией крупных палочковидных подвижных клеток Macromonas sp. с полярно расположенным жгутиком и Thiospira sp., спирилл с винтообразным движением (культуры предоставляет преподаватель).
В течение 30 — 60 мин наблюдают за образованием и накоплением серы внутри клеток Thiospira sp. при инкубации их в среде с полисульфидом. Для этого каплю суспензии клеток Thiospira sp., отделенных от культуральной жидкости центрифугированием (3 тыс. g, 10 мин), помещают на предметное стекло, добавляют каплю полисульфида (10 г Na2S • 9Н2О + 3 г S° + 15 мл Н2О), закрывают покровным стеклом и рассматривают под микроскопом с объективом 40 х. Наблюдение можно проводить в поляризованном свете микроскопа.
Определение биомассы и pH. За развитием культур следят по накоплению биомассы, изменению pH, помутнению среды (за счет выделения серы клетками тиобацилл). Биомассу определяют на примере Т. thioparus. Предварительно избавляются от выделившейся в среду серы путем центрифугирования 5 мл культуральной жидкости, отобранной из каждой колбы, при 2 тыс. g на настольной центрифуге в течение 2 мин. После осаждения серы оптическую плотность супернатанта измеряют в кювете с длиной оптического пути 0,5 см (контролем служит вода). Об изменении кислотности серы судят по измерению pH культуральной жидкости потенциометрически после определения в ней биомассы на нефелометре.
Определение количества образованного сульфата. В динамике развития культур A. thiooxidans, Т. thioparus и A. ferrooxidans определяют количество сульфатов, образованных при окислении тиосульфата и элементарной серы. В основе метода лежит свойство сульфатов образовывать нерастворимую в воде соль BaSO4, оптическую плотность суспензии которой определяют нефелометрически и по стандартной кривой пересчитывают на содержание сульфатов в определяемом растворе.
Ход определения. К 0,2 мл опытного образца (культуральная жидкость после отделения клеток и серы центрифугированием при 3 тыс. g, 15 мин), содержащего ионы SO4", прибавляют 3,8 мл 4%-й трихлоруксусной кислоты и 1 мл раствора ВаС12-желатин. Смесь тщательно перемешивают и через 10—15 мин величину рассеивания измеряют на ФЭК-56 при длине волны 560 нм (светофильтр № 6) в кювете с длиной оптического пути 0,5 см. Чувствительность метода — 100 мкг/мл. Стандартную кривую строят по Na2SO4 или MgSO4- 2Н2О при концентрациях сульфата 100, 500, 1500 и 2000 мкг/мл. Реактивы для определения: ТХУ — 4%-й раствор; ВаС12-желатин: 0,5 г желатина растворяют в 100 мл горячей воды (60 — 70 °C), выдерживают 6 ч; 0,5 г ВаС12 2 — 3 ч растворяют в желатиновом растворе, затем фильтруют в горячем состоянии, раствор стабилен в холодильнике 3—4 дня. На основании полученных данных строят калибровочную кривую: по оси абсцисс откладывают концентрацию SO4~ (мкг/мл), по оси ординат — оптическую плотность растворов. Данные определений заносят также в таблицу.
Оформление результатов. Рассчитанные значения количества SO4~, содержащиеся в культуральной жидкости, вносят в табл. 24.1.
Таблица 24.1
Образование сульфата тионовыми бактериями
Название штамма Возраст культуры, сут Оптическая плотность клеток, ед. Количество SO^“, мкг/мл Интенсивность процесса, мкг/мл ед. опт. пл.
Т. thioparus A. thiooxidans A. ferrooxidans 0 2 3 5 0 2 3 5 0 2 3 5
Делают вывод о проводимом культурами тиобацилл процессе и сравнивают скорости процесса у разных культур в пересчете на биомассу.
Материалы, оборудование, посуда, реактивы
Микроскоп; фазово-контрастное устройство; мерные колбы на 100 мл — 2; на 50 мл — 2; чашки Петри — 5; пипетки на 1—2 мл (стерильные) — 18; шпатель — 1; пробирки — 30; реактивы для приготовления сред и определения количества сульфата. ФЭК.
План выполнения задачи
На выполнение задачи требуется 25 — 30 ч.
Занятие 1. Приготовление и стерилизация сред; стерилизация посуды.
Занятие 2. Посев культур тионовых бактерий, отбор начальных точек, построение калибровочной кривой по сульфату, определение начальной биомассы и содержания сульфатов.
Занятия 3, 4, 5. Микроскопирование, определение pH, оптической плотности двух-, трех- и пятисуточной культуры, приготовление реактивов и определение сульфатов, оформление и обсуждение результатов.
ЗАДАЧА 2. ОКИСЛЕНИЕ СОЕДИНЕНИЙ АЗОТА.
ОПРЕДЕЛЕНИЕ ПРОДУКТОВ ЭНЕРГЕТИЧЕСКОГО МЕТАБОЛИЗМА НИТРИФИЦИРУЮЩИХ БАКТЕРИЙ
Нитрифицирующие бактерии относятся к группе хемолитоавтотрофов, получающих энергию для своего роста за счет окисления неорганических соединений азота. Хемолитоавтотрофы образуют клеточный материал из СО2, а необходимую для этого энергию и восстановительные эквиваленты в виде НАД(Ф)Н извлекают при окислении строго определенного для каждой группы субстрата.
Процесс нитрификации, который протекает в почве, навозе, воде и приводит к окислению аммиака до NO2 и затем до NO3, с давнего времени известен в сельскохозяйственной практике и в процессе приготовления селитры для изготовления пороха. До середины XIX в., т. е. до работ Л. Пастера, эти превращения объясняли чисто химическими явлениями. Пастер предположил участие в них микроорганизмов. К бактериям, осуществляющим I фазу нитрификации, относятся представители четырех родов: Nitrosomonas, Nitrosospira, Nitrosococcus, Nitrosolobus. Дальнейшее окисление NO2 до NO3, выполняют нитритокисляющие бактерии: Nitrospina, Nitrobacter, Nitrococcus. Бактерии обеих фаз представляют собой грамотрицательные, довольно мелкие микроорганизмы. Морфологическое изучение нитрификаторов проводится с использованием фазово-контрастной и электронной микроскопии.
Метаболизм нитрифицирующих бактерий характеризуется способностью бактерий использовать для конструктивных процессов СО2 как единственный источник углерода; ассимиляция углекислоты идет через фосфорилированные продукты рибулозоди-фосфатного цикла, синтез мономеров — через цикл трикарбоновых кислот, который у них, как и у многих облигатных хемоавтотрофов, не замкнут. Особенности регуляции обмена мономеров обусловливают токсическое действие ряда органических веществ на нитрифицирующие бактерии. Синтез НАДН и получение энергии, необходимой для ассимиляции СО2, происходят в результате окисления неорганических доноров электронов NH4 и NO2- При этом, поскольку место входа электронов в дыхательную цепь находится на уровне цитохрома с у аммонийокисляющих и цитохрома о, — нитрит-окисляющих бактерий, часть аккумулированной бактериями энергии используется на осуществление обратного транспорта электронов против термодинамического градиента для восстановления НАД+. Стандартный окислительно-восстановительный потенциал пары НАДН/НАД+-0,32 В, тогда как стандартные потенциалы пар NO£/NH2OH и NO3/NO7 равны соответственно +0,066 В и +0,42 В, поэтому для восстановления 1 моль НАД+ такими донорами с высоким окислительно-восстановительным потенциалом необходимо затратить энергию около 75 и 143 кДж — это может быть достигнуто за счет гидролиза 3 и 5 молекул АТФ соответственно. В связи с тем что в бактериях имеется всего один пункт генерирования энергии, значительная часть которой используется для восстановления НАД+ против термодинамического градиента, клетки нитрификаторов растут довольно медленно, минимальное время их удвоения — около 24 ч.
Пель работы: изучение процесса нитрификации путем определения продуктов энергетического метаболизма нитрифицирующих бактерий и наблюдение динамики роста клеток.
Ход выполнения задачи
Микроорганизмы и их культивирование. В работе используют двух представителей нитрификаторов I фазы — Nitrosomonas sp. и Nitrosospira sp. и одного представителя II фазы — Nitrobacter winogradskyi.
Энергетическим субстратом и источником азота для нитрифицирующих бактерий I фазы является NH4. Эти микроорганизмы культивируют в среде Виноградского (для I фазы — среда 1) или на среде Сориано и Уокера (среда 2).
Среда 1 (г/л): (NH4)2SO4 — 2,0; К2НРО4 — 1,0 (стерилизуют отдельно); MgSO4-7H2O — 0,5; NaCl — 2,0; FeSO4 — 0,05; СаСО3 — 5,0; среда 2 (г/л): (NH4)2SO4 — 0,5; КН2РО4 — 0,2 (стерилизуют отдельно); СаС12-2Н2О — 0,04; MgSO4 • 7Н2О — 0,04; Fe-лимоннокислый — 0,0005 (стерилизуют отдельно); 1 мл 0,05%-го раствора фенолового красного. Среды стерилизуют при 1 ати, после чего
pH в них доводят до 7,5—7,8 с помощью 5%-го раствора NaHCO3, а в среду 2 вносят также 1 мл стерильного раствора микроэлементов (по Пфеннигу и Липперту, см. приложение 4).
Как видно при сравнении сред 1 и 2, они включают одни и те же основные компоненты: фосфор, магний, железо, кальций, однако кальций, введенный в среду 1 в виде карбоната, определяет мутность этой среды (среда 2 прозрачна) и регулирует постоянно меняющийся в процессе культивирования pH. При выращивании нитрификаторов на среде Сориано и Уокера происходит быстрое подкисление среды за счет образования возрастающих количеств нитритов. Уменьшение pH в этом случае можно отметить с помощью универсального индикатора. Более того, добавленный при посеве индикатор феноловый красный вызывает при подкислении среды изменение цвета культуральной жидкости от сиреневатых до желтоватых оттенков. Аммонийокисляющие бактерии чувствительны к подкислению среды, поэтому для сохранения их дальнейшего роста на среде Сориано и Уокера следует с помощью 0,1 %-го NaHCO3 ежедневно поддерживать pH в нужных пределах (7,5—7,8).
Энергетическим субстратом и источником азота для возбудителя II фазы нитрификации является NO2. Для культивирования Nitrobacter Winogradsky7, представляющего эту группу микроорганизмов, используются также среда Виноградского (для II фазы — среда 3) и среда Ватсона и Вотербари (среда 4). Среда 3 (г/л): NaNO2 — 1,0; К2НРО4 — 0,5 (стерилизуют отдельно); MgSO4- 7Н2О — 0,5; NaCl - 0,5; FeSO4- 7Н2О - 0,005; Na2CO3 - 1,0. Среда 4 (г/л): NaNO2 - 0,07, MgSO4-7H2O — 0,1; СаС12-6Н2О — 0,0006; КН2РО4 — 0,02 (стерилизуют отдельно). Среды стерилизуют при 1 ати 30 мин, после стерилизации pH сред доводят до 7,5 с помощью 5%-го NaHCO3, а в среду 4 вносят 1 мл стерильного раствора микроэлементов (по Пфеннигу и Липперту). Клетки Nitrobacter winogradskyi, культивируемые в этих средах как представители II фазы нитрификации, в отличие от аммонийокисляющих бактерий не производят при росте заметного сдвига pH среды.
Приготовленные среды разливают по 100 мл в качал очные колбы на 750 мл, стерилизуют, вносят по 10 мл соответствующей бактериальной суспензии 5 — 7-суточного возраста и выращивают на качалке. Некоторое количество среды всех вариантов оставляют незасеянной для определения исходного количества субстратов.
Микроорганизмы культивируют на качалке при 30 °C в течение 5 — 7 сут. Жидкие культуры нитрифицирующих бактерий поддерживают в темноте при 15 °C и пересевают каждые 4—6 мес. Для аммонийокисляющих бактерий используют среду, содержащую 0,001%-й индикатор феноловый красный, 0,5 М HEPES-буфера (pH 7,8 —8,0) и соответствующие соли. Через 6 — 8 нед цвет среды изменится с красного на желтый, указывая на окисление большей части аммония, поэтому в среду вносят дополнительное количество аммония и доводят pH до нужного значении. При поддержании нитритокисляющих бактерий периодически анализируют содержание нитритов и при уменьшении их количества в среду вносят новую порцию NaNO2 до нужного уровня. Перед засевом в опыт культуру инкубируют при 25 °C в течение нескольких дней на свежей среде.
В процессе роста культур изучают динамик}' окисления бактериями неорганических энергодающих субстратов. Регистрируют изменение pH в случае нитрификаторов 1 фазы.
Определяют сначала наличие (качественные реакции), а при получении положительного ответа — количество NH4 и NO/ у бактерий I фазы окисления, у бактерий II фазы — NO/и NO/. Изучение показателей энергетического обмена нитрификаторов проводят в культуральной жидкости после отделения клеток центрифугированием при 6 тыс. g. Полученные данные сравнивают с исходными уровнями окисляемых субстратов, которые определяют в средах, не содержащих посевного материала.
Морфологическое изучение нитрифицирующих бактерий проводят с использованием фазово-контрастной микроскопии. Nitrosomonas sp. — это мелкая, овальная палочка, почти кокковидная; ее размеры 0,6—1 х 0,9 — 2 мкм, размножается делением пополам. Nitrosospira sp. — также представитель нитрификаторов I фазы, в чистой культуре имеет вид палочковидных клеток, которые, как показывают электронно-микроскопические исследования, представляют собой плотно свернутые спирали нитей диаметром 0,3 — 0,4 мкм.
Определение NO/. Количество NO/, образуемого клетками Nitrosomonas и Nitrosospira, определяют в 4-, 6- и 8-суточных культурах.
Качественная реакция на присутствие NO/. На белой пластинке 1 каплю реактива Грисса смешивают с 1 каплей исследуемой жидкости. При наличии NO2 развивается интенсивное малиновое окрашивание.
Колориметрический метод определения NO/ с сс-нафтиламином и сульфаниловой кислотой основан на диазатировании сульфаниловой кислоты присутствующими в пробе нитритами с образованием красно-фиолетового азокрасителя. Чувствительность метода 20— 100 мкг/мл. В анализируемой пробе не должны присутствовать сильные окислители и сильные восстановители.
Реактивы, сульфаниловая кислота, 0,6%-й раствор (растворяют 6 г сульфаниловой кислоты в 750 мл горячей дистиллированной воды; к полученному раствору добавляют 250 мл ледяной уксусной кислоты); а-нафтиламин, 0,6%-й раствор (растворяют 1,2 г а-нафтиламина в дистиллированной воде, прибавляют 50 мл ледяной уксусной кислоты и доводят до 200 мл водой).
Ход определения. Берут 1 мл исследуемого образца и доводят до 45 мл водой в мерной колбе на 50 мл. Прибавляют 1 мл раствора сульфаниловой кислоты и тщательно перемешивают. Через 5 мин прибавляют 1 мл свежеприготовленного раствора а-нафтиламина, смесь снова перемешивают и доводят до метки водой. Затем образцы колориметрируют на ФЭК — светофильтр зеленый, X = 520 нм, кювета 1,0 см. Полученная окраска нестойкая. Концентрацию NO2 определяют по формуле CNO_ (мкг/мл) = С- 50/К, где С — концентрация NO/ в мкг/мл, определенная по калибровочной кривой, составленной на основе различных концентраций NO/ (от 20 до 100 мкг/мл); V — объем пробы, взятой для определения (50 — объем, до которого разбавляется проба). Для определения количества NO/ применяют также модификацию этого метода, в которой вместо а-нафтиламина используют так называемую кислоту Клеве (1-нафтиламин-7-сульфоновую кислоту).
Определение NO/.
Колориметрическое определение NO/. Определение основано на образовании желтого соединения в результате реакции нитратов с фенолдисульфоновой кислотой. Чувствительность метода от 50 до 250 мкг/мл NO/.
Реактивы. Фенолдисульфоновая кислота: растворяют 25 г фенола (неокрашенного) в 150 мл конц. H2SO4. Прибавляют 75 мл дымящей H2SO4 (олеум с 15 % SO3), тщательно перемешивают и нагревают с обратным холодильником в течение 2 ч на кипящей водяной бане.
Ход определения. 2 мл образца досуха упаривают в фарфоровой чашке на водяной бане. После охлаждения прибавляют 1 мл фенолдисульфонового реактива и растирают осадок стеклянной палочкой. Через 5 мин прибавляют 15 мл 10%-го раствора NaOH, при этом жидкость окрашивается в желтый цвет. Полученный раствор помещают в
мерную колбу на 100 мл, доводят до метки дистиллированной водой и колориметрируют, светофильтр № 3 (А. = 410 нм, кювета 1 см). Концентрацию NO3 определяют по формуле CN0- (мкг/мл) = С- 100/К где С— концентрация NO3 в мкг/мл, определенная по калибровочной кривой, составленной на основе концентраций KNO3 (от 50 до 250 мкг/мл), 100 — объем окрашенной пробы, мл; V — объем пробы, взятой для анализа.
Количественное определение NO3 с помощью ион-селективного электрода. Ион-се -лективные электроды представляют собой электрохимические полуэлементы, для которых разность потенциалов на границе раздела фаз электродный материал — электролит зависит от активности (концентрации) определяемого в среде иона.
Основные принципы использования ион-селективных электродов. Величина активности иона а, измеряемая с помощью ион-селективного электрода, связана с его концентрацией С простым соотношением: а = у С, где = у — коэффициент активности иона. Он может быть экспериментально найден или вычислен по формуле
. A-Z2-y/S
-Igy =------
1 + Bbjs
где Z — заряд иона; S — ионная сила; S = 1/2 - ^C/Z^fC,- — концентрация /-иона с зарядом Z,); А, В, Ь — константы: для NOj А = 0,51; В = 0,33; Ь = 3 при 25 °C. Приближенные значения коэффициентов активности ионов в зависимости от ионной силы раствора приведены в табл. 24.2.
Погружение электрода в раствор, содержащий исследуемые ионы, вызывает на его поверхности обменную реакцию, в результате которой индикаторный электрод приобретает потенциал, обусловленный активностью данного иона в измеряемой среде. Для измерения активностей ионов в исследуемых объектах составляют электрическую цепь из индикаторного и вспомогательного электрода, а также измерительного прибора (pH-метра, ионометра, потенциометра), погружают оба электрода в раствор и измеряют ЭДС полученной цепи. Индикаторный электрод должен быть предварительно прокалиброван. Для этого измеряются ЭДС цепей с несколькими стандартными растворами, в которых известна активность измеряемого иона и строится график зависимости ЭДС от рХ, где рХ — отрицательный логарифм активности определяемого иона X. Если полученная зависимость имеет линейный характер, а изменение рХ на 1 соответствует скачку ЭДС на 59 мВ, то используемый электрод можно считать пригодным для работы. Скачок потенциала между электродом и средой в простейшем случае описывается уравнением Нернста
£= Ео'± 2,303^7" lgax- «Т1,
где Ео — потенциал электрода в среде с активностью ионов а = 1, т. е. стандартный потенциал электрода, мВ; R — универсальная газовая постоянная; Т — абсолютная
Таблица 24.2
Значения коэффициентов активности ионов в зависимости от ионной силы раствора
Ионнная сила раствора, М ю-4 2-10-4 5-Ю-4 io-3 2 -10-3 5-10-3 io-2 2-10-2 0,1 0,2
Коэффициент активности однозаряженного иона 0,99 0,98 0,97 0,96 0,94 0,92 0,90 0,85 0,75 0,70
температура среды, К; F— постоянная Фарадея; п— изменение заряда определяемого иона в результате электрохимической реакции; ах — активность иона Хв исследуемой среде.
Постоянный коэффициент уравнения Нернста 2,303 RT/F, обычно обозначаемый буквой V, при 25 "С равен 59 мВ. Если Igo,, обозначить рХ, уравнение примет вид: Е = Ео ±59/п рХ. Знак «+» имеет место при измерении активности катионов, знак «-» — анионов. Таким образом, теоретически при изменении активности иона в 10 раз потенциал электрода должен измениться на 59 мВ, если ион однозарядный, и на 29,5 мВ в случае двухзарядного иона. Потенциал индикаторного электрода зависит от величины рХ прямолинейно, что и позволяет использовать эти электроды в аналитической практике. Отечественная и зарубежная промышленность выпускают ион-селектив-ные электроды различных типов: твердые мембранные, жидкие мембранные, газочувствительные, на основе нейтральных переносчиков, биоактивные ион-селектив-ные и др.
Нитратный электрод, применяемый в данной задаче для определения концентрации ионов NO3, образуемых в процессе роста нитрификатором II фазы Nitrobacter Winogradskyi, имеет мембрану, насыщенную ионами NOj. Внутренняя полость электрода заполнена раствором, содержащим 0,1 М KNO3 и 0,005 М КС1, в который погружен внутренний электрод сравнения. В качестве внутреннего электрода сравнения чаще используют хлорсеребряный или каломельный электроды. Мембранным электродом (ЭМ-NOj-O.l) можно измерять активность ионов NO3 прямым потенциометрическим методом в пределах от 0,35 до 4,0 pNOj. Контролируемая среда не должна содержать ионов Г или Вг“. Электрод селективен в присутствии ионов СГ, НСО3, СН3СОО”, SO| и F~ при превышении их концентрации над концентрацией NO3 в 100, 500, 500, 1000 и 1000 раз соответственно. Перед употреблением новый нитратный электрод дважды промывают изнутри дистиллированной водой и дважды — раствором KNO3 и КС1 указанной выше концентрации, затем заливают в его полость 1,5 мл этого раствора и погружают в него внутренний электрод сравнения. Собранный электрод выдерживают в течение суток в 0,1 М растворе KNO3 и в дальнейшем хранят в этом растворе. После подготовки электрода проверяют зависимость его потенциала от величины pNO3. Для этого измеряют ЭДС цепи, состоящей из электрода сравнения и нитратного электрода, погруженного в стандартные растворы NaNO3 (табл. 24.3). Отсчет по шкале прибора производят через 5 мин после погружения электрода в раствор.
По результатам измерений ЭДС в 3 — 4 стандартных растворах NaNO3, близких по составу и ионной силе к испытываемому раствору, строят калибровочный график, наклон которого должен соответствовать изменению ЭДС цепи на 59 мВ при изменении величины pNO3 на I.
Аналогично проводят измерения ЭДС цепи с исследуемым раствором, по калибровочному графику находят величину pNO3 и затем рассчитывают концентрацию ионов NO3 в растворе.
Определение NH4.
Колориметрический метод определения NH4 с реактивом Несслера. Аммиак в щелочной среде образует с йодомеркуриатом калия осадок желто-коричневого цвета. Чувствительность метода от 10 до 50 мкг/мл NH4. Протеканию реакции мешают амины, хлорамины, альдегиды, ацетон, спирты и некоторые другие органические соединения.
Таблица 24.3
Растворы для проверки нитратной функции электрода
Концентрация раствора NaNO3 I м 10-' м 10~2 м 5- Ю~2 М 10-’ м IO4 м
pNO3 0,35 1,13 2,05 2,33 3,02 4,00
Реактивы. Реактив Plecc/iepa: растворяют 100 г Hgl2 и 70 г KI в небольшом количестве бидистиллята и смешивают с раствором NaOH (100 г NaOH в 500 мл Н2О). Смесь доводится бидистиллятом до 1 л и отстаивается 4 ч. Тартрат Na-K (сегнетова соль), 50%-й раствор: растворяют 50 г KNaC4H4O6'4Н2О в бидистилляте, доводят до 100 мл и добавляют 0,5 мл реактива Несслера. Раствор применяют после осветления.
Ход определения. К 1 мл образца в 25-миллилитровой мерной колбе прибавляют 0,2 мл 50%-го раствора сегнетовой соли, 10 мл воды, 2 мл реактива Несслера, доводят до метки и перемешивают. Через 10 мин колориметрируют; светофильтр фиолетовый, X = 400 — 425 нм, кювета 1 см. Концентрацию NH4 (мкг/мл) определяют по формуле
CNH;= С-25/V,
где С— концентрация NH4 (мкг/мл), определенная по калибровочной кривой, составленной на основе концентраций NH4C1 от 10 до 50 мкг/мл NH4; V— объем пробы, взятой для анализа, мл; 25 — объем пробы, мл.
Оформление результатов. На основании изучения динамики содержания соединений азота в среде культивируемых бактерий в совокупности с данными об автотрофности нитрификаторов (что следует из состава сред, в которых отсутствуют источники органического углерода) делается вывод об участии этих соединений в процессах получения энергии и восстановительных эквивалентов.
Материалы, оборудование, посуда, реактивы
ФЭК-56М; центрифуги настольные; потенциометр pH 121; нитратный ион-се -лективный электрод; фарфоровые чашки для выпаривания — 5; водяные бани, снабженные крышкой с отверстиями для фарфоровых чашек; обратный холодильник — 1; мерные колбы на 100 мл — 5; стеклянные палочки — 5; колбы качалочные на
750 мл — 3; пипетки стерильные на 10 мл —
Таблица 24.4
Образование нитратов и нитритов нитрификаторами
Исследуемые образцы Содержание в исследуемых образцах, мкг/мл
NO; NO; NH;
Культуральная жидкость: Nitrosomonas sp. Nitrosospira sp. Контрольдля нитрификаторов: 1 фазы II фазы Культуральная жидкость: Nitrobacter winogradskyi * * * * *
* Не определять.
13; колба на 500 мл — 1; мерные колбы на 50 мл — 7; пипетки на 1 мл — 5; колбы на 250 мл — 2; цилиндр на 250 мл — 1; цилиндр на 50 мл — 1; реактивы для приготовления сред и проведения анализов.
План выполнения задачи
Для выполнения задачи необходимо 18-20 ч.
Занятие 1. Приготовление и стерилизация сред, добавок и посуды. Посев бактерий в культуральные среды.
Занятие 2. Построение калибровочных кривых для определения NO2, NOj, NH4. Морфологическое изучение клеток посевной культуры с использованием фазовоконтрастной микроскопии.
Занятия 3, 4, 5. (Опыты повторяют, чтобы получить представление о динамике процесса нитрификации.) Отделение культуральной жидкости от клеток центрифугированием. Определение в культуральной жидкости числа клеток и содержания NO2“, NOJ, NH4. Оформление результатов.
ЗАДАЧА 3. ОКИСЛЕНИЕ СОЕДИНЕНИЙ ЖЕЛЕЗА ЖЕЛЕЗОБАКТЕРИЯМИ
В некоторых водоемах с пресной водой, где в высоких концентрациях содержатся восстановленные формы железа, железобактерии образуют природные колонии, покрытые коркой окиси железа. Биологическое значение окисления железа различно для разных организмов. Acidithiobacillus ferroxidans (по традиции и из таксономических соображений отнесенный к тионовым бактериям) обладает типичным литоавтотрофным обменом, окисляя двухвалентное железо в трехвалентное через цитохром с и цитохромоксидазу. Образующийся при этом АТФ служит для автотрофной фиксации углекислоты. A. ferroxidans способен окислять пирит (FeS2) с образованием трехвалентного железа и серной кислоты. Развиваясь в кислой среде (pH 1,4—6,0), где в растворе устойчив не только Fe2+, но и Fe3+, организм не образует оформленных отложений железа, но при этом идет реакция
4Fe2+ + 4Н' + О2 -> 4Fe3+ + 2Н2О
К собственно железобактериям относят организмы, образующие оформленные осадки железа. Типичными представителями железобактерий являются нитчатые, заключенные в чехол бактерии группы Sphaerotilus (у многих из них чехол инкрустирован окисью железа), а также представители родов Leptothrix, Toxothrix, Siderococcus. Их легко выращивать в средах, содержащих органические вещества и Fe2+. Такие организмы накапливают в чехлах окись железа, но его значение для физиологии этих бактерий и их способность развиваться как хемоавтотрофы не доказана. Более очевидной представляется способность к хемоавтотрофному росту для другой, имеющей характерную структуру железобактерии — Gallionella. Она растет на минеральной среде, содержащей в качестве минерального источника восстановленного железа осадок сульфида железа. Использование фактически нерастворимой соли железа сводит к минимуму трудности, связанные со спонтанным окислением железа при pH около 7,0. В этих культурах Gallionella образует на стенках сосуда хлопьевидные колонии. Они состоят в основном из окисла железа, а маленькие бобовидные клетки данной бактерии располагаются на разветвленных кончиках пропитанных гидроокисью железа стебельков.
Gallionella напоминает Siderococcus, чрезвычайно мелкий организм, едва отличимый от частиц железистого осадка. Электронно-микроскопические снимки показывают, что мелкие клетки Siderococcus несут нитевидный придаток, напоминающий отдельное волокно из «стебелька» Gallionella. Клетки Siderococcus объединяются по 8— 10 в подвижную колонию, которая активно плавает над поверхностью дна, хотя жгутиков не было найдено. В зоне развития клеток встречается обычно большое количество осадков железа, но сами клетки остаются свободными.
Leptothrix ochracea — наиболее широко распространенная железобактерия, которая образует обильные скопления окислов железа ржавого цвета в медленно текущих ручьях, на болотах, на выходе железистых источников. Несмотря на признанное участие в образовании болотных железных руд, относительно L. ochracea до сих пор нет твердой уверенности, что это самостоятельный организм, а не форма роста другой бактерии, например Sphaerotililus.
Leptothrix образует тонкие железистые трубки, представляющие собой влагалища, которые всегда бывают гладкими, неветвящимися, иногда слегка волнистыми и на всем своем протяжении сохраняют строго одинаковый диаметр (внутренний — порядка 1 мкм, внешний — порядка 2 — 3 мкм). Поверхность влагалища имеет тонкую сетчатую структуру из косо расположенных нитей, которую иногда удается наблюдать и при
помощи светового микроскопа. Влагалище L. ochracea, подобно таковому у бактерий группы Sphaerotilus, образовано цепочкой цилиндрических клеток, которые обладают склонностью выскальзывать из него, поэтому только на одном конце трубки можно найти живой организм, вся остальная часть представляет собой покинутое влагалище. Клетки L. ochracea напоминают клетки Sphaerotilus. В них также наблюдаются капли липидных включений, освободившиеся клетки подвижны благодаря наличию жгутиков.
Бактерии группы Toxothrix встречаются сравнительно редко в холодных железистых источниках, образуя отдельные железистые нити, не переплетенные между собой. Возможно, что ожелезнению подвергается выделяемая при движении клеток слизь.
Цель работы, наблюдение за процессом микробиологического окисления двухвалентного железа в трехвалентное и количественная оценка этого процесса.
Ход выполнения задачи
Микроорганизмы и их культивирование. Используют культуры Leptothrix sp. и Acidithiobacillus ferroxidans (окисляющие как соединения железа, так и соединения серы).
Leptothrix sp. выращивают в среде следующего состава (г/л): пептон — 4,0; глюкоза — 3,0; MgSO4 — 0,1; СаС12 — 0,05; MnSO4 — 0,01; FeCI3_ 0,001; тиамин — 0,2 мг; биотин — 0,02 мг; (МН4)2Рс-лимоннокислый (закисная соль) — 0,25; вода водопроводная — до 1 л. pH среды до стерилизации устанавливают 7,0 с помощью 10%-х растворов NaOH и H2SO4.
A. ferroxidans культивируют в среде следующего состава (г/л): I. (NH4)2SO4 — 3,0; К2НРО4 - 0,5; КС1 - 0,1; MgSO4-7H2O - 0,5; Ca(NO3)2 - 0,01; вода дистиллированная — до 700 мл. II. FeSO4- 2Н2О — 44,2; 1 мл 10 н. H2SO4; вода дистиллированная — до 300 мл.
Растворы I и П стерилизуют отдельно при 0,5 ати 30 мин. Перед посевом растворы сливают, стерильно доводят pH до 2,5 с помощью 10%-х H2SO4 или NaHCO3. Среду разливают по 100 мл в стерильные качалочные колбы.
В качестве посевного материала служат культуры в конце экспоненциальной фазы роста (3-суточные). Их вносят в количестве 10 % (по объему).
Выращивание культур ведут на качалках при температуре 28 — 30 °C в течение недели. Ежедневно отбирают пробы для микроскопирования клеток и определения количества окисного железа.
Микроскопирование. Для наблюдения за образованием железистых чехлов Leptothrix sp. готовят фиксированный препарат клеток, погружают его на 3 — 5 мин в 5%-й раствор желтой кровяной соли, затем без промывания — в 0,1 М раствор НС1. Осадок трехвалентного железа синеет, что видно при микроскопировании препарата. Чтобы определить количество клеток A. ferroxidans, готовят фиксированные препараты и подсчет ведут по методу Виноградского.
Определение количества Fe3+. Проводят дифференциальным спектрофотометрическим методом, основанным на взаимодействии Fe3+ с сульфосалициловой кислотой, при котором в зависимости от pH раствора образуется ряд комплексных соединений. В частности, при pH 1,8— 2,5 образуется моносульфосалицилат железа красно-фиолетового цвета, имеющий спектр поглощения с максимумом при 510 нм.
Ход определения. I мл культуральной жидкости с клетками помещают в пробирку, прибавляют 2 мл 10%-й сульфосалициловой кислоты, 0,1 мл 2 и. H2SO4, перемешива
ют и доводят объем раствора до 10 мл дистиллированной водой. Оптическую плотность смеси измеряют на ФЭК при X = 510 нм в кювете шириной 10 мм. Определение ведут по значениям стандартной кривой, которую строят в тех же условиях, используя навеску железоаммиачных квасцов с 0,1 — 1 мкг/мл Fe3+ в растворе.
Оформление результатов. Результаты вносят в таблицу и строят график зависимости окисления Fe2+ и появления в культуральной жидкости Fe3+ от времени выращивания культуры и количества образованной биомассы.
Материалы, оборудование, посуда, реактивы
Пипетки на 1 мл (стерильные) — 10; пробирки — 25; серная кислота (концентрированная); железоаммиачные квасцы; аммиак; сульфосалициловая кислота.
План выполнения задачи
На выполнение задачи требуется 16— 18 ч.
Занятие 1. Приготовление и стерилизация сред, посуды.
Занятие 2. Посев культуры, микроскопирование, приготовление реактивов для определения, построения стандартной кривой.
Занятие 3. Определение содержания Fe3+, числа клеток в культуральной жидкости, оформление результатов.
Глава 25
СУЛЬФИДОГЕНЫ. СУЛЬФАТНОЕ И СЕРНОЕ ДЫХАНИЕ
Восстановление серы и ее окисленных соединений возможно в двух различных процессах. Во-первых, многие микроорганизмы используют вышеуказанные соединения в качестве источников серы, необходимых для реакций биосинтеза. В этом случае говорят об ассимиляционном восстановлении серы, сульфита, тиосульфата, а также об ассимиляционной сульфатредукции, когда восстанавливается сульфат. Во-вторых, ряд бактерий и археи используют серу и ее окисленные соединения в качестве конечного акцептора электронов. Прокариот, образующих сульфид при восстановлении соединений серы разной степени окисленности (сульфата, сульфита, тиосульфата), а также молекулярной (элементной) серы, называют сулъфидогенами. Процессы восстановления соединений серы при использовании их в качестве конечного акцептора электронов называют анаэробным дыханием, выделяя сульфатное и серное дыхание.
Сульфатное дыхание, или диссимиляционная сульфатредукция — это использование сульфатов в качестве акцепторов электронов при окислении разных субстратов с образованием сероводорода. Восстановление сульфата в сульфид осуществляется комплексом ферментов:
SO*- + АТФ АТФ-сульфурилаза > дфС + ф_ф.
(аденил илсульфат)
Ф-Фн+ Н2О------пирофосфатаза > 2^.
(пирофосфат)
АФС + 2е----АФС-рсдуктаза > дМф + SQ2-
Дальнейшее восстановление сульфита в сульфид происходит при участии специфического фермента — альтернативной диссимиляционной сульфитредуктазы:
3SO2"+2e--------сульфитредукгаза g q2-
J •> о
(тритионат)
Два других фермента превращают тритионат в сульфид с высвобождением на каждом этапе сульфита:
S3O|- + 2е“........ тритионатрсдуктаза ^2- + gQ2-.
S20j- + 2е~--------тиосульфатрсдукгаза > g2 + g2-
Считают, что восстановление сульфита до сульфида может происходить и одноступенчато, при участии альтернативной диссимиляционной сульфитредуктазы. Прокариот, осуществляющих диссимиляционную сульфатредукцию, называют десульфатирующими или сульфатвосстанавливающими. Большинство из них способно также использовать при анаэробном дыхании в качестве акцепторов электронов сульфит и/или тиосульфат-ионы.
Сульфатредукторы относятся к двум доменам — Bacteria (около 20 родов) и Archaea (Archaeoglobus). Это чаще всего строгие анаэробы. Формы клеток: виброидные, сферические или палочковидные, у ряда видов имеющие жгутики. При восстановлении сульфата (тиосульфата, сульфита) до сероводорода донорами электронов для этих микроорганизмов служат молекулярный водород (он обеспечивает возможность их роста в хемолитоавтотрофных и/или хемолитоорганотрофных условиях) или некоторые органические соединения (лактат, формиат, пируват, жирные кислоты, спирты и др.), окисление которых бывает полным (до СО2 и воды) или неполным (обычно до ацетата и СО2). По типу энергетического обмена сульфатредукторы — окислители; для них единственным конечным акцептором электронов служат окисленные соединения серы, а донорами — Н2 или ряд органических соединений. Некоторые виды могут использовать в качестве акцепторов электронов и нитраты, восстанавливая их до нитритов, а затем до аммония; фумарат, восстанавливая его до сукцината, а также структурный аналог сульфата селенат, восстанавливая его до селенида. Кроме сульфатного дыхания многие десульфатирующие бактерии способны и к брожению.
Среди микроорганизмов-сульфатредукторов можно выделить три подгруппы. Первая подгруппа — представители р. Desulfotomaculum. Это грамположительные, образующие эндоспоры, как правило, подвижные бактерии. Все известные виды растут на синтетических средах простого состава, дрожжевой экстракт существенно стимулирует такой рост. Часть видов способна к автотрофии. Ко второй подгруппе относят не образующих спор грамотрицательных бактерий, способных окислять органические соединения с образованием ацетата и СО2. Это представители родов Desulfobulbus, Desulfomicrobium, Desulfovibrio, Thermodesulfobacterium. Третья подгруппа — представители значительного числа родов бактерий, например Desulfobacter, Desulfo bacterium, Desulfococcus и др., а также сульфатвосстанавливающие археи (р. Archaeoglobus), способные полностью окислять органические субстраты.
Типичные местообитания прокариот, осуществляющих процесс диссимиляционной сульфатредукции, — это бескислородные осадки и придонный слой в пресноводных, морских и сильно соленых водоемах, пластовые воды нефтяных месторождений. Сульфатредукторы — активные участники процесса межвидового переноса водорода. Они могут также создавать ассоциации с микроорганизмами на основе образования сероводорода и/или ацетата. Прокариоты, способные к сульфатному дыханию, встречаются в качестве симбионтов в желудочно-кишечном тракте ряда животных, а также в техногенных местообитаниях: в трубопроводах тепловой водной сети, в очистных сооружениях.
Особо следует отметить важную роль термофильных сульфатвосстанавливающих прокариот, обитающих в морских и наземных гидротермальных экосистемах, в геотер
мальных водах подземной гидросферы. К настоящему времени изучено около 20 видов термофилов-сульфатредукторов, относящихся к 5 родам бактерий (Desulfotomaculum, Thermodesulfobactetrium, Thennodesulfovibrio, Desulfacinum, Thermodesulforhabdus) и одному роду архей (Archaeoglobus). Они представляют собой филогенетически и физиологически гетерогенную группу, хотя все являются облигатными анаэробами, способными к литотрофному росту с молекулярным водородом. Эти микроорганизмы могут также окислять широкий спектр несбраживаемых субстратов. Значительное число их видов способно к автотрофии, следовательно, они играют роль продуцентов в гидротермальных экосистемах. В то же время представители родов Desulfotomaculum, Desulfacinum, Thermodesulforhabdus — обязательные участники процесса деструкции органического вещества гидротерм.
В термальных микробных сообществах еще более важную роль, чем сульфатредукто-ры, играют прокариоты, способные к серному дыханию — диссимиляционному восстановлению элементной серы и ее окисленных соединений (но не сульфата) с образованием сероводорода. Они являются наиболее значимыми первичными продуцентами органического вещества в анаэробных микробных сообществах гидротерм и его универсальными деструкторами (представители гипертермофильных архей родов Ther-moproteus и Pyrobaculum способны за счет серного дыхания осуществлять полную минерализацию органического вещества в анаэробных условиях).
Прокариоты, использующие элементную серу как акцептор электронов, представлены несколькими родами бактерий и 6 родами архей. Бактерии — в основном мезофильные (роды Desulfuromonas, Desulfovibrio, Desulfomicrobium) и умеренно термофильные (роды Desulfurella, Hippea), археи — гипертермофильные, оптимальная температура для роста большинства из них — 80—105 °C (например, Thermofilum, Pyrodictium, Acidianus и др.). Бактерии грамотрицательные, чаще всего встречаются палочки, вибрионы, иногда — со жгутиком. Морфология клеток архей — кокки, палочки, почкующиеся палочки, диски.
Среди серовосстанавливающих прокариот известны факультативные и строгие анаэробы, гетеротрофы и автотрофы (небольшое число хемолитоавтотрофных видов, например представители порядка Thermoproteales, и значительно большее — хемоорган отрофных, например представители порядка Thermococcales). Почти все термофилы — облигатные анаэробы. Гипертермофильные археи способны к автотрофному росту и полному окислению за счет сероредукции белков, полисахаридов, но не низкомолекулярных несбраживаемых субстратов (летучих жирных кислот, спиртов и др.), как это характерно для термофильных сульфатредукторов. Интересный пример: гипертермофильные археи Acidianus infemus, Desulfurolobus ambivalens способны расти не только в анаэробных условиях благодаря серному дыханию и использованию в качестве донора электронов Н2, но и в аэробных, окисляя элементную серу до сульфата.
Таким образом, прокариоты, осуществляющие сульфатное и серное дыхание, активно участвуют в круговороте серы в природе, образуя сероводород. Они участвуют также в круговороте углерода. Особенно важную роль играют эти микроорганизмы в глубоководных гидротермах, где существуют специфические термофильные прокариотные экосистемы — наиболее близкие аналоги древнейших биоценозов Земли.
ЗАДАЧА. ИЗУЧЕНИЕ АНАЭРОБНОГО ДЫХАНИЯ НА ПРИМЕРЕ СУЛЬФАТНОГО
Цель работы — знакомство с некоторыми представителями сульфатредуци-рующих бактерий, осуществляющих процесс сульфатного дыхания.
Ход выполнения задачи
Микроорганизмы и их культивирование. В работе используют несколько представителей сульфатредуцирующих бактерий: Desulfovibrio desulfuricans или D. baculatus, а также термофильный Desulfotomaculum nigrificans.
Среда культивирования (г/л): КН2РО4 — 0,5; NH4C1 — 1,0; (NH4)2SO4 — 7,0 или Na2SO4 — 4,5; СаС12- 6Н2О — 0,06; MgSO4- 7Н2О — 0,06; лактат Na — 6,0; лимоннокислый Na (трехзамещенный) — 0,3; FeSO4 — 0,01; NaCl — 5,0; Na2S • 9Н2О — 1 мМ (0,22 г/л) в качестве восстановителя; дрожжевой экстракт — 1,0; микроэлементы по Пфеннингу и Липперту — 1 мл/л. Отдельно от основной среды стерилизуют 0,1%-й раствор FeS04- 7Н2О в 1%-й НС1; 5%-й раствор Na2S • 9Н2О в 1%-м растворе NaHCO3; 5 %-й раствор NaHCO3; 5%-й раствор НС1; 10%-й раствор дрожжевого автолизата (стерилизуют при 0,5 ати, остальные компоненты сред стерилизуют при 1 ати, 30 мин). Раствор микроэлементов вводят в среду при посеве, стерилизуют его также при 0,5 ати. Раствор микроэлементов (по Пфеннингу и Липперту) содержит в 100 мл дистиллированной воды (мг): ЭДТА - 500; FeSO4-7H2O - 200; ZnSO4-7H2O - 10; МпС12-4Н2О - 3; СоС12-6Н2О - 20; Н3ВО3 - 30; NiCl2-6Н2О - 2; Na2MoO4-2H2O - 3,0; CuC14-2H2O - 1, 0.
После автоклавирования горячую среду охлаждают в токе азота и вносят в нее стерильно необходимые добавки в следующем порядке: 1) микроэлементы, 2) дрожжевой экстракт, 3) FeSO4; далее доводят pH до 7,0 —7,5 по бром-тимолблау с помощью стерильных 10%-х растворов бикарбоната или НО и вносят Na2S по каплям до образования сероватого оттенка в среде. В среды немедленно вводят посевной материал (5 % от объема), разливают под током азота в пробирки Хангейта на 18 мл по 12 мл, а оставшийся объем заполняют молекулярным азотом. Процесс культивирования ведут в стационарных условиях в термостате при 30 и 60 °C (для D. nigrificans).
Ход анализа. Ежедневно проводят микроскопирование в фазовом контрасте, следя за изменением морфологии клеток. Готовят препараты клеток для просвечивающей электронной микроскопии (негативное контрастирование). Для этого каплю суспензии клеток (по возможности, без осадка) наносят на сетки, покрытые формваровой пленкой. Через 30 с лишнюю жидкость убирают фильтровальной бумагой. Затем наносят на сетки на 1 мин 2%-й раствор натриевой соли фосфовольфрамовой кислоты (pH 7,0 —7,2), убирая лишнюю жидкость также с помощью полоски фильтровальной бумаги. Все препараты на сетках после окончания культивирования изучают с помощью трансмиссионного электронного микроскопа JEM —100В (фирма Jeol, Япония), отмечая изменение морфологии клеток, размер, число и расположение жгутиков. Подсчитывают число клеток в динамике роста культуры по Виноградскому. Количественный учет биомассы проводят также по приросту белка в культуре, определяемого по биуретовой реакции или по методу Брэдфорд.
Определение сульфидов. В процессе диссимиляционной сульфатредукции клетки выделяют сероводород, который, связываясь с присутствующим в среде железом, вызывает ее почернение. Определение образовавшегося сульфида в опытных и контрольных пробирках проводят по методу Трюпера. В пробирки Эппендорфа на 1,5 мл вносят 0,2 мл 2%-ного раствора ацетата пинка; 0,01 мл пробы и 0,3 мл Н2О, раствор перемешивают. Приливают 0,1 мл 0,2%-го раствора М^-диметилпарафенилендиамина в 20%-й H2SO4, перемешивают. Приливают 50 мкл 1%-го раствора железоаммиачных квасцов в 2%-й H2SO4 и 0,40 мл воды, перемешивают. Через 30 мин интенсивность окраски измеряют
на ФЭК при длине волны 802 нм. В качестве контроля вместо пробы берут 0,01 мл воды. Измерение проводят в кюветах с длиной оптического пути 3 мм.
Пересчет содержания сульфида ведут с использованием калибровочной кривой, построенной для растворов с содержанием 50—200 мкг S2" на 1 мл (в пересчете на серу).
Оформление результатов. Результаты оформляют в виде таблицы и графиков прироста биомассы и количества образованного культурами сульфида (мкг/мл) в зависимости от времени по каждой культуре. Рассчитывают интенсивность процесса по часам, делают вывод о различии скорости образования сульфида в зависимости от вида бактерий и условий выращивания клеток. Анализируют микроскопическую картину клеток.
Материалы, оборудование, посуда, реактивы
Термостаты на 30 и 60 °C, ФЭК, микроскоп с фазово-контрастным устройством; электронный микроскоп (лаборатория электронной микроскопии), пробирки Эппен-дорфа на 1,5 мл. Посуда для культивирования анаэробов согласно техники Хангейта. Предметные и покровные стекла; необходимые, ранее перечисленные реактивы, фуксин основной.
План выполнения задачи
На выполнение задачи необходимо 15—16 ч.
Занятие 1. Подготовка сред и посуды для стерилизации. Построение калибровочной кривой определения сульфида.
Занятие 2. Посев культур в опыт. Отбор проб. Изучение морфологии клеток, подсчет клеток по Виноградскому и определение белка по Брэдфорд или в биуретовой реакции.
Занятия 3—5. Отбор проб, определение образовавшегося сульфида, подсчет числа клеток, подготовка препаратов для электронной микроскопии, определение белка, расчет продуктивности клеток. Оформление результатов.
Глава 26
МЕТАБОЛИЧЕСКАЯ ВАРИАБЕЛЬНОСТЬ МИКРООРГАНИЗМОВ
Микроорганизмы обладают чрезвычайно высокой способностью приспосабливаться к часто меняющимся условиям окружающей среды. Среди микроорганизмов есть примеры бактерий, способных расти как гетеротрофно, так и автотрофно. Однако поистине удивительную приспособляемость демонстрирует Р. denitrificans, организм, дыхательная система которого чрезвычайно напоминает митохондриальную. Этот организм (или его древний предок) рассматривается в качестве возможного предшественника митохондрий в теории сим-биогенеза. Р. denitrificans способен расти, используя готовые органические вещества (гетеротрофно) аэробно и анаэробно (восстанавливая нитраты, нитриты и N2O до N2), автотрофно, окисляя водород кислородом воздуха и фиксируя СО2 через рибулозобисфосфатный цикл (цикл Кальвина), или на среде с водородом и нитратом анаэробно и, наконец, метилотрофно в аэробных и анаэробных (денитрифицирующих) условиях, используя метанол или формиат в качестве источника углерода и энергии. Широкая норма реакции генома
P. denitrificans на изменяющиеся условия существования позволяет этому организму легко приспосабливаться к появлению новых субстратов для роста, изменению условий аэрации, исчезновению органического вещества. Организм хорошо изучен в биоэнергетическом плане, его метаболизм известен, что дает возможность предсказать наличие тех или иных ферментативных активностей в клетках после смены условий выращивания культур.
ЗАДАЧА. ИЗУЧЕНИЕ МЕТАБОЛИЧЕСКОЙ ВАРИАБЕЛЬНОСТИ PARACOCCUS DENITRIFICANS
Цель работы, определить возможность фиксации клетками Р. denitrificans 14С-метанола и 14СО2 в зависимости от условий выращивания организма.
Ход выполнения задачи
Выращивание Р. denitrificans. Культуру Р. denitrificans АТСС 13543 или NCIB 8944 поддерживают на агаризованной среде с дрожжевым экстрактом (2 г/л), глутаматом и малатом (по 20 мМ). Пересевают каждые 6 нед. Суспензию можно хранить в 10 %-м глицероле при -15 °C в течение года. Клетки организма выращивают аэробно на качалке (200 об/мин, 37 °C, колбы на 250 мл со 100 мл среды) или анаэробно в бутыли на 1 л с 200—500 мл среды с использованием техники Хайгейта (см. гл. 16.2).
Состав сред. Среда 1 (гетеротрофная, аэробные условия) (мМ): маннит — 20 или сукцинат Na - 50; КН2РО4 - 30; Na2HPO4 - 15; NH4C1 - 30; MgSO4 -1; Na2MoO4 — 0,1; цитрат Fe(III) — 0,02; биотин — 4 мкг/мл, pH среды 7,5; стерилизация при 1 ати.
Среда 2 (гетеротрофная, анаэробные условия): та же, что и среда 1, с добавлением KNO3 — 20 мМ. Среду разлить по 500 мл в 1 л бутыли, прокипятить, охладить под током азота, закупорить анаэробно и автоклавировать при 1 ати.
Среда 3 (метилотрофная, аэробные условия): та же, что и среда 1, но источником углерода служит метанол, берется в концентрации ЮОмМ. Метанол стерилизуют отдельно в виде 50%-го раствора при 1 ати, добавляют при посеве.
Среда 4 (метилотрофная, анаэробные условия): та же, что среда 3, с добавлением KNO3 — 20 мМ. Готовить и разливать анаэробно, как и среду 2.
Среда 5 (автотрофная, аэробные условия): та же, что и среда 1, но источником углерода служит NaHCO3 — 0,05 %. Среду разливают по 200 мл в бутыли на 1 л, закупоривают герметично и заменяют газовую фазу на Н2/СО2/О2 в соотношении 70/10/20.
Среда 6 (автотрофная, анаэробные условия): та же, что и среда 5, но с добавлением KNO3 — 20 мМ и заменой газовой фазы на Н2/СО2 (80/20). Стерилизовать при 1 ати.
Каждый студент готовит по одной бутыли со средами 2, 4, 5 и 6 и по две колбы со средами 1 и 3.
Подготовка клеток для экспериментов. Выросшие культуры из середины экспоненциальной фазы роста (24 ч; оптическая плотность аэробных культур — около 4,0; число клеток ~109 кл/мл) центрифугируют в течение 20 мин (6200 g, 4 °C), отмывают
холодной свежей средой без источника углерода и суспендируют в соответствующей среде со всеми добавками аэробно или анаэробно в зависимости от происхождения клеток. Контрольные варианты суспендируют в среде без источника углерода или энергии.
Варианты опытов: гетеротрофно — аэробно/анаэробно; метилотрофно — аэробно / анаэробно; автотрофно — аэробно/анаэробно. В каждом варианте ставят контроли, в которые не вносят энергетический субстрат.
После инкубации во все варианты вносят меченые субстраты: |4С-метанол и |4С-СО2 — в гетеротрофные варианты, метанол и 14С-СО2 — в метилотрофные варианты, 14С-СО2 — в автотрофные варианты. Отбирают пробы в двух повторностях через каждые 30 мин в течение 2 ч инкубации (всего 5 точек, включая начальную). Пробы быстро фильтруют под тягой через ультрафильтры и активность на фильтрах просчитывают в сцинцилляционном счетчике (подробно процедура описана в гл. 27). Целесообразно, чтобы один и тот же студент отбирал пробы из одного или двух вариантов опыта.
Оформление результатов. Результаты оформляют в виде графиков нарастания радиоактивности, включившейся в клетки, осажденные на ультрафильтрах. Студенты обмениваются полученными графиками, с тем чтобы каждый студент имел графики фиксации углекислоты и метанола во всех вариантах опыта. Делают заключение об изменении способности клеток к фиксации углекислоты и/или метанола в зависимости от способа выращивания культур. Обсуждают возможные варианты развития клеток Р. denitrificans в природных экологических нишах или тех или иных консорциумах микроорганизмов.
Материалы, оборудование, посуда, реактивы
Орбитальная термостатированная качалка на 37 °C, колбы на 250 мл для вариантов культуры — 4; бутыли на 1 л с герметичными резиновыми пробками — 4; шприцы для отбора проб — 20; установка для ультрафильтрации — 1; реактивы для приготовления сред, pH-метр, ФЭК, центрифуга, меченные по углероду 14С-метанол и 14С-СО2.
План выполнения задачи
На проведение задачи необходимо 24 ч.
Занятие 1. Подготовка сред и посуды к стерилизации. Выращивание посевного материала Р. denitrificans.
Занятие 2. Засев клеток в опытные среды, отбор проб при посеве и определение оптической плотности культур, расчет вариантов постановки опыта и количеств вносимых меченых СО2 и метанола в различные варианты.
Занятие 3. Постановка экспериментов, отбор проб, определение динамики фиксации меченых субстратов.
Занятие 4. Расчет количества фиксированных субстратов в разных вариантах опыта. Построение кривых фиксации углерода клетками из разных вариантов. Написание отчета.
Глава 27
АССИМИЛЯЦИЯ УГЛЕКИСЛОТЫ ЦИАНОБАКТЕРИЯМИ
Цианобактерии — широко распространенная группа оксигенных фототрофных микроорганизмов. Подавляющее большинство цианобактерий растет в фотоавтотрофных условиях, ассимилируя углекислоту.
ЗАДАЧА. ФИКСАЦИЯ УГЛЕКИСЛОТЫ ЦИАНОБАКТЕРИЯМИ
Цель работы: определить светозависимый характер ассимиляции цианобактериями углекислоты по включению в клетки радиоактивного углерода из NaH14CO3.
Ход выполнения задачи
Микроорганизмы и их культивирование. Используют одноклеточные или нитчатые цианобактерии из родов Synechococcus, Synechocystis, Plectonema и др. Культуру цианобактерий выращивают на среде BG-11N или Кратца-Майерса (состав сред см. в приложении 4).
Среду разливают в колбы на 750 мл по 300 мл и стерилизуют при 1 ати. После стерилизации доводят значение pH до 7,5 —8,0, используя стерильный 5 %-й раствор NaHCO3.
В качестве посевного материала используют 4—5-суточную культуру цианобактерий, выросшую на среде того же состава. Посевной материал вносят в количестве 5 —10 % (по объему). Культивирование ведут в люминостате (3000 лк) при 25 — 28 °C в течение 4—6 сут.
Подготовка суспензии клеток. Для постановки опыта клетки из выросших культур осаждают центрифугированием (3000 g, 15 мин), отмывают и суспендируют в свежей опытной среде, содержащей трис- или Na-фосфатный буфер (pH 8,0, конечная концентрация 1 мМ) и NaHCO3 в качестве носителя (в концентрации 1 мМ). Суспензию разводят опытной средой до оптической плотности 0,5 (длина оптического пути 1 см, А = 750 нм) и разливают по 10 мл в две колбы Эрленмейера на 50 мл. Одну колбу помещают в люминостат (3000 лк), а другую выдерживают в темноте.
Проведение эксперимента. Через 10—15 мин после начала инкубации в каждую колбу вносят 100 мкл NaH14CO3 (конечная концентрация — 104 Бк/мл) и немедленно отбирают автоматической пипеткой (1,0 мл) начальную пробу (в двух повторностях). Последующие пробы того же объема отбирают в двух повторностях через 30, 60 и 90 мин после внесения меченого бикарбоната. Отбор проб производят под тягой. Пробы немедленно осаждают на мембранных фильтрах («Сынпор», N6) и промывают двумя порциями слегка подкисленной воды (pH 5) для удаления углекислоты, которая не успела фиксироваться клетками. Пробы фильтруют на фильтровальной установке, подключенной к водоструйному насосу. Затем фильтры высушивают на воздухе (под тягой или при подогреве лампой накаливания).
Сухие фильтры помещают в сцинтилляционные флаконы (биомассой вверх), наливают по 5 мл сцинтилляционной жидкости ЖС-106 и плотно закрывают пробками (работа проводится под тягой, сцинтилляционная жидкость отбирается с помощью дозатора). Радиоактивность клеток на фильтрах просчитывают на сцинтилляционном счетчике в течение 1 мин.
Оформление результатов. Результаты эксперимента представляют в виде ленты с цифрами радиоактивности, а также в виде графика фиксации цианобактериями углекислоты (в импульсах в минуту, срт) на свету и в темноте с течением времени. Делают заключение о светозависимом характере ассимиляции цианобактериями углекислоты.
Материалы, оборудование, посуда, реактивы
Люминостат; люксметр; водоструйный насос; ФЭК или спектрофотометр; мембранные фильтры; пипетки автоматические на 1 мл и 100 мкл; колбы на 750 мл; колбы Эрленмейера на 50 мл — 2; пипетки на 1—2 и 5 мл — по 3; соли для приготовления среды; NaHl4CO3; сцинтилляционная жидкость ЖС-106.
План проведения задачи
Для проведения задачи требуется 6—8 ч.
Занятие 1. Приготовление и стерилизация среды.
Занятие 2. Посев цианобактерий.
Занятие 3. Приготовление суспензии клеток, проведение эксперимента по фиксации NaH4CO3.
Занятие 4. Измерение радиоактивности фильтров (проводится оператором). Оформление результатов.
Глава 28
ТРАНСПОРТ СУБСТРАТОВ В КЛЕТКИ МИКРООРГАНИЗМОВ
Многие питательные вещества поступают в клетки путем простой, или облегченной, диффузии. Однако для многих быстрорастущих организмов такой механизм поступления веществ в клетку оказывается неприемлемым вследствие низкой скорости поступления субстрата. Многие микроорганизмы выработали специальные механизмы активного транспорта веществ в клетку, который позволяет накапливать субстрат роста против градиента концентрации, таким образом создавая внутриклеточные концентрации веществ гораздо более высокие по сравнению с окружающей клетки средой. К системам активного транспорта относятся симпорт, унипорт, антипорт и перенос групп — все они зависят от энергии и считаются системами вторичного транспорта. К первичному транспорту относят создание трансмембранного градиента протонов за счет протекания реакций химического окисления веществ или фотосинтетических процессов.
Изучение транспортных процессов проводят на чистых культурах микроорганизмов, взятых из середины или конца экспоненциальной фазы роста, отмывая клетки соответствующей средой или буфером и инкубируя их с меченными по углероду или азоту субстратами транспорта.
Хорошо изучена система транспорта иона аммония, для исследования которой часто использовали неметаболизируемый обычными клетками |4С-ме-тиламин — структурный аналог иона аммония. Однако имеется целый ряд микроорганизмов — метилотрофов, которые активно метаболизируют метиламин, используя его в качестве источника углерода и энергии, а часто и азота. Имеется, в частности, метилотрофная бактерия MethylobaciUus flagellatus КТ, которая использует для роста всего лишь два субстрата — метанол и метиламин, являясь облигатным организмом. Клетки М. flagellatus способны расти на средах с метанолом и аммонием (источники С и N), с метанолом (С) и метиламином (N), с метиламином (С) и аммонием (N) и метиламином (С и N). Во всех случаях клетки формируют разные в кинетическом отношении транспорт
ные системы для метиламина и метанола, отличающиеся по скорости процесса и Км для соответствующего субстрата.
ЗАДАЧА. ИЗУЧЕНИЕ ТРАНСПОРТНЫХ СИСТЕМ ОБЛИГАТНОГО МЕТИЛОТРОФА METHYLOBACILLUS FLAGELLATUS КТ
Цель работы: установить кинетические параметры транспортных систем для |4С-метанола и 14С-метиламина в клетки М. flagellatus КТ и выявить энергозависимость транспортных процессов для этих субстратов в клетки организма.
Ход выполнения задачи
Выращивание культуры и подготовка клеток для экспериментов. Бактерии культивируют в жидкой минеральной среде MMS (см. прил. 4) с метанолом (150 мМ) или метиламином (40 мМ) в качестве источников углерода и энергии. Клетки выращивают в нескольких вариантах:
Вариант 7: метанол (150 мМ) и метиламин (10 мМ) без аммония. Вариант 2\ метанол (150 мМ) с аммонием в составе среды. Вариант 3: метиламин (40 мМ) без аммония. Культивирование проводят на орбитальной качалке при 45 °C в течение суток (до достижения Л650 ~0,8—0,9). Клетки центрифугируют (6 тыс. g, 15 мин, 4 °C), отмывают три раза холодной средой MSL (без источника углерода) и суспендируют в холодной среде MSL до плотности 0,05 — 0,1 мг сух. массы клеток/мл. Колбы с суспензиями помещают на качалку при 45 °C на 30 — 40 мин для снижения эндогенного дыхания.
Проведение эксперимента. По 10 мл суспензии клеток из каждого варианта помещают в термостатируемую ячейку (45 °C) на магнитную мешалку и после установления нужной температуры добавляют 14С-метанол или 14С-метиламин до конечной концентрации 1 — 5 мМ. Специфическую радиоактивность субстратов подбирают на уровне 0,1 —0,2 мкКп/ммоль.
Из суспензий отбирают пробы с интервалом в 1 мин и немедленно осаждают клетки на мембранных фильтрах («Владипор», РФ; 0,45 мкм). Клетки на фильтре отмывают 10 мл 50 мМ К-фосфатного буфера (pH 7,4) при 20 °C и высушивают при 100 °C в сушильном шкафу в течение 10 мин. Радиоактивность просчитывают в сцинцилляционном счетчике Магк-2 (Nuclear Chicago, США) после помещения фильтров (биомассой вверх!) в сцинцилляционные стаканы и добавления 5 мл сцинцилляционной жидкости ЖС-105 в каждый.
Пробы отбирают в течение 10 мин и строят кривые нарастания радиоактивности в клетках метилотрофов (включение метки от субстратов).
Изучение влияния ингибиторов транспорта. В параллельных экспериментах изучают влияние ингибиторов дыхания и разобщителей на скорость транспорта веществ. Эксперимент начинают, как в опыте 28.2, однако после 5 мин инкубации добавляют цианид (10-3 М, конечная концентрация), азид (10'3 М) или СССР (carbonyl cyanide-3-chlorophenylhydrazone, 10 3 М). Пробы продолжают отбирать с интервалами 1 мин. Строят графики транспорта веществ и сравнивают их с графиками незаингибированного процесса.
Обработка результатов. Строят графики транспорта субстратов в клетки, выращенных в разных условиях, и сравнивают (рассчитывают) скорость транспорта для каждого случая. Делают заключение о влиянии условий выращивания клеток на скорость и специфичность транспортных процессов для метанола и метиламина. Делают выводы о влиянии ингибиторов на транспорт веществ у метилотрофов.
Материалы, оборудование, посуда, реактивы
Колбы для орбитальной качалки — 20; орбитальная качалка; ультратермостат; автоматические пипетки для отбора проб с носиками; фильтры с насадками для отбора проб; буфер К-фосфатный (50 мМ, pH 7,2); среда В (см. приложение 4) для выращивания бактерий; метанол; метиламин; 14С — метанол и метиламин; сушильный шкаф на 100 °C; фильтровальная бумага.
План выполнения задачи
Для проведения задачи необходимо 18 —20 ч (3 занятия).
Занятие 1. Посев микроорганизмов, подготовка сред и буферов для отбора проб, расчет количества меченых субстратов и ингибиторов.
Занятие 2. Центрифугирование культуры, подготовка клеток, постановка эксперимента, подсчет результатов.
Занятие 3. Обработка результатов, построение графиков, расчет скорости транспорта субстрата в клетки.
Глава 29
ОБРАЗОВАНИЕ МИКРООРГАНИЗМАМИ ФЕРМЕНТОВ
И МЕТОДЫ ИХ УЧЕТА
ЗАДАЧА 1. ОБРАЗОВАНИЕ ВНЕКЛЕТОЧНЫХ ФЕРМЕНТОВ И АНТИБИОТИКОВ
Микроорганизмы могут быть источником получения разных ферментов, имеющих важное значение для научных биологических исследований, медицины, сельского хозяйства, пищевой, легкой и других отраслей промышленности. Благодаря простоте культивирования, высокой скорости роста, возможности получения сверхпродуцентов путем селекции и применения генетических методов микроорганизмы более перспективны как источники ферментов по сравнению с растениями и животными. Кроме того, отдельные микроорганизмы образуют ферменты, которые ни растения, ни животные не синтезируют. К числу продуцентов практически важных ферментов относится ряд мицелиальных грибов, дрожжей и бактерий.
Ферменты микроорганизмов, которые уже получают промышленным путем, являются главным образом внеклеточными, т.е. ферментами, локализованными с внешней стороны клеточной мембраны. Преимущество получения внеклеточных ферментов (экзоферментов) состоит в легкости отделения от среды и очистки. Это делает их про
изводство более дешевым, чем получение эндоферментов. Функция большинства экзоферментов заключается в расщеплении высокомолекулярных субстратов с превращением их в форму, в которой они могут поглощаться клетками микроорганизмов. В качестве экзоферментов микроорганизмы чаще всего образуют гидролазы: протеазы (протеиназы и пептидазы), амилазы, декстраназы, целлюлазы и ряд других. Помимо ферментов многие микроорганизмы синтезируют антибиотики.
Цель работы: определить протеолитическую, амилазную и антибиотическую активность в культурах Streptomyces fradiae при росте на средах разного состава.
Ход выполнения задачи
Микроорганизмы и их культивирование. В работе используют стрептомицет Streptomyces fradiae (штамм № 25 из коллекции кафедры микробиологии МГУ). Этот штамм образует комплекс экзоферментов, который включает по крайней мере пять протеиназ, две пептидазы и сс-амилазу. Образование отдельных экзоферментов 5. fradiae зависит от условий роста этой бактерии. У. fradiae помимо экзоферментов выделяет в культуральную жидкость антибиотик тилозин, который активен против грамположительных и некоторых грамотрицательных бактерий, и применяется в животноводстве.
Культуру S. fradiae поддерживают на твердой среде следующего состава (г/л): кукурузный экстракт — 10,0; крахмал растворимый — 10,0; (NH4)2SO4 — 3,0; NaCl — 3,0; СаСО3 — 2,0; агар — 20,0; вода водопроводная — до 1 л. Навеску крахмала растворяют отдельно в небольшом объеме водопроводной воды при слабом нагревании и вносят в общий объем среды. Начальное значение pH доводят до 7,5 — 8,0 с помощью 1 н. NaOH и затем вносят навеску агара. Колбу с готовой средой ставят на кипящую водяную баню. После того как агар расплавится и среда станет гомогенной, ее разливают в пробирки (по 1 /3 их объема) и стерилизуют в автоклаве при 1,0 ати. Культуру S. fradiae выращивают на скошенном агаре в течение 5 сут в термостате при 30 °C и хранят в холодильнике при 4 °C.
Для получения посевного материала используют жидкую среду следующего состава (г/л): кукурузный экстракт — 10,0; крахмал растворимый — 15,0; КН2РО4 — 0,45; (NH4)2SO4 - 0,5; NaCl - 2,0; ZnSO4 - 0,02; MgSO4 - 0,9; CaCO3 - 5,0; вода водопроводная — до 1 л. Крахмал до внесения в среду готовят, как описано выше. Начальное значение pH среды доводят до 7,5 —8,0 с помощью 1 н. NaOH, после чего разливают по 100 мл в качалочные колбы объемом 750 мл, закрывают их ватными пробками и стерилизуют в автоклаве при 1 ати.
Посев проводят изогнутой под прямым углом бактериологической иглой (крючком), перенося стрептомицет из пробирки (хранящейся в холодильнике) вместе с блочком твердой среды в колбу с жидкой средой. Колбы помещают на качалку (200 об/мин) и выращивают культуру при 30 °C в течение 3 сут. Выросший посевной материал вносят в разные варианты опытной среды (см. ниже). В качестве посевного материала можно также использовать споры S. fradiae, полученные выращиванием стрептомицета в пробирках на твердой питательной среде в течение 5 сут. Споры смывают с поверхности питательной среды в пробирке стерильной водопроводной водой или жидкой питательной средой (3 — 5 мл), используемой для получения посевного материала, и вносят в опытные варианты питательных сред.
Для опыта используют среду следующего состава (г/л): кукурузный экстракт — 10,0; крахмал растворимый — 15,0; КН2РО4 — 0,45; (NH4)2SO4 — 0,5; NaCl — 2,0; ZnSO4 — 0,02; MgSO4 — 0,9; CaCO3 — 5,0. К основной среде (контроль) добавляют один из следующих субстратов (г/л): глюкоза — 10,0; соевая мука — 10,0; роговая мука— 5,0; рыбная мука — 5,0; фибрин — 5,0; шерсть — 5,0 (шерсть измельчают бритвой на мелкие фрагменты длиной около 1 мм); вода водопроводная — до 1 л. Крахмал до внесения в среду готовят, как описано выше. Начальное значение pH среды доводят до 7,5 — 8,0 с помощью 1 н. NaOH. Затем среду разливают по 100 мл в качал очные колбы объемом 750 мл, закрывают их ватными пробками и стерилизуют в автоклаве при 1,0 ати. При приготовлении среды с глюкозой этот субстрат стерилизуют отдельно в виде 50%-го водного раствора в автоклаве при 0,5 ати и добавляют к стерильной среде перед посевом в количестве 2 мл на 100 мл среды.
Каждый студент использует в работе колбу с одним вариантом опытной среды (т. е. с глюкозой, соевой, роговой, рыбной, мукой фибрином или шерстью) и в качестве контроля — вторую колбу с основной средой, не содержащей ни один из перечисленных субстратов. В колбы с опытной и основной средой вносят посевной материал, полученный на жидкой (5 % по объему) или твердой питательной среде (1 % по объему). После посева колбы помещают на качалку (200 об/мин) и выращивают культуры при 30 °C в течение 4 сут. В качестве тест-культуры при определении образования антибиотика тилозина S. fradiae используют культуру Bacillus subtilis, штамм № 6633, из коллекции кафедры микробиологии МГУ. В. subtilis выращивают на 2%-м МПА, pH 7,0— 7,2, смывают со скошенного агара дистиллированной водой и стерильно засевают данной суспензией клеток матрасы с 2%-м МПА. Бактерии выращивают при 37 °C в течение 5 — 7 сут. Делают смыв клеток с твердой среды матраца дистиллированной водой и полученную суспензию пастеризуют при 80 °C в течение 10 мин. Данную суспензию можно использовать на протяжении 6 мес при температуре хранения не более 5 °C.
Методы анализов
После инокуляции питательной среды (0 ч) и в процессе роста стрептоми-цета через 24, 48, 72 и 96 ч культивирования стерильно отбирают в пробирки пробы (по 7 мл). Отобранные пробы сразу же анализируют или ставят на хранение в холодильник при -5 °C. О росте стрептомицета судят по увеличению оптической плотности культуры. Биомассу отделяют центрифугированием при 10 тыс. g в течение 20 мин. Для определения синтеза антибиотика культуральную жидкость (2 мл) отделяют от клеток в стерильных центрифужных стаканах. В супернатанте (культуральной жидкости) определяют содержание растворимого белка, протеолитическую и а-амилазную активности.
Определение количества биомассы нефелометрическим методом. В основе метода лежит измерение количества света рассеянного суспензией клеток. С этой целью в пробирку наливают 5 мл 50%-го раствора (объем/объем) глицерина и 0,1 мл культуры S. fradiae, тщательно перемешивают. Глицерин применяют для предотвращения оседания клеток. Оптическую плотность при 450 нм измеряют в кюветах с длиной оптического пути 1 см на ФЭК или спектрофотометре. Количество биомассы в пробе определяют по калибровочной кривой с учетом разведений.
Для построения калибровочной кривой к 5 мл 50%-го раствора глицерина добавляют 0,1; 0,2; 0,3; 0,4; 0,5 мл жидкой культуры стрептомицета и после тщательного перемешивания измеряют оптическую плотность полученных суспензий, как указано выше. Затем каждую пробу фильтруют через бумажный фильтр, предварительно доведенный до постоянной массы при 105 °C, и промывают биомассу на фильтре физраствором. Фильтр с биомассой доводят до постоянной массы высушиванием в сушильном шкафу при 105 °C. Сухую биомассу рассчитывают по разности массы фильтра до и после фильтрования. Калибровочную кривую строят, откладывая на оси ординат показания спектрофотометра, а на оси абсцисс — количество биомассы (мг).
Определение растворимого белка в супернатанте. Определение белка проводят по методу Бредфорд. В качестве красителя используют кумасси ярко-голубой G-250 (Sigma, США). Содержание белка в пробе определяют по калибровочной кривой, которую строят по оптической плотности бычьего сывороточного альбумина (БСА). После построения калибровочной кривой проводят определение белка в пробе супернатанта. Для этого к раствору, содержащему от 30 до 500 мкг белка в 1 мл, добавляют 2,0 мл раствора красителя и тщательно перемешивают. Оптическую плотность при 595 нм измеряют в кюветах с длиной оптического пути 1 см не раньше, чем через 5 мин, и не позже, чем через 1 ч после смешивания реагентов. Для сравнения используют контрольную пробу, содержащую 0,1 мл 0,02 М трис-НС! буферного раствора (pH 7,8) и 2,0 мл раствора красителя.
Определение протеолитической активности. Метод основан на гидролизе окрашенного нерастворимого аналога белка протеолитическими ферментами, находящимися в исследуемом растворе, с последующим определением количества высвободившегося в результате гидролиза белка пигмента. В качестве окрашенного аналога белка применяют казеин, окрашенный римазолом ярко-голубым, азоказеин, азоколлаген и другие субстраты.
Для определения протеолитической активности с белком, окрашенным римазолом ярко-голубым, 500 мг этого субстрата помешают в фарфоровую ступку, растирают в 5—10 мл 0,02 М трис-НС! буфера (pH 7,5 —7,8) до получения однородного материала и постепенно, с растиранием, добавляют буфер до объема 40 — 50 мл. Затем содержимое переносят в мерную колбу на 100 мл, остатки белка смывают буфером со стенок фарфоровой ступки, количественно переносят в мерную колбу, доводят буфером объем до 100 мл и получают 0,5%-й раствор окрашенного белка. В пробирку наливают 1 мл раствора окрашенного белка, добавляют 0,5 мл 0,2 М трис-НС! буфера и 0,5 мл опытной пробы (культуральной жидкости), которая разводится в зависимости от содержащегося в ней белка. Пробирки инкубируют в ультратермостате при 37 °C 10 мин, после чего содержимое фильтруют через мембранный фильтр «Сынпор № 2» с размером пор 2,5 мкм. В фильтрате спектрофотометрически определяют количество высвободившегося в результате гидролиза белка красителя при X = 595 нм в кюветах с длиной оптического пути 1 см. Для сравнения используют контрольную пробу, содержащую 1 мл субстрата, 0,5 мл 0,2 М трис-НС! и 0,5 мл дистиллированной воды.
Для определения протеолитической активности с азоказеином готовят 0,5%-й раствор этого белка в 0,05 М трис-HCl буфере (pH 7,5 —7,8), как описано для белка, окрашенного римазолом. Затем в пробирку наливают 1 мл раствора азоказеина, добавляют 0,1 мл 0,1%-го СаС12 и 0,1 мл опытной пробы (культуральной жидкости) и инкубируют в ультратермостате при 37 °C 20 мин. Далее в пробирку вносят для остановки реакции и осаждения непрореагировавшего субстрата 2 мл 10%-й трихлоруксусной кислоты. Пробирку выдерживают 30 мин в ледяной бане, содержимое центрифугируют 15 мин при 10 тыс. g и к 2 мл супернатанта добавляют 2 мл 0,5 М NaOH. Количество высвободившегося в результате гидролиза белка красителя определяют на спектрофотометре при 440 нм в кюветах с длиной оптического пути 1 см. В контрольную пробу вместо опытной пробы добавляют 0,1 мл дистиллированной воды или буфера.
За единицу протеолитической активности (Д) принимают такое количество фермента, которое вызывает увеличение оптической плотности фильтрата на 0,01 ед. при измерении против контрольной пробы. Протеолитическую активность выражают (с учетом разведения культуральной жидкости) в Д/мин • мл и Д/мин • мг белка.
Определение амилолитической активности. Амилаза, образуемая S. fradiae, гидролизует 1,4-а-связи в амилозе и амилопектине, из которых состоит крахмал. Активность а-амилазы определяют по методу Смита и Роя в модификации Маннинг и Кемпбелл. Метод основан на фотометрическом определении динамики изменения окраски крахмала с йодом под действием фермента.
Реактивы. 0,1 н. ацетатный буфер (pH 5,0); свежеприготовленный 1%-й раствор растворимого крахмала (навеску крахмала растворяют на горячей водяной бане, после чего фильтруют) в 0,1 н. ацетатном буфере (pH 5,0); 0,5 М раствор СаС12; 1,0 н. раствор НС1; 0,3%-й раствор йода в 3%-м растворе К1.
В мерную пробирку А на 25 мл (опыт) наливают 0,2 мл 1%-го раствора субстрата (крахмала); 0,5 мл 0,5 М СаС12 и 1,5 мл 0,1 н. ацетатного буфера. Те же количества реактивов добавляют в контрольную пробирку Б. Обе пробирки выдерживают при 40 °C в течение 5 мин. Затем добавляют 1 мл раствора фермента (культуральной жидкости) в опытную пробирку А, быстро перемешивают и выдерживают 15 мин при 40 °C. В обе пробирки добавляют по 0,5 мл 1 н. НС1 (для остановки реакции). После этого в пробирку Б добавляют 1 мл раствора фермента (культуральную жидкость). Далее в обе пробирки добавляют 0,1 мл раствора йода. Объем жидкости в пробирках А и Б доводят до 25 мл дистиллированной водой и тщательно перемешивают.
Оптическую плотность измеряют на ФЭК (фильтр № 7 - оранжевый) или спектрофотометре при X = 610 нм в кюветах с длиной оптического пути 1 см. В качестве контроля используют дистиллированную воду.
Действие фермента учитывают по разнице окраски в контроле и опыте (Б—-А). Активность фермента (Е) выражают в условных единицах в 1 мин на 1 мл культуральной жидкости (к. ж) с учетом разведения (Е/мин мл к.ж) и на 1 мг белка культуральной жидкости в 1 мин с учетом разведения (Е/мин-мг белка к.ж):
Е (Б-А)25
15 мин
Определение антибиотической активности. Содержание антибиотика тилозина в культуральной жидкости в динамике развития A fradiae определяют биологическим методом, основанным на непосредственном биологическом действии антибиотика на используемый тест-организм В. subtilis, чувствительный к данному препарату. Точность биологического метода составляет ± 10 %.
Количество антибиотика в культуральной жидкости определяют методом последовательных разведений. Используют среду, благоприятную для развития выбранного тест-организма (прозрачный мясо-пептонный бульон). Из пропастеризованной и хранящейся в холодильнике суспензии B.subtilis готовят одномиллиардную суспензию по стандарту мутности. Эту суспензию в количестве 0,5 мл добавляют к 100 мл МПБ, перемешивают и разливают по 1 мл в 4 ряда стерильных пробирок по 15 пробирок в каждом ряду. Далее в первые пробирки трех рядов в МПБ с тест-культурой добавляют по 1 мл отцентрифугированной стерильной культуральной жидкости соответствующих отобранных проб (48, 96 и 144 ч) и таким образом данную пробу культуральной жидкости последовательно разводят (тест-культурой в МПБ) в 2, 4 и т.д. раз (табл. 29.1).
В первую пробирку с тест-культурой в МПБ четвертого ряда (стандарт) вносят стандартный раствор антибиотика (тилозина), который также последовательно разводят (тест-культурой в МПБ в 2, 4, 8, 16 и т.д. раз). Исходная концентрация стандартного раствора содержит 30 мкг/мл тилозина. Антибиотик сначала растворяют в 1 — 2 каплях спирта и далее доводят дистиллированной водой до нужного объема. Параллельно
Схема определения антибиотической активности
1 —3-й ряды пробирок Номер пробирки
1 2 3 4 5 15
100 мл МПБ + 0,5 мл спор В. subtilis 1 мл 1 мл 1 мл 1 мл 1 мл 1 МЛ
1 мл 48-часовой пробы культуральной жидкости добавляют в пробирку 1 и переносят последовательно в следующие пробирки 1 мл 1 мл 1 мл 1 мл 1 мл 1 мл
Полученные разведения культуральной жидкости (коэффициент разведения) 2 4 8 16 64 32 448 раз
ставят два контроля: 1 мл стерильного МПБ (контроль среды); 1 мл МПБ + тест-культура (контроль роста тест-культуры).
Пробирки помещают в термостат на 24 ч при 30 °C. Далее измеряют оптическую плотность тест-культуры при X = 650 нм в кюветах 1 см или визуально сравнивают рост тест-культуры в контроле (ряд разведений исходного стандартного раствора тилозина) и опыте (ряд разведений культуральной жидкости). Совпадение оптической плотности между пробирками разведений культуральной жидкости и стандартного раствора тилозина позволяет определить С (концентрацию антибиотика в культуральной жидкости) с учетом соответствующих разведений:
С(мкг/мл) = ~Сн).,
где Ккж — коэффициент разведения культуральной жидкости отобранной пробы, в которой отсутствует рост тест-культуры; Кст — коэффициент разведения стандартного раствора тилозина, в которой отсутствует рост тест-культуры; Сст — концентрация исходного стандартного раствора тилозина (мкг/мл) или С (мкг/мл) = LDKkjk, где LD — летальная доза стандартного растворатилозина (мкг/мл), вызывающая подавление роста тест-культуры.
Полученные результаты оформляют в виде таблицы (табл. 29.2) и графиков. На графиках должны быть представлены: кривая роста культуры, содержание белка в культуральной жидкости, удельная протеолитическая и амилолитическая активности, антибиотическая активность. Делают вывод о ферментативной и антибиотической активности S. fradiae в зависимости от фазы роста культуры и состава питательной среды.
Материалы, оборудование, посуда, реактивы
Спектрофотометр; центрифуга; ультратермостат; водоструйный насос; качал очные колбы — 2; пробирки обычные — 70; пробирки обычные стерильные — 8; пробирки мерные на 25 мл — 2; пипетки на 2 мл и на 10 мл — по 10; реактивы для приготовления сред, определения белка, протеолитической и амилолитической активности, тилозина (см. текст).
План выполнения задачи
Для выполнения работы необходимо 36 — 40 ч.
Занятие 1. Приготовление и стерилизация питательных сред и посуды. Подготовка посевного материала S. fradiae.
Занятие 2. Посев культуры S. fradiae в основную и опытную среды. Построение калибровочной кривой для определения белка в культуральной среде по методу Бред-
Протеолитическая, амилолитическая и антибиотическая активность S', fradiae при росте на среде с соевой (кукурузной, рыбной и т.д.) мукой
Показатель Время культивирования, ч
24 48 96 144
Биомасса, ед. опт. пл.
Содержание белка в культуральной жидкости, мг/мл
Протеолитическая активность культуральной жидкосш, Д/мин-мл
Удельная протеолитическая активность, Д/мин мг белка культуральной жидкости
Амилолитическая активность культуральной жидкости, Е/мин • мл
Удельная амилолитическая активность, Е/мин • мг белка культуральной жидкости
Количество антибиотика в культуральной жидкости, мкг/мл
Продуктивность культуры, мкг антибиотика/мг биомассы
форд. Отбор проб суспензии (по 7 мл) сразу после посева и через 24 ч культивирования (хранить в холодильнике).
Занятие 3. Отбор проб культуры через 48 ч роста. Определение в культуральной жидкости (пробы 0, 24 и 48 ч) белка по Бредфорд; протеазной и амилазной активности. Определение количества биомассы (по оптической плотности, пробы 0, 24 и 48 ч). Отбор проб культуры через 72, 96 и 120 ч роста. Подготовка мембранных фильтров для построения калибровочной кривой.
Занятие 4. Определение в культуральной жидкости (пробы 72, 96 и 120 ч) белка по Бредфорд, протеазной и амилазной активности. Определение количества биомассы (по оптической плотности, пробы 72, 96 и 120 ч). Построение калибровочной кривой для определения биомассы бактерий.
Занятие 5. Определение антибиотической активности культуральной жидкости (пробы 72, 96, 120 ч).
Занятие 6. Расчеты, оформление результатов, зачет.
ЗАДАЧА 2. РЕГУЛЯЦИЯ СИНТЕЗА ТРИПТОФАНАЗЫ ESCHERICHIA COLI*
Регуляция метаболизма позволяет управлять скоростью биохимических процессов в клетке и служит важнейшим механизмом приспособления микроор
* Задача разработана сотрудниками кафедры микробиологии биологического факультета МГУ М. Н. Пименовой и Н.Н.Гречушкиной; приводится здесь в переработанном и сокращенном варианте. Впервые текст задачи опубликован в кн.: Избранные задачи большого практикума по микробиологии. — М.: Изд-во МГУ, 1985.
ганизмов к окружающей среде. Регуляция может осуществляться на уровне синтеза и активности ферментов. Основными механизмами регуляции синтеза ферментов являются индукция и репрессия. Большое значение имеет так называемая катаболитная репрессия, при которой синтез фермента, разлагающего субстрат, подавляется при использовании клетками альтернативного, более выгодного источника энергии (чаще всего, глюкозы).
Примером фермента, синтез которого регулируется путем катаболитной репрессии, является триптофаназа (НФЛ. 1.99.1., L-триптофаниндоллиаза дезаминирующая)*, катализирующая реакцию разложения триптофана:
Z-триптофан + Н2О -> индол + пируват + NH3
Цель работы', определить влияние глюкозы на синтез триптофаназы Escherichia coli.
Ход выполнения задачи
Микроорганизм и его культивирование. В качестве объекта исследования используют штамм E.coli 85 из коллекции культур кафедры микробиологии МГУ. Полученную из музея культуру E.coli пересевают в пробирки со скошенным МПА и выращивают 1 — 2 сут при 30 °C. Затем петлю такой культуры переносят в 100 мл основной среды следующего состава (г/л): гидролизат казеина — 5,0; КН2РО4 -1,0; К2НРО4 - 1,0; NaCl - 1,0; MgSO4-7H2O - 0,7; (NH4)2SO4 - 4,0; цитрат натрия — 0,5; вода дистиллированная. Исходное значение pH среды 6,7 — 7,0. Среду готовят в качалочных колбах на 750 мл и стерилизуют при 1 ати. Культуру выращивают в стационарных условиях при 30 °C в течение 18 —24 ч и используют затем в качестве инокулята опытных сред.
При постановке опыта для культивирования Е. coli используют два варианта среды: 1) основную среду', 2) среду с глюкозой (2,0 г /100 мл основной среды).
Основную среду разливают по 200 мл в качалочные колбы и стерилизуют при 1 ати. Раствор глюкозы готовят отдельно и вносят в основную среду перед ее засевом. Для этого навеску глюкозы вносят в мерный цилиндр и доводят объем до 10 мл дистиллированной водой, затем полученный раствор глюкозы переливают в пробирку и стерилизуют при 0,5 ати. Отдельно в пробирке стерилизуют 10 мл дистиллированной воды, которую вносят затем в среду 1. Посевной материал вносят в количестве 2 % (по объему). Засеянные колбы помещают на качалку (200 — 300 об/мин). Культивирование проводят при 30 °C в течение 18 —24 ч.
Приготовление суспензии клеток. Из каждого варианта культуры отбирают 80— 100 мл и центрифугируют при 5—6 тыс. д в течение 10—15 мин. Клетки отмывают водой или физиологическим раствором (0,85 % NaCl) и суспендируют в 1 мл воды. Из каждой суспензии отбирают 0,1 мл для определения белка. Оставшуюся часть суспензии используют для определения активности триптофаназы.
Определение активности триптофаназы. Для проведения реакции к суспензии клеток в фосфатном буфере (реакционной смеси) добавляют /.-триптофан и инкубируют определенное время при 37 °C. Об активности триптофаназы судят по количеству индо
* Подробнее см.: Hart I. et al. Mechanism of catabolite repression of tryptophanase synthesis in Escherichia coli // Microbiology. — 1994. — V. 140. — P. 2125—2134; Konan К. V, Yanofsky C. Regulation of the Escherichia colitna operon: nascent leader peptide control at the tnaC stop codon // J.Bacteriol., 1997. — V. 179. - P. 1774—1779.
ла, образованного клетками за время реакции. В качестве контроля используют суспензию клеток без триптофана. При расчете активности фермента учитывают содержание в суспензии клеток белка.
Реакционные смеси готовят в пробирках с притертыми пробками. Для каждого варианта используют 6 пробирок (3 контрольных и 3 опытных). В каждую пробирку вносят 0,2 мл 0,2 М Na-фосфатного буфера (pH 7,8); 0,1 мл суспензии клеток и 1,6 мл дистиллированной воды. Смеси тщательно перемешивают и помещают пробирки в ультратермостат на 37 °C. Через 10 мин в опытные варианты вносят по 0,1 мл раствора триптофана (10 мг/мл).
Через 1, 20 и 60 мин пробирки вынимают из ультратермостата и останавливают реакцию, добавляя к реакционной смеси 0,2 мл свежеприготовленного раствора трихлоруксусной (ТХУ) кислоты. После этого в реакционной смеси определяют количество индола.
Определение индола. Индол экстрагируют из смеси толуолом и определяют колориметрически с реактивом Эрлиха. Для этого в пробирки с реакционной смесью добавляют по 2 мл толуола и перемешивают содержимое пробирки, вращая пробирки между ладонями. {Внимание! Работа с толуолом проводится под тягой!) Встряхивать пробирки не следует, так как образуется эмульсия, которая не расслаивается. После расслоения жидкости в пробирках отбирают сухой пипеткой 0,5 мл раствора индола в толуоле (верхний слой) и переносят его в сухую пробирку с притертой пробкой. Добавляют 1 мл реактива Эрлиха и 4,5 мл кислого спирта, тщательно перемешивают. Окраска развивается в течение 15 мин. Оптическую плотность раствором измеряют на ФЭКе или спектрофотометре при 540 нм в кювете с рабочим расстоянием 1 см. Измерение ведут против воды. Количество индола (мкг) пересчитывают по калибровочной кривой и вносят в табл. 29.3.
Для построения калибровочной кривой вместо реакционной смеси используют растворы, содержащие в 2 мл от 10 до 100 мгк индола. Для этого вначале готовят стандартный раствор с концентрацией индола 50 мкг/мл, а затем делают ряд разведений (табл. 29.4). К полученным разведениям добавляют толуол и проводят определение индола, как описано выше.
Необходимые для анализа реактивы готовят следующим образом. Для приготовления реактива Эрлиха 5 г и-диметиламинобензальдегида растворяют в 100 мл 96%-го этилового спирта. Для приготовления кислого спирта к 92 мл 96%-го этилового спирта добавляют 8 мл концентрированной НС1.
Определение белка. Определение белка в суспензии клеток осуществляют методом Лоури. Метод основан на биуретовой реакции пептидной связи и реакции на циклические аминокислоты тирозин и триптофан. Чувствительность — 20—150 мкг белка в 1 мл раствора. Метод применим только для растворимого в воде белка, поэтому клетки бактерий предварительно гидролизуют.
Таблица 29.3
Определение индола в реакции триптофаназы
| Вариант культуры Время инкубационной смеси, мин Оптическая плот-ность раствора Количество индола, мкг
в реакционной смеси в 1 мл суспензии клеток
1 1 20 60
2 1 20 60
Приготовление растворов индола для построения калибровочной кривой и их оптическая плотность
Показатель Номер разведения
0 1 2 3 4 5 6
Объем исходного раствора индола, мл 0 0,2 0,4 0,8 1,2 1,6 2,0
Объем Н2О, мл 2,0 1,8 1,6 1,2 0,8 0,4 0
Содержание индола, мкг 0 10 20 40 60 80 100
Оптическая плотность растворов
Для проведения гидролиза 0,1 мл тщательно перемешанной суспензии клеток и 2,9 мл 2 н. NaOH помешают в пробирку, закрывают ее резиновой пробкой и выдерживают 2 ч при 37 °C. После гидролиза к смеси добавляют 3 мл Н2О. Для анализа белка берут 0,05; 0,10; 0,15 и 0,20 мл тщательно перемешанного гидролизата, доводят объем каждой пробы в пробирках до 1 мл дистиллированной водой, добавляют при постоянном встряхивании 5 мл реактива В (см. ниже) и оставляют при комнатной температуре на 10 мин. Затем к смеси добавляют 0,1 мл реактива Фолина (см. Приложение 4), перемешивают и выдерживают 40 мин в темноте. Оптическую плотность раствора определяют на ФЭКе или спектрофотометре при 660 нм в кювете с длиной оптического пути 0,5 или 1 см. Для приготовления контрольного раствора используют те же реактивы, но вместо гидролизата белка берут Н2О.
При расчете содержания белка используют калибровочную кривую. Для построения калибровочной кривой готовят раствор, содержащий в 1 мл 200 мкг бычьего альбумина. Растворяют 10 мг альбумина в 50 мл Н2О. Затем из исходного раствора готовят разведения с содержанием белка от 20 до 160 мкг/мл (табл. 29.5). К разведениям добавляют реактивы В и Фолина и определяют оптическую плотность растворов, как указано выше.
Необходимые реактивы готовят следующим образом. Реактив В готовят в день проведения анализа. Его составляют из 50 мл раствора А (2% раствора Na2CO3 в 0,1 н. NaOH) и 1 мл раствора Б. Раствор Б готовят смешиванием равных объемов 1%-го раствора CuSO4 и 2%-го раствора К,№-виннокислого (сегнетовой соли).
Оформление результатов. Полученные результаты оформляют окончательно в виде сводной таблицы (табл. 29.6). Рассчитывают активность триптофана-зы (мг индола/мг белка) за 30 и 60 мин. Делают заключение о влиянии состава среды на синтез клетками триптофаназы.
Таблица 29.5
Приготовление растворов альбумина для построения калибровочной кривой и их оптическая плотность
Показатель Номер разведения
0 1 2 3 4 5 6 7 8
Объем исходного раствора белка, мл 0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8
Объем Н2О, мл 1,0 0,9 0,8 0,7 0,6 0,5 0,4 о,з 0,2
Содержание белка в растворе, мкг 0 20 40 60 80 100 120 140 160
Оптическая плотность раствора
Таблица 29.6
Активность триптофаназы в клетках E.coli, выросших на разных средах
Среда роста Время инкубации реакционной смеси, мин Количество образованного индола, мкг/мл суспензии Содержание белка, мкг/мл суспензии Активность фермента, мкг индола/(мг белка мин)
1
2
Материалы, оборудование, посуда, реактивы
Спектрофотометр или ФЭК; ультратермостат на 30 °C; центрифуга; качалочные колбы; пробирки обыкновенные и с притертыми пробками; соли и реактивы для приготовления сред и анализов.
План выполнения задачи
Для выполнения задачи требуется около 20 ч.
Занятие 1. Приготовление и стерилизация сред и посуды. Накануне следующего занятия пересеять культуру E.coli в пробирки со скошенным МПА и поместить в термостат на 30 °C.
Занятие 2. Посев культуры в основную среду для получения посевного материала. Построение калибровочных кривых. Накануне следующего занятия посеять культуру E.coli в опытные среды.
Занятие 3. Приготовление суспензии клеток, определение активности триптофаназы и содержания белка.
Занятие 4. Оформление результатов, обсуждение, зачет.
ЗАДАЧА 3. ОБРАЗОВАНИЕ L-АСПАРАГИНОВОЙ КИСЛОТЫ ИЗ ФУМАРАТА АММОНИЯ СВОБОДНЫМИ
И ИММОБИЛИЗОВАННЫМИ КЛЕТКАМИ ESCHERICHIA COLI*
Иммобилизация клеток — это закрепление их тем или иным способом на поверхности или внутри носителя. Основной целью иммобилизации является стабилизация и удлинение срока действия определенных ферментов. С помощью иммобилизованных клеток микроорганизмов можно получать некоторые аминокислоты, органические кислоты, антибиотики и другие практически важные соединения. В качестве носителя при иммобилизации клеток можно, например, использовать каррагинан — добываемый из морских красных водорослей полисахарид, состоящий из сульфата р-D-галактозы и 3,6-ангидро-а-7)-галактозы (мол. масса 100 — 800 кДа).
Производство Z-аспарагиновой кислоты с помощью иммобилизованных клеток Escherichia coli было разработано и внедрено в промышленность в Японии в 1974 г. Аспартат образуется в результате аминирования фумарата:
фумарат + NH4 -> аспартат.
* Задача разработана сотрудниками кафедры микробиологии биологического факультета МГУ Н. Н.Гречушкиной, М.Н. Пименовой, И.В.Авсюк. Впервые опубликована в кн.: Избранные задачи большого практикума по микробиологии. — М.: Изд-во МГУ, 1985. — С. 47 — 64.
Реакцию катализирует аспартаза (L-аспартат-аммиак-лиаза, НФ.4.3.1.1), кофакторами которой служат ионы магния или других двухвалентных металлов. Аспартаза обнаружена у ряда бактерий. Обычно фермент синтезируется при росте культур на богатых средах, содержащих мясной бульон, пептон, дрожжевой экстракт и т.д. Скорость выделения аспартата клетками можно повысить, увеличив их проницаемость, например, в результате обработки толуолом.
Цель работы, сравнить образование /.-аспарагиновой кислоты свободными и иммобилизованными в каррагинан клетками E.coli в отсутствие и в присутствии толуола.
Ход выполнения задачи
Микроорганизм и его культивирование. Объектом исследования служит штамм E.coli, имеющий высокую аспартазную активность. Полученный из музея штамм пересевают на скошенный МПА и выращивают в течение суток при 30 °C.
Для постановки опыта культуру бактерии выращивают в МПБ. В качалочные колбы на 750 мл вносят 150 мл МПБ и стерилизуют при 1 ати. В качестве посевного материала используют 1 петлю культуры, выросшей на МПА. Культивирование ведут на качалке при 30 °C в течение 19 — 20 ч.
Приготовление суспензии клеток. Для приготовления суспензии клеток используют не менее 120 мл культуры. Клетки отделяют от среды центрифугированием (3 — 4 тыс. g, 10—15 мин), супернатант сливают. Для удаления остатков супернатанта центрифужные стаканы с клетками ставят перевернутыми на фильтровальную бумагу. Затем клетки суспендируют в 3,5 —4,0 мл 0,85%-го раствора NaCl, переносят в пробирку и закрывают резиновой пробкой. Для определения содержания белка отбирают 0,1 мл суспензии клеток. Оставшуюся часть суспензии хранят до постановки опытов в холодильнике.
Содержание в клетках бактерий белка определяют методом Лоури (см. 29.2).
1. Образование аспарагиновой кислоты свободными клетками. Опыт проводят в двух вариантах, используя клетки, обработанные и не обработанные толуолом (рис. 29.1.). Реакционные смеси готовят в двух колбах на 50 мл. В каждую колбу вносят 9,5 мл 0,5 М фумарата аммония в 0,05 М калий-фосфатном буфере (pH 8,0) и 0,5 мл суспензии клеток. Затем в одну из колб добавляют 0,1 мл толуола. Колбы закрывают ватными пробками и помещают на качалку на 3 ч при 30 °C или на 2 ч при 37 °C.
По истечении указанного времени реакционные смеси центрифугируют в сухих центрифужных стаканчиках (3—4 тыс. g, 10—15 мин). В супернатанте определяют количество аспарагиновой кислоты и фумарата (см. ниже). Параллельно определяют содержание этих соединений в исходном растворе фумарата аммония в фосфатном буфере.
При определении концентрации фумарата аммония супернатант (или исходный раствор фумарата аммония) во всех вариантах опытов разводят в 2000 раз. Для определения исходного содержания аспарагиновой кислоты в растворе фумарата аммония отбирают пробы объемом 0,1; 0,2; 1,0 и 2,0 мл. Для определения количества аспарагиновой кислоты, образованной клетками, берут 0,1; 0,2 и 0,5 мл супернатанта. Супернатант смеси с толуолом предварительно разводят дистиллированной водой в 10—100 раз (в зависимости от активности клеток).
Рис. 29.1. Схема опыта со свободными клетками
На основании полученных результатов рассчитывают активность аспартазы (в мг образованной аспарагиновой кислоты/мг белка-ч), а также ее продуктивность (количество образованной аспарагиновой кислоты в процентах от внесенного фумарата). Сравнивают активность аспартазы обработанных и не обработанных толуолом клеток. Результаты вносят в табл. 29.7.
2. Образование аспарагиновой кислоты иммобилизованными клетками. Иммобилизация клеток. Иммобилизацию клеток E.coli осуществляют путем их включения в каррагинан (см. схему опыта, рис. 29.2). В химический стакан объемом 50 мл вносят 350 мг каррагинана, добавляют 2 мл дистиллированной воды, закрывают фольгой или часовым стеклом и выдерживают 15 — 30 мин при комнатной температуре для набухания. Добавляют б мл 0,85%-го раствора NaCl и, закрыв фольгой или часовым стеклом, помещают стакан на кипящую водяную баню. Смесь непрерывно помешивают покачиванием стакана (стеклянную палочку использовать не рекомендуется). После полного расплавления каррагинана стакан вынимают из бани, охлаждают его содержимое до 50 — 55 °C, вносят при непрерывном помешивании 1,5 мл суспензии клеток и быстро ставят стакан в сосуд со льдом (или в воду, налитую поверх ледяного слоя).
Таблица 29.7
Потребление фумарата и образование аспарагиновой кислоты свободными клетками Е. coll
Вариант опыта Содержание в реакционной смеси, г/мл Образовалось аспарагиновой кислоты Аспаразная активность клеток, мг мг1 белка Ч'1
белка фумарата аспара-гиновой кислоты % от фумарата мг/мг белка
исходное конечное использовано
-Толуол
+Толуол
350 мл каррагинана + 2 мл дистиллированной воды, 15—30 мин при комнатной температуре (набухание) + 6 мл 0,85%-го раствора NaCl
Расплавить каррагинан в кипящей водяной бане
Охладить на воздухе до 55—60 °C
Добавить 1,5 мл суспензии клеток при постоянном перемешивании и сразу поместить в сосуд со льдом
Перенести блок в стакан со 100 мл охлажденного 2%-го раствора КС1, где выдержать 20—30 мин
Протереть блок через сито в фарфоровую чашку с 8—10 мл 2%-го КС1.Смыть гранулы с сита 15—20 мл этого раствора
Перенести гранулы в растворе КС1 в цилиндр на 50 мл, отставить на 5—10 мин
--- ~
Промыть гранулы 2 раза по 20—25 мл 2%-м раствором
KCI и один раз 20—25 мл 0,1 М фосфатного буфера
Измерить объем гранул в фосфатном буфере и добавить равный объем 1 М раствора фумарата аммония с MgClj
Декантация (р-р КС1)
Промывные воды
Рис. 29-2. Схема опыта с иммобилизованными клетками
I
I
I
I
I
I
Застывший блок каррагинана с клетками переносят в стакан со 100 мл 2%-го раствора КС1 (отвердителя), охлажденного в холодильнике, и выдерживают 20—30 мин. Блок помещают в сито (диаметр пор около 1 мм), прозрачный слой КС1 удаляют, а осадок неиммобилизованных клеток вместе с небольшим объемом раствора переливают в мерный цилиндр на 100— 150 мл. Блок с клетками перетирают через сито фарфоровым пестиком в стакан или фарфоровую чашку, содержащую 8 — 10 мл 2%-го раствора КС1. Гранулы как можно полнее
смывают с сита 15 — 20 мл раствора КС1, переносят в цилиндр на 50 мл и отстаивают в течение 5 —10 мин. Затем раствор КО сливают декантацией в цилиндр с неиммобилизованными клетками. Последние порции жидкости над гранулами аккуратно отбирают пипеткой.
Гранулы с клетками промывают два раза 2%-м раствором КС1 и один раз 0,1 М К-фосфатным буфером. Для этого в цилиндр с гранулами вносят по 20— 25 мл соответствующего раствора, осторожно перемешивают стеклянной палочкой, отстаивают 5 — 10 мин и сливают раствор в цилиндр с неиммобилизованными клетками.
Общий объем промывных вод не должен превышать 100 мл. В промывных водах определяют белок неиммобилизованных клеток (см. ниже).
Проведение реакции. К смеси гранул в К-фосфатном буфере добавляют равный объем 1 М фумарата аммония, тщательно перемешивают, отбирают пипеткой с широким носиком 10 мл и переносят в реакционную колбу на 50 мл. Добавляют 0,1 мл толуола, закрывают ватной пробкой и колбу помещают на качалку (на 2 или 3 ч при 37 или 30 °C соответственно).
Оставшуюся часть суспензии гранул сразу отфильтровывают через бумажный фильтр в сухую химически чистую пробирку или центрифугируют (3—4 тыс. g, 10—15 мин). В фильтрате (супернатанте) определяют количество фумарата и аспарагиновой кислоты. Для определения аспарагиновой кислоты фильтрат разводят в 10—100 раз, объем проб: 0,2; 0,5 и 1,0 мл.
Чтобы найти содержание белка в иммобилизованных клетках, определяют белок отмывшихся (неиммобилизованных) клеток в промывных водах. Для этого к 2 мл тщательно перемешанных промывных вод добавляют 2 мл 2 н. NaOH и проводят гидролиз (2 ч при 37 °C). Для анализа берут 0,1; 0,4; 0,6 и 0,8 мл гидролизата. По разности в содержании белка в исходной суспензии клеток и промывных водах находят количество белка в иммобилизованных клетках, а затем в 1 мл реакционной смеси. Результаты вносят в табл. 29.8.
Результаты определений фумарата, аспарагиновой кислоты и белка в опыте с иммобилизованными клетками обобщают в табл. 29.9, которую строят по аналогии с табл. 29.7.
Таблица 29.8
Исходные данные и результаты определения белка иммобилизованных клеток
Показатель Цифровые данные
Объемы гидролизата белка, взятые для определения, мл 0,2 0,4 0,6 0,8
Показания ФЭК
Содержание белка, мкг: в испытуемых пробах, по калибровочной кривой в 4 мл гидролизата белка, т. е. в 2 мл промывных вод в общем объеме промывных вод в общем объеме промывных вод, среднее значение (А) в 1,5 мл суспензии клеток, введенных в каррагинан (Б) в иммобилизованных клетках, т.е. в смеси гранул с К-фос-фатным буфером (Б-А) в реакционной смеси в общем объеме в 1 мл
Потребление фумарата и образование аспарагиновой кислоты иммобилизованными клетками E.coli
Количество в реакционной смеси, мг/мл Образовалось аспарагиновой кислоты Аспартазная активность клеток, мг кислоты/(мг белка• ч)
белка клеток фумарата аспараги-новой кислоты % от внесенного фумарата мг/мг белка клеток
в начале опыта в конце опыта использовано
На основании полученных данных сравнивают активность и продуктивность аспартазы свободных и иммобилизованных клеток. Результаты оформляют в табл. 29.10.
Определение фумарата. Концентрацию фумаровой кислоты определяют спектрофотометрически по изменению поглощения при 240 нм. Коэффициент молярной экстинкции фумарата — 2530. Учитывая, что все пробы перед определением разводили в 2000 раз, концентрацию фумарата рассчитывают по формуле
л-М = д£- 2000/2530 = ЕЕ-0,7905,
где хМ — молярность исследуемого раствора; ЕЕ — оптическая плотность раствора. Зная концентрацию фумарата в молях и его молекулярную массу (133), рассчитывают содержание фумарата в мкг/мл.
Определение аспарагиновой кислоты. Содержание аспарагиновой кислоты определяют колориметрическим методом с использованием нингидрина. В пробирку с притертой пробкой вносят 2 мл пробы и 1 мл нингидринового реактива (отобрать с помощью дозатора или резиновой груши). Плотно закрытые пробирки выдерживают 3 мин (точно!) на кипящей водяной бане и затем немедленно охлаждают в сосуде с холодной водой. Оптическую плотность раствора измеряют при Х = 500 нм (зеленый светофильтр) в кювете с рабочим расстоянием 1 см. Окраска L-аспарагиновой кислоты с нингидрином стабильна в течение часа. Контрольный раствор получают используя вместо аспарагиновой кислоты воду. .
Количество аспарагиновой кислоты в объеме испытуемой пробы рассчитывают используя калибровочную кривую. Готовят раствор, 1 мл которого содержит 0,5 мкг L-аспарагиновой кислоты. Для этого взвешивают 50 мг аспарагиновой кислоты и растворяют в 100 мл дистиллированной воды (в мерной колбе) с небольшим подщелачиванием и осторожным нагреванием. Затем из исходного раствора готовят разведения
Таблица 29.10
Образование аспарагиновой кислоты свободными и иммобилизованными клетками
E.coli
Вариант опыта Аспарагиновая кислота Аспартазная активность клеток, мг аспарагиновой кислоты / (мг белка - ч)
% от внесенного фумарата мг/мг белка клеток
Свободные клетки -Толуол +Толуол
Иммобилизованные клетки
Схема приготовления стандартных растворов Z-аспарагиновой кислоты и их оптическая плотность
Показатель Номер пробирки
0 1 2 3 4 5 6 7 8 9
Объем исходного раствора, мл 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,0
Объем воды, мл 1,9 1,8 1,7 1,6 1,5 1,4 1,3 1,2 1,1 1,0
Содержание аспарагиновой кислоты, мкг 50 100 150 200 250 300 350 400 450 500
Оптическая плотность
(табл. 29.11) с содержанием аспарагиновой кислоты от 50 до 500 мкг (разведения готовить в пробирках с притертыми пробками). Определяют оптическую плотность стандартных растворов аспарагиновой кислоты с нингидрином. На основании полученных результатов строят калибровочную кривую, откладывая по оси абсцисс содержание аспарагиновой кислоты (мкг), а по оси ординат — оптическую плотность.
Нингидриновый реактив включает следующие компоненты: 2 г нингидрина; 20 мл ацетона; 0,5 г хлористого кадмия (CdCl2) в 77 мл дистиллированной воды; 3 мл ледяной уксусной кислоты. В химический стакан вносят нингидрин и заливают его ацетоном. Отмеряют необходимый объем воды и в большей его части растворяют хлористый кадмий. В раствор хлористого кадмия вносят ледяную уксусную кислоту и приливают к этому раствору нингидрин в ацетоне. Остатки нингидрина смывают неиспользованным объемом воды. Нельзя приливать кислоту к нингидрину!
Приготовление раствора фумарата аммония в К-фосфатном буфере. Для приготовления раствора фумарата аммония в К-фосфатном буфере смешивают 1 М раствор фумарата аммония с хлористым магнием с равным объемом 0,1 М К-фосфатного буфера (pH 8,0).
Фумарат аммония готовят из фумаровой кислоты и аммиака. 1 М раствор содержит 116,07 г фумаровой кислоты в 1 л. В мерный стакан помещают необходимую навеску фумаровой кислоты, добавляют около половины требуемого объема дистиллированной воды и затем осторожно, небольшими порциями вносят концентрированный аммиак (до pH 8,0 —8,5, не выше!). Удобно следить за изменением pH, пользуясь рН-метром, а стакан с фумаровой кислотой держать на магнитной мешалке. При подщелачивании раствора до pH выше 8,5 избыток щелочи нейтрализуют соляной кислотой. Смесь интенсивно перемешивают стеклянной палочкой, время от времени добавляя новые порции воды, до полного растворения соли. Когда соль растворится, в раствор фумарата добавляют необходимую навеску MgCI2 (из расчета, что в 1 л 0,001 М раствора хлористого магния должно быть 92,5 мг этой соли) и доводят объем до требуемого уровня. Магний стимулирует реакцию аспартазы.
Материалы, оборудование, посуда, реактивы
ФЭК; спектрофотометр; pH-метр; ультратермостат (качалка); водяная и ледяная бани; колбы качалочные на 750 и 50 мл; мерные цилиндры; пробирки с притертыми пробками; сито; фарфоровая ступка; химические стаканы на 50—200 мл; пробирки; пипетки.
План выполнения задачи
На выполнение задачи требуется 12—15 ч.
Занятие 1. Приготовление и стерилизация МПБ. Посев культуры E.coli в пробирки со скошенным МПА. Построение калибровочных кривых для белка и аспарагиновой
кислоты. Накануне следующего занятия пересеять суточную культуру E.coli со скошенного МПА в колбу с МПБ.
Занятие 2. Приготовление суспензии клеток. Опыт со свободными клетками. Гидролиз клеток и определение содержания белка в суспензии.
Занятие 3. Опыт с иммобилизованными клетками.
Занятие 4. Оформление результатов.
Глава 30
ОБРАЗОВАНИЕ И НАПРАВЛЕННЫЙ СИНТЕЗ
АНТИБИОТИКОВ-АКТИНОМИЦИНОВ
Антибиотики-актиномицины образуются некоторыми видами стрептоми-цетов и представляют собой хромопептиды, состоящие из неизменной хромофорной и варьируемой пептидной части молекулы. Вариации в структуре, сочетании, взаимном расположении аминокислотных остатков пептидных цепей создают многообразие группы актиномициновых антибиотиков. Продуценты актиномицинов, как правило, образуют смесь этих веществ, компонентный состав которой зависит от вида и штамма стрептомицета, а также условий его роста и биосинтеза антибиотиков. Направленный биосинтез — изменение не только количества образующегося антибиотика, но и вариаций его структуры и компонентного состава под влиянием факторов среды.
По механизму действия актиномицины относятся к антибиотикам, нарушающим синтез нуклеиновых кислот: молекула актиномицина образует комплекс с определенными участками ДНК, что ведет к подавлению транскрипции. Антибиотики данной группы являются сильными цитостатиками, что определяет их действие не только на микроорганизмы, но и на животные ткани, в том числе злокачественные новообразования.
ЗАДАЧА. ОБРАЗОВАНИЕ АНТИНОМИЦИНА И ИССЛЕДОВАНИЕ ПРОДУКТИВНОСТИ АНТИБИОТИКА КУЛЬТУРОЙ STREPTOMYCES
ANULATUS VAR. CHRYSOMALLUS
Цель работы. 1. Определение антибиотического спектра действия стрептомицета — продуцента актиномицинов. 2. Изучение образования антибиотиков и определение антибиотической продуктивности при росте стрептомицета на разных средах. 3. Выделение из культуры стрептомицета комплекса антибиотических веществ, их идентификация и определение его компонентного состава в зависимости от добавок к среде разных аминокислот.
Ход выполнения задачи
Микроорганизмы и их культивирование. В работе используют стрептомицет Streptomyces anulatus var. chrysomallus (штамм № 2 из коллекции кафедры мик
робиологии биологического факультета МГУ), который образует комплекс антибиотиков, состоящий в основном из трех актиномицинов: С,, С2 и С3. Для определения антимикробных свойств штамма используют следующий набор тест-микроорганизмов: Staphylococcus aureus, Escherichia coli, Proteus vulgaris, Bacillus cereus, Bacillus mycoides, Micrococcus luteus, Streptomyces anulatus var. griseus, Candida guilliermondii, Mycobacterium sp., Penicillium junicolosum, Rhodotorula aurantiaca. B. cereus используют также для определения количества образуемых актиномицинов методом диффузии антибиотиков в агаризованные среды и при биоавтографии антибиотика.
Состав используемых сред (г/л). Среда 1 для поддержания стрептомицета: гороховая мука — 50,0; агар — 20,0 и бульон Хоттингера — 15 мл; вода водопроводная до 1 л. Среду готовят в пробирках по 5 и 20 мл.Стерилизуют в автоклаве при 0,5 ати, pH 7,0—7,2. Среда 2 для определения антимикробных свойств штамма: глюкоза — 30,0; пептон — 1,0; KNO3 — 3,0; К2НРО4 — 0,2; MgSO4 — 0,5; NaCl — 1,0; ZnSO4 — 0,02; FeSO4 — 0,02; вода дистиллированная до 1 л. Среду готовят в пробирках по 20 мл и стерилизуют при 0,5 ати, pH 7,0 —7,2. Среда 5 для выращивания посевного мицелия стрептомицета: глицерин — 30,0; пептон — 5,0; KNO3 — 0,5; К2НРО4 — 0,4; MgSO4 — 1,0; NaCl — 0,1; вода водопроводная до 1 л. Среду готовят в качалочных колбах на 750 мл по 100 мл и стерилизуют при 0,5 ати, pH 6,5 —7,0. Среда 4 для глубинного выращивания стрептомицета и биосинтеза антибиотика: глюкоза (или глицерин) — 30,0; KNO3 - 4,0; К2НРО4 - 0,2; MgSO4 - 0,5; NaCl - 1,0; ZnSO4 - 0,02; FeSO4 -0,02; вода дистиллированная до 1 л. Среду готовят в качалочных колбах на 750 мл по 120 мл и стерилизуют при 0,5 ати, pH 6,5 —7,0. Среда 5 для определения количества актиномицинов методами диффузии и биоавтографии: Na2HPO4 — 3,0; агар — 20,0 и бульон Хоттингера — 500 мл; вода водопроводная до 1 л, pH 7,0 —7,2. Среду готовят в колбах по 250 мл и стерилизуют при 0,5 ати.
Растворы аминокислот', /.-валина, /.-изолейцина, /.-лейцина, /,-норвалина, готовят в пробирках, содержащих одну из аминокислот в количестве 100 мг/10 мл дистиллированной воды. Стерилизуют при 0,5 ати.
Методы анализов
1. Определение антимикробных свойств стрептомицета.
В стерильные чашки Петри наливают по 20 мл расплавленной среды 2. После ее застывания на поверхность агара изогнутой бактериологической петлей наносят по диаметру чашки полосу шириной 2 см воздушного мицелия стрептомицета из 6 — 7-суточной культуры, выросшей на среде 1. Полосу для более точной работы отмечают на внешней стороне чашки стеклографом. Чашки помещают на 5 —7 сут в термостат при 28 °C. После этого перпендикулярно к полосе выросшего стрептомицета на агар наносят штрихи из суспензий тест-микроорганизмов. Штрихи проводят бактериологической петлей от полосы стрептомицета, не касаясь его, к краю чашки.
Суспензии готовят в стерильной водопроводной воде из суточной культуры тест-микроорганизмов, выросших на среде 5. Названия микроорганизмов пишут на нижней стороне чашки Петри. Чашки ставят в термостат при 30 °C и просматривают через сутки. Чувствительность микроорганизмов к антибиотику, образуемому стрептомицетом,
определяют по величине зоны отсутствия роста и выражают в миллиметрах. Результаты представляют в табл. 30.1.
Таблица 30.1
Антимикробные свойства стрептомицета
Микроорганизм
Подавление роста, мм
2. Определение образования антибиотика при росте стрептомицета на средах разного состава. В этой части задачи в процессе роста стрептомицета определяют скорость нарастания биомассы мицелия, количество антибиотика, антибиотическую продуктивность в зависимости от возраста культуры и состава среды. Изучают морфологию стрептомицета.
Колбы со средой 3 засевают воздушным мицелием в количестве 1 см2 мицелия 6 — 7-дневной культуры стрептомицета, выросшего на среде 1, на колбу. Выращивание проводят на качалке при 28 °C в течение 36—48 ч. Из полученного посевного мицелия вносят по 5 мл перемешанной культуры в опытные колбы, содержащие по 120 мл среды 4, и ставят на качалку при 28 °C. Через 24 и 48 ч в отдельные колбы вносят растворы одной из аминокислот в количестве 100 мг на колбу и вновь ставят на качалку. Таким образом создаются условия для изменения компонентного состава актиномицинового комплекса.
Отбор проб. Опыт длится 96 ч, пробы отбирают через каждые 24 ч. Содержимое каждой опытной колбы тщательно перемешивают и стерильной пипеткой отбирают 3 мл культуры, из которых одну каплю наносят на предметное стекло, а остальное вносят в чистую сухую пробирку. К пробе в пробирке добавляют 3 мл этанола, пробирки закрывают резиновыми пробками с парафильмом и оставляют при комнатной температуре в темноте до конца опыта. Добавление этанола обеспечивает консервацию пробы, а также растворение актиномициновых антибиотиков, плохо растворимых в воде и находящихся в культуральной жидкости в виде взвеси мелких кристаллов и осадка на поверхности мицелия.
Микроскопирование. Каплю культуры стрептомицета с каплей воды распределяют тонким слоем по предметному стеклу, подсушивают на воздухе, фиксируют жидкостью Карнуа, красят 3 — 5 мин метиленовым синим по Леффлеру. Препарат осторожно промывают водой, подсушивают на воздухе и просматривают с иммерсией в световом микроскопе, отмечая: толщину, длину, ветвление периферических гиф мицелия, степень базофилии, дифференциацию мицелия (образование светлых участков, темной зернистости, капель волютина, глубинных спор), плотность микроколоний, распад на фрагменты, автолиз. Отмечают морфологические особенности мицелия в период активного синтеза антибиотика. Результаты записывают и зарисовывают.
Определение биомассы. Содержимое пробирок с отобранными и залитыми этанолом пробами после растворения актиномицинов (жидкость окрашивается в оранжевый цвет, осадок мицелия — бесцветный) фильтруют через пронумерованные, доведенные до постоянной массы бумажные фильтры. Фильтры с мицелием высушивают в сушильном шкафу при 100 °C, взвешивают и по разнице массы фильтров с мицелием и без него вычисляют величину биомассы мицелия, которую выражают в мг АСБ/мл культуральной жидкости.
Отфильтрованную в чистые сухие пробирки смесь культуральной жидкости и этанола (1:1) используют для определения количества антибиотика.
Колориметрическое определение количества антибиотика. Определение основано на хромофорных свойствах актиномицинов и зависимости поглощения света от количества антибиотика в растворе.
Из стандартного раствора антибиотика в этиловом спирте (500 мкг/мл) готовят серию разведений в дистиллированной воде с содержанием 50; 25; 12,5 и 6,2 мкг/мл антибиотика. Измеряют величину поглощения растворов на ФЭК при 400 нм в кюветах с длиной оптического пути 0,5 см и строят калибровочную кривую. В таких же условиях измеряют опытные образцы с обязательным точным разведением в случае высокого содержания антибиотика. Прямая зависимость величин экстинкции и концентрации вещества сохраняется при 5—50 мкг/мл актиномицина. Количество антибиотика рассчитывают по калибровочной кривой с учетом разведений (в том числе в два раза при отборе проб) и выражают в мкг/мл культуральной жидкости.
Биологическое определение количества антибиотика. Определение основано на диффузии антибиотика в агаризованную, содержащую тест-микроорганизм среду и учете подавления его роста. Величина диаметра зон подавления роста в определенных пределах пропорциональна логарифму концентрации антибиотика.
Из стандартного спиртового раствора актиномицина (500 мкг/мл) готовят в дистиллированной воде серию разведений с 50; 25; 12,5 и 6,2 мкг/мл антибиотика. Опытные образцы также разводят до приблизительного содержания антибиотика 5 — 50 мкг/мл. Расплавляют 250 мл среды 5 на водяной бане, охлаждают до 60 — 65 "С и вносят в среду 5 — 7 мл густой суспензии тест-микроорганизма В. cereus, выросшего в пробирках на среде 5. Среду перемешивают и быстро выливают на горизонтально расположенный лоток размером 30 х 30 см. После застывания в агаризованной среде стерильным пробочным сверлом делают лунки диаметром 5 мм на расстоянии 2 — 3 см друг от друга. В лунки вносят точное и одинаковое количество стандартных (четыре концентрации) и опытных (две концентрации) растворов антибиотика. Лоток помещают в термостат при 30 °C. Через 24 ч измеряют диаметры зон подавления роста тест-микроорганизма. На полулогарифмической сетке строят кривую зависимости величин зон подавления роста тест-микроорганизма от концентрации антибиотика в стандартных растворах. По калибровочной кривой рассчитывают содержание антибиотика в опытных образцах в мкг/мл культуральной жидкости. Калибровочная кривая строится для каждого используемого в работе лотка самостоятельно.
Расчет антибиотической продуктивности. Антибиотическую продуктивность мицелия — количество антибиотика, образуемое 1 мг биомассы за единицу времени (мкг/мг/ч), рассчитывают по формуле
Д2 — А ,
р---------/--=-- = X МКГ/МГ Ч,
где Ац и А,г — количество антибиотика, мкг/мл; Mh и Mt2 — количество биомассы, мг/мл; t — время роста стрептомицета, ч.
Результаты представляют в виде таблицы (табл. 30.2) и графика.
3. Выделение и идентификация антибиотиков-актиномицинов.
Проводят выделение сырца антибиотика из культуры стрептомицета и его хроматографирование. На хроматограммах проводят идентификацию антибиотика по Rf, химическим реакциям, биологическому действию, сравнивая с
Рост стрептомицета и образование антибиотика
Время, ч Биомасса, мг/мл Количество антибиотика, мкг/мл Антибиотическая продуктивность Морфология мицелия
колориметрический метод биологический метод
0
24
48
72
96
120
хроматограммой стандартного препарата актиномицина. Определяют количество и относительное содержание компонентов в образовавшемся актиномициновом комплексе, устанавливают зависимость этих величин от изменения среды, на которой растет стрептомицет, т. е. эффект направленного биосинтеза.
Выделение антибиотика. Оставшуюся после отбора 96-часовой пробы культуру стреп-томицета выливают в делительную воронку. Туда же добавляют 20 — 30 мл этилацетата и легким встряхиванием проводят экстракцию актиномицинов в этилацетат. Когда слой этилацетата (верхний слой) становится ярко-оранжевым и свободным от пузырьков воды, его осторожно выливают в фарфоровую чашку.
Необходимо очень тщательно следить за прозрачностью слоя этилацетата!
Экстракцию можно провести 2 — 3 раза. Работа ведется в вытяжном шкафу. Фарфоровые чашки с растворами актиномицинов в этилацетате оставляют в вытяжном шкафу до полного испарения эфира. Оставшийся на стенках чашки красный осадок представляет собой сырец актиномициновых антибиотиков. Перед хроматографированием осадок растворяют в 2—4 мл ацетона.
Хроматографирование. Актиномициновые антибиотики делят на отдельные компоненты, отличающиеся по аминокислотным остаткам хроматографированием на бумаге.
Система растворителей для хроматографии актиномицинов готовится в соотношении бутилацетат: дибутиловый эфир: 10%-й водный раствор салицилово-кислого натрия (1:1:2). Растворители встряхивают в делительной воронке и после разделения сливают в отдельные флаконы нижнюю водную и верхнюю органическую фазы.
Полосу хроматографической бумаги (ленинградская медленная) размером 20 х 50 см смачивают в нижней фазе растворителей, слегка подсушивают и на влажную бумагу с помощью капилляров наносят ацетоновые растворы полученных антибиотиков и стандартного препарата актиномицина. Каждый раствор наносят узкой полосой длиной 2 см, расстояние между полосами нанесения разных образцов — 3 см, от краев бумаги — 2 см. При высокой концентрации антибиотика в растворе достаточно нанести его на бумагу один раз, при низкой — раствор антибиотика после подсушивания на бумаге следует нанести несколько раз. Верхнюю часть бумаги помещают в органическую фазу растворителей и нисходящую хроматограмму оставляют в камере на ночь. Хроматограммы с разделившимися на компоненты антибиотиками высушивают в вытяжном шкафу и анализируют. Актиномицины — окрашенные вещества, поэтому специального проявления хроматограмм не требуется.
Анализ хроматограмм антибиотика. При хроматографировании стандартный препарат актиномицинов группы С делится на три компонента, различающихся по остаткам D-аминокислот во втором положении полипептидных цепей. Менее подвижный компонент С, содержит два валина, С2 — один валин и один изолейцин, более подвиж-
ный компонент С3 — два остатка изолейцина. Под действием экзогенных аминокислот изменяется количественное содержание названных компонентов или образуются новые, отделяющиеся на хроматограмме от известных. Подсчитывают количество компонентов (отдельных актиномицинов), на которые разделился в каждом случае образец антибиотика, измеряют пути прохождения и вычисляют Rf каждого компонента. За Rf актиномицинов принято считать отношение пути, пройденного данным актиномицином, к пути, пройденным актиномицином С2, ^которого принимают за 1. Сравнение ведется со стандартным препаратом актиномицина. Вырезают дорожку, пройденную каждым образцом антибиотика, и отрезают две узкие полоски, по 0,5 см шириной, по краям дорожки. Центральную широкую полосу используют для колориметрического измерения количества каждого из компонентов. Для этого вырезают каждое пятно, разрезают на мелкие полоски и помешают в пробирку с 5 мл этанола.
После экстракции веществ их количество измеряют на ФЭК при X = 400 нм, кювета 0,5 см. Сумму экстинкций всех компонентов принимают за 100%, по величине экстинкции каждого компонента вычисляют его процентное содержание в антибиотическом комплексе. Одну из отрезанных узких полосок хроматограммы используют для проверки антимикробных свойств методом биоавтографии. Для этого полоски хроматограмм опытных и стандартных актиномицинов накладывают на агаровую пластинку с тест-микроорганизмом (готовится, как описано в разделе «Биологическое определение количества антибиотика»). Пластинку ставят на сутки в термостат при 30 °C и затем измеряют зоны подавления роста тест-микроорганизма (мм). Вторую узкую полоску хроматограммы используют для проверки характерного для актиномицинов изменения окраски в зависимости от величины pH среды: в кислой среде — увеличение яркости окраски, в щелочной — обесцвечивание. Полоску хроматограммы последовательно держат над парами соляной кислоты и затем погружают в 5 — 10%-й раствор щелочи. Результаты представляют в виде табл. 30.3.
Из проведенных анализов делают выводы: 1) идентификация и свойства полученных антибиотиков; 2) компонентный состав антибиотиков; 3) влияние добавок аминокислот на состав антибиотиков.
Тонкослойная хроматография актиномицинов. Хроматографическое разделение актиномицинов также можно проводить на пластинках для тонкослойной хроматографии HP-TLC-Alufolien Kieselgel 60 фирмы Merck (ФРГ). Разделение ведут в восходящей системе растворителей бензол: ацетон (2:1). В стеклянный цилиндр высотой 20—25 см и диаметром 10 —15 см наливают слой растворителей высотой 0,5 см. На пластинку на расстоянии 1 см от края наносят капиллярами точки стандартного и исследуемых растворов актиномицинов. Диаметр
Таблица 30.3
Характеристика антибиотиков, образуемых при росте стрептомицета на разных средах
Показатель Образец антибиотика Стандарт актиномицинового антибиотика
Компоненты 1 2 3 4 5 с, С2 С3
Rf
Содержание, %
Антимикробная активность, мм
Окраска в кислой среде
Окраска в щелочной среде
пятна нанесения — 2 мм. Пластинки ставят в цилиндр с системой растворителей и сверху герметически закрывают. Хроматографическое разделение веществ происходит в течение 30 — 40 мин.
Материалы, оборудование, посуда, реактивы
Микроскоп; ФЭК; термостат; качалка; хроматографическая камера; хроматографическая бумага; делительные воронки; капилляры; фарфоровые чашки; чашки Петри; качалочные колбы; лотки для тест-организмов; предметные стекла; пробирки; пипетки; мерные колбы на 50 и 100 мл; этанол; ацетон; этилацетат; бутилацетат; дибутиловый эфир; салицилат Na; соляная кислота; 10%-й раствор NaOH; метиленовый синий по Леффлеру; фиксатор Карнуа; стандарт актиномицина 500 мкг/мл; реактивы для приготовления сред. Тест-организмы.
План выполнения задачи
Для проведения задачи необходимо 35 — 38 ч.
Занятие 1. Приготовление сред и посуды для стерилизации.
Посев стрептомицета в качалочные колбы со средой № 3 (посевной материал).
Занятие 2. Посев стрептомицета в качалочные колбы со средой 4. Отбор нулевой пробы. Приготовление чашек Петри со средой 2. Приготовление бумажных фильтров. Построение калибровочной кривой для стандартного раствора актиномицинов. Внесение стерильных растворов аминокислот в 24- и 48-часовую культуру стрептомицета и отбор проб из культуры.
Занятия 3 — 4. Отбор проб из культуры растущего стрептомицета. Посев стрептомицета штрихом на чашки Петри со средой 2. Взвешивание бумажных фильтров. Фиксация и окраска мицелия стрептомицета на предметных стеклах и просмотр препаратов под микроскопом. Экстракция антибиотика из 96-й часовой культуры стрептомицета.
Занятие 5. Фильтрование проб через доведенные до постоянного веса бумажные фильтры. Определение количества антибиотика колориметрическим и биологическим методами.
Занятие 6. Измерение зон подавления роста тест — микроорганизма на лотках; построение калибровочных кривых и расчет количества антибиотика. Взвешивание бумажных фильтров с мицелием. Посев тест-организмов к штриху стрептомицета. Постановка хроматограмм.
Занятие 7—8. Анализ хроматограмм. Определение количества компонентов антибиотиков. Биоавтография. Проверка чашек Петри со штрихом стрептомицета и тест-организмами.
Занятие 9. Анализ данных по биоавтографии. Оформление результатов, обсуждение, зачет.
Глава 31
МЕТОДЫ ИЗУЧЕНИЯ ОБРАЗОВАНИЯ ВИТАМИНОВ МИКРООРГАНИЗМАМИ
Витаминами называют низкомолекулярные органические соединения, обладающие высокой биологической активностью и поэтому необходимые живым организмам в очень небольших количествах.
Человеку в сутки необходимо витаминов от нескольких микрограмм до нескольких десятков миллиграмм. Многие микроорганизмы также нуждаются в витаминах для сво-
Количество различных витаминов (мкг/л питательной среды), необходимое для роста большинства микроорганизмов (по Н.Д. Иерусалимскому)
Название вещества Дозировка в 1 л среды, мкг
минимум оптимум
Тиамин (В,) 0,1-0,3 1-100
Рибофлавин (В2) 5 10-1000
Са-пантотенат (В3) 4 30-1000
Никотиновая кислота (РР, В5) 5-10 100-1000
Пиридоксин (В6) 0,1 10-1000
/-Инозит (Bg) 1000 3000-6000
Фолиевая кислота (В9) 0,02 0,05-0,5
р-Лминобензойпая кислота 0,01 0,02
Биотин (витамин Н) 0,002-0,01 0,1-10
Р-Аланин 200 500-1000
Холин 20 1000-2000
Пуриновые и пиримидиновые основания 1000 5000-10 000
его развития и роста. Витамины влияют на рост и размножение микроорганизмов, на процессы метаболизма, на строение клетки и другие свойства. Они служат кофакторами многих ферментов. Активность некоторых витаминов и витаминоподобных соединений, а также их свойства показаны в табл. 31.1, 31.2.
Таблица 31.2
Свойства различных витаминов и близких к ним веществ
Название вещества Разрушение нагреванием Адсорбция углем Разрушение светом Летучесть в вакууме в виде эфиров Растворимость
в щелочном растворе в растворах кислот при pH 3—4 при pH 7,0 В воде в спирте 70%-м в эфире, хлороформе
'Тиамин + - - ± - - + + -
Рибофлавин + - - + + - 4- т -
Са-пантотенат т + + - - - т + т
Р-Аланин - - - т - т + + -
Холин - - ? ? - т + т -
Фактор V + + ? ? - - ± - -
Никотиновая кислота - - + - - + + + -
Название вещества Разрушение нагреванием Адсорбция углем Разрушение светом Летучесть в вакууме в виде эфиров Растворимость
в щелочном растворе в растворах кислот при pH 3—4 при рн 7,0 В воде В спирте 70%-м в эфире, хлороформе
Пиридоксин - - — — ± + 4- 4- —
Биотин - - 4- + — +• + + —
i-Инозит - — — 4” — + 4- 4- —
Фолевая кислота + +- 4- - ± - + ? ?
/j-Аминобен-зойная кислота - + -1- — - + 4- 4- 4-
Гемин ? ? в щелочной среде
Пуриновые основания 4- + В кислой среде ±
Урацил - - 4- - - + 4- + +
В настоящее время в медицине, фармацевтической и пищевой промышленности широко применяют наряду с химическими также микробиологические методы определения различных витаминов. Как правило, химические методы определения витаминов трудоемки и требуют довольно большого количества исследуемого материала. Микробиологические методы обладают большой точностью и позволяют определить содержание витаминов практически в любом биологическом материале. Ниже приводятся задачи, знакомящие студентов с микробиологическими методами определения витамина В|2, никотиновой кислоты (витамина РР, или В5) и инозита (витамина В8).
ЗАДАЧА 1. ОПРЕДЕЛЕНИЕ ВИТАМИНА В12
Витамин В |2 обнаружен в растительных клетках, органах и тканях животных, но к самостоятельному синтезу этого соединения способны только микроорганизмы. Наиболее активными продуцентами витамина являются представители рода Propionibacterium, термофильные штаммы рода Bacillus, ряд штаммов рода Micromonospora, Pseudomonas denitrificans и метаногенные археи. Витамин В|2 применяют при лечении ряда заболеваний, а также для обогащения кисло-молочных продуктов. Для медицинских целей препараты витамина В12 получают из пропионово-кислых бактерий. В сельском хозяйстве используют препарат, приготовленный на основе биомассы метаногенов.
Цель работы: определить содержание витамина В|2 в клетках пропионовокислых бактерий тремя различными способами: 1) методом диффузии в агар; 2) пробирочным методом; 3) спектрофотометрическим методом.
1. ОПРЕДЕЛЕНИЕ КОЛИЧЕСТВА ВИТАМИНА В12 МЕТОДОМ ДИФФУЗИИ В АГАР
Ход выполнения задачи
Микроорганизм и его культивирование. Для проведения анализов необходимо 10— 15 мл культуры Propionibacteriumfreudenreichii. Состав и приготовление среды, подготовка посевного материала для культивирования и поддержания пропионовокислых бактерий описаны в задаче «Пропионовокислое брожение» (гл. 23), которую можно по времени совместить с настоящей задачей. По окончании культивирования биомассу пропионовокислых бактерий определяют нефелометрически. Пропионовокислые бактерии можно хранить в холодильнике в виде живой культуры 14—20 сут или в виде отделенной от культуральной жидкости биомассы в морозильной камере более месяца.
Метод основан на определении интенсивности роста тест-организма, мутантного штамма Escherichia coli 113-3, требующего витамин В!2, на агаризованной среде вокруг лунок, в которые внесен определенный объем экстракта витамина В|2> полученного из культуры пропионовокислых бактерий. Развитие тест-организма (в мм зон роста) зависит от количества витамина В|2 в экстракте.
Подготовка тест-организма. Культуру Е. coli 113-3 поддерживают на среде следующего состава (г/л): гидролизат казеина — 6,0; К2НРО4 — 0,2; FeSO4 — 0,005; MgSO4 — 0,2; дЬ-аспарагин — 0,2 (растворяют в 0,1 н НС1); глицерин — 2,0; витамин В]2— 100 мкг; агар — 15,0; вода дистиллированная; pH среды 6,8—7,0. Среду стерилизуют при 0,5 ати.
Перед определением витамина В|2 E.coli 113-3 пересевают на «голодную среду», содержащую (в г/л): пептон — 5,0; NaCl — 5,0; агар — 1,5; вода водопроводная; pH среды 7,0. Среду стерилизуют при 1 ати. E.coli культивируют при 37 °C в течение суток.
Экстракция витамина В12. Витамин В12 извлекают из клеток пропионовокислых бактерий в кислой среде при нагревании. Чтобы витамин В|2 не разрушился, его стабилизируют нитритом натрия (или цианистым калием или натрием). Для этого к 1 мл культуры в градуированной пробирке добавляют 3 — 4 мл дистиллированной воды, перемешивают и подкисляют 0,1 н. раствором НС1 до pH 4 — 5 (по метилрот-оранжево-крас-ная окраска). Пробы отбирают капилляром или стеклянной палочкой. Затем в пробирку вносят 2 мл 0,1%-го раствора NaNO2 и нагревают в кипящей водяной бане в течение 15 — 20 мин. После чего осторожно доводят pH раствора до 7,0 сухой содой по бромти-молблау или с помощью pH-метра. Содержимое пробирки доводят до 10 мл дистиллированной водой и используют для определения витамина.
Определение витамина Bj2 методом диффузии в агар. Готовят 250 мл среды следующего состава (г/л): глюкоза — 10,0; лактоза — 3,0; лимоннокислый натрий трехзамещенный — 3,0; NH4C1 — 3,0; К2НРО4 — 0,4; NaCl — 3,0; агар — 20,0; вода дистиллированная; pH среды 7,0. Среду стерилизуют при 0,5 ати в колбе на 500 — 700 мл. В пробирку с односуточной культурой тест-организма Е. coli 113-3 на «голодной среде» приливают 5 мл стерильной дистиллированной воды. Полученную суспензию в количестве 1 — 2 мл (суспензия не должна быть очень густой!) вносят в предварительно расплавленную и охлажденную до 45 °C среду. Среду, инокулированную Е. coli 113-3, тщательно перемешивают и сразу же выливают на дно лотка размером 35 х 30 см. Борта лотка сделаны из
нержавеющей стали, алюминия или дерева, а дном и крышкой служат стеклянные пластины. Лоток предварительно стерилизуют спиртом и обжигают пламенем горелки. Среда должна застыть ровным слоем, поэтому лотки помещают на строго горизонтальную поверхность (по уровню).
После того как агар застынет, лоток ставят на трафарет и пробочным сверлом (диаметр около 8 мм), простерилизованным спиртом и пламенем, вырезают в среде лунки. Расстояние между лунками 5 — 8 см. В один ряд лунок закапывают стандартные растворы витамина В,2, а в другой — экстракт витамина В,2 из пропионовокислых бактерий.
Стандартные растворы витамина В|2 готовят из стерильного раствора определенной концентрации (аптечный препарат), находящегося в ампуле (концентрация витамина в 1 мл отмечена на ампуле). Исходный раствор витамина В ^должен содержать 0,2 мкг/мл. Для приготовления такого раствора ампулу осторожно вскрывают, необходимое количество раствора витамина переносят в чистую мерную колбу и растворяют в дистиллированной воде. Из этого раствора готовят ряд разведений, содержащих витамин В12 в концентрациях 0,05; 0,025; 0,0125 и 0,006125 мкг/мл.
Все приготовленные стандартные растворы витамина (включая и исходный) закапывают в 10 лунок. Каждый раствор вносят в две лунки. Экстракт витамина В|2 из пропионовых бактерий закапывают в две лунки без разведения, в две лунки с разведением в 2 раза и в две лунки — с разведением в 4 раза. В каждую лунку вносят 50 мкл раствора или экстракта. Стандартные и опытные растворы закапывают в лунки на одном лотке, который затем выдерживают 2 ч при комнатной температуре для диффузии витамина В12 в агар, а потом помещают в термостат на 37 °C.
Диаметр зон роста Е. coli 113-3 вокруг лунок измеряют через сутки после внесения витамина. На основании данных со стандартными растворами вычерчивают, пользуясь специальной полулогарифмической шкалой, график зависимости роста Е. coli от количества витамина. По результатам, полученным с опытными растворами, определяют, пользуясь стандартной прямой, количество витамина В12в исследуемом экстракте пропионовокислых бактерий и пересчитывают его на 1 мл и на 1 л культуры, а также на 1 г абсолютно сухой биомассы (АСБ). При этом учитывают, что при приготовлении экстракта культуру разводили в 10 раз, а также все последующие разведения. Для построения стандартной кривой и определения витамина используют средние результаты двух повторностей. Полученные данные о содержании витамина В]2 в биомассе пропионовокислых бактерий вносят в табл. 31.3.
Материалы, оборудование, посуда, реактивы
Пробирки — 10; пипетки на 1 —2 мл —5; колба на 500 мл; цилиндр мерный; лоток для определения витамина методом диффузии в агар; баня водяная; штатив для пробирок; пробочное сверло; трафарет, расчерченный на квадраты; NaNO2; полулогарифмическая миллиметровая бумага; раствор витамина В12 в ампуле; реактивы для приготовления сред.
Таблица 31.3
Количество витамина В12, образованное пропионовокислыми бактериями
Повторность Биомасса, г/л Витамин В|2 Витамин В12, мкг/г АСБ
1
2
План выполнения задачи
Для выполнения задачи необходимо 18 — 20 ч.
Занятие 1. Приготовление и стерилизация среды для определения витамина В,2 с помощью Е. coli 113-3. Подготовка «голодной среды» для Е. coli 113-3. Пересев тест-культуры Е. coli 113-3 на «голодную среду» за сутки до занятия 2.
Занятие 2. Определение витамина В|2 методом диффузии в агар.
Занятие 3. Оформление результатов задачи. Зачет.
2. ОПРЕДЕЛЕНИЕ КОЛИЧЕСТВА ВИТАМИНА В12 ПРОБИРОЧНЫМ МЕТОДОМ
Ход выполнения задачи
Микроорганизмы и их культивирование. Для проведения анализов необходимо 10 — 15 мл культуры Р. freudenreichii. Через 48 ч после внесения посевного материала в растущую культуру Р. freudenreichiii вносят предшественник синтеза витамина В,2 — 5,6-диметилбензимидазол (5,6-ДМБ) в количестве 2 мг/л и культивирование продолжают еще 48 ч. Исходный раствор 5,6-ДМБ готовят на дистиллированной воде, подкисленной НС1. По окончании культивирования биомассу пропионовокислых бактерий определяют нефелометрически.
Тест-культуру Е. coli 113-3 поддерживают, как описано выше.
Метод основан на определении интенсивности роста в жидкой среде в пробирках мутанта Е. coli 113-3, требующего для роста витамин В12. Чувствительность метода — 0,002 — 0,004 мкг витамина в образце.
В качестве опытной используют среду следующего состава (г/л): К2НРО4 — 7,0; КН2РО4 — 3,0; натрий лимоннокислый — 0,5; MgSO4-7H2O — 0,1; (NH4)2SO4 — 1,0; глюкоза — 2,0; NaCl — 0,5; КС1 — 1 мг; вода бидистиллированная; pH среды 6,8 —7,0; режим стерилизации — 0,5 ати. КО добавляют в виде 0,02%-го раствора. Используют идеально чистую посуду, вымытую хромпиком, и реактивы «осч», а раствор глюкозы предварительно обрабатывают активированным углем.
Посевной материал Е. coll 113-3 готовят в пробирках за сутки до засева опытной среды. Для этого в 2 пробирки наливают по 5 мл опытной среды и по 5 мл стандартного раствора, содержащего в 1 мл 0,00005 мкг витамина В,2. Этот же раствор используют для приготовления стандартной кривой. Содержимое пробирок стерилизуют при 0,5 ати в течение 20 мин и после охлаждения засевают культурой Е. coli 113-3 с агаризованной среды. Засеянные пробирки ставят в термостат при 37 °C на 16 — 20 ч. Затем отбирают 0,1 мл выросшей культуры и переносят в пробирку с 10 мл стерильного 0,9%-го раствора NaCl. Полученной суспензией Е. coli 113-3 засевают опытную среду в пробирках.
Для получения калибровочной кривой применяют водный раствор витамина В|2, в 1 мл которого содержится 0,1 мкг витамина. В день опыта его разводят до содержания 0,00005 мкг/мл, т.е. в 2000 раз, и используют для анализа.
Ход определения. В широкие пробирки (1,7 х 18 см) вносят по 5 мл опытной среды, добавляют раствор витамина В|2 (0,5; 1,0; 1,5 и 2,0 мл) и доводят объем в каждой пробирке до 10 мл дистиллированной водой. На каждую концентрацию витамина и контрольную пробу (без витамина) берут по 3 параллельные пробирки (всего 15 пробирок). При анализе опытных образцов вместо стандартного раствора витамина в пробирки вносят экстракт витамина В|2 в количестве 0,5; 1,0; 1,5; 3,0 и 4,0 мл и также доводят общий объем до 10 мл дистиллированной водой. Стандартные и испытуемые
образцы автоклавируют 20 мин при 0,5 ати, охлаждают и засевают среды в пробирках одной каплей инокулята Е. coli 113-3. Засеянные пробирки инкубируют 20 — 24 ч при 37 °C.
По окончании инкубации содержимое пробирок перемешивают. Рост Е. coli 113-3 определяют нефелометрически при 400 нм. По полученным данным строят калибровочную кривую, откладывая на оси ординат среднее значение мутности для каждой концентрации стандартного раствора, а на оси абсцисс — абсолютное количество витамина В12, содержащееся в данной пробирке.
Содержание витамина определяют отдельно в каждой пробирке испытуемого образца. По данным, которые уложились в стандартную кривую, определяют количество кобаламинов в испытуемом образце. При этом учитывают, что при приготовлении экстракта культуру пропионовокислых бактерий разводили в 10 раз. Для определения содержания витамина необходимо рассчитывать не менее 2 — 3 его разведений. Полученные данные о содержании витамина В12в биомассе пропионовокислых бактерий вносят в табл. 31.3.
Материалы, оборудование, посуда, реактивы
Пробирки — 30; пипетки на 1 —2 мл —5; колбы на 500 мл; цилиндр мерный на 0,5 л; баня водяная; ФЭК; центрифуга; штатив для пробирок; раствор витамина В,2 в ампуле; NaNO2; реактивы для приготовления сред.
План выполнения задачи
Для выполнения задачи необходимо 20 ч.
Занятие 1. Приготовление и стерилизация среды для определения витамина В|2. Подготовка среды для Е. coli 113-3. Пересев тест-культуры Е. coli 113-3 на среду за сутки до занятия 2.
Занятие 2. Определение витамина В|2 пробирочным методом.
Занятие 3. Оформление результатов задачи. Зачет.
3. ОПРЕДЕЛЕНИЕ КОЛИЧЕСТВА ВИТАМИНА В12 СПЕКТРОФОТОМЕТРИЧЕСКИМ МЕТОДОМ
Ход выполнения задачи
Микроорганизм и его культивирование. Для проведения анализов необходимо 100—150 мл культуры Р. freudenreichii (см. с. 365).
Метод заключается в отделении и промывке клеток бактерий с переводом кобаламинов в водный раствор путем гидролиза, воздействии светом на полученный гидролизат для перевода кобаламина в оксикобаламин и определении оптической плотности при длине волны 530 нм. Оптическая плотность раствора пропорциональна содержанию кобаламина. Чувствительность метода — 10 — 100 мкг в пробе. Метод требует высокого содержания витамина в образце. Необходимо, чтобы в клетках содержалось не менее 0,1 — 0,2 % витамина на сухое вещество.
Ход определения. Для анализа берут 100—150 мл культуры Р. freudenreichii, центрифугируют при 5 тыс. g 20 мин. Супернатант удаляют, клетки промывают 3 — 4 объемами 366
Количество витамина В12, образованное пропионовокислым бактериями, мкг/г биомассы
Повторность Биомасса, г/л Метод
ЛОТКОВ пробирочный спектрофотометрический
1
2
дистиллированной воды, отделяя промывные воды центрифугированием в том же режиме. Промытую влажную биомассу взвешивают и переносят количественно в колбу Эрленмейера с 30-кратным объемом дистиллированной воды, подкисленной 0,1 н. НС1 до pH 4,6 — 5,0.
Полученную суспензию гидролизуют на кипящей водяной бане в течение 40 мин. После охлаждения гидролизат центрифугируют, определяют значение pH и объем супернатанта. Гидролизат должен иметь pH 4,6 — 5,0. В случае подщелачивания его подкисляют 0,1 н. НС1. После этого гидролизат помещают под лампу (60 Вт) на расстоянии 25 см и освещают в течение 30 мин. В случае выпадения осадка гидролизат фильтруют через бумажный фильтр.
Оптическую плотность раствора определяют на спектрофотометре при X = 530 нм относительно дистиллированной воды. Содержимое кобаламинов (мкг/мл) в культуральной жидкости рассчитывают по формуле
С = А5Х-104- И./56-И2,
где Л53о — оптическая плотность фильтрата, измеренная при длине волны 530 нм; И, _ объем гидролизата, мл; 56 — удельный показатель (коэффициент экстинкции) для оксикобаламина при 530 нм; V2 — количество культуральной жидкости, взятой для анализа, мл.
Полученные данные о содержании витамина В|2в биомассе пропионовокислых бактерий вносят в табл. 31.3. Если образец исследуют несколькими методами, то результаты вносят также в общую табл. 31.4.
Материалы, оборудование, посуда, реактивы
Пробирки — 10; пипетки на 1 — 2 мл —5; 0,5 л; колба на 500мл; баня водяная; спектрофотометр; центрифуга; штатив для пробирок; лампа (60 Вт); раствор витамина В|2 в ампуле; реактивы для приготовления сред.
План выполнения задачи
Для проведения задачи необходимо 6 —7 ч.
Занятие 1. Определение витамина В|2 спектрофотометрическим методом.
Занятие 2. Оформление результатов задачи. Зачет.
ЗАДАЧА 2. ОПРЕДЕЛЕНИЕ НИКОТИНОВОЙ КИСЛОТЫ
Никотиновая кислота (витамин РР, витамин В5, никотинамид, ниацин) является пиридин-3-карбоновой кислотой, а никотинамид — ее амидом. Оба соединения легко превращаются друг в друга и поэтому обладают одинаковой витаминной активностью по отношению к живым организмам. Суточная по
требность в витамине РР для человека составляет 20 — 25 мг. Источником витамина являются животные (мясо, печень) и многие растительные продукты — рис (не полированный), гречка, хлеб, картофель.
Цель работы', определение содержания никотиновой кислоты в одном из продуктов питания (рис, гречка, мясо) микробиологическим методом.
Ход выполнения задачи
Микроорганизм и его культивирование. В работе используют тест-культуру Kluyveromyces marxianus 734. Культуру дрожжей поддерживают на среде следующего состава: 7 °Б сусло, агар 2%-й и водопроводная вода. Культивирование проводят в течение 48 ч при температуре 28 —30 °C. Хранят дрожжи при 4— 6 °C, пересевая не реже 1 раза в месяц.
Метод основан на определении интенсивности роста тест-организма — К. marxianus 734, требующего для роста никотиновую кислоту, в жидкой среде. Интенсивность роста культуры зависит от количества никотиновой кислоты в образце. Для выявления витамина в исследуемом образце пищевого продукта необходимо предварительно подвергнуть этот образец экстракции. Для этого 1 г хорошо измельченного образца (рис, гречка, мясо) смешивают с 30 мл 1 н H2SO4 и автоклавируют при 1 ати в течение 15 мин. После этого гидролизат охлаждают, подщелачивают с помощью 1 н. р-ра NaOH до значения pH 5,5 —5,8 и доводят объем до 100 мл в мерной колбе дистиллированной водой. Содержимое мерной колбы фильтруют и при необходимости разбавляют до содержания никотиновой кислоты в экстракте приблизительно до 0,3 — 0,5 мкг/мл.
Перед определением витамина готовят посевной материал тест-культуры. Для этого небольшое количество биомассы суточной культуры К. marxianus вносят петлей в 10 мл физиологического раствора. Мутность полученной суспензии должна быть незначительной.
Ход определения. Определение ведут в двух повторностях. Для этого в штативе расставляют 2 ряда пробирок по 6 в каждом ряду. В каждую из пробирок разливают по 2 мл дистиллированной воды. Затем в первые пробирки ряда вносят по 2 мл раствора образца или стандартного раствора никотиновой кислоты при построении калибровочной кривой. В каждом ряду пробирок делают 5 последовательных двукратных разведений,
Таблица 31.5
Количество никотиновой кислоты в образце
Повторность Количество витамина Разведения образца в пробирках
1 2 3 4 5 6
1 По калибровочной кривой, мкг/мл
мкг/г образца
2 По калибровочной кривой, мкг/мл
мкг/г образца
Среднее по двум повторностям мкг/г образца
перенося по 2 мл раствора из первой пробирки ряда в последующие. После этого во все пробирки стерильно добавляют по 2 мл питательной среды
Состава среды (г/л): сахароза — 40,0; (NH4)2SO4 — 6,0; MgSO4 — 1,4; NaCl — 1,0; КН2РО4 — 2,0; К2НРО4 — 0,2; тиамин хлорид — 0,002; пантотенат кальция — 0,002; пиридоксин — 0,0002; биотин — 0,000002. Вода дистиллированная, pH среды 5,5 —5,7. Сахарозу и соли, используемые для приготовления питательной среды, обрабатывают активированным углем. Для этого к 100 мл 20%-го раствора сахарозы или солей добавляют 5 г измельченного угля, помещают на круговую качалку на 40 мин и фильтруют через бумажный фильтр.
Конечный объем раствора в каждой из пробирок должен составлять 4 мл. В качестве контроля используют пробирку, содержащую 2 мл дистиллированной воды и 2 мл питательной среды. Растворы в пробирках стерилизуют при 0,5 ати 15 мин, охлаждают и вносят посевной материал дрожжей по 1 капле в пробирку. Засеянные пробирки инкубируют 48 ч при 28 — 30 °C. Интенсивность роста тест-культуры в пробирках со стандартными и исследуемыми растворами измеряют на ФЭК при длине волны 540 нм в кювете с длиной оптического пути 0,3 см.
Для построения калибровочной кривой готовят стандартный раствор никотиновой кислоты. Для этого 0,01 г витамина растворяют в 20 %-м этаноле в мерной колбе на 100 мл. Из полученного раствора, содержащего 100 мкг/мл, готовят рабочий раствор, содержащий 0,4 мкг/мл никотиновой кислоты. Содержание витамина в ряду пробирок для калибровочной кривой должно составлять 0,1; 0,05; 0,025; 0,0125; 0,00625; 0,0031 мкг/мл. В качестве контроля используют дистиллированную воду. Полученные результаты вносят в табл. 31.5. Делают выводы о наличии никотиновой кислоты в образце.
Материалы, оборудование, посуда, реактивы
Пробирки — 40; пипетки на 1 — 2 мл —15; колбы на 500 мл; цилиндр мерный на 100 мл; ФЭК; штатив для пробирок; никотиновая кислота; активированный уголь; реактивы для приготовления сред.
План проведения задачи
Для проведения задачи необходимо 10—12 ч.
Занятие 1. Приготовление и стерилизация среды для определения никотиновой кислоты. Гидролиз исследуемого образца. Подготовка среды для тест-организма — К. marxianus 734. Пересев тест-культуры за 36 ч до занятия 2.
Занятие 2. Определение никотиновой кислоты
Занятие 3. Оформление результатов задачи. Зачет.
ЗАДАЧА 3. ОПРЕДЕЛЕНИЕ ИНОЗИТА
Инозит, или витамин В8, — шестиатомный циклический спирт, хорошо растворимый в воде. В продуктах животного происхождения инозит находится преимущественно в связанной форме, главным образом в виде фосфолипидов, а в растениях — в виде эфира фитина, обладающего витаминными свойствами. Суточная потребность в инозите для человека составляет 1,0—1,5 г. Этот витамин находится практически во всех продуктах животного и растительного происхождения, но особенно много его в печени, мясе, хлебе, картофеле, зеленом горошке.
Цель работы: определение содержания инозита (витамин В8) в одном из продуктов питания (мясо, картофель, хлеб, зеленый горошек) микробиологическим методом.
Ход выполнения задачи
Микроорганизм и его культивирование. В работе используют тест-культуру дрожжей Kluyveromyces thermotolerans\-53>3. Дрожжи выращивают на среде состоящей из 7 °Б сусла и 2%-го агара, в течение 48 ч при 28 °C и хранят в холодильнике.
Метод основан на определении интенсивности роста штамма дрожжей К. ther-motolerans Y-533, не способного синтезировать инозит, в жидкой среде. Интенсивность роста тест-культуры зависит от количества инозита в образце.
Для выявления витамина в исследуемом образце пищевого продукта необходимо предварительно подвергнуть этот образец экстракции путем гидролиза. Для этого 1 г хорошо измельченного образца смешивают с 30 мл 6 н. раствора соляной кислоты и автоклавируют в течение 1 ч при 1 ати. После этого экстракт охлаждают, подщелачивают с помощью 1 н. р-ра NaOH до значения pH 5,5 —5,8 и доводят объем до 100 мл в мерной колбе дистиллированной водой.
Приблизительная концентрация инозита в образце должна быть 5—6 мкг/мл. При необходимости делают разведения, как это описано для никотиновой кислоты. Использующийся для построения калибровочной кривой стандартный образец инозита подвергается аналогичной обработке. Содержание инозита в ряду стандартных растворов, использующихся для построения калибровочной кривой, должно соответствовать 1,2; 0,6; 0,3; 0,15; 0,075 мкг/мл. В качестве контроля используется дистиллированная вода.
Для определения инозита используют среду Ридер, содержащую (г/л): глюкоза — 40,0; (NH4)2SO4 - 6,0; MgSO4 — 1,4; NaCl - 1,0; КН2РО4 - 2,0; К2НРО4 - 0,2; пантотенат кальция — 0,005. Вода дистиллированная, pH среды до стерилизации 5,5 —5,7. Перед доведением значения pH среду обрабатывают активированным углем из расчета 5 г угля на 100 мл 20%-го раствора глюкозы и фильтруют через бумажный фильтр.
Ход определения. Определение ведут в двух повторностях. Для этого в штативе расставляют 2 ряда пробирок по 6 в каждом ряду. В каждую пробирку разливают по 2 мл дистиллированной воды. Затем в первые пробирки ряда вносят по 2 мл раствора образца или стандартного раствора инозита при построении калибровочной кривой. В обоих рядах пробирок делают 5 последовательных двукратных разведений, перенося по 2 мл раствора из первой пробирки ряда в последующие. После этого во все пробирки стерильно добавляют по 2 мл питательной среды. В качестве контроля используют пробирку, содержащую 2 мл дистиллированной воды и 2 мл питательной среды. Растворы в пробирках стерилизуют при 0,5 ати 15 мин, охлаждают и вносят посевной материал по 1 капле в пробирку. В 1 мл суспензии дрожжей, используемой в качестве посевного материала, должно находиться около 1,6 • 103 клеток. Засеянные пробирки инкубируют 48 ч при 28 — 30 °C. Интенсивность роста тест-культуры в пробирках со стандартными и исследуемыми растворами измеряют на ФЭК при длине волны 540 нм в кювете с длиной оптического пути 0,3 см.
Количество инозита в образце определяют по калибровочной кривой. Полученные результаты вносят в табл. 31.6.
Материалы, оборудование, посуда, реактивы
Пробирки — 40; пипетки на 1 — 2 мл —15; колбы на 500 мл; цилиндр мерный на 100 мл; ФЭК; штатив для пробирок; инозит; активированный уголь; реактивы для приготовления сред.
Количество инозита в образце
Повторность Количество витамина Разведение образца в пробирках
1 2 3 4 5 6
1 По калибровочной кривой, мкг/мл
мкг/г образца
2 По калибровочной кривой, мкг/мл
мкг/г образца
i Среднее по двум j повторностям мкг/г образца
План проведения задачи
Для проведения задачи необходимо 10—12 ч.
Занятие 1. Приготовление и стерилизация среды для определения инозита. Гидролиз исследуемого образца. Подготовка среды для тест-организма —дрожжей К. thermotolerans Y-533. Пересев тест-культуры за 36 ч до занятия 2.
Занятие 2. Определение инозита.
Занятие 3. Оформление результатов задачи. Зачет.
Глава 32
ПОЛИФУНКЦИОНАЛЬНЫЕ БЕЛКИ БАКТЕРИЙ
У все большего числа белков обнаруживают способность проявлять не одну, а две или несколько различных активностей, причем с помощью различающихся механизмов (для чего в молекуле белка должно быть несколько активных центров). Такое множественное проявление биологической активности белков называют полифункционалъностъю. Полифункциональность белков находится в соответствии с принципом экономии генома и вполне обоснована с точки зрения современных знаний об эволюции белков, их доменной организации.
Полифункциональные белки синтезируют как эукариоты, так и прокариоты. Например, лизоцимы, образуемые множеством животных, не только гидролизуют пептидогликан бактериальных клеток благодаря своей ферментативной активности, но и разрушают их цитоплазматическую мембрану, используя еще до конца не известный механизм. Мембранотропная активность лизоцима сохраняется даже после потери им ферментативной активности, например в результате кипячения. Два белка, выделенные с поверхности клеток азотфиксирующей бактерии Bacillus polymyxa, проявляют бактерицидную, протеолитическую активности, а также адгезивные свойства, т. е. являются одновременно антибиотиками, ферментами и лектинами. Лектины — это белки, которые способны обратимо и избирательно связываться с углеводами. Лектины (агглютинины) могут осаждать углеводсодержащие соединения, агглютинировать клетки и определенным образом влиять на их метаболизм, например активируя автолизины этих клеток. Известно, что лектины принимают участие в специфической адгезии бактерий к клеткам корней и других органов растений, а также к клеткам животных. Лектины псевдомонад, ризобий, азоспирилл могут выступать и в качестве бактериоцинов.
Агглютинины прокариот играют важную роль при образовании различных симбиозов. Наиболее изучены лектины ряда патогенных бактерий, являющиеся токсинами, а в ряде случаев проявляющие различную антибиотическую активность. У бактерий, патогенных для растений и животных, лектины могут быть тесно связанными с фимбриями, а также находиться на поверхности клетки или экскретироваться из нее. Например, типичным лектином считают экзотоксин Vibrio cholerae, обладающий протеолитической активностью.
Полифункциональны Cry-белки Bacillus thuringiensis — высокоспецифичные токсины некоторых насекомых и других беспозвоночных (нематод, клещей), проявляющие также свойства антибиотиков, бактериоцинов, лектинов. Такие белки образуют в клетках В. thuringiensis параспоральные кристаллы, способные длительное время сохраняться во внешней среде наряду со спорами. В состав кристаллов некоторых подвидов В. thuringiensis кроме С iy-белков одного или нескольких классов могут входить также цитолитические Cyt -белки и некоторые малоизученные белки. При попадании в организм чувствительного беспозвоночного вместе с пищей кристаллы растворяются, Cry-белки разрушают мембрану клеток кишечника жертвы. В результате этого жертва погибает, и внутри ее тела начинают прорастать споры бациллы. На первых этапах развития популяции клеток В. thuringiensis из проросших спор антибиотическое действие Сгу-белков, а также других белков параспоральных кристаллов защищает бактерий В. thuringiensis от конкурирующей микрофлоры, пока сама молодая популяция этих бактерий не достигнет кворума и не станет способной синтезировать различные антибиотические соединения. Способность Cry-белков связываться с углеводами важна как для их токсического, так и для антибиотического эффекта.
Гены некоторых полифункциональных белков введены в состав рекомбинантных организмов. Так, например, Cry-белки нескольких подвидов В. thuringiensis синтезируются картофелем, кукурузой, томатами и другими трансгенными растениями, защищая последние от вредных насекомых. В связи с этим изучение всех проявлений биологической активности полифункциональных белков, а особенно их антибиотического действия на микроорганизмы (важные компоненты природных биоценозов, прежде всего в почвах), необходимо для анализа и предотвращения возникновения возможных нежелательных экологических последствий расширяющегося внедрения трансгенных организмов в природные биоценозы.
ЗАДАЧА. ИЗУЧЕНИЕ ПОЛИФУНКЦИОНАЛЬНЫХ БЕЛКОВ БАКТЕРИЙ
НА ПРИМЕРЕ CRY-БЕЛКОВ ПАРАСПОРАЛЬНЫХ КРИСТАЛЛОВ BACILLUS THURINGIENSIS
Цель работы: изучение множественного проявления активности полифункциональных белков на примере Cry-белков параспоральных кристаллов В. thuringiensis.
Ход выполнения задачи
Микроорганизмы и их культивирование. В работе используют штамм энто-мопатогенной бактерии В. thuringiensis subsp. alesti, в состав параспоральных кристаллов которой входит один CrylA-белок. В работе может также быть выделена кристаллообразующая бактерия из природного материала — почвы, трупа насекомого и т.п. Свойства белков ее кристаллов можно в таком случае сравнить со свойствами CrylA-белка подвида alesti, и на основании такого сравнения
сделать предположение о природе белков, образующих параспоральные кристаллы выделенной бактерии.
Для выделения кристаллообразующих споровых бактерий используют метод ацетатной селекции, сущность которого состоит в следующем. Образец природного материала помещают в 0,1 М раствор ацетата Na, пастеризуют для убивания вегетативных клеток, затем помещают в термостат (30 °C) на сутки в питательном бульоне, например МПБ, на 0,1 н. ацетате (pH 6,8) для прорастания всех спор, кроме спор кристаллообразующих бактерий, которые не прорастают в растворе ацетата. Затем убивают вегетативные клетки, образовавшиеся из проросших спор, также путем пастеризации и проводят высев суспензии, содержащей непроросшие споры кристаллообразующих бактерий, на агаризованную картофельную среду (состав среды дан в приложении 4).
Отбирают среди выросших колоний колонию кристаллообразующих бактерий с помощью фазово-контрастной микроскопии или микроскопии окрашенных (для выявления кристаллов) красителем аналиновым черным или амидошварцем (см. приложение 6) препаратов. Затем выращивают выделенных кристаллообразующих бактерий так же, как и alesti, на жидкой картофельной среде с добавлением (г/л): MnSO4 — 0,05; СаС12 — 0,08; MgSO4 — 0,1; в 3 колбах на 750 мл с 200 мл среды на качалке при 220 об/мин и 30 °C в течение 2 сут до завершения образования спор и кристаллов.
В качестве тест-культур используют Micrococcus luteus шт. 140 и Streptomyces chrysomallus шт. 257. Выращивание микрококков, а также определение их чувствительности к антибиотическому действию белков кристаллов методом диффузии в агар проводят на агаризованной среде следующего состава (г/л): К2НРО4 — 7,0; КН2РО4 — 2,0; цитрат натрия — 0,4; MgSO4 — 0,05; (NH4)2SO4 — 1,0; Н2О дистиллированная; pH 7,4. К среде добавляют 1 % трипказина («Reanal», Венгрия) и 1,5 % агара Bitek (Difco, США). Эту же среду, но без добавления агара используют при определении антибиотической активности белков методом микроразведений. Чувствительность стрептомицетов к антибиотическому действию белков кристаллов определяют на агаризованной среде Чапека с 2 % глюкозы.
Выделение белков из кристаллов бактерий. Суспензию полученных кристаллов и спор отделяют центрифугированием (6000 g, 15 мин), промывают водой, 1 М NaCl, 50 %-м ацетоном и затем трижды водой, каждый раз центрифугируя. Кристаллы растворяют в течение 1 часа в 0,02 н. NaOH при комнатной температуре при перемешивании. Супернатант отделяют центрифугированием (8000 g, 15 мин) и оставляют при комнатной температуре на 16—24 ч для активации протоксинов протеиназами, связанными с кристаллами. После этого белки кристаллов осаждают ледяной уксусной кислотой при pH 4—5, немедленно центрифугируют с охлаждением (8000 g, 25 мин). Cry-белки сразу растворяют в стерильном 0,01М трис-НС\ буферном растворе, pH 8,2 с 0,2 М КС1 (буфер К) и определяют концентрацию белка в растворе спектрофотометрически при 280 нм (е280). При этом учитывают проведенные для ряда Сгу-белков подсчеты, которые показали, что растворы этих белков с концентрацией 1 мг/мл имеют е280 = 1,6.
Приготовление растворов белков для определения различных проявлений их биологической активности в присутствии и отсутствие различных сахаров. Готовят по 5 мл растворов CrylA-белка, а также белков из кристаллов выделенных бактерий в стерильном буфере К с концентрациями 400, 200, 100, 50 мкг/мл.
Для этого каждый из исходных растворов белков с определенной концентрацией разбавляют буфером К. Этот же буфер используют для приготовления всех разведений белков.
Готовят по 4 мл 0,5 М растворов D-глюкозы, D-галактозы, N-ацетилглюко-замина. N-ацетилгалактозамина в буфере К и смешивают их по 0,5 мл в соотношении 1:1с растворами белков кристаллов в этом же буфере так, чтобы конечная концентрация белка в полученных растворах составляла 50, 100, 200 и 400 мкг/мл, а каждого сахара — 0,25 М. Оставляют при комнатной температуре на 1 ч, а затем используют для определения антибиотической, лектиноподобной, цитолитической активности этих растворов.
Подготовка стерильных суспензий клеток тест-микрооргапизмов. Микрококков и стрептомицетов выращивают в течение суток на агаризованных вышеуказанных средах в 3 и 5 чашках Петри соответственно. Затем снимают стерильной петлей клетки с поверхности агара на этих чашках, помещают в стерильные центрифужные стаканы с крышками на 50 мл с 30 мл стерильной воды, тщательно гомогенизируют путем взбалтывания и оставляют при 4 °C на 1 — 2 сут. После этого клетки отделяют центрифугированием при 6 тыс. g, в течение 15 мин и готовят суспензии клеток в стерильных питательных средах для определения чувствительности к антибиотическому действию изучаемых белков. Для всех определений в этой задаче требуются химически чистые реактивы и высоко-очищенный агар производства фирм «Difco» (США) или «Fenik» (Германия).
Определение антибиотической активности полифункциональных белков. Определение антибиотической активности белков методом микроразведений проводят в стерильных иммунологических плашках. В каждую из углублений (лунок) в плашке вносят по 80 мкл суспензии клеток микрококков в жидкой среде (примерно 105 клеток/мл), указанной выше, смешивают с изучаемыми растворами белков и белков с сахарами так, чтобы конечная концентрация белков составила в жидкой суспензии 5, 10, 20, 50 и 100 мкг/мл, а конечный объем жидкости во всех лунках был одинаковым — 100 мкл. Немедленно измеряют поглощение клеточной суспензии с растворами белков, а также с соответствующими контролями (буфер К, растворы сахаров в буфере К), на многоканальном плашечном спектрофотометре Uniflana (или другом подобном приборе) при X = 600 нм. Затем (через 2 ч при комнатной температуре) плашки помещают на 30 °C и вновь измеряют изменение поглощения клеточной суспензии на плашечном спектрофотометре через 2, 5 и, если необходимо, 20 ч.
Минимальной ингибирующей концентрацией при всех определениях антибиотической активности является наименьшая концентрация белка, подавляющая рост тест-микроорганизмов.
Для сравнения определяют антибиотическую активность изучаемых растворов методом диффузии в агаре. Газоны тест-микроорганизмов в вышеуказанных средах готовят общепринятым способом, разливая агаризованную среду с суспензией клеток на лотки тонким ровным слоем. Делают в агаре лунки диаметром 6 см специальным сверлом. Агаризованные среды с клетками подсушивают в ламинаре при продувке в течение 10—15 мин и немедленно вносят в лунки по 20 мкл всех приготовленных растворов белков, белков с сахарами и соответствующие контроли, оставляют закрытые лотки при комнатной температуре для диффузии растворов на 12 —20 ч. Затем помещают лотки в термостат на 30 °C и измеряют зоны подавления роста микрококков через сутки, а стрептомицетов — через 2 — 3 сут.
Определение агглютинирующей способности белков кристаллов в присутствии и в отсутствие сахаров. Гемагглютинирующую активность изучаемых растворов белков определяют реакцией гемагглютинации с самопроизвольным оседанием эритроцитов. Метод основан на явлении осаждения эритроцитов со скоростью, пропорциональной концентрации агглютининов. Степень агглютинации оценивают нефелометрически измерением плотности слоя неосевших эритроцитов при X = 600 нм. Дополнительно контролируют под световым микроскопом вид осевших эритроцитов. Используют ранее подготовленную 2%-ю суспензию трипсинизированных эритроцитов в 0,05 М K-Na-фосфатном буферном растворе, pH 7,4, приготовленном на физиологическом растворе — 0,9%-й NaCl (буфер Ф). В иммунологические плашки вносят по 100 мкл такой суспензии и по 100 мкл изучаемых растворов, а также, где необходимо, буфера К, чтобы конечная концентрация белков в смеси составила 5, 10, 20, 50 и 100 мкг/мл. Эти разведения делают из исходных разведений с наибольшей концентрацией белков — 400 мкг/мл, чтобы в инкубационной смеси преобладал буфер Ф. Измеряют при X = 600 нм исходную плотность суспензии эритроцитов на плашечном спектрофотометре, а также через 20 и 60 мин стояния плашки при комнатной температуре на строго горизонтальной поверхности. Затем микропипеткой осторожно собирают жидкость над осевшими эритроцитами (по 100 мкл) и измеряют ее поглощение при К = 600 нм. Если в жидкости появляется раствор гемоглобина, это значит, что часть эритроцитов лизировалась — свидетельствует о присутствии в изучаемых растворах Cyt-белка с литической активностью.
Минимальной агглютинирующей концентрацией является та концентрация белка, которая вызвала реакцию оседания эритроцитов.
Электронно-микроскопическое изучение влияния полифункциональных белков кристаллов на эритроциты и клетки микрококков. Отбирают контрольные клетки микрококков, а также эритроциты до начала их использования в опыте, а также эти клетки в каком-либо варианте после опыта, где наглядно определена антибиотическая и лектинподобная активности изучаемых белков. Готовят препараты этих клеток для просвечивающей электронной микроскопии (негативное контрастирование). Для этого каплю суспензии клеток наносят на сетки, покрытые формваровой пленкой, через 30 с лишнюю жидкость убирают фильтровальной бумагой. Затем наносят на сетки на 1 мин 2%-й раствор натриевой соли фосфовольфрамовой кислоты (pH 7,0—7,2), убирая лишнюю жидкость также с помощью полоски фильтровальной бумаги. Все препараты на сетках просматривают в трансмиссионном электронном микроскопе, например JEM —100В (фирма «Jeol», Япония).
Оформление результатов. Результаты оформляют в виде таблиц значений антибиотической и лектинподобной активности изучаемых растворов белков в присутствии разных сахаров. Проводят сравнительный анализ влияния сахаров на активность растворов в отношении эритроцитов и клеток прокариот. Анализируют микроскопическую картину разрушения клеток под действием растворов изучаемых белков.
Материалы, оборудование, посуда, реактивы
Реактивы, указанные в методической части; ламинар; световой и электронный микроскопы (лаборатория электронной микроскопии); плашечный спектрофотометр; посуда; микропипетки; пробирки Эппендорфа; плашки стерильные.
План выполнения задачи
Для выполнения задачи требуется примерно 7 занятий.
Занятие 1. Подготовка посуды, питательных сред. Ацетатная селекция кристаллообразующих бактерий.
Занятие 2. Посев кристаллообразующих и тест-культур. Подготовка реактивов.
Занятие 3. Выделение белков из кристаллов. Подготовка суспензий клеток тест-микроорганизмов.
Занятие 4. Постановка опытов по определению антибиотической и агглютинирующей активности изучаемых белков.
Занятие 5. Определение антибиотической активности белков методом диффузии в агар. Предварительный анализ полученных результатов.
Занятие 6. Электронная микроскопия клеток.
Занятие 7. Оформление результатов, их анализ и обсуждение.
Глава 33
АНАЭРОБНАЯ БИОДЕГРАДАЦИЯ АЗОКРАСИТЕЛЕЙ И ИХ ПРОИЗВОДНЫХ
Сброс огромного количества бытовых и промышленных отходов создает избыточную нагрузку на природные экосистемы, в основном из-за созданных человеком соединений, которые в обычных условиях не разрушаются. Эти чужеродные вещества — ксенобиотики — имеют уникальную биологическую активность даже в небольших концентрациях. Многие из них обладают значительной стабильностью, и для их полного разложения требуются столетия. Происходит непрерывный перенос таких вешеств по пищевым цепям и их накопление на конечных этапах, к которым относится и человек. Огромное число ксенобиотиков имеет техногенное происхождение и многие чрезвычайно токсичны, проявляют мутагенную и канцерогенную активность.
Самоочищение природных экосистем связано прежде всего с огромным потенциалом микробной активности. Производственные процессы очистки и.переработки отходов деятельности человека также основаны на понятии биодеградации, т. е разрушении вещества с помощью биологической активности, большую часть которой проявляют именно микроорганизмы. К биодеградации относят реакции незначительных изменений молекулы {трансформацию), распад сложной молекулы до более простых соединений {фрагментацию) и разложение сложного вещества до самых простых (СО2, Н2О, NH3, СН4 и т.д.), или полную минерализацию. Использование биологической активности для удаления загрязняющих вешеств из окружающей среды называют биоремедиацией. Для успеха таких процессов необходимо сначала найти соответствующую активность, а затем разными методами ее стимулировать и создать по возможности оптимальные условия для проявления. Только микроорганизмы могут помочь справиться с антропогенным загрязнением, так как у них огромный потенциал и лабильный обмен, позволяющий воздействовать даже на необычные, искусственно созданные субстанции, да при этом еще давать полезные продукты (газы, металлы, растворители и т. д.). Здесь огромное поле деятельности для работы скорее не индивидуальных микроорганизмов, а микробных сообществ (например, метаногенных).
В таком сообществе микроорганизмов возникают прочные трофические связи, и часто субстрат может быть расщеплен до простых веществ именно группой микроорга
низмов, а не отдельными ее членами, которые в виде чистых культур этот субстрат вообще не могут использовать из-за энергетических барьеров. При таких условиях возникает синтрофия — полная взаимозависимость друг от друга в пищевых потребностях, которая более облигатна в анаэробных условиях. Как правило, для нормального функционирования анаэробной пищевой цепи необходим межвидовой перенос некоторых интермедиатов, обычно молекулярного водорода, иногда ацетата, формиата, так как эндэргонические реакции начальной фазы деградации требуют отвода Н2. Обычно эти ассоциации оформлены структурно в виде гранул, хлопьев, сложных биопленок, что позволяет микроорганизмам, ответственным за каждую стадию деградации сложных соединений, находиться пространственно близко друг к другу для облегчения межвидового переноса интермедиатов. Структурно оформленные сообщества называют консорциумами.
Еще 15 — 20 лет назад считалось, что органические соединения, содержащие ароматические и конденсированные циклические структуры, в анаэробных условиях микроорганизмами не используются. В настоящее время убедительно показана способность анаэробных микроорганизмов к деградации таких ксенобиотиков. Когда процесс проводит не индивидуальный микроорганизм, а структурированная микробная ассоциация, это позволяет увеличить эффективность и глубину деградации сложных органических соединений в анаэробных условиях, а также получить полезные конечные продукты (например, биогаз в метаногенном сообществе). Пока механизмы этих процессов, видовой состав сообществ и взаимодействие микроорганизмов в них изучены мало, поэтому массовое применение биоремедиации весьма ограничено. Однако разнообразие метаболических путей в микробных сообществах позволяет надеяться, что в перспективе возможно создать ассоциации, способные разрушать полный набор синтезированных человеком соединений, тем самым возвращая в глобальные циклы элементов активные формы углерода, азота и т.д.
Химическая и нефтехимическая промышленность — основной источник токсичных и устойчивых к биоразложению загрязнений. Одной из наиболее распространенных и широко используемых групп таких загрязнений являются красители — синтетические органические вещества, проявляющие большое разнообразие структур и используемые для различных целей в текстильной, косметической, бумажной, пищевой и фармакологической промышленности. Самую большую группу синтетических красителей составляют азокрасители. Благодаря своему химическому строению азокрасители поглощают свет в видимой области спектра. Химическая структура азокрасителей характеризуется наличием одной или более азогрупп (-N = N-). Азогруппа соединена с бензольным или нафталиновыми кольцами, которые могут содержать большое количество различных заместителей, таких, как хлор (-С1), метил (-СН3), нитрогруппу (-NO2), аминогруппу (-NH2), гидроксил (-ОН) и карбоксильную группу (-СООН). В азокрасителях в качестве заместителя часто встречается сульфогруппа (-SO3H). Азокрасители, содержащие такой заместитель, называются сульфоазокрасителями. В процессе производства и использования азокрасителей примерно 10 — 50% попадает в окружающую среду. Попадая в окружающую среду вместе со сточными водами текстильного, красильного, отделочного производств, они накапливаются в биосфере. Даже небольшой концентрации красителя достаточно, чтобы сточные воды были сильно окрашены. Помимо негативного эстетического эффекта некоторые красители и многие продукты их неполной трансформации, особенно в аэробных условиях, оказывают сильное токсичное и/или канцерогенное влияние на живые организмы.
Деградация красителей химическими и физико-химическими методами является дорогостоящей и имеет массу других недостатков.
Существенным недостатком аэробных технологий, особенно при обработке концентрированных сточных вод, являются высокие энергозатраты на аэрацию и проблемы, связанные с обработкой и утилизацией больших количеств образующегося избыточного ила, имеющего очень низкую водоотдающую способность. Использование повсеместно применяемой технологии его длительной естественной сушки на иловых
Рис. 33.1. Механизм восстановления азосвязи в анаэробных условиях
бензоат резорцин
Рис. 33.2. Основные интермедиаты распада ароматических соединений в анаэробных условиях
площадках приводит к отчуждению значительных территорий, а также к ухудшению экологической обстановки. Аэробные методы обработки сточных вод, загрязненных азокрасителями, крайне неэффективны.
В то же время азокрасители могут подвергаться анаэробному восстановлению с помощью неспецифических азоредуктаз различных групп микроорганизмов. Среди бактерий, проводящих такой процесс, — молочнокислые бактерии, Proteus sp., Enterococcus sp., Aeromonas hydrophilia, Streptococcus faecalis, Bacillus subtilis, B. pyocyaneus, B. cereus, Pseudomonas sp., Caprococcus catus, Fusobacteriumsp., Bac-teroides thetaitaomicron, Peptostreptococcus productus, Eubacterium biforme, Bifidobacterium infantis, Citrobacter sp. и т.д.
При расщеплении азосвязи нарушается структура хромофорной части красителя, что приводит к его обесцвечиванию.
В результате восстановительного расщепления азокрасителей в анаэробных условиях образуются соответствующие ароматические амины (рис. 33.1), которые часто более токсичны, чем исходные соединения. В анаэробных условиях при отсутствии такого окислителя, как кислород, разрушение ароматических веществ происходит более сложно, в
несколько этапов. Основные этапы процесса биодеградации — активирование бензольного кольца, его разрыв и образование С,- и С2-соединений. Активирование кольца может быть результатом реакций карбоксилирования, анаэробного гидроксилирования и образования КоА-тиоэфиров ароматических кислот. Существуют три различных пути конверсии ароматических соединений в анаэробных условиях, отличающиеся последними ароматическими интермедиатами, такими, как бензоат, резорцин и флороглюцин (рис. 33. 2).
В настоящее время наиболее распространенным среди микроорганизмов считается бензоил-КоА-путь биодеградации ароматических веществ (через образование КоА-тио-эфира бензоата). В этой реакции участвуют растворимые, неспецифичные, индуци-бельные КоА-лигазы или КоА-трансферазы. При наличии заместителя на бензольном
кольце далее может следовать восстановительное или гидролитическое его удаление. Бензоил-КоА затем подвергается серии последовательных восстановлений под действием бензоил-КоА-редуктазы и гидролитическому расщеплению образовавшегося производного циклогексана. Полученный неароматический продукт в конечном счете окисляется с образованием ацетил-КоА.
Микроорганизмы способны использовать широкий набор ароматических субстратов в нитрат-, сульфат-, железо- и карбонат-восстанавливающих условиях (рис. 33.3).
Использование в качестве акцептора электронов Н+ требует присутствия в системе синтрофных партнеров, потребляющих молекулярный водород (метаногенов, сульфи-догенов или гомоацетогенов).
На сегодняшний день из микроорганизмов, способных к биодеструкции ароматических соединений, в виде чистых культур выделены Pseudomonas sp., Thauera aromatica, T chlo-robenzoica, Desulfobacterium anilini, Azoarcus evansii, Magnetospirillum sp., Delftia acidovorans, Rhodopseudomonaspalustris, Syntrophus gentinae и S. buswellii. Значительно более успешно биодеградацию таких соединений осуществляют анаэробные микробные сообщества. В метаногенных условиях конечные стадии биодеструкции представлены археями родов Methano-sarcina, Methanospirillum, Methanosaeta и Methanobacterium.
Рис. 33.3. Схема анаэробной деградации ароматического вещества
Конверсия любого сложного органического вещества с получением метана требует наличия как минимум бактерий, обладающих гидролазами, микроорганизмов, осуществляющих брожение с образованием в конечном счете ацетата, углекислоты и водорода, и метаногенов.
При деградации трудноразлагаемых соединений следует учитывать такое явление, как кометабализм (в аэробных условиях — соокислениё), когда проявление способности к преобразованию сложной молекулы обусловливается наличием доступного источника энергии для поддержания жизнедеятельности, так как сам ксенобиотик не может использоваться микроорганизмом в этих целях. Как правило, процесс комета-болизма характеризуется тем, что культура, трансформирующая чужеродное соединение, при этом не размножается и скорость процесса остается постоянно низкой, а продукты трансформации могут накапливаться в окружающей среде. Применительно к микробному сообществу именно такая стадия может быть лимитирующей для общего процесса полной минерализации ксенобиотика, и тогда внесение дополнительного источника углерода и энергии может существенно повлиять на скорость биодеградации и время формирования стабильной высокоактивной ассоциации микроорганизмов.
ЗАДАЧА. АНАЭРОБНАЯ БИОДЕГРАДАЦИЯ АЗОКРАСИТЕЛЯ ACID ORANGE 7 МИКРООРГАНИЗМАМИ АКТИВНОГО ИЛА
Основными способами удаления красителей из сточных вод служат ряд физико-химических методов (коагуляция известью или солями алюминия, использование мембран, адсорбция на угольной колонке, обратный осмос, химическое и электрохимическое окисление), а также аэробная и анаэробная биологическая обработка, причем наиболее эффективны именно микробиологические методы. Метаногенное сообщество активного ила способно минерализовать очень устойчивые вещества, содержащие в молекуле ароматические кольца. Используемый в задаче азокраситель Acid Orange 7 (рис. 33.4) производится в мире в промышленном масштабе, широко применяется в различных отраслях и поэтому Попадает в окружающую среду в значительных количествах. Предлагаемая задача состоит из четырех опытов, каждый их которых может быть выполнен как самостоятельное исследование. В качестве разрушаемого вещества можно использовать практически любой ксенобиотик, однако следует учитывать, что в этом случае период адаптации микроорганизмов активного ила к данному веществу бывает очень длительным (до нескольких лет).
Рис. 33.4. Характеристика азокрасителя Acid Orange 7 и его формула
Цель работы', изучение процесса биодеградации азокрасителя в анаэробных условиях и влияния на него ряда факторов (токсичности ксенобиотика, внесения дополнительного источника углерода, ингибирования ацетокластическо-го метаногенеза).
Ход выполнения задачи
Микробные ассоциации. В работе используют метаногенные анаэробные осадки любого происхождения (предпочтительно — активный ил очистных сооружений, обрабатывающих бытовые и промышленные стоки).
Среды и растворы. Для всех опытов применяют минеральную среду следующего состава (мг/л): NH4C1 - 280; СаС12-2Н2О - 10; К2НРО4 - 250; MgSO4- 7Н2О — 100; ЭДТА — 1; NaHCO3 — 5000; дрожжевой экстракт — 100, концентрированный раствор микроэлементов — 1 мл.
Такой раствор содержит (мг/мл): Н3ВО3 — 0,05; FeCl3- 4Н2О — 2; ZnCl2 — 0,05; МпС12-4Н2О - 0,05; СлС12-2Н2О - 0,03; (NH4)2SeO3 • 5Н2О - 0,05; А1С13- 6Н2О - 2; NiCl2- 6Н2О - 0,05; Na2SeO3- 5Н2О - 0,1.
Минеральную среду кипятят для избавления от кислорода, затем охлаждают до комнатной температуры и разливают под током азота для предотвращения диффузии кислорода в среду. Среду разливают по 40—50 мл во флаконы объемом 120 мл, закрывают резиновыми пробками и зажимают алюминиевыми колпачками. Пузырьки со средой продувают азотом. Конечное давление во флаконах должно составить ~ 1,6 ат. Пузырьки автоклавируют при 0,5 ати, pH среды после стерилизации равен 7,0. Все субстраты и добавки готовят в виде стерильных анаэробных концентрированных растворов и вносят по мере необходимости с помощью стерильных шприцев с соблюдением условий анаэробиоза.
Для приготовления концентрированного раствора азокрасителя его взвешивают в асептических условиях в рассчитанном количестве, переносят в стерильный пузырек, закрывают стерильной резиновой пробкой и зажимают алюминиевым колпачком. Во флакон добавляют стерильную дистиллированную воду до достижения нужной концентрации, продувают азотом и оставляют на 1—2 сут при комнатной температуре. Концентрированные растворы красителей могут образовывать осадок. Перед использованием такие растворы нагревают на водяной бане до полного растворения осадка, на
что требуется от 2 до 10 мин. Концентрированные растворы ацетата, пирувата и бром-этансульфоновой кислоты готовят на дистиллированной воде, продувают азотом и стерилизуют при 0,5 ати.
Для разведения культуральной жидкости перед спектрофотометрическими измерениями используют нестерильный 50 мМ К-фосфатный буфер (pH 7,0), приготовленный стандартным способом.
Методы определения параметров процесса
Перед всеми измерениями культуральную жидкость освобождают от клеток центрифугированием при 15 тыс. g в течение 10 мин.
Определение концентраций азокрасителя и наличия его ароматических производных. Оптимум поглощения красителя Acid Orange 7—490 нм. Резкое снижение высоты основного пика в сочетании с появлением и увеличением высот пиков, соответствующих ароматическим составляющим красителя, свидетельствует о разрыве азосвязи в молекуле красителя и появлении в среде более простых ароматических соединений, которые в свою очередь также могут быть подвержены дальнейшему расщеплению микроорганизмами активного ила. Фрагментация азокрасителя визуально выражается в постепенном обесцвечивании культуральной жидкости.
Спектрофотометрическое сканирование проводят в диапазоне длин волн 200 — 600 нм. Для анализа берут 100 мкл пробы жидкой фазы и доводят объем до 2 мл 50 мМ К-фосфатным буфером (pH 7,0). Измерения осуществляют на спектрофотометре «Shimadzu UV-1202» (Япония), или аналогичном, в кварцевой кювете с 1= 0,5 см. Расчеты истинной концентрации С (мМ) азокрасителя проводят в соответствии с уравнением, полученным путем построения калибровочной кривой при содержании вещества от 10 до 500 мг/л:
с DN
ХМ’
где D — оптическая плотность данной пробы при 490 нм; N— разведение; X— тангенс угла наклона калибровочной кривой; М — молекулярная масса азокрасителя (350,32).
Определение количества метана, углекислоты и молекулярного водорода. Метан и углекислота являются конечными продуктами биодеградации азокрасителя, а молекулярный водород — промежуточный продукт, регистрирующийся только на начальных стадиях деструкции ксенобиотика, пока в микробном сообществе не сбалансирован обмен веществ.
Концентрации газов определяют на газовом хроматографе ЛХМ 8 МД (модель 3, СССР) с катарометром, газ-носитель — аргон (скорость потока газа 20 мл/мин). Колонки длиной 2 м заполнены порапаком QS. Предварительно измеряют давление в атмосферах во флаконах с помощью манометра, на входном отверстии которого укреплена игла от шприца. Для каждого анализа с помощью газового шприпа на 0,5 —1,0 мл берут пробу газа 0,2 мл. При температуре термостата колонок 50 °C пик водорода регистрируется через 65 с, метана — через 112 с, СО2 — через 185 с.
Количество газа (моль/л) рассчитывают следующим образом:
C=D/VM-P/P0-T0/T- К/Иж,
где D — содержание данного газа в газовой фазе, об. %; VM — объем 1 моля газа; Р — давление в сосуде, ат; Ро — давление при нормальных условиях (1 ат); То — температура при нормальных условиях (273 К); Т — температура культивирования, К; Vr — объем газовой фазы в сосуде; V* — объем жидкой фазы в сосуде. Величину D рассчитывают по высотам хроматографических пиков, так как они довольно узки (обычно расчеты делают по площадям поверхности под пиком): высоту пика для данного газа (мм) делят на 100 и на тангенс угла наклона калибровочной кривой. При проведении расчетов следует учитывать чувствительность прибора при измерении данной пробы.
Калибровочные кривые получают следующим образом: флаконы емкостью 10 мл заполняют аргоном и закрывают их герметично резиновыми пробками, предусматривающими отбор проб газа. В эти флаконы газовым шприцем добавляют нужный объем анализируемого газа, чтобы его содержание составляло 0,5 — 96 % для метана, 0,5—100 % — для водорода и 1 — 100 % — для углекислого газа. Из каждого флакона отбирают 3 пробы по 0,2 мл и фиксируют высоту хроматографического пика для анализируемого газа. Полученные данные усредняют и осуществляют построение калибровочной кривой в координатах: содержание газа (об. %) соответствует высоте пика (мм).
Определение ацетата и других летучих жирных кислот (ЛЖК) и спиртов. Интермедиатами анаэробного распада азокрасителей и ароматических аминов являются ЛЖК и спирты с длиной углеводородного скелета не более 6 атомов, концентрацию которых определяют на газовом хроматографе «Chrom-5» с пламенно-ионизационным детектором, на колонке 1 м, заполненной хромосорбом 101. Скорость газа-носителя (аргона) — 30 мл/мин. Для каждого анализа берут пробу жидкой фазы 1 мкл, подкисленную соляной кислотой. При температуре термостата колонок 200 °C происходит четкое разделение продуктов, идентифицируемых по временам удержания соответствующих соединений в стандартных растворах. Расчет концентрации каждого вещества (мМ) проводят по высоте пика, отнесенной к высоте пика этого вещества в стандартном растворе:
h N С
х-1 _ 'TipJ * пр^ст
“ h N ' "ст-* * ст
где h — высота пика в пробе или стандартном растворе; N — чувствительность прибора при соответствующем измерении; С — концентрация вещества в соответствующем растворе.
Определение концентрации ароматических интермедиатов и продуктов их дальнейшего преобразования. Исходя из химической формулы азокрасителя, возможного пути его фрагментации и максимумов поглощения пиков продуктов при спектрофотометрическом сканировании, подбирают в качестве стандартов соответствующие амины и другие ароматические соединения. Идентификацию веществ и определение их концентрации проводят при помощи метода высокоэффективной жидкостной хроматографии (ВЭЖХ) на хроматографе Du Pont (США), используя колонку с обращенной фазой (Диасорб 130-С16Т, 6 мкм) в системе уксусная кислота (1 %) — метанол (в соотношении 85 : 15 по объему). В качестве детектора используют спектрофотометр, на
строенный на длину волны 225 нм (средняя длина волны поглощения ароматических соединений). Скорость подачи элюента 1 мл/мин, давление 148 ат. Объем вводимой пробы 20 мкл. До проведения анализа отобранные пробы могут храниться при -20 °C в течение 30 сут в закрытых пластиковых пробирках Эппен-дорф. Ароматические вещества определяют по соответствию их времен удержания временам удержания стандартных растворов, а расчет концентрации компонентов смеси ведут по площадям пиков, отнесенным к площадям пиков веществ в стандартных растворах.
Определение концентрации иона аммония. Концентрацию ионов аммония определяют по методу Фоссета и Скотта. Для этого готовят реактив Фоссета, состоящий из двух растворов. Раствор А (г/л): фенол — 10; нитропруссид натрия — 0,05. Раствор Б (г/л): Na2HPO4 — 37; NaOH — 10, 1%-й раствор гипохлорита натрия (NaOCl) — 10 мл.
Для определения концентрации аммония в жидкой фазе 200 мкл исследуемого раствора помещают в мерную пробирку, разбавляют водой до 400 мкл, затем добавляют 0,8 мл раствора А и 0,8 мл раствора Б и интенсивно перемешивают. Растворы инкубируют 30 мин при 37 °C, а затем сразу измеряют оптическую плотность при 578 нм и / = 1 см. В качестве контроля используют раствор, не содержащий аммония. По калибровочной кривой (от 1 до 15 мг/л NH+) определяют содержание аммония в исследуемом растворе. При необходимости растворы А и Б можно хранить в сосудах из темного стекла не более 2 мес.
Определение концентрации сульфида. Исходный азокраситель содержит в своем составе сульфогруппу, поэтому можно ожидать появления иона сульфида в процессе анаэробной биодеструкции. Концентрацию сульфид-иона определяют по методу Трю-пера и Шлегеля с использованием трех растворов, содержащих в 100 мл каждого раствора: раствор А', ацетат цинка — 4 г, ледяная уксусная кислота — 0,05 мл; раствор В: 2-диметил-л-фенилендиаминсульфат — 0,2 г; H2SO4 (конц.)— 20 г; раствор С: Fe(NHSO4)2 - 10 г, H2SO4 (конц.) - 2 г.
К 100 мкл раствора А в пластиковой пробирке Эппендорф быстро добавляют 10 мкл пробы и 785 мкл дистиллированной воды. Затем в смесь вносят 100 мкл раствора В и 5 мкл раствора С. Через 10 мин измеряют поглощение образовавшегося хромогенного комплекса при 660 нм и / = 1 см. По калибровочной кривой (от 100 до 500 мкг/мл S2 ) определяют содержание сульфида в исследуемом растворе. В качестве контроля используют воду.
Определение белка. Прирост биомассы микроорганизмов оценивают по общему содержанию белка в среде, которое определяют методом Бредфорд (см. гл. 14). Для анализа отбирают пробу жидкой фазы объемом 0,3 мл, центрифугируют в течение 15 мин при 12 тыс. g, удаляют надосадочную жидкость. К осадку добавляют 0,3 мл щелочи (3 N NaOH) и выдерживают при 30 °C в течение 12 ч. При определении содержания белка в пробирки вносят по 200 мкл пробы, добавляют по 2 мл реактива Бредфорд, перемешивают и через 20 мин измеряют оптическую плотность при 7. = 595 нм. Пользуясь калибровочной кривой, определяют концентрацию белка в исследуемых растворах. Калибровочную кривую строят, используя бычий сывороточный альбумин, высушенный в эксикаторе до постоянного веса, из которого готовят растворы с концентрацией белка от 10 до 100 мкг/мл. Затем растворы обрабатывают, как было описано выше, и строят зависимость оптической плотности от концентрации белка.
Микроскопия. Для анализа изменений микроскопической картины микробного сообщества готовят фиксированные окрашенные препараты и просматривают их с иммерсионной системой под световым микроскопом.
Условия проведения экспериментов. На предварительном этапе работы рассчитывают необходимые количества реактивов для приготовления концентрированных растворов. При этом следует учитывать, что итоговая концентрация
субстратов и добавок в среде должна включать: пируват натрия — 3 мМ; ацетат натрия — 6 мМ; бромэтансульфоновая кислота — 45 мМ и азокраситель в разных вариантах опыта — 0,7; 1,4; 2,1; 2,8 и 3,5 мМ. Каждая добавка должна быть внесена в объеме, не превышающем 1 мл.
Снимают полный спектр раствора азокрасителя (концентрация 300 мг/л) в диапазоне длин волн 200 — 600 нм (рис. 33.4). Затем осуществляют построение калибровочных кривых для всех измеряемых соединений.
В пузырьки со средой стерильно вносят активный ил из расчета 10 % по объему и необходимые добавки. В качестве контрольных используют варианты:
• пузырьки без активного ила с добавлением азокрасителя в концентрации 0,7 мМ для регистрации возможных физико-химических преобразований субстрата в минеральной среде (без участия биологической активности);
• пузырьки без красителя, но с активным илом для учета выделения метана за счет лизиса инокулята;
• флаконы, содержащие ил, предварительно инактивированный автоклавированием при 0,5 ати, для выявления роли адсорбции азокрасителя на матрикс ила.
Полный набор вариантов опыта представлен на рис. 33.5. Каждый вариант ставят в двух повторностях.
Культивирование во всех вариантах проводят в стационарных условиях (без перемешивания) в темноте при 30 °C. Отбор проб жидкой (0,8 мл) и газовой (0,2 мл) фаз осуществляют в нулевой точке и каждые 24 ч (48 ч) стерильными шприцами, без нарушения анаэробиоза.
В эксперименте по биоразлагаемости сравнивают изменение концентраций азокрасителя, промежуточных (Н2, ароматических аминов, ацетата и других ЛЖК и спиртов) и конечных (метана, углекислоты, аммония и сульфида) продуктов, а также прирост биомассы в опытном и трех контрольных вариантах. Выявленные ароматические интермедиаты определяют с помощью ВЭЖХ и по максимумам поглощения при спектрофотометрии. Описывают характерные культуральные свойства и микроскопическую картину сообществ из разных вариантов. Полученные данные представляют в виде таблиц, спектров азокрасителя и ароматических производных, а также графиков зависимости содержания всех вышеперечисленных веществ (мМ) и биомассы (мг/л) от времени (сут). Делают выводы о биологической природе процесса, роли адсорбции, последовательности появления и химической структуре ароматических интермедиатов, а также о возможных механизмах конверсии азокрасителя. Анализируют соответствие различных фаз процесса биодеструкции азокрасителя изменениям в микробном составе анаэробной ассоциации.
В эксперименте по изучению влияния дополнительного источника углерода (пирувата) сравнивают те же параметры, что и в предыдущем эксперименте, но во флаконах с активным илом и азокрасителем с пируватом или без него. Данные представляют в виде таблиц и графиков, делают выводы об изменении хода процесса биодеструкции красителя в присутствии легкодоступного субстрата (увеличении скорости, изменении состава и соотношения продуктов, приросте биомассы, изменении микроскопической картины активного ила).
Для накопления и определения интермедиатов распада азокрасителя проводят эксперимент по элиминированию последней стадии биодеградации в анаэробных условиях — метанообразования. Во флаконы с активным илом и азокрасителем, содержащие и не содержащие дополнительный источник уг-
2. Эксперимент по влиянию дополнительного источника углерода
1. Эксперимент по биоразлагаемости
(0,7 ммоль/л)
АИ + АК -(0,7 ммоль/л)
«Убитый» АИ + АК (0,7 ммоль/л)
АИ + АК (0,7 ммоль/л)
(0,7 ммоль/л)
+ П (3 ммоль/л) + БЭСК (45 ммоль/л)
АИ + Ац
АИ + Ац (6 ммоль/л) + АК (0,7 ммоль/л)
АИ +Ац (6 ммоль/л) + АК (2,1 ммоль/л)
АИ + Ац (6 ммоль/л) + АК (2,8 ммоль/л)
(0,7 ммоль/л)
БЭСК (45 ммоль/л)
АИ + Ац (6 ммоль/л) + АК (1,4 ммоль/л)
4. Эксперимент по токсичности азокрасителя
3. Эксперимент по ингибированию метаногенеза
АИ +Ац (6 ммоль/л) + АК (3,5 ммоль/л)
Рис. 33.5. Варианты опыта:
АИ — активный ил; АК — азокраситель; Ац — ацетат; П — пируват; БЭСК — бромэтансуль-фоновая кислота. Скобки и рамки объединяют варианты, учитываемые в различных экспериментах
лерода пируват, вносят ингибитор метанообразования БЭСК в концентрации 45 мМ. За процессом следят по виду спектра культуральной жидкости, хроматограммы ВЭЖХ, накоплению интермедиатов в жидкой фазе. Определяют морфологические и культуральные изменения в разных вариантах. Результаты представляют в виде таблиц и графиков. Уточняют данные о химической структуре кольцевых и линейных интермедиатов. Делают выводы о влиянии метаногенной стадии на скорость процесса биодеградации азокрасителя в условиях, когда он является единственным источником углерода и энергии или имеется также другой легкодоступный субстрат.
Исследование по токсичности азокрасителя проводят на фоне стандартного метода определения ацетокластической метаногенной активности анаэробной биомассы. Во все пузырьки вносят раствор ацетата натрия до конечной концентрации 6 мМ в качестве основного субстрата и азокраситель с конечной концентрацией 0,7; 1,4; 2,1; 2,8 и 3,5 мМ. В качестве контрольных используют пузырьки без добавления азокрасителя. Через каждые 24 ч (48 ч) измеряют концентрацию метана в газовой фазе, а полученные результаты представляют в виде графика зависимости количества метана от времени. Измерения сопровождают приготовлением фиксированных окрашенных препаратов культуральной жидкости. Значение ацетокластической активности анаэробного ила рассчитывают по тангенсу угла наклона линейного участка этой кривой. За 100 % активности принимают тангенс угла наклона линейного участка кривой зависимости концентрации метана от времени в эксперименте без добавления азокрасителя. Поскольку скорость разложения самого азокрасителя в этих условиях существенно ниже скорости ацетокластической реакции, его вкладом в образование метана можно пренебречь. Результаты представляют в виде графиков зависимости ацето пластического метаногенеза от времени для разных концентраций красителя, а также кривой зависимости образования метана (%) от концентрации азокрасителя (мМ). Определяют ЛД50 для данного азокрасителя и делают вывод о его токсичности по отношению к метаногенной активности анаэробного ила. Анализируют изменение микроскопической картины в зависимости от концентрации красителя и делают вывод об изменении микробного состава активного ила при увеличении «давления» ксенобиотика.
Материалы, оборудование, посуда, реактивы
Спектрофотометр «Shimadzu UV-1202» (Япония) или аналогичный, кювета кварцевая с /= 0,5 см и кювета стеклянная с / = 1,0 см; газовый хроматограф ЛХМ 8 МД (модель 3, СССР) с катарометром; газовый хроматограф «Chrom-5» (ЧССР) или аналогичный с пламенно-ионизационным детектором; хроматограф ВЭЖХ Du Pont (США) или аналогичный; световой микроскоп с масляным иммерсионным объективом 90Х; баллон с аргоном (азотом); щипцы для зажимания алюминиевых колпачков; центрифуга с гнездами для пробирок Эппендорф; манометр с иглой. Флаконы на 120 мл с герметичными резиновыми пробками и алюминиевыми колпачками — 26; пробирки химические мерные с притертыми пробками на 5 —10 мл — 90 (45); пластиковые пробирки Эппендорф на 2 мл с крышечками или аналогичные — 300 (150); пипетки на 1 — 2 мл — 540 (270), на 0,1 мл — 25 (13); шприцы на 1—2 мл (лучше одноразовые) — 300 (150); газовый шприц на 0,5—1,0 мл — 1; предметные стекла для микроскопии — 50 (25). Минеральная среда — 1100 мл; концентрированные растворы: азокрасителя — 50 мл, пирувата натрия — 10 мл, ацетата натрия — 20 мл, бромэтансульфоновой кислоты — 10 мл; 50 мМ К-фосфатный буфер (pH = 7,0) — 100 мл; растворы А и Б для определения аммония — по 30 мл; растворы А, В, С для определения сульфида —30, 30 и 1,5 мл соответственно; 3 N NaOH — 10 мл; реактив Бредфорд — 100 мл; краситель метиленовый синий для окрашивания микроскопических препаратов ~30 мл; иммерсионное масло кедровое ~5 мл.
План выполнения задачи
Занятие 1. Подготовка и стерилизация минеральной среды. Расчет необходимых количеств веществ для приготовления концентрированных растворов азокрасителя и добавок. Приготовление и стерилизация растворов добавок и инструментов (4 ч). Приготовление концентрированного раствора азокрасителя (48 ч).
Занятие 2. Приготовление реактивов, растворов и стандартов для определения различных веществ (см. с. 381). Построение калибровочных кривых и снятие спектра раствора азокрасителя (4 ч).
Занятие 3. Засев ({маконов активным илом, внесение субстрата и необходимых добавок. Отбор проб в нулевой точке и проведение всех необходимых определений. Опи
сание культуральных признаков и приготовление фиксированных окрашенных препаратов (4 ч). Инкубация посевов (20 сут).
Занятия 4—23 (4—13). Отбор проб каждые 24 ч (48 ч). Описание культуральных признаков и приготовление фиксированных окрашенных препаратов. Проведение необходимых определений. Занесение результатов в таблицы. Закладка на хранение проб для ВЭЖХ (4 ч).
Занятие 24 (14). Отбор и приготовление стандартов для ВЭЖХ. Проведение ВЭЖХ для всех хранящихся проб (8ч).
Занятие 25 (15). Оформление результатов (4 ч).
Глава 34
ИЗУЧЕНИЕ ЯВЛЕНИЯ ДИССОЦИАЦИИ У МИКРООРГАНИЗМОВ
Диссоциация — это явление морфофизиологической гетерогенности популяции микроорганизмов, которая визуально выражается в развитии морфологически разных колоний при рассеве чистой культуры.
На самом деле такая гетерогенность микробной популяции затрагивает не только морфологические, но и физиолого-биохимические, и генетические признаки. В отличие от спонтанных мутаций эти изменения имеют постоянный обратимый характер и более высокую частоту (~10“4). Традиционно по морфологии различают три вида колоний: R — шероховатые, S — гладкие, М — слизистые. Механизм этого явления еще не раскрыт полностью, и, более того, вопрос этот на протяжении многих лет остается дискуссионным, однако в практике научных исследований с ним сталкивается практически каждый, кто работает с микроорганизмами. Первоначально явление диссоциации было отмечено у патогенных микроорганизмов Salmonella, Shigella, Е. coli и в связи с разными фазами протекания болезни было названо «вариацией фаз» (S — вирулентный вариант, преобладает в эпидемической фазе, R — в постэпидемической). Диссоциация, безусловно, связана с изменениями в геноме.
Итак, если после выделения чистой культуры из отдельной клетки в дальнейшем вырастают разные колонии, то это диссоциация. Колонии по диаметру различаются, но клеток в колониях одинаковое количество, и они по-разному упакованы. При этом в каждой колонии уже есть клетки всех трех типов. В природе обычно какой-то из диссоциантов преобладает, но присутствуют все три варианта.
Различия в виде колоний обычно выявляются только на богатой углеводами среде. Различия на клеточном уровне лежат в способе расхождения делящихся клеток (образуются либо цепочки, либо «палисадные» V-образные формы). У диссоциантов одна из главных первопричин вариаций — различия в клеточных оболочках (капсула+клеточная стенка), в частности разная толщина и химический состав капсул (например, переход S-варианта в R-вариант связан с потерей О-антигена). Клеточная стенка R-варианта в 1,5 — 2 раза толще, чем S-варианта. Морфологические особенности деления можно связать с химическим составом и электрозаряженностью вещества капсул (при отрицательных зарядах возникает отталкивание оболочек). У диссоциантов разное количество ненасыщенных липидов в мембранах, что влияет на ее текучесть (у R-варианта обнаруживается больше липидов, содержащих больше насыщенных кислот).
Различия в клеточных оболочках приводят к разной скорости поглощения и экскреции веществ, активности мембранных ферментов, что в свою очередь влияет на изменение скорости роста, выделения продуктов, устойчивости к токсическим веществам.
Свойство диссоциации важно учитывать при проведении процессов в микробиологических производствах. При культивировании медленно растущие варианты вытесня-
Таблица 34.1
Селективные преимущества (большая устойчивость) диссоциантов ряда мезофильных грамположительных и грамотрицательных бактерий при воздействии различных факторов
Факторы R • S м
Температура, °C: 41 14 + - +
Ультрафиолет + - -
Антибиотики + — —
Высушивание + - —
Литические вещества + - -
NaCl (7 %) - - +
pH 4,0 - + -
pH 9,0 - — +
Аэрация + - —
КН2РО4 (0,5 %) - + -
ются быстрорастущими, но, как правило, менее активными диссоциантами. У диссоциантов различаются не только количество, но и состав биологически активных веществ. Условия культивирования оказывают существенное влияние на состав популяции. Если один диссоциант имеет преимущество, то к концу цикла состав популяции может полностью поменяться. Табл. 34.1 дает представление о селективных преимуществах диссоциантов при воздействии различных факторов.
Таким образом, можно сказать, что диссоциация — это адаптация к меняющимся условиям среды на уровне популяции.
Показано, что в создании гетерогенности микробной популяции в результате процесса диссоциации играют роль все генетические процессы — трансформация, трансдукция, фаговая конверсия, мигрирующие генетические элементы (умеренные фаги, плазмиды, транспозоны), а при получении культуры из одной клетки — только мигрирующие генетические элементы генома одной клетки.
Для стабилизации синтеза биологически активных веществ в промышленности необходимо: 1) получить недиссоциирующий клон; 2) вести стабилизирующий отбор; 3) соблюдать оптимальные условия хранения; 4) соблюдать оптимальные условия роста высокопродуктивного диссоцианта. Знания о диссоциации позволяют прогнозировать течение процесса и дают возможность им управлять.
По систематическому положению диссоцианты определяются как один вид, так как в определителе дается признак по принципу «есть-нет», а диссоциация дает изменения по принципу «больше-меньше».
ЗАДАЧА. ДИССОЦИАЦИЯ У PSEUDOMONAS AERUGINOSA К-2
И ВЛИЯНИЕ УСЛОВИЙ КУЛЬТИВИРОВАНИЯ НА ЭТОТ ПРОЦЕСС
Бактерии рода Pseudomonas являются настоящими космополитами, обладают способностью к существованию в самых разнообразных условиях и встречаются на Земле повсеместно.
Разнообразие метаболитических возможностей, высокая скорость роста на простых средах и особенности организации генетического аппарата делают псевдомонад привлекательными и перспективными для биотехнологии объектами. Представители рода Pseudomonas используются для получения ряда индивидуальных химических веществ, в производстве препаратов для сельского хозяйства, полимеров, в биогидрометаллургии, в нефтедобыче — для повышения отдачи пластов, а также при переработке и удалении отходов и веществ, загрязняющих окружающую среду, в том числе и углеводородов.
При рассеве псевдомонад на плотные среды, как правило, обнаруживаются R-, S- и М-диссоцианты, различающиеся рядом физиолого-биохимических признаков, ультратонким строением клеток, морфологией колоний, скоростью роста, устойчивостью к действию ряда химических и физических факторов. Диссоцианты Р. aeruginosa К-2 различаются: 1) размером и строением колоний — самые большие и прозрачные колонии образует М-диссоциант, клетки которого обладают мощной капсулой; R-диссоциант растет в виде небольших плотных колоний, так как его клетки имеют тонкую капсулу; 2) потребностями в основных биогенных элементах (углероде, азоте и фосфоре) — R-клетки лучше растут на богатых средах, а М-клетки имеют селективные преимущества на средах с недостатком азота или фосфора; 3) путями использования глюкозы — у R-диссоцианта преобладает окислительный путь, а М-клетки наиболее активно осуществляют превращение сахара с образованием муравьиной кислоты, что приводит к резкому подкислению среды. S-диссоциант способен переключаться с одного из вышеуказанных способов использования глюкозы на другой.
Цель работы, изучение влияния состава питательных сред и условий культивирования на расщепление популяции Р. aeruginosa К-2 на диссоцианты и описание ряда морфофизиологических признаков полученных вариантов.
Ход выполнения задачи
Микроорганизм. В работе используют S-диссоциант непатогенного штамма Р. aeruginosa К-2, выделенного из пластовых вод сибирского нефтяного месторождения Ван-Еган.
Среды и растворы. В качестве богатой среды применяют бульон-сусло-агар (БСА), pH 7,0. Для приготовления среды к МПБ добавляют неохмеленное сусло (б°Б) и 1,5 % агара. Среду стерилизуют при 0,5 ати.
Минеральную среду применяют в двух модификациях: а) полноценная следующего состава (г/л): КС1 — 0,6; KNO3 — 5,0; NaH2PO4-2H2O — 0,5; MgSO4-7H2O — 0,2, pH 7,0; б) «голодная по фосфору» — NaH2PO4-2H2O — 0,05. Среду стерилизуют при 1,0 ати. После стерилизации вносят 2,0 % глюкозы в виде 40%-го раствора, простерилизованного при 0,5 ати.
Для приготовления суспензий клеток и десятичных разведений готовят стерильный физиологический раствор (0,85%-й NaCl в дистиллированной воде, стерилизация при 1,0 ати).
Общая схема эксперимента (рис. 34.1). Для получения исходной суточной культуры осуществляют посев петлей из выданных преподавателем пробирок с S-диссоциантом Р. aeruginosa К-2 на свежий скошенный БСА (выдаются лаборан-
том) и инкубируют засеянные пробирки при 30 °C в течение 24 ч. В стерильную узкую пробирку наливают пипеткой стерильный физраствор (~3 мл), куца осторожно переносят петлей выросшие бактерии, не захватывая БСА, энергично перемешивают и доводят плотность суспензии стерильным физраствором до концентрации 109 клеток в 1 мл, сравнивая ее с соответствующим стандартом мутности. Общее количество получившейся суспензии должно быть не менее 7 мл.
скошенном БСА
—|0,5мл|—-
Приготовление разведений до 10“7 и рассев по 0,1 мл в чашки с БСА
среды
Полноценная
«Голодная
минеральная по фосфору» среда минеральная среда
Инкубация при 30 °C на качалке 200 об/мин
2 сут 3 сут
Полноценная минеральная среда
I
Инкубация 1 сут при 37 °C на качалке 300 об/мин
1
Определение pH и оптической плотности культурной жидкости. Приготовление десятичных разведений и рассев суспензии по 0,1 мл в чашки с БСА из разведений 10-5—10-8
Инкубация 2 сут при 30 °C
Инкубация 2 сут при 30 °C
Рис. 34.1. Схема эксперимента по диссоциации S-варианта Р. aeruginosa
Из части суспензии (0,5 мл) готовят десятичные разведения в стерильном физрастворе до 10"7, вносят по 0,1 мл в чашки с БСА из разведений 10“5— 10 7 (в трех повторностях) и тщательно растирают стеклянными шпателями. Инкубируют засеянные чашки в течение 2 сут при 30 °C, после чего до подсчета колоний хранят в холодильнике до 5 сут.
Из оставшейся суспензии производят посев в жидкие минеральные среды в широких пробирках (по 10 мл) и круглодонных колбах (по 50 мл), внося по 3 % (по объему) посевного материала. Каждый вариант опыта ставят не менее чем в трех повторностях. Культуру инкубируют в колбах при 37 °C на качалке при 300 об/мин в течение суток, а в пробирках при 30 °C на качалке при 200 об/мин на полноценной среде — 2 сут, на «голодной по фосфору» — 3 сут. Готовят десятичные разведения усредненных проб в стерильном физрастворе до 10 s и вносят по 0,1 мл из трех последних разведений в чашки с БСА (в трех повторностях). Суспензию тщательно растирают по агару стеклянным шпателем. Засеянные чашки инкубирую'г при 30 °C в течение 48 ч. Культуры в жидких средах до рассева и чашки с выросшей культурой до подсчета колоний можно хранить в холодильнике до 5 сут.
В культуральной жидкости всех вариантов в усредненной пробе определяют оптическую плотность и pH. Результаты вносят в табл. 34.2.
На чашках подсчитывают общее число колоний и число колоний каждого морфотипа. Описывают имеющиеся типы колоний. Определяют долю диссоциантов в разных вариантах опыта. Результаты вносят в табл. 34.2.
Делают выводы о влиянии состава среды, температуры и аэрации на гетерогенность популяции исходной культуры. Соотносят наблюдаемый сдвиг pH с увеличением доли определенного диссоцианта.
Чашки с колониями М-диссоцианта оставляют на хранение при комнатной температуре на 1 мес. После этого колонию снимают петлей с агара, суспендируют в стерильном физрастворе, делают десятичные разведения до 10-8 и рас-севают обычным способом на чашки с БСА из четырех последних разведений (в двух повторностях). Инкубируют 2 сут при 30 °C. Подсчитывают общее число колоний и долю диссоциантов в популяции. Делают вывод о влиянии активности воды на расщепление культуры на диссоцианты.
Таблица 34.2
Состав популяции Р. aeruginosa К-2 при разных условиях культивирования
Номер Среда, условия pH Оптическая плотность, усл. ед. Число клеток в 1 мл, % от общего количества
S R М
1 БСА (исходная культура)
2 Полноценная минеральная среда, 30 °C, 200 об/мин
3 «Голодная по фосфору» среда, 30 °C, 200 об/мин
4 Полноценная минеральная среда, 37 °C, 300 об/мин
Материалы, оборудование, посуда, реактивы
Качалки 200 и 300 об/мин (30 и 37 °C); pH-метр; спектрофотометр; стандарт мутности (109 клеток/мл); счетчик колоний; микробиологическая петля. Чашки Петри — 55; пробирки узкие с ватными пробками — 40; пробирки широкие с ватными пробками — 10; стеклянные шпатели — 10; пипетки на 1 мл — 45; на 5 мл — 5; на 10 мл — 1; круглодонные колбы на 250 мл — 3. БСА — 1 л; жидкие минеральные среды: полноценная — 0,2 л в 3 широких пробирках по 10 мл и в 3 круглодонных колбах по 50 мл; «голодная по фосфору» — 50 мл в 3 широких пробирках по 10 мл; 40%-й раствор глюкозы — 50 мл, физиологический раствор — 0,3 л, скошенный БСА в пробирках — 2 шт. (готовится заранее лаборантом).
План проведения задачи
Занятие 1. Подготовка и стерилизация посуды и сред (4 ч). Посев бактерий на скошенный БСА и их выращивание (24 ч).
Занятие 2. Приготовление суспензии клеток бактерий и ее стандартизация до концентрации 109 (по стандарту мутности). Приготовление разведений и посев бактерий на чашки с БСА и жидкие минеральные среды в широких пробирках и колбах (4 ч). Инкубация посевов в разных условиях (24, 48 и 72 ч).
Занятие 3. Описание культуральных свойств вариантов культуры псевдомонад на жидких средах. Определение оптической плотности и pH культуральной жидкости усредненной пробы каждого варианта. Приготовление разведений усредненных проб из разных вариантов жидких сред и посев бактерий на чашки с БСА (4 ч). Инкубация посевов (48 ч).
Занятие 4. Подсчет колоний, выросших на чашках с БСА. Определение доли диссоциантов в разных вариантах опыта. Оформление результатов. Закладка на хранение чашек с М-диссоциантом при комнатной температуре (4 ч). Хранение чашек с М-диссо-циантом до подсыхания (1 мес).
Занятие 5. Приготовление десятичных разведений из колонии М-диссоцианта и посев бактерий на чашки с БСА (2 ч). Инкубация посевов (48 ч).
Занятие 6. Подсчет колоний, выросших на БСА, и определение доли диссоциантов. Оформление результатов (4 ч).
Глава 35
ГЕНЕТИКА МИКРООРГАНИЗМОВ
Наследственность обеспечивает материальную и функциональную преемственность между поколениями организмов.
Наследственная информация, необходимая для существования и воспроизведения организмов, заключена в генах, которые обладают тремя основными свойствами: они выполняют специфическую функцию, способны к точному самовоспроизведению, чрезвычайно стабильны. Гены расположены в линейном порядке на хромосомах, причем каждый ген занимает свое собственное место, или локус. Таким образом, основной структурой, обеспечивающей материальную основу наследственности, является хромосома — система линейно сцепленных генов, обеспечивающих хранение и передачу информации.
Прокариоты и эукариоты различаются генетической организацией. У эукариот наследственная информация находится в хромосомах, расположенных в ядре. Экстрахро-мосомальная ДНК у эукариот может быть представлена плазмидами, митохондриаль
ной ДНК, хлоропластной ДНК. Генетический аппарат прокариот представлен у большинства групп прокариот одной кольцевой молекулой ДНК. В определенных условиях в клетке может находиться несколько копий бактериальной хромосомы. ДНК содержится также во внехромосомных генетических элементах плазмидах, в большинстве случаев кольцевых, автономно реплицирующихся небольших молекулах.
Совокупность всех генов организма называют генотипом, а совокупность присущих организму признаков — фенотипом. Спецификой генетических исследований микроорганизмов является изучение свойств не отдельной особи (клетки), а популяции в целом (микробной культуры).
Изменчивость организмов может быть наследственной и ненаследственной. Ненас-ледственная изменчивость (модификационная, фенотипическая) является адаптивной, приспособительной к изменению факторов внешней среды, однако она генетически детерминирована и находится в пределах нормы реакции организма. Таким образом, фенотип микроорганизма часто в значительной степени определяется условиями его выращивания. При изменении внешних условий большинство клеток в популяции претерпевает изменения приспособительного характера, вызванные регуляцией клеточного метаболизма.
Наследственная изменчивость подразделяется на комбинативную и мутационную. В первом случае изменения обусловлены генетической рекомбинацией, т.е. перекомбина-цией уже имеющихся генов, участков хромосом и хромосом, несущих разные аллели. Комбинативная изменчивость определяет разнообразие потомков, получивших новые комбинации генов и хромосом, существовавших у родительских форм. В значительной степени эти'явления присущи организмам, имеющим половой процесс.
Несмотря на стабильность, гены обладают способностью подвергаться случайным внезапным изменениям, или мутациям, в результате чего появляются новые аллели, функционально отличные от исходного гена. Таким образом, мутационная изменчивость — это возникновение новых дискретных единиц генетического материала, прежде всего новых аллелей.
Основные понятия генетики микроорганизмов
Каждый вид микроорганизма может быть представлен рядом штаммов. В генетике микроорганизмов термином «штамм» обозначают генетически однородную культуру определенного вида, выделенную из одной клетки и отличающуюся от других штаммов происхождением, а часто и рядом признаков, несущественных для систематики. Штамм, выделенный из природного источника, называют диким типом.
Один из основных методов генетического анализа микроорганизмов — метод клонирования культуры. Клон — это генетически однородное потомство, полученное при размножении одной клетки (у прокариот) или вирусной частицы. У эукариотических микроорганизмов клон — потомство одной клетки (споры), делящейся митотически.
Получение отдельных клонов позволяет изучать свойства генетически однородной совокупности клеток (популяции). На практике клоны получают из отдельных колоний микроорганизмов, выросших на поверхности плотной питательной среды из отдельных клеток (принцип «одна клетка — одна колония»).
Признаки микробных культур можно подразделить на морфологические, физиологические и биохимические. Наследственной основой всех проявляемых признаков определенного штамма (фенотипа) служит его наследуемая генетическая организация (генотип).
Перед проведением генетических экспериментов необходимо убедиться в чистоте штаммов, отклонировать их, вырастить в небольшом объеме среды и соответствующим образом проверить их генотип. На практических занятиях по генетике микроорганизмов наиболее широко используемыми объектами из бактерий являются Escherichia coli и Bacillus subtilis, а из эукариот — некоторые виды грибов. Однако это не исключает
использование и других генетически хорошо изученных микроорганизмов. В данном практическом руководстве рассматриваемые методики относятся главным образом к Е. coli.
Мутагенез
Мутации являются первоисточником наследственной изменчивости и наряду с генетической рекомбинацией поставляют материал для эволюции, а также искусственного отбора. Они представляют собой ценный инструмент в генетических и биохимических исследованиях: 1) вызывая изменения в гене, а следовательно, и в фенотипе, мутации служат генетическими маркерами, позволяющими не только идентифицировать ген, но также локализовать его на хромосоме, плазмиде или другой молекуле ДНК в клетке с помощью методов генетического картирования; 2) наличие набора мутаций помогает исследовать процессы метаболизма и механизмы их генетического контроля; 3) исследование белков, измененных в результате мутаций, способсгвует установлению их структуры и функционирования; 4) мутации являются основой для селекции штаммов микроорганизмов с полезными свойствами (например, штаммов-продуцентов антибиотиков). По своему происхождению мутации могут быть спонтанными и индуцированными.
Спонтанные мутации возникают в естественных условиях в результате нормальных процессов в клетке или при взаимодействии клеток с окружающей средой. Мутагенным действием обладают неконтролируемые факторы внешней среды, например естественная радиация. Мутагенное действие могут оказывать также определенные компоненты питательных сред. Существенная роль в возникновении спонтанных мутаций принадлежит таким процессам, как рекомбинация, репликация и репарация. Спонтанные мутации в популяции клеток по разным генам возникают с очень низкой частотой (в пределах 10-5— 10'7). Однако если существует метод отбора (селекции) мутантного фенотипа, то оказывается возможным обнаружить даже крайне редкие спонтанные мутанты. При наличии таких методов отбора часто предпочтение отдается выделению именно спонтанных мутантов, поскольку многие мутагенные факторы потенциально опасны для человека, а химические мутагены, кроме того, загрязняют окружающую среду.
Индуцированные мутации происходят под влиянием определенных, специально примененных физических, химических или биологических агентов, называемых мутагенами. Мутагенным действием обладают ионизирующее излучение, УФ-лучи, ряд химических соединений, транспозирующиеся элементы и транспозон-подобные фаги (например, фаг Ми), а также мутации в определенных генах (гены-мутаторы).
Разные мутагены обладают различными механизмами действия и эффективностью в отношении конкретного организма. Выбор мутагена определяется типом мутации, которую желательно получить, т. е. замена основания, делеция, сдвиг рамки считывания. Например, делеционные мутанты всегда имеют ярко выраженный фенотип, не бывают температурозависимыми и условно летальными, истинные реверсии к дикому типу у них невозможны. Разные виды микроорганизмов могут требовать разных доз и условий для эффективного мутагенеза. С этой целью перед экспериментом следует построить кривую выживаемости клеток в зависимости от дозы мутагенного фактора или времени обработки. Некоторые мутагены, используемые для получения мутаций у микроорганизмов, приведены в табл. 35.1.
• УФ-лучи. Простым и удобным методом получения мутантов разного типа у ряда микроорганизмов является ультрафиолетовое облучение. Для этого используются любые источники света с максимумом испускания в коротковолновой области (около 254 нм), например бактерицидные лампы. Поскольку УФ-лучи являются относительно слабым мутагеном, то наилучшие результаты получают при низкой выживаемости клеток (0,1 — 1,0 %). Необходимо помнить, что УФ-лучи практически полностью поглощаются стек-
Свойства некоторых мутагенов, используемых для индукции мутаций у микроорганизмов
Мутаген Механизм действия Тип мутации Эффективность Характерные особенности
Радиация Рентгеновское излучение, быстрые нейтроны Преимущественно разрывы хромосомы Делеции, инверсии Средняя Требует специального оборудования
УФ-облучение Димеризация пиримидиновых оснований Транзиции, трансверсии, делеции Средняя Высокая частота индукции мутаций достигается только в условиях низкой выживаемости, фотореактивация должна быть предотвращена
Химические агенты Аналоги оснований: 2-аминопурин, 5-бромурацил Ошибки в репликации ДНК Транзиции Низкая Слабый мутаген
Г идроксиламин Дезаминирование цитозина Транзиции Низкая Слабый мутаген
Азотистая кислота Дезаминирование цитозина и аденина Транзиции, делеции Средняя Высокая частота индукции мутаций достигается только в условиях низкой выживаемости
Нитрозогуаниди н Алкилирование оснований в репликацион-ной вилке Транзиции, трансверсии, с низкой частотой делеции Очень высокая Опасен в обращении, высокая частота вторичных мутаций
Этилметансульфо-нат Алкилирование гуанидина Транзиции, трансверсии Высокая Опасен в обращении, по сравнению с нитрозогуанидином — меньше вторичных мутаций
Акридиновые красители, бромистый этидий Интеркаляция между основаниями во время репликации ДНК Мутации со сдвигом рамки, небольшие вставки, делеции Низкая Эффективен при излечивании клеток от плазмид
Биологические агенты Гены-мутаторы Нарушение процессов репликации и репарации ДНК Транзиции, трансверсии Средняя Необходимо генетическое конструирование штаммов
Мутаген Механизм действия Тип мутации Эффективность Характерные особенности
Транспонирующиеся элементы (IS-элементы, транспозоны, фаг Ми и др.) Встраивание в днк Вставки, изредка делеции Очень высокая Необходимо генетическое конструирование штаммов и/или векторов
лом, поэтому при облучении суспензию вегетативных клеток или спор помещают в открытый сосуд, например чашку Петри.
Чтобы исключить эффект экранирования, следует использовать суспензии с концентрацией не выше 5 • 108 клеток/мл и проводить облучение в буферных растворах. Летальный эффект УФ-лучей зависит от физиологического состояния клеток, прежде всего, от их возраста. Обычно в фазе экспоненциального роста культуры клетки более чувствительны к УФ-лучам, чем в стационарной фазе. Кроме того, облучение и дальнейшую инкубацию клеток микроорганизма следует проводить в условиях, исключающих фотореактивацию, т.е. в темноте.
• Нитрозогуанидин. ТЧ-метил-М-нитро-Г<-нитрозогуанидин (НГ) является одним из самых мощных и широко применяемых мутагенов. Исходные растворы НГ готовят непосредственно перед использованием, растворяя его в соответствующем буфере (например, 0,1 М цитратном буфере, pH 5,5) до концентрации 1 мг/мл. Рабочая концентрация НГ составляет обычно 50 — 500 мкг/мл. Клетки (обычно из 5 мл культуры в экспоненциальной фазе роста) осаждают центрифугированием, промывают равным объемом цитратного буфера, ресуспендируют в 5 мл буфера, добавляют НГ и инкубируют при 30—37 °C в течение определенного времени. Время обработки подбирают предварительно таким образом, чтобы выживаемость клеток составляла примерно 50%. Клетки обрабатывают НГ в конечной концентрации 50— 100 мкг/мл в течение 30 мин. Обработанные клетки осаждают центрифугированием, промывают 0,1 М К-фосфат-ным буфером (pH 7,0), чтобы удалить НГ. После этого клетки переносят в богатую питательную среду и инкубируют в течение ночи (12— 16 ч).
• Азотистая кислота. Раствор азотистой кислоты готовят непосредственно перед использованием, растворяя нитрит натрия в 0,1 М Na-ацетатном буфере (pH 4,6) до конечной концентрации 0,05 М.
Клетки (обычно из 5 мл культуры в экспоненциальной фазе роста) осаждают центрифугированием, промывают равным объемом Na-ацетатного буфера, ресуспендируют в 1 мл раствора азотистой кислоты и инкубируют при 30 — 37 °C в течение определенного времени. Время обработки подбирают предварительно таким образом, чтобы выживаемость клеток составляла примерно 0,01 —0,1%. Клетки обрабатывают азотистой кислотой в течение 10 — 20 мин. После окончания инкубации к ним добавляют 5 — 10 мл минимальной среды, чтобы остановить реакцию. Затем клетки осаждают центрифугированием, ресуспендируют в 10 мл богатой питательной среды и выращивают в течение ночи.
• Алкилирующие агенты. Из алкилирующих соединений в качестве мутагена наиболее широко используют этилметансульфонат (ЭМС). Клетки (обычно из 5 мл культуры в поздней экспоненциальной фазе роста) осаждают центрифугированием и ресуспендируют в 2 мл минимальной питательной среды без источника углерода, содержащей 0,2 М mpuc-W2\ (pH 7,5). К суспензии клеток добавляют 0,03 мл ЭМС, энергично перемешивают до его полного растворения и инкубируют при 30 — 37 °C в течение 1 — 2 ч (желательно на качалке). Затем к суспензии клеток добавляют 10 — 15 мл минимальной или богатой питательной среды и инкубируют в течение ночи.
Меры предосторожности при работе с мутагенами. Физические и химические мутагены не только увеличивают частоту мутаций, многие из них обладают канцерогенным
действием, способным вызвать опухоли у млекопитающих. Кроме того, химические мутагены при попадании на кожу или внутрь могут привести к ожогам и отравлению.
При работе с мутагенами следует строго соблюдать следующие правила:
• Любую работу с химическими мутагенами необходимо проводить в вытяжном шкафу, рабочая поверхность которого застелена фильтровальной бумагой.
• Руки должны быть защищены резиновыми или пластиковыми перчатками.
• Растворы мутагенов нельзя засасывать ртом, следует использовать автоматические пипетки или пипетки с резиновой грушей.
• Растворы мутагенов нельзя сливать в раковину, лучше пропитать ими соответствующие твердые материалы (такие, как вермикулит) и поместить в герметичный контейнер, подобно другим опасным отходам.
• При работе с источниками УФ-лучей следует защищать глаза стеклянными очками.
Экспрессия мутаций. Для проявления мутации необходимо, чтобы произошла репликация ДНК и изменение закрепилось в дочерней молекуле. Поскольку большинство мутаций являются рецессивными, а клетка имеет в экспоненциальной фазе роста несколько копий хромосомы, то мутация проявится фенотипически через определенное время (сегрегационный лаг-период), в течение которого образуются клетки, содержащие только мутантные хромосомы (или ядра у эукариот). Для фенотипического проявления мутации необходимо также прохождение транскрипции и трансляции. Микроорганизмы существуют не в виде отдельных особей, а в виде популяций, поэтому для появления нового признака должно произойти, как правило, несколько клеточных делений (фенотипический лаг-период). Например, для проявления мутации устойчивости к фагу у Е. coli необходимо, чтобы все рецепторы клеточной стенки заменились на мутантные. Это происходит только после 12 дополнительных делений. Следовательно, суспензию клеток микроорганизма, обработанную мутагеном, необходимо подрастить в течение определенного периода времени. Длительность фенотипического лат-периода зависит от скорости роста культуры и от особенностей ее развития. При склонности клеток к образованию устойчивых агрегатов (нитей, гроздьев и т.д.) его длительность увеличивается.
Таким образом, после подращивания клеток не все мутанты, которые обнаруживаются в культуре, являются независимыми, так как часть мутантных клеток происходит от одной исходной мутантной клетки в результате ее размножения. Поэтому для получения ряда независимых мутантов культуру после обработки мутагеном необходимо разделить на ряд субкультур и из каждой субкультуры отбирать только по одному мутанту.
Отбор мутантов
Прямой отбор мутантов. Метод прямого отбора заключается в высеве обработанной мутагеном суспензии на селективные среды и позволяет сразу получать колонии только мутантных микроорганизмов, так как немутировавшие микроорганизмы на таких средах не вырастут. Метод прямого отбора обладает высокой чувствительностью и позволяет отбирать клетки определенного фенотипа, заданного условиями культивирования на селективной среде. В случае приобретения мутантами устойчивости к какому-либо веществу (антибиотику, токсину и т.д.) селективная среда должна содержать это вещество, подавляющее рост клеток дикого типа. Мутанты, способные использовать необычный источник питательных элементов (например, ксенобиотик), могут быть выделены на селективных средах, в состав которых не входит традиционный для микроорганизмов дикого типа источник этого элемента. Селективный агент добавляют в среду в оптимальной концентрации, подавляющей рост клеток дикого типа, но позволяющей расти устойчивым к данному агенту мутантам. Для этого предварительно определяют зависимость выживаемости микроорганизма от концентрации селективного агента. При отборе обратных мутантов (истинных или супрессорных), у которых про
изошло восстановление свойств дикого типа, например, от ауксотрофности по какому-либо фактору роста к прототрофности, необходимо использовать селективные среды, в которых этот фактор роста отсутствует. Высокая чувствительность метода позволяет выявлять редкие мутантные клетки на фоне немутировавших клеток.
Непрямой отбор мутантов. Для непрямой селекции мутантов используют высевы на индикаторные среды и метод реплик (или отпечатков).
• Использование индикаторных сред. Индикаторные среды, т. е. среды, на которых колонии микроорганизма с разными фенотипами отличаются по внешнему виду, широко используются в генетике микроорганизмов, в частности для непрямой селекции мутантов. Они позволяют различать выросшие колонии с разными свойствами по их внешним признакам, чаще всего по изменению окраски.
Индикаторные среды бывают двух типов: с индикатором на использование субстрата и с хромогенным субстратом. Первый тип содержит определенный субстрат, использование которого приводит к изменению pH, и индикатор, меняющий свой цвет в зависимости от кислотности среды. Наиболее распространены среды, содержащие какой-либо углевод (например, лактозу) и индикатор изменения кислотности среды (хлорид трифенилтетразолия, эозин-метиленовый синий, индикатор Мак-Конки и др.).
На среде с лактозой и трифенилтетразолием клетки Е. coli, использующие лактозу, образуют кислоты и понижают pH. При низких значениях pH восстановления трифенилтетразолия не происходит и колонии имеют нейтральный белый цвет. Колонии, состоящие из клеток, не способных использовать лактозу, имеют ярко-красную окраску. Напротив, на среде с индикатором Мак-Конки, содержащей лактозу, колонии, клетки которых способны использовать ее, приобретают темно-красный цвет. Клетки, не способные использовать лактозу, образуют колонии белого цвета.
Применение индикаторных сред пригодно для поиска любых мутантов, если имеется возможность различать колонии мутантов по изменению их окраски. С помощью индикаторных сред можно, например, выявлять мутанты по фосфатазной, нуклеазной или протеолитической активности. Важной особенностью метода индикаторных сред является подбор оптимального разведения культуры при посеве на агаризованные среды. Количество колоний на чашке не должно быть слишком большим (обычно не более 100), иначе фенотипы соседних колоний (например, из-за подкисления среды) могут быть выражены нечетко.
Второй тип индикаторных сред содержит специальные хромогенные .субстраты, несущие в своей молекуле группировку, освобождающуюся при гидролизе и придающую окраску колонии, клетки которой обладают активностью по отношению к такому субстрату. Хромогенные субстраты либо просто добавляют в среду, либо опрыскивают ими среды в чашках после того, как на них выросли колонии. В качестве хромогенного субстрата широко используют X-gal (5-бром-4-хлор-3-индолил-р-7)-галактозид), с помощью которого обнаруживают наличие р-галактозидазы, одного из ферментов, необходимых для утилизации лактозы. Клетки, синтезирующие р-галактозидазу, расщепляют X-gal с образованием 5-бром-4-хлор-индиго, который окрашивает колонии в ярко-синий цвет.
• Метод отпечатков, или реплик, — один из широко используемых способов выявления мутантов. Он заключается в получении отпечатков колоний, выросших на полноценной агаризованной среде при посеве обработанной мутагеном суспензии микроорганизмов, на чашках с селективными средами. Это позволяет выявить колонии, растущие на полноценной среде, но не растущие на какой-либо селективной среде, которые и проверяют на наличие мутаций.
Наиболее удобно использовать тканевый репликатор (рис. 35.1). На цилиндрическую болванку (5) натягивается бархатная ткань (/) и закрепляется металлическим кольцом (2). Прикасаясь стерильным репликатором к колониям в исходной чашке, получают отпечатки колоний на ткани (7). Затем прижимают репликатор к поверхности разных селективных сред последовательно в 10 чашках, при этом сохраняется расположение колоний относительно друг друга.
Рис. 35.1. Тканевый репликатор (см. текст)
Рис. 35.2. Игольчатый репликатор
При использовании, игольчатого репликатора (рис. 35.2) сначала делают отпечатки его циллиндрических иголок на поверхности стерильной агаризованной полноценной среды, не повреждая агар. Затем с помощью стерильных спичек или заостренных полосок толстой бумаги переносят на следы иголок клетки из колоний, выросших из суспензии культуры, обработанной мутагеном. Когда на следах иголок образуются колонии, с помощью того же репликатора делают отпечатки на ряд селективных сред. При отсутствии репликаторов можно осуществлять перенесение клеток из колоний, выросших при рассеве обработанной мутагеном культуры, стерильными спичками или заостренными полосками бумаги на агаризованные среды в чашках, дно которых расчерчено по трафарету на пронумерованные квадраты. Одной спичкой (полоской) прикасаются к нужной колонии, а затем последовательно к поверхности агара в чашках с селективной и полноценной средой в квадратах с одним номером. Этот способ, конечно, более трудоемкий и занимает больше времени, однако позволяет достичь аналогичного результата без применения специальных инструментов.
Пенициллиновый метод обогащения мутантов. Возникновение мутаций даже при их индукции сильными мутагенами — относительно редкое событие. При прямых методах селекции это обстоятельство не играет существенной роли, поскольку с помощью селективных сред элиминируются все немутантные клетки. Фактором, ограничивающим разрешающую способность непрямых методов отбора, является низкая частота возникновения мутаций (даже когда они индуцированы сильным мутагеном), так как поиск мутантов приходится проводить на значительном фоне колоний дикого типа. Для повышения доли мутантных клеток в популяции используют метод обогащения, элиминирующий значительное количество немутантных клеток.
При работе с бактериями чаще всего используют пенициллиновый метод. Как известно, этот антибиотик подавляет синтез пептидогликана клеточной стенки, что приводит к гибели активно растущих клеток. Если обработанную мутагеном культуру выращивать на минимальной среде, то размножаться и расти будут только клетки дикого типа (прототрофы). Именно они и подвергнутся воздействию внесенного в такую среду пенициллина и погибнут, а количество мутантных (ауксотрофных) нерастущих клеток не изменится. Таким образом, доля мутантов в популяции увеличится, т.е. произойдет обогащение популяции мутантными клетками. Для успешного проведения процедуры обогащения обрабатывать пенициллином следует культуру, проинкубированную в минимальной среде в течение 3—4 генераций для истощения у мутантных клеток эндогенных метаболитов, за счет которых возможны их рост в среде с пенициллином и обусловленная этим гибель. Обрабатываемая антибиотиком суспензия не должна быть густой (до 107 клеток/мл), так как продукты лизиса погибших от пенициллина клеток могут поддерживать рост мутантных клеток и тогда они также подвергнутся действию этого антибиотика. Если придерживаться оптимальных условий, то можно достичь 1000-кратного обогащения популяции мутантами.
Перенос генетической информации у бактерий
Наследственную изменчивость у прокариотических микроорганизмов вызывают рекомбинации генетического материала трех основных типов: трансформация, конъюгация и трансдукция, широко используемые в генетических экспериментах.
Трансформацией называют процесс переноса генетической информации, в результате которого экзогенная ДНК проникает в реципиентную клетку и вызывает ее наследственные изменения. При этом ДНК может представлять весь донорный геном либо его часть. Передачу в клетку целого генома фага, приводящую к образованию в ней зрелых фаговых частиц, называют трансфекцией. По способу переноса ДНК в бактериальную клетку этот процесс является трансформацией, однако в отличие от последней в результате трансфекции происходит лизис клетки, а не ее наследственное изменение.
Конъюгация — процесс переноса генетической информации из клетки-донора в клетку-реципиент, который осуществляется при их непосредственном контакте между собой.
Трансдукция — перенос генетической информации из клетки-донора в клетку-реципиент, осуществляемый фагом.
Общим свойством всех трех процессов является их однонаправленность, т. е. одна клетка всегда выступает в роли донора ДНК, а другая — в роли реципиента. Взаимного (реципрокного) обмена генетической информацией у бактерий не происходит.
В экспериментальной работе наиболее часто используют методы, основанные на процессах трансформации и конъюгации.
Т рансформация
Трансформация может осуществляться как хромосомной, так и плазмидной ДНК. Реципиентная клетка, в которой происходит экспрессия генетического материала донора, называется трансформантом. Важное условие способности клетки к трансформации — развитие у нее особого физиологического состояния — компетентности. По развитию состояния компетентности микроорганизмы можно разделить на три группы: 1) компетентность возникает лишь в определенной фазе роста культуры (например, стрептококки, бациллы); 2) клетки компетентны в любой фазе роста (например, гонококки, менингококки); 3) естественная компетентность отсутствует, и клетки становятся компетентными только после специальной обработки (например, клетки Е. coli становятся компетентными после обработки на холоду хлористым кальцием).
Посев трансформированных клеток на селективные среды необходимо производить после определенного времени их инкубации в богатой питательной среде. В период такой инкубации в клетках-трансформантах происходит экспрессия полученных ими новых маркеров (генов) и они приобретают новый фенотип.
У ряда микроорганизмов с естественной компетентностью внутрь клетки проникает только однонитевая ДНК, поэтому трансформация плазмидными ДНК, которые находятся в ковалентно-замкнутой суперскрученной форме, не осуществляется или осуществляется с низкой эффективностью. Чтобы увеличить эффективность трансформации, следует предварительно получить протопласты клеток.
При проведении трансформации у бактерий с естественной компетентностью необходимо учитывать факторы, влияющие на такое состояние: температуру, состав среды и ее pH, наличие определенных концентраций двухвалентных катионов.
В последнее время распространение получил метод трансформации, названный электропорацией. Электропорация — введение ДНК в клетки с помощью электрических импульсов, создаваемых специальной аппаратурой. Электропорация является перспективным методом, поскольку позволяет вводить ДНК в клетки микроорганизмов, для которых системы трансформации неизвестны или осуществляются с низкой эффективностью.
Для различных микроорганизмов, у которых описан процесс трансформации, разработаны свои методы получения компетентных клеток и их трансформации. Ниже приведена методика приготовления компетентных клеток Е. coli штамма С600 и их трансформации плазмидными ДНК с высокой эффективностью.
Выделение плазмидной ДНК из клеток Е. coli. Имеется большое количество методов выделения препаратов плазмидной ДНК, пригодной для трансформации клеток Е. coli без ее дальнейшей очистки. Одним из широко распространенных методов является метод выделения плазмидных ДНК с помощью лизиса клеток при кипячении. Модифицированный вариант указанного метода приведен ниже.
Клетки из ночной культуры Е. coli (1,5 — 5 мл) осаждают центрифугированием при 15 тыс. g в течение 3 мин в пробирках Эппендорф. Супернатант тщательно сливают, а осадок ресуспендируют для лизиса в 350 мкл STET-буфера (8 %-я сахароза, 5 %-й тритон Х-100, 50 мМ ЭДТА-№2, 50 мМ mpuc-HCV, pH 8,0). Добавляют 25 мкл раствора лизоцима в дистиллированной воде (10 мг/мл), перемешивают и инкубируют смесь при комнатной температуре в течение 5 мин. Пробирки переносят в кипящую водяную баню и инкубируют в течение 40 с. Затем полученные лизаты центрифугируют при 15 тыс. g в течение 30 мин и образовавшийся осадок (лизированные клетки, денатурированные белки и денатурированная хромосомная ДНК) удаляют чистой спичкой или переносят супернатант в чистую пробирку. В пробирку к супернатанту добавляют 174 мкл 7,5 М раствора ацетата аммония и 300 мкл изопропилового спирта, перемешивают и инкубируют при -20 °C в течение 15 мин. В этих условиях происходит преципитация ДНК, тогда как неденатурированные белки остаются в растворе. Преципитат осаждают центрифугированием при 15 тыс. g в течение 15 мин. Осадок дважды промывают 1,5 мл 70 %-го этанола, охлажденного до -20 °C (этанол следует аккуратно добавлять в пробирку, не перемешивая осадка; центрифугирование проводят, как указано выше, в течение 10 мин). Полученный осадок (беловатый налет на дне пробирки) высушивают струей воздуха (можно использовать фен) и растворяют в 50 мкл ТЕ-буфера (10 мМ mpuc-HCl, 1 мМ ЭДТА-Ка2, pH 7,5). При необходимости раствор ДНК следует хранить при -20 °C.
Получение компетентных клеток Е. coli. Ночную культуру Е. coli штамма С600, выращенную без перемешивания, разводят в 20 раз теплой средой LB (см. приложение 4) и инкубируют на качалке при 37 °C в течение 90—100 мин. Клетки осаждают центрифугированием на холоде (6 тыс. g, 10—15 мин), ресуспендируют в ’/2 объема культуры 10 мМ раствора хлористого кальция при 4 °C, осаждают ценрифугированием на холоду (6 тыс. g, 10—15 мин) и ресуспендируют в 100 мМ растворе хлористого кальция при 4 °C (’/^ исходного объема).
Приготовленные таким образом клетки Е. coli сохраняют компетентность при их инкубации во льду в течение суток. Компетентность утрачивается даже при незначительном и кратковременном повышении температуры. Для более длительного хранения к компетентным клеткам добавляют глицерин до конечной концентрации 15%, разливают аликвоты по 0,2 мл, замораживают и хранят при -20...-80°C.
Трансформация компетентных клеток Е. coli. К 0,2 мл компетентных клеток Е. coli добавляют 1 — 10 мкл плазмидной ДНК (1— 100 нг) и инкубируют во льду в течение 30 — 45 мин. Затем клетки подвергают тепловому шоку 5— 10 мин
при 42 °C. Добавляют к клеткам 0,5 мл среды LB и инкубируют с аэрацией при 37 °C в течение 2 ч. После инкубации высевают по 0,1 мл культуры на чашки с селективной средой и инкубируют в течение 16—24 ч.
Конъюгация
Процесс конъюгации у бактерий обусловлен наличием в клетках-донорах определенных плазмид, названных конъюгативными. Конъюгативные плазмиды содержат в своем составе tra-гены. Эти гены кодируют формирование половых волосков-пилей, за счет которых образуются скрещивающиеся (конъюгирующие) пары, обеспечивающие конъ-югативный перенос плазмидной или хромосомной ДНК в клетку-реципиент. Плазмиды, не способные к конъюгативному переносу, называются неконъюгативными. Первой описанной конъюгативной плазмидой, обеспечивающей конъюгацию у Е. coli К-12, является половой фактор F (fertility). Фактор F, существующий в автономном состоянии, способен с высокой эффективностью передаваться из клеток-доноров реципиентам. При интеграции полового фактора в хромосому Е. coll образуются штаммы Hfr (high frequency of recombination), способные переносить в реципиентные клетки хромосомную ДНК. Реципиентные клетки, в которых происходит экспрессия генетического материала донора, называются трансконъюгантами.
При проведении экспериментов по конъюгации необходимо учитывать следующее. 1. Перенос конъюгативной плазмиды и хромосомы донора в случае Hfr-штаммов начинается с определенной точки на плазмиде, называемой опТ. 2. Перенос хромосомы донора происходит ориентированно, т. е. гены переносятся в той последовательности, в которой они расположены на хромосоме. Начало переноса связано с местом интеграции конъюгативной плазмиды в хромосому донорного штамма и ее ориентацией. 3. Скорость переноса хромосомных маркеров зависит от температуры, но в стандартных условиях скрещивания ее величина постоянная. Каждый генетический детерминант донора попадает в реципиентную клетку через определенное время после начала скрещивания. 4. Перенос является частичным. Во время передачи хромосомы происходят ее спонтанные разрывы, поэтому возникающие зиготы неполные, т. е. содержат только часть генома донора (мерозиготы).
На использовании конъюгации основаны три метода генетического анализа у бактерий.
1. Определение частот рекомбинации между генетическими маркерами. Из-за очень высокой частоты кроссинговера маркеры, удаленные друг от друга на расстояние более 3 мин переноса, ведут себя как несцепленные. Данный метод обладает высокой разрешающей способностью при анализе тесно сцепленных генов и при внутривенном картировании.
2. Определение последовательности генов на хромосоме по градиенту их передачи.
3. Локализация генов по времени их проникновения в реципиентную клетку — картирование методом прерывания конъюгации.
Мобилизация неконъюгативных плазмид. Определенные неконъюгативные плазмиды способны передаваться в реципиентные клетки в присутствии определенных конъ-югативных плазмид. Данное явление получило название мобилизации неконъюгативных плазмид. Известны два основных механизма этого процесса. Во-первых, перенос неконъюгативных плазмид осуществляется за счет продуктов tra-генов конъюгатив-ных плазмид. Перенос по первому типу требует функционирования собственных генов mob и начинается с собственного сайта ori Т. По такому механизму происходит, например, мобилизация плазмид ColEl и RSF1010. При этом механизме мобилизации перенос конъюгативной плазмиды в реципиентную клетку может и не происходить. Во-вторых, перенос неконъюгативных плазмид в реципиентные клетки может осуществляться вместе с конъюгативными плазмидами. В этом случае они составляют единую молекулу ДНК, объединяющую два или более репликонов. По такому меха
низму, например, происходит мобилизация широко используемых векторных плазмид серии pBR.
Интеграция полового фактора в хромосому, приводящая к образованию Hfr-штам-мов, также является примером формирования коинтегратов. В данном случае в единую молекулу ДНК объединены два репликона — плазмида (половой фактор) и хромосома.
Мобилизация неконъюгативных плазмид широко используется в практике генетических экспериментов. Этот метод менее трудоемок, чем трансформация, и, кроме того, позволяет вводить неконъюгативные плазмиды в клетки микроорганизмов, для которых не разработаны системы трансформации.
Для мобилизации неконъюгативных плазмид можно использовать трехродительские скрещивания, когда мобилизуемая и мобилизующая плазмиды находятся в разных донорных штаммах. Трехродительские скрещивания обладают рядом преимуществ по сравнению с двуродительскими. Прежде всего отпадает необходимость конструировать донорный штамм, содержащий две плазмиды. Кроме того, трехродительские скрещивания позволяют преодолеть барьер несовместимости, когда мобилизуемая и мобилизующая плазмиды относятся к одной группе несовместимости.
Постановка скрещивания. Существует несколько способов скрещивания бактериальных штаммов при помощи конъюгации: в жидкой среде, на поверхности питательного агара, на мембранных фильтрах. Выбор способа скрещивания определяется типом конъюгативной плазмиды и целью эксперимента.
Самым простым способом является скрещивание в жидкой среде. В этих условиях с высокой эффективностью происходит скрещивание, обусловленное половым фактором F и родственными ему плазмидами. Однако этот метод имеет ограниченное применение, поскольку многие плазмиды детерминируют образование коротких пилей (например, плазмиды P-группы несовместимости), которые в условиях жидкой среды не обеспечивают тесного контакта между клетками. Для создания такого контакта скрещивание проводят непосредственно в чашке с агаризованной питательной средой или на мембранном фильтре, помещенном на поверхность агаризованной питательной среды. Последний способ более удобен в случае, когда необходимо провести одновременно несколько скрещиваний. Для скрещивания используют мембранные фильтры, задерживающие микроорганизмы, т.е. с диаметром пор 0,45 или 0,22 мкм.
Детальное описание основанных на конъюгации экспериментов для генетического анализа у бактерий можно найти в специальной литературе. Ниже приведена общая методика постановки конъюгации на примере мобилизации неконъюгативной плазмиды RSF1010 конъюгативной плазмидой RP1.
Родительские штаммы микроорганизмов, используемые в скрещивании, должны различаться между собой генетическими маркерами, например маркерами ауксотрофности или устойчивости к антибиотикам. В случае Е. coli одним штаммом может быть, например, С600 (thr-, loir, thi-), а другим — J53 (pro-, met-). Допустим, что один из штаммов С600 содержит конъюгативную плазмиду RP1 (кодирующую устойчивость к ампициллину, тетрациклину и канамицину), а второй штамм С600 содержит неконъюгативную плазмиду RSF1010 (кодирующую устойчивость к сульфаниламидам и стрептомицину), которую нужно передать в штамм J53.
Для скрещивания используют молодые, активно растущие культуры, причем концентрация реципиентных клеток должна значительно (примерно в 10 раз) превышать концентрацию донорных клеток. С этой целью ночные культуры
всех трех штаммов Е. coli (выращивать можно без аэрации) разводят примерно в 5 раз теплой средой LB и подращивают в течение 90—100 мин. Затем в пробирке (удобно использовать пробирки Эппендорф) смешивают по 20 мкл донорных культур, т.е. штаммов с плазмидами RP1 и RSF1010, и 0,1 мл культуры реципиента. Растирают смесь шпателем по поверхности агаризованной среды LB и чашку инкубируют при 37 °C в течение 3 ч. По окончании скрещивания клетки смывают шпателем с поверхности агара 2 мл физраствора и делают десятикратные разведения конъюгационной смеси в физрастворе до 10“5. Затем пробы по 0,1 мл из разведений 10“3 и 10~5 высевают на чашки с селективными средами. Одна чашка должна содержать селективную среду с одним из антибиотиков для отбора трансконъюгантов, получивших плазмиду RP1 (канамицин — 50 мкг/мл, тетрациклин — 20 мкг/мл, ампициллин — 100 мкг/мл), другая — со стрептомицином (50 мкг/мл) для отбора трансконъюгантов, получивших плазмиду RSF1010. В качестве основы для селективной среды используют минимальную среду А (с добавками пролина и метионина), на которой могут расти только клетки штамма J53. В качестве контроля на селективные чашки засевают по 0,1 мл культур родительских штаммов, чтобы убедиться в неспособности клеток этих штаммов образовывать колонии на селективных средах. Засеянные чашки помещают в термостат при 37 °C на 48 ч. По окончании инкубации проводят подсчет трансконъюгантов в чашках с селективными средами. Частота мобилизации неконъюгативной плазмиды RSF1010 определяется как отношение количества трансконъюгантов, получивших маркер неконъюгативной плазмиды, к количеству трансконъюгантов, получивших маркер конъюгативной плазмиды.
ЗАДАЧА 1. ЛЕТАЛЬНОЕ И МУТАГЕННОЕ ДЕЙСТВИЕ УЛЬТРАФИОЛЕТОВЫХ ЛУЧЕЙ (УФЛ)
НА КЛЕТКИ ESCHERICHIA COLI
Ультрафиолет в зависимости от длины волны и дозы может вызывать как летальный, так и мутагенный эффект. При этом прежде всего повреждаются молекулы ДНК. В них возникают тиминовые димеры, ингибирующие репликацию. Повреждения могут быть устранены двумя путями: фотореактивацией и темновой репарацией. В первом случае поврежденное место репарируется ферментом, активируемым синим (видимым) светом, который исправляет структуру ДНК, расщепляя связи между тиминовыми основаниями в димерах. Во втором случае свет не нужен, повреждения устраняются с помощью ферментов: эндонуклеазы (вырезает поврежденный участок), полимеразы (синтезирует правильную структуру по комплементарной цепи) и лигазы (сшивает синтезированные последовательности).
Ультрафиолет с X = 325 — 400 нм также вреден для микроорганизмов, поскольку наряду с образованием тиминовых димеров разрушается триптофан и образуются его токсичные фотопродукты, которые действуют как химические мутагены. Наибольший эффект УФЛ наблюдается при длине волны 260 нм, совпадающей с максимумом поглощения ДНК клеток. Выживаемость клеток экспоненциально зависит от дозы УФЛ.
Облучать клетки следует в буферных растворах, так как вещества питательного бульона интенсивно поглощают УФЛ, снижая тем самым дозу, получаемую клетками. В то же время образующиеся в бульоне под действием облучения токсичные пере-
киси увеличивают летальное действие УФЛ на клетки. Все манипуляции следует осуществлять в условиях, исключающих воздействие видимого света, чтобы процесс фотореактивации не исказил результаты.
Для характеристики мутагенного действия УФЛ часто используют обратные мутации. Несмотря на то что часть реверсий обусловлена супрессорными мутациями, этот тест удобен, так как позволяет использовать прямые методы отбора мутантов с помощью селективных сред и таким образом увеличить размер анализируемой популяции. Концентрацию и частоту обратных мутаций по одному гену при этом выражают в числе мутантов на определенное количество жизнеспособных клеток (например, 1 106).
Цель работы: изучение летального и мутагенного действия УФЛ на клетки Е. coli и определение зависимости выживаемости клеток и частоты мутаций по каждому гену от дозы облучения.
Ход выполнения задачи
Микроорганизм. В работе используют штамм Е. coli АВ 1157 thr-, leu-, argE-, proA-, thi-, lac-, gal-, ara, xyl, str-r, t°-s, нуждающийся в треонине, лейцине, аргинине, пролине и тиамине (В]), а также лактозо- и галактозоотрицательный, способный использовать арабинозу и ксилозу, стрептомицинустойчи-вый и температурочувствительный.
Среды и растворы. В качестве полноценной среды применяют мясопептонный бульон (МПБ) с pH 7,0. Для приготовления твердой среды к МПБ добавляют 2% агара (МПА). Среду стерилизуют при 1,0 ати. После стерилизации вносят 2 % глюкозы в виде 40 %-го раствора, простерилизованного при 0,5 ати.
В качестве минимальной применяют среду Гловера следующего состава (г/л): NH4C1 - 5,0; NH4NO3 - 1,0; Na2SO4 _ 2,0; К2НРО4 - 3,0; КН2РО4 - 1,0; MgSO4• 7Н2О — 0,1; pH 7,0. Для получения твердой среды добавляют 2 % агара. Среду стерилизуют при 1,0 ати. После стерилизации вносят 1,5 —2 % глюкозы в виде 40 %-го раствора, простерилизованного при 0,5 ати.
Для выявления обратных мутаций по локусам thr и leu используют селективные среды LAPB] и ТАРЕ, соответственно. Селективную среду LAPB] получают добавлением лейцина, аргинина, пролина и тиамина в минимальную среду Гловера, а среду ТАРЕ, — добавлением треонина, аргинина, пролина и тиамина. Аминокислоты и витамин стерилизуют отдельно в виде концентрированных растворов при 0,5 ати и вносят в стерильные среды до конечных концентраций 100 мг/л и 10 мг/л соответственно.
Для отмывки и ресуспендирования клеток, а также для приготовления десятичных разведений суспензии готовят 50 мМ К-фосфатный буфер, pH 7,0.
Общая схема эксперимента приведена на рис. 35.3.
Для получения исходной суспензии клеток Е. coli проводят посев культуры со скошенной агаризованной полноценной среды в 50 мл МПБ в круглодонных колбах на 250 мл (1 полную петлю). Колбы помещают на круговую качалку и инкубируют 18 ч при 37 °C и 200 об/мин. Выросшую культуру (15 мл суспензии клеток на 1 студента) стерильно центрифугируют 10—15 мин при 6 тыс. g, предварительно пометив уровень жидкости в попарно уравновешенных вместе с пробками центрифужных стаканах. После осаждения клеток надосадочную жидкость сливают в емкость с дезраствором, а осадок перемешивают стерильной стеклянной палочкой до гомогенного состояния и заливают 50 мМ К-фосфатным
буфером (pH 7,0) до прежнего уровня и снова центрифугируют в том же режиме. Осажденные клетки ресуспендируют вышеприведенным способом в буфере и содержимое обоих центрифужных стаканов сливают в стерильную пробирку. Полученная суспензия должна содержать примерно 2 109 клеток в мл. Суспензию разливают по 3 мл в стерильные чашки Петри по числу вариантов опыта (контрольный вариант остается в пробирке). Все варианты опыта следует оберегать от попадания света.
Суспензию бактерий облучают с помощью установки, состоящей из двух ламп БУФ-15, смонтированных параллельно на специальном стенде. Используются дозы в 1, 2, 3 и 4 тыс. эрг/мм2, которым на установке соответствуют экспозиции в 8, 16, 24 и 32 с на расстоянии 5 см от ламп. Облучение бактерий проводят в открытых чашках Петри (диаметр 100—110 мм). Для каждой дозы
Посев со скошенного МПА (одна полная
Рис. 35.3. Схема опыта по определению действия ультрафиолета на клетки Е coli
используют отдельную чашку с 3 мл суспензии (вариант опыта). Во время облучения чашку постоянно покачивают для перемешивания суспензии. Время контролируют по секундомеру. После облучения чашки закрывают крышками и помещают в темное место.
Для определения количества жизнеспособных клеток из облученных и контрольной суспензий берут по 0,5 мл и делают ряд десятичных разведений в 50 мМ К-фосфатном буфере, pH 7,0. Конечное разведение каждого варианта опыта зависит от дозы облучения:
Вариант опыта Экспозиция, с Конечное разведение
К 0 ю-8
1 8 ю-7
2 16 io-6
3 24 10-5
4 32 IO4
Из пяти последних разведений каждого варианта отбирают микропипеткой по 0,1 мл, вносят на поверхность двух чашек Петри с МПА и тщательно растирают стеклянными шпателями. Чашки помещают крышками вниз в термостат (37 °C) на 24 ч (возможно выращивание при 30 °C, но более длительное время).
Для определения концентрации обратных мутантов по 0,1 мл суспензии каждого варианта высевают без разведения на селективные среды (5 чашек со средой LAPP, и 5 чашек со средой ТАРВ,). Засеянные чашки с селективными средами помещают крышками вниз в термостат (37 °C) на 48 ч.
По истечении времени инкубации подсчитывают число колоний на чашках. При подсчете колоний на МПА выбирают чашки, Где выросло не более 100 колоний (наиболее удачное разведение суспензии), рассчитывают количество жизнеспособных клеток в 1 мл суспензии каждого варианта и выражают выживаемость облученных УФЛ клеток в процентах от контроля. Данные вносят в табл. 35.2.
Вычерчивают кривые выживаемости в арифметической и логарифмической шкалах в зависимости от дозы облучения.
Подсчет колоний на селективных средах (число жизнеспособных клеток в этих вариантах уже известен: см. 4-й столбец табл. 35.2) позволяет определить
Таблица 35.2
Летальное действие УФЛ на клетки Е. coli АВ 1157
Вариант опыта Разведение Количество колоний на чашке Число жизнеспособных клеток в 1 мл суспензии Выживаемость, %
К 100
1
2
3
4
Мутагенное действие УФЛ на клетки Е. coli АВ 1157
Вариант опыта Число жизнеспособных клеток в 1 мл суспензии* Количество жизнеспособных клеток в 1 мл суспензии на средах Концентрация обратных мутантов на 108 клеток по локусам
LAP В, ТАРВ, thr leu
к
1
2
3
4
* Данные берутся из табл. 35.2.
концентрацию обратных мутаций по локусам thr и leu в разных вариантах опыта. Данные вносят в табл. 35.3.
Для каждого локуса строят кривую зависимости частоты мутаций от дозы облучения.
После обработки каждым студентом собственных результатов определяется средняя по всей группе выживаемость и мутабильность по двум локусам для каждой дозы УФЛ. Результаты каждого студента принимаются за повторность опыта всей группы.
Материалы, оборудование, посуда, реактивы
Центрифуга; источник УФЛ; секундомер; термостат на 37 °C; счетчик колоний; микробиологическая петля. Чашки Петри — ПО; пробирки с ватными пробками — 35; стеклянные палочки — 2; стеклянные центрифужные стаканчики с пробками — 2; стеклянные шпатели — 35; пипетки на 1 мл — 30; на 0,1 мл — 30; на 5 мл — 5; на 10 мл — 3. МПБ — 50 мл; МПА — 1 л; селективные среды — по 0,5 л; 50 мМ К-фосфатный буфер (pH 7,0) — 0,2 л.
План проведения задачи
Занятие 1. Подготовка и стерилизация посуды и сред (4 ч). Посев бактерий в МПБ и их выращивание (18 ч).
Занятие 2. Приготовление суспензии клеток бактерий. .Облучение, приготовление разведений и посев бактерий на МПА и селективные среды (4 ч). Инкубация посевов (24 и 48 ч).
Занятие 3. Подсчет колоний, выросших на МПА и селективных средах. Оформление результатов (4 ч).
ЗАДАЧА 2. ВЫДЕЛЕНИЕ И ИДЕНТИФИКАЦИЯ АУКСОТРОФНЫХ МУТАНТОВ ESCHERICHIA COLI
Термин «биохимические мутанты» обычно используют для обозначения мутантов с измененными метаболическими процессами.
Среди биохимических мутантов наиболее изучены ауксотрофные мутанты, которые утратили способность к синтезу необходимых для их роста соединений (аминокис-
лот, витаминов, азотистых оснований и т.д.) в отличие от прототрофных микроорганизмов, способных к такому синтезу. Для развития ауксотрофного организма в питательную среду необходимо добавлять дополнительные факторы роста.
Частота спонтанного возникновения мутаций любого типа обычно не превышает 10~3. Для ее повышения применяют различные мутагенные факторы, наиболее эффективные из них нитрозосоединения. При обработке популяции клеток такими химическими мутагенами удается получить до 10 —20 % биохимических мутантов.
Мутагенный эффект химических соединений, как правило, связан с возникновением точечных мутаций, в то время как физические мутагены вызывают в большей степени аберрации и поломки хромосом. При использовании химических мутагенов большое значение имеют условия обработки, концентрация вещества, продолжительность экспозиции, pH среды и др. При проведении эксперимента необходимо учитывать задержку в проявлении мутантных микроорганизмов, которая связана с сегрегационным и фенотипическим лаг-периодами.
Выделение мутантов определенного фенотипа или мутантов по одному гену из популяции прототрофов довольно трудоемко, так как их содержание даже после обработки химическим мутагеном очень мало. В этих случаях применяют специальные методы обогащения для повышения относительного количества ауксотрофных мутантов в популяции прототрофов.
Пель работы', получение и идентификация ауксотрофных мутантов Е. coli под действием нитрозосоединений с последующей обработкой пенициллином.
Ход выполнения задачи
Микроорганизм. В работе используют один из штаммов Е. coli К-12 thr (Hfr 5, 109 или 85).
Среды и растворы. В качестве полноценной среды применяют МПБ и МПА, pH 7,0; в качестве минимальной применяют среду Гловера (как в предыдущей задаче).
Для приготовления селективных сред в минимальную среду Гловера вносят необходимые соединения в следующих количествах (мг/л): DL-аминокислоты — 100, L-аминокислоты — 50, витамины — 10, азотистые основания и нуклеозиды — 20. Растворы этих веществ готовят на дистиллированной воде и стерилизуют при 0,5 ати или пропусканием через бактериальный фильтр.
Для приготовления десятичных разведений суспензии перед посевами и раствора пенициллина готовят 50 мМ К-фосфатный буфер, pH 7,0.
При использовании в качестве химического мутагена нитрозоэтилмочеви-ны (НЭМ) для ресуспендирования клеток и приготовления раствора мутагена готовят 50 мМ К-фосфатный буфер, pH 6,0 (Точное значение! Проверять по рН-метру!).
Если для индуцированного мутагенеза применяют нитрозогуанидин (НГ), то готовят 50 мМ цитратный буфер, pH 5,5 (Точное значение!Проверять порН-метру!).
Общая схема эксперимента приведена на рис. 35.4.
Культуру Е. coli выращивают на МПБ в колбах на качалке (200 об/мин) в течение 18 ч при 37 °C. Плотность культуры должна соответствовать 2-109 кле-ток/мл. Выросшую культуру, взятую в количестве 2 мл, стерильно центрифугируют при 6 тыс. g в течение 15 мин, бульон сливают в дезраствор, осадок тща-тельно растирают стерильной стеклянной палочкой и ресуспендируют в исход-
Посев Е. coli с агаризованной среды (1 полная петля)
Центрифугирование 15 мин при 6 тыс.^ и ресуспендирование в 2 мл К-фосфат-ного буфера, pH 6,0 в случае НЭМ или цитрат -
при 200 об/мин и 37 °C 2 мл суспензии на одного студента случае НГ 2 мл суспензии
Круглодонная колба на
250 мл с 50 мл МПБ
1 мл суспензии + 1 мл 2%-го раствора НЭМ в К-фосфатном буфере, pH 6,0, или 0,02%-го раствора НГ в цитратном буфере, pH 5,5
1 мл суспензии + 1 мл К-фосфатного буфера, pH 6,0, (НЭМ) или цитратного буфера, pH 5,5 (НГ) (контроль)
Инкубация 30 мин при 37 °C на качалке
0,5 мл для приготовления разведений в К-фосфатнрм буфере, pH 7,0, до 10~°
0,5 мл для приготовления разведений в К-фосфатном буфере, pH 7,0, до 10"7
0,05 мл из
Г~ разведения 10~2
Посев из разведений по 0,1 мл в чашки с МПА
Широкая пробирка с 5 мл МПБ
Из всех разведений в двух повторностях
Вариант 2
Инкубация па качалке 24 ч
Рис. 35.4. Схема опыта по получению ауксотрофных мутантов
ном объеме буфера. При работе с НЭМ в качестве мутагена используют 50 мМ К-фосфатный буфер, pH 6,0, а для НГ применяют 50 мМ цитратный буфер, pH 5,5.
Полученную суспензию делят на две порции: 1 мл смешивают с 1 мл раствора мутагена (2 %-го раствора НЭМ в 50 мМ К-фосфатном буфере, pH 6,0 или 0,02%-го раствора НГ в 50 мМ цитратном буфере, pH 5,5), а оставшийся 1 мл суспензии — с 1 мл соответствующего буфера без мутагена (контроль). Обе порции инкубируют на качалке 30 мин при 37°С. Затем делают десятичные разведения смесей в 50 мМ К-фосфатном буфере, но уже pH 7,0. Начинать следует с варианта, обработанного мутагеном, что позволяет остановить его действие. Вариант с мутагеном разводят до 10“6 и производят высевы из исходной суспензии и из всех разведений по 0,1 мл на чашки с МПА в двух повторностях. Эту серию посевов обозначают как вариант 2. Контрольный вариант разводят до 10“7 и высевают по 0,1 мл из четырех последних разведений на чашки с МПА в двух повторностях. Эту серию посевов обозначают как вариант 1. Все засеянные чашки с МПА инкубируют 24 ч при 37 °C (или 48 ч при 30 °C), а затем хранят в холодильнике до использования.
Из второго разведения 0,05 мл обработанной мутагеном суспензии переносят в пробирку с 5 мл МПБ для подращивания и помещают на качалку на 24 ч при 37 °C. Затем 0,05 мл выросшей культуры переносят в 5 мл теплой среды Гловера с тиамином и глюкозой и выдерживают на качалке 3 ч при 37 °C для истощения эндогенных запасов питательных веществ в клетках. После инкубации из пробирки отбирают 0,5 мл культуры для приготовления десятичных разведений до 1 СТ6 в К-фосфатном буфере, pH 7,0. Из четырех последних раз-ведений высевают по 0,1 мл на чашки с МПА в двух повторностях и инкубируют 24 ч при 37 °C (или 48 ч при 30 °C), а затем хранят в холодильнике до использования. Эту серию посевов обозначают как вариант 3.
К оставшимся в пробирке со средой Гловера 4,5 мл суспензии приливают 0,5 мл раствора пенициллина в том же буфере, так чтобы конечная концентрация антибиотика составила 2000 ед./мл. Смесь оставляют на качалке при 37 °C на 1 ч, а затем готовят разведения до 10~5 и высевают из них по 0,1 мл на чашки с МПА в двух повторностях. Все засеянные чашки с МПА инкубируют 24 ч при 37 °C (или 48 ч при 30 °C), а затем хранят в холодильнике до использования. Эту серию посевов обозначают как вариант 4.
Таблица 35.4
Определение летального эффекта нитрозоэтилмочевины (или нитрозогуанидина) и пенициллина
Вариант опыта Разведение Количество колоний на чашках Количество жизнеспособных клеток в 1 мл суспензии Выживаемость, %
ЦК) 100
2
3 100
4
Таблица 35.5
Определение количества ауксотрофных мутантов в разных вариантах опыта (количество отсеянных колоний равно 100)
Вариант опыта Количество ауксотрофных колоний
абсолютное %
ЦК)
2
3
4
[-* 50 колоний
1 чашка со средой Гловера
Чашки с МПА
100 колоний —
----------- Г"~ Гловера 50 колоний — ,___________
1 чашка со средой
Рис. 35.5. Схема рассева колоний из каждого варианта для определения ауксотрофов
В каждой серии посевов отбирают чашки из наиболее удачных разведений и делают подсчет колоний для определения числа жизнеспособных клеток в 1 мл суспензии каждого варианта. Подсчет ведут, не раскрывая чашек, поскольку в дальнейшем из них проводится отсев колоний. Результаты вносят в табл. 35.4, определяя летальные эффекты мутагена (при сравнении вариантов 7 и 2) и пенициллина (при сравнении вариантов 3 и 4).
Для определения ауксотрофных колоний проверяют способность к росту на минимальной среде у 100 колоний из каждого варианта опыта (рис. 35.5). Для этого готовят восемь пар чашек: с полноценной (МПА) и минимальной (среда Гловера с тиамином и глюкозой) средами. Наружную сторону дна каждой чашки расчерчивают на 50 пронумерованных квадратов. Затем каждую из 100 колоний высевают на две чашки (с минимальной и полноценной средой) в квадраты с одинаковыми номерами. Это удобно делать кончиками стерильных спичек или заостренными концами полосок плотной бумаги. Для переноса каждой колонии используют свою спичку или полоску — концом спички прикасаются к выбранной колонии, затем к поверхности агара в соответствующем квадрате чашки с минимальной средой, а затем к поверхности МПА в квадрате с таким же номером.
Засеянные чашки инкубируют 24 ч при 37 °C. В парных чашках сравнивают рост колоний в квадратах с одинаковыми номерами {чашки с МПА не открывать!). Колонии ауксотрофов распознают по отсутствию роста на минимальной среде. Результаты подсчета вносят в табл. 35.5.
Для идентификации ауксотрофов отбирают 10 колоний (желательно из варианта 2) и каждую высевают штрихом на один сектор в каждой из 12 чашек (рис. 35.6).
Каждая чашка содержит минимальную среду Гловера с одной из комбинаций ростовых веществ. Комбинации ростовых веществ представлены в табл. 35.6.
Колонии мутантов обычно малы, поэтому колонию, снятую с МПА, суспендируют в небольшом количестве буфера, и для посева на каждой из 12 чашек с комбинациями делают штрих зеркальцем петли, опущенной в эту суспензию. Засеянные чашки инкубируют 24—48 ч при 37 °C.
Рис. 35.6. Схема посева 10 ауксотрофов на чашку со средой Гловера и одной из комбинаций ростовых веществ
Комбинация ростовых веществ для идентификации ауксотрофов
Номер комбинации 1 2 3 4 5 6
7 Аденин Биотин Фенилаланин Аланин Аргинин Лейцин
8 Гипоксантин Фолиевая кислота Серин Цистеин Орнитин Глицин
9 Цитозин Пантотеновая кислота Триптофан Треонин Аспарагиновая кислота Изолейцин
10 Гуанин Пиридоксин Тирозин Тиосульфат натрия Пролин Гистидин
11 Тимин Тиамин Пара-Амтю-бензойная кислота Метионин Глутаминовая кислота Лизин
12 Урацил Рибофлавин Никотиновая кислота Холин Инозит Валин
При просмотре результатов посева отмечают, на каких комбинациях веществ растет данный мутант. Специфические потребности выделенных мутантов определяю!' следующим образом: если мутант растет только на двух каких-либо комбинациях веществ, то он нуждается в соединении, которое входит в состав обеих комбинаций. Например, если мутант дает рост на среде с комбинациями № 3 и № 9, то он нуждается в триптофане. Однако следует учитывать, что мутанты могут быть дефектны не по одному, а по нескольким метаболитам (мутант будет расти на одной комбинации или более чем на двух комбинациях, или на нескольких комбинациях, не имеющих общих компонентов).
Материалы, оборудование, посуда, реактивы
Центрифуга, счетчик колоний, микробиологическая петля. Чашки Петри — 75; пробирки обычные с ватными пробками — 30; пробирки широкие — 2; стеклянные палочки — 2; стеклянные центрифужные стаканчики с пробками — 2; стеклянные шпатели — 5; пипетки на 1 мл — 30; на 0,1 мл — 5; на 5 мл — 5; спички без серных головок — 400. МПБ — 100 мл; МПА — 1 л; минимальная среда Гловера с глюкозой и тиамином — 5 мл; агаризованная минимальная среда Гловера с глюкозой и тиамином — 200 мл; селективные среды с разными комбинациями ростовых веществ — по 30 мл; 50 мМ К-фосфатный буфер (pH 7,0) — 0,2 л; 50 мМ К-фосфатный буфер (pH 6,0) или 50 мМ цитратный буфер (pH 5,5) — 10 мл; раствор мутагена — 1 мл; раствор пенициллина — 0,5 мл.
План проведения задачи
Занятие 1. Приготовление и стерилизация сред, растворов и посуды (4 ч). Посев и инкубация бактериальной культуры (18 ч).
Занятие 2. Обработка бактериальных клеток мутагеном. Приготовление десятичных разведений и посев на чашки с МПА. Посев обработанной мутагеном культуры в МПБ на подращивание (4 ч). Инкубация засеянных чашек и пробирок (24 ч).
Занятие 3. Посев обработанной мутагеном культуры после роста на МПБ на минимальную среду Гловера и инкубация для истощения эндогенных запасов питательных веществ (3 ч). Обработка клеток пенициллином (1 ч). Приготовление десятичных разве-
дений культуры со среды Гловера и после пенициллинового обогащения и посев на чашки с МПА (1ч). Инкубация засеянных чашек (24 ч).
Занятие 4. Подсчет колоний на чашках МПА во всех вариантах опыта и определение летального действия мутагена и пенициллина (1 ч). Пересев колоний из каждого варианта опыта на пары чашек с полноценной и минимальной средами для выявления ауксотрофных мутантов (3 ч). Инкубация засеянных чашек (24 ч).
Занятие 5. Определение содержания ауксотрофных мутантов в каждом варианте опыта. Пересев мутантов на селективные среды с комбинациями ростовых веществ (4 ч). Инкубация засеянных чашек (24—48 ч).
Занятие 6. Идентификация ауксотрофных мутантов и оформление результатов (4 ч).
ЗАДАЧА 3. КОНЪЮГАЦИЯ У ESCHERICHIA COLI.
КАРТИРОВАНИЕ ГЕНОМА ПО ГРАДИЕНТУ ПЕРЕДАЧИ МАРКЕРОВ
Конъюгация предполагает непосредственный контакт клетки-донора и клетки-реципиента. Клетка-донор должна обладать плазмидой, или F-фактором, который может быть автономен или интегрирован в хромосому. F-фактор обусловливает способность донорной клетки вступать в контакт с реципиентом, формировать половые F-пили и передавать генетический материал. При интеграции F-фактора в хромосому такая передача осуществляется с высокой частотой (штаммы Hfr). Перенос генетического материала строго ориентирован: разрыв копии хромосомы и передача ДНК происходит, начиная с локуса ori в пределах полового фактора. Скорость переноса в одинаковых условиях для определенного штамма постоянна. Обычно всей хромосоме не удается перейти в клетку-реципиент, так как контакт клеток очень нестабилен и часто прерывается до завершения перехода. Реципиенту первым передается всегда один и тот же, специфичный для каждого штамма, участок хромосомы, поэтому частота передачи стоящих следом за ним генов позволяет расположить их по отношению к этому локусу и составить генетическую карту хромосомы.
Цель работы', определить порядок расположения четырех локусов на хромосоме Е. coli К-12 по градиенту их передачи рекомбинантам.
Ход выполнения задачи
Микроорганизмы. В скрещивании используются штаммы Escherichia coli К-12, различающиеся по резистентности к стрептомицину и зависимости от ростовых факторов: в качестве донора — Hfr Н thi- str-s (тиаминзависимый, стрептомицинчувствительный), в качестве реципиента — J-62 thi-, F-, pro', try-, his-, lac-, str-r (тиамин-, пролин-, триптофан- и гистидинзависимый, лактозоотрицательный, стрептомицинустойчивый).
Среды и растворы. В качестве полноценной среды применяют МПБ и МПА, pH 7,0, а в качестве минимальной — среду Гловера (как в предыдущих задачах).
Для отбора рекомбинантов применяют следующие агаризованные селективные среды'. SBi TH + глюкоза — среда со стрептомицином, тиамином, триптофаном, гистидином и глюкозой; ВВЦН + глюкоза — среда со стрептомицином, тиамином, пролином, гистидином и глюкозой; SB{ ТР + глюкоза — среда со стрептомицином, тиамином, пролином, триптофаном и глюкозой; SB{ ТРИ + лактоза — среда со стрептомицином, тиамином, пролином, триптофаном, гистидином
и лактозой. Для приготовления селективных сред в минимальную среду Гловера вносят необходимые соединения в следующих количествах (мг/л): DL-аминокислоты — 100; Z-аминокислоты — 50; витамин — 10. Растворы этих веществ готовят на дистиллированной воде и стерилизуют при 0,5 ати или пропусканием через бактериальный фильтр. Лактозу вносят в той же концентрации и тем же способом, что и глюкозу. Стрептомицин добавляют из его концентрированного раствора в К-фосфатном буфере до конечной концентрации 100 мг/мл.
Для отмывки и ресуспендирования клеток, а также для приготовления десятичных разведений суспензии готовят 50 мМ К-фосфатный буфер, pH 7,0.
Культуры донорного и реципиентного штаммов выращивают таким образом, чтобы к началу скрещивания иметь молодые клетки. Для этого петлей снимают одну колонию каждого штамма и помещают их в колбы объемом 250 мл с 50 мл МПБ. Колбы инкубируют в течение ночи (—18 ч) на качалке при 200 об/мин и 30 °C. Утром по 5 мл выросших культур засевают в такие же колбы со свежим МПБ и помешают их на качалку при 37 °C на 3 ч. Полученные таким образом суспензии штамма-донора и штамма-реципиента вводят в скрещивание в соответствии со схемой эксперимента (рис. 35.7).
Проводят скрещивание двух штаммов Е. coli (Hfr и J-62), несущих разные генетические маркеры. В результате скрещивания возникают рекомбинанты, сочетающие признаки исходных родительских штаммов. Для проведения скрещивания 2 мл суспензии клеток J-62 и 1 мл суспензии клеток Hfr вносят в стерильную колбу на 50 мл. Колбу помещают на термостатированную качалку, производящую мягкое покачивание (перемешивание) суспензии (~30 об/мин). Скрещивание проходит 1,5 ч при 37 °C.
Во время скрещивания определяют максимально возможное количество конъюгативных пар, которое способно образоваться в данной смеси. Оно равно числу жизнеспособных клеток штамма-донора (Hfr), поскольку клетки штамма-реципиента берутся заведомо в избытке. Для определения количества жизнеспособных клеток штамма Hfr готовят десятичные разведения в 50 мМ К-фосфатном буфере и высевают по 0,1 мл суспензии из разведений 10 4—10“' на чашки Петри с МПА. Засеянные чашки инкубируют 24 — 48 ч при 30 °C.
Чтобы убедиться в неспособности обоих штаммов (Hfr и J-62) расти на используемых селективных средах, исходные суспензии родительских штаммов центрифугируют и ресуспендируют в К-фосфатном буфере до получения разбавленных суспензий. Затем на разные селективные среды петлей наносят прямые штрихи из суспензий, используя для каждого штамма свою половину чашки.
По окончании скрещивания с помощью селективных сред производят отбор рекомбинантов по каждому из четырех анализируемых генетических маркеров и определяют частоту образования рекомбинантов в процентах от числа живых клеток донорного штамма. Для этого готовят десятичные разведения конъюгационной смеси до 10“5. На каждую селективную среду в двух повторностях высевают по 0,1 мл из разведений 10~2— 10 5. Засеянные чашки инкубируют при 37 °C в течение 24—48 ч.
Подсчитывают число колоний штамма-донора на МПА и количество колоний рекомбинантов на селективных средах. Определяют концентрацию клеток Hfr и рекомбинантов pro+str-r, try+str-r, his+str-r, lac+str-r в конъюгационной смеси. Частоту образования рекомбинантов выражают в процентах от числа клеток штамма-донора, введенных в скрещивание. Результаты вносят в табл. 35.7.
Суспензия Hfr (донор)
Суспензия J-62 (реципиент)
1 мл суспензии штамма-донора
Посев по 0,1 мл из разведений 10-4— 10~7 на МПА в двух повторностях
0,5 мл для приготовления десятичных разведений в буфере до 10~7
Инкубация при 30 °C 24 — 48 ч
Центрифугирование 15 мин при 6 ’1ыс.д и ресуспендирование в буфере (разбавленная суспензия)
Посев петлей штрихом на каждую селективную среду
15 мл
2 мл суспензии штамма-реципиента! з мл
Центрифугирование 6 тыс. g 15 мин и ресуспендирование в 3 мл теплого МПБ
Скрещивание 1,5 ч на качалке 30 об/мин при 37 °C
0,5 мл для приготовления десятичных разведений в фосфатном буфере (pH 7,0)
I мл
Центрифугирование 15 мин при 6 тыс. g и ресуспендирование в буфере (разбавленная суспензия)
Посев по 0,1 мл из разведений 10~Н0~5 на все селективные среды в двух повторностях
Рис. 35.7. Схема опыта по конъюгации
Инкубация при 37 °C 24-48 ч
Посев петлей штрихом на каждую селективную среду
На основании полученных данных определяют последовательность анализируемых маркеров на хромосоме Е. coli К-12. Представляют схему расположения этих маркеров по отношению к начальной точке оп.
Материалы, оборудование, посуда, реактивы
Центрифуга; счетчик колоний; микробиологическая петля. Чашки Петри — 50 шт.; пробирки обычные с ватными пробками — 15; стеклянные палочки — 3; стеклянные центрифужные стаканчики с пробками — 4; стеклянные шпатели — 10; пипетки на 1 мл — 20; на 0,1 мл — 5; на 5 мл — 5; на 10 мл — 2; колбы на 250 мл — 4; на 50 мл — 1.
Анализ конъюгационного скрещивания Е. coli К-12 Hfr х J-62 по градиенту передачи маркеров донора рекомбинантам
Селективная среда Отбираемые рекомбинанты Разведение суспензии Количество колоний рекомбинантов на чашках Концентрация клеток в конъ-югационной смеси Содержание рекомбинантов, %
SB,TH + глюкоза
SI^PH + глюкоза
SB,TP + глюкоза
SB,TPH + лактоза
МПА Штамм Hfr 100
МПБ — 250 мл; МПА — 200 мл; селективные среды — по 200 мл; 50 мМ К-фосфатный буфер (pH 7,0) — 100 мл.
План выполнения задачи
Занятие 1. Приготовление и стерилизация сред, растворов и посуды (4 ч). Подготовка бактериальных культур, вводимых в скрещивание (22 ч).
Занятие 2. Проведение скрещивания, приготовление разведений и посевы на селективные среды и МПА (4 ч). Инкубация засеянных чашек (24 — 48 ч).
Занятие 3. Подсчет колоний на чашках и оформление результатов (4 ч).
ЗАДАЧА 4. ИЗУЧЕНИЕ ИНДУЦИРОВАННОГО ХИМИЧЕСКОГО МУТАГЕНЕЗА С ПРИМЕНЕНИЕМ ТЕСТА ЭЙМСА
Известно, что определенные виды рака человека и животных вызываются химическими веществами, называемыми канцерогенами. Поэтому важно иметь быстрый и дешевый способ определения потенциальной опасности большего количества химических соединений, имеющих контакт с пищей, входящих в состав новых продуктов, или веществ, попадающих в окружающую среду в результате хозяйственной деятельности человека.
Проверка каждого химического соединения на его канцерогенность (способность вызывать раковые заболевания) с лабораторными животными занимает около 2—3 лет, и стоимость проверки одного вещества доходит до 100 000 долл. США. Учитывая тысячи соединений, которые необходимо проверить, и стоимость каждого теста, можно убедиться, что проверки на животных слишком долговременны и дороги для массовых испытаний многих веществ. Поскольку 90 % от уже проверенных канцерогенов являются также мутагенами, вызывающими хромосомные мутации (это предполагает возникновение некоторых видов рака животных при соответствующих мутационных изменениях клеточных ДНК), Брюс Эймс с коллегами из Калифорнийского университета (США) разработали тест, в котором использованы специальные мутантные штаммы бактерий, чрезвычайно чувствительные к мутагенам. С разработкой этого метода появилась возможность проверять множество химических соединений на мутагенность и потенциальную канцерогенность быстро и относительно недорого.
Соединения, оказавшиеся сильными мутагенами, в дальнейшем уже на животных проверяют на канцерогенность. Тест Эймса занимает несколько дней, и проверка одного соединения стоит лишь несколько сотен долларов.
Природа мутагенов Эймса
Дикий тип энтеробактерий Salmonella typhimurium, будучи прототрофом, растет на средах без добавок аминокислот и других факторов роста. Мутанты Эймса требуют наличия в питательных средах гистидина, поскольку они ауксотрофны по этой аминокислоте. Мутанты получены путем одиночной вставки или делении основания в последовательность генов, кодирующих синтез одного из ферментов пути биосинтеза гистидина. В результате такой мутации происходит сдвиг рамки считывания нуклеотидов и возникают «нонсенс-мутанты» по одному из генов, которые, однако, легко могут быть превращены в прототрофные штаммы вставкой или делецией одного нуклеотида в нарушенную последовательность. Такие мутанты, не способные расти на средах, в которые не добавлен гистидин, называют гистидинзависимыми, или hys^-мутачтами.
Кроме того что мутанты Эймса являются делеционными, они также лишены репарационных систем, которые обычно исправляют такого рода делеционные мутации. Возвращение к прототрофному состоянию для такого рода мутантов без систем репарации ДНК невозможно без дополнительных мутаций, поэтому такие штаммы довольно стабильны, т. е. дают небольшой процент спонтанных ревертантов, что важно для проведения теста. Все, что нужно клетке Ays’-мутанта для возвращения в прото-трофное состояние, — вставить выпавший нуклеотид в место делении, а это может произойти только в результате инсерционного мутагенеза, когда клетка будет способна снова синтезировать гистидин и станет прототрофной по этому соединению (реверсия, обратная мутация).
Поскольку мутагены увеличивают частоту (скорость) мутаций, возникающих в популяции клеток без видимых причин (спонтанных мутаций), они будут также увеличивать частоту обратных мутаций (реверсий) мутантов Эймса. Чем сильнее действует на ДНК мутаген, тем выше будет число ревертантов. Таким образом, количество реверсий служит мерой мутагенности проверяемого соединения. Сильные мутагены очень часто являются и потенциальными канцерогенами.
Отбор ревертантов
Для определения числа ревертантов, появившихся в популяции клеток, подвергнутых действию проверяемого потенциального мутагена, необходимо иметь способ дифференцировки таких мутантов от спонтанно образуемых ревертантов. В тесте Эймса для этой цели применяют синтетические среды с определенного состава. Одним из способов проведения эксперимента является нанесение клеток-мутантов Эймса на поверхность чашки Петри с плотной синтетической средой и немедленное введение в контакт с проверяемым соединением. Культуру микроорганизмов инкубируют до появления видимых колоний ревертантов.
Питательные среды, используемые в тесте Эймса, содержат следовые количества гистидина, позволяющие клеткам мутантов разделиться несколько раз, однако недостаточные для формирования колоний нормального размера. Это необходимо для того, чтобы испытываемые мутагены смогли бы подействовать и на стадии репликации ДНК (известно, что многие мутагены действуют на клетку именно на этой стадии ее развития). Таким образом, в синтетических средах с минимальным количеством гистидина клетки даже ЛуУ-мутантов Эймса претерпевают несколько циклов редупликации ДНК, а потенциальные мутации, если они происходят, представлены в «+» и «-» цепях делящейся ДНК. На чашках со средой после инкубации виден рост Ays “-мутантных клонов в виде слабого сплошного газона с образованием муарового налета, на этом фоне проявляются крупные колонии ревертантов (Лу5+-колонии).
Проведение теста Эймса
Тесты, проводимые в специальных лабораториях, должны строго соответствовать методике, приведенной Эймсом с коллегами, во избежание разночтения результатов экспериментов. Постановка тестов в стандартных условиях дает возможность сравнения результатов, полученных в разных лабораториях и в разное время, что важно для их независимой оценки и/или возможной перепроверки. Тест Эймса по полной схеме включает проведение экспериментов с различными мутантными штаммами и предполагает наличие многочисленных контролей при различных концентрациях испытываемых соединений.
Цель задачи: знакомство студентов с теоретическими предпосылками и общими методами проведения эксперимента.
Задача не включает обучение методике тестирования по полной программе. В ней рассматривается только один мутант, тогда как тест Эймса предусматривает использование нескольких мутантов 5. typhimurium по гистидиновому опе-рону, полученных с помощью разных типов мутаций. В то же время все мутации имеют одну природу — это точечные мутации со сдвигом рамки считывания или мутации замены основания нуклеотидов. Каждый мутант предназначен для проверки различных мутагенов, вызывающих обратные мутации, которые приводят к появлению клеток дикого типа (ревертантов). Полная программа теста Эймса описана в специальном руководстве (G. Gerhard, 1994).
Многие химические соединения не являются мутагенными (или канцерогенными) до тех пор, пока не будут подвергнуты действию микросомальных ферментных комплексов, находящихся в печени животных. Химические превращения веществ в канцерогенную форму нередко являются частью нормального процесса, происходящего в печени в связи с индукцией ферментов, ответственных за детоксикацию вредных соединений, которые могут быть опасными для организма. Некоторые химические вещества или их производные реагируют с детоксифицирующими ферментами (оксигеназами) с образованием эпоксидов, связывающихся с ДНК клетки. Таким образом, организм может превратить относительно невредное соединение в потенциальный мутаген (канцероген). По этой причине в тесте Эймса перед добавлением химических веществ к мутантам Эймса все проверяемые вещества обрабатывают индуцированными препаратами печени крыс. Для образования детоксифицирующих ферментов в печени крыс животным делают инъекцию препарата Aroclor, имеющегося в продаже. Затем крыс забивают, печень удаляют и гомогенезируют, а гомогенаты используют в тесте Эймса.
В задаче студенты испытывают класс химических соединений, известных как ншп-роканцерогены. Эти вещества относительно безвредны сами по себе, однако в гастроэн-теральном тракте животных они под действием ферментов и ферментных систем энтеробактерий превращаются в нитрозамины — потенциальные канцерогены (мутагены). Поскольку мутанты Эймса (A. typhimurium) также принадлежат к энтеробактериям, а в задаче исследуются только нитроканцерогены, то в данном случае можно исключить стадию обработки испытываемых соединений ферментными препаратами печени крыс.
Общая схема проведения теста Эймса. Каждый тестируемый препарат обрабатывают гомогенатом печени животного (обычно крысы). Аликвоты суспензий каждого мутанта Эймса распределяют по чашкам Петри с плотной агаризованной средой со следами гистидина. Затем в центр чашки помещают диски стерильной фильтровальной бумаги, смоченной дистиллированной водой (контроль) или растворами различных концентраций тестируемого соединения после его контакта с «активирующей» смесью ферментов из печени. Проверяемое соединение диффундирует в среду, формируя градиент концентрации. Если тестируемое вещество является мутагеном или преврашается в
него под действием ферментов печени, то некоторые мутантные клетки, реагируя на него, будут образовывать колонии. Чем сильнее мутагенный эффект тест-соединения, тем больше колоний образуется на чашке вблизи диска, и тем дальше от диска они будут встречаться. Слабые мутагены вызывают образование меньшего числа колоний ревертантов, и они появляются ближе к диску. Другими словами, частота реверсий зависит от потенциальной мутагенной способности и концентрации того или иного соединения. Чашки с дисками, смоченными в дистиллированной воде, дают очень небольшое число спонтанных обратных мутаций; они служат отрицательным контролем эксперимента.
Недостатком тестирования с помощью градиентной концентрации является то, что количества действующего вещества достаточно трудно определить. Более точные результаты по количественным характеристикам силы мутагена можно получить другим способом. В чашки Петри поверх застывшей агаризованной среды (минимальный агар Эймса) заливают еще один слой полужидкой среды с минимальной концентрацией гистидина, куда вносят определенную аликвоту клеток (в мл) и известную концентрацию испытываемого соединения.
Тест Эймса был применен для испытаний тысяч химических веществ, таких, как промышленные химикаты, косметические препараты, краски для волос, пищевые добавки и пестициды. В каждой категории этих веществ были обнаружены потенциальные канцерогены. Высокая частота реверсий не всегда свидетельствует о канцерогенности проверяемого соединения. Точные доказательства могут быть получены только в опытах на животных. В то же время только несколько из более чем 300 известных канцерогенов животных не вызывали повышенной частоты реверсии в тесте Эймса.
Техника безопасности при работе с мутагенами (потенциальными канцерогенами).
• Исходные {концентрированные) растворы мутагена студентам не выдаются, аликвоты этих растворов в расплавленный и остуженный агар вносит преподаватель или лаборант. Исходный раствор мутагена не должен быть доступен для студентов.
• Всю работу проводят в резиновых перчатках для исключения попадания мутагена на кожу.
• Вся лабораторная посуда, находившаяся в контакте с мутагеном, должна быть сложена в специальную емкость для последующей обработки и дезактивации.
• Чашки Петри, в которые вносили мутаген, после эксперимента должны быть также дезактивированы лаборантом.
• Перед началом практической работы студенты должны изучить технику безопасности по работе с мутагенами, после чего расписаться в специальном журнале.
Ход выполнения задачи
Для выращивания мутантов Эймса готовят: 1) минимальный агар Эймса для использования в качестве подстилающей среды в чашках Петри при проведении тестирования химических соединений на мутагенность {среда 7); 2) полужидкий агар верхнего слоя Эймса в чашках Петри с минимальной концентрацией гистидина {среда 2).
Состав среды 1 (количество компонентов на 1 л): глюкоза (40 %-й раствор) — 50,0 мл; раствор солей Vogel-Bonner — 20 мл; агар — 15,0 г. Раствор солей Vogel-Bonner (г/л): MgSO4-7H2O — 10,0; лимонная кислота (моногидрат) — 100,0 г; К2НРО4 (б/в) - 500,0; NaHNH4PO4-4H2O - 175,0.
Для приготовления раствора необходимо подогреть около 1,5 л дистиллированной воды до 45 °C. Растворить компоненты раствора в указанном порядке один за другим в 600 мл подогретой дистиллированной воды, довести до 1 л, разлить по сосудам с навинчивающимися пробками и простерилизовать при
1 ати с полуоткрытыми пробками для уравнивания образующегося внутри сосудов давления. После автоклавирования и охлаждения раствора завернуть крышки плотно. Хранить раствор при комнатной температуре.
Для приготовления агаровой среды простерилизовать 1,5 %-й агар на дистиллированной воде при 1 ати, затем расплавить его и инкубировать при 50 °C на водяной бане. К 930 мл расплавленной агаризованной среды прилить 50 мл нагретого до той же температуры раствора стерильной 40 %-й глюкозы (стерилизация автоклавированием при 0,5 ати) и 20 мл нагретого раствора Vogel-Bonner. Среду хорошо перемешать и разлить при температуре 50 °C по стерильным чашкам Петри по 30 мл в каждую.
Среду 2 получают смешением нагретых до температуры 50 °C агаровой смеси (100 мл) и 10 мл раствора биотина с гистидином. Агаровую смесь готовят растворением 5 г NaCl в 500 мл дистиллированной воды, доведением объема до 1 л, расплавлением в нем б г агара и автоклавированием по 100 мл в колбах на 250 мл при 1 ати. Раствор биотина с гистидином готовят растворением 1,24 г £>-биотина и 9,6 г Z-гистидина в 10 мл дистиллированной воды, стерилизуют при 0,5 ати и хранят при 4 °C в холодильнике. Агаровая смесь (г/л): агар — 6,0; NaCl — 5,0. Раствор биотина и гистидина (мг/л): биотин — 124,0; гистидин — 96,0.
Перед началом экспериментом в смесь агаровой среды (0,6 %) с раствором биотин-гистидина добавляют суспензию тест-организма, стерильную дистиллированную воду (контроль) или растворы испытываемого вещества и разливают по чашкам с застывшим подстилающим слоем агара (1,5 %).
Преподаватель добавляет в расплавленный полужидкий агар стерильную дистиллированную воду (контроль) и стерильные растворы мутагенов в разных концентрациях. Содержимое пробирок тщательно перемешивают. Пробирки до внесения их на чашки с твердым агаром следует держать при 50 °C на водяной бане.
Каждый студент заливает 4 чашки Петри расплавленной в пробирках подстилающей средой Эймса и каждую чашку аккуратно подписывает: имя, дата, контроль, опыт I, 2 или 3 (разные химические соединения соответственно). Для эксперимента каждый студент имеет четыре пробирки с полужидким расплавленным агаром, помеченные контролем (дистиллированная вода) или опытами 1, 2 или 3. Преподаватель сообщает студентам названия испытываемых соединений, которые они записывают в лабораторную тетрадь. Далее студенты асептически добавляют в каждую из пробирок с расплавленным агаром по 0,1 мл суспензии клеток S. typhimurium ТА98, тщательно и быстро перемешивают содержимое вращением пробирки в ладонях и немедленно асептически вносят в чашки с застывшим подстилающим агаром под соответствующими номерами. Покачивая чашку с закрытой крышкой, добиваются равномерного распределения верхнего агарового слоя по поверхности нижнего подстилающего слоя и оставляют чашку на столе для затвердевания агара.
Для контроля готовят на группу студентов 4 чашки с триптозным соевым агаром (ТС-агаром) в качестве подстилающего слоя и разливают сверху содержимое из 4 пробирок (культура V. typhimurium ТА98 и внесенные тестируемые вещества). Этот опыт продемонстрирует студентам необходимость использования минимального агара Эймса в качестве подстилающего слоя в чашках Петри.
Чашка Проверяемое соединение Количество больших колоний
Минимальный агар ТС-агар
Контроль н |2 3 Нет
Чашки инкубируют, не переворачивая, при 30 °C в течение 48 — 72 ч. После окончания инкубации выбирают чашки с максимальным числом больших колоний и подсчитывают их. Если число колоний на чашке слишком велико (более 300 на чашку), то делят поверхность на 2 или 4 равные части и просчитывают число обратных мутантов по секторам, умножая затем на число секторов чашки. Колонии просчитывают на чашках с минимальным агаром и ТС-агаром. Результаты подсчетов заносят в таблицу (табл. 35.8).
После подсчетов чашки с добавленными мутагенами необходимо сложить в специальный лоток и подвергнуть обработке для дезактивации мутагенов (по инструкциям преподавателя).
Интерпретация результатов эксперимента. Эймс-мутант S. typhimurium не способен синтезировать гистидин. На основной синтетической среде без гистидина (подстилающий слой) клетки мутанта не растут, однако верхний слой полужидкого агара содержит минимальную концентрацию этой аминокислоты, достаточную для роста клеток в течение 2—3 генераций. После инкубации мутанты дадут сплошной рост в верхнем, полужидком слое агара, что будет проявляться в виде небольшого равномерного помутнения среды, на фоне которого отчетливо видны крупные колонии ревертантов к дикому типу.
Сильные мутагены стимулируют возвращение Эймс-мутантов к их прото-трофному состоянию (реверсии к родительскому фенотипу), которые уже не требует экзогенно добавленного гистидина для формирования крупных колоний. Поэтому появление каждой крупной колонии на фоне газона остаточного роста в чашках свидетельствует о возникновении обратной мутации под воздействием мутагена или произошедшей спонтанно.
Число колоний на чашке с добавленной в виде контроля дистиллированной водой вместо испытываемого мутагена определяет уровень спонтанного мутагенеза культуры в данных условиях. Если тестируемое соединение дает уровень мутагенеза превышающий спонтанный мутагенез в два раза, то оно считается мутагеном и поэтому потенциальным канцерогеном для человека.
Глава 36
ИССЛЕДОВАНИЯ БАКТЕРИОФАГОВ
Бактериальные вирусы {бактериофаги) представляют относительно простую биологическую систему и являются удобной моделью для изучения основных проблем молекулярной биологии и генетики.
Значительные успехи в изучении механизмов репликации нуклеиновых кислот, исследований тонкой структуры генетического материала, молекулярного механизма мутаций, регуляции белкового синтеза и многие другие достижения современной биологии связаны с применением бактериофагов как удобных модельных объектов.
Бактериофаги заражают и реплицируются в самых различных бактериях, однако наиболее изучены бактериофаги кишечной палочки (Escherichia coli). В связи с этим серия последующих задач посвящена освоению методов работы с бактериофагами этой группы.
ЗАДАЧА 1. ВЫРАЩИВАНИЕ И ХРАНЕНИЕ БАКТЕРИАЛЬНЫХ КУЛЬТУР
Питательные среды. Выращивание клеток Е. coli можно проводить как на синтетической среде, так и на питательном бульоне. Питательная синтетическая среда, состоящая из NH4C1, MgSO4, КН2РО4, NaHPO4 и глюкозы, способна поддерживать рост кишечной палочки, одновременно обеспечивая буферные свойства раствора (реакция среды сохраняется на уровне, близком к нейтральному). Для ускорения процесса роста клетки Е. coli культивируют не на этой простой среде, а на питательном бульоне, содержащем в своем составе аминокислоты, азотистые основания и сахара.
Следует отметить, что клетки Е. coli принадлежат к классу так называемых факультативных анаэробов, которые при наличии необходимых питательных веществ размножаются и в отсутствие кислорода. Поскольку размножение Е. coli в аэробных условиях протекает значительно быстрее, через питательную среду пропускают ток воздуха либо интенсивно встряхивают ее на качалке в целях аэрации.
Выращивание бактерий можно проводить и на твердых питательных средах, получаемых добавлением к питательному бульону 1,5% агара.
Хранение бактериальных культур. Бактериальные штаммы поддерживают путем пересева клеток с помощью металлической петли на «скошенный» питательный агар (1,5 %). Для получения чистой бактериальной культуры выращивают отдельные колонии этих клеток, рассеивая бактерии штрихами на чашках с питательным агаром.
Выращивание бактерий. Бактериальные клетки в процессе размножения в жидкой питательной среде проходят несколько фаз: лаг-период, логарифмическую фазу (период активного деления) и стационарную фазу. Деление клеток прекращается из-за истощения запасов питательных веществ и выделения в среду токсических продуктов жизнедеятельности клеток. Хорошо аэрируемые культуры Е. coli, растущие в простой синтетической среде или на питательном бульоне, вступают в стационарную фазу, когда концентрация бактерий достигает (2 - 5)-109 клеток на 1 мл. Эффективное воспроизводство фагового потомства имеет место в период наиболее активного деления бактериальных клеток, т.е. в логарифмической фазе роста.
Бактериальные культуры в логарифмической фазе можно условно разделить на две категории: собственно логарифмические культуры, соответствующие середине логарифмической фазы, и так называемые «ночные культуры», где бактерии находятся в самом конце логарифмической фазы. Для получения «ночной культуры» в пробирку с 5 мл питательного бульона вносят посевной материал из отдельной колонии. Пробирку инкубируют при 37 °C в течение ночи без аэрации. К этому времени концентрация клеток достигает 1-Ю9 в 1 мл. Для получения культуры в логарифмической фазе роста «ночную культуру» разводят в 10—100 раз питательным бульоном и инкубируют при 37 °C,
встряхивая на качалке для лучшей аэрации. Инкубацию продолжают до тех пор, пока плотность культуры не достигнет (2 - 3) 10s клеток в 1 мл. При разведении в 10 раз для подращивания клеток до указанной концентрации требуется 1,5 ч, а при разведении в 100 раз — 2,5 ч.
Определение концентрации бактерий. Распространенный способ определения концентрации бактерий — метод подсчета бактериальных колоний на чашках Петри. Пробы (0,5 мл) соответствующих разведений бактериальной суспензии распределяют стеклянным шпателем по поверхности питательного агара и после инкубации подсчитывают число колоний. Поскольку каждая бактериальная клетка дает начало отдельной колонии клеток, число колоний на чашке (с учетом разведения и объема наносимой суспензии) будет характеризовать исходную концентрацию бактерий в культуре. Например, если при нанесении на чашку 0,1 мл культуры, разведенной в 100 000 раз, образуется 100 колоний, то количество бактериальных клеток в 1 мл исследуемой суспензии составляет 100- 10-105, т.е. 1 108.
В качестве экспресс-метода определения концентрации бактерий в питательной среде можно рекомендовать измерение мутности суспензии. Для этого необходимо иметь калибровочную кривую зависимости мутности от количества клеток в конкретных условиях выращивания, и естественно, что при изменении условий требуется построение новой кривой.
Цель работы', получить «ночную культуру» Е. coli и (в процессе ее выращивания) построить кривую роста клеток во времени для условий, обычно используемых при выращивании бактериофагов.
Ход выполнения задачи
Чашки Петри с питательным агаром для посева бактерий готовят в день опыта или накануне. Расплавленный агар разливают в чашки по 20 — 30 мл, что соответствует % объема чашки. После застывания агара чашки выдерживают для просушивания в термостате при 37 °C в течение ночи в перевернутом положении с закрытыми крышками. Избыток конденсационной воды можно удалить, выдерживая чашки в перевернутом положении с открытыми крышками в термостате в течение 1 ч. При необходимости чашки с агаром можно хранить в холодильнике в течение нескольких дней.
Для получения «ночной культуры» Е. coli в стерильную пробирку с питательным бульоном (5 мл) вносят петлей посевной материал из отдельной колонии на чашке. Смесь инкубируют в течение ночи без аэрации.
В стерильный флакон на 250 мл вносят 30 мл питательного бульона и 3 мл ночной бульонной культуры Е. coli. Инкубационную смесь ставят на качалку при 37 °C и аэрируют в течение 2 ч. Через 60, 90 и 120 мин стерильно отбирают по 3 мл суспензии. В каждой пробе определяют мутность и количество бактериальных клеток. О мутности судят, определяя интенсивность светорассеивания бактериальной суспензии при длине волн 600 нм.
Перед высевом проб из чашки Петри суспензию клеток разводят серией десятикратных разведений в стерильном физиологическом растворе. Для получения оптимального количества колоний (около 100 на 1 чашку) необходимо сделать 5 десятикратных разведений исходной суспензии. Наносят на поверх
ность чашки Петри с 1,5% питательным агаром в двух повторностях 0,1 мл последнего (пятого) разведения каждой пробы и растирают шпателем.
После инкубации чашек в течение ночи при 37 °C подсчитывают количество образовавшихся колоний, определяют концентрацию бактерий в 1 мл в каждой пробе и строят кривую зависимости мутности суспензии от концентрации бактериальных клеток в данных условиях.
ЗАДАЧА 2. ВЫРАЩИВАНИЕ Т-ЧЕТНЫХ БАКТЕРИОФАГОВ
Получение фаговой суспензии с высоким титром требует определенного опыта работы с системой «фаг—клетка». При внесении небольшого количества фага в культуру чувствительных бактерий, активно размножающихся в жидкой питательной среде, частицы бактериофага адсорбируются на бактериальных клетках и после латентного периода лизируют их. Освободившееся в среду потомство частиц бактериофага заражает клетки, не вовлеченные в процесс инфекции, вызывая второй инфекционный цикл. Таким образом, последовательно заражаются все клетки суспензии, и процесс продолжается асинхронно до тех пор, пока не произойдет лизис всех чувствительных бактерий. Теоретически выход фага равен численности популяций бактерий, умноженной на величину среднего выхода фага при лизисе клетки. Действительный выход часто меньше, так как некоторые из освободившихся вирусных частиц утрачиваются в результате повторной адсорбции на инфицированных, но не лизировавшихся бактериях или на обломках бактерий.
Период времени, необходимый для лизиса всех клеток, зависит главным образом от продолжительности латентного периода и числа введенных фаговых частиц. На процесс фаголизиса влияют также температура, степень аэрации, состав питательной среды, реакция среды и физиологическое состояние бактерий. Так, например, заражение бактериальной культуры в стационарной фазе не приводит к появлению фагового потомства.
В конце логарифмической фазы роста концентрация клеток в питательном бульоне составляет около 1 • 109 в 1 мл. Если такую культуру заразить достаточным количеством фага (1Т09 фаговых частиц в 1 мл), то все бактерии инфицируются почти одновременно и лизис культуры наступит через 1 — 2 ч. При этом можно ожидать, что 109 инфицированных бактерий в 1 мл могут дать 10" фаговых частиц в 1 мл, поскольку средний выход (для Т-четных фагов) составляет 100 — 200 фаговых частиц.
Для получения высокоочищенных фаговых суспензий применяют синтетические среды. С этой целью обычно используют аэрируемые колбы или бутыли, а также всевозможные ферментеры.
Цель работы: получение суспензий фага Т4 при использовании бактерий Е. coli в качестве хозяина.
Ход выполнения задачи
В стерильный флакон на 250 мл вносят 30 мл питательного бульона и 3 мл ночной бульонной культуры Е. coli. Инкубационную смесь ставят на качалку при 37 °C и аэрируют в течение 1,5 ч. После этого берут пробу в количестве 3 мл для определения мутности культуры и установления точной концентрации бактерий по кривой, полученной в задаче 1.
Бактериальную суспензию заражают внесением суспензии фага с известным титром при множественности заражения 0,1 (отношение числа фаговых
частиц к числу бактериальных клеток). Аэрацию инфицированных фагом клеток продолжают в течение 2 ч, затем добавляют несколько капель хлороформа для разрушения нелизированных бактериальных клеток и продолжают встряхивание в течение 15 мин.
Полученный лизат центрифугируют при 3 — 4 тыс. g в течение 20 мин для удаления обломков клеток и фагоустойчивых бактерий и хранят в холодильнике для дальнейшей работы.
ЗАДАЧА 3. ТИТРОВАНИЕ ФАГА МЕТОДОМ АГАРОВЫХ СЛОЕВ
Активность бактериофага определяют по его способности вызывать лизис бактериальной культуры в жидких или твердых средах и численно выражают степенью максимального разведения, в котором испытуемый фаг проявил свое литическое действие. Весьма точно активность бактериофага оценивается количеством инфекционных активных единиц в единице объема, т. е. определением титра бактериофага.
Для этой цели обычно пользуются методом агаровых слоев. Сущность метода состоит в том, что культуру индикаторных бактерий в «мягком» (0,8%-м) агаре при 48 °C заражают фагом в соответствующем разведении. Эту смесь выливают на поверхность застывшего 1,5%-го питательного агара, дают застыть верхнему слою агара и ставят на инкубацию в термостат. В течение 6 —12 ч бактерии размножаются внутри мягкого слоя агара в виде множества мелких колоний, получая питание из нижнего слоя 1,5%-го агара, который применяют как основу. Низкая концентрация верхнего слоя агара создает пониженную вязкость, что способствует хорошей диффузии фаговых частиц. В результате фаговые частицы заражают бактериальные клетки, находящиеся в непосредственном соседстве с ними, размножаются и лизируют их. Фаговое потомство инфицирует затем соседние бактерии, которые в свою очередь подвергаются лизису в результате появления второго поколения фага, и т.д. В конечном итоге на растущем бактериальном газоне образуются прозрачные зоны отсутствия роста бактерий — негативные колонии, стерильные пятна, или «бляшки». Образование каждого стерильного пятна может быть вызвано единственной частицей бактериофага. Следовательно, число негативных колоний может служить количественным показателем содержания инфекционных фаговых частиц в исследуемом растворе.
Пель работы: определить титр фага, т.е. содержание активных фаговых частиц в 1 мл лизатов, полученных ранее при выполнении предыдущей задачи (см. задачу в гл. 36).
Ход выполнения задачи
В случае, если титр фага предположительно составляет 1О10 фаговых частиц в 1 мл, необходимо провести восемь десятикратных разведений исходной суспензии в стерильном физрастворе.
Агар для верхнего слоя (0,8 %), предварительно разлитый по 5 мл в пробирки, расплавляют и остужают в водяной бане до 48 °C. В таком состоянии агар может храниться в водяной бане ~20 мин, так как при более продолжительном хранении он приобретает гелеобразную консистенцию. Готовят 2 пробирки (№ 1 и № 2). В каждую пробирку вносят 0,1 мл ночной бульонной культуры Е. coli.
В обе пробирки с мягким агаром и индикаторной культурой добавляют соответственно по 0,1 мл седьмого и восьмого разведений фаговой суспензии,
тщательно перемешивают и выливают содержимое на чашку Петри с 1,5 %-м питательным агаром, равномерно распределяя по поверхности агара. После застывания верхнего слоя агара (10—15 мин) чашки переворачивают вверх дном и оставляют в термостате при 37 °C на ночь.
На следующий день считают количество негативных колоний на каждой чашке и определяют титр фага в исходном препарате с учетом фактора разведения и объема вносимого материала. Например, если при нанесении на чашку 0,1 мл фаговой суспензии при разведении 1-107 на этой чашке образовалось 150 стерильных пятен, то титр фаголизата будет равен (150 10) • 107 = 1,5 • 1010 частиц в 1 мл.
ЗАДАЧА 4. НЕЙТРАЛИЗАЦИЯ БАКТЕРИОФАГА СПЕЦИФИЧЕСКОЙ АНТИСЫВОРОТКОЙ
При внутривенном введении кроликам фаголизатов в сыворотке этих животных образуются специфические антитела, способные нейтрализовать инфекционность гомологичных фаговых частиц. Скорость реакции зависит от концентрации фага, активности сыворотки, температуры и свойств среды и подчиняется кинетическому уравнению первого порядка. При постоянной температуре и постоянстве свойств среды уравнение имеет следующий вид:
К= 2,3(r/t)lg(P0/P),
где Ро — количество фага при времени 0 мин; Р — количество фага при времени t мин; D — конечное разведение сыворотки в смеси «сыворотка+бактериофаг». Величина К, называемая константой скорости нейтрализации, — показатель активности данной конкретной антифаговой сыворотки. Совпадение или близость значений К в опытах по перекрестной нейтрализации дает возможность установить степень серологического родства бактериофагов. Предварительно найденная величина Жданной антисыворотки позволяет оценить оптимальную степень разведения сыворотки, применяемой для инактивации данного препарата фага.
Различные фаги существенно отличаются скоростью реакций с гомологичными антисыворотками. Значения вскорости нейтрализации колеблются от 10 до 103 в зависимости от вида фага. Эта величина зависит также от ряда факторов, не поддающихся контролю (техника иммунизации и индивидуальных особенностей животных).
Цель работы, определить значения К для антисывороток к фагу Т4.
Ход выполнения задачи
Разведение антисыворотки. Готовят три разведения антифаговой сыворотки (1:10, 1:100, 1:1000) физиологическим раствором и инкубируют в водяной бане при 37 °C в течение 10 мин.
Разведение фага. Фаговую суспензию разводят до титра 1 • 107.
Постановка реакции. В каждое разведение антисыворотки (всего 3 пробирки) объемом 0,9 мл добавляют по 0,1 мл разведенной суспензии бактериофага и ставят на инкубацию при 37 °C на 5 мин.
Остановка реакции. Через 5 мин к 0,1 мл каждой смеси «фаг—сыворотка» добавляют 9,9 мл физиологического раствора. На чашки высевают методом
агаровых слоев 0,1 мл каждой разведенной смеси, используя в качестве индикаторной культуры Е. coli. Высев проводят для каждого разведения антисыворотки. В качестве контроля берут смесь, состоящую из 0,1 мл суспензии бактериофага и 0,9 мл соответствующего растворителя (вместо разведений антисыворотки).
На следующий день подсчитывают количество негативных колоний на всех чашках. Если фаг не был нейтрализован, то после инкубации чашек обнаруживается около 1000 стерильных пятен. При инактивации 90 % фага их количество составит 100 и т.д. Например, если сыворотка в разведении 1:100 инактивирует 90 % фага в течение 5 мин, то величина К= 2,3-100/5 lg(100/10), т.е. 46.
Материалы и оборудование
Термостат на 37 °C; водяная баня на 48 °C; качалка; сушильный шкаф.
Стерильная посуда: флакон на 250 мл — 1 шт; пробирки — 20; пипетки на 1 — 2 мл — 10; 5 мл — 5; чашки Петри — 4.
Стерильные растворы: физиологический раствор (0,15 М NaCl) — 100 мл; питательный бульон — 30 мл; 1,5%-й питательный агар на питательном бульоне — 100 мл; антифаговая сыворотка — 0,5 мл; хлороформ.
Культура Е. coli, суспензия бактериофага с известным титром.
РАЗДЕЛ V
НЕКОТОРЫЕ АСПЕКТЫ ПРИКЛАДНОЙ МИКРОБИОЛОГИИ (ЭКОЛОГИЯ, БИОТЕХНОЛОГИЯ, САНИТАРНАЯ И МЕДИЦИНСКАЯ
МИКРОБИОЛОГИЯ)
Глава 37
ЭКОЛОГИЯ И ТРОФИЧЕСКИЕ ЦЕПИ
Выделение микроорганизмов из природных местообитаний и описание их физиолого-биохимических свойств, безусловно, относится к одному из важнейших методов микробной экологии. Вместе с тем давно установлено, что при высеве на питательные среды удается получить рост лишь очень незначительной части природной микробной популяции (по разным оценкам, не более 0,1— 1,0 %). И хотя в результате введения новых сред и усовершенствования методов культивирования число выделяемых видов микроорганизмов постоянно увеличивается, тем не менее информация о составе микробной популяции, получаемая при анализе роста на питательных средах, заведомо неполная. Это хорошо подтверждает сравнение результатов количественного учета микроорганизмов методами высева на питательные среды и подсчета клеток под микроскопом, а также сравнение с результатами посева данных, полученных с использованием ДНК-зондов. В связи с этим для экологии микроорганизмов большое значение имеют методы, позволяющие, насколько возможно вести наблюдения за микроорганизмами непосредственно в их природных местообитаниях. Многие из подобных методов разработаны давно, но с развитием техники созданы их новые, усовершенствованные модификации.
37.1. МЕТОД СТЕКОЛ ОБРАСТАНИЯ
Выдающимся микробиологом Н. Г.Холодным (1882—1953) были разработаны такие методы прямого микроскопического изучения почвенных микроорганизмов, как метод стекол обрастания, метод почвенных камер, метод проращивания почвенной пыли на стекле. Эти методы подробно описаны в книге Д, М. Новогрудского «Почвенная микробиология»*. Наиболее широкое распространение получил метод стекол обрастания по Холодному—Росси, позволяющий увидеть «микробный пейзаж». Суть метода заключается в микроскопическом изучении зафиксированных и окрашенных микробных обрастаний на предметных (или, по Росси, покровных) стеклах, инкубируемых в почве.
* Новогрудский Д.М. Почвенная микробиология. — Алма-Ата: Изд-во Каз. ССР, 1953. — С. 135 — 139.
При постановке опыта в природных условиях (в лесу, поле) в исследуемой почве острым ножом делают вертикальный разрез. В него вставляют стерильное предметное стекло, плотно прижимают к почве и слегка надавливают ладонью на почву, чтобы ее плотность по возможности не изменялась. Стекло оставляют в почве на разные сроки — от нескольких дней до недель и месяцев. Аналогичные опыты могут проводиться в лабораторных условиях, в наполненных почвой эксикаторах.
Во время инкубации стекло покрывается почвенным раствором, и на его влажной поверхности начинают размножаться различные микроорганизмы, образуя, в конце концов, характерные для данной почвы микробиоценозы.
По окончании опыта стекло осторожно извлекают из почвы, не допуская скользящего движения. Одну сторону стекла (нижнюю) тщательно вытирают. Стекло фиксируют в пламени горелки, помещают в кювету с водопроводной водой и осторожно отмывают препарат от прилипших крупных частиц почвы, затем препарат высушивают и окрашивают. Рекомендуется окрасить стекло 1%-м карболовым эритрозином (время окраски 1 ч), промыть дистиллированной водой, высушить и микроскопировать с иммерсионным объективом. При микроскопировании отмечают взаимное расположение микроорганизмов, плотность обрастания, доминирующие формы.
Вместо эритрозина можно использовать краситель акридин оранжевый (водный раствор, 1:10000, окраска 2 мин), затем промыть препарат в непроточной водопроводной воде (10 мин) и исследовать с помощью люминесцентной микроскопии. Клетки бактерий, светящиеся зеленоватым светом, будут четко видны на красноватом фоне почвенных частиц. Подсчет числа бактериальных микроколоний, выросших на стекле за время инкубации в почве, и учет числа клеток (2, 4, 8, 16 и т.д.) в микроколониях позволяют с помощью соответствующего математического аппарата определить скорость миграции клеток в почве и время генерации бактерий. Для этого имеются и специальные компьютерные программы, разработанные на кафедре биологии почв МГУ.
Существуют различные модификации метода. Например, перед помещением в почву стекла покрывают какой-нибудь питательной средой или веществом, защищающим их от повреждений (фильтровальной бумагой, тканью и т.п.).
Следует отметить, что метод стекол обрастания давно используется не только в почвенной, но и в водной микробиологии; в лимнологических исследованиях он был впервые применен известным гидробиологом Э.Науманном (под названием «пластин обрастания») при изучении микроорганизмов ила.
37.2. МЕТОД КАПИЛЛЯРОВ ПЕРФИЛЬЕВА
Метод, позволяющий выявить «микробный пейзаж» в природных субстратах, был разработан Б. В. Перфильевым*. Для наблюдения за живыми микроорганизмами он предложил использовать капилляры с плоскопараллельными стенками (рис. 37.1). Благодаря плоским стеклам и узкому прямоугольному просвету каналов капилляры с попавшими в них клетками можно рассматривать под микроскопом, причем при большом увеличении и с иммерсией. Создание подобных капилляров стало возможным благодаря разработке специальной капиллярной микростеклотехники, основанной на шлифовке и спекании листовых стеклянных пластин с их последующим растяжением.
Для наблюдения за развитием микроорганизмов в илах Б. В. Перфильевым были сконструированы специальные приборы, представляющие собой наборы нескольких плоских капилляров, закрепленных на стеклянной палочке или специальном держателе, и были названы пелоскопы. Пелоскоп помещают вертикально в стакан, куда ранее внесен ил из водоема. Иловой раствор проникает в капилляры и распределяется, обра-
* Перфильев Б.В., Бабе Д.Р. Капиллярные методы изучения микроорганизмов. — М.-Л.: Изд-во АН СССР, 1961. — 534 с.
Рис. 37.1. Капилляры Перфильева
(Б. В. Перфильев, Д.Р. Габе, 1961): а — наборы капиллярных ячеек; б — пелоскоп; в — педоскоп
зуя микрозоны с различными физико-химическими условиями, аналогично тому, как это имеет место в образце ила. Вместе с иловым раствором по соответствующим микрозонам распределяются и микроорганизмы, воспроизводя так называемый вторичный диагенетический бактериальный иловой профиль. Через некоторое время (от 2— 3 дней до нескольких месяцев) капилляры извлекают из ила, тщательно протирают снаружи и микроскопируют, отмечая состав и характер распределения микроорганизмов. Использование пелоскопа позволило Перфильеву исследовать микрозональное строение ила и происходящие в нем процессы минералообразования, а также обнаружить и описать новые необычные микроорганизмы, которые не удавалось выделить на питательных средах. Многие из них характеризуются необычной морфологией и сложным циклом развития.
На аналогичных принципах основаны и другие сконструированные Б. В. Перфильевым приборы: педоскоп — для анализа микробного пейзажа в почвах и грунтах, крено-скоп — для изучения микроорганизмов грунтовых и подземных вод, перифитонометр — для микроскопического исследования водных обрастаний. Использование этих установок позволило значительно расширить наши представления о разнообразии микроорганизмов, их взаимоотношениях и геологической деятельности. Разработанная Перфильевым капиллярная техника может быть использована и в других направлениях микробиологических исследований, например для выделения культур из отдельной клетки
(капиллярные методы одноклеточной изоляции, см. гл.6.), культивирования микроорганизмов (методы капиллярных микрокультур), а также для их количественного учета (капиллярный метод прямого счета микробных тел, см. гл. 7).
Другими примерами методов микроскопического анализа, часто используемых в микробной экологии, могут служить и методы люминесцентно-микроскопического наблюдения микроорганизмов в почвенных монолитах по Звягинцеву, электронномикроскопические исследования почвенных микроорганизмов, методы исследования адсорбции почвенных микроорганизмов с помощью люминесцентной микроскопии. Многие из них разработаны сотрудниками кафедры биологии почв МГУ и подробно описаны в руководствах по почвенной биологии.
37.3. КОЛОНКА ВИНОГРАДСКОГО
Колонка Виноградского (рис. 37.2), названная по имени крупнейшего русского ученого-микробиолога Сергея Николаевича Виноградского (1856—1953), представляет собой один из способов получения накопительной культуры фототрофных бактерий*. Кроме того (и это особенно важно), она является моделью, позволяющей представить взаиморасположение в водоеме разных групп микроорганизмов, прежде всего связанных с циклами углерода и серы.
При постановке колонки Виноградского используют высокий цилиндр объемом 500 мл, на дно которого помещают ил из исследуемого водоема (20—25 г, примерно до '/5 высоты). Ил должен быть как можно более свежим, желательно отобрать его в день постановки колонки. В цилиндр вносят также немного (примерно) 0,5 г CaSO4 и небольшое количество органического вещества — это может быть тонко нарезанная фильтровальная бумага, разлагающиеся растительные остатки и т.д. (В случае соленого водоема добавляют также соответствующее количество NaCl.) Затем цилиндр доверху заполняют водой (хорошо отстоявшейся водопроводной или иногда профильтрованной водой из исследуемого водоема), закрывают ватной пробкой или колпачком из фольги и помещают на свет. Полученную таким образом колонку выдерживают в течение 2 — 3 мес при комнатной температуре в условиях естественного освещения (на окне), но можно инкубировать ее и в люминостате. Во время инкубации за колонкой регулярно ведут наблюдения, отмечая происходящие в ней визуально заметные изменения. Периодически длинной пастеровской пипеткой с разной глубины отбирают пробы воды и ила, которые затем микроскопируют и/или рассевают в питательные среды для выделения разных групп микроорганизмов (пробы отбирают осторожно, не перемешивая содержимое колонки). Результаты наблюдений зарисовывают и записывают.
Важнейшей закономерностью, которую можно выявить, наблюдая за развитием колонки Виноградского, является возникновение вертикального градиента окислительно-восстановительных условий (от анаэробных в нижней части к аэробным в верхней) и создание многочисленных экологических ниш для разных групп бактерий.
В типичном случае в первые дни после постановки колонки в толще ила начинается анаэробное разложение органического вещества, катализируемое группировкой мик-роорганизмов-гидролитиков и первичных анаэробов (бродилыциков). Продукты брожения (Н2, органические кислоты, в частности ацетат; спирты) используются вторичными анаэробами, в том числе сульфатредуцируюшими бактериями. Продукты метаболизма последних (сульфид и СО2) диффундируют в среду и служат субстратами для роста фототрофных аноксигенных (пурпурных и зеленых) и хемолитотрофных (тионовых, бесцветных серных) бактерий. Аноксигенные фототрофные бактерии развиваются в анаэробной зоне осадка и воды, образуя окрашенные слои пли пятна на обращен-
* Шлегель Г. Общая микробиология. — М.: Мир, 1987. — С. 382 — 383. Шлегель Г. История микробиологии. — М.: УРСС, 2002. — С.74—94.
Рис. 37.2. Колонка Виноградского (по Г. Шлегелю, 2002):
1 — пурпурные и зеленые серобактерии; 2 — цианобактерии и водоросли
ной к свету стороне цилиндра. В верхней части колонки появляются цианобактерии и водоросли, выделяющие на свету О2. Тионовые и нитчатые бесцветные серные бактерии могут развиваться в условиях одновременного присутствия H2S, диффундирующего снизу, и О2, поступающего сверху.
В разные периоды времени в разных зонах колонки создаются условия и для развития других многочисленных групп микроорганизмов, входящих в состав экосистемы водоема. Полнота их учета зависит от тщательности наблюдений и времени инкубации.
Колонка Виноградского имитирует события, происходящие в природном водоеме, и, как и в природной системе, многие параметры в ней являются неконтролируемыми, не зависящими от исследователя. Вместе с тем, меняя некоторые условия (количество добавленного ила, при
роду вносимого органического вещества, температуру инкубации, освещенность, спектральный состав света и т.п.), можно добиться преимущественно-
го развития определенных групп микроорганизмов.
Примечание. Целесообразно совместить постановку задачи с проведением практики по экологии микроорганизмов и включить результаты анализа в отчет по практике.
Глава 38
ЦИКЛ АЗОТА И МИКРООРГАНИЗМЫ В НЕМ УЧАСТВУЮЩИЕ
В природе цикл азота состоит из нескольких этапов, основную роль в которых играют микроорганизмы, преимущественно бактерии. При этом только бактерии могут выполнять все реакции цикла. В этом круговороте участвует молекулярный азот и его связанные соединения — минеральные и органические (рис. 38.1).
38.1. БИОЛОГИЧЕСКАЯ ФИКСАЦИЯ АТМОСФЕРНОГО АЗОТА
Процессом фиксации атмосферного азота лимитированы все остальные звенья превращения азота. По значению этот процесс можно сравнить с фотосинтезом: установлено, что общая продукция микробной фиксации азота составляет до 330 млн т/год.
Способность к биологическому связыванию молекулярного азота присуща только прокариотам. Азотфиксаторы (диазотрофы) известны среди 100 родов бактерий, среди которых такие функциональные группы, как анаэробные клостридии, сульфатредуци-рующие бактерии, энтеробактерии, фотосинтезирующие анаэробы, метанотрофы, спириллы, псевдомонады, актиномицеты, цианобактерии и др.
Различают симбиотическую и ассоциативную азотфиксацию. Последняя осуществляется свободноживущими бактериями и в связи с этим распространена значительно
Азотсодержащие органические соединения (аминокислоты, пептиды) микроорганизмов, растений, животных
шире, а ее суммарный вклад в баланс азота на Земле существенно выше, чем у симбиотической. Для осуществления азотфиксации необходимо большое количество энергии, поэтому процесс активно идет в тех случаях, когда микроорганизмы находятся в тесном контакте с растением, которое обеспечивает их источником энергии.
Клубеньковые бактерии, относящиеся к роду Rhizobium, образуют симбиотическое сообщество с бобовыми растениями. При внедрении этих мелких, не более 3 мкм в длину, подвижных грамотрицательных палочек в корень происходит разрастание ткани с образованием опухолевидных клубеньков. Через корневые волоски прорастающие семена растений выделяют специальные сигнальные молекулы белковой природы — лектины. В свою очередь бактерии синтезируют экзополисахариды, чье взаимодействие с лектинами и определяет специфичность взаимодействия между разными видами растений и бактерий.
У разных видов бобовых растений клубеньки имеют свои морфологические особенности: у клевера они продолговатые и очень мелкие, у гороха и вики округлые и более крупные, у сои и фасоли их диаметр равен примерно 1 см, а на корнях нута эти разрастания имеют многочисленные выросты толщиной в несколько сантиметров. Со строением клубеньковых бактерий можно познакомиться, если приготовить препарат из тонких срезов клубенька или, лучше, срезав бритвой кусочек клубенька, выдавить из него каплю сока на предметное стекло, прибавить 1 каплю воды и высушить. После термической фиксации препарата окрасить его генциановым фиолетовым или метиленовой синью. Очень чистые препараты получаются при окраске их 3%-м эритрозином в 5%-м феноле. Хорошая окраска получается в случае применения смеси равных частей фуксина и метиленовой сини в 1%-й уксусной кислоте. Окрашивать препарат по последнему методу следует 3 — 5 с, иначе он перекрашивается и перестает быть видна внутренняя структура бактерий. На препарате в синий цвет окрашена ткань клубенька, в красный — сами бактерии. Размеры клеток Rhizobium колеблются не только в зависимости от вида и условий выращивания, но даже и в пределах одного клубенька. Клубеньковые бактерии подвижны. В клубеньках можно встретить удлиненные, а у некоторых форм ветвистые клетки — так называемые бактероиды.
Для выделения клубеньковых бактерий пользуются следующими методами.
• Свежие клубеньки отмывают от почвы в стерильной воде. Затем в стерильных же условиях их растирают в небольшом количестве воды, и взвесь петлей вносят в пробирки с бобовым отваром. На 2 — 4-й день 1 мл взвеси, находящейся в пробирке, подвергают разведению в 6 —8 пробирках. Из последнего разведения с помощью шпателя производят высев на плотную среду. Колонии мелкие, чаще белые, слизистые.
• Клубеньки тщательно отмывают от почвы (для этого удобны жесткие кисточки и зубные щетки). Затем их погружают на 1 мин в этиловый спирт, далее на 5 мин в 0,1%-й водный раствор сулемы, а для удаления сулемы их вновь погружают на 1 мин в спирт. После этого клубеньки тщательно промывают стерильной водой, переносят в чашку Петри или в баночку с небольшим количеством (1 — 1,5 мл) воды.
Для стерилизации клубеньков удобен тигель Гуча с пористым дном, который прокаленным пинцетом последовательно переносят в большие закрытые чашки Петри или в тигли со стерилизующими растворами. Промывают клубеньки над колбой Бунзена 0,5 —0,7 л стерильной воды и быстро переносят в маленькую стерильную баночку или пузырек из-под пенициллина. Клубенек раздавливают пинцетом и взвесь бактерий вносят в охлажденный до 40 °C 5%-й отвар белой фасоли или бобов, содержащий 2 % сахарозы; 0,1 % К2НРО4; 0,03 % MgSO4- 7Н20; 1 % красителя конго в разведении 1:400 и 1,5 % агара, перемешивают и вливают в чашку Петри. Можно заражать поверхность питательного агара, растирая взвесь бактерий одним и тем же шпателем последовательно в 3 чашках. Особенно аккуратно надо проводить выделение долгорастущих форм, какими, например, являются клубеньковые бактерии люпина, обнаруживающиеся лишь через 6 —9 дней после посева. Из большинства клубеньков клетки прорастают на 3—5-й день.
• При выделении клубеньковых бактерий из почвы пользуются и другим методом. Из почвы делают взвесь в стерильной воде в соотношении 1:1000. У капилляра пипетки Пастера с питательной средой (маннит — 20,0 г; К2НРО4; MgSO4; NaCl — по 0,2 г; K2SO4 — 0,1 г; СаСО3 — 5,0 г; кристаллический фиолетовый — 0,01 г; воды 1000 мл) отламывают стерильно кончик и опускают его на 2 ч в почвенную взвесь. Клубеньковые бактерии под действием хемотаксиса проникают в капилляр. Затем содержимое капилляров переносят в агаризованную среду того же состава, где через 2 — 3 дня разовьются колонии клубеньковых бактерий, в частности Rhizobium.
Клубеньковые бактерии обладают избирательностью по отношению к растению-хозяину. Из 16 групп клубеньковых бактерий назовем основные группы (по растениям-хозяевам): 1) гороха, вики, чечевицы, чины, русских бобов; 2) клевера красного, белого, пунцового; 3) люцерны, донника; 4) фасоли; 5) люпина, сераделлы; 6) сои; 7) коровьего гороха, арахиса; 8) нута; 9) эспарцета; 10) маша. Бактерии этих групп отличаются друг от друга и рядом морфофизиологических культуральных признаков (см. определители).
Для накопления клубеньковых бактерий можно воспользоваться одной из следующих сред:
• К2НРО4 — 0,5 г; MgS04- 7Н20 и NaCl — по 0,2 г; CaSO4 — 0,1 г; дрожжевая вода — 100 мл (10 г дрожжей выдерживают 2 сут в 100 мл воды при 37 °C); маннит — 10 г; MnSO4 — 0,005 г; (NH4)6Mo7O24 — 0,002 г в 1000 мл воды.
• Клубеньковые бактерии хорошо развиваются и в условиях глубинного культивирования с аэрацией на различных средах из семян или сена бобовых и на средах с дрожжевым отваром при температуре 24—26 °C. Исходный титр при посеве — не менее 1 млн на 1 мл засеваемой среды. Уже через 24 ч титр культуральной жидкости для быстрорастущих форм достигает 8 • 108—2 • 1010 клеток. У медленно размножающихся (люпин) через 2 сут титр увеличивается примерно до 4 • 108 кл.
Выделенными бактериями можно заразить семена соответствующих бобовых растений и затем наблюдать за образованием и развитием на них клубеньков, а также за развитием самого бобового растения. Для этого поступают следующим образом. В высокие колбы или цилиндры наливают среду: MgS04 — 0,1 %; К2НРО4 — 0,1 %; Са3(РО4)2 — 0,002 %; FeSO4 — следы; агар — 0,8— 1,0 %. Агар наливают в колбу слоем в 1 см, а цилиндры заполняют наполовину. Посев проводят стерильными семенами, так как с не стерильными семенами в среду могут попасть другие микроорганизмы, в том числе и клубеньковые с места репродукции семян. Для этого семена тщательно моют, затем на 3 — 4 с погружают в спирт, далее на 10 мин в 0,1 %-й раствор сулемы. При отсутствии сулемы можно простерилизовать семена 1 %-м бромом в течение 15 мин, после чего семена следует тщательно промыть стерильной водой. В стерильную среду асептически вносят клубеньковые бактерии данного вида растения или почвенную взвесь в соответствующем разведении. Приготовленные таким образом колбы с семенами закрывают ватными пробками и помещают в хорошо освещаемое место с искусственным или естественным освещением.
В том случае, если испытуемые бактерии были активны, через 2 — 4 недели на корнях растений появятся видимые невооруженным глазом клубеньки. При контроле корневых волосков под микроскопом образование клубеньков можно выявить и раньше, на 8— 12-й день. Если ввести в несколько сосудов почвенную взвесь в различных разведениях, то по образованию клубеньков можно сделать заключение о присутствии в исследуемых взвесях клубеньковых бактерий (метод предельных разведений). Этим методом пользуются для количественного учета клубеньковых бактерий в почве.
При изучении развития клубеньков у клевера выяснилось, что на 3 —4-й день развития на корнях красного клевера появляются корневые волоски, вокруг которых можно заметить скопления бактерий. Отмыв корни с помощью мягкой кисточки, их опускают на 30—40 с в 1%-й раствор метиленовой сини и затем отмывают в воде до тех пор, пока протоплазма корневых волосков не обесцветится. После этого в корневых волосках можно заметить окрашенные мелкие короткие палочки бактерий.
Со старением культур клубеньковых бактерий появляются бактероиды, форма которых характерна для каждого вида бактерий; у гороха, чины, вики — ветвистые, у клевера — часто шарообразные, у сераделлы, фасоли, люпина — мало отличающиеся от обычных палочек. Даже в пределах вида в зависимости от условий форма бактероидов может меняться. Бактероиды усиленно образуются при добавлении к среде солей органических кислот: янтарной — 0,5 г на 1 л среды, а также винной и яблочной в количестве 1 г/л. Для получения бактероидов в питательные среды добавляют фосфорнокислые соли, сернокислый магний, нитраты, вытяжки из бобовых растений. Все эти изменения клубеньковых бактерий можно проследить при микроскопировании в динамике развития культур и бактероидной ткани клубеньков.
Соли азота и навоз задерживают развитие клубеньков у растения: нитраты — при концентрации 1:10 000, аммонийные соли — при концентрации 1: 2000. Поэтому при посевах растений вносят солей азота не более 10% от нормы, т.е. столько, сколько необходимо на первые 20 дней, до развития клубеньков. К органическому азоту клу
беньковые бактерии менее чувствительны, а почвенный гумус стимулирует их развитие и образование клубеньков. Для развития клубеньковых бактерий, а также для образования клубеньков у растений необходимы фосфаты, соли калия и кальция. При отсутствии в среде бора и молибдена клубеньки не образуются — бактерии вырождаются. Оптимальные концентрации этих элементов — 0,5 — 1,0 мг на 1 кг среды.
Клубеньковые бактерии развиваются в пределах 6—36 °C, оптимальной для них является температура 20—28°С, погибают они при 60—70°С. Легко переносят низкие температуры: -15°...-19’С. Границы pH широкие: развитие в среднем происходит в пределах 4,3 — 11,0, оптимум pH около 7. При плохом доступе воздуха клубеньки образуются плохо и фиксация азота ничтожна. Для клубеньковых бактерий оптимальная влажность составляет 60— 80 % от полной влагоемкости почвы. Вирулентность и активность клубеньковых бактерий тесно связана с развитием растения. Наиболее активны клубеньки, выделенные в фазы бутонизации и цветения.
Клубеньки образуются только на свету. При недостатке освещения активные расы клубеньковых бактерий, находящиеся в уже развившихся клубеньках, приобретают паразитические свойства: недостаток углеводов заставляет их разрушать протоплазму клеток растения-хозяина. Клубеньковые бактерии в почве остаются в активном состоянии от 3 до 15 лет.
Для выделения свободноживущих аэробных азотфиксаторов используют среды Эшби и Федорова в модификации Калининской (состав см. в приложении 4) Для получения накопительной культуры диазотрофов агаризованную среду разливают в стерильные чашки Петри и дают остыть на горизонтальной поверхности. Затем, пользуясь стеклянным капилляром, смоченным в воде, раскладывают на поверхность среды в чашке (по трафарету) 15 — 25 комочков почвы величиной с булавочную головку. Вместо агара в качестве уплотнителя среды можно также использовать силикагель. В этом случае готовят концентрированную среду Эшби, концентрацию всех соединений в которой увеличивают в 10 раз. На поверхность чашки наносят 2—3 мл этой среды и подсушивают в сушильном шкафу (10—15 мин при 60 °C). Затем, как описано выше, раскладывают комочки почвы. Продолжительность культивирования бактерий — 7— 10 сут. Получение накопительной культуры азотфиксаторов проверяют по образованию слизистых колоний (часто окрашенных при длительном росте в коричневый цвет) вокруг комочков почвы на чашках. При микроскопировании культуры обнаруживают обычно представителей родов Azotobacter или Beijerinckia. Для выявления внеклеточных капсул используют метод негативного контрастирования (см. гл. 5).
При выделении накопительной культуры свободноживущих анаэробных азотфик-сирующих бактерий рода Clostridium используют среду Виноградского (состав см. в приложении 4). В высокие пробирки вносят небольшое (0,5—1,0 г) количество почвы, наливают среду Виноградского, оставляя воздушное пространство в 1 см между пробкой и средой, и пастеризуют в течение 10 мин при 80 °C (в водяной бане). Пробирки помещают в термостат (30 °C) на 5 — 7 сут. В таких условиях обычно вырастает С. pas-teurianum. После окончания культивирования отмечают рост азотфиксаторов (помутнение среды, образование газа, появление запаха масляной кислоты) или его отсутствие. При микроскопировании культуры в препаратах «раздавленная капля» и «висячая капля» отмечают наличие подвижных палочек, образующих споры. В клетках бактерий выявляют характерные гранулы крахмалоподобного вещества гранулезы. Для этого к капле суспензии клеток на предметном стекле добавляют каплю раствора Люголя. Включения гранулезы окрашиваются в синий цвет.
Для работы с чистыми культурами анаэробных диазотрофов среду Виноградского стерилизуют при 0,5 ати, заливают в стерильные высокие пробирки до пробки и прогревают на кипящей водяной бане в течение 20 мин для удаления растворенного в среде кислорода. Затем среду в пробирках быстро охлаждают под струей холодной воды и проводят посев суспензией микроорганизмов пипеткой на дно пробирки.
Диазотрофы обнаруживаются как в ассоциях с растениями, так и с животными. Доказана способность к азотфиксации у бактерий кишечной группы рода Klebsiella, живущих в качестве комменсалов в пищеварительном тракте человека.
Главную роль в азотфиксации играет нитрогеназа, представляющая собой комбинацию из двух белков. Молекула одного из них содержит два атома молибдена и около 30 атомов железа, молекула другого — только железо. В активации молекулы азота участвует молибден, а у некоторых бактерий — заменяющий его ванадий. Соединения железа используются как переносчики электронов. Процесс требует присутствия восстановителя с низким окислительно-восстановительным потенциалом, ионов магния и АТФ, энергия распада которой используется для восстановления молибдена. Источниками энергии в почве могут быть гидролизуемые микроорганизмами растительные и животные остатки, а также почвенное органическое вещество.
Нитрогеназная система катализирует восстановление азота до аммония, который включается в молекулу глутаминовой кислоты в реакции, катализируемой глутаминсинтетазой:
глутаминовая кислота + NH4 + АТФ глутамин + АДФ + Фн.
Затем с помощью глутаматсинтазы осуществляется перенос амидной группы на молекулу а-кетоглутарата:
а-кетоглутарат + глутамин -> 2 глутаминовая кислота.
Азот, накопленный биологическим путем, находится в органических соединениях, главным образом в белке в аминной форме. Активность нитрогеназы снижается и подавляется при наличии более выгодного источника азота, чем атмосферный азот, например ионов аммония (switch-off-effect).
Нитрогеназная система азотфиксаторов чувствительна к кислороду. В снижении ингибирующего действия кислорода определенную роль играют морфолого-цитологические и физиолого-биохимические особенности клеток, в частности капсулы и дополнительные клеточные покровы, увеличение скорости дыхания (многие из диазотрофов имеют разветвленную дыхательную цепь). Нитчатые цианобактерии для разобщения процессов азотфиксации и фотосинтеза формируют специальные дифференцированные клетки — гетероцисты. Представители рода Gloeothece (одноклеточные цианобактерии) способны к аэробной азотфиксации за счет наличия мощного чехла, окружающего клетки. Возможны конформационные изменения нитрогеназной системы. Поступление молекулярного кислорода в бактероиды контролируется леггемоглоби-ном — цитоплазматическим гемопротеином, в синтезе которого участвуют и растение, и бактерии-симбионты.
В экспериментах фиксация атмосферного азота прокариотами легче выявляется при низком парциальном давлении кислорода и в анаэробных условиях. Вместе с тем недавно был выделен микроорганизм Streptomyces thennoautotrophicus, нитрогеназная система которого не чувствительна к кислороду, а сам актиномицет способен к окислению СО. Другие особенности нитрогеназной системы этого микроорганизма — независимость от АТФ и неспособность восстанавливать ацетилен, в то время как «обычные» нитрогеназы восстанавливают ацетилен, азид и цианид.
38.2. АММОНИФИКАЦИЯ
Аммонификация — это отщепление аминогруппы от аминокислоты с выделением свободного аммиака в процессе дезаминирования. Аммонификация производных белка — пептидов и аминокислот, представляет собой наиболее динамичное звено в цикле азота, хотя этому процессу подвержены и другие азотсодержащие органические соединения — производные нуклеиновых кислот, пуриновые и пиримидиновые основания, мочевина и мочевая кислота, азотсодержащий полисахарид хитин, гуминовые кислоты и др.
Внеклеточные протеазы микроорганизмов гидролизуют высокомолекулярные белки до пептидов различных размеров и аминокислот. В дальнейшем аминокислоты могут участвовать в синтезе белка и других полимеров, а также подвергаться переаминиро-ванию, дезаминированию или декарбоксилированию. Процесс отщепления аминогруппы с высвобождением аммиака может протекать как внутри клетки, так и внекле-точно.
Гидролитическое разложение белков с одновременной аммонификацией продуктов такого гидролиза легко выявить, если чашку с МПЖ заразить маленькими частичками почвы или навоза. Можно сделать и укол в столбик МПЖ иглой, которой предварительно прикасаются к почве. Уже через 1 — 2 сут в местах заражения можно наблюдать развитие гнилостных микробов (в просторечии процесс разложения белоксодер-жащих веществ называется гниением) и разжижение желатина. По месту, где происходит разжижение желатина (сверху или в средней части или у дна пробирки), можно сказать, протекает ли этот процесс под влиянием аэробов или анаэробов (см. также разд. 14.12).
При разложении белка кроме аммиака образуются СО2, сероводород, жирные и ароматические кислоты, спирты, индол, скатол, метилмеркаптан и другие вещества.
Наиболее активные аммонификаторы — это представители грамположительных споровых бактерий из рода Bacillus. Из не образующих эндоспоры форм к аммонификаторам относятся виды грамположительных бактерий родов Micrococcus, Arthrobacter, Mycobacterium, грамотрицательные виды рода Proteus, Pseudomonas, а также многие микромицеты.
Роль аммонификаторов в природе значительна, поскольку доля белка в тканях умерших растений и животных велика, а аммонифицирующие микроорганизмы осуществляют минерализацию белков, разлагая их в конечном счете до СО2, NH3 и H2S. Образующийся при микробном разложении азотсодержащих органических соединений аммиак частично адсорбируется в почве, потребляется как источник азота в процессе метаболизма почвенных микроорганизмов и в аммонийной форме растениями (иммобилизуется), выделяется в атмосферу, а также окисляется в нитриты и нитраты в процессе нитрификации.
Для выделения аммонификаторов используют мясопептонный бульон, который в количестве 30 мл наливают в колбы на 100 мл, куда затем добавляют 0,3 г почвы. Колбы закрывают ватными пробками. Для обнаружения над средой аммиака, выделяющегося в процессе аммонификации, в колбе между стенкой и пробкой закрепляют лакмусовую бумажку, смоченную дистиллированной водой (бумажка не должна касаться среды). Сверху колбочку закрывают пергаментной бумагой или полиэтиленом и ставят в термостат (30 °C) на 4 — 6 сут. При образовании аммиака красный лакмус синеет. Выделение и условия культивирования аммонифицирующих бактерий описаны также в разд. 14.1.
38.3. НИТРИФИКАЦИЯ
Нитрификация является единственным в цикле азота процессом, ведущим к образованию окисленных форм азотистых соединений. Все известные хемо-автотрофные нитрифицирующие бактерии, окисляющие восстановленные формы азота, отнесены к семейству Nitrobacteriaceae. Все они представлены грамотрицательными одноклеточными микроорганизмами, облигатными аэробами или микроаэрофилами. Они различаются по форме и размерам, способам размножения, типам жгутикования подвижных форм и другим характеристикам. В результате окисления аммиака нитрификаторы получают энергию для автотрофной ассимиляции СО2 (см. также разд. 24.3).
Нитрификаторы представлены двумя группами, каждая из которых проводит один из двух этапов окисления азота. Бактерии, окисляющие аммоний до нитритов, принадлежат к родам Nitrosomonas, Nitrosococcus, Nitrosolobus, Nitrosospira, Nitrosovibrio. Из бактерий, осуществляющих вторую фазу нитрификации (перевод нитритов в нитраты), лучше других изучены представители родов Nitrobacter, Nitrococcus, Nitrospira, Nitrospina.
Процессу нитрификации предшествуют поглощение NH4 и перенос его через ЦПМ с помощью медьсодержащей транслоказы. Окисление NH4 происходит на клеточных мембранах. Первую фазу нитрификации можно выразить уравнением:
NH4 + 1,5 О2 -> NO2- + Н2О + 2Н+, AG°' = -272 кДж/моль, вторую фазу'—
NO2- + 0,5 О2 -> NO3-, Дб°' = -73 кДж/моль.
Результаты многочисленных исследований позволяют считать, что субстратом для нитрификации первой фазы, видимо, является не аммоний, а аммиак, который превращается в нитрит в результате двух последовательных реакций. В первой из них участвует Cu-содержащая, мембраносвязанная монооксигеназа:
NH3 + О2 + НАДН + Н+ -> NH2OH + Н2О + НАД+
Гидроксиламин далее ферментативно окисляется до нитрита:
NH2OH + О2 -> NO2- + Н2О + Н +
Полагают, что сначала гидроксиламин превращается в нитроксил (HNO), а также оксид азота в связанной форме:
NH2OH -> (HNO) -> (NO) -> NO2-
Окисление гидроксиламина сопряжено с функционированием дыхательной элект-ронтранспортной цепи (ЭТЦ), в которую входят цитохромы типов с и с, а также терминальная оксидаза. Конечным акцептором служит молекулярный кислород.
Действие дыхательной системы приводит к синтезу АТФ. Часть энергии Дцн+, видимо, расходуется на образование НАДН, необходимого для ряда биосинтетических реакций в результате обратного переноса электрона, поскольку редокс-потенциал гид-роксиламина недостаточен для его прямого восстановления.
Реакцию окисления нитрита в нитрат катализирует Mo-зависимая мембраносвязанная нитритоксидоредуктаза. В состав ЭТЦ нитрификаторов второй фазы входят цитохромы ah ct и аа3. В условиях высокого содержания в среде молекулярного кислорода Функцию терминальной оксидазы может выполнять цитохром о.
Автотрофные нитрификаторы распространены в почве, а также различных пресных соленых водоемах, в сточных водах, на горных породах, на каменных и железобетонных строениях. В своей деятельности нитрификаторы развиваются совместно с микро-эрганизмами, осуществляющими минерализацию (аммонификацию) азотсодержащих
органических веществ и приводящими к накоплению исходного субстрата нитрификации — аммиака.
Для выделения накопительной культуры нитрифицирующих бактерий используют среду Виноградского для нитрификаторов (состав см. в гл. 51), которую разливают в колбы объемом 50 мл по 15 мл в каждую (высота слоя среды должна быть 1,0 — 1,5 см). В каждую колбу вносят на кончике шпателя небольшое количество СаСО3. Посевным материалом служит почва. Колбы помешают в термостат на 30 °C. Через 1 —3 недели о развитии нитрификаторов судят по появлению в среде нитритов и (или) нитратов. Для их обнаружения проводят качественные реакции на нитриты — с реактивом Грисса или с помощью крахмал-йодной реакции — и на нитраты с дифениламином. Рост нитрифицирующих бактерий ведет также к подкислению среды.
Качественные реакции на нитриты.
• Реакция на нитриты с реактивом Грисса основана на образовании в кислой среде в присутствии нитритов и ароматических аминов (сульфаниловой кислоты и ct-нафтиламина) азотсоединения, окрашенного в краснорозовый цвет. Для обнаружения нитритов к капле реактива Грисса на керамической пластинке добавляют каплю выросшей жидкой культуры или культуральной жидкости. Появление красного окрашивания свидетельствует о присутствии нитритов.
• Крахмал-йодная реакция основана на том, что нитриты в кислой среде окисляют йодистый цинк с выделением йода, присутствие которого обнаруживают с крахмалом. Для проведения реакции к капле выросшей жидкой культуры на керамической пластинке добавляют каплю раствора, содержащего ZnCl2, KI, крахмал и каплю 5%-го раствора НС1. При наличии в среде нитритов появляется синее окрашивание.
Качественная реакция на нитраты.
• На белой керамической пластинке несколько кристаллов дифениламина растворяют в капле концентрированной серной кислоты и к смеси добавляют каплю исследуемой жидкой культуры. При наличии нитратов возникает синее окрашивание. Контролем служит исходная стерильная среда. Следует иметь в виду, что пробу на нитраты можно проводить только в отсутствие нитритов, так как последние тоже дают синее окрашивание с дифениламином.
Изменение значения pH среды определяют с помощью бромтимолблау (в качестве контроля используют исходную стерильную среду).
Накопительную культуру микроскопируют в препарате «раздавленная капля» и отмечают морфологические особенности выросших бактерий и их подвижность.
Анаммокс-процесс. В конце 90-х годов XX в. было экспериментально выявлен процесс анаэробного окисления аммония, фактически анаэробная нитрификация. Возможность такого процесса предсказывалась еще в 80-х годах Е. Бродой. В настоящее время анаммокс-процесс обнаружен в сточных водах, а бактерии, осуществляющие его, получили название «анаммокс-бактерий». В чистые культуры эти микроорганизмы пока еще не выделены, однако двум из организмов уже были даны предварительные названия Candidatus «Brocadia anammoxidans» и «Kuenenia stuttgartiensis» (KS). Суммарно анаммокс-процесс можно представить в следующем виде:
NHt + NO2- + Н2О -> N2 + 2Н2О
Таблица 38.1
Сравнительная характеристика процессов аэробного и анаэробного окисления аммония*
Параметр Нитрификация (например, Nitrosomonas sp.) Анаммокс процесс (например, Brocadia sp.) Единицы
Реакция Свободная энергия Выход биомассы Скорость процесса: в аэробных условиях в анаэробных условиях: Р(тах) g Ks (NH3 KJNH4) Ks (О2) NH4+ 1,5O2->NO2 + + Н2О + 2Н+ -275 0,08 200-600 2 0,04 (аэробиоз) 0,73 5-2600 10-50 NH4 + NO2 + Н2О -> -> N2 + 2Н2О -357 0,07 0 60 0,003 (анаэробиоз) 10,6 5 < 5 кДж/моль МОЛЬС/МОЛЬ ]\Ц-Ц нмоль/мин мг белка нмоль/мин мг белка ч-1 сут мкМ мкМ мкМ
* Киепеп J. G., Jetten M.S.M. Extraordinary anaerobis ammonium-oxidizing bacteria. — 2001. ASM News. — V. 67(9). — P. 456—463.
Процесс анаэробной нитрификации протекает как минимум на порядок медленнее аэробного процесса. Сравнительные характеристики классической нитрификации и анаммокс процесса приведены в табл. 38.1.
Гетеротрофная нитрификация. Многие хемоорганогетеротрофные бактерии, принадлежащие к родам Arthrobacter, Flavobacterium, Xanthomonas, Pseudomonas, Alcaligenes, Nocardia, Streptomyces, и некоторые другие способны окислять аммиак и другие азотсодержащие органические вещества, образуя при этом гидроксиламин, нитриты, нитраты, гидроксамовые кислоты, оксид и закись азота, а также другие продукты. Гетеротрофная нитрификация, однако, не обеспечивает эти микроорганизмы энергией. Одна из предполагаемых схем процесса выглядит следующим образом:
RNH2 -> RNHOH -э RNO -> RNO2 -> RNO3
Возможно, этот процесс связан с разрушением перекиси водорода, которая образуется микроорганизмами под действием пероксидазы. Образующийся при этом активный кислород окисляет NH3 до NO3.
38.4. ДЕНИТРИФИКАЦИЯ
Денитрификация — это процесс восстановления нитратов до нитритов и далее до какой-либо из газообразных форм азота (окиси азота, закиси азота и молекулярного азота):
NO3 -> NO2 -> NO -> N2O -> N2
Способность к денитрификации обнаружена у многих почвенных и водных прокариот, среди них фото- и хемотрофы. Наиболее часто она встречается у грамположительных бактерий из родов Bacillus и Micrococcus, а также у грамотрицательных бактерий, принадлежащих к роду Pseudomonas. Денитрификаторы — факультативные ана
эробы, переключающиеся на денитрификацию только в отсутствие кислорода. В аэробных условиях эти микроорганизмы осуществляют процесс кислородного дыхания.
В анаэробных условиях денитрификаторы используют нитраты и нитриты как конечные акцепторы электронов при окислении органических субстратов для получения энергии. Серная бактерия Thiobacillus denitrificans восстанавливает нитраты в процессе окисления серосодержащих соединений.
В процессе денитрификации каждый из четырех восстановительных этапов катализируется специфической мембраносвязанной редуктазой. Электрон-транспортные цепи денитрификаторов в анаэробных условиях содержат флавопротеины, хиноны, цитохромы типов b и с (цитохромоксидазы в этих условиях не синтезируются). Выход энергии при денитрификации только на 10% ниже, чем при кислородном дыхании.
Иногда денитрификацию называют анаэробным нитратным дыханием или диссимиляционной нитратредукцией, хотя это не совсем верно. Так же как и денитрификация, диссимиляционная нитратредукция осуществляется в анаэробных условиях, а клетка получает энергию за счет использования NO3 как конечного акцептора электронов. Однако образования восстановленных газообразных форм азота в этом процессе не происходит, а восстановление нитрата заканчивается образованием нитрита. Нитратное дыхание широко распространено среди прокариот, встречаясь у представителей более чем 70 родов.
Восстановление нитратов может происходить также и химическим путем за счет водорода, образующегося в анаэробных условиях.
Денитрификация — одна из причин обеднения почв азотом и неполного использования растениями вносимых в почву азотных удобрений. В то же время этот процесс имеет большое экологическое значение в связи с тем, что он восстанавливает баланс азота в атмосфере, являясь единственным источником атмосферного азота, а также предохраняет водоемы от чрезмерного накопления в них нитратов, вымываемых из почв.
Денитрифицирующие бактерии широко распространены в почвах, особенно во влажных и богатых неразлсжившимися органическими остатками, в ризосферах, а также в илах.
Для получения накопительной культуры денитрифицирующих микроорганизмов используют среду Гилътея. Раствор Т. KNO3 — 2 г; аспарагин — 1 г, дистиллированной воды — 250 мл. Раствор II: натрий лимоннокислый — 2,5 г; КН2РО4 — 2,0 г; MgSO4- 7 Н2О — 2,0 г; СаС12 — 0,2 г; FeCl3 — следы; дистиллированная вода — 500 мл. Оба раствора сливают и доводят объем среды до 1000 мл. По индикатору бромтимоловому синему устанавливают pH 6,8 — 7,2. Среду стерилизуют при 1 ати и разливают в высокие пробирки, оставляя воздушное пространство в 1 см между пробкой и средой. Предварительно в каждую пробирку помещают стеклянный поплавок (запаенным концом вверх) так, чтобы он был полностью заполнен средой. После заражения почвой или илом пробирки помещают в термостат (25 —30 °C). Среду в одной пробирке оставляют незасеянной для контроля. Обильное газообразование, исчезновение нитратов с появлением нитритов, возможно и аммиака, свидетельствует об идущей денитрификации. О наличии или отсутствии нитратов судят по качественной реакции с дифениламином (см. 38.3).
Происходит подщелачивание среды, которое определяют с помощью бром-тимолблау. Накопительную культуру микроскопируют в препарате «раздавленная капля» и делают фиксированный, окрашенный фуксином, препарат.
Для количественного определения денитрификаторов пользуются МПА, содержащим 0,1 % KNO3 и 1 % агара. Посев проводят методом предельных разведений. О содержании денитрифицирующих бактерий в посевном материале судят по пузырькам и
разрывам в агаре, образовавшимся вследствие выделения денитрификаторами газа. Для обнаружения этих микроорганизмов пользуются средой Омелянского: KNO3 — 5,0 г; крахмал растворимый — 5,0 г; пептон — 1,0 г; К2НРО4 — 0,5 г; MgSO4-7H2O — 0,3 г; NaCl — 0,3 г; KI — 0,5 г; агар-агар — 20,0 г; дистиллированная вода —1000 мл. Около выросших колоний вырезают кусочки агара и опускают их в раствор 5%-й соляной кислоты: в присутствии нитритов агар посинеет. Выделение денитрификаторов производится на одной из вышеуказанных жидких сред, к которым прибавляют агар. Для создания анаэробных условий используют методы, описанные выше.
Для выделения денитрификаторов, окисляющих серу в серную кислоту, пользуются средой Бейеринкст. серный цвет — 100 г; KNO3 — 0,5г; Na2CO3 — 0,2 г; СаСО3 — 20,0 г; К2НРО4 — 0,2 г; дистиллированная вода — 1000 мл. Посевным материалом является почва. При 30 °C через 5 — 6 сут уже заметно восстановление нитратов и накопление серной кислоты (газ, SO42- и др.). Повторный пересев производят на ту же среду с дистиллированной водой и 0,01 % хлористого магния. Выделение развившейся перетрихиальной палочки Acidithiobacillus denitrificans производят на агаризованной среде в анаэробных условиях.
Для накопления Sulphomonas denitrificans (мелкие палочки с монотрихаль-ным типом жгутикования) используют среду Лиске: Na2S2O3 — 5,0 г, KNO3 — 5,0 г; Na(HCO3)2 - 1,0 г; К2НРО4 - 0,2 г; MgCl2 - 0,1 г; СаС12 и FeCl3 -следы; вода — 1000 мл.
Для создания анаэробных условий колбочку заполняют средой до резиновой пробки. Для выхода газов в пробку вставляют трубку, которая отходит в сосуд с той же средой. Стерилизуют среду текучим паром. Исходными субстратами для засева могут быть почва, стоячая вода или ил. Температурный оптимум 30—35 °C.
38.5. АССИМИЛЯЦИОННАЯ НИТРАТРЕДУКЦИЯ
Восстановление нитрата в реакциях конструктивного метаболизма, не приводящее к получению энергии, называется ассимиляционной ншпратредукцией.
Способность к ассимиляции нитрата характерна для растений, некоторых грибов и бактерий. В целом процесс можно охарактеризовать следующими реакциями, которые катализируются редуктазами:
NO3 NO2 -> HNO -> NN2OH -> NH4 -> аминокислоты.
Амонийный и нитратный азот, поглощенный микробными клетками, включается в органические азотсодержащие полимеры клеточных компонентов и временно выводятся из круговорота азота, т.е. происходит их иммобилизация.
Глава 39
ЛАБОРАТОРНЫЕ ОПЫТЫ С РАСТЕНИЯМИ
В микробиологической практике растительные объекты используют, в частности, для изучения влияния микроорганизмов на рост растений и в биологических тестах при обнаружении активности фитогормонов, синтезируемых
микроорганизмами. Чаще всего тест-объектами являются сельскохозяйственные культуры: пшеница, просо, овес, ячмень, рапс, кукуруза, фасоль, горох, лук, помидоры, огурцы. Для экспериментов отбирают одинаковые по размерам и внешнему виду семена одного сорта с хорошей всхожестью.
При исследовании способности микроорганизмов колонизировать корни прорастающих семян и влияния микроорганизмов на рост и развитие растений (за счет синтеза фитогормонов, изменения минерального и водного питания, фиксации атмосферного азота и воздействия на фитопатогены) проводят лабораторные, вегетационные и полевые опыты. В лабораторных опытах растения выращивают в широких пробирках, цилиндрах или других флаконах, подходящих для целей эксперимента. Опыты необходимо проводить в стерильных условиях, для чего посуду стерилизуют в сухожаровых шкафах или в автоклавах. На начальных стадиях проращивания семян используют чашки Петри с влажной фильтровальной бумагой диаметром 10 см. Смачивание проводят стерильной водопроводной водой, раствором минеральных солей по Хогланду (см. приложение 4) или их разведением в 2—4 раза, а также средой культивирования или культуральной жидкостью (КЖ) изучаемых микроорганизмов.
В качестве субстратов можно использовать 1,5%-й водный агар или смоченные водой почву и кварцевый песок, для стерилизации которых применяют длительный режим автоклавирования. На стерильные субстраты помещают семена или 4 — 10-дневные проростки растений, пророщенные в чашках Петри.
Существует несколько способов стерилизации семян. Чем плотнее оболочка семян, тем более жестким должен быть режим стерилизации, причем необходимо учитывать, что семена бывают заражены не только с поверхности, но и изнутри. К примеру, перед стерилизацией семена бобовых можно в течение 20 мин выдерживать в мыльном растворе, после чего их 1 ч промывают в проточной водопроводной воде. Стерилизация семян облегчается, если они предварительно набухнут в стерильной водопроводной воде.
В настоящее время почти не применяют стерилизацию 1%-м раствором брома (необходимо проводить в вытяжном шкафу) и сильными кислотами (соляной, серной, азотной). Наиболее популярны следующие методы:
• обработка 0,1 — 0,5%-м раствором сулемы (HgCl2) в течение 2 — 5 мин;
• 96%-м или 70%-м этанолом в течение 1 — 5 или 2— 15 мин соответственно;
• 1 — 5%-м раствором гипохлорита кальция в течение 5 — 20 мин или 5 — 15%-м отбеливающим средством (АСЕ, Procter and Gamble) в течение 20 мин;
• 3%-м раствором перекиси водорода в течение 5—20 мин;
• 5%-м хлорамином 1 — 20 мин;
• если семена заражены грибами, их протравливают 5 мин 1%-м формалином;
• применяют также комбинированную стерилизацию, например смесью перекиси водорода и этанола (1: 1), или последовательную обработку 96%-м этанолом в течение 1 мин, а затем одним из вышеперечисленных веществ.
После стерилизации семена отмывают от 3 до 5 раз стерильной дистиллированной водой. Концентрации используемых растворов и время стерилизации могут зависеть от физиологических особенностей тест-объекта и целей эксперимента.
Стерильные семена заражают изучаемыми микроорганизмами через 1 —2 сут после прорастания семян в сосудах для проращивания или после засева в субстрат. Бактеризацию проводят с помощью суспензии бактерий в концентрации 106—107 кл/мл. Засеянные сосуды выставляют на естественный или искусственный (люминостат с фотопериодизмом) свет. Для предохранения корневых систем и микроорганизмов от света и перегрева прикрывают бумагой ту часть сосуда, где должна развиваться корневая система.
Другой сферой использования растений в микробиологии являются биотесты. Они основаны на ростовых реакциях различных частей растений, специфически реагирующих на стимуляторы роста растений — фитогормоны, которые могут содержаться в КЖ микроорганизмов. Фитогормоны — это органические, сравнительно низкомолекулярные соединения, образующиеся в растениях в малых количествах и стимулирующие в них ростовые и формообразующие процессы. Микроорганизмы также способны синтезировать основные из известных фитогормонов — ауксины, цитокинины, гиббереллины, этилен, абсцизовую кислоту и некоторые другие фитогормонподобные вещества.
В этом разделе рассматриваются несколько способов определения биологически активных ауксинов, гиббереллинов и цитокининов, содержащихся в культуральной жидкости микроорганизмов. Однако для получения более полной информации о механизмах действия, структуре, функциях и методах определения активности фитогормонов необходимо обращаться к специальной литературе по физиологии растений.
Определение влияния ауксинов на рост отрезков колеоптилей злаков
Для проведения этого биотеста используют колеоптили злаков в фазе растяжения, но не в фазе активного роста. Продолжительность деления и растяжения клеток в колеоптиле различается в зависимости от условий выращивания и проведения опыта, от выбранного тест-объекта и его сортовой принадлежности. В связи с этим необходимо проведение предварительных опытов по установлению временных границ фаз роста у изучаемых объектов, для чего измеряют длину колеоптилей в динамике, а также определяют сырую и сухую массы колеоптилей.
Для биотеста отбирают семена пшеницы, кукурузы или овса, обрабатывают их 70 %-м этанолом в течение 5 мин и замачивают в воде в течение 3 —4 ч при 22—25 °C. Набухшие семена раскладывают в кюветы (или специальные камеры) на стекло, обернутое влажной фильтровальной бумагой, и помещают в темноту в термостат на 26 °C. Отобранные проростки злаков в фазе «чистого» растяжения, выращенные в течение 3 дней до длины колеоптилей 25 — 30 мм, укладывают на линейку. Отступая от верхнего кончика 4 мм, отрезают лезвием сегмент длиной 5 мм, из которого капилляром удаляют первичный лист. Отрезки помещают на 2 ч в чашки Петри, наполненные дистиллированной водой со следами сульфата магния, после чего их переносят в стерильные чашки Петри с 20 мл стерильной культуральной жидкостью микроорганизмов (для экономии можно использовать одноразовые чашки Петри диаметром 3,5 см и 2 мл культуральной жидкости). В качестве контроля используют неинокулиро-ванную микроорганизмами питательную среду, а также растворы индолилук-сусной кислоты (ИУК) — 10, 50 и 100 мкг/мл на 5 мМ калийфосфатном буфере, pH 6,0. Инкубацию проводят в темноте во влажной камере в течение 20—35 ч при температуре 25 °C. Измеряют длину отрезков колеоптилей и определяют среднюю величину для каждой группы колеоптилей, взятой в эксперимент. Для получения статистически верных результатов эксперимент необходимо проводить в 3 — 5 повторностях, отбирая для каждой группы не менее 10 отрезков колеоптилей.
Определение влияния стимуляторов роста на корнеобразование у черенков фасоли
В этом биотесте используют черенки фасоли обыкновенной (Phaseolus vulgaris). Для этого сортовые семена фасоли выдерживают в мыльном растворе в течение 20 мин, промывают в проточной водопроводной воде 1 ч и раскладывают в кювету на стекло, обернутое влажной фильтровальной бумагой. В кювету наливают воду до уровня стекла и помещают в термостат при 20—24 °C. Проросшие семена переносят в сосуды на специальные пластиковые круги с отверстиями так, чтобы наклюнувшиеся корешки оказались в воде. Инкубацию проводят при естественном или искусственном (люминостат с фотопериодизмом) освещении при температуре 20—24 °C. В эксперименте используют черенки фасоли длиной 12 —14 см с первой парой листьев, у которых под водой отрезают базальные части на расстоянии 0,5 —1,0 см выше корневой шейки. Такие черенки в количестве не менее 5 штук в одном варианте опыта погружают нижними концами на 6 ч во флаконы с культуральной жидкостью микроорганизмов.
Контролем служат черенки, помещенные в стерильную водопроводную воду и стерильную неинокулированную среду. Также можно использовать растворы кинетина (цитокинина) в концентрации 2 мкг/мл, ИУК в концентрации 10, 50 и 100 мкг/мл и их комбинацию. Черенки должны быть погружены в растворы на 3 — 4 см. После того как черенки вынимают из опытных растворов, их ополаскивают водопроводной водой и помещают в колбы или иные флаконы с водопроводной водой, которую меняют каждый день на протяжении эксперимента. Колбы держат в условиях естественного освещения при комнатной температуре или в люминостате. Показателями активности ростовых стимуляторов служат время появления корней на черенках, высота стебля, на котором закладываются корни, длина и количество образуемых корней. Последние три характеристики учитывают через 1 — 2 нед после постановки опыта. Эксперимент проводят в трех-пяти повторностях.
Определение влияния гиббереллина на рост гипокотилей салата
Биотест проводят на гипокотилях (подсемядольное колено) проросших семян салата (Lactuca sativa). Для этого семена выдерживают в кювете на стекле, обернутом влажной фильтровальной бумагой при 25 °C в темноте в течение двух дней. Затем отбирают проростки с одинаковыми по длине корешками (6 — 8 мм) и помещают их в стерильные чашки Петри на диск фильтровальной бумаги, смоченный 6 мл КЖ микроорганизмов, а также раствором гиббереллиновой кислоты (GA3) в концентрации 25 мг/л. В качестве контроля используют стерильную неинокулированную среду. Для каждой пробы берут по 10 проростков. Инкубацию проводят, располагая опытные кюветы на 15 см ниже постоянного источника света — ламп дневного освещения, с температурой в этом положении около 25 °C. Измерение длины гипокотилей проводят в течение 5 сут. Другим вариантом опыта является проращивание семян салата при постоянном освещении и измерение длины гипокотилей через 3 сут.
В этом случае свет увеличивает ингибирование собственного роста гипокотилей салата, но не изменяет их чувствительность к гиббереллиновой кислоте.
Определение влияния цитокининов на сохранение хлорофилла в листовых отрезках ячменя
Для биотеста, разработанного О. Н. Кулаевой, используют листья первого яруса сортового ячменя, который выращивают в комнатных условиях в сосудах или ящиках с землей. В возрасте 10—15 сут отбирают сходные по размеру и цвету листья первого яруса. Из них вырезают отрезки длиной 2 см, отступив на 2 см от нижнего края листа. Сегменты раскладывают в стерильные чашки Петри на фильтровальную бумагу диаметром 10 см, смоченную 3 мл КЖ микроорганизмов. В каждую чашку помещают до 9 листовых отрезков. Чашки оставляют под стеклом в кювете' при искусственном освещении лампами дневного света и температуре 20—25 °C. В качестве контроля используют стерильную неинокулированную среду, дистиллированную воду, а также растворы кинетина в концентрациях 10 — 20 мг/л. Навеску растворяют в дистиллированной воде, добавляя по каплям 0,1 н. NaOH (или КОН), перемешивают и нагревают на водяной бане до полного растворения. Сохранение хлорофилла в сегментах листьев ячменя оценивают через 7—10 сут инкубации визуально (для качественного определения) или экспериментально. Для этого из отобранных трех отрезков вырезают пробочным сверлом по два диска диаметром 5 мм. Диски объединяют и растирают с 80%-м этанолом в фарфоровой чашке для упаривания, добавляя на кончике ланцета кварцевый песок и СаСО3.
Полученный раствор количественно переносят в центрифужную пробирку и отделяют осадок центрифугированием в течение 5 мин при 2 тыс. g. Осадок отбрасывают, а объем надосадочной жидкости доводят до 5 мл 80%-м этанолом. Оптическую плотность раствора определяют на спектрофотометре при 665 нм против контроля, которым является раствор, полученный путем объединения экстрактов хлорофилла из отрезков листьев на воде или неинокулированной среде, а также 80%-й этанол. Результаты выражают в оптических показателях спектрофотометра, умноженных на 1000. Для одной чашки Петри проводят три определения содержания хлорофилла в отрезках (всего 9 листовых сегментов на чашку), в эксперимент берут 3 чашки Петри.
Для построения калибровочной кривой используют отрезки листьев ячменя, которые раскладывают на фильтровальную бумагу, смоченную растворами кинетина разных концентраций от 1 до 50 мг/л. Контролем служат отрезки листьев на дистиллированной воде.
Результаты биотестов зависят от ряда причин, в частности от чувствительности выбранного тест-объекта к ростовым стимуляторам, содержащимся в культуральной жидкости микроорганизмов, а также от их концентрации. Если их количество будет ниже или выше действующей концентрации, то может быть отмечено угнетающее действие вещества. В ряде случаев, если микроорганизм-продуцент синтезирует значительные количества фитогормона, культуральную жидкость необходимо разводить. Кроме того, в культуральной жидкости помимо необходимого ростового стимулятора может содержаться значительное количество других биологически активных веществ, которые также влияют на растительные объекты и, таким образом, на конечный результат эксперимента. Биотесты дают ответ на вопрос о биологической активности фитогор
монов, содержащихся в культуральной жидкости, но для определения их количеств и концентрации необходимо параллельное проведение экспериментов по идентификации фитогормонов с помощью колориметрического метода с реактивом Сальковского (определение ауксинов), а также хроматографического и иммуноферментного методов. При проведении тонкослойных хроматограмм не обработанные химическими реагентами зоны, соответствующие по показателям ^идентифицированным фитогормонам, вырезают и элюируют из адсорбента действующее вещество. Подобные экстракты можно использовать в вышеописанных биотестах.
Глава 40
БАКТЕРИИ, МИНЕРАЛИЗУЮЩИЕ СОЕДИНЕНИЯ ФОСФОРА
Фосфор содержится в почве, как правило, в виде труднодоступных минеральных и органических соединений, которые становятся доступными для растений только после их минерализации микроорганизмами. Минеральные вещества представлены главным образом ортофосфатами кальция (нейтральные и щелочные почвы), железа и алюминия (кислые почвы). Органические соединения фосфора встречаются в виде инозитфосфатов, фосфорсодержащих белков, нуклеиновых кислот, фосфолипидов и др. Около половины органического фосфора почвы содержится в наиболее устойчивых к действию микроорганизмов фосфатах инозита (фитатах). Особенно широко распространен инозитолгексафосфат (фитин). Микроорганизмы, синтезирующие фосфатазу, способны отщеплять от органических фосфатов фосфорную кислоту, которая, взаимодействуя с катионами, переходит в соли фосфорной кислоты, доступные для растений.
40.1. МЕТОДЫ УЧЕТА МИКРООРГАНИЗМОВ, РАСТВОРЯЮЩИХ ОРТОФОСФАТЫ КАЛЬЦИЯ, АЛЮМИНИЯ И ЖЕЛЕЗА
Одним из самых удобных методов выделения фосфатрастворяющих микроорганизмов является чашечный, когда активные организмы отбирают из колоний на агаровой пластинке в зонах растворения фосфата. Эти зоны формируются четкими в случае, когда среда достаточно и равномерно мутна, а количество нерастворимого фосфата в ней сравнительно невелико. Поэтому наилучший эффект дает внесение в среду свежеосажденных солей.
Для выделения из почвы бактерий, растворяющих ортофосфаты кальция, используют глюкозоаспарагиновую среду Муромцева состава (г/л): глюкоза —10; аспарагин — 1; K2SO4 — 0,2; MgSO4 — 0,2; агар выщелоченный — 20; кукурузный экстракт — 0,02; вода водопроводная, pH 6,8. Стерилизация при 0,5 ати 25 мин.
Фосфаты кальция Са3(РО4)2 вносят в питательную среду методом осаждения. Для этого в стерильную расплавленную агаризованную среду вносят (из расчета на 1 л) 3,4 г СаС12-6Н2О, а после его растворения добавляют 3,8 г Na3PO412H2O. Соли вносят в сухом виде, что позволяет избежать быстрого гидролиза Na3PO4B растворе и обеспечивает постепенное взаимодействие СаС12-бН2О и Na3PO4 по мере их растворения. На 1 л
среды образуется 1,5 г свежеосажденного Са3(РО4)2. Приготовленную таким образом питательную среду разливают в чашки Петри и после застывания агара засевают поверхностно 0,1 мл почвенной суспензии из разведений 10-4—10-7, которые готовят из 1 г почвы и 100 мл стерильной водопроводной воды (разведение 10~2). Чашки инкубируют при 28 — 30 °C в течение 3 — 9 сут. По образованию зон просветления вокруг колоний активных микроорганизмов судят о растворении фосфата кальция. Наряду с внесением в среду свежеосажденных солей можно пользоваться и готовыми препаратами фосфатов, но зоны растворения получатся менее отчетливыми. Готовые фосфаты в агаризованную среду вносят в следующих количествах (г/л): Са3(РО4)2 — 4 — 5; фосфорит — 6 — 7; апатит —8—10. Порошок фосфата всыпают в горячий расплавленный агар и дают отстояться для осаждения крупных частиц. Среду сразу же разливают в чашки и засевают почвенной суспензией.
Для выделения из почвы микроорганизмов, способных к растворению ортофосфатов Fe и А1, используют свежеосажденные фосфаты, которые получают из солей алюминия и железа. С этой целью для получения А1РО4 или FePO4 смешивают по 10 мл стерильных растворов FePO4 и А1С13 или Na3PO4 и FeCl3 соответственно. Стерилизацию растворов проводят, пропуская их через фильтр Зейтца. Осажденный фосфат алюминия или железа после тщательного перемешивания вносят в расплавленную глюкозоаспарагиновую среду Муромцева (см. выше). На 1 л среды берут 570 мг Na3PO4- 12Н2О и 362 мг А1С13- 6Н2О или 405 мг FeCl3 • 6Н2О. Готовая среда содержит 264 мг А1РО4 • ЗН2О или 280 мг FePO4- 2Н2О в 1 л. Значение pH среды доводят до 6,8 10%-м раствором NaOH. По 0,1 мл почвенной суспензии из разведений 10'4—10~7 высевают на чашки со средой. После инкубации при 28 — 30 °C в течение 3 — 10 сут вокруг колоний выросших микроорганизмов видны зоны просветления.
В почве не обнаружены специфичные микроорганизмы, растворяющие ортофосфаты кальция, железа и алюминия. В этом процессе бактерии участвуют лишь косвенно. Так, многие микроорганизмы в процессе дыхания выделяют углекислоту, нитрифицирующие бактерии, окисляя аммоний, образуют азотистую и азотную кислоты, серобактерии, окисляя сероводород и серу, продуцируют серную кислоту. Эти кислоты взаимодействуют с фосфатами и образуют кислые соли, доступные растениям:
Са3(РО4)2 + 2СО2 + 2Н2О = 2СаНРО4 + Са(НСО3)2;
Са3(РО4)2 + 2H2SO4 = Са(Н2РО4)2 + 2CaSO4;
Са3(РО4)2 + 4HNO3 = Са(Н2РО4)2 + 2Ca(NO3)2.
Сходным образом действуют на фосфаты и бактерии, образующие органические кислоты в процессе ассимиляции углеводов.
40.2. МЕТОДЫ УЧЕТА МИКРООРГАНИЗМОВ, РАСТВОРЯЮЩИХ ФИТАТЫ КАЛЬЦИЯ, АЛЮМИНИЯ И ЖЕЛЕЗА
Органические соединения фосфора в почве, органофосфаты, представлены в нейтральных и щелочных почвах в основном фитатами Са и Mg, а в кислых — фитатами Fe и А1. Для обнаружения в почве бактерий, способных растворять фитаты, используют глюкозоаспарагиновый агар с соответствующим фитатом. В качестве фитата Са — Mg используют препарат фитин, представляющий собой кальций-магниевую соль инозитфосфорной кислоты. Фитин предварительно растворяют и переосаждают. Для этого 0,5 г препарата растворяют в 10 мл 1 н. НС1 и добавляют 1 н. раствора NaOH до pH 7,0. Выпавший осадок фитата Са—
Mg отстаивают, отделяют от надосадочной жидкости и вносят в 500 мл глюко-зоаспарагиновой среды. Посевы разведений почвенной суспензии инкубируют 10—12 сут при 28 — 30 °C.
Соли Fe и А1 получают из фитина. Для этого препарат растворяют в 2%-й НО и осаждают фосфат концентрированным водным раствором FeCl3. Обильный осадок фитата Fe отделяют на воронке Бюхнера и промывают соляной кислотой до тех пор, пока смывной фильтрат не перестанет мутнеть при нейтрализации его NaOH до pH 7,0. Фитат А1 получают таким же методом с помощью раствора А1С13, однако перед внесением хлористого алюминия pH раствора фитина в 2%-й НС1 предварительно доводят с помощью NaOH до значения 3,5. Осадок фитата А1 отделяют на воронке Бюхнера и обрабатывают аналогично фитату Fe. Полученные таким образом фитаты Fe и А1 вносят в горячий глюкозоаспарагиновый агар в количестве 0,5 г на 500 мл среды, размешивают до получения равномерного помутнения и, доведя pH до 7,0, разливают по чашкам Петри. Посевы разведений почвенной суспензии инкубируют 10—12 сут при 28 — 30 °C и подсчитывают количество колоний, вокруг которых образовались зоны растворения органофосфатов.
Микробиологическое растворение фитатов в основном имеет характер неферментативного и неспецифического, как это имеет место в случае растворения ортофосфатов кальция, алюминия и железа. Однако обнаружены и микроорганизмы, способные к ферментативному дефосфорилированию органических соединений фосфора с помощью фосфатазы. Это некоторые штаммы B.megaterium и Pseudomonas.
Глава 41
МИКРООРГАНИЗМЫ, УЧАСТВУЮЩИЕ В РАЗЛОЖЕНИИ СИЛИКАТОВ
Микроорганизмы, разрушающие горные породы, давно известны исследователям. По способу питания это гетеротрофы. Они участвуют в процессах выветривания косвенным образом, образуя кислые продукты своего метаболизма — органические или неорганические кислоты, и переводя нерастворимые компоненты скальных пород в растворимые соединения, вымываемые впоследствии дождевыми осадками. Лучшего развития такие микроорганизмы достигают на средах с полевым шпатом и биотитом, мобилизуя фосфор и калий из этих минералов для биосинтетических целей.
Имеется целый ряд свидетельств выделения из почв так называемых силикатных бактерий, которые растворяют алюмосиликаты с высвобождением водорастворимых калиевых соединений, усваиваемых растениями. Силикатные бактерии встречаются на поверхности скальных пород, лишайников, мхов и водорослей. В почвах такие бактерии концентрируются на поверхности корней, получая основной поток углерода в виде корневых экссудатов органической природы. В почве количество силикатных бактерий может достигать 106 кл/г, такое же высокое число обнаруживают и на поверхности водорослей.
Выделение силикатных бактерий проводят на среде с алюмосиликатами и крахмалом (г/л): крахмал — 20,0; алюмосиликат калия — 2,0; СаСО3 — 2,0; Са3(РО4)2 - 1,5; MgSO4-7H2O - 0,15; NaCl - 0,15; MnSO4 - 0,05; FeSO4 -
0,05; агар — 15,0; вода водопроводная — до 1 л. Стерилизациия при 1 атм; pH около 7,0. Алюмосиликат калия можно заменить толченой слюдой или толченым калиевым стеклом.
Для выделения силикатных бактерий можно использовать и пластинки силикагеля (приготовление см. в гл. 3), пропитанные средой Эшби, используя в качестве посевного материала комочки почвы или почвенные суспензии в соответствующих разведениях. Для активации силикатных бактерий исследуемую почву выдерживают 2 сут при влажности 60 — 80 % от полной влагоемкости при 25 °C. Через 2—3 сут в условиях культивирования при 28 — 30 °C на поверхности среды появляются выпуклые, прозрачные, часто слизистые (экзополи-сахариды) сливающиеся колонии силикатных бактерий. На поверхности среды при прибавлении к ней 0,1 % (NH4)2SO4 развиваются мелкие несливаюгциеся колонии спорообразуюших силикатных бактерий. Клетки колоний окрашивают фуксином и метиленовой синью; капсулы выявляют негативным контрастированием с тушью (см. гл. 5).
Среда (г/л) для длительного хранения спорообразующих силикатных бактерий, которые часто применяют в качестве бактериального удобрения: крахмал — 10,0; КН2РО4 - 0,3; NaCl - 0,5; MgCO3 - 1,0; KNO3 - 1,0; агар - 20,0. Культуры выращивают на скошенном агаре и после окончания процесса спорообразования заливают стерильным вазелиновым маслом. Хранят культуры при 4—6 ° С. В таких условиях споры сохраняют жизнеспособность в течение 2—3 лет.
Г л а в а 42
БАКТЕРИАЛЬНЫЕ УДОБРЕНИЯ
Многие почвенные микроорганизмы улучшают плодородие почвы, минерализуя органические и неорганические соединения, повышая эффективность использования минеральных и органических удобрений, обогащая почву молекулярным азотом атмосферы и синтезируя ряд полезных для растений веществ — полисахаридов, белков, витаминов, гормонов роста, антибиотиков-фунгицидов и т. п. В результате резко повышается урожайность растений. В практике сельского хозяйства нашли применение несколько типов бактериальных удобрений, которые обогащают почву и корневую систему растений полезными микроорганизмами: нитрагин, ризоторофин, азотобактерин, фосфобак-терин.
42.1. НИТРАГИН И РИЗОТОРФИН
С давних пор в сельском хозяйстве практиковалось восстановление и улучшение почв путем выращивания на них бобовых растений. Еще в 1820 г. в России появилось Общество сельского хозяйства, пропагандирующее выращивание бобовых. Роль микроорганизмов в питании растений была выяснена через много лет. Растениями лучше усваиваются аммонийный и нитратный азот, чем азот органических соединений, за исключением аспарагина, глутамина и мочевины — соединений, дающих аммоний
ный азот. Поэтому большое значение для питания растений имеют почвенные организмы, которые минерализуют содержащийся в почве органический азот, превращая его в аммиак, который используется растениями для синтеза аминокислот и белков.
Нитрагин представляет собой бактериальное удобрение, основой которого являются жизнеспособные клетки клубеньковых бактерий рода Rhizobium. Нитрагин применяют для повышения урожая различных бобовых растений — фасоли, гороха, сои, клевера, люцерны, люпина, эспарцета, вики и др.
Чистая культура клубеньковых бактерий была выделена М.Бейеринком в 1888 г. Клубеньковые бактерии — это грамотрицательные аэробные подвижные палочки размером 0,5 —0,9 х 1,2 —3,0 мкм. При росте в жидкой среде с перемешиванием культуры достигают стационарной фазы роста через 2 — 3 сут инкубации. Среду с минеральными солями и маннитом или другими углеводами при росте эти бактерии подкисляют. Их рост на среде с углеводами обычно сопровождается обильным образованием внеклеточной слизи полисахаридной природы. Источниками азота могут служить соли аммония, нитрат, нитрит и большинство аминокислот.
Характерной особенностью клубеньковых бактерий является их способность проникать в корневые волоски бобовых растений семейства Leguminosae умеренного пояса, а также некоторых представителей бобовых тропического пояса, вызывая образование корневых клубеньков, внутри которых эти бактерии присутствуют как внутриклеточные симбионты. В корневых волосках бактерии образуют бактероиды — специфические формы бактерий рода Rhizobium, окруженные мембранами растительного происхождения. Живущие в почве бактерии рода Rhizobium прикрепляются к корневым волоскам растения-хозяина, вызывают их деформацию, скручивание и формирование инфекционной нити. Оболочка, образовавшаяся из инфекционной нити и окружающая цитоплазму клетки Rhizobium, называется перибактероидной мембраной. Именно такие формы ризобий называют бактероидами. Дальнейший рост и дифференциация бактероидов приводят к развитию крупных клеток неправильной формы — симбиосом, которые окружены симбиосомной мембраной. Они отличаются от клубеньковых бактерий, развивающихся вне растений, более крупными размерами, высоким содержанием гликогена и жира, большим количеством волютиновых гранул (внутриклеточный резерв фосфата) и активной фиксацией молекулярного азота. Бактероиды и симбиосо-мы теряют способность к существованию в форме свободноживущих клеток Rhizobium из-за нарушения у них так называемого «генетического пула». Клубеньковые бактерии в форме бактероидов образуют нитрогеназный ферментный комплекс, который восстанавливает молекулярный азот до аммиака. Аммиак вовлекается в ряд ферментативных реакций, приводящих к образованию аминокислот, идущих на биосинтез белка.
Различают активные и неактивные культуры клубеньковых бактерий. Под активностью бактерий понимают способность их в симбиозе с бобовым растением фиксировать атмосферный азот. Клубеньки, образованные активными штаммами Rhizobium, отличаются от клубеньков, сформированных неактивными штаммами. Первые окрашены в розовый цвет благодаря наличию в них пигмента леггемоглобина, вторые имеют зеленоватую окраску и не способны фиксировать азот.
Кроме критерия активности в характеристике штаммов клубеньковых бактерий применяют критерий вирулентности — способность вступать в симбиоз с бобовым растением путем быстрого проникновения в корень через корневые волоски и вызывать образование клубеньков. В симбиотическом комплексе растение—бактерии растение обеспечивает бактерии необходимыми питательными веществами и создает для них оптимальные условия роста. Бактерии, находящиеся в клубеньках, снабжают растение азотом. Накопление азота в почве при участии клубеньковых бактерий может достигать до 300 кг на 1 га в год. Следует отметить, что бактерии без участия растения in vivo практически не способны фиксировать азот. Возможность незначительной фик
сации N2 чистыми культурами клубеньковых бактерий показана в лабораторных условиях при низком парциальном давлении кислорода.
Клубеньковые бактерии различных видов обладают индивидуальной избирательной способностью в отношении инфицирования бобовых растений. Эта способность связана с их вирулентностью. Так, для типового вида R. leguminosarum растениями-хозяевами являются горох, вика, чина, чечевица, кормовые бобы, клевер, фасоль, для R. meliloti — люцерна, донник и тригонелла, а для R.loti — только лядвенец. Важно, что вирулентность и видовая избирательность взаимосвязаны и не являются устойчивыми признаками штаммов клубеньковых бактерий. Промышленную селекцию ризобий осуществляют по обоим этим признакам.
Практическое использование клубеньковых бактерий фактически началось еще в середине XIX в., когда почву, на которой до того выращивали бобовые культуры, в количестве 3 — 5 т/га разбрасывали по полям, где также планировали посевы бобовых и где они раньше не выращивались. Однако в конце XIX — начале XX в. стали применять более эффективный метод: клубеньки с корней бобовых подсушивали, тонко измельчали и размешивали с наполнителями (тальк, бентонит). Такими препаратами, получившими название нитрагин, обрабатывали семена бобовых перед посевом в США (1986), Германии (1896), Венгрии (1898), Англии (1906), России (1907). В настоящее время препараты клубеньковых бактерий производят и применяют во многих странах.
Самыми распространенными видами нитрагина являются почвенный, сухой и торфяной (ризоторфин). Почвенный нитрагин представляет собой культуру клубеньковых бактерий, выращенную в стерильной садовой почве. Стерильную почву, обычно в стеклянных сосудах, инокулируют жидкой культурой клубеньковых бактерий и выращивают в термостате при 28 °C. В 1 г такого препарата должно содержаться не менее 300 млн клеток. Препаратом обрабатывают семена бобовых перед посевом. Однако такая технология производства препарата очень трудоемкая и дорогостоящая, а сам он неудобен в применении.
Более совершенна технология производства сухого нитрагина — порошка высушенных клеток бактерий рода Rhizobium в смеси с наполнителем (каолин, бентонит, мел). В 1 г такого порошка при влажности 5 —7 % содержится не менее 10 млрд жизнеспособных клеток бактерий. Исходную культуру клубеньковых бактерий выращивают на одной из агаризованных сред, например, содержащей (г/л): отвар гороха — 100; сахароза — 15; агар — 20; pH 6,8 —7,0. На этапах промышленного производства бактерии выращивают в ферментерах на жидкой среде с кукурузным экстрактом или мелассой, (NH4)2SO4, KH2PO4j К2НРО4 MgSO4 и СаСО3. Значение pH поддерживается в диапазоне 6,5 — 7,0 при интенсивности аэрации в соотношении — 1 объем воздуха на 1 объем среды в 1 мин. Суспензию клеток из ферментера, содержащую около 10 млрд клеток в 1 мл, сепарируют. Биомассу в виде пасты с влажностью 70 — 80 % смешивают с защитной средой, содержащей, например, 20 % мелассы и 1 % мочевины, и высушивают в вакуум-сушильных установках или в распылительных сушилках. В порошке определяют количество жизнеспособных бактерий в 1 г, смешивают с наполнителями, фасуют и герметизируют в полиэтиленовые пакеты по 0,2—1,0 кг, которые хранят при температуре 8 —12 °C в течение 6 мес.
Клетки клубеньковых бактерий, как и других неспоровых микроорганизмов, очень чувствительны к потере воды, поэтому даже при строгом соблюдении технологических параметров большая часть клеток в процессе высушивания погибает. Этого удается избежать в торфяном нитрагине — ризоторфине.
Для получения ризотрофина торф освобождают от корней, высушивают до 25 — 30 % влажности и размалывают так, чтобы размер частиц не превышал 0,1 мм. Размолотый торф увлажняют до 35 — 40%, прибавляют СаСО3 в таком количестве, чтобы довести pH до 6,8 — 7,0, и расфасовывают в тонкие полиэтиленовые мешки по 100 — 500 г. Запаянные пакеты с торфом стерилизуют радиационным способом с использованием мощных гамма-установок. В пакеты со стерильным торфом в стерильном авто
матизированном боксе специальной иглой вносят жидкую посевную культуру определенного вида клубеньковых бактерий в количестве 80 мл на каждые 250 г торфа. Норму засева выбирают так, чтобы исходный титр бактерий в инокулированном торфе составлял около 1 млрд клеток на 1 г препарата влажностью 50 — 60 %. До инокуляции в жидкую культуру вносят стерильные растворы мелассы, декстрина или молочной сыворотки в таком количестве, чтобы их количество составляло в итоге около 3 % к массе сухого торфа. Если необходимо быстро получить готовый препарат и использовать его, инокулированные пакеты помещают на 5 —7 сут в термостат при 20 —22 °C. Для длительного хранения в течение 6 мес пакеты сразу же ставят на хранение при 12— 15 °C. Содержание клеток клубеньковых бактерий в ризоторфине через 6 мес после его изготовления должно быть не ниже 3 — 4 млрд в 1 г препарата.
Наиболее распространенный метод применения нитрагина и ризоторфина — это предпосевная обработка семян в день их посева из расчета 200 г сухого нитрагина и 500 г ризоторфина на 1 га. Применение фосфорно-калиевых и органических удобрений повышает эффективность препаратов клубеньковых бактерий. Нитрагин и ризоторфин увеличивают урожайность бобовых на полях, где они выращиваются впервые, в некоторых случаях до 100 %.
42.2. ЭКСПРЕСС-МЕТОД ОПРЕДЕЛЕНИЯ ЖИЗНЕСПОСОБНОСТИ КЛЕТОК КЛУБЕНЬКОВЫХ БАКТЕРИЙ В СУХОМ НИТРАГИНЕ
В 99 мл стерильной водопроводной воды разводят 1 г сухого нитрагина и выдерживают на качалке в течение 2 ч при 200 об/мин. Затем суспензию клеток помещают в пробирку и окрашивают раствором примулина (конечное разведение 1:1000). Через 15 мин 0,01 мл окрашенных клеток наносят на предметное стекло, накрывают покровным стеклом 18x18 мм, края которого заливают расплавленным парафином. Под люминесцентным микроскопом МЛ-2 препарат просматривают и подсчитывают общее количество клеток и процент живых клеток в 1 мл суспензии по методу Виноградского—Шульгиной (см. гл. 7). Примулин практически не проникает в живые клетки, но накапливается в мертвых. Благодаря этому при просмотре препарата в свете люминесценции при освещении объекта через объектив живые клетки в поле зрения тускло люминесцируют, а мертвые светятся ярким зеленовато-желтым светом. Общее количество клеток можно определить в том же поле зрения также при фазовоконтрастном освещении объекта снизу, через конденсор. Количество жизнеспособных клеток в 1 г сухого нитрагина определяют с учетом того, что для приготовления микроскопического препарата используют разведение нитрагина 10“2.
42.3. АЗОТОБАКТЕРИН
Бактерии рода Azotobacter широко распространены в природе. Наиболее известен Azotobacter chroococcum, повсеместно встречающийся в богатых органическим веществом, хорошо дренированных и увлажненных почвах. Азотобактер — строгий аэроб, в несимбиотическом состоянии фиксирует не менее 10 мг N2 в расчете на 1 г потребленного углевода (глюкозы). Клетки азотобактера грамотрицательные, овальной формы, диаметром 1,5 — 2,0 мкм, плейоморф-
ные, от палочковидных до кокковидных. Для азотфиксации азотобактер нуждается в молибдене, который может быть частично заменен ванадием. В качестве источников азота используют соли аммония, азотной кислоты, мочевину, некоторые аминокислоты. Азотобактер фиксирует азот только в среде, обедненной или вообще лишенной связанного азота. Эта бактерия требует наличия в среде высокого содержания фосфора в виде органических и неорганических соединений. Недостаток фосфора замедляет рост бактерий и снижает азотфик-сирующую способность. Показано, что нитрогеназная система, осуществляющая фиксацию азота, представляет собой сложный мультиферментный комплекс, содержащий не связанное с гемом железо, молибден и SH-группы.
Азотобактер уже с начала 1920-х годов используется для изготовления на его основе бактериального удобрения — азотобактерина. При выращивании овощных культур — томатов, салата, огурцов удавалось получить значительный эффект. Однако позднее было показано, что действие азотобактера на растения обусловлено его способностью синтезировать биологически активные вещества — никотиновую и пантотеновую кислоты, пиридоксин, биотин, гетероауксин, гиббереллин, фунгицидные вещества и др. Этот комплекс соединений стимулирует прорастание семян растений, ускоряет их рост, защищает от микроскопических грибов, многие из которых угнетают рост растений. В то же время азот, фиксируемый азотобактером, существенно не влияет на урожайность растений.
Сухой азотобактерин производят по технологии, во многом сходной с технологией производства сухого нитрагина. Культуру бактерий выращивают методом глубинного культивирования на среде, содержащей практически те же компоненты, что и при культивировании клеток Rhizobium, но с добавлением сульфата железа и сложной соли молибденовой кислоты. Значение pH поддерживается в диапазоне 6,1 —6,5 при интенсивности аэрации в соотношении — 1 объем воздуха на 1 объем среды в 1 мин. Для того чтобы клетки не теряли способности фиксировать азот и сохраняли биологически активные вещества, их выращивание ведут до середины логарифмической фазы роста. После сепарации и высушивания клеток в вакуум-сушильных установках или в распылительных сушилках вместе с защитными средами в порошке определяют количество жизнеспособных бактерий в 1 г, стандартизуют смешиванием с наполнителями (каолин, бентонит, мел), фасуют в полиэтиленовые пакеты по 0,2— 1,0 кг и хранят при температуре до 12 °C не более 3 мес. В I г такого порошка при влажности 5 — 6 % в конце срока хранения должно содержаться не менее 0,5 млрд жизнеспособных клеток азотобактера.
Торфяной азотобактерин представляет собой торф с нейтральной реакцией среды, содержащий в 1 г не менее 50 млн жизнеспособных клеток. Для его приготовления размолотый торф увлажняют до 40%, прибавляют 0,1 % суперфосфата, минеральные соли, 3% мелассы и СаСО3 в таком количестве, чтобы довести pH до 6,5 —7,0, и расфасовывают в тонкие полиэтиленовые пакеты по 100 — 500 г. Запаянные пакеты с торфом стерилизуют радиационным способом. Культуру азотобактера выращивают методом глубинного культивирования и добавляют в пакеты со стерильным торфом с таким расчетом, чтобы в 1 г препарата содержалось 40 — 50 млн жизнеспособных клеток. Мешки помещают в термостат и подращивают в них культуру азотобактера при 25 —27 °C в течение 4—6 сут до содержания не менее 180 — 200 млн клеток азотобактера в 1 г препарата. Затем мешки герметично запаивают и хранят в холодильнике при 7 — 12 °C. Полученные препараты сохраняют свою активность в течение 3 — 4 мес.
Препараты на основе азотобактера целесообразно использовать лишь в плодородных почвах, содержащих фосфор и микроэлементы, особенно молибден, ванадий и
бор, б сочетании с внесением органических и минеральных удобрений. Способ применения зависит от особенностей обрабатываемого посевного материала. Семена зерновых обрабатывают сухим азотобактерином механизированным способом из расчета 100 млрд жизнеспособных клеток на одну гектарную порцию семян. Клубни картофеля, предназначенные для посадки на 1 га, смачивают водной суспензией бактерий из расчета 300 млрд клеток на 15 л воды.
42.4. МЕТОД ОПРЕДЕЛЕНИЯ ЖИЗНЕСПОСОБНОСТИ КЛЕТОК АЗОТОБАКТЕРА В ТОРФЯНОМ АЗОТОБАКТЕРИНЕ
Для десорбции клеток азотобактера с торфяных частиц 10 г торфяного азотобактерина разводят в 90 мл стерильного 0,2%-го раствора пирофосфата натрия, предварительно растерев препарат в небольшом количестве пирофосфата в ступке до пастообразного состояния. Затем сосуд с суспензией выдерживают на круговой качалке в течение 2 ч при 200 об/мин. Из полученной суспензии готовят разведения IO-3—10-9. Из последних 3 разведений помещают по 1 мл суспензии в стерильные чашки Петри и заливают слоем расплавленной и охлажденной до 45 °C агаризованной среды Эшби (см. гл. 51). Среду в чашках перемешивают с суспензией бактерий и выдерживают в термостате при 28 °C в течение 3 — 4 сут. Численность посторонней микрофлоры в азотобактерине определяют микроскопированием суспензии и высевом разведений 10 3— 10-9на чашки с МПА.
42.5. ФОСФОБАКТЕРИН
Фосфобактерин — это препарат в виде порошка, содержащий в 1 г не менее 8—10 млрд жизнеспособных спор культуры Bacillus megaterium var. phosphaticum, способной превращать сложные фосфорорганические соединения и минеральные фосфаты в доступную для растений форму. Промышленное производство бактерий осуществляют в ферментерах в строго асептических условиях на одной из сред, например, содержащей (г/л): кукурузный экстракт — 20; мелассу — 15; фосфат калия двухзамещенный — 0,1; сульфат аммония — 1; мел — 10 при температуре 30 °C и pH 6,8 —7,3.
Полученную биомассу клеток отделяют центрифугированием и высушивают в распылительной сушилке при 65 —75 °C до влажности 3 — 4%. Полученные высушенные споры В. megaterium смешивают с наполнителем (каолин, бентонит, мел) и расфасовывают в герметичные полиэтиленовые пакеты. В I г препарата должно быть не менее 8 — 10 млрд жизнеспособных спор. Препарат стабилен не менее 1 года при комнатной температуре.
Основными проблемами производства фосфобактерина являются фаголи-зис культуры и непрорастаемость спор. Фаголизис связан с тем, что промышленные штаммы бацилл чувствительны к действию бактериофагов, попадающих в виде инфекции извне, а также сами содержат в себе профаг, который в определенных условиях активизируется. В целях борьбы с фаголизисом поддерживают асептические условия на всех стадиях производства, проводят селекцию устойчивых к фагам штаммов, вводят в среду для культивирования ба
цилл соли органических кислот в количестве до 0,1 %. Непрорастаемость спор связана с нарушением соотношения в среде культивирования фосфатов и сульфатов из-за возможных нестандартных источников сырья.
Фосфобактерин применяют для повышения урожайности картофеля, сахарной свеклы, зерновых на черноземных почвах, отличающихся большим количеством фосфорорганических соединений. На гектарную порцию зерновых требуется 100 млрд спор В. megaterium, а на 1 га посадочного материала картофеля — 150 — 200 млрд спор бактерий.
Глава 43
МИКРООРГАНИЗМЫ — ВРЕДИТЕЛИ ПРОИЗВОДСТВА
43.1. ХЛЕБОПЕЧЕНИЕ
Хлеб является одним из основных продуктов питания. Изготовление его представляет собой сложный цикл микробиологических и биохимических процессов, происходящих в тесте с момента смешивания муки с водой и заканчивая его выпечкой. В состав муки, используемой для выпечки пшеничного и ржаного хлеба, входят компоненты, необходимые для развития многих микроорганизмов. Кроме крахмала в муке имеется до 2 % сбраживаемых сахаров — глюкозы, фруктозы, мальтозы, сахарозы, раффинозы. Мука сильных сортов пшеницы содержит до 14 % белка. Азотсодержащие вещества муки представлены разнообразными группами белков — альбуминами, глобулинами и др. Мука содержит до 2 % жиров и жироподобных веществ и до 2 % минеральных веществ, в том числе микроэлементы.
Решающую роль в приготовлении хлеба, наряду с ферментами муки, играет жизнедеятельность микроорганизмов. Мука всегда содержит значительное количество различных микроорганизмов. Число их зависит от степени загрязненности зерна и способов его очистки. Вносят микроорганизмы и с добавками к тесту в процессе приготовления хлеба. Однако из всего разнообразия микроорганизмов теста наиболее важное значение имеют дрожжи и молочнокислые бактерии, для развития которых в тесте есть все необходимые условия — влажность 40 — 50 %, незначительное содержание молекулярного кислорода и наличие питательных веществ. Ведущая роль в формировании качества хлеба принадлежит дрожжам. Их главная функция при выпечке хлеба состоит в заквашивании теста, в результате чего оно поднимается под действием выделяющегося при брожении углекислого газа. Микробиологические процессы и связанные с ними биохимические изменения в тесте определяют пористость, окраску и сохранение свежести хлеба, придают ему вкус и аромат, несколько повышают его питательную ценность.
Дрожжи Saccharomyces cerevisiae различных рас являются главными разрыхлителями теста. Для хлебопечения особенно важны такие свойства дрожжей, как высокая потенциальная активность гликолитических ферментов, стойкость при хранении, способность переносить высокие концентрации сахаров и NaCl в среде. В хлебопечении чаще всего используют быстрорастущие расы сахаромицетов верхового брожения. У полноценных рас дрожжей подъемная сила, т.е. длительность подъема теста на стандартную высоту 70 мм в стандартной форме, должна быть не более 45 мин, зимазная и мальтазная
активности — до 50 и 75 мин соответственно. Зимазная и мальтазная активности выражаются временем, необходимым для выделения 10 мл СО2 при сбраживании 20 мл 5%-го раствора глюкозы дрожжами, которые вносятся в количестве 2,5 % к объему среды (2 • 10s клеток). Дрожжи также должны иметь 100%-ю устойчивость к мелассе и скорость роста не менее 0,2 ч_|. На хлебозаводах применяют прессованные и сухие дрожжи.
Схема приготовления жидких дрожжей из пресованных и сухих включает в зависимости от сорта хлеба заквашивание осахаренной и охлажденной до 50 °C мучной заварки молочнокислыми бактериями, например Lactobacillus delbrueckii. Они ускоряют процесс брожения в тесте и обладают высокой протеолитической активностью, которая позволяет накапливать в тесте значительное количество аминного азота, положительно влияющего на скорость роста и подъемную силу дрожжей. Наряду с сахаромицетами в тесто спонтанно могут попадать дрожжи родов Candida и Pichia — С. krusei, C.utilis, C.guilliermondii, Р. xylosa. Р. vini. В пшеничных и ржаных заквасках теста преобладают молочнокислые бактерии, а не дрожжи.
43.1.1. Определение подъемной силы дрожжей
В мерный стакан на 100 мл вносят 3,2 г прессованных дрожжей и немного воды. Стеклянной палочкой дрожжи хорошо растирают до исчезновения комков, затем доливают до метки воду (температура 30 °C) и вновь хорошо размешивают. В фарфоровую чашку переносят 5 мл полученной дрожжевой суспензии, прибавляют 8 г пшеничной муки второго сорта и замешивают тесто, придав ему форму шарика. Приготовление теста и шарика следует делать быстро, не более 4 мин. Шарик опускают в стакан на 200 мл, наполненный водой температурой 30 °C. Стакан с шариком оставляют стоять в термостате или на водяной бане при температуре 30 °C. Отмечают время, прошедшее с момента погружения шарика на дно стакана, до момента его всплывания на поверхность воды. Очень хорошие дрожжи имеют подъемную силу 10—15 мин, хорошие дрожжи — 15—20 мин.
41.1.2. Микроорганизмы, вызывающие болезни хлеба
Готовый выпеченный хлеб при определенных условиях хранения, таких, как повышенная влажность, температура, недостаточное соблюдение чистоты помещения, служит хорошим субстратом для развития в нем посторонних микроорганизмов. Они быстрее поражают мякиш хлеба, обладающий необходимой для них влажностью.
Картофельная болезнь. Болезнь получила свое название от ее возбудителя — так называемой «картофельной палочки» Bacillus subtilis var. mesentericus. Появляется она на пшеничном хлебе большей частью в летнее время. При интенсивном развитии бактерий внешние признаки мякиша изменяются, в нем появляются тонкие серебристые нити, которые при разломе мякиша тянутся в длину («тянущийся хлеб»). Влажность мякиша повышается и, кроме того, он становится липким. Равномерная пористость хлеба в мякише нарушается и часто образуются провалы в результате расщепления крахмала. Цвет мякиша меняется, появляются желто-бурые пятна. Развившаяся болезнь усиливает неприятный гнилостный запах хлеба, к которому иногда примешивается также сладковатый запах.
Наличие этих изменений позволяет без особых затруднений констатировать картофельную болезнь. Для обнаружения в муке Б. subtilis var. mesentericus применяют метод посева мучной болтушки на чашки с мясопептонным агаром и в пробирки с бульоном. Перед посевом мучную болтушку, приготовленную путем смешивания 1 г муки с 100 мл стерильной воды, пастеризуют на кипящей водяной бане в течение 30 мин. На чашках картофельная палочка образует серовато-белые, морщинистые колонии, а на бульоне дает характерную складчатую пленку. Под микроскопом обнаруживаются тонкие длинные и короткие палочки 0,5 — 0,6 х 3—10 мкм, одиночные или соединенные в длинные нити, а также эндоспоры.
Кроме микробиологического метода определения картофельной палочки успешно применяются также биохимические методы, в частности определение в муке активности каталазы (см. гл. 13): в муке, зараженной этими бактериями, ее концентрация увеличивается.
Хлеб, пораженный картофельной палочкой, употреблять в пищу запрещается. Методы борьбы с картофельной болезнью хлеба сводятся к поддержанию в помещениях для хранении хлеба температуры не выше 12 °C и необходимой влажности, подкислению теста путем внесения в него молочной кислоты. Заводское помещение должно содержаться в чистоте с регулярным мытьем полов водой с 1 % уксусной кислоты. Мука должна проверяться на наличие спор картофельной палочки.
«Красный хлеб». Возбудителем этого вида заболевания является широко распространенная в природе бактерия Serratia marcescens. Попадая из окружающей среды в хлеб и при благоприятной температуре 20 — 25 °C развиваясь в нем, бактерии образуют на белом фоне пшеничного мякиша сначала розовые, а с усилением процесса ярко-красные пятна пигмента продигиозина. Он выделяется клетками в среду и является производным трипиррола. Продигиозин не растворим в воде, хорошо растворяется в спирте, ксилоле, хлороформе. Бактерии в хлебе вызывают разжижение клейковины и расщепление крахмала. Хлеб, пораженный 5. marcescens, употреблять в пищу запрещено.
Выделение бактерий производят из зараженного хлеба. Для этого кусочки окрашенного в красный цвет мякиша хорошо взбалтывают в колбе со стерильной водой. Полученную суспензию высевают на чашки с мясо-пептонным бульоном с добавлением 1 % глюкозы и выдерживают в термостате при 23 — 25 °C 3 сут. На чашках вырастают красные колонии S. marcescens. Под микроскопом обнаруживаются грамотрицатель-ные, подвижные прямые палочки (0,5—-0,8) х (0,9 —2,0) мкм.
Меры борьбы с бактериальным заражением сводятся главным образом к поддержанию помещений в чистоте, мойке пола горячей водой с мылом, обработке посуды кипятком. Не образующие спор клетки S. marcescens при этом погибают.
«Меловая болезнь». Внешним признаком болезни является белый мучнистый налет, который при старении становится похожим на мел. Он образуется на мякише белого и черного хлеба. Налет образуется мицелием дрожжеподобного гриба Endomyces flbuliger. Микроорганизм хорошо растет на агаризованной среде с суслом, образуя на воздухе мицелий со спорами. В глубоких слоях этой среды образуются характерные дрожжевидные клетки. Хлеб, зараженный меловой болезнью, считается безвредным.
Плесневелый хлеб. Большое распространение плесневых грибов и высокая устойчивость их спор способствует порче хлеба за счет развития в нем этих микроорганизмов. Наиболее важным фактором при этом является повышенная влажность помещения, где хранится хлеб. В сухом помещении даже при значительном обсеменении спорами развития плесневых грибов на хлебе не происходит. Хорошо пропеченный целый хлеб заражается труднее, чем хлеб мало пропеченный или хлеб с нарушенной коркой, через которую попадают споры и заражают мякиш. Чаще всего возбудителями плесени являются грибы родов Penicillium и Aspergillus. Эти грибы содержат целый комплекс ферментов, под действием которых изменяется химический состав хлеба, а также ухудшаются его пищевые качества, вкус и аромат. Часто эти грибы являются причиной появления в хлебе неприятного запаха. Хлеб, пораженный плесенью, употреблять в пищу запрещается.
«Пьяный хлеб». Недоброкачественный хлеб, вызывающий после его употребления тошноту, рвоту, головокружение, получил название «пьяный хлеб». В отличие от указанных выше болезней хлеба «пьяный хлеб» внешне ничем не отличается от доброкачественного. Его вредные свойства обусловлены заражением зерна, из которого приготовлена мука и выпечен хлеб. Поэтому правильнее считать, что это заболевание зерна, а не хлеба. Возбудителем является гриб Fusarium graminearum, заражающий зерна пшеницы, ржи, овса, ячменя. В колосках образуется сначала розовый налет, принимающий затем вид темных скоплений — мицелий. Мицелий проникает в ткани зерна, разлагает их с выделением аммиака и токсических веществ. Для нейтрализации вред-пых веществ зараженное зерно перед помолом нагревают при температуре 95 —100 °C. Хлеб, приготовленный из муки, полученной из прогретого зерна, не имеет свойств «пьяного хлеба».
Спорынья. Заболевание зерна, известное как спорынья, вызывается грибом Claviceps purpurea. Чаще всего спорынья поражает рожь, затем пшеницу и ячмень. Мука, полученная из зараженного зерна, и выпеченный из нее хлеб обладают токсическими свойствами. Во время цветения злаков аскоспоры спорыньи заражают завязь. Мицелий разрастается в завязи и превращает ее в белую мягкую массу с желобками на поверхности. В этих желобках на тесно прижатых друг к другу кончиках гиф образуется много мелких бесцветных конидий. Конидии, взвешенные в сладком соке — медвяной росе, разносятся насекомыми. Вскоре все это образование высыхает и пронизанная гифами завязь превращается в твердый, как рог, склероций. Ко времени созревания злака склероции (маточные рожки) отпадают и перезимовывают в почве, а весной прорастают, на них образуются сидящие на ножках головчатые стромы с плодовыми телами — перитециями, в которых содержатся аски с аскоспорами. Склероции содержат сильнодействующие алкалоиды — эргобазины, эрготоксины, эрготамины и др. Алкалоиды поражают центральную нервную систему, вызывая галлюцинации, поражают мышцы конечностей человека, вызывая в тяжелых случаях их омертвение. В то же время некоторые из этих веществ, например эргометрин и эрготамин, применяются в определенных дозах и в качестве лечебных средств. Проявление тех или иных признаков отравления обусловлено соотношением указанных веществ в хлебе. При обнаружении спорыньи зерно должно быть подвергнуто очистке на специальных машинах.
43.2. ПРОИЗВОДСТВО СИЛОСА
В силосовании кормов ведущую роль играют молочно-кислые бактерии, обитающие на растениях, так как в основе силосования лежит молочнокислое брожение. Молочнокислые бактерии сбраживают сахара силосующихся растений в молочную и частично уксусную кислоты, подавляющие развитие гнилостных, маслянокислых и других бактерий, снижающих качество корма. Молочнокислые бактерии снижают pH корма до 4,2—4,0. Если значение pH силоса по тем или иным причинам повышается до 4,5 —4,7, то создаются условия, благоприятствующие жизнедеятельности микроорганизмов, ухудшающих хранение корма. В нем накапливаются масляная кислота, амины, аммиак и др. Чтобы обеспечить нормальное развитие молочнокислых бактерий в процессе силосования, необходимо достаточное содержание сахара в силосующихся растениях и создание анаэробных условий. Плесневые грибы переносят сильное подкисление, но являются строгими аэробами, поэтому в хорошо спресованном, укрытом, заквашенном корме размножаться не могут. Кислотность растительной массы при силосовании зависит также от буферности растительной массы, связывающей часть молочной кислоты. В первый период после закваски
силоса бурно развивается микрофлора смешанной фазы брожения, представленная на поверхности здоровых растений, — гнилостные бактерии, бактерии группы кишечной палочки, маслянокислые бактерии, уксуснокислые бактерии, дрожжи и др. При подкислении корма в анаэробных условиях их сменяют молочнокислые лактококки, а затем более кислотоустойчивые молочнокислые палочки. Микробиологические процессы заканчиваются в силосе в основном через 14—20 дней.
Хороший силос характеризуется оливково-зеленым цветом, запахом моченых яблок, pH 4,0—4,2, влажностью 70%. Микрофлора такого силоса представлена молочнокислыми палочками Lactobacillus delbrueckii L.xylosus и молочнокислыми лактококками Lactococcus plantarum, L.lactis. Часто в небольших количествах встречаются дрожжи Saccharomyces cerevisiae и др. Дрожжи образуют эфиры, придающие силосу приятный запах и обогащающие корм белком и витаминами. Однако в больших количествах дрожжи ухудшают качество силоса, повышая значение pH, поскольку являются конкурентами молочнокислых бактерий в потреблении сахаров.
Определение качественного состава микрофлоры силоса. Кусочек свежего силоса пинцетом плотно прижимают к предметному стеклу без добавления воды так, чтобы на стекле остался отпечаток. Препарат сушат на воздухе, фиксируют на пламени, окрашивают метиленовым синим в течение 2—3 мин и микроскопируют с иммерсионной системой. Под микроскопом видны неспорообразующие палочки, варьирующие в размере (молочнокислые бактерии), и молочнокислые лактококки. Среди них обычно преобладают L. delbrueckii и L. plantarum. Встречаются и клетки почкующихся дрожжей сахаромицетов. Споровые формы бактерий наблюдаются редко. В некачественном силосе на препарате видны спорообразующие палочки — маслянокислые бактерии, аэробные гнилостные бактерии, а также плесневые грибы.
Количественный учет микроорганизмов в силосе. Навеску силоса (5 г) помещают в колбу с 50 мл стерильной водопроводной воды и 3 г песка. Колбу взбалтывают круговыми вращательными движениями 10 мин. Из полученной вытяжки готовят разведения 10~2— 10-7. Из разведений делают высевы на элективные среды: 1 мл суспензии при глубинном посеве и 0,05 мл при поверхностном. Посевы инкубируют в термостате при 28 °C.
Определение количества молочно-кислых бактерий проводят на сусло-агаре с мелом (30 г стерильного мела на 1 л сусло-агара). Вокруг колоний молочнокислых бактерий (подсчитывают на 6-й день) вследствие накопления молочной кислоты образуются зоны растворения мела. Количество микроскопических грибов и дрожжей определяется на сусло-агаре со стрептомицином поверхностным посевом. В расплавленный сусло-агар перед разливом его в чашки Петри добавляют стрептомицин из расчета 100 ЕД на 1 мл среды. Подсчет колоний ведут на 6 —7-е сут. Титр маслянокислых бактерий определяют на картофельной среде с мелом. В пробирки вносят стерильный мел (на кончике шпателя) и 8—10 картофельных кубиков величиной 2—3 мм, заливают 10 мл водопроводной воды и стерилизуют при 1 ати 30 мин. Учет роста ведут по интенсивности выделения газа (кусочки картофеля всплывают). Для определения количества спор маслянокислых бактерий делают посев на картофельную среду с мелом из суспензии после ее пастеризации в течение 15 мин при 80 °C. Титр маслянокислых бактерий или их спор устанавливают методом предельных разведений по таблице Мак-Креди. Споры аэробных гнилостных бацилл обнаруживают на МПА с сусло-агаром (БСА).
43.3. ВИНОДЕЛИЕ
Созревание и старение вина происходят на стадиях бочковой (резервуарной) и бутылочной выдержки. При созревании вино становится стабильным (разливостойким), приобретает соответствующие его типу аромат и вкус. Ста
дия старения вина происходит в процессе выдержки в анаэробных условиях. При этом улучшаются его органолептические качества и оно приобретает свойства, присущие выдержанным винам. Вино является благоприятной средой для развития многих микроорганизмов.
Дрожжевые помутнения. Дрожжи, вызывающие инфицирование вина, относятся к различным группам. Помутнения красных и белых вин после разлива в бутылки чаще всего вызывается развитием в вине дрожжей родов Candida, Hansenula, Pichia и др. Благоприятными условиями их роста является присутствие остаточного недобродившего сахара, кислорода и некоторого количества живых дрожжевых клеток. Даже полное отсутствие сахара в сухих винах не исключает возможности возникновения этой болезни, так как некоторые дрожжевые организмы могут развиваться, используя спирт и органические кислоты вина, которые они хорошо усваивают.
Кроме микроскопического метода обнаружения в вине указанных микроорганизмов применяют также метод определения стойкости вина посевом на жидкую среду: стерильное вино, содержащее 10 % сахара, около 10 % (объемных) спирта. В пробирку с 10 мл среды [’.носят 0,5 мл исследуемого вина и выдерживают при 25 °C в течение 6 сут. Пробирки ежедневно проверяют, отмечая время появления помутнения среды, образование и характер роста пленки.
Цвель вина. Пленкообразующие дрожжи встречаются преимущественно в молодом вине до фильтрации, они вызывают так называемую цвель вина — образование пленки на его поверхности. Развитие болезни наблюдается при свободном доступе воздуха. На поверхности вина образуется пленка мучнисто-белого цвета. Вначале она тонкая и гладкая, а с возрастом утолщается и выглядит морщинистой. Пленка рыхлая, легко разрывается и, падая на дно, вызывает помутнение вина. Вкус вина ухудшается, появляется неприятный запах. Сильно развившаяся цвель вызывает полную утрату вкуса и букета вина. Этой болезнью поражаются белые и красные вина с содержанием спирта не выше 12 %. Возбудитель болезни Candida ту coderm а снижает в вине содержание спирта с одновременным накоплением уксусной кислоты, ацетальдегида и ацетоина. Для предупреждения заболевания вина цвелью одним из первых условий является прекращение доступа воздуха путем частых доливок вина в сосуд до уровня пробки или хранение его в анаэробных условиях.
Молочнокислое скисание. Существенный вред виноделию при нарушении технологического режима производства и хранения вина могут приносить молочнокислые бактерии Lactobacillus delbrueckii, L.carnis, L.mali, L.xylosus, L.divergens, L.piscicola, Lactococcus plantarum, L.lactis, L.piscium. Развиваясь в винах, они сбраживают сахар и вызывают их заболевание — скисание, помутнение, превращение лимонной, винной кислот и глицерина в молочную, уксусную кислоты и углекислый газ. При восстановлении этими бактериями фруктозы в маннит вино приобретает неприятный кисло-сладкий вкус. Молочнокислые бактерии рода Leuconostoc вызывают осклизнение вин, при этом из сахара образуется декстран.
Уксуснокислое скисание. Эта болезнь широко распространена и очень опасна для вин. Вызывают ее уксуснокислые бактерии Acetobacter xylinum, A. aceti, которые в присутствии воздуха образуют на поверхности вина пленку. Поражаются вина даже с высоким содержанием спирта — 15 % и более. Чтобы предупредить развитие в вине уксуснокислых бактерий, не допускают снижения кислотности вина. Хранить вино следует при температуре не выше 10—12 °C.
Предупреждения болезней вина. Источниками инфицирования вина чаще всего являются микроорганизмы, находящиеся на оборудовании или в помещениях. Много их в винных подвалах, на древесине наружной поверхности бочек, в шлангах, на бетонных стенках и т. п.
Для предупреждения и лечения болезней вина применяют различные методы. Борьбу с бактериями ведут при помощи сульфитации — насыщения вин сернистым ангидридом в количестве 80—120 мг/л. Для стабилизации вина применяют его пастериза
цию, УФ- и гамма-облучение, стерилизующую фильтрацию и центрифугирование. Биологическую стабилизацию осуществляют также посредством добавления антисептиков, например сорбиновой кислоты.
43.4. ПИВОВАРЕНИЕ
При производстве пива могут развиваться посторонние виды дрожжей и бактерий. Они ухудшают процесс брожения и осветления пива, вызывают его помутнение, скисание, осклизнение, появление осадка, придают посторонний вкус и запах.
Очаги инфицирования посторонними дрожжами возникают на недостаточно продезинфицированной аппаратуре, а также при различных нарушениях режима ведения производственных дрожжей. Источником инфекции может быть и хмель. В настоящее время описаны десятки видов дрожжей, инфицирующих пиво. По мнению некоторых исследователей, многие «виды» Saccharomyces, описанные в качестве инфекции пивоварения, в большинстве случаев, видимо, являются мутантами культурных производственных дрожжей. Часть из них рассматривается как синоним Saccharomyces cerevisiae. Среди дрожжей-несахаромицетов, вызывающих инфицирование пива, чаще всего обнаруживают представителей родов Candida, Pichia, Hansenula, Rhodotonda.
Ряд таких пороков пива, как помутнение, скисание, осклизнение, могут вызывать молочнокислые бактерии — Lactobacillus delbrueckii, L.xylosus, L.casei, Lactococcus plantarum и бактерии рода педиококкус — Pediococcus acidilactici, P.dextrinicus, P. damnosus, P.pentosaceus и др. В результате накопления педиококками диацетила и мукополисахаридов в пиве появляются слизь и осадок, оно приобретает нежелательный привкус и запах.
Для стабилизации пива применяют его стерилизующую фильтрацию, пастеризацию, УФ- и гамма-облучение, центрифугирование.
43.5. ПРОИЗВОДСТВО МОЛОКА И МОЛОЧНЫХ ПРОДУКТОВ
Молоко — благоприятная среда для развития микроорганизмов, поэтому при соответствующих температурных условиях микроорганизмы в молоке бурно размножаются. В молоке содержатся белки, свободные аминокислоты, небольшое количество пептона, жиры, сахара, витамины (А, Е, D, С, группы В) и др. Попадание микроорганизмов в молоко происходит при дойке с поверхности вымени, с кожи животного, посуды, оборудования, кормов, рук и одежды доилыцика. При соблюдении санитарных правил в молоке преобладают микрококки, молочнокислые бактерии, стрептококки, сарцины. Загрязненное молоко содержит значительное количество бактерий группы кишечной палочки, а также гнилостных и маслянокислых бактерий. Во время хранения молока изменяется количество содержащихся в нем бактерий и соотношение между отдельными их видами. Характер этих изменений зависит от продолжительности, температуры хранения и состава микрофлоры в молоке в начале его хранения.
При микробиологическом анализе молока определяют общую численность бактерий, титр кишечной палочки и проводят пробу на редуктазу.
Проба на редуктазу. Количество редуктазы в молоке является показателем его бактериальной обсемененности. В молоке содержатся различные ферменты, в том числе
редуктаза — анаэробная дегидрогеназа, передающая водород от окисляемого субстрата любому ненасыщенному соединению, но неспособная передавать его кислороду воздуха. Редуктаза накапливается в молоке главным образом при размножении в нем микроорганизмов, поэтому ее количество в молоке является показателем его бактериальной обсемененности. Обнаруживается редуктаза по обесцвечиванию метиленовой сини или резазурина. При применении резазурина в стерильные пробирки наливают 10 мл анализируемого молока, 1 мл 0,0005%-го водного раствора резазурина, тщательно перемешивают и помещают в термостат на 38 — 40 °C. Изменение окраски учитывают через 20 мин и через 1 ч (табл. 43.1).
Определение микробного числа (общего количества бактерий в 1 мл) в молоке. Готовят разведения молока в стерильной водопроводной воде или физрастворе 10-1 —10“9. Из каждого разведения по 1 мл вносят в стерильные чашки Петри, которые затем заливают расплавленным и охлажденным до 45 —50 °C МПА (глубинный посев). Засеянные чашки помещают в термостат при 30 °C. Выросшие колонии подсчитывают на 3-й (бактерии) и 4—5-й (дрожжи, плесени) день. С учетом разведений, применяя не менее трех повторностей опыта, определяют число клеток в 1 мл молока. По санитарным нормативам для пастеризованного молока группы А и группы Б допускается микробное число 0,5 • 105 и 105 соответственно.
Определение титра бактерий группы кишечной палочки (коли-титр). Бактерии этой группы указывают на фекальное загрязнение молока. Коли-титр — наименьший объем молока, в котором обнаружены бактерии группы кишечной палочки (БГКП) — представители родов Escherichia, Citrobacter, Enterobacter.
Для определения коли-титра молоко засевают в пробирки со средой Кесслера состава (г): пептона — 10; желчи — 50; лактоза — 2,5; кристаллвиолет (0,075%-й водный раствор) — 2,0 мл; вода водопроводная — до 1 л. Значение pH среды до стерилизации доводят до 7,0. Стерилизацию проводят при 0,5 ати 30 мин.
В три пробирки со средой Кесслера вносят по 1 мл молока, а в другие три — по 0,1 мл молока. Посевы инкубируют при 42 °C в течение суток, после чего из забродивших проб делают посевы на среду Эндо (см. приложение 4) и инкубируют при 37 °C. Если бродильная проба положительная и культура на среде Эндо дает ярко-красные колонии, то в исследуемом молоке с большой долей вероятности присутствуют БГКП. Однако для полной идентификации БГКП из выросших колоний красного цвета готовят мазки, окрашивают их по Граму, микроскопируют и делают из этих же колоний бактериологической петлей посев в пробирки со средой Козера ив 1%-ю пептонную воду с 1 % глюкозы (среда с глюкозой)-. 10 г пептона; 10 г глюкозы и 2 мл 1,6%-го спиртового раствора бромтимолблау на 1 л воды; поплавки, pH 7,0; стерилизация при 0,5 ати. Среда Козера содержит (г): цитрат натрия — 2,5; сульфат магния — 0,2; фосфат калия однозамещенный — 1,0; фосфат аммония — 1,5; дистиллированная вода — 1 л; pH 7,0. Стерилизация при 0,5 ати.
Таблица 43.1
Классификация молока по редуктазной пробе с применением резазурина
Качество молока Количество бактерий, млн/мл Продолжительность изменения цвета, мин Окраска молока
Хорошее Менее 0,5 60 Сине-стальная
Удовлетворительное 0,6-4,0 60 Сине-фиолетовая или сиреневая
Плохое 4,0-20,0 60 Розовая или белая
Очень плохое Более 20,0 20 Белая
Пробирки с посевами на среде Козера инкубируют при 37 °C, а на среде с глюкозой — при 43 °C в течение суток. При оценке результата на среде с глюкозой учитывают варианты, где наблюдается брожение глюкозы с образованием газа и кислоты (среда желтеет), а на цитратной среде Козера — где нет роста. На основании определения коли-титра молоко по качеству подразделяют на классы. По санитарным нормативам для пастеризованного молока группы А и группы Б допускаются коли-титры 3 и 0,3 мл соответственно.
Аналогичным образом исследуют на редуктазу, микробное число и коли-титр сливки, молочнокислые продукты, мороженое.
Глава 44
МОЛОЧНОКИСЛЫЕ БАКТЕРИИ
Молочнокислые бактерии широко распространены в природе. Вопрос о естественной среде их обитания обсуждается в различных работах, авторы которых придерживаются двух точек зрения: одни считают, что это молоко и молочные продукты, другие склонны назвать прверхность растений, ризосферу и прикорневую зону. Вместе с растениями молочнокислые бактерии попадают в желудочно-кишечный тракт человека и животных, составляя его микрофлору. Элективной средой является молоко и молочные продукты. Бактерии рода Bifidobacterium преобладают в кишечнике грудных детей, особенно вскармливаемых грудью, поскольку эти бактерии нуждаются в углеводах, содержащих N-ацетилглюкозамин, который найден только в женском молоке. Обнаружены они и в кишечной флоре взрослых людей, а также в гниющем иле.
Основным свойством молочнокислых бактерий, по которому их объединяют в отдельную обширную группу микроорганизмов, является способность образовывать в качестве главного продукта брожения молочную кислоту. Молочнокислое брожение осуществляют бактериальные организмы, гетерогенные по морфологии, относящиеся к родам Lactobacillus, Leuconostoc, Bifidobacterium, Pediococcus, Streptoccoccus, Lactococcus. Следует отметить, что по морфологии молочнокислые кокки — типичные стрептококки серологической группы N, кроме Streptococcus thermophilus, относящийся к группе D. Однако недавно по систематическому положению они выделены из группы микроорганизмов рода Streptococcus, включающего и патогенные формы, и под новым названием Lactococcus отнесены к категории «GRAS», куда относятся микроорганизмы, не вызывающие инфекционных заболеваний человека и животных. В этот род отнесены следующие виды и подвиды: L. lactis — подвиды cremoris, hordniae, lactis', L. garvieac, L. pischlunr, L. plantarum', L. raffinolactis.
Молочнокислые бактерии по характеру продуктов сбраживания гексоз (глюкоза, фруктоза, манноза, галактоза), дисахаридов (лактоза, мальтоза, сахароза) и полисахаридов (декстрин, крахмал), согласно терминологии, предложенной А. И. Клюйвером и Г.Л.Донкером в 1925 г., относят к гомоферментативным и гетероферментативным.
Гомоферментативные молочнокислые бактерии
Гомоферментативное молочнокислое брожение представляет собой эволюционно самый древний и примитивный метаболический путь. Последовательность биохимических реакций, лежащих в основе этого брожения, осуществляется по гликолитиче
ской схеме Эмбдена—Мейергофа—Парнаса — по именам исследователей, внесших большой вклад в изучение этого процесса. Основной энергетический субстрат для молочнокислых бактерий, осуществляющих гомоферментативное молочнокислое брожение, — моносахара (в первую очередь, глюкоза) и дисахара (лактоза, мальтоза). Распад отдельных, существенных для бактерий углеводов завершается, как правило, образованием трех главных моносахаров — глюкозы, фруктозы и галактозы, которые служат исходным материалом для осуществления различных реакций обмена веществ.
Гомоферментативное молочнокислое брожение интересно тем, что в нем процесс сбраживания углевода наименее связан с конструктивным обменом, в результате чего 98 % углевода превращается в молочную кислоту. Это объясняется тем, что углевод или совсем не расходуется, или используется в незначительном количестве, так как конструктивный обмен, за счет которого образуется масса бактерий, основан на использовании аминокислот среды. Молекулярный кислород не влияет на процесс молочнокислого брожения, и клетки способны расти в его присутствии. Гомоферментативные молочнокислые бактерии являются факультативными анаэробами. В их клетках найдены флавиновые ферменты, с помощью которых кислород постепенно восстанавливается до перекиси водорода. Накопление перекиси замедляет окисление глюкозы, ингибирует рост.
Лишь небольшая часть пирувата подвергается декарбоксилированию с превращением в уксусную кислоту, этиловый спирт, углекислоту и следы ацетоина. В процессе гомоферментативного молочнокислого брожения большая часть пирувата восстанавливается до лактата лактатдегидрогеназой. Оптическая активность образуемого лактата различна у разных бактерий и зависит от стереоспецифичности этого фермента, а также от того, содержит ли клетка лактатрацемазу, превращающую D-лактат в i-форму.
Гомоферментативные молочнокислые бактерии включают морфологически разные группы — кокки (роды Lactococcus, Streptococcus, Pediococcus) и палочковидные формы (род Lactobacillus). Молярное содержание ГЦ в ДНК в кокках находится в пределах 33 — 44%, а в лактобациллах — 35 — 51 %. При сбраживании глюкозы или глюконата образуют D-, L- или Di-молочную кислоту без образования газа, не сбраживают рибозу и не нуждаются в тиамине. Представители гомоферментативных молочнокислых кокков рода Lacioccoccus образуют в основном молочную кислоту в конфигурации D-, а рода Pediococcus— в конфигурации DL-. Гомоферментативные молочнокислые бактерии рода Lactobacillus образуют разные формы молочной кислоты: D-молочную кислоту — Lb. delbruekii, Lb. leichemani, Lb.jensenii, Lb. lactis, Lb. bulgaricus-, Di-молочную кислоту — Lb. helveticus, Lb. acidophilus. Бактерии Lb. salivarus образуют главным образом i-молочную кислоту, но есть и следы D-молочной кислоты. Лактобациллы Lb. casei subsp. easel,.Lb. casei subsp. tolerans, Lb. casei subsp rhamnosus, Lb.lactosus, Lb. xylosus образуют i-молочную кислоту, однако другие виды (Lb. casei subsp. pseudoplantarum, Lb. curvatus) образуют Di-молочную кислоту и сбраживают рибозу.
Гетероферментативные молочнокислые бактерии
Гетероферментативные молочнокислые бактерии образуют лактат иным путем. Процесс гетероферментативного молочнокислого брожения более сложный, чем гомоферментативного типа. Последняя стадия, восстановление пирувата, такая же, как и при гомоферментативном молочнокислом брожении, но пируват образуется при расщеплении глюкозы исключительно по гексозомонофосфатному пути, а из ксилулозо-5-фосфата образуются 3-фосфоглицериновый альдегид и ацетилфосфат. Затем 3-фос-фоглицериновый альдегид превращается в пировиноградную кислоту, а уже из нее образуется как молочная кислота, так и этанол. Ацетилфосфат отдает фосфат на АДФ, превращаясь в уксусную кислоту:
В процессе брожения из одной молекулы глюкозы образуется 1 молекула АТФ. Итак, в процессе гетероферментативного молочнокислого брожения кроме лактата образуются значительные количества уксусной кислоты, этилового спирта, углекислого газа
и иных побочных нейтральных продуктов (диацетила, ацетоина), на это используются до 50 % сбраживаемых гексоз. Известен другой тип гетероферментативного молочнокислого брожения, когда бактерии вместо этанола образуют другие спирты, например маннит {Leuconostoc mesenteroides) или глицерин.
Представителями гетероферментативных молочнокислых бактерий являются некоторые палочковидные лактобациллы из родов Lactobacillus и Streptobacterium, вариабельные по форме бифидобактерии из рода Bifidobacterium и кокковые — род Leuconostoc.
При изучении молочнокислого брожения в динамике развития культур было установлено, что химические превращения в среде изменяются по ходу развития. При сбраживании углеводов ясно можно различить две фазы. В первый период (логарифмическая фаза роста бактерий) идет интенсивный синтез белков и других веществ клетки, более восстановленных, чем углеводы. В среде накапливаются более окисленные продукты обмена. Вторая фаза характеризуется замедлением биосинтеза и постепенным снижением окислительно-восстановительного потенциала в культуре.
44.1. МОРФОЛОГИЯ КЛЕТОК МОЛОЧНОКИСЛЫХ БАКТЕРИЙ
Для микроскопических исследований морфологии молочнокислых бактерий, выросших на молоке или молочных продуктах, готовят фиксированные окрашенные препараты. Для этого бактериологическую петлю вводят в молочный сгусток и размазывают его по поверхности предметного стекла тонким слоем. Препарат сушат на воздухе, затем фиксируют смесью спирта с эфиром в соотношении 1:1, несколько раз нанося смесь на мазок. Обработка препарата эфиром приводит к извлечению и удалению жира из мазка. Фиксированный препарат рекомендуют окрашивать метиленовым синим в течение 2—3 мин, поскольку именно этот краситель слабо окрашивает основной фон препарата (казеин молока) и хорошо прокрашивает клетки, что обеспечивает четкость препарата.
Молочнокислые бактерии неподвижны, не образуют спор, капсул, положительно окрашиваются по Граму. По форме клеток молочнокислые бактерии представляют собой кокки сферической или эллипсовидной формы и палочки разной длины и формы. Форма и длина клеток у различных культур одних и тех же видов молочнокислых бактерий часто зависит от состава среды, присутствия кислорода и способа культивирования. Бактерии рода Lactococcus — круглые или слегка овальные клетки, расположенные единично, парами или цепочками. Типичный представитель этого рода L. lactis svbsp.lactis некоторые считают палочковидной формой, поскольку его клетки больше в длину, чем в ширину, а в сброженном молочном сгустке преобладают сочетания в виде диплококков. Сливочный лактококк L.lactis subsp. cremoris отличается от предыдущего тем, что его клетки располагаются в виде длинных цепочек. Большинство штаммов термофильных стрептококков Streptococcus thermophilus также образуют длинные цепочки.
Род Lactobacillus по морфологии характеризуются большим разнообразием — от коротких коккообразных до нитевидных палочек длиной от 0,7— 1,1 до 3,0—8,0 мкм, расположенных единично или собранных в цепочки. Значительное увеличение длины клеток лактобацилл наблюдается при недостатке витамина В12, культивировании в средах с высоким содержанием этилового спирта, антибиотиков, при воздействии на клетки лизоцима и ионизирующего излучения. Группа молочнокислых палочек по сво
им свойствам разделяется на термофильные, мезофильные и газообразующие. Мезофильные палочки из подрода Streptobactreium мельче и тоньше термофильных, а газообразующие бетабактерии (Betabacterium) по морфологии сходны с мезофильными стрептобактериями.
Молочнокислые бактерии размножаются делением перегородкой. Если клетки делятся в одной плоскости, это приводит к образованию цепочек; когда же деление идет в двух плоскостях, образуются так называемые тетрады, характерные для молочнокислых бактерий рода Pediococcus. Клетки бактерий рода Leuconostoc сферические, часто чечевицеобразные, делятся в двух плоскостях, образуя пары или тетрады. Клетки рода Lactococcus делятся в одной плоскости, в результате чего образуются пары и цепочки кокков. Бактерии Bifidobacterium bifidum представляют собой палочки, изогнутые, булавовидные и разветвленные, расположенные поодиночке, парами или цепочками, V-образно, палисадом или розеткой, иногда встречаются раздутые кокковидные формы.
На агаровых средах бактерии формируют колонии сферические, часто чечевицеобразные, выпуклые белые или слегка кремового цвета. Форму колоний исследуют в глубине агара после засева его уколом или внесения в него небольшой петлей культуры, выросшей в жидкой среде. Термофильные молочнокислые палочки образуют шероховатые колонии, мезофильные, как правило, гладкие, округлой формы. Lb.bulgaricus на плотных средах образует мелкие характерные колонии в виде комочков ваты.
Молочнокислые бактерии под влиянием факторов внешней среды часто подвержены диссоциации популяции на R-, S- и М-варианты, отличающиеся по генетическим, морфологическим и физиолого-биохимическим признакам.
Ультраструктура клеток молочнокислых бактерий и ряда других грамположительных бактерий во многом похожа. Внутренняя структура клеток может быть различной у отдельных видов. Иногда палочковидные формы содержат зерна метахроматина, который может образовывать в них крупные включения. Нахождение таких включений в клетках характерно для определенных видов рода Thermobacterium. Например, Lactobacillus helveticus и Lb. jugurti можно отличить от Lb. bulgaricus и L. lactis по присутствию у последних метахроматиновых гранул, которые обнаруживаются при выращивании бактерий в молоке с глюкозой и дрожжевым автолизатом.
44.2. ФИЗИОЛОГО-БИОХИМИЧЕСКИЕ СВОЙСТВА МОЛОЧНОКИСЛЫХ БАКТЕРИЙ
Простота внешней формы молочнокислых бактерий и строения их клетки делают чисто морфологическую классификацию молочнокислых бактерий затруднительной. Поэтому культуральные и физико-биохимические признаки, наряду с морфологическими, занимают важное место. Из культуральных признаков используют характер роста на МПБ (пленка, муть, осадок), тип колоний на МПА, рост в присутствии 4%-й NaCl (осмотолерантность). По отношению к молекулярному кислороду бактерии являются факультативными анаэробами, но Bifidobacterium bifidum — строгие анаэробы. Молочнокислые бактерии каталазо- и оксидазо-отрицательные, не образуют пигмент, кроме Leuconostoc citreum, образующего капсулы и желтый пигмент, не восстанавли
вают нитраты в нитриты. Однако описаны штаммы, по некоторым свойствам отличные от типичных. Они подвижны, восстанавливают нитраты в нитриты, образуют споры, дают положительную реакцию на каталазу.
Некоторые молочнокислые бактерии способны разлагать образующуюся при использовании определенных субстратов перекись водорода благодаря наличию у них каталазной активности, осуществляемой так называемой псевдокаталазой. Псевдокаталаза обнаружена у штаммов Pediococcuspentosaceus, Leuconostoc mesenteroides, Lactobacillus plantarum при выращивании их на питательном агаре с 0,5 % глюкозы. Второй фермент, разрушающий перекись, каталаза, найден у ряда молочнокислых бактерий только при выращивании их на среде, содержащей прогретую кровь или гематин.
Физиологической особенностью молочнокислых бактерий является их кислотоустойчивость, как следствие характерного для них энергетического обмена. Кокковые формы могут развиваться в нейтральных и щелочных средах, большинство же палочковидных форм не способны расти в среде с pH выше 6,0; бифидобактерии не растут при pH выше 8,2. Рост всех молочнокислых бактерий продолжается до тех пор, пока в процессе брожения углеводов pH в среде не снизится до 5,0 и ниже. В результате жизнедеятельности гомофермен-тативных лактококков накапливается до 1 %, a Lb.bulgaricus — до 3,5 % молочной кислоты.
Молочнокислые бактерии обладают определенной протеолитической активностью, обусловливаемой действием внеклеточных и внутриклеточных протеиназ и пептидаз. Протеолитическую активность проявляют как кокковые формы, так и лактобактерии, включая термофилов. Молочнокислые палочковидные бактерии обладают большей протеолитической активностью, чем кокковые формы. Например, L. lactis subsp. casei могут переводить до 25 — 30% казеина в растворимую форму, тогда как L. lactis subsp cremoris и L. subsp lactis — 15 — 17 %. Гидролиз молочнокислыми бактериями большинства пептидов (например, DZ-аланилглицина и глицил-£>£-аланина) сильно стимулируется ионами кобальта при pH 5,5 —6,0.
Молочнокислые бактерии либо вовсе не обладают липолитической активностью, либо проявляют ее в определенной степени при воздействии на некоторые субстраты. Штаммы многих видов лактококков (£. lactis, L. cremoris, L. diacetilactis, Streptococcus thennophilus, а также Leuconostoc mesenteroides, Pediococcus cerevisiae) и лактобацилл (Lb. helveticus, Lb. delbrueckii, Lb. casei, Lb. pentosusvt Lb. brevis) в качестве субстрата используют некоторые триглицериды.
Следует отметить, что молочнокислые бактерии явились объектом, с помощью которого были открыты некоторые новые жирные кислоты. Изучая природу жирных кислот у отдельных лактобацилл, установили, что «свободные» и «связанные» липиды, извлеченные из Lb. arabinosus, содержат пальмитиновую, стеариновую, а также ранее неизвестную жирную кислоту — лактобацилловую (С19 Н36 О2).
Отдельные виды лактобацилл (Lb. fermenti, Lb. casei, Lb. salivarius, Lb. viridescens) способны образовывать внеклеточные нуклеазы при выращивании на агаре, содержащем ДНК или РНК. Судя по величине диаметра светлых зон вокруг колоний на агаре с ДНК (зоны до 3—4 мм) некоторые штаммы Lb. fermenti характеризуются значительной ДНКазной активностью. Иногда лактобациллы синтезируют одновременно и внеклеточные ДНКазы и РНКазы.
Если способность к образованию ацетоина достаточно распространена у молочнокислых бактерий, то штаммы, образующие диацетил в значительных количествах, встречаются достаточно редко. Все известные штаммы, синтезирующие диацетил, образуют и ацетоин, однако, если штамм образует ацетоин, это еще не значит, что при этом синтезируется и диацетил.
В основном молочнокислые бактерии культивируют на средах с шестиуглеродными сахарами, но они могут также сбраживать некоторые пятиуглеродные сахара, сахароспирты, органические кислоты и полисахариды.
Отличительным признаком молочнокислых бактерий является высокая потребность в сложных питательных веществах, определенных аминокислотах, витаминах, особенно группы В. Наиболее важным источником энергии для молочнокислых бактерий являются моно- и дисахариды — глюкоза, лактоза, сахароза, мальтоза, но одновременно они не сбраживают трисахарид рафинозу и пентозу ксилозу. Используют в качестве источника энергии и в конструктивном обмене также органические кислоты, такие, как лимонную, яблочную, пировиноградную, фумаровую и др. Свойство сбраживать различные сахара и органические кислоты, в частности рибозу, цитрат или ацетат, лежит в основе отличительных тестов для идентификации молочнокислых бактерий.
44.2.1. Определение количества молочной кислоты
Количество молочной кислоты определяют методом титрования и по разности между объемами 0,1 н. раствора NaOH, пошедшего на титрование среды культивирования до и после роста бактерий. Для титрования берут 10 мл культуральной жидкости или обрата, помещают в колбу Эрленмейера, добавляют 20 мл дистиллированной воды, 1 — 2 капли фенолфталеина и титруют 0,1 н. раствором NaOH при постоянном взбалтывании до появления устойчивой слаборозовой окраски. Кислотность молока или культуральной жидкости выражают в градусах Тернера (°Т) или процентах молочной кислоты. Так, 1 °Т соответствует 1 мл 0,1 н. раствора NaOH, пошедшего на титрование 100 мл исследуемой среды. Общее содержание молочной кислоты выражается в процентах. Поскольку известно, что молекулярная масса молочной кислоты составляет 90, то для приготовления 1 л 1 н. раствора требуется 90 г кислоты, значит, в 1 мл 0,1 н. раствора содержится 0,009 г кислоты, что соответствует 1 °Т.
44.2.2. Качественная реакция на молочную кислоту
Принцип реакции состоит в переводе молочной кислоты в уксусный альдегид в кислой среде при температуре кипения в присутствии перманганата КМпО4 и взаимодействия альдегида с аммиачным серебром, дающего реакцию «серебряного зеркала». Для проведения реакции культуральную жидкость или сброженный обрат фильтруют через бумажный складчатый фильтр. Берут 5 мл фильтрата, помещают в колбу Эрленмейера, добавляют 2 мл крепкой серной кислоты и, взбалтывая, нагревают на асбестовой сетке до начала кипения. Затем, продолжая помешивание, по каплям пипеткой добавляют 5 мл 5 %-го раствора КМпО4, который в процессе реакции обесцвечивается. При этом молочная кис
лота переходит в уксусный альдегид- Для проведения характерной реакции быстро покрывают горлышко колбы фильтровальной бумагой, смоченной аммиачным раствором нитрата серебра AgNO3. Фильтровальную бумагу аккуратно прижимают к краям колбы, продолжая нагревание. Уксусный альдегид улетучивается и, вступая в реакцию с аммиачным раствором AgNO3, вызывает почернение бумаги серебристыми разводами (выделяется металлическое серебро).
Аммиачный раствор нитрата серебра AgNO3 готовят следующим образом: к 1 — 2 мл 10%-го раствора AgNO3 в пробирке приливают по каплям аммиак, что приводит к выпадению осадка Ag2O, постепенно растворяющегося далее в избытке аммиака.
44.3. ВЫДЕЛЕНИЕ И ИДЕНТИФИКАЦИЯ МОЛОЧНОКИСЛЫХ БАКТЕРИЙ
Молочнокислые бактерии, обитающие в природных и производственных субстратах, обычно не занимают доминирующего положения, над ними превалируют различные сапрофитные микроорганизмы, такие, как грамотрицательные бактерии, споровые и бесспоровые формы, кокки, дрожжи, плесени. Поэтому для их выделения используют элективные питательные среды, способствующие их росту и угнетающие рост сопутствующей микрофлоры. В качестве ингибиторов роста посторонней микрофлоры в среды добавляют сорбиновую и уксусную кислоты, спирт, азид натрия и др.
Источниками для выделения лактококков служат кисломолочные продукты (сметана, простокваша, сыры, кефир и др.), лактобацилл — силос и травы, пищевые продукты и фекалии. Для определения присутствия и учета молочнокислых бактерий навеску испытуемого материала разводят в физиологическом растворе и высевают в жидкие накопительные среды или же сразу на твердые агаровые среды для подсчета их количества.
Известна высокая питательная ценность сред из гидролизата молока, пригодных для учета и выделения молочнокислых бактерий. Для приготовления гидролизата берут 1 л пастеризованного при 85 °C и охлажденного до 45 °C обезжиренного молока, доводят активную кислотность 40%-м водным раствором NaOH до pH 7,4 —7,6, добавляют к нему 1 г порошка панкреатина и 5 мл хлороформа и помещают в термостат на 40 °C на 72 ч. Затем гидролизат фильтруют, устанавливают pH, равным 7,0—7,2, и стерилизуют при 1 атм в течение 10 мин. Для приготовления агаризованных сред гидролизат разводят в два раза водопроводной водой, добавляют 2,5 % дрожжевого автолизата и 2,0 % агар-агара. Учет количества бактерий облегчается при добавления в среду инкубирования 2 мл 0,1 %-го раствора индикатора бромкрезолового пурпурного или мела (100 мл 3%-й суспензии углекислого кальция на 1 л), образующих при глубинном инкубировании прозрачную зону просветления вокруг колоний.
Для выделения ароматообразующих лактококков L. lactis subsp. diacetilactis из исследуемых образцов в гидролизат молока добавляют 1 % лимоннокислого натрия. После культивирования бактерий в этой накопительной среде в течение 18 —24 ч при 30 °C суспензию высевают глубинным посевом на агаризован-ную среду того же состава. По пузырькам газа, выделяющегося при использовании цитрата, находят колонии этих лактококков. Для выделения слизеобразующих бактерий Leuconostoc mesenteroides в эту же среду добавляют 1 % сахарозы.
При разделении смешанных культур лактококков рекомендуют среду Редди с аргинином следующего состава (%): триптон — 0,5; СаСО3 — 0,3: КМЦ — 0,6
(карбоксиметилцеллюлоза для суспендирования СаСО3); К2НРО4 — 0,1; дрожжевой экстракт — 0,5; £-аргинин солянокислый — 0,5; агар-1,5; pH после стерилизации при 1 атм в течение 15 мин должен быть 6,3. Кроме расплавленной и остуженной до 45 — 50 °C основы добавляют 5 мл стерильного обезжиренного молока и бромкрезоловый пурпурный или мел в количествах, как указано выше. На этой среде L.lactis subsp. cremortis, не гидролизующий аргинин, образует желтые колонии, a L. subsp. lactis и L. subsp. diacetilactis — белые.
В состав сред для выращивания лактобацилл входят ингредиенты, богатые аминокислотами, витаминами. Добавляют томатный сок, дрожжевой экстракт или автолизат как источник пуринов и пиримидинов, необходимых для роста. Среда PC для выделения бактерий рода Lactobacillus и Lactococcus plantarum содержит (%): пептон — 1,0; глюкозу — 2,0; печеночный экстракт — 1,0; дрожжевой экстракт — 0,2; полисорбат — 0,8; К2НРО4 — 0,2; MgSO4 — 0,2; MnSO4 — 0,05; триаммониевый цитрат — 0,2; ацетат натрия — 0,5; твин-80 — 0,01. Среду разливают в стерильные пробирки и стерилизуют при 1 атм в течение 15 мин. Устанавливают pH 6,2. Для повышения селективности в среду перед засевом добавляют 1 мл 0,4%-го щелочного раствора сорбиновой кислоты. Испытуемый материал засевают вначале в жидкую среду, а затем наслаивают на нее «голодный агар» (столбик высотой до 2 см) для создания анаэробных условий, инкубируют в течение 5 сут при 30 °C или 3 сут 37 °C. При наличии мути и газообразования инокулят высевают на агаризованную среду этого же состава, инкубируют при заданной температуре и производят подсчет лактобацилл через 48 ч. Для создания анаэробных условий на застывшую в чашках Петри среду с инокулятом наливают второй слой расплавленной и остуженной голодной агаровой среды в количестве 5 см3.
Для выделения бактерий рода Bifidobacterium используют кукурузо-лактозную среду. Для приготовления среды берут 25 г агара из расчета на 1 л, расплавляют его в небольшом количестве воды, а к остальному количеству дистиллированной воды добавляют 10 г пептона; 40 лит водного раствора кукурузного экстракта, разбавленного в соотношении 1:1; 6,6 г натрия лимоннокислого трехзамещенного; 0,12 г MgSO4; 2 г К2НРО4 Смесь нагревают до 80 °C и соединяют с расплавленным агаром, затем добавляют 10 г лактозы, 0,15 г цистеина солянокислого и 0,5 г аскорбиновой кислоты. Предварительно цистеин растворяют в небольшом количестве дистиллированной воды и устанавливают pH 8,45 раствором гидроокиси натрия. Всю смесь доводят до 1 л водой и устанавливают pH среды 7,0. Среду разливают в пробирки высоким столбиком по 10 мл и стерилизуют при 0,5 атм в течение 30 мин.
Исследуемый образец разводят в физиологическом растворе, нейтрализуют 10%-м раствором гидрокарбоната натрия и засевают из серии разведений в пробирки со средой, предварительно прокипяченной в течение 15 мин для снижения в ней содержания растворенного кислорода. В момент посева температура среды должна быть 38 °C. Пробирки с посевами инкубируют при 37 °C в течение 72 ч, но просматривают посевы и через 24—48 ч. По окончании учитывают колонии, выросшие в последних разведениях, где выросли типичные колонии для бифидобактерий — в виде «гвоздиков», «вытянутых веретен», иногда в виде «полос», расположенных вдоль пробирки. На плотных средах колонии имеют вид «дисков» или «гречишных зерен». Выросшие колонии подсчитывают с учетом разведения.
Для культивирования бактерий рода Bifidobacterium известна также среда, приготовленная на водопроводной воде, содержащая (г/л): гидролизат казеина — 10,0; мясной экстракт — 5,0; дрожжевой экстракт — 5,0; глюкоза — 10,0; К2НРО4 — 3,0; твин-80 — 1,0 мл, pH 6,8. После стерилизации при 0,5 атм в среду добавляют 1,0 % аскорбата натрия и 0,05 % цистеина солянокислого.
Принадлежность каждой отобранной колонии к определенным микроорганизмам определяют по отношению к окраске по Граму, по подвижности и другим морфологическим признакам. Культуры, образующие псевдокаталазу, относят к Pediococcus, как микроорганизмы, не имеющие цитохромов, но дающие положительную реакцию с бензидином. При идентификации бактерий рода Leuconostoc препарат окрашивают по Бурри для выявления капсул. Отсут-ствие роста в МПБ при pH 9,6 и с 6,5%-м NaCl характерно для лактококков и Streptococcus thermophilus. Причем, инкубируя последние на питательной среде при 45 °C в течение 3 сут, можно отделить мезофильные кокки. После выделения чистую культуру пересевают в обрат и наблюдают характер и время образования молочного сгустка. Лактококки L. lactis subsp. lactis образуют плотный сгусток за 6—10 ч с максимальной кислотностью 100—125 °Т; L. lactis subsp. cremoris дает сметанообразный сгусток за это же время; L. lactis subsp. diacetilacis свертывает молоко за 18—48 ч. Активные расы термофильных лактобацилл при оптимальной температуре сбраживают молоко за 10—12 ч, причем предельная кислотность достигает 300 — 450 °Т, мезофильные стрептобактерии образуют плотный сгусток через 3 — 5 сут с кислотностью 200 — 250 °Т, а газообразующий тип лактобацилл не образует сгустка.
Молочнокислые бактерии синтезируют различные ингибиторные вещества, например: кислоты, ацетон, перекиси, а также низкомолекулярные антимикробные белки — бактериоцины, которые подавляют рост сопутствующей микрофлоры.
44.4. ОБРАЗОВАНИЕ БАКТЕРИОЦИНОВ МОЛОЧНОКИСЛЫМИ БАКТЕРИЯМИ
Бактериоцины составляют большое семейство полипептидов, которые подразделяют на различные классы, основываясь на их действии и их структуре. Все бактериоцины по биохимической природе являются белками. В пользу этого предположения свидетельствует тот факт, что большинство антибактериальных веществ, относящихся к бактериоцинам, разрушаются протеолитическими ферментами, но у разных бактериоцинов чувствительность к протеазам варьирует. Некоторые бактериоцины резистентны к действию протеаз. Устойчивые к протеазам бактериоцины, например низин и лактицин 481, образуемые L. lactis subsp. lactis., характеризуются наличием в их молекулах сульфидных колец, которые содержат мезо-лантионин и р-метиллантионин, а также необычные аминокислоты — дегидромасляную и D-дегидроаланин.
Бактериоцины имеют молекулярный вес от 3 до 10 кДа. Белок бактериоци-на связан с липополисахаридом клеточной оболочки, но именно белковая часть комплекса обладает антибактериальной активностью. Эти факты указывают на наличие в клетках определенных механизмов, регулирующих функции бактериоциногенных факторов. Биосинтез бактериоцинов — наследственная
особенность организмов, проявляющаяся в том, что каждый штамм способен образовывать один или несколько определенных, строго специфичных для него антибиотических веществ.
К числу общих свойств бактериоцинов относится их чувствительность к температурным воздействиям, хотя это свойство также варьирует в широких пределах. Одни из них разрушаются уже при температуре 48 —50 °C, другие кратковременно выдерживают температуру 60 —70 °C, а отдельные сохраняют активность даже при 100 °C, например, низин выдерживает кипячение до 120 °C без потери активности.
Существуют различные способы выявления антибиотических свойств микроорганизмов. Многие из них основаны на способности антибиотических веществ диффундировать в плотные агаризованные среды и образовывать зоны подавления роста тест-организма. Величина зоны отсутствия роста указывает на степень активности данного антибиотического вещества в отношении исследуемой тест-культуры и зависит от его концентрации и химической природы.
За единицу активности бактериоцина низина принимают такое его количество, которое подавляет рост Streptococcus agalactiae (стрептококка группы В, присутствующего в желудочно-кишечном тракте) в 1 мл питательного бульона за 16 ч инкубирования, учитывая, что 1 мг чистого низина содержит 40 000 международных единиц (ME).
Низинобразующую активность определяют микробиологическим методом с тест-культурой Bacillus coagulans — термофильной споровой кислотоустойчивой бактерией, которую выращивают в агаровой среде, приготовленной на бульоне Хоттингера с добавлением (г/л): NaCl — 2,0; агар — 20,0; пептон — 10,0; глюкоза — 10,0. Стандартом служат растворы очищенного коммерческого препарата Nisaplin («АррПп & Barrett, Ltd.», Великобритания) с активностью 5, 10, 15, 20, 40 МЕ/мл. Для титрования бактериоцина используют суспензию 17-часовой культуры бациллы с оптической плотностью 0,7 (при X = 540 нм, оптический путь 0,5 см), которой засевают среду, приготовленную на фосфатном буфере (pH 5,0) и содержащую (г/л): пептон — 20,0; глюкозу — 10,0; агар — 20,0. Экстракцию антибиотика из культуральной жидкости и стандартных растворов проводят смесью ацетон: уксусная кислота: вода (4:1:5) при 55 °C в течение 90 мин. Экстракты разводят фосфатным буфером (pH 5,0) в соотношении 1:10 и вносят в лунки на агаровой среде с тест-организмом, затем инкубируют при 55 °C в течение 17 ч. Количественное определение антибиотической активности проводят по измерению зон подавления роста В. coagulans с дальнейшим пересчетом по калибровочной кривой стандартных растворов низина.
Хранение молочнокислых бактерий
Традиционные методы поддержания культур молочнокислых бактерий сводятся к их выращиванию на богатых питательных средах с частыми пересевами. Элективной средой является обезжиренное молоко (обрат). Бактерии, выращенные в обрате, хранят в виде молочного сгустка в течение месяца. Однако частые периодические пересевы ведут к снижению выживаемости бактерий, изменению состава популяции, потере ряда физиолого-биохимических свойств. Гетероферментативные молочнокислые бактерии могут утратить способность к образованию летучих кислот, спирта и СО2 и перейти на гомоферментативный путь сбраживания гексоз. Известны способы хранения культур, выращенных на специфических для бактерий средах, в 25%-м растворе глицерина или под слоем вазелинового масла при -20 °C, в лиофильном состоянии, высушенных с использованием защитных сред в качестве криопротекторов, изменяющих осмотическое давление в среде и снижающих величину активности воды. В качестве защитных сред используют водные растворы различных компонентов (%): 1) сахароза, 10,0 + желатин, 1,0; 2) сахароза, 22,0 + желатин, 11,0 + глутамат натрия, 4,4 + цитрат
натрия трехзамещенный, 5,0; 3) сахароза, 22,0 + глутамат натрия, 4,4. Лиофилизация обеспечивает длительную жизнеспособность культур молочнокислых бактерий. Подбор условий лиофилизации и выбор защитной среды специфичен для разных типов бактерий. Способ хранения под вазелиновым маслом обеспечивает высокий уровень выживаемости для лактобацилл и не эффективен для гомоферментативных лактококков.
Использование молочнокислых бактерий
Молочнокислые бактерии уже более 100 лет привлекают к себе возрастающее внимание исследователей. Классические работы Л. Пастера открыли первые страницы в изучении этой важной группы микроорганизмов.
Развитие микробиологии резко расширило и усовершенствовало области применения молочнокислых бактерий. На основе их использования возникли крупные промышленные производства. Промышленным источником углеводного компонента при молочнокислом брожении являются меласса, патока, молочная сыворотка.
Многие молочнокислые бактерии изначально присутствуют в молоке и вызывают его спонтанное сквашивание. Молочная кислота в молоке отщепляет кальций от казеина, белок превращается параказеин и выпадает в осадок, что вызывает свертывание молока, образование молочного сгустка. Заквашивание молока представляет собой своеобразный способ консервации этого скоропортящегося продукта. При изготовлении молочных продуктов, таких, как масло, сыр, йогурт, широко используются молочнокислые бактерии.
Изготовление сыра и других продуктов посредством створаживания молока возникли также как способ консервации молока. При изготовлении сыра первичное молочнокислое брожение осуществляется гомоферментативными лактококками L.lactis subsp. cremoris и lactis, а затем в процесс включаются термофильные молочнокислые бактерии рода Lactobacillus или микрорганизмы других таксономических групп, например пропионовые. Часто при изготовлении сыров используют молочнокислые бактерии в симбиозе с грибами, плесенями. Некоторые обменные процессы лактобактерий обусловливают специфические функции, прямо или опосредованно участвующие в процессах созревания сыра: сбраживание лактозы, восстановление окислительно-восстановительного потенциала сыра, сбраживание цитрата, расщепление казеина, действие ряда протеиназ, пептидаз и аминопептидаз. В результате гидролиза казеина образуются свободные аминокислоты, а также пептиды различной молекулярной массы. Таким образом, казеин служит источником незаменимых и ростовых аминокислот, используемых лактококками.
Выявлен ряд бактериоцинов, образуемых молочнокислыми бактериями. Некоторые из них с успехом применяются в пищевой промышленности, а также в медицине. Например, бактериоцин низин впервые был использован в промышленном производстве как консервант в Великобритании около 50 лет назад для сохранения сыров и пресервов. Низин обладает бактерицидным действием на грамположительные микроорганизмы: стрептококки различных серологических групп, стафилококки, пневмококки, микобактерии, споровые аэробные и анаэробные бактерии. Обладая способностью адсорбироваться на поверхности спор чувствительных к нему спорообразующих микроорганизмов, низин нарушает проницаемость мембран, снижает термоустойчивость спор. Вследствие этого низин способствует сохранению качества продуктов при снижении режимов стерилизации в процессах производства консервов и пресервов.
Качество молочнокислых продуктов зависит от способности микроорганизмов, входящих в состав закваски, образовывать вкусовые и ароматические вещества. К ним относятся ацетоин и диацетил. Диацетил, продуцируемый Lb. bulgaricus, отвечает за «масляный» аромат и важен в создании вкусовых оттенков кислосливочного масла. В состав закваски для изготовления йогурта входят Lb. bulgaricus и L. lactis subsp. diacetilactis. Биойогурт получают при сквашивании молока бактериями Lb. acidophilus и Streptococcus
thermophilus. Известен ряд кисломолочных продуктов, полученных в результате смешанного молочнокислого и спиртового брожения. Например, при приготовлении кефира используют кефирные зерна — комплексную закваску на основе L. lactis. subsp. lactis, Lb. bulgaricus и дрожжей, сбраживающих лактозу. Известны национальные напитки, такие, как мацун, лебен, приготовленные на коровьем молоке. Широкую известность как лечебного напитка для лечения туберкулеза, заболеваний желудочно-кишечного тракта и сердечно-сосудистой системы получил кумыс. Готовят кумыс из кобыльего молока, близкого по составу женскому, посредством сквашивания его лактобациллами и Saccharomyces lactis. Лечебное назначение зависит от крепости кумыса и содержания в нем спирта, что определяется длительностью процесса брожения. Из верблюжьего молока подобным образом готовят шубат.
Осознание роли, которую играют микроорганизмы в производстве продуктов питания, привело к развитию особой ветви микробиологии — технической микробиологии пищевых продуктов. С помощью молочнокислых бактерий Lb. casei, Lb. delbrukii, Lb. leichemanii и др. в промышленности получают молочную кислоту. Преимущество микробиологического способа получения молочной кислоты перед дорогостоящим и технически сложным химическим состоит в возможности направленного синтеза определенного изомера кислоты (например, /.-формы).
Декстран, продуцируемый некоторыми видами лейконостоков, например Leuconostoc mesenteroides, нашел широкое применение в пищевой и медицинской промышленности. Это один из первых плазмозаменителей крови, на его основе получены ферригли-кин, реогликин и другие препараты, используемые при различных патологических состояниях людей. Сульфатированные декстраны применяют как антикоагулянты крови при сердечно-сосудистых заболеваниях. Сефадексы — нерастворимые сшитые декстраны, широко применяются в технике разделения полимерных веществ.
Огромна роль молочнокислых бактерий в мясной промышленности. Попадая извне, они могут вызвать различные виды порчи мяса и мясных продуктов, но при целенаправленном применении стартовые культуры (наиболее используемые бактерии родов Pediococcus и Lactococcus) при размножении в фарше вытесняют сопутствующую микрофлору и благоприятно влияют на консистенцию мясного фарша, способствуют сохранению цвета колбасных изделий, созданию их аромата. Подобную роль выполняют молочнокислые бактерии, например Lb. plantarum, Lb. coryniformis и др. в хлебопечении. Лактобактерии с успехом применяют в рыбной, спиртовой промышленностях, при приготовлении кислых спиртных напитков (кваса, белого пива), при консервировании овощей, квашении капусты. Одна из основных задач техники силосования кормов состоит в создании благоприятных условий для жизнедеятельности молочнокислых бактерий, обитающих на растениях, и подавления сопутствующей вредной микрофлоры.
Глава 45
ВЫДЕЛЕНИЕ УГЛЕВОДОРОДОКИСЛЯЮЩИХ МИКРООРГАНИЗМОВ
Среди различных видов техногенных нарушений природы одним из наиболее серьезных и трудно устраняемых является нефтезагрязнение. Микроорганизмы, способные использовать нефть, распространены очень широко и могут быть выделены из любой полевой, лесной или луговой почвы. Кроме того, способность использовать нефть в качестве источника энергии присуща не единичным специализированным формам, а многим группам микроорганизмов углеводородокисляющих (УВОМ). В их числе бактерии из родов Acinetobacter,
Arthrobacter, Bacillus, Cytophaga, Clostridium, Corynebacterium, Flavobacterium, Methanobacterium, Micrococcus, Mycobacterium, Nocardia, Rhodococcus, Pseudomonas, Vibrio, мицелиальные грибы родов Aspergillus, Penicillium, Mucor, Fusarium, Trichoderma, дрожжи — Candida, Endomyces, Rhodotorula, Saccharomyces, Torulopsis.
45.1. ПОЛУЧЕНИЕ НАКОПИТЕЛЬНОЙ КУЛЬТУРЫ УВОМ
Накопительные культуры получают следующим образом: почвенную суспензию в соотношении 1:100 (1 г почвы вносят в 99 мл стерильной жидкой среды) переносят в колбы объемом 250 мл, добавляют стерильную нефть, керосин, дизельное топливо или соляровое масло, культивируют на качалке при 190 — 220 об/мин в течение 5 — 10 сут. Среда содержит (г/л): NaNO3 — 3,0; К2НРО4- ЗН2О - 1,0; MgSO4 - 7Н2О - 0,5; КС1 - 0,5; FeSO4- 7Н2О - 0,01; вода водопроводная. Стерилизуют при 1 ати, pH после стерилизации 6,5 —7,0. Стерильную нефть и ее легкие фракции вносят в стерильные среды в количестве 1 % (по объему).
Нефть, керосин, дизельное топливо и соляровое масло в лабораторных условиях стерилизуют в пенициллиновых флакончиках, закрытых резиновыми пробками с тефлоновым покрытием, закрепленными металлическими колпачками, в автоклаве при 1 ати.
Рост оценивают визуально по степени изменения эмульсии в сравнении с контролем, микроскопированием и по изменению белка биомассы.
45.2. ПОЛУЧЕНИЕ ЧИСТОЙ КУЛЬТУРЫ УВОМ
Чистые культуры получают рассевом на минеральные агаризованные среды (содержание агара — 1,7 %), на поверхность которых наносят стерильную нефть или ее фракции. Готовят разведения накопительной культуры, чтобы получить изолированные колонии после инкубации колоний на чашке. В качестве исходной суспензии используют 1 мл накопительной культуры в 100 мл стерильной среды. Наносят 0,1 мл разведенной культуры на поверхность среды в чашках Петри и затем равномерно распределяют шпателем по чашке, стараясь покрыть всю ее поверхность. Чашки инкубируют при комнатной температуре (18 — 22 °C).
Выросшие изолированные колонии отсевают петлей на поверхность скошенной плотной минеральной среды с углеводородами, в пробирки или на агаризованные среды в чашках Петри. В качестве единственного источника углерода и энергии микроорганизмы при этом используют углеводороды нефти.
45.3. ОПРЕДЕЛЕНИЕ УГЛЕВОДОРОДОКИСЛЯЮЩЕЙ АКТИВНОСТИ БАКТЕРИЙ РОДА CYTOPHAGA
Известна способность углеводородокисляющих бактерий рода Cytophaga разжижать поверхность агаризованной среды, поэтому для доказательства углево-дородокисляющей активности производят посев этих культур на среду с силикагелем в качестве уплотнителя.
Культивирование бактерий рода Cytophaga на среде с силикагелем. Силикагель готовят следующим образом: к соляной кислоте с плотностью d = 1,1 добавляют при перемешивании равный объем раствора жидкого стекла (Na2SiO3 или K2SiO3) той же плотности. Смесь разливают в чашки Петри по 20 — 30 мл в каждую и оставляют чашки на горизонтальной поверхности на несколько часов до образования кремнекислого геля. Затем открытые чашки Петри помещают в стеклянный или эмалированный сосуд и промывают в течение 2 — 3 сут проточной водой для удаления хлоридов, а потом несколько раз горячей дистиллированной водой. Об отсутствии хлоридов судят по качественной пробе промывных вод с 1%-м раствором азотнокислого серебра: при наличии хлоридов образуется белый осадок AgCl2. Чашки с силикагелевыми пластинками до употребления сохраняют под водой. Отмытые от хлорида пластинки пропитывают 2 мл концентрированной минеральной среды, содержание компонентов в которой в 7,5 раз выше, чем в соответствующей среде. Затем чашки с гелевыми пластинками помещают открытыми в сушильный шкаф и подсушивают при 50 — 60 °C, следя за тем, чтобы гель не растрескался и его поверхность осталась влажной. Перед посевом чашки заворачивают в бумагу и стерилизуют в автоклаве при 0,5 ати 15 мин.
Посев микроорганизмов осуществляют обычным образом: на поверхность силикагелевой пластинки, пропитанной концентрированным раствором минеральной среды, наносят субстрат в виде нефти или ее легких фракций и засевают культурой.
Процесс подготовки силикагелевых пластинок является трудоемким и не предусматривает их длительного хранения, поэтому на кафедре микробиологии биологического факультета МГУ был разработан оригинальный способ моделирования плотной среды с использованием поролона, пропитанного минеральной средой с сырой нефтью. Этот материал был выбран по следующим причинам: поролон не используется известными бактериями; является гидрофобным материалом, что обеспечивает абсорбцию нефти или ее легких фракций; он обладает пористой структурой, а это способствует хорошему водно-солевому и воздушному обмену, выдерживает стерилизацию в автоклавах.
Из поролона толщиной 3 — 4 мм готовят диски диаметром 5— 10 см и стерилизуют автоклавированием при 1 атм во влажном состоянии. Затем диск помещают в чашку Петри, пропитывают стерильной минеральной средой, равномерно наносят нефть и засевают углеводородокисляющие микроорганизмы с помощью бактериологической петли. На поверхности и в толще поролона наблюдают рост бактерий.
Культивирование выделенных углеводородокислякицих микроорганизмов в жидкой среде. Для доказательства углеводородокисляющей активности бактерий рода Cytophaga (помимо культивирования на силикагеле и поролоне) культуры высевают на жидкие минеральные среды с сырой нефтью, соляровым маслом, дизельным топливом и керосином (1 — 2%} в качестве единственного источника углерода и энергии.
По мере накопления биомассы и уменьшения субстрата (сырой нефти, Керосина, дизельного топлива или солярового масла) судят о способности выделенных культур окислять углеводороды.
Среднюю удельную скорость роста К за определенный промежуток (6 - г0) вычисляют по формуле
К = 2,303 (log/??! - Logw0)/A - 4),
где т0 и тх — начальная и конечная масса клеток, связанная обратной зависимостью со временем генерации (g = 0,693/А').
45.4. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ НЕФТИ В ПОЧВЕ
Анализ содержания нефти и нефтепродуктов в почве осуществляют гравиметрическим методом после экстракции хлороформом. Остаточную нефть и продукты ее окисления трижды экстрагируют из образцов хлороформом в соотношении 1:3 (по объему). Экстракты смешивают и во флаконах с известным весом помещают для испарения хлороформа под тягу на 24 ч, взвешивают и сравнивают с контролем.
45.5. ОЦЕНКА МИКРОБНОЙ ДЕСТРУКЦИИ НЕФТИ И НЕФТЕПРОДУКТОВ
Микробную деградацию нефти и нефтепродуктов оценивают по изменению их концентраций после разделения отдельных фракций на хроматографической колонке. Проводят определение группового фракционного состава нефти и нефтепродуктов до и после развития бактерий.
Групповой фракционный состав нефти или нефтепродуктов (керосина, дизельного топлива, солярового масла) определяют методом жидкостно-адсорбционной колоночной хроматографии с использованием двойного сорбента. Нефть или нефтепродукт (около 0,4 г в порции) разделяют на колонке (диаметром 8 мм) с двойным сорбентом (внизу — окись алюминия, высота 2 см; вверху — силикагель фирмы «Сйетаро!» (Чехия), 40/100 мкм, высота 1,5 см). Колонку набивают сухим способом и уравновешивают гексаном, субстрат наносят в виде раствора в 1 мл гексана. В качестве элюента используют последовательно смеси: 5 мл гексана; 5 мл смеси гексан; бензол (1: 1 по объему); 8 мл бензола; 5,5 мл смеси бензол:этанол (10:1); 200 мл смеси бензолгэтанол (1: 1). Скорость элюции — 20 мл/ч. Фракции собирают и анализируют на регистрирующем УФ-спектро-фотометре 200 — 20 (Hitachi, Япония), в кюветах с длиной оптического пути 10 мм в диапазоне длин волн 190 — 380 нм и выражают в процентах убыли субстрата.
Глава 46
САНИТАРНАЯ МИКРОБИОЛОГИЯ
Санитарная микробиология изучает микрофлору окружающей среды (воды, воздуха, почвы, продуктов питания и предметов обихода) и ее влияние на здоровье человека.
Эта наука представляет собой смежную с эпидемиологией и гигиеной область медицинской микробиологии. Основные санитарно-микробиологические методы включают в себя как оригинальные методики, так и методы общей и медицинской микробиологии, и направлены на определение общей микробной обсемененности (общее микробное число); обнаружение и титрование санитарно-показательных микроорганизмов (СПМ); выявление патогенных микроорганизмов и их метаболитов; определение степени недоброкачественности изучаемых объектов, обусловленной наличием микроорганизмов.
Для оценки санитарно-эпидемического состояния внешней среды в настоящее время используют методы прямого обнаружения патогенных микроорганизмов (посев на пи
тательные среды, гибридизация нуклеиновых кислот, полимеразная цепная реакция, серологические, биологические и др. методы), а также методы, позволяющие получить косвенную оценку возможного присутствия возбудителя во внешней среде (определение общего микробного числа и содержания СПМ).
Прямое обнаружение патогенов — наиболее точный показатель неблагополучности исследуемого объекта. Однако выделение патогенных микроорганизмов из объектов окружающей среды затруднено из-за наличия в них большого количества сапрофитов, которые препятствуют росту патогенной микрофлоры на питательных средах. Серьезно осложняет процесс анализа неравномерное распределение патогенов в окружающей среде. Кроме того, в условиях внешней среды происходят генетические процессы (мутации, трансформация, трансдукция и др.), которые приводят к изменению исходных и формированию новых признаков патогенных микроорганизмов. К сожалению, даже современные методы не гарантируют успеха при незначительном содержании патогенов в исследуемых объектах. В связи с этим исследования на патогенную микрофлору в санитарной микробиологии проводят только по эпидемическим показаниям, и санитарно-микробиологические исследования, как правило, имеют целью не обнаружение патогенов, а поиск косвенных показателей неблагополучия объектов внешней среды.
Для санитарной оценки микробной обсемененности применяется определение общего микробного числа (ОМЧ). Под ОМЧ понимают количество колоний, вырастающих на МПА в чашках Петри после определенного срока инкубации при той или иной температуре из 1 мл (для твердых субстратов 1 г) исследуемого материала. Таким образом, чаще всего учитываются мезофильные аэробные и факультативно-анаэробные микроорганизмы (МАФАМ), способные расти на МПА. Предполагается, что чем больше объект загрязнен органическими веществами, тем выше ОМЧ. Определение ОМЧ широко используется для санитарной оценки воды и почвы, хотя при этом значительное количество почвенных и водных микроорганизмов не учитывается (выпадают из учета анаэробы, грибы, серные, азотфиксирующие, нитрифицирующие и другие бактерии). При необходимости для учета этих микроорганизмов используют соответствующие питательные среды. Общее количество микроорганизмов также можно определить путем прямого подсчета под микроскопом или с помощью специальных электронных счетчиков.
Определение санитарно-показательных микроорганизмов (СПМ) — специфический метод санитарной микробиологии. К СПМ относят представителей облигатной микрофлоры организма человека и теплокровных животных, обитаюших в кишечнике или воздушно-дыхательных путях. В качестве СПМ могут выступать не все представители нормальной микрофлоры человека и животных, а лишь те, которые удовлетворяют следующим требованиям (А. И. Коротяев, С.А. Бабичев, 1998; А.З. Байчурина и др., 1998; О. К. Поздеев, 2001):
• постоянно присутствуют в выделениях человека и теплокровных животных и попадают в окружающую среду в больших количествах;
• не имеют иного природного резервуара, кроме организма человека или животного;
• не размножаются активно во внешней среде (исключая пищевые продукты);
• выживают во внешней среде несколько больше, чем патогенные микроорганизмы:
• не имеют во внешней среде «двойников» или аналогов, поэтому исключена путаница;
• не изменяют сколько-нибудь значительно свои биологические свойства в окружающей среде;
• растут на питательных средах независимо от влияния других присутствующих микроорганизмов;
• распределены в объекте внешней среды по возможности равномерно (плотные объекты при исследовании подвергаются гомогенизации);
• могут быть обнаружены, идентифицированы и количественно учтены современными, простыми и легко доступными методами;
• встречаются в организме хозяина и во внешней среде в значительно больших количествах по сравнению с соответствующими патогенными микроорганизмами.
Предполагается, что чем больше объект загрязнен экскрементами человека и животных, тем больше будет СПМ и тем вероятнее присутствие патогенов.
СПМ, которые обычно обнаруживаются в исследуемых объектах, приведены в табл. 46.1.
СПМ чаще всего выявляют методом посева на определенные питательные среды. Содержание СПМ оценивают по двум показателям — титру и индексу. Титром называется выраженное в миллилитрах (для твердых тел в граммах) наименьшее количество исследуемого материала, в котором обнаруживаются данные бактерии. Индексом называется количество СПМ, обнаруженных в 1 л жидкости (1 г плотных веществ, 1 м3 воздуха). Индекс — величина, обратная титру.
К СПМ относятся представители разных систематических групп бактерий. Наиболее важными СПМ во всем мире считаются бактерии группы кишечной палочки (БГКП). Под этим общим понятием объединяют бактерии семейства Enterobacteriaceae родов Escherichia, Citrobacter, Enterobacter, Klebsiella. К категории БГКП относят грамотрица-тельные, не образующие спор и не обладающие оксидазной активностью палочки, сбраживающие лактозу и глюкозу до кислоты и газа при 37 °C в течение 24 ч. Эти бактерии выделяются в окружающую среду только с фекалиями человека и теплокровных животных. Среди БГКП выделяют также колиформные палочки, которые сбраживают только лактозу при 37 °C с образованием кислоты и газа, и фекальные кишечные палочки (ФКП), сбраживающие лактозу при 44,5 °C. Из фекальных кишечных палочек к E.coli относятся только бактерии, не способные расти на среде, содержащей в качестве единственного источника углерода и энергии цитрат. Эти бактерии являются показателями свежего фекального загрязнения.
Бактерии рода Enterococcus также являются постоянными обитателями кишечника человека. Энтерококки отмирают во внешней среде значительно раньше, чем Е. coli, поэтому они всегда свидетельствуют о свежем фекальном загрязнении.
Большое санитарное значение имеют представители рода Proteus. Р. vilgaris свидетельствует о загрязнении объекта органическими веществами (его чаше обнаруживают в гниющих остатках), а Р. mirabilis — о фекальном загрязнении (его чаще обнаруживают в фекалиях). Обнаружение протеев в пищевых продуктах свидетельствует о гнилостном распаде.
Таблица 46.1
Санитарно-показательные микроорганизмы окружающей среды и пищевых продуктов (по Л. Б. Борисову и др., 1993)
Объект Характер загрязнения Санитарно-показательные микроорганизмы
Вода Фекальное Бактерии группы кишечных палочек Е. coli, Citrobacterfreundii, Enterobacter aerogenes, Streptococcus faecalis
Почва Фекальное Те же бактерии и клостридии (Clostridium perfringens, Cl. sporogenes и др.)
Промышленно-бытовое (разлагающиеся отбросы) Термофильные бактерии {Lactobacillus lactis, Streptococcus thermophilus и др.), Proteus vulgaris
Пищевые продукты Фекальное Бактерии группы кишечных палочек, Streptococcus faecalis, Р. vulgaris
Орально-капельное Staphylococcus aureus
Воздух Орально-капельное S. aureus, S. pyogenes
Вода, почва, воздух Промышленное Производственные штаммы микроорганизмов
Обнаружение Clostridium perfringens свидетельствует о некогда имевшем место фекальном загрязнении (эти бактерии образуют споры, что позволяет им длительно сохраняться в окружающей среде). В отечественной практике количественный учет клостридий предусмотрен при исследованиях почвы, лечебных грязей, воды открытых водоемов. Определение этого микроорганизма проводят и в некоторых пищевых продуктах, но уже как возможного возбудителя пищевых отравлений.
Термофильные микроорганизмы, имеющие санитарное значение, представлены преимущественно спорообразующими бактериями, способными размножаться при 50 — 60 °C. Их выделяют из объектов внешней среды (загрязненных компостом или навозом), реже из кишечника человека и животных. В отечественной практике этот показатель применяют при исследовании почвы. Резкое увеличение количества этих бактерий в почве может свидетельствовать о загрязнении разлагающимися отбросами. Анализ на содержание термофилов проводят также при исследовании консервов для индикации эффективности автоклавирования.
Обнаружение в объектах окружающей среды бактерий рода Salmonella всегда свидетельствует о фекальном загрязнении. Эти микроорганизмы являются наиболее распространенными возбудителями острых кишечных заболеваний и поэтому могут быть индикаторами возможного присутствия других возбудителей инфекций с аналогичными патогенезом и эпидемиологией.
Staphylococcus aureus — факультативный обитатель носоглотки, зева и кожных покровов человека. Присутствие этого микроорганизма в воздухе помещений или на находящихся там предметах указывает на орально-капельное загрязнение. Наличие Staphylococcus aureus в пищевых продуктах может привести к тяжелым пищевым интоксикациям.
Гемолитические стрептококки, являясь транзитными обитателями носоглотки и зева, выделяются с капельками слизи орально-капельным путем. Сроки их выживания во внешней среде аналогичны таковым для большинства других возбудителей воздушнокапельных инфекций. Следовательно, обнаружение этих бактерий в воздухе помещений свидетельствует о возможном загрязнении патогенами.
Санитарно-показательное значение имеют также бактериофаги. Они, как правило, сопутствуют тем бактериям, к которым адаптированы, однако бактериофаги могут длительно сохраняться и переживать соответствующих бактерий. Это, тем не менее, не исключает возможности использовать бактериофаги кишечных бактерий для гигиенической оценки внешней среды.
На результаты санитарно-микробиологических анализов могут повлиять различные факторы, поэтому при проведении таких исследований необходимо соблюдать следующие принципы: правильный отбор проб; серийность проводимых анализов; применение только стандартных и унифицированных методов исследования; использование комплекса тестов (прямых и косвенных); проведение оценки объектов по совокупности полученных результатов (с учетом результатов, полученных с помощью органолептических, химических, физических и других методов).
Приводимые ниже задачи (1 — 6) непосредственно знакомят студентов с методами санитарной микробиологии.
Цель работы, получить общее представление о методах, используемых при проведении санитарно-микробиологических анализов воды, воздуха, почвы, предметов обихода и пищевых продуктов.
ЗАДАЧА 1. САНИТАРНО-МИКРОБИОЛОГИЧЕСКОЕ ИССЛЕДОВАНИЕ ВОДЫ
Вода водоемов содержит большое количество микроорганизмов, относящихся к автохтонной микрофлоре, а также попадающих в нее с осадками,
сточными водами и другими загрязнениями — аллохтонных микроорганизмов. Количество и видовой состав микрофлоры воды в значительной степени зависят от условий среды (наличие питательных веществ, температура, аэрация, окислительно-восстановительные условия, pH и др., а также вещества, поступающие в водоем). Основным путем микробного загрязнения водоемов является попадание неочищенных бытовых и промышленных отходов и сточных вод. Микрофлора сточных вод содержит обитателей кишечника человека и животных, включая представителей нормальной и условно-патогенной флоры. Патогенные бактерии слабо приспособлены к существованию в воде, где на них оказывают неблагоприятное воздействие солнечный свет и различные другие факторы, включая конкурентную водную микрофлору, тем не менее многие из них могут сохраняться в воде достаточно долго.
Микробиологические методы исследования воды сводятся к определению общего количества микроорганизмов в 1 мл воды и выявлению патогенных микроорганизмов (сальмонелл, холерных вибрионов, лептоспир, шигелл и энтеровирусов). Последний анализ проводится по эпидемическим показаниям. Кроме того, поскольку прямое выделение патогенных бактерий из воды требует специальных исследований, существуют косвенные методы, позволяющие дать количественную оценку степени фекального загрязнения воды (выявление БГКП, Е. coll, энтерококков, стафилококков).
Исследованию подлежит вода: централизованного водоснабжения, колодцев, открытых водоемов, плавательных бассейнов, сточные жидкости.
Цель работы, провести оценку санитарно-бактериологического состояния воды, включающую 1) определение микробного числа водопроводной воды и воды открытых водоемов, 2) определение коли-титра и коли-индекса воды.
Ход выполнения задачи
Подготовка сред и материалов. Подготовить к стерилизации (в расчете на одного студента): чашки Петри — 10 шт.; пробирки — 10; пипетки на 1 — 2 мл — 10; на 5—10 мл — 3; бутылка или флакон (объем 500 мл) — 1—2 шт.
Приготовить (состав и приготовление сред — см. в прил. 4): 0,85%-й раствор NaCl стерильный (или вода водопроводная стерильная) — 150 — 200 мл; МПА стерильный — 150 мл; полужидкая среда с глюкозой — 30 мл (разлить в 3 пробирки по 7—10 мл); среда Эндо — 50 мл (сразу после приготовления разлить в 2—3 стерильных чашки Петри, дать застыть, завернуть чашки в бумагу и оставить в холодильнике); среда КОДА или среда Кесслера — 33 мл концентрированной (х10) среды (разлить по 10 мл в 3 колбы на 150 — 250 мл и по 1 мл в 3 обычные пробирки), в каждую колбу и пробирку поместить по поплавку или комочку ваты; 60 мл среды КОДА или Кесслера нормальной концентрации (разлить по 10 мл в 6 обычных пробирок), в каждую пробирку поместить по поплавку или комочку ваты.
Отбор проб воды. Воду для санитарно-бактериологического исследования отбирают в количестве 500 мл в бутылки или флаконы, предварительно про-стерилизованные в бумажных пакетах, с ватно-марлевой пробкой. К горлышку бутылки привязывают пакетик с завернутой в него запасной пробкой.
Пробы воды из открытых водоемов, бассейнов, баков и т.п. отбирают с глубины 10— 15 см, а при малой глубине — не менее 10—15 см от дна. Пробы
Рис. 46.1. Батометр (прибор для взятия проб воды)
из проруби берут на глубине 10—15 см от нижней поверхности льда. Пробы воды из желаемого водного горизонта открытых водоемов (колодцев, бассейнов, озер, рек и пр.) отбирают с помощью батометров (рис. 46.1). После наполнения бутылки водой с заданной глубины ее извлекают из батометра и закрывают стерильной пробкой, сверху надевают бумажный колпачок.
Из водопроводных кранов воду отбирают следующим образом. Кран протирают тампоном, смоченным в спирте, и обжигают, после чего 10—15 мин спускают воду. Затем отбирают приблизительно 400 мл воды. Заполненный флакон плотно закрывают стерильной резиновой или корковой пробкой, а сверху надевают бумажный колпачок. При проведении анализа хлорированной воды во флакон для отбора проб (емкость 500 мл) перед стерилизацией вносят дехлоратор — 10 мг гипосульфита натрия.
Бактериологическое исследование отобранных проб воды должно производиться не позднее 2 ч с момента отбора. В случае невозможности соблюдения этих сроков допускается проведение анализа воды не позднее чем через 6 ч при хранении пробы при температуре от 1 до 6 °C.
Определение микробного числа воды. С флаконов, содержащих пробу воды, снимают бумажные колпачки, вынимают пробки, горлышки фламбируют. Воду тща-
тельно перемешивают осторожным продуванием воздуха через стерильную пи-
петку.
Из каждой пробы делают посев не менее двух различных объемов, отобранных с таким расчетом, чтобы число выросших на чашках колоний колебалось от 30 до 300 (табл. 46.2).
Для посева 0,1 мл и меньших объемов исследуемую воду разводят стерильной водой. Готовят последовательно десятикратные разведения, используя для каждого разведения стерильную пипетку. По 1 мл каждого разведения вносят в две стерильные чашки Петри, после чего их заливают 10—15 мл расплавленного и остуженного до 45 — 50 °C МПА, который тщательно, круговыми движениями перемешивают. Среде дают застыть на строго горизонтальной поверх-
Таблица 46.2
Рекомендуемые объемы воды для определения микробного числа
Тип исследуемой воды Объем воды, мл
Водопроводная 1
Чистая 1 и 0,1
Более загрязненная 0,01 и 0,001
Сильно загрязненная 0,0001 и 0,00001
ности. Посевы выращивают в течение суток при 37 °C. Воду из открытых водоемов засевают параллельно на две серии чашек, одну из которых инкубируют при 37 °C в течение суток, а другую — 2 сут при 20 °C. Затем подсчитывают количество выросших на поверхности и в глубине среды колоний (видимых невооруженным глазом и при увеличении в 2 — 5 раз) и вычисляют микробное число воды — количество микроорганизмов в 1 мл. Питьевая вода считается хорошей, если общее количество бактерий в 1 мл не более 100; сомнительной — 100—150; загрязненной — 500 и более.
Определение коли-титра и коли-индекса воды. Коли-титр — минимальное количество воды (мл), в котором обнаруживаются БГКП. Коли-индекс — количество БГКП, содержащихся в 1 л исследуемой воды (по международному и европейскому стандарту — в 100 мл). Эти показатели определяют двухэтапным бродильным (титрационным) методом или методом мембранных фильтров.
Бродильный метод — основан на посеве определенных объемов анализируемой воды и подращивании при 37 °C в средах накопления с последующим высевом бактерий на плотную среду Эндо, дифференцировании выросших бактерий и определении наибольшего вероятного числа бактерий группы кишечных палочек (БГКП) в 1 л воды по таблицам.
При исследовании воды открытых водоемов, а также воды на этапах очистки и обеззараживания засевают 100,0; 10,0; 1,0 и 0,1 мл. Для исследования водопроводной воды засевают три объема по 100,0 мл, три объема по 10,0 мл и три объема по 1,0 мл.
Указанные объемы воды помещают во флаконы или пробирки со средой Кесслера или КОДА, снабженные поплавками или комочками ваты, погруженными на дно сосуда. Посев 100,0 и 10,0 мл воды производят во флаконы и пробирки соответственно с 10,0 и 1,0 мл концентрированной среды; посев 1,0 и 0,1 мл воды в пробирки с 10,0 мл среды нормальной концентрации. Посевы инкубируют 24 ч при 37 °C. Отсутствие помутнения и образования кислоты и газа во флаконах и пробирках позволяет получить отрицательный результат и закончить исследование.
Из каждого флакона или пробирки, в которых замечены помутнение, кислота, газ, делают посев петлей на поверхность среды Эндо, разделенной на 3—4 сектора. Посевной материал следует брать с таким расчетом, чтобы получить изолированные колонии. Чашки инкубируют при 37 °C в течение 16— 18 ч. При отсутствии роста, а также при наличии колоний, не характерных для бактерий группы кишечной палочки (пленчатых, губчатых, с неровными краями и поверхностью, плесневых и др.), считают результат отрицательным. Из выросших на среде Эндо красных, розовых, бледно-розовых колоний с металлическим блеском или без него (лактозоположительные колонии) делают мазки, окрашивают по Граму и ставят оксидазный тест, позволяющий дифференцировать БГКП от грамотрицательных бактерий семейства Pseudomonadaceae и других оксидазоположительных бактерий, обитающих в воде. С этой целью 2— 3 изолированные колонии снимают с поверхности среды стеклянной палочкой, наносят штрихом на фильтрованную бумагу, смоченную раствором диметил-и-фенилендиамина. При отрицательном оксидазном тесте цвет бумаги не изменяется, при положительном она окрашивается в синий цвет в течение 1 мин.
Наличие грамотрицательных палочек в мазках и отрицательный оксидазный тест позволяют немедленно дать положительный ответ о наличии БГКП
при анализе воды на этапах очистки и воды открытых водоемов. При исследовании водопроводной воды грамотрицательные палочки, не образующие оксидазу, вновь исследуют в бродильном тесте, внося петлей посевной материал в полужидкий питательный агар с 0,5 % глюкозы, и инкубируют при 37 °C в течение суток. При наличии кислоты и газа получают положительный результат.
Результат анализа выражают в виде коли-индекса, величину которого определяют по табл. 46.3 и 46.4. Зная коли-индекс, рассчитывают коли-титр по формуле
1000 коли-титр =------------.
коли-индекс
Таблица 46.3
Определение коли-индекса при исследовании воды
Объем исследуемой воды, мл Коли-индекс Коли-титр
100 10 1,0 0,1
— __ __ Менее 9 Более 111
— — + __ 9 Ill
__ + — — 10 105
+ — — — 23 43
+ — + 94 10
4- + — — 230 4
+ + —- -1- 960 1
+ + + — 2380 0,4
+ + + -1- Более 2380 Менее 0,4
Таблица 46.4
Определение коли-индекса при исследовании питьевой воды
Количество положительных результатов анализов воды Индекс
из трех флаконов по 100 мл из трех пробирок по 10 мл из трех пробирок по 1 мл
0 0 0 Менее 3
0 0 1 3
0 1 0 3
1 0 0 4
1 0 1 7
1 1 0 7
1 1 1 11
1 2 0 11
2 0 0 9
Окончание табл. 46.4
Количество положительных результатов анализов воды Индекс
из трех флаконов по 100 мл из трех пробирок по 10 мл из трех пробирок по 1 мл
2 0 1 14
2 1 0 15
2 1 1 20
2 2 0 21
2 2 1 28
3 0 0 23
3 0 1 39
3 0 2 64
3 1 0 43
3 1 1 75
3 1 2 120
3 2 0 93
3 2 1 150
3 2 2 210
3 3 0 240
3 3 1 460
3 3 2 1100
3 3 3 Более 1100
Метод мембранных фильтров. Сущность метода заключается в концентрировании бактерий из определенного объема анализируемой воды на мембранный фильтр, выращивании их при 37 °C на среде Эндо, дифференцировании выросших колоний и подсчете бактерий группы кишечных палочек в 1 л воды.
Воду из водопроводной сети и воду из артезианских скважин фильтруют в объеме 333 мл . Чистую воду открытого водоема фильтруют в объеме 100; 10; 1 и 0,1 мл, более загрязненную — 0,1; 0,01 и 0,001 мл.
Для фильтрования вода используют мембранные фильтры №3 (диаметр пор 0,7 мкм), помещают их в стеклянный стакан с подогретой до 80 °C дистиллированной водой и ставят на слабый огонь до закипания. Кипячение фильтров производят трижды по 10 мин. После первого и второго кипячения воду сливают. После последнего кипячения воду не сливают и фильтры оставляют в ней до употребления.
Перед работой фильтровальный аппарат Зейтца протирают спиртом. Фильтр помещают в воронку фильтровального аппарата, который присоединен к вакуум-насосу. Начинают исследования с больших разведений. При посевах объемов 1 мл и меньше их разводят в 10 мл стерильной водопроводной воды, а затем фильтруют. После окончания процесса фильтры укладывают на поверхность среды Эндо в чашках Петри фильтрующей поверхностью вверх. На одну чашку можно помещать 3 — 4 фильтра. Чашки с фильтрами выдерживают 18 —24 ч при 37 °C. Для подсчета выбирают фильтры с числом колоний не менее 10 и не более 50. Учету подлежат все красные или темно-красные
колонии с металлическим блеском или без него. В дальнейшем схема подтверждения принадлежности выделенных бактерий к группе кишечных палочек аналогична описанной для бродильного метода.
Материалы, посуда, реактивы, оборудование
Стерильные чашки Петри; стерильные пробирки; стерильные пипетки; стерильные бутылки или флаконы емкостью 500 мл; стаканчики стеклянные термостойкие; колбы или флаконы емкостью 150 — 250 мл; палочки стеклянные; пинцеты; петли бактериологические; лупа; барометр; фильтровальная установка; фильтры мембранные № 3; бумага фильтровальная; вата; микроскопы; стекла предметные и покровные; термостаты на 30 и 37 °C. Стерильный 0,85%-й раствор NaCl (или вода водопроводная стерильная); среда Эндо; среда Кесслера или КОДА; МПА; полужидкая среда с глюкозой, свежеприготовленный раствор диметил-//-фенилендиамина; свежеприготовленный 3%-й раствор КОН; гипосульфит натрия; реактивы для окраски по Граму; фуксин; 70%-й этанол.
ЗАДАЧА 2. САНИТАРНО-МИКРОБИОЛОГИЧЕСКОЕ ИССЛЕДОВАНИЕ ПОЧВЫ
Попавшие в почву представители нормальной микрофлоры человека и животных, а также патогенные микроорганизмы обычно длительно не выживают. Однако многие представители нормальной микрофлоры человека способны включаться в состав биоценоза почвы, а отдельные виды остаются постоянными обитателями. На сроки выживания патогенных бактерий в почве влияют состав и тип почвы, температура, влажность, атмосферные осадки, степень загрязненности, а также ее характер (органическое, микробное или химическое загрязнение). Патогенные микроорганизмы, обнаруживаемые в почве, разделяют на три группы (А. 3. Байчурина и др., 1998):
1. Патогенные микроорганизмы, постоянно обитающие в почве (например, Clostridium botulinum, возбудители подкожных микозов, виды Actinomyces, некоторые возбудители микотоксикозов).
2. Спорообразующие патогенные микроорганизмы {Bacillus anthracis, Clostridium tetani, виды Clostridium, вызывающие анаэробные инфекции). Бактерии попадают в почву с выделениями человека и животных, а также с трупами погибших животных. При благоприятных условиях они могут размножаться и сохраняться в виде спор длительное время.
3. Патогенные микроорганизмы, попадающие в почву с выделениями человека и животных и сохраняющиеся сравнительно недолго (в течение нескольких недель или месяцев). Это различные неспоровые бактерии (виды Salmonella, Shigella, Vibrio, Brucella, Francisella, Mycobacterium, Leptospira, Pseudomonas, энтеровирусы, вирус ящура).
Оценку санитарного состояния почв проводят с учетом комплекса показателей: подсчитывают общее количество сапрофитных микроорганизмов и определяют наличие санитарно-показательных микроорганизмов (БГКП, Clostridium perfringens и др.). Высокая численность сапрофитной микрофлоры свидетельствует об органическом загрязнении, при микробной контаминации преобладают санитарно-показательные микроорганизмы. При необходимости также исследуют состав нитрифицирующих и аммонифицирующих бактерий, актиномицетов, грибов, целлюлозолитических микроорганизмов и др.
Цель работы, провести санитарно-бактериологическую оценку состояния почвы по результатам определения общего микробного числа, коли-титра. перфрингенс-титра и количества термофильных бактерий.
Ход выполнения задачи
Подготовка сред и материалов. Подготовить к стерилизации (в расчете на одного студента): чашки Петри — 12—15 шт.; пробирки — 15 шт.; пипетки на 1 — 2 мл — 10 шт., на 5 — 10 мл — 3 шт.; колба на 300—500 мл — 1 шт.; банка или пакет (вместимостью -1000 мл) — 1 шт. Приготовить (состав и приготовление сред см. в приложении 4): вода водопроводная стерильная — 350 — 400 мл (из них 270 мл отдельно в колбе на 500 мл); МПА стерильный — 150— 200 мл; стерильное обезжиренное молоко — 30 мл (разлить в пробирки по 5 мл) или среда Вильсона—Блера 60 мл; полужидкая среда с глюкозой — 30 мл (разлить в 3 пробирки по 7—10 мл); среда КОДА или среда Кесслера (нормальной концентрации) — 60 мл (разлить по 10 мл в 6 обычных пробирок), в каждую пробирку поместить по поплавку или комочку ваты; среда Эндо — 20—60 мл (сразу после приготовления разлить в стерильные чашки Петри, дать застыть, завернуть в бумагу и оставить в холодильнике).
Отбор проб и определение микробного числа. На обследуемой территории до 1000 м2 выделяют два участка по 25 м2. Один должен быть расположен вблизи источника загрязнения (выгребные ямы, мусорные ящики и пр.), а другой — в отдалении от них. На каждом участке намечают 5 точек — четыре по углам и одну в центре участка. Таким образом, отбирают 10 проб (из разных мест исследуемой территории). Почву берут стерильным ножом на глубине 10—15 см, образец массой 200—300 г помещают в стерильную банку или пакет и хорошо перемешивают. Следовательно, смешанный образец с каждого из двух выбранных участков должен иметь массу не менее 1 кг. Отобранные пробы анализируют в тот же день. Допускается хранение образцов почвы в лаборатории не более 12 —18 ч при температуре 1 — 5 °C.
Из проб готовят навеску (30 г), которую вносят в колбу со стерильной водой (270 мл), и тщательно встряхивают в течение 10 мин. Из полученной суспензии готовят 5 разведений: от 10“3 до 10-7. По 1 мл из двух последних разведений вносят в две стерильные чашки Петри, после чего их заливают 10—15 мл расплавленного и остуженного до 45 —50 °C МПА, который тщательно, круговыми движениями перемешивают. Среде дают застыть на строго горизонтальной поверхности. Посевы выращивают в течение суток при 37 °C. Затем подсчитывают число выросших колоний и определяют микробное число.
Определение коли-титра, перфрингенс-титра и количества термофильных бактерий. Для определения коли-титра различные разведения почвенной суспензии засевают по 1 мл в пробирки с 5 мл среды Кесслера или КОДА и инкубируют при 37 °C в течение 24—48 ч. В дальнейшем анализ проводят по схеме, применяемой для определения коли-титра воды (см. 46.1). Для посевов пользуются различными разведениями (чистую почву засевают от 1 г до разведения Ю-3, загрязненную — от 10~3 до I О6). При анализе загрязненных почв можно провести прямой поверхностный посев почвенной суспензии в количестве 0,1 — 0,5 мл на среду Эндо (используют разведения до 10-6).
Для определения перфрингенс-титра также пользуются разведениями почвенной взвеси (чистую почву засевают в разведении 10 ' —10 3, загрязненную — от 10“4 до 10“5). По 1 мл из выбранных разведений засевают в пробирки с 5 мл стерильного обезжиренного молока или с железосульфитной средой Вильсона— Блера, приготовленной ex tempore. Посевы инкубируют при 43 °C в течение
Таблица 46.5
Оценка санитарного состояния почвы по основным микробиологическим показателям
Характеристика почвы Коли-титр Перфрингенс-титр Количество термофильных бактерий в 1 г почвы
Чистая 1,0 и выше 0,01 и выше 102—103
Загрязненная 0,9-0,01 0,009-0,0001 От 103до 105
Сильно загрязненная 0,009 и ниже 0,00009 и ниже От 105 до 4 106
24—48 ч, после чего учитывают результаты по характерному свертыванию молока (образование губчатого сгустка в верхней части пробирки и просветление сыворотки) или по образованию колоний Clostridium perfringens в агаровом столбике среды Вильсона—Блера (колонии черного цвета различной интенсивности окраски, имеющие форму двояковыпуклой линзы, комочка ваты или «самолетика»). Из колоний делают мазки, окрашивают по Граму, микроскопируют и вычисляют перфрингенс-титр. Клетки С. perfringens — подвижные (реже неподвижные) короткие толстые палочки с закругленными концами. Образуют овальные или круглые эндоспоры, расположенные центрально и придающие клеткам веретенообразную форму (споры также могут располагаться терминально и субтерминально). В мазках расположены одиночно, попарно, в виде цепочек или штакетообразных (параллельно друг к другу) скоплений, грамположительны, каталазоотрицательны. Предельное разведение почвенной суспензии, которое дает на молочной среде размножение С. perfringens, означает титр этого микроорганизма в почве.
Для определения количества термофильных бактерий разведения полученной суспензии (10-1, 10“2 и ПГ3) по 1 мл вносят в чашки Петри, заливают из расплавленным и остуженным МПА, тщательно перемешивают. Посевы инкубируют при 60 °C в течение суток, а затем подсчитывают количество выросших колоний и пересчитывают на 1 г почвы.
Санитарно-микробиологическую оценку почвы проводят по комплексу показателей, из которых наиболее важным является степень фекального загрязнения (табл. 46.5).
Материалы, оборудование, посуда, реактивы
Стерильные чашки Петри; стерильные пробирки; стерильные пипетки; стерильные банки или пакеты; нож, совок, колбы емкостью 500 мл; палочки стеклянные; пинцеты; петли бактериологические; вата; весы; микроскопы; стекла предметные и покровные; термостаты на 30 и 37 °C.
Вода водопроводная стерильная; среды: Эндо, Кесслера или КОДА, МПА, полужидкая с глюкозой, стерильное обезжиренное молоко или среда Вильсона — Блера; свежеприготовленный раствор диметил-и-фенилендиамина, свежеприготовленный 3%-й раствор КОН; реактивы для окраски по Граму; фуксин; 70%-й этанол.
ЗАДАЧА 3. АНАЛИЗ МИКРОФЛОРЫ ВОЗДУХА
Атмосферный воздух и воздух закрытых помещений значительно различаются по количественному и качественному составу микроорганизмов. Бактериальная обсеме-
ненность жилых помещений выше плотности бактерий в атмосферном воздухе, в том числе и патогенными видами, попадающими в воздух от больных людей и животных (капельным путем в составе аэрозоля, образующегося при разговоре, кашле, чихании, со слущивающимся эпителием кожных и шерстных покровов, с пылью загрязненного постельного белья и зараженной почвы).
Микрофлора воздуха формируется в основном за счет почвенных микроорганизмов, отличающихся большой устойчивостью к неблагоприятным факторам внешней среды, таким, как высушивание, ультрафиолетовые лучи солнечного света, колебания температуры и пр. Обычно из воздуха выделяют представителей рода Micrococcus, Bacillus subtilis, В. cereus var. mycoides, В. mesentericus, различные виды Actinomyces, Penicillium, Aspergillus, Mucor и др.
При оценке санитарного состояния закрытых помещений в зависимости от задач исследования определяют общее микробное число, наличие санитарнопоказательных микроорганизмов (стафилококков, а- и р-гемолитических стрептококков, являющихся показателями контаминации микрофлорой носоглотки человека). Кроме того, например, при исследовании воздуха медицинских стационаров (хирургические клиники, родильные дома) основное внимание направлено на выявление патогенных стафилококков, а также синегнойных палочек и других грамотрицательных условнопатогенных бактерий — возбудителей внутрибольничных инфекций. На предприятиях микробиологической промышленности выявляют наличие и содержание микроорганизмов-продуцентов (Candida на гидролизно-дрожжевых заводах и заводах по производству БВК, Aspergillus и споровые бактерии на ферментных заводах, Bacillus thuringiensis и сальмонеллы — при производстве бактериальных средств зашиты растений и борьбы с грызунами и т.д.).
Определение тех или иных патогенных или условно-патогенных микроорганизмов из воздуха проводят на специальных дифференциально-диагностических средах.
Для исследования микрофлоры воздуха используют различные методы.
Седиментационный метод (метод Коха, 1881) — основан на оседании бактериальных частиц и капель под действием силы тяжести на поверхности питательной среды открытой чашки Петри. Обычно производят перерасчет по Омелянскому: на поверхность 100 см2 плотной среды оседает за 5 мин такое количество бактерий, которое содержится в 10 л воздуха. Однако позднее было определено, что эти показатели занижены в 3 раза. Метод не точен и абсолютно не пригоден для атмосферного воздуха, где имеют место большие колебания в скорости его движения. Он может быть использован в тех случаях, когда отсутствуют более совершенные приборы или нет электроэнергии.
Фильтрационный метод (воздух продувается через жидкость) — основан на улавливании бактерий в жидкости, которая затем может быть использована для посева на различные среды — прибор Дьяконова (1925) (рис. 46.2) и др.
Методы, основанные на осаждении микробных аэрозолей паром или распыленной жидкостью, — прибор Речменского (рис. 46.3) и пр.
Методы, основанные на принципе ударно-прибивного действия воздушной струи с использованием специальных приборов, например прибора Кротова (1951) (рис. 46.4). Струя воздуха приходит через узкую клиновидную щель и с большой скоростью ударяется о влажную поверхность питательной среды. В результате удара находящиеся в воздухе аэрозоли, в том числе содержащие бактерии пылевые частицы и капли, прибиваются к поверхности МПА или элективных сред. Производительность такого прибора составляет от 20 до 40 л/мин. Подобные методы наиболее надежны и точны.
Рис. 46.2. Прибор Дьяконова в модификации Шафира
2 3 7 81
Рис. 46.3. Прибор Речменского:
I — стеклянный цилиндр; 2 — входное отверстие; 3, 4 — капилляры; 5 — резервуар для улавливающей жидкости; 6 — резиновая трубка; 7 — отверстие для забора улавливающей жидкости; 8 — стеклянная лопатка
Рис. 46.4. Аппарат Кротова:
1 — цилиндрический корпус; 2 — основание корпуса; 3 — электромотор; 4 — центробежный вентилятор; 5 — восьмилопастная крыльчатка; 6 — диск; 7 —- пружины; 8 — чашка Петри; 9 — крышка прибора; 10 — накидные замки; 11 — диски из плексигласа; 12 — клиновидная щель;
13 — разрезное кольцо; 14 — штуцер с диафрагмой; 15 — выводная трубка
Цель работы', провести санитарно-бактериологическую оценку состояния воздуха по результатам определения микробного числа с использованием седиментационного метода.
Ход выполнения задачи
Подготовка сред и материалов. Подготовить к стерилизации (в расчете на одного студента): чашки Петри — 2 шт. Приготовить МПА стерильный — 40— 50 мл.
Проведение анализа. Две чашки Петри со стерильным МПА оставляют открытыми в течение 60 мин (чашки располагают на высоте, соответствующей уровню дыхания сидящего или стоящего человека), после чего посевы инкубируют при 37 °C в течение 1 сут (в этих условиях развивается бактериальная флора). Затем чашки держат при 25 °C, что позволяет развиться бактериям, требующим для своего роста более низких температур, и плесневым грибам. Подсчитывают суммарное число колоний, выросших на обеих чашках: при наличии менее 250 колоний воздух считается чистым; 250—500 колоний — загрязненным в средней степени, при количестве колоний более 500 — загрязненным.
Материалы и оборудование, посуда, реактивы
Стерильные чашки Петри — 2 шт.; термостат на 37 °C; МПА стерильный.
ЗАДАЧА 4. САНИТАРНО-БАКТЕРИОЛОГИЧЕСКОЕ ИССЛЕДОВАНИЕ ПИЩЕВЫХ ПРОДУКТОВ
Многие пищевые продукты являются благоприятной средой для размножения микроорганизмов. Кроме того, технологические процессы производства ряда продуктов питания (молочнокислые продукты, квас, пиво, хлеб и пр.) предусматривают внесение и развитие специфической микрофлоры, которая полезна, или по крайней мере не представляет опасности для человека.
Обычно пищевые продукты подвергаются целому комплексу исследований, которые направлены на установление соответствия того или иного продукта существующему для него стандарту, определение доброкачественности, отсутствия вредных и ядовитых веществ, степени бактериальной обсемененности и наличия условно-патогенных и патогенных микроорганизмов (табл. 46.6 — 46.8).
Установленные медико-биологическими требованиями и санитарными нормами качества продовольственного сырья и пищевых продуктов критерии безопасности включают определение следующих групп микроорганизмов.
• Санитарно-показательные микроорганизмы. Определяют мезофильные аэробные и факультативно анаэробные микроорганизмы (их число выражают количеством колониеобразующих единиц (КОЕ) в 1 г или 1 см3 продукта) и «бактерии группы кишечной палочки» (БГКП).
• Потенциально-патогенные микроорганизмы. Определяют коагулазоположительные Staphylococcus aureus, Bacillus cereus, сульфитредуцирующие клостридии, бактерии рода Proteus, Pseudomonas aeruginosa, Vibrio parahaemolyticus.
Разведения для определения общего числа бактерий при посеве молока и некоторых молочных продуктов
Продукт Засеваемые разведения
Молоко и сливки сырые ю-4- -ПН
Молоко и сливки пастеризованные 10'- - 1О-3
Мороженое, коктейли молочные 10-'- -10-3
Молоко сгущенное с сахаром, какао и кофе со сгущенным молоком 10-'- -10-3
Плавленый сыр 10-'- -10-2
Таблица 46.7
Санитарно-бактериологические нормативы для молока и некоторых молочных продуктов (СанПиН 2.3.2.560-96)
Продукт Микробное число — допустимое коли-чество микроорганизмов (КОЕ в 1 мл или 1 г), не более Масса продукта (г), в которой не допускаются Другие показатели
БГКП (коли-формы) патогенные, в том числе сальмонеллы 5. aureus
Молоко сырое (высший сорт) 3 • 105 — 25 — —
Молоко пастеризованное в бутылках и пакетах: гр. А гр. Б 5-Ю4 1-Ю5 1,0 0,01 25 25 1,0 од —
Молоко пастеризованное: во флягах в цистернах 2-105 3 • 105 0,01 0,01 25 25 — —
Сливки пастеризованные в бутылках и пакетах 1-Ю5 0,1 25 1,0 —
Кисломолоч н ые напитки (кефир, простокваша и пр.) (наличие только микрофлоры закваски) 0,01 25 1,0 *—
Ряженка — 1,0 25 1,0 —
Сметана Сметана с термической обработкой — 0,001 0,01 25 25 1,0 1,0 —
Творог и творожные изделия, вырабатываемые без термообработки — 0,001 25 0,1 —
Продукт Микробное число — допустимое количе-ство микроорганизмов (КОЕ в 1 мл или 1 г), не более Масса продукта (г), в которой не допускаются Другие показатели
БГКП (коли-формы) патогенные, в том числе сальмонеллы 5. aureus
Десерты сливочные — 0,01 25 0,1 —
Молоко сгущенное с сахаром 2,5-104 1,0 25 — —
Какао и кофе со сгущ. молоком и сахаром 3,5-104 1,0 25 — —
Сыры сычужные твердые и мягкие — 0,001 25 < 1000 КОЕ/г —
Сыр Российский — 0,001 25 < 500 КОЕ/г —
Сыры плавленые: без наполнителей с наполнителями (овощи, грибы и т.д.) 5-Ю3 1 • 104 0,1 0,1 25 25 — Плесени, дрожжи < 50 < 50 < 100 < 100 (КОЕ/г)
Мороженое 1-Ю5 0,01 25 1,0 —
Таблица 46.8
Некоторые санитарно-бактериологические нормативы для мяса, колбасных изделий и мясных продуктов (СанПиН 2.3.2.560-96)
Продукт Микробное число — допустимое количе-ство микроорганизмов (КОЕ в 1 мл или 1 г), не более Масса продукта (г), в которой не допускаются Другие показатели
БГКП (коли-формы) патогенные, в том числе сальмонеллы 5. aureus
Мясо замороженное (все виды убойных животных) Мясо в отрубах Телятина, свинина куском 1- 104 5-105 0,01 0,01 25 25 —
Фарш говяжий 5-106 0,0001 25 —
Колбасы с/к и с/к изделия из мяса убойных животных 0,1 25 1 Сульфит-восстанав-ливающие клостридии в 0,01 г не доп.
Продукт Микробное число — допустимое количе-ство микроорганизмов (КОЕ в 1 мл или 1 г), не более Масса продукта (г), в которой не допускаются Другие показатели
БГКП (коли-формы) патогенные, в том числе сальмонеллы 5. aureus
Колбасы п/к 1,0 25 1,0 Сульфит-восстанав-ливающие клостридии в 0,01 г не доп.
Колбасные изделия вареные (колбасы, сардельки, сосиски): высший и первый сорта второй сорт 1 • 103 2,5-103 1,0 1,0 25 25 1,0 1,о Тоже
Птица охладженная и замороженная 1 105 — 25 — —
• Патогенные микроорганизмы, в том числе сальмонеллы, Listeria monocytogenes.
• Показатели микробиологической стабильности продукта — включают дрожжи, плесени и молочнокислые бактерии.
Микробиологические исследования пищевых продуктов проводят в соответствии с ГОСТами, санитарными правилами и нормами, методическими указаниями, инструкциями и другими нормативными документами.
Цель работы, провести санитарно-бактериологическую оценку состояния различных продуктов питания.
Ход выполнения задачи
Подготовка сред и материалов. Подготовить к стерилизации (из расчета на одного студента): чашка Петри — 10—15 шт.; пробирки — 15; пипетки на 1 — 2 мл — 20, на 5 —10 мл — 5 шт. Приготовить (состав и приготовление сред см. в приложении 4): 0,85%-й раствор NaCl стерильный (или вода водопроводная стерильная) — 200 мл; МПА стерильный — 100 мл; желточно-солевой (или молочно-солевой) агар — 40—60 мл; среда Эндо — 60 мл (сразу после приготовления разлить в стерильные чашки Петри, дать застыть, завернуть в бумагу и оставить в холодильнике); среда Сабуро — 60 мл; 6,5%-й солевой бульон — 20 мл; полужидкая среда с глюкозой — 10 мл; среда Кесслера или КОДА — 15 — 20 мл; среда Козера — 10 мл; среда Клиглера — 10 мл; селенитовый бульон — 125 мл; висмут-сульфит агар — 20 мл; среда Вильсона—Блера — 20 мл.
Подготовка проб пищевых продуктов к исследованию. Перед исследованием пробы вначале подготавливают навеску, которая должна охарактеризовать всю исследуемую пробу. Навески продукта берут стерильно из разных мест пробы — с поверхности и из глубины. Отбирают навеску 10 г, взвешивая ее на весах,
растирают в стерильной фарфоровой ступке, постепенно добавляя 90 мл изотонического раствора хлорида натрия, и оставляют при комнатной температуре на несколько минут. Затем стерильной пипеткой с широким носиком отбирают взвесь для посевов и разведений. Принимается, что 1 мл приготовленной взвеси содержит 0,1 г исходного продукта.
Продукты жидкой консистенции засевают и разводят для посевов без предварительной подготовки. Продукты, имеющие кислую реакцию (pH 4,0 —6,0), перед исследованием нейтрализуют стерильным 10%-м раствором двууглекислого натрия до слабощелочной реакции (pH 7,2 —7,4). Реакцию среды проверяют по универсальной индикаторной бумаге.
Для исследования на наличие сальмонелл из усредненной пробы отбирается отдельная навеска массой 25 г.
Приготовление разведений пищевых продуктов для посева. Для продуктов жидкой консистенции разведения готовят следующим образом: отбирают стерильной пипеткой 1 мл продукта и вносят в пробирку, содержащую 9 мл стерильного изотонического раствора хлорида натрия, тщательно перемешивают. Полученное разведение (10-1) подвергают таким же образом дальнейшему разведению в необходимое количество раз (кратное 10).
При исследовании плотных продуктов в качестве первого разведения используют 10%-ю взвесь, полученную способом, описанным выше.
Определение общей обсемененности. Выбранные для посева разведения вносят по 1 мл (каждого разведения) в стерильные чашки Петри, после чего их заливают 10—15 мл расплавленного и осужденного до 45 —50 °C МПА, который тщательно, круговыми движениями перемешивают. Среде дают застыть на строго горизонтальной поверхности. Посевы выращивают в течение суток при 37 °C, после чего подсчитывают число выросших колоний. Определяют общее количество бактерий в 1 мл или 1 г продукта.
Определение наличия плесеней и дрожжей. Выбранные для посева разведения вносят по 1 мл (каждого разведения) в стерильные чашки Петри, после чего их заливают 10—15 мл расплавленной и остуженной до 45 —50 °C среды Сабуро, которую тщательно, круговыми движениями перемешивают. Среде дают застыть на строго горизонтальной поверхности. Посевы выращивают в течение 5 сут при 25 °C, после чего подсчитывают число выросших колоний. Определяют общее количество дрожжей и плесеневых грибов в 1 мл или 1 г продукта.
Определение наличия БГКП. Для посева используется то количество продукта, в котором в соответствии с установленными нормами предусматривается отсутствие БГКП. Из выбранных разведений исследуемых образцов продуктов берут по 1 мл и засевают в пробирки со средой Кесслера (в двух повторностях). Засеянные пробирки инкубируют при 37 °C 24 ч. При отсутствии признаков роста (газообразования или изменения цвета среды) делают заключение о соответствии исследуемого продукта нормативу. При наличии признаков роста производят высев на среду Эндо. Посевы инкубируют 18 —20 ч при 37 °C. При просмотре посевов отмечают колонии, подозрительные или типичные для БГКП (образуют красные, розовые, бледнорозовые колонии с металлическим блеском или без него). Из них делают препараты живых и фиксированных клеток, окрашивают по Граму, микроскопируют. Обнаружение грамотри-цательных, не образующих спор палочек с закругленными концами, свидетельствует о возможном наличии БГКП. При необходимости проводят окси
дазный тест и высев на полужидкую среду с глюкозой, срезу Козера. БГКП оксидазоотрицательны, большинство видов сбраживают глюкозу с образованием органических кислот, некоторые виды также выделяют газ, цитрат не утилизируют. Сопоставляя полученные данные, делают вывод о наличии БГКП в исследуемом образце.
Определение наличия Salmonella. Определение сальмонелл проводят в навеске продукта 25 г. Производят посев навески в 100 мл селенитового бульона. Посев инкубируют 24 ч при 37 °C. Затем производят высев истощающим штрихом на поверхность висмут-сульфит агара, инкубируют 24 ч при 37 °C. Изолированные колонии, характерные для бактерий рода Salmonella (черные, с характерным металлическим блеском, с прокрашиванием участка среды под колонией в черный цвет; некоторые серовары образуют нежные светло-зеленые или крупные серовато-зеленые колонии), пересевают на среду Клиглера штрихом по скошенной поверхности и уколом в столбик. Посевы помешают на 12— 16 ч в термостат с температурой 37 °C. Все Salmonella сбраживают глюкозу с образованием кислоты и газа, подавляющее большинство не сбраживают лактозу (кроме представителей двух видов). Для дальнейшей предварительной идентификации готовят препараты живых и фиксированных клеток, окрашивают по Граму. К Salmonella относят палочки с закругленными концами, в большинстве подвижные, не образующие спор и капсул, грамотрицательные, оксидазоотрицательные.
Делают вывод о возможном наличии сальмонелл в исследуемом образце. Подробно полная схема идентификации сальмонелл описана в специальных руководствах по медицинской микробиологии.
Определение наличия Staphylococcus aureus. Из выбранных разведений исследуемых образцов продуктов берут по 1 мл и засевают в пробирки с 5 мл 6,5%-го солевого бульона. Кроме того, основную 10%-ю взвесь продукта в количестве 0,2 мл наносят на поверхность желточно-солевого или молочносолевого агара и тщательно растирают шпателем. Посевы инкубируют 18 — 20 ч при 37 °C. Чашки с плотными средами оставляют еще на сутки при комнатной температуре (для проявления пигмента).
Просматривают посевы на плотных средах. Из подозрительных колоний делают препараты, окрашивают по Граму. Проверяют наличие каталазы. Staphylococcus aureus — неподвижные кокки, расположенные одиночно, парами или гроздьями. Грамположительны, каталазоположительны, обычно оксидазоотрицательны. Из пробирок с солевым бульоном делают высев на желточно-солевой (колонии имеют форму плоских дисков (диаметр 2—4 мм) белого, желтого, кремового, лимонного, золотистого цвета с ровными краями; вокруг колоний образуются радужное кольцо и зона помутнения среды) или молочносолевой агар (мутные, круглые, ровные, выпуклые колонии белого, кремового, желтого или оранжевого цвета 2—4 мм в диаметре), проводят предварительную идентификацию, как описано выше. Делают вывод о возможном наличии Staphylococcus aureus в исследуемом образце. Полную схему идентификации золотистого стафилококка можно найти в специальных руководствах.
Определение наличия Proteus. Для выделения бактерий рода Proteus производят посев по методу Шукевича: 0,5 мл взвеси продукта засевают в конденсационную воду свежескошенного МПА. Посевы инкубируют 18 —20 ч при 37 °C. Если на скошенном МПА наблюдается вползающий нежный вуалеобразный
рост, сопровождающийся специфическим запахом, то из верхнего края выросшей культуры готовят препараты, окрашивают по Граму, выявляют подвижность. К Proteus относят прямые, подвижные (иногда встречаются неподвижные формы, лишенные жгутиков) палочки с закругленными концами, не образующие спор и капсул. Клетки склонны к полиморфизму, наблюдаются кокковидные и нитевидные формы. Грамотрицательны, каталазоположительны, оксидазоотрицательны. Делают вывод о возможном наличии бактерий рода Proteus в исследуемом образце.
Определение наличия сульфитвосстанавливающих клостридий. Из выбранных разведений исследуемых образцов продуктов берут по 1 мл и засевают в пробирки с 9 мл расплавленной и охлажденной до 45 °C средой Вильсона — Блера, тщательно перемешивают. Посевы инкубируют 18—20 ч при 37 °C. Появление в среде черных колоний указывает на присутствие сульфитвосстанавливающих клостридий. Из подозрительных колоний готовят препараты, окрашивают по Граму. Проверяют наличие каталазы с помощью раствора перекиси водорода (30 г/дм3). Делают вывод о возможном наличии сульфитвосстанавливающих клостридий в исследуемом образце.
Материалы, оборудование, приборы, реактивы
Стерильные чашки Петри; стерильные пробирки; стерильные пипетки; петли бактериологические; скальпель; ступка фарфоровая с пестиком; лупа; бумага фильтровальная; вата; микроскопы; стекла предметные и покровные; весы; микроскоп; термостаты на 25, 30 и 37 °C.
Стерильный 0,85%-й раствор NaCl (или вода водопроводная стерильная); среды: Эндо, Кесслера (или КОДА), МПА, Сабуро, желточно-солевой (или молочно-солевой) агар, солевой бульон, среды Клиглера и Козера, селенитовый бульон, висмут-сульфит агар, среда Вильсона—Блера, полужидкая среда с глюкозой, свежеприготовленный раствор диметил-/7-фенилендиамина; свежеприготовленный 3%-й раствор КОН; раствор перекиси водорода (30 г/дм3); реактивы для окраски по Граму; фуксин; 70%-й этанол; бумага индикаторная универсальная.
ЗАДАЧА 5. САНИТАРНО-БАКТЕРИОЛОГИЧЕСКОЕ ИССЛЕДОВАНИЕ ПРЕДМЕТОВ ОБИХОДА И РУК ПЕРСОНАЛА
Изучение бактериальной загрязненности рук и различных предметов обихода производится в целях оценки санитарно-гигиенического состояния исследуемого объекта, установления путей распространения инфекции при эпидемиологических заболеваниях, лабораторного контроля эффективности обработки кожи рук.
В зависимости от цели проводимого исследования и характера контролируемых объектов в полученных смывах определяют;
• общую микробную обсемененность (обычно с пересчетом на 1 см2 исследуемой поверхности);
• наличие бактерий группы кишечной палочки, как показателей фекального загрязнения;
• наличие патогенных бактерий кишечной группы, обнаружение которых является безусловным показателем эпидемического неблагополучия;
• наличие стафилококков и других микроорганизмов.
Цель работы, определить степень микробной загрязненности кожи рук и предметов обихода.
Ход выполнения задачи
Подготовка сред и материалов. Подготовить к стерилизации (в расчете на I студента): чашки Петри — 10 шт.; пробирки — 10; пипетки на 1 — 2 мл — 5, на 5 —10 мл — 3; салфетка марлевая 5 х 5 см (или ватный тампон на палочке) — 1 шт. Приготовить (состав и приготовление сред см. в приложении 4): 0,85%-й раствор NaCl стерильный (или вода водопроводная стерильная) — 100 мл; МПА стерильный — 100 мл.; МПБ с 7,5%-м NaCl стерильный — 10 мл; желточно-солевой (или молочно-солевой) агар — 20—40 мл; полужидкая среда с глюкозой — 30 мл (разлить в 3 пробирки по 7—10 мл); среда КОДА или среда Кесслера (нормальной концентрации) — 60 мл (разлить по 10 мл в 6 обычных пробирок), в каждую пробирку поместить по поплавку или комочку ваты; среда Эндо — 20—60 мл (сразу после приготовления разлить в стерильные чашки Петри, дать застыть, завернуть в бумагу и оставить в холодильнике); висмут-сульфит агар — 20 мл; селенитовый бульон — 10 мл.
Приготовление смывов. Для получения смывов пользуются стерильными ватными тампонами или марлевыми салфетками. В день взятия смыва в каждую пробирку с тампоном наливают 2 мл стерильной водопроводной воды или изотонического раствора хлорида натрия так, чтобы ватный тампон находился над уровнем жидкости. Марлевые салфетки 5 х 5 см заворачивают по одной в бумагу. К каждой салфетке готовят заранее пробирку с 2 мл стерильной воды для смачивания. Непосредственно перед взятием смыва пробирку наклоняют, увлажняя находящийся в ней тампон. В случае применения салфеток их захватывают кончиком стерильного пинцета, прокаленного над пламенем горелки, и увлажняют стерильной водой из пробирки.
Увлажненный тампон или салфетку дают в руки обследуемому, предлагая протереть им сначала кожу левой, а затем правой руки (от участков с меньшей к участкам с большей загрязненностью): тыл кисти, поверхность ладони, межпальцевые пространства, ногтевые ложа. По окончании процедуры салфетки или тампоны помещают в пробирку, в которой они находились.
При взятии смывов с предметов, имеющих большую поверхность, исследуемый участок ограничивают рамкой-трафаретом площадью 25, 50 или 100 см2. Трафарет, изготовленный из проволоки, после каждого смыва и перед употреблением прожигают над пламенем горелки. При исследовании мелких предметов смыв делают со всей поверхности.
Исследование смывов. Для определения общего числа микроорганизмов в исследуемом смыве к 2 мл воды, которая была использована для увлажнения тампона, прибавляют еще 8 мл стерильной воды. Тампон тщательно в течение 2—3 мин отмывают, получая исходное разведение. Из него готовят рад последовательных десятикратных разведений. Затем из различных разведений смыва берут по I мл, вносят в стерильные чашки Петри, заливают расплавленным и остуженным МПА. Посевы выдерживают 24 ч при 37 °C и 24 ч при комнатной температуре, после чего производят подсчет выросших колоний.
Устанавливают количество микроорганизмов в 1 мл исходного разведения смыва (для этого подсчитывают число колоний в чашке и полученную величину умножают на степень разведения смыва).
Определяют количество микроорганизмов в 10 мл смыва, соответствующее общему числу микроорганизмов, находящихся на той площади, с которой произведен смыв (для этого величину, соответствующую количеству микроорганизмов в 1 мл смыва, умножают на 10).
Определяют количество микроорганизмов на 1 см2 исследуемой поверхности (для этого величину, характеризующую количество микроорганизмов в 1 мл смыва, делят на число квадратных сантиметров, с которых сделан смыв).
Для выявления бактерий группы кишечной палочки можно пользоваться прямым посевом исследуемого материала на чашки со средой Эндо. Материал, находящийся на тампоне, втирают в поверхность питательной среды Эндо. Кроме того, используют среды обогащения (среда Кесслера, КОДА и др.) — в этом случае тампон, снятый с палочки, помещают в соответствующую жидкую среду. Посевы на плотных средах инкубируют при 37 °C, на жидких средах — при 43 °C в течение 18 —24 ч. На второй день из флаконов с признаками роста производят высев на чашки со средой Эндо. Дальнейший ход исследования полностью соответствует стандартной схеме выявления бактерий группы кишечной палочки (см. разд. 46.1).
Для выявления патогенных бактерий кишечной группы содержащийся на тампоне материал тщательно втирают в поверхность плотных питательных сред (висмут-сульфит агар и др.). После произведенного посева тампон и оставшуюся в пробирке жидкость-смыв помещают в среду обогащения (селенитовый бульон). Через 24—48 ч инкубации при 37 °C просматривают посевы, сделанные на плотные среды, выявляя колонии, характерные для патогенных бактерий кишечной группы. Идентификация выделенных микроорганизмов с бактериями группы сальмонелл производится по методам, изложенным выше (см. разд. 46.4).
Для выявления стафилококка производят посев 0,5 — 1 мл смыва на молочно-солевой или желточно-солевой агар и в среду накопления (солевой бульон — МПБ, содержащий 6,5 % NaCl). Колонии с признаками, характерными для сатфилококков, изучают, как описано выше (см. разд. 46.4).
Материалы, оборудование, посуда, реактивы
Стерильные чашки Петри; стерильные пробирки; стерильные пипетки; стерильные ватные тампоны или марлевые салфетки; палочки стеклянные; пинцеты; петли бактериологические; вата; микроскопы; стекла предметные и покровные. Вода водопроводная стерильная; среды: Эндо, Кесслера или КОДА, МПА, полужидкая с глюкозой, молочно-солевой или желточно-солевой агар, солевой бульон, висмут-сульфит агар, селенитовый бульон, свежеприготовленный раствор диметил-л-фенилендиамина; свежеприготовленный 3%-й раствор КОН; реактивы для окраски по Граму; фуксин; 70%-й этанол.
ЗАДАЧА 6. МИКРОБИОТА ЧЕЛОВЕКА. МИКРОФЛОРА ПОЛОСТИ РТА
Полость рта является исключительно благоприятной средой для существования и размножения самых различных видов микроорганизмов. Это обуслов
лено постоянным наличием питательных веществ, оптимальной для размножения многих микроорганизмов температурой (37 °C), влажностью и pH (около 6,9 —7,0). Микрофлора полости рта подразделяется на постоянную и случайную. Обычными представителями нормальной микрофлоры ротовой полости являются стрептококки: факультативно анаэробные (в том числе Streptococcus salivarius, S. mitis, кариесогенные — S. mutans и др.) и облигатные анаэробы {Peptostreptococcus), а также грамположительные нитевидные микроорганизмы {Actinomyces viscosus). В составе микрофлоры полости рта можно обнаружить также лактобациллы, грамотрицательные кокки, относящиеся к родам Neisseria и Veiionella, а также грамотрицательные бактерии, строгие анаэробы из семейства Bacteroidaceae {Bacteroides, Fusobacterium, Leptotrichia'). В состав постоянной микрофлоры полости рта входят различные виды спирохет (Treponema denticola, Т. orale, Т macrodenticum, Borrelia buccalis), микоплазмы, нокардии и др. К случайной микрофлоре полости рта относятся комменсалы, обитающие на других слизистых оболочках и коже, сапрофиты внешней среды и различные патогенные микроорганизмы, которые попадают в полость рта в результате аэрогенного или алиментарного заражения от больных или бактерионосителей. Очень высокой способностью адаптироваться к существованию в ротовой полости обладают стафилококки, стрептококки, коринебактерии, грибы Candida, вирусы герпеса, эпидемического паротита и др.
Цель работы: описать микрофлору зубного налета, слизистой оболочки зева на основании данных микроскопического исследования; выделить и произвести количественный учет некоторых групп микроорганизмов из слизистой оболочки зева.
Ход выполнения задачи
Подготовка сред и материалов. Подготовить к стерилизации (шт.): чашки Петри — 5; пробирки — 5; пипетки на 1 — 2 мл — 5 и 5—10 мл — 3; ватный
тампон на палочке — 1. Приготовить (состав и приготовление сред см. в приложении 4): 0,85%-й раствор NaCl стерильный (или вода водопроводная стерильная) — 100 мл; МПА стерильный — 20 — 60 мл; желточно-солевой (или молочно-солевой) агар — 20 мл; среда Эндо — 20 мл (сразу после приготовле-
ния разлить в стерильные чашки Петри, дать застыть, завернуть в бумагу и оставить в холодильнике); среда Сабуро — 20 мл.
Проведение анализа. Из зубного налета, смывов со слизистой оболочки зева готовят препараты-мазки, окрашивают их и микроскопируют (рис. 46.5). Из зубного налета так-
Рис. 46.5. Микрофлора зубного налета (по Л. В. Борисову и др., 1993):
/ — Leptotrichia buccalis: 2 — Lactobacillus sp.; 3, 4 — Treponema denticola, Treponema, sp.', 5, 6— стрептококки и диплококки (Streptococcus salivarius, S. mitis, S. sanguis, Veiionella sp.); 7 — Fusobacterium nucleatum
же готовят препарат «раздавленная капля» для обнаружения подвижных микроорганизмов.
Для посевов берут определенный объем исследуемого материала (например, смыв из зева или зубной налет), разводят в стерильной воде (в зависимости от предполагаемой концентрации) и делают посевы из соответствующих разведений на питательные среды: для выявления стафилококка — на молочно-солевой или желточно-солевой агар; для выявления бактерий группы кишечной палочки — на среду Эндо; для выявления дрожжей рода Candida — на среду Сабуро. Ориентировочную идентификацию микроорганизмов проводят по характеру выросших колоний, морфологии клеток, окраске по Граму.
Материалы, оборудование, посуда, реактивы
Стерильные чашки Петри — 5 шт., стерильные пробирки; стерильные пипетки; петли бактериологические; вата; микроскопы; стекла предметные и покровные. Вода водопроводная стерильная; среды: Эндо, молочно-солевой или желточно-солевой агар, Сабуро, МПА, свежеприготовленный раствор диметил-н-фенилендиамина; свежеприготовленный 3%-й раствор КОН; реактивы для окраски по Граму; фуксин; 70%-й этанол.
План проведения задачи
На выполнение всей задачи требуется около 18 ч.
Занятие 1. Подготовка посуды и приготовление сред.
Занятие 2. Анализ воды. Анализ воздуха.
Занятие 3. Продолжение анализа воды и воздуха. Анализ почвы.
Занятие 4. Продолжение анализа воды и почвы. Анализ пишевых продуктов. Анализ микрофлоры рук (или предметов обихода). Микрофлора человека (зубной налет, мазок из зева).
Занятое 5. Продолжение анализа почвы, пищевых продуктов и пр.
Занятие 6. Обработка результатов, подведение итогов.
Г л а в а 47
ОСНОВЫ МЕДИЦИНСКОЙ МИКРОБИОЛОГИИ
ЗАДАЧА 1. ДЕМОНСТРАЦИЯ ПОСТУЛАТОВ КОХА В ЭКСПЕРИМЕНТАХ С РАСТЕНИЯМИ
Великий немецкий микробиолог Роберт Кох (1843—1910) начал свои исследования в то время, когда роль микроорганизмов в возникновении инфекционных заболеваний подвергалась серьезным сомнениям.
Для доказательства этой роли требовались четкие критерии, которым должны удовлетворять эксперименты, подтверждающие причинную связь между данным микроорганизмом и определенной болезнью. Эти критерии были сформулированы примерно за 30 лет до работ Коха другим немецким микробиологом — Ф. Генле, но вошли в историю под названием «постулаты Коха», поскольку именно Р. Кох впервые применил их в эксперименте, изучая бактериальную этиологию сибирской язвы. Суть постулатов заключалась в следующем:
1) один и тот же микроорганизм — предполагаемый возбудитель болезни, должен обнаруживаться во всех случаях данного заболевания и не выделяться при других болезнях и от здоровых лиц;
2) микроб-возбудитель должен быть выделен из организма больного и поддерживаться в чистой культуре;
3) чистая культура данного микроорганизма должна вызывать у восприимчивого хозяина (экспериментально зараженных животных) заболевание с клинической и патологоанатомической картиной, аналогичной заболеванию человека, из организма которого выделена чистая культура;
4) микроб-возбудитель должен быть снова выделен из организма экспериментально зараженного животного и его свойства должны быть идентичны первому изоляту.
Роль микроорганизмов в возникновении инфекционных заболеваний была доказана Кохом только после того, как им был разработан и начал активно использоваться в медицине метод выделения чистых культур микроорганизмов на плотных питательных средах из отдельной колонии (см. гл. 6).
Практика показала, что постулаты Коха имеют в известной мере относительное значение, поскольку не всегда удается выделить возбудителя болезни в чистой культуре и вызвать у подопытных животных заболевание, свойственное человеку. Кроме того, болезнетворные микроорганизмы в некоторых случаях могут быть найдены и в организмах здоровых людей, обладающих иммунитетом, особенно после перенесенного ими ранее инфекционного заболевания. Однако на ранних этапах формирования медицинской микробиологии, когда из организма больных выделяли многие микроорганизмы, не имеющие отношения к данной болезни, постулаты Коха сыграли важную роль для установления истинного возбудителя заболевания. Например, Кох окончательно доказал, что ранее обнаруженный у животных, больных сибирской язвой, микроорганизм отвечает требованиям постулатов и является истинным возбудителем данного заболевания и у людей. Хотя Кох работал с животными, его постулаты можно продемонстрировать и с использованием растений.
Цель работы, подтвердить в эксперименте основные постулаты Коха.
Ход выполнения задачи
Микроорганизмы и их культивирование. В работе используют яблоко или клубень картофеля с гладкой неповрежденной кожицей, подверженные гниению в результате спонтанного развития гнилостной микрофлоры на начальном этапе процесса.
Основным методом работы является метод выделения чистой культуры микроорганизма на плотной питательной среде.
Для выделения чистой культуры из гниющего картофеля используют мясопептонный агар, а из гниющего яблока — среду Саборадо состава (г/л): глюкоза — 20,0; пептон — 10,0; агар — 18,0; вода водопроводная — до 1 л. Значение pH среды Саборадо до стерилизации доводят до 5,6 —5,8. Стерилизацию проводят при 0,5 ати 30 мин.
Питательную среду, предназначенную для работы по выделению чистой культуры, расплавляют и разливают в стерильные чашки Петри из расчета 12 чашек (около 250 мл) на один объект изучения.
Описание характера поражения. Перед выделением из яблока (или клубня картофеля) культуры микроорганизма, вызывающего гниение, объект осторожно моют водой с мылом и характеризуют внешние признаки поражения — отмечают изменения цвета и консистенции пораженных микроорганизмами участков
поверхности кожицы. Затем яблоко (или клубень) протирают ватой, смоченной 70%-м этиловым спиртом, и помещают в стерильный стеклянный сосуд.
Выделение культуры возбудителя процесса гниения. Для выделения культуры возбудителя процесса гниения стерильной иглой или скальпелем делают в кожице яблока или клубня в месте поражения отверстие диаметром 3 — 4 мм. Из этого отверстия стерильно, с помощью бактериологической петли берут небольшой комочек гниющей ткани и переносят в пробирку с 6 — 7 мл стерильной водопроводной воды. Содержимое пробирки энергично перемешивают встряхиванием. Выделение культуры возбудителя процесса гниения проводят с помощью бактериологической петли методом истощающего штриха. Используют 3 чашки Петри с соответствующей стерильной средой. Кроме того, для получения чистой культуры используют стеклянные шпатели Дригальского. С этой целью петлю («зеркальце») суспензии гнилостных микроорганизмов наносят на поверхность стерильной среды в чашке Петри, а затем распределяют микроорганизмы стерильным шпателем и, не обжигая шпатель в пламени горелки, распределяют материал по 2-й и 3-й чашкам Петри со стерильной средой. Засеянные чашки инкубируют в термостате при 30 °C в течение 2—3 сут (картофель) и 5 —7 сут (яблоко). Выросшие колонии описывают и проводят изучение морфологии клеток возбудителя процесса гниения, используя препарат «раздавленная капля» и объектив *40 при работе с яблоком и, кроме того, окрашенный фуксином препарат (объектив хЮО), при работе с картофелем.
Экспериментальное заражение восприимчивого «хозяина». Здоровое яблоко или клубень картофеля моют с мылом, протирают 70%-м спиртом и помещают в простерилизованный сттртом сосуд. Затем раскаленной бактериологической иглой делают на поверхности здорового объекта 2 колотых отверстия глубиной около 1 — 2 см. Иглу опять стерилизуют в пламени и с ее помощью заражают оба отверстия здорового яблока или клубня картофеля чистой культурой, выделенной из гниющих яблока или клубня соответственно. Зараженный объект инкубируют в стеклянном сосуде при 30 °C в течение 5 — 7 сут (яблоко) или 2—4 сут (картофель).
Изучение культуры возбудителя процесса гниения из организма экспериментально зараженного объекта. Характер экспериментального поражения поверхности яблока или клубня описывают и сравнивают с таковым у спонтанно гниющих яблока или клубня. Для подтверждения четвертого постулата Коха можно провести повторное выделение культуры, вызывающей гниение, из экспериментально зараженного объекта. Выросшие колонии описывают и изучают морфологию клеток возбудителя процесса гниения под микроскопом. Однако достаточно провести только микроскопическое изучение гниющих тканей яблока или клубня и сравнить результаты с данными, полученными при выделении культуры возбудителя процесса гниения.
Материалы, оборудование, посуда, реактивы (на 1 студента)
Пробирки — 1 шт.; колба на 500 мл; реактивы для приготовления сред; 12 чашек Петри; гниющие и здоровые яблоки или клубни картофеля; микроскоп; покровные и предметные стекла; фуксин.
План выполнения задачи (12 ч)
Занятие 1. Приготовление и стерилизация питательных сред, пипеток и шпателей.
Занятие 2. Подготовка объекта, описание характера поражения. Выделение культуры возбудителя процесса гниения.
Занятие 3. Описание колонии и микроскопическое изучение выделенной культуры возбудителя процесса гниения. Экспериментальное заражение восприимчивого «хозяина».
Занятие 4. Изучение культуры возбудителя процесса гниения из организма экспериментально зараженного объекта методом микроскопии.
ЗАДАЧА 2. МОДЕЛИРОВАНИЕ ПРОЦЕССА ВОЗНИКНОВЕНИЯ ЭПИДЕМИИ НА ПРИМЕРЕ КУЛЬТУРЫ ПЕКАРСКИХ ДРОЖЖЕЙ
Эпидемия — относительно кратковременное, быстрое и непрерывное распространение инфекционного заболевания в пределах какой-то совокупности организмов в определенном регионе. При этом регистрируется заболеваемость намного выше обычно существующего уровня. Распространение инфекционного заболевания людей или животных на территории целой страны, континента или всей суши называется пандемией. Инфекционное заболевание, или инфекция (от лат. infectio — заражение), представляет собой совокупность физиологических и патологических адаптационных и репарационных реакций, которые возникают и развиваются в макроорганизме в процессе взаимодействия с патогенными микроорганизмами.
Формы инфекции разнообразны и носят различные наименования в зависимости от природы возбудителя, его локализации в макроорганизме и путей распространения. Так, экзогенная инфекция возникает в результате заражения патогенными микроорганизмами, поступившими в организм из окружающей среды с пищей, водой, воздухом, почвой, выделениями больного человека или микробоносителя. Часто распространение инфекционных болезней происходит при несоблюдении элементарных правил гигиены.
Цель работы, получение экспериментальной модели возникновения эпидемии.
Ход выполнения задачи
Микроорганизмы и их культивирование. В работе используют культуры пекарских дрожжей Saccharomyces cerevisiae. Дрожжи выращивают в пробирках с сусло-агаром в течение 3 сут при 30 °C и хранят в холодильнике при 4 °C.
Основным методом работы является получение культуры дрожжей с поверхности ладоней и пальцев человека методом смыва с последующим высевом смытой суспензии на плотную питательную среду.
Для опытов используют 3-суточную культуру 5. cerevisiae, полученную на сусло-агаре при 30 °C.
Группу студентов разделяют на подгруппы по 4 человека в каждой. На подгруппу готовится 4 чашки Петри с сусло-агаром. Каждую чашку нумеруют. Все студенты подгруппы тщательно моют руки с мылом и стерилизуют их 70%-м этанолом.
Контрольный тест для доказательства чистоты рук. Чтобы убедиться в отсутствии дрожжевых клеток на руках студентов, участвующих в эксперименте, делают смыв с ладоней и пальцев их правой руки. Первый студент с помощью стерильного ватного тампона на деревянной палочке, смоченного в стерильной водопроводной воде, делает смыв с поверхности ладони и пальцев правой
руки второго студента и проводит тампоном штрих по поверхности среды в чашке Петри 2а с сусло-агаром (делает контрольный высев). В свою очередь второй студент осуществляет аналогичный смыв с ладони и пальцев правой руки первого студента и делает контрольный высев в чашку 1а. Третий и четвертый студенты производят смыв ладоней и пальцев правой руки друг другу по такой же схеме и делают контрольные высевы в чашки с сусло-агаром За и 4а соответственно.
Моделирование процесса возникновения эпидемии. Делают суспензию из 3-суточной культуры дрожжей 5. cerevisiae, выросшей при температуре 30 °C. Для этого в пробирку с выросшей культурой дрожжей, содержащую скошенный сусло-агар, стерильно вносят 3 — 4 мл стерильной водопроводной воды и с помощью бактериологической петли осторожно суспендируют дрожжевую массу с поверхности агара. Полученную суспензию переносят в стерильную пробирку. Студенты, входящие в экспериментальную подгруппу, еще раз моют руки с мылом, протирают их 70%-м этанолом и дают высохнуть. Затем студент-инструктор или преподаватель протирает с помощью ватного тампона на палочке, смоченного суспензией 5. cerevisiae, поверхность ладони и пальцев правой руки первого студента. Он обменивается энергичным рукопожатием правой рукой со вторым студентом. Тот пожимает правую руку третьего студента, а он в свою очередь обменивается рукопожатием с четвертым студентом.
Выделение культуры возбудителя «инфекционного процесса». Для выделения культуры дрожжей с поверхности ладоней и пальцев правой руки студентов используют стерильные ватные тампоны на деревянной палочке, слегка смоченные стерильной водопроводной водой. Студенты — первый и второй, третий и четвертый — делают смывы ладоней и пальцев правых рук друг друга и высевают смывы на чашки 16, 26, 36 и 46 соответственно. Для получения отдельных колоний дрожжей, смытых тампонами с рук, штрих на чашке Петри с сусло-агаром распределяют по всей поверхности чашки бактериологической петлей методом «истощающего штриха». Делают контрольный высев на сусло-агар суспензии дрожжей, использованной в эксперименте, бактериологической петлей методом «истощающего штриха». Засеянные чашки со средой инкубируют в течение 3 сут при температуре 30 ”С. Выросшие колонии на чашках просматривают, определяют интенсивность роста — скудный, умеренный или обильный. При возможности подсчитывают количество колоний на чашках. Делают препараты «раздавленная капля» из колоний дрожжей в чашках 16, 26, 36 и 46, а также дрожжей рабочей суспензии и просматривают их с объективом 40 х. Делают выводы о способах возникновения и путях распространения экзогенной инфекции.
Материалы, оборудование, посуда, реактивы (на 4 студентов)
Пробирки — 2 шт.; колба на 500 мл; сусло-агар; 10 чашек Петри; микроскоп; покровные и предметные стекла; пинцет; стерильные ватные тампоны на деревянной палочке — 10.
План выполнения задачи (9 ч)
Занятие 1. Приготовление и стерилизация питательной среды и воды. Засев дрожжей в пробирку.
Занятие 2. Контрольный тест для доказательства чистоты рук. Моделирование процесса возникновения эпидемии. Выделение культуры возбудителя «инфекционного процесса».
Занятие 3. Обработка результатов работы по выделению культуры возбудителя «инфекционного процесса». Зачет.
ЗАДАЧА 3. ПРЯМОЕ ВЫДЕЛЕНИЕ ПАТОГЕНА
Известно, что не существует такой питательной среды или такого сочетания физико-химических условий, в которых могли бы расти все разнообразные типы микроорганизмов, встречающиеся в природе. Следовательно, любая среда, подходящая для роста какого-то определенного организма или группы организмов, будет для них в некоторой степени селективной, или избирательной, потому что в ней развитие всех других видов будет невозможно или в значительной степени подавлено: в одних случаях — в связи с нехваткой необходимых компонентов, а в других — с наличием ингибирующих веществ. Если все пищевые потребности организма известны, то можно подобрать условия, наиболее благоприятные для развития именно этого организма, что позволит выделить его из такой смешанной популяции, в которой он представлен лишь незначительным количеством особей. Из природных местообитаний — почва, вода, воздух, организм человека или животного — можно избирательно получать те или иные виды микроорганизмов путем прямого выделения либо путем обогащения (см. гл. 6).
Прямое выделение проводят методом рассева смешанной популяции микроорганизмов по поверхности твердой питательной среды. В результате распределения по поверхности среды конкуренция между клетками различных видов за питательные вещества существенно уменьшается, и в этих условиях даже сравнительно медленно растущие организмы могут формировать колонии. Прямым высевом материала из природных местообитаний на чашки с селективной средой пользуются в тех случаях, когда хотят выделить сравнительно много видов микроорганизмов, способных расти в данных условиях.
Для получения культур из смешанной популяции путем обогащения используют посев материала в жидкую селективную (элективную) среду, в которой различные организмы постоянно конкурируют друг с другом за питательные вещества. В результате в таких накопительных культурах из всех членов внесенной в среду популяции, способных расти в данных условиях, постепенно отбираются те виды, которые растут в данных условиях с наибольшей скоростью.
Получение патогенных или условно патогенных микроорганизмов непосредственно из организма или из выделений, инфицированных болезнетворными микроорганизмами, часто проводят с применением метода прямого выделения. Используют большое количество различных видов селективных сред.
Цель работы: получение условно-патогенных бактерий из организма человека методом прямого выделения с использованием плотных селективных сред.
Ход выполнения задачи
Микроорганизмы и их культивирование. В работе используют бактерии Staphylococcus aureus. Этот микроорганизм обитает в носоглотке человека и счи
тается условно-патогенным. 5. aureus выделяет энтеротоксин, является причиной некоторых видов пищевых отравлений, вызывает фурункулезы и т.п. Для получения бактерий из организма человека методом прямого выделения используют плотные селективные среды — агаризованную среду с маннитом и NaCl и стафилококковую среду ПО.
Агаризованная среда с маннитом и NaCl содержит (г/л): пептон — 10,0; питательный бульон — 1,0; D-маннит — 10,0; NaCl —75,0; феноловый красный — 0,025; агар — 15,0. Хлористый натрий ингибирует рост многих других форм бактерий. Об образовании кислоты из маннита свидетельствует желтый цвет, появляющийся в присутствии индикатора фенолового красного.
Селективный эффект стафилококковой среды 110 также основывается на действии 7,5%-го хлористого натрия, разжижении желатины и сбраживании маннита. Среда ПО содержит (г/л): триптон — 10,0; дрожжевой экстракт — 2,5; желатина — 30; лактоза — 2,0; D-маннит — 10,0; NaCl —75,0; К2НРО4 — 5,0; агар — 15,0.
Отсев S. aureus для хранения в лаборатории не производится. Все использованные питательные среды с посевами, предметные стекла и т.п. тщательно контролируются и после выполнения работы заливаются дезраствором и затем убиваются методом автоклавирования.
Основным методом работы является метод выделения чистой культуры микроорганизма на плотной питательной среде. Для получения опытного материала используют студентов учебной группы.
Выделение X. aureus из носоглотки человека. Готовят ватные тампоны на деревянной палочке, заворачивают их в бумагу и стерилизуют в сушильном шкафу. В чашки Петри разливают питательные среды — агаризованную среду с маннитом и NaCl и стафилококковую среду ПО. С помощью ватного тампона из горла или внутренней полости носа извлекают исследуемый материал и сразу же проводят тампоном полосу (штрих) по поверхности агаризованной среды с маннитом и NaCl. Используя второй тампон, еще раз извлекают исследуемый материал и проводят тампоном полосу по поверхности второй среды — стафилококковой среды 110. Полосу проводят тампоном не по центру чашек, а ближе к их краю. Стерильной бактериологической петлей распределяют исследуемый материал из полосы по всей поверхности каждой среды на чашке методом истощающего штриха. Используя вторую чашку с соответствующей средой и стерильную бактериологическую петлю, рассев материала истощающим штрихом продолжают по поверхности среды и в этой чашке. Засеянные чашки — по 2 с каждой средой, инкубируют в термостате при 35 “С в течение 48 ч. Далее чашки извлекают из термостата и оставляют еще на 24 ч в лаборатории на свету при комнатной температуре для дополнительного биосинтеза в колониях стафилококков бактериальных пигментов.
Обнаружение характерных колоний S. aureus. После экспонирования выросших колоний в течение 24 ч в лаборатории на свету при комнатной температуре чашки с каждой из сред исследуют на наличие характерных колоний S. aureus.
Агаризованная среда с маннитом и NaCl. На этой среде условно-патогенные стафилококки S. aureus образуют крупные колонии, окруженные желтым кольцом (зоной), образовавшимся в результате реакции синтезированных при брожении маннита кислот с индикатором феноловым красным. Другие виды стафилококков образуют на агаризованной среде с маннитом и NaCl колонии
меньшего размера, окруженные пурпурной или красной зонами. Клетки из типичных колоний, окруженных желтым кольцом, красят по Граму. Положительное окрашивание клеток по Граму подтверждает принадлежность исследуемых бактерий к стафилококкам.
Стафилококковая среда ПО. На среде ПО условно-патогенные формы стафилококков образуют оранжевые колонии, в то время как непатогенные формы формируют белые колонии. Типичные пигментированные колонии пересевают в пробирки с агаризованной средой ПО и через 24 ч культивирования при 35 °C красят клетки по Граму. Положительное окрашивание подтверждает, что исследуемые бактерии относятся к стафилококкам. На остаток типичной оранжевой колонии на чашке, из которой произвели пересев на скошенный агар, наносят каплю 0,25%-го спиртового раствора индикатора крезолового пурпурного. Изменение цвета колонии на желтый свидетельствует о процессе сбраживания бактериями маннита с образованием кислот. В качестве контроля наносят каплю крезолового пурпурного на участок агаризованной среды на чашке, свободный от выросших колоний стафилококков. Для обнаружения процесса разжижения желатины, вызванного действием протеолитических ферментов, на типичную оранжевую колонию наносят каплю насыщенного раствора сульфата аммония. Выдерживают чашку 10 мин при 30 °C. О положительной реакции свидетельствует появление вокруг колонии зоны просветления.
Материалы, оборудование, посуда, реактивы (на 1 студента')
Пробирки — 2 шт.; реактивы для приготовления сред; чашки Петри — 4; микроскоп; предметные стекла; ватные тампоны на деревянной палочке в пробирках — 2; реактивы для окрашивания клеток по Граму.
План выполнения задачи (15—18 ч)
Занятие 1. Приготовление и стерилизация питательных сред, чашек Петри, тампонов.
Занятие 2. Выделение 5. aureus из носоглотки человека.
Занятие 3. Обнаружение характерных колоний 5. aureus. Зачет.
47.1. ИЗУЧЕНИЕ ИММУННОЙ СИСТЕМЫ
Иммунная система людей и животных обеспечивает специфическую защиту организма от генетически чужеродных молекул и клеток — бактерий, грибов, простейших, вирусов. В кровеносной и лимфатической системах постоянно циркулируют компоненты иммунной системы — лейкоциты разных типов и производимые ими антитела. Задачей иммунной системы является распознавание и уничтожение чужеродных антигенов инфекционных агентов и химических веществ, попадающих в организм. Это обеспечивает постоянство внутренней среды организма.
Реакции иммунной системы обладают несколькими характерными особенностями: • память — после того как впервые возникает иммунная реакция на определенный чужеродный фактор, иммунная система организма «запоминает» этот фактор; повторные попадания в организм данного фактора переносятся легче, этим объясняется то,
что многими болезнями, например такими, как свинка, корь и оспа, человек болеет обычно один раз в жизни;
• специфичность — каждая иммунная реакция направлена против определенного чужеродного агента;
• толерантность — специфическая инертность, отсутствие реакций на клетки и продукты жизнедеятельности своего организма, в отличие от выраженной реакции на чужеродные клетки и их продукты.
Организм «узнает» чужеродный фактор по реакции между антигенами этого фактора и антителами, имеющимися в этом организме. Антигеном является любое вещество, в состав которого обычно входит какой-либо белок, способное вызвать иммунную реакцию. Антитела, или иммуноглобулины, — это белки плазмы крови, которые по своему химическому составу относятся к гликопротеидам, поскольку их молекула обычно состоит из белка и олигосахарида. Антитела синтезируются клетками иммунной системы и способны за счет формы и распределения электрического заряда по их поверхности связывать антигены, комплементарные им по форме и распределению заряда. Центральные органы иммунной системы человека — костный мозг и вилочковая железа, в которых образуются иммунокомпетентные клетки — Т- и В-лимфоциты. В организме человека содержится более 70% Т-лимфоцитов, около 15% В-лимфоцитов. Маркерами, по которым отличают Т- и В-лимфоциты, являются, иммуноглобулиновые рецепторы, находящиеся на поверхности В-лимфоцитов, и рецепторы к эритроцитам барана, которыми обладают Т-лимфоциты.
Основной функцией В-лимфоцитов является образование иммуноглобулинов. Связывание антигена с гомологичными рецепторами на мембране В-лимфоцита является сигналом для его роста, в результате чего образуется иммунологически однородный клон В-лимфоцитов, обладающий идентичными иммуноглобулиновыми рецепторами. Далее однородный клон В-лимфоцитов под влиянием клеточных медиаторов дифференцируется на два типа клеток — клеток иммунологической памяти и клеток, образующих антитела.
Клетки иммунологической памяти появляются тогда, когда определенный антиген впервые попадает в организм. Иммунный ответ, возникающий при этом, называют первичной иммунной реакцией. После того как организм успешно справился с этим антигеном, число В-лимфоцитов, производящих специфические к данному антигену антитела, в крови постепенно уменьшается, однако некоторое их количество остается в лимфатических узлах в течение многих месяцев или лет. Эти клетки выполняют роль иммунологической памяти организма и готовы начать борьбу, если тот же чужеродный агент снова попадет в организм. Повторное попадание того же антигена вызывает вторичную иммунную реакцию. Она развивается быстрее, более активна и сохраняется дольше первичной. По этой причине люди редко болеют дважды в сезон гриппом, вызываемым одним и тем же штаммом вируса. Иммунная память на многие болезни — оспу, свинку, корь и некоторые другие — сохраняется на всю жизнь, ими редко болеют в жизни дважды. Разработаны вакцины против очень многих бактериальных и вирусных инфекций. Введение в организм вакцинируемого антигена в виде вакцины — убитого или ослабленного возбудителя болезни, вызывает выработку иммунной системой антител и лейкоцитов, которые обеспечивают защиту в случае проникновения инфекционного агента.
Клетки второго типа В-лимфоцитов, образующих антитела, увеличиваются в размерах, прекращают рост и начинают образовывать антитела — иммуноглобулины классов IgA, IgG, IgM, IgD и др. Эти В-лимфоциты называются плазматическими клетками, срок их существования составляет всего несколько дней. За это время они успевают образовать большое количество специфических к данному антигену антител.
Т-лимфоциты дифференцируются на четыре типа: Т-хелперы, Т-супрессоры, Т-киллеры и Т-эффекторы. Т-хелперы, или помощники, и Т-супрессоры, или ингибиторы, выполняют регуляторные функции. Т-хелперы узнают специфический анти
ген на мембране макрофага и активируют при помощи медиаторов В-лимфоциты и Т-эффекторы. Т-супрессоры угнетают Т-хелперы, В-лимфоциты или плазматические клетки, что задерживает образование антител. Т-эффекторы синтезируют лимфокины — клеточные медиаторы, обеспечивающие клеточный специфический иммунитет. Т-киллеры уничтожают клетки, несущие чужеродный антиген.
47.1.1. Группы крови
Одна из первых реакций антиген—антитело была обнаружена при переливании крови от одного человека (донора) к другому (реципиенту). В плазме крови каждого человека содержатся антитела против антигенов, содержащихся на мембранах эритроцитов чужеродной крови. При переливании крови антитела крови реципиента будут связываться с антигенами эритроцитов донорской крови и образовывать скопления слипшихся эритроцитов крови донора. Образование таких сгустков эритроцитов крови донора называется реакцией агглютинации. Слипшиеся эритроциты донора препятствуют циркуляции крови в мелких кровеносных сосудах реципиента и могут вызвать смертельный исход. Поэтому перед переливанием крови следует обязательно провести тест на совместимость крови донора и реципиента. Для этого смешивают по капле кровь донора и реципиента на предметном стекле и наблюдают, не произойдет ли реакция агглютинации — появление зернистых скоплений эритроцитов реципиента. Группы крови человека системы АВО с вариантами возможного переливания крови от донора к реципиенту представлены в табл. 47.1. Например, если реципиенту с группой 0(1) переливают кровь донора группы А(П), то антитела реципиента (анти-А) связываются с антигеном А, находящимся на поверхности эритроцитов группы крови А(П), вызывая их агглютинацию. Скопления эритроцитов донора — группы А(П), слипшихся при участии антител анти-А, осаждаются и закупоривают мелкие сосуды реципиента.
Группы крови системы резус-фактора (Rh-фактора) отличаются от групп системы АВО тем, что антитела против Rh-фактора образуются в большом количестве только при попадании в организм чужеродных Rh-антигенов. Людей, у которых эритроциты несут Rh-антиген, называют Rh-положителъными (Rh+), а тех, у кого этот антиген отсутствует, — ВА\-отрицателъными (Rh“). Если Rlr-реципиенту перелить кровь Rh'-донора, то организм такого реципиента в течение 2 — 4 мес будет вырабатывать антитела, специфичные к Rh-
Таблица 47.1
Группы крови человека системы АВО
Группа крови Антигены мембраны эритроцитов Антитела плазмы крови Кому можно переливать От кого можно переливать
0(1) Нет Анти-А, Анти-В Всем группам Только от 0(1)
А(П) А Анти-В А(П) и AB(IV) А(П) и 0(1)
В(Ш) В Анти-А В(Ш) и AB(IV) В(П1) и 0(1)
AB(IV) А и В Нет AB(IV) От всех групп
фактору (Rh-антитела). Если затем этомуже реципиенту снова перелить кровь Rh'-донора, то Rh-антитела реципиента вызовут агглютинацию RlT-эритро-цитов донора, и эти скопления будут закупоривать кровеносные сосуды реципиента.
47.1.2. Специфичность антител
Лимфоциты человека вырабатывают миллионы различных антител. На ранних стадиях развития зародыша человека лейкоциты содержат такой же генетический материал, как и любая клетка организма, в том числе некоторое количество участков ДНК, потенциально представляющих собой части генов, ответственных за синтез антител. По мере возникновения специализированных клеток, образующих антитела, некоторые из этих участков ДНК перестраиваются и «сшиваются» друг с другом, образуя полноценный функционирующий ген. После этого лейкоцитарная клетка начинает вырабатывать антитела в соответствии с содержащейся в этом гене информацией. Выбор участков ДНК, которые войдут в состав такого полноценно функционирующего гена, варьирует в различных типах лейкоцитов. К концу развития зародыша человека организм содержит лимфоциты, производящие в малых количествах миллионы разных антител, хотя этот организм никогда еще не сталкивался с соответствующими антигенами. Однако иммунная система ребенка становится способной к полноценному иммунному ответу только через несколько месяцев жизни. Все это время на защите ребенка стоит так называемый пассивный иммунитет, возникающий во время внутриутробного развития плода, когда некоторое количество антител из крови матери попадает в кровь плода. Этот пассивный иммунитет защищает новорожденного от инфекций до вступления в действие собственной иммунной системы.
Лимфоцитарная клетка, вырабатывающая антитела, производит антитела только одного типа. Эта клетка несет на своей мембране образец этих антител, где он связывается с чужеродными антигенами, попадающими в организм. Антитела каждого типа способны связываться, как правило, только с определенным антигеном, к которому оно специфично и подходит, как ключ к замку.
Чтобы в случае необходимости реагировать на внедрение чужеродного агента, иммунная система должна распознавать все клетки и вещества собственного организма. Не только эритроциты, но и большинство других клеток организма вырабатывают антигены, размещающиеся на их клеточной мембране. Состав антигенов клеточной поверхности у человека кодируется рядом генов. У монозиготных близнецов набор антигенов клеточных мембран полностью идентичен, а у близких родственников он различается незначительно. У чужих индивидуумов набор антигенов их организмов различается и является специфичным.
47.1.3. Иммунный ответ и вакцинация
Чужеродный антиген вызывает в организме ряд реакций, называемых иммунным ответом. Различают несколько типов иммунного ответа, каждый из которых обеспечивается функционированием лейкоцитов определенного типа. Гуморальный ответ осуществляется за счет специфических антител —иммуно
глобулинов, выделяющихся лимфоцитами в кровь. При клеточном ответе происходят сложные взаимодействия между различными лейкоцитами иммунной системы: накапливается большое количество активных Т-лимфоцитов, действующих либо непосредственно, либо через выделяемые ими вещества — лим-фокины (Т-эффекторы). Иммунологическая память и иммунологическая толерантность также считаются типами иммунного ответа. Любой тип иммунного ответа — клеточный, гуморальный, иммунологическая память и иммунологическая толерантность — это результат функционирования и взаимодействия разновидностей лейкоцитов — макрофагов (macro—большой; phage —пожиратель), Т- и В-лимфоцитов. Макрофаги поглощают мертвые клетки и агрегаты из слипшихся антигенов и антител, и организм очищается от чужеродного антигена.
Примером гуморального иммунного ответа могут служить прививки против инфекционных болезней, осуществляемые с помощью вакцин. Так, вакциной против дифтерии служит дифтерийный анатоксин — ослабленный действием высокой температуры экзотоксин, выделяемый возбудителем дифтерии Corynebacterium diphterie. Являющиеся антигенами молекулы анатоксина связываются со специфичными для них антителами, находящимися на поверхности лимфоцитов организма. Реакция связывания служит для лимфоцитов, производящих антитела, сигналом к их росту, делению, образованию и выделению в кровь огромного количества антител определенного типа. Процесс синтеза специфических антител при вакцинации является сложным процессом и обеспечивается по крайней мере несколькими типами лейкоцитов — антиген-представляющими макрофагами, Т-хелперами и В-лимфоцитами.
Клеточный иммунный ответ лежит в основе воспалительных процессов, противовирусного, противоопухолевого и трансплантационного иммунитетов. Главную роль при клеточном иммунном ответе играют цитотоксические лимфоциты — Т-киллеры. В развитии клеточной реакции иммунного воспаления, представляющей собой сложную цепь межклеточных взаимодействий, участвуют макрофаги, Т-хелперы и Т-эффекторы и другие типы клеток. Иммунный ответ в основном клеточного типа наблюдается при отторжении органа, например кожи, пересаженной от одного человека к другому. При пересадке кожи антигены донорских клеток связываются с антителами клеток реципиента. Клетки реципиента, связавшиеся с клетками донора, выделяют вещество интерлейкин, привлекающее к месту пересадки кожи макрофагов. Кроме того, клетки реципиента, связавшись с клетками донора, начинают усиленно расти, делиться и производить все новые антитела против чужеродного антигена. В результате пересаженный участок кожи разрыхляется, под ним скапливается гной и пересаженная ткань отмирает.
Во время внутриутробного развития некоторое количество антител из крови матери попадает в кровь плода и возникает пассивный иммунитет. Своеобразным вариантом создания пассивного иммунитета является введение больному антисыворотки, например в случае попадании в организм большой дозы антигена в виде яда при укусе ядовитой змеи. Змеиная антисыворотка готовится из крови какого-нибудь животного, чаще всего лошади, периодически получающей змеиный яд в малых дозах. Организм лошади вырабатывает при этом антитела, которые при введении их человеку в виде препарата антисыворотки способны обезвредить антиген — змеиный яд.
47.1.4. Аллергия
Иммунный ответ организма может быть не только механизмом защиты. Он может также служить причиной патологических процессов.
Аллергией называют неадекватную иммунную реакцию на безвредные вещества, содержащиеся в окружающей среде, пищевых продуктах и лекарственных препаратах. Это может быть пыльца растений, молоко, шоколад, некоторые антибиотики и многое другое. Антиген, способный вызвать сенсибилизацию организма и индуцировать в нем аллергическую реакцию, называют аллергеном. Обычно впервые попавший в организм аллерген не вызывает болезненных симптомов, но запускает иммунную реакцию, так что при повторном его проникновении развивается вторичная реакция. Аллергическая реакция может быть очень сильной и сопровождаться повреждением различных тканей организма. В зависимости, от преимущественной роли тех или иных патологических механизмов в развитии аллергических реакций их делят на 4 основных типа. Одними из самых распространенных являются аллергические реакции типа 1, или анафилактические. Их обычно вызывают экзогенные аллергены — пыльца цветущих растений, пищевые аллергены и др. Такие аллергены при первом контакте с организмом активируют особую субпопуляцию специфических Т-хелперов, которые в свою очередь активируют В-лимфоциты, вырабатывающие антитела IgE.
Для развития анафилактических реакций большое значение имеет наследственная предрасположенность к гиперпродукции антител IgE в ответ на сенсибилизацию определенным аллергеном, поэтому у близких родственников часто бывает аллергия на один и тот же фактор. Лабораторная диагностика аллергий проводится с учетом механизмов аллергических реакций. Прежде всего определяют тип аллергии и идентифицируют аллерген. С этой целью, например, при реакциях анафилактического типа ставят кожно-аллергические пробы с аллергенами из стандартных наборов — пищевых, лекарственных, пылевых, пыльцовых и др. В месте кожной пробы через 25 — 40 мин возникает покраснение. Выраженность этой реакции зависит от степени сенсибилизации организма. Дальнейшая диагностика при реакциях типа I заключается в выявлении антител, относящихся к классу IgE и содержащихся в сыворотке крови.
47.1.5. СЕРОЛОГИЧЕСКИЕ РЕАКЦИИ
Реакции иммунитета легли в основу лабораторных методов диагностики инфекционных и неинфекционных заболеваний. Так, они используются для идентификации возбудителей болезней и других антигенов, для выявления специфических антител, определения групп крови, подбора адекватного донора при пересадке органов и тканей.
Серологические реакции, или реакции между антителами и антигенами in vitro, применяют для выявления неизвестного антигена или антитела с помощью известного антитела или антигена соответственно.
В диагностических целях серологические реакции применяются для серодиагностики инфекционных заболеваний — выявления антител с помощью набора известных антигенов и для определения вида или серовара микроорганизма (антигена) с помощью диагностических антисывороток.
Серологические реакции дают возможность судить о динамике накопления антител в процессе болезни и об активности (напряженности) иммунитета, возникающего после профилактических прививок, например вакцинаций.
Серологические реакции характеризуются специфичностью и чувствительностью. Специфичность — это способность антигенов реагировать только с гомологичными антителами. Чувствительность измеряется минимальным количеством антигенов или антител, которое может выявить данная реакция.
Основными методами оценки иммунного статуса человека являются: оценка функциональной активности фагоцитирующих клеток; титрование лизоцима в слюне; титрование комплемента сыворотки крови; определение спонтанного розеткообразования с эритроцитами барана (Е-РОК) и с эритроцитами барана, обработанными антителами и комплементом (ЕАС-РОК); иммунофлюо-ресцентная идентификация Т-хелперов и Т-супрессоров; радиальная иммунодиффузия по Манчини. Так, для титрованием лизоцима в слюне готовят ряд последовательных разведений слюны в пробирках и в каждую из них вносят по 1 мл суспензии суточной культуры Micrococcus luteus, содержащей 1 млрд кле-ток/мл. После инкубации в термостате в течение 3 сут определяют титр лизоцима по последнему разведению, в котором наблюдается полный лизис бактерий в виде просветления. Более точную оценку активности лизоцима можно проводить, измеряя спектрофотометрическим методом оптическую плотность микробной взвеси после инкубации ее с разведениями лизоцима. Пользуясь калибровочными кривыми, активность лизоцима можно выразить в мкг/мл. Уровень содержания лизоцима в слюне здоровых людей составляет 50—70 мкг/мл, в сыворотке крови — от 5 до 20 мкг/мл.
Для выявления и идентификации специфических антигенов пользуются различными вариантами реакций агглютинации, преципитации, иммуноэлектрофорезом, иммунофлюоресцентным и иммуноферментным методами, радио-иммунологическим анализом. Так, применение иммунофлюоресцентного метода обнаружения бактериальных и вирусных антигенов в инфекционных материалах основано на использовании флюоресцирующих антител (сывороток). Данный метод относится к экспресс-диагностике. Приготовление сывороток основано на способности некоторых флюорохромов (например, изотиоцианата флюоресцеина) вступать в химическую связь с сывороточными белками, не нарушая их иммунологической специфичности. При прямом методе Кумбса специфические флюоресцирующие антитела образуют комплексы с микробными антигенам, которые светятся при люминесцентной микроскопии препаратов.
Недостатком прямого метода является необходимость приготовления широкого набора флюоресцирующих специфических сывороток против каждого изучаемого антигена. Непрямой метод предусматривает использование только одной универсальной антиглобулиновой сыворотки, содержащей антитела только против кроличьих глобулинов. Диагностические антисыворотки, полученные путем иммунизации кроликов, также содержат кроличьи глобулины, поэтому они в качестве антигенов реагируют с флюоресцирующей антиглобулиновой сывороткой и вместе с тем в качестве антител соединяются со специфическими изучаемыми антигенами микроорганизмов. При этом на образующемся комплексе «специфические антитела + исследуемый антиген» фиксиру
ются антиглобулиновые антитела, вызывая его свечение при люминесцентной микроскопии.
В целях выявления специфических антител применяют реакции агглютинации, непрямой гемагглютинации, преципитации в геле, лизиса, связывания комплемента, реакцию Кумбса, флоккуляции, торможения гемагглютинации, антистрептолизиновую реакцию и иммуноферментный анализ. Последний метод является одним из наиболее чувствительных для выявления специфических антител. Специфический антиген адсорбируют на поверхности лунок из полистирола. Фиксированный в лунках антиген инкубируют с испытуемыми человеческими сыворотками. После отмывания от несвязавшихся белков связанные иммобилизованными антигенами иммуноглобулины выявляют с помощью антивидовой (античеловеческой) антииммуноглобулиновой сыворотки, к антителам которой ковалентно прикреплен фермент — пероксидаза. После инкубации с субстратом (пероксидом водорода) и индикатором (диаминобензидином) оценивают ферментативную активность по интенсивности окраски. Интенсивность окраски, пропорциональную количеству сорбированных на антигене антител, можно оценить визуально или на спектрофотометре. В данном методе удобна универсальность меченого реагента, позволяющая выявлять антитела к разным антигенам.
ПРИЛОЖЕНИЯ
ПРИЛОЖЕНИЕ 1
ПРИМЕРНАЯ ПРОГРАММА ПРАКТИЧЕСКИХ ЗАНЯТИЙ ПО МИКРОБИОЛОГИИ ДЛЯ СТУДЕНТОВ БИОЛОГИЧЕСКИХ ФАКУЛЬТЕТОВ УНИВЕРСИТЕТОВ
Программа разработана для проведения практических занятий по микробиологии для студентов всех специальностей биологических факультетов и вузов. Программа включает следующие темы для изучения:
Тема 1. Микроскопические методы исследования микроорганизмов (занятия 1 — 3).
Тема 2. Приготовление питательных сред и методы стерилизации (занятие 4).
Тема 3. Получение накопительных и чистых культур микроорганизмов (занятия 5—6).
Тема 4. Идентификация микроорганизмов (занятия 7—11).
Тема 5. Методы количественного учета микроорганизмов (занятие 12).
Студенты, выполняющие практические работы по предлагаемой программе, знакомятся с основами микробиологии на так называемом «малом» практикуме, который обязателен для всех специальностей студентов-биологов любых высших учебных заведений.
Программа рассчитана на 11 практических занятий (по 3 академических часа каждое) и 3 семинарских занятия (по 2 академических часа каждое), т.е. на период одного семестра.
На некоторых занятиях студенты изучают две темы. На практических занятиях студенты овладевают техникой микробиологической работы в лаборатории и готовят теоретические разделы программы для участия в семинарских занятиях. На семинарах студентам целесообразно предлагать выступить с докладами на означенные темы или на другие темы по выбору преподавателя.
ТЕМА 1. МИКРОСКОПИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ МИКРООРГАНИЗМОВ (ЗАНЯТИЯ 1—3)
Занятие 1
Цель занятий 1—3: знакомство с морфологией и цитологией разных микроорганизмов. Для этого студентам необходимо изучить устройство микроскопа, ознакомиться с особенностями разных видов микроскопии (светлопольной, темнопольной, фазово-контрастной, люминесцентной) и научиться готовить препараты микроорганизмов.
1. ПРИГОТОВЛЕНИЕ ПРЕПАРАТОВ ЖИВЫХ МИКРООРГАНИЗМОВ И ЗНАКОМСТВО С ИХ МОРФОЛОГИЕЙ
ВВЕДЕНИЕ. ПРАВИЛА РАБОТЫ В МИКРОБИОЛОГИЧЕСКОЙ ЛАБОРАТОРИИ
Препараты готовят на обезжиренных предметных стеклах. Живые микроорганизмы рассматривают в препаратах «раздавленная капля», «висячая капля» и «отпечаток».
Для приготовления препарата «раздавленная капля» на предметное стекло наносят каплю водопроводной воды и помещают в нее с помощью простерилизованной в пламени горелки бактериологической петли небольшое количество культуры изучаемого микроорганизма, выросшей на твердой питательной среде, размешивают и накрывают покровным стеклом. Каплю культуры, выращенной в жидкой питательной среде, наносят на предметное стекло стерильной пипеткой. Капля исследуемого материала должна быть настолько мала, чтобы после прижимания ее покровным стеклом не было избытка жидкости, выступающего из-под него. Избыток жидкости удаляют фильтровальной бумагой, которую затем опускают в дезинфицирующий раствор.
Препараты живых клеток рассматривают с «сухими системами» микроскопа (8х и 40х).
Задание. Приготовить препараты «раздавленная капля» микроорганизмов с различной морфологией: кокки {Micrococcus luteus)-, стрептококки {Leuconostoc mesenteroides)', палочковидные бактерии {Lactobacillus delbrueckii)', извитые формы {Rhodospirillum rubrum)', нитчатые формы {Oscillatoria sp., Spirulina sp., Anabaena variabilis, Fischerella muscicola). Просмотреть и зарисовать микроорганизмы, отметить форму и сочетание клеток.
Примечание'. Вместо указанных видов могут быть использованы другие организмы с соответствующей морфологией. Часть микроорганизмов может быть продемонстрирована в виде готовых препаратов.
Задание на долг, с. 24—30, 56 — 61, 69 — 73.
Занятие 2
2. ПРИГОТОВЛЕНИЕ ФИКСИРОВАННЫХ ПРЕПАРАТОВ МИКРООРГАНИЗМОВ И ЗНАКОМСТВО С ИХ МОРФОЛОГИЕЙ
На обезжиренное предметное стекло помещают каплю водопроводной воды и вносят в нее бактериологической петлей культуру с твердой среды. Каплю жидкой культуры наносят на предметное стекло пипеткой. Равномерно размазывают петлей каплю с микроорганизмами тонким слоем, высушивают на воздухе и фиксируют.
Цели фиксации', убить микроорганизмы; обеспечить лучшее прилипание клеток к стеклу; сделать микроорганизмы более восприимчивыми к окраске, так как мертвые клетки окрашиваются лучше живых.
Самым распространенным способом фиксации микроорганизмов является термическая обработка. Для этого высушенный препарат обычно трижды проводят через наиболее горячую часть пламени горелки, держа предметное стекло мазком вверх. При исследовании строения микроорганизмов часто прибегают к фиксации клеток различными химическими веществами (спирт, осмиевая кислота, формалин и др.). Фиксирующую жидкость наливают на высушенный препарат, либо препарат на определенное время (3— 15 мин) погружают в стакан с фиксатором.
Для окраски фиксированных микроорганизмов чаще всего используют анилиновые красители. Существуют простые и дифференциальные способы окрашивания микроорганизмов. При простой окраске прокрашивается вся клетка так, что становится хо
рошо видна ее форма и размеры. Дифференциальная окраска предполагает окрашивание не всей клетки, а только определенных ее структур или включений. Для окрашивания клеток фиксированный препарат помещают на параллельные стеклянные рейки, лежащие над кюветой, и заливают красителем на 1 — 15 мин. Следят за тем, чтобы во время окрашивания раствор красителя на мазке не подсыхал. В случае необходимости на мазок наливают новые порции красителя.
По окончании окраски препарат промывают водой, высушивают, помещают на окрашенный мазок каплю иммерсионного масла и просматривают с объективами 90х или 100х.
Задание. Приготовить фиксированные препараты и окрасить фуксином клетки следующих микроорганизмов: палочковидные бактерии — Pseudomonas sp.; изогнутые и ветвящиеся бактерии — Mycobacterium flavescens, Nocardia sp.; мицелиальные формы, образующие экзоспоры — Streptomyces sp.; бактерии, образующие выросты — Caulobacter crescentus (стебельки), и простеки — Rhodomicrobium vannielii. Просмотреть и зарисовать микроорганизмы, отметить форму и сочетание клеток.
Задание на дом: с. 74—81.
Занятие 3
3. ОБНАРУЖЕНИЕ КАПСУЛ МЕТОДОМ НЕГАТИВНОГО КОНТРАСТИРОВАНИЯ
Небольшое количество клеток Azotobacter chroococcum с твердой питательной среды помещают петлей на предметное стекло в каплю водопроводной воды, смешивают с каплей туши, накрывают покровным стеклом и просматривают с объективом 40х. На сером фоне препарата хорошо видны бесцветные капсулы, окружающие клетки.
4. ОКРАСКА ЭНДОСПОР
Небольшое количество клеток Bacillus cereus или другого вида спорообразующих бактерий с твердой среды помещают петлей на предметное стекло в каплю водопроводной воды и делают мазок. Мазок высушивают на воздухе, фиксируют в пламени горелки и заливают раствором метиленового синего по Леффлеру. Краситель доводят до кипения, держа предметное стекло пинцетом над пламенем горелки. По мере испарения красителя добавляют новые его порции. Продолжительность окраски трехкратная, по 10—15 с. Затем предметное стекло охлаждают, препарат тщательно промывают над эксикатором водой, после чего клетки в течение 30 с докрашивают 0,5%-м водным раствором сафранина. Краситель сливают, препарат промывают водой, сушат и просматривают с иммерсионной системой (90х — ЮОх). При правильном окрашивании вегетативные клетки имеют красный, а споры синий цвет. Вместо метиленового синего можно использовать другой краситель, например малахитовый зеленый.
В этом случае после фиксации препарат заливают на 7—10 мин 7,5%-м раствором малахитового зеленого. Окрашивание проводят с подогревом, помещая препарат над сосудом с кипяшей водой, и докрашивают клетки 0,25%-м водным раствором сафранина в течение 1 — 2 мин. Споры окрашиваются в зеленый цвет, а вегетативные клетки в розовый.
Эндоспоры можно также обнаружить с помощью фазово-контрастной микроскопии или методом простого окрашивания (например, фуксином). Они обладают многослойными труднопроницаемыми для красителей оболочками, поэтому обнаруживаются в клетке в виде бесцветных включений.
5. ВЫЯВЛЕНИЕ ВКЛЮЧЕНИЙ ПОЛИФОСФАТОВ (СИНОНИМЫ: ВОЛЮТИН, МЕТАХРОМАТИН)
Метод окраски основан на свойстве метахромазии — изменении цвета некоторых красителей (например, метиленового синего) при взаимодействии с определенным компонентом клетки. Готовят тонкий мазок клеток Lactobacillus delbrueckii subsp. bulgaricus на предметном стекле, высушивают на воздухе, фиксируют в пламени горелки, заливают раствором метиленового синего по Леффлеру и окрашивают в течение 10—15 мин. Краситель сливают, препарат промывают водой, высушивают на воздухе и просматривают с иммерсионной системой (90— ЮОх). При этом клетки окрашиваются в голубой цвет, а гранулы полифосфатов в фиолетово-красный.
6. ВЫЯВЛЕНИЕ ВКЛЮЧЕНИЙ ГЛИКОГЕНОПОДОБНЫХ ПОЛИСАХАРИДОВ
К капле слегка подкисленной суспензии клеток дрожжей Saccharomyces cerevisiae на предметном стекле добавляют каплю раствора Люголя. Препарат накрывают покровным стеклом и микроскопируют с объективом 40*. Включения гликогеноподобного полисахарида окрашиваются в красно-коричневый цвет.
7. ВЫЯВЛЕНИЕ ВКЛЮЧЕНИЙ СЕРЫ
Благодаря двойному светопреломлению включения серы хорошо заметны в клетках Beggiatoa sp., Chromatium minutissimum или некоторых других бактерий без специального окрашивания, для чего необходимо приготовить препарат «раздавленная капля» и про-микроскопировать его, пользуясь объективом 40х.
Задание на дом: с. 31—36, 45 — 56.
ТЕМА 2. ПРИГОТОВЛЕНИЕ ПИТАТЕЛЬНЫХ СРЕД И МЕТОДЫ СТЕРИЛИЗАЦИИ
Занятие 4
Цель занятия: приготовление питательной среды для выращивания микроорганизмов, знакомство с методами стерилизации сред, посуды и инструментов.
Задание 1. Приготовить по 150 — 200 мл питательной среды (БСА), содержащей мясопептонный бульон (МПБ) и 4 °Б (с концентрацией сахара 4 градуса, определенной с помощью ареометра Баллинга) неохмеленное пивное сусло (в соотношении 1:1) с 2% агара. Среду готовят в колбе объемом 300 — 700 мл. Колбу с готовой средой ставят в кипящую водяную баню. После того, как агар расплавится и среда станет гомогенной, ее разливают в 7 —8 широких пробирок (по 20 мл) и в 5 —8 узких пробирок (по 5 мл). Пробирки с питательной средой закрывают ватными пробками и стерилизуют в автоклаве при 0,5 ати. Среду после стерилизации используют при дальнейшей работе с микроорганизмами.
Задание 2. Налить водопроводную воду в 4—5 узких пробирок (по 10 мл). Пробирки закрывают ватными пробками и стерилизуют в автоклаве при 1 ати. Стерильную воду используют затем для приготовления суспензий микроорганизмов.
Задание 3. Подготовить к стерилизации сухим жаром:
чашки Петри — 9 шт. (по 3 в пачку); пипетки на 1 —2 мл — 4 — 5; на 5 мл — 1—2 шт.; пробирки обычные — 4 шт.; стеклянные шпатели (Дригальского) — 3 шт. Посуду и инструменты стерилизуют в сушильном шкафу при 165—170° С в течение 2 ч.
Задание 4. Сделать ватно-марлевые пробки для нескольких пробирок.
Задание 5. Познакомиться с устройством автоклава.
Задание на дом: с. 36 — 45, 93—101, 159—167.
ТЕМА 3. ПОЛУЧЕНИЕ НАКОПИТЕЛЬНЫХ И ЧИСТЫХ КУЛЬТУР МИКРООРГАНИЗМОВ
Занятия 5—6
Цель занятий 5—6: получение накопительной и чистой культуры микроорганизмов.
1. ПОЛУЧЕНИЕ НАКОПИТЕЛЬНОЙ КУЛЬТУРЫ МИКРООРГАНИЗМОВ
1.1. Свободноживущие аэробные азотфиксирующие бактерии
Занятие 5. Используют среду Эшби, не содержащую соединений азота, (г/л): маннит - 20,0; К2НРО4 - 0,2; MgSO4 - 0,2; NaCl - 0,2; K2SO4 - 0,1; СаСО3 - 5,0; агар -20,0. Вода дистиллированная. Среду в колбе расплавляют на водяной бане, разливают в стерильные чашки Петри и дают остыть на горизонтальной поверхности. Затем, пользуясь стеклянным капилляром, смоченным в воде, раскладывают на поверхность среды в чашке (по трафарету) 15 — 25 комочков почвы величиной с булавочную головку. Чашку Петри закрывают и ставят в термостат (30 °C) на 3 — 5 сут. Вместо агара в качестве уплотнителя среды можно также использовать силикагель. В этом случае готовят 10-кратно концентрированную среду Эшби. Этой средой пропитывают пластинки силикагеля (2—3 мл на чашку), подсушивают в сушильном шкафу (10—15 мин при 60 °C) и раскладывают комочки почвы.
Занятие 6. Проверяют получение накопительной культуры азотфиксирующих бактерий по образованию слизистых колоний (при длительном росте часто окрашенных в коричневый цвет) вокруг комочков почвы на чашках.
Микроскопируют накопительную культуру с использованием метода негативного контрастирования (см. занятие 3). Обнаруживают клетки бактерий (обычно представителей рода Azotobacter или Beijerinckia), окруженные слизистой капсулой.
1.2. Свободноживущие анаэробные азотфиксирующие бактерии
Занятие 5. Используют среду Виноградского следующего состава (г/л): глюкоза — 20,0; К2НРО4 -1,0; MgSO4 - 0,5; NaCl - 0,5; MnSO4 и FeSO4 следы; СаСО3 - 20,0. Вода, водопроводная. В высокие пробирки вносят небольшое (0,5—1,0 г) количество почвы, наливают среду Виноградского, оставляя воздушное пространство в 1 см между пробкой и средой, и пастеризуют в течение 10 мин при 80 °C (в водяной бане). Пробирки помешают в термостат (30 °C) на 7 сут. На этой среде, как правило, вырастают клостридии, обычно Clostridium pasteurianum.
Занятие 6. Проверяют получение накопительной культуры азотфиксирующих клостридий по газовыделению и запаху масляной кислоты (контролем служит исходная стерильная среда).
Микроскопируют накопительную культуру. Отмечают наличие подвижных палочек, образующих споры. В клетках бактерий выявляют характерные гранулы крахмалоподобного вещества (гранулезы). Для этого к капле суспензии клеток на предметном стекле добавляют каплю раствора Люголя. Препарат накрывают покровным стеклом и микроскопируют с объективом 40 х. Включения гранулезы окрашиваются в синий цвет.
1.3. Аэробные спорообразующие бактерии
Вариант 1. Занятие 5. Нарезают неочищенный клубень картофеля на мелкие ломтики, помещают их в колбу объемом 100— 150 см, добавляют небольшое количество СаСО3 (на кончике шпателя), заливают ломтики водопроводной водой (с таким расчетом, чтобы вода покрывала их примерно наполовину), закрывают колбочку ватной пробкой и прогревают в кипящей водяной бане 10—15 мин. При этом вегетативные клетки бактерий, находящихся на поверхности клубней картофеля, погибают, а жизнеспособными обычно остаются лишь термостабильные споры. Колбы помещают в термостат (30 °C) на 5 —7 сут.
Занятие 6. Проверяют получение накопительной культуры спорообразующих бактерий (обычно рода Bacillus) по появлению на поверхности среды морщинистой пленки.
Для микроскопирования готовят препараты фиксированных окрашенных клеток. Отмечают наличие эндоспор. Для этого препарат фиксированных клеток окрашивают фуксином в течение 2 — 3 мин. Цитоплазма окрашивается в розово-красный цвет, а спора не окрашивается фуксином и видна в клетке в виде бесцветного включения. Можно также провести дифференциальную окраску спор и цитоплазмы (занятие 3).
Вариант 2. Занятие 5. Помещают 1 г сена в колбочку объемом 100—150 мл и заливают 20 — 30 мл водопроводной воды, нагретой до 40 — 50°С. Сено настаивают в течение 30 мин. За это время из сена экстрагируются вещества, которые служат питательным субстратом для бактерий. Через 30 мин сенной настой отделяют от сена декантацией, разливают в 2 пробирки (примерно по 5 — 7 мл), закрывают их ватными пробками и прогревают в кипящей водяной бане 15 — 20 мин. При этом лишь споры некоторых бактерий остаются жизнеспособными. Колбы помещают в термостат (30 °C) на 7 сут.
Занятие 6. Проверяют получение накопительной культуры спорообразующих бактерий (обычно Bacillus subtilis) по наличию в накопительном материале эндоспор методом микроскопирования (см. вариант 1).
1.4. Уробактерии
Занятие 5. К 25 мл мясопептонного бульона (МПБ) добавляют 2,5 г (10%) мочевины. Полученную среду разливают в обычные пробирки (по 5 — 6 мл) и инокулируют небольшим количеством навоза. В такой среде развиваются бактерии (обычно Bacillus pasteurii, Sporosarcina игеае или Proteus vulgaris), способные разлагать мочевину с помощью уреазы согласно уравнению: CO(NH2)2 + Н2О £> СО2 + 2NH3 В результате pH среды повышается до тех значений (9—10), при которых сохраняют способность расти преимущественно уробактерии. Большинство других бактерий из-за высокого значения pH не растут. Для получения накопительной культуры уробактерий пробирки помещают в термостат (30 °C) на 7 сут.
Занятие 6. Проверяют рост накопительной культуры уробактерий по выделению аммиака и подщелачиванию среды (контролем служит исходная стерильная среда).
Микроскопируют накопительную культуру с фазово-контрастным устройством в препарате «раздавленная капля». Отмечают морфологические особенности выросших бактерий, их подвижность, наличие в клетках спор (см. занятие 3).
1.5. Метилотрофы
Занятие 5. Метилотрофы, использующие метанол, легко выделяются из сточных вод и других источников, где этот субстрат часто присутствует. Для получения накопительной культуры метилотрофных бактерий используют синтетическую среду Канеда следующего состава (г/л): КН2РО4 — 2,0; (NH4)2SO4 — 2,0; NaCl — 0,5; MgSO4 — 0,025; FeSO4 — 0,01. Вода водопроводная. В качалочную колбу объемом 750 мл помещают 100 мл среды, 4 капли 10%-го дрожжевого экстракта и 0,3%-го (по объему) метанола. С помощью 10%-х растворов NaOH и НС1 доводят значение pH среды до 7,0. Затем в колбу вносят 3 — 4 мл сточной жидкости или другой образец природного материала. Колбу помещают на качалку (200 об/мин) и оставляют при 30 °C на несколько суток.
Занятие 6. Проверяют получение накопительной культуры метилотрофных бактерий по помутнению и часто появляющемуся розовому окрашиванию среды (контролем служит исходная стерильная среда).
Микроскопируют накопительную культуру в препарате «раздавленная капля». Отмечают морфологические особенности выросших бактерий, их подвижность.
1.6. Нитрифицирующие бактерии
Занятие 5. Используют среду Виноградского следующего состава (г/л): (NH4)4SO4 — 2,0; К2НРО4 —1,0; MgSO4 — 0,5; FeSO4 — 0,4; NaCl — 2,0. Вода водопроводная. Среду разливают в колбы объемом 50 мл по 15 мл в каждую (высота слоя среды должна быть 1,0— 1,5 см). В каждую колбу вносят на кончике шпателя небольшое количество СаСО3. Посевным материалом служит почва. Колбы помешают в термостат на 30 °C.
Через 1 — 3 недели проверяют получение накопительной культуры нитрификаторов по появлению в среде нитритов и (или) нитратов. Для их обнаружения проводят качественные реакции на нитриты с реактивом Грисса или с помощью крахмал-йодной реакции и на нитраты с дифениламином. Развитие нитрифицирующих бактерий приводит также к подкислению среды.
• Качественные реакции на нитриты. Для обнаружения нитритов к капле реактива Грисса на керамической пластинке добавляют каплю выросшей жидкой культуры. Появление красного окрашивания свидетельствует о присутствии нитритов.
• Крахмал-йодная реакция основана на том, что нитриты в кислой среде окисляют йодистый цинк с выделением йода, присутствие которого обнаруживают с крахмалом. Для проведения реакции к капле выросшей жидкой культуры на керамической пластинке добавляют каплю раствора, содержащего ZnCI2, KI, крахмал и каплю 5%-го раствора НС1. При наличии в среде нитритов появляется синее окрашивание.
• Качественная реакция на нитраты. На белой керамической пластинке несколько кристаллов дифениламина растворяют в капле концентрированной серной кислоты и к смеси добавляют каплю исследуемой жидкой культуры. При наличии нитратов возникает синее окрашивание. Контролем служит исходная стерильная среда. Следует иметь в виду, что пробу на нитраты можно проводить только в отсутствие нитритов, так как последние тоже дают синее окрашивание с дифениламином.
Изменение значения pH среды определяют с помощью бромтимолблау (контролем служит исходная стерильная среда).
Микроскопируют накопительную культуру в препарате «раздавленная капля». Отмечают морфологические особенности выросших бактерий и их подвижность.
1.7. Денитрифицирующие бактерии
Занятие 5. Используют среду следующего состава (г/л): лимоннокислый калий или натрий (трехзамещенный) — 5,0; KNO3 — 2,0; КН2РО4 — 2,0; MgSO4 — 2,0; СаСО3 — 0,2;
аспарагин —1,0; FeCI3 — следы, вода водопроводная. Исходное значение pH среды — 6,8 —7,2. Среду разливают в высокие пробирки, оставляя воздушное пространство в 1 см между пробкой и средой. Предварительно в каждую пробирку помещают стеклянный поплавок (запаянным концом вверх) так, чтобы он был полностью заполнен средой. Посевной материал — почва. Среду в одной пробирке оставляют незасеянной для контроля. Пробирки помещают в термостат (30 °C) на 1 — 2 нед.
Занятие 6. Проверяют рост накопительной культуры денитрифицирующих бактерий по газовыделению (пузырек воздуха в поплавке), отсутствию в среде нитратов, подщелачиванию среды (контролем служит исходная стерильная среда). О наличии или отсутствии нитратов судят по качественной реакции с дифениламином (см. получение накопительной культуры нитрифицирующих бактерий). Изменение pH среды определяют с помощью бромтимолблау.
Микроскопируют накопительную культуру в препарате «раздавленная капля». Отмечают морфологические особенности выросших бактерий и их подвижность.
1.8. Сульфатредуцирующие бактерии
Занятие 5. Используют среду следующего состава (г/л): КН2РО4 — 0,5; NH4C1 — 1,0; (NH4)2SO4 — 7,0 или Na2SO4 — 4,5; СаС12 — 0,06; лактат натрия — 6,0; лимоннокислый натрий (трехзамещенный) — 0,3; NaCl — 5,0. Для удаления из среды растворенного кислорода ее кипятят в колбе в водяной бане в течение 5 мин и затем осторожно охлаждают водой. После этого в среду вносят остальные компоненты в следующем порядке (мл/л):
1) 0,1 %-й раствор FeSO4 в 1%-м растворе НО — 10,0;
2) дрожжевой экстракт — 10,0;
3) раствор микроэлементов (по Пфеннигу) — 1,0.
Далее доводят значение pH до 7,0 —7,5 с помощью 5%-х растворов NaHCO3 и НО и добавляют Na2S 9Н2О (5%-й раствор в 1%-м растворе NaHCO3) по каплям до образования сероватого оттенка в среде. После этого в среду немедленно вносят посевной материал (почву), разливают доверху в пробирки и закрывают их резиновыми пробками. Пробирки помещают в термостат (30 °C) на 7 сут.
Занятие 6. Проверяют получение накопительной культуры сульфатредуцирующих бактерий по выделению сероводорода, который, связываясь с присутствующим в среде железом, вызывает ее почернение (контролем служит исходная стерильная среда).
Микроскопируют накопительную культуру в препарате «раздавленная капля». Отмечают морфологические особенности выросших бактерий, наличие спор (см. занятие 3) и подвижность.
1.9. Фототрофные пурпурные бактерии
Занятие 5. Пурпурные бактерии могут быть выделены из различных пресных и соленых водоемов. Для выделения несерных пурпурных бактерий готовят среду следующего состава (г/л): NH4C1 — 1,0; КН2РО4 — 0,5; MgCl2 — 0,5; микроэлементы по Пфеннигу — 1 мл. К основному фону среды добавляют (г/л): NaHCO3 _ 2,0; Na2S — 0,15; ацетат, лактат или малат натрия — 2,0; дрожжевой экстракт — 0,1. Исходное значение pH среды доводят до 7,0 с помощью 10%-х стерильных растворов NaOH и Н3РО4.
В пробирку (или колбу) с притертой пробкой вносят 5 —10 % (по объему) посевного материала (воду или ил из различных водоемов) и доливают до пробки средой так, чтобы между средой и пробкой не оставалось воздушного пространства. Пробирки (или колбы) помещают в люминостат (800—1000 люкс, 28 —30°C) на 1 — 2 нед.
Занятие 6. Проверяют получение накопительной культуры пурпурных бактерий по окрашиванию среды в розовый, красный или коричневатый цвет, благодаря наличию большого количества каротиноидов.
Микроскопируют накопительную культуру в препарате «раздавленная капля». Отмечают морфологические особенности выросших бактерий, их подвижность.
1.10. Микроорганизмы, использующие углеводороды
Занятие 5. Используют среду следующего состава (г/л): KNO3 — 4,0; КН2РО4 — 0,6; Na2HPO4 — 1,4; MgSO4 — 0,8; вода водопроводная. Исходное значение pH среды доводят до 6,8 —7,2 растворами NaOH и НС1 (проверяют по бромтимолблау). В две качалочные колбы объемом 750 мл помещают по 100 мл среды. В одну из колб вносят около 1 г почвы, отобранной у бензоколонки, и 2 мл керосина, а в другую — такое же количество почвы и 2 мл вазелинового масла. Колбы закрывают ватными пробками и помещают на качалку (220 об/мин) при 30 °C на 5 — 7 сут.
Занятие 6. Проверяют получение накопительной культуры углеводородокисляю-щих микроорганизмов по помутнению среды и наличию пленки на ее поверхности (контролем служит исходная стерильная среда).
Микроскопируют накопительную культуру в препаратах «раздавленная капля» и фиксированных окрашенных (фуксином) клеток. Отмечают морфологические особенности выросших бактерий, их подвижность.
1.11. Аммонифицирующие бактерии
Занятие 5. Для получения накопительной культуры используют мясо-пептонный бульон. 30 мл среды наливают в колбу емкостью 100 мл, добавляют 0,3 г почвы и закрывают ватной пробкой. Для обнаружения аммиака над средой в колбе между стенкой и пробкой подвешивают красную лакмусовую бумажку, смоченную дистиллированной водой (бумажка не должна касаться среды). Сверху колбочку закрывают пергаментной бумагой и ставят в термостат (30 °C) на 4—6 сут.
Занятие 6. Проверяют получение накопительной культуры аммонифицирующих бактерий по посинению лакмусовой бумажки и запаху сероводорода.
Микроскопируют накопительную культуру в препаратах «раздавленная капля» и фиксированных окрашенных (фуксином) клеток. Отмечают морфологические особенности выросших бактерий, их подвижность.
Свойства всех полученных накопительных культур бактерий (1.1 —1.10) обобщают в табл. П.1.
Таблица П.1
Свойства накопительной культуры бактерий
Среда Культуральные признаки Морфология клеток
2. ВЫДЕЛЕНИЕ ЧИСТЫХ КУЛЬТУР МИКРООРГАНИЗМОВ
Для выделения чистых культур микроорганизмов из накопительных обычно используют метод Коха. Принцип его заключается в получении чистой культуры из отдельной колонии. Выделение чистых культур аэробных и факультативно анаэробных микроорганизмов проводят, рассевая накопительную культуру на поверхность твердой
питательной среды. В зависимости от свойств накопительных культур микроорганизмов для получения отдельных колоний используют разные среды:
• споровые аэробные бактерии — БСА (МПБ с суслом, 1: 1, + 2% агара);
• денитрифицирующие бактерии — БСА;
• уробактерии — МПА с 2 % мочевины;
• метилотрофные бактерии— агаризованную среду с метанолом;
• углеводородокисляющие бактерии — БСА;
• азотфиксирующие аэробные бактерии — агаризованную среду Эшби;
• аммонифицирующие бактерии — МПА.
Занятие 5. Посев проводят бактериологической петлей методом «истощающего штриха». Чашки с посевами бактерий (крышками вниз) помешают в термостат (30 °C) на 4 —6 сут.
Занятие 6. Выбирают и описывают 2 изолированные колонии микроорганизмов, выросшие на поверхности питательных сред.
Расплавляют на кипящей водяной бане соответствующие стерильные среды в пробирках (по 1—2 пробирки каждой среды) для разных бактерий (см. 2.1).
Скашивают среды и делают посев в них из двух описанных колоний микроорганизмов.
Посев проводят штрихом бактериологической петлей, простерилизованной в пламени горелки. Пробирки помещают в термостат (30 °C) на 3 — 5 сут.
После того, как культуры в пробирках вырастут, проверяют чистоту выделенных культур визуально (однородный рост по всему штриху) и микроскопированием (чистые культуры многих бактерий морфологически однородны, допустимо лишь некоторое варьирование размеров клеток).
Необходимо также провести рассев на твердую среду (БСА) в чашки Петри. Однородность колоний и совпадение их признаков с описанными ранее — свидетельство чистоты культуры.
Следует иметь в виду, что некоторые виды бактерий могут образовывать колонии разной морфологии в результате процесса диссоциации.
Задание на дом: с. 114—117, 132—142.
ТЕМА 4. ИДЕНТИФИКАЦИЯ МИКРООРГАНИЗМОВ (ЗАНЯТИЯ 7—10)
Цель занятий 7—10: знакомство с основными принципами определения микроорганизмов. В процессе выполнения практической работы каждый студент изучает свойства бактерий, необходимые для описания бактериального штамма и идентификации его до уровня рода.
Занятие 7
1. ОПРЕДЕЛЕНИЕ ЧИСТОТЫ ИДЕНТИФИЦИРУЕМОЙ БАКТЕРИИ И ИЗУЧЕНИЕ МОРФОЛОГИИ КЛЕТОК
Для проведения работы по идентификации микроорганизмов каждый студент получает одну культуру бактерий (на скошенной агаризованной среде в пробирке), которую затем проверяет на чистоту. Это осуществляют несколькими способами: визуально, высевом на питательные среды и микроскопией.
Просматривают характер роста полученной бактерии по штриху на поверхности скошенной агаризованной среды. Если рост по штриху неоднороден, то культура загрязнена. Затем культуру отсевают в пробирку на скошенную среду (МПА, БСА) для использования в дальнейшей работе, а также делают рассев на поверхность твердой среды в чашке Петри методом истощающего штриха для проверки на чистоту (по однородности выросших колоний). Засеянные пробирки и чашки помещают в термостат (30 °C) на 2—3 сут. Остаток исходной культуры бактерии в пробирке используют для проверки на чистоту методом микроскопии (по морфологической однородности популяции), а также изучения формы, взаимного расположения, подвижности клеток и их размеров. Микроскопируют культуру с использованием препаратов «раздавленная капля» и препарата фиксированных, окрашенных фуксином клеток (см. занятия 1 и 2). Результаты вносят в табл. П.2.
Таблица П.2
Свойства идентифицируемой бактерии
Признаки Результаты
Описание колонии
Форма
Размер, мм
Цвет
Край
Блеск
Поверхность
Профиль
Рисунок
Морфология клеток и цитология
Форма и расположение клеток
Подвижность
Наличие эндоспор
Окраска по Граму
Окраска на кислотоустойчивость
Физиолого-биохимические свойства
Рост на среде с глюкозой
Протеолитическая активность: рост на среде с желатином рост на среде с молоком
Амилазная активность: рост на среде с крахмалом Тест на каталазу Тест на оксидазу
Чувствительность к антибиотикам
2. КУЛЬТУРАЛЬНЫЕ СВОЙСТВА
На следующем занятии просматривают чашку Петри, засеянную суспензией идентифицируемой бактерии. Критерием чистоты культуры (при отсутствии диссоциации) является однородность выросших колоний. Описывают культуральные (макро-морфологические) свойства бактериальных колоний и результаты вносят в табл. П.2.
Задание на дом: с. 76, 118—129.
Занятие 8
3. ИЗУЧЕНИЕ ЦИТОЛОГИЧЕСКИХ СВОЙСТВ ИДЕНТИФИЦИРУЕМЫХ БАКТЕРИЙ
3.1. Наличие эндоспор
Наличие эндоспор в клетках определяют, как описано выше (см. занятие 3).
3.2. Окраска по Граму
На обезжиренном предметном стекле в каплях воды делают мазки трех микроорганизмов: в центре готовят мазок клеток исследуемой бактерии, а слева и справа — контрольных бактерий. Клетки одной из контрольных бактерий должны быть грампо-ложительными, например Micrococcus luteus, другой — грамотрицательными, например Escherichia coli.
Мазки следует готовить тонкими, чтобы клетки равномерно распределялись по поверхности стекла и не образовывали скоплений. Препараты высушивают на воздухе, фиксируют над пламенем горелки и окрашивают в течение 1 — 2 мин карболовым ген-циановым или кристаллическим фиолетовым. Затем краситель сливают и мазки обрабатывают 1 — 2 мин раствором Люголя до почернения. Сливают раствор Люголя, препарат обесцвечивают 0,5— 1,0 мин 96%-м этиловым спиртом, быстро промывают водой и дополнительно окрашивают 1 — 2 мин водным фуксином. Краситель сливают, препарат промывают водой, высушивают и микроскопируют с иммерсионной системой. При правильном окрашивании грамположительные бактерии имеют сине-фиолетовый, грамотрицательные — розово-красный цвет.
Для получения достоверных результатов необходимо готовить мазки для окраски по Граму из молодых, активно растущих (обычно односуточных) культур, так как клетки из старых культур иногда дают неустойчивую реакцию по Граму. Грамотрицательные бактерии могут выглядеть как грамположительные, если бактериальная пленка (мазок) слишком толста и обесцвечивание спиртом не проведено до конца. Грамположительные бактерии могут выглядеть как грамотрицательные, если мазок переобесцве-чен спиртом.
Для сравнения можно использовать ускоренный тест для определения принадлежности бактерий к грамположительным или грамотрицательным видам. Для этого исследуемую культуру бактерий с помощью петли переносят с твердой среды на предметное стекло в каплю 3%-го раствора КОН и тщательно перемешивают. Через 10 с петлю поднимают над каплей. Для грамотрицательных бактерий характерно образование слизи ДНК, которая тянется за петлей на 0,5— 1,0 см. Если образование слизи не наблюдается, бактерии относятся к грамположительным. Образование слизи происходит в результате разрушения клеточных стенок грамотрицательных бактерий и выхода из них нуклеиновых кислот.
3.3. Окраска на кислотоустойчивость
На обезжиренном предметном стекле в каплях воды готовят два мазка: исследуемой бактерии и кислотоустойчивых микобактерий (Mycobacterium flavescens). Препараты высушивают на воздухе и фиксируют над пламенем горелки. На мазки помещают фильтровальную бумагу, заливают препараты карболовым фуксином Циля и 2—3 раза подогревают их до появления паров, держа предметное стекло с помощью пинцета высоко над пламенем горелки. За появлением паров наблюдают, глядя на мазок сбоку, и при их появлении тотчас отставляют препарат в сторону. Дают препаратам остыть, снимают фильтровальную бумагу, сливают краситель и мазки промывают водой. Затем клетки обесцвечивают 5%-м раствором H2SO4. Для этого предметное стекло погружают 2—3 раза в стакан с серной кислотой, не задерживая его в ней. Препарат вновь тщательно промывают водой и докрашивают 3 — 5 мин метиленовым синим (по Леффлеру). Краску сливают, препарат промывают водой, высушивают и рассматривают с иммерсионной системой. При правильном окрашивании кислотоустойчивых бактерий клетки имеют красный цвет, а некислотоустойчивые — синий.
4. ИЗУЧЕНИЕ ФИЗИОЛОГО-БИОХИМИЧЕСКИХ СВОЙСТВ ИДЕНТИФИЦИРУЕМЫХ БАКТЕРИЙ
4.1. Рост на среде с глюкозой и пептоном
Культуру вносят стерильной петлей в жидкую среду, содержащую (г/л): пептон — 5,0; К2НРО4 — 1,0; глюкоза — 10,0; бромтимолблау — 2 мл (1,6 % спиртового раствора), вода дистиллированная, разлитую в пробирки (по 8—10 мл) с поплавками. Продолжительность культивирования 7 сут в термостате на 30 °C. Рост микроорганизмов или его отсутствие определяют по помутнению среды, образованию пленки или осадка. Изменение цвета индикатора (бромтимолблау) указывает на образование кислых (желтая окраска среды) или щелочных (синяя окраска среды) продуктов метаболизма. Об образовании газа свидетельствует накопление его в поплавке. Результаты наблюдений сравнивают со стерильной средой.
4.2. Рост на среде с желатиной
Активность у микроорганизмов внеклеточных протеолитических ферментов определяют, используя в качестве субстрата желатин, казеин или другие белки. Среда с желатином состоит из МПБ и 10—15% желатина (МПЖ). Посев проводят уколом. Бактериологической иглой стерильно отбирают клетки микроорганизмов с косяка и вводят иглу в толщу столбика МПЖ до дна пробирки.
Продолжительность культивирования 7—10 сут при комнатной температуре. Разжижение желатины или его отсутствие отмечают визуально.
4.3. Рост на среде с молоком
Высев на «молочный агар» в чашки Петри производят для определения способности бактерий разлагать казеин молока. Среда состоит из равных частей стерильного обезжиренного молока и стерильного 3%-го водного агар-агара. Бактерии высевают петлей, проводя штрих по диаметру чашки или по центру сектора, на которые разделена чашка. Продолжительность культивирования бактерий в термостате при 30 °C 7 сут. Гидролиз казеина обнаруживают по зоне просветления среды вокруг колоний или вырос -
шей по штриху культуры микроорганизмов. Особенно четко зона видна после обработки среды с выросшими бактериями раствором 5%-й трихлоруксусной кислоты. Зону гидролиза казеина измеряют в мм от края штриха или колонии до границы светлой зоны. Чем больше диаметр светлой зоны, тем выше казеинолитическая активность бактерий.
4.4. Рост на среде с крахмалом
Высев на агаризованную среду с крахмалом (в чашки Петри), содержащую (г/л): пептон — 10,0; КН2РО4 — 5,0; растворимый крахмал — 2,0; агар — 15,0; pH 6,8 —7,0 производят для определения образования микроорганизмами амилазы. Бактерии высевают петлей, проводя штрих по диаметру чашки или по центру сектора, на которые разделена чашка. Продолжительность культивирования бактерий 7 сут в термостате при 30 °C. Гидролиз крахмала обнаруживают после обработки среды с выросшими бактериями раствором Люголя. Для этого на поверхность среды наливают 3 — 5 мл раствора Люголя. Среда, содержащая крахмал, окрашивается в синий цвет, а зона гидролиза остается бесцветной или приобретает красно-бурую окраску, если крахмал гидролизовался до декстринов. Зону гидролиза крахмала измеряют от края штриха (колонии) до границы светлой зоны (мм). Чем больше диаметр светлой зоны, тем выше активность амилазы.
4.5. Тест на каталазу
Часть выросшей культуры суспензируют с помощью бактериологической петли в капле 3 % перекиси водорода на предметном стекле. О наличии каталазы свидетельствует образование пузырьков газа, наблюдаемое через 1 —5 мин после внесения бактерий невооруженным глазом или в микроскопе при малом увеличении. Можно нанести несколько капель перекиси водорода непосредственно на колонию или на культуру, выросшую на скошенном агаре, и наблюдать выделение молекулярного кислорода.
4.6. Тест на оксидазу
Наносят несколько капель 1%-го свежеприготовленного раствора солянокислого тетраметил-л-фенилендиамина на отрезок фильтровальной бумаги. Суточную культуру бактерий распределяют по поверхности увлажненной этим раствором бумаги. При положительной реакции через 10—20 с развивается фиолетовая или пурпурная окраска.
Задание на дом: с. 129—132, 159—167.
Занятие 9
5. УЧЕТ РЕЗУЛЬТАТОВ
Учесть результаты опытов по изучению физиологических и биохимических свойств бактерий, проведенных на предыдущем занятии, и внести в табл. П. 2
6. ОПРЕДЕЛЕНИЕ ЧУВСТВИТЕЛЬНОСТИ БАКТЕРИИ К АНТИБИОТИКАМ
Определение чувствительности микроорганизмов к антибиотикам можно проводить методом агаровых блочков и с помощью бумажных дисков, пропитанных разны
ми антибиотиками. Для этого готовят в стерильной водопроводной воде густую суспензию изучаемого микроорганизма путем смыва клеток водой с поверхности твердой питательной среды. Работая около пламени горелки, вносят по 3 — 5 капель полученной суспензии в пробирку с предварительно расплавленной и остуженной до 45 — 50 °C питательной средой (БСА). Содержимое пробирки тщательно перемешивают и переливают в стерильную чашку Петри.
В качестве продуцентов антибиотиков используют бактерии рода Streptomyces (табл. П.З). Продуценты антибиотиков предварительно выращивают на агаризованных средах в чашках Петри сплошным газоном. Стерильным пробочным сверлом (диаметр 6 — 8 мм) вырезают блочки с газона микроорганизма и переносят их на поверхность застывшей среды, засеянной изучаемой бактерией. Бумажные диски, пропитанные разными антибиотиками (выдает лаборант), помещают на поверхность засеянной бактерией среды на равном расстоянии друг от друга и на 1,5 —2,0 см от края чашки.
Чашки Петри, не переворачивая, помещают в термостат (30 °C) на 24 ч. Через сутки отмечают образование зон подавление роста исследуемых микроорганизмов вокруг агаровых блочков и дисков. Если исследуемая бактерия чувствительна к определенным антибиотикам, то вокруг блочков или дисков обнаруживаются зоны отсутствия роста культуры. Диаметр зоны подавления роста измеряют миллиметровой линейкой и записывают результаты в табл. П.З. Зона более 30 мм свидетельствует о высокой чувствительности микроорганизмов к антибиотику, а менее 12 мм — о слабой чувствительности.
Таблица П.З
Действие антибиотиков на рост бактерии
Антибиотик Диаметр зон подавления роста, мм
Диск с пенициллином
Диск с левомицетином
Блок с 5. violaceus (мицетин)
Блок с У anulatus var. griseus (стрептомицин)
Блок с .S'. anulatus var. sphaeroides (новобиоцин)
Блоке .S’. anulatus var. chrysomallus (актиномицин)
Задание на дом: с. 101 — 113.
Занятия 10—11
7. ПОДВЕДЕНИЕ ИТОГОВ
Оформить сводную таблицу о свойствах изучаемой бактерии.
Провести идентификацию изучаемой бактерии по определителю Берджи. Результаты определения и таблицу со свойствами бактерии сдать преподавателю.
ТЕМА 5. МЕТОДЫ КОЛИЧЕСТВЕННОГО УЧЕТА МИКРООРГАНИЗМОВ
Цель занятия: сравнить разные методы количественного учета микроорганизмов.
1. МЕТОД КОХА
Расплавляют в кипящей водяной бане 20 мл ранее приготовленной стерильной среды (БСА) в трех пробирках и разливают ее в три стерильные чашки Петри около пламени горелки. Когда среда застынет, делают высев из полученной от преподавателя культуры дрожжей Saccharomyces cerevisiae для определения числа живых клеток. С этой целью готовят разведения исходной культуры в десять и сто раз в стерильной водопроводной воде. Для этого стерильной пипеткой 1 мл исходной культуры вносят в пробирку с 9 мл стерильной водопроводной воды. Суспензию этого разведения (1: 10) тщательно перемешивают с помощью новой стерильной пипетки, вбирая в пипетку и выпуская из нее полученную взвесь. Эту процедуру повторяют 3 — 5 раз, что обеспечивает перемешивание суспензии и уменьшает адсорбцию клеток на стенках пипетки. Затем новой стерильной пипеткой берут 1 мл суспензии полученного разведения и переносят во вторую пробирку получая второе разведение 1:100. Производят посев из исходной культуры и полученных разведении путем нанесения каждый раз новой стерильной пипеткой по 0,1 мл суспензий (предварительно тщательно размешанной) на поверхность агаровых пластинок в чашках Петри. Посевной материал тщательно распределяют по поверхности агаризованной среды стерильными шпателями Дригаль-ского. Для исходной культуры и полученных разведений берут каждый раз новый стерильный шпатель.
Чашки Петри и пробирки, засеянные микроорганизмами, помещают в термостат (30 °C) на 3 — 5 сут. Чашки Петри инкубируют в термостате крышками вниз.
На следующем занятии подсчитывают число колоний S. cerevisiae в чашках Петри, и делают пересчет для определения общего числа живых клеток в 1 мл суспензии по формуле
где М — количество клеток в 1 мл; а — среднее число колоний при высеве данного разведения; 10 — коэффициент разведения; п — порядковый номер разведения, из которого сделан высев; V — объем суспензии, взятый для посева.
2. НЕФЕЛОМЕТРИЧЕСКИЙ МЕТОД
Плотность клеток 5. cerevisiae в жидкой среде определяют нефелометрически (ФЭК), используя кювету с длиной оптического пути 0,5 см и зеленый светофильтр (h = X 540 нм). Для получения достоверных результатов плотность клеток должна быть в пределах 0,1 — 0,6. При концентрациях клеток выше 0,6 происходит вторичное рассеяние света, что приводит к получению заниженных результатов. Поэтому суспензии больших плотностей перед измерением светопропускания следует разводить водой в 2, 4, 6, 8 и 10 раз. Результаты измерений оптической плотности каждой суспензии (контролем служит вода) записывают в табл. П.4.
3. ПОДСЧЕТ КЛЕТОК В КАМЕРЕ ГОРЯЕВА-ТОМА
Для перехода от показаний фотоэлектроколориметра (ФЭК) к количеству клеток микроорганизмов в среде подсчитывают их число, пользуясь счетной камерой Горяева-Тома и разведениями суспензии дрожжей, которые использовали для определения их плотности нефелометрическим методом. Клетки подсчитывают в больших квадратах сетки, пользуясь объективом 8х. Общее число подсчитанных клеток должно быть не
менее 600. Количество клеток в 1 мл соответствующей суспензии рассчитывают по формуле
М =
1000-а•п h-S
где М — число клеток в 1 мл суспензии; а— среднее число клеток в большом квадрате сетки; h — глубина камеры (*/10 мм); S— площадь большого квадрата сетки ('/25 мм2); п — степень разведения суспензии; 1000 мм3 — 1 мл. Полученные результаты, млн клеток в 1 мл (записывают в табл. П.4).
Таблица П.4
Количество клеток дрожжей в суспензиях, установленное с помощью камеры Горяева, и соответствующие показания ФЭК
Показатель Номер суспензии
1 2 3 4 5 6 И т. д.
Количество клеток, млн/мл (М)
Показания ФЭК
4. ОФОРМЛЕНИЕ РЕЗУЛЬТАТОВ
Построить калибровочную кривую на основании данных табл. П.4, откладывая на оси абсцисс количество клеток дрожжей, а на оси ординат оптическую плотность соответствующей суспензии. Калибровочную кривую строят для быстрого определения количества клеток определенного вида микроорганизмов по показаниям ФЭК.
Сравнить количество клеток дрожжей в 1 мл. Определенное подсчетом в камере Горяева (табл. П.4) и методом Коха. Сделать вывод о соотношении живых и мертвых клеток дрожжей в исходной суспензии.
5. ЗАЧЕТ ПО КУРСУ ПРАКТИЧЕСКИХ ЗАНЯТИЙ {ТЕМЫ 1—5)
ПЛАН СЕМИНАРСКИХ ЗАНЯТИЙ
Практикум рекомендуется дополнить семинарскими занятиями. Примерные вопросы для обсуждения следующие:
ЗАНЯТИЕ 1
Тема. Цитология и систематика микроорганизмов
1. Строение прокариотической клетки.
2. Клеточные стенки прокариот.
3. Дифференцированные клетки бактерий. Спорообразование.
4. Основные филогенетические группы организмов: археи, бактерии, эукарии.
5. Теория симбиогенеза.
Тема. Метаболизм микроорганизмов
1. Анаболизм и катаболизм. Типы питания микроорганизмов. Общая схема обмена веществ у микроорганизмов. Особенности метаболизма аэробов и анаэробов.
2. Дыхание и брожение. Виды анаэробного дыхания.
3. Хемосинтез.
4. Фотосинтез у прокариот.
5. Физиологические группы микроорганизмов.
6. Жизнь микроорганизмов в экстремальных условиях.
ЗАНЯТИЕ 3
Тема. Микроорганизмы в биосфере
1. Участие микроорганизмов в циклах С, N, S и других элементов.
2. Взаимодействие микроорганизмов. Трофическая цепь.
3. Симбиозы.
4. Нормальная микрофлора человека.
5. Санитарно-бактериологический анализ пищевых продуктов, воды и воздуха. Микробное число, коли-титр, коли-индекс.
6. Участие микроорганизмов в приготовлении и порче пищевых продуктов.
7. Участие микроорганизмов в процессах очищения окружающей среды.
На занятии можно также просмотреть препараты микроорганизмов, приготовленные из квашеной капусты, кисломолочных продуктов, чайного гриба и других субстратов.
ПРИЛОЖЕНИЕ 2
УСТРОЙСТВО ПРИБОРОВ И ПРИНЦИПЫ РАБОТЫ НА НИХ
ОСНОВЫ ЦЕНТРИФУГИРОВАНИЯ
Центрифугирование может заменить фильтрование и применяется для разделения смесей, где один из компонентов — жидкость. Центрифугирование позволяет избежать многих трудностей, связанных с фильтрованием, например засорения пор фильтра мелкодисперсными частицами, адсорбции частиц на фильтре, дает возможность разделять смеси веществ, портящихся от соприкосновения с фильтром, и значительно повышает скорость разделения смесей. Методом дифференциального центрифугирования в биологии достигается разделение содержимого клеток на компоненты: ядра, митохондрии, рибосомы. После разрушения клеток микроорганизмов или тканей животных и растений получаются гомогенаты, содержащие все клеточные компоненты, различающиеся между собой относительной молекулярной массой. В основе метода центрифугирования лежит принцип отделения крупных частиц с большей плотностью
при низких скоростях. При повышении скорости центрифугирования можно осадить все более и более мелкие частицы.
Подбирая свойства жидкости в смеси и силу осаждения, можно направленно выделять отдельные клеточные компоненты. Особенно это важно при так называемом градиентном центрифугировании, когда осаждение ведется в смеси, где жидкость имеет самую низкую удельную плотность у верха центрифужной пробирки, а самую высокую — в ее нижней части. Наслаивая на поверхность жидкости в пробирке клеточные гомогенаты, подвергаемые разделению в процессе длительного центрифугирования, можно получить картину распределения клеточных компонентов слоями, которые соответствуют плотности жидкости на данной высоте пробирки. Аналитическое центрифугирование применяется для определения коэффициента седиментации и связанной с ним молекулярной массы различных веществ, например белков.
Во время вращения на пробирку действует центробежная сила (А), которая выражается уравнением R = 1,117/-№ -10-5, где г — радиус вращения, см; N — число оборотов в минуту. В отечественной литературе относительную центробежную силу часто называют фактором разделения g (кратно силе притяжения). Для расчета g необходимо знать скорость вращения центрифуги и радиус (см) от оси вращения до дна центрифужной пробирки. Например, если дно пробирки удалено от оси вращения на 15 см, то при скорости вращения ротора центрифуги 6 тыс. об/мин относительная центробежная сила у дна пробирки равна:
g= 1,117 -15 - 60002-10 5 = 6000.
КОНСТРУКЦИИ ЦЕНТРИФУГ И ПОРЯДОК РАБОТЫ НА НИХ
Центрифуга угловая малогабаритная настольная ЦУМ-1
Центрифуга ЦУМ-L (рис. П.1) предназначена для разделения неоднородных жидких систем с удельной массой не более 2 г/см3 (для пробирок из полимерных материалов) и не более 1,5 г/см3 (для стеклянных пробирок). Максимальный объем центри-фугата — 180 мл при оборотах от 2000 до 8000 через каждую 1000, максимальный фактор разделения (g) — 6000. Время разгона ротора не более 10 мин, время остановки — 8 мин. Питание центрифуги происходит от сети переменного тока 220 В при частоте 50 Гц. Потребляемая мощность до 280 Вт, время непрерывной работы не должно превышать 60 мин с последующим 15-минутным перерывом. Габариты 275 х 235 х 315 мм, масса 16 кг.
Рис. П.1. Внешний вид центрифуги ЦУМ-1:
1 — крышка, прикрывающая ротор; 2 — кнопка включения и выбора режима работы («с часами» и «без часов»);
3 — электрочасы; 4 — переключатель частоты вращения ротора; 5 — контрольная лампа включения центрифуги
Подготовка и порядок работы. Открыть крышку центрифуги и плотно насадить ротор на вал привода. Перед началом работы с пробирками из стекла вложить резиновые амортизаторы под каждую стеклянную пробирку. Разместить попарно уравновешенные пробирки (разбаланс допускается не более 0,1 г) в диаметрально противоположные гнезда ротора, закрыть крышку центрифуги и установить переключатель частоты вращения ротора в положение «0».
При работе с часами ручкой переключателя электрочасов установить требуемое время центрифугирования с учетом времени разгона ротора. Установить тумблер в положение «с часами». Подключить центрифугу к источнику питания, при этом загорится сигнальная лампа. Для работы центрифуги на частоте вращения 2000—6000 об/ мин ручку переключателя вначале необходимо установить на две ступени выше требуемой частоты вращения, а через 0,5 — 2 мин перевести ее в нужное положение, причем переключение в сторону увеличения частоты вращения ротора необходимо производить с выдержкой на каждой ступени не менее 10 с, а в сторону уменьшения — 2-3 с.
По истечении заданного времени сработают электрочасы, сигнальная лампа погаснет, центрифуга автоматически остановится, после чего необходимо поставить ручку переключателя оборотов в положение «0». После остановки ротора открыть крышку центрифуги, отвинтить крышку ротора и вынуть пробирки. При работе без учета времени тумблер устанавливают в положение «без часов», все дальнейшие операции по разгону и остановке центрифуги производят так же.
Меры безопасности. Запрещается работать на незаземленной центрифуге, включать центрифугу без ротора, работать с открытыми крышками ротора и центрифуги, задавать частоту вращения ротора более 4000 об/мин при работе со стеклянными пробирками, заземлять центрифугу последовательно с заземлением других приборов.
Центрифуга J-6B (Beckman)
Операционные системы центрифуги. Крышка центрифуги закрывается электромагнитным замком на правой части крышки. При открывании крышки на 3/4 включается в действие газовая пружина, которая открывает дверь полностью. (Придерживайте дверь рукой до полного ее открытия!) Дверь находится в открытом виде без дополнительной поддержки. На внутреннюю сторону двери в открытом состоянии навешивается воздушный щит при разгрузке-загрузке ротора. Замок двери срабатывает после нажатия кнопки START. Замок будет оставаться закрытым, если внезапно отключится электричество. В таком случае крышку центрифуги можно будет открыть, нажав на упор замка тонким твердым инструментом (шилом). Если приводной ремень центрифуги оборвется во время открутки, крышка центрифуги не откроется. Когда ремень будет восстановлен, крышка снова откроется.
Привод ротора закреплен на отдельной плате для уменьшения вибраций. Однако небольшие вибрации могут возникать при прохождении точки критической скорости (между 600 и 800 об/мин). Такая вибрация считается нормальной. Избыточная вибрация включит датчик несбалансированности ротора и центрифуга отключится. Центрифуга подключается к сети 220В, 50 Гц и выдерживает перепады напряжения от 214 до 224 В. Центрифуга снабжена двумя собственными предохранителями по 25 А каждый. Контроль температуры осуществляется автоматически, и ее величина поддерживается в пределах ±1°С от заданного уровня. Центрифуга отключится, если температура в камере превысит 45 °C или стрелочный индикатор зайдет за верхний предел, ограниченный красным датчиком OVERTEMP. Компрессор во время работы охлаждается автоматически воздухом, который втягивается в задний нижней части центрифуги и выходит через заднюю верхнюю часть. Забор воздуха осуществляется через фильтр, который периодически необходимо очищать пылесосом.
Контрольная панель (рис. П.2) содержит ручку установления скорости торможения (BRAKE) с позициями от 0 до 10 (шкала нелинейна). При очень сильном торможении может произойти взмучивание осадка. Лампа состояния щеток двигателя загорается, когда щетки необходимо поменять. Необходима быстрая замена щеток после включения лампы. Длительная эксплуатация центрифуги с горящей лампой состояния щеток может привести к выходу из строя двигателя центрифуги. Лампа IMBAL зажигается, если ротор не сбалансирован, и горит все время до полной остановки центрифуги (тормоз в этом случае не действует). Лампа выключается автоматически при включении кнопки START. Балансировка противоположных стаканов ротора должна быть в пределах ±0,1 г. Кнопка OVERTEMP включается, показывая, что центрифуга отключилась для того, чтобы препятствовать повышению температуры выше установленной красным ограничителем на температурном датчике. Оператор вручную устанавливает верхний предел температуры при данном центрифугировании, чтобы избежать перегрева образцов. Если желательная температура OVERTEMP выбрана ниже комнатной, ее устанавливают два раза: первый — при запуске центрифуги (выше комнатной), затем — при охлаждении ротора до температуры центрифугирования (желаемое значение).
Кнопка включения и лампа включения центрифуги расположены на нижней левой стороне панели. Выключение тумблера ON выключает электромагнитный замок и позволяет открыть крышку центрифуги. Выключение центрифуги блокирует замок дверцы, и чтобы этого избежать, дверцу держат открытой. Кнопка включения запускает индикаторную лампу, систему охлаждения и всю электронику центрифуги. Выключатель автоматически выключает центрифугу в случае нарушения электроснабжения или короткого замыкания в сети. Включите центрифугу только один раз после отключения. Если она не включится, нужно вызывать механика. Кнопка установки скорости вращения ротора (не выше 5 тыс. об/мин) позволяет установить желаемую скорость вращения ротора, от скорости вращения ротора зависит и его температура. При включении таймера с нажатием кнопки START начинается разгон ротора. Кнопка светится зеленым светом до тех пор, пока не включится торможение (от таймера или от нажатия кнопки STOP). Кнопка STOP перестает светится красным светом после полной остановки ротора. Таймер контролирует время движения ротора. Переключение таймера на «0» активирует тормозную систему. Таймер нужно включать на HOLD и контролировать время работы центрифуги вручную, если время открутки превышает 36 мин.
Установка параметров центрифугирования. 1. Установить значение OVERTEMP гораздо выше комнатной температуры. 2. Включить тумблер ON. 3. Выставить значение желаемой температуры ротора согласно специальной таблице (кнопка SET). 4. Установить желаемую скорость торможения кнопкой BRAKE (обычно 4—6). 5. Охладить ротор в течение 20 мин при 1000 об/мин. Крышку воздушного щита не устанавливать! 6. Поста-
Рис. П.2. Внешний вид панели центрифуги J-6B
вить в ротор охлажденные стаканы с пробами (±7 °C от желаемой температуры центрифугирования!). 7. Поставить желаемое значение OVERTEMP (обычно на 3 — 5 °C выше желаемой температуры ротора). 8. Выбрать скорость вращения ротора. 9. Поставить таймер (не более 36мин!). 10. Надеть крышку воздушного шита. 11. Включить кнопку START. Таймер может быть переустановлен в любое время центрифугирования в сторону уменьшения или увеличения времени. При повторном центрифугированиях повторяют шаги 6—10, следя за тем, чтобы крышка центрифуги открывалась на максимально короткие промежутки времени (для препятствия намерзания конденсата на стенки камеры центрифуги).
Остановка центрифуги может быть произведена после полного времени срабатывания таймера, при ручной постановке таймера на «0», при нажатии кнопки STOP (лампа кнопки зажжется и будет гореть до тех пор, пока ротор полностью не остановится). После выключения лампы STOP можно открыть крышку центрифуги. Крышка воздушного щита должна висеть на открытой двери. После того как лед на стенках камеры растает, необходимо убрать конденсационную воду губкой и насухо вытереть стенки и дно камеры.
Уход за центрифугой. Центрифугу можно мыть нейтральным детергентом (шампунем) и дезинфицировать 70%-м этанолом. Раз в 1 — 3 мес необходимо очищать фильтр забора воздуха с помощью пылесоса. Один раз в год необходимо промывать фильтр проточной водой.
Установка ротора. Ротор устанавливается в центрифугу в специальные гнезда и фиксируется контролирующей гайкой с отверстием, в которое продевается специальная металлическая палочка для усиления фиксации. После установки на ротор надевают воздушный щит с отверстием, в которое входит центральный шток ротора. Периодически необходимо проверять плотность закрутки фиксирующей гайки ротора. С установленным в центрифуге ротором JS-5.2 запрещается разгонять ротор до более 5000 об/мин. При такой скорости ускорение будет составлять 6330 g у дна центрифужного стакана (/• = 226 мм).
Уход за ротором. Ротор и его стаканы должны быть немедленно вымыты с мягким (нейтральным) детергентом в случае попадания на него жидкости при. центрифугировании. Затем ротор споласкивают дистиллированной водой и сразу вытирают досуха. При нормальной работе стаканы необходимо мыть раз в неделю. Стаканы ротора можно дезинфицировать 70%-м этанолом.
Центрифужные стаканы для ротора JS-5.2 сделаны из прочного полиэтилена и их необходимо устанавливать в ротор, используя специальные синие прокладки. Поместите стакан в прокладку и затем установите в ротор.
Центрифуга J2-21 (Beckman)
Конструкция центрифуги. Центрифуга предназначена для высокоскоростного осаждения материала из жидкостей с удельной плотностью не выше 1,2 г/мл. Роторы центрифуги сделаны из высокопрочного алюминия с анодированным покрытием. Охлаждение центрифуги происходит за счет работы компрессора, который включается автоматически при запуске центрифуги. Контроль температуры осуществляется термистором, расположенным на дне центрифужной камеры. Крышка центрифуги закрыта электромагнитной защелкой, которая закрывает ее при включении кнопки START. Для открытия крышки, закрытой при внезапном отключении напряжения, см. действия, аналогичные открытию центрифуги J-6B.
Тумблер включения центрифуги расположен на левой нижней части панели управления (рис. П.З). Включение тумблера выключает замок крышки центрифуги, загорается лампа общего включения, начинает работать компрессор и вся электронная система. В случае внепланового выключения центрифуги включайте тумблер ON только один раз.
Рис. П.З. Внешний вид панели центрифуги J2-21
Если центрифуга не включится, необходим вызов механика. Селектор выбора скорости позволяет установить желаемую скорость вращения ротора — от 1 до 20 тыс. об/мин. Таймер контролирует включение мотора центрифуги и работу вакуумного насоса. Поворот кнопки таймера включает вакуумный насос, если крышка центрифуги закрыта. Выключение таймера приводит к остановке вакуумного насоса, мотора привода и включает тормоз. Необходимо устанавливать таймер на HOLD при времени работы более 190 мин. Таймер включает систему вакууммирования, понижая давление в камере до 0,5 атм, тем самым уменьшая трение воздуха. Оптимальное значение для вакуума достигнуто, когда стрелка вакуумметра находится в пределах зеленой зоны. Откачка воздуха из камеры происходит через центральное отверстие в крышке центрифуги. Вакуум должен достигать оптимума в течение одной минуты после включения таймера и закрытия крышки. Селектор BRAKE устанавливает значение силы торможения от нуля до максимума (10 позиций) в нелинейном режиме. Выбор значения торможения определяется весом ротора и проб, а также скоростью вращения и плотностью формируемого осадка.
Селектор температуры разделен шкалой со значением деления 2 °C. Красный индикатор устанавливает предел температуры (кнопка OVERTEMP), зеленый индикатор — нужную температуру ротора (кнопка SET). Черная стрелка указывает температуру ротора, если кнопкой СОМР установлена соответствующая поправка (по таблицам). При запуске центрифуги красный индикатор устанавливают выше комнатной температуры и затем, после охлаждения камеры, устанавливают на значении на 5 —8 °C выше температуры ротора. Для разных роторов центрифуга J2-21 устанавливают разные значения температурной компенсации (кнопка СОМР) в зависимости от скорости вращения ротора и температуры центрифугирования (по таблице).
Кнопка START приводит в действие двигатель центрифуги и закрывает замок крышки после включения таймера. Лампа кнопки будет гореть зеленым светом, пока не включится торможение (кнопка STOP загорится красным светом) по истечении времени, заданного таймером. Лампа STOP погаснет, когда ротор полностью остановится. Нажатие кнопки STOP включает тормоз, но не выключает вакуумный насос. Поэтому центрифугу лучше останавливать, выставляя кнопку таймера на «0». Если ротор несбалансирован, то датчик баланса отключит двигатель, и ротор будет останавливаться без торможения (при этом будет мигать лампа дисбаланса). Необходимо сбалансировать ротор (пробирки) и запустить центрифугу кнопкой START. Светодиод BRUSH загорается, когда необходимо заменить щетки электродвигателя. Эксплуатировать центрифугу с включенной лампочкой BRUSHES запрещается!
Запуск центрифуги. 1. Охладить ротор до желаемой температуры в холодильнике, поставить в него охлажденные пробирки. (Внимание! Запрещается охлаждать ротор в центрифуге!) 2. Выставить кнопкой OVERTEMP желаемую предельную температуру. 3. Включить тумблер ON. 4. Установить температурную компенсацию ротора кнопкой
COMP (по таблице). 5. Установить нужную скорость торможения (ручка BRAKE, от О до 10). 6. Установить ротор с пробирками, закройте крышку ротора и центрифуги. Пробирки должны быть уравновешены с точностью +10 мг! 7. Переставить красный индикатор температуры на 5 °C выше температуры ротора. 8. Выбрать и установить нужную скорость вращения ротора. 9. Выставить таймер. 10. Следует дождаться, пока указатель вакуума дойдет до зеленой зоны. 11. Нажать кнопку START. После прохождения времени, заданного таймером, центрифуга автоматически отключится (зажжется кнопка STOP и будет гореть до полной остановки ротора). После этого электромагнитный замок крышки откроется. Для открытия крышки центрифуги на нее нужно немного нажать и поднять вверх, придерживая рукой все время, пока она не дойдет до упора. Удалить ротор из центрифуги и отключить тумблер (OFF). После разморозки стенок центрифуги удалить воду губкой и насухо протереть камеру.
Меры предосторожности. В случае появления неожиданной вибрации отключить центрифугу и вызвать механика. В период загрузки/разгрузки центрифуги следует отключать кнопку OFF. Перед установкой смазать посадочное место ротора на шпинделе силиконовой вакуумной смазкой. Ставить и снимать роторы осторожно, медленно и строго вертикально во избежание деформации струны шпинделя.
Периодически необходимо прочищать фильтры забора охлаждающего воздуха пылесосом (раз в 2 мес), мыть и очищать поверхность центрифуги нейтральными мылами.
Подготовка ротора. Ротор перед установкой в центрифугу должен быть охлажден до нужной температуры (±1°С). Необходимо также смазать тонким слоем силиконовой вакуумной смазки уплотняющие резиновые кольца ротора и его крышки, а также посадочное место ротора на шпинделе. После установки в ротор пробирок крышка ротора должна быть плотно завернута.
Ротор JA-20 предназначен для осаждения при скорости вращения до 18 тыс. об/мин (39 000 gy дна пробирки). Пробирки перед постановкой в ротор должны быть сбалансированны до ±10 мг. Запрещается оставлять ротор в центрифуге после работы! Ротор должен быть вымыт теплой водой с нейтральным мылом раз в неделю. После каждого использования ротор необходимо сполоснуть дистиллированной водой, вытереть и хранить при комнатной температуре в перевернутом состоянии.
Ротор JA-14 предназначен для осаждения при скорости вращения до 12 тыс. об/мин (21 000 g у дна пробирки). Пробирки перед постановкой в ротор должны быть сбалансированны до ±100 мг. Запрещается оставлять ротор в центрифуге после работы! Ротор должен быть вымыт теплой водой с нейтральным мылом раз в неделю. После каждого использования ротор необходимо сполоснуть дистиллированной водой, вытереть и хранить при комнатной температуре в перевернутом состоянии.
Ультрацентрифуга L5-65B (Beckman)
Центрифуга предназначена для осаждения образцов при оборотах ротора от 1 до 65 000/мин (в зависимости от типа ротора). Число оборотов контролируется в пределах ± 500 об/мин. Температура центрифугирования может быть задана в пределах от 0 до 40 °C, температура ротора автоматически поддерживается в пределах ± 1 °C. Центрифугу необходимо устанавливать в проветриваемом помещении с температурой от 15 до 40 °C, учитывая при этом, что она производит нагрев помещения при работе с мощностью 1,2 кВт-ч.
Принципы работы. Модель L5-65B — охлаждаемая ультрацентрифуга с прямым приводом на ротор, вращающийся в вакууме с прецизионным контролем температуры. Диффузионный насос с вакуумным насосом обеспечивают достижение вакуума до 5 мкм. Число оборотов и температура контролируются электроникой. Неуравновешенность ротора контролируется специальным датчиком, соленоидный замок препятствует открыванию крышки камеры во время включения ротора.
Temperature Gauge
Tachometer
Confidence Lights
DRY CYCLE PROGRAM Buton
Circuit
ACCELERATION Control
Vacuum Guage
Revolution Counter
Timer
Speed Selector
STOP Button
Zonal Switch
Start Button
AUTOMATIC OPERATION Cluster
Рис. П.6. Внешний вид центрифуги L5-65B
Описание контрольной панели центрифуги (рис. П.6). Тахометр — стрелочный индикатор вращения ротора (тыс. об/мин). Температурный датчик показывает температуру' ротора (°C). Черная стрелка — указатель температуры. Два красных индикатора устанавливают границы изменения температуры ротора (с помощью двух кнопок — Temp Set и Temp Limit), выходя за которые центрифуга останавливается автоматически.
Примечание. Компрессор центрифуги включен постоянно при наборе вакуума или вращении ротора.
Счетчики оборотов показывают в накопительном режиме общее количество оборотов ротора (тыс.). Датчик вакуума показывает степень вакуума в камере (мк). Кнопка выбора скорости ротора позволяет выбрать нужную скорость (тыс. об/мин). Таймер устанавливает время вращения ротора (до 300 мин). Если датчик установлен в положение HOLD, ротор будет вращаться, пока не нажата кнопка STOP. Кнопка STOP прекращает вращение ротора и горит, пока ротор полностью не остановится. Ключ включения для зональных роторов — специальный ключ с замком при открутках с зональными роторами, позволяет держать крышку камеры открытой до 2000 об/мин в позиции ALJTO и до 5000 об/мин в позиции MANUAL. Когда ключ повернут в нормальное положение, ротор не будет вращаться до закрытия двери камеры. Кнопка Start позволяет включить центрифугу для выбора частоты вращения ротора. Кнопка будет гореть до тех пор, пока ротор вращается. Лампочки подтверждения указывают на штатный режим работы центрифуги (давления масла, воды, баланса ротора и числа его оборотов). Если лампочка начинает мигать, это указывает на проблемы в соответствующем блоке. Ротор будет останавливаться автоматически.
Кнопки автоматического оперирования — вакуум, торможение и способ открутки, находятся в двух положениях по желанию оператора: в ручном или автоматическом. Способ открутки (Mode): если включен в AUTO-положении (AUTO-лампа включена), то вакуумный насос включится автоматически при нажатии кнопки START, а камера будет автоматически проветрена в конце открутки (если кнопка «вакуум» — в AUTO-положении). Если кнопка Mode находится в положении ручное управление (MANUAL), вакуумный насос должен быть включен вручную до начала раскрутки ротора, а камера должна быть проветрена в конце открутки. Торможение (Brake): при включении кнопки в положение NORMAL, тормоз будет останавливать ротор до скорости 50 об/мин и далее до полной его остановки. Если Brake находится в положении SLOW, тормоз будет останавливать ротор до 1000 об/мин, далее ротор будет останавливаться по инерции. Кнопка вакуума. если она в AUTO-положении, то вакуумный насос включается
одновременно с нажатием кнопки Start, и камера автоматически проветрится при остановке ротора, когда его скорость упадет до 50 об/мин. Если кнопка вакуума в положении ОМ (и крышка камеры закрыта), вакуумный насос будет включен постоянно до того, как кнопка будет переведена в положение AUTO. Диффузионный насос включается автоматически по достижении вакуума в камере в 750 мкм (за исключением нажатия кнопки STOP).
Примечание. Кнопки вакуума и Mode должны быть в положении AUTO для автоматического контроля работы вакуумного насоса.
Кнопка сухого цикла предназначена для включения программы осушки камеры от влаги или льда до начала открутки. Нажатие кнопки приводит в действие программу нагрева стенок камеры до 40 °C с одновременным включением вакуумного насоса. Камера автоматически проветривается, когда вакуум достигнет 750 мкм, показывая, что камера осушена. Камера может быть просушена в любое время, для прекращения действия программы необходимо повторно нажать кнопку.
Кнопка ускорения позволяет менять скорость раскрутки ротора. Максимальный разгон в 2,5 раза превышает минимальный. Например, если какой-то ротор разгоняется за 10 мин при полном повороте кнопки по часовой стрелке, то тот же ротор будет разгоняться за 23 мин, если кнопка повернута полностью против часовой стрелки. Торможение также зависит от положения кнопки (если кнопка «тормоз» в положении NORMAL). Скорость разгона/торможения особенно важна при работе с градиентами: быстрое торможение может смешать установившиеся слои. Кнопки общего включения и лампочки расположены слева внизу контрольной панели центрифуги. При включении кнопки загораются две лампы на 120 В, показывая, что напряжение включено.
Обычное центрифугирование
До открывания дверцы камеры необходимо включить кнопки общего включения (на щитке приборов и на панели инструментов). Ротор должен быть предохлажден до значения не более ±1 °C от желаемой температуры (в холодильнике, не в центрифуге}). Оботрите влагу с ротора до постановки его в камеру. Проверьте датчик снабжения маслом. Для осушки замерзшей или влажной камеры включите программу сухого цикла. Ротор при этом может быть установлен в камере, но не крутиться, иначе программа не включится.
Ручное управление. 1. Включить кнопку POWER ON и подачу воды. 2. Установить кнопку MODE в положение MANUAL. 3. Установить предохлажденный ротор в камеру и закрыть крышку. 4. Убедиться, что ключ зонального центрифугирования в положении «обычное». 5. Выставить скорость, границы температуры и желаемую температуру. 6. Установить время открутки. 7. Выставить кнопку ускорения. 8. Переключить кнопку вакуума в положение ON. 9. Выбрать положение NORMAL или SLOW на кнопке тормоза. 10. Нажать кнопку START. Когда компрессор включится, ротор начнет разгоняться. Счетчик оборотов начнет отсчет. Скорость ротора не превысит 2000 об/мин, пока вакуум не опустится до 750 мкм, затем ротор разгонится до нужной скорости, и только тогда таймер начнет отсчет времени. Торможение ротора начнется, когда таймер дойдет до нуля или будет нажата кнопка STOP. При загорании лампы STOP необходимо нажать кнопку вакуума для снятия вакуума в камере. Охлаждение отключится, и тогда можно открыть люк и вынуть ротор из камеры.
Автоматическое центрифугирование. 1. Включить кнопку POWER ON и подачу воды. 2. Выбрать положение AUTO на кнопке MODE. 3. Поставить ротор в камеру и закрыть крышку. 4. Следует убедиться, что ключ зонального центрифугирования в положении «обычное». 5. Выставить скорость, границы температуры и нужную температуру. 6. Установить время открутки. 7. Выставить кнопку ускорения. 8. Нажать кнопку START.
Компрессор и вакуумный насос включатся с нажатием кнопки START. Начнется разгон ротора до 2000 об/мин, дальнейший разгон происходит по достижении разрежения в 750 мкм. Центрифугирование прекратится, когда таймер дойдет до нуля или будет нажата кнопка STOP. При загорании кнопки ротор будет останавливаться в за
висимости от выбора позиции на кнопке тормоза. Если кнопка вакуума в позиции AUTO, камера автоматически проветрится, когда ротор остановится до 50 об/мин. При положении переключателя в положении ON камера должна быть проветрена нажатием кнопки вакуума. Компрессор выключится, когда камера будет проветрена. Открыть крышку и вынуть ротор из камеры.
Зональное центрифугирование
Предварительно готовят центрифугу и ротор так же, как для обычного центрифугирования.
Ручное управление. (Если предполагается использовать таймер центрифуги, то центрифугирование лучше проводить в автоматическом режиме).
Загрузка. 1. Включить центрифугу в сеть, включить тумблер ON. 2. Выбрать MANUAL на включателе MODE. 3. Повернуть ключ зонального центрифугирования в позицию ZONAL. 4. Выбрать ротор и установить его в камеру согласно инструкции. 5. Выбрать температуру и установить ее границы. 6. Выбрать скорость загрузки (<5000 об/мин, рекомендуемая скорость — 2000 об/мин). 7. Поставить таймер в положение HOLD. 8. Поставить включатель вакуума в положение ON. 9. Включить кнопку START (включится компрессор, ротор разгонится до заданной скорости). 10. Загрузить и закрыть ротор. 11. После загрузки ротора закрыть дверцу камеры. Вакуумный насос включится автоматически. Выбрать скорость вращения и установить таймер. Ротор разгоняется до -5000 об/мин и затем будет разгоняться до выбранной скорости после достижения вакуума в камере 750 мкм. Оператор выключает центрифугу (кнопка STOP в положении таймера HOLD) или центрифуга выключится автоматически (таймер).
Разгрузка. 1. Выбрать скорость разгрузки (<5000 об/мин, рекомендуемая скорость — 2000 об/мин). Ротор будет останавливаться до выбранной скорости при позиции таймера HOLD. 2. Поставить кнопку вакуума в положение AUTO для выключения вакуумного насоса. [Примечание: при переключении кнопки вакуума в положение AUTO вакуум в камере отключится на 5000 об/мин]. 3. Открыть дверцу камеры и разгрузить ротор. 4. Нажать кнопку STOP для остановки ротора.
Автоматический режим. В таком режиме загрузка и разгрузка ротора происходит при 2000 об/мин.
Примечание. Замок крышки камеры не функционирует в зональном режиме.
Загрузка. 1. Включить выключатель центрифуги в положение ON. 2. Выбрать положение AUTO на кнопке MODE. 3. Выбрать зональное положение ключа-переключателя. 4. Собрать ротор и загрузить его согласно инструкции. 5. Выбрать температуру, ее пределы и скорость. 6. Поставить таймер. 7. Включить кнопку вакуума в положение AUTO. 8. Нажать кнопку STAR!' — включится компрессор и ротор разгонится до 2000 об/мин для загрузки. 9. Загрузить и закрыть ротор. 10. Закрыть дверцу камеры. Вакуумный насос включится, ротор будет вращаться со скоростью 2000 об/мин, пока вакуум не достигнет 750 мкм, затем разгонится до выбранной скорости.
Разгрузка. 1. После окончания центрифугирования ротор замедлится до 2000 об/мин и будет вращаться с этой скоростью. Вакуум сбросится автоматически.
Примечание. Не нажимайте кнопку STOP в этом режиме, иначе ротор полностью остановится.
2. Когда вакуум будет сброшен, открыть камеру и разгрузить ротор. 3. Нажать кнопку STOP для полной остановки ротора.
Особые замечания о работе на центрифуге
1. Никогда не включайте центрифугу без ротора. 2. Для быстрой установки температуры охладите ротор до постановки в камеру в пределах ±1 °C от желаемой. 3. Не оставляйте камеру под вакуумом с выключенной сетью. 4. Не оставляйте дверь камеры открытой между открутками: вода будет конденсироваться на холодных стенках камеры, препятствуя достижению нужного вакуума. 5. Держите в чистоте полиэтиленовую плен
ку радиометра. 6. Держите линзы источника света для стробоскопа чистыми. 7. Держите камеру центрифуги чистой и сухой. 8. Всякий раз перед центрифугированием убеждайтесь в наличии достаточного количества масла.
Что важно помнить о роторах
1. Никогда не запускайте ротор без правильно установленного стробоскопа. 2. Чаще проверяйте ротор на наличие царапин, сколов, трещин или коррозии.
Центрифуга должна содержаться в чистоте. Периодически (раз в 3 — 4 мес) необходимо протирать уплотнительное резиновое кольцо крышки камеры 70%-м этанолом, затем смазывать тонким слоем силиконовой смазки. Необходимо (раз в 3 мес) очищать воздушный фильтр (сзади на центрифуге) с помощью пылесоса, влажной тряпки или вентилятора.
Общие рекомендации по использованию роторов и стаканов для центрифугирования
Для центрифугирования критическим моментом является правильная балансировка роторов. По этой причине роторы должны быть загружены стаканами симметрично, причем следует использовать лишь один тип стаканов и крышки соответствующего типа при каждом центрифугировании. Тонкостенные нитратцеллюлозные или полиалломерные стаканы необходимо заполнять максимально. Перед заполнением пробирки проверяют на наличие повреждений. Целлюлозные и полиалломерные пробирки сначала заполняют на 2/3 объема. Внешние поверхности пробирок должны оставаться сухими при заполнении. Пробирку закрывают крышкой, фиксируют ее неплотно закрученной гайкой и убеждаются в том, что верхняя часть крышки плотно соприкасается с верхом пробирки. Затянув гайку специальным торцовым ключом, пробирки заполняют доверху с помощью шприца. Пробирки следует уравновесить с точностью до 1 мг на аналитических весах и затянуть крепежные винты. Убедиться в том, что пробирки и пробки не протекают, нажав на каждую пробирку двумя пальцами. Если пробирки подтекают, то надо обнаружить место утечки и ликвидировать его, добавив смазки и крепче затянув винты крепления крышки. Перед сборкой крышки необходимо смазать резьбу тонким слоем силиконовой смазки. Резиновые прокладки и поверхности, с которыми они соприкасаются, должны быть сухими перед сборкой крышки. Перед каждым центрифугированием необходимо убедиться в том, что крышки пробирок не повреждены.
Под действием центрифужных сил гнезда ротора становятся эллиптическими, что удерживает пробирки в основном в двух местах. Это ведет к преждевременному износу нижней части коронки крышки, изменению формы крышки и может повлечь за собой поломку ротора. В дополнение к визуальному контролю, проведите кончиком пальца по поверхности коронки и убедитесь в том, что они плоские: не вогнутые и не выпуклые. Осмотрев нижнюю часть крышки, убедитесь в том, что на ней нет тонких трещин. Любая крышка, даже с самыми мелкими трещинами, к работе не пригодна.
Красные анодированные крышки с резиновыми прокладками используют для больших пробирок (до 38 мл) и ротора Ti60. Они должны закрываться (закручиваться) с помощью торцевого ключа с торсионом до определенного значения приложенной силы. В них смазка резьбы особенно важна для препятствия трению при использовании торсионного ключа. Первые четыре закрутки должны быть сделаны с усилием 21 кг/см, в дальнейшем усилие снижают до 18 кг/см. Закручивание следует производить короткими и непрерывными движениями.
При заполнении пробирок доверху с помощью шприца необходимо учитывать, что: 1) тонкостенные пробирки заполняют доверху, толстостенные пробирки могут содержать до 13 мм воздуха; 2) если пробирки (толстостенные полиалломерные) заполнены частично (но не менее 2/3 объема), убеждаются, что все они заполнены до одного уровня. Пробирки герметизируют с помощью фиксирующего винта, используя тот же ключ, которым пробирки достают из ротора (см. ниже). Винт закручивают пол
ностью, т.е. до тех пор, пока он не войдет в плотный контакт с нейлоновой прокладкой. Чтобы убедиться в герметичности тонкостенных нитроцеллюлозных и полиалломерных пробирок, их надо сжать с двух сторон.
Заполнение ротора
Гнезда ротора должны быть чистыми и сухими. Любая влага между стенками пробирки и гнездом ротора вызывает появление вакуумного присасывания при центрифугировании и приводит к растрескиванию нитратцеллюлозных и полиалломерных пробирок. Пробирки в ротор закладывают симметрично, друг против друга. При разбалансировке ротора центрифуга выйдет из строя. Если нужна еще одна пробирка для баланса ротора, ее заполняют материалом той же специфической плотности, что и материал, используемый в работе. Резьбу ротора смазывают силиконовой смазкой и небольшое количество вакуумной смазки наносят на резиновые кольца крышки ротора и его ручки. Крышку ротора и ручку следует плотно завернуть, но не до вхождения в контакт с металлическими частями.
Бакетпые роторы. Пробирки заполняют раствором градиента, помещая более плотные слои градиента вниз (нитроцеллюлозные пробирки). Полиалломерные пробирки начинают заполнять с «легкого» конца градиента, так как их стенки несмачиваемые. После заполнения пробирки легкие слои замещаются тяжелыми. Заполнение ведут с помощью тонкой полиэтиленовой трубки, надетой на иглу шприца и опущенной на дно пробирки. Пробирки помещают в бакеты до нанесения образца. Пробирки должны быть заполнены правильно (3 мм от края для нитроцеллюлозных и полиалломерных пробирок) и равномерно (для уравновешивания). На резьбу крышки бакета наносят тонкий слой силиконовой смазки и плотно закручивают крышку двумя пальцами. Поверхности бакетов для нитроцеллюлозных и полиалломерных пробирок должны быть сухими, как и поверхности самих пробирок, поскольку даже самые малые количества воды быстро повреждают пробирки.
При подвеске на ротор всегда помещайте каждый бакет на предназначенное ему место (бакеты и их места на роторе размечены). Убедитесь, что противоположные бакеты (1 и 4, 2 и 5, 3 и 6) уравновешены с точностью ±10 мг. При установке ротора в центрифугу убедитесь, что ножки на шпинделе и выступы на роторе не совпадают, иначе при раскрутке центрифуги ротор может соскочить и повредить центрифугу. После установки бакетного ротора в центрифугу проверьте правильность (вертикальность) положения бакетов в роторе с помощью зеркала. Все бакеты должны свободно и вертикально висеть на роторе. Убедитесь также, что каждый бакет легко поворачивается в гнезде на 90° в одну и в другую сторону.
Максимальная скорость ротора. Чрезвычайно важно вести аккуратную регистрацию каждого использования ротора, записывая показатели счетчика оборотов в конце каждой открутки, температуру, скорость, время и уровень масла в центрифуге. Эта информация необходима для контроля сроков использования ротора и его гарантий: после определенного срока все бакетные и алюминевые роторы должны эксплуатироваться с 10%-м уменьшением числа оборотов относительно максимального.
Удаление пробирок из ротора производят с помощью специального ключа, винт которого вворачивается в крышку пробирки. Пробирки с пластиковыми крышками удаляют особым ключом с иголками. При извлечении пробирки ее необходимо слегка повернуть для снятия вакуумного прилипания, затем вынуть пробирку из ротора. Для разборки крышки и высвобождения пробирки служит специальная подставка. [Примечание: не выкручивайте болтик из крышки до ее съема, иначе крышка может провалиться в пробирку]. Пробирки удаляют из бакетов щипцами или пинцетом.
Удаление содержимого пробирок. В зависимости от целей эксперимента применяют декантирование, удаление супернатанта через заливное отверстие в крышке или удаление слоя из градиента с помощью щприца, протыкая иглой боковую часть тонкостенной пробирки.
Средства ухода за пробирками, крышками и роторами. Пробирки поставляются с завода нестерильными. Их можно промыть дистиллированной водой или 1%-м раствором нейтрального детергента и высушить теплым воздухом. Нитроцеллюлозные пробирки автоклавировать нельзя во избежание взрыва.
Все пробирки, за исключением нитроцеллюлозных и старых поликарбонатных пробирок, можно стерилизовать автоклавированием. Полиалломерные пробирки при автоклавировании могут деформироваться, поэтому для них рекомендуются низкотемпературные способы стерилизации.
Крышки для очистки разбирают и промывают в теплой воде с 1%-м нейтральным мылом. Для очистки внутренней поверхности крышек используют мягкие, неабразивные инструменты, например очиститель курительных трубок. Затем крышки замачивают в горячей дистиллированной воде на 10 мин для удаления прилипшего материала, после чего их промывают дистиллированной водой и высушивают теплым воздухом.
Примечание. Осматривайте резиновые прокладки и части крышек после каждого использования, заменяя лопнувшие или деформированные прокладки.
Роторы следует регулярно осматривать для своевременного обнаружения потертостей или трещин. Промывают роторы теплой дистиллированной водой в конце работы, и сушат феном. При попадании на ротор корродирующих или солевых растворов его немедленно промывают под струей горячей воды, а затем для очистки гнезд в 1%-м нейтральном мыльном растворе ершиком для мытья пробирок. Промывают немедленно дистиллированной водой и высушивают горячим воздухом. Хранят роторы в некорродирующей атмосфере в перевернутом состоянии со снятыми крышками. Резьбовые соединения ротора периодически смазывают силиконовой смазкой. Резиновые прокладки из ротора удаляют, очищают (70%-м этанолом) и покрывают тонким слоем вакуумной смазки через каждые три цикла работы.
После каждых 100 ч откруток необходимо проверять роторы на наличие трещин, особенно в гнездах. Если трещина или изменение цвета становятся видимыми, не используйте ротор.
ЛАБОРАТОРНЫЕ ВЕСЫ И ПРАВИЛА ВЗВЕШИВАНИЯ
Весы лабораторные технические квадрантные (ВЛТК-500)
Весы лабораторные технические квадрантные, модели ВЛТК-500 (рис. П.7), предназначены для лабораторных определений массы веществ при производстве технических анализов в различных отраслях промышленности и сельского хозяйства.
Рис. П.7. Схема весов ВЛТК-500:
1 — чашка весов; 2 — тумблер включения освещения шкалы; 3 — наклонный экран шкалы; 4, 5, 6 — отклоняющие зеркала; 7 — рукоятка управления механизмом снятия гирь
Основные характеристики весов: наибольшая нагрузка до 500 г, цена деления оптической шкалы равновесия квадранта 100 мг, диапазон измерения по оптической шкале 100 г, погрешность взвешивания не более ± 0,08 г, вариация показаний не более 0,08 г, время успокоения колебаний квадранта 10 с, габариты 315 х 200 х 350 мм, масса 8 кг.
Масса груза, находящегося на чашке, определяется путем непосредственного отсчета в граммах по изображению шкалы, встроенной в маятниковый противовес (квадрант) на экране весов, и счетчику. До 100 г отсчет производится по оптической шкале. Например, по счетчику взят отсчет 1, а по оптической шкале — 27,3 г. Это будет соответствовать 127,3 г.
Весы лабораторные аналитические (ВЛА-200 г-М)
Весы (рис. П.8) предназначены для точных определений массы при лабораторном анализе. Наибольшая измеряемая масса ограничена 200 г, цена деления весов 0,1 мг, диапазон измерения весов по оптической шкале ± 10 мг, весы позволяют взвешивать груз массой от 0,2 до 200 г. Время успокоения указателя равновесия коромысла не более 10 с. Габариты 420 х 420 х 470 мм, масса 14 кг.
В основу конструкции весов заложен принцип взвешивания на равноплечем коромысле. Для регулировки нулевого положения на коромысле имеются тарировочные винты с гайками; центр тяжести регулируется верхними гайками, расположенными на вертикальном винте. Конец стрелки с отсчетной шкалой (рис. П.8, 10) располагается перед микрообъективом оптической системы, в которую входит также осветитель, конденсор и отсчетное устройство с экраном (рис. П.8, 11). Лампа осветителя подключается к сети 220 В через трансформатор, понижающий напряжение до 6,3 В. Микровыключатель, вмонтированный в цепь лампы, включает лампу только при введении весов в рабочее состояние. Включение и выключение весов производится ручкой (рис. П.8,74), надетой на валик изолирующего механизма, вынесенного на переднюю стенку основания. Весы необходимо заземлить через корпус трансформатора.
Помещение, в котором устанавливаются весы, должно быть сухим (относительная влажность воздуха 30 — 80 %), изолированным от проникновения вредно действующих паров и газов. Стены, пол и потолок не должны подвергаться сотрясениям. Весы рекомендуется устанавливать на специальный фундамент или на кронштейн, встроенный в капитальную стену. Следует избегать одностороннего нагревания или охлаждения весов.
Перед началом работы дверцы весов оставляют открытыми не менее чем на 30 мин. В нерабочем положении коромысло весов изолируется без нагрузок на чашках. Взвеши-
Рис. П.8. Внешний вид аналитических весов:
1 — основание; 2 — колонка крепления коромысла весов; 3— воздушные успокоители (демпферы); 4— штанга крепления чашек; 5 — балансир; 6 — регулировочный винт; 7 — защитное стекло; 8 — большой лимб (сотни миллиграммов); 9 — малый лимб (десятки миллиграммов); 10 — стрелки с отсчетной шкалой; 11 — отсчетное устройство с экраном; 12 — чашки с дужками; 13 — ножки регулировочные для установки весов по уровню; 14 — ручка включения и арретирования
весов; 75 — ручка нониуса
ваемый объект и разновесы разрешается помещать на чашки и снимать их только при закрытом изолире. Открывать и закрывать изолир нужно осторожно, путем плавного вращения ручки. Взвешивание следует производить всегда в чистой посуде. Вещества, выделяющие газы, надо взвешивать в закрытой посуде. Взвешивание объектов необходимо проводить после принятия ими температуры окружающей среды.
Весы следует регулярно осматривать и очищать от пыли. Ее удаляют мягкой фланелевой тряпочкой с наружных и внутренних поверхностей витрины, с основания весов и чашек, которые предварительно вынимают из витрины.
Весы аналитические электронные ER-60A
Основные характеристики весов (рис. П.9): наибольшая измеряемая масса ограничена 60 г, цена деления весов 0,1 мг, стандартное отклонение 0,1 мг, погрешность
взвешивания ±0,2 мг. Время успокоения колебаний чаши около 5 с. Габариты: 195 х 411 к 266 мм, масса 11 кг, диаметр чаши весов 85 мм.
Весы должны быть установлены на столе жесткой конструкции, в углу комнаты, на удалении от дверей и окон. Необходимо исключить воздействие на них прямых солнечных лучей, а также приборов, содержащих магниты или являющихся источниками электромагнитного излучения. Весы следует устанавливать горизонтально и заземлять, а с взвешиваемых объектов снимать электростатический заряд.
Перед началом работы убеждаются, что весы установлены горизонтально и на их чаше нет загрязнений. Включив весы в сеть и нажав кнопку on/off, дают прогреться в течение 30 мин. Циф-
Рис. П.9. Схема аналитических
ровой индикатор весов должен показывать весов
«0,0000».
При первоначальной установке прибора, а
также при его перемещении весы необходимо откалибровать. Кроме того, калибровка производится один раз в день перед началом работы. Калибровка осуществляется автоматически, через внутренний стандарт массы.
Калибровка
1. Когда на цифровом индикаторе устанавливается надпись «0,0000», нажать кнопку «CAL» (включение режима калибровки).
2. На цифровом индикаторе появится надпись «CAL in» и через несколько секунд «CAL». Несколько секунд спустя эта надпись сменится на «CAL dn».
3. После появления надписи «CAL dn» необходимо медленно и аккуратно опустить вниз рычажок, расположенный на правой боковой стенке.
4. На цифровом индикаторе появится надпись «CAL».
5. Через некоторое время появится «CAL ир» и тогда надо медленно перевести рычажок в исходное положение.
6. На цифровом индикаторе появится «С4£», затем через несколько секунд появится надпись «CAL end», которая сменится на «0,0000». Калибровка окончена.
7. Если в процессе калибровки на цифровом индикаторе появится «CAL по», следует выключить весы кнопкой on/off и включить их снова.
Взвешивание
1. Поместите тару на чашку весов.
2. Нажмите кнопку «TARE» для приведения веса тары к нулю.
3. Поместите взвешиваемый образец в таре на чашку весов и прочитайте на индикаторе показания чистого веса образца.
4. Снимите тару с образцом с чашки. Для приведения показаний к нулю нажмите « TARE».
УЛЬТРАТЕРМОСТАТ U-10 (ФРГ)
При помощи термостата U-10 (рис. П.10) можно поддерживать постоянную температуру от -60 °C до +300 °C. Этот прибор обладает всеми свойствами, предъявляемыми к термостатам, работающим по принципу циркуляции жидкости. С помощью термостата можно привести в циркуляцию жидкость, предназначенную для регулировки температуры закрытых и открытых потребителей. Управление температурой осуществляется посредством заменяемых контактных термометров.
Технические характеристики прибора: точность регулировки ±0,02 °C; объем водяной бани 12 л; мощность насоса 8 л/мин; мощность обогрева до 1200 Вт; напряжение питания 220 В; 50 Гц. Габариты 400 к 370 х 530 мм, масса 18 кг.
Перед работой на ультратермостате необходимо вставить нагнетательный насос с мотором в отверстие верхней плиты прибора и зажать его при помощи быстродействующих защелок. Штепсель электродвигателя вставить в штепсельную розетку, помещенную на стороне реле. Патрубки насоса (4, 5) соединить с потребителем, температуру которого следует регулировать. При непосредственной работе в бане термостата нужно замкнуть накоротко насос термостата при помощи шланга.
Наполнить термостат жидкостью. Предельный уровень заполнения контролируют по указателю уровня жидкости (2). Следует применять дистиллированную воду, так как водопроводная вода образует осадок и покрывает налетом присоединенные измерительные приборы. Для пониженных температур до -60 °C можно применять метиловый или этиловый спирты или их смеси с водой в различных соотношениях и сухой лед; при температурах от 80 до 160 °C — глицерол (чистый или смешанный с водой в соответствующем соотношении) или очищенное минеральное масло с точкой воспламенения выше 180 °C. При работе с более высокими температурами (160 — 300 °C) применяют цилиндровое масло для перегретого пара с высокой точкой воспламенения.
Рис. П.10. Внешний вид ультратермостата U-10:
1 — корпус прибора; 2 — указатель уровня жидкости; 3 — откидные ручки для переноски прибора; 4, 5 — патрубки для заполнения и слива жидкости; 6 — кнопка управления сливом жидкости; 7— опорная плита; 8 — переставляемая подъемная платформа; 9 — зажимные винты платформы; 10 — кольца водяной бани; 11 — крышка; 12 — холодильный змеевик; 13, 14 — шланговые втулки; 75 — контрольный термометр; 16 — контактный термометр; 17— защитная трубка контактного термометра; 18 —поворотный магнит контактного термометра; 19 — релейный кабель контактного термометра; 20 — электронное реле
В верхнюю плиту прибора вставить контрольный термометр (15) со шлифом и контактный термометр (16) с защитной трубкой. Перед пуском прибора проверить контактный термометр. Специальным кабелем присоединить контактный термометр к двухполюсной штепсельной розетке, расположенной на реле прибора слева вверху. По контактному термометру (верхний край ходовой гайки) установить нужную температуру. При рабочих температурах ниже 20 °C следует соединить с водопроводом холодильный змеевик.
Подсоединить прибор к сети. Прибор включают, управляя ручкой выключателя электронного реле (20). Возможны следующие положения выключателя:
0 — выключено; НО — включены реле и электродвигатель;
Н1 — включен нагреватель 270 Вт;
Н2 - 400 Вт;
НЗ - 800 Вт;
Н4 — 1200 Вт. Целесообразно выбрать оптимальную мощность обогрева термостата при существующих рабочих условиях. В этом случае время работы термостата и время его отключения будут приблизительно равны. При этом следует учитывать, что чем ниже рабочая температура, тем меньше должна быть мощность нагревательных элементов, и наоборот.
ФОТОЭЛЕКТРОКОЛОРИМЕТР-НЕФЕЛОМЕТР (ФЭК-56М)
Колориметр-нефелометр фотоэлектрический ФЭК-56М (рис. П.11) предназначен для определения концентрации растворов различных веществ колориметрическим (фотометрическим) методом, а также для измерения коэффициента пропускания или оптической плотности веществ. Прибор позволяет также производить измерение свето-рассеивания взвесей, эмульсий и коллоидных растворов в проходящем свете. Для определения концентрации растворов производится предварительная градуировка прибора по набору стандартных растворов с известной концентрацией.
Эксплуатировать прибор следует в чистом помещении при температуре окружающей среды 20 ± 5 °C и относительной влажности воздуха не более 80 %. Погрешность прибора при измерении коэффициента пропускания не превышает ±1 абс.%, вариация показаний прибора не превышает 0,3 абс.%. Прибор обеспечивает измерение све-топропускания от 100 до 5 % (оптическая плотность от 0 до 1,3). Участок шкалы от 5 до 0,1 % по светопропусканию (по оптической плотности от 1,3 до 3) служит для ориентировочных измерений. Цена деления шкалы коэффициента светопропускания (%) на участках от 100 до 10 % — 1; от 10 до 2 % — 0,5; от 2 до 1 % — 0,2.
Рис. П.11. Общий вид фотоэлектроколориметра ФЭК-56М:
1 — крышка прибора; 2 — рукоятка шторки; 3 — рукоятка для перемещения кювет; 4 — измерительный барабан;
5 — компенсационынй барабан; 6 — микроамперметр
Прибор снабжен набором кювет объемом от 0,5 до 20 мл. В качестве источников света в приборе применяются лампа накаливания 8 В, 35 Вт (СЦ-98) и ртутно-кварцевая лампа сверхвысокого давления (СВД-120А) мощностью 120 Вт. Время непрерывной работы прибора с включенной лампой СЦ-98 не более 8 ч, с включенной лампой СВД-120А не более 4 ч. С двумя лампами возможна работа в диапазоне длин волн от 315 до 630 нм. Лампу СВД-120А применяют при измерении в ультрафиолетовой области спектра. С этой же лампой можно измерять и в видимой области, если имеется необходимость пользоваться очень узкими спектральными линиями спектра. В приборе используется излучение лампы СВД-120А следующих длин волн, нм: 577/9, 564, 436, 405/8, 365, 313. Приемниками световой энергии в приборе служат два сурьмяно-цези-евых фотоэлемента Ф-4 и Ф-26, включенных по дифференциальной схеме через усилитель на микроамперметр. Потребляемая мощность с лампой СЦ-98 не более 50 Вт, с лампой СВД-120А не более 280 Вт. Питание прибора производится от сети переменного тока напряжением 220 + 22 В частотой 50 ± 5 Гц. Габариты прибора 382 к 270 х 187 мм, блока питания 315 х 210 х 141 мм. Масса прибора (общая) 22,5 кг.
Устройство и работа прибора
Принцип измерения светопропускания состоит в том, что на фотоэлементы направляют поочередно световые потоки — полный и пропущенный через исследуемую среду, и определяют их соотношение. Оптическая схема фотоэлектроколориметра ФЭК-56М изображена на рис. П.12. Световой пучок от источника света (7) падает на призму (2), которая делит его на два пучка — левый и правый. Отразившись от зеркала (3) и пройдя через линзы, пучки света идут параллельно, проходят через кюветы (4) и попадают на фотоэлементы (6). Последние соединены с гальванометром. При равенстве интенсивности световых пучков, падающих на фотоэлемент, стрелка гальванометра не отклоняется. Если же на пути одного из параллельно падающих лучей помещена кювета с раствором, рассеивающим свет, то интенсивность света, прошедшего через нее, становится меньше интенсивности света, прошедшего через кювету с растворителем, и стрелка гальванометра отклоняется. Для выравнивания интенсивности световых потоков в правых и левых барабанах имеются раздвижные диафрагмы (5).
Правый световой поток — измерительный, левый — компенсационный. Обозначив полный световой поток буквой Фо, а пропущенный через среду Ф, получим значение коэффициента светопропускания (%) в виде соотношения
Ф-100
т =------
Фо
Рис. П.12. Оптическая схема фотоэлектроколориметра. Обозначения см. в тексте
На приборе это отношение определяется следующим образом. В правый световой пучок помещают кювету с исследуемым раствором. Раздвижная диафрагма правого плеча полностью открыта (отсчет 10 по шкале светопропускания), что соответствует полному световому потоку Фо. Диафрагма левого плеча тоже должна быть открытой. Вследствие поглощения или рассеяния света раствором на правый фотоэлемент будет падать световой поток меньшей интенсивности, чем на левый, и стрелка микроамперметра будет отклоняться. Вращая барабан левой раздвижной диафрагмы, уравнивают интенсивности обоих световых потоков и стрелку микроамперметра устанавли
вают на «О». Затем кювету с раствором в правом плече заменяют кюветой с растворителем, по отношению к которой производится измерение раствора. Фотометрическое равновесие вновь нарушается, так как растворитель прозрачнее, и интенсивность светового потока, падающего на правый фотоэлемент, увеличивается.
Вращая правый барабан, уменьшают интенсивность светового потока до первоначальной, т.е. стрелка микроамперметра опять должна быть на «О». Полученный по шкале правого барабана отсчет будет соответствовать световому потоку Ф; этот отсчет дает коэффициент светопропускания промеренного раствора (%), так как
•100
100
В момент установки стрелки микроамперметра на «0» освещенность в измерительном (правом) фотоэлементе остается неизменной как в начале каждого измерения, так и в конце, что исключает ошибки, связанные с нелинейностью характеристик фотоэлементов.
Использование прибора для нефелометрических определений основано на принципе измерения направленного светопропускания исследуемого раствора. Другими словами, определение светорассеяния осуществляется косвенно по светопропусканию мутного раствора по отношению к прозрачному растворителю или к воде. Светорассеяние растворов со слабой мутностью, светопропускание которых незначительно отличается от светопропускания растворителя, на данном приборе определять нельзя.
В корпус прибора вмонтированы: узел светофильтров, узел кюветодержателя, измерительные диафрагмы с отсчетными барабанами, узел зеркал и фотоэлементов, микроамперметр, усилитель и стабилизатор. Девять стеклянных светофильтров вмонтированы в диск, укрепленный на задней стенке корпуса прибора (табл. П.1).
Светофильтр в световой пучок включается рукояткой. Цифры на шкале показывают, какие светофильтры включены. Рабочее положение каждого светофильтра фиксируется. При необходимости замены светофильтра открывают крышку со стороны осветителя и с помощью прилагаемого к прибору ключа вынимают светофильтр, заменяя
его другим.
В правом кюветодержателе устанавливают две кюветы. Их переключение производят поворотом рукоятки до упора. Рукоятка скреплена с червяком, по которому перемещается винт, несущий на себе кю-ветодержатель. В левом пучке устанавливают при необходимости одну кювету. К прибору прилагаются четыре комплекта кювет. Каждый комплект состоит из семи кювет с нижеуказанными расстояниями между рабочими гранями (мм): 50, 30, 20, 10, 5, 3, 1.
На правый барабан нанесены две шкалы: черная — шкала светопропускания (процентные отношения Ф/Фо) и красная, соответствующая оптической плотности (£>) образца, под которой понимается десятичный логарифм величины, обратной коэффициенту пропускания (см. табл. П.5). Таким образом, если при данной установке барабана отсчет по шкале светопропускания равен т, то по шкале оптической плотности D = -1g т.
Таблица П.5
Светофильтры прибора ФЭК-56М
Маркировка светофильтра Длина волны, соответствующая максимуму пропускания, нм Полуширина пропускания, нм
1 315+5 35+15
2 364+5 25+10
3 400+5 45+10
4 440±10 40+15
5 490+10 35+10
6 540+10 25+10
7 580+10 30+10
8 597+10 —
9 630+10 —
На наклонной плоскости корпуса прибора укреплен микроамперметр. В момент равенства фототоков стрелка микроамперметра устанавливается на «О». Узел шторки расположен на верхней части прибора. Открытие и закрытие светового окна осуществляют переключением рукоятки.
Блок питания с прибором соединяется через штепсельный разъем. Все основные узлы блока питания смонтированы на массивном основании и закрыты кожухом, на боковых стенках которого имеются вентиляционные окна для охлаждения рабочих частей блока питания. Тумблер служит для переключения ламп СЦ-98 и СВД-120А. На вилке, посредством которой блок питания включается в сеть 220 В, выведен контакт с гайкой. Перед началом работы к этому контакту подсоединяется заземляющий проводник, в результате чего заземляются прибор и блок питания. Блок питания снабжен предохранителем на 5А и выключателем сетевого напряжения.
Методика определения концентрации вещества в окрашенных и мутных растворах одинакова, поэтому все дальнейшие указания по работе прибора являются общими для колориметрических и нефелометрических измерений. Измерения на приборе можно начинать спустя 30 мин после включения блока питания и лампы СЦ-98 фотометрической. В течение этого времени электросхема прибора будет прогрета, и наступает достаточно стабильный режим ее работы. При работе с ртутной лампой ее включают за 10—15 мин до начала измерения, но так же после 30-минутного предварительного прогрева электросхемы с лампой СЦ-98. Нельзя оставлять без надобности включенной ртутную лампу прибора, так как это сокращает срок службы лампы. Кроме того, лампа разогревает светофильтры прибора, что также нежелательно. В случае перерыва в работе более 20 мин ртутную лампу следует выключать.
При работе с ртутной лампой и лампой накаливания с некоторыми светофильтрами поступающий на фотоэлементы световой поток оказывается чрезмерно высоким, что приводит к нестабильной работе прибора и выражается в колебании стрелки микроамперметра. В таких случаях необходимо уменьшить чувствительность прибора электрически, повернув рукоятку чувствительности по часовой стрелке, либо, если нестабильность остается высокой, ввести в пучок лучей нейтральные светофильтры, приложенные к прибору. Нейтральные светофильтры устанавливаются в световые окна в кюветном отделении.
Рабочие поверхности кювет перед каждым измерением следует тщательно протирать. При установке кювет в кюветодержателе нельзя касаться пальцами рабочих участков поверхностей (ниже уровня жидкости в кювете). Наличие загрязнений или капель раствора на рабочих поверхностях кюветы искажает результаты измерений.
Если в процессе работы светофильтр, с которым производилось измерение, меняется на другой, то измерения рекомендуется производить не ранее чем через 5 мин после смены светофильтра.
Измерение светопропускания или оптической плотности раствора
Устанавливают «электрический нуль» прибора, перекрывая световые пучки шторкой. Рукояткой нулевого тока стрелку микроамперметра подводят к нулю. В левом световом пучке на все время измерения помещают кювету с растворителем. Если растворитель не окрашен, можно ставить кювету с дистиллированной водой. Это делают, чтобы исключить возможность разогревания левого фотоэлемента теплом светового пучка. Известно, что вода и водные растворы очень хорошо поглощают тепловые лучи. В ряде случаев кювету в левый пучок не ставят. В правый пучок света помещают кювету с исследуемым раствором. Индекс правого барабана выводят на отсчет 100 по шкале светопропускания. Вращением левого измерительного барабана добиваются установки стрелки микроамперметра на «0».
Если левым измерительным барабаном установить «0» не удается, то в правый световой пучок прибора вводят нейтральный светофильтр, который обеспечит установку
«О» левым барабаном. Затем поворотом рукоятки кювета с раствором в правом пучке заменяется кюветой с растворителем, при этом стрелка микроамперметра смещается от нулевого положения. Вращением правого измерительного барабана стрелку возвращают в положение «О» и отсчитывают по шкале этого барабана величину светопропускания или оптической плотности замеренного раствора.
Чтобы избежать ошибок, которые могут возникать в процессе измерения, рекомендуется не ограничиваться одним измерением. При измерениях барабан измерительной диафрагмы необходимо подводить к индексу с одной стороны — для исключения люфта в механизме и достижения вариаций не более 0,3 %.
Выбор светофильтра. Наличие в приборе комплекта светофильтров и набора кювет позволяет подобрать такое их сочетание, при котором ошибка в определении концентрации будет наименьшей. При работе со светофильтрами № 1 и 2 рекомендуется применять лампу СВД-120А. Максимум пропускания светофильтров № 3, 4, 6, 7 примерно совпадает со спектральными линиями ртути (405, 436, 546, 578 нм), что позволяет использовать эти светофильтры при работе с ртутной лампой.
Выбор светофильтра производят следующим образом. Раствор наливают в кювету (о выборе размера кювет см. ниже) и производится определение оптической плотности для всех светофильтров (№ 1 — 9) на двух лампах. По полученным данным строят кривую, откладывая по горизонтали длины волн, соответствующие максимуму светопропускания светофильтров, указанные в описании прибора, а по вертикальной оси — соответствующие значения оптической плотности раствора. Отмечают тот участок кривой, для которого выполняются следующие условия:
1) оптическая плотность имеет значительную величину;
2) ход кривой примерно параллелен горизонтальной оси, т.е. оптическая плотность мало зависит от длины волн.
Светофильтр для работы выбирается так, чтобы длина волны, соответствующая его максимуму светопропускания, приходилась на отмеченный выше участок спектральной кривой испытуемого раствора. Если эти условия выполняются для нескольких светофильтров, то выбирают тот из них, для которого чувствительность прибора выше.
Выбор кюветы. Как выше указывалось, абсолютная ошибка измерения коэффициента светопропускания не превышает 1 %. Относительная ошибка определения концентрации раствора будет различной при работе на разных участках шкалы прибора и достигает минимума при значении оптической плотности 0,4. Поэтому при работе на приборе рекомендуется путем соответствующего выбора кювет работать вблизи указанного значения оптической плотности. Предварительный выбор кювет производится визуально, соответственно интенсивности окраски раствора. Если раствор интенсивно окрашен (темный), следует пользоваться кюветами с малой рабочей длиной (1 — 3 мм). В случае слабоокрашенных растворов рекомендуется использовать кюветы с большой рабочей длиной (30 — 50 мм).
В предварительно подобранную кювету наливают раствор и измеряют его оптическую плотность, включив в ход лучей соответствующий для данного раствора светофильтр. При измерении ряда растворов кювету заполняют раствором средней концентрации. Если полученное значение оптической плотности составляет примерно 0,3 —0,5, данную кювету выбирают для работы с этим раствором. Если D > 0,5, берут кювету с меньшей рабочей длиной: при D < 0,3 следует выбрать кювету с большей рабочей длиной.
Определение концентрации вещества в растворе. При определении концентрации вещества в растворе следует соблюдать определенную последовательность в работе:
выбор светофильтра;
выбор кюветы;
построение калибровочной кривой для данного вещества;
измерение оптической плотности исследуемого раствора и определение концентрации вещества в растворе.
Построение калибровочной кривой производится следующим образом. Готовят ряд растворов данного вещества с известными концентрациями, охватывающими область
возможных изменений концентраций этого вещества в исследуемом растворе. Измеряют оптические плотности всех растворов и строят калибровочную кривую, откладывая на горизонтальной оси известные концентрации, а на вертикальной — соответствующие им значения оптической плотности. По калибровочной кривой в дальнейшем определяют неизвестную концентрацию вещества в исследуемых растворах. Для этого раствор наливают в ту же кювету, для которой построена калибровочная кривая, и, включив тот же светофильтр, определяют оптическую плотность раствора. Затем по калибровочной кривой находят концентрацию, соответствующую измеренному значению оптической плотности.
В целях получения точных результатов измерений, а также во избежание повреждений прибора необходимо все оптические детали прибора и лампочки оберегать от запыления.
РН-МЕТР — МИЛЛИВОЛЬТМЕТР РН-121 И ПРАВИЛА РАБОТЫ НА НЕМ
Прибор (рис. П.13) предназначен для измерения активности ионов водорода (pH) и окислительно-восстановительного потенциала (/:),) водных растворов, а также для использования в качестве высокоомного нуль-индикатора и милливольтметра. Применяя электроды, селективные к одновалентным катионам, с помощью этого прибора можно измерять активности этих катионов (рХ+), а работа с блоком автоматического титрования дает возможность использовать прибор для массового однотипного титрования. Прибором pH-121 можно производить измерения как методом отбора проб, так и непрерывно в лабораторных установках.
Диапазоны измерения величины pH преобразователем следующие: -1+14; -1+4; 4 + 9; 9+14. Пределы измерения величины pH (рХ+) определяются типом применяемых электродов. Пределы измерения ЭДС от +100 до ±1400 мВ с диапазонами:
-1004-1400 или 100-^-1400 мВ;
-1004-400 или 1004—400 мВ;
4004-900 или -4004--900 мВ;
9004-1400 или -900 л- -1400 мВ.
Минимальный объем дозы измерения 0,5 мл. Потребляемая мощность не более 50 Вт. Габаритные размеры преобразователя pH-121 не более 365 к 230 к 260 мм; размеры подставки не более 160 х 190 х 500 мм. Масса прибора pH-121 не превышает 15 кг.
Работа pH-метра основана на преобразовании ЭДС электродной системы, состоящей из измерительного и вспомогательного электродов, в постоянный ток, пропорциональный измеряемой величине. Преобразование ЭДС электродной системы в постоянный ток осуществляется высокоомным преобразователем с автокомпенсаци-онным принципом действия. Преобразователь состоит из усилителя, измерительного блока, позволяющего подстраиваться под реальную электродную систему, и блока питания.
На шкале показывающего прибора имеются следующие оцифровки: -14-14 — используется при измерении на широком диапазоне; -14-4, 44-9, 94-14 — при измерении на соответствующем узком диапазоне; 0+100 — для установки ручного термокомпенсатора («температура раствора») в положение, соответствующее температуре контролируемого раствора.
К органам оперативного управления относятся: ручки переменных резисторов «калибровка», «pH,,», «крутизна», «НИ» (при нажатой кнопке «0; I» и отжатой кнопке «НИ» устанавливают ручку «температура раствора» в положение, соответствующее температуре контролируемого раствора, а при нажатой кнопке «НИ» ручкой «НИ» устанавливают 0 нуль-индикатора) и ручка «температура раствора»; 4 кнопки выбора рода работы: «pH», «+mv», «-mv», «0; Г»; кнопка выбора подключения высокоомного гнезда «изм. 1» или « изм. 2»; -«изм. 1»/«изм. 2»; 5 кнопок выбора диапазона измерения: «НИ»,
7
8
Рис. П.13. рН-метр — милливольтметр pH-121:
1 — нижняя планка; 2 — ручки оперативного управления прибором; 3 — цанговый зажим; 4 — корректор нуля; 5 — лицевая панель; 6 — показывающий прибор; 7 — кнопка выбора рода работы; 8 — глазок индикации включения; 9 — кнопки выбора диапазона измерения
«-1-5-14», «-1-5-4», «4-5-9», «9-5-14»; корректор показывающего прибора; ось переменного резистора R25 — «Д, грубо», которая расположена под нижней планкой.
Для предотвращения случайного поворачивания ручки переменных резисторов «калибровка», «рНи», «НИ» и «крутизна» после настройки фиксируются гайками цанговых зажимов. Оси этих резисторов снабжены удлинителями, которые могут быть сняты при нагреве паяльником и приклеены на новый резистор клеем БФ-2 или подобным.
Резисторы «Е1( грубо», «калибровка», «крутизна», «рНи» служат для настройки прибора на данную электродную систему. Резистор R9 «НИ» служит для настройки прибора при использовании его в качестве нуль-индикатора. Ось переменного резистора «Г,, грубо» фиксируется цанговым зажимом.
Подставка прибора предназначена для крепления электродов и установки сосудов с контролируемым раствором. На подставке располагаются два кронштейна, высоту их установки можно регулировать в зависимости от вида измерений (в стакане, в термостатированной ячейке или в ячейке для микроизмерений). Кронштейн, на котором закрепляется столик или упор, поворачивается в горизонтальной плоскости. На кронштейне расположен держатель, который имеет отверстия для установки электродов, термометра и автоматического термокомпенсатора.
Подготовка прибора и порядок работы. Включить прибор в сеть 220 В, 50 Гц, прогреть его не менее 30 мин. Прибор готов к работе. Налить дистиллированную воду в стакан. Отвести столик в сторону. Погрузив электроды в стакан, подвести под него столик. Отрегулировать положение кронштейнов таким образом, чтобы электроды при погружении в стакан не доходили до дна на 4—6 мм, закрепить кронштейны винтами. Электроды перед погружением в буферный или контролируемый раствор необходимо тщательно промыть дистиллированной водой, остатки воды с электродов удалить фильтровальной бумагой. Необходимо иметь в виду, что буферные и контрольные растворы при многократном применении могут изменять значение pH. Прежде чем производить корректировку прибора с помощью ручки «Калибровка», следует убедиться в том, что
погрешность измерения вызвана изменением настройки прибора, а не изменением pH буферного или контрольного раствора. Изменение настройки прибора может быть обнаружено проверкой по свежеприготовленному буферному или контрольному раствору.
По окончании работы с прибором электроды для измерения pH должны оставаться погруженными в воду или в 0,1 М раствор соляной кислоты, а электроды для измерения рХ других ионов — в соответствующие растворы, указанные в паспортах на эти электроды.
В первые несколько дней эксплуатации прибора или нового стеклянного электрода проверку прибора по буферным или контрольным растворам следует производить каждый день, так как характеристики стеклянного электрода могут изменяться. При последующей работе это можно делать реже (до 1 раза в 3 дня).
Ручной температурной компенсацией рекомендуется пользоваться в случае измерения pH (рХ) растворов, имеющих постоянную температуру. При этом переключатель рода термокомпенсации установить в положение «Руч», на переключателе рода работ нажать кнопку «0; и на переключателе диапазонов — кнопку любого диапазона, кроме нуль-индикатора (НИ). Рукояткой «Температура раствора» выставить на верхней шкале показывающего прибора измеренное значение температуры контролируемого раствора, руководствуясь оцифровкой 0... 100. В случае измерения pH (рХ) растворов, температура которых меняется, рекомендуется применять автоматическую температурную компенсацию. Переключатель рода термокомпенсации установить в положение «авт», в держатель установить автоматический термокомпенсатор, который подключить в розетку «термокомпенсатор». Отсчет по шкале прибора следует производить после того, как его показания установятся. Обычно время установления показаний не превышает 3 мин.
В случае измерения pH сильнокислых и сильнощелочных растворов при температурах, близких к нулю, или измерениях pH растворов с очень маленькой буферной емкостью время установления показаний может значительно возрасти (до 10 мин).
При замене контролируемого раствора и по окончании измерений должна быть нажата кнопка «0; Г». Для обеспечения надежного контактирования рекомендуется производить 3 — 4 включения — переключения каждой кнопки переключателей. При работе в одном из узких диапазонов (-1<-4, 4-г 9, 9^ 14) отсчет показаний необходимо производить по верхней шкале показывающего прибора, руководствуясь оцифровкой, соответствующей выбранному диапазону измерения; при нажатии кнопки «-1 -н 14» отсчет производят по нижней шкале.
Буферные растворы и их приготовление. Буферные растворы готовят из реактивов квалификации «для pH-метрии», которые выпускаются в виде фиксаналов, рассчитанных на приготовление 1000 мл буферного раствора каждого наименования. Для приготовления буферных растворов применяют дистиллированную воду, предварительно прокипяченную в течение 30 — 40 мин, чтобы удалить растворенную углекислоту.
Не следует производить проверку pH-метра по растворам, приготовленным из случайно имеющихся реактивов, так как при этом возможны значительные ошибки в величинах pH приготовленных растворов.
Настройка приборов по буферным растворам. Настройку прибора рекомендуется производить по буферному раствору со значениями pH, расположенными в диапазоне, на котором производятся измерения. Настраивание приборов для измерения pH растворов с постоянной температурой рекомендуется производить по буферным растворам при этой температуре. При настройке и в процессе измерения рекомендуется использовать один и тот же вид температурной компенсации.
Настройка производится по буферным растворам с температурой 20 °C и по буферному раствору с температурой, близкой к предельной температуре контролируемого раствора в следующем порядке. При ручной температурной компенсации нажать кнопку «0; Г» (кнопка «НИ» должна быть отжата), ручкой «Температура раствора» установить стрелку показывающего прибора против отметки, соответствующей 20 °C. Поместить электроды в буферный раствор с температурой 20 °C. Нажать кнопку выбора ди
апазона измерения, а затем кнопку «pH». Ручкой «Калибровка» установить стрелку показывающего прибора на отметку, равную значению pH буферного раствора при 20 °C. Проверить показания прибора в буферном растворе со значением pH, наиболее удаленном от первоначального. В случае необходимости откорректировать показания прибора ручкой «Крутизна». Ручку «Температура раствора» установить в положение, соответствующее выбранной температуре буферного раствора. Поместить электроды в раствор. Нажать кнопку выбора диапазона и кнопку «pH». Ручкой «рНи» установить показания прибора, соответствующие значению pH буферного раствора при данной температуре. При использовании автоматической температурной компенсации в растворы помещается также автоматический температурный автокомпенсатор.
Меры безопасности. К работе с прибором допускаются лица, изучившие паспорт прибора, а также действующие правила по эксплуатации электроустановок и правила работы с химическими растворами. Прибор и мешалка в процессе работы должны быть надежно заземлены. Не разрешается работать с прибором, у которого не светится глазок индикации включения.
ПРИБОР ДЛЯ СЧЕТА КОЛОНИЙ
Рис. П. 14. Общий вид прибора для счета колоний:
1 — столик для чашки Петри; 2 — перо с пружинным устройством; 3 — показатель счетчика; 4 — тумблер для импульсного счетчика; 5 — тумблер для включения лампы освещения столика
Прибор предназначен для счета колоний бактерий в чашках Петри (рис. П.14).
Устройство прибора. Напряжение питающей сети 220/127 В; частота 50 Гц; потребляемая мощность 30 Вт; количество цветных светофильтров 4; габаритные размеры 300 х 240 х 180 мм; масса 4,5 кг. Прибор состоит из корпуса, диска со светофильтрами, электропера, лупы, импульсного счетчика, источника освещения. На диске по окружности расположены пять отверстий, в четырех из них имеются светофильтры разных цветов. Диск со светофильтрами вращается вокруг вертикальной оси, установленной в скобе. На передней панели укреплены тумблеры для включения лампочки освещения и импульсного счетчика. В раструбе верхней крышки установлены два стекла: термоизоляционное (предохраняющее бактерии от теплового воздействия лучей) и молочное с растровой сеткой. Электроперо состоит из кожуха, в котором установлены подвижная и неподвижная втулки. Между втулками находится пружина. В пазах неподвижной втулки установлены пластинчатые контакты. В подвижную втулку ввинчены с одной стороны авторучка, а с другой — винт для регулировки зазоров между контактами. Свет от лампочки проходит через светофильтр (цвет которого подбирается для лучшей контрастности), затем через термозащитное стекло, создавая равномерную освещенность.
Чашку Петри с колониями бактерий устанавливают вверх дном на молочном стекле. Растровая сетка на стекле предназначена для систематизации при зондировании колоний электропером. Для подсчета колоний бактерий электропером на дно чашки наносят точки в тех местах, где находится каждая колония, при этом контакты электоропе-ра замыкаются, и поступающий к счетчику электрический импульс производит отсчет. Для удобства работы прибор снабжен авторучками с чернилами различного цвета. В случае необходимости
вместо молочного стекла в приборе можно использовать темный экран с растровой сеткой. Для установки прибора в наклонное положение имеется откидная скоба.
Работа с прибором. Включить прибор в сеть. Включить тумблер освещения (загорается лампочка). Чашку Петри поместить на растровое стекло вверх дном. Установить лупу так, чтобы хорошо просматривались зондируемые колонии и растровая сетка. Поворачивая диск, подобрать нужный светофильтр. Взять авторучку с нужным цветом чернил и ввернуть ее в корпус электропера. Включить тумблер импульсного счетчика. Нажав на перо, проверить его работу. При каждом нажатии показания импульсного счетчика должны увеличиваться на единицу. Отсчет количества колоний бактерий производится путем вычисления разности показаний импульсного счетчика до начала отсчета и после его окончания.
УСТРОЙСТВО ПОЛЯРОГРАФА И ПОЛЯРОГРАФИЧЕСКИЕ ИЗМЕРЕНИЯ СОДЕРЖАНИЯ КИСЛОРОДА В СРЕДЕ
Платиновый электрод полярографического датчика (рис. П.15) при напряжении от -0,5 до -0,6 В потребляет кислород согласно уравнению: О2 + 2Н2О + 4ё = 4ОН-. Из уравнения следует, что возникает ток к электроду. Этот ток, протекая через сопротивление, наводит потенциал на его концах. Если электрод соединить соляным мостиком с каломельным или Ag—AgCl-электродом, то потенциал между ними может быть измерен. Альтернативно этому можно измерять ток гальванометром.
Платиновые электроды бывают стационарные, вращающиеся или колеблющиеся для снижения градиента концентрации кислорода, возникающего у поверхности электрода. Такой электрод, однако, обычно загрязняется в биологических экспериментах. Для снижения загрязнения электрод закрывают газопроницаемыми мембранами (тефлон) подобно «закрытому» электроду Кларка. Ток, возникающий на электроде, пропорционален скорости диффузии кислорода через мембрану. При такой конструкции электрода возникает градиент через мембрану до нулевой концентрации кислорода у поверхности электрода, где кислород восстанавливается. Скорость диффузии кислорода через мембрану линейно зависит от его концентрации или парциального давления. Величина тока зависит также от величины поверхности электрода.
График увеличения тока с увеличением напряжения от 0 до 2 В (полярограмма) имеет область плато между -0,5 и -0,8 В. Именно при этих значениях на электроде восстанавливается растворенный кислород. Для работы выбирают напряжение, соответствующее среднему значению области плато (-0,65 — 0,70 В). Увеличение тока в ответ на увеличение приложенного напряжения — основное свойство кислородного электрода. Ток, возникающий на электроде, чрезвычайно чувствителен к изменению температуры, так
Рис. П.15. Схема внешнего вида кислородного электрода «закрытого» типа:
1 — проводники для подключения к полярографу; 2 — уплотнительные гайки; 3 — корпус электрода; 4 — выступающий платиновый электрод, покрытый газопроницаемой мембраной; 5— отверстие
для добавок; 6 — ячейка с внутренним объемом 1,5 мл
как материал мембраны имеет высокий температурный коэффициент (около 3 — 5 % на градус). Кроме того, приходится учитывать разницу в растворимости кислорода при различных температурах. По этой причине необходимо строго контролировать температуру растворов и вводить соответствующие поправки в измерения.
Калибровка и расчеты. Калибровка прибора основана на известной растворимости кислорода в воде при определенных условиях. Нулевую концентрацию кислорода и остаточный ток определяют при измерении с дитионитом натрия. Ток при 100%-м насыщении раствора кислородом регистрируется с использованием дистиллированной воды, насыщенной кислородом воздуха при 25 °C. Для большинства экспериментов точная концентрация кислорода в стандартных растворах не нужна, необходимы лишь растворы с постоянно воспроизводимыми концентрациями для каждодневной юстировки прибора, поскольку в полярографических измерениях кислорода, поглощаемого или выделяемого биологическими объектами, важны относительные измерения; сравнение всегда идет с контролем.
Содержание кислорода в стандартных растворах зависит от температуры, концентрации солей и атмосферного давления. Изменение атмосферного давления может вызвать изменение содержания кислорода в стандартных растворах на +3 %. Если необходимо, то коррекцию на истинное атмосферное давление вводят по уравнению:
истинное атмосферное давление: 760 х растворимость кислорода в растворе при 760 мм рт.ст. = истинное содержание кислорода в растворе.
Измерение концентрации кислорода в пробе. Перед началом экспериментов электрод (рис. П.15) должен быть проверен, имеет ли он «плато-область» в районе -0,5...0,8 В. Для этого включают развертку напряжения полярографа от 0 до -0,9 В с одновременной регистрацией изменения тока на самописце. В области -0,5...0,8 В у хорошо работающего кислородного электрода не должно быть изменения тока. Проверяют также быстроту ответа (установление тока) электрода на ступенчатое изменение напряжения, подаваемого не него. Отсутствие горизонтального плато электрода свидетельствует о загрязненности его поверхности. Установка тока для нормально работающего электрода должна происходить в течение 1 —1,5 мин. Возникающий шум на самописце может свидетельствовать о плохих контактах в электрической цепи или недостаточно хорошем заземлении. Шум может быть связан с мембраной электрода (складки, дырки, кристаллы КС1 или высыхание) или с загрязнением серебряного электрода (в этом случае электрод чистят аммиаком).
В хорошо отлаженном электроде дрейф пера самописца не превышает 2 % в час (от 100%-й шкалы насыщения кислородом). Дрейф может быть связан с изменением атмосферного давления или с загрязнением органическим материалом, поэтому необходимо тщательно оберегать электрод от загрязнений и между экспериментами хорошо отмывать его поверхность. Иногда воспроизводимые результаты получаются лишь после промывки электрода буфером, используемым в экспериментах. Следует оберегать мембрану электрода от повреждений. Запрещается прикасаться к ней предметами или руками. Допускается осторожное снятие капли воды или буфера фильтровальной бумагой.
Устройство полярографа типа ОН-Ю2 (ВНР)
Полярограф ОН-102 — это настольный прибор, предназначенный для проведения экспериментов по определению концентрации различных ионов в водных растворах с использованием ртутного капающего или кислородного электрода. Передняя панель прибора показана на рис. П.16. Полярограф ОН-102 включается в сеть 220 В, 50 Гц и заземляется через клемму на нижней панели прибора.
Проведение эксперимента. Перед началом эксперимента необходимо прогреть полярограф в течение 15 мин с включенным электродом, который опущен в дистиллиро-
Рис. П.16. Схема передней панели полярографа ОН-102 (ВНР):
1 — тумблер включения прибора; 2 — гнезда подключения электрода; 3 — самописец; 4 — трехходовый тумблер включения самописца; 5 — механизм изменения скорости движения ленты самописца; 6 — тумблер включения автоматической развертки потенциометра: 7 — потенциометр; 8 — ручка выбора напряжения на потенциометре; 9, 10 — ручки компенсатора остаточного тока электрода; 11 — ручка выбора начального напряжения потенциометра; 12 — ручка выбора размаха шкалы потенциометра; 13 — ручка выбора чувствительности (тонко); 14 — ручка выбора вида работ: 15 — ручка управления нулевой линией самописца: 16 — ручка выбора чувствительности (грубо); 17 — ручка компенсации диффузного тока (грубо); 18 —
ручка компенсации диффузного тока (тонко)
ванную воду, перемешиваемую магнитной мешалкой. После прогрева включают самописец и записывают уровень тока прибора, соответствующий 100%-му насыщению дистиллированной (недеионизированной.) воды кислородом воздуха. При нормальном давлении и температуре воды 25 °C в 1 мл воды растворено 586 н-атомов О2.
Затем переносят электрод в среду без кислорода (вода с дитионитом натрия) и регистрируют остаточный ток прибора в воде, не содержащей кислород. Разница между 100%-м и нулевым содержанием кислорода в растворе, зарегистрированная на приборе, составит рабочую шкалу, на которой удобно проводить эксперименты. Выбирают такую чувствительность полярографа, чтобы изменения тока от 100 % О2 до 0 % О-укладывались на шкале самописца прибора или были чуть меньше ее.
После тщательной отмывки электрод вводят в полярографическую ячейку с микроорганизмами, суспендированными в среде или буфере без субстрата дыхания. В течение некоторого времени записывают уровень дыхания клеток в отсутствие субстрата (эндогенное дыхание). Затем через отверстие в полярографической ячейке шприцем вводят субстрат дыхания. Концентрации клеток и субстрата подбирают экспериментально с тем, чтобы скорость дыхания суспензии не была слишком низкой (увеличивает время эксперимента) и слишком высокой (занижается уровень дыхания клеток и часто не хватает времени для внесения последующих добавок). Выбирают концентрацию клеток и скорость движения ленты самописца, чтобы изменения тока шли примерно под углом 45° к оси абсцисс.
В течение одного эксперимента можно внести последовательно субстрат дыхания, его ингибитор, другой субстрат дыхания и ингибитор цитохром-оксидазы (например, сукцинат, ротенон, аскорбат и тетраметилпарафенилендиамин, затем цианид или азид).
Внимание'. Ингибиторы дыхания являются сильнейшими ядами и работать с ними нужно, соблюдая все правила техники безопасности.
По окончании каждого эксперимента следует тщательно отмывать электрод дистиллированной водой, не менее трех раз меняя воду и используя магнитную мешалку.
Расчет результатов. Полярографическая ячейка с вмонтированным электродом имеет внутренний объем 1,5 мл. Из этого объема следует исходить при подготовке растворов субстратов дыхания, которые вносят в конечной концентрации 10 '3— КГ2 М. При приготовлении свежей пробы в пробирку вносят суспензию микроорганизмов в буфере или минеральной среде без источника углерода. Отмеряют 3 мл суспензии и наливают ее в ячейку до образования высокого мениска. Осторожно вставляют электрод, следя за тем, чтобы внутри ячейки не оставалось пузырьков воздуха. На дне ячейки должен находится маленький магнитик. Ставят ячейку с укрепленным электродом на магнитную мешалку и включают ее. Порядок внесения добавок определяется в каждом конкретном случае в зависимости от целей эксперимента.
ФЕРМЕНТЕР И ПРИНЦИПЫ РАБОТЫ НА НЕМ
Для изучения принципов проточного культивирования подходит любой ферментер, в котором при соблюдении стерильности проводимого процесса имеется возможность автоматически регулировать температуру, скорость перемешивания, количество подаваемого стерильного воздуха, pH и скорость подачи стерильной среды в ферментер.
Описание ферментера. Стенд «Микроферм» МА-114 (фирмы Ныо-Браунсвик, США) (рис. П.17) представляет собой компактный настольный прибор, предназначенный для массового культивирования микроорганизмов в периодическом и непрерывном режиме. Используя этот прибор, можно выращивать большое число различных видов микроорганизмов в строго контролируемых условиях. Ферментер полностью оборудован приборами для поддержания и контролирования температуры, скорости перемешивания, аэрации и pH.
Клетки культивируют в толстостенном стеклянном сосуде, выдерживающем многократное автоклавирование. Сосуд фиксируется на стенде с помощью двух пружинных зажимов с винтами, позволяющими быстро отделять культиватор от стенда. Чтобы уменьшить возможность занесения в ферментер посторонней микрофлоры, мешалка соединена с приводом мотора при помощи двух постоянных магнитов. Лопасти мешалки обеспечивают эффективное перемешивание содержимого ферментера в радиальном направлении и могут быть легко приспособлены к любому уровню среды в сосуде. Высокоточный мотор с калиброванным электрическим тахометром позволяет устанавливать скорость вращения мешалки от 100 до 2000 об/мин. Контролирующий прибор поддерживает скорость вращения на заданном уровне независимо от скачков напряжения в сети или вязкости культуры с помощью механизма, работающего по типу обратной связи. Стенд снабжен газометром, позволяющим учитывать и регулировать количество подаваемого в прибор воздуха или другого газа (СО2, аргона).
Регулирование температуры жидкости в ферментере осуществляется путем подачи горячей воды из термостата прибора через замкнутую систему погруженных в культуральную жидкость трубок. Охлаждение происходит водопроводной водой. Измерение температуры ведется с помощью термопары, помещенной внутрь сосуда.
Кислотность среды регулируется автоматически специальным прибором для контроля pH. Два электрода (измерительный и сравнения) монтируются на верхней крышке сосуда (рис. П.15) перед стерилизацией и затем с помощью проводов соединяются с соответствующими гнездами прибора. Устройство электродов данного прибора позволяет стерилизовать их в автоклаве при избыточном давлении 1 ати, однако для других типов электродов может быть с успехом применена стерилизация спиртом. Регулирование pH осуществляется подачей щелочи или кислоты через два перистальтических насоса, вмонтированных в прибор. Следует подобрать заранее и простерилизо-вать вместе с сосудом силиконовые шланги подачи кислоты и щелочи нужного диаметра и необходимой длины.
Подготовка сосуда к стерилизации. Перед стерилизацией стеклянный сосуд надо чисто вымыть, ополоснуть дистиллированной водой, залить средой для культивирования. В верхнюю металлическую часть ферментера (см. рис. П.16) вкрутить измерительный и сравнительный электроды контроля pH (перед каждой стерилизацией следует проверять уровень жидкости внутри электродов и при необходимости доливать свежий раствор электролита). Перед тем как соединить стеклянный сосуд ферментера с металлической верхней частью, нужно с двух сторон смазать уплотнительное резиновое кольцо вакуумной смазкой. Крышка стеклянного сосуда фиксируется с помощью шести винтов — «барашков». К отверстиям для ввода кислоты и щелочи присоединяют силиконовые шланги, в свободные концы которых вставляют ватные пробки и фольгу. В шланги, подающие и отводящие воздух, помещают воздушные фильтры, наполненные стеклянной ватой (воздух через фильтры должен проходить свободно). Устройство прибора
Рис. П.17. Схема ферментера «Микроферм МА-114» фирмы Нью-Браунсвик (США):
А — вид спереди: / — стенд прибора; 2 — сосуд ферментера на 14 л; 3 — привод мешалки; 4 — фильтр подачи воздуха; 5 — фильтр выхода воздуха: 6 — линия подачи воды из термостата; 7 — кронштейн крепления сосуда с зажимами; 8 — тумблер и контрольная лампочка включения прибора; 9 — тумблер и контрольная лампочка включения нагревателя; 10— тумблер и контрольная лампочка включения перемешивания; 11 — тумблер и контрольная лампочка включения охлаждения; 12 — регулятор температуры; 13 — регулятор числа оборотов мешалки; 14 — тахометр скорости вращения мешалки; 15 — автоматический регистратор температуры: 16 — указатель расхода воздуха; 17 — воздушный манометр; 18 — регулятор давления воздуха. Б —
вид слева: 19 — гнездо заземления; 20 — гнезде
датчика пеногашения; 21 — гнездо датчика уровня жидкости в сосуде; 22 — гнездо термистора: 23 — гнездо термометра. В — верхняя крышка сосуда: 24 — конденсатор водяных паров; 25 — выход воздуха; 26— вход воздуха; 27 — отверстие для датчика пеногашения; 28 — отверстие для датчика уровня жидкости; 29 — отверстие для термистора; 30 — линия подачи воды; 31 — отверстия для добавок; 32 — патрубок для добавок; 33— патрубок слива жидкости; 34 — отверстие для термометра; 35 — отверстие для подачи кислоты, щелочи и пеногасителя; 36 — засев-ной лючок; 37 — отверстие для добавок; 38 — отверстие для электродов измерения pH; 39 —
заземление
дает возможность использовать для аэрации магистральный сжатый воздух, который стерилизуется, проходя через нагретые до 130 °C фильтры со стеклянной ватой. Воздух, выходящий из ферментера, также автоматически стерилизуется с помощью фильтра. При подготовке ферментера к стерилизации необходимо убедиться, что все трубки, опущенные ниже уровня среды, заполняющей его, были плотно закрыты зажимами.
чтобы избежать вытеснения среды из сосуда при стерилизации. Кроме того, необходимо следить за тем, чтобы отверстия, до которых среда не доходит, были свободны для выхода пара во время стерилизации (их затыкают ватными пробками). Ферментер стерилизуют в вертикальном положении в автоклаве при давлении до 0,5 ати от 30 мин до 1 ч в зависимости от состава и количества среды в сосуде. После стерилизации во избежание вспенивания содержимого необходимо убедиться, что избыточное давление в автоклаве полностью отсутствует. Альтернативно можно простерилизовать сосуд и среду отдельно.
Закончив стерилизацию, сосуд укрепляют на стенд и зажимают прижимными планками. С помощью специальных шлангов подводят нагревающую/охлаждающую воду, вставляют металлические фильтры подачи воздуха в предназначенные для них отверстия, подсоединяют шланги конденсационной воды, вставляют два терморезистора (термопары) контроля температуры, подсоединяют pH-электроды к прибору контроля pH, а шланги подачи кислоты и щелочи пропускают через перистальтические насосы и опускают (стерильно, возле газовой горелки'.) в стерильные растворы кислоты и щелочи. Подсоединят мешалку ферментера к мотору и включают прибор. После достижения заданной температуры производят посев культуры, используя специальное отверстие (лючок).
Необходимо соблюдать максимум асептики: протирать лючок спиртом до и после посева с обжигом его газовой горелкой; посев производить с использованием стерильной воронки и как можно быстрее. При посеве мешалка должна быть отключена.
ПРИЛОЖЕНИЕ 3
РАБОТА С ИЗОТОПАМИ
ОБЩИЕ ПРИНЦИПЫ
В биологии в основном применяются изотопы с р- и у-излучениями. Выбор изотопа определяется применяемым методом, его удельной активностью, энергией, периодом полураспада и другими факторами, от которых зависит эксперимент. Период полураспада выбранного изотопа и его энергия определяют вид выбранного для подсчета активности инструмента и скорость засвечивания фотопленки, так же как и необходимость коррекции удельной активности радиоактивной метки в течение опыта. р-Излу-чение (поток электронов) от трития и |4С можно измерять в газовой камере, присоединенной к электрометру, счетчиком Гейгера или с помощью сцинтилляционной техники. В последнее время жидкостные сцинтилляционные счетчики благодаря их удобству и чувствительности заменяют все основные виды подсчета в биологии. В нижеприведенной таблице даются основные свойства некоторых изотопов (табл. П.6).
Для подсчета излучения 1311 и 1251 используют гамма-счетчики.
ТЕХНИКА БЕЗОПАСНОСТИ
Лаборатория, в которой проводится работа с изотопами, оборудованная вытяжными шкафами и химическими столами, должна быть принята комиссией по технике безопасности. Перед работой с изотопами необходимо пройти медицинскую комиссию и сдать зачет по технике безопасности для работы с радиоактивностью.
Таблица П.6
Характеристика изотопов, применяемых в биологических исследованиях
Изотоп Период полураспада (Т'/2) Тип распада Энергия МэВ Специфическая активность
Теоретический максимум, ТБк/атом Обычное содержание в соединениях. МБк/моль
Тритий (3Н, Т) 12,26 лет ₽ 0,018 846,0 102—104
Углерод-14 (|4С) 5568 лет ₽ 0,155 2,3 1 -102
Сера-35 (35S) 87,2 сут ₽ 0,167 55,5 1 — ю2
Хлор-36 (36С1) 3 • 105 лет ₽. 0,714 4,4- 102 10-103
Фосфор-32 (32Р) 14,3 сут ₽ 1,710 340,8- 103 10-Ю3
Иод-131 (,3,1) 8,06 сут 7 0,810 599,0- 103 102—104
Иод-125 (1251) 60 сут Y 0,035 80,6 • 103 102-104
Основные требования по технике безопасности при работе с радиоактивными изотопами.
1. Лаборатория должна быть приспособлена для работы с требуемым видом изотопа. Работать с летучими веществами (3Н2О) следует только в вытяжном шкафу. Необходимо контролировать используемое количество изотопа и его изменение. Для отбросов надо иметь готовые контейнеры. Для мытья рабочего места применяют детергенты.
2. В изотопной лаборатории запрещается есть, пить и курить.
3. Запрещается набирать жидкости в пипетки ртом.
4. Необходимо носить рабочий халат, перчатки и защитные очки.
5. Все склянки с изотопами должны быть помечены знаком «радиоактивность» с датой приготовления.
6. По возможности следует использовать одноразовую пластмассовую посуду (пипетки, пробирки, сцинтилляционные стаканы) и после использования складывать их в соответствующий контейнер для радиоактивных отходов.
7. После опыта всю использованную лабораторную посуду следует промыть и дезактивировать для дальнейшего использования.
8. Необходимо вести строгий учет всех использованных изотопов в специальной книге.
9. После работы следует убрать рабочее место и перед уходом из лаборатории тщательно вымыть руки.
10. В случае любых инцидентов, связанных с разбрызгиванием или разлитием изотопа, надо сообщить всем работающим в комнате и преподавателю.
Тритий и 14С в количествах, используемых в экспериментальной микробиологии, не представляю! радиационной опасности. Однако, учитывая очень длительный период полураспада 14С (более 5 тыс. лет), он может надолго задержаться в организме, включаясь, например, в ДНК, и поэтому представляет собой опасность для здоровья.
ПРИЛОЖЕНИЕ 4
СРЕДЫ ДЛЯ КУЛЬТИВИРОВАНИЯ РАЗЛИЧНЫХ МИКРООРГАНИЗМОВ
Среды стерилизуют при 1,0 или 0,5 ати в зависимости от наличия в них витаминов или других термолабильных факторов. Сахара стерилизуют отдельно при 0,5 ати в течение 15 — 30 мин в виде концентрированных растворов (40 %) и добавляют в среды при посеве.
СРЕДЫ, ОБЕСПЕЧИВАЮЩИЕ РОСТ РАЗЛИЧНЫХ ХЕМООРГАНОГЕТЕРОТРОФНЫХ БАКТЕРИЙ
1. Мясопептонный бульон (МПБ).
Основой для приготовления МПБ служит мясная вода, которую готовят следующим образом: мясо освобождают от костей, жира и сухожилий, мелко нарезают или пропускают через мясорубку. 500 г полученного мясного фарша заливают 1 л водопроводной воды и оставляют для экстракции при комнатной температуре на 12 ч или в термостате при 30 °C на 6 ч, или при температуре 37 — 39 °C на 2 ч, а при 50 °C — на 1 ч. За это время из мяса экстрагируются различные вещества, в том числе водорастворимые витамины. Затем мясо отжимают через марлю, и полученный настой кипятят 30 мин. При этом свертываются белки. Остывшую массу фильтруют через ватный фильтр и доливают водой до первоначального объема. К полученной мясной воде добавляют 1 % пептона и 0,5 % NaCl.
МПБ — богатая питательная среда, но она почти не содержит углеводов. В случае необходимости их добавляют к МПБ чаще всего в количестве 1 — 2 г на 100 мл.
МПБ стерилизуют при 1 ати 20 — 30 мин.
2. Солодовое (неохмеленное пивное) сусло.
Очень хорошая среда для развития некоторых молочнокислых и уксуснокислых бактерий, дрожжей, мицелиальных грибов и других представителей гетеротрофных микроорганизмов. Сусло содержит до 16 % сахаров, преимущественно мальтозу (80 %), и частично глюкозу, декстрины, азотистые вещества (до 6 —7 % от общей массы сухого остатка, витамины (преимущественно группы В), минеральные соли и органические кислоты.
Зерна ячменя замачивают в холодной воде и проращивают при 35 °C. После того, как ростки станут вдвое больше длины зерна, последние высушивают до воздушносухого состояния (можно при слабом подогреве) и получают солод.
Сусло готовят следующим образом: 250 г размолотого солода заливают 1 л водопроводной воды, нагревают до 48 — 50 °C и поддерживают эту температуру в течение 30 мин, непрерывно помешивая смесь, чтобы избежать образования комков. Затем температуру поднимают до 55 —58 °C и поддерживают ее на этом уровне до полного осахаривания крахмала, т. е. до тех пор, пока реакция остывшей пробы с йодом не будет отрицательной. При указанном режиме происходит также гидролиз белков до аминокислот и пептидов. Полученный экстракт отфильтровывают через вату или бумажный фильтр. В фильтрате определяют концентрацию сахаров, пользуясь ареометром Баллинга, градусы (°Б) которого примерно соответствуют процентному содержанию сахара в растворе. До требуемой концентрации сахара сусло доводят водопроводной водой.
Для культивирования микроскопических грибов чаще всего используют 3—4 °Б сусло, для дрожжей — 6 —8 °Б, для уксуснокислых бактерий 4—6 °Б, а для наиболее требова
тельных молочнокислых бактерий — 8—12 °Б сусло. Сусло стерилизуют при 0,5 ати 20 — 30 мин.
3. Среда бульон —сусло—агар (БСА).
Мясо-пептонный бульон смешивают с равным объемом 6 °Б сусла и добавляют 2 г/л агара. Среду стерилизуют при 0,5 ати 20 — 30 мин.
4. Цельное молоко.
Молоко содержит все питательные вещества, необходимые для развития гетеротрофных микроорганизмов: лактозы — около 4,5%, белков — 5,0%, минеральных соединений — 1 %, витамины. В молоке без добавок содержится примерно 0,01 % свободных аминокислот, что составляет менее, чем 20 % аминокислот, которые содержатся в средах, обеспечивающих оптимальный рост бактерий. Для того чтобы добиться нормального роста на среде с казеином молока в качестве основного источника азота, у организмов должна быть определенная способность к протеолизу.
Молоко нагревают до кипения, наливают в цилиндры и ставят на 10 —20 ч в холодильник. По истечении этого времени сливают сливки, разливают частично обезжиренное таким образом молоко по пробиркам, закрывают их ватными пробками и стерилизуют при 0,5 ати 30 мин.
5. Обезжиренное молоко.
Для приготовления питательных сред часто используют обезжиренное молоко (так называемый обрат), поскольку жир в молоке может неблагоприятно влиять на рост некоторых микроорганизмов.
Цельное молоко сепарируют и далее поступают так же, как при использовании цельного молока. Обезжиренное молоко можно также получить путем центрифугирования (15 мин при 700—1500 g) или кипячения с последующим отстаиванием в холодильнике в течение 2 сут.
6. Гидролизованное молоко (по Богданову).
Берут 1 л прокипяченного и остуженного до 45 °C обезжиренного молока, устанавливают pH 7,6 —7,8, добавляют 0,5 г порошка панкреатина (предварительно разведенного в небольшом количестве теплой воды) и через несколько минут 5 мл хлороформа. После этого сосуд тщательно встряхивают и помещают на 3 сут в термостат при температуре 40 °C при ежедневном перемешивании жидкости. По истечении указанного срока для удаления хлороформа бутыль открывают, жидкость фильтруют, разбавляют в 2 — 3 раза водопроводной водой и доводят pH до 7,0 —7,2.
Среду стерилизуют при 0,5 ати 30 мин.
7. Молочно-пептонная сыворотка.
Обезжиренное молоко, нагретое до 40 °C, свертывают сычужным ферментом (на 1 ~ вносят 0,1 г препарата, растворенного в 10 мл воды) и выдерживают при указанной температуре в течение 1 ч. Затем сгусток разбивают стеклянной палочкой и жидкость нагревают до 80 °C. Сыворотку процеживают через редкий холст и добавляют 1 % пептона. Для осаждения белков сыворотку нагревают в автоклаве 20 мин при 0,5 ати и горячей отфильтровывают через ватный фильтр. Среду стерилизуют при 0,5 ати 15 мин.
8. Гидролизат казеина.
Первый способ'. 20 г казеина заливают 200 мл воды, добавляют 10 мл концентрированной H2SO4 и выдерживают в автоклаве 4 ч при 1,5 ати.
Второй способ: к 200 г казеина добавляют 280 мл 6 н раствора НС1 и смесь кипятят с обратным холодильником 18 ч. Полученный гидролизат нейтрализуют 50%-м раствором NaOH до pH 7,0, фильтруют через бумажный фильтр и стерилизуют при 0,5 ати 30 мин. Содержание аминного азота в гидролизате, полученном таким способом, составляет 700 —1000 мг на 100 мл. Его добавляют к средам в таком количестве, чтобы концентрация по аминному азоту составляла 10 —30 мг на 100 мл. Следует иметь ь виду, что при кислотном гидролизе полностью разрушается триптофан, в достаточна большой степени цистеин и незначительно серин и треонин. Имеется готовый препарат гидролизата казеина. Его вносят в среды в количестве от 0,1 до 1,0 г на 100 мл а зависимости от потребностей микроорганизмов.
9. Дрожжевая среда.
Используется для культивирования различных представителей гетеротрофных микроорганизмов. Основа дрожжевой среды — дрожжевая вода. Для ее приготовления 70 — 100 г свежих прессованных дрожжей или 7 —10 г сухих дрожжей кипятят 30 мин в 1 л водопроводной воды. Затем массе дают отстояться в высоком цилиндре на холоду, жидкость декантируют и фильтруют через вату. К фильтрату добавляют 1 л воды, еще раз кипятят 30 мин и вновь фильтруют. К полученной дрожжевой воде добавляют К2НРО4 (0,1 %) и NaCl (0,5 %), а в случае необходимости углеводы (1 —2 %). Доводят pH полученной среды до 6,8—7,2. Среду стерилизуют при 0,5 ати 20 — 30 мин.
10. Дрожжевой автолизат.
Дрожжи Saccharomyces cerevisiae содержат 40 — 60 % азотистых веществ, а также ряд ферментов, способствующих их самоперевариванию. При автолизе дрожжей образуются продукты, характерные для триптического переваривания животных белков.
Для приготовления дрожжевого автолизата следует выбирать емкость с таким расчетом, чтобы автолизат занимал около '/5 объема посуды. 40 г свежих прессованных дрожжей (или 10 г сухих дрожжей) разводят в 100 мл воды, добавляют несколько кристаллов тимола или 1 — 2 мл хлороформа. Смесь выдерживают в термостате при 50—55 °C трое суток. По мере разжижения дрожжей смесь встряхивают для равномерного прогревания (1 — 2 раза в сутки). За это время клетки дрожжей отмирают, а ферменты остаются активными и гидролизуют белки, а также другие биополимеры. Конец автолиза характеризуется полным разжижением дрожжей. Автолизат должен иметь коричневый оттенок и приятный запах. Полученный автолизат хорошо перемешивают и кипятят 20 мин на слабом огне (для свертывания нерасщепившихся белков и лучшего фильтрования), а затем фильтруют через бумажный фильтр.
Автолизат стерилизуют при 0,5 ати 15 мин и сохраняют в холодильнике.
11. Картофельная среда.
Используют в основном для культивирования спорообразующих бактерий, представителей рода Caulobacter и некоторых других хемоорганотрофных бактерий. В сухом веществе картофеля содержится 5 — 13 % азотистых веществ и около 75 % углеводов, он богат соединениями калия и фосфора, рядом витаминов. На картофельных средах хорошо развиваются микроорганизмы, образующие амилолитические ферменты, но чаще других на них выращивают представителей рода Clostridium.
Клубни картофеля, отобранные для приготовления этой среды, тщательно моют, очищают от кожуры, глазков и испорченных мест и снова моют. 200 г мелко нарезанного картофеля заливают 1 л водопроводной воды и кипятят 30 мин. Отвар фильтруют через вату, доводят объем фильтрата до 1 л и разливают в сосуды для культивирования.
Среду стерилизуют 1 ч при 1 ати или 30 мин при 1,5 ати. Необходимость длительной стерилизации обусловлена тем, что на картофеле всегда много спорообразующих почвенных форм микроорганизмов.
12. Почвенная вытяжка (почвенный экстракт).
Используется для выделения и культивирования многих почвенных бактерий.
500 г плодородной почвы заливают 1,5 л водопроводной воды и автоклавируют 30 мин при 1 ати. Полученный экстракт фильтруют через бумажный фильтр, добавляют к горячему фильтрату 0,5 г СаСО3, тщательно перемешивают и через 5 — 7 мин фильтруют вновь. К экстракту, как правило, добавляют 0,2 г К2НРО4 и доводят объем до 1 л, pH 6,8 —7,0. Среду стерилизуют при 1,5 ати 30 мин.
13. Пептон-дрожжевая среда с глюкозой.
Среда состава (г): пептон — 10,0; дрожжевой экстракт — 1,0; глюкоза — 1,0 —4,0; вода дистиллированная — 1000 мл; pH 6,8 —7,0. Среду стерилизуют при 0,5 ати 20 — 30 мин.
14. Бобовый отвар.
50 г бобов (белой фасоли или гороха) заливают 1 л водопроводной воды и варят до набухания и растрескивания кожуры, но не до полного разваривания (необходимо
следить, чтобы крахмал не превратился в клейстер). Горячий отвар фильтруют через вату или несколько слоев марли и доводят объем до 1 л.
15. Крахмало-аммиачная среда (КАА).
Среда состава (г): растворимый крахмал — 10,0; (NH4)2SO4 — 2,0; К2НРО4 — 1,0; MgSO4-7H2O — 1,0; NaCl — 1,0; СаСО3 — 3,0; агар — 15,0; вода водопроводная — 1000 мл. Крахмал предварительно заваривают отдельно в небольшом количестве горячей воды и затем приливают к остальной среде. Среду стерилизуют при 1 ати 20 — 30 мин.
16. Среда Чапека.
Среда состава (г): сахароза — 30,0 или глюкоза — 20,0; NaNO3 — 2,0; К2НРО4 — 1,0: MgSO4- 7Н2О — 0,5; КС1 — 0,5; FeSO4- 7Н2О — 0,1; вода дистиллированная — 1000 мл. Среду стерилизуют при 0,5 ати 20 —30 мин.
СРЕДЫ ДЛЯ САХАРОЛИТИЧЕСКИХ КЛОСТРИДИЙ
17. Картофельная среда с мелом.
После стерилизации к среде № 11 добавляют стерильный мел в количестве 1—2 г/ л. Мел стерилизуют в муфельных печах при 450 —500 °C 20 — 30 мин или в сушильном шкафу при 160 °C в течение 5 —6 ч.
18. Среда с глюкозой и пептоном.
Среда состава (г): глюкоза — 10,0; пептон — 10,0; К2НРО4 — 1,0; СаСО3 — 3,0 — 5,0: вода водопроводная — 1000 мл. Мел стерилизуют отдельно (см. среду № 17) и добавляют к стерильной среде.
СРЕДЫ ДЛЯ МОЛОЧНОКИСЛЫХ БАКТЕРИЙ
19. Сусло с дробиной.
8—10 °Б сусло (среда № 2) с дробиной. К стерильной среде добавляется в избытке (10 г/л) стерильный мел (см. среду № 17).
20. Обрат.
Обезжиренное молоко (обрат) — среда № 5. Часто его разбавляют водой в соотношении 2: 1.
21. Среда MRS.
Среда состава (г): гидролизат казеина — 10,0; мясной экстракт — 10,0; дрожжевой экстракт — 5,0; глюкоза — 20,0; ацетат натрия — 5,0; цитрат аммония (двузамещенный) — 2,0; твин 80—1,0; К2НРО4 — 2,0; MgSO4-7H2O — 0,2; MnSO4-4 Н2О — 0,05: вода дистиллированная — 1000 мл; pH 6,2 —6,5. Среду стерилизуют при 0,5 ати 20 — 30 мин.
СРЕДЫ ДЛЯ САПРОТРОФНЫХ БАКТЕРИЙ
22. Среда для сапротрофных коринебактерий.
Среда состава (г): гидролизат казеина — 10,0; дрожжевой экстракт — 5,0; глюкоза — 5,0; NaCl — 5,0; вода дистиллированная — 1000 мл; pH 7,2 —7,4. Среду стерилизуют при 0,5 ати 20 — 30 мин.
23. Среда для сапротрофных микобактерий и нокардий.
Среда состава (г): мясо-пептонный бульон (среда № 1) — 100 мл; 3 —4°Б сусло (среда № 2) — 100 мл; агар — 3,0 г. Среду стерилизуют при 0,5 ати 20 —30 мин.
СРЕДЫ ДЛЯ ПРОПИОНОВОКИСЛЫХ БАКТЕРИЙ
Жирные кислоты, образуемые пропионовокислыми бактериями при использовании глюкозы, необходимо ежедневно нейтрализовать стерильным 10%-м раствором NaHCO3.
24. Среда I.
Среда состава (г): глюкоза — 20,0; дрожжевой экстракт — 4,0; гидролизат казеина — 10,0; (NH4)2SO4 — 3,0; СоС12- 6Н2О — 0,005; MnSO4- 4Н2О — 0,005; вода дистиллированная — 1000 мл; pH 6,8 — 7,0. Глюкозу стерилизуют отдельно и вносят в стерильную среду перед посевом.
25. Среда II.
Среда состава (г): лактат Na или Са — 10,0; дрожжевой экстракт — 10,0; гидролизат казеина — 10,0; КН2РО4 — 2,5; вода дистиллированная — 1000 мл; pH 6,8 —7,0.
26. Среда III.
Среда состава (г): глюкоза — 20,0; кукурузный экстракт — 20,0; (NH4)2SO4 — 3,0; СоС12-6Н2О — 0,005; вода дистиллированная — 1000 мл; pH 6,8 —7,0. Глюкозу стерилизуют отдельно и вносят в стерильную среду перед посевом.
27. Среда IV.
Среда состава (г): глюкоза — 20,0; (NH4)2SO4 — 3,0; КН2РО4 — 1,0; MgSO4- 7Н2О — 0,2; СоС12-6Н2О — 0,005; биотин — 1,0 мкг; пантотенат Са — 1000 мкг; тиамин — 200 мкг; вода дистиллированная — 1000 мл; pH 6,8 —7,0. Глюкозу и витамины стерилизуют отдельно и вносят в стерильную среду перед посевом.
СРЕДЫ ДЛЯ АКТИНОМИЦЕТОВ
28. Овсяный агар (ISP — 3).
Среду применяют для хранения и определения культуральных, а также морфологических признаков. Овсяная мука (или хлопья) — 20,0 г; агар — 20,0 — 25,0 г; вода дистиллированная — 1000 мл; следы солей — FeSO4 — 0,1 г; МпС12 — 0,1 г; ZnSO4 — 0,1 г. Вместо дистиллированной воды и солей можно использовать водопроводную воду. Для приготовления среды овсяную муку (или хлопья) варят в 1 л воды 20 мин, фильтруют и доводят объем до 1 л. Среду стерилизуют при 0,5 ати 20 —30 мин.
29. Среда Красильникова № 1.
Среда состава (г): глюкоза — 20,0; KNO3 — 1,0; К2НРО4 — 0,5; MgSO4- 7Н2О — 0,5; NaCl — 0,5; СаСО3 — 1,0; FeSO4- 7Н2О — следы; вода водопроводная — 1000 мл; pH 7,0 —7,2. Среду стерилизуют при 0,5 ати 20 — 30 мин.
30. Среда Красильникова № 6.
Среда состава (г): крахмал растворимый — 10,0; кукурузный экстракт — 10,0; (NH4)2SO4 — 3,0; NaCl — 3,0; СаСО3 — 3,0; вода водопроводная 1000 мл; pH 7,0 —7,2. Среду стерилизуют при 0,5 ати 20 — 30 мин.
31. Среда Красильникова № 71.
Среда состава (г): крахмал растворимый — 15,0; кукурузный экстракт — 10,0; (NH4)2SO4 — 4,0; К2НРО4 — 2,0; СаСО3 — 3,0; вода водопроводная 1000 мл; pH 6,6 —7,2. Среду стерилизуют при 0,5 ати 20 — 30 мин.
32. Среда Гаузе № 1.
Среда состава (г): крахмал растворимый — 20,0; KNO3 — 1,0; К2НРО4 — 0,5; MgSO4-7H2O — 0,5; NaCl — 0,5; FeSO4-7H2O — следы; вода водопроводная 1000 мл; pH 7,2 —7,4. Среду стерилизуют при 0,5 ати 20 — 30 мин.
33. Среда Ваксмана.
Среда состава (г): глицерин — 3,0; К2НРО4 — 1,0; NaNO3 — 2,0; MgSO4- 7Н2О — 0,5; КС1 — 0,5; FeSO4 • 7Н2О — 0,01; вода водопроводная 1000 мл; pH 7,0. Среду стерилизуют при 0,5 ати 20 — 30 мин.
СРЕДЫ ДЛЯ МИКРООРГАНИЗМОВ, ИСПОЛЬЗУЮЩИХ ЦЕЛЛЮЛЮЗУ
34. Среда Хетчинсона и Клейтона для аэробных бактерий.
Целлюлюза — кусочки фильтровальной бумаги (10 г/л); NaNO3 — 2,5 г; К2НРО4 — 1,0 г; MgSO4 • 7Н2О - 0,3 г; NaCl - 0,1 г; СаС12 • 4Н2О - 0,1 г; FeCl3 • 6Н2О — 0,01 г; вода дистиллированная — 1000 мл; pH 7,2—7,3. Среду стерилизуют при 1,0 ати 20 — 30 мин.
35. Среда Имшенецкого для анаэробных бактерий.
Среда состава (г): фильтровальная бумага — 15,0; NaNH4HPO4 — 1,5; КН2РО4 — 0,5; К2НРО4 — 0,5; MgSO4-7H2O — 0,4; NaCl — 0,1; пептон - 5,0; MnSO4-4H2O -следы; FeSO4-7H2O — следы; СаСО3 — 2,0; вода дистиллированная — 1000 мл; pH 7,0 — 7,4. Среду стерилизуют при 0,5 ати 20 —30 мин.
СРЕДЫ ДЛЯ УКСУСНОКИСЛЫХ БАКТЕРИЙ
36. Сусло с этанолом.
4 — 6°Б сусло (среда № 2) — 100 мл; этанол (ректификат) — 4 — 5 мл; pH 6,5 —6,8. Среду стерилизуют при 0,5 ати 20 — 30 мин.
37. Среда с глюкозой.
Среда состава (г): глюкоза — 100,0; дрожжевой экстракт — 10,0; СаСО3 — 20,0; агар — 15,0; вода дистиллированная — 1000 мл; pH 6,8. Среду стерилизуют при 0,5 ати 20 — 30 мин.
38. Среда для бактерий рода Gluconobacter.
Среда состава (г): дрожжевой экстракт — 5,0; пептон —3,0; маннит — 25,0; агар — 15,0; вода дистиллированная — 1000 мл; pH 6,7 —6,8. Среду стерилизуют при 0,5 ати 20 — 30 мин.
СРЕДЫ ДЛЯ БАКТЕРИЙ РОДА CAULOBACTER
39. Картофельный агар.
Готовят картофельную среду (среда № 11) с добавлением 20 г/л агара.
40. Среда Красильникова и Беляева.
Среда состава (г): пептон —1,0; дрожжевой автолизат (среда № 10) — 1.0 мл: MgSO4- 7Н2О — 0,2; агар — 15,0; вода дистиллированная — 1000 мл. Среду стерилизуют при 0,5 ати 20 —30 мин.
СРЕДЫ ДЛЯ МЕТИЛОТРОФОВ
41. Среда Хирш и Конти.
Среда состава (г): (NH4)2SO4 — 0,5 г; КН2РО4 —1,36 г; Na2HPO4-7H2O — 2,13 г; MgSO4-7H2O - 0,2 г; СаС12-2Н2О - 10,0 мг; MnSO4-4H2O - 2,5 мг; FeSO4-7H2O -5,0 мг; Na2MoO4-2H2O — 2,5 мг; гидрохлорид метиламина — 6,75 мг; агар — 18,0 г; вода дистиллированная — 1000 мл; pH 7,2. Непосредственно в питательную среду добавляют 4 г/л метанола. В качестве источника азота вместо (NH4)2SO4 можно использовать KNO3 или мочевину в концентрации 100 мМ.
42. Среда Мевиуса.
Среда состава (г): NaNO3 — 1,0; К2НРО4 — 1,0; NaCl — 0,5; MgSO4-7H2O — 0,5: вода водопроводная — 1000 мл. К 20 мл стерильной среды добавляют несколько капель метанола или после посева колбы помещают в эксикатор, в который ставят открытую пробирку с метанолом.
43. Среда Канеда.
Среда состава (г): КН2РО4 — 2,0; (NH4)2SO4 — 2,0; NaCl — 0,5; MgSO4-7H2O — 0,025; FeSO4- 7Н2О — 0,01; вода дистиллированная — 1000 мл. Исходное значение pH среды 7,5. Среду стерилизуют при 1,0 ати 20 — 30 мин. После стерилизации в среду вносят 0,3 % (по объему) стерильного метанола (стерилизуют фильтрованием).
44. Среда MMS.
Среда содержит (г/л): К2НРО4 — 1,2; КН2РО4 — 0,62; СаС12’6Н2О — 0,05; MgSO4-7H2O — 0,20; NaCl — 0,10. Микроэлементы (мкг/л, конечная концентрация в среде): FeCl2-6H2O - 1000,0; (NH4)2SO4 - 0,5; CuSO4-5H2O — 5,0; MnSO4-5H2O -10,0; Na2MoO4-2H2O —10,0; H3BO3 — 10,0; ZnSO4-7H2O - 70,0; CoCl2-6H2O - 5,0. Стерилизация — при 1 ати. В стерильную среду добавляют стерильный (фильтрация) метанол до 0,1 —0,2 %, pH среды — 7,0. В агаризованную среду добавляют 0,5 —2,0 % очищенного агара (выщелоченного). Метилобактерий не требуют витаминов.
СРЕДЫ ДЛЯ АЗОТФИКСА ТОРОВ
45. Среда Эшби для азотобактера и олигонитрофилов.
Среда состава (г): сахароза или маннит — 20,0; К2НРО4 — 0,2; MgSO4- 7Н2О — 0,2; NaCl — 0,2; K2SO4 — 0,1; СаСО3 — 5,0; вода дистиллированная — 1000 мл. В среду рекомендуется вносить смесь микроэлементов — 1 мл/л.
Раствор микроэлементов (по Федорову) (г): Н3ВО3 — 5,0; (NH4)2 МоО4-2Н2О — 5,0; ZnSO4’ 7Н2О — 0,2; К1 и NaBr — по 0,5; A12(SO4)3- 18Н2О — 0,3; вода дистиллированная — 1000 мл.
46. Среда Виноградского для анаэробных азотфиксаторов из рода Clostridium.
Среда состава (г): глюкоза — 20,0; К2НРО4 — 1,0; MgSO4 7Н2О — 0,5; СаСО3 — 20,0 (для нейтрализации кислот, образующихся при брожении); NaCl — 0,5, MnSO4, FeSO4 — следы; вода дистиллированная — 1000 мл. В среду рекомендуется добавлять дрожжевой экстракт — 10 мг/л и раствор микроэлементов по Федорову (см. среду № 45) — 1 мл/л.
47. Среда Федорова в модификации Калининской для выделения азотфиксаторов.
Среда состава (г): глюкоза — 10,0—15,0; К2НРО4 — 1,74; КН2РО4 — 0,91; MgSO4-7H2O — 0,3; СаС12-6Н2О — 0,1; NaCl — 0,5; FeCl3-6H2O — 0,01; дрожжевой экстракт — 0,015; раствор микроэлементов по Федорову (см. среду № 45) — 1 мл; вода дистиллированная — 1000 мл.
48. Бобовый агар для клубеньковых бактерий.
Среда состава (г): бобовый отвар (среда № 14) — 1000 мл; сахароза — 2,0; КН2РО4 — 1,0; MgSO4- 7Н2О — 0,3; агар — 15,0; pH 7,0 —7,2. Среду стерилизуют при 0,5 ати 20 — 30 мин.
49. Маннитно-дрожжевая среда для клубеньковых бактерий.
Среда состава (г): маннит — 10,0; дрожжевой экстракт — 1,0; К2НРО4 — 0,5; MgSO4-7H2O — 0,2; NaCl — 0,1 — 0,2; FeCl3-6 Н2О — 0,002; агар — 15,0; вода дистиллированная — 1000 мл; pH 6,8 —7,0. Среду стерилизуют при 0,5 ати 20 —30 мин.
СРЕДЫ ДЛЯ ТИОНОВЫХ БАКТЕРИЙ
50. Среда Траутвейна для Thiobacillus denitrificans.
Среда состава (г): Na2S2O3-5H2O — 2,0; NH4C1 — 0,1; Na2HPO4-2H2O — 0,1; MgCl2- 6H2O — 0,1; KNO3 — 1,0; NaHCO3 — 0,1; раствор микроэлементов по Древсу — 5 мл; вода дистиллированная — 1000 мл; pH 7,0. Среду стерилизуют при 1,0 ати 20—30 мин.
Раствор микроэлементов по Древсу (Drews, 1976) (мг): ЭДТА Na — 800; МпС12- 4Н2О — 10; СоС12- 6Н2О - 4; CuSO4 - 1; Na2MoO4-2H2O - 3; ZnCl2 - 2; LiCl - 0,5; SnCl2- 2H2O -
0,5; Н3ВО3 —1; KBr — 2; KJ — 2; BaCl2 — 0,5; вода дистиллированная — 1000 мл; pH 6,0. На 1 л среды добавляют от 5 до 10 мл этого раствора.
51. Среда Сильвермана и Люндгрена 9К для Thiobacillus ferrooxidans.
1-й раствор: в 700 мл дистиллированной воды растворяют (г): (NH4)2SO4 — 3,0; К2НРО4 - 0,5; КС1 - 0,1; MgSO4-7H2O - 0,5; Ca(NO3)2-4Н2О - 0,01.
2-й раствор: в 300 мл дистиллированной воды растворяют 44,2 г FeSO4-7H2O и добавляют 1 мл Юн серной кислоты. Растворы стерилизуют (при 1,0 ати 20 —30 мин) отдельно и смешивают перед посевом; pH среды 2,5.
52. Среда Ваксмана для Thiobacillus thiooxidans.
Среда состава (г): сера (серный цвет) — 10,0 или Na2S2O3- 5Н2О — 5,0; (NH4)2SO4 — 0,3; КН2РО4 - 3,0; СаС12-6Н2О - 0,25; MgSO4-7H2O - 0,5; FeSO4-7H2O - 0,01; раствор микроэлементов по Древсу (см. среду № 50) — 10 мл; вода дистиллированная — 1000 мл; pH 4,0— 5,0. Среду стерилизуют при 1,0 ати 20 — 30 мин. Серу и Na2S2O3 • 5Н2О стерилизуют отдельно и вносят в стерильную среду перед посевом. Серу стерилизуют текучим паром или спиртом 2 ч, затем спирт испаряют в сушильном шкафу при 50 °C.
53. Среда Старки для Thiobacillus thioparus.
Среда состава (г): Na2S2O3- 5Н2О - 5,0; (NH4)2SO4 - 0,2; КН2РО4 - 3,0; СаС12- 6Н2О -0,25; MgSO4- 7Н2О — 0,5; FeSO4- 7Н2О —0,01; раствор микроэлементов по Древсу (см. среду № 50) — 10 мл; вода дистиллированная — 1000 мл; pH 7,5 —8,0. Na2S2O3-5H2O стерилизуют отдельно и вносят в стерильную среду перед посевом.
СРЕДА ДЛЯ ВОДОРОДОКИСЛЯЮЩИХ БАКТЕРИЙ
54. Среда Казерера.
Среда состава (г): NH4C1 — 1,0; К2НРО4 — 0,5; MgSO4- 7Н2О — 0,2; NaHCO3 — 0,5; раствор микроэлементов по Древсу (см. среду № 50) — 10 мл; вода дистиллированная — 1000 мл; pH 7,0. Среду стерилизуют при 1,0 ати 20 — 30 мин. После посева колбы помещают в эксикатор, заполненный газовой смесью состава (%): СО2 — 10; О2 — 5 — 30; Н2 - 60-85.
Все газовые смеси для водородных бактерий взрывоопасны, поэтому при работе необходимо строго соблюдать правила техники безопасности'.
СРЕДЫ ДЛЯ ДЕНИТРИФИЦИРУЮЩИХ БАКТЕРИЙ
55. Среда Гильтея.
Среда состава (г): цитрат Na или К (трехзамещенный) — 5,0; KNO3 — 2,0; аспарагин - 1,0; КН2РО4 - 2,0; MgSO4- 7Н2О - 2,0; СаС12- 6Н2О - 0,2; FeCl3- 6Н2О - следы; вода дистиллированная — 1000 мл; pH 6,8 —7,2. Среду стерилизуют при 0,5 ати 20 — 30 мин.
56. Среда Березовой.
Среда состава (г): цитрат Na или К (трехзамещенный) — 20,0; KNO3 — 1,0; КН2РО4 — 1,0; К2НРО4 - 1,0; MgSO4- 7Н2О - 2,0; СаС12- 6Н2О - 0,2; FeCl3- 6Н2О - следы; вода дистиллированная — 1000 мл. pH 6,8 —7,2. Среду стерилизуют при 0,5 ати 20 —30 мин.
СРЕДЫ ДЛЯ СУЛЬФАТРЕДУЦИРУЮЩИХ БАКТЕРИЙ
57. Среда Баар.
Среда состава (г): лактат Na — 3,5 мл; NH4C1 — 1,0; CaSO4- Н2О — 1,0; КН2РО4 — 0,5; MgSO4-7H2O — 2,0; (NH4)2Fe(SO4)2- 6Н2О (соль Мора) — 0,5; 1%-й раствор Na2S в 1%-м растворе NaHCO3 — 5 — 8 мл; вода дистиллированная — 1000 мл; pH 7,0 —7,5.
Среду стерилизуют при 0,5 ати 20—30 мин. 1 %-й раствор Na2S в 1 %-м растворе NaHCO3 стерилизуют отдельно и добавляют в стерильную среду перед посевом.
58. Среда Постгейта В.
Среда состава (г): лактат Na — 3,5 мл; дрожжевой экстракт — 1,0; NH4C1 — 1,0; CaSO4H2O - 1,0; КН2РО4 - 0,5; MgSO4-7H2O - 2,0; FeSO4-7H2O - 0,5; раствор микроэлементов по Пфеннигу (см. среду № 63) — 1 мл; 1%-й раствор Na2S в 1%-м растворе NaHCO3 — 5 — 8 мл; вода дистиллированная — 1000 мл; pH 7,0 — 7,5. 1%-й раствор Na2S в 1%-м растворе NaHCO3 стерилизуют отдельно и добавляют в стерильную среду перед посевом. Сернокислое железо растворяют в 1%-й НС1, стерилизуют отдельно и добавляют в стерильную среду перед посевом.
СРЕДЫ ВИНОГРАДСКОГО ДЛЯ НИТРИФИКАТОРОВ
К средам рекомендуется добавлять раствор микроэлементов по Пфеннигу (см. среду № 63) — 1 мл на 1 л среды.
59. I фаза нитрификации.
Среда состава (г): (NH4)2SO4 — 2,0; К2НРО4 — 1,0; MgSO4- 7Н2О — 0,5; NaCl — 2,0; FeSO4- 7Н2О — 0,05; СаСО3 — 5,0; вода водопроводная — 1000 мл. Среду стерилизуют при 1,0 ати 20 —30 мин.
60. II фаза нитрификации.
Среда состава (г): NaNO2 — 1,0; К2НРО4 — 0,5; MgSO4-7H2O — 0,5; NaCl — 0,5; FeSO4- 7Н2О — 0,4; Na2CO3 — 1,0; вода водопроводная 1000 мл. Среду стерилизуют при 1,0 ати 20 —30 мин.
СРЕДЫ ДЛЯ ВЫДЕЛЕНИЯ БАКТЕРИЙ, ОКИСЛЯЮЩИХ ЖЕЛЕЗО И МАРГАНЕЦ
61. Среда Лиске для железобактерий.
Среда состава (г): (NH4)2SO4 — 1,5; К2НРО4 — 0,05; КО — 0,05; MgSO4-7H2O — 0,05; Ca(NO3)2- 4Н2О — 0,01; вода дистиллированная — 1000 мл; pH 7,0. Среду стерилизуют при 1,0 ати 20 —30 мин. Среду после стерилизации оставляют стоять на 2 — 3 дня для насыщения кислородом и углекислотой, затем добавляют стерильную железную проволоку или свежеосажденное сернистое железо.
62. Среда Дубининой для Metallogenium.
Крахмал гидролизованный — 1 г; нормальная лошадиная сыворотка — 1 мл; ДНК — 10 мкг; свежеосажденный МпСО3 — избыток; вода дистиллированная — 1000 мл. Среду стерилизуют при 0,5 ати 20 — 30 мин.
СРЕДЫ ДЛЯ ФОТОТРОФНЫХ БАКТЕРИЙ
63. Среда для зеленых и пурпурных серобактерий.
Среда состава (г): NH4C1 — 1,0; NaHCO3 — 2,0 —5,0; КН2РО4 — 1,0; MgCl2- 6Н2О — 0,5; СаС12- 2Н2О — 0,1; Na2S • 9Н2О — 1,0; витамин В12 — 1 мкг; раствор микроэлементов (по Пфеннигу) — 1 мл; к средам для морских штаммов добавляют NaCl; вода дистиллированная — 1000 мл; pH 7,0 —8,0. Среду стерилизуют при 1,0 ати 20 — 30 мин.
Важно! Растворы NaHCO3, Na2S • 9Н2О и витамина В12 стерилизуют отдельно и вносят в среду перед посевом.
Раствор микроэлементов по Пфеннигу (Pfennig, 1965) (мг): ЭДТА — 500; FeSO2-7H2O - 200; ZnSO4-7H2O - 10; MnCl2-4H2O - 3; Н3ВО3 - 30; СоС12- 2Н2О -
20; Си Cl2- 2Н2О — 1; NiCl2- 6Н2О — 2; Na2MoO4- 2Н2О — 3; вода дистиллированная — 1000 мл; pH 6,0. На 1 л среды добавляют от 1 до 10 мл этого раствора.
64. Среда для пурпурных несерных бактерий.
Среда состава (г): NH4C1 — 0,4; дрожжевой экстракт — 0,2; соединение углерода — 1,0; MgSO4-7H2O — 0,2; NaCl — 0,4; витамин В12 (1 мг/100мл) — 1 мл; цитрат Fe (0,1 г/100 мл) - 5 мл; СаС2- 2Н2О - 0,05; КН2РО4 - 0,5; NaHCO3 - 2,0; раствор СаС12 микроэлементов по Пфеннигу (см. среду № 63) — 1 мл; вода дистиллированная — 1000 мл; pH 6,8—7,3.
Важно! Растворы NaHCO3, витамина В12 и цитрата Fe стерилизуют отдельно и вносят в среду перед посевом.
65. Среда DGN для зеленых несерных бактерий.
Среда состава (г); ЭДТА — 0,02; CaSO4-2H2O — 0,06; MgSO4-7H2O — 0,10; NaCl — 0,008; Na2HPO4 — 0,11; NH4C1 — 0,20; FeCl3 раствор (100 мг/л) — 3,0 мл; глицил-глицин — 0,8; раствор микроэлементов — 0,5 мл; витамин В12 — 200 мкг; ацетат натрия — 1,0 или NaHCO3 — 1,0; дрожжевой экстракт — 0,1; гидролизат казеина — 0,1; вода дистиллированная — 1000 мл; pH 7,8 —8,5. В качестве источника электронов при автотрофном росте используют Na2S • 9Н2О — 0,5 г/л или молекулярный водород (в этом случае после посева микроорганизмы культивируют в атмосфере Н2). Раствор микроэлементов (г): MnSO4- Н2О — 2,28; Zn SO4- 7Н2О — 0,5; Н3ВО3 — 0,5; CuSO4- 5Н20 — 0,025; Na2MoO4- 2Н2О — 0,025; СоС12 • 6Н2О — 0,045; H2SO4 (кони.) — 0,5 мл; вода дистиллированная — 1000 мл. В ряде случаев перед посевом в среду вносят (1мл/л) простерили-зованный при 0,5 ати раствор витаминов, содержащий (мг/100 мл): тиамин НО — 10,0; биотин — 0,5; рибофлавин — 3,0; фолиевую кислоту — 5,0.
Важно! Растворы NaHCO3, витамина В,2, ацетата натрия, дрожжевого экстракта, гидролизата казеина и Na2S • 9Н2О стерилизуют отдельно (при 0,5 ати) и вносят в среду перед посевом.
66. Среда Кратца и Мейерса для цианобактерий.
Среда состава (г): Ca(NO3)2 • 4Н2О — 0,025; KNO3 — 1,0; MgSO4 • 7Н2О 0,25; К2НРО4 — 1,0; Na2CO3 — 1,5; FeNH4 цитрат — 1,0; FeCl3- 6 Н2О — 0,001; раствор микроэлементов — 1 мл; вода дистиллированная — 1000 мл; pH 6,8 —7,0. Среду стерилизуют при 0,5 ати 20 — 30 мин.
Состав раствора микроэлементов (г): Н3ВО3 — 2,86; МпС12-4Н2О — 1,81; ZnSO4- 7Н2О — 0,222; Na2MoO4- 2Н2О — 0,03; CuSO4- 5Н20 — 0,079; вода дистиллированная — 1000 мл.
66а. Среда BG-11.
Среда состава (г/л): NaNO3 - 1,5; К2НРО4-ЗН2О - 0,04; MgSO4-7H2O - 0,075; СаС12 — 0,036; лимонная кислота — 0,006; цитрат Fe (II) — 0,006; ЭДТА — 0,001; Na2CO3 — 0,02; раствор микроэлементов А5 + Со — 1 мл/л; вода дистиллированная — 1000 мл; pH среды — 7,4. Среду стерилизуют при 1,0 ати 20 —30 мин. Среда используется в двух вариантах: BGN-11 (с NaNO3) и BGO-11 (без NaNO3).
Состав раствора микроэлементов А5 + Со (г): Н3ВО3 — 2,86; МпС12-4Н2О — 1,81; ZnSO4- 7Н2О — 0,222; Na2MoO4- 2Н2О - 0,390; CuSO4- 5Н20 - 0,079; Co(NO3)2- 6Н2О -0,0494; вода дистиллированная — 1000 мл.
666. Среда ASN-Ш.
Среда состава (г): NaCl - 25,0; MgCl2-6H2O - 2,0; КС1 - 0,5; NaNO3 - 0,75; K2HPO4-3H2O — 0,02; MgSO4-7H2O — 3,5; СаС12-2Н2О — 0,2; лимонная кислота — 0,003; цитрат Fe (II) — 0,003; ЭДТА — 0,0005; Na2CO3 — 0,02; раствор микроэлементов А5 + Со (см. среду № 66а) — 1 мл/л; вода дистиллированная — 1000 мл; pH среды — 7,5. Среду стерилизуют при 1,0 ати 20 — 30 мин.
66в. Среда Громова № 6.
Среда состава (г): KNO3 — 1,0; К2НРО4 • ЗН2О — 0,2; MgSO4-7Н2О — 0,2; СаС12-2Н2О — 0,15; NaHCO3 — 0,2; раствор микроэлементов — 1 мл; вода дистиллированная — 1000 мл; pH среды — 7,0. Среду стерилизуют при 1,0 ати 20 — 30 мин.
Раствор микроэлементов (г): ZnSO4- 7Н2О — 0,22; MnSO4- Н2О — 1,81; CuSO4- 5Н2О — 0,079; NaBO3- 4Н2О - 2,63; (NH4)6Mo7O24- 4Н2О - 1,0; FeSO4- Н2О - 9,3; СаС12 - 1,2; Co(NO3)2- Н2О — 0,02; ЭДТА — 0,010. Вода дистиллированная — 1000 мл.
66г. Среда Громова № 16.
Среда состава (г): NaHCO3 — 16,8; К2НРО4 — 0,5; NaNO3 — 2,5; K2SO4 — 1,0; NaCl — 1,0; MgSO4-7H2O - 0,2; СаС12-2Н2О - 0,04; FeSO4 - 6,01; ЭДТА - 0,08; раствор микроэлементов — 1 мл; вода дистиллированная — 1000 мл; pH среды — 7,0. Среду стерилизуют при 1,0 ати 20 — 30 мин.
Раствор микроэлементов (г): Н3ВО3 — 2,86; МпС12-4Н2О — 1,81; Zn SO4-7H2O — 0,22; МоО3 — 0,015; CuSO4.- 5Н20 — 0,08; вода дистиллированная — 1000 мл.
СРЕДЫ ДЛЯ ДРОЖЖЕЙ
67. Сусло для дрожжей.
Используют 6—8 °Б сусло (среда N° 2). При необходимости в среду вносят агар — 20 г/л.
68. Глюкозо-пептонпая среда (среда Сабуро).
Среда состава (г): глюкоза — 40,0; пептон — 10,0; агар — 18,0 — 20,0; вода водопроводная — 1000 мл. Среду стерилизуют при 0,5 ати 20 —30 мин. Иногда для обогащения факторами роста к среде добавляют дрожжевой или мясной экстракты в количестве 2—5 г/л.
69. Глюкозо-аммонийная среда.
Среда состава (г): глюкоза — 20,0; (NH4)2SO4 — 5,0; КН2РО4 — 0,85; К2НРО4 — 0,15; MgSO4- 7Н2О — 0,5; NaCl — 0,1; СаС12-4Н2О — 0,1; вода дистиллированная — 1000 мл.
В ряде случаев для обогащения факторами роста к среде добавляют дрожжевой или мясной экстракты в количестве 2 — 5 г/л. Среду стерилизуют при 0,5 ати 20—30 мин.
СРЕДЫ ДЛЯ МИЦЕЛИАЛЬНЫХ ГРИБОВ
70. Сусло и сусло-агар для мицелиальных грибов.
Используют 3 —4°Б сусло (среда № 2). При необходимости в среду вносят агар — 20 г/л. Иногда перед посевом в расплавленный сусло-агар стерильно добавляют 0,4 % (по объему) концентрированной молочной кислоты. Среду стерилизуют при 0,5 ати 20 — 30 мин.
71. Пентонно-декстрозный агар Ваксмана.
Среда состава (г): глюкоза — 10,0; пептон — 5,0; КН2РО4 — 1,0; MgSO4-7H2O — 0,5; агар— 15,0—20,0; вода водопроводная— 1000 мл. Среду стерилизуют при 0,5 ати 20—30 мин.
72. Среда Чапека с добавками микроэлементов и дрожжевого экстракта.
Среда состава (г): сахара (глюкоза или сахароза) — 30,0; NaNO3 — 3,0; КН3РО4 — 0,3; MgSO4-7H2O - 0,25; КС! - 0,25; FeSO4 - 0,01; ZnSO4 - 0,04, CuSO4-5H2O — 0,005; дрожжевой экстракт — 50 мл; агар — 20,0; вода дистиллированная — 1000 мл. Среду стерилизуют при 0,5 ати 20 — 30 мин.
73. Среда Чапека — Докса.
Среда состава (г): сахароза — 30,0; NaNO3 — 3,0; К2НРО4 — 1,0; MgSO4-7H2O — 0,5; КС! — 0,5; FeSO4-7H2O — 0,01; агар — 15,0; вода дистиллированная — 1000 мл. Среду стерилизуют при 0,5 ати 20 —30 мин.
74. Картофельно-декстрозный агар.
Среда состава (г): картофельный экстракт 0,23 л (см. среду № 11); глюкоза — 20,0 г; водопроводная вода — 0,77 л или potato-dextrose agar — PDA (Difco).
75. Среда Мартина.
Среда состава (г): глюкоза — 10; КН2РО4 — 5; MgSO4 — 0,5; пептон — 5; агар — 20; водопроводная вода — 1000 мл; pH — 5,5. Среду стерилизуют при 0,5 ати 20 — 30 мин.
76. Среда Ролэна.
Среда состава (г): сахароза — 50; NH4NO3 — 3; КН2РО4 — 1; MgSO4-7H2O — 1; CuSO4-5H2O — 0,02; агар — 20; дистиллированная вода — 1000 мл.
77. Среда Частухина, видоизмененная Захаровым.
Среда состава (г): целлюлоза — 10,0; (NH4)2SO4 — 0,75; КН2РО4 — 0,5; MgSO4- 7Н2О — 0,25; FeSO4 — следы; MnSO4 — следы; агар — 20,0; вода дистиллированная 1000 мл.
78. YpSS агар.
Среда состава (г): дрожжевой экстракт — 4,0, КН2РО4 — 1,0; MgSO4-7H2O — 0,5; растворимый крахмал — 15; агар — 20,0; вода (3/4 водопроводной, */4 дистиллированной) — 1000 мл.
79. Овсяный агар по Эмерсону.
Среда состава (г): овсяная мука — 50; агар — 20; водопроводная вода — 1000 мл.
80. Среда Сабуро с глицерином.
среда состава (г): пептон — 16; глюкоза — 20; глицерин — 5; агар — 12,5; дистиллированная вода — 1000 мл.
81. Глюкозо-минеральная среда для выделения дрожжей.
Среда состава (г): глюкоза — 20; (NH4)2SO4 — 5,0; КН2РО4 — 0,85; К2НРО4 — 0,15; MgSO4 7Н2О — 0,5; NaCl — 0,1; агар — 20; дистиллированная вода — 1000 мл; pH 4,5.
82. Среда Эшби.
Среда состава (г): сахароза (или манит, сорбит, крахмал) — 20,0; КН2РО4 — 0,2; К2НРО4 — 0,1; MgSO4 7Н2О — 0,2; NaCl — 0,2; K2SO4 — 0,1; агар — 20; стрептомицин сульфат 80 мг/л; дистиллированная вода — 1000 мл.
83. Среда Юргенсена и Даниелсона для выделения липомицетов.
Среда состава (г): глюкоза — 5,0; КН2РО4 — 1,0; MgSO4 7Н2О — 0,5; FeCl3 — следы; агар — 20,0; азот — имидазол — 0,25 или тимин — 0,48; pH — 5,2; циклогексимид — 200 мг/л; стрептомицин сульфат — 100 мг/л или ауреомицин — 50 мг/л; дистиллированная вода — 1000 мл.
84. Среда для фитопатогенов на основе сока V-8.
Среда состава: V-8 (нефильтрованный) — 0,2 л; СаСО3 — 3 г; агар — 9 г; вода — 0,8 л; pH 7,3.
85. Среда для выделения хитридиомицетов рода Allomyces.
Среда состава: тиамин НО — 0,15 мг; DL-метионин — 0,1 г; глюкоза — 5,0 г; MgCl2 — 0,005 М; КН2РО4 - 0,005 М; (NH4)2HPO4 - 0,005 М; СаС12 - 0,0005 М; МпС12 - 0,5 ppm; ZnSO4 — 0,1 ppm; CuSO4 — 0,1 ppm; FeCl3 — 1,0 ppm; (NH4)6MO7O2 — 0,2 ppm; H3BO3 — 0,5 ppm; CaCl2 — 0,2 ppm. Глюкозу и растворы СаС12 и MgCl2 стерилизуют отдельно.
86. Среда для выделения фитофторовых грибов Phytophthora cactorum.
Среда состава (г): сахароза — 5,0; кукурузное масло — 0,1 мл; DL-треонин — 1,0; КН2РО4 — 1,0; MgSO4-7H2O — 0,1; CaSO4-2H2O — 0,1; тиамин-гидрохлорид — 0,02; тергитол — 0,1 мл; ванкомицин — 200 мг; пимарицин — 10 мг; полимиксин В — 175 000 ЕД; агар — 20 г; вода дистиллированная — 1000 мл.
87. Среда Хагема для выделения эктомикоризных грибов.
Среда состава (г): солодовый экстракт — 5,0; глюкоза — 5,0; NH4C1 — 0,5; КН2РО4 — 0,5; MgSO4 • 7Н2О — 0,5; 1%-й раствор FeC13 — 0,5 мл; агар — 15,0; дистиллированная вода — 1000 мл.
88. Среда Мелина-Норканса для выделения эктомикоризных грибов.
Среда состава (г): сахароза — 2,5; КН2РО4 — 0,5, MgSO4-7H2O — 0,15; (NH4)2HPO4 — 0,25; NaCl — 0,025; СаС12 —0,1%-й раствор FeCl3 — 0,5 мл; агар — 15,0; дистиллированная вода — 1000 мл.
89. Среда Ван-Итерсона для грибов, использующих целлюлозу.
Двойной кружок фильтровальной бумаги сворачивают в виде конуса и ставят острием вверх в колбу Эрленмейера емкостью 100 мл с 50 мл водопроводной воды, со-580
держащей 0,75 г/л КН2РО4 и 0,1 г/л MgSO4. Устанавливают pH 7,0 —7,4 и стерилизуют при 1,0 ати.
СРЕДА ДЛЯ ВЫДЕЛЕНИЯ ГАЛОФИЛОВ
90. Среда Сегала.
Жидкая среда (г/л): гидролизат казеина — 7,5; дрожжевой автолизат — 10,0; 3-заме-щенный цитрат натрия — 3,0; КО — 2,0; MgSO4-7H2O —20,0; FeCl3 — 0,023; NaCl — 250,0; вода дистиллированная — 1000 мл. Компоненты среды последовательно растворяют в 800 мл воды, доводят pH до 7,5—7,8 с помощью 1 М КОН, затем смесь автоклавируют при 1 ати 5 мин. Среду охлаждают и фильтруют от осадка. Доводят pH до 7,4 с помощью 1 М НО, а объем до 1000 мл и стерилизуют автоклавированием при 0,5 ати. Если необходима твердая среда, то к жидкой среде после первого автоклавирования и доведения pH добавляют 20 г/л агара, нагревают до его расплавления, а затем автоклавируют. Среду разливают в чашки Петри при 60—70 °C, чтобы предотвратить преждевременное затвердение.
СРЕДЫ ДЛЯ ГЕНЕТИЧЕСКИХ ЭКСПЕРИМЕНТОВ С Е. COLI
91. Минимальная среда А.
Среда состава (г): К2НРО4 — 10,5; КН2РО4 — 4,5; (NH4)2SO4 — 0,5; вода дистиллированная — 1000 мл. Среду стерилизуют при 1,0 ати 20 —30 мин.
После автоклавирования при посеве добавляют 1 мл/л 1 М стерильного раствора MgSO4-7H2O и 10 мл/л 20%-го стерильного раствора глюкозы. Аминокислоты добавляют до конечной концентрации 40 мкг/мл (в случае D-, L-формы) и 20 мкг/мл (в случае L-формы). Витамины добавляют до конечной концентрации 1 мкг/мл.
92. Среда LB (Лурия-Бертрани).
Среда состава (г): триптон — 10,0; дрожжевой экстракт — 5,0; NaCl — 5,0; вода дистиллированная — 1000 мл. Среду стерилизуют при 0,5 ати 20 —30 мин.
НЕКОТОРЫЕ СРЕДЫ, ИСПОЛЬЗУЕМЫЕ В САНИТАРНОЙ
МИКРОБИОЛОГИИ
93. Среда Эндо (селективная среда для энтеробактерий).
100 мл стерильного МПА (pH 7,4) расплавляют на водяной бане и охлаждают до 70 °C, прибавляют 1 г лактозы (х.ч.), предварительно растворенной в стерильной пробирке в небольшом количестве дистиллированной воды и прокипяченной. В отдельных пробирках готовят: 2—3 мл спиртового насыщенного раствора фуксина и 10 мл 10% водного раствора Na2SO3 В стерильную пробирку вносят 1 мл фуксина и прибавляют раствор сульфита натрия до обесцвечивания фуксина (бледно-розовый цвет). Приготовленную смесь вливают в расплавленный МПА с лактозой, хорошо перемешивают и разливают по стерильным чашкам. Правильно приготовленная среда в горячем виде имеет бледно-розовый цвет, а при застывании становится бесцветной. Среду используют сразу, без стерилизации. Среда выпускается в сухом виде. Ее готовят по прописи на этикетке.
94. Среда Кесслера (среда для выделения бактерий группы кишечной палочки).
К 1 л водопроводной воды добавляют 10 г пептона и 50 мл бычьей желчи, кипятят смесь при помешивании 20 — 30 мин на водяной бане и фильтруют через вату. Прибавляют 2,5 г лактозы (х.ч.), доводят объем водой до 1 л, устанавливают pH 7,4 —7,6, после чего добавляют 2 мл 1 % водного раствора генцианового фиолетового для подавления роста грамположительных бактерий, разливают (по 5 или 10 мл) в пробирки с
поплавками или комочками ваты и стерилизуют 10 мин при 1 ати. Среда выпускается в сухом виде. Ее готовят по прописи на этикетке.
95. Среда КОДА (среда для выделения бактерий группы кишечной палочки).
Выпускается в сухом виде. Состоит из пептона, гидролизата рыбы, додецилсульфата натрия, хлорида натрия, лактозы, бромтимолового синего. Среду готовят по прописи на этикетке.
96. Полужидкая среда с глюкозой.
В 1 л дистиллированной воды растворяют 10 г пептона; 5 г NaCl; 4 — 5 г агар; доводят до кипения, устанавливают pH (7,2—7,4), добавляют 1 мл 1,6 % спиртового раствора бромтимолового синего. В расплавленную среду вносят 5 г глюкозы, нагревают до кипения, разливают в пробирки столбиком высотой около 3 см и стерилизуют 15 мин при 0,5 ати.
97. Среда Козера.
В 1 л дистиллированной воды растворяют 1,5 г фосфата натрий-аммония [Na(NH4)HPO4], 0,2 г сульфата магния, 3 г нейтрального цитрата натрия, 1 г фосфата калия однозамещенного. Раствор разливают в пробирки по 5 мл и стерилизуют 30 мин при 1 ати. Среда прозрачная, при росте цитрат-ассимилирующих бактерий становится мутной.
98. Среда Клиглера.
К 1 л МПА добавляют 10 г лактозы, 10 г сахарозы и 1 г глюкозы. После растворения углеводов добавляют по 10 мл 2 % раствора сульфата железа, 0,8 % раствора тиосульфата натрия (Na2S2O3-7Н2О) и 0,4% раствора сульфита натрия (Na2SO3). Эта смесь служит реактивом на присутствие сероводорода. Добавляют 24 мл 1 % водно-спиртового раствора фенолового красного (pH 7,4). Среду разливают в пробирки по 5 мл и стерилизуют 20 — 30 мин при 0,5 ати. Среда выпускается в сухом виде. Ее готовят по прописи на этикетке.
99. Селенитовая среда (селенитовый бульон) — среда для накопления сальмонелл.
Среда состава (г): однозамещенный кислый селенит натрия (NaHSeO3) — 4,0; пептон — 5,0; Na2HPO4 — 7,0; NaH2PO4 — 3,0; лактоза — 4,0; вода дистиллированная 1000 мл. Для приготовления среды используют два раствора.
Раствор 1: к 950 мл раствора фосфатов добавляют пептон и лактозу. Среду разливают во флаконы и стерилизуют при 0,5 ати. При приготовлении среду подтитровывают, чтобы при добавлении пептона и селенита натрия готовая среда имела pH 6,9 —7,1. Раствор хранят при 4—10 °C 1 —2 мес.
Раствор 2. однозамещенный кислый селенит натрия растворяют в 40 мл стерильной дистиллированной воды. Раствор готовят непосредственно перед приготовлением среды.
В каждый флакон с 50 мл раствора 1 добавляют по 2 мл раствора 2 и перемешивают. Среда выпускается в сухом виде. Ее готовят по прописи на этикетке.
100. Висмут—сульфит агар (ВСА) — селективная среда для сальмонелл.
Выпускается в сухом виде. Состоит из агара, гидролизата рыбы, глюкозы, цитрата висмута, сульфита натрия, соли Мора, фосфата натрия, бриллиантового зеленого. Среду готовят по прописи на этикетке. Готовая среда имеет зеленоватую окраску.
101. Солевой бульон (среда для накопления стафилококков).
В 1000 мл МПБ (среда № 1) растворяют 65 г NaCl, устанавливают pH 7,4 и стерилизуют при 1 ати.
102. Молочно-солевой агар (среда для культивирования стафилококков).
Непосредственно перед использованием к 100 мл расплавленного и охлажденного до 60—70°C стерильного МПА (pH 7,4), содержащего 6,5 г NaCl, добавляют 10 мл стерильного обезжиренного молока (среда № 5), перемешивают и разливают по стерильным чашкам Петри.
103. Желточно-солевой агар Чистовича (среда для культивирования стафилококков).
Непосредственно перед использованием к 100 мл расплавленного и охлажденного до 60—70 °C стерильного МПА (pH 7,2), содержащего 10 г NaCl, добавляют 10 — 20 мл желточной взвеси, перемешивают и разливают по стерильным чашкам Петри. Для при
готовления желточной взвеси желток асептически извлекают из яйца, взбалтывают с 200 мл стерильного изотонического раствора NaCl (0,85 % раствор NaCl, pH 6,9 —7,0).
104. Среда Вильсона—Блера (среда для культивирования мезофильных анаэробов).
Непосредственно перед использованием к 100 мл расплавленного и охлажденного до 60—70 °C стерильного МПА (pH 7,2), содержащего 1 % глюкозы, добавляют 10 мл 20 % раствора сульфита натрия и 1 мл 8 % раствора хлорида железа (FeCl2). Оба раствора готовят на стерильной дистиллированной воде.
СРЕДЫ ДЛЯ КУЛЬТИВИРОВАНИЯ ПРОСТЕЙШИХ
Иногда для сравнения рекомендуется употреблять несколько сред одновременно, а также помещать пробы почвы в стерильную дождевую или дистиллированную воду. Однако в такой воде численность простейших гораздо меньше, чем в питательной среде, и они быстро исчезают или инцистируются.
Агаровые среды разливают по чашкам Петри, стерилизуют, затем заражают исследуемой почвой и помещают в термостат при температуре 20 — 25 °C. В течение ряда дней среды исследуют для обнаружения развившихся простейших. Некоторые простейшие появляются очень быстро, но скоро инцистируются или погибают, другие виды появляются не сразу и развиваются в течение нескольких недель.
105. Настой из навоза.
Приготавливают 5%-й настой из лошадиного или коровьего навоза, оставляют на сутки, затем разбавляют до 1 — 2 %, фильтруют и стерилизуют.
106. Сенной отвар (среда для культивирования Paramecium).
15 г измельченного сена кипятят 15 мин в дистиллированной воде, затем фильтруют. Среду наливают в чашки Коха (высотой 6 см, диаметром 10 см) или в стеклянные банки. В каждый сосуд вносят несколько прокипяченных стебельков сена. Сосуды оставляют на ночь открытыми, чтобы в среду могли попасть бактерии. Затем их закрывают неплотно прилегающей крышкой. Через 1 — 2 дня вносят парамеций.
107. Почвенный экстракт.
Берут огородную или садовую почву, высушивают на воздухе и затем помещают в колбу с водой из расчета 10% по весу или даже 1 кг на литр воды. Суспензию кипятят 15 мин, нейтрализуют, раствор отжимают через тряпку, затем разбавляют и фильтруют, после чего стерилизуют. Почвенную вытяжку употребляют иногда вместе с 3—4%-м сенным настоем.
108. Настои из овощей.
Овощи (картофель, салат, морковь) кипятят до разваривания и готовят 1 — 5%-й настой, который употребляют после стерилизации.
109. Рисовая среда.
Несколько зерен риса помещают в чашку Петри со стерильной водой. Через 2—3 сут в чашку вносят пробу почвы.
ПО. Плотные среды для жгутиконосцев.
Используют агаризованные (15 — 20 г/л агара) среды Виноградского (см. среды № 59 и 60) и Омелянского для нитрифицирующих бактерий.
111. Плотная среда для амеб и мелких инфузорий.
Среда состава: маннит — 10 г; К2НРО4 — 1 г; почвенная вытяжка — 1000 мл; агар — 15 г.
112. Мясной агар.
Среда состава (г): мясной экстракт — 0,3 —0,5; NaCl — 0,5; агар — 20,0. Вода — 1000 мл.
113. Среда Чокли для хищных амеб.
Среда состава (мг): NaCl — 80; NaHCO3 — 4; КС1 — 4; СаС12 — 4; СаН4(РО4)2- 2Н2О — 1,6. Вода бидистиллированная — 1000 мл. Приблизительно 2 раза в неделю к культурам необходимо добавлять кормовые организмы (как правило, таких инфузорий, как Paramecium и Colpidium).
fPS. (Е^еда. для автотрофных протестов (jEugfena и др.).
Среда состава (мл): жидкое удобрение для комнатных растений — 1; вода бидистил-лированная — 1000 мл.
115. Среда для аксенического культивирования инфузорий {Paramecium, Tetrahymena).
Среда состава (г): протеозопептон — 10,0; NaCl — 5,0; декстроза — 2,0; дрожжевой экстракт — 8,0; вода бидистиллированная — 1000 мл. Среду стерилизуют 20 — 30 мин при 0,5 ати.
СРЕДА ДЛЯ ВЫРАЩИВАНИЯ РАСТЕНИЙ В ЛАБОРАТОРНЫХ УСЛОВИЯХ
116. Среда Хогланда.
Маточный раствор (г/л): КН2 РО4 — 136,12; KNO3 — 101,11; Ca(NO3)2-4Н2О — 236,16; MgSO4 — 247,0; FeEDTA — 6,92; раствор микроэлементов (г/л): КС1 — 3,726, Н3ВО3 - 1,546; MnSO4-H2O - 0,338; ZnSO4-7H2O - 0,575; CuSO4-5H2O - 0,125; Н2МоО4 — 0,081. Для приготовления среды Хогланда в 1 л дистиллированной воды растворяют 0,5 мл маточного раствора КН2РО4, 2,5 мл раствора KNO3, 2,5 мл Ca(NO3)2-4H2O, 1 мл MgSO4, 0,5 мл FeEDTA и 0,5 раствора микроэлементов. Среду стерилизуют 30 мин при 1,0 ати.
ПРИЛОЖЕНИЕ 5
РЕЦЕПТЫ КРАСИТЕЛЕЙ И ИНДИКАТОРОВ
1. Фуксин основной (насыщенный спиртовой раствор).
Фуксин основной — 10,0 г; этанол 96° — 100 мл.
2. Фуксин основной карболовый (фуксин Циля).
5%-й водный раствор свежеперегнанного фенола — 100 мл; насыщенный спиртовой раствор фуксина основного — 10 мл. Приготовленную смесь через 48 ч отфильтровывают. Краситель отличается устойчивостью.
3. Фуксин основной, водный раствор.
Карболовый фуксин Циля — 1 мл; вода дистиллированная — 9 мл. Водный фуксин готовят непосредственно перед употреблением, так как он нестоек.
4. Метиленовый синий, насыщенный раствор.
Метиленовый синий — 3,0 г; этанол 96° — 100 мл. Раствор оставляют на 2 —3 дня, несколько раз перемешивают (взбалтывают), затем фильтруют. Раствор устойчив.
5. Метиленовый синий 1:40.
Насыщенный спиртовой раствор метиленового синего — 1 мл; вода дистиллированная — 40 мл.
6. Метиленовый синий (по Леффлеру).
Насыщенный спиртовой раствор метиленового синего —30 мл; вода дистиллированная — 100 мл; КОН, 1%-й водный раствор — 1 мл.
7. Генциановый фиолетовый карболовый.
Раствор 1: генциановый фиолетовый — 1,0 г; этанол 96° — 10 мл.
Раствор 2. 5%-й водный раствор свежеперегнанного фенола — 100 мл.
После полного растворения генцианового фиолетового растворы смешивают.
8. Кристаллический фиолетовый, водный раствор.
Кристаллический фиолетовый — 20 мг; вода дистиллированная — 100 мл.
9. Эритрозин карболовый.
Эритрозин — 3,0 г; вода дистиллированная — 100 мл; фенол свежеперегнанный — 5,0 г.
После растворения эритрозина и фенола смеси дают отстояться.
10. Сафранин, водный раствор.
2,5%-й раствор сафранина в 96 °-м этаноле — 10 мл; вода дистиллированная — 100 мл.
11. Реактивы для окраски жгутиков.
Протрава. 12 г таннина растворяют при нагревании в 48 мл дистиллированной воды, к раствору прибавляют 30 мл насыщенного водного раствора железного купороса (FeSO4) и 6 мл насыщенного спиртового раствора фуксина. Раствор отфильтровывают и сохраняют в банке с притертой пробкой. Протрава бывает готовой к употреблению через несколько дней после приготовления и может храниться в течение нескольких месяцев.
Краситель. Карболовый фуксин Циля и вода дистиллированная в соотношении 1:1.
Протраву и карболовый фуксин Циля готовят заранее. Разбавленный фуксин следует готовить незадолго до употребления. Непосредственно перед работой протраву и разбавленный фуксин необходимо профильтровать через складчатый бумажный фильтр, так как осадки красок, легко образующиеся на стенке сосуда при наличии даже небольших количеств белка, будут сильно мешать окрашиванию и рассмотрению объекта.
12. Реактивы для окраски жгутиков по методу Фонтана.
Протрава, таннин — 5,0 г; фенол кристаллический — 1,0 г; вода дистиллированная — 100 мл.
Реактивы для серебрения', серебро азотнокислое — 5,0 г; вода дистиллированная — 100 мл. Раствор серебра следует готовить без фильтрования и хранить в темной склянке. К 3 — 4 мл 5%-го раствора азотнокислого серебра по каплям прибавляют раствор аммиака до помутнения и образования осадка, а затем осторожно — до растворения осадка. После этого вновь прибавляют раствор азотнокислого серебра до появления легкой опалесценции. Полученный раствор аммиачного серебра разводят дистиллированной водой в 10 раз.
13. Анилиновый черный.
Анилиновый черный — 1,5 г; этанол 96° — 50 мл; уксусная кислота 80%-я — 10 мл; вода дистиллированная — 40 мл. После растворения краску фильтруют и через 3 дня она готова к употреблению.
14. Красители для выявления липидов.
1. Судан III — 0,5 г; молочная кислота концентрированная — 100 мл.
2. Судан черный В — 0,3 г; спирт этиловый, 70%-й горячий — 100 мл.
Раствор выдерживают в течение нескольких часов при 60 °C в закупоренной склянке, затем охлаждают и фильтруют.
15. Малахитовый зеленый, водный раствор.
Малахитовый зеленый (малахитгрюн) — 7,5 г; вода дистиллированная — 100 мл.
16. Раствор Люголя в модификации Грама.
Йод кристаллический — 1,0 г; калий йодистый — 2,0 г; вода дистиллированная — 300 мл. В ступку емкостью 30—50 мл помешают навеску йода и йодистого калия, растирают смесь пестиком, добавляют 1 мл дистиллированной воды и, продолжая растирать кристаллы, добавляют еще 5 мл воды. Йод растворяется в йодистом калии. Раствор количественно переносят в склянку и доводят общий объем до 300 мл. Срок годности раствора не более 30 дней; хранят его в темной посуде.
17. Раствор Люголя для выявления гликогена и гранулезы.
Йод кристаллический — 1,0 г; калий йодистый — 3,0 г; вода дистиллированная — 300 мл. Раствор готовят так же, как и предыдущий.
18. Приготовление туши для негативного контрастирования.
Тушь черная — 10 мл; вода дистиллированная — 30 мл. Разведенную тушь центрифугируют, надосадочный слой разливают в пробирки и стерилизуют при 0,5 ати. Сохранить тушь можно также, добавляя к ней раствор тимерсола (1: 1000) в соотношении 1:2 и твин-80 (1: 100) — 1 капля на 100 мл раствора.
19. Бумага, пропитанная раствором уксуснокислого свинца.
Нарезают полоски фильтровальной бумаги (0,2 —0,4x5 —6 см), погружают их в 5%-й водный раствор уксуснокислого свинца РЬ(СН3СОО)2, высушивают на воздухе и стерилизуют в чашке Петри автоклавированием при 0,5 ати. Перед употреблением бумага бесцветна, а при наличии сероводорода она приобретает черно-бурый цвет.
20. Реактивы для определения белка по Лоури.
Раствор А. 2%-й раствор Na2CO3 в 0,1 н. NaOH.
Раствор В: готовится в день определения. 0,1 г CuSO4- 5Н2О растворяют в 20 мл 1%-го раствора K-Na тартрата (сегнетовой соли). Этот раствор сохраняется не более 30 мин\ 1 мл полученного раствора смешивают с 50 мл раствора А. Даже едва мутный раствор не пригоден для использования!
Реактив Фолина. В колбу объемом 1500 мл вносят 700 мл дистиллированной воды, 100 г Na2WO4-2H2O, 25 г Na2MoO4-2Н2О, 50 мл 85%-й Н3РО4 и 100 мл конц. НС1. Полученную смесь кипятят на слабом огне в течение 10 ч с обратным холодильником (можно дробно). К охлажденной смеси добавляют 150 г сернокислого лития (LiSO4), 50 мл дистиллированной воды и 15 капель брома. Избыток брома удаляют кипячением в течение 15 мин без холодильника. Раствор охлаждают. Он должен быть желтым. Если раствор зеленый, добавляют еще несколько капель брома и вновь выпаривают избыток последнего. Объем раствора доводят до 1000 мл и сохраняют в холодильнике в темной склянке с притертой пробкой. Основной раствор обычно бывает 2 н. Белок определяют с помощью 1 н. раствора Фолина. Для этого титрованием 0,1 н. NaOH с фенолфталеином определяют концентрацию основного раствора и разбавляют его дистиллированной водой до 1 н. в продаже имеется готовый реактив Фолина.
21. Реактив Эрлиха.
Лс/дг-диметиламинобензальдегид — 1,0 г; этанол 96° — 95 мл; НС1 концентрированная — 20 мл.
Модифицированный реактив Эрлиха: 5 г n-диметиламинобензальдегида растворяют в 100 мл кислого спирта. Кислый спирт: к 92 мл этилового спирта 96° добавляют 8 мл концентрированной НС1.
22. Реактив Эрлиха в модификации Ковача.
Пара-диметиламинобензальдегид — 1,0 г; амиловый или изоамиловый спирт — 15 мл; НС1 концентрированная — 10 мл.
23. Реактив Несслера.
KI - 70,0 г; Hgl2 - 100,0 г; КОН - 100,0 г.
Раствор 1. Навески KI и Hgl2 растворяют в 400 мл дистиллированной воды.
Раствор 2. Навеску КОН растворяют в 500 мл дистиллированной воды.
Растворы 1 и 2 смешивают и доводят общий объем до 1 л. В случае образования осадка верхний прозрачный слой декантируют. Раствор хранят в темной стеклянной посуде. В продаже имеется готовый реактив Несслера.
24. Реактив Грисса.
Раствор 1: 0,5 г сульфаниловой кислоты растворяют в 30 мл ледяной уксусной кислоты. Добавляют 100 мл дистиллированной воды и фильтруют. Раствор сульфаниловой кислоты устойчив в течение месяца.
Раствор 2: 0,1 г а-нафтиламина растворяют в 100 мл кипящей дистиллированной воды. Остужают и добавляют 30 мл ледяной уксусной кислоты. Раствор а-нафтиламина фильтруют и хранят не более недели. Непосредственно перед употреблением смешивают равные объемы этих растворов.
25. Реактивы для крахмало-йодной пробы на нитриты.
Крахмал — 0,4 г; ZnCl2 — 2,0 г; вода дистиллированная — 100 мл.
ZnCl2 растворяют в 10 мл воды, кипятят и добавляют крахмал. Доводят объем до 100 мл и оставляют стоять на неделю. Затем раствор фильтруют и добавляют равный объем 0,2%-го раствора К1.
Раствор соляной кислоты. НО концентрированная — 16 мл; вода дистиллированная — 84 мл.
РЕЦЕПТЫ РАСТВОРОВ
1. Реактивы для выявления оксидазы
• Свежеприготовленный 1%-й водный раствор солянокислого тетраметил-и-фе-ннлендиамина.
• 30 — 40 мг а-нафтола растворяют в 2,5 мл этилового спирта-ректификата, добавляют 7,5 мл дистиллированной воды и растворяют 40 — 60 мг диметил-л-фе-нилендиамина.
2. Фиксирующие жидкости
• Этиловый спирт 96°; время фиксации — 10—15 мин.
• Метиловый спирт безводный; время фиксации — 3 — 5 мин.
• Смесь Никифорова: этиловый спирт и серный эфир в равных объемах; время фиксации — 5—10 мин.
• Спиртформол: 40%-й формалин — 5 мл; этиловый спирт 96° — 95 мл; время фиксации — 5—15 мин.
• 1—2%-й водный раствор осмиевой кислоты; фиксация, в парах 3 — 5 мин. Раствор осмиевой кислоты сохраняют в темном флаконе с притертой пробкой. При работе следует соблюдать осторожность, так как пары осмия могут повредить глаза\
• 40%-й формалин; фиксация в парах несколько секунд.
• Фиксатор Карнуа: этиловый спирт 96° — 60 мл; хлороформ — 30 мл; ледяная уксусная кислота — 10 мл; время фиксации — 15 мин.
• Жидкость Руге: 40%-й формалин — 20 мл; ледяная уксусная кислота — 1 мл; вода дистиллированная — 100 мл; время фиксации — 5 мин. Рекомендуется для фиксации клеток, выращенных в жидких средах неопределенного состава.
• Фосфорномолибденовая кислота, 5%-й водный раствор; время фиксации — 5 мин.
• Смесь Шаудина: насыщенный раствор сулемы — 2 части; этиловый спирт 96° или абсолютный — 1 часть; на мазок наносят каплю фиксатора и медленно подсушивают на воздухе.
3. Чернила по стеклу
Раствор J: 1,0 г фуксина основного растирают в ступке с 15 мл этилового спирта.
Раствор 2: к 2,0 г таннина добавляют 15 мл воды и нагревают до кипения.
Растворы 1 и 2 смешивают в равных объемах.
4. Перегонка фенола
Фенол перегоняют из колбы Вюрца, закрытой корковой пробкой. Колбу помещают непосредственно на сетку. В качестве приемника используют заранее взвешенную сухую фарфоровую чашку. В нее отгоняют некоторое количество фенола и, когда он застынет, определяют его массу, исходя из которой, готовят соответствующий объем раствора или необходимого реактива. Перегонку’ фенола осуществляют в вытяжном шкафу. При работе с фенолом следует соблюдать большую осторожность, так как он вызывает сильные ожогиХ
5. Физиологический раствор
Готовят 0,85%-й раствор NaCl в дистиллированной воде. При необходимости стерилизуют 30 мин при 1 ати.
6. Подготовка предметных и покровных стекол для приготовления препаратов.
Предметные и покровные стекла считаются чистыми, когда капля воды растекается по их поверхности. Новые стекла обычно кипятят в 1%-ном растворе соды, затем промывают дистиллированной водой, слабым раствором соляной кислоты и затем опять дистиллированной водой. Стекла, бывшие в употреблении, кипятят в мыльной воде и
затем не менее суток выдерживают в растворе хромовой смеси. От бихромата стекла отмывают дистиллированной водой. Чистые стекла хранят в 96-градусном этаноле.
7. Обработка предметных стекол по Цеттнову
• Стекла кипятят 10 мин в следующем растворе: бихромата калия — 20,0 г; дистиллированная вода — 200 мл; концентрированная серная кислота — 20 мл.
• Промывают в течение 5 мин слабым раствором едкого натра.
• Тщательно промывают водой (10 раз).
• Промывают спиртом (3 раза).
• Быстрый способ обезжиривания предметных стекол
• При отсутствии заранее приготовленных обезжиренных стекол можно быстро приготовить стекла, натирая их в сухом виде хозяйственным мылом и очищая затем чистой хлопчатобумажной тканью.
8. Хромовые смеси для мытья посуды.
• В концентрированную серную кислоту добавляют около 5 % (от объема серной кислоты) размельченного в порошок кристаллического К2Сг2О7 и осторожно нагревают в фарфоровой чашке на водяной бане до его растворения.
• Двухромовокислый калий растворяют в воде, затем в раствор осторожно добавляют серную кислоту. Смесь готовят из расчета: вода — 100 мл; двухромовокислый калий — 6,0 г; серная кислота (плотность 1,84) — 100 мл.
После многократного употребления темно-оранжевый цвет хромовой смеси меняется на темно-зеленый. Такая смесь уже не обладает моющими свойствами. Хромовой смесью не следует мыть посуду, загрязненную парафином, керосином, минеральными маслами и другими продуктами перегонки нефти.
Вниманий. Хромовая смесь сильно разрушает ткани животного и растительного происхождения, поэтому работать с ней следует осторожно. Если она попала на руки или одежду, то пораженное место следует немедленно обмыть большим количеством воды, затем разбавленным раствором аммиака или соды, а затем снова водой.
9. Спиртовой раствор КОН для мытья посуды.
40—50 г КОН растворяют в 500 мл воды. После остывания раствора к нему добавляют спирт-сырец в таком количестве, чтобы общий объем составил 1 л.
10. Свойства важнейших кислотно-основных индикаторов (по Лурье, 1967).
Индикатор Концентрация, % Растворитель Интервал перехода pH Окраска индикатора
Метиловый фиолетовый (метил виолет, 1 -й переход) 0,10 и 0,05 Вода 0,13-0,5 Желтая — зеленая
Метиловый зеленый 0,05 Вода 0,1-2,0 Желтая — зелено-голубая
Метиловый фиолетовый (2-й переход) 0,10 Вода 1,0-1,5 Зеленая — синяя
лгКрезоловый пурпурный (1-й переход) 0,04 Спирт 20° 1,2-2,8 Красная — желтая
Тимоловый синий (ти-молблау, 1-й переход) 0,10 а) Спирт 20°. б) Вода с добавлением 4,3 мл 0,05 н. NaOH на 100 мг индикатора 1,2 -2,8 Красная — желтая
Индикатор Концентрация, % Растворитель Интервал перехода pH Окраска индикатора
Метиловый фиолетовый (3-й переход) 0,10 Вода 2,0 -3,0 Синяя — фиолетовая
Метиловый оранжевый (метилоранж, оранж. III, геллантин) 0,10 Вода 3,1-4,4 Красная — оранжевожелтая
Бромфеноловый синий (бромфенол блау) 0,10 а) Спирт 20°. б) Вода с добавлением 3 мл 0,05 н. NaOH на 100 мг индикатора
Конго-красный (конгорот) 0,10 и 1,00 Вода 3,0-5,2 Сине-фиолетовая — крас-ная
Бромкрезоловый синий (бромкрезолгрюн, бром-крезолблау, бромкрезоловый зеленый) 0,10 а) Спирт 20°. б) Вода с добавлением 2,9 мл 0,05 н. NaOH на 100 мг индикатора 3,8-5,4 Желтая — синяя
Метиловый красный (метилрот) 0,10 и 0,02 Спирт 60° 4,2-6,4 Красная — желтая
Бромфеноловый красный (бромфенолрот, дибромфенолсульфоф-талеин) 0,10 и 0,04 а) Спирт 20°. б) Вода с добавлением 3,9 мл 0,05 н. NaOH на 100 мг индикатора 5,0-6,8 Желтая — красная
Бромкрезоловый пурпурный (бромкрезол-пурпур, дибром -о-кре-золсульфофталеин) 0,10 а) Спирт 20°. б) Вода с добавлением 3,7 мл 0,05 н. NaOH на 100 мг индикатора 5,2-6,8 Желтая — пурпурная
Бромтимоловый синий (бромтимолблау, дибром-тимолсульфофт-алеин) 0,05 и 0,10 а) Спирт, 20°. б) Вода с добавлением 3,2 мл 0,05 н. NaOH на 100 мг индикатора 6,0-7,6 Желтая — синяя
Нейтральный красный (нейтральрот) 0,10 Спирт 60° 6,8-8,4 Красная — ян-тарно-же-лтая
Феноловый красный (фенолрот, фенолсульфофталеин) 0,10 и 0,05 а) Спирт 20°. б) Вода с добавлением 5,7 мл 0,05 н. NaOH на 100 мг индикатора 6,8-8,4 Желтая — красная
Индикатор Концентрация, % Растворитель Интервал перехода pH Окраска индикатора
Крезоловый красный (крезолрот, о-крезол-сульфофталеин) 0,10 а) Спирт 50°. б) Вода с добавлением 5,3 мл 0,05 н. NaOH на 100 мг индикатора 7,2-8,8 Янтарно-желтая — пурпурно-красная
м-Крезоловый пурпурный (2-й переход) 0,04 Спирт 20° 7,4-9,0 Желтая — пурпурная
Тимоловый синий (2-й переход) 0,10 а) Спирт 20°. б) Вода с добавлением 4,3 мл 0,05 н. NaOH на 100 мг индикатора 8,0-9,6 Желтая — синяя
Фенолфталеин 0,10 и 1,00 Спирт 60° 8,2-10,0 Бесцветная — пурпурная
Тимолфталеин 0,10 и 0,04 Спирт 90° 9,3-10,5 Бесцветная — синяя
Оранжевый 0,10 Вода 11,5-14,0 Желтая — красная
Примечание. Вода дистиллированная.
11. Буферные растворы для приготовления и разведения суспензий микроорганизмов.
Схема приготовления буферных растворов
Буферный раствор Компонент А Компонент Б
Фосфатный 0,1 М Na2HPO4- 2Н2О 17,8 г/л 0,1 М NaH2PO4-2H2O 15,6 г/л
Трис — НО 0,1 М НС1 8,5 мл 36%-й НС1 + 991,5 мл Н2О 0,1 М трис (оксиметиламиометан) 12,1 г/л
pH Количество компонента А, мл
Фосфатный буфер Трис — НО буфер
6,0 61,5 —
6,2 92,5 —
6,4 132,5 —
6,6 187,5 —
6,8 245,0 —
7,0 305,0 —
7,2 360,0 221,0
7,4 405,0 207,0
7,6 435,0 192,0
Буферный раствор Компонент А Компонент Б
7,8 457,5 162,5
8,0 473,5 134,0
Количество компонента Б, мл 500-А 500
Довести до объема дисти-лированной водой 1 л 1 л
Физические свойства некоторых буферных растворов
Наименование p.K (20 “C) pK«/°c Молярная масса (основание) Молярность насыщенного раствора при 0 °C
MES 6,15 -0,011 195,23 0,65
ADA 6,60 -0,011 212,15
BIS—TRIS PROPANE 6,80 -0,016 282,35 2,30
PIPES 6,80 -0,009 342,26 1,40
ACES 6,90 -0,020 182,20 0,22
MOPS 7,20 -0,006 209,26 3,00
TES 7,50 -0,200 229,25 2,60
HEPES 7,55 -0,014 238,31 2,30
HEPPS 8,00 -0,007 252,33 2,50
TRICINE 8,15 -0,021 179,18 0,80
TRIS 8,30 -0,310 121,13 2,40
BICINE 8,35 -0,018 163,17 1,10
Глицил—глициновый 8,40 -0,028 132,13 1,10
CHES 9,50 -0,009 207,30 0,85
CAPS 10,40 -0,009 221,32 0,85
ПРИЛОЖЕНИЕ 7
НЕАНГЛИЙСКИЕ ВЫРАЖЕНИЯ И СОКРАЩЕНИЯ, ВСТРЕЧАЮЩИЕСЯ В НАУЧНОЙ ЛИТЕРАТУРЕ
ab initio
ab ovo
а. с. (anni currentis)
A.D. (Anno Domini)
с начала
с самого начала текущего года нашей эры
а. т. (ante meridiem) ad hoc
ad infinitum
до полудня для данной цели, к этому случаю до бесконечности
ad interium в промежутке
ad libitum как угодно, по желанию, на выбор
addendum добавление
a posteriori на основании опы-
a priori та
независимо от опыта, заранее
B.C. (before Christ) до нашей эры
c. (circa) приблизительно, около
c. p. (ceteris paribus) при прочих равных условиях
cf. (confer) сравни
corrigenda список ошибок
cum включая, с чем-то или кем-то еще
e. g. (exempli gratia) например
e. r. (en route) в пути
erratum (pl. errata) опечатка, опечатки
et al. и другие
etc. (et cetera) и так далее
et seq. (et sequentia) и последующие
ex situ не на месте, не в данном месте
f. v. (folio verso) на обороте листа
ib., ibid, (ibidem) в том же месте, там же
id. (idem) то же самое, тот же, так же
i. e. (id est) то есть
in ex. (in extenso) полностью, довольно
in loc (in loco) на своем месте
in loc cit (in loco citato) в цитируемом месте
in parvo в незначительной мере
in re относительно, по вопросу
in situ в данном месте, на месте
in toto в целом
in vitro в лабораторном сосуде
in vivo в живом организме, в естественных условиях
int. al. (inter alia) между прочим
ipso facto в силу очевидности, самим фактом
loc. cit. указанное сочинение
locus (pl. loci) место, месторасположение, места
m. p. (manu propria) собственноручно
med. (medium) середина, центр
memo (memorandum) меморандум
modus operandi способ действия
mutatis mutandis сделав соответствующие изменения
N. B., n. b. (nota bene) примечание, отметка
op. cit. (opus citatum) цитируемое произведение
par example например
par excellence преимущественно, по преимуществу
pari passu попутно
per capita на душу населения
per se сам по себе, по существу
p. ni. (post meridiem) после полудня
prima facie на первый взгляд
pro et con. (pro et contra) за и против
pro forma формально, для вида
pro rata пропорциональный, пропорционально
pro tem (pro tempore) временно
quantum libet сколько угодно
quod vide смотри там-то
re, in re по вопросу, по делу
s. a. (sine anno) без указания года (издания)
s. a. e. 1. (sine anno без указания
et loco) года и места (издания)
s. d. (sine die) без указания срока или даты, на неопределенный срок
s. s. (sensu stricto) в буквальном смысле
s. 1. (sensu lato) в широком смысле
sic! буквально так! (указывает на важность или подлинность данного места в тексте или на ошибочность
sui generis
tabula rasa
terra incognita
приведенных слов)
своего рода, своеобразный
буквально «чистая дощечка», нечто нетронутое незнакомая область
u. i. (ut infra)
u. s. (ut sup, ut supra) vs., vers, (versus)
v. v. (vice versa)
v. i. (vide infra)
v. s. (vide supra) viz. (videlicet) vulgo
как указано ниже как указано выше против, в сравнении
наоборот
смотри ниже смотри выше
а именно, то есть обычно
СПИСОК ЛИТЕРАТУРЫ
Основная литература
Бабъева И.П., Зенова Г.М. Биология почв. — М.: Изд-во МГУ, 1989.
Большой практикум по микробиологии / Под ред. Г.Л.Селибера. — М.: Высшая школа, 1962.
Борисов Л. Б., Козьмин-Соколов Б.Н., Фрейдлин И. С. Руководство к лабораторным занятиям по медицинской микробиологии, вирусологии и иммунологии. — М.: Медицина, 1993.
Гусев М.В., Минеева Л. А. Микробиология. — М.: Изд-во «Академия», 2003.
Заварзин Г.А., Колотилова Н.Н. Введение в природоведческую микробиологию. — М.: Книжный дом «Университет», 2001.
Зенова Г.М., Степанов А.Л., Лихачева Н.А., Манучарова Н.А. Практикум по биологии почв. — М.: Изд-во МГУ, 2002.
Избранные задачи большого практикума по микробиологии. — М.: Изд-во МГУ, 1991.
Кондратьева Е.П. Автотрофные прокариоты. — М.: Изд-во МГУ, 1996.
Метаболизм микроорганизмов / Под ред. Н. С. Егорова. — М: Изд-во МГУ, 1986.
Методы почвенной микробиологии и биохимии / Под ред. Д. Г. Звягинцева. — М.: Изд-во МГУ, 1991.
Методы общей бактериологии: В 3 т. / Под ред. Ф. Герхардта. — М.: М ир, 1984.
Определитель бактерий Берджи: В 2 т. / Под ред. Дж.Хоулта, Н. Крига, П.Снита, Дж. Стейли, С. Уильямса. — М.: Мир, 1997.
Плакунов В. Г. Основы энзимологии. — М.: Логос, 2002.
Практикум по микробиологии / Под ред. Н. С. Егорова. — М.: Изд-во МГУ, 1976.
Промышленная микробиология/ Под ред. Н.С. Егорова. — М.: Высшая школа, 1989.
Руководство к практическим занятиям по микробиологии / Под ред. Н.С.Егорова. — М.: Изд-во МГУ, 1995.
Стейниер Р. и др. Мир микробов: В 3 т. — М.: Мир, 1979.
Теппер Е.З., Шильникова В.К., Переверзева К. И. Практикум по микробиологии. — М.: Колос, 1994.
Шлегель Г. Общая микробиология. — М.: Мир, 1986.
Bergey’s Manual of Systematic Bacteriology. 1st. ed. / Eds. A.Balows et al. — V. 1 —4. Baltimore, 1984, 1986, 1989.
Bergey’s Manual of Systematic Bacteriology / L.R. Boone, R.W.Castenholz (eds). — Vol. 1, New York: Springer-Verlag, 2001. — P. 485 — 487.
Methods for general and molecular bacteriology / P.Gerhardt et al.(eds.). ASM, Waschington, USA, 1994.
The Prokaryotes. A handbook on the biology of bacteria: ecophysiology, Isolation, Identification, Applications. 2nd ed./ Eds. A.Balows et al. Berlin, New York: Springer-Verlag, 1992.-V. 1-4.
Seeley H.W., VanDemark P.J., Lee J.J. Microbes in action. A laboratory manual of microbiology. New York: W.Y.Freeman and Company, 1991.
Рекомендуемая литература
К главе 5
Уикли Б. Электронная микроскопия для начинающих. — М.: Мир, 1975.
Волков О. В., Шахламов В.А., Миронов А. А. Атлас сканирующей электронной микроскопии клеток, тканей, органов, — М.: Медицина, 1987. — С. 5 — 33.
К главе 7
Bunthof С., Bloemen К., Breeuwer Р. et. al. Flow cytometric assessment of viability of lactic bacteria //Appl. Environm. Microbiol, 2001. — V. 67(5). — P. 2326—2335.
Davey H.M., Kell D.B. Flow cytometry and cell sorting of heterogenous microbial populations: the importance of single cell analyses // Microbiol. Rev. — 1996. — V. 60. 641 — 696.
Lloyd D. (ed.). Flow cytometry in microbiology. Berlin: Springer-Verlag, 1993.
К главе 8
Егоров H. С. Основы учения об антибиотиках. — М.: Изд-во МГУ, 2003.
К главе 9
Goodfellow М., O'Donnell A. G. Handbook of new bacterial systematics. — Acad. Press, 1993.
К главе 10
DeLong E.F., Taylor L.T., Marsh T., Preston C.M. Visualization and Enumeration of Marine planktonic archaea and bacteria by using polyribonucleotide probes and Fluorescent in situ hybridization. Apl.& Environm, 1999.
DNA Microarrays. A Practical Approach. Edd. by M.Schena. Oxford Univ. Press, 1999.
Muyzer G., Brmkhoff N., Nubel U., Swntegoeds C., Schafter H., Wawer C. Denaturing gradient gel electrohhoresis (DGGE) in microbial ecology. In.: Molecular Microbial Ecology Manual. Kluver Academic Publishers. The Netherlands, 1998.
Van Diepeningen A.D., De Vos O., Zelenev V. V., Semenov A. M. and Van Bruggen A. H. C. Patterns of microbial community compositions along wheat roots. Appl. &Environm. Microbiol, 2004.
К главе 11
Милько E.C., Егоров Н. С. Гетерогенность популяции бактерий и процесс диссоциации. - М.: Изд-во МГУ, 1991. - С. 82-90.
К главе 13
Кочетов Г. А. Практическое руководство по энзимологии / Под ред. С. Е. Северина. — М.: Высшая школа, 1971.
Fawcett J. К., Scott J. Е. A rapid and precise method for the determination of urea // J. Clin. Path. - 1960. - V. 13. - P. 156-159.
Miller G. L. Use of dinitrosalicylic acid reagent for determination of reducing sugar // Anal. Chem. 1959. — V. 31. - P. 427.
К главе 14
Грачева И.М., Кривова А.Ю. Технология ферментных препаратов. — М.: «Элевар», 2000.-С. 227-255.
Звягинцев Д.Г., Зенова ГМ. Экология актиномицетов. — М.: Геде, 2001.
Прист Ф. Внеклеточные ферменты микроорганизмов. — М.: «Мир», 1987.
Cuppels D., Kelman A. Evaluation and Selective Media for isolation of Soft-Rot Bacteria from Soil and Plant Tissue // Phytopatholgy. — 1974. — V. 64. — P. 468 — 475.
Stackebrandt E., Rainey F., Ward-Rainey N. Proposal for a new hierarchic classification system, Actinobacteria classis nov. // Int. J. Syst. Bacteriol, 1997. — V. 47. — № 2. — P. 479—491.
К главе 16
Das A., LJungdahl L.G. Acetogenesis and acetogenic bacteria// Encyclopedia of Microbiology. 2 ed. — V. 1. San Diego: Acad. Press, 2000. — P. 18 — 27.
Sowers K.R. Methanogenesis. // Encyclopedia of Microbiology. 2 ed. — V. 3. San Diego: Acad. Press, 2000. — P. 204—225.
К главе 17
Владимирова М.Г, Семененко В. Е. Интенсивная культура одноклеточных водорослей. — М.: Изд-во АН СССР, 1962.
Зенова Г.М., Штина Э.А. Почвенные водоросли. — М.: Изд-во МГУ, 1990.
Кондратьева Е.Н. Фотосинтезирующие бактерии. — М.: Изд-во АН СССР, 1963. — С. 25-37.
Культивирование коллекционных штаммов водорослей / Под ред. Б. Б. Громова. — Л.: Изд-во ЛГУ, 1983.
Методы физиолого-биохимического исследования водорослей в гидробиологической практике. — Киев: Наукова думка, 1975.
Успенская В. И. Экология и физиология питания пресноводных водорослей. — М.: Изд-во МГУ, 1966.
К главе 18
Кураков А. В. Методы выделения и характеристики структуры комплексов микроскопических грибов наземных экосистем. — М.: МАКС Пресс, 2001.
Методы экспериментальной микологии / Под ред. В. И. Билай. — Киев. Наукова Думка. 1982.
Мирчинк Т.Г. Почвенная микология. — М.: Изд-во МГУ, 1988.
Мэгарран Э. Экологическое разнообразие и его измерение. — М.: Мир, 1992.
Сэги Й. Методы почвенной микробиологии. — М.: Колос, 1983.
Gams W., E.S. Hoekstra and A.Aptroot (eds). CBS course of mycology. Fourth edition. Centraalbureau voor Schimmelcultures. AG Baarn. Ponsen & Looyen BV. Wageningen. The Netherlands, 1998.
Parkinson D. Filamentous fungi.. Methods of Soil Analysis. Part 2. Chemical and Microbiological properties-Agronomy Monograth no.9 (2nd Edition, Page A.L. et al.), ASA-SSSA, Madison, WI, USA, 1982.
К главе 19
Алимов А. Ф. Протесты: Руководство по зоологии. — СПб.: Наука, 2000. — Т. 1.
Хаусман К. Протозоология. — М.: Мир, 1988.
К главе 21
Варфоломеев С.Д., Калюжный С. В. Биотехнология. Кинетические основы микробиологических процессов. — М.: Высшая школа, 1990.
Перт С.Дж. Основы культивирования микроорганизмов и клеток. — М.: Мир, 1978.
К главе 22
Берри Д. Биология дрожжей. — М.: Мир, 1985.
Канопкайте С. Кобаламины. — Вильнюс: Мокслас, 1978.
Рыжкова (Иордан) Е. П. Множественные функции корриноидов в биологии прокариотических организмов // Прикл. биохим. и микробиол, 2003. — Т. 39. — № 2. — С. 133—159.
Cummins C.S., Johnson J.L. The Genus «Propionibacterium» // The Prokaryotes. Second edition. Balows et al. (eds.). — NY: Springer-Verlag, 1992. — V. 2. — P. 834—849.
К главе 25
Слободкин А.И., Заварзина Д.Г., Соколова Т.Г., Бонч-Осмоловская Е.А. Диссимиляционное восстановление неорганических акцепторов электронов термофильными анаэробными прокариотами // Микробиология, 1999. — Т. 65. — № 5. — С. 600—622.
К главе 26
Kooijmon S.A.L.M., Muller Е.В, Stouthamer A.U. Microbial growth dynamics on the basis of individual budgets. Ant. van Leeuw., Int. J. Gen. Microbiol, 1992. — V. 60. — № 3 — 4. — P. 159-174.
Stouthamer A. U. Metabolic pathways in Paracoccus denitrificans and closely related bacteria in relation to the phylogeny of procaryotes. Ant. van Leeuw., Int. J. Gen. Mol. Microbiol, 1992. — V.61.—№ l.-P. 1-34.
К главе 27
Романова А.К. Биохимические методы изучения автотрофии у микроорганизмов. — М.: Наука, 1980. - С. 22-33.
К главе 28
Borisov N., Netrusov A. One-carbon compounds transport switches in the obligate methylotroph//Arch. Microbiol, 1989. — V. 152. — P. 201 — 205.
К главе 29
Баев А. А. (ред.). Биотехнология. — M.: Наука, 1984.
Готтшалк Г. Метаболизм бактерий. — М.: Мир, 1982.
Прист Ф. Внеклеточные ферменты микроорганизмов. — М.: Мир, 1987.
Hart I. et al. Mechanism of catabolite repression of tryptophanase synthesis in Escherichia coliЦ Microbiology, 1994. — V. 140. — P. 2125 — 2134.
Konan К. V., Yanofsky C. Regulation of the Escherichia coli tna operon: nascent leader peptide control at the tnaCstop codon // J.Bacteriol, 1997. — V. 179. — P. 1774— 1779.
К главе 30
Егоров H. С. Основы учения об антибиотиках. — М.: Изд-во МГУ, 2003.
Кузнецова В. С., Орлова Т.И., Силаев А. Б. Актиномицины из Actinomyces sp. № 2, продуцента актиномициновый комплекс аурантин // Антибиотики и их продуценты / Под ред. А. Б. Силаева. — М.: Наука, 1975. — С. 40—53.
Нефелова М.В. Биологическая модификация структуры актиномицинов //Антибиотики и их продуценты / Под ред. А. Б. Силаева. — М.: Наука. 1975. — С. 12 — 23.
Филиппова М. С., Нефелова М.В. Морфологические и физиолого-биохимические свойства актиномицета, продуцирующего актиномициновый комплекс аурантин // Антибиотики и их продуценты / Под ред. А. Б. Силаева. — М.: Наука, 1975. — С. 24—38.
Морозкина Т. С., Мойсенок А. Г. Витамины. — Минск: ООО «Асар», 2002.
К главе 32
Карпунина Л. В., Мельникова У. 10., Суслова Ю. В. и др. Бактерицидные свойства лектинов азотфиксирующих бацилл // Микробиология, 2003. — Т. 72. — № 3. — С. 343 — 347.
Юдина Т.Г., Егоров Н.С. Антимикробная активность белковых включений различных бактерий // Доклады РАН, 1996. — Т. 349. — № 2. — С. 283 — 286.
К главе 33
Калюжный С. В., Данилович Д. А., Ножевникова А. Н. Анаэробная биологическая очистка сточных вод // Итоги науки и техники. Сер. «Биотехнология». — М., 1991. — Т. 29.
Савельева О. В., Емашова Н.А., Котова И. Б. и др. Анаэробная биоконверсия аминоароматических соединений // Усп. совр. биол., 2003. — Т. 123. — № 4 — С. 336— 349.
HeiderJ., Fuchs G. Microbial anaerobic aromatic methabolism // Anaerobe, 1997. — V. 3. — P. 1-22.
Razo-Flores E., Luijten M., Donlon B. et al. Biodegradation of selected azo dyes under methanogenic conditions //Wat. Sci. Technol, 1997. — V.3, № 6/7. — P. 65 — 72.
К главе 34
Милько E. С. Нестабильность синтеза практически ценных веществ и процесс диссоциации И Прикл. биохим. и микробиол., 1990. — Т. 26 — Вып. 6 — С. 732 — 742.
Милько Е. С., Егоров Н. С. Гетерогенность популяции бактерий и процесс диссоциации. — М.: Изд-во МГУ, 1991.
Милько Е. С., Мартынкина Л.П. Морфологические и физиолого-биохимические особенности диссоциантов Р. aeruginosa // Микробиология, 1996. — Т. 65, № 3. — С. 352 — 356.
К главе 37
Кожевин П.А. Микробные популяции в природе. — М.: Изд-во МГУ, 1989.
К главе 38
Умаров М. М. Ассоциативная азотфиксация. — М.: Изд-во МГУ, 1986.
Gadkari D., Morsdorf G., Meyer О. Chemolithoautotrophic assimilation of dinitrogen by Streptomycces thermoautotrophicus UBT1: identification of an unusual N2-fixing system // J.Bacterio!, 1992. Nov. — P. 6840 — 6843.
К главе 39
Кулаева O.H. Цитокинины, их структура и функция. — М.: Наука, 1973.
Рубин Б. А. Большой практикум по физиологии растений. — М.: Высшая школа. 1978.
Frankland В., Wareing F.A. Effect of gibberellic acid on hypocotyls growth of lettuce seedlings // Nature, 1960. — V. 185. — P. 255.
Thakur M.S., Eyas KM. Production of plant growth regulators by some Fusarium species // Folia Microbiol, 1983. — V. 28. — P. 124—129.
К главе 40
Сэги Й. Методы почвенной микробиологии. — М.: Колос, 1983. — С. 159—162.
Мосичев М.С., Складнее А.А., Котов В.Б. Общая технология микробиологических производств. — М.: Легкая и пищевая промышленность, 1982. — С.172—187.
К главе 43
Квасников Е.И., Щелокова И.Ф. Дрожжи. Биология. Пути использования. — Киев.: Наукова думка, 1991. — С. 171—232.
Борисов Л.Б. и др. Руководство к лабораторным занятиям по медицинской микробиологии, вирусологии и иммунологии. — М.: Медицина, 1993. — С. 69 — 74.
К главе 44
Квасников Е.И., Нестеренко О. А. Молочнокислые бактерии и пути их использования. — М.: Наука, 1975.
Gasson M.J., de Vos W. M. Genetics and biotechnology of lactic acid bacteria. — Ldn.: Acad. Pres., 1994.
McAuliffe O., Ross R.P., Hill C. Lantibiotics: structure, biosynthesis and mode of action // FEMS Microbiol. Revs, 2001. - V. 25. — P. 285—308.
Yother J., Trieu-Cuot P., Klaenhammer T.R., de Vos И7. M. Genetics of streptococci, lactococci and enterococci: review of the 6th International Conference // J. Bacteriol, 2002. — V. 184.-P. 6085-6092.
К главе 46
Байчурина А.З. и др. В кн.: Медицинская микробиология / Гл. ред. В. И. Покровский, О.К.Поздеев. — М.: ГЭОТАР-Медицина, 1998.
Вольпе И.М., Кучеренко В.Д. Практическое руководство по санитарной микробиологии. — М.: Изд-во МГУ, 1970.
Коротяев А. И., Бабичев С. А. Медицинская микробиология, иммунология и вирусология. — СПб.: Специальная Литература, 1998.
Лабинская А. С. Микробиология с техникой микробиологических исследований. — М.: Медицина, 1978.
Поздеев О. К. Медицинская микробиология. — М.: ГЭОТАР-Медицина, 2001.
К главе 47
Борисов Л. Б. и др. Медицинская микробиология, вирусология, иммунология. — М.: Медицина, 1994.
К приложению 7
Михеева А. В. и др. Словарь-минимум для чтения научной литературы на английском языке. — М.: Наука, 1979.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
А
Автоклав, автоклавирование 46
Агар-агар 35
Агглютинация 514
Адаптация 388
Азокрасители 377
Азотобактерин 456
Активный ил 379
Актиномицеты 200, 354
Актиномицины 354
Алкогольоксидаза 193
Аллергены 517
Аллергия 517
Амилаза 188
Амилолитическая активность (выявление) 341, 530, 533
Анаэробное восстановление 327, 378
Анаэробное дыхание 327
----карбонатное 219
----серное 327
----сульфатное 329
Анаэробные микробные сообщества 376
— микроорганизмы 41, 216, 219
Анаэробный бокс 218, 221
— донный осадок 217, 219
Анаэростат 41, 218
Антибиотики 129, 337
Антибиотическая активность 129, 341
Антиген 513
Антитело 513
Антисыворотка 516
Ауксотрофы 32, 408
Ацето пластический метаногенез 217
Аэробные микроорганизмы 38
Б
БГКП 483
Бактериальные удобрения 453
Бактериоцины 475, 477
Бактероиды 435, 454
Белок (методы определения) 167-169, 340
Бинарная номенклатура 133
Биоавтография 359
Биогаз 217
Биодеградация 376
Биоремедиация 376
Биотесты 447
Болезни вина 464
— хлеба 460
Брожение 214
— ацетоно-бутиловое 216
— гомоацетатное 219
— маслянокислое 216
— молочнокислое 214, 467
— пропионовокислое 215, 307
В
Вакцины 516
Вид микроорганизмов 133
Виноделие 463
Витамин В|2 307, 361
Витамины 360
Внеклеточные ферменты 126, 337
Время генерации 285
Г
Генотип микроорганизмов 138, 393
Гены 392
Гетерогенность микробной популяции 159, 387
Гидролизат молока 473
Глутаматоксидаза 193
Глюкозооксидаза 193
Гомоацетогены 219
Графоаналитический метод расчета параметров 291
Группы крови 514
Д
Действительное (узаконенное) опубликование таксона микроорганизмов 135
Дендрограмма 138
Диазотрофы 434
Диссоциация 387, 470
Дрожжевой автолизат 33, 473
— экстракт 33, 474
Дрожжи 215, 299, 459, 464
Ж
Желатин 35, 128, 440
И
Идентификация микроорганизмов 132, 134, 473
— прокариот 135
Иммунная память 512
— система 512
Иммунный ответ 515
Иммуноглобулины 515
Инвертаза 188
Индол 122
Инозит (В8) 361
Интенсивность аэрации 293, 294
Инфекционные заболевания 508
Инфекция 508
Инфицирование вина 464
— пива 465
К
Канцерогены 418, 420
Капельный метод Линднера 99
Капилляры Перфильева 100, 431
Каррагинан 35
«Картофельная болезнь» 460
Каталаза 195, 533
Классификация микроорганизмов 132
Клубеньковые бактерии 435, 437, 454
Коли-титр и коли-индекс воды 487
— молока 466
— почвы 491
Коллекции микроорганизмов 133
Колонка Виноградского 228, 433
Кометаболизм 379
Компетентность 400
Консорциум 377
Контрастирование 86, 87
Концентрация биомассы 288
Конъюгация 400
Коэффициент скорости деления 285
«Красный хлеб» 461
Кремнекислый гель (силикагель) 35
Ксенобиотики 376
Кукурузный экстракт 33
Культуральные свойства микроорганизмов 115, 136
Л
Лактобациллы 463, 469, 470
Лактококки 463, 469
Ламинарный бокс 24
Леггемоглобин 454
Лейкоциты 512
Лектины 435
Лизоцим 371
Лимфоциты 513, 516
Лиофилизация 162, 476, 477
Липаза 187
Липолитическая активность (выявление) 129, 473
Локус 392
М
Мальтаза 190
«Меловая болезнь» 461
Метаногены 216
Метанококки 218
Метод агаровых блочков 130, 534
Метод микроразведений 373
Метод перпендикулярных штрихов 130
Метод предельных разведений 108
Микробиологический анализ молока 465 Микробиологическое растворение соединений фосфора 450
Микробное число 466, 482
--- воды 486
---воздуха 495
--- почвы 491
Микроманипулятор 99
Микроселектор Перфильева 100
Микроскопия 56, 520
— конфокальная лазерная сканирующая 56, 67
— люминесцентная 56, 66
— просвечивающая (трансмиссионная) электронная 83
— с фазово-контрастным устройством 63
— светлопольная 56
— сканирующая туннельная 92
---электронная 88
— темнопольная 61
Многоканальный планшетный спектрофотометр 374
Молочнокислое скисание вина 464
Молочнокислые бактерии 467
Молярный экономический коэффициент 287
Морфологическая характеристика микроорганизмов 54, 136, 469
Мочевина (использование микроорганизмами) 122
Мутационная изменчивость 393
Н
Накопительная культура микроорганизмов 93
Наследственная и ненаследственная изменчивость 393
«Некультивируемый» микроорганизм 140
Непрерывное культивирование микроорганизмов 44, 284, 288
Низин 475
Никотиновая кислота (В5) 361
Нитрагин 453, 454
Нитраты 437
— реакция с дифениламином 125, 442
Нитриты
— реакция с реактивом Грисса 126, 442, 526
— крахмал-йодная реакция 125, 442, 526
Номенклатура микроорганизмов 132
Нумерическая таксономия 138
О
Общая обсемененность пищевых продуктов 499
Оксидаза 194
Описания штаммов бактерий 133
Определение способности микроорганизмов к аэробному дыханию 124 --------брожению 126 --------денитрификации 125, 443
Определение способности микроорганизмов использовать азот минеральных солей 122
--------молекулярный азот 123, 434, 439
--------органические кислоты 120
--------органические азотсодержащие вещества 121
-------- углеводороды 120
--------углеводы и спирты 118
Определение чувствительности микроорганизмов к антибиотикам 131, 341, 533
Органические соединения фосфора 451
П
Пандемия 508
Параметры микробного роста 285
Пассивный иммунитет 515, 516
Педоскоп 432
Пектиновая кислота 197, 198
Пектинэстераза 198, 199
Пектолитические ферменты 197, 198
Пелоскоп 432
Перибактероидная мембрана 454
Периодическое культивирование микро-
организмов 44, 284
Пероксидаза 194
Перфрингенс-титр почвы 491
Пивоварение 465
Плазмиды 392
Плодородие почвы 453
Подъемная сила дрожжей 460
Полифункциональные белки 371
----cry-белки Bacillus thuringiensis 372
Постулаты Коха 505
Почвенный нитрагин 455
Приготовление смывов 503, 509
Продуктивность периодического процесса 290
Производство силоса 462
Пропионовокислые бактерии 307, 363
Простейшие (методы изучения) 255
Протеазы 190, 338
Протеолиз 532
— желатины 128, 532
— казеина 128, 532
Протеолитическая активность 128, 340
Протопектин 197
Прототрофы 32
Прямое выделение патогенов 510
«Пьяный хлеб» 462
Р
Реактив Рейнольдса 88
Реактив Фоссета 383
Реакция агглютинации 514
Редуктаза (проба на редуктазу) 465
Резус-фактор 514
Репликон 402
Ризоторфин 453
Рубцовая жидкость 217, 219
С
Санитарно-показательные микроорганизмы 466, 482
Сбалансированный рост микроорганизмов 285
Селективные преимущества 388
Сероводород (обнаружение) 121
Серологические реакции 517
Сибирская язва 505
Симбиосомы 454
Синтрофия 377
Систематика микроорганизмов 132
Скорость разбавления 44, 288, 290
Соокисление 379
Спорынья 464
Среды
— дифференциально-диагностические (индикаторные) 34
— жидкие 34
— натуральные (естественные) 34
— осветленные 36
— селективные 95, 474
— синтетические 34
— сыпучие 34
— твердые 34
Стабилизация пива 465
Стандарт мутности 113, 390
Стекла обрастания 430
Степень аэрации 38, 293
Стрептомицеты 338
Сульфитация вина 464
Сульфитное число 39
Сульфоазокрасители 377
Сухой азотобактерин 457
Сухой нитрагин 455
Счетная камера Горяева—Тома 101
Т
Таблицы Мак-Креди 108
Таксон микроорганизмов 132
Таксономическая категория 132
Таксономия микроорганизмов 132
Термическая стерилизация 45
Тест-культура 366
Тест-организмы 130, 360, 363
Тиндализация (дробная стерилизация) 49
Типовой штамм микроорганизмов 133
Торфяной азотобактерин 453
— нитрагин (ризоторфин) 455
Трансдукция 400
Трансформация 376, 400
Триптофаназа 342
Турбидостат 44, 285
У
Удельная скорость роста 287
Уксуснокислое скисание вина 464
Уравнение материального баланса 289
Уреаза 190
Уробактерии 526
Условно патогенные микроорганизмы 511
Ф
Фазы развития культуры 284
Фенотип 393
Ферментер 285, 294
Физиолого-биохимические свойства мик-
роорганизмов 118, 137
Фиксация азота 434, 454
Фосфобактерин 454
X
Хемостат 44, 285
Холодная стерилизация 45
Хранение микроорганизмов 159, 476
Хромосомы 392
Ц
Цвель вина 464
Целлюлаза 192
Ч
Чистая культура микроорганизмов 93
Ш
Штамм микроорганизмов 133, 393
Э
Экономический коэффициент 288, 302
Элективные условия 93, 473
Электропорация 400
Эпидемия 508
Эффективное опубликование микроорганизмов 135